РОДСТВЕННЫЕ ЗАЯВКИ
Согласно 35 U.S.C. § 119(e) по настоящей заявке испрашивается приоритет предварительной патентной заявки U.S.S.N. 61/937,031, поданной 7 февраля, 2014, которая включена в настоящий документ в качестве ссылки.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Свертывание крови является первой линией обороны против потери крови после травмы. "Каскад" системы свертывания крови включает в себя некоторое количество проферментов, циркулирующих сериновых протеаз, регуляторные кофакторы и ингибиторы. Каждый фермент, после получения из профермента, специфически расщепляет следующий профермент в каскаде с получением активной протеазы. Этот процесс повторяется до тех пор, пока наконец тромбин не расщепит фибринопептиды от фибриногена с образованием фибрина, который полимеризуется с образованием сгустка крови. Хотя эффективное свертывание ограничивает потери крови на месте травмы, он также создает риск системного свертывания крови, что приводит к массовому тромбозу. В норме гемостаз поддерживает баланс между формированием тромба (коагуляция) и растворением тромба (фибринолиза). Однако, при некоторых заболеваниях, таких как острый инфаркт миокарда и нестабильная стенокардия, отрыв существующей атеросклеротической бляшки приводит к патологическому тромбообразованию в сосудах коронарных артерий.
Заболевания, которые основаны на свертывании крови, такие как инфаркт миокарда, нестабильная стенокардия, фибрилляция предсердий, инсульт, эмболия легочной артерии и тромбоз глубоких вен, являются одной из ведущих причин смерти в развитых странах. Существующая антикоагулянтная терапия, такая как нефракционированный и низкомолекулярный (LMW) гепарин, вводимый путем инъекции, и варфарин (кумадин), вводимый перорально, несет риск возникновения кровотечений и показывает различие среди пациентов, что приводит к необходимости строгого наблюдения и титрования терапевтических доз. Следовательно, существует значительная медицинская потребность в новых антикоагулянтных препаратах, которые не вызывают некоторые или все эти побочные эффекты препаратов, существующих в настоящее время.
Фактор XIa является привлекательной терапевтической мишенью, вовлеченной в путь, связанный с этими заболеваниями. Повышенные уровни активности Фактора XIa или Фактора XIa наблюдали при некоторых тромбоэмболических нарушениях, включая тромбоз вен (Meijers et al., N. Engl. J. Med. 342:696, 2000), острый инфаркт миокарда (Minnema et al., Arterioscler Thromb Vasc Biol 20:2489, 2000), острый коронарный синдром (Butenas et al., Thromb Haemost 99:142, 2008), болезнь коронарных артерий (Butenas et al., Thromb Haemost 99:142, 2008), хроническое обструктивное заболевание легких (Jankowski et al., Thromb Res 127:242, 2011), стеноз аорты (Blood Coagul Fibrinolysis, 22:473, 2011), острая ишемия сосудов мозга (Undas et al., Eur J Clin Invest, 42:123, 2012) и систолическая сердечная недостаточность в результате ишемической кардиомиопатии (Zabcyk et al., Pol Arch Med Wewn. 120:334, 2010). У пациентов, у которых отсутствует Фактор XI, в результате генетической недостаточности Фактора XI, наблюдаются единичные случаи, если вообще наблюдаются, ишемического инсульта (Salomon et al., Blood, 111:4113, 2008). В то же время, потеря активности Фактора XIa, который отсутствует в одном из путей, которые инициируют первоначальную коагуляцию, не нарушает гомеостаз. В организме человека дефицит Фактора XI может привести к нарушению свертываемости крови, от легкой до умеренной, особенно в тканях с высоким уровнем местной фибринолитической активности, таких как мочевыводящие пути, нос, ротовая полость и миндалины. Кроме того, у мышей с дефицитом фактора XI гомеостаз практически не нарушается (Gailani, Blood Coagul Fibrinolysis, 8:134, 1997). Следовательно, соединения, которые ингибируют Фактор XIa, обладают возможностью профилактики или лечения широкого спектра тромбоэмболических нарушений, при этом не вызывая побочных эффектов и терапевтических трудностей, которые вызывают противотромбозные препараты, которые ингибируют другие компоненты пути коагуляции. Кроме того, из-за ограниченной эффективности и неблагоприятных побочных эффектов, некоторые существующие лекарственные средства для ингибирования нежелательного тромбоза (например, тромбоз глубоких вен и инсульт), для профилактики или лечения нежелательного тромбоза необходимы более совершенные соединения и способы (например, соединения и способы, которые связаны с Фактором XIa).
Еще одной терапевтической мишенью является фермент калликреин. Калликреин плазмы человека представляет собой сериновую протеазу, которая может отвечать за активацию нескольких факторов в последующих стадиях пути свертывания (например, брадикинин и плазмин), которые являются важными для коагуляции и контроля, например, артериального давления, воспаления и боли. Каллекреины экспрессируются, например, в предстательной железе, эпидермисе и центральной нервной системе (ЦНС) и могут участвовать, например, в регуляции сжижения семенной жидкости, расщеплении белков клеточной адгезии и пластичности нейронов в ЦНС. Кроме того, каллекреины могут быть вовлечены в опухолеобразование и развитие рака, а также в ангионевротический отек, например, в наследственный ангионевротический отек. Повышенная активность калликреин-кининового пути может привести к ряду нарушений, включая ангионевротический отек, например, наследственный ангионевротический отек (Schneider et al., J. Allergy Clin. Immunol. 120:2, 416, 2007). На настоящий момент, существует ограниченное число вариантов лечения HAE (например, WO2003/076458). Таким образом, существует необходимость в лекарственных средствах для профилактики или лечения этих заболеваний.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к соединениям, которые ингибируют Фактор XIa или калликреин, и к способам профилактики или лечения нежелательного тромбоза или ангионевротического отекa (например, наследственный ангионевротический отек) путем введения одного или нескольких этих соединений самостоятельно или в комбинации с другими молекулами млекопитающему. Настоящее изобретение также относится к способам разработки или выбора дополнительных ингибиторов Фактора XIa или калликреина, используя эти структуры. Желательно, эти соединения имеют определенные структурные, физические и пространственные характеристики, позволяющие соединениям взаимодействовать с конкретным остатком на активном сайте Фактора XIa или калликреина.
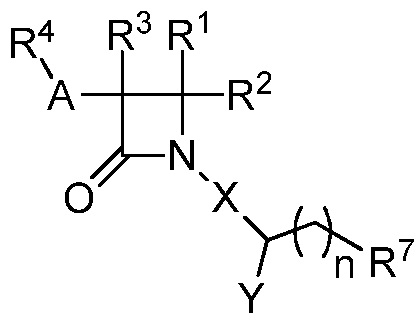
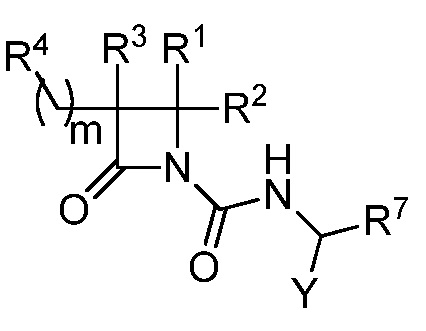
В одном из аспектов настоящее изобретение относится к соединению формулы (I):
 (I),
(I),
или к его фармацевтически приемлемой соли, где R1 представляет собой H или -C1-6 алкил; R2 представляет собой Н, -C1-6 алкил, -CO2R5, -C(O)NR9R10, -CN, -SOqR5, -OR5, -CHN(OR5) или гетероарил; R3 представляет собой Н или -C1-6 алкил; A представляет собой связь, C1-6 алкилен, C2-6 алкинилен или C2-6 алкинилен; R4 представляет собой циклоалкил, арил, гетероарил или гетероциклил (например, пиперидонил, пиперидинил, пиридонил, бензодиоксолил, например, дифторбензодиоксолил), каждый из которых замещен 0-3 R6; каждый R5 независимо представляет собой H, -C1-6 алкил, аралкил или арил, замещенный 0-3 -NH2 или R6; каждый R6 независимо представляет собой галоген, гидрокси, циано, нитро, -C1-6 алкил (например, метил, этил, галогеналкил(например, -CF3)), -C1-6 алкокси (например, галогеналкокси (например, -OCF3)), -NHR10, -NR9R10, -C(O)R11, -C(O)OR11, -C(O)NR9R10, -C(NR8)(N(R8)2), -SOqR11, -SO2NR9R10, -NHC(O)OR11, -NHC(O)R11, --OC(O)R11, арил, гетероарил, аралкил, циклоалкил, гетероаралкил, гетероциклил или гетероциклилалкил (например, 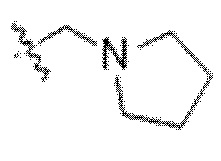 ), или две R6 группы вместе с атомами, к которым они присоединены образуют 5-7-членное кольцо (например,
), или две R6 группы вместе с атомами, к которым они присоединены образуют 5-7-членное кольцо (например, 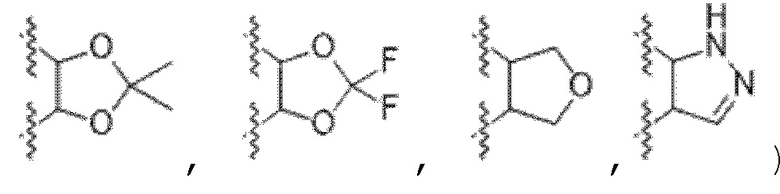 ); X представляет собой -C(O)O-, -OC(O)-, -C(O)S(O)2-, -S(O)2C(O)-, - C(O)N(R5)- или -N(R5)C(O)-; Y представляет собой -C1-6 алкил, циклоалкил (например, 3-8-членный циклоалкил, например, 5-7-членный циклоалкил), арил, гетероарил или гетероциклил (например, 3-8-членный гетероциклил, например, 5-7-членный гетероциклил), каждый из которых замещен 0-3 -NH2 или R6; R7 представляет собой Н, -C1-6 алкил (например, галогеналкил(например, -CF3)), циклоалкил, арил, гетероарил или гетероциклил, каждый из которых замещен 0-3 -NH2 или R6; каждый R8 независимо представляет собой H, -C1-6 алкил (например, галогеналкил(например, -CF3)), -C(O)R5, -C(O)OR5, арил, гетероарил, аралкил, гетероаралкил, гетероциклил или гетероциклилалкил; каждый R9 и R10 независимо представляет собой -C1-6 алкил, циклоалкил, гетероциклил, арил или гетероарил, или R9 и R10 вместе образуют необязательно замещенное 5-7-членное кольцо (например,
); X представляет собой -C(O)O-, -OC(O)-, -C(O)S(O)2-, -S(O)2C(O)-, - C(O)N(R5)- или -N(R5)C(O)-; Y представляет собой -C1-6 алкил, циклоалкил (например, 3-8-членный циклоалкил, например, 5-7-членный циклоалкил), арил, гетероарил или гетероциклил (например, 3-8-членный гетероциклил, например, 5-7-членный гетероциклил), каждый из которых замещен 0-3 -NH2 или R6; R7 представляет собой Н, -C1-6 алкил (например, галогеналкил(например, -CF3)), циклоалкил, арил, гетероарил или гетероциклил, каждый из которых замещен 0-3 -NH2 или R6; каждый R8 независимо представляет собой H, -C1-6 алкил (например, галогеналкил(например, -CF3)), -C(O)R5, -C(O)OR5, арил, гетероарил, аралкил, гетероаралкил, гетероциклил или гетероциклилалкил; каждый R9 и R10 независимо представляет собой -C1-6 алкил, циклоалкил, гетероциклил, арил или гетероарил, или R9 и R10 вместе образуют необязательно замещенное 5-7-членное кольцо (например, 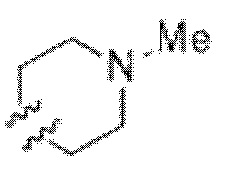 ); каждый R11 независимо представляет собой H, -C1-6 алкил (например, замещенный алкил (например,
); каждый R11 независимо представляет собой H, -C1-6 алкил (например, замещенный алкил (например, 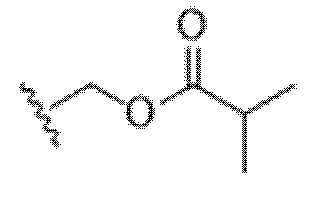
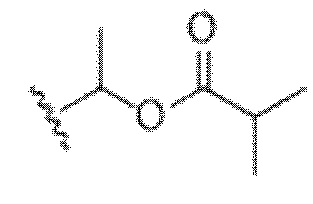 )), аралкил или арил; q равно целому числу от 0 до 2; и n равно целому числу от 0 до 2.
)), аралкил или арил; q равно целому числу от 0 до 2; и n равно целому числу от 0 до 2.
В некоторых вариантах осуществления R1 представляет собой H.
В некоторых вариантах осуществления R2 представляет собой -CO2R5, и R5 представляет собой Н, -C1-6 алкил, аралкил или арил, замещенный 1 -NH2 или R6. В некоторых вариантах осуществления R5 представляет собой Н, метил, этил, изопропил или бензил, замещенный 1 R6. В некоторых вариантах осуществления R5 представляет собой H. В некоторых вариантах осуществления R5 представляет собой этил.
В некоторых вариантах осуществления A представляет собой C1-6 алкилен (например, этилен или пропилен).
В некоторых вариантах осуществления R4 представляет собой арил или гетероарил. В некоторых вариантах осуществления R4 представляет собой фенил с 0 R6. В некоторых вариантах осуществления R4 представляет собой фенил, замещенный 1-2 R6. В некоторых вариантах осуществления R6 представляет собой галоген, -C1-6 алкокси или -C(NR8)(N(R8)2). В некоторых вариантах осуществления R6 представляет собой -C(NR8)(N(R8)2), и каждый R8 представляет собой H. В некоторых вариантах осуществления R6 представляет собой -C(NR8)(N(R8)2), и каждый R8 независимо представляет собой H или -C(O)OR5. В некоторых вариантах осуществления R6 представляет собой -C(NR8)(N(R8)2), и R5 представляет собой -C1-6 алкил (например, гексил). В некоторых вариантах осуществления R4 представляет собой гетероарил (например, 6-членный гетероарил или 5-членный гетероарил), замещенный 0-3 R6. В некоторых вариантах осуществления R4 представляет собой 6-членный гетероарил (например, пиридил), замещенный 0-3 R6. В некоторых вариантах осуществления R4 представляет собой азот-содержащий гетероарил (например, пиридил). В некоторых вариантах осуществления R4 представляет собой пиридил, замещенный 1-2 R6. В некоторых вариантах осуществления R6 представляет собой галоген (например, хлор, бром, фтор). В некоторых вариантах осуществления R6 представляет собой -NHR10, и R10 представляет собой -C1-6 алкил. В некоторых вариантах осуществления R6 представляет собой -NHC(O)OR11, и R11 представляет собой -C1-6 алкил (например, замещенный алкил (например, 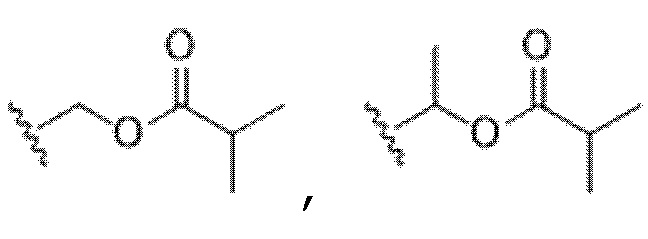 )).
)).
В некоторых вариантах осуществления X представляет собой -C(O)N(R5)- или -N(R5)C(O)-. В некоторых вариантах осуществления X представляет собой -C(O)N(R5)-, и R5 представляет собой H.
В некоторых вариантах осуществления n равно 0.
В некоторых вариантах осуществления R7 представляет собой -C1-6 алкил (например, метил, этил, пропил, -CF3).
В некоторых вариантах осуществления Y представляет собой циклоалкил (например, циклогексил). В некоторых вариантах осуществления Y представляет собой арил или гетероарил, замещенный 0-3 R6. В некоторых вариантах осуществления Y представляет собой фенил, замещенный 0 R6. В некоторых вариантах осуществления Y представляет собой фенил, замещенный 1 R6. В некоторых вариантах осуществления Y представляет собой фенил, замещенный 2 R6.В некоторых вариантах осуществления Y представляет собой арил или гетероарил, и R6 представляет собой галогеналкокси (например, -OCF3). В некоторых вариантах осуществления Y представляет собой арил или гетероарил, и две R6 группы, взятые вместе с атомами, к которым они присоединены, образуют 5-7 членное кольцо. В некоторых вариантах осуществления Y представляет собой арил или гетероарил, и две R6 группы, взятые вместе с атомами, к которым они присоединены, образуют 5-7 членное кольцо, выбранное из: 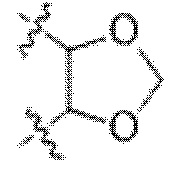 и
и 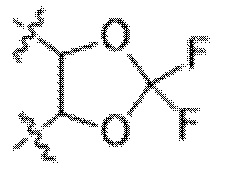 . В некоторых вариантах осуществления Y представляет собой фенил, и R6 представляет собой галогеналкокси (например, -OCF3). В некоторых вариантах осуществления Y представляет собой фенил, и две R6 группы, взятые вместе с атомами, к которым они присоединены, образуют 5-7 членное кольцо. В некоторых вариантах осуществления Y представляет собой фенил, и две R6 группы, взятые вместе с атомами, к которым они присоединены, образуют 5-7 членное кольцо, выбранное из: и .
. В некоторых вариантах осуществления Y представляет собой фенил, и R6 представляет собой галогеналкокси (например, -OCF3). В некоторых вариантах осуществления Y представляет собой фенил, и две R6 группы, взятые вместе с атомами, к которым они присоединены, образуют 5-7 членное кольцо. В некоторых вариантах осуществления Y представляет собой фенил, и две R6 группы, взятые вместе с атомами, к которым они присоединены, образуют 5-7 членное кольцо, выбранное из: и .
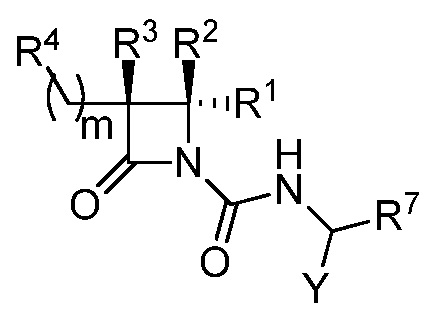
В некоторых вариантах осуществления соединение формулы (I) выбрано из соединения формулы (Ia):
 (Ia),
(Ia),
где R1, R2, R3, R4, R7 и Y такие, как описано для формулы (I), и m равно целому числу от 1 до 6.
В некоторых вариантах осуществления соединение формулы (Ia) выбрано из соединения формулы (Ib):
 (Ib),
(Ib),
где R1, R2, R3, R4, R7, Y и m такие, как описано для формулы (Ia).
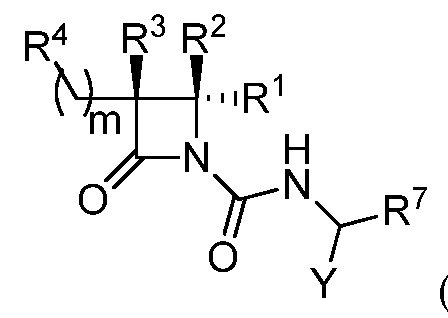
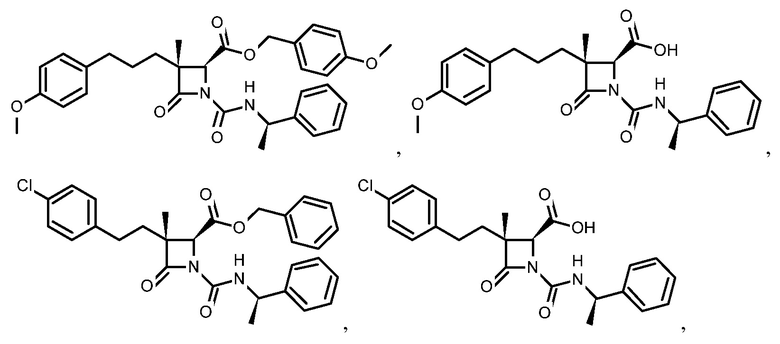
В некоторых вариантах осуществления соединение формулы (Ib) представляет собой:
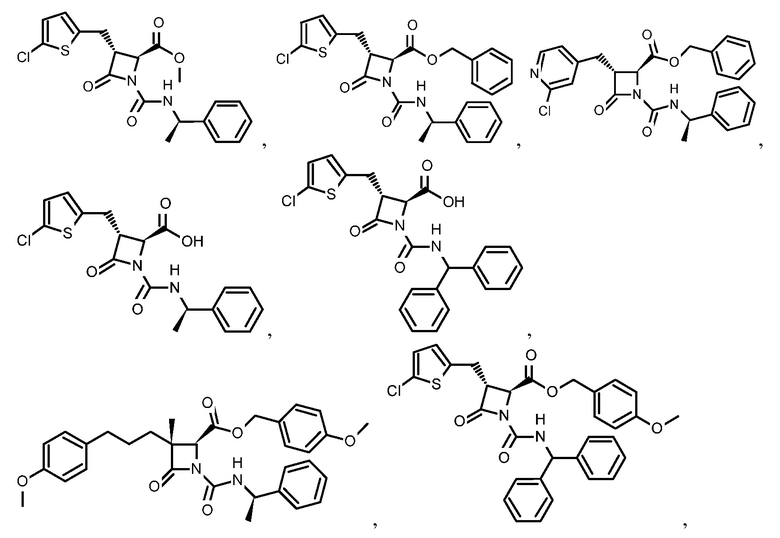
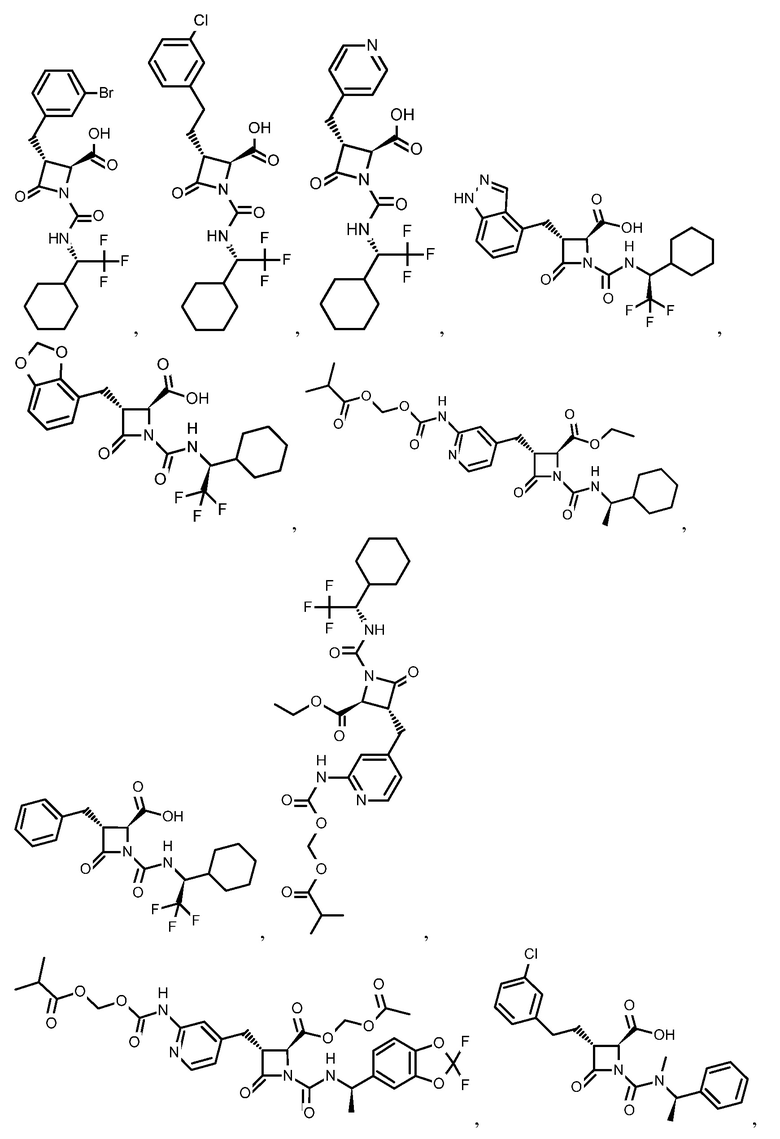
 или
или 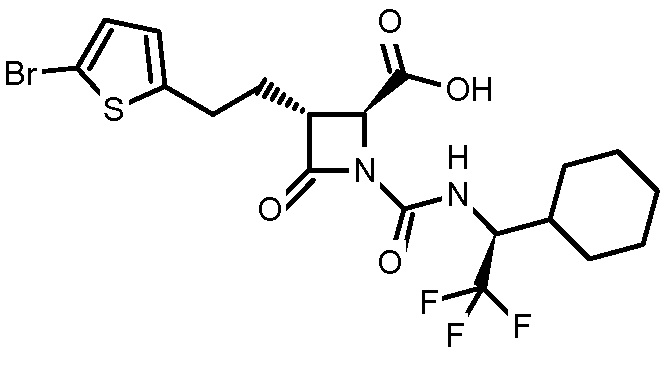 .
.
В одном из аспектов настоящее изобретение относится к соединению формулы (II):
 (II),
(II),
или к его фармацевтически приемлемой соли, где R1 представляет собой Н иои-C1-6 алкил; R2 представляет собой Н, -C1-6 алкил, -CO2R5, -C(O)NR9R10, -CN, -SOqR5, -OR5, -CHN(OR5) или гетероарил; R3 представляет собой Н или-C1-6 алкил; A представляет собой связь, C1-6 алкилен, C2-6 алкинилен или C2-6 алкинилен; R4 представляет собой циклоалкил, арил, гетероарил или гетероциклил, каждый из которых замещен 0-3 -NH2 или R6; каждый R5 независимо представляет собой H, -C1-6 алкил, аралкил или арил, замещенный 0-3 -NH2 или R6; каждый R6 независимо представляет собой галоген, гидрокси, циано, нитро, C1-6 алкил (например, метил, этил, галогеналкил (например, -CF3)), C1-6 алкокси (например, галогеналкокси (например, -OCF3)), -NR9R10, -NHR10, -C(O)R11, -C(O)OR11, -C(O)NR9R10, -C(NR8)(N(R8)2), -SOqR11, -SO2NR9R10, -NHC(O)OR11, -NHC(O)R11, арил, гетероарил, аралкил, циклоалкил, гетероаралкил, гетероциклил или гетероциклилалкил, или две R6 группы, вместе с атомами, к которым они присоединены, образуют 5-7-членное кольцо (например, 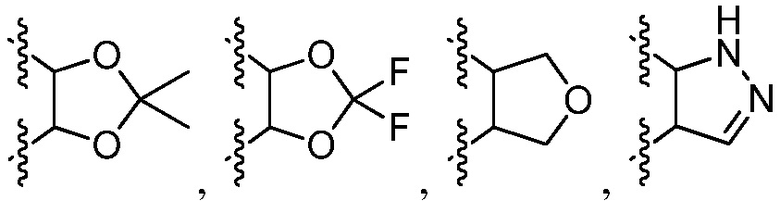 ); Х представляет собой -C(O)O-, -OC(O)-, -C(O)S(O)2-, -S(O)2C(O)-, -C(O)N(R5)- или -N(R5)C(O) -; Y представляет собой циклоалкил (например, 3-8-членный циклоалкил, например, 5-7-членный циклоалкил), гетероарил или гетероциклил (например, 3-8-членный гетероциклил, например, 5-7-членный гетероциклил), каждый из которых замещен 0-3 -NH2 или R6; или замещенный-C1-6 алкил, или замещенный арил (например, замещенный 1-3 R6); R7 представляет собой Н, -C1-6 алкил (например, метил, галогеналкил (например, -CF3)), циклоалкил, арил, гетероарил или гетероциклил, каждый из которых замещен 0-3 -NH2 или R6; каждый R8 независимо представляет собой H, -C1-6 алкил (например, галогеналкил (например, -CF3)), -C(O)R5, -C(O)OR5, арил, гетероарил, аралкил, гетероаралкил, гетероциклил или гетероциклилалкил; каждый из R9 и R10 независимо представляет собой -C1-6 алкил, циклоалкил, гетероциклил, арил или гетероарил, или R9 и R10 вместе образуют необязательно замещенное 5-7-членное кольцо; каждый R11 независимо представляет собой H, -C1-6 алкил (например, замещенный алкил (например,
); Х представляет собой -C(O)O-, -OC(O)-, -C(O)S(O)2-, -S(O)2C(O)-, -C(O)N(R5)- или -N(R5)C(O) -; Y представляет собой циклоалкил (например, 3-8-членный циклоалкил, например, 5-7-членный циклоалкил), гетероарил или гетероциклил (например, 3-8-членный гетероциклил, например, 5-7-членный гетероциклил), каждый из которых замещен 0-3 -NH2 или R6; или замещенный-C1-6 алкил, или замещенный арил (например, замещенный 1-3 R6); R7 представляет собой Н, -C1-6 алкил (например, метил, галогеналкил (например, -CF3)), циклоалкил, арил, гетероарил или гетероциклил, каждый из которых замещен 0-3 -NH2 или R6; каждый R8 независимо представляет собой H, -C1-6 алкил (например, галогеналкил (например, -CF3)), -C(O)R5, -C(O)OR5, арил, гетероарил, аралкил, гетероаралкил, гетероциклил или гетероциклилалкил; каждый из R9 и R10 независимо представляет собой -C1-6 алкил, циклоалкил, гетероциклил, арил или гетероарил, или R9 и R10 вместе образуют необязательно замещенное 5-7-членное кольцо; каждый R11 независимо представляет собой H, -C1-6 алкил (например, замещенный алкил (например,  )), аралкил, или арил; q равно целому числу от 0 до 2; и n равно целому числу от 0 до 2.
