Изобретение относится к вышеуказанному способу, как изложено в ограничительной части п. 1 формулы изобретения.
В уровне техники известно большое количество процессов полимеризации, например для ММА (метилметакрилат) или ПММА (полиметилметакрилат). Здесь можно отметить, в частности, WO 2004/072131, DE 10 2005 001 802 А1 или ЕР 1 590 075 А1. Эти документы приведены в качестве примера.
В этом документе, в частности, однако также только в качестве примера, рассматривают ПЛА (полилактид). ПЛА представляет собой очень перспективный полимер, так как его получают из сырья НЕ-нефтяного происхождения, однако в принципе, по свойствам его можно сравнить со сложным полиэфиром (СПЭ). Проведенные до настоящего времени эксперименты показали, что процесс полимеризации идет хорошо, однако завершающий этап, основанный на равновесной реакции (деполимеризации), еще не является достаточно оптимальным. Кроме того, в существующих способах пытаются преодолеть проблемы изменения цвета, обусловленные температурой.
Задачей изобретения является разработка предпочтительно двухступенчатого способа вышеуказанного типа, при котором полимеризацию, и в особенности удаление летучих, или завершающий этап, в совокупности осуществляют оптимальным образом.
Указанная задача решается посредством способа осуществления процесса полимеризации, где на первой стадии осуществляют (со)полимеризацию мономера(ов), а на второй стадии осуществляют разделение продукта и мономеров, олигомеров, продуктов реакции, а также добавок и растворителей, характеризующегося тем, что перед и/или на второй стадии, т.е. на завершающем этапе, в реакционную смесь добавляют вещество, с помощью которого осуществляют остановку полимеризации и, в качестве второй операции, отпарку и/или температурное воздействие, причем остановку полимеризации и/или удаление избыточного мономера с помощью отпаривающего агента осуществляют с использованием одного и того же вещества в конечной секции, и при использовании для удаления мономера, вещество добавляют в конечную секцию в виде жидкости в одном месте или нескольких местах, распределяя его по всей длине секции, и используемое вещество представляет собой спирт или его производные, такой как октанол, изопетнанол, циклогексанол, этиленгликоль, изоамиловый спирт.
В результате добавления вещества (веществ) равновесие реакции может смещаться в сторону полимеров, и/или скорость реакции с образованием мономера может замедляться или реакция может останавливаться.
С помощью вещества, используемого для удаления мономера, можно осуществлять отпарку мономера из процесса посредством снижения парциального давления, при этом парциальное давление мономера при данной температуре может находиться ниже такового при температуре кипения мономера.
Вещество, используемое для удаления мономера, может служить для транспортировки мономера из конечной секции.
Вещество, используемое для удаления мономера, в условиях технологического процесса может испаряться на поверхности реакционной смеси, действуя таким образом в качестве агента для испарительного охлаждения.
Вещество, используемое для удаления мономера, в концентрации от 0,01 до 10%, предпочтительно, от 0,2 до 2%, может частично оставаться в полимере и служить в качестве ингибитора полимеризации.
Вещество, используемое для удаления мономера, в концентрации от 0,01 до 10%, предпочтительно, от 0,2 до 2%, может частично оставаться в полимере и действовать в качестве функционализирующего агента.
Температура полимера в конечной секции, благодаря добавленному веществу, может постоянно сохраняться в диапазоне от 175°C до 225°C, предпочтительно, от 180°C до 195°C.
Смесь мономера и отпаривающего агента можно выпускать из конечной секции в виде парообразного потока, и последующее разделение мономера и отпаривающего агента может быть проведено, например, за счет разности температур кипения, или посредством осаждения мономера в отпаривающем агенте.
Очищенный отпаривающий агент можно повторно использовать в процессе на завершающем этапе.
Остановку полимеризации и удаление мономера можно осуществлять с помощью одного и того же вещества в дегазационном смесителе. Смеситель может иметь один или более валов. При смешивании посредством более чем одного вала, указанные валы могут вращаться в разных или одном направлении, а также при различном и одинаковом числе оборотов.
Смесители можно эксплуатировать в непрерывном режиме.
Смесители могут обеспечивать интенсивное обновление наружной поверхности.
Смеситель можно эксплуатировать при рабочем давлении от 100 до 200000 Па абс.(от 1 до 2000 мбар абс.), предпочтительно, от 500 до 50000 Па абс.(от 5 до 500 мбар абс).
Вещество можно добавлять в полимер в конечной секции в соотношении от 0,1 к 1 до 1 к 1.
Вещество при добавлении в дегазационный смеситель можно распределять по его длине.
В настоящее время известны новые сведения о веществах, применяемых прежде всего на завершающем этапе производства каучуков, чувствительных к температуре, которые обеспечивают турбоэффект, в результате чего радикально уменьшается время дегазации, и одновременно резко снижается остаточное содержание растворителя.
