Настоящая заявка испрашивает приоритет на основании предварительной заявки на патент США №61/394136, поданной 18 октября 2010 года, и предварительной заявки на патент США №61/444212, поданной 18 февраля 2011 года, содержание каждой из которых включено в настоящую заявку по всей полноте посредством ссылки.
Область техники
В изобретении предложены соединения, композиции и способы лечения или предотвращения аномальных состояний у субъекта.
Уровень техники
Холестерин, циркулирующий в организме человека, переносится липопротеинами плазмы, которые представляют собой частицы со сложным липидным и белковым составом, осуществляющие транспорт липидов в крови. Двумя типами липопротеинов плазмы, которые переносят холестерин, являются липопротеины низкой плотности (ЛПНП) и липопротеины высокой плотности (ЛПВП). Полагают, что частицы ЛПНП отвечают за доставку холестерина из печени (где происходит его синтез или получение из пищевых источников) к внепеченочным тканям организма. С другой стороны, полагают, что частицы ЛПВП способствуют транспорту холестерина из внепеченочных тканей к печени, где холестерин подвергается катаболизму и выводится из организма. Указанный транспорт холестерина из внепеченочных тканей к печени называют «обратным транспортом холестерина».
Путь обратного транспорта холестерина («ОТХ») имеет три основных стадии: (i) выход холестерина, т.е. начальное удаление холестерина из различных групп периферических клеток; (ii) этерификация холестерина под действием лецитин:холестерин-ацилтрансферазы (ЛХАТ), с предотвращением тем самым повторного поступления выведенного холестерина в клетки; и (iii) захват холестерилового эфира ЛПВП и доставка комплекса ЛПВП-холестериловый эфир к клеткам печени.
Путь ОТХ опосредован частицами ЛПВП. Недавно был описан путь в печени с участием F1-АТФазы и рецептора P2Y13 (\l "_ENREF_24" \o "Martinez, 2003 #2324), который регулирует удаление ЛПВП-холестерина. Также недавно была описана нуклеотидазная активность субъединицы F1-АТФазы на клеточной поверхности гепатоцитов, обеспечивающая возможность гидролиза АТФ до АДФ, что, в свою очередь, стимулирует рецептор P2Y13, приводя к захвату ЛПВП клетками ((\l "_ENREF_17" \o "Jacquet, 2005 #5373). Позже Фабр с соавторами (\l "_ENREF_12" \o "Fabre, 2010 #5462) подтвердили наличие связи между P2Y13r и обратным транспортом холестерина у мышей.
Краткое описание изобретения
В одном из вариантов реализации в настоящем изобретении предложены соединения следующей Формулы (I)
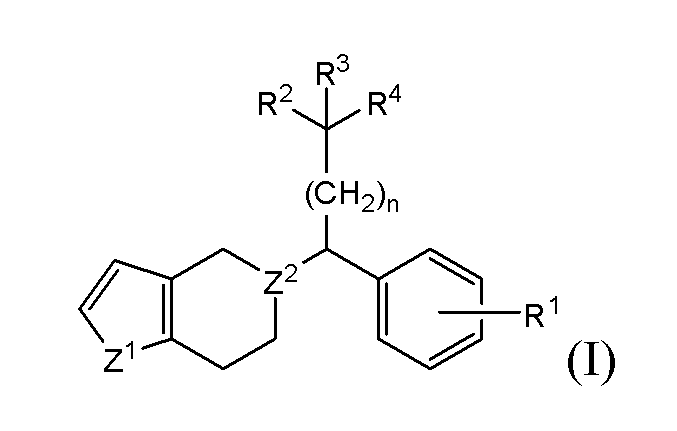
и их фармацевтически приемлемые соли, где
каждый из R1, R2 и R3 независимо представляет собой H, -OH, -NH2, -NH(алкил), -N(алкил)(алкил), гидрокарбил, -O-гидрокарбил, арил, -O-арил, аралкил, -O-аралкил, гетероарил, -O-гетероарил, гетероциклил, -O-гетероциклил, галоген, -OCF3, -C(O)O(алкил), -OC(O)(алкил), -C(O)NH2, -C(O)NH(алкил), -C(O)N(алкил)(алкил), -NHC(O)(алкил), N(алкил)C(O)(алкил), -OC(O)O(алкил), -OC(O)NH2, -OC(O)NH(алкил), -OC(O)N(алкил)(алкил), -CHNH, -CHN(алкил) или -SO2NH2;
R4 представляет собой -H, -OH, -COOH, -NH2, -NH(алкил), -N(алкил)(алкил), -гидрокарбил, -O-гидрокарбил, -арил, -O-арил, -аралкил, -O-аралкил, -гетероарил, -O-гетероарил, -гетероциклил, -O-гетероциклил, -галоген, -C(O)O(алкил), -OC(O)(алкил), -C(O)NH2, -C(O)NH(алкил), -C(O)N(алкил)(алкил), -NHC(O)(алкил), N(алкил)C(O)(алкил), -OC(O)O(алкил), -OC(O)NH2, -OC(O)NH(алкил) или -OC(O)N(алкил)(алкил);
Z1 представляет собой CH2, S, O, NH, N-гидрокарбил, N-арил, N-гетероарил или N-гетероциклил;
Z2 представляет собой СН или N; и
n представляет собой целое число от 1 до 6.
В другом варианте реализации в изобретении предложены соединения следующей Формулы (II)
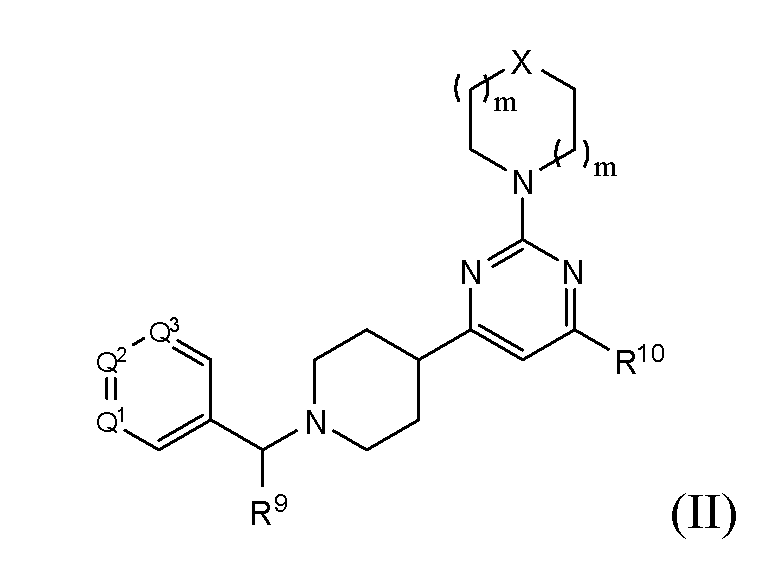
и их фармацевтически приемлемые соли, где
каждый R9 независимо представляет собой -Н, -гидрокарбил, -арил, -аралкил, -гетероарил, -гетероциклил, -C(O)O(алкил), -OC(O)(алкил), -C(O)NH2, -C(O)NH(алкил), -C(O)N(алкил)(алкил), -NHC(O)(алкил), N(алкил)C(O)(алкил) или -SO2NH2;
каждый R10 независимо представляет собой -H, -OH, -NH2, -NH(алкил), -N(алкил)(алкил), гидрокарбил, -O-алкил, -O-алкенил, арил, -O-арил, аралкил, -O-аралкил, гетероарил, -O-гетероарил, гетероциклил, -O-гетероциклил, галоген, -OCF3, -C(O)O(алкил), -OC(O)(алкил), -C(O)NH2, -C(O)NH(алкил), -C(O)N(алкил)(алкил), NHC(O)(C2-C10-алкил), N(алкил)C(O)(алкил), -OC(O)O(алкил), -OC(O)NH2, -OC(O)NH(алкил), -OC(O)N(алкил)(алкил), -CHNH, -CHN(алкил) или -SO2NH2;
каждый из Q1, Q2 и Q3 независимо представляет собой CR10 или N;
Х представляет собой CHR10, S, O или NR9; и
каждый m независимо представляет собой целое число от 0 до 3.
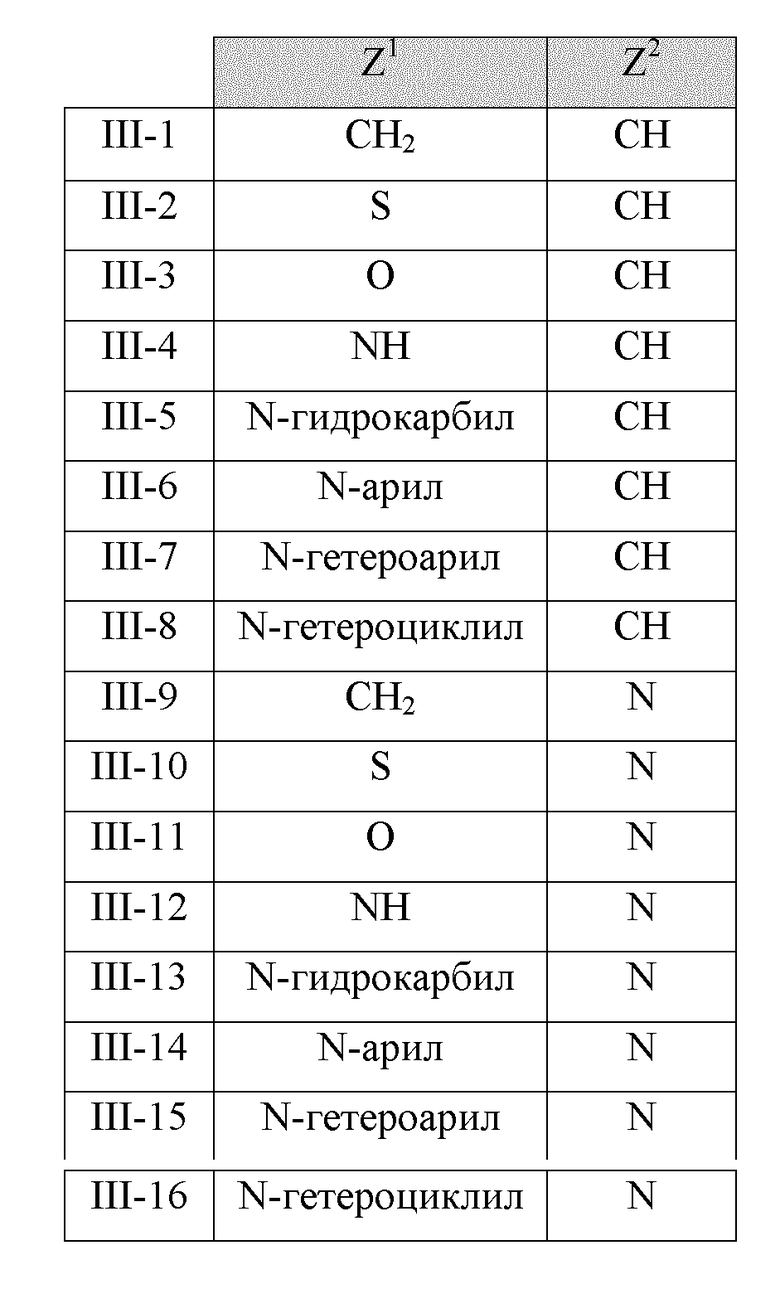
В другом варианте реализации в изобретении предложены соединения следующей Формулы III:
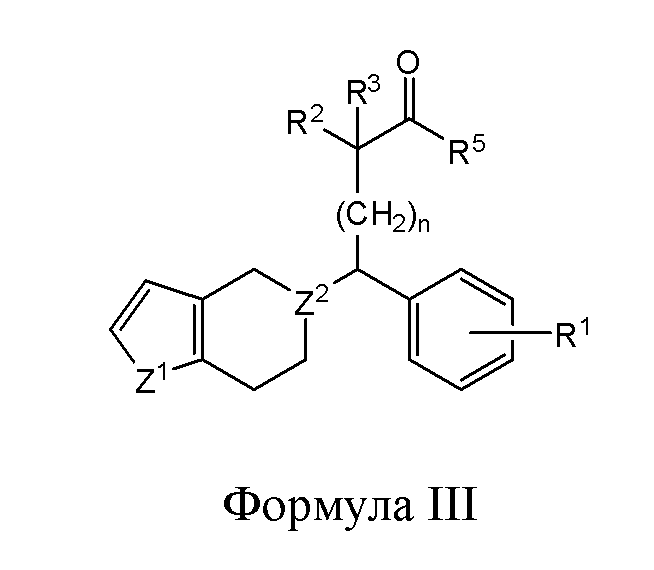
и их фармацевтически приемлемые соли, где
каждый из R1, R2 и R3 независимо представляет собой H,-OH, -NH2, -NH(алкил), -N(алкил)(алкил), гидрокарбил, -O-гидрокарбил, арил, -O-арил, аралкил, -O-аралкил, гетероарил, -O-гетероарил, гетероциклил, -O-гетероциклил, галоген, -OCF3, -C(O)O(алкил), -OC(O)(алкил), -C(O)NH2, -C(O)NH(алкил), -C(O)N(алкил)(алкил), -NHC(O)(алкил), N(алкил)C(O)(алкил), -OC(O)O(алкил), -OC(O)NH2, -OC(O)NH(алкил), -OC(O)N(алкил)(алкил), -CHNH, -CHN(алкил) или -SO2NH2;
R5 представляет собой -H, -OH, -NH2, -NH(алкил), -N(алкил)(алкил), гидрокарбил, -O-гидрокарбил, арил, -O-арил, аралкил, -O-аралкил, гетероарил, -O-гетероарил, гетероциклил или -O-гетероциклил;
Z1 представляет собой CH2, S, O, NH, N-гидрокарбил, N-арил, N-гетероарил или N-гетероциклил;
Z2 представляет собой СН или N; и
n представляет собой целое число от 1 до 6.
В другом варианте реализации в изобретении предложены соединения следующей Формулы IV:
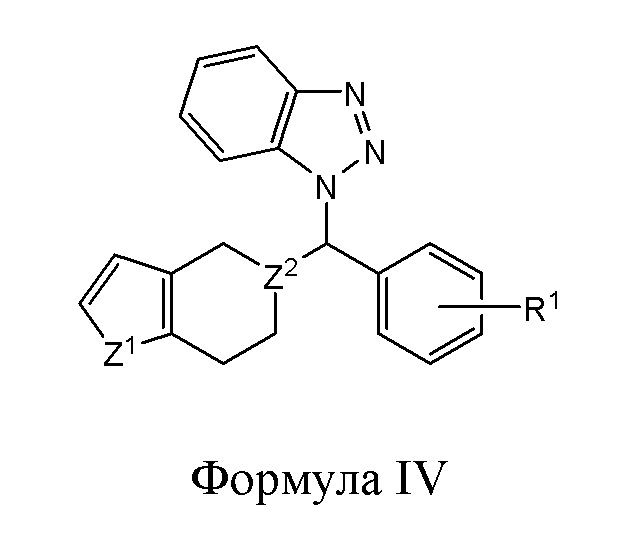
и их фармацевтически приемлемые соли, где
R1 представляет собой H, -OH, -NH2, -NH(алкил), -N(алкил)(алкил), гидрокарбил, -O-гидрокарбил, арил, -O-арил, аралкил, -O-аралкил, гетероарил, -O-гетероарил, гетероциклил, -O-гетероциклил, галоген, -OCF3, -C(O)O(алкил), -OC(O)(алкил), -C(O)NH2, -C(O)NH(алкил), -C(O)N(алкил)(алкил), -NHC(O)(алкил), N(алкил)C(O)(алкил), -OC(O)O(алкил), -OC(O)NH2, -OC(O)NH(алкил), -OC(O)N(алкил)(алкил), -CHNH, -CHN(алкил) или -SO2NH2;
Z1 представляет собой CH2, S, O, NH, N-гидрокарбил, N-арил, N-гетероарил или N-гетероциклил; и
Z2 представляет собой СН или N.
В другом варианте реализации в изобретении предложены соединения следующей формулы V:
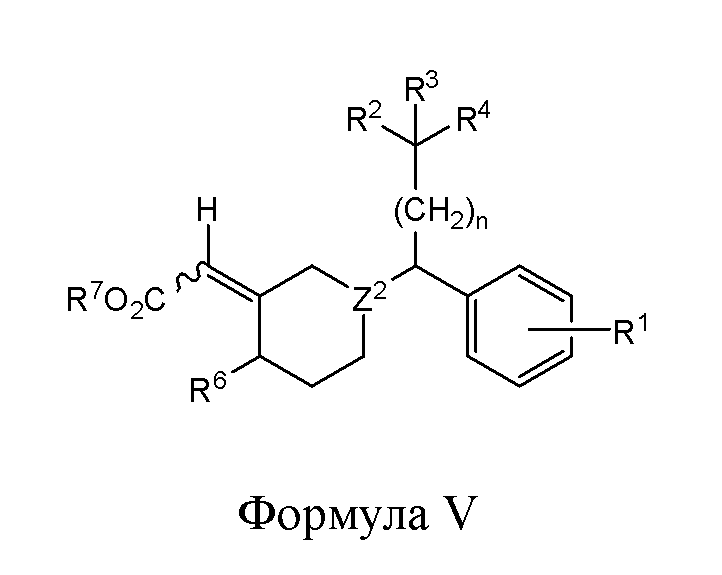
и их фармацевтически приемлемые соли, где
каждый из R1, R2 и R3 независимо представляет собой H, -OH, -NH2, -NH(алкил), -N(алкил)(алкил), гидрокарбил, -O-гидрокарбил, арил, -O-арил, аралкил, -O-аралкил, гетероарил, -O-гетероарил, гетероциклил, -O-гетероциклил, галоген, OCF3, -C(O)O(алкил), -OC(O)(алкил), -C(O)NH2, -C(O)NH(алкил), -C(O)N(алкил)(алкил), -NHC(O)(алкил), N(алкил)C(O)(алкил), -OC(O)O(алкил), -OC(O)NH2, -OC(O)NH(алкил), -OC(O)N(алкил)(алкил), -CHNH, -CHN(алкил) или -SO2NH2;
R4 представляет собой -H, -OH, -COOH, -NH2, -NH(алкил), -N(алкил)(алкил), гидрокарбил, -O-гидрокарбил, арил, -O-арил, аралкил, -O-аралкил, гетероарил, -O-гетероарил, гетероциклил, -O-гетероциклил, галоген, -C(O)O(алкил), -OC(O)(алкил), -C(O)NH2, -C(O)NH(алкил), -C(O)N(алкил)(алкил), -NHC(O)(алкил), N(алкил)C(O)(алкил), -OC(O)O(алкил), -OC(O)NH2, -OC(O)NH(алкил) или -OC(O)N(алкил)(алкил);
R6 представляет собой -H, -OH, -SH, -S-гидрокарбил, -COOH, -NH2, -NH(алкил), -N(алкил)(алкил), гидрокарбил, -O-гидрокарбил, арил, -O-арил, аралкил, -O-аралкил, гетероарил, -O-гетероарил, гетероциклил, -O-гетероциклил, галоген, -C(O)O(алкил), -OC(O)(алкил), -C(O)NH2, -C(O)NH(алкил), -C(O)N(алкил)(алкил), -NHC(O)(алкил), N(алкил)C(O)(алкил), -OC(O)O(алкил), -OC(O)NH2, -OC(O)NH(алкил) или -OC(O)N(алкил)(алкил);
R7 представляет собой Н, гидрокарбил, арил, аралкил, гетероарил или гетероциклил;
Z2 представляет собой СН или N; и
n представляет собой целое число от 1 до 6.
В одном из вариантов реализации в изобретении предложены соединения следующей Формулы VI:
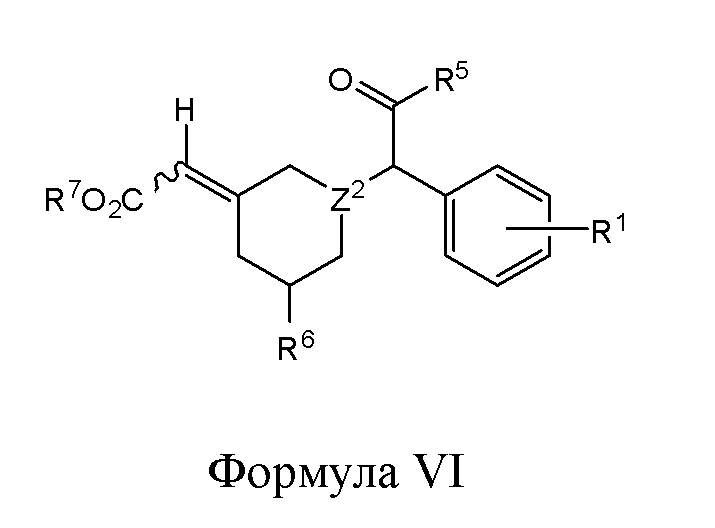
и их фармацевтически приемлемые соли, где
R1 представляет собой H, -OH, -NH2, -NH(алкил), -N(алкил)(алкил), гидрокарбил, -O-гидрокарбил, арил, -O-арил, аралкил, -O-аралкил, гетероарил, -O-гетероарил, гетероциклил, -O-гетероциклил, галоген, -OCF3, -C(O)O(алкил), -OC(O)(алкил), -C(O)NH2, -C(O)NH(алкил), -C(O)N(алкил)(алкил), -NHC(O)(алкил), N(алкил)C(O)(алкил), -OC(O)O(алкил), -OC(O)NH2, -OC(O)NH(алкил), -OC(O)N(алкил)(алкил), -CHNH, -CHN(алкил) или -SO2NH2;
R5 представляет собой -H, -OH, -NH2, -NH(алкил), -N(алкил)(алкил), гидрокарбил, -O-гидрокарбил, арил, -O-арил, аралкил, -O-аралкил, гетероарил, -O-гетероарил, гетероциклил, -O-гетероциклил или галоген;
R6 представляет собой -H, -OH, -SH, -S-гидрокарбил, -COOH, -NH2, -NH(алкил), -N(алкил)(алкил), гидрокарбил, -O-гидрокарбил, арил, -O-арил, аралкил, -O-аралкил, гетероарил, -O-гетероарил, гетероциклил, -O-гетероциклил, галоген, -C(O)O(алкил), -OC(O)(алкил), -C(O)NH2, -C(O)NH(алкил), -C(O)N(алкил)(алкил), -NHC(O)(алкил), N(алкил)C(O)(алкил), -OC(O)O(алкил), -OC(O)NH2, -OC(O)NH(алкил) или -OC(O)N(алкил)(алкил);
R7 представляет собой Н, гидрокарбил, арил, аралкил, гетероарил или гетероциклил; и
Z2 представляет собой СН или N.
В одном из вариантов реализации в изобретении предложены соединения следующей Формулы VII:
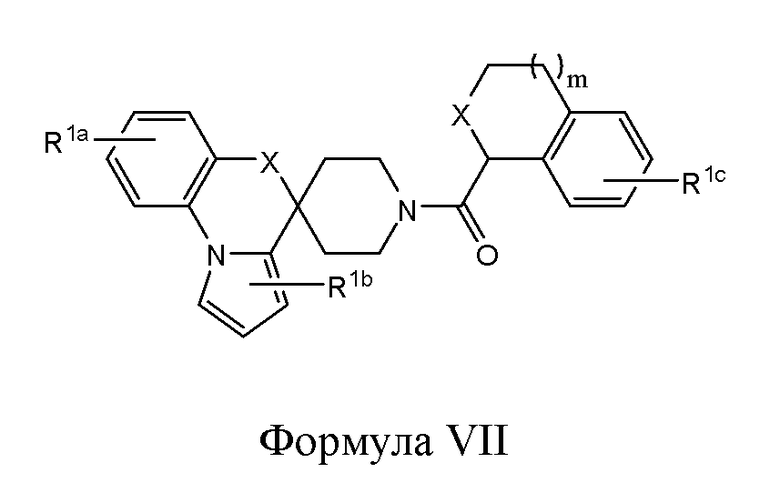
и их фармацевтически приемлемые соли, где
каждый из R1a, R1b и R1c независимо представляет собой -H, -OH, -NH2, -NH(алкил), -N(алкил)(алкил), гидрокарбил, -O-гидрокарбил, арил, -O-арил, аралкил, -O-аралкил, гетероарил, -O-гетероарил, гетероциклил, -O-гетероциклил, галоген, -OCF3, -C(O)O(алкил), -OC(O)(алкил), -C(O)NH2, -C(O)NH(алкил), -C(O)N(алкил)(алкил), -NHC(O)(алкил), N(алкил)C(O)(алкил), -OC(O)O(алкил), -OC(O)NH2, -OC(O)NH(алкил), -OC(O)N(алкил)(алкил), -CHNH, -CHN(алкил) или -SO2NH2;
каждый Х независимо представляет собой CHR10, S, O или NR9;
каждый R9 независимо представляет собой H, гидрокарбил, арил, аралкил, гетероарил, гетероциклил, -C(O)O(алкил), -OC(O)(алкил), -C(O)NH2, -C(O)NH(алкил), -C(O)N(алкил)(алкил), -NHC(O)(алкил), N(алкил)C(O)(алкил) или -SO2NH2;
каждый R10 независимо представляет собой -H, -OH, -NH2, -NH(алкил), -N(алкил)(алкил), гидрокарбил, -O-гидрокарбил, арил, -O-арил, аралкил, -O-аралкил, гетероарил, -O-гетероарил, гетероциклил, -O-гетероциклил, галоген, -OCF3, -C(O)O(алкил), -OC(O)(алкил), -C(O)NH2, -C(O)NH(алкил), -C(O)N(алкил)(алкил), -NHC(O)(алкил), N(алкил)C(O)(алкил), -OC(O)O(алкил), -OC(O)NH2, -OC(O)NH(алкил), -OC(O)N(алкил)(алкил), -CHNH, -CHN(алкил) или -SO2NH2; и
m представляет собой целое число от 0 до 3.
В другом варианте реализации в изобретении предложены соединения следующей Формулы VIII:
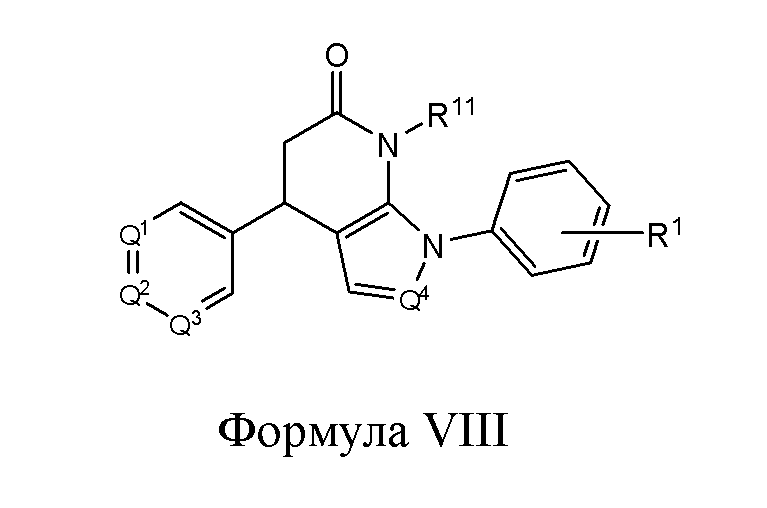
и их фармацевтически приемлемые соли, где
R1 представляет собой -H, -OH, -NH2, -NH(алкил), -N(алкил)(алкил), гидрокарбил, -O-гидрокарбил, арил, -O-арил, аралкил, -O-аралкил, гетероарил, -O-гетероарил, гетероциклил, -O-гетероциклил, галоген, -OCF3, -C(O)O(алкил), -OC(O)(алкил), -C(O)NH2, -C(O)NH(алкил), -C(O)N(алкил)(алкил), -NHC(O)(алкил), N(алкил)C(O)(алкил), -OC(O)O(алкил), -OC(O)NH2, -OC(O)NH(алкил), -OC(O)N(алкил)(алкил), -CHNH, -CHN(алкил) или -SO2NH2;
R11 представляет собой H, гидрокарбил, арил, аралкил, гетероарил, гетероциклил, -C(O)O(алкил), -OC(O)(алкил), -C(O)NH2, -C(O)NH(алкил), -C(O)N(алкил)(алкил), -NHC(O)(алкил), N(алкил)C(O)(алкил) или -SO2NH2;
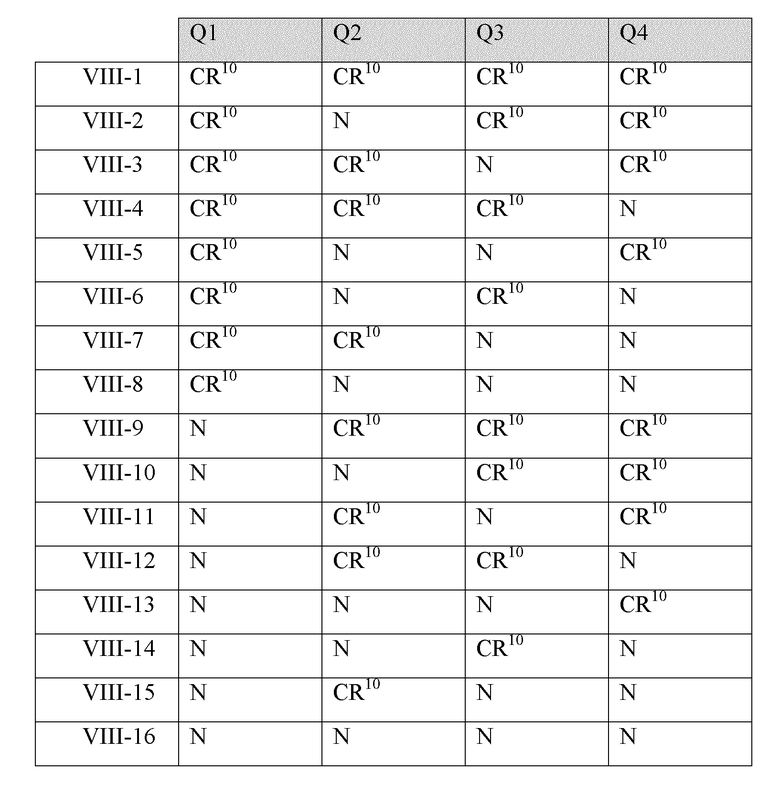
каждый из Q1, Q2, Q3 и Q4 независимо представляет собой CR10 или N; и
каждый R10 независимо представляет собой -H, -OH, -NH2, -NH(алкил), -N(алкил)(алкил), гидрокарбил, -O-гидрокарбил, арил, -O-арил, аралкил, -O-аралкил, гетероарил, -O-гетероарил, гетероциклил, -O-гетероциклил, галоген, -OCF3, -C(O)O(алкил), -OC(O)(алкил), -C(O)NH2, -C(O)NH(алкил), -C(O)N(алкил)(алкил), -NHC(O)(алкил), N(алкил)C(O)(алкил), -OC(O)O(алкил), -OC(O)NH2, -OC(O)NH(алкил), -OC(O)N(алкил)(алкил), -CHNH, -CHN(алкил) или -SO2NH2.
В другом варианте реализации в изобретении предложены соединения следующей Формулы IX:
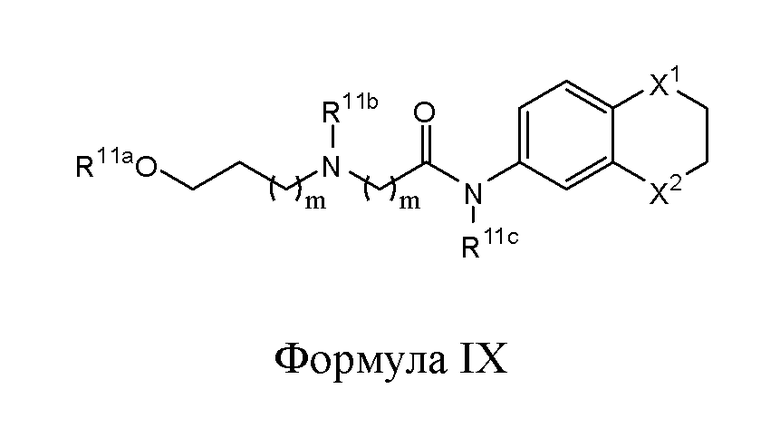
и их фармацевтически приемлемые соли, где
каждый из R11a, R11b и R11c независимо представляет собой H, гидрокарбил, арил, аралкил, гетероарил, гетероциклил, -C(O)(алкил), -C(O)O(алкил), -C(O)NH2, -C(O)NH(алкил), -C(O)N(алкил)(алкил) или -SO2NH2;
каждый из Х1 и Х2 независимо представляет собой CHR10, S, O, NR9 или N-ацил;
каждый R9 независимо представляет собой H, гидрокарбил, арил, аралкил, гетероарил, гетероциклил, -C(O)O(алкил), -OC(O)(алкил), -C(O)NH2, -C(O)NH(алкил), -C(O)N(алкил)(алкил), -NHC(O)(алкил), N(алкил)C(O)(алкил), -OC(O)O(алкил) или -SO2NH2;
каждый R10 независимо представляет собой -H, -OH, -NH2, -NH(алкил), -N(алкил)(алкил), гидрокарбил, -O-гидрокарбил, арил, -O-арил, аралкил, -O-аралкил, гетероарил, -O-гетероарил, гетероциклил, -O-гетероциклил, галоген, -OCF3, -C(O)O(алкил), -OC(O)(алкил), -C(O)NH2, -C(O)NH(алкил), -C(O)N(алкил)(алкил), -NHC(O)(алкил), N(алкил)C(O)(алкил), -OC(O)O(алкил), -OC(O)NH2, -OC(O)NH(алкил), -OC(O)N(алкил)(алкил), -CHNH, -CHN(алкил) или -SO2NH2; и
каждый m независимо представляет собой целое число от 1 до 3.
В другом варианте реализации в изобретении дополнительно предложены соединения следующей Формулы Х:
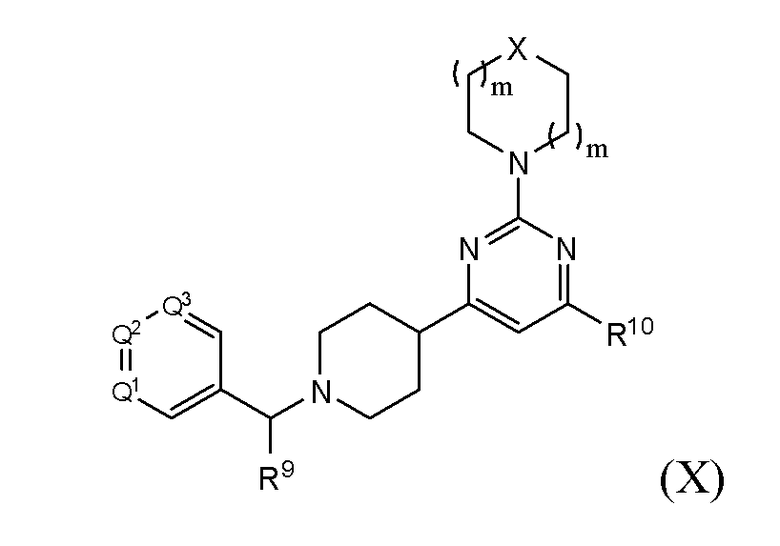
и их фармацевтически приемлемые соли, где
каждый R9 независимо представляет собой -H, -гидрокарбил, -арил, -аралкил, -гетероарил, -гетероциклил, -C(O)O(алкил), -OC(O)(алкил), -C(O)NH2, -C(O)NH(алкил), -C(O)N(алкил)(алкил), -NHC(O)(алкил), N(алкил)C(O)(алкил) или -SO2NH2;
каждый R10 независимо представляет собой -H, -OH, -NH2, -NH(алкил), -N(алкил)(алкил), гидрокарбил, -O-гидрокарбил, арил, -O-арил, аралкил, -O-аралкил, гетероарил, -O-гетероарил, гетероциклил, -O-гетероциклил, галоген, -OCF3, -C(O)O(алкил), -OC(O)(алкил), -C(O)NH2, -C(O)NH(алкил), -C(O)N(алкил)(алкил), -NHC(O)(алкил), N(алкил)C(O)(алкил), -OC(O)O(алкил), -OC(O)NH2, -OC(O)NH(алкил), -OC(O)N(алкил)(алкил), -CHNH, -CHN(алкил) или -SO2NH2;
каждый из Q1, Q2 и Q3 независимо представляет собой CR10 или N;
Х представляет собой CHR10, S, O или NR9; и
каждый m независимо представляет собой целое число от 0 до 3.
В другом варианте реализации в изобретении дополнительно предложены соединения следующей Формулы XI:
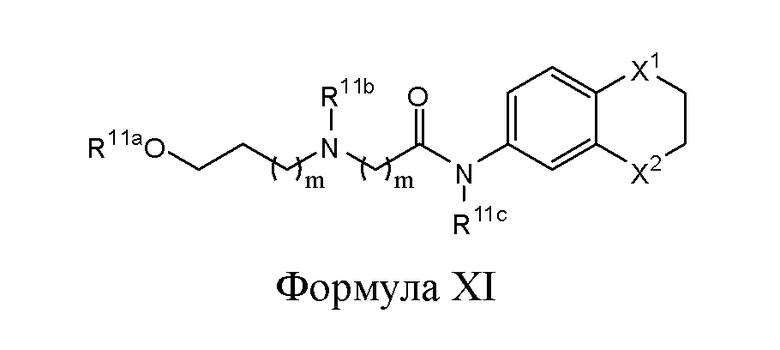
и их фармацевтически приемлемые соли, где
каждый из R11a, R11b и R11c независимо представляет собой H, гидрокарбил, арил, аралкил, гетероарил, гетероциклил, -C(O)(алкил), -C(O)O(алкил), -C(O)NH2, -C(O)NH(алкил), -C(O)N(алкил)(алкил) или -SO2NH2;
каждый из Х1 и Х2 независимо представляет собой CHR10, S, O, NR9 или N-ацил;
каждый R9 независимо представляет собой H, гидрокарбил, арил, аралкил, гетероарил, гетероциклил, -C(O)O(алкил), -OC(O)(алкил), -C(O)NH2, -C(O)NH(алкил), -C(O)N(алкил)(алкил), -NHC(O)(алкил), N(алкил)C(O)(алкил), -OC(O)O(алкил) или -SO2NH2;
каждый R10 независимо представляет собой -H, -OH, -NH2, -NH(алкил), -N(алкил)(алкил), гидрокарбил, -O-гидрокарбил, арил, -O-арил, аралкил, -O-аралкил, гетероарил, -O-гетероарил, гетероциклил, -O-гетероциклил, галоген, -OCF3, -C(O)O(алкил), -OC(O)(алкил), -C(O)NH2, -C(O)NH(алкил), -C(O)N(алкил)(алкил), -NHC(O)(алкил), N(алкил)C(O)(алкил), -OC(O)O(алкил), -OC(O)NH2, -OC(O)NH(алкил), -OC(O)N(алкил)(алкил), -CHNH, -CHN(алкил) или -SO2NH2; и
каждый m независимо представляет собой целое число от 0 до 3.
В другом варианте реализации в изобретении предложены соединения следующей Формулы (XII)
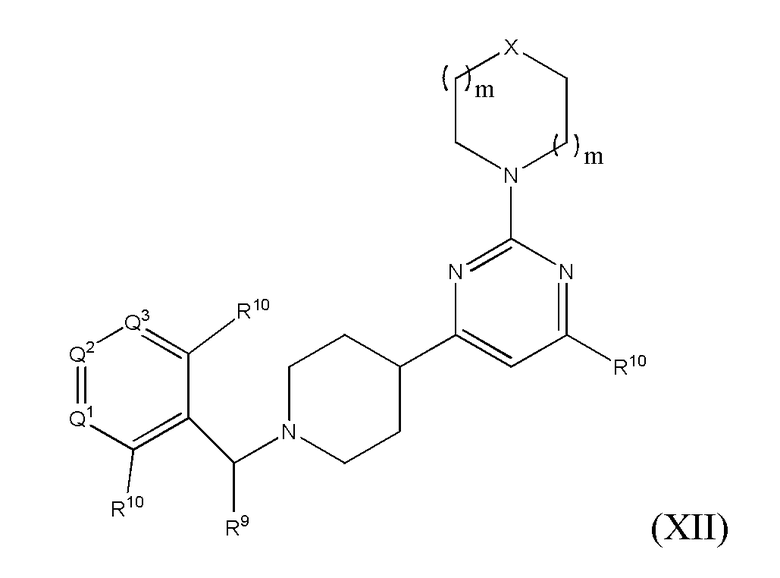
и их фармацевтически приемлемые соли, где
каждый R9 независимо представляет собой -H, -гидрокарбил, -арил, -аралкил, -гетероарил, -гетероциклил, -C(O)O(алкил), -OC(O)(алкил), -C(O)NH2, -C(O)NH(алкил), -C(O)N(алкил)(алкил), -NHC(O)(алкил), N(алкил)C(O)(алкил) или -SO2NH2;
каждый R10 независимо представляет собой -H, -OH, -NH2, -NH(алкил), -N(алкил)(алкил), гидрокарбил, -O-алкил, -O-алкенил, арил, -O-арил, аралкил, -O-аралкил, гетероарил, -O-гетероарил, гетероциклил, -O-гетероциклил, галоген, -OCF3, -C(O)O(алкил), -OC(O)(алкил), -C(O)NH2, -C(O)NH(алкил), -C(O)N(алкил)(алкил), NHC(O)(C2-C10-алкил), N(алкил)C(O)(алкил), -OC(O)O(алкил), -OC(O)NH2, -OC(O)NH(алкил), -OC(O)N(алкил)(алкил), -CHNH, -CHN(алкил), -SO2NH2, -S-алкил, -S-арил, -S-гетероарил, -S-гетероцикл или -S-гидрокарбил;
каждый из Q1, Q2 и Q3 независимо представляет собой CR10 или N;
Х представляет собой CHR10, S, O или NR9; и
каждый m независимо представляет собой целое число от 0 до 3.
В другом варианте реализации в изобретении предложены соединения следующей Формулы XIII:
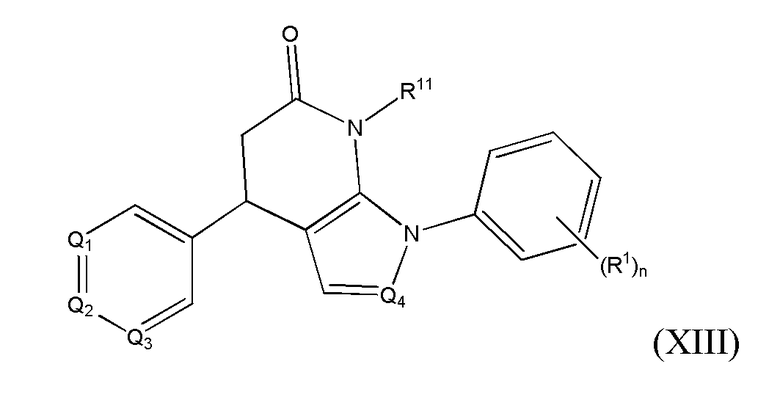
и их фармацевтически приемлемые соли, где
R1 независимо представляет собой -H, -OH, -NH2, -NH(алкил), -N(алкил)(алкил), гидрокарбил, -O-гидрокарбил, арил, -O-арил, аралкил, -O-аралкил, гетероарил, -O-гетероарил, гетероциклил, -O-гетероциклил, галоген, -OCF3, -C(O)O(алкил), -OC(O)(алкил), -C(O)NH2, -C(O)NH(алкил), -C(O)N(алкил)(алкил), -NHC(O)(алкил), N(алкил)C(O)(алкил), -OC(O)O(алкил), -OC(O)NH2, -OC(O)NH(алкил), -OC(O)N(алкил)(алкил), -CHNH, -CHN(алкил) или -SO2NH2;
R11 представляет собой H, гидрокарбил, арил, аралкил, гетероарил, гетероциклил, -C(O)O(алкил), -OC(O)(алкил), -C(O)NH2, -C(O)NH(алкил), -C(O)N(алкил)(алкил), -NHC(O)(алкил), N(алкил)C(O)(алкил) или -SO2NH2;
каждый из Q1, Q2, Q3 и Q4 независимо представляет собой CR10 или N;
n представляет собой целое число от 1 до 4; и
каждый R10 независимо представляет собой -H, -OH, -NH2, -NH(алкил), -N(алкил)(алкил), гидрокарбил, -O-гидрокарбил, арил, -O-арил, аралкил, -O-аралкил, гетероарил, -O-гетероарил, гетероциклил, -O-гетероциклил, галоген, -OCF3, -C(O)O(алкил), -OC(O)(алкил), -C(O)NH2, -C(O)NH(алкил), -C(O)N(алкил)(алкил), -NHC(O)(алкил), N(алкил)C(O)(алкил), -OC(O)O(алкил), -OC(O)NH2, -OC(O)NH(алкил), -OC(O)N(алкил)(алкил), -CHNH, -CHN(алкил) или -SO2NH2.
Соединение или фармацевтически приемлемая соль соединения любой из Формул (I)-(XIII) представляют собой «соединение согласно настоящему изобретению».
В другом варианте реализации в изобретении предложены композиции, содержащие эффективное количество соединения согласно настоящему изобретению и фармацевтически приемлемый наполнитель или носитель.
В другом варианте реализации в изобретении предложены способы лечения или предотвращения нарушения метаболизма липопротеинов, нарушения метаболизма глюкозы, сердечнососудистого нарушения или родственного сосудистого нарушения, нарушения, связанного с аномальной модуляцией С-реактивного белка, или родственного нарушения, возрастных заболеваний, болезни Альцгеймера, болезни Паркинсона, панкреатита, воспаления поджелудочной железы или аномальной выработки желчи (каждое из которых называют «Состояние»), включающие введение субъекту, нуждающемуся в этом, эффективного количества соединения согласно настоящему изобретению.
Согласно другому варианту реализации изобретение охватывает способы отслеживания хода лечения сердечнососудистого нарушения у субъекта, включающие:
а. определение уровня свободного холестерина липопротеинов высокой плотности в крови субъекта;
b. введение субъекту соединения согласно настоящему изобретению;
с. отслеживание уровня свободного холестерина в крови субъекта в течение периода времени после введения соединения; и
d. оценку уровня улучшения состояния субъекта на основании сравнения результатов, полученных на стадиях а. и с.
Согласно другому варианту реализации изобретение охватывает способы определения уровня активности P2Y13 у субъекта, включающие:
а. определение уровня свободного холестерина липопротеинов высокой плотности в крови субъекта;
b. введение субъекту соединения согласно настоящему изобретению;
с. отслеживание уровня свободного холестерина в крови субъекта в течение периода времени после введения соединения; и
d. оценку уровня активности P2Y13 у субъекта на основании сравнения результатов, полученных на стадиях а. и с.
Краткое описание чертежей
ФИГУРА 1: Активность соединения Va (черный треугольник; верхний график), соединения Ih (R-изомер) (черный треугольник; средний график) и соединения Ih (S-изомер) (черный треугольник; нижний график) в отношении клеток 1321 N1, трансфицированных рецептором P2Y13. Нетрансфицированные клетки 1321 N1 использовали в качестве отрицательного контроля (черные круги). Каждое значение соответствует среднему значению, полученному для трех лунок.
ФИГУРА 2: Активность иллюстративных соединений согласно настоящему изобретению (черные треугольники) в отношении клеток 1321 N1, трансфицированных рецептором P2Y13, по сравнению с положительным контролем с использованием соединения сравнения. Нетрансфицированные клетки использовали в качестве отрицательного контроля (черные круги). Каждое значение соответствует среднему значению, полученному для трех лунок.
ФИГУРА 3: Зависимость действия выбранных соединений на интернализацию ЛПВП клетками HepG2 от дозы. Клетки инкубировали в течение 10 минут при 37°С с 75 мкг/мл 3Н холестерина, холестерилолеат [холестерил-1,2-3Н(N)] ЛПВП, соединением сравнения (100 нМ) и иллюстративными соединениями согласно настоящему изобретению (1 мкМ). Данные выражали в виде изменения в процентах радиоактивности после интернализации относительно контрольного значения (принятого за 0). *p<0,005, **p<0,0001.
ФИГУРА 4: Зависимость интернализации ЛПВП клетками HepG2 от дозы выбранных соединений. Клетки инкубировали в течение 10 минут при 37°С с 75 мкг/мл 3Н холестерина, холестерилолеат [холестерил-1,2-3Н(N)] ЛПВП, соединением сравнения (100 нМ) и выбранными соединениями с увеличивающимися концентрациями. А: Соединения IIa и IIc (0,1, 1, 10, 100 и 1000 нМ). В: Соединение Ih (S-изомер) и соединение Ih (R-изомер) (1, 10, 100 и 1000 нМ). Данные выражены в виде изменения в процентах радиоактивности после интернализации относительно контрольного значения (принятого за 0).
ФИГУРА 5: Зависимость действия иллюстративных соединений согласно настоящему изобретению на секрецию желчных кислот от дозы IV инъекции. Мышам прекращали доступ к пище на 2 часа, а затем в хвостовую вену вводили инъекцию выбранных соединений (10 нмоль/кг) в растворе PBS (100 мкл). Через 4 часа анализировали состав желчи. А. Данные выражены в виде концентрации желчных кислот. В. Данные выражены в виде общего содержания желчных кислот в организме мыши (*p<0,05, **p<0,005, ***p<0,0001). Группа животных, n=5.
ФИГУРА 6: Зависимость действия соединения IIa на уровень секреции желчных кислот, фосфолипидов в желчь и холестерина в желчь от дозы. Мышам прекращали доступ к пище на 2 часа, а затем принудительно перорально вводили соединение IIa с увеличивающимися концентрациями (0,003, 0,03 и 0,3 мг/кг). Через 6 часов анализировали состав желчи. А. Данные выражены в виде концентрации желчных кислот. В. Данные выражены в виде общего содержания желчных кислот, отнесенного к массе тела мыши. С. Данные выражены в виде концентрации холестерина в желчи. D. Данные выражены в виде общего содержания холестерина в желчи, отнесенного к массе тела мыши. Е. Данные выражены в виде концентрации фосфолипидов в желчи. F. Данные выражены в виде общего содержания фосфолипидов в желчи, отнесенного к массе тела мыши. Группа животных, n=10. (*p<0,05, **p<0,005).
ФИГУРА 7: Зависимость действия соединения XIa на уровень секреции желчных кислот, фосфолипидов в желчь и холестерина в желчь от дозы. Мышам прекращали доступ к пище на 2 часа, а затем принудительно перорально вводили соединение XIa с увеличивающимися концентрациями (0,003, 0,03 и 0,3 мг/кг). Через 6 часов анализировали состав желчи. А. Данные выражены в виде концентрации желчных кислот. В. Данные выражены в виде общего содержания желчных кислот, отнесенного к массе тела мыши. С. Данные выражены в виде концентрации холестерина в желчи. D. Данные выражены в виде общего содержания холестерина в желчи, отнесенного к массе тела мыши. Е. Данные выражены в виде концентрации фосфолипидов в желчи. F. Данные выражены в виде общего содержания фосфолипидов в желчи, отнесенного к массе тела мыши. Группа животных, n=10. (*p<0,05, **p<0,005, ***p<0,0005, ****p<0,0001).
ФИГУРА 8: Зависимость действия соединения VIIa на уровень секреции желчных кислот, фосфолипидов в желчь и холестерина в желчь от дозы. Мышам прекращали доступ к пище на 2 часа, а затем принудительно перорально вводили соединение VIIa с увеличивающимися концентрациями (0,003, 0,03 и 0,3 мг/кг). Через 6 часов анализировали состав желчи. А. Данные выражены в виде концентрации желчных кислот. В. Данные выражены в виде общего содержания желчных кислот, отнесенного к массе тела мыши. С. Данные выражены в виде концентрации холестерина в желчи. D. Данные выражены в виде общего содержания холестерина в желчи, отнесенного к массе тела мыши. Е. Данные выражены в виде концентрации фосфолипидов в желчи. F. Данные выражены в виде общего содержания фосфолипидов в желчи, отнесенного к массе тела мыши. Группа животных, n=10. (*p<0,05, **p<0,01, ***p<0,005).
ФИГУРА 9: Зависимость действия соединения IIb на уровень секреции желчных кислот, фосфолипидов в желчь и холестерина в желчь от дозы. Мышам прекращали доступ к пище на 2 часа, а затем принудительно перорально вводили соединение IIb с увеличивающимися концентрациями (0,003, 0,03 и 0,3 мг/кг). Через 6 часов анализировали состав желчи. А. Данные выражены в виде концентрации желчных кислот. В. Данные выражены в виде общего содержания желчных кислот, отнесенного к массе тела мыши. С. Данные выражены в виде концентрации холестерина в желчи. D. Данные выражены в виде общего содержания холестерина в желчи, отнесенного к массе тела мыши. Е. Данные выражены в виде концентрации фосфолипидов в желчи. F. Данные выражены в виде общего содержания фосфолипидов в желчи, отнесенного к массе тела мыши. Группа животных, n=10. (*p<0,05, **p<0,005, ***p<0,0005).
ФИГУРА 10: Зависимость действия соединения VIIIa на уровень секреции желчных кислот, фосфолипидов в желчь и холестерина в желчь от дозы. Мышам прекращали доступ к пище на 2 часа, а затем принудительно перорально вводили соединение VIIIa с увеличивающимися концентрациями (0,003, 0,03 и 0,3 мг/кг). Через 6 часов анализировали состав желчи. А. Данные выражены в виде концентрации желчных кислот. В. Данные выражены в виде общего содержания желчных кислот, отнесенного к массе тела мыши. С. Данные выражены в виде концентрации холестерина в желчи. D. Данные выражены в виде общего содержания холестерина в желчи, отнесенного к массе тела мыши. Е. Данные выражены в виде концентрации фосфолипидов в желчи. F. Данные выражены в виде общего содержания фосфолипидов в желчи, отнесенного к массе тела мыши. Группа животных, n=10. (*p<0,05, **p<0,01, ***p<0,005, ****p<0,001, *****p<0,0005).
ФИГУРА 11: Зависимость действия соединения IIc на уровень секреции желчных кислот, фосфолипидов в желчь и холестерина в желчь от дозы. Мышам прекращали доступ к пище на 2 часа, а затем принудительно перорально вводили соединение IIc с увеличивающимися концентрациями (0,003, 0,03 и 0,3 мг/кг). Через 6 часов анализировали состав желчи. А. Данные выражены в виде концентрации желчных кислот. В. Данные выражены в виде общего содержания желчных кислот, отнесенного к массе тела мыши. С. Данные выражены в виде концентрации холестерина в желчи. D. Данные выражены в виде общего содержания холестерина в желчи, отнесенного к массе тела мыши. Е. Данные выражены в виде концентрации фосфолипидов в желчи. F. Данные выражены в виде общего содержания фосфолипидов в желчи, отнесенного к массе тела мыши. Группа животных, n=10. (*p<0,05, **p<0,005, ***p<0,0005).
ФИГУРА 12: Действие соединения Ih (рацемат) на секрецию желчных кислот, фосфолипидов в желчь и холестерина в желчь. Мышам прекращали доступ к пище на 2 часа, а затем принудительно перорально вводили соединение Ih (рацемат) (3, 30 и 100 мг/кг). Через шесть часов анализировали состав желчи. А: данные выражены в виде концентрации желчных кислот. В: данные выражены в виде общего содержания желчных кислот, отнесенного к массе тела мыши. С: данные выражены в виде концентрации холестерина в желчи. D: данные выражены в виде общего содержания холестерина в желчи, отнесенного к массе тела мыши. Е: данные выражены в виде концентрации фосфолипидов в желчи. F: данные выражены в виде общего содержания фосфолипидов в желчи, отнесенного к массе тела мыши. Группа животных, n=10. (*p<0,05, **p<0,005, ***p<0,0001).
ФИГУРА 13: Зависимость действия соединения IIa на секрецию желчных кислот от дозы после введения в течение одной недели. Мышам ежедневно принудительно перорально вводили соединение IIa (0,03 мг/кг). В день умерщвления мышам прекращали доступ к пище на 3 часа, затем принудительно перорально вводили соединение (в той же концентрации). Через 4 часа анализировали состав желчи. А: данные выражены в виде концентрации желчных кислот. В: данные выражены в виде общего содержания желчных кислот, отнесенного к массе тела мыши (*p<0,05). Группа животных, n=10.
ФИГУРА 14: Нокдаун P2Y13 в клетках Hepa 1-6. Клеткам Нера 1-6 трансдуцировали лентивирусные частицы, кодирующие кшРНК P2Y13 (MOI 40), индуцируемые путем добавления доксициклина. Клетки обрабатывали в течение 72 часов доксициклином (конечная концентрация 10 мкМ) или не обрабатывали и измеряли экспрессию иРНК P2Y13 путем КПЦР. *p<0,0001.
ФИГУРА 15: Захват ЛПВП в клетки Hepa 1-6 с нокдауном P2Y13. Клетки Нера 1-6, трансдуцированные лентивирусными частицами, кодирующими кшРНК P2Y13, индуцировали путем добавления доксициклина в течение 72 часов. Клетки инкубировали в течение 10 минут при 37°С с 75 мкг/мл 3Н холестерилового эфира ЛПВП и соединением IIa (1 мкМ). Данные выражены в виде изменения в процентах радиоактивности после интернализации относительно контрольного значения (принятого за 0). *p<0,05, **p<0,005.
ФИГУРА 16: Увеличение секреции желчи после активации пути рецептора P2Y13 у мышей. Мышам C57Bl/6J (n=10) прекращали доступ к пище на 2 часа, а затем вводили внутривенную инъекцию Cangrelor® или соединения IIa (10 нмоль/кг). Через 4 часа желчные пузыри удаляли и анализировали: Содержание желчных кислот (А); содержание холестерина в желчи (В). Мышам C57Bl/6J (n=10) прекращали доступ к пище на 2 часа, затем перорально вводили одну дозу агониста P2Y13, соединения IIa, в концентрациях 3, 30 или 300 мкг/кг. Через шесть часов содержание желчных кислот (С); содержание холестерина в желчи (D) и содержание желчных кислот в печени (Е) оценивали путем ВЭЖХ и с использованием ферментных наборов для желчных пузырей. Образцы плазмы собирали в момент времени 0 и через 6 часов после введения агониста, затем анализировали: общее содержание холестерина (▲), содержание неэтерифицированного холестерина (●), этерифицированного холестерина (■) и холестерина ЛПВП (▼) при помощи ферментных наборов или Lipoprint® в случае холестерина ЛПВП (F). Мышам C57Bl/6J (n=5) вводили внутривенную инъекцию меченого [3Н]-холестерина ЛПВП мыши (G), меченого [3Н]-холестерилолеата ЛПВП мыши (Н) или меченого [3Н]-холестерина ЛПНП мыши (I) и Cangrelor® или соединения IIa (10 нмоль/кг). Радиоактивность в печени определяли через 2 часа после определения уровня ЛПВП или через 5 часов после ЛПНП. Содержание холестерина в экскрементах животных (100 мкг/кг) определяли путем детектирования заряженных аэрозолей (J). *p<0,05, **p<0,01.
ФИГУРА 17: Зависимость действия агониста P2Y13 на прогрессирование атеросклеротических бляшек у мышей ApoE-/- от дозы. Мышам АроЕ-/- лигировали верхнюю часть левой сонной артерии. В день проведения хирургии животных помещали на Западную диету и принудительно перорально вводили носитель или соединение IIa с увеличивающимися дозами. Экстрагировали липиды из лигированных сонных артерий смесью 2:1 хлороформ/метанол. А: концентрации неэтерифицированного холестерина (темные столбцы) и общего холестерина (расположенные выше серые столбцы) измеряли путем ВЭЖХ. Е и Н: гематоксилин-эозиновое окрашивание продольного сечения сонных артерий мышей АроЕ-/-. F, I: окрашивание Oil Red O продольного сечения сонных артерий мышей АроЕ-/-. G и J: окрашивание антителом CD-68 продольного сечения сонных артерий мышей АроЕ-/-. В: количественная оценка соотношения интима/медиа (n=10). С: количественная оценка положительного окрашивания Oil Red O (n=10). D: количественная оценка окрашивания антителом CD68 (n=10). Мышам в течение 2 недель вводили носитель (Е, F и G) или соединение IIa в концентрации 100 мкг/кг (Н, I и J).
ФИГУРА 18: Влияние сайленсинга P2Y13 на прогрессирование атеросклеротических бляшек у мышей АроЕ-/-. Мышей АроЕ-/- (n=10) инфицировали 5х109 аденовирусными частицами, кодирующими пустой вектор (обычные), или вектор, кодирующий кшРНК рецептора P2Y13, за 3 дня до лигирования левой сонной артерии. В день проведения хирургии животных помещали на Западную диету, а также принудительно перорально вводили носитель или соединение IIa в концентрации 100 мкг/кг один раз в день в течение 2 недель. А: Вестерн-блот анализ гомогенатов печени, меченых антителами к P2Y13r или P2Y1r. В: Липиды экстрагировали из лигированных сонных артерий смесью 2:1 хлороформ/метанол. Концентрации неэтерифицированного холестерина (темные столбцы) и общего холестерина (расположенные выше серые столбцы) измеряли путем ВЭЖХ. С: концентрации этерифицированного (темные столбцы) и общего холестерина (расположенные выше серые столбцы) в плазме мышей измеряли при помощи ферментного набора. D, E и F: содержание в плазме холестерина ЛПОНП, холестерина ЛПНП и холестерина ЛПВП определяли путем ВЭЖХ на колонке Superose 6 с применением потокового детектирования ферментов. Темные столбцы соответствуют содержанию неэтерифицированного холестерина, серые столбцы - этерифицированного холестерина. *p<0,05, ** p<0,001.
ФИГУРА 19: Действие агониста P2Y13 на прогрессирование атеросклеротических бляшек в аортах мышей ароЕ-/-. Мышей АроЕ-/- (n=20) помещали на Западную диету на 8 недель, затем в течение 4 недель принудительно перорально вводили носитель или 100 мкг/кг соединения IIa. Липиды экстрагировали из аорт (n=10) смесью 2:1 хлороформ/метанол. А: общую концентрацию холестерина измеряли путем ВЭЖХ и ГХ/МС и сравнивали с исследуемой параллельно группой мышей ароЕ-/- (n=10), которую помещали на нормальную диету для грызунов, используемой в качестве исходного уровня. В: количественная оценка соотношения интима/медиа (n=10). С: количественная оценка окрашивания антителом F4/80 (n=10). D: количественная оценка положительного окрашивания Oil Red O (n=10). Е: количественная оценка окрашивания сириусом красным (n=10). F: количественная оценка окрашивания антителом к VCAM1 (n=10). Количества выражали в виде процентного соотношения площади окрашенной зоны и суммы площадей интимы и медиа. *p<0,05. G-L: слайды с типовыми примерами окрашивания аорт, используемые для количественной оценки. G и J: гематоксилин-эозиновое окрашивание поперечного сечения аорт мышей ароЕ-/-. Н и К: окрашивание Oil Red O поперечного сечения аорт мышей ароЕ-/-. I и L: окрашивание антителом к VCAM1 поперечного сечения аорт мышей ароЕ-/-.
ФИГУРА 20: Регрессия бляшек у кроликов, которым вводили соединение IIa. Новозеландским кроликам (n=15) с атеросклеротическими бляшками, развивавшимися в результате 2-месячной диеты с высоким содержанием холестерина, принудительно перорально вводили соединение IIa в концентрациях 30, 100 и 300 мкг/кг один раз в день в течение 4 недель. А: анализировали концентрацию холестерина в липидах, экстрагированных из аорт, путем ГХ/МС. Темные столбцы, неэтерифицированный холестерин; расположенные выше серые столбцы, общий холестерин. *p<0,05. В, С и D: Содержание в плазме холестерина ЛПОНП, холестерина ЛПНП и холестерина ЛПВП определяли согласно описанию Фигуры 2. Результаты выражены в виде изменения в процентах относительно момента до введения дозы. Е: концентрацию ароВ в плазме определяли путем Вестерн-блот анализа. F: содержание триглицеридов в плазме определяли с применением набора Biolabo. G: концентрации желчных кислот в печени определяли с применением ферментного набора. Изображения Н и К: гематоксилин/эозиновое окрашивание аорт кроликов, которым вводили носитель (Н) или соединение IIa в концентрации 300 мкг/кг (К). Изображения I и L: окрашивание клеток гладких мышц аорт кроликов, которым вводили носитель (I) или соединение IIa в концентрации 300 мкг/кг (L). Изображения J и М: окрашивание макрофагов и моноцитов аорт кроликов, которым вводили носитель (J) или соединение IIa в концентрации 300 мкг/кг (М).
ФИГУРА 21: Функциональное исследование плазмы кроликов, которых помещали на диету с высоким содержанием холестерина, которым вводили соединение IIa. А: концентрацию ApoA-I в плазме измеряли при помощи анализа SELDI-TOF. Чистый ApoA-I homo sapiens (MMSELDI=28083 Да) использовали в качестве примера сравнения для определения концентрации ApoA-I кролика (MMSELDI=27838 Да). Результаты выражены в виде изменения в процентах относительно момента после введения дозы. В: уровень иРНК ApoA-I определяли при помощи КПЦР. Необходимо отметить общее снижение размера частиц ЛПВП у животных, которым проводили лечение. С: ЛПВП из плазмы кролика разделяли по группам в соответствии с размером различных частиц ЛПВП с применением системы Lipoprint®. Данные выражены в виде изменения в процентах каждой подгруппы ЛПВП относительно содержания ЛПВП у животных после введения дозы. Количественно оценивали две основные подгруппы частиц ЛПВП (с высокой и промежуточной плотностью, черные и серые столбцы, соответственно). D: пример профилей липопротеинов кроликов, которым вводили носитель (серая линия) и соединение IIa (300 мкг/кг, черная линия), полученных с применением способа разделения Lipoprint®. Е и F: определение выхода холестерина в плазме и ЛПВП кролика, соответственно, с применением предварительно введенных макрофагов [3Н]-холестерин-окЛПНП. Результаты выражены в виде изменения в процентах выхода холестерина. *p<0,05, **p<0,005.
Подробное описание изобретения
I. Определения
Следующие определения используют в изобретении, описанном в настоящей заявке:
Термин «алкил», используемый в настоящем описании, если отсутствуют иные определения, относится к линейной, разветвленной или циклической насыщенной группе, полученной в результате удаления атома водорода в алкане. Типичные линейные алкильные группы включают -метил, -этил, -н-пропил, -н-бутил, -н-пентил и -н-гептил. Типичные разветвленные алкильные группы включают -изопропил, -втор-бутил, -изобутил, -трет-бутил, -изопентил, -неопентил, 1-метилбутил, 2-метилбутил, 3-метилбутил, 1,1-диметилпропил и 1,2-диметилпропил. Типичные циклические алкильные группы включают циклогексил, циклопентил и циклопропил.
Термин «алкенил» относится к линейной, разветвленной или циклической углеводородной группе, содержащей по меньшей мере одну двойную связь. Типичные алкенильные группы включают, но не ограничиваются ими, этилен, пропилен, 1-бутилен, 2-бутилен, изобутилен, втор-бутилен, 1-пентен, 2-пентен, изопентен, 1-гексен, 2-гексен, 3-гексен и изогексен.
Термин «алкинил» относится к линейному или разветвленному углеводороду, содержащему по меньшей мере одну тройную связь. Типичные алкинильные группы включают, но не ограничиваются ими, ацетилен, пропин, 1-бутин, 2-бутин, изобутин, втор-бутин, 1-пентин, 2-пентин, изопентин, 1-гексин, 2-гексин, 3-гексин и изогексин.
Термин «гидрокарбил», используемый в настоящем описании, если отсутствуют иные определения, относится к заместителю, полученному в результате удаления атома водорода от молекулы углеводорода. Неограничивающие примеры гидрокарбилов включают алкил, алкенил, алкинил; циклические группы, состоящие из атомов водорода и углерода, такие как арил, описанный в настоящей заявке, включая ароматические и неароматические группы, описанные в настоящей заявке; или аралкил, описанный в настоящей заявке.
Термин «арил», используемый в настоящем описании, если отсутствуют иные определения, относится к ароматической группе. Неограничивающие примеры арилов включают фенил, нафтил, пиридил, фенантрил, антрил, фуранил, азолил, имидазолил и индолил. В одном из вариантов реализации арильная группа замещена одной или более следующими группами: -галоген, -O-(C1-C6 алкил), -OH, -CN, -COOR', -OC(O)R', -N(R')2, -NHC(O)R' или -C(O)NHR', где каждый R' независимо представляет собой -H или незамещенный -C1-C6 алкил. Если конкретно не указано иное, арил является незамещенным.
Термин «гетероарил», используемый в настоящем описании, если отсутствуют иные определения, относится к ароматической группе, где ароматическая группа содержит по меньшей мере один атом в кольце, отличный от углерода. Неограничивающие примеры гетероарилов включают пиридил, фуранил, азолил, имидазолил, тиофенил и индолил. В одном из вариантов реализации арильная группа замещена одной или более следующими группами: -галоген, -O-(C1-C6 алкил), -OH, -CN, -COOR', -OC(O)R', -N(R')2, -NHC(O)R' или -C(O)NHR', где каждый R' независимо представляет собой -H или незамещенный -C1-C6 алкил. Если конкретно не указано иное, гетероарил является незамещенным.
Термин «аралкил», используемый в настоящем описании, если отсутствуют иные определения, относится к алкильной группе, которая замещена арильной группой. Неограничивающие примеры аралкильных групп включают бензил, пиколил, нафтилметил.
Термин «гетероциклил», используемый в настоящем описании, если отсутствуют иные определения, относится к циклической группе, где циклическая группа содержит по меньшей мере один атом в кольце, отличный от углерода. Типичные примеры гетероциклильных групп включают, но не ограничиваются ими, фуранил, фуразанил, имидазолидинил, имидазолинил, имидазолил, изотиазолил, изоксазолил, морфолинил, оксадиазолил, оксазолидинил, оксазолил, оксазолидинил, пиримидинил, фенантридинил, фенантролинил, пиперазинил, пиперидинил, пиранил, пиразинил, пиразолидинил, пиразолинил, пиразолил, пиридазинил, пиридооксазол, пиридоимидазол, пиридотиазол, пиридинил, пиримидинил, пирролидинил, пирролинил, хинуклидинил, тетрагидрофуранил, тиадиазинил, тиадиазолил, тиенил, тиенотиазолил, тиенооксазолил, тиеноимидазолил, тиоморфолинил, тиофенил, триазинил, триазолил. В одном из вариантов реализации арильная группа замещена одной или более следующими группами: -галоген, -O-(C1-C6 алкил), -OH, -CN, -COOR', -OC(O)R', -N(R')2, -NHC(O)R' или -C(O)NHR', где каждый R' независимо представляет собой -H или незамещенный -C1-C6 алкил. Если конкретно не указано иное, гетероцикл является незамещенным.
Термин «алкокси», используемый в настоящем описании, если отсутствуют иные определения, относится к группе -О-(алкил), где алкил такой, как определено выше. Типичные примеры С1-С6 алкокси включают, но не ограничиваются ими, -OCH3, -OCH2CH3, -OCH2CH2CH3, -OCH(CH3)CH3, -OCH2CH2CH2CH3, -OCH2CH(CH3)CH3, -OCH(CH3)CH2CH3, -OC(CH3)3, -OCH2CH2CH2CH2CH3, -OCH2CH(CH3)CH2CH3, -OCH2CH2CH2CH2CH2CH3 и -OCH2CH2CH(CH3)CH2CH3.
Термины «галоген-» и «галоген», используемые в настоящем описании, если отсутствуют иные определения, относятся к -F, -Cl, -Br или -I.
Термин «субъект», используемый в настоящем описании, если отсутствуют иные определения, представляет собой млекопитающее, например, человека, мышь, крысу, морскую свинку, собаку, кошку, лошадь, корову, свинью или отличного от человека примата, такого как мартышка, шимпанзе или бабуин. В одном из вариантов реализации субъект представляет собой человека.
Термин «фармацевтически приемлемая соль», используемый в настоящем описании, если отсутствуют иные определения, представляет собой соль основной группы, такой как аминогруппа, или кислой группы, такой как карбоксильная группа, соединений согласно настоящему изобретению. Иллюстративные соли основной группы включают, но не ограничиваются ими, сульфат, цитрат, ацетат, оксалат, хлорид, бромид, йодид, нитрат, бисульфат, фосфат, кислый фосфат, изоникотинат, лактат, салицилат, кислый цитрат, тартрат, олеат, таннат, пантотенат, битартрат, аскорбат, сукцинат, малеат, гентизинат, фумарат, глюконат, глюкаронат, сахарат, формиат, бензоат, глутамат, метансульфонат, этансульфонат, бензолсульфонат, п-толуолсульфонат, камфорсульфонат и памоат (т.е. 1,1'-метилен-бис-(2-гидрокси-3-нафтоат)). Иллюстративные соли кислой группы включают, но не ограничиваются ими, соли лития, натрия, калия, кальция, магния, алюминия, хрома, железа, меди, цинка, кадмия, аммония, гуанидиния, пиридиния и органические аммонийные соли.
Термины «гидрат» и «сольват», используемые в настоящем описании, если отсутствуют иные определения, описывают соединения согласно настоящему изобретению или их соли, которые дополнительно содержат стехиометрическое или нестехиометрическое количество воды или другого растворителя, связанного посредством нековалентных межмолекулярных связей.
Термин «обратный транспорт холестерина» (ОТХ), используемый в настоящем описании, если отсутствуют иные определения, описывает транспорт холестерина от внепеченочных тканей в печень, где он подвергается катаболизму и выводится из организма. Частицы ЛПВП могут играть основную роль в процессе обратного транспорта, выступая в качестве поглотителей холестерина, содержащегося в тканях.
Термин «изменение метаболизма липидов», используемый в настоящем описании, если отсутствуют иные определения, означает значительное (поддающееся измерению) изменение по меньшей мере одного аспекта метаболизма липидов, включая, но не ограничиваясь ими, общее содержание липидов в крови, содержание в крови холестерина ЛПВП, содержание в крови холестерина ЛПНП, содержание в крови холестерина ЛПОНП, содержание ТГ в крови, содержание Lp(a) в крови, содержание Apo A-I в крови, содержание Аро Е в крови и содержание НЭЖК в крови.
Термин «изменение метаболизма глюкозы», используемый в настоящем описании, если отсутствуют иные определения, означает значительное (поддающееся измерению) изменение по меньшей мере одного аспекта метаболизма глюкозы, включая, но не ограничиваясь ими, общее содержание глюкозы в крови, содержание инсулина в крови, соотношении содержания инсулина и глюкозы в крови, чувствительность к инсулину и потребление кислорода.
«Эффективное количество», используемое в отношении соединения согласно настоящему изобретению, представляет собой количество, которое является эффективным для лечения или предотвращения состояния, описанного в настоящей заявке.
«Эффективное количество», используемое в отношении другого терапевтического агента, представляет собой количество, которое является эффективным для лечения или предотвращения состояния в комбинации с соединением согласно настоящему изобретению. Термин «в комбинации с» включает введение в виде одной композиции или в отдельных композициях, причем в последнем случае другой терапевтический агент является эффективным для лечения или предотвращения состояния с учетом того, что соединение согласно настоящему изобретению уже осуществляет профилактическое или терапевтическое действие, и наоборот.
Формулировка «по существу не содержит соответствующий противоположный энантиомер» означает содержание не более примерно 10 мол.%, согласно другому варианту реализации не более примерно 5 мол.%, согласно другому варианту реализации не более примерно 2 мол.%, согласно другому варианту реализации не более примерно 1 мол.%, согласно другому варианту реализации не более примерно 0,5 мол.%, согласно другому варианту реализации не более примерно 0,1 мол.% соответствующего противоположного энантиомера.
Формулировка «по существу не содержит другой стереоизомер» означает содержание не более примерно 10 мол.%, согласно другому варианту реализации не более примерно 5 мол.%, согласно другому варианту реализации не более примерно 2 мол.%, согласно другому варианту реализации не более примерно 1 мол.%, согласно другому варианту реализации не более примерно 0,5 мол.%, согласно другому варианту реализации не более примерно 0,1 мол.% другого стереоизомера.
Термин «примерно», используемый в отношении указанного числового значения, означает указанное числовое значение плюс-минус 10% указанного числового значения. Например, формулировка «примерно 50» охватывает диапазон от 45 до 55.
Используемый в настоящем описании термин «пожилой человек» относится к человеку в возрасте 65 лет или старше.
Используемый в настоящем описании термин «взрослый человек» относится к человеку в возрасте 18 лет или старше.
Используемый в настоящем описании термин «ребенок» относится к человеку в возрасте от 1 года до 18 лет.
Используемый в настоящем описании термин «ребенок ясельного возраста» относится к человеку в возрасте от 1 года до 3 лет.
Используемый в настоящем описании термин «младенец» относится к человеку, начиная с рождения и до 1 года.
Используемый в настоящем описании термин «недоношенный ребенок» относится к человеку, рожденному менее чем через 37 недель беременности.
Используемый в настоящем описании термин «Apo(a)» относится к аполипопротеину (а).
Используемый в настоящем описании термин «Apo A-I» относится к аполипопротеину A-I.
Используемый в настоящем описании термин «Apo B» относится к аполипопротеину В.
Используемый в настоящем описании термин «Аро Е» относится к аполипопротеину Е.
Используемый в настоящем описании термин «ЖК» относится к жирным кислотам.
Используемый в настоящем описании термин «ЛПВП» относится к липопротеину высокой плотности.
Используемый в настоящем описании термин «ЛППП» относится к липопротеину промежуточной плотности.
Используемый в настоящем описании термин «ИЗСД» относится к инсулинозависимому сахарному диабету.
Используемый в настоящем описании термин «ЛДГ» относится к лактатдегидрогеназе.
Используемый в настоящем описании термин «ЛПНП» относится к липопротеину низкой плотности.
Используемый в настоящем описании термин «Lp(a)» относится к липопротеину (а).
Используемый в настоящем описании термин «ИНЗСД» относится к инсулинонезависимому сахарному диабету.
Используемый в настоящем описании термин «НЭЖК» относится к неэтерифицированным жирным кислотам.
Используемый в настоящем описании термин «P2Y13» относится к рецептору GPCR.
Используемый в настоящем описании термин «P2Y13r» относится к рецептору P2Y13.
Используемый в настоящем описании термины «P2Y13r» и «рецептор P2Y13» используют взаимозаменяемо.
Используемый в настоящем описании термин «RXR» относится к ретиноидному рецептору Х.
Используемый в настоящем описании термин «ТГ» относится к триглицеридам.
Используемый в настоящем описании термин «ЛПОНП» относится к липопротеину очень низкой плотности.
II. Соединения согласно настоящему изобретению
В одном из вариантов реализации в изобретении предложены соединения следующей Формулы I:
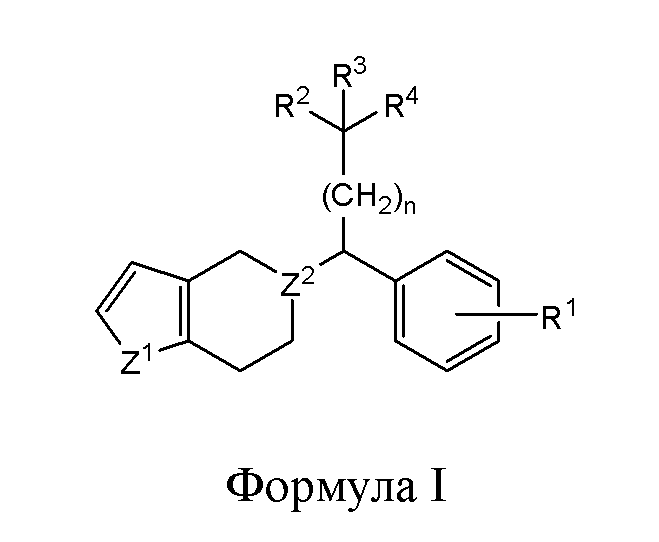
и их фармацевтически приемлемые соли, где
каждый из R1, R2 и R3 независимо представляет собой H, -OH, -NH2, -NH(алкил), -N(алкил)(алкил), гидрокарбил, -O-гидрокарбил, арил, -O-арил, аралкил, -O-аралкил, гетероарил, -O-гетероарил, гетероциклил, -O-гетероциклил, галоген, -OCF3, -C(O)O(алкил), -OC(O)(алкил), -C(O)NH2, -C(O)NH(алкил), -C(O)N(алкил)(алкил), -NHC(O)(алкил), N(алкил)C(O)(алкил), -OC(O)O(алкил), -OC(O)NH2, -OC(O)NH(алкил), -OC(O)N(алкил)(алкил), -CHNH, -CHN(алкил) или -SO2NH2;
R4 представляет собой -H, -OH, -COOH, -NH2, -NH(алкил), -N(алкил)(алкил), -гидрокарбил, -O-гидрокарбил, -арил, -O-арил, -аралкил, -O-аралкил, -гетероарил, -O-гетероарил, -гетероциклил, -O-гетероциклил, -галоген, -C(O)O(алкил), -OC(O)(алкил), -C(O)NH2, -C(O)NH(алкил), -C(O)N(алкил)(алкил), -NHC(O)(алкил), N(алкил)C(O)(алкил), -OC(O)O(алкил), -OC(O)NH2, -OC(O)NH(алкил) или -OC(O)N(алкил)(алкил);
Z1 представляет собой CH2, S, O, NH, N-гидрокарбил, N-арил, N-гетероарил или N-гетероциклил;
Z2 представляет собой СН или N; и
n представляет собой целое число от 1 до 6.
В некоторых вариантах реализации соединения Формулы I представляют собой соединения, где Z2 представляет собой N. В других вариантах реализации Z2 представляет собой СН.
В некоторых вариантах реализации R1 представляет собой галоген. В других вариантах реализации R1 представляет собой хлор. В других вариантах реализации R1 представляет собой 2-галоген. В других вариантах реализации R1 представляет собой 2-хлор. В других вариантах реализации R1 представляет собой Н.
В некоторых вариантах реализации Z1 представляет собой S. В других вариантах реализации Z1 представляет собой О. В других вариантах реализации Z1 представляет собой NH. В других вариантах реализации Z1 представляет собой N-алкил.
В некоторых вариантах реализации каждый из R2 и R3 независимо представляет собой H или алкил. В других вариантах реализации каждый из R2 и R3 представляет собой H. В других вариантах реализации каждый из R2 и R3 представляет собой алкил. В других вариантах реализации каждый из R2 и R3 представляет собой метил.
В некоторых вариантах реализации R4 представляет собой -OH, -COOH, -C(O)O(алкил) или -OC(O)(алкил). В других вариантах реализации R4 представляет собой -OH. В других вариантах реализации R4 представляет собой -COOH. В других вариантах реализации R4 представляет собой -C(O)O(алкил) или -OC(O)(алкил). В других вариантах реализации R4 представляет собой -COOEt. В других вариантах реализации R4 представляет собой -COOMe.
В некоторых вариантах реализации n представляет собой целое число от 1 до 6. В других вариантах реализации n равен 1. В других вариантах реализации n равен 2. В других вариантах реализации n равен 3. В других вариантах реализации n равен 4. В других вариантах реализации n равен 5. В других вариантах реализации n равен 6.
В некоторых вариантах реализации каждый из R2 и R3 представляет собой алкил, а R4 представляет собой -C(O)O-алкил. В других вариантах реализации каждый из R2 и R3 представляет собой метил, а R4 представляет собой -C(O)O-алкил. В других вариантах реализации каждый из R2 и R3 представляет собой алкил, а R4 представляет собой -C(O)OEt. В других вариантах реализации каждый из R2 и R3 представляет собой метил, а R4 представляет собой -C(O)OEt.
В некоторых вариантах реализации каждый из R2 и R3 представляет собой Н, а R4 представляет собой -C(O)O-алкил. В других вариантах реализации каждый из R2 и R3 представляет собой Н, а R4 представляет собой -C(O)OEt.
