Настоящее изобретение относится к собственно магнитному гидроксиапатиту и его применениям в восстановлении костной и костно-хрящевой ткани в качестве носителя для биологических веществ и/или лекарственных средств и в качестве контрастного агента в диагностике.
Гидроксиапатит (HA), Ca10(PO4)6(OH)2, является основным минеральным компонентом костной ткани. Принимая во внимание его высокую биосовместимость и природную аффинность к биологическим веществам, гидроксиапатит обычно используют для замены/регенерации костной и костно-хрящевой ткани и в качестве носителя для белков, генов, стволовых клеток, факторов роста, активных веществ и т.д.
Известно, что гидроксиапатит имеет гексагональную кристаллическую решетку, содержащую фосфат-ионы, гидроксильные ионы и ионы кальция, причем последние с гексавалентной или тетравалентной координацией (положения 6h и 4f).
Известно также, что структура гидроксиапатита допускает различные типы ионных замещений в позициях фосфат-иона, гидроксильного иона и иона кальция без разрушения структуры.
Другими словами, гидроксиапатит является веществом, которое может быть легировано разными типами ионов, не вызывая его фазовую деструкцию.
Помимо легирования гидроксиапатита, осуществляемого с целью усиления его биомиметических свойств в отношении минеральной фазы, составляющей костную ткань, выполнялись многочисленные замещения ионами, способными придавать магнитные свойства, такими как Fe, Co, Mn и La.
В частности, Mayer et al. (Journal of Inorganic Biochemistry 1992, 45, 129-133) сообщили о синтезе гидроксиапатита, легированного ионами трехвалентного железа (Fe3+HA) с использованием Fe(NO3)3 в качестве реагента. Согласно сообщению этих авторов ионы трехвалентного железа не встраивались в решетку апатита, а присутствовали в самом апатите в форме FeOOH. Wu et al. (Nanotechnology 2007, 18, 165601-10) сообщили о синтезе гидроксиапатита, легированного ионами двухвалентного железа (Fe2+HA) с использованием 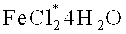
Ming Jang et al. (Review, Condensed Matter and Materials Physics, 2002, 66, 224107-224115) легировали гидроксиапатит ионами Fe2+ и Fe3+, добавляя по каплям растворы Ca(NO3)2 и Fe(NO3)2 в раствор фосфата аммония. В этой статье нет никаких сведений, указывающих на возможную собственную намагниченность гидроксиапатита.
Одно из самых существенных ограничений, связанных с использованием каркасных конструкций для регенерации костной или костно-хрящевой ткани, относится к трудности контролирования развития и скорости протекания процессов клеточной дифференциации и ангиогенеза в месте имплантации каркасной конструкции.
Этим процессам способствует быстрота миграции факторов роста костной ткани и факторов васкуляризации в место имплантации.
Контроль над миграцией специфических факторов в участок имплантации в соответствии с тем, что необходимо пациенту, и в течение длительного периода времени имеет огромное значение для способствования остеоинтеграции протеза и регенерации костной ткани и, следовательно, для выздоровления пациента.
Следовательно, в этой области существует весьма ощутимая потребность в системе доставки и высвобождения биологических веществ и лекарственных средств, которая обеспечивает контроль миграции факторов роста, факторов васкуляризации или других биологических веществ, способных ускорять остеоинтеграцию и регенерацию костной ткани. Ощущается также потребность в системе доставки лекарственного средства, которой можно точно и правильно управлять, чтобы лекарственное средство высвобождалось непосредственно, селективным образом и в соответствии с реальными качественными и количественными требованиями, только в том месте, где имеются патологические изменения.
