ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к липосомальной композиции длительного действия с контролируемым высвобождением, которая содержит действующий компонент, например лекарственное средство.
УРОВЕНЬ ТЕХНИКИ
В последние годы, сопровождающиеся прогрессирующим старением общества, увеличивается число пациентов, нуждающихся в персонале, осуществляющем уход, например, пациенты, страдающие слабоумием, заболеваниями мозга, болезнью Паркинсона и т.п. Такие пациенты могут забыть принять лекарственное средство или могут испытывать сложности с принятием лекарственного средства из-за затруднения глотания. Таким образом, этим пациентам сложно самим выполнять действия, связанные с приемом лекарств. Поэтому существует потребность в способе введения, отличающемся от перорального введения. Кроме того, например, в случаях пациентов с психическими заболеваниями их состояние осложняется появлением симптомов непосредственно после окончания действия лекарственного средства. В таких случаях, в силу вышесказанного, введение лекарственного препарата следует повторять несколько раз в день, прежде чем закончится действие лекарственного препарата. Частое введение лекарственного препарата является тяжелой нагрузкой для пациента. Следовательно, препараты с продолжительным контролируемым высвобождением востребованы при заболеваниях всех систем.
Большинство препаратов с контролируемым высвобождением, которые рассматривались до сих пор в качестве препаратов для подкожного введения или внутримышечного введения, представляют собой микросферы на основе применения сополимера полимолочной кислоты и полигликолевой кислоты (PLGA). Например, существует препарат микрокапсул Лейплин (зарегистрированная торговая марка Leuplin), в котором лейпрорелин, в качестве канцеростатического средства, инкапсулирован в сшитую матрицу PLGA. Известно, что микросферы с применением PLGA высвобождают лекарственное средство непосредственно после введения (начальный “взрыв”), и его концентрация в крови немедленно увеличивается выше эффективной концентрации. Это приводит к возможности появления побочных эффектов. Кроме того, в случае применения PLGA, сложно инкапсулировать лекарственное средство в высокой концентрации и с высокой эффективностью. С клинической точки зрения, с другой стороны, существует ограничение в отношении дозы, в которой можно ввести лекарственное средство. В силу вышесказанного, кроме того, существуют проблемы, которые необходимо решить в отношении увеличения количества инкапсулированного лекарственного средства. Кроме того, в случае применения PLGA в процессе получения препарата применяют органический растворитель при условии обязательного удаления органического растворителя из препарата. Такое удаление растворителя часто сложно осуществить при производстве препарата в промышленных масштабах. Кроме того, согласно сообщениям, применение PLGA связано с серьезной проблемой, поскольку местная активация кислого вспомогательного вещества при гидролизе вызывает воспаление в месте введения.
Созданы другие способы, отличающиеся от указанных выше способов, в которых бупивакаин инкапсулируют в липосомы из многослойных мембран с помощью метода “бесконтактной” загрузки. Однако в соответствующих документах не прослеживалась взаимосвязь между диаметром частицы и свойствами контролируемого высвобождения и не получены данные в отношении оптимального диаметра частиц в препаратах с контролируемым высвобождением. Кроме того, показатели времени контролируемого высвобождения, раскрытые в документах, нельзя считать удовлетворительными с клинической точки зрения (Anesthesiology 101 (2004) 133-137, International Society for Anaesthetic Pharmacology 110 (2004) 1018-1023). Кроме того, мультивезикулярная липосома (MVL), описанная в JP-T-2001-522870, была разработана в качестве лекарственной поддержки на липидной основе с контролируемым высвобождением для местной или системной доставки лекарств. Однако данный способ также не отвечает требованиям и характеризуется проблемами, требующими решения, связанными с количеством инкапсулированного лекарственного средства и временем контролируемого высвобождения.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Техническая проблема
Целью настоящего изобретения является предоставление липосомальной композиции, позволяющей получить липосомальный препарат, характеризующийся свойствами продолжительного контролируемого высвобождения, с помощью которого лекарственное средство можно вводить в клинически необходимом количестве и в небольшом объеме и, кроме того, позволяющей устойчиво поддерживать клинически эффективную концентрацию лекарственного средства в крови в течение длительного периода времени.
Техническое решение
Авторы настоящего изобретения обнаружили, что, когда лекарственное средство инкапсулируют в липосомальной композиции, которая содержит в себе однородную внутреннюю водную фазу внутри многослойной мембраны по меньшей мере с 12-слойной структурой и в которой образуется ионный градиент между внутренней водной фазой и внешней водной фазой липосом (в дальнейшем также обозначаемых как пустые липосомы), можно получить липосомальный препарат, обладающий свойствами продолжительного контролируемого высвобождения. Настоящее изобретение осуществлено на основе полученных данных.
Указанная выше цель достигается с помощью настоящего изобретения, которое распространяется на следующее.
(1) Липосомальная композиция, полученная путем смешивания водорастворимого органического раствора, в котором содержатся фосфолипид и холестерин в общей концентрации от 100 до 200% масс./об. в водорастворимом органическом растворителе, с раствором первой водной фазы в объемном отношении к водорастворимому органическому раствору от 3/1 до 12/1, с получением эмульсии, в которой общая концентрация фосфолипида и холестерина в полученной смешанной фазе составляет от 15 до 50% масс./об., с последующей заменой внешнего раствора эмульсии раствором, представляющим собой вторую водную фазу, при этом образуется ионный градиент между водной фазой внутренней области липосомальной мембраны, где водная фаза внутренней области содержит раствор первой водной фазы, и водной фазой внешней области липосомальной мембраны, где водная фаза внешней области содержит раствор второй водной фазы.
(2) Липосомальная композиция, как описано в приведенном выше параграфе (1), где ионный градиент представляет собой протонный градиент pH и липосомальная композиция характеризуется градиентом pH, таким что значение pH водной фазы внутренней области липосомы (в дальнейшем также обозначаемой как внутренняя водная фаза) ниже, чем значение pH водной фазы внешней области липосомы (в дальнейшем также обозначаемой как внешняя водная фаза).
(3) Липосомальная композиция, как описано в приведенном выше параграфе (1) или (2), где липосома содержит водную фазу внутри многослойной мембраны, имеющей водную фазу внутренней области внутри многослойной мембраны, характеризующейся средним диаметром частиц в терминах внешнего диаметра, составляющим 2,5 мкм или больше, и имеющей по меньшей мере 12-слойную структуру.
(4) Липосомальный препарат, содержащий лекарственное средство, введенное в водную фазу во внутренней области в липосомальной композиции, как описано в любом из приведенных выше параграфов (1)-(3), путем применения ионного градиента, где лекарственное средство содержится в липосоме в молярном отношении, равном 0,08 (моль) или больше, вычисленном по общему содержанию липидов (моль).
(5) Липосомальный препарат, как описано в приведенном выше параграфе (4), где липосомальный препарат обладает либо свойствами системного контролируемого высвобождения, так что эффективная концентрация в крови лекарственного средства, инкапсулированного во внутренней области, поддерживается в течение четырех или более дней, либо свойствами местного контролируемого высвобождения, так что эффективная местная концентрация лекарственного средства, инкапсулированного во внутренней области, поддерживается в течение четырех или более дней.
(6) Липосомальный препарат, как описано в приведенном выше параграфе (4) или (5), в котором лекарственное средство представляет собой амфипатическое слабоосновное соединение.
(7) Набор препарата с продолжительным контролируемым высвобождением, включающий липосомальный препарат по любому из параграфов (4)-(6) и инъекционную иглу, характеризующуюся по меньшей мере размером одного калибра, выбранного из калибров от 27 до 33.
Липосомальная композиция настоящего изобретения может содержать другую липосому, отличающуюся от липосомы настоящего изобретения в липосомальной композиции. Примеры других липосом, отличающихся от липосомы настоящего изобретения, включают одномембранные небольшие липосомы и мультивезикулярные липосомы. Липосома настоящего изобретения предпочтительно составляет не меньше чем 50 масс.%, более предпочтительно не меньше чем 60 масс.% и также предпочтительно не меньше чем 80 масс.% относительно предшествующих липосом.
Полезный эффект изобретения
Липосомальная композиция настоящего изобретения позволяет инкапсулировать лекарственное средство с высокой эффективностью, когда липиды присутствуют в установленной общей концентрации липидов, в результате чего можно получить липосомальный препарат с высокой концентрацией лекарственного средства. Кроме того, липосомальная композиция настоящего изобретения, в которой водная фаза внутренней области представлена внутри многослойной мембраны, позволяет гарантировать, что при инкапсулировании лекарственного средства в липосомальной композиции для получения липосомального препарата лекарственное средство может устойчиво высвобождаться непрерывным образом в течение длительного периода времени, вместе с тем сохраняя клинически эффективную концентрацию лекарственного средства в крови и без начального “взрывного” высвобождения. Причем можно избежать вероятности возникновения побочных эффектов и сократить количество введений лекарственного средства. В силу вышесказанного липосомальный препарат является очень полезным. Кроме того, согласно липосомальному препарату настоящего изобретения необходимое количество лекарственного средства можно вводить в небольшом объеме. В целом, препараты для подкожного и внутримышечного введения следует вводить с помощью тонкой иглы калибра 19-21. С другой стороны, липосомальный препарат настоящего изобретения можно вводить даже с помощью игл небольшого диаметра с большими номерами калибров. Таким образом, липосомальный препарат настоящего изобретения является полезным липосомальным препаратом, с помощью которого можно значительно улучшить QOL пациентов.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Фиг.1 представляет собой фотографии, полученные при электронно-микроскопическом исследовании реплик с замороженного среза липосомального препарата донепезила, полученного в примере получения 2, где фиг.1a представляет собой изображение трещины, сфотографированной на внешней поверхности липосомального препарата, и фиг.1b представляет собой изображение трещины, сфотографированной по существу в центре липосомального препарата.