В некоторых вариантах реализации каждый из R2 и R3 представляет собой алкил, а R4 представляет собой -C(O)OH. В других вариантах реализации каждый из R2 и R3 представляет собой метил, а R4 представляет собой -C(O)OH.
В некоторых вариантах реализации каждый из R2 и R3 представляет собой Н, а R4 представляет собой -C(O)OH.
В некоторых вариантах реализации каждый из R2 и R3 представляет собой алкил, а R4 представляет собой -OH. В других вариантах реализации каждый из R2 и R3 представляет собой метил, а R4 представляет собой -OH.
В некоторых вариантах реализации каждый из R2 и R3 представляет собой H, а R4 представляет собой -OH.
В некоторых вариантах реализации Z1 и Z2 в соединениях Формулы I имеют следующие значения:
В некоторых вариантах реализации соединение Формулы I имеет структуру:
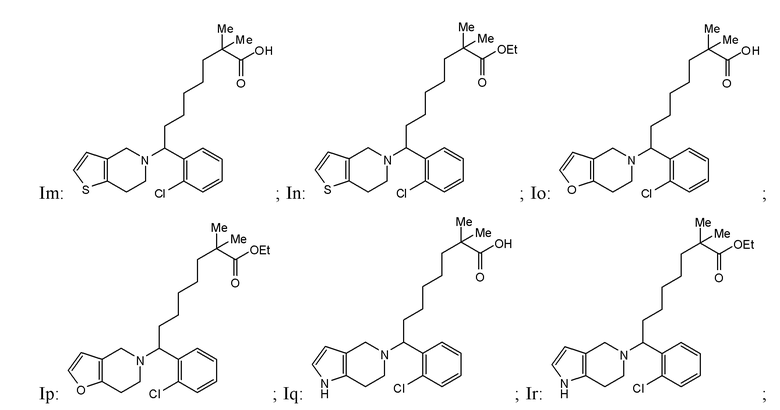
или Is:  или фармацевтически приемлемой соли любого из приведенных выше соединений.
или фармацевтически приемлемой соли любого из приведенных выше соединений.
В другом варианте реализации в изобретении предложены соединения следующей Формулы II:
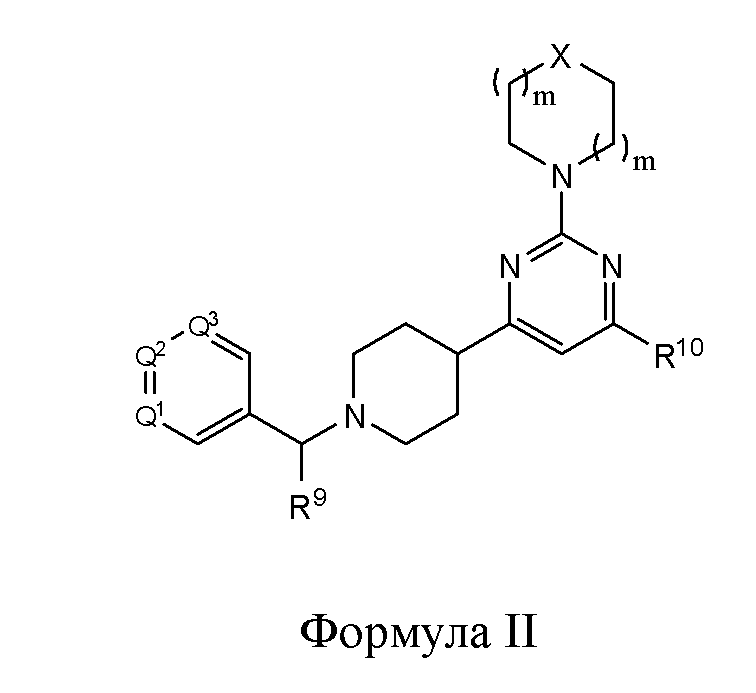
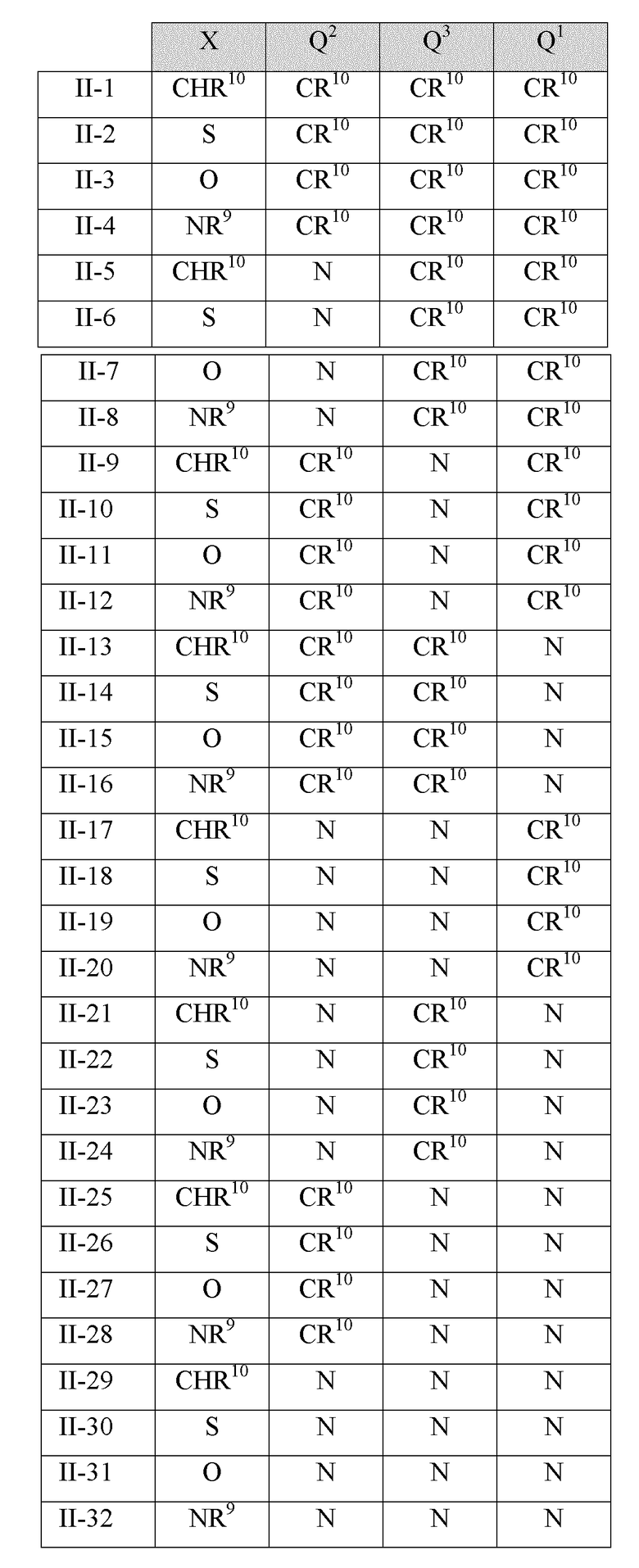
и их фармацевтически приемлемые соли, где
каждый R9 независимо представляет собой -H, -гидрокарбил, -арил, -аралкил, -гетероарил, -гетероциклил, -C(O)O(алкил), -OC(O)(алкил), -C(O)NH2, -C(O)NH(алкил), -C(O)N(алкил)(алкил), -NHC(O)(алкил), N(алкил)C(O)(алкил) или -SO2NH2;
каждый R10 независимо представляет собой -H, -OH, -NH2, -NH(алкил), -N(алкил)(алкил), гидрокарбил, -O-алкил, -O-алкенил, арил, -O-арил, аралкил, -O-аралкил, гетероарил, -O-гетероарил, гетероциклил, -O-гетероциклил, галоген, -OCF3, -C(O)O(алкил), -OC(O)(алкил), -C(O)NH2, -C(O)NH(алкил), -C(O)N(алкил)(алкил), NHC(O)(C2-C10-алкил), N(алкил)C(O)(алкил), -OC(O)O(алкил), -OC(O)NH2, -OC(O)NH(алкил), -OC(O)N(алкил)(алкил), -CHNH, -CHN(алкил) или -SO2NH2;
каждый из Q1, Q2 и Q3 независимо представляет собой CR10 или N;
Х представляет собой CHR10, S, O или NR9; и
каждый m независимо представляет собой целое число от 0 до 3.
В некоторых вариантах реализации соединения Формулы II представляют собой соединения, где R9 представляет собой H или гидрокарбил. В других вариантах реализации R9 представляет собой Н. В других вариантах реализации R9 представляет собой гидрокарбил. В других вариантах реализации R9 представляет собой алкил. В других вариантах реализации R9 представляет собой метил. В других вариантах реализации R9 представляет собой этил. В других вариантах реализации R9 представляет собой фенил.
В некоторых вариантах реализации соединения Формулы II представляют собой соединения, где R10 представляет собой H, -OH или гидрокарбил. В других вариантах реализации R10 представляет собой H. В других вариантах реализации R10 представляет собой -OH. В других вариантах реализации R10 представляет собой -OMe. В других вариантах реализации R10 представляет собой -OEt. В других вариантах реализации R10 представляет собой -NH2. В других вариантах реализации R10 представляет собой -NHMe. В других вариантах реализации R10 представляет собой -NMe2. В других вариантах реализации R10 представляет собой гидрокарбил. В других вариантах реализации R10 представляет собой алкил. В других вариантах реализации R10 представляет собой метил. В других вариантах реализации R10 представляет собой этил. В других вариантах реализации R10 представляет собой фенил.
В другом варианте реализации R9 представляет собой H, а R10 представляет собой -OH.
В некоторых вариантах реализации соединения Формулы II представляют собой соединения, где каждый из Q1, Q2 и Q3 представляет собой N. В других вариантах реализации каждый из Q1, Q2 и Q3 представляет собой CR10. В других вариантах реализации каждый из Q1, Q2 представляет собой N, а Q3 представляет собой CR10.
В некоторых вариантах реализации соединения Формулы II представляют собой соединения, где Х представляет собой СН2. В других вариантах реализации Х представляет собой О. В других вариантах реализации Х представляет собой NH. В других вариантах реализации Х представляет собой NMe. В других вариантах реализации Х представляет собой N-бензил.
В некоторых вариантах реализации соединения Формулы II представляют собой соединения, где каждый m независимо представляет собой целое число от 0 до 3. В других вариантах реализации каждый m независимо представляет собой целое число от 1 до 3. В других вариантах реализации m равен 0. В других вариантах реализации m равен 1. В других вариантах реализации m равен 2. В других вариантах реализации m равен 3.
В других вариантах реализации Q1 и Q3 представляют собой N, Q2 представляет собой CR10, а R10 представляет собой -N(алкил)(алкил). В других вариантах реализации Q1 и Q3 представляют собой N, Q2 представляет собой CR10, а R10 представляет собой -N(H)(алкил). В других вариантах реализации Q1 и Q3 представляют собой N, Q2 представляет собой CR10, а R10 представляет собой -N(CH3)2. В других вариантах реализации Q1 и Q3 представляют собой N, Q2 представляет собой CR10, а R10 представляет собой -N(H)(CH3).
В других вариантах реализации каждый m равен 1, Х представляет собой NR9, а R9 представляет собой H. В других вариантах реализации каждый m равен 1, а X представляет собой O.
В некоторых вариантах реализации Q1, Q2, Q3 и X в соединениях Формулы II имеют следующие значения:
В некоторых вариантах реализации соединение Формулы II существует в виде единственного таутомера или смеси таутомеров. Специалистам в данной области будут очевидны структуры, для которых возможно существование таутомерных форм, а также будет понятно, что изображение единственного таутомера охватывает структуру всех возможных таутомерных форм. В некоторых вариантах реализации R10 представляет собой ОН, а соединение Формулы II существует в одной, двух или трех таутомерных формах Формулы II, что проиллюстрировано в Уравнении I.
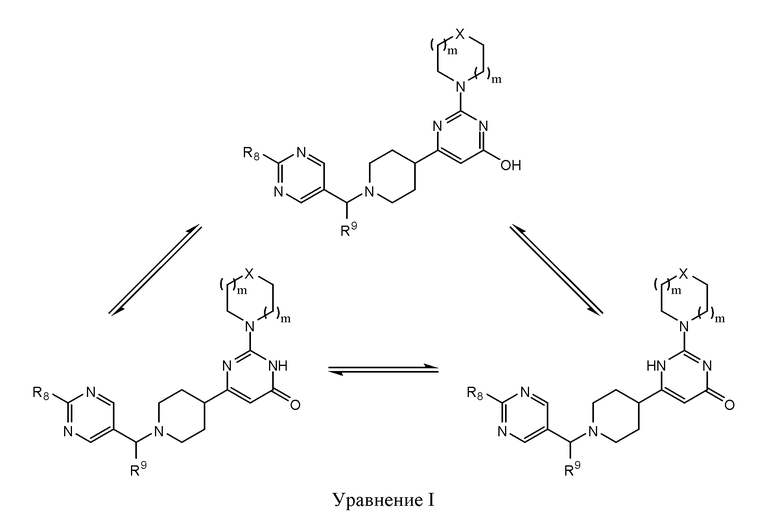
В некоторых вариантах реализации соединение Формулы II имеет структуру:
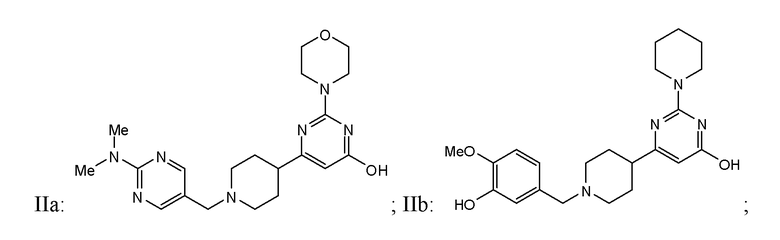
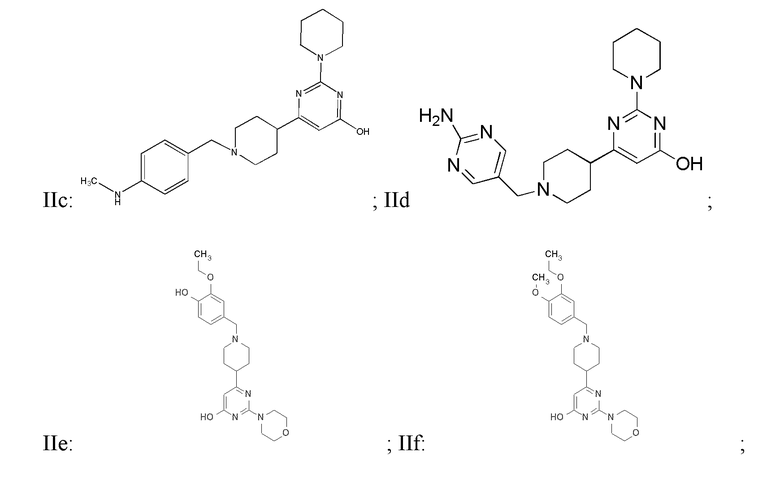
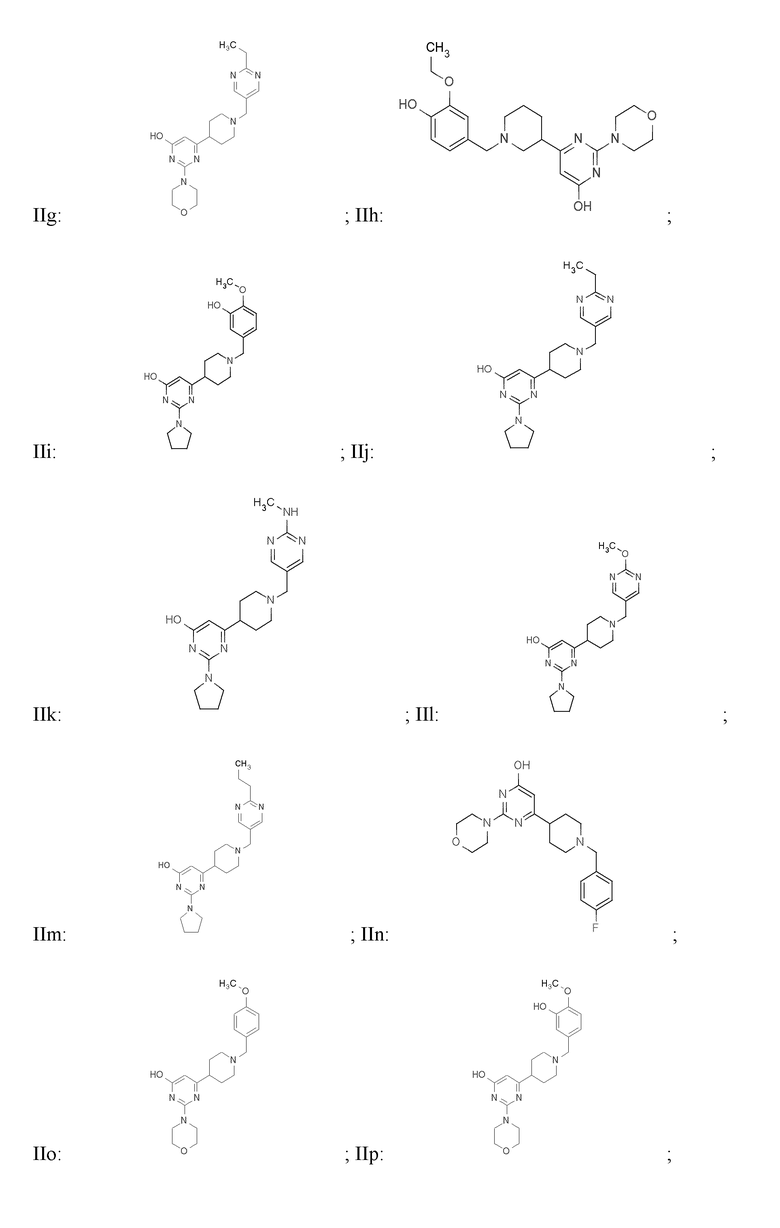
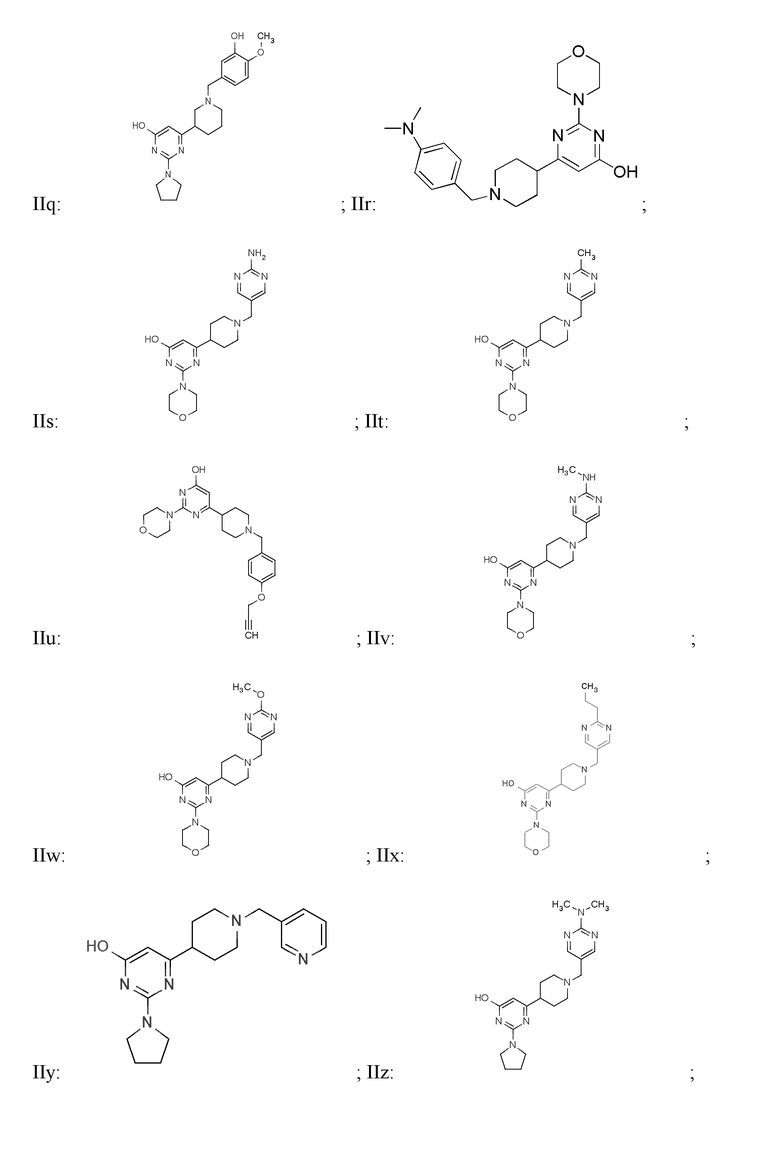
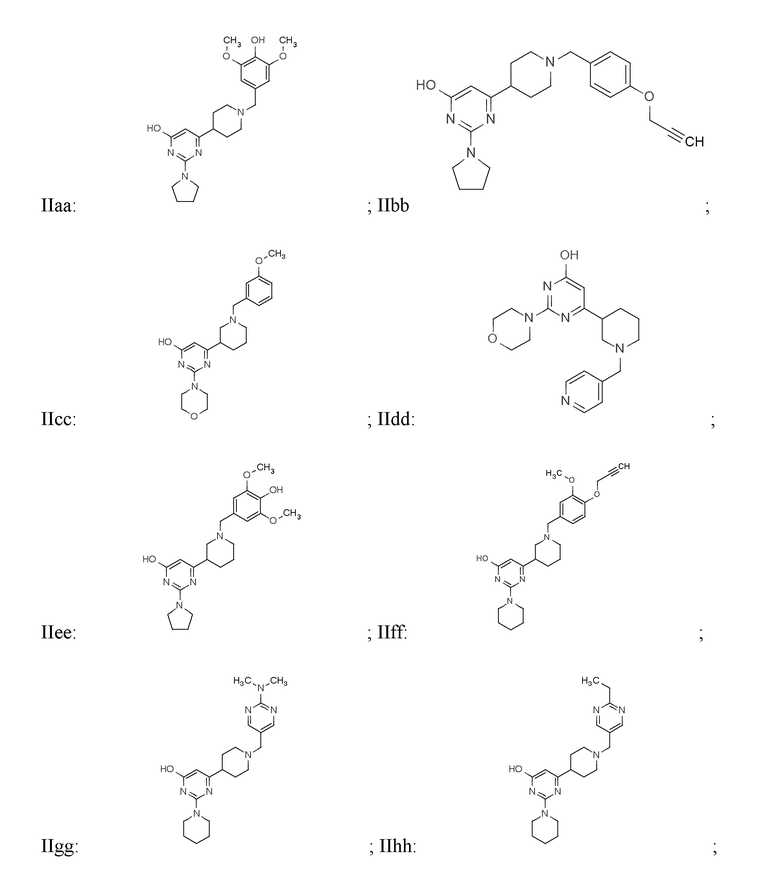
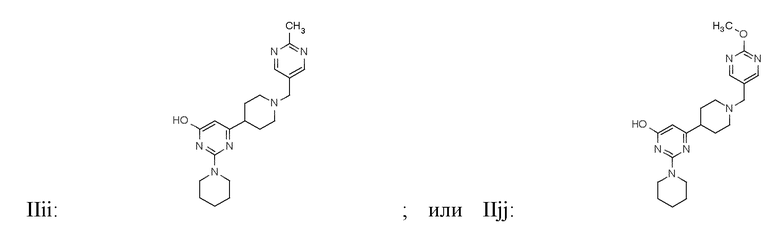
или фармацевтически приемлемой соли любого из приведенных выше соединений.
В другом варианте реализации изобретение охватывает соединения следующей Формулы III:
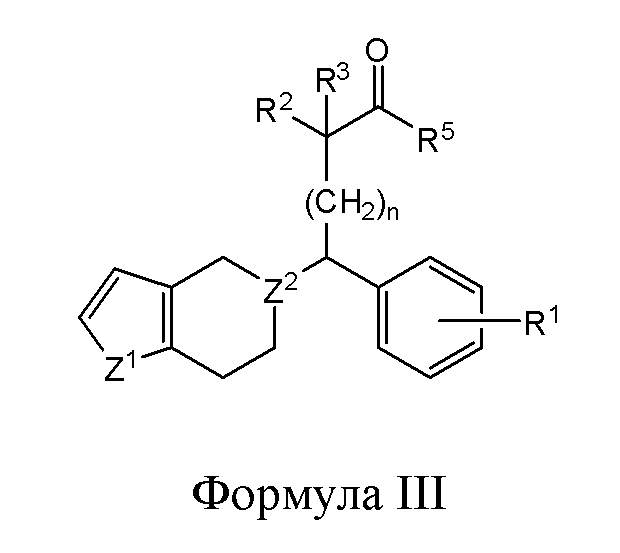
и их фармацевтически приемлемые соли, где
каждый из R1, R2 и R3 независимо представляет собой H, -OH, -NH2, -NH(алкил), -N(алкил)(алкил), гидрокарбил, -O-гидрокарбил, арил, -O-арил, аралкил, -O-аралкил, гетероарил, -O-гетероарил, гетероциклил, -O-гетероциклил, галоген, -OCF3, -C(O)O(алкил), -OC(O)(алкил), -C(O)NH2, -C(O)NH(алкил), -C(O)N(алкил)(алкил), -NHC(O)(алкил), N(алкил)C(O)(алкил), -OC(O)O(алкил), -OC(O)NH2, -OC(O)NH(алкил), -OC(O)N(алкил)(алкил), -CHNH, -CHN(алкил) или -SO2NH2;
R5 представляет собой -H, -OH, -NH2, -NH(алкил), -N(алкил)(алкил), гидрокарбил, -O-гидрокарбил, арил, -O-арил, аралкил, -O-аралкил, гетероарил, -O-гетероарил, гетероциклил или -O-гетероциклил;
Z1 представляет собой CH2, S, O, NH, N-гидрокарбил, N-арил, N-гетероарил или N-гетероциклил;
Z2 представляет собой СН или N; и
n представляет собой целое число от 1 до 6.
В некоторых вариантах реализации соединения Формулы III представляют собой соединения, где Z2 представляет собой N. В других вариантах реализации Z2 представляет собой СН.
В некоторых вариантах реализации R1 представляет собой галоген. В других вариантах реализации R1 представляет собой хлор. В других вариантах реализации R1 представляет собой 2-галоген. В других вариантах реализации R1 представляет собой 2-хлор. В других вариантах реализации R1 представляет собой H.
В некоторых вариантах реализации Z1 представляет собой S. В других вариантах реализации Z1 представляет собой O. В других вариантах реализации Z1 представляет собой NH. В других вариантах реализации Z1 представляет собой N-алкил.
В некоторых вариантах реализации каждый из R2 и R3 независимо представляет собой H или алкил. В других вариантах реализации каждый из R2 и R3 представляет собой H. В других вариантах реализации каждый из R2 и R3 представляет собой метил.
В некоторых вариантах реализации R5 представляет собой -OH, -O-алкил или -O-аралкил. В других вариантах реализации R5 представляет собой -OH. В других вариантах реализации R5 представляет собой -O-бензил. В других вариантах реализации R5 представляет собой -OEt. В других вариантах реализации R5 представляет собой -OMe.
В некоторых вариантах реализации n представляет собой целое число от 1 до 6. В других вариантах реализации n равен 1. В других вариантах реализации n равен 2. В других вариантах реализации n равен 3. В других вариантах реализации n равен 4. В других вариантах реализации n равен 5. В других вариантах реализации n равен 6.
В некоторых вариантах реализации Z1 и Z2 в соединениях Формулы III имеют следующие значения:
В другом варианте реализации в изобретении предложены соединения следующей Формулы IV:
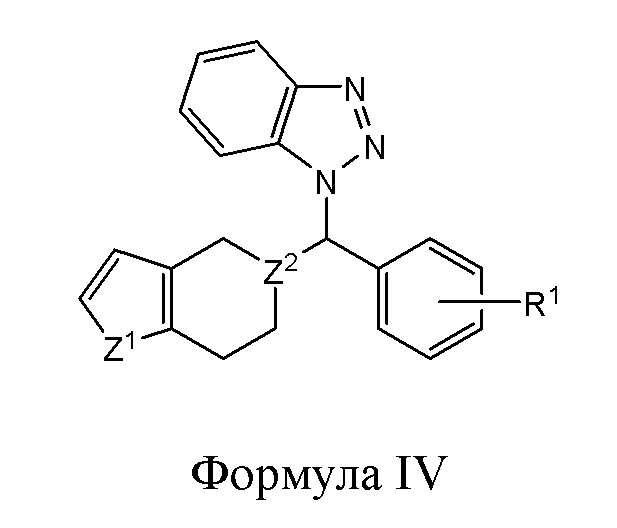
и их фармацевтически приемлемые соли, где
R1 представляет собой H, -OH, -NH2, -NH(алкил), -N(алкил)(алкил), гидрокарбил, -O-гидрокарбил, арил, -O-арил, аралкил, -O-аралкил, гетероарил, -O-гетероарил, гетероциклил, -O-гетероциклил, галоген, -OCF3, -C(O)O(алкил), -OC(O)(алкил), -C(O)NH2, -C(O)NH(алкил), -C(O)N(алкил)(алкил), -NHC(O)(алкил), N(алкил)C(O)(алкил), -OC(O)O(алкил), -OC(O)NH2, -OC(O)NH(алкил), -OC(O)N(алкил)(алкил), -CHNH, -CHN(алкил) или -SO2NH2;
Z1 представляет собой CH2, S, O, NH, N-гидрокарбил, N-арил, N-гетероарил или N-гетероциклил; и
Z2 представляет собой СН или N.
В некоторых вариантах реализации соединения Формулы IV представляют собой соединения, где Z2 представляет собой N. В других вариантах реализации Z2 представляет собой СН.
В некоторых вариантах реализации R1 представляет собой галоген. В других вариантах реализации R1 представляет собой хлор. В других вариантах реализации R1 представляет собой 2-галоген. В других вариантах реализации R1 представляет собой 2-хлор. В других вариантах реализации R1 представляет собой Н.
В некоторых вариантах реализации Z1 представляет собой S. В других вариантах реализации Z1 представляет собой O. В других вариантах реализации Z1 представляет собой NH. В других вариантах реализации Z1 представляет собой N-алкил.
В некоторых вариантах реализации Z1 и Z2 в соединениях Формулы IV имеют следующие значения:
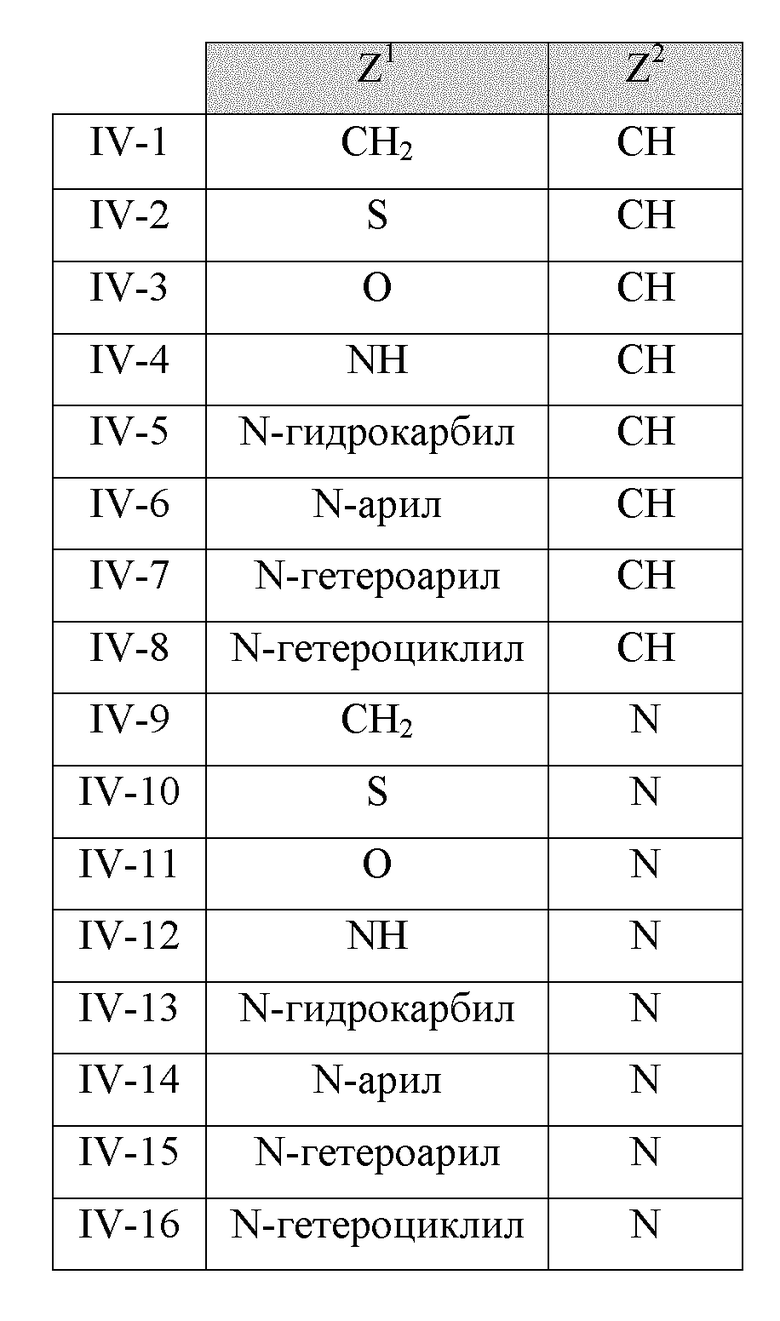
В некоторых вариантах реализации соединение Формулы IV имеет структуру IVa: 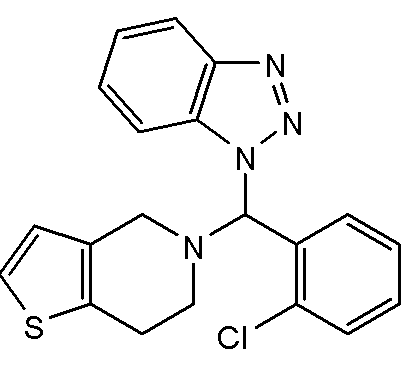 или ее фармацевтически приемлемой соли.
или ее фармацевтически приемлемой соли.
В одном из вариантов реализации изобретение охватывает соединения следующей Формулы V:
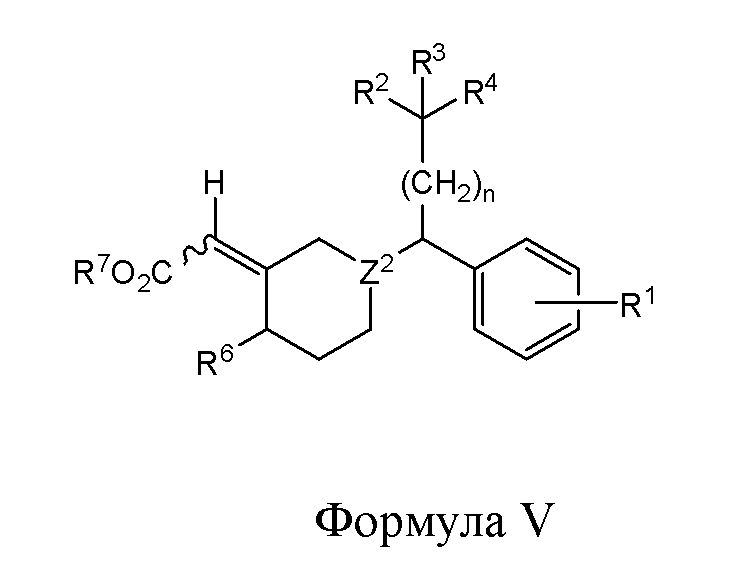
и их фармацевтически приемлемые соли, где
каждый из R1, R2 и R3 независимо представляет собой H, -OH, -NH2, -NH(алкил), -N(алкил)(алкил), гидрокарбил, -O-гидрокарбил, арил, -O-арил, аралкил, -O-аралкил, гетероарил, -O-гетероарил, гетероциклил, -O-гетероциклил, галоген, OCF3, -C(O)O(алкил), -OC(O)(алкил), -C(O)NH2, -C(O)NH(алкил), -C(O)N(алкил)(алкил), -NHC(O)(алкил), N(алкил)C(O)(алкил), -OC(O)O(алкил), -OC(O)NH2, -OC(O)NH(алкил), -OC(O)N(алкил)(алкил), -CHNH, -CHN(алкил) или -SO2NH2;
R4 представляет собой -H, -OH, -COOH, -NH2, -NH(алкил), -N(алкил)(алкил), гидрокарбил, -O-гидрокарбил, арил, -O-арил, аралкил, -O-аралкил, гетероарил, -O-гетероарил, гетероциклил, -O-гетероциклил, галоген, -C(O)O(алкил), -OC(O)(алкил), -C(O)NH2, -C(O)NH(алкил), -C(O)N(алкил)(алкил), -NHC(O)(алкил), N(алкил)C(O)(алкил), -OC(O)O(алкил), -OC(O)NH2, -OC(O)NH(алкил) или -OC(O)N(алкил)(алкил);
R6 представляет собой -H, -OH, -SH, -S-гидрокарбил, -COOH, -NH2, -NH(алкил), -N(алкил)(алкил), гидрокарбил, -O-гидрокарбил, арил, -O-арил, аралкил, -O-аралкил, гетероарил, -O-гетероарил, гетероциклил, -O-гетероциклил, галоген, -C(O)O(алкил), -OC(O)(алкил), -C(O)NH2, -C(O)NH(алкил), -C(O)N(алкил)(алкил), -NHC(O)(алкил), N(алкил)C(O)(алкил), -OC(O)O(алкил), -OC(O)NH2, -OC(O)NH(алкил) или -OC(O)N(алкил)(алкил);
R7 представляет собой Н, гидрокарбил, арил, аралкил, гетероарил или гетероциклил;
Z2 представляет собой СН или N; и
n представляет собой целое число от 1 до 6.
В некоторых вариантах реализации соединения Формулы V представляют собой соединения, где Z2 представляет собой N. В других вариантах реализации Z2 представляет собой СН.
В некоторых вариантах реализации R1 представляет собой галоген. В других вариантах реализации R1 представляет собой хлор. В других вариантах реализации R1 представляет собой 2-галоген. В других вариантах реализации R1 представляет собой 2-хлор. В других вариантах реализации R1 представляет собой Н.
В некоторых вариантах реализации каждый из R2 и R3 независимо представляет собой Н или алкил. В других вариантах реализации каждый из R2 и R3 представляет собой Н. В других вариантах реализации каждый из R2 и R3 представляет собой алкил. В других вариантах реализации каждый из R2 и R3 представляет собой метил.
В некоторых вариантах реализации R4 представляет собой -OH, -COOH, -C(O)O(алкил) или -OC(O)(алкил). В других вариантах реализации R4 представляет собой -OH. В других вариантах реализации R4 представляет собой -COOH. В других вариантах реализации R4 представляет собой -C(O)O(алкил) или -OC(O)(алкил). В других вариантах реализации R4 представляет собой -COOEt. В других вариантах реализации R4 представляет собой -COOMe.
В некоторых вариантах реализации R6 представляет собой -OH, -O-алкил, -SH, -S-алкил или алкил. В других вариантах реализации R6 представляет собой -OH. В других вариантах реализации R6 представляет собой -OMe. В других вариантах реализации R6 представляет собой H. В других вариантах реализации R6 представляет собой метил. В других вариантах реализации R6 представляет собой SH. В других вариантах реализации R6 представляет собой -SMe.
В некоторых вариантах реализации R7 представляет собой H. В других вариантах реализации R7 представляет собой метил. В других вариантах реализации R7 представляет собой этил. В других вариантах реализации R7 представляет собой бензил.
В некоторых вариантах реализации n представляет собой целое число от 1 до 6. В других вариантах реализации n равен 1. В других вариантах реализации n равен 2. В других вариантах реализации n равен 3. В других вариантах реализации n равен 4. В других вариантах реализации n равен 5. В других вариантах реализации n равен 6.
В некоторых вариантах реализации соединение Формулы V имеет структуру Va: 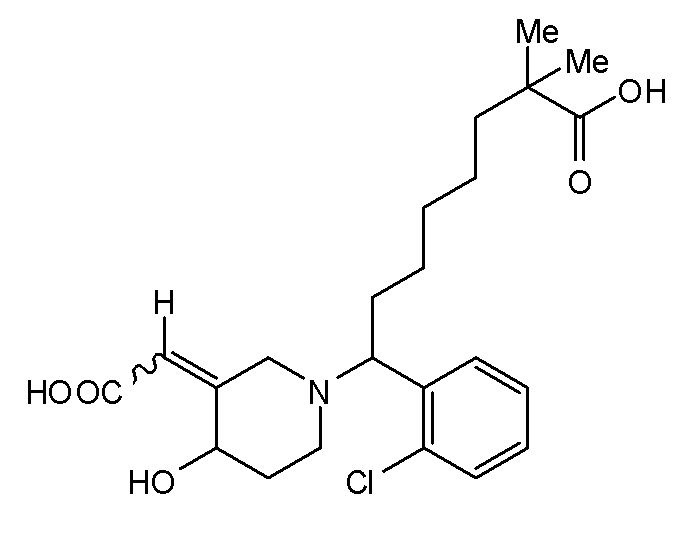 или ее фармацевтически приемлемой соли.
или ее фармацевтически приемлемой соли.
В одном из вариантов реализации изобретение охватывает соединения следующей Формулы VI:
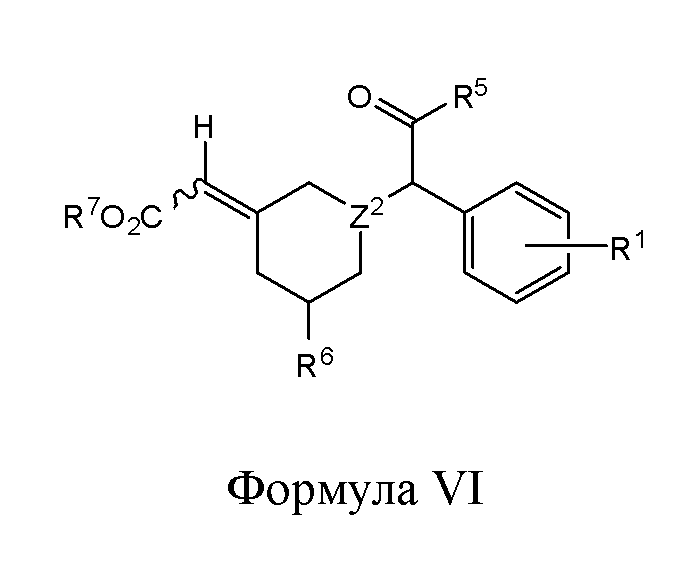
и их фармацевтически приемлемые соли, где
R1 представляет собой H, -OH, -NH2, -NH(алкил), -N(алкил)(алкил), гидрокарбил, -O-гидрокарбил, арил, -O-арил, аралкил, -O-аралкил, гетероарил, -O-гетероарил, гетероциклил, -O-гетероциклил, галоген, -OCF3, -C(O)O(алкил), -OC(O)(алкил), -C(O)NH2, -C(O)NH(алкил), -C(O)N(алкил)(алкил), -NHC(O)(алкил), N(алкил)C(O)(алкил), -OC(O)O(алкил), -OC(O)NH2, -OC(O)NH(алкил), -OC(O)N(алкил)(алкил), -CHNH, -CHN(алкил) или -SO2NH2;
R5 представляет собой -H, -OH, -NH2, -NH(алкил), -N(алкил)(алкил), гидрокарбил, -O-гидрокарбил, арил, -O-арил, аралкил, -O-аралкил, гетероарил, -O-гетероарил, гетероциклил, -O-гетероциклил или галоген;
R6 представляет собой -H, -OH, -SH, -S-гидрокарбил, -COOH, -NH2, -NH(алкил), -N(алкил)(алкил), гидрокарбил, -O-гидрокарбил, арил, -O-арил, аралкил, -O-аралкил, гетероарил, -O-гетероарил, гетероциклил, -O-гетероциклил, галоген, -C(O)O(алкил), -OC(O)(алкил), -C(O)NH2, -C(O)NH(алкил), -C(O)N(алкил)(алкил), -NHC(O)(алкил), N(алкил)C(O)(алкил), -OC(O)O(алкил), -OC(O)NH2, -OC(O)NH(алкил) или -OC(O)N(алкил)(алкил);
R7 представляет собой Н, гидрокарбил, арил, аралкил, гетероарил или гетероциклил; и
Z2 представляет собой СН или N.
В некоторых вариантах реализации соединения Формулы VI представляют собой соединения, где Z2 представляет собой N. В других вариантах реализации Z2 представляет собой СН.
В некоторых вариантах реализации R1 представляет собой галоген. В других вариантах реализации R1 представляет собой хлор. В других вариантах реализации R1 представляет собой 2-галоген. В других вариантах реализации R1 представляет собой 2-хлор. В других вариантах реализации R1 представляет собой Н.
В некоторых вариантах реализации R5 представляет собой OH, O-алкил или O-аралкил. В других вариантах реализации R5 представляет собой OH. В других вариантах реализации R5 представляет собой O-бензил. В других вариантах реализации R5 представляет собой OEt. В других вариантах реализации R5 представляет собой OMe.
В некоторых вариантах реализации R6 представляет собой -OH, -O-алкил, -SH, -S-алкил или алкил. В других вариантах реализации R6 представляет собой -OH. В других вариантах реализации R6 представляет собой -OMe. В других вариантах реализации R6 представляет собой -H. В других вариантах реализации R6 представляет собой метил. В других вариантах реализации R6 представляет собой -SH. В других вариантах реализации R6 представляет собой -SMe.
В некоторых вариантах реализации R7 представляет собой H. В других вариантах реализации R7 представляет собой метил. В других вариантах реализации R7 представляет собой этил. В других вариантах реализации R7 представляет собой бензил.
В некоторых вариантах реализации соединение Формулы VI имеет структуру
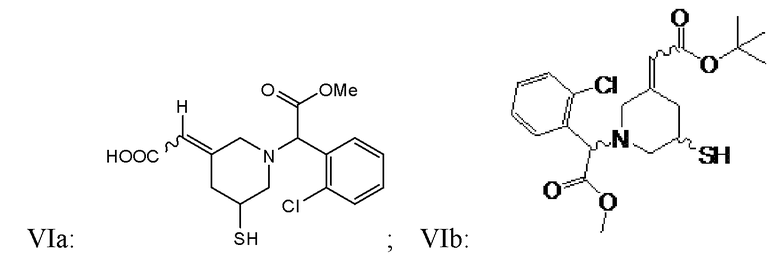
или их фармацевтически приемлемых солей.
В одном из вариантов реализации изобретение охватывает соединения следующей Формулы VII:
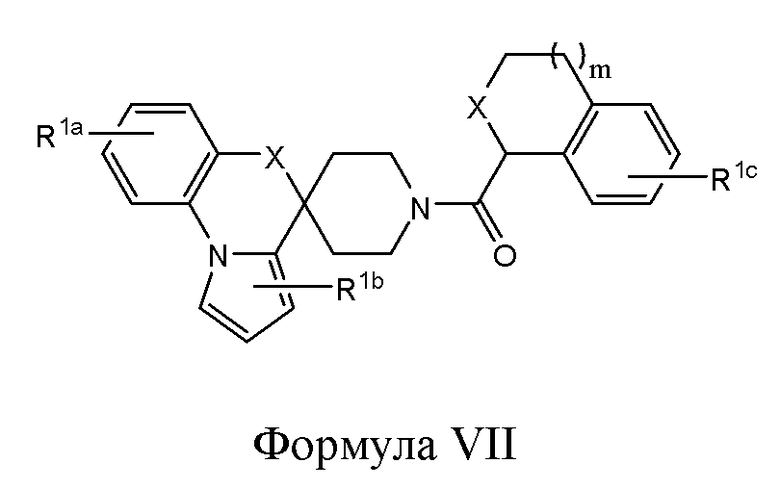
и их фармацевтически приемлемые соли, где
каждый из R1a, R1b и R1c независимо представляет собой -H, -OH, -NH2, -NH(алкил), -N(алкил)(алкил), гидрокарбил, -O-гидрокарбил, арил, -O-арил, аралкил, -O-аралкил, гетероарил, -O-гетероарил, гетероциклил, -O-гетероциклил, галоген, -OCF3, -C(O)O(алкил), -OC(O)(алкил), -C(O)NH2, -C(O)NH(алкил), -C(O)N(алкил)(алкил), -NHC(O)(алкил), N(алкил)C(O)(алкил), -OC(O)O(алкил), -OC(O)NH2, -OC(O)NH(алкил), -OC(O)N(алкил)(алкил), -CHNH, -CHN(алкил) или -SO2NH2;
каждый Х независимо представляет собой CHR10, S, O или NR9;
каждый R9 независимо представляет собой H, гидрокарбил, арил, аралкил, гетероарил, гетероциклил, -C(O)O(алкил), -OC(O)(алкил), -C(O)NH2, -C(O)NH(алкил), -C(O)N(алкил)(алкил), -NHC(O)(алкил), N(алкил)C(O)(алкил) или -SO2NH2;
каждый R10 независимо представляет собой -H, -OH, -NH2, -NH(алкил), -N(алкил)(алкил), гидрокарбил, -O-гидрокарбил, арил, -O-арил, аралкил, -O-аралкил, гетероарил, -O-гетероарил, гетероциклил, -O-гетероциклил, галоген, -OCF3, -C(O)O(алкил), -OC(O)(алкил), -C(O)NH2, -C(O)NH(алкил), -C(O)N(алкил)(алкил), -NHC(O)(алкил), N(алкил)C(O)(алкил), -OC(O)O(алкил), -OC(O)NH2, -OC(O)NH(алкил), -OC(O)N(алкил)(алкил), -CHNH, -CHN(алкил) или -SO2NH2; и
m представляет собой целое число от 0 до 3.
В некоторых вариантах реализации один или более из R1a, R1b и R1c представляет собой галоген. В других вариантах реализации один или более из R1a, R1b и R1c представляет собой хлор. В других вариантах реализации каждый из R1a, R1b и R1c представляет собой H.
В некоторых вариантах реализации Х представляет собой СН2. В других вариантах реализации Х представляет собой О. В других вариантах реализации Х представляет собой NH. В других вариантах реализации Х представляет собой NMe. В других вариантах реализации Х представляет собой N-бензил.
В некоторых вариантах реализации R9 представляет собой H или гидрокарбил. В других вариантах реализации R9 представляет собой H. В других вариантах реализации R9 представляет собой гидрокарбил. В других вариантах реализации R9 представляет собой алкил. В других вариантах реализации R9 представляет собой метил. В других вариантах реализации R9 представляет собой этил. В других вариантах реализации R9 представляет собой фенил.
В некоторых вариантах реализации R10 представляет собой H, -OH или гидрокарбил. В других вариантах реализации R10 представляет собой H. В других вариантах реализации R10 представляет собой -OH. В других вариантах реализации R10 представляет собой -OMe. В других вариантах реализации R10 представляет собой -OEt. В других вариантах реализации R10 представляет собой -NH2. В других вариантах реализации R10 представляет собой -NHMe. В других вариантах реализации R10 представляет собой -NMe2. В других вариантах реализации R10 представляет собой гидрокарбил. В других вариантах реализации R10 представляет собой алкил. В других вариантах реализации R10 представляет собой метил. В других вариантах реализации R10 представляет собой этил. В других вариантах реализации R10 представляет собой фенил.
В некоторых вариантах реализации m представляет собой целое число от 0 до 3. В других вариантах реализации m представляет собой целое число от 1 до 3. В других вариантах реализации m равен 0. В других вариантах реализации m равен 1. В других вариантах реализации m равен 2. В других вариантах реализации m равен 3.
В некоторых вариантах реализации изобретение охватывает соединения следующей формулы VII-1:
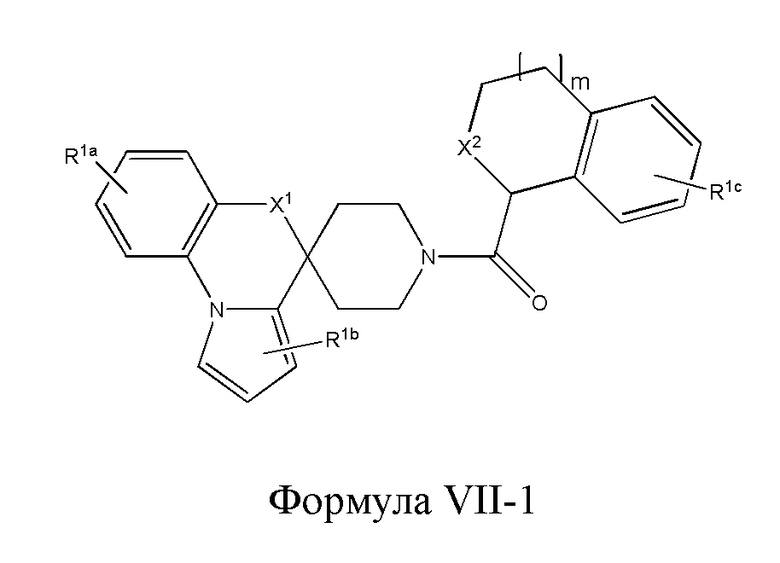
где R1a, R1b, R1c, m, R9 и R10 такие, как определено выше, а X1 и X2 имеют приведенное выше определение для Х.
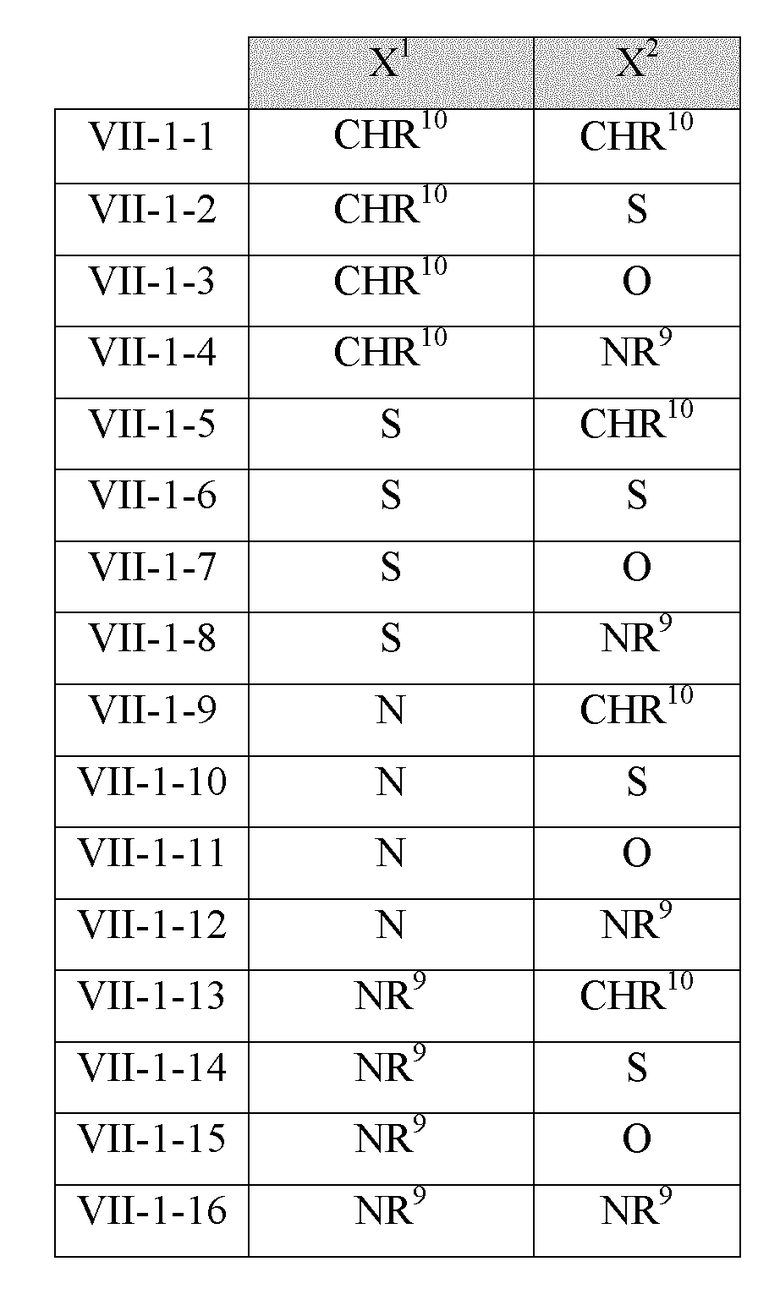
В некоторых вариантах реализации Х1 и Х2 в соединениях Формулы VII-1 имеют следующие значения:
В некоторых вариантах реализации соединение Формулы VII имеет структуру
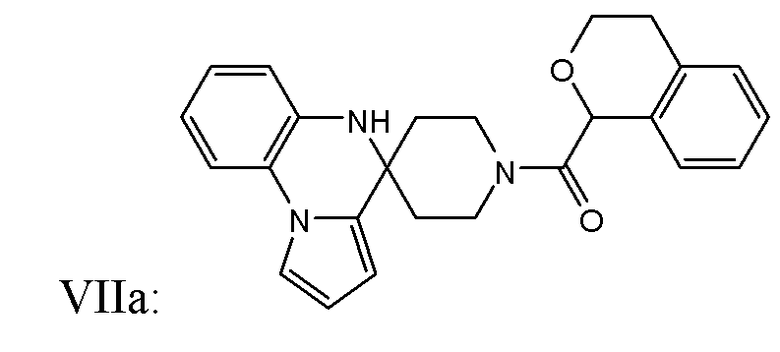 или ее фармацевтически приемлемой соли.
или ее фармацевтически приемлемой соли.
В другом варианте реализации изобретение охватывает соединения следующей Формулы VIII:
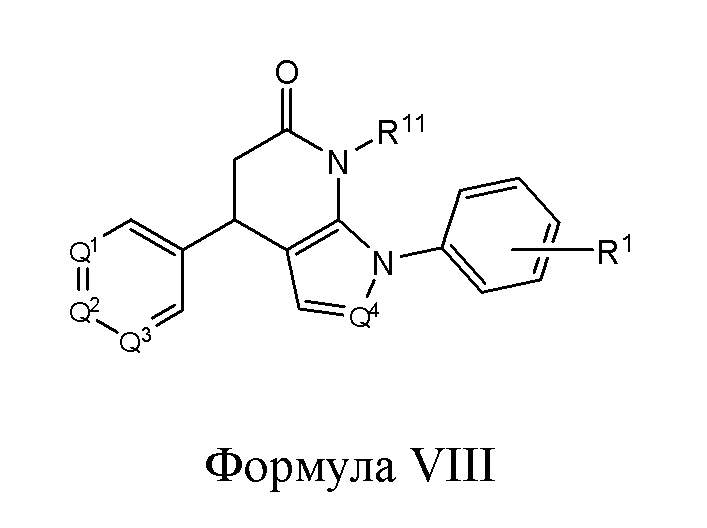
или их фармацевтически приемлемые соли, где
R1 представляет собой -H, -OH, -NH2, -NH(алкил), -N(алкил)(алкил), гидрокарбил, -O-гидрокарбил, арил, -O-арил, аралкил, -O-аралкил, гетероарил, -O-гетероарил, гетероциклил, -O-гетероциклил, галоген, -OCF3, -C(O)O(алкил), -OC(O)(алкил), -C(O)NH2, -C(O)NH(алкил), -C(O)N(алкил)(алкил), -NHC(O)(алкил), N(алкил)C(O)(алкил), -OC(O)O(алкил), -OC(O)NH2, -OC(O)NH(алкил), -OC(O)N(алкил)(алкил), -CHNH, -CHN(алкил) или -SO2NH2;
R11 представляет собой H, гидрокарбил, арил, аралкил, гетероарил, гетероциклил, -C(O)O(алкил), -OC(O)(алкил), -C(O)NH2, -C(O)NH(алкил), -C(O)N(алкил)(алкил), -NHC(O)(алкил), N(алкил)C(O)(алкил) или -SO2NH2;
каждый из Q1, Q2, Q3 и Q4 независимо представляет собой CR10 или N; и
каждый R10 независимо представляет собой -H, -OH, -NH2, -NH(алкил), -N(алкил)(алкил), гидрокарбил, -O-гидрокарбил, арил, -O-арил, аралкил, -O-аралкил, гетероарил, -O-гетероарил, гетероциклил, -O-гетероциклил, галоген, -OCF3, -C(O)O(алкил), -OC(O)(алкил), -C(O)NH2, -C(O)NH(алкил), -C(O)N(алкил)(алкил), -NHC(O)(алкил), N(алкил)C(O)(алкил), -OC(O)O(алкил), -OC(O)NH2, -OC(O)NH(алкил), -OC(O)N(алкил)(алкил), -CHNH, -CHN(алкил) или -SO2NH2.
В некоторых вариантах реализации R1 представляет собой алкил. В других вариантах реализации R1 представляет собой 2-алкил.
В других вариантах реализации Q1 представляет собой H, Q2 представляет собой CR10, и R10 представляет собой -OH, а Q3 представляет собой CR10, и R10 представляет собой O-алкил. В других вариантах реализации Q1 представляет собой CR10, и R10 представляет собой -O-алкил, Q2 представляет собой CR10 , и R10 представляет собой -OH, а Q3 представляет собой H.
В других вариантах реализации Q4 представляет собой N. В других вариантах реализации Q4 представляет собой CR10. В других вариантах реализации Q4 представляет собой С(Н).
В некоторых вариантах реализации R10 представляет собой H, -OH или гидрокарбил. В других вариантах реализации R10 представляет собой H. В других вариантах реализации R10 представляет собой -OH. В других вариантах реализации R10 представляет собой -OMe. В других вариантах реализации R10 представляет собой -OEt. В других вариантах реализации R10 представляет собой -NH2. В других вариантах реализации R10 представляет собой -NHMe. В других вариантах реализации R10 представляет собой -NMe2. В других вариантах реализации R10 представляет собой гидрокарбил. В других вариантах реализации R10 представляет собой алкил. В других вариантах реализации R10 представляет собой метил. В других вариантах реализации R10 представляет собой этил. В других вариантах реализации R10 представляет собой фенил.
В некоторых вариантах реализации R11 представляет собой H. В других вариантах реализации R11 представляет собой метил. В других вариантах реализации R11 представляет собой этил. В других вариантах реализации R11 представляет собой бензил.
В некоторых вариантах реализации Q1, Q2, Q3 и Q4 в соединениях Формулы VIII имеют следующие определения:
В некоторых вариантах реализации соединение Формулы VIII имеет структуру:
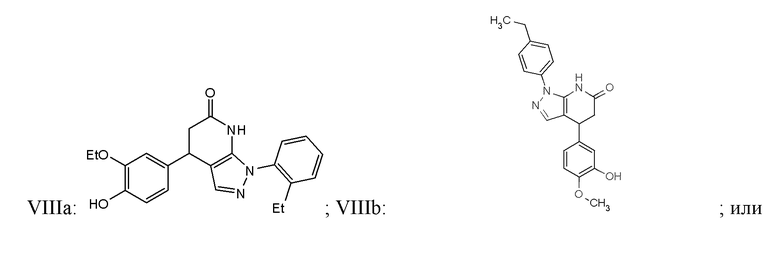
VIIIc: 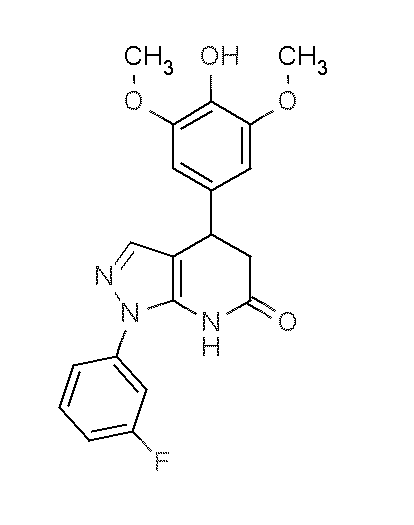 ; или их фармацевтически приемлемых солей.
; или их фармацевтически приемлемых солей.
В другом варианте реализации изобретение охватывает соединения следующей Формулы IX:
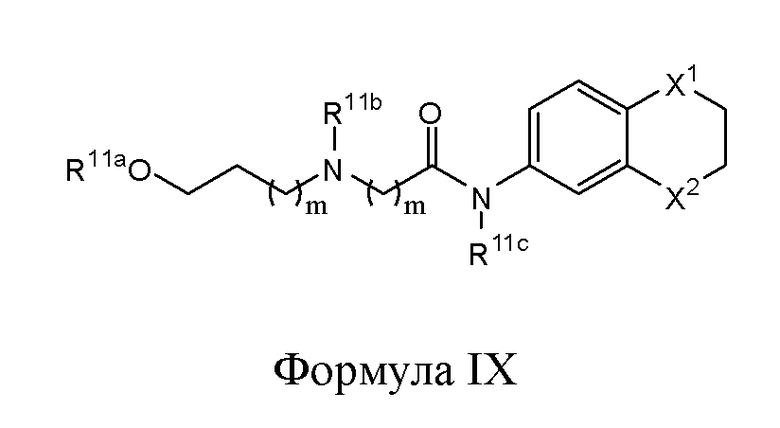
и их фармацевтически приемлемые соли, где
каждый из R11a, R11b и R11c независимо представляет собой Н, гидрокарбил, арил, аралкил, гетероарил, гетероциклил, -C(O)(алкил), -C(O)O(алкил), -C(O)NH2, -C(O)NH(алкил), -C(O)N(алкил)(алкил) или -SO2NH2;
каждый из X1 и X2 независимо представляет собой CHR10, S, O, NR9 или N-ацил;
каждый R9 независимо представляет собой H, гидрокарбил, арил, аралкил, гетероарил, гетероциклил, -C(O)O(алкил), -OC(O)(алкил), -C(O)NH2, -C(O)NH(алкил), -C(O)N(алкил)(алкил), -NHC(O)(алкил), N(алкил)C(O)(алкил), -OC(O)O(алкил) или -SO2NH2;
каждый R10 независимо представляет собой -H, -OH, -NH2, -NH(алкил), -N(алкил)(алкил), гидрокарбил, -O-гидрокарбил, арил, -O-арил, аралкил, -O-аралкил, гетероарил, -O-гетероарил, гетероциклил, -O-гетероциклил, галоген, -OCF3, -C(O)O(алкил), -OC(O)(алкил), -C(O)NH2, -C(O)NH(алкил), -C(O)N(алкил)(алкил), -NHC(O)(алкил), N(алкил)C(O)(алкил), -OC(O)O(алкил), -OC(O)NH2, -OC(O)NH(алкил), -OC(O)N(алкил)(алкил), -CHNH, -CHN(алкил) или -SO2NH2; и
каждый m независимо представляет собой целое число от 1 до 3.
В некоторых вариантах реализации каждый из X1 и X2 представляет собой CH2. В других вариантах реализации каждый из X1 и X2 представляет собой O. В других вариантах реализации каждый из X1 и X2 представляет собой NH. В других вариантах реализации каждый из X1 и X2 представляет собой NMe. В других вариантах реализации каждый из X1 и X2 представляет собой N-бензил.
В некоторых вариантах реализации R9 представляет собой Н или гидрокарбил. В других вариантах реализации R9 представляет собой Н. В других вариантах реализации R9 представляет собой гидрокарбил. В других вариантах реализации R9 представляет собой алкил. В других вариантах реализации R9 представляет собой метил. В других вариантах реализации R9 представляет собой этил. В других вариантах реализации R9 представляет собой фенил.
В некоторых вариантах реализации R10 представляет собой H, -OH или гидрокарбил. В других вариантах реализации R10 представляет собой H. В других вариантах реализации R10 представляет собой -OH. В других вариантах реализации R10 представляет собой -OMe. В других вариантах реализации R10 представляет собой -OEt. В других вариантах реализации R10 представляет собой -NH2. В других вариантах реализации R10 представляет собой -NHMe. В других вариантах реализации R10 представляет собой -NMe2. В других вариантах реализации R10 представляет собой гидрокарбил. В других вариантах реализации R10 представляет собой алкил. В других вариантах реализации R10 представляет собой метил. В других вариантах реализации R10 представляет собой этил. В других вариантах реализации R10 представляет собой фенил.
В некоторых вариантах реализации каждый из R11a, R11b и R11c представляет собой Н. В других вариантах реализации один или более из R11a, R11b и R11c представляют собой метил. В других вариантах реализации один или более из R11a, R11b и R11c представляют собой этил. В других вариантах реализации один или более из R11a, R11b и R11c представляют собой бензил. В других вариантах реализации один или более из R11a, R11b и R11c представляют собой ацетил. В других вариантах реализации один или более из R11a, R11b и R11c представляют собой бензоил.
В некоторых вариантах реализации каждый m независимо представляет собой целое число от 1 до 3. В других вариантах реализации m равен 1. В других вариантах реализации m равен 2. В других вариантах реализации m равен 3.
В некоторых вариантах реализации Х1 и Х2 в соединениях Формулы IX имеют следующие значения:
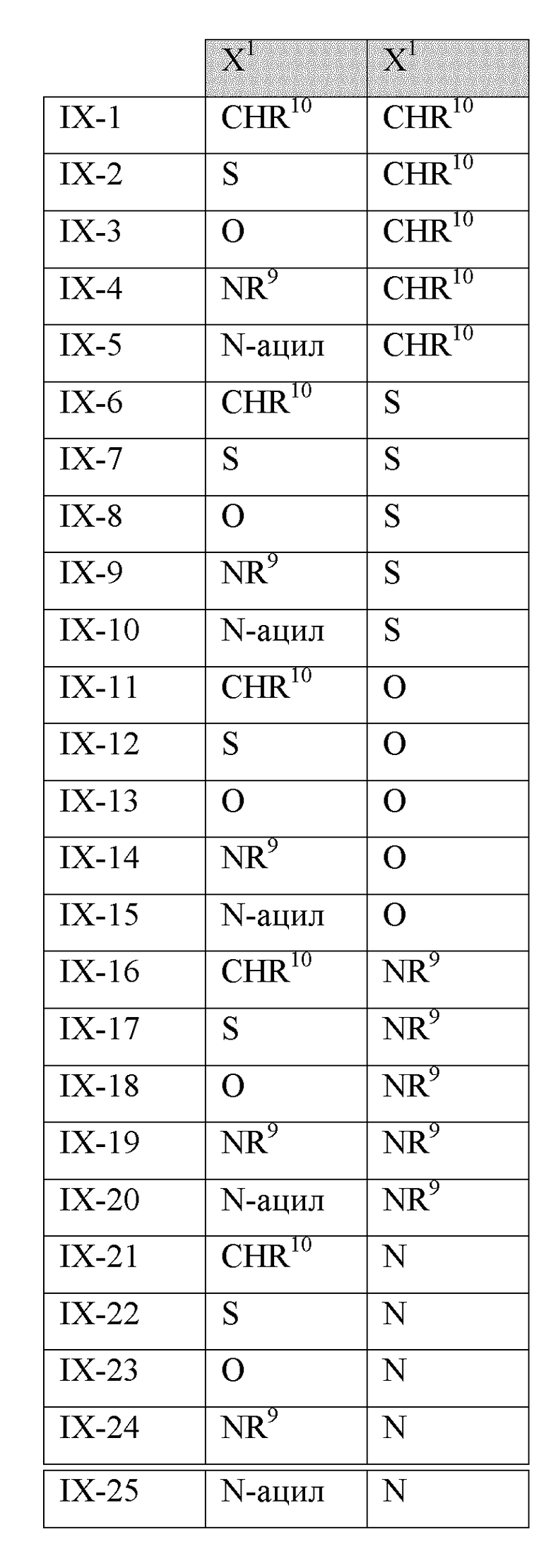
Соединения, описанные в настоящей заявке, могут содержать один или более хиральных центров и/или двойных связей и, таким образом, могут существовать в виде стереоизомеров, таких как геометрические изомеры, энантиомеры или диастереомеры. Изображения соединений согласно настоящему изобретению охватывают все возможные изомеры в виде смесей или очищенных форм. Очищенные формы могут представлять собой формы, обогащенные геометрическим изомером, энантиомерно обогащенные формы, диастереомерно обогащенные формы, формы, обогащенные оптическим изомером, геометрически чистые формы, энантиомерно чистые формы, диастереомерно чистые формы или оптически чистые формы. Смеси могут включать любые комбинации изомеров, такие как энантиомерные, диастереомерные, рацемические и стереоизомерные смеси. В одном из вариантов реализации соединения согласно настоящему изобретению существуют в виде единственного стереоизомера, по существу не содержащего другие стереоизомеры. В другом варианте реализации соединения согласно настоящему изобретению существуют в виде единственного энантиомера, по существу не содержащего соответствующий противоположный энантиомер. В другом варианте реализации соединения согласно настоящему изобретению существуют в виде рацематов.
Соединения согласно настоящему изобретению можно получать, выделять или очищать с получением рацематов, соединений, по существу не содержащих другие стереоизомеры, или по существу не содержащих соответствующий противоположный энантиомер, при помощи способов, известных специалистам в данной области техники, включая хиральную высокоэффективную жидкостную хроматографию, селективную кристаллизацию и взаимодействие с хиральным разделяющим агентом или хиральным вспомогательным веществом.
В одном из вариантов реализации соединение согласно настоящему изобретению представляет собой соединение или фармацевтически приемлемую соль соединения следующей Формулы Х:
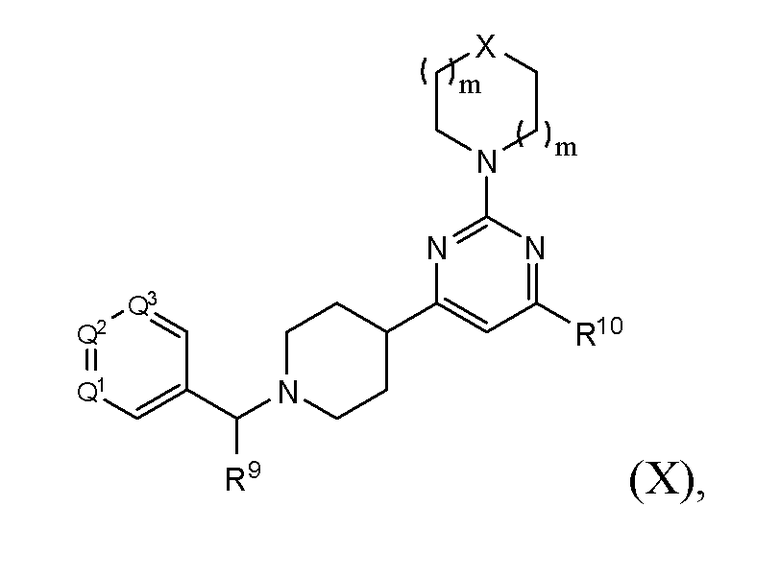
где
каждый R9 независимо представляет собой -H, -гидрокарбил, -арил, -аралкил, -гетероарил, -гетероциклил, -C(O)O(алкил), -OC(O)(алкил), -C(O)NH2, -C(O)NH(алкил), -C(O)N(алкил)(алкил), -NHC(O)(алкил), N(алкил)C(O)(алкил) или -SO2NH2;
каждый R10 независимо представляет собой -H, -OH, -NH2, -NH(алкил), -N(алкил)(алкил), гидрокарбил, -O-гидрокарбил, арил, -O-арил, аралкил, -O-аралкил, гетероарил, -O-гетероарил, гетероциклил, -O-гетероциклил, галоген, -OCF3, -C(O)O(алкил), -OC(O)(алкил), -C(O)NH2, -C(O)NH(алкил), -C(O)N(алкил)(алкил), -NHC(O)(алкил), N(алкил)C(O)(алкил), -OC(O)O(алкил), -OC(O)NH2, -OC(O)NH(алкил), -OC(O)N(алкил)(алкил), -CHNH, -CHN(алкил) или -SO2NH2;
каждый из Q1, Q2 и Q3 независимо представляет собой CR10 или N;
Х представляет собой CHR10, S, O или NR9; и
каждый m независимо представляет собой целое число от 0 до 3.
В некоторых вариантах реализации Q1, Q2, Q3 и X в соединениях Формулы Х имеют следующие значения:
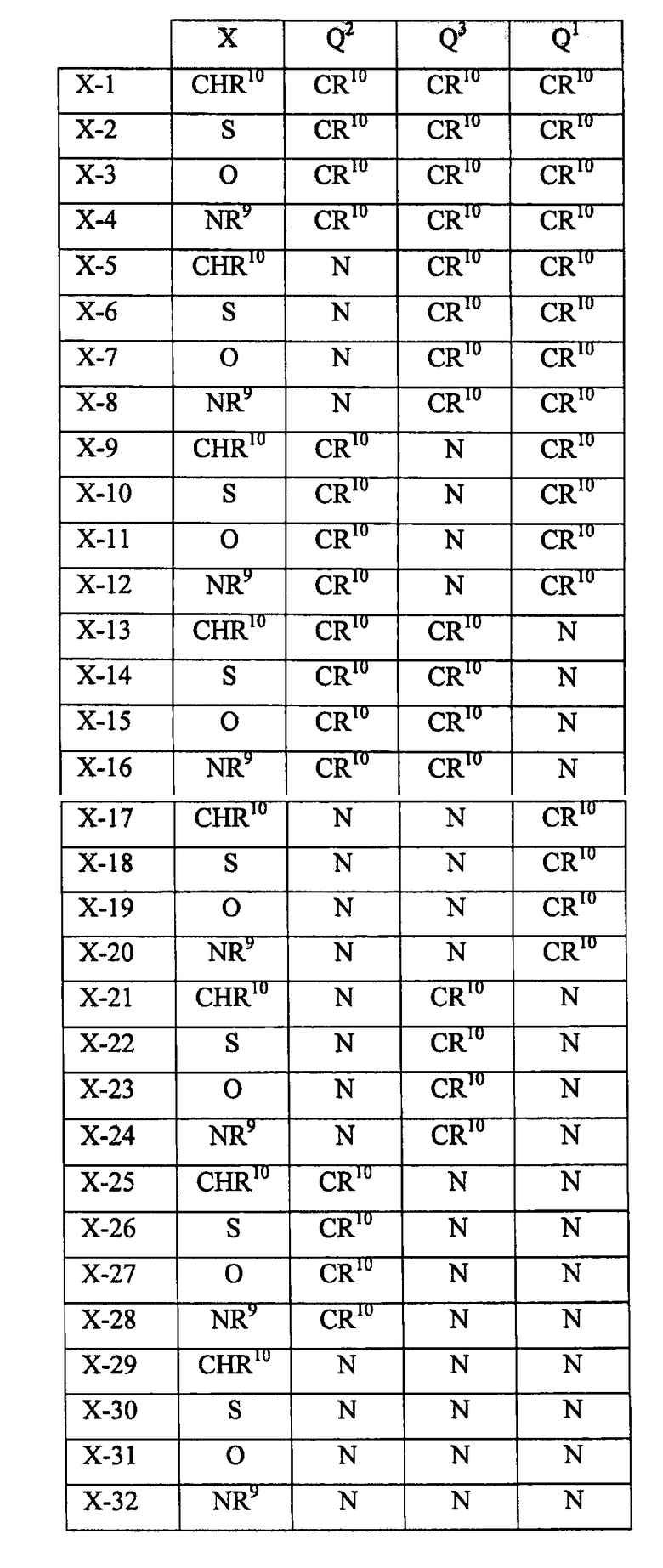
В одном из вариантов реализации соединение согласно настоящему изобретению представляет собой соединение или фармацевтически приемлемую соль соединения следующей Формулы XI:
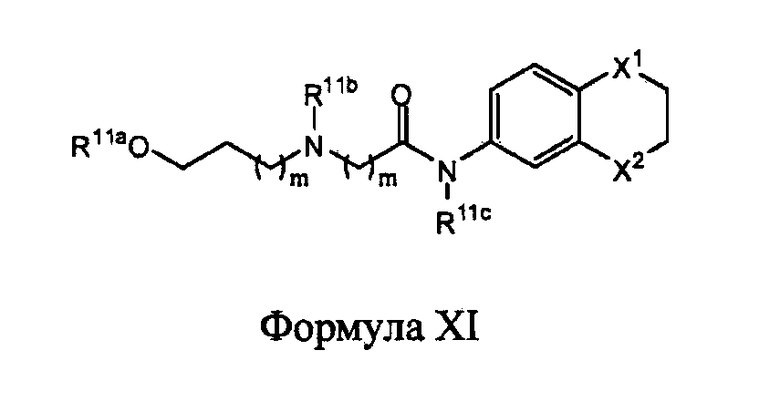
где
каждый из R11a, R11b и R11c независимо представляет собой H, гидрокарбил, арил, аралкил, гетероарил, гетероциклил, -C(O)(алкил), -C(O)O(алкил), -C(O)NH2, -C(O)NH(алкил), -C(O)N(алкил)(алкил) или -SO2NH2;
каждый из X1 и X2 независимо представляет собой CHR10, S, O, NR9 или N-ацил;
каждый R9 независимо представляет собой H, гидрокарбил, арил, аралкил, гетероарил, гетероциклил, -C(O)O(алкил), -OC(O)(алкил), -C(O)NH2, -C(O)NH(алкил), -C(O)N(алкил)(алкил), -NHC(O)(алкил), N(алкил)C(O)(алкил), -OC(O)O(алкил) или -SO2NH2;
каждый R10 независимо представляет собой -H, -OH, -NH2, -NH(алкил), -N(алкил)(алкил), гидрокарбил, -O-гидрокарбил, арил, -O-арил, аралкил, -O-аралкил, гетероарил, -O-гетероарил, гетероциклил, -O-гетероциклил, галоген, -OCF3, -C(O)O(алкил), -OC(O)(алкил), -C(O)NH2, -C(O)NH(алкил), -C(O)N(алкил)(алкил), -NHC(O)(алкил), N(алкил)C(O)(алкил), -OC(O)O(алкил), -OC(O)NH2, -OC(O)NH(алкил), -OC(O)N(алкил)(алкил), -CHNH, -CHN(алкил) или -SO2NH2; и
каждый m независимо представляет собой целое число от 0 до 3.
