ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
[0001] Изобретение относится к способу получения фармацевтических композиций, содержащих форму I 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамидо)-3-метилпиридин-2-ил)бензойной кислоты (соединения 1) и твердую дисперсию, содержащую по существу аморфный N-(5-гидрокси-2,4-дитрет-бутил-фенил)-4-оксо-1H-хинолин-3-карбоксамид (Соединение 2), к связанным с ними способам лечения, способам введения и наборам.
УРОВЕНЬ ТЕХНИКИ, ПРЕДШЕСТВУЮЩИЙ ИЗОБРЕТЕНИЮ
[0002] Кистозный фиброз (CF) представляет собой рецессивное генетическое заболевание, поражающее приблизительно 30000 детей и взрослых в Соединенных Штатах Америки и приблизительно 30000 детей и взрослых в Европе. Несмотря на прогресс в лечении CF, не существует способов его излечения.
[0003] У пациентов с CF, мутации в CFTR, эндогенно экспрессированном в эпителии дыхательных путей, приводят к уменьшенной апикальной секреции анионов, вызывающей дисбаланс в транспорте ионов и жидкостей. Полученное уменьшение транспорта анионов способствует усиленному накоплению слизи в легком и сопутствующим микробным инфекциям, которые в конечном итоге вызывают смерть у пациентов с CF. В дополнение к респираторному заболеванию, пациенты с CF, как правило, страдают от желудочно-кишечных проблем и недостаточности поджелудочной железы, которая при отсутствии лечения приводит к смерти. Кроме того, большинство мужчин с кистозным фиброзом являются бесплодными, и фертильность является уменьшенной среди женщин с кистозным фиброзом. В отличие от серьезных эффектов двух копий ассоциированного c CF гена, индивидуумы с единственной копией ассоциированного с CF гена обладают увеличенной устойчивостью к холере и дегидратации, происходящей в результате диареи, что, возможно, объясняет относительно высокую частоту гена CF в популяции.
[0004] Анализ последовательности гена CFTR хромосом при CF выявил разнообразие вызывающих это заболевание мутаций (Cutting, G. R. Et al. (1990) Nature 346:366-369; Dean, M. et al. (1990) Cell 61:863:870; и Kerem, B-S. et al. (1989) Science 245:1073-1080; Kerem, B-S et al. (1990) Proc. Natl. Acad. Sci. USA 87:8447-8451). К настоящему времени идентифицированы более 1000 вызывающих заболевание мутаций в гене CF (http://www.genet.sickkids.on.ca/cftr/). Наиболее преобладающей мутацией является делеция фенилаланина в положении 508 аминокислотной последовательности CFTR, и она имеет общепринятое обозначение ΔF508-CFTR. Эта мутация встречается приблизительно в 70% случаев кистозного фиброза и ассоциирована с тяжелым заболеванием.
[0005] Делеция остатка 508 в ΔF508-CFTR препятствует точной укладке образующегося белка. Это приводит к неспособности мутантного белка выходить из ER и мигрировать к плазматической мембране. В результате, число каналов, присутствующих в мембране, намного меньше, чем число каналов, наблюдаемое в клетках, экспрессирующих CFTR дикого типа. Кроме нарушенной миграции, мутация приводит к дефектному пропусканию ионных каналов. Вместе это уменьшенное число каналов в мембране и дефектное пропускание приводят к уменьшенному транспорту через эпителий, приводя к дефектному транспорту ионов и жидкости (Quinton, P. M. (1990), FASEB J. 4: 2709-2727). Исследования показали, однако, что уменьшенные количества ΔF508-CFTR в мембране являются функциональными, хотя в меньшей степени, чем CFTR дикого типа (Dolmans et al. (1991), Nature Lond. 354: 526-528; Denning et at., выше; Pasyk and Foskett (1995), J. Cell. Biochem. 270: 12347-50). В дополнение к ΔF508-CFTR, другие вызывающие заболевания мутации в CFTR, которые приводят к дефектной миграции, дефектному синтезу и/или пропусканию ионных каналов, могут подвергаться повышающей или понижающей регуляции для изменения секреции анионов и модификации прогрессирования и/или тяжести заболевания.
[0006] Соединение 1 в форме соли описано в Международной публикации PCT WO2007056341 и Патенте Соединенных Штатов No. 7741321 в качестве индуктора активности CFTR и таким образом, в качестве применимого лекарственного средства для опосредованных CFTR заболеваний, таких как кистозный фиброз. Форма I соединения 1, которая является по существу кристаллической и бессолевой формой, описана в Международной публикации PCT WO2009073757 и Патенте Соединенных Штатов No. 8507534. Соединение 2 описано в Международной публикации PCT WO2006002421 и Патенте Соединенных Штатов No. 7495103 в качестве индуктора активности CFTR и таким образом, в качестве применимого лекарственного средства для опосредованных CFTR заболеваний, таких как кистозный фиброз. Твердая дисперсия, содержащая по существу аморфное соединение 2, описана в Международной публикации PCT WO2010019239 и опубликованной Патентной заявке Соединенных Штатов No. US20100074949. Полное содержание всех вышеуказанных заявок и патентов приведено в настоящем документе в качестве ссылки.
[0007] Независимо показано, что соединения, являющиеся усилителями CFTR, такие как соединение 2, и соединения, являющиеся корректорами CFTR, такие как соединение 1, обладают полезностью для лечения связанных с CFTR заболеваний, таких как кистозный фиброз.
[0008] Соответственно, существует необходимость в новых лекарственных средствах против опосредованных CFTR заболеваний, включающих в себя соединения - корректоры и усилители CFTR.
[0009] В частности, существует необходимость в способах комбинированной терапии для лечения опосредованных CFTR заболеваний, таких как кистозный фиброз, включающих в себя соединения -усилители и корректоры CFTR.
[0010] Более конкретно, существует необходимость в способах комбинированной терапии для лечения опосредованных CFTR заболеваний, таких как кистозный фиброз, включающих в себя соединения - усилители CFTR, такие как по существу аморфное соединение 2, в комбинации с соединениями - корректорами CFTR, такими как форма I соединения 1.
[0011] Соединение 1 в качестве части комбинации с соединением 2 признано принципиально новым лекарственным средством Управлением по контролю качества пищевых продуктов и лекарственных (FDA) для лечения кистозного фиброза, одно из только двух таких признанных средств на время подачи этой заявки (где другое представляет собой соединение 2). Это указывает на значительную неудовлетворенную необходимость в эффективном лечении причины кистозного фиброза вместо симптоматического лечения. Дополнительно, общераспространенной проблемой лекарственных средств, одобренных FDA, является отсутствие время от времени доступности лекарственного средства для нуждающихся в этом пациентов. Соответственно, существует значительная неудовлетворенная необходимость в описанных в настоящее время составах соединения 1 и соединения 2, и способах их получения непрерывным и контролируемым образом.
[0012] Дополнительно, соблюдение пациентом схемы лечения с расписанием лечения и величиной доз в большой степени зависит от простоты введения лекарственного средства. Фармацевтическая композиция, содержащая фиксированную величину доз корректора CFTR и усилителя CFTR, где твердые формы указанного корректора и усилителя являются стабильными, является значительным прорывом для лечения опосредованных CFTR заболеваний, таких как кистозный фиброз.
СУЩНОСТЬ
[0013] Изобретение относится к способу получения фармацевтических композиций, содержащих 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамидо)-3-метилпиридин-2-ил)бензойную кислоту, форму I соединения 1, обладающую структурой ниже:
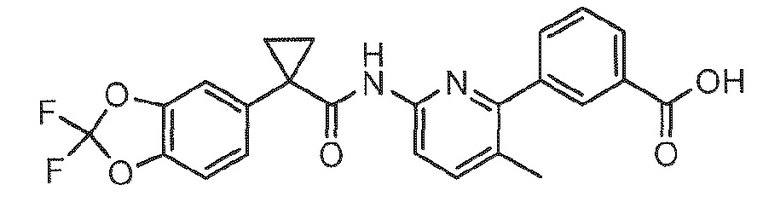
Соединение 1; и
твердую дисперсию по существу аморфного N-(5-гидрокси-2,4-дитрет-бутил-фенил)-4-оксо-1H-хинолин-3-карбоксамида, соединения 2, обладающего структурой ниже:
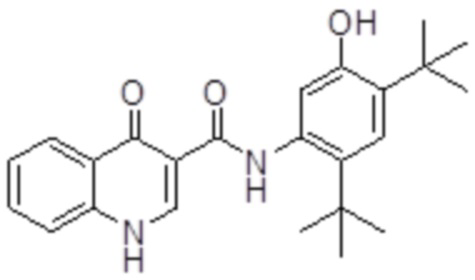
Соединение 2;
к связанным с ними способам лечения, способам введения и наборам.
[0014] В одном из аспектов настоящее изобретение относится к способу получения фармацевтической композиции, содержащей:
a. форму I соединения 1;
b. твердую дисперсию, содержащую по существу аморфное соединение 2;
c. наполнитель;
d. дезинтегрирующее средство;
e. поверхностно-активное вещество; и
f. связывающее средство;
обозначенной PC-I.
[0015] В одном из вариантов осуществления способ получения фармацевтических композиций по настоящему изобретению включает в себя 30-55 процентов по массе формы I соединения 1, и 10-45 процентов по массе твердой дисперсии, содержащей по существу аморфное соединение 2.
[0016] В одном из вариантов осуществления наполнитель выбран из целлюлозы, модифицированной целлюлозы, карбоксиметилцеллюлозы натрия, этилцеллюлозы гидроксиметилцеллюлозы, гидроксипропилцеллюлозы, ацетата целлюлозы, микрокристаллической целлюлозы, гидрофосфата кальция, сахарозы, лактозы, кукурузного крахмала, картофельного крахмала или любого их сочетания. В другом варианте осуществления наполнитель представляет собой микрокристаллическую целлюлозу и присутствует в количестве в диапазоне от 10 до 20 процентов по массе.
[0017] В одном из вариантов осуществления дезинтегрирующее средство выбрано из агар-агара, альгинов, карбоната кальция, карьоксиметилцеллюлозы, целлюлозы, гидроксипропилцеллюлозы, низкозамещенной гидроксипропилцеллюлозы, глин, кроскармеллозы натрия, кросповидона, камедей, алюмосиликата магния, метилцеллюлозы, полакрилина калия, альгината натрия, крахмалгликолята натрия, кукурузного крахмала, картофельного крахмала, тапиокового крахмала или любого их сочетания. В другом варианте осуществления дезинтегрирующее средство представляет собой кроскармеллозу натрия и присутствует в количестве в диапазоне от 1 до 3 процентов по массе.
[0018] В одном из вариантов осуществления поверхностно-активное вещество выбрано из лаурилсульфата натрия, стеарилфумарата натрия, моноолеата полиоксиэтилен-20-сорбитана или любого их сочетания. В другом варианте осуществления поверхностно-активное вещество представляет собой лаурилсульфат натрия и присутствует в количестве в диапазоне от 0,5 до 2 процентов по массе.
[0019] В одном из вариантов осуществления связывающее средство выбрано из поливинилпирролидона, гидрофосфата кальция, сахарозы, кукурузного крахмала, модифицированной целлюлозы или любого их сочетания. В другом варианте осуществления связывающее средство представляет собой поливинилпирролидон и присутствует в количестве в диапазоне от 0 до 5 процентов по массе.
[0020] В одном из вариантов осуществления настоящее изобретение относится к способу получения фармацевтической композиции, обладающей следующим составом:
обозначенной как PC-II.
[0021] В другом аспекте настоящее изобретение относится к способу получения фармацевтической композиции, содержащей:
a. форму I соединения 1;
b. твердую дисперсию, содержащую по существу аморфное соединение 2;
c. наполнитель;
d. дезинтегрирующее средство;
e. поверхностно-активное вещество;
f. связывающее средство; и
g. смазочное средство;
обозначенной как PC-III.
[0022] В одном из вариантов осуществления способ получения фармацевтической композиции по настоящему изобретению включает в себя приблизительно от 100 до 250 мг формы I соединения 1 и приблизительно от 100 до 150 мг по существу аморфного соединения 2. В другом варианте осуществления фармацевтические композиции по настоящему изобретению содержат приблизительно 200 мг формы I соединения 1 и приблизительно 125 мг по существу аморфного соединения 2. В другом варианте осуществления фармацевтические композиции по настоящему изобретению содержат приблизительно 150 мг формы I соединения 1 и приблизительно 125 мг по существу аморфного соединения 2.
[0023] В одном из вариантов осуществления способ получения фармацевтических композиций по настоящему изобретению включает в себя 25-50 процентов по массе формы I соединения 1, и 15-35 процентов по массе твердой дисперсии, содержащей по существу аморфное соединение 2.
[0024] В одном из вариантов осуществления наполнитель выбран из целлюлозы, модифицированной целлюлозы, карбоксиметилцеллюлозы натрия, этилцеллюлозы гидроксиметилцеллюлозы, гидроксипропилцеллюлозы, ацетата целлюлозы, микрокристаллической целлюлозы, гидрофосфата кальция, сахарозы, лактозы, кукурузного крахмала, картофельного крахмала или любого их сочетания. В другом варианте осуществления наполнитель представляет собой микрокристаллическую целлюлозу и присутствует в количестве в диапазоне от 20 до 30 процентов по массе.
[0025] В одном из вариантов осуществления дезинтегрирующее средство выбрано из агар-агара, альгинов, карбоната кальция, карьоксиметилцеллюлозы, целлюлозы, гидроксипропилцеллюлозы, низкозамещенной гидроксипропилцеллюлозы, глин, кроскармеллозы натрия, кросповидона, камедей, алюмосиликата магния, метилцеллюлозы, полакрилина калия, альгината натрия, крахмалгликолята натрия, кукурузного крахмала, картофельного крахмала, тапиокового крахмала или любого их сочетания. В другом варианте осуществления дезинтегрирующее средство представляет собой кроскармеллозу натрия и присутствует в количестве в диапазоне от 3 до 10 процентов по массе.
[0026] В одном из вариантов осуществления поверхностно-активное вещество выбрано из лаурилсульфата натрия, стеарилфумарата натрия, моноолеата полиоксиэтилен-20-сорбитана или любого их сочетания. В другом варианте осуществления поверхностно-активное вещество представляет собой лаурилсульфат натрия и присутствует в количестве в диапазоне от 0,5 до 2 процент по массе.
[0027] В одном из вариантов осуществления связывающее средство выбрано из поливинилпирролидона, гидрофосфата кальция, сахарозы, кукурузного крахмала, модифицированной целлюлозы или любого их сочетания. В другом варианте осуществления связывающее средство представляет собой поливинилпирролидон и присутствует в количестве в диапазоне от 0 до 5 процентов по массе.
[0028] В одном из вариантов осуществления смазочное средство выбрано из стеарата магния, стеарата кальция, стеарата цинка, стеарата натрия, стеариновой кислоты, стеарата алюминия, лейцина, глицерилбегената, гидрогенизированного растительного масла или любого их сочетания. В другом варианте осуществления смазочное средство представляет собой стеарат магния и присутствует в количестве в диапазоне от 0,5 до 2 процентов по массе.
[0029] В одном из вариантов осуществления настоящее изобретение относится к способу получения фармацевтической композиции, обладающей следующим составом:
обозначенной как PC-в/в.
[0030] В одном из вариантов осуществления способ получения фармацевтических композиций по настоящему изобретению дополнительно включает в себя краситель и, необязательно, воск. В другом варианте осуществления краситель присутствует в количестве в диапазоне от 2 до 4 процентов по массе. В другом варианте осуществления воск представляет собой карнаубский воск и присутствует в количестве в диапазоне от 0 до 0,020 процентов по массе.
[0031] В одном из вариантов осуществления способ получения фармацевтических композиций по настоящему изобретению относится к твердым пероральным фармацевтическим композициям. В другом варианте осуществления твердые пероральные фармацевтические композиции представляют собой гранулированную фармацевтическую композицию или таблетку.
[0032] В одном из вариантов осуществления представлен способ получения гранулированной фармацевтической композиции по настоящему изобретению, обладающей следующим составом:
обозначенной как PC-V.
[0033] В одном из вариантов осуществления представлен способ получения гранулированной фармацевтической композиции по настоящему изобретению, обладающей следующим составом:
обозначенной как PC-VI.
[0034] В одном из вариантов осуществления представлен способ получения гранулированной фармацевтической композиции по настоящему изобретению, обладающей следующим составом:
обозначенной как PC-VII.
[0035] В одном из вариантов осуществления представлен способ получения таблеток по настоящему изобретению, обладающих следующим составом:
обозначенных как PC-VIII.
[0036] В одном из вариантов осуществления представлен способ получения таблеток по настоящему изобретению, обладающих следующим составом:
обозначенных как PC-IX.
[0037] В одном из вариантов осуществления представлен способ получения таблеток по настоящему изобретению, обладающих следующим составом:
обозначенных как PC-X.
[0038] В одном из вариантов осуществления представлен способ получения таблеток по настоящему изобретению, обладающих следующим составом:
обозначенных как PC-XI.
[0039] В одном из вариантов осуществления представлен способ получения таблеток по настоящему изобретению, обладающих следующим составом:
обозначенных как PC-XII.
[0040] В одном из вариантов осуществления представлен способ получения таблеток по настоящему изобретению, обладающих следующим составом:
обозначенных как PC-XIII.
[0041] В одном из вариантов осуществления представлен способ получения таблеток по настоящему изобретению, обладающих следующим составом:
обозначенных как PC-XIV.
[0042] В одном из вариантов осуществления представлен способ получения таблеток по настоящему изобретению, обладающих следующим составом:
обозначенных как PC-XV.
[0043] В одном из вариантов осуществления представлен способ получения таблеток по настоящему изобретению, обладающих следующим составом:
обозначенных как PC-XVI.
[0044] В одном из вариантов осуществления представлен способ получения таблеток по настоящему изобретению, обладающих следующим составом:
обозначенных как PC-XVII.
[0045] В одном из вариантов осуществления представлен способ получения таблеток по настоящему изобретению, обладающих следующим составом:
обозначенных как PC-XVIII
[0046] В одном из вариантов осуществления представлен способ получения таблеток по настоящему изобретению, обладающих следующим составом:
обозначенных как PC-XIX.
[0047] В одном из вариантов осуществления представлен способ получения таблеток по настоящему изобретению, обладающих следующим составом:
обозначенных как PC-XX.
[0048] В одном из вариантов осуществления представлен способ получения таблеток по настоящему изобретению, обладающих следующим составом:
обозначенных как PC-XXI.
[0049] В одном из вариантов осуществления представлен способ получения таблеток по настоящему изобретению, обладающих следующим составом:
обозначенных как PC-XXII.
[0050] В одном из вариантов осуществления представлен способ получения таблеток по настоящему изобретению, обладающих следующим составом:
обозначенных как PC-XXIII.
[0051] В одном из вариантов осуществления представлен способ получения таблеток по настоящему изобретению, обладающих следующим составом:
обозначенных как PC-XXIV.
[0052] В одном из вариантов осуществления представлен способ получения таблеток по настоящему изобретению, обладающих следующим составом:
обозначенных как PC-XXV.
[0053] В одном из аспектов настоящее изобретение относится к способу лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающему в себя введение пациенту эффективного количество фармацевтической композиции, гранулированной фармацевтической композиции или таблетки по настоящему изобретению.
[0054] В одном из вариантов осуществления настоящее изобретение относится к способу лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающему в себя введение пациенту эффективного количества фармацевтической композиции, гранулированной фармацевтической композиции или таблетки любого одного из составов PC-I - PC-XXV.
[0055] В одном из вариантов осуществления пациент обладает мутацией ΔF508 CFTR. В другом варианте осуществления пациент является гомозиготным по ΔF508. В другом варианте осуществления пациент является гетерозиготным по ΔF508. В другом варианте осуществления две таблетки вводят пациенту в сутки.
[0056] В одном из аспектов настоящее изобретение относится к способу получения гранулированной фармацевтической композиции, включающему в себя влажную грануляцию следующих компонентов:
a. форма I соединения 1;
b. твердая дисперсия, содержащая по существу аморфное соединение 2;
c. наполнитель;
d. дезинтегрирующее средство;
e. поверхностно-активное вещество; и
f. связывающее средство.
[0057] В одном из аспектов настоящее изобретение относится к способу получения таблетки, включающему в себя прессование:
i) множества гранулированных фармацевтических композиций, содержащих следующие компоненты:
a. форма I соединения 1;
b. твердая дисперсия, содержащая по существу аморфное соединение 2;
c. наполнитель;
d. дезинтегрирующее средство;
e. поверхностно-активное вещество; и
f. связывающее средство;
ii) дезинтегрирующего средства;
iii) наполнителя; и
iv) смазочного средства.
[0058] В одном из аспектов настоящее изобретение относится к набору, содержащему фармацевтические композиции, гранулированные фармацевтические композиции или таблетки по настоящему изобретению и отдельное терапевтическое средство или его фармацевтическую композицию.
[0059] В одном из вариантов осуществления фармацевтические композиции, гранулированные фармацевтические композиции или таблетки по настоящему изобретению и отдельное терапевтическое средство или его фармацевтическая композиция находятся в отдельных контейнерах. В другом варианте осуществления отдельные контейнеры представляют собой бутыли. В другом варианте осуществления отдельные контейнеры представляют собой флаконы. В другом варианте осуществления отдельные контейнеры представляют собой блистерные упаковки.
[0060] В другом аспекте изобретение относится к непрерывному или полунепрерывному способу получения фармацевтических композиций, описываемых в настоящем документе посредством способа двухшнековой влажной грануляции, включающего в себя просеивание и взвешивание соединения 1, соединения 2 и наполнителей; смешивание соединения 1, соединения 2 и наполнителей в смесителе и загрузку смеси в гранулятор непрерывного действия с добавлением в то же время гранулирующей жидкости, содержащей поверхностно-активное вещество и связывающее средство с подходящей скорости в течение подходящего количества времени и нарезку смеси на гранулы; сушку гранул; смешивание гранул с экстрагранулированными наполнителями в течение подходящего количества времени; прессование смеси в таблетки; покрытие таблеток; и, необязательно, печать монограммы на одной или обеих сторонах таблетки.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
[0061] Фигура 1 представляет собой картину рентгенодифракции, рассчитанную для монокристаллической структуры формы I соединения 1.
[0062] Фигура 2 представляет собой фактическую картину порошковой рентгенодифракции формы I соединения 1.
[0063] Фигура 3 представляет собой график, изображающий профили растворения соединения 1 в градиенте pH для таблеток, изготовленных способом грануляции с большим усилием сдвига (HSG) и способом двухшнековой влажной грануляции (TSWG) (LOD обозначает потери при сушке, измерение для определения количества воды в порошке/гранулах).
[0064] Фигура 4 представляет собой график, изображающий стабильность по существу аморфной формы соединения 2 в составе таблетки PC-XVII при 50°C после предварительного уравновешивания при относительной влажности 60% посредством демонстрации только небольшой степени кристалличности с течением времени.
[0065] Фигура 5 представляет собой график, изображающий стабильность по существу аморфной формы соединения 2 в составе таблетки PC-XVII при 60°C после предварительного уравновешивания при относительной влажности 60% посредством демонстрации только небольшой степени кристалличности с течением времени.
[0066] Фигура 6 представляет собой график, изображающий стабильность по существу аморфной формы соединения 2 в составе таблетки PC-XX при 60°C после предварительного уравновешивания при относительной влажности 60% посредством демонстрации только небольшой степени кристалличности с течением времени.
[0067] Фигура 7 представляет собой график, изображающий стабильность по существу аморфной формы соединения 2 в составе таблетки PC-XX при 50°C после предварительного уравновешивания при относительной влажности 60% посредством демонстрации только небольшой степени кристалличности с течением времени.
[0068] Фигура 8 представляет собой спектр 1HЯМР соединения 1.
[0069] Фигура 9 представляет собой спектр 1HЯМР соли соединения 1 с HCl.
[0070] Фигура 10 представляет собой запись дифференциальной сканирующей калориметрии (DSC) формы I соединения 1.
[0071] Фигура 11 представляет собой изображение конформации формы I соединения 1 на основании монокристаллического рентгенографического анализа.
[0072] Фигура 12 представляет собой схематическое изображение аналитической технологии процесса (PAT), обеспечивающей непрерывный процесс, где на стадии 1) подачи/смесителя один, PAT1 NIR измеряет характеристики материала в ходе скрининга сырья; на стадии 2) двухшнекового гранулятора, PAT2 NIR измеряет состав и BU; на стадии 3) сушилки с псевдоожиженным слоем, PAT 3a NIR измеряет однородность гранул, LOD, форму твердого состояния и физические характеристики гранул, PAT 3b лазерная дифракция измеряет распределение размера частиц; на стадии 4) размола, PAT4 NIR измеряет состав и BU; на стадии 5) подачи/смесителя два, спектр Рамана PAT 5a измеряет анализ и CU, PAT 5b массу, жесткость, густоту; на стадии 6) прессования, спектр Рамана PAT6 измеряет толщину покрытия; и на стадии 7) покрытие.
[0073] Фигура 13 представляет собой схематическое изображение, показывающее PAT Sentronics NIR в процессе производства, локализованную после смесителя один, мельницы для гранул и дополнительного смесителя для гранул. Каждый тест имеет 7 точек, которые меняются в циклической последовательности для максимизации отбора образцов, и NIR с мультиплексором-NIR, обеспечивающие надежный и всеобъемлющий отбор образцов посредством контролируемого потока порошка через тестирующие оптические приборы.
[0074] Фигура 14 представляет собой изображение NIR в текущем порошке.
[0075] Фигура 15 представляет собой спектр Рамана Kaiser формы I соединения 1 и формы II соединения 1 (форма II соединения 1 представляет собой отличный полиморф, описанный в US 201131588, полное содержание которого приведено в настоящем документе в качестве ссылки), взятых после прессования таблеток. Спектрометр Рамана Kaiser монтирован на оборудовании для тестирования таблеток Kraemer UTS.
[0076] Фигура 16 представляет собой график, показывающий хорошую корреляцию между предсказанным и эталонным отбором вне процесса производства образцов NIR гранул соединения 2.
[0077] Фигура 17 представляет собой серии спектров NIR, измеряющих содержание воды в образцах гранул соединения 1.
[0078] Фигура 18 представляет собой серии спектров NIR, измеряющих диапазон композиций, содержащих различные соотношения формы I соединения 1 и твердых дисперсий, содержащих по существу аморфное соединение 2 слева, и спектр после предварительной обработки справа, изображающий диапазон A для идентифицированной формы I соединения 1 и диапазон B для идентифицированного аморфного соединения 2.
[0079] На фигуре 19 изображена калибровочная кривая для предсказанного содержания формы I соединения 1 по сравнению с эталонным (фактическим) содержанием формы I соединения 1 с использованием способов дробных наименьших квадратов (PLS).
[0080] На фигуре 20 изображены фактические результаты для неизвестных образцов, содержащих различное содержание формы I соединения 1 (эталонное Y) по сравнению с предсказанным содержанием с использованием калибровочной кривой, рассчитанной из фигуры 19 (предсказанное Y).
[0081] На фигуре 21 изображен процент трансмиссии при измерении лазерной дифракции в ответ на изменения линейной скорости (скорости потока) для композиции, содержащей форму I соединения 1 и твердые дисперсии, содержащие по существу аморфное соединение 2, показывающий ожидаемое уменьшение процента трансмиссии как увеличение линейной скорости.
[0082] На фигуре 22 изображены измерения лазерной дифракции для частиц, содержащих форму I соединения 1 и твердые дисперсии, содержащие по существу аморфное соединение 2, при различных линейных скоростях, показывающие, что на средний размер частиц (Dv(50)) не влияет линейная скорость.
[0083] На фигуре 23 изображены измерения лазерной дифракции для частиц, содержащих форму I соединения 1 и твердые дисперсии, содержащие по существу аморфное соединение 2, при различных параметрах переработки, показывающие, что измерения размера частиц являются чувствительными к таким изменениям.
[0084] На фигуре 24 изображены предсказательные возможности моделей аналитической технологии процесса с использованием спектроскопии Рамана, как прерывистой, так и непрерывной, для мониторирования идентичности твердой формы соединения 1 в таблетке.
[0085] На фигуре 25 изображены предсказательные возможности моделей аналитической технологии процесса с использованием спектроскопии Рамана, как прерывистой, так и непрерывной, для мониторирования идентичности твердой формы соединения 2 в таблетке.
ПОДРОБНОЕ ОПИСАНИЕ
ОПРЕДЕЛЕНИЯ
[0086] Как используют в настоящем документе, «CFTR» обозначает регулятор трансмембранной проводимости при кистозном фиброзе.
[0087] Как используют в настоящем документе, «мутация ΔF508» или «мутация F508-del» представляет собой специфическую мутацию в белке CFTR. Мутация представляет собой делецию трех нуклеотидов, содержащих кодон для аминокислоты фенилаланина в положении 508, что в результате приводит к белку CFTR, лишенному остатка фенилаланина.
[0088] Как используют в настоящем документе, пациент, который является «гомозиготным» по конкретной мутации, например, ΔF508, обладает одинаковой мутацией в каждом аллеле.
[0089] Как используют в настоящем документе, пациент, который является «гетерозиготным» по конкретной мутации, например, ΔF508, обладает этой мутацией в одном аллеле, и отличной мутацией в другом аллеле.
[0090] Как используют в настоящем документе, термин «корректор CFTR» относится к соединению, которое увеличивает количество функционального белка CFTR на клеточной поверхности, что приводит в результате к усилению транспорта ионов.
[0091] Как используют в настоящем документе, термин «усилитель CFTR» относится к соединению, которое увеличивает канальную активность белка CFTR, локализованного на клеточной поверхности, что приводит в результате к усилению транспорта ионов.
[0092] Как используют в настоящем документе, термин «активный фармацевтический ингредиент» или «API» относится к биологически активному соединению.
[0093] Как используют в настоящем документе, термин «PAT» обозначает аналитическую технологию процесса.
[0094] Как используют в настоящем документе, термин «CU» обозначает однородность состава.
[0095] Термины «твердая форма», «твердые формы» и родственные термины, при использовании в настоящем документе относятся к соединению 1 или соединению 2, в конкретной твердой форме, например, кристаллах, аморфных состояниях и т.п.
[0096] Как используют в настоящем документе, термин «по существу аморфный» относится к твердому материалу, обладающему небольшим или отсутствующим дальним порядком в положении его молекул. Например, по существу аморфные материалы обладают менее, чем приблизительно 15% кристалличностью (например, менее, чем приблизительно 10% кристалличностью или менее, чем приблизительно 5% кристалличностью). Также следует отметить, что термин «по существу аморфное» включает в себя характеристику «аморфное», которая обозначает материалы, не обладающие (0%) кристалличностью.
[0097] Как используют в настоящем документе, термин «по существу кристаллический» (как во фразе по существу кристаллическая форма I соединения 1, относится к твердому материалу, преимущественно обладающему дальним порядком в положении его молекул. Например, по существу кристаллические материалы обладают более чем приблизительно 85% кристалличностью (например, более чем приблизительно 90% кристалличностью или более чем приблизительно 95% кристалличностью). Также следует отметить, что термин «по существу кристаллический» представляет собой характеристику «кристаллический», который обозначает материалы, обладающие 100% кристалличностью.
[0098] Термин «кристаллический» и родственные термины, используемые в настоящем документе, при использовании для описания вещества, компонента, продукта или формы, означают, что вещество, компонент или продукт являются по существу кристаллическими, как определено посредством рентгенодифракции. (см., например, Remington: The Science and Practice of Pharmacy, 21st Ed., Lippincott Williams & Wilkins, Baltimore, Md. (2003); Фармакопею Соединенных Штатов, 23-е издание, 1843-1844 (1995)).
[0099] Как используют в настоящем документе, «наполнитель» включает в себя функциональные и нефункциональные ингредиенты в фармацевтической композиции.
[00100] Как используют в настоящем документе, «дезинтегрирующее средство» представляет собой наполнитель, который гидратирует фармацевтическую композицию и способствует дисперсии таблетки. Как используют в настоящем документе, «разбавитель» или «наполнитель» представляет собой наполнитель, добавляющий объем фармацевтической композиции.
[00101] Как используют в настоящем документе, «поверхностно-активное вещество» представляет собой наполнитель, придающий фармацевтическим композициям улучшенную растворимость и/или смачиваемость.
[00102] Как используют в настоящем документе, «связывающее средство» представляет собой наполнитель, придающий фармацевтическим композициям усиленную когезию или сопротивление растяжению (например, жесткость).
[00103] Как используют в настоящем документе, «способствующее скольжению средство» представляет собой наполнитель, придающий фармацевтическим композициям улучшенные реологические свойства.
[00104] Как используют в настоящем документе, «краситель» представляет собой наполнитель, придающей фармацевтической композиции, например, таблетке, желательную окраску. Примеры красителей включают в себя коммерчески доступные пигменты, такие как как FD&C синий # 1 алюминиевый лак, FD&C синий #2, другие синие красители FD&C, диоксид титана, оксид железа и/или их сочетания. В одном из вариантов осуществления таблетка, предоставленная по изобретению, является розовой.
[00105] Как используют в настоящем документе, «смазочное средство» представляет собой наполнитель, который добавляют к фармацевтическим композициям, которые прессуют в таблетки. Смазочное средство способствует уплотнению гранул в таблетки и извлечению таблетки фармацевтических композиций из штамповочного пресса.
[00106] Как используют в настоящем документе, «кубический сантиметр» и «см3» используют взаимозаменяемо для обозначения единицы объема. Следует отметить, что 1 см3=1 мл.
[00107] Как используют в настоящем документе, «килофунт» и «кф» используют взаимозаменяемо, и они относятся к измерению силы, где кф=приблизительно 9,8 Ньютон.
[00108] Как используют в настоящем документе, «устойчивость к истиранию» относится к свойству таблетки оставаться интактной и сохранять свою форму несмотря на внешнее усилие давления. Устойчивость к истиранию можно оценивать количественно с использованием математического выражения, представленного в уравнении 1:
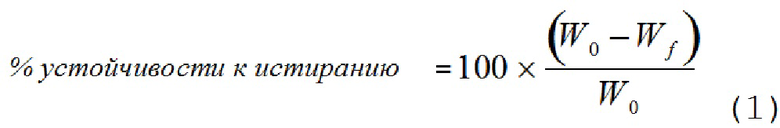 ,
,
где W0 представляет собой исходную массу таблетки, и Wf представляет собой конечную массу таблетки после того, как ее пропустили через фриабилятор. Устойчивость к истиранию измеряют с использованием стандартного устройства для тестирования USP, которое переворачивает экспериментальные таблетки в течение 100 или 400 оборотов. Некоторые таблетки по изобретению обладают устойчивостью к истиранию менее 5,0%. В другом варианте осуществления устойчивость к истиранию составляет менее 2,0%. В другом варианте осуществления намеченная устойчивость к истиранию составляет менее 1,0% после 400 оборотов.
[00109] Как используют в настоящем документе, «средний диаметр частиц» представляет собой средний диаметр частиц, как измерено с использованием таких способов, как лазерное светорассеяние, анализ изображений или анализ на ситах. В одном из вариантов осуществления гранулы, использованные для получения фармацевтических композиций, представленных по изобретению, обладают средним диаметром частиц менее 1,0 мм.
[00110] Как используют в настоящем документе, «объемная плотность» представляет собой массу частиц материала, деленную на общий объем, занимаемый частицами. Общий объем обозначает объем частиц, объем пустот между частицами и внутренний объем пор. Объемная плотность не является присущим материалу свойством; она может меняться в зависимости от того, как перерабатывают материал. В одном из вариантов осуществления гранулы, использованные для получения фармацевтических композиций, представленных по изобретению, обладают объемной плотностью приблизительно 0,5-0,7 г/см3.
[00111] «Эффективное количество» или «терапевтически эффективное количество» соединения по изобретению может меняться в соответствии с такими факторами, как состояние болезни, возраст и масса субъекта, и способность соединения по изобретению вызывать желательный ответ у субъекта. Режимы дозирования можно корректировать для обеспечения оптимального терапевтического ответа. Эффективное количество также представляет собой количество, при котором терапевтически положительное воздействие превосходит любые токсические или неблагоприятные эффекты (например, побочные эффекты) соединений по изобретению.
[00112] Как используют в настоящем документе, и если не указано иначе, термины «терапевтически эффективное количество» и «эффективное количество» соединения обозначают количество, достаточное для обеспечения терапевтического преимущества в лечении или управлении течением заболевания или нарушения, или для задержки или минимизации одного или нескольких симптомов, ассоциированных с заболеванием или нарушением. «Терапевтически эффективное количество» и «эффективное количество» соединения обозначают количество терапевтического средства, отдельно или в комбинации с одним или несколькими другими средством(средствами), обеспечивающее терапевтическое преимущество в лечении или управлении течением заболевания или нарушения. Термины «терапевтически эффективное количество» и «эффективное количество» могут включать в себя количество, которое улучшает терапию в целом, уменьшает или устраняет симптомы или причины заболевания или нарушения, или усиливает терапевтическую эффективность другого терапевтического средства.
[00113] «По существу чистое», как используют в фразе «по существу чистая форма I соединения 1», обозначает чистоту, превышающую приблизительно 90%. В другом варианте осуществления по существу чистое обозначает чистоту, превышающую приблизительно 95%. В другом варианте осуществления по существу чистое обозначает чистоту, превышающую приблизительно 98%. В другом варианте осуществления по существу чистое обозначает чистоту, превышающую приблизительно 99%.
[00114] В отношении формы I соединения 1 или твердой дисперсии, содержащей по существу аморфное соединение 2, термины «около» и «приблизительно», при использовании по отношению к дозам, количествам или проценту по массе ингредиентов композиции или лекарственной формы, обозначают дозу, количество или процент по массе, которое специалист в данной области признает обеспечивающим фармакологический эффект, эквивалентный эффекту, полученному от указанных дозы, количества или процент по массе. Конкретно термин «около» или «приблизительно» обозначает приемлемую ошибку для конкретного значения, как определено специалистом в данной области, которая частично зависит от того, как значение измеряют или определяют. В определенных вариантах осуществления термин «около» или «приблизительно» означает в пределах 1, 2, 3 или 4 стандартных отклонений. В определенных вариантах осуществления термин «около» или «приблизительно» означает в пределах 30%, 25%, 20%, 15%, 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1%, 0,5%, 0,1% или 0,05% от данного значения или диапазона.
ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ
[00115] Изобретение относится к фармацевтическим композициям, содержащим форму I соединения 1 и твердую дисперсию, содержащую по существу аморфное соединение 2. В некоторых вариантах осуществления этого аспекта, количество формы I соединения 1, присутствующее в фармацевтической композиции, составляет 100 мг, 125 мг, 150 мг, 200 мг, 250 мг, 300 мг или 400 мг. В некоторых вариантах осуществления этого аспекта, процент по массе формы I соединения 1, присутствующий в фармацевтической композиции, составляет от 10 до 75 процентов. В этих и других вариантах осуществления, форма I соединения 1 присутствует как по существу чистая форма I соединения 1. В некоторых вариантах осуществления этого аспекта, количество по существу аморфного соединения 2, присутствующее в фармацевтической композиции, составляет 100 мг, 125 мг, 150 мг, 200 мг или 250 мг. В некоторых вариантах осуществления этого аспекта, процент по массе по существу аморфного соединения 2, присутствующий в фармацевтической композиции, составляет от 10 до 75 процентов. В этих и других вариантах осуществления, по существу аморфное соединение 2 присутствует как по существу чистое и аморфное соединение 2. «По существу чистое» означает чистоту, превышающую девяносто процентов; предпочтительно, чистоту, превышающую 95 процентов; более предпочтительно, чистоту, превышающую 99,5 процентов.
[00116] Таким образом, в одном из аспектов изобретение относится к фармацевтической композиции, содержащей:
a. форму I соединения 1;
b. твердую дисперсию по существу аморфного соединения 2;
c. наполнитель;
d. дезинтегрирующее средство;
e. поверхностно-активное вещество; и
f. связывающее средство.
[00117] В одном из вариантов осуществления этого аспекта, фармацевтическая композиция содержит 25 мг формы I соединения 1. В другом варианте осуществления этого аспекта, фармацевтическая композиция содержит 50 мг формы I соединения 1. В другом варианте осуществления этого аспекта, фармацевтическая композиция содержит 100 мг формы I соединения 1. В другом варианте осуществления этого аспекта, фармацевтическая композиция содержит 125 мг формы I соединения 1. В другом варианте осуществления этого аспекта, фармацевтическая композиция содержит 150 мг формы I соединения 1. В другом варианте осуществления этого аспекта, фармацевтическая композиция содержит 200 мг формы I соединения 1. В другом варианте осуществления этого аспекта, фармацевтическая композиция содержит 250 мг формы I соединения 1. В другом варианте осуществления этого аспекта, фармацевтическая композиция содержит 400 мг формы I соединения 1.
[00118] В одном из вариантов осуществления этого аспекта, фармацевтическая композиция содержит 25 мг по существу аморфного соединения 2. В другом варианте осуществления этого аспекта, фармацевтическая композиция содержит 50 мг по существу аморфного соединения 2. В другом варианте осуществления этого аспекта, фармацевтическая композиция содержит 100 мг по существу аморфного соединения 2. В другом варианте осуществления этого аспекта, фармацевтическая композиция содержит 125 мг по существу аморфного соединения 2. В другом варианте осуществления этого аспекта, фармацевтическая композиция содержит 150 мг по существу аморфного соединения 2. В другом варианте осуществления этого аспекта, фармацевтическая композиция содержит 200 мг по существу аморфного соединения 2. В другом варианте осуществления этого аспекта, фармацевтическая композиция содержит 250 мг по существу аморфного соединения 2.
[00119] В некоторых вариантах осуществления фармацевтические композиции содержат форму I соединения 1, где Форма I соединения 1 присутствует в количестве по меньшей мере 15% масс. (например, по меньшей мере 20% масс., по меньшей мере 30% масс., по меньшей мере 40% масс., по меньшей мере 50% масс., или по меньшей мере 60% масс.) по массе композиции.
[00120] В некоторых вариантах осуществления фармацевтические композиции содержат по существу аморфное соединение 2, где по существу аморфное соединение 2 присутствует в количестве по меньшей мере 15% масс. (например, по меньшей мере 20% масс., по меньшей мере 30% масс., по меньшей мере 40% масс., по меньшей мере 50% масс., или по меньшей мере 60% масс.) по массе композиции.
[00121] В некоторых вариантах осуществления фармацевтическая композиция содержит форму I соединения 1, твердую дисперсию, содержащую по существу аморфное соединение 2, наполнитель, дезинтегрирующее средство, поверхностно-активное вещество и связывающее средство. В этом варианте осуществления композиция содержит от приблизительно 25% масс. до приблизительно 55% масс. (например, приблизительно 30-50% масс.) формы I соединения 1 по массе композиции, и более типично, от 40% масс. до приблизительно 45% масс. формы I соединения 1 по массе композиции. В этом варианте осуществления композиция содержит от приблизительно 15% масс. до приблизительно 40% масс. (например, приблизительно 20-35% масс.) по существу аморфного соединения 2 по массе композиции, и более типично, от 25% масс. до приблизительно 30% масс. по существу аморфного соединения 2 по массе композиции.
[00122] Концентрация формы I соединения 1 и по существу аморфного соединения 2 в композиции зависит от нескольких факторов, таких как количество фармацевтической композиции, необходимое для обеспечения желательного количества формы I соединения 1 и по существу аморфного соединения 2, и желательного профиля растворения фармацевтической композиции.
[00123] В другом варианте осуществления фармацевтическая композиция содержит форму I соединения 1, где форма I соединения 1 в ее твердой форме обладает средним диаметром частиц, измеренным посредством светорассеяния (например, с использованием Malvern Mastersizer, доступного из Malvern Instruments в Англии), от 0,1 микрон до 10 микрон. В другом варианте осуществления размер частиц формы I соединения 1 составляет от 1 микрона до 5 микрон. В другом варианте осуществления форма I соединения 1 обладает размером частиц D50 2,0 микрон.
[00124] Как указано, в дополнение к форме I соединения 1 и твердой дисперсии по существу аморфного соединения 2, в некоторых вариантах осуществления изобретения фармацевтическ композиции, представляющие собой пероральные составы, содержат также один или несколько наполнителей, таких как наполнители, дезинтегрирующее средства, поверхностно-активные вещества, разбавители, связывающее средства, способствующие скольжению средства, смазочные средства, красители или ароматизаторы и любое их сочетание.
[00125] Наполнители, пригодные по изобретению, являются совместимыми с ингредиентами фармацевтической композиции, т.е., они значительно не уменьшают растворимость, жесткость, химическую стабильность, физическую стабильность или биологическую активность фармацевтической композиции. Иллюстративные наполнители включают в себя: целлюлозы, модифицированные целлюлозы, (например, карбоксиметилцеллюлозу натрия, этилцеллюлозу гидроксиметилцеллюлозы, гидроксипропилцеллюлозу), ацетат целлюлозы, микрокристаллическую целлюлозу, фосфаты кальция, гидрофосфат кальция, крахмалы (например, кукурузный крахмал, картофельный крахмал), сахара (например, сорбит) лактозу, сахарозу или т.п.), или любое их сочетание.
[00126] Таким образом, в одном из вариантов осуществления фармацевтическая композиция содержит по меньшей мере один наполнитель в количестве по меньшей мере 5% масс. (например, по меньшей мере приблизительно 20% масс., по меньшей мере приблизительно 30% масс., или по меньшей мере приблизительно 40% масс.) по массе композиции. Например, фармацевтическая композиция содержит от приблизительно 10% масс. до приблизительно 60% масс. (например, от приблизительно 20% масс. до приблизительно 55% масс., от приблизительно 25% масс. до приблизительно 50% масс., или от приблизительно 27% масс. до приблизительно 45% масс.) наполнителя, по массе композиции. в другом примере, фармацевтическая композиция содержит по меньшей мере приблизительно 20% масс. (например, по меньшей мере 30% масс. или по меньшей мере 40% масс.) микрокристаллической целлюлозы, например, MCC Avicel PH102, по массе композиции. В другом примере, фармацевтическая композиция содержит от приблизительно 10% масс. до приблизительно 60% масс. (например, от приблизительно 20% масс. до приблизительно 55% масс. или от приблизительно 25% масс. до приблизительно 45% масс.) микроцеллюлозы, по массе композиции.