)), аралкил, или арил; q равно целому числу от 0 до 2; и n равно целому числу от 0 до 2.
В некоторых вариантах осуществления R1 представляет собой H.
В некоторых вариантах осуществления R2 представляет собой -CO2R5, и R5 представляет собой Н, -C1-6 алкил, аралкил или арил, замещенный 1 -NH2 или R6. В некоторых вариантах осуществления R5 представляет собой Н, метил, этил, изопропил или бензил, замещенный 1 R6. В некоторых вариантах осуществления R5 представляет собой H. В некоторых вариантах осуществления R5 представляет собой этил.
В некоторых вариантах осуществления A представляет собой C1-6 алкилен (например, этилен или пропилен).
В некоторых вариантах осуществления R4 представляет собой арил или гетероарил. В некоторых вариантах осуществления R4 представляет собой фенил, замещенный 1 R6. В некоторых вариантах осуществления R6 представляет собой галоген, C1-6 алкокси или -C(NR8)(N(R8)2). В некоторых вариантах осуществления R6 представляет собой -C(NR8)(N(R8)2), и каждый R8 представляет собой H. В некоторых вариантах осуществления R8 независимо представляет собой H или -CO2R5. В некоторых вариантах осуществления R5 представляет собой -C1-6 алкил (например, гексил). В некоторых вариантах осуществления R4 представляет собой гетероарил (например, 6-членный гетероарил или 5-членный гетероарил), замещенный 0-3 -NH2 или R6. В некоторых вариантах осуществления R4 представляет собой 6-членный гетероарил (например, пиридил), замещенный 0-3 -NH2 или R6. В некоторых вариантах осуществления R4 представляет собой азот-содержащий гетероарил (например, пиридил), замещенный 0-3 -NH2 или R6. В некоторых вариантах осуществления R4 представляет собой пиридил, замещенный 1 R6. В некоторых вариантах осуществления R6 представляет собой галоген (например, хлор, бром, фтор). В некоторых вариантах осуществления R4 представляет собой пиридил, замещенный 1 -NH2. В некоторых вариантах осуществления R6 представляет собой -NHR10, и R10 представляет собой -C1-6 алкил. В некоторых вариантах осуществления R6 представляет собой -NHC(O)OR11, и R11 представляет собой -C1-6 алкил (например, замещенный алкил (например,  )).
)).
В некоторых вариантах осуществления X представляет собой -C(O)N(R5)- или -N(R5)C(O)-. В некоторых вариантах осуществления X представляет собой -C(O)N(R5)-, и R5 представляет собой H.
В некоторых вариантах осуществления n равно 0.
В некоторых вариантах осуществления R7 представляет собой -C1-6 алкил (например, метил, этил, пропил, -CF3). В некоторых вариантах осуществления R7 представляет собой метил. В некоторых вариантах осуществления R7 представляет собой -CF3.
В некоторых вариантах осуществления Y представляет собой циклоалкил, гетероарил или гетероциклил, каждый из которых замещен 0-3 -NH2 или R6, или замещенный арил. В некоторых вариантах осуществления Y представляет собой циклоалкил (например, циклогексил). В некоторых вариантах осуществления Y представляет собой гетероарил (например, пиридил, пиразинил, пиримидинил, хинолинил, изохинолинил, тиазолил, индазолил). В некоторых вариантах осуществления Y представляет собой замещенный арил (например, замещенный фенил, нафтил). В некоторых вариантах осуществления Y представляет собой фенил, замещенный 1-2 R6. В некоторых вариантах осуществления Y представляет собой фенил, замещенный 1 R6. В некоторых вариантах осуществления Y представляет собой фенил, и R6 представляет собой галогеналкокси (например, -OCF3). В некоторых вариантах осуществления Y представляет собой фенил, и R6 представляет собой галоген (например, хлор, бром, фтор). В некоторых вариантах осуществления Y представляет собой фенил, замещенный 2 R6.
В некоторых вариантах осуществления R6 представляет собой галогеналкокси (например, -OCF3). В некоторых вариантах осуществления две R6 группы, взятые вместе с атомами, к которым они присоединены, образуют 5-7 членное кольцо. В некоторых вариантах осуществления две R6 группы, взятые вместе с атомами, к которым они присоединены, образуют 5-7 членное кольцо, и кольцо выбрано из: 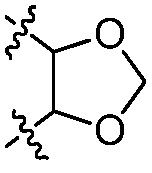 и
и  .
.
В некоторых вариантах осуществления соединение формулы (II) выбрано из соединения формулы (IIa):
 (IIa),
(IIa),
где R1, R2, R3, R4, R7 и Y такие, как описано для формулы (II), и m равно целому числу от 1 до 6.
В некоторых вариантах осуществления соединение формулы (IIa) выбрано из соединения формулы (IIb):
 (IIb),
(IIb),
где R1, R2, R3, R4, R7, Y и m такие, как описано для формулы (IIa).
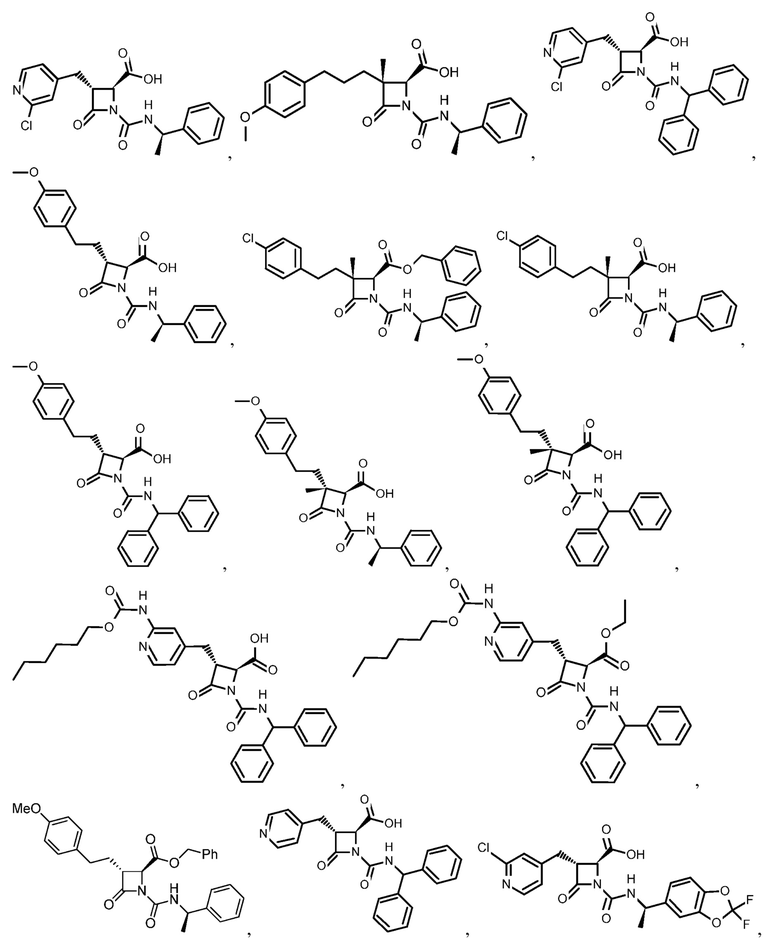
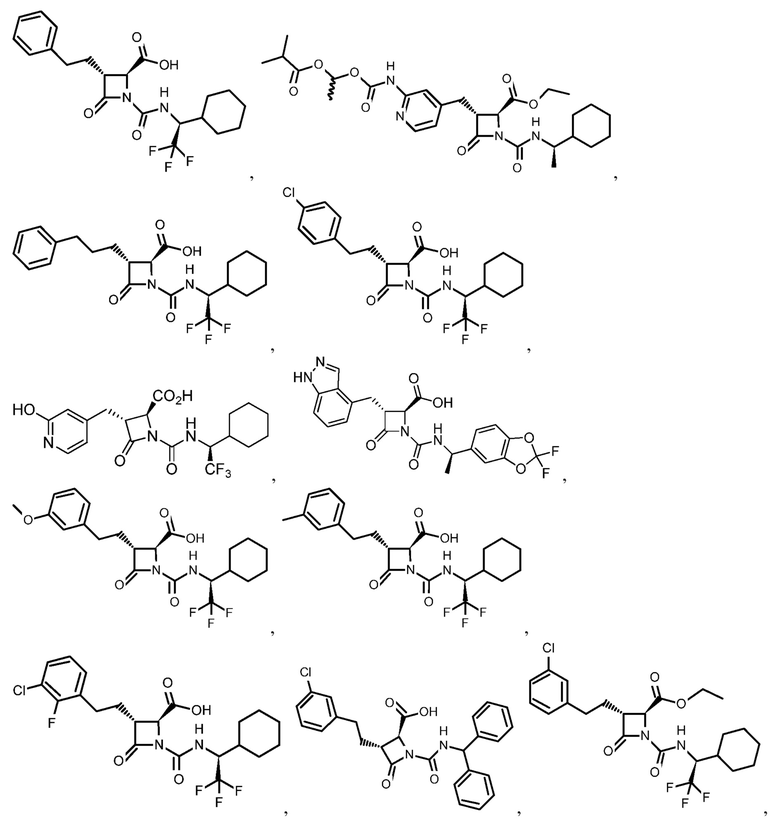
В некоторых вариантах осуществления соединение формулы (IIb) представляет собой:
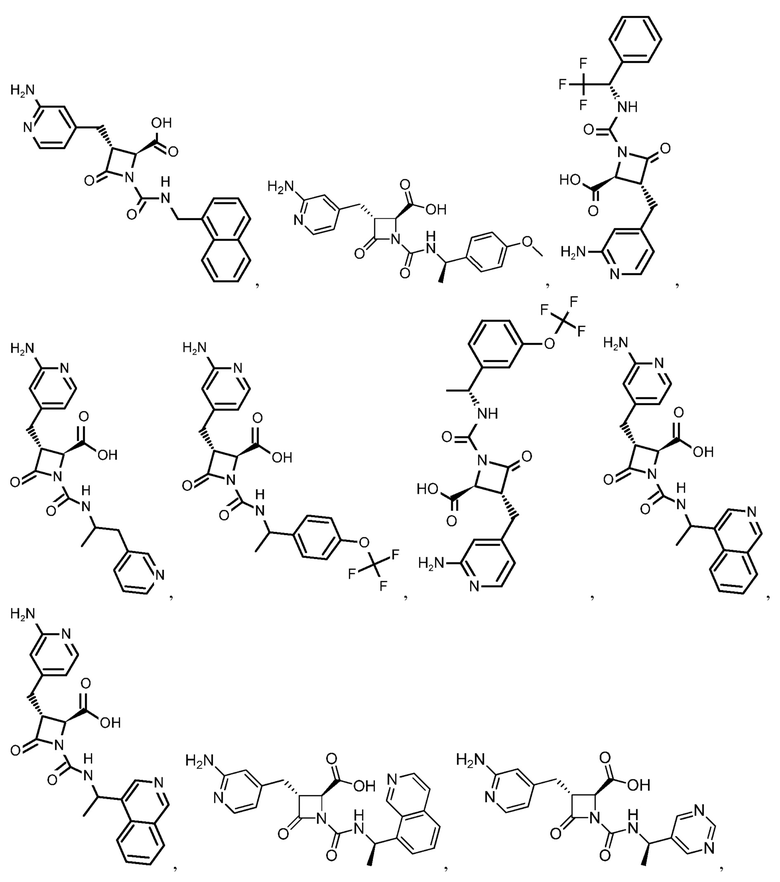
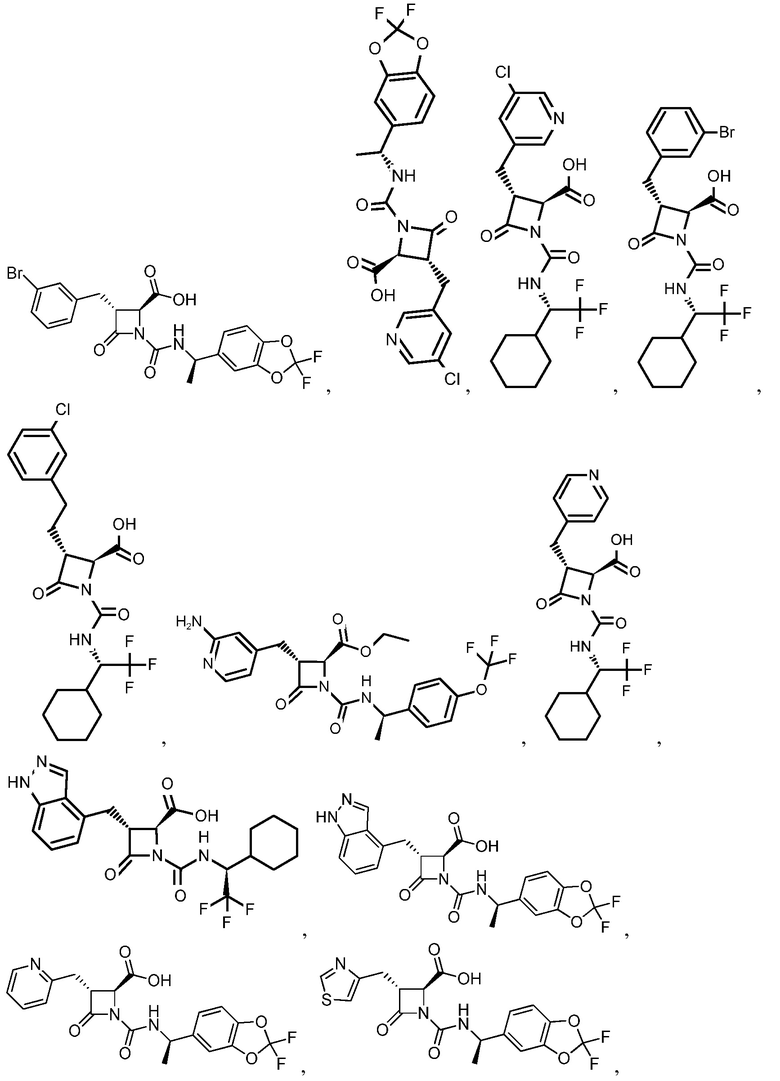
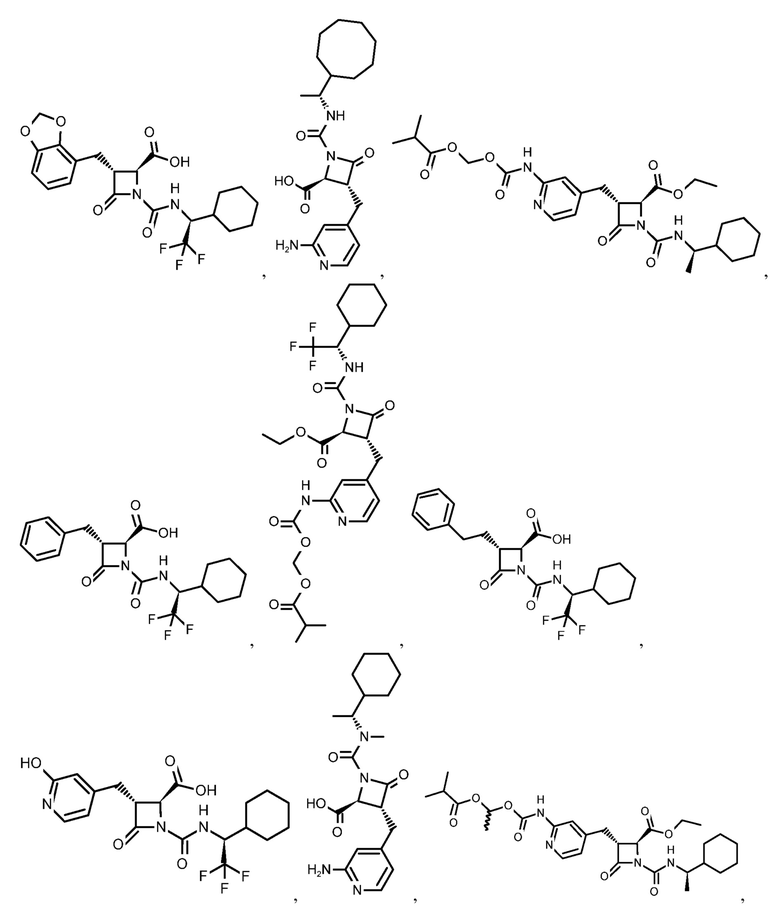
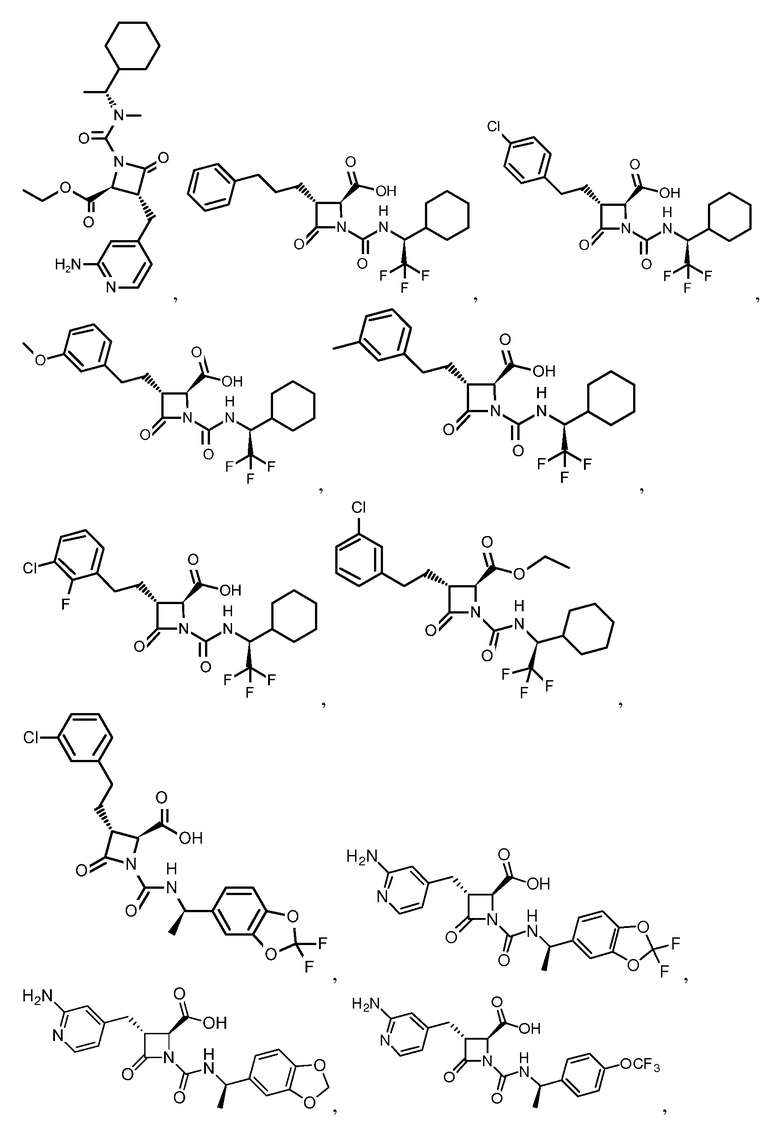
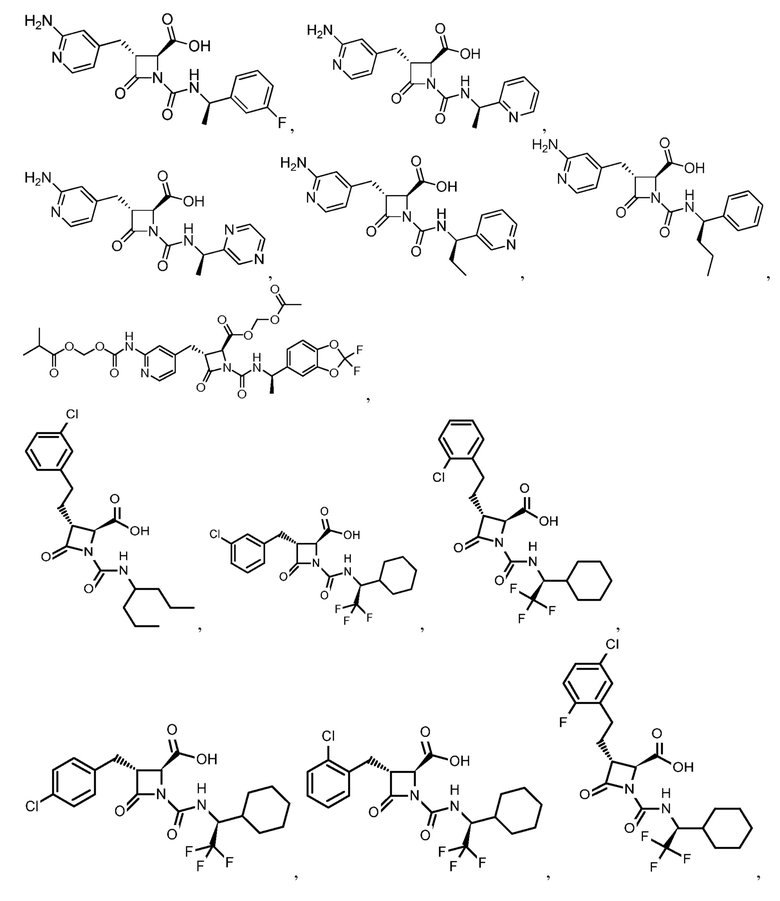
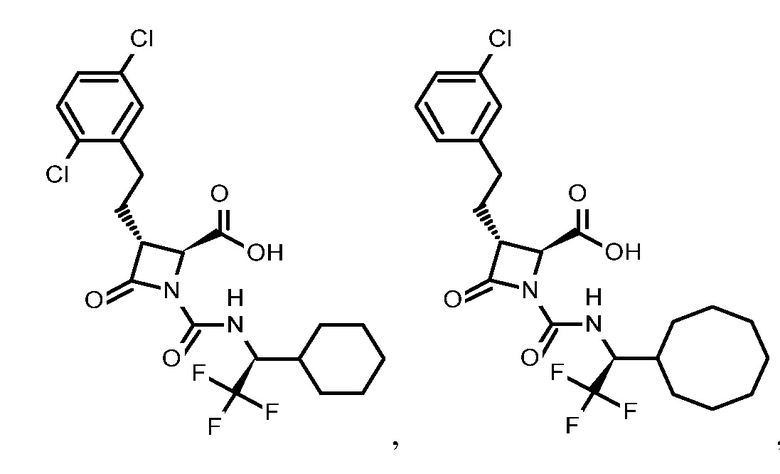 или
или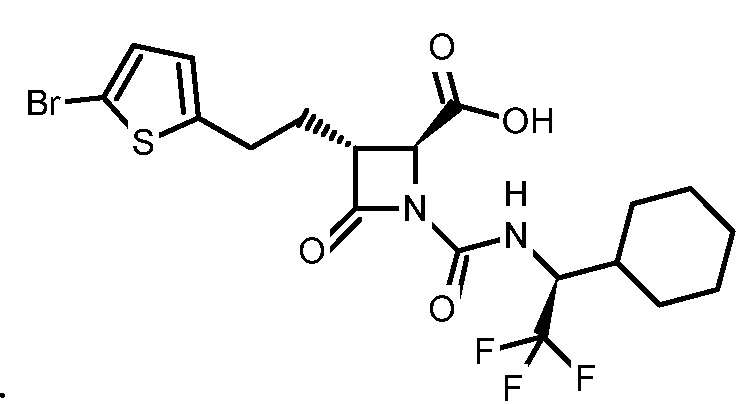 .
.
В некоторых вариантах осуществления соединение формулы (IIb) выбрано из соединения формулы (IIc):
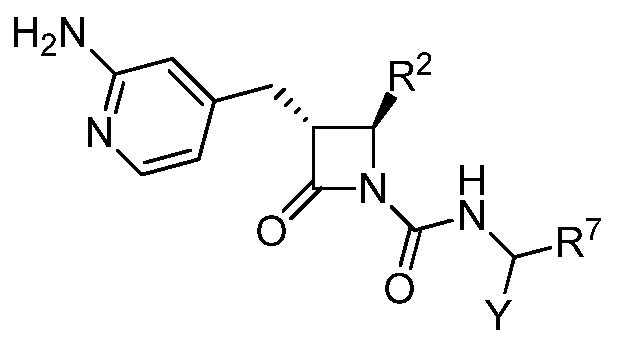 (IIc).
(IIc).
В одном из аспектов настоящее изобретение относится к соединению формулы (III):
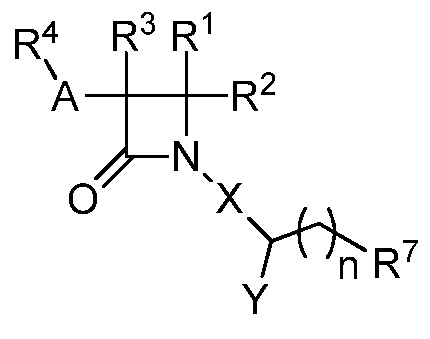 (III),
(III),
или к его фармацевтически приемлемой соли, где R1 представляет собой Н или -C2-6 алкил; R2 представляет собой Н, -C2-6 алкил, галогеналкил, -CO2R12, -C(O)NH2, -CN, -SOqR5, -OR5, -CHN(OR5), или гетероарил; R3 представляет собой Н или -C1-6 алкил; A представляет собой связь, C1-6 алкилен, C2-6 алкинилен или C2-6 алкинилен; R4 представляет собой циклоалкил, арил, гетероарил или гетероциклил, каждый из которых замещен 0-3 -NH2 или R6; каждый R5 независимо представляет собой H, -C1-6 алкил, аралкил или арил, замещенный 0-3 -NH2 или R6; каждый R6 независимо представляет собой галоген, гидрокси, циано, нитро, -C1-6 алкил, -C1-6 алкокси, -NHR10, -NR9R10, -C(O)R11, -C(O)OR11, -C(O)NR9R10, -C(NR8)(N(R8)2), -SOqR11, -SO2NR9R10, -NHC(O)OR11, -NHC(O)R11, арил, гетероарил, аралкил, циклоалкил, гетероаралкил, гетероциклил или гетероциклилалкил, или две R6 группы, вместе с атомами, к которым они присоединены, образуют 5-7-членное кольцо; Х представляет собой -C(O)O-, -OC(O)-, -C(O)S(O)2-, -S(O)2C(O)-, -C(O)N(R5)- или -N(R5)C(O)-; Y представляет собой -C1-6 алкил, циклоалкил, арил, гетероарил, или гетероциклил, каждый из которых замещен 0-3 -NH2 или R6; R7 представляет собой Н, -C1-6 алкил, циклоалкил, арил, гетероарил или гетероциклил, каждый из которых замещен 0-3 -NH2 или R6; каждый R8 независимо представляет собой H, -C1-6 алкил, -C(O)R5, -C(O)OR5, арил, гетероарил, аралкил, гетероаралкил, гетероциклил, или гетероциклилалкил; каждый из R9 и R10 независимо представляет собой -C1-6 алкил, циклоалкил, гетероциклил, арил или гетероарил, или R9 и R10 вместе образуют необязательно замещенное 5-7-членное кольцо; каждый R11 независимо представляет собой H, -C1-6 алкил, аралкил или арил; каждый R12 независимо представляет собой галогеналкил, необязательно замещенный -C3-6 алкил или аралкил; q равно целому числу от 0 до 2; и n равно целому числу от 0 до 2.
В некоторых вариантах осуществления R1 представляет собой H.