В некоторых вариантах реализации X1 и X2 в соединениях Формулы XI имеют следующие значения:
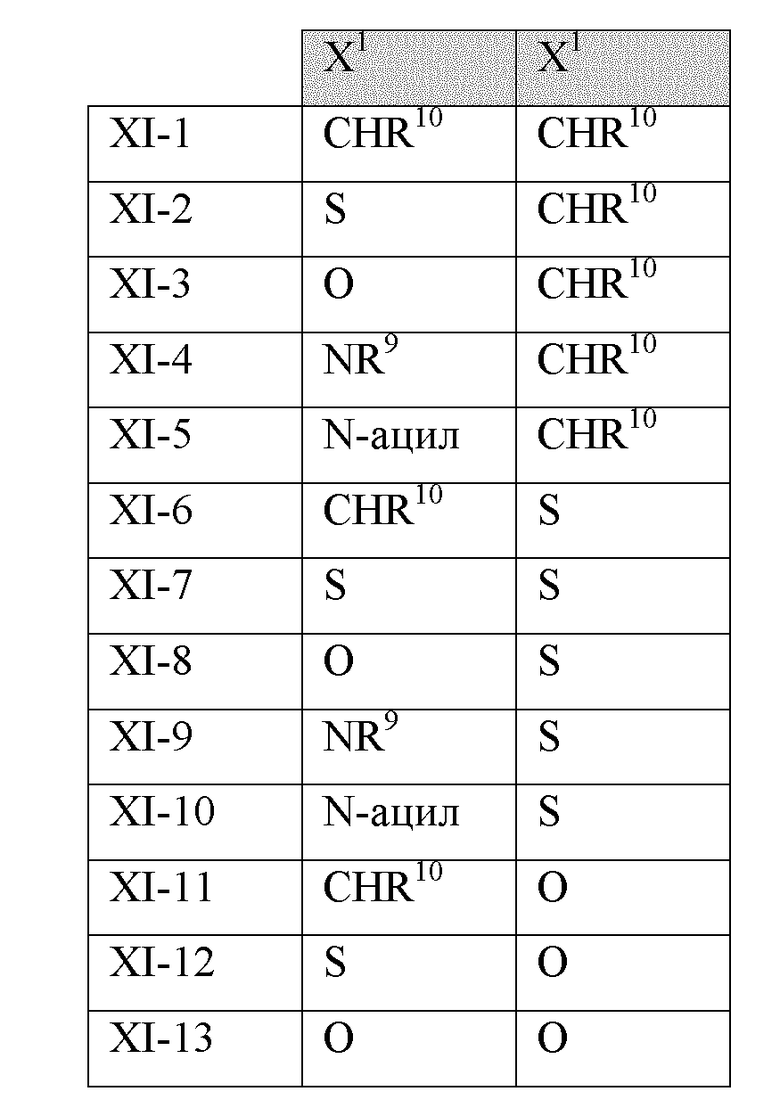
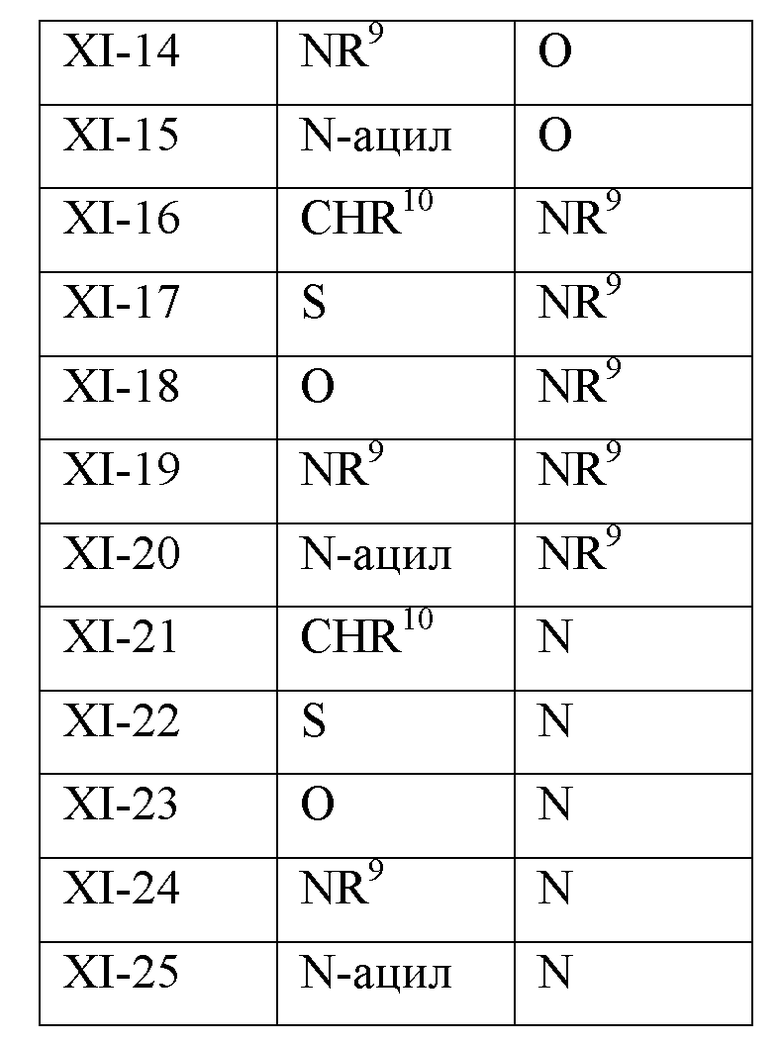
В одном из вариантов реализации соединение Формулы XI имеет структуру XIa:
 или ее фармацевтически приемлемой соли.
или ее фармацевтически приемлемой соли.
В другом варианте реализации в изобретении предложены соединения следующей Формулы XII:
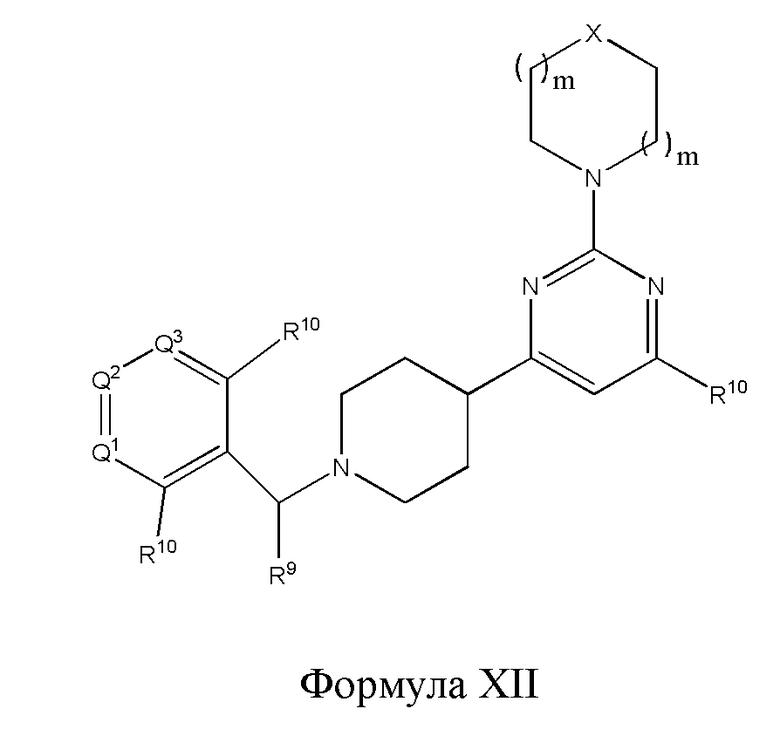
и их фармацевтически приемлемые соли, где
каждый R9 независимо представляет собой -H, -гидрокарбил, -арил, -аралкил, -гетероарил, -гетероциклил, -C(O)O(алкил), -OC(O)(алкил), -C(O)NH2, -C(O)NH(алкил), -C(O)N(алкил)(алкил), -NHC(O)(алкил), N(алкил)C(O)(алкил) или -SO2NH2;
каждый R10 независимо представляет собой -H, -OH, -NH2, -NH(алкил), -N(алкил)(алкил), гидрокарбил, -O-алкил, -O-алкенил, арил, -O-арил, аралкил, -O-аралкил, гетероарил, -O-гетероарил, гетероциклил, -O-гетероциклил, галоген, -OCF3, -C(O)O(алкил), -OC(O)(алкил), -C(O)NH2, -C(O)NH(алкил), -C(O)N(алкил)(алкил), NHC(O)(C2-C10-алкил), N(алкил)C(O)(алкил), -OC(O)O(алкил), -OC(O)NH2, -OC(O)NH(алкил), -OC(O)N(алкил)(алкил), -CHNH, -CHN(алкил), -SO2NH2, -S-алкил, -S-арил, -S-гетероарил, -S-гетероцикл или -S-гидрокарбил;
каждый из Q1, Q2 и Q3 независимо представляет собой CR10 или N;
Х представляет собой CHR10, S, O или NR9; и
каждый m независимо представляет собой целое число от 0 до 3.
В некоторых вариантах реализации соединения Формулы XII представляют собой соединения, где R9 представляет собой H или гидрокарбил. В других вариантах реализации R9 представляет собой H. В других вариантах реализации R9 представляет собой гидрокарбил. В других вариантах реализации R9 представляет собой алкил. В других вариантах реализации R9 представляет собой метил. В других вариантах реализации R9 представляет собой этил. В других вариантах реализации R9 представляет собой фенил.
В некоторых вариантах реализации соединения Формулы XII представляют собой соединения, где R10 представляет собой H, -OH или гидрокарбил. В других вариантах реализации R10 представляет собой H. В других вариантах реализации R10 представляет собой -OH. В других вариантах реализации R10 представляет собой -OMe. В других вариантах реализации R10 представляет собой -OEt. В других вариантах реализации R10 представляет собой -NH2. В других вариантах реализации R10 представляет собой -NHMe. В других вариантах реализации R10 представляет собой -NMe2. В других вариантах реализации R10 представляет собой гидрокарбил. В других вариантах реализации R10 представляет собой алкил. В других вариантах реализации R10 представляет собой метил. В других вариантах реализации R10 представляет собой этил. В других вариантах реализации R10 представляет собой фенил. В других вариантах реализации R10 представляет собой -S-алкил.
В другом варианте реализации R9 представляет собой H, а R10 представляет собой -OH.
В некоторых вариантах реализации соединения Формулы XII представляют собой соединения, где каждый из Q1, Q2 и Q3 представляет собой N. В других вариантах реализации каждый из Q1, Q2 и Q3 представляет собой CR10. В других вариантах реализации каждый из Q1, Q2 представляет собой N, а Q3 представляет собой CR10.
В некоторых вариантах реализации соединения Формулы XII представляют собой соединения, где Х представляет собой СН2. В других вариантах реализации Х представляет собой О. В других вариантах реализации Х представляет собой NH. В других вариантах реализации Х представляет собой NMe. В других вариантах реализации Х представляет собой N-бензил.
В некоторых вариантах реализации соединения Формулы XII представляют собой соединения, где каждый m независимо представляет собой целое число от 0 до 3. В других вариантах реализации каждый m независимо представляет собой целое число от 1 до 3. В других вариантах реализации m равен 0. В других вариантах реализации m равен 1. В других вариантах реализации m равен 2. В других вариантах реализации m равен 3.
В других вариантах реализации Q1 и Q3 представляют собой N, Q2 представляет собой CR10, а R10 представляет собой -N(алкил)(алкил). В других вариантах реализации Q1 и Q3 представляют собой N, Q2 представляет собой CR10, а R10 представляет собой -N(H)(алкил). В других вариантах реализации Q1 и Q3 представляют собой N, Q2 представляет собой CR10, а R10 представляет собой -N(CH3)2. В других вариантах реализации Q1 и Q3 представляют собой N, Q2 представляет собой CR10, а R10 представляет собой -N(H)(CH3).
В других вариантах реализации каждый m равен 1, Х представляет собой NR9, а R9 представляет собой H. В других вариантах реализации каждый m равен 1, а X представляет собой O.
В некоторых вариантах реализации Q1, Q2, Q3 и X в соединениях Формулы XII имеют следующие значения:
В некоторых вариантах реализации соединение Формулы XII существует в виде единственного таутомера или смеси таутомеров. Специалистам в данной области техники будут очевидными структуры, для которых возможно существование таутомерных форм, а также будет понятно, что изображение единственного таутомера охватывает структуры всех возможных таутомерных форм. В некоторых вариантах реализации R10 представляет собой ОН, и соединение Формулы XII существует в одной, двух или трех таутомерных формах Формулы XII, что проиллюстрировано в Уравнении II.
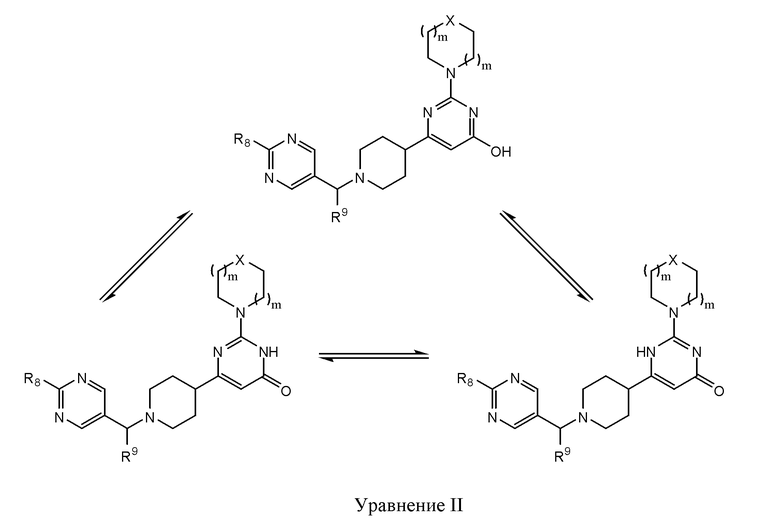
В конкретных вариантах реализации соединение Формулы XII имеет структуру формулы II, и иллюстративные соединения Формулы XII включают соединения IIa-IIjj или их фармацевтически приемлемые соли.
В некоторых вариантах реализации соединение Формулы XII имеет структуру
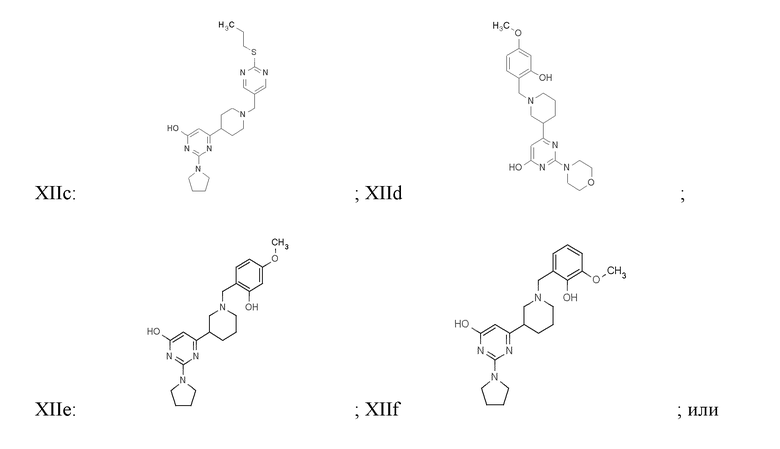
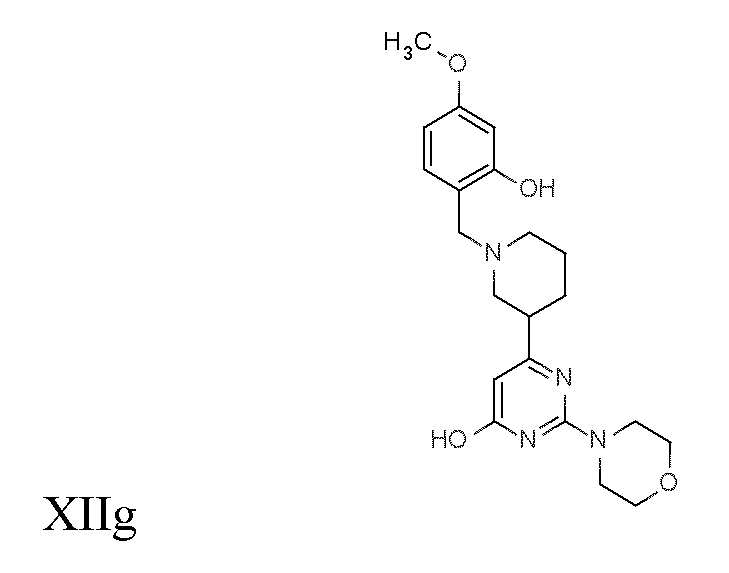 или их фармацевтически приемлемых солей.
или их фармацевтически приемлемых солей.
В другом варианте реализации в изобретении предложены соединения следующей Формулы XIII:
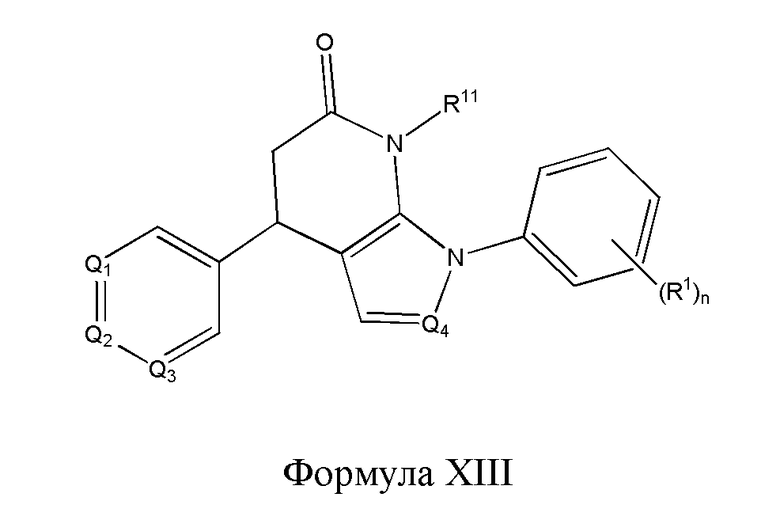
и их фармацевтически приемлемые соли, где
R1 независимо представляет собой -H, -OH, -NH2, -NH(алкил), -N(алкил)(алкил), гидрокарбил, -O-гидрокарбил, арил, -O-арил, аралкил, -O-аралкил, гетероарил, -O-гетероарил, гетероциклил, -O-гетероциклил, галоген, -OCF3, -C(O)O(алкил), -OC(O)(алкил), -C(O)NH2, -C(O)NH(алкил), -C(O)N(алкил)(алкил), -NHC(O)(алкил), N(алкил)C(O)(алкил), -OC(O)O(алкил), -OC(O)NH2, -OC(O)NH(алкил), -OC(O)N(алкил)(алкил), -CHNH, -CHN(алкил) или -SO2NH2;
R11 представляет собой H, гидрокарбил, арил, аралкил, гетероарил, гетероциклил, -C(O)O(алкил), -OC(O)(алкил), -C(O)NH2, -C(O)NH(алкил), -C(O)N(алкил)(алкил), -NHC(O)(алкил), N(алкил)C(O)(алкил) или -SO2NH2;
каждый из Q1, Q2, Q3 и Q4 независимо представляет собой CR10 или N;
n представляет собой целое число от 1 до 4; и
каждый R10 независимо представляет собой -H, -OH, -NH2, -NH(алкил), -N(алкил)(алкил), гидрокарбил, -O-гидрокарбил, арил, -O-арил, аралкил, -O-аралкил, гетероарил, -O-гетероарил, гетероциклил, -O-гетероциклил, галоген, -OCF3, -C(O)O(алкил), -OC(O)(алкил), -C(O)NH2, -C(O)NH(алкил), -C(O)N(алкил)(алкил), -NHC(O)(алкил), N(алкил)C(O)(алкил), -OC(O)O(алкил), -OC(O)NH2, -OC(O)NH(алкил), -OC(O)N(алкил)(алкил), -CHNH, -CHN(алкил) или -SO2NH2.
В некоторых вариантах реализации R1 представляет собой алкил. В других вариантах реализации R1 представляет собой 2-алкил.
В других вариантах реализации Q1 представляет собой H, Q2 представляет собой CR10 , и R10 представляет собой -OH, а Q3 представляет собой CR10, и R10 представляет собой O-алкил. В других вариантах реализации Q1 представляет собой CR10, и R10 представляет собой -O-алкил, Q2 представляет собой CR10, и R10 представляет собой -OH, а Q3 представляет собой H.
В других вариантах реализации Q4 представляет собой N. В других вариантах реализации Q4 представляет собой CR10. В других вариантах реализации Q4 представляет собой C(H).
В некоторых вариантах реализации R10 представляет собой H, -OH или гидрокарбил. В других вариантах реализации R10 представляет собой H. В других вариантах реализации R10 представляет собой -OH. В других вариантах реализации R10 представляет собой -OMe. В других вариантах реализации R10 представляет собой -OEt. В других вариантах реализации R10 представляет собой -NH2. В других вариантах реализации R10 представляет собой -NHMe. В других вариантах реализации R10 представляет собой -NMe2. В других вариантах реализации R10 представляет собой гидрокарбил. В других вариантах реализации R10 представляет собой алкил. В других вариантах реализации R10 представляет собой метил. В других вариантах реализации R10 представляет собой этил. В других вариантах реализации R10 представляет собой фенил.
В некоторых вариантах реализации R11 представляет собой H. В других вариантах реализации R11 представляет собой метил. В других вариантах реализации R11 представляет собой этил. В других вариантах реализации R11 представляет собой бензил.
В некоторых вариантах реализации соединения Формулы XIII представляют собой соединения, где n независимо представляет собой целое число от 1 до 4. В других вариантах реализации n равен 1. В других вариантах реализации n равен 2. В других вариантах реализации n равен 3. В других вариантах реализации n равен 4.
В некоторых вариантах реализации Q1, Q2, Q3 и Q4 в соединениях Формулы XIII имеют следующие определения:
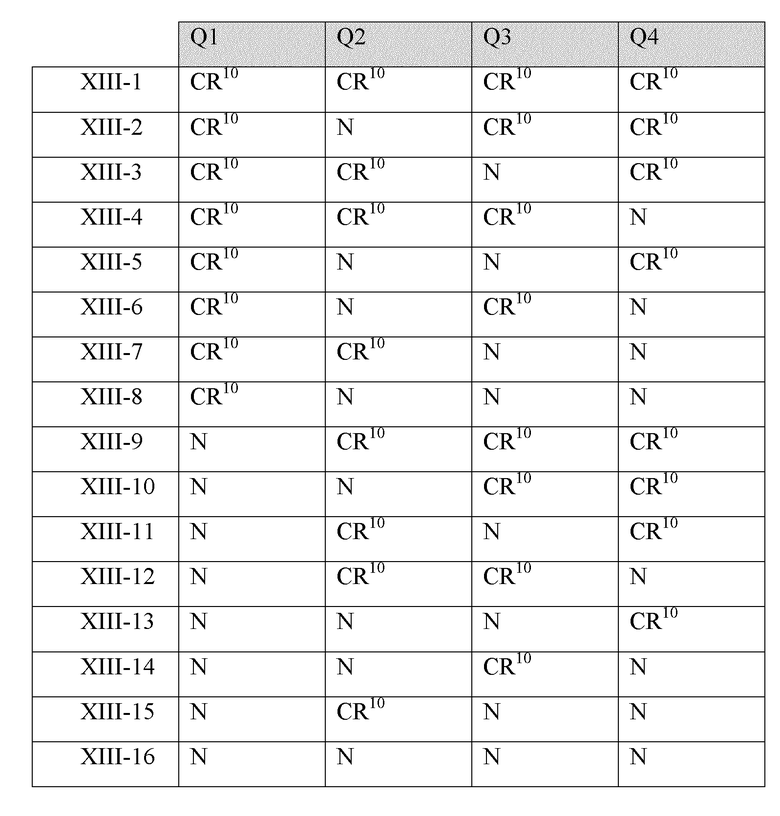
В конкретных вариантах реализации соединение Формулы XIII имеет структуру формулы VIII, и иллюстративные соединения Формулы XIII включают соединения VIIIa, VIIIc или их фармацевтически приемлемые соли.
В некоторых вариантах реализации соединение Формулы XIII имеет структуру XIIIa:
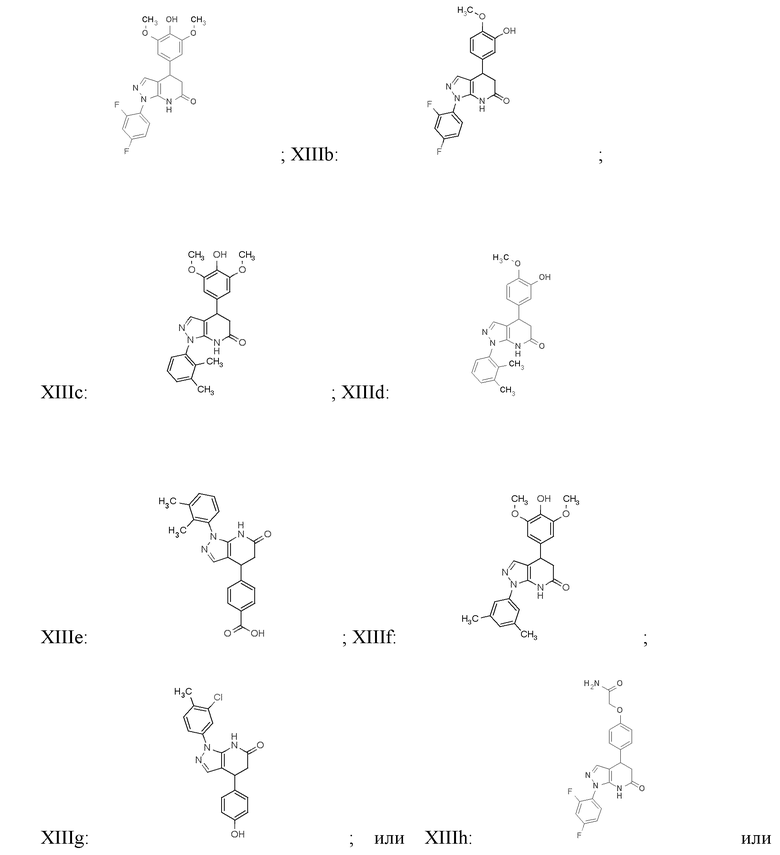
их фармацевтически приемлемых солей.
III. Лечение или предотвращение состояния с применением соединений согласно настоящему изобретению
Согласно настоящему изобретению соединения согласно настоящему изобретению подходят для лечения или предотвращения одного или более состояний.
В одном из вариантов реализации в изобретении предложены способы лечения или предотвращения одного или более состояний, включающие введение субъекту, нуждающемуся в этом, эффективного количества соединения согласно настоящему изобретению.
В другом варианте реализации в изобретении предложено применение одного или более соединений согласно настоящему изобретению для получения лекарственного средства, подходящего для лечения или предотвращения одного или более состояний.
В другом варианте реализации в изобретении предложены одно или более соединений согласно настоящему изобретению для лечения или предотвращения одного или более состояний.
Неограничивающие примеры состояний, которые поддаются лечению или предотвратимы в результате введения одного или более соединений согласно настоящему изобретению, включают: (i) нарушение метаболизма липопротеинов, включая дислипидемию, дислипопротеинемию, сверхсинтез или дефицит липопротеинов, повышение уровня общего холестерина, повышение концентрации липопротеинов низкой плотности, повышение концентрации триглицеридов, выделение липидов в желчь, нарушение метаболизма, выделение фосфолипидов в желчь, выделение оксистерола в желчь и нарушения, связанные с рецептором, активируемым пролифератором пероксисом; (ii) нарушение метаболизма глюкозы, включая инсулинорезистентность, нарушенную переносимость глюкозы, аномальный уровень глюкозы в крови натощак, сахарный диабет, липодистрофию, центральное ожирение, периферическую липоатрофию, диабетическую нефропатию, диабетическую ретинопатию, заболевания почек и септицемию; (iii) сердечнососудистое нарушение или связанное нарушение сосудов, включая гипертензию, ишемическую болезнь сердца, инфаркт миокарда, аритмию, мерцание предсердий, заболевания сердечного клапана, сердечную недостаточность, кардиомиопатию, перикардит и импотенцию; (iv) нарушение, вызванное аномальное модуляцией С-реактивного белка, или родственное нарушение, включая воспаление, ишемический некроз, рак толстой кишки и тромботические нарушения; и (v) возрастные заболевания, болезнь Альцгеймера, болезнь Паркинсона, панкреатит, воспаление поджелудочной железы и аномальную выработку желчи.
В изобретении также предложены способы определения биомаркера одного или более состояний, включающие введение эффективного количества соединения согласно настоящему изобретению субъекту, нуждающемуся в этом, и измерение уровня неэтерифицированного холестерина в крови субъекта. В одном из вариантов реализации состояние представляет собой нарушение сердечнососудистой системы. В другом варианте реализации биомаркером является наличие обратного транспорта холестерина от артерий в печень или выведение холестерина в желчные кислоты, или оба указанных маркера.
В изобретении дополнительно предложено применение свободного холестерина в качестве биомаркера активации P2Y13, которая приводит к функционализации ЛПВП, т.е. к повышению их активности в качестве агентов обратного транспорта холестерина. Соответственно, в изобретении дополнительно предложены способы определения уровня активации P2Y13, включающие введение эффективного количества соединения согласно настоящему изобретению субъекту, нуждающемуся в этом, и измерение содержания свободного холестерина в крови субъекта.
Соединения согласно настоящему изобретению и композиции, содержащие указанные соединения, можно вводить перорально. Соединения согласно настоящему изобретению и композиции, содержащие указанные соединения, также можно вводить при помощи любого другого удобного способа, например, путем внутривенной инфузии или инъекции болюса, всасывания через эпителиальную выстилку или поверхность кожи или слизистую оболочку (например, через слизистую оболочку полости рта, прямой кишки и кишечника и т.д.), а также можно вводить совместно с другим биологически активным агентом. Введение может быть системным или местным. Известны различные системы доставки, например, инкапсулирование в липосомы, микрочастицы, микрокапсулы, капсулы и т.д., и их можно применять для доставки соединения согласно настоящему изобретению. В конкретных вариантах реализации субъекту можно вводить более одного соединения согласно настоящему изобретению. Способы введения включают, но не ограничиваются ими, внутрикожное, внутримышечное, интраперитонеальное, внутривенное, подкожное, интраназальное, эпидуральное, пероральное, подъязычное, интраназальное, внутримозговое, внутривагинальное, чрескожное, ректальное введение, ингаляцию или местное введение, в частности, в уши, нос, глаза или на кожу. Способ введения определяется выбором врача и зависит, в частности, от участка, в котором возникло медицинское состояние. В большинстве случаев введение приводит к высвобождению соединений согласно настоящему изобретению в кровоток.
В конкретных вариантах реализации может быть желательным местное введение одного или более соединений согласно настоящему изобретению в участок, в котором необходимо лечение. Местное введение можно проводить, в качестве примера, но не ограничения, путем местной инфузии во время хирургии, местного нанесения, например, при зашивании раны после хирургии, путем инъекции, с применением катетера, с применением суппозитория, или путем имплантации, где имплант может представлять собой пористый, беспористый или гелеобразный материал, включая мембраны, такие как сиаластические мембраны, или волокна. В одном из вариантов реализации введение можно проводить путем прямой инъекции в участок (или бывший участок) атеросклеротической бляшки в ткани.
В конкретных вариантах реализации соединение согласно настоящему изобретению можно вводить в центральную нервную систему при помощи любого подходящего способа, включая внутрижелудочковую, интратекальную и эпидуральную инъекцию. Внутрижелудочковую инъекцию можно облегчать путем применения внутрижелудочкового катетера, например, присоединенного к резервуару, такому как резервуар Оммайя.
Также можно применять внутрилегочное введение, например, с применением ингалятора или небулайзера, состава с аэрозолем или путем перфузии во фторуглеродный или синтетический легочный сурфактант. В конкретных вариантах реализации соединения согласно настоящему изобретению можно вводить в состав суппозиториев совместно с традиционными связывающими веществами и наполнителями, такими как триглицериды.
В другом варианте реализации соединения и композиции, содержащие соединения согласно настоящему изобретению, можно доставлять в виде везикул, в частности, липосом (см., Langer, 1990, Science 249:1527-1533; Treat et al., in Liposomes in the Therapy of Infectious Disease and Cancer, Lopez-Berestein and Fidler (eds.), Liss, New York, pp. 353-365 (1989); Lopez-Berestein, там же., pp. 317-327; общий обзор см. там же).
В другом варианте реализации соединения и композиции, содержащие соединения согласно настоящему изобретению, можно доставлять в системе с контролируемым высвобождением. В одном из вариантов реализации можно применять насос (см., Лангер (Langer), выше; Sefton, 1987, CRC Crit. Ref. Biomed. Eng. 14:201; Buchwald et al., 1980, Surgery 88:507 Saudek et al., 1989, N. Engl. J. Med. 321:574). В другом варианте реализации можно применять полимерные материалы (см., Medical Applications of Controlled Release, Langer and Wise (eds.), CRC Pres., Boca Raton, Florida (1974); Controlled Drug Bioavailability, Drug Product Design and Performance, Smolen and Ball (eds.), Wiley, New York (1984); Ranger and Peppas, 1983, J. Macromol. Sci. Rev. Macromol. Chem. 23:61; также см. Levy et al., 1985, Science 228:190; During et al., 1989, Ann. Neurol. 25:351; Howard et al., 1989, J. Neurosurg. 71:105). В другом варианте реализации систему с контролируемым высвобождением можно помещать в непосредственной близости от целевого участка, требующего лечения, например, печени, и, таким образом, требуется введение только части дозы, которую необходимо использовать с случае системного введения (см., например, Goodson, in Medical Applications of Controlled Release, выше, vol. 2, pp. 115-138 (1984)). Можно применять другие системы с контролируемым высвобождением, обсуждаемые в обзоре Лангера (Langer, 1990, Science 249:1527-1533).
Композиции согласно настоящему изобретению содержат терапевтически эффективное количество соединения согласно настоящему изобретению, возможно более чем одного соединения согласно настоящему изобретению, совместно с подходящим количеством фармацевтически приемлемого носителя, и имеют форму, подходящую для введения субъекту.
Композиции согласно настоящему изобретению могут иметь форму растворов, суспензий, эмульсий, таблеток, пилюль, драже, капсул, содержащих жидкость, порошков, составов с замедленным высвобождением, суппозиториев, эмульсий, аэрозолей, спреев, суспензий или любую другую форму, подходящую для применения. В одном из вариантов реализации фармацевтически приемлемый носитель представляет собой капсулу (см., например, патент США №5698155). Другие примеры подходящих фармацевтических носителей описаны в «Remington's Pharmaceutical Sciences» под ред. Е.У. Мартина (E.W. Martin), содержание которой включено посредством ссылки для описания фармацевтических композиций и способов их введения.
В некоторых вариантах реализации соединения и композиции, содержащие соединения согласно настоящему изобретению, вводят в состав фармацевтической композиции, предназначенной для внутривенного введения человеку, в соответствии с традиционными способами. Соединения и композиции, содержащие соединения согласно настоящему изобретению, подходящие для внутривенного введения, могут представлять собой растворы в стерильном изотоническом водном буфере. Композиции также могут содержать вещество, увеличивающее растворимость. Композиции для внутривенного введения могут содержать местный анестетик, такой как лигнокаин. Ингредиенты можно вводить отдельно или смешивать в стандартной лекарственной форме, например, в виде сухого лиофилизированного порошка или не содержащего воду концентрата в герметичном контейнере, таком как ампула или сашет. Если соединение согласно настоящему изобретению необходимо вводить путем внутривенной инфузии, то его можно вводить, например, при помощи бутыли для инфузии, содержащей стерильную воду или солевой раствор для фармацевтических целей. Если соединение согласно настоящему изобретению вводят путем инъекции, то можно применять ампулу, содержащую стерильную воду для инъекций или солевой раствор, и, таким образом, ингредиенты можно смешивать перед введением.
Соединения и композиции, содержащие соединения согласно настоящему изобретению, подходящие для пероральной доставки, могут находиться в форме таблеток, пастилок, водных или масляных суспензий, гранул, порошков, эмульсий, капсул, сиропов или эликсиров. Соединения и композиции, содержащие соединения согласно настоящему изобретению, подходящие для пероральной доставки, также могут быть приготовлены в форме пищевых продуктов и смесей. Перорально вводимые композиции могут содержать один или более дополнительных агентов, например, подсластителей, таких как фруктоза, аспартам или сахарин; ароматизаторов, таких как перечная мята, винтергриновое масло или вишневый ароматизатор; красителей; и консервантов, для получения фармацевтического препарата, привлекательного для потребителя. Композиции могут быть покрыты оболочкой для задержки распадания и всасывания в желудочно-кишечном тракте, что, таким образом, обеспечивает длительное действие в течение продолжительного периода времени. Селективно проницаемые мембраны, расположенные вокруг соединения, доставляемого под действием осмоса, также подходят для перорального введения соединений и композиций, содержащих соединения согласно настоящему изобретению. Вещества, задерживающие высвобождение, такие как глицерилмоностеарат или глицерилстеарат, также можно применять. Пероральные композиции могут содержать традиционные носители, такие как маннит, лактоза, крахмал, стеарат магния, сахаринат натрия, целлюлоза и карбонат магния.
Количество соединения согласно настоящему изобретению, которое является эффективным для лечения конкретного состояния, описанного в настоящей заявке, может зависеть от природы состояния, и его можно определять при помощи стандартных клинических способов. Для определения оптимального диапазона дозировки можно использовать in vitro и in vivo исследования. Точная доза композиции также может зависеть от способа введения или тяжести состояния и может быть выбрана в соответствии с оценкой лечащего врача и с учетом медицинского состояния каждого субъекта. Тем не менее, подходящий диапазон дозировок для перорального введения, как правило, составляет примерно от 0,001 мг до 2000 мг соединения согласно настоящему изобретению на килограмм массы тела. В некоторых вариантах реализации пероральная доза составляет от 0,01 мг до 100 мг на кг массы тела, от 0,1 мг до 50 мг на кг массы тела, от 0,5 мг до 20 мг на кг массы тела или от 1 мг до 10 мг на кг массы тела. В некоторых вариантах реализации пероральная доза составляет 5 мг соединения согласно настоящему изобретению на кг массы тела. Дозируемые количества, описанные в настоящей заявке, относятся к общему вводимому количеству, то есть, если вводят более одного соединения согласно настоящему изобретению, то дозировка соответствует общему количеству вводимых соединений согласно настоящему изобретению. Пероральные композиции могут содержать от 10% до 95% активного ингредиента по массе.
В конкретных вариантах реализации соединения или композиции, содержащие соединения согласно настоящему изобретению, вводят субъекту, такому как человек, в качестве профилактической или превентивной меры для состояния, описанного в настоящей заявке. Композиции согласно настоящему изобретению можно вводить в качестве превентивной меры субъекту, имеющему генетическую предрасположенность к состоянию, такому как сердечнососудистое заболевание, дислипидемия, дислипопротеинемия, нарушение метаболизма глюкозы, болезнь Альцгеймера, синдром Х, связанное с P2Y13 нарушение, септицемия, тромботическое нарушение, ожирение, панкреатит, гипертензия, заболевание почек, рак, воспаление или импотенция. Примеры указаных генетических предрасположенностей включают, но не ограничиваются ими, аллель ε4 аполипопротеина Е, который повышает вероятность болезни Альцгеймера; потерю функции или нулевую мутацию кодирующей области или промотора гена липопротеинлипазы (например, мутации кодирующих областей, приводящие к заменам D9N и N291S; для обзора генетических мутаций гена липопротеинлипазы, которые повышают риск сердечнососудистых заболеваний, дислипидемии и дислипопротеинемии см. Hayden and Ma, 1992, Mol. Cell Biochem. 113:171-176); и семейную комбинированную гиперлипидемию и семейную гиперхолестеринемию.
Соединения или композиции, содержащие соединения согласно настоящему изобретению, можно вводить в качестве превентивной меры субъекту, имеющему негенетическую предрасположенность к состоянию, такому как сердечнососудистое заболевание, дислипидемия, дислипопротеинемия, нарушение метаболизма глюкозы, болезнь Альцгеймера, синдром Х, нарушение, связанное с P2Y13, септицемия, тромботическое нарушение, ожирение, панкреатит, гипертензия, заболевание почек, рак, воспаление или импотенция. Примеры указанных негенетических предрасположенностей включают, но не ограничиваются ими, коронарное шунтирование сердца и чрескожную транслюминальную коронарную ангиопластику, которые могут приводить к рестенозу, ускоренную форму атеросклероза; диабет у женщин, который может приводить к поликистозу яичников; и сердечнососудистые заболевания, которые могут приводить к импотенции. Соответственно, композиции, содержащие соединения согласно настоящему изобретению, можно применять для предотвращения одного заболевания или нарушения и одновременного лечения другого (например, для предотвращения поликистоза яичников, сопровождающегося лечением диабета; предотвращения импотенции, сопровождающегося лечением сердечнососудистого заболевания).
В настоящем изобретении предложены способы лечения или предотвращения сердечнососудистого заболевания или его симптомов, включающие введение субъекту, нуждающемуся в этом, эффективного количества соединения согласно настоящему изобретению. В одном из вариантов реализации изобретения соединение согласно настоящему изобретению входит в состав композиции, которая дополнительно содержит фармацевтически приемлемый носитель. Используемый в настоящем описании термин «сердечнососудистое заболевание» относится к заболеванию сердца и системы кровообращения. Сердечнососудистое заболевание может быть связано с дислипопротеинемией или дислипидемией, или с обоими факторами. Сердечнососудистые заболевания включают, но не ограничиваются ими, артериосклероз; атеросклероз; инсульт; ишемию; болезнь периферических сосудов (БПС); транзисторную ишемическую атаку (ТИА); скоротечный атеросклероз; атеросклероз трансплантированных органов; дисфункции эндотелия, в частности, дисфункции, воздействующие на эластичность кровяных сосудов; заболевания периферических сосудов; коронарную болезнь сердца; инфаркт миокарда; церебральный инфаркт и рестеноз. Неограничивающие примеры симптомов сердечнососудистых заболеваний включают стенокардию, одышку, головокружение, тошноту, усталость, нерегулярное сердцебиение и импотенцию. В некоторых вариантах реализации лечение сердечнососудистого заболевания излечивает один или более симптомов сердечнососудистого заболевания. В некоторых вариантах реализации лечение сердечнососудистого заболевания излечивает импотенцию.
В настоящем изобретении предложены способы лечения или предотвращения дислипидемии, включающие введение субъекту, нуждающемуся в этом, эффективного количества соединения согласно настоящему изобретению. В одном из вариантов реализации соединение согласно настоящему изобретению входит в состав композиции, которая дополнительно содержит фармацевтически приемлемый носитель.
Дислипидемии включают, но не ограничиваются ими: гиперлипидемию и низкое содержание в крови холестерина липопротеинов высокой плотности (ЛПВП). В конкретных вариантах реализации гиперлипидемия представляет собой семейную гиперхолестеринемию; семейную комбинированную гиперлипидемию; снижение или дефицит уровня или активности липопротеинлипазы, включая снижение и дефицит, возникающие в результате мутаций липопротеинлипазы; гипертриглицеридемию; гиперхолестеринемию; высокое содержание в крови кетоновых тел (например, β-ОН-масляной кислоты); высокое содержание в крови холестерина Lp(a); высокое содержание в крови холестерина липопротеинов низкой плотности (ЛПНП); высокое содержание в крови холестерина липопротеинов очень низкой плотности (ЛПОНП) и высокое содержание в крови неэтерифицированных жирных кислот.
В настоящем изобретении дополнительно предложены способы изменения метаболизма липидов у субъекта, например, снижения уровня ЛПНП в крови субъекта, снижения уровня свободных триглицеридов в крови субъекта, повышения соотношения ЛПВП и ЛПНП в крови субъекта и ингибирования синтеза омыленных или неомыленных жирных кислот, включающие введение субъекту, нуждающемуся в этом, эффективного количества соединения согласно настоящему изобретению. В одном из вариантов реализации соединение согласно настоящему изобретению входит в состав композиции, которая дополнительно содержит фармацевтически приемлемый носитель.
В настоящем изобретении предложены способы лечения или предотвращения дислипопротеинемии, включающие введение субъекту, нуждающемуся в этом, эффективного количества соединения согласно настоящему изобретению. В одном из вариантов реализации соединение согласно настоящему изобретению входит в состав композиции, которая дополнительно содержит фармацевтически приемлемый носитель.
Дислипопротеинемия относится к нарушениям, которые вызывают нарушение уровня липопротеинов в кровотоке или обусловлены указанным нарушением. Если содержание липопротеинов в крови является аномально высоким, соединение согласно настоящему изобретению вводят субъекту для восстановления нормального уровня. С другой стороны, если содержание липопротеинов в крови является аномально низким, соединение согласно настоящему изобретению вводят субъекту для восстановления нормального уровня. Нормальный уровень липопротеинов хорошо известен специалистам в данной области техники.
Дислипопротеинемии включают, но не ограничиваются ими: высокое содержание ЛПНП в крови; высокое содержание аполипопротеина В (аро В) в крови; высокое содержание Lp(a) в крови; высокое содержание аро(а) в крови; высокое содержание ЛПОНП в крови; низкое содержание ЛПВП в крови; снижение или дефицит уровня или активности липопротеинлипазы, включая снижение или дефицит, возникающие в результате мутаций липопротеинлипазы; гипоальфалипопротеинемию; аномалии липопротеинов, связанные с диабетом; аномалии липопротеинов, связанные с ожирением; аномалии липопротеинов, связанные с болезнью Альцгеймера; и семейную комбинированную гиперлипидемию.
В настоящем изобретении дополнительно предложены способы снижения уровня аро С-II в крови субъекта; снижения уровня аро C-III в крови субъекта; повышения уровня белков, связанных с ЛПВП, включающих, но не ограничивающихся ими, аро А-I, аро A-II, apo A-IV и аро Е, в крови субъекта; повышения уровня аро Е в крови субъекта или ускорения выведения триглицеридов из крови субъекта, включающие введение субъекту, нуждающемуся в этом, эффективного количества соединения согласно настоящему изобретению. В одном из вариантов реализации соединение согласно настоящему изобретению входит в состав композиции, которая дополнительно содержит фармацевтически приемлемый носитель.
В настоящем изобретении предложены способы лечения или предотвращения нарушения метаболизма глюкозы, включающие введение субъекту, нуждающемуся в этом, эффективного количества соединения согласно настоящему изобретению. В одном из вариантов реализации соединение согласно настоящему изобретению входит в состав композиции, которая дополнительно содержит фармацевтически приемлемый носитель.
Нарушения метаболизма глюкозы могут включать нарушения хранения и/или потребления глюкозы. Если один или более показателей метаболизма глюкозы (т.е. содержание инсулина в крови, уровень глюкозы в крови) являются аномально высокими, соединение согласно настоящему изобретению вводят субъекту для восстановления нормального уровня. С другой стороны, если один или более показателей метаболизма глюкозы являются аномально низкими, соединение согласно настоящему изобретению вводят субъекту для восстановления нормального уровня. Нормальные значения показателей метаболизма глюкозы хорошо известны специалистам в данной области техники.
Нарушения метаболизма глюкозы включают, но не ограничиваются ими: нарушенную переносимость глюкозы; диабетическую ретинопатию, диабетическую нефропатию, инсулинорезистентность; связанный с инсулинорезистентностью рак, такой как рак груди, толстой кишки или простаты; диабет, включая, но не ограничиваясь ими, инсулинонезависимый сахарный диабет (ИНСЗД), инсулинозависимый сахарный диабет (ИЗСД), гестационный сахарный диабет (ГСД) и сахарный диабет взрослого типа у молодых (MODY); панкреатит; гипертензию; поликистоз яичников; и повышенное содержание в крови инсулина или глюкозы, или обоих этих веществ.
В настоящем изобретении дополнительно предложены способы изменения метаболизма глюкозы у субъекта, например, повышения чувствительности к инсулину или потребления кислорода субъектом, включающие введение субъекту, нуждающемуся в этом, эффективного количества соединения согласно настоящему изобретению. В одном из вариантов реализации соединение согласно настоящему изобретению входит в состав композиции, которая дополнительно содержит фармацевтически приемлемый носитель.
В настоящем изобретении предложены способы лечения или предотвращения связанного с P2Y13 нарушения, включающие введение субъекту, нуждающемуся в этом, эффективного количества соединения согласно настоящему изобретению. В одном из вариантов реализации соединение согласно настоящему изобретению входит в состав композиции, которая дополнительно содержит фармацевтически приемлемый носитель.
Примеры связанных с P2Y13 нарушений включают, но не ограничиваются ими, ревматоидный артрит; рассеянный склероз; псориаз; воспалительную болезнь кишечника; рак груди, толстой кишки или простаты; низкий уровень ЛПВП в крови; низкий уровень аро Е в крови, лимфе и/или спинномозговой жидкости; низкий уровень аро A-I в крови, лимфе и/или спинномозговой жидкости; высокий уровень ЛПОНП в крови; высокий уровень ЛПНП в крови; высокий уровень триглицеридов в крови; высокий уровень аро В в крови; высокий уровень аро С-III в крови и сниженное соотношение постгепариновой активности печеночной липазы и липопротеинлипазы. Уровень ЛПВП может быть повышен в лимфе или спинномозговой жидкости, или в обеих средах.
В настоящем изобретении предложены способы лечения или предотвращения стеатоза печени, включая алкогольный и неалкогольный стеатоз печени, включающие введение субъекту, нуждающемуся в этом, эффективного количества соединения согласно настоящему изобретению. В одном из вариантов реализации соединение согласно настоящему изобретению входит в состав композиции, которая дополнительно содержит фармацевтически приемлемый носитель.
В настоящем изобретении предложены способы лечения или предотвращения заболевания почек, включающие введение субъекту, нуждающемуся в этом, эффективного количества соединения согласно настоящему изобретению. В одном из вариантов реализации соединение согласно настоящему изобретению входит в состав композиции, которая дополнительно содержит фармацевтически приемлемый носитель.
Заболевания почек включают: гломерулярные заболевания (включая, но не ограничиваясь ими, острый и хронический гломерулонефрит, быстро прогрессирующий гломерулонефрит, нефротический синдром, фокальный пролиферативный гломерулонефрит, гломерулярные поражения, связанные с системными заболеваниями, такими как системная красная волчанка, синдром Гудпасчера, множественная миелома, диабет, неоплазия, серповидноклеточная болезнь и хронические воспалительные заболевания), заболевания канальцев (включая, но не ограничиваясь ими, острый некроз канальцев и острую почечную недостаточность, поликистоз медуллярной спонгиозной почки, медуллярный кистоз, нефрогенный диабет и почечный канальцевый ацидоз), тубулоинтерстициальные заболевания (включая, но не ограничиваясь ими, пиелонефрит, вызванные лекарствами и токсинами тубулоинтерстициальный нефрит, гиперкальциемическую нефропатию и гиперкалиемическую нефропатию), острую и быстро прогрессирующую почечную недостаточность, хроническую почечную недостаточность, нефролитиаз или опухоли (включая, но не ограничиваясь ими, почечноклеточную карциному и нефробластому). В другом варианте реализации заболевание почек представляет собой сосудистое заболевание, включая, но не ограничиваясь ими, гипертензию, нефросклероз, микроангиопатическую гемолитическую анемию, атероэмболическую болезнь почек, диффузный кортикальный некроз и инфаркт почки.
В настоящем изобретении предложены способы лечения или предотвращения нейродегенеративного заболевания или нарушения, болезни Паркинсона, болезни Альцгеймера, синдрома Х, септицемии, тромботических нарушений, ожирения, панкреатита, гипертензии, воспаления или импотенции, включающие введение субъекту, нуждающемуся в этом, эффективного количества соединения согласно настоящему изобретению. В одном из вариантов реализации соединение согласно настоящему изобретению входит в состав композиции, которая дополнительно содержит фармацевтически приемлемый носитель.
Лечение или предотвращение болезни Альцгеймера также могут включать лечение или предотвращение одной или более аномалий липопротеинов, связанных с болезнью Альцгеймера.
Лечение или предотвращение синдрома Х или метаболического синдрома также могут включать лечение или предотвращение их симптомов, включая, но не ограничиваясь ими: нарушенную переносимость глюкозы, гипертензию и дислипидемию или дислипопротеинемию.
Лечение или предотвращение септицемии также могут включать лечение или предотвращение септического шока.
Лечение или предотвращение тромботического нарушения также могут включать лечение или предотвращение высокого содержания в крови фибриногена или промотирование фибринолиза.
Соединения согласно настоящему изобретению также подходят для ускорения снижения массы тела субъекта.
Соединения согласно настоящему изобретению подходят для медицинских применений, заключающихся в лечении или предотвращении ряда заболеваний и нарушений, включая, но не ограничиваясь ими, заболевания и нарушения, такие как сердечнососудистые заболевания, инсульт и заболевания периферических сосудов; дислипидемия; гиперхолестеринемия, атеросклероз, болезнь периферических сосудов (БПС), инсульт, ТИА, скоротечный атеросклероз, атеросклероз трансплантированных органов, дислипопротеинемия; нарушение метаболизма глюкозы; диабетическая нефропатия, диабетическая ретинопатия, инсулинорезистентность, метаболический синдром (например, синдром Х); нарушение, связанное с рецептором, активируемым пролифератором пероксисом; септицемия; тромботическое нарушение; ожирение; панкреатит; гипертензия; заболевание почек; воспаление; воспалительные заболевания мышечной ткани, такие как ревматическая полимиалгия, полимиозит, миопатия и фиброз; воспалительные нарушения, такие как астма, васкулит, язвенный колит, болезнь Крона, болезнь Кавасаки, гранулематоз Вегенера, ревматоидный артрит (РА), системная красная волчанка (СКВ), рассеянный склероз (РС) и аутоиммунный хронический гепатит; артрит, такой как ревматоидный артрит, ювенильный ревматоидный артрит и остеоартрит; остеопороз, ревматизм мягких тканей, такой как тендинит; бурсит; аутоиммунные заболевания, такие как системная волчанка и системный эритематоз; склеродермия; анкилозирующий спондилит; подагра; псевдоподагра; инсулинонезависимый сахарный диабет; поликистоз яичников; гиперлипидемии, такие как семейная гиперхолестеринемия (СГ), семейная комбинированная гиперлипидемия (СКГЛ); дефицит липопротеинлипазы, такой как гипертриглицеридемия, гипоальфалипопротеинемия и гиперхолестеринемия; аномалии липопротеинов, связанные с диабетом; аномалии липопротеинов, связанные с ожирением. Соединения и композиции согласно настоящему изобретению подходят для лечения или предотвращения повышенного содержания триглицеридов в крови, повышенного содержания холестерина липопротеинов низкой плотности, повышенного содержания аполипопротеина В, повышенного содержания холестерина липопротеинов Lp(a), повышенного содержания холестерина липопротеинов очень низкой плотности, повышенного содержания фибриногена, повышенного содержания инсулина, повышенного содержания глюкозы и пониженного содержания холестерина липопротеинов высокой плотности. Соединения и композиции согласно настоящему изобретению также применяют для лечения инсулинонезависимого сахарного диабета (ИНЗСД), которое не приводит к набору массы тела. Соединения согласно настоящему изобретению также можно применять для снижения содержания жира в мясе домашнего скота и снижения содержания холестерина в яйцах.
В изобретении предложены новые соединения, особенно подходящие для лечения или предотвращения ряда заболеваний и состояний, которые включают, но не ограничиваются ими, возрастные заболевания, болезнь Альцгеймера и аномалии липопротеинов, связанные с болезнью Альцгеймера, болезнь Паркинсона, рак, сердечнососудистые заболевания, диабетическую нефропатию, диабетическую ретинопатию, нарушение метаболизма глюкозы, дислипидемию, дислипопротеинемию, повышенную выработку желчи, гипертензию, импотенцию, воспаление, инсулинорезистентность, выделение липидов в желчь, модуляцию С-реактивного белка, ожирение, выделение оксистерола в желчь, панкреатит, воспаление поджелудочной железы, импотенцию; заболевания желудочно-кишечного тракта; синдром раздраженного кишечника; воспалительную болезнь кишечника; нарушение, связанное с рецептором, активируемым пролифератором пероксисом, выделение фосфолипидов в желчь, заболевания почек, септицемию, метаболический синдром (например, синдром Х) и тромботические нарушения.
Сердечнососудистые заболевания, такие как атеросклероз, могут требовать применения хирургических процедур, таких как ангиопластика. Ангиопластику можно дополнять размещением прочной металлической трубкообразной структуры, известной как «стент», в поврежденной коронарной артерии. В случае более серьезных состояний, может требоваться проведение хирургической операции на открытом сердце, такой как коронарное шунтирование. Эти хирургические процедуры можно проводить с применением инвазивных хирургических устройств или имплантатов, что связано с повышенным риском рестеноза и тромбоза. Соответственно, соединения согласно настоящему изобретению являются подходящими покрытиями для хирургических устройств (например, катетеров) или имплантатов (например, стентов), снижающими риск рестеноза и тромбоза, связанных с инвазивными процедурами, применяемыми для лечения сердечнососудистых заболеваний. Соответственно, в настоящем изобретении дополнительно предложены хирургические устройства или имплантаты, которые имеют оболочку, которая содержит эффективное количество соединения согласно настоящему изобретению.
Соединение согласно настоящему изобретению можно вводить животному, отличному от человека, для ветеринарного применения для лечения или предотвращения заболевания или нарушения, описанного в настоящей заявке.
В конкретном варианте реализации животное, отличное от человека, представляет собой домашнее животное. В другом конкретном варианте реализации животное, отличное от человека, представляет собой домашний скот. В другом варианте реализации животное, отличное от человека, представляет собой млекопитающее, например, корову, лошадь, овцу, свинью, кошку, собаку, мышь, крысу, кролика или морскую свинку. В другом варианте реализации животное, отличное от человека, представляет собой домашнюю птицу, такую как курица, индейка, утка, гусь или перепел.
В дополнение к ветеринарным применениям соединения и композиции согласно настоящему изобретению подходят для снижения содержания жира в организме домашнего скота для получения мяса с низким содержанием жира. В качестве альтернативы соединения и композиции согласно настоящему изобретению подходят для снижения содержания холестерина в яйцах в результате введения соединений курице, перепелке или утке. В случае применения для животных, отличных от человека, соединения и композиции согласно настоящему изобретению можно вводить животным с кормом или перорально в виде густой жидкой композиции (drench composition). Соответственно, в настоящем изобретении дополнительно предложены способы снижения содержания жира в организме домашнего скота или содержания холестерина в яйцах, включающие введение эффективного количества соединения согласно настоящему изобретению субъекту, нуждающемуся в этом.
А. Лечение или предотвращение рака
Соединения согласно настоящему изобретению подходят для лечения или предотвращения рака. Соответственно, в изобретении предложены способы лечения или предотвращения рака, включающие введение эффективного количества соединения согласно настоящему изобретению субъекту, нуждающемуся в этом. В одном из вариантов реализации способы дополнительно включают введение эффективного количества другого противоракового агента. Примеры раковых заболеваний, для лечения или предотвращения которых подходят соединения согласно настоящему изобретению, включают, но не ограничиваются ими, раковые заболевания, описанные ниже в Таблице 1, а также их метастазы.
В одном из вариантов реализации раковое заболевание представляет собой рак легкого, рак груди, колоректальный рак, рак простаты, лейкемию, лимфому, неходжкинскую лимфому, рак кожи, рак мозга, рак центральной нервной системы, рак яичников, рак матки, рак желудка, рак поджелудочной железы, рак пищевода, рак почки, рак печени или рак головы и шеи. В другом варианте реализации раковое заболевание представляет собой метастазирующее раковое заболевание.
В другом варианте реализации раковое заболевание представляет собой рак мозга или меланому. В одном из вариантов реализации рак мозга представляет собой метастазирующий рак мозга или глиому. В одном из вариантов реализации глиома представляет собой пилоцитарную астроцитому, астроцитому, анапластическую астроцитому или мультиформную глиобластому. В одном из вариантов реализации раковое заболевание представляет собой опухоли с дефицитом гомологичной рекомбинации, например, с дефицитом BRCA-I или BRCA-2 или с дефицитом одного или более белков семейства Фанкони. В одном из вариантов реализации дефицит вызван генетической мутацией. В другом варианте реализации фенотип, возникающий в результате дефицита, обусловлен аномальной низкой экспрессией белков BRCA-I или BRCA-2. В другом варианте реализации фенотип, возникающий в результате дефицита, обусловлен аномально низкой экспрессией одного или более белков семейства Фанкони.
В другом варианте реализации раковое заболевание представляет собой лейкемию, включая, но не ограничиваясь ими: острую лейкемию, острую лимфоцитарную лейкемию, острые миелоцитарные лейкемии, такие как миелобластная, промиелоцитарная, миеломоноцитарная, моноцитарная и эритролейкемическая лейкемия и миелодиспластический синдром; хроническую лейкемию, включая, но не ограничиваясь ими, хроническую миелоцитарную (гранулоцитарную) лейкемию, хроническую лимфоцитарную лейкемию, лейкемию ворсистых клеток; истинную полицитемию; лимфомы, включая, но не ограничиваясь ими, болезнь Ходжкина, неходжкинскую лимфому; множественные миеломы, включая, но не ограничиваясь ими, тлеющую множественную миелому, несекретирующую миелому, остеосклеротическую миелому, плазмаклеточную лейкемию, изолированную плазмоцитому и экстрамедуллярную плазмоцитому; макроглобулинемию Вальденстрема; моноклональную гаммапатию неизвестной этиологии; доброкачественную моноклональную гаммапатию; болезнь тяжелых цепей; рак дендритных клеток, включая рак плазмацитоидных дендритных клеток, NK-клеточную бластную лимфому (также известную как кожная NK/T-клеточная лимфома и агранулярные (CD4+/CD56+) дерматологические новообразования); базофильную лейкемию; саркомы кости и соединительной ткани, включая, но не ограничиваясь ими, саркому кости, остеосаркому, хондросаркому, саркому Юинга, злокачественную гигантоклеточную опухоль, фибросаркому кости, хордому, периостальную саркому, саркомы мягких тканей, ангиосаркому (гемангиосаркому), фибросаркому, саркому Капоши, лейомиосаркому, липосаркому, лимфангиосаркому, неврилеммому, рабдомиосаркому, синовиальную саркому; опухоли мозга, включая, но не ограничиваясь ими, глиому, астроцитому, глиому ствола головного мозга, эпендимому, олигодендроглиому, неглиальные опухоли, акустическую невриному, краниофарингиому, медуллобластому, менингиому, пинеоцитому, пинеобластому, первичную лимфому мозга; рак груди, включая, но не ограничиваясь ими, дуктальную карциному, аденокарциному, лобулярную (мелкоклеточную) карциному, интрадуктальную карциному, медуллярный рак груди, слизеобразующий рак груди, тубулярный рак груди, папиллярный рак груди, болезнь Педжета и воспалительный рак груди; рак надпочечников, включая, но не ограничиваясь ими, феохромоцитому и адренокортикальную карциному; рак щитовидной железы, включая, но не ограничиваясь ими, папиллярный или фолликулярный рак щитовидной железы, медуллярный рак щитовидной железы и анапластический рак щитовидной железы; рак поджелудочной железы, включая, но не ограничиваясь ими, инсулиному, гастриному, глюкагоному, випому, соматостатин-секретирующую опухоль и карциноидную опухоль или опухоль островков поджелудочной железы; рак гипофиза, включая, но не ограничиваясь ими, болезнь Кушинга, пролактин-секретирующую опухоль, акромегалию и несахарных диабет; рак глаз, включая, но не ограничиваясь ими, окулярную меланому, такую как меланома радужки, меланома хороидеи и меланома ресничного тела, и ретинобластому; вагинальный рак, такой как плоскоклеточная карцинома, аденокарцинома и меланома; рак влагалища, такой как плоскоклеточная карцинома, меланома, аденокарцинома, базальноклеточная карцинома, саркома и болезнь Педжета; рак шейки матки, включая, но не ограничиваясь ими, плоскоклеточную карциному и аденокарциному; рак матки, включая, но не ограничиваясь ими, эндометриальную карциному и саркому матки; рак яичников, включая, но не ограничиваясь ими, карциному эпителия яичников, пограничную опухоль, эмбрионально-клеточные опухоли и стромальные опухоли; рак пищевода, включая, но не ограничиваясь ими, плоскоклеточный рак, аденокарциному, аденоидную кистозную карциному, мукоэпидермоидную карциному, аденоплоскоклеточную карциному, саркому, меланому, плазмоцитому, бородавчатую карциному и овсяно-клеточную (мелкоклеточную) карциному; рак желудка, включая, но не ограничиваясь ими, аденокарциному, грибовидную (полиповидную), язвенную, поверхностную, диффузную, злокачественную лимфому, липосаркому, фибросаркому и карциносаркому; рак толстой кишки; рак прямой кишки; рак печени, включая, но не ограничиваясь ими, печеночноклеточную карциному и гепатобластому; рак желчного пузыря, такой как аденокарцинома; холангиокарциномы, включая, но не ограничиваясь ими, папиллярную, узловую и диффузную формы; рак легкого, такой как немелкоклеточный рак легкого, плоскоклеточная карцинома (эпидермоидная карцинома), аденокарцинома, крупноклеточная карцинома и мелкоклеточный рак легкого; рак яичек, включая, но не ограничиваясь ими, эмбрионально-клеточную опухоль, семиному, анапластическую, классическую (типичную), сперматоцитарные несеминомные опухоли, эмбриональную карциному, тератому, хориокарциному (карциному желточного мешка), рак простаты, включая, но не ограничиваясь ими, простатическую интраэпителиальную неоплазию, аденокарциному, лейомиосаркому и рабдомиосаркому; рак полового члена; рак полости рта, включая, но не ограничиваясь ей, плоскоклеточную карциному; базальный рак; рак слюнных желез, включая, но не ограничиваясь ими, аденокарциному, мукоэпидермоидную карциному и аденокистозную карциному; рак глотки, включая, но не ограничиваясь ими, плоскоклеточный рак и бородавчатый рак; рак кожи, включая, но не ограничиваясь ими, базальноклеточную карциному, плоскоклеточную карциному и меланому, поверхностную меланому, узловую меланому, пигментную злокачественную меланому, акральную лентигинозную меланому; рак почки, включая, но не ограничиваясь ими, почечноклеточную карциному, аденокарциному, гипернефрому, фибросаркому, переходно-клеточный рак (почечной лоханки или мочеточника); опухоль Вильмса; рак мочевого пузыря, включая, но не ограничиваясь ими, переходно-клеточную карциному, плоскоклеточный рак, аденокарциному, карциносаркому. В дополнение, раковые заболевания включают миксосаркому, остеогенную саркому, эндотелиосаркому, лимфангиоэндотелиосаркому, мезотелиому, синовиому, гемангиобластому, эпителиальную карциному, цистаденокарциному, бронхогенную карциному, карциному потовых желез, карциному сальной железы, папиллярную карциному и папиллярные аденокарциномы (для обзора указанных нарушений см. Fishman et al., 1985, Medicine, 2d Ed., J.B. Lippincott Co., Philadelphia и Murphy et al., 1997, Informed Decisions: The Complete Book of Cancer Diagnosis, Treatment, and Recovery, Viking Penguin, Penguin Books U.S.A., Inc., United States of America).
Более конкретно, в указанном варианте реализации раковое заболевание представляет собой заболевание, которое связано с расщеплением Notch γ-секретазой, включая, но не ограничиваясь ими, лейкемию, немелкоклеточный рак легкого, рак яичников, рак груди или рак мозга.
В некоторых вариантах реализации раковое заболевание представляет собой рак толстой кишки. В некоторых вариантах реализации раковое заболевание представляет собой рак толстой кишки, груди, легкого или меланому.
В другом варианте реализации субъект, нуждающийся в лечении, проходил ранее или в настоящее время проходит курс лечения от рака. Способы лечения включают, но не ограничиваются ими, химиотерапию, радиационную терапию, хирургию или иммунотерапию, такую как введение вакцины против рака. В некоторых вариантах реализации соединения согласно настоящему изобретению применяют для предотвращения метастазов и инвазии протекавшего ранее ракового заболевания.
Соединения согласно настоящему изобретению также подходят для лечения или предотвращения ракового заболевания, вызванного вирусом. Указанные вирусы включают вирус папилломы человека, который может приводить к раку шейки матки (см., например, Hernandez- Avila et al., Archives of Medical Research (1997) 28:265-271); вирус Эпштейна-Барр (ВЭБ), который может приводить к лимфоме (см., например, Herrmann et al., J. Pathol. (2003) 199(2):140-5); вирус гепатита В или С, который может приводить к карциноме печени (см., например, El-Serag, J. Clin. Gastroenterol. (2002) 35(5 Suppl. 2):S72-8); вирус Т-клеточной лейкемии человека (HTLV)-I, который может приводить к Т-клеточной лейкемии (см., например, Mortreux et al., Leukemia (2003) 17(l):26-38); герпесвирусную инфекцию человека 8 типа, которая может приводить к саркоме Капоши (см., например, Kadow et al., Curr. Opin. Investig. Drugs (2002) 3(11): 1574-9); и вирусную инфекцию иммунодефицита человека (ВИЧ), который может приводить к раку, вызванному иммунодефицитом (см., например, Dal Maso et al., Lancet Oncol (2003) 4(2): 110-9). Каждый из указанных источников включен в настоящую заявку посредством ссылки.
Соединения согласно настоящему изобретению также подходят для предотвращения рака или предотвращения прогрессирования рака, включающего, но не ограничивающегося ими, раковые заболевания, перечисленные в Таблице 1. Указанное профилактическое применение включает применение для состояний, при которых происходит рост неопухолевых клеток, такой как гиперплазия, метаплазия или более конкретно дисплазия. В качестве альтернативы или в дополнение к аномальному росту клеток, характеризуемому гиперплазией, метаплазией или дисплазией, наличие одного или более признаков трансформированного фенотипа или фенотипа злокачественной опухоли, определяемых in vivo или in vitro из клеточной пробы субъекта, может свидетельствовать о желательности проведения профилактического или терапевтического введения соединения согласно настоящему изобретению. Указанные параметры трансформированного фенотипа включают изменения морфологии, присоединение свободных субстратов, снижение контактного торможения, снижение зависимости от культуральной подложки, высвобождение протеазы, повышенный транспорт сахара, ухудшение параметров состава сыворотки, экспрессию фентальных антигенов, элиминация белка клеточной поверхности с массой 250000 дальтон и т.д. В конкретном варианте реализации лейкоплакия, доброкачественное гиперпластическое или диспластическое поражение эпителия, или болезнь Боуэна, карцинома in situ, поддаются лечению или предотвратимы в соответствии со способами согласно настоящему изобретению.
В другом варианте реализации фиброкистозные заболевания (кистозная гиперплазия, дисплазия молочной железы, в частности, аденоз (доброкачественная гиперплазия эндотелия)) поддаются лечению или предотвратимы в соответствии со способами согласно настоящему изобретению.
В других вариантах реализации субъект имеет один или более следующих факторов предрасположенности к развитию злокачественных опухолей, которые поддаются лечению путем введения эффективного количества соединения согласно настоящему изобретению: хромосомную транслокацию, связанную со злокачественной опухолью (например, филадельфийскую хромосому в случае хронической миелогенной лейкемии; t(14;18) в случае фолликулярной лимфомы); семейный полипоз или синдром Гарднера; доброкачественную моноклональную гаммапатию; родство первой степени с персонами, страдающими от ракового или предракового заболевания, наследуемого по менделевскому (генетическому) типу (например, от семейного полипоза толстой кишки, синдрома Гарднера, наследственного экзостоза, полиэндокринного аденоматоза, медуллярной карциномы щитовидной железы, сопровождающейся выработкой амилоида и феокхромоцитомой, синдрома Пейтца-Джигерса, нейрофиброматоза фон Реклингхаузена, ретинобластомы, опухоли каротидного тельца, кожной меланокарциномы, внутриглазной меланокарциномы, пигментной ксеродермы, атаксии-телеангиэктазии, синдрома Чедиака-Хигаси, альбинизма, апластической анемии Фанкони и синдрома Блума); и воздействие карциногенов (например, курение, пассивное курение и вдыхание или контакт с определенными химическими веществами).
Согласно одному из аспектов способы лечения или предотвращения рака согласно настоящему изобретению могут дополнительно включать введение другого противоракового агента.
В одном из вариантов реализации в настоящем изобретении предложены способы лечения или предотвращения рака, включающие введение эффективного количества соединения согласно настоящему изобретению и другого противоракового агента субъекту, нуждающемуся в этом. Соединение согласно настоящему изобретению и другой противораковый агент можно вводить параллельно. В указанном варианте реализации соединение согласно настоящему изобретению и другой противораковый агент можно вводить в составе одной композиции или различных композиций при помощи одного или различных способов введения. В другом варианте реализации соединение согласно настоящему изобретению вводят в то время, когда другой противораковый агент уже осуществляет свое профилактическое или терапевтическое действие, или наоборот.
В другом варианте реализации соединение согласно настоящему изобретению или другой противораковый агент вводят в дозах, обычно используемых в случаях, если указанные агенты применяют в виде единственной терапии для лечения рака.
В одном из вариантов реализации соединение согласно настоящему изобретению или другой противораковый агент вводят в меньших дозах по сравнению с дозами, которые обычно используют в случаях, если указанные агенты применяют в виде единственной терапии для лечения рака.
В другом варианте реализации соединение согласно настоящему изобретению и другой противораковый агент оказывают синергическое действие, и их вводят в меньших дозах по сравнению с дозами, которые обычно используют в случаях, если укзаанные агенты применяют в виде единственной терапии для лечения рака. Дозировка вводимого соединения согласно настоящему изобретению или другого противоракового агента, а также схема дозирования, могут зависеть от ряда параметров, включая, но не ограничиваясь ими, раковое заболевание, подвергающееся лечению, общее состояние здоровья субъекта и выбор лечащего врача. Соединение согласно настоящему изобретению можно вводить до (например, за 5 минут, 15 минут, 30 минут, 45 минут, 1 час, 2 часа, 4 часа, 6 часов, 12 часов, 24 часа, 48 часов, 72 часа, 96 часов, 1 неделю, 2 недели, 3 недели, 4 недели, 5 недель, 6 недель, 8 недель или 12 недель), во время или после (например, через 5 минут, 15 минут, 30 минут, 45 минут, 1 час, 2 часа, 4 часа, 6 часов, 12 часов, 24 часа, 48 часов, 72 часа, 96 часов, 1 неделю, 2 недели, 3 недели, 4 недели, 5 недель, 6 недель, 8 недель или 12 недель) введения другого противоракового агента субъекту, нуждающемуся в этом. В различных вариантах реализации соединение согласно настоящему изобретению и другой противораковый агент вводят с интервалом в 1 минуту, 10 минут, 30 минут, менее 1 часа, 1 час, от 1 часа до 2 часов, от 2 часов до 3 часов, от 3 часов до 4 часов, от 4 часов до 5 часов, от 5 часов до 6 часов, от 6 часов до 7 часов, от 7 часов до 8 часов, от 8 часов до 9 часов, от 9 часов до 10 часов, от 10 часов до 11 часов, от 11 часов до 12 часов, не более 24 часов или не более 48 часов. В одном из вариантов реализации соединение согласно настоящему изобретению и другой противораковый агент вводят с интервалом не более 3 часов. В другом варианте реализации соединение согласно настоящему изобретению и другой противораковый агент вводят с интервалом от 1 минуты до 24 часов.
В одном из вариантов реализации эффективное количество соединения согласно настоящему изобретению и эффективное количество другого противоракового агента входят в состав одной композиции. В одном из вариантов реализации указанная композиция подходит для перорального введения, в другом варианте реализации указанная композиция подходит для внутривенного введения.
В одном из вариантов реализации композиции содержат количества соединения согласно настоящему изобретению и другого противоракового агента, которые совместно являются эффективными для лечения или предотвращения рака.
В другом варианте реализации композиции содержат эффективное количество темозоломида, прокарбазина, дакарбазина, интерлейкина-2, иринотекана или доксорубицина, физиологически приемлемый носитель, разбавитель, наполнитель или среду-носитель и эффективное количество соединения согласно настоящему изобретению.
В одном из вариантов реализации количество соединения согласно настоящему изобретению и другого противоракового агента составляет по меньшей мере примерно 0,01% комбинации химиотерапевтических агентов от массы композиции. В случае перорального введения это количество может изменяться от примерно 0,1% до примерно 80% от массы композиции. Некоторые пероральные композиции могут содержать от примерно 4% до примерно 50% суммарного количества соединения согласно настоящему изобретению и другого противоракового агента от массы композиции. Другие композиции согласно настоящему изобретению получают таким образом, что стандартная пероральная лекарственная форма содержит от примерно 0,01% до примерно 2% комбинации от массы композиции.
Раковые заболевания, которые можно лечить или предотвращать путем введения соединения согласно настоящему изобретению и другого противоракового агента, включают, но не ограничиваются ими, заболевания, перечисленные выше в Таблице 1.
В одном из вариантов реализации раковое заболевание представляет собой рак мозга. В конкретных вариантах реализации рак мозга представляет собой пилоцитарную астроцитому, астроцитому, анапластическую астроцитому, мультиформную глиобластому или метастазирующую опухоль головного мозга.
В одном из вариантов реализации раковое заболевание представляет собой меланому. В конкретном варианте реализации меланома представляет собой метастазирующую меланому.
Соединение согласно настоящему изобретению и другой противораковый агент могут оказывать дополняющее или синергическое действие. Синергическая комбинация соединения согласно настоящему изобретению и другого противоракового агента может позволять применять меньшие дозировки одного или обоих агентов или реже вводить агенты субъекту, страдающему от рака. Возможное использование меньших дозировок соединения согласно настоящему изобретению или другого противоракового агента, или обоих агентов, или менее частое введение агентов могут снижать токсичность, связанную с введением агентов субъекту, не снижая эффективность агентов для лечения рака. В дополнение, синергическое действие может приводить к повышению эффективности указанных агентов для лечения рака и/или к снижению любых отрицательных или нежелательных побочных эффектов, связанных с применением каждого агента по отдельности.
В одном из вариантов реализации введение эффективного количества соединения согласно настоящему изобретению и эффективного количества другого противоракового агента подавляет резистентность ракового заболевания к другому противораковому агенту. В одном из вариантов реализации раковое заболевание представляет собой опухоль.
Другие противораковые агенты, подходящие для применения в способах и композиция согласно настоящему изобретению, включают, но не ограничиваются ими, темозоломид, ингибитор топоизомеразы I, прокарбазин, дакарбазин, гемцитабин, капецитабин, метотрексат, таксол, таксотер, меркаптопурин, тиогуанин, гидроксимочевину, цитарабин, циклофосфамид, ифосфамид, нитрозомочевины, цисплатин, карбоплатин, митомицин, дакарбазин, прокарбизин, этопозид, тенипозид, камптотецины, блеомицин, доксорубицин, идарубицин, даунорубицин, дактиномицин, пликамицин, митоксантрон, L-аспарагиназу, доксорубицин, эпирубицин, 5-фторурацил, таксаны, такие как доцетаксел и паклитаксел, лейковорин, левамизол, иринотекан, эстрамустин, этопозид, азотистые иприты, БХНМ, нитрозомочевины, такие как кармустин и ломустин, винкаалкалоиды, такие как винбластин, винкристин и винорелбин, комплексы платины, такие как цисплатин, карбоплатин и оксалиплатин, иматиниба мезилат, гексаметилмеламин, топотекан, ингибиторы тирозинкиназы, тирфосфтины, гербимицин А, генистеин, эрбстатин и лавендустин А.
В одном из вариантов реализации другой противораковый агент представляет собой, но не ограничивается ими, лекарственные средства, перечисленные в Таблице 2.
дегид
(BPD-MA)
(IP-10)
Другие дополнительные противораковые агенты, которые подходят для применения в композициях и способах согласно настоящему изобретению, включают, но не ограничиваются ими: ацивицин; акларубицин; акодазола гидрохлорид; акронин; адозелесин; альдеслейкин; алтретамин; амбомицин; аметантрона ацетат; аминоглутетимид; амсакрин; анастрозол; антрамицин; аспарагиназу; асперлин; азацитидин; азетепа; азотомицин; батимастат; бензодепа; бикалутамид; бисантрена гидрохлорид; биснафида димезилат; бизелесин; блеомицина сульфат; бреквинар натрия; бропиримин; бусульфан; кактиномицин; калустерон; карацемид; карбетимер; карбоплатин; кармустин; карубицина гидрохлорид; карзелесин; цедефингол; хлорамбуцил; циролемицин; цисплатин; кладрибин; криснатола мезилат; циклофосфамид; цитарабин; дакарбазин; дактиномицин; даунорубицина гидрохлорид; децитабин; дексормаплатин; дезагуанин; дезагуанина мезилат; диазиквон; доцетаксел; доксорубицин; доксорубицина гидрохлорид; дролоксифен; дролоксифена цитрат; дромостанолона пропионат; дуазомицин; эдатрексат; эфлорнтина гидрохлорид; эльзамитруцин; энлоплатин; энпромат; эпипропидин; эпирубицина гидрохлорид; эрбулозол; эзорубицина гидрохлорид; эстрамустин; натриевая соль эстрамустина фосфата; этанидазол; этопозид; этопозида фосфат; этоприн; фадрозола гидрохлорид; фазарабин; фенретинид; флоксуридин; флударабина фосфат; фторурацил; фторцитабин; фосквидон; фостриецин натрия; гемцитабина гидрохлорид; гидроксимочевину; идарубицина гидрохлорид; ифосфамид; илмофозин; интерлейкин-2 (включая рекомбинантный интерлейкин-2 или rIL2), интерферон альфа-2α; интерферон альфа-2β; интерферон альфа-nl; интерферон альфа-n3; интерферон бета-Iα; интерферон γ-Iβ; ипроплатин; иринотекана гидрохлорид; ланреотида ацетат; летрозол; лейпролида ацетат; лиарозола гидрохлорид; лометрексол натрия; ломустин; лозоксантрона гидрохлорид; мазопрокол; майтанзин; мехлорэтамина гидрохлорид; мегестрола ацетат; меленгестрола ацетат; мелфалан; меногарил; меркаптопурин; метотрексат; метотрексат натрия; метоприн; метуредепа; митиндомид; митокарцин; митокромин; митогиллин; митомальцин; митомицин; митоспер; митотан; митоксантрона гидрохлорид; микофеноловую кислоту; нокодазол; ногаламицин; ормаплатин; оксисуран; паклитаксел; пегаспаргазу; пелиомицин; пентамустин; пепломицина сульфат; перфосфамид; пипобромин; пипосульфан; пироксантрона гидрохлорид; пликамицин; пломестан; порфимер натрия; порфиромицин; преднимустин; прокарбазина гидрохлорид; пуромицин; пуромицина гидрохлорид; пиразофурин; рибоприн; роглетимид; сафингол; сафингола гидрохлорид; семустин; симтразен; спарфозат натрия; спарсомицин; спирогермания гидрохлорид; спиромустин; спироплатин; стрептонигрин; стрептозоцин; сулофенур; тализомицин; текогалан натрия; тегафур; телоксантрона гидрохлорид; темопорфин; тенипозид; тероксирон; тестолактон; тиамиприн; тиогуанин; тиотепа; тиазофурин; тирапазамин; торемифена цитрат; трестолона ацетат; трицирибина фосфат; триметрексат; триметрексата глюкуронат; трипторелин; тубулозола гидрохлорид; урациловый иприт; уредепа; вапреотид; вертепорфин; винбластина сульфат; винкристина сульфат; виндесин; виндесина сульфат; винепидина сульфат; винглицината сульфат; винлейрозина сульфат; винорелбина тартрат; винросидина сульфат; винзолидина сульфат; ворозол; зениплатин; зиностатин; и зорубицина гидрохлорид.
