ТЕХНИЧЕСКАЯ ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к соединениям, пригодным в качестве ингибиторов фосфатидилинозитол-3-киназы (PI3K). Также изобретение относится к фармацевтически приемлемым композициям, содержащим соединения по изобретению, и к способам применения композиций для лечения различных нарушений.
УРОВЕНЬ ТЕХНИКИ
PI3K представляют собой семейство киназ липидов, которые катализируют фосфорилирование мембранного липида фосфатидилинозитола (PI) по 3'-OH кольца инозитола с образованием PI 3-фосфата [PI(3)P, PIP], PI 3,4-бифосфата [PI(3,4)P2, PIP2] и PI 3,4,5-трифосфата [PI(3,4,5)P3, PIP3]. PI(3,4)P2 и PI(3,4,5)P3 действуют в качестве центров для привлечения различных белков внутриклеточной передачи сигнала, которые, в свою очередь, образуют комплексы передачи сигнала для трансляции внеклеточных сигналов на цитоплазматическую сторону плазматической мембраны.
До настоящего времени идентифицировано восемь PI3K млекопитающих, включая четыре PI3K класса I. Класс Ia включает PI3Κα, PI3Κβ и PI3Κδ. Все из ферментов класса Ia представляют собой гетеродимерные комплексы, содержащие каталитическую субъединцу (p110a, p110β или p110δ), связанную с содержащей SH2-домен адаптерной субъединицей p85. PI3K класса Ia активируются через передачу сигнала тирозинкиназы и вовлечены в пролиферацию и выживание клеток. PI3Kα и PI3Kβ также вовлечены в образование опухоли при различных злокачественных опухолях человека. Таким образом, фармакологические ингибиторы PI3Kα и PI3Kβ пригодны для лечения различных типов злокачественной опухоли.
PI3Kγ, единственный представитель PI3K класса Ib, состоит из каталитической субъединицы p110γ, которая связана с регуляторной субъединицей p101. PI3Kγ регулируется сопряженными с G-белком рецепторами (GPCR) через связь с субъединицами βγ гетеротримерных G-белков. PI3Kγ экспрессируется, главным образом, в кроветворных клетках и кардиомиоцитах и вовлечена в воспаление и функционирование тучных клеток. Таким образом, фармакологические ингибиторы PI3Kγ пригодны для лечения различных воспалительных заболеваний, аллергии и сердечно-сосудистых заболеваний.
Хотя был разработан ряд ингибиторов PI3K, остается потребность в дополнительных соединениях для ингибирования PI3K для лечения различных нарушений и заболеваний, особенно нарушений и заболеваний, поражающих центральную нервную систему (ЦНС). Таким образом, было бы желательно разработать дополнительные соединения, которые являются пригодными в качестве ингибиторов PI3K, которые проникают через гематоэнцефатический барьер (ГЭБ).
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Было открыто, что соединения по этому изобретению и их фармацевтически приемлемые композиции являются эффективными в качестве ингибиторов PI3K, в частности PI3Kγ. Таким образом, изобретение относится к соединениям, имеющим общую формулу:
или их фармацевтически приемлемым солям, где каждый из A, B, C, D, E, X1, X2, R1, R2, R3 и R4 являются такими, как определено в настоящем описании.
Также изобретение относится к фармацевтическим композициям, которые включают соединение формулы I и фармацевтически приемлемый носитель, адъювант или наполнитель. Эти соединения и фармацевтические композиции пригодны для лечения или снижения тяжести различных нарушений, включая аутоиммунные заболевания и воспалительные заболевания ЦНС.
Соединения и композиции, предусматриваемые в рамках настоящего изобретения, также пригодны для исследования PI3K в биологических и патологических явлениях; исследованиях внутриклеточных каскадов передачи сигнала, опосредуемых такими киназами; и для сравнительной оценки новых ингибиторов киназ.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Определения и общая терминология
Как используют в рамках изобретения, применяются следующие определения, если нет иных указаний. Для целей настоящего изобретения, химические элементы указаны в соответствии с периодической таблицей элементов, версия CAS, Handbook of Chemistry and Physics, 75th Ed. Кроме того, основные принципы органической химии описаны в "Organic Chemistry", Thomas Sorrell, University Science Books, Sausalito: 1999, и "March's Advanced Organic Chemistry", 5th Ed., Ed.: Smith, M.B. and March, J., John Wiley & Sons, New York: 2001, полное содержание которых включено в настоящее описание в качестве ссылки.
Соединения, которые изображены с определенными стереохимическими центрами, являются стереохимически чистыми, но, тем не менее, с не определенной абсолютной стереохимией. Такие соединения могут иметь либо R-конфигурацию, либо S-конфигурацию. В тех случаях, когда такое абсолютное распределение определено, хиральный центр(ы) обозначен на изображении как R или S.
Как описано в настоящем описании, соединения по настоящему изобретению необязательно могут быть замещены одним или несколькими заместителями, такими как представлены, главным образом, выше, или как проиллюстрировано конкретными классами, подклассами и типами по изобретению. Будет понятно, что выражение "необязательно замещенный" используют взаимозаменяемо с выражением "замещенный или незамещенный". Как правило, термин "замещенный", которому либо предшествует, либо не предшествует термин "необязательно", относится к замене водородных радикалов в данной структуре радикалом конкретного заместителя. Если нет иных указаний, необязательно замещенная группа может обладать заместителем в каждом поддающемся замещению положению группы. Когда более одного положения в данной структуре может быть замещено более чем одним заместителем, выбранным из указанной группы, заместители могут быть либо одинаковыми в каждом положении, либо они могут отличаться.
Как описано в настоящем описании, когда термин "необязательно замещенный" предшествует перечню, указанный термин относится ко всем последующим поддающимся замещению группам в этом перечне. Например, если X представляет собой галоген; необязательно замещенный C1-3алкил или фенил; X может представлять собой либо необязательно замещенный алкил, либо или необязательно замещенный фенил. Аналогично, если термин "необязательно замещенный" следует после перечня, указанный термин также относится ко всем поддающимся замещению группам в предшествующем перечне, если нет иных указаний. Например: если X представляет собой галоген, C1-3алкил или фенил, где X необязательно замещен JX, тогда как C1-3алкил, так и фенил, может быть необязательно замещен JX. Как будет понятно специалисту в данной области, группы, такие как H, галоген, NO2, CN, NH2, OH или OCF3 не включены, поскольку они не являются поддающимися замещению группами. Если радикал заместителя или структура не идентифицированы или не определены как "необязательно замещенные", радикал заместителя или структура являются незамещенными.
Комбинации заместителей, предусматриваемых этим изобретением, предпочтительно представляют собой комбинации, которые приводят к стабильным или химически допустимым соединениям. Термин "стабильный", как используют в рамках изобретения, относится к соединениям, которые по существу не изменяются под воздействием условий, обеспечивающих их продукцию, детекцию и предпочтительно их выделение, очистку и применение для одной или нескольких целей, описанных в настоящем описании.
Термин "алифатический" или "алифатическая группа", как используют в рамках изобретения, означает прямую (т.е. неразветвленную) или разветвленную, замещенную или незамещенную углеводородную цепь, которая является полностью насыщенной или которая содержит один или несколько элементов ненасыщенности. Если нет иных указаний, алифатические группы содержат 1-20 атомов углерода. В некоторых вариантах осуществления алифатические группы содержат 1-10 атомов углерода. В других вариантах осуществления алифатические группы содержат 1-8 атомов углерода. В других вариантах осуществления алифатические группы содержат 1-6 атомов углерода, и в других вариантах осуществления алифатические группы содержат 1-4 атомов углерода. Пригодные алифатические группы включают, но не ограничиваются ими, линейные или разветвленные, замещенные или незамещенные алкильные, алкенильные или алкинильные группы. Дополнительные примеры алифатических групп включают метил, этил, пропил, бутил, изопропил, изобутил, винил и втор-бутил. Термины "алкил" и приставка "алк-", как используют в рамках изобретения, включают как неразветвленную, так и разветвленную насыщенную углеродную цепь. Термин "алкилен", как используют в рамках изобретения, соответствует насыщенной двухвалентной прямой или разветвленной углеводородной группе, и его примерами являются метилен, этилен, изопропилен и т.п. Термин "алкилиден", как используют в рамках изобретения, соответствует двухвалентной неразветвленной алкильной соединительной группе. Термин "алкенил", как используют в рамках изобретения, соответствует одновалентной прямой или разветвленной углеводородной группе, содержащий одну или несколько углерод-углеродных двойных связей. Термин "алкинил", как используют в рамках изобретения, соответствует одновалентной прямой или разветвленной углеводородной группе, содержащей одну или несколько углерод-углеродных тройных связей.
Термин "циклоалифатический" (или "карбоцикл") относится к моноциклическому C3-C8углеводороду или бициклическому C8-C12углеводороду, который является полностью насыщенным или который содержит один или несколько элементов ненасыщенности, но который не является ароматическим, и который имеет одну точку присоединения к остальной части молекулы, и где любое отдельное кольцо в указанной бициклической кольцевой системе имеет 3-7 членов. Пригодные циклоалифатические группы включают, но не ограничиваются ими, циклоалкил, циклоалкенил и циклоалкинил. Следующие примеры алифатических групп включают циклопентил, циклопентенил, циклогексил, циклогексенил, циклогептил и циклогептенил.
Термин "гетероцикл", "гетероциклил", "гетероциклоалифатический" или "гетероциклический", как используют в рамках изобретения, относится к моноциклической, бициклической или трициклической кольцевой системе, в которой по меньшей мере одно кольцо в системе содержит один или несколько гетероатомов, которые являются одинаковыми или отличаются, и которая является полностью насыщенной или которая содержит один или несколько элементов ненасыщенности, но которая не является ароматической, и которая имеет одну точку присоединения к остальной части молекулы. В некоторых вариантах осуществления "гетероцикл", "гетероциклил", "гетероциклоалифатическая" или "гетероциклическая" группа имеет от трех до четырнадцати членов в кольце, где один или несколько членов кольца представляет собой гетероатом, независимо выбранный из кислорода, серы, азота или фосфора, и каждое кольцо в системе содержит от 3 до 8 членов в кольце.
Примеры гетероциклических колец включают, но не ограничиваются ими, следующие моноциклы: 2-тетрагидрофуранил, 3-тетрагидрофуранил, 2-тетрагидротиофенил, 3-тетрагидротиофенил, 2-морфолино, 3-морфолино, 4-морфолино, 2-тиоморфолино, 3-тиоморфолино, 4-тиоморфолино, 1-пирролидинил, 2-пирролидинил, 3-пирролидинил, 1-тетрагидропиперазинил, 2-тетрагидропиперазинил, 3-тетрагидропиперазинил, 1-пиперидинил, 2-пиперидинил, 3-пиперидинил, 1-пиразолинил, 3-пиразолинил, 4-пиразолинил, 5-пиразолинил, 1-пиперидинил, 2-пиперидинил, 3-пиперидинил, 4-пиперидинил, 2-тиазолидинил, 3-тиазолидинил, 4-тиазолидинил, 1-имидазолидинил, 2-имидазолидинил, 4-имидазолидинил, 5-имидазолидинил; и следующие бициклы: 3-1H-бензимидазол-2-он, 3-(1-алкил)бензимидазол-2-он, индолинил, тетрагидрохинолинил, тетрагидроизохинолинил, бензотиолан, бензодитиан и 1,3-дигидроимидазол-2-он.
Термин "гетероатом" означает один или несколько из кислорода, серы, азота, фосфора или кремния, включая любую окисленную форму азота, серы или фосфора; кватернизованную форму любого основного азота; или поддающийся замещению азот гетероциклического кольца, например N (как в 3,4-дигидро-2H-пирролиле), NR+ (как в пирролидиниле) или NR+ (как в N-замещенном пирролидиниле).
Термин "ненасыщенный", как используют в рамках изобретения, означает, что группа имеет один или несколько элементов ненасыщенности.
Термин "алкокси" или "тиоалкил", как используют в рамках изобретения, относится к алкильной группе, как определено выше, связанной с основной углеродной цепью через атом кислорода ("алкокси") или серы ("тиоалкил").
Термины "галогеналкил", "галогеналкенил" и "галогеналкокси" означают алкил, алкенил или алкокси, в зависимости от обстоятельств, замещенный одним или несколькими атомами галогена. Термин "галоген" означает F, CI, Br или I.
Термин "арил", используемый отдельно или в качестве части более крупной группы, как в "аралкиле", "аралкокси" или "арилоксиалкиле", относится к моноциклической, бициклической или трициклической карбоциклической кольцевой системе, имеющей всего от шести до четырнадцати членов в кольце, где указанная кольцевая система имеет одну точку присоединения к остальной части молекулы, по меньшей мере одно кольцо в системе является ароматическим, и где каждое кольцо в системе содержит от 3 до 7 членов кольца. Термин "арил" можно использовать взаимозаменяемо с термином "арильное кольцо". Примеры арильных колец включают фенил, нафтил и антрацен.
Термин "гетероарил", используемый отдельно или в качестве части более крупной группы, как в "гетероаралкиле" или "гетероарилалкокси" относится к моноциклической, бициклической и трициклической кольцевой системе, имеющей всего от пяти до четырнадцати членов в кольце, где указанная кольцевая система имеет одну точку присоединения к остальной части молекулы, по меньшей мере одно кольцо в системе является ароматическим, по меньшей мере одно кольцо в системе содержит один или несколько гетероатомов, независимо выбранных из азота, кислорода, серы или фосфора, и где каждое кольцо в системе содержит от 3 до 7 членов в кольце. Термин "гетероарил" можно использовать взаимозаменяемо с термином "гетероарильное кольцо" или термином "гетероароматический".
Дополнительные примеры гетероарильных колец включают следующие моноциклы: 2-фуранил, 3-фуранил, N-имидазолил, 2-имидазолил, 4-имидазолил, 5-имидазолил, 3-изоксазолил, 4-изоксазолил, 5-изоксазолил, 2-оксазолил, 4-оксазолил, 5-оксазолил, N-пирролил, 2-пирролил, 3-пирролил, 2-пиридил, 3-пиридил, 4-пиридил, 2-пиримидинил, 4-пиримидинил, 5-пиримидинил, пиридазинил (например, 3-пиридазинил), 2-тиазолил, 4-тиазолил, 5-тиазолил, тетразолил (например, 5-тетразолил), триазолил (например, 2-триазолил и 5-триазолил), 2-тиенил, 3-тиенил, пиразолил (например, 2-пиразолил), изотиазолил, 1,2,3-оксадиазолил, 1,2,5-оксадиазолил, 1,2,4-оксадиазолил, 1,2,3-триазолил, 1,2,3-тиадиазолил, 1,3,4-тиадиазолил, 1,2,5-тиадиазолил, пиразинил, 1,3,5-триазинил, и следующие бициклы: бензимидазолил, бензофурил, бензотиофенил, индолил (например, 2-индолил), пуринил, хинолинил (например, 2-хинолинил, 3-хинолинил, 4-хинолинил) и изохинолинил (например, 1-изохинолинил, 3-изохинолинил или 4-изохинолинил).
В некоторых вариантах осуществления арильная (включая аралкил, аралкокси, арилоксиалкил и т.п.) или гетероарильная (включая гетероаралкил, гетероарилалкокси и т.п.) группа может содержать один или несколько заместителей. Пригодные заместители на ненасыщенном атоме углерода арильной или гетероарильной группы включают: галоген; C1-4алифатическую группу, -OH; -OR°; -SH°; -SR°; 1,2-метилендиокси; 1,2-этилендиокси; фенил (Ph); -O(Ph); -(CH2)1-2(Ph); -CH=CH(Ph); -NO2; -CN; -NH2; -NH(R°); -N(R°)2; -NHC(O)R°; -NR°C(O)R°; -NHC(S)R°; -NR°C(S)R°; -NHC(O)NH2; -NHC(O)NH(R°); -NHC(O)N(R°)2; -NR°C(O)NH(R°); -NR°C(O)N(R°)2; -NHC(S)NH2; -NHC(S)N(R°)2; -NHC(S)NH(R°); -NR°C(S)NH(R°); -NR°C(S)N(R°)2; -NHC(O)OR°; -NR°C(O)OR°; -C(O)OH; -C(O)OR°; -C(O)R°; -C(S)R°; -C(O)NH2; -C(O)NH(R°); -C(O)N(R°)2; -C(S)NH2; -C(S)NH(R°); -C(S)N(R°)2; -OC(O)NH2; -OC(O)NH(R°); -OC(O)N(R°)2; -OC(O)R°; -C(NOR°)H; -C(NOR°)R°; -S(O)2R°; -S(O)3R°; -S(O)3H; -S(O)2NH2; -S(O)2NH(R°); -S(O)2N(R°)2; -S(O)R°; -NHS(O)2R°; -NR°S(O)2R°; -N(OR°)R°; -(CH2)0-2NHC(O)R°; -1-R°; -1-N(R°)2; -1-SR°; -1-OR°; -1-(C3-10циклоалифатическую группу), -1-(C6-10арил), -1-(5-10-члченный гетероарил), -1-(5-10-членный гетероциклил), оксо, C1-4галогеналкокси, C1-4галогеналкил, -1-NO2, -1-CN, -1-OH, -1-CF3; или два заместителя, на одном атоме углерода или на различных атомах углерода, вместе с атомом углерода или промежуточными атомами углерода, с которыми они связаны, образуют 5-7-членное насыщенное, ненасыщенное или частично насыщенное кольцо, где L представляет собой группу C1-6алкилена, в которой вплоть до трех элементов метилена заменены -NH-, -NR°-, -O-, -S-, -C(O)O-, -OC(O)-, -C(O)CO-, -C(O)-, -C(O)NH-, -C(O)NR°-, -C(=N-CN), -NHCO-, -NR°CO-, -NHC(O)O-, -NR°C(O)O-, -S(O)2NH-, -S(O)2NR°-, -NHS(O)2-, -NR°S(O)2-, -NHC(O)NH-, -NR°C(O)NH-, -NHC(O)NR°-, -NR°C(O)NR°, -OC(O)NH-, -OC(O)NR°-, -NHS(O)2NH-, -NR°S(O)2NH-, -NHS(O)2NR°-, -NR°S(O)2NR°-, -S(O)- или -S(O)2-, и где каждый встречающийся R° независимо выбран из необязательно замещенной C1-6алифатической группы, незамещенного 5-6-членного гетероарильного или гетероциклического кольца, фенила или -CH2(Ph), или два независимо встречающихся R°, на одном заместителе или на различных заместителях, взятые вместе с атомом(ами), с которыми связана каждая группа R°, образуют 5-8-членное гетероциклильное, арильное или гетероарильное кольцо или 3-8-членное циклоалкильное кольцо, где указанное гетероарильное или гетероциклильное кольцо имеет от 1 до 3 гетероатомов, независимо выбранных из азота, кислорода или серы. Неограничивающие необязательные заместители на алифатической группе R° включают -NH2, -NH(C1-4алифатическую группу), -N(C1-4алифатическую группу)2, галоген, C1-4алифатическую группу, -OH, -O(C1-4алифатическую группу), -NO2, -CN, -C(O)OH, -C(O)O(C1-4алифатическую группу), -O(галогенC1-4алифатическую группу) или галогенC1-4алифатическую группу, где каждая из указанных выше C1-4алифатических групп в R° является незамещенной.
В некоторых вариантах осуществления алифатическая или гетероалифатическая группа или неароматическое гетероциклическое кольцо могут содержать один или несколько заместителей. Пригодные заместители на насыщенном атоме углерода алифатической или гетероалифатической группы или неароматического гетероциклического кольца выбирают из заместителей, приведенных выше для ненасыщенного атома углерода арильной или гетероарильной группы и, кроме того, включают следующие: =O, =S, =NNHR*, =NN(R*)2, =NNHC(O)R*, =NNHC(O)O(алкил), =NNHS(O)2(алкил) или =NR*, где каждый R* независимо выбран из водорода или необязательно замещенной C1-8алифатической группы. Необязательные заместители на алифатической группе R выбраны из -NH2, -NH(C1-4алифатической группы), -N(C1-4алифатической группы)2, галогена, C1-4алифатической группы, -OH, -O(C1-4алифатической группы), -NO2, -CN, -C(O)OH, -C(O)O(C1-4алифатической группы), -C(O)NH2, -C(O)NH(C1-4алифатической группы), -C(O)N(C1-4алифатической группы)2, -O(галоген-C1-4алифатической группы) и галоген(C1-4алифатической группы), где каждая из указанных выше C1-4алифатических групп R* является незамещенной; или два R* на одном азоте, взятые вместе с азотом, образуют 5-8-членное гетероциклильное или гетероарильное кольцо, имеющее 1-3 гетероатомов, независимо выбранных из азота, кислорода и серы.
В некоторых вариантах осуществления необязательные заместители на атоме азота неароматического гетероциклического кольца включают -R+, -N(R+)2, -C(O)R+, -C(O)OR+, -C(O)C(O)R+, -C(O)CH2C(O)R+, -S(O)2R+, -S(O)2N(R+)2, -C(=S)N(R+)2, -C(=NH)-N(R+)2 или -NR+S(O)2R+; где R+ представляет собой водород, необязательно замещенную C1-6алифатическую группу, необязательно замещенный фенил, необязательно замещенный -O(Ph), необязательно замещенный -CH2(Ph), необязательно замещенный -(CH2)1-2(Ph); необязательно замещенный -CH=CH(Ph); или незамещенное 5-6-членное гетероарильное или гетероциклическое кольцо, имеющее от одного до четырех гетероатомов, независимо выбранных из кислорода, азота или серы, или два независимо встречающихся R+, на одном заместителе или различных заместителях, взятые вместе с атомом(ами), с которыми связана группа R+, образуют 5-8-членное гетероциклильное, арильное или гетероарильное или 3-8-членное циклоалкильное кольцо, где указанное гетероарильное или гетероциклильное кольцо имеет 1-3 гетероатомов, независимо выбранных из азота, кислорода или серы. Необязательные заместители на алифатической группе или фенильном кольце R+ выбраны из -NH2, -NH(C1-4алифатической группы), -N(C1-4алифатической группы)2, галогена, C1-4алифатической группы, -OH, -O(C1-4алифатической группы), -NO2, -CN, -C(O)OH, -C(O)O(C1-4алифатической группы), -O(галоген(C1-4алифатической группы)), или галоген(C1-4алифатической группы), где каждая из указанных выше C1-4алифатических групп в R+ является незамещенной.
Как подробно описано выше, в некоторых вариантах осуществления два независимо встречающихся R° (или R+, или любая другая переменная, сходным образом определенная в настоящем описании), могут, взятые вместе с атомом(ами), с которым каждая из переменных связана, образовывать 5-8-членное гетероциклильное, арильное или гетероарильное кольцо или 3-8-членное циклоалкильное кольцо. Иллюстративные кольца, которые образуются, когда два независимо встречающихся R° (или R+, или любые другие переменные, сходным образом определенные в настоящем описании), взятые вместе с атомом(ами), с которыми каждая из переменных связана, включают, но не ограничиваются ими, следующие: a) два независимо встречающихся R° (или R+, или любые другие переменные, сходным образом определенные в настоящем описании), которые связаны с одним атомом и, взятые с этим атомом, образуют кольцо, например N(R°)2, где оба встречающихся R°, взятые вместе с атомом азота, образуют пиперидин-1-ильную, пиперазин-1-ильную или морфолин-4-ильную группу; и b) два независимо встречающихся R° (или R+, или любые другие переменные, сходным образом определенные в настоящем описании), которые связаны с различными атомами и, взятые вместе с обоими из этих атомов, образуют кольцо, например, где фенильная группа замещена двумя встречающимися OR° , эти два встречающихся R°, взятые вместе с атомами кислорода, с которыми они связаны, образуют конденсированное 6-членное кислородсодержащее кольцо . Будет понятно, что может образовываться множество других колец, когда два независимо встречающихся R° (или R+, или любые переменные, сходным образом определенные в настоящем описании), взятые вместе с атомом(ами), с которым каждая из переменных связана, и что подразумевается, что примеры, подробно описанные выше, не являются ограничивающими.
В некоторых вариантах осуществления элемент метилена алкильной или алифатической цепи необязательно заменен другим атомом или группой. Примеры таких атомов или групп включают, но не ограничиваются ими, -NR°-, -O-, -S-, -C(O)O-, -OC(O)-, -C(O)CO-, -C(O)-, -C(O)NR°-, -C(=N-CN), -NR°CO-, -NR°C(O)O-, -S(O)2NR°-, -NR°S(O)2-, -NR°C(O)NR°-, -OC(O)NR°-, -NR°S(O)2NR°-, -S(O)- или -S(O)2-, где R° является таким, как определено в настоящем описании. Если нет иных указаний, необязательные замены образуют химически стабильное соединение. Необязательная замена атома или группы может встречаться в цепи или на любом конце цепи; т.е. в точке присоединения и/или также на конце. Две необязательных замены также могут быть соседними друг с другом в цепи, при условии, что это приводит к химически стабильному соединению. Если нет иных указаний, если замена встречается на конце, заменяющий атом связан с H на конце. Например, если один элемент метилена -CH2CH2CH3 необязательно заменен -O-, полученное соединение может представлять собой -OCH2CH3, -CH2OCH3 или -CH2CH2OH.
Как описано в настоящем описании, связь, изображенная от заместителя к центру одного кольца во множественной кольцевой системе (как представлено ниже) отражает замещение заместителем в поддающемся замещению положении любого из колец во множественной кольцевой системе. Например, структура a отражает возможное замещение в любом из положений, представленных в структуре b.
Это также применимо к множественным кольцевым системам, конденсированным с необязательными кольцевыми системами (которые изображаются пунктирными линиями). Например, в структуре c, X представляет собой необязательный заместитель как для кольца A, так и для кольца B.
Структура с
Однако, если каждое из двух колец во множественной кольцевой системе имеет отличающиеся заместители, изображенные от центра каждого кольца, тогда, если нет иных указаний, каждый заместитель отражает только замещение на кольце, к которому он присоединен. Например, на структуре d, Y представляет собой необязательный заместитель только для кольца A, и X представляет собой необязательный заместитель только для кольца B.
Структура d
Термин "защитная группа", как используют в рамках изобретения, обозначает группы, предназначенные для защиты функциональной группы, например, такой как спирт, амин, карбоксил, карбонил и т.д., от нежелательных реакций в процессе синтеза. Обычно используемые защитные группы описаны в Greene and Wuts, Protective Groups In Organic Synthesis, 3rd Edition (John Wiley & Sons, New York, 1999), которая включена в настоящий документ в качестве ссылки. Примеры азот-защитных групп включают ацильную, ароильную или карбамильную группы, такие как формил, ацетил, пропионил, пивалоил, трет-бутилацетил, 2-хлорацетил, 2-бромацетил, трифторацетил, трихлорацетил, фталил, o-нитрофеноксиацетил, α-хлорбутирил, бензоил, 4-хлорбензоил, 4-бромбензоил, 4-нитробензоил и хиральные вспомогательные группы, такие как защищенные или незащищенные D-, L- или D,L-аминокислоты, такие как аланин, лейцин, фенилаланин и т.п.; сульфонильные группы, такие как бензолсульфонил, п-толуолсульфонил и т.п.; карбаматные группы, такие как бензилоксикарбонил, п-хлорбензилоксикарбонил, п-метоксибензилоксикарбонил, п-нитробензилоксикарбонил, 2-нитробензилоксикарбонил, п-бромбензилоксикарбонил, 3,4-диметоксибензилоксикарбонил, 3,5-диметоксибензилоксикарбонил, 2,4-диметоксибензилоксикарбонил, 4-метоксибензилоксикарбонил, 2-нитро-4,5-диметоксибензилоксикарбонил, 3,4,5-триметоксибензилоксикарбонил, 1-(п-бифенилил)-1-метилэтоксикарбонил, α,α-диметил-3,5-диметоксибензилоксикарбонил, бензгидрилоксикарбонил, трет-бутилоксикарбонил, диизопропилметоксикарбонил, изопропилоксикарбонил, этоксикарбонил, метоксикарбонил, аллилоксикарбонил, 2,2,2,-трихлорэтоксикарбонил, феноксикарбонил, 4-нитрофеноксикарбонил, флуоренил-9-метоксикарбонил, циклопентилоксикарбонил, адамантилоксикарбонил, циклогексилоксикарбонил, фенилтиокарбонил и т.п., арилалкильные группы, такие как бензил, трифенилметил, бензилоксиметил и т.п., и силильные группы, такие как триметилсилил и т.п. Предпочтительными N-защитными группами являются формил, ацетил, бензоил, пивалоил, трет-бутилацетил, аланил, фенилсульфонил, бензил, трет-бутилоксикарбонил (Boc) и бензилоксикарбонил (Cbz).
Термин "пролекарство", как используют в рамках изобретения, обозначает соединение, которое трансформируется in vivo в соединение формулы I или соединение, приведенное в таблице 1. Такая трансформация может обеспечиваться, например, гидролизом в крови или ферментативной трансформацией пролекарственной формы в исходную форму в крови или ткани. Пролекарства соединений по изобретению могут представлять собой, например, сложные эфиры. Сложные эфиры, которые можно использовать в качестве пролекарств в рамках настоящего изобретения, представляют собой фениловые сложные эфиры, алифатические (C1-C24) сложные эфиры, ацилоксиметиловые сложные эфиры, карбонаты, карбаматы и сложные эфиры аминокислот. Например, соединение по изобретению, которое содержит OH-группу, может быть ацилированным в этом положении в его форме пролекарства. Другие формы пролекарств включают фосфаты, например, такие как фосфаты, образующиеся при фосфонировании OH-группы на исходном соединении. Подробное обсуждение пролекарств предоставлено в T. Higuchi and V. Stella, Pro-drugs as Novel Delivery Systems, Vol. 14 of the A.C.S. Symposium Series, Edward B. Roche, ed., Bioreversible Carriers in Drug Design, American Pharmaceutical Association and Pergamon Press, 1987, и Judkins et al., Synthetic Communications 26(23):4351-4367, 1996, каждая из которых включена в настоящий документ в качестве ссылки.
Если нет иных указаний, структуры, представленные в настоящем описании, также включают все изомерные (например, энантиомерные, диастереомерные и геометрические (или конформационные)) формы структуры; например, R- и S-конфигурации для каждого асимметричного центра, изомеры по двойной связи (Z) и (E), и конформационные изомеры (Z) и (E). Таким образом, в объем изобретения входят единичные стереохимические изомеры, а также энантиомерные, диастереомерные и геометрические (или конформационные) смеси соединений по настоящему изобретению.
Если нет иных указаний, в объем изобретения входят все таутомерные формы соединений по изобретению. Кроме того, если нет иных указаний, структуры, изображенные в настоящем описании, также включают соединения, которые отличаются только присутствием одной или нескольких изотопно обогащенных форм. Например, в объем настоящего изобретения входят соединения, имеющие структуры по настоящему изобретению, за исключением замены водорода дейтерием или тритием, или замены углерода 13C- или 14C-обогащенным углеродом. Такие соединения пригодны, например, в качестве аналитических инструментов, зондов в биологических анализах, или в качестве ингибиторов PI3K с улучшенным терапевтическим профилем.
Описание соединений по изобретению
В одном аспекте изобретение относится к соединениям формулы I:
или их фармацевтически приемлемой соли, где:
X1 представляет собой N или CH;
X2 представляет собой N, CH или C-CH3;
R1 выбран из фенильного кольца, 5-6-членного гетероарильного кольца, кольца пиридона, или 9-10-членной конденсированной бициклической гетероарильной или гетероциклической кольцевой системы, где каждое из указанных колец или кольцевых систем необязательно замещено 1 или 2 независимо встречающимися R1a, и каждое из указанных гетероарильных или гетероциклических колец имеет 1, 2 или 3 гетероатома, выбранных из азота, кислорода или серы;
R1a представляет собой хлор, фтор, C1-8алифатическую группу, -(CH2)0-2C3-6циклоалифатическую группу, -(CH2)0-2-5-6-членную гетероциклическую группу, имеющую вплоть до двух гетероатомов, выбранных из азота, кислорода или серы, -CN, -C(O)C1-4алифатическую группу, -C(O)NH(C1-4алифатическую группу), -C(O)N(C1-4алифатическую группу)2, -C(O)OC1-4алифатическую группу, -S(O)2NH(C1-4алифатическую группу), -S(O)2N(C1-4алифатическую группу)2 или -S(O)2C1-4алифатическую группу, где вплоть до 3 несоседних атомов углерода указанной алифатической или циклоалифатической группы R1a могут быть замещены -O-, -S- или -N(R1b)-, и где каждая из алифатической, циклоалифатической или гетероциклической групп R1a необязательно и независимо замещена вплоть до 4 встречающимися JR;
каждый JR независимо представляет собой фтор, оксо, -(CH2)0-2CN, -(CH2)0-2CF3, -C(O)R1b, -C(O)N(R1b)2, -C(O)O(R1b), -N(R1b)2, -N(R1b)C(O)R1b, -(CH2)0-2OR1b, фенил или 5-6-членный гетероарил, 4-6-гетероциклил, или 9-11-членный конденсированный бициклический гетероарил или гетероциклил, причем каждое из указанных гетероарильных или гетероциклильных колец имеет вплоть до 3 атомов, выбранных из азота, кислорода или серы, где каждая из указанных циклоалифатических, фенильных, гетероарильных или гетероциклильных групп необязательно замещена вплоть до 2 R1c;
каждый R1b независимо выбран из водорода, C1-8алифатической группы, -(CH2)0-1C3-6циклоалифатической группы, -(CH2)0-1C4-6гетероциклической группы, имеющей вплоть до двух гетероатомов, выбранных из N или O, или две R1b, вместе с атомом, с которым они связаны, образуют 5-6-членное гетероциклическое кольцо, где каждое алифатическое, циклоалифатическое или гетероциклическое кольцо необязательно замещено посредством вплоть до трех атомов F или вплоть до двух -OH, -C1-2алкильных или -OC1-2алкильных групп;
каждый R1c независимо представляет собой фтор, хлор, C1-4алифатическую группу, -(CH2)0-2ОН, -CN, -C(O)C1-4алифатическую группу или -C(O)OC1-4алифатическую группу;
R2 представляет собой водород, F, Cl, CF3, C1-2алифатическую группу, C3-4циклоалифатическую группу, -N(CH3)2, -N(CH2)3, -OCF3, -OCHF2 или -OC1-2алифатическую группу;
R3 представляет собой водород, C1-6алифатическую группу, C3-6циклоалифатическую группу, C4-7гетероциклильную группу, имеющую 1 или 2 атома, выбранных из N или O, -(CH2)0-1CF3, -OH, -OC1-6алифатическую группу, -OC3-6циклоалифатическую группу, -OC3-6гетероциклильную группу, имеющую один атом кислорода, -O(CH2)2OC1-2алифатическую группу или -OC1-2алкилC(O)OC1-3алифатическую группу, или бензил; и
R4 представляет собой водород или C1-6алкил; или R3 и R4 вместе с атомом углерода, с которым они связаны, образуют 3-6-членное циклоалифатическое кольцо, 3-6-членное гетероциклическое кольцо, имеющее вплоть до двух атомов, выбранных из N или O, или C2алкенил, где каждая из указанных алифатических, циклоалифатических или гетероциклильных групп R3, R4, или R3 и R4 вместе необязательно замещена посредством вплоть до трех атомов F или вплоть до двух C1-2алкильных, -C(O)C1-4алкильных, -C(O)OC1-4алкильных, -OH, или -OC1-2алкильных групп;
A представляет собой N или CRA;
B представляет собой N или CRB, или A=B представляет собой атом серы;
C представляет собой N или CRC;
D представляет собой N или CRD;
E представляет собой N или CRE, где не более двух из A, B, C, D или E представляют собой N;
RA представляет собой водород, CH3 или OCH3;
RB представляет собой водород, F, Cl, C1-3алифатическую группу, -(CH2)0-1CF3, -(CH2)0-1CHF2 или -O(CH2)0-1CF3;
RC представляет собой водород, F, Cl, C1-3алифатическую группу, -(CH2)0-1CF3, -(CH2)0-1CHF2, N(R1b)2, -OH, -O(CH2)0-1CF3 или -OC1-8алифатическую группу, где вплоть до 2 несоседних атомов углерода указанной алифатической группы могут быть замещены -O-;
RD представляет собой водород, фтор, хлор, C1-4алифатическую группу, -C(O)OH, -C(O)OC1-4алифатическую группу, -C(O)N(R1b)2, -CN, -C(RD1)=N-OR1b, -N(R1b)2, -N(RD1)C(O)C1-4алифатическую группу, -N(RD1)C(O)фенил, -N(RD1)S(O)2C1-4алифатическую группу, -N(RD1)S(O)2N(R1b)2, -N(RD1)S(O)2фенил-OH, -OC1-8алифатическую группу, -O(CH2)0-1C3-6циклоалифатическую группу, -SC1-4алифатическую группу, -S(O)C1-4алифатическую группу, -S(O)2C1-4алифатическую группу или -S(O)2N(R1b)2; где вплоть до 2 несоседних атомов углерода указанной алифатической, циклоалифатической или гетероциклической группы RD может быть замещен -O-, и каждый из указанной алифатической группы, циклоалифатической группы или фенила RD могут быть замещены посредством вплоть до 5 атомов фтор; или RD и RC, вместе с атомами, к которым они присоединены, образуют фенильное или пиридильное кольцо;
каждый RD1 независимо представляет собой водород или C1-2алкил; и
RE представляет собой водород, F, Cl, -NHC(O)C1-8алифатическую группу, -OH, -OC1-2алифатическую группу, -(CH2)0-1CF3, -(CH2)0-1CHF2, C1-3алифатическую группу, C3-4циклоалифатическую группу, N(R1b)2, азетидин-1-ил.
В одном варианте осуществления соединения имеют формулу I и RD представляет собой водород, фтор, хлор, C1-4алифатическую группу, -(CH2)0-1CF3, -C(O)N(R1b)2, -CN, -N(R1b)2, -NHC(O)C1-8алифатическую группу, -OH, -O(CH2)0-1CF3, -O(CH2)0-1CHF2, -O(CH2)0-1CH2F, -OC1-8алифатическую группу, -O(CH2)0-1C3-6циклоалифатическую группу, -SC1-8алифатическую группу, -S(O)2C1-8алифатическую группу, -S(O)2N(R1b)2; где вплоть до 2 несоседних атомов углерода указанной алифатической, циклоалифатической или гетероциклической группы RD могут быть замещены -O-, или RD и RC вместе с атомами, к которым они присоединены, образуют фенильное или пиридильное кольцо; R3 представляет собой водород, C1-6алкил, C3-6циклоалкил, -(CH2)0-1CF3, -OH, -OC1-6алкил, -OC3-6циклоалкил, -OC3-6гетероциклил, имеющий один атом кислорода, -O(CH2)2OC1-2алкил, или -OC1-2алкилC(O)OC1-3алкил или бензил; и R4 представляет собой водород или C1-6алкил; или R3 и R4 вместе с атомом углерода, с которым они связаны, образуют 3-6-членное циклоалкильное кольцо, 3-6-членное гетероциклическое кольцо, имеющее один атом кислорода, где каждый из указанного алкила, циклоалкила или гетероциклила R3, R4, или R3 и R4 вместе необязательно замещен посредством вплоть до двух F, C1-2алкилов или -OC1-2алкилов.
В другом варианте осуществления соединения имеют формулу I и каждый R1b независимо выбран из водорода, C1-4алифатической группы или C3-6циклоалифатической группы; RB представляет собой водород, F, Cl, -OCF3, -OC1-2алифатическую группу, -CF3 или C1-2алифатическую группу; RC представляет собой водород, F, Cl, C1-3алифатическую группу, -(CH2)0-1CF3, -N(R1b)2, -OH, -OCF3 или -OC1-8алифатическую группу; RD представляет собой водород, фтор, хлор, C1-4алифатическую группу, (CH2)0-1CF3, -C(O)NHC1-8алифатическую группу, -CN, -N(R1b)2, -NHC(O)C1-8алифатическую группу, -OH, -OCF3, -OCHF2, -OC1-8алифатическую группу, -O(CH2)0-1C3-6циклоалифатическую группу, -SC1-8алифатическую группу, -S(O)2C1-8алифатическую группу, -S(O)2N(R1b)2; где вплоть до 2 несоседних атомов углерода указанной алифатической или циклоалифатической группы RD могут быть замещены -O-, или RD и RC, вместе с атомами, к которым они присоединены, образуют фенильное или пиридильное кольцо; RE представляет собой водород, F, Cl, -NHC(O)C1-8алифатическую группу, -OH, -OCF3, -OC1-2алифатическую группу, CF3, C1-2алифатическую группу, C3-4циклоалифатическую группу, N(CH3)2, азетидин-1-ил; R2 представляет собой водород, F, Cl, CF3, C1-2алифатическую группу, C3-4циклоалифатическую группу, -N(CH3)2, -N(CH2)3, -OCF3 или -OC1-2алифатическую группу; и R3 представляет собой водород, C1-2алкил, -OH, -OC1-2алкил, -O(CH2)2OC1-2алкил или -OC1-2алкилC(O)OC1-2алкил; и R4 представляет собой водород или C1-2алкил.