Важно, чтобы применяемый химически активный ингибитор полимеризации одновременно действовал как агент для испарительного охлаждения, так и как отпаривающий агент, однако также мог оставаться в малых концентрациях в ПЛА, являлся доступным по цене и известным пользователям, а также допущенным для использования с продуктами питания (допуск Управления по контролю за пищевыми продуктами и лекарственными средствами (FDA) для других сопоставимых способов применения).
Полимеризация ПЛА является равновесной реакцией, которая при реализации данного изобретения может быть доведена до чрезвычайно высокого выхода от 90 до 100%, обычно, 91-95%, например 93% (в существующих способах выход составляет от 50 до 80%). Для достижения требуемого для применения остаточного содержания мономера (лактида) 2000 частей на миллион (ppm) или менее, следует проводить завершение процесса. Для демономеризации (на завершающем этапе) до сих пор использовали самый глубокий возможный вакуум (100 Па абс.(1 мбар абс.)), тем не менее требуемого значения 2000 ppm достигнуть не удавалось или удавалось с большим трудом.
Основная идея настоящего изобретения заключается в том, чтобы, на основе равновесной реакции между полимером и мономером, использовать перед завершающим этапом химический ингибитор полимеризации, называемый также «агентом, блокирующим концевые группы». Такими «агентами, блокирующими концевые группы» являются высокомолекулярные спирты, которые одновременно служат для функционализации.
Задача заключается в том, чтобы найти агент, блокирующий концевые группы, который обеспечит достижение целей данного изобретения. После изучения литературы и сопоставления способов получения и свойств получаемого сложного полиэфира (СПЭ) был выбран гликоль, поскольку он имеет привлекательные температуры кипения и представлен такими соединениями, как моно-, ди- и триэтиленгликоль, и, таким образом, пригоден в качестве функционализирующего агента. Благодаря температурам кипения, составляющим 157°C при 25 кПа (250 мбар), 177°C при 50 кПа (500 мбар) и 198°C при атмосферном давлении, этиленгликоль идеально подходит для того, чтобы поддерживать температуру продукта приблизительно 190°C (+/-10 K) на завершающем этапе. Однако могут также представлять интерес и другие полиспирты от С6 до С10, такие как октанол.
Из-за низкого давления пара мономера (лактида), в дополнение к технологическому давлению, следует создать очень низкое парциальное давление лактида, чтобы обеспечить перепад, достаточный для дегазации. Для этого необходимо провести испытания с необходимой аналитической корреляцией параметров. На основании экспериментальных данных для каучука известно, что необходимо примерно 0,2-0,3 кг отпаривающего агента на кг конечного полимера.
На основании экспериментальных данных для каучука также известно, что небольшое количество отпаривающего агента остается в полимере. Поэтому был проведен поиск сведений в литературе, касающихся остаточного содержания этиленгликоля в СПЭ. Содержание диэтиленгликоля в СПЭ обычно составляет 2 масс. %. Однако для высокосортных СПЭ это значение составляет 0,6 масс. % (6000 ppm!). Целевое содержание мономера в ПЛА для лактида составляет менее 2000 ppm, предпочтительно, менее 1500 ppm, или даже 1000 ppm. Так как эти значения однозначно ниже требований и эффективных значений для СПЭ, способ согласно данному изобретению представляет собой очень привлекательное решение.
Открытым вопросом остается только взаимосвязь между температурой продукта, необходимым вакуумом и достигнутыми при этом значениями содержания лактида и гликоля, что необходимо выяснить и понять на основании экспериментов.
Большим преимуществом данного способа по сравнению с известными способами является возможность соблюдать точный температурный режим на низком уровне, в результате чего равновесная реакция смещается в сторону полимера, и деградация, а вместе с ней изменение цвета, могут быть минимизированы или полностью предотвращены. Благодаря более низким температурам можно при меньшем количестве катализатора получить более стабильный ПЛА с более длинной цепью, т.е. более качественный продукт.
Другими возможными применениями являются полимеризация акриловой кислоты в полиакриловую кислоту и получение сополимеров ММА и акриловой кислоты, используемых в качестве клеев без растворителей. В последнем случае предложенный способ позволяет избежать смещения мономерного состава.
Полимеризация с получением эластомеров, например бутадиена в полибутадиеновый каучук (PBR), стирола и бутадиена в стирол-бутадиеновый каучук (SBR), а также изопрена в полиизопреновый каучук (ИК) или изопрена и изобутилена в бутилкаучук (ИИК) может быть осуществлена с высокой эффективностью, и, следовательно, в том же процессе может протекать деполимеризация.
Кроме того, успешно испытана полимеризация янтарной кислоты (янтарная кислота, сукцинол или бутандиовая кислота) в полимеры на основе биосырья.