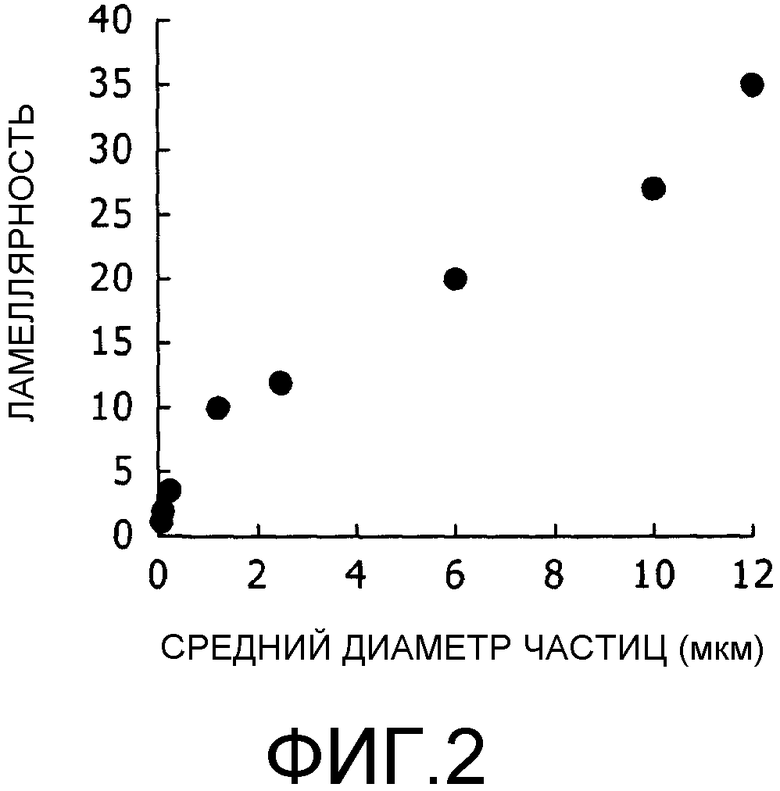
Фиг.2 представляет собой график, на котором показана взаимосвязь между средним диаметром частицы и числом слоев мембран (ламеллярностью).
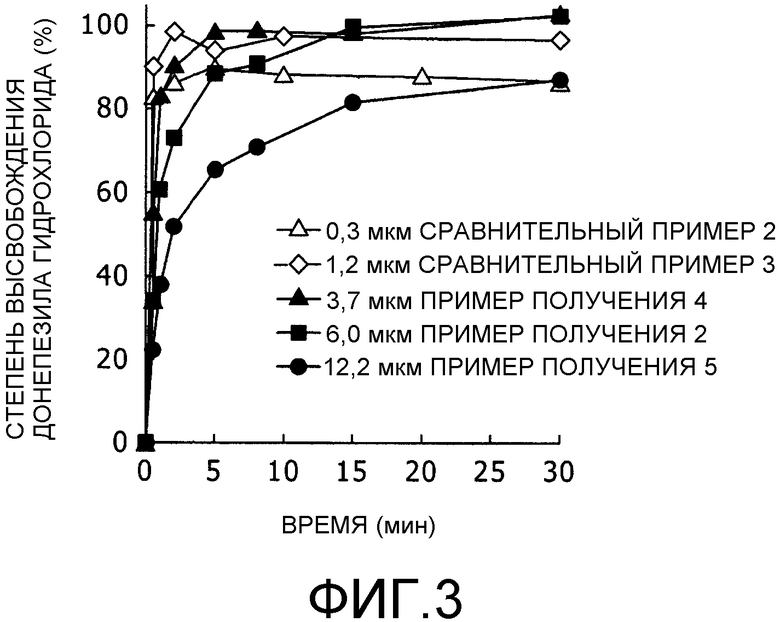
Фиг.3 представляет собой график, на котором показаны характеристики высвобождения (результаты измерения, полученные с помощью метода проверки высвобождения in vitro с применением сульфата аммония) из липосомальных препаратов, характеризующихся различными диаметрами частиц.
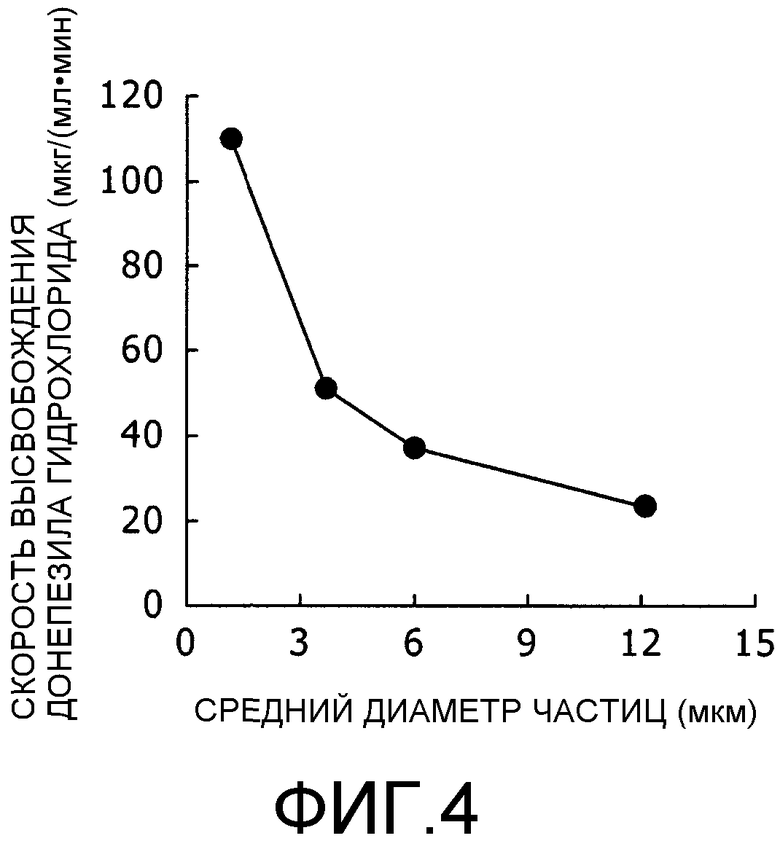
Фиг.4 представляет собой график, на котором показана взаимосвязь между диаметром частицы и скоростью высвобождения лекарственного средства.
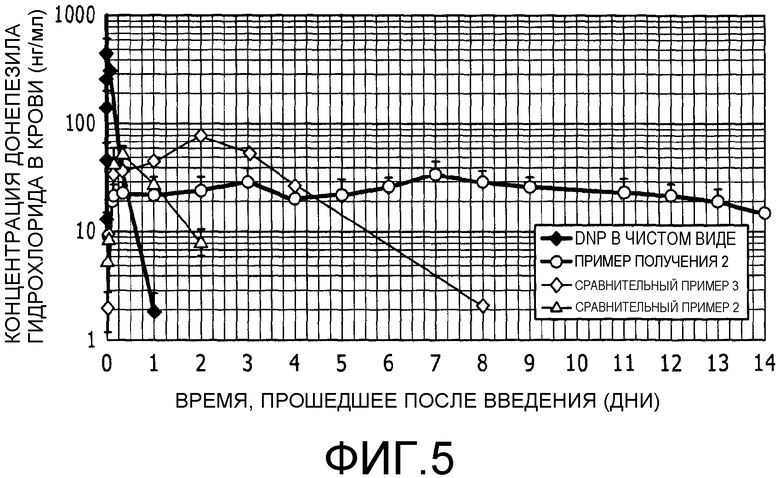
Фиг.5 представляет собой график, на котором показана концентрация донепезила гидрохлорида в крови после подкожного введения только одного донепезила и липосомальных препаратов.
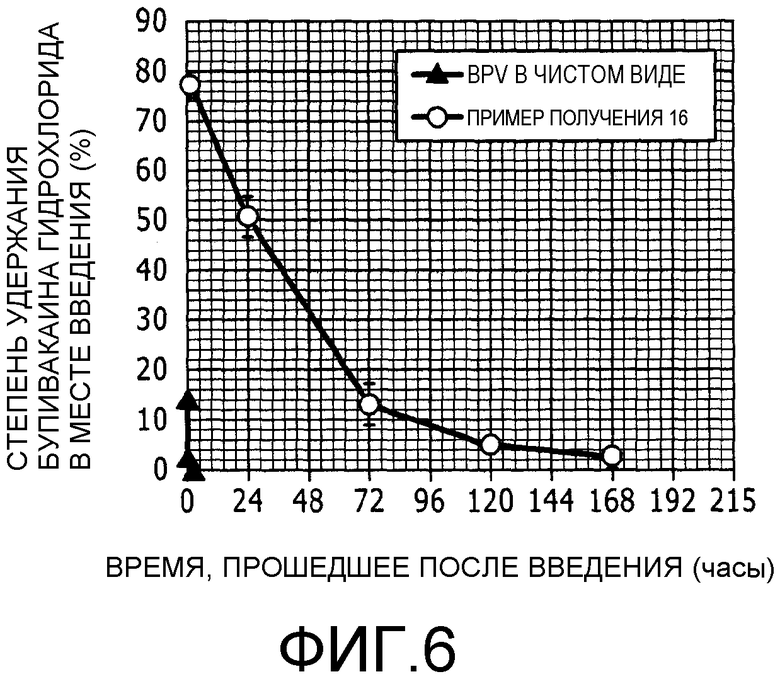
Фиг.6 представляет собой график, на котором показан процент удержания (масс.%) бупивакаина гидрохлорида в месте введения после внутримышечного введения бупивакаина гидрохлорида отдельно и в составе липосомального препарата.
СПОСОБ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Липосома в настоящем изобретении имеет некоторое количество липидных бислоев, имеющих установленный диаметр частиц (внешний диаметр липосомальных частиц), и содержит одну внутреннюю водную фазу в нем. Средний диаметр частиц липосом в настоящем изобретении составляет предпочтительно от 2,5 до 12 мкм. Средний диаметр частиц составляет более предпочтительно 3,7-10 мкм, также предпочтительно 5-10 мкм и особенно предпочтительно 6-10 мкм. Липосомы в настоящем изобретении предпочтительно имеют средний диаметр частиц, равный 2,5 мкм или больше. Диаметр частиц липосом в настоящем изобретении можно измерить как средний диаметр частиц (среднее значение внешнего диаметра) с помощью устройства для измерения распределения частиц по размерам (например, анализатор для измерения распределения частиц по размерам на основе светорассеяния, например, Beckman Coulter LS230).