Другие противораковые агенты, которые подходят для применения в способах и композициях согласно настоящему изобретению, включают, но не ограничиваются ими: 20-эпи-l,25 дигидроксивитамин D3; 5-этинилурацил; абиратерон; акларубицин; ацилфульвен; адеципенол; адозелесин; альдеслейкин; антагонисты ALL-TK; алтретамин; амбамустин; амидокс; амифостин; аминолевулиновую кислоту; амрубицин; амсакрин; анагрелид; анастрозол; андрографолид; ингибиторы ангиогенеза; антагонист D; антагонист G; антареликс; антидорсальный морфогенетический белок 1; антиандрогенную терапию карциномы простаты; антиэстрогенную терапию; антинеопластоны; античувствительные олигонуклеотиды; афидиколина глицинат; генные модуляторы апоптоза; регуляторы апоптоза; апуриновую кислоту; ara-CDP-DL-PTBA; аргининдеаминазу; асулакрин; атаместан; атримустин; аксинастатин 1; аксинастатин 2; аксинастатин 3; азасетрон; азатоксин; азатирозин; производные баккатина III; баланол; батимастат; антагонисты BCR/ABL; бензохлорины; бензоилстауроспорин; производные бета-лактама; бета-алетин; бетакламицин B; бетулиновую кислоту; ингибитор оФРФ; бикалутамид; бисантрен; бисазиридинилспермин; биснафид; бистратен A; бизелесин; брефлат; бропиримин; будотитан; бутионина сульфоксимин; кальципотриол; калфостин C; производные камптотецина; канарипокс IL-2; карбоксамидаминотриазол; карбоксиамидотриазол; CaRest M3; CARN 700; хрящевой ингибитор; карзелесин; ингибиторы казеинкиназы (ICOS); кастаноспермин; цекропин B; цетрореликс; хлорины; хлорхиноксалина сульфонамид; цикапрост; цис-порфирин; кладрибин; аналоги кломифена; клотримазол; коллисмицин A; коллисмицин B; комбрестатин A4; аналог комбрестатина; конагенин; крамбесцидин 816; криснатол; криптофицин 8; производные криптофицина A; курацин A; циклопентантрахиноны; циклоплатам; ципемицин; цитарабина окфосфат; цитолитический фактор; цитостатин; дакликсимаб; децитабин; дегидродидемнин B; деслорелин; дексаметазон; дексифосфамид; дексразоксан; дексверапамил; диазиквон; дидемнин B; дидокс; диэтилнорспермин; дигидро-5-ацитидин; дигидротаксол; диоксамицин; дифенилспиромустин; доцетаксел; докозанол; доласетрон; доксифлуридин; дролоксифен; дронабинол; дуокармицин SA; эбселен; экомустин; эдельфозин; эдреколомаб; эфлорнитин; элемен; эмитефур; эпирубицин; эпристерид; аналог эстримустина; агонисты эстрогенов; антагонисты эстрогенов; этанидазол; этопозида фосфат; экземестан; фадрозол; фазарабин; фенретинид; филграстим; финастерид; флавопиридол; флезеластин; флуастерон; флударабин; фтордаунорубицина гидрохлорид; форфенимекс; форместан; фостриецин; фотемустин; гадолиния тексафирин; галлия нитрат; галоцитабин; ганиреликс; ингибиторы желатиназы; гемцитабин; ингибиторы глутатиона; гепсульфам; герегулин; гексаметилена бисацетамид; гиперицин; ибандроновую кислоту; идарубицин; идоксифен; идрамантон; илмофозин; иломастат; имидазоакридоны; имиквимод; иммуностимулирующие пептиды; ингибитор рецептора инсулиноподобного фактора роста 1; агонисты интерферона; интерфероны; интерлейкины; иобенгуан; йоддоксорубицин; 4-ипомеанол; ироплакт; ирсогладин; изобенгазол; изогомогаликондрин B; итасетрон; ясплакинолид; кагалалид F; ламелларина-N триацетат; ланреотид; лейнамицин; ленограстим; лентинана сульфат; лептолстатин; летрозол; фактор ингибирования лейкемии; лейкоцитарный интерферон альфа; лейпролид+эстроген+прогестерон; лейпрорелин; левамизол; лиарозол; аналог линейного полиамина; липофильный дисахаридный пептид; липофильные комплексы платины; лиссоклинамид 7; лобаплатин; ломбрицин; лометрексол; лонидамин; лосоксантрон; ловастатин; локсорибин; луртотекан; лютеция тексафирин; лизофиллин; литические пептиды; майтанзин; манностатин A; маримастат; мазопрокол; маспин; ингибиторы матрилизина; ингибиторы матричных металлопротеиназ; меногарил; мербарон; метерелин; метиониназы; метоклопрамид; ингибитор МИФ; мифепристон; милтефозин; миримостим; некомплементарную двухцепочечную РНК; митогуазон; митолактол; аналоги митомицина; митонафид; митотоксин; фактор роста фибробластов-сапорин; митоксантрон; мофаротен; молграмостим; моноклональное антитело, человеческий хорионический гонадотропин; монофосфориллипид A+клеточную стенку миобактерий sk; мопидамол; ингибитор гена множественной лекарственной резистентности; терапию, направленную на супрессор множественных опухолей 1; ипритистые противораковые агенты; микапероксид B; экстракт клеточной стенки миобактерий; мириапорон; N-ацетилдиналин; N-замещенные бензамиды; нафарелин; нагрестип; налоксон+пентазоцин; напавин; нафтерпин; нартограстим; недаплатин; неморубицин; неридроновую кислоту; нейтральную эндопептидазу; нилутамид; низамицин; модуляторы синтеза закиси азота; антиоксидант, действующий на нитроксидные радикалы; нитруллин; О-6-бензилгуанин; октреотид; окиценон; олигонуклеотиды; онапристон; ондансетрон; ондансетрон; орацин; пероральный индуктор цитокинов; ормаплатин; осатерон; оксалиплатин; оксауномицин; паклитаксел; аналоги паклитаксела; производные паклитаксела; палауамин; пальмитоилризоксин; памидроновую кислоту; панакситриол; паномифен; парабактин; пазеллиптин; пегаспаргазу; пелдестин; пентозана полисульфат натрия; пентостатин; пентрозол; перфлуброн; перфосфамид; периллиловый спирт; феназиномицин; фенилацетат; ингибиторы фосфатазы; пицибанил; пилокаприна гидрохлорид; пирарубицин; пиритрексим; плацетин A; плацетин B; ингибитор активатора плазминогена; комплекс платины; комплексы платины; комплекс платина-триамин; порфимер натрия; порфиромицин; преднизон; пропил-бис-акридон; простагландин J2; ингибиторы протеасомы; иммуномодулятор на основе белка А; ингибитор протеинкиназы С; микроскопические водоросли-ингибиторы протеинкиназы С; белковые ингибиторы тирозинфосфатазы; ингибиторы фосфорилазы пуриновых нуклеозидов; пурпурины; пиразолоакридин; конъюгат пиридоксилированного гемоглобина и полиоксиэтилена; антагонисты raf; ралтитрексед; рамостерон; ингибиторы ras-фарнезилпротеинтрансферазы; ингибиторы ras; ингибитор ras-GAP; деметилированный ретеллиптин; рения Re 186 этидронат; ризоксин; рибозимы; ретинамид RII; роглетимид; рохитукин; ромуртид; роквинимекс; рубигинон Bl; рабоксил; сафингол; саинтопин; SarCNU; саркофитол A; саграмостим; миметики Sdi 1; семустин; производное ингибитора старения 1; смысловые олигонуклеотиды; ингибиторы сигнальной трансдуции; модуляторы сигнальной трансдукции; белок, связывающий одноцепочечный антиген; сизофиран; собузоксан; натрия борокаптат; натрия фенилацетат; солверол; соматомединсвязывающий белок; сонермин; спарфозовую кислоту; спикамицин D; спиромустин; спленопентин; спонгистатин 1; скваламин; ингибитор стволовых клеток; ингибиторы деления стволовых клеток; стипиамид; ингибиторы стромелезина; сульфинозин; суперактивный антагонист вазоактивного кишечного пептида; сурадиста; сурамин; свайнсонин; синтетические гликозаминогликаны; таллимустин; тамоксифена метиотид; тауромустин; тазаторен; текогалан натрия; тегафур; теллураперилий; ингибиторы теломеразы; темопорфин; темозоломид; тенипозид; тетрахлордекаоксид; тетразомин; талибластин; тиокоралин; тромбопоэтин; миметический тромбопоэтин; тималфастин; агонист рецептора тимопоэтина; тимотринан; гормон, стимулирующий щитовидную железу; этиопурпурин этилолова; тирапазамин; титаноцена бихлорид; топсентин; торемифен; фактор тотипотентных стволовых клеток; ингибиторы трансляции; третиноин; триацетилуридин; трицирибин; триметрексат; трипторелин; трописетрон; туростерид; ингибиторы тирозинкиназы; тирфостины; ингибиторы UBC; убенимекс; ингибирующий фактор роста урогенитального синуса; антагонисты рецептора урокиназы; вапреотид; вариолин B; векторную систему, эритроцитарную генную терапию; веларезол; верамин; вердины; вертепорфин; винорелбин; винксалтин; витаксин; ворозол; занотерон; зениплатин; зиласкорб; и зиностатин стималамер.
В другом варианте реализации другой противораковый агент представляет собой интерферон-α. В другом варианте реализации другой противораковый агент представляет собой интерлейкин-2. В одном из вариантов реализации другой противораковый агент представляет собой алкилирующий агент, такой как азотистый иприт, нитрозомочевину, алкилсульфонат, триазен или платиносодержащий агент. В одном из вариантов реализации другой противораковый агент представляет собой триазеновый алкилирующий агент. В одном из вариантов реализации другой противораковый агент представляет собой О-6-бензилгуанин. В другом варианте реализации другой противораковый агент представляет собой О-6-бензилгуанин и темозоломид. В другом варианте реализации другой противораковый агент представляет собой О-6-бензилгуанин и прокарбазин. В другом варианте реализации другой противораковый агент представляет собой О-6-бензилгуанин и дакарбазин.
Соединения согласно настоящему изобретению можно вводить субъекту, которому проводили или проводят один или более дополнительных курсов противораковой терапии, включая, но не ограничиваясь ими, хирургию, радиационную терапию или иммунотерапию, такую как вакцинирование против рака.
В одном из вариантов реализации в изобретении предложены способы лечения или предотвращения рака, включающие введение субъекту, нуждающемуся в этом, эффективного количества (1) соединения согласно настоящему изобретению и (2) проведение другой противораковой терапии, включая, но не ограничиваясь ими, хирургию, радиационную терапию или иммунотерапию, такую как вакцинирование против рака.
В одном из вариантов реализации другая противораковая терапия представляет собой радиационную терапию. В другом варианте реализации другая противораковая терапия представляет собой хирургию. В другом варианте реализации другая противораковая терапия представляет собой иммунотерапию.
В конкретном варианте реализации способы лечения или предотвращения рака согласно настоящему изобретению включают введение эффективного количества соединения согласно настоящему изобретению и проведение радиационной терапии. Радиационную терапию можно проводить во время, до или после введения соединения согласно настоящему изобретению, согласно одному из вариантов реализации по меньшей мере с интервалом, равным часу, пяти часам, 12 часам, дню, неделе, месяцу, согласно другому варианту реализации с интервалом, равным нескольким месяцам (например, не более чем трем месяцам) до или после введения соединения согласно настоящему изобретению. Если другая противораковая терапия представляет собой радиационную терапию, то радиационную терапию можно проводить согласно любому протоколу в зависимости от типа ракового заболевания, подвергающегося лечению. В качестве примера, но не ограничения, можно применять рентгеновское излучение; в частности, облучение частицами высокой энергии (облучение более 1 МэВ) можно проводить в случае опухолей внутренних органов, а облучение пучком электронов и ортовольтажное рентгеновоское облучение можно проводить в случае раковых заболеваний кожи. Радиоизотопы, испускающие гамма-лучи, такие как радиоактивные изотопы радия, кобальта и других элементов, также можно вводить.
Кроме того, в изобретении предложены способы лечения рака, включающие введение соединения согласно настоящему изобретению в качестве альтернативы химиотерапии или радиационной терапии в случаях, когда химиотерапия или радиационная терапия приводят к возникновению негативных побочных эффектов у субъекта, подвергающегося лечению. Субъекту, подвергающемуся лечению, можно проводить другую противораковую терапию, такую как хирургия, радиационная терапия или иммунотерапия.
Соединения согласно настоящему изобретению также можно вводить in vitro или ex vivo, например, для лечения определенных раковых заболеваний, включая, но не ограничиваясь ими, лейкемии и лимфомы, где указанное лечение проводят с применением трансплантатов аутологичных стволовых клеток. Лечение может включать процесс, при котором аутологичные гематопоэтические стволовые клетки субъекта выращивают и очищают от всех раковых клеток, оставшуюся популяцию клеток костного мозга субъекта затем уничтожают путем введения соединения согласно настоящему изобретению или радиации, или проведения обоих видов терапии, и проводят инфузию полученных стволовых клеток субъекту. Затем можно проводить поддерживающую терапию во время восстановления функции костного мозга субъекта.
В. Лечение или предотвращение нейродегенеративного заболевания
В изобретении предложены способы лечения или предотвращения нейродегенеративного заболевания, включающие введение эффективного количества соединения согласно настоящему изобретению субъекту, нуждающемуся в этом. В одном из вариантов реализации соединение входит в состав композиции, которая дополнительно содержит фармацевтически приемлемый носитель.
Примеры нейродегенеративных заболеваний включают, но не ограничиваются ими, болезнь Александера, болезнь Альпера, болезнь Альцгеймера, боковой амиотрофический склероз (БАС), атаксию-телеангиэктазию, болезнь Баттена (также известную как болезнь Шпильмейера-Фогта-Шегрена-Баттена), губчатую энцефалопатию крупного рогатого скота, болезнь Канавана, синдром Коккейна, кортикобазальную дегенерацию, болезнь Крейтцфельда-Якоба, болезнь Хантингтона, связанное с ВИЧ слабоумие, болезнь Кеннеди, болезнь Краббе, деменцию с тельцами Леви, болезнь Мачадо-Джозефа (спинально-церебеллярную атаксию 3 типа), рассеянный склероз (РС), множественную системную атрофию, нарколепсию, нейроборрелиоз, болезнь Паркинсона, болезнь Пелицеуса-Мерцбахера, болезнь Пика, первичный латеральный склероз, прионные болезни, прогрессирующий надъядерный паралич, болезнь Рефсума, болезнь Сандхоффа, болезнь Шильдера, подострую сочетанную дегенерацию спинного мозга, вызванную пернициозной анемией, спинально-церебеллярную атаксию, спинальную мышечную атрофию, болезнь Стила-Ричардсона-Ольшевского и сухотку спинного мозга. В одном из вариантов реализации нейродегенеративное заболевание представляет собой болезнь Альцгеймера. Другие примеры нейродегенеративных заболеваний включают, но не ограничиваются ими, болезнь диффузных телец Леви, мультисистемную дегенерацию (синдром Шая-Дрейджера), заболевания мотонейрона, включая боковой амиотрофический склероз, дегенеративные атаксии, кортикобазальную дегенерацию, комплекс острова Гуам (БАС-болезнь Паркинсона-деменция), подострый склерозирующий панэнцефалит, болезнь Хантингтона, синуклеинопатии, первичную прогрессирующую афазию, стриатонигральную дегенерацию, болезнь Мачадо-Джозефа/спинально-церебральную атаксию 3 типа и оливопонтоцеребеллярные дегенерации, болезнь Жиля де ля Туррета, бульбарный и всевдобульбарный паралич, спинальную и бульбарную мышечную атрофию (болезнь Кеннеди), первичный латеральный склероз, семейную спастическую параплегию, болезнь Верднига-Гоффмана, болезнь Кугельберга-Веландера, болезнь Тея-Сакса, болезнь Сандхоффа, семейную спастическую болезнь, болезнь Вольфарта-Кугельберга-Веландера, спастический парапарез, прогрессирующую мультифокальную лейкоэнцефалопатию, прионные болезни (включая болезнь Крейтцфельда-Якоба, болезнь Герстманна-Штреусслера-Шейнкера, куру и фатальную семейную бессонницу), старческое слабоумие и другие состояния, сопровождающиеся потерей памяти, такие как сосудистая деменция, диффузное поражение белого вещества (болезнь Бинсвангера), деменция эндокринной или метаболической природы, деменция, вызванная травмой головы и диффузным повреждением мозга, деменция боксеров и лобно-височная деменция, ишемию или инфаркт головного мозга, включая эмболическую окклюзию и тромботическую окклюзию, а также внутричерепное кровотечение любого типа (включая, но не ограничиваясь ими, эпидуральное, субдуральное, субарахноидальное и внутримозговое) и внутричерепные и межпозвоночные повреждения (включая, но не ограничиваясь ими, контузию, проникающее повреждение, смещение, сжатие и разрыв).
Согласно одному из аспектов способы лечения или предотвращения нейродегенеративного заболевания согласно настоящему изобретению могут дополнительно включать введение другого агента против нейродегенеративного заболевания.
В одном из вариантов реализации в настоящем изобретении предложены способы лечения или предотвращения нейродегенеративного заболевания, включающие введение эффективного количества соединения согласно настоящему изобретению и другого агента против нейродегенеративного заболевания субъекту, нуждающемуся в лечении или предотвращении нейродегенеративного заболевания. Соединение согласно настоящему изобретению и другой агент против нейродегенеративного заболевания можно вводить раздельно. Соединение согласно настоящему изобретению и другой агент против нейродегенеративного заболевания также можно вводить одновременно. В указанном варианте реализации соединение согласно настоящему изобретению и другой агент против нейродегенеративного заболевания можно вводить в одной композиции или можно вводить в различных композициях при помощи одного или различных способов введения. В другом варианте реализации соединение согласно настоящему изобретению вводят в то время, когда другой агент против нейродегенеративного заболевания уже оказывает свое профилактическое или терапевтическое действие, или наоборот.
В другом варианте реализации соединение согласно настоящему изобретению или другой агент против нейродегенеративного заболевания вводят в дозах, используемых обычно в случаях, когда указанные агенты применяют в качестве единственной терапии для лечения нейродегенеративного заболевания.
В одном из вариантов реализации соединение согласно настоящему изобретению или другой агент против нейродегенеративного заболевания вводят в меньших дозах по сравнению с дозами, используемыми обычно в случаях, если указанные агенты применяют в качестве единственной терапии для лечения нейродегенеративного заболевания.
В другом варианте реализации соединение согласно настоящему изобретению и другой агент против нейродегенеративного заболевания оказывают синергическое действие, и их вводят в меньших дозах по сравнению с дозами, используемыми обычно в случаях, если указанные агенты применяют в качестве единственной терапии для лечения нейродегенеративного заболевания. Дозировка вводимого соединения согласно настоящему изобретению или другого агента против нейродегенеративного заболевания, а также схема дозирования могут зависеть от ряда параметров, включая, но не ограничиваясь ими, нейродегенеративное заболевание, подвергающееся лечению, общее состояние здоровья субъекта и выбор лечащего врача. Соединение согласно настоящему изобретению можно вводить до (например, за 5 минут, 15 минут, 30 минут, 45 минут, 1 час, 2 часа, 4 часа, 6 часов, 12 часов, 24 часа, 48 часов, 72 часа, 96 часов, 1 неделю, 2 недели, 3 недели, 4 недели, 5 недель, 6 недель, 8 недель или 12 недель), во время или после (например, через 5 минут, 15 минут, 30 минут, 45 минут, 1 час, 2 часа, 4 часа, 6 часов, 12 часов, 24 часа, 48 часов, 72 часа, 96 часов, 1 неделю, 2 недели, 3 недели, 4 недели, 5 недель, 6 недель, 8 недель или 12 недель) введения другого агента против нейродегенеративного заболевания субъекту, нуждающемуся в лечении или предотвращении нейродегенеративного заболевания. В ряде вариантов реализации соединение согласно настоящему изобретению и другой агент против нейродегенеративного заболевания вводят с интервалом в 1 минуту, 10 минут, 30 минут, менее 1 часа, 1 час, от 1 часа до 2 часов, от 2 часов до 3 часов, от 3 часов до 4 часов, от 4 часов до 5 часов, от 5 часов до 6 часов, от 6 часов до 7 часов, от 7 часов до 8 часов, от 8 часов до 9 часов, от 9 часов до 10 часов, от 10 часов до 11 часов, от 11 часов до 12 часов, не более 24 часов или не более 48 часов. В одном из вариантов реализации соединение согласно настоящему изобретению и другой агент против нейродегенеративного заболевания вводят с интервалом не более 3 часов. В другом варианте реализации соединение согласно настоящему изобретению и другой агент против нейродегенеративного заболевания вводят с интервалом, равным от 1 минуты до 24 часов.
В одном из вариантов реализации эффективное количество соединения согласно настоящему изобретению и эффективное количество другого агента против нейродегенеративного заболевания входят в состав одной композиции. В одном из вариантов реализации указанная композиция подходит для перорального введения. В другом варианте реализации указанная композиция подходит для внутривенного введения.
В одном из вариантов реализации композиции содержат количества соединения согласно настоящему изобретению и другого агента против нейродегенеративного заболевания, которые совместно являются эффективными для лечения или предотвращения нейродегенеративного заболевания.
Соединения согласно настоящему изобретению и другой агент против нейродегенеративного заболевания могут оказывать дополняющее или синергическое действие. Синергическая комбинация соединения согласно настоящему изобретению и другого агента против нейродегенеративного заболевания может позволять применять меньшие дозировки одного или обоих агентов и/или реже вводить агенты субъекту, страдающему от нейродегенеративного заболевания. Возможное использование меньших дозировок соединения согласно настоящему изобретению или другого агента против нейродегенеративного заболевания, или обоих агентов, и/или менее частое введение агентов могут снижать токсичность, связанную с введением агентов субъекту, не снижая эффективность агентов для лечения нейродегенеративного заболевания. В дополнение, синергическое действие может приводить к повышению эффективности указанных агентов для лечения нейродегенеративного заболевания и/или к снижению любых отрицательных или нежелательных побочных эффектов, связанных с применением каждого агента по отдельности.
В одном из вариантов реализации введение эффективного количества соединения согласно настоящему изобретению и эффективного количества другого агента против нейродегенеративного заболевания подавляет резистентность нейродегенеративного заболевания к другому агенту против нейродегенеративного заболевания.
Другие агенты против нейродегенеративного заболевания, подходящие для применения в способах и композициях согласно настоящему изобретению, включают, но не ограничиваются ими: агенты против болезни Альцгеймера, такие как ингибиторы холинэстеразы (например, такрин, донезепила гидрохлорид, ривастигмин или галантамин) или неполные антагонисты глутаматных рецепторов (например, мемантин); агенты против болезни Паркинсона, такие как леводопа, карбидопа, толкапон, бромкриптин, перголид, прамипексол, ропинирол, селегилин или амантадин; агенты против БАС, такие как рилузол; и агенты против РС, такие как интерферон бета-1а, интерферон бета-1b, глатирамера ацетат, митоксантрон или натализумаб.
С. Комбинированная терапия
Дополнительные агенты, которые можно применять в комбинации с соединениями согласно настоящему изобретению для лечения или предотвращения состояния, например, заболевания, связанного с активностью γ-секретазы, или предотвращения заболеваний, связанных с активностью γ-секретазы, включают, но не ограничиваются ими, небольшую молекулу, синтетическое лекарственное средство, пептид (включая циклический пептид), полипептид, белок, нуклеиновую кислоту (например, нуклеотид ДНК и РНК, включая, но не ограничиваясь ими, антисмысловую последовательность нуклеотидов, тройную спираль, РНКи и нуклеотидную последовательность, кодирующую биологически активный белок, полипептид или пептид), антитело, синтетическую или природную неорганическую молекулу, миметический агент и синтетическую или природную органическую молекулу. Конкретные примеры указанных агентов включают, но не ограничиваются ими, иммуномодулирующий агент (например, интерферон), противовоспалительный агент (например, адренокортикоид, кортикостероид (например, беклометазон, будезонид, флунизолид, флутиказон, триамцинолон, метилпреднизолон, преднизолон, преднизон, гидрокортизон), глюкокортикоид, стероид и нестероидный противовоспалительный агент (например, аспирин, ибупрофен, диклофенак и ингибитор СОХ-2), обезболивающее, антагонист лейкотриеновых рецепторов (например, монтелукаст, метилксантин, зафирлукаст и зилейтон), бета2-агонист (например, албутерол, битерол, фенотерол, изоэтарин, метапротеренол, пирбутерол, салбутанол, тербуталин, формотерол, салметерол и сальбутанол тербуталин), антихолинэргический агент (например, ипратропия бромид и окситропия бромид), сульфалазин, пеницилламин, дапсон, антигистаминный агент, противомалярийный агент (например, гидроксихлорохин), противовирусный агент (например, аналог нуклеозида (например, зидовудин, ацикловир, гангцикловир, видарабин, идоксуридин, трифлуридин и рибавирин), фоскарнет, амантадин, римантадин, саквинавир, индинавир, ритонавир и AZT) и антибиотик (например, дактиномицин (бывший актиномицин), блеомицин, эритромицин, пенициллин, митрамицин и антрамицин (АМС)).
V. Терапевтическое или профилактическое введение и композиции согласно настоящему изобретению
Вследствие своей активности соединения согласно настоящему изобретению обладают преимуществами, подходящими для ветеринарных препаратов и препаратов для человека.
При введении субъекту соединения согласно настоящему изобретению можно вводить в качестве компонента композиции, которая содержит фармацевтически приемлемый носитель, разбавитель, наполнитель или среду-носитель. Композиции согласно настоящему изобретению, которые содержат соединение согласно настоящему изобретению, можно вводить перорально. Соединения согласно настоящему изобретению также можно вводить при помощи любого другого удобного способа, например, путем инфузии или инъекции болюса, всасывания через эпителиальную выстилку или поверхность кожи или слизистую оболочку (например, через слизистую оболочку полости рта, прямой кишки и кишечника), а также можно вводить совместно с другим биологически активным агентом. Введение может быть системным или местным. Известны различные системы доставки, например, инкапсулирование в липосомы, микрочастицы, микрокапсулы и капсулы.
Способы введения включают, но не ограничиваются ими, внутрикожное, внутримышечное, интраперитонеальное, внутривенное, подкожное, интраназальное, эпидуральное, пероральное, подъязычное, внутримозговое, внутривагинальное, чрескожное, ректальное, введение путем ингаляции или местное введение, в частности, в уши, нос, глаза или на кожу. В некоторых случаях введение приводит к высвобождению соединения согласно настоящему изобретению в кровоток.
В одном из вариантов реализации соединения согласно настоящему изобретению вводят перорально. В других вариантах реализации может быть желательным местное введение соединений согласно настоящему изобретению, которое можно проводить в качестве примера, но не ограничения, путем местной инфузии во время хирургии, местного нанесения при зашивании раны после хирургии, путем инъекции, с применением катетера, с применением суппозитория или клизмы, или путем имплантации, где имплант может представлять собой пористый, беспористый или гелеобразный материал, включая мембраны, такие как сиаластические мембраны, или волокна.
В конкретных вариантах реализации может быть желательным введение соединений согласно настоящему изобретению в центральную нервную систему или желудочно-кишечный тракт при помощи любого подходящего способа, включая внутрижелудочковую, интратекальную и эпидуральную инъекцию и клизму. Внутрижелудочковую инъекцию можно облегчать путем применения внутрижелудочкового катетера, например, присоединенного к резервуару, такому как резервуар Оммайя.
Также можно применять внутрилегочное введение, например, с применением ингалятора или небулайзера, состава, содержащего аэрозольный агент, или путем перфузии во фторуглеродный или синтетический легочный сурфактант. В конкретных вариантах реализации соединения согласно настоящему изобретению могут входить в состав суппозитория совместно с традиционными связующими веществами и наполнителями, такими как триглицериды.
В другом варианте реализации соединения согласно настоящему изобретению можно доставлять в виде везикулы, в частности, липосомы (см., Langer, Science 249:1527-1533 (1990) и Liposomes in Therapy of Infectious Disease and Cancer 317-327 and 353-365 (1989)).
В другом варианте реализации соединения согласно настоящему изобретению можно доставлять при помощи системы с контролируемым высвобождением или системы с замедленным высвобождением (см., например, Goodson, in Medical Applications of Controlled Release, выше, vol. 2, pp. 115-138 (1984)). Можно применять другие системы с контролируемым или замедленным высвобождением, обсуждаемые в обзоре Лангера (Langer, Science 249: 1527-1533 (1990)). В одном из вариантов реализации можно применять насос (Langer, Science 249: 1527- 1533 (1990); Sefton, CRC Crit. Ref. Biomed. Eng. 14:201 (1987); Buchwald et al, Surgery 88:507 (1980); и Saudek et al., N. Engl. J Med. 321:574 (1989)). В другом варианте реализации можно применять полимерные материалы (см., Medical Applications of Controlled Release (Langer and Wise eds., 1974); Controlled Drug Bioavailability, Drug Product Design and Performance (Smolen and Ball eds., 1984); Ranger and Peppas, J. Macromol. Sd. Rev. Macromol. Chem. 2:61 (1983); Levy et al, Science 228:190 (1935); During et al, Ann. Neural. 25:351 (1989); и Howard et al, J. Neurosurg. 71:105 (1989)).
В другом варианте реализации систему с контролируемым или замедленным высвобождением можно помещать в непосредственной близости от мишени соединений согласно настоящему изобретению, например, позвоночника, мозга, кожи, легкого или желудочно-кишечного тракта, и, таким образом, требуется введение только части дозы, которую необходимо использовать с случае системного введения.
Композиции согласно настоящему изобретению могут содержать подходящее количество фармацевтически приемлемого наполнителя для получения формы, подходящей для введения субъекту.
Указанные фармацевтические наполнители могут представлять собой жидкости, такие как вода и масла, включая масла минеральной, животной, растительной или синтетической природы, такие как арахисовое масло, соевое масло, минеральное масло, кунжутное масло и т.д. Фармацевтические наполнители могут представлять собой солевой раствор, аравийскую камедь, желатин, крахмальную пасту, тальк, кератин, коллоидный оксид кремния, мочевину и т.д. В дополнение, можно применять вспомогательные агенты, стабилизаторы, загустители, смазки и красители. В одном из вариантов реализации фармацевтически приемлемые наполнители являются стерильными при введении субъекту. Вода является подходящим наполнителем в случае внутривенного введения соединения согласно настоящему изобретению. Солевые растворы и водные растворы декстрозы и глицерина также можно применять в качестве жидких наполнителей, в частности в случае инъецируемых растворов. Подходящие фармацевтические наполнители также включают крахмал, глюкозу, лактозу, сахарозу, желатин, солод, рис, муку, карбонат кальция, силикагель, стеарат натрия, глицерилмоностеарат, тальк, хлорид натрия, сухое обезжиренное молоко, глицерин, пропиленгликоль, воду, этанол и т.д. Композиции согласно настоящему изобретению при желании также могут содержать незначительные количества увлажнителей или эмульгаторов или буферных агентов.
Композиции согласно настоящему изобретению могут иметь форму растворов, суспензий, эмульсий, таблеток, пилюль, драже, капсул, капсул, содержащих жидкость, порошков, составов с замедленным высвобождением, суппозиториев, эмульсий, аэрозолей, спреев, суспензий или любую другую форму, подходящую для применения. В одном из вариантов реализации композиция находится в форме капсулы (см., например, патент США №5698155). Другие примеры фармацевтически приемлемых наполнителей описаны в Remington 's Pharmaceutical Sciences 1447-1676 (Alfonso R. Gennaro eds., 19th ed. 1995), содержание которой включено в настоящую заявку посредством ссылки.
В одном из вариантов реализации соединения согласно настоящему изобретению в соответствии с традиционными способами вводят в состав композиции, предназначенной для перорального введения человеку. Композиции для пероральной доставки, например, могут иметь форму таблеток, пастилок, водных или масляных суспензий, гранул, порошков, эмульсий, капсул, сиропов или эликсиров. Композиции для перорального введения могут содержать один или более агентов, например, подсластителей, таких как фруктоза, аспартам или сахарин; ароматизаторов, таких как перечная мята, винтергриновое масло или вишневый ароматизатор; красителей; и консервантов, для получения фармацевтического препарата, привлекательного для потребителя. Более того в случае таблеток или пилюль композиции могут быть покрыты оболочкой для задержки распадания и всасывания в желудочно-кишечном тракте, что, таким образом, обеспечивает продолжительное действие в течение длительного периода времени. Селективно проницаемые мембраны, расположенные вокруг соединения согласно настоящему изобретению, доставляемого под действием осмоса, также подходят для перорального введения композиций. В указанных последних лекарственных формах жидкая среда, окружающая капсулу, поглощается доставляемым соединением, которое набухает, в результате чего агент или композиция, содержащая агент, вытесняются через мембрану. Указанные платформы доставки могут обеспечивать профиль доставки нулевого порядка в отличие от пикообразного профиля составов с немедленным высвобождением. Вещество, задерживающее высвобождение, такое как глицерилмоностерат или глицерилстеарат, также можно применять. Пероральные композиции могут содержать стандартные наполнители, такие как маннит, лактоза, крахмал, стеарат магния, сахаринат натрия, целлюлозу и карбонат магния. В одном из вариантов реализации наполнители имеют фармацевтическую степень чистоты.
В другом варианте реализации соединения согласно настоящему изобретению можно вводить в состав для внутривенного введения. Как правило, композиции для внутривенного введения содержат стерильный изотонический водный буфер. При необходимости композиции также могут содержать агент, увеличивающий растворимость. Композиции для внутривенного введения могут содержать местный анестетик, такой как лигнокаин, для снижения боли в месте инъекции.
В целом, ингредиенты вводят по отдельности или смешивают в стандартной лекарственной форме, например, в виде сухого лиофилизированного порошка или безводного концентрата, в герметичном контейнере, таком как ампула или сашет, с указанием количества активного агента. Если соединения согласно настоящему изобретению необходимо вводить путем инфузии, их можно помещать, например, в бутыль для инфузии, содержащую стерильную воду или солевой раствор фармацевтической степени чистоты. Если соединения согласно настоящему изобретению вводят путем инъекции, то можно применять ампулу, содержащую стерильную воду для инъекций или солевой раствор, и, таким образом, ингредиенты можно смешивать перед введением.
Соединения согласно настоящему изобретению можно вводить при помощи средств контролируемого или замедленного высвобождения или с применением устройств доставки, хорошо известных специалистам в данной области техники. Примеры включают, но не ограничиваются ими, устройства и средства, описанные в патентах США №3845770; 3916899; 3536809; 3598123; 4008719; 5674533; 5059595; 5591767; 5120548; 5073543; 5639476; 5354556; и 5733556, содержание каждого из которых включено в настоящую заявку во всей полноте посредством ссылки. Указанные лекарственные формы могут обеспечивать контролируемое или замедленное высвобождение одного или более активных ингредиентов с применением в различных соотношениях, например, гидроксипропилметилцеллюлозы, других полимерных матриц, гелей, проницаемых мембран, осмотических систем, многослойных оболочек, микрочастиц, липосом, микросфер или их комбинации, для достижения желаемого профиля высвобождения. Можно легко выбирать составы с контролируемым или замедленным высвобождением, известные специалистам в данной области техники, включая составы, описанные в настоящей заявке, подходящие для применения активных ингредиентов Формулы I-XI. Таким образом, в изобретении предложены стандартные лекарственные формы, подходящие для перорального введения, включая, но не ограничиваясь ими, таблетки, капсулы, желатиновые капсулы и каплеты, которые предназначены для контролируемого или замедленного высвобождения.
Контролируемое или замедленное высвобождение активного ингредиента могут стимулироваться различными условиями, включая, но не ограничиваясь ими, изменения рН, изменения температуры, концентрацию или доступность ферментов, концентрацию или доступность воды, или другие физиологические состояния или соединения. Количество соединений согласно настоящему изобретению, которое является эффективным для лечения или предотвращения нейродегенеративного заболевания, можно легко определять при помощи стандартных клинических способов. В дополнение, in vitro или in vivo исследования можно использовать для определения оптимальных диапазонов дозировки. Точная доза также может зависеть от способа введения, тяжести состояния, подвергающегося лечению, и может быть определена практикующим специалистом на основании особенностей субъекта, например, с учетом опубликованных клинических исследований. Подходящие эффективные дозируемые количества, тем не менее, находятся в диапазоне примерно от 10 микрограммов до 5 граммов примерно раз в 4 часа, как правило, составляют примерно 500 мг или менее раз в 4 часа. В одном из вариантов реализации эффективная дозировка составляет примерно 0,01 мг, 0,5 мг, примерно 1 мг, примерно 50 мг, примерно 100 мг, примерно 200 мг, примерно 300 мг, примерно 400 мг, примерно 500 мг, примерно 600 мг, примерно 700 мг, примерно 800 мг, примерно 900 мг, примерно 1 г, примерно 1,2 г, примерно 1,4 г, примерно 1,6 г, примерно 1,8 г, примерно 2,0 г, примерно 2,2 г, примерно 2,4 г, примерно 2,6 г, примерно 2,8 г, примерно 3,0 г, примерно 3,2 г, примерно 3,4 г, примерно 3,6 г, примерно 3,8 г, примерно 4,0 г, примерно 4,2 г, примерно 4,4 г, примерно 4,6 г, примерно 4,8 г и примерно 5,0 г раз в 4 часа. Эквивалентные дозировки можно вводить с различными интервалами, включая, но не ограничиваясь ими, примерно раз в 2 часа, примерно раз в 6 часов, примерно раз в 8 часов, примерно раз в 12 часов, примерно раз в 24 часа, примерно раз в 36 часов, примерно раз в 48 часов, примерно раз в 72 часа, примерно раз в неделю, примерно раз в две недели, примерно раз в три недели, примерно раз в месяц и примерно раз в два месяца. Эффективные дозируемые количества, описанные в настоящей заявке, относятся к общему вводимому количеству; то есть, если вводят более одного соединения согласно настоящему изобретению, то эффективные дозируемые количества соответствуют общему вводимому количеству.
Композиции можно получать согласно традиционным способам смешения гранулирования или нанесения оболочки, соответственно, и композиции согласно настоящему изобретению могут содержать, в соответствии с одним из вариантов реализации, от примерно 0,1% до примерно 99%; в соответствии с другим вариантом реализации, от примерно 1% до примерно 70% соединения согласно настоящему изобретению по массе или объему.
Схему дозирования соединения согласно настоящему изобретению можно выбирать с учетом ряда факторов, включая тип, особенности, возраст, массу тела, пол и медицинское состояние субъекта; тяжесть состояния, подвергающегося лечению; способ введения; функцию почек и печени субъекта; и конкретное применяемое соединение согласно настоящему изобретению. Соединение согласно настоящему изобретению можно вводить один раз в день; или ежедневную дозу можно разделять на дозы, которые вводят два, три или четыре раза в день. Кроме того, соединение согласно настоящему изобретению можно вводить в нос с применением носителей, подходящих для местного введения в нос, или путем чрескожного введения с применением чрескожных пластырей, хорошо известных специалистам в данной области техники. В случае системы чрескожной доставки введение должно быть непрерывным, а не периодическим. Другие иллюстративные местные препараты включают кремы, мази, лосьоны, аэрозоли, спреи и гели, где концентрация соединения согласно настоящему изобретению находится в диапазоне от примерно 0,1% до примерно 15% (масс./масс. или масс./об.). Соединения согласно настоящему изобретению можно исследовать in vitro или in vivo для определения желаемой терапевтической или профилактической активности перед применением у человека. Животные модельные системы можно использовать для подтверждания безопасности и эффективности.
В конкретных вариантах реализации соединение согласно настоящему изобретению или фармацевтическую композицию, содержащую указанное соединение, вводят человеку в возрасте в диапазоне от примерно 0 месяцев до примерно 6 месяцев, от примерно 6 до примерно 12 месяцев, от примерно 6 до примерно 18 месяцев, от примерно 18 до примерно 36 месяцев, от примерно 1 до примерно 5 лет, от примерно 5 до примерно 10 лет, от примерно 10 до примерно 15 лет, от примерно 15 до примерно 20 лет, от примерно 20 до примерно 25 лет, от примерно 25 до примерно 30 лет, от примерно 30 до примерно 35 лет, от примерно 35 до примерно 40 лет, от примерно 40 до примерно 45 лет, от примерно 45 до примерно 50 лет, от примерно 50 до примерно 55 лет, от примерно 55 до примерно 60 лет, от примерно 60 до примерно 65 лет, от примерно 65 до примерно 70 лет, от примерно 70 до примерно 75 лет, от примерно 75 до примерно 80 лет, от примерно 80 до примерно 85 лет, от примерно 85 до примерно 90 лет, от примерно 90 до примерно 95 лет или от примерно 95 до примерно 100 лет.
В некоторых вариантах реализации соединение согласно настоящему изобретению или фармацевтическую композицию, содержащую указанное соединение, вводят младенцу. В других вариантах реализации соединение согласно настоящему изобретению или фармацевтическую композицию, содержащую указанное соединение, вводят ребенку ясельного возраста. В других вариантах реализации соединение согласно настоящему изобретению или фармацевтическую композицию, содержащую указанное соединение, вводят ребенку. В других вариантах реализации соединение согласно настоящему изобретению или фармацевтическую композицию, содержащую указанное соединение, вводят взрослому человеку. В других вариантах реализации соединение согласно настоящему изобретению или фармацевтическую композицию, содержащую указанное соединение, вводят пожилому человеку.
В конкретных вариантах реализации соединение согласно настоящему изобретению или композицию, содержащую указанное соединение, вводят субъекту с нарушенным иммунитетом или ослабленным иммунитетом или субъекту с повышенным риском нарушения или ослабления иммунитета. В конкретных вариантах реализации соединение согласно настоящему изобретению или фармацевтическую композицию, содержащую указанное соединение, вводят субъекту, которому проводят иммуносупрессивную терапию, или субъекту, который восстанавливается после иммуносуппрессивной терапии.
В некоторых вариантах реализации соединение согласно настоящему изобретению или фармацевтическую композицию, содержащую указанное соединение, вводят пациенту, который подвержен побочным реакциям, вызванным традиционными терапиями γ-секретазы. В некоторых вариантах реализации ингибитор γ-секретазы или фармацевтическую композицию, содержащую ингибитор, вводят пациенту, который невосприимчив к терапиям γ-секретазы, отличным от введения ингибиторов γ-секретазы, но не к указанной терапии. В число таких пациентов входят невосприимчивые пациенты и молодые пациенты, которым нельзя проводить традиционную терапию.
В некоторых вариантах реализации субъекту, которому вводят соединение согласно настоящему изобретению или фармацевтическую композицию, содержащую указанное соединение, не проводили терапию до введения соединения согласно настоящему изобретению или композиции, содержащей указанное соединение.
IV. Наборы, содержащие соединение согласно настоящему изобретению
В изобретении предложены наборы, которые могут упрощать введение соединения согласно настоящему изобретению субъекту.
Типовой набор согласно настоящему изобретению содержит соединение согласно настоящему изобретению, например, в стандартной лекарственной форме. В одном из вариантов реализации стандартная лекарственная форма представляет собой контейнер, который может быть стерильным, и содержит эффективное количество соединения согласно настоящему изобретению и фармацевтически приемлемый носитель, разбавитель, наполнитель или среду-носитель. Набор может дополнительно содержать этикетку или печатную инструкцию по применению соединения согласно настоящему изобретению для лечения или предотвращения состояния. Набор дополнительно может содержать другой профилактический или терапевтический агент, например, в стандартной лекарственной форме, такой как контейнер, содержащий эффективное количество другого профилактического или терапевтического агента. В одном из вариантов реализации набор содержит контейнер, содержащий эффективное количество соединения согласно настоящему изобретению и эффективное количество другого профилактического или терапевтического агента. Примеры других профилактических или терапевтических агентов включают, но не ограничиваются ими, агенты, перечисленные выше.
Каждый документ, отмеченный в настоящем описании, включен в настоящую заявку по всей полноте посредством ссылки.
Синтез соединений согласно изобретению
Соединения формулы I
Сложные галогенэфиры типа 3 могут быть получены путем проведения реакции между соединениями 2, где E представляет собой подходящую уходящую группу, и соединениями 1, где R2 и R3 являются такими, как определено в Формуле I. Подходящие уходящие группы хорошо известны в данной области техники и включают, но не ограничиваются ими, галогениды, такие как хлорид, бромид и йодид; арил- или алкилсульфонилокси, замещенные арилсульфонилокси (например, тозилокси или мезилокси); замещенные алкилсульфонилокси (например, галогеналкилсульфонилокси); (C6)арилокси или замещенные (C6)арилокси и ацилоксигруппы. Соединения 2 являются коммерчески доступными (например, от компании Aldrich Chemical Co., Милуоки, Висконсин) или могут быть получены с помощью хорошо известных методов, таких как галогенирование или сульфонирование бутандиола. Соединения 1 также являются коммерчески доступными (например, от компании Aldrich Chemical Co., Милуоки, Висконсин) или могут быть получены с помощью хорошо известных методов, таких как методы, перечисленные в источнике Larock Comprehensive Organic Transformations; Wiley-VCH: New York, 1999, pp. 1754-1755 and 1765. Обзор реакций алкилирования сложных эфиров типа 1 приведен в источнике J. Mulzer in Comprehensive Organic Functional Transformations, Pergamon, Oxford 1995, pp. 148-151, и примеры синтетических процессов для проведения реакций между соединениями 1 и соединениями 2 описаны в патенте США № 5,648,387, графа 6, и источнике Ackerly, et al., J. Med. Chem. 1995, pp. 1608. Реакция может протекать в присутствии подходящего основания. Согласно конкретным вариантам реализации изобретения, pKa подходящего основания составляет более чем примерно 25, согласно другому варианту реализации изобретения, pKa подходящего основания составляет более чем примерно 30. Подходящие основания включают, но не ограничиваются ими, алкилметаллические основания, такие как диизопропиламид лития, метиллитий, н-бутиллитий, трет-бутиллитий, втор-бутиллитий, фениллитий, фенилнатрий и фенилкалий; металламидные основания, такие как амид лития, амид натрия, амид калия, тетраметилпиперидид лития, диэтиламид лития, дициклогексиламид лития, гексаметилдисилазид натрия и гексаметилдисилазид лития; гидридные основания, такие как гидрид натрия и гидрид калия. В частности, могут использоваться металламидные основания, такие как диизопропиламид лития. Согласно конкретным вариантам реализации изобретения, для проведения реакций между соединениями 1 и соединениями 2 к перемешиваемому раствору, содержащему сложный эфир 1 и подходящий органический растворитель, можно добавлять раствор, содержащий от примерно 1 до примерно 2 эквивалентов подходящего основания, в инертной атмосфере. Полученный раствор поддерживается при постоянной температуре в диапазоне от примерно -95°C до примерно комнатной температуры, согласно конкретным вариантам реализации изобретения, при температуре от примерно -78°C до примерно -20°C. Согласно некоторым вариантам реализации изобретения, основание можно разводить в подходящем органическом растворителе перед его добавлением. Согласно некоторым вариантам реализации изобретения, основание можно добавлять со скоростью примерно 1,5 моля в час. Органические растворители, подходящие для реакции между соединениями 1 и соединениями 2, включают, но не ограничиваются ими, дихлорметан, диэтиловый эфир, тетрагидрофуран, диметилформамид, диметилсульфоксид, бензол, толуол, ксилол, углеводородные растворители (например, пентан, гексан и гептан) и их смеси. После добавления основания реакционной смеси можно позволять перемешиваться в течение от примерно 1 до примерно 2 часов. Соединение 2, которое может быть растворено в подходящем органическом растворителе, согласно конкретным вариантам реализации изобретения, добавляют со скоростью, позволяющей поддерживать температуру реакционной смеси в пределах от примерно одного до двух градусов от исходной температуры указанной реакционной смеси. После добавления соединения 2 температуру реакционной смеси можно доводить до температуры в пределах диапазона от примерно -20°C до примерно комнатной температуры, включая примерно комнатную температуру, и реакционной смеси можно позволять перемешиваться до тех пор, пока не наблюдается фактическое завершение реакции по данным подходящего аналитического метода, такого как тонкослойная хроматография (ТСХ) или высокоэффективная жидкостная хроматография (ВЭЖХ). Затем реакцию гасят, и соединения 3 могут быть выделены посредством обработки.
Далее соединения 3 можно подвергать реагированию с замещенными альдегидами посредством реакции Гриньяра с получением спиртов 4. Примеры способов проведения реакций Гриньяра см. в источнике March, J. Advanced Organic Chemistry; Reactions Mechanisms, and Structure, 4th ed., 1992, pp. 920-929. Согласно конкретным вариантам реализации изобретения, соединения 3 обрабатывают Zn или Mg в диэтиловом эфире или тетрагидрофуране, как описано в источнике Drake, N. L.; Cooke, G. B. Org. Synth. Coll. Vol. II, 1943, 406. Галогениды 5 можно синтезировать с использованием различных способов, начиная со спиртов 4. Один из указанных способов включает превращение спирта в уходящую группу, такую как сложный сульфоновый эфир, включая, но не ограничиваясь ими, например, тозилат, брозилат, мезилат или нозилат. Указанное промежуточное соединение затем можно обрабатывать источником X-, где X- представляет собой I-, Br- или Cl-, в растворителе, таком как ТГФ или эфир. Общий способ для превращения винил- и фенилспиртов в тиолы включает первоначальное превращение спирта в уходящую группу (например, тозилат) с последующей обработкой галоген-нуклеофилом. Примеры указанных способов описаны в источниках Forrest, O. A.; Gregory, C. F. J. Am. Chem. Soc, 2005, 127, 10482-10483 (NBS/THF), и Possel, O.; van Leusen, A. M. Tetrahedron. Lett,1977, 48, 4229-4232 (HCl/MeOH).
Галогениды 5 можно подвергать сочетанию с различными гетероциклами с получением сложных эфиров 6. В частности, 4,5,6,7-тетрагидротиено[3,2-c]пиридином, где Z1=S, в присутствии карбоната калия, как описано в источнике Aubert, D.; Touch, P. D.; Ferrand, C.; Ramonville, S.; Maffrand, J. United States Patent 4,529,596. 1985. Для получения соединений согласно изобретению, где Z1=CH2, галогениды 5 подвергают сочетанию с 6, 7-дигидро-5H-[1]пиридин-3-карбоновой кислотой, как описано в источнике Godar E.M., Mariella R.P., J. Org. Chem., 1960, 25, 557-559. Для получения сложных эфиров 6, где Z1=O, можно проводить реакцию между галогенидами 5 и 4,5,6,7-тетрагидрофуро[3,2-c]пиридином, как описано в источнике Koike, H.; Asai, F.; Sugidachi, Kimure, T.; Inoue, T.; Nishino, S.; Tsuzaki, Y. United States Patent 5,288,726, 1994. Подобным образом, можно проводить реакцию между галогенидами 5 и сложным трет-бутиловым эфиром пирроло[3,2-c]пиридин-1-карбоновой кислоты, как описано в источнике Krichevskii, E. S.; Alekseeva, L. M.; Granik, V. G. Chem. Hetercycl. Compd. 1990, 1235-1238, с получением соединений согласно изобретению, где Z1=N.
Сложные эфиры 6 можно подвергать последовательным реакциям функционализирования, как описано, например, в источнике Carey, F.; Giuliano, R. Organic Chemistry, McGraw-Hill Science/Engineering/Math; 8 edition (2010), Chapter 20. В качестве конкретного примера, кислоты типа 7 получали путем последовательного гидролиза групп сложного эфира в присутствии гидроксида калия (общий метод см. в источнике Vogel's Practical Organic Chemistry, 4th Edition, Longman Inc.: New York 1978, pp 491).
На Схеме 2 представлен общий способ синтеза соединений согласно изобретению. Металлы Рике 1 являются коммерчески доступными, или они могут быть получены и подвергаться реакции с замещенными бензальдегидами, как описано в источнике Zhu L., Wehmeyer R.M., Rieke R.D., J. Org. Chem., 1991, 56, 1445-1453. Кетоновое промежуточное соединение 2 можно восстанавливать с использованием боргидрида натрия с получением спиртов 3. Указанный спирт можно обрабатывать подходящей уходящей группой в ходе подготовки к последующей реакции сочетания, как описано выше для синтеза производного 6 на Схеме 1. Подходящие уходящие группы хорошо известны в данной области техники и включают, но не ограничиваются ими, галогениды, такие как хлорид, бромид и йодид; арил- или алкилсульфонилокси, замещенные арилсульфонилокси (например, тозилокси или мезилокси); замещенные алкилсульфонилокси (например, галоалкилсульфонилокси); (C6)арилокси или замещенные (C6)арилокси; и ацилоксигруппы. Согласно конкретным вариантам реализации изобретения, соединение типа 4 обрабатывают метансульфонилхлоридом при низких температурах, например, 0-5°C, в присутствии избытка триэтиламина с получением мезилата. Мезилат 4 далее подвергают сочетанию с подходящим гетероциклом путем нагревания в ТГФ с избытком триэтиламина с получением производного 5. Производные нитрила 5 подвергаются превращениям с получением различных других функциональных групп посредством хорошо известных нормативных реакций или путем взаимопревращений производных карбоновой кислоты.
Соединения формулы II
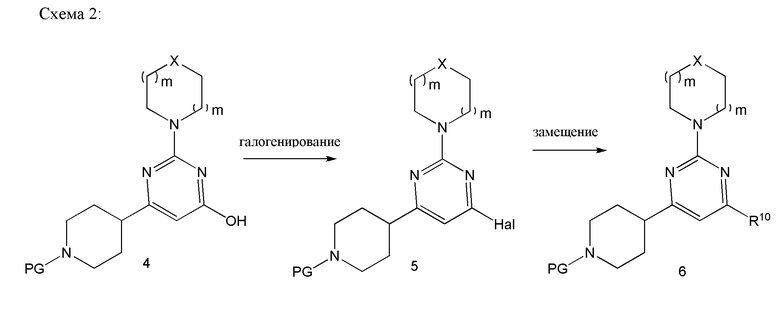
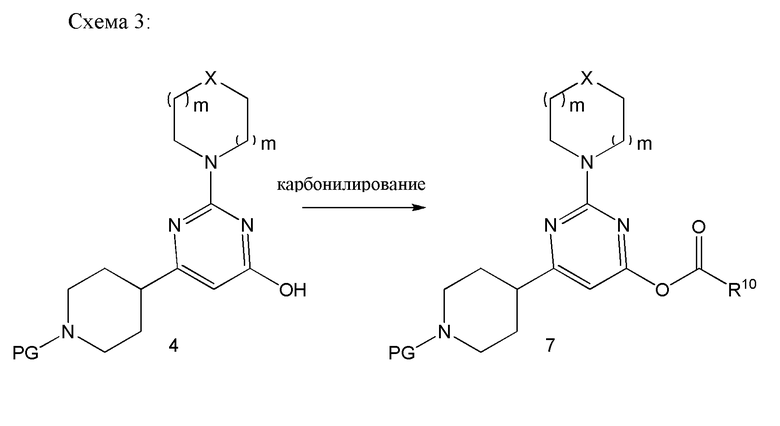
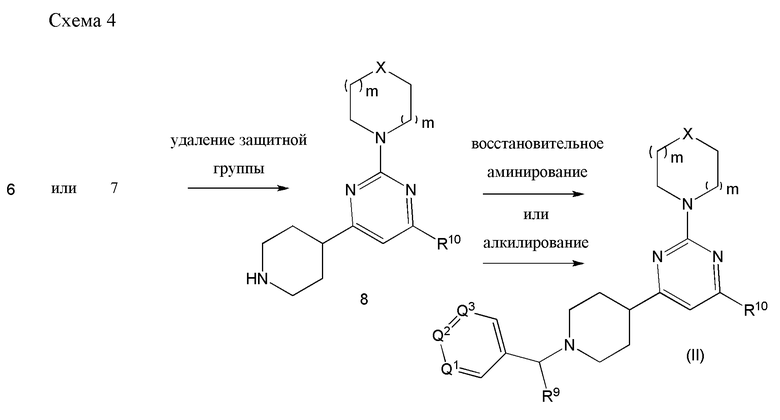
На Схеме 1 показан синтез пиримидинов формулы 4. Синтез можно осуществлять путем конденсации сложных кетоэфиров формулы 2 и амидинов формулы 3. Реакцию конденсации можно проводить, как описано в источнике Hull R. et al., 1946, Journal of the Chemical Society pp. 357-362. Сложные кетоэфиры формулы 2 можно получать из коммерчески доступных сложных эфиров формулы 1 (например, от компании Matrix Scientific, Колумбия, Южная Каролина) с помощью методов, описанных в источнике Raimundo, Brian C., et al., 2004, Journal of Medicinal Chemistry 47(12), pp. 3111-3131, или соответствующей кислоты (например, от компании Sigma-Aldrich Corp., Сент-Луис, Миссури) с помощью метода, описанного в источнике Bashford K. E. et al., 2003, Tetrahedron Letters 44, pp. 1627-1629 and Oikawa Y. et al., 1978, Journal of Organic Chemistry 43(10), pp. 2087-2088. Присутствующий амин обычно содержит защитную группу, включая, но не ограничиваясь ими, t-Boc, CBZ или бензильную защитную группу. Амидины формулы 3 (где m представляет собой целое число от 0 до 3) в целом являются коммерчески доступными (например, от компании Sigma-Aldrich Corp., Сент-Луис, Миссури), но также могут быть получены из соответствующего амина путем проведения реакции с сульфатом 2-метил-2-тиопсевдомочевины, который также является коммерчески доступным (например, от компании Sigma-Aldrich Corp., Сент-Луис, Миссури). Один из таких неограничивающих способов описан в источнике Bonafoux D. et al., 2009, Bioorganic & Medicinal Chemistry Letters 19(3), pp. 912-916.
На схеме 2 описан процесс превращения пиримидинов формулы 4 в соответствующие галогенированные соединения формулы 5 с использованием галогенирующих агентов, включая, но не ограничиваясь ими, PCl5, PBr5, оксихлорид фосфора и оксибромид фосфора. Пример указанного превращения описан в источнике Altenbach R. J., et al., 2008, Journal of Medicinal Chemistry 51(20), pp. 6571-6580. Можно проводить реакцию между галогенированными соединениями формулы 5 и рядом углеродных, азотных, кислородных или серных нуклеофилов для замещения галогена с получением более сложных аналогов пиримидина формулы 6. Агенты, используемые для замещения, являются коммерчески доступными (например, от компании Sigma-Aldrich Corp., Сент-Луис, Миссури) и, в целом, требуют присутствия дополнительного основания, включая, но не ограничиваясь ими, триэтиламин, диизопропилэтиламин, трет-бутоксид калия или натрия, карбонат калия или натрия и гидрид натрия. Примеры реакций замещения указанного типа с азотными нуклеофилами (как описано в источнике Ghoneim K. M. et al., 1986, Journal of the Indian Chemical Society, 63(10, pp. 914-917)), кислородными нуклеофилами (как описано в источнике Dubey P. K. et al., 2006, Indian Journal of Hetercyclic Chemistry 15(4), pp. 405-406), углеродными нуклеофилами (как описано в источнике Gillespie R.J. et al, 2009, Bioorganic & Medicinal Chemistry 17(18), pp. 6590-6605) и серными нуклеофилами (как описано в источнике Backer H.J. et al, 1942, Recueil des Travaux Chimiques des Pays-Bas et de la Belgique 61, pp. 291-298) хорошо известны в литературе.
На Схеме 3 описано превращение пиримидинов формулы 4 в соответствующие O-карбонилпиримидины формулы 7. O-карбонилпиримидины формулы 7 могут быть получены из пиримидинов формулы 4 путем добавления соответствующего хлорангидрида в присутствии основания, включая, но не ограничиваясь ими, триэтиламин, диизопропилэтиламин, трет-бутоксид калия или натрия, карбонат калия или натрия и гидрид натрия. Хлорангидриды являются коммерчески доступными (например, от компании Sigma-Aldrich Corp., Сент-Луис, Миссури) или могут быть получены путем нагревания коммерчески доступных кислот с хлорирующими агентами, которые включают, но не ограничиваются указанными, тионилхлорид или оксалилхлорид. Один пример способа карбонилирования пиримидинов описан в источнике Shabbir S. et al., 2010, Tetrahedron 66(35), pp. 7204-7212.
На Схеме 4 показано превращение пиримидинов формулы 6 и формулы 7 с получением конечных структур формулы (II). Сначала можно удалять защитную группу с получением пиримидинов формулы 8 с помощью стандартных способов удаления используемой защитной группы (способы удаления обычных защитных групп см. в источнике Greene T. W. and Wuts P. G., 1999, Protective Groups in Organic Synthesis 3rd edition, Wiley-Interscience, Нью-Йорк). Пиримидины затем можно алкилировать с использованием коммерчески доступных агентов (например, от компании Sigma-Aldrich Corp., Сент-Луис, Миссури) в присутствии основания, включая, но не ограничиваясь ими, триэтиламин, диизопропилэтиламин, трет-бутоксид калия или натрия, карбонат калия или натрия и гидрид натрия. Процедуры, описанные в источнике Levy D. E. et al., 2008, Bioorganic & Medicinal Chemistry Letters, 18(7), pp. 2395-2398, могут использоваться в реакциях указанного типа алкилирования. Второй общий способ, который может использоваться в реакциях превращения пиримидинов формулы 8 с получением конечных структур формулы (II), представляет собой восстановительное аминирование. Пиримидины формулы 8 можно обрабатывать коммерчески доступными альдегидами или кетонами (например, от компании Sigma-Aldrich Corp., Сент-Луис, Миссури) в присутствии слабой кислоты (включая, но не ограничиваясь ими, уксусную кислоту, трифторуксусную кислоту или соляную кислоту) и восстанавливающего реагента (включая, но не ограничиваясь ими, борогидрид натрия, цианоборогидрид натрия и триацетоксиборогидрид натрия) с получением желаемых конечных структур формулы (II). Процедуры, предложенные в источниках Taibakhsh M. et al, 2011, Synthesis, pp. 490-496, и Abdel-Magid A. F. et al., 1996, Journal Organic Chemistry, 61, pp. 3849-3862, описывают способы, которые могут использоваться в реакциях восстановительного аминирования.
Кроме того, соединения формулы II можно синтезировать с помощью методов, описанных в следующих источниках: Abdel-Magid A. F. et al., 1996, “Reductive Amination of Aldehydes and Ketones with Sodium Triacetoxyborohydride. Studies on Direct and Indirect Reductive Amination Procedures(1)”, Journal Organic Chemistry, 61, pp. 3849-3862; Altenbach R. J., et al., 2008, “Structure-Activity Studies on a Series of a 2-Aminopyrimidine-Containing Histamine H4 Receptor Ligands”, Journal of Medicinal Chemistry 51(20), pp. 6571-6580; Backer H. J. et al, 1942, “Several Sulfanilamido-4-methylpyrimidines”,Recueil des Travaux Chimiques des Pays-Bas et de la Belgique 61, pp. 291-298; Bashford K. E. et al., 2003, “The Bohlmann-Ratz Route to Functionalised Pyridine Scaffolds and Their Use in Library Synthesis', Tetrahedron Letter, 44, pp. 1627-1629; Bonafoux D. et al., 2009, “2-Aminoimidazoles Inhibitors of TGF-Beta Receptor 1”, Bioorganic & Medicinal Chemistry Letters 19(3), pp. 912-916; Dubey P. K. et al., 2006, “Synthesis of Threobromine Incorporated Pyrimidine”,Indian Journal of Hetercyclic Chemistry 15(4), pp. 405-406; Ghoneim K. M. et al., 1986, “Synthesis and Evaluation of some 2-, 4- and 2,4-di-substituted-6-Methylpyrimidine Derivatives for Antimicrobial Activity”, Journal of the Indian Chemical Society, 63(10, pp. 914-917; Gillespie R. J. et al, 2009, “Preparation of Pyrimidine Carboxamides as Purine Receptor, Particularly Adenosine Receptor Antagonists”, Bioorganic & Medicinal Chemistry 17(18), pp. 6590-6605; Greene T. W. and Wuts P. G., 1999, Protective Groups in Organic Synthesis, 3rd edition, Wiley-Interscience, New York; Hull R. et al., 1946, “Synthetic Antimalarials. III. Some Derivatives of Mono- and Dialkylpyrimidines”, Journal of the Chemical Society pp. 357-362; Levy D. E. et al., 2008, “Aryl-indolyl Maleimides as Inhibitors of CaMKII-Delta. Part 2.: SAR of the Amine Tether”, Bioorganic & Medicinal Chemistry Letters, 18(7), pp. 2395-2398; Oikawa Y. et al., 1978, “ Meldrum's Acid in Organic Synthesis. 2. Q General and Versatile Synthesis of Beta-Keto Esters”, Journal of Organic Chemistry 43(10), pp. 2087-2088; Raimundo, Brian C. et al, 2004, “Integrating Fragment Assembly and Biophysical Methods in the Chemical Advancement of Small-Molecule Antagonists of IL-2: An Approach for Inhibiting Protein-Protein Interactions”, Journal of Medicinal Chemistry, 47(12), pp.3111-3130; Shabbir S. et al., 2010, “Pyrimidine Based Carboxylic Acid Terminated Aromatic and Semiaromatic Hyperbranched Polyamide-esters: Synthesis and Characterization”, Tetrahedron 66(35), pp. 7204-7212; Taibakhsh M. et al, 2011, “Catalyst-Free One-Pot Reductive Alkylation of Primary and Secondary Amines and N,N-Dimethylation of Amino Acids Using Sodium Borohydride in 2,2,2-Trifluoroethanol”, Synthesis, pp. 490-496.
Соединения формулы III
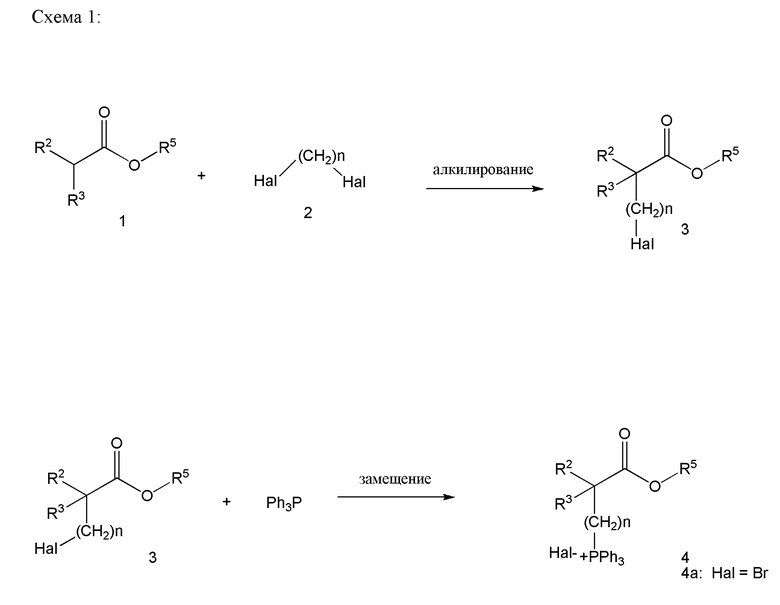
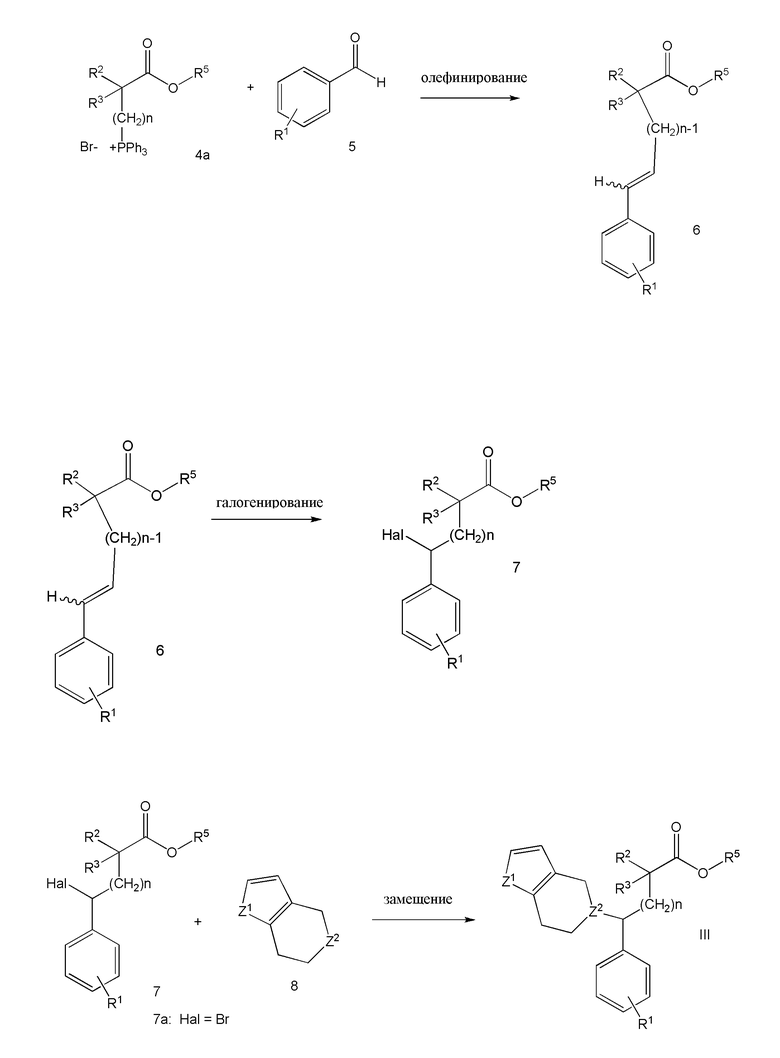
На Схеме 1 представлена общая последовательность синтеза для получения соединений формулы III. Коммерчески доступные сложные эфиры, амиды или кислоты формулы 1 (например, от компании Sigma-Aldrich Corp., Сент-Луис, Миссури) можно обрабатывать основанием (включая, но не ограничиваясь ими, диизопропиламид лития, гексаметилдисилиламид натрия, трет-бутоксид натрия или калия) и алкилировать с помощью коммерчески доступных дигалогенированных алканов формулы 2 (например, от компании Sigma-Aldrich Corp., Сент-Луис, Миссури). Способы, изложенные в источнике Mueller R. et al, 2004, Journal of Medicinal Chemistry, 47(24), pp. 6082-6099, описывают реакцию алкилирования. Алкилированные продукты формулы 3 можно нагревать в присутствии трифенилфосфина с получением солей фосфония формулы 4 таким образом, как описано в источнике Parikka K. et al., 2009, Beilstein Journal of Organic Chemistry, 5(22), pp. 1-5. Соли фосфония формулы 4 можно обрабатывать основанием (включая, но не ограничиваясь ими, гидроксид натрия или калия, трет-бутоксид калия или натрия, карбонат калия или натрия и гидрид натрия) в присутствии коммерчески доступных альдегидов формулы 5 (например, от компании Sigma-Aldrich Corp., Сент-Луис, Миссури) с получением олефинов формулы 6 таким образом, как описано в источнике Le Bigot Y. et al., 1988, Tetrahedron 44(4), pp. 1057-1072, в виде смеси цис- и транс-изомеров. Смесь цис- и транс-изомеров формулы 6 можно галогенировать путем конденсации с безводным газообразным галогеноводородом (например, газообразным бромоводородом) в уксусной кислоте при низкой температуре с получением галогенированного соединения формулы 7 таким образом, как описано в источнике Crispi G. et al, 1982, Synthesis 9, pp. 787-788. Конечные продукты формулы III могут быть получены путем нагревания галогенидов формулы 7 (например, бромида 7a) в присутствии основания (включая, но не ограничиваясь ими, гидроксид натрия или калия, трет-бутоксид калия или натрия, карбонат калия или натрия и гидрид натрия) вместе с коммерчески доступными циклическими соединениями формулы 8 (например, от компании Acc Corporation, Сан Диего, Калифорния, и компании Ryan Scientific, Маунт Плезант, Южная Каролина). Продукты замещения могут быть получены с помощью способов, описанных в литературе для получения подобных соединений (например, Cheng D. et al., 2008, Chinese Chemical Letters 19(6), pp. 689-692).
Кроме того, соединения формулы III можно синтезировать с помощью методов, описанных в следующих источниках: Cheng D. et al., 2008, “Synthesis and Activity Evaluation of Some Novel Derivatives of 4,5,6,7-Tetrahydrothiono[3,2-c]-pyridine”, Chinese Chemical Letters 19(6), pp. 689-692; Crispi G. et al, 1982, “Enamine; 42. A simple Synthesis of 3,4-Diaminobiphenyls”, Synthesis (9), pp. 787-788; Le Bigot Y. et al., 1988, “Reactions in a Slightly Hydrated Solid-Liquid Heterogenous Medium: the Wittig Reaction in Alkaline Hydroxide-Aprotic Organic Solvent System”, Tetrahedron 44(4), pp. 1057-1072; Mueller R. et al, 2004, “Long Hydrocarbon Chain Keto Diols and Diacids that Favorably Alter Lipid Disorders in Vivo”, Journal of Medicinal Chemistry, 47(24), pp. 6082-6099; Parikka K. et al., 2009, “An Expedient Synthsis of 5-n-Alkylresorcinols and Novel 5-n-Alkylresorcinols Hapatens”, Beilstein Journal of Organic Chemistry, 5(22) pp. 1-5; Cheng D. et al., 2008, “Synthesis and Activity Evaluation of Some Novel Derivatives of 4,5,6,7-Tetrahydrothiono[3,2-c]-pyridine”, Chinese Chemical Letters 19(6), pp. 689-692; Crispi G. et al, 1982, “Enamine; 42. A simple Synthesis of 3,4-Diaminobiphenyls”, Synthesis (9), pp. 787-788; Le Bigot Y. et al., 1988, “Reactions in a Slightly Hydrated Solid-Liquid Heterogenous Medium: the Wittig Reaction in Alkaline Hydroxide-Aprotic Organic Solvent System”, Tetrahedron 44(4), pp. 1057-1072; Mueller R. et al, 2004, “Long Hydrocarbon Chain Keto Diols and Diacids that Favorably Alter Lipid Disorders in Vivo”, Journal of Medicinal Chemistry, 47(24), pp. 6082-6099; Parikka K. et al., 2009, “An Expedient Synthsis of 5-n-Alkylresorcinols and Novel 5-n-Alkylresorcinols Hapatens”, Beilstein Journal of Organic Chemistry, 5(22) pp. 1-5.
Соединения формулы IV
На схеме 1 описан синтетический путь для получения соединений формулы IV. Согласно конкретным вариантам реализации изобретения, получение включает перемешивание коммерчески доступных альдегидов формулы 1 (например, от компании Sigma-Aldrich Corp., Сент-Луис, Миссури), коммерчески доступных циклических соединений формулы 2 (например, Acc Corporation, Сан Диего, Калифорния, и Ryan Scientific, Маунт Плезант, Южная Каролина) и безводного бензотриазола 3 в подходящем растворителе, включая, но не ограничиваясь ими, диэтиловый эфир, тетрагидрофуран, диоксан и толуол. Экспериментальные реакции можно проводить с нагреванием или без нагревания, и можно использовать сухие молекулярные сита для удаления воды из указанных реакций. Продукты формулы 4 затем можно очищать путем растирания, кристаллизации или колоночной хроматографии с использованием соответствующего наполнителя (например, силикагеля или оксида алюминия). Примеры указанного способа приведены в источнике Katritzky A. et al., 1989, “A General Method for the Preparation of Beta-Amino Esters”, Synthesis (10), pp. 747-751.
Соединения формулы V
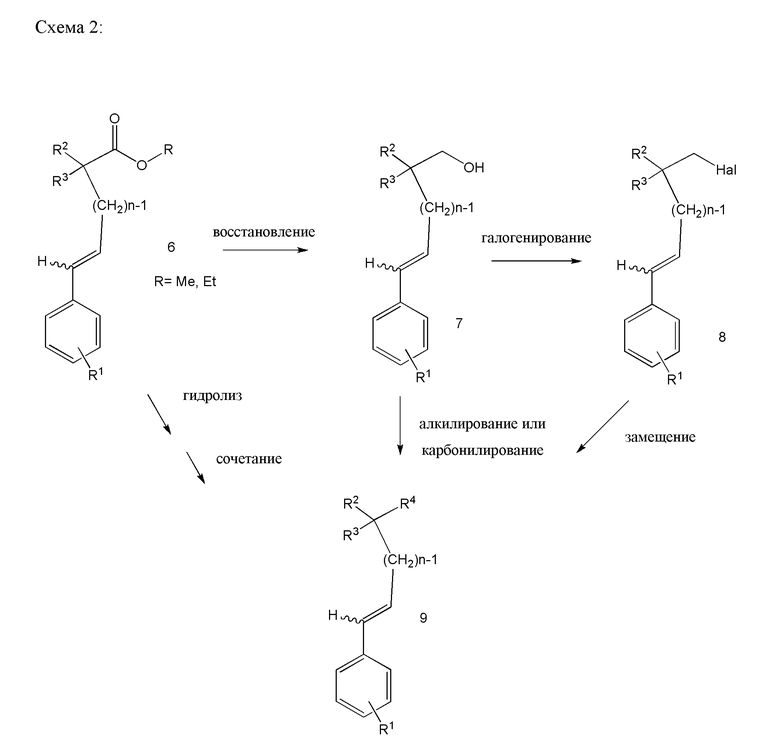
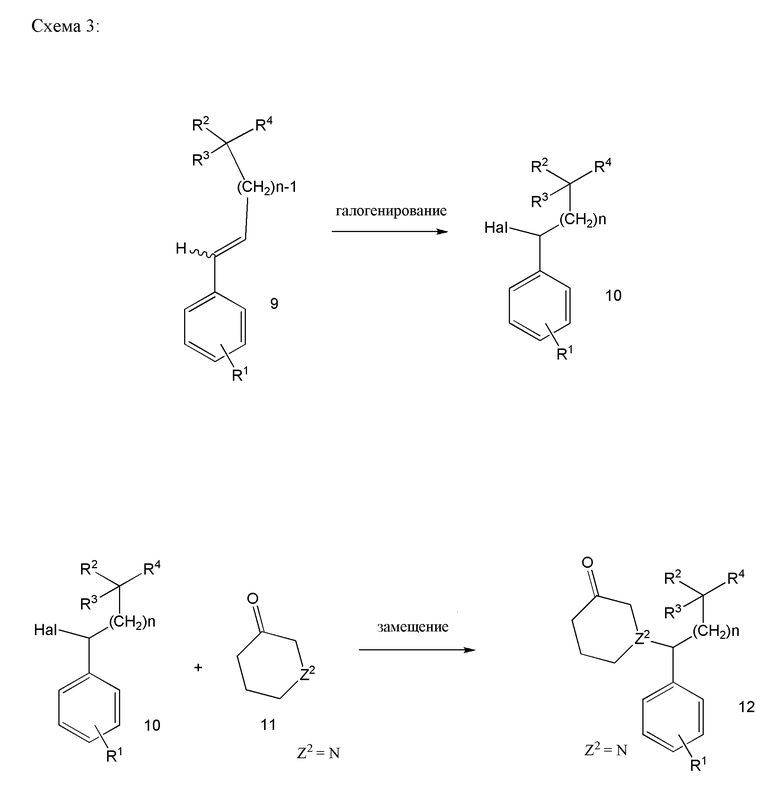
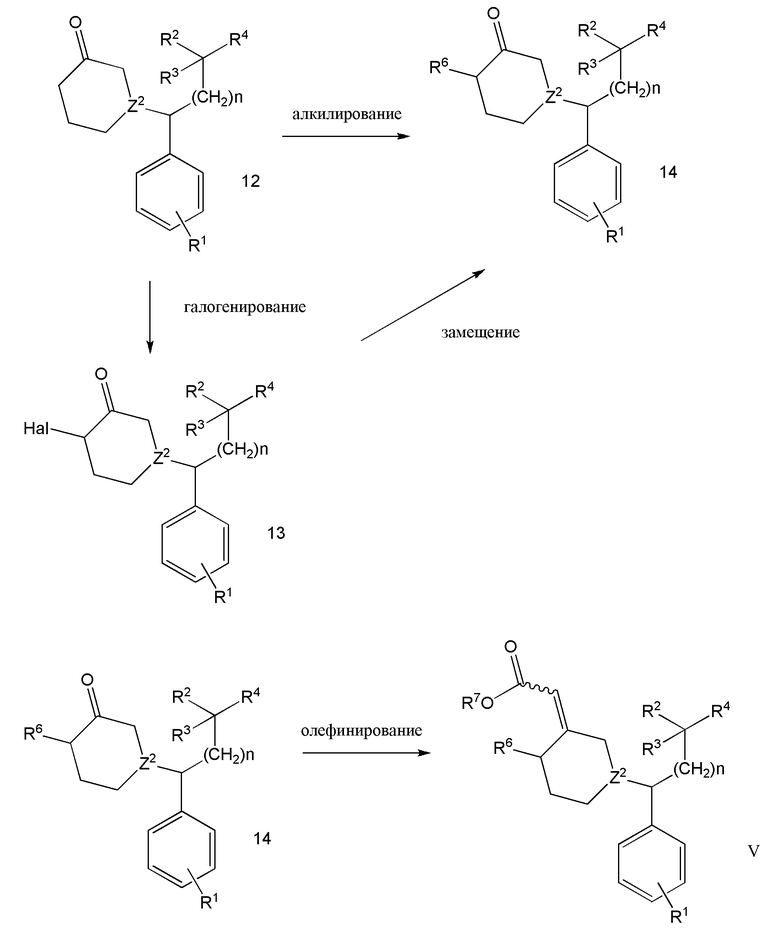
На Схеме 1 представлена общая последовательность синтеза для получения соединений формулы V. Коммерчески доступные сложные эфиры, амиды или кислоты формулы 1 (например, от компании Sigma-Aldrich Corp., Сент-Луис, Миссури) можно обрабатывать основанием (включая, но не ограничиваясь ими, диизопропиламид лития, гексаметилдисилиламид натрия, трет-бутоксид натрия и калия) и алкилировать с использованием коммерчески доступных дигалогенированных алканов формулы 2 (например, от компании Sigma-Aldrich Corp., Сент-Луис, Миссури). В способах, приведенных в источнике Mueller R. et al, 2004, Journal of Medicinal Chemistry, 47(24), pp. 6082-6099, описаны реакции алкилирования. Алкилированные продукты формулы 3 можно нагревать в присутствии трифенилфосфина с получением солей фосфония формулы 4 таким образом, как описано в источнике Parikka K. et al., 2009, Beilstein Journal of Organic Chemistry, 5(22), pp. 1-5. Соли фосфония формулы 4 можно обрабатывать основанием (включая, но не ограничиваясь ими, гидроксид натрия или калия, трет-бутоксид калия или натрия, карбонат калия или натрия и гидрид натрия) в присутствии коммерчески доступных альдегидов формулы 5 (например, от компании Sigma-Aldrich Corp., Сент-Луис, Миссури) с получением олефинов формулы 6 в виде смеси цис- и транс-изомеров таким образом, как описано в источнике Le Bigot Y. et al., 1988, Tetrahedron 44(4), pp. 1057-1072.
На Схеме 2 показано превращение сложных эфиров формулы 6 в галогенированные соединения формулы 9. Спирты формулы 7 могут быть получены путем восстановления группы сложного эфира в соединениях формулы 6. Широкое разнообразие реагентов доступно для восстановления таких сложных эфиров до спиртов, например, см. M. Hudlicky, Reductions in Organic Chemistry, 2nd ed., 1996 pp. 212-217, специально включенный в настоящую заявку посредством ссылки. Согласно конкретным вариантам реализации изобретения, восстановление можно осуществлять с использованием восстанавливающего агента типа гидридов, например, алюмогидрида лития, борогидрида лития, триэтилборогидрида лития, гидрида диизобутилалюминия, гидрида триметоксиалюминия лития или гидрида бис(2-метокси)алюминия натрия. Неограничивающие примеры способов восстановления сложных эфиров до спиртов см. в следующих источниках: Nystrom et al., 1947, J. Am. Chem. Soc. 69:1197; Moffet et al., 1963, Org. Synth., Collect. 834(4), с использованием алюмогидрида лития; Brown et al.,1965, J. Am. Chem. Soc. 87:5614, с использованием гидрида триметоксиалюминия лития; Cerny et al., 1969, Collect. Czech. Chem. Commun. 34:1025, с использованием гидрида бис(2-метокси)алюминия натрия; Nystrom et al., 1949, J. Am. Chem. 71:245, с использованием борогидрида лития; и Brown et al., 1980, J. Org. Chem. 45:1, с использованием триэтилборогидрида лития. Спирт, присутствующий в соединении формулы 7, может быть превращен в галогенид с получением соединений формулы 8 (наглядное описание различных способов превращения спиртов в галогениды см. в источнике March, J. Advanced Organic Chemistry; Reactions Mechanisms, and Structure, 4th ed., 1992, pp. 431-433). Соединения формулы 9 могут быть непосредственно получены из сложного эфира 6 путем гидролиза указанного сложного эфира с помощью основания с последующим осуществлением стандартных способов сочетания (с использованием DCC (N,N'-дициклогексилкарбодиимида), EDC (1-этил-3-(3-диметиламинопропил)карбодиимида), HBTU (гексафторфосфата O-(бензотриазол-1-ил)-N,N,N′,N′-тетраметилурония), например, с коммерчески доступными спиртами, аминами и тиолами. Соединения формулы 9 также можно получать из соединений формулы 7 путем алкилирования или карбонилирования с помощью коммерчески доступных алкилирующих и ацилирующих реагентов. Соединения формулы 9 также можно получать путем замещения галогенида, присутствующего в соединении формулы 8, коммерчески доступными спиртами, аминами и тиолами.