В одном варианте осуществления соединения представляют собой соединения формулы II:
или их фармацевтически приемлемые соли, где:
X1 представляет собой CH или N;
R1 выбран из фенильного кольца, 5-членного гетероарильного кольца, 6-членного гетероарильного кольца, или 9- или 10-членной конденсированной бициклической гетероарильной или гетероциклической кольцевой системы, где каждое из указанных колец или кольцевых систем необязательно замещено 1 или 2 независимо встречающимися R1a, и каждый из указанных гетероарильных или гетероциклических колец имеет 1, 2 или 3 гетероатома, выбранных из азота, кислорода или серы;
R1a представляет собой хлор, фтор, C1-6алифатическую группу, C3-6циклоалифатическую группу, -CN, -C(O)R1b, -C(O)N(R1b)2, -C(O)O(R1b) или -OR1b, где каждая из указанных алифатических или циклоалифатических групп необязательно замещена посредством вплоть до 3 встречающихся JR;
каждый JR независимо представляет собой фтор, оксо, -CN, -C(O)R1b, -C(O)N(R1b)2, -C(O)O(R1b), -N(R1b)2, -N(R1b)C(O)R1b, -OR1b, или 5-членный гетероарил или гетероциклил, имеющий вплоть до 3 атомов, выбранных из азота, кислорода или серы;
каждый R1b независимо выбран из водорода, C1-4алифатической группы или C3-6циклоалифатической группы;
R2 представляет собой водород, F, Cl, CF3 или CH3;
B представляет собой N;
C представляет собой CRC, где каждый RC представляет собой водород, фтор, хлор, C1-3алифатическую группу, CF3, -OCF3 или -OC1-2алифатическую группу; и
D представляет собой CRD, где RD представляет собой фтор, хлор, C1-3алифатическую группу, CF3, -OCF3 или -OC1-2алифатическую группу.
В одном варианте осуществления соединений формулы II, X1 представляет собой N.
В одном варианте осуществления соединений формулы II, R2 представляет собой CH3.
В следующем варианте осуществления представляет собой замещенный пиридин-3-ил.
В другом аспекте изобретение относится к соединениям формулы I-A:
или их фармацевтически приемлемым солям, где R1 представляет собой
где
R1a представляет собой -C1-4алкил, необязательно и независимо замещенный посредством -CN, вплоть до трех атомов F или вплоть до двух CH3, -OC1-2алкильной или -OH группами;
R2 представляет собой C1-2алкил;
R3 представляет собой водород, -OH, -OC1-4алкил или C1-4алкил, необязательно замещенный посредством вплоть до двух -OH групп;
R4 представляет собой водород или CH3, или R3 и R4 вместе образуют C3-6циклоалкильное кольцо, необязательно замещенной посредством вплоть до двух OH-групп, или 4-6-членное гетероциклическое кольцо, имеющее один атом кислорода или азота, необязательно замещенный C1-4алкилом, -C(O)C1-4алкилом или C(O)OC1-4алкилом;
RC представляет собой водород, F, C1-2алкил или -OC1-2алкил; и
RD представляет собой -ORD1, -C(O)N(RD1)RD2, -S(O)2N(RD1)RD2, -S(O)1-2RD2, -N(RD1)S(O)2RD2 или -N(RD1)S(O)2N(RD1)RD2, где
RD1 представляет собой водород или C1-2алкил, и RD2 представляет собой C1-4алкил, -(CH2)0-1C3-6циклоалкил или -(CH2)0-1C4-6гетероциклил, имеющий вплоть до двух атомов кислорода или азота, причем каждый алкил, циклоалкил или гетероциклил необязательно замещен посредством вплоть до трех атомов F или вплоть до двух -OH групп.
В одном варианте осуществления R1a представляет собой C1-2алкил, необязательно замещенный посредством вплоть до 3 атомов фтора.
В другом варианте осуществления R1a представляет собой C1-4алкил, необязательно замещенный CN.
В другом варианте осуществления R2 представляет собой CH3.
В другом варианте осуществления по меньшей мере один из R3 и R4 не является водородом.
В следующем варианте осуществления каждый из R3 и R4 представляет собой CH3.
В другом варианте осуществления R3 и R4 вместе образуют 4-6-членное гетероциклическое кольцо, имеющее один атом кислорода или азота, необязательно замещенный C1-4алкилом, -C(O)C1-4алкилом или -C(O)OC1-4алкилом.
В другом варианте осуществления для любого из соединений формул I, II или I-A, R1 представляет собой 5-членное гетероарильное кольцо, имеющее 1-3 гетероатома, выбранных из N, O или S, и необязательно замещенное 1 или 2 группами R1a. Примеры включают необязательно замещенную пиразол-4-ильную, пиразол-3-ильную, имидазол-4-ильную, 1,2,3-триазол-4-ильную, 1,2,4-триазол-3-ильную, 1,2,5-триазол-3-ильную, 1,3-тиазол-4-ильную, 1,3-тиазол-2-ильную, 1,2-тиазол-5-ильную, 1,2-изоксазол-3-ильную кольцевые системы.
В другом варианте осуществления R1 выбран из
В другом варианте осуществления R1 представляет собой
В другом варианте осуществления R1 представляет собой
В другом варианте осуществления R1 выбран из
В другом варианте осуществления R1 представляет собой
В одном варианте осуществления для любого из соединений формулы I, II или I-A,
R1 представляет собой
R2 представляет собой CH3;
R3 представляет собой водород, C1-2алкил, OH или OCH3;
R4 представляет собой водород или CH3;
RC представляет собой водород; и
RD представляет собой -OC1-2алкил или -OC3-5циклоалкил, каждый из которых необязательно замещен посредством вплоть до 3 атомов фтора.
В следующем варианте осуществления R1 представляет собой 1-(2,2-дифторэтил)-1H-пиразол-4-ил или 1-(2,2,2-трифторэтил)-1H-пиразол-4-ил.
В одном варианте осуществления для любого из соединений формул I, II или I-A, R1 представляет собой 6-членное гетероарильное кольцо, имеющее 1-3 атома азота и необязательно замещенное 1 или 2 группами R1a. В следующем варианте осуществления R1 представляет собой необязательно замещенное пиридильное кольцо.
В следующем варианте осуществления
R1 представляет собой
R2 представляет собой CH3;
R3 представляет собой водород, C1-2алкил, OH или OCH3;
R4 представляет собой водород или CH3;
RC представляет собой водород, F, Cl, C1-3алифатическую группу, (CH2)0-1CF3, -OCF3 или -OC1-8алифатическую группу; и
RD представляет собой -C(O)NHC1-8алифатическую группу.
В другом варианте осуществления R1 выбран из
В одном варианте осуществления для любого из соединений формул I, II или I-A, RD представляет собой -C(O)OH, -C(O)N(R1b)2, -CN, -S(O)2C1-8алифатическую группу или -S(O)2N(R1b)2.
В другом варианте осуществления каждый из RC и RD независимо представляет собой водород, фтор, хлор, C1-3алифатическую группу, CF3, -OCF3, -OCHF2 или -OC1-2алифатическую группу, где по меньшей мере один из RC и RD не является водородом.
В другом варианте осуществления RC представляет собой водород, и RD представляет собой -OC1-3алкил, необязательно замещенный посредством вплоть до 3 атомов F. В следующем варианте осуществления RC представляет собой водород, и RD представляет собой -OCH3, -OCH2CH3, -OCF3, -OCH2CF3, OCHF2 или OCH2CHF2.
В другом варианте осуществления каждый из RC и RD представляет собой -OCH3.
В одном варианте осуществления выбран из:
В другом варианте осуществления R3 и R4 вместе с промежуточным атомом углерода образуют 4-6-членное гетероциклическое кольцо, имеющее один атом, выбранный из N или O.
В другом варианте осуществления изобретение относится к соединению, выбранному из группы соединений, приведенных в таблице 1.
Также изобретение относится к фармацевтической композиции, содержащей соединение по изобретению и фармацевтически приемлемый носитель, адъювант или наполнитель.
В одном варианте осуществления композиция включает терапевтическое средство, выбранное из средства для лечения рассеянного склероза, противовоспалительного средства, иммуномодулирующего средства или иммунодепрессивного средства. Примеры таких дополнительных терапевтических средств включают бета-интерферон, глатирамир, натализумаб или митоксантрон.
В другом варианте осуществления изобретение относится к способу ингибирования активности PI3K-киназы у пациента путем введения пациенту соединения формулы I, II или I-A, или его фармацевтической композиции. В следующем варианте осуществления PI3K-гамма селективно ингибируется среди PI3K-альфа, PI3K-бета или PI3K-гамма. В следующем варианте осуществления PI3K-гамма селективно ингибируется среди PI3K-альфа, PI3K-бета и PI3K-гамма.
В другом варианте осуществления изобретение относится к способу лечения или уменьшения тяжести заболевания или состояния, выбранного из аутоиммунного заболевания или воспалительного заболевания головного мозга или спинного мозга, выбранного из рассеянного склероза, эпилепсии, болезни Паркинсона, болезни Альцгеймера, болезни Гентингтона или бокового амиотрофического склероза у пациента путем введения пациенту соединения формулы I, II или I-A, или его фармацевтической композиции.
В следующем варианте осуществления заболевание или нарушение представляет собой рассеянный склероз.
В другом варианте осуществления способ лечения включает введение пациенту соединения или композиции по изобретению и дополнительного терапевтического средства, где дополнительное терапевтическое средство пригодно для подвергаемого лечению заболевания и его вводят вместе с соединением или композицией в качестве единичной дозированной формы или отдельно в качестве части множественной дозированной формы. Примерами таких дополнительных терапевтических средств являются средства, пригодные для лечения рассеянного склероза, такие как бета-интерферон, глатирамир, натализумаб или митоксантрон.
Также изобретение относится к нетерапевтическому способу ингибирования активности киназы PI3K-гамма в биологическом образце in vitro, включающему контактирование указанного биологического образца с соединением формулы I, II или I-A, или композицией, содержащей указанное соединение.
Композиции, составы и введение соединений по изобретению
В другом варианте осуществления изобретение относится к фармацевтической композиции, содержащей соединение согласно любой из формул или классов, описанных в настоящем описании. В следующем варианте осуществления изобретение относится к фармацевтической композиции, содержащей соединение, представленное в таблице 1. В следующем варианте осуществления композиция, кроме того, содержит дополнительное терапевтическое средство.
Согласно другому варианту осуществления изобретение относится к композиции, содержащей соединение по изобретению или его фармацевтически приемлемое производное и фармацевтически приемлемый носитель, адъювант или наполнитель. В одном варианте осуществления количество соединения в композиции по изобретению является таким, что оно является эффективным для подающегося измерению ингибирования PI3K, в частности PI3Kγ, в биологическом образце или у пациента. В другом варианте осуществления количество соединения в композициях по изобретению является таким, что оно является эффективным для поддающегося измерению ингибирования PI3Kα. В одном варианте осуществления композиция по изобретению изготовлена для введения пациенту, нуждающемуся в такой композиции. В следующем варианте осуществления композиция по изобретению изготовлена для перорального введения пациенту.
Термин "пациент", как используют в рамках изобретения, означает животного, предпочтительно млекопитающего, и наиболее предпочтительно человека.
Также следует понимать, что определенные соединения по настоящему изобретению могут существовать в свободной форме для лечения, или, когда это целесообразно, в качестве их фармацевтически приемлемых производных. В соответствии с настоящим изобретением, фармацевтически приемлемое производное включает, но не ограничивается ими, фармацевтически приемлемые пролекарства, соли, сложные эфиры, соли таких сложных эфиров, или любой другой аддукт или производное, которые при введении пациенту, нуждающемуся в этом, способны предоставлять, прямо или непрямо, соединение, описанное в настоящем описании, или метаболит или его остаток. Как используют в рамках изобретения, термин "активный в отношении ингибирования метаболит или его остаток" означает, что метаболит или его остаток также является ингибитором PI3K.
Как используют в рамках изобретения, термин "фармацевтически приемлемая соль" относится к солям, которые являются пригодными, с медицинской точки зрения, для применения в контакте с тканями людей и низших животных без излишней токсичности, раздражения или аллергического ответа и т.п.
Фармацевтически приемлемые соли хорошо известны в данной области. Например, S. M. Berge et al., описывают подробно фармацевтически приемлемые соли в работе J. Pharmaceutical Sciences, 66: 1-19, 1977, которая включена в настоящий документ в качестве ссылки. Фармацевтически приемлемые соли соединений по изобретению включают соли, образованные из пригодных неорганических и органических кислот и оснований. Примерами фармацевтически приемлемых, нетоксичных кислотно-аддитивных солей являются соли аминогруппы, образованные с неорганическими кислотами, такими как хлористоводородная кислота, бромистоводородная кислота, фосфорная кислота, серная кислота и перхлорная кислота или с органическими кислотами, такими как уксусная кислота, щавелевая кислота, малеиновая кислота, виннокаменная кислота, лимонная кислота, янтарная кислота или малоновая кислота, или с использованием других способов, используемых в данной области, таких как ионный обмен. Другие фармацевтически приемлемые соли включают: адипаты, альгинаты, аскорбаты, аспартаты, бензолсульфонаты, бензоаты, бисульфаты, бораты, бутираты, камфораты, камфорсульфонаты, цитраты, циклопентанпропионаты, диглюконаты, додецилсульфаты, этансульфонаты, формиаты, фумараты, глюкогептаноаты, глицерофосфаты, глюконаты, гемисульфаты, гептаноаты, гексаноаты, гидройодиды, 2-гидроксиэтансульфонаты, лактобионаты, лактаты, лаураты, лаурилсульфаты, малаты, малеаты, малонаты, метансульфонаты, 2-нафталинсульфонаты, никотинаты, нитраты, олеаты, оксалаты, пальмитаты, памоаты, пектинаты, персульфаты, 3-фенилпропионаты, фосфаты, пикраты, пивалаты, пропионаты, стеараты, сукцинаты, сульфаты, тартраты, тиоцианаты, п-толуолсульфонаты, ундеканоаты, валераты и т.п. Соли, происходящие пригодных оснований, включают соли щелочных металлов, щелочноземельных металлов, аммония и N+(C1-4алкил)4-соли. Также изобретение предусматривает кватернизацию любой их основных азотсодержащих групп соединений, описанных в настоящем описании. Путем такой кватернизации можно получать растворимые или диспергируемые в воде или масле продукты. Типичные соли щелочных или щелочноземельных металлов включают соли натрия, лития, калия, кальция, магния и т.п. Другие фармацевтически приемлемые соли включают, когда это является пригодным, нетоксические соли аммония, четвертичного аммония и катионов аминов, образованных с использованием противоионов, такие как галогенид, гидроксид, карбоксилат, сульфат, фосфат, нитрат, C1-8сульфонат и арилсульфонат.
Как описано выше, фармацевтически приемлемые композиции по настоящему изобретению, кроме того, содержат фармацевтически приемлемый носитель, адъювант или наполнитель, который, как используют в рамках изобретения, включает любые и все растворители, разбавители или другие жидкие носители, дисперсионные или суспензионные добавки, поверхностно-активные вещества, обеспечивающие изотоничность вещества, загустители или эмульгаторы, консерванты, твердые связующие вещества, смазывающие вещества и т.п., пригодные для конкретной желаемой дозированной формы. В Remington: The Science и Practice of Pharmacy, 21st edition, 2005, ed. D.B. Troy, Lippincott Williams & Wilkins, Philadelphia, and Encyclopedia of Pharmaceutical Technology, eds. J. Swarbrick and J. C. Boylan, 1988-1999, Marcel Dekker, New York, содержание каждой из которых включено в настоящий документ в качестве ссылки, описаны различные носители, используемые для изготовления фармацевтически приемлемых композиций и известные способы их получения. За исключением случаев, когда какой-либо общепринятый носитель несовместим с соединениями по изобретению, например, вследствие возникновения какого-либо нежелательного биологического эффекта или в ином случае взаимодействия неблагоприятным образом с другим компонентом(ами) фармацевтически приемлемой композиции, его применение предусмотрено в объеме этого изобретения.
Некоторые примеры материалов, которые могут служить в качестве фармацевтически приемлемых носителей, включают, но не ограничиваются ими, ионообменные вещества, оксид алюминия, стеарат алюминия, лецитин, сывороточные белки, такие как сывороточный альбумин человека, буферные вещества, такие как фосфаты, глицин, сорбиновая кислота или сорбат калия, смеси неполных глицеридов насыщенных растительных жирных кислот, воду, соли или электролиты, такие как сульфат протамина, гидрофосфат натрия, дигидрофосфат калия, хлорид натрия, соли цинка, коллоидный диоксид кремния, трисалицилат магния, поливинилпирролидон, полиакрилаты, воски, блок-сополимеры полиэтилен-полиоксипропилен, ланолин, сахара, такие как лактоза, глюкоза и сахароза; крахмалы, такие как кукурузный крахмал и картофельный крахмал; целлюлозу и ее производные, такие как карбоксиметилцеллюлоза натрия, этилцеллюлоза и ацетат целлюлозы; порошковый трагакант; солод; желатин; тальк; эксципиенты, такие как масло какао и воски для суппозиториев; масла, такие как арахисовое масло, хлопковое масло; сафлоровое масло; кунжутное масло; оливковое масло; кукурузное масло и соевое масло; гликоли, такие как пропиленгликоль или полиэтиленгликоль; сложные эфиры, такие как этилолеат и этиллаурат; агар; буферные средства, такие как гидроксид магния и гидроксид алюминия; альгиновую кислоту; не содержащую пирогенов воду; изотонический солевой раствор; раствор Рингера; этиловый спирт и фосфатные буферные растворы, а также другие нетоксичные совместимые смазывающие вещества, такие как лаурилсульфат натрия и стеарат магния, а также красители, вещества для высвобождения, вещества для покрытия, подсластители, вкусовые добавки и отдушки, консерванты и антиоксиданты, также могут присутствовать в композиции в соответствии с мнением лица, составляющего композицию.
Композиции по настоящему изобретению можно вводить перорально, парентерально, с помощью ингаляционного аэрозоля, местно, ректально, назально, буккально, вагинально или через имплантированный резервуар. Термин "парентеральный", как используют в рамках изобретения, включает подкожный, внутривенный, внутримышечный, внутрисуставной, внутрисиновиальный, внутригрудинный, интратекальный, внутриглазеной, внутрипеченочный, проводимый внутрь очага повреждения, эпидуральный, внутриспинномозговой и внутричерепной способы инъекции или инфузии. Предпочтительно, композиции вводят перорально, внутрибрюшинно или внутривенно. Стерильные инъецируемые формы композиций по изобретению могут представлять собой водную или маслянистую суспензию. Эти суспензии могут быть изготовлены в соответствии со способами, известными в данной области, с использованием пригодных диспергирующих или смачивающих веществ и суспендирующих веществ. Стерильный инъецируемый препарат также может представлять собой стерильный инъецируемый раствор или суспензию в нетоксичном парантерально приемлемом разбавителе или растворителе, например раствор в 1,3-бутандиоле. Среди приемлемых наполнителей и растворителей, которые можно использовать, находятся вода, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, в качестве растворителя или суспендирующей среды традиционно используют стерильные жирные масла.
Для этой цели можно использовать любое легкое жирное масло, включая синтетические моно- или диглицериды. Жирные кислоты, такие как олеиновая кислота и ее глицеридные производные, пригодны для получения инъецируемых препаратов, также как природные фармацевтически приемлемые масла, такие как оливковое масло или касторовое масло, особенно в их полиоксиэтилированных версиях. Эти масляные растворы или суспензии также могут содержать разбавитель или диспергирующее вещество на основе спирта длинной цепи, такое как карбоксиметилцеллюлоза или сходные диспергирующие вещества, которые обычно используют при изготовлении фармацевтически приемлемых дозированных форм, включая эмульсии и суспензии. Также для изготовления состава можно использовать другие широко используемые поверхностно-активные вещества, такие как Tween, Span и другие эмульгаторы или усилители биодоступности, которые обычно используются при изготовлении фармацевтически приемлемых твердых, жидких или иных дозированных форм.
Фармацевтически приемлемые композиции по изобретению можно вводить перорально в любой перорально приемлемой дозированной форме, включая, но не ограничиваясь ими, капсулы, таблетки, водные суспензии или растворы. В случае таблеток для перорального применения, обычно используемые носители включают лактозу и кукурузный крахмал. Также обычно добавляют смазывающие вещества, такие как стеарат магния. Для перорального введения в форме капсулы, пригодные разбавители включают лактозу и сухой кукурузный крахмал. Когда для перорального применения требуются водные суспензии, активный ингредиент комбинируют с эмульгирующими и суспендирующими веществами. Если желательно, также можно добавлять определенные подсластители, вкусовые добавки или красители.
Альтернативно фармацевтически приемлемые композиции по изобретению можно вводить в форме суппозиториев для ректального введения. Их можно получать путем смешения средства с пригодным не вызывающим раздражения эксципиентом, который является твердым при комнатной температуре, но жидким при ректальной температуре и, таким образом, плавится в прямой кишке, высвобождая терапевтическое средство. Такие материалы включают масло какао, пчелиный воск и полиэтиленгликоли.
Фармацевтические композиции по этому изобретению также можно вводить местно, особенно когда мишень для лечения включает области или органы, легкодоступные при местном применении, включая заболевания глаза, кожи или нижнего отдела кишечника. Пригодные составы для местного введения легко получают для каждой из этих областей или органов.
Местное применение для нижнего отдела кишечника можно осуществлять с помощью состава ректального суппозитория (см. выше) или в пригодном составе для применения с помощью клизмы. Также можно использовать местные чрескожные пластыри.
Для местного применения фармацевтически приемлемые композиции можно изготавливать в форме пригодной мази, содержащей активный компонент, суспендированный или растворенный в одном или нескольких носителях. Носители для местного введения соединений по изобретению включают, но не ограничиваются ими, минеральное масло, жидкий вазелин, белый вазелин, пропиленгликоль, полиоксиэтилен, соединение полиоксипропилена, эмульгирующий воск и воду. Альтернативно, фармацевтические композиции можно изготавливать в виде пригодного лосьона или крема, содержащего активные компоненты, суспендированные или растворенные в одном нескольких фармацевтически приемлемых носителях. Пригодные носители включают, но не ограничиваются ими, минеральное масло, моностеарат сорбитана, полисорбат 60, воск на основе цетиловых сложных эфиров, цетеариловый спирт, 2 октилдодеканол, бензиловый спирт и воду.
Для офтальмологического применения, фармацевтически приемлемые композиции могут быть изготовлены в форме микронизированных суспензий в изотоническом стерильном физиологическом растворе со скорректированным значением pH или в другом водном растворе, или, предпочтительно, в качестве растворов в изотоническом стерильном физиологическом растворе со скорректированным значением pH или другом водном растворе, либо с консервантом, таким как хлорид бензалкония, либо без него. Альтернативно, для офтальмологического применения фармацевтические композиции можно изготавливать в форме мази, такой как вазелин. Фармацевтически приемлемые композиции по изобретению также можно вводить при помощи назального аэрозоля или ингаляции. Такие композиции получают способами, хорошо известными в области изготовления фармацевтических препаратов, и их можно получать в форме растворов в физиологическом растворе с использованием бензилового спирта или других пригодных консервантов, усиливающих всасывание средств для повышения биодоступности, фторуглеродов и/или других общепринятых растворяющих или диспергирующих веществ.
Наиболее предпочтительно, фармацевтически приемлемые композиции по изобретению изготавливают для перорального введения.
Жидкие дозированные формы для перорального введения включают, но не ограничиваются ими, фармацевтически приемлемые эмульсии, микроэмульсии, растворы, суспензии, сиропы и эликсиры. В дополнение к активным соединениям, жидкие дозированные формы могут содержать инертные разбавители, обычно используемые в данной области, например, такие как вода или другие растворители, солюбилизирующие вещества и эмульгаторы, такие как этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, диметилформамид, масла (в частности, хлопковое, арахисовое, кукурузное масла, масло из зародышей, оливковое, касторовое и кунжутное масла), глицерин, тетрагидрофурфуриловый спирт, полиэтиленгликоли и сложные эфиры жирных кислот и сорбитана, и их смеси. Помимо инертных разбавителей, пероральные композиции также могут включать адъюванты, такие как смачивающие вещества, эмульгирующие и суспендирующие вещества, подсластители, вкусовые добавки и отдушки.
Инъецируемые препараты, например, стерильные инъецируемые водные или масляные суспензии можно изготавливать известными способами с использованием пригодных диспергирующих или смачивающих веществ и суспендирующих веществ. Стерильный инъецируемый препарат также может представлять собой стерильный инъецируемый раствор, суспензию или эмульсию в нетоксичном парентерально приемлемом разбавителе или растворителе, например, раствор в 1,3-бутандиоле. Среди приемлемых носителей и растворителей, которые можно использовать, находятся вода, раствор Рингера, U.S.P. и изотонический раствор хлорида натрия. Кроме того, в качестве растворителя или суспендирующей среды традиционно используют стерильные жирные масла. Для этой цели можно использовать любое легкое жирное масло, включая синтетические моно- или диглицериды. Кроме того, для получения инъецируемых препаратов используют жирные кислоты, такие как олеиновая кислота.
Инъецируемые составы можно стерилизовать, например, фильтрацией через сдерживающий бактерии фильтр, или путем включения стерилизующих средств в форме стерильных твердых композиций, которые могут быть растворены или диспергированы в стерильной воде или другом стерильном носителе для инъекций перед применением.
Для пролонгирования эффекта соединения по настоящему изобретению часто желательно замедлить всасывание соединения из области подкожной или внутримышечной инъекции. Это можно осуществлять с использованием жидкой суспензии кристаллического или аморфного материала с плохой растворимостью. В этом случае уровень всасывания соединения зависит от скорости растворения, которая, в свою очередь, может зависеть от размера кристаллов и кристаллической формы. Альтернативно растворение или суспендирование соединения в масляном носителе обеспечивает замедленное всасывание парентерально вводимой формы соединения. Инъецируемые депо-формы получают путем формирования микроинкапсулированных матриц соединения в биодеградируемые полимеры, такие как полилактид-полигликолид. В зависимости от соотношения соединения и полимера и характеристик конкретного используемого полимера, можно контролировать скорость высвобождения соединения. Примеры других биодеградируемых полимеров включают сложные поли(ортоэфиры) и поли(ангидриды). Инъецируемые депо-составы также получают путем заключения соединения в липосомы или микроэмульсии, которые совместимы с тканями организма.
Композиции для ректального или вагинального введения предпочтительно представляют собой суппозитории, которые можно получать смешением соединений по изобретению с пригодными не вызывающими раздражения эксципиентами или носителями, такими как масло какао, полиэтиленгликоль или воск для суппозиториев, которые являются твердыми при температуре окружающей среды, но жидкими при температуре тела и, таким образом, плавятся в прямой кишке или вагинальной полости и высвобождают активное соединение.
Твердые дозированные формы для перорального введения включают капсулы, таблетки, пилюли, порошки и гранулы. В таких твердых дозированных формах активное соединение смешивают по меньшей мере с одним инертным фармацевтически приемлемым эксципиентом или носителем, таким как цитрат натрия или дикальций фосфат и/или a) наполнителями или сухими разбавителями, такими как крахмалы, лактоза, сахароза, глюкоза, маннит и кремниевая кислота, b) связующими веществами, например, такими как карбоксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидинон, сахароза и гуммиарабик, c) смачивающими веществами, такими как глицерин, д) дезинтегрирующими веществами, такими как агар-агар, карбонат кальция, картофельный или маниоковый крахмал, альгиновая кислота, определенные силикаты и карбонат натрия, e) удерживающими раствор веществами, такими как парафин, f) ускоряющими всасывание веществами, такими как четвертичные соединения аммония, g) смачивающими веществами, например, такими как цетиловый спирт и моностеарат глицерина, h) абсорбентами, такими как каолин и бентонитовая глина, и i) смазывающими веществами, такими как тальк, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, лаурилсульфат натрия и их смеси. В случае капсул, таблеток и пилюль, дозированная форма также содержит буферные агенты.
Твердые композиции сходного типа также можно использовать в качестве наполнителей в мягких и твердых заполненных желатиновых капсулах с использованием таких эксципиентов, как лактоза или молочный сахар, а также высокомолекулярные полиэтиленгликоли и т.п. Твердые дозированные формы в виде таблеток, драже, капсул, пилюль и гранул можно получать с покрытиями и оболочками, такими как желудочно-резистентные оболочки и другие покрытия, хорошо известные в области изготовления фармацевтических составов. Они необязательно могут содержать замутняющие вещества и также могут иметь такую композицию, чтобы высвобождать активный ингредиент(ы) только, или предпочтительно, в определенной части желудочно-кишечного тракта, необязательно, замедленным образом. Примеры композиций для покрытия, которые можно использовать, включают полимерные вещества и воски. Твердые композиции сходного типа также можно использовать в качестве наполнителей в мягких и твердых заполненных желатиновых капсулах с использованием таких эксципиентов, как лактоза или молочный сахар, а также высокомолекулярные полиэтиленгликоли и т.п.
Активные соединения также могут быть в микроинкапсулированной форме с одним или несколькими эксципиентами, как указано выше. Твердые дозированные формы таблеток, драже, капсул, пилюль и гранул можно получать с покрытиями и оболочками, такими как желудочно-резистинтные покрытия, контролирующие высвобождение покрытия и другие покрытия, хорошо известные в области изготовления фармацевтических составов. В таких твердых дозированных формах активное соединение можно смешивать по меньшей мере с одним инертным разбавителем, таким как сахароза, лактоза или крахмал. Такие дозированные формы также могут содержать, согласно обычной практике, дополнительные вещества, отличные от инертных разбавителей, например, смазывающие вещества для таблетирования и другие добавки для таблетирования, такие как стеарат магния и микрокристаллическая целлюлоза. В случае капсул, таблеток и пилюль, дозированные формы также могут содержать буферные средства. Они необязательно могут содержать замутняющие вещества и также могут иметь композицию, чтобы высвобождать активный ингредиент(ы) только, или предпочтительно, в определенной части кишечного тракта, необязательно, замедленным образом. Примеры композиций покрытия, которые можно использовать, включают полимерные вещества и воски.
Дозированные формы для местного или чрескожного введения соединения по изобретению включают мази, пасты, кремы, лосьоны, гели, порошки, растворы, спреи, ингалируемые средства или пластыри. Активный компонент смешивают в стерильных условиях с фармацевтически приемлемым носителем и любыми необходимыми консервантами или буферами, если требуется. Также в объем изобретения входят офтальмологические составы, ушные капли и глазные капли. Кроме того, настоящее изобретение охватывает применение чрескожных пластырей, которые имеют дополнительное преимущество обеспечения контролируемой доставки соединения в организм. Такие дозированные формы можно получать путем растворения или диспергирования соединения в надлежащей среде. Также можно использовать усиливающие всасывание вещества для увеличения потока соединения через кожу. Скорость можно контролировать либо путем предоставления контролирующей скорость мембраны, либо путем диспергирования соединения в полимерной матрице или геле.
Соединения по изобретению предпочтительно изготавливают в единичной дозированной форме для простоты введения и единообразия дозировки. Выражение "единичная дозированная форма", как используют в рамках изобретения, относится к физически дискретной единице вещества, пригодной для пациента, подлежащего лечению. Однако понятно, что общее суточное применение соединений и композиций по настоящему изобретению будет определяться лечащим врачом, в соответствии с медицинской точкой зрения. Конкретный уровень эффективной дозы для любого конкретного пациента или организма, зависят от различных факторов, включающих нарушение, подвергаемое лечению и тяжесть нарушения; активность конкретного используемого соединения; конкретной используемой композиции; возраста, массы тела, общего состояния здоровья, пола и режима питания пациента; времени введения, пути введения и скорости экскреции конкретного используемого соединения; длительности лечения; терапевтических средств, используемых в комбинации или по совпадению с конкретным используемым соединением, и сходных факторов, хорошо известных в области медицины.
Количество соединений по настоящему изобретению, которое можно комбинировать с материалами носителей для получения композиции в единичной дозированной форме, может варьировать, в зависимости от хозяина, подвергаемого лечению, или конкретного способа введения. Предпочтительно, композиции должны быть изготовлены так, чтобы дозировку 0,01-100 мг/кг массы тела/сутки можно было вводить пациенту, получающему эти композиции.
В зависимости от конкретного состояния или заболевания, подлежащего лечению или профилактике, также в композициях по изобретению могут присутствовать дополнительные терапевтические средства, которые обычно вводят для лечения или профилактики состояния. Как используют в рамках изобретения, дополнительные терапевтические средства, которые обычно вводят для лечения или профилактики конкретного заболевания или состояния, известны как "пригодные для заболевания или состояния, подлежащего лечению". Примеры дополнительных терапевтических средств предоставлены ниже.
Количество дополнительного терапевтического средства, присутствующего в композициях по изобретению, не превышает количество, которое обычно вводят в композиции, содержащей это терапевтическое средство в качестве единственного активного вещества. Предпочтительно количество дополнительного терапевтического средства в композициях по настоящему изобретению находится в диапазоне приблизительно от 50% до 100% от количества, обычно присутствующего в композиции, содержащей это средство в качестве единственного терапевтически активного средства.
Применения соединений и композиций по изобретению
В одном аспекте изобретения изобретение относится к способу лечения или снижения тяжести опосредуемого PI3K состояния или заболевания в головном мозге или спинном мозге пациента. Термин "опосредуемое PI3K заболевание", как используют в рамках изобретения, означает любое заболевание или другое вредоносное состояние, известное тем, что при нем изоформа PI3K играет роль. В одном варианте осуществления изоформа PI3K представляет собой PI3Kγ.
Таким образом, в одном варианте осуществления изобретение относится к способу лечения опосредуемого PI3Κγ заболевания центральной нервной системы. Такие состояния включают, но не ограничиваются ими, воспалительные заболевания и связанные с аутоиммунными процессами заболевания центральной нервной системы. Таким образом, изобретение относится к способу лечения или снижения тяжести заболевания или состояния, выбранного из аутоиммунного заболевания или воспалительного заболевания центральной нервной системы пациента, включающему введение указанному пациенту соединения или композиции по изобретению.
В следующем варианте осуществления соединение по изобретению является селективным в отношении ингибирования PI3Κγ-изоформы. В одном варианте осуществления соединения по изобретению является более селективными в отношении ингибирования изоформы PI3K-гамма относительно изоформы PI3K-альфа в анализе in vitro по меньшей мере в 20 раз. В другом варианте осуществления селективные в отношении PI3Κγ соединения по изобретению ингибируют гамма-изоформу относительно каждой из альфа-, бета- и дельта-изоформ в анализе in vitro по меньшей мере в 3 раза. В другом варианте осуществления PI3Κγ-селективные соединения по изобретению ингибируют гамма-изоформу относительно каждой из альфа-, бета- и дельта-изоформ в анализе in vitro по меньшей мере в 5 раз. В другом варианте осуществления PI3Κγ-селективные соединения по изобретению ингибируют гамма-изоформу относительно каждой из альфа-, бета- и дельта-изоформ в анализе in vitro по меньшей мере в 10 раз.
В другом варианте осуществления изобретение относится к способу лечения или снижения тяжести воспалительного или аутоиммунного заболевания или нарушения центральной нервной системы. В другом варианте осуществления изобретение относится к способу лечения или снижения тяжести симптома воспалительного или аутоиммунного заболевания или нарушения центральной нервной системы. В следующем варианте осуществления изобретение относится к способу лечения нейровоспаления. Такие заболевания или нарушения включают, но не ограничиваются ими, рассеянный склероз, поперечный миелит, прогрессирующую многоочаговую лейкоэнцефалопатию, менингит, энцефалит, миелит, энцефаломиелит, внутричерепной или внутрипозвоночный абсцесс, флебит или тромбофлебит внутричерепных венозных синусов, болезнь Паркинсона, болезнь Альцгеймера, болезнь Гентингтона, болезнь Пика, боковой амиотрофический склероз, связанная с ВИЧ типа I деменция, лобно-височная деменция, или травматическое повреждение головного мозга или спинного мозга.
Также было описано, что PI3Κγ является важной при заболеваниях или нарушениях, характеризующихся активацией резидентных клеток, таких как микроглия. См. Jin et al., 2010, Biochemical and Biophysical Research Communications 399:458-464. Микроглия может усиливать нарушение целостности гематоэнцефалического барьера (BBB) и ставить под угрозу выживание нейронов вследствие высвобождения активных форм кислорода или провоспалительных цитокинов и других нейротоксинов. Ингибирование активации микроглии может защитить головной мозг после ишемического инсульта, а также других нервно-сосудистых нарушений, таких как рассеянный склероз.
Соединения или композиции по изобретению можно вводить с одним или несколькими дополнительными терапевтическими средствами, где дополнительное терапевтическое средство пригодно для подвергаемого лечению заболевания и дополнительное терапевтическое средство вводят вместе с соединением или композицией по изобретению в качестве единой дозированной формы или отдельно от соединения или композиции в качестве части множественной дозированной формы. Дополнительное терапевтическое средство можно вводить в то же время, что и соединение по изобретению или в другое время. В последнем случае введение может быть разделено, например, периодом 6 часов, 12 часов, 1 сутки, 2 суток, 3 суток, 1 неделя, 2 недели, 3 недели, 1 месяц или 2 месяца.
Неограничивающие примеры химиотерапевтических средств или других антипролиферативных средств, которые можно комбинировать с соединениями по изобретению, включают таксаны, ингибиторы ароматазы, антрациклины, нацеленные на микротрубочки лекарственные средства, лекарственные средства, направленные против топоизомеразы, нацеленные моноклональные или поликлональные антитела, ингибиторы молукулярной мишени или фермента (например, ингибитор киназы), или аналоги цитидина. В одном варианте осуществления дополнительное химиотерапевтическое средство представляет собой амсакрин, анастрозол, аспарагиназа, AvastinTM (бевацизумаб) азатиоприн, бикалутамид, блеомицин, камптотецин, кармустин, хлорамбуцил, циклофосфамид, цитарабин (araC), даунонибицин, дактиномицин, доксорубицин (адриамицин), эпирубицин, эпотилон, этопозид, экземестан, флударабин, 5-фторурацил (5-FU), флутамид, GemzarTM (гемцитабин), GleevecTM (иматаниб), HerceptinTM (трастузумаб), идарубицин, ифосфамид, интерферон, интерлейкин, иринотекан, лектрозол, леупролид, ломустин, ловастатин, мехлорэтамин, магестрол, мелфалан, 6-меркаптопурин, метотрексат (MTX), минозин, митомицин, митоксантрон, навелбин, нокодазол, производные платины, такие как цисплатин, карбоплатин и оксалиплатин, ралоксифен, тамоксифен, TaxotereTM (доцетаксел), TaxolTM (паклитаксел), тенипозид, топотекан, фактор некроза опухоли (TNF), винбластин, винкристсин, виндезин, винорелбин или ZoladexTM (гозерелин). Другое химиотерапевтическое средство также может представлять собой цитокин, такой как G-CSF (гранулоцитарный колониестимулирующий фактор). В другом варианте осуществления соединение по настоящему изобретению, или его фармацевтически приемлемая соль, пролекарство, метаболит, аналог или производное, можно вводить в комбинации с хирургической операцией, лучевой терапией или со стандартными химиотерапевтическим комбинациями, такими как, но не ограничиваясь ими, CMF (циклофосфамид, метотрексат и 5-фторурацил), CAF (циклофосфамид, адриамицин и 5-фторурацил), AC (адриамицин и циклофосфамид), FEC (5-фторурацил, эпирубицин, и циклофосфамид), ACT или ATC (адриамицин, циклофосфамид и паклитаксел), или CMFP (циклофосфамид, метотрексат, 5- фторурацил и преднизон).
Дополнительные терапевтические средства также включают средства, пригодные для лечения рассеянного склероза (MS), например, такие как бета-интерферон (например, Avonex® и Rebif®), глатирамир (Copaxone®), Tysabri® (натализумаб), Betaseron® (IFN-бета) и митоксантрон.
Изобретение относится к способу ингибирования активности PI3K-киназы в биологическом образце, который включает контактирование биологического образца с соединением или композицией по изобретению. Термин "биологический образец", как используют в рамках изобретения, означает образец вне живого организма и включает, но не ограничивается ими, клеточные культуры и их экстракты; биоптат, полученный от млекопитающего, или его экстракты; и кровь, слюну, мочу, фекалии, семенную жидкость, слезную жидкость или другие жидкости организма или их экстракты. Ингибирование активности киназы, в частности, активности PI3K-киназы, в биологическом образце, пригодно для различных целей, известных специалисту в данной области. Примеры таких целей включают, но не ограничиваются ими, хранение биологического образца и биологические анализы. В одном варианте осуществления способ ингибирования активности PI3K-киназы в биологическом образце ограничивается нетерапевтическими способами.
Получение соединений по изобретению
Как используют в рамках изобретения, все сокращения, символы и условия согласуются с теми, которые используются в современной научной литературе. См., например, Janet S. Dodd, ed., The ACS Style Guide: A Manual for Authors и Editors, 2nd Ed., Washington, D.C.: American Chemical Society, 1997. Следующие определения описывают термины и сокращения, используемые в настоящем описании:
Общие методики синтеза
Главным образом, соединения по изобретению можно получать способами, описанными в настоящем описании, или другими способами, известными специалистам в данной области.