Описанный процесс согласно изобретению например применяется для полимеризации бутадиена с получением полибутадиенового каучука (PBR). Процесс выполняется под давлением 3-4 бар абс. при температурах до 80°C и конверсии мономеров до 80%. В последующей демономеризации используется изобретательский замысел с обновлением поверхности, останавливающего вещества и охлаждения испарением, так что полимер не деградирует, но сохраняет молекулярно-массовое распределение, полученное при полимеризации. Мономерный бутадиен должен быть удален до остаточного содержания меньше 10 ppm, для чего используются различные низко- и высококипящие вещества.
Процесс согласно изобретению успешно протестирован для полимеризации акриловой кислоты в полиакриловую кислоту, сополимеризации метилметакрилата (ММА) и акриловой кислоты, полимеризации бутадиена в полибутадиен, сополимеризации бутадиена и стирола, сополимеризации изопрена и изобутилена и полимеризации янтарной кислоты.
Далее изобретение проиллюстрировано конкретными примерами его осуществления. Представленные примеры являются только иллюстрацией получения полимера, не ограничивающей изобретение.
Газохроматографическое определение дилактида, этиленгликоля и октанола в образцах полилактида (PLA)
1. Постановка задачи
Для определения в PLA остаточного мономера дилактида, образцы PLA растворяли в хлороформе и осаждали изопропанолом. В экстрактах определяли количественное содержание дилактида методом газовой хроматографии. Кроме того, образцы проверяли на присутствие этиленгликоля и октанола.
2. Экспериментальная часть
2.1. Образцы
19 растворов из 9 образцов PLA
2-1
2-2
2-3
3-1
3-2
3-3
4-1
5-1
PLA Nature Works 4042 D
7
2.2. Реактивы.
Дилактид, первоначальный раствор (внутр.)
этиленгликоль, безводный, 99,8%, Fa. Aldrich
н-октанол, 99%, Fa. Sigma-Aldrich
хлороформ, высшей очистки, J.T. Baker
изопропанол, высшей очистки, J.T. Baker
2.3. ГХ система
Газовый хроматограф для работы капиллярных колонок HP 6890 с автоматическим пробоотборником 7673 в сочетании с масс-селективным детектором MSD HP 5973 с электронно-ударной ионизацией, Agilent.
Управление ПК, Chemstation.


2.4. Приготовление образцов
Растворы были отфильтрованы через 0,45 мкм нейлоновый тканевый фильтр и помещены в 1,5 мл флаконы.
2.5. Калибровка
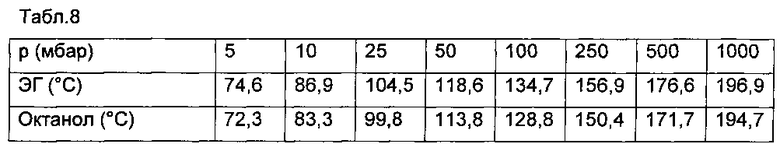
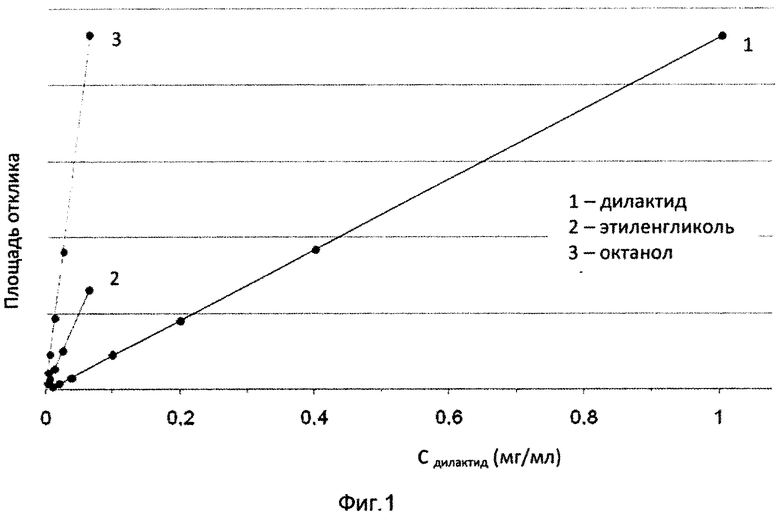
В мерные колбы объемом 50 мл поместили навески примерно 50 мг дилактида или примерно 30 мг этиленгликоля и примерно 30 мг октанола и растворили с помощью 10 мл хлороформа. Затем мерные колбы были заполнены до калибровочной метки изопропанолом. Калибровочные стандарты приготовлены путем разбавления аликвот до 50 мл. Каждый стандарт был введен три раза. Калибровочные линии (дилактид: R2=0,99996; этиленгликоль: R2=0,99971 и октанол: R2=0,99986) и соответствующие данные калибровки приведены на фиг. 1 и в табл. 3-5.