На Схеме 3 смесь цис- и транс-изомеров формулы 9 можно галогенировать путем конденсации с безводным газообразным галогенводородом (например, газообразным бромоводородом) в уксусной кислоте при низкой температуре с получением галогенированного соединения формулы 10 таким образом, как описано в источнике Crispi G. et al, 1982, Synthesis 9, pp. 787-788. Соединения формулы 11 (Z2=N), которые могут быть получены, как описано в источнике Taillier C. et al., 2007, Tetrahedron 63(21), pp. 3589-3592, можно подвергать сочетанию с галогенидами фигуры 10 в присутствии основания (включая, но не ограничиваясь ими, гидроксид натрия или калия, трет-бутоксид калия или натрия, карбонат калия или натрия и гидрид натрия) с получением соединений формулы 12. Продукты замещения можно получать таким же образом, как описано для подобных соединений в литературе (см., например, Cheng D. et al., 2008, Chinese Chemical Letters 19(6), pp. 689-692). Кетоны формулы 12 можно обрабатывать основанием и алкилировать с использованием ряда коммерчески доступных алкилирующих (или ацилирующих) реагентов с получением соединений формулы 14. Кетоны формулы 12 также можно галогенировать с получением соединений формулы 13 с помощью способов, описанных в источнике Newman M.S. et al., 1945, Organic Synthesis 25 and Mellegaard-Waetzig S. R. et al., 2006, Tetrahedron, pp. 7191-7198. Галогенированные соединения формулы 13 затем можно подвергать реакциям замещения с использованием коммерчески доступных соединений, содержащих азотные, кислородные или серные нуклеофилы, с получением кетонов формулы 14. Желаемые соединения формулы V можно получать путем олефинирования кетонов формулы 14 с использованием коммерчески доступных фосфонатов или солей фосфония в присутствии основания.
Кроме того, соединения формулы V можно синтезировать с помощью методов, описанных в следующих источниках: Cheng D. et al., 2008, “Synthesis and Activity Evaluation of Some Novel Derivatives of 4,5,6,7-Tetrahydrothiono[3,2-c]-pyridine”, Chinese Chemical Letters 19(6), pp. 689-692; Crispi G. et al, 1982, “Enamine; 42. A simple Synthesis of 3,4-Diaminobiphenyls”, Synthesis (9), pp. 787-788; M. Hudlicky, Reductions in Organic Chemistry, 2nd ed., 1996 pp. 212-217; Le Bigot Y. et al., 1988, “Reactions in a Slightly Hydrated Solid-Liquid Heterogenous Medium: the Wittig Reaction in Alkaline Hydroxide-Aprotic Organic Solvent System”, Tetrahedron 44(4), pp. 1057-1072; March, J. Advanced Organic Chemistry; Reactions Mechanisms, and Structure, 4th ed., 1992, pp. 431-433; Mellegaard-Waetzig S. R. et al., 2006, “Selenium-Catalyzed Oxidative halogenations”, Tetrahedron, pp. 7191-7198; Mueller R. et al, 2004, “Long Hydrocarbon Chain Keto Diols and Diacids that Favorably Alter Lipid Disorders in Vivo”, Journal of Medicinal Chemistry, 47(24), pp. 6082-6099; Newman M.S. et al., 1945, “2-chlorocyclohexanone”, Organic Synthesis 25; Parikka K. et al., 2009, “An Expedient Synthsis of 5-n-Alkylresorcinols and Novel 5-n-Alkylresorcinols Hapatens”, Beilstein Journal of Organic Chemistry, 5(22) pp. 1-5; Taillier C. et al., 2007, “Synthesis of 3-Oxooxa and 2-Oxoazacycloak-4-enes by Ring-Closing Metathesis. Application to the Synthesis of an Inhibitor of Cathepsin K”, Tetrahedron 63(21), pp. 3589-3592.
Соединения формулы VI
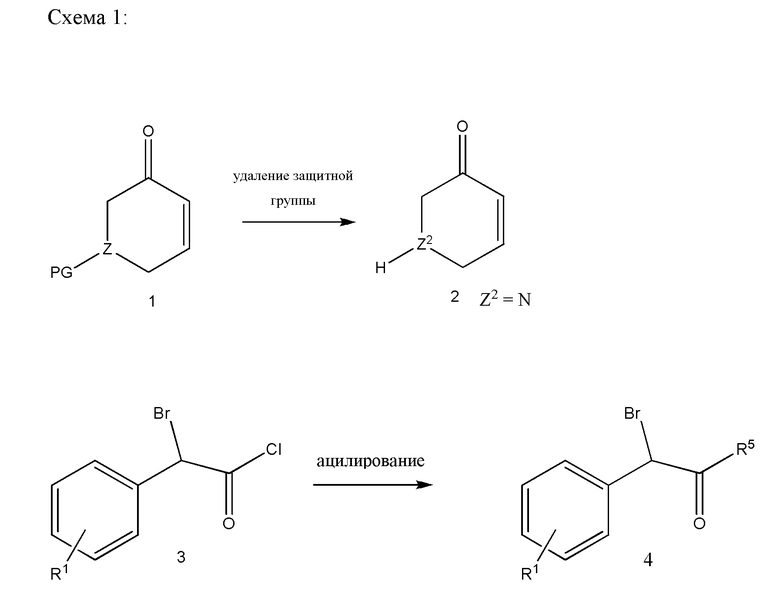
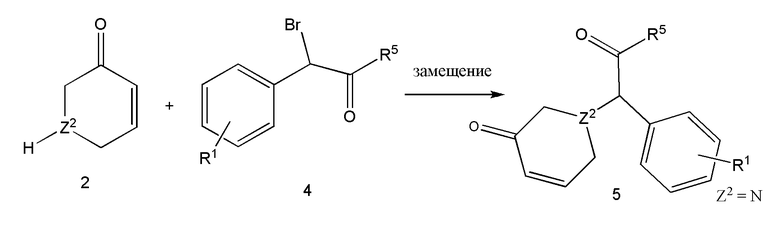
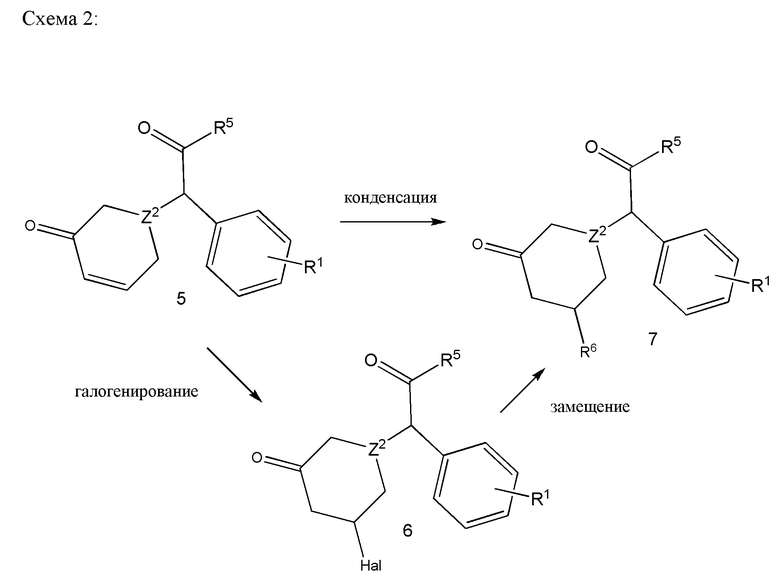
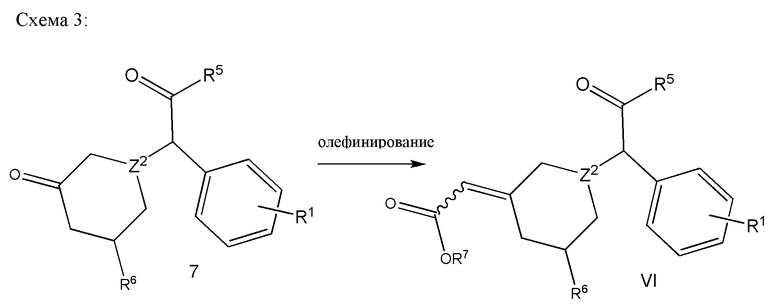
На Схеме 1 описан путь синтеза для получения соединений формулы VI. Указанное получение может включать конденсацию соединений формулы 2 и формулы 4. Соединения формулы 1 (Z2=N) могут быть получены с защитной группой boc, как описано в источнике Taillier C. et al., 2007, Tetrahedron 63(21), pp. 3589-3592. Защитная группа может быть удалена посредством перемешивания указанного соединения в трифторуксусной кислоте с получением соединения формулы 2. Соединения формулы 4 являются коммерчески доступными (например, от компании Sigma-Aldrich Corp., Сент-Луис, Миссури) или могут быть получены из коммерчески доступных соединений формулы 3 путем ацилирования в присутствии пространственно-затрудненного основания (например, диизопропилэтиламина). Эквимолярные количества соединений формулы 2 и формулы 4 можно нагревать в полярных апротонных растворителях (например, ДМФА или ацетонитриле) с образованием продуктов замещения формулы 5. Подобные неограничивающие реакции замещения описаны в литературе (например, Cheng D. et al., 2008, Chinese Chemical Letters 19(6), pp. 689-692).
На Схеме 2 описано получение аналогов формулы 7 из соединений формулы 5. Аналоги формулы 7 могут быть получены путем прямой конденсации коммерчески доступных соединений (например, от компании Sigma-Aldrich Corp., Сент-Луис, Миссури), содержащих серные и азотные нуклеофилы, как описано в источнике Kall A. et al., 2010, Synthetic Communications 40(12), pp. 1730-1735, с соединениями формулы 5. Кроме того, соединения формулы 5 можно галогенировать с помощью метода, описанного в источнике Marx J. et al., 1983, Tetrahedron 39(9), pp. 1529-1531, с получением соединений формулы 6. Соединения формулы 6 можно нагревать с коммерчески доступными соединениями (например, от компании Sigma-Aldrich Corp., Сент-Луис, Миссури), содержащими серные, азотные или кислородные нуклеофилы, с получением соединений формулы 7 путем замещения.
На Схеме 3 описано превращение соединений формулы 7 в желаемые соединения формулы VI путем олефинирования. Коммерчески доступные соли ацилфосфония или ацилфосфонаты (например, от компании Sigma-Aldrich Corp., Сент-Луис, Миссури) можно обрабатывать основанием (включая, но не ограничиваясь ими, гидроксид натрия или калия, трет-бутоксид калия или натрия, карбонат калия или натрия, диизопропиламид лития и гидрид натрия) в присутствии соединений формулы 7 с получением конечного соединения формулы VI. Неограничивающий пример подобного способа описан в источнике Lombardo L. et al., 1987, Synthetic Communications 8(7), pp. 463-468.
Кроме того, соединения формулы VI можно синтезировать с помощью методов, описанных в следующих источниках: Cheng D. et al., 2008, “Synthesis and Activity Evaluation of Some Novel Derivatives of 4,5,6,7-Tetrahydrothiono[3,2-c]-pyridine”, Chinese Chemical Letters 19(6), pp. 689-692; Kall A. et al., 2010, “Microwave-Induced Aza-Michael reaction in Water. A Remakably Simple Procedure”, Synthetic Communications 40(12), pp. 1730-1735; Lombardo L. et al., 1987, “An Improved Procedure for the Conversion of Carbonyl Compounds to Alpha,Beta-Unsaturated Carboxylic Acids”, Synthetic Communications 8(7), pp. 463-468; Marx J. et al., 1983, “A Simple and Convenient Synthesis of Beta-Halo Ketones”, Tetrahedron 39(9), pp. 1529-1531; Taillier C. et al., 2007, “Synthesis of 3-Oxooxa and 2-Oxoazacycloak-4-enes by Ring-Closing Metathesis. Application to the Synthesis of an Inhibitor of Cathepsin K”, Tetrahedron 63(21), pp. 3589-3592.
Соединения формулы VII
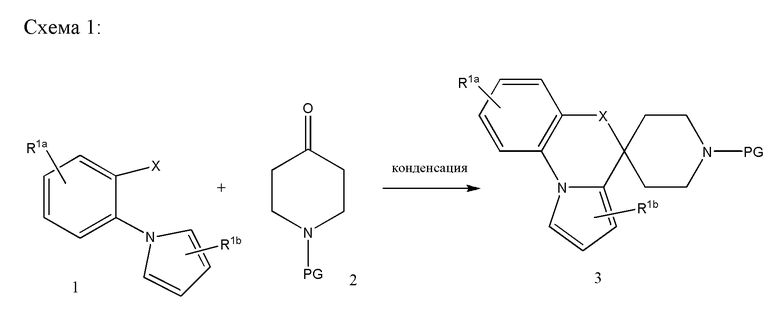
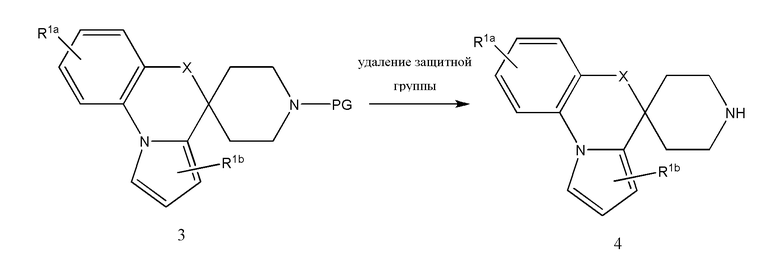
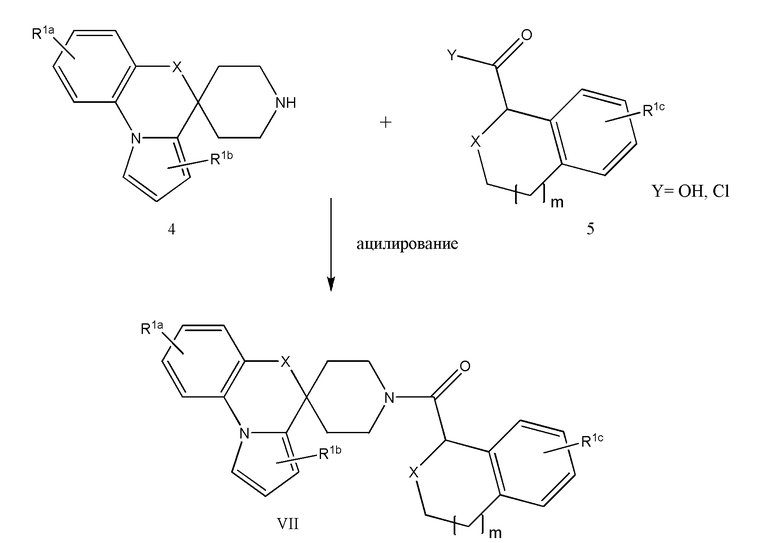
На Схеме 1 показан синтез соединений формулы VII. Коммерчески доступные аналоги формулы 1 (например, от компании Sigma-Aldrich Corp., Сент-Луис, Миссури) можно конденсировать с содержащим подходящие защитные группы коммерчески доступным 4-пиперидоном (например, от компании Sigma-Aldrich Corp., Сент-Луис, Миссури) в присутствии каталитического количества кислоты (например, п-толуолсульфоновой кислоты) при нагревании, как описано в способе в источнике Raines S. et al., 1976, Journal of Heterocyclic Chemistry 13(4), pp. 711-716. Защитные группы можно удалять соответствующим образом (способы удаления обычных защитных групп см. в источнике Greene T. W. and Wuts P. G., 1999, Protective Groups in Organic Synthesis 3rd edition, Wiley-Interscience, Нью-Йорк) с образованием аминов формулы 4. Указанные амины формулы 4 можно подвергать сочетанию с коммерчески доступными хлорангидридами (например, от компании Sigma-Aldrich Corp., Сент-Луис, Миссури) в присутствии дополнительного основания формулы 5 (включая, но не ограничиваясь ими, триэтиламин, диизопропилэтиламин, трет-бутоксид калия или натрия, карбонат калия или натрия и гидрид натрия) с образованием желаемых амидов формулы VII. Кроме того, желаемые амиды формулы VII можно получать посредством стандартных реакций сочетания амидов (включая использование DCC (N,N'-дициклогексилкарбодиимида), EDC (1-этил-3-(3-диметиламинопропил)карбодиимида) и HBTU (гексафторфосфата O-(бензотриазол-1-ил)-N,N,N′,N′-тетраметилурония), но не ограничиваясь ими) с аминами формулы 4 и коммерчески доступными карбоновыми кислотами формулы 5 (например, от компании Sigma-Aldrich Corp., Сент-Луис, Миссури).
Кроме того, соединения формулы VII можно синтезировать с помощью методов, описанных в следующих источниках: Greene T. W. and Wuts P. G., 1999, Protective Groups in Organic Synthesis, 3rd edition, Wiley-Interscience, New York; Raines S. et al., 1976, “Mannich Reactions. Synthesis of 4,5-Dihydropyrrolo[1,2-a]quinoxalines, 2,3,4,5-Tetrahydro-1H-pyrrolo[1,2-a][1,4]diazapines and 5,6-Dihydropyrrolo[1,2-a][1,4]benzodiazapines”, Journal of Heterocyclic Chemistry 13(4), pp. 711-716.
Соединения формулы VIII
На Схеме 1 показан синтез амидов формулы VIII. Для получения промежуточных соединений формулы 3 коммерчески доступные амины и фенилгидразины формулы 1 (например, от компании Sigma-Aldrich Corp., Сент-Луис, Миссури) можно конденсировать с 2-хлоракрилонитрилом 2 путем нагревания в присутствии избытка кислоты (например, серной кислоты). В современной литературе встречается несколько неограничивающих примеров указанного типа конденсации, включая способ, описанный в источнике Ege G. et al., 1982, Journal of Heterocyclic Chemistry 19(6), pp. 1265-1266. Промежуточные соединения формулы 3 можно конденсировать с коммерчески доступной кислотой Мельдрума 4 (например, от компании Sigma-Aldrich Corp., Сент-Луис, Миссури) и коммерчески доступными альдегидами формулы 5 (например, от компании Sigma-Aldrich Corp., Сент-Луис, Миссури) с получением амидов формулы 6 таким же образом, как описано в источнике Quiroga J. et al., 1998, Journal of Heterocyclic Chemistry 35(2), pp. 409-412. Амиды формулы 6 можно алкилировать с использованием ряда коммерчески доступных алкилирующих агентов (например, от компании Sigma-Aldrich Corp., Сент-Луис, Миссури), например, алкилгалогенидов, ацилхлоридов, хлорсульфонатов, в присутствии основания (включая, но не ограничиваясь ими, гидроксид натрия или калия, трет-бутоксид калия или натрия, карбонат калия или натрия, диизопропиламид лития и гидрид натрия) для получения желаемых соединений формулы VIII.
Кроме того, соединения формулы VIII можно синтезировать с помощью методов, описанных в следующих источниках: Ege G. et al., 1982, “Aminopyrazoles. III. Novel One-Flask Preparations of 1=Phenylpyrazol-3-amine”, Journal of Heterocyclic Chemistry 19(6), pp. 1265-1266. Quiroga J. et al., 1998, “Reactions of 5-Amino-1-aryl-3-methylpyrazoles with Benzylidene Derivatives of Meldrum's Acid: Synthesis and Characterization of Pyrazolo[3,4-b]pyridinones”, Journal of Heterocyclic Chemistry 35(2), pp. 409-412.
Соединения формулы IX
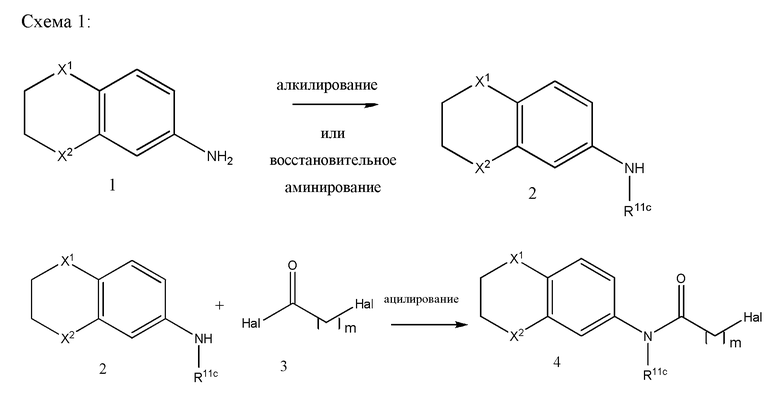
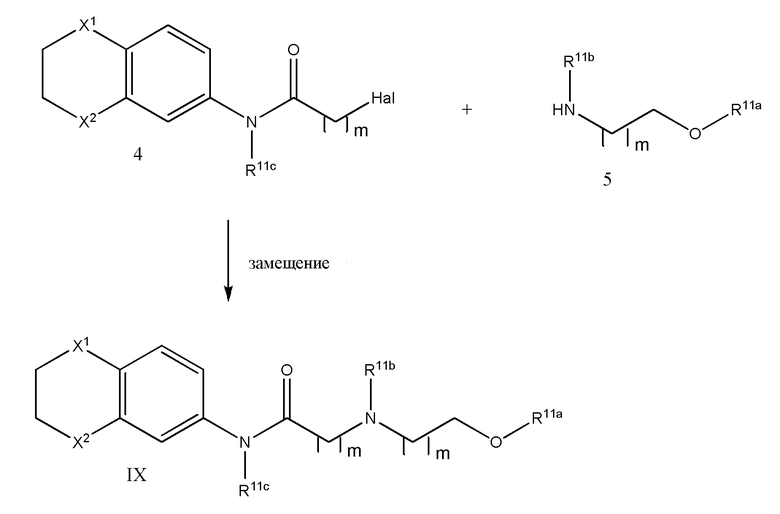
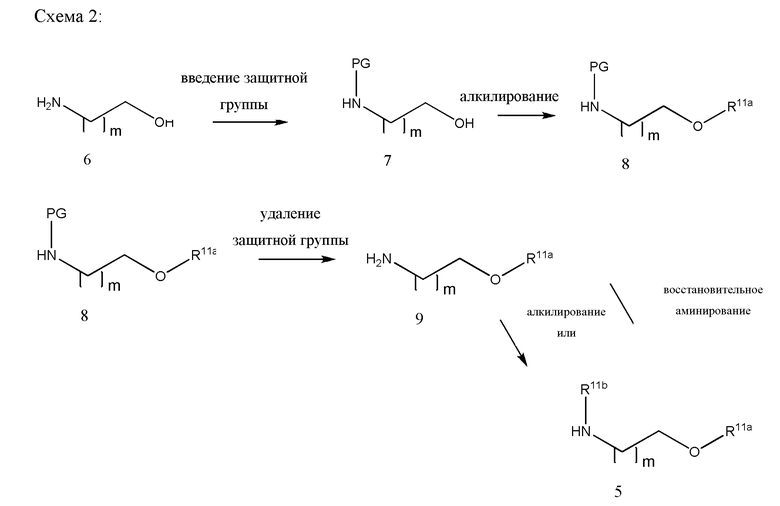
На Схеме 1 показан синтез амидов формулы IX. Для получения аминов фигуры 2 можно проводить реакцию между коммерчески доступными аминами фигуры 1 (например, от компании Sigma-Aldrich Corp., Сент-Луис, Миссури) и любыми алкилирующими агентами (например, алкилгалогенидами, ацилхлоридами, хлоркарбаматами или хлорсульфонатами). Амины фигуры 2 также могут быть получены путем перемешивания аминов фигуры 1 с коммерчески доступными альдегидами (например, алкилгалогенидами, ацилхлоридами, хлорсульфонатами) в присутствии восстанавливающего реагента (включая, но не ограничиваясь ими, борогидрид натрия, цианоборогидрид натрия и триацетоксиборогидрид натрия). Обычно используется способ восстановительного аминирования, описанный в источнике Levy D. E. et al., 2008, Bioorganic & Medicinal Chemistry Letters, 18(7), pp. 2395-2398. Амиды формулы 4 можно получать путем ацилирования структур формулы 1 или формулы 2 с использованием коммерчески доступных галогенидов формулы 3 (например, от компании Sigma-Aldrich Corp., Сент-Луис, Миссури) в присутствии пространственно-затрудненного основания (например, триэтиламина, диизопропилэтиламина, дифенилэтиламина), как описано в источнике Renzi L. et al., 1956, Gazzetta Chimica Italiana 86, pp. 1362-1366. Желаемые соединения формулы IX могут быть получены посредством реакции замещения между аминами формулы 5 и соединениями формулы 4 в присутствии основания, включая, но не ограничиваясь ими, триэтиламин, диизопропилэтиламин, трет-бутоксид калия или натрия, DMAP, карбонат калия или натрия, или гидрид натрия, таким образом, как описано в источнике Demchenko A. M. et al., 2003, Russian Journal of Organic Chemistry 39(7), pp. 1025-1028. Амины фигуры 5 могут быть получены из коммерческих источников (например, от компании Sigma-Aldrich Corp., Сент-Луис, Миссури) или таким образом, как показано на Схеме 2.
На Схеме 2 показан общий способ синтеза аминов формулы 5. Можно селективно вводить подходящую защитную группу в положение атома азота коммерчески доступных аминоспиртов формулы 6 (например, от компании Sigma-Aldrich Corp., Сент-Луис, Миссури) (boc, бензил или CBZ) с использованием общих известных из литературы способов. Атом кислорода, присутствующий в промежуточных соединениях формулы 7, можно алкилировать с помощью подходящих алкилирующих агентов (например, алкилгалогенидов, ацилхлоридов, хлоркарбаматов или хлорсульфонатов) и коммерчески доступных алкилирующих агентов (например, от компании Sigma-Aldrich Corp., Сент-Луис, Миссури) с получением промежуточных соединений формулы 8. Защитные группы затем можно удалять с использованием стандартных методов (см. Greene T. W. and Wuts P. G., 1999, Protective Groups in Organic Synthesis, 3rd edition, Wiley-Interscience, Нью-Йорк) для получения промежуточных соединений формулы 9. Амины формулы 5 можно получать из аминов формулы 9 либо путем алкилирования, либо восстановительного аминирования, из коммерчески доступных реагентов, как описано выше.
Кроме того, соединения формулы IX можно синтезировать с помощью методов, описанных в следующих источниках: Demchenko A. M. et al., 2003, “Synthesis of N5-(Arylcarbonyl)methyl Derivatives of Spinaceamine and 2-Azaspinacaceamine”, Russian Journal of Organic Chemistry 39(7), pp. 1025-1028; Greene T. W. and Wuts P. G., 1999, Protective Groups in Organic Synthesis, 3rd edition, Wiley-Interscience, New York; Levy D. E. et al., 2008, “Aryl-indolyl Maleimides as Inhibitors of CaMKII-Delta. Part 2.: SAR of the Amine Tether”, Bioorganic & Medicinal Chemistry Letters, 18(7), pp. 2395-2398; and Renzi L. et al., 1956, “The 1,4-Benzodioxan Series V. Some Derivatives of 7-Aminobenzodioxan”, Gazzetta Chimica Italiana 86, pp. 1362-1366.
Изобретение далее описано со ссылкой на следующие примеры.
Примеры
ПРИМЕР 1. Гидрохлорид 5-(2-хлорфенил)-5-(6,7-дигидро-4H-тиено[3,2-c]пиридин-5-ил)-2,2-диметилпентановой кислоты. (Гидрохлорид Соединения Ia)
Стадия 1: Синтез сложного этилового эфира 5-(2-хлорфенил)-2,2-диметилпент-4-еновой кислоты.
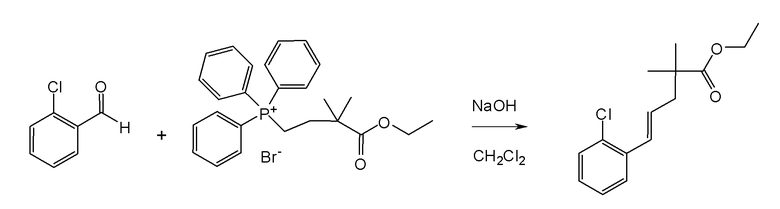
Соль фосфония (15,0 г, 30,9 ммоль) и 2-хлорбензальдегид (4,3 г, 30,6 ммоль) растворяли в дихлорметане (100 мл) и интенсивно перемешивали. Добавляли по каплям раствор гидроксида натрия (24 г, 50%) в течение 5 минут. Смеси позволяли перемешиваться в течение 2 часов при комнатной температуре. Добавляли воду (100 мл) и слои разделяли. Фракцию дихлорметана сушили над сульфатом натрия, фильтровали и концентрировали. Оставшееся оранжевое масло (8,3 г) очищали посредством колоночной хроматографии на силикагеле (150 г) с использованием в качестве элюента 5% этилацетата в гептане с получением сложного этилового эфира 5-(2-хлорфенил)-2,2-диметилпент-4-еновой кислоты (4,02 г, выход 55%) в виде бесцветного масла, которое представляло собой смесь транс/цис изомеров. 1H ЯМР (300 МГц, CDCl3/ТМС): δ=(основная фракция; смесь транс/цис изомеров 60:40) 7,46 (дд, 0,6H, J=7,5, 1,5 Гц), 7,35 (дд, 0,4H, J=7,2, 1,5 Гц), 7,32-7,08 (м, 4H), 6,77 (д, 0,6H, J=15,5 Гц), 6,57 (д, 0,4H, J=11,7 Гц), 6,13 (дт, 0,6H, J=15,5, 7,5 Гц), 5,74 (дт, 0,4H, J=11,7, 7,5 Гц), 4,11 (м, 2H), 2,45 (м, 2H), 1,26-1,15 (м, 9H). 13C ЯМР (75 МГц, CDCl3/ТМС): δ=(основная фракция; смесь транс/цис изомеров 60:40) 177,16, 177,12, 135,55, 133,82, 133,62, 132,60, 130,55, 129,53, 129,38, 129,31, 129,26, 129,15, 128,59, 128,15, 126,82, 126,75, 126,22, 60,52, 44,12, 42,79, 42,40, 38,66, 25,19, 25,08, 14,46, 14,34. МС (GC-EI): Рассчитано: C15H19O2Cl (MH)+: 267,1146, обнаружено: 267,1143.
Стадия 2: Синтез сложного этилового эфира 5-бром-5-(2-хлорфенил)-2,2-диметилпентановой кислоты.
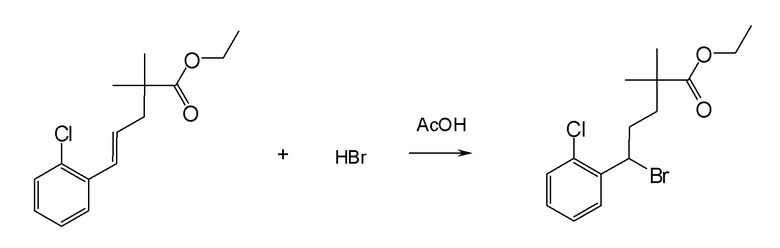
Сложный этиловый эфир 5-(2-хлорфенил)-2,2-диметилпент-4-еновой кислоты (4,0 г, 15,0 ммоль) растворяли в уксусной кислоте (40 мл). Колбу охлаждали на ледяной/водяной бане и через указанный раствор барботировали газообразный бромоводород в течение пяти часов по мере нагревания колбы до комнатной температуры. Через пять часов прекращали подачу газообразного бромоводорода и колбу хранили в морозильной камере в течение ночи (0ºC). Через 20 часов инкубации при 0ºC раствор выливали в смесь льда с водой (200 г). Продукт экстрагировали дихлорметаном (2 x 75 мл). Фракции дихлорметана объединяли и промывали насыщенным раствором бикарбоната натрия (200 мл) и водой (100 мл). Дихлорметан сушили над сульфатом натрия, фильтровали и концентрировали. Оставшееся масло (5,13 г) очищали посредством хроматографии на силикагеле (200 г) с использованием в качестве элюента 5% этилацетата в гептане с получением сложного этилового эфира 5-бром-5-(2-хлорфенил)-2,2-диметилпентановой кислоты (3,31 г, выход 63,5%) в виде бесцветного масла. 1H ЯМР (300 МГц, CDCl3/ТМС): δ=7,57 (дд, 1H, J=7,5, 1,5 Гц), 7,34-7,16 (м, 3H), 5,39 (т, 1H, J=7,5 Гц), 4,11 (кв, 2H, J=7,2 Гц), 2,29-2,04 (м, 2H), 1,77 (дт, 1H, J=12,0, 4,5 Гц), 1,52 (дт, 1H, J=12,0, 4,2 Гц), 1,23 (т, 3H, J=7,2 Гц), 1,18 (с, 3H), 1,17 (с, 3H). 13C ЯМР (75 МГц, CDCl3/ТМС): δ=177,05, 139,06, 132,67, 129,65, 129,31, 128,70, 127,43, 60,49, 50,25, 41,87, 38,78, 34,85, 25,64, 25,08, 14,42. МСВР (CI-TOF): Рассчитано: C15H20O2BrCl (MH+): 347, обнаружено: 267,1146. (теряется в ходе МС).
Стадия 3: Синтез сложного этилового эфира 5-(2-хлорфенил)-5-(6,7-дигидро-4H-тиено[3,2-c]пиридин-5-ил)-2,2-диметилпентановой кислоты. (Соединение Ib)
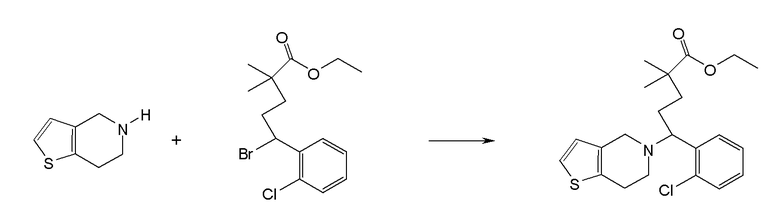
4,5,6,7-Тетрагидротиено[3,2-c]пиридин (0,65 г, 4,61 ммоль) и сложный этиловый эфир 5-бром-5-(2-хлорфенил)-2,2-диметилпентановой кислоты (1,42 г, 4,08 ммоль) растворяли в ТГФ (5 мл) и триэтиламине (3 мл) с содержанием небольшого количества кристаллов диметиламинопиридина (DMAP). Раствор нагревали в течение ночи до 85ºC с использованием масляной бани. Через 20 часов указанный раствор охлаждали до комнатной температуры и добавляли раствор бикарбоната натрия (20 мл). После перемешивания продукт экстрагировали дихлорметаном (2×25 мл). Объединенные фракции дихлорметана сушили над сульфатом натрия, фильтровали и концентрировали. Оставшееся желтое масло очищали посредством жидкостной хроматографии среднего давления (ЖХСД) на хроматографической установке Companion на силикагеле (40 г) при использовании в качестве элюента градиента 100% гептан - 10% этилацетат в течение 20 минут с последующим использованием второго градиента (до 100% этилацетата) в течение 40 минут. Вторую широкую фракцию собирали и концентрировали с получением сложного этилового эфира 5-(2-хлорфенил)-5-(6,7-дигидро-4H-тиено[3,2-c]пиридин-5-ил)-2,2-диметилпентановой кислоты (Соединение Ib, 1,11 г, выход 67,2%) в виде светло-желтого масла. 1H ЯМР (300 МГц, CDCl3/ТМС): δ=7,46 (дд, 1H, J=7,80, 1,5 Гц), 7,38(дд, 1H, J=7,80, 1,5 Гц), 7,30-7,15 (м, 2H), 7,04 (д, 1H, J=5,1 Гц), 6,70 (д, 1H, J=5,1 Гц), 4,18 (дд, 1H, J=8,7, 4,5 Гц), 4,08 (кв, 2H, J=7,2 Гц), 3,79 (д, 1H, J=14,4 Гц), 3,45 (д, 1H, J=14,4 Гц), 2,90-2,62 (м, 4H), 2,0-1,85 (м, 1H), 1,82-1,68 (м, 1H), 1,49 (дт, 1H, J=13,5, 4,2 Гц), 1,30 (дт, 1H, J=13,5, 4,5 Гц), 1,20 (т, 3H, J=7,2 Гц), 1,10 (с, 6H). 13C ЯМР (75 МГц, CDCl3/ТМС): δ=177,59, 138,99, 134,95, 134,25, 133,59, 129,64, 129,11, 128,07, 126,73, 125,43, 122,61, 63,81, 60,38, 50,63, 47,95, 42,22, 36,43, 28,22, 26,17, 25,52, 25,38, 14,47. МС (MMI-TOF): Рассчитано: C22H28NO2ClS (MH+): 406,1602; обнаружено: 406,1576.
Стадия 4: Гидрохлорид 5-(2-хлорфенил)-5-(6,7-дигидро-4H-тиено[3,2-c]пиридин-5-ил)-2,2-диметилпентановой кислоты.
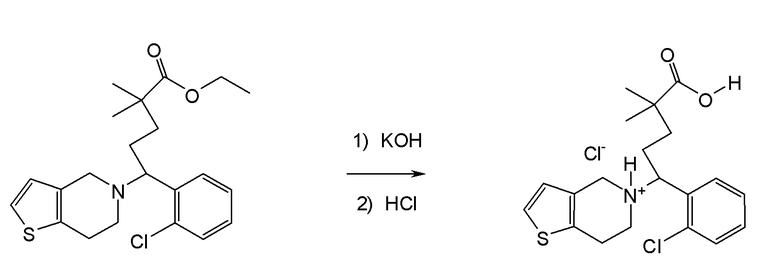
Сложный этиловый эфир 5-(2-хлорфенил)-5-(6,7-дигидро-4H-тиено[3,2-c]пиридин-5-ил)-2,2-диметилпентановой кислоты (1,0 г, 2,46 ммоль) растворяли в воде (5 мл) и этаноле (5 мл), содержащем гидроксид калия (1 г). Смесь нагревали до обратной конденсации в течение 6 часов. После охлаждения до комнатной температуры этанол удаляли при пониженном давлении. Добавляли воду (25 мл) и раствор подкисляли до pH=5-6 концентрированной соляной кислотой. Продукт экстрагировали дихлорметаном (2x25 мл). Объединенные экстракты дихлорметана сушили над сульфатом натрия, фильтровали и концентрировали. Неочищенный продукт (1,06 г) очищали посредством колоночной хроматографии на силикагеле (20 г) с использованием в качестве элюента смеси гептан/этилацетат (1:1). Фракции, содержащие продукт, объединяли, концентрировали и растворяли в дихлорметане (25 мл). Раствор дихлорметана подкисляли соляной кислотой (2 н) в диэтиловом эфире (6 мл). Растворители удаляли при пониженном давлении. Оставшееся твердое вещество сушили до достижения постоянной массы при комнатной температуре в высоком вакууме с получением гидрохлорида 5-(2-хлорфенил)-5-(6,7-дигидро-4H-тиено[3,2-c]пиридин-5-ил)-2,2-диметилпентановой кислоты (гидрохлорид Соединения Ia, 0,71 г, выход 69,6%) в виде твердого вещества не совсем белого цвета. 1H ЯМР (300 МГц, DMS-d6/ТМС): δ=12,20 (уш.с, 1H), 11,83 (уш.с, 0,5H), 11,61 (уш.с, 0,5H), 8,10 (м, 1H), 7,77-7,41 (м, 4H), 6,97 (д, 1H, J=4,5 Гц), 6,81 (д, 1H, J=4,5 Гц), 4,90-4,70 (м, 1,5H), 4,42-4,37 (м, 0,5H), 4,11-4,01 (м, 1H), 3,90-3,80 (м, 0,5H), 3,51 (м, 0,5H), 3,20-2,84 (м, 2H), 2,37 (м, 1H), 2,14 (м, 1H), 1,28 (м, 1H), 1,04 (с, 3H), 1,02 (с, 3H), 0,90 (м, 1H). (смесь ротаизомеров). 13C ЯМР (75 МГц, DMS-d6/ТМС): δ=177,64, 134,50, 131,23, 130,89, 129,72, 129,38, 128,02, 127,33, 125,06, 124,68, 64,62, 63,52, 54,63, 49,73, 48,54, 47,93, 47,20, 40,72, 35,21, 25,90 (d), 24,78, 24,37, 21,79, 21,31 (d), 14,99. МС (MMI-TOF): Рассчитано: C20H25NO2Cl2S (MH+): 378,1289, обнаружено: 378,1271. CHN-анализ: Рассчитано; 57,97 C, 6,08 H, 3,38 N, 17,11 Cl, обнаружено; 56,07 C, 6,25 H, 3,12 N, 18,55 Cl. Оптимальное соотношение CHN: C20H25Cl2NO2S + 0,65 H2O+0,05 HCl
ПРИМЕР 2. Гидрохлорид 6-(2-хлорфенил)-6-(6,7-дигидро-4H-тиено[3,2-c]пиридин-5-ил)-2,2-диметилкапроновой кислоты. (Гидрохлорид Соединения Ic)
Стадия 1: Синтез бромида (4-этоксикарбонил-4-метил)трифенилфосфония.
К раствору сложного этилового эфира 5-бром-2,2-диметилпентановой кислоты (6 г, 25,3 ммоль) в толуоле (55 мл) добавляли трифенилфосфин (6,6 г, 25,3 ммоль). Раствор нагревали до обратной конденсации (масляная баня, 122°C) в течение 24 часов. Толуол выпаривали и осадок промывали гептаном (20 мл) и диэтиловым эфиром (20 мл). Оставшееся твердое вещество сушили в высоком вакууме с получением бромида (4-этоксикарбонил-4-метил)трифенилфосфония (11 г, 87,3%) в виде порошка не совсем белого цвета (температура плавления 245-250ºC). 1H ЯМР (Поле: 300 МГц, Растворитель: CDCl3/ТМС) δ (ч/млн): 7,94-7,63 (м, 15H), 3,99 (кв, 2H, J=6,9 Гц), 3,75 (м, 2H), 1,91 (м, 2H), 1,61 (м, 2H), 1,10 (м, 9H). 13C ЯМР (Поле: 75 МГц, Растворитель: CDCl3/ТМС) δ (ч/млн): 176,79, 134,81, 133,29 (д, J=10 Гц), 130,22 (д, J=12 Гц), 117,79 (д, J=85 Гц), 60,14, 41,94, 40,60 (д, J=16 Гц), 24,95, 22,98 (д, J=50 Гц), 18,37, 14,09. МС (FIA-ESI-TOFM): Рассчитано: C27H32BrOP (MH)+: 419,2134; обнаружено: 419,2138.
Стадия 2: Синтез сложного этилового эфира 6-(2-хлорфенил)-2,2-диметилгекс-5-еновой кислоты.
Бромид (4-этоксикарбонил-4-метил)трифенилфосфония (7 г, 14 ммоль) и 2-хлорбензальдегид (1,96 г, 14 ммоль) в CH2Cl2 (21 мл) перемешивали по возможности максимально интенсивно и добавляли по каплям 50% раствор NaOH (8 мл). После этого продолжали перемешивать в течение 1,5 часов. Смесь переносили в сепаратор и добавляли дихлорметан (70 мл) и воду (70 мл). После перемешивания водный слой удаляли и экстрагировали дихлорметаном (3×70 мл). Объединенные органические экстракты промывали солевым раствором (50 мл), сушили над Na2SO4, концентрировали и очищали посредством колоночной хроматографии (силикагель, этилацетат/гептан = 1/10-1/6) с получением сложного этилового эфира 6-(2-хлорфенил)-2,2-диметилгекс-5-еновой кислоты (2,81 г, 72,2%) в виде желтого масла. 1H ЯМР (Поле: 300 МГц, Растворитель: CDCl3/ТМС) δ (ч/млн): 7,35-7,08 (м, 4H), 6,46 (д, 1H, J=11,7 Гц), 5,76-5,68 (м, 1H), 4,04 (кв, 2H, J=7,2 Гц), 2,23-2,08 (м, 2H), 1,74-1,61 (м, 2H), 1,20 (т, 3H, J=7,2 Гц), 1,13 (с, 6H). (смесь цис- и транс-изомеров, представлен основной/цис-изомер). 13C ЯМР (Поле: 75 МГц, Растворитель: CDCl3/ТМС) δ (ч/млн): 177,34, 135,54, 133,49, 130,34, 129,30, 128,03, 126,40, 126,14, 60,32, 42,13, 40,53, 29,06, 25,33, 14,30. (смесь цис- и транс-изомеров, представлен основной/цис-изомер). МС (GC-CI): Рассчитано: C16H21ClO2 (MH)+: 281,1303; обнаружено: 282,1324
Стадия 3: Синтез сложного этилового эфира 6-бром-6-(2-хлорфенил)-2,2-диметилкапроновой кислоты.
Сложный этиловый эфир 6-(2-хлорфенил)-2,2-диметилгекс-5-еновой кислоты (15 г, 53,6 ммоль) растворяли в ледяной уксусной кислоте (150 мл). Полученный раствор охлаждали на ледяной бане (примерно 15°C) по мере барботирования сухого бромоводорода через указанный раствор в течение 8 часов. Реакционную смесь выливали в смесь лед/вода (250 мл) и экстрагировали этилацетатом (3×100 мл). Объединенные органические экстракты промывали насыщенным раствором NaHCO3 (100 мл), сушили над Na2SO4 и концентрировали при пониженном давлении. Неочищенный продукт (18 г) очищали посредством колоночной хроматографии (силикагель, этилацетат/гептан = 1/20-1/10) с получением сложного этилового эфира 6-бром-6-(2-хлорфенил)-2,2-диметилкапроной кислоты (15,5 г, 80,14%) в виде желтого масла. 1H ЯМР (Поле: 300 МГц, Растворитель: CDCl3/ТМС) δ (ч/млн):7,58 (д, 1H, J=7,8 Гц), 7,34 (т, 1H, J=7,8 Гц), 7,27 (д, 1H, J=7,2 Гц), 7,21 (т, 1H, J=7,2 Гц), 5,45 (т, 1H, J=8,1 Гц), 4,09 (кв, 2H, J=6,9 Гц), 2,19 (м, 2H), 1,55 (м, 2H), 1,24 (т, 3H, J=6,9 Гц), 1,14 (с, 6H). 13C ЯМР (Поле: 75 МГц, Растворитель: CDCl3/ТМС) δ (ч/млн): 177,45, 139,25, 132,64, 129,64, 129,27, 128,75, 127,43, 60,36, 49,97, 42,17, 39,84, 39,51, 25,41, 25,20, 23,67, 14,43. МС (GC-CI): Рассчитано: C16H22BrClO2 (MH)+: 361,0564; обнаружено: 361,0547.
Стадия 4: Синтез сложного этилового эфира 6-(2-хлорфенил)-6-(6,7-дигидро-4H-тиено[3,2-c]пиридин-5-ил)-2,2-диметилкапроновой кислоты. (Соединение Ie)
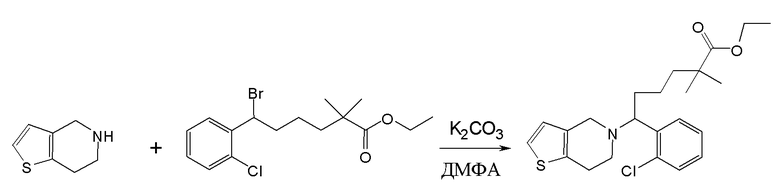
Гидрохлорид 4,5,6,7-тетрагидро-тиено[3,2-c]пиридина (2,5 г, 8,8 ммоль) смешивали с гидроксидом натрия (2,7 г) в воде (80 мл). Соответствующий свободный амин экстрагировали дихлорметаном (3×20 мл), который сушили над Na2SO4, фильтровали и концентрировали с получением 4,5,6,7-тетрагидротиено[3,2-c]пиридина (1,22 г). Указанный 4,5,6,7-тетрагидротиено[3,2-c]пиридин (1,22 г, 8,8 ммоль) и сложный этиловый эфир 6-бром-6-(2-хлорфенил)-2,2-диметилкапроновой кислоты (2,5 г, 8,8 ммоль) объединяли в ДМФА (90 мл) и добавляли карбонат калия (1,82 г, 13,2 ммоль). Смесь нагревали до 70°C в течение 1 часа и затем до 60°C в течение ночи. Смесь охлаждали до комнатной температуры и добавляли воду (100 мл). Продукт экстрагировали диэтиловым эфиром (3×150 мл). Объединенные экстракты эфира промывали водой (3×80 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали посредством колоночной хроматографии (силикагель, смесь этилацетат/гептан = 1/10) с получением сложного этилового эфира 6-(2-хлорфенил)-6-(6,7-дигидро-4H-тиено[3,2-c]пиридин-5-ил)-2,2-диметилкапроновой кислоты (Соединение Ie, 1,26 г, 42,04%) в виде желтого масла. 1H ЯМР (Поле: 300 МГц, Растворитель: CDCl3/ТМС) δ (ч/млн):7,49 (д, 1H, J=7,8 Гц), 7,37 (д, 1H, J=8,1 Гц), 7,26-7,14 (м, 2H), 7,05 (д, 1H, J=5,4 Гц), 6,70 (д, 1H, J=5,4 Гц), 4,19 (дд, 1H, J=8,7, 4,5 Гц), 4,02 (кв, 2H, J=7,2 Гц), 3,81 (д, 1H, J=14,4 Гц), 3,47 (д, 1H, J=14,4 Гц), 2,88-2,66 (м, 4H), 1,95-1,74 (м, 2H), 1,52-1,46 (м, 2H), 1,13 (т, 3H, J=7,2 Гц), 1,15-1,08 (м, 2H), 1,07 (с, 6H). 13C ЯМР (Поле: 75 МГц, Растворитель: CDCl3/ТМС) δ (ч/млн): 177,63, 139,16, 134,91, 134,21, 133,55, 129,53, 129,03, 128,03, 126,81, 124,46, 122,65, 63,47, 60,33, 50,73, 48,04, 42,31, 41,04, 33,48, 26,14, 25,58, 25,12, 21,07, 14,44. МС (GC-CI): Рассчитано: C23H30ClNO2S (MH)+: 420,1758; обнаружено: 420,1725.
Стадия 5: Синтез гидрохлорида 6-(2-хлорфенил)-6-(6,7-дигидро-4H-тиено[3,2-c]пиридин-5-ил)-2,2-диметилкапроновой кислоты.
Сложный этиловый эфир 6-(2-хлорфенил)-6-(6,7-дигидро-4H -тиено[3,2-c]пиридин-5-ил)-2,2-диметилкапроновой кислоты (2,5 г, 7,33 ммоль) добавляли к раствору этанола (100 мл) и гидроксида натрия (1,8 г, 43,8 ммоль) в воде (32 мл). Смесь нагревали до обратной конденсации в течение 6,5 часов. Раствор выпаривали при пониженном давлении и к осадку добавляли воду (100 мл). Водный раствор экстрагировали смесью этилацетат/гептан = 1/10 (100 мл). Органические слои отбирали и pH водного раствора доводили 10 н. раствором соляной кислоты до pH=6. Продукт экстрагировали дихлорметаном (3×100 мл), который сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением 6-(2-хлорфенил)-6-(6,7-дигидро-4H-тиено[3,2-c]пиридин-5-ил)-2,2-диметилкапроновой кислоты (1,6 г, 68,7%) в виде желтого масла. Указанную 6-(2-хлорфенил)-6-(6,7-дигидро-4H-тиено[3,2-c]пиридин-5-ил)-2,2-диметилкапроновую кислоту (3,97 г, 10,2 ммоль) растворяли в 36% растворе HCl (0,95 мл) и воде (59 мл). После перемешивания в течение 10 минут водный раствор экстрагировали смесью этилацетат/гептан (20/80, 110 мл). Органический экстракт отбирали и водную фракцию лиофилизировали с получением гидрохлорида 6-(2-хлорфенил)-6-(6,7-дигидро-4H-тиено[3,2-c]пиридин-5-ил)-2,2-диметилкапроновой кислоты (гидрохлорид Соединения Ic, 3,48 г, выход 79,8%, чистота 99,7%, ВЭЖХ) в виде твердого вещества белого цвета, которое плавилось при температуре 136-138°C. 1H ЯМР (Поле: 300 МГц, Растворитель: CDCl3/ТМС) δ (ч/млн):7,69 (с, 1H), 7,47-7,38 (м, 3H), 7,21 (д, 1H, J=3,9 Гц), 6,71 (д, 1H, J=3,9 Гц), 4,92 (м, 1H), 4,31-4,15 (м, 2H), 3,15-3,05 (м, 4H), 2,24-2,14 (м, 2H), 1,47-1,36 (м, 2H), 1,08-1,06 (м, 2H), 0,99 (с, 6H). (смесь ротаизомеров, ЯМР). 13C ЯМР (Поле: 75 МГц, Растворитель: CDCl3/ТМС) δ (ч/млн): 181,10, 137,13, 132,80, 132,77, 132,04, 131,79, 130,48, 129,73, 128,73, 126,62, 126,14, 66,14, 51,52, 42,87, 40,92, 32,04, 25,88, 25,47, 23,45, 22,50. МС (FIA-ESI-TOF): Рассчитано: C21H26ClNO2S (MH)+: 392,1446; обнаружено: 392,1453. CHN-анализ: Рассчитано; 58,87 C, 6,35 H, 3,27 N, 16,55 Cl, 7,48 S, обнаружено:; 57,80 C, 6,44 H, 3,15 N, 16,14 Cl, 7,15 S, Оптимальное соотношение CHN: C21H27Cl2NO2S + 0,55 H2O
ПРИМЕР 3. 1 2-(2-Хлорфенил)-2-(6,7-дигидро-4H-тиено[3,2-c]пиридин-5-ил)этанол. (Соединение Is)
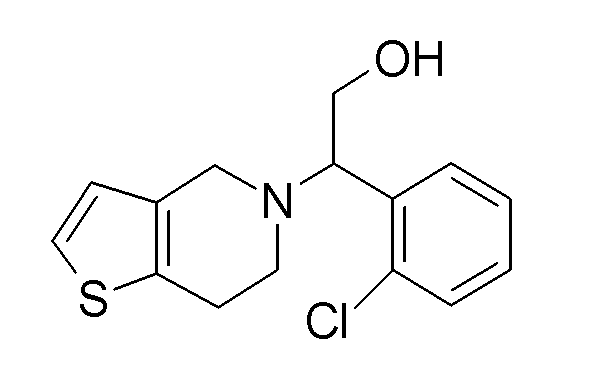
К раствору гидросульфата клопидогреля (10 г, 23,8 ммоль) в деионизированной воде (300 мл) малыми порциями добавляли раствор бикарбоната натрия (4 г, 47,6 ммоль). После перемешивания добавляли трет-бутилметиловый эфир (200 мл) и раствор перемешивали в течение одного часа. Слои разделяли и водный слой снова экстрагировали трет-бутилметиловым эфиром (2×100 мл). Объединенные органические экстракты промывали солевым раствором (100 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Оставшееся коричневое масло сушили в высоком вакууме до постоянной массы с получением клопидогреля в виде свободного основания (8,24 г, выход 106%, со следовым количеством МТБЭ). К раствору клопидогреля в виде свободного основания (3,22 г, 10 ммоль) в безводном диэтиловом эфире (4 мл) в атмосфере азота добавляли по каплям раствор алюмогидрида лития (0,46 г, 12 ммоль) в безводном диэтиловом эфире (8 мл). Алюмогидрид лития добавляли со скоростью, позволяющей поддерживать медленный процесс обратной конденсации эфира. Добавление завершали и смесь перемешивали при температуре обратной конденсации в течение дополнительных 30 минут. Через 30 минут раствор охлаждали до 0°C на ледяной/водяной бане. Избыток LiAlH4 удаляли с помощью этилацетата (2 мл, добавляли медленно), затем добавляли 6 н. раствор HCl (20 мл, добавляли медленно при интенсивном перемешивании). Смесь переносили в делительную воронку. Водную фракцию отделяли и экстрагировали эфиром (30 мл, который отбирали). Водную фракцию доводили до pH=7 6 н. раствором NaOH (20 мл) и экстрагировали эфиром (4×15 мл). Объединенные экстракты эфира промывали солевым раствором (20 мл), сушили над MgSO4 и концентрировали в вакууме с получением 2-(2-хлорфенил)-2-(6,7-дигидро-4H-тиено[3,2-c]пиридин-5-ил)этанола (Соединение Is, 2,12 г, выход 75,4%) в виде твердого вещества белого цвета. 1H ЯМР (Поле: 300 МГц, Растворитель: CDCl3/ТМС) δ (ч/млн): 7,45-7,40 (м, 2H), 7,23 (м, 2H), 7,04 (д, 1H, J=5,1 Гц), 6,70 (д, 1H, J=5,1 Гц), 4,47 (дд, 1H, J=6,9, 5,8 Гц), 3,97-3,91 (м, 1H), 3,81-3,74 (м, 2H), 3,62 (д, 1H, J=14,4 Гц), 3,01-2,95 (м, 1H), 2,84 (м, 2H) 2,76-2,68 (м, 1H). 13C ЯМР (Поле: 75 МГц, Растворитель: CDCl3/ТМС) δ (ч/млн): 135,31, 134,92, 133,81, 133,26, 130,03, 129,35, 128,85, 126,72, 125,34, 122,82, 64,53, 61,55, 50,20, 47,67, 26,15. МС (HR, DIP-CI): Рассчитано: C15H17ClNOS (MH)+: 294,0714, обнаружено: 294,0715.
ПРИМЕР 4. 4-(2-Хлорфенил)-4-(6,7-дигидро-4H-тиено[3,2-c]пиридин-5-ил)бутан-1-ол. (Соединение Id)
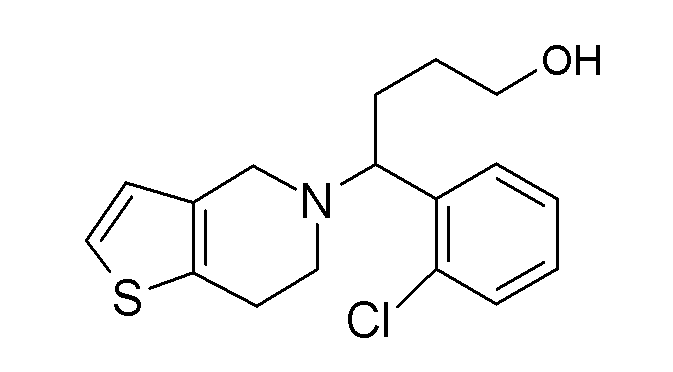
Стадия 1. Синтез 5-[2-хлор-1-(2-хлорфенил)этил]-4,5,6,7-тетрагидротиено[3,2-c]пиридина.
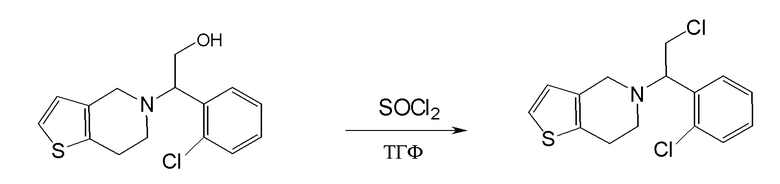
К раствору 2-(2-хлорфенил)-2-(6,7-дигидро-4H-тиено[3,2-c]пиридин-5-ил)этанола (9,7 г, 33,1 ммоль) в ТГФ (280 мл) добавляли по каплям тионилхлорид (7,9 г, 66,2 ммоль). Раствор перемешивали при комнатной температуре в течение 1,5 часов (ходреакции контролировали посредством ТСХ). Избыток тионилхлорида выпаривали при пониженном давлении. Осадок растворяли в CH2Cl2 (200 мл), промывали насыщенным раствором NaHCO3 (до pH=7), сушили над Na2SO4 и концентрировали в вакууме с получением 5-[2-хлор-1-(2-хлорфенил)этил]-4,5,6,7-тетрагидротиено[3,2-c]пиридина (10,1 г, 98,1%) в виде светло-желтого масла. 1H ЯМР (Поле: 300 МГц, Растворитель: CDCl3/ТМС) δ (ч/млн): 7,63 (д, J=6,3 Гц, 1H), 7,36-7,22 (м, 3H), 7,05 (д, J=5,1 Гц, 1H), 6,70 (д, J=5,1 Гц, 1H), 5,62 (т, J=6,3, 1H), 3,71 (с, 2H), 3,10-3,07 (м, 2H), 2,95-2,91 (м, 2H), 2,86-2,84 (м, 2H). 13C ЯМР (Поле: 75 МГц, Растворитель: CDCl3/ТМС) δ (ч/млн): 137,72, 133,43, 132,79, 129,56, 129,47, 129,00, 127,43, 125,31, 122,70, 64,37, 56,60, 53,24, 50,98, 25,24. МС (HR, GC-CI): Рассчитано: C15H15Cl2NS (MH)+: 312,0375, обнаружено: 312,0349.
Стадия 2. Синтез сложного диэтилового эфира 2-[2-хлорфенил)-2-(6,7-дигидро-4H-тиено[3,2-c]пиридин-5-ил)этил]малоновой кислоты.
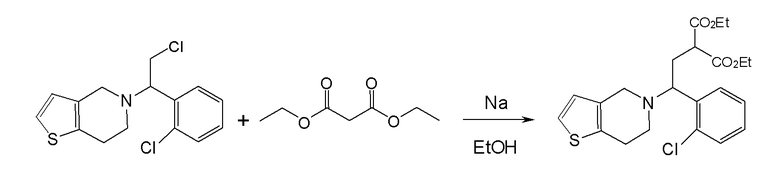
К этанолу (38 мл) в атмосфере азота добавляли натрий (1,03 г, 44,9 ммоль) маленькими кусочками. Полученный раствор этоксида натрия охлаждали и добавляли по каплям диэтилмалонат (7,4 г, 46,2 ммоль). Реакционную смесь перемешивали в течение 5 минут при комнатной температуре и затем добавляли по каплям 5-[2-хлор-1-(2-хлорфенил)этил]-4,5,6,7-тетрагидротиено[3,2-c]пиридин (14 г, 44,9 ммоль) в этаноле (30 мл). Смесь нагревали до обратной конденсации в течение 2 часов. Через два часа растворитель удаляли при пониженном давлении и добавляли воду (100 мл). Продукт экстрагировали CH2Cl2 (3×200 мл) и объединенные экстракты сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением неочищенного сложного диэтилового эфира 2-[2-хлорфенил)-2-(6,7-дигидро-4H-тиено[3,2-c]пиридин-5-ил)этил]малоновой кислоты (17 г, 86,7%) в виде коричневого масла. 1H ЯМР (Поле: 300 МГц, Растворитель: CDCl3/ТМС) δ (ч/млн): 7,36 (д, J=7,8 Гц, 1H), 7,15 (м, 3H), 7,02 (д, J=5,1 Гц, 1H), 6,69 (д, J=5,1 Гц, 1H), 4,45-4,37 (м, 1H), 3,96-3,74 (м, 5H), 3,6 (д, J=14,4 Гц, 1H), 3,48 (д, J=14,4 Гц, 1H), 3,00-2,95 (м, 1H), 2,81-2,60 (м, 4H), 1,05 (т, J=7,2 Гц, 3H), 0,94 (т, J=7,2 Гц, 3H). 13C ЯМР (Поле: 75 МГц, Растворитель: CDCl3/ТМС) δ (ч/млн): 168,01, 167,77, 138,09, 134,28, 133,92, 133,34, 129,81, 127,98, 127,76, 126,88, 125,05, 122,38, 61,57, 61,50, 61,23, 56,56, 53,25, 50,72, 40,02, 25,21, 13,88, 13,81. МС (HR, GC-CI): Рассчитано: C22H27ClNO4S (MH)+: 436,1344, обнаружено: 436,1318
Стадия 3. Синтез сложного этилового эфира 4-(2-хлорфенил)-4-(6,7-дигидро-4H-тиено[3,2-c]пиридин-5-ил)масляной кислоты. (Соединение If)
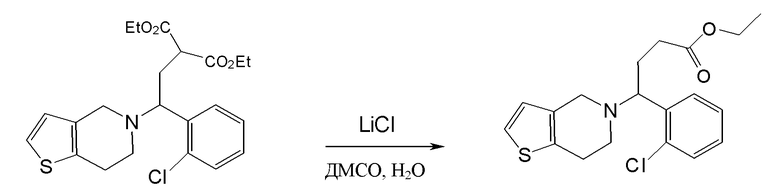
Смесь сложного диэтилового эфира 2-[2-хлорфенил)-2-(6,7-дигидро-4H-тиено[3,2-c]пиридин-5-ил)этил]малоновой кислоты (17,5 г, 40,2 ммоль) и хлорида лития (6,8 г, 161,9 ммоль) в ДМСО (230 мл) и воде (3,9 мл) нагревали в течение 4 часов до обратной конденсации (масляная баня, примерно 190°C) при перемешивании. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой (150 мл) и экстрагировали этилацетатом (3×150 мл). Объединенные органические экстракты промывали водой (3×80 мл), солевым раствором (80 мл) и сушили над MgSO4. Продукт фильтровали, концентрировали при пониженном давлении и сушили в высоком вакууме. В результате указанной процедуры получали неочищенный продукт (15,4 г). Неочищенный продукт очищали посредством колоночной хроматографии (силикагель, смесь этилацетат/гептан 1:3) с получением сложного этилового эфира 4-(2-хлорфенил)-4-(6,7-дигидро-4H-тиено[3,2-c]пиридин-5-ил)масляной кислоты (Соединение If, 11,1 г, выход 76,1%) в виде светло-желтого масла, 1H ЯМР (Поле: 300 МГц, Растворитель: CDCl3/ТМС) δ (ч/млн): 7,34 (д, J=7,8 Гц, 1H), 7,24-7,10 (м, 3H), 7,03 (д, J=5,1 Гц, 1H), 6,71 (д, J=5,1 Гц, 1H), 4,15-4,02 (м, 1H), 3,92 (кв, J=7,2 Гц, 2H), 3,67 (д, J=14,4 Гц, 1H), 3,51 (д, J=14,4 Гц, 1H), 2,97-2,80 (м, 4H), 2,75-2,53 (м, 4H), 1,06 (т, J=7,2 Гц, 3H). 13C ЯМР (Поле: 75 МГц, Растворитель: CDCl3/ТМС) δ (ч/млн): 172,25, 139,97, 134,09, 133,99, 133,46, 129,77, 127,76, 127,60, 127,01, 125,22, 122,46, 62,68, 60,32, 53,43, 51,14, 38,41, 36,49, 25,59, 14,20. МС (HR, GC-CI): Рассчитано: C19H23ClNO2S (MH)+ 364,1133, обнаружено: 364,1152.
Стадия 4. Синтез 4-(2-хлорфенил)-4-(6,7-дигидро-4H-тиено[3,2-c]пиридин-5-ил)бутан-1-ола.
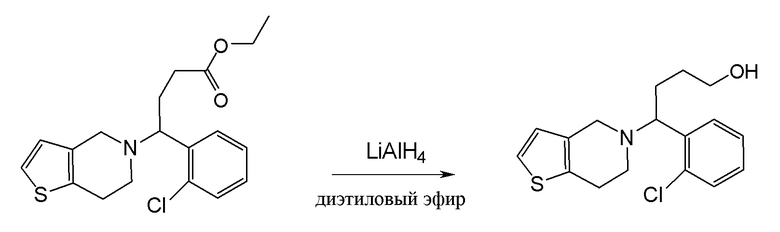
К раствору алюмогидрида лития (0,13 г, 3,31 ммоль) в безводном диэтиловом эфире (7 мл) добавляли по каплям сложный этиловый эфир 4-(2-хлорфенил)-4-(6,7-дигидро-4H-тиено[3,2-c]пиридин-5-ил)масляной кислоты (1,0 г, 2,75 ммоль) в безводном диэтиловом эфире (3 мл) в атмосфере азота. Алюмогидрид лития добавляли со скоростью, позволяющей поддерживать процесс медленной конденсации эфира. После завершения добавления смесь перемешивали при температуре обратной конденсации в течение дополнительных 30 минут. Реакционную смесь охлаждали до 0°C на бане лед/вода. Избыток LiAlH4 удаляли с помощью этилацетата (1 мл, медленно добавляли). Затем медленно добавляли 6 н. раствор HCl (10 мл) при интенсивном перемешивании. Воду отделяли и экстрагировали эфиром (30 мл), которые затем отбирали. Водную фракцию нейтрализовали 6 н. раствором NaOH (15 мл, до pH=7) и экстрагировали эфиром (3×20 мл). Объединенные органические слои промывали солевым раствором (15 мл), сушили над MgSO4 и концентрировали при пониженном давлении с получением 4-(2-хлорфенил)-4-(6,7-дигидро-4H-тиено[3,2-c]пиридин-5-ил)бутан-1-ола (Соединение Id, 0,77 г, выход 87,2%) в виде желтого вязкого масла. 1H ЯМР (Поле: 300 МГц, Растворитель: CDCl3/ТМС) δ (ч/млн): 7,36 (д, 1H, J=7,5 Гц), 7,25-7,12 (м, 3H), 7,07 (д, 1H, J=5,1 Гц), 6,72 (д, 1H, J=5,1 Гц), 6,55 (уш.с, 1H), 3,82-3,54 (м, 5H), 3,2-3,13 (м, 1H), 2,95 (уш.т, 2H , J=4,8 Гц), 2,82-2,75 (м, 1H), 2,70-2,60 (м, 2H), 2,06-1,90 (м, 2H). 13C ЯМР (Поле: 75 МГц, Растворитель: CDCl3/ТМС) δ (ч/млн): 142,33, 133,14, 133,06, 132,63, 129,70, 127,75, 127,68, 127,28, 125,16, 123,11, 63,80, 62,19, 53,41, 51,01, 39,78, 39,43, 24,88. МС (HR, DIP-CI): Рассчитано: C17H21ClNOS (MH)+: 322,1027, обнаружено: 322,1006.
ПРИМЕР 5. 1-[(2-Хлорфенил)-(6,7-дигидро-4H-тиено[3,2-c]пиридин-5-ил)-метил]-1H-бензотриазол. (Соединение IVa)
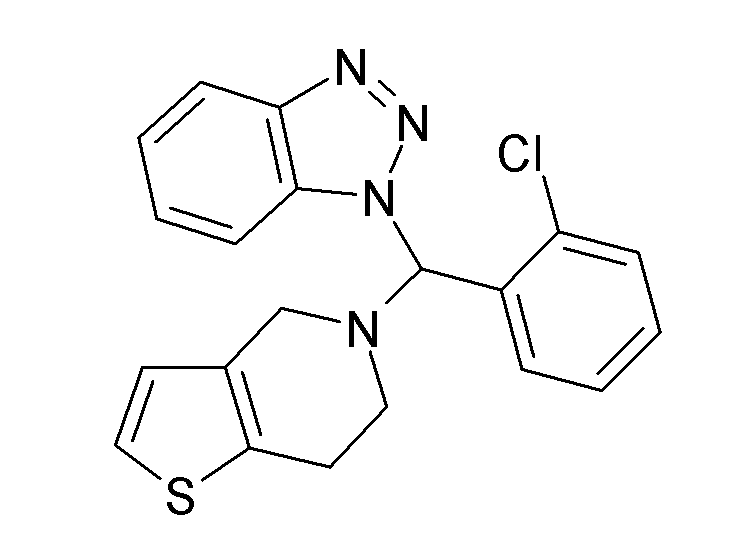
4,5,6,7-Тетрагидро-тиено[3,2-c]пиридин (0,5 г, 3,59 ммоль), бензотриазол (428 мг, 3,59 ммоль) и 2-хлорбензальдегид (504 мг, 3,59 ммоль) растворяли в диэтиловом эфире и перемешивали в течение 3 дней в присутствии молекулярных сит (4 Å) в атмосфере аргона при комнатной температуре. Через 3 дня эфир фильтровали и промывали насыщенным раствором бикарбоната натрия (20 мл). Эфир сушили над сульфатом натрия, фильтровали и концентрировали. Оставшуюся бесцветную пену растворяли в смеси гептан/диэтиловый эфир (5 мл/1 мл) и хранили в морозильной камере (-10ºC) в течение ночи. Через 20 часов при -10ºC гептан декантировали и оставшееся масло сушили в высоком вакууме при комнатной температуре до достижения постоянной массы с получением 1-[(2-хлорфенил)-(6,7-дигидро-4H-тиено[3,2-c]пиридин-5-ил)метил]-1H-бензотриазола (Соединение IVa, 0,84 г, выход 61,7%) в виде бесцветной пены, которая по результатам ЯМР представляла собой смесь изомеров (80:20). 1H ЯМР (300 МГц, CDCl3/ТМС): δ=8,08 (д, 1H, J=8,1 Гц), 7,91-7,79 (м, 2H), 7,55 (д, 1H, J=8,4 Гц), 7,53-7,24 (м, 4H), 7,11 (с, 1H), 7,04 (д, 1H, J=5,1 Гц), 6,61 (д, 1H, J=5,1 Гц), 3,90 (д, 1H, J=14,4 Гц), 3,69 (д, 1H, J=14,4 Гц), 3,21-2,95 (м, 2H), 2,88 (м, 2H). (основной изомер). 13C ЯМР (75 МГц, CDCl3/ТМС): δ=145,48, 133,78, 133,45, 132,94, 132,65, 130,10, 129,40, 127,50, 126,98, 126,56, 124,97, 124,00, 122,79, 119,80, 110,39, 84,85, 49,59, 47,28, 25,50. (основной изомер). МС (HR, DIP-CI): Рассчитано: (MH+): 379,0779, обнаружено: 379,0752.
ПРИМЕР 6. Гидрохлорид 7-(2-хлорфенил)-7-(6,7-дигидро-4H-тиено[3,2-c]пиридин-5-ил)-2,2-диметилгептановой кислоты. (Гидрохлорид Соединения Ig)
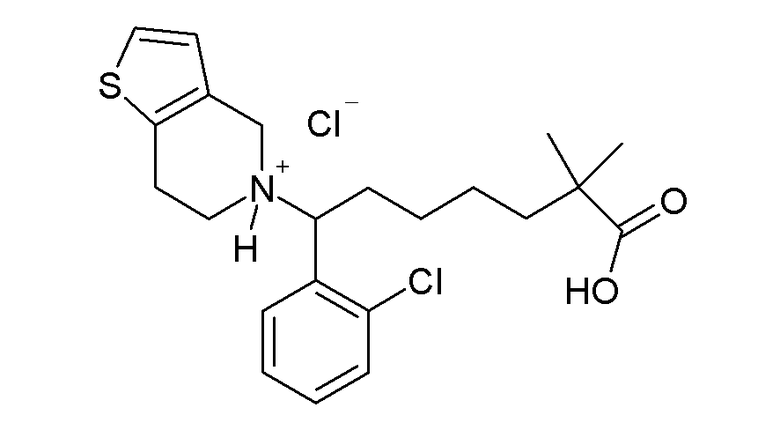
Стадия 1. Синтез бромида (5-этоксикарбонил-5-метилгексил)трифенилфосфония.
Сложный этиловый эфир 6-бром-2,2-диметилкапроновой кислоты (38,0 г, 0,15 моль) и трифенилфосфин (40,0 г, 0,15 моль) растворяли в толуоле (100 мл) и нагревали до обратной конденсации в течение 24 часов в атмосфере аргона. После нагревания в течение 24 часов колбе позволяли охлаждаться до комнатной температуры и реакционную смесь перемешивали в течение дополнительных 24 часов при комнатной температуре. Через 48 часов толуол удаляли при пониженном давлении. Оставшийся материал растворяли в дихлорметане (100 мл) и добавляли по каплям к трет-бутилметиловому эфиру (600 мл). Эфир отделяли от осадка, который сушили до достижения постоянной массы в высоком вакууме с получением бромида (5-этоксикарбонил-5-метилгексил)трифенилфосфония (71,5 г, выход 93,1%) в виде светло-желтой пены. 1H ЯМР (300 МГц, CDCl3/ТМС): δ=7,87-7,57 (м, 15H), 4,05 (1, 2H, J=7,2 Гц), 3,67 (м, 2H), 1,78-1,45 (м, 6H), 1,20 (т, 3H, J=7,2 Гц), 1,12 (с, 6H). 13C ЯМР (75 МГц, CDCl3/ТМС): δ=177,02, 134,56 (д, 3C, J=2,4 Гц), 132,94 (д, 6C, J=10,1 Гц), 129,99 (д, 6C, J=12,6 Гц), 117,44 (д, 3C, J=85,4 Гц), 59,78, 41,47, 39,12, 25,44 (д, 1C, J=15,5 Гц), 24,62, 22,59 (д, 1C, J=4,1 Гц), 21,96, 13,83. МСВР (ESI-TOF): Рассчитано: (M+): 433,2291, обнаружено: 433,2330.
Стадия 2. Синтез сложного этилового эфира 7-(2-Хлорфенил)-2,2-диметилгепт-6-еновой кислоты.
Бромид (5-этоксикарбонил-5-метилгексил)трифенилфосфония (35,0 г, 0,0619 моль) растворяли в дихлорметане (100 мл), содержащем 2-хлорбензальдегид (8,70 г, 0,0619 моль). Порциями добавляли раствор гидроксида натрия (10 г, 0,25 моль) в воде (10 мл) в течение десяти минут. Смесь перемешивали в течение 2 часов при комнатной температуре. Через два часа слои разделяли и фракцию дихлорметана промывали водой (2×150 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Оставшееся коричневое масло (18,99 г) фильтровали через силикагель (200 г) с использованием в качестве элюента смеси этилацетат/гептан (1:9). Фракции, содержащие продукт, объединяли и концентрировали при пониженном давлении. Оставшееся желтое масло (10,5 г) содержало примесь 2-хлорбензальдегида (30-40%). Материал повторно обрабатывали путем растворения в свежем дихлорметане (100 мл), содержащем дополнительную порцию соли фосфония (25 г, 0,048 моль). Гидроксид натрия (10 г, 0,25 моль) порциями добавляли в воду (10 мл) и полученной смеси позволяли перемешиваться в течение 4 часов при комнатной температуре. Слои разделяли и фракцию дихлорметана промывали водой (2×100 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Оставшееся коричневое масло очищали посредством колоночной хроматографии на силикагеле (200 г) с использованием в качестве элюента смеси этилацетат/гептан (1:9). Фракции, содержащие продукт, объединяли и концентрировали при пониженном давлении с получением сложного этилового эфира 7-(2-хлорфенил)-2,2-диметилгепт-6-еновой кислоты (12,0 г, выход 60%) в виде светло-желтой жидкости. 1H ЯМР (300 МГц, CDCl3/ТМС): δ=7,47 (д, 0,4 H, J=7,5 Гц), 7,36-7,07 (м, 3,6 H), 6,74 (д, 0,4 H, J=15,6 Гц), 6,50 (д, 0,6 H, J=11,4 Гц), 6,16 (дт, 0,4 H, J=15,6, 7,2 Гц), 5,75 (дт, 0,6 H, J=11,4, 7,2 Гц), 4,10 (м, 2H), 2,26-2,12 (м, 2H), 1,61-1,34 (м, 4H), 1,29-1,17 (м, 3H), 1,14 (с, 6H). (смесь цис/транс изомеров). 13C ЯМР (75 МГц, CDCl3/ТМС): δ=177,59, 135,63 (2 пика), 133,67, 133,45, 133,31, 132,38, 130,31, 129,41, 129,19, 127,84, 127,73, 126,57, 126,47, 126,24, 126,00, 60,16, 42,12, 42,07, 40,22, 33,56, 28,83, 25,21, 25,16, 24,69, 14,30. (смесь цис/транс изомеров). МСВР (MMI-TOF): Рассчитано: C22H29Cl2NO2S (MH+): 406,1202, обнаружено: 406,1594.
Стадия 3. Синтез сложного этилового эфира 7-бром-7-(2-хлорфенил)-2,2-диметилгептановой кислоты.
Сложный этиловый эфир 7-(2-хлорфенил)-2,2-диметилгепт-6-еновой кислоты (11,75 г, 0,040 моль) растворяли в уксусной кислоте (80 мл) в атмосфере аргона. Колбу охлаждали на ледяной/водяной бане. Через раствор медленно пропускали газообразный бромоводород в течение 3 часов по мере медленного нагревания указанного раствора до комнатной температуры. Добавляли лед (250 г) и после перемешивания продукт экстрагировали этилацетатом (2×100 мл). Экстракты этилацетата объединяли и промывали насыщенным раствором бикарбоната натрия (3×75 мл) и водой (100 мл). Раствор сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Оставшееся масло (14,06 г) очищали посредством колоночной хроматографии на силикагеле (250 г) с использованием в качестве элюента смеси гептан/этилацетат в отношении 3:1 (с последующим использованием указанной смеси в отношении 2:1). Фракции, содержащие продукт, объединяли и концентрировали при пониженном давлении с получением сложного этилового эфира 7-бром-7-(2-хлорфенил)-2,2-диметилгептановой кислоты (12,9 г, выход 86%) в виде прозрачного масла. 1H ЯМР (300 МГц, CDCl3/ТМС): δ=7,57 (д, 1H, J=7,50 Гц), 7,33-7,15 (м, 3H), 5,43 (т, 1H, J=6,9 Гц), 4,08 (кв, 2H, J=7,2 Гц), 2,30-2,21 (м, 2H), 1,55-1,42 (м, 3H), 1,34-1,19 (м, 5H), 1,13 (с, 6H). 13C ЯМР (75 МГц, CDCl3/ТМС): δ=177,72, 139,41, 132,69, 129,67, 129,28, 128,92, 127,48, 60,36, 50,17, 42,26, 40,57, 39,19, 28,59, 25,38, 25,34, 24,45, 14,50. МС (HR, DIP-CI): Рассчитано: (MH+): 375,0726, обнаружено: 375,0726.
Стадия 4. Синтез сложного этилового эфира 7-(2-хлорфенил)-7-(6,7-дигидро-4H-тиено[3,2-c]пиридин-5-ил)-2,2-диметилгептановой кислоты. (Соединение Ih)
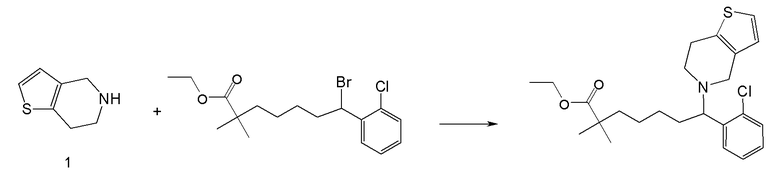
Гидрохлорид 4,5,6,7-тетрагидротиено[3,2-c]пиридина (3,2 г, 18,9 ммоль) растворяли в воде (120 мл) и добавляли 50% раствор гидроксида натрия (10 мл). Соответствующее свободное основание экстрагировали дихлорметаном (2×25 мл), который сушили над сульфатом натрия, фильтровали и концентрировали. Оставшееся масло (2,58 г, 18,9 ммоль) растворяли в ДМФА (10 мл) в атмосфере аргона. Добавляли карбонат калия (5 г) и сложный этиловый эфир 7-бром-7-(2-хлорфенил)-2,2-диметилгептановой кислоты (7,0 г, 18,63 ммоль) и полученную смесь перемешивали в течение 44 часов при 75ºC. Колбу охлаждали до комнатной температуры и добавляли воду (50 мл). Продукт экстрагировали дихлорметаном (2×75 мл), который сушили над сульфатом натрия, фильтровали и концентрировали. Оставшееся коричневое масло (8,94 г) очищали посредством колоночной хроматографии на силикагеле (140 г) с использованием в качестве элюента смеси гептан/этилацетат (4:1-2:1). Фракции, содержащие продукт, объединяли, концентрировали и сушили до достижения постоянной массы в высоком вакууме при комнатной температуре с получением сложного этилового эфира 7-(2-хлорфенил)-7-(6,7-дигидро-4H-тиено[3,2-c]пиридин-5-ил)-2,2-диметилгептановой кислоты (Соединение Ih, 3,85 г, выход 49%) в виде прозрачного масла. 1H ЯМР (300 МГц, CDCl3/ТМС): δ=7,53 (дд, 1H, J=7,5, 1,2 Гц), 7,40 (дд, 1H, J=7,8, 1,2 Гц), 7,31-7,18 (м, 2H), 7,08 (д, 1H, J=5,1 Гц), 6,74 (д, 1H, J=5,1 Гц), 4,22 (дд, 1H, J=8,7, 4,5 Гц), 4,12 (кв, 2H, J=7,2 Гц), 3,83 (д, 1H, J=14,1 Гц), 3,49 (д, 1H, J=14,1 Гц), 2,92-2,68 (м, 4H), 2,08-1,73 (м, 2H), 1,46 (м, 2H), 1,30-1,19 (м, 7H), 1,15 (с, 6H). 13C ЯМР (75 МГц, CDCl3/ТМС): δ=178,00, 139,41, 134,99, 134,26, 133,62, 129,53, 128,13, 128,03, 126,87, 125,51, 122,67, 63,69, 60,37, 50,79, 48,15, 42,33, 40,84, 33,11, 26,19, 25,40, 14,55. МСВР (DIP-CI): Рассчитано: (M+H+): 434,1921, обнаружено: 434,1871.