Липосома в настоящем изобретении имеет стенку, состоящую из липидных бислоев. При условии, что одна слоевая структура (ламеллярность) состоит из одного липидного бислоя, липосома в настоящем изобретении имеет конфигурацию, при которой внешняя оболочка в виде стенки состоит из определенного числа слоевых структур. В частности, липосома в настоящем изобретении имеет слоевую структуру из 12-35 слоев (12-слойная - 35-слойная, или 12-слоистая - 35-слоистая) ((средний диаметр частиц) липосомы, если она имеет слоевую структуру, составляющую 2,5-12 мкм). Предпочтительно, липосома имеет слоевую структуру из 14-27 слоев (14-слойная - 27-слойная) (внешний диаметр (средний диаметр частиц), если она имеет слоевую структуру, составляющую 3,7-10 мкм). Указанные слоевые структуры расположены так, что слоевые структуры, прилегающие друг к другу, соприкасаются поочередно внутренними и наружными сторонами, при этом между ними содержится очень небольшое количество воды. Число слоевых структур можно определить путем проведения исследования сканирующей электронной микроскопии (SEM) и подсчета числа слоевых структур в условиях, когда часть стенки липосомы настоящего изобретения сломана.
Липосома настоящего изобретения содержит внутри себя внутреннюю водную фазу. Внутренняя водная фаза заключена во внутренней области внутренней поверхности слоевой структуры, представленной на самой внутренней стороне липосомы. Толщина одной слоевой структуры (липидного бислоя) составляет приблизительно 10 нм, и толщина стенки липосомы формируется в соответствии с числом представленных липидных бислоев. В случае, когда слоевая структура липидных бислоев имеет 20 слоев, 20 слоев толщиной 10 нм располагаются на обеих сторонах внутренней водной фазы, так что толщина обеих стенок составляет 0,4 мкм. Путем вычитания 0,4 мкм из внешнего диаметра липосомы, состоящей из 20 слоев, можно рассчитать внутренний диаметр внутренней водной фазы. Внутренний диаметр внутренней водной фазы липосомальной композиции и препарата согласно настоящему изобретению составляет предпочтительно от 2,2 мкм (12 слоев со средним диаметром частиц 2,5 мкм) до 11,3 мкм (35 слоев со средним диаметром частиц 12 мкм), например, и более предпочтительно от 3,4 мкм (14 слоев со средним диаметром частиц 3,7 мкм) до 9,4 мкм (27 слоев со средним диаметром частиц 10 мкм). Липосома в настоящем изобретении имеет, например, слоевую структуру из 12 слоев, если средний диаметр частиц составляет 2,5 мкм. Аналогичным образом, липосома имеет, например, слоевую структуру из 14 слоев, если средний диаметр частиц составляет 3,7 мкм. Например, слоистая структура имеет 27 слоев, если средний диаметр частиц составляет 10 мкм, и слоистая структура имеет 35 слоев, если средний диаметр частиц составляет 12 мкм.
Липосома, полученная таким образом, позволяет гарантировать, что могут быть получены полезные свойства контролируемого высвобождения и что однородная липосомальная композиция может быть создана.
Липосома в настоящем изобретении представляет собой липосому, полученную с помощью следующих стадий и образует липосомальную композицию согласно настоящему изобретению при диспергировании в водной фазе внешней области.
Липосома в настоящем изобретении представляет собой липосому, полученную путем смешивания водорастворимого органического раствора, в котором фосфолипид и холестерин содержатся в общей концентрации (в дальнейшем также обозначаемой как общая концентрация липидов) от 100 до 200% масс./об. (100-200 г в 100 мл) в водорастворимом органическом растворителе с раствором первой водной фазы в объемном отношении с водорастворимым органическим раствором от 3/1 до 12/1, с получением эмульсии, в которой общая концентрация фосфолипида и холестерина в получаемой смешанной фазе составляет от 15 до 50% масс./об. (15-50 г в 100 мл), с последующей заменой внешнего раствора эмульсии раствором второй водной фазы.
В описании "водорастворимый органический растворитель" представляет собой любой из спиртов, таких как метанол, этанол, изопропиловый спирт, и бутанол, из которых предпочтительным является этанол.
"Водорастворимый органический раствор (в дальнейшем также обозначаемой как спиртовой раствор)" представляет собой раствор, в котором фосфолипид и холестерин содержатся в спирте в общей концентрации от 100 до 200% масс./об. (100-200 г в 100 мл).
"Смешанная фаза" представляет собой смешанную фазу, которую получают при смешивании водорастворимого органического раствора с раствором первой водной фазы в объемном отношении к водорастворимому органическому раствору от 3/1 до 12/1 и которая характеризуется общей концентрацией липидов от 15 до 50% масс./об. (15-50 г в 100 мл).
"Первая водная фаза", также называемая водной фазой внутренней области или внутренней водной фазой, представляет собой водную фазу во внутренней области, представленную равномерно внутри многослойной мембраны липосомы в настоящем изобретении.
"Вторая водная фаза", также называемая водной фазой внешней области или внешней водной фазой, представляет собой водную фазу, которая представлена с внешней стороны многослойной мембраны липосомы в настоящем изобретении и которая образует ионный градиент вместе с водной фазой внутренней области.
Далее будет описан каждый из компонентов.
Фосфолипид
Фосфолипид, который является одним из главных компонентов липидной мембраны липосомальной композиции согласно настоящему изобретению, представляет собой основную структурную составляющую биомембраны и обычно является амфипатическим веществом, которое содержит в своей молекуле как гидрофобную группу, состоящую из длинноцепочечной алкильной группы, так и гидрофильную группу, состоящую из фосфатной группы. Предпочтительные примеры фосфолипида включают: глицерофосфорные кислоты, такие как фосфатидилхолин (=лецитин), фосфатидилглицерин, фосфатидная кислота, фосфатидилэтаноламин, фосфатидилсерин, фосфатидилинозит и т.д.; сфингофосфолипиды, такие как сфннгомиелин (SM), и т.д.; натуральные или синтетические дифосфатидилфосфолипиды, такие как кардиолипин, и т.д. и их производные; продукты гидрогенизации указанных фосфолипидов, такие как гидрогенизованный фосфатидилхолин соевых бобов (HSPC), гидрогенизованный фосфатидилхолин яичного желтка, дистеарилфосфатидилхолин, дипальмитоилфосфатидилхолин, и димиристоилфосфатидилхолин. Фосфолипиды можно использовать либо по отдельности, либо в комбинации нескольких из них.
Другие вспомогательные вещества, кроме фосфолипида
Мембранный липид в липосомальной композиции согласно настоящему изобретению может содержать другие мембранные компоненты наряду с упомянутым выше главным компонентом. Например, липосомальная композиция может содержать другие липиды, кроме фосфолипидов или производные других липидов, мембранные стабилизаторы, антиоксиданты и т.п. в соответствии с требованиями. Другие липиды, кроме фосфолипидов, представляют собой липиды, имеющие гидрофобную группу, такую как длинноцепочечная алкильная группа в молекуле, но не содержащие фосфатную группу в молекуле, и не специально ограничиваются. Примеры других липидов включают глицерогликолипиды, сфингогликолипиды, производные стерола, например холестерин, и их производные, такие как продукты их гидрирования. Примеры производных холестерина включают такие стеролы, которые имеют циклопентанпергидрофенантреновое кольцо. Из них предпочтительно холестерин содержится в липосомальной композиции настоящего изобретения. Примеры антиоксидантов включают аскорбиновую кислоту, мочевую кислоту, и гомологи токоферола или витамин E. Несмотря на то, что токоферол имеет четыре изомера, а именно α-, β-, γ- и δ-токоферолы, любой из них можно использовать в настоящем изобретении.
В настоящем изобретении общая сумма фосфолипида и холестерина также может обозначаться как общее содержание липидов.
В связи с этим липосомальная композиция настоящего изобретения может быть получена как предпочтительная липосомальная композиция с помощью отбора, как указано далее, композиции с общим содержанием липидов, что означает с содержанием фосфолипида и холестерина.
1) Липидная мембрана липосомы состоит только из фосфолипида, содержащего ацильную цепь насыщенной жирной кислоты длиной 16-18.
2) В случае, когда липидная мембрана липосомы включает фосфолипид, имеющий ацильную цепь насыщенной жирной кислоты длиной 14-18, и холестерин в качестве главных компонентов, молярное отношение обоих компонентов составляет от 80:20 до 50:50.
3) В случае когда липидная мембрана липосомы включает фосфолипид, имеющий ацильную цепь ненасыщенной жирной кислоты длиной 16-18, и холестерин в качестве главных компонентов, молярное отношение обоих компонентов составляет от 60:40 до 50:50.
В описании длина ацильной цепи означает число атомов углерода в ацильной цепи.
Насыщенная жирная кислота с числом атомов углерода в ацильной цепи 14 представляет собой миристиновую кислоту, насыщенная жирная кислота с числом атомов углерода в ацильной цепи 15 представляет собой пентадекановую кислоту, насыщенная жирная кислота с числом атомов углерода в ацильной цепи 16 представляет собой пальмитиновую кислоту (тривиальные названия: цетиловая кислота и гексидециловая кислота, системное название: гексадекановая кислота), насыщенная жирная кислота с числом атомов углерода в ацильной цепи 17 представляет собой гептадекановую кислоту, и насыщенные или ненасыщенные жирные кислоты с числом атомов углерода в ацильной цепи 18 включают стеариновую кислоту (системное название: октадекановая кислота), олеиновую кислоту, линолевую кислоту, и линоленовую кислоту.
Раствор внутренней водной фазы (первой водной фазы) липосомы
В настоящем изобретении для раствора внутренней водной фазы липосомы, который используют для инкапсулирования амфипатического слабоосновного лекарственного средства в липосоме с высокой эффективностью и стабильностью, большое значение имеет выбор противоиона, который инкапсулируют в липосоме вместе с амфипатическим слабоосновным соединением. Липосомальная композиция согласно настоящему изобретению предпочтительно содержит ионы сульфатов для инкапсулирования лекарственного средства с высокой эффективностью и получения свойств контролируемого высвобождения, устойчивых в течение длительного времени. Несмотря на то, что обычно используют сульфат аммония в качестве соединения для получения сульфатных ионов, соединение для данной цели также можно выбрать из других соединений, таких как сульфат декстрана и сульфат хондроитина. Кроме того, другие примеры противоиона включают неорганические или органические анионы, такие как гидроксид, фосфат, глюкуронат, цитрат, карбонат, бикарбонат, нитрат, цианат, ацетат, бензоат, бромид и ионы хлорида, а также анионные полимеры.