[00127] Дезинтегрирующее средства, пригодные по изобретению, улучшают диспергирование фармацевтической композиции и являются совместимыми с ингредиентами фармацевтической композиции, т.е., они значительно не уменьшают химическую стабильность, физическую стабильность, жесткость или биологическую активность фармацевтической композиции. Иллюстративные дезинтегрирующие средства включают в себя кроскармеллозу натрия, крахмалгликолят натрия или их сочетание.
[00128] Таким образом, в одном из вариантов осуществления фармацевтическая композиция содержит дезинтегрирующее средство в количестве приблизительно 10% масс. или менее (например, приблизительно 7% масс. или менее, приблизительно 6% масс. или менее, или приблизительно 5% масс. или менее) по массе композиции. Например, фармацевтическая композиция содержит от приблизительно 1% масс. до приблизительно 10% масс. (например, от приблизительно 1,5% масс. до приблизительно 7,5% масс. или от приблизительно 2,5% масс. до приблизительно 6% масс.) дезинтегрирующего средства, по массе композиции. В другом примере, фармацевтическая композиция содержит приблизительно 10% масс. или менее (например, 7% масс. или менее, 6% масс. или менее, или 5% масс. или менее) кроскармеллозы натрия, по массе композиции. В другом примере, фармацевтическая композиция содержит от приблизительно 1% масс. до приблизительно 10% масс. (например, от приблизительно 1,5% масс. до приблизительно 7,5% масс. или от приблизительно 2,5% масс. до приблизительно 6% масс.) кроскармеллозы натрия, по массе композиции. В некоторых примерах, фармацевтическая композиция содержит от приблизительно 0,1% до приблизительно 10% масс. (например, от приблизительно 0,5% масс. до приблизительно 7,5% масс. или от приблизительно 1,5% масс. до приблизительно 6% масс.) дезинтегрирующего средства, по массе композиции. В других примерах, фармацевтическая композиция содержит от приблизительно 0,5% до приблизительно 10% масс. (например, от приблизительно 1,5% масс. до приблизительно 7,5% масс. или от приблизительно 2,5% масс. до приблизительно 6% масс.) дезинтегрирующего средства, по массе композиции.
[00129] Поверхностно-активные вещества, пригодные по изобретению, улучшают смачиваемость фармацевтической композиции и являются совместимыми с ингредиентами фармацевтической композиции, т.е., они значительно не уменьшают химическую стабильность, физическую стабильность, жесткость или биологическую активность фармацевтической композиции. Иллюстративные поверхностно-активные вещества включают в себя лаурилсульфат натрия (SLS), стеарилфумарат натрия (SSF), моноолеат полиоксиэтилен-20-сорбитана (например, Tween™), любое их сочетание или т.п.
[00130] Таким образом, в одном из вариантов осуществления фармацевтическая композиция содержит поверхностно-активное вещество в количестве приблизительно 10% масс. или менее (например, приблизительно 5% масс. или менее, приблизительно 2% масс. или менее, приблизительно 1% масс. или менее, приблизительно 0,8% масс. или менее, или приблизительно 0,6% масс. или менее) по массе композиции. Например, фармацевтическая композиция содержит от приблизительно 10% масс. до приблизительно 0,1% масс. (например, от приблизительно 5% масс. до приблизительно 0,2% масс. или от приблизительно 2% масс. до приблизительно 0,3% масс.) поверхностно-активного вещества, по массе композиции. В другом примере, фармацевтическая композиция содержит 10% масс. или менее (например, приблизительно 5% масс. или менее, приблизительно 2% масс. или менее, приблизительно 1% масс. или менее, приблизительно 0,8% масс. или менее, или приблизительно 0,6% масс. или менее) лаурилсульфата натрия, по массе композиции. В другом примере, фармацевтическая композиция содержит от приблизительно 10% масс. до приблизительно 0,1% масс. (например, от приблизительно 5% масс. до приблизительно 0,2% масс. или от приблизительно 2% масс. до приблизительно 0,3% масс.) лаурилсульфата натрия, по массе композиции.
[00131] Связывающие средства, пригодные по изобретению, усиливают прочность таблеток из фармацевтической композиции и являются совместимыми с ингредиентами фармацевтической композиции, т.е., они значительно не уменьшают химическую стабильность, физическую стабильность или биологическую активность фармацевтической композиции. Иллюстративные связывающие средства включают в себя поливинилпирролидон, гидрофосфат кальция, сахарозу, кукурузный (маисовый) крахмал, модифицированную целлюлозу (например, гидроксиметилцеллюлозу), или любое их сочетание.
[00132] Таким образом, в одном из вариантов осуществления фармацевтическая композиция содержит связывающее средство в количестве по меньшей мере приблизительно 0,1% масс. (например, по меньшей мере приблизительно 1% масс., по меньшей мере приблизительно 3% масс., по меньшей мере приблизительно 4% масс., или по меньшей мере приблизительно 5% масс.) по массе композиции. Например, фармацевтическая композиция содержит от приблизительно 0,1% масс. до приблизительно 10% масс. (например, от приблизительно 1% масс. до приблизительно 10% масс. или от приблизительно 2% масс. до приблизительно 7% масс.) связывающего средства, по массе композиции. в другом примере, фармацевтическая композиция содержит по меньшей мере приблизительно 0,1% масс. (например, по меньшей мере приблизительно 1% масс., по меньшей мере приблизительно 2% масс., по меньшей мере приблизительно 3% масс., или по меньшей мере приблизительно 4% масс.) поливинилпирролидона, по массе композиции. В другом примере, фармацевтическая композиция содержит способствующее скольжению средство в количестве в диапазоне от приблизительно 0,1% масс. до приблизительно 10% масс. (например, от приблизительно 1% масс. до приблизительно 8% масс. или от приблизительно 2% масс. до приблизительно 5% масс.) поливинилпирролидона, по массе композиции.
[00133] Разбавители, пригодные по изобретению, могут добавлять необходимый объем составу для получения таблеток желательного объема, и являются в основном совместимыми с ингредиентами фармацевтической композиции, т.е., они значительно не уменьшают растворимость, жесткость, химическую стабильность, физическую стабильность или биологическую активность фармацевтической композиции. Иллюстративные разбавители включают в себя: сахара, например, кондитерский сахар, прессуемый сахар, декстраты, декстрин, декстроза, лактозу, маннит, сорбит, целлюлозу и модифицированные целлюлозы, например, порошкообразную целлюлозу, тальк, фосфат кальция, крахмал или любое их сочетание.
[00134] Таким образом, в одном из вариантов осуществления фармацевтическая композиция содержит разбавитель в количестве 40% масс. или менее (например, 35% масс. или менее, 30% масс. или менее, или 25% масс. или менее, или 20% масс. или менее, или 15% масс. или менее, или 10% масс. или менее) по массе композиции. Например, фармацевтическая композиция содержит от приблизительно 40% масс. до приблизительно 1% масс. (например, от приблизительно 35% масс. до приблизительно 5% масс. или от приблизительно 30% масс. до приблизительно 7% масс., от приблизительно 25% масс. до приблизительно 10% масс., от приблизительно 20% масс. до приблизительно 15% масс.) разбавителя, по массе композиции. В другом примере, фармацевтическая композиция содержит 40% масс. или менее (например, 35% масс. или менее, 25% масс. или менее, или 15% масс. или менее) маннита, по массе композиции. В другом примере, фармацевтическая композиция содержит от приблизительно 35% масс. до приблизительно 1% масс. (например, от приблизительно 30% масс. до приблизительно 5% масс. или от приблизительно 25% масс. до приблизительно 10% масс.) маннита, по массе композиции.
[00135] Способствующие скольжению средства пригодные по изобретению, улучшают реологические свойства фармацевтической композиции и являются совместимыми с ингредиентами фармацевтической композиции, т.е., они значительно не уменьшают растворимость, жесткость, химическую стабильность, физическую стабильность или биологическую активность фармацевтической композиции. Иллюстративные способствующие скольжению средства включают в себя коллоидный диоксид кремния, тальк или их сочетание.
[00136] Таким образом, в одном из вариантов осуществления фармацевтическая композиция содержит способствующее скольжению средство в количестве 2% масс. или менее (например, 1,75% масс., 1,25% масс. или менее, или 1,00% масс. или менее) по массе композиции. Например, фармацевтическая композиция содержит от приблизительно 2% масс. до приблизительно 0,05% масс. (например, от приблизительно 1,5% масс. до приблизительно 0,07% масс. или от приблизительно 1,0% масс. до приблизительно 0,09% масс.) способствующего скольжению средства, по массе композиции. В другом примере, фармацевтическая композиция содержит 2% масс. или менее (например, 1,75% масс., 1,25% масс. или менее, или 1,00% масс. или менее) коллоидного диоксида кремния, по массе композиции. В другом примере, фармацевтическая композиция содержит от приблизительно 2% масс. до приблизительно 0,05% масс. (например, от приблизительно 1,5% масс. до приблизительно 0,07% масс. или от приблизительно 1,0% масс. до приблизительно 0,09% масс.) коллоидного диоксида кремния, по массе композиции.
[00137] В некоторых вариантах осуществления фармацевтическая композиция может включать в себя пероральную твердую фармацевтическую лекарственную форму, которая может содержать смазочное средство, которое может предотвращать адгезию смеси гранулята-бусин к поверхности (например, поверхности чаши смесителя, штампа и/или пуансона для прессования). Смазочное средство может также уменьшать трение между частицами внутри гранулята и улучшать прессование и извлечение прессованных фармацевтических композиций из штамповочного пресса. Смазочное средство также является совместимым с ингредиентами фармацевтической композиции, т.е., оно значительно не уменьшает растворимость, жесткость или биологическую активность фармацевтической композиции. Иллюстративные смазочные средства включают в себя стеарат магния, стеарат кальция, стеарат цинка, стеарат натрия, стеариновую кислоту, стеарат алюминия, лейцин, глицерилбегенат, гидрогенизированное растительное масло или любое их сочетание. В одном из вариантов осуществления фармацевтическая композиция содержит смазочное средство в количестве 5% масс. или менее (например, 4,75% масс., 4,0% масс. или менее, или 3,00% масс. или менее, или 2,0% масс. или менее) по массе композиции. Например, фармацевтическая композиция содержит от приблизительно 5% масс. до приблизительно 0,10% масс. (например, от приблизительно 4,5% масс. до приблизительно 0,5% масс. или от приблизительно 3% масс. до приблизительно 1% масс.) смазочного средства, по массе композиции. В другом примере, фармацевтическая композиция содержит 5% масс. или менее (например, 4,0% масс. или менее, 3,0% масс. или менее, или 2,0% масс. или менее, или 1,0% масс. или менее) стеарата магния, по массе композиции. В другом примере, фармацевтическая композиция содержит от приблизительно 5% масс. до приблизительно 0,10% масс. (например, от приблизительно 4,5% масс. до приблизительно 0,15% масс. или от приблизительно 3,0% масс. до приблизительно 0,50% масс.) стеарата магния, по массе композиции.
[00138] Фармацевтические композиции по изобретению могут, необязательно, содержать один или несколько красителей, вкусоароматических добавок и/или ароматизаторов для улучшения внешнего вида, вкуса и/или запаха композиции. Подходящие красители, вкусоароматические добавки или ароматизаторы являются совместимыми с ингредиентами фармацевтической композиции, т.е., они значительно не уменьшают растворимость, химическую стабильность, физическую стабильность, жесткость или биологическую активность фармацевтической композиции. В одном из вариантов осуществления фармацевтическая композиция содержит краситель, вкусоароматическую добавку и/или ароматизатор. В одном из вариантов осуществления фармацевтические композиции, представленные по изобретению, являются пурпурными.
[00139] В некоторых вариантах осуществления фармацевтическая композиция включает в себя таблетки или может быть изготовлена в форме таблеток, и таблетки могут являться покрытыми красителем и необязательно, меченными логотипом, другим изображением и/или текстом с использованием подходящих чернил. В других вариантах осуществления фармацевтическая композиция включает в себя таблетки или может быть изготовлена в форме таблеток, и таблетки могут являться покрытыми красителем, покрытыми воском и необязательно, меченными логотипом, другим изображением и/или текстом с использованием подходящих чернил. Подходящие красители и чернила являются совместимыми с ингредиентами фармацевтической композиции, т.е., они значительно не уменьшают растворимость, химическую стабильность, физическую стабильность, жесткость или биологическую активность фармацевтической композиции. Подходящие красители и чернила могут быть любого цвета и на водной основе или на основе растворителя. В одном из вариантов осуществления таблетки, изготовленные из фармацевтической композиции, покрывают красителем, а затем метят логотипом, другим изображением и/или текстом с использованием подходящих чернил. Например, таблетки, содержащие фармацевтическую композицию, как описано в настоящем документе, можно покрывать приблизительно 3% масс. (например, менее приблизительно 6% масс. или менее приблизительно 4% масс.) пленочного покрытия, содержащего краситель. Окрашенные таблетки можно метить логотипом и текстом, указывающим концентрацию активного ингредиента в таблетке, с использованием подходящих чернил. В другом примере, таблетки, содержащие фармацевтическую композицию, как описано в настоящем документе, можно покрывать приблизительно 3% масс. (например, менее приблизительно 6% масс. или менее приблизительно 4% масс.) пленочного покрытия, содержащего краситель.
[00140] В другом варианте осуществления таблетки, изготовленные из фармацевтической композиции, покрывают красителем, покрывают воском, а затем метят логотипом, другим изображением и/или текстом с использованием подходящих чернил. Например, таблетки, содержащие фармацевтическую композицию, как описано в настоящем документе, можно покрывать приблизительно 3% масс. (например, менее приблизительно 6% масс. или менее приблизительно 4% масс.) пленочного покрытия, содержащего краситель. Окрашенные таблетки можно покрывать воском с помощью порошка карнаубского воска, взвешенного в количестве приблизительно 0,01% масс./масс. от исходной массы сердцевины таблетки. Покрытые воском таблетки можно метить логотипом и текстом, указывающим концентрацию активного ингредиента в таблетке, с использованием подходящих чернил. В другом примере, таблетки, содержащие фармацевтическую композицию, как описано в настоящем документе, можно покрывать приблизительно 3% масс. (например, менее приблизительно 6% масс. или менее приблизительно 4% масс.) пленочного покрытия, содержащего краситель Окрашенные таблетки можно покрывать воском с помощью порошка карнаубского воска, взвешенного в количестве приблизительно 0,01% масс./масс. от исходной массы сердцевины таблетки. Покрытые воском таблетки можно метить логотипом и текстом, указывающим концентрацию активного ингредиента в таблетке, с использованием чернил фармацевтической квалификации, таких как черные чернила (например, Opacode® S-1-17823, чернила на основе растворителя, коммерчески доступные из Colorcon, Inc., West Point, PA.).
[00141] Одна иллюстративная фармацевтическая композиция содержит от приблизительно 15% масс. до приблизительно 70% масс. (например, от приблизительно 15% масс. до приблизительно 60% масс., от приблизительно 15% масс. до приблизительно 50% масс., или от приблизительно 20% масс. до приблизительно 70% масс., или от приблизительно 30% масс. до приблизительно 70% масс.) формы I соединения 1, по массе композиции; и от приблизительно 15% масс. до приблизительно 40% масс. (например, приблизительно 20-35% масс.) по существу аморфного соединения 2 по массе композиции, и более типично, от 25% масс. до приблизительно 30% масс. по существу аморфного соединения 2 по массе композиции. Указанные выше композиции могут также включать один или несколько фармацевтически приемлемых наполнителей, например, от приблизительно 20% масс. до приблизительно 50% масс. наполнителя; от приблизительно 1% масс. до приблизительно 5% масс. дезинтегрирующего средства; от приблизительно 2% масс. до приблизительно 0,3% масс. поверхностно-активного вещества; и от приблизительно 0,1% масс. до приблизительно 5% масс. связывающего средства.
[00142] Другая иллюстративная фармацевтическая композиция содержит от приблизительно 15% масс. до приблизительно 70% масс. (например, от приблизительно 15% масс. до приблизительно 60% масс., от приблизительно 15% масс. до приблизительно 50% масс., или от приблизительно 15% масс. до приблизительно 40% масс. или от приблизительно 20% масс. до приблизительно 70% масс., или от приблизительно 30% масс. до приблизительно 70% масс., или от приблизительно 40% масс. до приблизительно 70% масс., или от приблизительно 50% масс. до приблизительно 70% масс.) формы I соединения 1 по массе композиции, от приблизительно 15% масс. до приблизительно 40% масс. (например, приблизительно 20-35% масс.) по существу аморфного соединения 2 по массе композиции, и более типично, от 25% масс. до приблизительно 30% масс. по существу аморфного соединения 2 по массе композиции, и один или несколько наполнителей, например, от приблизительно 20% масс. до приблизительно 50% масс. наполнителя; от приблизительно 1% масс. до приблизительно 5% масс. дезинтегрирующего средства; от приблизительно 2% масс. до приблизительно 0,3% масс. поверхностно-активного вещества; от приблизительно 0,1% масс. до приблизительно 5% масс. связывающего средства; и от приблизительно 2% масс. до приблизительно 0,1% масс. смазочного средства.
[00143] Другая иллюстративная фармацевтическая композиция содержит от приблизительно 15% масс. до приблизительно 70% масс. (например, от приблизительно 15% масс. до приблизительно 60% масс., от приблизительно 15% масс. до приблизительно 50% масс., или от приблизительно 15% масс. до приблизительно 40% масс. или от приблизительно 20% масс. до приблизительно 70% масс., или от приблизительно 30% масс. до приблизительно 70% масс., или от приблизительно 40% масс. до приблизительно 70% масс., или от приблизительно 50% масс. до приблизительно 70% масс.) формы I соединения 1 по массе композиции, от приблизительно 15% масс. до приблизительно 40% масс. (например, приблизительно 20-35% масс.) по существу аморфного соединения 2 по массе композиции, и более типично, от 25% масс. до приблизительно 30% масс. по существу аморфного соединения 2 по массе композиции, и один или несколько наполнителей, например, от приблизительно 20% масс. до приблизительно 50% масс. наполнителя; от приблизительно 1% масс. до приблизительно 5% масс. дезинтегрирующего средства; от приблизительно 2% масс. до приблизительно 0,3% масс. поверхностно-активного вещества; от приблизительно 0,1% масс. до приблизительно 5% масс. связывающего средства; от приблизительно 2% масс. до приблизительно 0,1% масс. смазочного средства; от приблизительно 2% масс. до приблизительно 4% масс. красителя; и от приблизительно 0,005% масс. до приблизительно 0,015% масс. воска.
[00144] В одном из вариантов осуществления изобретение относится к гранулированной фармацевтической композиции, содержащей:
a. приблизительно 43% масс. формы I соединения 1 по массе композиции;
b. приблизительно 34% масс. твердой дисперсии, содержащей по существу аморфное соединение 2 по массе композиции;
c. приблизительно 17% масс. микрокристаллической целлюлозы по массе композиции;
d. приблизительно 2% масс. кроскармеллозы натрия по массе композиции;
e. приблизительно 1% масс. лаурилсульфата натрия по массе композиции; и
f. приблизительно 3% масс. поливинилпирролидона по массе композиции.
[00145] В одном из вариантов осуществления изобретение относится к таблетке, содержащей:
a. приблизительно 35% масс. формы I соединения 1 по массе композиции;
b. приблизительно 28% масс. твердой дисперсии, содержащей по существу аморфное соединение 2 по массе композиции;
c. приблизительно 26% масс. микрокристаллической целлюлозы по массе композиции;
d. приблизительно 6% масс. кроскармеллозы натрия по массе композиции;
e. приблизительно 3% масс. поливинилпирролидона по массе композиции;
f. приблизительно 1% масс. лаурилсульфата натрия по массе композиции; и
g. приблизительно 1% масс. стеарата магния по массе композиции.
[00146] В одном из вариантов осуществления изобретение относится к таблетке, содержащей:
a. приблизительно 34% масс. формы I соединения 1 по массе композиции;
b. приблизительно 27% масс. твердой дисперсии, содержащей по существу аморфное соединение 2 по массе композиции;
c. приблизительно 26% масс. микрокристаллической целлюлозы по массе композиции;
d. приблизительно 6% масс. кроскармеллозы натрия по массе композиции;
e. приблизительно 2% масс. поливинилпирролидона по массе композиции
f. приблизительно 1% масс. лаурилсульфата натрия по массе композиции;
g. приблизительно 1% масс. стеарата магния по массе композиции;
h. приблизительно 3% масс. красителя по массе композиции; и
i. приблизительно 0,010% масс. воска по массе композиции.
[00147] Другая таблетка по изобретению содержит:
a. от приблизительно 150 до 250 мг формы I соединения 1;
b. от приблизительно 100 до 150 мг по существу аморфного соединения 2;
c. от приблизительно 125 до 175 мг микрокристаллической целлюлозы;
d. от приблизительно 20 до 40 мг кроскармеллозы натрия;
e. от приблизительно 10 до 20 мг поливинилпирролидона;
f. от приблизительно 2 до 6 мг лаурилсульфата натрия; и
g. от приблизительно 3 до 7 мг стеарата магния.
[00148] Другая таблетка по изобретению содержит:
a. приблизительно 200 мг формы I соединения 1;
b. приблизительно 125 мг по существу аморфного соединения 2;
c. приблизительно 150 мг микрокристаллической целлюлозы;
d. приблизительно 34 мг кроскармеллозы натрия;
e. приблизительно 15 мг поливинилпирролидона;
f. приблизительно 4 мг лаурилсульфата натрия; и
g. приблизительно 6 мг стеарата магния.
[00149] Другая таблетка по изобретению содержит:
a. приблизительно 200 мг формы I соединения 1;
b. приблизительно 125 мг по существу аморфного соединения 2;
c. приблизительно 150 мг микрокристаллической целлюлозы;
d. приблизительно 34 мг кроскармеллозы натрия;
e. приблизительно 15 мг поливинилпирролидона;
f. приблизительно 4 мг лаурилсульфата натрия;
g. приблизительно 6 мг стеарата магния;
h. приблизительно 17 мг красителя; и
i. приблизительно 0,06 мг воска.
[00150] В одном из вариантов осуществления изобретение относится к гранулированной фармацевтической композиции, содержащей:
a. приблизительно 38% масс. формы I соединения 1 по массе композиции;
b. приблизительно 40% масс. твердой дисперсии, содержащей по существу аморфное соединение 2 по массе композиции;
c. приблизительно 16% масс. микрокристаллической целлюлозы по массе композиции;
d. приблизительно 2% масс. кроскармеллозы натрия по массе композиции;
e. приблизительно 1% масс. лаурилсульфата натрия по массе композиции; и
f. приблизительно 3% масс. поливинилпирролидона по массе композиции.
[00151] В одном из вариантов осуществления изобретение относится к таблетке, содержащей:
a. приблизительно 31% масс. формы I соединения 1 по массе композиции;
b. приблизительно 32% масс. твердой дисперсии, содержащей по существу аморфное соединение 2 по массе композиции;
c. приблизительно 26% масс. микрокристаллической целлюлозы по массе композиции;
d. приблизительно 6% масс. кроскармеллозы натрия по массе композиции;
e. приблизительно 3% масс. поливинилпирролидона по массе композиции
f. приблизительно 1% масс. лаурилсульфата натрия по массе композиции;
g. приблизительно 1% масс. стеарата магния по массе композиции; и
h. приблизительно 3% масс. красителя по массе композиции.
[00152] Другая таблетка по изобретению содержит:
a. от приблизительно 100 до 200 мг формы I соединения 1;
b. от приблизительно 100 до 150 мг по существу аморфного соединения 2;
c. от приблизительно 100 до 150 мг микрокристаллической целлюлозы;
d. от приблизительно 20 до 40 мг кроскармеллозы натрия;
e. от приблизительно 10 до 20 мг поливинилпирролидона;
f. от приблизительно 2 до 6 мг лаурилсульфата натрия; и
g. от приблизительно 3 до 7 мг стеарата магния.
[00153] Другая таблетка по изобретению содержит:
a. приблизительно 150 мг формы I соединения 1;
b. приблизительно 125 мг по существу аморфного соединения 2;
c. приблизительно 129 мг микрокристаллической целлюлозы;
d. приблизительно 29 мг кроскармеллозы натрия;
e. приблизительно 13 мг поливинилпирролидона;
f. приблизительно 4 мг лаурилсульфата натрия;
g. приблизительно 5 мг стеарата магния; и
h. приблизительно 15 мг красителя.
[00154] Фармацевтические композиции по изобретению можно перерабатывать в форму таблетки, форму капсулы, форму саше, форму таблетки-леденца или другую твердую форму, подходящую для перорального введения. Таким образом в некоторых вариантах осуществления фармацевтические композиции находятся в форме таблетки.
[00155] В другом аспекте изобретение относится к фармацевтическому составу, состоящему из таблетки, содержащей форму I соединения 1, твердую дисперсию, содержащую по существу аморфное соединение 2, и наполнители (например, наполнитель, дезинтегрирующее средство, поверхностно-активное вещество, связывающее средство, краситель, смазочное средство или любое их сочетание), каждое из которых описано выше и в примерах ниже, где таблетка обладает растворением по меньшей мере приблизительно 50% (например, по меньшей мере приблизительно 60%, по меньшей мере приблизительно 70%, по меньшей мере приблизительно 80%, по меньшей мере приблизительно 90%, или по меньшей мере приблизительно 99%) за приблизительно 30 минут.
[00156] В одном примере, фармацевтическая композиция состоит из таблетки, содержащей форму I соединения 1 в количестве в диапазоне от 25 мг до 400 мг, например, 25 мг, или 50 мг, или 75 мг, или 100 мг, или 150 мг, 200 мг, 250 мг, 300 мг или 400 мг, по существу аморфное соединение 2 в количестве в диапазоне от 25 мг до 250 мг, например, 25 мг, или 50 мг, или 75 мг, или 100 мг, или 150 мг, 200 мг, 250 мг, и один или несколько наполнителей (например, наполнитель, дезинтегрирующее средство, поверхностно-активное вещество, связывающее средство, краситель, смазочное средство или любое их сочетание), каждое из которых описано выше и в примерах ниже, где таблетка обладает растворением от приблизительно 50% до приблизительно 100% (например, от приблизительно 55% до приблизительно 95% или от приблизительно 60% до приблизительно 90%) за приблизительно 30 минут.
[00157] Растворение можно измерять с помощью стандартного устройства USP типа II, в котором используют среду для растворения из 0,1% CTAB, растворенного в 900 мл DI воды, забуференного до pH 6,8 с помощью 50 мМ одноосновного фосфата калия, перемешиваемую при приблизительно 50-75 об./мин. при температуре приблизительно 37°C. Одну экспериментальную таблетку тестируют в каждом тестовом сосуде устройства. Растворение можно также измерять с помощью стандартного устройства USP типа II, в котором используют среду для растворения из 0,7% лаурилсульфата натрия, растворенного в 900 мл буфера 50 мМ фосфата натрия (pH 6,8), перемешиваемую при приблизительно 65 об./мин. при температуре приблизительно 37°C. Одну экспериментальную таблетку тестируют в каждом тестовом сосуде устройства. Растворение можно также измерять с помощью стандартного устройства USP типа II, в котором используют среду для растворения из 0,5% лаурилсульфата натрия, растворенного в 900 мл буфера 50 мМ фосфата натрия (pH 6,8), перемешиваемую при приблизительно 65 об./мин. при температуре приблизительно 37°C. Одну экспериментальную таблетку тестируют в каждом тестовом сосуде устройства.
СПОСОБЫ ПОЛУЧЕНИЯ ФОРМЫ I СОЕДИНЕНИЯ 1 И ТВЕРДОЙ ДИСПЕРСИИ, СОДЕРЖАЩЕЙ ПО СУЩЕСТВУ АМОРФНОЕ СОЕДИНЕНИЕ 2
Соединение 1
[00158] Соединение 1 используют в качестве исходной точки для формы I соединения 1, и его можно получать посредством присоединения к группе хлорангидрида аминогруппы в соответствии со схемами 1-4.
Схема 1. Синтез группы хлорангидрида.
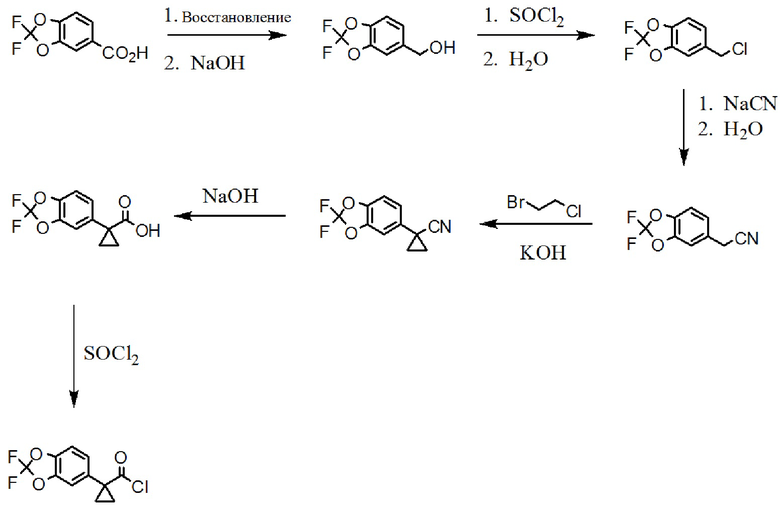
[00159] На схеме 1 изображено получение хлорида 1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбонила, который используют в схеме 3 для получения амидной связи соединения 1.
[00160] Исходное вещество, 2,2-дифторбензо[d][1,3]диоксол-5-карбоновая кислота, является коммерчески доступной из Saltigo (филиала Lanxess Corporation). Посредством восстановления группы карбоновой кислоты в 2,2-дифторбензо[d][1,3]диоксол-5-карбоновой кислоте до первичного спирта, с последующим переводом в соответствующий хлорид с использованием тионилхлорида (SOCl2), получают 5-(хлорметил)-2,2-дифторбензо[d][1,3]диоксол, который затем переводят в 2-(2,2-дифторбензо[d][1,3]диоксол-5-ил)ацетонитрил с использованием цианида натрия. Посредством обработки 2-(2,2-дифторбензо[d][1,3]диоксол-5-ил)ацетонитрила основанием и 1-бром-2-хлорэтаном получают 1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбонитрил. Группу нитрила в 1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбонитриле переводят в карбоновую кислоту с использованием основания для получения 1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоновой кислоты, которую переводят в желательный хлорангидрид с использованием тионилхлорида.
Схема 2. Альтернативный синтез группы хлорангидрида.
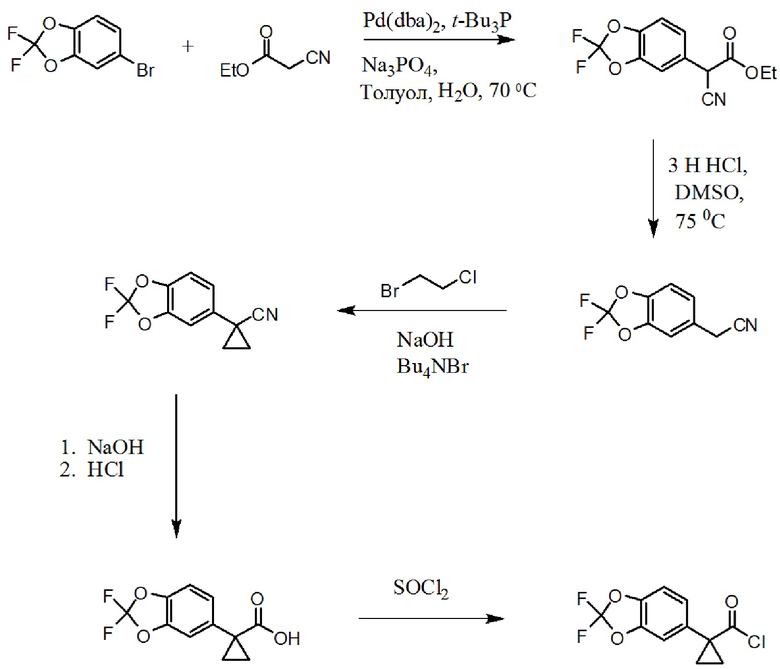
[00161] На схеме 2 изображен альтернативный синтез необходимого хлорангидрида. К 5-бромметил-2,2-дифтор-1,3-бензодиоксолу присоединяют этилцианоацетат в присутствии палладиевого катализатора для формирования соответствующего альфа-цианоэтилового сложного эфира. Посредством сапонификации группы сложного эфира до карбоновой кислоты получают соединение цианоэтила. Посредством алкилирования соединения цианоэтила с помощью 1-бром-2-хлорэтана в присутствии основания получают соединение цианоциклопропила. Посредством обработки соединения цианоциклопропила основанием получают соль карбоксилат, которую переводят в карбоновую кислоту посредством обработки кислотой. Затем осуществляют перевод карбоновой кислоты в хлорангидрид с использованием хлорирующего средства, такого как тионилхлорид или т.п.
Схема 3. Синтез аминогруппы.
[00162] На схеме 3 изображено получение необходимого трет-бутил 3-(6-амино-3-метилпиридин-2-ил)бензоата, который соединяют с 1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбонилхлоридом на схеме 3 для получения соединения 1. Посредством катализируемого палладием содинения 2-бром-3-метилпиридина с 3-(трет-бутоксикарбонил)фенилбороновой кислотой получают трет-бутил-3-(3-метилпиридин-2-ил)бензоат, который затем переводят в желательное соединение.
Схема 4. Получение кислой соли 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамидо)3-метилпиридин-2-ил)бензойной кислоты.
[00163] На схеме 4 изображено соединение 1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбонилхлорида с трет-бутил-3-(6-амино-3-метилпиридин-2-ил)бензоатом с использованием триэтиламина и 4-диметиламинопиридина для исходного получения трет-бутилового сложного эфира соединения 1.
Форма I соединения 1
[00164] Форму I соединения 1 получают посредством диспергирования или растворения формы соли, такой как соль с HCl, соединения 1 в подходящем растворителя в течение эффективного количества времени. Посредством обработки трет-бутилового сложного эфира кислотой, такой как HCl, получают соль с HCL соединения 1, которая, как правило, представляет собой кристаллическое твердое вещество. Форму I соединения 1 также можно получать непосредственно из предшественника трет-бутилового сложного эфира посредством обработки подходящей кислотой, такой как муравьиная кислота.
[00165] Соль с HCl 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамидо)-3-метилпиридин-2-ил)бензойной кислоты можно использовать для получения формы I посредством диспергирования или растворения соли с HCl 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамидо)-3-метилпиридин-2-ил)бензойной кислоты в подходящем растворителе в течение эффективного количества времени. Можно использовать другие соли 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамидо)-3-метилпиридин-2-ил)бензойной кислоты, например, такие как соли, полученные из других неорганических или органических кислот. Другие соли приводят в результате к опосредованному кислотой гидролизу группы трет-бутилового сложного эфира. Соли, полученные из других кислот, могут включать в себя, например, соли азотной, серной, фосфорной, борной, уксусной, бензойной и малоновой кислот. Эти формы соли 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамидо)-3-метилпиридин-2-ил)бензойной кислоты могут являться или не являться растворимыми, в зависимости от используемого растворителя, однако отсутствие растворимости не мешает формированию формы I соединения 1. Например, в одном из вариантов осуществления соответствующий растворитель может представлять собой воду или смесь спирт/вода, такую как смесь 50% метанол/вода, даже несмотря на то, что форма соли с HCl 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил) циклопропанкарбоксамидо)-3-метилпиридин-2-ил)бензойной кислоты является только умеренно растворимой в воде. В одном из вариантов осуществления соответствующий растворитель представляет собой воду.
[00166] Эффективное количество времени для формирования формы I соединения 1 из соли 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамидо)-3-метилпиридин-2-ил)бензойной кислоты может представлять собой любой период времени между 2 и 24 часами или более. Установлено, что необходимое количество времени является обратно пропорциональным температуре. То есть, чем выше температура, тем меньше время, необходимое, чтобы вызвать диссоциацию кислоты для формирования формы I соединения 1. Когда растворитель представляет собой воду, посредством перемешивания дисперсии в течение приблизительно 24 часов при комнатной температуре получают форму I соединения 1 с выходом приблизительно 98%. Если раствор соли 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил) циклопропанкарбоксамидо)-3-метилпиридин-2-ил)бензойной кислоты является желательным для целей процесса, можно использовать повышенную температуру. После перемешивания раствора в течение эффективного количества времени при повышенной температуре, посредством перекристаллизации при охлаждении получают по существу чистую форму I соединения 1. В одном из вариантов осуществления по существу чистый обозначает чистоту, превышающую приблизительно 90%. В другом варианте осуществления по существу чистое обозначает чистоту, превышающую приблизительно 95%. В другом варианте осуществления по существу чистое обозначает чистоту, превышающую приблизительно 98%. В другом варианте осуществления по существу чистое обозначает чистоту, превышающую приблизительно 99%. Выбранная температура частично зависит от используемого растворителя, и ее определение полностью находится в компетенции специалиста в данной области. В одном из вариантов осуществления температура находится между комнатной температурой и приблизительно 80°C. В другом варианте осуществления температура находится между комнатной температурой и приблизительно 40°C. В другом варианте осуществления температура составляет от приблизительно 40°C и приблизительно 60°C. В другом варианте осуществления температура составляет между приблизительно 60°C и приблизительно 80°C.
[00167] Форму I соединения 1 также можно формировать непосредственно из 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил) циклопропанкарбоксамидо)-3-метилпиридин-2-ил)-трет-бутилбензоата (см. схему 3), которое представляет собой предшественник соли соединения 1. Таким образом, 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамидо)-3-метилпиридин-2-ил)трет-бутилбензоату позволяют подвергаться реакции с соответствующей кислотой, например, такой как муравьиная кислота, в подходящих условиях реакции для получения формы I соединения 1.
[00168] Форму I соединения 1 можно далее очищать посредством перекристаллизации из органического растворителя. Органические растворители в качестве неограничивающих примеров включают в себя, толуол, кумол, анизол, 1-бутанол, изопропилацетат, бутилацетат, изобутилацетат, метил-трет-бутиловый простой эфир, метилизобутилкетон и смеси 1-пропанол-вода. Температура может являться такой, как описано выше. Например, форму I соединения 1 растворяют в 1-бутаноле при 75°C, пока она полностью не растворится. Посредством охлаждения раствора до 10°C со скоростью 0,2°C/мин получают кристаллы формы I соединения 1, которые можно выделять посредством фильтрации.
[00169] В одном из вариантов осуществления Форма I соединения 1 характеризуется одним или несколькими пиками при 15,2-15,6 градусах, 16,1-16,5 градусах и 14,3-14,7 градусах при рентгеновской порошковой дифракции, полученных с использованием излучения Cu K альфа. В другом варианте осуществления Форма I соединения 1 характеризуется одним или несколькими пиками при 15,4, 16,3 и 14,5 градусах. В другом варианте осуществления форма I соединения 1 дополнительно характеризуется пиком при 14,6 до 15,0 градусах. В другом варианте осуществления форма I соединения 1 дополнительно характеризуется пиком при 14,8 degrees. В другом варианте осуществления форма I соединения 1 дополнительно характеризуется пиком при 17,6 до 18,0 градусах. В другом варианте осуществления форма I соединения 1 дополнительно характеризуется пиком при 17,8 градусах. В другом варианте осуществления форма I соединения 1 дополнительно характеризуется пиком при 16,4 до 16,8 градусах. В другом варианте осуществления форма I соединения 1 дополнительно характеризуется пиком при 16,4 до 16,8 градусах. В другом варианте осуществления Форма I соединения 1 дополнительно характеризуется пиком при 16,6 градусах. В другом варианте осуществления форма I соединения 1 дополнительно характеризуется пиком при 7,6 до 8,0 градусах. В другом варианте осуществления форма I соединения 1 дополнительно характеризуется пиком при 7,8 градусах. В другом варианте осуществления форма I соединения 1 дополнительно характеризуется пиком при 25,8-26,2 градусах. В другом варианте осуществления форма I соединения 1 дополнительно характеризуется пиком при 26,0 градусах. В другом варианте осуществления форма I соединения 1 дополнительно характеризуется пиком при 21,4-21,8 градусах. В другом варианте осуществления форма I соединения 1 дополнительно характеризуется пиком при 21,6 градусах. В другом варианте осуществления форма I соединения 1 дополнительно характеризуется пиком при 23,1-23,5 градусах. В другом варианте осуществления форма I соединения 1 дополнительно характеризуется пиком при 23,3 градусах. В некоторых вариантах осуществления форма I соединения 1 характеризуется картиной дифракции, по существу сходной с картиной дифракции на фигуре 1. В некоторых вариантах осуществления форма I соединения 1 характеризуется картиной дифракции, по существу сходной с картиной дифракции на фигуре 2.
[00170] В некоторых вариантах осуществления распределение размера частиц D90 составляет приблизительно 82 мкм или менее для формы I соединения 1. В некоторых вариантах осуществления распределение размера частиц D50 составляет приблизительно 30 мкм или менее для формы I соединения 1.
Соединение 2
[00171] Соединение 2 является исходной точкой для твердой дисперсии, содержащей по существу аморфное соединение 2, и его можно получать посредством соединения группы 4-оксо-дигидрохинолинкарбоновой кислоты с аминогруппой в соответствии со схемами 5-7.
Схема 5: Синтез группы 4-оксодигидрохинолинкарбоновой кислоты.
Схема 6: Синтез аминогруппы.
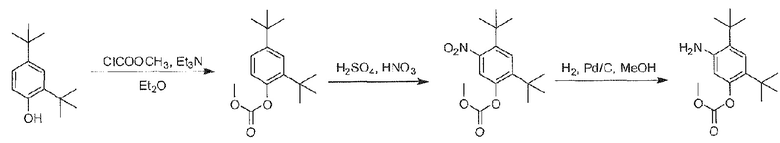
Схема 7: Соединение группы 4-оксо-дигидрохинолинкарбоновой кислоты с аминогруппой.
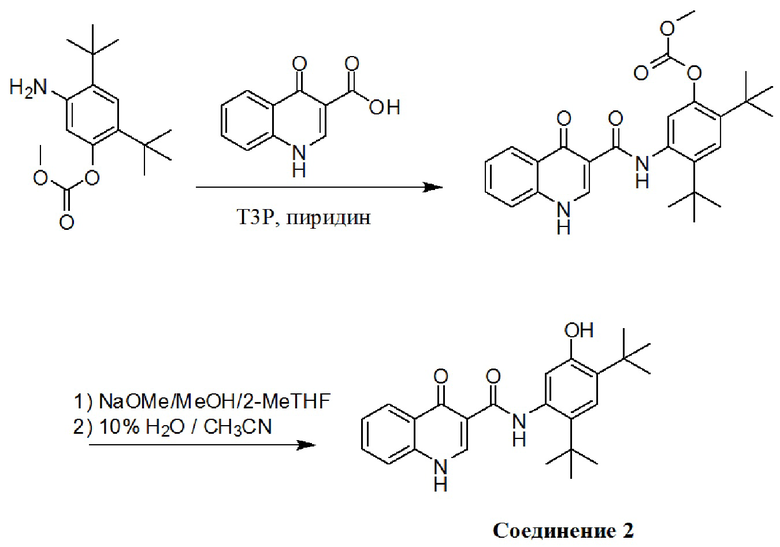
Твердая дисперсия, содержащая по существу аморфное соединение 2
[00172] Начиная с соединения 2, аморфную форму соединения 2 можно получать способами сушки распылением. Сушка распылением представляет собой процесс, который переводит жидкое сырье в форму высушенных частиц. Необязательно, процесс вторичной сушки, такой как сушка в псевдоожиженном слое или вакуумная сушка, можно использовать для уменьшения количества остаточных растворителей до фармацевтически приемлемых уровней. Как правило, сушка распылением включает в себя контакт высокодиспергированной жидкой суспензии или раствора, и достаточного объема горячего воздуха для получения испарения и высушивания жидких капель. Препарат, подлежащий сушке распылением, может представлять собой любой раствор, грубую суспензию, взвесь, коллоидную дисперсию или пасту, которые можно распылять с использованием выбранного устройства для сушки распылением. В общепринятом способе, препарат распыляют в потоке теплого фильтрованного воздуха, который выпаривает растворитель и переносит высушенный продукт в коллектор (например, a циклон). Использованный воздух затем истощают по растворителю, или альтернативно, использованный воздух направляют в конденсатор для захвата и потенциальной рециркуляции растворителя. Коммерчески доступные типы устройств можно использовать для проведения сушки распылением. Например, коммерческие сушилки для сушки распылением изготавливают в Buchi Ltd. и Niro (например, линия PSD сушилок для сушки распылением, изготовленных в Niro) (см., US 2004/0105820; US 2003/0144257).
[00173] Сушка распылением, как правило, включает в себя загрузку твердых материалов от приблизительно 3% до приблизительно 30% по массе, (т.е., лекарственного средства и наполнителей), например, от приблизительно 4% до приблизительно 20% по массе, предпочтительно, по меньшей мере приблизительно 10%. Как правило, верхний предел загрузки твердых материалов продиктован вязкостью (например, способностью к перекачиванию насосом) полученного раствора и растворимостью компонентов в растворе. Как правило, вязкость раствора может определять размер частиц в полученном порошкообразном продукте.