В данной области существует также потребность в протезе для регенерации костной и костно-хрящевой ткани, который одновременно является биосовместимым и может быть управляемым и фиксируемым в конкретной позиции имплантации in vivo с помощью системы контроля, находящейся вне организма пациента, тем самым исключая использование существующих в настоящее время необходимых инвазивных фиксирующих систем.
Эту задачу решают собственно магнитный гидроксиапатит и способ его получения, которые определены в прилагаемой формуле изобретения.
Подробное описание изобретения приведено ниже и со ссылкой на прилагаемые графические материалы, в которых:
- Фиг.1 иллюстрирует морфологию образца HA, замещенного ионами железа;
- на Фиг.2 представлены результаты анализа РДА (дифракция рентгеновских лучей) образца НА, синтезированного в присутствии ионов Fe3+ при 40°C (соотношение Fe/Ca=0,20);
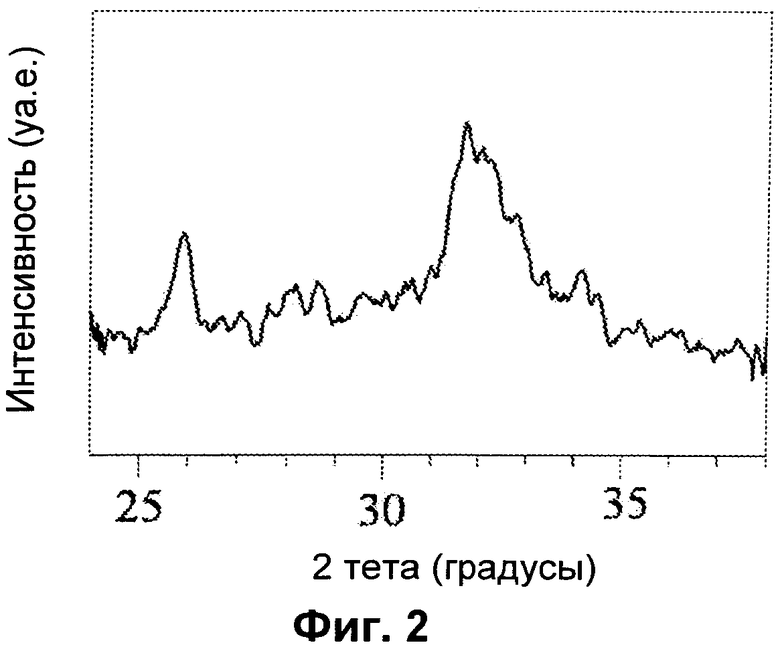
на Фиг.3 представлены рентгеновские дифрактограммы нелегированного HA и HA, синтезированного с добавлением ионов Fe2+(Fe/Ca=0,20) при разных температурах [D=25°C; E=40°C; F=60°C]; пики, отмеченные символом •, соответствуют фазе магнетита (Fe3O4);
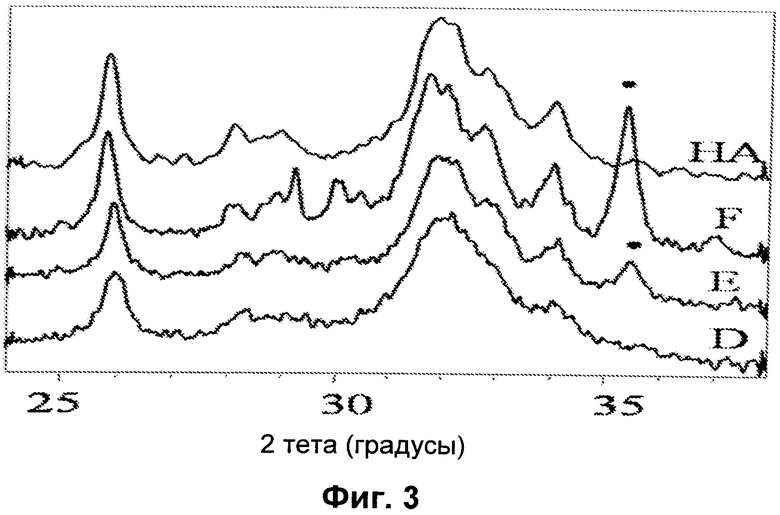
- на Фиг.4 представлена картина РДА нелегированного HA и HA, полученного с добавлением ионов Fe3+ и Fe2+(Fe/Ca=0,20), синтезированного при 40°C (H);
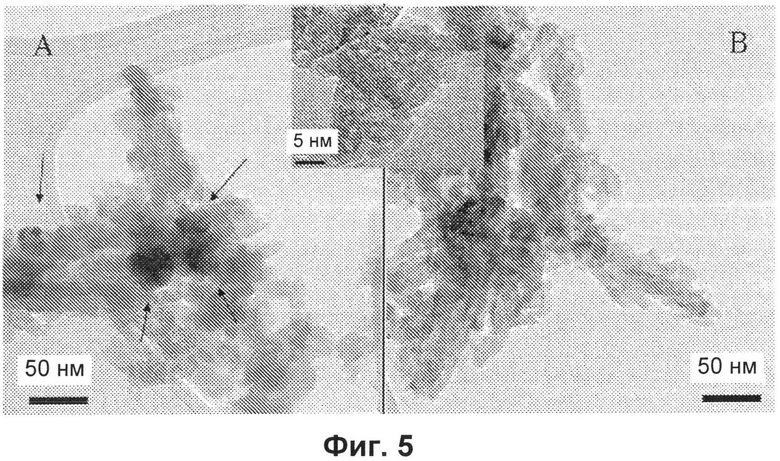
- на Фиг.5A и 5B представлены ТЭМ-изображения (изображения, полученные методом трансмиссионной электронной микроскопии) образца E (черные стрелки указывают частицы магнетита) и образца H соответственно; общее прямоугольное окно представляет собой усиленное ТЭМ-изображение, демонстрирующее наличие нанопустот в этих двух образцах;
- на Фиг.6 представлена ТЭМ-микрофотография высокого разрешения, демонстрирующая репрезентативную частицу образца E или H, наблюдаемую в направлении осей [2,0,1] HA; черная стрелка указывает направление с-оси решетки HA, полученной в результате Фурье-преобразования изображения (прямоугольное окно), а белая стрелка указывает аморфную зону частицы;
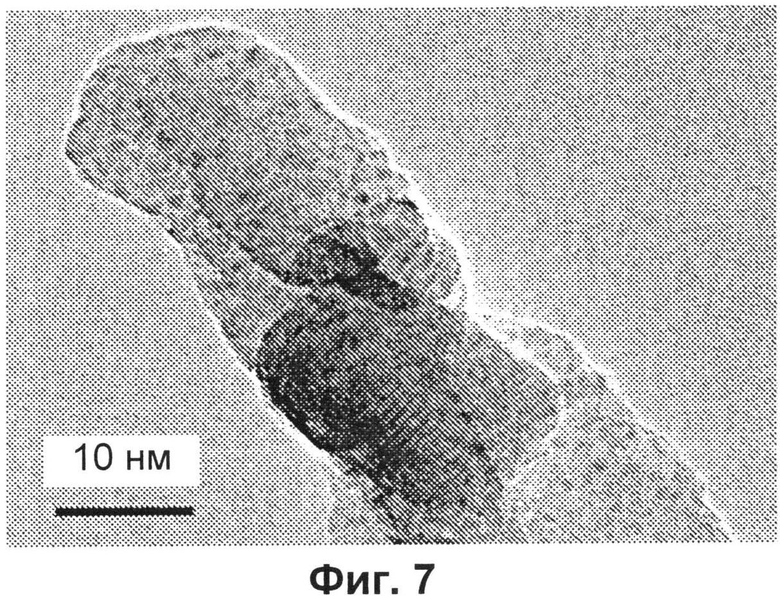
- на Фиг.7 представлено ТЭМ-изображение частицы образца E после радиационного повреждения; более темные области изображения соответствуют нанофазе, богатой железом;
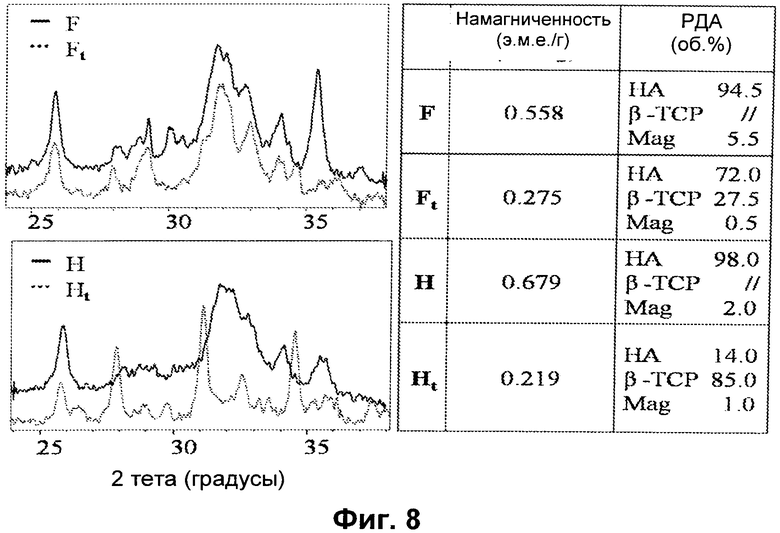
- Фиг.8 иллюстрирует соотношение значений намагниченности и картин РДА, полученных для образцов E и H;
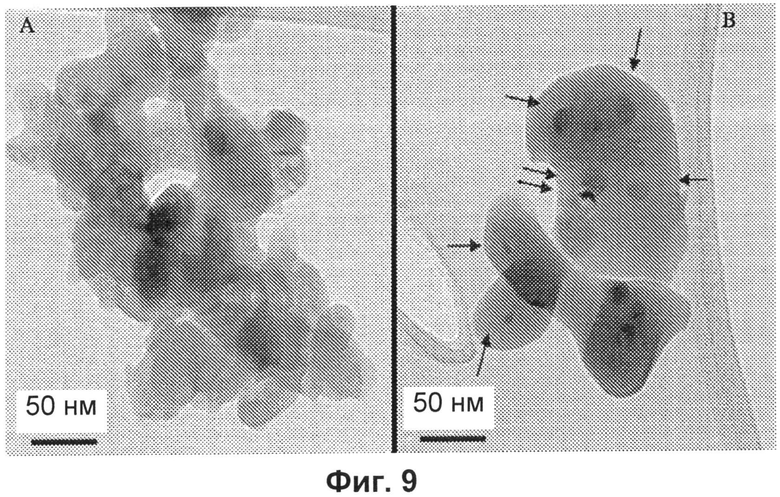
- на Фиг.9A и 9B представлены ТЭМ-изображения низкого разрешения веществ, обработанных при 700°C: A) образец Et; B) образец Ht (стрелки указывают фазу, богатую железом);
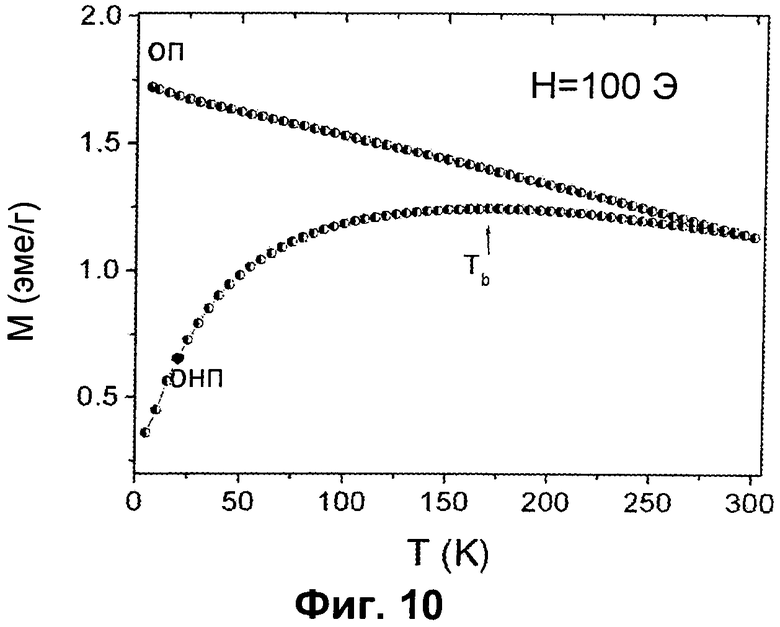
- на Фиг.10 представлены магнитные ОНП-ОП кривые для образца H в магнитном поле 100 Э; стрелка указывает температуру блокировки (Тв);
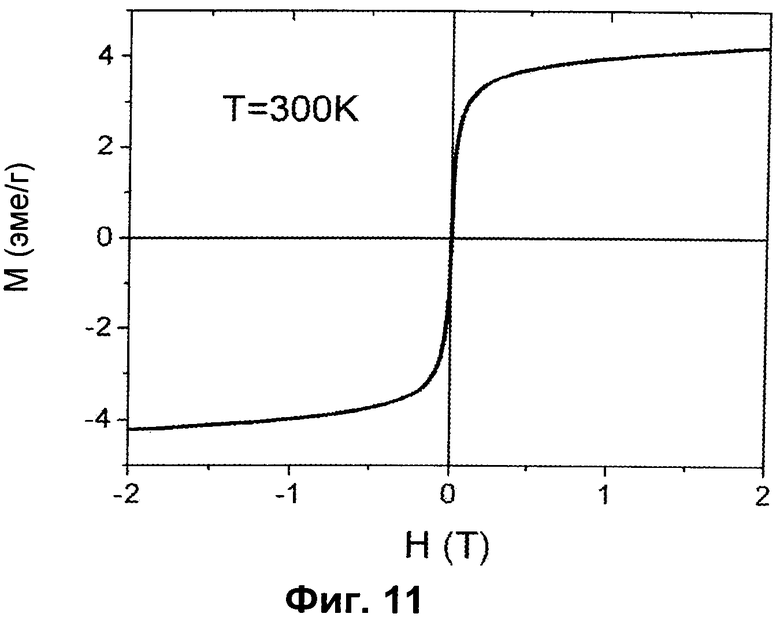
- Фиг.11 иллюстрирует магнетизм как функцию магнитного поля, приложенного при 300К, для образца H.
Настоящее изобретение относится к собственно магнитному гидроксиапатиту, характеризующемуся степенью намагниченности в пределах от 0,05 до 8 эме/г (эме=электромагнитная единица), предпочтительно от 0,1 до 5 эме/г, которая зарегистрирована в магнитном поле 34 Э.
В частности, гидроксиапатит по изобретению легирован ионами Fe2+ и Fe3+, которые частично замещают кальций в кристаллической решетке.
Соотношение Fe3+/Fe2+ в решетке гидроксиапатита находится в пределах от 1 до 4, предпочтительно от 2 до 3,5. Гидроксиапатит по изобретению является собственно магнитным, т.е. он наделен магнитными свойствами, по существу за счет легирования решетки ионами Fe2+ и Fe3+, которые замещают (частично) кальций в количестве, в соответствующих положениях, в соотношении состояние окисления/положение в решетке и в конкретных координационных состояниях, таких, при которых возникает магнетизм.
Без связи с какой-либо теорией предполагается, что магнетизм является следствием существования очень маленьких структурных доменов в решетке легированного HA, которые имеют сходство со структурой магнетита и способны активировать механизмы, ответственные за суперпарамагнитное свойство.