Стадия 5. Синтез гидрохлорида 7-(2-хлорфенил)-7-(6,7-дигидро-4H-тиено[3,2-c]пиридин-5-ил)-2,2-диметилгептановой кислоты.
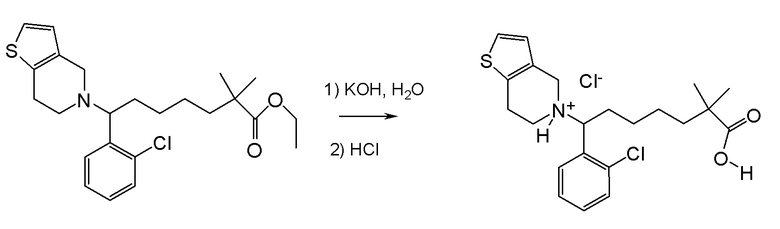
Сложный этиловый эфир 7-(2-хлорфенил)-7-(6,7-дигидро-4H-тиено[3,2-c]пиридин-5-ил)-2,2-диметилгептановой кислоты (2,80 г, 7,23 ммоль) растворяли в этаноле (40 мл) и воде (25 мл), содержащей гидроксид калия (10 г, 250 ммоль). Смесь нагревали до обратной конденсации в течение 24 часов. Через 24 часа колбу охлаждали до комнатной температуры и реакционную смесь концентрировали. Добавляли воду (50 мл) и экстрагировали 10% этилацетатом в гептане. Водную фракцию подкисляли (до pH=6) концентрированной соляной кислотой и продукт экстрагировали дихлорметаном (2×100 мл). Экстракты дихлорметана объединяли, сушили над сульфатом натрия, фильтровали и концентрировали. Оставшееся масло рыжеватого цвета (3,0 г) очищали посредством хроматографии на силикагеле (100 г) с использованием в качестве элюента смеси гептан/этилацетат (2:1) с получением 7-(2-хлорфенил)-7-(6,7-дигидро-4H-тиено[3,2-c]пиридин-5-ил)-2,2-диметилгептановой кислоты в виде твердого вещества белого цвета (2,1 г, выход 71,6%). Продукт растворяли в воде (класса ASTM 1, 200 мл), содержащей соляную кислоту (1%) и ацетон (25 мл). Основную часть ацетона и избыток соляной кислоты удаляли при пониженном давлении. Оставшуюся воду (200 мл) лиофилизировали с получением гидрохлорида 7-(2-хлорфенил)-7-(6,7-дигидро-4H-тиено[3,2-c]пиридин-5-ил)-2,2-диметилгептановой кислоты (гидрохлорид Соединения Ig, 2,2 г, температура плавления 97-100ºC, чистота 99,47%, ВЭЖХ) в виде твердого вещества белого цвета. 1H ЯМР (300 МГц, ДМСО-d6): δ=12,25 (уш.с, 0,5H), 12,12 (уш.с, 0,5H), 8,24 (уш.с, 1H), 7,60-7,38 (м, 4H), 6,94 (д, 1H, J=4,5 Гц), 6,77 (д, 1H, J=4,5 Гц), 4,90-4,68 (м, 1,5H), 4,38 (дд, 0,5H, J=14,4, 5,4 Гц), 4,01 (м, 1H), 3,80 (дд, 0,5H, J=14,4, 5,1 Гц), 3,60-3,30 (м, 1,5H), 3,20-2,85 (м, 2H), 2,42 (м, 1H), 2,23 (м, 1H), 1,40-1,0 (м, 5H), 1,01 (с, 6H), 0,79 (м, 1H). (смесь ротаизомеров). 13C ЯМР (75 МГц, ДМСО-d6): δ=178,41, 134,70, 134,60, 131,83, 131,49, 131,23, 131,05, 129,93, 129,75, 128,38, 128,35, 128,10, 125,43, 125,26, 124,87, 124,86, 64,35, 63,17, 49,87, 48,70, 48,00, 47,13, 41,16, 30,78, 30,30, 30,12, 25,77, 25,02, 24,14, 21,78, 21,37. (смесь ротаизомеров). МСВР (MMI-TOF): Рассчитано: C22H29Cl2NO2S (MH+): 406,1202, обнаружено: 406,1594.
ПРИМЕР 7. Гидрохлорид 8-(2-хлорфенил)-8-(6,7-дигидро-4H-фуро[3,2-c]пиридин-5-ил)-2,2-диметилоктановой кислоты. (Гидрохлорид Соединения Io)
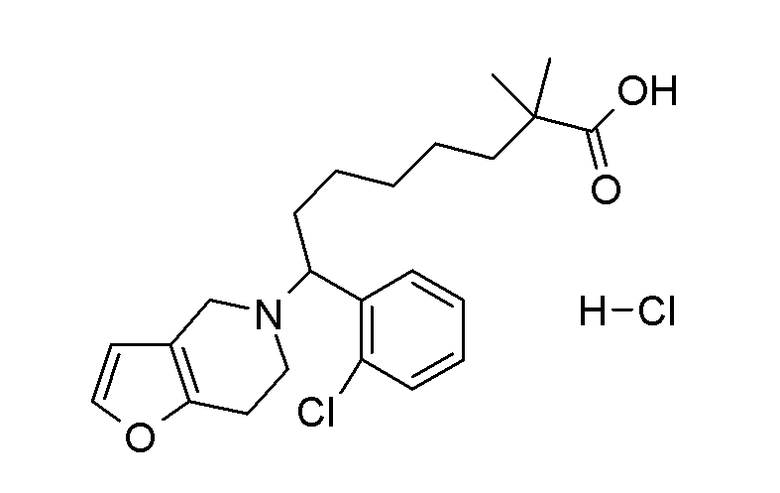
Стадия 1. Синтез 2-(2-нитровинил)фурана.
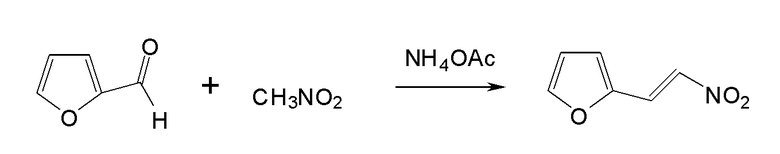
К нитрометану (169 мл) добавляли 2-фуральдегид (18,2 г, 190 ммоль) и ацетат аммония (13 г, 169 ммоль) и полученную смесь нагревали до обратной конденсации в течение 45 минут. Раствор концентрировали и добавляли дихлорметан (250 мл). Полученный раствор дихлорметана промывали водой (2×250 мл), сушили над сульфатом натрия и фильтровали с получением неочищенного продукта. В результате очистки посредством колоночной хроматографии (силикагель, смесь этилацетат/гептан = 1/10-2/3) получали чистый 2-(2-нитровинил)фуран (14 г, 53,0%) в виде желтого порошка (температура плавления 71-74°C). 1H ЯМР (Поле: 300 МГц, Растворитель: CDCl3/ТМС) δ (ч/млн): 7,78 (д, 1H, J=13,2 Гц), 7,59 (м, 1H), 7,52 (д, 1H, J=13,5 Гц), 6,90 (д, 1H, J=3,6 Гц), 6,58 (м, 1H). 13C ЯМР (Поле: 75 МГц, Растворитель: CDCl3/ТМС) δ (ч/млн): 146,90, 146,67, 134,93, 125,51, 120,12, 113,46.
Стадия 2: Синтез 2-фуран-2-илэтиламина.
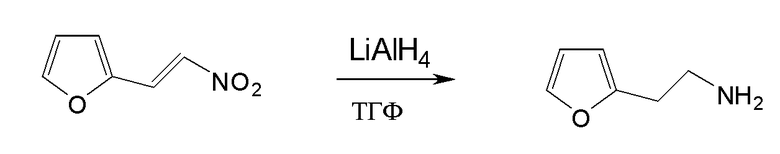
К смеси алюмогидрида лития (13,4 г, 353 ммоль) в безводном ТГФ (400 мл) добавляли по каплям 2-(2-нитровинил)фуран (14 г, 101 ммоль) в ТГФ (100 мл) в течение 30 минут при 0°C в атмосфере аргона. Охлаждение прекращали и колбу нагревали при температуре обратной конденсации в течение дополнительных 3,5 часов. Реакционную смесь охлаждали до комнатной температуры, медленно добавляли воду (13 мл) при интенсивном перемешивании. После завершения добавления воды добавляли 15% раствор гидроксида натрия (13 мл), затем снова добавляли воду (36 мл). После интенсивного перемешивания в течение 10 мин твердое вещество фильтровали и промывали диэтиловым эфиром (3×150 мл). Объединенные фильтраты сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением 2-фуран-2-илэтиламина (11 г, выход 85,6%) в виде коричневого масла. 1H ЯМР (Поле: 300 МГц, Растворитель: CDCl3/ТМС) δ (ч/млн): 7,31 (м, 1H), 6,29 (д, 1H, J=2,1 Гц), 6,05 (д, 1H, J=2,1 Гц), 2,96 (т, 2H, J=6,6 Гц,), 2,76 (т, 2H, J=6,6 Гц), 1,84 (уш., 2H). 13C ЯМР (Поле: 75 МГц, Растворитель: CDCl3/ТМС) δ (ч/млн): 153,82, 141,21, 110,15, 106,00, 40,79, 32,35.
Стадия 3: Синтез сложного трет-бутилового эфира 6,7-дигидро-4H-фуро[3,2-c]пиридин-5-карбоновой кислоты.
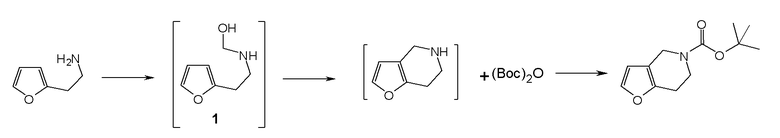
К чистому 2-фуран-2-ил-этиламину (5,5 г, 43,3 ммоль) добавляли по каплям формалин (37% водный раствор формальдегида, 4 г, 49,2 ммоль) и смеси позволяли перемешиваться в течение 30 минут при комнатной температуре. Неочищенное промежуточное вещество 1 экстрагировали диэтиловым эфиром (3×80 мл). Экстракты диэтилового эфира объединяли, сушили над сульфатом натрия, фильтровали и концентрировали. ДМФА (35 мл) насыщали газообразным хлороводородом путем барботирования хлороводорода через указанный раствор в течение часа. Оставшееся масло растворяли в ДМФА (10 мл) и добавляли к раствору ДМФА/HCl. После перемешивания при комнатной температуре в течение 3 часов ДМФА удаляли в высоком вакууме. Добавляли метил-трет-бутиловый эфир и следы ДМФА удаляли посредством экстрагирования водой (100 мл), pH которой доводили до значения pH=11 насыщенным раствором бикарбоната натрия. Раствор эфира сушили над сульфатом натрия, фильтровали и концентрировали. Оставшееся неочищенное промежуточное соединение 4,5,6,7-тетрагидрофуро[3,2-c]пиридин (0,87 г, 7,1 ммоль) в дихлорметане (50 мл) добавляли по каплям к ди-трет-бутилдикарбонату (1,6 г, 7,4 ммоль) в CH2Cl2 (50 мл) при комнатной температуре. Реакционную смесь перемешивали при такой же температуре в течение 1,5 часов, ход реакции контролировали посредством ТСХ. Растворитель выпаривали при пониженном давлении и неочищенный продукт очищали посредством колоночной хроматографии (силикагель, смесь этилацетат/гептан = 1/9) с получением чистого сложного трет-бутилового эфира 6,7-дигидро-4H-фуро[3,2-c]пиридин-5-карбоновой кислоты (0,56 г, 35,4%) в виде желтого масла. 1H ЯМР (Поле: 300 МГц, Растворитель: CDCl3/ТМС) δ (ч/млн): 7,19 (с, 1H), 6,13 (д, 1H, J=1,5 Гц), 4,25 (с, 2H), 3,63 (м, 2H), 2,59 (д, m), 1,44 (с, 9H). 13C ЯМР (Поле: 75 МГц, Растворитель: CDCl3/ТМС) δ (ч/млн): 154,78, 146,85, 141,23, 108,19, 85,03, 79,82, 41,58, 40,05, 28,52, 23,90.
Стадия 4: Синтез гидрохлорида 4,5,6,7-тетрагидрофуро[3,2-c]пиридина.
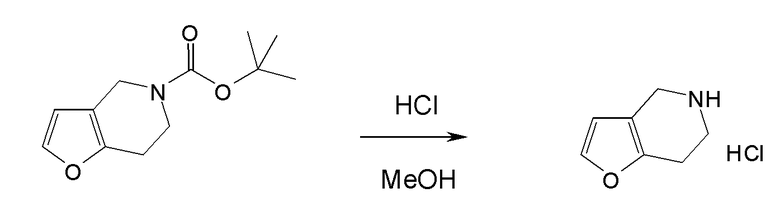
К сложному трет-бутиловому эфиру 6,7-дигидро-4H-фуро[3,2-c]пиридин-5-карбоновой кислоты (0,56 г, 2,51 ммоль) в метаноле (50 мл) добавляли концентрированный раствор соляной кислоты (37%, 1,4 мл). Смесь перемешивали при комнатной температуре в течение 5,5 часов, ход реакции контролировали посредством ТСХ. Метанол выпаривали при пониженном давлении с получением гидрохлорида 4,5,6,7-тетрагидрофуро[3,2-c]пиридина (0,5 г, выход 100%) в виде желтого порошка. 1H ЯМР (Поле: 300 МГц, Растворитель: CD3OD/ТМС) δ (ч/млн): 7,47 (с, 1H), 6,42 (с, 1H), 4,20 (с, 2H), 3,57 (м, 2H), 3,03 (м, 2H). 13C ЯМР (Поле: 75 МГц, Растворитель: CDCl3/ТМС) δ (ч/млн): 147,23, 143,89, 112,23, 109,50, 43,10, 42,37, 21,70.
Стадия 5: Сложный этиловый эфир 8-(2-хлорфенил)-8-(6,7-дигидро-4H-фуро[3,2-c]пиридин-5-ил)-2,2-диметилоктановой кислоты. (Соединение Ip)
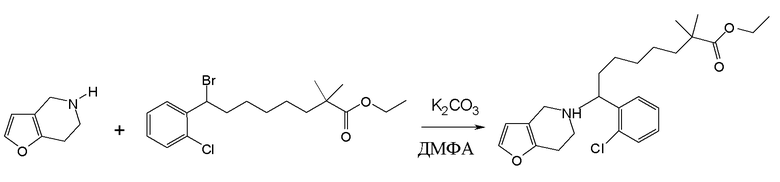
Сложный этиловый эфир 4,5,6,7-тетрагидрофуро[3,2-c]пиридина (0,47 г, 3,82 ммоль), 8-бром-8-(2-хлорфенил)-2,2-диметилоктановую кислоту (1,47 г, 3,78 ммоль) в ДМФА (36 мл) и карбонат калия (0,77 г, 5,58 ммоль) объединяли в атмосфере аргона при комнатной температуре. Смесь нагревали до 60°C в течение 3 дней в атмосфере аргона, ход реакцию контролировали посредством ТСХ. ДМФА выпаривали при пониженном давлении и осадок растворяли в диэтиловом эфире (120 мл), промывали водой (3×30 мл), солевым раствором (30 мл) и сушили над Na2SO4. Неочищенное масло очищали посредством колоночной хроматографии (силикагель, смесь этилацетат/гептан 1/9) с получением чистого сложного этилового эфира 8-(2-хлорфенил)-8-(6,7-дигидро-4H-фуро[3,2-c]пиридин-5-ил)-2,2-диметилоктановой кислоты (Соединение Ip, 0,50 г, выход 30,3%) в виде желтого масла. 1H ЯМР (Поле: 300 МГц, Растворитель: CD3OD/ТМС) δ (ч/млн): 7,48 (д, 1H, J=7,5 Гц), 7,36 (д, 1H, J=7,8 Гц), 7,32-7,15 (м, 3H), 6,17 (с, 1H), 4,20 (дд, 1H, J=9,0, 4,8 Гц), 4,08 (кв, 2H, J=7,2 Гц), 3,62 (д, 1H, J=13,5 Гц), 3,11 (д, 1H, J=13,5 Гц), 2,88-2,58 (м, 4H), 1,95-1,70 (м, 2H), 1,45-1,40 (м, 2H), 1,30-1,0 (м, 15H). 13C ЯМР (Поле: 75 МГц, Растворитель: CDCl3/ТМС) δ (ч/млн): 178,01, 149,01, 140,94, 139,50, 135,00, 129,57, 129,04, 128,03, 126,85, 115,92, 108,90, 63,43, 60,36, 47,65, 47,54, 42,36, 40,91, 33,27, 30,57, 25,67, 25,41, 25,06, 24,73, 14,55. МСВР (HR, DART-TOF): Рассчитано: (M+H)+: 432,2300, обнаружено: 432,2316.
Стадия 6: Синтез гидрохлорида 8-(2-хлорфенил)-8-(6,7-дигидро-4H-фуро[3,2-c]пиридин-5-ил)-2,2-диметилоктановой кислоты
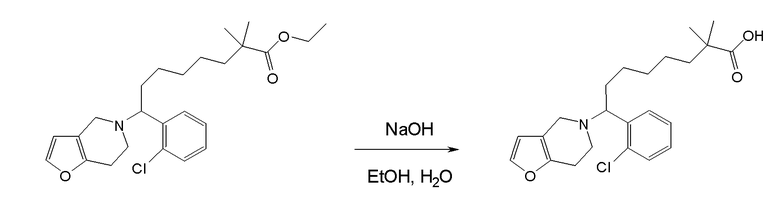
Сложный этиловый эфир 8-(2-хлорфенил)-8-(6,7-дигидро-4H-фуро[3,2-c]пиридин-5-ил)-2,2-диметилоктановой кислоты (0,5 г, 1,16 ммоль) добавляли к смеси этанола (20 мл) и гидроксида натрия (0,32 г, 8,0 ммоль) в воде (6,6 мл). Смесь нагревали до обратной конденсации в течение 6,5 часов. Растворитель выпаривали при пониженном давлении и к осадку добавляли воду (20 мл). Водный раствор промывали смесью этилацетат/гептан 1/10 (10 мл) и экстракт отбирали. Водную фракцию подкисляли концентрированной соляной кислотой до pH=6 и продукт экстрагировали дихлорметаном (3×20 мл). Объединенные экстракты дихлорметана сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением 8-(2-хлорфенил)-8-(6,7-дигидро-4H-фуро[3,2-c]пиридин-5-ил)-2,2-диметилоктановой кислоты (0,25 г, 46,9%) в виде желтого масла. Материал растворяли в диэтиловом эфире (5 мл) и добавляли к раствору соляной кислоты (2 н. HCl в диэтиловом эфире, 0,34 мл). Добавляли воду (12 мл) и после перемешивания водную фазу отделяли и лиофилизировали с получением гидрохлорида 8-(2-хлорфенил)-8-(6,7-дигидро-4H-фуро[3,2-c]пиридин-5-ил)-2,2-диметилоктановой кислоты (гидрохлорид Соединения Io, 0,16 г, светло-желтый порошок, выход 59,3%). 1H ЯМР (Поле: 300 МГц, Растворитель: CD3OD /ТМС) δ (ч/млн): 7,77 (уш.с, 1H), 7,63-7,48 (м, 5H), 6,37 (уш.с, 1H), 5,05 (м, 1H), 4,18 (м, 1H), 3,62 (м, 1H), 3,02 (м, 2H), 2,36-2,24 (м, 2H), 1,42-1,40 (м, 2H), 1,32 (м, 2H), 1,20-1,18 (м, 4H), 1,11 (с, 6H), 1,08-0,92 (м, 1H). 13C ЯМР (Поле: 75 МГц, Растворитель: CD3OD/ТМС) δ (ч/млн): 181,70, 147,11, 144,55, 137,40, 132,15, 131,89, 130,08, 128,21, 129,87, 112,37, 109,71, 66,34, 52,10, 50,05, 43,07, 41,60, 35,10, 31,67, 30,56, 26,64, 25,85, 22,31. МСВР (HR, DIP-CI): Рассчитано: C23H30ClNO3 (M+H)+: 404,1987, обнаружено: 404,2009. CHN-анализ: Рассчитано; 62,73 C, 7,09 H, 3,18 N, 16,10 Cl, обнаружено:; 59,13 C, 7,04 H, 3,03 N, 13,58 Cl.
ПРИМЕР 8. Гидрохлорид 8-(2-хлорфенил)-2,2-диметил-8-(1,4,6,7-тетрагидропирроло[3,2-c]пиридин-5-ил)-октановой кислоты. (Гидрохлорид Соединения Iq)
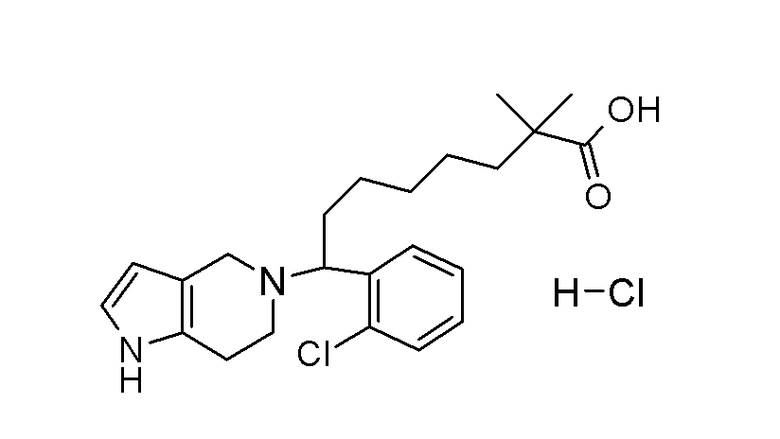
Стадия 1: Синтез сложного трет-бутилового эфира пирроло[3,2-c]пиридин-1-карбоновой кислоты.
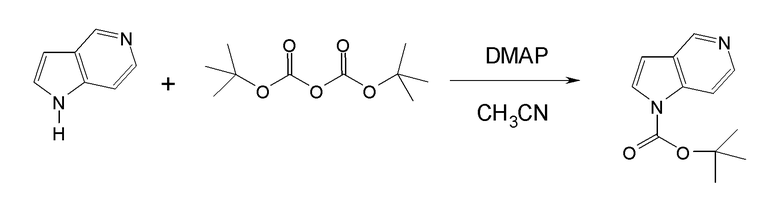
Диметиламинопиридин (DMAP) (2,08 г, 16,9 ммоль) в ацетонитриле (20 мл) добавляли по каплям к 5-азаиндолу (2,0 г, 16,9 ммоль) в ацетонитриле (70 мл) при комнатной температуре. После перемешивания смеси в течение 2 часов порцией добавляли ди-трет-бутилдикарбонат (3,68 г, 16,9 ммоль) при такой же температуре. Через 2,5 часа растворитель выпаривали при пониженном давлении и осадок (5,4 г) очищали посредством колоночной хроматографии (силикагель, смесь этилацетат/гептан 1/10 - 1/2) с получением сложного трет-бутилового эфира пирроло[3,2-c]пиридин-1-карбоновой кислоты (2,72 г, выход 92,4%) в виде ярко-желтого масла. 1H ЯМР (Поле: 300 МГц, Растворитель: CD3OD/ТМС) δ (ч/млн): 8,87 (с, 1H), 8,48 (д, 1H, J=5,7 Гц), 7,98 (д, 1H, J=5,1 Гц), 7,60 (д, 1H, J=3,3 Гц), 6,63 (д, 1H, J=3,0 Гц), 1,68 (с, 9H). 13C ЯМР (Поле: 75 МГц, Растворитель: CDCl3/ТМС) δ (ч/млн): 148,82, 143,75, 143,51, 139,50, 126,69, 109,83, 105,46, 84,60, 28,05.
Стадия 2: Синтез бромида 1-трет-бутоксикарбонил-5-[1-(2-хлорфенил)-7-метилоктил]-1H-пирроло[3,2-c]пиридин-5-ия.
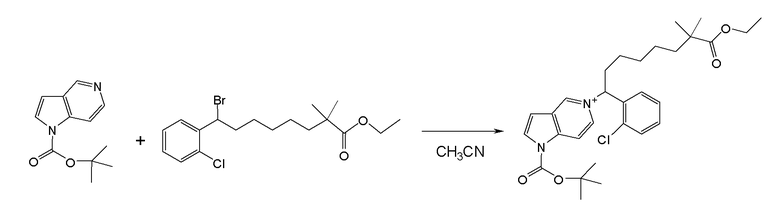
Смесь сложного трет-бутилового эфира пирроло[3,2-c]пиридин-1-карбоновой кислоты (0,81 г, 3,73 ммоль), сложного этилового эфира 8-бром-8-(2-хлорфенил)-2,2-диметилоктановой кислоты (1,44 г, 3,73 ммоль) и ацетонитрила (25 мл) перемешивали при 45°C в течение 52 часов. Растворитель выпаривали при пониженном давлении и осадок промывали диэтиловым эфиром (3×20 мл) с получением бромида 1-трет-бутоксикарбонил-5-[1-(2-хлорфенил)-7-метилоктил]-1H-пирроло[3,2-c]пиридин-5-ия (1,37 г, выход 60,6%) в виде желтого масла. 1H ЯМР (Поле: 300 МГц, Растворитель: CD3OD/ТМС) δ (ч/млн): 10,52 (с, 1H), 8,84 (д, 1H, J=7,2 Гц), 8,23 (д, 1H, J=7,8 Гц), 7,91 (т, 1H, J=3,6 Гц), 7,54-7,31 (м, 4H), 7,39 (с, 1H), 6,95 (с, 1H), 6,42 (т, 1H, J=7,2 Гц), 4,09 (кв, 2H, J=7,2 Гц), 2,67 (м, 1H), 1,70 (с, 9H), 1,42-1,19 (м, 8H), 1,21 (т, 3H, J=6,9 Гц), 1,10 (с, 6H). 13C ЯМР (Поле: 75 МГц, Растворитель: CDCl3/ТМС) δ (ч/млн): 178,00, 147,44, 140,17, 136,60, 133,19, 131,97, 131,45, 130,42, 130,03, 128,64, 113,10, 108,61, 88,15, 71,55, 60,34, 42,23, 40,54, 34,54, 29,69, 28,13, 26,42, 25,28, 24,92, 14,46. МСВР (HR, DIP-CI): Рассчитано: C30H40ClN2O2 (M+H)+: не обнаружено (разрушение в ходе анализа).
Стадия 3: Синтез сложного трет-бутилового эфира 5-[1-(2-хлорфенил)-7-этоксикарбонил-7-метилоктил]-4,5,6,7-тетрагидропирроло[3,2-c]пиридин-1-карбоновой кислоты.
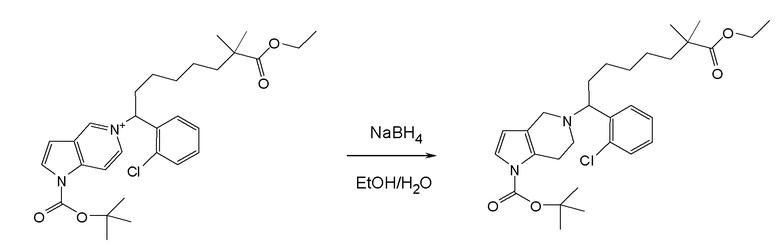
К раствору бромида 1-трет-бутоксикарбонил-5-[1-(2-хлорфенил)-7-метилоктил]-1H-пирроло[3,2-c]пиридин-5-ия (1,3 г, 2,14 ммоль) в 70% EtOH (30 мл) добавляли порциями борогидрид натрия (0,16 г, 4,28 ммоль) при комнатной температуре в течение 30 минут. После завершения добавления смесь перемешивали в течение 0,5 часов. Растворитель выпаривали при пониженном давлении и добавляли воду (60 мл). Продукт экстрагировали дихлорметаном (3×80 мл). Объединенные органические экстракты сушили над сульфатом натрия, фильтровали и концентрировали. Полученный третичный амин очищали посредством колоночной хроматографии (силикагель, этилацетат/гептан = 1/10-1/3) с получением сложного трет-бутилового эфира 5-[1-(2-хлорфенил)-7-этоксикарбонил-7-метилоктил]-4,5,6,7-тетрагидропирроло[3,2-c]пиридин-1-карбоновой кислоты (0,53 г, 46,9%) в виде желтого масла. 1H ЯМР (Поле: 300 МГц, Растворитель: CD3OD/ТМС) δ (ч/млн): 7,52 (д, 1H, J=7,8 Гц), 7,37 (д, 1H, J=7,8 Гц), 7,27 (т, 1H, J=6,6 Гц), 7,19 (т, 1H, J=7,5 Гц), 7,12 (д, 1H, J=3,3 Гц), 5,95 (д, 1H, J=3,3 Гц), 4,16-4,13 (м, 1H), 4,09 (кв, 2H, J=7,2 Гц), 3,31 (д, 1H, J=13,8 Гц), 2,85-2,59 (м, 4H), 1,95-1,76 (м, 2H), 1,55 (с, 9H), 1,45-1,40 (м, 2H), 1,28-1,14 (м, 6H), 1,21 (т, 3H, J=7,2 Гц), 1,11 (с, 6H). 13C ЯМР (Поле: 75 МГц, Растворитель: CDCl3/ТМС) δ (ч/млн): 178,00, 149,47, 139,70, 134,90, 129,45, 129,15, 127,89, 127,60, 126,80, 120,94, 119,76, 109,18, 83,24, 63,80, 60,30, 48,78, 42,32, 40,88, 33,20, 30,56, 28,31, 26,45, 25,44, 25,38, 25,03, 14,52. МСВР (HR): Рассчитано: C30H43ClN2O2 (M+H)+: 531,2990, обнаружено: 531,2933.
Стадия 4: Синтез сложного этилового эфира 8-(2-хлорфенил)-2,2-диметил-8-(1,4,6,7-тетрагидропирроло[3,2-c]пиридин-5-ил)октановой кислоты. (Соединение Ir)
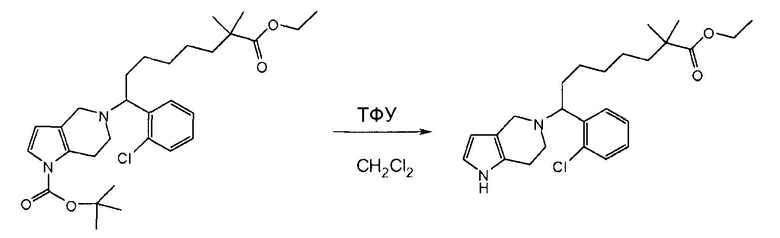
Сложный трет-бутиловый эфир 5-[1-(2-хлорфенил)-7-этоксикарбонил-7-метилоктил]-4,5,6,7-тетрагидропирроло[3,2-c]пиридин-1-карбоновой кислоты (0,12 г, 0,23 ммоль) растворяли в дихлорметане (5 мл) при перемешивании в атмосфере аргона. К раствору добавляли ТФУ (0,48 мл, 4,75 ммоль). Через 1 час посредством ТСХ наблюдали исчезновение материала. Смесь выливали в холодную воду (50 мл) и экстрагировали дихлорметаном (3×20 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали посредством колоночной хроматографии (силикагель, этилацетат/гептан = 1/9-1/3) с получением сложного этилового эфира 8-(2-хлорфенил)-2,2-диметил-8-(1,4,6,7-тетрагидропирроло[3,2-c]пиридин-5-ил)октановой кислоты (Соединение Ir, 65 мг, выход 65,9 %) в виде желтого масла. 1H ЯМР (Поле: 300 МГц, Растворитель: CD3OD/ТМС) δ (ч/млн): 7,80 (с, 1H), 7,54 (д, 1H, J=7,5 Гц), 7,37 (д, 1H, J=7,8 Гц), 7,26-7,16 (м, 2H), 6,60 (с, 1H), 5,94 (с, 1H), 4,17 (м, 1H), 4,08 (кв, 2H, J=7,2 Гц), 3,75 (д, 1H, J=13,2 Гц), 3,35 (д, 1H, J=13,2 Гц), 2,83-2,57 (м, 4H), 2,01-1,56 (м, 4H), 1,45-1,40 (м, 2H), 1,21 (т, 3H, J=7,2 Гц), 1,24-1,11 (м, 4H), 1,11 (с, 6H). 13C ЯМР (Поле: 75 МГц, Растворитель: CDCl3/ТМС) δ (ч/млн): 178,14, 140,07, 134,95, 129,43, 129,25, 127,82, 126,79, 125,12, 116,43, 115,93, 105,80, 63,89, 60,37, 48,79, 48,32, 42,37, 40,94, 33,34, 30,64, 25,55, 25,41, 25,08, 24,07, 14,56. МСВР (HR, DART-TOF): Рассчитано: C25H35ClN2O2 (M+H)+: 431,2460, обнаружено: 431,2476.
Стадия 5: Синтез 8-(2-хлорфенил)-2,2-диметил-8-(1,4,6,7-тетрагидропирроло[3,2-c]пиридин-5-ил)октановой кислоты (Соединение Iq).
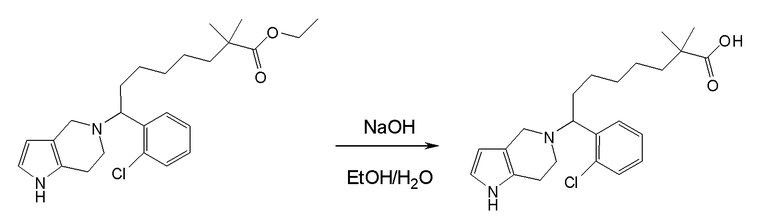
К смеси этанола (15 мл), воды (1,35 мл) и гидроксида натрия (0,07 г, 1,75 ммоль) добавляли сложный этиловый эфир 8-(2-хлорфенил)-2,2-диметил-8-(1,4,6,7-тетрагидропирроло[3,2-c]пиридин-5-ил)октановой кислоты (0,11 г, 0,25 ммоль). Указанную смесь нагревали до обратной конденсации (95-98°C на масляной бане) в течение 11 часов. После охлаждения до комнатной температуры растворитель выпаривали при пониженном давлении. К осадку добавляли воду (10 мл) и доводили значение pH до pH=6 10 н. раствором соляной кислоты. Продукт экстрагировали дихлорметаном (3×10 мл). Объединенные органические слои промывали водой (10 мл), солевым раствором (10 мл), сушили над Na2SO4. После фильтрования раствор дихлорметана концентрировали при пониженном давлении с получением неочищенного продукта. Указанный неочищенный продукт очищали посредством колоночной хроматографии (силикагель, этилацетат/гептан = 3/7) с получением 8-(2-хлорфенил)-2,2-диметил-8-(1,4,6,7-тетрагидропирроло[3,2-c]пиридин-5-ил)октановой кислоты (Соединение Iq, 0,06 г, выход 59,8%) в виде желтого масла. 1H ЯМР (Поле: 300 МГц, Растворитель: CD3OD/ТМС) δ (ч/млн): 7,60 (д, 1H, J=7,5 Гц), 7,48 (д, 1H, J=7,8 Гц), 7,41-7,30 (м, 2H), 6,58 (д, 1H, J=2,7 Гц), 5,89 (д, 1H, J=2,7 Гц), 4,56-4,52 (м, 1H), 3,96 (д, 1H, J=13,5 Гц), 3,65 (д, 1H, J=13,5 Гц), 2,97-2,65 (м, 4H), 2,00-1,87 (м, 2H), 1,43-1,39 (м, 2H), 1,28-1,18 (м, 6H), 1,11 (с, 6H). 13C ЯМР (Поле: 75 МГц, Растворитель: CD3OD /ТМС) δ (ч/млн) 183,09, 136,97, 136,77, 131,03, 130,05, 130,31, 128,80, 124,29, 118,35, 113,24, 105,71, 65,49, 50,79, 49,81, 43,48, 42,01, 33,13, 30,39, 26,74, 26,19, 25,98, 23,34.
Стадия 6: Синтез гидрохлорида 8-(2-хлорфенил)-2,2-диметил-8-(1,4,6,7-тетрагидропирроло[3,2-c]пиридин-5-ил)октановой кислоты.
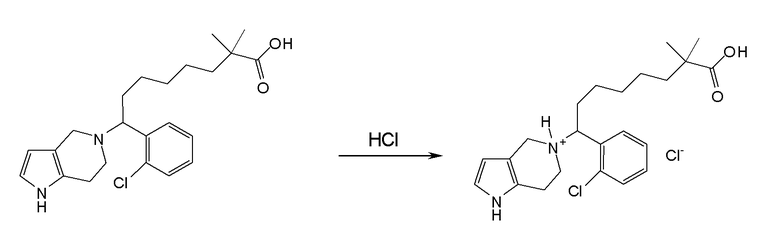
8-(2-Хлорфенил)-2,2 диметил-8-(1,4,6,7-тетрагидропирроло[3,2-c]пиридин-5-ил)октановую кислоту (0,09 г, 0,22 ммоль) растворяли в диэтиловом эфире (5 мл) и добавляли к раствору HCl (2 н. раствор HCl в диэтиловом эфире). Добавляли воду (5 мл) и после перемешивания отбирали органический слой. Водный раствор лиофилизировали с получением гидрохлорида 8-(2-хлорфенил)-2,2-диметил-8-(1,4,6,7-тетрагидро-пирроло[3,2-c]пиридин-5-ил)октановой кислоты (гидрохлорид Соединения Iq, 0,09 г, выход 86,5%) в виде порошка светло-желтого цвета. 1H ЯМР (Поле: 300 МГц, Растворитель: CD3OD/ТМС) δ (ч/млн): 10,28 (уш., 1H), 7,61 (уш.с, 1H), 7,48-7,40 (м, 3H), 6,57 (д, 1H, J=10,8 Гц), 5,86-5,70 (2с, 1H), 4,79-4,52 (м, 3H), 4,19-3,86 (м, 2H), 3,45-2,77 (м, 4H), 2,22-2,06 (м, 2H), 1,26-0,65 (м, 8H), 0,97 (с, 6H). (смесь конформационных изомеров, ЯМР). 13C ЯМР (Поле: 75 МГц, Растворитель: CDCl3/ТМС) δ (ч/млн) 181,66, 137,12, 132,64, 132,39, 129,68, 122,62, 119,72, 109,61, 105,95, 105,76, 66,09, 51,79, 50,73, 43,06, 41,57, 37,95, 31,80, 30,52, 26,59, 25,78, 21,67. Масс-спектрометрия высокого разрешения, МСВР (HR, DIP-CI): Рассчитано: C23H31ClN2O2 (M+H)+: 403,2147, обнаружено: 403,2158. CHN-анализ: Рассчитано; 62,87 C, 7,34 H, 6,37 N, 16,14 Cl, обнаружено:; 60,04 C, 7,05 H, 5,92 N, 15,88 Cl.
ПРИМЕР 9. Сложный метиловый эфир (3-карбоксиметилен-5-меркаптопиперидин-1-ил)-(2-хлорфенил)уксусной кислоты, гидрохлорид. (Гидрохлорид Соединения VIa)
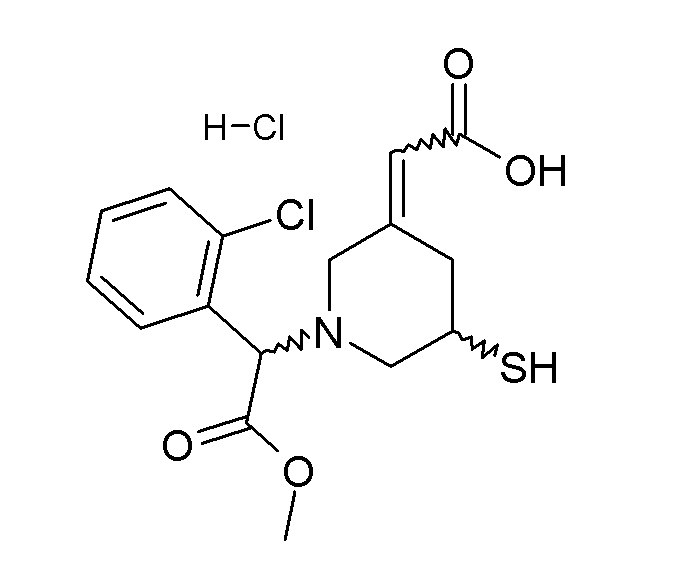
Стадия 1: Синтез сложного трет-бутилового эфира аллил(1-гидроксиаллил)карбаминовой кислоты.
К аллиламину (2, 16 мл) и воде (1 мл) добавляли моноксид бутадиена (1, 5,0 г, 71,3 ммоль) при охлаждении при 15°C. Смесь нагревали до обратной конденсации (100°C) в течение 6 часов. После охлаждения до комнатной температуры летучие вещества удаляли при пониженном давлении при комнатной температуре. Оставшееся масло, содержащее аллиламин, растворяли в диоксане (100 мл) и воде (20 мл). Колбу охлаждали на водяной бане и добавляли 1М раствор гидроксида натрия (80 мл). Добавляли ди-трет-бутилдикарбонат (17,95 г, 82,2 ммоль) и раствору позволяли перемешиваться в течение ночи при комнатной температуре. Через 18 часов основную часть диоксана удаляли при пониженном давлении. Оставшийся раствор вода/диоксан (40 мл) экстрагировали диэтиловым эфиром (2×100 мл). Эфир сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Оставшееся масло (19,0 г) очищали посредством колоночной хроматографии на силикагеле (250 г) с использованием в качестве элюента смеси гептан/этилацетат (4:1-1:1) с получением сложного трет-бутилового эфира аллил(1-гидроксиаллил)карбаминовой кислоты (10,5 г, выход 65%) в виде прозрачного масла. 1H ЯМР (300 МГц, CDCl3/ТМС): δ=5,82-5,67 (м, 2H), 5,28-5,02 (м, 4H), 4,25 (м, 1H), 3,80-3,65 (м, 2H), 3,22 (м, 1H), 1,39 (с, 9H). 13C ЯМР (75 МГц, CDCl3/ТМС): δ=138,52, 135,38, 133,88, 117,42, 116,34, 115,58, 80,30, 72,53, 63,42, 61,04, 53,41, 51,77, 28,50.
Стадия 2: Синтез сложного трет-бутилового эфира 3-гидрокси-3,6-дигидро-2H-пиридин-1-карбоновой кислоты.
Через сложный трет-бутиловый эфир аллил(1-гидроксиаллил)карбаминовой кислоты (5,0 г, 21,9 ммоль) в дихлорметане (350 мл) барботировали газообразный аргон в течение 5 минут. Колбу помещали в атмосферу аргона и добавляли катализатор «Grubb» I (0,48 г). Смесь перемешивали в течение ночи в атмосфере аргона при комнатной температуре. Через 18 часов раствор концентрировали и оставшееся масло очищали посредством колоночной хроматографии на силикагеле (150 г) с использованием в качестве элюента смеси гептан/этилацетат (4:1 - 1:1). Фракции, содержащие продукт, объединяли, концентрировали при пониженном давлении и сушили до достижения постоянной массы в высоком вакууме при комнатной температуре с получением сложного трет-бутилового эфира 3-гидрокси-3,6-дигидро-2H-пиридин-1-карбоновой кислоты (4,20 г, выход 96%) в виде густого коричневого масла. 1H ЯМР (300 МГц, CDCl3/ТМС): δ=5,85-5,65 (м, 2H), 4,13 (м, 1H), 3,80 (с, 2H), 3,66 (м, 1H), 3,26 (м, 1H), 1,39 (с, 9H). 13C ЯМР (75 МГц, CDCl3/ТМС): δ=155,06, 128,81, 126,79, 80,11, 67,53, 66,83, 63,60, 47,38, 43,36, 28,56.
Стадия 3: Синтез сложного трет-бутилового эфира 3-оксо-3,6-дигидро-2H-пиридин-1-карбоновой кислоты.
Сложный трет-бутиловый эфир 3-гидрокси-3,6-дигидро-2H-пиридин-1-карбоновой кислоты (3,1 г, 17,3 ммоль) растворяли в дихлорметане (30 мл) в атмосфере аргона. Порциями добавляли хлорхромат пиридиния (5 г, 23 ммоль) в течение 1 часа. Раствор дихлорметана фильтровали через силикагель (100 г) с использованием в качестве элюента дихлорметана. Неочищенное твердое вещество коричневого цвета (2,84 г) очищали посредством ЖХСД (Companion) на картридже, заполненном силикагелем (40 г), с использованием в качестве элюента 100% гептана и последующим использованием градиента этилацетат/гептан (0-60%). Фракции, содержащие продукт, объединяли, концентрировали и сушили в высоком вакууме в течение 1 часа при комнатной температуре с получением сложного трет-бутилового эфира 3-оксо-3,6-дигидро-2H-пиридин-1-карбоновой кислоты (2,1 г, выход 61%) в виде прозрачного масла, которое затвердевало при отстаивании при низкой температуре. 1H ЯМР (300 МГц, CDCl3/ТМС): δ=6,95 (м, 1H), 6,08 (д, 1H, J=10,5 Гц), 4,15 (м, 2H), 4,02 (уш.с, 2H), 1,39 (с, 9H). 13C ЯМР (75 МГц, CDCl3/ТМС): δ=193,11, 154,02, 147,17, 127,40, 80,89, 51,97, 42,69, 28,41.
Стадия 4: Синтез соли трифторуксусной кислоты 1,6-дигидро-2H-пиридин-3-она.
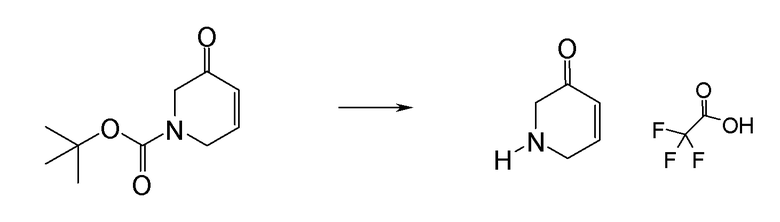
Сложный трет-бутиловый эфир 3-оксо-3,6-дигидро-2H-пиридин-1-карбоновой кислоты (0,42 г, 2,13 ммоль) растворяли в дихлорметане (5 мл) в атмосфере аргона. Добавляли трифторуксусную кислоту (1,5 мл) и смесь перемешивали в течение 4 часов при комнатной температуре. Раствор ТФУ/дихлорметан концентрировали при пониженном давлении и сушили в высоком вакууме в течение 1 часа при комнатной температуре. Оставшееся коричневое масло, содержащее соль трифторуксусной кислоты 1,6-дигидро-2H-пиридин-3-она (0,44 г, выход 97%), непосредственно использовали на следующей стадии. 1H ЯМР (300 МГц, CDCl3-CD3OD/ТМС): δ=7,12 (д, 1H, J=10,5 Гц), 6,8 (уш., 2H), 6,32 (д, 1H, J=10,5 Гц), 4,06 (м, 2H), 3,90 (уш.с, 2H). 13C ЯМР (75 МГц, CDCl3-CD3OD/ТМС): δ=187,85, 160,79 (кв, J=38 Гц), 132,63, 128,31, 115,71 (кв, J=284 Гц), 49,32, 41,11.
Стадия 5: Синтез сложного метилового эфира бром-(2-хлорфенил)уксусной кислоты.
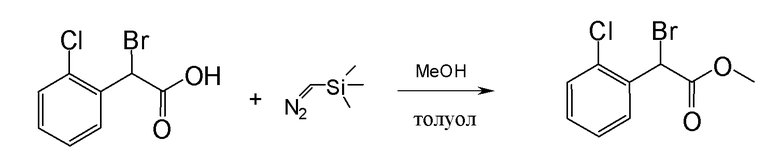
К толуолу (10 мл) добавляли метанол (1 мл) в атмосфере аргона при комнатной температуре. Колбу охлаждали на водяной бане и добавляли 2М (триметилсилил)диазометан (5 мл, 10 ммоль) с последующим добавлением порциями α-бром-2-хлорфенилуксусной кислоты (2,2 г, 8,81 ммоль) в течение 5 минут. Еще через 10 минут смесь толуол/метанол удаляли при пониженном давлении. Неочищенное масло очищали посредством ЖХСД (Companion) на картридже, заполненном силикагелем (40 г), с использованием градиента этилацетата в гептане (10-50%) в течение 20 минут. Фракции, содержащие продукт, объединяли, концентрировали и сушили в высоком вакууме в течение 1 часа при комнатной температуре с получением сложного метилового эфира бром-(2-хлорфенил)уксусной кислоты (2,0 г, выход 86%) в виде прозрачной жидкости, которая затвердевала при низких температурах (-10ºC). 1H ЯМР (300 МГц, CDCl3/ТМС): δ=7,75 (д, 1H, J=6,9 Гц), 7,39-7,26 (м, 3H), 5,91 (с, 1H), 3,80 (с, 3H). 13C ЯМР (75 МГц, CDCl3/ТМС): δ=168,29, 133,82, 133,30, 130,93, 130,47, 129,83, 127,66, 53,80, 43,08.
Стадия 6: Синтез сложного метилового эфира (2-хлорфенил)-(3-оксо-3,6-дигидро-2H-пиридин-1-ил)уксусной кислоты.
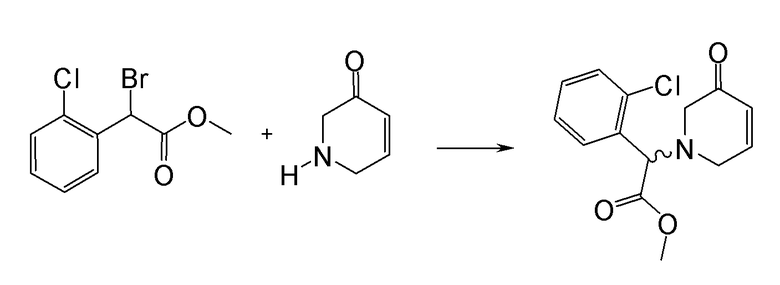
Соль трифторуксусной кислоты 1,6-дигидро-2H-пиридин-3-она, (1,27 г, 6,0 ммоль) и сложный метиловый эфир бром-(2-хлорфенил)уксусной кислоты (1,50 г, 6,15 ммоль) растворяли в ДМФА (5 мл) в атмосфере аргона при комнатной температуре. Добавляли карбонат калия (2 г, 14,5 ммоль) и смесь перемешивали в течение 3 часов при комнатной температуре. Добавляли воду (25 мл) и продукт экстрагировали дихлорметаном (2×25 мл). Экстракты дихлорметана объединяли, сушили над сульфатом натрия, фильтровали и концентрировали. Оставшееся масло очищали на силикагеле (30 г) с использованием в качестве элюента смеси гептан/этилацетат (3:1). Фракции, содержащие продукт, объединяли, концентрировали при пониженном давлении и сушили в высоком вакууме в течение 2 часов при комнатной температуре с получением сложного метилового эфира (2-хлорфенил)-(3-оксо-3,6-дигидро-2H-пиридин-1-ил)уксусной кислоты (0,50 г, выход 29,7%) в виде светло-желтого масла. 1H ЯМР (300 МГц, CDCl3/ТМС): δ=7,50-7,39 (м, 2H), 7,30-7,26 (м, 2H), 7,02-6,97 (м, 1H), 6,09 (д, 1H, J=10,5 Гц), 4,94 (с, 1H), 3,70 (с, 3H), 3,49 (м, 2H), 3,39 (д, 1H, J=15,9 Гц), 3,31 (д, 1H, J=15,9 Гц). 13C ЯМР (75 МГц, CDCl3/ТМС): δ=194,81, 170,41, 148,28, 134,74, 132,10, 129,95, 129,73, 129,69, 127,44, 126,99, 66,70, 57,89, 52,14, 49,16. МСВР (GC-CI): Рассчитано: (M+H+): 280,0740, обнаружено: 280,0727.
Стадия 7: Синтез сложного метилового эфира (2-хлорфенил)-(3-меркапто-5-оксопиперидин-1-ил)уксусной кислоты.
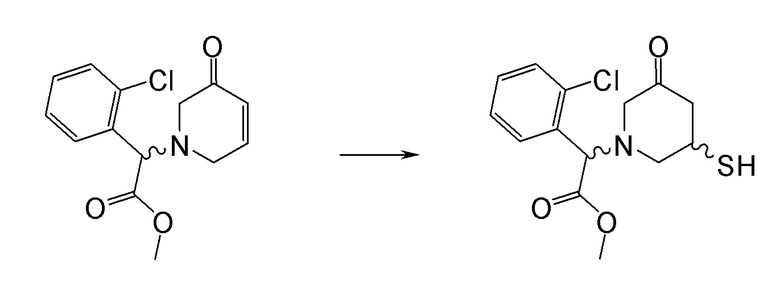
Сложный метиловый эфир (2-хлорфенил)-(3-оксо-3,6-дигидро-2H-пиридин-1-ил)уксусной кислоты (0,45 г, 1,6 ммоль) растворяли в метаноле (100 мл) в атмосфере аргона. Добавляли несколько капель триэтиламина и через указанный раствор пропускали газообразный аргон в течение 5 минут. Прекращали подачу аргона и через указанный раствор барботировали сероводород в течение 45 минут. Прекращали барботирование сероводорода и раствор помещали в атмосферу аргона на 30 дополнительных минут. Избыток сероводорода удаляли путем барботирования аргона через указанный раствор в течение 10 минут. Метанол удаляли при пониженном давлении и неочищенный сложный метиловый эфир (2-хлорфенил)-(3-меркапто-5-оксопиперидин-1-ил)уксусной кислоты (0,48 г, выход 96%, желтое стекло) использовали непосредственно на следующей стадии. 1H ЯМР (300 МГц, CDCl3/ТМС): δ=7,40-7,20 (м, 4H), 4,88 (3×с, 1H), 3,70 (с, 3H), 3,40-3,00 (м, 4H), 2,90-2,60 (м, 2H), 2,45-2,15 (м, 1H). 13C ЯМР (75 МГц, CDCl3/ТМС): δ=203,04, 170,75, 135,07, 132,49, 130,30, 129,96, 129,85, 127,04, 67,06, 60,38 (4 пика), 57,19 (2 пика), 52,29, 45,88 (3 пика), 39,06 (4 пика). МСВР (GC-CI): Рассчитано: (M+H+): 314,0618, обнаружено: 314,0588.
Стадия 8: Синтез сложного метилового эфира 3-трет-бутоксикарбонилметилен-5-меркаптопиперидин-1-ил)-(2-хлорфенил)уксусной кислоты. (Соединение VIb)
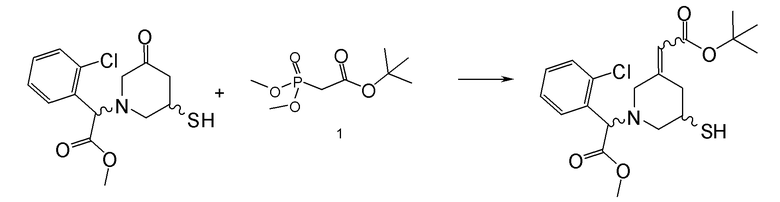
Неочищенный сложный метиловый эфир (2-хлорфенил)-(3-меркапто-5-оксопиперидин-1-ил)уксусной кислоты (0,36 г, 1,14 ммоль) растворяли в ТГФ (5 мл), через который барботировали аргон, и добавляли к смеси трет-бутил-P,P-диметилфосфоноацетата (0,50 г, 2,23 ммоль) и 60% гидрида натрия (0,075 г, 1,87 ммоль) в ТГФ (4 мл), через который барботировали аргон, при комнатной температуре в атмосфере аргона. Через 20 минут добавляли дихлорметан (25 мл), через который барботировали аргон, и смесь промывали водой (25 мл), через которую барботировали аргон. Раствор дихлорметана сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Оставшееся желтое масло очищали посредством колоночной хроматографии на силикагеле (15 г) с использованием в качестве элюента смеси гептан/этилацетат (4:1), через которую барботировали аргон, с получением сложного метилового эфира 3-трет-бутоксикарбонилметилен-5-меркаптопиперидин-1-ил)-(2-хлорфенил)уксусной кислоты (0,28 г, выход 59%) в виде прозрачного геля, который хранили в атмосфере аргона при низкой температуре для предотвращения окисления до дисульфида. 1H ЯМР (300 МГц, CDCl3/ТМС): δ=7,60-7,15 (м, 4H), 5,55 (м, 1H), 4,77 (м, 1H), 3,67 (с, 3H), 3,20-2,00 (м, 7H), 1,41 (м, 9H). 13C ЯМР (75 МГц, CDCl3/ТМС): δ=170,63 (2×), 165,26 (2×), 151,09 (3×), 134,64, 133,11 (4×), 129,83 (5×), 127,21, 118,35 (2×), 80,32 (2×), 67,57 (5×), 57,28 (6×), 52,28, 42,49-40,14 (8×), 34,54, 28,38 (2×). МСВР (DIP-CI): Рассчитано: (M+H+): 412,1349, обнаружено: 412,1312.
Стадия 9: Синтез гидрохлорида сложного метилового эфира (3-карбоксиметилен-5-меркаптопиперидин-1-ил)-(2-хлорфенил)уксусной кислоты.
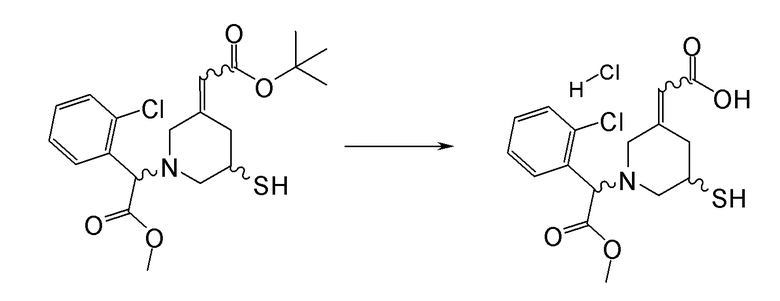
Сложный метиловый эфир 3-трет-бутоксикарбонилметилен-5-меркаптопиперидин-1-ил)-(2-хлорфенил)уксусной кислоты (0,18 г, 0,43 ммоль) растворяли в смеси дихлорметана и трифторуксусной кислоты (1:1, 6 мл), через которую барботировали аргон. Раствор перемешивали в течение 3 часов в атмосфере аргона при комнатной температуре. Добавляли дихлорметан (25 мл) и затем промывали 5% раствором бикарбоната натрия (2×25 мл), через который барботировали аргон. Дихлорметан сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Добавляли дихлорметан (5 мл), через который барботировали аргон, и получали соль путем добавления 2 н. раствора соляной кислоты в диэтиловом эфире (2 мл), через который барботировали аргон. Добавляли дополнительную порцию диэтилового эфира (25 мл), через которые барботировали аргон, и твердый осадок фильтровали и сушили в высоком вакууме в течение 3 часов при комнатной температуре с получением гидрохлорида сложного метилового эфира (3-карбоксиметилен-5-меркаптопиперидин-1-ил)-(2-хлорфенил)уксусной кислоты (гидрохлорид Соединения VIa, 0,12 г, выход 70%) в виде твердого вещества бледно-розового цвета. 1H ЯМР (300 МГц, ДМСО/ТМС): δ=7,65-7,25 (м, 4H), 5,71 (м, 1H), 4,91 (м, 1H), 3,66 (с, 3H), 3,40-2,00 (м, 7H). (смесь 8 изомеров). 13C ЯМР (75 МГц, ДМСО/ТМС): δ=169,60-165,99 (6 пиков), 194,52 (4 пика), 133,46-127,01 (13 пиков), 119,50-117,50 (6 пиков), 66,94-65,45 (7 пиков), 56,74-49,17 (12 пиков), 33,31 (уш.). МСВР (DIP-CI): Рассчитано: (M+H+): 356,0723, обнаружено: 356,0712. CHN-анализ: Рассчитано; 48,99 C, 4,88 H, 3,51 N, обнаружено: 49,25 C, 5,00 H, 3,57 N.
ПРИМЕР 10. 8-(2-Хлорфенил)-8-(6,7-дигидро-4H-тиено[3,2-c]пиридин-5-ил)-2,2-диметилоктановая кислота. (Соединение Im)
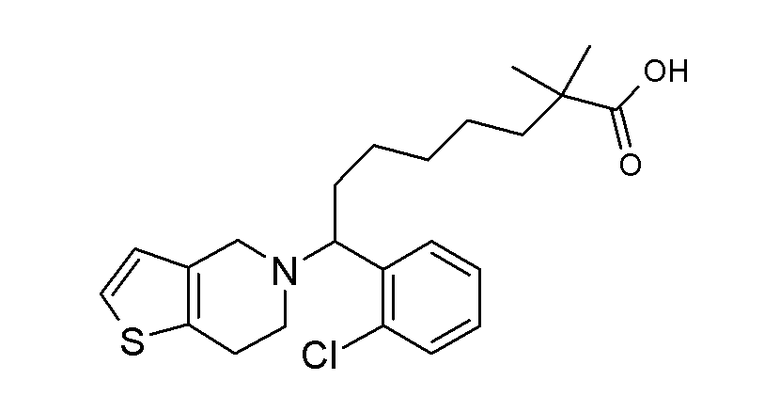
Стадия 1: Синтез бромида 6-этоксикарбонил-6-метилгептил)трифенилфосфония.
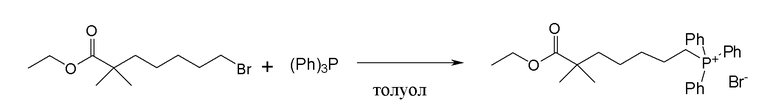
К раствору сложного этилового эфира 7-бром-2,2-диметилгептановой кислоты (15,0 г, 56,6 ммоль) в толуоле (110 мл) добавляли трифенилфосфин (14,8 г, 56,6 ммоль). Раствор нагревали до обратной конденсации (масляная баня, 122°C) в течение 24 часов. Толуол выпаривали и осадок промывали гептаном (2×60 мл), диэтиловом эфиром (2×60 мл) и сушили в высоком вакууме до достижения постоянной массы с получением бромида 6-этоксикарбонил-6-метилгептил)трифенилфосфония (24,39 г, выход 81,8%) в виде порошка не совсем белого цвета (температура плавления 165-170°C). 1H ЯМР (Поле: 300 МГц, Растворитель: CDCl3/ТМС) δ (ч/млн): 7,88-7,82 (м, 9H), 7,75-7,72 (м, 6H), 4,06 (кв, 2H, J=6,9 Гц), 3,76 (м, 2H), 1,64 (м, 4H), 1,46-1,41 (м, 2H), 1,20 (т, 5H, J=6,9 Гц), 1,10 (с, 6H). 13C ЯМР (Поле: 75 МГц, Растворитель: CDCl3/ТМС) δ (ч/млн): 177,82, 134,99, 133,69 (д, J=10 Гц), 130,57 (д, J=12 Гц), 117,69 (д, J=85 Гц), 60,29, 42,12, 40,09, 30,94 (д, J=16 Гц), 25,26 (д, J=41 Гц), 23,18 (д, J=41 Гц), 14,40. МСВР (FIA-ESI-TOFM)): Рассчитано: C29H36BrO2P (M+H)+ 447,2447, обнаружено: 447,2446.
Стадия 2: Синтез сложного этилового эфира 8-(2-хлорфенил)-2,2-диметилокт-7-еновой кислоты.
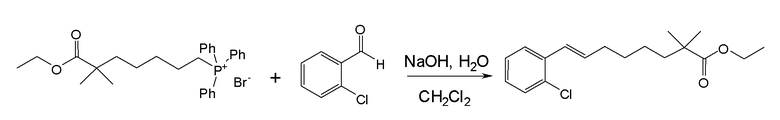
Бромид 6-этоксикарбонил-6-метилгептил)трифенилфосфония (24 г, 45,5 ммоль) и 2-хлорбензальдегид (6,38 г, 45,5 ммоль) в CH2Cl2 (60 мл) перемешивали по возможности максимально интенсивно и добавляли по каплям 50% раствор NaOH (24 мл). После завершения добавления смесь продолжали перемешивать в течение 3 часов. Смесь переносили в сепаратор и промывали дихлорметаном (200 мл) и водой (200 мл). Водную фазу экстрагировали дихлорметаном (3×150 мл). Объединенные экстракты дихлорметана промывали солевым раствором (150 мл), сушили над Na2SO4, концентрировали и очищали посредством колоночной хроматографии (силикагель, этилацетат/гептан = 1/10-1/6) с получением сложного этилового эфира 8-(2-хлорфенил)-2,2-диметилокт-7-еновой кислоты (10 г, выход 71,6%) в виде желтого масла. 1H ЯМР (Поле: 300 МГц, Растворитель: CDCl3/ТМС) δ (ч/млн): 7,46 (дд, 1H, J=7,5, 1,5 Гц), 7,36-7,06 (м, 3H), 6,49 (д, 1H, J=11,4 Гц), 5,74 (м, 1H), 4,09 (м, 2H), 2,20 (м, 2H), 1,58-1,36 (м, 4H), 1,21 (м, 5H), 1,13 (с, 6H). (основной изомер). 13C ЯМР (Поле: 75 МГц, Растворитель: CDCl3/ТМС) δ (ч/млн): 177,76, 135,78, 134,00, 133,69, 130,48, 129,52, 127,94, 126,69, 126,27, 60,25, 42,26, 40,68, 33,14, 30,27, 28,46, 25,28, 24,73, 14,32. (основной изомер). МСВР (FIA-ESI-TOFM): Рассчитано: C18H25ClO2 (M+Na): 331,1435, обнаружено: 331,1446.
Стадия 3: Синтез сложного этилового эфира 8-бром-8-(2-хлорфенил)2,2-диметилоктановой кислоты.
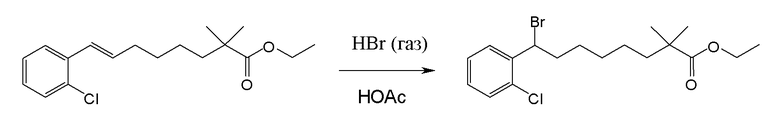
Сложный этиловый эфир 8-(2-хлорфенил)-2,2-диметилокт-7-еновой кислоты (7 г, 22,8 ммоль) растворяли в ледяной уксусной кислоте (60 мл). Раствор охлаждали на ледяной бане (примерно 15°C) по мере барботирования сухого бромоводорода через указанный раствор в течение 8 часов. Реакционную смесь выливали в смесь лед/вода (130 мл) и экстрагировали этилацетатом (3×50 мл). Объединенные органические слои промывали насыщенным раствором NaHCO3 (100 мл), сушили над Na2SO4 и концентрировали при пониженном давлении. Неочищенный продукт (10 г) очищали посредством колоночной хроматографии (200 г силикагеля, с использованием в качестве элюента смеси этилацетат/гептан = 1/20-1/10) с получением сложного этилового эфира 8-бром-8-(2-хлорфенил)2,2-диметилоктановой кислоты (7,46 г, выход 81,8%) в виде желтого масла. 1H ЯМР (Поле: 300 МГц, Растворитель: CDCl3/ТМС) δ (ч/млн):7,59 (д, 1H, J=7,8 Гц), 7,34 (т, 1H, J=7,5 Гц), 7,26 (д, 1H, J=6,9 Гц), 7,19 (т, 1H, J=7,2 Гц), 5,45 (т, 1H, J=7,5 Гц), 4,09 (кв, 2H, J=7,2 Гц), 2,26-2,09 (м, 4H), 1,52-1,46 (м, 2H), 1,41-1,31 (м, 2H), 1,28-1,18 (м, 2H), 1,20 (т, 3H, J=7,2 Гц), 1,14 (с, 6H). 13C ЯМР (Поле: 75 МГц, Растворитель: CDCl3/ТМС) δ (ч/млн): 177,80, 139,43, 132,65, 129,64, 129,24, 128,88, 127,45, 60,28, 50,30, 42,25, 39,21, 29,48, 27,98, 25,36, 24,88, 14,49. МСВР (GC-Cl): Рассчитано: C18H26BrClO2 (M+H)+ 389,0883, обнаружено: 389,0867.
Стадия 4: Синтез сложного этилового эфира 8-(2-хлорфенил)-8-(6,7-дигидро-4H-тиено[3,2-c]пиридин-5-ил)-2,2-диметилоктановой кислоты. (Соединение In)
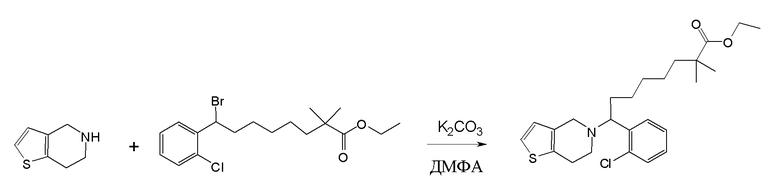
К гидроксиду натрия (1,38 г) в воде (86 мл) добавляли гидрохлорид 4,5,6,7-тетрагидротиено[3,2-c]пиридина (1,30 г, 7,19 ммоль) и экстрагировали дихлорметаном (3×20 мл). Дихлорметан сушили над сульфатом натрия, фильтровали и концентрировали с получением свободного основания (1,0 г). 4,5,6,7-Тетрагидротиено[3,2-c]пиридин в виде свободного основания (1,0 г, 7,19 ммоль) и сложный этиловый эфир 8-бром-8-(2-хлорфенил)2,2-диметилоктановой кислоты (2,8 г, 7,19 ммоль) растворяли в ДМФА (75 мл), содержащем карбонат калия (1,49 г, 10,79 ммоль). Смесь нагревали до 65-70°C в течение ночи. Через 18 часов смесь охлаждали до комнатной температуры и добавляли воду (50 мл). Продукт экстрагировали диэтиловым эфиром (3×50 мл). Объединенные органические экстракты промывали водой (3×50 мл), сушили над сульфатом натрия и концентрировали при пониженном давлении. Неочищенный продукт очищали посредством колоночной хроматографии на силикагеле с использованием в качестве элюента смеси этилацетат/гептан (1/10) с получением сложного этилового эфира 8-(2-хлорфенил)-8-(6,7-дигидро-4H-тиено[3,2-c]пиридин-5-ил)-2,2-диметилоктановой кислоты (Соединение In, 1,4 г, 43,5%) в виде желтого масла. 1H ЯМР (Поле: 300 МГц, Растворитель: CDCl3/ТМС) δ (ч/млн):7,49 (дд, 1H, J=7,5, 1,5 Гц), 7,35 (дд, 1H, J=8,1, 1,2 Гц), 7,24 (дт, 1H, J=7,5, 1,0 Гц), 7,16 (дт, 1H, J=7,8, 1,5 Гц), 7,02 (д, 1H, J=5,0 Гц), 6,68 (д, 1H, J=5,0 Гц), 4,18 (дд, 1H, J=8,7, 4,2 Гц), 4,08 (кв, 2H, J=6,9 Гц), 3,82 (д, 1H, J=14,1 Гц), 3,48 (д, 1H, J=14,1 Гц), 2,88-2,66 (м, 4H), 1,96-1,76 (м, 2H), 1,46-1,41 (м, 2H), 1,26-1,18 (м, 6H), 1,21 (т, 3H, J=6,9 Гц), 1,11 (с, 6H). 13C ЯМР (Поле: 75 МГц, Растворитель: CDCl3/ТМС) δ (ч/млн): 177,92, 134,92, 134,21, 133,53, 129,48, 129,06, 127,94, 126,77, 125,44, 122,60, 63,67, 60,27, 50,73, 48,08, 42,29, 40,85, 33,02, 30,52, 26,13, 25,39, 25,01, 14,51. МСВР (FIA-ESI-TOFM): Рассчитано: C25H34ClNO2S (M+H)+: 448,2072, обнаружено: 448,2067.
Стадия 5: Синтез 8-(2-хлорфенил)-8-(6,7-дигидро-4H-тиено[3,2-c]пиридин-5-ил)-2,2-диметилоктановой кислоты (Соединение Im).
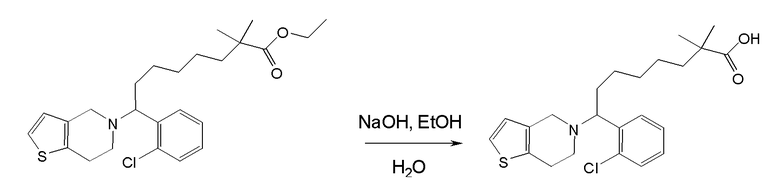
Сложный этиловый эфир 8-(2-хлорфенил)-8-(6,7-дигидро-4H-тиено[3,2-c]пиридин-5-ил)-2,2-диметилоктановой кислоты (0,86 г, 1,92 ммоль) добавляли к раствору этанола (32 мл) и гидроксида натрия (0,53 г, 13,4 ммоль) в воде (10,4 мл). Смесь нагревали до обратной конденсации в течение 6,5 часов. Раствор концентрировали при пониженном давлении и к осадку добавляли воду (43 мл). Весь исходный материал экстрагировали смесью этилацетат/гептан (1/10, 43 мл). Экстракт гептана отбирали. Значение pH оставшегося водного раствора доводили до pH=6 10 н. раствором соляной кислоты. Продукт экстрагировали CH2Cl2 (3×40 мл), сушили над Na2SO4, концентрировали и очищали посредством колоночной хроматографии (силикагель, этилацетат/гептан = 1/10-1/3) с получением 8-(2-хлорфенил)-8-(6,7-дигидро-4H-тиено[3,2-c]пиридин-5-ил)-2,2-диметилоктановой кислоты (Соединение Im, 0,35 г, выход 43,8%, чистота 99,6%, ВЭЖХ) в виде желтого масла. 1H ЯМР (Поле: 300 МГц, Растворитель: CDCl3/ТМС) δ (ч/млн):10,43 (уш., 1H), 7,52 (д, 1H, J=7,50 Гц), 7,39 (д, 1H, J=8,10 Гц), 7,28-7,15 (м, 2H), 7,03 (д, 1H, J=5,0 Гц), 6,69 (д, 1H, J=5,0 Гц), 4,24 дд, 1H, J=9,3, 4,2 Гц), 3,83 (д, 1H, J=14,40 Гц), 3,51 (д, 1H, J=14,40 Гц), 2,92-2,73 (м, 4H), 1,99-1,81 (м, 2H), 1,46-1,41 (м, 2H), 1,28-1,15 (м, 6H), 1,13 (с, 6H). (т, 3H, J=7,2 Гц), 1,15-1,08 (м, 2H), 1,07 (с, 6H). 13C ЯМР (Поле: 75 МГц, Растворитель: CDCl3/ТМС) δ (ч/млн): 184,02, 138,85, 135,16, 133,87, 133,42, 129,60, 129,17, 128,17, 126,92, 125,52, 122,74, 63,53, 50,54, 47,90, 42,29, 40,70, 32,95, 30,52, 25,75, 25,64, 25,26, 24,96. МСВР (FIA-ESI): Рассчитано: C23H30ClNO2S (M+H)+: 420,1759, обнаружено: 420,1779. CHN-анализ: Рассчитано; 65,77 C, 7,20 H, 3,33 N, обнаружено:; 60,77 C, 6,68 H, 3,11 N. Оптимальное соотношение CHN: C23H30ClNO2S+HCl
ПРИМЕР 11. Гидрохлорид 7-(2-хлорфенил)-7-(6,7-дигидро-4H-фуро[3,2-c]пиридин-5-ил)-2,2-диметилгептановой кислоты. (Гидрохлорид Соединения Ii)
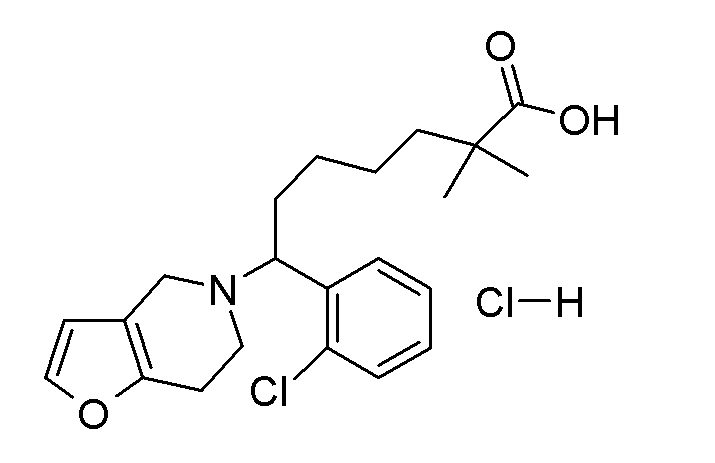
Стадия 1: Синтез сложного этилового эфира 7-(2-хлорфенил)-7-(6,7-дигидро-4H-фуро[3,2-c]пиридин-5-ил)-2,2-диметилгептановой кислоты. (Соединение Ij)
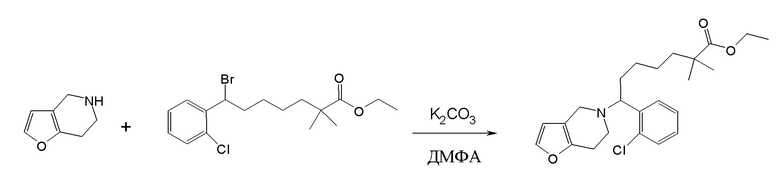
4,5,6,7-Тетрагидрофуро[3,2-c]пиридин (0,16 г, 1,3 ммоль), сложный этиловый эфир 7-бром-7-(2-хлорфенил)-2,2-диметилгептановой кислоты (0,49 г, 1,3 ммоль) в ДМФА (13 мл) и карбонат калия (0,27 г, 1,95 ммоль) объединяли в атмосфере аргона при комнатной температуре. Полученную смесь нагревали до 65°C в течение ночи в атмосфере аргона. Реакцию продолжали при 65°C в течение 48 часов. После охлаждения до комнатной температуры и концентрирования при пониженном давлении, неочищенный продукт (0,28 г) очищали посредством колоночной хроматографии (силикагель, смесь этилацетат/гептан = 1/20-1/5) с получением сложного этилового эфира 7-(2-хлорфенил)-7-(6,7-дигидро-4H-фуро[3,2-c]пиридин-5-ил)-2,2-диметилгептановой кислоты (Соединение Ij, 0,26 г, выход 48,1%) в виде желтого масла. 1H ЯМР (Поле: 300 МГц, Растворитель: CDCl3/ТМС) δ (ч/млн): 7,42 (д, 1H, J=7,5 Гц), 7,29 (д, 1H, J=7,2 Гц), 7,17 (т, 1H, J=8,7 Гц), 7,14 (с, 1H), 7,09 (т, 1H, J=8,1 Гц), 6,08 (с, 1H), 4,12 (т, 1H, J=4,2 Гц), 4,01 (кв, 2H, J=7,2 Гц), 3,98 (д, 1H, J=14,4 Гц), 3,26 (д, 1H, J=13,5 Гц), 2,79-2,53 (м, 4H), 1,90-1,73 (м, 2H), 1,35-1,32 (м, 2H), 1,20-1,06 (м, 7H), 1,04 (с, 6H). 13C ЯМР (Поле: 75 МГц, Растворитель: CDCl3/ТМС) δ (ч/млн): 177,91, 148,97, 140,89, 139,39, 134,93, 129,50, 129,00, 128,02, 126,84, 115,82, 108,84, 63,33, 60,54, 47,61, 47,51, 42,28, 40,78, 33,21, 26,23, 25,35, 24,65, 14,50.
Стадия 2: Синтез гидрохлорида 7-(2-хлорфенил)-7-(6,7-дигидро-4H-фуро[3,2-c]пиридин-5-ил)-2,2-диметилгептановой кислоты.
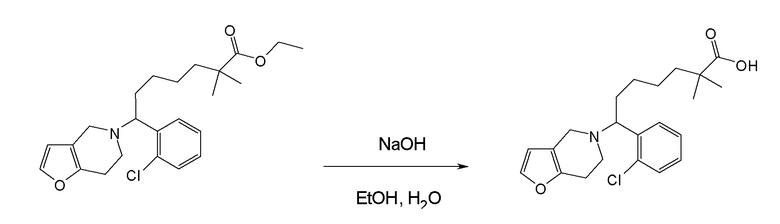
Сложный этиловый эфир 7-(2-хлорфенил)-7-(6,7-дигидро-4H-фуро[3,2-c]пиридин-5-ил)-2,2-диметилгептановой кислоты (0,26 г, 0,62 ммоль) добавляли к смеси раствора этанола (10 мл) и гидроксида натрия (0,17 г, 4,4 ммоль) в воде (3,3 мл), смесь нагревали до обратной конденсации в течение 6,5 часов, после чего посредством ТСХ наблюдали исчезновение материала. Этанол удаляли при пониженном давлении и к осадку добавляли дополнительную порцию воды (15 мл). Водную фазу промывали смесью этилацетат/гептан 1/10 (10 мл), которую затем отбирали. pH водной фракции доводили до значения pH=6 концентрированной соляной кислотой. Продукт экстрагировали дихлорметаном (3×15 мл). Объединенные слои дихлорметана сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Получали продукт, представляющий собой кислоту (0,17 г, выход 70,1%, чистота 97,23% по данным ВЭЖХ) в виде желтого масла. Порцию материала (0,15 г, 0,38 ммоль) растворяли в диэтиловом эфире (5 мл) и добавляли к 2 н. раствору HCl в эфире (0,21 мл). Твердый осадок экстрагировали водой (10 мл) и лиофилизировали с получением 7-(2-хлорфенил)-7-(6,7-дигидро-4H-фуро[3,2-c]пиридин-5-ил)-2,2-диметилгептановой кислоты (гидрохлорид Соединения Ii, 0,11 г, выход 68,8%, чистота 99,08%, ВЭЖХ) в виде порошка светло-желтого цвета. 1H ЯМР (Поле: 300 МГц, Растворитель: CD3OD/ТМС) δ (ч/млн): 7,80 (уш.с, 1H), 7,61-7,48 (м, 4H), 6,43 (с, 0,5H), 6,29 (с, 0,5H), 5,07 (м, 1H), 4,68 (д, 0,5H, J=13,8 Гц), 4,30 (д, 0,5H, J=13,8 Гц), 4,25-3,95 (м, 1H), 3,64-3,40 (м, 1H), 3,09-2,95 (м, 2H), 2,40-2,27 (м, 2H), 1,46-0,87 (м, 6H), 1,11 (с, 6H), 1,09 (уш.с, 1H). (смесь ротаизомеров). 13C ЯМР (Поле: 75 МГц, Растворитель: CD3OD/ТМС) δ (ч/млн): 181,47, 147,11, 141,52, 137,12, 132,84, 132,09 131,86, 130,50, 129,75, 112,35, 109,69, 66,20 (уш.), 43,03, 41,40, 33,10, 31,71, 30,18, 27,36, 25,92, 25,70, 25,63, 23,80, 22,26, 14,50. (смесь ротаизомеров). МСВР (DIP-CI): Рассчитано: C22H28ClNO3 (M+H)+: 390,1830, обнаружено: 390,1792. CHN-анализ: Рассчитано: C22H29NCl2O3S: 61,97 C, 6,86 H, 3,28 N, 16,61 Cl; обнаружено: 60,28 C, 6,88 H, 3,13 N, 16,24 Cl.