В некоторых вариантах осуществления R2 представляет собой галогеналкил (например, -CH2F, -CHF2, -CF3), -CO2R12, -C(O)NH2, -CN, -SOqR5 (например, -SO2R5), -OR5, -CHN(OR5) или гетероарил (например, триазолил, тетразолил, необязательно замещенный оксазолил, необязательно замещенный изоксазолил). В некоторых вариантах осуществления R12 представляет собой галогеналкил, пропил или аралкил (например, бензил). В некоторых вариантах осуществления R5 представляет собой Н, -C1-6 алкил (например, метил, этил, пропил) или арил (например, фенил).
В некоторых вариантах осуществления A представляет собой C1-6 алкилен (например, этилен или пропилен).
В некоторых вариантах осуществления R4 представляет собой арил или гетероарил. В некоторых вариантах осуществления R4 представляет собой фенил, замещенный 1 R6. В некоторых вариантах осуществления R6 представляет собой C1-6 алкокси или -C(NR8)(N(R8)2). В некоторых вариантах осуществления R6 представляет собой -C(NR8)(N(R8)2), и каждый R8 представляет собой H. В некоторых вариантах осуществления R8 независимо представляет собой H или -C(O)OR5. В некоторых вариантах осуществления R5 представляет собой -C1-6 алкил (например, гексил).
В некоторых вариантах осуществления R4 представляет собой гетероарил (например, 6-членный гетероарил или 5-членный гетероарил), замещенный 0-3 -NH2 или R6. В некоторых вариантах осуществления R4 представляет собой 6-членный гетероарил (например, пиридил), замещенный 0-3 -NH2 или R6. В некоторых вариантах осуществления R6 представляет собой галоген (например, хлор). В некоторых вариантах осуществления R4 представляет собой пиридил, замещенный 1 -NH2.
В некоторых вариантах осуществления X представляет собой -C(O)N(R5)- или -N(R5)C(O)-. В некоторых вариантах осуществления X представляет собой -C(O)N(R5)-, и R5 представляет собой H.
В некоторых вариантах осуществления n равно 0.
В некоторых вариантах осуществления R7 представляет собой -C1-6 алкил (например, метил). В некоторых вариантах осуществления R7 представляет собой арил (например, фенил).
В некоторых вариантах осуществления Y представляет собой циклоалкил, арил, гетероарил или гетероциклил, каждый из которых замещен 0-3 -NH2 или R6. В некоторых вариантах осуществления Y представляет собой фенил, замещенный 0 R6. В некоторых вариантах осуществления Y представляет собой фенил, замещенный 1 R6. В некоторых вариантах осуществления R6 представляет собой -C1-6 алкокси.
В некоторых вариантах осуществления соединение формулы (III) выбрано из соединения формулы (IIIa):
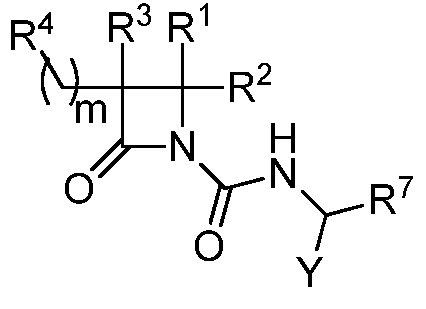 (IIIa),
(IIIa),
где R1, R2, R3, R4, R7 и Y такие, как описано для формулы (III), и m равно целому числу от 1 до 6.
В некоторых вариантах осуществления соединение формулы (IIIa) выбрано из соединения формулы (IIIb):
 (IIIb),
(IIIb),
где R1, R2, R3, R4, R7, Y и m такие, как описано для формулы (IIIa).
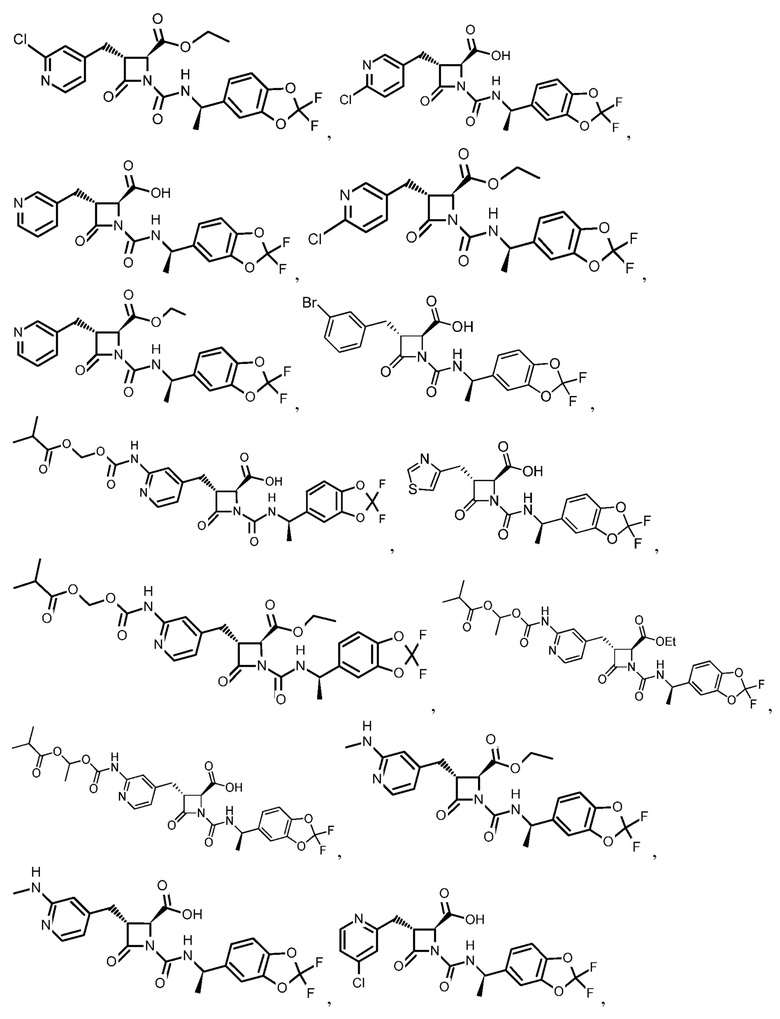
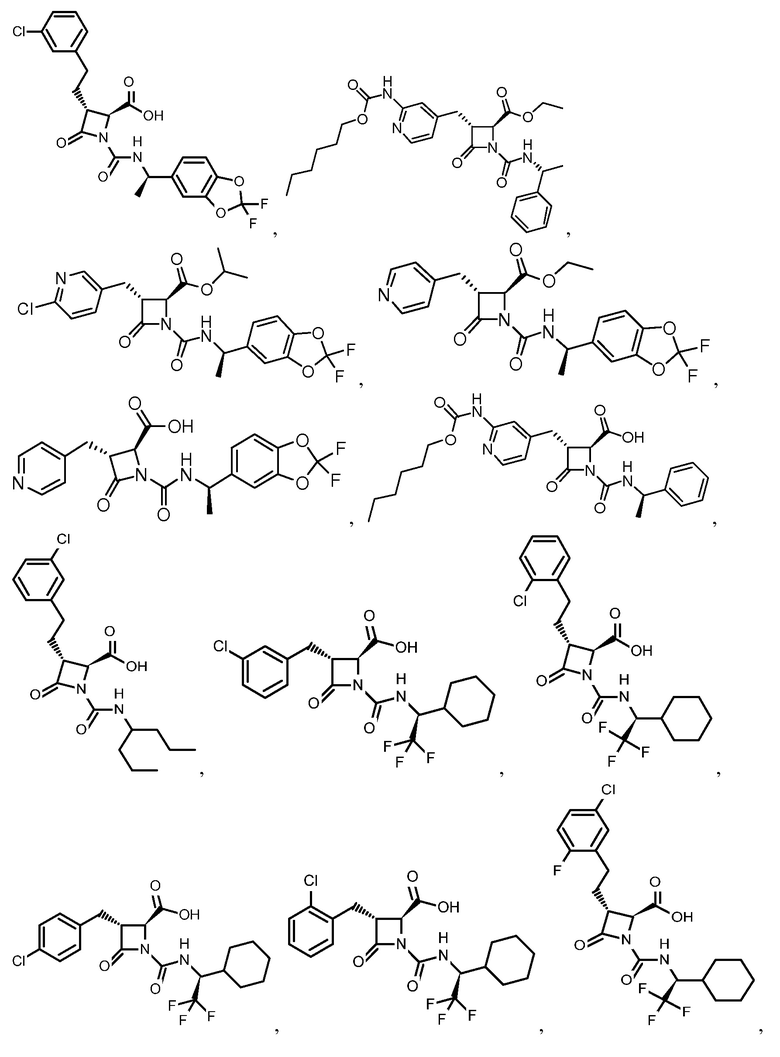
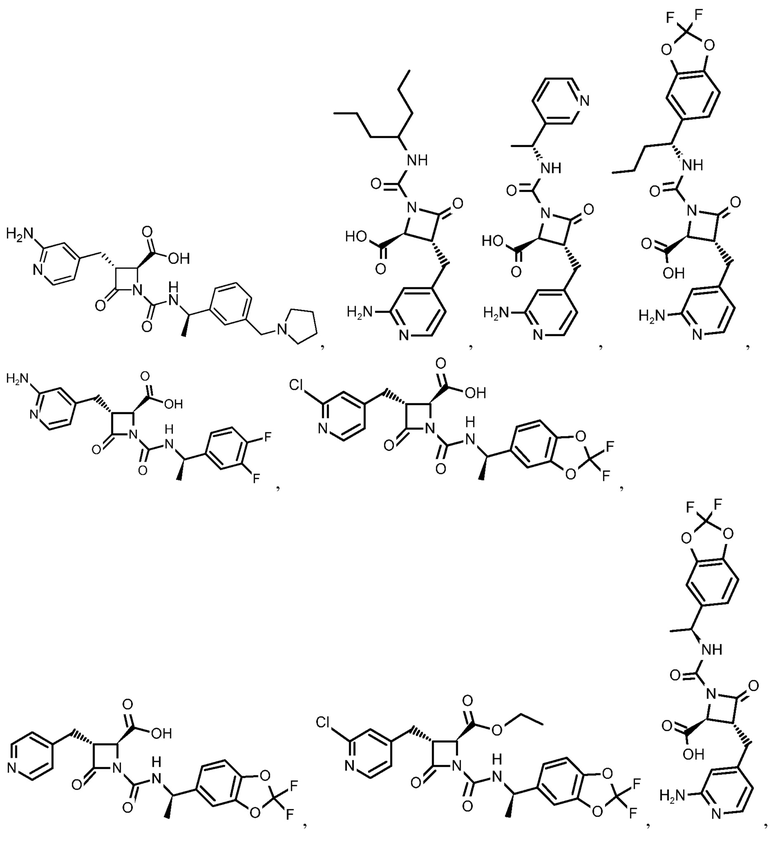
В некоторых вариантах осуществления соединение формулы (IIIb) представляет собой:
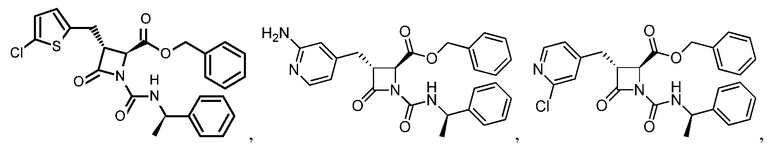


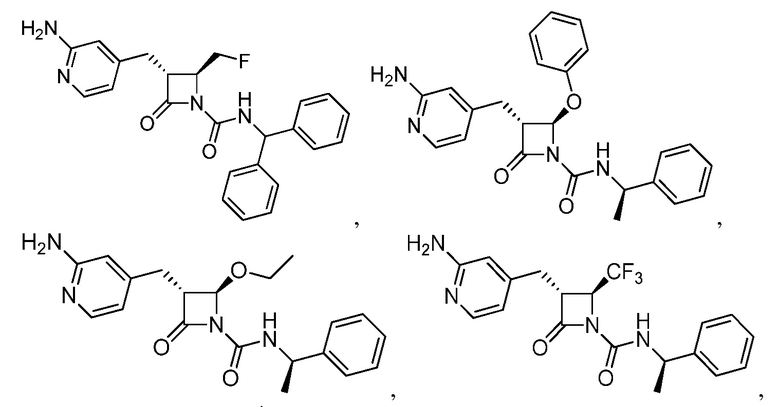
или  .
.
В одном из аспектов настоящее изобретение относится к соединению формулы (IV):
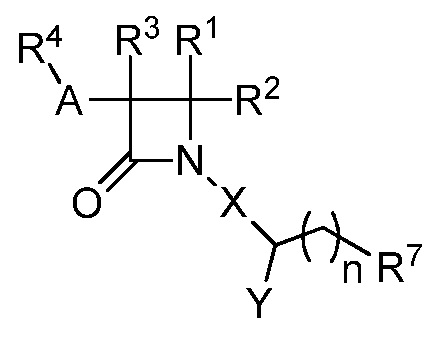 (IV),
(IV),
или к его фармацевтически приемлемой соли, где R1 представляет собой Н или -C1-6 алкил; R2 представляет собой Н, -C1-6 алкил, -CO2R5, -C(O)NR9R10, -CN, -SOqR5, -OR5, -CHN(OR5) или гетероарил; R3 представляет собой Н или -C1-6 алкил; A представляет собой связь, C1-6 алкилен, C2-6 алкинилен или C2-6 алкинилен; R4 представляет собой циклоалкил, арил, гетероарил или гетероциклил, каждый из которых замещен 0-3 -NH2 или R6; каждый R5 независимо представляет собой H, -C1-6 алкил, аралкил или арил, замещенный 0-3 -NH2 или R6; каждый R6 независимо представляет собой галоген, -C1-6 алкил, -C1-6 алкокси, -NR9R10, -C(O)R11, -C(O)OR11, -C(O)NR9R10, -C(NR8)(N(R8)2), -SOqR11, -SO2NR9R10, -NHC(O)OR11, арил, гетероарил, аралкил, циклоалкил, гетероаралкил, гетероциклил или гетероциклилалкил, или две R6 группы, вместе с атомами, к которым они присоединены, образуют 5-7-членное кольцо; Х представляет собой -C(O)O-, -OC(O) -, -C(O)S(O)2-, -S(O)2C(O)-, -C(O)N(R5)- или -N(R5)C(O)-; Y представляет собой Н, -C1-6 алкил, циклоалкил, арил, гетероарил или гетероциклил, каждый из которых замещен 0-3 -NH2 или R6; R7 представляет собой Н, -C2-6 алкил, галогеналкил, циклоалкил, гетероарил или гетероциклил, каждый из которых замещен 0-3 -NH2 или R6, или замещенный арил; каждый R8 независимо представляет собой H, -C1-6 алкил, -C(O)R5, -C(O)OR5, арил, гетероарил, аралкил, гетероаралкил, гетероциклил или гетероциклилалкил; каждый из R9 и R10 независимо представляет собой -C1-6 алкил, циклоалкил, гетероциклил, арил или гетероарил, или R9 и R10 вместе образуют необязательно замещенное 5-7-членное кольцо; каждый R11 независимо представляет собой H, -C1-6 алкил, аралкил или арил; q равно целому числу от 0 до 2; и n равно целому числу от 0 до 2.
В некоторых вариантах осуществления R1 представляет собой H.
В некоторых вариантах осуществления R2 представляет собой -CO2R5, где R5 представляет собой Н или -C1-6 алкил (например, этил).
В некоторых вариантах осуществления A представляет собой C1-6 алкилен (например, этилен или пропилен).
В некоторых вариантах осуществления R4 представляет собой арил или гетероарил. В некоторых вариантах осуществления R4 представляет собой фенил, замещенный 1 R6. В некоторых вариантах осуществления R6 представляет собой галоген, C1-6 алкокси или -C(NR8)(N(R8)2). В некоторых вариантах осуществления R6 представляет собой -C(NR8)(N(R8)2), и каждый R8 представляет собой H. В некоторых вариантах осуществления R4 представляет собой гетероарил (например, 6-членный гетероарил или 5-членный гетероарил), замещенный 0-3 -NH2 или R6. В некоторых вариантах осуществления R4 представляет собой 6-членный гетероарил (например, пиридил), замещенный 0-3 -NH2 или R6. В некоторых вариантах осуществления R6 представляет собой галоген (например, хлор, бром, фтор). В некоторых вариантах осуществления R6 представляет собой -NHC(O)OR11, и R11 представляет собой -C1-6 алкил (например, замещенный алкил (например, 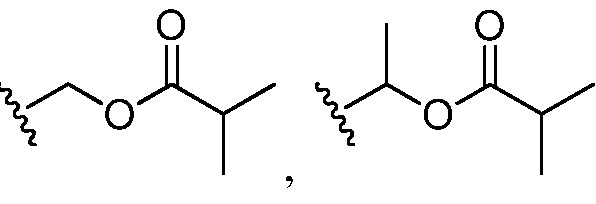 , )).
, )).
В некоторых вариантах осуществления X представляет собой -C(O)N(R5)- или -N(R5)C(O)-. В некоторых вариантах осуществления X представляет собой -C(O)N(R5)-, и R5 представляет собой H.
В некоторых вариантах осуществления R7 представляет собой Н, -C2-6 алкил, галогеналкил, циклоалкил или замещенный арил.
В некоторых вариантах осуществления n равно 0.
В некоторых вариантах осуществления Y представляет собой Н, -C1-6 алкил, циклоалкил, арил, гетероарил или гетероциклил, каждый из которых замещен 0-3 -NH2 или R6. В некоторых вариантах осуществления Y представляет собой арил (например, арил, нафтил). В некоторых вариантах осуществления Y представляет собой фенил, замещенный 0 R6. В некоторых вариантах осуществления Y представляет собой фенил, замещенный 1 R6. В некоторых вариантах осуществления R6 представляет собой галогеналкокси (например, -OCF3) или галоген (например, хлор). В некоторых вариантах осуществления Y представляет собой арил или гетероарил, и две R6 группы, взятые вместе с атомами, к которым они присоединены, образуют 5-7 членное кольцо, выбранное из: 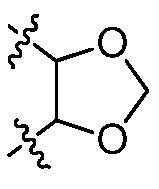 и
и 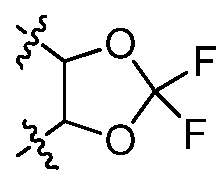 . В некоторых вариантах осуществления Y представляет собой фенил, и две R6 группы, взятые вместе с атомами, к которым они присоединены, образуют 5-7 членное кольцо. В некоторых вариантах осуществления Y представляет собой фенил, и две R6 группы, взятые вместе с атомами, к которым они присоединены, образуют 5-7 членное кольцо, выбранное из: и . В некоторых вариантах осуществления Y представляет собой гетероарил (например, пиридил, пиразинил, пиримидинил, хинолинил, изохинолинил, тиазолил, индазолил), замещенный 0-3 R6. В некоторых вариантах осуществления Y представляет собой циклоалкил (например, циклогексил). В некоторых вариантах осуществления Y представляет собой -C1-6 алкил, замещенный 0 R6. В некоторых вариантах осуществления Y представляет собой-C1-6 алкил, замещенный 1-3 R6, и R6 представляет собой арил. В некоторых вариантах осуществления Y представляет собой -C1-6 алкил, замещенный 2 R6, и R6 представляет собой арил (например, фенил).
. В некоторых вариантах осуществления Y представляет собой фенил, и две R6 группы, взятые вместе с атомами, к которым они присоединены, образуют 5-7 членное кольцо. В некоторых вариантах осуществления Y представляет собой фенил, и две R6 группы, взятые вместе с атомами, к которым они присоединены, образуют 5-7 членное кольцо, выбранное из: и . В некоторых вариантах осуществления Y представляет собой гетероарил (например, пиридил, пиразинил, пиримидинил, хинолинил, изохинолинил, тиазолил, индазолил), замещенный 0-3 R6. В некоторых вариантах осуществления Y представляет собой циклоалкил (например, циклогексил). В некоторых вариантах осуществления Y представляет собой -C1-6 алкил, замещенный 0 R6. В некоторых вариантах осуществления Y представляет собой-C1-6 алкил, замещенный 1-3 R6, и R6 представляет собой арил. В некоторых вариантах осуществления Y представляет собой -C1-6 алкил, замещенный 2 R6, и R6 представляет собой арил (например, фенил).
В некоторых вариантах осуществления соединение формулы (IV) выбрано из соединения формулы (IVa):
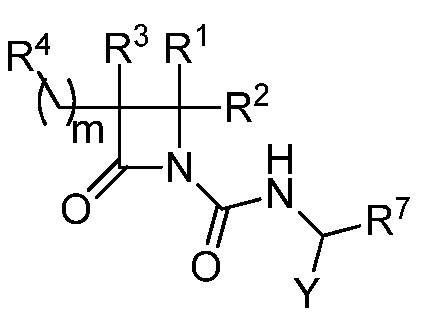 (IVa),
(IVa),
где R1, R2, R3, R4, R7 и Y такие, как описано для формулы (IV), и m равно целому числу от 1 до 6.
В некоторых вариантах осуществления соединение формулы (IVa) выбрано из соединения формулы (IVb):
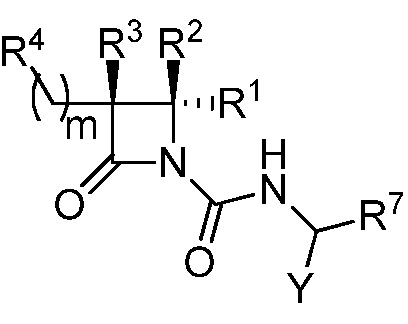 (IVb),
(IVb),
где R1, R2, R3, R4, R7, Y и m такие, как описано для формулы (IVa).
