Перекрестная ссылка на родственные заявки
Настоящая заявка испрашивает приоритет согласно предварительной заявке на патент США №61/721622, поданной 2 ноября 2012 г., 61/728328, поданной 20 ноября 2012 г.; 61/770668, поданной 28 февраля 2013 г.; 61/824005, поданной 16 мая 2013 г., и 61/840668, поданной 28 июня 2013 г., содержание всех заявок полностью включено в настоящую заявку посредством ссылки.
Область техники, к которой относится настоящее изобретение
Изобретение относится к фармацевтическим композициям, включающим 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамидо)-3-метилпиридин-2-ил)бензойную кислоту (Соединение 1) Формы I и твердую дисперсию, включающую по существу аморфный N-(5-гидрокси-2,4-дитрет-бутил-фенил)-4-оксо-1Н-хинолин-3-карбоксамид (Соединение 2), способам лечения, способам производства, способам введения и их наборам.
Предшествующий уровень техники
Кистозный фиброз (КФ) представляет собой рецессивное генетическое заболевание, которое поражает приблизительно 30000 детей и взрослых в США и приблизительно 30000 детей и взрослых в Европе. Несмотря на прогресс в лечении КФ, вылечить заболевание не удается.
У пациентов с КФ мутация в CFTR (трансмембранный регулятор кистозного фиброза), эндогенно экспрессирующемся в дыхательном эпителии, приводит к снижению апикальной секреции анионов, вызывая нарушение баланса транспорта ионов и жидкости. Снижение транспорта анионов в результате этого способствует увеличению накопления слизи в легких и сопутствующих микробных инфекций, которые неизбежно приводят к смерти пациентов с КФ. Наряду с респираторными заболеваниями пациенты с КФ обычно страдают от проблем с желудочно-кишечным трактом и недостаточности поджелудочной железы, которые, без лечения приводят к смерти. Кроме того, большинство мужчин с кистозным фиброзом являются стерильными, а фертильность женщин с кистозным фиброзом понижена. В отличие от тяжелых эффектов наличия двух копий гена, ассоциированного с кистозным фиброзом, у людей с одной копией гена, ассоциированного с кистозным фиброзом, наблюдается повышенная устойчивость к холере и дегидратации по причине диареи, что, возможно, объясняет относительно высокую частоту гена КФ в популяции.
Анализ последовательности гена CFTR на КФ хромосомах выявил множество мутаций, вызывающих заболевание (Cutting, G.R. et al. (1990) Nature 346:366-369; Dean, M. et al. (1990) Cell 61:863:870; и Kerem, B-S. et al. (1989) Science 245:1073-1080; Kerem, B-S et al. (1990) Proc. Natl. Acad. Sci. USA 87:8447-8451). На сегодняшний день было идентифицировано более 1000 мутаций в гене КФ, вызывающих заболевание (http://www.genet.sickkids.on.ca/cftr/app). Наиболее часто встречающейся мутацией является делеция фенилаланина в положении 508 аминокислотной последовательности CFTR, обычно обозначаемая ΔF508-CFTR. Данная мутация встречается приблизительно в 70% случаях кистозного фиброза и ассоциируется с тяжелой формой заболевания.
Делеция остатка 508 в ΔF508-CFTR препятствует правильному сворачиванию образующегося белка. Это приводит к неспособности мутантного белка покинуть ЭР (эндоплазматический ретикулум) и транспортироваться к плазматической мембране. В результате количество находящихся в мембране каналов гораздо ниже такового, наблюдаемого в клетках, экспрессирующих CFTR дикого типа. Наряду с нарушенным транспортом мутация вызывает дефекты воротного механизма канала. Вместе уменьшенное количество каналов в мембране и дефект воротного механизма приводят к снижению анионного транспорта через эпителий, приводя к нарушению транспорта ионов и жидкости. (Quinton, P.M. (1990), FASEB J. 4: 2709-2727). Однако исследования показали, что сниженные количества ΔF508-CFTR в мембране являются функциональными, хотя и менее функциональными, чем CFTR дикого типа. (Dalemans et al. (1991), Nature Lond. 354: 526-528; Denning et al., supra; Pasyk and Foskett (1995), J. Cell. Biochem. 270: 12347-50). Помимо ΔF508-CFTR другое заболевание, вызывающее мутации в CFTR, которое приводит к нарушенному транспорту, синтезу и/или воротному механизму канала, можно было бы регулировать либо путем активации, либо ингибирования для изменения анионной секреции и модификации прогрессирования и/или тяжести заболевания.
Соединение 1 в форме соли раскрыто в Международной публикации РСТ WO 2007056341 и в патенте США №7741321 в качестве индуктора активности CFTR и, таким образом, в качестве подходящего лечения заболеваний, опосредованных CFTR, таких как кистозный фиброз. Соединение 1 Форма 1, которое главным образом представляет собой кристаллическую и обессоленную форму, раскрыто в Международной публикации РСТ WO 2009073757 и в патенте США №8507534. Соединение 2 раскрыто в Международной публикации РСТ WO 2006002421 и в патенте США №7495103 в качестве индуктора активности CFTR и, таким образом, в качестве подходящего лечения заболеваний, опосредованных CFTR, таких как кистозный фиброз. Твердая дисперсия, включающая по существу аморфное Соединение 2, раскрыта в Международной публикации РСТ WO 2010019239 и в опубликованной патентной заявке США № US 20100074949. Все перечисленные вше заявки и патенты полностью включены в данный документ посредством ссылки.
Независимо было показано, что соединения, которые потенцируют CFTR, такие как Соединение 2, и соединения, которые корректируют CFTR, такие как Соединение 1, применимы в лечении связанных с CFTR заболеваний, таких как кистозный фиброз.
Соответственно, существует необходимость в новом лечении CFTR-опосредованных заболеваний, которые включают корректирующие и потенцирующие соединения.
В частности, существует необходимость в комбинированной терапии для лечения CFTR-опосредованных заболеваний, таких как кистозный фиброз, которые включают корректирующие и потенцирующие соединения.
В более частном случае, существует необходимость в комбинированной терапии для лечения CFTR-опосредованных заболеваний, таких как кистозный фиброз, которые включают потенцирующие.соединения, такие как по существу аморфное Соединение 2, в комбинации с корректирующими соединениями, такими как Соединение 1 Форма I.
Соединение 1 как часть комбинации с Соединением 2 было отмечено Признанием нового препарата в качестве терапии прорыва Управления по контролю качества пищевых продуктов, медикаментов и косметических средств (FDA, Food and Drug Administration) для лечения кистозного фиброза, одним из всего лишь двух таких грантов на момент подачи данной заявки (второй был для Соединения 2). Это показывает, что существует значительная неудовлетворенная потребность в эффективном лечении причины кистозного фиброза, а не в симптоматическом лечении. Кроме того, основной сложностью, связанной с одобренными FDA лекарственными препаратами, является возникающая время от времени недоступность лекарственного препарата для нуждающихся в нем пациентов. Соответственно, существует неудовлетворенная потребность в раскрытых здесь лекарственных формах Соединения 1 и Соединения 2 и процессе их приготовления непрерывным и контролируемым способом.
Кроме того, способность пациента соблюдать графики лечения и величину дозы по существу зависит от простоты введения лекарственного препарата. Фармацевтическая композиция, включающая фиксированные величины доз корректирующего и потенцирующего CFTR вещества, в которой твердые формы названного корректирующего и потенцирующего вещества стабильны, является значимым прорывом в лечении CFTR-опосредованных заболеваний, таких как кистозный фиброз.
Краткое содержание изобретения
Изобретение содержит фармацевтические композиции, включающие 3-(6-(1-(2,2-дифторбензол[d][1,3]диоксол-5-ил)циклопропанкарбоксамидо)-3-метилпиридин-2-ил)бензойную кислоту, Соединение 1 Форму I, со структурой, приведенной ниже:
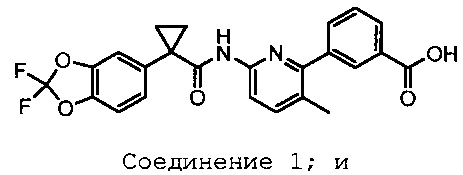
твердую дисперсию по существу аморфного N-(5-гидрокси-2,4-дитрет-бутил-фенил)-4-оксо-1Н-хинолин-3-карбоксамида, Соединение 2, со структурой, приведенной ниже:
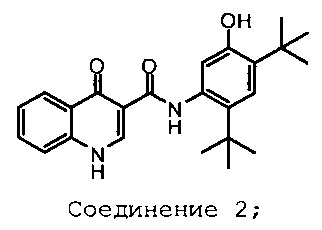
способы лечения, способы производства, способы введения и наборы реактивов с ними.
В одном аспекте настоящее изобретение содержит фармацевтическую композицию, включающую:
а. Соединение 1 Форма I;
б. твердую дисперсию по существу аморфного Соединения 2;
в. наполнитель;
г. разрыхлитель;
д. сурфактант; и
е. связывающее вещество;
называемую PC-I.
В одном варианте применения изобретения фармацевтические композиции, являющиеся предметом настоящего изобретения, включают от 30 до 55 процентов по массе Соединения 1 Формы I и от 10 до 45 процентов по массе твердой дисперсии, включающей по существу аморфное Соединение 2.
В одном варианте применения изобретения наполнитель выбирают из целлюлозы, модифицированной целлюлозы, натриевой карбоксиметилцеллюлозы, этилцеллюлозы, гидроксиметилцеллюлозы, гидроксипропилцеллюлозы, ацетата целлюлозы, микрокристаллической целлюлозы, двузамещенного фосфата кальция, сахарозы, лактозы, кукурузного крахмала, картофельного крахмала или любой их комбинации. В другом варианте применения изобретения наполнитель является микрокристаллической целлюлозой и присутствует в количестве, варьирующем в интервале от 10 до 20 процентов по массе.
В одном варианте применения изобретения разрыхлитель выбирают из агар-агара, альгинов, карбоната кальция, карбоксиметилцеллюлозы, целлюлозы, гидроксипропилцеллюлозы, гидроксипропилцеллюлозы с низкой степенью замещения, глин, кроскармеллозы натрия, кросповидона, камедей, алюмосиликата магния, метилцеллюлозы, полакрилина калия, альгината натрия, натрия крахмалгликолята, кукурузного крахмала, картофельного крахмала, тапиокового крахмала или любой их комбинации. В другом варианте применения изобретения разрыхлитель является кроскармеллозой натрия и присутствует в количестве, варьирующем в интервале от 1 до 3 процентов по массе.
В одном варианте применения изобретения сурфактант выбирают из лаурилсульфата натрия, стеарилфумарата натрия, полиоксиэтилен 20 сорбитан моноолеата или любой их комбинации. В другом варианте применения изобретения сурфактант является лаурилсульфатом натрия и присутствует в количестве, варьирующем в интервале от 0,5 до 2 процентов по массе.
В одном варианте применения изобретения связывающее вещество выбирают из поливинилпирролидона, двузамещенного фосфата кальция, сахарозы, кукурузного крахмала, модифицированной целлюлозы или любой их комбинации. В другом варианте применения изобретения связывающее вещество является поливинилпирролидоном и присутствует в количестве, варьирующем в интервале от 0 до 5 процентов по массе.
В одном варианте применения изобретения настоящее изобретение содержит фармацевтическую композицию следующего состава:
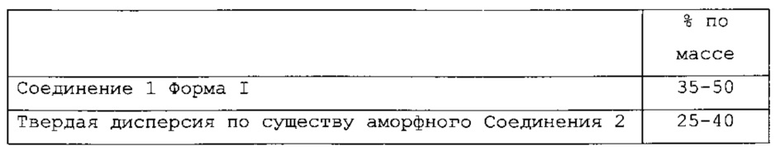
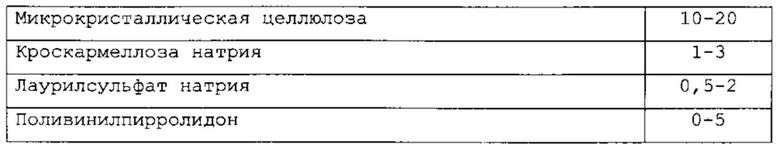
называемую PC-II.
В другом аспекте настоящее изобретение содержит фармацевтическую композицию, включающую:
а. Соединение 1 Форма I;
б. твердую дисперсию, включающую по существу аморфного Соединения 2;
в. наполнитель;
г. разрыхлитель;
д. сурфактант;
е. связывающее вещество; и
ж. смазывающее вещество;
называемую PC-III.
В одном варианте применения изобретения фармацевтические композиции, являющиеся предметом настоящего изобретения, включают приблизительно от 100 до 250 мг Соединения 1 Формы I и приблизительно от 100 до 150 мг по существу аморфного Соединения 2. В другом варианте применения изобретения фармацевтические композиции, являющиеся предметом настоящего изобретения, включают около 200 мг Соединения 1 Формы I и около 125 мг по существу аморфного Соединения 2. В другом варианте применения изобретения фармацевтические композиции, являющиеся предметом настоящего изобретения, включают около 150 мг Соединения 1 Формы I и около 125 мг по существу аморфного Соединения 2.
В одном из вариантов применения изобретения фармацевтические композиции, являющиеся предметом настоящего изобретения, включают от 25 до 50 процентов по массе Соединения 1 Формы I и от 15 до 35 процентов по массе твердой дисперсии, включающей по существу аморфное Соединение 2.
В одном варианте применения изобретения наполнитель выбирают из целлюлозы, модифицированной целлюлозы, натриевой карбоксиметилцеллюлозы, этилцеллюлозы, гидроксиметилцеллюлозы, гидроксипропилцеллюлозы, ацетата целлюлозы, микрокристаллической целлюлозы, двузамещенного фосфата кальция, сахарозы, лактозы, кукурузного крахмала, картофельного крахмала или любой их комбинации. В другом варианте применения изобретения наполнитель является микрокристаллической целлюлозой и присутствует в количестве, варьирующем в интервале от 20 до 30 процентов по массе.
В одном варианте применения изобретения разрыхлитель выбирают из агар-агара, альгинов, карбоната кальция, карбоксиметилцеллюлозы, целлюлозы, гидроксипропилцеллюлозы, гидроксипропилцеллюлозы с низкой степенью замещения, глин, кроскармеллозы натрия, кросповидона, камедей, алюмосиликата магния, метилцеллюлозы, полакрилина калия, альгината натрия, натрия крахмалгликолята, кукурузного крахмала, картофельного крахмала, тапиокового крахмала или любой их комбинации. В другом варианте применения изобретения разрыхлитель является кроскармеллозой натрия и присутствует в количестве, варьирующем в интервале от 3 до 10 процентов по массе.
В одном варианте применения изобретения сурфактант выбирают из лаурилсульфата натрия, стеарилфумарата натрия, полиоксиэтилен 20 сорбитан моноолеата или любой их комбинации. В другом варианте применения изобретения сурфактант является лаурилсульфатом натрия и присутствует в количестве, варьирующем в интервале от 0,5 до 2 процентов по массе.
В одном варианте применения изобретения связывающее вещество выбирают из поливинилпирролидона, двузамещенного фосфата кальция, сахарозы, кукурузного крахмала, модифицированной целлюлозы или любой их комбинации. В другом варианте применения изобретения связывающее вещество является поливинилпирролидоном и присутствует в количестве, варьирующем в интервале от 0 до 5 процентов по массе.
В одном варианте применения изобретения смазывающее вещество выбирают из стеарата магния, стеарата кальция, стеарата цинка, стеарата натрия, стеариновой кислоты, стеарата алюминия, лейцина, глицерилбегената, гидрогенизированного растительного масла или любой их комбинации. В другом варианте применения изобретения смазывающее вещество является стеаратом магния и присутствует в количестве, варьирующем в интервале от 0,5 до 2 процентов по массе.
В одном варианте применения изобретения настоящее изобретение содержит фармацевтическую композицию следующего состава:

называемую PC-IV.
В одном варианте применения изобретения фармацевтические композиции, являющиеся предметом настоящего изобретения, далее включают краситель или, при желании, воск. В другом варианте применения изобретения краситель присутствует в количестве, варьирующем в интервале от 2 до 4 процентов по массе. В другом варианте применения изобретения воск является карнаубским воском, присутствующим в количестве, варьирующем в интервале от 0 до 0,020 процентов по массе.
В одном варианте применения изобретения фармацевтические композиции, являющиеся предметом настоящего изобретения, являются твердыми пероральными фармацевтическими композициями. В другом варианте применения изобретения твердые пероральные фармацевтические композиции являются гранулярной фармацевтической композицией или таблеткой.
В одном варианте применения изобретения гранулярные фармацевтические композиции, являющиеся предметом настоящего изобретения, имеют следующий состав:

и называются PC-V.
В одном варианте применения изобретения гранулярные фармацевтические композиции, являющиеся предметом настоящего изобретения, имеют следующий состав:

и называются PC-VI.
В одном варианте применения изобретения гранулярные фармацевтические композиции, являющиеся предметом настоящего изобретения, имеют следующий состав:

и называются PC-VII.
В одном варианте применения изобретения таблетки, являющиеся предметом настоящего изобретения, имеют следующий состав:

и называются PC-VTII.
В одном варианте применения изобретения таблетки, являющиеся предметом настоящего изобретения, имеют следующий состав:

и называются PC-IX.
В одном варианте применения изобретения таблетки, являющиеся предметом настоящего изобретения, имеют следующий состав:
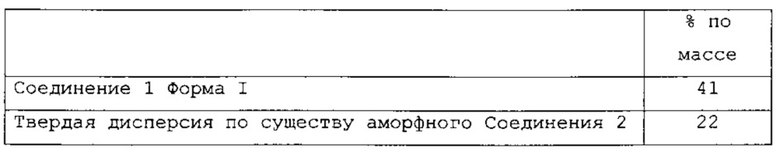
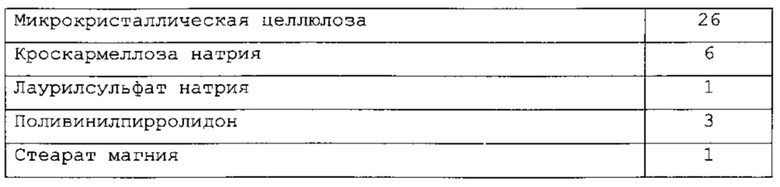
и называются РС-Х.
В одном варианте применения изобретения таблетки, являющиеся предметом настоящего изобретения, имеют следующий состав:

и называются PC-XI.
В одном варианте применения изобретения таблетки, являющиеся предметом настоящего изобретения, имеют следующий состав:

и называются PC-XII.
В одном варианте применения изобретения таблетки, являющиеся предметом настоящего изобретения, имеют следующий состав:

и называются PC-XIII.
В одном варианте применения изобретения таблетки, являющиеся предметом настоящего изобретения, имеют следующий состав:

и называются PC-XIV.
В одном варианте применения изобретения таблетки, являющиеся предметом настоящего изобретения, имеют следующий состав:


и называются PC-XV.
В одном варианте применения изобретения таблетки, являющиеся предметом настоящего изобретения, имеют следующий состав:

и называются PC-XVI.
В одном варианте применения изобретения таблетки, являющиеся предметом настоящего изобретения, имеют следующий состав:

и называются PC-XVII.
В одном варианте применения изобретения таблетки, являющиеся предметом настоящего изобретения, имеют следующий состав:

и называются PC-XVIII.
В одном варианте применения изобретения таблетки, являющиеся предметом настоящего изобретения, имеют следующий состав:

и называются PC-XIX.
В одном варианте применения изобретения таблетки, являющиеся предметом настоящего изобретения, имеют следующий состав:


и называются РС-ХХ.
В одном варианте применения изобретения таблетки, являющиеся предметом настоящего изобретения, имеют следующий состав:

и называются PC-XXI.
В одном варианте применения изобретения таблетки, являющиеся предметом настоящего изобретения, имеют следующий состав:

и называются PC-XXII.
В одном варианте применения изобретения таблетки, являющиеся предметом настоящего изобретения, имеют следующий состав:
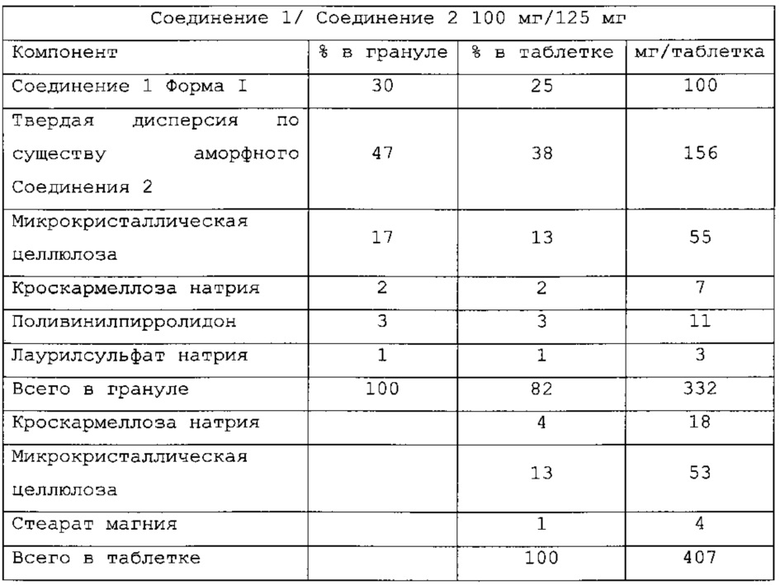
и называются PC-XXIII.
В одном варианте применения изобретения таблетки, являющиеся предметом настоящего изобретения, имеют следующий состав:
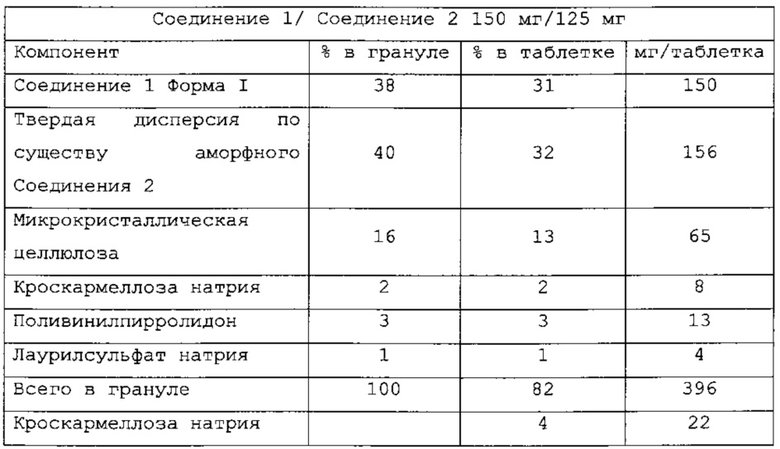