[00174] Технологии и способы для сушки распылением можно найти в Perry's Chemical Engineering Handbook, 6th Ed., R. H. Perry, D. W. Green & J. O. Maloney, eds.), McGraw-Hill book co. (1984); и Marshall «Atomization and Spray-Drying» 50, Chem. Eng. Prog. Monogr. Series 2 (1954). Как правило, сушку распылением проводят при температуре на входе от приблизительно 60°C до приблизительно 200°C, например, от приблизительно 95°C до приблизительно 185°C, от приблизительно 110°C до приблизительно 182°C, от приблизительно 96°C до приблизительно 180 °C, например, приблизительно 145°C. Сушку распылением, как правило, проводят при температуре на выходе от приблизительно 30°C до приблизительно 90°C, например, от приблизительно 40°C до приблизительно 80°C, от приблизительно 45°C до приблизительно 80°C например, приблизительно 75°C. Скорость потока при распылении, как правило, составляет от приблизительно 4 кг/час до приблизительно 12 кг/час, например, от приблизительно 4,3 кг/час до приблизительно 10,5 кг/час, например, приблизительно 6 кг/час или приблизительно 10,5 кг/час. Скорость потока при загрузке, как правило, составляет от приблизительно 3 кг/час до приблизительно 10 кг/час, например, от приблизительно 3,5 кг/час до приблизительно 9,0 кг/час, например, приблизительно 8 кг/час или приблизительно 7,1 кг/час. Коэффициент измельчения, как правило, составляет от приблизительно 0,3 до 1,7, например, от приблизительно 0,5 до 1,5, например, приблизительно 0,8 или приблизительно 1,5.
[00175] Удаление растворителя может требовать последующей стадии сушки, такой как сушка на лотках, сушка в псевдоожиженном слое (например, от приблизительно комнатной температуры до приблизительно 100°C), вакуумная сушка, индукционная сушка, сушка во вращающемся барабане или сушка в двухконусной вакуумной сушилке (например, от приблизительно комнатной температуры до приблизительно 200°C).
[00176] В одном из вариантов осуществления дисперсию после сушки распылением высушивают в сушилке с псевдоожиженным слоем.
[00177] В одном способе, растворитель включает в себя летучий растворитель, например, растворитель, обладающий температурой кипения менее приблизительно 100°C. В некоторых вариантах осуществления растворитель включает в себя смесь растворителей, например, смесь летучих растворителей или смесь летучих и нелетучих растворителей. Когда используют смеси растворителей, смесь может включать в себя один или несколько нелетучих растворителей, например, где нелетучий растворитель присутствует в смеси при менее приблизительно 15%, например, менее приблизительно 12%, менее приблизительно 10%, менее приблизительно 8%, менее приблизительно 5%, менее приблизительно 3% или менее приблизительно 2%.
[00178] Предпочтительными растворителями являются те растворители, в которых соединение 2 обладает растворимостью по меньшей мере приблизительно 10 мг/мл, (например, по меньшей мере приблизительно 15 мг/мл, 20 мг/мл, 25 мг/мл, 30 мг/мл, 35 мг/мл, 40 мг/мл, 45 мг/мл, 50 мг/мл или более). Более предпочтительные растворители включают в себя те, в которых соединение 2 обладает растворимостью по меньшей мере приблизительно 20 мг/мл.
[00179] Иллюстративные растворители, которые можно тестировать, включают в себя ацетон, циклогексан, дихлорметан, N,N-диметилацетамид (DMA), N,N-диметилформамид (DMF), 1,3-диметил-2-имидазолидинон (DMI), диметилсульфоксид (DMSO), диоксан, этилацетат, этиловый эфир, ледяную уксусную кислоту (HAc), метилэтилкетон (MEK), N-метил-2-пирролидинон (NMP), метил-трет-бутиловый простой эфир (MTBE), тетрагидрофуран (THF), пентан, ацетонитрил, метанол, этанол, изопропиловый спирт, изопропилацетат и толуол. Иллюстративные сорастворители включают в себя ацетон/DMSO, ацетон/DMF, ацетон/воду, MEK/воду, THF/воду, диоксан/воду. В системе двух растворителей, растворители могут присутствовать при от приблизительно 0,1% до приблизительно 99,9%. В некоторых предпочтительных вариантах осуществления вода является сорастворителем с ацетоном, где вода присутствует при от приблизительно 0,1% до приблизительно 15%, например, от приблизительно 9% до приблизительно 11%, например, приблизительно 10%. В некоторых предпочтительных вариантах осуществления вода является сорастворителем с MEK, где вода присутствует при от приблизительно 0,1% до приблизительно 15%, например, от приблизительно 9% до приблизительно 11%, например, приблизительно 10%. В некоторых вариантах осуществления раствор растворителя содержит три растворителя. Например, ацетон и воду можно смешивать с третьим растворителем, таким как DMA, DMF, DMI, DMSO или HAc. В случае, когда по существу аморфное соединение 2 является компонентом твердой дисперсии, предпочтительные растворители растворяют как соединение 2, так и полимер. Подходящие растворители включают в себя растворители, описанные выше, например, MEK, ацетон, воду, метанол и их смеси.
[00180] Размер частиц и диапазон температур сушки можно модифицировать для получения оптимальной высушенной распылением дисперсии. Как понятно специалистам в данной области, малый размер частиц приводит к улучшенному удалению растворителя. Однако, заявители обнаружили, что более мелкие частицы могут приводить к рыхлым частицам, которые в некоторых условиях не обеспечивают оптимальных высушенных распылением дисперсий для переработки для выделения и очистки конечного продукта, например, изготовления таблеток. При более высоких температурах могут происходить кристаллизация или химическая деградация по существу аморфного соединения 2. При более низких температурах, может не произойти удаление достаточного количества растворителя. Способы в настоящем документе обеспечивают оптимальный размер частиц и оптимальную температуру сушки.
[00181] Как правило, размер частиц является таким, что D10 (мкм) составляет менее приблизительно 5, например, менее приблизительно 4,5, менее приблизительно 4,0, или менее приблизительно 3,5, D50 (мкм), как правило, составляет менее приблизительно 17, например, менее приблизительно 16, менее приблизительно 15, менее приблизительно 14, менее приблизительно 13, и D90 (мкм), как правило, составляет менее приблизительно 175, например, менее приблизительно 170, менее приблизительно 170, менее приблизительно 150, менее приблизительно 125, менее приблизительно 100, менее приблизительно 90, менее приблизительно 80, менее приблизительно 70, менее приблизительно 60, или менее приблизительно менее приблизительно 50. Как правило, объемная плотность высушенных распылением частиц составляет от приблизительно 0,08 г/см3 до приблизительно 0,20 г/см3, например, от приблизительно 0,10 до приблизительно 0,15 г/см3, например, приблизительно 0,11 г/см3 или приблизительно 0,14 г/см3. Насыпная плотность после утряски высушенных распылением частиц, как правило, лежит в диапазоне от приблизительно 0,08 г/см3 до приблизительно 0,20 г/см3, например, от приблизительно 0,10 до приблизительно 0,15 г/см3, например, приблизительно 0,11 г/см3или приблизительно 0,14 г/см3, для 10 встряхиваний; от 0,10 г/ см3 до приблизительно 0,25 г/см3, например, от приблизительно 0,11 до приблизительно 0,21 г/см3, например, приблизительно 0,15 г/см3, приблизительно 0,19 г/см3, или приблизительно 0,21 г/см3 для 500 встряхиваний; 0,15 г/см3 до приблизительно 0,27 г/см3, например, от приблизительно 0,18 до приблизительно 0,24 г/см3, например, приблизительно 0,18 г/см3, приблизительно 0,19 г/см3, приблизительно 0,20 г/см3, или приблизительно 0,24 г/см3 для 1250 встряхиваний; и от 0,15 г/ см3 до приблизительно 0,27 г/см3, например, от приблизительно 0,18 до приблизительно 0,24 г/см3, например, приблизительно 0,18 г/см3, приблизительно 0,21 г/ см3, приблизительно 0,23 г/см3, или приблизительно 0,24 г/ см3 для 2500 встряхиваний.
[00182] Полимеры
[00183] Высушенные распылением дисперсии, содержащие аморфное соединение 2 и полимер (или носитель в твердом состоянии), также включены в настоящий документ. Например, соединение 2 присутствует в форме аморфного соединения как компонент твердой аморфной дисперсии. Твердая аморфная дисерсия, как правило, содержит по существу аморфное соединение 2 и полимер. Иллюстративные полимеры включают в себя целлюлозные полимеры, такие как HPMC или HPMCAS, и содержащие пирролидон полимеры, такие как PVP/VA. В некоторых вариантах осуществления твердая аморфная дисерсия содержит один или несколько дополнительных наполнителей, таких как поверхностно-активное вещество.
[00184] В одном из вариантов осуществления полимер является способным к растворению в водной среде. Растворимость полимеров может являться независимой от pH или зависимой от pH. Последняя включает в себя один или несколько растворимых в кишечнике полимеры. Термин «растворимый в кишечнике полимер» относится к полимеру, который предпочтительно является растворимым в менее кислой среде кишечника относительно более кислой среды желудка, например, полимер, который является нерастворимым в кислой водной среде, но растворимым, когда pH составляет выше 5-6. Подходящий полимер должен является химически и биологически инертным. Для улучшения физической стабильности высушенных распылением дисперсий, температура стеклования (Tg) полимера должна являться настолько высокой, насколько возможно. Например, предпочтительные полимеры обладают температурой стеклования, по меньшей мере равной или большей, чем температура стеклования лекарственного средства (т.е., соединения 2). Другие предпочтительные полимеры обладают температурой стеклования в диапазоне от приблизительно 10 до приблизительно 15°C от температуры стеклования лекарственного средства (т.е., соединения 2). Примеры подходящей температуры стеклования полимеров включают в себя по меньшей мере приблизительно 90°C, по меньшей мере приблизительно 95°C, по меньшей мере приблизительно 100°C, по меньшей мере приблизительно 105°C, по меньшей мере приблизительно 110 °C, по меньшей мере приблизительно 115 °C, по меньшей мере приблизительно 120°C, по меньшей мере приблизительно 125°C, по меньшей мере приблизительно 130°C, по меньшей мере приблизительно 135°C, по меньшей мере приблизительно 140°C, по меньшей мере приблизительно 145°C, по меньшей мере приблизительно 150°C, по меньшей мере приблизительно 155°C, по меньшей мере приблизительно 160°C, по меньшей мере приблизительно 165°C, по меньшей мере приблизительно 170°C, или по меньшей мере приблизительно 175°C (как измерено в сухом состоянии). Без желания быть связанными теорией, полагают, что лежащий в основе механизм представляет собой то, что полимер с более высокой Tg, как правило, обладает меньшей молекулярной подвижностью при комнатной температуре, что может являться критическим фактором в стабилизации физической стабильности аморфной высушенной распылением дисперсии.
[00185] Дополнительно, гигроскопичность полимеров должна являться настолько низкой, как, например, менее приблизительно 10%. Для целей сравнения в этой заявке, гигроскопичность полимера или композиции характеризуют при приблизительно 60% относительной влажности. В некоторых предпочтительных вариантах осуществления полимер обладает поглощением воды менее приблизительно 10%, например, менее приблизительно 9%, менее приблизительно 8%, менее приблизительно 7%, менее приблизительно 6%, менее приблизительно 5%, менее приблизительно 4%, менее приблизительно 3%, или менее приблизительно 2% поглощением воды. Гигроскопичность может также влиять на физическую стабильность высушенных распылением дисперсий. Как правило, влажность, адсорбированная полимерами, может сильно уменьшать Tg полимеров, а также полученных высушенных распылением дисперсий, что может далее уменьшать физическую стабильность высушенных распылением дисперсий, как описано выше.
[00186] В одном из вариантов осуществления полимер представляет собой один или несколько водорастворимых полимера(полимеров) или частично водорастворимых полимера(полимеров). Водорастворимые или частично водорастворимые полимеры в качестве неограничивающих примеров включают в себя производные целлюлозы (например, гидроксипропилметилцеллюлоза (HPMC), гидроксипропилцеллюлоза (HPC)) или этилцеллюлоза; поливинилпирролидоны (PVP); полиэтиленгликоли (PEG); поливиниловые спирты (PVA); акрилаты, так как полиметакрилат (например, Эудражит® E); циклодекстрины (например, β-циклодекстрин), и их сополимеры и производные, включая, например, PVP-VA (поливинилпирролидон-винилацетат).
[00187] В некоторых вариантах осуществления полимер представляет собой гидроксипропилметилцеллюлозу (HPMC), такую как HPMCAS, HPMC E50, HPMCE15 или HPMC60SH50).
[00188] Как обсуждают в настоящем документе, полимер может представлять собой растворимый в кишечнике в зависимости от pH полимер. Такие растворимые в кишечнике в зависимости от pH полимеры в качестве неограничивающих примеров включают в себя производные целлюлозы (например, ацетатфталат целлюлозы (CAP)), фталаты гидроксипропилметилцеллюлозы (HPMCP), гидроксипропилметилацетат-сукцинат целлюлозы (HPMCAS), карбоксиметилцеллюлоза (CMC) или их соль (например, натриевую соль, так как (CMC-Na)); тримеллитат ацетилцеллюлозы (CAT), гидроксипропилацетатфталат целлюлозы (HPCAP), гидроксипропилметил-ацетатфталат целлюлозы (HPMCAP) и метилацетатфталат целлюлозы (MCAP), или полиметакрилаты (например, эудражит® S). В некоторых вариантах осуществления полимер представляет собой гидроксипропилметилацетат-сукцинат целлюлозы (HPMCAS). В некоторых вариантах осуществления полимер представляет собой гидроксипропилметилацетат-сукцинат целлюлозы квалификации HG (HPMCAS-HG).
[00189] В еще одном варианте осуществления полимер представляет собой сополимер поливинилпирролидона, например, сополимер винилпирролидона/винилацетата (PVP/VA).
[00190] В вариантах осуществления, где соединение 2 образует высушенную распылением дисперсию с полимером, например, с полимером HPMC, HPMCAS или PVP/VA, количество полимера относительно общей массы высушенной распылением дисперсии лежит в диапазоне от приблизительно 0,1% до 99% по массе. Если не указано иначе, проценты лекарственного средства, полимера и других наполнителей, как описано в дисперсии, приведены в массовых долях. Количество полимера, как правило, составляет по меньшей мере приблизительно 20% и предпочтительно, по меньшей мере приблизительно 30%, например, по меньшей мере приблизительно 35%, по меньшей мере приблизительно 40%, по меньшей мере приблизительно 45%, или приблизительно 50% (например, 49,5%). Количество составляет как правило приблизительно 99% или менее, и предпочтительно приблизительно 80% или менее, например, приблизительно 75% или менее, приблизительно 70% или менее, приблизительно 65% или менее, приблизительно 60% или менее, или приблизительно 55% или менее. В одном из вариантов осуществления полимер присутствует в количестве вплоть до приблизительно 50% от общей массы дисперсии (и даже более конкретно, от приблизительно 40% и 50%, например, приблизительно 49%, приблизительно 49,5%, или приблизительно 50%). HPMC и HPMCAS являются доступными в диапазоне квалификаций из ShinEtsu, например, HPMCAS является доступным в ряде вариантов, включая AS-LF, AS-MF, AS-HF, AS-LG, AS-МГ, AS-HG. В каждой из этих квалификаций меняется процент замещения ацетата и сукцината.
[00191] В некоторых вариантах осуществления по существу аморфное соединение 2 и полимер присутствуют в приблизительно равных количествах, например, каждый из полимера и лекарственного средства составляет приблизительно половину процентного содержания по массе дисперсии. Например, полимер присутствует в приблизительно 49,5%, и лекарственное средство присутствует в приблизительно 50%.
[00192] В некоторых вариантах осуществления по существу аморфное соединение 2 и полимер вместе представляют от 1% до 20% масс./масс. общего содержания твердого вещества из не высушенной распылением дисперсии перед сушкой распылением. В некоторых вариантах осуществления по существу аморфное соединение 2 и полимер вместе представляют от 5% до 15% масс./масс. общего содержания твердого вещества из не высушенной распылением дисперсии перед сушкой распылением. В некоторых вариантах осуществления по существу аморфное соединение 2 и полимер вместе представляют приблизительно 11% масс./масс. общего содержания твердого вещества из не высушенной распылением дисперсии перед сушкой распылением.
[00193] В некоторых вариантах осуществления дисперсия дополнительно содержит другие ингредиенты в незначительных количествах, таких как поверхностно-активное вещество (например, SLS). В некоторых вариантах осуществления поверхностно-активное вещество присутствует в менее приблизительно 10% из дисперсии, например, менее приблизительно 9%, менее приблизительно 8%, менее приблизительно 7%, менее приблизительно 6%, менее приблизительно 5%, менее приблизительно 4%, менее приблизительно 3%, менее приблизительно 2%, приблизительно 1% или приблизительно 0,5%.
[00194] В вариантах осуществления, включающих в себя полимер, полимер должен присутствовать в количестве, эффективном для стабилизации высушенной распылением дисперсии. Стабилизация включает в себя ингибирование или предотвращение кристаллизации по существу аморфного соединения 2. Такая стабилизация может ингибировать переход соединения 2 из аморфной в кристаллическую форму. Например, полимер может предотвращать по меньшей мере для части (например, приблизительно 5%, приблизительно 10%, приблизительно 15%, приблизительно 20%, приблизительно 25%, приблизительно 30%, приблизительно 35%, приблизительно 40%, приблизительно 45%, приблизительно 50%, приблизительно 55%, приблизительно 60%, приблизительно 65%, приблизительно 70%, приблизительно 75% или более) соединения 2 переход из аморфной в кристаллическую форму. Стабилизацию можно измерять, например, посредством измерения температуры стеклования высушенной распылением дисперсии, измерения скорости релаксации аморфного материала, или посредством измерения растворимости или биодоступности соединения 2.
[00195] Подходящие полимеры для использования в комбинации с соединением 2, например, в форме высушенной распылением дисперсии, такой как аморфная высушенная распылением дисперсия, должны обладать одним или несколькими из следующих свойств:
[00196] Температура стеклования полимера должна составлять температуру не менее, чем приблизительно на 10-15°C ниже, чем температура стеклования по существу аморфного соединения 2. Предпочтительно, температура стеклования полимера составляет более температуры стеклования по существу аморфного соединения 2, и как правило, по меньшей мере на 50°C выше, чем желательная температура хранения продукта лекарственного средства. Например, по меньшей мере приблизительно 100°C, по меньшей мере приблизительно 105°C, по меньшей мере приблизительно 105°C, по меньшей мере приблизительно 110°C, по меньшей мере приблизительно 120°C, по меньшей мере приблизительно 130°C, по меньшей мере приблизительно 140°C, по меньшей мере приблизительно 150°C, по меньшей мере приблизительно 160°C, по меньшей мере приблизительно 160°C или более.
[00197] Полимер должен является относительно негигроскопичным. Например, полимер должен, при хранении в стандартных условиях, поглощать менее приблизительно 10% воды, например, менее приблизительно 9%, менее приблизительно 8%, менее приблизительно 7%, менее приблизительно 6%, или менее приблизительно 5%, менее приблизительно 4%, или менее приблизительно 3% воды. Предпочтительно полимер должен, при хранении в стандартных условиях, являться по существу свободным от абсорбированной воды.
[00198] Полимер должен обладать сходной или лучшей растворимостью в растворителях, подходящих для способов сушки распылением, относительно растворимости соединения 2. В предпочтительных вариантах осуществления полимер может растворяться в одном или нескольких из тех же растворителей или систем растворителей, что и соединение 2. Предпочтительно, чтобы полимер являлся растворимым по меньшей мере в одном не содержащем гидроксигруппы растворителе, таком как метиленхлорид, ацетон или их сочетание.
[00199] Полимер, при комбинации с по существу аморфным соединением 2, например, в высушенной распылением дисперсии или в жидкой суспензии, должен увеличивать растворимость соединения 2 в водной и родственной физиологической среде либо относительно растворимости соединения 2 в отсутствие полимера, либо относительно растворимости соединения 2 при комбинации с эталонным полимером. Например, полимер может увеличивать растворимость аморфного соединения 2 посредством уменьшения количества аморфного соединения 2, которое переходит в кристаллическое соединение 2, либо из твердой аморфной дисперсии, либо из жидкой суспензии.
[00200] Полимер должен уменьшать скорость релаксации аморфного вещества.
[00201] Полимер должен увеличивать физическую и/или химическую стабильность по существу аморфного соединения 2
[00202] Полимер должен улучшать технологичность по существу аморфного соединения 2.
[00203] Полимер должен улучшать одно или несколько из свойств обработки, введения или хранения по существу аморфного соединения 2.
[00204] Полимер не должен вступать в неблагоприятные взаимодействия с другими фармацевтическими компонентами, например, наполнителями.
[00205] Пригодность кандидата - полимера (или другого компонента) можно тестировать с использованием способов сушки распылением (или других способов), описываем в настоящем документе для формирования аморфной композиции. Композицию-кандидат можно сравнивать в отношении стабильности, устойчивости к формированию кристаллов или других свойств, и сравнивать с эталонным препаратом, например, препаратом чистого аморфного соединения 2 или кристаллического соединения 2. Например, композицию-кандидат можно обрабатывать для определения того, замедляет ли она время до начала опосредованной растворителем кристаллизации, или уменьшает процент перехода за данное время в контролируемых условиях, по меньшей мере на 50%, 75%, 100% или 110% а также эталонный препарат, или композицию-кандидат можно тестировать для определения того, обладает ли она улучшенной биодоступностью или растворимостью относительно кристаллического соединения 2.
[00206] Поверхностно-активные вещества
[00207] Высушенная распылением дисперсия может содержать поверхностно-активное вещество. Поверхностно-активное вещество или смесь поверхностно-активных веществ в общем уменьшает междуфазное натяжение между высушенной распылением дисперсией и водной средой. Подходящее поверхностно-активное вещество или смесь поверхностно-активных веществ может также улучшать растворимость в воде и биодоступность соединения 2 из высушенной распылением дисперсии. Поверхностно-активные вещества для применения в связи с настоящим изобретением в качестве неограничивающих примеров включают в себя сложные эфиры сорбитана и жирной кислоты (например, виды Span®), сложные эфиры полиоксиэтиленсорбитана и жирной кислоты (например, виды Tween®), лаурилсульфат натрия (SLS), додецилбензолсульфонат натрия (SDBS) диоктилсульфосукцинат натрия (докузат), натриевую соль диоксихолевой кислоты (DOSS), моностеарат сорбитана, тристеарат сорбитана, бромид гексадецилтриметиламмония (HTAB), N-лауроилсаркозин натрия, олеат натрия, миристат натрия, стеарат натрия, пальмитат натрия, гелюцир 44/14, этилендиаминтетрауксусную кислоту (ЭДТА), сукцинат d-альфа-токоферилполиэтиленгликоля 1000 витамина E (TPGS), лецитин, MW 677-692, моногидрат глутаминовой кислоты мононатрия, лабразол, PEG 8 каприловый/каприновый глицериды, транскутол, моноэтиловый простой эфир диэтиленгликоля, солютол HS-15, полиэтиленгликоль/гидроксистеарат, таурохолевую кислоту, плюроник F68, плюроник F108 и плюроник F127 (или любые другие сополимеры полиоксиэтилена-полиоксипропилена (плюроники®) или насыщенные полигликолизированные глицериды (геликуры®)). Конкретные примеры таких поверхностно-активных веществ, которые можно использовать в связи с этим изобретением, включают в себя, но без ограничения, Span 65, Span 25, Tween 20, каприол 90, плюроник F108, лаурилсульфат натрия (SLS), Витамин E TPGS, плюроники и сополимеры. SLS, как правило, является предпочтительным.
[00208] Количество поверхностно-активного вещества (например, SLS) относительно общей массы высушенной распылением дисперсии может составлять между 0,1-15%. Предпочтительно, оно составляет от приблизительно 0,5% до приблизительно 10%, более предпочтительно, от приблизительно 0,5 до приблизительно 5%, например, от приблизительно 0,5 до 4%, от приблизительно 0,5 до 3%, от приблизительно 0,5 до 2%, от приблизительно 0,5 до 1%, или приблизительно 0,5%.
[00209] В определенных вариантах осуществления количество поверхностно-активного вещества относительно общей массы высушенной распылением дисперсии составляет по меньшей мере приблизительно 0,1%, предпочтительно, приблизительно 0,5%. В этих варианты осуществления, поверхностно-активное вещество присутствует в количестве не более приблизительно 15%, и предпочтительно не более приблизительно 12%, приблизительно 11%, приблизительно 10%, приблизительно 9%, приблизительно 8%, приблизительно 7%, приблизительно 6%, приблизительно 5%, приблизительно 4%, приблизительно 3%, приблизительно 2% или приблизительно 1%. Вариант осуществления, где поверхностно-активное вещество присутствует в количестве приблизительно 0,5% по массе, является предпочтительным.
[00210] Кандидаты - поверхностно-активные вещества (или другие компоненты) можно тестировать по пригодности для применения по изобретению способом, сходным со способом, описанным для тестируемых полимеров.
СПОСОБЫ ПОЛУЧЕНИЯ ФАРМАЦЕВТИЧЕСКИХ КОМПОЗИЦИЙ
[00211] Фармацевтические композиции по изобретению можно получать посредством влажной грануляции, уплотнения или прессования смеси или композиции, например, порошка или гранул, под давлением для формирования стабильной трехмерной формы (например, таблетки). Как используют в настоящем документе, «таблетка» включает в себя прессованные фармацевтические стандартные лекарственные формы всех форм и размеров, покрытые или непокрытые.
[00212] Термин «таблетка», как используют в настоящем документе, относится к физически дискретной единице средства, подходящей для пациента, подлежащего лечению. Как правило, уплотненная смесь обладает плотностью более, чем смеси до уплотнения. Доза в форме таблетки по изобретению может обладать почти любой формой, включая вогнутые и/или выпуклые поверхности, закругленные или угловатые края, и от круглой до прямолинейной формы. В некоторых вариантах осуществления прессованные таблетки по изобретению включают в себя закругленную таблетку, обладающую плоскими поверхностями. Таблетки по изобретению можно получать посредством любого способа уплотнения и прессования, известного специалистам в области получения прессованных твердых фармацевтических форм дозирования. В конкретных вариантах осуществления составы, представленные в настоящем документе, можно получать с использованием общепринятых способов, известных специалистам в области получения фармацевтических составов, как описано, например, в соответствующих руководствах. См., например, Remington: The Science and Practice of Pharmacy, 21st Ed., Lippincott Williams & Wilkins, Baltimore, Md. (2003); Ansel et al., Pharmaceutical Dosage Forms And Drug Delivery Systems, 7th Edition, Lippincott Williams & Wilkins, (1999); The Handbook of Pharmaceutical Excipients, 4th edition, Rowe et al., Eds., American Pharmaceuticals Association (2003); Gibson, Pharmaceutical Preformulation And Formulation, CRC Press (2001), полное содержание этих ссылок, таким образом, включено в настоящий документ в качестве ссылки.
Грануляция и прессование
[00213] В некоторых вариантах осуществления ингредиенты взвешивают в соответствии с формулой, указанной в настоящем документе. Затем все интрагранулярные ингредиенты просеивают и хорошо перемешивают. Ингредиенты можно смазывать подходящим смазочным средством, например, стеаратом магния. Следующая стадия может включать в себя уплотнение/набивку порошкообразной смеси и ингредиентов необходимого размера. Затем уплотненные или набитые смеси размалывают в гранулы и просеивают для получения желательного размера. Затем гранулы можно дополнительно смазывать, например, стеаратом магния. Затем гранулированную композицию по изобретению можно прессовать на подходящих штампах в различные фармацевтические составы по изобретению. Необязательно, таблетки можно покрывать пленкой, красителем или другим покрытием.
[00214] В другом аспекте изобретение относится к способу получения фармацевтической композиции, включающему в себя получение смеси композиции, содержащей форму I соединения 1, твердую дисперсию, содержащую по существу аморфное соединение 2 и один или несколько наполнителей, выбранных из: наполнителя, разбавителя, связывающего средства, поверхностно-активного вещества, смазочного средства, дезинтегрирующего средства, и прессование композиции в таблетки, обладающие растворением по меньшей мере приблизительно 50% за приблизительно 30 минут.
[00215] В другом варианте осуществления способ влажной грануляции осуществляют для получения фармацевтического состава по изобретению из смеси порошкообразных и жидких ингредиентов. Например, фармацевтическую композицию, содержащую смесь композиции, содержащей форму I соединения 1, твердую дисперсию, содержащую по существу аморфное соединение 2, и один или несколько наполнителей, выбранных из: наполнителя, связывающего средства, поверхностно-активного вещества или дезинтегрирующего средства, взвешивают в соответствии с формулой, указанной в настоящем документе. Затем все интрагранулярные ингредиенты просеивают и смешивают в грануляторе с высоким или низким усилием сдвига с использованием воды или воды с поверхностно-активным веществом, или воды с связывающим средством, или воды с поверхностно-активным веществом и связывающим средством для грануляции порошкообразной смеси. Жидкость, отличную от воды, также можно использовать в присутствии или в отсутствие поверхностно-активного вещества и/или связывающего средства для грануляции порошкообразной смеси. Затем, влажные гранулы можно, необязательно, размалывать с использованием подходящей мельницы. Затем воду можно, необязательно, удалять из смеси посредством сушки ингредиентов любым подходящим способом. Затем высушенные гранулы можно, необязательно, размалывать до равного размера. Затем, можно добавлять экстрагранулярные наполнители посредством смешивания (например, наполнитель, разбавитель и дезинтегрирующее средство). Затем гранулы необходимого размера можно дополнительно смазывать стеаратом магния и дезинтегрирующим средством, например, кроскармеллозой натрия. Затем гранулярную композицию по изобретению можно просеивать в течение времени, достаточного для получения правильного размера, а затем прессовать в подходящих штампах в различные фармацевтические составы по изобретению. Необязательно, таблетки можно покрывать пленкой, красителем или другим покрытием. Неожиданно, влажную грануляцию можно проводить без значительной потери твердого состояния формы I соединения 1 или по существу аморфного соединения 2.
[00216] В особенно предпочтительном варианте осуществления, фармацевтические композиции по настоящему изобретению получают посредством способа непрерывной двухшнековой влажной грануляции (TSWG). Посредством непрерывного изготовления получают продукт высокого качества и высокого соответствия с мониторированием и контролем в процессе производства. Непрерывное изготовление также способствует качеству посредством разработки пространства проектных решений «для интенсивной обработки данных» и более простого понимания вклада вышележащих переменных в нижележащий процесс и качество конечного продукта. Кроме того, можно рано завершать окончательную разработку фармацевтических композиций по настоящему изобретению на оборудовании промышленного масштаба, что исключает риски масштабирования и изменения состава при более поздней разработке. Наконец, непрерывное изготовление обладает преимуществами коммерческого изготовления, такими как улучшенный контроль процесса, уменьшение манипуляций с продуктом и контроль эффективности при выпуске в режиме реального времени. Общим результатом является более надежный, контролируемый и поддающийся масштабированию процесс, обладающий меньшим количеством производственных проверок, что в результате приводит к увеличенному качеству продукта, и таким образом, к большей безопасности для пациента. Эти преимущества снимают опасения Janet Woodcock (директора Центра оценки и изучения лекарственных средства (CDER)), что химические реакции, изготовление и контроль (CMC) неспособны находиться на одном уровне с быстрой клинической разработкой высоко эффективных лекарственных средств («авторы настоящего изобретения наблюдали, что часто ограничивающей скорость стадией может являться изготовление», 24 июля 2013 года Friends of Cancer провели в ходе конгресса брифинг «Answering a Compelling Need: Expediting Life-Saving Treatments to Patients» для обсуждения получения статуса принципиально нового лекарственного средства от Управления по контролю пищевых продуктов и лекарственных средств).
[00217] Например, для грануляции с большим усилием сдвига (HSG), общепринятого способа грануляции, хорошо известен риск избыточной грануляции и плохой контроль процесса. Масштабирование этого процесса является очень сложным и вовлекает значительный риск. Замена процесса HSG на непрерывный процесс TSWG позволяет масштабирование с использованием одного и того же оборудования для получения различных размеров партий, посредством функционирования в течение более длительного времени. Это исключает риск масштабирования, обычно возникающий в других процессах грануляции. Дополнительно обнаружено, что процесс TSWG более надежен, являясь менее чувствительным к избыточной грануляции. Как можно видеть на фигуре 3 для таблеток соединения 1, для процесса HSG показано значительное замедление растворения с увеличением содержания воды, в то время как для процесса TSWG не показано изменения в сходном диапазоне добавления воды. Неожиданно, не обнаружено изменений производительности для составов таблеток, содержащих между 45 и 55 процентами по массе соединения 1, и составов таблеток, содержащих между 60 и 70 процентами по массе соединения 1, при использовании способа двухшнековой влажной грануляции. Это отличалось от случая с процессом HSG. Дополнительно, этот непрерывный и увеличивающий качество продукта процесс снимает общераспространенной опасение FDA относительно утраты доступности лекарственного средства для нуждающихся в этом пациентов.
[00218] В одном из вариантов осуществления непрерывный процесс начинается с загрузки индивидуальных наполнителей, соединения 1 и соединения 2 в непрерывный встроенный смеситель посредством подачи с уменьшением массы. Из этого смесителя материал непрерывно подают и перерабатывают посредством двухшнековой влажной грануляции, сушки, помола, добавления экстрагранулярного наполнителя, смешивания, прессования и покрытия пленкой.
[00219] Например, в одном из вариантов осуществления таблетки, содержащие соединение 1 и соединение 2, можно получать непрерывно в соответствии с блок-схемой ниже.
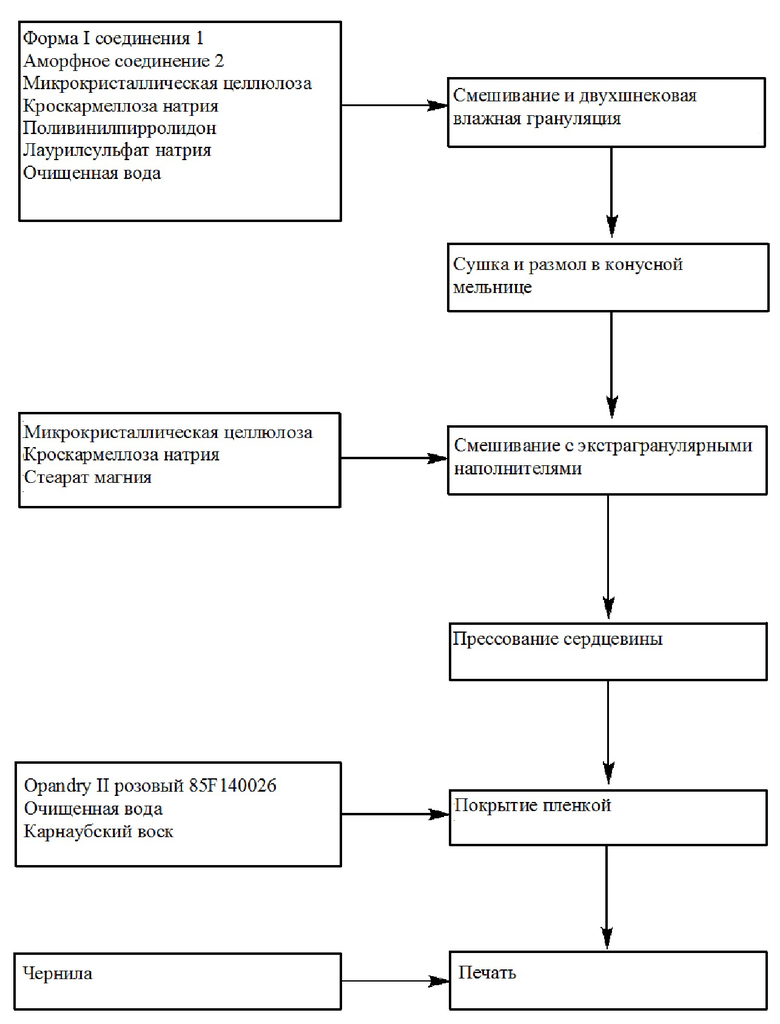
[00220] Каждый из ингредиентов этой иллюстративной смеси описан выше и в примерах ниже. Кроме того, смесь может содержать необязательные добавки, такие как один или несколько красителей, одна или несколько вкусоароматических добавок, и/или один или несколько ароматизаторов, как описано выше и в примерах ниже. В некоторых вариантах осуществления относительные концентрации (например,% масс.) каждого из этих ингредиентов (и любых необязательных добавок) в смеси также представлены выше и в примерах ниже. Ингредиенты, составляющие смесь, можно предоставлять последовательно или в любой комбинации добавлений; и ингредиенты или комбинацию ингредиентов можно предоставлять в любом порядке. В одном из вариантов осуществления смазочное средство является последним компонентом, добавляемым в смесь.
[00221] В другом варианте осуществления смесь содержит композицию формы I соединения 1, твердой дисперсии по существу аморфного соединения 2, и любых одного или нескольких из наполнителей; связывающего средства, поверхностно-активного вещества, разбавителя, смазочного средства, дезинтегрирующего средства и наполнителя, где каждый из этих ингредиентов представлен в форме порошка (например, представлен в форме частиц, обладающих средним или усредненным диаметром, измеренным по светорассеянию как 250 мкм или менее (например, 150 мкм или менее, 100 мкм или менее, 50 мкм или менее, 45 мкм или менее, 40 мкм или менее, или 35 мкм или менее)).
[00222] В другом варианте осуществления прессование смеси в таблетки проводят посредством заполнения формы (например, отливки) смесью и приложения давления к смеси. Это можно проводить с использованием штамповочного пресса или другого сходного устройства. В некоторых вариантах осуществления смесь формы I соединения 1, твердой дисперсии, содержащей по существу аморфное соединение 2, и наполнителей можно сначала перерабатывать в гранулярную форму. Гранулы можно затем доводить до необходимого размера и прессовать в таблетки или составлять для инкапсуляции в соответствии с известными способами в области фармацевтики. Также следует отметить, что приложение давления к смеси в форме можно повторять с использованием одинакового давления в ходе каждого прессования или с использованием различного давления в ходе прессований. В другом примере, смесь порошкообразных ингредиентов или гранул можно прессовать с использованием штамповочного пресса, прилагающего достаточное давление для формирования таблетки, обладающей растворением приблизительно 50% или более за приблизительно 30 минут (например, приблизительно 55% или более за приблизительно 30 минут или приблизительно 60% или более за приблизительно 30 минут). Например, смесь прессуют с использованием штамповочного пресса для получения жесткости таблетки по меньшей мере приблизительно 5 кф (49 Ньютон) (по меньшей мере приблизительно 5,5 кф (53,9 Ньютон), по меньшей мере приблизительно 6 кф (58,8 Ньютон), по меньшей мере приблизительно 7 кф (68,6 Ньютон), по меньшей мере приблизительно 10 кф (98 Ньютон), или по меньшей мере 15 кф (147 Ньютон)). В некоторых случаях смесь прессуют для получения жесткости таблетки между приблизительно 5 и 20 кф (49 и 196 Ньютон).
[00223] В некоторых вариантах осуществления таблетки, содержащие фармацевтическую композицию, как описано в настоящем документе, можно покрывать приблизительно 3,0% масс. пленочного покрытия, содержащего краситель, по массе таблетки. В конкретных случаях, суспензия или раствор красителя, используемые для покрытия таблеток, содержат приблизительно 20% масс./масс. твердых веществ по массе суспензии или раствора красителя. В других случаях, покрытые таблетки можно метить с помощью логотипа, другого изображения или текста.
[00224] В другом варианте осуществления способ получения фармацевтической композиции включает в себя получение смеси твердых форм, например, смеси порошкообразных форм, и/или жидких ингредиентов, где смесь содержит форму I соединения 1, твердую дисперсию, содержащую по существу аморфное соединение 2, и один или несколько наполнителей, выбранных из: связывающего средства, разбавителя, поверхностно-активного вещества, смазочного средства, дезинтегрирующего средства и наполнителя; перемешивание смеси, пока смесь не станет по существу гомогенной, и прессование или уплотнение смеси в гранулярную форму. Затем гранулярную композицию, содержащую форму I соединения 1 и твердую дисперсию, содержащую по существу аморфное соединение 2, можно прессовать в таблетки или составлять в капсулы, как описано выше или в примерах ниже. Альтернативно, способы получения фармацевтической композиции включают в себя получение смеси формы I соединения 1, твердой дисперсии, содержащей по существу аморфное соединение 2, и одного или нескольких наполнителей, например, связывающего средства, разбавителя, поверхностно-активного вещества, смазочного средства, дезинтегрирующего средства и наполнителя; перемешивание смеси, пока смесь не станет по существу гомогенной, и прессование/уплотнение смеси в гранулярную форму с использованием способа уплотнения влажных гранул с большим усилием сдвига, как указано в примерах ниже. Фармацевтические составы, например, таблетки, как описано в настоящем документе, можно получать с использованием полученных гранул, включающих форму I соединения 1 и твердую дисперсию, содержащую по существу аморфное соединение 2, в дополнение к избранным наполнителям, описываемым в настоящем документе.
[00225] В некоторых вариантах осуществления смесь перемешивают посредством смешивания, измельчения, встряхивания или т.п. с использованием смешивания вручную, смесителя, измельчителя, любого их сочетания или т.п. Когда ингредиенты или комбинации ингредиентов добавляют последовательно, смешивание может происходить между последовательными добавлениями, непрерывно на всем протяжении добавления ингредиента, после добавления всех ингредиентов или комбинаций ингредиентов, или в любом их сочетании. Смесь перемешивают, пока она не будет обладать по существу гомогенным составом.
[00226] В другом варианте осуществления настоящее изобретение относится к размолу в струйной мельнице фармацевтической композиции, содержащей форму I соединения 1 и твердую дисперсию, содержащую по существу аморфное соединение 2 в подходящем, общепринятом размалывающем устройстве с использованием давления воздуха, подходящего для получения частиц, обладающих значительной фракцией размера частиц между 0,1 микрон и 50 микрон. В другом варианте осуществления размер частиц составляет между 0,1 микрон и 20 микрон. В другом варианте осуществления размер частиц составляет между 0,1 микрон и 10 микрон. В другом варианте осуществления размер частиц составляет между 1,0 микрон и 5 микрон. В другом варианте осуществления, фармацевтическая композиция обладает размером частиц D50 2,0 микрон.
[00227] Составы по настоящему изобретению обеспечивают фиксированную дозу двух API для эффективного лечения кистозного фиброза, комбинацию, получившую один из только двух статусов принципиально нового лекарственного средства от FDA, и получившую его с неожиданной стабильностью, как измерено по небольшой потере аморфной твердой формы соединения 2. На фигуре 4 изображен небольшой уровень кристалличности соединения 2 с течением времени в PC-XVII при 50°C после предварительного уравновешивания при относительной влажности 60%. Даже после около 1000 часов в этих условиях, менее 5% по массе соединения 2 кристаллизовалось. На фигуре 5 показано для PC-XVII, что даже при более высокой температуре 60°C после предварительного уравновешивания при относительной влажности 60%, при около 1000 часов в этих условиях, менее 10% по массе соединения 2 кристаллизовалось. На фигурах 6 и 7 показаны сходные результаты для PC-XIX. Настоящие составы, таким образом, обеспечивают удобство фиксированной дозы двух принципиально новых API в неожиданно стабильной фармацевтической композиции. Такие составы увеличивают соблюдение пациентом схемы лечения, что непосредственно относится к эффективному лечению заболеваний.
[00228] Лекарственные формы, полученные, как выше, можно подвергать оценке растворения in vitro в соответствии с тестом 711 «Растворение» Фармакопеи Соединенных Штатов 29, United States Pharmacopeial Convention, Inc., Rockville, Md., 2005 («USP»), для определения скорости, с которой активное вещество высвобождается из лекарственной формы. Содержание активного вещества и уровни примесей в целях удобства измеряют такими способами, как высокоэффективная жидкостная хроматография (ВЭЖХ).
[00229] В некоторых вариантах осуществления изобретение включает в себя использование упаковочных материалов, таких как контейнеры и герметичные емкости из полиэтилена высокой плотности (HDPE), полиэтилена низкой плотности (LDPE) и/или полипропилена, и/или стекла, вощеной фольги, алюминиевые пакеты и блистеры или полоски, состоящие из алюминия или поливинилхлорида высокой плотности (PVC), необязательно, включая десикант, полиэтилен (PE), поливинилидендихлорид (PVDC), PVC/PE/PVDC, и т.п. Эти упаковочные материалы можно использовать для хранения различных фармацевтических композиций и составов стерильным образом после соответствующей стерилизации упаковки и ее содержимого с использованием способов химической или физической стерилизации, общепринятых в области фармацевтики.
СПОСОБЫ ВВЕДЕНИЯ ФАРМАЦЕВТИЧЕСКИХ КОМПОЗИЦИЙ
[00230] В одном из аспектов фармацевтические композиции по изобретению можно вводить пациенту один раз в сутки или приблизительно каждые двадцать четыре часа. Альтернативно, фармацевтические композиции по изобретению можно вводить пациенту дважды в сутки. Альтернативно, фармацевтическую композицию по изобретению можно вводить приблизительно каждые двенадцать часов. Эти фармацевтические композиции вводят как пероральные составы, содержащие приблизительно 25 мг, 50 мг, 100 мг, 125 мг, 150 мг, 200 мг, 250 мг, 300 мг, или 400 мг формы I соединения 1; и приблизительно 25 мг, 50 мг, 100 мг, 125 мг, 150 мг, 200 мг, или 250 мг по существу аморфного соединения 2. В этом аспекте, в дополнение к форме I соединения 1 и по существу аморфному соединению 2, фармацевтические композиции содержат наполнитель; дезинтегрирующее средство; поверхностно-активное вещество; связывающее средство; и смазочное средство (в зависимости от того, представляет собой фармацевтическая композиция гранулу или таблетку). Например, доза 400 мг формы I соединения 1 может содержать две таблетки по изобретению, каждая из которых содержит 200 мг формы I соединения 1. Доза 250 мг по существу аморфного соединения 2 может содержать две таблетки по изобретению, каждая из которых содержит 125 мг по существу аморфного соединения 2.