РДА-анализы и компьютерное моделирование на структурных моделях четко показывают, что оба вида ионов Fe находятся в положениях кальция, замещая его, и не находятся в междоузлиях в решетке HA.
Положение Ca(2) с координационным числом 6 и положение Ca(1) с координационным числом 4 заняты взаимообразно, наделяя порошки магнитной восприимчивостью.
Исследования методом ТЭМ, а также магнитные измерения подтвердили присутствие новой магнитной фазы HA вместе с нанокластерами, подобными магнетиту. Эта новая фаза представляет собой деформированный/разупорядоченный гидроксиапатит с Fe2+ в его номинальном двухвалентном состоянии и Fe3+, который имеет отклонение от его номинального трехвалентного состояния, организованные на уровне поверхности и объема и координированные для создания магнетизма в самом HA.
Гидроксиапатит по изобретению может содержать вторичные магнитные фазы (например, вторичные фазы типа магнетита) в количестве ниже примерно 3% по объему. Предпочтительно, количество вторичных магнитных фаз составляет ≤2% по объему.
Небольшое количество вторичных фаз, присутствующих совместно с гидроксиапатитом по изобретению, указывает на то, что большая часть ионов железа, использованных для легирования, замещает кальций в решетке HA, и только небольшая их часть участвует в образовании оксидов железа (подобных магнетиту), которые обладают магнитными свойствами.
Благодаря небольшому количеству вторичных магнитных фаз, например магнетита, легированный гидроксиапатит по изобретению сохраняет хорошие характеристики биосовместимости, типичные для нелегированного гидроксиапатита. Фактически, чем выше количество вторичных магнитных фаз, тем ниже биосовместимость гидроксиапатита.
Гидроксиапатит по изобретению предпочтительно имеет соотношение (Fe+Ca)/P в пределах от 1,5 до 1,9. Такие значения близки к соотношению Ca/P в не легированном HA. Значения соотношения (Fe+Ca)/P в пределах от 1,5 до 1,9 указывают на то, что легирование ионами железа в качестве заместителей кальция не влечет за собой больших изменений в химической структуре этого вещества, которое даже после легирования сохраняет химические и структурные характеристики, типичные для не легированного гидроксиапатита.
Легированный гидроксиапатит по изобретению предпочтительно находится в форме частиц (или наночастиц), имеющих размеры от 5-10 нм до 20-30 нм в ширину и вплоть до 80-150 нм в длину. Частицы могут содержать сферические пустоты 2-5 нм.
Собственно магнитный гидроксиапатит по изобретению синтезируют способом, включающим следующие стадии:
а) добавление раствора, содержащего ионы Fe(II) и Fe(III) в суспензию/раствор, содержащую(ий) ионы Ca(II);
б) нагревание суспензии со стадии (а) до температуры в диапазоне от 15°C до 80°C;
в) добавление раствора фосфат-ионов в суспензию/раствор со стадии
(а);
г) отделение осадка от маточных растворов.
Предпочтительно, источниками указанных ионов Fe(II) и Fe(III) являются соответственно FeCl2 и FeCl3.
Предпочтительно, источниками указанных ионов Ca(II) являются гидроксид кальция, нитрат кальция, ацетат кальция, карбонат кальция и/или другие кальциевые соли.
Раствор фосфат-ионов добавляют в суспензию/раствор, содержащую(ий) ионы кальция и ионы железа в течение периода времени 1-3 часа, предпочтительно при нагревании и перемешивании суспензии. Предпочтительно, источником фосфат-ионов является фосфорная кислота и/или ее растворимые соли.
Количество используемых ионов железа является таким, чтобы достигалось молярное соотношение Fe/Ca в пределах от 5 до 30, предпочтительно от 10 до 20 мол.%.
После завершения этой стадии суспензия может быть выдержана при постоянном перемешивании в течение 1-2 часов и затем оставлена стоять без перемешивания или нагревания в течение 12-36 часов.