Пример 1. Общий способ получения соединений формулы I
Соединения формулы I можно получать, как показано на схеме 1, пути A-K, где каждый из A, B, C, D, E, X1, X2, R1 и R2 является таким, как описано в настоящем описании, и каждый из R3 и R4 представляет собой H. Как показано на пути A схемы, для соединений формулы I, где X представляет собой CH, арилгалогенид формулы A1 обрабатывают N-бромсукцинимидом (NBS) с получением бромалкильных соединений формулы A2. Последующая реакция бромалкильных соединений с аминами формулы A3 дает 5-галогенизоиндолиноны формулы A4. Реакция соединения формулы A4 с бороновой кислотой или боронатом формулы A5 в присутствии соответствующего палладиевого катализатора дает соединения формулы I, где каждый из X1 и X2 представляет собой CH. Методики получения бороната или бороновой кислоты из арил- или гетероарилгалогенидов описаны в Boronic Acids, ISBN: 3-527-30991-8, Wiley-VCH, 2005 (Dennis G. Hall, редактор). В одном примере галоген представляет собой бром и боронат получают путем реакции арил- или гетероарилбромида с 4,4,5,5-тетраметил-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,3,2-диоксабороланом. В последующих реакциях сочетания боронаты или бороновые кислоты, полученные таким образом, можно подвергать реакции с арил- или гетероарилгалогенидами в присутствии 1'1-бис(дифенилфосфино)ферроцендихлорпалладий-дихлорметана (dppfPdCl2).
Как показано на пути B, последовательность стадий, указанную выше, можно изменять так, чтобы соединения формулы A1 сначала реагировали с соединениями A5, с последующим образованием алкилбромидов A7, а затем реакцией алкилбромидов с аминами формулы A3 с получением соединений формулы I, где X представляет собой CH, и R5 представляет собой водород.
Схема 1, Пути А и В
Как показано на пути C, для получения соединений формулы I, где X1 представляет собой N, соединения формул A8 можно подвергать реакции с 1,3,5-трихлор-1,3,5-триазинан-2,4,6-трионом с образованием хлоралкильных соединений формулы A9. Затем соединения формулы A9 обрабатывают окислителями, такими как мета-хлорпербензойная кислота, с образованием N-оксидов формулы A10, которые затем обрабатывают оксихлоридом фосфора с получением пиридилхлоридов формулы A11. Реакция соединения формулы A11 с амином формулы A3 дает 5-хлоразаизоиндолинон формулы A12. Реакция соединения формулы A12 с бороновой кислотой или боронатом формулы A5 в присутствии соответствующего палладиевого катализатора дает соединение формулы I, где X1 представляет собой N, и X2 представляет собой CH или C-CH3.
Схема 1, Путь С
Соединения формулы I, где X1 представляет собой CH или N, X2 представляет собой N, и каждый из R3 и R4 представляет собой водород, также можно получать аналогично тому, как описано для пути C, как показано на пути D.
Схема 1, Путь D
Соединения формулы I, где X1 представляет собой CH, X2 представляет собой N, и каждый из R3 и R4 представляет собой водород, можно получать, как показано на пути E. Таким образом, амины формулы подвергают реакции с галогенметилацетиленом с получением соединений формулы A18, которые затем подвергают реакции с карбоксиацетиленами с получением соединений формулы A19. Реакция с этилкарбонизоцианитидатом в присутствии хлор(пентаметилциклопентадиенил)(циклооктадиен)рутения(II) дает соединения формулы A20. Кислотный гидролиз и декарбоксилирование сложного этоксиэфира с последующим образованием хлорпиридина через оксихлорид фосфора дает соединения формулы A21. Реакция с боронатами формулы A4 в каталитических условиях кросс-сочетания дает соединения формулы I.
Схема 1, Путь Е
Соединения формулы I, где арил- или гетероарилгалогенид используют для внесения R1, можно получать, как показано на пути F. Таким образом, амиды формулы A23 (полученные аминолизом соединений формулы A22) подвергают реакции с арилхлоридами, арилбромидами, арилйодидами или арилтрифлатами в присутствии медного или палладиевого катализатора с получением соединений формулы I.
Схема 1, Путь F
Как показано на пути G, соединения формулы I, где R1 представляет собой замещенный пиразол, также можно получать реакцией соединений формулы A25 в основных условиях алкилирующим агентом формулы R1a-X, где X представляет собой галогенидную или сульфонатную уходящую группу.
Схема 1, Путь G
Как показано на пути H, реакция пирролопиридиндионов формулы A26 с восстановителем, таким как Zn/уксусная кислота, дает спирты формулы A27. Эти спирты затем можно алкилировать алкилирующим агентом (например, L представляет собой уходящую группу, такую как сульфонат или галогенид, и R3' представляет собой алифатическую группу, циклоалифатическую группу или гетероциклическую группу) в присутствии основания с получением соединений формулы A28. Затем соединения формулы A28 обрабатывают окислителями, такими как мета-хлорпербензойная кислота, с получением N-оксидов формулы A29, которые затем обрабатывают оксихлоридом фосфора с получением пиридилхлоридов формулы A30. Реакция с боронатами или бороновыми кислотами формулы A5 в опосредуемой Pd реакции кросс-сочетания дает соединения формулы I, где R3 представляет собой связанную через простой эфир группу и R4 представляет собой водород. Когда присутствует смесь энантиомеров или диастереомеров, такие смеси можно разделять способами разделения, такими как сверхкритическая жидкостная хроматография.
Схема 1, Путь Н
Как показано на пути I, реакция пирролопиридиндионов формулы A26 с реагентом Гриньяра, с последующей дегидратацией и гидрогенизацией промежуточного соединения формулы A31 дает соединения формулы A32. Затем соединения формулы A32 обрабатывают окислителями, такими как мета-хлорпербензойная кислота, с образованием N-оксидов формулы A33, которые затем обрабатывают оксихлоридом фосфора с получением пиридилхлоридов формулы A34. Реакция с боронатами или бороновыми кислотами формулы A5 в опосредуемой Pd реакции кросс-сочетания дает соединения формулы I в качестве смеси энантиомеров. Такие смеси можно разделять способами разделения, такими как сверхкритическая жидкостная хроматография.
Схема 1, Путь I
Как показано на пути J, соединения формулы I, где ни R3, ни R4 не представляют собой водород, можно получать обработкой соединений формулы A35 сильным ненуклеофильным основанием с последующей реакцией по меньшей мере с двумя эквивалентами алкилирующего агента.
Схема 1, Путь J
Как представлено на схеме 1, путь K, соединения формулы I, где R3 и R4 и промежуточный углерод образуют кольцо, можно получать обработкой соединений формулы A35 сильным ненуклеофильным основанием с последующей реакцией с эквивалентом бис-алкилирующего средства.
Схема 1, Путь K
В следующих типичных примерах предоставлено подробное описание получения соединений по изобретению.
Пример 2. Получение 3-этокси-2-метокси-5-бромпиридина (соединение 2004)
Как показано на стадии 2-i схемы 2, к суспензии NaH (4,0 г, 60% в минеральном масле, 0,1 моль) в 100 мл DMF добавляли 10 мл раствора абсолютного этилового спирта (4,6 г, 0,1 моль)/DMF при К.Т. После выделения газообразного водорода реакционную смесь перемешивали при К.Т. в течение 30 минут и полученный раствор этоксида переносили в раствор 3,5-дибромпиридина (11,84 г, 0,05 моль, полученный от Aldrich Chemical Co.) в 100 мл DMF при 60°C. Реакционную смесь перемешивали при 60°C в течение 4 часов, а затем позволяли достигнуть К.Т. Добавляли рассол и этилацетат, и органические вещества распределяли, сушили над MgSO4, фильтровали и летучие вещества удаляли при пониженном давлении. Полученный неочищенный материал очищали хроматографией на силикагеле, элюируя желаемый продукт смесью 20% этилацетат/гексан с получением 3-бром-5-этоксипиридина (соединение 2001, 4,25 г, выход 42%): 1H ЯМР (CDCl3) δ 8,3 (дд, 2H), 7,4 (д, 1H), 4,12 (кв., 2H), 1,45 (т, 3H). 3-Бензилокси-5-бромпиридин получали по аналогичной методике: 1H ЯМР (CDCl3) δ 8,33 (д, 2H), 7,5-7,35 (м, 6H), 5,15 (с, 2H).
Альтернативно, как показано на стадии 2-ii схемы 2, 3-бром-5-гидроксипиридин (100 мг, 0,57 ммоль, полученный от Aldrich Chemical Co.) разбавляли DMF (3 мл). Добавляли карбонат калия (158,8 мг, 1,15 ммоль), а затем добавляли бромэтан (62,6 мг, 42,6 мл, 0,57 ммоль). Смесь нагревали до 60°C и перемешивали в течение ночи. После охлаждения смесь растворяли в этилацетате и промывали 2 M NaOH, а затем водой. Органические фазы сушили над сульфатом натрия, фильтровали и летучие вещества удаляли при пониженном давлении. Полученный неочищенный 3-бром-5-этоксипиридин (соединение 2001) использовали без дальнейшей очистки. Следующие соединения получали по аналогичным методикам: 3-бром-5-пропоксипиридин, ESMS (M+H) 218,19/216,19; 3-бром-5-бутилпиридин, ESMS (M+H) 230,22/232,22; 3-бром-5-(циклогексилметокси)пиридин, ESMS (M+H) 270,2/272,22; 3-(2-фторэтокси)-5-бромпиридин, ESMS (M+H) 220,14/222,14; 3-(2,2-дифторэтокси)-5-бромпиридин; и 3-(2-этилбутокси)-5-бромпиридин, ESMS (M+H) 258,33/256,33.
Как показано на стадии 2-iii схемы 2, 3-хлорпероксибензойную кислоту (9,426 г, 42,06 ммоль) добавляли к 3-бром-5-метоксипиридину (4,25 г, 21 ммоль) в 200 мл DCM при К.Т. Реакционную смесь перемешивали в течение ночи и смесь промывали 200 мл 2н NaOH и 2×200 мл рассола. Органическую фазу сушили над MgSO4, фильтровали и летучие вещества удаляли при пониженном давлении с получением 3-бром-5-этоксипиридина, 1-оксида (соединение 2002, 4,4 г): 1H ЯМР (CDCl3): δ 8,05 (с, 1H), 7,9 (с, 1H), 7,0 (с, 1H), 4,12 (кв., 2H), 1,45 (т, 3H).
Как показано на стадии 2-iv схемы 2, оксихлорид фосфора (48,02 г, 403,6 ммоль) добавляли к 3-бром-5-этоксипиридин-1-оксиду (4,4 г, 20,18 ммоль) в 700 мл DCM при К.Т. Реакционную смесь перемешивали при К.Т. в течение ночи. После добавления рассола органические слои распределяли, сушили над MgSO4, фильтровали и фильтрат концентрировали при пониженном давлении. Продукт очищали фильтрацией концентрата через слой силикагеля и элюированием слоя этилацетатом. Летучие вещества удаляли при пониженном давлении с получением 5-бром-2-хлор-3-этоксипиридина (соединение 2003, 4,3 г, 85,6%): 1H ЯМР (CDCl3) δ 8,1 (с, 1H), 7,32 (с, 1H), 4,15 (кв., 2H), 1,6 (т, 3H).
Как показано на стадии 2-v схемы 2, 40,51 мл раствора 25% MeONa/MeOH добавляли к 5-бром-2-хлор-3-этоксипиридину (4,3 г, 17,27 ммоль). Реакционную смесь кипятили с обратным холодильником в течение 2 часов. После охлаждения к смеси добавляли этилацетат и рассол. Органическую фазу сушили MgSO4, фильтровали и упаривали при пониженном давлении. После очистки хроматографией на силикагеле получали 5-бром-3-этокси-2-метоксипиридин (соединение 2004, 2,1 г, выход 50%): 1H ЯМР (CDCl3) δ 7,8 (с, 1H), 7,15 (с, 1H), 4,1 (кв., 2H), 4,0 (с, 3H), 1,5 (т, 3H). Следующие соединения синтезировали по аналогичной методике: 5-бром-3-изопропокси-2-метоксипиридин: 1H ЯМР (CDCl3) δ 7,7 (с, 1H), 7,1 (с, 1H), 4,55-4,5 (м, 1H), 3,9 (с, 3H), 1,3 (д, 6H); 5-бром-2-этокси-3-метоксипиридин: ESMS (M+H) 232, 234; 5-бром-3-метокси-2-пропоксипиридин: ESMS (M+H) 246, 248; 5-бром-2-изопропокси-3-метоксипиридин: ESMS (M+H) 246, 248; 5-бром-2-(2,2-дифторэтокси)-3-метоксипиридин: ESMS (M+H) 268, 270; 5-бром-2,3-диэтоксипиридин: ESMS (M+H) 246, 248; 5-бром-2-(2,2-дифторэтокси)-3-этоксипиридин: ESMS (M+H) 282, 284; 5-бром-3-этокси-2-пропоксипиридин: ESMS (M+H) 260, 262; 5-бром-3-этокси-2-изопропоксипиридин: ESMS (M+H) 260, 262; 5-бром-3-(2-фторэтокси)-2-метоксипиридин: ESMS (M+H) 250, 252; 5-бром-2-метокси-3-пропоксипиридин: ESMS (M+H) 246, 248; 5-бром-2-метокси-3-(2-метоксиэтокси)пиридин: ESMS (M+H) 262, 264; 5-бром-3-(2,2-дифторэтокси)-2-метоксипиридин: 1H ЯМР (CDCl3) δ 7,9 (д, 1H), 7,2 (д, 1H), 6,1 (тт, 1H), 4,4 (кв., 2H), 4,2 (тд, 2H), 1,4 (т, 3H); 5-бром-2-этокси-3-изопропоксипиридин: 1H ЯМР (CDCl3) δ 7,7 (д, 1H), 7,1 (д, 1H), 4,4 (м, 1H), 4,3 (кв., 2H), 1,3 (м, 9H); 5-бром-3-бутокси-2-метоксипиридин: ESMS (M+H) 260, 262; 5-бром-2-метокси-3-(2,2,2-трифторэтокси)пиридин: ESMS (M+H) 286, 288; и 5-бром-2-этокси-3-(2,2,2-трифторэтокси)пиридин: ESMS (M+H) 300, 302.
Также с помощью методики, аналогичной методике, представленной на схеме 2, получали 5-метокси-3-бромпиридин, 5-дифторметокси-3-бромпиридин, 2-амино-3-дифторметокси-5-бромпиридин, 2,3-диметокси-5-бромпиридин, 2-этокси-3-метокси-5-бромпиридин, 2-(2-метоксиэтокси)-3-метокси-5-бромпиридин, 2-(2-этоксиэтокси)-3-метокси-5-бромпиридин, 2-(2-пропилоксиэтокси)-3-метокси-5-бромпиридин, 2-(2-этоксибутокси)-3-метокси-5-бромпиридин, 2-(2-(2-метоксиэтокси)этокси)-3-метокси-5-бромпиридин, 2,3-диэтокси-5-бромпиридин, 2-метокси-3-пропокси-5-бромпиридин, 2-метокси-3-(2 -метоксиэтокси)-5-бромпиридин, 2-метокси-3-(2-бутоксиэтокси)-5-бромпиридин, 2-метокси-3-(3-метоксипропилокси)-5-бромпиридин, 2-метокси-3-(тетрагидро-2H-пиран-4-илокси)-5-бромпиридин и 2-метокси-3-(тетрагидро-2H-пиран-4-илокси)-5-бромпиридин.
Схема 2
Пример 3(a). Получение 5-бром-3-(дифторметокси)-2-метоксипиридина (соединение 2010)
Как показано на стадии 3(a)-i схемы 3(a), 2-хлор-3-гидроксипиридин (соединение 2005, 2,0 г, 15,4 ммоль, полученный от Aldrich Chemical Co.) растворяли в 40 мл DMF и 5,0 мл воды вместе с хлордифторацетатом натрия (4,71 г, 30,9 ммоль, полученный от Lancaster Synthesis, Inc.) и безводным карбонатом калия (2,56 г; 18,5 ммоль). Реакционную смесь нагревали на масляной бане при 100°C в течение 2 часов. Добавляли другой эквивалент хлордифторацетата натрия и 1,2 экв. карбоната калия и нагревание продолжали в течение дополнительных 2,0 часов. После этого реакционную смесь охлаждали и летучие вещества удаляли при пониженном давлении. Осадок распределяли между рассолом и этилацетатом и органические слои промывали еще один раз рассолом, сушили над Na2SO4, фильтровали, и летучие вещества удаляли при пониженном давлении. Продукт очищали хроматографией на силикагеле, элюируя градиентом от смеси гексан/DCM до DCM, с получением 2-хлор-3-(дифторметокси)пиридина в виде белого твердого вещества (соединение 2006, 2,0 г, выход 72%): ESMS (M+H) 180; 1H ЯМР (CDCl3) δ 8,05 (м, 1H), 7,45 (м, 1H), 6,90 (м, 1H), 6,60 (т, 1H; J=75 Гц), 4,01 (с, 3H).
Как показано на стадии 3(a)-ii схемы 3(a), избыток металлического натрия растворяли в 20 мл безводного метанола и добавляли 2-хлор-3-(дифторметокси)пиридин (2,0 г, 11,1 ммоль) в безводном метаноле добавляли. Реакционную смесь перемешивали в закрытой емкости при 100°C в течение 6 часов. Летучие вещества удаляли при пониженном давлении и осадок распределяли между EtOAc и рассолом. Рассол экстрагировали EtOAc и объединенные органические фазы сушили над Na2SO4, фильтровали и летучие вещества удаляли при пониженном давлении. Продукт очищали хроматографией на силикагеле (DCM) с получением 3-(дифторметокси)-2-метоксипиридина в виде бесцветного масла (соединение 2007, 1,1 г, выход 56%: ESMS (M+H) 176.
Как показано на стадии 3(a)-iii схемы 3(a), 3-(дифторметокси)-2-метоксипиридин (270 мг, 1,54 ммоль) растворяли в 5 мл DCM и добавляли BBr3 (540 мкл 1275 мг; 4,10 ммоль) в гептане. Реакционную смесь перемешивали в течение 10 минут при К.Т. в атмосфере азота, доводили до кипения с обратным холодильником, а затем перемешивали в течение дополнительных 4 часов. Смесь охлаждали и добавляли воду для гашения реакции. pH доводили до 7-8 бикарбонатом натрия, органические слои распределяли, и водный слой насыщали NaCl и экстрагировали еще два раза DCM. Объединенные органические фазы сушили над Na2SO4, фильтровали и летучие вещества удаляли при пониженном давлении. Продукт очищали хроматографией на силикагеле (градиент от DCM до 5% MeOH/DCM) с получением 3-(дифторметокси)пиридин-2-ола в виде белого твердого вещества (соединение 2008, 986 мг, выход 97%): ESMS (M+H) 162.
Как показано на стадии 3(a)-iv схемы 3(a), 3-(дифторметокси)пиридин-2-ол (986 мг; 6,12 ммоль) растворяли в 25 мл ледяной уксусной кислоты и добавляли ацетат натрия (79 мг; 9,6 ммоль). Смесь охлаждали на ледяной бане и добавляли бром (780 мкл; 1,63 г; 10,22 ммоль) в 10 мл ледяной уксусной кислоты в течение 10 минут. Реакционную смесь перемешивали в течение 30 минут при 10-15°C. Летучие вещества удаляли при пониженном давлении и осадок распределяли между рассолом/насыщенным раствором карбоната натрия и этилацетатом. После прекращения выделения газа органический и водный слои разделяли и водный раствор экстрагировали три дополнительных раза EtOAc. Объединенные органические фазы сушили над Na2SO4, фильтровали и летучие вещества удаляли при пониженном давлении. Осадок очищали два раза хроматографией на силикагеле (сначала градиент от DCM до 10% MeOH/DCM, затем 1:1 EtOAc/гексан) с получением 5-бром-3-(дифторметокси)пиридин-2-ола в виде светло-желтого порошка (соединение 2009, 810 мг, выход 55%): ESMS (M+H) 241,9/243,9; 1H NMR (CDCl3) δ 13,2 (шир.м, 1H), 7,44 (д, 1H, J=2,1 Гц), 7,18 (д, 1H, J=2,1 Гц), 6,92 (т, 1H, J=75 Гц).
Как показано на стадии 3(a)-v схемы 3(a), 5-бром-3-(дифторметокси)пиридин-2-ол (300 мг; 1,25 ммоль) растворяли в 5 мл хлороформа. Добавляли карбонат серебра (690 мг; 2,5 ммоль) и метилйодид (780 мкл; 1,77 г; 12,5 ммоль) и смесь перемешивали при К.Т. в течение ночи. Реакционную смесь фильтровали через диатомитовую землю, которую промывали дополнительным CHCl3. Фильтраты концентрировали при пониженном давлении с получением масла, которое очищали хроматографией на силикагеле с получением 5-бром-3-(дифторметокси)-2-метоксипиридина в виде белого твердого вещества (соединение 2010, 250 мг, выход 78%): ESMS (M+H) 254/256; 1H ЯМР (CDCl3) δ 8,08 (д, 1H, J=2,1 Гц), 7,56 (д, 1H, J=2,1 Гц), 6,60 (т, 1H, J=75 Гц), 3,98 (s,3H).
Схема 3(а)
Пример 3(b). Получение 5-бром-2-этокси-3-метоксипиридина (соединение 2011) и 5-бром-2,3-диметоксипиридина (соединение 2012)
Как показано на стадии 3(b)-i схемы 3(b), 5-бром-2-хлор-3-метоксипиридин (1,0 г, 4,5 ммоль, полученный аналогично тому, как соединение 2003 в примере 2, начиная с 3-бром-5-метоксипиридина) обрабатывали раствором этоксид натрия/этанол (5,05 мл, 21% масс./об., 13,5 ммоль) и реакционную смесь обрабатывали микроволновым излучением при 100°C в течение 20 минут. Добавляли воду и этанол выпаривали при пониженном давлении. Полученный водный раствор экстрагировали DCM и простым эфиром, а затем объединенные экстракты сушили над MgSO4. После фильтрации удаление летучих веществ при пониженном давлении дало 5-бром-2-этокси-3-метоксипиридин (соединение 2011), 0,72 г, выход 69%): ESMS (M+H) 232,32/234,23. Как показано на стадии 3(b)-ii схемы 3(b), соединение 2012 (ESMS (M+H) 218,32/220,23) получали аналогично тому, как и соединение 2011, с использованием метоксида натрия в метаноле вместо этоксида натрия в этаноле.
Схема 3(b)
Пример 4(a). Получение 3-этокси-2-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридина (соединение 2014)
Как показано на стадии 4(a)-i схемы 4(a), йодэтан (13,69 г, 7,021 мл, 87,75 ммоль) добавляли к 5-бром-2-метилпиридин-3-олу (5,5 г, 29,25 ммоль) и K2CO3 (12,13 г, 87,75 ммоль) в 200 мл DMF. Смесь перемешивали при 70°C в течение ночи и к смеси добавляли насыщенный NaHCO3. Смесь экстрагировали EtOAc (3x) и объединенные органические фазы промывали водой (3x) и рассолом. После высушивания над сульфатом натрия смесь фильтровали и концентрировали при пониженном давлении. Осадок очищали хроматографией на силикагеле среднего давления (30% EtOAc в гексане) с получением 5-бром-3-этокси-2-метилпиридина (соединение 2013, 4,6 г, выход 65%): ESMS (M+H) 216,18; 1H ЯМР (CDCl3) δ 8,14 (д, J= 1,9 Гц, 1H), 7,20 (д, J= 1,8 Гц, 1H), 4,03 (кв., J= 7,0 Гц, 2H), 2,43 (с, 3H), 1,47 (т, J= 7,0 Гц, 3H).
Как показано на стадии 4(a)-ii схемы 4(a), 5-бром-3-этокси-2-метилпиридин (4,16 г, 19,25 ммоль), 4,4,5,5-тетраметил-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,3,2-диоксаборолан (5,866 г, 23,10 ммоль) и KOAc (5,668 г, 57,75 ммоль) смешивали в 200 мл диоксане. Смесь дегазировали в течение 1 ч посредством N2, затем добавляли 1,1'-бис(дифенилфосфино)ферроцендихлорпалладий-дихлорметан (162,6 мг, 0,1925 ммоль) и смесь нагревали при 80°C в атмосфере N2 в течение 16 часов. После охлаждения до К.Т. к смеси добавляли MTBE, которую затем пропускали через Florisil®. Растворитель удаляли при пониженном давлении с получением серого твердого вещества, которое растирали с гексаном с получением 3-этокси-2-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридина (соединение 2014, 4,05 г, выход 80%): 1H ЯМР (CDCl3) δ 8,43 (д, J= 1,1 Гц, 1H), 7,40 (с, 1H), 4,03 (д, J= 7,0 Гц, 2H), 2,51 (с, 3H), 1,46 (т, = 7,0 Гц, 3H), 1,37 (с, 12H).
Схема 4(а)
Пример 4(b). Получение N-этил-2-метокси-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)никотинамида (соединение 2016)
Как показано на стадии 4(b)-i схемы 4(b), HATU (8,194 г, 2,55 ммоль) и DIPEA (5,570 г, 7,507 мл, 43,10 ммоль) добавляли к раствору 5-бром-2-метоксипиридин-3-карбоновой кислоты (5 г, 21,55 ммоль) в DMF (50 мл). Полученный раствор перемешивали в течение 10 минут, а затем добавляли этанаминхлористоводородную кислоту (1,757 г, 2,196 мл, 21,55 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 5 часов. К реакционной смеси добавляли воду (100 мл) и этилацетат (100 мл). Органический слой отделяли и сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Неочищенный осадок очищали хроматографией на силикагеле (градиент 0-2% метанола в дихлорметане) с получением 5-бром-N-этил-2-метоксиникотинамида в виде не совсем белого твердого вещества (соединение 2015, 3,4 г): 1H ЯМР (DMSO-d6) δ 8,41 (д, J=2,5 Гц, 1H), 8,31 (с, 1H), 8,20-8,13 (м, 1H), 3,95 (с, 3H), 3,35-3,23 (м, 2H), 1,11 (т, J=7,2 Гц, 3H).
Как показано на стадии 4(b)-ii схемы 4(b), в закрытую пробирку помещали бис(дипинаколато)дибор (3,332 г, 13,12 ммоль), дихлорбис(трициклогексилфосфоранил)палладий (484,2 мг, 0,6560 ммоль), KOAc (3,863 г, 39,36 ммоль) и 2-метилтетрагидрофуран (52,46 мл). Смесь дегазировали в течение 10 мин, а затем нагревали на масляной бане в течение 12 ч при 125°C. Завершение реакции определяли с помощью ВЭЖХ. Затем проводили фильтрацию через florisil и концентрирование в вакууме. Затем желтое масло растирали с гексаном, что вызывало осаждение не совсем белого твердого вещества. Его собирали вакуумной фильтрацией и сушили в высоком вакууме до постоянной массы (4,7 г). 1H ЯМР (300 МГц, DMSO) δ 8,43 (д, J=2,0 Гц, 1H), 8,27 (д, J=2,0 Гц, 1H), 8,23 (д, J=5,5 Гц, 1H), 3,98 (с, 3H), 3,35-3,25 (м, 2H), 1,26 (с, 12H), 1,12 (т, J=7,2 Гц, 3H).
Схема 4(b)
Пример 5. Получение 2-[4-[5-(5,6-диметокси-3-пиридил)-1-оксоизоиндолин-2-ил]пиразол-1-ил]ацетонитрила (соединение 3)
Как показано на стадии 5-i схемы 5, метил-4-бром-2-(бромметил)бензоат (соединение 2018, 2,08 г, 6,75 ммоль; полученное реакцией 1-(4-бром-2-метилфенил)этанона с NBS), 1H-пиразол-4-амин (561 мг, 6,75 ммоль) и DIEA (873 мг, 1,18 мл, 6,75 ммоль) объединяли в DMF (7,78 мл) и нагревали при 110°C в течение 90 мин. Реакционную смесь разбавляли MeOH (60 мл) и полученное белое кристаллическое твердое вещество собирали фильтрацией и сушили в вакууме с получением 5-бром-2-(1H-пиразол-4-ил)изоиндолин-1-она (соединение 2019, 1,21 г, 4,35 ммоль, выход 64%): ESMS (M+H) 279,99.
Как показано на стадии 5-ii схемы 5, 5-бром-2-(1H-пиразол-4-ил)изоиндолин-1-он (1,2 г, 4,32 ммоль) объединяли с карбонатом цезия (1,69 г, 5,18 ммоль) в DMF (10 мл) в закрытой пробирке и через раствор барботировали газообразный азот в течение 5 минут. Добавляли 2-йодацетонитрил (1,08 г, 468 мл, 6,47 ммоль) и пробирку закрывали и нагревали до 110°C на масляной бане в течение 18 часов. Добавляли дополнительно йодацетонитрил (0,5 мл) и реакционную смесь нагревали в течение дополнительных 24 часов. Реакционную смесь выливали в H2O/EtOAc и полученное темно-коричневое твердое вещество собирали фильтрацией. Твердое вещество промывали MeOH, а затем диэтиловым эфиром с получением 2-[4-(5-бром-1-оксо-изоиндолин-2-ил)пиразол-1-ил]ацетонитрила (соединение 2020, 920 мг, 2,9 ммоль, выход 67%): ESMS (M+H) 319,04; 1H ЯМР (DMSO-d6) δ 8,35 (с, 1H), 7,92 (м, 2H), 7,70 (м, 2H), 5,53 (с, 2H), 4,88 (с, 2H).
Как показано на стадии 5-iii схемы 5, 2,3-диметокси-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин [соединение 2021, 376 мг, 1,42 ммоль; полученный реакцией 5-бром-2,3-диметоксипиридина с 4,4,5,5-тетраметил-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,3,2-диоксабороланом], 2-[4-(5-бром-1-оксо-изоиндолин-2-ил)пиразол-1-ил]ацетонитрил (450 мг, 1,42 ммоль) и карбонат цезия (925 мг, 2,84 ммоль) отбирали в DMSO (7,5 мл) в закрывающуюся пробирку. Через раствор барботировали газообразный раствор в течение 5 минут, добавляли dppfPdCl2 (141 мг, 0,17 ммоль) и емкость закрывали. Реакционную смесь нагревали до 100°C в течение 50 мин. После охлаждения смесь выливали в EtOAc/H2O, полученный темный твердый материал отфильтровывали и органические слои пропускали через слой florisil. Фильтрат концентрировали до твердого состояния при пониженном давлении, осадок суспендировали в MeOH, и твердое вещество собирали фильтрацией с получением 2-[4-[5-(5,6-диметокси-3-пиридил)-1-оксоизоиндолин-2-ил]пиразол-1-ил]ацетонитрила в виде твердого вещества (соединение 3, 323 мг, выход 57%): ESMS (M+H) 376,28; 1H NMR (DMSO-d6) δ 8,37 (с, 1H), 8,11 (д, J=1,8 Гц, 1H), 7,99 (с, 1H), 7,95 (с, 1H), 7,84 (м, 2H), 7,64 (д, J=1,7 Гц, 1H), 5,55 (с, 2H), 4,93 (с, 2H), 3,93 (с, 3H), 3,91 (с, 3H).
Схема 5
Пример 6. Получение 2-(4-(5-(5,6-диметоксипиридин-3-ил)-7-метил-1-оксоизоиндолин-2-ил)-1H-пиразол-1-ил)ацетонитрила (соединение 48)
Как показано на стадии 6-i схемы 6, 2,3-диметокси-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин (соединение 2021, 920 мг, 3,47 ммоль), метил-4-бром-2,6-диметилбензоат (844 мг, 3,47 ммоль) и Cs2CO3 (2,26 г, 6,94 ммоль) отбирали в DMSO (12 мл) в закрывающуюся пробирку. Через раствор барботировали газообразный азот в течение 5 минут, добавляли dppfPdCl2 (141 мг, 0,174 ммоль) и емкость закрывали. Реакционную смесь нагревали при 90°C в течение 60 минут в атмосфере азота. После охлаждения смесь выливали в смесь EtOAc/вода. Органические слои промывали водой, рассолом, пропускали через слой Florisil® и концентрировали при пониженном давлении с получением твердого вещества. Твердое вещество суспендировали в MeOH и собирали фильтрацией с получением метил-4-(5,6-диметокси-3-пиридил)-2,6-диметилбензоата (соединение 2022, 310 мг). Фильтрат концентрировали и очищали хроматографией на силикагеле (0-50% EtOAc/hex) с получением дополнительных 400 мг соединения 2022 (общий выход 710 мг, 2,4 ммоль, выход 68%). Это соединение использовали в последующих реакциях как есть.
Как показано на стадии 6-ii схемы 6, соединение 2022 (710 мг, 2,36 ммоль) растворяли в CCl4 (20 мл) и добавляли K2CO3 (651 мг, 4,71 ммоль), NBS (461 мг, 2,59 ммоль) и бензоилпероксид (57 мг, 0,24 ммоль). Реакционную смесь нагревали до температуры кипячения с обратным холодильником в течение 4 часов. После охлаждения до комнатной температуры реакционную смесь фильтровали и твердое вещество промывали CCl4. Фильтрат концентрировали до масла при пониженном давлении и очищали хроматографией на силикагеле с получением метил-2-(бромметил)-4-(5,6-диметокси-3-пиридил)-6-метилбензоата (соединение 2023, 685 мг, чистота приблизительно 70%). Это соединение использовали в последующих реакциях как есть.
Как показано на стадии 6-iii схемы 6, 1H-пиразол-4-амин (149 мг, 1,79 ммоль), метил-2-(бромметил)-4-(5,6-диметокси-3-пиридил)-6-метилбензоат (680 мг, 1,79 ммоль) и DIEA (231 мг, 312 мкл, 1,79 ммоль) объединяли в DMF (5 мл), нагревали до 90°C в течение 6 часов и позволяли охладиться до комнатной температуры в течение 16 часов. Реакционную смесь отбирали в смесь EtOAc/вода и органический слой промывали водой, рассолом, сушили и концентрировали при пониженном давлении с получением пены. Пену перекристаллизовывали в DCM/MeOH. Полученное твердое вещество собирали фильтрацией, промывали DCM и сушили в вакууме с получением 5-(5,6-диметокси-3-пиридил)-7-метил-2-(1H-пиразол-4-ил)изоиндолин-1-она (соединение 2024, 115 мг). Фильтрат после перекристаллизации концентрировали при пониженном давлении и осадок очищали хроматографией на силикагеле (20-100% EtOAc/гексан) с получением дополнительных 88 мг соединения 2024 (общий выход 203 мг, 0,66 ммоль, выход 32%): ESMS (M+H) 351,26; 1H ЯМР (DMSO-d6) δ 12,83 (с, 1H), 8,10 (м, 2H), 7,88 (с, 1H), 7,74 (с, 1H), 7,61 (с, 2H), 4,83 (с, 2H), 3,92 (с, 3H), 3,91 (с, 3H), 2,72 (с, 3H).
Как показано на стадии 6-iv схемы 6, 5-(5,6-диметокси-3-пиридил)-7-метил-2-(1H-пиразол-4-ил)изоиндолин-1-он (88 мг, 0,25 ммоль) растворяли в DMF (2 мл) с Cs2CO3 (123 мг, 0,38 ммоль). Добавляли 2-бромацетонитрил (45 мг, 0,38 ммоль) и реакционную смесь перемешивали в течение ночи при комнатной температуре. Реакционную смесь разбавляли MeOH/H2O/EtOAc и полученное белое твердое вещество собирали фильтрацией; промывали последовательно водой, MeOH и Et2O; и сушили в вакууме с получением 2-(4-(5-(5,6-диметоксипиридин-3-ил)-7-метил-1-оксоизоиндолин-2-ил)-1H-пиразол-1-ил)ацетонитрила (Соединение 48, 30 мг, 0,077 ммоль, выход 29%): ESMS (M+H) 390,29; 1H ЯМР (DMSO-d6) δ 8,34 (с, 1H), 8,10 (д, J=1,8 Гц, 1H), 7,95 (с, 1H), 7,76 (с, 1H), 7,68-7,53 (м, 2H), 5,53 (с, 2H), 4,85 (с, 2H), 3,92 (с, 3H), 3,91 (с, 3H), 2,72 (с, 3H).
Схема 6
Пример 7. Получение 2-(4-(7-хлор-5-(5,6-диметоксипиридин-3-ил)-1-оксоизоиндолин-2-ил)-1H-пиразол-1-ил)ацетонитрила (соединение 58)
Как показано на стадии 7-i схемы 7, 2,3-диметокси-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин (503 мг, 1,9 ммоль), метил 4-бром-2-хлор-6-метил-бензоат (500 мг, 1,9 ммоль) и Cs2CO3 (1,24 г, 3,8 ммоль) объединяли в DMSO (7 мл) в закрывающейся пробирке. Через раствор барботировали газообразный азот в течение 5 минут, добавляли dppfPdCl2 (78 мг, 0,1 ммоль) и емкость закрывали. Реакционную смесь нагревали при 90°C в 60 минут в атмосфере азота. После охлаждения смесь выливали в смесь EtOAc/вода. Органический слой промывали водой, рассолом, пропускали через слой Florisil®, и концентрировали при пониженном давлении. Полученное масло очищали хроматографией на силикагеле (0-50% EtOAc/гексан) с получением метил-2-хлор-4-(5,6-диметокси-3-пиридил)-6-метилбензоата (соединение 2025, 367 мг, 1,14 ммоль, выход 60%): ESMS (M+H) 322,12.
Как показано на стадии 7-ii схемы 7, метил 2-хлор-4-(5,6-диметокси-3-пиридил)-6-метилбензоат (365 мг, 1,13 ммоль) растворяли в CCl4 (20 мл) и добавляли NBS (162 мг, 0,907 ммоль). Реакционную смесь нагревали до температуры кипячения с обратным холодильником 3 часов, после чего добавляли K2CO3 (150 мг) и дополнительный NBS (60 мг). Реакционную смесь нагревали в течение дополнительных 6 часов. В это время анализ ВЭЖХ показал, что приблизительно 30% исходного материала (соединение 2025) конвертировалось в соединение 2026. Добавляли дополнительный NBS (60 мг) и реакционную смесь кипятили с обратным холодильником в течение дополнительных 6 часов, а затем смеси позволяли стоять при комнатной температуре в течение ночи. После этого анализ ВЭЖХ показал, что приблизительно 50% исходного материала конвертировалось в продукт. Реакционную смесь фильтровали, промывали CCl4, и фильтрат концентрировали при пониженном давлении с получением неочищенного 2-(4-(7-хлор-5-(5,6-диметоксипиридин-3-ил)-1-оксоизоиндолин-2-ил)-1H-пиразол-1-ил)ацетонитрила (соединение 2026) в виде масла, которое использовали в последующих реакциях как есть.
Как показано на стадии 7-iii схемы 7, соединение 2026, полученное на стадии 7-ii, растворяли в DMF (5 мл) и добавляли 2-(4-аминопиразол-1-ил)ацетонитрил (100 мг, 0,82 ммоль) и DIEA (226 мг, 304 мкл, 1,75 ммоль). Реакционную смесь нагревали до 80°C в течение 4 часов, охлаждали до комнатной температуры и позволяли стоять в течение ночи. Смесь выливали в смесь EtOAc/вода (1:1 160 мл). Органический слой промывали водой, сушили и концентрировали при пониженном давлении с получением осадка, который застывал после обработки MeOH (20 мл). Твердое вещество собирали фильтрацией, отбирали в EtOAc (30 мл) и нагревали до температуры кипячения с обратным холодильником. После охлаждения до комнатной температуры кристаллический продукт собирали фильтрацией и сушили в вакууме с получением 2-(4-(7-хлор-5-(5,6-диметоксипиридин-3-ил)-1-оксоизоиндолин-2-ил)-1H-пиразол-1-ил)ацетонитрила (соединение 58, 68 мг, 0,17 ммоль, выход 15%): ESMS (M+H) 410,3; 1H ЯМР (DMSO-d6) δ 8,36 (с, 1H), 8,16 (д, J=1,8 Гц, 1H), 7,97 (м, 2H), 7,92 (с, 1H), 7,69 (с, 1H), 5,56 (с, 2H), 4,90 (с, 2H), 3,93 (с, 3H), 3,92 (с, 3H).
Схема 7
Пример 8. Получение 2-(4-(2-(5,6-диметоксипиридин-3-ил)-5-оксо-5H-пирроло[3,4-b]пиридин-6(7H)-ил)-1H-пиразол-1-ил)ацетонитрила (соединение 25)
Как показано на стадии 8-i схемы 8, согласно методике из публикации международной патентной заявки № WO2006/095159, смесь этил-2-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата (5,92 г, 32,6 ммоль) в оксихлориде фосфора (45 мл) нагревали при 90°C в течение 1 часа. После охлаждения реакционную смесь концентрировали при пониженном давлении и к осадку добавляли ледяную воду, а затем добавляли 28% гидроксид аммония для доведения pH до 7. Полученное белое твердое вещество собирали фильтрацией, промывали водой и сушили в высоком вакууме с получением этил-6-хлор-2-метилникотината (соединение 2027, 6,18 г, выход 94,7%): ESMS (M+1) 200,19; 1H ЯМР (CDCl3) δ 8,18 (д, J=8,2 Гц, 1H), 7,27 (д, J=8,2 Гц, 1H), 4,40 (кв., J=7,1 Гц, 2H), 2,84 (с, 3H), 1,42 (т, J=7,4, 3H).