и называются PC-XXIV.
В одном варианте применения изобретения таблетки, являющиеся предметом настоящего изобретения, имеют следующий состав:
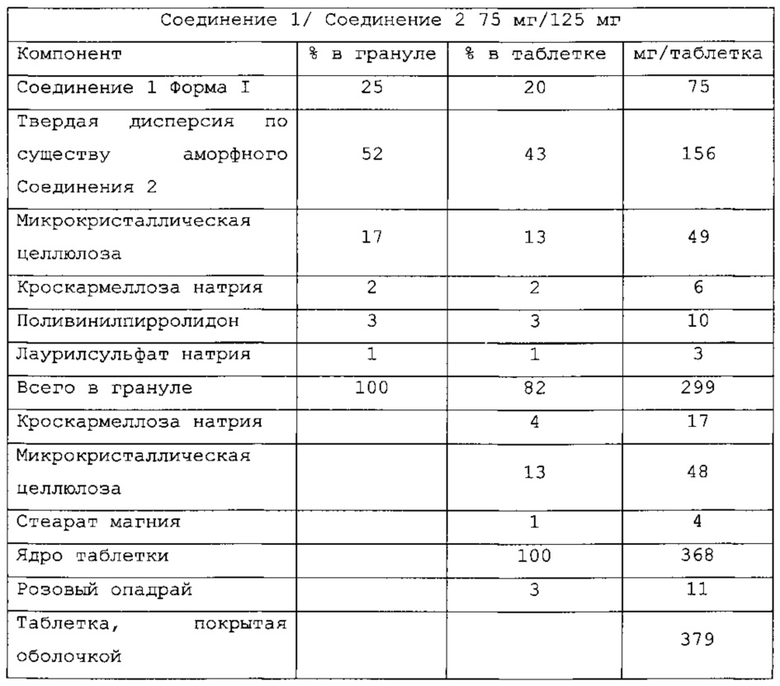
и называются PC-XXV.
В одном аспекте настоящее изобретение содержит способ лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включая введение пациенту эффективного количества фармацевтической композиции, гранулярной фармацевтической композиции или таблеток, являющихся предметом настоящего изобретения.
Во варианте применения изобретения настоящее изобретение содержит способ лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включая введение пациенту эффективного количества фармацевтической композиции, гранулярной фармацевтической композиции или таблеток любого состава от PC-I до PC-XXV.
В одном варианте применения изобретения у пациента есть ΔF508 мутация CFTR. В другом варианте применения изобретения пациент гомозиготен по ΔF508. В другом варианте применения изобретения пациент гетерозиготен по ΔF508. В другом варианте применения изобретения пациенту вводят две таблетки в день.
В одном аспекте настоящее изобретение содержит способ изготовления гранулярной фармацевтической композиции, включающий влажное гранулирование следующих компонентов:
а. Соединения 1 Формы I;
б. твердой дисперсии, включающей по существу аморфное Соединение 2;
в. наполнителя;
г. разрыхлителя;
д. сурфактанта; и
е. связывающего вещества.
В одном аспекте настоящее изобретение содержит способ изготовления таблетки, включающий прессование:
i) множества гранулярных фармацевтических композиций, включающих следующие компоненты:
а. Соединение 1 Формы I;
б. твердую дисперсию, включающую по существу аморфное Соединение 2;
в. наполнитель;
г. разрыхлитель;
д. сурфактант; и
е. связывающее вещество;
ii) разрыхлитель;
iii) наполнитель; и
iv) смазывающее вещество.
В одном аспекте настоящее изобретение содержит набор реактивов, включающий фармацевтические композиции, гранулярные фармацевтические композиции или таблетки, являющиеся предметом настоящего изобретения, и отдельный терапевтический агент или его фармацевтическую композицию.
В одном варианте применения фармацевтические композиции, гранулярные фармацевтические композиции или таблетки, являющиеся предметом настоящего изобретения, и отдельный терапевтический агент или его фармацевтическая композиция находятся в отдельных контейнерах. В другом варианте применения изобретения отдельные контейнеры являются бутылями. В другом варианте применения изобретения отдельные контейнеры являются флаконами. В другом варианте применения изобретения отдельные контейнеры являются блистерными упаковками.
В другом аспекте изобретение представляет непрерывный или полунепрерывный процесс изготовления фармацевтических композиций, описанных здесь, посредством двухшнекового процесса влажной грануляции, включающий стадии скрининга и взвешивания Соединения 1, Соединения 2 и вспомогательных веществ, смешивания Соединения 1, Соединения 2 и вспомогательных веществ в смесителе и подачи смеси в непрерывный гранулятор при добавлении гранулирующей жидкости, включающей сурфактант и связывающее вещество, при подходящей скорости в течение подходящего количества времени и перемалывания смеси в гранулы; сушку гранул; перемешивание гранул с экстрагранулярными вспомогательными веществами в течение подходящего количества времени; прессование смеси в таблетки; покрытие таблеток оболочкой; и, при желании, печать монограммы на одной или двух сторонах таблетки.
Краткое описание чертежей
Фиг. 1 является дифракционной рентгенограммой, рассчитанной по монокристаллической структуре Соединения 1 Формы I.
Фиг. 2 является порошковой дифракционной рентгенограммой Соединения 1 Формы I.
Фиг. 3 является графиком, показывающим профили растворимости в градиенте рН Соединения 1 для таблеток, изготовленных посредством процесса грануляции с большим усилием сдвига (HSG, high shear granulation) и двухшнекового процесса влажной грануляции (TSWG, twin screw wet granulation) (LOD соответствует потере при сушке, мере определения количества воды в порошке/грануле).
Фиг. 4 является графиком, показывающим стабильность по существу аморфной формы Соединения 2 в форме таблеток PC-XVII при 50°С после предварительного уравновешивания при 60% относительной влажности, демонстрируя лишь невысокую степень кристаллизации в течение времени.
Фиг. 5 является графиком, показывающим стабильность по существу аморфной формы Соединения 2 в форме таблеток PC-XVII при 60°С после предварительного уравновешивания при 60% относительной влажности, демонстрируя лишь невысокую степень кристаллизации в течение времени.
Фиг. 6 является графиком, показывающим стабильность по существу аморфной формы Соединения 2 в форме таблеток РС-ХХ при 60°С после предварительного уравновешивания при 60% относительной влажности, демонстрируя лишь невысокую степень кристаллизации в течение времени.
Фиг. 7 является графиком, показывающим стабильность по существу аморфной формы Соединения 2 в форме таблеток РС-ХХ при 50°С после предварительного уравновешивания при 60% относительной влажности, демонстрируя лишь невысокую степень кристаллизации в течение времени.
Фиг. 8 является 1Н-ЯМР спектром Соединения 1.
Фиг. 9 является 1Н-ЯМР спектром HCl соли Соединения 1.
Фиг. 10 является кривой дифференциальной сканирующей калориметрии Соединения 1 Формы I.
Фиг. 11 является конформационным изображением Соединения 1 Формы I на основе монокристаллического рентгенографического анализа.
Фиг. 12 является картой производственного процесса, описывающей получение таблетки из Соединения 1 и Соединения 2 непрерывными способами.
Подробное описание изобретения
ОПРЕДЕЛЕНИЯ
''CFTR'' в настоящей заявке означает регулятор трансмембранной проводимости кистозного фиброза.
''ΔF508 мутация'' или ''F508-del мутация'' в настоящей заявке является специфичной мутацией в белке CFTR. Эта мутация представляет собой делецию трех нуклестидов, которые включают ко дон аминокислоты фенилаланина в положении 508, приводящую к нехватке этого остатка фенилаланина в белке CFTR.
В настоящей заявке пациент, ''гомозиготный'' по специфичной мутации, например, ΔF508, имеет одинаковую мутацию в каждой аллели.
В настоящей заявке пациент, ''гетерозиготный'' по специфичной мутации, например, ΔF508, имеет эту мутацию в одной аллели и другую мутацию в другой аллели.
В настоящей заявке термин ''корректирующее CFTR вещество'' относится к соединению, которое увеличивает количество функционального белка CFTR на поверхности клетки, приводя к увеличению ионного транспорта.
В настоящей заявке термин ''потенцирующее CFTR вещество'' относится к соединению, которое увеличивает канальную активность белка CFTR, расположенного на поверхности клетки, приводя к увеличению ионного транспорта.
В настоящей заявке термин ''активный фармацевтический ингредиент'' или ''АФИ'' относится к биологически активному соединению.
Термин ''твердая форма'', ''твердые формы'' и связанные термины в настоящей заявке относятся к Соединению 1 или Соединению 2, в специальной твердой форме, например, кристаллах, аморфных состояниях и подобном.
В настоящей заявке термин ''по существу аморфный'' относится к твердому материалу, имеющему малый или не дальний порядок расположения молекул. Например, по существу аморфные материалы обладают степенью кристаллизации менее 15% (например, степенью кристаллизации менее 10% или степенью кристаллизации менее 5%). Также отмечается, что термин ''по существу аморфный'' включает определение ''аморфный'', которое относится к материалу без кристаллической структуры (степень кристаллизации 0%).
В настоящей заявке термин ''по существу кристаллический'' (как во фразе ''по существу кристаллическое Соединение 1 Форма I'') относится к твердому материалу, обладающему по существу дальним порядком расположения молекул. Например, по существу кристаллические материалы обладают степенью кристаллизации более 85% (например, степенью кристаллизации более 90% или степенью кристаллизации более 95%). Также отмечается, что термин ''по существу кристаллический'' включает определение ''кристаллический'', которое относится к материалу со степенью кристаллизации 100%.
Термин ''кристаллический'' и связанные термины в настоящей заявке при применении для описания вещества, компонента, продукта или формы, означает, что вещество, компонент или продукт является по существу кристаллическим, согласно определению с помощью рентгенодифракции (См., например, Remington: The Science and Practice of Pharmacy, 21st Ed., Lippincott Williams & Wilkins, Baltimore, Md. (2003); The United States Pharmacopeia, 23rd ed., 1843-1844 (1995)).
В настоящей заявке термин ''вспомогательное вещество'' включает функциональные и нефункциональные ингредиенты фармацевтической композиции.
В настоящей заявке термин ''разрыхлитель'' является вспомогательным веществом, которое гидратирует фармацевтическую композицию и способствует диспергированию таблетки. В настоящей заявке ''растворитель'' или ''наполнитель'' является вспомогательным веществом, которое добавляет объемность фармацевтической композиции.
В настоящей заявке термин ''сурфактант'' является вспомогательным веществом, которое придает фармацевтическим композициям повышенную растворимость и/или способность к увлажнению.
В настоящей заявке термин ''связывающее вещество'' является вспомогательным веществом, которое придает фармацевтической композиции повышенную когезионную прочность и повышенный предел прочности на разрыв (например, твердость).
В настоящей заявке термин ''вещество, способствующее скольжению'' является вспомогательным веществом, которое придает фармацевтическим композициям свойства повышенной текучести.
В настоящей заявке термин ''краситель'' является вспомогательным веществом, которое придает фармацевтической композиции, например, таблетке, желаемый цвет. Примеры красителей включают коммерчески доступные пигменты, такие как FD&C Blue # 1 Aluminum Lake, FD&C Blue #2, другие красители FD&C Blue, оксид титана, оксид железа и/или их комбинации. В одном варианте применения изобретения таблетка, представляемая изобретением, розовая.
В настоящей заявке термин ''смазывающее вещество'' является вспомогательным веществом, которое добавляют в фармацевтические композиции, которые прессуют в таблетки. Смазывающее вещество способствует компактизации гранул в таблетки и выходу таблетки фармацевтической композиции из штамповочного пресса.
В настоящей заявке ''кубический сантиметр'' и ''см3'' являются взаимозаменяемыми и применяются для представления единиц объема. Следует отметить, что 1 см3 = 1 мл.
В настоящей заявке ''килоПонд'' и ''кП'' являются взаимозаменяемыми и применяются для измерения силы, где кП = приблизительно 9,8 Ньютонов.
В настоящей заявке ''прочность таблеток на истирание'' относится к свойству таблетки оставаться интактной и сохранять форму под давлением внешней силы. Прочность таблеток на истирание можно оценить количественно, применяя математическую формулу, представленную в уравнении 1
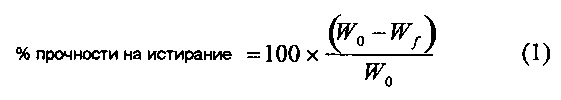
где W0 является исходным весом таблетки и Wf является конечным весом таблетки после пропускания через фриабилятор. Прочность таблеток на истирание измеряют с помощью аппарата для стандартного теста Фармакопеи США, который испытывает экспериментальные таблетки в барабане при 100 или 400 оборотах. У некоторых таблеток, являющихся предметом изобретения, прочность на истирание менее 5,0%. В другом варианте применения изобретения прочность на истирание менее 2,0%. В другом варианте применения изобретения целевая прочность на истирание менее 1,0% после 400 оборотов.
В настоящей заявке ''средний диаметр частиц'' является средним диаметром частиц, измеренным с помощью таких техник, как рассеяние лазерного излучения, анализ изображений или ситовый анализ. В одном варианте применения изобретения средний размер частиц гранул, применяемых для приготовления фармацевтических композиций, представленных изобретением, составляет менее 1,0 мм.
В настоящей заявке ''насыпная плотность'' является массой частиц материала, деленной на общий объем, занимаемый частицами. Общий объем включает объем частиц, свободный объем между частицами и объем внутренних пор. Насыпной объем не является внутренним свойством материала; он может изменяться в зависимости от обработки материала. В одном варианте применения изобретения насыпная плотность гранул, применяемых для изготовления фармацевтических композиций, представленных изобретением, составляет около 0,5-0,7 г/см3.
''Эффективное количество'' или ''терапевтически эффективное количество'' соединения, являющегося предметом изобретения, может варьировать в соответствии с такими факторами, как состояние, возраст и вес субъекта, и со способностью соединения, являющегося предметом изобретения, вызывать желаемый ответ у субъекта. Режимы дозирования можно подстраивать для обеспечения оптимального терапевтического ответа. Эффективное количество также является количеством, при котором положительные терапевтические эффекты превосходят любые токсические или отрицательные эффекты (например, побочные эффекты) соединения, являющегося предметом настоящего изобретения.
В настоящей заявке и если иное не оговорено, термины ''терапевтически эффективное количество'' и ''эффективное количество'' соединения означают количество, достаточное для обеспечения терапевтической пользы при лечении или контроле заболевания или для отсрочки или минимизации одного или более симптомов, ассоциированных с заболеванием или нарушением. В настоящей заявке и если иное не оговорено, термины ''терапевтически эффективное количество'' и ''эффективное количество'' соединения означают количество терапевтического агента, в индивидуальном виде или в комбинации с одним или более другими агентами, которое обеспечивает терапевтическую пользу при лечении или контроле заболевания или нарушения. Термины ''терапевтически эффективное количество'' и ''эффективное количество'' могут включать количество, которое улучшает терапию в целом, снижает или предотвращает симптомы или причины заболевания или нарушения, или усиливает терапевтическую эффективность другого терапевтического агента.
''По существу чистый'' как применяется во фразе ''по существу чистое Соединение 1 Формы I'' означает более чем 90% чистоту. В другом варианте применения изобретения ''по существу чистый'' относится к более чем 95% чистоте. В другом варианте применения изобретения ''по существу чистый'' относится к более чем 98% чистоте. В другом варианте применения изобретения ''по существу чистый'' относится к более чем 99% чистоте.
В отношении Соединения 1 Формы I или твердой дисперсии, включающей по существу аморфное Соединение 2 термины ''около'' и ''приблизительно'', применяемые отношении процента дозы, количества или веса ингредиентов композиции или лекарственной формы, означают процент дозы, количества или веса, который, по мнению среднего специалиста в данной области техники, обеспечивает фармакологический эффект, эквивалентный эффекту от специально установленного процента дозы, количества или веса. В частности, термин ''около'' и ''приблизительно'' означает приемлемую ошибку для определенного значения, определяемую средним специалистом в данной области техники, которая частично зависит от того, как измеряют или определяют значение. В определенных вариантах применения изобретения термин ''около'' и ''приблизительно'' означает в пределах 1, 2, 3 или 4 стандартных отклонений. В определенных вариантах применения изобретения термин ''около'' и ''приблизительно'' означает внутри 30%, 25%, 20%, 15%, 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1%, 0,5%, 0,1% или 0,05% заданного значения или интервала.
ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ
Изобретение представляет фармацевтические композиции, включающие Соединение 1 Форму I и твердую дисперсию, включающую по существу аморфное Соединение 2. В некоторых вариантах применения изобретения в данном аспекте количество Соединения 1 Формы I, присутствующего в фармацевтической композиции, составляет 100 мг, 125 мг, 150 мг, 200 мг, 250 мг, 300 мг или 400 мг. В некоторых вариантах применения изобретения в данном аспекте процент по массе Соединения 1 Формы I, присутствующего в фармацевтической композиции, составляет от 10 до 75 процентов. В этих и других вариантах применения изобретения Соединение 1 Формы I присутствует в виде по существу чистого Соединения 1 Формы I. В некоторых вариантах применения изобретения в данном аспекте количество по существу аморфного Соединения 2, присутствующего в фармацевтической композиции, составляет 100 мг, 125 мг, 150 мг, 200 мг или 250 мг. В некоторых вариантах применения изобретения в данном аспекте процент по массе по существу аморфного Соединения 2, присутствующего в фармацевтической композиции, составляет от 10 до 75 процентов. В этих и других вариантах применения изобретения по существу аморфное Соединение 2 присутствует в виде по существу чистого и аморфного Соединения 2. ''По существу чистое'' означает более чистое более чем на девяносто процентов; предпочтительно более чем 95% чистое; более предпочтительно 99,5% чистое.
Таким образом, в одном аспекте изобретение представляет фармацевтическую композицию, включающую:
а. Соединение 1 Форма I;
б. твердую дисперсию по существу аморфного Соединения 2;
в. наполнитель;
г. разрыхлитель;
д. сурфактант; и
е. связывающее вещество.
В одном варианте применения изобретения в данном аспекте фармацевтическая композиция включает 25 мг Соединения 1 Формы I. В другом варианте применения изобретения в данном аспекте фармацевтическая композиция включает 50 мг Соединения 1 Формы I. В другом варианте применения изобретения в данном аспекте фармацевтическая композиция включает 100 мг Соединения 1 Формы I. В другом варианте применения изобретения в данном аспекте фармацевтическая композиция включает 125 мг Соединения 1 Формы I. В другом варианте применения изобретения в данном аспекте фармацевтическая композиция включает 150 мг Соединения 1 Формы I. В другом варианте применения изобретения в данном аспекте фармацевтическая композиция включает 200 мг Соединения 1 Формы I. В другом варианте применения изобретения в данном аспекте фармацевтическая композиция включает 250 мг Соединения 1 Формы I. В другом варианте применения изобретения в данном аспекте фармацевтическая композиция включает 400 мг Соединения 1 Формы I.
В одном варианте применения изобретения в данном аспекте фармацевтическая композиция включает 25 мг по существу аморфного Соединения 2. В другом варианте применения изобретения в данном аспекте фармацевтическая композиция включает 50 мг по существу аморфного Соединения 2. В другом варианте применения изобретения в данном аспекте фармацевтическая композиция включает 100 мг по существу аморфного Соединения 2. В другом варианте применения изобретения в данном аспекте фармацевтическая композиция включает 125 мг по существу аморфного Соединения 2. В другом варианте применения изобретения в данном аспекте фармацевтическая композиция включает 150 мг по существу аморфного Соединения 2. В другом варианте применения изобретения в данном аспекте фармацевтическая композиция включает 200 мг по существу аморфного Соединения 2. В другом варианте применения изобретения в данном аспекте фармацевтическая композиция включает 250 мг по существу аморфного Соединения 2.
В некоторых вариантах применения изобретения фармацевтическая композиция включает Соединение 1 Форму I, где Соединение 1 Форма I присутствует в количестве, составляющем по меньшей мере 15% по массе от композиции (например, по меньшей мере 20% по массе, по меньшей мере 30% по массе, по меньшей мере 40% по массе, по меньшей мере 50% по массе или по меньшей мере 60% по массе).
В некоторых вариантах применения изобретения фармацевтическая композиция включает по существу аморфное Соединение 2, где по существу аморфное Соединение 2 присутствует в количестве, составляющем по меньшей мере 15% по массе от композиции (например, по меньшей мере 20% по массе, по меньшей мере 30% по массе, по меньшей мере 40% по массе, по меньшей мере 50% по массе или по меньшей мере 60% по массе).
В некоторых вариантах применения изобретения фармацевтическая композиция включает Соединение 1 Форму I, твердую дисперсию, включающую по существу аморфное Соединение 2, наполнитель, разрыхлитель, сурфактант и связывающее вещество. В данном варианте применения изобретения композиция включает приблизительно от 25% по массе до 55% по массе (например, около 30-50% по массе) Соединения 1 Формы I по массе композиции, и более часто, от 40% по массе приблизительно до 45% по массе Соединения 1 Формы I по массе от композиции. В данном варианте применения изобретения фармацевтическая композиция включает приблизительно от 15% по массе приблизительно до 40% по массе (например, около 20-35% по массе) по существу аморфного Соединения 2 по массе от композиции, и более часто, от 25% по массе приблизительно до 30% по массе по существу аморфного Соединения 2 по массе от композиции.
Концентрация Соединения 1 Формы I и по существу аморфного Соединения 2 в композиции зависит от нескольких факторов, таких как количество фармацевтической композиции, необходимой для обеспечения желаемого количества Соединения 1 Формы I и по существу аморфного Соединения 2, и желаемого профиля растворимости фармацевтической композиции.
В другом варианте применения изобретения фармацевтическая композиция включает Соединение 1 Форму I, в котором Соединение 1 Формы I в твердой форме имеет средний диаметр частиц, измеренный с помощью светового рассеяния (например, с помощью Malvern Mastersizer производства Malvern Instruments в Англии), равный от 0,1 микрон до 10 микрон. В другом варианте применения изобретения размер частиц Соединения 1 Формы I составляет от 1 микрона до 5 микрон. В другом варианте применения изобретения размер частиц D50 Соединения 1 Формы I составляет 2,0 микрона.
Как указано, в дополнение к Соединению 1 Формы I и твердой дисперсии по существу аморфного Соединения 2 в некоторых вариантах применения изобретения фармацевтические композиции в форме для перорального применения также включают одно или более вспомогательные вещества, такие как наполнители, разрыхлители, сурфактанты, растворители, связывающие вещества, вещества, способствующие скольжению, смазывающие вещества, красители или ароматизаторы и любые их комбинации.
Наполнители, подходящие для изобретения, совместимы с ингредиентами фармацевтической композиции, т.е. они не снижают в значительной степени растворимость, твердость, химическую стабильность, физическую стабильность или биологическую активность фармацевтической композиции. Примеры наполнителей включают: целлюлозы, модифицированные целлюлозы (например, натриевую карбоксиметилцеллюлозу, этилцеллюлозу, гидроксиметилцеллюлозу, гидроксипропилцеллюлозу), ацетат целлюлозы, микрокристаллическую целлюлозу, фосфат кальция, двузамещенный фосфат кальция, крахмалы (например, кукурузный крахмал, картофельный крахмал), сахара (например, сорбитол, лактозу, сахарозу или подобные) или любую их комбинации.
Таким образом, в одном варианте применения изобретения фармацевтическая композиция включает по меньшей мере один наполнитель в количестве равном по меньшей мере 5% по массе от композиции (например, по меньшей мере 20% по массе, по меньшей мере 30% по массе или по меньшей мере 40% по массе). Например, фармацевтическая композиция включает приблизительно от 10% по массе приблизительно до 60% по массе наполнителя (например, приблизительно от 20% по массе приблизительно до 55% по массе, приблизительно от 25% по массе приблизительно до 50% по массе или приблизительно от 27% по массе приблизительно до 45% по массе), по массе от композиции. В другом примере фармацевтическая композиция включает по меньшей мере около 20% по массе (например, по меньшей мере 30% по массе или по меньшей мере 40% по массе) микрокристаллической целлюлозы, например МСС Avicel PH102, по массе от композиции. В еще одном примере фармацевтическая композиция включает приблизительно от 10% по массе приблизительно до 60% по массе (например, приблизительно от 20% по массе приблизительно до 55% по массе до или приблизительно от 25% по массе приблизительно до 45% по массе) микроцеллюлозы по массе от композиции.
Разрыхлители, подходящие для изобретения, усиливают дисперсность фармацевтической композиции и являются совместимыми с ингредиентами фармацевтической композиции, т.е. они не снижают в значительной степени химическую стабильность, физическую стабильность, твердость или биологическую активность фармацевтической композиции. Примеры разрыхлителей включают кроскармеллозу натрия, натрия крахмалгликолят или их комбинацию.
Таким образом, в одном варианте применения изобретения фармацевтическая композиция включает разрыхлитель в количестве около 10% по массе или менее (например, около 7% по массе или менее, около 6% по массе или менее, или около 5% по массе или менее) по массе от композиции. Например, фармацевтическая композиция включает приблизительно от 1% по массе приблизительно до 10% по массе (например, приблизительно от 1,5% по массе приблизительно до 7,5% по массе или приблизительно от 2,5% по массе приблизительно до 6% по массе) разрыхлителя, по массе от композиции. В другом примере фармацевтическая композиция включает приблизительно от 10% по массе или менее (например, 7% по массе или менее, 6% по массе или менее или 5% по массе или менее) кроскармеллозы натрия, по массе от композиции. В еще одном примере фармацевтическая композиция включает приблизительно от 1% по массе приблизительно до 10% по массе (например, приблизительно от 1,5% по массе приблизительно до 7,5% по массе или приблизительно от 2,5% по массе приблизительно до 6% по массе) кроскармеллозы натрия, по массе от композиции. В некоторых примерах фармацевтическая композиция включает приблизительно от 0,1% по массе приблизительно до 10% по массе (например, приблизительно от 0,5% по массе приблизительно до 7,5% по массе или приблизительно от 1,5% по массе приблизительно до 6% по массе) разрыхлителя, по массе от композиции. В других примерах фармацевтическая композиция включает приблизительно от 0,5% по массе приблизительно до 10% по массе (например, приблизительно от 1,5% по массе приблизительно до 7,5% по массе или приблизительно от 2,5% по массе приблизительно до 6% по массе) разрыхлителя, по массе от композиции.
Сурфактанты, подходящие для изобретения, усиливают способность к увлажнению фармацевтической композиции и являются совместимыми с ингредиентами фармацевтической композиции, т.е. они не снижают в значительной степени химическую стабильность, физическую стабильность, твердость или биологическую активность фармацевтической композиции. Примеры сурфактантов включают лаурилсульфат натрия, стеарилфумарат натрия, полиоксиэтилен 20, сорбитан моноолеат (например, Tween™), любую их комбинацию или подобные.
Таким образом, в одном варианте применения изобретения фармацевтическая композиция включает сурфактант в количестве около 10% по массе или менее (например, около 5% по массе или менее, около 2% по массе или менее, около 1% по массе или менее, около 0,8% по массе или менее или около 0,6% по массе или менее) по массе от композиции. Например, фармацевтическая композиция включает приблизительно от 10% по массе приблизительно до 0,1% по массе (например, приблизительно от 5% по массе приблизительно до 0,2% по массе или приблизительно от 2% по массе приблизительно до 0,3% по массе) сурфактанта, по массе от композиции. В другом примере фармацевтическая композиция включает приблизительно от 10% по массе или менее (например, около 5% по массе или менее, около 2% по массе или менее, около 1% по массе или менее, около 0,8% по массе или менее или около 0,6% по массе или менее) лаурилсульфата натрия, по массе от композиции. В еще одном примере фармацевтическая композиция включает приблизительно от 10% по массе приблизительно до 0,1% по массе (например, приблизительно от 5% по массе приблизительно до 0,2% по массе или приблизительно от 2% по массе приблизительно до 0,3% по массе) лаурилсульфата натрия, по массе от композиции.
Связывающие вещества, подходящие для изобретения, усиливают силу действия таблеток фармацевтической композиции и являются совместимыми с ингредиентами фармацевтической композиции, т.е. они не снижают в значительной степени химическую стабильность, физическую стабильность или биологическую активность фармацевтической композиции. Примеры сурфактантов включают поливинилпирролидон, двузамещенный фосфат кальция, сахарозу, кукурузный (маисовый) крахмал, модифицированную целлюлозу (например, гидроксиметилцеллюлозу) или любую их комбинацию.
Таким образом, в одном варианте применения изобретения фармацевтическая композиция включает связывающее вещество в количестве по меньшей мере около 0,1% по массе (например, по меньшей мере около 1% по массе, по меньшей мере около 3% по массе, по меньшей мере около 4% по массе или по меньшей мере около 5% по массе) по массе от композиции. Например, фармацевтическая композиция включает приблизительно от 0,1% по массе приблизительно до 10% по массе (например, приблизительно от 1% по массе приблизительно до 10% по массе или приблизительно от 2% по массе приблизительно до 7% по массе) связывающего вещества, по массе от композиции. В другом примере фармацевтическая композиция включает по меньшей мере около 0,1% по массе (например, по меньшей мере около 1% по массе, по меньшей мере около 2% по массе, по меньшей мере около 3% по массе или по меньшей мере около 4% по массе) поливинилпирролидона, по массе от композиции. В еще одном примере фармацевтическая композиция включает вещество, способствующее скольжению, в количестве, варьирующем приблизительно от 0,1% по массе приблизительно до 10% по массе (например, приблизительно от 1% по массе приблизительно до 8% по массе или приблизительно от 2% по массе приблизительно до 5% по массе) поливинилпирролидона, по массе от композиции.
Растворители, подходящие для изобретения, могут добавлять необходимый объем составу для изготовления таблеток желаемого размера и являются в целом совместимыми с ингредиентами фармацевтической композиции, т.е. они не снижают в значительной степени растворимость, твердость, химическую стабильность, физическую стабильность или биологическую активность фармацевтической композиции. Примеры растворителей включают сахара, например, кондитерский сахар, прессованный сахар, декстраты, декстрин, декстрозу, лактозу, маннитол, сорбитол, целлюлозу и модифицированные целлюлозы, например, порошковую целлюлозу, тальк, фосфат кальция, крахмал или любую их комбинацию.
Таким образом, в одном варианте применения изобретения фармацевтическая композиция включает растворитель в количестве 40% по массе или менее (например, 35% по массе или менее, 30% по массе или менее или 25% по массе или менее или 20% по массе или менее или 15% по массе или менее или 10% по массе или менее) по массе от композиции. Например, фармацевтическая композиция включает приблизительно от 40% по массе приблизительно до 1% по массе (например, приблизительно от 35% по массе приблизительно до 5% по массе или приблизительно от 30% по массе приблизительно до 7% по массе, приблизительно от 25% по массе приблизительно до 10% по массе или приблизительно от 20% по массе приблизительно до 15% по массе) растворителя, по массе от композиции. В другом примере фармацевтическая композиция включает приблизительно 40% по массе или менее (например, 35% по массе или менее, 25% по массе или менее или 15% по массе или менее) маннитола, по массе от композиции. В еще одном примере фармацевтическая композиция включает приблизительно от 35% по массе приблизительно до 1% по массе (например, приблизительно от 30% по массе приблизительно до 5% по массе или приблизительно от 25% по массе приблизительно до 10% по массе) маннитола, по массе от композиции.
Вещества, способствующие скольжению, подходящие для изобретения, усиливают свойства текучести фармацевтической композиции и являются совместимыми с ингредиентами фармацевтической композиции, т.е. они не снижают в значительной степени растворимость, твердость, химическую стабильность, физическую стабильность или биологическую активность фармацевтической композиции. Примеры веществ, способствующих скольжению, включают коллоидный диоксид кремния, тальк или их комбинацию.
Таким образом, в одном варианте применения изобретения фармацевтическая композиция включает вещество, способствующее скольжению, в количестве 2% по массе или менее (например, 1,75% по массе, 1,25% по массе или менее или 1,00% по массе или менее) по массе от композиции. Например, фармацевтическая композиция включает приблизительно от 2% по массе приблизительно до 0,05% по массе (например, приблизительно от 1,5% по массе приблизительно до 0,07% по массе или приблизительно от 1,0% по массе приблизительно до 0,09% по массе) вещества, способствующего скольжению, по массе от композиции. В другом примере фармацевтическая композиция включает приблизительно 2% по массе или менее (например, 1,75% по массе, 1,25% по массе или менее или 1,00% по массе или менее) коллоидного диоксида кремния по массе от композиции. В еще одном примере фармацевтическая композиция включает приблизительно от 2% по массе приблизительно до 0,05% по массе (например, приблизительно от 1,5% по массе приблизительно до 0,07% по массе или приблизительно от 1,0% по массе приблизительно до 0,09% по массе) коллоидного диоксида кремния по массе от композиции.
В некоторых вариантах применения изобретения фармацевтическая композиция может включать пероральную твердую фармацевтическую форму выпуска, которая может включать смазывающее вещество, которое может предотвращать связывание смеси гранул и шариков с поверхностью (например, с поверхностью посуды для смешивания, пресс-формы или пуансона). Смазывающее вещество также может снижать трение между частицами внутри гранулята и улучшать прессование и высвобождение прессованных фармацевтических композиций из штамповочного пресса. Смазывающее вещество также является совместимым с ингредиентами фармацевтической композиции, т.е. они не снижают в значительной степени растворимость, твердость или биологическую активность фармацевтической композиции. Примеры растворителей включают стеарат магния, стеарат кальция, стеарат цинка, стеарат натрия, стеариновую кислоту, стеарат алюминия, лейцин, глицерилбегенат, гидрогенезированное растительное масло или любую их комбинацию. В одном варианте применения изобретения фармацевтическая композиция включает смазывающее вещество в количестве 5% по массе или менее (например, 4,75% по массе, 4,0% по массе или менее или 3,00% по массе или менее, или 2,0% по массе или менее) по массе от композиции. Например, фармацевтическая композиция включает приблизительно от 5% по массе приблизительно до 0,10% по массе (например, приблизительно от 4,5% по массе приблизительно до 0,5% по массе или приблизительно от 3% по массе приблизительно до 1% по массе) смазывающего вещества по массе от композиции. В другом примере фармацевтическая композиция включает приблизительно 5% по массе или менее (например, 4,0% по массе или менее, 3,0% по массе или менее, или 2,0% по массе или менее, или 1,0% по массе или менее) стеарата магния по массе от композиции. В еще одном примере фармацевтическая композиция включает приблизительно от 5% по массе приблизительно до 0,10% по массе (например, приблизительно от 4,5% по массе приблизительно до 0,15% по массе или приблизительно от 3,0% по массе приблизительно до 0,50% по массе) стеарата магния по массе от композиции.
Фармацевтические композиции, являющиеся предметом изобретения, могут, при желании, включать один или более красителей, вкусовых и/или ароматических добавок для улучшения внешнего вида, усиления вкуса и/или запаха композиции. Подходящие красители, вкусовые или ароматические добавки совместимы с ингредиентами фармацевтической композиции, т.е. они не снижают в значительной степени растворимость, химическую стабильность, физическую стабильность, твердость или биологическую активность фармацевтической композиции. В одном варианте применения изобретения фармацевтическая композиция включает краситель, вкусовую и/или ароматическую добавку. В одном варианте применения изобретения фармацевтические композиции, представленные изобретением, пурпурного цвета.
В некоторых вариантах применения изобретения фармацевтическая композиция включает или может производиться в виде таблеток, и таблетки могут быть покрыты красителем и, при желании, отмечены логотипом, другим изображением и/или текстом с помощью подходящей краски. Подходящие красители и краски совместимы с ингредиентами фармацевтической композиции, т.е. они не снижают в значительной степени растворимость, химическую стабильность, физическую стабильность, твердость или биологическую активность фармацевтической композиции. Подходящие красители и краски могут быть любого цвета, на водной основе или на основе растворителя. В одном варианте применения изобретения таблетки, сделанные из фармацевтической композиции, покрыты красителем, а затем отмечены логотипом, другим изображением и/или текстом с помощью подходящей краски. Например, таблетки, включающие фармацевтическую композицию, как описано в данной заявке, могут быть покрыты 3% по массе (например, менее чем приблизительно 6% по массе или менее чем приблизительно 4% по массе) пленочным покрытием, содержащим краситель. Цветные таблетки могут быть отмечены логотипом и текстом, показывающим силу действующего вещества в таблетке, с помощью подходящей краски. В другом примере таблетки, включающие фармацевтическую композицию, как описано в данной заявке, могут быть покрыты 3% по массе (например, менее чем приблизительно 6% по массе или менее чем приблизительно 4% по массе) пленочным покрытием, содержащим краситель.
В другом варианте применения изобретения таблетки, сделанные из фармацевтической композиции, покрыты красителем, воском и затем отмечены логотипом, другим изображением и/или текстом с помощью подходящей краски. Например, таблетки, включающие фармацевтическую композицию, как описано в данной заявке, могут быть покрыты 3% по массе (например, менее чем приблизительно 6% по массе или менее чем приблизительно 4% по массе) пленочным покрытием, содержащим краситель. Окрашенные таблетки могут быть покрыты воском с помощью порошка карнаубского воска, взвешенного в количестве, составляющем около 0,01% вес/вес от исходного веса ядра таблетки. Покрытые воском таблетки могут быть отмечены логотипом и текстом, показывающим силу действующего вещества в таблетке, с помощью подходящей краски. В другом примере таблетки, включающие фармацевтическую композицию, как описано в данной заявке, могут быть покрыты 3% по массе (например, менее чем приблизительно 6% по массе или менее чем приблизительно 4% по массе) пленочным покрытием, содержащим краситель. Окрашенные таблетки могут быть покрыты воском с помощью порошка карнаубского воска, взвешенного в количестве, составляющем около 0,01% вес/вес от исходного веса ядра таблетки. Покрытые воском таблетки могут быть отмечены логотипом и текстом, показывающим силу действующего вещества в таблетке, с помощью краски фармацевтического качества, такой как черная краска (например, Opacode® S-1-17823, краска на основе растворителя, коммерчески доступного в Colorcon, Inc. Вест Пойнт, штат Филадельфия).
Один пример фармацевтической композиции включает приблизительно от 15% по массе приблизительно до 70% по массе (например, приблизительно от 15% по массе приблизительно до 60% по массе, приблизительно от 15% по массе приблизительно до 50% по массе, или приблизительно от 20% по массе приблизительно до 70% по массе, или приблизительно от 30% по массе приблизительно до 70% по массе) Соединения 1 Формы I по массе от композиции; и приблизительно от 15% приблизительно до 40% по массе (например, приблизительно 20-35% по массе) по существу аморфного Соединения 2 по массе от композиции, и чаще от 25% по массе приблизительно до 30% по массе по существу аморфного Соединения 2 по массе от композиции. Вышеупомянутые композиции также могут включать один или более фармацевтически приемлемые вспомогательные вещества, например, приблизительно от 20% по массе приблизительно до 50% по массе наполнителя; приблизительно от 1% по массе приблизительно до 5% по массе разрыхлителя; приблизительно от 2% по массе приблизительно до 0,3% по массе сурфактанта; и приблизительно от 0,1% по массе приблизительно до 5% по массе связывающего вещества.
Другой пример фармацевтической композиции включает приблизительно от 15% по массе приблизительно до 70% по массе (например, приблизительно от 15% по массе приблизительно до 60% по массе, приблизительно от 15% по массе приблизительно до 50% по массе, или приблизительно от 15% по массе приблизительно до 40% по массе, или приблизительно от 20% по массе приблизительно до 70% по массе, или приблизительно от 30% по массе приблизительно до 70% по массе, или приблизительно от 40% по массе приблизительно до 70% по массе, или приблизительно от 50% по массе приблизительно до 70% по массе) Соединения 1 Формы I, по массе от композиции; приблизительно от 15% по массе до 40% по массе (например, около 20-35% по массе) по существу аморфного Соединения 2 по массе от композиции, и чаще от 25% по массе приблизительно до 30% по массе по существу аморфного Соединения 2 по массе от композиции и одно или более вспомогательные вещества, например, приблизительно от 20% по массе до 50% по массе наполнителя, от 1% по массе приблизительно до 5% по массе разрыхлителя; приблизительно от 2% по массе приблизительно до 0,3% по массе сурфактанта; приблизительно от 0,1% по массе приблизительно до 5% по массе связывающего вещества; и приблизительно от 2% по массе приблизительно до 0,1% по массе смазывающего вещества.
Другой пример фармацевтической композиции включает приблизительно от 15% по массе приблизительно до 70% по массе (например, приблизительно от 15% по массе приблизительно до 60% по массе, приблизительно от 15% по массе приблизительно до 50% по массе, или приблизительно от 15% по массе приблизительно до 40% по массе, или приблизительно от 20% по массе приблизительно до 70% по массе, или приблизительно от 30% по массе приблизительно до 70% по массе, или приблизительно от 40% по массе приблизительно до 70% по массе, или приблизительно от 50% по массе приблизительно до 70% по массе) Соединения 1 Формы I по массе от композиции, приблизительно от 15% по массе приблизительно до 40% по массе (например, около 20-35% по массе) по существу аморфного Соединения 2 по массе от композиции, и чаще от 25% по массе приблизительно до 30% по массе по существу аморфного Соединения 2 по массе от композиции и одно или более вспомогательные вещества, например, приблизительно от 20% по массе приблизительно до 50% по массе наполнителя, от 1% по массе приблизительно до 5% по массе разрыхлителя; приблизительно от 2% по массе приблизительно до 0,3% по массе сурфактанта; приблизительно от 0,1% по массе приблизительно до 5% по массе связывающего вещества; и приблизительно от 2% по массе приблизительно до 0,1% по массе смазывающего вещества; приблизительно от 2% по массе приблизительно до 4% по массе красителя; и приблизительно от 0,005% по массе приблизительно до 0,015% по массе воска.
В одном варианте применения изобретение является гранулярной фармацевтической композицией, включающей:
а. около 43% по массе Соединения 1 Формы I по массе от композиции;
б. около 34% по массе твердой дисперсии, включающей по существу аморфное Соединение 2 по массе от композиции;
в. около 17% по массе микрокристаллической целлюлозы по массе от композиции;
г. около 2% по массе кроскармеллозы натрия по массе от композиции;
д. около 1% по массе лаурилсульфата натрия по массе от композиции; и
е. около 3% по массе поливинилпирролидона по массе от композиции.
В одном варианте применения изобретение является таблеткой, включающей:
а. около 35% по массе Соединения 1 Формы I по массе от композиции;
б. около 28% по массе твердой дисперсии, включающей по существу аморфное Соединение 2 по массе от композиции;
в. около 26% по массе микрокристаллической целлюлозы по массе от композиции;
г. около 6% по массе кроскармеллозы натрия по массе от композиции;
д. около 3%% по массе поливинилпирролидона по массе от композиции;
е. около 1% по массе лаурилсульфата натрия по массе от композиции; и
ж. около 1% по массе стеарата магния по массе от композиции.
В одном варианте применения изобретение является таблеткой, включающей:
а. около 34% по массе Соединения 1 Формы I по массе от композиции;
б. около 27% по массе твердой дисперсии, включающей по существу аморфное Соединение 2 по массе от композиции;
в. около 26% по массе микрокристаллической целлюлозы по массе от композиции;
г. около 6% по массе кроскармеллозы натрия по массе от композиции;
д. около 2% по массе поливинилпирролидона по массе от композиции
е. около 1% по массе лаурилсульфата натрия по массе от композиции;
ж. около 1% по массе стеарата магния по массе от композиции;
з. около 3% по массе красителя по массе от композиции; и
и. около 0,010% по массе воска по массе от композиции.
Другая таблетка, являющаяся предметом изобретения, включает:
а. около 150 до 250 мг Соединения 1 Формы I;
б. около 100 до 150 мг по существу аморфного Соединения 2;
в. около 125 до 175 мг микрокристаллической целлюлозы;
г. около 20 до 40 мг кроскармеллозы натрия;
д. около 10 до 20 мг поливинилпирролидона;
е. около 2 до 6 мг лаурилсульфата натрия; и
ж. около 3 до 7 мг стеарата магния.
Другая таблетка, являющаяся предметом изобретения, включает:
а. около 200 мг Соединения 1 Формы I;
б. около 125 мг по существу аморфного Соединения 2;
в. около 150 мг микрокристаллической целлюлозы;
г. около 34 мг кроскармеллозы натрия;
д. около 15 мг поливинилпирролидона;
е. около 4 мг лаурилсульфата натрия; и
ж. около 6 мг стеарата магния.
Другая таблетка, являющаяся предметом изобретения, включает:
а. около 200 мг Соединения 1 Формы I;
б. около 125 мг по существу аморфного Соединения 2;
в. около 150 мг микрокристаллической целлюлозы;
г. около 34 мг кроскармеллозы натрия;
д. около 15 мг поливинилпирролидона;
е. около 4 мг лаурилсульфата натрия;
ж. около 6 мг стеарата магния;
з. около 17 мг красителя; и
и. около 0,06 мг воска.
В одном варианте применения изобретение является гранулярной фармацевтической композицией, включающей:
а. около 38% по массе Соединения 1 Формы I по массе от композиции;
б. около 40% по массе твердой дисперсии, включающей по существу аморфное Соединение 2 по массе от композиции;
в. около 16% по массе микрокристаллической целлюлозы по массе от композиции;
г. около 2% по массе кроскармеллозы натрия по массе от композиции;
д. около 1% по массе лаурилсульфата натрия по массе от композиции; и
е. около 3% по массе поливинилпирролидона по массе от композиции.
В одном варианте применения изобретение является таблеткой, включающей:
а. около 31% по массе Соединения 1 Формы I по массе от композиции;
б. около 32% по массе твердой дисперсии, включающей по существу аморфное Соединение 2 по массе от композиции;
в. около 26% по массе микрокристаллической целлюлозы по массе от композиции;
г. около 6% по массе кроскармеллозы натрия по массе от композиции;
д. около 3% по массе поливинилпирролидона по массе от композиции
е. около 1% по массе лаурилсульфата натрия по массе от композиции;
ж. около 1% по массе стеарата магния по массе от композиции; и
з. около 3% по массе красителя по массе от композиции.
Другая таблетка, являющаяся предметом изобретения, включает:
а. около 100 до 200 мг Соединения 1 Формы I;
б. около 100 до 150 мг по существу аморфного Соединения 2;
в. около 100 до 150 мг микрокристаллической целлюлозы;
г. около 20 до 4 0 мг кроскармеллозы натрия;
д. около 10 до 20 мг поливинилпирролидона;
е. около 2 до 6 мг лаурилсульфата натрия; и
ж. около 3 до 7 мг стеарата магния.
Другая таблетка, являющаяся предметом изобретения, включает:
а. около 150 мг Соединения 1 Формы I;
б. около 125 мг по существу аморфного Соединения 2;
в. около 129 мг микрокристаллической целлюлозы;
г. около 29 мг кроскармеллозы натрия;
д. около 13 мг поливинилпирролидона;
е. около 4 мг лаурилсульфата натрия;
ж. около 5 мг стеарата магния; и
з. около 15 мг красителя.
Фармацевтические композиции, являющиеся предметом изобретения, можно производить в форме таблеток, в форме капсул, в форме саше, в форме пастилок для рассасывания или в другой твердой форме, которая подходит для перорального введения. Таким образом, в некоторых вариантах применения изобретения фармацевтические композиции выпускают в форме таблеток.
Другой аспект изобретения представляет фармацевтическую композицию, состоящую из таблеток, которые включают Соединение 1 Формы I, твердую дисперсию, включающую по существу аморфное Соединение 2, и вспомогательные вещества (например, наполнитель, разрыхлитель, сурфактант, связывающее вещество, краситель, смазывающее вещество или любую их комбинацию), каждое из которых описано выше и в Примерах ниже, где растворимость таблетки составляет по меньшей мере около 50% (например, по меньшей мере около 60%, по меньшей мере около 70%, по меньшей мере около 80%, по меньшей мере около 90% или по меньшей мере около 99%) в течение приблизительно 30 минут.
В одном примере фармацевтическая композиция состоит из таблеток, которые включают Соединение 1 Формы I в количестве, варьирующем от 25 мг до 400 мг, например, 25 мг или 50 мг, или 75 мг, или 100 мг, или 150 мг, 200 мг, 250 мг, 300 мг, или 400 мг, по существу аморфное Соединение 2 в количестве, варьирующем от 25 мг до 250 мг, например, 25 мг или 50 мг, или 75 мг, или 100 мг, или 150 мг, 200 мг, 250 мг и одно или более вспомогательные вещества (например, наполнитель, разрыхлитель, сурфактант, связывающее вещество, краситель, смазывающее вещество или любую их комбинацию) каждое из которых описано выше и в Примерах ниже, где растворимость таблетки составляет по меньшей мере приблизительно от 50% приблизительно до 100% (например, приблизительно от 55% приблизительно до 95% или приблизительно от 60% приблизительно до 90%) в течение приблизительно 30 минут.
Растворимость можно измерить с помощью стандартного аппарата Фармакопеи США типа II, в котором применяют среду для растворения, состоящую из 0,1% СТАВ (цетилтриметиламмоний бромид), растворенного в 900 мл деионизированной воды, в буфере из 50 мМ однозамещенного фосфата кальция рН 6,8, при перемешивании при 50-75 об/мин при температуре около 37°С. Одну экспериментальную таблетку тестируют в каждом экспериментальном сосуде аппарата. Растворимость также можно измерить с помощью стандартного аппарата Фармакопеи США типа II, в котором применяют среду для растворения, состоящую из 0,7% лаурилсульфата натрия, растворенного в 900 мл 50 мМ натрийфосфатного буфера (рН 6,8), при перемешивании при 65 об/мин при температуре около 37°С. Одну экспериментальную таблетку тестируют в каждом экспериментальном сосуде аппарата. Растворимость также можно измерить с помощью стандартного аппарата Фармакопеи США типа II, в котором применяют среду для растворения, состоящую из 0,5% лаурилсульфата натрия, растворенного в 900 мл 50 мМ натрийфосфатного буфера (рН 6,8), при перемешивании при 65 об/мин при температуре около 37°С. Одну экспериментальную таблетку тестируют в каждом экспериментальном сосуде аппарата.
СПОСОБЫ ИЗГОТОВЛЕНИЯ СОЕДИНЕНИЯ 1 ФОРМЫ I И ТВЕРДОЙ ДИСПЕРСИИ, ВКЛЮЧАЮЩЕЙ ПО СУЩЕСТВУ АМОРФНОЕ СОЕДИНЕНИЕ 2
Соединение 1
Соединение 1 применяют как исходное вещество для Соединения 1 Формы I, и его можно приготовить посредством связывания функциональной группы хлорангидрида с функциональной группой амина в соответствии со Схемами 1-4.
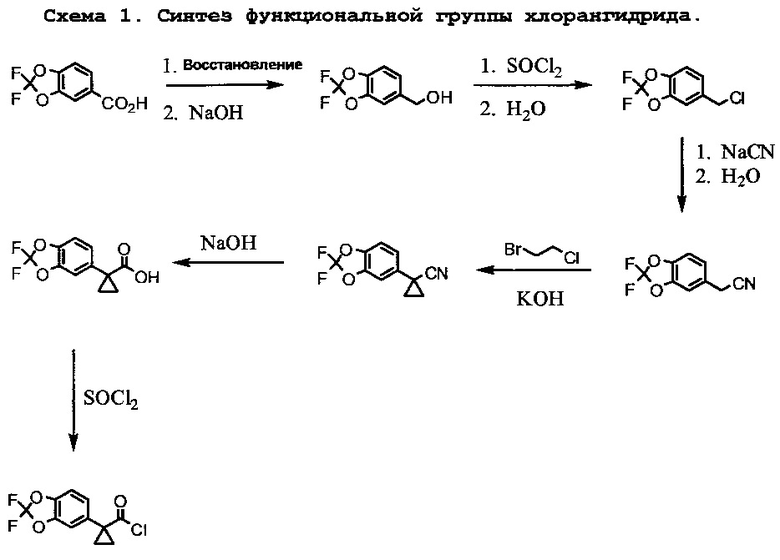
Схема 1 иллюстрирует получение 1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбонилхлорида, который применяют в Схеме 3 для образования амидной связи Соединения 1.
Исходный материал, 2,2-дифторбензо[d][1,3]диоксол-5-карбоновая кислота, является коммерчески доступной в Saltigo (дочернее предприятие Lanxess Corporation). Восстановление функциональной группы карбоновой кислоты в 2,2-дифторбензо[d][1,3]диоксол-5-карбоновой кислоте в первичном спирте с последующим превращением в соответствующий хлорид с помощью тионилхлорида (SOCl2) образует 5-(хлорметил)-2,2-дифторбензо[d][1,3]диоксол, который впоследствии превращается в 2-(2,2-дифторбензо[d][1,3]диоксол-5-ил)ацетонитрил с помощью цианида натрия. Обработка 2-(2,2-дифторбензо[d][1,3]диоксол-5-ил)ацетонитрила основанием и 1-бром-2-хлорэтаном образует 1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбонитрил. Нитрильная функциональная группа в 1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбонитриле превращается в карбоксильную группу с помощью основания с образованием 1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоновой кислоты, которая превращается в искомый хлорангидрид с помощью тионилхлорида.
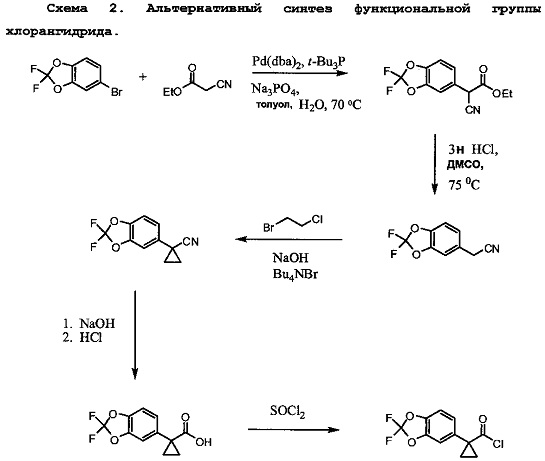
Схема 2 показывает альтернативный синтез необходимого хлорангидрида. 5-бромметил-2,2-дифтор-1,3-бензодиоксол связывают с этилцианоацетатом в присутствии палладиевого катализатора для образования соответствующего альфа-цианоэтилового эфира. Омыление эфирной группы до карбоновой кислоты приводит к образованию цианоэтилового соединения. Алкилирование цианоэтилового соединения 1-бром-2-хлорэтаном в присутствии основания приводит к образованию цианоциклопропилового соединения. Обработка цианоциклопропилового соединения основанием приводит к образованию соли карбоновой кислоты, которая превращается в карбоновую кислоту при обработке кислотой. Превращение карбоновой кислоты в хлорангидрид впоследствии завершается с помощью хлорирующего агента, такого как тионилхлорид или подобных.
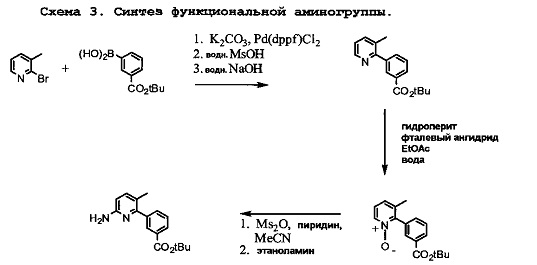
Схема 3 показывает приготовление необходимого третбутил-3-(6-амино-3-метилпиридин-2-ил)бензоата, который связывают с 1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбонилхлоридом на Схеме 3 для образования Соединения 1. Катализируемое палладием связывание 2-бром-3-метилпиридина с 3-(третбутоксикарбонил)фенилбороновой кислотой приводит к образованию третбутил-3-(3-метилпиридин-2-ил)бензоата, который затем превращается в искомое соединение.
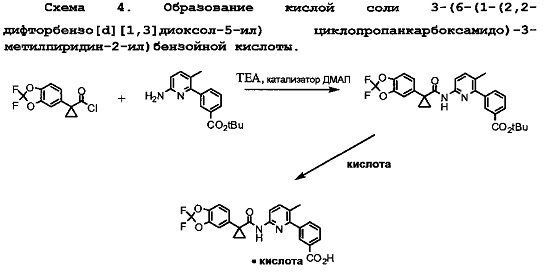
Схема 4 показывает связывание 1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбонилхлорида с третбутил-3-(6-амино-3-метилпириин-2-ил)бензоатом с помощью триэтиламина и 4-диметиламинопиридина для образования третбутилового эфира Соединения 1.
Соединение 1 Форма I
Соединение 1 Форму I готовят посредством диспергирования или растворения солевой формы, такой как соли HCl, Соединения 1 в подходящем растворителе в течение эффективного количества времени. Соединение 1 Форму I также можно приготовить напрямую из t-бутилэфирного предшественника посредством обработки соответствующей кислотой, такой как муравьиная кислота.
HCl соль 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамидо)-3-метилпиридин-2-ил)бензойной кислоты можно применять для образования Формы I посредством диспергирования или растворения HCl соли 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамидо)-3-метилпиридин-2-ил)бензойной кислоты в подходящем растворителе в течение эффективного количества времени. Можно применять другие соли 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамидо)-3-метилпиридин-2-ил)бензойной кислоты, такие как, например, соли, полученные из других неорганических или органических кислот. Другие соли образуются в результате кислотного гидролиза функциональной группы t-бутилового эфира. Соли, полученные из других кислот, могут включать, например, соли азотной, серной, фосфорной, борной, уксусной, бензойной и малоновой кислот. Эти солевые формы 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамидо)-3-метилпиридин-2-ил)бензойной кислоты могут быть или не быть растворимыми в зависимости от применяемого растворителя, но отсутствие растворимости не препятствует образованию Соединения 1 Формы I. Например, в одном варианте применения изобретения подходящий растворитель может быть водой или смесью спирта/воды, такой как смесь 50% метанола/воды, хотя форма HCl соли 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамидо)-3-метилпиридин-2-ил)бензойной кислоты слабо растворима в воде. В одном варианте применения изобретения подходящим растворителем является вода.
Эффективное количество времени для образования Соединения 1 Формы I из соли 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамидо)-3-метилпиридин-2-ил)бензойной кислоты может быть любым временем от 2 до 2 4 часов или более. Признано, что необходимое количество времени обратно пропорционально температуре. То есть чем выше температура, тем меньше требуется времени для диссоциации кислоты с образованием Соединения 1 Формы I. Когда растворителем является вода, перемешивание дисперсии в течение приблизительно 24 часов при комнатной температуре приводит к образованию Соединения 1 Формы I с приблизительно 98% выходом. Если для технологических нужд требуется раствор соли 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамидо)-3-метилпиридин-2-ил)бензойной кислоты, можно применять повышенную температуру. После перемешивания раствора в течение эффективного количества времени при повышенной температуре перекристаллизация при охлаждении приводит к образованию по существу чистого Соединения 1 Формы I. В одном варианте применения изобретения ''по существу чистый'' как применяют во фразе ''по существу чистое Соединение 1 Формы I'' означает более чем 90% чистоту. В другом варианте применения изобретения ''по существу чистый'' относится к более чем 95% чистоте. В другом варианте применения изобретения ''по существу чистый'' относится к более чем 98% чистоте. В другом варианте применения изобретения ''по существу чистый'' относится к более чем 99% чистоте. Выбранная температура частично зависит от применяемого растворителя и находится в пределах возможностей определения среднего специалиста в данной области техники. В одном варианте применения изобретения температура находится в пределах от комнатной температуры приблизительно до 80°С. В другом варианте применения изобретения температура находится в пределах от комнатной температуры приблизительно до 40°С. В другом варианте применения изобретения температура находится в пределах от 40°С и приблизительно до 60°С. В другом варианте применения изобретения температура находится в пределах от 60°С и приблизительно до 80°С.
Соединение 1 Форма I также может быть образовано напрямую из 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамидо)-3-метилпиридин-2-ил)-t-бутилбензоата (ср. со Схемой 3), который является предшественником соли Соединения 1. Таким образом, 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамидо)-3-метилпиридин-2-ил)-t-бутилбензоат может реагировать с соответствующей кислотой, такой как, например, муравьиная кислота, в подходящих условиях реакции с образованием Соединения 1 Формы I.
Соединение 1 Форму I можно далее очистить посредством перекристаллизации из органического растворителя. Примеры органических растворителей включают, но не ограничиваются толуолом, кумолом, анизолом, 1-бутанолом, изопропилацетатом, бутилацетатом, изобутилацетатом, метил t-бутиловым эфиром, метилизобутилкетоном и смесями 1-пропанол-вода. Температура может быть такой, как описанная выше. Например, Соединение 1 Форма I растворяется в 1-бутаноле при 75°С до полного растворения. Охлаждение раствора до 10°С со скоростью 0,2°С/мин приводит к образованию кристаллов Соединения 1 Формы I, которые можно выделить фильтрацией.
В одном варианте применения изобретения Соединение 1 Форма I характеризуется одним или более пиками в интервале от 15,2 до 15,6 градусов, от 16,1 до 16,5 градусов и от 14,3 до 14,7 градусов рентгеновской порошковой дифрактометрии, полученной с помощью Cu K альфа излучения. В другом варианте применения изобретения Соединение 1 Форма I характеризуется одним или более пиками при 15,4, 16,3 и 14,5 градусах. В другом варианте применения изобретения Соединение 1 Форма I далее характеризуется пиком в интервале от 14,6 до 15,0 градусов. В другом варианте применения изобретения Соединение 1 Форма I далее характеризуется пиком при 14,8 градусах. В другом варианте применения изобретения Соединение 1 Форма I далее характеризуется пиком в интервале от 17,6 до 18,0 градусов. В другом варианте применения изобретения Соединение 1 Форма I далее характеризуется пиком при 17,8 градусах. В другом варианте применения изобретения Соединение 1 Форма I далее характеризуется пиком в интервале от 16,4 до 16,8 градусов. В другом варианте применения изобретения Соединение 1 Форма I далее характеризуется пиком в интервале от 16,4 до 16,8 градусов. В другом варианте применения изобретения Соединение 1 Форма I далее характеризуется пиком при 16,6 градусах. В другом варианте применения изобретения Соединение 1 Форма I далее характеризуется пиком в интервале от 7,6 до 8,0 градусов. В другом варианте применения изобретения Соединение 1. Форма I далее характеризуется пиком при 7,8 градусах. В другом варианте применения изобретения Соединение 1 Форма I далее характеризуется пиком в интервале от 25,8 до 26,2 градусов. В другом варианте применения изобретения Соединение 1 Форма I далее характеризуется пиком при 26,0 градусах. В другом варианте применения изобретения Соединение 1 Форма I далее характеризуется пиком в интервале от 21,4 до 21,8 градусов. В другом варианте применения изобретения Соединение 1 Форма I далее характеризуется пиком при 21,6 градусах. В другом варианте применения изобретения Соединение 1 Форма I далее характеризуется пиком в интервале от 23,1 до 23,5 градусов. В другом варианте применения изобретения Соединение 1 Форма I далее характеризуется пиком при 23,3 градусах. В некоторых вариантах применения изобретения Соединение 1 Форма I характеризуется дифракционной картиной по существу схожей с таковой на фиг. 1. В некоторых вариантах применения изобретения Соединение 1 Форма I характеризуется дифракционной картиной по существу схожей с таковой на фиг. 2.
В некоторых вариантах применения изобретения распределение размера частиц D90 составляет около 82 мкм или менее для Соединения 1 Формы I. В некоторых вариантах применения изобретения распределение размера частиц D50 составляет около 30 мкм или менее для Соединения 1 Формы I.
Соединение 2
Соединение 2 является исходным соединением для твердой дисперсии, включающей по существу аморфное Соединение 2, и может быть приготовлено при связывании функциональной группы 4-оксо-дигидрохинолинкарбоновой кислоты с аминогруппой в соответствии со Схемами 5-7.