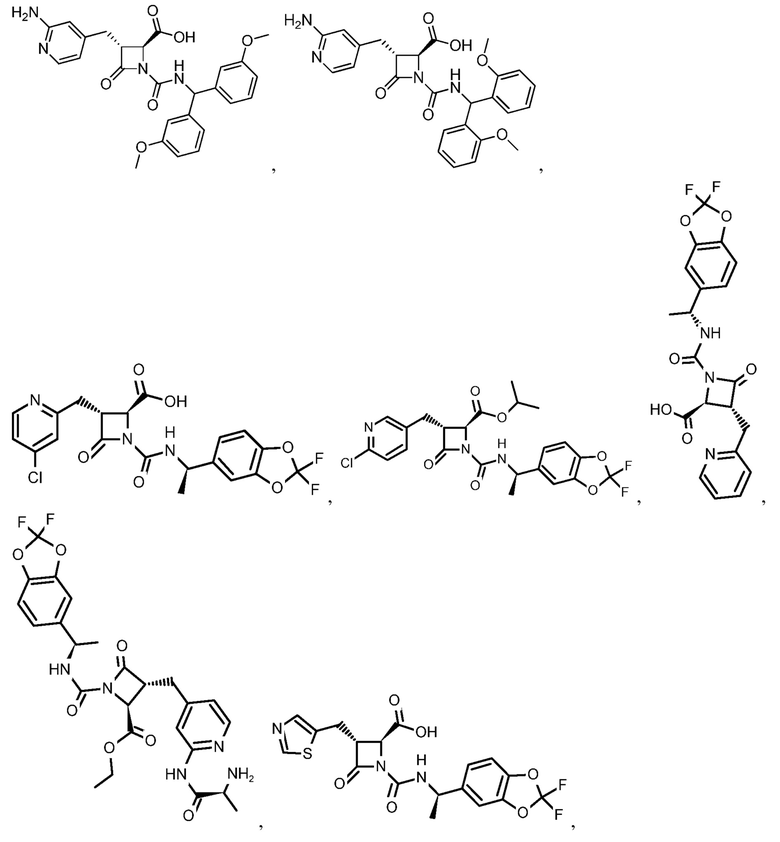
В некоторых вариантах осуществления соединение формулы (IV) представляет собой:
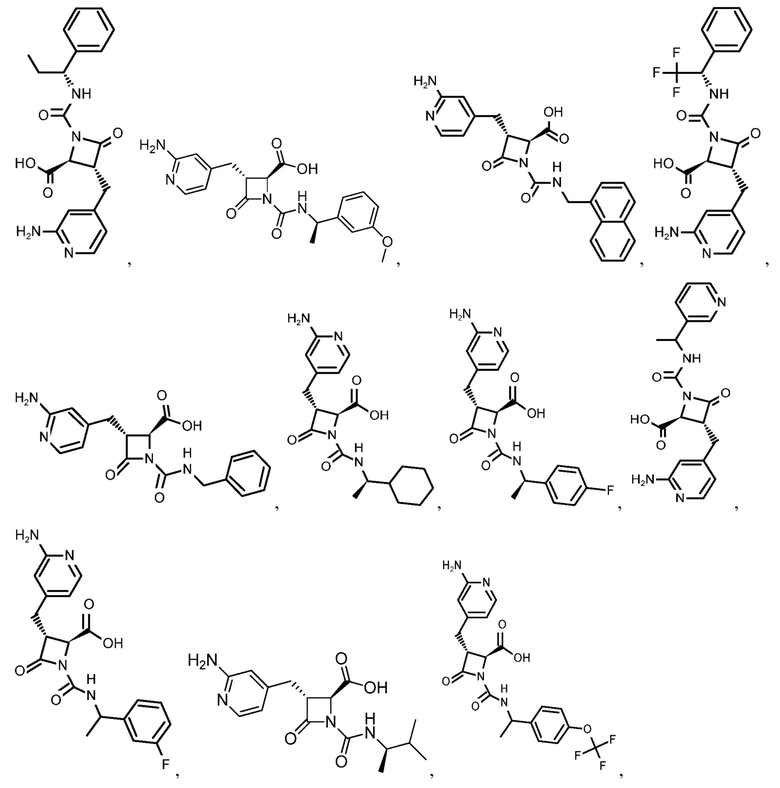
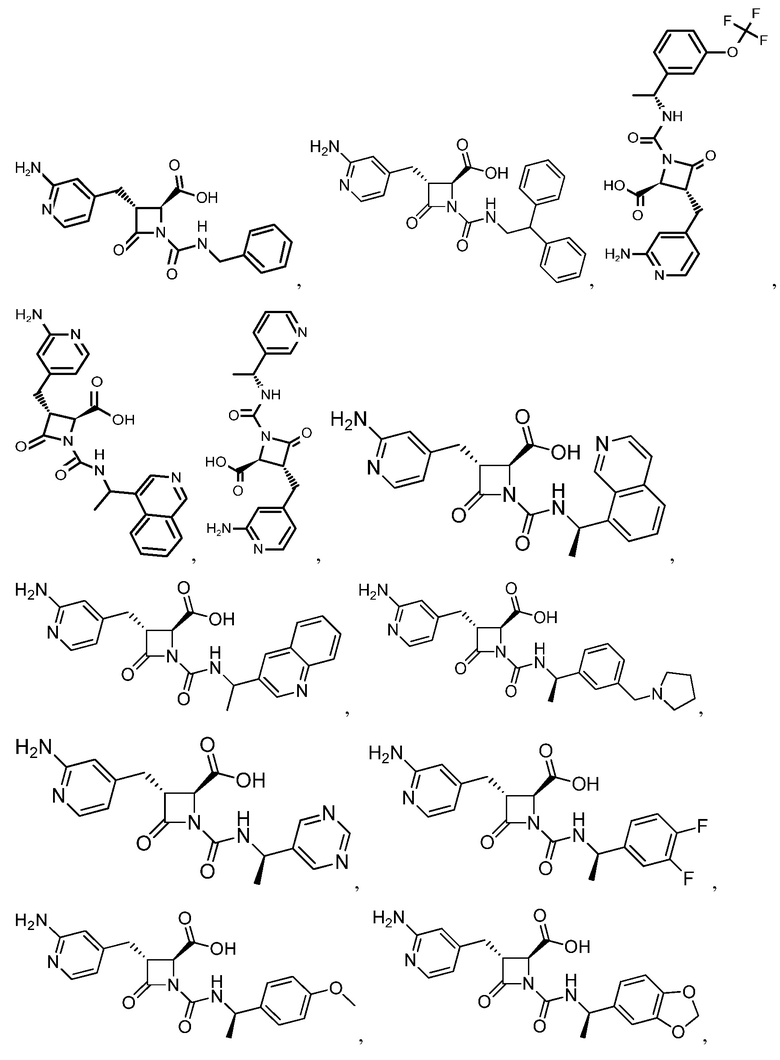
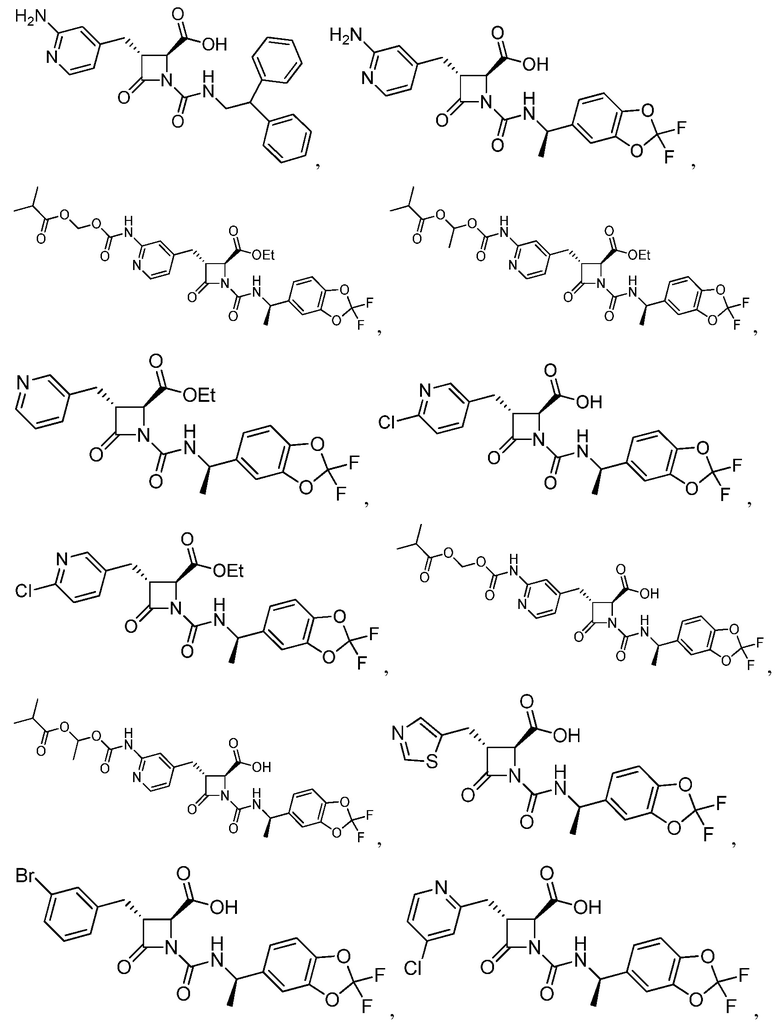
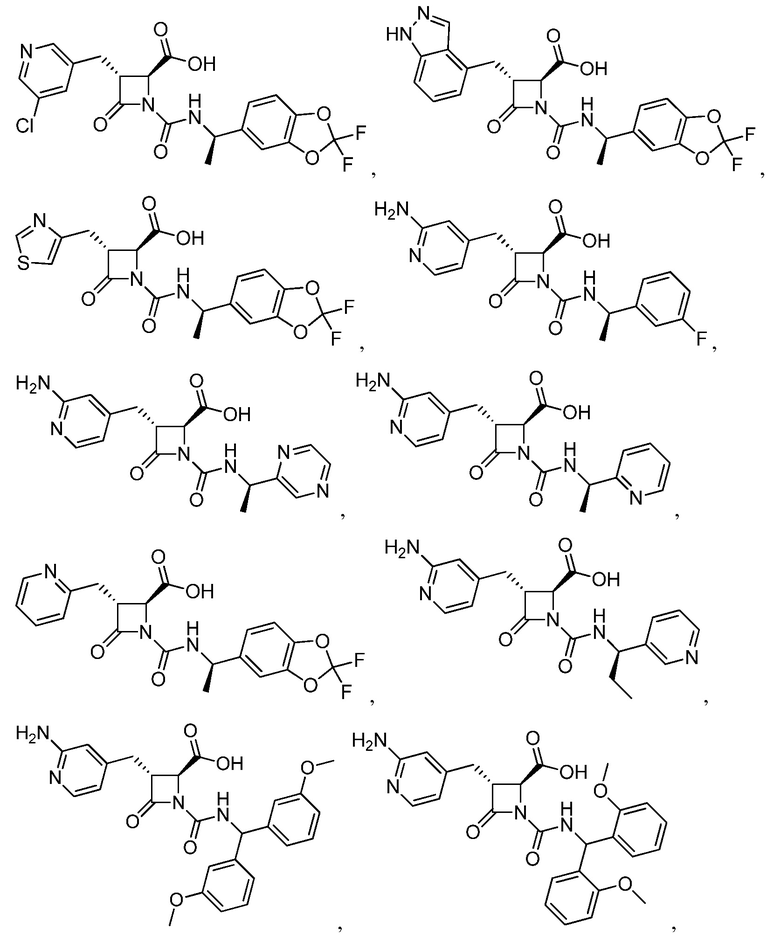


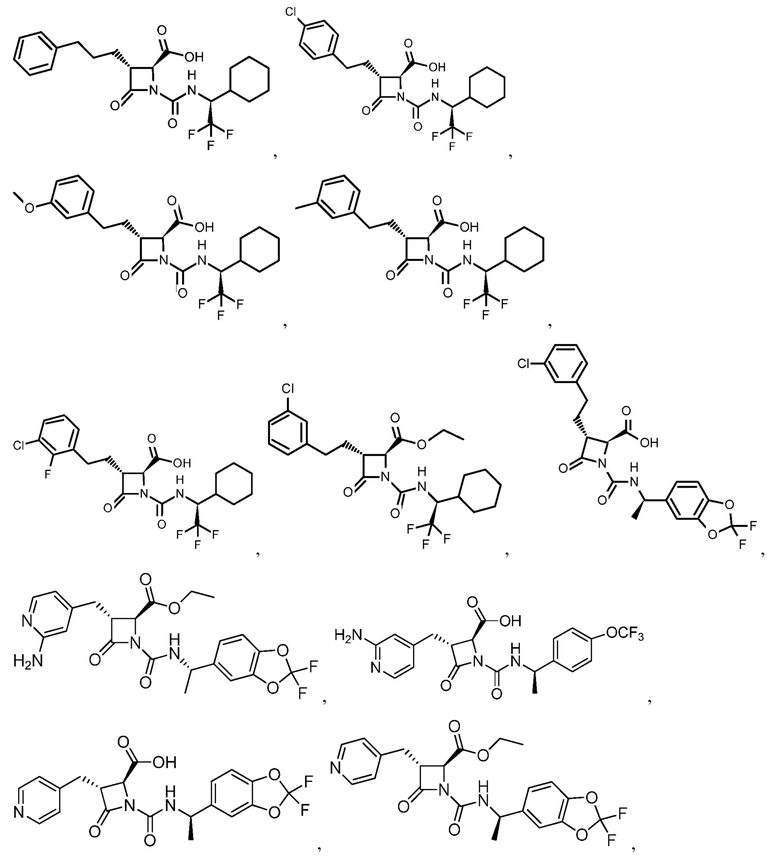
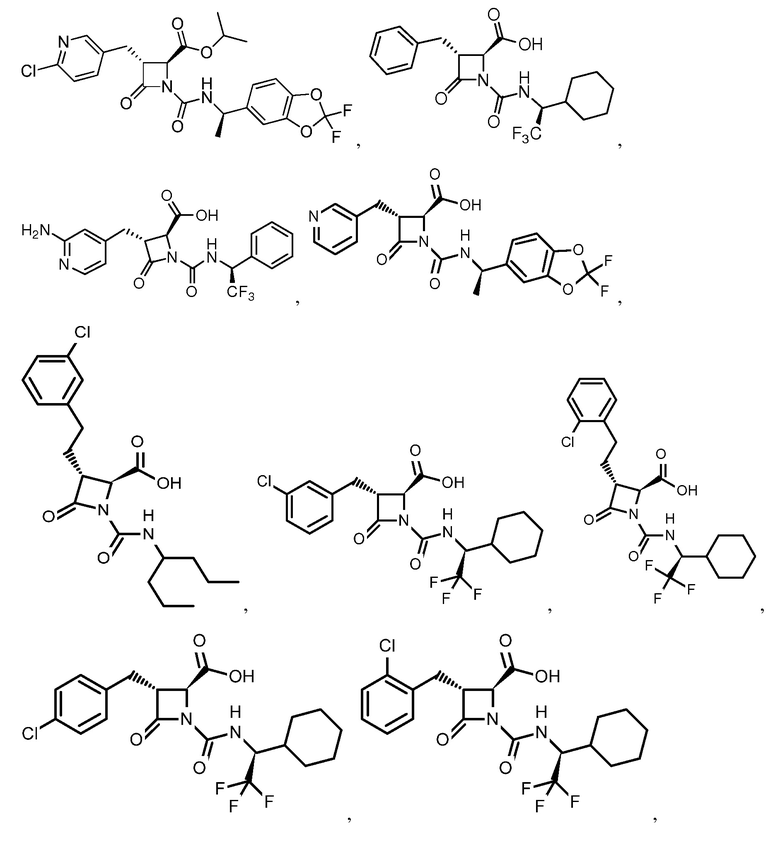
 или
или 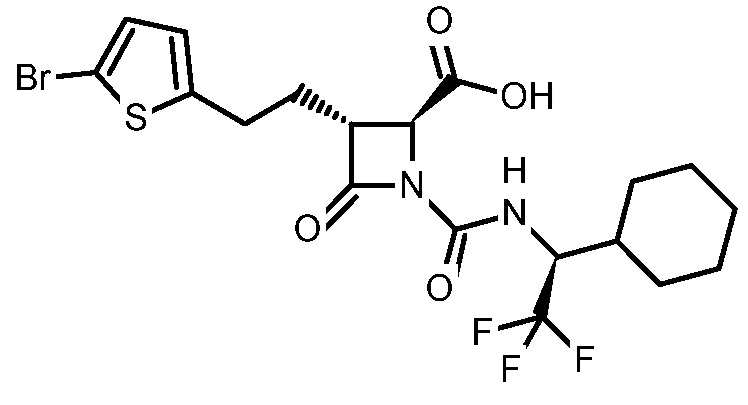 .
.
В одном из аспектов настоящее изобретение относится к соединению формулы (V):
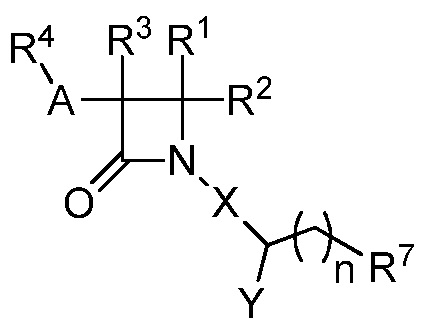 (V),
(V),
или к его фармацевтически приемлемой соли, где R1 представляет собой H или -C1-6 алкил; R2 представляет собой H, -C1-6 алкил, -CO2R5, -C(O)NR9R10, -CN, -SOqR5, -OR5, -CHN(OR5) или гетероарил; R3 представляет собой Н или -C1-6 алкил; A представляет собой C2-6 алкилен, C2-6 алкинилен или C2-6 алкинилен; R4 представляет собой циклоалкил, арил, гетероарил или гетероциклил, каждый из которых замещен 0-3 -NH2 или R6; каждый R5 независимо представляет собой H, -C1-6 алкил, аралкил или арил, замещенный 0-3 -NH2 или R6; каждый R6 независимо представляет собой галоген, гидрокси, циано, нитро, -C1-6 алкил, -C1-6 алкокси, -NHR10, -NR9R10, -C(O)R11, -C(O)OR11, -C(O)NR9R10, -C(NR8)(N(R8)2), -SOqR11, -SO2NR9R10, -NHC(O)OR11, -NHC(O)R11, арил, гетероарил, аралкил, циклоалкил, гетероаралкил, гетероциклил или гетероциклилалкил, или две R6 группы, вместе с атомами, к которым они присоединены, образуют 5-7-членное кольцо; Х представляет собой -C(O)O-, -OC(O)-, -C(O)S(O)2-, -S(O)2C(O)-, -C(O)N(R5)- или -N(R5)C(O)-; Y представляет собой -C1-6 алкил, циклоалкил, арил, гетероарил или гетероциклил, каждый из которых замещен 0-3 -NH2 или R6; R7 представляет собой Н, -C1-6 алкил, циклоалкил, арил, гетероарил или гетероциклил, каждый из которых замещен 0-3 -NH2 или R6; каждый R8 независимо представляет собой H, -C1-6 алкил, -C(O)R5, -C(O)OR5, арил, гетероарил, аралкил, гетероаралкил, гетероциклил или гетероциклилалкил; каждый из R9 и R10 независимо представляет собой -C1-6 алкил, циклоалкил, гетероциклил, арил, или гетероарил, или R9 и R10 вместе образуют необязательно замещенное 5-7-членное кольцо; каждый R11 независимо представляет собой H, -C1-6 алкил, аралкил или арил; q равно целому числу от 0 до 2; и n равно целому числу от 0 до 2.
В некоторых вариантах осуществления R1 представляет собой H.
В некоторых вариантах осуществления R2 представляет собой -CO2R5, где R5 представляет собой Н, -C1-6 алкил (например, этил), или аралкил (например, бензил).
В некоторых вариантах осуществления A представляет собой C2-6 алкилен (например, этилен или пропилен).
В некоторых вариантах осуществления R4 представляет собой арил или гетероарил. В некоторых вариантах осуществления R4 представляет собой арил. В некоторых вариантах осуществления R4 представляет собой фенил, замещенный 1 R6. В некоторых вариантах осуществления R6 представляет собой -C1-6 алкил, галоген (например, хлор, бром, фтор), галогеналкокси (например, -OCF3) или -C(NR8)(N(R8)2). В некоторых вариантах осуществления R6 представляет собой -C(NR8)(N(R8)2), и каждый R8 представляет собой Н. В некоторых вариантах осуществления R6 представляет собой -C(NR8)(N(R8)2), и каждый R8 представляет собой Н. В некоторых вариантах осуществления R8 представляет собой Н или -C(O)OR5. В некоторых вариантах осуществления R5 представляет собой -C1-6 алкил (например, гексил).
В некоторых вариантах осуществления R4 представляет собой гетероарил (например, 6-членный гетероарил или 5-членный гетероарил), замещенный 0-3 -NH2 или R6. В некоторых вариантах осуществления R4 представляет собой 6-членный гетероарил (например, пиридил), замещенный 0-3 -NH2 или R6. В некоторых вариантах осуществления R6 представляет собой галоген (например, хлор).
В некоторых вариантах осуществления X представляет собой -C(O)N(R5)- или -N(R5)C(O)-. В некоторых вариантах осуществления X представляет собой -C(O)N(R5)-, и R5 представляет собой H.
В некоторых вариантах осуществления n равно 0.
В некоторых вариантах осуществления R7 представляет собой -C1-6 алкил (например, метил, -CF3).
В некоторых вариантах осуществления Y представляет собой -C1-6 алкил (например, этил, пропил), циклоалкил, арил, гетероарил или гетероциклил, каждый из которых замещен 0-3 -NH2 или R6. В некоторых вариантах осуществления Y представляет собой циклоалкил (например, циклогексил). В некоторых вариантах осуществления Y представляет собой фенил, замещенный 0 R6. В некоторых вариантах осуществления Y представляет собой фенил, замещенный 1 R6. В некоторых вариантах осуществления R6 представляет собой -C1-6 алкил, галоген (например, хлор, бром, фтор) или галогеналкокси (например, -OCF3). В некоторых вариантах осуществления Y представляет собой фенил, замещенный 2 R6. В некоторых вариантах осуществления две R6 группы, взятые вместе с атомами, к которым они присоединены, образуют 5-7 членное кольцо. В некоторых вариантах осуществления две R6 группы, взятые вместе с атомами, к которым они присоединены, образуют 5-7 членное кольцо, и кольцо выбрано из: 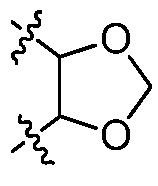 и
и 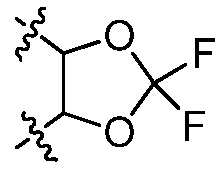 .
.
В некоторых вариантах осуществления соединение формулы (V) выбрано из соединения формулы (Va):
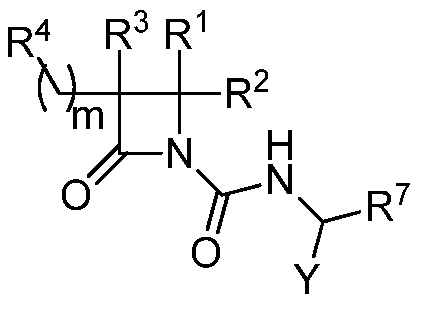 (Va),
(Va),
где R1, R2, R3, R4, R7 и Y такие, как описано для формулы (V), и m равно целому числу от 2 до 6.
В некоторых вариантах осуществления соединение формулы (Va) выбрано из соединения формулы (Vb):
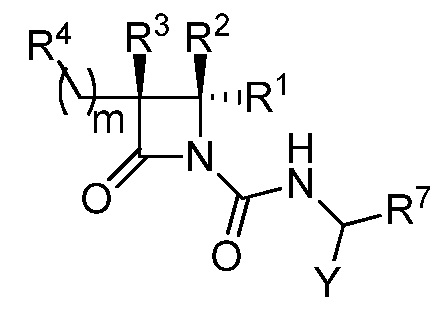 (Vb),
(Vb),
где R1, R2, R3, R4, R7, Y и m такие, как описано для формулы (Va).
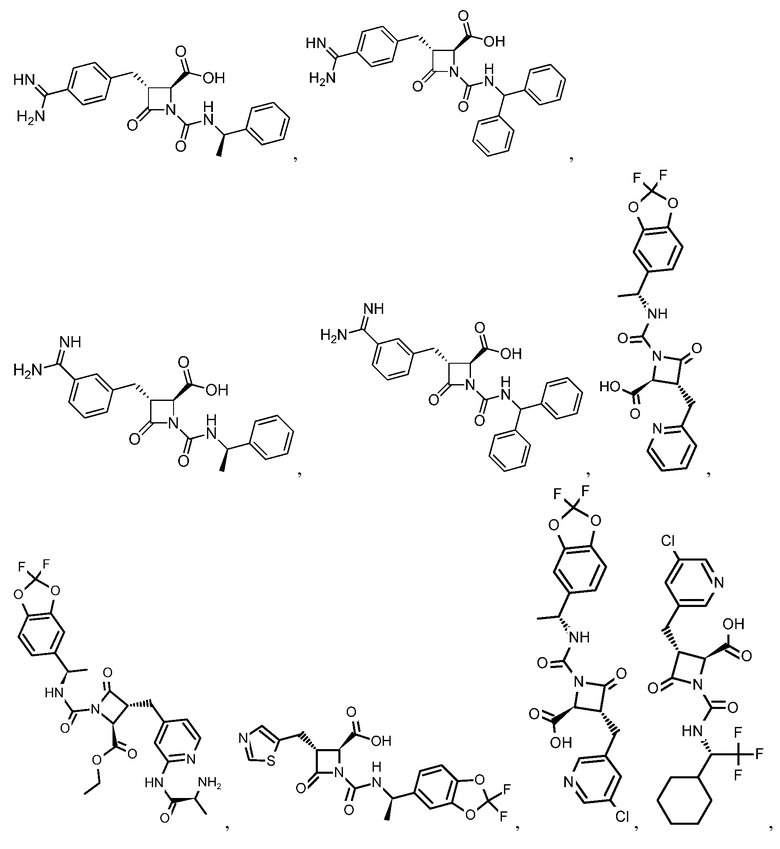
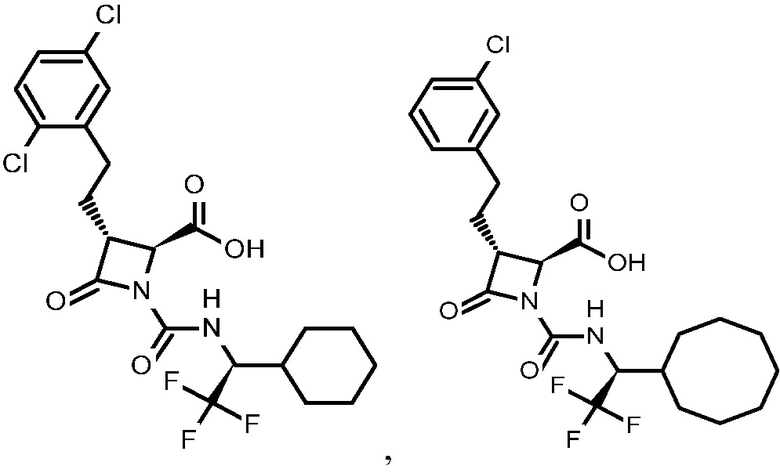
В некоторых вариантах осуществления соединение формулы (Vb) представляет собой:


 или
или  .
.
В одном из аспектов настоящее изобретение относится к соединению формулы (VI):
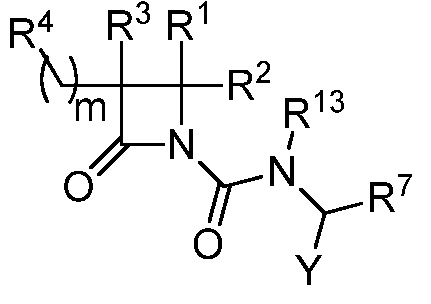 (VI),
(VI),
или к его фармацевтически приемлемой соли, где R1 представляет собой Н или -C1-6 алкил; R2 представляет собой Н, -C1-6 алкил, -CO2R5, -C(O)NR9R10, -CN, -SOqR5, -OR5, -CHN(OR5) или гетероарил; R3 представляет собой Н или -C1-6 алкил; R4 представляет собой циклоалкил, арил, гетероарил или гетероциклил, каждый из которых замещен 0-3 -NH2 или R6; каждый R5 независимо представляет собой H, -C1-6 алкил, аралкил или арил, замещенный 0-3 -NH2 или R6; каждый R6 независимо представляет собой галоген, гидрокси, циано, нитро, -C1-6 алкил, -C1-6 алкокси, -NHR10, -NR9R10, -C(O)R11, -C(O)OR11, -C(O)NR9R10, -C(NR8)(N(R8)2), -SOqR11, -SO2NR9R10, -NHC(O)OR11, -NHC(O)R11, арил, гетероарил, аралкил, циклоалкил, гетероаралкил, гетероциклил или гетероциклилалкил, или две R6 группы, вместе с атомами, к которым они присоединены, образуют 5-7-членное кольцо; Y представляет собой -C1-6 алкил, циклоалкил, арил, гетероарил, или гетероциклил, каждый из которых замещен 0-3 -NH2 или R6; R7 представляет собой Н, -C1-6 алкил, циклоалкил, арил, гетероарил или гетероциклил, каждый из которых замещен 0-3 -NH2 или R6; каждый R8 независимо представляет собой H, -C1-6 алкил, -C(O)R5, -C(O)OR5, арил, гетероарил, аралкил, гетероаралкил, гетероциклил или гетероциклилалкил; каждый из R9 и R10 независимо представляет собой -C1-6 алкил, циклоалкил, гетероциклил, арил или гетероарил, или R9 и R10 вместе образуют необязательно замещенное 5-7-членное кольцо; каждый R11 независимо представляет собой H, -C1-6 алкил, аралкил или арил; R13 представляет собой -C1-6 алкил; q равно целому числу от 0 до 2; и m равно целому числу от 1 до 6.
В некоторых вариантах осуществления R1 представляет собой H.
В некоторых вариантах осуществления R2 представляет собой -CO2R5, где R5 представляет собой Н или -C1-6 алкил (например, этил).
В некоторых вариантах осуществления A представляет собой C1-6 алкилен (например, этилен или пропилен).
В некоторых вариантах осуществления R4 представляет собой арил или гетероарил. В некоторых вариантах осуществления R4 представляет собой фенил, замещенный 1 R6. В некоторых вариантах осуществления R6 представляет собой галоген (например, хлор).
В некоторых вариантах осуществления R4 представляет собой гетероарил (например, 6-членный гетероарил или 5-членный гетероарил), замещенный 0-3 -NH2 или R6. В некоторых вариантах осуществления R4 представляет собой 6-членный гетероарил (например, пиридил), замещенный 0-3 -NH2 или R6. В некоторых вариантах осуществления R6 представляет собой галоген (например, хлор). В некоторых вариантах осуществления R4 представляет собой пиридил, замещенный 1 -NH2.
В некоторых вариантах осуществления X представляет собой -C(O)N(R5)- или -N(R5)C(O)-. В некоторых вариантах осуществления X представляет собой -C(O)N(R5)-, и R5 представляет собой H.
В некоторых вариантах осуществления n равно 0.
В некоторых вариантах осуществления R7 представляет собой -C1-6 алкил (например, метил).
В некоторых вариантах осуществления Y представляет собой циклоалкил, арил, гетероарил или гетероциклил, каждый из которых замещен 0-3 -NH2 или R6. В некоторых вариантах осуществления Y представляет собой циклоалкил (например, циклогексил). В некоторых вариантах осуществления Y представляет собой фенил, замещенный 0 R6. В некоторых вариантах осуществления Y представляет собой фенил, замещенный 1 R6.
В некоторых вариантах осуществления R13 представляет собой метил.
В некоторых вариантах осуществления соединение формулы (VI) выбрано из соединения формулы (VIa):
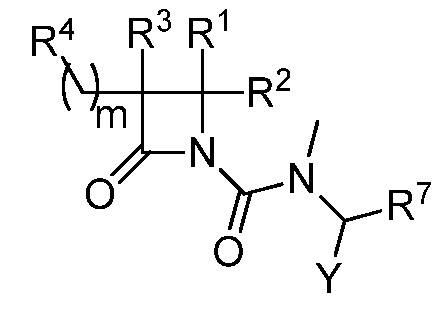 (VIa),
(VIa),
где R1, R2, R3, R4, R7, и Y такие, как описано для формулы (VI), и m равно целому числу от 1 до 6.
В некоторых вариантах осуществления соединение формулы (VIa) выбрано из соединения формулы (VIb):
 (VIb),
(VIb),
где R1, R2, R3, R4, R7, Y и m такие, как описано для формулы (VIa).
В некоторых вариантах осуществления соединение формулы (VIb) представляет собой:
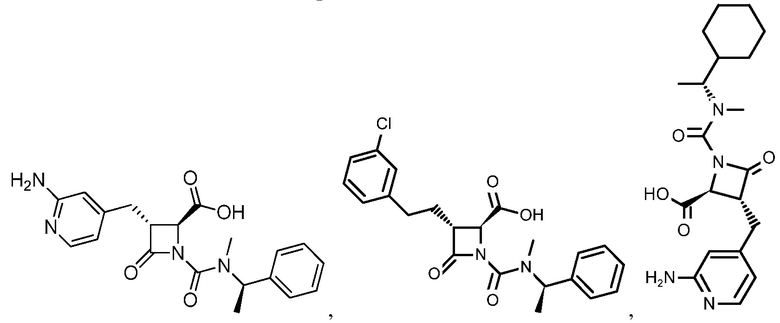 или
или 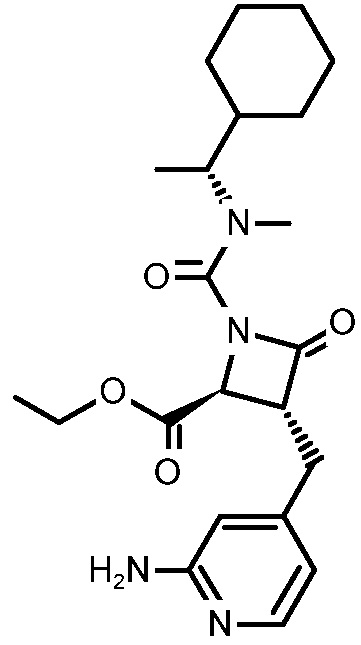 .
.