Как показано на стадии 8-ii схемы 8, смесь этил-6-хлор-2-метилпиридин-3-карбоксилата (соединение 2027, 4,0 г, 20 ммоль), 2,3-диметокси-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридина (соединение 2021, 5,84 г, 22 ммоль), Pd(PPh3)4 (1,15 г, 1 ммоль), и карбоната натрия (6,37 г, 60 ммоль) в смеси ацетонитрил/вода (3:1, 90 мл) нагревали при 90°C в атмосфере азота в течение 4 часов. После охлаждения летучие вещества удаляли при пониженном давлении и осадок растворяли в DCM. После промывания водой органическую фазу сушили (Na2SO4), фильтровали и концентрировали при пониженном давлении. Осадок очищали хроматографией на силикагеле (0-20% EtOAc/гексан) с получением этил-5',6'-диметокси-6-метил-2,3'-бипиридин-5-карбоксилата (соединение 2028, 5,8 г, выход 95,7%): ESMS (M+1) 303,41; 1H ЯМР (CDCl3) δ 8,27 (д, J=1,8 Гц, 1H), 8,17 (д, J=8,2 Гц, 1H), 7,83 (д, J=1,8 Гц, 1H), 7,52 (д, J=8,3 Гц, 1H), 4,32 (кв., J=7,1 Гц, 2H), 4,01 (с, 2H), 3,93 (с, 3H), 2,83 (с, 3H), 1,34 (т, J=7,1 Гц, 3H).
Как показано на стадии 8-iii схемы 8, к раствору соединения 2029 (4,4 г, 14,6 ммоль) в CCl4 (75 мл) добавляли 2,2'-азобис(изобутиронитрил) (AIBN, 239 мг, 1,46 ммоль) и NBS (1,7 г, 9,55 ммоль). Смесь перемешивали при 80°C в течение 1,5 часов в атмосфере азота. Реакционную смесь фильтровали и концентрировали. Осадок очищали хроматографией на силикагеле (20% EtOAc/гексан) с получением смеси исходного материала и этил-5',6'-диметокси-6-бромметил-2,3'-бипиридин-5-карбоксилата (соединение 2029, 4,44 г, чистота приблизительно 60%): ESMS (M+1) 381,4, 383,19. Продукт использовали как есть при последующих реакциях.
Как показано на стадии 8-iv схемы 8, раствор указанной выше смеси (соединение 2029, 2,2 г, чистота приблизительно 60%) в DMF (40 мл) при 0°C капельно добавляли в течение 2 часов к суспензии 2-(4-амино-1H-пиразол-1-ил)ацетонитрила (844 мг, 6,9 ммоль) и карбоната натрия (732 мг, 6,9 ммоль) в DMF (20 мл). После завершения добавления реакционную смесь перемешивали при 0°C в течение 2 часов и нагревали при 80°C в течение 15 часов. Добавляли дополнительный карбонат натрия (732 мг) и реакционную смесь нагревали при 90°C в течение дополнительных 7 часов. После охлаждения смесь выливали в воду, и образовывался осадок. Твердое вещество собирали фильтрацией, промывали метил-трет-бутиловым эфиром и сушили в высоком вакууме с получением 2-(4-(2-(5,6-диметоксипиридин-3-ил)-5-оксо-5H-пирроло[3,4-b]пиридин-6(7H)-ил)-1H-пиразол-1-ил)ацетонитрила (соединение 25, 450 мг).
Схема 8
Пример 9. Получение 2-(5,6-диметоксипиридин-3-ил)-4-метокси-6-(1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)-6,7-дигидро-5H-пирроло[3,4-b]пиридин-5-она (соединение 88)
Как показано на стадии 9-i схемы 9, этил-4,6-дигидрокси-2-метилпиридин-3-карбоксилат (Accla Biochem Inc.) суспендировали в 50 мл POCl3 и нагревали до 90°C в атмосфере азота в течение 5 часов. Реакционную смесь охлаждали и концентрировали при пониженном давлении. К темному маслу при перемешивании добавляли лед, а затем добавляли этилацетат и воду. Органические слои промывали водой, рассолом и сушили над сульфатом натрия. После фильтрации летучие вещества удаляли при пониженном давлении и неочищенный продукт очищали хроматографией на силикагеле с получением этил-4,6-дихлор-2-метилникотината в виде светло-желтого масла (соединение 2030, выход 62%): ESMS (M+H) 234/236/238; 1H ЯМР (CDCl3) δ 7,27 (с, 1H), 4,4 (кварт., 2H), 2,55 с, 3H), 1,41 (т, 3H).
Как показано на стадии 9-ii схемы 9, соединение 2030 (500 мг, 2,14 ммоль) и 2,3-диметокси-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин (соединение 2021, 566 мг, 2,14 ммоль) растворяли в 10 мл DME и продували азотом в течение 5 минут. Добавляли тетракистрифенилфосфинпалладий(0) (250 мг, 0,2136 ммоль) при продолжении потока азота. Добавляли раствор насыщенного водного 2 M K2CO3 (2,2 мл) (после продувания азотом) и смесь нагревали до 70°C в течение 1,5 часов. Летучие вещества удаляли при пониженном давлении и осадок растирали с водой и 2 мл 1,0н HCl. Образовывался осадок, который распределяли между EtOAc и водой. Органические слои промывали водой, рассолом, сушили над сульфатом натрия, и летучие вещества удаляли при пониженном давлении. Неочищенный продукт очищали хроматографией на силикагеле (DCM - 25% EtOAc/DCM) с получением этил-4-хлор-5',6'-диметокси-6-метил-2,3'-бипиридин-5-карбоксилата (соединение 2031, 650 мг, выход 90%) светло-бежевого твердого вещества: ESMS (M+1) 337/339; 1H ЯМР (CDCl3) δ 8,26 (д, 1H J=2 Гц), 7,77 (д, 1H, J=2 Гц), 7,56 (с, 1H), 4,48 (кварт., 2H), 4,08 (с, 3H), 4,00 (с, 3H), 2,63 (с, 3H), 1,43 (т, 3H).
Как показано на стадии 9-iii схемы 9, соединение 2031 (650 мг, 1,93 ммоль) растворяли в безводном метаноле и к нему добавляли 3,0 свежеприготовленного 2,54 M метоксида натрия. Реакционную смесь нагревали в атмосфере азота при 60°C в течение 16 часов. После охлаждения до комнатной температуры летучие вещества удаляли при пониженном давлении и осадок распределяли между водой и EtOAc. Органические слои промывали водой, рассолом, сушили над сульфатом натрия, фильтровали и летучие вещества удаляли при пониженном давлении. Очистка хроматографией на силикагеле дала этил-4,5',6'-триметокси-6-метил-2,3'-бипиридин-5-карбоксилат (соединение 2032, 200 мг, выход 32%) в виде не совсем белого твердого вещества: ESMS (M+1) 319; 1H ЯМР (CDCl3) δ 8,23 (д, 1H, J=2Гц), 7,8 (д, 1H, J=2Гц), 7,06 (с, 1H), 4,07 (с, 3H), 3,99 (с, 3H), 3,94 (с, 3H).
Как показано на стадии 9-iv схемы 9, соединение 2032 (200 мг, 0,628 ммоль) и NBS (112 мг, 0,63 ммоль) добавляли к 15 мл CCl4 и раствор продували азотом в течение 5 минут. Добавляли бензоилпероксид (20 моль%) и реакционную смесь нагревали при 65°C в атмосфере азота в течение 16 часов. Добавляли дополнительный эквивалент NBS и 0,3 эквивалента бензоилпероксида и нагревание продолжали в течение дополнительного часа. Добавляли карбонат калия (1,0 г) в качестве реагента для удаления кислоты и нагревание продолжали в течение дополнительных 24 часов. Реакционную смесь охлаждали и фильтровали через слой ваты и летучие вещества удаляли при пониженном давлении. Осадок отбирали в DCM и пропускали через слой силикагеля, а затем элиюровали дополнительным DCM. Элюирование EtOAc привело к неочищенному продукту, который далее очищали хроматографией на силикагеле (DCM - 1:1 EtOAc/DCM) с получением этил 6-(бромметил)-4,5',6'-триметокси-2,3'-бипиридин-5-карбоксилата (соединение 2033, 87 мг белого твердого вещества) ESMS (M+1) 397/399.
Как показано на стадии 9-v схемы 9, соединение 2033 (100 мг, 0,252 ммоль), 1-(2,2,2-трифторэтил)-4-аминопиразол (42 мг, 0,25 ммоль) и DIEA (65 мг, 0,5 ммоль) растворяли в 4 мл DMF и нагревали в течение 3 часов при 110°C. Реакционную смесь охлаждали, разбавляли водой и экстрагировали EtOAc. Органические слои промывали водой, рассолом, сушили над сульфатом натрия и растворитель удаляли при пониженном давлении. Неочищенный продукт пропускали через слой силикагеля, а затем элюировали 5% EtOH/EtOAc. Очистка фильтрата хроматографией на силикагеле (5% MeOH/DCM - 7,5% MeOH/DCM) дала 2-(5,6-диметоксипиридин-3-ил)-4-метокси-6-(1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)-6,7-дигидро-5H-пирроло[3,4-b]пиридин-5-он (соединение 88, 11 мг, выход 9%) в виде бежевого твердого вещества: ESMS (M+1) 450.
Схема 9
Пример 10. Получение 4-хлор-2-(5,6-диметоксипиридин-3-ил)-6-(1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)-6,7-дигидро-5H-пирроло[3,4-b]пиридин-5-она (соединение 82) и 2-(5,6-диметоксипиридин-3-ил)-4-этил-6-(1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)-6,7-дигидро-5H-пирроло[3,4-b]пиридин-5-она (соединение 182)
Как показано на стадиях 10-i и 10-ii схемы 10, соединение 2031 конвертировали в соединение 82 аналогично конвертированию соединения 2032 в соединение 88.
Как показано на стадии 10-iii схемы 10, 4-хлор-2-(5,6-диметоксипиридин-3-ил)-6-(1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)-6,7-дигидро-5H-пирроло[3,4-b]пиридин-5-он (соединение 82, 75 мг, 0,165 ммоль) и дегазированный раствор 2 M водного раствора карбоната натрия (826 мкл, 1,65 ммоль) суспендировали в 10 мл толуола. Реакционную смесь продували азотом в течение 5 минут и добавляли тетракистрифенилфосфинпалладий(0) (38 мг, 0,033 ммоль), а затем добавляли 2 M раствор триэтилбората в THF (992 мкл, 1,0 ммоль). Реакционную емкость закрывали и нагревали в течение 14 часов при 80°C. Реакционную смесь охлаждали и летучие вещества удаляли при пониженном давлении. Осадок растворяли в EtOAc/DCM 1:1 и фильтровали. Фильтрат концентрировали при пониженном давлении. Продукт очищали препаративной тонкослойной хроматографией с получением 2-(5,6-диметоксипиридин-3-ил)-4-этил-6-(1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)-6,7-дигидро-5H-пирроло[3,4-b]пиридин-5-она (соединение 182, 1,1 мг): ESMS (M+1) 448.
Схема 10
Пример 11. Получение 2-(5,6-диметоксипиридин-3-ил)-3-метил-6-(1H-пиразол-4-ил)-6,7-дигидро-5H-пирроло[3,4-b]пиридин-5-она (соединение 238)
Как показано на стадии 11-i схемы 11, 2,5-диметилникотиновую кислоту (соединение 2034, 519 мг, 3,43 ммоль) растворяли в 1,1,1-триэтоксиэтане (5,57 г, 6,29 мл, 34,3 ммоль) в емкости для обработки микроволновым излучением. Реакционную смесь нагревали до 150°C в течение 5 минут. После разбавления 30 мл DCM, органические слои промывали 10 мл насыщенного NaHCO3. Органический слой пропускали через фазовый разделитель, а затем концентрировали при пониженном давлении с получением этил-2,5-диметилникотината (соединение 2035, 390 мг, выход 63%) в виде желтого масла: ESMS (M+1) 179,89; 1H ЯМР (300 МГц, DMSO-d6) δ 8,46 (д, J= 1,9 Гц, 1H), 7,98 (д, J=1,9 Гц, 1H), 4,31 (кв., J= 7,1 Гц, 2H), 2,65 (с, 3H), 2,31 (с, 3H), 1,32 (т, J=7,1 Гц, 3H).
Как показано на стадии 11-ii схемы 11, соединение 2035 (354 мг, 1,98 ммоль) и 1,3,5-трихлор-1,3,5-триазинан-2,4,6-трион (689 мг, 2,96 ммоль) объединяли в DCM (1,8 мл). После перемешивания в течение 18 часов при комнатной температуре смесь разбавляли насыщенным карбонатом натрия и дихлорметаном, по 20 мл каждого. Органические слои разделяли, промывали насыщенным карбонатом натрия, промывали рассолом, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением этил-2-(хлорметил)-5-метилникотината (соединение 2036, 465 мг) в виде светло-желтого масла: ESMS (M+1) 213,86.
Как показано на стадии 11-iii схемы 11, соединение 2036 (465 мг, 2,18 ммоль) помещали в емкость объемом 40 мл, разбавляли DCM (4,35 мл) и добавляли 3-хлорбензолкарбопероксоевую кислоту (m-CPBA, 551 мг, 2,39 ммоль) при комнатной температуре при перемешивании. Через 18 часов смесь разбавляли 30 мл DCM, промывали насыщенным карбонатом натрия (3×5 мл) и промывали рассолом. Органические слои пропускали через фазовый разделитель и концентрировали до сухого состояния при пониженном давлении с получением этил-2-(хлорметил)-3-(этоксикарбонил)-5-метилпиридин-1-оксида (соединение 2037, 318 мг, 1,38 ммоль, выход 63%): ESMS (M+1) 230,14. Этот материал использовали как есть в последующих реакциях.
Как показано на стадии 11-iv схемы 11, соединение 2037 (318 мг, 1,385 ммоль) растворяли в оксихлориде фосфора (4,25 г, 2,58 мл, 27,7 ммоль). Реакционную смесь нагревали до 90°C в атмосфере азота в течение 18 часов. Смесь концентрировали до сухого состояния при пониженном давлении, разбавляли 5 мл DCM, и промывали 5 мл воды. Органические слои пропускали через фазовый разделитель и летучие вещества удаляли при пониженном давлении. Очистка хроматографией на силикагеле дала этил-6-хлор-2-(хлорметил)-5-метилникотинат (соединение 2038, 78 мг, 0,314 ммоль, 22,7%): ESMS (M+1) 248,17; 1H ЯМР (300 МГц, DMSO-d6) δ 8,29 (с, 1H), 5,00 (с, 2H), 4,51-4,23 (м, 2H), 2,38 (с, 3H), 1,35 (т, J= 7,1 Гц, 3H).
Как показано на стадии 11-v схемы 11, соединение 2038 (76 мг, 0,306 ммоль) растворяли в 1 мл DMF и капельно добавляли к перемешиваемому раствору 1H-пиразол-4-амина (63,6 мг, 0,766 ммоль) и DIEA (59,4 мг, 801 мкл, 0,46 ммоль) в 1 мл DMF. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, а затем нагревали в течение ночи при 80°C. После добавления 10 мл метанола смеси позволяли охладиться с получением твердого вещества. Твердое вещество собирали фильтрацией и промывали 3 мл метанола. Твердое вещество сушили в течение ночи в высоком вакууме с получением 2-хлор-3-метил-6-(1H-пиразол-4-ил)-6,7-дигидро-5H-пирроло[3,4-b]пиридин-5-она (соединение 2039, 35 мг, 0,141 ммоль, выход 46%). ESMS (M+1) 249,08.
Как показано на стадии 11-vi схемы 11, 2,3-диметокси-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин (Соединение 2028, 45 мг, 0,169 ммоль), 1 M карбонат натрия (281 мкл, 0,281 ммоль) и соединение 2039 (35 мг, 0,141 ммоль) отбирали в 3 мл DMF в виде взвеси. Смесь дегазировали азотом в течение 30 минут и добавляли Pd(PPh3)4 (32,5 мг, 0,028 ммоль). Смесь дегазировали азотом в течение дополнительных 5 минут, а затем нагревали в течение 18 часов при 80°C в закрытой емкости. Добавляли дополнительный метанол, а затем разбавляли DCM. Органические слои промывали насыщенным раствором Na2CO3, пропускали через фазовый разделитель и концентрировали при пониженном давлении до сухого состояния. Продукт очищали обращенно-фазовой ВЭЖХ (10-90% ацетонитрил/вода) с получением 2-(5,6-диметоксипиридин-3-ил)-3-метил-6-(1H-пиразол-4-ил)-6,7-дигидро-5H-пирроло[3,4-b]пиридин-5-она (соединение 238, 18,8 мг, 0,052 ммоль, выход 37%); ESMS (M+1) 352,26; 1H ЯМР (300 МГц, CDCl3) δ 8,10 (д, J=11,8 Гц, 3H), 7,97 (д, J=1,9 Гц, 1H), 7,34 (д, J=1,8 Гц, 1H), 4,83 (с, 2H), 4,10 (с, 3H), 3,96 (с, 3H), 2,54 (с, 3H).
Схема 11
Пример 12. Получение 2-(5,6-диметоксипиридин-3-ил)-4-метил-6-(1H-пиразол-4-ил)-6,7-дигидро-5H-пирроло[3,4-b]пиридин-5-она (соединение 135)
Как показано на стадии 12-i схемы 12, к дегазированной смеси соединения 2021 (111 мг, 0,42 ммоль), карбоната натрия (97 мг, 0,91 ммоль) и 2-хлор-4-метил-6-(1H-пиразол-4-ил)-6,7-дигидро-5H-пирроло[3,4-b]пиридин-5-она (соединение 2040, 104 мг, 0,42 ммоль; полученного из этил-6-хлор-2-(хлорметил)-4-метилникотината аналогично получению соединения 2039 на стадии 11-v примера 11) в смеси DMF/ацетонитрил/вода (1:1:0,5) добавляли Pd(PPh3)4 (50 мг, 0,04 ммоль). Реакционную смесь нагревали в закрытой пробирке при 90°C в течение 48 часов. Добавляли воду (5 мл) и смесь перемешивали при К.Т. в течение 30 минут. После фильтрации собранное твердое вещество промывали MeOH и EtOAc, обрабатывали ультразвуком в EtOAc, а затем собирали фильтрацией с получением 2-(5,6-диметоксипиридин-3-ил)-4-метил-6-(1H-пиразол-4-ил)-6,7-дигидро-5H-пирроло[3,4-b]пиридин-5-она (соединение 135, 100 мг, выход 66%) в виде светло-зеленого твердого вещества: ESMS (M+H) 352,4; 1H ЯМР (300 МГц, DMSO-d6) δ 12,88 (с, 1H), 8,54 (д, J=1,9 Гц, 1H), 8,29-7,66 (м, 4H), 4,90 (с, 2H), 3,93 (д, J=10,5 Гц, 6H), 2,72 (с, 3H).
Схема 12
Пример 13. Получение 2-(5,6-диметоксипиридин-3-ил)-4-метил-6,7-дигидро-5H-пирроло[3,4-b]пиридин-5-она (соединение 2043)
Как показано на стадии 13-i схемы 13, этил-6-хлор-2-(хлорметил)-4-метилникотинат (соединение 2041, 5,11 г, 20,6 ммоль; полученное из 2,5-диметилникотиновой кислоты аналогично получению соединения 2038 в примере 11), растворяли в метаноле (30,6 мл). Добавляли 7 M аммиак/MeOH (21,3 мл, 149 ммоль), а затем добавляли гидроксид аммония (18,7 г, 20,8 мл, 533 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре, и осадок, который образовывался, собирали фильтрацией и сушили в высоком вакууме с получением 2-хлор-4-метил-6,7-дигидро-5H-пирроло[3,4-b]пиридин-5-она (соединение 2042, 2,6 г, 14,2 ммоль, выход 69%): ESMS (M+1) 183,29; 1H NMR (300 МГц, CDCl3) δ 8,74 (с, 1H), 7,44 (д, J=0,5 Гц, 1H), 4,35 (с, 2H), 2,60 (с, 3H).
Как показано на стадии 13-ii схемы 13, 2-хлор-4-метил-6,7-дигидропирроло[3,4-b]пиридин-5-он (2,54 г, 13,91 ммоль), 1 M карбонат натрия (27,82 ммоль) и 2,3-диметокси-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин (4,43 г, 16,7 ммоль) суспендировали в DMF (216 мл). Реакционную смесь продували азотом в течение 30 минут. Добавляли Pd(PPh3)4 (1,607 г, 1,391 ммоль) и продувание азотом продолжали в течение дополнительных 5 минут. Затем реакционную смесь нагревали при 80°C в течение 16 часов. Смесь разбавляли 1 л этилацетата и 350 мл насыщенного NaHCO3. В делительной воронке образовывался осадок, который собирали фильтрацией и промывали EtOAc, водой и этиловым эфиром. Высушивание твердого вещества в течение ночи в высоком вакууме дало 2-(5,6-диметокси-3-пиридил)-4-метил-6,7-дигидропирроло[3,4-b]пиридин-5-он (соединение 2043, 3,82 г, 13,38 ммоль, выход 96%): ESMS (M+H) 286,29; 1H ЯМР (300 МГц, DMSO-d6) δ 8,63 (с, 1H), 8,49 (д, J=1,9 Гц, 1H), 8,06-7,82 (м, 2H), 4,41 (с, 2H), 3,94 (с, 3H), 3,90 (с, 3H), 2,67 (с, 3H).
Схема 13
Пример 14. Получение 2-(5,6-диметоксипиридин-3-ил)-6-(5-метилтиофен-2-ил)-6,7-дигидро-5H-пирроло[3,4-b]пиридин-5-она (соединение 252)
Как показано на стадии 14-i схемы 14, 2-(5,6-диметокси-3-пиридил)-6,7-дигидропирроло[3,4-b]пиридин-5-он (соединение 2044, 100 мг, 0,37 ммоль; полученный аминолизом соединения 2029, как показано на стадии 13-i примера 13), 2-йод-5-метилтиофен (99 мг, 54 мкл, 0,44 ммоль) и карбонат цезия (240 мг, 0,737 ммоль) отвешивали в небольшую пробирку с навинчивающейся крышкой. Реакционную смесь продували азотом в течение 15 минут. Добавляли CuI (14,0 мг, 0,074 ммоль) и N,N'-диметилэтан-1,2-диамин (6,5 мг, 7,8 мкл, 0,073 ммоль) и продувание азотом продолжали в течение дополнительных 5 минут. Пробирку закрывали и содержимое нагревали при 100°C в течение 18 часов. После охлаждения до комнатной температуры реакционную смесь разбавляли 20 мл воды и осадок собирали фильтрацией. Твердое вещество промывали водой, промывали метанолом, а затем отбирали в 6 мл DMSO. Очистка обращенно-фазовой ВЭЖХ дала 2-(5,6-диметоксипиридин-3-ил)-6-(5-метилтиофен-2-ил)-6,7-дигидро-5H-пирроло[3,4-b]пиридин-5-он (соединение 252, 31,6 мг, 0,084 ммоль, выход 23%): ESMS (M+H) 368,01; 1H ЯМР (300 МГц, DMSO-d6) δ 8,56 (д, J=2,0 Гц, 1H), 8,30-8,12 (м, 2H), 7,99 (д, J=2,0 Гц, 1H), 6,73 (д, J=3,7 Гц, 1H), 6,67 (дд, J=3,7, 1,1 Гц, 1H), 5,09 (с, 2H), 3,96 (с, 3H), 3,92 (с, 3H), 2,42 (д, J=0,7 Гц, 3H).
Схема 14
Пример 15. Получение 2-(5,6-диметоксипиридин-3-ил)-7,7-диметил-6-(1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)-6,7-дигидро-5H-пирроло[3,4-b]пиридин-5-она (соединение 311)
Как показано на стадии 15-i схемы 15, к раствору 2-(5,6-диметоксипиридин-3-ил)-6-(1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)-6,7-дигидро-5H-пирроло[3,4-b]пиридин-5-она (соединение 115, 20 мг, 0,047 ммоль, полученного реакцией соединения 2029 с 1 -(2,2,2-трифторэтил)-1H-пиразол-4-амином аналогично тому, как показано в примере 8) в DMF (500 мкл) добавляли йодметан (17 мг, 0,119 ммоль), а затем NaH (6 мг, 60% масс./масс. в минеральном масле). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, гасили насыщенным водным раствором NaHCO3 (1 мл) и экстрагировали DCM (3×2 мл). Органические слои концентрировали и неочищенный осадок очищали препаративной тонкослойной хроматографией на силикагеле (100% EtOAc) с получением 2-(5,6-диметоксипиридин-3-ил)-7,7-диметил-6-(1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)-6,7-дигидро-5H-пирроло[3,4-b]пиридин-5-она (соединение 346, 11,2 мг, выход 50%) в виде белого твердого вещества: ESMS (M+H) 447,87; 1H ЯМР (300 МГц, CDCl3) δ 8,45 (д, J= 1,9 Гц, 1H), 8,24-8,08 (м, 2H), 7,84 (дд, J= 19,0, 4,1 Гц, 3H), 4,75 (кв., J= 8,3 Гц, 2H), 4,08 (т, J= 17,3 Гц, 6H), 1,72 (с, 6H).
Схема 15
Пример 16. Получение (5)-2-(6-этокси-5-метоксипиридин-3-ил)-7-метил-6-(1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)-6,7-дигидро-5H-пирроло[3,4-b]пиридин-5-она (соединение 354) и (R)-2-(6-этокси-5-метоксипиридин-3-ил)-7-метил-6-(1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)-6,7-дигидро-5H-пирроло[3,4-b]пиридин-5-она (соединение 355)
Как показано на стадии 16-i схемы 16, к раствору 2,2,2-трифторэтанола (26,54 г, 19,33 мл, 265 ммоль) и пиридина (20,99 г, 21,46 мл, 265 ммоль) в DCM (120 мл) при 0°C добавляли раствор трифторметилсульфонилтрифторметансульфоната (74,85 г, 44,6 мл, 265 ммоль) в DCM (150 мл) через капельную воронку в течение 45 минут. Реакционную смесь перемешивали в течение дополнительных 15 минут после завершения добавления, а затем гасили водой (400 мл). Органические слои промывали водой (400 мл), сушили над MgSO4 и фильтровали с получением 2,2,2-трифторэтилтрифторметансульфоната (соединение 2045), который использовали как есть. Как показано на стадии 16-ii схемы 16, раствор соединения 2045 добавляли в течение 25 мин к раствору 4-нитро-1H-пиразола (25 г, 221,1 ммоль) в DMF (200 мл) с K2CO3 (61,11 г, 442,2 ммоль), охлажденному на ледяной бане. После завершения добавления охлаждающую баню удаляли и реакционную смесь перемешивали при 23°C в течение 16 часов. Органические слои промывали водой (500 мл) и водный слой экстрагировали DCM (150 мл). Объединенные органические фазы сушили над MgSO4, фильтровали и концентрировали при пониженном давлении. Полученный содержащий DMF концентрат разбавляли 1:1 EtOAc:гексан (500 мл), промывали водой (3×250 мл), рассолом (200 мл), сушили (MgSO4), фильтровали и концентрировали при пониженном давлении с получением 4-нитро-1-(2,2,2-трифторэтил)пиразола (соединение 2046, 40,4 г, 207,1 ммоль, выход 93,65%) в виде коричневато-желтого твердого вещества: 1H ЯМР (CDCl3, 300 МГц) δ 8,31 (с, 1H), 8,17 (с, 1H), 4,79 (кв., J=9 Гц, 2H).
Как показано на стадии 16-iii схемы 16, к раствору соединения 2046 (40,4 г, 207,1 ммоль) в EtOH (600 мл) в колбе Парра добавляли палладий (10 г, 9,397 ммоль) (Pd/C, 10 масс.% в расчете на сухую массу, влажный, типа Degussa). Смесь помещали под давление 50 фунт/кв. дюйм (0,34 МПа) газообразного водорода и встряхивали при 23°C в течение 40 минут. Реакционную смесь пропускали через 0,22-мкм PES-мембрану Corning и фильтрат концентрировали с получением 1-(2,2,2-трифторэтил)пиразол-4-амина (соединение 2047, 33,94 г, 205,6 ммоль, выход 99,24%) в виде прозрачного красноватого масла: 1H ЯМР (CDCl3, 300 МГц) δ 7,26 (с, 1H), 7,10 (с, 1H), 4,59 (кв., J=9 Гц, 2H), 2,95 (шир.с, 2H). ESMS (M+H) 165,97.
Как показано на стадии 16-iv схемы 16, к соединению 2047 (7,16 г, 43,36 ммоль) в THF (204,6 мл) при 23°C добавляли фуро[3,4-b]пиридин-5,7-дион (6,465 г, 43,36 ммоль), а затем добавляли DMAP (52,97 мг, 0,4336 ммоль). Реакционную смесь перемешивали при 50°C. Через 3 часа добавляли уксусный ангидрид (8,853 г, 8,182 мл, 86,72 ммоль) и реакционную смесь нагревали при 70°C в течение дополнительных 1,5 часов. После охлаждения реакционную смесь концентрировали и осадок распределяли между DCM и насыщенным водным раствором NaHCO3 (по 100 мл каждый). Водный слой экстрагировали DCM (50 мл) и объединенные органические фазы промывали насыщенным водным раствором NaHCO3 (100 мл), сушили (MgSO4), фильтровали и концентрировали при пониженном давлении. Осадок перекристаллизовывали из горячей смеси EtOAc/гексан с получением 6-(1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)-5H-пирроло[3,4-b]пиридин-5,7(6H)-диона (соединение 2048, 6,07 г, 20,49 ммоль, 47,27%) в виде желтых игл: ESMS (M+H) 297,23; 1H ЯМР (CDCl3, 300 МГц) δ 9,05 (м, 1H), 8,32 (с, 1H), 8,31 (с, 1H), 8,26 (м, 1H), 7,70 (м, 1H), 4,79 (кв., J=9 Гц, 2H).
Как показано на стадии 16-v схемы 16, к соединению 2048 (5,69 г, 19,21 ммоль) в THF (500 мл) при -78°C в атмосфере азота медленно добавляли метиловый бромид магния (16,95 г, 16,46 мл 1,4 M раствора в 1:3 THF:толуол, 23,05 ммоль). После перемешивания в течение 1 часа при -78°C реакционную смесь нагревали до 0°C и перемешивали в течение дополнительного 1 часа. Реакционную смесь гасили насыщенным водным раствором NH4Cl (100 мл). После перемешивания в течение 15 минут смесь частично концентрировали при пониженном давлении и распределяли между водой (150 мл) и EtOAc (200 мл). Органические слои промывали рассолом (150 мл), сушили (MgSO4), фильтровали и концентрировали с получением 7-гидрокси-7-метил-6-(1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)-6,7-дигидро-5H-пирроло[3,4-b]пиридин-5-она (соединение 2049, 5,77 г, 18,48 ммоль, выход 96,2%) в виде желтого твердого вещества: ESMS (M+H) 313,23; 1H ЯМР (CDCl3, 300 МГц) δ 8,73 (м, 1H), 8,10 (с, 1H), 8,01 (с, 1H), 8,00 (м, 1H), 7,40 (м, 1H), 4,73 (кв., J=9 Гц, 2H), 1,84 (с, 3H).
Как показано на стадии 16-vi схемы 16, к соединению 2049 (5,77 г, 18,48 ммоль) в DCM (170 мл) при 23°C добавляли Et3N (7,48 г, 10,30 мл, 73,9 ммоль), а затем метансульфонилхлорид (MsCl, 3,18 г, 2,15 мл, 27,7 ммоль). После перемешивания в течение 20 минут добавляли EtOH (6 мл) и перемешивание продолжали в течение 10 минут, чтобы погасить какой-либо избыток MsCl. Реакционную смесь распределяли между насыщенным водным раствором NaHCO3 (300 мл) и DCM (50 мл). Водный слой экстрагировали DCM (150 мл) и объединенные органические фазы сушили (MgSO4), фильтровали и объединяли с EtOH (200 мл). Полученный раствор (содержавший 7-метилен-6-(1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)-6,7-дигидро-5H-пирроло[3,4-b]пиридин-5-он, соединение 2050) концентрировали при пониженном давлении до приблизительно 100 мл и разбавляли дополнительным EtOH (250 мл). Добавляли Pd/C (10 масс.% в расчете на сухую массу, влажный, типа Degussa, 2,85 г) и реакционную смесь перемешивали в атмосфере водорода в течение 1 часа, как показано на стадии 16-vii схемы 16. Анализ показал неполное конвертирование исходного материала в продукт, так что смесь фильтровали, обрабатывали свежим катализатором (3,0 г), затем перемешивали в атмосфере водорода в течение 90 минут при 23°C. Катализатор удаляли фильтрацией и полученный раствор концентрировали при пониженном давлении с получением 7-метил-6-(1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)-6,7-дигидро-5H-пирроло[3,4-b]пиридин-5-она (соединение 2051, 5,532 г, 18,67 ммоль, выход 100%): ESMS (M+H) 297,23; 1H ЯМР (CDCl3, 300 МГц) δ 8,80 (м, 1H), 8,28 (с, 1H), 8,19 (м, 1H), 7,76 (с, 1H), 7,47 (м, 1H), 4,98 (кв., J=6 Гц, 1H), 4,76 (кв., J=9 Гц, 2H), 1,70 (д, J=6 Гц, 3H). Анализ ЯМР показал небольшое количество перевосстановленного материала, но неочищенный продукт использовали как есть в последующих реакциях.
Как показано на стадии 16-viii схемы 16, к раствору соединения 2051 (5,532 г, 18,67 ммоль) в CHCl3 (58,20 мл) добавляли m-CPBA (6,903 г, 28,00 ммоль). Реакционную смесь перемешивали при 23°C в течение 2 суток. Реакционную смесь распределяли между насыщенным водным NaHCO3 и DCM (по 100 мл каждого) и водный слой экстрагировали DCM (100 мл). Объединенные органические фазы сушили (MgSO4), фильтровали и концентрировали при пониженном давлении. Осадок очищали хроматографией на силикагеле среднего давления (0-15% MeOH в DCM) с получением 7-метил-5-оксо-6-(1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)-6,7-дигидро-5H-пирроло[3,4-b]пиридин-1-оксида (соединение 2052, 3,47 г, 11,11 ммоль, выход 59,5%) в виде белого твердого вещества: ESMS (M+H) 313,23; 1H ЯМР (CD3OD, 300 МГц) δ 8,50 (дд, J=3,6 Гц, 1H), 8,30 (с, 1H), 7,97 (с, 1H), 7,94 (д, J=9 Гц, 1H), 7,69 (т, J=6 Гц, 1H), 5,36 (кв., J=6 Гц, 1H), 5,00 (кв., J=9 Гц, 2H), 1,74 (д, J=6 Гц, 3H).
Как показано на стадии 16-ix схемы 16, к соединению 2052 (3,47 г, 11,11 ммоль) в CHCl3 (10 мл) при 85°C добавляли POCl3 (17,04 г, 10,36 мл, 111 ммоль). После 2,5 часов при 85°C, реакционную смесь обрабатывали толуолом (30 мл), а затем концентрировали с получением темно-фиолетового прозрачного масла, которое распределяли между DCM и насыщенным водным NaHCO3 (по 300 мл каждого). Наблюдали нерастворимый темный материал. Водный слой экстрагировали DCM (3×150 мл) и объединенные органические фазы сушили (MgSO4), фильтровали и концентрировали при пониженном давлении. Осадок очищали хроматографией на силикагеле среднего давления (0-80% EtOAc в гексане) с получением 2-хлор-7-метил-6-(1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)-6,7-дигидро-5H-пирроло[3,4-b]пиридин-5-она (соединение 2053, 1,20 г, 3,64 ммоль, выход 32,8%) в виде желто-коричневого твердого вещества: ESMS (M+H) 331,19; 1H ЯМР (CDCl3, 300 МГц) δ 8,26 (с, 1H), 8,13 (д, J=9 Гц, 1H), 7,74 (с, 1H), 7,50 (д, J=9 Гц, 1H), 4,95 (кв., J=6 Гц, 1H), 4,75 (кв., J=9 Гц, 2H), 1,70 (д, J=6 Гц, 3H).
Как показано на стадии 16-x схемы 16, к соединению 2053 (366 мг, 1,107 ммоль), 2-этокси-3-метокси-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридину (371 мг, 1,328 ммоль), Na2CO3 (352 мг, 3,32 ммоль) и Pd(PPh3)4 (64 мг, 0,055 ммоль) добавляли DMF (12 мл), а затем воду (3 мл). Из реакционной емкости выкачивали воздух, помещали ее в атмосферу водорода и нагревали до 100°C (песочная баня). Через 18 часов реакционную смесь распределяли между EtOAc и водой (по 100 мл каждого). Органические слои промывали водой (2×80 мл), рассолом (80 мл) сушили (MgSO4), фильтровали и концентрировали при пониженном давлении. Осадок растворяли в горячем EtOAc (20 мл), а затем обрабатывали гексаном (20 мл). После стояния при 23°C в течение 2,5 ч полученный осадок собирали фильтрацией и сушили в вакууме с получением смеси (S)-2-(6-этокси-5-метоксипиридин-3-ил)-7-метил-6-(1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)-6,7-дигидро-5H-пирроло[3,4-b]пиридин-5-она (соединение 354) и (R)-2-(6-этокси-5-метоксипиридин-3-ил)-7-метил-6-(1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)-6,7-дигидро-5H-пирроло[3,4-b]пиридин-5-она (соединение 355) (347 мг, 0,7583 ммоль, 68,50%) в виде не совсем белых игл: ESMS (M+H) 448,39; 1H ЯМР (CDCl3, 300 МГц) δ 8,44 (д, J=3 Гц, 1H), 8,31 (с, 1H), 8,22 (д, J=9 Гц, 1H), 7,93 (д, J=3 Гц, 1H), 7,86 (д, J=9 Гц, 1H), 7,77 (с, 1H), 5,03 (кв., J=6 Гц, 1H), 4,77 (кв., J=9 Гц, 2H), 4,61 (кв., J=6 Гц, 2H), 4,04 (с, 3H), 1,76 (д, J=6 Гц, 3H), 1,52 (т, J=6 Гц, 3H). Эти два соединения разделяли сверхкритической жидкостной хроматографией на колонке Whelk-O-1® (Regis Technologies, Inc.) с использованием DMF в качестве модификатора с получением отдельных энантиомеров.
Схема 16
Пример 17. Получение 2-(5,6-диметоксипиридин-3-ил)-7-метокси-6-(1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)-6,7-дигидро-5H-пирроло[3,4-b]пиридин-5-она (соединение 256)
Как показано на стадии 17-i схемы 17, к 6-(1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)-5H-пирроло[3,4-b]пиридин-5,7(6H)-диону (соединение 2048, 2,32 г, 7,832 ммоль) в AcOH (30 мл) при 23°C добавляли Zn (2,561 г, 39,16 ммоль). После перемешивания в течение 20 минут при 23°C реакционную смесь фильтровали через стеклообразную фритту, и фильтрат концентрировали. Осадок растворяли/суспендировали в горячем EtOH (40 мл). Полученную смесь охлаждали, обрабатывали Et2O (50 мл). Полученный осадок собирали фильтрацией и маточную жидкость концентрировали при пониженном давлении и полученное твердое вещество перекристаллизовывали из горячего EtOH (20 мл) и Et2O с получением дополнительного 7-гидрокси-6-(1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)-6,7-дигидро-5H-пирроло[3,4-b]пиридин-5-она (соединение 2055, всего 1,61 г) в виде желтого твердого вещества: ESMS (M+H) 299,26; 1H ЯМР (DMSO-d6, 300 МГц) δ 8,83 (дд, J=3, 6 Гц, 1H), 8,32 (с, 1H), 8,15 (дд, J=3,9 Гц, 1H), 7,94 (с, 1H), 7,61 (дд, J=6, 9 Гц, 1H), 7,13 (д, J=9 Гц, 1H), 6,21 (д, J=9 Гц, 1H), 5,20 (кв., J=9 Гц, 2H).
Как показано на стадии 17-ii схемы 17, к раствору/суспензии соединения 2055 (1,16 г, 3,890 ммоль) в DCM (20 мл) и THF (10 мл) при 23°C добавляли TEA (1,58 г, 2,17 мл, 15,56 ммоль), а затем MsCl (668 мг, 452 мкл, 5,84 ммоль). Исходный материал переходил в раствор в течение 10 минут. Через 1 час добавляли метанол (10 мл). После перемешивания в течение дополнительных 2 часов, смесь концентрировали при пониженном давлении. Осадок распределяли между DCM и насыщенным водным NaHCO3 (по 100 мл каждого) и водный слой экстрагировали DCM (50 мл). Объединенные органические фазы сушили (MgSO4), фильтровали и концентрировали при пониженном давлении. Осадок очищали хроматографией на силикагеле среднего давления (0-100% EtOAc в гексане) с получением 7-метокси-6-(1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)-6,7-дигидро-5H-пирроло[3,4-b]пиридин-5-она (соединение 2056, 0,82 г, 2,63 ммоль, выход 67,5%) в виде белого твердого вещества: ESMS (M+H) 313,29; 1H ЯМР (CDCl3, 300 МГц) δ 8,89 (дд, J=3, 6 Гц, 1H), 8,26 (с, 1H), 8,19 (дд, J=3,9 Гц, 1H), 8,05 (с, 1H), 7,54 (дд, J=6,9 Гц, 1H), 6,15 (с, 1H), 4,76 (кв., J=9 Гц, 2H), 3,12 (с, 3H).