ПРИМЕР 12. Гидрохлорид 7-(2-хлорфенил)-2,2-диметил-7-(1,4,6,7-тетрагидропирроло[3,2-c]пиридин-5-ил)гептановой кислоты. (Гидрохлорид Соединения Ik)
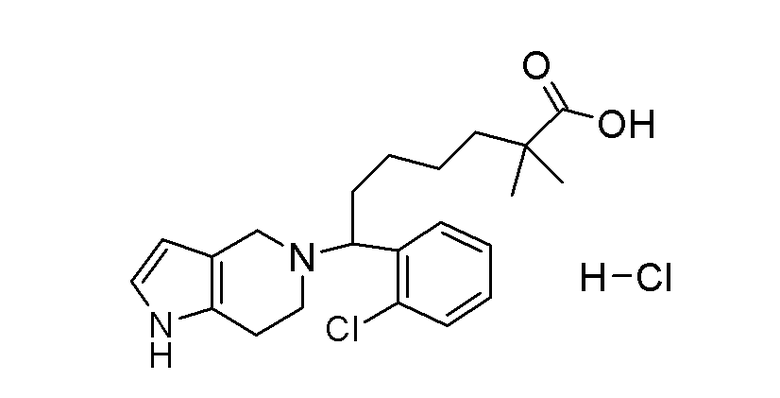
Стадия 1: Синтез бромида 1-трет-бутоксикарбонил-5-[1-(2-хлорфенил)-6-метилгептил]-1 H-пирроло[3,2-c]пиридин-5-ия.
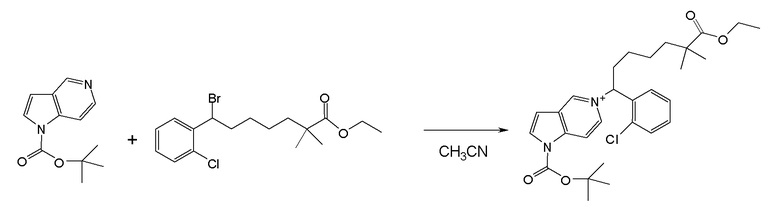
Смесь трет-бутил-1H-пирроло[3,2-c]пиридин-1-карбоксилата (0,81 г, 3,73 ммоль), сложного этилового эфира 7-бром-7-(2-хлорфенил)-2,2-диметилоктановой кислоты (1,40 г, 3,73 ммоль) в ацетонитриле (25 мл) перемешивали и нагревали до 45°C. Через 52 часа реакция завершалась. Растворитель выпаривали при пониженном давлении, осадок промывали диэтиловым эфиром (3×20 мл) с получением бромида 1-трет-бутоксикарбонил-5-[1-(2-хлорфенил)-6-метилгептил]-1 H-пирроло[3,2-c]пиридин-5-ия (1,47 г, выход 66,5%) в виде желтого масла. 1H ЯМР (Поле: 300 МГц, Растворитель: CD3OD/ТМС) δ (ч/млн): 10,54 (с, 1H), 8,84 (д, 1H, J=6,9 Гц), 8,23 (д, 1H, J=7,8 Гц), 7,72 (с, 1H), 7,53 (т, 1H, J=4,2 Гц), 7,45 (т, 1H, J=7,2 Гц), 7,39 (д, 1H, J=3,6 Гц), 7,31 (с, 1H), 6,96 (с, 1H), 6,47 (т, 1H, J=7,2 Гц), 4,10 (кв, 2H, J=5,7 Гц), 2,72 (д, 2H, J=6,6 Гц), 2,40 (м, 1H), 1,69 (с, 9H), 1,50-1,32 (м, 4H), 1,21 (т, 3H, J=3,3 Гц), 1,12 (с, 6H). (неочищенная смесь. МСВР (DIP-CI): данные не получены (разрушение в ходе анализа).
Стадия 2: Синтез сложного трет-бутилового эфира 5-[1-(2-хлорфенил)-6-этоксикарбонил-6-метилгептил]-4,5,6,7-тетрагидро-пирроло[3,2-c]пиридин-1-карбоновой кислоты.
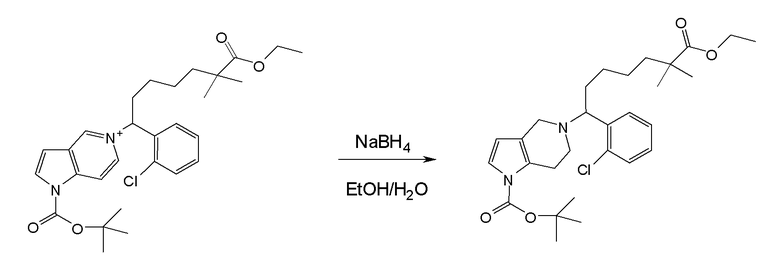
К раствору бромида 1-трет-бутоксикарбонил-5-[1-(2-хлорфенил)-6-метилгептил]-1 H-пирроло[3,2-c]пиридин-5-ия (2,4 г, 4,0 ммоль) в 70% EtOH (60 мл) порциями добавляли борогидрид натрия (0,30 г, 8,0 ммоль) при комнатной температуре и интенсивном перемешивании. После завершения добавления борогидрида натрия прекращали интенсивное перемешивание и полученную смесь перемешивали в течение 0,5 часов. Растворитель выпаривали при пониженном давлении и добавляли воду (60 мл). Продукт экстрагировали дихлорметаном (3 × 150 мл) и объединенные органические экстракты сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Неочищенный осадок очищали посредством колоночной хроматографии (силикагель, смесь этилацетат/гептан = 1/10-1/3) с получением сложного трет-бутилового эфира 5-[1-(2-хлорфенил)-6-этоксикарбонил-6-метилгептил]-4,5,6,7-тетрагидропирроло[3,2-c]пиридин-1-карбоновой кислоты (1,45 г, выход 60,9%) в виде желтого масла. 1H ЯМР (Поле: 300 МГц, Растворитель: CD3OD/ТМС) δ (ч/млн): 7,50 (дд, 1H, J=7,5, 1,2 Гц), 7,37 (д, 1H, J=8,1, 1,5 Гц), 7,27-7,10 (м, 3H), 5,94 (д, 1H, J=3,3 Гц), 4,16-4,13 (м, 1H), 4,09 (кв, 2H, J=6,9 Гц), 3,62 (д, 1H, J=14,1 Гц), 3,28 (д, 1H, J=13,5 Гц), 2,85-2,60 (м, 4H), 1,98-1,76 (м, 2H), 1,55 (с, 9H), 1,42 (т, 2H, J=6,9 Гц), 1,28-1,16 (м, 4H), 1,21 (т, 3H, J=7,2 Гц), 1,11 (с, 6H). 13C ЯМР (Поле: 75 МГц, Растворитель: CDCl3/ТМС) δ (ч/млн): 177,98, 149,49, 139,66, 134,90, 129,44, 129,15, 127,93, 127,60, 126,84, 120,92, 119,77, 109,20, 83,26, 63,76, 60,33, 48,81, 48,19, 42,30, 40,80, 33,23, 32,13, 28,31, 26,07, 25,35, 22,96, 14,52. МСВР (DART-TOF): Рассчитано: C29H41ClN2O2 (M+H)+: 517,2828, обнаружено: 517,2833.
Стадия 3: Синтез сложного этилового эфира 7-(2-хлорфенил)-2,2 диметил-7-(1,4,6,7-тетрагидро-пирроло[3,2-c]пиридин-5-ил)гептановой кислоты. (Соединение Il)
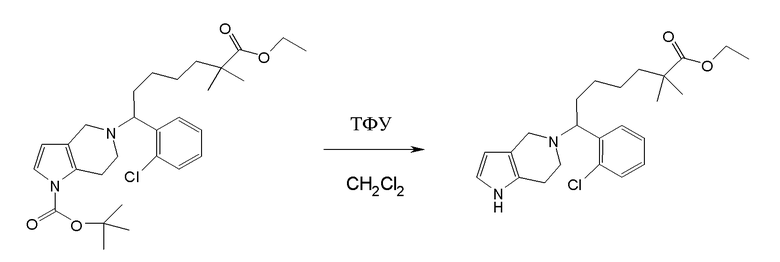
Сложный трет-бутиловый эфир 5-[1-(2-хлорфенил)-6-этоксикарбонил-6-метилгептил]-4,5,6,7-тетрагидропирроло[3,2-c]пиридин-1-карбоновой кислоты (1,0 г, 1,94 ммоль) растворяли в дихлорметане (40 мл) в атмосфере аргона. К раствору добавляли трифторуксусную кислоту (4,0 мл, 40,7 ммоль). Через 2 часа указанную смесь выливали в смесь лед/вода (90 мл) и экстрагировали дихлорметаном (3×20 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали посредством колоночной хроматографии (силикагель, этилацетат/гептан = 1/9-1/3) с получением сложного этилового эфира 7-(2-хлорфенил)-2,2-диметил-7-(1,4,6,7-тетрагидропирроло[3,2-c]пиридин-5-ил)гептановой кислоты (Соединение Il, 330 мг, выход 41,2%) в виде желтого масла. 1H ЯМР (Поле: 300 МГц, Растворитель: CDCl3/ТМС) δ (ч/млн): 7,93 (уш., 1H), 7,53 (д, 1H, J=7,5 Гц), 7,35 (д, 1H, J=7,8 Гц), 7,26-7,13 (м, 2H), 6,58 (с, 1H), 5,93 (с, 1H), 4,16 (дд, 1H, J=9,3, 3,9 Гц), 4,10 (кв, 2H, J=7,2 Гц), 3,75 (д, 1H, J=13,5 Гц), 3,39 (д, 1H, J=13,2 Гц), 2,80-2,56 (м, 4H), 2,04-1,78 (м, 2H), 1,44-1,39 (м, 2H), 1,21 (т, 3H, J=6,9 Гц), 1,26-1,16 (м, 4H), 1,11 (с, 6H). 13C ЯМР (Поле: 75 МГц, Растворитель: CDCl3/ТМС) δ (ч/млн): 178,01, 139,87, 134,94, 129,40, 129,22, 127,86, 126,81, 125,01, 116,45, 115,70, 105,69, 63,80, 60,37, 48,78, 48,29, 42,31, 40,81, 33,29, 26,15, 25,36, 23,95, 14,52. МСВР (DART-TOF): Рассчитано: C24H33ClN2O2 (M+H)+: 717,2303, обнаружено: 417,2295.
Стадия 4: Синтез 7-(2-хлорфенил)-2,2-диметил-7-(1,4,6,7-тетрагидропирроло[3,2-c]пиридин-5-ил)гептановой кислоты. (Соединение Ik)
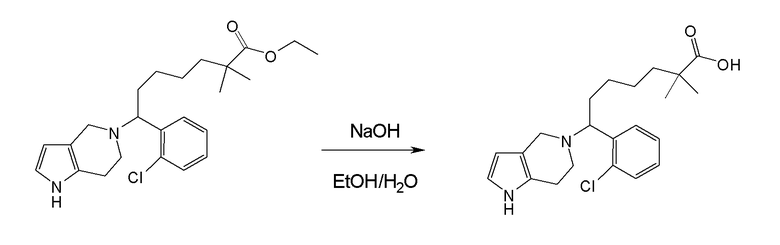
Сложный этиловый эфир 7-(2-хлорфенил)-2,2-диметил-7-(1,4,6,7-тетрагидропирроло[3,2-c]пиридин-5-ил)гептановой кислоты (0,21 г, 0,51 ммоль) добавляли к смеси этанола (10 мл), воды (2,70 мл) и гидроксида натрия (0,14 г, 3,54 ммоль). Полученную смесь нагревали до обратной конденсации (95-98°C, масляная баня) в течение 16 часов. Через 26 часов колбу охлаждали до комнатной температуры и растворитель выпаривали при пониженном давлении. К осадку добавляли воду (15 мл) и продукт экстрагировали диэтиловым эфиром (15 мл). Экстракт отбирали и pH водной фракции доводили до pH=6 10 н. раствором соляной кислоты. Продукт экстрагировали дихлорметаном (3×15 мл) и объединенные слои дихлорметана промывали водой (20 мл) и солевым раствором (20 мл). Раствор дихлорметана сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали посредством колоночной хроматографии (силикагель, MeOH/CH2Cl2=1/9) с получением 7-(2-хлорфенил)-2,2 диметил-7-(1,4,6,7-тетрагидропирроло[3,2-c]пиридин-5-ил)гептановой кислоты (Соединение Ik, 0,11 г, выход 57,9%) в виде желтого масла. 1H ЯМР (Поле: 300 МГц, Растворитель: ДМСО/ТМС) δ (ч/млн): 12,10 (уш, 1H), 10,30 (уш, 1H), 7,53 (м, 1H), 7,43 (д, 1H, J=7,2 Гц), 7,36-7,25 (м, 2H), 6,50 (с, 1H), 5,71 (с, 1H), 4,10 (м, 1H), 3,39 (м, 5H), 2,75 (м, 1H), 1,14 (м, 2H), 1,34 (м, 2H), 1,23-1,07 (м, 4H), 1,03 (с, 6H). 13C ЯМР (Поле: 75 МГц, Растворитель: ДМСО /ТМС) δ (ч/млн) 178,45, 178,97, 133,83, 129,07, 128,23, 126,93, 123,97, 115,83, 113,99, 104,19, 63,11, 48,27, 47,60, 41,19, 40,06, 31,59, 25,84, 25,03, 24,96, 24,55, 23,34.
Стадия 5: Синтез гидрохлорида 7-(2-хлорфенил)-2,2-диметил-7-(1,4,6,7-тетрагидропирроло[3,2-c]пиридин-5-ил)гептановой кислоты.
7-(2-Хлорфенил)-2,2-диметил-7-(1,4,6,7-тетрагидро-пирроло[3,2-c]пиридин-5-ил)гептановую кислоту (0,26 г, 0,67 ммоль) растворяли в диэтиловом эфире (15 мл) и добавляли раствор соляной кислоты (2 н. HCl в диэтиловом эфире). Раствор эфира экстрагировали водой (15 мл) и органический слой отбирали. Водный раствор лиофилизировали с получением гидрохлорида 7-(2-хлорфенил)-2,2-диметил-7-(1,4,6,7-тетрагидропирроло[3,2-c]пиридин-5-ил)гептановой кислоты (гидрохлорид Соединения Ik, 0,19 г, выход 61,9%) в виде порошка светло-желтого цвета (чистота 97,33%, ВЭЖХ, температура плавления 168-172°C). 1H ЯМР (Поле: 300 МГц, Растворитель: CD3OD/ТМС) δ (ч/млн): 10,26 (уш.с, 1H), 7,65 (уш.с, 1H), 7,45-7,38 (м, 4H), 6,54-6,50 (2с, 1H), 5,84 (с, 0,5H), 5,66 (с, 0,5H), 4,79 (м, 1H), 4,51 (м, 1H), 4,14 -3,82 (м, 2H), 3,43-2,76 (м, 4H), 2,43-2,09 (м, 2H), 1,25-1,14 (м, 4H), 1,39-0,65 (м, 2H), 0,96 (с, 6H). (смесь конформационных изомеров). 13C ЯМР (Поле: 75 МГц, Растворитель: CDCl3/ТМС) δ (ч/млн) 181,52, 137,17, 132,64, 132,32, 131,75, 130,09, 129,79, 122,85, 119,88, 109,70, 106,00, 65,79, 51,79, 51,10, 42,98, 41,43, 31,84, 27,28, 25,096, 25,76, 21,78. МС (HR, DIP-CI): Рассчитано: C22H30Cl2N2O2 (M+H)+: 389,1994, обнаружено: 389,1996. CHN-анализ: Рассчитано; 62,12 C, 7,11 H, 6,59 N, 16,67Cl, обнаружено:; 59,23 C, 7,04 H, 6,26 N, 16,93 Cl.
ПРИМЕР 13. 6-[1-(2-Диметиламинопиримидин-5-илметил)пиперидин-4-ил]-2-морфолин-4-ил-пиримидин-4-ол, Соединения IIa
Стадия 1: Синтез сложного трет-бутилового эфира 4-(2-этоксикарбонилацетил)пиперидин-1-карбоновой кислоты.
Сложный 4-этиловый эфир 1-трет-бутилового эфира пиперидин-1,4-дикарбоновой кислоты (0,50 г, 1,94 ммоль) в ДМФА (5 мл) смешивали с этилацетатом (0,38 мл, 3,88 ммоль) и трет-бутоксидом калия (0,33 г, 2,92 ммоль). Смесь нагревали до 50ºC в течение 20 часов. После охлаждения до комнатной температуры добавляли воду (50 мл) и продукт экстрагировали диэтиловым эфиром. Экстракт эфира сушили над сульфатом натрия, фильтровали и концентрировали. Неочищенный продукт очищали посредством колоночной хроматографии на силикагеле с использованием в качестве элюента смеси гептан/этилацетат с получением сложного трет-бутилового эфира 4-(2-этоксикарбонилацетил)пиперидин-1-карбоновой кислоты (0,27 г, выход 47%). 1H ЯМР (300 МГц, CDCl3/ТМС): δ=4,20 (кв, 2H, J=7,2 Гц), 4,18-4,05 (м, 2H), 3,50 (с, 2H), 2,86-2,70 (м, 2H), 2,68-2,55 (м, 1H), 1,90-1,78 (м, 2H), 1,60-1,50 (м, 2H), 1,45 (с, 9H), 1,28 (т, 3H, J=7,2 Гц). 13C ЯМР (75 МГц, CDCl3/ТМС): δ=204,21, 167,20, 154,67, 79,91, 61,70, 48,93, 47,56, 43,41, 28,73, 27,56, 14,46.
Стадия 2: Синтез сложного трет-бутилового эфира 4-(6-гидрокси-2-морфолин-4-илпиримидин-4-ил)пиперидин-1-карбоновой кислоты.
Гидробромид морфолин-4-карбоксамидина (0,267 г) и сложный трет-бутиловый эфир 4-(2-этоксикарбонилацетил)пиперидин-1-карбоновой кислоты (0,38 г, 1,27 ммоль) перемешивали в этаноле (10 мл) и добавляли диазобициклоундекан (285 мкл). Реакционную смесь перемешивали в течение 18 часов при комнатной температуре. Удаляли этанол при пониженном давлении и добавляли воду (25 мл). Раствор подкисляли (до pH=4) уксусной кислотой. Продукт экстрагировали дихлорметаном (4×30 мл). Дихлорметан удаляли и неочищенный продукт дважды очищали на силикагеле (Combiflash), с использованием в качестве элюента 10% метанола в дихлорметане с получением сложного трет-бутилового эфира 4-(6-гидрокси-2-морфолин-4-илпиримидин-4-ил)пиперидин-1-карбоновой кислоты (0,15 г, выход 40,5%) в виде твердого вещества белого цвета. 1H ЯМР (300 МГц, CDCl3/ТМС): δ=5,63 (с, 1H), 4,25-4,05 (м, 2H), 3,75 (м, 8H), 2,75-2,65 (м, 2H), 2,47-2,35 (м, 1H), 1,00-1,75 (м, 2H), 1,70-1,50 (м, 2H), 1,46 (с, 9H). 13C ЯМР (75 МГц, CDCl3/ТМС): δ=173,35, 166,87, 154,96, 154,06, 98,88, 79,70, 66,72, 45,13, 44,24, 30,64, 28,82, 27,58.
Стадия 3: Синтез гидрохлорида 2-морфолин-4-ил-6-пиперидин-4-ил-пиримидин-4-ола.
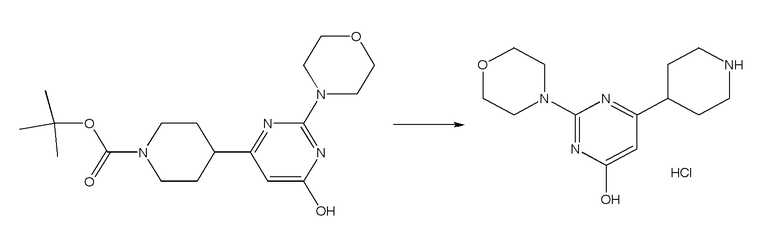
К 2 н. раствору соляной кислоты в диэтиловом эфире добавляли сложный трет-бутиловый эфир 4-(6-гидрокси-2-морфолин-4-илпиримидин-4-ил)пиперидин-1-карбоновой кислоты (0,19 г, 0,52 ммоль) и перемешивали в течение 18 часов при комнатной температуре. Полученный продукт выделяли посредством фильтрования и промывали эфиром с получением гидрохлорида 2-морфолин-4-ил-6-пиперидин-4-ил-пиримидин-4-ола (0,16 г, выход 91%) в виде твердого вещества белого цвета. 1H ЯМР (300 МГц, CD3OD/ТМС): δ=6,17 (с, 1H), 3,90-3,78 (м, 8H), 3,60-3,50 (м, 2H), 3,26-3,1 (м, 3H), 2,32-2,20 (м, 2H), 2,00-1,80 (м, 2H). 13C ЯМР (75 МГц, CD3OD/ТМС): δ=171,60, 164,86, 155,10, 97,63, 66,93, 45,13, 46,92, 44,81, 38,29, 28,41.
Стадия 4: Синтез 6-[1-(2-диметиламинопиримидин-5-илметил)пиперидин-4-ил]-2-морфолин-4-илпиримидин-4-ола.
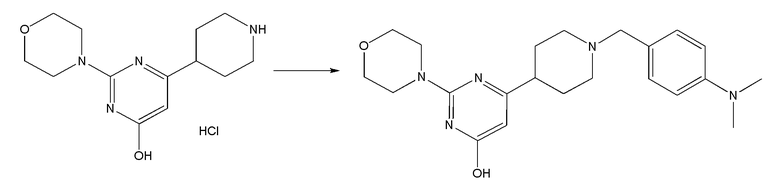
Гидрохлорид 2-морфолин-4-ил-6-пиперидин-4-ил-пиримидин-4-ола (0,16 г, 0,47 ммоль), 2-диметиламинопиримидин-5-карбоксальдегид (86 мг, 0,57 ммоль) и одну каплю уксусной кислоты перемешивали в безводном дихлорметане (10 мл) в течение 3 часов при комнатной температуре. Добавляли цианоборогидрид натрия (88 мг, 1,42 ммоль) и смесь перемешивали в течение 3 дней. Смесь выливали в насыщенный раствор бикарбоната натрия и экстрагировали дихлорметаном (3×50 мл). Материал очищали на силикагеле с использованием в качестве элюента 10% метанола в дихлорметане с получением 6-[1-(2-диметиламинопиримидин-5-илметил)пиперидин-4-ил]-2-морфолин-4-илпиримидин-4-ола (Соединение IIa, 0,11 г, выход 69%) в виде твердого вещества белого цвета. CHN-анализ: Рассчитано: C20H29N7O2: 60,13 C, 7,32 H, 24,54 N; обнаружено: 57,31 C, 7,11 H, 23,68 H. Оптимальное соотношение C20H29N7O2 + 1H2O; 57,54 C, 7,48 H, 23,48 N. 1H ЯМР (300 МГц, CDCl3/CD3OD/ТМС): δ=8,26 (с, 2H), 5,65 (с, 1H), 3,85-3,60 (м, 8H), 3,42 (м, 2H), 3,19 (с, 6H), 3,15-2,85 (м, 3H), 2,40-2,00 (м, 2H), 2,18-1,60 (м, 4H). 13C ЯМР (75 МГц, CDCl3/CD3OD/ТМС): δ=173,66, 161,63, 158,91, 153,90, 116,63, 98,76, 66,56, 57,62, 53,12, 44,94, 43,65, 37,48, 30,26.
ПРИМЕР 14. Получение N-(2,3-дигидробензо[1,4]диоксин-6-ил)-2-(2-метоксиэтиламино)ацетамида, Соединения XIa
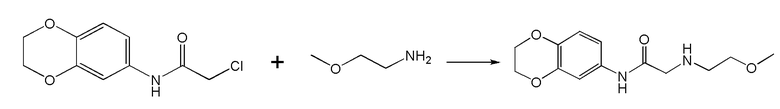
Смесь 2-хлор-N-(2,3-дигидро-1,4-бензодиоксин-6-ил)ацетамида (2 ммоль), карбоната калия (10 ммоль), 2-метоксиэтиламина (2 ммоль) и безводного ДМФА (4 мл) перемешивали в течение 2 часов при 100ºC (ход реакции контролировали посредством ТСХ). Смесь охлаждали до комнатной температуры, обрабатывали холодной водой (30 мл). Осадок собирали посредством фильтрования, промывали диэтиловом эфиром и сушили с получением N-(2,3-дигидробензо[1,4]диоксин-6-ил)-2-(2-метокси-этиламино)ацетамида (Соединение XIa, 96 мг, 36%). 1H ЯМР (300 МГц, CDCl3/ТМС): δ=9,29 (с, 1H), 7,24 (д, 1H, J=2,4 Гц), 6,97 (дд, 1H, J=8,7, 2,4 Гц), 6,78 (д, 1H, J=8,7 Гц), 4,32-4,15 (м, 4H), 3,46 (т, 2H, J=4,8 Гц), 3,36 (с, 3H), 3,34 (с, 2H), 2,82 (т, 2H, J=4,8 Гц), 2,17 (с, 1H). 13C ЯМР (75 МГц, CDCl3/CD3OD/ТМС): δ=169,48, 143,16, 139,87, 131,43, 116,84, 112,74, 108,89, 71,41, 64,28, 64,12, 58,68, 52,57, 49,35.
ПРИМЕР 15. Получение 6-[1-(3-Гидрокси-4-метоксибензил)-пиперидин-4-ил]-2-пиперидин-1-ил-пиримидин-4-ола, Соединения IIb
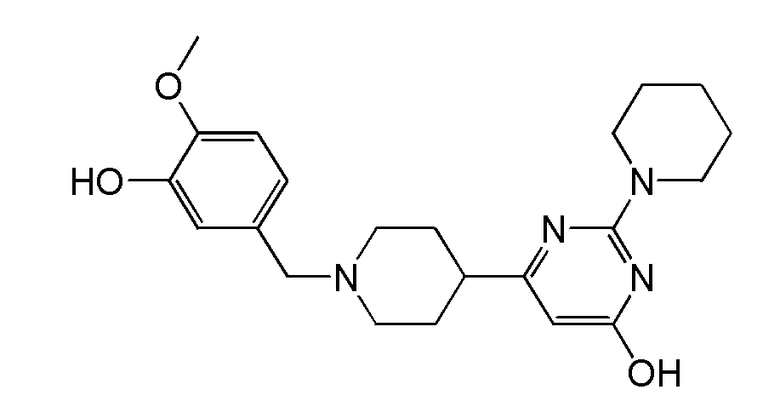
Стадия 1: Синтез сложного трет-бутилового эфира 4-(2-этоксикарбонилацетил)пиперидин-1-карбоновой кислоты.
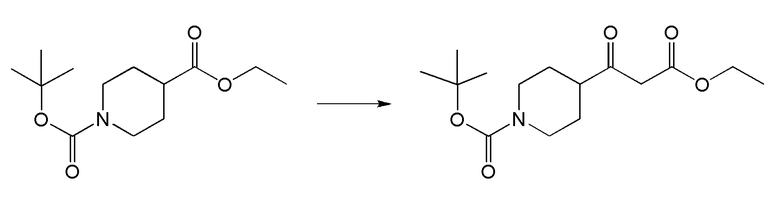
Сложный 4-этиловый эфир 1-трет-бутилового эфира пиперидин-1,4-дикарбоновой кислоты (0,50 г, 1,94 ммоль) в ДМФА (5 мл) смешивали с этилацетатом (0,38 мл, 3,88 ммоль) и трет-бутоксидом калия (0,33 г, 2,92 ммоль). Смесь нагревали до 50ºC в течение 20 часов. После охлаждения до комнатной температуры добавляли воду (50 мл) и продукт экстрагировали диэтиловым эфиром. Экстракт эфира сушили над сульфатом натрия, фильтровали и концентрировали. Неочищенный продукт очищали посредством колоночной хроматографии на силикагеле с использованием в качестве элюента смеси гептан/этилацетат с получением сложного трет-бутилового эфира 4-(2-этоксикарбонилацетил)пиперидин-1-карбоновой кислоты (0,27 г, выход 47%).
Стадия 2: Синтез сложного трет-бутилового эфира 4-(6-гидрокси-2-пиперидин-1-илпиримидин-4-ил)-пиперидин-1-карбоновой кислоты.
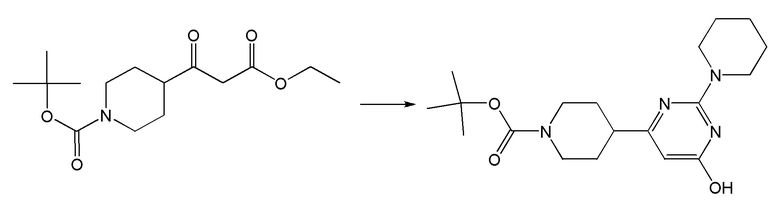
Натрий (1 моль) растворяли в безводном этаноле (400 мл). К полученному раствору порциями осторожно добавляли пиперидин-1-карбоксамидин (0,5 моль). Затем добавляли по каплям сложный трет-бутиловый эфир 4-(2-этоксикарбонилацетил)пиперидин-1-карбоновой кислоты (0,5 моль) и смесь перемешивали при температуре обратной конденсации в течение 4-6 часов (ход реакции контролировали посредством ТСХ), охлаждали до комнатной температуры, концентрировали, разбавляли водой (300 мл) и подкисляли уксусной кислотой до pH-4. Образовавшийся осадок собирали посредством фильтрования, промывали водой и сушили с получением сложного трет-бутилового эфира 4-(6-гидрокси-2-пиперидин-1-илпиримидин-4-ил)пиперидин-1-карбоновой кислоты (89 г, 49%).
Стадия 3: Синтез гидрохлорида 6-пиперидин-4-ил-2-пиперидин-1-илпиримидин-4-ола.
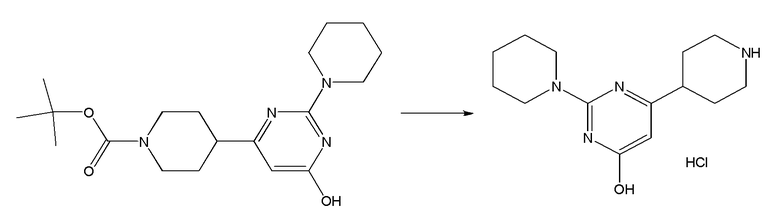
Суспензию сложного трет-бутилового эфира 4-(6-гидрокси-2-пиперидин-1-илпиримидин-4-ил)пиперидин-1-карбоновой кислоты (0,05 моль) в 15% HCl в диоксане (100 мл) перемешивали при температуре обратной конденсации в течение 2 часов. После завершения реакции смесь охлаждали, осадок фильтровали, промывали сухим эфиром и сушили с получением гидрохлорида 6-пиперидин-4-ил-2-пиперидин-1-илпиримидин-4-ола (12 г, 81%).
Стадия 4: Синтез 6-[1-(3-гидрокси-4-метоксибензил)пиперидин-4-ил]-2-пиперидин-1-ил-пиримидин-4-ола.
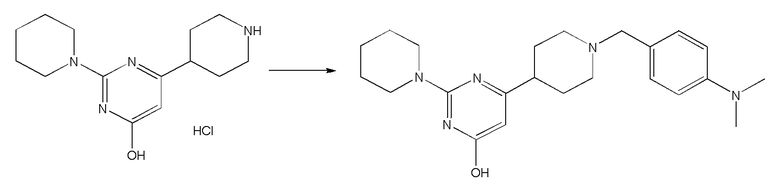
Смесь гидрохлорида 6-пиперидин-4-ил-2-пиперидин-1-ил-пиримидин-4-ола (2,0 ммоль), 3-гидрокси-4-метоксибензальдегида (2,6 ммоль), триэтиламина (4,0 ммоль) и 3 капли уксусной кислоты в безводным дихлорметане (20 мл) перемешивали при комнатной температуре в течение 3 часов. Затем порциями добавляли триацетоксиборогидрид натрия (6,0 ммоль) и продолжали перемешивать в течение 48 часов (ход реакции контролировали посредством ТСХ). Реакцию гасили насыщенным водным раствором бикарбоната натрия (20 мл) и продукт экстрагировали дихлорметаном (2×10 мл). Экстракты промывали солевым раствором и сушили над сульфатом натрия. Растворитель выпаривали в вакууме с получением неочищенного продукта. В результате очистки посредством колоночной хроматографии (силикагель, смесь этилацетат/гексан) получали 6-[1-(3-гидрокси-4-метоксибензил)пиперидин-4-ил]-2-пиперидин-1-ил-пиримидин-4-ол (Соединение IIb, 542 мг, 68%).
ПРИМЕР 16. Получение 6-(1-(4-(метиламино)бензил)пиперидин-4-ил)-2-(пиперидин-1-ил)пиримидин-4-ола, Соединения IIc
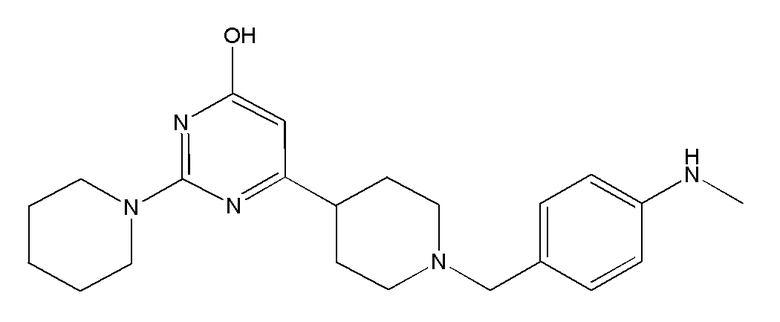
Стадия 1: Синтез сложного трет-бутилового эфира 4-(2-этоксикарбонилацетил)пиперидин-1-карбоновой кислоты.
Сложный 4-этиловый эфир 1-трет-бутилового эфира пиперидин-1,4-дикарбоновой кислоты (0,50 г, 1,94 ммоль) в ДМФА (5 мл) смешивали с этилацетатом (0,38 мл, 3,88 ммоль) и трет-бутоксидом калия (0,33 г, 2,92 ммоль). Смесь нагревали до 50ºC в течение 20 часов. После охлаждения до комнатной температуры добавляли воду (50 мл) и продукт экстрагировали диэтиловым эфиром. Экстракт эфира сушили над сульфатом натрия, фильтровали и концентрировали. Неочищенный продукт очищали посредством колоночной хроматографии на силикагеле с использованием в качестве элюента смеси гептан/этилацетат с получением сложного трет-бутилового эфира 4-(2-этоксикарбонилацетил)пиперидин-1-карбоновой кислоты (0,27 г, выход 47%).
Стадия 2: Синтез трет-бутил-4-(6-гидрокси-2-(пиперидин-1-ил)пиримидин-4-ил)пиперидин-1-карбоксилата
Натрий (1 моль) растворяли в безводном этаноле (400 мл). К полученному раствору порциями осторожно добавляли пиперидин-1-карбоксамидин (0,5 моль). Затем добавляли по каплям сложный трет-бутиловый эфир 4-(2-этоксикарбонил-ацетил)пиперидин-1-карбоновой кислоты (0,5 моль) и смесь перемешивали при температуре обратной конденсации в течение 4-6 часов (ход реакции контролировали посредством ТСХ), охлаждали до комнатной температуры, концентрировали, разбавляли водой (300 мл) и подкисляли уксусной кислотой до pH-4. Образовавшийся осадок собирали посредством фильтрования, промывали водой и сушили с получением трет-бутилового эфира 4-(6-гидрокси-2-пиперидин-1-ил-пиримидин-4-ил)-пиперидин-1-карбоновой кислоты (80 г, 44%).
Стадия 3: Синтез гидрохлорида 2-(пиперидин-1-ил)-6-(пиперидин-4-ил)пиримидин-4-ола.
Суспензию сложного трет-бутилового эфира 4-(6-гидрокси-2-пиперидин-1-илпиримидин-4-ил)пиперидин-1-карбоновой кислоты (0,05 моль) в 15% HCl в диоксане (100 мл) перемешивали при температуре обратной конденсации в течение 2 часов. После завершения реакции смесь охлаждали, осадок фильтровали, промывали безводным эфиром и сушили с получением гидрохлорида 2-морфолин-4-ил-6-пиперидин-4-ил-2-пиперидин-1-ил-пиримидин-4-ола (12 г, 82%).
Стадия 4: Синтез 6-(1-(4-(метиламино)бензил)пиперидин-4-ил)-2-(пиперидин-1-ил)пиримидин-4-ола.
Смесь гидрохлорида 6-пиперидин-4-ил-2-пиперидин-1-ил-пиримидин-4-ола (2,0 ммоль), 2-метиламинопиримидин-5-карбальдегида (2,6 ммоль), триэтиламина (4,0 ммоль) и 3 капли уксусной кислоты в безводном дихлорметане (20 мл) перемешивали при комнатной температуре в течение 3 часов. Затем порциями добавляли триацетоксиборогидрид натрия (6,0 ммоль) и продолжали перемешивать в течение 48 часов (ход реакции контролировали посредством ТСХ). Реакцию гасили насыщенным водным раствором бикарбоната натрия (20 мл) и продукт экстрагировали дихлорметаном (2×10 мл). Экстракты промывали солевым раствором и сушили над сульфатом натрия. Растворитель выпаривали в вакууме с получением неочищенного продукта. В результате очистки посредством колоночной хроматографии (силикагель, смесь этилацетат/гексан) получали 6-[1-(2-метиламинопиримидин-5-илметил)пиперидин-4-ил]-2-пиперидин-1-ил-пиримидин-4-ол (Соединение IIc, 637 мг, 83%).
ПРИМЕР 17. Получение изохроман-1-ил(5'H-спиро[пиперидин-4,4'-пирроло[1,2-a]хиноксалин]-1-ил)метанона, Соединения VIIa
Стадия 1: Синтез сложного трет-бутилового эфира 4,5-дигидропирроло[1,2-a]хиноксалин-спиро-4-пиперидин-1-карбоновой кислоты.
2-Пиррол-1-илфениламин (0,05 моль) и N-Boc-пиперидон (0,05 моль) растворяли в этаноле (50 мл), в качестве катализатора добавляли моногидрат п-толуолсульфоновой кислоты, перемешиваемую реакционную смесь нагревали до обратной конденсации в течение 2-3 часов в атмосфере аргона. Ход реакции контролировали посредством ТСХ. Образовавшийся продукт фильтровали, промывали холодным этанолом и сушили с получением сложного трет-бутилового эфира 4,5-дигидропирроло[1,2-a]хиноксалин-спиро-4-пиперидин-1-карбоновой кислоты (7 г, 44%). 1H ЯМР (300 МГц, CDCl3/ТМС) δ 7,30 (д, J=7,5 Гц, 1H), 7,18-7,12 (м, 1H), 6,97 (т, J=7,4 Гц, 1H), 6,84 (т, J=7,6 Гц, 1H), 6,78 (д, J=8,1 Гц, 1H), 6,30 (т, J=3,0 Гц, 1H), 6,07-6,02 (м, 1H), 4,21 (с, 1H), 3,95-3,77 (м, 2H), 3,30-3,17 (м, 2H), 2,15-1,90 (м, 2H), 1,90-1,78 (м, 2H), 1,47 (с, 9H). 13C ЯМР (75 МГц, CDCl3/ТМС) δ 154,55, 133,87, 133,03, 125,53, 124,59, 119,45, 116,06, 114,49, 114,26, 109,83, 102,62, 79,69, 51,10, 39,58, 35,50, 28,38.
Стадия 2: Синтез свободного амина.
Сложный трет-бутиловый эфир 4,5-дигидропирроло[1,2-a]хиноксалин-спиро-4-пиперидин-1-карбоновой кислоты (0,1 моль) растворяли в изопропаноле (100 мл) и нагревали до обратной конденсации. К интенсивно перемешиваемой смеси добавляли по каплям 30-40 мл HCl (14-16%) в диоксане. Наблюдали выделение газа. Образовывался продукт, представляющий собой гидрохлорид, в виде осадка белого цвета. Смесь нагревали до обратной конденсации в течение 30-40 минут для завершения реакции. В результате фильтрования получали гидрохлорид 4,5-дигидропирроло[1,2-a]хиноксалин-спиро-4-пиперидина. Указанный гидрохлорид 4,5-дигидропирроло[1,2-a]хиноксалин-спиро-4-пиперидина растворяли в воде (50 мл) и реакцию гасили твердым карбонатом натрия до достижения pH 7-8. Осадок собирали посредством фильтрования с получением 4,5-дигидропирроло[1,2-a]хиноксалинспиро-4-пиперидина (22 г 94%) в виде свободного основания. 1H ЯМР (300 МГц, CDCl3/ТМС) δ 7,28 (д, J=7,8 Гц, 1H), 7,15-7,11 (м, 1H), 6,96 (т, J=7,5 Гц, 1H), 6,86-6,76 (м, 2H), 6,29 (т, J=3,0 Гц, 1H), 6,08-6,04 (м, 1H), 4,39 (с, 1H), 3,69 (с, 1H), 3,07-2,85 (м, 4H), 2,03-1,90 (м, 2H), 1,90-1,79 (м, 2H). 13C ЯМР (75 МГц, CDCl3/ТМС) δ 134,13, 134,01, 125,45, 124,49, 119,11, 115,88, 114,41, 114,02, 109,77, 102,53, 66,96, 51,26, 42,07, 36,54.
Стадия 3: Синтез изохроман-1-ил(5'H-спиро[пиперидин-4,4'-пирроло[1,2-a]хиноксалин]-1-ил)метанона.
Смесь 4,5-дигидропирроло[1,2-a]хиноксалин-спиро-4-пиперидина (2,0 ммоль), триэтиламина (2 ммоль), изохроман-1-карбоновой кислоты (2 ммоль) и гексафторфосфата бензотриазол-1-илокси-трис(диметиламино)фосфония (2 ммоль) в дихлорметане (10 мл) перемешивали при комнатной температуре в течение 16 часов. Реакцию гасили насыщенным водным раствором бикарбоната натрия (10 мл), перемешивали в течение 2 часов и продукт экстрагировали дихлорметаном (2×10 мл). Объединенные органические слои сушили над сульфатом натрия и концентрировали при пониженном давлении с получением неочищенного продукта. В результате очистки посредством колоночной хроматографии (силикагель, смесь этилацетат/гексан) получали изохроман-1-ил(5'H-спиро[пиперидин-4,4'-пирроло[1,2-a]хиноксалин]-1-ил)метанон. 1H ЯМР (300 МГц, CDCl3/ТМС) δ 7,31-7,05 (м, 6H), 6,96 (дд, J=13,6, 6,1 Гц, 1H), 6,87-6,71 (м, 2H), 6,30 (т, J=3,1 Гц, 0,5H), 6,25 (т, J=3,1 Гц, 0,5H), 6,07 (д, J=2,1 Гц, 0,5H), 5,88 (д, J=2,1 Гц, 0,5H), 5,52 (с, 1H), 4,28-4,03 (м, 3H), 3,91-3,68 (м, 2H), 3,48-3,12 (м, 2H), 3,10-2,96 (м, 1H), 2,12-1,78 (м, 2,5H), 1,68-1,54 (м, 2H), 1,44-1,36 (м, 0,5H). 13C ЯМР (75 МГц, CDCl3/ТМС) δ 168,30, 133,82, 132,75, 132,61, 132,31, 132,18, 129,01, 127,24, 126,29, 125,68, 124,63, 119,64, 116,30, 114,50, 114,36, 109,91, 102,92, 102,61, 79,59, 79,39, 64,55, 51,36, 41,63, 41,50, 38,67, 36,30, 35,69, 35,52, 28,00.
К раствору (2-этилфенил)гидразина 1 (35 ммоль) и этилендиаминтетраацетата динатрия (20 мг) в метаноле (60 мл) добавляли по каплям 2-хлоракрилонитрил 2 (105 ммоль) при 60°C и полученную смесь перемешивали при температуре обратной конденсации в течение 8 часов. Затем добавляли концентрированную серную кислоту (94 ммоль) и смесь нагревали в течение дополнительных 6 часов. После нагревания при комнатной температуре реакцию гасили безводным карбонатом натрия (105 ммоль) и растворитель удаляли при пониженном давлении. Осадок обрабатывали водой (100 мл) и продукт экстрагировали этилацетатом (3*100 мл). Объединенные органические экстракты сушили над безводным сульфатом магния и растворитель удаляли при пониженном давлении. Осадок очищали посредством колоночной хроматографии с получением (28 ммоль) 2-(2-этилфенил)-2H-пиразол-3-иламина 3. 1H ЯМР (300 МГц, CDCl3/ТМС) δ 7,44-7,35 (м, 3H), 7,30-7,25 (м, 2H), 5,57 (д, J=2,1 Гц, 1H), 3,57 (уш.с, 2H), 2,49 (кв, J=7,6 Гц, 2H), 1,10 (т, J=7,6 Гц, 3H). 13C ЯМР (75 МГц, CDCl3/ТМС) δ 145,20, 142,60, 139,62, 135,93, 129,47, 129,41, 127,88, 126,55, 88,70, 24,04, 14,34.
К 2-(2-этилфенил)-2H-пиразол-3-иламину 3 (25,9 ммоль) в EtOH (150 мл) добавляли 3-этокси-4-гидрокси-бензальдегид 5 (28,5 ммоль) с последующим добавлением кислоты Мельдрума 4 (28,5 ммоль). Реакционную смесь нагревали до 75°C, затем через 2 часа указанную реакционную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. Осадок обрабатывали водой (100 мл) и продукт экстрагировали дихлорметаном (200 мл). Органический экстракт сушили над безводным сульфатом магния и растворитель удаляли при пониженном давлении. Осадок очищали посредством колоночной хроматографии с получением (11 ммоль) 4-(3-этокси-4-гидроксифенил)-1-(2-этилфенил)-1,4,5,7-тетрагидропиразоло[3,4-b]пиридин-6-она 6. 1H ЯМР (300 МГц, CDCl3/ТМС) δ 7,44-7,35 (м, 3H), 7,30-7,25 (м, 2H), 5,57 (д, J=2,1 Гц, 1H), 3,57 (уш.с, 2H), 2,49 (кв, J=7,6 Гц, 2H), 1,10 (т, J=7,6 Гц, 3H); 13C ЯМР (75 МГц, CDCl3/ТМС) δ 145,20, 142,60, 139,62, 135,93, 129,47, 129,41, 127,88, 126,55, 88,70, 24,04, 14,34.
ПРИМЕР 17. Получение 6-[1-(2-амино-пиримидин-5-илметил)пиперидин-4-ил]-2-пиперидин-1-илпиримидин-4-ола, Соединения IId
К смеси этил-N-Boc-пиперидин-4-карбоксилата (0,5 моль) и этилацетата (3 моль) несколькими порциями добавляли t-BuOK(1,5 моль) при 0ºC. Смесь перемешивали при комнатной температуре в течение 3 часов (ход реакции контролировали посредством ТСХ), концентрировали до половины объема, разбавляли водой (200 мл) и экстрагировали эфиром. Органический слой сушили над сульфатом натрия и выпаривали в вакууме. В результате очистки посредством колоночной хроматографии (силикагель, смесь этилацетат/гексан) получали сложный трет-бутиловый эфир 4-(2-этоксикарбонил-ацетил)пиперидин-1-карбоновой кислоты (78 г, 52 %)
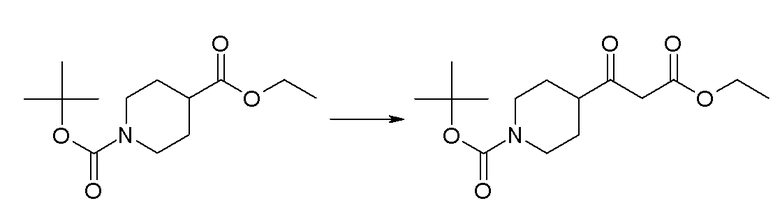
Натрий (1 моль) растворяли в безводном этаноле (400 мл). К полученному раствору порциями осторожно добавляли пиперидин-1-карбоксамидин (0,5 моль). Затем добавляли по каплям сложный трет-бутиловый эфир 4-(2-этоксикарбонилацетил)пиперидин-1-карбоновой кислоты (0,5 моль) и смесь перемешивали при температуре обратной конденсации в течение 4-6 часов (ход реакции контролировали посредством ТСХ), охлаждали до комнатной температуры, концентрировали, разбавляли водой (300 мл) и подкисляли уксусной кислотой до pH-4. Образовавшийся осадок собирали посредством фильтрования, промывали водой и сушили с получением сложного трет-бутилового эфира 4-(6-гидрокси-2-пиперидин-1-ил-пиримидин-4-ил)-пиперидин-1-карбоновой кислоты (82 г, 45%).
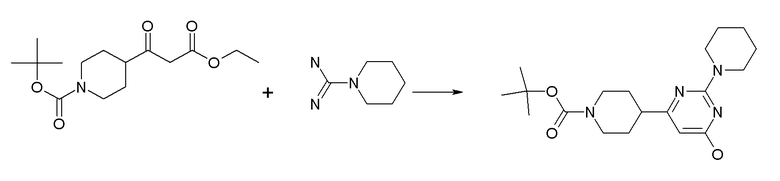
Суспензию сложного трет-бутилового эфира 4-(6-гидрокси-2-пиперидин-1-ил-пиримидин-4-ил)-пиперидин-1-карбоновой кислоты (0,05 моль) в 15% HCl в диоксане (100 мл) перемешивали при температуре обратной конденсации в течение 2 часов. После завершения реакции смесь охлаждали, осадок фильтровали, промывали сухим эфиром и сушили с получением 6-пиперидин-4-ил-2-пиперидин-1-илпиримидин-4-ола в виде гидрохлорида (12 г, 82%).
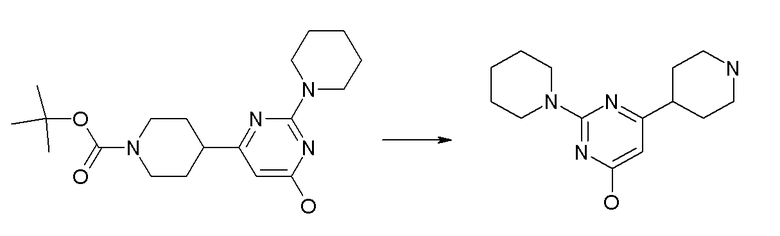
Смесь 6-пиперидин-4-ил-2-пиперидин-1-ил-пиримидин-4-ола (2,0 ммоль), 2-аминопиримидин-5-карбальдегида (2,6 ммоль), триэтиламина (4,0 ммоль) и 3 капли уксусной кислоты в безводном дихлорметане (20 мл) перемешивали при комнатной температуре в течение 3 часов. Затем порциями добавляли триацетоксиборогидрид натрия (6,0 ммоль) и продолжали перемешивать в течение 48 часов (ход реакции контролировали посредством ТСХ). Реакцию гасили насыщенным водным раствором бикарбоната натрия (20 мл) и продукт экстрагировали дихлорметаном (2x10 мл). Экстракты промывали солевым раствором и сушили над сульфатом натрия. Растворитель выпаривали в вакууме с получением неочищенного продукта. В результате очистки посредством колоночной хроматографии (силикагель, смесь этилацетат/гексан) получали 6-[1-(2-аминопиримидин-5-илметил)-пиперидин-4-ил]-2-пиперидин-1-ил-пиримидин-4-ол (606 мг, 82%).
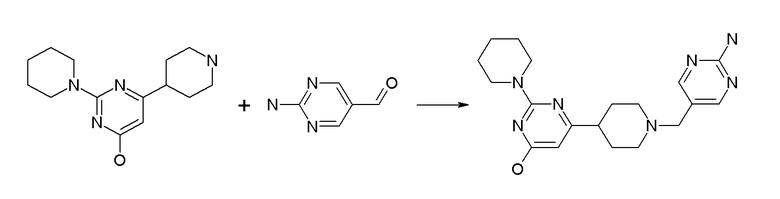
ПРИМЕР 18. Получение изохроман-1-ил(5'H-спиро[пиперидин-4,4'-пирроло[1,2-a]хиноксалин]-1-ил)метанона, Соединения VIIa
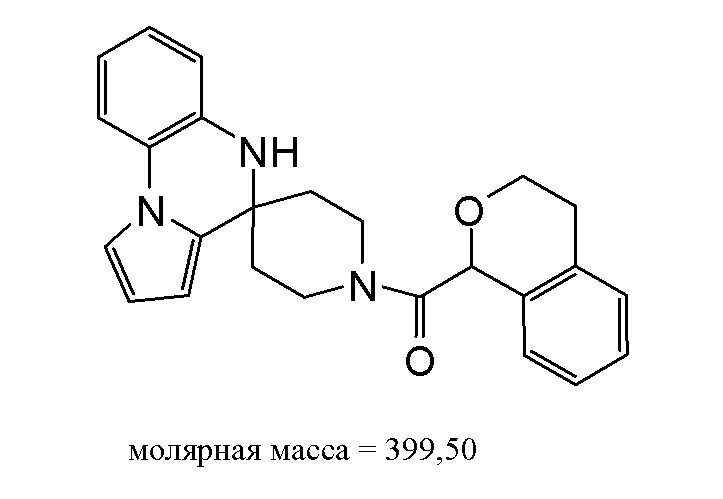
Стадия 1: Синтез сложного трет-бутилового эфира 4,5-дигидропирроло[1,2-a]хиноксалин-спиро-4-пиперидин-1-карбоновой кислоты.
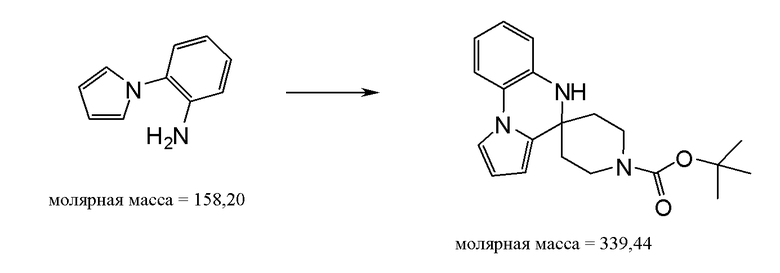
2-Пиррол-1-ил-фениламин (0,05 моль) и N-Boc-пиперидон (0,05 моль) растворяли в этаноле (50 мл), в качестве катализатора добавляли моногидрат п-толуолсульфоновой кислоты. Перемешиваемую реакционную смесь нагревали до обратной конденсации в течение 2-3 часов в атмосфере аргона. Ход реакции контролировали посредством ТСХ. Образовавшийся продукт фильтровали, промывали холодным этанолом и сушили с получением сложного трет-бутилового эфира 4,5-дигидро-пирроло[1,2-a]хиноксалин-спиро-4-пиперидин-1-карбоновой кислоты (7 г, 44%).
Стадия 2: Синтез свободного амина.
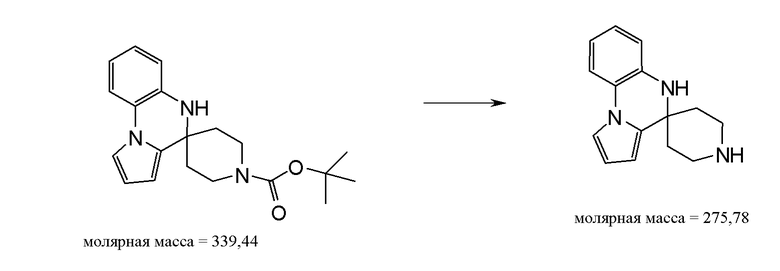
Сложный трет-бутиловый эфир 4,5-дигидро-пирроло[1,2-a]хиноксалин-спиро-4-пиперидин-1-карбоновой кислоты (0,1 моль) растворяли в изопропаноле (100 мл) и нагревали до обратной конденсации. К интенсивно перемешиваемой смеси добавляли по каплям 30-40 мл HCl (14-16%) в диоксане. Наблюдали выделение газа. Образовывался продукт, представляющий собой гидрохлорид, в виде осадка белого цвета. Смесь нагревали до обратной конденсации в течение 30-40 минут для завершения реакции. В результате фильтрования получали гидрохлорид 4,5-дигидропирроло[1,2-a]хиноксалин-спиро-4-пиперидина. Указанный гидрохлорид 4,5-дигидропирроло[1,2-a]хиноксалин-спиро-4-пиперидина растворяли в воде (50 мл) и реакцию гасили твердым карбонатом калия до достижения pH 7-8. Осадок собирали посредством фильтрования с получением 4,5-дигидропирроло[1,2-a]хиноксалинспиро-4-пиперидина (22 г 94%) в виде свободного основания.
Стадия 3: Синтез изохроман-1-ил(5'H-спиро[пиперидин-4,4'-пирроло[1,2-a]хиноксалин]-1-ил)метанона.
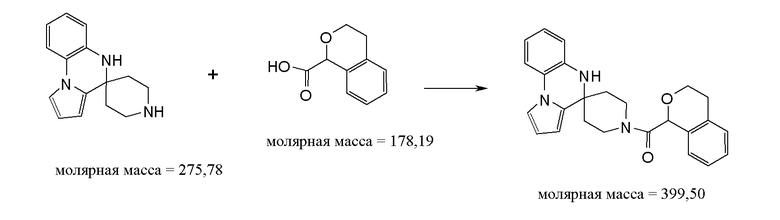
Смесь 4,5-дигидропирроло[1,2-a]хиноксалин-спиро-4-пиперидина (2,0 ммоль), триэтиламина (2 ммоль), изохроман-1-карбоновой кислоты (2 ммоль) и гексафторфосфата бензотриазол-1-илокси-трис(диметиламино)фосфония (2 ммоль) в дихлорметане (10 мл) перемешивали при комнатной температуре в течение 16 часов. Реакцию гасили насыщенным водным раствором бикарбоната натрия (10 мл), смесь перемешивали в течение 2 часов и продукт экстрагировали дихлорметаном (2×10 мл). Объединенные органические слои сушили над сульфатом натрия и концентрировали при пониженном давлении с получением неочищенного продукта. В результате очистки посредством колоночной хроматографии (силикагель, смесь этилацетат/гексан) получали изохроман-1-ил(5'H-спиро[пиперидин-4,4'-пирроло[1,2-a]хиноксалин]-1-ил)метанон (Соединение VIIa, 527 мг, 66%).
БИОЛОГИЧЕСКИЕ ПРИМЕРЫ
ПРИМЕР 19. Агонистическая активность иллюстративных соединений согласно настоящему изобретению на примере клеточной линии, трансфицированной рецептором P2Y13
Исследовали агонистическую активность соединений в отношении клеток, трансфицированных GPCR P2Y13, при помощи исследования тока Ca++ (детектирование с использованием флуоресцирующего красителя) с использованием соединения сравнения в качестве положительного контроля. Две клеточные линии, 1321N1-P2Y13, устойчиво экспрессирующую рецептор P2Y13 человека, и родительскую клеточную линию 1321N1 в качестве контроля, применяли в этом исследовании. Родительскую линию 1321N1 и 1321N1-P2Y13 помещали в колбы Т25 в количестве 3 миллиона клеток на колбу, соответственно. Клетки инкубировали при 37°С и 5% СО2 в течение ночи. На следующий день клетки трансфицировали белком Gqi5 с применением трансфекционного реагента Fugene (реагент Fugene, производство Roche).
После 24-часовой трансфекции клетки собирали из колб и помещали в 384-луночные планшеты по 8000 клеток на лунку. Исследование проводили через 48 часов после трансфекции Gqi5.
Способы: Ток Ca++ определяли в соответствии с протоколом производителя (Molecular Devices FLIPR Calcium 4 Assay kit (R8142)). Вкратце, среду для клеточной культуры удаляли из лунок и заменяли 25 мкл буфера Хэнка или PBS. 25 мкл красителя Ca++ добавляли в каждую лунку. Планшет инкубировали в течение 1 часа при 37°С и 5% СО2. После инкубации планшет переносили в FlexStation III (FlexStation III (Molecular Devices)). Соединение автоматически впрыскивали в каждую лунку.
Результаты: Получали значения EC50 для иллюстративных соединений согласно настоящему изобретению. Результаты приведены на Фигуре 1 и в Таблице 3. Значения ЕС50 также сравнивали со значениями, полученными для соединения сравнения.
Исследовали агонистическую активность дополнительных иллюстративных соединений согласно настоящему изобретению в отношении клеток, трансфицированных рецептором P2Y13, результаты представлены на Фигуре 2, значения ЕС50 приведены в Таблице 4.
Значения EC50 типовых соединений в отношении активности рецептора P2Y13, полученные по данным Фигуры 1
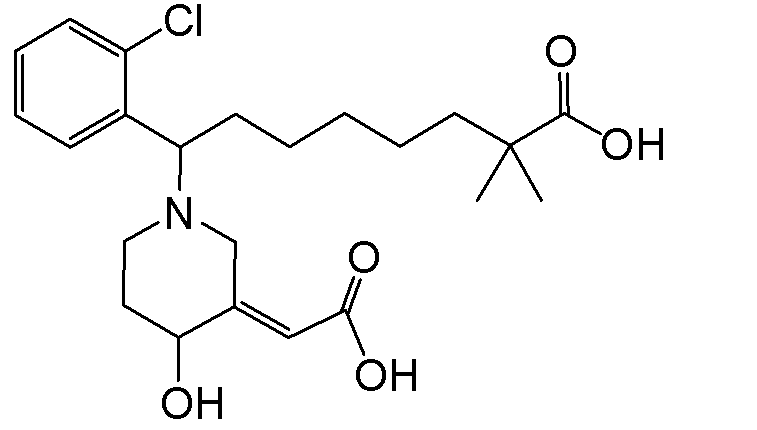
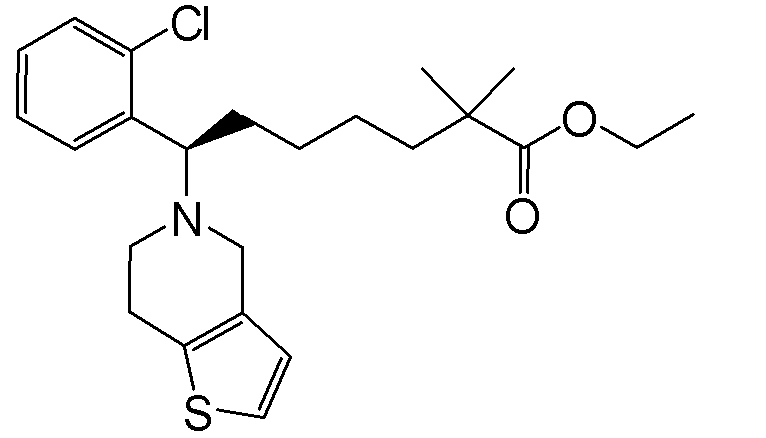
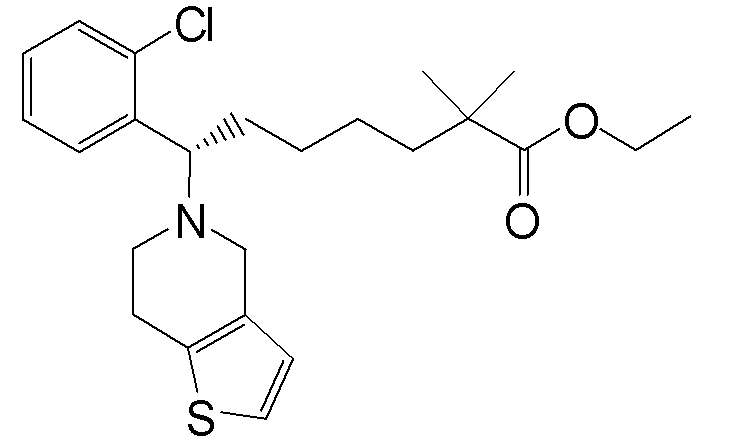
Значения EC50 в отношении активности рецептора P2Y13, полученные по данным Фигуры 2
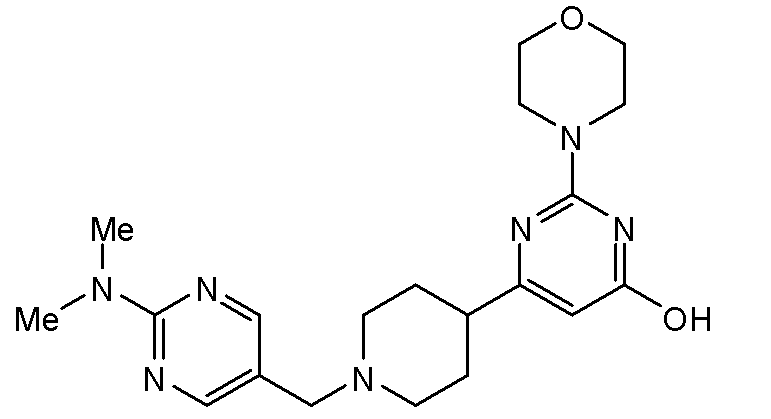
Химическое название соединения сравнения: дихлор-(((((2R,3S,4R,5R)-3,4-дигидрокси-5-(6-(2-(метилтио)этиламино)-2-(3,3,3-трифторпропилтио)-9H-пурин-9-ил)тетрагидрофуран-2-ил)метокси)(гидрокси)фосфорилокси)(гидрокси)фосфорил)-метилфосфоновая кислота. Соединение сравнения также известно как Cangrelor®. Химическая структура изображена ниже:
Интернализация ЛПВП в гепатоцитах человека (HepG2) in vitro
Действие иллюстративных соединений согласно настоящему изобретению на интернализацию ЛПВП определяли при помощи исследования клеток HepG2 in vitro (Фигура 3). 3Н холестерин, холестерилолеат [холестерил-1,2-3Н(N)-меченый ЛПВП] (доступный в PerkinElmer, http://www.perkinelmer.com/Catalog/Product/ID/NET746L001MC) добавляли в клетки HepG2 в присутствии иллюстративных соединений согласно настоящему изобретению (конечная концентрация 1 мкМ). Ранее сообщалось, что соединение сравнения увеличивает захват ЛПВП клетками HepG2 in vitro (Jacquet S, Malaval C, Martinez LO, Sak K, Rolland C, Perez C, Nauze M, Champagne E, Tercé F, Gachet C, Perret B, Collet X, Boeynaems JM, Barbaras R. The nucleotide receptor P2Y13 is a key regulator of hepatic high-density lipoprotein (HDL) endocytosis. Cell Mol Life Sci. 2005 Nov;62(21):2508-15), его применяли в качестве положительного контроля захвата ЛПВП клетками HepG2. Определили, что соединения XIa, IIc, IIa и Ih (R-изомер) оказывают сильное воздействие на захват ЛПВП клетками HepG2; Соединения IIb, VIIa и VIIIa оказывают незначительное воздействие на захват ЛПВП клетками HepG2.
Влияние внутривенной инъекции выбранных соединений на физиологию желчных кислот
Желчные кислоты получаются из холестерина, и их синтез является основным путем катаболизма холестерина. Поглощение ЛПВП в печени непосредственно связано с секрецией желчных кислот и выведением холестерина из желчи. Для подтверждения действия иллюстративных соединений согласно настоящему изобретению на физиологию желчи вводили инъекцию соединений в хвост мышей C57Bl6, через 4 часа мышей умерщвляли и анализировали состав желчи (Фигура 5). Наблюдали повышение концентрации желчных кислот, а также объема желчи. Результаты для всех соединений были одинаковыми с результатами, полученными для контрольного соединения (соединения сравнения), или даже превосходили их.
Влияние дозы выбранных соединений на физиологию желчи
Определяли зависимость доза-ответ после принудительного перорального введения соединений согласно настоящему изобретению. Исследовали дозы 0,003, 0,03 и 0,3 мг/кг. Выбранные соединения согласно настоящему изобретению принудительно перорально вводили мышам, через 6 часов мышей умерщвляли и анализировали состав желчи (Фигуры 6-11). Наблюдали зависящее от дозы повышение содержания желчных кислот, холестерина в желчи и фосфолипидов в желчи для всех концентраций всех соединений. Действие соединений IIa, VIIIa, IIc и XIa проявлялось в концентрации 0,003 мг/кг; а действие соединений IIb и VIIa проявлялось в концентрации 0,3 мг/кг.
Действие введения соединения IIa один раз в день в течение одной недели на физиологию желчных кислот
В приведенных выше примерах иллюстративные соединения согласно настоящему изобретению вводили один раз и действие на физиологию желчи определяли через 6 часов. Соединение IIa принудительно перорально вводили один раз в день в течение недели для определения действия на физиологию желчи, вызванного продолжительным лечением. В день умерщвления условия были аналогичны условиям, используемым в экспериментах по определению зависимости доза-ответ. Определяли итоговое действие соединения IIa на секрецию желчи. Объем желчи также значительно увеличивался по сравнению с введением носителя.
Нокдаун P2Y13 в клетках Hepa 1-6
Для определения связи между специфическим захватом ЛПВП P2Y13 и иллюстративными соединениями согласно настоящему изобретению исследовали нокдаун P2Y13 клеточной линии печени мышей (Нера 1-6). Применяли лентивирусные частицы, кодирующие кшРНК P2Y13 мыши и индуцируемые путем добавления доксициклина в клетки (вектор pTRIPZ, производство Open Biosystem). В присутствии доксициклина наблюдали снижение иРНК P2Y13 (Фигура 11). Захват ЛПВП данной клеточной линией определяли в присутствии и в отсутствии соединения IIa и доксициклина (Фигура 12). Значительное снижение захвата ЛПВП (8,5%) наблюдали в случае, когда клетки обрабатывали доксициклином (относительно группы, которой вводили носитель, p<0,05). Снижение также наблюдали в присутствии соединения IIa (17,5% - p<0,005), это, тем самым, подтверждает, что мишенью соединения IIa является P2Y13.
Материалы и методы
Набор определения состава желчных кислот приобретали в Diazyme; наборы определения уровня фосфолипидов и общего холестерина приобретали в Biolabo; мышей C57Bl6 приобретали в Janvier. кшРНК (мкРНК) P2Y13 лентивируса pGIPZ мышей (RMM4431-98723221) приобретали в Openbiosystem и субклонировали в pTRIZ (индуцируемый доксициклином вектор). 1 мл препарата лентивирусных частиц получали в Vectalys с применением 1,2Е8 трансдуцирующих единиц на мл. Праймеры P2Y13 мышей (Mm 00546978-m1), праймеры ГАФДГ (Mm03302249-g1) и праймеры рибосомального гена 18S (Mm02601777-g1) для ПЦР в реальном времени приобретали в Applied. Клетки HepG2 (HB-8065) и Hepa 1-6 (CRL-1830) приобретали в АТСС.
Получение липопротеинов
Липопротеины выделяли из плазмы мышей путем последовательного ультрацентрифугирования в градиенте плотности KBr. Получали ЛПВП с плотностью d=1,21. Очищенные ЛПВП подвергали диализу с PBS и применяли в экспериментах по определению захвата.
Введение метки в ЛПВП
Раствор 3Н холестерина холестерилолеата [холестерил-1,2-3Н(N)] (PerkinElmer) (250 мкл) сушили и суспендировали в ацетоне (250 мкл). Раствор смешивали с липопротеинами делипидизированной сыворотки (20 мл в концентрации 40 мг/мл) и ДТНБ (конечная концентрация 0,4 мкМ). ЛПВП (7 мг) добавляли и инкубировали в течение ночи при 4°С при осторожном перемешивании. ЛПВП очищали в градиенте плотности KBr согласно приведенному выше описанию.
Захват ЛПВП клетками HepG2 и Hepa 1-6, трансдуцированными кшP2Y13
Трансдуцированные клетки Hepa 1-6 помещали в количестве 300000 клеток/лунку в 6х-луночные планшеты. Клетки обрабатывали каждый день доксициклином (10 мкМ) в течение 3 дней (для подавления P2Y13) или не обрабатывали (контроль «утечки» плазмид). В день определения захвата клетки промывали один раз DMEM и инкубировали в течение 1 часа в DMEM при 37°С. 75 мкг радиомеченого ЛПВП (6500 dpm/мкг ЛПВП3) добавляли в среду и инкубировали в течение 10 минут при 37°С. Клетки промывали один раз DMEM (2 мл) и инкубировали в течение 1,5 часов в DMEM при 4°С, после чего происходила диссоциация. Затем клетки промывали еще раз холодной DMEM (2 мл) и захваченный холестериловый эфир выделяли из клеток HepG2 путем добавления раствора гексан:изопропанол (3:2, 1 мл).
Захват ЛПВП
Гепатоциты помещали в количестве 300000 клеток/лунку в 6х-луночные планшеты. Через два дня клетки промывали один раз DMEM и инкубировали в течение 1 часа в DMEM при 37°С. 75 мкг радиомеченого ЛПВП (6500 dpm/мкг) добавляли в среду и инкубировали в течение 10 минут при 37°С. Клетки промывали один раз DMEM (2 мл) и инкубировали в течение 1,5 часов в DMEM при 4°С, после чего происходила диссоциация. Затем клетки промывали еще раз холодной DMEM (2 мл) и захваченный холестериловый эфир выделяли из клеток HepG2 путем добавления раствора гексан:изопропанол (3:2, 1 мл).
Выход холестерина
Величину емкости выхода холестерина количественно оценивали в соответствии с существующим описанием (Ванг с соавторами (Wang, et al.), 2007) в образцах плазмы крови кроликов, собранных перед введением дозы в день 0 и после 4-недельного введения соединения IIa. Вкратце, окисленные ЛПНП (25 мкг/мл), меченые [3Н]-холестерином (2 мкКи/мл; PerkinElmer), добавляли в макрофаги J774, которые выращивали в течение 24 часов в 2,5% сывороточной среде. Высвобождение [3Н]-холестерина измеряли через 6 часов инкубирования 2,5% (об./об.) плазмы кролика или плазмы, не содержащей β-липопротеины (набор РТА - Biolabo, France). Все исследования проводили в трех повторностях. Плазму кролика (калибратор) вводили в каждый планшет, и значения нормировали путем деления значений, полученных для образцов плазмы, на значения, полученные для калибраторной пробы, для определения нормированной емкости выхода холестерина в образцах.
Протокол исследования животных
Содержание и уход за животными соответствовали рекомендациям консульской директивы 86/609/ЕЕС, протоколы одобрены в институциональных этических комитетах.
«Модель остановки кровотока» у мышей АроЕ-/-: Левые сонные артерии мышей ароЕ-/- лигировали и мышей помещали на западную диету. Мышам вводили соединение IIa один раз в день в дозировке 100 мкг/кг (0,5% СМС, 0,2% Tween80) в течение 2 недель.
«Модель диеты с высоким содержанием холестерина» ApoE-/-: Мышей на два месяца помещали на западную диету (0,2% холестерина, 21% пахта), затем вводили соединение IIa один раз в день в дозировке 100 мкг/кг (0,5% СМС, 0,2% Tween80) в течение еще 4 недель. Мышей умерщвляли, собирали аорты для биохимической и иммуногистологической оценки. Одну группу аорт (n=10) отделяли у основания сердца и помещали в стеклянную пробирку, содержащую 3 мл смеси хлороформ-метанол (2:1, об./об.) и стигмастерол в качестве внутреннего стандарта. Затем проводили описанный выше анализ. Другую группу аорт (n=10) сначала тщательно промывали PBS, затем 4% параформальдегидом (500 мкл) и, наконец, реагентом Tissue-Tek OCT (500 мкл). Затем аорты помещали в OCT и замораживали при -20°С. Разделяли замороженные срезы на три части по 10 мкм и отделяли друг от друга, обрабатывали и окрашивали Oil Red O (ORO), сириусом красным и гематоксилин/эозином. Макрофаги детектировали при помощи первичного моноклонального антитела крыс F4/80 (Abcam), VCAM детектировали при помощи крысиного антимышиного CD106 (AbDSerotec).
Новозеландские кролики: После 2 месячной диеты с высоким содержанием холестерина (0,5% холестерина) и 2-недельного периода вымывания животным вводили (принудительно перорально) один раз в день соединение IIa в дозировке 30, 100 и 300 мкг/кг в течение 4 недель. Образцы крови собирали до введения дозы в день 0 и после 4-недельного введения соединения IIa. После отбора последней пробы крови собирали образцы печени, желчного пузыря, сердца, торакальной и брюшной аорт от сердца (левый желудочек) до подвздошных артерий.
Макрофаги и окрашивание сонных артерий мышей ароЕ-/- Oil Red O. Левую сонную артерию, зафиксированную формалином, помещали в ОСТ и замораживали при -20°С. Разделяли замороженные срезы на четыре части и отделяли их друг от друга, обрабатывали и окрашивали Oil Red O (ORO). Макрофаги детектировали при помощи крысиного моноклонального антитела к CD-68 (Abcam).
Окрашивание аорт кроликов. Аорты кроликов фиксировали нейтральным формалиновым буфером и помещали в парафин. Продольные 3 мкм разрезы окрашивали гематоксилин-эозин-шафрановым для гистологического анализа. Срезы тканей депарафинировали и инкубировали в безбелковом блокирующем растворе для ингибирования неспецифического связывания первичных моноклональных антител, применяемых для иммуноокрашивания моноцитов и макрофагов кроликов (RAM11, производство Dako Corp., разбавление 1:100) и клеточного актина гладких мышц (HHF35, производство Dako Corp., разбавление 1:100). Применяли вторичное биотинилированное антикозье антитело, разбавленное 1:300 (Dako).
Трансдукция Нера 1-6
Лентивирусный вектор для НЕК 293Т и права на лентивирусный вектор получали в Vectalys. Клетки Нера 1-6 помещали в количестве 75000 клеток в 24-луночный планшет за день до проведения трансдукции. Через 24 часа после трансдукции определяли число клеток. Среду из других лунок удаляли и заменяли 1 мл 10% сыворотки в DMEM, содержащей 8 мкг/мл полибрена. Проводили трансдукцию клеток путем добавления 40 трансдуцирующих единиц вирусного вектора (1,2Е8 TU/мл) на клетку. Через 17 часов после трансдукции среду заменяли и устанавливали число трансдуцированных клеток Нера.
КПЦР
Для количественной ПЦР РНК очищали от выращиваемых клеток при помощи набора RiboPure™ (АМ1924 Ambion). Проводили обратную транскрипцию РНК и одноцепочечной кДНК при помощи набора High Capacity RNA-to-cDNA (4387406 Applied Biosystems). КПЦР проводили в формате Taqman согласно инструкции производителя (Applied Biosystems).
Анализ
Желчные кислоты. Желчные кислоты разбавляли в PBS (1:5000) и концентрацию измеряли в соответствии с протоколом производителя с незначительными модификациями. Концентрации желчных кислот на калибровочной кривой изменяли от 50 до 200 мМ, и ферментную реакцию проводили в течение 20 минут. 4 мкл желчи, разбавленной 1:5000, применяли для определения концентрации.
Фосфолипиды желчи. Уровень фосфолипидов в желчи измеряли в соответствии с протоколом производителя. Вкратце, концентрация стандартов составляла от 0 до 20 мкг/мл, в исследовании проводили измерение в 4 мкл образце желчи. Реакцию проводили в течение 5 минут при 37°С.
Холестерин желчи. Уровень холестерина в желчи измеряли в соответствии с протоколом производителя. Вкратце, концентрация стандартов составляла от 0 до 60 мкг/мл, в исследовании проводили измерение в 10 мкл образце желчи (разбавление 1:10). Реакцию проводили в течение 5 минут при 37°С.
ПРИМЕР 20. ДЕЙСТВИЕ СОЕДИНЕНИЯ IIa НА ПУТЬ P2Y13r
Соединение IIa оказывало значительное действие на подавление прогрессирования атеросклероза у мышей за счет стимуляции пути ЛПВП-P2Y13r. Также была показана специфичность к пути P2Y13r in vivo с применением аденовируса, содержащего кшРНК P2Y13r, у инфицированных мышей ароЕ-/-. Более того, соединение IIa приводило к уменьшению существовавших атеросклеротических бляшек в аортах кроликов, которым давали корм с высоким содержанием холестерина. Эти результаты показывают, что P2Y13r играет ключевую роль для метаболизма ЛПВП и атеросклероза, а также, что P2Y13r является подходящей терапевтической мишенью для исследования биологии атеросклероза.
Применяли гетероциклические соединения, разработанные в качестве перорально активных агонистов рецептора P2Y13. Эти соединения выбирали с учетом специфического связывания и активности в отношении клеток 1321N1, экспрессирующих рецепторы P2Y13 (не показано) \l "_ENREF_8" \o "Kim, 2005 #5501). Также исследовали способность соединений стимулировать захват [3Н]-холестерин-меченых-ЛПВП in vitro гепатоцитами мышей (Нера) и клетками гепатомы человека (HepG2). Специфичность захвата также подтверждали при помощи зонда миРНК P2Y13 (не показано). Предположили, что введение агонистов P2Y13r животным может повышать обратный транспорт холестерина, т.е. захват ЛПВП в печени, и, следовательно, увеличивать секрецию желчных кислот и холестерина в желчь. Во-первых, через 4 часа после внутривенного введения мышам C57Bl/6J соединения IIa (10 нмоль/кг) наблюдали увеличение секреции желчных кислот (ВА) и свободного холестерина в желчь, которое незначительно превышало эффект, достигаемый при введении Cangrelor (The Medicines Company), известного агониста P2Y13r (Фигуры 16А и В) (Jacquet, S. et al. The nucleotide receptor P2Y13 is a key regulator of hepatic high-density lipoprotein (HDL) endocytosis. Cell Mol Life Sci 62, 2508-2515 (2005)). Подтверждали стимуляцию секреции желчных кислот и холестерина в желчь животных путем принудительного перорального введения агониста P2Y13r в дозах, составляющих всего 30 мкг/кг (Фигуры 16С и D). Указанное повышение секреции желчных кислот, наблюдаемое при введении агонистов P2Y13r, может быть вызвано секрецией ВА из внутреннего пространства печени в желчные протоки (Schwartz, C. C., Halloran, L. G., Vlahcevic, Z. R., Gregory, D. H. & Swell, L. Preferential utilization of free cholesterol from high-density lipoproteins for biliary cholesterol secretion in man. Science 200, 62-64 (1978)) или синтезом ВА de novo в печени после стимуляции пути захвата ЛПВП P2Y13r. Было явно показано повышение содержания желчных кислот в печени при введении 30 мкг/кг агониста P2Y13r, соответствующее стимуляции захвата ЛПВП в печени, которая в свою очередь приводит к повышению синтеза ВА (Фигура 16Е). Необходимо отметить, что уровень холестерина и ЛПВП в плазме значительно не изменялся (Фигура 16F) через 4 часа после введения за исключением уровня неэтерифицированного холестерина, изменение которого можно объяснить описанным ранее специфическим захватом холестерина ЛПВП в печени (Schwartz, C. C., Halloran, L. G., Vlahcevic, Z. R., Gregory, D. H. & Swell, L. Preferential utilization of free cholesterol from high-density lipoproteins for biliary cholesterol secretion in man. Science 200, 62-64 (1978)). Чтобы дополнительно подчеркнуть участие ЛПВП в повышение секреции желчи после введения мышам соединения IIa проводили in vivo анализ захвата в печени [3Н]-холестерин-меченых ЛПВП у мышей (Фигура 16G), меченого [3Н]-холестерилового эфира ЛПВП у мышей (Фигура 16Н) и [3Н]-холестерин-меченых ЛПНП у мышей (Фигура 16I). Внутривенное введение 10 нмоль/кг агониста P2Y13r животным приводило к специфической стимуляции захвата ЛПВП по сравнению с ЛПНП, что подтверждает гипотезу о стимуляции обратного транспорта холестерина, опосредованного ЛПВП.