Значение pH внутренней водной фазы отличается в зависимости от технологии способа “бесконтактной загрузки”. Например, в том случае, когда используют лимонную кислоту, необходимо предварительно создать градиент pH между внутренней водной фазой и внешней водной фазой. В таком случае разница pH между внутренней водной фазой и внешней водной фазой предпочтительно составляет не меньше чем 3. Кроме того, в том случае, когда используется сульфат аммония, градиент pH создается с помощью химического равновесия, так что нет необходимости специально учитывать градиент pH.
Кроме того, при создании липосомальной композиции настоящего изобретения водорастворимый органический раствор, содержащий липид в водорастворимом растворителе, смешивают с раствором первой водной фазы в объемном отношении с водорастворимым органическим раствором от 3/1 до 12/1. Объемное отношение раствора первой водной фазы (внутренняя водная фаза) к водорастворимому органическому раствору предпочтительно находится в диапазоне от 3 до 9, более предпочтительно от 3 до 5,6.
Раствор внешней водной фазы (вторая водная фаза) липосомы
В качестве внешней водной фазы используют водный раствор, в котором концентрация иона, формирующего упомянутый выше ионный градиент, ниже, чем в растворе внутренней водной фазы. Конкретные примеры водного раствора включают раствор HEPES, раствор NaCl и водные растворы сахаридов, например глюкозы и сахарозы. Значение pH внешней водной фазы по желанию контролируют с помощью буфера. Принимая во внимание химическое разложение липида и разрыв значений pH во время введения живой организм, значение pH предпочтительно контролируют в пределах 5,5-8,5, более предпочтительно в пределах 6,0-7,5. Что касается показателей осмотического давления внутренней водной фазы и внешней водной фазы липосомы, они особенно не ограничиваются, поскольку они контролируются в таких пределах, что липосома не будет повреждена вследствие разницы осмотического давления между указанными водными фазами. Учитывая физическую стабильность липосомы, меньшая разница осмотического давления является более желательной.
Липосомальный препарат
Лекарственное средство включают в липосомальную композицию настоящего изобретения методом “бесконтактной” загрузки, в результате чего получают липосомальный препарат согласно настоящему изобретению.
Метод “бесконтактной” загрузки
Метод “бесконтактной” загрузки представляет собой метод введения лекарственного средства в липосомы путем создания пустых липосом без какого-либо лекарственного средства, инкапсулированного в них, и добавления лекарственного средства во внешнюю жидкость, а именно жидкость вне липосом. В методе “бесконтактной” загрузки лекарственное средство, добавленное к внешней жидкости, активно переносится в липосомы для заключения внутри липосом. В качестве движущей силы для активного переноса используется градиент растворимости, ионный градиент, градиент pH или т.п. Например, обычно используется метод, в котором лекарственное средство вводится внутрь липосом с применением ионного градиента, образованного по разные стороны липосомальной мембраны. В качестве конкретного примера, существует технология, в которой лекарственное средство добавляют к предварительно сформированным липосомам с помощью метода “бесконтактной” загрузки на основе градиента концентрации Na+/K+ (смотрите патент Японии № 2847065).
В методе “бесконтактной” загрузки, основанном на ионном градиенте, наиболее широко используется градиент концентрации протонов. Например, можно применить способ, в котором градиент pH такой, что значение pH водной фазы (внутренняя водная фаза) во внутренней области липосомальной мембраны ниже, чем значение pH водной фазы (внешняя водная фаза) во внешней области липосомальной мембраны, создают с применением лимонной кислоты. Кроме того, градиент pH можно создать с помощью градиента концентрации иона аммония и/или градиента концентрации органического соединения, содержащего аминогруппу, которая может присоединять протон (смотрите патент Японии № 2659136). Кроме того, в последние годы раскрыт метод, в котором “бесконтактная” нагрузка осуществляется путем введения ионофора в мембрану липосомы (смотрите патент США № 4885172, патент США № 5059421, патент США № 5171578, и патент США № 5837282).
Лекарственное средство, которое заключают в липосомы
Лекарственное средство, которое содержится в липосомальном препарате согласно настоящему изобретению, особенно не ограничивается в той мере, в какой лекарственное средство может удерживаться внутри липосом с помощью метода ионного градиента; однако предпочтительно лекарственное средство представляет собой амфипатическое слабое основание. Кроме того, с точки зрения эффекта лекарственное средство предпочтительно представляет собой лекарственное средство, для которого желательны свойства продолжительного контролируемого высвобождения при местном применении. Особенно предпочтительные примеры лекарственного средства представляют собой лекарственные препараты, используемые при церебрально-васкулярном нарушении, болезни Паркинсона, психическом заболевании, слабоумии и т.д., и болеутоляющие средства. Примеры таких лекарств включают донепезил, рисперидон, ривастигмин, галантамин, физостигмин, гептилфизостигмин, фенсерин, толсерин, цимсерин, тиатолсерин, тиацимсерин, неостигмин, гиперзин, такрин, метрифонат, миноциклин, фазудила гидрохлорид, нимодин, морфин, бупивакаин, ропивакаин, левобупивакаин, трамадол и лидокаин. Другие примеры включают допамин, L-DOPA, серотонин, эпинефрин, кодеин, мепередин, метадон, морфин, атропин, дицикломин, метиксен, пропантелин, имипрамин, амитриптилин, доксепин, дезипрамин, хинидин, пропранолол, хлорпромазин, прометазин и перфеназин.
Способ введения
Способ введения липосомального препарата настоящего изобретения особенно не ограничен; предпочтительно, однако, липосомальный препарат вводят не перорально и местно. Например, можно выбрать подкожное, внутримышечное, интраперитонеальное, межоболочечное, экстрадуральное или внутрижелудочковое введение. Подходящий способ введения можно выбрать в соответствии со значимым симптомом. Более предпочтительным способом введения является подкожное или внутримышечное введение. В качестве конкретного способа введения липосомальный препарат можно вводить с помощью шприца или распылительного устройства.
Кроме того, местное введение может быть осуществлено с помощью катетера, введенного в живой организм, например в просвет в организме, например в кровеносный сосуд, и проведенного до места повреждения.
Липосомальный препарат согласно настоящему изобретению обладает свойствами системного контролируемого высвобождения, так что эффективная концентрация в крови лекарственного средства, инкапсулированного во внутренней области липосомального препарата сохраняется в течение по меньшей мере четырех дней. Липосомальный препарат настоящего изобретения обладает свойствами местного контролируемого высвобождения, так что эффективная местная концентрация лекарственного средства, инкапсулированного во внутренней области липосомального препарата сохраняется по меньшей мере в течение четырех дней. Липосомальный препарат настоящего изобретения обладает свойствами контролируемого высвобождения, так что свойства контролируемого высвобождения лекарственного средства сохраняются в течение по меньшей мере трех дней, предпочтительно по меньшей мере четырех дней, более предпочтительно по меньшей мере пяти дней и также предпочтительно по меньшей мере семи дней после введения.
В описании “концентрация в крови” означает концентрацию, определяемую путем забора образца крови из кровеносного сосуда и определения концентрации лекарственного средства в плазме, например, с помощью высокоэффективной жидкостной хроматографии, которая описана в примерах ниже.
“Местная концентрация” обозначает значение, полученное при отборе образцов определенной части ткани, органа или т.п. и измерение концентрации лекарственного средства в супернатанте, полученном из гомогената указанной части ткани, органа и т.п. “Эффективная концентрация” обозначает минимальную концентрацию, которую считают терапевтически эффективной и определяют в соответствии с заболеванием и видом лекарственного средства.
Набор препарата с продолжительным контролируемым высвобождением согласно настоящему изобретению включает упомянутый выше липосомальный препарат настоящего изобретения и инъекционную иглу по меньшей мере одного размера, выбранного из размеров игл с 27 калибра по 33 калибр. Количество инъекционных игл может составлять одну иглу или больше чем одну иглу, и размеры инъекционных игл могут быть либо одинаковыми, либо представлять собой комбинацию двух или больше размеров игл. Липосомальный препарат согласно настоящему изобретению можно вводить даже с помощью тонкой иглы, такой как игла калибра 27, 30 или 33, поэтому пациент, которому вводят препарат, испытывает незначительную нагрузку. Кроме того, липосомальный препарат, который вводят, обладает хорошими свойствами контролируемого высвобождения. Таким образом, набор препарата с продолжительным контролируемым высвобождением настоящего изобретения является очень полезным.
ПРИМЕРЫ
Далее настоящее изобретение описывается более подробно путем представления примеров, но изобретение не ограничивается указанными примерами.
Концентрацию и диаметр частиц каждой из нагруженных лекарственным средством липосом, приготовленных в примерах, определяли следующим образом.
Концентрация фосфолипида (мг/мл).
Концентрация фосфолипида в липосомальной суспензии, которую определяют с помощью высокоэффективной жидкостной хроматографии или определения содержания фосфолипида.
Концентрация холестерина (мг/мл).
Концентрация холестерина в суспензии липосом, которую определяют с помощью высокоэффективной жидкостной хроматографии.
Общая концентрация липидов (моль/л): общая молярная концентрация (мМ) липидов в виде компонентов мембраны, которая представляет собой общую сумму, которая представляет собой общую сумму упомянутых выше концентрации фосфолипида и концентрации холестерина.
Концентрации лекарственного средства (мг/мл)
(1) Концентрация донепезила гидрохлорида (мг/мл). Липосомальную композицию разводили водой RO (вода, очищенная с помощью обратного осмоса), так чтобы общая концентрация липидов препарата составляла приблизительно 20-30 мг/мл. Затем разведенную липосомальную композицию дополнительно разводили метанолом в 20 раз, и липосома разрушалась. Определяли абсорбцию полученного раствора при 315 нм с помощью высокоэффективной жидкостной хроматографии с использованием абсорбциометра для УФ и видимой областей, с получением значения концентрации донепезила гидрохлорида в препарате.