Как показано на стадии 17-iii схемы 17, к раствору соединения 2056 (0,82 г, 2,63 ммоль) в CHCl3 (10 мл) добавляли mCPBA (777 мг, 3,15 ммоль). Реакционную смесь перемешивали при 23°C в течение 5 минут, нагревали до 59°C (песочная баня), перемешивали при 59°C в течение 24 часов, а затем при 23°C в течение дополнительных 3 суток. Очистка хроматографией на силикагеле среднего давления (0-15% MeOH в DCM) дала 7-метокси-5-оксо-6-(1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)-6,7-дигидро-5H-пирроло[3,4-b]пиридин-1-оксид (соединение 2057, 628 мг, 1,91 ммоль, выход 73%) в виде белого твердого вещества: ESMS (M+H) 329,23; 1H ЯМР (CDCl3, 300 МГц) δ 8,49 (дд, J=2,6 Гц, 1H), 8,31 (с, 1H), 7,89 (с, 1H), 7,71 (м, 2H), 6,52 (с, 1H), 5,21 (кв., J=9 Гц, 2H), 3,18 (с, 3H).
Как показано на стадии 17-iv схемы 17, к соединению 2057 (620 мг, 1,89 ммоль) добавляли CHCl3 (3,5 мл), а затем добавляли POCl3 (5,79 г, 3,52 мл, 37,8 ммоль). Реакционную смесь нагревали до 90°C (песочная баня). Через 1,8 часов добавляли толуол (10 мл), а затем раствор концентрировали при пониженном давлении для удаления избыточного POCl3. Осадок распределяли между DCM и насыщенным водным раствором NaHCO3 (по 100 мл каждого) и водный слой экстрагировали DCM (50 мл). Объединенные органические фазы сушили (MgSO4), фильтровали, концентрировали при пониженном давлении и осадок очищали хроматографией на силикагеле среднего давления (0-65% EtOAc в гексане) с получением 2-хлор-7-метокси-6-(1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)-6,7-дигидро-5H-пирроло[3,4-b]пиридин-5-она (соединение 2058, 275 мг, 0,79 ммоль, выход 42%) в виде прозрачного масла: ESMS (M+H) 347,18; 1H ЯМР (CDCI3, 300 МГц) δ 8,23 (с, 1H), 8,13 (д, J=9 Гц, 1H), 8,02 (с, 1H), 7,57 (д, J=9 Гц, 1H), 6,08 (с, 1H), 4,76 (кв., J=9 Гц, 2H), 3,18 (с, 3H).
Как показано на стадии 17-v схемы 17, к соединению 2058 (293 мг, 0,85 ммоль), 2,3-диметокси-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридину (269 мг, 1,01 ммоль), Na2CO3 (179 мг, 1,69 ммоль) и Pd(PPh3)4 (98 мг, 0,085 ммоль) добавляли DMF (10 мл), а затем добавляли воду (2 мл). Из реакционной емкости откачивали воздух, помещали в атмосферу азота, затем нагревали до 110°C (песочная баня). Через 16 часов реакционную смесь распределяли между EtOAc и водой (по 100 мл каждого). Органические слои промывали водой (70 мл), рассолом (70 мл), сушили (MgSO4), фильтровали и концентрировали при пониженном давлении. Осадок растворяли/суспендировали в EtOAc (7 мл) и нагревали при вихревом перемешивании на водяной бане при 50°C в течение 40 минут. Полученную смесь обрабатывали гексаном (5 мл) и подвергали вихревому перемешиванию в течение дополнительных 5 минут при 50°C. После охлаждения до 23°C, полученное твердое вещество собирали фильтрацией и промывали 1:1 (EtOAc:гексан, 5 мл) с получением 2-(5,6-диметоксипиридин-3-ил)-7-метокси-6-(1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)-6,7-дигидро-5H-пирроло[3,4-b]пиридин-5-она (соединение 256) в виде смеси энантиомеров: ESMS (M+H) 450,44; 1H ЯМР (CDCl3, 300 МГц) δ 8,57 (д, J=3 Гц, 1H), 8,35 (с, 1H), 8,27 (с, 2H), 7,99 (д, J=3 Гц, 1H), 7,93 (с, 1H), 6,40 (с, 1H), 5,22 (кв., J=9 Гц, 2H), 3,96 (с, 3H), 3,93 (с, 3H), 3,13 (с, 3H).
Схема 17
Пример 18. Получение 2-(5,6-диметоксипиридин-3-ил)-7-(2-метоксиэтокси)-6-(1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)-6,7-дигидро-5H-пирроло[3,4-b]пиридин-5-она (соединение 336)
Как показано на стадии 18-i схемы 18, к 7-гидрокси-6-(1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)-6,7-дигидро-5H-пирроло[3,4-b]пиридин-5-ону (соединение 2055, 4,35 г, 14,6 ммоль) в CHCl3 (80 мл) и MeOH (40 мл) добавляли mCPBA (5,39 г, 21,9 ммоль). После перемешивания в течение 24 часов добавляли дополнительный mCPBA (1,26 г, 7,30 ммоль). После перемешивания в течение дополнительных 16 часов, полученный осадок собирали фильтрацией и промывали DCM (10 мл) и Et2O (20 мл) с получением 7-гидрокси-5-оксо-6-(1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)-6,7-дигидро-5H-пирроло[3,4-b]пиридин-1-оксида (соединение 2059, 2,49 г) в виде белого твердого вещества: ESMS (M+H) 315,25; 1H ЯМР (DMSO-d6, 300 МГц) δ 8,44 (д, J=6 Гц, 1H), 8,31 (с, 1H), 7,89 (с, 1H), 7,65 (м, 2H), 7,43 (д, J=9 Гц, 1H), 6,41 (д, J=9 Гц, 1H), 5,20 (кв., J=9 Гц, 2H).
Как показано на стадии 18-ii схемы 18, к соединению 2059 (312 мг, 0,99 ммоль) в MeCN (10 мл) добавляли K2CO3 (686 мг, 4,96 ммоль), а затем добавляли POCl3 (761 мг, 463 мкл, 4,96 ммоль). Реакционную смесь кипятили с обратным холодильником в атмосфере азота в течение 24 часов, а затем фильтровали через стеклообразную фритту. Фильтрат концентрировали при пониженном давлении, а затем распределяли между DCM (60 мл), водой (10 мл) и насыщенным водным NaHCO3 (50 мл). Водный слой экстрагировали DCM (50 мл) и объединенные органические фазы сушили (MgSO4), фильтровали и концентрировали при пониженном давлении. Осадок очищали хроматографией на силикагеле среднего давления (0-60% EtOAc в гексане) с получением 2,7-дихлор-6-(1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)-6,7-дигидро-5H-пирроло[3,4-b]пиридин-5-она (соединение 2060, 150 мг, 0,43 ммоль, 43%) в виде белого твердого вещества: 1H ЯМР (CDCl3, 300 МГц) δ 8,14 (с, 1H), 8,07 (д, J=9 Гц, 1H), 7,89 (с, 1H), 7,51 (д, J=9 Гц, 1H), 6,50 (с, 1H), 4,69 (кв., J=9 Гц, 2H).
Как показано на стадии 18-ii схемы 18, к соединению 2060 (180 мг, 0,5127 ммоль) в DMF (6 мл) добавляли 2,3-диметокси-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин (соединение 2021, 163 мг, 0,62 ммоль), Na2CO3 (217 мг, 2,05 ммоль), Pd(PPh3)4 (30 мг, 0,026 ммоль) и 2-метоксиэтанол (1,5 мл, 19,0 ммоль). Из реакционной емкости откачивали воздух, помещали ее в атмосферу азота, а затем нагревали до 100°C (песочная баня). Через 16 часов реакционную смесь распределяли между EtOAc и водой (по 100 мл каждого). Органические слои концентрировали при пониженном давлении и осадок растворяли в DMSO (5 мл) и очищали обращенно-фазовой ВЭЖХ (10-90% водный MeCN с 0,1% буфером TFA) с получением 2-(5,6-диметоксипиридин-3-ил)-7-(2-метоксиэтокси)-6-(1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)-6,7-дигидро-5H-пирроло[3,4-b]пиридин-5-она (соединение 336, 55 мг) в виде белого лиофилизата: ESMS (M+H) 494,39; 1H ЯМР (CD3OD, 300 МГц) δ 8,50 (д, J=3 Гц, 1H), 8,33 (с, 1H), 8,22 (д, J=9 Гц, 1H), 8,13 (д, J=9 Гц, 1H), 8,08 (д, J=3 Гц, 1H), 8,00 (с, 1H), 6,29 (с, 1H), 4,99 (кв., J=9 Гц, 2H), 4,03 (с, 3H), 3,97 (с, 3H), 3,66 (м, 1H), 3,53 (м, 3H), 3,29 (с, 3H).
Схема 18
Пример 19. Получение метил-2-(2-(5,6-диметоксипиридин-3-ил)-5-оксо-6-(1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)-6,7-дигидро-5H-пирроло[3,4-b]пиридин-7-ил)ацетата (соединение 307)
Как показано на стадии 19-i схемы 19, к 7-гидрокси-6-(1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)-6,7-дигидро-5H-пирроло[3,4-b]пиридин-5-ону (соединение 2055, 1,75 г, 5,87 ммоль) и метил-2-трифенилфосфоранилиденацетату (2,06 г, 6,16 ммоль) добавляли толуол (23 мл) и THF (12 мл). Реакционную смесь нагревали до температуры кипячения с обратным холодильником и удерживали при этой температуре в течение 2,5 часов. После охлаждения реакционную смесь концентрировали при пониженном давлении и осадок очищали хроматографией на силикагеле среднего давления (0-7,5% EtOH в DCM) с получением метил-2-(5-оксо-6-(1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)-6,7-дигидро-5H-пирроло[3,4-b]пиридин-7-ил)ацетата (соединение 2061, 2,25 г, 6,35 ммоль) в виде прозрачного масла: ESMS (M+H) 355,29. Продукт содержал небольшое количество оксида трифенилфосфина, однако его использовали в последующих реакциях как есть.
Как показано на стадии 19-ii схемы 19, к соединению 2061 (2,08 г, 5,89 ммоль) в CHCl3 (32 мл) добавляли mCPBA (2,17 г, 8,80 ммоль) и реакционную смесь кипятили с обратным холодильником в течение 2 часов, охлаждали и концентрировали при пониженном давлении. Полученный осадок очищали хроматографией на силикагеле среднего давления (0-12% EtOH в DCM) с получением 7-(2-метокси-2-оксоэтил)-5-оксо-6-(1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)-6,7-дигидро-5H-пирроло[3,4-b]пиридин-1-оксида (соединение 2062, 1,31 г, 3,55 ммоль, выход 60%) в виде белого твердого вещества: ESMS (M+H) 371,35; 1H ЯМР (DMSO-d6, 300 МГц) δ 8,48 (д, J=6 Гц, 1H), 8,30 (с, 1H), 7,91 (с, 1H), 7,75 (д, J=6 Гц, 1H), 7,63 (т, J=3 Гц, 1H), 5,63 (м, 1H), 5,21 (кв., J=9 Гц, 2H), 3,56 (дд, J=3,15 Гц, 1H), 3,36 (с, 3H), 3,16 (дд, J=6,15 Гц, 1H).
Как показано на стадии 19-iii схемы 19, к соединению 2062 (1,06 г, 2,86 ммоль) добавляли POCl3 (13,17 г, 8,00 мл, 85,9 ммоль). Реакционную смесь нагревали при 90°C (песочная баня) в течение 2,5 часов, а затем POCl3 удаляли при пониженном давлении. Осадок распределяли между насыщенным водным раствором NaHCO3 и DCM (по 100 мл каждого) и водный слой экстрагировали DCM (50 мл). Объединенные органические фазы сушили (MgSO4), фильтровали и концентрировали при пониженном давлении. Осадок очищали хроматографией на силикагеле среднего давления (0-100% EtOAc в гексане с получением метил-2-(2-хлор-5-оксо-6-(1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)-6,7-дигидро-5H-пирроло[3,4-b]пиридин-7-ил)ацетата (соединение 2063, 378 мг, 0,97 ммоль, выход 34%) в виде белого твердого вещества: ESMS (M+H) 389,33; 1H ЯМР (CDCl3, 300 МГц) δ 8,13 (д, J=9 Гц, 1H), 8,12 (с, 1H), 7,75 (с, 1H), 7,51 (д, J=9 Гц, 1H), 5,30 (м, 1H), 4,76 (кв., J=9 Гц, 2H), 3,62 (с, 3H), 3,10 (м, 2H).
Как показано на стадии 19-iv схемы 19, к соединению 2063 (375 мг, 0,965 ммоль), 2,3-диметокси-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридину (соединение 2021, 307 мг, 1,16 ммоль), Na2CO3 (205 мг, 1,93 ммоль), Pd(PPh3)4 (112 мг, 0,0965 ммоль) добавляли DMF (12 мл), а затем добавляли воду (2,5 мл). Из реакционной емкости откачивали воздух, помещали ее в атмосферу азота, а затем нагревали до 110°C (песочная баня). Через 18 часов реакционную смесь охлаждали и распределяли между EtOAc и водой (по 100 мл каждого). Органические слои промывали рассолом (50 мл), сушили (MgSO4) фильтровали и концентрировали при пониженном давлении. Осадок суспендировали в горячем EtOAc (20 мл) и подвергали вихревому перемешиванию в течение 45 мин при 60°C до получения однородной суспензии, который позволяли стоять при 23°C в течение 24 часов. Полученное твердое вещество собирали фильтрацией, растворяли в теплом DMSO (50 мл) и фильтровали через 0,45-мкм мембрану PTFE (фильтр в виде шприца). Фильтрат обрабатывали водой (5 мл) и полученный осадок собирали фильтрацией, промывали водой (10 мл), суспендировали в теплом MeCN (5 мл) и обрабатывали водой (5 мл). Полученную суспензию замораживали и лиофилизировали с получением метил-2-(2-(5,6-диметоксипиридин-3-ил)-5-оксо-6-(1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)-6,7-дигидро-5H-пирроло[3,4-b]пиридин-7-ил)ацетата (соединение 307, 195 мг, 0,38 ммоль, выход 39%) в виде белого твердого вещества: ESMS (M+H) 491,86; 1H ЯМР (DMSO-d6, 300 МГц) δ 8,56 (д, J=3 Гц, 1H), 8,30 (с, 1H), 8,23 (д, J=9 Гц, 1H), 8,19 (д, J=9 Гц, 1H), 7,99 (д, J=3 Гц, 1H), 7,89 (с, 1H), 5,51 (м, 1H), 5,21 (кв., J=9 Гц, 2H), 3,96 (с, 3H), 3,92 (с, 3H), 3,46 (с, 3H), 3,26 (дд, J=3,15 Гц, 1H), 3,03 (дд, J=6,15 Гц, 1H).
Схема 19
Пример 20. Получение 6-(1-(2-(1H-пиразол-1-ил)этил)-1H-пиразол-4-ил)-2-(5,6-диметоксипиридин-3-ил)-6,7-дигидро-5H-пирроло[3,4-b]пиридин-5-она (соединение 171)
Как показано на стадии 20-i схемы 20, 2-(5,6-диметоксипиридин-3-ил)-6-(1H-пиразол-4-ил)-6,7-дигидро-5H-пирроло[3,4-b]пиридин-5-он [соединение 70, 100 мг, 0,2964 ммоль, полученный из 2-хлор-6-(1H-пиразол-4-ил)-6,7-дигидро-5H-пирроло[3,4-b]пиридин-5-она и 2,3-диметокси-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридина (соединение 2021) аналогично получению соединения 135, как представлено в примере 12], и карбонат цезия (193 мг, 0,593 ммоль) отвешивали в коническую колбу для обработки микроволновым излучением, оборудованную магнитной мешалкой. Добавляли DMF (1,05 мл), а затем добавляли 1-(2-хлорэтил)пиразол (77 мг, 0,593 ммоль). Колбу закрывали и нагревали при 120°C в течение 15 минут. Добавляли воду (3 мл) и полученный осадок собирали фильтрацией и промывали 5 мл воды. Фильтрат концентрировали при пониженном давлении. Как собранное твердое вещество, так и осадок после концентрирования фильтрата разбавляли DMSO до солюбилизации и очищали обращенно-фазовой ВЭЖХ с получением 6-(1-(2-(1H-пиразол-1-ил)этил)-1H-пиразол-4-ил)-2-(5,6-диметоксипиридин-3-ил)-6,7-дигидро-5H-пирроло[3,4-b]пиридин-5-она (соединение 171, 22 мг, 0,05 ммоль, выход 17%): ESMS (M+H) 432,0; 1H ЯМР (DMSO-d6, 300 МГц) δ 8,47 (д, J=2,0 Гц, 1H), 8,10 (с, 2H), 7,95-7,87 (м, 2H), 7,75 (с, 1H), 7,45 (д, J=2,2 Гц, 1H), 7,40 (д, J=1,3 Гц, 1H), 6,12 (т, J=2,0 Гц, 1H), 4,84 (с, 2H), 4,50 (с, 4H), 3,88 (с, 3H), 3,85 (с, 3H).
Схема 20
Пример 21. Получение 6-(5,6-диметоксипиридин-3-ил)-4-метил-2-(1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)-1H-пирроло[3,4-c]пиридин-3(2H)-она (соединение 243)
Как показано на стадии 21-i схемы 21, в круглодонную колбу, содержащую 1-(2,2,2-трифторэтил)пиразол-4-амин (2,01 г, 12,2 ммоль) и карбонат калия (3,364 г, 24,34 ммоль) в атмосфере азота добавляли DMF (15 мл), а затем добавляли 3-бромпроп-1-ин (1,45 г, 1,09 мл, 12,2 ммоль). Реакционную смесь перемешивали при К.Т. в течение 18 часов. Добавляли воду и EtOAc и водный слой экстрагировали EtOAc. Объединенные органические фазы промывали рассолом, водой, сушили (сульфат натрия), фильтровали и концентрировали при пониженном давлении. Очитка хроматографией на силикагеле (петролейный эфир:EtOAc, 1:1) дала N-(проп-2-инил)-1-(2,2,2-трифторэтил)-1H-пиразол-4-амин (соединение 2064, 1,19 г, выход 48%) в виде оранжевого твердого вещества: 1H ЯМР (CDCl3, 300 МГц) δ 7,32 (1H, с), 7,16 (1H, с), 4,62 (2H, кв.), 3,81 (2H, дд), 3,20 (1H, т), 2,27 (1H, т).
Как показано на стадии 21-ii схемы 21, к раствору соединения 2064 (1,19 г, 5,88 ммоль) в DCM (20 мл) добавляли DIEA (2,28 г, 3,07 мл, 17,6 ммоль), бут-2-иновую кислоту (544 мг, 6,47 ммоль) и DMAP (36 мг, 0,29 ммоль). Реакционную смесь охлаждали на ледяной бане, добавляли EDCI (1,05 г, 6,76 ммоль) и холодную баню удаляли через 3 минуты. После перемешивания реакционной смеси в течение 18 часов при К.Т. добавляли воду и DCM и слои разделяли. Водный слой экстрагировали DCM и объединенные органические фазы промывали рассолом, водой, сушили (сульфат натрия), фильтровали и концентрировали при пониженном давлении. Очистка хроматографией на силикагеле (петролейный эфир:EtOAc, 1:1) дала N-(проп-2-инил)-N-(1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)бут-2-инамид (соединение 2065, 650 мг, выход 41%) в виде белого твердого вещества: 1H ЯМР (CDCl3, 300 МГц) δ 8,18 (1H, с), 7,77 (1H, с), 7,71 (1H, с), 7,69 (1H, с), 4,78 (2H, д), 4,76-4,63 (2H, м), 4,46 (2H, д), 2,40 (1H, т), 2,27 (1H, т), 2,13 (2H, с), 1,83 (2H, с).
Как показано на стадии 21-iii схемы 21, к раствору этил-N-(оксометилен)карбамата (385 мг, 345 мкл, 3,34 ммоль) и Cp*RuCl(cod) (21 мг, 0,056 ммоль) в сухом 1,2-дихлорэтане (3 мл) добавляли раствор соединения 2065 (303 мг, 1,11 ммоль) в 1,2-дихлорэтане (6 мл) в течение 25 мин в атмосфере азота при К.Т. Реакционную смесь нагревали при 65°C в течение 1 ч, затем концентрировали при пониженном давлении. Очистка хроматографией на силикагеле (петролейный эфир:EtOAc, градиент от 1/1 до 0/1) дала этил-4-метил-3,6-диоксо-2-(1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)-2,3-дигидро-1H-пирроло[3,4-c]пиридин-5(6H)-карбоксилат (соединение 2066, 179 мг, выход 41%) в виде белого твердого вещества: 1H ЯМР (DMSO-d6, 300 МГц) δ 8,28 (1H, с), 7,84 (1H, д), 6,52 (1H, с), 5,18 (2H, кв.), 4,75 (2H, с), 4,51 (2H, кв.), 2,67 (2H, с), 1,36 (2H, т).
Как показано на стадии 21-iv схемы 21, к раствору соединения 2066 (614 мг, 1,58 ммоль) в THF (10 мл) добавляли HCl (6 M, 10 мл) при К.Т. Реакционную смесь нагревали при кипячении с обратным холодильником в течение ночи, а затем концентрировали при пониженном давлении. К осадку добавляли оксихлорид фосфора (15 мл, 161 ммоль) и реакционную смесь нагревали при 95°C в течение 3 часов в атмосфере азота. После охлаждения и концентрирования смеси при пониженном давлении добавляли лед, а затем добавляли EtOAc через 30 минут. Водный слой экстрагировали EtOAc и объединенные органические фазы промывали рассолом, сушили (сульфат натрия), фильтровали и концентрировали при пониженном давлении. Очистка хроматографией на силикагеле (петролейный эфир:EtOAc, 1:1) дала 6-хлор-4-метил-2-(1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)-1H-пирроло[3,4-c]пиридин-3(2H)-он (соединение 2067, 356 мг, 68%) в виде белого твердого вещества: ESMS (M+H) 330,90.
Как показано на стадии 21-iv схемы 21, соединение 2067 (129 мг, 0,390 ммоль) и соединение 2021 (134 мг, 0,51 ммоль) отбирали в DMF и карбонат натрия (1 M, 0,780 ммоль) и через раствор пропускали азот в течение 30 минут. Добавляли тетракистрифенилфосфинпалладий(0) (23 мг, 0,020 ммоль) и реакционную смесь продували азотом в течение дополнительных 5 мин, затем нагревали при 110°C в течение ночи. После охлаждения реакционной смеси до комнатной температуры добавляли EtOAc и воду. Водный слой экстрагировали EtOAc и объединенные органические фазы сушили (сульфат натрия), фильтровали через диатомитовую землю и концентрировали при пониженном давлении. Очистка хроматографией на силикагеле (EtOAc) дала 6-(5,6-диметоксипиридин-3-ил)-4-метил-2-(1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)-1H-пирроло[3,4-c]пиридин-3(2H)-он (соединение 243, 137 мг; выход 77%) в виде белого твердого вещества.
Схема 21
Пример 22. Получение 2-(5,6-диметоксипиридин-3-ил)-7-метил-6-(1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)-6,7-дигидро-5H-пирроло[3,4-b]пиридин-5-она (соединение 254)
Как показано на стадии 22-i схемы 22, в 1-л круглодонную колбу, оборудованную конденсатором, помещали 2,6-дихлорпиридин-3-карбоновую кислоту (10,0 г, 52,1 ммоль), 2,3-диметокси-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин (соединение 2021, 13,81 г, 52,1 ммоль), Pd(PPh3)4 (3,01 г, 2,60 ммоль), Na2CO3 (16,56 г, 156 ммоль), диоксан (250 мл) и воду (100 мл). Из колбы откачивали воздух в течение 1 минуты и смесь помещали в атмосферу N2. Смесь нагревали при 110°C в течение 16 часов, после чего образовывался осадок. Реакционную смесь охлаждали и переносили в делительную воронку. Добавляли Na2CO3 (200 мл 10 масс.%, водный), а затем воду (100 мл) и EtOAc (500 мл). Оставался осадок/эмульсия, главным образом, расположенные в водном слое. Водный слой отделяли, промывали EtOAc (300 мл), а затем осторожно подкисляли концентрированной HCl (~50 мл) до pH 2. Полученный осадок собирали фильтрацией и промывали водой (50 мл). Влажное твердое вещество переносили в 1-л колбу с добавлением EtOH (200 мл), затем упаривали до сухого состояния. Твердый осадок растворяли/суспендировали в EtOAc (120 мл), а затем обрабатывали гексаном (120 мл). Полученное твердое вещество собирали фильтрацией, промывали гексаном (50 мл) и сушили при пониженном давлении с получением 2-хлор-6-(5,6-диметокси-3-пиридил)пиридин-3-карбоновой кислоты (соединение 2068, 11,99 г, выход 78%) в виде не совсем белого твердого вещества: ESMS (M+H) 295,27; 1H ЯМР (DMSO-d6, 300 МГц) δ 13,72 (с, 1H), 8,49 (д, J=1,98, 1H), 8,30 (д, J=8,08, 1H), 8,15 (д, J=8,11, 1H), 7,88 (д, J=1,98, 1H), 3,95 (с, 3H), 3,91 (с, 3H).
Как показано на стадии 22-ii схемы 22, к раствору 1-(2,2,2-трифторэтил)пиразол-4-амина (соединение 2047, 5,78 г, 35,0 ммоль), 2-хлор-6-(5,6- диметокси-3-пиридил)пиридин-3-карбоновой кислоты (соединение 2068, 9,38 г, 31,8 ммоль) и HBTU (13,28 г, 35,0 ммоль) в DMF (150 мл) при 23°C добавляли DIEA (12,35 г, 16,64 мл, 95,52 ммоль). Реакционную смесь перемешивали в течение 2 ч, а затем распределяли между EtOAc (400 мл) и водой (400 мл). Органический слой отделяли, промывали водой (400 мл), 10% водным раствором Na2CO3 (300 мл), рассолом (300 мл), комбинированным рассолом и 2н HCl (300 мл, 20 мл) и рассолом (300 мл). Затем органический слой разбавляли EtOAc (150 мл) и EtOH (70 мл) и нагревали 75°C при вихревом перемешивании с получением прозрачного раствора. Раствор обрабатывали MgSO4 и фильтровали, пока он еще был теплым. После концентрирования при пониженном давлении осадок растворяли/суспендировали EtOAc (200 мл) и центрифугировали при 80°C в течение 1 ч с получением однородной суспензии. Добавляли гексан (200 мл) и полученную суспензию оставляли стоять при 23°C в течение 14 ч. Осадок собирали фильтрацией, промывали гексаном (100 мл) с получением 2-хлор-6-(5,6-диметокси-3-пиридил)-N-[1-(2,2,2-трифторэтил)пиразол-4-ил]пиридин-3-карбоксамида (соединение 2069, 11,32 г, выход 80%) в виде белого твердого вещества после высушивания в вакууме: ESMS (M+H) 442,50; 1H ЯМР (DMSO-d6, 300 МГц) δ 10,86 (с, 1H), 8,49 (д, J=1,89, 1H), 8,25 (с, 1H), 8,16 (м, 2H), 7,88 (д, J=1,89, 1H), 7,66 (с, 1H), 5,16 (кв., J=9,10, 2H), 3,95 (с, 3H), 3,92 (с, 3H).
Как показано на стадии 22-iii схемы 22, в 250-мл колбу Парра помещали 2-хлор-6-(5,6-диметокси-3-пиридил)-N-[1-(2,2,2-трифторэтил)пиразол-4-ил]пиридин-3-карбоксамид (соединение 2069, 5,00 г, 11,32 ммоль), PdCl2(CH3CN)2 (147 мг, 0,566 ммоль) и дициклогексил-[2-(2,4,6-триизопропилфенил)фенил]фосфан (378 мг, 0,792 ммоль). Через смесь барботировали азот в течение 3 мин, а затем добавляли триэтиламин (5,727 г, 7,888 мл, 56,60 ммоль), а затем этинил(триметил)силан (3,34 г, 4,80 мл, 34,0 ммоль). Емкость закрывали и нагревали до 100°C. Через 14 часов реакционную смесь охлаждали до 23°C и распределяли между EtOAc (300 мл) и водой (300 мл). Органический слой отделяли, промывали водой (300 мл), насыщенным водным NaHCO3 (200 мл), рассолом (300 мл), сушили (MgSO4), фильтровали и концентрировали при пониженном давлении. Осадок очищали хроматографией на силикагеле среднего давления: 0-100% EtOAc в гексане с получением 3,7 г желтовато-коричневого твердого вещества. Твердое вещество растворяли в EtOAc (15 мл, горячий, 70°C), затем обрабатывали гексаном (30 мл). Полученную суспензию центрифугировали и охлаждали на ледяной бане в течение 40 мин, затем осадок собирали фильтрацией, промывали гексаном (30 мл) с получением 6-(5,6-диметокси-3-пиридил)-N-[1-(2,2,2-трифторэтил)пиразол-4-ил]-2-(2-триметилсилилэтинил)пиридин-3-карбоксамида (соединение 2070, 3,14 г, 55%) в виде светло-желтого твердого вещества: ESMS (M+H) 504,63; 1H ЯМР (CDCl3, 300 МГц) δ 9,69 (с, 1H), 8,54 (д, J=8,44, 1H), 8,36 (д, J=1,96, 1H), 8,28 (с, 1H), 7,87 (д, J=1,93, 1H), 7,83 (д, J=8,48 Гц, 1H), 7,62 (с, 1H), 4,71 (кв., J=8,33, 2H), 4,11 (с, 3H), 4,03 (с, 3H).
Как показано на стадии 22-iv схемы 22, к раствору 6-(5,6-диметокси-3-пиридил)-N-[1-(2,2,2-трифторэтил)пиразол-4-ил]-2-(2-триметилсилилэтинил)пиридин-3-карбоксамида (соединение 2070, 540 мг, 1,07 ммоль) в EtOH (18,5 мл) при 23°C капельно добавляли EtONa (165 мкл 1,3 M раствора в EtOH, 0,214 ммоль). Через 25 мин полученную взвесь охлаждали до 0°C и, после перемешивания в течение 10 мин при 0°C, взвесь фильтровали и собранное твердое вещество промывали ледяным EtOH (3×10 мл) с получением 2-(5,6-диметокси-3-пиридил)-7-метилен-6-[1-(2,2,2-трифторэтил)пиразол-4-ил]пирроло[3,4-b]пиридин-5-она (соединение 2071, 436 мг, 93%) в виде светло-желтого твердого вещества после высушивания в вакууме: ESMS (M+H) 432,52; 1H ЯМР (CDCl3, 300 МГц) δ 8,38 (д, J=1,96, 1H), 8,15 (д, J=8,13, 1H), 7,88 (д, J=1,98, 1H), 7,84 (с, 1H), 7,82 (д, J=8,16 Гц, 1H), 7,74 (с, 1H), 5,86 (д, J=1,89, 1H), 5,11 (д, J=1,89, 1H), 4,72 (кв., J=8,32, 2H), 4,05 (с, 3H), 3,96 (с, 3H).
Как показано на стадии 22-v схемы 22, к раствору 2-(5,6-диметокси-3-пиридил)-7-метилен-6-[1-(2,2,2-трифторэтил)пиразол-4-ил]пирроло[3,4-b]пиридин-5-она (соединение 2071, 436 мг, 1,01 ммоль) в THF (20 мл) добавляли Pd/C (200 мг, 10 масс.% в расчете на сухую массу, влажный, типа Degussa). Из реакционной емкости откачивали воздух, а затем помещали в атмосферу H2 (баллон). После перемешивания в течение 2,5 ч, реакционную смесь фильтровали через слой кремнизема и промывали THF (80 мл). Полученный фильтрат концентрировали при пониженном давлении и осадок обрабатывали EtOAc (6 мл). После нагревания до температуры кипячения с обратным холодильником до получения однородной суспензии добавляли гексан (10 мл). Полученную суспензию охлаждали до 0°C (баня с ледяной водой), держали при 0°C в течение 5 мин, и осадок собирали фильтрацией с получением 2-(5,6-диметокси-3-пиридил)-7-метил-6-[1-(2,2,2-трифторэтил)пиразол-4-ил]-7H-пирроло[3,4-b]пиридин-5-она (соединение 254, 300 мг, 68%) в виде светло-желто-коричневого твердого вещества: ESMS (M+H) 434,44; 1H ЯМР (DMSO-d6, 300 МГц) δ 8,57 (д, J=3 Гц, 1H), 8,39 (с, 1H), 8,23 (д, J=9 Гц, 1H), 8,18 (д, J=9 Гц, 1H), 8,00 (д, J=3 Гц, 1H), 7,97 (с, 1H), 5,22 (м, 3H), 3,96 (с, 3H), 3,93 (с, 3H), 1,60 (д, J=6 Гц, 3H).
Схема 22
Пример 23. Получение 2'-(5-метоксипиридин-3-ил)-4'-метил-6'-(1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)спиро[циклопропан-1,7'-пирроло[3,4-b]пиридин]-5'(6'H)-она (соединение 651)
Как показано на стадии 23-i схемы 23, 1,2-дибромэтан (369,3 мг, 169,4 мкл, 1,966 ммоль) добавляли к перемешиваемому раствору 2-хлор-4-метил-6-[1-(2,2,2-трифторэтил)пиразол-4-ил]-7H-пирроло[3,4-b]пиридин-5-она (500 мг, 1,512 ммоль, полученный реакцией 1-(2,2,2-трифторэтил)-4-аминопиразола с соединением 2041) в DMF (12 мл) при К.Т., а затем добавляли NaH (133 мг, 3,326 ммоль, 60 масс.% дисперсия в минеральном масле). Реакционную смесь перемешивали в течение 30 мин при К.Т., охлаждали до 0°C и гасили насыщенным NaHCO3 (10 мл). Реакционную смесь экстрагировали DCM (3x10 мл) и объединенные органические фазы сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Неочищенный осадок очищали хроматографией на силикагеле среднего давления (0-50% EtOAc/гексан) с получением 2'-хлор-4'-метил-6'-(1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)спиро[циклопропан-1,7'-пирроло[3,4-b]пиридин]-5'(6'H)-она (соединение 2073, 250 мг, выход 47%): ESMS (M+H) 358,0.
Как показано на стадии 23-ii схемы 23, ацетат калия (20,64 мг, 0,2103 ммоль) и Pd(PPh3)4 (16,20 мг, 0,01402 ммоль) добавляли к раствору соединения 2073 (50 мг, 0,1402 ммоль) и 3-метокси-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридина (соединение 2074, 49,44 мг, 0,2103 ммоль) в DMF (383,6 мкл) и H2O (127,9 мкл). Раствор дегазировали, а затем нагревали до 100°C с помощью микроволнового излучения в течение 1 часа. Реакционную смесь концентрировали и осадок очищали хроматографией на силикагеле среднего давления (0-100% EtOAc/гексан) с получением 2'-(5-метоксипиридин-3-ил)-4'-метил-6'-(1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)спиро[циклопропан-1,7'-пирроло[3,4-b]пиридин]-5(6'H)-она (соединение 651, 30 мг, выход 47%) в виде белого твердого вещества: ESMS (M+H) 430,59.
С использованием соответствующих промежуточных соединений, защищенных при необходимости, также получали соединения 832, 833, 957 и 966 по сходной методике. Эту методику также использовали в качестве альтернативы методике, описанной в примере 15 для получения соединения 311.
Схема 23
Пример 24. Получение 2-(5-метокси-3-пиридил)-4-метил-6-[1-(2,2,2-трифторэтил)пиразол-4-ил]спиро[пирроло[3,4-b]пиридин-7,3'-тетрагидрофуран]-5-она (соединение 972)
Как показано на стадии 24-i схемы 24, триметилсилилцианид (4,241 г, 5,700 мл, 42,75 ммоль) добавляли к раствору 1-(2,2,2-трифторэтил)пиразол-4-амина (7,059 г, 42,75 ммоль) и тетрагидрофуран-3-она (3,68 г, 42,75 ммоль) в AcOH (45 мл) при 0°C через шприц в течение 30 секунд. Реакционную смесь медленно нагревали до 23°C. После перемешивания в течение 16 часов смесь добавляли к смеси 1:1 гидроксид аммония:лед (200 мл) и экстрагировали DCM (2×200 мл). Органические слои сушили (сульфат магния) фильтровали и концентрировали. Осадок очищали хроматографией на силикагеле среднего давления (0-100% EtOAc в гексане с получением 3-[[1-(2,2,2-трифторэтил)пиразол-4-ил]амино]тетрагидрофуран-3-карбонитрила (соединение 2075, 3,66 г, 14,07 ммоль, выход 32,91%) в виде коричневого масла: ESMS (M+H) 261,32; 1H ЯМР (CDCl3, 300 МГц) δ 7,47 (с, 1H), 7,46 (с, 1H), 4,68 (кв., J=9,2H), 4,06 (м, 4H), 2,42 (м, 3H).
Как показано на стадии 24-ii схемы 24, к раствору бут-2-иновой кислоты (833,6 мг, 9,915 ммоль) в DCM (12 мл) при 0°C добавляли 1-хлор-N,N,2-триметилпроп-1-ен-1-амин (1,325 г, 1,312 мл, 9,915 ммоль). После перемешивания в течение 40 минут реакционную смесь охлаждали до -78°C и добавляли раствор 3-[[1-(2,2,2-трифторэтил)пиразол-4-ил]амино]тетрагидрофуран-3-карбонитрила (соединение 2075, 1,72 г, 6,610 ммоль) и DIEA (2,563 г, 3,454 мл, 19,83 ммоль) в DCM (12 мл). Реакционную смесь нагревали до 0°C (ледяная баня) и через 1 час смесь нагревали до 23°C. После 30 мин при 23°C реакционную смесь распределяли между водой и EtOAc (по 100 мл каждого). Органические слои отделяли (присутствовал нерастворимый осадок), промывали водой, затем рассолом (по 100 мл каждого), сушили (сульфат магния), фильтровали и концентрировали при пониженном давлении. Осадок очищали хроматографией на силикагеле среднего давления (0-80% EtOAc/гексан) с получением N-(3-цианотетрагидрофуран-3-ил)-N-[1-(2,2,2-трифторэтил)пиразол-4-ил]бута-2,3-диенамид (соединение 2076, 1,48 г) в виде желтого масла: ESMS (M+H) 327,20; 1H ЯМР (CDCl3, 300 МГц) δ 7,70 (с, 1H), 7,63 (с, 1H), 5,57 (т, J=6, 1H), 5,19 (д, J=6, 2H), 4,77 (кв., J=9, 2H), 4,00 (м, 4H), 2,50 (м, 1H), 2,30 (м, 1H).
Как показано на стадии 24-iii схемы 24, к раствору дитрет-бутилпропандиоата (3,579 г, 3,705 мл, 16,55 ммоль) в THF (50 мл) при 23°C добавляли NaH (496,4 мг, 12,41 ммоль). После перемешивания в течение 20 минут добавляли раствор соединения 2076 (2,70 г, 8,275 ммоль) в THF (50 мл). Через 1 час реакционную смесь гасили насыщенным водным раствором хлорида аммония (100 мл) и распределяли между водой и EtOAc (по 150 мл каждого). Органические слои отделяли, промывали рассолом (200 мл), содержащим 1н HCl (10 мл водн.), промывали рассолом (150 мл), сушили (сульфат магния), фильтровали и концентрировали при пониженном давлении. Осадок очищали хроматографией на силикагеле среднего давления (0-100% EtOAc в гексане) с получением промежуточного соединения трет-бутил-4'-метил-2',5'-диоксо-6'-(1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)-1',2',4,5,5',6'-гексагидро-2H-спиро[фуран-3,7'-пирроло[3,4-b]пиридин]-3'-карбоксилата (2,03 г, 4,334 ммоль) в виде светло-желтого твердого вещества: ESMS (M+H) 469,31; 1H ЯМР (DMSO-d6, 300 МГц) δ 12,80 (шир.с, 1H), 8,16 (с, 1H), 7,80 (с, 1H), 5,17 (кв., J=9, 2H), 4,18 (д, J=12, 1H), 3,90 (м, 2H), 3,75 (м, 1H), 2,45 (м, 1H), 2,44 (с, 3H), 2,33 (м, 1H), 1,52 (с, 9H).
Как показано на стадии 24-iv схемы 24, трет-бутил-4'-метил-2',5'-диоксо-6'-(1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)-1',2',4,5,5',6'-гексагидро-2H-спиро[фуран-3,7'-пирроло[3,4-b]пиридин]-3'-карбоксилат (2,00 г, 4,270 ммоль) отбирали в DCM (25 мл) при 23°C и добавляли TFA (25 мл). Через 30 минут реакционную смесь обрабатывали толуолом (80 мл) и концентрировали при пониженном давлении с получением 4'-метил-2',5'-диоксо-6'-(1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)-1',2',4,5,5',6'-гексагидро-2H-спиро[фуран-3,7'-пирроло[3,4-b]пиридин]-3'-карбоновой кислоты (соединение 2077, 1,776 г, 4,307 ммоль, 52,37%) общий выход) в виде не совсем белого твердого вещества: ESMS (M+H) 413,32; 1H ЯМР (DMSO-d6, 300 МГц) δ 13,50 (шир.с, 1H), 8,16 (с, 1H), 7,79 (с, 1H), 5,17 (кв., J=9, 2H), 4,15 (д, J=12, 1H), 3,99 (м, 2H), 3,85 (м, 1H), 2,63 (с, 3H), 2,55 (м, 1H), 2,35 (м, 1H).