Твердая дисперсия, включающая по существу аморфное Соединение 2
Исходя из Соединения 2, аморфную форму Соединения 2 можно приготовить с помощью способа высушивания распылением. Высушивание распылением представляет собой процесс, который превращает жидкое сырье в форму сухих микрочастиц. При желании можно применять процесс вторичного высушивания, такого как высушивание в псевдосжиженном слое или вакуумное высушивание, для снижения остаточного содержания растворителей до фармацевтически приемлемых уровней. Обычно высушивание распылением включает контакт высокодисперсной жидкой суспензии или раствора с достаточным объемом горячего воздуха для образования и высушивания капель жидкости. Препарат, предназначенный для высушивания распылением, может быть любым раствором, грубой суспензией, суспензией, коллоидной дисперсии или пастой, которую можно размельчить с помощью выбранного аппарата для высушивания распылением. В стандартной процедуре препарат распыляют в потоке теплого фильтрованного воздуха, который испаряет растворитель и подает высушенный продукт в коллектор (например, циклонный уловитель). Отработанный воздух затем выходит вместе с растворителем или, в качестве альтернативы, отработанный воздух отправляют в конденсатор для сбора и возможной рециркуляции растворителя. Коммерчески доступные типы аппаратов можно применять для проведения высушивания распылением. Например, коммерческие приборы для высушивания распылением производят Buchi Ltd. и Niro (например, приборов для высушивания распылением линии PSD производства Niro) (см., US 2004/0105820; US 2003/0144257).
В высушивании распылением обычно применяют нагрузку твердыми частицами материала равную приблизительно от 3% приблизительно до 30% по массе, (т.е., лекарственного препарата и вспомогательных веществ), например, приблизительно от 4% приблизительно до 20% по массе, предпочтительно по меньшей мере около 10%. В общем случае верхний предел нагрузки твердыми частицами определяется вязкостью (например, способностью к нагнетанию) образующегося в результате раствора и растворимостью компонентов в растворе. В общем случае вязкость раствора может определять размер частиц в образующемся в результате порошковом продукте.
Техники и способы высушивания распылением можно найти в Perry's Chemical Engineering Handbook, 6th Ed., R.H. Perry, D.W. Green & J.O. Maloney, eds., McGraw-Hill book со. (1984); и Marshall ''Atomization and Spray-Drying'' 50, Chem. Eng. Prog. Monogr. Series 2 (1954). В общем случае высушивание распылением проводят при температуре на входе приблизительно от 60°С приблизительно до 200°С, например, приблизительно от 95°С приблизительно до 185°С, приблизительно от 110°С приблизительно до 182°С, приблизительно от 96°С приблизительно до 180°С, например, около 145°С. Высушивание распылением обычно проводят при температуре на выходе приблизительно от 30°С приблизительно до 90°С, например, приблизительно от 40°С приблизительно до 80°С, приблизительно от 45°С приблизительно до 80°С например, около 75°С. Скорость потока распыления обычно составляет приблизительно от 4 кг/ч приблизительно до 12 кг/ч, например, приблизительно от 4,3 кг/ч приблизительно до 10,5 кг/ч, например, около 6 кг/ч или около 10,5 кг/ч. Скорость подаваемого потока обычно составляет приблизительно от 3 кг/ч приблизительно до 10 кг/ч, например, приблизительно от 3,5 кг/ч приблизительно до 9,0 кг/ч, например, приблизительно 8 кг/ч или приблизительно 7,1 кг/ч. Степень распыления обычно составляет приблизительно от 0,3 до 1,7, например, приблизительно от 0,5 до 1,5, например, около 0,8 или приблизительно 1,5.
Удаление растворителя может потребовать последующей стадии высушивания, такой как высушивание на подложке, высушивание в псевдосжиженном слое (например, приблизительно от комнатной температуры приблизительно до 100°С), вакуумное высушивание, микроволновое высушивание, высушивание во вращающемся барабане или биконическое вакуумное высушивание (например, приблизительно от комнатной температуры приблизительно до 200°С).
В одном варианте применения изобретения высушенная распылением дисперсия является высушенной в псевдосжиженном слое.
В одном процессе растворитель включает летучий растворитель, например, растворитель с точкой кипения приблизительно менее 100°С. В некоторых вариантах применения изобретения растворитель включает смесь растворителей, например, смесь летучих растворителей или смесь летучих и нелетучих растворителей. При применении растворителей смесь может включать один или более нелетучие растворители, например, где нелетучий растворитель присутствует в смеси · в количестве менее приблизительно 15%, например, менее приблизительно 12%, менее приблизительно 10%, менее приблизительно 8%, менее приблизительно 5%, менее приблизительно 3% или менее приблизительно 2%.
Предпочтительными растворителями являются растворители, в которых Соединение 2 имеет растворимость равную по меньшей мере приблизительно 10 мг/мл (например, по меньшей мере приблизительно 15 мг/мл, 20 мг/мл, 25 мг/мл, 30 мг/мл, 35 мг/мл, 40 мг/мл, 45 мг/мл, 50 мг/мл или более). Более предпочтительные растворители включают такие растворители, в которых Соединение 2 имеет растворимость равную по меньшей мере приблизительно 20 мг/мл.
Примеры растворителей, которые можно исследовать, включают ацетон, циклогексан, дихлорметан, N,N-диметилацетамид (ДМА), N,N-диметилформамид (ДМФ), 1,3-диметил-2-имидазолидинон (ДМИ), диметилсульфоксид (ДМСО), диоксан, этилацетат, этиловый эфир, ледяную уксусную кислоту, метилэтилкетон (МЭК), N-метил-2-пирролидон (NMP), метил-трет-бутиловый эфир (МТБЭ), тетрагидрофуран (ТГФ), пентан, ацетонитрил, метанол, этанол, изопропиловый спирт, изопропилацетат и толуол. Примеры вспомогательных растворителей включают ацетон/ДМСО, ацетон/ДМФ, ацетон/воду, МЭК/воду, ТГФ/воду, диоксан/воду. В системе двух растворителей растворители могут присутствовать в количестве приблизительно от 0,1% приблизительно до 99,9%. В некоторых вариантах применения изобретения вода является вспомогательным растворителем вместе с ацетоном, где вода присутствует в количестве приблизительно от 0,1% приблизительно до 15%, например, приблизительно от 9% приблизительно до 11%, например, около 10%. В некоторых предпочтительных вариантах применения изобретения вода является вспомогательным растворителем вместе с МЭК, где вода присутствует в количество приблизительно от 0,1% приблизительно до 15%, например, приблизительно от 9% приблизительно до 11%, например, около 10%. В некоторых вариантах применения изобретения раствор растворителя включает три растворителя. Например, ацетон и вода могут быть смешаны с третьи растворителем, таким как ДМА, ДМФ, ДМИ, ДМСО или с ледяной уксусной кислотой. В случаях, когда по существу аморфное Соединение 2 является компонентом твердой дисперсии, предпочтительные растворители растворяют как Соединение 2, так и полимер. Подходящие растворители включают описанные выше, например, МЭК, ацетон, воду, метанол и их смеси.
Размер частиц и температурный интервал высушивания можно модифицировать для приготовления оптимальной высушенной распылением дисперсии. Специалистам в данной области техники ясно, что частицы малого размера будут обеспечивать улучшенное удаление растворителя. Однако заявители обнаружили, что частицы меньшего размера будут приводить к образованию рыхлых частиц, которые в определенных условиях не могут обеспечить образование высушенной распылением дисперсии оптимальной для последующей обработки, такой как таблетирование. При более высоких температурах может произойти кристаллизация или химическая деградация по существу аморфного Соединения 2. При более низких температурах может быть удалено недостаточное количество растворителя. Способы в данной заявке представляют оптимальный размер частиц и оптимальную температуру высушивания.
В общем случае размер частиц таков, что D10 (мкм) составляет приблизительно менее 5, например, приблизительно менее 4,5, приблизительно менее 4,0 или приблизительно менее 3,5, D50 (мкм) в общем случае составляет приблизительно менее 17, например, приблизительно менее 16, приблизительно менее 15, приблизительно менее 14, приблизительно менее 13, и D90 (мкм) в общем случае составляет приблизительно менее 175, например, приблизительно менее 170, приблизительно менее 170, приблизительно менее 150, приблизительно менее 125, приблизительно менее 100, приблизительно менее 90, приблизительно менее 80, приблизительно менее 70, приблизительно менее 60 или приблизительно менее 50. В общем случае объемная площадь высушенных распылением частиц составляет приблизительно от 0,08 г/см3 приблизительно до 0,20 г/см3, например, приблизительно от 0,10 приблизительно до 0,15 г/см3, например, приблизительно 0,11 г/см3 или приблизительно 0,14 г/см3. Плотность утряски высушенных распылением частиц составляет приблизительно от 0,08 г/см3 приблизительно до 0,20 г/см3, например, приблизительно от 0,10 приблизительно до 0,15 г/см3, например, приблизительно 0,11 г/см3 или приблизительно 0,14 г/см3 для 10 встряхиваний; 0,10 г/см3 приблизительно до 0,25 г/см3, например, приблизительно от 0,11 приблизительно до 0,21 г/см3, например, приблизительно 0,15 г/см3, приблизительно 0,19 г/см3 или приблизительно 0,21 г/см3 для 500 встряхиваний; 0,15 г/см3 приблизительно до 0,27 г/см3, например, приблизительно от 0,18 приблизительно до 0,24 г/см3, например, приблизительно 0,18 г/см3, приблизительно 0,19 г/см3, приблизительно 0,20 г/см3 или приблизительно 0,24 г/см3 для 1250 встряхиваний; и 0,15 г/см3 приблизительно до 0,27 г/см3, например, приблизительно от 0,18 приблизительно до 0,24 г/см3, например, приблизительно 0,18 г/см3, приблизительно 0,21 г/см3, приблизительно 0,23 г/см3 или приблизительно 0,24 г/см3 для 2500 встряхиваний.
Полимеры
Высушенные распылением дисперсии, включая аморфное Соединение 2 и полимер (или твердый носитель), также включены в данную заявку. Например, Соединение 2 присутствует в виде аморфного соединения как компонент твердой аморфной дисперсии. Твердая аморфная дисперсия в общем случае включает по существу аморфное Соединение 2 и полимер. Примеры полимеров включают целлюлозные полимеры, такие как гидроксипропилметилцеллюлоза (НРМС) или ацетат сукцинат гидроксипропилметилцеллюлозы (HPMCAS) и пирролидон-содержащие полимеры, такие как поливинилпирролидон/винилацетат (PVP/VA). В некоторых вариантах применения изобретения твердая аморфная дисперсия включает одно или более вспомогательные вещества, такие как сурфактант.
В одном варианте применения изобретения полимер способен растворяться в водной среде. Растворимость полимеров может быть рН-независимой или рН-зависимой. Последние включают один или более растворимые в кишечнике полимеры. Термин ''растворимый в кишечнике полимер'' относится к полимеру, который предпочтительно растворяется в менее кислой среде кишечника по сравнению с более кислой средой желудка, например, полимер, не растворимый в кислой водной среде, но растворимый при рН выше 5-6. Подходящий полимер должен быть химически и биологически инертным. Для улучшения физической стабильности высушенных распылением дисперсий температура стеклования (Тс) полимера должна быть максимально возможной. Например, предпочтительные полимеры имеют температуру стеклования по меньшей мере равную или превышающую температуру стеклования лекарственного препарата (т.е. Соединения 2). Другие предпочтительные полимеры имеют температуру стеклования в интервале приблизительно от 10 приблизительно до 15°С лекарственного препарата (т.е., Соединения 2). Примеры подходящих температур стеклования полимеров включают по меньшей мере приблизительно 90°С, по меньшей мере приблизительно 95°С, по меньшей мере приблизительно 100°С, по меньшей мере приблизительно 105°С, по меньшей мере приблизительно 110°С, по меньшей мере приблизительно 115°С, по меньшей мере приблизительно 120°С, по меньшей мере приблизительно 125°С, по меньшей мере приблизительно 130°С, по меньшей мере приблизительно 135°С, по меньшей мере приблизительно 140°С, по меньшей мере приблизительно 145°С, по меньшей мере приблизительно 150°С, по меньшей мере приблизительно 155°С, по меньшей мере приблизительно 160°С, по меньшей мере приблизительно 165°С, по меньшей мере приблизительно 170°С или по меньшей мере приблизительно 175°С (при измерении в сухих условиях). Не желая быть связанными соответствием какой-либо теории, авторы полагают, что лежащий в основе механизм заключается в том, что полимер с более высокой Тс в общем случае обладает меньшей молекулярной подвижностью при комнатной температуре, которая может быть важнейшим фактором в стабилизации физической стабильности аморфной высушенной распылением дисперсии.
Кроме того, гигроскопичность полимеров должна быть как можно ниже, например, приблизительно менее 10%. С целью сравнения в данной заявке гигроскопичность полимера или композиции характеризуется приблизительно 60% относительной влажностью. В некоторых предпочтительных вариантах применения изобретения полимер обладает приблизительно менее 10% гигроскопичностью, например, приблизительно менее 9%, приблизительно менее 8%, приблизительно менее 7%, приблизительно менее 6%, приблизительно менее 5%, приблизительно менее 4%, приблизительно менее 3% или приблизительно менее 2% гигроскопичностью. Гигроскопичность также может влиять на физическую стабильность высушенных распылением дисперсий. В общем случае влага, адсорбированная полимерами, может значительно снизить Тс полимеров, а также образующихся в результате высушенных распылением дисперсий, что может далее снижать физическую стабильность высушенных распылением дисперсий, как описано выше.
В одном варианте применения изобретения полимер является одним или более водорастворимыми полимерами или частично водорастворимыми полимерами. Водорастворимые или частично водорастворимые полимеры включают, но не ограничиваются производными целлюлозы (например, гидроксипропилметилцеллюлозой (НРМС), гидроксипропилцеллюлозой (НРС)) или этилцеллюлозой; поливинилпирролидонами (PVP); полиэтиленгликолями (ПЭГ); поливиниловыми спиртами (PVA); акрилатами, такими как полиметакрилат (например, Eudragit® Ε); циклодекстринами (например, β-циклодекстрином) и их сополимерами и производными, включая, например PVP-VA (поливинилпирролидонвинилацетат).
В некоторых вариантах применения изобретения полимер является гидроксипропилметилцеллюлозой (НРМС), такой как HPMCAS, НРМС Е50, НРМСЕ15 или HPMC60SH50.
Как обсуждается к данной заявке, полимер может быть рН-зависимым растворимым в кишечнике полимером. Такие рН-зависимые растворимые в кишечнике полимеры включают, но не ограничиваются производными целлюлозы (например, ацетатфталатом целлюлозы (САР)), фталатами гидроксипропилметилцеллюлозы (НРМСР), ацетатсукцинатом гидроксипропилметилцеллюлозы (HPMCAS), карбоксиметилцеллюлозой (CMC) или их солями (например, натриевой солью, такой как (CMC-Na)); ацетаттримеллитатом целлюлозы (CAT), ацетатфталатом гидроксипропилцеллюлозы (НРСАР), ацетатфталатом гидроксипропилметилцеллюлозы (НРМСАР) и ацетатфталатом метилцеллюлозы (МСАР) или полиметакрилатами (например, Eudragit® S). В некоторых вариантах применения изобретения полимер является ацетатсукцинатом гидроксипропилметилцеллюлозы (HPMCAS). В некоторых вариантах применения изобретения полимер является ацетатсукцинатом гидроксипропилметилцеллюлозы марки HG (в виде гранул, растворяющийся при высоком рН) (HPMCAS-HG).
В еще одном варианте применения изобретения полимер является поливинилпирролидоновым сополимером, например, авинилпирролидон/винилацетатным сополимером (PVP/VA).
В вариантах применения изобретения, где Соединение 2 образует высушенную распылением дисперсию с полимером, например, с НРМС, HPMCAS или PVP/VA полимером, количество полимера относительно общего веса высушенной распылением дисперсии варьирует приблизительно от 0,1% до 99% по массе. Если иное не оговорено, процентное содержание лекарственного препарата, полимера и других вспомогательных веществ в дисперсии дано как процентное содержание по массе. Количество полимера обычно составляет по меньшей мере приблизительно 20% и предпочтительно приблизительно по меньшей мере приблизительно 30%, например, по меньшей мере приблизительно 35%, по меньшей мере приблизительно 40%, по меньшей мере приблизительно 45% или приблизительно 50% (например, 49,5%). Количество обычно составляет около 99% или менее, и предпочтительно около 80% или менее, например, около 75% или менее, около 70% или менее, около 65% или менее, около 60% или менее или около 55% или менее. В одном варианте применения изобретения полимер присутствует в количестве, составляющем вплоть до приблизительно 50% от общего веса дисперсии (и точнее приблизительно между 40% и 50%, в частности около 49%, около 49,5% или около 50%). НРМС и HPMCAS доступны в виде различных марок производства ShinEtsu, например, HPMCAS доступна в виде нескольких разновидностей, включая AS-LF (ацетатсукцинат в виде мелкозернистого порошка, растворяющийся при низком рН), AS-MF (ацетатсукцинат в виде мелкозернистого порошка, растворяющийся при среднем рН), AS-HF (ацетатсукцинат в виде мелкозернистого порошка, растворяющийся при высоком рН), AS-LG (ацетатсукцинат в виде гранул, растворяющийся при низком рН), AS-MG (ацетатсукцинат в виде гранул, растворяющийся при среднем рН), AS-HG (ацетатсукцинат в виде гранул, растворяющийся при высоком рН). Каждая из этих марок отличается по проценту замещения ацетата и сукцината.
В некоторых вариантах применения изобретения аморфное Соединение 2 и полимер присутствуют в приблизительно равных количествах, например, и полимер и лекарственный препарат составляют около половины процентного содержания (по массе) дисперсии. Например, полимер присутствует в количестве около 49,5%, а лекарственный препарат присутствует в количестве около 50%.
В некоторых вариантах применения изобретения комбинация по существу аморфного Соединения 2 и полимера составляет от 1% до 20% вес/вес от общего твердого содержимого не высушенной распылением дисперсии перед высушиванием распылением. В некоторых вариантах применения изобретения комбинация по существу аморфного Соединения 2 и полимера составляет от 5% до 15% вес/вес от общего твердого содержимого не высушенной распылением дисперсии перед высушиванием распылением. В некоторых вариантах применения изобретения комбинация по существу аморфного Соединения 2 и полимера составляет около 11% вес/вес от общего твердого содержимого не высушенной распылением дисперсии перед высушиванием распылением.
В некоторых вариантах применения изобретения дисперсия далее включает другие присутствующие в незначительном количестве ингредиенты, такие как сурфактант (например, лаурилсульфат натрия). В некоторых вариантах применения изобретения сурфактант присутствует в количестве, составляющем приблизительно менее 10% дисперсии, например, приблизительно менее 9%, приблизительно менее 8%, приблизительно менее 7%, приблизительно менее 6%, приблизительно менее 5%, приблизительно менее 4%, приблизительно менее 3%, приблизительно менее 2%, приблизительно 1% или приблизительно 0,5%.
В вариантах применения изобретения, включающих полимер, полимер должен присутствовать в эффективном количестве для стабилизации высушенной распылением дисперсии. Стабилизация включает ингибирование или предотвращение кристаллизации по существу аморфного Соединения 2. Такая стабилизация ингибирует превращение Соединения 2 из аморфной в кристаллическую форму. Например, полимер будет препятствовать превращению по меньшей мере части (например, приблизительно 5%, приблизительно 10%, приблизительно 15%, приблизительно 20%, приблизительно 25%, приблизительно 30%, приблизительно 35%, приблизительно 40%, приблизительно 45%, приблизительно 50%, приблизительно 55%, приблизительно 60%, приблизительно 65%, приблизительно 70%, приблизительно 75% или более) Соединения 2 из аморфной формы в кристаллическую. Стабилизацию можно измерить, например, измеряя температуру стеклования высушенной распылением дисперсии, измеряя уровень релаксации аморфного материала или измеряя растворимость или биодоступность Соединения 2.
Подходящие полимеры для применения в комбинации с Соединением 2, например, для образования высушенной распылением дисперсии, такой как аморфной высушенной распылением дисперсии, должны обладать одним или более следующими свойствами:
Температура стеклования полимера должна быть приблизительно не менее чем на 10-15°С ниже температуры стеклования по существу аморфного Соединения 2. Предпочтительно, чтобы температура стеклования полимера была выше температуры стеклования по существу аморфного Соединения 2 и в общем случае была по меньшей мере на 50°С больше, чем желаемая температура хранения лекарственного препарата. Например, по меньшей мере около 100°С, по меньшей мере около 105°С, по меньшей мере около 105°С, по меньшей мере около 110°С, по меньшей мере около 120°С, по меньшей мере около 130°С, по меньшей мере около 140°С, по меньшей мере около 150°С, по меньшей мере около 160°С, по меньшей мере около 160°С или более.
Полимер должен быть относительно негигроскопичным. Например, полимер при хранении в стандартных условиях должен адсорбировать приблизительно менее 10% воды, например, приблизительно менее 9%, приблизительно менее 8%, приблизительно менее 7%, приблизительно менее 6% или приблизительно менее 5%, приблизительно менее 4% или приблизительно менее 3% воды. Предпочтительно, чтобы полимер при хранении в стандартных условиях был по существу свободным от адсорбированной воды.
Полимер должен обладать схожей или лучшей растворимостью в растворителях, подходящих для процессов высушивания распылением, по сравнению с Соединением 2. В предпочтительных вариантах применения изобретения полимер будет растворяться в одном или более таких же растворителях или системах растворителей, как и Соединение 2. Предпочтительно, чтобы полимер растворялся по меньшей мере в одном не содержащем гидрокси-группу растворителе, таком как хлорид метилена, ацетон или их комбинации.
Полимер при комбинации с по существу аморфным Соединением 2, например, в высушенной распылением дисперсии или в жидкой суспензии, должен повышать растворимость Соединения 2 в водной и физиологически родственной среде относительно растворимости Соединения 2 в отсутствие полимера или относительно растворимости Соединения 2 в комбинации с полимером сравнения. Например, полимер может увеличивать растворимость аморфного Соединения 2 посредством снижения количества аморфного Соединения 2, которое превращается в кристаллическое Соединение 2 либо из твердой аморфной дисперсии, либо из жидкой суспензии.
Полимер должен снижать уровень релаксации аморфного вещества.
Полимер должен повышать физическую и/или химическую стабильность по существу аморфного Соединения 2.
Полимер должен улучшать возможности производства по существу аморфного Соединения 2.
Полимер должен улучшать одно или более свойства обращения, введения или хранения по существу аморфного Соединения 2.
Полимер не должен вступать в нежелательные взаимодействия с другими фармацевтическими компонентами, например, вспомогательными веществами.
Пригодность кандидатного полимера (или другого компонента) для образования аморфной композиции можно проверить с помощью способов высушивания распылением (или других способов), описанных в данной заявке. Кандидатную композицию можно сравнивать. с точки зрения стабильности, сопротивлению образованию кристаллов или других свойств и сравнивать с препаратом сравнения, например, препаратом строго аморфного Соединения 2 или кристаллического Соединения 2. Например, кандидатную композицию можно исследовать, чтобы определить, замедляет ли она время до начала опосредованной растворителем кристаллизации, или уменьшает ли процент конверсии в заданное время в контролируемых условиях, на по меньшей мере 50%, 75%, 100% или 110%, так же как и препарат сравнения, или кандидатную композицию можно исследовать, чтобы определить, обладает ли она повышенной биодоступностью или растворимостью относительно кристаллического Соединения 2.
Сурфактанты
Высушенная распылением дисперсия может включать сурфактант. Сурфактант или смесь сурфактантов будет в общем случае снижать натяжение на границе раздела высушенной распылением дисперсии и водной среды. Соответствующий сурфактант или смесь сурфактантов может также усиливать водорастворимость и биодоступность Соединения 2 из высушенной распылением дисперсии. Сурфактанты для применения в связи с настоящим изобретением включают, но не ограничиваются эфирами сорбитана и жирной кислоты (например, Spans®), полиоксиэтиленовыми эфирами сорбитана и жирной кислоты (например, Tweens®), лаурилсульфатом натрия (SLS), додецилбензенсульфонатом натрия (SDBS), сульфосукцинатом диоктилнатрия (Docusate), натриевой солью диоксихолиновой кислоты (DOSS), сорбитанмоностеаратом, сорбитантристеаратом, гексадецилтриметиламмонийбромидом (НТАВ), N-лауроилсаркозином натрия, олеатом натрия, миристатом натрия, стеаратом натрия, пальмиатом натрия, Gelucire 44/14, этилендиаминтетрауксусной кислотой (ЭДТА), полиэтиленгликоль 1000 сукцинатом витамина Ε d-альфа токоферола (TPGS), лецитином, мол. вес 677-692, мононатриевым моногидратом глутаминовой кислоты, Labrasol, ПЭГ 8 каприловыми/каприновыми глицеридами, Transcutol, моноэтиловым эфиром диэтиленгликоля, Solutol HS-15, полиэтилгликоль/гидроксистеаратом, таурохолевой кислотой, Pluronic F68, Pluronic F108 и Pluronic F127 (или другими полиоксиэтилен-полиоксипропиленовыми сополимерами (Pluronics®) или насыщенными полигликозилированными глицеридами (Gelucirs®)). Специфичные примеры таких сурфактантов, которые можно применять в связи с настоящим изобретением, включают, но не ограничиваются Span 65, Span 25, Tween 20, Capryol 90, Pluronic F108, лаурилсульфатом натрия (SLS), полиэтиленгликоль 1000 сукцинатом витамина Ε d-альфа токоферола, плюрониками и сополимерами. В общем случае лаурилсульфат натрия является предпочтительным.
Количество сурфактанта (например, лаурилсульфата натрия) относительно общего веса высушенной распылением дисперсии может быть в пределах 0,1-15%. Предпочтительно, чтобы оно составляло приблизительно от 0,5% приблизительно до 10%, более предпочтительно, приблизительно от 0,5 приблизительно до 5%, например, приблизительно от 0,5 до 4%, приблизительно от 0,5 до 3%, приблизительно от 0,5 до 2%, приблизительно от 0,5 до 1% или приблизительно 0,5%.
В определенных вариантах применения изобретения количество сурфактанта относительно общего веса высушенной распылением дисперсии составляет по меньшей мере приблизительно 0,1%, предпочтительно около 0,5%. В данных вариантах применения изобретения сурфактант будет присутствовать в количестве, не превышающем приблизительно 15%, и предпочтительно, не превышающем приблизительно 12%, приблизительно 11%, приблизительно 10%, приблизительно 9%, приблизительно 8%, приблизительно 7%, приблизительно 6%, приблизительно 5%, приблизительно 4%, приблизительно 3%, приблизительно 2% или приблизительно 1%. Вариант применения изобретения, в котором сурфактант содержится в количестве, равном приблизительно 0,5% по массе, является предпочтительным.
Кандидатные сурфактанты (или другие компоненты) можно исследовать на соответствие применению в изобретении способом, схожим с описанным для исследования полимеров.
СПОСОБЫ СОЗДАНИЯ ФАРМАЦЕВТИЧЕСКИХ КОМПОЗИЦИЙ
Фармацевтические композиции, являющиеся предметом изобретения, можно производить посредством влажной грануляции, компактизации или прессования смеси или композиции, например, порошка или гранул, под давлением для образования стальной трехмерной формы (например, таблетки). Как применяется в данной заявке, ''таблетка'' включает спрессованные фармацевтические стандартные лекарственные формы всех форм и размеров, покрытые оболочкой или нет.
Термин ''таблетка'', как применяется в данной заявке, относится к физически дискретной единице агента, подходящей для пациента, которому требуется лечение. В общем случае компактизованная смесь обладает большей плотностью, чем плотность смеси перед компактизацией. Доза в форме таблетки, являющейся предметом настоящего изобретения, может обладать практически любой формой, включая вогнутую и/или выпуклую стороны, закругленные или острые углы и округлую или прямую форму. В некоторых вариантах применения изобретения спрессованные таблетки, являющиеся предметом изобретения, включают округлую таблетку с плоскими сторонами. Таблетки, являющиеся предметом изобретения, можно приготовить любым способом компактизации и прессования, известным среднему специалисту в данной области техники как способ образования прессованных твердых фармацевтических лекарственных форм. В частных случаях применения изобретения формы выпуска, представленные в данной заявке, можно приготовить с помощью традиционных способов, известных специалистам в области техники, относящейся к фармацевтическому составу, как описано, например, в учебниках на соответствующую тему. См., например, Remington: The Science and Practice of Pharmacy, 21st Ed., Lippincott Williams & Wilkins, Baltimore, Md. (2003); Ansel et al., Pharmaceutical Dosage Forms And Drug Delivery Systems, 7th Edition, Lippincott Williams & Wilkins, (1999); The Handbook of Pharmaceutical Excipients, 4th edition, Rowe et al., Eds., American Pharmaceuticals Association (2003); Gibson, Pharmaceutical Preformulation And Formulation, CRC Press (2001), эти ссылки в полном объеме включены в данную заявку посредством ссылки.
Грануляция и прессование
В некоторых вариантах применения изобретения ингредиенты взвешивают в соответствии с заданной здесь формулой. Затем все интрагранулярные ингредиенты просеивают и тщательно перемешивают. Ингредиенты можно смазать подходящим смазывающим веществом, например, стеаратом магния. Следующая стадия может включать компактизацию/брикетирование порошковой смеси и откалиброванных по размеру ингредиентов. Затем компактизованные или брикетированные смеси перемалывают в гранулы и просеивают для получения желаемого размера. Затем гранулы можно далее смазать, например, с помощью стеарата магния. Затем гранулярную композицию, являющуюся предметом изобретения, можно спрессовать с помощью подходящих пуансонов в разные фармацевтические формы в соответствии с изобретением. При желании таблетки можно покрыть пленочной оболочкой, красителем или другим покрытием.
Другой аспект изобретения представляет способ производства фармацевтической композиции, включающий образование смеси композиции, включающей Соединение 1 Формы I, твердой дисперсии, включающей по существу аморфное Соединение 2, и один или более вспомогательные вещества, выбранные из: наполнителя, растворителя, связывающего вещества, сурфактанта, смазывающего вещества, разрыхлителя, и прессование композиции в таблетки с растворимостью, равной по меньшей мере около 50% в течение 30 минут.
В другом варианте применения изобретения процесс влажной грануляции проводят для получения фармацевтической формы, являющейся предметом настоящего изобретения, из смеси порошковых и жидких ингредиентов. Например, фармацевтическую композицию, включающую смесь композиции, включающей Соединение 1 Формы I, твердую дисперсию, включающую по существу аморфное Соединение 2 и один или более вспомогательные вещества, выбранные из: наполнителя, связывающего вещества, сурфактанта или разрыхлителя, взвешивают в соответствии с заданной здесь формулой. Затем интрагранулярные ингредиенты просеивают и перемешивают в грануляторе с большим или малым усилием сдвига с помощью воды или воды с сурфактантом, или воды со связывающим веществом, или воды с сурфактантом и со связывающим веществом для грануляции порошковой смеси. Также можно применять жидкость, отличную от воды, содержащую или не содержащую сурфактант и/или связывающее вещество, для грануляции порошковой смеси. Затем влажные гранулы можно при желании перемолоть с помощью подходящего измельчителя. Затем при желании из смеси можно удалить воду посредством высушивания ингредиентов любым подходящим способом. Затем высушенные гранулы при желании можно перемолоть до заданного размера. Затем посредством перемешивания можно добавить экстрагранулярные вспомогательные вещества (например, наполнитель, растворитель и разрыхлитель). Затем откалиброванные по размеру гранулы можно далее смазать стеаратом магния и разрыхлителем, например, кроскармеллозой натрия. Затем гранулярную композицию, являющуюся предметом настоящего изобретения, можно просеять в течение достаточного количества времени для получения правильного размера и затем спрессовать с помощью подходящих пуансонов в разные фармацевтические формы в соответствии с изобретением. При желании таблетки можно покрыть пленочным покрытием, красителем или другим покрытием. Неожиданно оказалось, что влажную грануляцию можно провести без значительной потери твердых форм Соединения 1 Формы I или по существу аморфного Соединения 2.
В особенно предпочтительном варианте применения изобретения фармацевтические композиции, являющиеся предметом изобретения, готовят посредством непрерывного двухшнекового процесса влажной грануляции (TSWG). Непрерывное производство обеспечивает изготовление высококачественного и унифицированного продукта с текущим наблюдением и контролем. Непрерывное производство также улучшает качество за счет разработки плана с плотной сетью данных проектного поля и облегченным пониманием вклада вышестоящих переменных в последующие процессы и качество конечного продукта. Кроме того, фармацевтические композиции, являющиеся предметом настоящего изобретения, можно выпустить в окончательном виде на ранней стадии с помощью оборудования промышленного масштаба, что избавляет от масштабных рисков и изменения состава на поздних стадиях разработки. Наконец, непрерывное производство обладает коммерческими преимуществами производства, такими как улучшенный процесс контроля, сокращенное время перемещения продукта и эффективность выпуска в реальном времени. Общим результатом является более отлаженный, контролируемый и масштабируемый процесс с меньшим числом функциональных проверок, обеспечивающий в результате повышенное качество продукта и, следовательно, большую безопасность для пациента. Эти преимущества распространяются на вопросы Janet Woodcock (директора Центра Оценки и исследования Лекарственных препаратов (Center for Drug Evaluation and Research (CDER))) о том, что химические свойства, производство и контроль не смогут соответствовать быстрому клиническому развитию высокоэффективных видов терапии (''What we are seeing is that often the rate limiting step is going to be manufacturing,'' July 24, 2013 Friends of Cancer hosted congressional briefing ''Answering a Compelling Need: Expediting Life-Saving Treatments to Patients'' to discuss the Food and Drug Administration's Breakthrough Therapy Designation).
Например, известно, что грануляция с большим усилием сдвига (HSG, high shear granulation), являющаяся обычной техникой грануляции, характеризуется риском чрезмерной грануляции и плохим контролем процесса. Масштабирование данного процесса очень сложно и включает значительный риск. Замена процесса HSG непрерывным процессом двухшнековой влажной грануляции (TSWG, twin screw wet granulation) позволяет масштабировать с помощью того же оборудования для производства партий разного объема за счет проведения процесса в течение более длительного времени. Это устраняет риск масштабирования, обычно связанный с другими процессами грануляции. Кроме того, было обнаружено, что процесс TSWG является более основательным, будучи менее чувствительным к чрезмерной грануляции. Как можно видеть на фиг. 3 для таблетки Соединения 1, процесс HSG демонстрировал значительное замедление растворимости с увеличением содержания воды, в то время как процесс TSWG не показывал изменения в том же диапазоне добавления воды. Неожиданно не было обнаружено изменения рабочих характеристик форм выпуска в виде таблеток, включающих Соединение 1 в интервале 45-55 процентов по массе, и форм выпуска в виде таблеток, включающих Соединение 1 в интервале 60-70 процентов по массе, при применении двухшнекового процесса влажной грануляции. Такого не наблюдали в процессе HSG. Кроме того, данный непрерывный процесс производства продукта повышенного качества разрешает претензию Управления по контролю качества пищевых продуктов, медикаментов и косметических средств (FDA) в отношении недоступности лекарственного препарата для нуждающихся в нем пациентов.
В одном варианте применения изобретения непрерывный процесс начинается с заполнения последовательно расположенного смесителя индивидуальными вспомогательными веществами, Соединением 1 и Соединением 2 через весовой питатель непрерывного действия. Из этого смесителя материал непрерывно передают и обрабатывают посредством процесса двухшнековой влажной грануляции, высушивания, перемалывания, добавления экстрагранулярного вспомогательного вещества, перемешивания, компрессии и покрытия пленочной оболочкой.
Например, в одном варианте применения изобретения таблетку, включающую Соединение 1 и Соединение 2, можно приготовить непрерывно в соответствии с картой производственного процесса, представленной на Фиг. 12.
Каждый ингредиент данного примера смеси описан выше и в Примерах ниже. Более того, смесь может включать необязательные добавки, такие как один или более красители, один или более усилители вкуса и/или один или более ароматизаторы, как описано выше и в Примерах ниже. В некоторых вариантах применения изобретения относительные концентрации (например, % по массе) каждого из этих ингредиентов (и любых необязательных добавок) в смеси также представлены выше и в Примерах ниже. Ингредиенты, входящие в состав смеси, могут быть представлены последовательно или в любой комбинации с добавками; и ингредиенты или комбинации ингредиентов могут быть представлены в любом порядке. В одном варианте применения изобретения смазывающее вещество является последним компонентом, добавленным к смеси.
В другом варианте применения изобретения смесь включает композицию из Соединения 1 Формы I, твердой дисперсии по существу аморфного Соединения 2 и любого одного или более вспомогательных веществ; связывающего вещества, сурфактанта, растворителя, смазывающего вещества, разрыхлителя и наполнителя., где каждый из этих ингредиентов представлен в форме порошка (например, представлен в виде частиц, имеющих средний или усредненный диаметр, измеренный с помощью светорассеяния, равный 250 мкм или менее (например, 150 мкм или менее, 100 мкм или менее, 50 мкм или менее, 45 мкм или менее, 40 мкм или менее или 35 мкм или менее)).
В другом варианте применения изобретения прессование смеси в таблетки проводят, наполняя форму (например, пресс-форму) смесью и создавая давление на смесь. Это можно осуществить с помощью штамповочного пресса или другого похожего аппарата. В некоторых вариантах применения изобретения смесь, состоящая из Соединения 1 Формы I, твердой дисперсии по существу аморфного Соединения 2 и вспомогательных веществ, сначала можно превратить в гранулярную форму. Можно откалибровать гранулы по размеру и спрессовать их в таблетки или выпустить в форме капсул в соответствии со способами, известными в области фармацевтики. Также было замечено, что давление на смесь в форме можно повторить, применяя то же давление при каждом прессовании или разное давление во время прессования. В другом примере смесь порошковых ингредиентов или гранул можно спрессовать с помощью штамповочного пресса, который прикладывает достаточное давление для формирования таблетки с растворимостью около 50% или более в течение приблизительно 30 минут (например, около 55% или более в течение 30 минут или около 60% или более в течение 30 минут). Например, смесь прессуют с помощью штамповочного пресса для получения таблетки с твердостью равной по меньшей мере приблизительно 5 кП (по меньшей мере приблизительно 5,5 кП, по меньшей мере приблизительно 6 кП, по меньшей мере приблизительно 7 кП, по меньшей мере приблизительно 10 кП или по меньшей мере 15 кП). В ряде случаев смесь прессуют для получения таблетки с твердостью в интервале приблизительно от 5 до 20 кП.
В некоторых вариантах применения изобретения таблетки, включающие фармацевтическую композицию, как описано в данной заявке, могут быть покрыты пленочным покрытием, составляющим 3,0% по массе, включающим краситель по массе от таблетки. В определенных случаях суспензия или раствор красителя, применяемая для покрытия таблеток, включает около 20% вес/вес твердой фазы суспензии или раствора красителя по массе. В других случаях покрытые таблетки можно пометить логотипом, другим изображением или текстом.
В другом варианте применения изобретения способ производства фармацевтической композиции включает производство смеси твердых форм, например, смеси порошковых и/или жидких ингредиентов, смеси, включающей Соединение 1 Формы I, твердой дисперсии, включающей по существу аморфное Соединение 2 и одно или более вспомогательные вещества, выбираемые из: связывающего вещества, растворителя, сурфактанта, смазывающего вещества, разрыхлителя и наполнителя; перемешивание смеси, пока смесь не станет по существу гомогенной, и прессование или компактизацию смеси в гранулярную форму. Затем гранулярную композицию, включающую Соединение 1 Форму I и твердую дисперсию, включающую по существу аморфное Соединение 2, можно спрессовать в таблетки или выпустить в виде капсул, как описано выше или в Примерах ниже. В качестве альтернативы способы производства фармацевтической композиции включают производство смеси Соединения 1 Формы I, твердой дисперсии по существу аморфного Соединения 2 и одного или более вспомогательных веществ, например, связывающего вещества, растворителя, сурфактанта, смазывающего вещества, разрыхлителя и наполнителя; перемешивание смеси, пока смесь не станет по существу гомогенной, и прессование/компактизацию смеси в гранулярную форму с помощью процесса влажной грануляции с большим усилием сдвига, как изложено в Примерах ниже. Фармацевтические формы выпуска, например, таблетки, как описано в данной заявке, можно сделать с помощью приготовленных гранул, включающих Соединение 1 Форму I и твердую дисперсию по существу аморфного Соединения 2 в дополнение к выбранным вспомогательным веществам, описанным здесь.
В некоторых вариантах применения изобретения смесь перемешивают посредством взбалтывания, смешивания, встряхивания или подобного с помощью перемешивания вручную, миксера, смесителя, любой их комбинации или подобного. При последовательном добавлении ингредиентов или комбинаций ингредиентов перемешивание можно осуществить между последовательными добавлениями, непрерывно на протяжении добавления ингредиентов, после добавления всех ингредиентов или комбинаций ингредиентов или любой из комбинации. Смесь перемешивают, пока композиция не станет по существу гомогенной.
В другом варианте применения изобретения настоящее изобретение включает размол на струйной мельнице фармацевтической композиции, включающей Соединение 1 Форму I и твердую дисперсию, включающую по существу аморфное Соединение 2, в подходящем стандартном приборе для размалывания с помощью давления воздухом, подходящем для производства частиц, значительная фракция которых имеет размер между 0,1 микрон и 50 микрон. В другом варианте применения изобретения размер частиц составляет от 0,1 микрон до 20 микрон. В другом варианте применения изобретения размер частиц составляет от 0,1 микрон до 10 микрон. В другом варианте применения изобретения размер частиц составляет от 1,0 микрон до 5 микрон. В еще одном варианте применения изобретения фармацевтическая композиция имеет размер частиц D50 равный 2,0 микрон.
Формы выпуска, являющиеся предметом настоящего изобретения, представляют фиксированную величину дозы двух активных фармацевтических ингредиентов для эффективного лечения кистозного фиброза, комбинации, которая была отмечена одним из всего лишь двух Признаний нового препарата в качестве терапии прорыва Управления по контролю качества пищевых продуктов, медикаментов и косметических средств (FDA), и соответствует этому с удивительной стабильностью, о чем свидетельствуют измерения малых потерь аморфной твердой формы Соединения 2. Фиг. 4 показывает малую степень кристаллизации Соединения 2 в течение времени в PC-XVII при 50°С после предварительного уравновешивания при 60% относительной влажности. Даже после приближения к 1000 часам в этих условиях, менее 5% по массе Соединения 2 кристаллизовалось. Фиг. 5 показывает для PC-XVII, что даже при повышенной температуре равной 60°С после предварительного уравновешивания при 60% относительной влажности при приближении времени к 1000 часам в этих условиях, менее 10% по массе Соединения 2 кристаллизовалось. Фиг. 6 и 7 показывают схожие результаты для PC-XIX. Настоящие формы выпуска, таким образом, представляют удобство фиксированной дозировки двух инновационных активных фармацевтических ингредиентов в удивительно стабильной фармацевтической композиции. Такие формы выпуска увеличивают способность пациента следовать схеме лечения, что напрямую связано с эффективным лечением заболеваний.
Готовые лекарственные формы, приготовленные, как описано выше, можно подвергнуть оценке растворимости in vitro в соответствии с Тестом 711 ''Растворение'' Фармакопеи США 29, Фармакопейная конвенция США (United States Pharmacopeial Convention, Inc.), Роквилл, штат Мэдисон, 2005 (''USP''), чтобы определить скорость, с которой действующее вещество высвобождается из готовых лекарственных форм. Содержание действующего вещества и уровни примесей обычно измеряют с помощью таких техник, как высокоэффективная жидкостная хроматография (ВЭЖХ).
В некоторых вариантах применения изобретение включает применение упаковочных материалов, таких как контейнеры и крышки из полиэтилена высокой плотности, полиэтилена низкой плотности и/или полипропилена и/или стекла, глассиновой фольги, алюминиевого саше и блистеров или стрипов, состоящих из алюминия или поливинилхлорида высокой плотности (ПВХ), при желании, включающих высушивающее вещество, полиэтилен (ПЭ), поливинилидендихлорид (ПВДХ), ПВХ/ПЭ/ПВДХ и подобные. Эти упаковочные материалы можно применять для стерильного хранения разных фармацевтических композиций и составов после соответствующей стерилизации упаковки и ее содержимого с помощью техник химической или физической стерилизации, обычно применяемых в фармацевтической области техники.
СПОСОБЫ ВВЕДЕНИЯ ФАРМАЦЕВТИЧЕСКИХ КОМПОЗИЦИЙ
В одном аспекте фармацевтические композиции, являющиеся предметом изобретения, можно вводить пациенту один раз в день или приблизительно раз в двадцать четыре часа. В качестве альтернативы фармацевтические композиции, являющиеся предметом изобретения, можно вводить пациенту два раза в день. В качестве альтернативы фармацевтические композиции, являющиеся предметом изобретения, можно вводить каждые двенадцать часов. Эти фармацевтические композиции вводят в форме для перорального введения, содержащих около 25 мг, 50 мг, 100 мг, 125 мг, 150 мг, 200 мг, 250 мг, 300 мг или 400 мг Соединения 1 Формы I; и около 25 мг, 50 мг, 100 мг, 125 мг, 150 мг, 200 мг или 250 мг по существу аморфного Соединения 2. В этом аспекте в дополнение к Соединению 1 Форме I и по существу аморфному Соединению 2 фармацевтические композиции включают наполнитель, разрыхлитель, сурфактант, связывающее вещество и смазывающее вещество (в зависимости от того, является ли фармацевтическая композиция гранулой или таблеткой). Например, доза 400 мг Соединения 1 Формы I может включать две таблетки, являющиеся предметом изобретения, каждая из которых содержит 200 мг Соединения 1 Формы I. Доза 250 mg по существу аморфного Соединения 2 может включать две таблетки, являющиеся предметом изобретения, каждая из которых содержит 125 мг по существу аморфного Соединения 2.
Следует принимать во внимание, что соединение и фармацевтически приемлемые композиции и составы, являющиеся предметом изобретения, можно применять в комбинации терапий; иными словами Соединение 1 Форму I и твердую дисперсию по существу аморфного Соединения 2 и их фармацевтически приемлемые композиции можно вводить совместно с, перед или после одного или более других необходимых терапевтических средств или медицинских процедур.
В одном варианте применения изобретения дополнительный терапевтический агент выбирают из муколитического агента, бронходилататора, антибиотика, противоинфекционного агента, противовоспалительного агента, соединения, которое индуцирует активность CFTR, отличного от Соединения 1 Формы I и по существу аморфного Соединения 2, или питательного вещества.
В одном варианте применения изобретения дополнительный агент является (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1Н-индол-5-ил)циклопропанкарбоксамидом. В другом варианте применения изобретения дополнительный агент является 4-(3-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамидо)изохинолин-1-ил)бензойной кислотой. В другом варианте применения изобретения дополнительный агент выбирают из Таблицы 1:
В другом варианте применения изобретения дополнительный агент является любой комбинацией вышеупомянутых агентов. Например, комбинация может включать фармацевтическую композицию или таблетку, являющуюся предметом настоящего изобретения, включающую Соединение 1 Форму I и твердую дисперсию по существу аморфного Соединения 2, а дополнительный терапевтический агент является (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1Н-индол-5-ил)циклопропанкарбоксамидом. В другом примере комбинация может включать фармацевтическую композицию или таблетку, являющуюся предметом настоящего изобретения, включающую Соединение 1 Форму I и твердую дисперсию по существу аморфного Соединения 2, а дополнительный терапевтический агент является 4-(3-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамидо)изохинолин-3-ил)бензойной кислотой. В другом примере комбинация может включать фармацевтическую композицию или таблетку, являющуюся предметом настоящего изобретения, включающую Соединение 1 Форму I и твердую дисперсию по существу аморфного Соединения 2, а дополнительный терапевтический агент является любым соединением из Таблицы 1, т.е. соединениями от 1 до 14 из Таблицы 1 или любой их комбинацией.
В другом варианте применения изобретения дополнительный агент выбирают из Таблицы 1:



В другом варианте применения изобретения дополнительный агент выбирают из Таблицы 2:


В одном варианте применения изобретения дополнительный терапевтический агент является антибиотиком. Примеры антибиотиков, применимые в данной заявке, включают тобрамицин, включая тобрамицин в виде ингаляционного порошка, азитромицин, кайстон, азтреонам, включая аэрозольную форму азтреонама, амикацин, включая его липосомные формы, ципрофлоксацин, включая его формы, подходящие для введения посредством ингаляции, левофлаксацин, включая его аэрозольные формы, и комбинации двух антибиотиков, например, фосфомицин и тобрамицин.
В другом варианте применения изобретения дополнительный агент является муколитиком. Примеры муколитиков, применимые в данной заявке, включают Pulmozyme®.
В другом варианте применения изобретения дополнительный агент является бронходилататором. Примеры бронходилататоров включают альбутерол, метапротенеролсульфат, пирбутеролацетат, сальметерол или тетрабулинсульфат.
В другом варианте применения изобретения дополнительный агент является эффективным в отношении восстановления жидкости на поверхности дыхательных путей в легких. Такие агенты улучшают движение соли внутрь клетки и наружу, обеспечивая усиленную гидратацию слизи в дыхательных путях в легких и таким образом ее облегченную очистку. Примеры таких агентов включают гипертонический солевой раствор, денуфузол тетранатрия ([[(3S,5R)-5-(4-амино-2-оксопиримидин-1-ил)-3-гидроксиоксолан-2-ил]метокси-гидроксифосфорил][[[(2R,3S,4R,5R)-5-(2,4-диоксопиримидин-1-ил)-3,4-дигидроксиоксолан-2-ил]метокси-гидроксифосфорил]окси-гидроксифосфорил]гидрофосфат) или бронхитол (ингаляционную форму маннитола).
В другом варианте применения изобретения дополнительный агент является противовоспалительным агентом, т.е. агентом, который может уменьшать воспаление в легких. Примеры таких агентов, применимых в данной заявке, включают ибупрофен, докосагексаноевую кислоту (ДГА), силденафил, ингаляционный глутатион, пиоглитазон, гидроксихлорохин или симавастатин.
В другом варианте применения изобретения дополнительный агент является соединением, которое увеличивает или индуцирует активность CFTR, отличным от Соединения 1 Формы I или твердой дисперсии, включающей по существу аморфное Соединение 2, т.е. агентом, который оказывает эффект индукции или увеличения активности CFTR. Примеры таких агентов включают аталурен (''РТС124®''; 3-[5-(2-фторфенил)-1,2,4-оксадиазол-3-ил]бензойную кислоту), синапултид, ланковутид, депелестат (человеческий рекомбинантный ингибитор эластазы нейтрофилов) и кобипростон (7-{(2R,4aR,5R,7aR)-2-[(3S)-1,1-дифтор-3-метилпентил]-2-гидрокси-6-оксооктагидроциклопента[b]пиран-5-ил}гептановая кислота).
В другом варианте применения изобретения дополнительный агент является питательным веществом. Примеры питательных веществ включают панкрелипазу (заместитель панкреатического фермента), включая Pancrease®, Pancreacarb®, Ultrase® или Creon®, Liprotomase® (ранее известный как Trizytek®), Aquadeks® или глутатион в форме для ингаляции. В одном варианте применения изобретения дополнительное питательное вещество является панкрелипазой.
В другом варианте применения изобретения дополнительный агент является соединением, выбираемым из гентамицина, куркумина, циклофосфамида, 4-фенилбутирата, миглустата, фелодипина, нимодипина, Филоксина В, генестеина, Апигенина, цАМФ/цГМФ усилителей или индукторов, таких как ролипрам, силденафил, милринон, тадалафил, амринон, изопротеренол, альбутерол и алметерол, дезоксиспергуалин, ингибиторов HSP 90, ингибиторов HSP 70, протеасомных ингибиторов, таких как эпоксомицин, лактацистин и т.п.
В другом варианте применения изобретения дополнительный агент является соединением, выбираемым из (3,3,3-трифтор-2-гидрокси-2-метил-пропил)-амида 3-амино-6-(4-фторфенил)-5-трифторметил-пиридин-2-карбоновой кислоты; (3,3,3-трифтор-2-гидрокси-2-метил-пропил)-амида 5-амино-6'-метил-3-трифторметил-[2,3]бипиридинил-6-карбоновой кислоты; 3-амино-6-циклопропил-N-(3,3,3-трифтор-2-гидрокси-2-метилпропил)-5-трифторметил)пиколинамида; 3-амино-6-метокси-N-(3,3,3-трифтор-2-гидрокси-2-(трифторметил)пропил)-5-(трифторметил)пиколинамида; ((S)-3,3,3-трифтор-2-гидрокси-2-метил-пропил)-амида 3-амино-6-(4-фторфенил)-5-трифторметил-пиридин-2-карбоновой кислоты; ((S)-3,3,3-трифтор-2-гидрокси-2-метил-пропил)-амида 3-амино-6-метокси-5-трифторметил-пиридин-2-карбоновой кислоты; ((R)-3,3,3-трифтор-2-гидрокси-2-метил-пропил)-амида 3-амино-6-метокси-5-трифторметил-пиридин-2-карбоновой кислоты; ((S)-3,3,3-трифтор-2-гидрокси-2-метил-пропил)-амида 3-амино-6-(2,4-дихлорфенил)-5-трифторметил-пиридин-2-карбоновой кислоты; ((R)-3,3,3-трифтор-2-гидрокси-2-метил-пропил)-амида 3-амино-6-(2,4-дихлорфенил)-5-трифторметил-пиридин-2-карбоновой кислоты; (2-гидрокси-2-метил-пропил)-амида 3-амино-6-(4-фторфенил)-5-трифторметил-пиридин-2-карбоновой кислоты; ((S)-3,3,3-трифтор-2-гидрокси-2-метил-пропил)-амида 3-амино-5,6-бис-трифторметил-пиридин-2-карбоновой кислоты; ((R)-3,3,3-трифтор-2-гидрокси-2-метил-пропил)-амида 3-амино-5,6-бис-трифторметил-пиридин-2-карбоновой кислоты; (S)-3-амино-6-этокси-N-(3,3,3-трифтор-2-гидрокси-2-метилпропил)-5-(трифторметил)пиколинамида; ((S)-3,3,3-трифтор-2-гидрокси-2-метил-пропил)-амида 3-амино-6-метокси-5-трифторметил-пиридин-2-карбоновой кислоты; ((R)-3,3,3-трифтор-2-гидрокси-2-метил-пропил)-амида 3-амино-6-метокси-5-трифторметил-пиридин-2-карбоновой кислоты; (3,3,3-трифтор-2-гидрокси-2-метил-пропил)-амид 3-амино-6-(4-фторфенил)-5-трифторметил-пиридин-2-карбоновой кислоты; ((S)-3,3,3-трифтор-2-гидрокси-2-метил-пропил)-амида 3-амино-5,6-бис-трифторметил-пиридин-2-карбоновой кислоты; ((R)-3,3,3-трифтор-2-гидрокси-2-метил-пропил)-амида 3-амино-5,6-бис-трифторметил-пиридин-2-карбоновой кислоты или их фармацевтически приемлемые соли. В другом варианте применения изобретения дополнительный агент является соединением, раскрытым в Патенте США №8247436 и в Международной публикации согласно РСТ WO 20111138 94, полностью включенных в данный документ посредством ссылки.
В другом варианте применения изобретения дополнительный агент может быть регулятором эпителиального натриевого канала (ENac), раскрытым в публикациях согласно РСТ WO 2012035158, WO 2009074575, WO 2011028740, WO 2009150137, WO 2011079087 или WO 2008135557, полностью включенных в данный документ посредством ссылки.
В других вариантах применения изобретения дополнительный агент является соединением, раскрытым в WO 2004028480, WO 2004110352, WO 2005094374, WO 2005120497 или WO 2006101740, полностью включенных в данный документ посредством ссылки. В другом варианте применения изобретения дополнительный агент является производным бензо[с]хинолизиниума, которое обладает индуцирующей или усиливающей CFTR активностью, или производным бензопирана, которое обладает индуцирующей или усиливающей CFTR активностью. В другом варианте применения изобретения дополнительный агент является соединением, раскрытым в Патенте США №7202262, в Патенте США №6992096, US 20060148864, US 20060148863, US 20060035943, US 20050164973, WO 2006110483, WO 2006044456, WO 2006044682, WO 2006044505, WO 2006044503, WO 2006044502 или WO 2004091502, полностью включенных в данный документ посредством ссылки. В другом варианте применения изобретения дополнительный агент является соединением, раскрытым в WO 2004080972, WO 2004111014, WO 2005035514, WO 2005049018, WO 2006099256, WO 2006127588 или WO 2007044560, полностью включенных в данный документ посредством ссылки.
В одном варианте применения изобретения 400 мг Соединения 1 Формы I и 250 мг по существу аморфного Соединения 2 можно вводить нуждающемуся субъекту. В данных вариантах применения изобретения величину дозы можно достичь посредством введения одной или более таблеток, являющихся предметом изобретения. Например, введения 4 00 мг Соединения 1 Формы I и 250 мг по существу аморфного Соединения 2 можно достичь посредством введения двух таблеток, каждая из которых содержит 200 мг Соединения 1 Формы I и 125 мг по существу аморфного Соединения 2. Введение можно продолжать до улучшения состояния при заболевании или до совета терапевта субъекта, например, продолжительность введения может составлять менее недели, 1 неделю, 2 недели, 3 недели, четыре недели (28 дней) или месяц, или более. В одном варианте применения изобретения в день пациенту можно вводить две таблетки, каждая из которых содержит 200 мг Соединения 1 Формы I и 125 мг по существу аморфного Соединения 2. В дальнейшем варианте применения изобретения можно вводить две таблетки в одно и то же время или в разное время в течение дня. В дальнейшем варианте применения изобретения вводят одну таблетку каждые 12 часов.
В одном варианте применения изобретения 400 мг Соединения 1 Формы I и 500 мг по существу аморфного Соединения 2 можно вводить нуждающемуся субъекту. В данных вариантах применения изобретения величину дозы можно достичь посредством введения двух таблеток, каждая из которых содержит 200 мг Соединения 1 Формы I и 250 мг по существу аморфного Соединения 2. В одном варианте применения изобретения одну таблетку вводят раз в 12 часов. В другом варианте применения изобретения величину дозы можно достичь посредством введения двух таблеток, каждая из которых содержит 100 мг Соединения 1 Формы I и 125 мг по существу аморфного Соединения 2, каждые 12 часов. В другом варианте применения изобретения величину дозы также можно достичь посредством введения Соединения 1 Формы I по существу аморфного Соединения 2 в виде отдельных таблеток. Например, величину дозы можно достичь посредством введения двух таблеток, содержащих 200 мг Соединения 1 Формы I, и четырех таблеток, содержащих 125 мг по существу аморфного Соединения 2, или двух таблеток, содержащих 150 мг по существу аморфного Соединения 2, и двух таблеток, содержащих 100 мг по существу аморфного Соединения 2. Введение можно продолжать до улучшения состояния при заболевании или до совета терапевта субъекта, например, продолжительность введения может составлять менее недели, 1 неделю, 2 недели, 3 недели, четыре недели (28 дней) или месяц, или более. В одном варианте применения изобретения в день пациенту можно вводить две таблетки, содержащие 200 мг Соединения 1 Формы I, и четыре таблетки, содержащие 125 мг по существу аморфного Соединения 2. В одном варианте применения изобретения в день пациенту можно вводить две таблетки, содержащие 200 мг Соединения 1 Формы I, и дважды в день пациенту можно вводить две таблетки, содержащие 150 мг и 100 мг по существу аморфного Соединения 2. В дальнейшем варианте применения изобретения две таблетки вводят в одно и то же время или в разное время в течение дня. В дальнейшем варианте применения изобретения одну таблетку, содержащую 200 мг Соединения 1, вводят каждые 12 часов, и две таблетки, содержащие 150 мг и 100 мг по существу аморфного Соединения 2, вводят каждые 12 часов.
В одном варианте применения изобретения 300 мг Соединения 1 Формы I и 2 50 мг по существу аморфного Соединения 2 можно вводить нуждающемуся субъекту. В данных вариантах применения изобретения величину дозы можно достичь посредством введения одной или более таблеток, являющихся предметом изобретения. Например, введения 300 мг Соединения 1 Формы I и 250 мг по существу аморфного Соединения 2 можно достичь посредством введения двух таблеток, каждая из которых содержит 150 мг Соединения 1 Формы I и 125 мг по существу аморфного Соединения 2. Введение можно продолжать до улучшения состояния при заболевании или до совета терапевта субъекта, например, продолжительность введения может составлять менее недели, 1 неделю, 2 недели, 3 недели, четыре недели (28 дней) или месяц, или более. В одном варианте применения изобретения в день пациенту можно вводить две таблетки, каждая из которых содержит 150 мг Соединения 1 Формы I и 125 мг по существу аморфного Соединения 2. В дальнейшем варианте применения изобретения можно вводить две таблетки в одно и то же время или в разное время в течение дня. В дальнейшем варианте применения изобретения вводят одну таблетку каждые 12 часов.
В одном варианте применения изобретения 600 мг Соединения 1 Формы I и 500 мг по существу аморфного Соединения 2 можно вводить нуждающемуся субъекту. В данных вариантах применения изобретения величину дозы можно достичь посредством введения одной или более таблеток, являющихся предметом настоящего изобретения. Например, введения 600 мг Соединения 1 Формы I и 500 мг по существу аморфного Соединения 2 можно достичь посредством введения двух таблеток, каждая из которых содержит 150 мг Соединения 1 Формы I и 125 мг по существу аморфного Соединения 2, каждые 12 часов. Введение можно продолжать до улучшения состояния при заболевании или до совета терапевта субъекта, например, продолжительность введения может составлять менее недели, 1 неделю, 2 недели, 3 недели, четыре недели (28 дней) или месяц, или более. В одном варианте применения изобретения в день пациенту можно вводить четыре таблетки, каждая из которых содержит 150 мг Соединения 1 Формы I и 125 мг по существу аморфного Соединения 2. В дальнейшем варианте применения изобретения можно вводить четыре таблетки в одно и то же время или в разное время в течение дня. В дальнейшем варианте применения изобретения вводят две таблетки каждые 12 часов.
В одном варианте применения изобретения 800 мг Соединения 1 Формы I и 500 мг по существу аморфного Соединения 2 можно вводить нуждающемуся субъекту. В данных вариантах применения изобретения величину дозы можно достичь посредством введения одной или более таблеток, являющихся предметом настоящего изобретения. Например, введения 800 мг Соединения 1 Формы I и 500 мг по существу аморфного Соединения 2 можно достичь посредством введения четырех таблеток, каждая из которых содержит 200 мг Соединения 1 Формы I и 125 мг по существу аморфного Соединения 2. Введение можно продолжать до улучшения состояния при заболевании или до совета терапевта субъекта, например, продолжительность введения может составлять менее недели, 1 неделю, 2 недели, 3 недели, четыре недели (28 дней) или месяц, или более. В одном варианте применения изобретения в день пациенту можно вводить четыре таблетки, каждая из которых содержит 200 мг Соединения 1 Формы I и 125 мг по существу аморфного Соединения 2. В дальнейшем варианте применения изобретения можно вводить четыре таблетки в одно и то же время или в разное время в течение дня. В дальнейшем варианте применения изобретения вводят две таблетки за один прием, и в день проводят два приема. В дальнейшем варианте применения изобретения 800 мг Соединения 1 и 500 мг Соединения 2 вводят пациенту посредством введения двух таблеток, каждая из которых содержит 200 мг Соединения 1 и 125 мг Соединения 2, дважды в день. В дальнейшем варианте применения изобретения 800 мг Соединения 1 и 500 мг Соединения 2 вводят пациенту посредством введения двух таблеток, каждая из которых содержит 200 мг Соединения 1 и 125 мг Соединения 2, каждые 12 часов.
В одном варианте применения изобретения 600 мг Соединения 1 Формы I и 250 мг по существу аморфного Соединения 2 можно вводить нуждающемуся субъекту. В данных вариантах применения изобретения величину дозы можно достичь посредством введения одной или более таблеток, являющихся предметом настоящего изобретения. Например, введения 600 мг Соединения 1 Формы I и 250 мг по существу аморфного Соединения 2 можно достичь посредством введения трех таблеток, каждая из которых содержит 200 мг Соединения 1 Формы I и 83,3 мг по существу аморфного Соединения 2. Введение можно продолжать до улучшения состояния при заболевании или до совета терапевта субъекта, например, продолжительность введения может составлять менее недели, 1 неделю, 2 недели, 3 недели, четыре недели (28 дней) или месяц, или более. В одном варианте применения изобретения в день пациенту можно вводить три таблетки, каждая из которых содержит 200 мг Соединения 1 Формы I и 83,3 мг по существу аморфного Соединения 2. В дальнейшем варианте применения изобретения можно вводить три таблетки в одно и то же время или в разное время в течение дня. В дальнейшем варианте применения изобретения вводят три таблетки в одно и то же время.
В одном варианте применения изобретения 600 мг Соединения 1 Формы I и 500 мг по существу аморфного Соединения 2 можно вводить нуждающемуся субъекту. В данных вариантах применения изобретения величину дозы можно достичь посредством введения одной или более таблеток, являющихся предметом настоящего изобретения. Например, введения 600 мг Соединения 1 Формы I и 500 мг по существу аморфного Соединения 2 можно достичь посредством введения трех таблеток, каждая из которых содержит 200 мг Соединения 1 Формы I и 83,3 мг по существу аморфного Соединения 2, а затем двух дополнительных таблеток, каждая из которых содержит 125 мг Соединения 2. Введение можно продолжать до улучшения состояния при заболевании или до совета терапевта субъекта, например, продолжительность введения может составлять менее недели, 1 неделю, 2 недели, 3 недели, четыре недели (28 дней) или месяц, или более. В одном варианте применения изобретения 600 мг Соединения 1 можно вводить ежедневно и 250 мг Соединения 2 можно вводить дважды в день посредством введения трех таблеток, каждая из которых содержит 200 мг Соединения 1 Формы I и 83,3 мг по существу аморфного Соединения 2, ежедневно и двух таблеток, каждая из которых содержит 125 мг Соединения 2, каждые 12 часов. В одном варианте применения изобретения 600 мг Соединения 1 можно вводить ежедневно и 250 мг Соединения 2 вводить каждые 12 часов посредством введения трех таблеток, каждая из которых содержит 200 мг Соединения 1 Формы I и 83,3 мг по существу аморфного Соединения 2, ежедневно и двух таблеток, каждая из которых содержит 125 мг Соединения 2, каждые 12 часов.
Эти комбинации полезны для лечения заболеваний, описанных в данной заявке, включая кистозный фиброз. Эти комбинации также полезны в наборах реактивов, описанных в данной заявке. В другом аспекте настоящее изобретение содержит набор реактивов, включающий фармацевтическую композицию или таблетку, являющуюся предметом настоящего изобретения, включающую Соединение 1 Форму I и твердую дисперсию, включающую по существу аморфное Соединение 2, и отдельный дополнительный терапевтический агент или его фармацевтическую композицию. В другом варианте применения изобретения фармацевтическая композиция или таблетка, являющиеся предметом настоящего изобретения, отдельный дополнительный терапевтический агент или его фармацевтическая композиция находятся в отдельных контейнерах. В другом варианте применения изобретения отдельные контейнеры являются бутылями. В другом варианте применения изобретения отдельные контейнеры являются флаконами. В другом варианте применения изобретения отдельные контейнер являются блистерными упаковками.
Количество дополнительного терапевтического агента, присутствующего в композициях, являющихся предметом настоящего изобретения, не превысит количество, которое обычно вводят в составе композиции, включающей терапевтический агент в качестве единственного активного агента. Предпочтительно, чтобы количество дополнительного терапевтического агента в раскрытых здесь композициях варьировало в интервале приблизительно от 50% до 100% количества обычно присутствующего в композиции, содержащей этот агент в качестве единственного терапевтически активного агента.
ТЕРАПЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ КОМПОЗИЦИИ
В одном аспекте изобретение также представляет способ лечения, уменьшения тяжести или симптоматического лечения заболевания пациента, способ, включающий введение эффективного количества фармацевтической композиции или таблетки, являющихся предметом настоящего изобретения, пациенту, предпочтительно млекопитающему, в котором заболевание выбирают из кистозного фиброза, астмы, вызванного курением ХОБЛ, хронического бронхита, риносинусита, запора, панкреатита, недостаточности поджелудочной железы, мужского бесплодия, вызванного врожденным билатеральным отсутствием семявыносящего протока, умеренно выраженной болезни легких, идиопатического панкреатита, аллергического бронхолегочного аспергиллеза, заболевания печени, наследственной энфиземы, наследственного гемохроматоза, нарушений коагуляции и фибринолиза, таких как недостаток протеина С, наследственного ангионевротического отека типа 1, нарушений переработки липидов, таких как фамильная гиперхолестеролемия, хиломикронемия типа 1, абеталипопротеинемия, лизосомных болезней накопления, таких как болезнь I-клеток/болезнь Дери, мукополисахаридозов, болезни Сандхоффа/Тай-Сахса, болезни Криглера-Найяра типа II, полиэндокринопатий/гиперинсулинемии, сахарного диабета, карликовости Ларона, миелопероксидазной недостаточности, первичной недостаточности функции околощитовидных желез, меланомы, гликаноза CDG типа 1, врожденного гипертиреоза, несовершенного остеогенеза, наследственной гиперфибриногенемии, недостатка ACT (альфа 1-антихимотрипсина), несахарного диабета, несахарного нейрогипофизарного диабета, несахарного почечного диабета, болезни Шарко-Мари-Тута, болезни Пелицеуса-Мерцбахера, нейродегенеративных заболеваний, таких как болезнь Альцгеймера, болезнь Паркинсона, боковой амиотрофический склероз, прогрессирующий надъядерный паралич, болезнь Пика, некоторых полиглутаматных неврологических нарушений, таких как болезнь Хантингтона, спинально-церебеллярная атаксия типа I, спинальная и бульбарная мышечная атрофия, дентато-рубро-паллидо-льюисова атрофия и миотоническая дистрофия, а также губчатой энцефалопатии, такой как наследственная болезнь Крейцфельда-Якоба (из-за нарушения процессинга прионового белка), болезни Фабри, синдрома Штреусслера-Шейнкера, ХОБЛ, сухости глаз, синдрома Шегрена, остеопороза, остеопении, срастания перелома и роста кости (включая восстановление кости, регенерацию кости, уменьшения резорбции кости и увеличения отложения костной ткани), синдрома Горэма, нарушений хлорных каналов, таких как врожденная миотония (форм Томсона и Беккера), синдрома Бартера типа III, болезни Дента, гиперэкплексии, эпилепсии, лизосомных болезней накопления, синдрома Ангельмана и первичной цилиарной дискинезии, термина, обозначающего наследственные заболевания структуры и/или функций ресничек, включая первичную цилиарную дискинезию с транспозицией внутренних органов (также известную как синдром Картагенера), первичную цилиарную дискинезию без транспозиции внутренних органов и цилиарную аплазию.
В одном аспекте изобретение также представляет способ лечения, уменьшения тяжести или симптоматического лечения заболевания пациента, включающий введение эффективного количества фармацевтической композиции или таблетки, являющихся предметом настоящего изобретения, пациенту, предпочтительно млекопитающему, в котором заболевание выбирают из генерализованной эпилепсии с фебрильными судорогами плюс, генерализованной эпилепсии с фебрильными и афебрильными судорогами, миотонии, врожденной парамиотонии, миотонии, усиленной калием, семейного гиперкалиемического периодического паралича, синдрома удлиненного интервала QT/синдрома Бругада, аутосомно-доминантного синдрома удлиненного интервала QT с глухотой, аутосомно-рецессивного синдрома удлиненного интервала QT, синдрома удлиненного интервала QT с дизморфическими признаками, врожденного или приобретенного синдрома удлиненного интервала QT, синдрома Тимоти, персистирующей гиперинсулинемической гипогликемии младенцев, дилатационной кардиомиопатии, аутосомно-доминантного синдрома удлиненного интервала QT, болезни Дента, остеопорозз, синдрома Бартера типа III, болезни центральных волокон, злокачественной гипертермии и катехоламинергической полиморфной тахикардии.
В одном аспекте настоящее изобретение направлено на способ лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающий введение эффективного количества фармацевтической композиции или таблетки, являющихся предметом изобретения, пациенту, предпочтительно млекопитающему, в котором у пациента есть генетическая мутация CFTR N1303K, ΔΙ507 или R560T.
В одном аспекте настоящее изобретение направлено на способ лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающий введение эффективного количества фармацевтической композиции или таблетки, являющихся предметом изобретения, пациенту, предпочтительно млекопитающему, в котором у пациента есть генетическая мутация CFTR G551D. В другом варианте применения изобретения пациент гомозиготен по G551D. В другом варианте применения изобретения пациент гетерозиготен по G551D, и другая генетическая мутация CFTR является любой из ΔF508, G542X, N1303K, W1282X, R117H, R553X, 1717-1G->A, 621+1G->T, 2789+5G->A, 3849+10kbC->T, R1162X, G85E, 3120+1G->A, ΔΙ507, 1898+1G->A, 3659delC, R347P, R560T, R334W, A455E, 2184delA или 711+1G->T.
В одном аспекте настоящее изобретение направлено на способ лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающий введение эффективного количества фармацевтической композиции или таблетки, являющихся предметом изобретения, пациенту, предпочтительно млекопитающему, в котором у пациента есть генетическая мутация CFTR ΔF508. В другом варианте применения изобретения пациент гомозиготен по ΔF508. В другом варианте применения изобретения пациент гетерозиготен по ΔF508, и другая генетическая мутация CFTR является любой из G551D, G542X, N1303K, W1282X, R117H, R553X, 1717-1G->A, 621+1G->T, 2789+5G->A, 3849+10kbC->T, R1162X, G85E, 3120+1G->A, ΔΙ507, 1898+1G->A, 3659delC, R347P, R560T, R334W, A455E, 2184delA или 711+1G->T.
В одном аспекте настоящее изобретение направлено на способ лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающий введение эффективного количества фармацевтической композиции или таблетки, являющихся предметом настоящего, пациенту, предпочтительно млекопитающему, в котором у пациента есть генетическая мутация CFTR, выбираемая из G178R, G551S, G970R, G1244E, S1255P, G1349D, S549N, S549R, S1251N, E193K, F1052V, G1069R, R117C, D110H, R347H, R352Q, E56K, P67L, L206W, А455Е, D579G, S1235R, S945L, R1070W, F1074L, D110E, D1270N, D1152H, 1717-1G->A, 621+1G->T, 3120+1G->A, 1898+1G->A, 711+1G->T, 2622+1G->A, 405+1G->A, 406-1G->A, 4005+1G->A, 1812-1G->A, 1525-1G->A, 712-1G->T, 1248+1G->A, 1341+1G->A, 3121-1G->A, 4374+1G->T, 3850-1G->A, 2789+5G->A, 3849+10kbC->T, 3272-26A->G, 711+5G->A, 3120G->A, 1811+1.6kbA->G, 711+3A->G, 1898+3A->G, 1717-8G->A, 1342-2A->C, 405+3A->C, 1716G/A, 1811+1G->C, 1898+5G->T, 3850-3T->G, IVS14b+5G->A, 1898+1G->T, 4005+2T->C и 621+3A->G.
В одном аспекте настоящее изобретение направлено на способ лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающий введение эффективного количества фармацевтической композиции или таблетки, являющихся предметом изобретения, пациенту, предпочтительно млекопитающему, в котором у пациента есть генетическая мутация CFTR, выбираемая из G178R, G551S, G970R, G1244E, S1255P, G1349D, S549N, S549R, S1251N, E193K, F1052V и G1069R. В одном варианте применения изобретения в данном аспекте изобретение представляет способ лечения CFTR, включающий введение Соединения 1 пациенту с мутацией человеческого CFTR, выбираемой из G178R, G551S, G970R, G1244E, S1255P, G1349D, S549N, S549R и S1251N. В одном аспекте настоящее изобретение направлено на способ лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающий введение эффективного количества фармацевтической композиции или таблетки, являющихся предметом изобретения, пациенту, предпочтительно млекопитающему, в котором у пациента есть генетическая мутация CFTR, выбираемая из E193K, F1052V и G1069R. В некоторых вариантах применения изобретения в данном аспекте способ приводит к более чем 10-кратному увеличению транспорта хлорида относительно исходного уровня транспорта хлорида.
В одном аспекте настоящее изобретение направлено на способ лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающий введение эффективного количества фармацевтической композиции или таблетки, являющихся предметом изобретения, пациенту, предпочтительно млекопитающему, в котором у пациента есть генетическая мутация CFTR, выбираемая из R117C, D110H, R347H, R352Q, E56K, P67L, L206W, А455Е, D579G, S1235R, S945L, R1070W, F1074L, D110E, D1270N и D1152H. В одном варианте применения изобретения в данном аспекте способ приводит к увеличению транспорта хлорида, превышающему исходный уровень транспорта хлорид на 10% или более.
В одном аспекте настоящее изобретение направлено на способ лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающий введение эффективного количества фармацевтической композиции или таблетки, являющихся предметом изобретения, пациенту, предпочтительно млекопитающему, в котором у пациента есть генетическая мутация CFTR, выбираемая из 1717-1G->A, 621+1G->T, 3120+1G->A, 1898+1G->A, 711+1G->T, 2622+1G->A, 405+1G->A, 406-1G->A, 4005+1G->A, 1812-1G->A, 1525-1G->A, 712-1G->T, 1248+1G->A, 1341+1G->A, 3121-1G->A, 4374+1G->T, 3850-1G->A, 2789+5G->A, 3849+10kbC->T, 3272-26A->G, 711+5G->A, 3120G->A, 1811+1.6kbA->G, 711+3A->G, 1898+3A->G, 1717-8G->A, 1342-2A->C, 405+3A->C, 1716G/A, 1811+1G->C, 1898+5G->T, 3850-3T->G, IVS14b+5G->A, 1898+1G->T, 4005+2T->C и 621+3A->G. В одном аспекте настоящее изобретение направлено на способ лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающий введение эффективного количества фармацевтической композиции или таблетки, являющихся предметом изобретения, пациенту, предпочтительно млекопитающему, в котором у пациента есть генетическая мутация CFTR, выбираемая из 1717-1G->A, 1811+1.6kbA->G, 2789+5G->A, 3272-26A->G и 3849+10kbC->T. В одном аспекте настоящее изобретение направлено на способ лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающий, введение эффективного количества фармацевтической композиции или таблетки, являющихся предметом изобретения, пациенту, предпочтительно млекопитающему, в котором у пациента есть генетическая мутация CFTR, выбираемая из 2789+5G->A и 3272-26A->G.
В одном аспекте настоящее изобретение направлено на способ лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающий введение эффективного количества фармацевтической композиции или таблетки, являющихся предметом изобретения, пациенту, предпочтительно млекопитающему, в котором у пациента есть генетическая мутация CFTR, выбираемая из G178R, G551S, G970R, G1244E, S1255P, G1349D, S549N, S549R, S1251N, E193K, F1052V, G1069R, R117C, D110H, R347H, R352Q, E56K, P67L, L206W, А455Е, D579G, S1235R, S945L, R1070W, F1074L, D110E, D1270N, D1152H, 1717-1G->A, 621+1G->T, 3120+1G->A, 1898+1G->A, 711+1G->T, 2622+1G->A, 405+1G->A, 406-lG->A, 4005+1G->A, 1812-1G->A, 1525-1G->A, 712-1G->T, 1248+1G->A, 1341+1G->A, 3121-1G->A, 4374+1G->T, 3850-lG->A, 2789+5G->A, 3849+10kbC->T, 3272-26A->G, 711+5G->A, 3120G->A, 1811+1.6kbA->G, 711+3A->G, 1898+3A->G, 1717-8G->A, 1342-2A->C, 405+3A->C, 1716G/A, 1811+1G->C, 1898+5G->T, 3850-3T->G, IVS14b+5G->A, 1898+1G->T, 4005+2T->C и 621+3A->G, и мутация человеческого CFTR, выбираемая из ΔF508, R117H и G551D.
В одном аспекте настоящее изобретение направлено на способ лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающий введение эффективного количества фармацевтической композиции или таблетки, являющихся предметом изобретения, пациенту, предпочтительно млекопитающему, в котором у пациента есть генетическая мутация CFTR, выбираемая из G178R, G551S, G970R, G1244E, S1255P, G1349D, S549N, S549R, S1251N, E193K, F1052V и G1069R, и мутация человеческого CFTR, выбираемая из ΔF508, R117H и G551D. В одном аспекте настоящее изобретение направлено на способ лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающий введение эффективного количества фармацевтической композиции или таблетки, являющихся предметом изобретения, пациенту, предпочтительно млекопитающему, в котором у пациента есть генетическая мутация CFTR, выбираемая из G178R, G551S, G970R, G1244E, S1255P, G1349D, S549N, S549R и S1251N, и мутация человеческого CFTR, выбираемая из ΔF508, R117H и G551D. В одном аспекте настоящее изобретение направлено на способ лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающий введение эффективного количества фармацевтической композиции или таблетки, являющихся предметом изобретения, пациенту, предпочтительно млекопитающему, в котором у пациента есть генетическая мутация CFTR, выбираемая из E193K, F1052V и G1069R, и мутация человеческого CFTR, выбираемая из ΔF508, R117H и G551D. В некоторых вариантах применения изобретения в данном аспекте способ приводит к более чем 10-кратному увеличению транспорта хлорида относительно исходного уровня транспорта хлорида.
В одном аспекте настоящее изобретение направлено на способ лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающий введение эффективного количества фармацевтической композиции или таблетки, являющихся предметом изобретения, пациенту, предпочтительно млекопитающему, в котором у пациента есть генетическая мутация CFTR, выбираемая из R117C, D110H, R347H, R352Q, E56K, P67L, L206W, А455Е, D579G, S1235R, S945L, R1070W, F1074L, D110E, D1270N и D1152H, и мутация человеческого CFTR, выбираемая из ΔF508, R117H и G551D. В одном варианте применения изобретения в данном аспекте способ приводит к увеличению транспорта хлорида, превышающему исходный уровень транспорта хлорид на 10% или более.
В одном аспекте настоящее изобретение направлено на способ лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающий введение эффективного количества фармацевтической композиции или таблетки, являющихся предметом изобретения, пациенту, предпочтительно млекопитающему, в котором у пациента есть генетическая мутация CFTR, выбираемая из 1717-1G->A, 621+1G->T, 3120+1G->A, 1898+1G->A, 711+1G->T, 2622+1G->A, 405+1G->A, 406-lG->A, 4005+1G->A, 1812-1G->A, 1525-1G->A, 712-1G->T, 1248+1G->A, 1341+1G->A, 3121-1G->A, 4374+1G->T, 3850-lG->A, 2789+5G->A, 3849+10kbC->T, 3272-26A->G, 711+5G->A, 3120G->A, 1811+1.6kbA->G, 711+3A->G, 1898+3A->G, 1717-8G->A, 1342-2A->C, 405+3A->C, 1716G/A, 1811+1G->C, 1898+5G->T, 3850-3T->G, IVS14b+5G->A, 1898+1G->T, 4005+2T->C и 621+3A->G, и мутация человеческого CFTR, выбираемая из ΔF508, R117H hG551D. В одном аспекте настоящее изобретение направлено на способ лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающий введение эффективного количества фармацевтической композиции или таблетки, являющихся предметом изобретения, пациенту, предпочтительно млекопитающему, в котором у пациента есть генетическая мутация CFTR, выбираемая из 1717-1G->A, 1811+1.6kbA->G, 2789+5G->A, 3272-26A->G и 3849+10kbC->T, и мутация человеческого CFTR, выбираемая из ΔF508, R117H и G551D. В одном аспекте настоящее изобретение направлено на способ лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающий введение эффективного количества фармацевтической композиции или таблетки, являющихся предметом изобретения, пациенту, предпочтительно млекопитающему, в котором у пациента есть генетическая мутация CFTR, выбираемая из 2789+5G->A и 3272-26A->G, и мутация человеческого CFTR, выбираемая из ΔF508, R117H.
В одном аспекте настоящее изобретение направлено на способ лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающий введение эффективного количества фармацевтической композиции или таблетки, являющихся предметом изобретения, пациенту, предпочтительно млекопитающему, в котором у пациента есть генетическая мутация CFTR, выбираемая из G178R, G551S, G970R, G1244E, S1255P, G1349D, S549N, S549R, S1251N, E193K, F1052V, G1069R, R117C, D110H, R347H, R352Q, E56K, P67L, L206W, А455Е, D579G, S1235R, S945L, R1070W, F1074L, D110E, D1270N, D1152H, 1717-1G->A, 621+1G->T, 3120+1G->A, 1898+1G->A, 711+1G->T, 2622+1G->A, 405+1G->A, 406-lG->A, 4005+1G->A, 1812-1G->A, 1525-1G->A, 712-1G->T, 1248+1G->A, 1341+1G->A, 3121-1G->A, 4374+1G->T, 3850-lG->A, 2789+5G->A, 3849+10kbC->T, 3272-26A->G, 711+5G->A, 3120G->A, 1811+1.6kbA->G, 711+3A->G, 1898+3A->G, 1717-8G->A, 1342-2A->C, 405+3A->C, 1716G/A, 1811+1G->C, 1898+5G->T, 3850-3T->G, IVS14b+5G->A, 1898+1G->T, 4005+2T->C и 621+3A->G, и мутация человеческого CFTR, выбираемая из ΔF508, R117H и G551D.
В одном аспекте настоящее изобретение направлено на способ лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающий введение эффективного количества фармацевтической композиции или таблетки, являющихся предметом изобретения, пациенту, предпочтительно млекопитающему, в котором у пациента есть генетическая мутация CFTR, выбираемая из G178R, G551S, G970R, G1244E, S1255P, G1349D, S549N, S549R, S1251N, E193K, F1052V и G1069R. В одном аспекте настоящее изобретение направлено на способ лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающий введение эффективного количества фармацевтической композиции или таблетки, являющихся предметом изобретения, пациенту, предпочтительно млекопитающему, в котором у пациента есть генетическая мутация CFTR, выбираемая из G178R, G551S, G970R, G1244E, S1255P, G1349D, S549N, S549R и S1251N. В одном аспекте настоящее изобретение направлено на способ лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающий введение эффективного количества фармацевтической композиции или таблетки, являющихся предметом изобретения, пациенту, предпочтительно млекопитающему, в котором у пациента есть генетическая мутация CFTR, выбираемая из E193K, F1052V и G1069R. В некоторых вариантах применения изобретения в данном аспекте способ приводит к более чем 10-кратному увеличению транспорта хлорида относительно исходного уровня транспорта хлорида.
В одном аспекте настоящее изобретение направлено на способ лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающий введение эффективного количества фармацевтической композиции или таблетки, являющихся предметом изобретения, пациенту, предпочтительно млекопитающему, в котором у пациента есть генетическая мутация CFTR, выбираемая из R117C, D110H, R347H, R352Q, E56K, P67L, L206W, А455Е, D579G, S1235R, S945L, R1070W, F1074L, D110E, D1270N и 1152Н. В одном варианте применения изобретения в данном аспекте способ приводит к увеличению транспорта хлорида, превышающему исходный уровень транспорта хлорид на 10% или более.
В одном аспекте настоящее изобретение направлено на способ лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающий введение эффективного количества фармацевтической композиции или таблетки, являющихся предметом изобретения, пациенту, предпочтительно млекопитающему, в котором у пациента есть генетическая мутация CFTR, выбираемая из 1717-1G->A, 621+1G->T, 3120+1G->A, 1898+1G->A, 711+1G->T, 2622+1G->A, 405+1G->A, 406-lG->A, 4005+1G->A, 1812-1G->A, 1525-1G->A, 712-1G->T, 1248+1G->A, 1341+1G->A, 3121-1G->A, 4374+1G->T, 3850-lG->A, 2789+5G->A, 3849+10kbC->T, 3272-26A->G, 711+5G->A, 3120G->A, 1811+1.6kbA->G, 711+3A->G, 1898+3A->G, 1717-8G->A, 1342-2A->C, 405+3A->C, 1716G/A, 1811+1G->C, 1898+5G->T, 3850-3T->G, IVS14b+5G->A, 1898+1G->T, 4005+2T->C и 621+3A->G. В одном аспекте настоящее изобретение направлено на способ лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающий введение эффективного количества фармацевтической композиции или таблетки, являющихся предметом изобретения, пациенту, предпочтительно млекопитающему, в котором у пациента есть генетическая мутация CFTR, выбираемая из 1717-1G->A, 1811+1.6kbA->G, 2789+5G->A, 3272-26A->G и 3849+10kbC->T. В одном аспекте настоящее изобретение направлено на способ лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающий введение эффективного количества фармацевтической композиции или таблетки, являющихся предметом изобретения, пациенту, предпочтительно млекопитающему, в котором у пациента есть генетическая мутация CFTR, выбираемая из 2789+5G->A и 3272-26A->G.
В одном аспекте настоящее изобретение направлено на способ лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающий введение эффективного количества фармацевтической композиции или таблетки, являющихся предметом изобретения, пациенту, предпочтительно млекопитающему, в котором у пациента есть генетическая мутация CFTR, выбираемая из G178R, G551S, G970R, G1244E, S1255P, G1349D, S549N, S549R, S1251N, E193K, F1052V, G1069R, R117C, D110H, R347H, R352Q, E56K, P67L, L206W, А455Е, D579G, S1235R, S945L, R1070W, F1074L, D110E, D1270N, D1152H, 1717-1G->A, 621+1G->T, 3120+1G->A, 1898+1G->A, 711+1G->T, 2622+1G->A, 405+1G->A, 406-lG->A, 4005+1G->A, 1812-1G->A, 1525-1G->A, 712-1G->T, 1248+1G->A, 1341+1G->A, 3121-1G->A, 4374+1G->T, 3850-lG->A, 2789+5G->A, 3849+10kbC->T, 3272-26A->G, 711+5G->A, 3120G->A, 1811+1.6kbA->G, 711+3A->G, 1898+3A->G, 1717-8G->A, 1342-2A->C, 405+3A->C, 1716G/A, 1811+1G->C, 1898+5G->T, 3850-3T->G, IVS14b+5G->A, 1898+1G->T, 4005+2T->C и 621+3A->G, и мутация человеческого CFTR, выбираемая из ΔF508, R117H и G551D, и одна или более мутации человеческого CFTR, выбираемые из ΔF508, R117H и G551D.
В одном аспекте настоящее изобретение направлено на способ лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающий введение эффективного количества фармацевтической композиции или таблетки, являющихся предметом изобретения, пациенту, предпочтительно млекопитающему, в котором у пациента есть генетическая мутация CFTR, выбираемая из G178R, G551S, G970R, G1244E, S1255P, G1349D, S549N, S549R, S1251N, E193K, F1052V и G1069R, и одна или более мутации человеческого CFTR, выбираемые из ΔF508, R117H и G551D. В одном аспекте настоящее изобретение направлено на способ лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающий введение эффективного количества фармацевтической композиции или таблетки, являющихся предметом изобретения, пациенту, предпочтительно млекопитающему, в котором у пациента есть генетическая мутация CFTR, выбираемая из G178R, G551S, G970R, G1244E, S1255P, G1349D, S549N, S549R и S1251N, и одна или более мутации человеческого CFTR, выбираемые из ΔF508, R117H и G551D. В одном аспекте настоящее изобретение направлено на способ лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающий введение эффективного количества фармацевтической композиции или таблетки, являющихся предметом изобретения, пациенту, предпочтительно млекопитающему, в котором у пациента есть генетическая мутация CFTR, выбираемая из E193K, F1052V и G1069R, и одна или более мутации человеческого CFTR, выбираемые из ΔF508, R117H и G551D. В некоторых вариантах применения изобретения в данном аспекте способ приводит к более чем 10-кратному увеличению транспорта хлорида относительно исходного уровня транспорта хлорида.
В одном аспекте настоящее изобретение направлено на способ лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающий введение эффективного количества фармацевтической композиции или таблетки, являющихся предметом изобретения, пациенту, предпочтительно млекопитающему, в котором у пациента есть генетическая мутация CFTR, выбираемая из R117C, D110H, R347H, R352Q, E56K, P67L, L206W, А455Е, D579G, S1235R, S945L, R1070W, F1074L, D110E, D1270N и D1152H, и одна или более мутации человеческого CFTR, выбираемые из ΔF508, R117H и G551D. В одном варианте применения изобретения в данном аспекте способ приводит к увеличению транспорта хлорида, превышающему исходный уровень транспорта хлорид на 10% или более.
В одном аспекте настоящее изобретение направлено на способ лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающий введение эффективного количества фармацевтической композиции или таблетки, являющихся предметом изобретения, пациенту, предпочтительно млекопитающему, в котором у пациента есть генетическая мутация CFTR, выбираемая из 1717-1G->A, 621+1G->T, 3120+1G->A, 1898+1G->A, 711+1G->T, 2622+1G->A, 405+1G->A, 406-lG->A, 4005+1G->A, 1812-1G->A, 1525-1G->A, 712-1G->T, 1248+1G->A, 1341+1G->A, 3121-1G->A, 4374+1G->T, 3850-lG->A, 2789+5G->A, 3849+10kbC->T, 3272-26A->G, 711+5G->A, 3120G->A, 1811+1.6kbA->G, 711+3A->G, 1898+3A->G, 1717-8G->A, 1342-2A->C, 405+3A->C, 1716G/A, 1811+1G->C, 1898+5G->T, 3850-3T->G, IVS14b+5G->A, 1898+1G->T, 4005+2T->C и 621+3A->G, и одна или более мутации человеческого CFTR, выбираемые из ΔF508, R117H и G551D. В одном аспекте настоящее изобретение направлено на способ лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающий введение эффективного количества фармацевтической композиции или таблетки, являющихся предметом изобретения, пациенту, предпочтительно млекопитающему, в котором у пациента есть генетическая мутация CFTR, выбираемая из 1717-1G->A, 1811+1.6kbA->G, 2789+5G->A, 3272-26A->G и 3849+10kbC->T, и одна или более мутации человеческого CFTR, выбираемые из ΔF508, R117H и G551D. В одном аспекте настоящее изобретение направлено на способ лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающий введение эффективного количества фармацевтической композиции или таблетки, являющихся предметом изобретения, пациенту, предпочтительно млекопитающему, в котором у пациента есть генетическая мутация CFTR, выбираемая из 2789+5G->A и 3272-26A->G и одна или более мутации человеческого CFTR, выбираемые из ΔF508, R117H и G551D.
В некоторых вариантах применения изобретения фармацевтически приемлемая композиция или таблетка, являющиеся предметом настоящего изобретения, включающие Соединение 1 Форму I и твердую дисперсию по существу аморфного Соединения 2, применимы для лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациентов с остаточной активностью CFTR в апикальной мембране респираторного и нереспираторного эпителия. Наличие остаточной активности CFTR на поверхности эпителия можно легко детектировать с помощью способов, известных в данной области техники, например, стандартных электрофизиологических, биохимических или гистохимических техник. Такие способы определяют активность CFTR с помощью in vivo или ex vivo электрофизиологических техник, измерения концентраций Cl- в потовой жидкости или в слюне или ex vivo биохимических или гистохимических техник для определения поверхностной плотности клеток. Применяя такие способы, можно легко определить остаточную активность CFTR у пациентов, гетерозиготных или гомозиготных по множеству разных мутаций, включая пациентов, гомозиготных или гетерозиготных по наиболее частой мутации ΔF508, а также по другим мутациям, таким как мутация G551D или мутация R117H. В определенных вариантах применения изобретения фармацевтически приемлемые композиции или таблетки, включающие Соединение 1 Форму I и твердую дисперсию по существу аморфного Соединения 2, применимы для лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациентов с малой остаточной активностью CFTR или без остаточной активности CFTR. В определенных вариантах применения изобретения фармацевтически приемлемые композиции или таблетки, включающие Соединение 1 Форму I и твердую дисперсию по существу аморфного Соединения 2, применимы для лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациентов без остаточной активности CFTR в апикальной мембране респираторного эпителия.
В другом варианте применения изобретения соединения и композиции, являющиеся предметом настоящего изобретения, применимы для лечения или уменьшения тяжести кистозного фиброза у пациентов, у которых остаточная активность CFTR была индуцирована или увеличена с помощью фармакологических способов. В другом варианте применения изобретения соединения и композиции, являющиеся предметом настоящего изобретения, применимы для лечения или уменьшения тяжести кистозного фиброза у пациентов, у которых остаточная активность CFTR была индуцирована или увеличена с помощью генной терапии. Такие способы увеличивают количество CFTR, присутствующего на клеточной поверхности, индуцируя таким образом отсутствующую до этого активность CFTR у пациента или повышая имеющийся уровень остаточной активности CFTR у пациента.
В одном варианте применения изобретения фармацевтические композиции или таблетки, являющиеся предметом настоящего изобретения, включающие Соединение 1 Форму I и твердую дисперсию, включающую по существу аморфное Соединение 2, как описано в данной заявке, применимы для лечения или уменьшения тяжести кистозного фиброза у пациентов с определенным генотипом, с остаточной активностью CFTR, например, с мутациями класса I (отсутствие синтеза), мутацией класса II (неправильное сворачивание), мутациями класса III (нарушение регуляции или воротного механизма канала), мутациями класса IV (изменение проводимости) или мутациями класса V (пониженный уровень синтеза).
В одном варианте применения изобретения фармацевтические композиции или таблетки, являющиеся предметом настоящего изобретения, включающие Соединение 1 Форму I и твердую дисперсию, включающую по существу аморфное Соединение 2, как описано в данной заявке, применимы для лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациентов с определенным клиническим фенотипом, например, от слабо до умеренно выраженного клинического фенотипа, который обычно коррелирует с количеством остаточной активности CFTR в апикальной мембране эпителия. Такие фенотипы включают пациентов с нормальной функцией поджелудочной железы.
В одном варианте применения изобретения фармацевтические композиции или таблетки, являющиеся предметом настоящего изобретения, включающие Соединение 1 Форму I и твердую дисперсию, включающую по существу аморфное Соединение 2, как описано в данной заявке, применимы для лечения, уменьшения тяжести или симптоматического лечения пациентов с нормальной функцией поджелудочной железы., идиопатическим панкреатитом и врожденным билатеральным отсутствием семявыносящего протока или с умеренно выраженной болезнью легких, когда пациент имеет остаточную активность CFTR.
В одном варианте применения изобретения фармацевтические композиции или таблетки, являющиеся предметом настоящего изобретения, включающие Соединение 1 Форму I и твердую дисперсию, включающую по существу аморфное Соединение 2, как описано в данной заявке, применимы для лечения, уменьшения тяжести или симптоматического лечения пациентов с нормальной функцией поджелудочной железы., идиопатическим панкреатитом и врожденным билатеральным отсутствием семявыносящего протока или с умеренно выраженной болезнью легких, когда у пациента есть CFTR дикого типа.
В дополнение к кистозному фиброзу регуляция активности CFTR может быть полезной при других заболеваниях, которые напрямую не вызываются мутациями в CFTR, таких как секреторные болезни и другие болезни, связанные с нарушением сворачивания белка, опосредованные CFTR. Такие заболевания включают, но не ограничиваются хронической обструктивной болезнью легких (ХОБЛ), сухостью глаз, синдромом Шегрена. ХОБЛ характеризуется снижением поступления воздуха, которое прогрессирует и не является полностью обратимым. Снижение поступления воздуха происходит из-за гиперсекреции слизи, энфиземы и бронхиолита. Активаторы мутантного CFTR или CFTR дикого типа представляют собой потенциальное лечение гиперсекреции слизи и нарушенного мукоцилиарного клиренса, обычных при ХОБЛ. В частности, увеличивающаяся секреция анионов через CFTR может способствовать выходу жидкости в жидкость на поверхности дыхательных путей для гидратации слизи и улучшения вязкости перицилиарной жидкости. Это приведет к улучшению мукоцилиарного клиренса и уменьшению симптомов, связанных с ХОБЛ. Сухость глаз характеризуется уменьшением образования слезной жидкости и нарушенными липидными, белковыми и муциновыми профилями слезной пленки. Существует множество причин сухости глаз, некоторые из которых включают возраст, лазерную кератопластику in situ, артрит, лекарственные препараты, химические/термические ожоги, аллергии и заболевания, такие как кистозный фиброз и синдромом Шегрена. Увеличивающаяся секреция анионов через CFTR увеличит транспорт жидкости из корнеальных эндотелиоцитов и секреторных желез, окружающих глаз, для усиления гидратации роговицы. Это поможет уменьшить симптомы, ассоциированные с сухостью глаз. Синдром Шегрена является аутоиммунным заболеванием, при котором иммунная система атакует увлажняющие железы во всем теле, включая глаз, рот, кожу, респираторную ткань, печень, влагалище и кишечник. Симптомы включают сухость глаз, рта и влагалища, а также болезнь легких. Заболевание также ассоциировано с ревматоидным артритом, системной волчанкой, системным склерозом и полимиозитом/дерматомиозитом. Считается, что нарушенный транспорт белков вызывает заболевание, возможности лечения которого ограничены. Усилители или индукторы активности CFTR могут гидратировать разные органы, пораженные заболеванием, и способствовать улучшению ассоциированных симптомов.
В одном варианте применения изобретение относится к способу увеличения или индукции активность анионного канала in vitro или in vivo, включающему контакт канала с любой из фармацевтических композиций с PC-I по PC-XXV. В другом варианте применения изобретения анионный канал является хлоридным каналом или бикарбонатным каналом. В другом варианте применения изобретения анионный канал является хлоридным каналом.
Точное необходимое количество будет варьировать от субъекта к субъекту в зависимости от видовой принадлежности, возраста и общего состояния субъекта, степени тяжести инфекции, конкретного агента, способа его введения и подобного. Соединения, являющиеся предметом изобретения, предпочтительно выпускать в виде дозированной лекарственной формы для облегченного введения и однородности единиц дозирования. Выражение ''дозированная лекарственная форма'' в данной заявке относится к физически дискретной единице агента, подходящего для пациента, который нуждается в лечении. Однако следует иметь в виду, что общее суточное потребление соединений и композиций, являющихся предметом изобретения, будет определять наблюдающий терапевт по результатам медицинской оценки. Специфичная эффективная величина дозы для любого отдельного пациента или организма будет зависеть от множества факторов, включая заболевание, подлежащее лечению, и степень тяжести заболевания; активность конкретного применяемого соединения; конкретной применяемой композиции; возраста, веса тела, общего состояния здоровья, пола и диеты пациента; времени введения, способа введения и скорость выведения конкретного применяемого соединения; продолжительности лечения; лекарственных препаратов, применяемых в комбинации или совместно с конкретным применяемым соединением и подобных факторов, хорошо известных в медицинских областях техники. Термин ''пациент'' в данной заявке обозначает животное, предпочтительно млекопитающее и наиболее предпочтительно человека.
В любом месте настоящей заявки, где название соединения может некорректно описывать структуру соединения, структура заменяет название и имеет преимущественное значение.
ПРИМЕРЫ
XRPD (рентгеновская порошковая дифрактометрия)
Данные рентгеновской дифракции (XRD) Соединения 1 Формы I получали на порошковом дифрактометре Bruker D8 DISCOVER с 2-мерным детектором HI-STAR и плоским графитовым монохроматором. Запаянную Cu трубку с Κα излучением применяли при 40 кВ, 35 мА. Образцы помещали на кремниевые пластины с нулевым фоном при 25°С. Для каждого образца снимали две рамки данных, каждую за 120 секунд при 2 разных углах θ: 8° и 26°. Данные интегрировали посредством программного обеспечения GADDS и проводили наложение с помощью программного обеспечения DIFFRACTplusEVA. Погрешность положений пиков составляет ± 0,2 градуса.
Дифференциальная сканирующая калориметрия (ДСК)
Данные дифференциальной сканирующей калориметрии Соединения 1 Формы I собирали с помощью DSC Q100 V9.6 Build 290 (ТА Instruments, Нью-Касл, штата Делавэр). Температуру калибровали с помощью индия, а теплоемкость калибровали сапфиром. Образцы по 3-6 мг взвешивали в алюминиевых чашах, закрытых крышками с 1 точечным отверстием. Образцы сканировали в диапазоне от 25°С до 350°С при скорости нагревания равной 1,0°С/мин и с продувкой азотом при 50 мл/мин. Данные собирали с помощью программного обеспечения Thermal Advantage Q SeriesTM версии 2.2.0.248 и анализировали с помощью программного обеспечения Universal Analysis версии 4. ID (ТА Instruments, Нью-Касл, штата Делавэр). Показанные числа представляют единичные анализы.
Определение монокристаллической структуры Соединения 1 Формы I
Дифракционные данные получали на дифрактометре Bruker Apex II, снабженном K-альфа источником в запаянной Cu трубке и ПЗС-детектором Apex II. Разрешение и уточнение структуры проводили с помощью программы SHELX (Sheldrick, G.M., Acta Cryst., (2008) А64, 112-122). На основании статистики систематического отсутствия сигнала и интенсивности сигнала разрешали и уточняли структуру в P21/n группе симметрии.
Vitride® (бис(2-метоксиэтокси)алюмогидрид натрия [или NaAlH2 (ОСН2СН2ОСН3)2], 65% по массе раствора в толуоле) покупали в Aldrich Chemicals.
2,2-дифтор-1,3-бензодиоксол-5-карбоновую кислоту покупали в Saltigo (дочернее предприятие Lanxess Corporation).