Этим способом получают осадок, который отделяют от маточных растворов, предпочтительно центрифугированием. Отделенный осадок впоследствии диспергируют в дистиллированной воде и центрифугируют по меньшей мере три раза.
Предпочтительно осадок промывают по меньшей мере три раза, подвергают сублимационной сушке и просеивают.
Изобретение также относится к собственно магнитному гидроксиапатиту, предпочтительно в форме собственно магнитных наночастиц, полученному способом синтеза, описанным выше.
Собственно магнитный гидроксиапатит по изобретению является веществом, которое сохраняет биосовместимость, вполне сравнимую с биосовместимостью нелегированного гидроксиапатита, и поэтому его можно применять для разных целей в клинической и/или диагностической областях.
В частности, собственно магнитный гидроксиапатит можно применять в качестве средства для доставки и высвобождения биологических факторов или лекарственных средств, в качестве контрастного агента в диагностике или еще в качестве биоактивного магнитного заменителя для регенерации костной и костно-хрящевой ткани.
Что касается его применения в области диагностики, собственно магнитный гидроксиапатит по изобретению можно применять в качестве контрастного агента, например, в магнитно-резонансной визуализации (MRI). В этом случае магнитный гидроксиапатит сразу после его соответствующей функционализации (например, специфическими антителами, которые сами способны локализоваться в предопределенных участках организма, например в опухоли) вводят пациенту, и путем приложения внешнего магнитного поля подходящей напряженности можно локализовать частицы и, следовательно, обнаруживать, имеются или нет какие-либо патологические изменения.
Еще одним применением гидроксиапатита по изобретению является его применение в качестве средства для доставки и высвобождения активных веществ, например противоопухолевых лекарственных средств и/или антибиотиков, для селективного лечения в патологическом участке. В этом случае сразу после введения частицы магнитного гидроксиапатита могут быть направлены путем приложения подходящего магнитного поля в патологический участок, где они будут высвобождать активный ингредиент, который они несут на себе.
Таким образом, создана система доставки и высвобождения лекарственных средств контролируемым образом в смысле скорости и селективности высвобождения.
При лечении опухолей частицы магнитного гидроксиапатита по изобретению можно использовать для локального увеличения температуры (магнетотермия или гипертермия). С практической точки зрения, сразу после введения пациенту частицы гидроксиапатита (не содержащие никаких активных веществ) могут быть направлены путем приложения магнитного поля к месту локализации опухоли. После того, как они достигли этого места, температура может быть увеличена в подходящем магнитном поле таким образом, чтобы происходил некроз опухолевых клеток.
Еще одним применением частиц гидроксиапатита по изобретению является его применение в качестве системы доставки и высвобождения биологических агентов, в частности белков, генов, стволовых клеток, факторов роста и факторов васкуляризации. Эта система может быть направлена, путем приложения внешнего магнитного поля, в имплантированный магнитный заменитель кости (например, изготовленный из того же самого магнитного гидроксиапатита) или в немагнитную имплантированную каркасную конструкцию.
Таким путем можно влиять на скорость остеоинтеграции имплантированного костного и/или костно-хрящевого заменителя и регенерации ткани, увеличивая, если это необходимо для индивидуального пациента, количество факторов роста и васкуляризации в том участке, где выявлены затруднения в процессе заживления. Увеличение количества таких факторов достигается путем приложения внешнего магнитного поля, которое обеспечивает направленную доставку частиц по изобретению в участок, где регенерация костной или костно-хрящевой ткани особенно затруднительна.
Объектом изобретения, следовательно, является собственно магнитный гидроксиапатит, имеющий вышеописанные физико-химические характеристики, нагруженный биологическими веществами, выбранными из следующих: белки, гены, стволовые клетки, факторы роста, факторы васкуляризации и активные вещества или лекарственные средства. Указанный гидроксиапатит предпочтительно находится в форме собственно магнитных наночастиц.
Все вышеописанные применения основаны на принципе способности контролировать внутреннее распределение системы доставки и высвобождения из удаленного местоположения путем прикладывания внешнего магнитного поля. Таким образом, частицы собственно магнитного гидроксиапатита могут быть определены как наноустройства для доставки и высвобождения биологических и фармакологических веществ.
Основное преимущество гидроксиапатита по изобретению заключается в том, что он имеет высокую биосовместимость, сравнимую с биосовместимостью нелегированного гидроксиапатита и превышающую биосовместимость систем, состоящих из гидроксиапатита плюс вторичные магнитные фазы. Система доставки и высвобождения по изобретению поэтому не требует, в отличие от магнитных систем доставки и высвобождения, известных в данной области техники, никаких дополнительных модификаций (например, нанесения покрытий), способствующих повышению его свойств биосовместимости.
В действительности, известные магнитные частицы имеют магнитное ядро, защищенное различными монослоями инертного вещества, например диоксида кремния. Альтернативно, могут быть использованы также органические/биологические вещества; они могут быть адсорбированы на поверхность магнитных частиц для образования биосовместимого покрытия.
Примеры органических/биологических покрытий включают антитела и биополимеры (такие как коллаген) или монослои органических молекул, которые делают магнитные частицы биосовместимыми. Кроме того, вещества, которые нужно доставлять, должны быть связаны с известными магнитными частицами посредством линкера с реакционно-способными группами на обоих концах. Одна функциональная группа служит для связывания линкера с поверхностью частиц, а вторая функциональная группа используется для связывания с молекулами, которые требуется доставлять.
Преимущество частиц гидроксиапатита по изобретению заключается в том, что они являются собственно магнитными и собственно биосовместимыми, не требующими нанесения дополнительных слоев органического/неорганического вещества для повышения этого свойства. Более того, вещества, предназначенные для доставки, можно наносить непосредственно на гидроксиапатит, не используя вещество-линкер.
Еще одним применением магнитного гидроксиапатита по изобретению является его применение для получения трехмерных биомиметических конструкций, предназначенных для использования в качестве костных или костно-хрящевых заменителей в применениях для регенерации костной или костно-хрящевой ткани.
Эти магнитные биомиметические каркасные конструкции могут быть зафиксированы в заданной позиции in vivo за счет использования подходящей напряженности магнитного поля, прилагаемого извне. Более того, такими каркасными конструкциями можно управлять in situ, прикладывая подходящее внешнее магнитное поле, которое дает возможность направлять другие частицы магнитного гидроксиапатита по изобретению, нагруженные факторами роста факторами васкуляризации, стволовыми клетками, лекарственными средствами или, в любом случае, биологическими агентами, в магнитное устройство, так, чтобы вышеупомянутые вещества высвобождались in situ в соответствии с качественными и количественными и временными требованиями для пациента.
Таким образом, изобретение также относится к 3D биомиметическому устройству, в частности, устройству для использования в регенерации костной и костно-хрящевой ткани, содержащему частицы, предпочтительно наночастицы, собственно магнитного гидроксиапатита по изобретению.
Экспериментальная часть
Порошки магнитного и биомиметического гидроксиапатита (HA) получали с использованием FeCl2 и FeCl3 в качестве источников легирующих ионов Fe2+ и Fe3+. Сравнивали три разных способа синтеза из вышеуказанных солей. Эти способы подробно описаны в примерах 1-3.
Химический состав, структура и свойства синтезированных порошков Fe-НА определяли описанными ниже методами.