(2) Концентрация бупивакаина гидрохлорида (мг/мл). Липосомальную композицию разводили водой RO (вода, очищенная с помощью обратного осмоса), так чтобы общая концентрация липидов составляла приблизительно 20-30 мг/мл. Затем разведенную липосомальную композицию дополнительно разводили метанолом в 20 раз, и липосома разрушалась. Определяли абсорбцию полученного раствора при 263 нм с помощью высокоэффективной жидкостной хроматографии с использованием абсорбциометра для УФ и видимой областей, с получением значения концентрации бупивакаина гидрохлорида в препарате.
(3) Концентрация ропивакаина гидрохлорида (мг/мл). Липосомальную композицию разводили водой RO (вода, очищенная с помощью обратного осмоса), так чтобы общая концентрация липидов составляла приблизительно 20-30 мг/мл. Затем разведенную липосомальную композицию дополнительно разводили метанолом в 20 раз, и липосома разрушалась. Определяли абсорбцию полученного раствора при 263 нм с помощью высокоэффективной жидкостной хроматографии с использованием абсорбциометра для УФ и видимой областей, с получением значения концентрации ропивакаина гидрохлорида в препарате.
(4) Количество содержащегося в липосомах лекарственного средства (молярное отношение лекарственного средства к общему содержанию липидов). Молярное отношение лекарственного средства к общему содержанию липидов рассчитывали как отношение концентрации лекарственного средства, а именно донепезила гидрохлорида, или бупивакаина гидрохлорида, или ропивакаина гидрохлорида, заключенного в липосомах, к упомянутой выше общей концентрации липидов.
(5) Концентрация донепезила гидрохлорида в плазме (мг/мл). Отобранную плазму обрабатывали, и для супернатанта, полученного в результате центрифугирования, определяли флуоресценцию при длине волны возбуждения (Ex) 322 нм и длине волны детектирования (Em) 385 нм с применением высокоэффективной жидкостной хроматографии с использованием флуориметра, измеряя таким образом концентрацию донепезила гидрохлорида в плазме.
(6) Концентрация бупивакаина гидрохлорида в ткани (мг/мл). Отобранный образец ткани обрабатывали, и для супернатанта, полученного в результате с помощью центрифугирования, определяли абсорбцию при 210 нм с помощью высокоэффективной жидкостной хроматографии с использованием абсорбциометра для УФ и видимой областей, измеряя таким образом концентрацию бупивакаина гидрохлорида в ткани.
(7) Средний диаметр частиц (мкм). Средний диаметр частиц, который измеряют с помощью анализатора для определения распределения частиц по размеру на основе светорассеяния, Beckman Coulter LS230.
Сокращенные названия и показатели молекулярного веса используемых компонентов описаны ниже.
HSPC: гидрогенизированный соевый фосфатидилхолин (молекулярный вес 790, SPC3 производства Lipoid GmbH)
Chol: холестерин (молекулярный вес 388,66, производства Solvay S.A.)
DNP: донепезила гидрохлорид (в дальнейшем также обозначаемой как DNP) (молекулярный вес 415,95 производства UINAN CHENGHUI-SHUANFDA Chemical Co., Ltd.)
Бупивакаина гидрохлорид (молекулярный вес 324,89 производства JINAN CHENGHUI-SHUANGDA Chemical Co., Ltd.)
Ропивакаина гидрохлорид (молекулярный вес 310,88 производства JINAN CHENGHUI-SHUANGDA Chemical Co., Ltd.)
Сравнение количеств лекарств, заключенных в липосомальных композициях, полученных в различных контролируемых условиях.
Примеры получения 1-3
Получали липосомы с концентрацией липида в этаноле 150% масс./об. или 200% масс./об. и вводили лекарственное средство в липосомы с помощью градиента pH для создания липосомальных препаратов.
(1) Получение пустых липосом
HSPC и Chol взвешивали в количествах, которые указаны в таблице 1, и добавляли к ним безводный этанол, так чтобы концентрация липидов в безводном этаноле составляла 150% масс./об. или 200% масс./об., с последующим растворением при нагревании приблизительно при 70°С. Затем раствор липидов в этаноле, полученный при растворении, смешивали с раствором внутренней водной фазы (150 мМ водный раствор сульфата аммония), в качестве первой водной фазы, в количестве 4/1 или 3/1 (об./об.) в объемном отношении к раствору липидов в этаноле. Смесь перемешивали при нагревании с постоянной скоростью вращения в течение 10 минут с получением пустых липосом. После завершения нагревания липосомы немедленно охлаждали на льду.
(2) Создание градиента рН
Проводили замену внешней жидкости для охлажденных на льду пустых липосом с применением центрифугирования для создания градиента pH между внутренней водной фазой и внешней водной фазой липосом. Затем липосомы диспергировали приблизительно в 10-кратном количестве внешней водной фазы (20 мМ HEPES/0,9% хлорид натрия (pH 7,5)) с последующим центрифугированием при 3500 об/мин в течение 15 минут для осаждения липосом. Затем удаляли супернатант и впоследствии липосомы диспергировали в растворе 20 мМ HEPES/0,9% хлорид натрия, pH 7,5, добавленном к ним, с последующим центрифугированием таким же образом, как описано выше. Данную стадию повторяли три раза с последующим повторным диспергированием в растворе 20 мМ HEPES/0,9% хлорид натрия, pH 7,5, для создания градиента pH.
(3) Введение лекарственного средства с помощью градиента pH
После создания градиента pH определяли количества HSPC и холестерина липосом и рассчитывали общую концентрацию липидов. На основе рассчитанной таким образом общей концентрации липидов рассчитывали количество донепезила гидрохлорида (DNP, молекулярный вес 415,95) для получения соотношения DNP/общее содержание липидов (моль/моль), равного 0,16. После взвешивания требуемого количества DNP готовили раствор DNP (раствор лекарственного средства) с концентрацией 20 мг/мл с применением воды RO.
Заданное количество раствора DNP, предварительно нагретого до 65°С, добавляли к раствору липосом, нагретому до 65°С, с последующим нагреванием и перемешиванием при 65°С в течение 60 минут для осуществления введения лекарственного средства. После введения лекарственного средства липосомы немедленно охлаждали на льду.
(4) Удаление не включенного в липосомы лекарственного средства
После введения лекарственного средства липосомы диспергировали в растворе внешней водной фазы (20 мМ HEPES/0,9% раствор гидрохлорида натрия (pH 7,5)) добавленном к ним, с последующим центрифугированием при 3500 об/мин в течение 15 минут, для осаждения липосом. Затем супернатант удаляли и затем липосомы диспергировали в 20 мМ растворе HEPES/0,9% хлорид натрия (pH 7,5), добавленном к ним, с последующим центрифугированием таким же образом, как указано выше. Данную стадию повторяли три раза для удаления неинкапсулированного лекарственного средства.
В случае липосомальных препаратов примеров получения 1-3, созданных с помощью способа изготовления согласно настоящему изобретению, который упоминался выше, концентрация липидов в этаноле, объемное отношение внутренней водной фазы к липидсодержащему раствору этанола, количество содержащегося в липосомах лекарственного средства (молярное отношение лекарственного средства к общему содержанию липидов) и диаметр частиц при получении пустых липосом приведены в таблице 2. Выявили, что высокий показатель количества содержащегося в липосомах лекарственного средства, соответствующий молярному отношению лекарственное средство/(общее содержание липидов) не меньше чем 0,08 моль/моль, можно получить, если липосомальный препарат приготовлен в условиях, когда концентрация липидов в растворе этанола и объемное отношение внутренней водной фазы к раствору этанола являются такими, как указано в таблице 2.
Фиг.1 представляет собой фотографию, полученную при исследовании с помощью просвечивающего электронного микроскопа (TEM) среза липосомального препарата после введения лекарственного средства, произведенного в примере получения 2 в данном примере настоящего изобретения. Липосомальный препарат, представленный на фиг.1a, имеет трещину на внешней поверхности липосомального препарата. Липосомальный препарат, представленный на фиг.1b, имеет трещину по существу в центре липосомального препарата. На основании результатов электронно-микроскопического исследования можно видеть, что липосомальный препарат согласно настоящему изобретению обладает структурой, которая характеризуется одной гомогенной внутренней водной фазой, имеющей внешнюю мембрану, состоящую из нескольких липидных бислоев, как показано на фиг.1. Кроме того, из результатов измерения числа липидных бислоев (слоевая структура, ламеллярность) части липосомы, отличающейся высокой сложностью (часть, имеющая слоевую структуру), при оценке невооруженным глазом электронной микрофотографии и если считать, что один ряд липидного бислоя соответствует однослойной структуре, ламеллярность липосомального препарата со средним диаметром частиц 6 мкм, полученного в настоящем изобретении, составляла приблизительно 20. Кроме того, ламеллярность липосомального препарата со средним диаметром частиц 2,5 мкм составляла приблизительно 12. С другой стороны, предпочтительный средний диаметр частиц для проявления свойств продолжительного контролируемого высвобождения препарата, полученного согласно настоящему изобретению, составляет приблизительно 3 мкм или больше. Соответственно, предполагают, что подходящие свойства контролируемого высвобождения можно получить, если ламеллярность липосомального препарата составляет 12 или больше (предпочтительно 12-27).
В данном случае липосомальный препарат со средним диаметром частиц 6 мкм, который измеряли в данном примере, представляет собой липосомальный препарат, в котором липосомы размером 3 мкм или меньше представлены в пропорции менее чем приблизительно 10% (по объему или по количеству), липосомы размером 10 мкм или больше представлены в пропорции приблизительно 10% и липосомы размером 4-7 мкм представлены в пропорции, составляющей приблизительно 50% или больше.
Примеры получения 4 и 5
DNP-нагруженные липосомальные препараты получали таким же образом, как в примере получения 2, за исключением того, что контролировали скорость вращения суспензии липосом во время получения пустых липосом, в результате чего контролировали диаметр частиц. Результаты описаны в таблице 2. Как видно из таблицы, получали липосомальные препараты с разными значениями среднего диаметра частиц 3,7 мкм и 12,2 мкм. Кроме того, можно получить липосомальные препараты с высокой эффективностью включения и высоким показателем количества содержащегося в липосомах лекарственного средства таким же образом, как в примерах получения 1-3.