Коммерчески доступную 2,2-дифтор-1,3-бензодиоксол-5-карбоновуюя кислоту (1,0 экв.) суспендировали в толуоле (10 об.). Vitride® (2 экв.) добавляли через капельную воронку со скоростью, поддерживающей температуру на уровне 15-25°С. В конце добавления температуру повышали до 40°С в течение 2 часов (ч), затем аккуратно добавляли 10% (вес/вес) водный (водн.) NaOH (4,0 экв.) через капельную воронку, поддерживая температуру на уровне 40-50°С. После перемешивания в течение дополнительных 30 минут (мин) давали слоям разделиться при 40°С. Органическую фазу охлаждали до 20°С, затем промывали водой (2×1,5 об.), высушивали (Na2SO4), фильтровали и концентрировали до получения неочищенного (2,2-дифтор-1,3-бензодиоксол-5-ил)-метанола, который применяли напрямую на следующей стадии.

(2,2-дифтор-1,3-бензодиоксол-5-ил)-метанол (1/0 экв.) растворяли в МТВЕ (5 об.). Добавляли каталитическое количество 4-(N,N-диметил)аминопиридина (ДМАП) (1 молярный %) и добавляли SOCl2 (1,2 экв.) через капельную воронку. SOCl2 добавляли со скоростью, поддерживающей температуру в реакторе на уровне 15-25°С. Температуру повышали до 30°С в течение 1 ч, а затем остужали до 20°С. Воду (4 об.) добавляли через капельную воронку, поддерживая температуру ниже 30°С. После перемешивания в течение дополнительных 30 мин давали слоям разделиться. Органический слой перемешивали и добавляли 10% (вес/объем) водн. NaOH (4,4 об.). После перемешивания в течение от 15 до 20 мин, давали слоям разделиться. Затем органическую фазу высушивали (Na2SO4), фильтровали и концентрировали до получения неочищенного 5-хлорметил-2,2-дифтор-1,3-бензодиоксола, который применяли напрямую на следующей стадии.
Раствор 5-хлорметил-2,2-дифтор-1,3-бензодиоксола (1 экв.) в ДМСО (1,25 об.) добавляли к суспензии NaCN (1,4 экв.) в ДМСО (3 об.), поддерживая температуру между 30-40°С. Затем перемешивали смесь в течение 1 ч, а затем добавляли воду (6 об.), а потом метил-трет-бутиловый эфир (МТБЭ) (4 об.). После перемешивания в течение 30 мин. слои разделялись. Водный слой экстрагировали МТБЭ (1,8 об.). Объединенные органические слои промывали водой (1,8 об.), высушивали (Na2SO4), фильтровали и концентрировали до получения неочищенного (2,2-дифтор-1,3-бензодиоксол-5-ил)-ацетонитрила (95%), который применяли напрямую на следующей стадии.

Реактор продували азотом и заполняли 900 мл толуола. Растворитель дегазировали распылением азота в течение не менее 16 ч. Затем заполняли реактор Na3PO4 (155,7 г, 949,5 ммоль), а затем - бис (дибензилиденацетон) палладием(0) (7,28 г, 12,66 ммоль). 10% вес/вес раствором трет-бутилфосфина в гексане (51,23 г, 25,32 ммоль) заполняли в течение 10 мин при 23°С из капельной воронки, продутой азотом. Затем смесь перемешивали в течение 50 мин, после чего добавляли 5-бром-2,2-дифтор-1,3-бензодиоксол (75 г, 316,5 ммоль) в течение 1 мин. После перемешивания в течение дополнительных 50 мин в смесь добавляли этилцианоацетат (71,6 г, 633,0 ммоль) в течение 5 мин, а затем воду (4,5 мл) одной порцией. Смесь нагревали до 70°С в течение 40 мин и анализировали с помощью ВЭЖХ каждые 1-2 ч на предмет процента превращения реагирующего вещества в продукт. После полного превращения (обычно 100% превращение после 5-8 ч), смесь охлаждали до 20-25°С и фильтровали через целитную подложку. Целитную подложку споласкивали толуолом (2×450 мл) объединенные органические вещества концентрировали до 300 мл в вакууме при 60-65°С. В концентрат добавляли 225 мл ДМСО и концентрировали в вакууме при 70-80°С до прекращения активной ректификации растворителя. Раствор охлаждали до 20-25°С и разбавляли до 900 мл с помощью ДМСО в рамках подготовки к стадии 2. 1Н ЯМР (500 МГц, CDCl3) δ 7,16-7,10 (м, 2Н), 7,03 (д, J=8,2 Гц, 1Н), 4,63 (с, 1Н), 4,19 (м, 2Н), 1,23 (т, J=7,1 Гц, 3Н).
В раствор (2,2-дифтор-1,3-бензодиоксол-5-ил)-1-этилацетат-ацетонитрила в ДМСО, описанный выше добавляли 3н HCl (617,3 мл., 1,85 моль) в течение 20 мин, поддерживая внутреннюю температуру <40°С. Затем смесь нагревали до 75°С в течение 1 ч и анализировали с помощью ВЭЖХ каждые 1-2 ч для определения % превращения. При превращении >99% (обычно после 5-6 ч) реакцию охлаждали до 20-25°С и экстрагировали МТБЭ (2×525 мл) в течение времени, достаточного для завершения разделения фаз при экстракции. Объединенные органические экстракты промывали 5% NaCl (2×375 мл). Затем раствор переносили в прибор, подходящий для вакуумной перегонки при 1,5-2,5 торр, снабженный охлаждаемой колбой-приемником. Раствор концентрировали в вакууме при <60°С для удаления растворителей. (2,2-дифтор-1,3-бензодиоксол-5-ил)-ацетонитрил затем очищали от образующегося масла при 125-130°С (температура печи) и 1,5-2,0 торр. (2,2-дифтор-1,3-бензодиоксол-5-ил)-ацетонитрил выделяли в виде чистого масла с 66% выходом из 5-бром-2,2-дифтор-1,3-бензодиоксола (2 стадии) и с ВЭЖХ чистотой равной 91,5% AUC (соответствует 95% вес/вес). 1Н ЯМР (500 МГц, ДМСО) δ 7,44 (шир.с, 7,43 (д, J=8,4 Гц, 1Н), 7,22 (дд, J=8,2, 1,8 Гц, 1Н), 4,07 (с, 2Н).