Химический анализ осуществляли методом оптической эмиссионной спектрометрии с индуктивно-связанной плазмой (ИСП-ЭС/ICP-OES; Liberty 200, Varian, Clayton South, Australia) следующим образом: 20 мг порошка растворяли в 2 мл HNO3, и объем раствора доводили до 100 мл путем добавления деионизированной воды. Количество Fe2+ подтверждали посредством УФ-Vis спектрофотометрического анализа (спектрофотометрический анализ в УФ видимой области), используя способность ионов Fe2+ образовывать комплексы с ортофенантролином, детектируемые при 510 нм.
Количество Fe3+ определяли, вычисляя разность между общим количеством железа (определенным методом ИСП) и количеством Fe2+ (определенным методом УФ-Vis).
Морфологическую оценку порошков осуществляли с использованием сканирующего электронного микроскопа (СЭМ/SEM; Stereoscan 360, Leica, Cambridge, UK).
Определение с электронами обратного рассеяния (BSE) использовали для качественной визуализации распределения Fe в порошках. ЭДС/EDS (энергодисперсионная спектроскопия, Link Oxford) использовали для полуколичественного химического анализа.
Анализ дифракции рентгеновских лучей (CuKα излучение; Rigaku Geigerflex, Tokyo Japan) использовали для определения присутствующих кристаллических фаз и оценки степени кристалличности порошка.
Трансмиссионный электронный микроскоп (JEOL ТЭМ 3010-UHR, Japan, 300kV) использовали для исследования характеристик вещества на наноуровне.
Магнитные измерения порошков выполняли в слабом поле (34 Э) с использованием измерителя магнитной восприимчивости YSZ 01C/02C (Sartorius Mechatronics, Italy).
Магнитные измерения также осуществляли с использованием магнетометра со сверхпроводящим квантовым интерференционным датчиком (SQUID) (Quantic Design), способным работать при температуре от 1,8K до 350K в максимальном прилагаемом магнитном поле H=5T (50000 Э).
В этом случае выполняли измерения на нескольких миллиграммах образца в виде порошка (20 мг) при температуре от 5К до 300К в магнитном поле H=100Э для построения кривых зависимости M (намагниченность) от T (температура), а для построения кривых зависимости M от H (напряженность магнитного поля) выполняли измерения за один цикл изменения магнитного поля от 2T до -2T (+/- 20000 Э) при Т=300К.
Пример синтеза 1 (для сравнения)
При соблюдении общей методики синтеза, описанной ранее, один способ синтеза заключается в добавлении только FeCl3 в качестве источника ионов Fe3+, которые впоследствии частично восстанавливают до ионов Fe2+, чтобы получить соотношение Fe3+/Fe2+, равное примерно 3.
Порошки, синтезированные путем добавления ионов Fe3+, применяя начальное молярное соотношение Fe/Ca 0,20 и температуру 40°C, демонстрируют значительную деформацию решетки НА, что выявлено дифрактометрическим анализом РДА; однако никаких вторичных фаз обнаружено не было (Фиг.2).
Синтезированный порошок, не подвергнутый восстановлению, не продемонстрировал никаких признаков намагниченности, как и ожидалось с учетом того, что железо присутствует только в его наивысшем состоянии окисления.
Порошок затем подвергали восстановлению. Процесс восстановления проводили в закрытом автоклаве (Parr, Alloy C276), используя H2 (4%) в 96% Ar в качестве газа-восстановителя при разных давлениях. В Таблице 1 суммированы экспериментальные условия восстановления в автоклаве для порошка Fe3+HA.
Характеристики восстановленных порошков приведены в Таблице 2, которая демонстрирует взаимосвязь между намагниченностью и молярным соотношением Fe3+/Fe2+, вычисленным для восстановленного образца Fe3+HA.
Очень низкие значения намагниченности были зарегистрированы для всех синтезированных порошков. Фактически, можно выдвинуть гипотезу преимущественного восстановления ионов Fe3+, присутствующих на поверхности, сопровождаемого негомогенным восстановлением всего количества, что приводит к образованию избыточного количества ионов Fe2+. Применяемый способ восстановления, по-видимому, характеризуется реакционными условиями, которые являются слишком экстремальными для получения сбалансированного распределения ионов Fe3+/Fe2+ в решетке HA.
Фактически, концентрация Fe2+, определенная УФ-Vis спектрофотометрическим анализом, в котором используется способность ионов Fe2+ образовывать комплекс с ортофенантролином, оказалась очень высокой, что чрезмерно снижает соотношение Fe3+/Fe2+ (Таблица 2).
Пример синтеза 2 (для сравнения)
При соблюдении общей методики синтеза, описанной ранее, альтернативный способ синтеза предусматривает добавление только FeCl2 в качестве источника ионов Fe2+. Решетка подвергается спонтанному окислению до ионов Fe3+ в определенных реакционных условиях.
Порошки синтезировали путем добавления ионов Fe2+, используя начальное молярное соотношение Fe/Ca 0,20 и меняя температуру синтеза (25°C, 40°C, 60°C).
Экспериментальные реакционные условия и свойства полученных в результате порошков приведены в Таблице 3.
График РДА на Фиг.3 показывает пик магнетита при 20 примерно 36° в образцах E и F, тогда как характер разрешения в спектре образца D снижается, как и ожидалось, поскольку он был получен при более низкой температуре. Как показано в Таблице 3, характеристики порошков сильно зависят от способа синтеза.
Образование магнетита происходит предпочтительно при более высокой температуре (60° и 40°C), и в этих условиях значение намагниченности увеличивается, в основном благодаря вкладу магнетита, который образуется совместно с Fe-замещенным HA. При 25°C образование магнетита минимизировано, и ионы железа встраиваются в решетку в большем количестве, что продемонстрировано искажением сигнала (Фиг.3). Общее количество ионов железа, определенное методом ИСП (Таблица 3), соответствует номинальной концентрации FeCl2, введенного во все образцы. Соотношение Fe3+/Fe2+ вычисляют, используя количество Fe2+, определенное УФ-методом, и количество Fe3+, полученное в результате вычитания количества Fe2+ из общего содержания железа, определенного методом ИСП, в обоих случаях после вычитания вклада ионов Fe2+ и Fe3+, которые образуют магнетит. Как показано в Таблице 3, соотношение Fe3+/Fe2+ является очень высоким для образцов D и E. По мере увеличения реакционной температуры (образец F), количество магнетита в виде вторичной фазы также увеличивается, а количество Fe3+, доступное для встраивания в решетку HA, снижается.
Значение намагниченности образца D является очень низким, поскольку 1) магнетит в виде вторичной фазы отсутствует; 2) оба вида ионов железа входят в решетку, но соотношение Fe3+/Fe2+является очень высоким, и распределение ионов Fe2+ и Fe3+ и их координационные состояния не являются подходящими. В образцах E и F высокое значение намагниченности по существу связано с концентрацией магнетита.
Пример синтеза 3 (по изобретению)
FeCl2 и FeCl3 добавляют одновременно в суспензию гидроксида кальция в соотношении Fe3+:Fe2+1:1.
Раствор фосфорной кислоты (Aldrich 85 мас.%) добавляют в суспензию, содержащую диспергированный гидроксид кальция (Aldrich 95 мас.%) и ионы железа, в течение периода времени примерно 1-2 часа при постоянном нагревании и перемешивании. Реакцию синтеза проводят при 40°C. Количества хлорида железа являются такими, которые обеспечивают начальные молярные соотношения Fe/Ca=0,10 и 0,20.
Продукт реакции выдерживают в виде суспензии при постоянном перемешивании и нагревании в течение 1 часа и затем оставляют стоять на 24 часа без нагревания или перемешивания.
Полученный осадок коричневого цвета отделяют от маточных растворов центрифугированием и затем промывают и центрифугируют три раза.
Осадок затем подвергают сублимационной сушке и просеивают при 150 мкм.