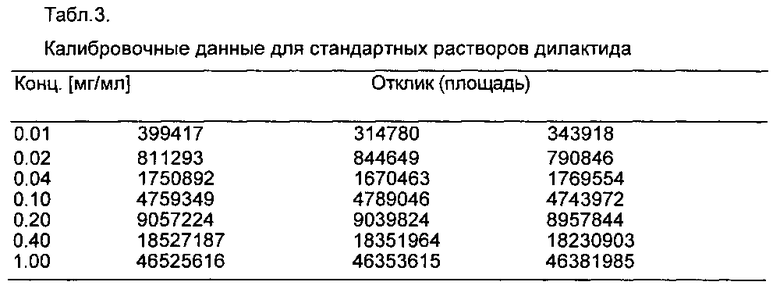
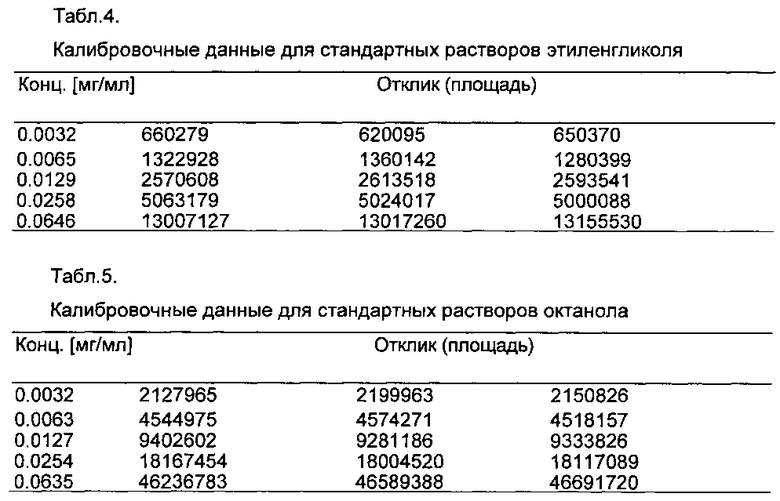
На фиг. 1 представлены калибровочные линии для стандартных растворов дилактида, этиленгликоля и октанола.
3. Результаты.
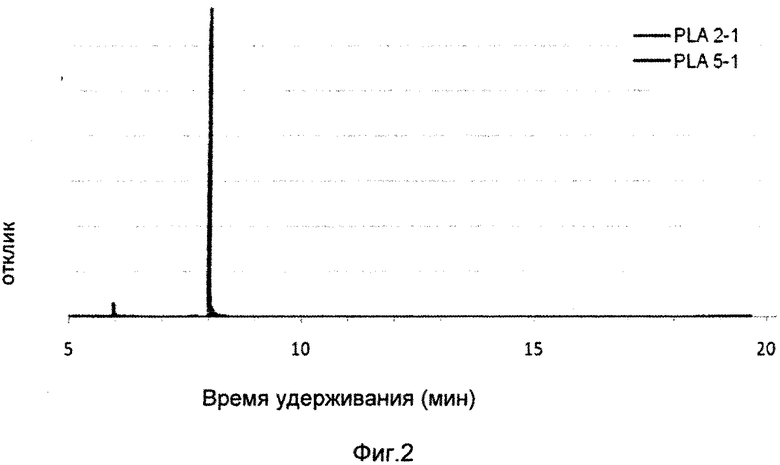
Каждый образец раствора также вводили по три раза. Была достигнута очень хорошая воспроизводимость. В тестах растворов было обнаружено, что концентрации дилактида составили от 0,3-3%, с точностью определения дилактида в образцах НОАК, будучи приблизительно ±0,1%. Концентрации дилактида в растворах экстрактов, определяемые с помощью ГХ-МС, и концентрации дилактида в пробах полимера, рассчитываемые из них, приведены в табл. 6. На фиг. 2 показан пример хроматограмм растворов с относительно высоким и низким уровнем дилактида. В дополнение к дилактиду, образец PLA 5-1 также содержит еще одно вещество (время удержания около 6 мин) - 2-октанол.
Для проведения количественного определения этиленгликоля и октанола разделительные колонки пришлось заменить ввиду неблагоприятных условий разделения и связанных с ними неблагоприятных форм пиков и чувствительности. В образце PLA 2-1 определить этиленгликоль или октанол было невозможно. Образцы 2-2, 2-3, 3-1, 3-2, 3-3 и 4-1 содержат этиленгликоль с концентрацией в диапазоне 0,01-0,7% (табл. 7). Образец PLA 5-1 содержит 0,09% 2-октанола.