Как показано на стадии 24-v схемы 24, к суспензии соединения 2077 (1,77 г, 4,293 ммоль) в MeCN (10 мл) добавляли LiOH дигидрат (270,2 мг, 6,440 ммоль), а затем воду (10 мл). После перемешивания в течение 5 минут, добавляли NBS (802,4 мг, 4,508 ммоль). После в общем 40 мин после добавления NBS реакционную смесь распределяли между EtOAc и водой (по 100 мл каждого). Водный слой обрабатывали 1н водн. HCl (8 мл) с получением белого осадка. Полученную суспензию концентрировали и осадок расщепляли в горячем EtOH (25 мл) с получением суспензии, которую обрабатывали водой (25 мл) и оставляли стоять при 23°C в течение 2 ч. Затем осадок собирали фильтрацией и промывали водой (20 мл). Высушивание твердого вещества при пониженном давлении дало 3'-бром-4'-метил-6'-(1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)-4,5-дигидро-2H-спиро[фуран-3,7'-пирроло[3,4-иридин]-2',5'(1'H,6'H)-дион (соединение 2078, 1,65 г, 3,690 ммоль, выход 85,94%) в виде не совсем белого твердого вещества: ESMS (M+H) 447,17; 1H ЯМР (DMSO-d6, 300 МГц) δ 13,20 (шир.с, 1H), 8,14 (с, 1H), 7,78 (с, 1H), 5,17 (кв., J=9, 2H), 4,14 (д, J=9, 1H), 3,98 (м, 2H), 3,83 (м, 1H), 2,59 (с, 3H), 2,49 (м, 1H), 2,32 (м, 1H).
Как показано на стадии 24-vi схемы 24, к суспензии соединения 2078 (1,65 г, 3,690 ммоль) в EtOH (50 мл) при 23°C добавляли триэтиламин (1,120 г, 1,543 мл, 11,07 ммоль), а затем добавляли Pd/C (430 мг, 0,4041 ммоль) (10 масс.% в расчете на сухую массу, типа Degussa, влажный). Из реакционной емкости откачивали воздух и газообразную среду реакционной смеси заменяли газообразным водородом. После перемешивания в течение 16 часов реакционную смесь обрабатывали MeOH и DCM (по 50 мл каждого), а затем фильтровали через диатомитовую землю, которую затем промывали 4:1 DCM:MeOH (100 мл). Объединенный фильтрат концентрировали при пониженном давлении с получением 4-метил-6-[1-(2,2,2-трифторэтил)пиразол-4-ил]спиро[1H-пирроло[3,4-b]пиридин-7,3'-тетрагидрофуран]-2,5-диона в виде белого твердого вещества (соединение 2079, 2 г, примеси триэтиламина), которое использовали при последующих реакциях: ESMS (M+H) 369,30; 1H ЯМР (DMSO-d6, 300 МГц) δ 8,16 (с, 1H), 7,80 (с, 1H), 6,34 (шир.с, 1H), 5,17 (кв., J=9, 2H), 4,13 (д, J=12, 1H), 3,93 (м, 3H), 2,45 (с, 3H), 2,30 (м, 2H).
Как показано на стадии 24-vii схемы 24, к раствору/суспензии соединения 2079 (1,359 г, 3,69 ммоль, оцененная масса исходного материала, из расчета 100% конвертирования на стадии 24-vi) в DCM (30 мл) добавляли DIEA (1,431 г, 1,929 мл, 11,07 ммоль), а затем добавляли N-(5-хлор-2-пиридил)-1,1,1-трифтор-N-(трифторметилсульфонил)метансульфонамид (реагент Коммина, 1,594 г, 4,059 ммоль). Реакционная смесь становится однородной в течение <10 минут. После перемешивания в течение 3 ч, добавляли дополнительный реагент Коммина (400 мг) и реакционную смесь перемешивали в течение дополнительных 90 минут. Смесь концентрировали, наносили непосредственно на колонку для хроматографии с силикагелем в DCM (15 мл), и очищали хроматографией на силикагеле среднего давления (0-50% EtOAc в гексане). Выделенный продукт содержал примеси реагента Коммина, так что его растворяли в DCM (100 мл), промывали насыщенным водным раствором бикарбоната натрия (100 мл), сушили (сульфат магния), фильтровали и концентрировали при пониженном давлении с получением 4'-метил-5'-оксо-6'-(1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)-4,5,5',6'-тетрагидро-2H-спиро[фуран-3,7'-пирроло[3,4-b]пиридин]-2'-илтрифторметансульфоната (соединение 2080, 1,757 г, 3,436 ммоль, 93,14%) в виде желтого масла: ESMS (M+H) 501,24; 1H ЯМР (DMSO-d6, 300 МГц) δ 8,29 (с, 1H), 7,90 (с, 1H), 7,64 (с, 1H), 5,22 (кв., J=9, 2H), 4,24 (д, J=12, 1H), 4,07 (м, 1H), 3,90 (м, 2H), 2,74 (с, 3H), 2,39 (м, 2H).
Как показано на стадии 24-viii схемы 24, в емкость для обработки микроволновым излучением помещали соединение 2080, 3-метокси-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин (121,4 мг, 0,5164 ммоль), карбонат натрия (136,8 мг, 1,291 ммоль), DMF (5 мл) и воду (2,5 мл). Через реакционную смесь пропускали поток азота в течение 5 мин, затем добавляли Pd(PPh3)4 (24,87 мг, 0,02152 ммоль). Через смесь барботировали газообразный азот в течение дополнительных 3 минут, а затем реакционную емкость закрывали мембраной и нагревали до 105°C (песочная баня). После 16 часов при этой температуре реакционную смесь распределяли между EtOAc и водой (по 100 мл каждого). Органические слои отделяли, промывали рассолом (50 мл), сушили (сульфат магния), фильтровали и концентрировали. Осадок очищали хроматографией на силикагеле среднего давления (0-8% EtOH в DCM) с получением неочищенного продукта, который растворяли/суспендировали в горячем EtOAc (2 мл), обрабатывали гексаном (3 мл) и оставляли стоять при 23°C в течение 30 минут. Полученный осадок собирали фильтрацией и сушили при пониженном давлении с получением 2-(5-метокси-3-пиридил)-4-метил-6-[1-(2,2,2-трифторэтил)пиразол-4-ил]спиро[пирроло[3,4-b]пиридин-7,3'-тетрагидрофуран]-5-она (соединение 972, 87 мг, 0,1866 ммоль, выход 43%) в виде не совсем белого твердого вещества: ESMS (M+H) 460,35; 1H ЯМР (DMSO-d6, 300 МГц) δ 8,93 (д, J=3, 1H), 8,36 (д, J=3, 1H), 8,26 (с, 1H), 8,04 (с, 1H), 8,01 (дд, J=3, 3, 1H), 7,88 (с, 1H), 5,16 (кв., J=9, 2H), 4,19 (д, J=9, 1H), 4,05 (м, 3H), 3,88 (с, 3H), 2,66 (с, 3H), 2,36 (м, 2H).
С использованием соответствующих промежуточных соединений, получали соединения 978 и 989 по сходной методике.
Схема 24
В таблице 1 предоставлены данные по аналитической охарактеризации соединений формулы I (пустые ячейки указывают на то, что тест не проводили)
(M+H)
(300 МГц, если нет иных указаний
2,72 (с, 3H)
Биологический анализ соединений по изобретению
Пример 25. Анализ ингибирования PI3K
С использованием Biomek FX от Beckman Coulter, 1,5 мкл каждого из десяти 2,5-кратных серийных разведений соединения по изобретению в 100% DMSO добавляли в отдельные лунки (далее "тестируемые лунки") в 96-луночном планшете из полистирола [Corning, Costar, номер № 3697]. Одна тестируемая лунка также содержала 1,5 мкл DMSO без соединения. Другая лунка содержала ингибитор в DMSO в концентрации, о которой известно, что она полностью ингибирует фермент (далее "фоновая лунка"). С использованием Titertek Multidrop, в каждую лунку добавляли 50 мкл реакционной смеси [100 мМ HEPES pH 7,5, 50 мМ NaCl, 10 мМ DTT, 0,2 мг/мл BSA, 60 мкл фосфатидилинозитол(4,5)бифосфата diC16(PI(4,5)P2; Avanti Polar Lipids, каталожный номер № 840046P) и представляющую интерес изоформу PI3K (см. таблицу 3 для концентраций изоформ)]. Для инициации реакции в каждую лунку добавляли 50 мкл смеси ATP Mix [20 мМ MgCl2, 6 мкл ATP (100 мкКи/мкмоль 33P-ATP)], а затем лунки инкубировали в течение 30 мин при 25°C. Конечные концентрации в каждой лунке составляли 50 мМ HEPES 7,5, 10 мМ MgCl2, 25 мМ NaCl, 5 мМ DTT, 0,1 мг/мл BSA, 30 мкМ PI(4,5)P2, 3 мкМ ATP, и указанную концентрацию представляющей интерес изоформы PI3K (см. таблицу 2). Конечные концентрации соединения в каждой лунке находились в диапазоне от 10 мкМ до 1 нМ.
После инкубации реакционные смеси в каждой лунке гасили добавлением 50 мкл стоп-раствора [30% TCA/вода, 10 мМ ATP]. Затем каждую реакционную смесь после гашения переносили в 96-луночный планшет со стекловолоконным фильтром [Corning, Costar номер № 3511]. Планшеты фильтровали в вакууме и промывали три раза 150 мкл смеси 5% TCA/вода в модифицированном автоматическом устройстве для промывания планшетов Bio-Tek Instruments ELX-405 Auto Plate Washer. В каждую лунку добавляли 50 мкл сцинтилляционной жидкости и проводили считывание планшета на жидкостном сцинтилляционном счетчике Perkin-Elmer TopCountTM NXT с получением количества импульсов 33P, отражающих величины ингибирования.
Величину для фоновой лунки вычитали из величины, полученной для каждой тестируемой лунки, и данные приводили в соответствие с уравнением для Ki конкурентного прочного связывания, описанным Morrison and Stone, Comments Mol. Cell Biophys. 2: 347-368, 1985.