[00231] Следует также понимать, что соединение и фармацевтически приемлемые композиции и составы по изобретению можно использовать в способах комбинированной терапии; то есть, форму I соединения 1 и твердую дисперсию по существу аморфного соединения 2 и их фармацевтически приемлемые композиции можно вводить во время, до или после введения одного или нескольких других желательных терапевтических средств или медицинских процедур.
[00232] В одном из вариантов осуществления дополнительное терапевтическое средство выбрано из муколитического средства, бронхорасширяющего средства, антибиотика, противоинфекционного средства, противовоспалительного средства, соединения, индуцирующего активность CFTR, отличного от формы I соединения 1 и по существу аморфного соединения 2, или питательной добавки.
[00233] В одном из вариантов осуществления дополнительное средство представляет собой (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1H-индол-5-ил)циклопропанкарбоксамид. В другом варианте осуществления дополнительное средство представляет собой 4-(3-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамидо)изохинолин-1-ил)бензойную кислоту. В другом варианте осуществления дополнительное средство выбрано из таблицы 1:
Таблица 1
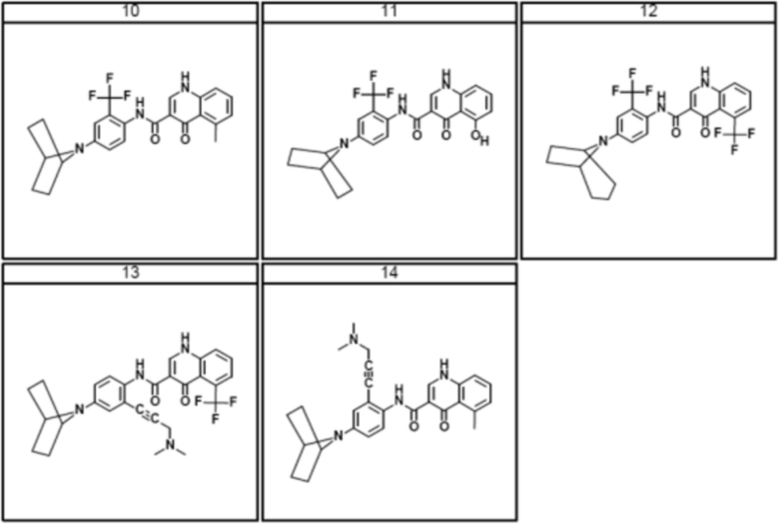
[00234] В другом варианте осуществления дополнительное средство представляет собой любую комбинацию вышеуказанных средств. Например, комбинация может содержать фармацевтическую композицию или таблетку по настоящему изобретению, содержащую форму I соединения 1 и твердую дисперсию по существу аморфного соединения 2, и дополнительное терапевтическое средство представляет собой (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1H-индол-5-ил)циклопропанкарбоксамид. в другом примере, комбинация может содержать фармацевтическую композицию или таблетку по настоящему изобретению, содержащую форму I соединения 1 и твердую дисперсию по существу аморфного соединения 2, и дополнительное терапевтическое средство представляет собой 4-(3-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамидо)изохинолин-1-ил)бензойную кислоту. В другом примере, комбинация может содержать фармацевтическую композицию или таблетку по настоящему изобретению, содержащую форму I соединения 1 и твердую дисперсию по существу аморфного соединения 2, и дополнительное терапевтическое средство представляет собой любое из соединений из таблицы 1, т.е. соединения 1-14 из таблицы 1, или любое их сочетание.
[00235] В другом варианте осуществления дополнительное средство выбрано из таблицы 1:
[00236] В другом варианте осуществления дополнительное средство выбрано из таблицы 2:
[00237] В одном из вариантов осуществления дополнительное терапевтическое средство представляет собой антибиотик. Иллюстративные антибиотики, которые можно использовать в настоящем документе, включают в себя тобрамицин, включая порошок тобрамицина для ингаляций (TIP), азитромицин, кейстон, азтреонам, включая аэрозольные формы азтреонама, амикацин, включая его липосомные составы, ципрофлоксацин, включая его составы, подходящие для введения посредством ингаляции, левофлоксацин, включая его аэрозольные составы, и комбинации двух антибиотиков, например, фосфомицина и тобрамицина.
[00238] В другом варианте осуществления дополнительное средство представляет собой муколитическое средство. Иллюстративные муколитические средства, которые можно использовать в настоящем документе, включают в себя пульмозим®.
[00239] В другом варианте осуществления дополнительное средство представляет собой бронхорасширяющее средство. Иллюстративные бронхорасширяющие средства включают в себя альбутерол, сульфат метапротенерола, ацетат пиребутерола, салметерол или сульфат тетрабулина.
[00240] В другом варианте осуществления дополнительное средство является эффективным для восстановления жидкости поверхности легочных дыхательных путей. Такие средства улучшают перемещение соли в клетки и из клеток, позволяя слизи в легочных дыхательных путях являться более гидратированной и, таким образом, более легко выводимой. Примеры таких средств включают в себя гипертонический солевой раствор, денуфозол-тетранатрий ([[(3S,5R)-5-(4-амино-2-оксопиримидин-1-ил)-3-гидроксиоксолан-2-ил]метокси-гидроксифосфорил][[[(2R,3S,4R,5R)-5-(2,4-диоксопиримидин-1-ил)-3,4-дигидроксиоксолан-2-ил]метоксигидроксифосфорил]оксигидроксифосфорил]гидрофосфат) или бронхитол (состав маннита для ингаляции).
[00241] В другом варианте осуществления дополнительное средство представляет собой противовоспалительное средство, т.е., средство, которое может уменьшать воспаление в легких. Примеры таких средств, которые можно использовать в настоящем документе, включают в себя ибупрофен, докозагексановую кислоту (DHA), силденафил, глутатион для ингаляции, пиоглитазон, гидроксихлороквин или симвастатин.
[00242] В другом варианте осуществления дополнительное средство представляет собой соединение, усиливающее или индуцирующее активность CFTR, отличное от формы I соединения 1 или твердой дисперсии, содержащей по существу аморфное соединение 2, т.е., средство, оказывающее эффект индукции или усиления активности CFTR. Примеры таких средств включают в себя аталурен («PTC124®»; 3-[5-(2-фторфенил)-1,2,4-оксадиазол-3-ил]бензойную кислоту), синапультид, ланковутид, депелестат (рекомбинантный нейтрофильный ингибитор эластазы человека) и кобипростон (7-{(2R,4aR,5R,7aR)-2-[(3S)-1,1-дифтор-3-метилпентил]-2-гидрокси-6-оксооктагидроциклопента[b]пиран-5-ил}гептановую кислоту).
[00243] В другом варианте осуществления дополнительное средство представляет собой питательные добавки. Примеры питательных добавок включают в себя панкрелипазу (заменитель панкреатического фермента), включая панкреазу®, панкреакарб®, ультразу® или креон®, липротомазу® (ранее трицитек®), аквадекс® или ингаляцию глутатиона. В одном из вариантов осуществления дополнительная питательная добавка представляет собой панкрелипазу.
[00244] В другом варианте осуществления дополнительное средство представляет собой соединение, выбранное из гентамицина, куркумина, циклофосфамида, 4-фенилбутирата, миглустата, фелодипина, нимодипина, филоксина B, генестеина, апигенина, усилителей или индукторов цАМФ/цГМФ, таких как ролипрам, силденафил, милринон, тадалафил, амринон, изопротеренол, альбутерол и альметерол, дезоксиспергуалина, ингибиторов HSP 90, ингибиторов HSP 70, ингибиторов протеосом, таких как эпоксомицин, лактацистин и т.д.
[00245] В другом варианте осуществления дополнительное средство представляет собой соединение, выбранное из (3,3,3-трифтор-2-гидрокси-2-метилпропил)амида 3-амино-6-(4-фтор-фенил)-5-трифторметилпиридин-2-карбоновой кислоты; (3,3,3-трифтор-2-гидрокси-2-метилпропил)амида 5-амино-6′-метил-3-трифторметил-[2,3]бипиридинил-6-карбоновой кислоты; 3-амино-6-циклопропил-N-(3,3,3-трифтор-2-гидрокси-2-метилпропил)-5-(трифторметил)пиколинамида; 3-амино-6-метокси-N-(3,3,3-трифтор-2-гидрокси-2-(трифторметил)пропил)-5-(трифторметил)пиколинамида; ((S)-3,3,3-трифтор-2-гидрокси-2-метилпропил)амида 3-амино-6-(4-фтор-фенил)-5-трифторметилпиридин-2-карбоновой кислоты; ((S-3,3,3-трифтор-2-гидрокси-2-метилпропил)амида 3-амино-6-метокси-5-трифторметилпиридин-2-карбоновой кислоты; ((R)-3,3,3-трифтор-2-гидрокси-2-метилпропил)амида 3-амино-6-метокси-5-трифторметилпиридин-2-карбоновой кислоты; ((S)-3,3,3-трифтор-2-гидрокси-2-метилпропил)амида 3-амино-6-(2,4-дихлорфенил)-5-трифторметилпиридин-2-карбоновой кислоты; ((R)-3,3,3-трифтор-2-гидрокси-2-метилпропил)-амида 3-амино-6-(2,4-дихлорфенил)-5-трифторметилпиридин-2-карбоновой кислоты; (2-гидрокси-2-метил-пропил)амида 3-амино-6-(4-фторфенил)-5-трифторметилпиридин-2-карбоновой кислоты; ((S)-3,3,3-трифтор-2-гидрокси-2-метил-пропил)амида 3-амино-5,6-бис-трифторметилпиридин-2-карбоновой кислоты; ((R)-3,3,3-трифтор-2-гидрокси-2-метилпропил)амида 3-амино-5,6-бис-трифторметилпиридин-2-карбоновой кислоты; (S)-3-амино-6-этокси-N-(3,3,3-трифтор-2-гидрокси-2-метилпропил)-5-(трифторметил)пиколинамида; ((S)-3,3,3-трифтор-2-гидрокси-2-метилпропил)амида 3-амино-6-метокси-5-трифторметил-пиридин-2-карбоновой кислоты; ((R)-3,3,3-трифтор-2-гидрокси-2-метилпропил)амида 3-амино-6-метокси-5-трифторметилпиридин-2-карбоновой кислоты; (3,3,3-трифтор-2-гидрокси-2-метилпропил)-амида 3-амино-6-(4-фторфенил)-5-трифторметилпиридин-2-карбоновой кислоты; ((S)-3,3,3-трифтор -2-гидрокси-2-метилпропил)амида 3-амино-5,6-бис-трифторметилпиридин-2-карбоновой кислоты; ((R)-3,3,3-трифтор-2-гидрокси-2-метилпропил)амида 3-амино-5,6-бис-трифторметилпиридин-2-карбоновой кислоты или их фармацевтически приемлемых солей. В другом варианте осуществления дополнительное средство представляет собой соединение, описанное в Патенте Соединенных Штатов No. 8247436 и Международной публикации PCT WO 2011113894, полное содержание которых включено в настоящий документ в качестве ссылки.
[00246] В другом варианте осуществления дополнительное средство может представлять собой модулятор эпителиального натриевого канала (ENac), описанный в публикациях PCT WO2012035158, WO2009074575, WO2011028740, WO2009150137, WO2011079087 или WO2008135557, полное содержание которых включено в настоящий документ в качестве ссылки.
[00247] В других вариантах осуществления дополнительное средство представляет собой соединение, описанное в WO 2004028480, WO 2004110352, WO 2005094374, WO 2005120497 или WO 2006101740, полное содержание которых включено в настоящий документ в качестве ссылки. В другом варианте осуществления дополнительное средство представляет собой производное бензо[c]хинолизина, обладающее активностью индукции или усиления CFTR, или производное бензопирана, обладающее активностью индукции или усиления CFTR. В другом варианте осуществления дополнительное средство представляет собой соединение, описанное в Патенте США No. 7202262, Патенте США No. 6992096, US20060148864, US20060148863, US20060035943, US20050164973, WO2006110483, WO2006044456, WO2006044682, WO2006044505, WO2006044503, WO2006044502, или WO2004091502, полное содержание которых включено в настоящий документ в качестве ссылки. В другом варианте осуществления дополнительное средство представляет собой соединение, описанное в WO2004080972, WO2004111014, WO2005035514, WO2005049018, WO2006099256, WO2006127588, или WO2007044560, полное содержание которых включено в настоящий документ в качестве ссылки.
[00248] В одном из вариантов осуществления 400 мг формы I соединения 1 и 250 мг по существу аморфного соединения 2 можно вводить нуждающемуся в этом субъекту. В этих вариантах осуществления величины доз можно достигать посредством введения одной или нескольких таблеток по изобретению. Например, введения 400 мг формы I соединения 1 и 250 мг по существу аморфного соединения 2 можно достигать посредством введения двух таблеток, каждая из которых содержит 200 мг формы I соединения 1 и 125 мг по существу аморфного соединения 2. Длительность введения может продолжаться до достижения улучшения состояния заболевания или по рекомендации лечащего врача субъекта, например, длительность введения может составлять менее недели, 1 неделю, 2 недели, 3 недели, четыре недели (28 суток), или месяц или дольше. В одном из вариантов осуществления две таблетки, каждая из которых содержит 200 мг формы I соединения 1 и 125 мг по существу аморфного соединения 2, можно вводить пациенту в сутки. В дополнительном варианте осуществления две таблетки можно вводить одновременно или в различные периоды времени в течение суток. В дополнительном варианте осуществления одну таблетку вводят каждые 12 часов.
[00249] В одном из вариантов осуществления 400 мг формы I соединения 1 и 500 мг по существу аморфного соединения 2 можно вводить нуждающемуся в этом субъекту. В этих вариантах осуществления величины доз можно достигать посредством введения двух таблеток, каждая из которых содержит 200 мг формы I соединения 1 и 250 мг по существу аморфного соединения 2. В одном из вариантов осуществления таблетку вводят раз в каждые 12 часов. В другом варианте осуществления величины дозы также можно достигать посредством введения двух таблеток, каждая из которых содержит 100 мг формы I соединения 1 и 125 мг по существу аморфного соединения 2, каждые 12 часов. В другом варианте осуществления величины доз также можно достигать посредством введения формы I соединения 1 и по существу аморфного соединения 2 в отдельных таблетках. Например, величины доз можно достигать посредством введения двух таблеток, содержащих 200 мг формы I соединения 1, и четырех таблеток, содержащих 125 мг по существу аморфного соединения 2 или двух таблеток, содержащих 150 мг по существу аморфного соединения 2 и двух таблеток, содержащих 100 мг по существу аморфного соединения 2. Длительность введения может продолжаться до достижения улучшения состояния заболевания или по рекомендации лечащего врача субъекта, например, длительность введения может составлять менее недели, 1 неделю, 2 недели, 3 недели, четыре недели (28 суток), или месяц или дольше. В одном из вариантов осуществления две таблетки, содержащие 200 мг формы I соединения 1, и четыре таблетки, содержащие 125 мг по существу аморфного соединения 2, можно вводить пациенту в сутки. В одном из вариантов осуществления две таблетки, содержащие 200 мг формы I соединения 1, можно вводить пациенту в сутки, и две таблетки, содержащие 150 мг и 100 мг по существу аморфного соединения 2, можно вводить пациенту два раза в сутки. В дополнительном варианте осуществления две таблетки можно вводить одновременно или в различные периоды времени в течение суток. В дополнительном варианте осуществления одну таблетку, содержащую 200 мг соединения 1, вводят каждые 12 часов, и две таблетки, содержащие 150 мг и 100 мг по существу аморфного соединения 2, вводят каждые 12 часов.
[00250] В одном из вариантов осуществления 300 мг формы I соединения 1 и 250 мг по существу аморфного соединения 2 можно вводить нуждающемуся в этом субъекту. В этих вариантах осуществления величины доз можно достигать посредством введения одной или нескольких таблеток по изобретению. Например, введения 300 мг формы I соединения 1 и 250 мг по существу аморфного соединения 2 можно достигать посредством введения двух таблеток, каждая из которых содержит 150 мг формы I соединения 1 и 125 мг по существу аморфного соединения 2. Длительность введения может продолжаться до достижения улучшения состояния заболевания или по рекомендации лечащего врача субъекта, например, длительность введения может составлять менее недели, 1 неделю, 2 недели, 3 недели, четыре недели (28 суток), или месяц или дольше. В одном из вариантов осуществления две таблетки, каждая из которых содержит 150 мг формы I соединения 1 и 125 мг по существу аморфного соединения 2, можно вводить пациенту в сутки. В дополнительном варианте осуществления две таблетки можно вводить одновременно или в различные периоды времени в течение суток. В дополнительном варианте осуществления одну таблетку вводят каждые 12 часов.
[00251] В одном из вариантов осуществления 600 мг формы I соединения 1 и 500 мг по существу аморфного соединения 2 можно вводить нуждающемуся в этом субъекту. В этих вариантах осуществления величины доз можно достигать посредством введения одной или нескольких таблеток по изобретению. Например, введения 600 мг формы I соединения 1 и 500 мг по существу аморфного соединения 2 можно достигать посредством введения двух таблеток, каждая из которых содержит 150 мг формы I соединения 1 и 125 мг по существу аморфного соединения 2, каждые 12 часов. Длительность введения может продолжаться до достижения улучшения состояния заболевания или по рекомендации лечащего врача субъекта, например, длительность введения может составлять менее недели, 1 неделю, 2 недели, 3 недели, четыре недели (28 суток), или месяц или дольше. В одном из вариантов осуществления четыре таблетки, каждая из которых содержит 150 мг формы I соединения 1, и 125 мг по существу аморфного соединения 2, можно вводить пациенту в сутки. В дополнительном варианте осуществления четыре таблетки можно вводить одновременно или в различные периоды времени в течение суток. В дополнительном варианте осуществления две таблетки вводят каждые 12 часов.
[00252] В одном из вариантов осуществления 800 мг формы I соединения 1 и 500 мг по существу аморфного соединения 2 можно вводить нуждающемуся в этом субъекту. В этих вариантах осуществления величины доз можно достигать посредством введения одной или нескольких таблеток по изобретению. Например, введения 800 мг формы I соединения 1 и 500 мг по существу аморфного соединения 2 можно достигать посредством введения четырех таблеток, каждая из которых содержит 200 мг формы I соединения 1 и 125 мг по существу аморфного соединения 2. Длительность введения может продолжаться до достижения улучшения состояния заболевания или по рекомендации лечащего врача субъекта, например, длительность введения может составлять менее недели, 1 неделю, 2 недели, 3 недели, четыре недели (28 суток), или месяц или дольше. В одном из вариантов осуществления четыре таблетки, каждая из которых содержит 200 мг формы I соединения 1, и 125 мг по существу аморфного соединения 2 можно вводить пациенту в сутки. В дополнительном варианте осуществления четыре таблетки можно вводить одновременно или в различные периоды времени в течение суток. В дополнительном варианте осуществления две таблетки вводят на прием дозирования, и присутствуют два приема дозирования в сутки. В дополнительном варианте осуществления 800 мг соединения 1 и 500 мг соединения 2 вводят пациенту посредством введения двух таблеток, каждая из которых содержит 200 мг соединения 1 и 125 мг соединения 2, дважды в сутки (BID). В дополнительном варианте осуществления 800 мг соединения 1 и 500 мг соединения 2 вводят пациенту посредством введения двух таблеток, каждая из которых содержит 200 мг соединения 1 и 125 мг соединения 2 каждые 12 часов (q12 час).
[00253] В одном из вариантов осуществления 600 мг формы I соединения 1 и 250 мг по существу аморфного соединения 2 можно вводить нуждающемуся в этом субъекту. В этих вариантах осуществления величины доз можно достигать посредством введения одной или нескольких таблеток по изобретению. Например, введения 600 мг формы I соединения 1 и 250 мг по существу аморфного соединения 2 можно достигать посредством введения трех таблеток, каждая из которых содержит 200 мг формы I соединения 1 и 83,3 мг по существу аморфного соединения 2. Длительность введения может продолжаться до достижения улучшения состояния заболевания или по рекомендации лечащего врача субъекта, например, длительность введения может составлять менее недели, 1 неделю, 2 недели, 3 недели, четыре недели (28 суток), или месяц или дольше. В одном из вариантов осуществления три таблетки, каждая из которых содержит 200 мг формы I соединения 1, и 83,3 мг по существу аморфного соединения 2, можно вводить пациенту в сутки. В дополнительном варианте осуществления три таблетки можно вводить одновременно или в различные периоды времени в течение суток. В дополнительном варианте осуществления три таблетки вводят одновременно.
[00254] В одном из вариантов осуществления 600 мг формы I соединения 1 и 500 мг по существу аморфного соединения 2 можно вводить нуждающемуся в этом субъекту. В этих вариантах осуществления величины доз можно достигать посредством введения одной или нескольких таблетки по изобретению. Например, введения 600 мг формы I соединения 1 и 500 мг по существу аморфного соединения 2 можно достигать посредством введения трех таблеток, каждая из которых содержит 200 мг формы I соединения 1 и 83,3 мг по существу аморфного соединения 2, а затем двух дополнительных таблеток, каждая из которых содержит 125 мг соединения 2. Длительность введения может продолжаться до достижения улучшения состояния заболевания или по рекомендации лечащего врача субъекта, например, длительность введения может составлять менее недели, 1 неделю, 2 недели, 3 недели, четыре недели (28 суток), или месяц или дольше. В одном из вариантов осуществления 600 мг соединения 1 можно вводить ежесуточно (qd), и 250 мг соединения 2 вводить дважды в сутки (bid) посредством введения трех таблеток, каждая из которых содержит 200 мг формы I соединения 1 и 83,3 мг по существу аморфного соединения 2, ежесуточно (qd) и двух таблеток, каждая из которых содержит 125 мг соединения 2, каждые 12 часов (q12 час). В одном из вариантов осуществления 600 мг соединения 1 можно вводить ежесуточно (qd), и 250 мг соединения 2 вводить каждые 12 часов (q12 час) посредством введения трех таблеток, каждая из которых содержит 200 мг формы I соединения 1 и 83,3 мг по существу аморфного соединения 2, ежесуточно (qd) и двух таблеток, каждая из которых содержит 125 мг соединения 2, каждые 12 часов (q12 час).
[00255] Эти комбинации можно использовать для лечения заболеваний, описываемых в настоящем документе, включая кистозный фиброз. Эти комбинации можно использовать также в наборах, описываемых в настоящем документе. В другом аспекте настоящее изобретение относится к набору, содержащему фармацевтическую композицию или таблетку по настоящему изобретению, содержащие форму I соединения 1 и твердую дисперсию, содержащую по существу аморфное соединение 2, и отдельное дополнительное терапевтическое средство или его фармацевтическую композицию. В другом варианте осуществления фармацевтическая композиция или таблетка по настоящему изобретению, отдельное дополнительное терапевтическое средство или его фармацевтическая композиция находятся в отдельных контейнерах. В другом варианте осуществления отдельные контейнеры представляют собой бутыли. В другом варианте осуществления отдельные контейнеры представляют собой флаконы. В другом варианте осуществления отдельные контейнеры представляют собой блистерные упаковки.
[00256] Количество дополнительного терапевтического средства, присутствующего в композициях по этому изобретению, составляет не более, чем количество, которое в норме вводят в композиции, содержащей это терапевтическое средство в качестве единственного активного средства. Предпочтительно, количество дополнительного терапевтического средства в обсуждаемых в настоящее время композициях лежит в диапазоне от приблизительно 50% до 100% от количества, в норме присутствующего в композиции, содержащей это средство в качестве единственного терапевтически активного средства.
ТЕРАПЕВТИЧЕСКИЕ ПРИМЕНЕНИЯ КОМПОЗИЦИИ
[00257] В одном из аспектов изобретение также относится к способу лечения, уменьшения тяжести или симптоматического лечения заболевания у пациента, где способ включает в себя введение эффективного количества фармацевтической композиции или таблетки по изобретению пациенту, предпочтительно, млекопитающему, где заболевание выбрано из кистозного фиброза, астмы, индуцированного курением COPD, хронического бронхита, риносинусита, запора, панкреатита, недостаточности поджелудочной железы, мужского бесплодия, вызванного врожденным билатеральным отсутствием семявыводящих протоков (CBAVD), заболевания легких умеренной тяжести, идиопатического панкреатита, аллергического бронхолегочного аспергиллеза (ABPA), заболевания печени, наследственной эмфиземы, наследственного гемохроматоза, недостаточности свертывания-фибринолиза, такой как недостаточность белка C, наследственного ангионевротического отека типа 1, недостаточности процессинга липидов, такой как семейная гиперхолестеринемия, хиломикронемия типа 1, абеталипопротеинемия, болезней лизосомного накопления, таких как болезнь клеточных включений/псевдосиндром Гурлера, мукополисахаридозов, болезни Сандхоффа/Тэя-Сакса, болезни Криглера-Найяра типа II, полиэндокринопатии/гиперинсулемии, сахарного диабета, карликовости лароновского типа, недостаточности миелопероксидазы, первичного гипопаратиреоза, меланомы, гликаноза CDG типа 1, врожденного гипертиреоза, несовершенного остеогенеза, наследственной гипофибриногенемии, недостаточности ACT, несахарного диабета (DI), нейрофизеального DI, нефрогенного DI, синдрома Шарко-Мари-Тута, болезни Перлицеуса-Мерцбахера, нейродегенеративных заболеваний, таких как болезнь Альцгеймера, болезнь Паркинсона, боковой амиотрофический склероз, прогрессирующий надъядерный паралич, болезнь Пика, несколько полиглутаминовых неврологических нарушений, таких как болезнь Гентингтона, спиноцеребеллярная атаксия типа I, спинальная и бульбарная мышечная атрофия, дентато-рубро-паллидо-льюисова атрофия и миотоническая дистрофия, а также губчатые энцефалопатии, такие как наследственная болезнь Крейтцфельдта-Якоба (обусловленная дефектом процессинга прионовых белков), болезнь Фабри, синдром Штаусслера-Шейнкера, COPD, заболевание сухих глаз или заболевание Шегрена, остеопороз, остеопению, нарушения срастания костей и роста костей (включая репарацию кости, регенерацию кости, уменьшение резорбции кости и увеличение наслоения кости), синдром Горхема, хлоридные каналопатии, такие как врожденная миотония (формы Томсона и Беккера), синдром Барттера типа III, болезнь Дента, гиперэкплексию, эпилепсию, заболевание лизосомного накопления, синдром Ангельмана и первичную цилиарную дискинезию (PCD), термин для наследственных нарушений структуры и/или функции ресничек, включая PCD с транспозицией органов (также известную как синдром Картагенера), PCD без транспозиции органов и цилиарной аплазии.
[00258] В одном из аспектов изобретение также относится к способу лечения, уменьшения тяжести или симптоматического лечения заболевания у пациента, включающему в себя введение эффективного количества фармацевтической композиции или таблетки по изобретению пациенту, предпочтительно, млекопитающему, где заболевание выбрано из генерализованной эпилепсии с фебрильными судорогами плюс (GEFS+), генерализованной эпилепсии с фебрильными и афебрильными судорогами, миотонии, врожденной парамиотонии, миотонии с агрегацией калия, гиперкалиемического периодического паралича, LQTS, LQTS/ синдрома Бругады, аутосомно-доминантного LQTS с глухотой, аутосомно-рецессивного LQTS, LQTS с дисморфическими признаками, наследственного и приобретенного LQTS, синдрома Тимоти, персистирующей гиперинсулинемической гипогликемии раннего возраста, дилатационной кардиомиопатии, аутосомно-доминантного LQTS, болезни Дента, остеопетроза, синдрома Бартера типа III, болезни центральных волокон, злокачественной гипертермии и катехоламинергической полиморфной тахикардии.
[00259] В одном из аспектов настоящее изобретение относится к способу лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающему в себя введение эффективного количества фармацевтической композиции или таблетки по изобретению пациенту, предпочтительно млекопитающему, где пациент обладает генетической мутацией CFTR N1303K, ΔI507, или R560T.
[00260] В одном из аспектов настоящее изобретение относится к способу лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающему в себя введение эффективного количества фармацевтической композиции или таблеток по изобретению пациенту, предпочтительно млекопитающему, где пациент обладает генетической мутацией CFTR G551D. В другом варианте осуществления пациент является гомозиготным по G551D. В другом варианте осуществления пациент является гетерозиготным по G551D, где другая генетическая мутация CFTR представляет собой любую из ΔF508, G542X, N1303K, W1282X, R117H, R553X, 1717-1 G->A, 621+1 G->T, 2789+5 G->A, 3849+10kbC->T, R1162X, G85E, 3120+1 G->A, ΔI507, 1898+1 G->A, 3659delC, R347P, R560T, R334W, A455E, 2184delA, или 711+1 G->T.
[00261] В одном из аспектов настоящее изобретение относится к способу лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающему в себя введение эффективного количества фармацевтической композиции или таблеток по изобретению пациенту, предпочтительно млекопитающему, где пациент обладает генетической мутацией CFTR ΔF508. В другом варианте осуществления пациент является гомозиготным по ΔF508. В другом варианте осуществления пациент является гетерозиготным по ΔF508, где другая генетическая мутация CFTR представляет собой любую из G551D, G542X, N1303K, W1282X, R117H, R553X, 1717-1 G->A, 621+1 G->T, 2789+5 G->A, 3849+10kbC->T, R1162X, G85E, 3120+1 G->A, ΔI507, 1898+1 G->A, 3659delC, R347P, R560T, R334W, A455E, 2184delA, или 711+1 G->T.
[00262] В одном из аспектов настоящее изобретение относится к способу лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающему в себя введение эффективного количества фармацевтической композиции или таблеток по изобретению пациенту, предпочтительно млекопитающему, где пациент обладает генетической мутацией CFTR, выбранной из G178R, G551S, G970R, G1244E, S1255P, G1349D, S549N, S549R, S1251N, E193K, F1052V, G1069R, R117C, D110H, R347H, R352Q, E56K, P67L, L206W, A455E, D579 G, S1235R, S945L, R1070W, F1074L, D110E, D1270N, D1152H, 1717-1 G->A, 621+1 G->T, 3120+1 G->A, 1898+1 G->A, 711+1 G->T, 2622+1 G->A, 405+1 G->A, 406-1 G->A, 4005+1 G->A, 1812-1 G->A, 1525-1 G->A, 712-1 G->T, 1248+1 G->A, 1341+1 G->A, 3121-1 G->A, 4374+1 G->T, 3850-1 G->A, 2789+5 G->A, 3849+10kbC->T, 3272-26A->G, 711+5 G->A, 3120 G->A, 1811+1,6kbA->G, 711+3A->G, 1898+3A->G, 1717-8 G->A, 1342-2A->C, 405+3A->C, 1716 G/A, 1811+1 G->C, 1898+5 G->T, 3850-3T->G, IVS14b+5 G->A, 1898+1 G->T, 4005+2T->C и 621+3A->G.
[00263] В одном из аспектов настоящее изобретение относится к способу лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающему в себя введение эффективного количества фармацевтической композиции или таблеток по изобретению пациенту, предпочтительно млекопитающему, где пациент обладает генетической мутацией CFTR, выбранной из G178R, G551S, G970R, G1244E, S1255P, G1349D, S549N, S549R, S1251N, E193K, F1052V и G1069R. В одном из вариантов осуществления этого аспекта, изобретение относится к способу лечения CFTR, включающему в себя введение соединения 1 пациенту, обладающему мутацией CFTR человека, выбранной из G178R, G551S, G970R, G1244E, S1255P, G1349D, S549N, S549R и S1251N. В одном из аспектов настоящее изобретение относится к способу лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающему в себя введение эффективного количества фармацевтической композиции или таблеток по изобретению пациенту, предпочтительно млекопитающему, где пациент обладает генетической мутацией CFTR, выбранной из E193K, F1052V и G1069R. В некоторых вариантах осуществления этого аспекта, способом получают более чем 10-кратное увеличение транспорта хлорида относительно исходного транспорта хлорида.
[00264] В одном из аспектов настоящее изобретение относится к способу лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающему в себя введение эффективного количества фармацевтической композиции или таблеток по изобретению пациенту, предпочтительно млекопитающему, где пациент обладает генетической мутацией CFTR, выбранной из R117C, D110H, R347H, R352Q, E56K, P67L, L206W, A455E, D579G, S1235R, S945L, R1070W, F1074L, D110E, D1270N и D1152H. В одном из вариантов осуществления этого аспекта, способом получают увеличение транспорта хлорида, большее или равное на 10% превышающему исходный транспорт хлорида.
[00265] В одном из аспектов настоящее изобретение относится к способу лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающему в себя введение эффективного количества фармацевтической композиции или таблеток по изобретению пациенту, предпочтительно млекопитающему, где пациент обладает генетической мутацией CFTR, выбранной из 1717-1 G->A, 621+1 G->T, 3120+1 G->A, 1898+1 G->A, 711+1 G->T, 2622+1 G->A, 405+1 G->A, 406-1 G->A, 4005+1 G->A, 1812-1 G->A, 1525-1 G->A, 712-1 G->T, 1248+1 G->A, 1341+1 G->A, 3121-1 G->A, 4374+1 G->T, 3850-1 G->A, 2789+5 G->A, 3849+10kbC->T, 3272-26A->G, 711+5 G->A, 3120 G->A, 1811+1,6kbA->G, 711+3A->G, 1898+3A->G, 1717-8 G->A, 1342-2A->C, 405+3A->C, 1716 G/A, 1811+1 G->C, 1898+5 G->T, 3850-3T->G, IVS14b+5 G->A, 1898+1 G->T, 4005+2T->C и 621+3A->G. В одном из аспектов настоящее изобретение относится к способу лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающему в себя введение эффективного количества фармацевтической композиции или таблеток по изобретению пациенту, предпочтительно млекопитающему, где пациент обладает генетической мутацией CFTR, выбранной из 1717-1 G->A, 1811+1,6kbA->G, 2789+5 G->A, 3272-26A->G и 3849+10kbC->T. В одном из аспектов настоящее изобретение относится к способу лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающему в себя введение эффективного количества фармацевтической композиции или таблеток по изобретению пациенту, предпочтительно млекопитающему, где пациент обладает генетической мутацией CFTR, выбранной из 2789+5 G->A и 3272-26A->G.
[00266] В одном из аспектов настоящее изобретение относится к способу лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающему в себя введение эффективного количества фармацевтической композиции или таблеток по изобретению пациенту, предпочтительно млекопитающему, где пациент обладает генетической мутацией CFTR, выбранной из G178R, G551S, G970R, G1244E, S1255P, G1349D, S549N, S549R, S1251N, E193K, F1052V, G1069R, R117C, D110H, R347H, R352Q, E56K, P67L, L206W, A455E, D579 G, S1235R, S945L, R1070W, F1074L, D110E, D1270N, D1152H, 1717-1 G->A, 621+1 G->T, 3120+1 G->A, 1898+1 G->A, 711+1 G->T, 2622+1 G->A, 405+1 G->A, 406-1 G->A, 4005+1 G->A, 1812-1 G->A, 1525-1 G->A, 712-1 G->T, 1248+1 G->A, 1341+1 G->A, 3121-1 G->A, 4374+1 G->T, 3850-1 G->A, 2789+5 G->A, 3849+10kbC->T, 3272-26A->G, 711+5 G->A, 3120 G->A, 1811+1,6kbA->G, 711+3A->G, 1898+3A->G, 1717-8 G->A, 1342-2A->C, 405+3A->C, 1716 G/A, 1811+1 G->C, 1898+5 G->T, 3850-3T->G, IVS14b+5 G->A, 1898+1 G->T, 4005+2T->C и 621+3A->G, и мутацией CFTR человека, выбранной из ΔF508, R117H и G551D.
[00267] В одном из аспектов настоящее изобретение относится к способу лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающему в себя введение эффективного количества фармацевтической композиции или таблеток по изобретению пациенту, предпочтительно млекопитающему, где пациент обладает генетической мутацией CFTR, выбранной из G178R, G551S, G970R, G1244E, S1255P, G1349D, S549N, S549R, S1251N, E193K, F1052V и G1069R, и мутацией CFTR человека, выбранной из ΔF508, R117H, и G551D. В одном из аспектов настоящее изобретение относится к способу лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающему в себя введение эффективного количества фармацевтической композиции или таблеток по изобретению пациенту, предпочтительно млекопитающему, где пациент обладает генетической мутацией CFTR, выбранной из G178R, G551S, G970R, G1244E, S1255P, G1349D, S549N, S549R и S1251N, и мутацией CFTR человека, выбранной из ΔF508, R117H, и G551D. В одном из аспектов настоящее изобретение относится к способу лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающему в себя введение эффективного количества фармацевтической композиции или таблеток по изобретению пациенту, предпочтительно млекопитающему, где пациент обладает генетической мутацией CFTR, выбранной из E193K, F1052V и G1069R, и мутацией CFTR человека, выбранной из ΔF508, R117H, и G551D. В некоторых вариантах осуществления этого аспекта, способом получают более чем 10-кратное увеличение транспорта хлорида относительно исходного транспорта хлорида.
[00268] В одном из аспектов настоящее изобретение относится к способу лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающему в себя введение эффективного количества фармацевтической композиции или таблеток по изобретению пациенту, предпочтительно млекопитающему, где пациент обладает генетической мутацией CFTR, выбранной из R117C, D110H, R347H, R352Q, E56K, P67L, L206W, A455E, D579 G, S1235R, S945L, R1070W, F1074L, D110E, D1270N и D1152H, и мутацией CFTR человека, выбранной из ΔF508, R117H, и G551D. В одном из вариантов осуществления этого аспекта, способом получают увеличение транспорта хлорида, большее или равное на 10% превышающему исходный транспорт хлорида.
[00269] В одном из аспектов настоящее изобретение относится к способу лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающему в себя введение эффективного количества фармацевтической композиции или таблеток по изобретению пациенту, предпочтительно млекопитающему, где пациент обладает генетической мутацией CFTR, выбранной из 1717-1 G->A, 621+1 G->T, 3120+1 G->A, 1898+1 G->A, 711+1 G->T, 2622+1 G->A, 405+1 G->A, 406-1 G->A, 4005+1 G->A, 1812-1 G->A, 1525-1 G->A, 712-1 G->T, 1248+1 G->A, 1341+1 G->A, 3121-1 G->A, 4374+1 G->T, 3850-1 G->A, 2789+5 G->A, 3849+10kbC->T, 3272-26A->G, 711+5 G->A, 3120 G->A, 1811+1,6kbA->G, 711+3A->G, 1898+3A->G, 1717-8 G->A, 1342-2A->C, 405+3A->C, 1716 G/A, 1811+1 G->C, 1898+5 G->T, 3850-3T->G, IVS14b+5 G->A, 1898+1 G->T, 4005+2T->C и 621+3A->G, и мутацией CFTR человека, выбранной из ΔF508, R117H и G551D. В одном из аспектов настоящее изобретение относится к способу лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающему в себя введение эффективного количества фармацевтической композиции или таблеток по изобретению пациенту, предпочтительно млекопитающему, где пациент обладает генетической мутацией CFTR, выбранной из 1717-1 G->A, 1811+1,6kbA->G, 2789+5 G->A, 3272-26A->G и 3849+10kbC->T, и мутацией CFTR человека, выбранной из ΔF508, R117H, и G551D. В одном из аспектов настоящее изобретение относится к способу лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающему в себя введение эффективного количества фармацевтической композиции или таблеток по изобретению пациенту, предпочтительно млекопитающему, где пациент обладает генетической мутацией CFTR, выбранной из 2789+5 G->A и 3272-26A->G, и мутацией CFTR человека, выбранной из ΔF508, R117H.
[00270] В одном из аспектов настоящее изобретение относится к способу лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающему в себя введение эффективного количества фармацевтической композиции или таблеток по изобретению пациенту, предпочтительно млекопитающему, где пациент обладает генетической мутацией CFTR, выбранной из G178R, G551S, G970R, G1244E, S1255P, G1349D, S549N, S549R, S1251N, E193K, F1052V, G1069R, R117C, D110H, R347H, R352Q, E56K, P67L, L206W, A455E, D579 G, S1235R, S945L, R1070W, F1074L, D110E, D1270N, D1152H, 1717-1 G->A, 621+1 G->T, 3120+1 G->A, 1898+1 G->A, 711+1 G->T, 2622+1 G->A, 405+1 G->A, 406-1 G->A, 4005+1 G->A, 1812-1 G->A, 1525-1 G->A, 712-1 G->T, 1248+1 G->A, 1341+1 G->A, 3121-1 G->A, 4374+1 G->T, 3850-1 G->A, 2789+5 G->A, 3849+10kbC->T, 3272-26A->G, 711+5 G->A, 3120 G->A, 1811+1,6kbA->G, 711+3A->G, 1898+3A->G, 1717-8 G->A, 1342-2A->C, 405+3A->C, 1716 G/A, 1811+1 G->C, 1898+5 G->T, 3850-3T->G, IVS14b+5 G->A, 1898+1 G->T, 4005+2T->C и 621+3A->G, и мутацией CFTR человека, выбранной из ΔF508, R117H, и G551D.
[00271] В одном из аспектов настоящее изобретение относится к способу лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающему в себя введение эффективного количества фармацевтической композиции или таблеток по изобретению пациенту, предпочтительно млекопитающему, где пациент обладает генетической мутацией CFTR, выбранной из G178R, G551S, G970R, G1244E, S1255P, G1349D, S549N, S549R, S1251N, E193K, F1052V и G1069R. В одном из аспектов настоящее изобретение относится к способу лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающему в себя введение эффективного количества фармацевтической композиции или таблеток по изобретению пациенту, предпочтительно млекопитающему, где пациент обладает генетической мутацией CFTR, выбранной из G178R, G551S, G970R, G1244E, S1255P, G1349D, S549N, S549R и S1251N. В одном из аспектов настоящее изобретение относится к способу лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающему в себя введение эффективного количества фармацевтической композиции или таблеток по изобретению пациенту, предпочтительно млекопитающему, где пациент обладает генетической мутацией CFTR, выбранной из E193K, F1052V и G1069R. В некоторых вариантах осуществления этого аспекта, способом получают более чем 10-кратное увеличение транспорта хлорида относительно исходного транспорта хлорида.
[00272] В одном из аспектов настоящее изобретение относится к способу лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающему в себя введение эффективного количества фармацевтической композиции или таблеток по изобретению пациенту, предпочтительно млекопитающему, где пациент обладает генетической мутацией CFTR, выбранной из R117C, D110H, R347H, R352Q, E56K, P67L, L206W, A455E, D579 G, S1235R, S945L, R1070W, F1074L, D110E, D1270N и D1152H. В одном из вариантов осуществления этого аспекта, способом получают увеличение транспорта хлорида, большее или равное на 10% превышающему исходный транспорт хлорида.
[00273] В одном из аспектов настоящее изобретение относится к способу лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающему в себя введение эффективного количества фармацевтической композиции или таблеток по изобретению пациенту, предпочтительно млекопитающему, где пациент обладает генетической мутацией CFTR, выбранной из 1717-1 G->A, 621+1 G->T, 3120+1 G->A, 1898+1 G->A, 711+1 G->T, 2622+1 G->A, 405+1 G->A, 406-1 G->A, 4005+1 G->A, 1812-1 G->A, 1525-1 G->A, 712-1 G->T, 1248+1 G->A, 1341+1 G->A, 3121-1 G->A, 4374+1 G->T, 3850-1 G->A, 2789+5 G->A, 3849+10kbC->T, 3272-26A->G, 711+5 G->A, 3120 G->A, 1811+1,6kbA->G, 711+3A->G, 1898+3A->G, 1717-8 G->A, 1342-2A->C, 405+3A->C, 1716 G/A, 1811+1 G->C, 1898+5 G->T, 3850-3T->G, IVS14b+5 G->A, 1898+1 G->T, 4005+2T->C и 621+3A->G. В одном из аспектов настоящее изобретение относится к способу лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающему в себя введение эффективного количества фармацевтической композиции или таблеток по изобретению пациенту, предпочтительно млекопитающему, где пациент обладает генетической мутацией CFTR, выбранной из 1717-1 G->A, 1811+1,6kbA->G, 2789+5 G->A, 3272-26A->G и 3849+10kbC->T. В одном из аспектов настоящее изобретение относится к способу лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающему в себя введение эффективного количества фармацевтической композиции или таблеток по изобретению пациенту, предпочтительно млекопитающему, где пациент обладает генетической мутацией CFTR, выбранной из 2789+5 G->A и 3272-26A->G.
[00274] В одном из аспектов настоящее изобретение относится к способу лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающему в себя введение эффективного количества фармацевтической композиции или таблеток по изобретению пациенту, предпочтительно млекопитающему, где пациент обладает генетической мутацией CFTR, выбранной из G178R, G551S, G970R, G1244E, S1255P, G1349D, S549N, S549R, S1251N, E193K, F1052V, G1069R, R117C, D110H, R347H, R352Q, E56K, P67L, L206W, A455E, D579 G, S1235R, S945L, R1070W, F1074L, D110E, D1270N, D1152H, 1717-1 G->A, 621+1 G->T, 3120+1 G->A, 1898+1 G->A, 711+1 G->T, 2622+1 G->A, 405+1 G->A, 406-1 G->A, 4005+1 G->A, 1812-1 G->A, 1525-1 G->A, 712-1 G->T, 1248+1 G->A, 1341+1 G->A, 3121-1 G->A, 4374+1 G->T, 3850-1 G->A, 2789+5 G->A, 3849+10kbC->T, 3272-26A->G, 711+5 G->A, 3120 G->A, 1811+1,6kbA->G, 711+3A->G, 1898+3A->G, 1717-8 G->A, 1342-2A->C, 405+3A->C, 1716 G/A, 1811+1 G->C, 1898+5 G->T, 3850-3T->G, IVS14b+5 G->A, 1898+1 G->T, 4005+2T->C и 621+3A->G, и мутацией CFTR человека, выбранной из ΔF508, R117H, и G551D, и одной или несколькими мутациями CFTR человека, выбранными из ΔF508, R117H, и G551D.