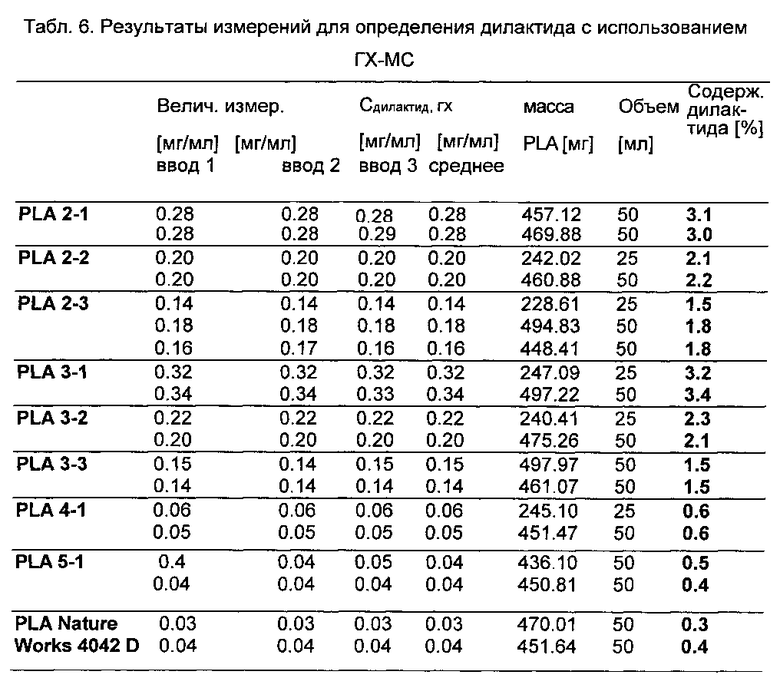
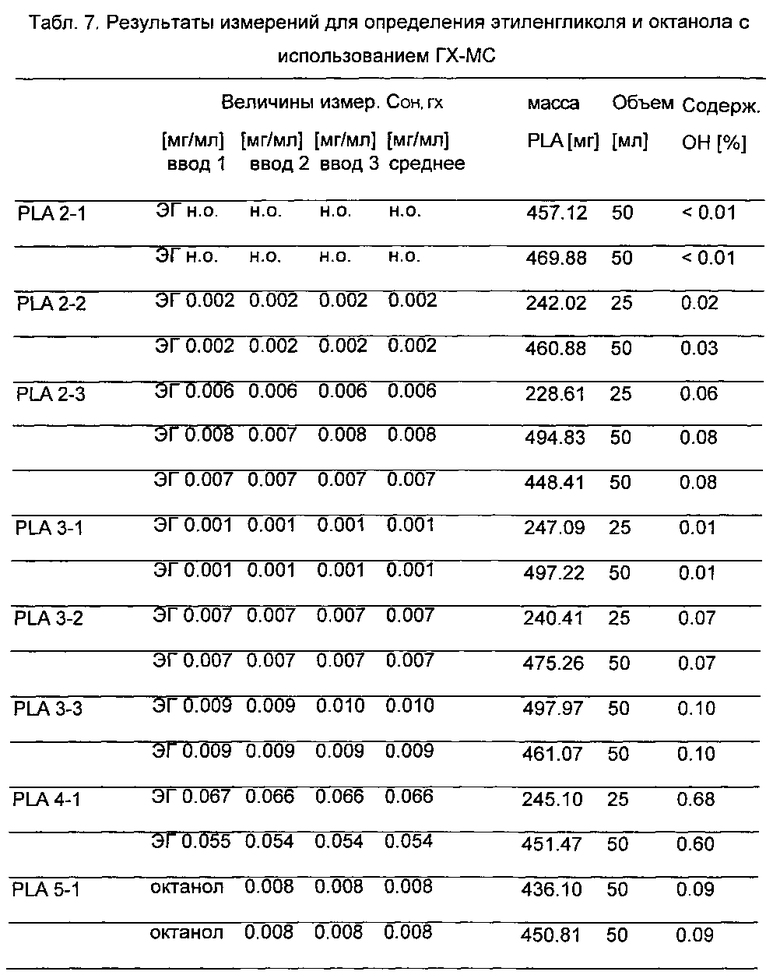
н.о. = не определено
В образце Nature Works 4042 D невозможно определить этиленгликоль и октанол (содержание <0.01%).Результаты непрерывной дегазации PLA от мономера лактида с использованием этиленгликоля и октанола в качестве отгоночного вещества
Давление/вакуум: 10-250 мбар абс.