В одном из аспектов настоящее изобретение относится к соединению формулы (VII):
 (VII),
(VII),
или к его фармацевтически приемлемой соли, где R1 представляет собой Н или -C1-6 алкил; R2 представляет собой Н, -C1-6 алкил, -CO2R5, -C(O)NR9R10, -CN, -SOqR5, -OR5, -CHN(OR5) или гетероарил; R3 представляет собой -C1-6 алкил; A представляет собой связь, C1-6 алкилен, C2-6 алкинилен или C2-6 алкинилен; R4 представляет собой циклоалкил, арил, гетероарил или гетероциклил, каждый из которых замещен 0-3 -NH2 или R6; каждый R5 независимо представляет собой H, -C1-6 алкил, аралкил, или арил, замещенный 0-3 -NH2 или R6; каждый R6 независимо представляет собой галоген, гидрокси, циано, нитро, -C1-6 алкил, -C1-6 алкокси, -NHR10, -NR9R10, -C(O)R11, -C(O)OR11, -C(O)NR9R10, -C(NR8)(N(R8)2), -SOqR11, -SO2NR9R10, -NHC(O)OR11, -NHC(O)R11, арил, гетероарил, аралкил, циклоалкил, гетероаралкил, гетероциклил или гетероциклилалкил, или две R6 группы, вместе с атомами, к которым они присоединены, образуют 5-7-членное кольцо; Х представляет собой -C(O)O-, -OC(O)-, -C(O)S(O)2-, -S(O)2C(O)-, -C(O)N(R5)- или -N(R5)C(O)-; Y представляет собой Н, -C1-6 алкил, циклоалкил, арил, гетероарил или гетероциклил, каждый из которых замещен 0-3 -NH2 или R6; R7 представляет собой Н, -C1-6 алкил, циклоалкил, арил, гетероарил или гетероциклил, каждый из которых замещен 0-3 -NH2 или R6; каждый R8 независимо представляет собой H, -C1-6 алкил, -C(O)R5, -C(O)OR5, арил, гетероарил, аралкил, гетероаралкил, гетероциклил или гетероциклилалкил; каждый из R9 и R10 независимо представляет собой -C1-6 алкил, циклоалкил, гетероциклил, арил или гетероарил, или R9 и R10 вместе образуют необязательно замещенное 5-7-членное кольцо; каждый R11 независимо представляет собой H, -C1-6 алкил, аралкил или арил; q равно целому числу от 0 до 2; и n равно целому числу от 0 до 2.
В некоторых вариантах осуществления R1 представляет собой H.
В некоторых вариантах осуществления R2 представляет собой -CO2R5, где R5 представляет собой Н, -C1-6 алкил или аралкил (например, бензил).
В некоторых вариантах осуществления R3 представляет собой метил.
В некоторых вариантах осуществления A представляет собой C1-6 алкилен (например, этилен или пропилен).
В некоторых вариантах осуществления R4 представляет собой арил или гетероарил. В некоторых вариантах осуществления R4 представляет собой фенил, замещенный 1 R6. В некоторых вариантах осуществления R6 представляет собой галоген (например, хлор) или C1-6 алкокси.
В некоторых вариантах осуществления R4 представляет собой гетероарил (например, 6-членный гетероарил или 5-членный гетероарил), замещенный 0-3 -NH2 или R6. В некоторых вариантах осуществления R4 представляет собой 6-членный гетероарил (например, пиридин), замещенный 0-3 -NH2 или R6. В некоторых вариантах осуществления R6 представляет собой галоген (например, хлор).
В некоторых вариантах осуществления X представляет собой -C(O)N(R5)- или -N(R5)C(O)-. В некоторых вариантах осуществления X представляет собой -C(O)N(R5)-, и R5 представляет собой H.
В некоторых вариантах осуществления n равно 0.
В некоторых вариантах осуществления R7 представляет собой -C1-6 алкил (например, метил) или арил (например, фенил).
В некоторых вариантах осуществления Y представляет собой арил, гетероарил или гетероциклил, каждый из которых замещен 0-3 -NH2 или R6. В некоторых вариантах осуществления Y представляет собой арил. В некоторых вариантах осуществления Y представляет собой фенил, замещенный 0 R6. В некоторых вариантах осуществления Y представляет собой фенил, замещенный 1 R6.
В некоторых вариантах осуществления соединение формулы (VII) выбрано из соединения формулы (VIIa):
 (VIIa),
(VIIa),
где
R1, R2, R3, R4, R7 и Y такие, как описано для формулы (VII), и
m равно целому числу от 1 до 6.
В некоторых вариантах осуществления соединение формулы (VIIa) выбрано из соединения формулы (VIIb):
 (VIIb),
(VIIb),
где R1, R2, R3, R4, R7, Y и m такие, как описано для формулы (VIIa).
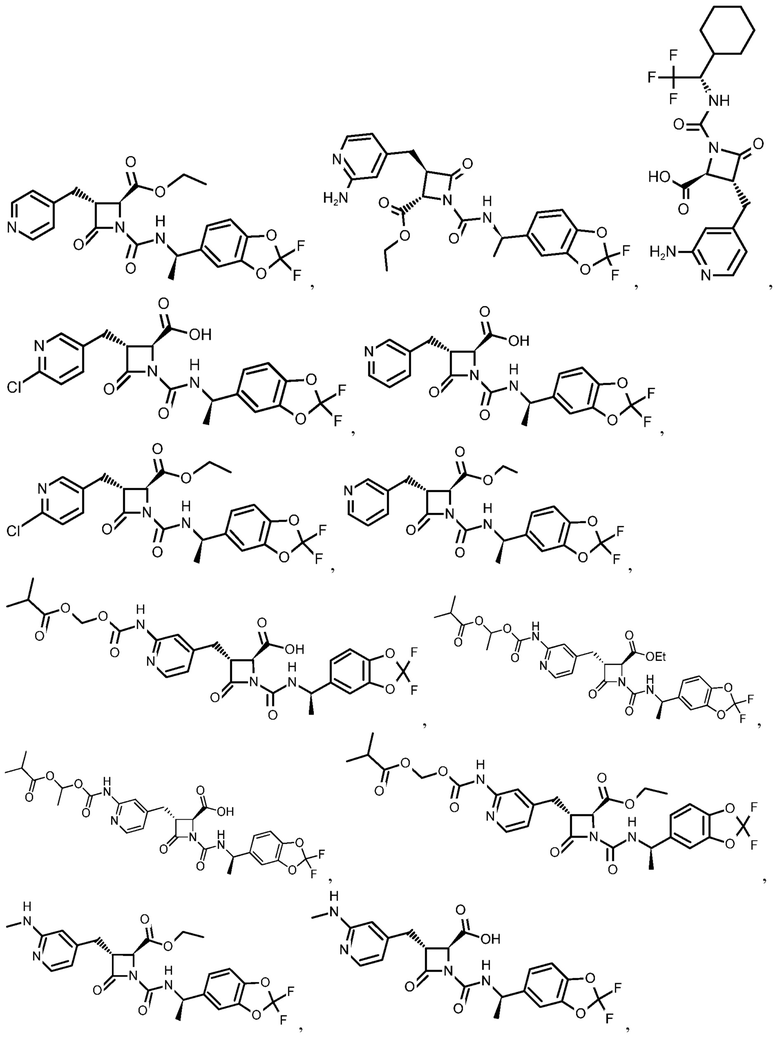
В некоторых вариантах осуществления соединение формулы (VIIb) представляет собой:
 или
или  .
.
В некоторых вариантах осуществления соединение формулы (VIIb) выбрано из соединения формулы (VIIc):
 (VIIc),
(VIIc),
где R1, R2, R3, R7, Y и m такие, как описано для формулы (VIIb), и R4 представляет собой циклоалкил, арил, гетероарил или гетероциклил, каждый из которых замещен 0-3 R6.
В одном из аспектов настоящее изобретение относится к фармацевтической композиции, содержащей соединение формулы (I)-(VII), или его фармацевтически приемлемую соль, и один или несколько фармацевтически приемлемых эксципиентов.
В некоторых вариантах осуществления композиция предоставлена в виде раствора.
В одном из аспектов настоящее изобретение относится к способу снижения риска инсульта (например, ишемия, например, транзиторная ишемическая атака) у индивида, который страдает ишемическим расстройством (например, транзиторная ишемическая атака), включающему введение индивиду эффективного количества соединения формулы (I)-(VII), или его фармацевтически приемлемой соли, или композиции, описанной в настоящем документе (например, композиции, содержащей соединение формулы (I)-(VII).
В некоторых вариантах осуществления введение снижает риск инсульта у индивида по сравнению с индивидом, которому это соединение не вводили.
В одном из аспектов настоящее изобретение относится к способу снижения системной эмболии, не связанной с центральной нервной системой (например, ишемия, например, транзиторная ишемическая атака) у индивида, который страдает ишемическим расстройством (например, транзиторная ишемическая атака), включающему введение индивиду эффективного количества соединения формулы (I)-(VII), или его фармацевтически приемлемой соли, или композиции, описанной в настоящем документе (например, композиции, содержащей соединение формулы (I)-(VII).
В некоторых вариантах осуществления введение снижает системную эмболию, не связанную с центральной нервной системой, у индивида по сравнению с индивидом, которому это соединение не вводили.
В одном из аспектов настоящее изобретение относится к способу лечения глубокого тромбоза вен, включающий введение индивиду, который страдает ишемическим нарушением (например, транзиторная ишемическая атака), эффективного количества соединения формулы (I)-(VII) или его фармацевтически приемлемой соли, или композиции, описанной в настоящем документе (например, композиции, содержащей соединение формулы (I)-(VII)).
В одном из аспектов настоящее изобретение относится к способу снижения риска рецидива глубокого тромбоза вен, включающий введение индивиду, который страдает глубоким тромбозом вен, эффективного количества соединения формулы (I)-(VII) или его фармацевтически приемлемой соли, или композиции, описанной в настоящем документе (например, композиции, содержащей соединение формулы (I)-(VII)). В некоторых вариантах осуществления введение снижает риск рецидива глубокого тромбоза вен у индивида по сравнению с индивидом, которому это соединение не вводили.
В одном из аспектов настоящее изобретение относится к способу снижения риска рецидива легочной эмболии (например, симптоматическая (!) легочная эмболия), включающий введение индивиду, который страдает легочной эмболией, эффективного количества соединения формулы (I)-(VII) или его фармацевтически приемлемой соли, или композиции, описанной в настоящем документе (например, композиции, содержащей соединение формулы (I)-(VII)).
В некоторых вариантах осуществления введение снижает риск рецидива легочной эмболии у индивида по сравнению с индивидом, которому это соединение не вводили.
В одном из аспектов настоящее изобретение относится к профилактики легочной эмболии у индивида, который страдает легочной эмболией, включающий введение индивиду эффективного количества соединения формулы (I)-(VII) или его фармацевтически приемлемой соли, или композиции, описанной в настоящем документе (например, композиции, содержащей соединение формулы (I)-(VII)).
В одном из аспектов настоящее изобретение относится к способу лечения индивида с ишемической атакой (например, транзиторная ишемическая атака), включающему: введение соединения формулы (I)-(VII) или его фармацевтически приемлемой соли, или композиции, описанной в настоящем документе (например, композиции, содержащей соединение формулы (I)-(VII)) индивиду. В некоторых вариантах осуществления соединение вводят индивиду в течение 24 часов или менее, например, 12, 10, 9, 8, 7, 6 часов или менее, после начала ишемической атаки у индивида.
В одном из аспектов настоящее изобретение относится к способу лечения индивида с ишемической атакой (например, транзиторная ишемическая атака), включающему: введение соединения формулы (I)-(VII) или его фармацевтически приемлемой соли, или композиции, описанной в настоящем документе (например, композиции, содержащей соединение формулы (I)-(VII)) индивиду. В некоторых вариантах осуществления соединение вводят индивиду в течение более 2 часов до 12 часов, например, более 2 часов до 10 часов или менее, более 2 часов до 8 часов или менее, после начала ишемической атаки у индивида.
В одном из аспектов настоящее изобретение относится к способу ингибирования Фактора XIa у индивида, включающий введение индивиду, который страдает ишемией, эффективного количества соединения формулы (I)-(VII) или его фармацевтически приемлемой соли, или композиции, описанной в настоящем документе (например, композиции, содержащей соединение формулы (I)-(VII)).
В некоторых вариантах осуществления индивидом является млекопитающее (например, человек). В некоторых вариантах осуществления индивиду было проведено хирургическое вмешательство (например, хирургическая замена коленного сустава, хирургическая замена тазобедренного сустава). В некоторых вариантах осуществления индивидом является индивид с «неклапанной» фибрилляцией предсердий. В некоторых вариантах осуществления индивид имеет один или более следующих факторов риска инсульта: предшествующий инсульт (например, ишемический, геморрагический, неизвестной природы), транзиторная ишемическая атака или системная эмболия, не связанная с ЦНС. В некоторых вариантах осуществления имеет один или более следующих факторов риска инсульта: возраст 75 лет и старше, гипертония, сердечная недостаточность или сниженная фракция выброса левого желудочка (например, меньше или равная 35%) или сахарный диабет.
В некоторых вариантах осуществления соединение вводят с помощью перорального или парентерального (например, внутривенное) введения.
В некоторых вариантах осуществления соединение вводят до начала ишемического приступа (например, индивиду с риском ишемического приступа).
В некоторых вариантах осуществления соединение вводят после ишемического приступа (например, транзиторная ишемическая атака).
В некоторых вариантах осуществления соединение вводят приблизительно через 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13 или 14 дней, или более после ишемического приступа (например, транзиторная ишемическая атака).
В некоторых вариантах осуществления соединение вводят приблизительно через 1, 2, 3, 4, 5, 6, 7 или 8 недель, или более после ишемического приступа (например, транзиторная ишемическая атака).
В некоторых вариантах осуществления, где соединение вводят в комбинации с дополнительным терапевтическим средством. В некоторых вариантах осуществления дополнительно терапевтическое средство вводят после введения соединения. В некоторых вариантах осуществления дополнительное терапевтическое средство вводят перорально. В некоторых вариантах осуществления дополнительное терапевтическое средство вводят по меньшей мере через 1, 2, 3, 4, 5, 6, 7, 8, 10, 12, 14, 16, 18, 20 или 24 часа, или более после введения соединения. В некоторых вариантах осуществления дополнительное терапевтическое средство вводят по меньшей мере через 1, 2, 3, 4, 5, 6, 7, 14, 21 или 28 дней, или более после введения соединения. В некоторых вариантах осуществления дополнительное терапевтическое средство вводят приблизительно через 1 день, приблизительно через 2 дня, приблизительно через 3 дня, приблизительно через 4 дня, приблизительно через 5 дней, приблизительно через 6 дней, приблизительно через 7 дней или более после введения соединения.
В некоторых вариантах осуществления где дополнительное терапевтическое средство вводят постоянно (например, в течение приблизительно 1 дня, приблизительно 2 дней, приблизительно 3 дней, приблизительно 4 дней, приблизительно 5 дней, приблизительно 6 дней, приблизительно 7 дней, приблизительно 8 дней, приблизительно 9 дней, приблизительно 10 дней, приблизительно 11 дней, приблизительно 12 дней, приблизительно 13 дней или приблизительно 14 дней, или более) после введения соединения.
В некоторых вариантах осуществления дополнительное терапевтическое средство действует на побочный эффект (например, активное патологическое кровотечение или тяжелые аллергические реакции (например, анафилактические реакции), спинномозговую или эпидуральную гематому, желудочно-кишечные нарушения (например, боль в верхней части живота, диспепсия, зубная боль), общие нарушения и состояния в месте введения (например, усталость), инфекции и инвазии (например, синусит, инфекция мочевыводящих путей), заболевания костно-мышечной и соединительной ткани (например, боли в спине, остеоартрит), дыхательные нарушения, нарушения в грудной клетке и средостении (например, боли в полости рта и глотке), травмы, отравления и осложнения после процедур (например, секреция раны), заболевания костно-мышечной и соединительной ткани (например, боли в конечностях, спазм мышц), расстройства нервной системы (например, обморок), кожные заболевания и заболевания подкожной клетчатки (например, зуд, пузыри), нарушения крови и лимфатической системы (например, агранулоцитоз), желудочно-кишечные нарушения (например, забрюшинное кровоизлияние), гепатобилиарные расстройства (например, желтуха, холестаз, цитолитический гепатит), нарушения иммунной системы (например. гиперчувствительность, анафилактическая реакция, анафилактический шок, ангионевротический отек), нарушения нервной системы (например, кровоизлияние в мозг, субдуральная гематома, эпидуральная гематома, гемипарез), заболевания кожи и подкожной клетчатки (например, синдром Стивенса-Джонсона).
В некоторых вариантах осуществления дополнительное терапевтическое средство представляет собой НПВП (например, аспирин, напроксен), ингибитор агрегации тромбоцитов (например, клопидогрель) или антикоагулянт (например, варфарин, эноксапарин).
В некоторых вариантах осуществления дополнительное терапевтическое средство оказывает аддитивный терапевтический эффект.
В некоторых вариантах осуществления дополнительное терапевтическое средство оказывает синергетический терапевтический эффект.
В еще одном аспекте настоящее изобретение относится к фармацевтической композиции, содержащей соединение, описанное в настоящем документе (например, соединение формулы (I)-(VII)) и фармацевтически приемлемый эксципиент.
В еще одном аспекте изобретение относится к способу модуляции (например, ингибирования) Фактора XIa у пациента. Способ включает стадию введения эффективного количества соединения, описанного в настоящем документе (например, соединения формулы (I)-(VII)) пациенту, при необходимости, таким образом модулируя (например, ингибируя) Фактор XIa.
В еще одном аспекте изобретение относится к способу лечения тромбоэмболического расстройства у индивида, при необходимости. Способ включает введение индивиду терапевтически эффективного количества соединения, описанного в настоящем документе (например, соединения формулы (I)-(VII)).
Тромбоэмболические расстройство может быть артериальным сердечно-сосудистым тромбоэмболическим нарушением, венозным сердечно-сосудистым тромбоэмболическим нарушением и тромбоэмболическим нарушением в камерах сердца; включая нестабильную стенокардию, острый коронарный синдром, первый инфаркт миокарда, повторный инфаркт миокарда, ишемию (например, внезапная смерть от ишемии или транзиторная ишемическая атака), инсульт, атеросклероз, окклюзионные заболевания периферических артерий, венозный тромбоз, тромбоз глубоких вен, тромбофлебит, артериальная эмболия, тромбоз коронарных артерий, тромбоз церебральных артерий, церебральная эмболия, эмболия почек, легочная эмболия и тромбоз в результате (а) протезов клапанов или других имплантантов, (b) катетеров , (c) стентов, (d) экстракорпорального кровообращения, (е) гемодиализа или (f) других процедур, при которых кровь контактирует с искусственной поверхностью, что способствует тромбозу.
В другом аспекте изобретение относится к способу лучения индивида, у которого обнаружен риск инсульта или тромбоза, таким образом сокращая вероятность инсульта или тромбоза у индивида. В некоторых вариантах осуществления у индивида дополнительно обнаружен риск кровотечения (например, чрезмерное кровотечение) или сепсис. В некоторых вариантах осуществления лечение является эффективным, не вызывая кровотечение. В некоторых вариантах осуществления лечение эффективно для поддержания проходимости инфузионных портов и линий. Кроме того, описываемые в настоящем документе соединения (например, соединения формулы (I)-(VII)) могут использоваться для лечения и профилактики других заболеваний, в которые вовлечена продукция тромбина в качестве физиологической роли. Например, тромбин вовлечен в заболеваемость и смертность при хронических и дегенеративных заболеваниях, таких как рак, артрит, атеросклероз, сосудистая деменция и болезнь Альцгеймера, за счет его способности регулировать множество различных типов клеток посредством специфического расщепления и активации рецептора тромбина на клеточной поверхности, митогенных эффектов, различных клеточных функций, таких как пролиферация клеток, например, патологическая пролиферация клеток сосудов, приводящая к рестенозу или ангиогенезу, высвобождение PDGF и синтез ДНК. Ингибирование Фактора XIa эффективно блокирует образование тромбина и, таким образом, нейтрализует любые физиологические эффекты тромбина на различные типы клеток. Характерные показания, которые обсуждались выше, включают некоторые, но не все, возможные клинические ситуации, поддающиеся лечению с помощью ингибитора Фактор XIa.
В еще одном из аспектов настоящее изобретение относится к способу лечения индивида с отеком (например, ангионевротический отек, например, наследственный ангионевротический отек), включающему введение соединения формулы (I)-(VII) или его фармацевтически приемлемой соли, или композиции, описанной в настоящем документе (например, композиции, содержащей соединение формулы (I)-(VII)) индивиду.
В еще одном из аспектов настоящее изобретение относится к способу ингибирования калликреина у индивида, включающему введение индивиду с отеком (например, ангионевротический отек, например, наследственный ангионевротический отек) эффективного количества соединения формулы (I)-(VII) или его фармацевтически приемлемой соли, или композиции, описанной в настоящем документе (например, композиции, содержащей соединение формулы (I)-(VII)) индивиду.
Краткое описание фигур
На ФИГ. 1A-1M изображена Диаграмма A и синтез Структур A-8 и A-9.
На ФИГ. 2 изображена Таблица В и синтез Структуры В-5.
На ФИГ. 3 изображена Таблица C и D и синтез Структуры D-8.
На ФИГ. 4 изображена Таблица E и синтез Структуры E-6.
На ФИГ. 5 изображена Таблица F и синтез Структуры F-8.
На ФИГ. 6 изображена Таблица G и синтез Структуры G-4.
На ФИГ. 7 изображена Таблица H и синтез Структуры H-9.
На ФИГ. 8 изображена Таблица I и синтез Структур I-4 - I-7.
На ФИГ. 9A-9B изображена Таблица J и синтез Структур J-3 и J-4.
На ФИГ. 10A-10B изображена Таблица K и синтез Структуры K-9.
Подробное описание
Определения
Термин "алкил," сам по себе или как часть другого заместителя, означает, если не указано иного, прямую или разветвленную цепь, или циклический углеводородный радикал, или их комбинацию, который может быть полностью насыщенным, моно- или полиненасыщенным и может включать ди- и мультивалентные радикалы, имеющие указанное число атомов углерода (то есть, C1-C10 означает от одного до десяти углеродов). Примеры насыщенных углеводородных радикалов включают, но ими не ограничиваются, такие группы, как метил, этил, н-пропил, изопропил, н-бутил, т-бутил, изобутил, втор-бутил, циклогексил, (циклогексил)метил, циклопропилметил, гомологи и изомеры, например, н-пентила, н-гексила, н-гептила, н-октила и тому подобное. Ненасыщенная алкильная группа представляет собой группу, имеющую одну или несколько двойных связей, или тройных связей. Примеры ненасыщенных алкильных групп включают, но ими не ограничиваются, винил, 2-пропенил, кротил, 2-изопентенил, 2-(бутадиенил), 2,4-пентадиенил, 3-(1,4-пентадиенил), этинил, 1- и 3-пропинил, 3-бутинил, и высшие гомологи и изомеры. Термин "алкил," если не указано иного, также обозначает такие производные, алкила, определенного в деталях ниже, такого как "гетероалкил." Алкильные группы, которые ограничены углеводородными группами, названы "гомоалкил". Если не указано иного, в каждом случае алкильная группа независимо необязательно замещена, то есть, не замещена (“незамещенный алкил”) или замещена (“замещенный алкил”) с одним или несколькими заместителями; например, от 1 до 5 заместителей, от 1 до 3 заместителей или 1 заместитель. В некоторых вариантах осуществления алкильная группа представляет собой незамещенный C1-10 алкил (например, -CH3). В некоторых вариантах осуществления алкильная группа представляет собой замещенный C1-10 алкил. Обычные аббревиатуры алкила включают Me (-CH3), Et (-CH2CH3), iPr (-CH(CH3)2), nPr (-CH2CH2CH3), n-Bu (-CH2CH2CH2CH3) или i-Bu (-CH2CH(CH3)2).
Термин "алкилен", сам по себе или как часть другого заместителя, означает двухвалентный радикал, полученный из алкана, примером которого служит, но им не ограничивается, --CH2CH2CH2CH2--, и дополнительно включает группы, которые описаны ниже в качестве "гетероалкилена". Обычно, алкильная (или алкиленовая) группа может иметь от 1 до 24 атомов углерода, где группы, которые имеют 10 или менее атомов углерода, являются предпочтительными в настоящем изобретении. "Низший алкил" или "низший алкилен" является более короткой цепью алкильной, или алкиленовой группы, обычно имеющей восемь или менее атомов углерода.
Термин “алкенил” относится к прямой или разветвленной углеводродной цепи, содержащей 2-12 атомов углерода (если не указано иного) и имеющей одну или несколько двойных связей. Примеры алкенильных группы включают, но ими не ограничиваются, группы аллил, пропенил, 2-бутенил, 3-гексенил и 3-октенил. Один из углеродов двойной связи может необязательно быть местом присоединения заместителя алкенила.
Термин “алкинилен” относится к двухвалентном алкенилу, например -CH=CH-, -CH2-CH=CH- и-CH=CH-CH2-.
Термин “алкинил” относится к прямой или разветвленной углеводородной цепи, содержащей 2-12 атомов углерода (если не указано иного) и отличающейся тем, что имеет один или несколько тройных связей. Примеры алкинильных групп включают, но ими не ограничиваются, этинил, пропаргил и 3-гексинил. Один из углеродов тройной связи может необязательно быть местом присоединения заместителя заместитель алкинила.
Термин “алкинилен” относится к двухвалентному алкинилу, например, -CH≡CH-, -CH2-CH≡CH- и -CH≡CH-CH2-.
Термины "алкокси," "алкиламино" и "алкилтио" (или тиоалкокси) используются в своем обычном значении, и относятся к алкильным группам, которые присоединены к остальной молекуле посредством атома кислорода, аминогруппы или атома серы, соответственно.
Термины “циано” и “нитрил” относятся к радикалу -CN.
Термины "циклоалкил", "гетероциклоалкил" или “гетероциклил”, сами по себе или в комбинации с другими терминами, представляет собой, если не указано иного, циклические варианты "алкила" и "гетероалкила", соответственно. Кроме того, для гетероциклоалкила или гетероциклила, гетероатом может занимать положение, в котором гетероцикл присоединен к остальной молекуле. Примеры циклоалкила включают, но ими не ограничиваются, циклопропил, циклобутил, циклопентил, циклогексил, 1-циклогексенил, 3-циклогексенил, циклогептил, циклооктанил и тому подобное. Примеры гетероциклоалкила и гетероциклила включают, но ими не ограничиваются, 1-1,2,5,6-тетрагидропиридил), 1-пиперидинил, 2-пиперидинил, 3-пиперидинил, 4-мофолинил, 3-морфолинил, тетрагидрофуран-2-ил, тетрагидрофуран-3-ил, тетрагидротиен-2-ил, тетрагидротиен-3-ил, 1-пиперазинил, 2-пиперазинил и тому подобное.
Термин “гетероалкил,” как используется в настоящем документе, относится к алкильной группе, как определено в настоящем документе, которая дополнительно содержит 1 или более (например, 1, 2, 3 или 4) гетероатомов (например, кислород, сера, азот, бор, кремний, фосфор) в пределах родительской цепи, где один или несколько гетероатомов встроены между соседними атомами углерода в пределах родительской углеродной цепи, и/или один или несколько гетероатомов встроены между атомом углерода и родительской молекулой, то есть, между местом присоединения. В некоторых вариантах осуществления гетероалкильная группа относится к насыщенной группе, имеющей от 1 до 10 атомов углерода и 1, 2, 3 или 4 гетероатома (“гетероC1-10 алкил”). В некоторых вариантах осуществления гетероалкильная группа представляет собой насыщенную группу, имеющую от 1 до 9 атомов углерода и 1, 2, 3 или 4 гетероатома (“гетероC1-9 алкил”). В некоторых вариантах осуществления гетероалкильная группа представляет собой насыщенную группу, имеющую от 1 до 8 атомов углерода и 1, 2, 3 или 4 гетероатомов (“гетероC1-8 алкил”). В некоторых вариантах осуществления гетероалкильная группа представляет собой насыщенную группу, имеющую от 1 до 7 атомов углерода и 1, 2, 3 или 4 гетероатома (“гетероC1-7 алкил”). В некоторых вариантах осуществления гетероалкильная группа представляет собой группу, имеющую от 1 до 6 атомов углерода и 1, 2 или 3 гетероатома (“гетероC1-6 алкил”). В некоторых вариантах осуществления гетероалкильная группа представляет собой насыщенную группу, имеющую от 1 до 5 атомов углерода и 1 или 2 гетероатома (“гетероC1-5 алкил”). В некоторых вариантах осуществления гетероалкильная группа представляет собой насыщенную группу, имеющую от 1 до 4 атомов углерода и 1 или 2 гетероатома (“гетероC1-4 алкил”). В некоторых вариантах осуществления гетероалкильная группа представляет собой насыщенную группу, имеющую от 1 до 3 атомов углерода и 1 гетероатом (“гетероC1-3 алкил”). В некоторых вариантах осуществления гетероалкильная группа представляет собой насыщенную группу, имеющую от 1 до 2 атомов углерода и 1 гетероатом (“гетероC1-2 алкил”). В некоторых вариантах осуществления гетероалкильная группа представляет собой насыщенную группу, имеющую 1 атом углерода и 1 гетероатом (“гетероC1 алкил”). В некоторых вариантах осуществления гетероалкильная группа представляет собой насыщенную группу, имеющую от 2 до 6 атомов углерода и 1 или 2 гетероатома (“гетероC2-6 алкил”). Если не указано иного, в каждом случае гетероалкильная группа независимо не замещена (“незамещенный гетероалкил”) или замещена (“замещенный гетероалкил”) одним или несколькими заместителями. В некоторых вариантах осуществления гетероалкильная группа представляет собой незамещенный гетероC1-10 алкил. В некоторых вариантах осуществления гетероалкильная группа представляет собой замещенный гетероC1-10 алкил.