Смесь из (2,2-дифтор-1,3-бензодиоксол-5-ил)-ацетонитрила (1,0 экв.)л 50% по массе водного KOH (5,0 экв.) 1-бром-2-хлорэтана (1,5 экв.) и Oct4NBr (0,02 экв.) нагревали при 70°С в течение 1 ч. Реакционную смесь охлаждали, затем обрабатывали МТБЭ и водой. Органическую фазу промывали водой и солевым раствором. Растворитель удаляли для получения (2,2-дифтор-1,3-бензодиоксол-5-ил)-циклопропанкарбонитрила.
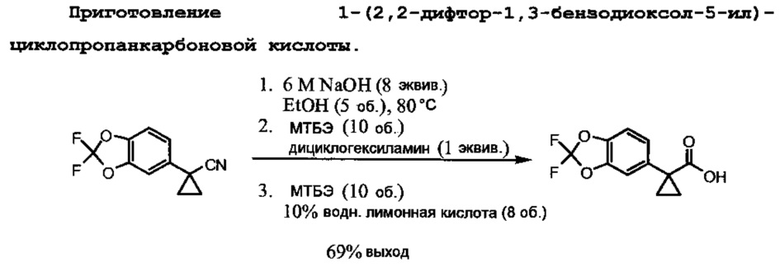
(2,2-дифтор-1,3-бензодиоксол-5-ил)-циклопропанкарбонитрил гидролизовали с помощью 6М NaOH (8 эквив.) в этаноле (5 об.) при 80°С в течение ночи. Смесь охлаждали до комнатной температуры и испаряли этанол в вакууме. К остатку добавляли воду и МТБЭ, 1М HCl и разделяли слои. Слой МТБЭ затем обрабатывали дициклогексиламином (ДЦГА) (0,97 эквив.). Суспензию охлаждали до 0°С, фильтровали и промывали гептаном для получения соответствующей соли ДЦГА. Соль погружали в МТБЭ и 10% лимонную кислоту и перемешивали до растворения твердых частиц. Слои разделяли и слой МТБЭ промывали водой и солевым раствором. Замещение растворителя гептаном и последующее фильтрование приводили к образованию 1-(2,2-диффтор-1,3-бензодиоксол-5-ил)-циклопропанкарбоновой кислоты после высушивания в вакуумном сушильном шкафу при 50°С в течение ночи.
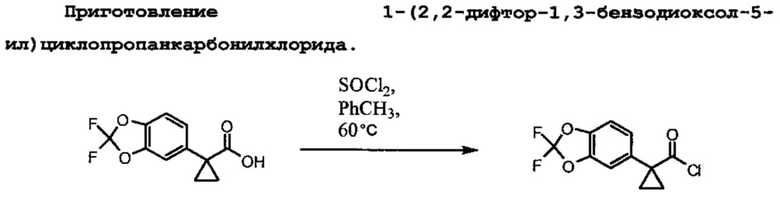
1-(2,2-диффтор-1,3-бензодиоксол-5-ил)-циклопропанкарбоновую кислоту (1,2 экв.) суспендировали в толуоле (2,5 об.), и смесь нагревали до 60°С. SOCl2 (1,4 экв.) добавляли через капельную воронку. Толуол и SOCl2 удаляли из реакционной смеси перегонкой через 30 минут. Дополнительно добавляли толуол (2,5 об.) и образующуюся в результате смесь снова перегоняли, получая продукт хлорангидрид в виде масла, который применяли без дальнейшей очистки.
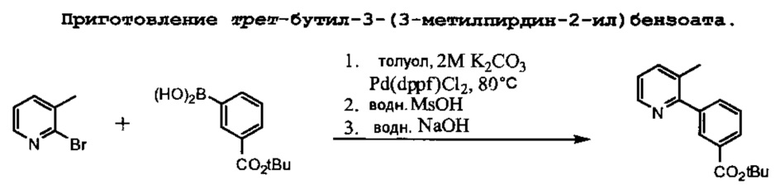
2-бром-3-метилпиридин (1,0 экв.) растворяли в толуоле (12 об.). Добавляли K2CO3 (4,8 экв.), затем воду (3,5 об.). Образованную в результате смесь нагревали до 65°С в потоке N2 в течение 1 часа. Затем добавляли 3-(t-бутоксикарбонил)фенилбороновую кислоту (1,05 экв.) и Pd(dppf)Cl2⋅CH2Cl2 (0,015 экв.) и смесь нагревали до 80°С. Через 2 часа нагрев выключали, добавляли воду (3,5 об.) и давали слоям разделиться. Затем органическую фазу промывали водой (3,5 об.) и экстрагировали 10% водной метансульфоновой кислотой (2 экв. MsOH, 7,7 об.). Водную фазу защелачивали 50% водным NaOH (2 экв.) и экстрагировали EtOAc (8 об.). Органический слой концентрировали для получения неочищенного трет-бутил-3-(3-метилпиридин-2-ил)бензоата (82%), который напрямую применяли на следующей стадии.
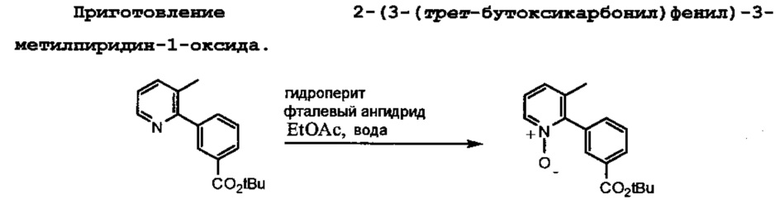
Трет-бутил-3-(3-метилпиридин-2-ил)бензоат (1,0 экв.) растворяли в EtOAc (6 об.). Добавляли воду (0,3 об.), в затем гидроперит (3 экв.). Затем к смеси порционно добавляли фталевый ангидрид (3 экв.) в твердом виде со скоростью, поддерживающей температуру в реакторе ниже 45°С. После завершения добавления фталевого ангидрида смесь нагревали до 45°С. После перемешивания в течение дополнительных 4 часов нагрев выключали. 10% вес/вес водный Na2SO3 (1,5 экв.) добавляли через капельную воронку. После завершения добавления Na2SO3 смесь перемешивали в течение дополнительных 30 мин, и слои разделялись. Органический слой перемешивали и добавляли 10% вес/вес водн. Na2CO3 (2 экв.). После перемешивания в течение 30 минут давали слоям разделиться. Органическую фазу промывали 13% вес/об. водн. NaCl. Затем органическую фазу фильтровали и концентрировали для получения неочищенного 2-(3-(трет-бутоксикарбонил)фенил)-3-метилпиридин-1-оксида (95%), который напрямую применяли на следующей стадии.
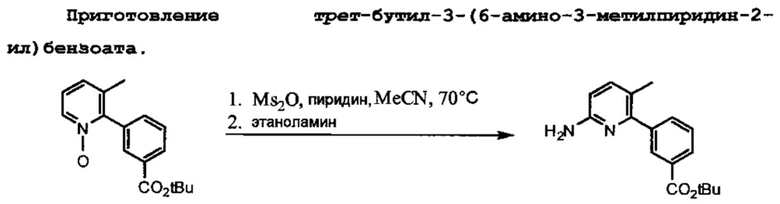
Раствор 2-(3-(трет-бутоксикарбонил)фенил)-3-метилпиридин-1-оксида (1 экв.) и пиридина (4 экв.) в ацетонитриле (8 об.) нагревали до 70°С. Добавляли раствор метансульфонового ангидрида (1,5 экв.) в MeCN (2 об.) в течение 50 мин через капельную воронку, поддерживая температуру ниже 75°С. Смесь перемешивали в течение дополнительных 0,5 часов после завершения добавления. Затем смесь оставляли остывать до комнатной температуры. Добавляли этаноламин (10 экв.) через капельную воронку. После перемешивания в течение 2 часов, добавляли воду (6 об.) и охлаждали смесь до 10°С. После перемешивания в течение 3 часов, твердую часть собирали фильтрацией и промывали водой (3 об.), 2:1 ацетонитрил/вода (3 об.), и ацетонитрилом (2×1,5 об.). Твердую часть высушивали до постоянного веса (<1% отличие) в вакуумном сушильном шкафу при 50°С при слабом пропускании N2 для получения трет-бутил-3-(6-амино-3-метилпиридин-2-ил)бензоата в виде красно-желтых твердых частиц (53% выход).
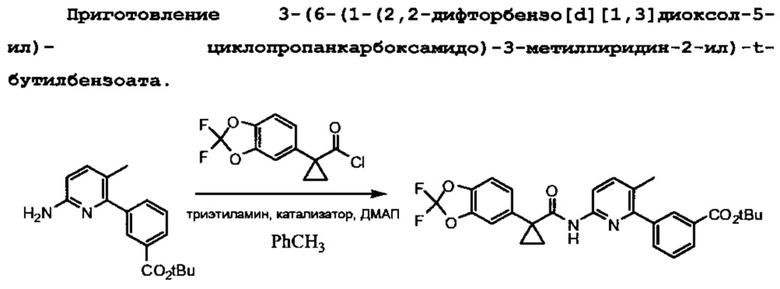
Неочищенный хлорангидрид, описанный выше, растворяли в толуоле (2,5 об. на основе хлорангидрида) и добавляли через капельную воронку к смеси трет-бутил-3-(6-амино-3-метилпиридин-2-ил)бензоата (1 экв.), ДМАП, (0,02 экв.) и триэтиламина (3,0 экв.) в толуоле (4 об. на основе трет-бутил-3-(6-амино-3-метилпиридин-2-ил)бензоата). Через 2 часа к реакционной смеси добавляли воду (4 об. на основе трет-бутил-3-(6-амино-3-метилпиридин-2-ил)бензоата). После перемешивания в течение 30 минут слои разделялись. Органическую фазу затем фильтровали и концентрировали для получения 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамидо)-3-метилпиридин-2-ил)-t-бутилбензоата (количественный выход неочищенного продукта) в виде вязкого масла. Добавляли ацетонитрил (3 об. на основе неочищенного продукта) и перегоняли до кристаллизации. Добавляли воду (2 об. на основе неочищенного продукта) и перемешивали смесь в течение 2 ч. Твердую часть собирали фильтрацией и промывали 1:1 (по объему) ацетонитрилом/водой (2×1 объема на основе неочищенного продукта) и частично высушивали на фильтре в вакууме. Твердую часть высушивали до постоянного веса (<1% отличие) в вакуумном сушильном шкафу при 60°С при слабом пропускании N2 для получения 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамидо)-3-метилпиридин-2-ил)-t-бутилбензоата в виде коричневых твердых частиц.
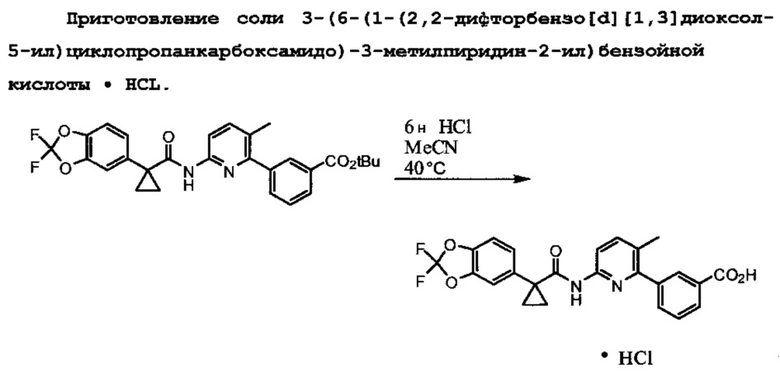
К суспензии 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамидо)-3-метилпиридин-2-ил)-t-бутилбензоата (1,0 экв.) в MeCN (3,0 об.) добавляли воду (0,83 об.), а затем концентрированную водную HCl (0,83 об.). Смесь нагревали до 45±5°С. После перемешивания в течение от 24 до 48 ч реакция завершалась и смесь оставляли остывать до комнатной температуры. Добавляли воду (1,33 об.) и перемешивали смесь. Твердую часть собирали фильтрацией и промывали водой (2×0,3 об.) и частично высушивали на фильтре в вакууме. Твердую часть высушивали до постоянного веса (<1% отличие) в вакуумном сушильном шкафу при 60°С при слабом пропускании N2 для получения соли 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил) циклопропанкарбоксамидо)-3-метилпиридин-2-ил)бензойной кислоты ⋅ HCl в виде беловатых твердых частиц.
1Н ЯМР спектр Соединения 1, показанный на фиг. 8 и фиг. 9, изображает 1Н ЯМР спектр Соединения 1 в виде соли HCl.
Таблица 2 ниже описывает данные 1Н ЯМР Соединения I.
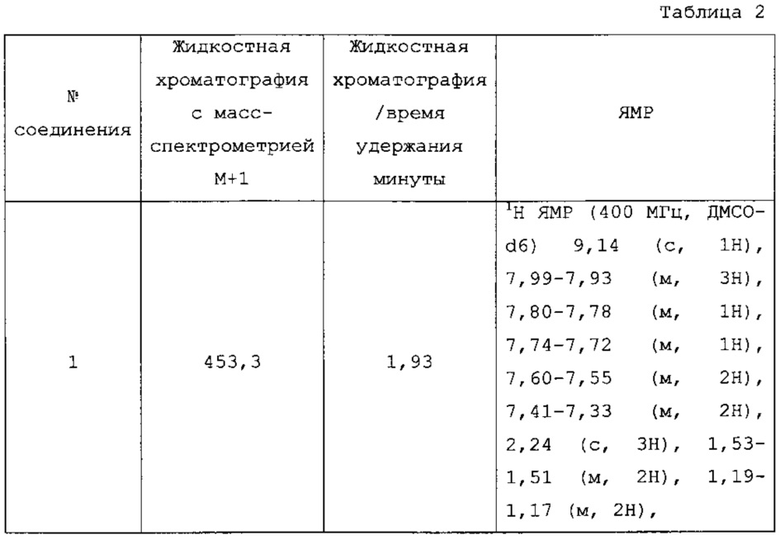
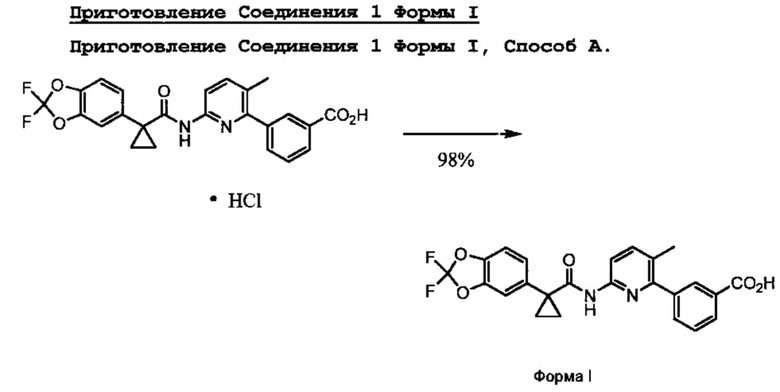
Суспензию 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамидо)-3-метилпиридин-2-ил)бензойной кислоты ⋅ HCl (1 экв.) в воде (10 об.) перемешивали при комнатной температуре. После перемешивания в течение 24 часов отбирали образец. Образец фильтровали и твердую часть промывали водой (2 раза). Твердый образец анализировали с помощью дифференциальной сканирующей калориметрии. Когда анализ с помощью дифференциальной сканирующей калориметрии показывал полное превращение в Форму I, твердую часть собирали фильтрацией и промывали водой (2×1,0 об.) и частично высушивали на фильтре в вакууме. Затем твердую часть высушивали до постоянного веса (<1% отличие) в вакуумном сушильном шкафу при 60°С при слабом пропускании N2 для получения Соединения 1 Формы I в виде беловатых твердых частиц (98% выход). 1Н ЯМР (400 МГц, ДМСО-d6) 9,14 (с, 1Н), 7,99-7,93 (м, 3Н), 7,80-7,78 (м, 1Н), 7,74-7,72 (м, 1Н), 7,60-7,55 (м, 2Н), 7,41-7,33 (м, 2Н), 2,24 (с, 3Н), 1,53-1,51 (м, 2Н), 1,19-1,17 (м, 2Н).
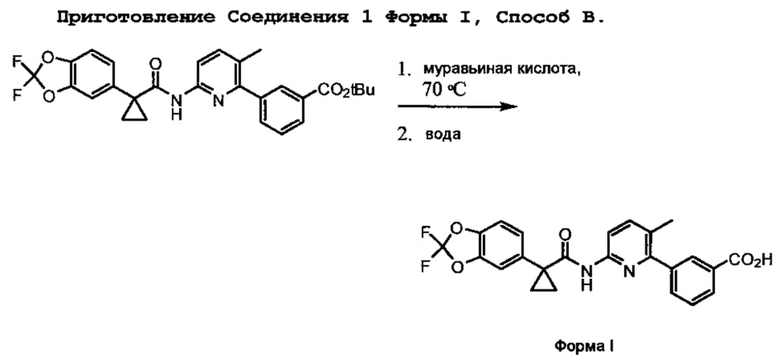
Раствор 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамидо)-3-метилпиридин-2-ил)-t-бутилбензоата (1,0 экв.) в муравьиной кислоте (3,0 об.) нагревали при помешивании до 70±10°С в течение 8 ч. Реакцию считали завершенной, когда оставалось не более 1,0% AUC при определении хроматографическими способами 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамидо)-3-метилпиридин-2-ил)-t-бутилбензоата. Смесь оставляли остывать до комнатной температуры. Раствор добавляли в воду (6 об.), нагревали при 50°С и смесь перемешивали. Затем смесь нагревали до 70±10°С, пока уровень 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамидо)-3-метилпиридин-2-ил)-t-бутилбензоата не превышал 0,8% (AUC). Твердую часть собирали фильтрацией и промывали водой (2×3 об.) и частично высушивали на фильтре в вакууме. Твердую часть высушивали до постоянного веса (<1% отличие) в вакуумном сушильном шкафу при 60°С при слабом пропускании N2 для получения Соединения 1 Формы I в виде беловатых твердых частиц.
След Соединения 1 Формы I, полученный с помощью дифференциальной сканирующей калориметрии, показан на фиг. 10. Плавление Соединения 1 Формы I происходит приблизительно при 204°С.
Дифракционную рентгенограмму рассчитывали по монокристаллической структуре Соединения 1 Формы I и показывали на фиг. 1. В Таблице 3 перечислены рассчитанные пики для Фигуры 1.
Действительная дифракционная порошковая рентгенограмма Соединения 1 Формы I показана на Фигуре 2. В Таблице 4 перечислены действительные пики для Фигуры 2.
Бесцветные кристаллы Соединения 1 Формы I получали охлаждением концентрированного раствора 1-бутанола с 75°С до 10°С со скоростью равной 0,2°С/мин. Отбирали кристалл с тремя измерениями равными 0,50×0,08×0,03 мм, очищали минеральным маслом, приклеивали на MicroMount и центрировали в системе Bruker APEX II. Получали три партии из 40 рамок, разделенных обратным пространством, для создания матрицы ориентации и исходных параметров ячеек. Конечные параметры ячеек получали и уточняли на основании полного набора данных.
Набор дифракционных данных обратного пространства получали с разрешением 0,82 Å с помощью шагов 0,5° с помощью 30 с экспозиции для каждой рамки. Данные собирали при 100 (2) K. Интеграцию интенсивностей и уточнение параметров ячеек проводили в помощью программного обеспечения APEXII. Наблюдения за кристаллом после сбора данных, не выявило признаков распада.
Конформационная картина Соединения 1 Формы I на основе монокристаллического рентгеновского анализа показана на Фигуре 11. Соединение 1 Формы I является моноклинным, Р21/n, со следующими постоянными решетки: а=4,9626(7) Å, b=12,299(2) Å, с=33,075(4) Å, β=93,938(9)°, V=2014,0 Å3, Z=4. Плотность Соединения 1 Формы I, рассчитанная на основании структурных данных, составляет 1,492 г/см3 при 100 K.
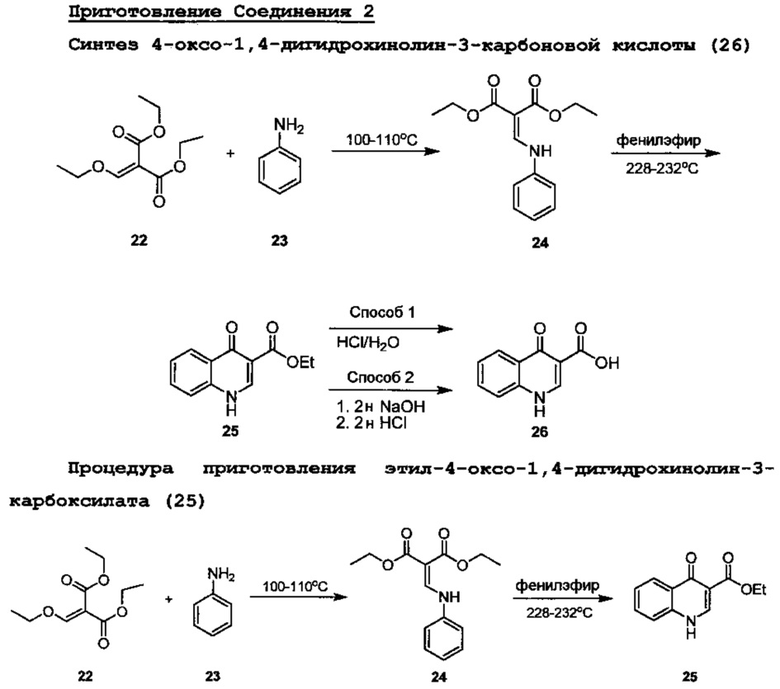
Соединение 23 (4,77 г, 47,7 ммоль) добавляли по каплям к соединению 22 (10 г, 46,3 ммоль) при подповерхностном потоке N2 для удаления этанола при температуре ниже 30°С в течение 0,5 часов. Затем раствор нагревали до 100-110°С и перемешивали в течение 2,5 часов. После охлаждения смеси до температуры ниже 60°С добавляли дифенилэфир. Образующийся в результате раствор по каплям добавляли к дифениловому эфиру, нагретому до 228-232°С в течение 1,5 часов при подповерхностном потоке N2 для удаления этанола. Смесь перемешивали при 228-232°С в течение еще 2 часов, охлаждали до температуры ниже 100°С и затем добавляли гептан для осаждения продукта. Образующуюся в результате суспензию перемешивали при 30°С в течение 0,5 часов. Затем твердые частицы фильтровали и уплотненный осадок на фильтре промывали гептаном и высушивали in vacuo для получения соединения 25 в виде коричневых твердых частиц. 1Н ЯМР (ДМСО-d6; 400 МГц) δ 12,25 (с), δ 8,49 (д), δ 8,10 (м), δ 7,64 (м), δ 7,55 (м), δ 7,34 (м), δ 4,16 (кв), δ 1,23 (т).
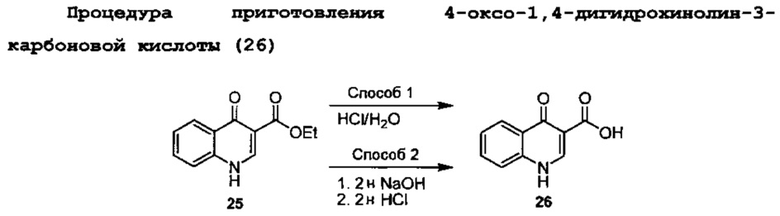
Соединение 25 (1,0 экв.) суспендировали в растворе HCl (10,0 экв.) и H2O (11,6 об.). Суспензию нагревали до 85-90°С, хотя альтернативные температуры также подходят для данной стадии гидролиза. Например, в качестве альтернативы гидролиз можно проводить при температуре приблизительно от 75 приблизительно до 100°С. В некоторых случаях гидролиз проводят при температуре приблизительно от 80 приблизительно до 95°С. В других случаях стадию гидролиза проводят при температуре приблизительно от 82 приблизительно до 93°С (например, приблизительно от 82,5 приблизительно до 92,5°С или приблизительно от 86 приблизительно до 89°С). После перемешивания при 85-90°С в течение приблизительно 6,5 часов, отбирали пробу для определения завершенности реакции. Перемешивание можно проводить при любой температуре, подходящей для гидролиза. Затем раствор охлаждали до 20-25°С и фильтровали. Реактор/уплотненный осадок на фильтре споласкивали H2O (2 об. × 2). Затем уплотненный осадок на фильтре промывали 2 об. H2O до достижения уровня рН≥3.0. Затем уплотненный осадок на фильтре высушивали в вакууме при 60°С для получения соединения 26.
Способ 2
Соединение 25 (11,3 г, 52 ммоль) добавляли к смеси 10% NaOH (водн.) (10 мл) и этанола (100 мл). Раствор нагревали для дефлегмации в течение 16 часов, охлаждали до 20-25°С, а затем подводили рН до 2-3 с помощью 8% HCl. Затем смесь перемешивали в течение 0,5 часов и фильтровали. Уплотненный осадок на фильтре промывали водой (50 мл), а затем высушивали in vacuo для получения соединения 26 в виде коричневых твердых частиц. 1Н ЯМР (ДМСО-d6; 400 МГц) δ 15,33 (с), δ 13,39 (с), δ 8,87 (с), δ 8,26 (м), δ 7,87 (м), δ 7,80 (м), δ 7,56 (м).
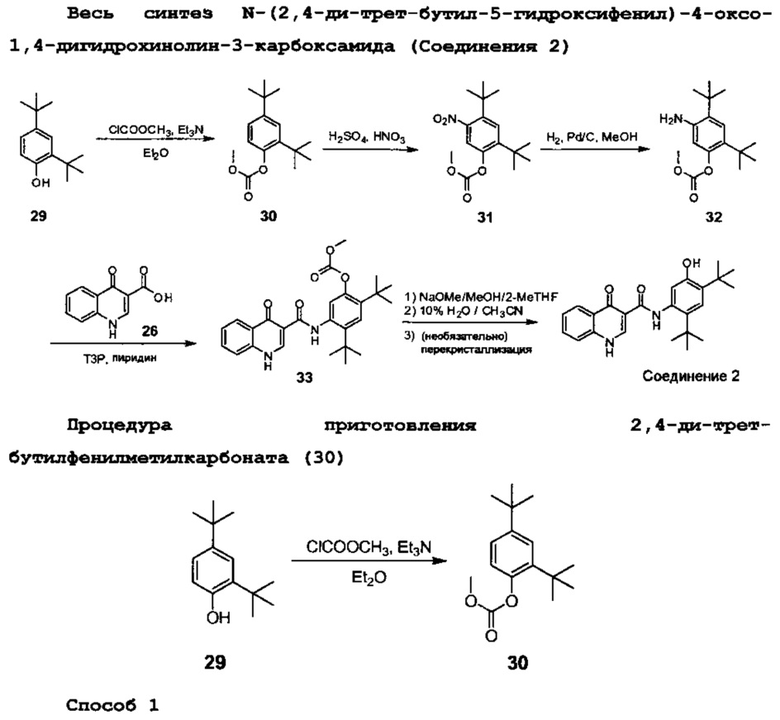
К раствору 2,4-ди-трет-бутилфенола, 29, (10 г, 48,5 ммоль) в диэтиловом эфире (100 мл) и триэтиламине (10,1 мл, 72,8 ммоль) по каплям добавляли метилхлорформиат (7,46 мл, 97 ммоль) при 0°С. Затем смесь оставляли нагреваться до комнатной температуры и перемешивали в течение дополнительных 2 часов. Затем добавляли дополнительные 5 мл триатиламина и 3,7 мл метилхлорформиата и перемешивали реакционную смесь в течение ночи. Затем реакционную смесь фильтровали, фильтрат охлаждали до 0°С и затем добавляли дополнительные 5 мл триэтиламина и 3,7 мл метилхлорформиата и смесь оставляли нагреваться до комнатной температуры, а затем перемешивали в течение дополнительного 1 часа. На этой стадии реакция была почти полностью завершена, и ее останавливали фильтрованием, затем промывкой водой (2х), а затем - солевым раствором. Затем раствор концентрировали для получения желтого масла и очищали с помощью колоночной хроматографии для получения соединения 30. 1Н ЯМР (400 МГц, ДМСО-d6) δ 7,35 (д, J=2,4 Гц, 1Н), 7,29 (дд, J=8,4, 2,4 Гц, 1Н), 7,06 (д, J=8,4 Гц, 1Н), 3,85 (с, 3Н), 1,30 (с, 9Н), 1,29 (с, 9Н).
Способ 2
В реакционный сосуд, заполненный 4-диметиламинопиридином (ДМАП, 3,16 г, 25,7 ммоль) и 2,4-дитрет-бутилфенолом (соединение 29, 103,5 г, 501,6 ммоль), добавляли хлорид метилена (415 г, 313 мл) и перемешивали раствор до полного растворения твердых частиц. Затем добавляли триэтиламин (76 г, 751 ммоль) и раствор охлаждали до 0-5°С. Затем по каплям добавляли метилхлорформиат (52 г, 550,3 ммоль) в течение 2,5-4 часов, поддерживая температуру раствора в интервале 0-5°С. Затем реакционную смесь медленно нагревали до 23-28°С и перемешивали в течение 20 часов. Затем реакционную смесь охлаждали до 10-15°С и добавляли 150 мл воды. Смесь перемешивали при 15-20°С в течение 35-45 минут и затем отделяли водный слой и экстрагировали 150 мл хлорида метилена. Органические слои объединяли и нейтрализовали 2,5% HCl (водн.) при температуре 5-20°С для получения конечного рН 5-6. Затем органический слой промывали водой и концентрировали in vacuo при температуре ниже 20°С до 150 мл для получения соединения 30 в хлориде метилена.
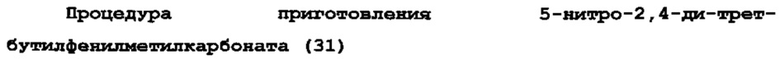
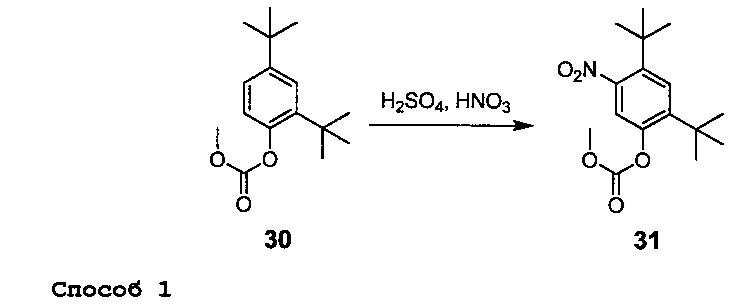
К перемешанному раствору соединения 30 (6,11 г, 25,6 ммоль) по каплям добавляли 6 мл смеси 1:1 серной кислоты и азотной кислоты при 0°С. Затем смесь оставляли нагреваться до комнатной температуры и перемешивали в течение дополнительного 1 часа. Продукт очищали с помощью жидкостной хроматографии (ISCO, 120 г, 0-7% EtOAc/гексаны, 38 мин) с образованием приблизительно 8:1-10:1 смеси региоизомеров соединения 31 в виде белых твердых частиц. 1Н ЯМР (400 МГц, ДМСО-d6) δ 7,63 (с, 1Н), 7,56 (с, 1Н), 3,87 (с, 3Н), 1,36 (с, 9Н), 1,32 (с, 9Н), ВЭЖХ время удержания 3,92 мин 10-99% CH3CN, 5 мин пробег; ESI-MS 310 m/z (МН)+.
Способ 2
К соединению 30 (100 г, 378 ммоль) добавляли дихлорметан (540 г, 408 мл). Смесь перемешивали до полного растворения твердых частиц, затем охлаждали до -5-0°С. Затем по каплям добавляли концентрированную серную кислоту (163 г), поддерживая исходную температуру реакционной смеси, и перемешивали смесь в течение 4,5 часов. Затем по каплям добавляли азотную кислоту (62 г) в течение 2-4 часов, поддерживая исходную температуру реакционной смеси, и затем перемешивали смесь при этой температуре в течение дополнительных 4,5 часов. Затем реакционную смесь медленно добавляли в холодную воду, поддерживая температуру ниже 5°С. охлажденную реакционную смесь затем нагревали до 25°С и удаляли водный слой и экстрагировали хлоридом метилена. Объединенные органические слои промывали водой, высушивали с помощью Na2SO4 и концентрировали до 124-155 мл. Добавляли гексан (48 г) и образующуюся в результате смесь вновь концентрировали до 124-155 мл. Впоследствии к смеси еще добавляли гексан (160 г). Затем смесь перемешивали при 23-27°С в течение 15,5 часов, а затем фильтровали. К уплотненному осадку на фильтре добавляли гексан (115 г), образующуюся в результате смесь нагревали до дефлегмации и перемешивали в течение 2-2,5 часов. Затем смесь охлаждали до 3-7°С, перемешивали в течение дополнительных 1-1,5 часов и фильтровали для получения соединения 31 в виде бледно-желтых твердых частиц.

Подходящий реактор для гидрогенизации наполняли 2,4-ди-трет-бутил-5-нитрофенилметилкарбонатом (1,00 экв.), а затем 5% Pd/C (2,50% по массе сухого вещества, Johnson-Matthey Туре 37). МеОН (15,0 об.) заполняли реактор и закрывали систему. Систему продували N2 (г) и нагнетали давление до 2,0 бар Н2 (г). Реакцию проводили при температуре реакции 25°С+/-5°С. После завершения реакционную смесь фильтровали, реактор/уплотненный осадок на фильтре промывали МеОН (4,00 об.). Образующийся в результате фильтрат перегоняли в вакууме при температуре не выше 50°С до 8,00 об. Добавляли воду (2,00 об.) при t 45°С+/-5°С. Образующуюся в результате суспензию охлаждали до 0°С+/-5°С. Суспензию держали при 0°С+/-5°С в течение не менее чем 1 часа, а затем фильтровали. Уплотненный осадок на фильтре промывали однократно с помощью 0°С+/-5°С МеОН/H2O (8:2) (2,00 об.). Уплотненный осадок на фильтре высушивали в вакууме (-0,90 бар и -0,86 бар) при 35°С-40°С для получения соединения 32. 1Н ЯМР (400 МГц, ДМСО-d6) δ 7,05 (с, 1Н), 6,39 (с, 1Н), 4,80 (с, 2Н), 3,82 (с, 3Н), 1,33 (с, 9Н), 1,23 (с, 9Н).
Сразу после окончания реакции образующуюся в результате смесь разводили с помощью от 5 до 10 объемов МеОН (например, приблизительно от 6 приблизительно до 9 объемов МеОН, приблизительно от 7 приблизительно до 8,5 объемов МеОН, приблизительно от 7,5 приблизительно до 8 объемов МеОН или приблизительно 7,7 объемов МеОН), нагревали до температуры около 35±5°С, фильтровали, промывали и высушивали, как описано выше.
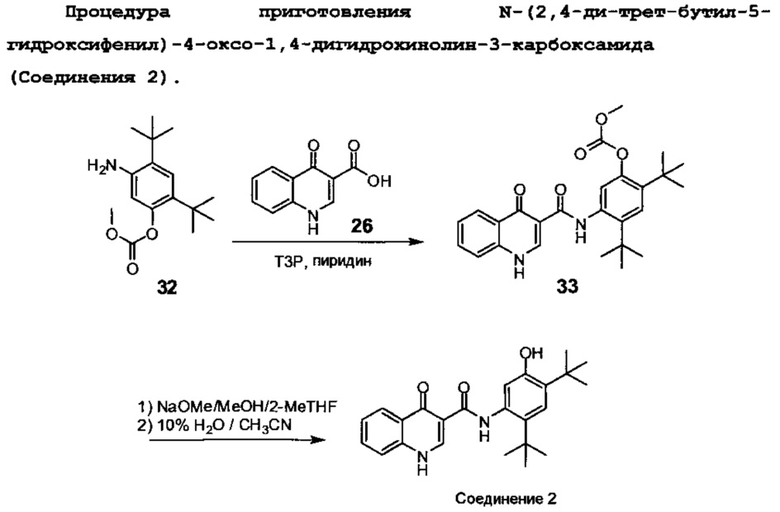
Реактор заполняли 4-оксо-1,4-дигидрохинолин-3-карбоновой кислотой, 26, (1,0 экв.) и 5-амино-2,4-ди-трет-бутилфенидметилкарбонатом, 32, (1,1 экв.). Добавляли 2-MeTHF (4,0 об., относительно кислоты), а затем - 50% раствор Т3Р® в 2-MeTHF (1,7 экв.). Заполненный Т3Р сосуд промывали 2-MeTHF (0,6 об.). Затем добавляли пиридин (2,0 экв.) и нагревали образующуюся в результате суспензию до 47,5+/-5,0°С и держали при этой температуре в течение 8 часов. Отбирали образец и проверяли степень завершенности реакции с помощью ВЭЖХ. После окончания реакции образующуюся в результате смесь охлаждали до 25,0°С+/-2,5°С. Для разведения смеси добавляли 2-MeTHF (12,5 об.). Реакционную смесь дважды промывали водой (10,0 об.). 2-MeTHF добавляли, чтобы довести общий объем реакции до 40,0 об. (~16,5 загруженного об.). К этому раствору добавляли NaOMe/MeOH (1,7 экв.) для проведения метанолиза. Реакцию перемешивали в течение не менее чем 1,0 часа и проверяли степень завершенности реакции с помощью ВЭЖХ. После завершения реакции реакцию останавливали 1 N HCl (10,0 об.) и промывали 0,1н HCl (10,0 об.). Органический раствор осветляли фильтрованием для удаления любых твердых частиц и помещали во второй реактор. Отфильтрованный раствор концентрировали при температуре не выше 35°С (температура в рубашке) и не ниже 8,0°С (температура внутренней реакции) под сниженным давлением до 20 об. CH3CN добавляли до 40 об. И концентрировали раствор при температуре не выше 35°С (температура в рубашке) и не ниже 8,0°С (температура внутренней реакции) до 20 об. Цикл добавления CH3CN и концентрации повторяли еще 2 раза, т.е. всего 3 добавления CH3CN и 4 концентрации до 20 об. После конечной концентрации до 20 об. добавляли 16,0 об. CH3CN, а затем 4,0 об. Н2О для получения конечной концентрации, составляющей 40 об. 10% Н2О/CH3CN относительно исходного количества кислоты. Эту суспензию нагревали до 78,0°С+/-5,0°С (дефлегмация). Затем перемешивали суспензию в течение не менее 5 часов. Суспензию охлаждали до 0,0°С+/-5°С в течение 5 часов и фильтровали. Уплотненный осадок на фильтре промывали 0,0°С+/-5,0°С CH3CN (5 об.) 4 раза. Образующееся в результате твердое вещество (Соединение 2) высушивали в вакуумном сушильном шкафу при 50,0°С+/-5,0°С. 1Н ЯМР (400 МГц, ДМСО-d6) δ 12,8 (с, 1Н), 11,8 (с, 1Н), 9,2 (с, 1Н), 8,9 (с, 1Н), 8,3 (с, 1Н), 7,2 (с, 1Н), 7,9 (т, 1Н), 7,8 (д, 1Н), 7,5 (т, 1Н), 7,1 (с, 1Н), 1,4 (с, 9Н), 1,4 (с, 9Н).

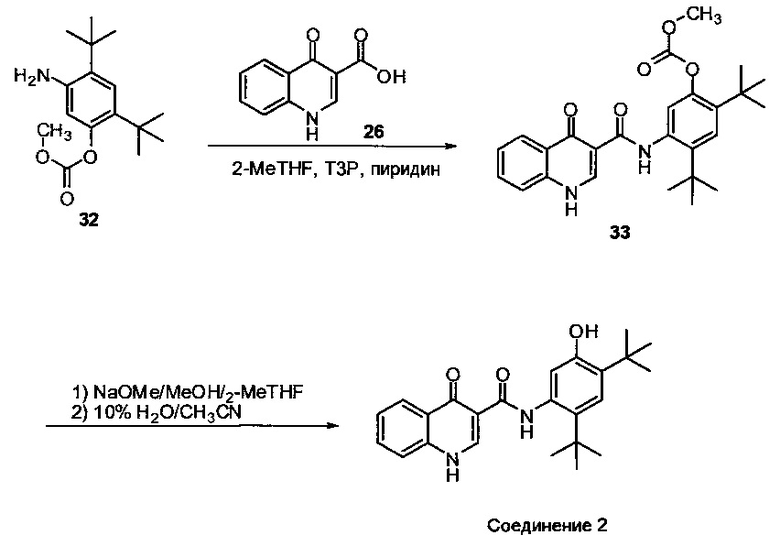
4-Оксо-1,4-дигидрохинолин-3-карбоновой кислотой, 26, (1,0 экв.) и 5-амино-2,4-ди-трет-бутилфенилметилкарбонатом, 32, (1,1 экв.) заполняли реактор. Добавляли 2-MeTHF (4,0 об., относительно кислоты), а затем 50% раствор Т3Р® в 2-MeTHF (1,7 экв.). Заполненный Т3Р сосуд промывали 2-MeTHF (0,6 об.). Затем добавляли пиридин (2,0 экв.) и нагревали образующуюся в результате суспензию до 47,5+/-5,0°С и держали при этой температуре в течение 8 часов. Отбирали образец и проверяли степень завершенности реакции с помощью ВЭЖХ. После окончания реакции образующуюся в результате смесь охлаждали до 25,0°С+/-2,5°С. Для разведения смесь добавляли 2-MeTHF (12,5 об.). Реакционную смесь дважды промывали водой (10,0 об.) и добавляли в реактор 2-MeTHF (16,5 об.). К этому раствору добавляли 30% вес/вес NaOMe/MeOH (1,7 экв.) для проведения метанолиза. Реакцию перемешивали при 25,0°С+/-5,0°С в течение не менее чем 1,0 часа и проверяли степень завершенности реакции с помощью ВЭЖХ. После завершения реакции реакцию останавливали 1,2н HCl/Н2О (10,0 об.) и промывали 0,1н HCl/H2O (10,0 об.). Органический раствор осветляли фильтрованием для удаления любых твердых частиц и помещали во второй реактор.
Отфильтрованный раствор концентрировали при температуре не выше 35°С (температура в рубашке) и не ниже 8,0°С (температура внутренней реакции) под сниженным давлением до 20 об. CH3CN добавляли до 40 об. и концентрировали раствор при температуре не выше 35°С (температура в рубашке) и не ниже 8,0°С (температура внутренней реакции) до 20 об. Цикл добавления CH3CN и концентрации повторяли еще 2 раза, т.е. всего 3 добавления CH3CN и 4 концентрации до 20 об. После конечной концентрации до 20 об. добавляли 16,0 об. CH3CN, а затем 4,0 об. H2O для получения конечной концентрации, составляющей 40 об. 10% H2O/CH3CN относительно исходного количества кислоты. Эту суспензию нагревали до 78,0°С+/-5,0°С (дефлегмация). Затем перемешивали суспензию в течение не менее 5 часов. Суспензию охлаждали до температуры от 20 до 25°С в течение 5 часов и фильтровали. Уплотненный осадок на фильтре 4 раза промывали CH3CN (5 об.), нагретым до температуры от 20 до 25°С. Образующееся в результате твердое вещество (Соединение 2) высушивали в вакуумном сушильном шкафу при 50,0°С+/-5,0°С. 1Н ЯМР (400 МГц, ДМСО-d6) δ 12,8 (с, 1Н), 11,8 (с, 1Н), 9,2 (с, 1Н), 8,9 (с, 1Н), 8,3 (с, 1Н), 7,2 (с, 1Н), 7,9 (т, 1Н), 7,8 (д, 1Н), 7,5 (т, 1Н), 7,1 (с, 1Н), 1,4 (с, 9Н), 1,4 (с, 9Н).
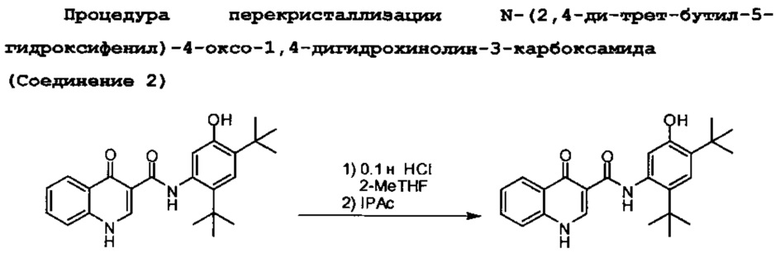
Реактор заполняли соединением 2 (1,0 экв.). Добавляли 2-MeTHF (20,0 об., а затем добавляли 0,1н HCl (5,0 об.). Двухфазный раствор перемешивали и разделяли, а верхнюю органическую фазу еще два раза промывали 0,1н HCl (5,0 об.). Органический раствор осветляли фильтрованием для удаления любых твердых частиц и помещали во второй реактор. Отфильтрованный раствор концентрировали при температуре не выше 35°С (температура в рубашке) и не ниже 8,0°С (температура внутренней реакции) под сниженным давлением до 10 об. Добавляли изопропилацетат (IPAc) (10 об.) и концентрировали раствор при температуре не выше 35°С (температура в рубашке) и не ниже 8,0°С (температура внутренней реакции) до 10 об. Добавления IPAc и концентрацию повторяли еще 2 раза, т.е. всего 3 добавления IPAc и 4 концентрации до 10 об. После конечной концентрации добавляли 10 об. IPAc и суспензию нагревали для дефлегмации и держали при этой температуре в течение 5 часов. Суспензию охлаждали до 0,0°С+/-5°С в течение 5 часов и фильтровали. Уплотненный осадок на фильтре один раз промывали IPAc (5 об.). Образующееся в результате твердое вещество высушивали в вакуумном сушильном шкафу при 50,0°С+/-5,0°С.
Приготовление твердой дисперсии, включающей по существу аморфное Соединение 2
Систему растворителей из МЭК и деионизированной воды, составленную в соответствии с соотношением 90% по массе МЭК/10% по массе деионизированной воды, нагревали до температуры 20-30°С в реакторе, снабженном магнитной мешалкой и системой подогрева. В эту систему растворителей добавляли полимер гипромеллозаацетатсукцината (HPMCAS)(в виде гранул, растворяющийся при высоком рН), лаурилсульфат натрия и Соединение 2 в соответствии с соотношением 19,5% по массе гипромеллозаацетатсукцината/0,5% по массе лаурилсульфата натрия/80% Соединения 2. Образующаяся в результате смесь содержала 10,5% по массе твердых частиц. Фактическое количество ингредиентов и растворителей, применяемых для получения этой смеси, указано в Таблице 5 ниже:


Температуру смеси регулировали в интервале 20-45°С и перемешивали до тех пор, пока смесь не становилась по существу гомогенной, а все компоненты по существу не растворялись.
Прибор для высушивания распылением, коммерческий прибор для высушивания распылением Niro PSD4, с форсункой (Spray Systems Maximum Passage серии SK-MFP с размером сопла/ядра 54/21), снабженной незабивающейся насадкой (anti-bearding cap), применяли в нормальном режиме высушивания распылением в соответствии с параметрами процесса высушивания распылением, перечисленными в Таблице 6 ниже.
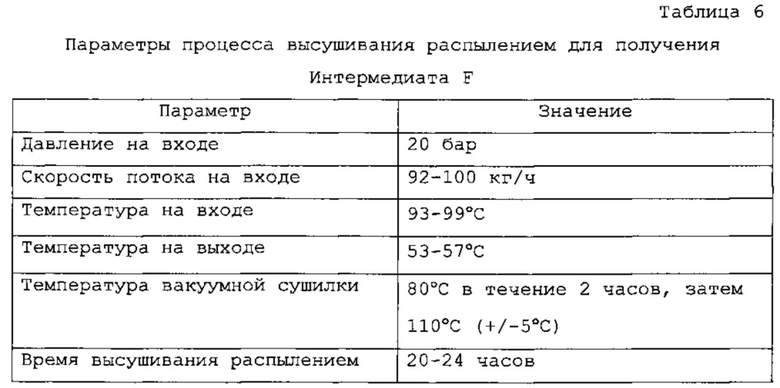
Высокоэффективный циклонный уловитель отделял влажный продукт от распыленного газа и паров растворителя. Влажный продукт содержал 8,5-9,7% МЭК и 0,56-0,83% воды, и средний размер его частиц составлял 17-19 мкм, а объемная плотность составляла 0,27-0,33 г/см3. Влажный продукт переносили в 4000-литровую двойную коническую сушилку из нержавеющей стали для снижения остаточного количества растворителей до уровня приблизительно менее 5000 м.д. для образования высушенной распылением дисперсии аморфного Соединения 2, содержащей <0,03% МЭК и 0,3% воды.
Образование таблеток в процессе полностью непрерывной влажной грануляции
Оборудование/Процесс
Оборудование
Полностью непрерывная установка разработки и запуска (DLR) или оборудование похожего типа.
Скрининг
Соединение 1 Форму I, твердую дисперсию, включающую по существу аморфное Соединение 2, и вспомогательные вещества можно диспергировать в отдельных промежуточных бункерных контейнерах (IBC). Эти материалы можно скринировать с помощью операции просеивания ''из контейнера в контейнер''. Подходящими размерами ячейки являются ячейки 20, ячейки 40 или ячейки 60.
Смешивание
Контейнеры IBC, содержащие откалиброванное Соединение 1 Форму I, твердую дисперсию, включающую по существу аморфное Соединение 2, и вспомогательные вещества можно присоединить к системе подачи, которая может подавать материалы контролируемым способом, например, с помощью объемного или гравиметрического весового питателя, в непрерывно работающий смеситель. Скорость подачи индивидуальных компонентов определяют по составу композиции и общей пропускной способности. Пропускная способность может составлять от 8 кг/ч до 30 кг/ч. Непрерывно работающий смеситель может иметь разную конфигурацию лезвий для обеспечения надлежащего смешивания, а скорость вращения этих лезвий может быть между 80 об/мин и 300 об/мин.
Влажная грануляция
Грануляционный раствор можно приготовить посредством растворения 48 г лаурилсульфата натрия и 159 г поливинилпирролидона в 1,626 г воды в контейнере из нержавеющей стали с помощью верхнеприводной мешалки со скоростью перемешивания равной 700 об/мин. Грануляционный раствор можно поместить в контейнер, из которого раствор можно нагнетать в двухшнековый гранулятор с помощью перистальтического насоса с массовым расходомером и регулятором, применяя подходящую для процесса скорость потока. Смесь можно гранулировать с помощью двухшнекового гранулятора, такого как гранулятор, являющийся частью DLR. Смесь можно добавить в дьухшнековый гранулятор с помощью весового питателя, такого как питатель K-Tron в DLR, со скоростью подачи от 8 кг/ч до 24 кг/ч. Двухшнековый гранулятор может работать при температуре емкости равной 25 градусов Цельсия и скорости вращения шнеков от 200 до 950 об/мин. Процесс грануляции можно проводить в течение трех минут для партий небольшого размера и в течение нескольких часов для партий большого размера.
Высушивание
Влажные гранулы можно напрямую подавать в сушилку с псевдосжиженным слоем, такую как сегментированная сушилка с псевдосжиженным слоем в DLR. Конечную точку высушивания можно выбрать при температуре продукта на выходе в интервале от 40 до 55 градусов Цельсия так, что в этой точке содержание воды в гранулах может составлять 2,1% вес/вес (''потеря при высушивании, LOD'') или менее. Время высушивания может составлять 12 минут или меньше, или больше для достижения желаемой конечной точки высушивания.
Перемалывание
Высушенные гранулы можно перемолоть для уменьшения размера гранул. Для этого можно применять коническую мельницу, такую как встроенная Quadro U10 CoMil.
Смешивание
Гранулы можно смешать с экстрагранулярными вспомогательными веществами, такими как наполнители и смазывающие вещества, с помощью весового питателя и непрерывного смешивания. Скорость смешивания может составлять 80-300 об/мин.
Прессование
Смесь для прессования можно спрессовать в таблетки с помощью односекционного или вращающегося таблеточного пресса, такого как Courtoy Modul Ρ пресс, являющийся частью системы DLR, с помощью соответствующим образом откалиброванного оборудования. Вес таблеток для дозы, состоящей из 200 мг Соединения 1 Формы I и 125 мг по существу аморфного Соединения 2, может составлять около 500 или 600 мг.
Покрытие пленочной оболочкой
Таблетки можно покрыть пленочной оболочкой с помощью инновационного дражировочного котла Omega, который является частью системы DLR. Этот дражировочный котел позволяет быстро нанести пленочную оболочку на подпартии от 1 до 4 кг, что обеспечивает непрерывное производство.
Печать
На покрытых пленочной оболочкой таблетках можно напечатать монограмму на одной или на обеих сторонах таблетки, например, с помощью наклонного принтера Ackley.
Образование таблеток в процессе двухшнековой влажной грануляции
Оборудование/Процесс
Оборудование
Двухшнековые влажные грануляторы: ConsiGma-1, ConsiGma-25 или Leistritz nano.
Скрининг/Взвешивание
Соединение 1 Форму I, твердую дисперсию, включающую по существу аморфное Соединение 2, и вспомогательные вещества можно скринировать до или после взвешивания. Подходящими размерами ячейки являются ячейки 20, ячейки 40 или ячейки 60. Соединение 1 Форму I и/или твердую дисперсию, включающую по существу аморфное Соединение 2, можно предварительно смешать с одним или более вспомогательными веществами для упрощения скрининга.
Смешивание
Соединение 1 Форму I, твердую дисперсию, включающую по существу аморфное Соединение 2, и вспомогательные вещества можно добавлять в смеситель в разном порядке. Смешивание можно проводить в смесителе Turbula, в v-образном смесителе или в бункерном смесителе. Компоненты можно смешивать в течение 10 минут.
Влажная грануляция
Грануляционный раствор можно приготовить посредством растворения 48 г лаурилсульфата натрия и 159 г поливинилпирролидона в 1,626 г воды в контейнере из нержавеющей стали с помощью верхнеприводной мешалки со скоростью перемешивания равной 700 об/мин. Смесь можно гранулировать с помощью двухшнекового гранулятора, такого как ConsiGma-1. Грануляционный раствор можно добавить в двухшнековый гранулятор с помощью перистальтического насоса, такого как насос в ConsiGma-1, со скоростью подачи равной 67 г/мин. Смесь можно добавить в двухшнековый гранулятор с помощью весового питателя, такого как питатель Brabender в ConsiGma-1, со скоростью подачи равной 10 кг/ч. Двухшнековый гранулятор может работать при температуре емкости равной 25 градусов Цельсия и скорости вращения шнеков равной 400 об/мин. Процесс грануляции можно проводить в течение четырех минут. Процесс грануляции можно проводить в течение более короткого или более длительного времени для получения партий с меньшим или большим количеством влажных гранул.
Высушивание
Влажные гранулы можно напрямую подавать в сушилку с псевдосжиженным слоем, такую как сушильная камера в ConsiGma-1 или сегментированная сушилка с псевдосжиженным слоем в CTL-25. Конечную точку высушивания можно выбрать при температуре продукта равной 43 градуса Цельсия так, что в этой точке содержание воды в гранулах может составлять 1,6% вес/вес (''потеря при высушивании, LOD''). Время высушивания может составлять 12 минут или меньше, или больше для достижения желаемой конечной точки высушивания. Высушивание можно проводить при скорости воздушного потока равной 59 м3/мин и температуре на входе равной 60 градусов Цельсия. В качестве альтернативы влажные гранулы, выходящие из двухшнекового гранулятора, можно собирать в бункер или контейнер в течение определенного периода времени, после которого влажные гранул переносят в отдельную изолированную сушилку с псевдосжиженным слоем, такую как Vector Multi 15.
Перемалывание
Высушенные гранулы можно перемолоть для уменьшения размера гранул. Для этого можно применять коническую мельницу, такую как Quadro 194 CoMil.
Смешивание
Гранулы можно смешать с экстрагранулярными вспомогательными веществами, такими как наполнители и смазывающее вещество, с помощью V-образного смесителя или бункерного смесителя. Время смешивания может составлять 5, 3 или 1 минуту.
Прессование
Семь для прессования можно спрессовать в таблетки с помощью односекционного или вращающегося таблеточного пресса, такого как Courtoy Modul Ρ пресс, применяя оборудование для производства овальной формы 0,55'×0,33'. Вес таблеток для дозы, состоящей из 200 мг Соединения 1 Формы I и 125 мг по существу аморфного Соединения 2, может составлять около 500 или 600 мг.
Покрытие пленочной оболочкой
Таблетки можно покрыть пленочной оболочкой с помощью дражировочного котла, такого как, например, прибор для нанесения покрытия Thomas Engineering Compu-Lab. Можно добавить следовые количества карнаубского воска для улучшения внешнего вида таблеток и возможностей процесса.
Печать
На покрытых пленочной оболочкой таблетках можно напечатать монограмму на одной или на обеих сторонах таблетки, например, с помощью принтера Hartnett Delta.
Образование таблеток в процессе непрерывной двухшнековой влажной грануляции
Оборудование/Процесс
Оборудование
Гранулятор: двухшнековый влажный гранулятор ConsiGma или Leistritz, или Thermo Fisher.
Скрининг/Взвешивание
Соединение 1 Форму I и вспомогательные вещества можно скринировать до или после взвешивания. Возможными размерами ячейки являются ячейки 20, ячейки 40 или ячейки 60. Соединение 1 можно предварительно смешать с одним или более вспомогательными веществами для упрощения скрининга.
Смешивание
Соединение 1 и вспомогательные вещества можно добавлять в смеситель в разном порядке. Смешивание можно проводить в смесителе Turbula, в v-образном смесителе, в бункерном смесителе или в непрерывно работающем смесителе. Компоненты можно смешивать в течение 10 минут в смесителе периодического действия или непрерывно в непрерывно работающем смесителе.
Проведение грануляции
Грануляционная жидкость - лаурилсульфат натрия и связывающее вещество добавляют к очищенной воде и перемешивают до растворения. Подходящее соотношение составляет 2,5% вес/вес лаурилсульфата натрия и 10,0% вес/вес ПВП K30 в воде.
Грануляция - смесь, содержащую Соединение 1 и вспомогательные вещества, можно добавлять дозами в двухшнековый гранулятор с помощью весового питателя со скоростью подачи равной 10 кг/ч. Грануляционную жидкость можно добавлять с помощью перистальтического насоса со скоростью подачи равной 3,5 кг/ч. Гранулятор может работать со скоростью равной 400 об/мин. Заметным преимуществом настоящего процесса двухшнековой влажной грануляции является применение грануляционной жидкости, которая включает как сурфактант, так и связывающее вещество для лучшей грануляции за счет повышенной способности к смачиванию. В одном варианте применения изобретения сурфактант является лаурилсульфатом натрия. Другим заметным преимуществом является то, что благодаря непрерывности процесса и тому, что в любой момент времени обрабатывается лишь ограниченное количество материала, процесс можно хорошо контролировать, что обеспечивает высокое качество продукции.
Перемалывание
Размер гранул можно уменьшить с помощью мельницы с ситом или конической мельницы либо до высушивания, либо после высушивания, либо в обоих вариантах.
Высушивание
Гранулы можно высушивать с помощью вакуумного сушильного шкафа, высушивания на подложке, двухконусной сушилки или сушилки с псевдосжиженным слоем.
Смешивание
Гранулы можно смешать с экстрагранулярными вспомогательными веществами. Гранулы смешивали с помощью 300-литрового бункерного смесителя при 60 оборотах.
Прессование
Смесь для прессования прессовали в таблетки с помощью вращающегося пресса Courtoy Modul P.
Покрытие пленочной оболочкой
Таблетки можно покрыть пленочной оболочкой с помощью дражировочного котла, такого как, например, О'Hara Labcoat.
Печать
На покрытых пленочной оболочкой таблетках можно напечатать монограмму на одной или на обеих сторонах таблетки, например, с помощью принтера Hartnett Delta.
ИССЛЕДОВАНИЯ
ПРОТОКОЛ 1
Исследования для обнаружения и измерения способности соединений потенцировать ΔF508-CFTR
Оптические способы измерения мембранного потенциала для исследования способности соединение модулировать ΔF508-CFTR
В исследовании применяют флуоресцентные чувствительные к напряжению красители для измерения изменений мембранного потенциала с помощью флуоресцентного планшетного спектрофотометра (например, FLIPR III, Molecular Devices, Inc.) как величины увеличения функциональных ΔF508-CFTR в клетках NIH 3Т3. Движущей силой ответа является создание градиента хлорид-ионов в связи с активацией канала посредством одной стадии добавления жидкости после предварительной обработки клеток соединениями и последующего нанесения чувствительного к напряжению красителя.
Идентификация потенцирующих соединений
Для идентификации потенцирующих ΔF508-CFTR соединений разработали формат высокопроизводительного скрининга с двукратным добавлением. В данном высокопроизводительном скрининге применяют флуоресцентные чувствительные к напряжению красители для измерения изменений мембранного потенциала на FLIPR III как меры увеличения активности воротного механизма канала (проводимости) ΔF508 CFTR в откорректированных температурой клетках ΔF508 CFTR NIH 3Т3. Движущей силой ответа является создание градиента Cl- ионов в связи с активацией канала форсколином в одну стадию добавления жидкости с помощью флуоресцентного планшетного спектрофотометра, такого как FLIPR III, после предварительной обработки клеток потенцирующими соединениями (или контроля растворителя ДМСО) и последующего нанесения перераспределяющегося красителя.
Растворы
Омывающий раствор №1: (в мМ) NaCl 160, KCl 4,5, CaCl2 2, MgCl2 1, HEPES 10, рН 7,4 с помощью NaOH.
Омывающий раствор без хлорида: Хлориды в Омывающем растворе №1 (выше) замещены глюконатными солями.
Культура клеток
Мышиные фибробласты NIH3T3, стабильно экспрессирующие ΔF508-CFTR, применяют для оптических измерений мембранного потенциала. Клетки держат при 37°С в 5% СО2 и 90% влажности в среде Игла, модифицированной по способу Дульбекко, с добавлением 2 мМ глутамина, 10% фетальной бычьей сыворотки, 1 Χ NEAA, β-меркаптоэтанола, 1 X пенициллин/стрептомицина и 25 мМ HEPES (4-(2-гидроксиэтил)-1-пиперазин этансульфоновой кислоты) в культуральных флаконах 175 см2. Для всех оптических исследований клетки рассаживали в количестве ~20000/лунка в 384-луночные покрытые матригелем плашки и культивировали в течение 2 ч при 37°С перед культивированием при 27°С в течение 24 ч. для исследования потенцирующих веществ. Для исследования коррекции клетки культивировали при 27°С или 37°С в присутствии или в отсутствие соединение в течение 16-24 часов.
Электрофизиологические исследования для изучения способности соединений модулировать ΔF508-CFTR.
Исследования в камере Уссинга
Эксперименты в камере Уссинга проводили на поляризованных эпителиальных клетках дыхательных путей, экспрессирующих ΔF508-CFTR, для дальнейшей характеристики усилителей или индукторов ΔF508-CFTR, идентифицированных в оптических исследованиях. Эпителий, пораженный и не пораженный кистозным фиброзом, выделяли из бронхиальной ткани, культивировали, как описано ранее (Galietta, L.J.V., Lantero, S., Gazzolo, Α., Sacco, О., Romano, L., Rossi, G.A., & Zegarra-Moran, O. (1998) In Vitro Cell. Dev. Biol. 34, 478-481) и помещали на фильтры Costar® Snapwell™, предварительно покрытые средой, кондиционированной NIH3T3 клетками. Через четыре дня апикальную среду удаляли и выращивали клетки на границе раздела водной и воздушной сред в течение >14 дней перед применением. Образующийся в результате монослой дифференцированных столбчатых реснитчатых клеток обладал свойствами, характерными для эпителия дыхательных путей. Не пораженный кистозным фиброзом бронхиальный эпителий человека выделяли из не курящих людей, не страдающих каким-либо известным заболеванием легких. Пораженный кистозным фиброзом бронхиальный эпителий человека выделяли из пациентов, гомозиготных по ΔF508.
Бронхиальный эпителий человека, растущий на вкладках Costar® Snapwell™ для клеточной культуры, заключали в камеру Уссинга (Physiologic Instruments, Inc., Сан-Диего, штат Калифорния) и измеряли трансэпителиальное сопротивление и ток короткого замыкания в присутствии базолатерально-апикального градиента Cl- (Isc) с помощью системы фиксации потенциала (Department of Bioengineering, University of Iowa, штат Айова). Кратко, бронхиальный эпителий человека исследовали в условиях регистрации фиксации потенциала (Vhold-0 мВ) при 37°С. Базолатеральный раствор содержал (в мМ) 145 NaCl, 0,83 K2HPO4, 3,3 KH2PO4, 1,2 MgCl2, 1,2 CaCl2, 10 глюкозы, 10 HEPES (рН, подведенный до 7,35 с помощью NaOH), а апикальный раствор содержал (в мМ) 145 глюконата натрия, 1,2 MgCl2, 1,2 CaCl2, 10 глюкозы, 10 HEPES (рН, подведенный до 7,35 с помощью NaOH).
Идентификация потенцирующих соединений
В типичном протоколе применяли базолатерально-апикальный мембранный градиент концентрации Cl-. Для формирования данного градиента применяли нормальный раствор Рингера для базолатеральной мембраны, в то время как апикальный NaCl заменяли эквимолярным глюконатом натрия (оттитрованный до рН 7,4 с помощью NaOH) для получения градиента больших концентраций Cl- через эпителий. Форсколин (10 мкМ) и все исследуемые соединения добавляли к апикальной стороне вкладок для клеточных культур. Эффективность возможных потенцирующих ΔF508-CFTR веществ сравнивали с эффективностью известного потенцирующего вещества генистеина.
Регистрация фиксации потенциала
Общий ток Cl- в клетках ΔF508-NIH3T3 исследовали с помощью регистрации в режиме перфорированной фиксации потенциала, как описано ранее (Rae, J., Cooper, K., Gates, P., & Watsky, M. (1991) J. Neurosci. Methods 37, 15-26). Регистрацию фиксации потенциала проводили при 22°С с помощью Axopatch 200 В амплификатора фиксации потенциала (Axon Instruments Inc., Фостер Сити, штат Калифорния). Раствор в пипетке содержал (в мМ) 150 N-метил-D-глюкамина (NMDG)-Cl, 2 MgCl2, 2 CaCl2, 10 ЭГТА, 10 HEPES и 240 мкг/мл амфотерицин-В (рН, подведенный до 7,35 с помощью HCl). Внеклеточная среда содержала (в мМ) 150 NMDG-Cl, 2 MgCl2, 2 CaCl2, 10 HEPES (рН, подведенный до 7,35 с помощью HCl). Генерацию импульсов, сбор данных и анализ проводили с помощью ПК с интерфейсом Digidata 1320 A/D вместе с Clampex 8 (Axon Instruments Inc.). Для активации ΔF508-CFTR добавляли 10 мкМ форсколина и 20 мкМ генистеина в резервуар и наблюдали за вольт-амперным соотношением каждые 30 сек.
Идентификация потенцирующих соединений
Способность потенцирующих ΔF508-CFTR веществ увеличивать макроскопический ΔF508-CFTR Cl- ток (IΔF508) в клетках NIH3T3, стабильно экспрессирующих ΔF508-CFTR, также исследовали с помощью техник регистрации в режиме перфорированной фиксации потенциала. Потенцирующие вещества, идентифицированные в оптических исследованиях, вызывали дозозависимое увеличение IΔF508 с той же активностью и эффективностью, что и наблюдаемые в оптических исследованиях. Во всех исследованных клетках реверсивный потенциал перед и во время действия потенцирующего вещества составлял около -30 мВ, что является расчетной ECl (-28 мВ).
Культура клеток
Мышиные фибробласты NIH3T3, стабильно экспрессирующие ΔF508-CFTR, применяют для регистрации фиксации потенциала во всей клетке. Клетки держат при 37°С в 5% CO2 и 90% влажности в среде Игла, модифицированной по способу Дульбекко, с добавлением 2 мМ глутамина, 10% фетальной бычьей сыворотки, 1 Χ NEAA, β-меркаптоэтанола, 1 X пенициллин/стрептомицина и 25 мМ HEPES в культуральных флаконах 175 см2. Для регистрации фиксации потенциала во всей клетке клетки 2500-5000 клеток рассаживали на покрытые поли-L-лизином покровные стекла и культивировали в течение 24-48 ч при 27°С перед применением для исследования активности потенцирующих веществ; и инкубировали в присутствии или в отсутствие корректирующего вещества при 37°С для изменения активности корректирующих веществ.
Регистрация в одном канале
Активность воротного механизма CFTR дикого типа и откорректированного температурой ΔF508-CFTR, экспрессирующегося в клетках NIH3T3, наблюдали, применяя регистрацию фиксации потенциала вырезанной вывернутой на изнанку мембраны, как описано ранее (Dalemans, W., Barbry, P., Champigny, G., Jallat, S., Dott, K., Dreyer, D., Crystal, R.G., Pavirani, Α., Lecocq, J-P., Lazdunski, M. (1991) Nature 354, 526-528) с помощью амплификатора фиксации потенциала Axopatch 200 В (Axon Instruments Inc.). Пипетка содержала (в мМ) 150 NMDG, 150 аспарагиновой кислоты, 5 CaCl2, 2 MgCl2 и 10 HEPES (рН, подведенный до 7,35 с помощью HCl). Резервуар содержал (в мМ) 150 NMDG-Cl, 2 MgCl2, 5 EGTA, 10 TES и 14 Триса в виде основания (рН, подведенный до 7,35 с помощью HCl). После вырезания и CFTR дикого типа, и ΔF508-CFTR активировали добавлением 1 мМ Mg-АТР, 75 нМ каталитической субъединицы цАМФ-зависимой протеинкиназы (РКА; Promega Corp. Мэдисон, штат Висконсин) и 10 мМ NaF для ингибирования протеинфосфатаз, которые препятствовали уменьшению тока. Потенциал в пипетке поддерживали на уровне 80 мВ. Активность канала анализировали в тонких слоях мембраны, содержащих ≤2 активных каналов. Максимальное число одновременных открываний определяло число активных каналов в ходе эксперимента. Для определения амплитуды тока одного канала данные, зарегистрированные от 120 сек активности ΔF508-CFTR, фильтровали в автономном режиме при 100 Гц, а затем применяли для построения гистограмм амплитуды во всех точках, которые соответствовали множественным гауссовым функциям с помощью программного обеспечения Bio-Patch Analysis (Bio-Logic Comp. Франция). Общий микроскопический ток и вероятность открывания (Po) определяли по 120 сек активности канала. Po определяли, применяя программное обеспечение Bio-Patch, или из отношения Po=I/i(N), где I = средний ток, i = амплитуда тока одного канала и N = число активных каналов в тонком слое.
Культура клеток
Мышиные фибробласты NIH3T3, стабильно экспрессирующие ΔF508-CFTR, применяют для регистрации фиксации потенциала вырезанной мембраны. Клетки держат при 37°С в 5% CO2 и 90% влажности в среде Игла, модифицированной по способу Дульбекко, с добавлением 2 мМ глутамина, 10% фетальной бычьей сыворотки, 1 X NEAA, β-меркаптоэтанола, 1 X пенициллин/стрептомицина и 25 мМ HEPES в культуральных флаконах 175 см2. Для регистрации в одном канале 2500-5000 клеток рассаживали на покрытые поли-L-лизином покровные стекла и культивировали в течение 24-48 ч при 27°С перед применением.
ПРОТОКОЛ 2
Исследования для определения и измерения корректирующих ΔF508-CFTR свойств соединений
Оптические способы измерения мембранного потенциала для исследования способности соединение модулировать ΔF508-CFTR.
В оптическом исследовании мембранного потенциала применяли чувствительные к напряжению сенсоры резонансного переноса энергии флуоресценции (FRET), описанные Gonzalez и Tsien (See Gonzalez, J.Ε. and R.Y. Tsien (1995) ''Voltage sensing by fluorescence resonance energy transfer in single cells'' Biophys J 69(4): 1272-80, and Gonzalez, J.E. and R.Y. Tsien (1997) ''Improved indicators of cell membrane potential that use fluorescence resonance energy transfer'' Chem Biol 4(4): 269-77) в комбинации с инструментами для измерения изменений флуоресценции, такими как Voltage/Ion Probe Reader (VIPR) (See, Gonzalez, J.E., K. Oades, et al. (1999) ''Cell-based assays and instrumentation for screening ion-channel targets'' Drug Discov Today 4(9): 431-439).
Эти чувствительные к напряжению исследования основаны на изменении резонансного переноса энергии флуоресценции (FRET) между растворимым в мембране чувствительным к напряжению красителем DiSBAC2(3) и флуоресцентным фосфолипидом CC2-DMPE, который присоединен к наружному слою плазматической мембраны и действует как донор FRET. Изменения мембранного потенциала (Vm) вызывают перераспределение отрицательно заряженного DiSBAC2(3) через плазматическую мембрану, и соответственно меняется количество перенесенной энергии от CC2-DMPE. Изменения испускания флуоресценции отслеживали с помощью VIPR™ II, который представляет собой интегрированную емкость для жидкости и детектор флуоресценции, сделанный так, чтобы проводить скрининг клеток в 96- или 384-луночных плашках для микротитрования.
Идентификация корректирующих соединений
Для идентификации малых молекул, которые корректируют нарушение транспорта, связанное с ΔF508-CFTR, был разработан формат высокоэффективного скрининга с однократным добавлением. Клетки инкубировали в бессывороточной среде в течение 16 ч при 37°С в присутствии или в отсутствие (отрицательный контроль) исследуемого соединения. В качестве положительного контроля клетки, рассаженные в 384-луночные плашки, инкубировали в течение 16 ч при 27°С, чтобы ''скорректировать с помощью температуры'' ΔF508-CFTR. Затем клетки споласкивали 3 раза в растворе Кребса-Рингера и добавляли чувствительные к напряжению красители. Для активации ΔF508-CFTR в каждую лунку добавляли 10 мкМ форсколина и потенцирующее CFTR вещество генистеин (20 мкМ) вместе с не содержащей Cl- средой. Добавление не содержащей Cl- среды способствовало выходу Cl- в ответ на активацию ΔF508-CFTR, и за происходящей в результате этого деполяризацией мембраны наблюдали оптическими способами, применяя чувствительные к напряжению красители на основе FRET.
Идентификация потенцирующих соединений
Для идентификации потенцирующих ΔF508-CFTR соединений был разработан формат высокоэффективного скрининга с двукратным добавлением. Во время первого добавления не содержащую Cl- среду, содержащую или не содержащую исследуемое соединение, добавляли в каждую лунку. Через 2 сек проводили второе добавление не содержащей Cl- среды, содержащей 2-10 мкМ форсколина, для активации ΔF508-CFTR. Концентрация внеклеточного Cl- после добавления обеих порций составляла 28 мМ, что способствовало выходу Cl- в ответ на активацию ΔF508-CFTR, и за происходящей в результате этого деполяризацией мембраны наблюдали оптическими способами, применяя чувствительные к напряжению красители на основе FRET.
Растворы
Омывающий раствор №1: (в мМ) NaCl 160, KCl 4,5, CaCl2 2, MgCl2 1, HEPES 10, рН 7,4 с помощью NaOH.
Омывающий раствор без хлорида: Хлориды в Омывающем растворе №1 (выше) замещены глюконатными солями.
C2-DMPE: Готовят в виде 10 мМ основного раствора в ДМСО и хранят при -20°С.
DiSBAC2(3): Готовят в виде 10 мМ основного раствора в ДМСО и хранят при -20°С.
Культура клеток
Мышиные фибробласты NIH3T3, стабильно экспрессирующие ΔF508-CFTR, применимы для оптических измерений мембранного потенциала. Клетки держат при 37°С в 5% CO2 и 90% влажности в среде Игла, модифицированной по способу Дульбекко, с добавлением 2 мМ глутамина, 10% фетальной бычьей сыворотки, 1 Χ NEAA, β-меркаптоэтанола, 1 X пенициллин/стрептомицина и 25 мМ HEPES в культуральных флаконах 175 см2. Для всех оптических исследований клетки рассаживали в количестве ~30000/лунка в 384-луночные покрытые матригелем плашки и культивировали в течение 2 ч при 37°С перед культивированием при 27°С в течение 24 ч для исследования потенцирующих веществ. Для исследований коррекции клетки культивировали при 27°С или 37°С в присутствии или в отсутствие соединение в течение 16-24 часов.
Электрофизиологические исследования для изучения способности соединений модулировать ΔF508-CFTR.
Исследования в камере Уссинга
Эксперименты в камере Уссинга проводили на поляризованных эпителиальных клетках, экспрессирующих ΔF508-CFTR, для дальнейшей характеристики усилителей или индукторов ΔF508-CFTR, идентифицированных в оптических исследованиях. Эпителиальные клетки FRT, растущие на вкладках для клеточных культур
Costar Snapwell, заключали в камеру Уссинга (Physiologic Instruments, Inc., Сан-Диего, штат Калифорния), и к монослоям непрерывно прикладывали ток короткого замыкания посредством системы фиксации потенциала (Department of Bioengineering, University of Iowa, штат Айова, и Physiologic Instruments, Inc., Сан-Диего, штат Калифорния). Трансэпителиальное сопротивление измеряли, прикладывая импульс 2 мВ. В этих условиях эпителий FRT показывал сопротивление равное 4 ΚΩ/ см2 или более. Раствор поддерживали при температуре 27°С и продували воздухом. Смещение потенциала электрода и сопротивление жидкости корректировали с помощью бесклеточной вкладки. В этих условиях ток отражает поток Cl- через ΔF508-CFTR, экспрессирующийся в апикальной мембране. Значения ISC получали в цифровой форме с помощью интерфейса МР100А-СЕ и программного обеспечения AcqKnowledge (v3.2.6; BIOPAC Systems, Санта-Барбара, штат Калифорния).
Идентификация корректирующих соединений
В типичном протоколе применяли базолатерально-апикальный мембранный градиент концентрации Cl-. Для формирования данного градиента применяли нормальный раствор Рингера для базолатеральной мембраны, в то время как апикальный NaCl заменяли эквимолярным глюконатом натрия (оттитрованным до рН 7,4 с помощью NaOH) для получения градиента больших концентраций Cl- через эпителий. Все эксперименты проводили с интактными монослоями. Для полной активации ΔF508-CFTR добавляли форсколин (10 мкМ) и ингибитор фосфодиэстеразы (PDE) IBMX (100 мкМ), а затем добавляли потенцирующее CFTR вещество генистеин (50 мкМ).
Как наблюдают в других типах клеток, инкубация клеток FRT, стабильно экспрессирующих ΔF508-CFTR, при низкой температуре повышает функциональную плотность CFTR в плазматической мембране. Для определения активности корректирующих веществ клетки инкубировали с 10 мкМ исследуемого соединения в течение 24 часов при 37°С, а затем 3 раза промывали перед снятием показаний. цАМФ- и генистеин-опосредованный ISC в обработанных соединением клетках нормализовали к 27°С и 37°С контролям и выражали в виде процента активности. Предварительная инкубация клеток с корректирующим веществом существенно повышала а цАМФ- и генистеин-опосредованный ISC по сравнению с 37°С контролями.
Идентификация потенцирующих соединений
В типичном протоколе применяли базолатерально-апикальный мембранный градиент концентрации Cl-. Для формирования данного градиента применяли нормальный раствор Рингера для базолатеральной мембраны и пермеабилизовали нистатином (360 мкг/мл), в то время как апикальный NaCl заменяли эквимолярным глюконатом натрия (оттитрованным до рН 7,4 с помощью NaOH) для получения градиента больших концентраций Cl- через эпителий. Все эксперименты проводили через 30 мин после пермеабилизации нистатином. Форсколин (10 мкМ) и все исследуемые соединения добавляли к обеим сторонам вкладок для клеточных культур. Эффективность возможных потенцирующих ΔF508-CFTR веществ сравнивали с эффективностью известного потенцирующего вещества генистеина.
Растворы
Базолатеральный раствор (в мМ): NaCl (135), CaCl2 (1,2), MgCl2 (1,2), K2HPO4 (2,4), KHPO4 (0,6), N-2-гидроксиэтилпиперазин-N'-2-этансульфоновая кислота (HEPES) (10) и декстроза (10). Раствор титровали до рН 7,4 с помощью NaOH.
Апикальный раствор (в мМ): такой же, как базолатеральный раствор с NaCl, замененным глюконатом Na (135).
Культура клеток
Крысиные эпителиальные клетки Фишера (FRT), экспрессирующие ΔF508-CFTR (FRTΔF508-CFTR), применяли в экспериментах в камере Уссинга для возможных усилителей или индукторов ΔF508-CFTR, идентифицированных в оптических исследованиях. Клетки культивировали на вкладках для клеточных культур Costar Snapwell и культивировали в течение пяти дней при 37°С в 5% CO2 и 90% влажности в среде Хэма F-12, модифицированной по способу Куна, с добавлением 5% фетальной бычьей сыворотки, 100 ед./мл пенициллина и 100 мкг/мл стрептомицина. Перед применением для характеристики потенцирующей активности соединений клетки инкубировали при 27°С в течение 16-48 ч для коррекции ΔF508-CFTR. Для определения активности корректирующих соединений клетки инкубировали при 27°С или 37°С в присутствии или в отсутствие соединение в течение 24 часов.
Регистрация фиксации потенциала во всей клетке
Макроскопический ΔF508-CFTR Cl- ток (IΔF508) в откорректированных температурой и исследуемыми соединениями клетках NIH3T3, стабильно экспрессирующих ΔF508-CFTR, исследовали с помощью регистрации в режиме перфорированной фиксации потенциала. Кратко, регистрацию фиксации потенциала IΔF508 проводили при комнатной температуре с помощью Axopatch 200 В амплификатора фиксации потенциала (Axon Instruments Inc., Фостер Сити, штат Калифорния). Все данные получали при частоте отбора проб равной 10 кГц и проводили низкочастотное фильтрование при 1 кГц. Сопротивление пипеток составляло 5-6 ΜΩ при заполнении внутриклеточным раствором. В этих условиях регистрации расчетный реверсивный потенциал для Cl- (ECl) при комнатной температуре составлял -28 мВ. У всех данных сопротивление уплотнения было >20 ΓΩ и добавочное сопротивление <15 ΜΩ. Генерацию импульсов, сбор данных и анализ проводили с помощью ПК с интерфейсом Digidata 1320 A/D вместе с Clampex 8 (Axon Instruments Inc.). Резервуар содержал <250 мкл физиологического раствора и его непрерывно перфузировали со скоростью 2 мл/мин посредством самотечной перфузионной системы.
Идентификация корректирующих соединений
Для определения активности корректирующих соединений, увеличивающей плотность функциональных ΔF508-CFTR в плазматической мембране, мы применяли описанные выше техники регистрации в режиме перфорированной фиксации потенциала для измерения плотности тока после 24-ч обработки корректирующими соединениями. Для полной активации ΔF508-CFTR к клеткам добавляли 10 мкМ форсколина и 20 мкМ генистеина. В наших условиях регистрации плотность тока после 24-ч инкубации при 27°С была выше наблюдаемой после 24-ч инкубации при 37°С. эти результаты соответствуют известным эффектам низкотемпературной инкубации на плотность ΔF508-CFTR в плазматической мембране. Для определения эффектов корректирующих соединений на плотность тока CFTR клетки инкубировали с 10 мкМ исследуемого соединения в течение 24 часов при 37°С и сравнивали плотность тока с 27°С и 37°С контролями (% активности). Перед регистрацией клетки 3 раза промывали внеклеточной средой для регистрации для удаления любых остатков исследуемого соединения. Предварительная инкубация с 10 мкМ корректирующих соединений значительно увеличивала цАМФ- и генистеин-зависимый ток по сравнению с 37°С контролями.
Идентификация потенцирующих соединений
Способность потенцирующих ΔF508-CFTR соединений увеличивать макроскопический ток Cl- ΔF508-CFTR (IΔF508) в клетках NIH3T3, стабильно экспрессирующих ΔF508-CFTR, также исследовали с помощью техник регистрации в режиме перфорированной фиксации потенциала. Потенцирующие вещества, идентифицированные в оптических исследованиях, вызывали дозозависимое увеличение IΔF508 с той же активностью и эффективностью, что и наблюдаемые в оптических исследованиях. Во всех исследованных клетках реверсивные потенциал перед и во время действия потенцирующего вещества составлял около -30 мВ, что является расчетной ECl (-28 мВ).
Растворы
Внутриклеточный раствор (в мМ): Cs-аспартат (90), CsCl (50), MgCl2 (1), HEPES (10) и 240 мкг/мл амфотерицина-В (рН, подведенный до 7,35 с помощью CsOH).
Внеклеточный раствор (в мМ): N-метил-D-глюкамин (NMDG)-Cl (150), MgCl2 (2), CaCl2 (2), HEPES (10) (рН, подведенный до 7,35 с помощью HCl).
Культура клеток
Мышиные фибробласты NIH3T3, стабильно экспрессирующие ΔF508-CFTR, применяют для регистрации фиксации потенциала во всей клетке. Клетки держат при 37°С в 5% CO2 и 90% влажности в среде Игла, модифицированной по способу Дульбекко, с добавлением 2 мМ глутамина, 10% фетальной бычьей сыворотки, 1 Χ ΝΕΑΆ, β-меркаптоэтанола, 1 X пенициллин/стрептомицина и 25 мМ HEPES в культуральных флаконах 175 см2. Для регистрации фиксации потенциала во всей клетке 2500-5000 клеток рассаживали на покрытые поли-L-лизином покровные стекла и культивировали в течение 24-48 ч при 27°С перед применением для исследования активности потенцирующих веществ; и инкубировали в присутствии или в отсутствие корректирующего вещества при 37°С для изменения активности корректирующих веществ.
Регистрация в одном канале
Одноканальную активность откорректированного температурой ΔF508-CFTR, стабильно экспрессирующегося в клетках NIH3T3, и активность потенцирующих соединений наблюдали, применяя регистрацию фиксации потенциала вырезанной вывернутой на изнанку мембраны. Кратко, регистрацию фиксации потенциала одноканальной активности проводили при комнатной температуре с помощью Axopatch 200 В амплификатора фиксации потенциала (Axon Instruments Inc.). Все данные получали при частоте отбора проб равной 10 кГц и проводили низкочастотное фильтрование при 400 Гц. Пипетки для фиксации потенциала были изготовлены из стекла Corning Kovar Sealing #7052 (World Precision Instruments, Inc., Сарасота, штат Флорида) и обладали сопротивлением 5-8 ΜΩ при заполнении внеклеточным раствором. ΔF508-CFTR активировали после вырезания добавлением 1 мМ Mg-АТФ, 75 нМ каталитической субъединицы цАМФ-зависимой протеинкиназы (РКА; Promega Corp. Мэдисон, штат Висконсин). После стабилизации активности канала тонкий слой мембраны перфузировали посредством самотечной микроперфузионной системы. Подачу жидкости осуществляли рядом с тонким слоем мембраны, что приводило к полному обмену раствора за 1-2 сек. Для поддержания активности ΔF508-CFTR во время быстрой перфузии добавляли неспецифичный ингибитор фосфатазы F- (10 мМ NaF) в омывающий раствор. В этих условиях регистрации активность канала оставалась постоянной на протяжении всей регистрации фиксации потенциала (вплоть до 60 мин). Токи, образованные положительными зарядами, перемещающимися из внутри-во внеклеточный раствор (анионами, движущимися в противоположном направлении), показаны как положительные токи. Потенциал в пипетке поддерживали на уровне 80 мВ.
Активность канала анализировали в тонких слоях мембраны, содержащих ≤2 активных каналов. Максимальное число одновременных открываний определяло число активных каналов в ходе эксперимента. Для определения амплитуды тока одного канала данные, зарегистрированные по 120 сек активности ΔF508-CFTR, фильтровали в автономном режиме при 100 Гц, а затем применяли для построения гистограмм амплитуды во всех точках, которые соответствовали множественным гауссовым функциям, с помощью программного обеспечения Bio-Patch Analysis (Bio-Logic Comp. Франция). Общий микроскопический ток и вероятность открывания (Po) определяли по 120 сек активности канала. Po определяли, применяя программное обеспечение Bio-Patch, или из отношения Po=I/i(N), где I = средний ток, i = амплитуда тока одного канала и N = число активных каналов в тонком слое.
Растворы
Внеклеточный раствор (в мМ): NMDG (150), аспарагиновая кислота (150), CaCl2 (5), MgCl2 (2) и HEPES (10) (рН, подведенный до 7,35 с помощью Триса в виде основания).
Внутриклеточный раствор (в мМ): NMDG-Cl (150), MgCl2 (2), EGTA (5), TES (10) и Трис в виде основания (14) (рН, подведенный до 7,35 с помощью HCl).
Культура клеток
Мышиные фибробласты NIH3T3, стабильно экспрессирующие ΔF508-CFTR, применяют для регистрации фиксации потенциала вырезанной мембраны. Клетки держат при 37°С в 5% CO2 и 90% влажности в среде Игла, модифицированной по способу Дульбекко, с добавлением 2 мМ глутамина, 10% фетальной бычьей сыворотки, 1 X NEAA, β-меркаптоэтанола, 1 X пенициллин/стрептомицина и 25 мМ HEPES в культуральных флаконах 175 см2. Для регистрации в одном канале 2500-5000 клеток рассаживали на покрытые поли-L-лизином покровные стекла и культивировали в течение 24-48 ч при 27°С перед применением.
Соединение 1 и Соединение 2, являющиеся предметом настоящего изобретения, применимы в качестве усилителей или индукторов активности CFTR. Таблица 7 ниже иллюстрирует ЕС50 и относительную эффективность Соединения 1 и Соединения 2. В Таблице 7 применяют следующие обозначения. ЕС50: ''+++'' означает <10 мкМ; ''++'' означает между 10 мкМ и 25 мкМ; ''+'' означает между 25 мкМ и 60 мкМ. % Эффективности: ''+'' означает <25%; ''++'' означает между 25% и 100%; ''+++'' означает >100%.
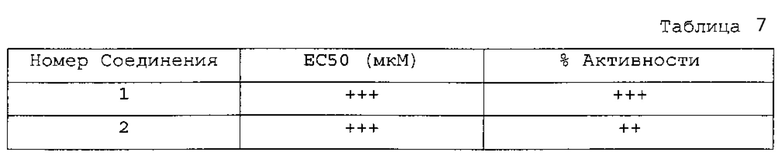
ДРУГИЕ ВАРИАНТЫ ПРИМЕНЕНИЯ ИЗОБРЕТЕНИЯ
Все публикации и патенты, относящиеся к данному раскрытию, включены в настоящую заявку посредством ссылки в том же объеме, что и при специальном и индивидуальном указании отдельных публикаций или патентных заявок для включения посредством ссылки. В том случае, если значения терминов в любом из патентов или публикаций, включенных посредством ссылки, противоречат значению терминов, применяемых в данном раскрытии, предполагается, что значение терминов в данном раскрытии является определяющим. Более того, вышеупомянутое обсуждение раскрывает и описывает лишь примерные варианты применения изобретения. Специалист в данной области техники легко распознает в этих обсуждениях и сопутствующих чертежах и формулах изобретения различные изменения, модификации и вариации, которые можно внести в них без отступления и существа объема изобретения, как определено в следующей формуле изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, ОПОСРЕДОВАННЫХ ТРАНСМЕМБРАННЫМ РЕГУЛЯТОРОМ КИСТОЗНОГО ФИБРОЗА CFTR | 2013 |
|
RU2827711C2 |
| СПОСОБ ПОЛУЧЕНИЯ ФАРМАЦЕВТИЧЕСКИХ КОМПОЗИЦИЙ ДЛЯ ЛЕЧЕНИЯ ОПОСРЕДОВАННЫХ CFTR ЗАБОЛЕВАНИЙ | 2014 |
|
RU2718044C2 |
| СПОСОБ ПРОВЕДЕНИЯ ВЫСОКОПРОИЗВОДИТЕЛЬНОЙ ТЕСТОВОЙ ВЫСОКОЭФФЕКТИВНОЙ ЖИДКОСТНОЙ ХРОМАТОГРАФИИ | 2015 |
|
RU2691136C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, ОПОСРЕДОВАННЫХ МУКОВИСЦИДОЗНЫМ ТРАНСМЕМБРАННЫМ РЕГУЛЯТОРОМ ПРОВОДИМОСТИ | 2015 |
|
RU2744460C2 |
| НОВОЕ ЛЕЧЕНИЕ | 2015 |
|
RU2688191C2 |
| ТВЕРДЫЕ ФОРМЫ (R)-1-(2,2-ДИФТОРБЕНЗО[d][1,3]ДИОКСОЛ-5-ИЛ)-N-(2,3-ДИГИДРОКСИПРОПИЛ)-6-ФТОР-2-(1-ГИДРОКСИ-2-МЕТИЛПРОПАН-2-ИЛ)-1H-ИНДОЛ-5-ИЛ)ЦИКЛОПРОПАНКАРБОКСАМИДА | 2011 |
|
RU2573830C2 |
| ТВЕРДЫЕ ФОРМЫ (R)-1-(2, 2-ДИФТОРБЕНЗО[d][1, 3]ДИОКСОЛ-5-ИЛ)-N-(1-(2, 3-ДИГИДРОКСИПРОПИЛ)-6-ФТОР-2-(1-ГИДРОКСИ-2-МЕТИЛПРОПАН-2-ИЛ)-1H-ИНДОЛ-5-ИЛ)ЦИКЛОПРОПАНКАРБОКСАМИДА | 2011 |
|
RU2711481C2 |
| СПОСОБ ПОЛУЧЕНИЯ МОДУЛЯТОРОВ РЕГУЛЯТОРА ТРАНСМЕМБРАННОЙ ПРОВОДИМОСТИ КИСТОЗНОГО ФИБРОЗА | 2010 |
|
RU2553989C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ МУКОВИСЦИДОЗА | 2019 |
|
RU2822220C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ЕЕ ВВЕДЕНИЕ | 2013 |
|
RU2802442C2 |
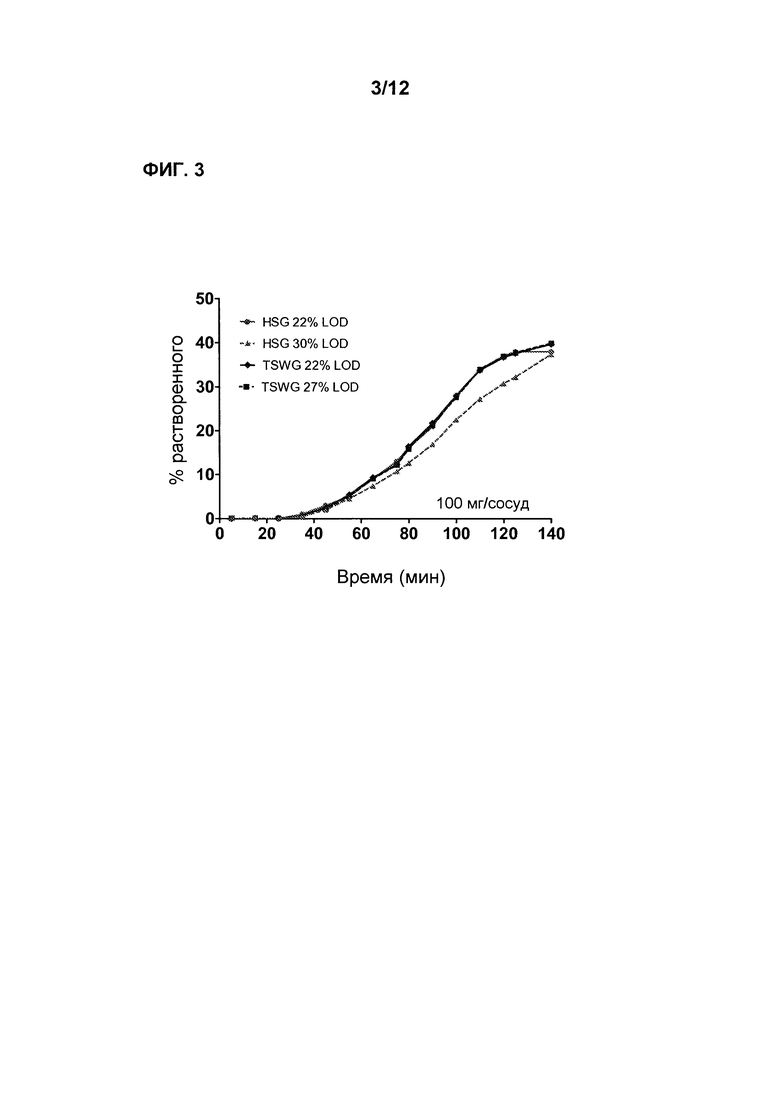
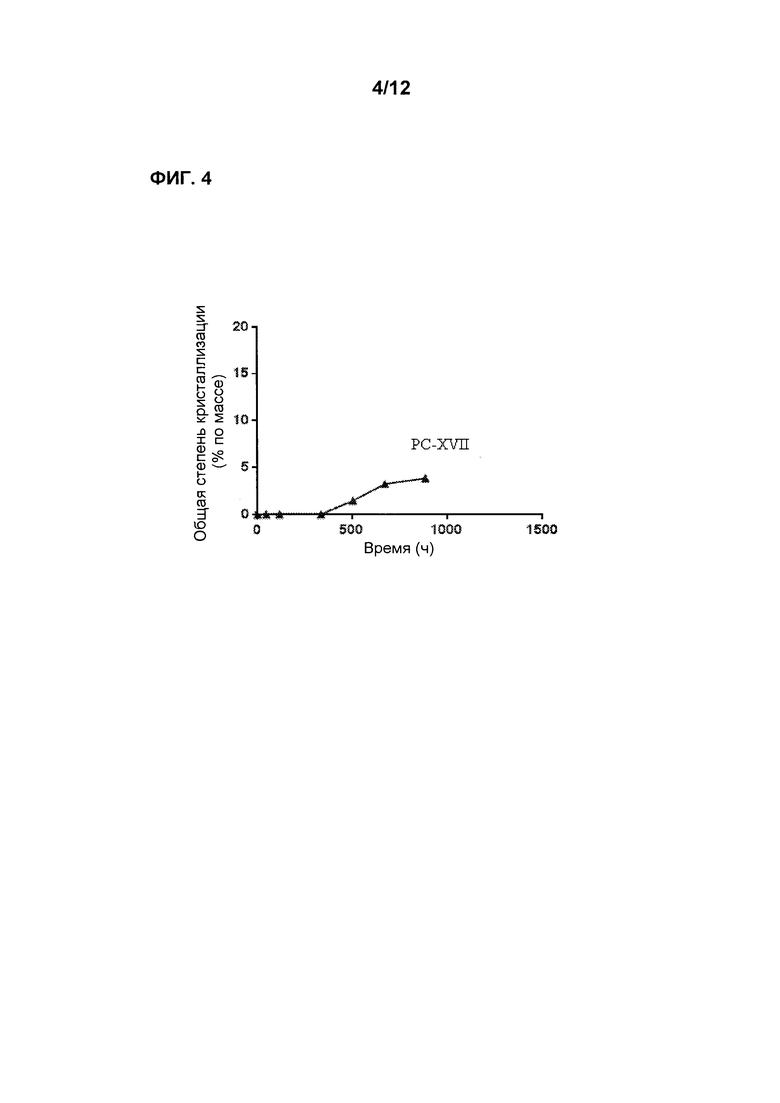
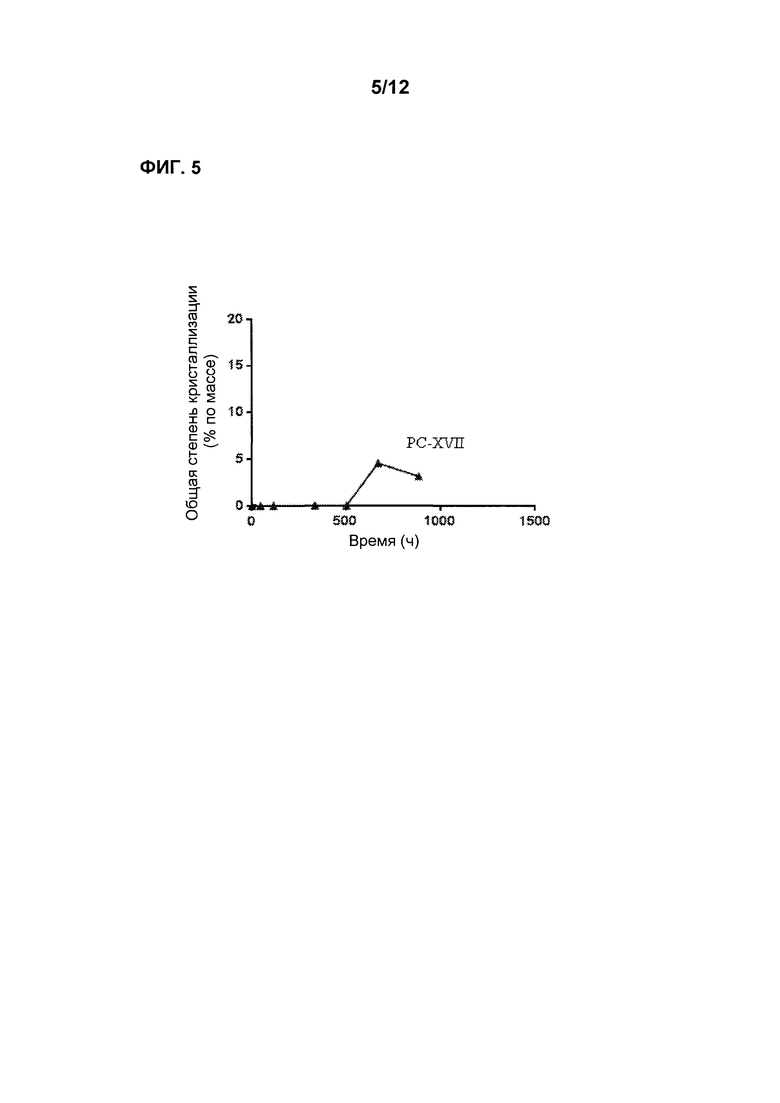
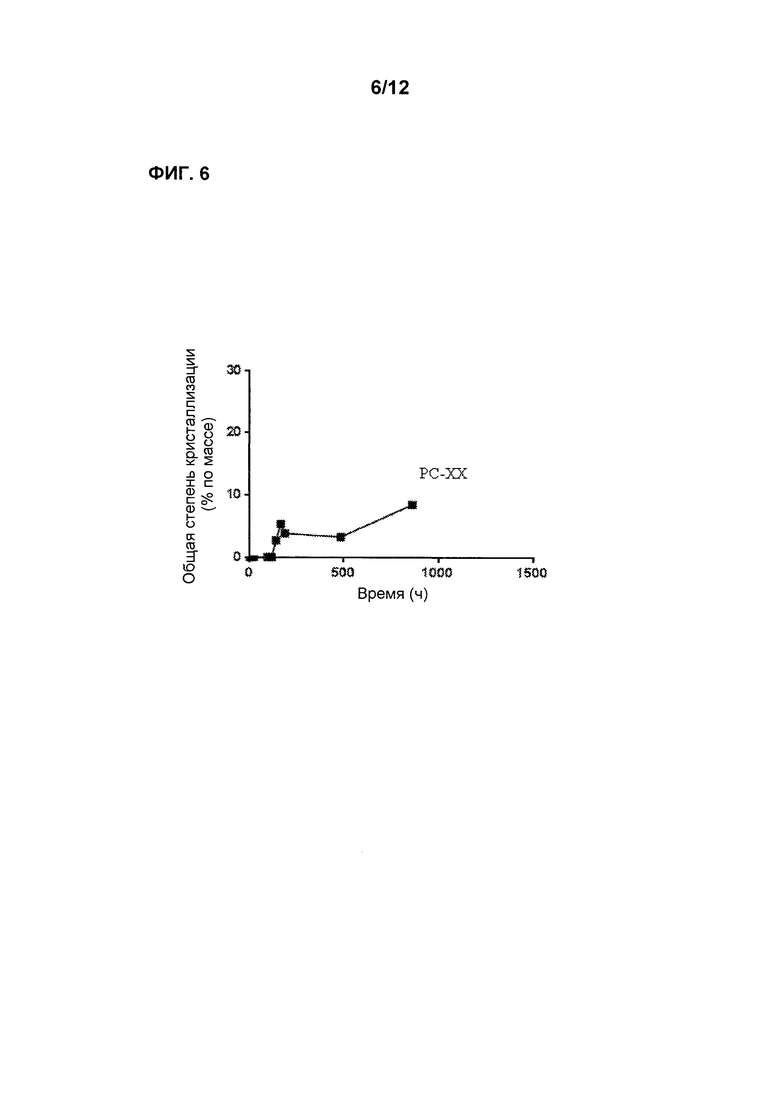
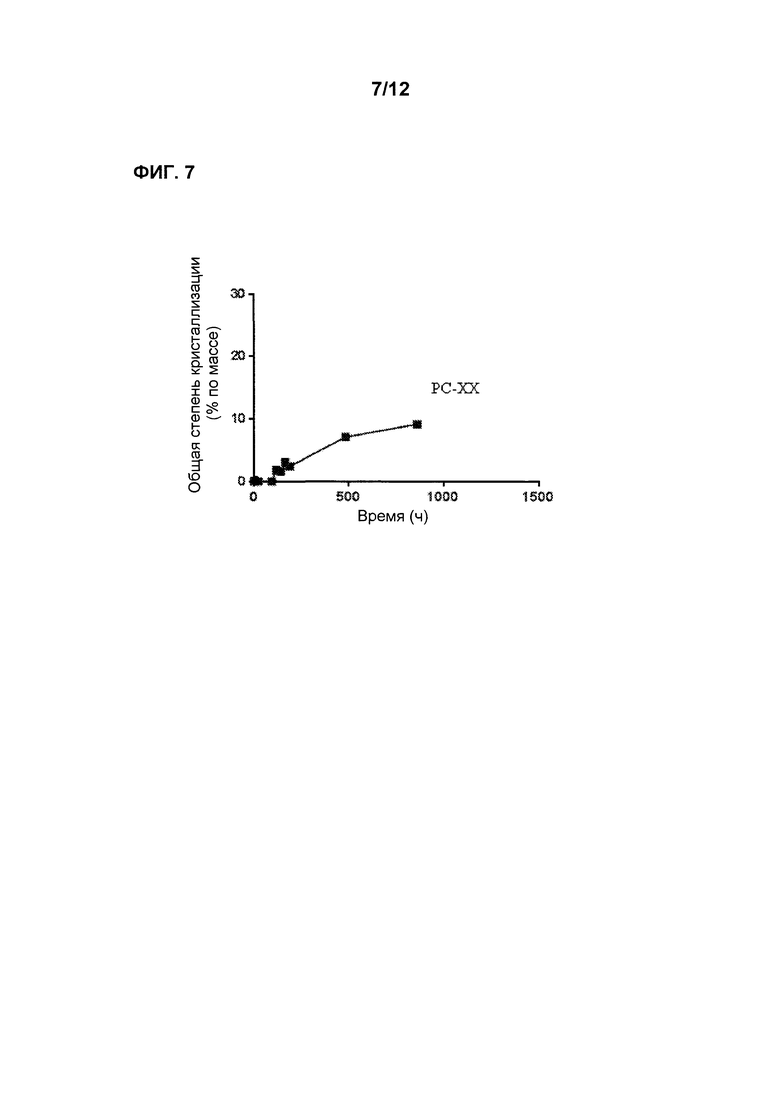
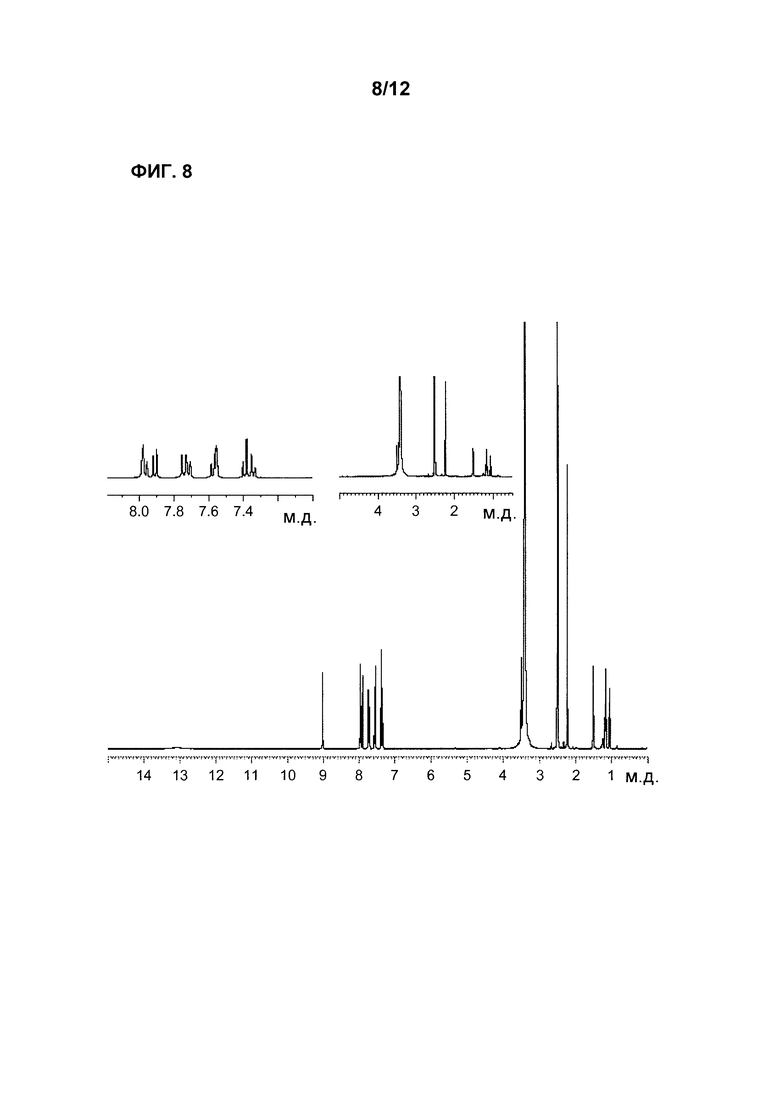
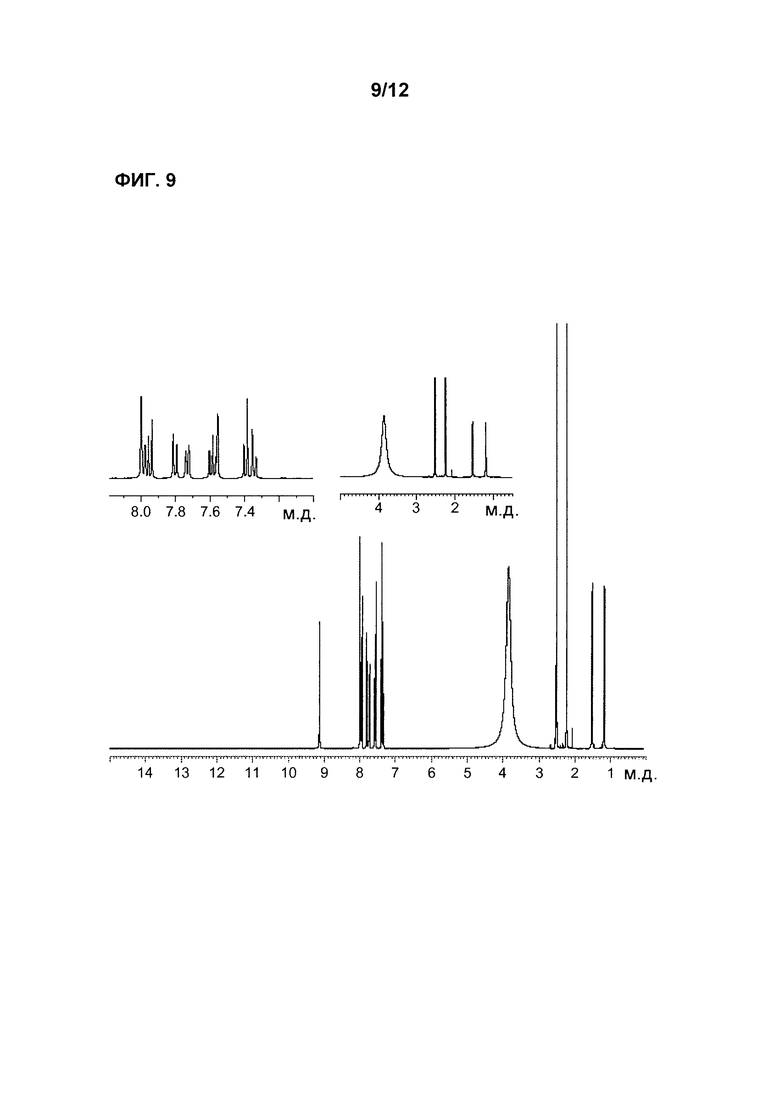
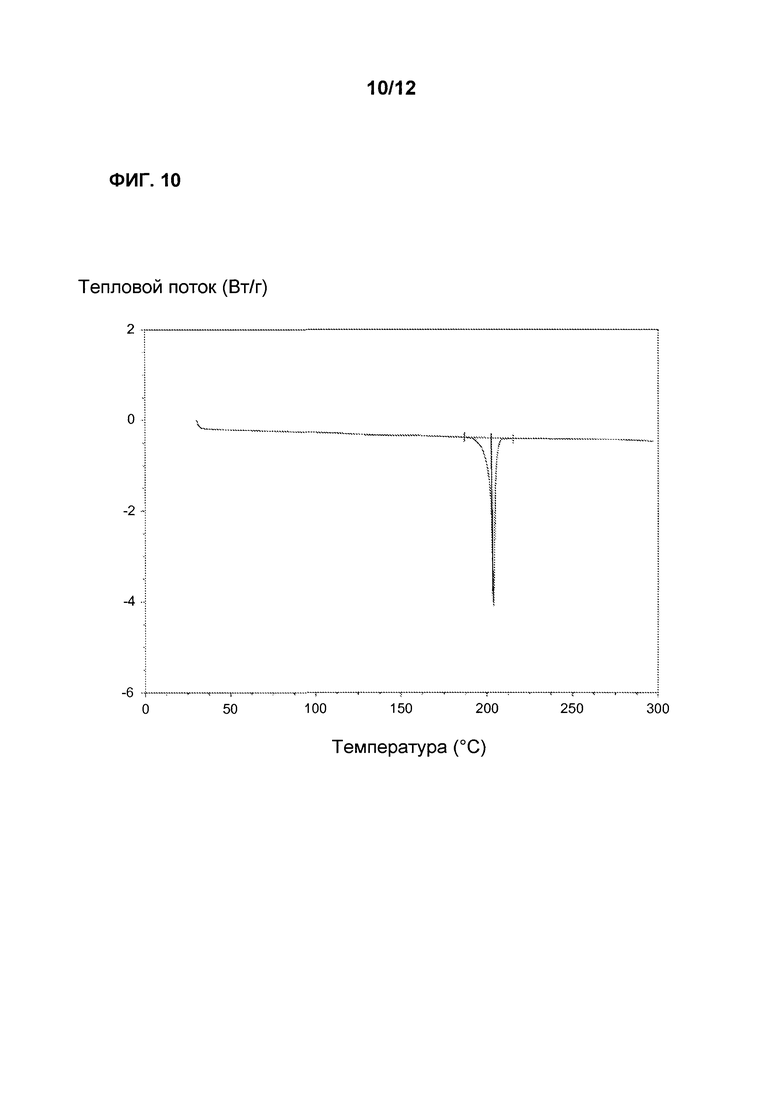