Средняя температура: 180-200°C
Среднее время пребывания: 0,15-0,5 ч
Продукт: 180-200°C
ΔНиспар. (ЭГ): 812 кДж/кг
ΔНиспар. (октанол): 410 кДж/кг
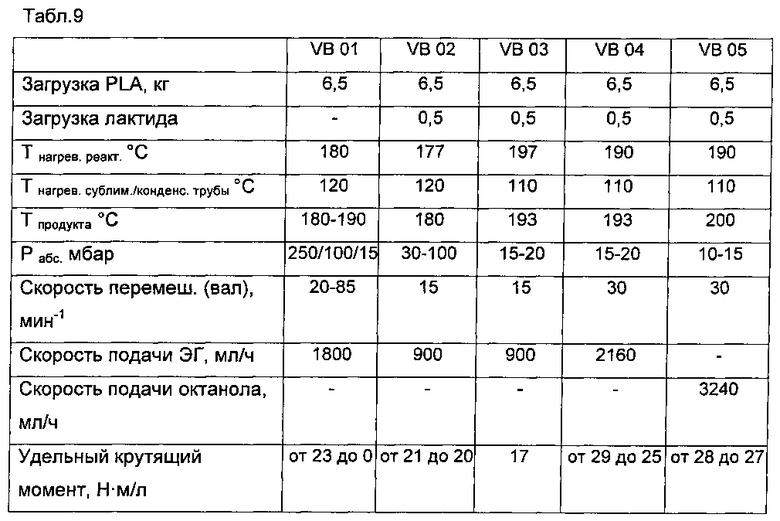
Продукт загружали в реактор CRP и дважды вакуумировали, стравливая затем азотом. Мешалки останавливали, когда крутящий момент в фазе расплава начинал расти, и позже вновь запускали.
Опыт 1 (без добавления лактида)
Атмосферное давление: после достижения температуры продукта 190°C через 15 мин отбирали первый образец - Образец 1-1.
250 мбар абс: при температуре продукта 190°C через 10 мин отбирали второй образец - Образец 1-2.
100 мбар абс: при температуре продукта 190°C через 10 мин отбирали третий образец - Образец 1-3.
15 мбар абс: при температуре продукта 190°C через 10 мин отбирали четвертый образец - Образец 1-4.
Затем начинали подавать этиленгликоль (1800 мл/ч) сверху на расплав (та же партия) при 250 мбар абс.
при температуре продукта 182°C через примерно 20-25 мин отбирали пятый образец - Образец 1-5.
при температуре продукта 176°C через примерно 20-25 мин отбирали шестой образец - Образец 1-6.
при температуре продукта 182°C через примерно 45 мин отбирали седьмой образец - Образец 1-7.
Скорость мешалки увеличивалась до конечной величины 80 об/мин ввиду потери удельного крутящего момента с течением времени. Последний образец 1-7 перерабатывали практически на холостом ходу CRP. Наиболее вероятно, продукт деградировал со временем.
Опыт 2 (с добавлением лактида / ЭГ=900 мл/ч)
После достижения температуры продукта 180°C (p=40 мбар абс.) отбирали «нулевой образец» - Образец 2-1.
Затем подавали ЭГ 900 мл/ч в течение 5 мин и отбирали второй образец (р абс. невозможно было поддерживать, оно составляло около 80 мбар ввиду частичной забивки выхода отходящего газа при десублимации) - Образец 2-2.
При той же температуре продукта 180°C еще через 5 мин (p~100 мбар абс.) отбирали третий образец - Образец 2-3.
Поддержание удельного крутящего момента на время отбора образцов на уровне 20-21 Н·м/мл.
Опыт 3 (с добавлением при более высокой температуре лактида / ЭГ=900 мл/ч /15-20 мбар абс.)
Опыт аналогичен опыту 2, но при температуре продукта 193°C.
Образец 3-1 («нулевой образец»).
Образец 3-2 (+5 мин).
Образец 3-3 (+5 мин) / Образец 3-4 (=Образец 3-3, но не охлажден в водяной бане после отбора).
В этом опыте удалось поддерживать давление 15-20 мбар.
Удельный крутящий момент можно было поддерживать постоянным (на уровне ~17 Н м/мл при более высокой температуре продукта).
При добавлении ЭГ после отбора проб и охлаждения было установлено, что продукт превращается в свободно сыпучий порошок при ~115°C, что облегчает чистку аппарата в конце опыта.
Опыт 4 (повторение опыта с добавлением при 193°C лактида, при двойной скорости 30 мин-1 / ЭГ=2160 мл/ч / 15-20 мбар абс.)
Чтобы не прерывать процесс для отбора проб, конечную пробу отбирали только после 30 мин суммарной отпарки.
Образец 4-1 (2 х конечная проба через 30 мин с охлаждением в воде).
Образец 4-2 (1 х конечная проба через 30 мин без охлаждения в воде).
Удельный крутящий момент можно было поддерживать на уровне 25-29 Н·м/мл в течение времени пребывания 30 мин.