Сравнительный пример 1
Липосома, полученная при концентрации липида в этаноле 100% масс./об.
HSPC и Chol взвешивали в количествах, указанных в таблице 3, и добавляли 1 мл безводного этанола, так чтобы концентрация липида в этаноле составляла 100% масс./об., с последующим растворением при нагревании приблизительно при 70°С. Затем раствор липидов в этаноле, полученный таким образом, смешивали с 9 мл раствора внутренней водной фазы (150 мМ раствор сульфата аммония) в качестве первой водной фазы, так чтобы объемное отношение к раствору этанола составляло 9/1, с последующим нагреванием и перемешиванием при постоянной скорости вращения в течение приблизительно 10 минут, для получения липосом. Общая концентрация липидов в смешанной фазе составляла 10% масс./об. После завершения нагревания пустые липосомы немедленно охлаждали на льду. Последующее образование градиента pH, введение лекарственного средства и удаление неинкапсулированного лекарственного средства осуществляли такими же способами, как в примерах получения 1-3, с получением нагруженного лекарственным средством липосомального препарата.
Общая концентрация липидов в этаноле и объемное отношение внутренней водной фазы к раствору этанола во время получения пустых липосом липосомальной композиции в сравнительном примере 1 описаны в таблице 4. Кроме того, количество содержащегося в липосомах лекарственного средства (молярное отношение лекарственного средства к общему содержанию липидов) и диаметр частиц полученной липосомальной композиции представлены в таблице 4. В результате выяснили, что хотя получили средний диаметр частиц, сравнимый с аналогичными показателями в Примерах получения 1-3, степень инкапсулирования лекарственного средства находится на более низком уровне, если общая концентрация липидов в смешанной фазе (раствор внутренней водной фазы + спиртовой раствор) является низкой (таблица 4). На основании вышеизложенного считают, что концентрация липида в этаноле и объемное отношение внутренней водной фазы к раствору этанола во время получения пустых липосом влияет на структуру липосом и, таким образом, влияет на количество инкапсулированного лекарственного средства.
Получение липосомального препарата донепезила гидрохлорида методом экструзии
Сравнительные примеры 2 и 3
HSPC и Chol взвешивали в количествах 0,71 г и 0,29 г, соответственно, и растворяли при нагревании в 1 мл безводного этанола, добавленного к ним. К 1 мл раствора липидов в этаноле, полученного таким образом, добавляли 9 мл раствора сульфата аммония (внутренняя водная фаза), нагретого приблизительно до 70°С, и полученную смесь перемешивали с помощью ультразвуковой установки при нагревании для получения первичной суспензии липосом. Первичную суспензию липосом последовательно пропускали через фильтр (производства Whatman pic), прикрепленный к экструдеру (экструдер T.10, производства Lipexbiomembranes Inc.), нагретому приблизительно до 70°С; а именно, пропускали пять раз через фильтр с диаметром пор 0,4 мкм или пять раз через фильтр с диаметром пор 2 мкм для получения пустых липосом с диаметром частиц приблизительно 300 нм. После завершения нагревания пустые липосомы немедленно охлаждали на льду. После охлаждения на льду проводили замену внешней жидкости с применением гель-фильтрации, с замещением в достаточной степени внешней водной фазой (20 мМ HEPES/0,9% раствор хлорида натрия (pH 7,5), для создания градиента pH. Затем осуществляли введение лекарственного средства путем добавления заданного количества раствора DNP таким образом, что лекарственное средство/общее содержание липидов (моль/моль)=0,16 и перемешивания при нагревании при 65°С в течение 60 минут. После введения лекарственного средства липосомы немедленно охлаждали на льду. Затем проводили удаление неинкапсулированного лекарственного средства с применением гель-фильтрации с замещением в достаточном объеме раствором 20 мМ HEPES/0,9% хлорид натрия (pH 7,5).
В таблице 4 представлены количества содержащегося в липосомах лекарственного средства и диаметры частиц липосомальных композиций, полученных в сравнительных примерах 2 и 3. В результате получены липосомальные препараты с относительно большими показателями количества содержащегося в липосомах лекарственного средства.
Оценка характеристик высвобождения in vitro при различных диаметрах частиц
В данном примере характеристики высвобождения лекарственного средства липосомальных композиций, полученных в Примерах получения 2, 4 и 5 и в сравнительных примерах 2 и 3, оценивали с помощью системы для проверки in vitro с применением сульфата аммония.
Взвешивали сульфат аммония в количестве, необходимом для получения концентрации 7,5 мМ, и добавляли к нему фосфатный буфер для приготовления раствора для теста высвобождения (7,5 мМ сульфат аммония/фосфатный буфер, при pH 7,4 и 300 мОсмоль). Каждую из липосомальных композиций, полученных в примерах получения 2, 4 и 5 и в сравнительных примерах 2 и 3, разводили раствором для теста высвобождения в 10 раз с последующим нагреванием при 37°С в течение заранее установленного времени. Через 0, 1, 2, 5, 8, 10, 15 и 30 минут после начала нагревания брали образцы, немедленно добавляли к образцу стоп-реагент и смесь охлаждали на льду для прекращения высвобождения лекарственного средства. Определение количества высвободившегося DNP осуществляли с помощью высокоэффективной жидкостной хроматографии согласно методу определения концентрации лекарственного средства, описанному выше.
В результате, установили, что высвобождение DNP (донепезила гидрохлорида) из липосом происходит тем быстрее, чем меньше диаметр частиц, как показано на фиг.3. На основе данных, показанных на фиг.3, рассчитывали значения угла наклона кривых высвобождения, нанесенных на график, в начальный период и рассчитывали скорость высвобождения для каждого среднего диаметра частиц. Результаты представлены на фиг.4. Скорость высвобождения составляла 23 (мкг/(мл·мин)) при среднем диаметре частиц 12,2 мкм; 37 ((мкм/(мл·мин)) при среднем диаметре частиц 6 мкм; 51 ((мкм/(мл·мин)) при среднем диаметре частиц 3,7 мкм; и 110 (мкг/(мл·мин)) при среднем диаметре частиц 1,2 мкм
Исходя из указанных результатов выяснили, что скорость высвобождения возрастает по мере того, как диаметр частиц уменьшается. В частности, когда средний диаметр частиц составлял 1,2 мкм, скорость высвобождения была особенно высокой.
На основании приведенных фактов предполагается, что если средний диаметр частиц составляет 1,2 мкм или меньше, лекарственное средство высвобождается за такое короткое время, что непрерывность высвобождения не может быть достигнута. Возможно, это связано с тем, что число липидных мембран (толщина слоистой структуры) уменьшается по мере того, как уменьшается диаметр частиц, в результате чего лекарственное средство легче распространяется за пределы липосом и высвобождение лекарственного средства увеличивается. Другая причина, как считают, заключается в том, что по мере уменьшения диаметра частиц, увеличивается искривление липидных мембран, что приводит к снижению упаковывающих свойств липидных мембран и лекарственное средство легче проникает через липидные мембраны.
Из приведенных результатов хорошо видно, что значение среднего диаметра частиц, при котором скорость высвобождения является очень высокой и нельзя ожидать непрерывности высвобождения, составляет 1,2 мкм или меньше и что средний диаметр частиц свыше 1,2 мкм является важным для осуществления непрерывности высвобождения.
Из приведенных выше данных можно видеть, что липосомальные композиции, полученные в сравнительных примерах 2 и 3, характеризуются высоким значением количества инкапсулированного лекарственного средства, но имеют заметно повышенную скорость высвобождения по сравнению с липосомальными композициями, полученными в примерах получения 2, 4 и 5. Поэтому сложно предположить, что в случае липосомальных композиций сравнительных примеров 2 и 3, что они будут высвобождаться in vivo в течение длительного периода времени продолжительностью по меньшей мере три дня. С другой стороны, липосомальные композиции примеров получения 2, 4 и 5 демонстрируют профили длительного высвобождения даже в тесте высвобождения in vitro; в частности выяснилось, что скорость высвобождения тем ниже, чем больше диаметр частиц.
Динамика концентрации лекарственного средства в крови в случае липосомального DNP
Липосомальные препараты донепезила, полученные в примере получения 2 и в сравнительных примерах 2 и 3, а также донепезил в чистом виде использовали в тесте определения динамики концентрации лекарственного средства в крови. Липосомальная композиция, приготовленная по способу получения настоящего изобретения, может предоставить в результате высокое количество инкапсулированного лекарственного средства и, соответственно, обеспечить введение лекарственного средства в небольшом объеме и с высокой концентрацией. Ввиду этого липосомальный препарат донепезила, полученный в примере получения 2, вводили подкожно в область спины крысы в количестве 50 мг/кг в пересчете на количество донепезила гидрохлорида. С другой стороны, липосомальные препараты, полученные в сравнительных примерах 2 и 3, имели ограничение по вводимому объему в зависимости от концентрации лекарственного средства в препарате; ввиду этого указанные липосомальные препараты вводили подкожно в область спины крысы в количествах 5 мг/кг и 25 мг/кг в пересчете на количество донепезила гидрохлорида. Кроме того, в целях сравнения донепезил, используемый в чистом виде, вводили подкожно в область спины крысы в количестве 5 мг/кг. Дозы донепезила гидрохлорида, используемого в чистом виде, и липосомальные препараты, полученные в сравнительных примерах 2 и 3 и в примере получения 2, описаны в таблице 5.
В случае донепезила, используемого в чистом виде, образцы крови получали из хвостовой вены через 0,5, 1, 5, 10, 30, 120, 480, 1440 и 2880 минут после введения. В случае липосомальных препаратов образцы крови получали из хвостовой вены через 0,5, 1, 3, 4, 8, 24, 48, 72, 96, 120, 144, 168, 192, 216, 264, 288, 312 и 336 часов после введения. Образцы крови центрифугировали (6000 об/мин, 10 минут, 4°С), при этом плазму получали дробно. Полученную таким образом плазму обрабатывали и измеряли интенсивность флуоресценции при длине волны возбуждения (Ex) 322 нм и длине волны детектирования (Em) 385 нм с применением высокоэффективной жидкостной хроматографии, определяя, таким образом, концентрацию донепезила гидрохлорида в каждом образце плазмы. Результаты представлены на фиг.5.