Термин "гетероциклил”, если используется в комбинации с другими терминами (например, гетероциклилалкил), включает гетероциклильные кольца, как определено выше. Так, термин "гетероциклилалкил” обозначает радикалы, у которых гетероциклильная группа присоединена к алкильной группе, включая такие алкильные группы, в которых атом углерода (например, метиленовая группа) замещен, например, атомом кислорода.
Термин "галоген", сам по себе или как часть другого заместителя, обозначает, если не указано иного, атом фтора, хлора, брома или йода.
Термин “галогеналкил,” как используется в настоящем документе, относится к алкильной группе, как определено в настоящем документе, которая дополнительно содержит 1 или более (например, 1, 2, 3 или 4) атомов галогена (например, фтор, хлор, бром или йод), где алкильная группа замещена одним или несколькими атомами галогена. В некоторых вариантах осуществления галогеналкильная группа относится к насыщенной группе, имеющей от 1 до 10 атомов углерода и 1, 2, 3 или 4 атома галогена (“галогенC1-10 алкил”). Дополнительно, термин "галогеналкил," означает моногалогеналкил и полигалогеналкил. Например, термин "галогеналкил" означает, но ими не ограничивается, трифторметил, 2,2,2-трифторэтил, 4-хлорбутил, 3-бромпропил и тому подобное.
Термины “галогеналкокси” или “галогеналкоксил”, как используется в настоящем документе, относятся к алкокси группе, как определено в настоящем документе, которая дополнительно содержит 1 или более (например, 1, 2, 3 или 4) атомов галогена (например, фтор, хлор, бром или йод), где алкокси группа замещена одним или несколькими атомами галогена.
Термин “гидрокси” относится к радикалу -OH.
Термин "арил" означает, если не указано иного, полиненасыщенный, ароматический, углеводородный заместитель, которой может быть единичным кольцом или множественными кольцами (предпочтительно, от 1 до 3 колец), которые конденсированы друг с другом или ковалентно связаны. Термин "гетероарил" относится к арильным группам (или кольцам), которые содержат от одного до четырех гетероатомов, выбранных из N, O и S, где атомы азота и серы необязательно окислены, а атом(ы) азота необязательно кватернизованы. Гетероарильная группа может быть присоединена к остальной молекуле посредством гетероатома. Неограничивающие примеры арильных и гетероарильных групп включают фенил, 1-нафтил, 2-нафтил, 4-бифенил, 1-пирролил, 2-пирролил, 3-пирролил, 3-пиразолил, 2-имидазолил, 4-имидазолил, пиразинил, 2-оксазолил, 4-оксазолил, 2-фенил-4-оксазолил, 5-оксазолил, 3-изоксазолил, 4-изоксазолил, 5-изоксазолил, 2-тиазолил, 4-тиазолил, 5-тиазолил, 2-фурил, 3-фурил, 2-тиенил, 3-тиенил, 2-пиридил, 3-пиридил, 4-пиридил, 2-пиримидил, 4-пиримидил, 5-бензотиазолил, пуринил, 2-бензимидазолил, 5-индолил, 1-изохинолил, 5-изохинолил, 2-хиноксалинил, 5-хиноксалинил, 3-хинолил и 6-хинолил. Заместители для каждого вышеуказанного арильной и гетероарильной кольцевой системы выбраны из группы приемлемых заместителей, описанных ниже.
Вкратце, термин "арил", когда используется в комбинации с другими терминами (например, арилокси, арилтиокси, арилалкил, аралкил, гетероаралкил) включает как арильное, так и гетероарильное кольцо, как определено выше. Таким образом, термины "арилалкил", “аралкил” и “гетероаралкил” включают такие радикалы, в которых арильная или гетероарильная группа представляет собой группу, присоединенную к алкильной группе (например, бензил, фенэтил, пиридилметил и тому подобное), включая такие алкильные группы, в которых атом углерода (например, группа метилена) заменена на, например, атом кислорода (например, феноксиметил, 2-пиридиоксиметил, 3-(1-нафтилокси) пропил и тому подобное).
Термин “нитро” относится к радикалу-NO2.
"Защитная группа", как используется в настоящем документе, относится к части заместителя, которая по существу стабильная при определенных реакционных условиях, но которая отщепляется от заместителя при других реакционных условиях. Защитная группа также может быть выбрана таким образом, что она осаждается при прямом окислении компонента ароматического кольца соединения по изобретению. Примеры, которые можно использовать в качестве защитных групп, см., например, Greene et al., Protective Groups in Organic Synthesis, John Wiley & Sons, New York, 1991.
Группы алкила, алкенила, алкинила, циклоалкила, гетероциклила, арила и гетероарила, как определено в настоящем документе, необязательно замещены (например, “замещенный” или “незамещенный” алкил, “замещенный” или “незамещенный” алкенил, “замещенный” или “незамещенный” алкинил, “замещенный” или “незамещенный” циклоалкил, “замещенный” или “незамещенный” гетероциклил, “замещенный” или “незамещенный” арил, или “замещенный” или “незамещенный” гетероарил). В целом, термин “замещенный”, если перед ним находится термин “необязательно” или не, означает, что по меньшей мере один водород, присутствующий на группе (например, атом углерода или азота) замещен возможным заместителем, например, заместителем, который при замещении дает стабильное соединение, например, соединение, которое не подвергается спонтанной трансформации, например, перестройке, циклизации, удалению или другой реакции. Если не указано иного, “замещенная” группа имеет заместитель в одной или нескольких положениях, способных к замещению, группы, и если больше одного положения в любой заданной структуре замещено, то заместитель либо одинаковый, либо различный в каждом положении. Термин “замещенный” следует рассматривать как предусматривающий замещение всеми возможными заместителями органических соединений, описанными в настоящем изобретении, что приводит к образованию стабильного соединения. Для целей изобретения гетероатомы, такие как азот, могут иметь водородные заместители и/или любой подходящий заместитель, как описано в настоящем документе, который удовлетворяет валентности гетероатомов и приводит к образованию стабильной группы.
Примеры заместителей атом углерода включают, но ими не ограничиваются, галоген, -CN, -NO2, -N3, -SO2H, -SO3H, -OH, -ORaa, -ON(Rbb)2, -N(Rbb)2, -N(Rbb)3+X-, -N(ORcc)Rbb, -SH, -SRaa, -SSRcc, -C(=O)Raa, -CO2H, -CHO, -C(ORcc)2, -CO2Raa, -OC(=O)Raa, -OCO2Raa, -C(=O)N(Rbb)2, -OC(=O)N(Rbb)2, -NRbbC(=O)Raa, -NRbbCO2Raa, -NRbbC(=O)N(Rbb)2, -C(=NRbb)Raa, -C(=NRbb)ORaa, -OC(=NRbb)Raa, -OC(=NRbb)ORaa, -C(=NRbb)N(Rbb)2, -OC(=NRbb)N(Rbb)2, -NRbbC(=NRbb)N(Rbb)2, -C(=O)NRbbSO2Raa, -NRbbSO2Raa, -SO2N(Rbb)2, -SO2Raa, -SO2ORaa, -OSO2Raa, -S(O)Raa -S(=O)Raa, -OS(=O)Raa, -Si(Raa)3, -OSi(Raa)3 -C(=S)N(Rbb)2, -C(=O)SRaa, -C(=S)SRaa, -SC(=S)SRaa, -SC(=O)SRaa, -OC(=O)SRaa, -SC(=O)ORaa, -SC(=O)Raa, -P(=O)2Raa, -OP(=O)2Raa, -P(=O)(Raa)2, -OP(=O)(Raa)2, -OP(=O)(ORcc)2, -P(=O)2N(Rbb)2, -OP(=O)2N(Rbb)2, -P(=O)(NRbb)2, -OP(=O)(NRbb)2, -NRbbP(=O)(ORcc)2, -NRbbP(=O)(NRbb)2, -P(Rcc)2, -P(Rcc)3, -OP(Rcc)2, -OP(Rcc)3, -B(Raa)2, -B(ORcc)2, -BRaa(ORcc), C1-10 алкил, C1-10 пергалогеналкил, C2-10 алкенил, C2-10 алкинил, C3-10 циклоалкил, 3-14-членный гетероциклил, C6-14 арил и 5-14-членный гетероарил, где каждый алкил, алкенил, алкинил, циклоалкил, гетероциклил, арил и гетероарил независимо замещен 0, 1, 2, 3, 4 или 5 группами Rdd;
В каждом случае Raa независимо выбран из C1-10 алкила, C1-10 пергалогеналкила, C2-10 алкенила, C2-10 алкинила, C3-10 циклоалкила, 3-14-членного гетероциклила, C6-14 арила и 5-14-членного гетероарила, или две Raa группы объединены с образованием 3-14-членного гетероциклила или 5-14-членного гетероарильного кольца, где каждый алкил, алкенил, алкинил, циклоалкил, гетероциклил, арил и гетероарил независимо замещен 0, 1, 2, 3, 4 или 5 Rdd группы;
В каждом случае Rbb независимо выбран из водорода, -OH, -ORaa, -N(Rcc)2, -CN, -C(=O)Raa, -C(=O)N(Rcc)2, -CO2Raa, -SO2Raa, -C(=NRcc)ORaa, -C(=NRcc)N(Rcc)2, -SO2N(Rcc)2, -SO2Rcc, -SO2ORcc, -SORaa, -C(=S)N(Rcc)2, -C(=O)SRcc, -C(=S)SRcc, -P(=O)2Raa, -P(=O)(Raa)2, -P(=O)2N(Rcc)2, -P(=O)(NRcc)2, C1-10 алкила, C1-10 пергалогеналкила, C2-10 алкенила, C2-10 алкинила, C3-10 циклоалкила, 3-14-членного гетероциклила, C6-14 арила, и 5-14-членного гетероарила, или две Rbb группы объединены с образованием a 3-14-членного гетероциклила или 5-14-членного гетероарильного кольца, где каждый алкил, алкенил, алкинил, циклоалкил, гетероциклил, арил, и гетероарил независимо замещен 0, 1, 2, 3, 4 или 5 Rdd группы;
В каждом случае Rcc независимо выбран из водорода, C1-10 алкила, C1-10 пергалогеналкила, C2-10 акленил, C2-10 алкинила, C3-10 циклоалкила, 3-14-членного гетероциклила, C6-14 арила и 5-14-членного гетероарила, или две группы Rcc объединены с образованием 3-14-членного гетероциклила или 5-14-членного гетероарильного кольца, где каждый алкил, алкенил, алкинил, циклоалкил, гетероциклил, арил и гетероарил независимо замещен 0, 1, 2, 3, 4 или 5 группами Rdd;
В каждом случае Rdd независимо выбран из галогена, -CN, -NO2, -N3, -SO2H, -SO3H, -OH, -ORee, -ON(Rff)2, -N(Rff)2, -N(Rff)3+X-, -N(ORee)Rff, -SH, -SRee, -SSRee, -C(=O)Ree, -CO2H, -CO2Ree, -OC(=O)Ree, -OCO2Ree, -C(=O)N(Rff)2, -OC(=O)N(Rff)2, -NRffC(=O)Ree, -NRffCO2Ree, -NRffC(=O)N(Rff)2, -C(=NRff)ORee, -OC(=NRff)Ree, -OC(=NRff)ORee, -C(=NRff)N(Rff)2, -OC(=NRff)N(Rff)2, -NRffC(=NRff)N(Rff)2,-NRffSO2Ree, -SO2N(Rff)2, -SO2Ree, -SO2ORee, -OSO2Ree, -S(=O)Ree, -Si(Ree)3, -OSi(Ree)3, -C(=S)N(Rff)2, -C(=O)SRee, -C(=S)SRee, -SC(=S)SRee, -P(=O)2Ree, -P(=O)(Ree)2, -OP(=O)(Ree)2, -OP(=O)(ORee)2, C1-6 алкила, C1-6 пергалогеналкила, C2-6 алкенила, C2-6 алкинила, C3-10 циклоалкила, 3-10-членного гетероциклила, C6-10 арила, 5-10-членного гетероарила, где каждый алкил, алкенил, алкинил, циклоалкил, гетероциклил, арил и гетероарил независимо замещен 0, 1, 2, 3, 4 или 5 группами Rgg, или два присоединенных к одному и тому же атому Rdd заместителя могут быть объединены вместе с образованием =O или =S;