Добавление обоих реагентов (FeCl2 и FeCl3) делает Fe2+ и Fe3+ одновременно доступными во время образования ядра HA: в этих условиях количество магнетита, которое образуется, меньше по сравнению со сравнительными способами, описанными выше. Спектр РДА на Фиг.4 демонстрирует, что одновременное добавление Fe2+ и Fe3+ вызывает сильное разупорядочение в решетке HA, что делает очень трудной оценку параметров решетки.
В Таблице 4 показана взаимосвязь, существующая между реакционными условиями и характеристиками Fe2+Fe3+HA.
Высокое значение намагниченности образца H (Таблица 4) нельзя объяснить только вкладом, привнесенным магнетитом. Следовательно, высокая намагниченность обусловлена оптимальным соотношением между ионами железа (Fe3+/Fe2+=3,15) и конкретным относительным положением и координацией двух состояний окисления железа.
Эти результаты свидетельствуют о существовании очень маленьких структурных доменов в решетке HA, которые имитируют структуру магнетита и способны активировать механизмы, ответственные за суперпарамагнитное свойство.
Анализ показал аморфную Ca-P фазу, которая содержит частицы оксида железа в очень низкой концентрации. Фаза под излучением является очень нестабильной, также вследствие высокой степени аморфности. Содержание железа, определенное с использованием ЭДС датчика, составляет 15-20%, и оно по всей вероятности находится в решетке HA или в очень маленьких кластерах менее 1-2 нм.
Микрофотографии образцов E и H представлены на Фиг.5. Частицы имеют в основном удлиненную морфологию, довольно однородную по размерам, от 5-10 нм до 20-30 нм в ширину и вплоть до 80-150 нм в длину, и могут содержать сферические пустоты 2-5 нм.
В случае образца E магнетит присутствует в форме наночастиц, имеющих размеры 10-30 нм.
ВР-ТЭМ анализ (анализ методом ТЭМ высокого разрешения) (Фиг.6) выявил, что часть вещества состоит как из аморфных доменов, так и кристаллических доменов HA, вытянутых в направлении оси C, которые также могут сосуществовать в одной и той же частице (смотри Фиг.6 и соответствующее подробное описание). Общее содержание железа, кальция и фосфора в фазе фосфата кальция измеряли для обоих порошков E и H с использованием метода ЭДС/ТЭМ (в случае образца E, тщательно выбирая участки, в которых магнетит отсутствует). Результаты в виде молярных соотношений между присутствующими элементами (Таблица 5) согласуются с результатами, полученными методами ИСП, РДА и УФ-Vis, которые описаны выше (Таблица 4).
В Таблице 5 представлены результаты количественного анализа методом ЭДС/ТЭМ образцов E и H и этих же образцов, обработанных при 700°C (Et и Ht).
Поскольку не было обнаружено никакого свидетельства присутствия богатой железом фазы, даже в порошке H (в котором начальное молярное соотношение Fe к кальцию составляет Feобщ/Ca (HA)=0,20), предполагается, что ионы железа распределены равномерно и с преобладанием замещения ионов Ca2+в решетке HA или в аморфной фазе в виде очень маленьких кластеров (<1 нм).
Непрямым доказательством равномерного распределения ионов железа может служить поведение образцов, подвергнутых воздействию относительно больших доз электронов. В этих условиях вещества фактически являются в высокой степени нестабильными: пустоты быстро обрушаются, и частицы претерпевают морфологические изменения, сопровождающиеся структурными перегруппировками, и часть вещества трансформируется в CaO.
Интересно отметить, что через несколько секунд образуются новые частицы диаметром 1-1,5 нм (смотри Фиг.7). Новая фаза, которая образуется, представляет собой богатое железом соединение, вероятно образующееся в результате коалесценции ионов железа или кластеров в процессе повреждения, которое происходит в результате бомбардировки электронами.
Затем осуществляют термообработку порошков E и H при 700°C в течение 1 часа в атмосфере Ar. Полученные спектры РДА представлены на Фиг.8.
Образец E, содержащий большие количества магнетита, оказался довольно стабильным, и после термообработки (Et) оставался HA в количестве примерно 72%.
Поскольку образуется вторичная фаза β-TCP Ca3(PO4)2 (26%), хотя магнетит почти исчезает, предполагается, что оба иона железа встраиваются в решетку β-TCP, образуя Ca9Fe(PO4)7 и CA9FeH(PO4)7.
В случае образца H (который, как предполагается, состоит из HA, чья решетка частично занята Fe2+ и Fe3+, которые образуют магнитно активные микродомены), термостабильность намного ниже. Это свидетельствует о сильном разупорядочении кристаллической структуры вследствие занятости одновременно обоими ионами железа (Ht).
Полученные методом ТЭМ микрофотографии образцов (Et) и (Ht) представлены на Фиг.9. Образец (Et) представляет собой частицы очень нерегулярной сфероидной формы шириной примерно 30-50 нм и различной длины, полученные в результате спекания первичных частиц исходных порошков.
Образец характеризуется однородным контрастом, что является показателем однородной дисперсии железа.
Анализ методом ВР-ТЭМ подтвердил, что фаза фосфата кальция является преимущественно фазой типа HA с некоторыми объемными фракциями β-TCP, и выявил, что узкая фракция вещества состоит из чистой фазы, которая демонстрирует аморфную природу, вероятно унаследованную от частиц магнетита, которые утеряли их структуру в результате термообработки.
Кроме того, в этом случае, даже если образец более устойчив к повреждению под воздействием излучения, после воздействия больших доз электронов на фосфат кальция появляются новые небольшие частицы, и это свидетельствует о присутствии очень маленьких кластеров ионов железа в неповрежденном HA или в решетке β-TCP. Интересно отметить, что измеренное содержание железа (Feобщ/Ca (HA)) в фосфате кальция составляет примерно 18±3% (смотри Таблицу 5), более чем в два раза больше, чем в исходных порошках. Это указывает на то, что термообработка способствует диффузии ионов железа из магнетита в фосфат кальция. Это в сочетании с аморфизацией магнетита объясняет исчезновение пиков магнетита, наблюдаемое на рентгеновской дифрактограмме (Фиг.8).
С другой стороны, частицы образца (Ht) являются более крупными и более сферическими (без пустот внутри) и демонстрируют очевидные колебания контраста (Фиг.9), связанные с присутствием богатой железом фазы, которая не наблюдается в исходных порошках.
Анализы методами ВР-ТЭМ и ЭДС совместно подтверждают, что фосфат кальция присутствует главным образом в форме β-TCP, замещенной железом, в которой инкапсулированы частицы оксида и аморфного железа.
Измеренное соотношение Fe/Ca (в участках, в которых отсутствует чистый оксид железа) составляет примерно 16±1%, т.е. ниже, чем в исходных порошках.
Определение физико-химических и магнитных характеристик
Полученные порошки были коричневого цвета и характеризовались агломератами с размерами примерно 30-60 мкм, как видно на СЭМ-изображении на Фиг.1.
Дифрактограммы РДА выглядят очень широкими, и это является показателем значительной деформации решетки HA; в любом случае вторичные фосфатные фазы не были обнаружены вблизи HA.
С помощью компьютерного моделирования на структурных моделях было получено четкое свидетельство, что ионы Fe находятся не в междоузлии в решетке гидроксиапатита, а скорее замещают ионы Ca (с небольшой разницей между тетра- и гексакоординированными положениями решетки 4f и 6h).
ICP-анализ подтвердил присутствие железа в порошке в количестве, равном номинально введенному количеству; соотношение Ca/P было ниже, чем теоретическое соотношение вследствие истощения кальция.