Раскрыты фармацевтическая композиция, включающая 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамидо)-3-метилпиридин-2-ил)бензойную кислоту (Соединение 1) в Форме I и твердую дисперсию, включающую по существу аморфный N-(5-гидрокси-2,4-дитрет-бутил-фенил)-4-оксо-1H-хинолин-3-карбоксамид (Соединение 2), способы лечения, уменьшения тяжести или симптоматического лечения заболеваний, опосредованных CFTR, таких как кистозный фиброз, способы производства, способы введения и их наборы. 7 н. и 36 з.п. ф-лы, 7 табл., 12 ил.
1. Способ лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающий введение пациенту 400 мг 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамидо)-3-метилпиридин-2-ил)бензойной кислоты (Соединение 1) Формы I и 250 мг по существу аморфного N-(5-гидрокси-2,4-дитрет-бутил-фенил)-4-оксо-1H-хинолин-3-карбоксамида (Соединение 2) каждые 12 часов;
где Соединение 1 Формы I характеризуется по меньшей мере одним пиком, имеющим значение 2θ в интервале, ±0,2 градуса, выбранном из 15,2-15,6 градусов; 16,1-16,5 градусов и 14,3-14,7 градусов в рентгеновской порошковой дифрактометрии,
где по существу аморфное Соединение 2 имеет степень кристаллизации менее 15% и
где пациент является гомозиготным по мутации ΔF508 в гене трансмембранного регулятора кистозного фиброза (CFTR).
2. Способ по п. 1, где 400 мг Соединения 1 Формы I и 250 мг по существу аморфного Соединения 2 вводятся двумя таблетками, каждая из которых содержит 200 мг Соединения 1 и 125 мг по существу аморфного Соединения 2.
3. Способ по п. 1 или 2, где Соединение 1 Формы I характеризуется пиком, имеющим значение 2θ в интервале, ±0,2 градуса, 16,1-16,5 градусов в рентгеновской порошковой дифрактометрии.
4. Способ по п. 3, где Соединение 1 Формы I характеризуется пиком, имеющим значение 2θ, ±0,2 градуса, при 16,3 градусах в рентгеновской порошковой дифрактометрии.
5. Способ по п. 1 или 2, где Соединение 1 Формы I характеризуется пиком, имеющим значение 2θ в интервале, ±0,2 градуса, 14,3-14,7 градусов в рентгеновской порошковой дифрактометрии.
6. Способ по п. 5, где Соединение 1 Формы I характеризуется пиком, имеющим значение 2θ, ±0,2 градуса, при 14,5 градусах в рентгеновской порошковой дифрактометрии.
7. Способ по п. 1 или 2, где Соединение 1 Формы I характеризуется пиком, имеющим значение 2θ в интервале, ±0,2 градуса, 15,2-15,6 градусов в рентгеновской порошковой дифрактометрии.
8. Способ по п. 7, где Соединение 1 Формы I характеризуется пиком, имеющим значение 2θ, ±0,2 градуса, при 15,4 градусов в рентгеновской порошковой дифрактометрии.
9. Способ по п. 1 или 2, где Соединение 1 Формы I дополнительно характеризуется пиком, имеющим значение 2θ в интервале, ±0,2 градуса, 17,6-18,0 градусов в рентгеновской порошковой дифрактометрии.
10. Способ по п. 1 или 2, где Соединение 1 Формы I дополнительно характеризуется пиком, имеющим значение 2θ в интервале, ±0,2 градуса, 7,6-8,0 градусов в рентгеновской порошковой дифрактометрии.
11. Способ по п. 1 или 2, где Соединение 1 Формы I характеризуется по меньшей мере одним пиком, имеющим значение 2θ, ±0,2 градуса, выбранным из 14,41 градусов, 14,64 градусов, 15,23 градусов, 16,11 градусов, 17,67 градусов, 19,32 градусов, 21,67 градусов, 23,40 градусов, 23,99 градусов, 26,10 градусов и 28,54 градусов в рентгеновской порошковой дифрактометрии.
12. Способ по п. 1 или 2, где Соединение Формы 1 характеризуется дифракционной рентгенограммой по существу схожей с Фиг. 1.
13. Способ по п. 1 или 2, где Соединение 1 Формы I характеризуется по меньшей мере одним пиком, имеющим значение 2θ, ±0,2 градуса, выбранным из 7,83 градусов, 14,51 градусов, 14,78 градусов, 15,39 градусов, 16,26 градусов, 16,62 градусов, 17,81 градусов, 21,59 градусов, 23,32 градусов, 24,93 градусов и 25,99 градусов в рентгеновской порошковой дифрактометрии.
14.Способ по п. 1 или 2, где Соединение Формы 1 характеризуется дифракционной рентгенограммой по существу схожей с Фиг. 2.
15. Способ лечения, уменьшения тяжести или симптоматического лечения кистозного фиброза у пациента, включающий введение пациенту 400 мг 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамидо)-3-метилпиридин-2-ил)бензойной кислоты (Соединение 1) Формы I и 250 мг по существу аморфного N-(5-гидрокси-2,4-дитрет-бутил-фенил)-4-оксо-1H-хинолин-3-карбоксамида (Соединение 2) каждые 12 часов;
где Соединение 1 Формы I характеризуется моноклинной кристаллической системой и P21/n группой симметрии и имеет следующие постоянные решетки:
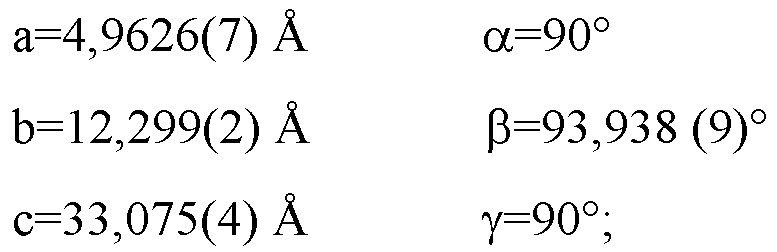
где по существу аморфное Соединение 2 имеет степень кристаллизации менее 15% и
где пациент является гомозиготным по мутации ΔF508 в гене трансмембранного регулятора кистозного фиброза (CFTR).
16. Способ по п. 15, где 400 мг Соединения 1 Формы I и 250 мг по существу аморфного Соединения 2 вводятся двумя таблетками, каждая из которых содержит 200 мг Соединения 1 и 125 мг по существу аморфного Соединения 2.
17. Фармацевтическая композиция для лечения или уменьшения тяжести кистозного фиброза, включающая 200 мг 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамидо)-3-метилпиридин-2-ил)бензойной кислоты (Соединение 1) Формы I и 125 мг по существу аморфного N-(5-гидрокси-2,4-дитрет-бутил-фенил)-4-оксо-1H-хинолин-3-карбоксамида (Соединение 2);
где Соединение 1 Формы I характеризуется по меньшей мере одним пиком, имеющим значение 2θ в интервале, ±0,2 градуса, выбранном из 15,2-15,6 градусов; 16,1-16,5 градусов и 14,3-14,7 градусов в рентгеновской порошковой дифрактометрии, и
где по существу аморфное Соединение 2 имеет степень кристаллизации менее 15%.
18. Фармацевтическая композиция для лечения или уменьшения тяжести кистозного фиброза, включающая 200 мг 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамидо)-3-метилпиридин-2-ил)бензойной кислоты (Соединение 1) Формы I и 125 мг по существу аморфного N-(5-гидрокси-2,4-дитрет-бутил-фенил)-4-оксо-1H-хинолин-3-карбоксамида (Соединение 2);
где Соединение 1 Формы I характеризуется моноклинной кристаллической системой и P21/n группой симметрии и имеет следующие постоянные решетки:
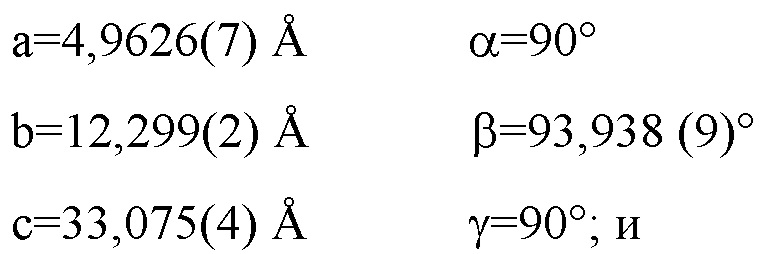
где по существу аморфное Соединение 2 имеет степень кристаллизации менее 15%.
19. Фармацевтическая композиция по п. 17 или 18, дополнительно содержащая:
микрокристаллическую целлюлозу в количестве 20-30 процентов по массе;
кроскармеллозу натрия в количестве 3-10 процентов по массе;
лаурилсульфат натрия в количестве 0,5-2 процентов по массе;
поливинилпирролидон в количестве 0-5 процентов по массе и
стеарат магния в количестве 0,5-2 процентов по массе.
20. Фармацевтическая композиция по любому из пп. 17-19, дополнительно включающая краситель или воск.
21. Фармацевтическая композиция по любому из пп. 17-19, где фармацевтическая композиция является твердой пероральной фармацевтической композицией.
22. Фармацевтическая композиция по п. 21, где твердая пероральная фармацевтическая композиция является таблеткой.
23. Фармацевтическая композиция по п. 22, где таблетка содержит:
Соединение 1 Формы I в количестве 35% по массе;
твердую дисперсию, включающую по существу аморфное Соединение 2, в количестве 28% по массе;
микрокристаллическую целлюлозу в количестве 26% по массе;
кроскармеллозу натрия в количестве 6% по массе;
лаурилсульфат натрия в количестве 1% по массе;
поливинилпирролидон в количестве 3% по массе и
стеарат магния в количестве 1% по массе.
24. Фармацевтическая композиция по п. 22, где таблетка содержит:
Соединение 1 Формы I в количестве 200 мг;
твердую дисперсию, включающую по существу аморфное Соединение 2 в количестве 156 мг;
микрокристаллическую целлюлозу в количестве 150 мг;
кроскармеллозу натрия в количестве 34 мг;
лаурилсульфат натрия в количестве 4 мг;
поливинилпирролидон в количестве 15 мг и
стеарат магния в количестве 6 мг.
25. Фармацевтическая композиция по п. 22, где таблетка содержит:
Соединение 1 Формы I в количестве 34% по массе;
твердую дисперсию, включающую по существу аморфное Соединение 2, в количестве 27% по массе;
микрокристаллическую целлюлозу в количестве 25% по массе;
кроскармеллозу натрия в количестве 6% по массе;
лаурилсульфат натрия в количестве 1% по массе;
поливинилпирролидон в количестве 3% по массе;
стеарат магния в количестве 1% по массе и
краситель в количестве 3% по массе.
26. Фармацевтическая композиция по п. 22, где таблетка содержит:
Соединение 1 Формы I в количестве 200 мг;
твердую дисперсию, включающую по существу аморфное Соединение 2 в количестве 156 мг;
микрокристаллическую целлюлозу в количестве 150 мг;
кроскармеллозу натрия в количестве 34 мг;
лаурилсульфат натрия в количестве 4 мг;
поливинилпирролидон в количестве 15 мг;
стеарат магния в количестве 6 мг и
краситель в количестве 17 мг.
27. Фармацевтическая композиция по п. 22, где таблетка содержит:
Соединение 1 Формы I в количестве 200 мг;
по существу аморфное Соединение 2 в количестве 125 мг;
микрокристаллическую целлюлозу в количестве 150 мг;
кроскармеллозу натрия в количестве 34 мг;
лаурилсульфат натрия в количестве 4 мг;
поливинилпирролидон в количестве 15 мг;
стеарат магния в количестве 6 мг и
краситель в количестве 17 мг.
28. Фармацевтическая композиция по п. 22, где таблетка содержит:
Соединение 1 Формы I в количестве 20-40% по массе;
твердую дисперсию, включающую по существу аморфное Соединение 2, в количестве 30-40% по массе;
микрокристаллическую целлюлозу в количестве 20-30% по массе;
кроскармеллозу натрия в количестве 1-10% по массе;
лаурилсульфат натрия в количестве 0,1-1% по массе;
поливинилпирролидон в количестве 1-5% по массе и
стеарат магния в количестве 0,5-1,5% по массе.
29. Фармацевтическая композиция по любому из пп. 17 или 19-28, где Соединение 1 Формы I характеризуется пиком, имеющим значение 2θ в интервале, ±0,2 градуса, 16,1-16,5 градусов в рентгеновской порошковой дифрактометрии.
30. Фармацевтическая композиция по п. 29, где Соединение 1 Формы I характеризуется пиком, имеющим значение 2θ, ±0,2 градуса, при 16,3 градусах в рентгеновской порошковой дифрактометрии.
31. Фармацевтическая композиция по любому из пп. 17 или 19-28, где Соединение 1 Формы I характеризуется пиком, имеющим значение 2θ в интервале, ±0,2 градуса, 14,3-14,7 градусов в рентгеновской порошковой дифрактометрии.
32. Фармацевтическая композиция по п. 31, где Соединение 1 Формы I характеризуется пиком, имеющим значение 2θ, ±0,2 градуса, при 14,5 градусах в рентгеновской порошковой дифрактометрии.
33. Фармацевтическая композиция по любому из пп. 17 или 19-28, где Соединение 1 Формы I характеризуется пиком, имеющим значение 2θ в интервале, ±0,2 градуса, 15,2-15,6 градусов в рентгеновской порошковой дифрактометрии.
34. Фармацевтическая композиция по п. 33, где Соединение 1 Формы I характеризуется пиком, имеющим значение 2θ, ±0,2 градуса, при 15,4 градусов в рентгеновской порошковой дифрактометрии.
35. Фармацевтическая композиция по любому из пп. 17 или 19-28, где Соединение 1 Формы I дополнительно характеризуется пиком, имеющим значение 2θ в интервале, ±0,2 градуса, 17,6-18,0 градусов в рентгеновской порошковой дифрактометрии.
36. Фармацевтическая композиция по любому из пп. 17 или 19-28, где Соединение 1 Формы I дополнительно характеризуется пиком, имеющим значение 2θ в интервале, ±0,2 градуса, 7,6-8,0 градусов в рентгеновской порошковой дифрактометрии.
37. Фармацевтическая композиция по любому из пп. 17 или 19-28, где Соединение 1 Формы I характеризуется по меньшей мере одним пиком, имеющим значение 2θ, ±0,2 градуса, выбранным из 14,41 градусов, 14,64 градусов, 15,23 градусов, 16,11 градусов, 17,67 градусов, 19,32 градусов, 21,67 градусов, 23,40 градусов, 23,99 градусов, 26,10 градусов и 28,54 градусов в рентгеновской порошковой дифрактометрии.
38. Фармацевтическая композиция по любому из пп. 17 или 19-28, где Соединение Формы 1 характеризуется дифракционной рентгенограммой по существу схожей с Фиг. 1.
39. Фармацевтическая композиция по любому из пп. 17 или 19-28, где Соединение 1 Формы I характеризуется по меньшей мере одним пиком, имеющим значение 2θ, ±0,2 градуса, выбранным из 7,83 градусов, 14,51 градусов, 14,78 градусов, 15,39 градусов, 16,26 градусов, 16,62 градусов, 17,81 градусов, 21,59 градусов, 23,32 градусов, 24,93 градусов и 25,99 градусов в рентгеновской порошковой дифрактометрии.
40. Фармацевтическая композиция по любому из пп. 17 или 19-28, где Соединение Формы 1 характеризуется дифракционной рентгенограммой по существу схожей с Фиг. 2.
41. Способ получения фармацевтической композиции по п. 17 или 18, включающий влажное гранулирование следующих компонентов:
а. Соединения 1 Формы I;
б. твердой дисперсии, включающей по существу аморфное Соединение 2;
в. наполнителя;
г.разрыхлителя;
д. сурфактанта и
е. связывающего вещества.
42. Способ получения фармацевтической композиции по п. 17 или 18, включающий прессование:
i) множества гранулярных фармацевтических композиций, включающих следующие компоненты:
а. Соединение 1 Формы I;
б. твердую дисперсию, включающую по существу аморфное Соединение 2;
в. наполнитель;
г. разрыхлитель;
д. сурфактант и
е. связывающее вещество;
ii) разрыхлитель;
iii) наполнитель и
iv) смазывающее вещество.
43. Непрерывный процесс получения фармацевтической композиции по п. 22, где приготовление таблетки включает стадии:
а) смешивания Соединения 1 Формы I, твердой дисперсии, включающей по существу аморфное Соединение 2, наполнителя и разрыхлителя в смесителе для образования смеси;
б) приготовления грануляционного раствора с водой, связывающим веществом и сурфактантом;
в) подачи смеси из а) в непрерывный двухшнековый гранулятор при добавлении грануляционного раствора из б) для образования гранул;
г) высушивания гранул из в) и их перемалывания;
д) перемешивания перемолотых гранул из г) с наполнителем, разрыхлителем и смазывающим веществом для образования смеси и
е) прессование смеси из д) в таблетку.
| WO 2010019239 A2, 18.02.2010 | |||
| WO 2009073757 A1, 11.06.2009 | |||
| US 2006074075 A1, 06.04.2006 | |||
| WO 2008137787 A2, 13.11.2008 | |||
| WO 2004047974 A1, 10.06.2004 | |||
| Андруз Дж | |||
| и др | |||
| Введение в химию окружающей среды, - М.: Мир, 1999 | |||
| Абрамзон А.А., Поверхностно-активные вещества: Свойства и применение, 2-е изд | |||
| - Л., 1981 | |||
| Харкевич Д.А | |||
| Фармакология: Учебник, 9-е издание, М.: ГЭОТАР-Медиа, 2006 | |||
| Van Goor F | |||
| et al | |||
| Способ выделения сульфокислот из нефтяных масел | 1913 |
|
SU508A1 |