Опыт 5 (сравнительный опыт с добавлением лактида при более высокой температуре продукта / октанол = 3240 мл/ч / 30 об/мин /10-15 мбар)
Вновь, чтобы не прерывать процесс для отбора проб, конечную пробу отбирали только после 30 мин суммарной отпарки.
Образец 5-1 (конечная проба через 30 мин с охлаждением в воде).
Образец 5-2 (1 х конечная проба через 30 мин без охлаждения в воде).
Ввиду более низкого ΔНиспар. октанола, подаваемое для отпарки количество необходимо увеличить.
Конечная температура продукта достигла 200°C (для снижения достигаемой конечной температуры количество октанола необходимо увеличить).
Удельный крутящий момент можно было поддерживать на уровне 28-29 Н·м/мл в течение времени пребывания 30 мин.
В целом удельный крутящий момент можно было поддерживать на уровне 20-30 Н·м/мл в течение разумного времени, необходимого для использования этиленгликоля и октанола в качестве отгоночного/испарительно-охладительного вещества.
Для лучшей обработки потока отходящего газа следует использовать фазовую диаграмму лактида (при более низком вакууме лактид теряет способность к конденсации и забивает трубу отходящего газа путем десублимации).
Далее проводили анализы содержания летучих веществ, возможной деструкции молекулярной массы и т.д.
| название | год | авторы | номер документа |
|---|---|---|---|
| Испытание качества полимеризуемой молочной кислоты и способ его осуществления | 2012 |
|
RU2631503C2 |
| СПОСОБ ПОЛУЧЕНИЯ СМЕСИ ПРОИЗВОДНЫХ ЛАКТИДОВ | 2009 |
|
RU2541567C2 |
| НЕПРЕРЫВНЫЙ СПОСОБ ПОЛУЧЕНИЯ СЛОЖНОГО ПОЛИЭФИРА ИЗ ЦИКЛИЧЕСКОГО СЛОЖНОЭФИРНОГО МОНОМЕРА | 2015 |
|
RU2707743C2 |
| СПОСОБ СТАБИЛИЗАЦИИ КОМПОЗИЦИИ КОНДЕНСИРОВАННОЙ ФАЗЫ, СОДЕРЖАЩЕЙ ЦИКЛИЧЕСКИЙ СЛОЖНЫЙ ЭФИР, В ПРОЦЕССЕ ПРОИЗВОДСТВА СЛОЖНОГО ПОЛИЭФИРА ИЗ ЛАКТИДА | 2015 |
|
RU2713408C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПОЛИГИДРОКСИКАРБОНОВОЙ КИСЛОТЫ | 2011 |
|
RU2575709C2 |
| ТЕРМИЧЕСКОЕ РАЗДЕЛЕНИЕ СМЕСЕЙ МАТЕРИАЛОВ С ПОМОЩЬЮ ОСНОВНОГО ИСПАРЕНИЯ И ДЕГАЗАЦИИ В ОТДЕЛЬНЫХ СМЕСИТЕЛЬНЫХ МАШИНАХ | 2010 |
|
RU2526548C2 |
| СПОСОБ ПОЛУЧЕНИЯ БИОРАЗЛАГАЕМЫХ МЕЖМОЛЕКУЛЯРНЫХ ЦИКЛИЧЕСКИХ СЛОЖНЫХ ДИЭФИРОВ АЛЬФА-ГИДРОКСИКАРБОНОВЫХ КИСЛОТ, СПОСОБ НЕПРЕРЫВНОГО ПОЛУЧЕНИЯ ПОЛИЛАКТИДА И ПРИМЕНЕНИЕ ЭТИХ СПОСОБОВ | 2008 |
|
RU2478098C2 |
| ЖЕВАТЕЛЬНАЯ РЕЗИНКА (ВАРИАНТЫ) И ГУММИОСНОВА ЖЕВАТЕЛЬНОЙ РЕЗИНКИ (ВАРИАНТЫ) | 2012 |
|
RU2600751C2 |
| СПОСОБ ОЧИСТКИ ТЕХНОЛОГИЧЕСКИХ ПОТОКОВ ПРИ ПРОИЗВОДСТВЕ ДИЛАКТИДА ИЛИ ПОЛИЛАКТИДА | 2008 |
|
RU2471791C2 |
| ЖЕВАТЕЛЬНАЯ РЕЗИНКА И ГУММИОСНОВА | 2010 |
|
RU2532049C2 |
Изобретение относится к способу осуществления процесса полимеризации. На первой стадии осуществляют (со)полимеризацию мономера(ов), а на второй стадии осуществляют разделение продукта и мономеров, олигомеров, продуктов реакции, а также добавок и растворителей. Перед и/или на второй стадии, т.е. на завершающем этапе, в реакционную смесь добавляют вещество, с помощью которого осуществляют отпарку и/или температурное воздействие, в результате чего равновесие реакции смещается в сторону полимеров, при этом замедляется скорость реакции с образованием мономера. Указанное вещество представляет собой спирт или его производные, такие как октанол, изопентанол, циклогексанол, этиленгликоль, изоамиловый спирт. Технический результат - разработка оптимального двухступенчатого способа полимеризации. 17 з.п. ф-лы, 9 табл., 2 ил.