В каждом случае Ree независимо выбран из C1-6 алкила, C1-6 пергалогеналкила, C2-6 алкенила, C2-6 алкинила, C3-10 циклоалкила, C6-10 арила, 3-10-членного гетероциклила и 3-10-членного гетероарила, где каждый алкил, алкенил, алкинил, циклоалкил, гетероциклил, арил и гетероарил независимо замещен 0, 1, 2, 3, 4 или 5 группами Rgg;
В каждом случае Rff независимо выбран из водорода, C1-6 алкила, C1-6 пергалогеналкила, C2-6 алкенила, C2-6 алкинила, C3-10 циклоалкила, 3-10-членного гетероциклила, C6-10 арила и 5-10-членного гетероарила, или две группы Rff объединены с образованием 3-14-членного гетероциклила, или 5-14-членного гетероарильного кольца, где каждый алкил, алкенил, алкинил, циклоалкил, гетероциклил, арил и гетероарил независимо замещен 0, 1, 2, 3, 4 или 5 группами Rgg; и
В каждом случае Rgg независимо представляет собой галоген, -CN, -NO2, -N3, -SO2H, -SO3H, -OH, -OC1-6 алкил, -ON(C1-6 алкил)2, -N(C1-6 алкил)2, -N(C1-6 алкил)3+X-, -NH(C1-6 алкил)2+X-, -NH2(C1-6 алкил) +X-, -NH3+X-, -N(OC1-6 алкил)(C1-6 алкил), -N(OH)(C1-6 алкил), -NH(OH), -SH, -SC1-6 алкил, -SS(C1-6 алкил), -C(=O)(C1-6 алкил), -CO2H, -CO2(C1-6 алкил), -OC(=O)(C1-6 алкил), -OCO2(C1-6 алкил), -C(=O)NH2, -C(=O)N(C1-6 алкил)2, -OC(=O)NH(C1-6 алкил), -NHC(=O)( C1-6 алкил), -N(C1-6 алкил)C(=O)( C1-6 алкил), -NHCO2(C1-6 алкил), -NHC(=O)N(C1-6 алкил)2, -NHC(=O)NH(C1-6 алкил), -NHC(=O)NH2, -C(=NH)O(C1-6 алкил), -OC(=NH)(C1-6 алкил), -OC(=NH)OC1-6 алкил, -C(=NH)N(C1-6 алкил)2, -C(=NH)NH(C1-6 алкил), -C(=NH)NH2, -OC(=NH)N(C1-6 алкил)2, -OC(NH)NH(C1-6 алкил), -OC(NH)NH2, -NHC(NH)N(C1-6 алкил)2, -NHC(=NH)NH2, -NHSO2(C1-6 алкил), -SO2N(C1-6 алкил)2, -SO2NH(C1-6 алкил), -SO2NH2,-SO2C1-6 алкил, -SO2OC1-6 алкил, -OSO2C1-6 алкил, -SOC1-6 алкил, -Si(C1-6 алкил)3, -OSi(C1-6 алкил)3 -C(=S)N(C1-6 алкил)2, C(=S)NH(C1-6 алкил), C(=S)NH2, -C(=O)S(C1-6 алкил), -C(=S)SC1-6 алкил, -SC(=S)SC1-6 алкил, -P(=O)2(C1-6 алкил), -P(=O)(C1-6 алкил)2, -OP(=O)(C1-6 алкил)2, -OP(=O)(OC1-6 алкил)2, C1-6 алкил, C1-6 пергалогеналкил, C2-6 алкенил, C2-6 алкинил, C3-10 циклоалкил, C6-10 арил, 3-10-членный гетероциклил, 5-10-членный гетероарил, или два присоединенных к одному и тому атому Rgg заместителя могут быть объединены с образованием =O или =S, где X' представляет собой противоионы.
Эти и другие примеры заместителей описаны более подробно в разделах «Подробное описание», «Примеры» и «Формула изобретения». Нет намерений каким-либо образом ограничить изобретение вышеуказанным перечнем примеров заместителей.
Соединения
В настоящем документе описаны соединения, которые ингибируют Фактор XIa или калликреин, например, соединения, описанные в настоящем документе (например, соединение формулы (I)-(VII)).
Примеры соединений включают, но ими не ограничиваются, соединения, раскрытые в Таблице 1 ниже:
Таблица 1
Примеры соединений по настоящему изобретению.

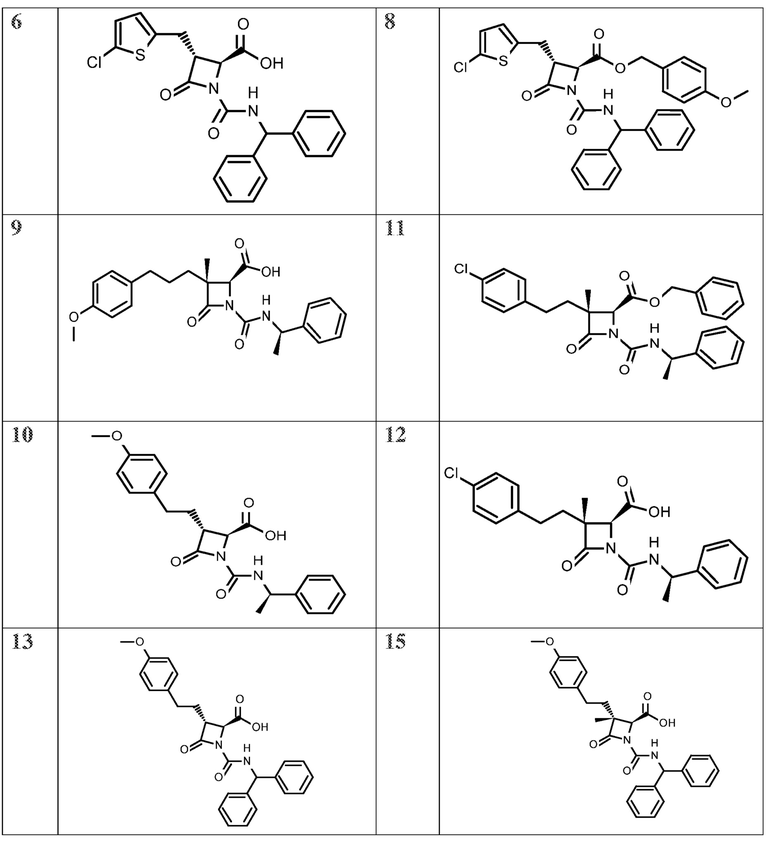

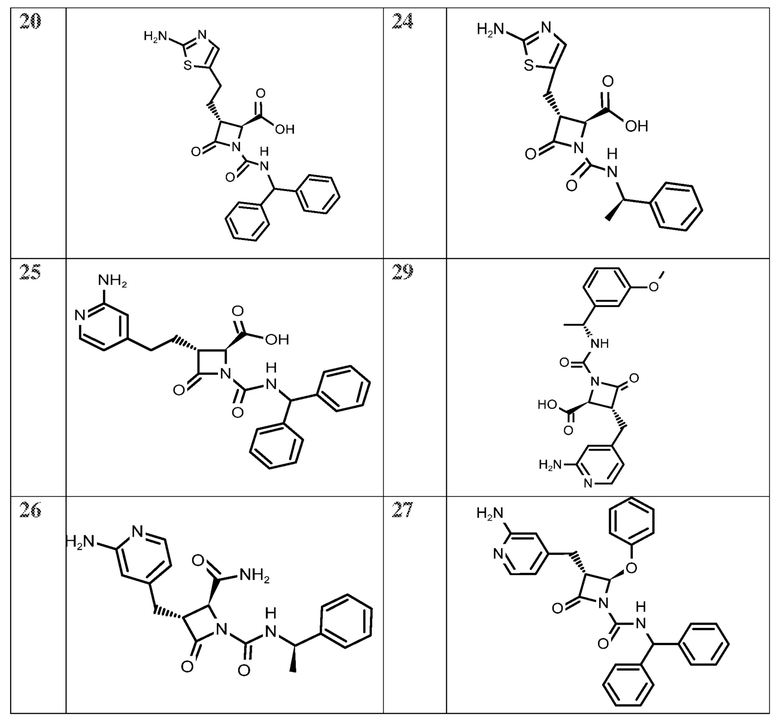
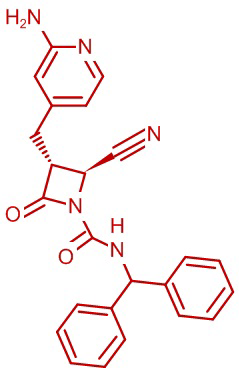


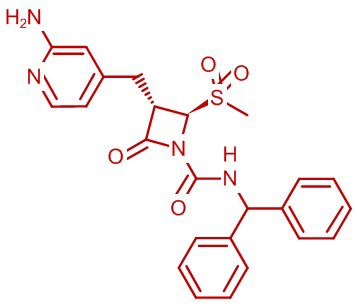

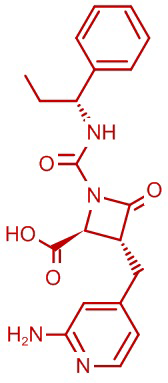
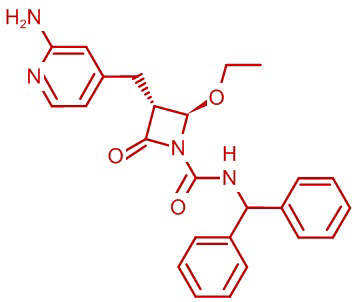

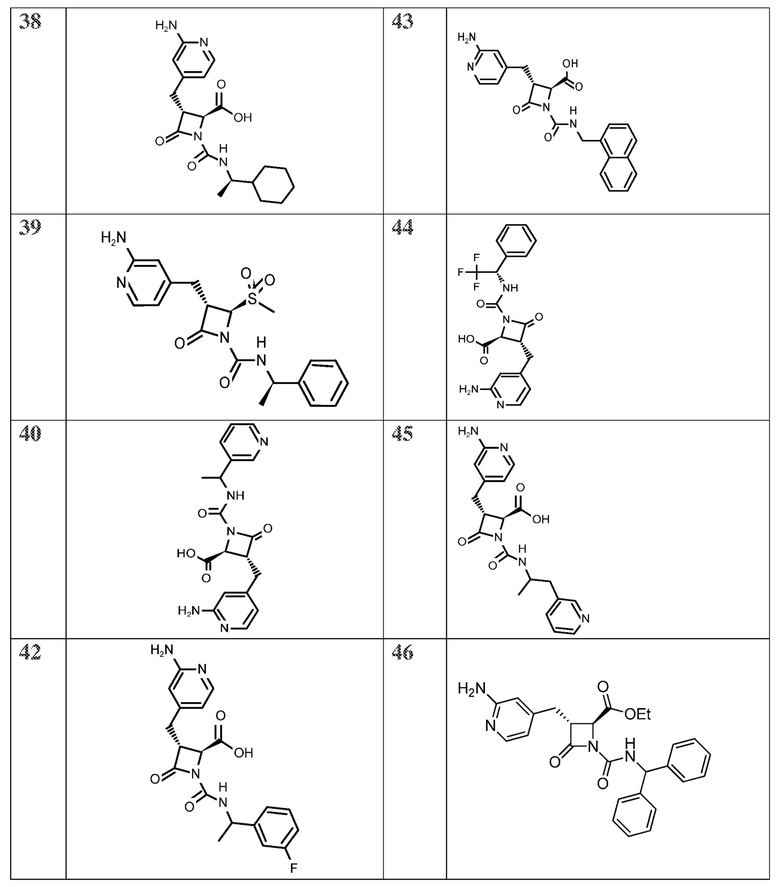
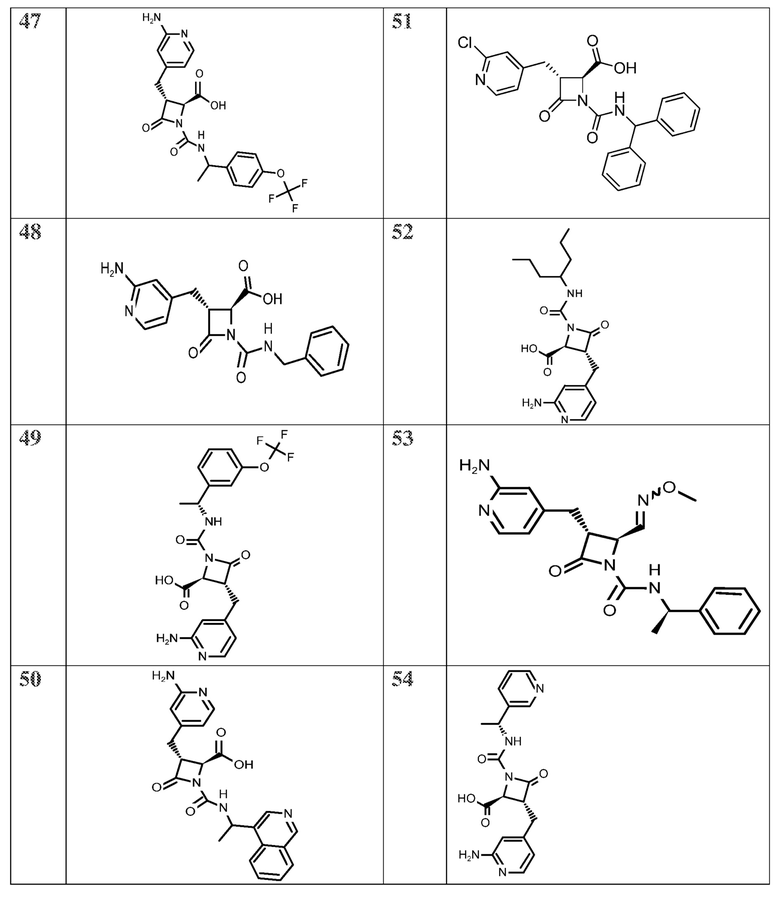
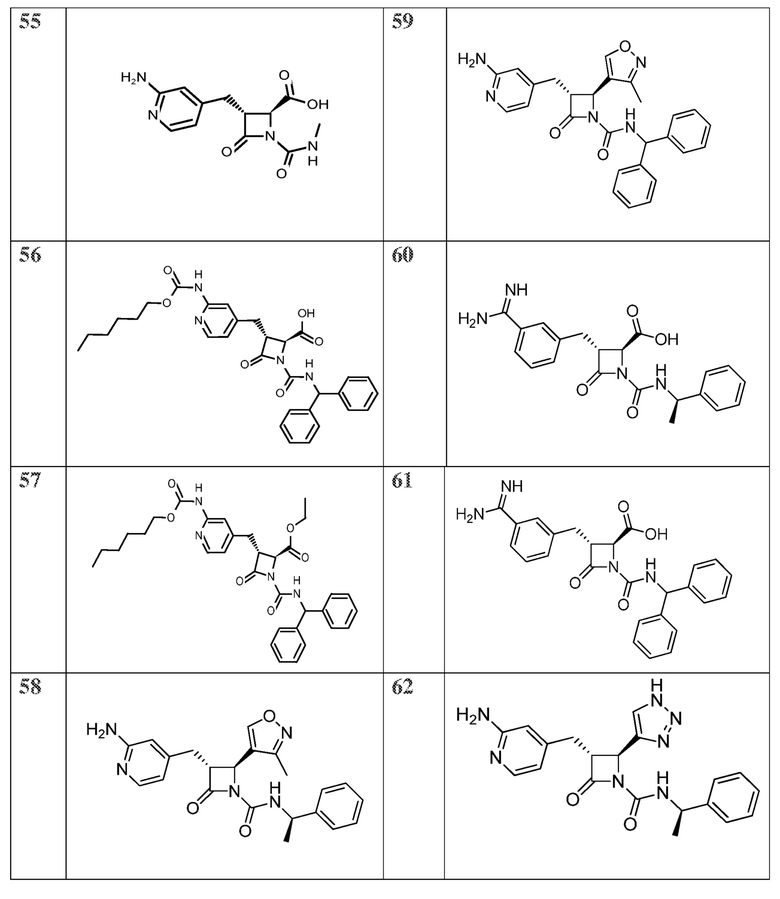
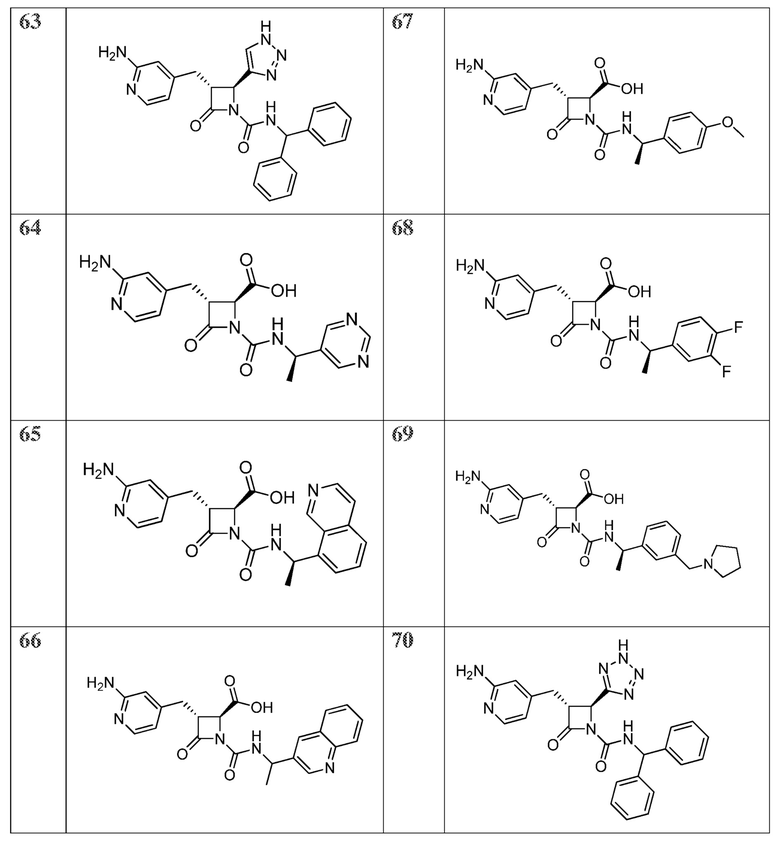
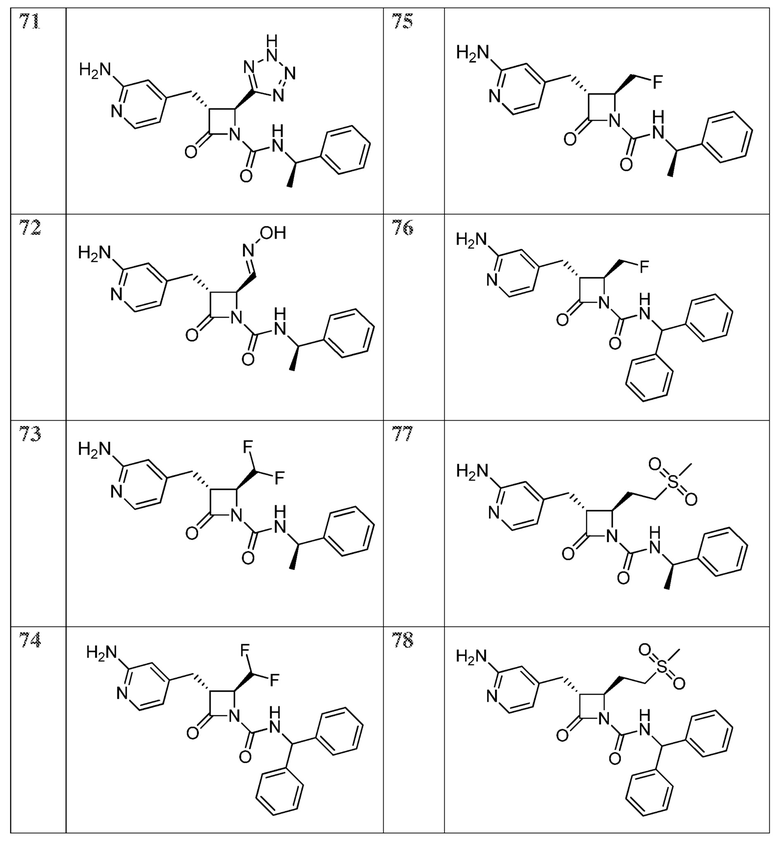
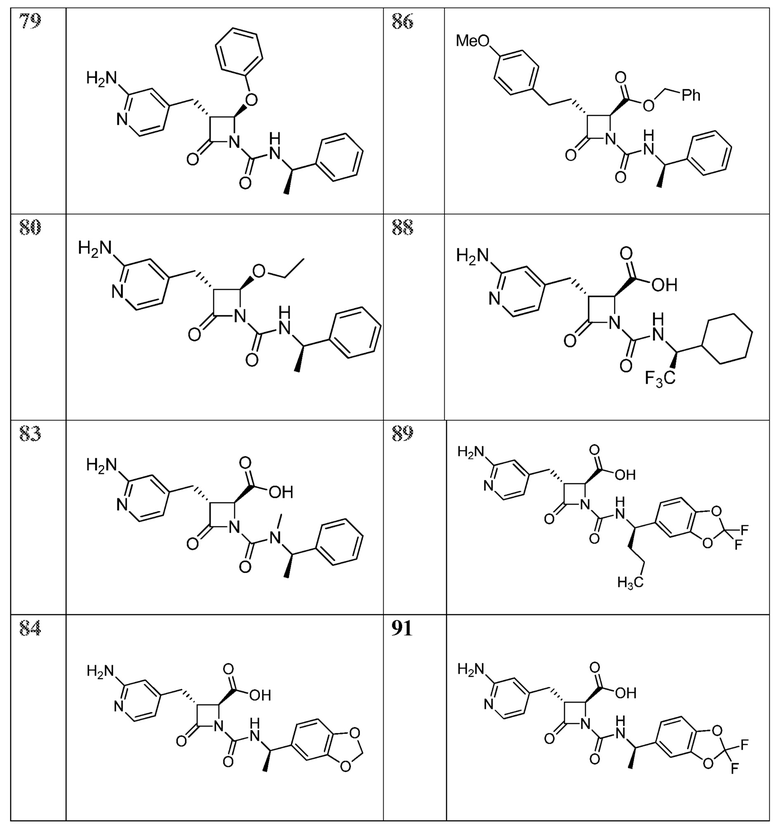
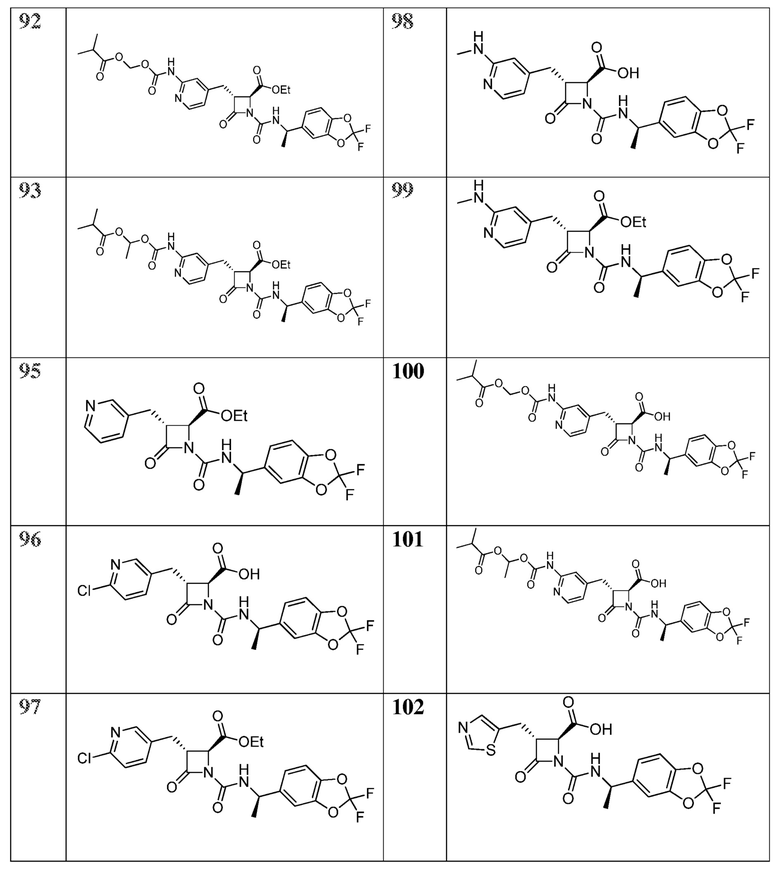
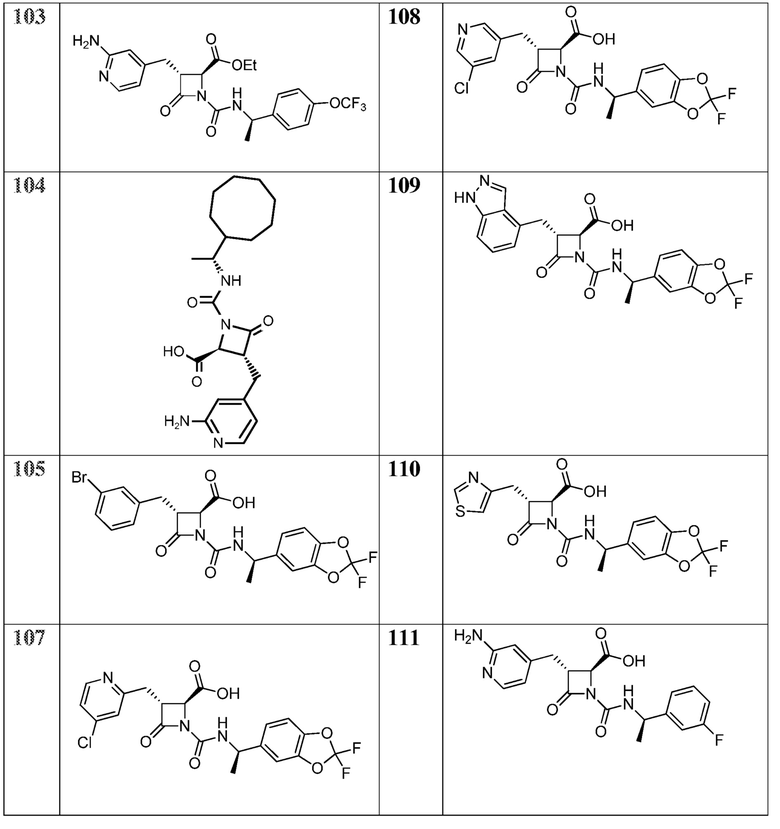
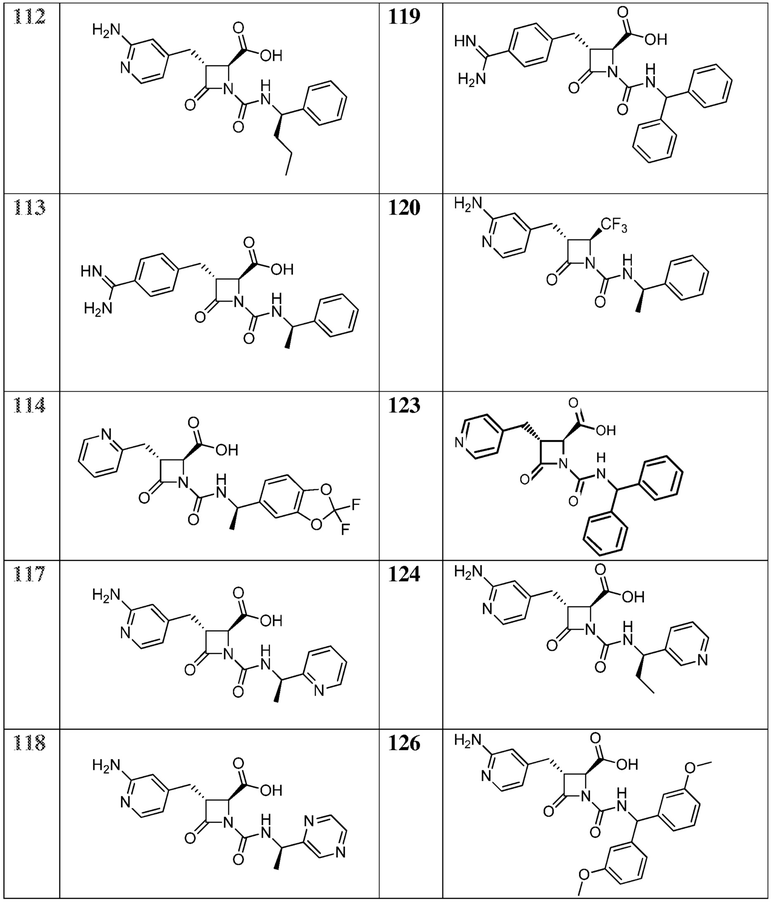
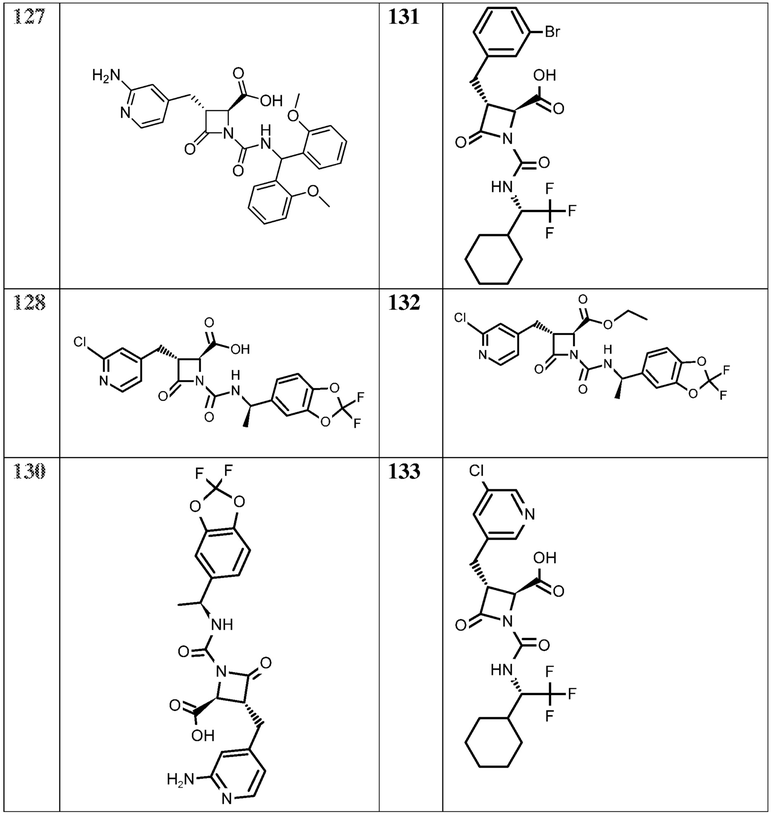


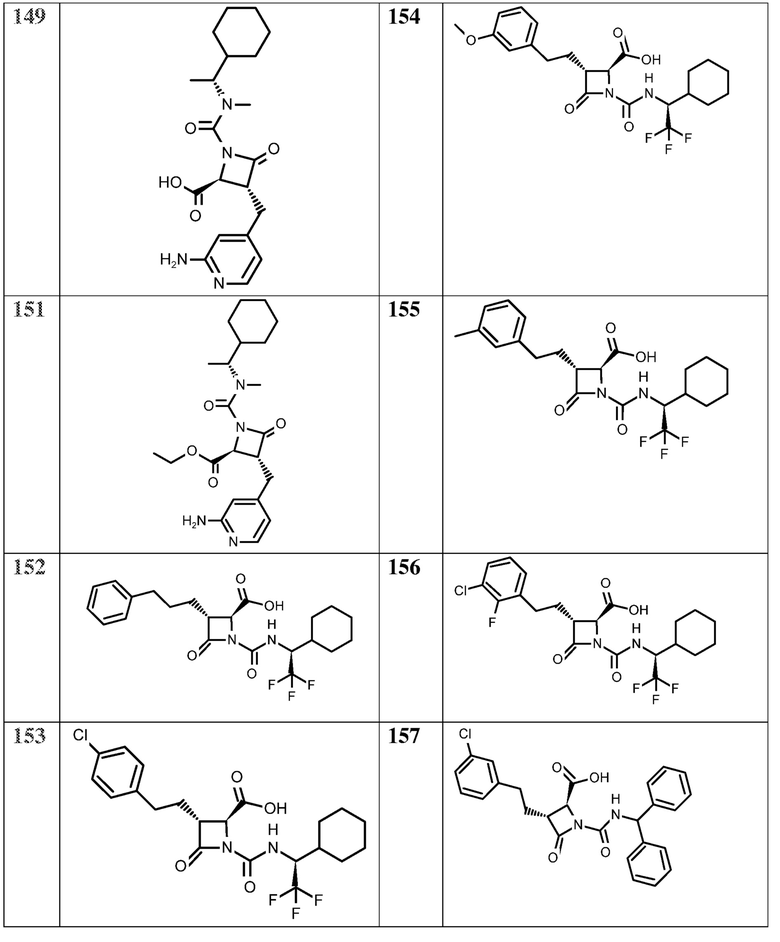
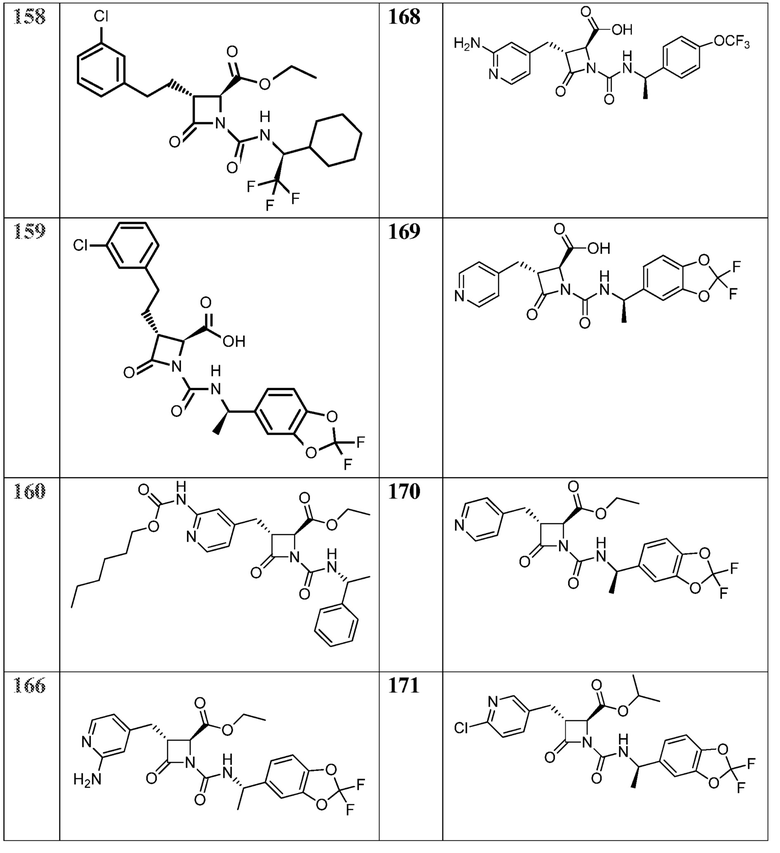
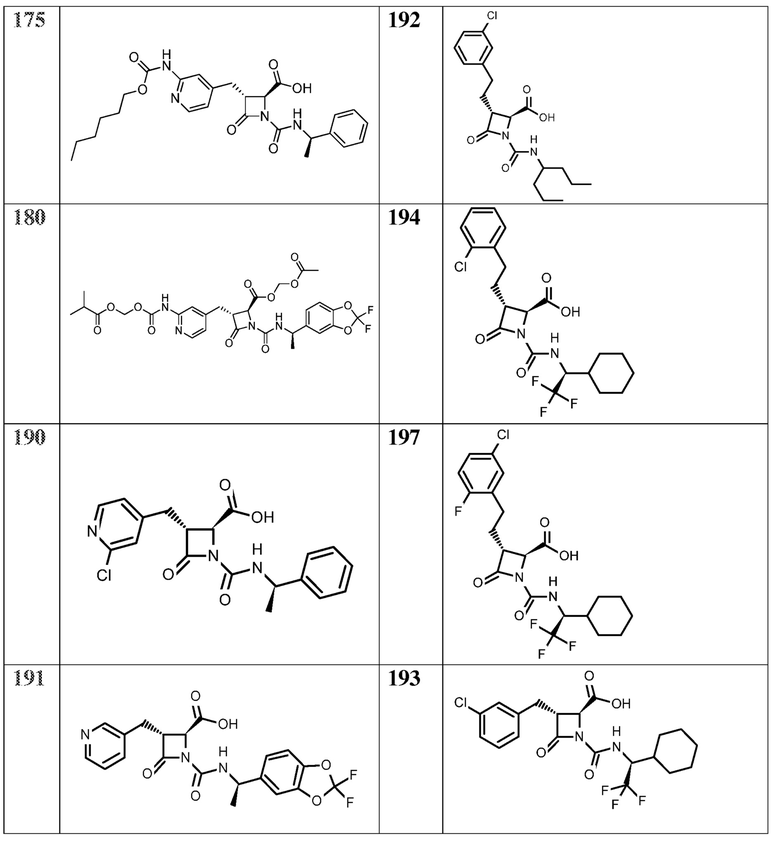
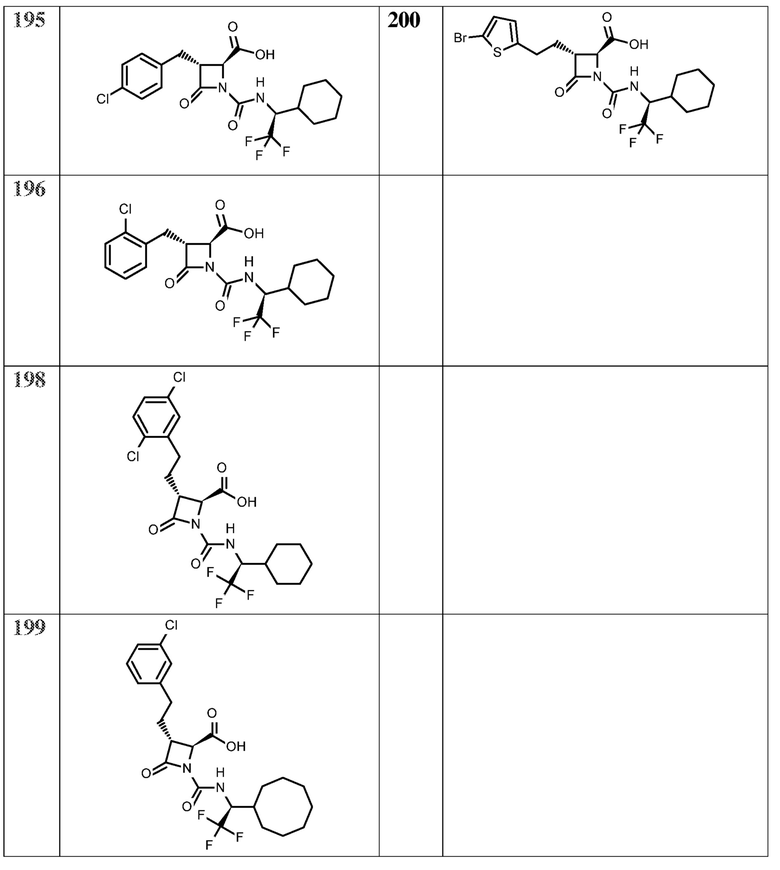
В некоторых вариантах осуществления соединения, описанное в настоящем документе, образует соль. Соединение, описанное в настоящем документе, может быть введено в виде свободной кислоты, цвиттериона или в виде соли. Соль также может быть образована между катионом и отрицательно заряженным заместителем на соединении, описанном в настоящем документе. Подходящие катионные противоионы включают ионы натрия, ионы калия, ионы магния, ионы кальция и ионы аммония (например, катион тетраалкил аммония, такой как ион тетраметиламмония). В соединениях, включая положительно заряженный заместитель или основной заместитель, соль может быть образована из аниона и положительно заряженного заместителя (например, аминогруппа) или основного заместителя (например, пиридила) на соединении, описанном в настоящем документе. Подходящие анионы включают хлорид, бромид, йодид, сульфат, нитрат, фосфат, цитрат, метансульфонат, трифторацетат и ацетат.
Фармацевтически приемлемые соли соединений, описанных в настоящем документе (например, соединение формулы (I)-(VII)), также включают соли, полученные из фармацевтически приемлемых неорганических и органических кислот и оснований. Примеры подходящих солей включают ацетат, адипат, альгинат, аспартат, бензоат, бензолсульфонат, бисульфат, бутират, цитрат, камфорат, камфорсульфонат, диглюконат, додецилсульфат, этансульфонат, формиат, фумарат, глюкогептаноат, гликолат, гемисульфат, гептаноат, гексаноат, гидрохлорид, гидробромид, гидройодид, 2-гидроксиэтансульфонат, лактат, малеат, малонат, метансульфонат, 2-нафталинсульфонат, никотинат, нитрат, пальмоат, пектинат, персульфат, 3-фенилпропионат, фосфат, пикрат, пивалат, пропионат, салицилат, сукцинат, сульфат, тартрат, тиоцианат, тозилат, трифторацетат и ундеканоат.
Соли, полученные из соответствующих оснований, включают соли щелочного металла (например, натрий), щелочно-земельного металла (например, магний), солей аммония и N-(алкил)4+. Настоящее изобретение также предусматривает кватернизацию любой основной азот-содержащей группы соединений, описанных в настоящем документе. Вода или масло-растворимые, или диспергируемые продукты могут быть получены с помощью такой кватернизации.
Как используется в настоящем документе соединение по настоящему изобретению, в том числе соединения формулы (I)-(VII), определены как включающие их фармацевтически приемлемые производные или пролекарства. "Фармацевтически приемлемое производное или пролекарство» означает любую фармацевтически приемлемую соль, сложный эфир, соль сложного эфира или другое производное соединение по настоящему изобретению, которое при введении реципиенту способно обеспечивать (напрямую или косвенно) соединение по настоящему изобретению. Особенно предпочтительными производными и пролекарствами являются производные и пролекарства, которые увеличивают биодоступность соединений по настоящему изобретению, при введении таких соединений млекопитающим (например, позволяя перорально вводимому соединению более легко абсорбироваться в кровь), или которые улучшают доставку исходного соединения в биологический компартмент (например, мозг или лимфатическая система) по сравнению с родительскими видами. Предпочтительные пролекарства включают производные, в которых группа, которая повышает растворимость в воде или активный транспорт через мембрану кишечника, присоединяется к структуре формул, как описано в настоящем документе.
Любая формула или соединение, описанное в настоящем документе, в данном случае также предназначена для включения не меченных форм, а также меченных изотопами форм соединений, изотопно-меченые соединения имеют структуры, изображенные с помощью формул, приведенных в настоящем документе, за исключением того, что один или несколько атомов заменены атомом, имеющим выбранное массовое число или массовый номер. Примеры изотопов, которые могут быть включены в соединения по изобретению, включают изотопы водорода, углерода, азота, кислорода, фосфора, фтора и хлора, такие как 2Н, 3Н, 11C, 13C, 14C, 15N, 18F 51P, 32P, 35S, 36Cl, 125I, соответственно. Изобретение включает различные меченные изотопами соединения, как определено в настоящем документе, например, соединения, в которых присутствуют радиоактивные изотопы, такие как 3Н, 13С и 14С. Такие меченные изотопами соединения, могут быть использованы в метаболических исследованиях (С14C), исследованиях кинетических реакций (в случае, например 'H или 3H), в способах визуализации или детекции, таких как позитронно-эмиссионная томография (PET) или однофотонная эмиссионная компьютерная томография (SPECT), включая лекарства или анализы распределения субстрата в тканях, или при радиоактивном лечении больных. В частности 18F или меченное соединение может быть особенно желательным для исследований PET или SPECT, соединения, меченные изотопами, по настоящему изобретению и их пролекарства могут,
как правило, быть получены путем проведения методик, описанных на схемах или в примерах, описанных ниже, путем замены реагентов, не меченных изотопами, на легко доступные соединения, меченные изотопами.