Каждое из нижеследующих соединений имеет Ki менее 0,1 микромоль для ингибирования PBK-гамма: 1, 3, 6, 10, 12, 16, 18-20, 24-26, 34-36, 38, 44, 46-48, 51-59, 64, 66-68, 70-73, 77-82, 85-109, 113-119, 122-124, 126, 128-133, 135-136, 138-141, 143-156, 158, 161, 165-166, 170, 172, 174-176, 178, 181-187, 194, 198-201, 204-219, 221-223, 225-227, 229-230, 234, 236-237, 239, 241, 243-247, 250-273, 275-285, 287-312, 315, 317, 320-322, 324-328, 330-332, 336-340, 342-347, 363, 365-366, 368-388, 388-389, 391-407, и 409-411, 412-424, 426-437, 439-455, 457-461, 463-464, 466, 468-472, 474-478, 480-481, 483-495, 497, 499-506, 508-573, 576-584, 587-607, 609, 612, 614-616, 618-623, 625-658, 661, 664, 666-672, 674-681, 683-685, 687-688, 690-695, 698-706, 708-710, 712-730, 732-740, 742-749, 754-758, 760-762, 764-770, 772-779, 781-787, 789-801, 802-803, 810-818, 822-825, 829-833, 835-838, 840, 843-871, 873, 875-893, 896, 899-900, 902-906, 909-911, 913-916, 918-933, 935-943, 945-950, 952-956, 958-984, 986-999.
Каждое из нижеследующих соединений имеет Ki от 0,1 микромоль до 0,49 микромоль для ингибирования PBK-гамма: 2, 5, 7, 9, 13-15, 17, 22, 23, 27-33, 37, 39-43, 45, 49-50, 60-63, 65, 69, 74-76, 83, 110, 112, 120, 125, 127, 134, 137, 142, 157, 159-160, 162, 164, 167-168, 171, 173, 177, 179-180, 188-193, 196-197, 202-203, 224, 228, 231-232, 240, 242, 248, 286, 313, 316, 318-319, 329, 333-335, 341, 348, 364, 367, 390, 408, 425, 456, 462, 465, 467, 473, 479, 482, 496, 507, 574-575, 585-586, 608, 611, 613, 617, 624, 660, 662, 686, 689, 696, 697, 707, 711, 731, 741, 750, 771, 780, 802, 809, 819, 820-821, 834, 839, 872, 874, 894-895, 897-898, 901, 907-908, 912, 934, 944, 951 и 957.
Каждое из нижеследующих соединений имеет Ki от 0,5 микромоль до 2,5 микромоль для ингибирования PBK-гамма: 4, 11, 21, 84, 111, 121, 163, 169, 195, 220, 233, 235, 238, 249, 274, 314, 323, 610, 659, 663, 665, 673, 682, 751, 759, 763, 826, 841, 842, 917 и 985.
Пример 26. Анализ активации микроглии
Самок мышей C57B1/6J (в возрасте 7 недель) приобретали от Jackson Laboratory (Maine, США). Животных подвергали акклиматизации в течение одной недели в стандартных лабораторных условиях (циклы с 12 ч на свету) со свободным доступом к корму для грызунов и воде. Все методики соблюдали в соответствии с National Institute of Health Guidelines for the care and Use of Laboratory Animals, и они были одобрены IACUCC. Эндотоксин липополисахарид (LPS) (E. coli 011:B4, каталожный номер № 437627) приобретали от Calbiochem. LPS растворяли в буфере PBS в концентрации 0,05 мг/мл и хранили замороженными аликвотами. В начале исследования мышам проводили внутрибрюшинную (i.p.) инъекцию LPS (0,5 мг/кг) в течение трех последовательных суток. Терапевтическое лечение соединениями VRT начинали вместе со 2-ым введением LPS и поддерживали на протяжении исследования. Соединение вводили два раза в сутки перорально с помощью принудительного питания всего с 4 дозами. Через 24 ч после последней инъекции LPS, и через 2 ч после последней дозы VRT, животных умерщвляли путем асфиксии с помощью CO2.
После умерщвления головной мозг быстро извлекали и фиксировали в течение ночи в 10% нейтральном забуференном формалине. Затем головной мозг обрабатывали для стандартной гистологии в автоматическом устройстве для обработки (Shandon Excelsior ES, Thermo Scientific) и погружали в парафин. Анализ IHC проводили на 5-мкм срезах в системе Ventana Benchmark System (Ventana Medical Systems Inc, Tucson, AZ) с использованием предварительно разбавленных антител к Iba1 (Wako chemical, США) при разведении 1:800. 3,3'-диаминобензидин (DAB) использовали в качестве хромогенного субстрата и предметные стекла контрастно окрашивали гематоксилином.
Цифровые изображения получали с использованием сканнера Aperio ScanScope Slide Scanner (Aperio Technologies, Vista, CA). Изображения получали при 20x оптическом увеличении и анализировали с использованием программного обеспечения Defmiens Developer XD. Алгоритмы создавали для подсчета активированных микроглиальных клеток, учитывая различные морфологические характеристики активированных клеток по сравнению с покоящейся микроглией. Эффективность соединения вычисляли в качестве процентного снижения количества активированной микроглии относительно контроля в виде носителя. Для соединения 271 наблюдали снижение 39 процентов при дозировании 10 мг/кг два раза в сутки. Для соединения 568 в трех отдельных экспериментах наблюдали снижение количества активированной микроглии в диапазоне от 44 до 60 процентов при дозировании 10 мг/кг два раза в сутки. Для соединения 410 в трех отдельных экспериментах наблюдали снижение количества активированной микроглии в диапазоне от 23 до 33 процентов при дозировании 5 мг/кг два раза в сутки.
Хотя представленное выше изобретение подробно описано с помощью иллюстраций и примеров для простоты понимания, специалистам в данной области будет очевидно с учетом идей настоящего изобретения, что определенные изменения и модификации можно вносить в него без отклонения от сущности или объема прилагаемой формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ ДНК-ПК | 2014 |
|
RU2676259C2 |
| ПЕСТИЦИДНЫЕ КОМПОЗИЦИИ | 2013 |
|
RU2616808C2 |
| ИНГИБИТОРЫ ДНК-ПК | 2013 |
|
RU2638540C1 |
| ПЕСТИЦИДНЫЕ КОМПОЗИЦИИ И СВЯЗАННЫЕ С НИМИ СПОСОБЫ | 2012 |
|
RU2597421C2 |
| ИНГИБИТОРНЫЕ СОЕДИНЕНИЯ | 2013 |
|
RU2673079C2 |
| ПЕСТИЦИДНЫЕ КОМПОЗИЦИИ И ОТНОСЯЩИЕСЯ К НИМ СПОСОБЫ | 2013 |
|
RU2651369C1 |
| ИНГИБИТОРЫ БРОМДОМЕНА | 2012 |
|
RU2671571C1 |
| ПЕСТИЦИДНЫЕ КОМПОЗИЦИИ И СВЯЗАННЫЕ С НИМИ СПОСОБЫ | 2013 |
|
RU2616812C2 |
| ПИРРОЛОПИРИДИНЫ, ПОЛЕЗНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗЫ | 2006 |
|
RU2435769C2 |
| ИНГИБИТОРЫ СИГМА-РЕЦЕПТОРА | 2005 |
|
RU2417987C2 |
Изобретение относится к соединениям, пригодным в качестве ингибиторов PI3K, в частности PI3Kγ. Также изобретение относится к фармацевтически приемлемым композициям, содержащим указанные соединения, и к способам применения композиций для лечения различных заболеваний, состояний или нарушений. 5 н. и 13 з.п. ф-лы, 2 табл., 26 пр.
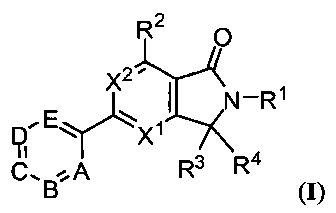
1. Соединение формулы:
или его фармацевтически приемлемая соль, где:
X1 представляет собой N;
X2 представляет собой СН;
R1 представляет собой пиразол-4-ил, необязательно замещенный 1 или 2 группами R1a;
R1a представляет собой хлор, фтор, С1-8алифатическую группу, -(СН2)0-2С3-6циклоалифатическую группу, -(СН2)0-2-5-6-членное гетероциклическое кольцо, -CN, -С(О)С1-4алифатическую группу, -С(О)OC1-4алифатическую группу, -S(О)2NH(С1-4алифатическую группу), -S(О)2N(С1-4алифатическую группу)2 или -S(О)2С1-4алифатическую группу, где вплоть до 3 несоседних атомов углерода указанной алифатической или циклоалифатической группы R1a могут быть замещены -О- или -N(R1b)-, где указанное гетероциклическое кольцо содержит вплоть до двух гетероатомов, выбранных из азота, кислорода или серы, и где каждая из алифатической, циклоалифатической групп или гетероциклическое кольцо R1a необязательно и независимо замещено вплоть до 4 группами JR;
каждый JR независимо представляет собой фтор, оксо, -(СН2)0-2CN, -(CH2)0-2CF3, -C(O)R1b, -C(O)N(R1b)2, -C(O)O(R1b), -N(R1b)2, -N(R1b)С(O)R1b, -(CH2)0-2OR1b, фенил, 5-6-членный гетероарил, 4-6-гетероциклил, 9-11-членный конденсированный бициклический гетероарил или 9-11-членный конденсированный бициклический гетероциклил, причем каждое из указанных гетероарильных или гетероциклильных колец имеет вплоть до 3 атомов, выбранных из азота, кислорода или серы, и где каждая из указанной циклоалифатической, фенильной, гетероарильной или гетероциклильной группы необязательно замещена вплоть до 2 R1c;
каждый R1b независимо выбран из водорода, С1-8алифатической группы, -(СН2)0-1С3-6циклоалифатической группы, -(СН2)0-1С4-6гетероциклической группы, имеющей вплоть до двух гетероатомов, выбранных из N или О, или две R1b, вместе с атомом, с которым они связаны, образуют 5-6-членное гетероциклическое кольцо, где каждое алифатическое, циклоалифатическое или гетероциклическое кольцо необязательно замещено посредством вплоть до трех атомов F или вплоть до двух -ОН, -С1-2алкильных или -ОС1-2алкильных групп;
каждый R1c независимо представляет собой фтор, хлор, С1-4алифатическую группу, -(СН2)0-2ОН, -CN, -С(О)С1-4алифатическую группу или -С(О)OC1-4алифатическую группу;
R2 представляет собой водород или С1-2алифатическую группу;
R3 представляет собой водород, С1-6алифатическую группу, С3-6циклоалифатическую группу, С4-7гетероциклильную группу, имеющую 1 или 2 атома, выбранных из N или О, -(СН2)0-1CF3, -ОН, -ОС1-6алифатическую группу, -ОС3-6циклоалифатическую группу, -OC3-6гетероциклильную группу, имеющую один атом кислорода, -О(СН2)2ОС1-2алифатическую группу, или -ОС1-2алкилС(О)ОС1-3алифатическую группу, или бензил; и