Представляется, что соответствующие количества железа в виде Fe3+, Fe2+ и (Fe3++Fe2+) замещают Ca, поскольку соотношение (Fe+Ca)/P почти равно теоретическому значению.
BSE-анализ подтвердил гомогенное распределение железа в порошке. Анализ микроструктуры также выявил такое же номинальное количество железа, добавленного во время реакции.
Магнитные свойства в сильном магнитном поле измеряли с использованием магнетометра SQUID (Quantum Design). Доказательство присутствия собственно магнитной фазы в решетке гидроксиапатита было обнаружено для образца H. На Фиг.10 представлена зависимость намагниченности от температуры в условиях охлаждения в нулевом поле, ОНП (zero-field-cooled, ZFC) и охлаждения в поле, ОП (field-cooled, FC) от 5K до 300K в магнитном поле 100 Э. Обе кривые показывают типичное поведение системы взаимодействующих магнитных частиц со средней температурой блокировки ТB=170К. ТB является в значительной степени высокой, вероятно, из-за присутствия агрегатов более мелких наночастиц магнетита (~10 нм), которые действуют как вторичная фаза (в соответствии с другими методами определения характеристик, которые подтверждают присутствие 2 об.% магнетита в виде вторичной фазы вне решетки гидроксиапатита) и отделяют состояние, блокированное суперпарамагнитным состоянием. Ниже Tв свободное движение магнитного момента частиц блокировано; выше Tв тепловая энергия индуцирует быстрые колебания магнитного момента всей частицы относительно времени наблюдения, что делает систему суперпарамагнитной. Tв четко коррелируется с дипольными взаимодействиями внутри частицы и увеличивается с увеличением дипольных взаимодействий.
Вследствие образования агрегатов локальная концентрация наночастиц и, следовательно, сила дипольных взаимодействий внутри частицы увеличивается, модифицируя энергетический барьер для магнитной релаксации и определяя общее магнитное поведение образцов.
На Фиг.11 представлены кривые намагничивания как функция приложенного магнитного поля, от -2 до 2 тесла, при 300K для образца H.
Можно явно видеть типичное суперпарамагнитное (SPM) поведение однодоменных магнитных наночастиц, что указывает на то, что размер частиц ниже диапазона размеров магнитных мультидоменных частиц (25-30 нм для магнетита). Это согласуется с полученными методом ТЭМ микрофотографиями, представленными на Фиг.5B, где можно видеть агрегаты магнетита примерно 5-10 нм. С другой стороны, намагниченность насыщения (Ms) образца является в значительной степени низкой (примерно 4-5 эме/г), и вторичная фаза, идентифицированная в форме наночастиц магнетита (которая составляет 2 об.%), не является достаточной, чтобы объяснить наблюдаемое значение намагниченности. Следовательно, два разных магнитных вклада ответственны за общую намагниченность вещества: один явно исходит от наночастиц магнетита, в другой от частиц гидроксиапатита, замещенных ионами Fe2+ и Fe3+.
В первом случае происхождение намагниченности известно, а в последнем случае намагниченность возникает в результате замещения конкретных сайтов Ca2+ в решетке HA магнитными ионами Fe2+ и Fe3+.
С целью нахождения структурной и магнитной фазы, ответственной за намагниченность в решетке гидроксиапатита, общую намагниченность системы наночастиц магнетита (диаметр примерно 10 нм, Ms примерно 60-80 эме/г) вычитали из общей приведенной намагниченности насыщения (Ms примерно 4-5 эме/г). Этим путем можно определить намагниченность, связанную только с HA, замещенным железом. В любом случае эта вычисленная Ms больше, чем прогнозируемая, также в том случае, когда суммарная масса Fe в веществе (6-7 мас.%) присутствует в виде магнетита.
Следовательно, обнаруженные магнитные данные подкрепляют предположение, что значение Ms обусловлено присутствием структурных магнитных доменов (подобных магнетиту и/или другим оксидам железа) в решетке гидроксиапатита.
Тесты in vitro
Предварительные тесты на клеточную адгезию и биосовместимость проводили на гранулированном HA и порошках магнитного HA с использованием мезенхимальных стволовых клеток (MSC), которые после определения характеристик культивировали в контролируемой атмосфере (5% CO2; T=37°C) в DMEM (среда Игла, модифицированная по Дульбекко) (Sigma, Milan, Italy), дополненной 10% сыворотки плода коровы (FBS), 1% не незаменимых аминокислот и антибиотиками. Клетки делили 1:2 с однонедельными интервалами и использовали между третьим и четвертным пассажем. Клетки из конфлюэнтных культур MSC отделяли с использованием 0,25% трипсина в 1 мл MEDTA и наносили в тройном повторе при плотности 5×104 и 1×104 клеток/см2 соответственно на гранулированные порошки, предназначенные для тестирования, и в 24-луночные полистирольные планшеты в качестве контроля.
Культуры в планшетах инкубировали при 37°C в течение 7 суток. После инкубирования культуральную среду удаляли; 200 мкл МТТ (3-диметилтиазол-2,5-дифенилтетразолия бромид для колориметрических тестов, Aldrich 135038) и 1,8 мл культуральной среды добавляли к индивидуальным клеточным слоям; многослойные планшеты инкубировали при 37°C в течение еще 3 часов.
После отделения надосадочной жидкости кристаллы формазана синего растворяли путем добавления 2 мл растворителя (4% HCI 1 н. в абсолютном изопропаноле) и спектрофотометрическим методом осуществляли количественное определение при 570 нм.
Что касается стехиометрического HA, порошки магнитного HA представляют собой подходящий субстрат с точки зрения клеточной адгезии и пролиферации для предшественников остеобластов (MSC).
Значение МТТ составляет 80% для магнитного HA, тогда как для HA оно составляет 82%. Морфологические анализы методом SEM, проведенные на культурах клеток, показали хорошую биосовместимость MSC со всеми протестированными порошками. Подобно их поведению на HA, на магнитном HA клетки показали диффузную "вытянутую" морфологию с некоторым количеством цитоплазматических удлинений в контакте с порошком.
В заключение, одновременное добавление FeCl2 и FeCl3 в качестве источников ионов для частичного замещения кальция в решетке HA и использование оптимизированных параметров синтеза сделало возможным поучение Fe3+/Fe2+-замещенного HA, имеющего соотношение Fe3+/Fe2+, равное примерно 3, и соотношение (Fe+Ca)/P, очень близкое к теоретическому соотношению.
Используя РДА и компьютерное моделирование на структурных моделях, получено четкое подтверждение, что оба вида ионов Fe находятся в положениях кальция, замещая его, а не в междоузлиях в решетке HA.
Положение Ca(2) с координационным числом 6 и положение Ca(1) с координационным числом 4 заняты взаимосвязанно, придавая порошкам магнитную восприимчивость, благодаря строго определенным параметрам синтеза.
Исследования методом ТЭМ, а также магнитные измерения подтверждают присутствие новой магнитной фазы HA вместе с нанокластерами, подобными магнетиту.
Эта новая фаза представляет собой деформированный/разупорядоченный гидроксиапатит с Fe2+ в его номинальном двухвалентном состоянии и Fe3+, который имеет отклонение от его номинального трехвалентного состояния, организованные на уровне поверхности и объема и координированные для создания намагниченности в самом HA.