[00275] В одном из аспектов настоящее изобретение относится к способу лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающему в себя введение эффективного количества фармацевтической композиции или таблеток по изобретению пациенту, предпочтительно млекопитающему, где пациент обладает генетической мутацией CFTR, выбранной из G178R, G551S, G970R, G1244E, S1255P, G1349D, S549N, S549R, S1251N, E193K, F1052V и G1069R, и одной или несколькими мутациями CFTR человека, выбранными из ΔF508, R117H, и G551D. В одном из аспектов настоящее изобретение относится к способу лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающему в себя введение эффективного количества фармацевтической композиции или таблеток по изобретению пациенту, предпочтительно млекопитающему, где пациент обладает генетической мутацией CFTR, выбранной из G178R, G551S, G970R, G1244E, S1255P, G1349D, S549N, S549R и S1251N, и одной или несколькими мутациями CFTR человека, выбранными из ΔF508, R117H, и G551D. В одном из аспектов настоящее изобретение относится к способу лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающему в себя введение эффективного количества фармацевтической композиции или таблеток по изобретению пациенту, предпочтительно млекопитающему, где пациент обладает генетической мутацией CFTR, выбранной из E193K, F1052V и G1069R, и одной или несколькими мутациями CFTR человека, выбранными из ΔF508, R117H, и G551D. В некоторых вариантах осуществления этого аспекта, способом получают более чем 10-кратное увеличение транспорта хлорида относительно исходного транспорта хлорида.
[00276] В одном из аспектов настоящее изобретение относится к способу лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающему в себя введение эффективного количества фармацевтической композиции или таблеток по изобретению пациенту, предпочтительно млекопитающему, где пациент обладает генетической мутацией CFTR, выбранной из R117C, D110H, R347H, R352Q, E56K, P67L, L206W, A455E, D579G, S1235R, S945L, R1070W, F1074L, D110E, D1270N и D1152H, и одной или несколькими мутациями CFTR человека, выбранными из ΔF508, R117H, и G551D. В одном из вариантов осуществления этого аспекта, способом получают увеличение транспорта хлорида, большее или равное на 10% превышающему исходный транспорт хлорида.
[00277] В одном из аспектов настоящее изобретение относится к способу лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающему в себя введение эффективного количества фармацевтической композиции или таблеток по изобретению пациенту, предпочтительно млекопитающему, где пациент обладает генетической мутацией CFTR, выбранной из 1717-1 G->A, 621+1 G->T, 3120+1 G->A, 1898+1 G->A, 711+1 G->T, 2622+1 G->A, 405+1 G->A, 406-1 G->A, 4005+1 G->A, 1812-1 G->A, 1525-1 G->A, 712-1 G->T, 1248+1 G->A, 1341+1 G->A, 3121-1 G->A, 4374+1 G->T, 3850-1 G->A, 2789+5 G->A, 3849+10kbC->T, 3272-26A->G, 711+5 G->A, 3120 G->A, 1811+1,6kbA->G, 711+3A->G, 1898+3A->G, 1717-8 G->A, 1342-2A->C, 405+3A->C, 1716 G/A, 1811+1 G->C, 1898+5 G->T, 3850-3T->G, IVS14b+5 G->A, 1898+1 G->T, 4005+2T->C и 621+3A->G, и одной или несколькими мутациями CFTR человека, выбранными из ΔF508, R117H, и G551D. В одном из аспектов настоящее изобретение относится к способу лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающему в себя введение эффективного количества фармацевтической композиции или таблеток по изобретению пациенту, предпочтительно млекопитающему, где пациент обладает генетической мутацией CFTR, выбранной из 1717-1 G->A, 1811+1,6kbA->G, 2789+5 G->A, 3272-26A->G и 3849+10kbC->T, и одной или несколькими мутациями CFTR человека, выбранными из ΔF508, R117H, и G551D. В одном из аспектов настоящее изобретение относится к способу лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающему в себя введение эффективного количества фармацевтической композиции или таблеток по изобретению пациенту, предпочтительно млекопитающему, где пациент обладает генетической мутацией CFTR, выбранной из 2789+5 G->A и 3272-26A->G, и одной или несколькими мутациями CFTR человека, выбранными из ΔF508, R117H, и G551D.
[00278] В определенных вариантах осуществления фармацевтически приемлемую композицию или таблетки по настоящему изобретению, содержащие форму I соединения 1 и твердую дисперсию по существу аморфного соединения 2, можно использовать для лечения, уменьшения тяжести или симтоматического лечения кистозного фиброза у пациентов, которые обладают остаточной активностью CFTR в апикальной мембране относящегося и не относящегося к дыхательным путям эпителия. Присутствие остаточной активности CFTR на эпителиальной поверхности можно легко детектировать известными в данной области способами, например, стандартными электрофизиологическими, биохимическими или гистохимическими способами. Такими способами идентифицируют активность CFTR с использованием in vivo или ex vivo электрофизиологических способов, измерения концентраций Cl- в поте или слюне, или ex vivo биохимических или гистохимических способов для мониторирования плотности клеточной поверхности. С использованием таких способов, остаточную активность CFTR можно легко детектировать у пациентов, гетерозиготных или гомозиготных по множеству различных мутаций, включая пациентов, гомозиготных или гетерозиготных по наиболее распространенной мутации, ΔF508, а также другим мутациям, таким как мутация G551D или мутация R117H. В определенных вариантах осуществления фармацевтически приемлемые композиции или таблетки, содержащие форму I соединения 1 и твердую дисперсию, содержащую по существу аморфное соединение 2, можно использовать для лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациентов, обладающих от небольшой до отсутствующей остаточной активностью CFTR. В определенных вариантах осуществления фармацевтически приемлемые композиции или таблетки, содержащие форму I соединения 1 и твердую дисперсию, содержащую по существу аморфное соединение 2 можно использовать для лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациентов, обладающих от небольшой до отсутствующей остаточной активностью CFTR в апикальной мембране эпителия дыхательных путей.
[00279] В другом варианте осуществления соединения и композиции по настоящему изобретению можно использовать для лечения или уменьшения тяжести кистозного фиброза у пациентов, обладающих остаточной активностью CFTR, индуцированной или усиленной с использованием фармакологических способов. В другом варианте осуществления соединения и композиции по настоящему изобретению можно использовать для лечения или уменьшения тяжести кистозного фиброза у пациентов, обладающих остаточной активностью CFTR, индуцированной или усиленной с использованием генотерапии. Такими способами увеличивают количество CFTR, присутствующее на клеточной поверхности, таким образом, индуцируя до этого момента отсутствующую активность CFTR у пациента или усиливая существующий уровень остаточной активности CFTR у пациента.
[00280] В одном из вариантов осуществления фармацевтические композиции и таблетки по настоящему изобретению, содержащие форму I соединения 1 и твердую дисперсию, содержащую по существу аморфное соединение 2, как описано в настоящем документе, можно использовать для лечения или уменьшения тяжести кистозного фиброза у пациентов с конкретными генотипами, обладающими остаточной активностью CFTR, например, мутации класса I (отсутствие синтеза), мутация класса II (неправильное сворачивание), мутации класса III (нарушенная регуляция или пропускание), мутации класса IV (измененная проводимость) или мутации класса V (уменьшенный синтез).
[00281] В одном из вариантов осуществления фармацевтические композиции и таблетки по настоящему изобретению, содержащие форму I соединения 1 и твердую дисперсию, содержащую по существу аморфное соединение 2, как описано в настоящем документе, можно использовать для лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациентов с конкретными клиническими фенотипами, например, с клиническим фенотипом от умеренного до слабого, которые, как правило, коррелируют с количеством остаточной активности CFTR в апикальной мембране эпителия. Такие фенотипы включают в себя пациентов, обладающих панкреатической недостаточностью.
[00282] В одном из вариантов осуществления фармацевтические композиции и таблетки по настоящему изобретению, содержащие форму I соединения 1 и твердую дисперсию, содержащую по существу аморфное соединение 2, как описано в настоящем документе, можно использовать для лечения, уменьшения тяжести или симптоматического лечения пациентов с диагнозом панкреатическая недостаточность, идиопатический панкреатит и врожденное билатеральное отсутствие семявыводящих протоков или заболевание легких умеренной тяжести, где пациент обладает остаточной активностью CFTR.
[00283] В одном из вариантов осуществления фармацевтические композиции и таблетки по настоящему изобретению, содержащие форму I соединения 1 и твердую дисперсию, содержащую по существу аморфное соединение 2, как описано в настоящем документе, можно использовать для лечения, уменьшения тяжести или симптоматического лечения пациентов с диагнозом панкреатическая недостаточность, идиопатический панкреатит и врожденное билатеральное отсутствие семявыводящих протоков или заболевание легких умеренной тяжести, где пациент обладает CFTR дикого типа.
[00284] В дополнение к кистозному фиброзу, модуляция активности CFTR может обеспечивать преимущество для других заболеваний, не вызванных непосредственно мутациями в CFTR, таких как секреторные заболевания и другие связанные со сворачиванием белка заболевания, опосредованные CFTR. Они включают в себя в качестве неограничивающих примеров, хроническое обструктивное заболевание легких (COPD), заболевание сухих глаз и синдром Шегрена. COPD характеризуется ограничением потока воздуха, которое является прогрессирующим и не полностью обратимым. Ограничение потока воздуха обусловлено гиперсекрецией слизи, эмфиземой и бронхиолитом. Активаторы мутантного CFTR или CFTR дикого типа предоставляют потенциальное лечение гиперсекреции слизи и нарушенного мукоцилиарного выведения, которое является общераспространенным при COPD. Конкретно, увеличение секреции анионов через CFTR может способствовать транспорту жидкости в жидкость поверхности дыхательных путей для гидратации слизи и оптимизации вязкости перицилиарной жидкости. Это может приводить к увеличенному мукоцилиарному выведению и уменьшению симптомов, ассоциированных с COPD. Заболевание сухих глаз характеризуется уменьшением продукции слезной жидкости и аномальными профилями липидов, белков и муцина в слезной пленке. Существует множество причин сухих глаз, некоторые из которых включают в себя возраст, лазерную кератопластику глаза in situ, артрит, лекарственные средства, химические/термические ожоги, аллергии и заболевания, такие как кистозный фиброз и синдром Шегрена. Увеличение секреции анионов посредством CFTR может усиливать транспорт жидкости из эндотелиальных клеток роговицы и секреторных желез, окружающих глаз, для увеличения гидратации роговицы. Это может помочь ослабить симптомы, ассоциированные с заболеванием сухих глаз. Синдром Шегрена представляет собой аутоиммунное заболевание, при котором иммунная система атакует продуцирующие жидкость железы во всем организме, включая глаза, полость рта, кожу, ткань дыхательных путей, печень, вагину и кишечник. Симптомы включают в себя сухость глаз, полости рта и вагины, а также заболевание легких. Заболевание ассоциировано также с ревматоидным артритом, системной красной волчанкой, системным склерозом и полимиозитом/дерматомиозитом. Считают, что дефектный транспорт белков вызывает это заболевание, для которого возможности лечения являются ограниченными. Усилители или индукторы активности CFTR могут вызывать гидратацию различных органов, пораженных этим заболеванием, и способствовать облегчению ассоциированных симптомов.
[00285] В одном из вариантов осуществления изобретение относится к способу усиления или индукции активности анионных каналов in vitro или in vivo, включающему в себя приведение канала в контакт с любой из фармацевтических композиций PC-I-PC-XXV. В другом варианте осуществления анионный канал представляет собой хлоридный канал или бикарбонатный канал. В другом варианте осуществления анионный канал представляет собой хлоридный канал.
[00286] Точное необходимое количество может меняться от субъекта к субъекту, в зависимости от вида, возраста и общего состояния здоровья субъекта, тяжести инфекции, конкретного средства, способа его введения, и т.п. Соединения по изобретению предпочтительно составляют в стандартной лекарственной форме для простоты введения и единообразия дозирования. Выражение «стандартная лекарственная форма», как используют в настоящем документе, относится к физически дискретной единице средства, подходящего для пациента, подлежащего лечению. Однако следует понимать, что решение об общем ежесуточном введении соединений и композиций по изобретению принимает лечащий врач в рамках медицинской оценки. Конкретный уровень эффективной дозы для любого конкретного пациента или организма зависит от множества факторов, включая нарушение, подлежащее лечению, и тяжесть нарушения; активность конкретного применяемого соединения; конкретную применяемую композицию; возраст, массу тела, общее состояние здоровья, пол и диету пациента; время введения, способ введения и скорость выведения конкретного применяемого соединения; длительность лечения; лекарственные средства, используемые в комбинации или совместимые с конкретным применяемым соединением, и подобные факторы, хорошо известные в области медицины. Термин «пациент», как используют в настоящем документе, обозначает животное, предпочтительно млекопитающее, и наиболее предпочтительно, человека.
[00287] В любом месте настоящей заявки, где наименование соединения может некорректно описывать структуру соединения, структура заменяет наименование и обладает преимуществом.
ПРИМЕРЫ
[00288] XRPD (Рентгеновская порошковая дифракция)
[00289] Данные рентгеновской порошковой дифракции (XRD) для формы I соединения 1 получали на рентгеновском порошковом дифрактометре Bruker D8 DISCOVER с 2-мерным детектором HI-STAR и плоским графитовым монохроматором. Герметично закрытую трубку с Cu с излучением Kα использовали при 40 кВ, 35 мА. Образцы помещали на силиконовые вафельные пластины с нулевым фоном при 25°C. Для каждого образца получали два пакета данных по 120 секунд каждый при 2 различных углах ϴ2: 8º и 26º. Данные интегрировали в программное обеспечение GADDS и объединяли с программным обеспечением DIFFRACTplusEVA. Неточности для зарегистрированных положений пиков составляют ± 0,2 градуса.
[00290] Дифференциальная сканирующая калориметрия (DSC)
[00291] Данные дифференциальной сканирующей калориметрии (DSC) для формы I соединения 1 получали с использованием DSC Q100 V9.6 Build 290 (TA Instruments, New Castle, DE). Температуру калибровали с помощью индия, и теплоемкость калибровали с помощью сапфира. Образцы по 3-6 мг взвешивали в алюминиевые барабаны, запрессованные с использованием крышек с 1 точечным отверстием. Образцы сканировали от 25°C до 350°C при скорости нагревания 1,0°C/мин и продували газообразным азотом при 50 мл/мин. Данные получали посредством программного обеспечения Thermal Advantage Q SeriesTM версии 2.2.0.248 и анализировали посредством программного обеспечения Universal Analysis версии 4.1D (TA Instruments, New Castle, DE). Зарегистрированные числа представляют отдельные анализы.
[00292] Определение монокристаллической структуры формы I соединения 1
[00293] Данные дифракции получали на дифрактометре Bruker Apex II, оборудованном источником Cu K-альфа в герметично закрытой трубке и детектором Apex II CCD. Структуру разрешали и уточняли с использованием программы SHELX (Sheldrick, G.M., Acta Cryst., (2008) A64, 112-122). На основании систематической статистики пропусков и интенсивности структуру разрешали и уточняли в пространственной группе P21/n.
[00294] Vitride® (гидрид натрия и бис(2-метоксиэтокси)алюминия [или NaAlH2(OCH2CH2OCH3)2], 65 масс.% раствор в толуоле) получали из Aldrich Chemicals.
[00295] 2,2-дифтор-1,3-бензодиоксол-5-карбоновую кислоту закупали из Saltigo (филиала Lanxess Corporation).
[00296] Получение соединения 1
[00297] Получение (2,2-дифтор-1,3-бензодиоксол-5-ил)метанола.
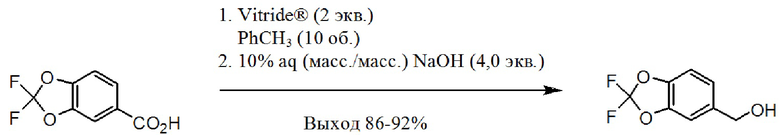
[00298] Коммерчески доступную 2,2-дифтор-1,3-бензодиоксол-5-карбоновую кислоту (1,0 экв.) суспендировали в толуоле (10 об.). Vitride® (2 экв.) добавляли через капельную воронку с такой скоростью, чтобы поддерживать температуру 15-25°C. В конце добавления температуру увеличивали до 40°C в течение 2 часов (час), затем 10% (масс./масс.) водного (aq) NaOH (4,0 экв.) осторожно добавляли через капельную воронку, поддерживая температуру при 40-50°C. После перемешивания в течение дополнительных 30 минут (мин), слоям позволяли разделяться при 40°C. Органическую фазу охлаждали до 20°C, затем промывали водой (2 × 1,5 об.), высушивали (Na2SO4), фильтровали и концентрировали для получения неочищенного (2,2-дифтор-1,3-бензодиоксол-5-ил)-метанола, который использовали непосредственно на следующей стадии.
[00299] Получение 5-хлорметил-2,2-дифтор-1,3-бензодиоксола.

[00300] (2,2-дифтор-1,3-бензодиоксол-5-ил)-метанол (1,0 экв.) растворяли в MTBE (5 об.). Добавляли каталитическое количество 4-(N,N-диметил)аминопиридина (DMAP) (1 моль%), и SOCl2 (1,2 экв.) добавляли через капельную воронку. SOCl2 добавляли с такой скоростью, чтобы поддерживать температуру в реакторе 15-25°C. Температуру увеличивали до 30°C в течение 1 час, а затем охлаждали до 20°C. Воду (4 об.) добавляли через капельную воронку при поддержании температуры менее 30°C. После перемешивания в течение дополнительных 30 мин, слоям позволяли разделяться. Органический слой перемешивали, и добавляли 10% (масс./об.) aq NaOH (4,4 об.). После перемешивания в течение 15-20 мин, слоям позволяли разделяться. Затем органическую фазу высушивали (Na2SO4), фильтровали и концентрировали для получения неочищенного 5-хлорметил-2,2-дифтор-1,3-бензодиоксола, который использовали непосредственно на следующей стадии.
[00301] Получение (2,2-дифтор-1,3-бензодиоксол-5-ил)ацетонитрила.

[00302] Раствор 5-хлорметил-2,2-дифтор-1,3-бензодиоксола (1 экв.) в DMSO (1,25 об.) добавляли к взвеси NaCN (1,4 экв.) в DMSO (3 об.), при поддержании температуры между 30 и 40°C. Смесь перемешивали в течение 1 час, а затем добавляли воду (6 об.), затем метил-трет-бутиловый простой эфир (MTBE) (4 об.). После перемешивания в течение 30 мин, слои разделяли. Водный слой экстрагировали с помощью MTBE (1,8 об.). Объединенные органические слои промывали водой (1,8 об.), высушивали (Na2SO4), фильтровали, и концентрировали для получения неочищенного (2,2-дифтор-1,3-бензодиоксол-5-ил)ацетонитрила (95%), который использовали непосредственно на следующей стадии.
[00303] Синтез (2,2-дифтор-1,3-бензодиоксол-5-ил)-1-этилацетатацетонитрила
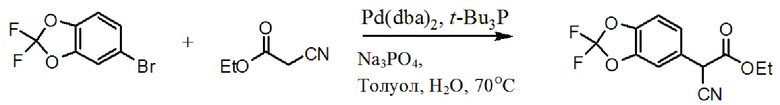
[00304] Реактор продували азотом и загружали 900 мл толуола. Растворитель дегазировали посредством орошения азотом в течение не менее 16 час. Затем в реактор загружали Na3PO4 (155,7 г, 949,5 ммоль), затем бис(дибензилиденацетон)палладий (0) (7,28 г, 12,66 ммоль). Раствор 10% масс./масс. трет-бутилфосфина в гексанах (51,23 г, 25,32 ммоль) загружали в течение 10 мин при 23°C из продутой азотом капельной воронки. Смеси позволяли перемешиваться в течение 50 мин, за это время 5-бром-2,2-дифтор-1,3-бензодиоксол (75 г, 316,5 ммоль) добавляли в течение 1 мин. После перемешивания в течение дополнительных 50 мин, в смесь загружали этилцианоацетат (71,6 г, 633,0 ммоль) в течение 5 мин, затем воду (4,5 мл) в один прием. Смесь нагревали до 70°C более 40 мин и анализировали посредством ВЭЖХ каждые 1-2 час по проценту перехода реагента в продукт. После наблюдения полного перехода (как правило, 100% перехода через 5-8 час), смесь охлаждали до 20-25°C и фильтровали через слой целита. Слой целита промывали толуолом (2 X 450 мл), и объединенные органические фазы концентрировали до 300 мл в вакууме при 60-65°C. В концентрат загружали 225 мл DMSO и концентрировали в вакууме при 70-80°C до завершения активной дистилляции растворителя. Раствор охлаждали до 20-25°C и разводили до 900 мл с помощью DMSO при подготовке для стадии 2. 1H ЯМР (500 МГц, CDCl3) δ 7,16-7,10 (m, 2H), 7,03 (d, J=8,2 Гц, 1H), 4,63 (s, 1H), 4,19 (m, 2H), 1,23 (t, J=7,1 Гц, 3H).
[00305] Синтез (2,2-дифтор-1,3-бензодиоксол-5-ил)ацетонитрила.
[00306] В раствор в DMSO (2,2-дифтор-1,3-бензодиоксол-5-ил)-1-этилацетатацетонитрила из вышеуказанного пункта загружали 3 Н HCl (617,3 мл, 1,85 моль) в течение 20 мин при поддержании внутренней температуры < 40°C. Затем смесь нагревали до 75°C более 1 час и анализировали посредством ВЭЖХ каждые 1-2 час по % перехода. После наблюдения перехода > 99% (как правило, через 5-6 час), реакционную смесь охлаждали до 20-25°C и экстрагировали MTBE (2 X 525 мл), в течение достаточного времени, чтобы позволить полное разделение фаз в ходе экстракций. Объединенные органические экстракты промывали 5% NaCl (2 X 375 мл). Затем раствор переносили в устроство, подходящее для вакуумной дистилляции при 1,5-2,5 торр (200,0-333,3 Па), оборудованное охлаждаемой колбой-приемником. Раствор концентрировали в вакууме при < 60°C для удаления растворителей. Затем (2,2-дифтор-1,3-бензодиоксол-5-ил)ацетонитрил отделяли дистилляцией от полученного в результате масла при 125-130°C (температура печи) и 1,5-2,5 торр (200,0-333,3 Па). (2,2-дифтор-1,3-бензодиоксол-5-ил)ацетонитрил отделяли в форме прозрачного масла от 5-бром-2,2-дифтор-1,3-бензодиоксола (2 стадии) с выходом 66% и с чистотой по ВЭЖХ 91,5% AUC (соответствует 95% по анализу масс./масс.). 1H ЯМР (500 МГц, DMSO) δ 7,44 (br s, 1H), 7,43 (d, J=8,4 Гц, 1H), 7,22 (dd, J=8,2, 1,8 Гц, 1H), 4,07 (s, 2H).
[00307] Получение (2,2-дифтор-1,3-бензодиоксол-5-ил)циклопропанкарбонитрила.

[00308] Смесь (2,2-дифтор-1,3-бензодиоксол-5-ил)-ацетонитрила (1,0 экв.), 50% масс. водного KOH (5,0 экв.), 1-бром-2-хлорэтана (1,5 экв.) и Oct4NBr (0,02 экв.) нагревали при 70°C в течение 1 час. Реакционную смесь охлаждали, затем доводили MTBE и водой. Органическую фазу промывали водой и насыщенным солевым раствором. Растворитель удаляли для получения (2,2-дифтор-1,3-бензодиоксол-5-ил)циклопропанкарбонитрила.
[00309] Получение 1-(2,2-дифтор-1,3-бензодиоксол-5-ил)циклопропанкарбоновой кислоты.

[00310] (2,2-дифтор-1,3-бензодиоксол-5-ил)циклопропанкарбонитрил гидролизовали с использованием 6 M NaOH (8 экв.) в этаноле (5 об.) при 80°C в течение ночи. Смесь охлаждали до комнатной температуры, и этанол выпаривали в вакууме. Остаток поглощали водой и MTBE, добавляли 1 M HCl, и слои разделяли. Затем слой MTBE обрабатывали дициклогексиламином (DCHA) (0,97 экв.). Взвесь охлаждали до 0°C, фильтровали и промывали гептаном для получения соответствующей соли DCHA. Соль поглощали MTBE и 10% лимонной кислотой и перемешивали до растворения всех твердых веществ. Слои разделяли, и слой MTBE промывали водой и насыщенным солевым раствором. Растворитель заменяли на гептан с последующей фильтрацией для получения 1-(2,2-дифтор-1,3-бензодиоксол-5-ил)циклопропанкарбоновой кислоты после сушки в вакуумной печи при 50°C в течение ночи.
[00311] Получение 1-(2,2-дифтор-1,3-бензодиоксол-5-ил)-циклопропанкарбонилхлорида.
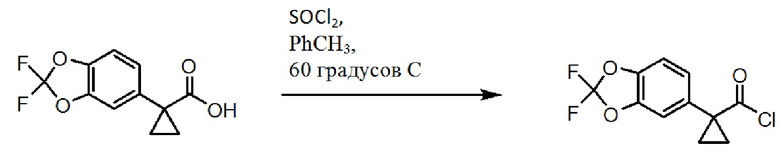
[00312] 1-(2,2-дифтор-1,3-бензодиоксол-5-ил)циклопропанкарбоновую кислоту (1,2 экв.) суспендировали в толуоле (2,5 об.), и смесь нагревали до 60°C. SOCl2 (1,4 экв.) добавляли через капельную воронку. Толуол и SOCl2 отделяли дистилляцией от реакционной смеси через 30 минут. Добавляли дополнительный толуол (2,5 об.), и полученную смесь снова подвергали дистилляции, оставляя продукт хлорангидрид в форме масла, которое использовали без дополнительной очистки.
[00313] Получение трет-бутил-3-(3-метилпиридин-2-ил)бензоата.
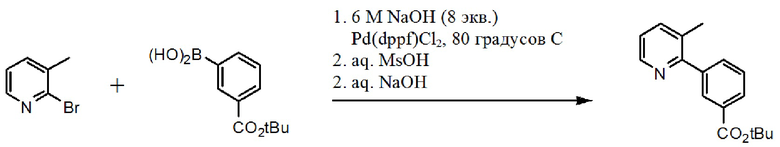
[00314] 2-бром-3-метилпиридин (1,0 экв.) растворяли в толуоле (12 об.). Добавляли K2CO3 (4,8 экв.), затем воду (3,5 об.). Полученную смесь нагревали до 65°C под струей N2 в течение 1 часа. Затем добавляли 3-(т-бутоксикарбонил)фенилбороновую кислоту (1,05 экв.) и Pd(dppf)Cl2•CH2Cl2 (0,015 экв.), и смесь нагревали до 80°C. Через 2 часа нагрев отключали, добавляли воду (3,5 об.), и слоям позволяли разделяться. Органическую фазу затем промывали водой (3,5 об.) и экстрагировали 10% водной метансульфоновой кислотой (2 экв. MsOH, 7,7 об.). Водную фазу делали более основной с помощью 50% водного NaOH (2 экв.) и экстрагировали EtOAc (8 об.). Органический слой концентрировали для получения неочищенного трет-бутил-3-(3-метилпиридин-2-ил)бензоата (82%), который использовали непосредственно на следующей стадии.
[00315] Получение 2-(3-(трет-бутоксикарбонил)фенил)-3-метилпиридин-1-оксида.

[00316] Трет-бутил-3-(3-метилпиридин-2-ил)бензоат (1,0 экв.) растворяли в EtOAc (6 об.). Добавляли воду (0,3 об.), затем мочевину-пероксид водорода (3 экв.). Фталевый ангидрид (3 экв.) затем добавляли в смесь порционно в виде твердого вещества с такой скоростью, чтобы поддерживать температуру в реакторе ниже 45°C. После завершения добавления фталевого ангидрида, смесь нагревали до 45 °C. После перемешивания в течение дополнительных 4 часов, нагрев отключали. 10% масс./масс. водного Na2SO3 (1,5 экв.) добавляли через капельную воронку. После завершения добавления Na2SO3, смесь перемешивали в течение дополнительных 30 мин, и слои разделяли. Органический слой перемешивали, и добавляли 10% масс./масс. водн. Na2CO3 (2 экв.). После перемешивания в течение 30 минут, слоям позволяли разделяться. Органическую фазу промывали 13% масс./об. aq NaCl. Органическую фазу затем фильтровали и концентрировали для получения неочищенного 2-(3-(трет-бутоксикарбонил)фенил)-3-метилпиридин-1-оксида (95%), который использовали непосредственно на следующей стадии.
[00317] Получение трет-бутил-3-(6-амино-3-метилпиридин-2-ил)бензоата.
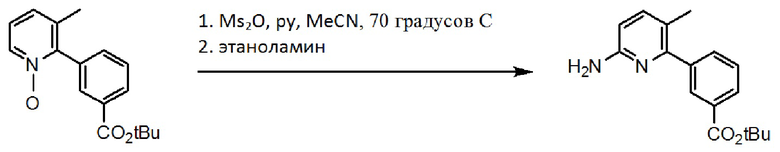
[00318] Раствор 2-(3-(трет-бутоксикарбонил)фенил)-3-метилпиридин-1-оксида (1 экв.) и пиридина (4 экв.) в ацетонитриле (8 об.) нагревали до 70°C. Раствор метансульфонового ангидрида (1,5 экв.) в MeCN (2 об.) добавляли в течение 50 мин через капельную воронку при поддержании температуры менее 75°C. Смесь перемешивали в течение дополнительных 0,5 часов после завершения добавления. Затем смеси позволяли охлаждаться до окружающей температуры. Этаноламин (10 экв.) добавляли через капельную воронку. После перемешивания в течение 2 часов, добавляли воду (6 об.), и смесь охлаждали 10°C. После перемешивания в течение 3 часов, твердое вещество собирали фильтрацией и промывали водой (3 об.), 2:1 ацетонитрилом/водой (3 об.) и ацетонитрилом (2×1,5 об.). Твердое вещество сушили до постоянной массы (различие <1%) в вакуумной печи при 50°C со слабым выпуском N2 для получения трет-бутил-3-(6-амино-3-метилпиридин-2-ил)бензоата в форме красно-желтого твердого вещества (выход 53%).
[00319] Получение 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамидо)-3-метилпиридин-2-ил)-трет-бутилбензоата.
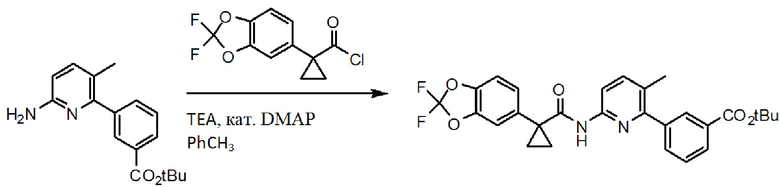
[00320] Неочищенный хлорангидрид, описанный выше, растворяли в толуоле (2,5 об. на основании хлорангидрида) и добавляли через капельную воронку к смеси трет-бутил-3-(6-амино-3-метилпиридин-2-ил)бензоата (1 экв.), DMAP, (0,02 экв.) и триэтиламина (3,0 экв.) в толуоле (4 об. на основании трет-бутил-3-(6-амино-3-метилпиридин-2-ил)бензоата). Через 2 часа, воду (4 об. на основании трет-бутил-3-(6-амино-3-метилпиридин-2-ил)бензоата) добавляли в реакционную смесь. После перемешивания в течение 30 минут, слои разделяли. Органическую фазу затем фильтровали и концентрировали для получения густого масла из 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамидо)-3-метилпиридин-2-ил)-трет-бутилбензоата (количественный выход неочищенного продукта). Добавляли ацетонитрил (3 об. на основании неочищенного продукта) и проводили дистилляцию до начала кристаллизации. Добавляли воду (2 об. на основании неочищенного продукта), и смесь перемешивали в течение 2 час. Твердое вещество собирали фильтрацией, промывали 1:1 (по объему) ацетонитрила/воды (2 × 1 объемов на основании неочищенного продукта), и частично высушивали на фильтре в вакууме. Твердое вещество сушили до постоянной массы (различие <1%) в вакуумной печи при 60°C со слабым выпуском N2 для получения 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамидо)-3-метилпиридин-2-ил)-трет-бутилбензоата в форме коричневого твердого вещества.
[00321] Получение соли 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамидо)-3-метилпиридин-2-ил)бензойной кислоты • HCl.
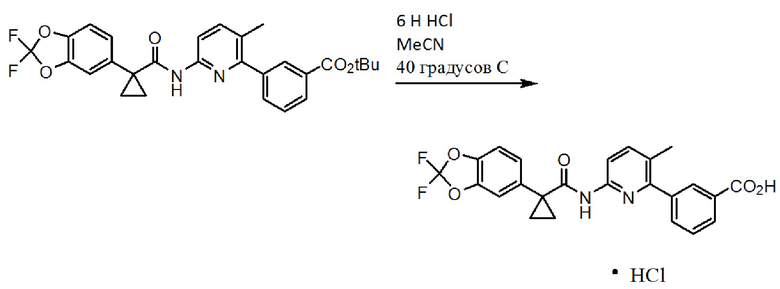
[00322] К взвеси 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамидо)-3-метилпиридин-2-ил)-трет-бутилбензоата (1,0 экв.) в MeCN (3,0 об.) добавляли воду (0,83 об.), затем концентрированную водную HCl (0,83 об.). Смесь нагревали до 45±5°C. После перемешивания в течение 24-48 час, реакцию завершали, и смеси позволяли охлаждаться до окружающей температуры. Добавляли воду (1,33 об.), и смесь перемешивали. Твердое вещество собирали фильтрацией, промывали водой (2 × 0,3 об.) и частично высушивали на фильтре в вакууме. Твердое вещество сушили до постоянной массы (различие <1%) в вакуумной печи при 60°C со слабым выпуском N2 для получения 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамидо)-3-метилпиридин-2-ил)бензойной кислоты • HCl в форме грязно-белого твердого вещества.
[00323] Спектр 1HЯМР соединения 1 изображен на фигуре 8, и на фигуре 9 изображен спектр 1HЯМР соединения 1 в форме соли с HCl.
[00324] В таблице 2 ниже приведены данные 1HNMR для соединения I.
M+1
минут
[00325] Получение формы I соединения 1
[00326] Получение формы i соединения 1, способ A.
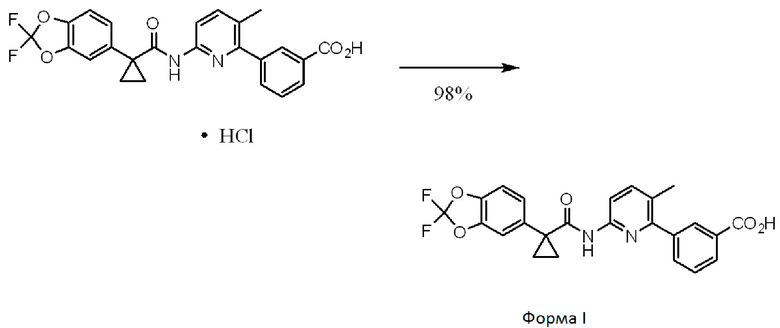
[00327] Взвесь 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил) циклопропанкарбоксамидо)-3-метилпиридин-2-ил)бензойной кислоты • HCl (1 экв.) в воде (10 об.) перемешивали при температуре окружающей среды. Образец отбирали после перемешивания в течение 24 час. Образец фильтровали, и твердое вещество промывали водой (2 раза). Твердый образец подвергали анализу DSC. Когда анализ DSC показывал полный переход в форму I, твердое вещество собирали фильтрацией, промывали водой (2 × 1,0 об.), и частично высушивали на фильтре в вакууме. Затем твердое вещество сушили до постоянной массы (различие <1%) в вакуумной печи при 60°C со слабым выпуском N2 для получения формы I соединения 1 в форме грязно-белого твердого вещества (выход 98%). 1H ЯМР (400 МГц, DMSO-d6) 9,14 (s, 1H), 7,99-7,93 (m, 3H), 7,80-7,78 (m, 1H), 7,74-7,72 (m, 1H), 7,60-7,55 (m, 2H), 7,41-7,33 (m, 2H), 2,24 (s, 3H), 1,53-1,51 (m, 2H), 1,19-1,17 (m, 2H).
[00328] Получение формы i соединения 1, способ B.
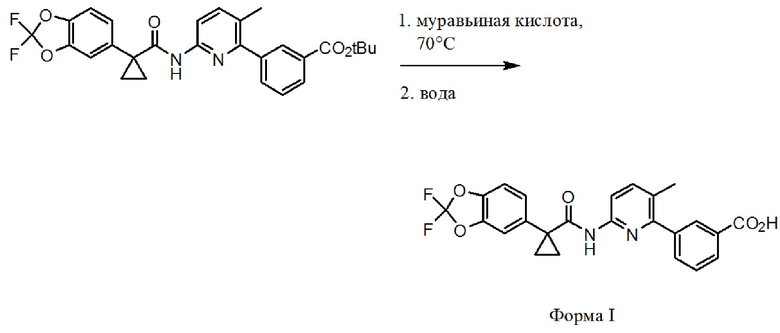
[00329] Раствор 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил) циклопропанкарбоксамидо)-3-метилпиридин-2-ил)-трет-бутилбензоата (1,0 экв.) в муравьиной кислоте (3,0 об.) нагревали при перемешивании до 70±10 °C, в течение 8 час. Реакцию считали завершенной, когда оставалось не более 1,0% AUC по хроматографическим способам 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамидо)-3-метилпиридин-2-ил)-трет-бутилбензоата. Смеси позволяли охлаждаться до окружающей температуры. Раствор добавляли в воду (6 об.), нагревали до 50°C, и смесь перемешивали. Затем смесь нагревали до 70±10°C, пока уровень 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамидо)-3-метилпиридин-2-ил)-трет-бутилбензоата не становился не более 0,8% (AUC). Твердое вещество собирали фильтрацией, промывали водой (2 × 3 об.), и частично высушивали на фильтре в вакууме. Твердое вещество сушили до постоянной массы (различие <1%) в вакуумной печи при 60°C со слабым выпуском N2 для получения формы I соединения 1 в форме грязно-белого твердого вещества.
[00330] Запись DSC формы I соединения 1 показана на фигуре 10. Плавление формы I соединения 1 происходит при приблизительно 204°C.
[00331] Картина рентгенодифракции рассчитана для монокристаллической структуры формы I соединения 1 и показана на фигуре 1. В таблице 3 перечислены рассчитанные пики для фигуры 1.
[00332] Фактическая картина порошковой рентгенодифракции формы I соединения 1 показана на фигуре 2. В таблице 4 перечислены фактические пики для фигуры 2.
[00333] Бесцветные кристаллы формы I соединения 1 получали посредством охлаждения концентрированного раствора в 1-бутаноле от 75°C до 10°C со скоростью 0,2 °C/мин. Кристаллы с измерениями 0,50 × 0,08 × 0,03 мм выбирали, очищали с помощью минерального масла, устанавливали в MicroMount и центрировали в системе Bruker APEX II. Получали три партии по 40 рамок, разделенных в обратном пространстве, для получения матрицы ориентации и начальных параметров ячейки. Конечные параметры ячейки получали и уточняли на основании полного набора данных.
[00334] Набор данных дифракции в обратном пространстве получали с разрешением 0,82 Å с использованием шагов по 0,5° с использованием экспозиции 30 сек для каждой рамки. Данные получали при 100 (2) K. Интеграцию интенсивностей и уточнение параметров ячейки проводили с использованием программного обеспечения APEXII. Обследование кристалла после получения данных не показало признаков разрушения.
[00335] Изображение конформации формы I соединения 1 на основании монокристаллического рентгенографического анализа показано на фигуре 11. Форма I соединения 1 является моноклинической, P21/n, со следующими измерениями единичной ячейки: a=4,9626(7) Å, b=12,299(2) Å, c=33,075 (4) Å, β=93,938(9)°, V=2014,0 Å3, Z=4. Плотность формы I соединения 1, рассчитанная по структурным данным, составляет 1,492 г/см3 при 100 K.
[00336] Получение соединения 2
[00337] Синтез 4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты (26)
[00338] Способ получения этил-4-оксо-1,4-дигидрохинолин-3-карбоксилата (25)
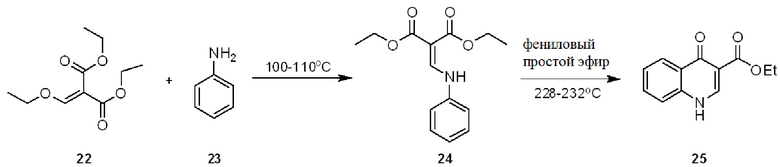
[00339] Соединение 23 (4,77 г, 47,7 ммоль) добавляли капельно к соединению 22 (10 г, 46,3 ммоль) c подповерхностным потоком N2 для вытеснения этанола при ниже 30°C в течение 0,5 часов. Затем раствор нагревали до 100-110°C и перемешивали в течение 2,5 часов. После охлаждения смеси до ниже 60°C, добавляли дифениловый простой эфир. Полученный раствор добавляли капельно к дифениловому простому эфиру, нагретому до 228-232°C, в течение 1,5 часов c подповерхностным потоком N2 для вытеснения этанола. Смесь перемешивали при 228-232°C в течение следующих 2 часов, охлаждали ниже 100°C, а затем гептан добавляли для преципитации продукта. Полученную взвесь перемешивали при 30°C в течение 0,5 часов. Затем твердые вещества фильтровали, и осадок промывали гептаном и высушивали в вакууме с получением соединения 25 в форме коричневого твердого вещества. 1H ЯМР (DMSO-d6; 400 МГц) δ 12,25 (s), δ 8,49 (d), δ 8,10 (m), δ 7,64 (m), δ 7,55 (m), δ 7,34 (m), δ 4,16 (q), δ 1,23 (t).
[00340] Способ получения 4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты (26)
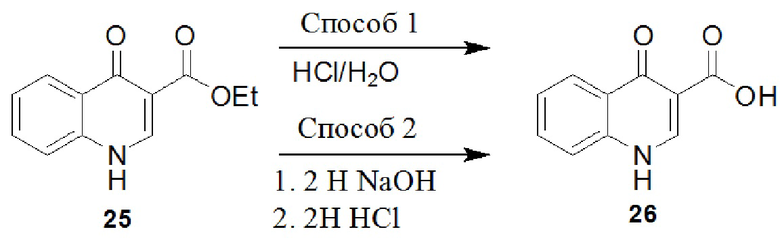
[00341] Соединение 25 (1,0 экв.) суспендировали в растворе HCl (10,0 экв.) и H2O (11,6 об.). Взвесь нагревали до 85-90°C, хотя альтернативные температуры также являются подходящими для этой стадии гидролиза. Например, гидролиз можно альтернативно проводить при температуре от приблизительно 75 до приблизительно 100°C. В некоторых случаях, гидролиз проводят при температуре от приблизительно 80 до приблизительно 95°C. В других случаях, стадию гидролиза проводят при температуре от приблизительно 82 до приблизительно 93°C (например, от приблизительно 82,5 до приблизительно 92,5°C или от приблизительно 86 до приблизительно 89 °C). После перемешивания при 85-90°C в течение приблизительно 6,5 часов, отбирали образец реакционной смеси для анализа завершения реакции. Перемешивание можно проводить при любой из температур, подходящих для гидролиза. Затем раствор охлаждали до 20-25°C и фильтровали. Реактор/осадок промывали H2O (2 об. x 2). Затем осадок промывали 2 об. H2O до pH≥3,0. Затем осадок высушивали в вакууме при 60°C для получения соединения 26.
Способ 2
[00342] Соединение 25 (11,3 г, 52 ммоль) добавляли в смесь 10% NaOH (aq) (10 мл) и этанола (100 мл). Раствор нагревали при кипячении с обратным холодильником в течение 16 часов, охлаждали до 20-25°C, а затем pH доводили до 2-3 с помощью 8% HCl. Затем смесь перемешивали в течение 0,5 часов и фильтровали. Осадок промывали водой (50 мл), а затем высушивали в вакууме с получением соединения 26 в форме коричневого твердого вещества. 1H ЯМР (DMSO-d6; 400 МГц) δ 15,33 (s), δ 13,39 (s), δ 8,87 (s), δ 8,26 (m), δ 7,87 (m), δ 7,80 (m), δ 7,56 (m).