1. Способ осуществления процесса полимеризации, где на первой стадии осуществляют (со)полимеризацию мономера(ов), а на второй стадии осуществляют разделение продукта и мономеров, олигомеров, продуктов реакции, а также добавок и растворителей, отличающийся тем, что перед и/или на второй стадии, т.е. на завершающем этапе, в реакционную смесь добавляют вещество, с помощью которого осуществляют остановку полимеризации и, в качестве второй операции, отпарку и/или температурное воздействие, причем остановку полимеризации и/или удаление избыточного мономера с помощью отпаривающего агента осуществляют с использованием одного и того же вещества в конечной секции, и при использовании для удаления мономера, вещество добавляют в конечную секцию в виде жидкости в одном месте или нескольких местах, распределяя его по всей длине секции, и используемое вещество представляет собой спирт или его производные, такой как октанол, изопентанол, циклогексанол, этиленгликоль, изоамиловый спирт.
2. Способ по п. 1, отличающийся тем, что в результате добавления вещества (веществ) равновесие реакции смещается в сторону полимеров, и/или скорость реакции с образованием мономера замедляется или реакция останавливается.
3. Способ по п. 1, отличающийся тем, что с помощью вещества, используемого для удаления мономера, осуществляют отпарку мономера из процесса посредством снижения парциального давления, при этом парциальное давление мономера при данной температуре находится ниже такового при температуре кипения мономера.
4. Способ по п. 1, отличающийся тем, что вещество, используемое для удаления мономера, служит для транспортировки мономера из конечной секции.
5. Способ по п. 1, отличающийся тем, что вещество, используемое для удаления мономера, в условиях технологического процесса испаряется на поверхности реакционной смеси, действуя таким образом в качестве агента для испарительного охлаждения.
6. Способ по п. 1, отличающийся тем, что вещество, используемое для удаления мономера, в концентрации от 0,01 до 10%, предпочтительно, от 0,2 до 2%, частично остается в полимере и служит в качестве ингибитора полимеризации.
7. Способ по п. 1, отличающийся тем, что вещество, используемое для удаления мономера, в концентрации от 0,01 до 10%, предпочтительно, от 0,2 до 2%, частично остается в полимере и действует в качестве функционализирующего агента.
8. Способ по п. 1, отличающийся тем, что температура полимера в конечной секции, благодаря добавленному веществу, постоянно сохраняется в диапазоне от 175°C до 225°C, предпочтительно, от 180°C до 195°C.
9. Способ по п. 1, отличающийся тем, что смесь мономера и отпаривающего агента выпускают из конечной секции в виде парообразного потока, и последующее разделение мономера и отпаривающего агента может быть проведено, например, за счет разности температур кипения, или посредством осаждения мономера в отпаривающем агенте.
10. Способ по п. 9, отличающийся тем, что очищенный отпаривающий агент можно повторно использовать в процессе на завершающем этапе.
11. Способ по п. 1, отличающийся тем, что остановку полимеризации и удаление мономера осуществляют с помощью одного и того же вещества в дегазационном смесителе.
12. Способ по п. 11, отличающийся тем, что смеситель имеет один или более валов.
13. Способ по п. 12, отличающийся тем, что при смешивании посредством более чем одного вала, указанные валы могут вращаться в разных или одном направлении, а также при различном и одинаковом числе оборотов.
14. Способ п. 11, отличающийся тем, что смесители эксплуатируют в непрерывном режиме.
15. Способ по п. 11, отличающийся тем, что смесители обеспечивают интенсивное обновление наружной поверхности.
16. Способ по п. 11, отличающийся тем, что смеситель эксплуатируют при рабочем давлении от 100 до 200000 Па абс.(от 1 до 2000 мбар абс.), предпочтительно, от 500 до 50000 Па абс.(от 5 до 500 мбар абс.).
17. Способ по любому из пп. 1-16, отличающийся тем, что вещество добавляют в полимер в конечной секции в соотношении от 0,1 к 1 до 1 к 1.
18. Способ по п. 11, отличающийся тем, что вещество при добавлении в дегазационный смеситель распределяют по его длине.
| DE 102005001802 A1 06.04.2006 | |||
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |
| US 6353087 B1 05.03.02002 | |||
| СПОСОБ ПОЛИМЕРИЗАЦИИ | 1996 |
|
RU2198896C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПОЛИЭТИЛЕНА | 2004 |
|
RU2255095C1 |