Эти результаты, наряду с биосовместимостью магнитного HA, обеспечивают регенеративную медицину новым семейством биосовместимых биомиметических веществ, которые можно контролировать и которыми можно управлять с помощью подходящих внешних магнитных полей. Эти вещества действительно можно использовать с целью получения 1) наночастиц для доставки и высвобождения биоактивных факторов и/или лекарственных средств, 2) наночастиц, используемых в диагностических целях (визуализация) и в целях лечения (гипертермия), 3) заменителей костной и костно-хрящевой ткани, которыми можно биологически управлять in situ (т.е. "перезагружать" или в любом случае стимулировать после имплантации in vivo специфическими факторами в соответствии с качественными - количественными и временными требованиями для пациента) и которые можно даже фиксировать в заданном положении in vivo (исключая зависимость от традиционных фиксирующих систем).
| название | год | авторы | номер документа |
|---|---|---|---|
| БИОСОВМЕСТИМЫЙ КОСТНОЗАМЕЩАЮЩИЙ МАТЕРИАЛ И СПОСОБ ПОЛУЧЕНИЯ ЕГО | 2012 |
|
RU2494721C1 |
| СПОСОБ ПОЛУЧЕНИЯ МАГНИТНОЙ ЖИДКОСТИ | 2007 |
|
RU2340972C2 |
| МАГНИТНЫЙ КОМПОЗИЦИОННЫЙ СОРБЕНТ | 2012 |
|
RU2547496C2 |
| СПОСОБ ПОЛУЧЕНИЯ НАНОРАЗМЕРНОГО ГИДРОКСИАПАТИТА В МИКРОВОЛНОВОМ ПОЛЕ С ИСПОЛЬЗОВАНИЕМ ВЫГОРАЮЩЕЙ ДОБАВКИ | 2014 |
|
RU2574455C1 |
| ПОРИСТЫЕ МИКРОСФЕРЫ НА ОСНОВЕ БИОФОСФАТОВ КАЛЬЦИЯ И МАГНИЯ С РЕГУЛИРУЕМЫМ РАЗМЕРОМ ЧАСТИЦ ДЛЯ РЕГЕНЕРАЦИИ КОСТНОЙ ТКАНИ | 2012 |
|
RU2497548C1 |
| СПОСОБ ПОЛУЧЕНИЯ НАНОКРИСТАЛЛИЧЕСКОГО КРЕМНИЙЗАМЕЩЕННОГО ГИДРОКСИАПАТИТА | 2012 |
|
RU2500840C1 |
| Способ получения катионзамещенного трикальцийфосфата | 2015 |
|
RU2607743C1 |
| СПОСОБ ПОЛУЧЕНИЯ НАНОКРИСТАЛЛИЧЕСКОГО МАГНИТНОГО ПОРОШКА ДОПИРОВАННОГО ОРТОФЕРРИТА ИТТРИЯ | 2013 |
|
RU2574558C2 |
| БИОМАТЕРИАЛЫ НА ОСНОВЕ ФОСФАТА КАЛЬЦИЯ | 2009 |
|
RU2501571C2 |
| СПОСОБ ПОЛУЧЕНИЯ СУБМИКРОННОЙ БИФАЗНОЙ КЕРАМИКИ НА ОСНОВЕ ТРИКАЛЬЦИЙФОСФАТА И ГИДРОКСИАПАТИТА | 2013 |
|
RU2555685C2 |


Группа изобретений относится к медицине, конкретно к гидроксиапатиту, легированному ионами Fe2+ и ионами Fe3+, которые частично замещают ионы кальция в кристаллической решетке. Гидроксиапатит характеризуется собственным магнетизмом от 0,05 до 8 эме/г, измеренным в приложенном магнитном поле 34Э, благодаря присутствию магнитных нанодоменов в кристаллической решетке НА, при наличии ограниченного количества магнитных вторичных фаз, составляющего менее примерно 3 об.%. Собственно магнитный гидроксиапатит может быть нагружен биологическими веществами, выбранными из группы, состоящей из белков, генов, стволовых клеток, факторов роста и факторов васкуляризации, активных веществ или лекарственных средств, под контролем внешнего магнитного поля, в качестве средства для доставки и высвобождения биологических веществ или лекарственных средств, в качестве контрастного агента в диагностике или для регенерации костной и костно-хрящевой ткани. Гидроксиапатит обеспечивает доставку и высвобождение лекарственных средств, обеспечивая контролируемую скорость и селективность высвобождения. 3 н. и 12 з.п. ф-лы, 11 ил., 5 табл., 3 пр.
1. Гидроксиапатит для медицинского или диагностического применения, содержащий ионы кальция и фосфат-ионы в кристаллической решетке, отличающийся тем, что он легирован ионами Fe2+и ионами Fe3+, которые частично замещают указанные ионы кальция в указанной кристаллической решетке, в количественном соотношении Fe3+/Fe2+от 1 до 4, имеет намагниченность от 0,05 до 8 эме/г, измеренную в приложенном магнитном поле 34 Э, благодаря присутствию магнитных нанодоменов в решетке гидроксиапатита, имеет соотношение (Fe+Ca)/P от 1,5 до 1,9 и содержит вторичные магнитные фазы в количестве менее примерно 3 об. %.
2. Гидроксиапатит по п. 1, где указанная намагниченность, зарегистрированная в приложенном магнитном поле 34 Э, составляет от 0,1 до 5 эме/г.
3. Гидроксиапатит по п. 1, где указанное соотношение Fe3+/Fe2+составляет от 2 до 3,5.
4. Гидроксиапатит по п. 1, содержащий вторичные магнитные фазы в количестве ≤2 об. %.
5. Гидроксиапатит по п. 1 в форме наночастиц, имеющих ширину от 5-10 нм до 20-30 нм и длину вплоть до 80-150 нм, или в форме агрегатов/гранул указанных наночастиц.
6. Гидроксиапатит по п. 5, где указанные наночастицы содержат сферические пустоты 2-5 нм.
7. Гидроксиапатит по п. 1, нагруженный биологическими веществами, выбранными из группы, состоящей из белков, генов, стволовых клеток, факторов роста и факторов васкуляризации, или нагруженный активными веществами или лекарственными средствами.
8. Гидроксиапатит по п. 1 для применения в качестве средства для доставки и высвобождения биологических веществ или лекарственных средств.
9. Биомиметический заменитель костной или костно-хрящевой ткани, изготовленный из гидроксиапатита по п. 1.
10. Биомиметический заменитель костной или костно-хрящевой ткани по п. 9 для регенерации костной и костно-хрящевой ткани.
11. Способ получения гидроксиапатита по п. 1, включающий следующие
стадии:
а) добавление раствора, содержащего ионы Fe(II) и Fe(III), источниками которых являются FeCl2 и FeCl3 соответственно, в суспензию/раствор, содержащую(ий) ионы Са(II);
б) добавление раствора фосфат-ионов в суспензию/раствор со стадии (а);
в) нагревание суспензии/раствора со стадии (б) до температуры от 15°C до 80°C, предпочтительно от 25°C до 60°C;
г) отделение осадка от маточных растворов.
12. Способ по п. 11, при котором указанный раствор фосфат-ионов добавляют в суспензию/раствор, содержащую(ий) ионы Са(II) и ионы железа, за период времени 1-3 часа, предпочтительно при нагревании и перемешивании суспензии.
13. Способ по п. 11, при котором после добавления фосфат-ионов количество ионов железа относительно ионов кальция корректируют путем дополнительного добавления раствора ионов железа до достижения молярного соотношения Fe/Ca от 5 до 30, предпочтительно от 10 до 20 мол. %.
14. Способ по п. 11, при котором источником ионов Са(II) является гидроксид кальция, нитрат кальция, ацетат кальция, карбонат кальция и/или другие соли кальция.
15. Способ по п. 11, при котором источником указанных фосфат-ионов является фосфорная кислота и/или ее растворимые соли.
| Колосоуборка | 1923 |
|
SU2009A1 |
| EP 0610333 A1, 17.08.1994 | |||
| Устройство для измерения постоянного тока | 1948 |
|
SU82764A1 |