[00343] Общий синтез N-(2,4-ди-трет-бутил-5-гидроксифенил)-4-оксо-1,4-дигидрохинолин-3-карбоксамида (соединения 2)
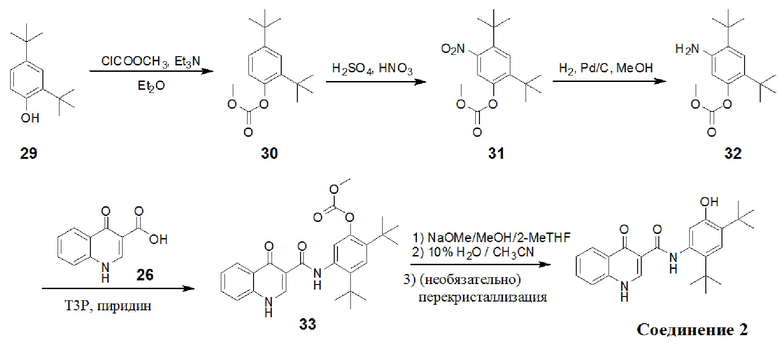
[00344] Способ получения 2,4-ди-трет-бутилфенилметилкарбоната (30)
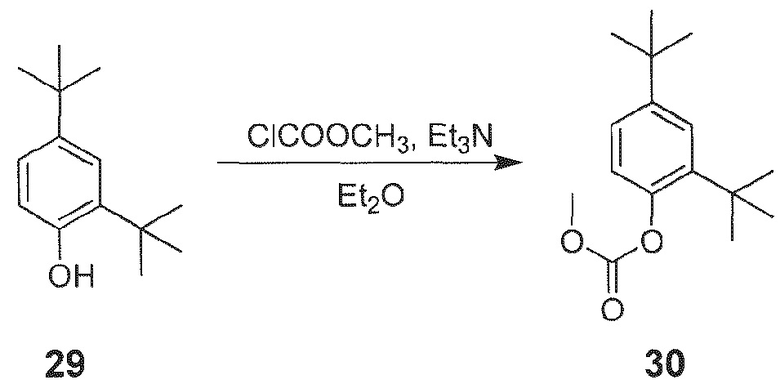
Способ 1
[00345] К раствору 2,4-ди-трет-бутил фенола, 29, (10 г, 48,5 ммоль) в простом диэтиловом эфире (100 мл) и триэтиламине (10,1 мл, 72,8 ммоль), добавляли метилхлорформиат (7,46 мл, 97 ммоль) капельно при 0°C. Затем смеси позволяли нагреться до комнатной температуры и перемешивали в течение дополнительных 2 часов. Затем добавляли дополнительно 5 мл триэтиламина и 3,7 мл метилхлорформиата, и реакционную смесь перемешивали в течение ночи. Затем реакционную смесь фильтровали, фильтрат охлаждали до 0°C, и затем добавляли дополнительно 5 мл триэтиламина и 3,7 мл метилхлорформиата, и реакционной смеси позволяли нагреться до комнатной температуры, а затем перемешивали в течение дополнительного 1 часа. На этой стадии реакция являлась почти завершенной, и реакционную смесь обрабатывали посредством фильтрации, затем промывки водой (2x), затем насыщенным солевым раствором. Затем раствор концентрировали до получения желтого масла и очищали с использованием хроматографии на колонке для получения соединения 30. 1H ЯМР (400 МГц, DMSO-d6) δ 7,35 (d, J=2,4 Гц, 1H), 7,29 (dd, J=8,4, 2,4 Гц, 1H), 7,06 (d, J=8,4 Гц, 1H), 3,85 (s, 3H), 1,30 (s, 9H), 1,29 (s, 9H).
Способ 2
[00346] В реакционный сосуд, загруженный 4-диметиламинопиридином (DMAP, 3,16 г, 25,7 ммоль) и 2,4-дитрет-бутилфенолом (соединение 29, 103,5 г, 501,6 ммоль), добавляли метиленхлорид (415 г, 313 мл), и раствор перемешивали до растворения всех твердых веществ. Затем добавляли триэтиламин (76 г, 751 ммоль) и раствор охлаждали до 0-5°C. Затем метилхлорформиат (52 г, 550,3 ммоль) добавляли капельно в течение 2,5-4 часов, при поддержании температуры раствора между 0 и 5°C. Затем реакционную смесь медленно нагревали до 23-28°C и перемешивали в течение 20 часов. Затем реакционную смесь охлаждали до 10-15°C и загружали 150 мл воды. Смесь перемешивали при 15-20°C в течение 35-45 минут, и затем водный слой отделяли и экстрагировали 150 мл метиленхлорида. Органические слои объединяли и нейтрализовывали с помощью 2,5% HCl (aq) при температуре 5-20°C для получения конечного pH 5-6. Органический слой затем промывали водой и концентрировали в вакууме при температуре ниже 20°C до 150 мл для получения соединение 30 в метиленхлориде.
[00347] Способ получения 5-нитро-2,4-ди-трет-бутилфенилметилкарбоната (31)
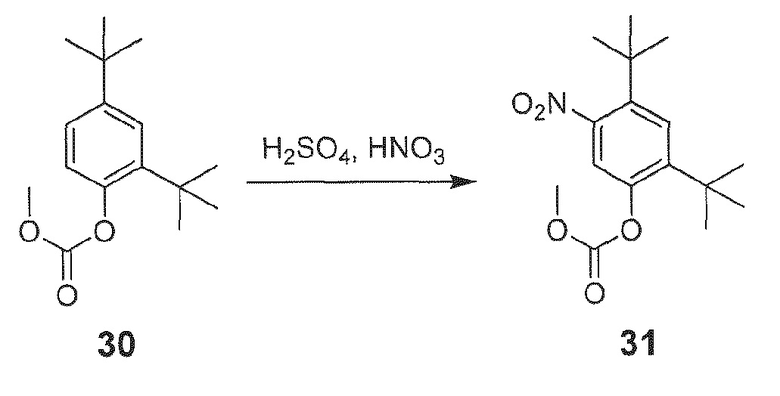
Способ 1
[00348] К перемешанному раствору соединения 30 (6,77 г, 25,6 ммоль) добавляли 6 мл смеси 1:1 серной кислоты и азотной кислоты при 0°C капельно. Смеси позволяли нагреться до комнатной температуры и перемешивали в течение 1 часа. Продукт очищали с использованием жидкостной хроматографии (ISCO, 120 г, 0-7% EtOAc/гексаны, 38 мин), получая смесь приблизительно 8:1-10:1 региоизомеров соединения 31 в форме белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ 7,63 (s, 1H), 7,56 (s, 1H), 3,87 (s, 3H), 1,36 (s, 9H), 1,32 (s, 9H). ВЭЖХ ret. time 3,92 мин 10-99% CH3CN, 5 мин run; ESI-MS 310 масса/заряд (MH)+.
Способ 2
[00349] К соединению 30 (100 г, 378 ммоль) добавляли DCM (540 г, 408 мл). Смесь перемешивали до растворения всех твердых веществ, а затем охлаждали до -5-0°C. Концентрированную серную кислоту (163 г) затем добавляли капельно, при поддержании начальной температуры реакционной смеси, и смесь перемешивали в течение 4,5 часов. Азотную кислоту (62 г) затем добавляли капельно в течение 2-4 часов при поддержании начальной температуры реакционной смеси, и затем перемешивали при этой температуре в течение дополнительных 4,5 часов. Реакционную смесь затем медленно добавляли в холодную воду, поддерживая температуру ниже 5°C. Смесь после остановки реакции затем нагревали до 25°C, и водный слой удаляли и экстрагировали метиленхлоридом. Объединенные органические слои промывали водой, высушивали с использованием Na2SO4, и концентрировали до 124-155 мл. Добавляли гексан (48 г), и полученную смесь снова концентрировали до 124-155 мл. Дополнительный гексан (160 г) затем добавляли в смесь. Затем смесь перемешивали при 23-27°C в течение 15,5 часов, и затем фильтровали. К осадку на фильтре добавляли гексан (115 г), Полученную смесь нагревали при кипячении с обратным холодильником и перемешивали в течение 2-2,5 часов. Смесь затем охлаждали до 3-7°C, перемешивали в течение дополнительных 1-1,5 часов и фильтровали для получения соединения 31 в форме бледно-желтого твердого вещества.
[00350] Способ получения 5-амино-2,4-ди-трет-бутилфенилметилкарбоната (32)
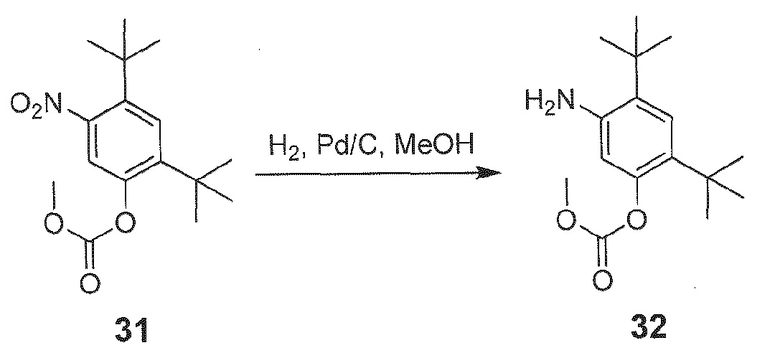
[00351] 2,4-ди-трет-бутил-5-нитрофенилметилкарбонат (1,00 экв.) загружали в подходящий реактор для гидрогенизации, затем 5% Pd/C (2,50% масс. на сухое вещество, Johnson-Matthey тип 37). MeOH (15,0 об.) загружали в реактор, и систему закрывали. Систему продували N2 (г), и затем подвергали давлению 2,0 бар (2,0 × 105 Па) с помощью H2 (г). Реакцию проводили при температуре реакции 25°C+/-5°C. После завершения реакции, реакционную смесь фильтровали, и реактор/осадок промывали MeOH (4,00 об.). Полученный фильтрат подвергали дистилляции в вакууме при не более, чем 50°C до 8,00 об. Воду (2,00 об.) добавляли при 45°C +/- 5 °C. Полученную взвесь охлаждали до 0°C +/- 5. Взвесь поддерживали при 0°C +/- 5°C в течение не менее 1 час, и фильтровали. Осадок промывали один раз 0°C +/- 5°C MeOH/H2O (8:2) (2,00 об.). Осадок сушили в вакууме (-0,90 бар (-0,90×105 Па) и -0,86 бар (-0,86×105 Па)) при 35 °C-40°C для получения соединения 32. 1H ЯМР (400 МГц, DMSO-d6) δ 7,05 (s, 1H), 6,39 (s, 1H), 4,80 (s, 2H), 3,82 (s, 3H), 1,33 (s, 9H), 1,23 (s, 9H).
[00352] После завершения реакции, полученную смесь разбавляли с помощью приблизительно от 5 до 10 объемов MeOH (например, от приблизительно 6 до приблизительно 9 объемов MeOH, от приблизительно 7 до приблизительно 8,5 объемов MeOH, от приблизительно 7,5 до приблизительно 8 объемов MeOH, или приблизительно 7,7 объемов MeOH), нагревали до температуры приблизительно 35±5°C, фильтровали, промывали и высушивали, как описано выше.
[00353] Получение N-(2,4-ди-трет-бутил-5-гидроксифенил)-4-оксо-1,4-дигидрохинолин-3-карбоксамида (соединения 2).
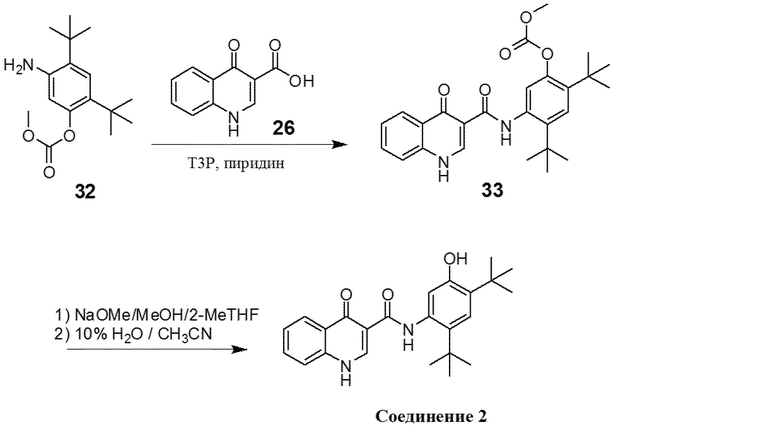
[00354] 4-оксо-1,4-дигидрохинолин-3-карбоновую кислоту, 26, (1,0 экв.) и 5-амино-2,4-ди-трет-бутилфенилметилкарбонат, 32, (1,1 экв.) загружали в реактор. Добавляли 2-MeTHF (4,0 об., относительно кислоты), затем 50% раствор T3P® в 2-MeTHF (1,7 экв.). Загруженный T3P сосуд промывали с помощью 2-MeTHF (0,6 об.). Затем добавляли пиридин (2,0 экв.), и полученную суспензию нагревали до 47,5 +/- 5,0°C и поддерживали при этой температуре в течение 8 часов. Образец отбирали и проверяли по завершению реакции посредством ВЭЖХ. После завершения реакции, полученную смесь охлаждали до 25,0°C +/- 2,5°C. Добавляли 2-MeTHF (12,5 об.) для разведения смеси. Реакционную смесь промывали водой (10,0 об.) 2 раза. 2-MeTHF добавляли для доведения общего объема реакционной смеси до 40,0 об. (~16,5 об. загружали). К этому раствору добавляли NaOMe/MeOH (1,7 экв.) для проведения метанолиза. Реакционную смесь перемешивали в течение не менее 1,0 час, и проверяли по завершению реакции посредством ВЭЖХ. После завершения, реакцию останавливали с помощью 1 Н HCl (10,0 об.), и промывали с помощью 0,1 Н HCl (10,0 об.). Органический раствор подвергали окончательной фильтрации для удаления всех частиц и помещали во второй реактор. Фильтрованный раствор концентрировали при не более, чем 35°C (температура рубашки) и не менее чем 8,0°C (внутренняя температура реакционной смеси) при пониженном давлении до 20 об. CH3CN добавляли до 40 об. и раствор концентрировали при не более, чем 35°C (температура рубашки) и не менее, чем 8,0°C (внутренняя температура реакционной смеси) до 20 об. Цикл добавления CH3CN и концентрации повторяли еще 2 раза всего за 3 добавления CH3CN и 4 концентрации до 20 об. После конечной концентрации до 20 об., добавляли 16,0 об. CH3CN, затем 4,0 об. H2O для получения конечной концентрации 40 об. 10% H2O/CH3CN относительно исходной кислоты. Эту взвесь нагревали до 78,0°C +/- 5,0°C (кипячение с обратным холодильником). Затем взвесь перемешивали в течение не менее 5 часов. Взвесь охлаждали до 0,0°C +/- 5°C в течение 5 часов и фильтровали. Осадок промывали с помощью 0,0°C +/- 5,0°C CH3CN (5 об.) 4 раза. Полученное твердое вещество (соединение 2) сушили в вакуумной печи при 50,0°C +/- 5,0 °C. 1H ЯМР (400 МГц, DMSO-d6) δ 12,8 (s, 1H), 11,8 (s, 1H), 9,2 (s, 1H), 8,9 (s, 1H), 8,3 (s, 1H), 7,2 (s, 1H), 7,9 (t, 1H), 7,8 (d, 1H), 7,5 (t, 1H), 7,1 (s, 1H), 1,4 (s, 9H), 1,4 (s, 9H).
[00355] Альтернативное получение N-(2,4-ди-трет-бутил-5-гидроксифенил)-4-оксо-1,4-дигидрохинолин-3-карбоксамида (соединения 2).
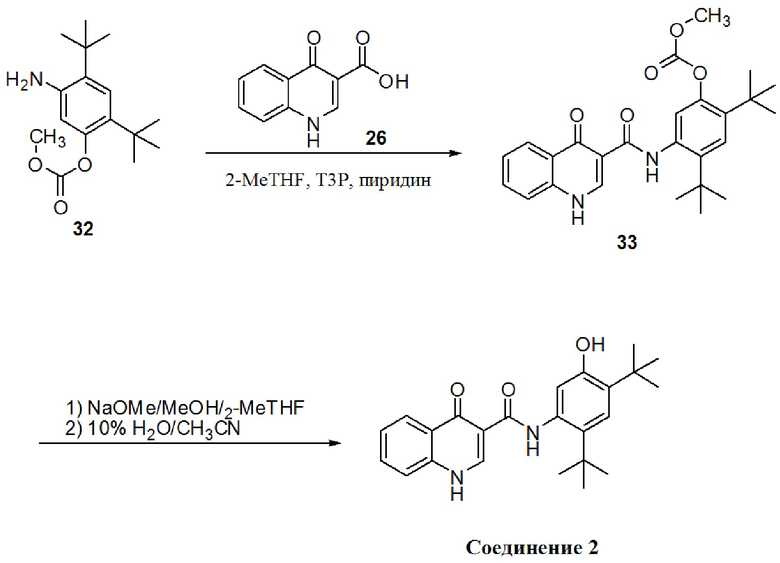
[00356] 4-оксо-1,4-дигидрохинолин-3-карбоновую кислоту, 26, (1,0 экв.) и 5-амино-2,4-ди-трет-бутилфенилметилкарбонат, 32, (1,1 экв.) загружали в реактор. Добавляли 2-MeTHF (4,0 об., относительно кислоты), затем 50% раствор T3P® в 2-MeTHF (1,7 экв.). Загруженный T3P сосуд промывали 2-MeTHF (0,6 об.). Затем добавляли пиридин (2,0 экв.), и полученную суспензию нагревали до 47,5 +/- 5,0°C и поддерживали при этой температуре в течение 8 часов. Образец отбирали и проверяли по завершению реакции посредством ВЭЖХ. После завершения реакции, полученную смесь охлаждали до 20°C +/- 5°C. Добавляли 2-MeTHF (12,5 об.) для разведения смеси. Реакционную смесь промывали водой (10,0 об.) 2 раза, и 2-MeTHF (16,5 об.) загружали в реактор. В этот раствор загружали 30% масс./масс. NaOMe/MeOH (1,7 экв.) для проведения метанолиза. Реакционную смесь перемешивали при 25,0°C +/- 5,0°C в течение не менее, чем 1,0 час, и проверяли по завершению реакции посредством ВЭЖХ. После завершения, реакцию останавливали с помощью 1,2 N HCl/H2O (10,0 об.), и промывали с помощью 0,1 Н HCl/H2O (10,0 об.). Органический раствор подвергали конечной фильтрации для удаления всех частиц и помещали во второй реактор.
[00357] Фильтрованный раствор концентрировали при не более, чем 35°C (температура рубашки) и не менее, чем 8,0°C (внутренняя температура реакционной смеси) при пониженном давлении до 20 об. CH3CN добавляли до 40 об., и раствор концентрировали при не более, чем 35°C (температура рубашки) и не менее, чем 8,0°C (внутренняя температура реакционной смеси) до 20 об. Цикл добавления CH3CN и концентрации повторяли еще 2 раза всего за 3 добавления CH3CN и 4 концентрации до 20 об. После конечной концентрации до 20 об., загружали 16,0 об. CH3CN, затем 4,0 об. H2O для доведения конечной концентрации 40 об. до 10% H2O/CH3CN относительно исходной кислоты. Эту взвесь нагревали до 78,0°C +/- 5,0°C (кипячение с обратным холодильником). Затем взвесь перемешивали в течение не менее 5 часов. Взвесь охлаждали до 20-25°C в течение 5 часов, и фильтровали. Осадок промывали с помощью CH3CN (5 об.), нагретого до 20-25°C 4 раза. Полученное твердое вещество (соединение 2) сушили в вакуумной печи при 50,0°C +/- 5,0 °C. 1H ЯМР (400 МГц, DMSO-d6) δ 12,8 (s, 1H), 11,8 (s, 1H), 9,2 (s, 1H), 8,9 (s, 1H), 8,3 (s, 1H), 7,2 (s, 1H), 7,9 (t, 1H), 7,8 (d, 1H), 7,5 (t, 1H), 7,1 (s, 1H), 1,4 (s, 9H), 1,4 (s, 9H).
[00358] Способ перекристаллизации N-(2,4-ди-трет-бутил-5-гидроксифенил)-4-оксо-1,4-дигидрохинолин-3-карбоксамида (соединения 2)
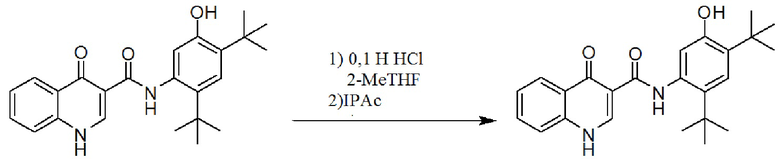
[00359] Соединение 2 (1,0 экв.) загружали в реактор. Добавляли 2-MeTHF (20,0 об.), затем 0,1 Н HCl (5,0 об.). Двухфазный раствор перемешивали и разделяли, и верхнюю органическую фазу дважды промывали дополнительно 0,1 Н HCl (5,0 об.). Органический раствор подвергали конечной фильтрации для удаления всех частиц и помещали во второй реактор. Фильтрованный раствор концентрировали при не более, чем 35°C (температура рубашки) и не более, чем 8,0°C (внутренняя температура реакционной смеси) при пониженном давлении до 10 об. Добавляли изопропилацетат (IPAc) (10 об.), и раствор концентрировали при не более, чем 35°C (температура рубашки) и не более, чем 8,0°C (внутренняя температура реакционной смеси) до 10 об. Добавление IPAc и концентрацию повторяли еще 2 раза всего за 3 добавления IPAc и 4 концентрации до 10 об. После конечной концентрации, загружали 10 об. IPAc, и взвесь нагревали до кипения с обратным холодильником и поддерживали при этой температуре в течение 5 часов. Взвесь охлаждали до 0,0°C +/- 5°C в течение 5 часов и фильтровали. Осадок промывали с помощью IPAc (5 об.) один раз. Полученное твердое вещество сушили в вакуумной печи при 50,0°C +/- 5,0 °C.
[00360] Получение твердой дисперсии, содержащей по существу аморфное соединение 2
[00361] Систему растворителя из MEK и DI воды составляли в соответствии с соотношением 90% масс. MEK/10% масс. DI вода, нагревали до температура 20-30°C в реакторе, оборудованном магнитной мешалкой и теплообменником. В эту систему растворителей добавляли полимер сукцинат ацетата гипромеллозы (HPMCAS)(квалификации HG), SLS, и соединение 2 в соответствии с соотношением 19,5% масс. сукцината ацетата гипромеллозы/0,5% масс. SLS/80% масс. соединения 2. Полученная смесь содержала 10,5% масс. твердых веществ. Фактические количества ингредиентов и растворителей, использованные для получения этой смеси, перечислены в таблице 5 ниже:
Ингредиенты твердой дисперсии для промежуточного соединения F
[00362] Температуру смеси доводили до диапазона 20-45°C и перемешивали, пока смесь не становилась по существу гомогенной, и все компоненты по существу не растворялись.
[00363] Сушилку для сушки распылением, коммерческую сушилку для сушки распылением Niro PSD4, оснащенную нагнетательным соплом (Spray Systems Maximum Passage серии SK-MFP, обладающую размером выходного отверстия/сердцевины 54/21), оборудованную крышкой против подтеков, использовали в нормальном режиме сушки распылением, следуя параметрам процесса сушки распылением, перечисленным в таблице 6 ниже.
Параметры способа сушки распылением, использованного для получения промежуточного соединения F
[00364] Высокоэффективный циклон отделял влажный продукт от газа для распыления и паров растворителя. Влажный продукт содержал 8,5-9,7% MEK и 0,56-0,83% воды, и обладал средним размером частиц 17-19 мкм и объемной плотностью 0,27-0,33 г/см3. Влажный продукт переносили в 4000 л двухконусную вакуумную сушилку из нержавеющей стали для сушки, для уменьшения количества остаточных растворителей до уровня менее приблизительно 5000 м.д. и для получения сухой высушенной распылением дисперсии аморфного соединения 2, содержащей <0,03% MEK и 0,3% воды.
[00365] Получение таблеток способом полностью непрерывной влажной грануляция
[00366] Оборудование/Способ
[00367] Оборудование
[00368] Устройство для полностью непрерывной переработки и выпуска (DLR) или оборудование сходного типа.
[00369] Просеивание
[00370] Форму I соединения 1, твердую дисперсию, содержащую по существу аморфное соединение 2, и наполнители можно распределять по отдельным промежуточным бункерным контейнерам (IBC). Эти материалы можно просеивать с использованием способа просеивания «из бункера в бункер». Подходящие размеры сит составляют меш 20, меш 40, или меш 60.
[00371] Смешивание
[00372] IBC, содержащие просеянные форму I соединения 1, твердую дисперсию, содержащую по существу аморфное соединение 2, и наполнители, можно пристыковывать к системе подачи, которая может подавать материалы контролируемым образом, например, с использованием волюметрических устройств для подачи или гравиметрических устройств для подачи с потерей массы, в непрерывный смеситель. Скорости подачи индивидуальных компонентов определяются составом композиции и общей линейной скоростью. Линейная скорость может составлять от 8 кг/час до 30 кг/час. Непрерывный смеситель может обладать различной конфигурацией лезвий, чтобы обеспечивать подходящее смешивание, и скорость вращения этих лезвий может составлять между 80 об./мин. и 300 об./мин.
[00373] Влажная грануляция
[00374] Раствор для грануляции можно получать посредством растворения 48 г лаурилсульфата натрия и 159 г поливинилпирролидона в 1626 г воды в контейнере из нержавеющей стали, с использованием верхнеприводной мешалки со скоростью перемешивания 700 об./мин. Раствор для грануляции можно помещать в контейнер, из которого раствор можно перекачивать в двухшнековый гранулятор с использованием перистальтического насоса с массовым расходомером и контролем, с использованием скорости потока, подходящей для способа. Смесь можно подвергать грануляции с использованием двухшнекового гранулятора, такого как гранулятор, являющийся частью DLR. Смесь можно добавлять в двухшнековый гранулятор с использованием устройства для подачи с потерей массы, такого как устройство для подачи K-Tron в DLR, со скоростью подачи от 8 кг/час до 24 кг/час. Двухшнековый гранулятор можно эксплуатировать с температурой барабана 25 градусов Цельсия и скоростью вращения шнека от 200 до 950 об./мин. Процесс грануляции можно проводить в течение трех минут для малых размеров партий или нескольких часов для больших размеров партий.
[00375] Сушка
[00376] Влажные гранулы можно подавать непосредственно в сушилку с псевдоожиженным слоем, такую как как сегментрованная сушилка с псевдоожиженным слоем в DLR. Конечную точку сушки можно выбирать при температуре продукта в ходе выгрузки в диапазоне от 40 до 55 градусов Цельсия, в точке, в которой содержание воды в гранулах может составлять 2,1% масс./масс. («потери при сушке, LOD») или менее. Время сушки может составлять 12 минут или короче, или длиннее, для достижения желательной конечной точки сушки.
[00377] Размол
[00378] Высушенные гранулы можно размалывать для уменьшения размера гранул. Конусную мельницу, такую как интегрированная Quadro U10 CoMil, можно использовать для этого.
[00379] Смешивание
[00380] Гранулы можно смешивать с экстрагранулярными наполнителями, такими как наполнители и смазочное средство, с использованием устройств для подачи с потерей массы и непрерывного смесителя. Скорость смешивания может составлять 80-300 об./мин.
[00381] Прессование
[00382] Смесь для прессования можно прессовать в таблетки с использованием отдельного или ротационного таблетировочного пресса, такого как пресс Courtoy Modul P, являющийся частью системы DLR, с использованием инструментов соответствующего размера. Масса таблеток для дозы 200 мг формы I соединения 1 и 125 мг по существу аморфного соединения 2 может составлять приблизительно 500 или 600 мг.
[00383] Покрытие пленкой
[00384] Таблетки можно покрывать оболочкой с использованием инновационного устройства для нанесения покрытий Omega film, являющегося частью системы DLR. Это устройство для нанесения покрытий позволяет быстрое покрытие пленкой подпартий от 1 до 4 кг для обеспечения непрерывного производства.
[00385] Печать
[00386] На покрытых оболочкой таблетках можно печатать монограмму на одной или обеих сторонах таблетки, например, с помощью наклонного принтера Ackley.
[00387] Непрерывный процесс, описанный выше в одном из вариантов осуществления, улучшают посредством технологий PAT, как описано в таблице 7. Существует 6 положений PВ, каждое из которых включает порт для ручного отбора образцов. Образцы можно получать в процессе производства для исследовательских целей, по мере необходимости, а также для поддержания, переноса и валидации модели PAT. Системы PAT можно использовать для тестирования выхода в режиме реального времени (RTRT) и также можно использовать для контроля в процессе (IPC) и контроля обратной/прямой связи.
[00388] Соответствие спецификациям можно осуществлять посредством RTRT, как описано в таблице 8.
[00389] Существует высокая вероятность детекции не соответствующего техническим требованиям материала. Например, если модельный критерий классификации устанавливают на минимум 95% соответствие, и 800 таблеток тестируют в ходе изготовления партии, 40 час функционирования со скоростью отбора образцов 1 таблетка каждые 3 минуты эквивалентны 800 таблеткам. Тогда вероятность пропуска несоответствующей партии является необычайно низкой: <(0,05)n-, где n=# образцов, таким образом, вероятность составляет <1,5×10-1041. Вероятность отсутствия детекции несоответствующих таблеток, возникающих в результате краткосрочного события (≥3 минут), является следующей: 1 таблетка (3-мин событие)→<0,05 (вероятность детекции >0,95); 2 таблетки (6-минутное событие)→<0,0025 (вероятность детекции >0,9975).
[00390] Измерения PAT могут служить заменой общепринятому конечному тестированию непосредственно путем комбинации измерений для выражения характеристик общепринятым образом (т.е. в виде анализа, CU, растворения и т.д.). Валидацию можно проводить с использованием ICH Q2 в качестве руководства. Последовательное развитие способа от способа вне процесса производства до способа в процессе производства позволяет оценку CQA экономным по отношению к материалу образом. В конечном итоге, RTRT может приводить к обеспечению качества продукта на более высоком уровне достоверности, чем общепринятое тестирование.
[00391] Получение таблеток способом двухшнековой влажной грануляции
[00392] Оборудование/Способ
[00393] Оборудование
[00394] Двухшнековые влажные грануляторы: ConsiGma-1, ConsiGma-25 или Leistritz nano.
[00395] Просеивание/Взвешивание
[00396] Форму I соединения 1, твердую дисперсию, содержащую по существу аморфное соединение 2, и наполнители можно просеивать до или после взвешивания. Подходящие размеры сит составляют 20 меш, 40 меш или 60 меш. Форму I соединения 1 и/или твердую дисперсию, содержащую по существу аморфное соединение 2, можно предварительно смешивать с одним или несколькими из наполнителей для упрощения просеивания.
[00397] Смешивание
[00398] Форму I соединения 1, твердую дисперсию, содержащую по существу аморфное соединение 2, и наполнители можно добавлять в смеситель в различном порядке. Смешивание можно проводить в смесителе Turbula, V-образном смесителе или в бункерном смесителе. Компоненты можно смешивать в течение 10 минут.
[00399] Влажная грануляция
[00400] Раствор для грануляции можно получать посредством растворения 48 г лаурилсульфата натрия и 159 г поливинилпирролидона в 1626 г воды в контейнере из нержавеющей стали, с использованием верхнеприводной мешалки со скоростью перемешивания 700 об./мин. Смесь можно подвергать грануляции с использованием двухшнекового гранулятора, такого как ConsiGma-1. Раствор для грануляции можно подавать в двухшнековый гранулятор с использованием перистальтического насоса, такого насос в ConsiGma-1, со скоростью подачи 67 г/мин. Смесь можно добавлять в двухшнековый гранулятор с использованием устройства для подачи с потерей массы, такого как устройство для подачи Brabender в ConsiGma-1, со скоростью подачи 10 кг/час. Двухшнековый гранулятор можно эксплуатировать с температурой барабана 25 градусов Цельсия и скоростью вращения шнека 400 об./мин. Процесс грануляции можно проводить в течение четырех минут. Процесс грануляции можно проводить в течение более короткого или более длительного времени для получения меньшего или большего количества влажных гранул.
[00401] Сушка
[00402] Влажные гранулы можно подавать непосредственно в сушилку с псевдоожиженным слоем, такую как как сушильная камера в ConsiGma-1 или сегментрованная сушилка с псевдоожиженным слоем в CTL-25. Конечную точку сушки можно выбирать при температуре продукта 43 градуса Цельсия, в точке, в которой содержание воды в гранулах может составлять 1,6% масс./масс. («потери при сушке, LOD»). Время сушки может составлять 12 минут или короче, или длиннее, для достижения желательной конечной точки сушки. Сушку можно проводить с помощью потока воздуха 59 м3/мин и температуры на входе 60 градусов Цельсия. Альтернативно, влажные гранулы, выходящие из двухшнекового гранулятора, можно собирать в бак или контейнер в течение определенного периода времени, после которого влажные гранулы переносят в отдельную сушилку с псевдоожиженным слоем, такую как Вектор Multi 15.
[00403] Размол
[00404] Высушенные гранулы можно размалывать для уменьшения размера гранул. Конусную мельницу, такую как Quadro 194 CoMil можно использовать для этого.
[00405] Смешивание
[00406] Гранулы можно смешивать с экстрагранулярными наполнителями, такими как наполнители и смазочное средство, с использованием V-образного смесителя или бункерного смесителя. Время смешивания может составлять 5, 3 или 1 минуту(минут).
[00407] Прессование
[00408] Смесь для прессования можно прессовать в таблетки с использованием отдельного или ротационного таблетировочного пресса, такого как пресс Courtoy Modul P, с использованием инструментов овальной формы 0,55'×0,33'. Масса таблеток для дозы 200 мг формы I соединения 1 и 125 мг по существу аморфного соединения 2 может составлять приблизительно 500 или 600 мг
[00409] Покрытие пленкой
[00410] Таблетки можно покрывать оболочкой с использованием бункерного устройства для нанесения покрытий, например, такого как устройство для нанесения покрытий Thomas Engineering Compu-Lab. Следовые количества карнаубского воска можно добавлять для улучшения внешнего вида таблеток и характеристик процесса.
[00411] Печать
[00412] На покрытых оболочкой таблетках можно печатать монограмму на одной или обеих сторонах таблетки, например, с помощью принтера Hartnett Delta.
[00413] Получение таблеток способом непрерывной двухшнековой влажной грануляции
[00414] Оборудование/Способ
[00415] Оборудование
Гранулятор: двухшнековый гранулятор ConsiGma или Leistritz, или Thermo Fisher.
[00416] Просеивание/Взвешивание
[00417] Соединение 1 и наполнители можно просеивать до или после взвешивания. Возможные размеры сит составляют 20 меш, 40 меш или 60 меш. Соединение 1 можно предварительно смешивать с одним или несколькими из наполнителей для упрощения просеивания.
[00418] Смешивание
[00419] Соединение 1 и наполнители можно добавлять в смеситель в различном порядке. Смешивание можно проводить в смесителе Turbula, V-образном смесителе, бункерном смесителе или непрерывном смесителе. Компоненты можно смешивать в течение 10 минут в случае смесителей периодического действия или непрерывно в случае непрерывного смесителя.
[00420] Операция грануляции
Жидкость для грануляции - SLS и связывающее средство добавляют в очищенную воду и перемешивают до растворения. Подходящее соотношение составляет 2,5% масс./масс. SLS и 10,0% масс./масс. PVP K30 в воде.
Грануляция - Смесь, содержащую соединение 1 и наполнители, можно дозированно подавать в двухшнековый гранулятор с использованием устройства для подачи с потерей массы со скоростью 10 кг/час. Жидкость для грануляции можно подавать с использованием перистальтического насоса со скоростью 3,5 кг/час. Гранулятор можно эксплуатировать со скоростью 400 об./мин. Заметным преимуществом настоящего способа двухшнековой влажной грануляции является использование жидкости для грануляции, содержащей как поверхностно-активное вещество, так и связывающее средство, для лучшей грануляции посредством увеличенной смачиваемости. В одном из вариантов осуществления поверхностно-активное вещество представляет собой SLS. Другим заметным преимуществом является то, что поскольку процесс является непрерывным и в любой момент времени перерабатывается только ограниченное количество материала, процесс можно хорошо контролировать, и он приводит в результате к продукту высокого качества.
[00421] Размол
[00422] Размер гранул можно уменьшать с использованием мельницы с ситами или конусной мельницы, либо до сушки, либо после сушки, либо в обоих случаях.
[00423] Сушка
[00424] Гранулы можно сушить с использованием вакуумной печи, полочной сушилки, двухконусной сушилки или сушилки с псевдоожиженным слоем.
[00425] Смешивание
[00426] Гранулы можно смешивать с экстрагранулярными наполнителями. Гранулы смешивают с использованием 300-литрового бункерного смесителя в течение 60 оборотов.
[00427] Прессование
[00428] Смесь для прессования прессуют в таблетки с использованием ротационного пресса Courtoy Modul P
[00429] Покрытие пленкой
[00430] Таблетки можно покрывать оболочкой с использованием бункерного устройства для нанесения покрытий, например, такого как O'Hara Labcoat.
[00431] Печать
[00432] На покрытых оболочкой таблетках можно печатать монограмму на одной или обеих сторонах таблетки, например, с помощью принтера Hartnett Delta.
АНАЛИЗЫ
[00433] ПРOТОКОЛ 1
[00434] Анализы для детекции и измерения свойств соединений как усилителей ΔF508-CFTR
Оптические способы измерения мембранного потенциала для анализа модулирующих ΔF508-CFTR свойств соединений
[00435] В анализе используют флуоресцентные потенциалочувствительные красители для измерения изменений в мембранном потенциале с использованием флуоресцентного планшетного спектрофотометра (например, FLIPR III, Molecular Devices, Inc.) в качестве считывания показаний увеличения функционального ΔF508-CFTR в клетках NIH 3T3. Движущей силой для этого ответа является создание градиента хлоридных ионов вместе с активацией каналов посредством единственной стадии добавления жидкости, после того как эти клетки были предварительно обработаны соединениями и затем нагружены потенциалочувствительным красителем.
Идентификация соединений-усилителей
[00436] Для идентификации усилителей ΔF508-CFTR, разработан формат анализа HTS с двойным добавлением. В этом анализе HTS используют флуоресцентные потенциалочувствительные красители для измерения изменений в мембранном потенциале на FLIPR III в качестве измерения для увеличения пропускания (проводимости) ΔF508 CFTR в клетках ΔF508 CFTR NIH 3T3 после коррекции температуры. Движущей силой для этого ответа является градиент ионов Cl- в комбинации с активацией каналов форсколином в единственной стадии добавления жидкости с использованием флуоресцентного планшетного спектрофотометра, такого как FLIPR III после того как эти клетки были предварительно обработаны соединениями-усилителями (или контрольным носителем DMSO) и затем нагружены перераспределяющим красителем.
Растворы
[00437] Раствор бани #1: (в мМ) NaCl 160, KCl 4,5, CaCl2 2, MgCl2 1, HEPES 10, pH 7,4 с NaOH
[00438] Не содержащий хлорида раствор бани: хлоридные соли в растворе бани #1 (выше) заменены глюконатными солями.
Культура клеток
[00439] Для оптических измерений мембранного потенциала используют фибробласты мыши NIH 3T3, стабильно экспрессирующие ΔF508-CFTR. Клетки поддерживают при 37 ºC в 5% CO2 и при влажности 90% в модифицированной Дульбекко среде Игла, дополненной 2 мМ глутамином, 10% эмбриональной телячьей сывороткой, 1 X NEAA, β-ME, 1 X пенициллином/стрептомицином и 25 мМ HEPES, в флаконах для культивирования 175 см2. Для всех оптических анализов, клетки высевают при ~20000/лунку в 384-луночные покрытые матригелем планшеты и культивируют в течение 2 час при 37°С перед культивированием при 27°C в течение 24 час. для анализа усилителя. Для анализов коррекции, клетки культивируют при 27°С или 37°С в присутствии и в отсутствие соединения в течение 16-24 часов.
[00440] Электрофизиологические анализы для исследования модулирующих ΔF508-CFTR свойств соединений.
Анализ в камере Уссинга
[00441] Эксперименты в камере Уссинга проводили на поляризованных эпителиальных клетках дыхательных путей, экспрессирующих ΔF508-CFTR, для дополнительной характеризации усилителей или индукторов ΔF508-CFTR, идентифицированных в оптических анализах. Эпителии дыхательных путей без CF и с CF выделяли из бронхиальной ткани, культивируемой, как описано ранее (Galietta, L.J.V., Lantero, S., Gazzolo, A., Saсм3o, O., Romano, L., Rossi, G.A., & Zegarra-Moran, O. (1998) In Vitro Cell. Dev. Biol. 34, 478-481), и высевали на фильтры Costar® SnapwellTM, которые предварительно покрывали кондиционированной NIH 3T3 средой. Через четверо суток апикальную среду удаляли, и клетки выращивали на поверхности раздела воздух-жидкость в течение >14 суток перед использованием. Это приводило к монослою полностью дифференцированных столбчатых клеток, которые являлись реснитчатыми, что является характерным признаком эпителия дыхательных путей. HBE без CF выделяли от некурящих субъектов, которые не страдали никаким известным заболеванием легких. CF-HBE выделяли от пациентов, гомозиготных по ΔF508.
[00442] HBE, выращенные на вставках для культивирования клеток Costar® Snapwell™, фиксировали в камере Уссинга (Physiologic Instruments, Inc., San Diego, CA), и трансэпителиальное сопротивление и ток короткого замыкания в присутствии базолатерального - апикального градиента Cl- (ISC) измеряли с использованием системы фиксации напряжения (Department of Bioengineering, University of Iowa, IA). Кратко, HBE исследовали в условиях регистрации с фиксацией напряжения (Vhold=0 мВ) при 37 oC. Базолатеральный раствор содержал (в мМ) 145 NaCl, 0,83 K2HPO4, 3,3 KH2PO4, 1,2 MgCl2, 1,2 CaCl2, 10 глюкозу, 10 HEPES (pH доводили до 7,35 с помощью NaOH), и апикальный раствор содержал (в мМ) 145 глюконат Na, 1,2 MgCl2, 1,2 CaCl2, 10 глюкозу, 10 HEPES (pH доводили до 7,35 с помощью NaOH).
Идентификация соединений-усилителей
[00443] В типичном протоколе использовали градиент концентрации Cl- от базолатеральной к апикальной мембране. Для установки этого градиента использовали обычные кольцеобразователи на базолатеральной мембране, в то время как апикальный NaCl заменяли эквимолярным глюконатом натрия (титрованным до pH 7,4 с помощью NaOH) для получения большого градиента концентрации Cl- поперек эпителия. Форсколин (10 мкМ) и все тестируемые соединения добавляли на апикальную сторону вставок для культивирования клеток. Эффективность предположительных усилителей ΔF508-CFTR сравнивали с эффективностью известного усилителя, генистеина.
Регистрация с фиксацией потенциала
[00444] Общий ток Cl- в клетках ∆F508-NIH 3T3 мониторировали с использованием регистрации конфигурации с перфорацией и фиксацией потенциала, как описано ранее (Rae, J., Cooper, K., Gates, P., & Watsky, M. (1991) J. Neurosci. Methods 37, 15-26). Регистрацию с фиксацией напряжения проводили при 22 ºC с использованием усилителя с фиксацией потенциала Axopatch 200B (Axon Instruments Inc., Foster City, CA). Раствор пипетки содержал (в мМ) 150 N-метил-D-глюкамин (NMDG)-Cl, 2 MgCl2, 2 CaCl2, 10 EGTA, 10 HEPES и 240 мкг/мл амфотерицина-B (pH доводили до 7,35 с помощью HCl). Внеклеточная среда содержала (в мМ) 150 NMDG-Cl, 2 MgCl2, 2 CaCl2, 10 HEPES (pH доводили до 7,35 с помощью HCl). Генерирование импульсов, получение и анализ данных проводили с использованием PC, оборудованного интерфейсом Digidata 1320 A/D в комбинации с Clampex 8 (Axon Instruments Inc.). Для активации ΔF508-CFTR, 10 мкМ форсколин и 20 мкМ генистеин добавляли в баню, и соотношение ток-напряжение мониторировали каждые 30 сек.
Идентификация соединений-усилителей
[00445] Способность усилителей ΔF508-CFTR увеличивать макроскопический ток Cl- для ΔF508-CFTR (IΔF508) NIH 3T3, стабильно экспрессирующих ΔF508-CFTR, исследовали также с использованием способов с перфорацией и фиксацией потенциала. Усилители, идентифицированные из оптических анализов, индуцировали зависимое от дозы увеличение IΔF508 с активностью и эффективностью, сходными с наблюдаемыми в оптических анализах. Во всех исследованных клетках, обратный потенциал до и во время введения усилителя составлял приблизительно -30 мВ, что представляет собой рассчитанный ECl (-28 мВ).
Культура клеток
[00446] Для регистраций цельных клеток используют фибробласты NIH 3T3 мыши, стабильно экспрессирующие ΔF508-CFTR. Клетки поддерживали при 37 ºC в 5% CO2 и при влажности 90% в модифицированной Дульбекко среде Игла, дополненной 2 мМ глутамином, 10% эмбриональной телячьей сывороткой, 1 X NEAA, β-ME, 1 X пенициллином/стрептомицином и 25 мМ HEPES, в флаконах для культивирования 175 см2. Для регистрации цельных клеток, 2500-5000 клеток высевали на покрытые поли-L-лизином стеклянные покровные стекла и культивировали в течение 24-48 час при 27°C перед использованием для тестирования активности усилителей; и инкубировали в присутствии или в отсутствие корректирующего соединения при 37°C для измерения активности корректоров.