Исследовали влияние стимуляции агонистами пути P2Y13r на атеросклеротические поражения у мышей ароЕ-/-, которые сильно подвержены атеросклерозу при краткосрочной западной (т.е. с высоким содержанием холестерина) диете. Для большего сродства с патологией человека площадь атеросклеротических поражений увеличивали при помощи методологии, описанной как «модель остановки кровотока» у мышей ароЕ-/- (Godin, D., Ivan, E., Johnson, C., Magid, R. & Galis, Z. S. Remodeling of carotid artery is associated with increased expression of matrix metalloproteinases in mouse blood flow cessation model. Circulation 102, 2861-2866 (2000); Ivan, E. et al. Expansive arterial remodeling is associated with increased neointimal macrophage foam cell content: the murine model of macrophage-rich carotid artery lesions. Circulation 105, 2686-2691 (2002); Lessner, S. M., Martinson, D. E. & Galis, Z. S. Compensatory vascular remodeling during atherosclerotic lesion growth depends on matrix metalloproteinase-9 activity. Arterioscler Thromb Vasc Biol 24, 2123-2129 (2004)). Вкратце, левую сонную артерию мыши лигировали для возникновения локального воспаления, что приводило к появлению выраженных атеросклеротических поражений через 2 недели западной диеты. Сопутствующее введение агониста P2Y13r в течение 2 недель наряду с диетой с высоким содержанием холестерина свидетельствовало о дозозависимом ингибировании прогрессирования атеросклеротических поражений по сравнению с контрольными животными с учетом содержания холестерина в пораженном участке ((Riedmüller K, Metz S, Bonaterra GA, Kelber O, Weiser D, Metz J, Kinscherf R. Cholesterol diet and effect of long-term withdrawal on plaque development and composition in the thoracic aorta of New Zealand White rabbits. Atherosclerosis 210 407-13(2010)) (Фигура 17А). Количественная оценка компонентов бляшки при помощи гистопатологического анализа сонных артерий, обработанных гематоксилин-эозином (Фигуры 17В, Е, Н), красителем Oil Red O (Фигуры 17С, F, I) и антителом CD68 (специфическим к макрофагам) (Фигуры 17D, G, J) является дополнительным подтверждением приведенного наблюдения. Окрашивание явно показывало снижение содержания липидов в сонных артериях животных, которым проводили лечение. Также наблюдали снижение отложений холестерина, вызванное действием агониста P2Y13r, в невоспаленных правых сонных артериях мышей ароЕ-/- в «модели остановки кровотока» (например, 18,2+/-3,6 и 7,5+/-3,4 нмоль/мг в ткани в случае носителя и 100 мкг/кг соединения IIa, соответственно).
Для подтверждения специфичности P2Y13r перед проведением лигирования сонной артерии и западной диеты мышей инфицировали аденовирусом, содержащим обычную кшРНК или специфическую кшРНК, направленную на P2Y13r, при этом проходил сильный нокдаун P2Y13r (Фигура 18А). Сопутствующее введение агонистов P2Y13r (0,1 мг/кг/день) в течение 2 недель наряду с диетой с высоким содержанием холестерина приводило к значительному снижению содержания холестерина (60% снижение) в лигированных сонных артериях в случае обычного аденовируса, что свидетельствовало о значительном ингибировании прогрессирования атеросклеротических поражений по сравнению с контрольным животными (Фигура 18В). Это наблюдение дополнительно подтверждали при помощи гистопатологического анализа сонных артерий, окрашенных гематоксилин-эозином и Oil Red O (не показан). Действие агониста P2Y13r на содержание холестерина в сонных артериях отсутствовало у мышей, которым вводили кшРНК, направленную на P2Y13r, что являлось еще одним доказательством специфичности P2Y13r в этой модели при нокдауне экспрессии P2Y13r (Фигура 18В). Специфичность P2Y13r также наблюдали при определении содержания холестерина в плазме (Фигура 18С), более конкретно, содержания холестерина ЛПНП и ЛПОНП (Фигуры 18Е и D, соответственно), основных классов липопротеинов для данной животной модели, при этом снижение наблюдали у мышей, которым вводили агонист P2Y13r и обычный аденовирус, но снижение отсутствовало у мышей, которым вводили агонист P2Y13r и кшРНК. Эти данные являются первым доказательством того, что стимуляция P2Y13r может оказывать положительное влияние на прогрессирование атеросклероза.
Предотвращение прогрессирования бляшек в аорте дополнительно оценивали у мышей ароЕ-/-, которых помещали на диету с высоким содержанием холестерина (Фигура 19). После 8-недельной диеты и последующего 4-недельного введения 100 мкг/кг/день соединения IIa, у животных, которым вводили соединение IIa, наблюдали значительное снижение размера бляшек (Фигуры 4В, 4G и 4J), содержания холестерина (Фигура 19А), экспрессии VCAM1, вызванной специфическим антителом CD106 (Фигуры 4F, 4I и 4L), снижение содержания макрофагов (антитело F4/80 - Фигура 19С) на стенках аорты и в бляшках и снижение окрашивания липидов (Фигуры 19D, Н и К). Содержание коллагена в стенках увеличивалось (Фигура 19Е).
Также рассматривали проблему влияния стимуляции пути P2Y13r на регрессию атеросклероза, вызванного диетой с высоким содержанием холестерина у кроликов. У этих животных после 2 месяцев на диете с высоким содержанием холестерина (0,5% холестерина в корме) и 3-недельного периода вымывания развивались атеросклеротические поражения в аорте. В дополнение, у кроликов экспрессировался белок переноса холестерилового эфира (БПХЭ, который не экспрессируется у мышей), а также наблюдался липопротеиновый профиль, схожий с профилем человека. После возникновения поражения и периода вымывания животным перорально вводили соединение IIa в дозах, составляющих не более 0,3 мг/кг, в течение 4 недель. Наблюдали значительное снижение размеров атеросклеротических бляшек в аорте (30% снижение) у животных, которым вводили агонист, определяемое по общему содержанию холестерина в артерии (Фигура 20А). Кроме того, иммуногистологический анализ различных отделов аорты (от проксимального до дистального) с применением гематоксилин-эозина, антитела HHF35 (специфического к клеткам гладких мышц) и антитела RAM11 (специфического к макрофагам) подтверждал снижение размера бляшек (не показан). Липопротеиновые профили, которые анализировали путем ВЭЖХ, показывали тенденцию увеличения уровня холестерина ЛПВП (Фигура 20D), а также ароА-I кролика, определенную при помощи анализа SELDI-TOF, при этом уровень ЛПОНП и ЛПНП снижался незначительно (Фигуры 20В и С, соответственно). Изменения экспрессии иРНК (измеренной при помощи КПЦР) рецепторами ЛПНП и SR-BI в печени кроликов, которым вводили соединение IIa, отсутствовали (не показано). Эти наблюдения свидетельствовали о том, что активация P2Y13r не имеет непосредственного влияния на метаболизм β-липопротеинов, что также подтверждается незначительными изменениями уровня ЛПОНП, ЛПНП, ароВ и триглицеридов в плазме. Как и в случае мышей (Фигура 16), содержание желчных кислот в печени повышалось у кроликов, которым вводили агонисты P2Y13r (Фигура 20G), что является еще одним доказательством влияния пути P2Y13r на наблюдаемые у кроликов явления. Относительно слабое влияние на уровень ЛПВП в плазме может быть вызван неосинтезом новых частиц ЛПВП, которые могут компенсировать захват зрелых ЛПВП в печени, вызванный действием соединения IIa. В качестве подтверждения, наблюдаемое повышение уровня ароА-I (Фигура 21А), связанное с повышением экспрессии иРНК ароА-I в печени (Фигура 21В), свидетельствует о поступлении новых частиц ЛПВП в кровоток.
В дополнение, вследствие эффективного обновления ЛПВП, опосредованного P2Y13r, и высокой активностью выведения холестерина из макрофагов, образующиеся частицы ЛПВП могут эффективно ускорять регрессию атеросклеротических бляшек.
Для исследования этой гипотезы размер частиц ЛПВП у животных, которым проводили лечение, анализировали при помощи техники Lipoprint®. Содержание ЛПВП у животных, которым проводили лечение, хорошо соответствовало общей закономерности, при которой наблюдалось специфическое снижение уровня крупных ЛПВП и повышение уровня частиц ЛПВП «промежуточного» размера по сравнению с контрольными животными (Фигуры 21С и D). Указанные частицы ЛПВП промежуточного размера рассматривают в качестве наиболее активных частиц, удаляющих холестерин из атеросклеротических бляшек за счет ускоренного вывода холестерина из макрофагов, присутствующих в поражении (Khera, A. V. et al. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N Engl J Med 364, 127-135 (2011); deGoma, E. M., deGoma, R. L. & Rader, D. J. Beyond high-density lipoprotein cholesterol levels evaluating high-density lipoprotein function as influenced by novel therapeutic approaches. J Am Coll Cardiol 51, 2199-2211 (2008)). Наконец, измерение выхода холестерина в макрофагах J774, содержащих [3Н]-холестерин, в плазме (Фигура 21Е) или в плазме с пониженным содержанием частиц ароВ (Фигура 21F) животных, которым проводили лечение, показало наличие дозозависимого выхода холестерина после введения агониста P2Y13r, подтверждая тем самым, что ЛПВП плазмы животных, которым проводили лечение, более активно выводят холестерин из макрофагов (Khera, A. V. et al. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N Engl J Med 364, 127-135 (2011)).
В заключении, впервые in vivo было показано, что рецепторы P2Y13 являются ключевыми участниками метаболизма ЛПВП, т.е. обратного транспорта холестерина, и их основной физиологической функции, т.е. защиты от атеросклероза. Эти данные подтверждают механизм, при котором стимуляция захвата ЛПВП или эндоцитоза в печени по пути P2Y13r ускоряет катаболизм холестерина в печени и его секрецию в желчь. По-видимому, захват крупных частиц ЛПВП в печени также стимулирует синтез новых частиц ЛПВП de novo и, тем самым, увеличивает емкость выхода сыворотки. Другими словами, было показано in vivo, что положительное прямое воздействие на функциональность ЛПВП, т.е. воздействие не только на уровень ЛПВП, может оказывать значительное влияние на патологию атеросклероза. Эти данные также подтверждают, что агонисты P2Y13r, такие как соединение IIa и другие соединения согласно настоящему изобретению, хорошо подходят в качестве фармакологических терапевтических средств для лечения атеросклероза или его осложнений.
| название | год | авторы | номер документа |
|---|---|---|---|
| (АЛЬФА-ЗАМЕЩЕННЫЕ АРАЛКИЛАМИНО- И ГЕТЕРОАРИЛАЛКИЛАМИНО)ПИРИМИДИНИЛ- И 1, 3, 5-ТРИАЗИНИЛБЕНЗИМИДАЗОЛЫ, ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И ИХ ПРИМЕНЕНИЕ В ЛЕЧЕНИИ ПРОЛИФЕРАТИВНЫХ ЗАБОЛЕВАНИЙ | 2012 |
|
RU2608742C2 |
| ПРОИЗВОДНЫЕ ПИРРОЛА КАК ЛЕКАРСТВЕННЫЕ ВЕЩЕСТВА | 2005 |
|
RU2470916C2 |
| N1-(4-(5-(ЦИКЛОПРОПИЛМЕТИЛ)-1-МЕТИЛ-1H-ПИРАЗОЛ-4-ИЛ)ПИРИДИН-2-ИЛ)ЦИКЛОГЕКСАН-1,4-ДИАМИНОВЫЕ ПРОИЗВОДНЫЕ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ СK1 И/ИЛИ IRAK1 ДЛЯ ЛЕЧЕНИЯ РАКА | 2018 |
|
RU2761457C2 |
| ПРОИЗВОДНЫЕ СПИРО(5.5)УНДЕКАНА | 2009 |
|
RU2515895C2 |
| СОДЕРЖАЩЕЕ ЗАМЕСТИТЕЛЬ, ПРЕДСТАВЛЯЮЩИЙ СОБОЙ БУТАН, В ГЕТЕРОЦИКЛИЧЕСКОМ КОЛЬЦЕ ПРОИЗВОДНОЕ ПИРИДОНА ДЛЯ ЛЕЧЕНИЯ ФИБРОЗА И ВОСПАЛИТЕЛЬНЫХ ЗАБОЛЕВАНИЙ | 2017 |
|
RU2738844C2 |
| ИНГИБИТОРЫ ОКСИДАЗЫ D-АМИНОКИСЛОТ И ИХ ТЕРАПЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2018 |
|
RU2795513C2 |
| ПРОИЗВОДНЫЕ ПИРРОЛО[2,3-b]ПИРИДИНА, ИНГИБИРУЮЩИЕ АКТИВНОСТЬ АНТИАПОПТИЧЕСКИХ БЕЛКОВ Bcl-2 | 2011 |
|
RU2567857C2 |
| ИНДУЦИРУЮЩИЕ АПОПТОЗ СРЕДСТВА ДЛЯ ЛЕЧЕНИЯ РАКА И ИММУННЫХ И АУТОИММУННЫХ ЗАБОЛЕВАНИЙ | 2010 |
|
RU2539587C2 |
| ТЕРАПЕВТИЧЕСКИЕ СОЕДИНЕНИЯ И КОМПОЗИЦИИ | 2015 |
|
RU2733405C2 |
| ГИДРОКСИМЕТИЛЦИКЛОГЕКСИЛАМИНЫ | 2009 |
|
RU2514192C2 |
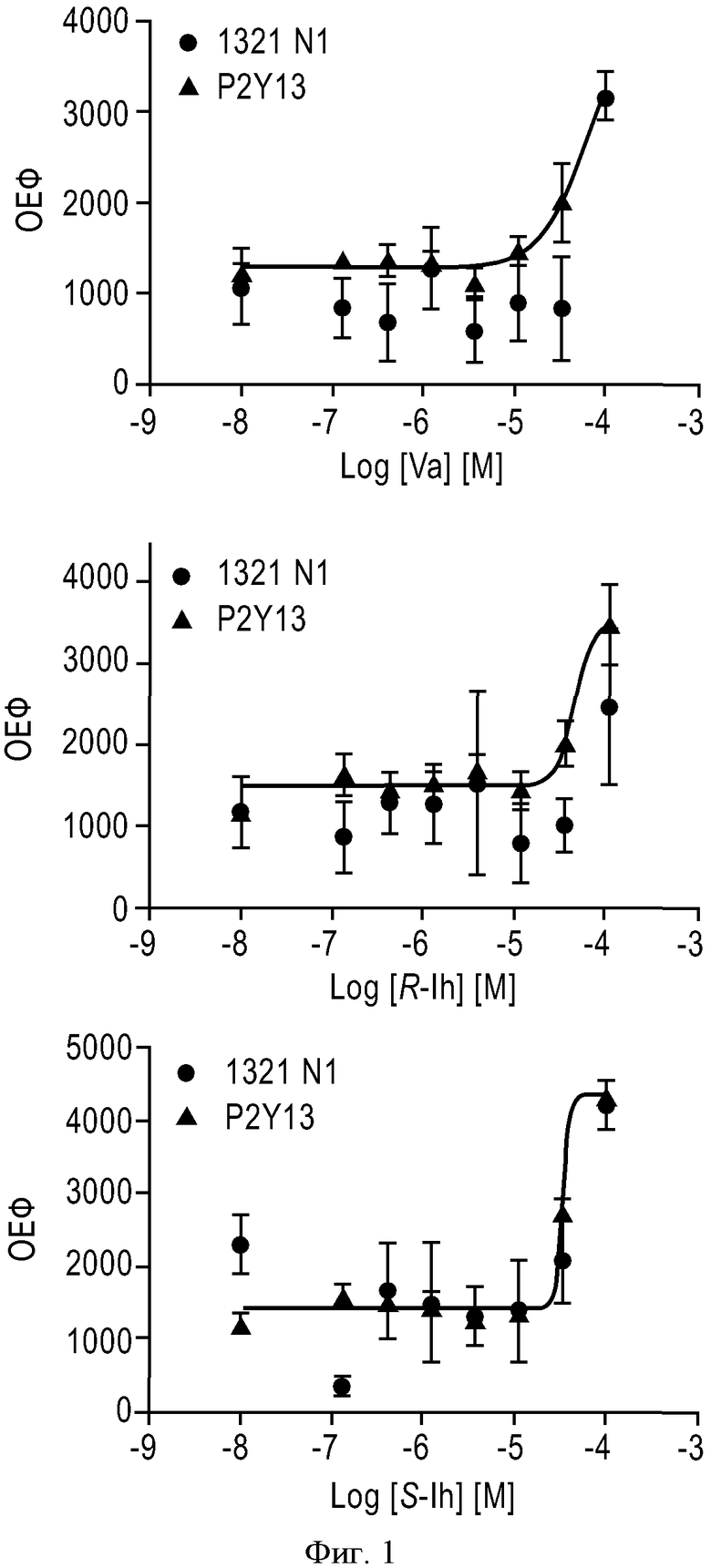
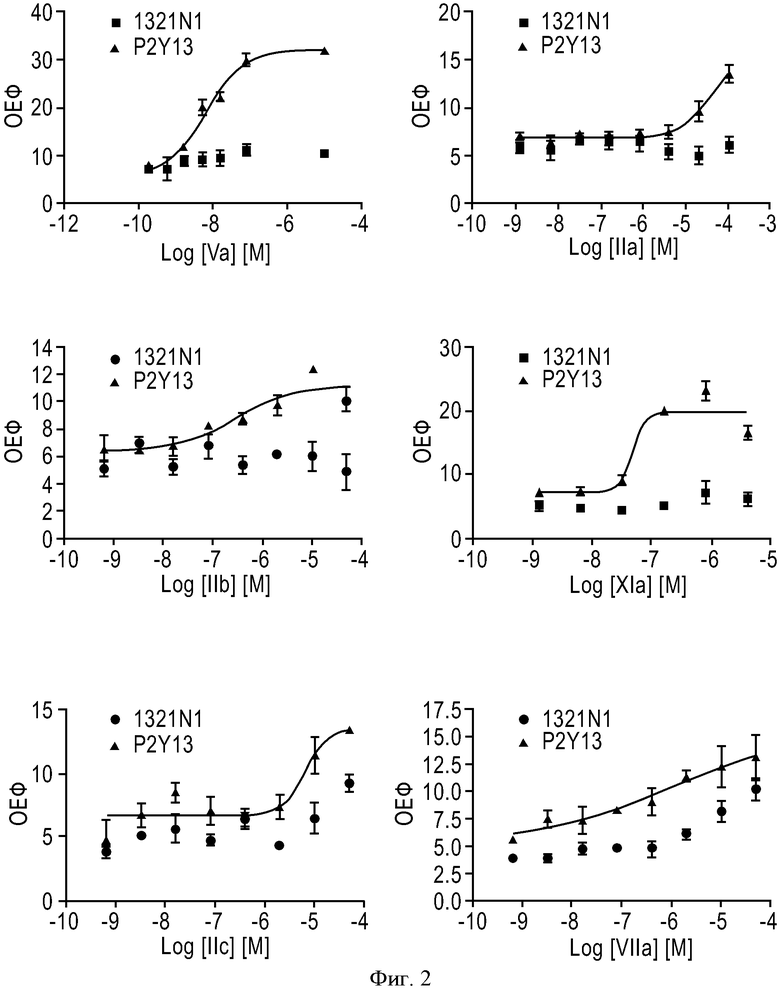
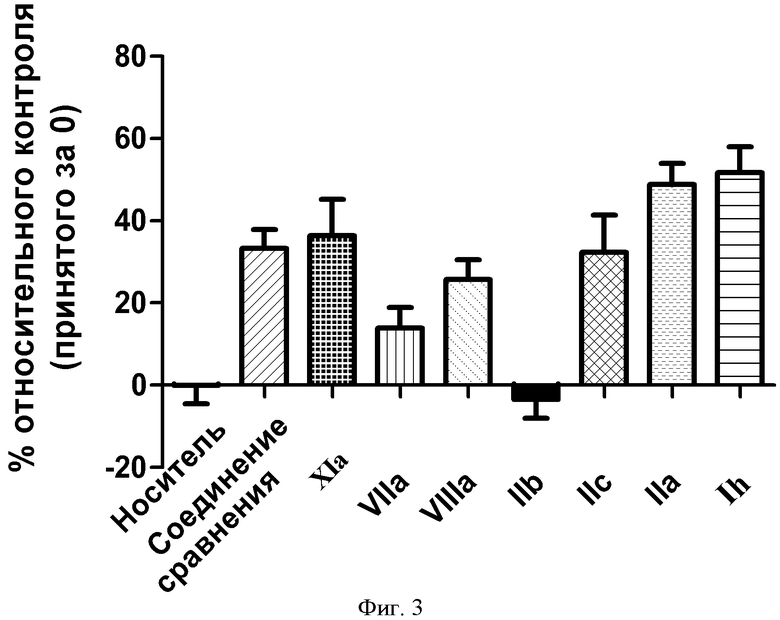
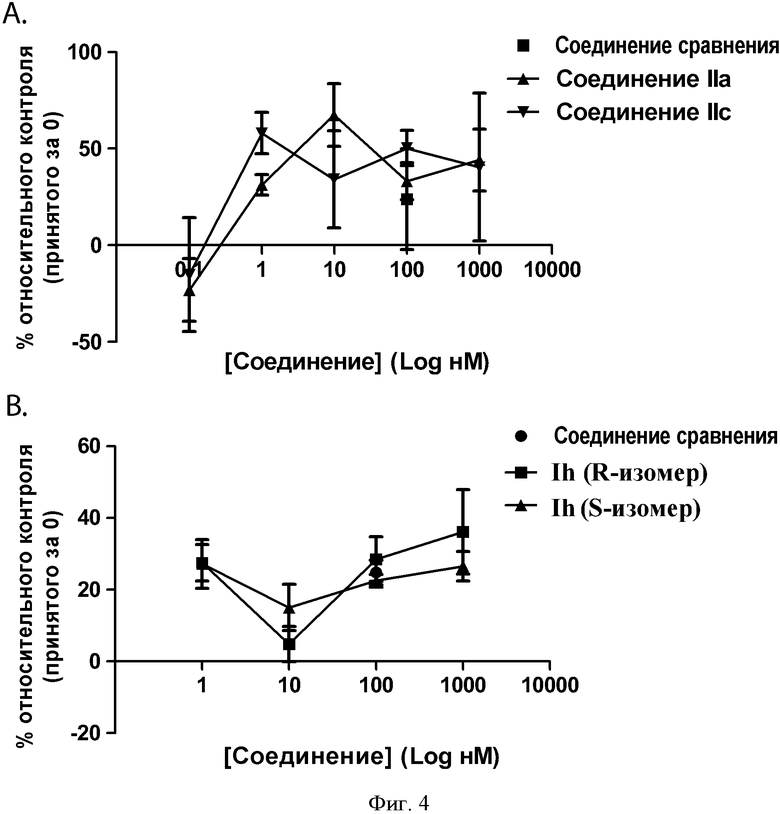
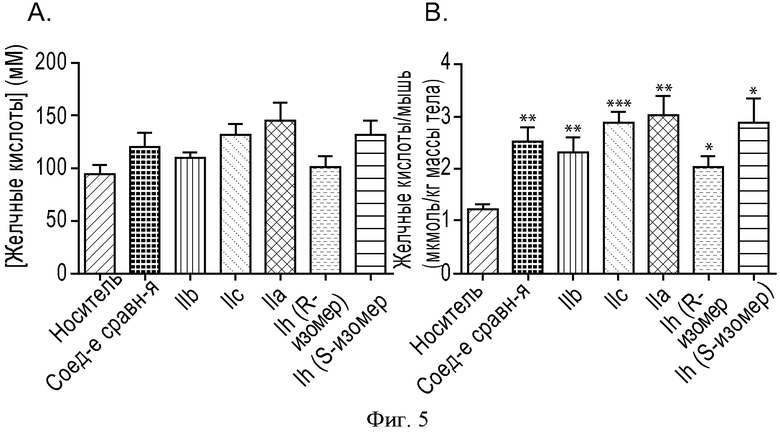
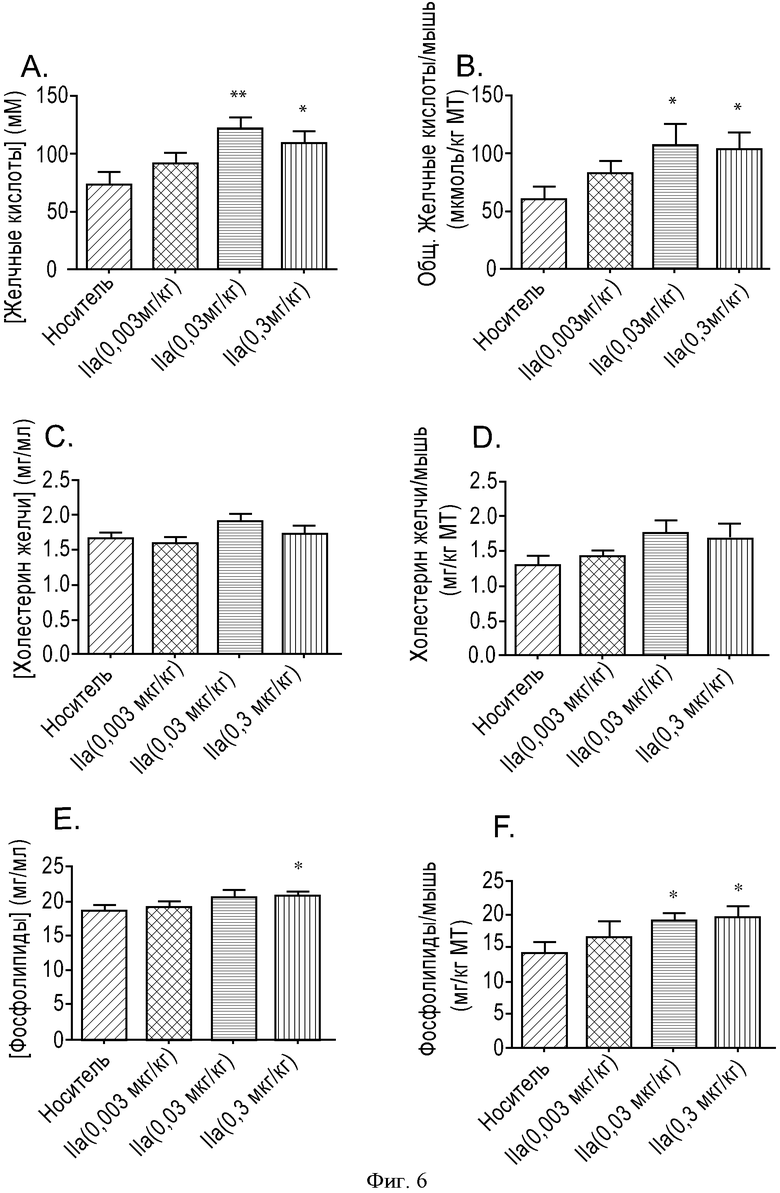
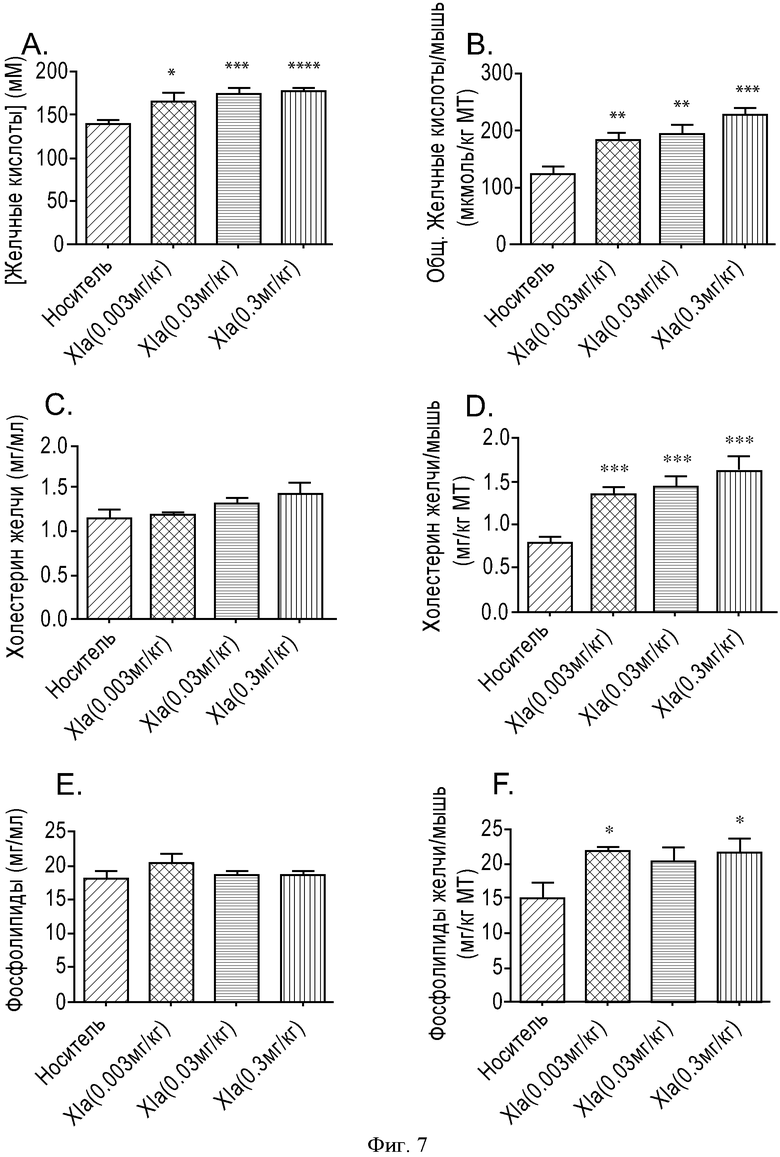
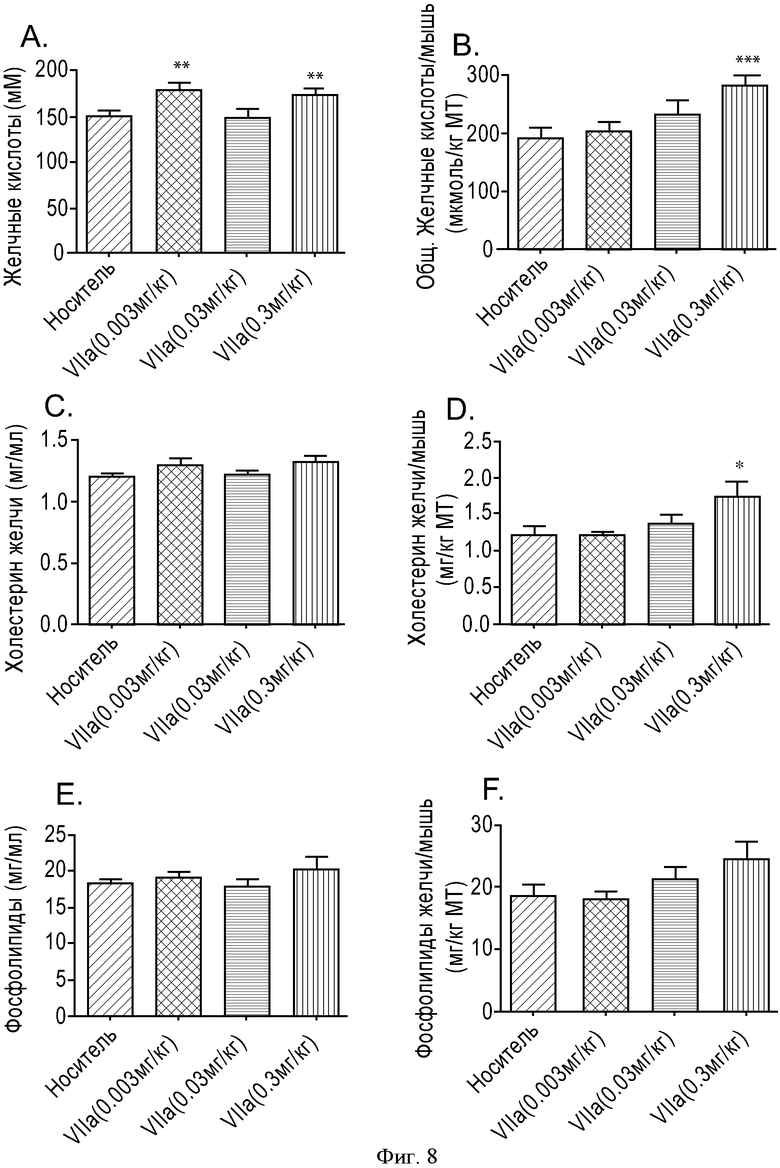
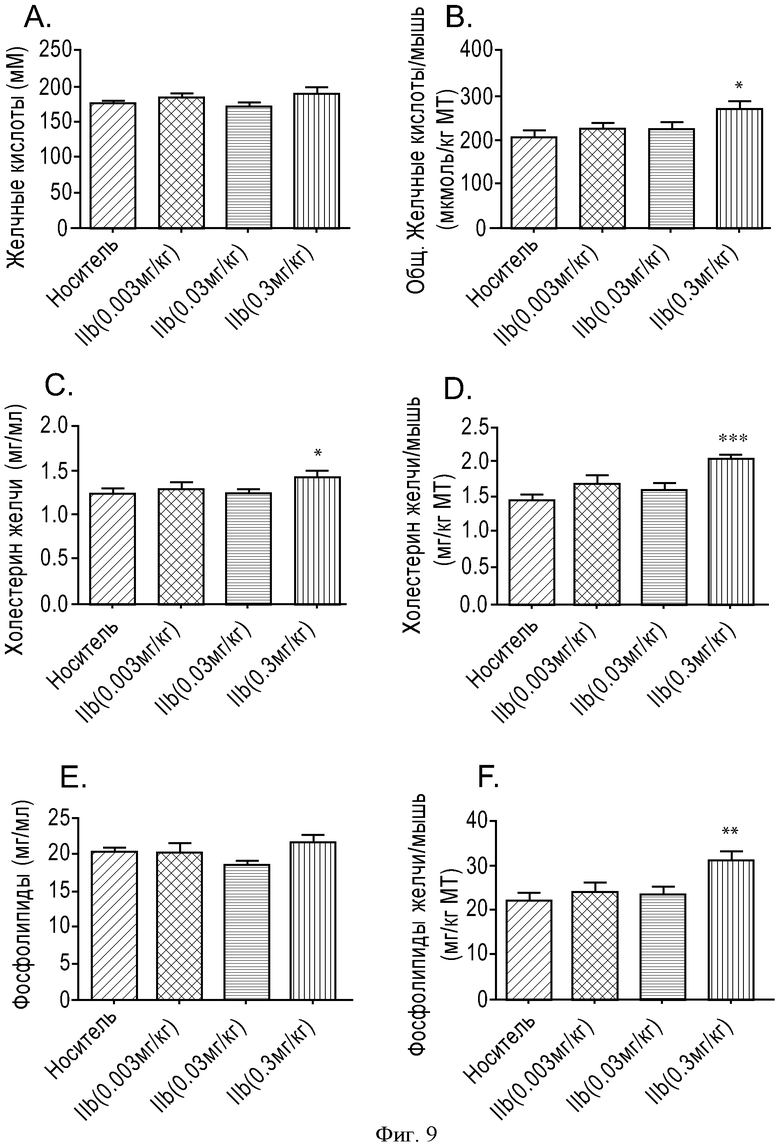
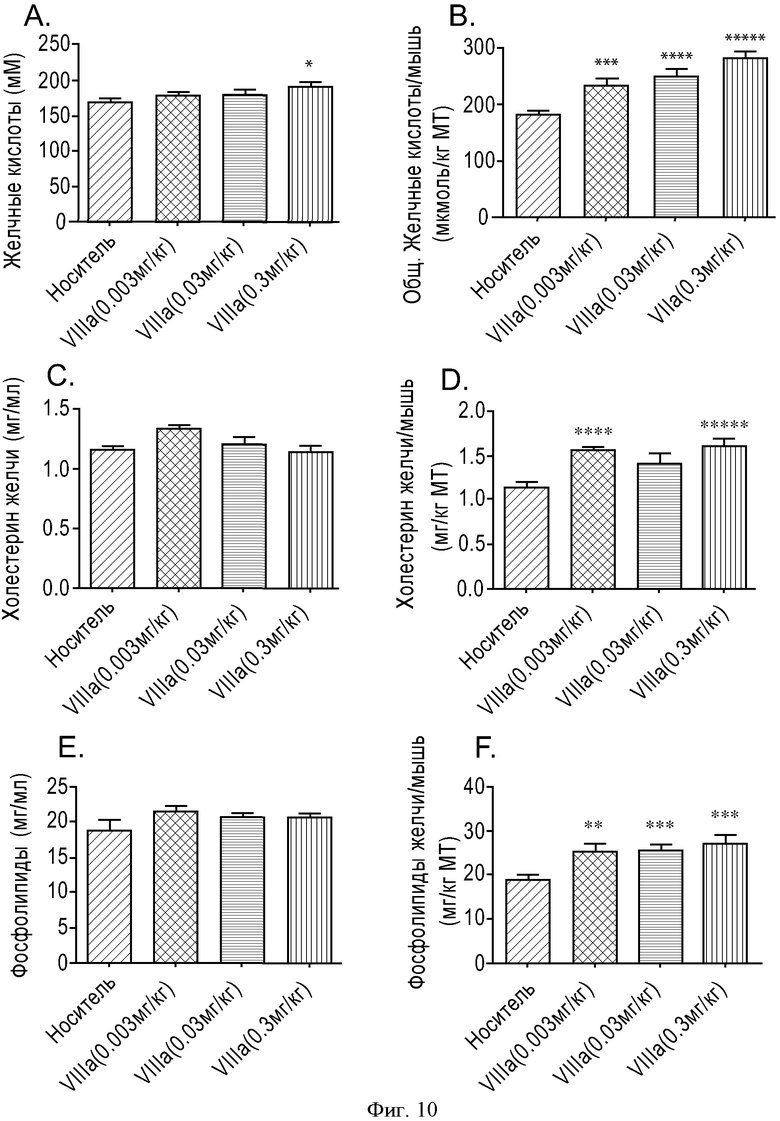
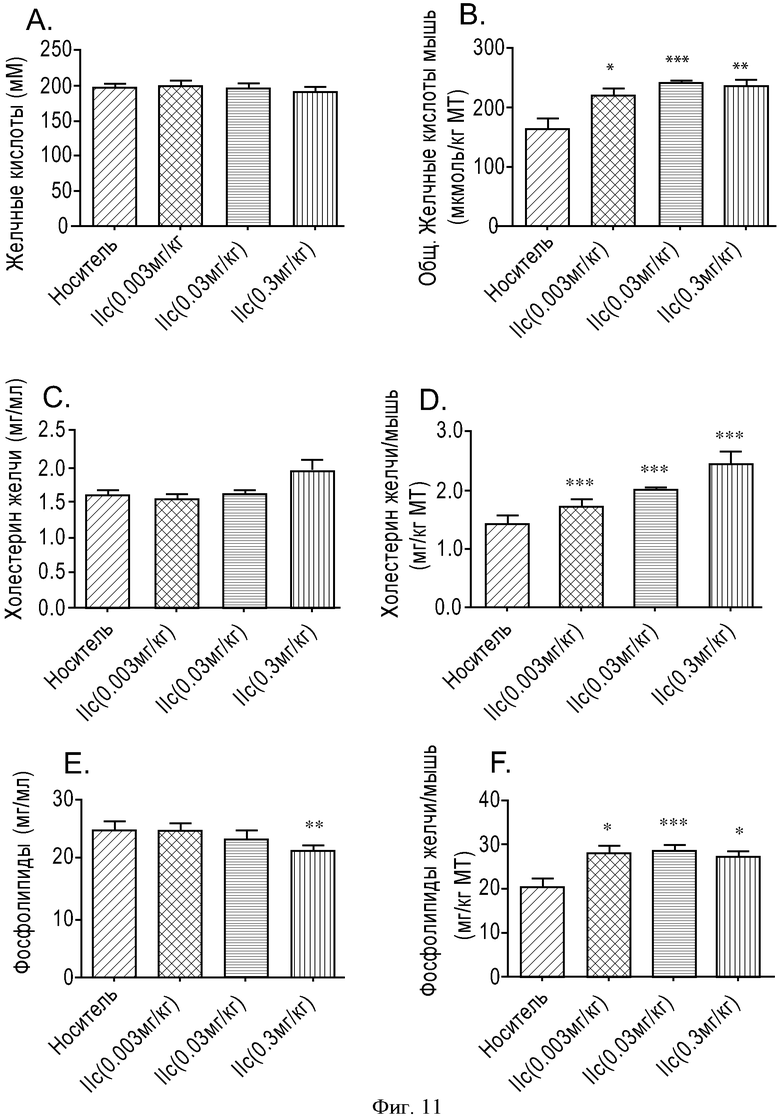
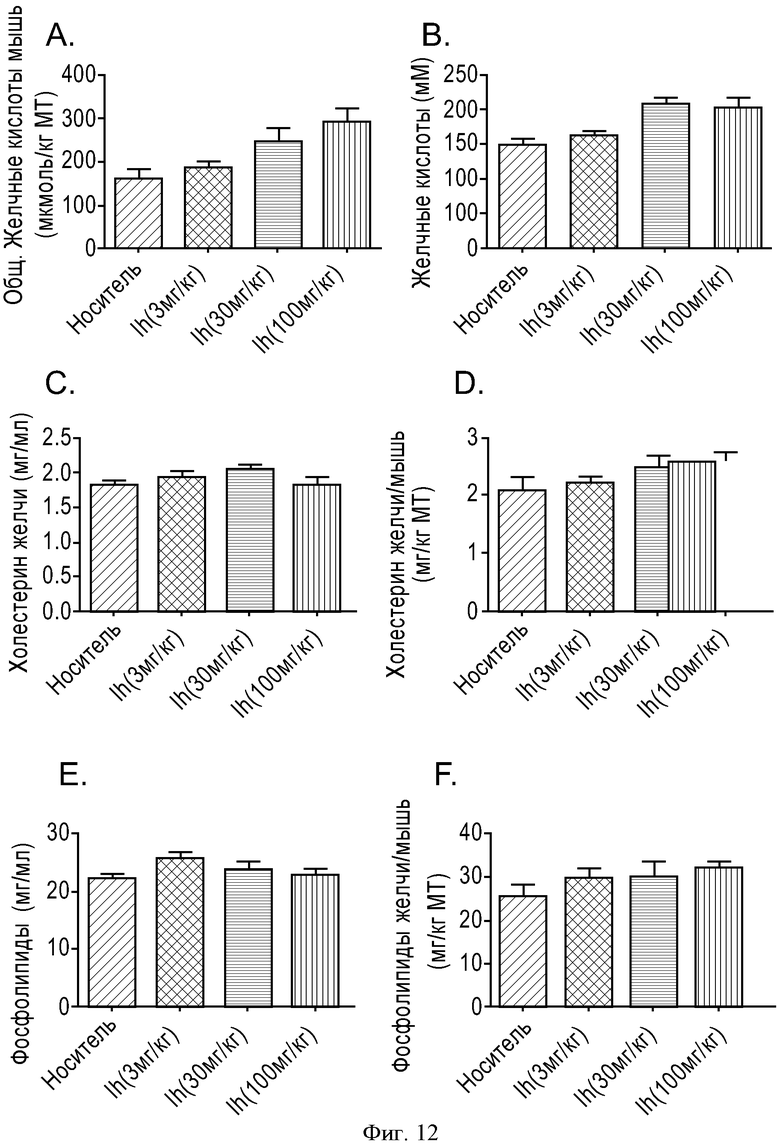
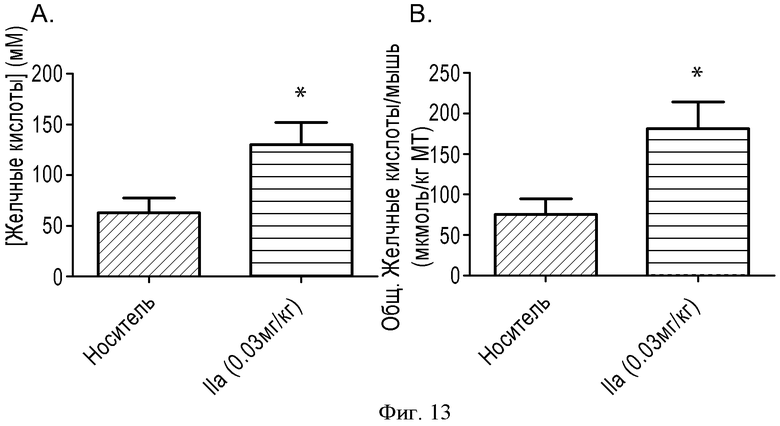
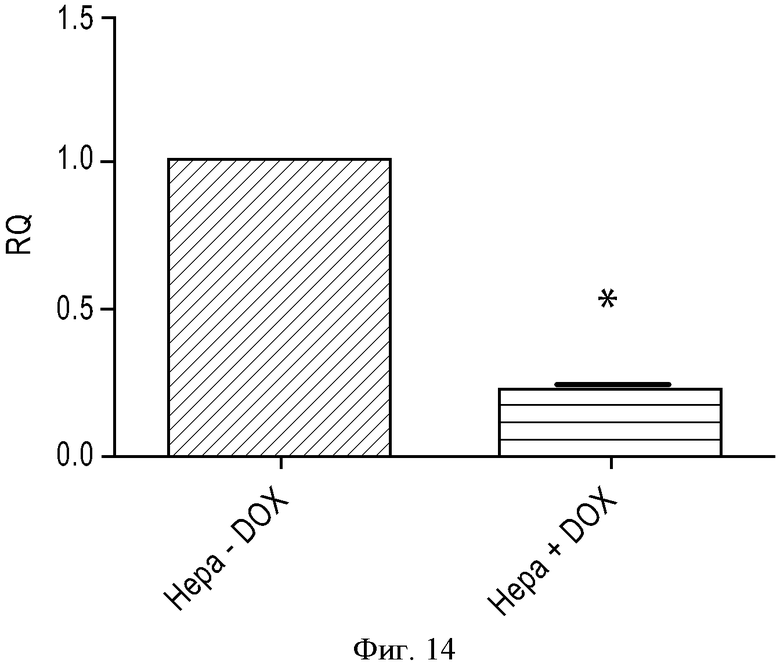
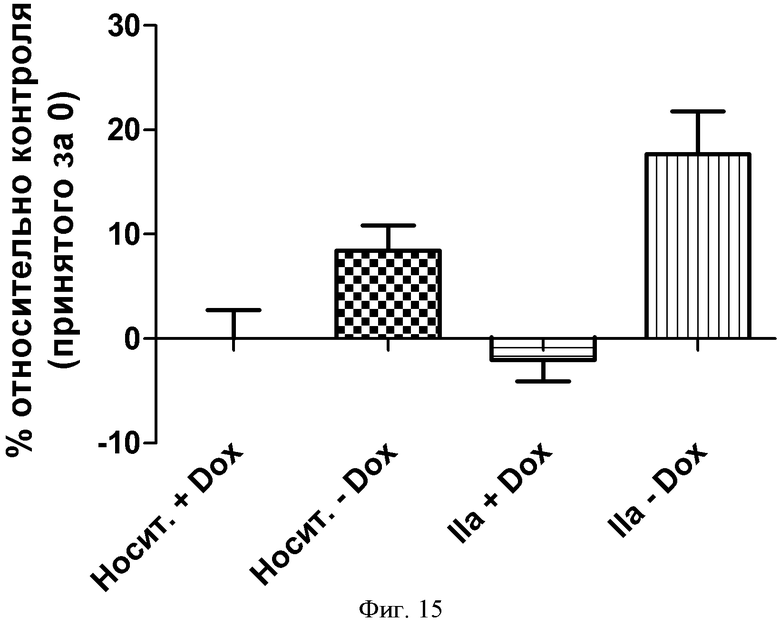
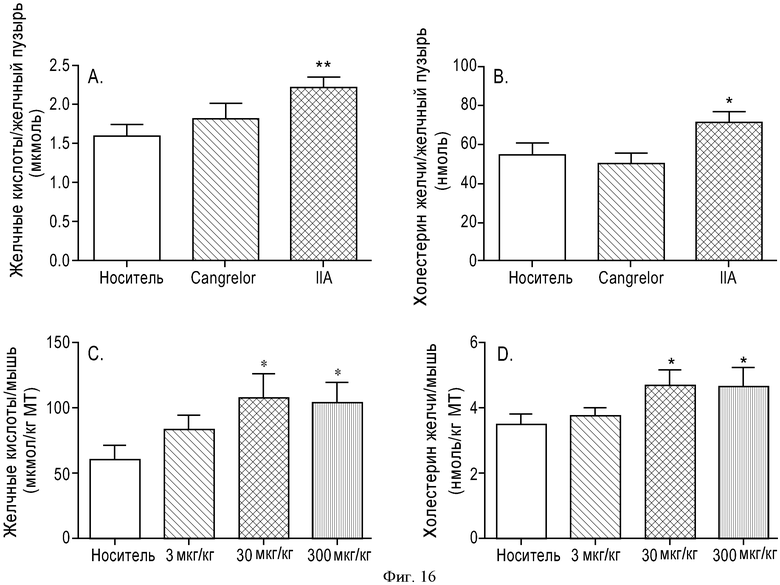
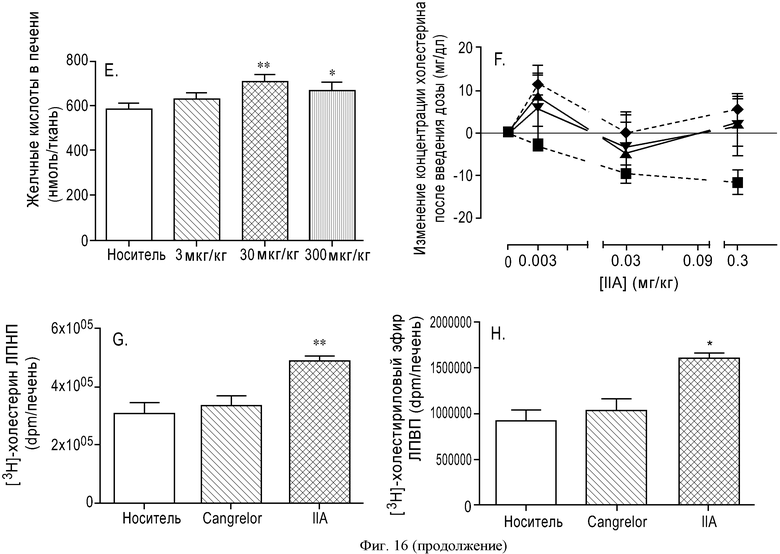
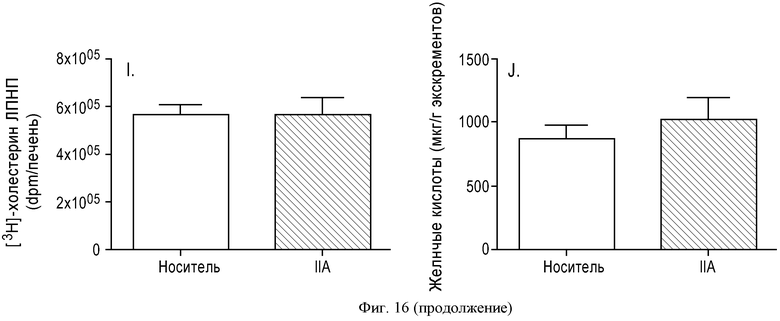
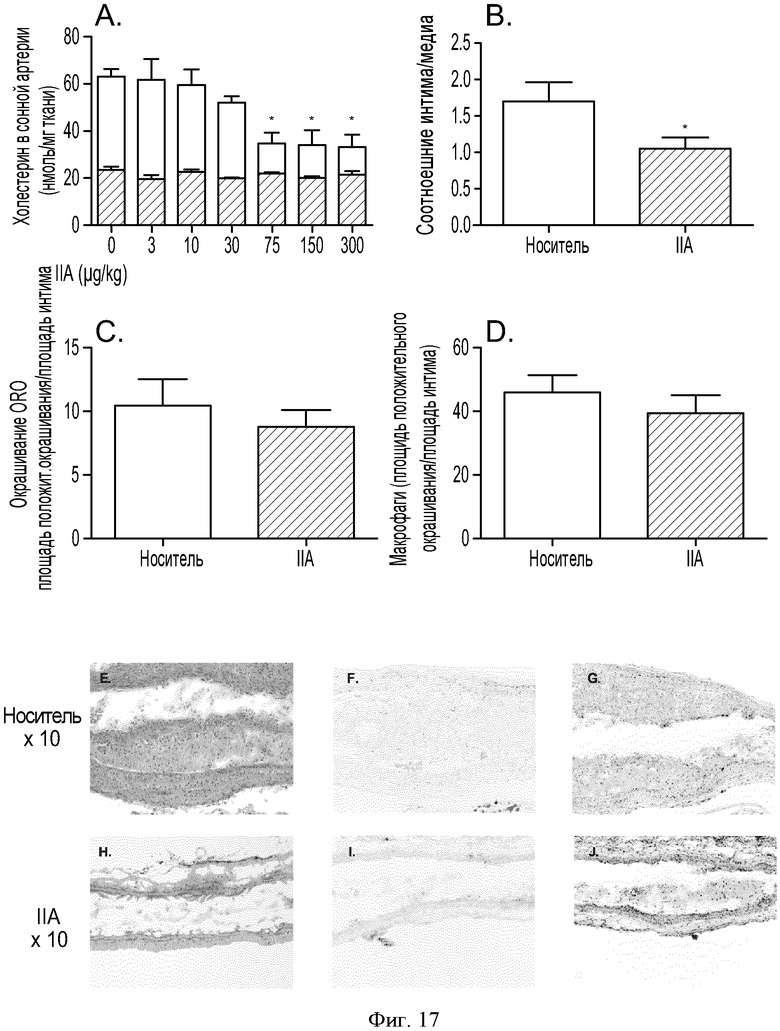
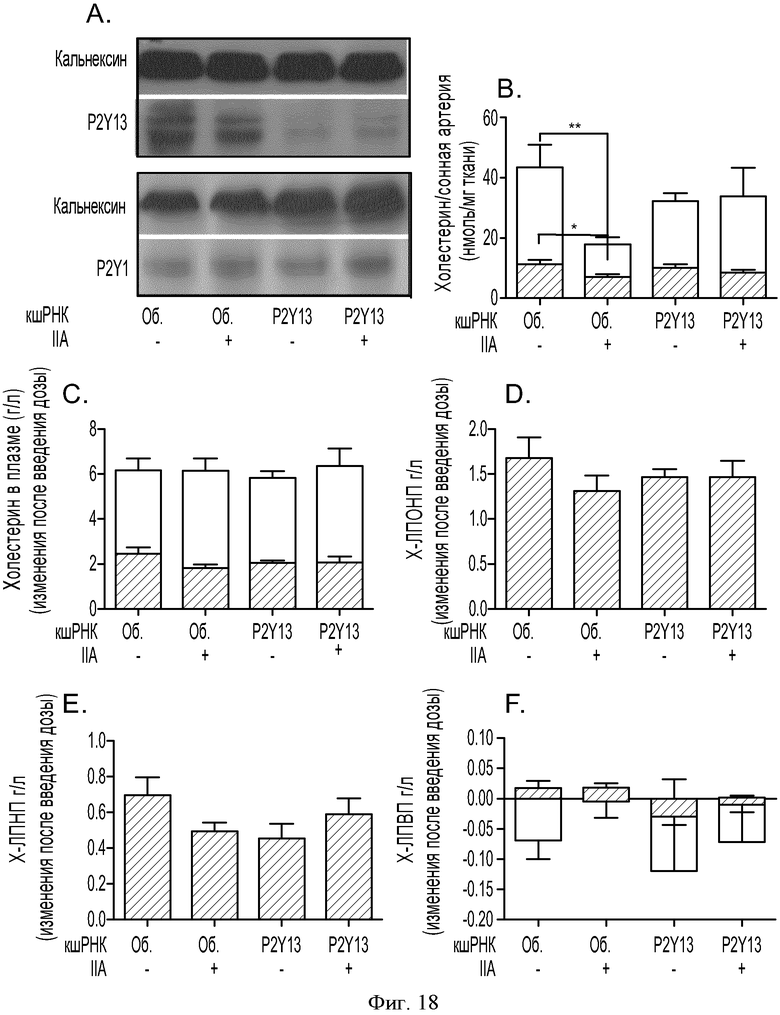
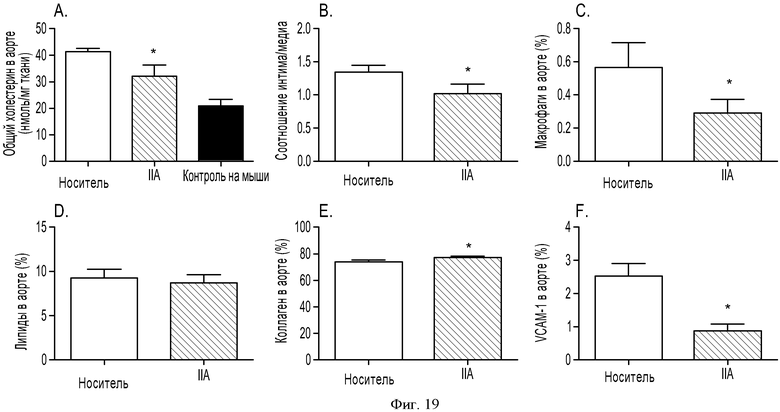
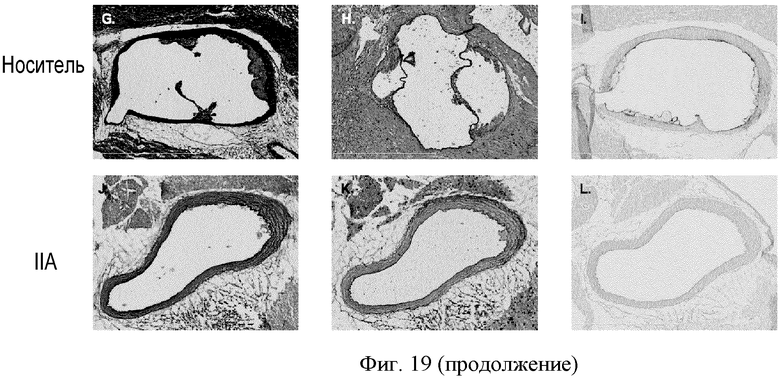
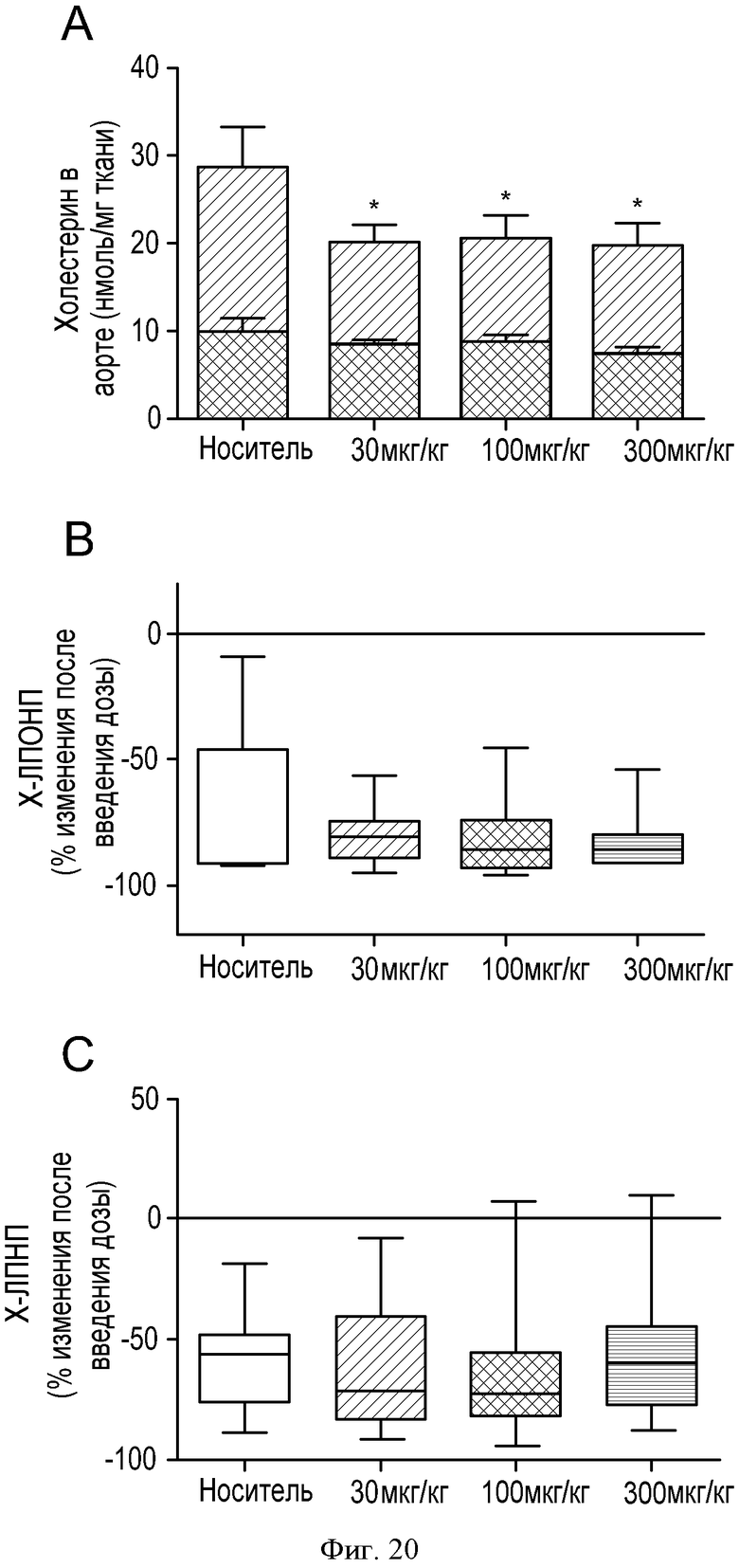
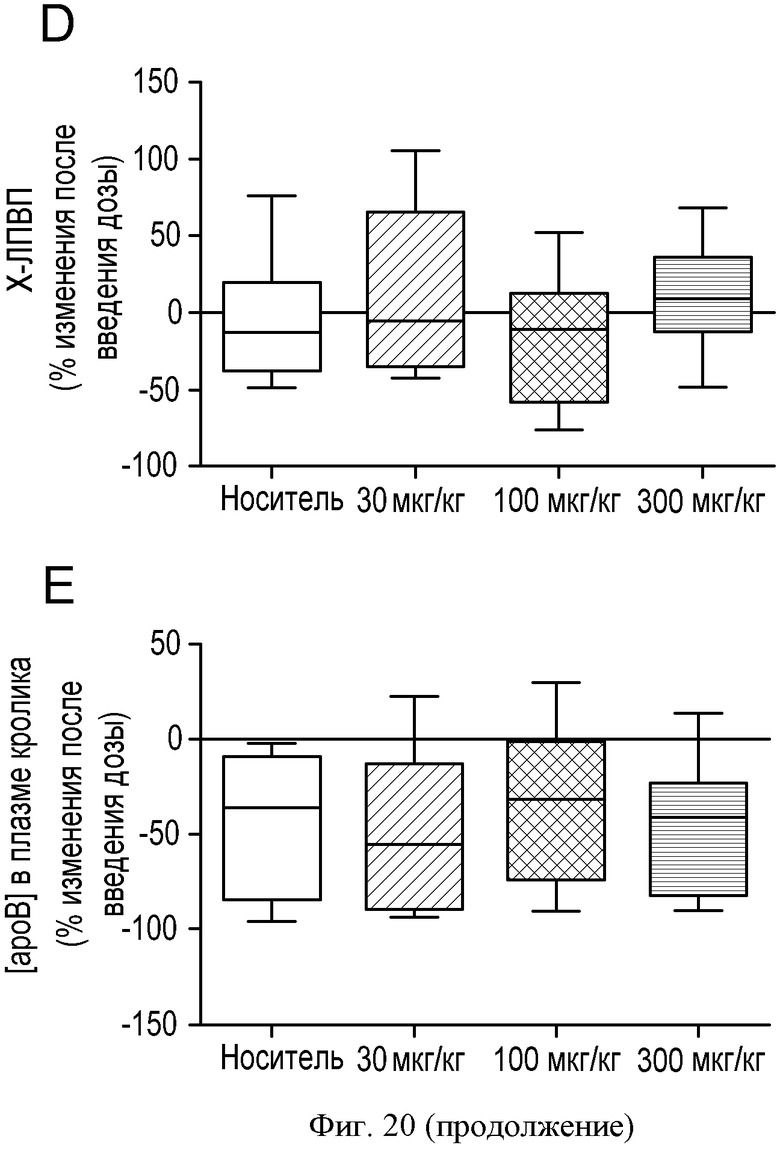
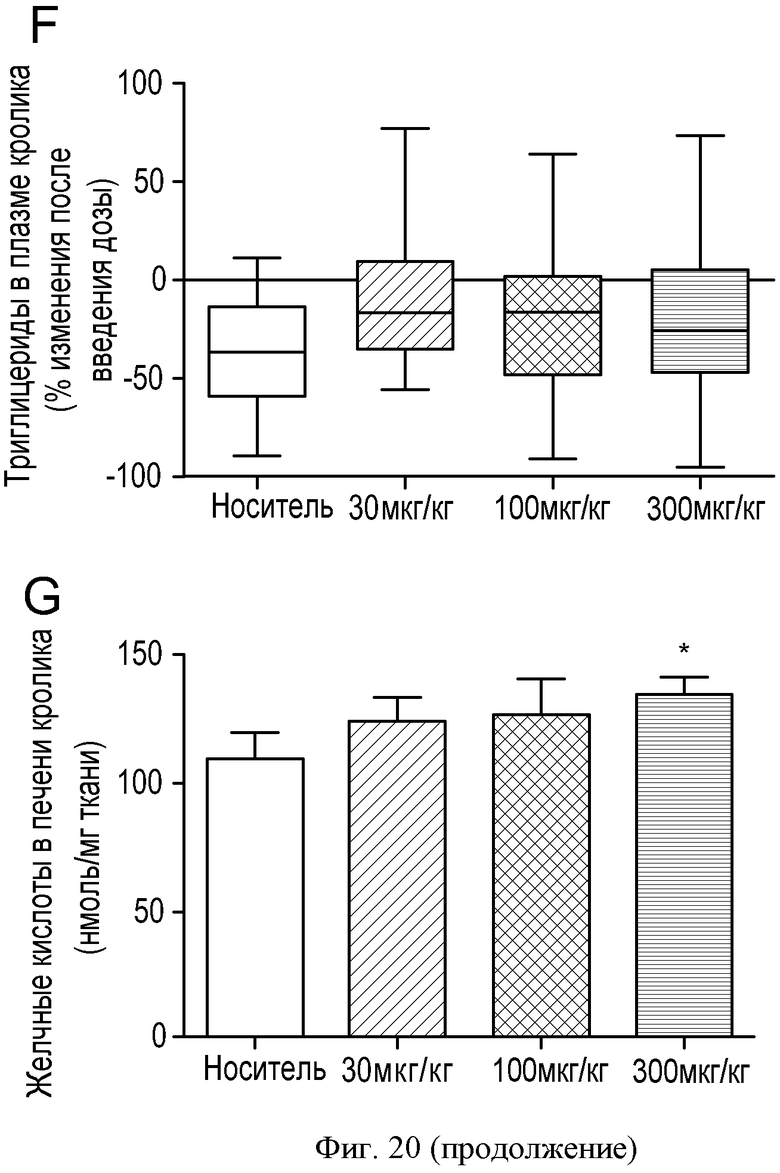
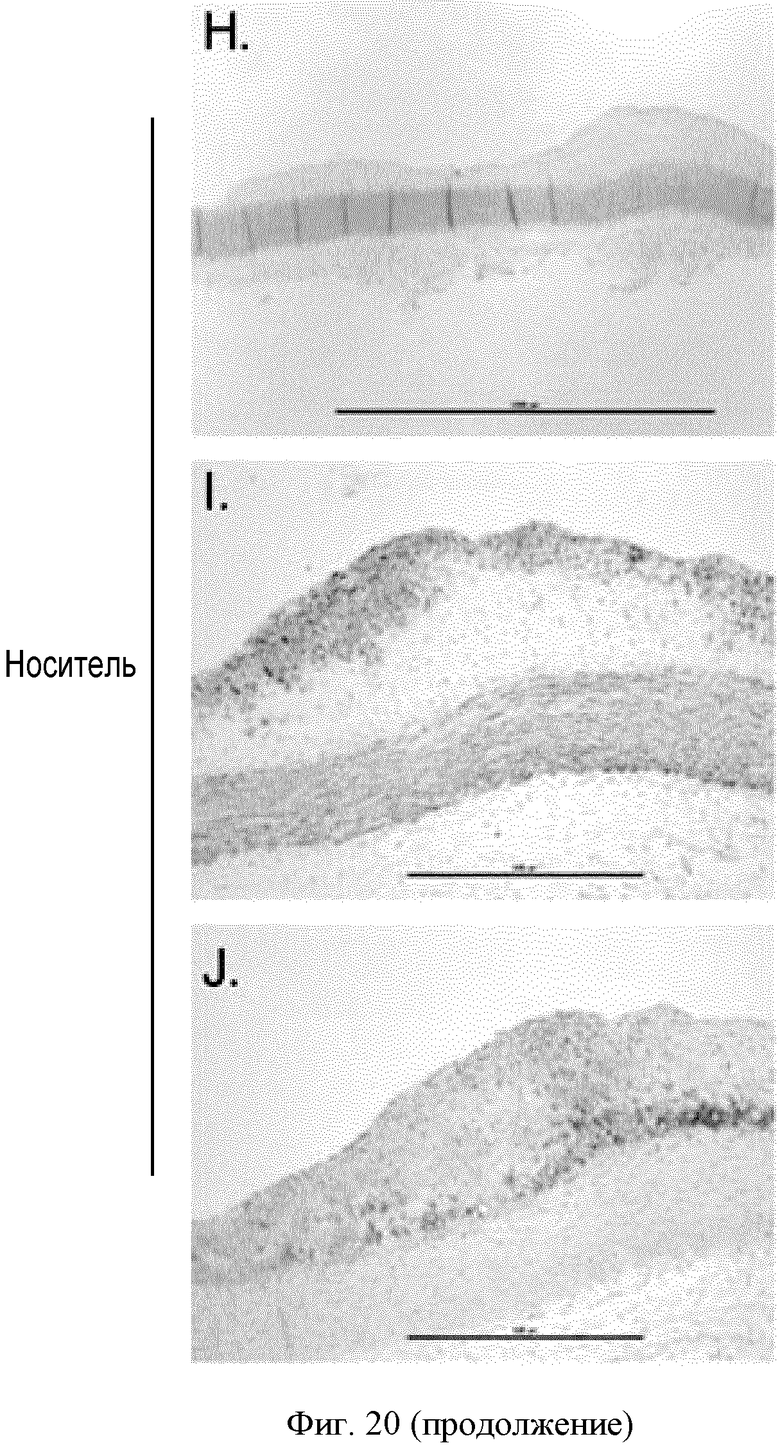
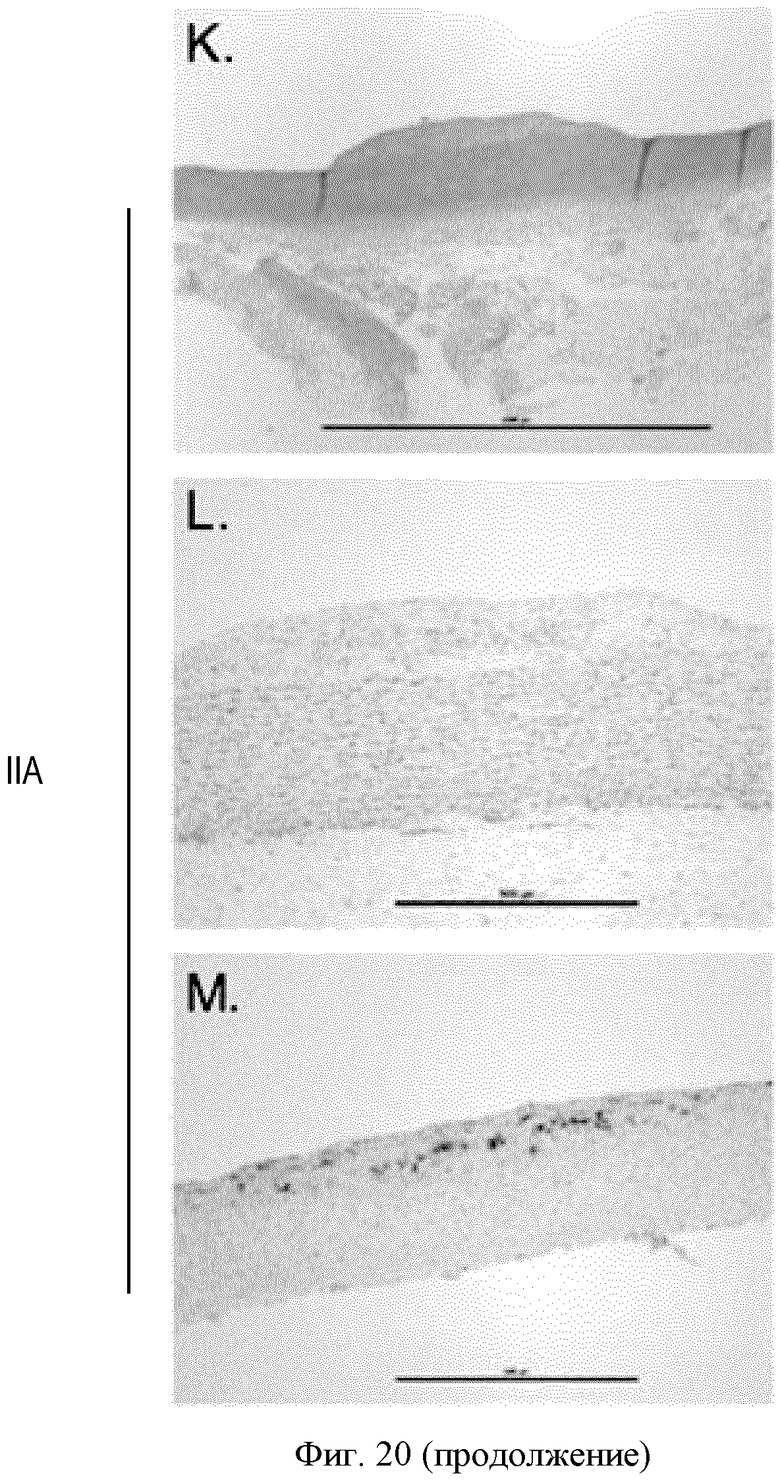
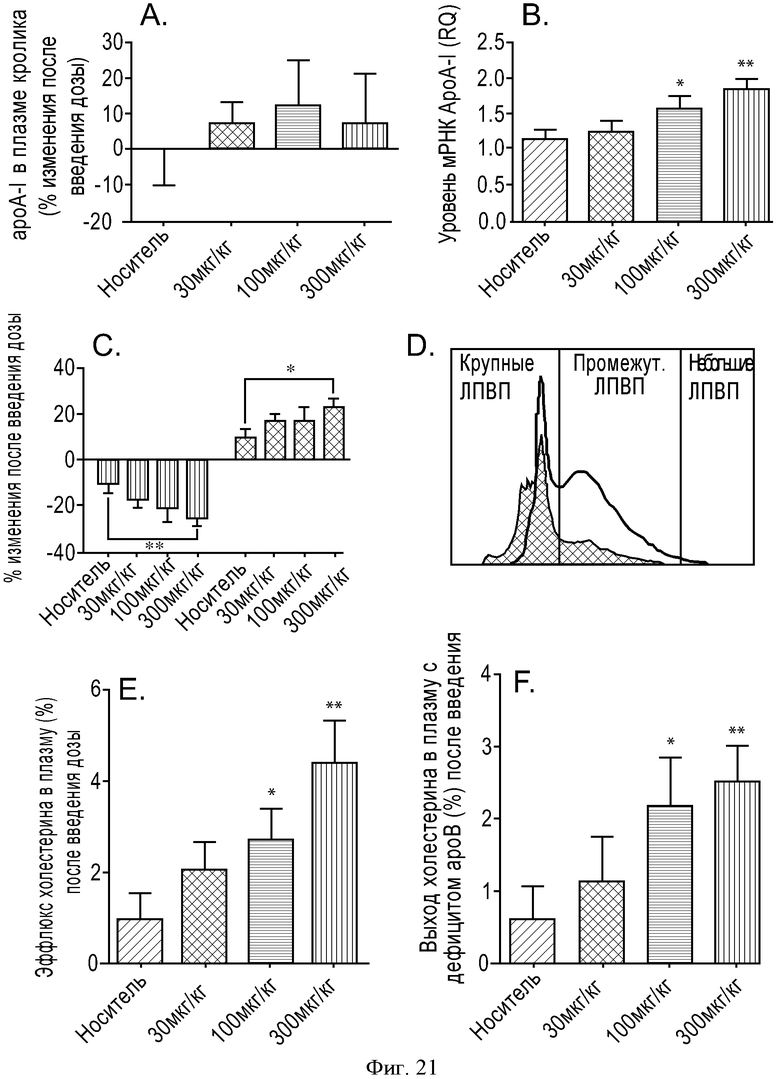
Изобретение относится к области органической химии, а именно к новому гетероциклическому соединению общей формулы (II) или к его фармацевтически приемлемой соли, где каждый R9 представляет собой -Н; каждый R10 независимо представляет собой -Н, -ОН, NH(метил), -N(метил)(метил) или -О-метил; каждый из Q1, Q2 и Q3 независимо представляет собой CR10 или N; X представляет собой CHR10 или О; и каждый m представляет собой 1. Также изобретение относится к фармацевтической композиции на основе соединения формулы (II), способу лечения, основанному на использовании соединения формулы (II), и способу определения уровня активности P2Y13 у субъекта, основанному на использовании соединения формулы (II). Технический результат: получены новые соединения, обладающие агонистической активностью в отношении рецептора P2Y13. 9 н. и 9 з.п. ф-лы, 28 ил., 4 табл., 20 пр.
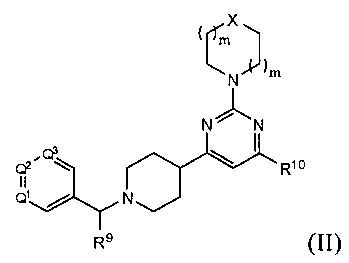
1. Соединение Формулы (II):
или его фармацевтически приемлемая соль, где
каждый R9 представляет собой -Н;
каждый R10 независимо представляет собой -Н, -ОН, NH(метил), -N(метил)(метил) или -О-метил;
каждый из Q1, Q2 и Q3 независимо представляет собой CR10 или N;
X представляет собой CHR10 или О; и
каждый m представляет собой 1.
2. Соединение или фармацевтически приемлемая соль по п.1, отличающиеся тем, что R9 представляет собой Н, a R10 представляет собой ОН.
3. Соединение по п.1, отличающееся тем, что соединение представляет собой:
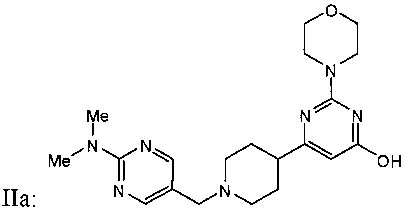 ;
; 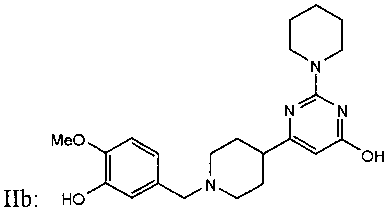 ;
;
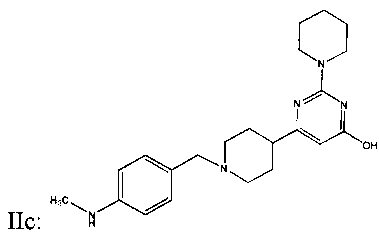 ;
;
или фармацевтически приемлемую соль любого из приведенных выше соединений.
4. Фармацевтическая композиция, обладающая агонистической активностью в отношении рецептора P2Y13, содержащая эффективное количество соединения или фармацевтически приемлемой соли соединения по п.1 и фармацевтически приемлемый наполнитель или носитель.
5. Фармацевтическая композиция по п.4, отличающаяся тем, что указанная композиция приготовлена в форме для перорального введения.
6. Фармацевтическая композиция по п.4, отличающаяся тем, что указанная композиция находится в форме таблетки или капсулы.
7. Фармацевтическая композиция по п.4, отличающаяся тем, что соединение или его соль присутствуют в количестве от примерно 1 мг до примерно 1000 мг.
8. Способ лечения нарушения метаболизма липопротеинов, включающий введение субъекту, нуждающемуся в этом, эффективного количества соединения или фармацевтически приемлемой соли соединения по п.1.
9. Способ по п.8, отличающийся тем, что нарушение метаболизма липопротеинов представляет собой дислипидемию, дислипопротеинемию, избыточную выработку или дефицит липопротеинов, повышение уровня общего холестерина, повышение концентрации липопротеинов низкой плотности, повышение концентрации триглицеридов, выведение липидов в желчь, выведение фосфолипидов в желчь, выведение оксистерола в желчь или нарушение, связанное с рецептором, активируемым пролифератором пероксисом.
10. Способ лечения нарушения метаболизма глюкозы, включающий введение субъекту, нуждающемуся в этом, эффективного количества соединения или фармацевтически приемлемой соли соединения по п.1.
11. Способ по п.10, отличающийся тем, что нарушение метаболизма глюкозы представляет собой инсулинорезистентность, нарушенную переносимость глюкозы, аномальный уровень глюкозы в крови натощак, сахарный диабет, липодистрофию, центральное ожирение, периферическую липоатрофию, диабетическую нефропатию, диабетическую ретинопатию, заболевания почек или септицемию.
12. Способ лечения атеросклероза, включающий введение субъекту, нуждающемуся в этом, эффективного количества соединения или фармацевтически приемлемой соли соединения по п.1.
13. Способ по п.12, отличающийся тем, что указанный способ обеспечивает ослабление по меньшей мере одного симптома атеросклероза.
14. Способ по п.13, отличающийся тем, что указанный симптом представляет собой импотенцию.
15. Способ лечения воспаления, включающий введение субъекту, нуждающемуся в этом, эффективного количества соединения или фармацевтически приемлемой соли соединения по п.1.
16. Способ лечения панкреатита, включающий введение субъекту, нуждающемуся в этом, эффективного количества соединения или фармацевтически приемлемой соли соединения по п.1.
17. Способ лечения аномальной выработки желчи, включающий введение субъекту, нуждающемуся в этом, эффективного количества соединения или фармацевтически приемлемой соли соединения по п.1.
18. Способ определения уровня активности P2Y13 у субъекта, включающий:
a. определение уровня свободного холестерина в липопротеинах высокой плотности в крови субъекта;
b. введение субъекту соединения по п.1;
c. отслеживание уровня свободного холестерина в крови субъекта в течение периода времени после введения соединения; и
d. оценку уровня активности P2Y13 у субъекта на основании сравнения результатов, полученных на стадиях а. и с.
| Кипятильник для воды | 1921 |
|
SU5A1 |
| AU 2006201262 A1, 27.04.2006 | |||
| Прибор для испытания цилиндрических дружин | 1929 |
|
SU13811A1 |