Как показано на фиг.5, концентрация донепезила гидрохлорида в крови в случае подкожного введения донепезила гидрохлорида в чистом виде достигала максимального значения в крови через 0,5 часа после введения и затем стремительно снижалась. Через 48 часов концентрация уже опускалась ниже предела обнаружения. Липосомальный препарат, полученный в сравнительном примере 2, не проявлял начального “взрывного” высвобождения в отличие от донепезила, используемого в чистом виде. Кроме того, хотя указанный препарат демонстрировал продолжительное контролируемое высвобождение вплоть до 48 часов, его концентрация в крови достигала максимального значения через восемь часов после введения, а потом быстро уменьшалась и опускалась ниже 10 нг/мл уже через 48 часов после введения. Липосомальный препарат, полученный в сравнительном примере 3, сохранял относительно высокую концентрацию лекарственного средства в крови в течение четырех дней после введения, но затем концентрация стремительно снижалась. Указанные препараты имеют относительно небольшой диаметр частиц 0,3 мкм и 1,2 мкм, соответственно, поэтому показатели высвобождения лекарственного средства из указанных липосомальных препаратов выше, чем показатели высвобождения из липосомальных препаратов с большим диаметром частиц. Кроме того, в данных случаях следует учитывать, что липосомы подвержены диффузии в области введения и они перемещаются в лимфу или в кровь, поэтому липосомы быстро теряются. По указанным причинам предполагается, что ожидаемые свойства продолжительного контролируемого высвобождения не могут быть достигнуты.
С другой стороны, липосомальный препарат, полученный в примере получения 2 согласно настоящему изобретению, не проявлял начального “взрыва” высвобождения и показывал заметное увеличение времени контролируемого высвобождения; таким образом, можно достичь свойств контролируемого высвобождения, сохраняющихся в течение длительного периода продолжительностью четыре дня. Рассматривается следующая причина. Наличие градиента pH позволяет надежно удерживать лекарственное средство во внутренней водной фазе. Кроме того, по мере того как диаметр частиц возрастает, число липидных бислоев увеличивается и слоевая структура становится толще, так что проникающая способность лекарственного средства через липидную мембрану подавляется. В результате можно добиться свойств контролируемого высвобождения, сохраняющихся в течение длительного периода времени.
Концентрация лекарственного средства в крови, полученная в данном примере, показывает эффективную концентрацию, которая является достаточной с клинической точки зрения. Отсутствие начального “взрыва” и способность устойчиво сохранять эффективную концентрацию в течение четырех дней гарантирует, что возможность появления побочных эффектов уменьшается и что QOL пациента может повыситься. Таким образом, можно выявить препарат, характеризующийся очень высокой способностью к контролируемому высвобождению.
Из вышеизложенного понятно, что липосомальная композиция, полученная по настоящему изобретению, имеет одну внутреннюю водную фазу с наружной мембраной, состоящей из нескольких липидных бислоев, и обеспечивает достаточное количество лекарственного средства, которое прочно удерживается во внутренней водной фазе методом градиента pH, таким образом позволяя заключать в липосомах лекарственное средство с высокой концентрацией. Кроме того, липосомальный препарат настоящего изобретения, в котором лекарственное средство заключают в липосомальной композиции настоящего изобретения, является превосходным препаратом с пролонгированным контролируемым высвобождением, который проявляет свойства контролируемого высвобождения, сохраняющиеся в течение длительного периода времени.
Получение липосомального препарата бупивакаина гидрохлорида согласно настоящему изобретению
Сравнительные примеры 6-13
HSPC и Chol взвешивали в соответствующих количествах, которые указаны в таблице 6, и добавляли к ним безводный этанол, так чтобы концентрация липида в безводном этаноле составляла 200, 150 и 100% масс./об. соответственно, с последующим растворением при нагревании приблизительно при 70°С. Затем каждый из растворов липидов в этаноле, полученный таким образом, смешивали с раствором внутренней водной фазы (раствор лимонной кислоты, pH 2,5) в объемных отношениях с раствором этанола 3/1, 4/1, 5,6/1 и 9/1 (об./об.) и полученные смеси перемешивали при нагревании с постоянной скоростью вращения в течение приблизительно 10 минут с получением липосом. После завершения нагревания липосомы немедленно охлаждали на льду. Соотношения этанола, внутренней водной фазы и концентрации липидов в условиях получения описаны в таблице 7.
Затем осуществляли замену внешней жидкости с помощью центрифугирования с применением раствора лимонной кислоты, pH 6,5, создавая таким образом градиент pH между внутренней областью и внешней областью липосом.
Затем осуществляли введение лекарственного средства таким же образом, как в примерах получения 1-3. В качестве лекарственного средства использовали бупивакаина гидрохлорид (BPV, молекулярный вес 324,89). После взвешивания необходимого количества бупивакаина гидрохлорида (BPV) к нему добавляли воду RO с получением 10 мг/мл раствора BPV (раствор лекарственного средства) и осуществляли введение лекарственного средства путем перемешивания при нагревании при 65°С в течение 60 минут. После введения лекарственного средства немедленно охлаждали липосомы на льду. Затем с использованием раствора лимонной кислоты, pH 6,5, проводили удаление неинкапсулированного лекарственного средства с помощью такой же процедуры, как в примерах получения 1-3. Для липосом бупивакаина гидрохлорида, полученных таким образом, молярное отношение (моль/моль) бупивакаина гидрохлорида к общему содержанию липидов представлено в таблице 8 в виде количества инкапсулированного лекарственного средства.
Таким образом, очень высокие значения количества инкапсулированного лекарственного средства, как показано в таблице 8, получали, когда концентрация липида в этаноле составляла 100-200% масс./об., внутреннюю водную фазу (раствор лимонной кислоты, pH 2,5) добавляли в объемном отношении 3/1, 4/1, 5,6/1 и 9/1 (об./об.) к раствору этанола и концентрация липидов составляла 15-50% масс./об., на основании общего раствора этанола и внутренней водной фазы (смешанная фаза), как показано в таблицах 2 и 7.
Примеры получения 14-17
Липосомы бупивакаина гидрохлорида получали таким же образом, как в примерах получения 6-9, за исключением того, что 150 мМ сульфат аммония использовали в качестве внутренней водной фазы. HSPC и Chol взвешивали в соответствующих количествах, которые указаны в таблице 9, и добавляли безводный этанол, так чтобы концентрация липидов в безводном этаноле составляла 200% масс./об., с последующим растворением при нагревании приблизительно при 70°С.
Затем, используя 150 мМ сульфат аммония в качестве внутренней водной фазы, добавляли раствор внутренней водной фазы (150 мМ сульфат аммония) к полученному ранее раствору липидов в этаноле в объемном отношении к раствору этанола 3/1, 4/1, 5,6/1 и 9/1 (об./об.), с последующим перемешиванием при нагревании при постоянной скорости вращения в течение приблизительно 10 минут для получения пустых липосом. После завершения нагревания липосомы немедленно охлаждали на льду.
Затем осуществляли создание градиента pH, введение бупивакаина гидрохлорида, и удаление неинкапсулированного лекарственного средства таким же образом, как в примерах получения 6-9 для создания липосомальных препаратов бупивакаина гидрохлорида. Результаты описаны в таблице 2. Также, в том случае, когда в качестве внутренней водной фазы использовали сульфат аммония, получали высокие показатели количества инкапсулированного лекарственного средства аналогичным образом, как и в том случае, когда применяли раствор лимонной кислоты, pH 2,5.
Сравнительные примеры 4-7
HSPC и Chol взвешивали в количествах, указанных в таблице 10, и добавляли безводный этанол, так чтобы концентрация липидов в безводном этаноле составляла 200, 150 и 100% масс./об. соответственно, с последующим растворением при нагревании приблизительно при 70 °С. Затем внутреннюю водную фазу (раствор лимонной кислоты, pH 2,5) добавляли к полученным ранее растворам липидов в этаноле в объемных отношениях к раствору этанола 1,5/1, 2/1 и 9/1 (об./об.), с последующим перемешиванием при нагревании с постоянной скоростью вращения в течение приблизительно 10 минут для получения липосом. После завершения нагревания липосомы немедленно охлаждали на льду.
Затем проводили замену внешней жидкости с применением раствора лимонной кислоты, pH 6,5 для создания градиента pH между внутренней водной фазой и внешней водной фазой липосом. Следующим шагом осуществляли введение лекарственного средства с использованием бупивакаина гидрохлорида.
Затем осуществляли удаление неинкапсулированного лекарственного средства с использованием раствора лимонной кислоты, pH 6,5, для получения липосомальных препаратов. Для полученных таким образом липосомальных препаратов количества инкапсулированного бупивакаина гидрохлорида представлены в таблицах 4 и 8.
В результате обнаружили, что, если концентрация липида в этаноле составляла 100-200% масс./об. и объемное отношение внутренней водной фазы (раствор лимонной кислоты, pH 2,5) к раствору липидов в этаноле составляло 2/1 или меньше, количество инкапсулированного лекарственного средства несколько уменьшалась, и количество неинкапсулированного лекарственного средства (отношение количества бупивакаина гидрохлорида во внешней жидкости к количеству бупивакаина гидрохлорида в препарате) было относительно высоким 6-9%. Приведенные выше результаты позволяют предположить, что в указанных выше условиях получения липосом достаточное количество лекарственного средства не может устойчиво удерживаться во внутренней водной фазе.