Регистрации отдельных каналов
[00447] Активность пропускания wt-CFTR и корректируемого температурой ∆F508-CFTR, экспрессируемого в клетках NIH 3T3 наблюдали с использованием регистраций вырезанных фрагментов вывернутой наружу мембраны, как описано ранее (Dalemans, W., Barbry, P., Champigny, G., Jallat, S., Dott, K., Dreyer, D., Crystal, R.G., Pavirani, A., Lecocq, J-P., Lazdunski, M. (1991) Nature 354, 526-528) Axopatch 200B (Axon Instruments Inc.). Пипетка содержала (в мМ): 150 NMDG, 150 аспарагиновой кислоты, 5 CaCl2, 2 MgCl2 и 10 HEPES (pH доводили до 7,35 с помощью Трис-основания). Баня содержала (в мМ): 150 NMDG-Cl, 2 MgCl2, 5 EGTA, 10 TES и 14 Трис-основания (pH доводили до 7,35 с помощью HCl). После вырезания, как wt-, так и ∆F508-CFTR, активировали добавлением 1 мМ Мg-АТФ, 75 нМ каталитической субъединицы зависимой от цАМФ протеинкиназы (PKA; Promega Corp. Madison, WI) и 10 мМ NaF для ингибирования протеинфосфатаз, которые предотвращали уменьшение тока. Потенциал пипетки поддерживали при 80 мВ. Активность канала анализировали по фрагментам мембраны, содержащим ≤2 активных каналов. Максимальное количество одновременных открываний определяло количество активных каналов в хода эксперимента. Для определения амплитуды тока одного канала, эти данные, зарегистрированные из 120-секундной активности ΔF508-CFTR, фильтровали «в автономном режиме» при 100 Гц, а затем использовали для построения гистограмм амплитуд для всех точек, которые приводили в соответствие с мультигауссовыми функциями с использованием программного обеспечения Bio-Patch Analysis (Bio-Logic Comp. France). Общий микроскопический ток и вероятность открытия (Po) определяли по 120 сек активности каналов. Po определяли с использованием программного обеспечения Bio-Patch или из отношения Po=I/i(N), где I=средний ток, i=амплитуда тока одного канала и N=количество активных каналов в фрагменте.
Культура клеток
[00448] Для регистраций вырезанных мембранных фрагментов с фиксацией потенциала используют фибробласты мыши NIH 3T3, стабильно экспрессирующие ΔF508-CFTR. Клетки поддерживали при 37ºC в 5% CO2 и при влажности 90% в модифицированной Дульбекко среде Игла, дополненной 2 мМ глутамиом, 10% эмбриональной телячьей сывороткой, 1 X NEAA, β-ME, 1 X пенициллином/стрептомицином и 25 мМ HEPES, в флаконах для культивирования 175 см2. Для регистрации отдельных каналов, 2500-5000 клеток высевали на покрытые поли-L-лизином стеклянные покровные стекла и культивировали в течение 24-48 час при 27°C перед использованием.
[00449] ПРOТОКОЛ 2
[00450] Анализы для детекции и измерения свойств соединений как корректоров ΔF508-CFTR
[00451] Оптические способы измерения мембранного потенциала для анализа модулирующих ΔF508-CFTR свойств соединений
[00452] В оптическом анализе мембранного потенциала используют чувствительные к напряжению сенсоры FRET, описанные в Gonzalez and Tsien (См. Gonzalez, J. E. and R. Y. Tsien (1995) «Voltage sensing by fluorescence resonance energy transfer in single cells» Biophys J 69(4): 1272-80, и Gonzalez, J. E. and R. Y. Tsien (1997) «Improved indicators of cell membrane potential that use fluorescence resonance energy transfer» Chem Biol 4(4): 269-77) в комбинации с устройствами для измерения изменений флуоресценции, таких как считыватель с вольтометровым/ионным зондом (VIPR) (См., Gonzalez, J. E., K. Oades, et al. (1999) «Cell-based assays and instrumentation for screening ion-channel targets» Drug Discov Discov Today 4(9): 431-439).
[00453] Эти чувствительные к напряжению анализы основаны на изменении резонансного переноса энергии флуоресценции (FRET) между растворимым в мембране, чувствительным к напряжению красителем, DiSBAC2(3), и флуоресцентным фосфолипидом, CC2-DMPE, который прикреплен к внешнему слою плазматической мембраны и действует как донор FRET. Изменения мембранного потенциала (Vm) заставляют отрицательно заряженный DiSBAC2(3) перераспределяться в плазматической мембране, и количество переноса энергии от СС2-DMPE изменяется соответственно. Изменения флуоресценции мониторировали с использованием VIPRTM II, который представляет собой объединенные жидкостный манипулятор и флуоресцентный детектор, разработанный для проведения скринингов на основе клеток в 96- или 384-луночных планшетах для микротитрования.
Идентификация соединений-корректоров
[00454] Для идентификации низкомолекулярных соединений, корректирующих дефект транспорта, ассоциированный с ΔF508-CFTR; разработан формат анализа HTS с одним добавлением. Клетки инкубировали в бессывороточной среде в течение 16 час при 37°C в присутствии или в отсутствие (отрицательный контроль) тестируемого соединения. В качестве положительного контроля, клетки, рассеянные в 384-луночные планшеты, инкубировали в течение 16 час при 27°C для «температурной коррекции» ΔF508-CFTR. Затем клетки промывали 3X раствором Кребса-Рингера и нагружали потенциалзависимыми красителями. Для активации ΔF508-CFTR, 10 мкМ форсколин и усилитель CFTR, генистеин (20 мкМ), добавляли вместе со свободной от Cl- средой в каждую лунку. Добавление свободной от Cl- среды стимулировало отток Cl- в ответ на активацию ΔF508-CFTR, и полученную деполляризацию мембраны оптически мониторировали с использованием чувствительных к напряжению красителей на основе FRET.
Идентификация соединений-усилителей
[00455] Для идентификации усилителей ΔF508-CFTR, разработан формат анализа HTS с двойным добавлением. В ходе первого добавления, свободную от Cl- среду в присутствии или в отсутствие тестируемого соединения добавляли в каждую лунку. Через 22 сек, второе добавление свободной от Cl- среды, содержащей 2-10 мкМ форсколин, добавляли для активации ΔF508-CFTR. Внеклеточная концентрация Cl- после обоих добавлений составляла 28 мМ, что стимулировало отток Cl- в ответ на активацию ΔF508-CFTR и полученную деполляризацию мембраны оптически мониторировали с использованием чувствительных к напряжению красителей на основе FRET.
Растворы
[00456]
[00457]
[00458]
[00459]
Культура клеток
[00460] Для оптических измерений мембранного потенциала используют фибробласты мыши NIH 3T3, стабильно экспрессирующие ΔF508-CFTR. Клетки поддерживают при 37 ºC в 5% CO2 и при влажности 90% в модифицированной Дульбекко среде Игла, дополненной 2 мМ глутамином, 10% эмбриональной телячьей сывороткой, 1 X NEAA, β-ME, 1 X пенициллином/стрептомицином и 25 мМ HEPES, в флаконах для культивирования 175 см2. Для всех оптических анализов, клетки высевают при 30000/лунку в 384-луночные покрытые матригелем планшеты и культивируют в течение 2 час при 37°C перед культивированием при 27°C в течение 24 час для анализа усилителя. Для анализов коррекции, клетки культивируют при 27°C или 37°C в присутствии и в отсутствие соединения в течение 16-24 часов.
[00461] Электрофизиологические анализы для исследования модулирующих ΔF508-CFTR свойств соединений
Анализ в камере Уссинга
[00462] Эксперименты в камере Уссинга проводили на поляризованных эпителиальных клетках, экспрессирующих ΔF508-CFTR, для дополнительной характеризации усилителей или индукторов ΔF508-CFTR, идентифицированных в оптических анализах. Эпителиальные клетки FRTΔF508-CFTR, выращенные на вставках для культивирования клеток Costar Snapwell, фиксировали в камере Уссинга (Physiologic Instruments, Inc., San Diego, CA), и монослои непрерывно подвергали воздействию короткого замыкания с использованием системы фиксации напряжения (Department of Bioengineering, University of Iowa, IA, и Physiologic Instruments, Inc., San Diego, CA). Трансэпителиальное сопротивление измеряли посредством приложения импульса 2-мВ. В этих условиях, для эпителия FRT показано сопротивление 4 KΩ/см2 или более. Растворы поддерживали при 27 ºC и продували воздухом. Смещение потенциалов и сопротивление в жидкости электродов корректировали с использованием бесклеточной вставки. В этих условиях, ток отражает поток Cl- через ΔF508-CFTR, экспрессированный на апикальной мембране. ISC получали в цифровой форме с использованием интерфейса MP100A-CE и программного обеспечения AcqKnowledge (v3.2.6; BIOPAC Systems, Santa Barbara, CA).
Идентификация соединений-корректоров
[00463] В типичном протоколе использовали градиент концентрации Cl- от базолатеральной к апикальной мембране. Для установки этого градиента использовали обычные кольцеобразователи на базолатеральной мембране, в то время как апикальный NaCl заменяли эквимолярным глюконатом натрия (титрованным до pH 7,4 с помощью NaOH) для получения большого градиента концентрации Cl- концентрация поперек эпителия. Все эксперименты проводили с интактными монослоями. Для полной активации ΔF508-CFTR, форсколин (10 мкМ) и ингибитор PDE, IBMX (100 мкМ), вводили с последующим добавлением усилителя CFTR, генистеина (50 мкМ).
[00464] Как наблюдали в других типах клеток, инкубация при низких температурах клеток FRT, стабильно экспрессирующих ΔF508-CFTR, увеличивает функциональную плотность CFTR в плазматической мембране. Для определения активности соединений-корректоров, клетки инкубировали с 10 мкМ тестируемого соединения в течение 24 часов при 37°C и затем промывали 3X перед регистрацией. Опосредованный ЦАМФ и генистеином ISC в обработанных соединением клетках нормализовали по контролю при 27°C и 37°C и выражали как процент активности. Предварительная инкубация клеток с соединением-корректором значительно увеличивала опосредованный цАМФ и генистеином ISC по сравнению с контролем при 37°C.
Идентификация соединений-усилителей
[00465] В типичном протоколе использовали градиент концентрации Cl- от базолатеральной к апикальной мембране. Для установки этого градиента использовали обычные кольцеобразователи на базолатеральной мембране и проводили пермеабилизацию с помощью нистатина (360 мкг/мл), в то время как апикальный NaCl заменяли эквимолярным глюконатом натрия (титрованным до pH 7,4 с помощью NaOH) для получения большого градиента концентрации Cl- поперек эпителия. Все эксперименты проводили через 30 мин после пермеабилизации нистатином. Форсколин (10 мкМ) и все тестируемые соединения добавляли на обе стороны вставок для культивирования клеток. Эффективность предположительных усилителей ΔF508-CFTR сравнивали с эффективностью известного усилителя, генистеина.
Растворы
[00466]
[00467]
Культура клеток
[00468] Эпителиальные клетки крыс Fisher (FRT), экспрессирующие ΔF508-CFTR (FRTΔF508-CFTR), использовали для экспериментов в камере Уссинга для предположительных усилителей или индукторов ΔF508-CFTR, идентифицированных в оптических анализах. Клетки культивировали на вставках для культивирования клеток Costar Snapwell, и культивировали в течение пяти суток при 37°C и 5% CO2 в модифицированной Куном среде Хэма F-12, дополненной 5% эмбриональной телячьей сывороткой, 100 Ед/мл пенициллина и 100 мкг/мл стрептомицина. Перед использованием для характеризации активности соединений-усилителей, клетки инкубировали при 27°C в течение 16-48 час для коррекции ΔF508-CFTR. Для определения активности соединений-корректоров, клетки инкубировали при 27°C или 37°C в присутствии и в отсутствие соединения в течение 24 часов.
Регистрация цельных клеток
[00469] Макроскопический ток ΔF508-CFTR (IΔF508) в корректированных с помощью температуры и тестируемого соединения клетках NIH 3T3, стабильно экспрессирующих ΔF508-CFTR, мониторировали с использованием регистрации цельных клеток с перфораций и фиксацией потенциала. Кратко, регистрации с фиксацией потенциала IΔF508 проводили при комнатной температуре с использованием усилителя с фиксацией потенциала Axopatch 200B (Axon Instruments Inc., Foster City, CA). Все регистрации получали с частотой получения образцов 10 кГц и фильтром низких частот при 1 кГц. Пипетки обладали сопротивлением 5-6 MΩ при заполнении внутриклеточным раствором В этих условиях регистрации, рассчитанный обратный потенциал для Cl- (ECl) при комнатной температуре составлял -28 мВ. Все регистрации обладали сопротивлением уплотнителя >20 ГΩ и последовательным сопротивлением <15 MΩ. Генерирование импульсов, получение и анализ данных проводили с использованием PC, оборудованного интерфейсом Digidata 1320 A/D в комбинации с Clampex 8 (Axon Instruments Inc.). Баня содержала < 250 мкл солевого раствора, и ее подвергали непрерывной перфузии со скоростью 2 мл/мин с использованием системы для гравитационной перфузии.
Идентификация соединений-корректоров
[00470] Для определения активности соединений-корректоров для увеличения плотности функционального ΔF508-CFTR на плазматической мембране, авторы настоящего изобретения использовали описанные выше способы регистрации с перфорацией и фиксацией потенциала для измерения плотности тока после 24-час обработки соединениями-корректорами. Для полной активации ΔF508-CFTR, 10 мкМ форсколин и 20 мкМ генистеин добавляли к клеткам. В условиях регистрации авторов настоящего изобретения, плотность тока после 24-час инкубации при 27°C превышала плотность тока, наблюдаемую после 24-час инкубации при 37°C. Эти результаты соответствуют известным эффектам инкубации при низкой температуре на плотность ΔF508-CFTR в плазматической мембране. Для определения эффектов соединений-корректоров на плотность тока CFTR, клетки инкубировали с 10 мкМ тестируемого соединения в течение 24 часов при 37°C, и плотность тока сравнивали с контрольной при 27°C и 37°C (% активности). Перед регистрацией, клетки промывали 3X внеклеточной средой для регистрации для удаления всего оставшегося тестируемого соединения. Предварительная инкубация с 10 мкМ соединениями-корректорами значительно увеличивала зависимый от цАМФ и генистеина ток по сравнению с контролем при 37°C.
Идентификация соединений-усилителей
[00471] Способность усилителей ΔF508-CFTR увеличивать макроскопический ток Cl- для ΔF508-CFTR (IΔF508) в клетках NIH 3T3, стабильно экспрессирующих ΔF508-CFTR, исследовали также с использованием способов с перфорацией и фиксацией потенциала. Усилители, идентифицированные из оптических анализов, индуцировали зависимое от дозы увеличение IΔF508 с активностью и эффективностью, сходными о наблюдаемыми в оптических анализах. Во всех исследованных клетках, обратный потенциал до и во время введения усилителя составлял приблизительно -30 мВ, что представляет собой рассчитанный ECl (-28 мВ).
Растворы
[00472]
[00473]
Культура клеток