R4 представляет собой водород или С1-6алкил; где по крайней мере один из R3 и R4 не является водородом; или R3 и R4 вместе с атомом углерода, с которым они связаны, образуют 3-6-членное циклоалифатическое кольцо, 3-6-членное гетероциклическое кольцо, имеющее вплоть до двух атомов, выбранных из N или О, или С2алкенил, где каждая из указанных алифатических, циклоалифатических или гетероциклильных групп R3, R4 или R3 и R4 вместе необязательно замещена посредством вплоть до трех атомов F или вплоть до двух С1-2алкильных, -С(О)С1-4алкильных, -C(O)OC1-4алкильных, -ОН или -ОС1-2алкильных групп;
А представляет собой CRA, где RA представляет собой водород;
В представляет собой N;
С представляет собой CRC;
D представляет собой CRD;
Е представляет собой CRE, где RE представляет собой водород;
RC представляет собой водород, F, Cl, С1-3алифатическую группу, -(CH2)0-1CF3, -(CH2)0-1CHF2, N(R1b)2, -ОН, -О(СН2)0-1CF3 или -OC1-8алифатическую группу, где вплоть до 2 несоседних атомов углерода указанной алифатической группы могут быть замещены -О-;
RD представляет собой водород, фтор, хлор, С1-4алифатическую группу, -С(О)ОН, -С(О)ОС1-4алифатическую группу, -С(О)N(R1b)2, -CN, -C(RD1)=N-OR1b, -N(R1b)2, -N(RD1)С(О)С1-4алифатическую группу, -N(RD1)С(О)фенил, -N(RD1)S(О)2С1-4алифатическую группу, -N(RD1)S(O)2N(R1b)2, -N(RD1)S(O)2фeнил, -ОН, -OC1-8алифатическую группу, -О(СН2)0-1С3-6циклоалифатическую группу, -SC1-4алифатическую группу, -S(О)С1-4алифатическую группу, -S(О)2С1-4алифатическую группу или -S(О)2N(R1b)2; где вплоть до 2 несоседних атомов углерода указанной алифатической, циклоалифатической или гетероциклической группы RD может быть замещен -О- и каждый из указанной алифатической группы, циклоалифатической группы или фенила RD может быть замещен посредством вплоть до 5 атомов фтора; или RD и RC, вместе с атомами, к которым они присоединены, образуют фенильное или пиридильное кольцо;
каждый RD1 независимо представляет собой водород или С1-2алкил.
2. Соединение по п. 1 или его фармацевтически приемлемая соль, где:
R1 представляет собой 1-(2,2-дифторэтил)-1Н-пиразол-4-ил или 1-(2,2,2-трифторэтил)-1Н-пиразол-4-ил.
3. Соединение по п. 1 или его фармацевтически приемлемая соль, где:
каждый RC и RD представляет собой -ОСН3.
4. Соединение, имеющее формулу:
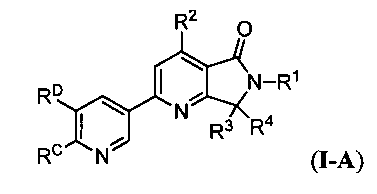
или его фармацевтически приемлемая соль, где
R1 представляет собой
где
R1a представляет собой -С1-4алкил, необязательно и независимо замещенный посредством -CN, вплоть до трех атомов F или вплоть до двух СН3, вплоть до двух -ОС1-2алкильных или вплоть до двух -ОН групп;
R2 представляет собой С1-2алкил;
R3 представляет собой водород, -ОН, -OC1-4алкил или С1-4алкил, необязательно замещенный посредством вплоть до двух -ОН групп;
R4 представляет собой водород или СН3, или R3 и R4 вместе образуют С3-6циклоалкильное кольцо, необязательно замещенное посредством вплоть до двух ОН групп, или R3 и R4 вместе образуют 4-6-членное гетероциклическое кольцо, имеющее один атом кислорода или один атом азота, необязательно замещенное С1-4алкилом, -С(О)С1-4алкилом или С(О)ОС1-4алкилом;
RC представляет собой водород, F, С1-2алкил или -ОС1-2алкил; и
RD представляет собой -ORD1, -С(О)N(RD1)RD2, -S(О)2N(RD1)RD2, -S(O)1-2RD2, -N(RD1)S(O)2RD2 или -N(RD1)S(О)2N(RD1)RD2, где
RD1 представляет собой водород или С1-2алкил, и RD2 представляет собой С1-4алкил, -(СН2)0-1С3-6циклоалкил или -(СН2)0-1С4-6гетероциклил, имеющий вплоть до двух атомов кислорода или атомов азота, и где каждый алкил RD1 и каждый алкил, циклоалкил или гетероциклил RD2 необязательно замещен посредством вплоть до трех атомов F или вплоть до двух -ОН групп.
5. Соединение по п. 4 или его фармацевтически приемлемая соль, где R1a представляет собой С1-2алкил, необязательно замещенный посредством вплоть до 3 атомов фтора.
6. Соединение по п. 4 или его фармацевтически приемлемая соль, где R1a представляет собой С1-4алкил, необязательно замещенный CN.
7. Соединение по п. 4 или его фармацевтически приемлемая соль, где R2 представляет собой СН3.
8. Соединение по п. 4 или его фармацевтически приемлемая соль, где каждый из R3 и R4 представляет собой СН3.
9. Соединение по п. 4 или его фармацевтически приемлемая соль, где R3 и R4 вместе образуют 4-6-членное гетероциклическое кольцо, имеющее один атом кислорода или атом азота необязательно, замещенный С1-4алкилом, -С(О)С1-4алкилом или -С(О)OC1-4алкилом.
10. Соединение по п. 7 или его фармацевтически приемлемая соль, где:
R1 представляет собой
R2 представляет собой СН3;
R3 представляет собой водород, С1-2алкил, ОН или ОСН3;
R4 представляет собой водород или СН3;
RC представляет собой водород; и
RD представляет собой -ОС1-2алкил или -ОС3-5циклоалкил, каждый из которых необязательно замещен посредством вплоть до 3 атомов фтора.
11. Соединение по п. 10 или его фармацевтически приемлемая соль, где R1 представляет собой 1-(2,2-дифторэтил)-1Н-пиразол-4-ил или 1-(2,2,2-трифторэтил)-1Н-пиразол-4-ил.
12. Соединение по п. 4 или его фармацевтически приемлемая соль, где каждый из RC и RD представляет собой -ОСН3.
13. Соединение по п. 4 или его фармацевтически приемлемая соль, где RD представляет собой -С(O)ОН, -С(О)N(R1b)2, -CN, -S(О)2С1-8алифатическую группу или -S(O)2N(R1b)2.
14. Соединение по п. 4 или его фармацевтически приемлемая соль, где каждый из RC и RD независимо представляет собой водород, фтор, хлор, С1-3алифатическую группу, CF3, -OCF3, -OCHF2 или -OC1-2алифатическую группу, где по меньшей мере один из RC и RD не является водородом.
15. Соединение по п. 14 или его фармацевтически приемлемая соль, где RC представляет собой водород, и RD представляет собой -ОС1-3алкил, необязательно замещенный посредством плоть до 3 атомов F.
16. Соединение, выбранное из:
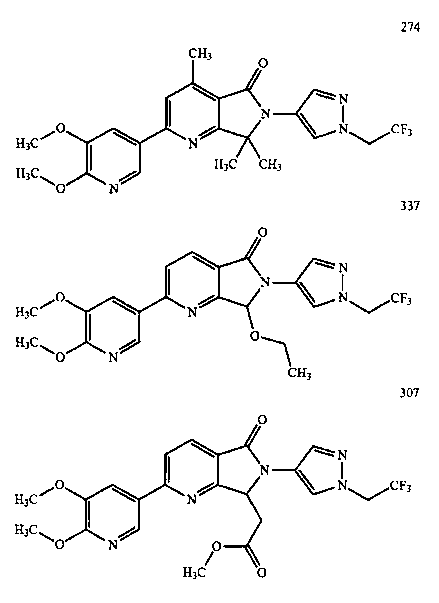
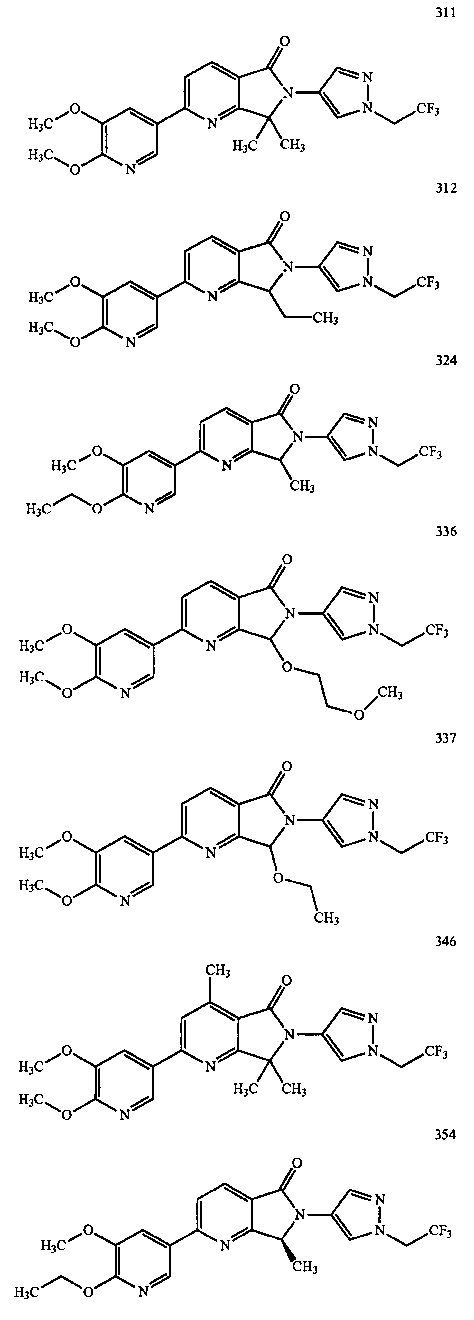
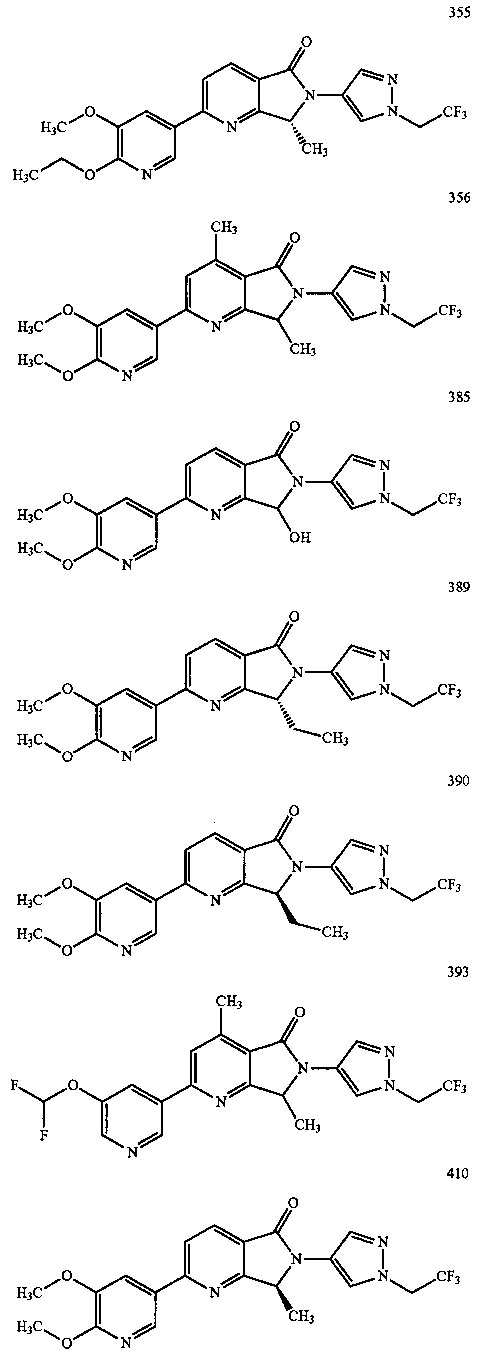
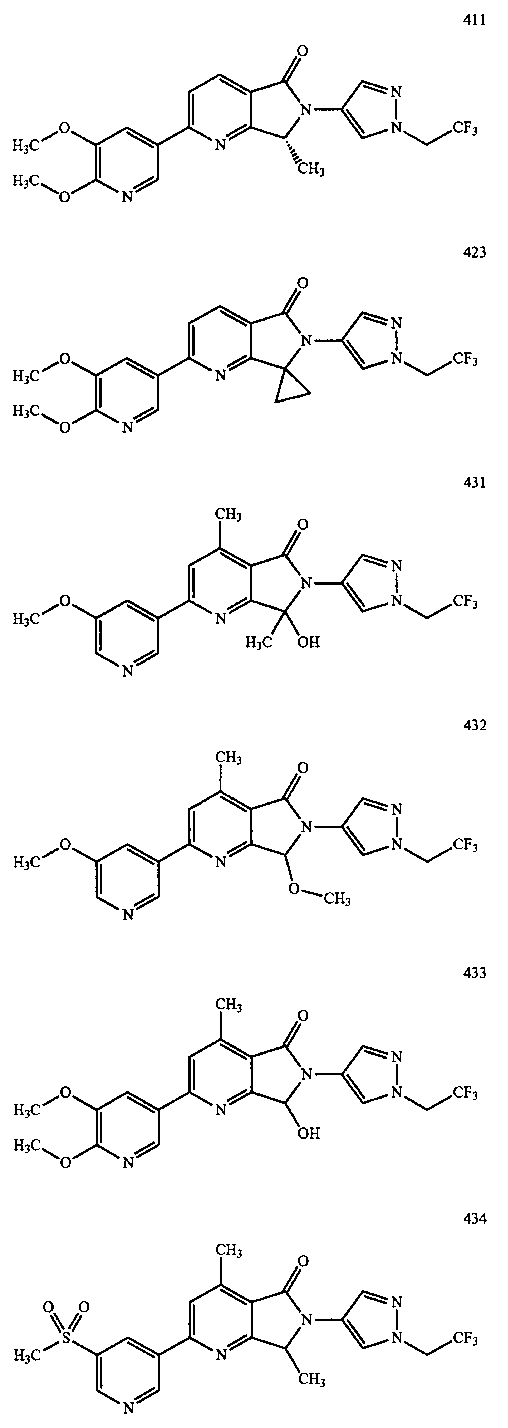
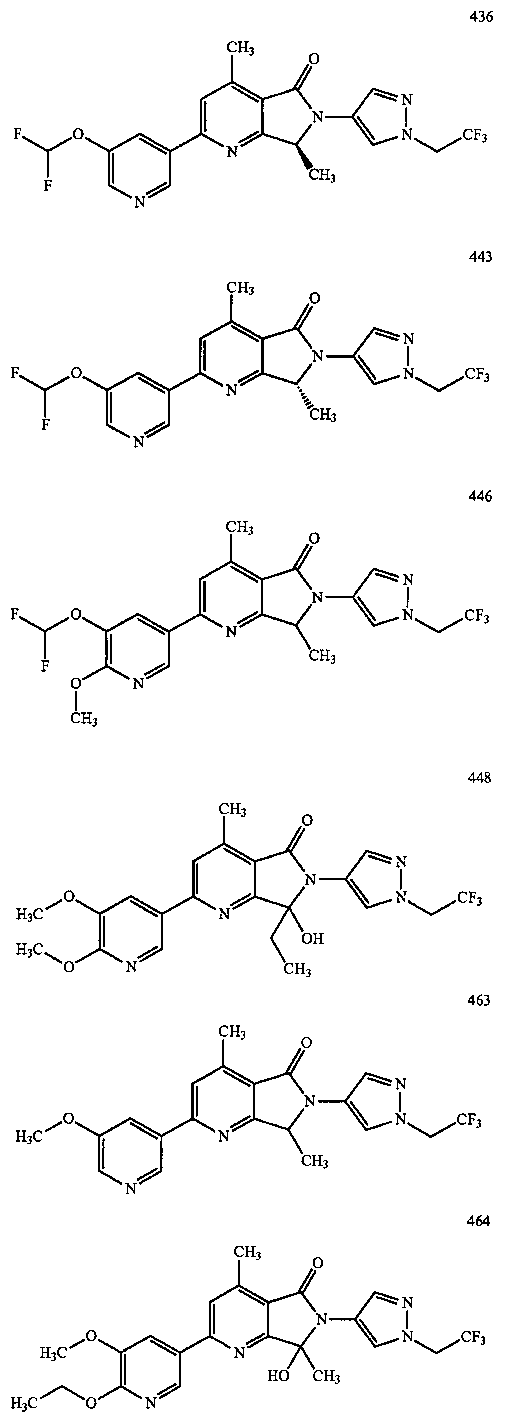
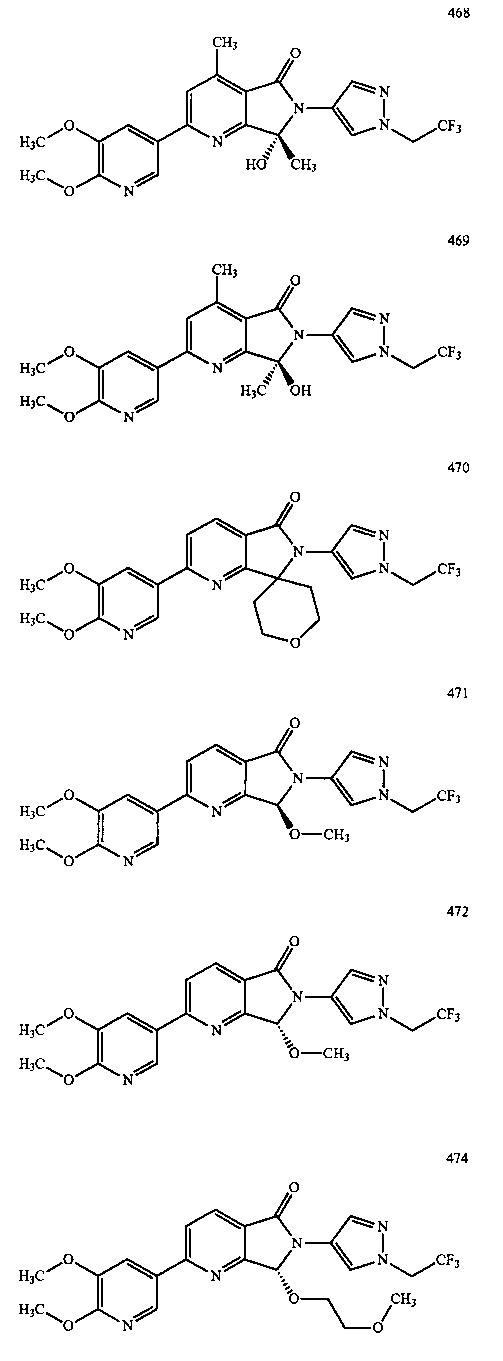
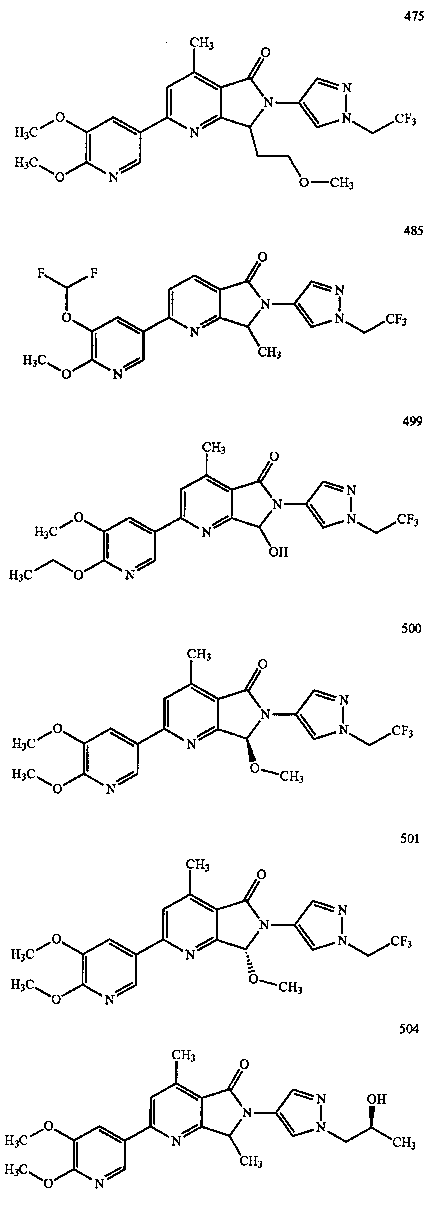
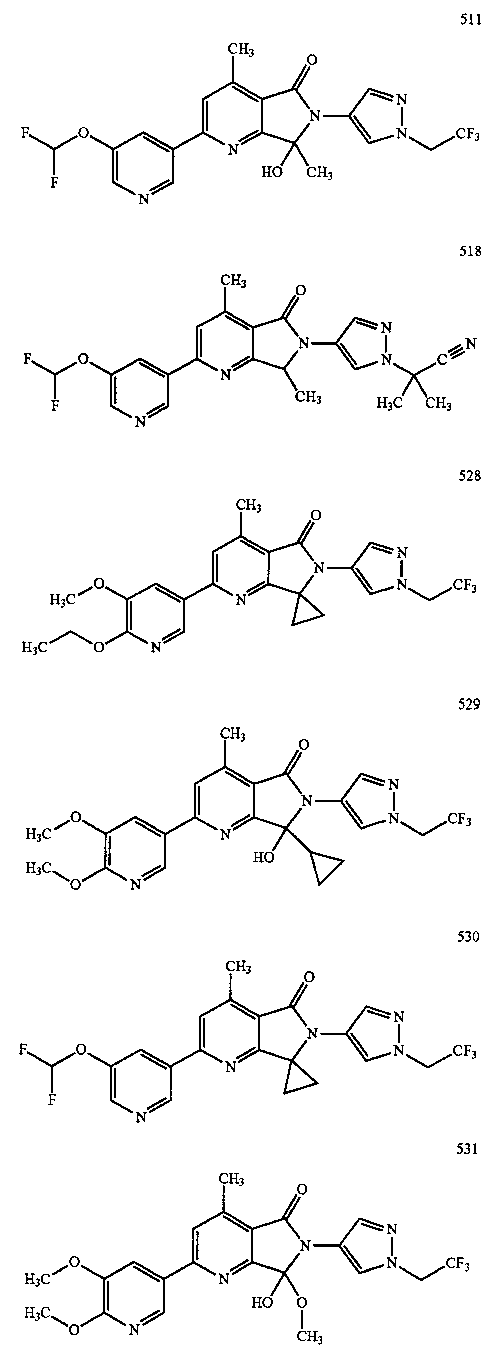
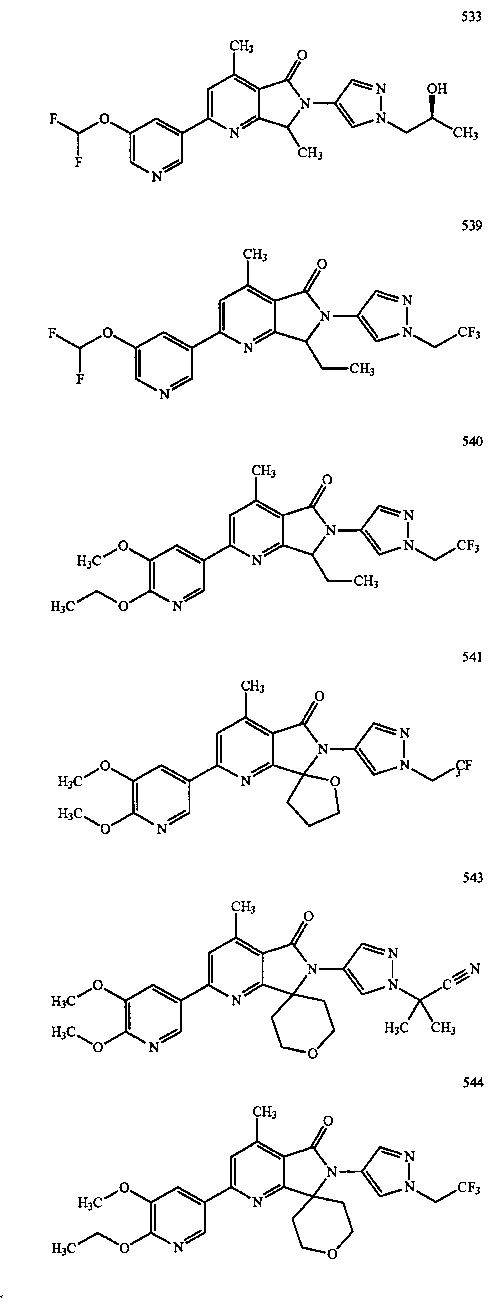
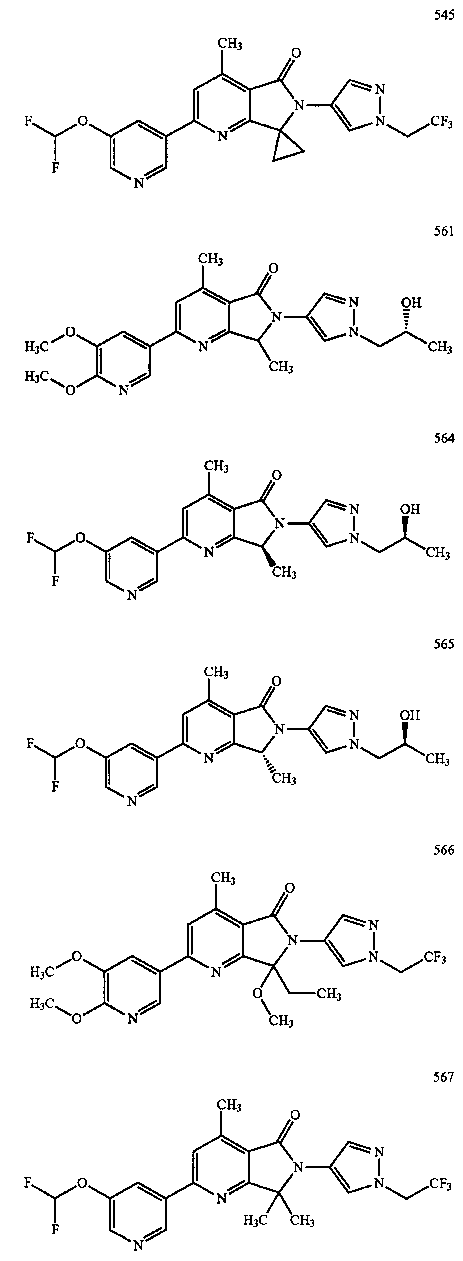
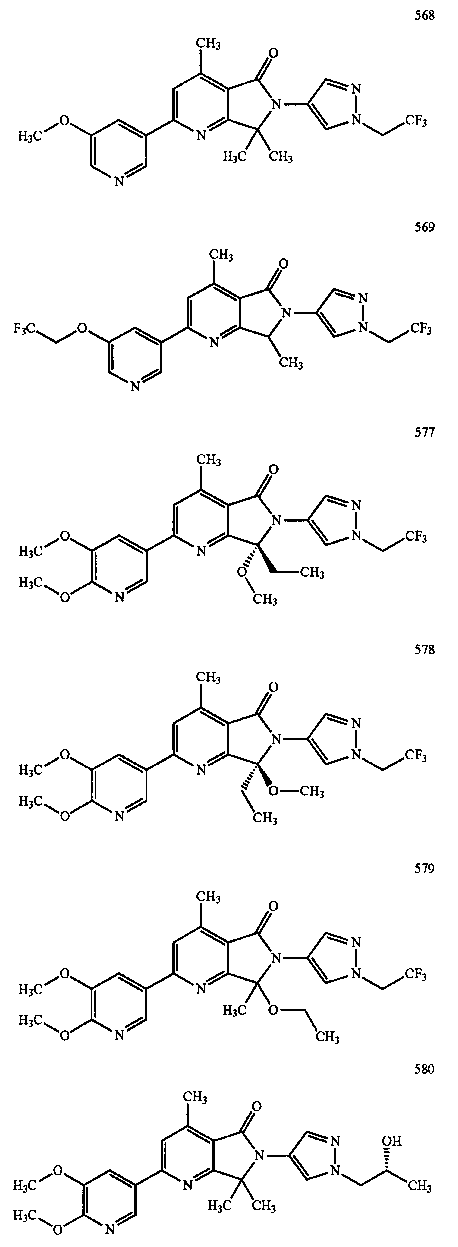
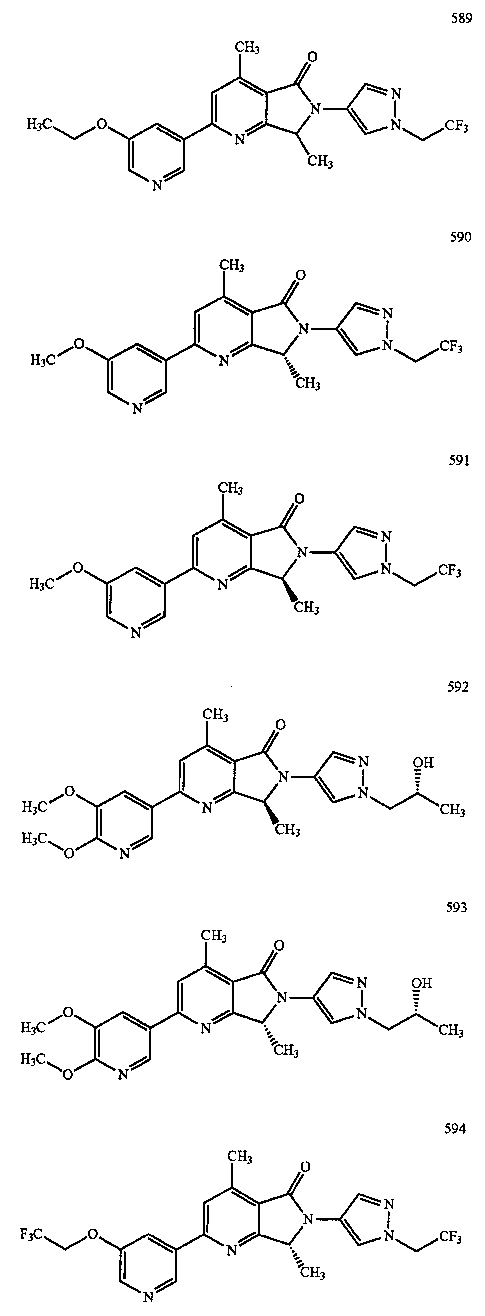
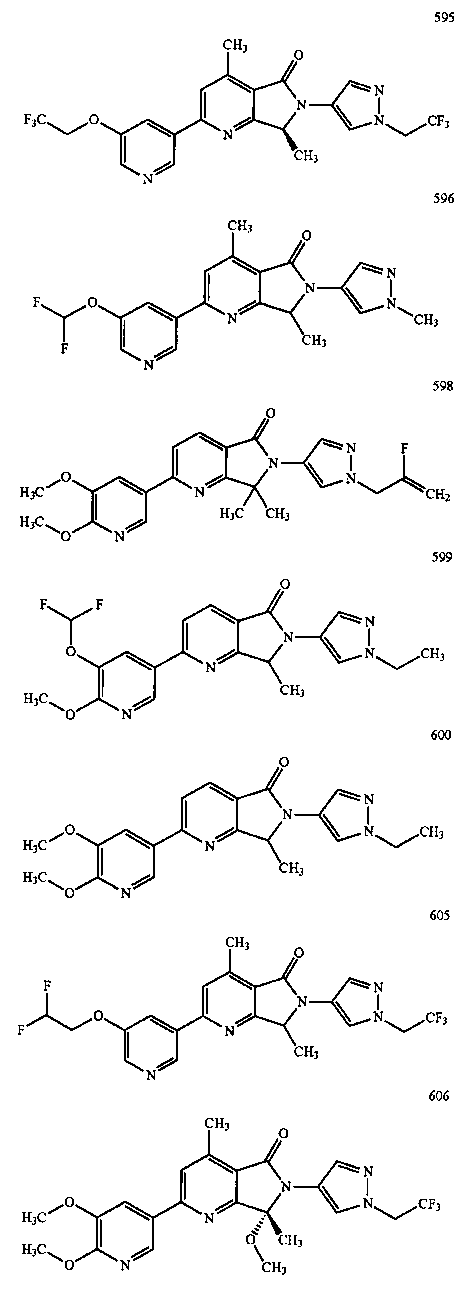
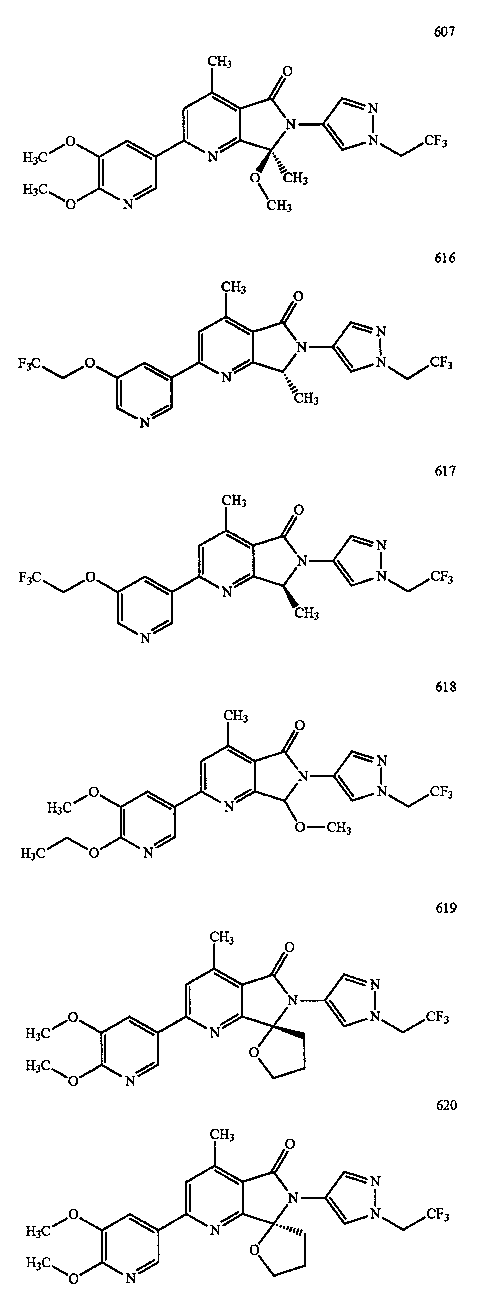
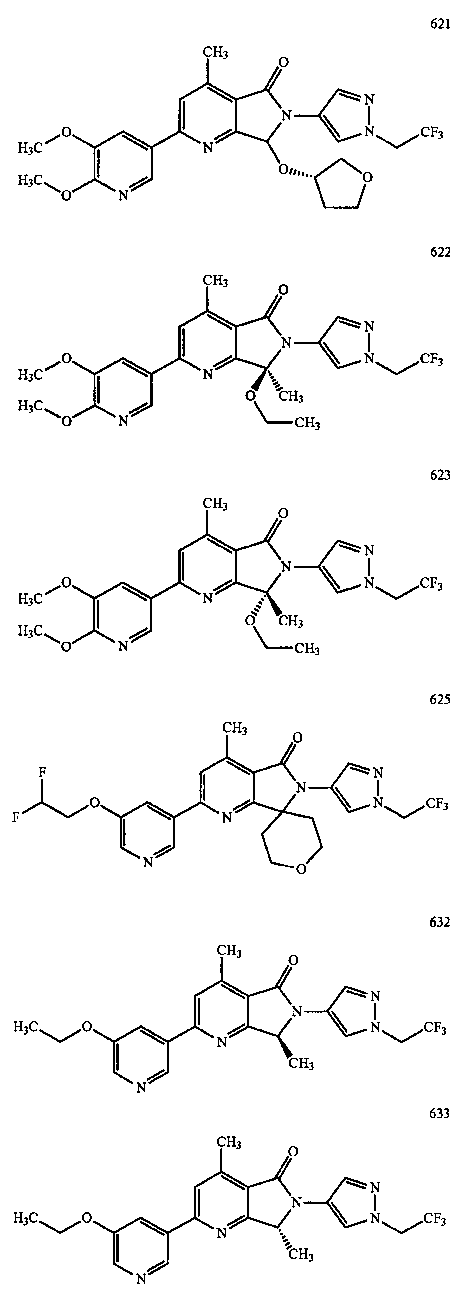
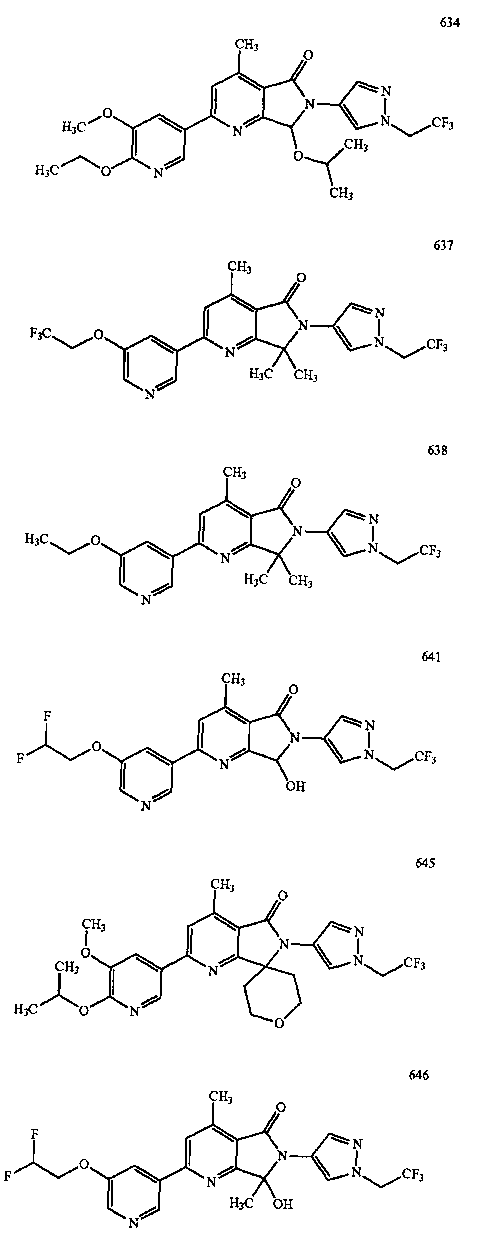
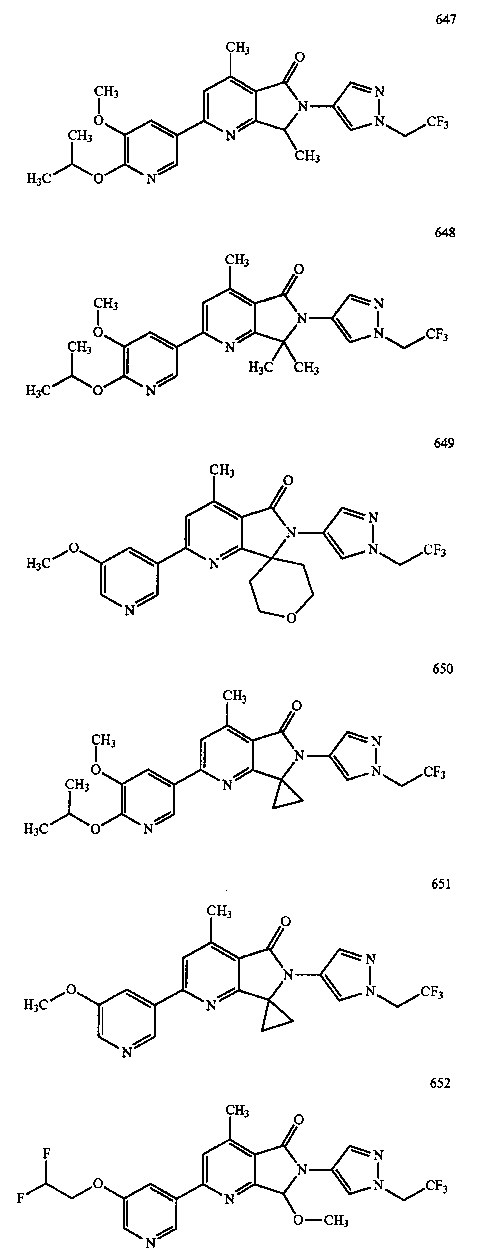
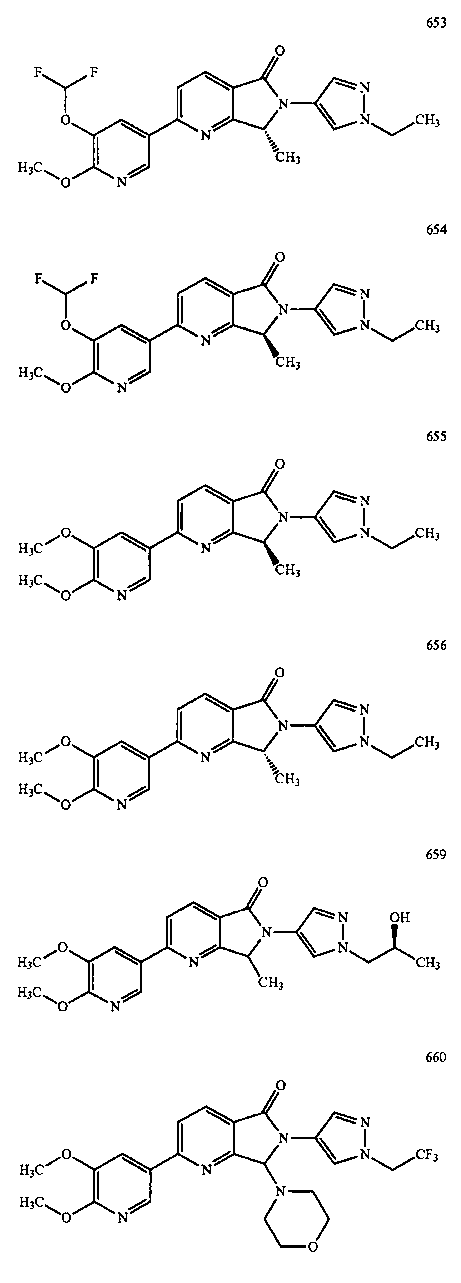
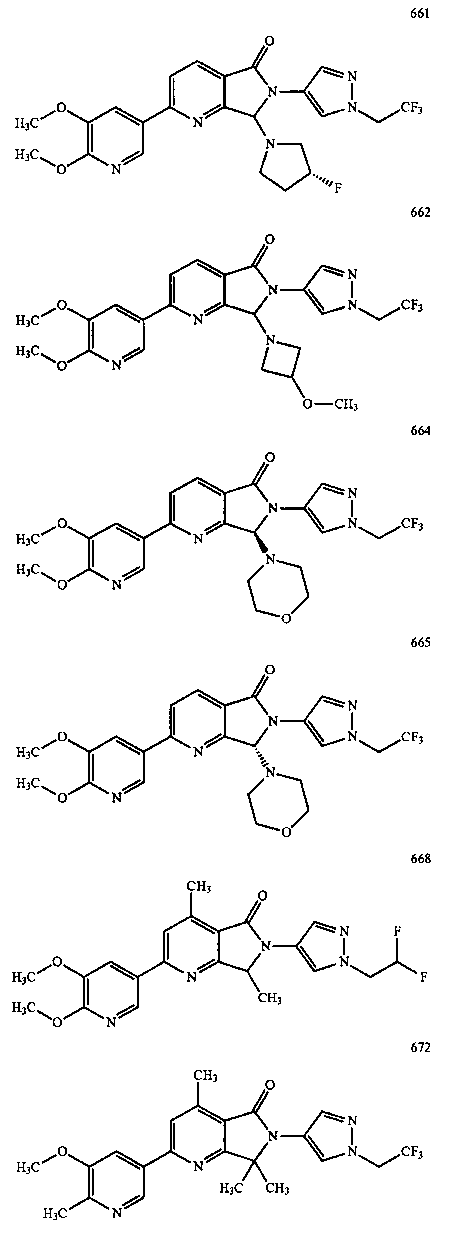
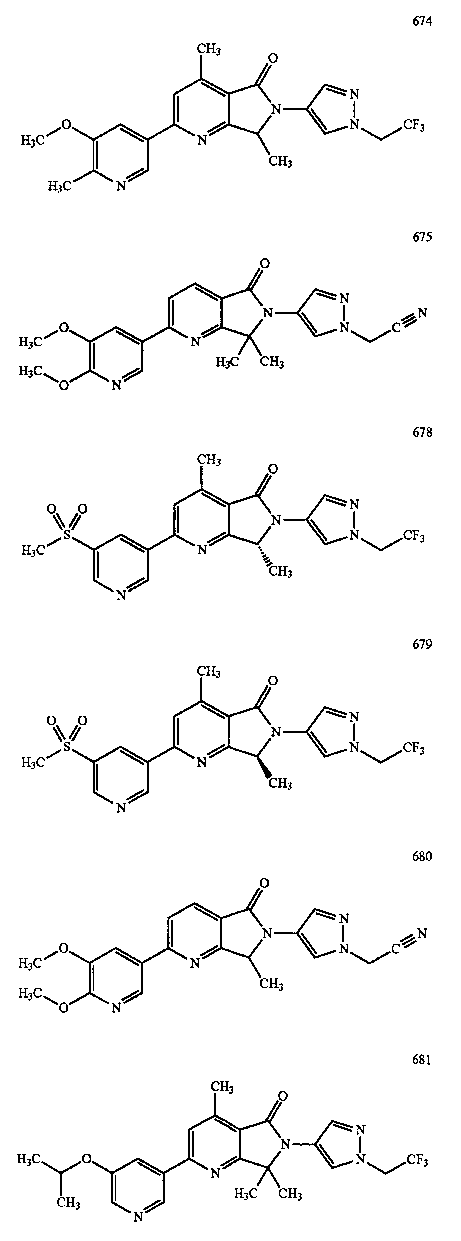
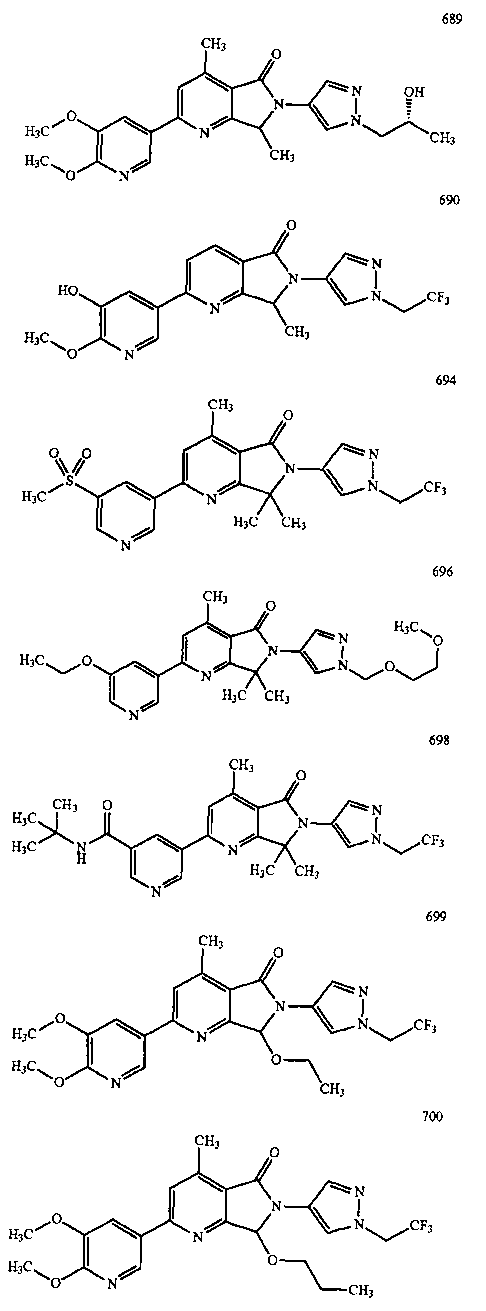
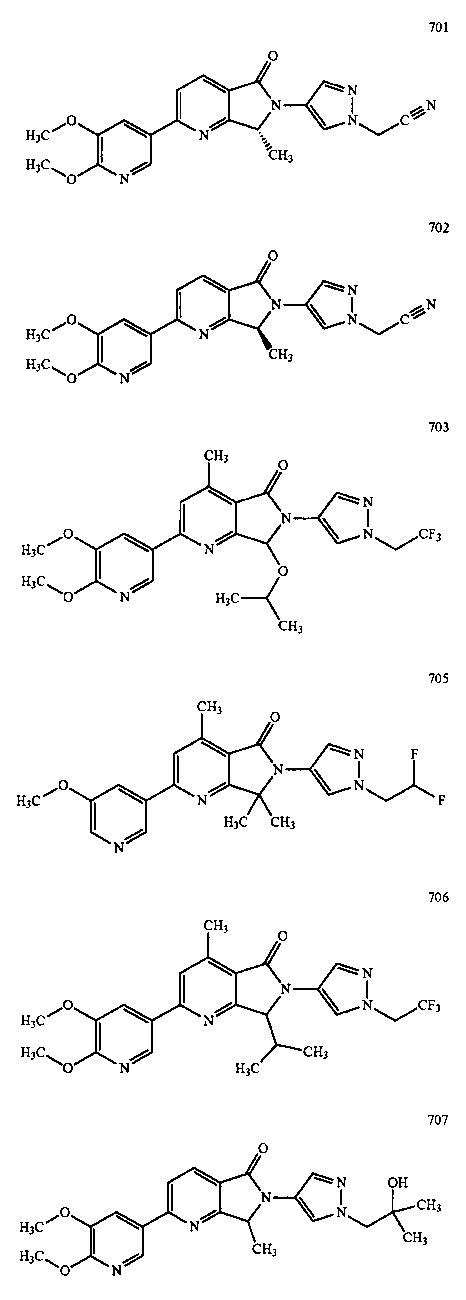
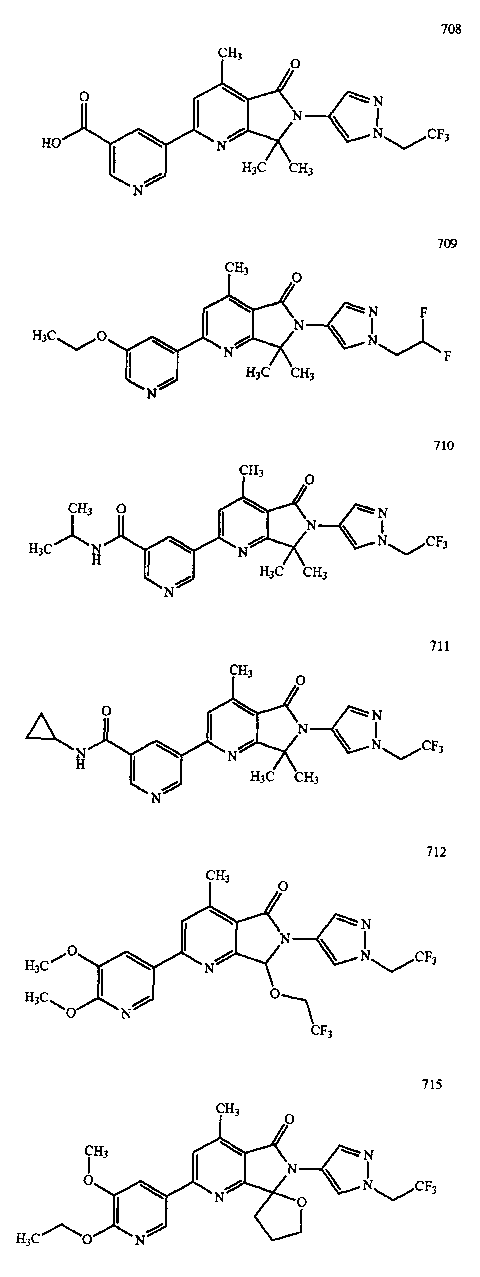
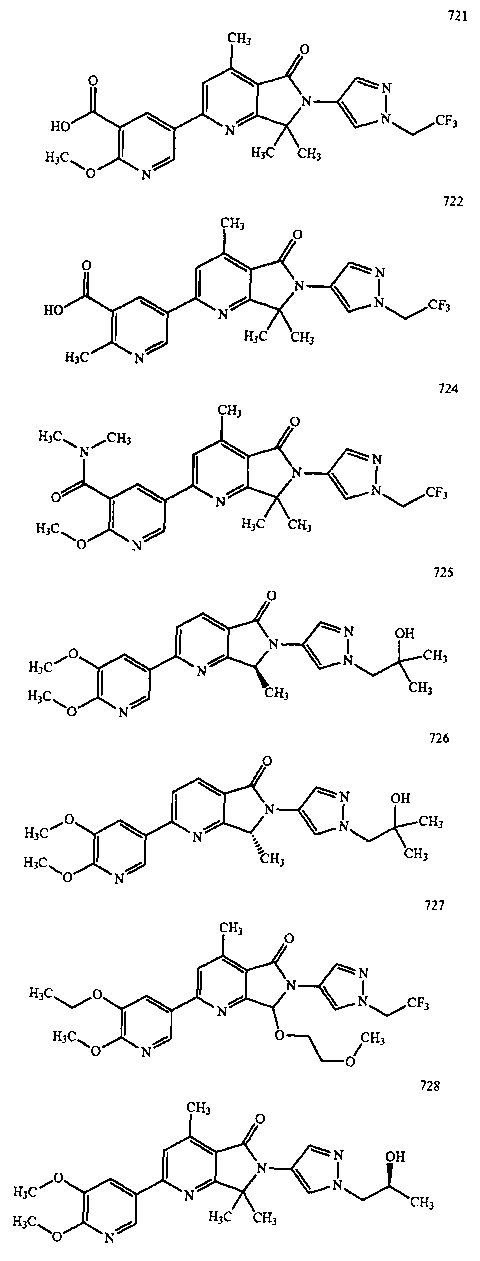
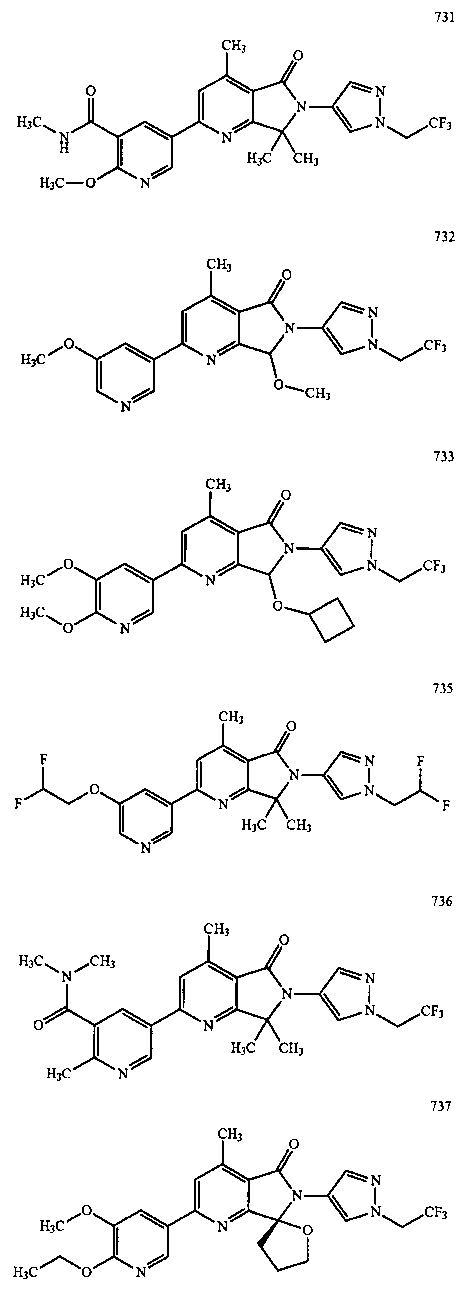
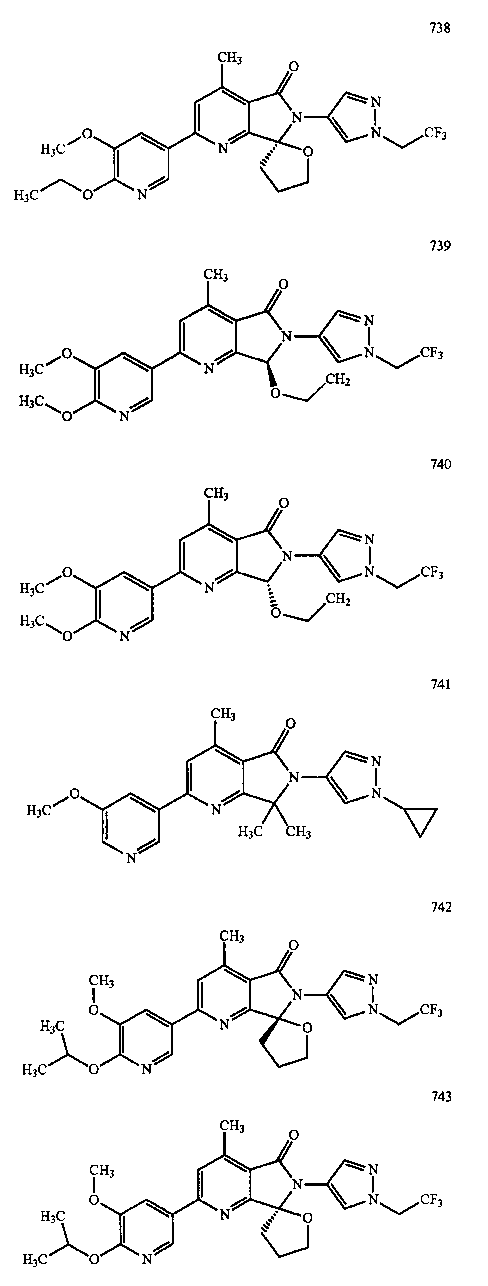
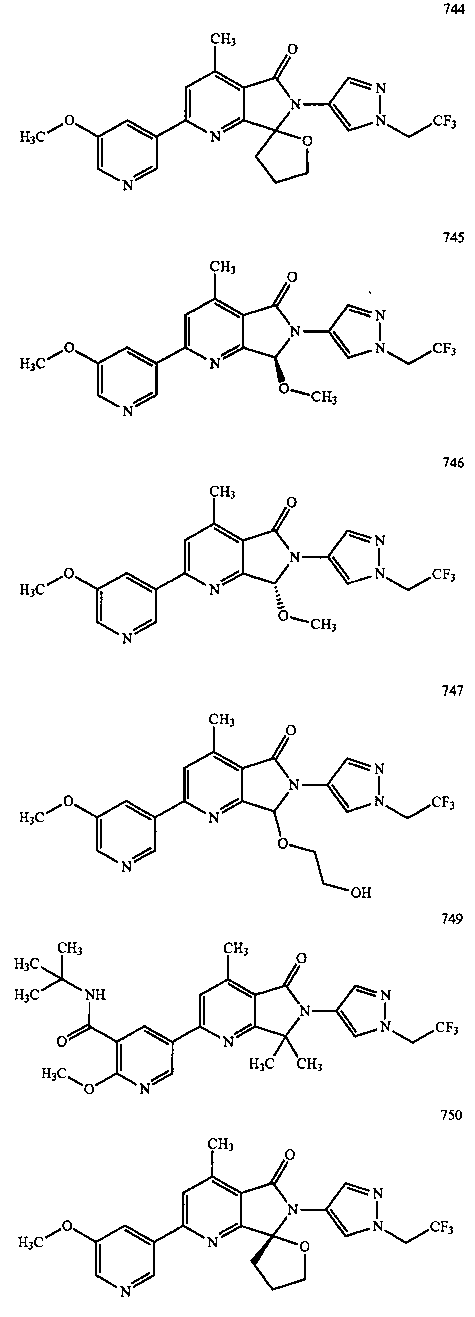
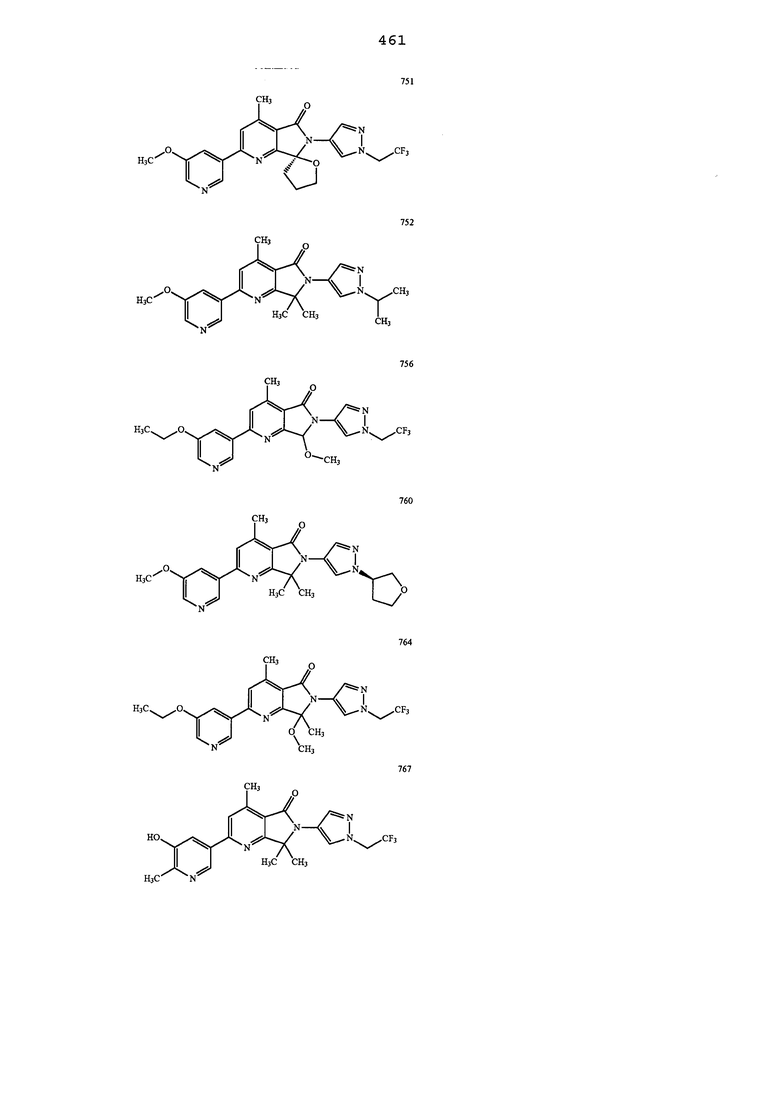
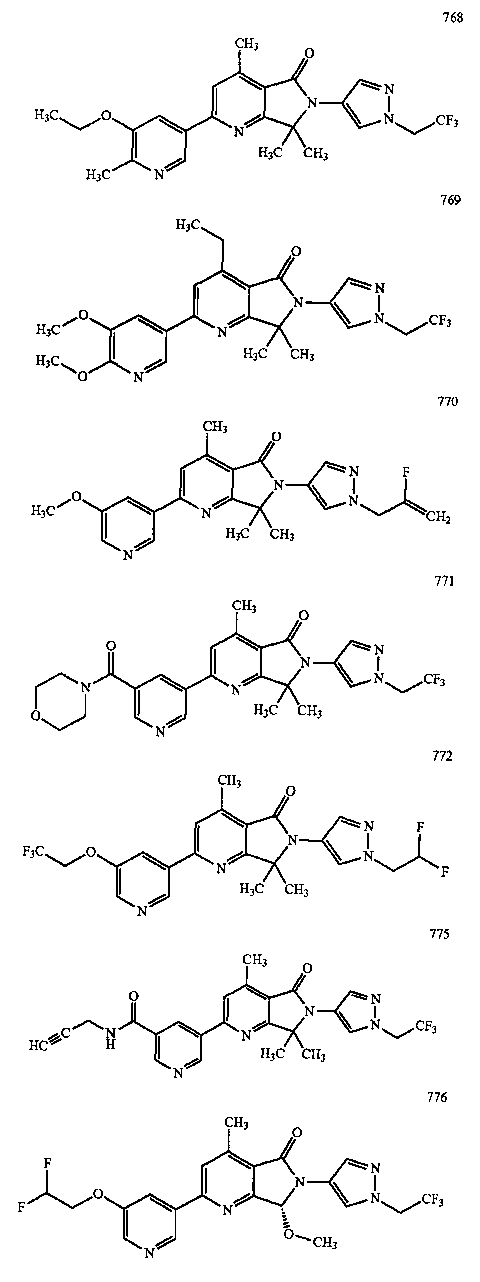
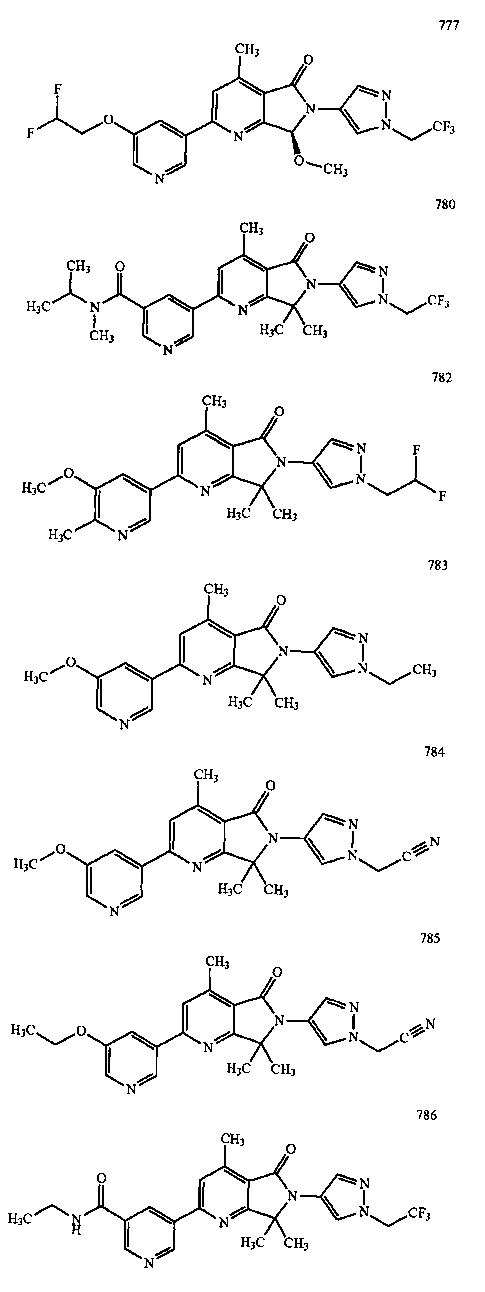
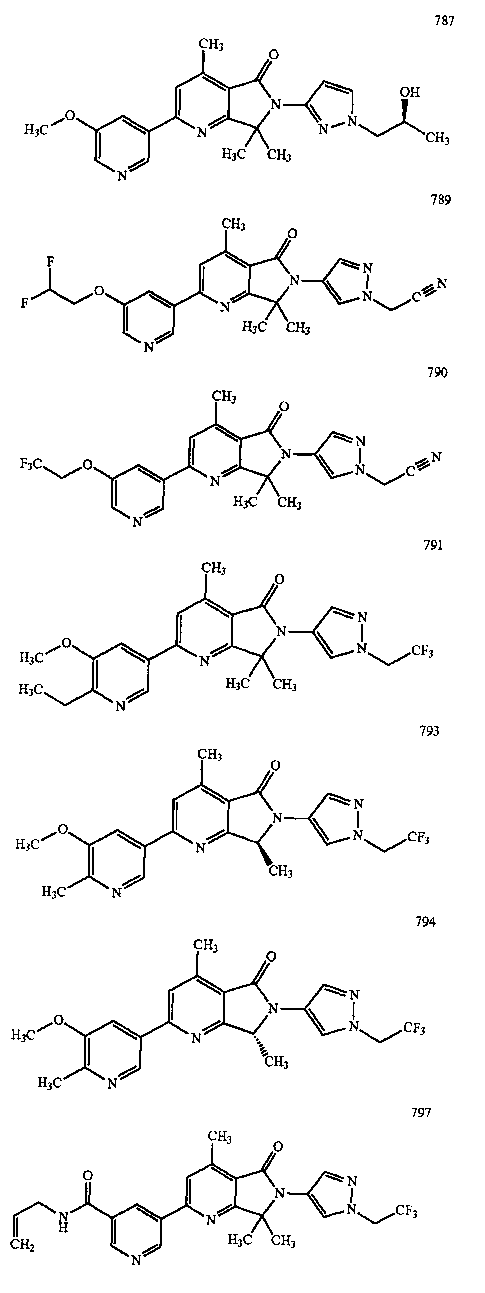
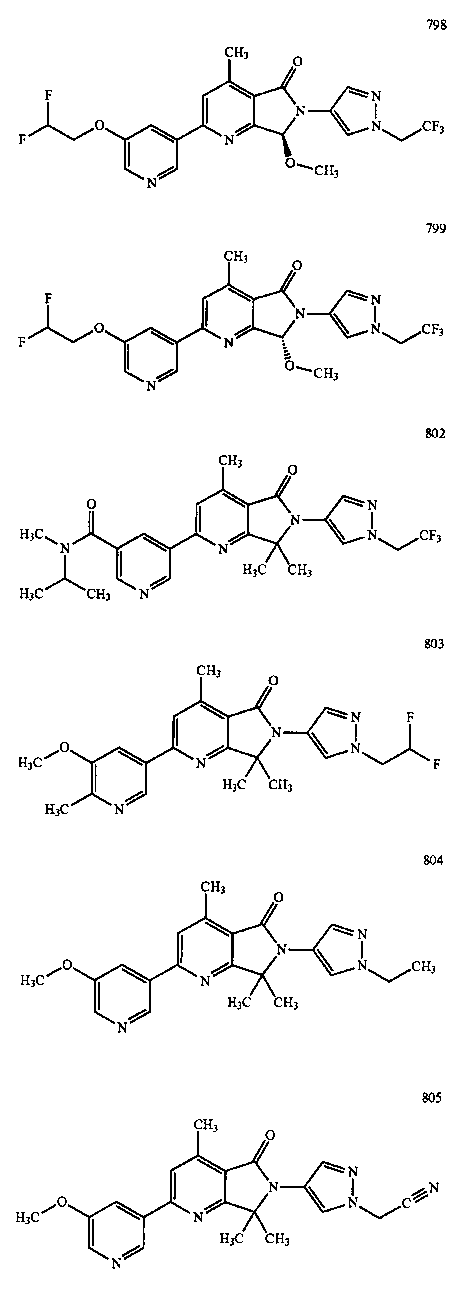
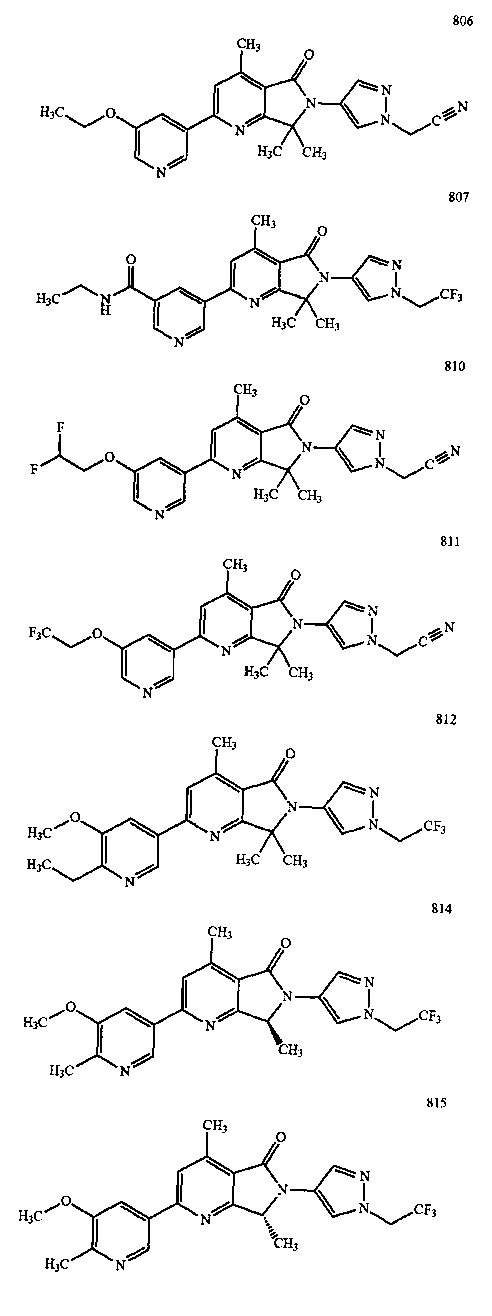
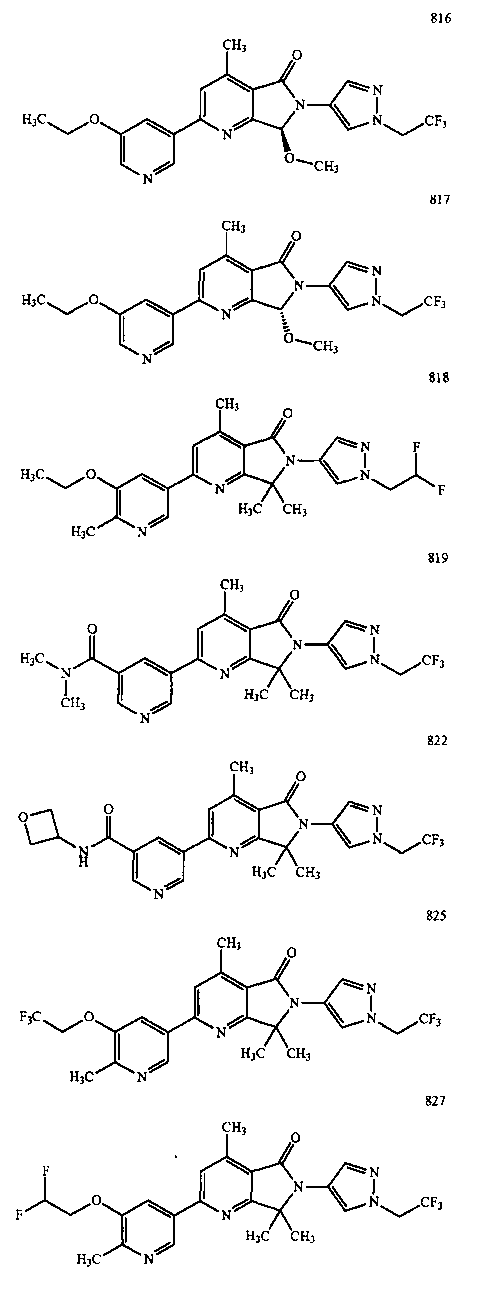
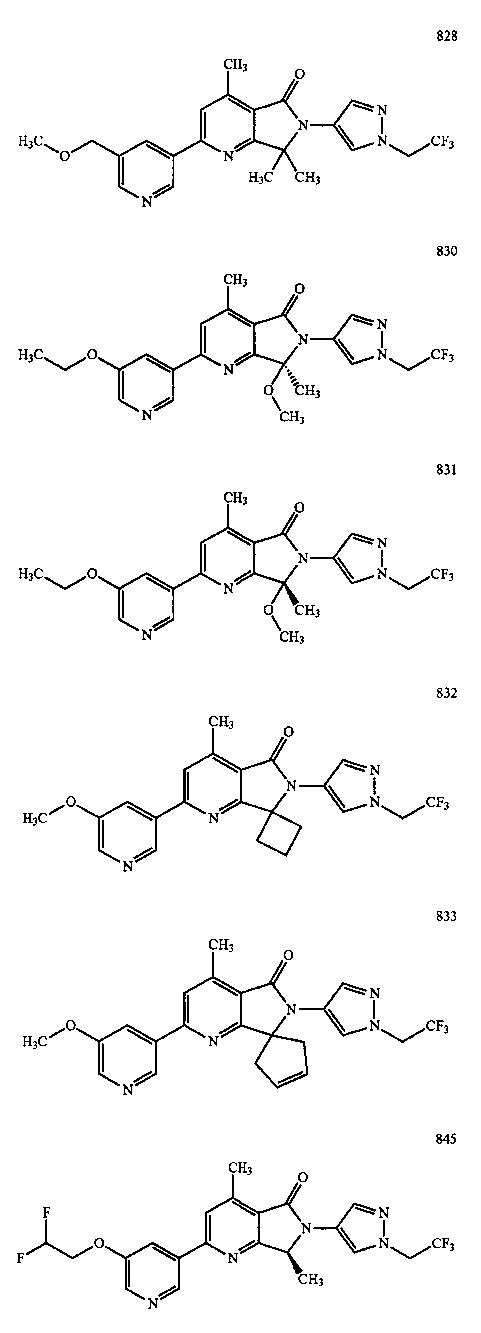
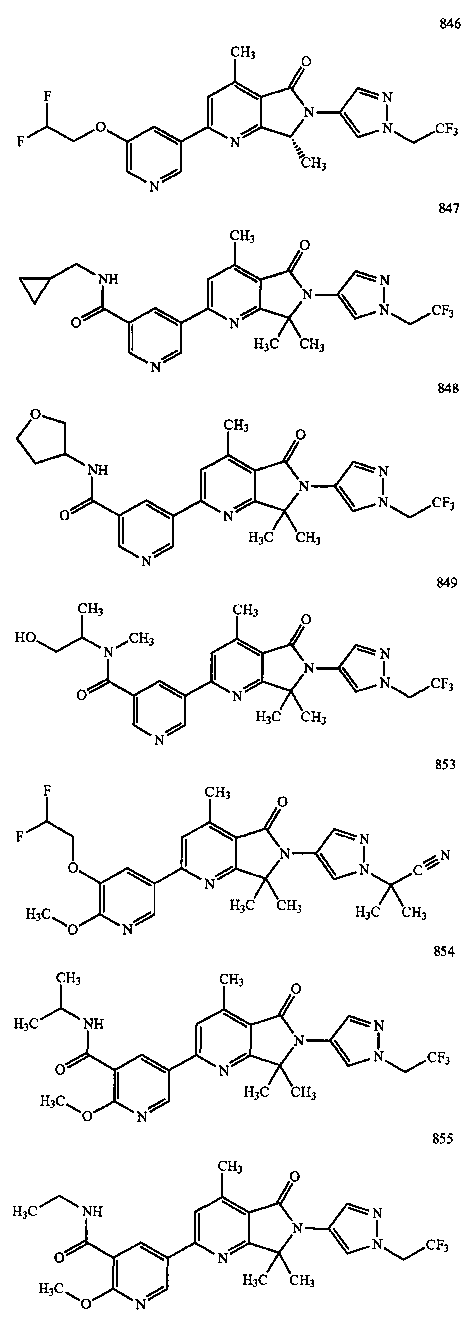
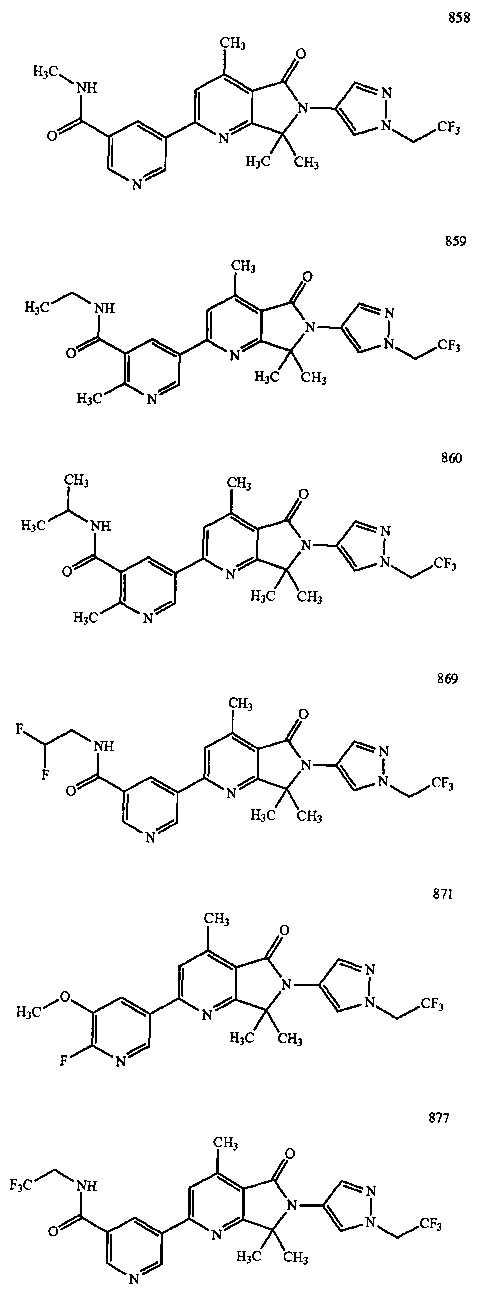
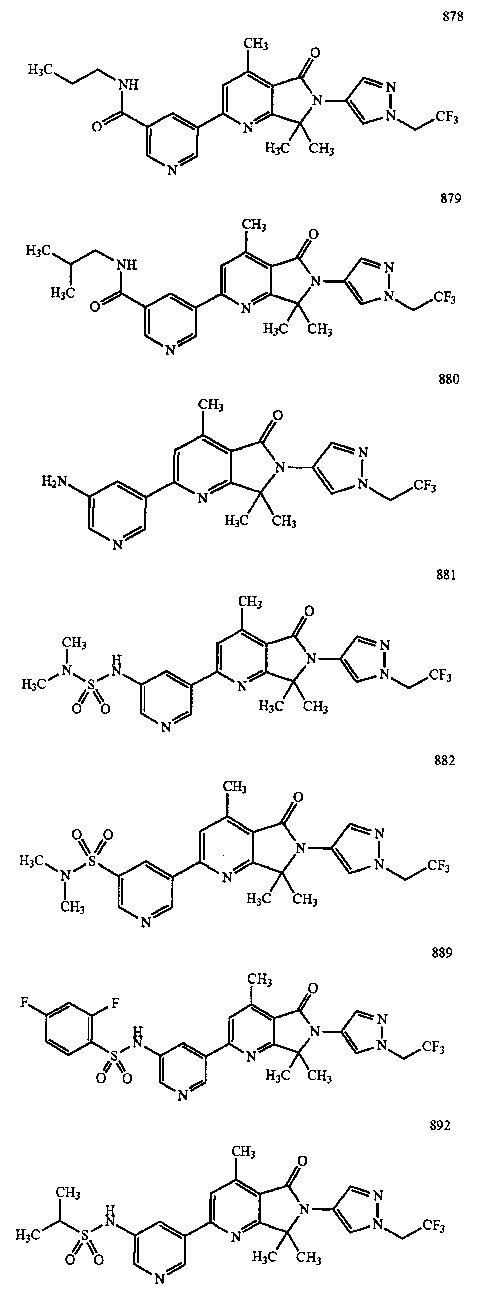
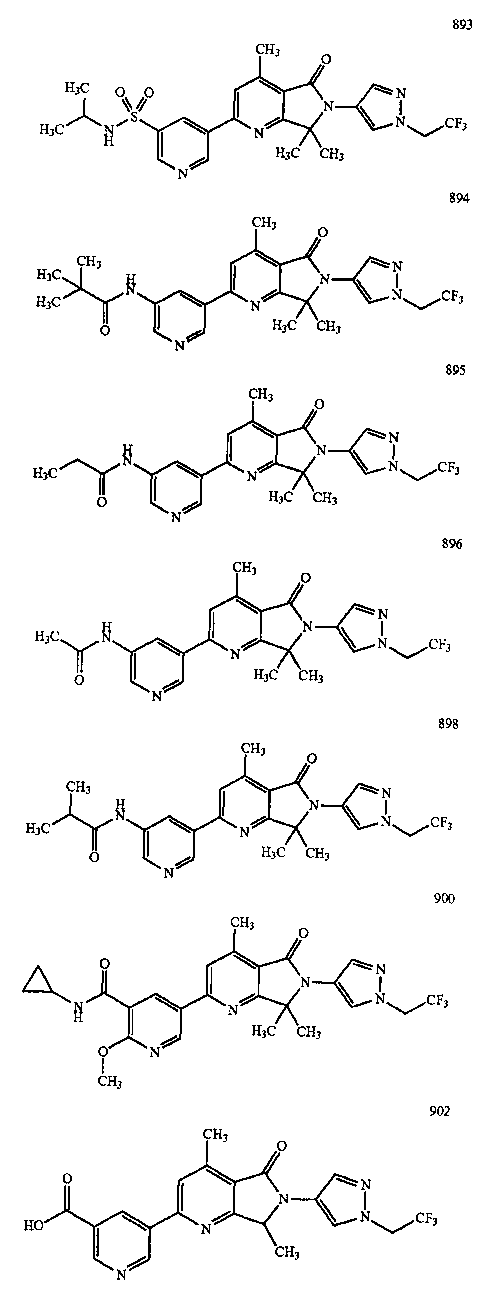
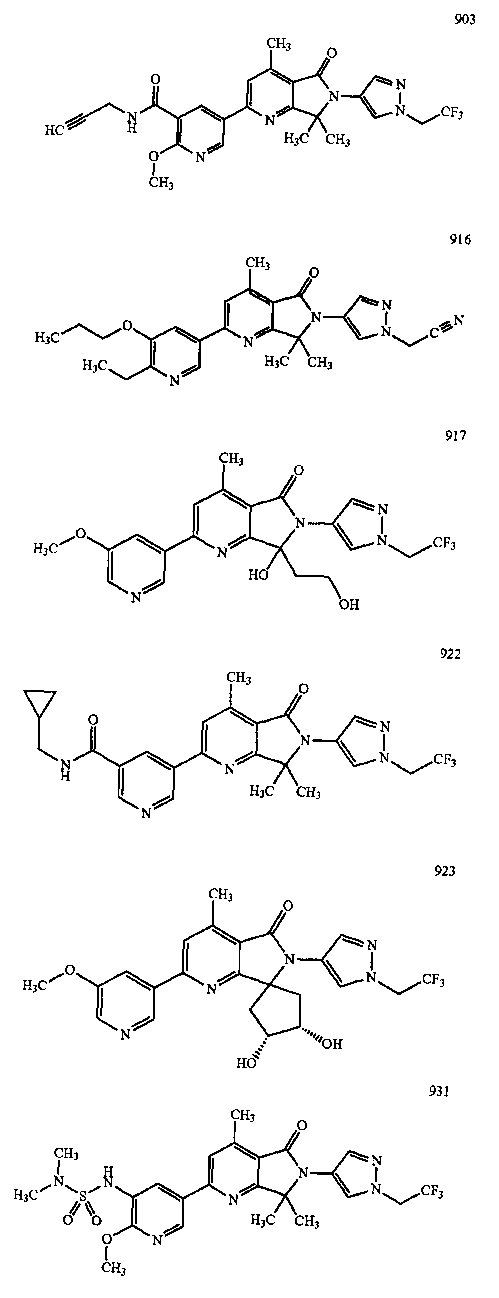
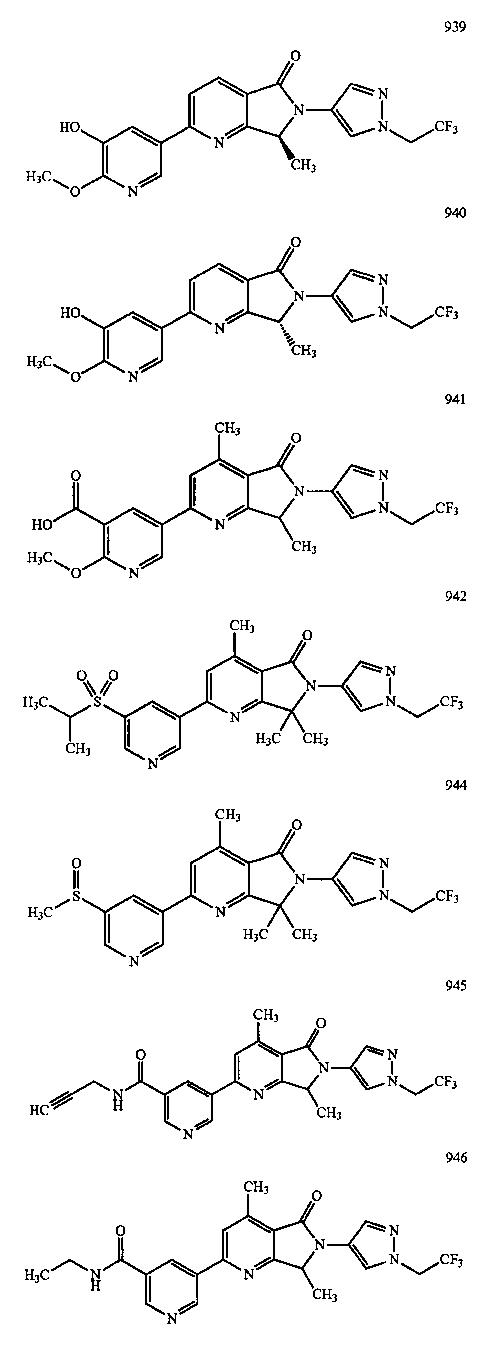

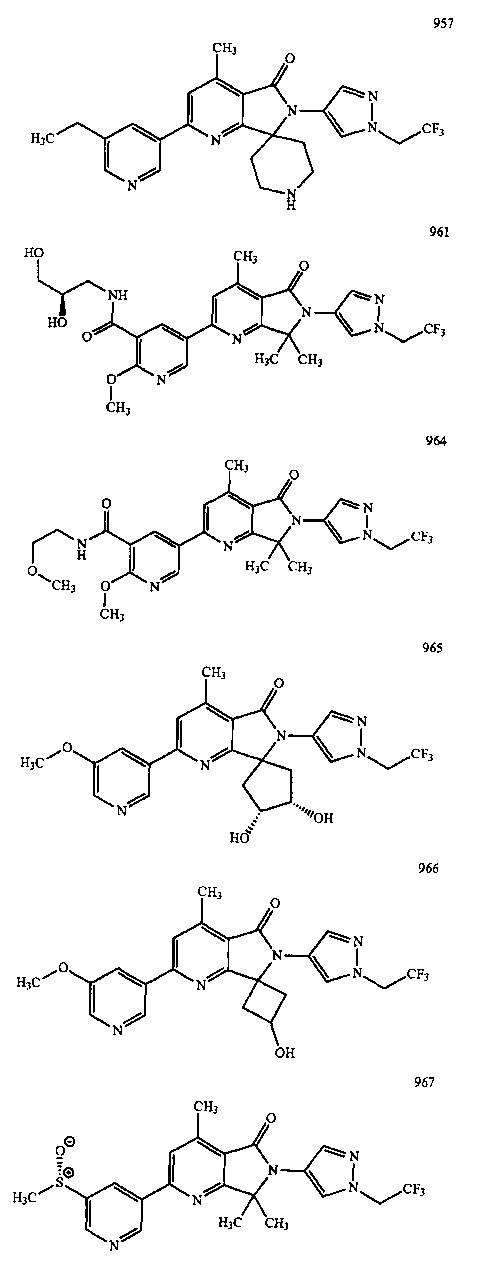
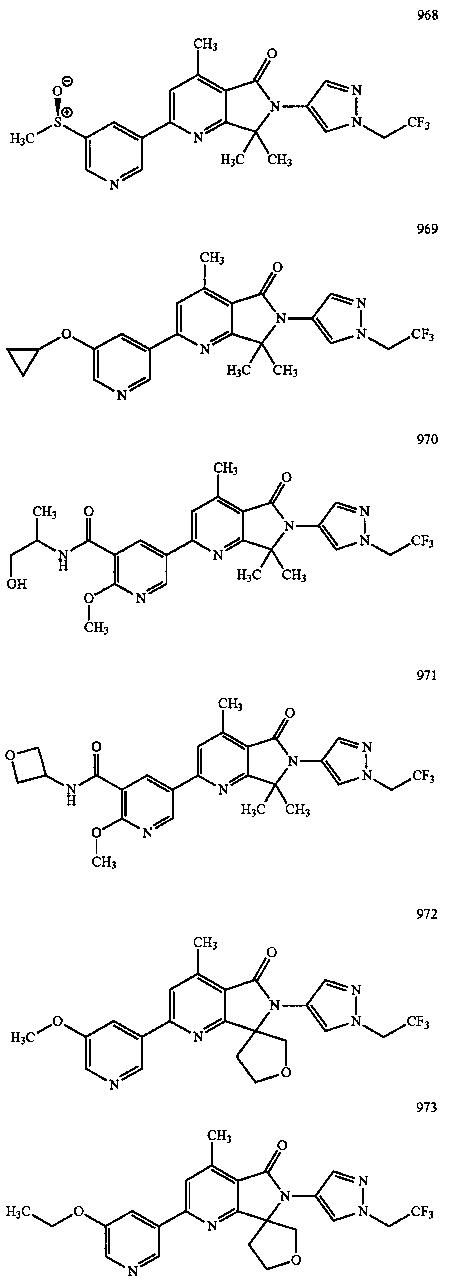
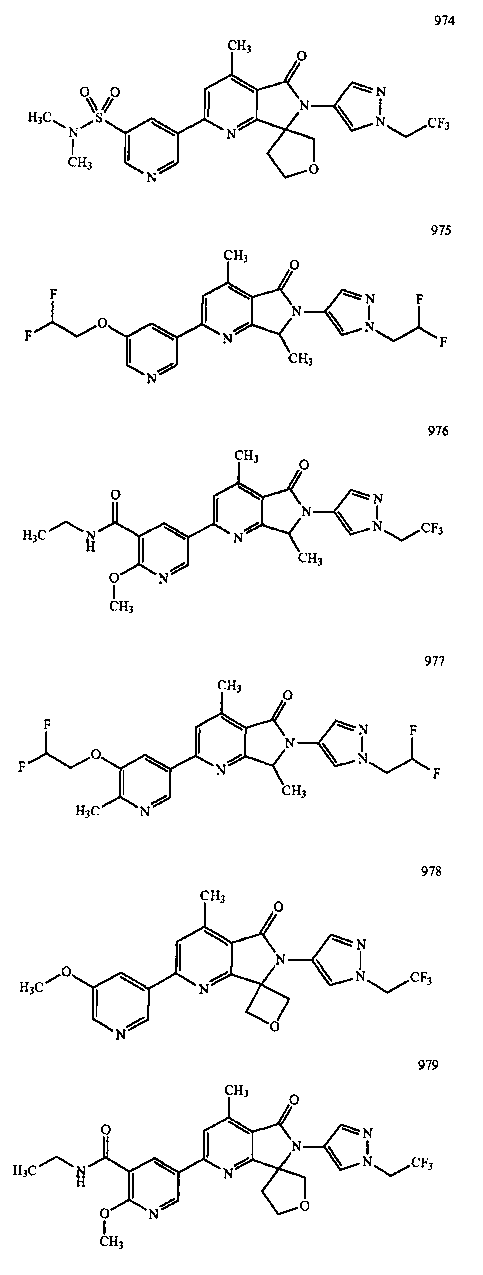
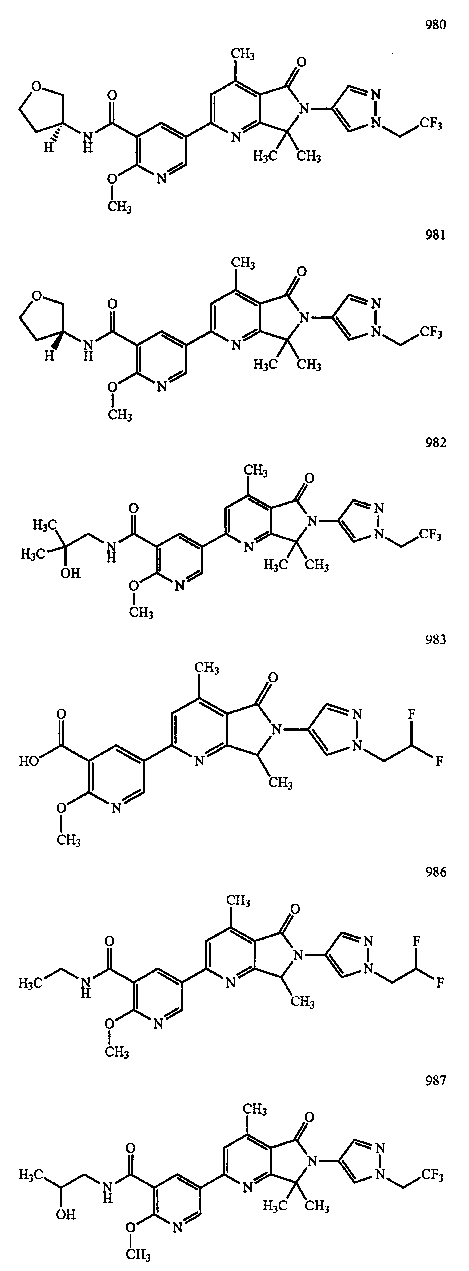
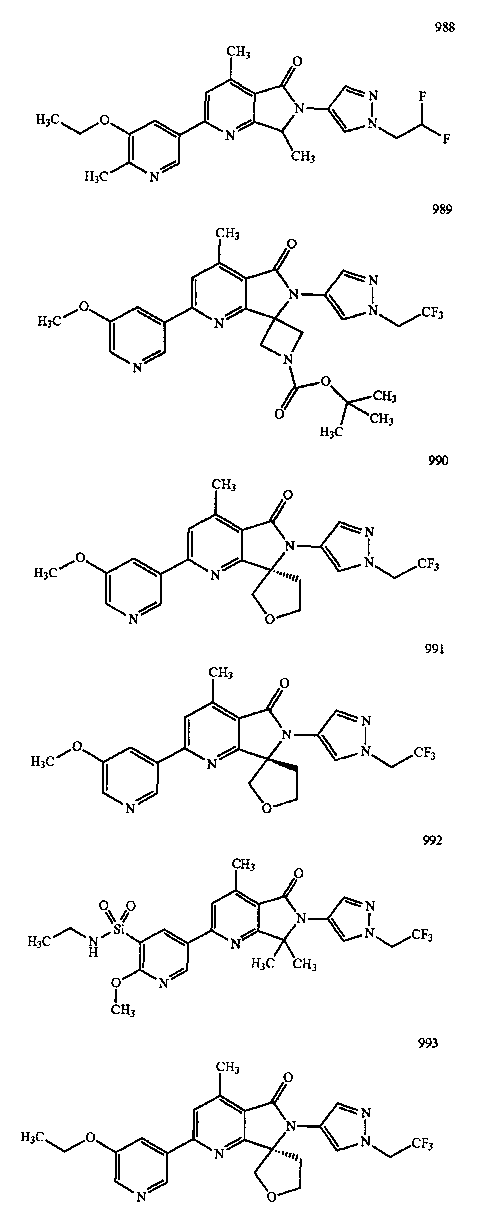
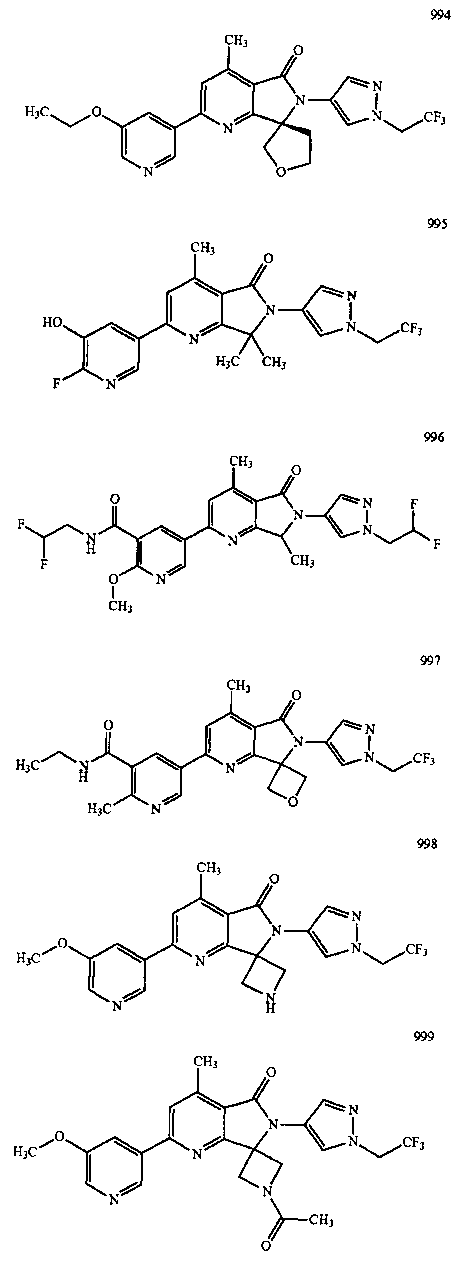

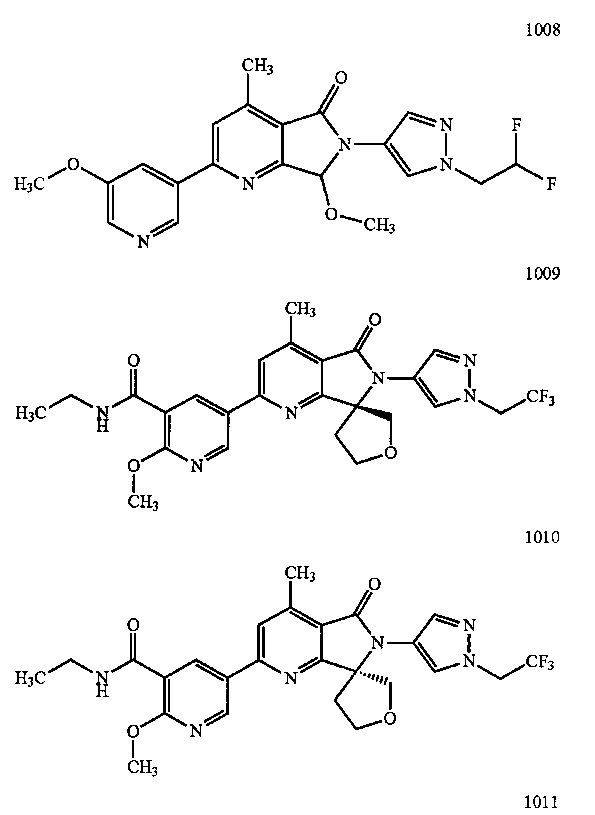
 или
или

или его фармацевтически приемлемая соль.
17. Фармацевтическая композиция, обладающая ингибирующей активностью в отношение киназы PI3K-гамма, содержащая соединение по п. 1 или 4 и фармацевтически приемлемый носитель, адъювант или наполнитель.
18. Способ ингибирования активности киназы PI3K-гамма в биологическом образце, включающий контактирование указанного биологического образца с соединением по п. 1 или 4 или композицией по п. 17.
| WO 2007095024 A1, 23.08.2007 | |||
| WO 2007000339 A1, 14.01.2007 | |||
| WO 2007109211 A2, 27.09.2007 | |||
| J | |||
| HURS, D | |||
| G | |||
| Машина для добывания торфа и т.п. | 1922 |
|
SU22A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| CHEM | |||
| SOC., 1962, pages 119-122 | |||
| WO 2004108672 A1, 16.12.2004 | |||
| EA 200970936 A1, 26.02.2010. | |||