Динамика концентрации лекарственного средства липосомального препарата бупивакаина гидрохлорида
Липосомальный препарат бупивакаина гидрохлорида, полученный в примере получения 16, и бупивакаина гидрохлорид, применяемый в чистом виде, использовали в тесте определения динамики концентрации. Липосомальный препарат и бупивакаина гидрохлорид, используемый в чистом виде, вводили подкожно в область спины крысы в соответствующих дозах, которые указаны в таблице 11, при этом дозы указаны в пересчете на количество бупивакаина гидрохлорида. Спустя 1, 24, 72, 120 и 168 часов после введения липосомальной композиции бупивакаина гидрохлорида и через 0,5, 4 и 24 часа после введения бупивакаина гидрохлорида, используемого в чистом виде, отбирали образец подкожной ткани из задней части в месте введения и гомогенизировали его. Затем обрабатывали раствор гомогената и образец раствора, полученный таким образом, использовали в анализе методом высокоэффективной жидкостной хроматографии для определения количества бупивакаина гидрохлорида, оставшегося в подкожной ткани в задней части в месте введения. Результаты представлены на фиг.6. Степень удержания бупивакаина гидрохлорида, используемого в чистом виде, в месте введения опускалась ниже 1% в течение четырех часов после введения. Указанный результат подтвердил, что бупивакаина гидрохлорид, используемый в чистом виде, исчезает из места введения в течение нескольких часов. С другой стороны, липосомальный препарат бупивакаина гидрохлорида имел профиль замедленного высвобождения из места введения с зависимостью от времени. В этом случае бупивакаина гидрохлорид сохранялся в месте введения в количестве приблизительно 10% на третий день, приблизительно 5% на пятый день и приблизительно 3% на седьмой день после введения. На основании полученных результатов выяснили, что введенные липосомы высвобождают бупивакаина гидрохлорид в месте введения продолжительным образом.
Исходя из вышеизложенного липосомальный препарат бупивакаина гидрохлорида, полученный по настоящему изобретению, обладает свойством продолжительного контролируемого высвобождения.
Получение липосомы ропивакаина гидрохлорида согласно настоящему изобретению
Пример получения 18
Липосомальную композицию создавали таким же образом, как в примере получения 15, за исключением применения ропивакаина гидрохлорида в качестве лекарственного средства, для получения липосом ропивакаина гидрохлорида.
Таким образом, как показано в таблице 1, в том случае, когда в качестве лекарственного средства использовали ропивакаина гидрохлорид, лекарственное средство также можно ввести методом градиента pH и получить высокий показатель количества инкапсулированного лекарственного средства. Кроме того, в отношении свойств высвобождения in vitro, описан профиль высвобождения, также сходный с профилями высвобождения липосомального препарата донепезила гидрохлорида и липосомального препарата бупивакаина гидрохлорида.
Подтверждение размера иглы для инъекций для введения липосомального препарата настоящего изобретения
Определяли наименьшую толщину иглы, с помощью которой можно ввести липосомальный препарат настоящего изобретения.
С использованием липосомального препарата настоящего изобретения, содержащего бупивакаина гидрохлорид, который получали в примере получения 16, осуществляли стадию набора препарата в шприц и стадию выпускания препарата из шприца с помощью инъекционных игл различного калибра. В результате установили, что липосомальный препарат настоящего изобретения легко можно набрать в шприц и выпустить из шприца с помощью любой из самых тонких инъекционных игл, таких как иглы калибра 27, калибра 30 и калибра 33.
Сравнительный эксперимент
В качестве сравнительного эксперимента для приведенного выше примера определяли, можно ли ввести препарат Risperdal Consta (производства Janssen Pharmaceutical K.K.) с помощью тонких инъекционных игл. Risperdal Consta является названием микросфер, состоящих из сополимера полимолочной кислоты и полигликолевой кислоты (PLGA). Конкретно, препарат представляет собой инъекцию длительного действия антипсихотического средства в форме комплексного продукта, предоставляемого вместе с иглой для инъекций калибра 20G. Средний диаметр частиц микросфер составляет 25-150 мкм. Процедуру набора указанных микросфер пробовали провести с применением инъекционных игл калибров 27-33. В результате установили, что инъекционные иглы с калибрами 27-33 не могут быть успешно использованы для набора микросфер. В отношении стадии выталкивания микросфер из шприца только толстые инъекционные иглы калибра 19-21 можно использовать для введения, тогда как тонкие инъекционные иглы калибра 27, 30 и 33 не могут быть успешно применены для выталкивания микросфер. Считают, что размер частиц микросфер является настолько большим, что микросферы нельзя ввести с помощью тонких игл.
Из вышеизложенного выявили, что нижний предел значения калибра иглы для введения микросфер, таких как липосомы, обычно составляет приблизительно 19-21, но липосомальный препарат настоящего изобретения можно ввести с помощью более тонких игл, таких как иглы калибра 27, 30 и 33.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЛИПОСОМНАЯ КОМПОЗИЦИЯ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2011 |
|
RU2577683C2 |
| ЛИПОСОМАЛЬНАЯ КОМПОЗИЦИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2018 |
|
RU2734900C1 |
| ЛИПОСОМАЛЬНАЯ КОМПОЗИЦИЯ | 2010 |
|
RU2476216C1 |
| ДВУСЛОЙНЫЕ ПРЕПАРАТЫ | 1995 |
|
RU2166332C2 |
| СРЕДСТВО ДЛЯ ЛЕЧЕНИЯ ОПУХОЛИ И НАБОР, СОДЕРЖАЩИЙ ЛИПОСОМАЛЬНУЮ КОМПОЗИЦИЮ ГЕМЦИТАБИНА | 2016 |
|
RU2761620C2 |
| ЛИПОСОМАЛЬНАЯ КОМПОЗИЦИЯ ДЛЯ ИСПОЛЬЗОВАНИЯ В ПЕРИТОНЕАЛЬНОМ ДИАЛИЗЕ | 2013 |
|
RU2609860C2 |
| СТАБИЛИЗИРУЮЩИЕ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ КАМПТОТЕЦИНА | 2016 |
|
RU2833053C2 |
| СТАБИЛИЗИРОВАННЫЕ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ КАМПТОТЕЦИНА | 2016 |
|
RU2732567C2 |
| СРЕДСТВО ДЛЯ ЛЕЧЕНИЯ ОПУХОЛИ И НАБОР, СОДЕРЖАЩИЙ ЛИПОСОМАЛЬНУЮ КОМПОЗИЦИЮ ГЕМЦИТАБИНА | 2016 |
|
RU2768178C2 |
| СРЕДСТВО ДЛЯ ЛЕЧЕНИЯ ОПУХОЛИ И НАБОР, СОДЕРЖАЩИЙ ЛИПОСОМАЛЬНУЮ КОМПОЗИЦИЮ ГЕМЦИТАБИНА | 2016 |
|
RU2738365C2 |

Группа изобретений относится к фармацевтике. Описывается липосомальная композиция, полученная путем смешивания водорастворимого органического раствора с раствором первой водной фазы и получением эмульсии. Водорастворимый органический раствор содержит фосфолипид и холестерин. Далее следует замена внешнего раствора эмульсии раствором, представляющим собой вторую водную фазу. При этом образуется ионный градиент между водной фазой внутренней области и водной фазой внешней области липосомальной мембраны. Полученная липосома характеризуется средним диаметром частиц в терминах внешнего диаметра, составляющим от 2,5 мкм до 12 мкм, и 12-слойной - 35-слойной структурой. Описан липосомальный препарат с продолжительным контролируемым высвобождением, содержащий терапевтический агент, а также набор препарата с продолжительным контролируемым высвобождением. Технический результат обеспечивает контролируемое высвобождение, при этом эффективная концентрация лекарственного средства в крови поддерживается в течение четырех дней. 3 н. и 3 з.п. ф-лы, 6 ил., 11 табл., 18 пр.
1. Липосомальная композиция, полученная путем смешивания водорастворимого органического раствора, в котором фосфолипид и холестерин содержатся в общей концентрации от 100 до 200% масс./об. (100-200 г в 100 мл) в водорастворимом органическом растворителе, с раствором первой водной фазы в количестве от 3/1 до 12/1 в объемном отношении с водорастворимым органическим раствором, с получением эмульсии, в которой общая концентрация фосфолипида и холестерина в получаемой смешанной фазе составляет от 15 до 50% масс./об. (15-50 г в 100 мл), с последующей заменой внешнего раствора эмульсии раствором, представляющим собой вторую водную фазу, при этом образуется ионный градиент между водной фазой внутренней области липосомальной мембраны, где водная фаза внутренней области содержит раствор первой водной фазы, и водной фазой внешней области липосомальной мембраны, где водная фаза внешней области содержит раствор второй водной фазы, где липосома содержит водную фазу внутренней области внутри многослойной мембраны, характеризующейся средним диаметром частиц в терминах внешнего диаметра, составляющим от 2,5 мкм до 12 мкм, и характеризующейся 12-слойной - 35-слойной структурой.
2. Липосомальная композиция по п.1, в которой ионный градиент представляет собой протонный градиент pH и липосомальная композиция характеризуется таким градиентом pH, что значение pH водной фазы внутренней области липосомы ниже, чем значение pH водной фазы внешней области липосомы.
3. Липосомальный препарат с продолжительным контролируемым высвобождением, содержащий лекарственное средство, введенное в водную фазу внутренней области в липосомальной композиции по любому из пп. 1-2 путем применения ионного градиента, где лекарственное средство содержится в липосоме в молярном отношении, составляющем от 0,08 (моль) до 0,16 (моль), вычисленном к общему содержанию липидов (моль), где липосома содержит водную фазу внутренней области внутри многослойной мембраны, характеризующейся 12-слойной - 35-слойной структурой.
4. Липосомальный препарат с продолжительным контролируемым высвобождением по п.3, где липосомальный препарат обладает либо свойствами системного контролируемого высвобождения, так что эффективная концентрация в крови лекарственного средства, инкапсулированного во внутренней области, поддерживается в течение четырех или более дней, либо свойствами местного контролируемого высвобождения, так что эффективная местная концентрация лекарственного средства, инкапсулированного во внутренней области, поддерживается в течение четырех или более дней.
5. Липосомальный препарат с продолжительным контролируемым высвобождением по п.3 или 4, в котором лекарственное средство представляет собой амфипатическое слабоосновное соединение.
6. Набор препарата с продолжительным контролируемым высвобождением, включающий липосомальный препарат по любому из пп.3-5 и инъекционную иглу, характеризующуюся по меньшей мере размером одного калибра, выбранного из калибров от 27 до 33.
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| HARAN G | |||
| et al | |||
| Transmembrane ammonium sulfate gradients in liposomes produce efficient and stable entrapment of amphipathic weak bases | |||
| Biochimica et Biophysica Acta, 1151 (1993) 201-215 | |||
| МНОГОСЛОЙНАЯ ВЕЗИКУЛЯРНАЯ КОМПОЗИЦИЯ | 2006 |
|
RU2385711C2 |
| US 6162462 A, 19.12.2000. | |||