[00474] Для регистраций цельных клеток используют фибробласты NIH 3T3 мыши, стабильно экспрессирующие ΔF508-CFTR. Клетки поддерживают при 37°C в 5% CO2 и при влажности 90% в модифицированной Дульбекко среде Игла, дополненной 2 мМ глутамином, 10% эмбриональной телячьей сывороткой, 1 X NEAA, β-ME, 1 X пенициллином/стрептомицином и 25 мМ HEPES, в флаконах для культивирования 175 см2. Для регистрации цельных клеток, 2500-5000 клеток высевали на покрытые поли-L-лизином стеклянные покровные стекла и культивировали в течение 24-48 час при 27°C перед использованием для тестирования активности усилителей; и инкубировали в присутствии или в отсутствие корректирующего соединения при 37°C для измерения активности корректоров.
Регистрации отдельных каналов
[00475] Активность отдельных каналов для корректируемого температурой ΔF508-CFTR, стабильно экспрессированного в клетках NIH 3T3, и активность соединения-усилителей наблюдали с использованием вырезанных фрагментов вывернутой наружу мембраны. Кратко, регистрацию с фиксацией потенциала для активности отдельных каналов проводили при комнатной температуре с использованием усилителя с фиксацией потенциала Axopatch 200B patch-clamp amplifier (Axon Instruments Inc.). Все регистрации получали с частотой получения образцов 10 кГц и фильтром низких частот при 400 Гц. Пэтч-пипетки были изготовлены из стекла Corning Kovar Sealing #7052 (World Precision Instruments, Inc., Sarasota, FL) и обладали сопротивлением 5-8 MΩ при заполнении внеклеточным раствором. ΔF508-CFTR активировали после вырезания, посредством добавления 1 мМ Мг-АТФ, и 75 нМ цАМФ-зависимой протеинкиназы, каталитической субъединицы (PKA; Promega Corp. Madison, WI). После стабилизации активности канала, фрагмент подвергали перфузии с использованием гравитационной системы для микроперфузии. Приток помещали вблизи фрагмента, получая в результате полную замену раствора в пределах 1-2 сек. Для поддержания активности ΔF508-CFTR во время быстрой перфузии, неспецифический ингибитор фосфатаз F- (10 мМ NaF) добавляли в раствор бани. В этих условиях регистрации, активность каналов оставалась постоянной на всем протяжении регистрации с фиксацией потенциала (вплоть до 60 мин). Токи, полученные посредством продвижения положительного заряда от внутриклеточных к внеклеточным растворам (при движении анионов в обратном направлении), показаны как положительные токи. Потенциал пипетки (Vp) поддерживали при 80 мВ.
[00476] Активность каналов анализировали для фрагментов мембраны, содержащих ≤2 активных каналов. Максимальное количество одновременных открытий определяет количество активных каналов в ходе эксперимента. Для определения амплитуды тока одного канала, эти данные, зарегистрированные из 120-секундной активности ΔF508-CFTR, фильтровали «в автономном режиме» при 100 Гц, а затем использовали для построения гистограмм амплитуд для всех точек, которые приводили в соответствие с мультигауссовыми функциями с использованием программного обеспечения Bio-Patch Analysis (Bio-Logic Comp. France). Общий микроскопический ток и вероятность открытия (Po) определяли по 120 сек активности каналов. Po определяли с использованием программного обеспечения Bio-Patch или из отношения Po=I/i(N), где I=средний ток, i=амплитуда тока одного канала и N=количество активных каналов в фрагменте.
Растворы
[00477]
[00478]
Культура клеток
[00479] Для регистраций вырезанных мембранных фрагментов с фиксацией потенциала используют фибробласты мыши NIH 3T3, стабильно экспрессирующие ΔF508-CFTR. Клетки поддерживают при 37°C в 5% CO2 и при влажности 90% в модифицированной Дульбекко среде Игла, дополненной 2 мМ глутамином, 10% эмбриональной телячьей сывороткой, 1 X NEAA, β-ME, 1 X пенициллином/стрептомицином и 25 мМ HEPES, в флаконах для культивирования 175 см2. Для регистрации отдельных каналов, 2500-5000 клеток высевали на покрытые поли-L-лизином стеклянные покровные стекла и культивировали в течение 24-48 час при 27°C перед использованием.
[00480] Соединение 1 и соединение 2 по изобретению можно использовать в качестве усилителей или индукторов активности CFTR. В таблице 9 ниже проиллюстрированы EC50 и относительная эффективность соединения 1 и соединения 2. В таблице 9 ниже, используют следующие обозначения. EC50: «+++» обозначает <10 мкМ; «++» обозначает между 10 мкМ и 25 мкМ; «+» обозначает между 25 мкМ и 60 мкМ. % эффективности: «+» обозначает <25%; «++» обозначает между 25% и 100%; «+++» обозначает >100%.
(мкМ)
ДРУГИЕ ВАРИАНТЫ ОСУЩЕСТВЛЕНИЯ
[00481] Содержание всех публикаций и патентов, на которые ссылаются в этом описании, включено в настоящий документ в качестве ссылки в той же степени, как если бы для каждой индивидуальной публикации или патентной заявки было конкретно и отдельно указано, что ее содержание включено в качестве ссылки. При конфликте значений терминов в любых из патентов или публикаций, включенных в качестве ссылки, со значением терминов, используемых в этом описании, значение терминов в этом описании предназначено, чтобы обладать преимуществом. Кроме того, в указанном выше обсуждении раскрыты и описаны только иллюстративные варианты осуществления изобретения. Специалисту в данной области ясно понятно из такого обсуждения и из сопутствующих чертежей и формулы изобретения, что различные изменения, модификации и варианты можно осуществлять для него без отклонения от сущности и объема изобретения, как определено в следующей формуле изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПРОВЕДЕНИЯ ВЫСОКОПРОИЗВОДИТЕЛЬНОЙ ТЕСТОВОЙ ВЫСОКОЭФФЕКТИВНОЙ ЖИДКОСТНОЙ ХРОМАТОГРАФИИ | 2015 |
|
RU2691136C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, ОПОСРЕДОВАННЫХ ТРАНСМЕМБРАННЫМ РЕГУЛЯТОРОМ КИСТОЗНОГО ФИБРОЗА CFTR | 2013 |
|
RU2827711C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, ОПОСРЕДОВАННЫХ ТРАНСМЕМБРАННЫМ РЕГУЛЯТОРОМ КИСТОЗНОГО ФИБРОЗА (CFTR) | 2013 |
|
RU2692676C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, ОПОСРЕДОВАННЫХ МУКОВИСЦИДОЗНЫМ ТРАНСМЕМБРАННЫМ РЕГУЛЯТОРОМ ПРОВОДИМОСТИ | 2015 |
|
RU2744460C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ МУКОВИСЦИДОЗА | 2019 |
|
RU2822220C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ЕЕ ВВЕДЕНИЕ | 2013 |
|
RU2802442C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ЕЕ ВВЕДЕНИЯ | 2013 |
|
RU2692779C2 |
| ТВЕРДЫЕ ФОРМЫ (R)-1-(2,2-ДИФТОРБЕНЗО[d][1,3]ДИОКСОЛ-5-ИЛ)-N-(2,3-ДИГИДРОКСИПРОПИЛ)-6-ФТОР-2-(1-ГИДРОКСИ-2-МЕТИЛПРОПАН-2-ИЛ)-1H-ИНДОЛ-5-ИЛ)ЦИКЛОПРОПАНКАРБОКСАМИДА | 2011 |
|
RU2573830C2 |
| ТВЕРДЫЕ ФОРМЫ (R)-1-(2, 2-ДИФТОРБЕНЗО[d][1, 3]ДИОКСОЛ-5-ИЛ)-N-(1-(2, 3-ДИГИДРОКСИПРОПИЛ)-6-ФТОР-2-(1-ГИДРОКСИ-2-МЕТИЛПРОПАН-2-ИЛ)-1H-ИНДОЛ-5-ИЛ)ЦИКЛОПРОПАНКАРБОКСАМИДА | 2011 |
|
RU2711481C2 |
| ВВЕДЕНИЕ ДЕЙТЕРИРОВАННЫХ УСИЛИТЕЛЕЙ CFTR | 2016 |
|
RU2761344C2 |
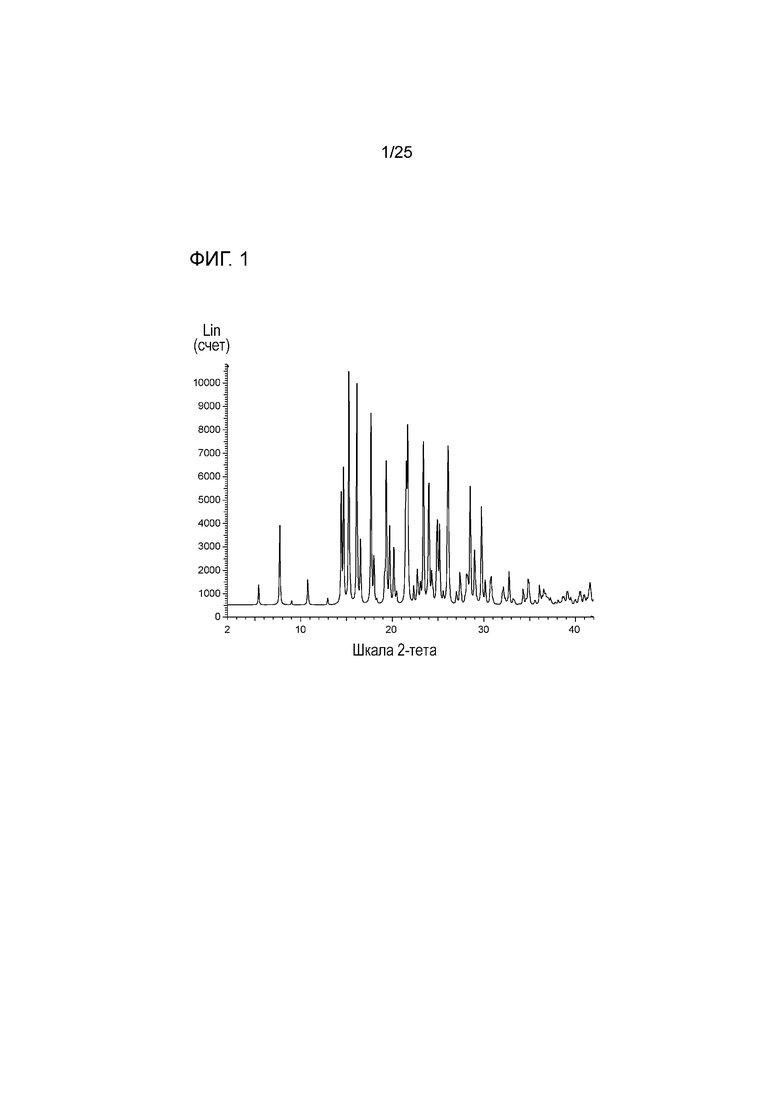
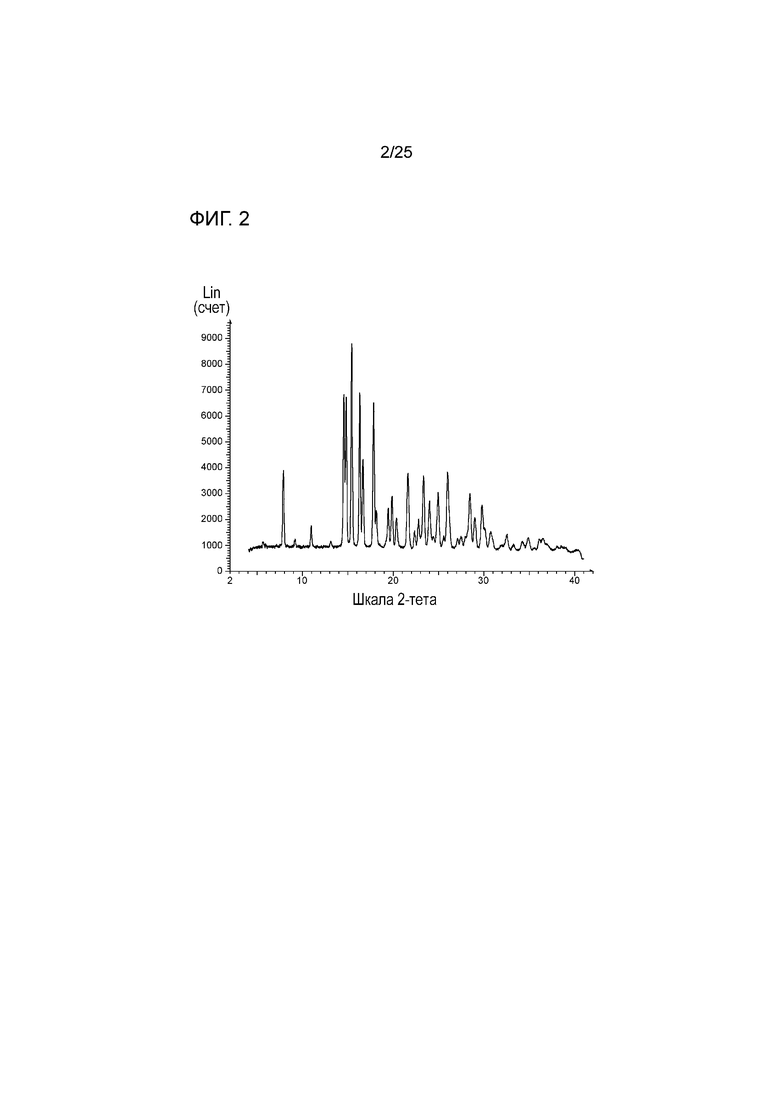
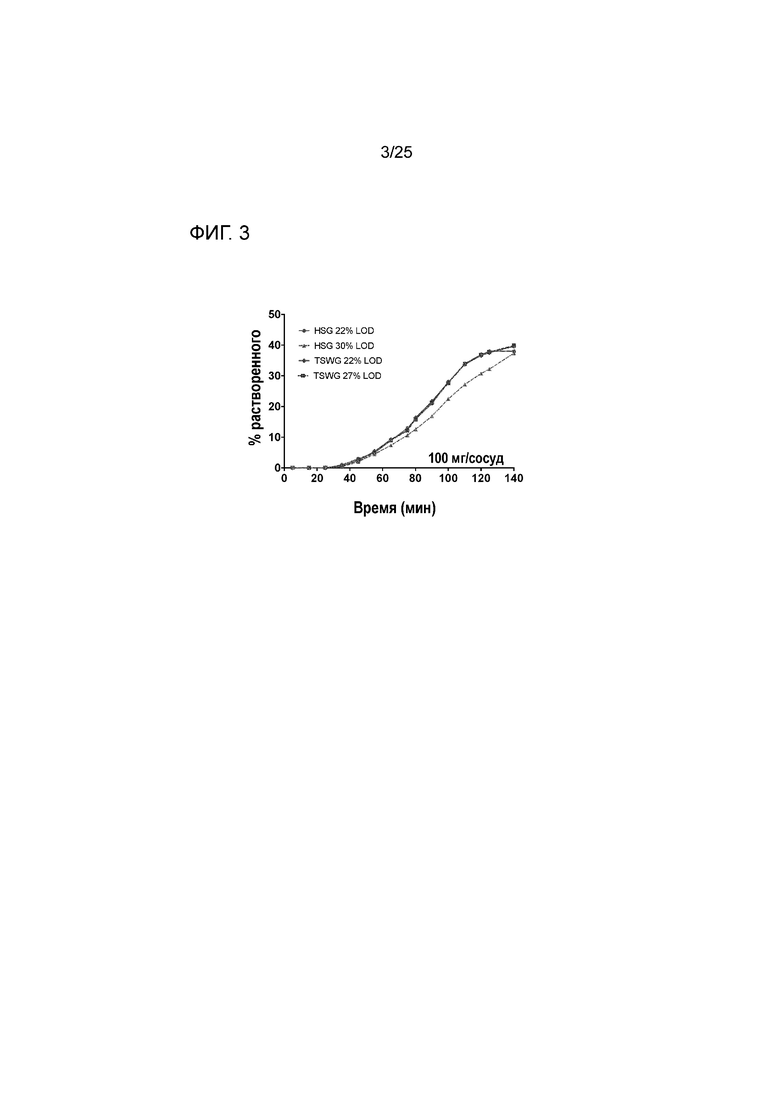
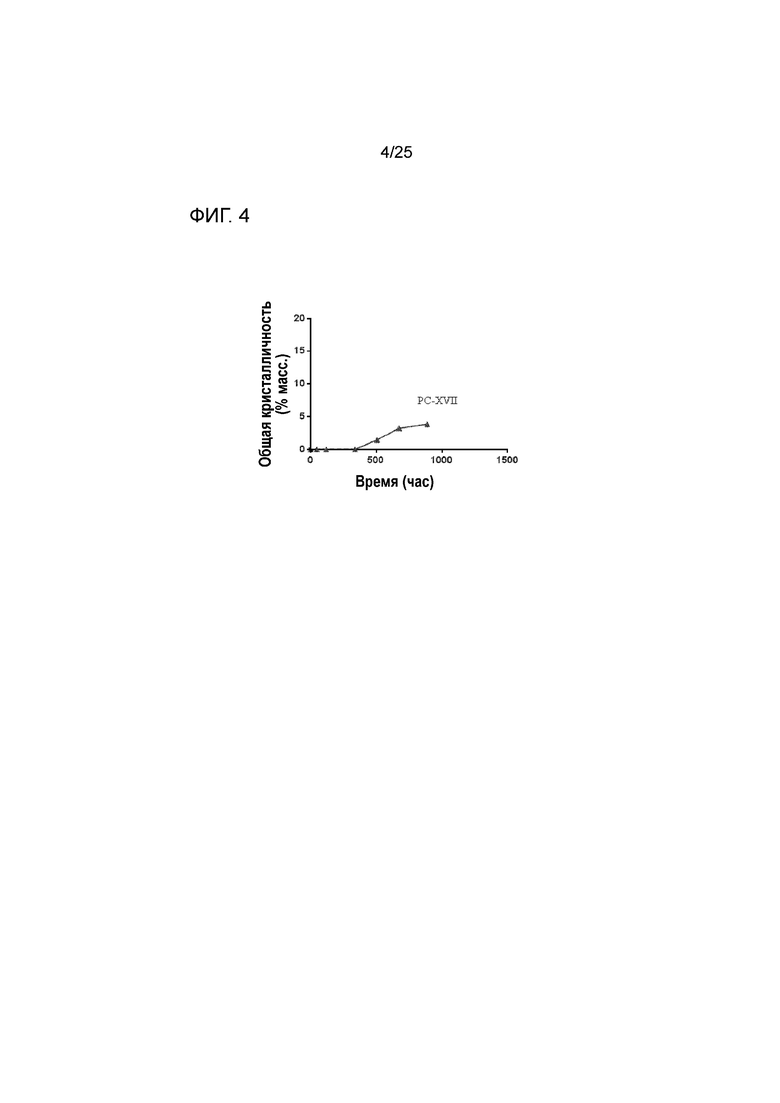
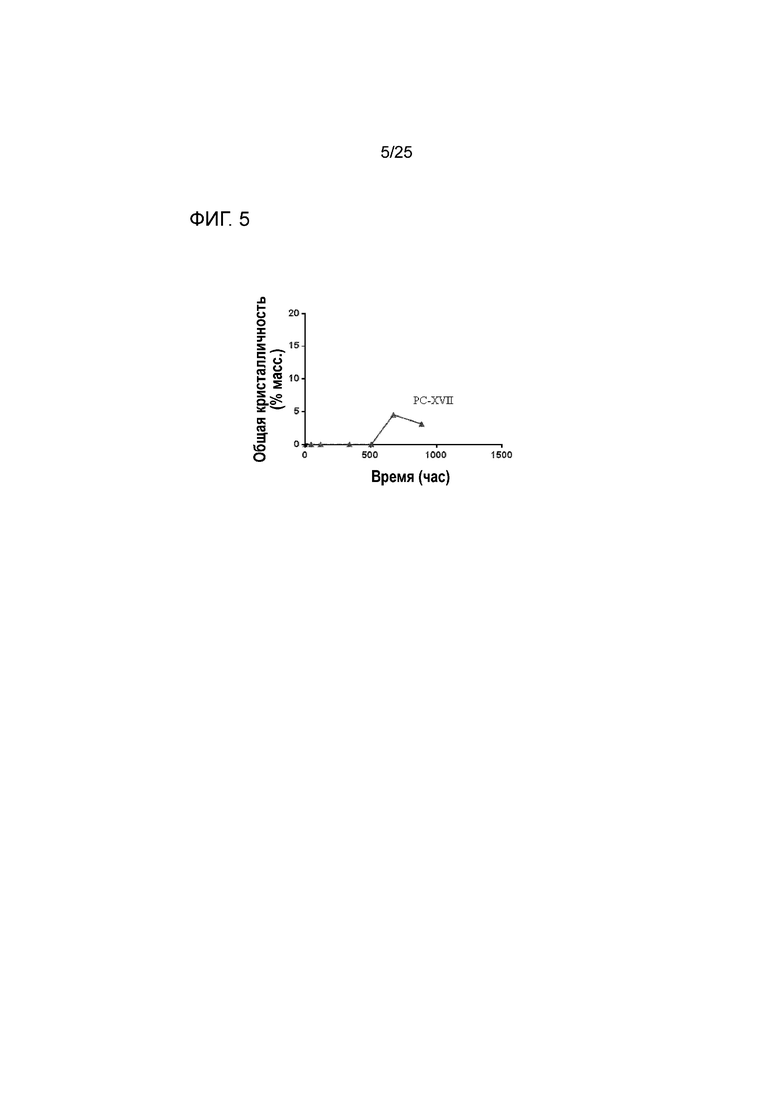
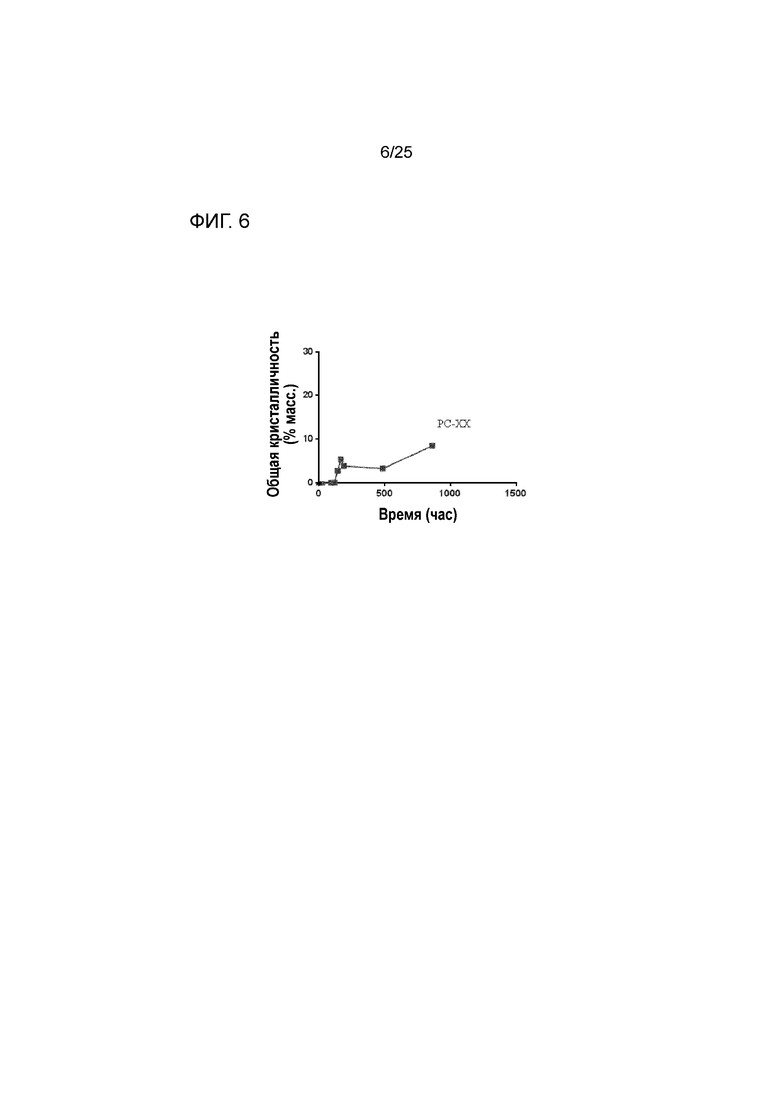
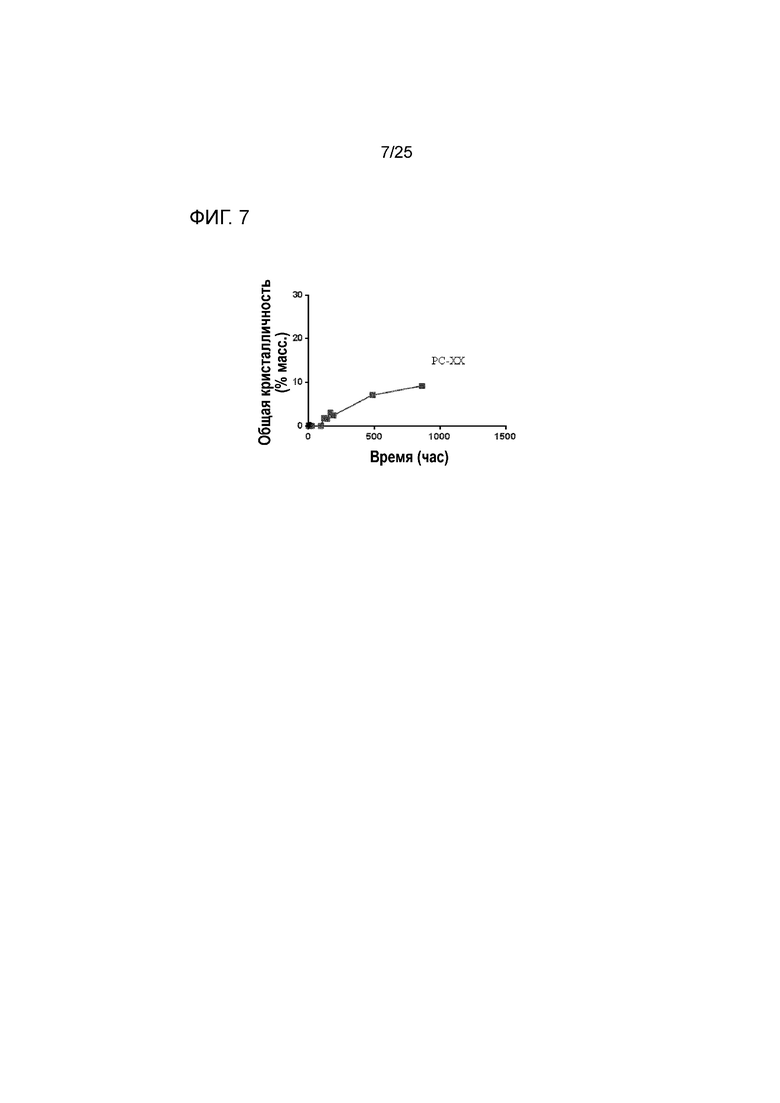
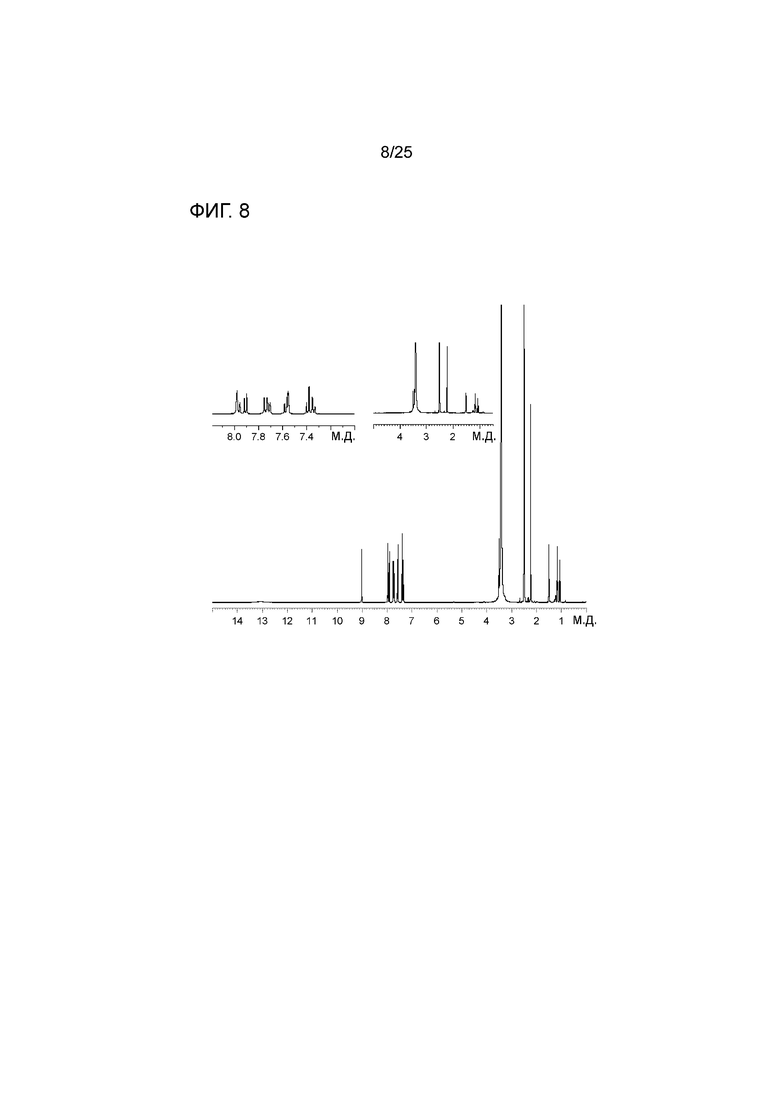
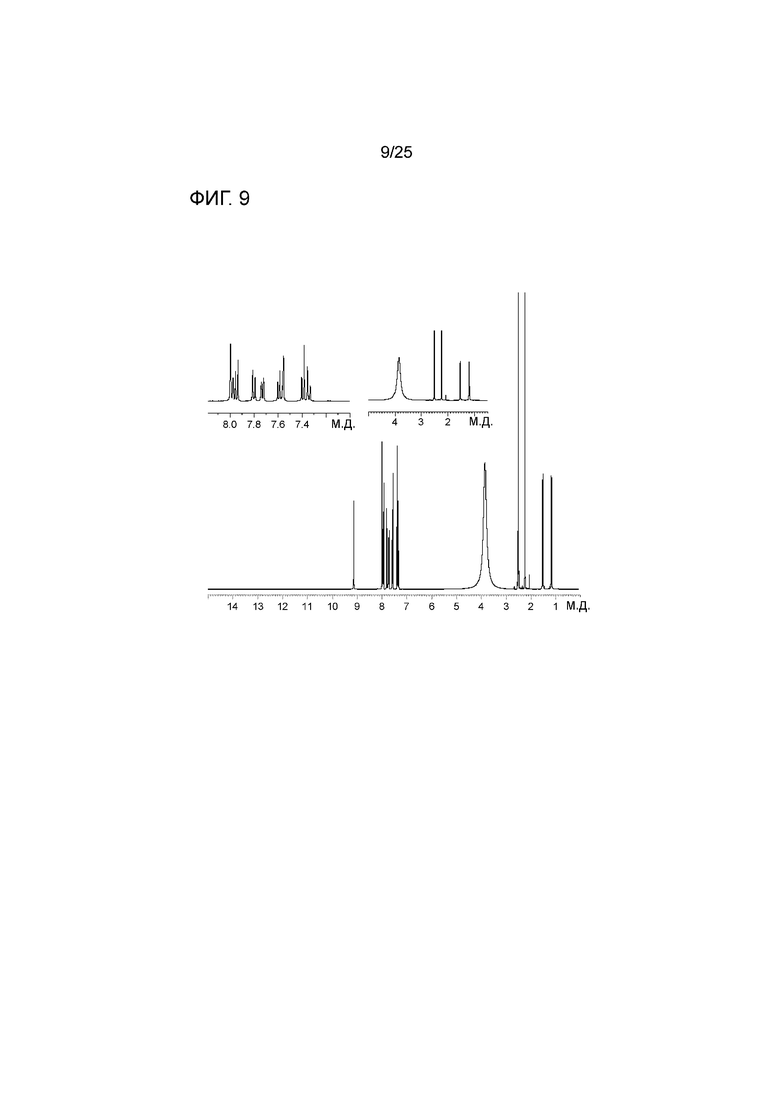
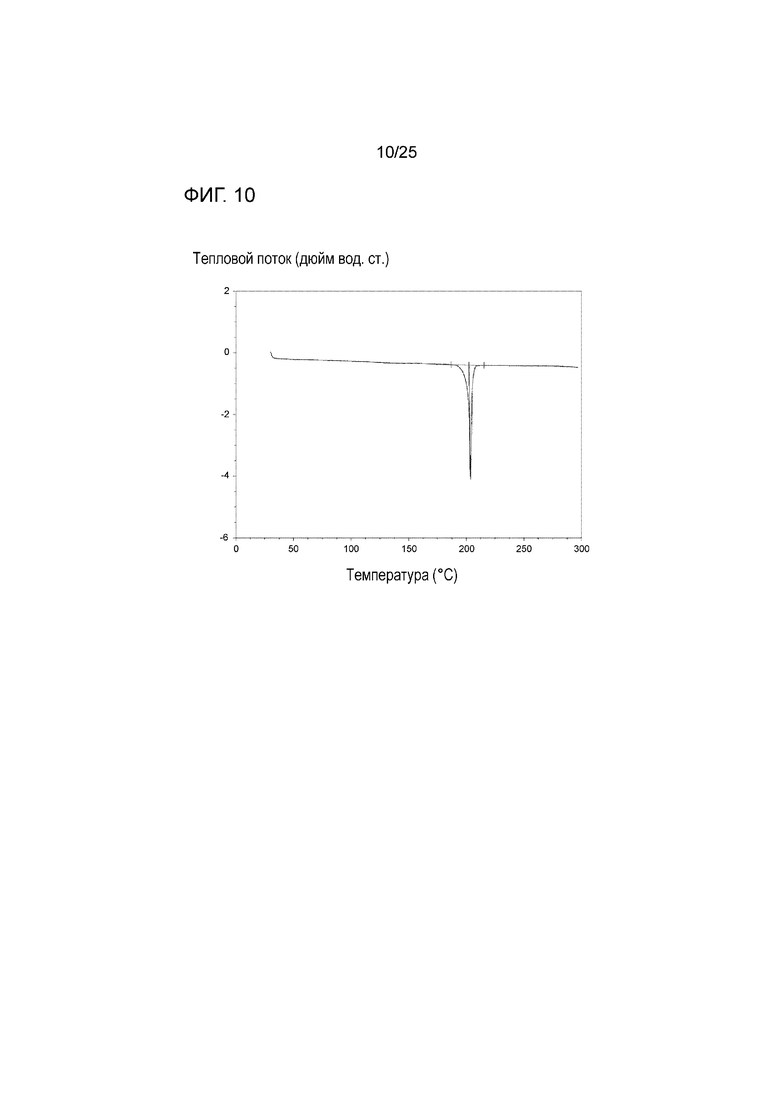
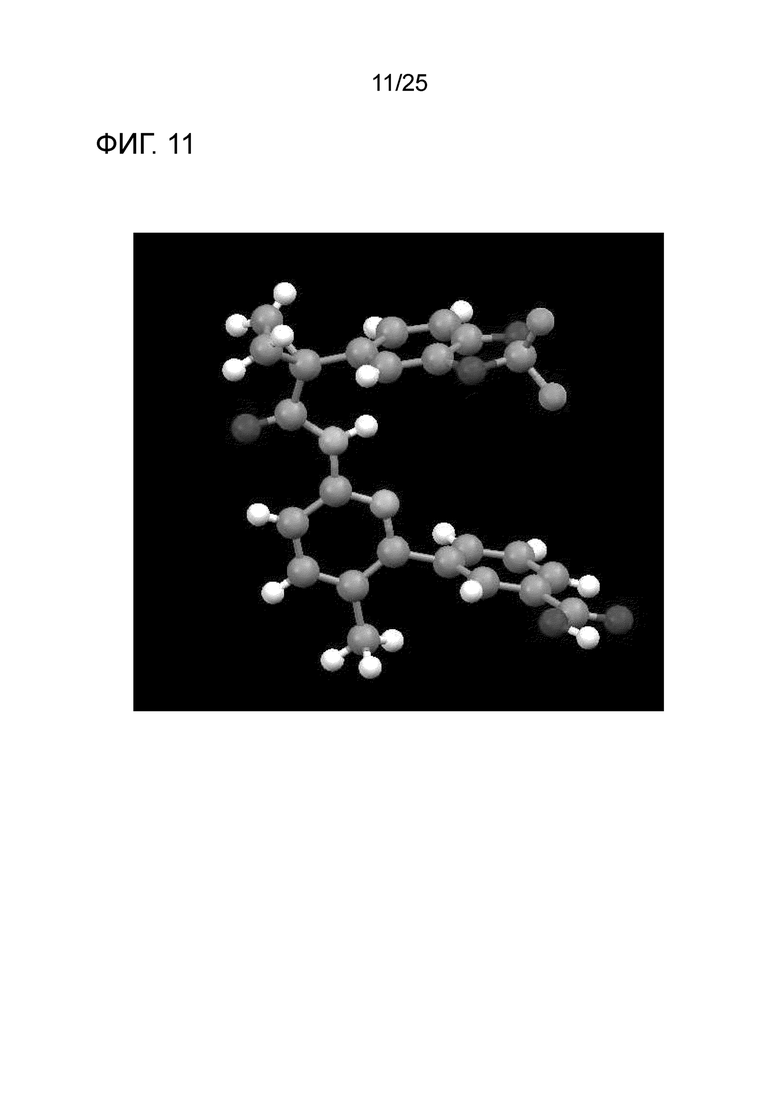
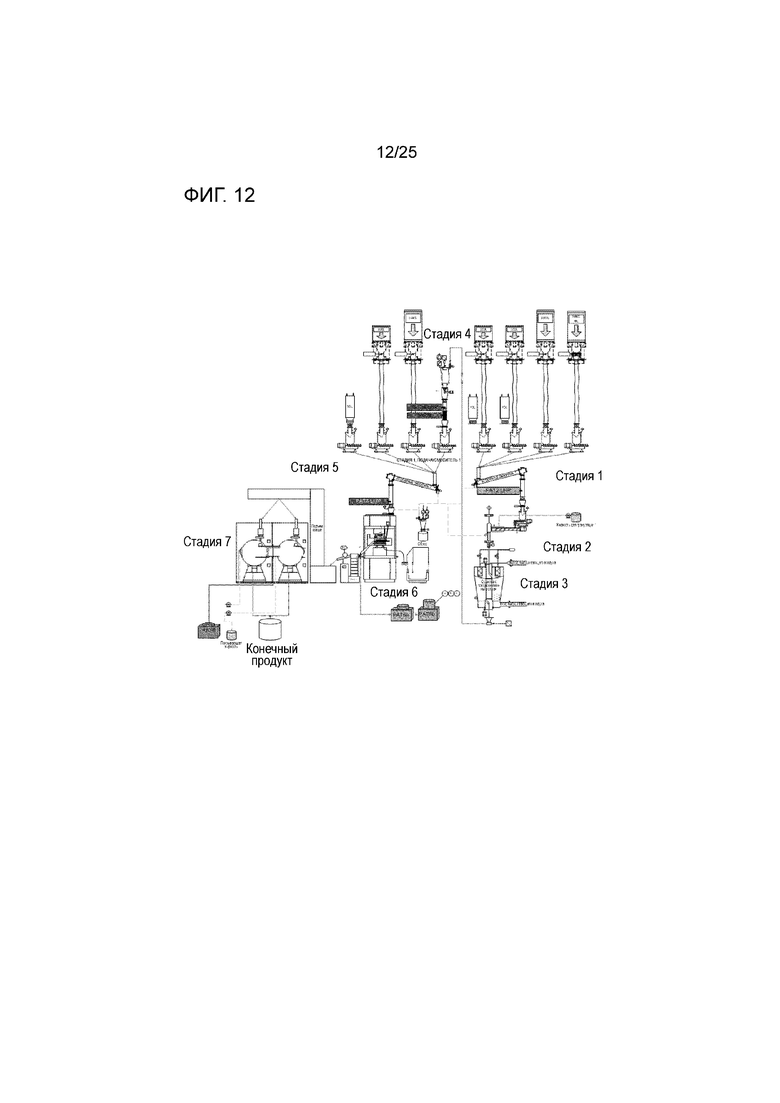
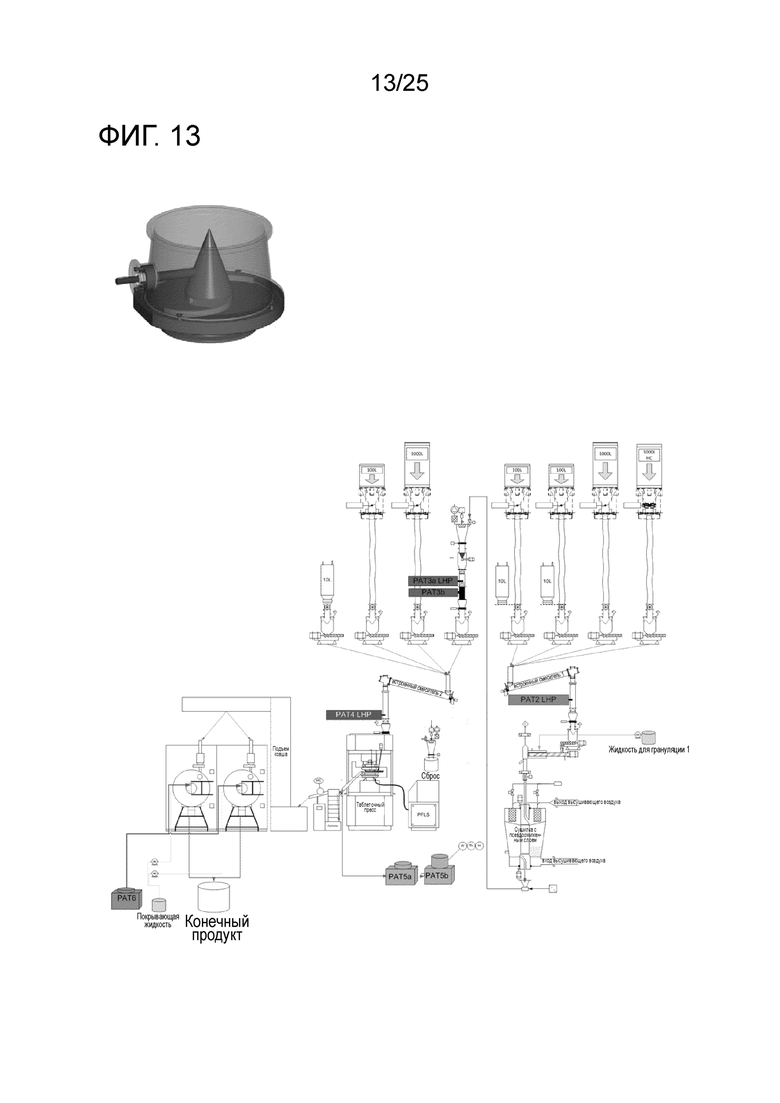
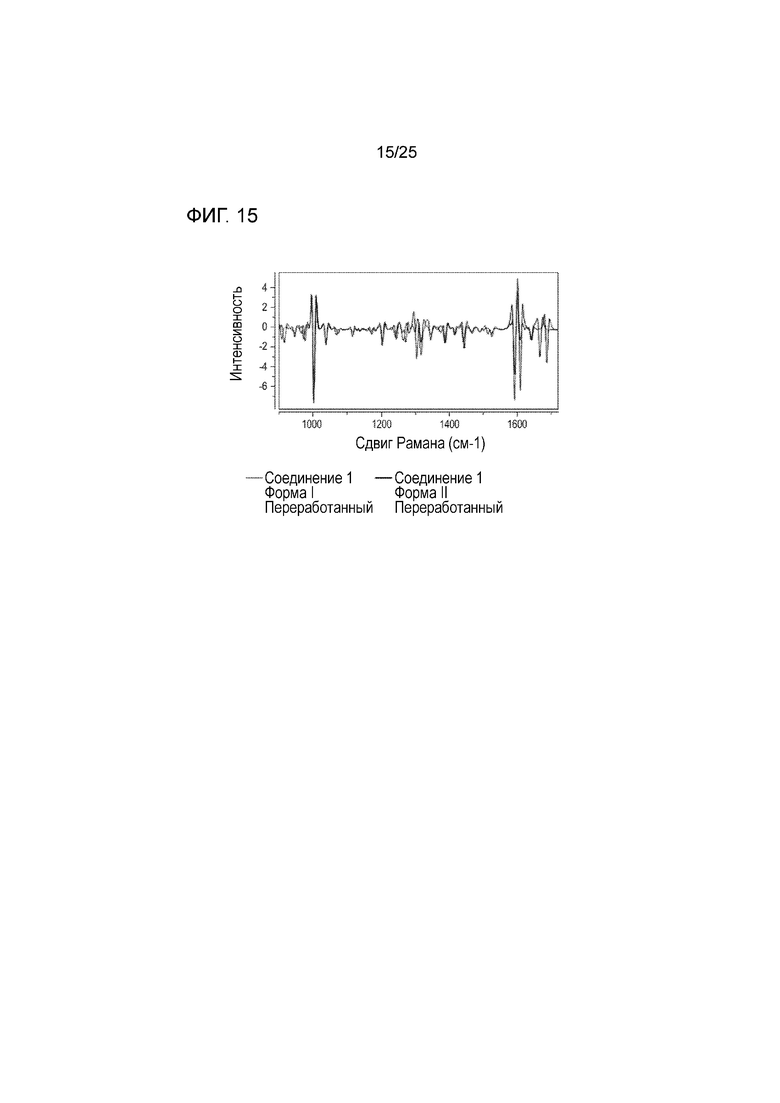
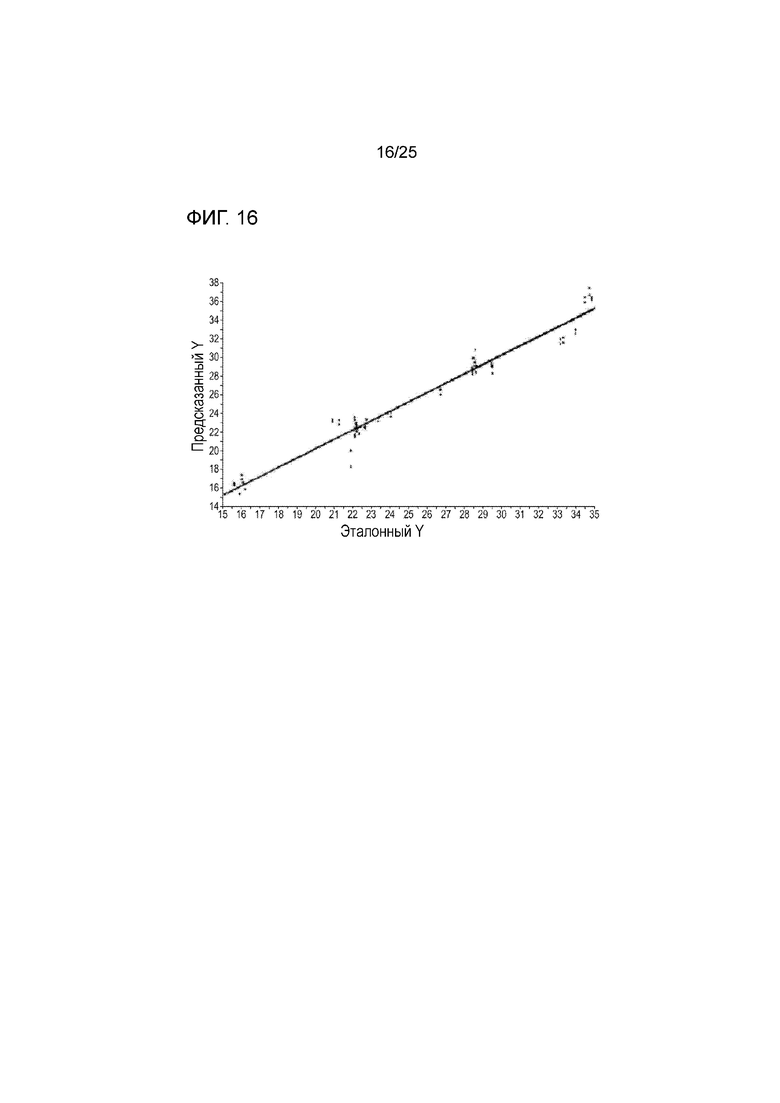
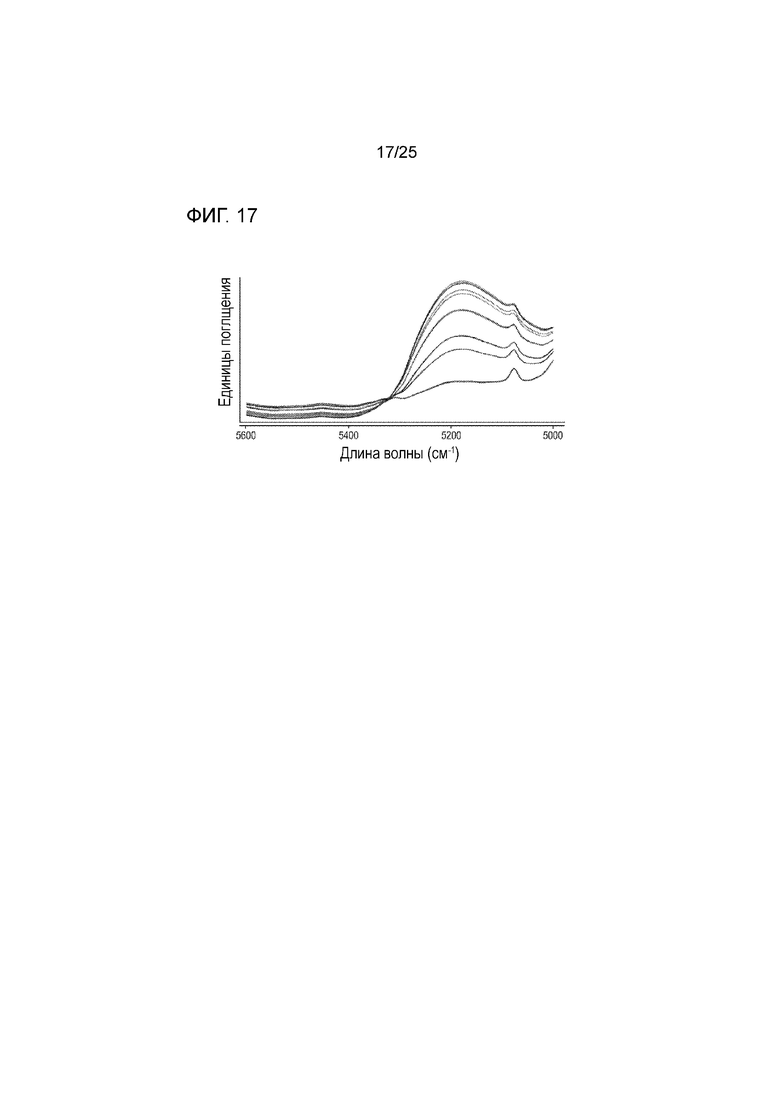
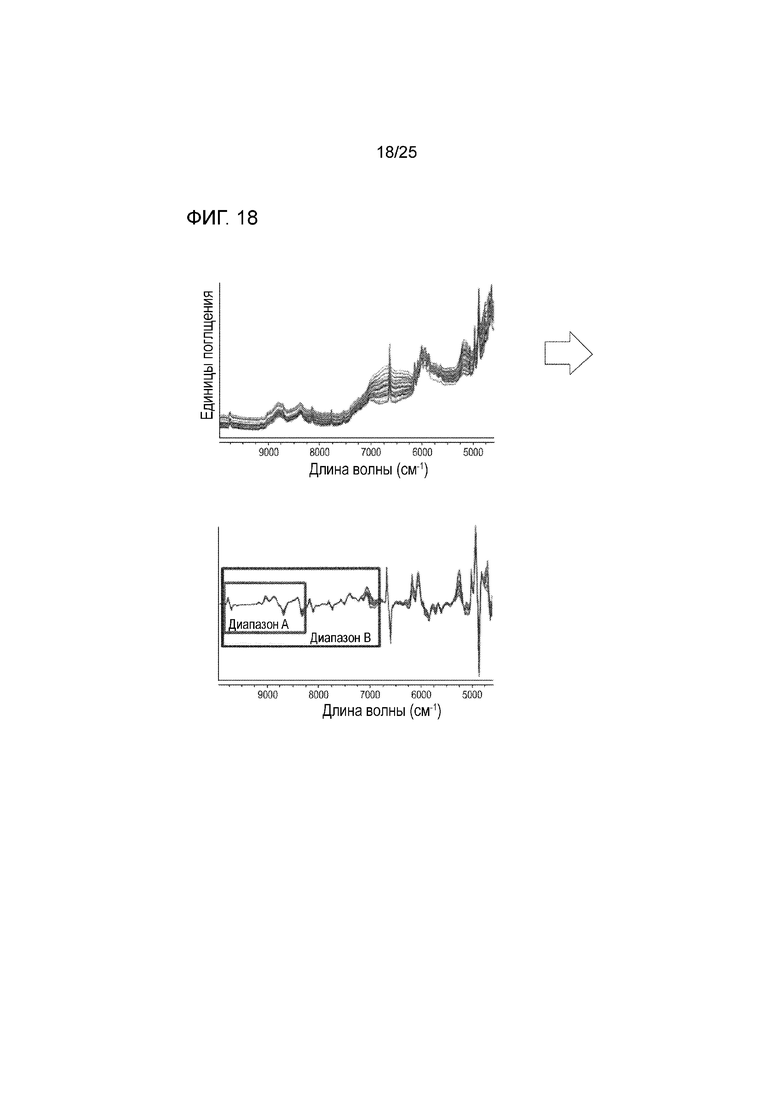

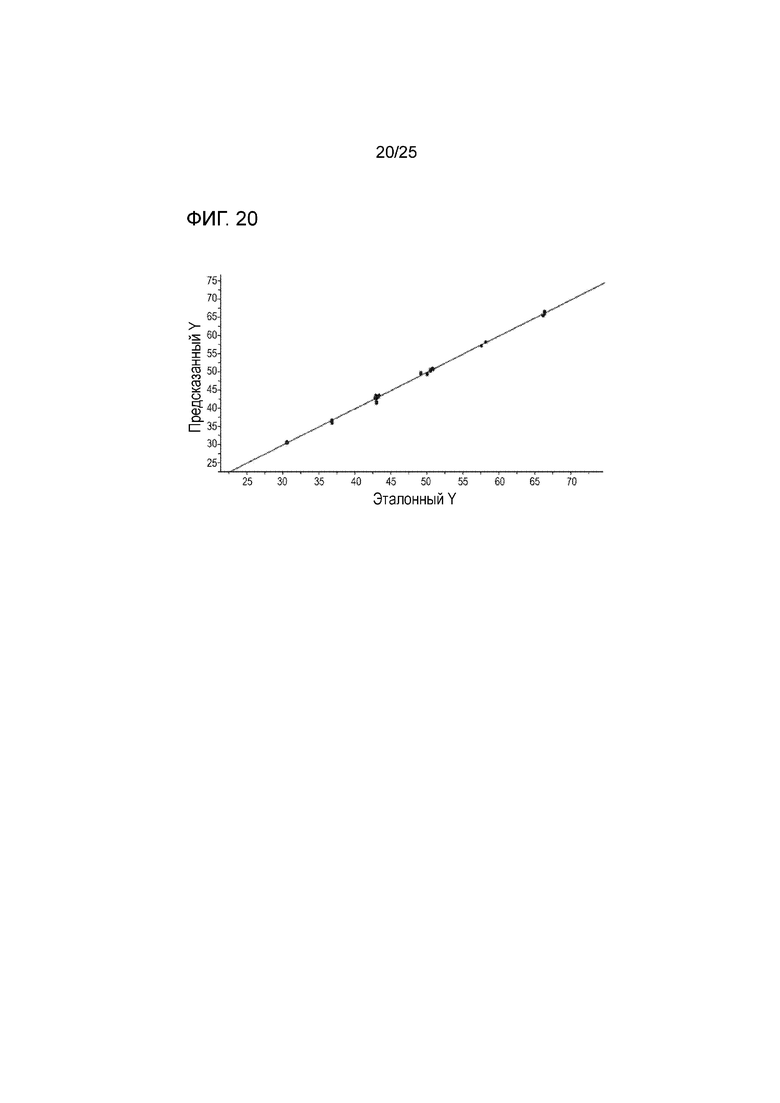
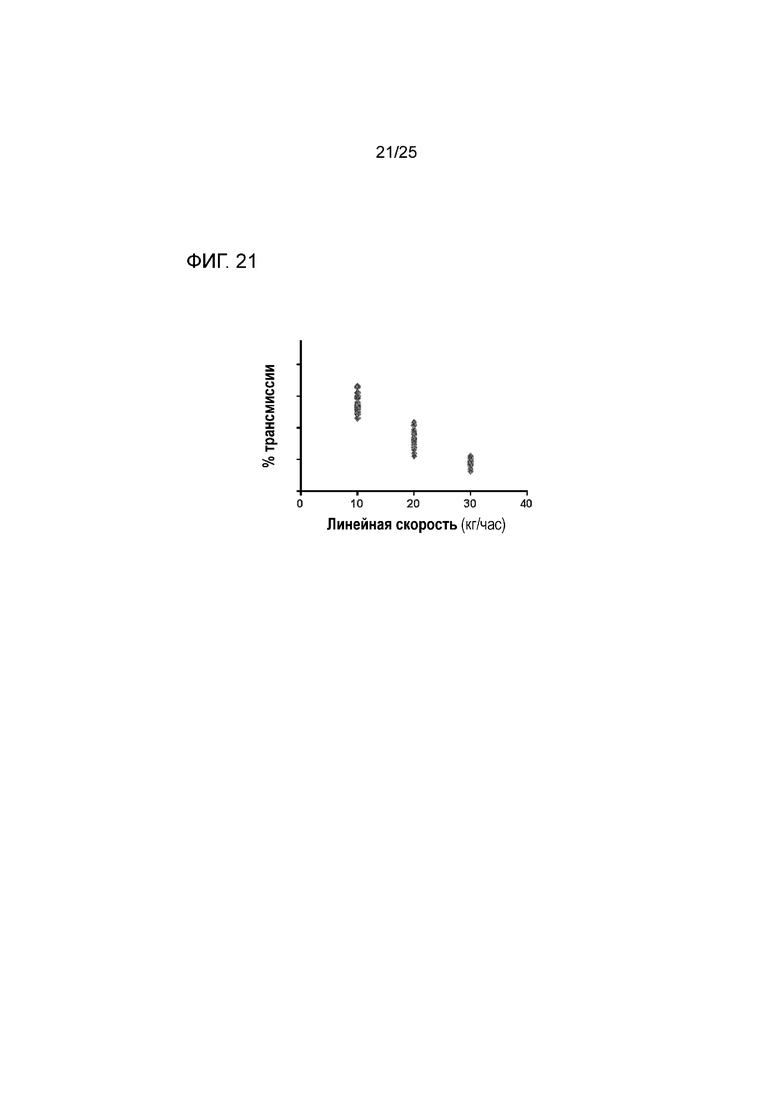
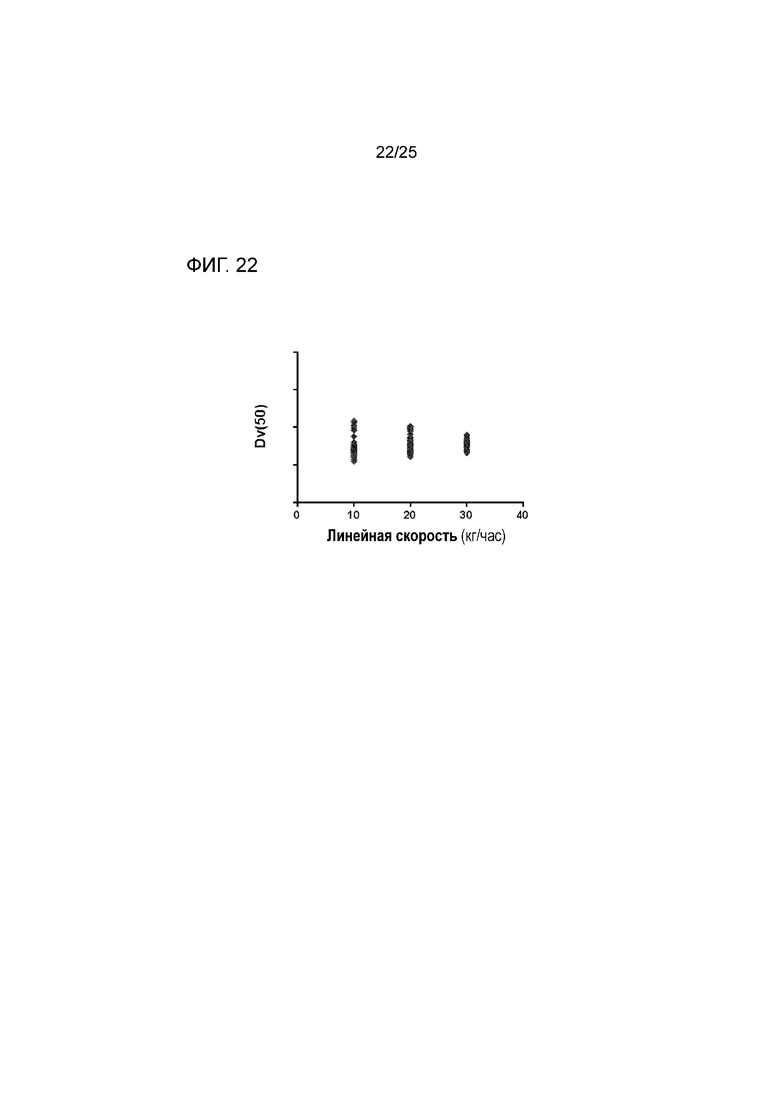
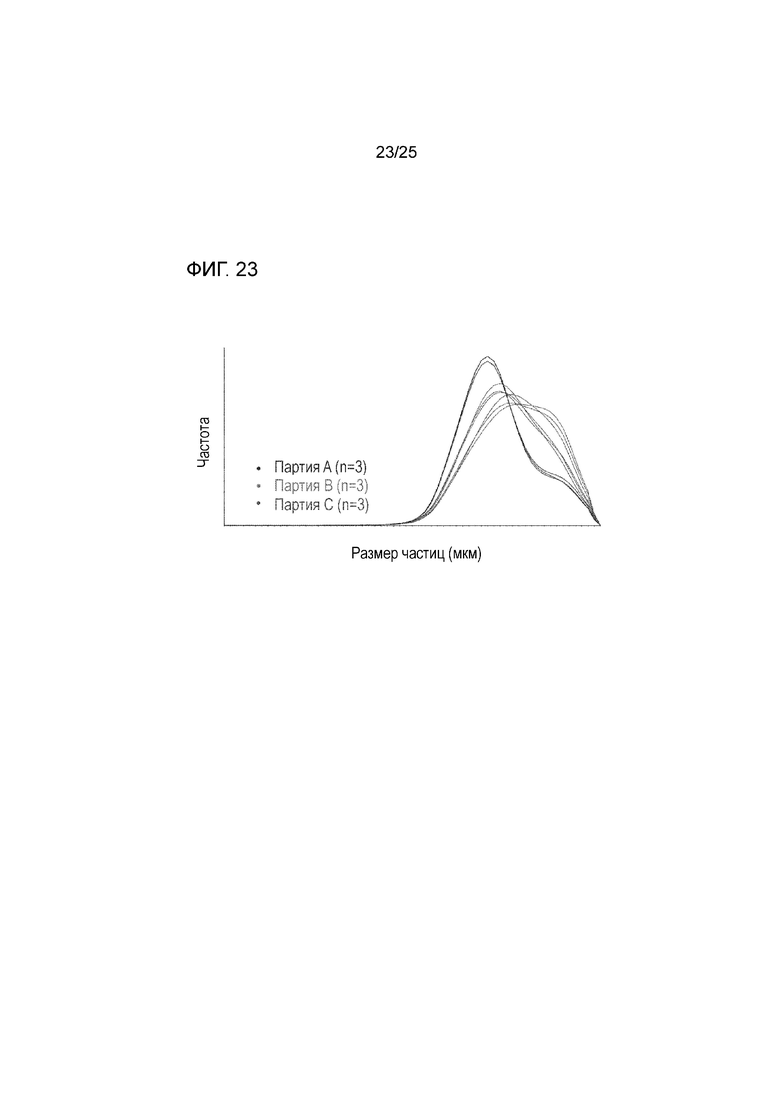
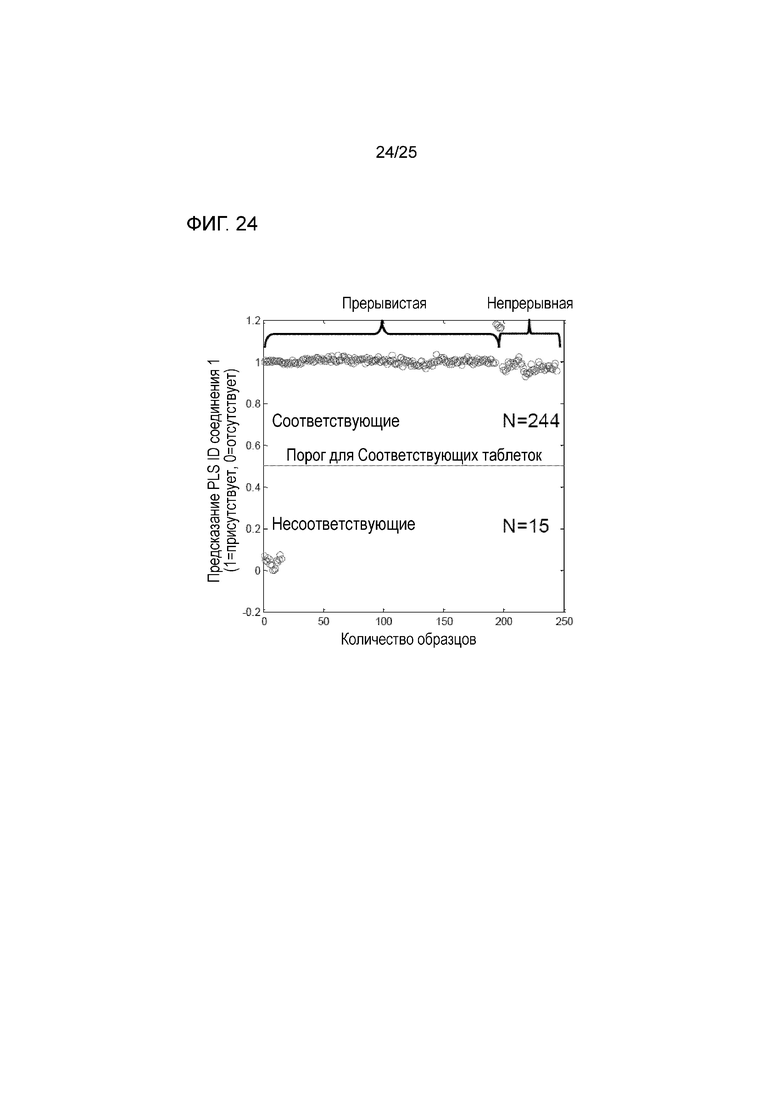
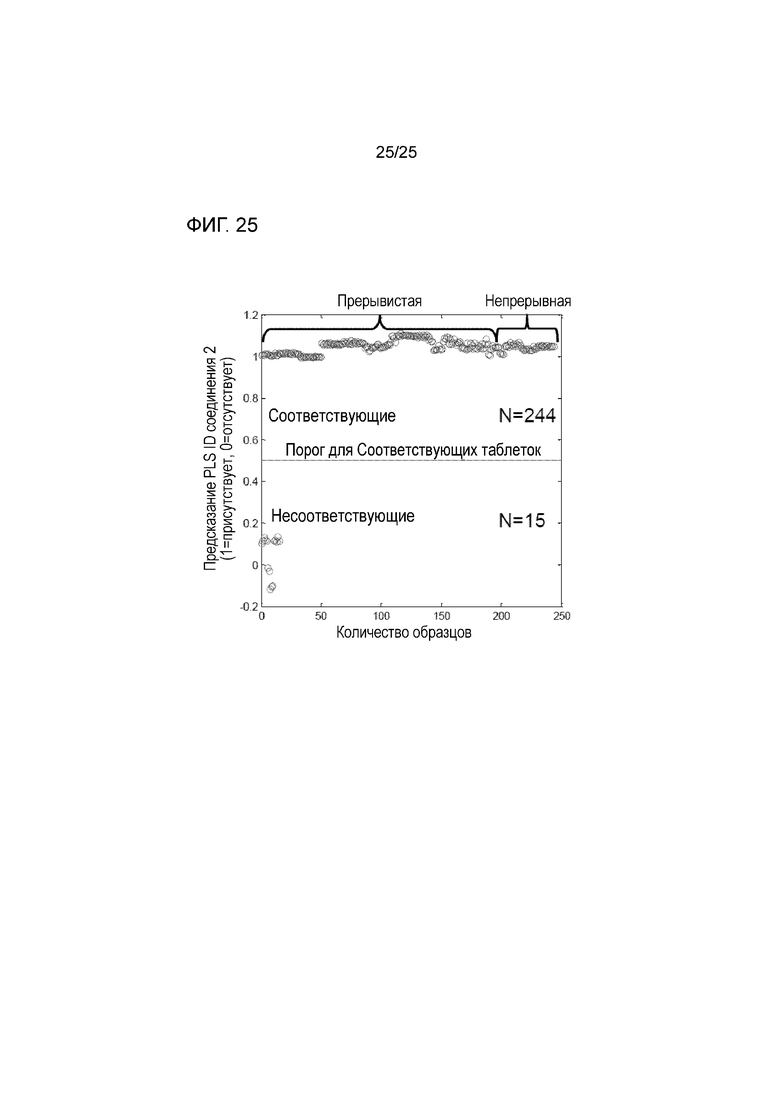
Описаны способы получения фармацевтических композиций, содержащих 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамидо)-3-метилпиридин-2-ил)бензойную кислоту (соединение 1) в форме I и твердую дисперсию, содержащую по существу аморфный N-(5-гидрокси-2,4-ди-трет-бутилфенил)-4-оксо-1H-хинолин-3-карбоксамид (соединение 2), связанные с ними способы лечения, уменьшения тяжести или симптоматического лечения опосредованных CFTR заболеваний, таких как кистозный фиброз, способы их введения и наборы с ними. 20 з.п. ф-лы, 34 табл., 25 ил.
1. Непрерывный способ получения таблеток, содержащих форму I соединения 1 и твердую дисперсию, содержащую по существу аморфное соединение 2, включающий в себя стадии:
a) смешивания формы I соединения 1, твердой дисперсии, содержащей по существу аморфное соединение 2, наполнителя и дезинтегрирующего средства в смесителе для получения смеси;
b) получения раствора для грануляции с водой, связывающим средством и поверхностно-активным веществом;
c) загрузки смеси со стадии a) в непрерывный двухшнековый гранулятор с добавлением в то же время раствора для грануляции со стадии b) для получения гранул;
d) сушки гранул со стадии c) и их размола;
e) смешивания размолотых гранул со стадии d) с наполнителем, дезинтегрирующим средством и смазочным средством для получения смеси; и
f) прессования смеси со стадии e) в таблетки, где по меньшей мере одна из вышеуказанных стадий включает в себя аналитическую технологию процесса;
где форма I соединения 1 представляет собой 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамидо)-3-метилпиридин-2-ил)бензойную кислоту, которая характеризуется по меньшей мере одним пиком, имеющим значение угла 2θ, выбранное из 15,4±0,2 градусов, 16,3±0,2 градусов и 14,5±0,2 градусов, полученным методом рентгеновской порошковой дифракции с использованием излучения CuK альфа;
где соединение 2 представляет собой N-(5-гидрокси-2,4-дитрет-бутил-фенил)-4-оксо-1H-хинолин-3-карбоксамид, и
где по существу аморфное соединение 2 обладает менее чем 15% кристалличностью.
2. Способ по п. 1, где аналитическая технология процесса включает применение спектроскопии в ближней инфракрасной области (NIR) лазерной дифракции и/или спектроскопии Рамана для мониторирования определенных стандартов, где определенный стандарт выбран из однородности смеси, однородности гранул, влажности, распределения размера частиц, идентичности твердой формы активного фармацевтического ингредиента, массы, густоты, жесткости и толщины покрытия.
3. Способ по п. 1, где стадия (f) включает мониторинг идентичности твердой формы соединения 1 и/или соединения 2 в таблетке с использованием спектроскопии Рамана.
4. Способ по п. 1, где стадия a) включает мониторирование однородности смеси с использованием NIR.
5. Способ по п. 1, где стадия c) включает мониторирование однородности гранул и/или влажности с использованием NIR.
6. Способ по п. 1, где стадия c) включает мониторирование распределения размера частиц с использованием лазерной дифракции.
7. Способ по п. 1, где стадия e) включает мониторирование однородности смеси и/или влажности с использованием NIR.
8. Способ по п. 1, где стадия f) дополнительно включает мониторирование массы, густоты и жесткости таблетки с использованием тестера таблеток.
9. Способ по п. 1, где форма I соединения 1 характеризуется пиком, имеющим значение угла 2θ 15,4±0,2 градусов, полученным методом рентгеновской порошковой дифракции с использованием излучения CuK альфа.
10. Способ по п. 9, где форма I соединения 1 характеризуется пиком, имеющим значение угла 2θ 15,4 градусов, полученным методом рентгеновской порошковой дифракции с использованием излучения CuK альфа.
11. Способ по п. 1, где форма I соединения 1 характеризуется пиком, имеющим значение угла 2θ 16,3±0,2 градусов, полученным методом рентгеновской порошковой дифракции с использованием излучения CuK альфа.
12. Способ по п. 11, где форма I соединения 1 характеризуется пиком, имеющим значение угла 2θ 16,3 градусов, полученным методом рентгеновской порошковой дифракции с использованием излучения CuK альфа.
13. Способ по п. 1, где форма I соединения 1 характеризуется пиком, имеющим значение угла 2θ 14,5±0,2 градусов, полученным методом рентгеновской порошковой дифракции с использованием излучения CuK альфа.
14. Способ по п. 13, где форма I соединения 1 характеризуется пиком, имеющим значение угла 2θ 14,5 градусов, полученным методом рентгеновской порошковой дифракции с использованием излучения CuK альфа.
15. Способ по п. 1, где форма I соединения 1 дополнительно характеризуется пиком, имеющим значение угла 2θ в диапазоне 17,6 до 18,0 градусов, полученным методом рентгеновской порошковой дифракции с использованием излучения CuK альфа.
16. Способ по п. 1, где форма I соединения 1 дополнительно характеризуется пиком, имеющим значение угла 2θ в диапазоне 7,6 до 8,0 градусов, полученным методом рентгеновской порошковой дифракции с использованием излучения CuK альфа.
17. Способ по любому одному из пп. 1-16, где форма I соединения 1 характеризуется по меньшей мере одним пиком, имеющим значение угла 2θ, выбранным из 14,41 градусов; 14,64 градусов; 15,23 градусов; 16,11 градусов; 17,67 градусов; 19,32 градусов; 21,67 градусов; 23,40 градусов; 23,99 градусов; 26,10 градусов и 28,54 градусов, полученным методом рентгеновской порошковой дифракции с использованием излучения CuK альфа.
18. Способ по любому одному из пп. 1-16, где форма I соединения 1 характеризуется по меньшей мере одним пиком, имеющим значение угла 2θ, выбранным из 7,83 градусов; 14,51 градусов; 14,78 градусов; 15,39 градусов; 16,26 градусов; 16,62 градусов; 17,81 градусов; 21,59 градусов; 23,32 градусов; 24,93 градусов и 25,99 градусов, полученным методом рентгеновской порошковой дифракции с использованием излучения CuK альфа.
19. Способ по любому одному из пп. 1-16, где форма I соединения 1 характеризуется картиной рентгеновской порошковой дифракции такой, которая изображена на Фиг. 1.
20. Способ по любому одному из пп. 1-16, где форма I соединения 1 характеризуется картиной рентгеновской порошковой дифракции такой, которая изображена на Фиг. 2.
21. Способ по любому одному из пп. 1-16, где форма I соединения 1 характеризуется тем, что имеет моноклиническую кристаллическую систему и P21/n пространственную группу и имеет следующие параметры единичной ячейки: a=4,9626(7) Å, b=12,299(2) Å, c=33,075 (4) Å, β=93,938(9)°.
| WO 2013130669 A1, 06.09.2013 | |||
| WO 2011133953 A1, 27.10.2011 | |||
| US 20120061869 A1, 15.03.2012 | |||
| EA 201170330 A1, 31.10.2011 | |||
| De Beer T | |||
| et al., Near infrared and Raman spectroscopy for the in-process monitoring of pharmaceutical production processes, International Journal of Pharmaceutics, Vol | |||
| Трубчатый паровой котел для центрального отопления | 1924 |
|
SU417A1 |