Дополнительно, замещение более тяжелыми изотопами, в частности дейтерием (то есть, 2H или D) может обеспечить определенные терапевтические преимущества, являющиеся результатом большей метаболической стабильности, например, увеличение периода полураспада in vivo или снижение дозировки, или улучшение терапевтического индекса. Понятно, что дейтерий в этом контексте рассматривается как заместитель соединения формулы, описанной в настоящем документе. Концентрация такого тяжелого изотопа, в частности, дейтерия, может быть определена с помощью изотопного коэффициента обогащения. Термин "изотопный фактор обогащения", как используется в настоящем документе, означает соотношение между изотопным составом и природным содержанием конкретного изотопа, если заместитель m соединения по настоящему изобретению обозначает дейтерий, то такое соединение имеет изотопный коэффициент обогащения для каждого указанного атома дейтерия по меньшей мере, 3500 (52,5% включения дейтерия в каждый указанный атом дейтерия), по меньшей мере 4000 (60% включения дейтерия), по меньшей мере 4500 (67,5% включения дейтерия), по меньшей мере 5000 (75% включения дейтерия), по меньшей мере 5500 (82,5% включения дейтерия), по меньшей мере 6000 (90% включения дейтерия), по меньшей мере 6333,3 (95% включения дейтерия), по меньшей мере 6466,7 (97% включения дейтерия), по меньшей мере 6600 (99% включения дейтерия) или по крайней мере 8633,3 (99,5% включения дейтерия).
Меченные изотопами соединения, описанные в настоящем документе, как правило, могут быть получены обычными способами, известными специалистам в данной области, или способами, аналогичными тем, которые описаны в сопроводительных разделах "Примеры" и "Лекарственные средства", используя соответствующие меченные изотопами реагенты вместо немеченых реагентов, использовавшихся ранее. Фармацевтически приемлемые сольваты в соответствии с изобретением, включают сольваты, в которых растворитель кристаллизации может быть изотопно замещенным, например, D2O, D2-ацетон, D2-DMSO.
Любой асимметричный атом (например, углерод или тому подобное) соединения (й) по настоящему изобретению может быть представлен в рацемической или энантиомерно обогащенной, например (R)-, (S)- или (RS)- конфигурации, в некоторых вариантах осуществления каждый асимметричный атом имеет по меньшей мере 50% энантиомерный избыток, по меньшей мере 60% энантиомерный избыток, по меньшей мере 70% энантиомерный избыток, по меньшей мере 80% энантиомерный избыток, по меньшей мере 90% энантиомерный избыток, по меньшей мере 95% энантиомерный избыток или по меньшей мере 99% энантиомерный избыток в (R)- или (S)- конфигурации. Заместители у атомов с ненасыщенными связями могут, если это возможно, присутствовать в цис- (Z)- или транс (Е)-форме.
Соответственно, как используется в настоящем документе, соединение по настоящему изобретению может быть в форме одного из возможных изомеров, ротамеров, атропоизомеров, таутомеров или их смесей, например, в виде по существу чистых геометрических (цис или транс) изомеров, диастереоизмоеров, оптических изомеров (антиподов), рацематов или смесей. Любые полученные смеси изомеров могут быть разделены на основе физико-химических различий составляющих, на чистые или по существу чистые геометрические или оптические изомеры, диастереоизомеры, рацематы, например, с помощью хроматографии или фракционной кристаллизации.
Любой полученный рецемат конечных продуктов или промежуточных соединений может быть разделен на оптические антиподы известными способами, например, путем разделения диастереомерных солей, полученных с оптически активной кислотой или основанием, и высвобождая оптически активное кислой или основное соединение. В частности, основная группа, таким образом, может быть использованы для разделения соединений по настоящему изобретению, на их оптические антиподы, например, путем фракционной кристаллизации соли, образованной с оптически активной кислотой, например, винная кислота, дибензоилвинная кислота, диацетилвинная кислота, (+)-O,O'-ди-п-толуол-D-винная кислота, миндальная кислота, яблочная кислота или камфора-10-сульфоновая кислота. Рацемические продукты также могут быть разделены с помощью хиральной хроматографии, например, высокоэффективная жидкостная хроматография (ВЭЖХ), используя хиральный адсорбент.
Соединения, описанные в настоящем документе (например, соединение формулы (I)-(VII)), также могут быть представлены в нескольких таутомерных формах, например, соединение формулы (I), (II), (III), (IV), (V), (VI) или (VII). В таких случаях, изобретение специально включает все таутомерные формы соединений, описанных в настоящем документе. Все кристаллические формы соединений, описанных в настоящем документе, непосредственно включены в настоящее изобретение.
Соединение, описанное в настоящем документе, (например, соединение формулы (I)-(VII)) можно оценить по его способности модулировать (например, ингибировать) Фактор XIa или калликреин.
Способы синтеза соединений
Соединения, описанные в настоящем документе, могут быть синтезированы обычными способами, используя коммерчески доступные исходные вещества и реагенты. Например, соединения могут быть синтезированы с использованием способов, указанных в патенте США 7501404 или, как описано в способах, описанных в настоящем документе.
Способы лечения
Соединения, описанные в настоящем документе (например, соединения формулы (I)-(VII)), могут ингибировать активность Фактора XIa или калликреина. В некоторых вариантах осуществления соединения, описанное в настоящем документе, может ингибировать как Фактор XIa, так и калликреин. В результате, эти соединения могут использоваться для лечения или профилактики расстройства, описанного в настоящем документе. Примеры нарушений включают тромботические нарушения, связанные с коронарной артерией и цереброваскулярными заболеваниями, тромбоз вен или артерий, нарушение коагуляции, ишемию и стенокардию (стабильная и нестабильная), тромбоз глубоких вен (DVT), диссеминированное внутрисосудистое свертывание, синдром Казабах-Мерритта, легочную эмболию, инфаркт миокарда, инфаркт мозга, тромбоз сосудов головного мозга, транзиторные ишемические атаки, фибрилляцию предсердий, церебральную эмболию, тромбоэмболические послеоперационные осложнения (например, замена тазобедренного сустава или колена, имплантация искусственных клапанов сердца и эндартерэктомия) и окклюзию периферических артерий, а также они могут использоваться для лечения или профилактики инфаркта миокарда, инсульта, стенокардии и других последствий разрыва атеросклеротической бляшки. Соединения по изобретению, обладающие ингибиторной активностью в отношении Фактора XIa или калликреина, могут также использоваться для профилактики тромбоза у больных раком, а также для профилактики тромбоэмболических нарушений или последующего восстановления проходимости сосудов с помощью тканевого активатора плазминогена или механического восстановления. Соединения по изобретению, обладающие ингибиторной активностью в отношении Фактора XIa или калликреина, могут также использоваться в качестве ингибиторов свертывания крови, например, во время получения, хранения и фракционирования цельной крови.
Ингибирование фактора XIa, в соответствии с настоящим изобретением, может быть более эффективным и безопасным способом ингибирования тромбоза по сравнению с ингибированием других сериновых протеаз свертывания, таких как тромбин или Фактор Ха. Введение низкомолекулярного ингибитора Фактора XIa должно быть эффективным для ингибирования синтеза тромбина и образования тромбов, не влияя, или практически не влияя, на время кровотечения и не нарушая, или незначительно нарушая, гемостаз. Эти результаты существенно отличаются от других ингибиторов протеаз свертывания крови "прямого действия" (например, ингибиторы активного сайта тромбина и Фактора Ха), которые увеличивают время кровотечения и их антитромботический эффект практически не отличается от эффекта удлинения времени кровотечения. Предпочтительным способом по изобретению включает введение млекопитающему фармацевтической композиции, содержащей по меньшей мере одно соединение по настоящему изобретению.
Соединения, описанные в настоящем документе (например, любое соединение формулы (I)-(VII)), могут ингибировать калликреин, например, соединения формулы (I), (II), (III), (IV), (V), (VI) или (VII). В результате, эти соединения могут использоваться для лечения или профилактики заболеваний, связанных с воспалением, таких как отек (например, отек головного мозга, отек желтого пятна и ангионевротический отек (например, наследственный ангионевротический отек)). В некоторых вариантах осуществления соединения по настоящему изобретению могут быть эффективны для лечения или профилактики наследственного ангионевротического отека. Соединения, описанные в настоящем документе (например, соединения формулы (I)-(VII)) также могут быть эффективны для лечения, например, инсульта, ишемии и периоперационной кровопотери, например, соединение формулы (I), (II), (III), (IV), (V), (VI) или (VII).
Способы по настоящему изобретению могут использоваться для лечения или профилактики таких состояний, которые связаны с активностью Фактора XIa или калликреина. Соответственно, способы по настоящему изобретению могут быть использованы для лечения последствий разрыва атеросклеротической бляшки, в том числе сердечно-сосудистых заболеваний, связанных с активацией каскада свертывания, при тромботических или тромбофилических состояниях. Как используются в настоящем документе термины "лечение" или "лечить", включают ответные или профилактики мероприятия, например, мероприятия, направленные на ингибирование или задержку развития заболевания, на полное или частичное снижение симптомов, или болезненного состояния, или на облегчение, уменьшение или излечение заболевания, или нарушения его симптомов.
Более конкретно, способы по настоящему изобретению могут быть использованы для лечения острого коронарного синдрома, такого как коронарная болезнь сердца, инфаркт миокарда, нестабильная стенокардия (в том числе стенокардия "крещендо"), ишемия (например, ишемия в результате окклюзии сосудов) и инфаркт головного мозга. Способы по настоящему изобретению могут быть также быть эффективны для лечения инсульта и связанных с ним заболеваний сосудов головного мозга (в том числе нарушении мозгового кровообращения, сосудистая деменция и транзиторная ишемическая атака); тромбоз вен и тромбоэмболия, например, тромбоз глубоких вен (DVT) и эмболия легочной артерии; тромбоз, связанный с фибрилляцией предсердий, гипертрофия желудочков, дилатационная сердечная миопатия или сердечная недостаточность; заболевание периферических артерий и перемежающаяся хромота; образование атеросклеротических бляшек и атеросклероз трансплантата; рестеноз после эндогенного повреждения (разрыв атеросклеротической бляшки) или экзогенного повреждения артерий (при инвазивных кардиологических процедурах, например, травмы стенок сосудов в результате ангиопластики); ДВС-синдром, синдром Казабаха-Мерритта, тромбоз сосудов головного мозга и эмболия сосудов головного мозга.
Кроме того, способы по настоящему изобретению могут использоваться для лечения тромбоэмболических последствий или осложнений, связанных с раком; с хирургическим вмешательством (например, замена тазобедренного сустава, эндартерэктомия, имплантация искусственных клапанов сердца, трансплантатов сосудов, механических органов и имплантация или трансплантация органов, тканей или клеток); с получением лекарственных средств (таких как активатор тканевого плазминогена или аналогичные средства и хирургическое восстановление проходимости кровеносных сосудов) у больных с инфарктом миокарда, инсультом, легочной эмболией и тому подобными состояниями; с получением лекарственных средств (например, пероральные контрацептивы, заместительная гормональная терапия и введение гепарина, например, для лечения гепарин-индуцированной тромбоцитопении); с сепсисом (например, сепсис, связанный с диссеминированным внутрисосудистым свертыванием); и с беременностью или родами. Способы по настоящему изобретению могут использоваться для лечения тромбоза при ограничении подвижности (то есть при иммобилизации, госпитализации, при постельном режиме, иммобилизации конечности, например, иммобилизирующими повязками и тому подобное).
Способы по настоящему изобретению также могут использоваться для поддержания проходимости кровеносных сосудов, например, у пациентов, перенесших коронарную ангиопластику, или в связи с хирургией сосудов, такой как шунтирование, реконструкция артерий, атеректомия, сосудистые трансплантаты, стенты и имплантация и трансплантация органов, тканей или клеток. Предлагаемые способы могут использоваться для ингибирования свертывания крови в связи с получением, хранением, фракционированием или использованием цельной крови. Например, способы по изобретению могут использоваться для поддержания цельной и фракционированной крови в жидкой фазе, как, например, требуется для аналитических и биологических исследований, например, для ex vivo исследований функции тромбоцитов и других клеток, биоаналитических процедур и количественных компонентов крови или для поддержания экстракорпорального кровообращения, как, например, при диализе или хирургическом вмешательстве (например, аортокоронарное шунтирование).
Кроме того, способы по настоящему изобретению могут использоваться для лечения и профилактики протромботическим осложнений рака. Эти способы могут использоваться для лечения роста опухоли, в качестве дополнения к химиотерапии, для профилактики ангиогенеза, а также для лечения рака, в частности, рака легких, простаты, толстой кишки, молочной железы, яичников и кости.
Ишемия
"Ишемия" или "ишемическое нарушение" является сосудистым заболеванием, обычно связанным с окклюзией сосудов или ограниченным кровоснабжением тканей. Ишемия может привести к кислородной недостаточности и недостаточности глюкозы, необходимых для клеточного метаболизма. Ишемию, как правило, вызывают нарушения в кровеносных сосудах, что приводит к повреждению или дисфункции ткани. Ишемия может также к относятся локальной потери крови или кислорода в той или иной части организма в результате застоя крови (например, при сужении сосуда, тромбозе или эмболии). Причины включают эмболию, тромбоз атеросклерозированных артерий, травма, венозные нарушения, аневризму, заболевания сердца (например, инфаркт миокарда, порок митрального клапана, хроническую артериальную фибрилляцию, кардиомиопатию и протезирование), травму или травматическое повреждение (например, в конечности с частичной или полной окклюзией сосуда), синдром грудного выхода, атеросклероз, гипогликемию, тахикардию, гипотензию, наружное сдавление кровеносных сосудов (например, опухолью), серповидно-клеточную анемию, локальное воздействие предельно низкой температуры (например, при обморожении), наложение кровеостанавливающего жгута , стимуляцию рецептора глутамата, артериовенозные пороки развития, разрыв значимых кровеносных сосудов, снабжающих ткани или органы, и анемию.
Транзиторная ишемическая атака, как правило относится к временному (например, кратковременному) эпизоду неврологической дисфункции, вызванной потерей кровотока (например, фокальные нарушения головного мозга, спинного мозга или сетчатки) без острого инфаркта (например, смерти ткани). В некоторых вариантах осуществления транзиторная ишемическая атака длится в течение менее чем 72 часов, 48 часов, 24 часов, 12 часов, 10 часов, 8 часов, 4 часа, 2 часа, 1 час, 45 минут, 30 минут, 20 минут, 15 минут, 10 минут, 5 минут, 4 минут, 3 минут, 2 минут или 1 минуты.
Ангионевротический отек
Ангионевротический отек является быстро распространяющимся отеком дермы, подкожной клетчатки, слизистой оболочки и подслизистых тканей. Ангионевротический отек обычно классифицируются как приобретенный или наследственный.
"Приобретенный ангионевротический отек" может быть иммунологическим, не иммунологическим или идиопатическим; вызванным, например, аллергией, как побочный эффект лекарственных средств, например, ингибитора ACE.
"Наследственный ангионевротический отек" или "HAE" относится к генетическим нарушениям, которые приводят к острым периодам отека (например, набухание), которые могут возникать почти во всех частях организма, в том числе на лице, конечностях, шее, горле, гортани, кистях и стопах, желудочно-кишечном тракте и половых органах. Атаки HAE часто могут быть опасными для жизни, тяжесть которых зависит от площади поражения, например, брюшные атаки могут привести к кишечной непроходимости, а отек гортани и верхних дыхательных путей может привести к асфиксии. Патогенез наследственного ангионевротического отека может быть связан с беспрепятственной активацией контактного пути посредством первоначального образования калликреина или факторов свертывания крови (например, Фактор XII).
Признаки и симптомы включают отек, например, лица, слизистой оболочки полости рта или горла, и языка. Зуд, боль, снижение чувствительности в пораженных областях, крапивница или стридор дыхательных путей также может быть признаком ангионевротического отека. Тем не менее, может и не быть зуда, или крапивницы, например, при наследственном ангионевротическом отеке. Больные HAE могут испытывать боль в животе (например, боль в животе продолжительностью от одного до пяти дней, брюшные атаки, увеличивающие число белых кровяных клеток у индивида), рвоту, слабость, водянистый понос или сыпь.
Брадикинин играет важную роль в ангионевротическом отеке, в частности, при наследственном ангионевротическом отеке. Брадикинин высвобождается различными типами клеток в ответ на многочисленные различные стимулы, и является медиатором боли. Вмешательство в продукцию или деградацию брадикинина может привести к ангионевротическому отеку. При наследственном ангионевротическом отеке непрерывная продукция фермента калликреина может способствовать образованию брадикинина. Ингибирование калликреина может влиять на продукцию брадикинина; и лечить или предотвращать ангионевротический отек.
Фармацевтические композиции
Композиции, описанные в настоящем документе, включают соединение, описанное в настоящем документе (например, соединение формулы (I)-(VII), например, соединение формулы (I), (II), (III), (IV), (V), (VI) или (VII)), а также дополнительные терапевтические средства, если они присутствуют, в количествах, эффективных для достижения лечения заболевания или симптомов заболевания (например, таких как заболевания, связанные с фактором XIa или калликреином).
Фармацевтически приемлемые носители, адъюванты и переносчики, которые могут быть использованы в фармацевтической композиции по настоящему изобретению, включают, но ими не ограничиваются, ионнообменники, глинозем, алюминий стеарат, лецитин, самоэмульгирующие системы доставки лекарственных средств (SEDDS), такие как d-α-токоферол полиэтиленгликоль 1000 сукцинат, поверхностно-активные вещества, используемые в лекарственных формах, такие как Tween или другие аналогичные полимерные матрицы для доставки, белки сыворотки крови, такие как сывороточный альбумин человека, буферные вещества, такие как фосфаты, глицин, сорбиновая кислота, сорбат калия, частичные смеси глицеридов, насыщенных растительных жирных кислот, вода, соли или электролиты, такие как протамин сульфат, динатрия гидрофосфат, калия гидрофосфат, хлорид натрия, соли цинка, коллоидный кремнезем, магния трисиликат, поливинил пирролидон, вещества на основе целлюлозы, полиэтиленгиколь, натрия карбоксиметилцеллюлоза, полиакрилаты, воски, полиэтилен-полиоксипропилен-блок полимеры, полиэтиленгликоль и жир шерсти. Циклодекстрины, такие как α-, β-, а также γ-циклодекстрин или химически модифицированные производные, такие как гидроксиалкилциклодекстрины, в том числе 2- и 3-гидроксипропил-β-циклодекстрины или другие растворенные производные, также могут быть эффективно использованы для повышения доставки соединений формул, описанных в настоящем документе.
Пути введения
Фармацевтические композиции, предлагаемые в настоящем документе, могут быть введены перорально, ректально или парентерально (например, внутривенная инфузия, внутривенная болюсная инъекция, ингаляцией, имплантацией). Термин парентеральный, как используется в настоящем документе, включает подкожный, внутрикожный, внутривенный (например, внутривенная инфузия, внутривенная болюсная инъекция), интраназальный, ингаляционный, пульмонарное, чрескожное, внутримышечное, внутрисуставное, внутриартериальное, интрастернальное, интратекальное, вводимый внутрь пораженных тканей и внутричерепную инъекцию или другие инфузионные методы. Фармацевтические композиции по настоящему изобретению могут содержать любые обычные нетоксичные фармацевтически приемлемые носители, адъюванты или переносчики. В некоторых случаях рН состава можно регулировать с фармацевтически приемлемыми кислотами, основаниями или буферами для повышения стабильности соединения, включенного в состав, или его формы доставки.
Фармацевтические композиции могут быть в форме стерильного инъекционного препарата, например, в виде стерильного водного или масляного раствора или суспензии, вводимой путем инъекции. Эта суспензия может быть получена в соответствии с методами, известными в данной области, используя подходящие диспергирующие или смачивающие средства (такие как, например, Tween 80) и суспендирующие средства. Стерильный препарат для инъекции также может представлять собой стерильный раствор или суспензию для инъекции в нетоксичном приемлемом для парентерального применения разбавителе или растворителе, например, в виде раствора в 1,3-бутандиоле. В числе приемлемых носителей и растворителей, которые могут быть использованы, находятся маннит, вода, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, стерильные нелетучие масла обычно используют в качестве растворителя или суспендирующей среды. С этой целью может быть использовано любое нелетучее масло, в том числе синтетические моно- или диглицериды. Жирные кислоты, такие как олеиновая кислота и производные глицеридов, являются эффективными для получения инъецируемых препаратов, как природные фармацевтически приемлемые масла, такие как оливковое масло или касторовое масло, особенно в полиоксиэтилиэтилированных вариантах. Эти масляные растворы или суспензии также могут содержать разбавитель или диспергатор длинноцепочечный спирт или карбоксиметилцеллюлозу или аналогичные диспергирующие средства, которые обычно используют в составе фармацевтически приемлемых лекарственных форм, например, эмульсии или суспензии. Другие, обычно используемые поверхностно-активные вещества, такие как Tweens или Spans, или другие аналогичные эмульгаторные агенты или усилители биодоступности, которые обычно используют для получения фармацевтически приемлемых твердых, жидких или других лекарственных форм, также могут быть использованы для целей получения состава.
Фармацевтические композиции по настоящему изобретению можно вводить перорально в любой перорально приемлемой лекарственной форме, включая, но ими не ограничиваясь, капсулы, таблетки, эмульсии и водные суспензии, дисперсии и растворы. В случае таблеток для перорального применения, носители, которые обычно используют, включают лактозу и кукурузный крахмал. Также, обычно добавляют смазывающие агенты, такие как стеарат магния. Для перорального введения в форме капсул, эффективные разбавители включают лактозу и высушенный кукурузный крахмал. Если водные суспензии или эмульсии вводят перорально, то активный ингредиент может быть суспендирован или растворен в масляной фазе, в сочетании с эмульгаторами или суспендирующими агентами. При желании могут быть добавлены некоторые подсластители или ароматизаторы, или красители, или агенты, корригирующие вкус и запах.
Соединения, описанные в настоящем документе, могут, например, быть введены путем инъекции, внутривенно (например, внутривенное введение, внутривенная болюсная инъекция), внутриартериально, внутрикожно, внутрибрюшинно, внутримышечно или подкожно; или перорально, трансбуккально, назально, через слизистую оболочку, местно, с дозировкой в диапазоне от приблизительно от 0,5, до приблизительно 100 мг/кг массы тела, в качестве альтернативы, в дозировке от 1 мг до 1000 мг/доза, в течение каждых 4-120 часов, или в соответствии с требованиями конкретного лекарственного препарата. В настоящем документе способы введения эффективного количества соединения или композиции соединения для достижения желаемого или заявленного эффекта. Обычно, фармацевтические композиции по настоящему изобретению вводят примерно от 1, до примерно 6 раз в день (например, путем внутривенной болюсной инъекции) или в качестве альтернативы, в виде непрерывной инфузии. Такое введение может быть использовано в качестве хронической или острой терапии. Количество активного ингредиента, которое можно комбинировать с веществами-носителями для получения разовой лекарственной формы, может изменяться в зависимости от получающего лечение индивида и конкретного способа введения. Характерный препарат может содержать от, примерно 5% до, примерно 95% активного соединения (масс./масс.). В качестве альтернативы, такие препараты содержат от, примерно 20% до, примерно 80% активного соединения.
Комбинации
При осуществлении способов по настоящему изобретению, может быть желательно вводить соединения по настоящему изобретению (фактор XIa или ингибиторы калликреина) в сочетании друг с другом, и с одним или несколькими другими средствами для достижения терапевтического эффекта, такими как антитромботическими или антикоагулянтыми средствами, антигипертензивными средствами, противоишемическими средствами и антиаритмическими средствами, ингибиторами функции тромбоцитов, и тому подобное. Например, способы по настоящему изобретению могут быть осуществлены путем введения низкомолекулярных ингибиторов фактора XIa или калликреина в сочетании с низкомолекулярным ингибитором фактора XIa или калликреина. Более конкретно, способы по настоящему изобретению могут быть осуществлены путем введения низкомолекулярных ингибиторов Фактор XIa или калликреина в сочетании с аспирином, клопидогрелем, тиклопидином или CS-747, варфарином, низкомолекулярными гепаринами (такими как LOVENOX), блокаторами GPIIb/GPIIIa, ингибиторами PAI-1, такими как XR-330 и T-686, антагонистами рецепторов P2Y1 и P2Y12; антагонистами рецептора тромбоксана (например, ифетробан), миметиками простациклина, ингибиторами тромбоксан A синтетазы (например, пикотамид), антагонистами рецептора серотонина-2 (например, кетансерин); соединениями, которые ингибируют другие факторы свертывания крови, такие как FVII, FVIII, FIX, FX, протромбин, TAFI и фибриноген или другие соединения, которые ингибируют FXI или калликреин; фибринолитиками, такими как TPA, стрептокиназа, ингибиторы PAI-1, и ингибиторами α-2-антиплазмина, такими как антитело против α-2-антиплазмина, антагонистами рецептора фибриногена, ингибиторами α-1-антитрипсина, гиполипидемическими средствами, такими как ингибиторы HMG-CoА-редуктазы (например, правастатин, симвастатин, аторвастатин, флувастатин, церивастатин, AZ4522 и итавастатин) и ингибиторами микросомального белка транспорта триглицерида (такие как раскрыты в патенте США 5739135, 5712279 и 5760246); гипотензивными средствами, такими как ингибиторы ангиотензин-превращающего фермента (например, каптоприл, лизиноприл или фозиноприл); антагонистами рецептора ангиотензина-II (например, ирбесартан, лозартан или валсартан); ингибиторами ACE/NEP (например, омапатрилат и гемопатрилат); или β-блокаторами (такими как пропранолол, надолол и карведилол). Способы по изобретению могут быть выполнены путем введения ингибиторов низкомолекулярного фактора XIa или калликреина в комбинации с антиаритмическими средствами, такими как средства при фибрилляции предсердий, например, амиодарон или дофетилид.
При осуществлении способов по настоящему изобретению, может быть желательно введение соединений по настоящему изобретению (ингибиторы фактора XIa или калликреина) в комбинации со средствами, которые повышают уровни сAMP или cGMP в клетках для терапевтического эффекта. Например, соединения по настоящему изобретению, могут иметь нежелательные эффекты при использовании в комбинации с ингибиторами фосфодиэстеразы, в том числе ингибиторами PDE1 (такими как описаны в журнале Medicinal Chemistry, Vol. 40, стр. 2196-2210 [1997]), ингибиторами PDE2, ингибиторами PDE3 (такими как ревизинон, пимобендан или олпринон), ингибиторами PDE 4 (такими как ролипрам, циломиласт или пикламиласт), ингибиторами PDE7 или другими ингибиторами PDE, такими как дипиридамол, цилостазол, силденафил, денбутилин, теофиллин (1,2-диметилксантин), арофилин (то есть, цис-4-циано-4-[3-(циклопентоксилокси)-4-метоксифенил]циклогексан-1-карбоновая кислота), арофилин, рофлумиласт, C-11294A, CDC-801, BAY-19 -8004, ципамфиллин, SCH351591, YM-976, PD-189659, мезиопрам, пумафентрин, CDC-998, IC-485 и KW-4490.
Способы по изобретению могут быть осуществлены путем введения соединения по настоящему изобретению в комбинации с протромболитическими средствами, такими как тканевый активатор плазминогена (природный или рекомбинантный), стрептокиназа, ретеплаза, активаза, ланотеплаза, урокиназа, проурокиназа, изолированный активатор комплекса стрептокиназа-плазминоген (ASPAC), активаторы празминогена слюнной железы животных и тому подобное.
Способы по изобретению могут быть осуществлены путем введения соединений по изобретению в комбинации с бета-адренергическими агонистами, такими как альбутерол, тербуталин, формотерол, сальметерол, битолтерол, пилбутерол или фенотерол; антихолинергическими средстами, такими как ипратропия бромид; противовоспалительными кортикостероидами, такими как беклометазон, триамцинолон, будесонид, флутиказон, флунизолид или дексаметазон; и противовоспалительными средствами, такими как кромолин, недокромил, теофиллин, зилейтон, зафирлукаст, монтелукаст и пранлукаст.
Низкомолекулярные ингибиторы фактора XIa или калликреина могут действовать синергически с одним или несколькими из вышеуказанных средств. Таким образом, можно использовать пониженные дозы тромболитических(ого) средств(а), таким образом получения преимущества от введения таких соединений при сведении к минимуму возможных геморрагических и других побочных эффектов.
Курс лечения
Композиции, описанные в настоящем изобретении, включают терапевтически эффективное количество соединения по настоящему изобретению (например, ингибитор Фактора XIa или калликреина) в комбинации с одним или несколькими другими средствами (например, дополнительное терапевтическое средство), такими как антитромботические или антикоагулянтые средства, антигипертензивные средства, анти-ишемические средства, антиаритмические средства, ингибиторы функции тромбоцитов и тому подобное для достижения терапевтического эффекта.
В некоторых вариантах осуществления дополнительное лекарственное средство вводят после введения соединения по настоящему изобретению (например, ингибитор Фактора XIa или калликреина). В некоторых вариантах осуществления дополнительное терапевтическое средство вводят через 15 минут, 30 минут, 1 час, 2 часа, 4 часа, 6 часов, 8 часов, 10 часов, 12 часов, 14 часов, 18 часов, 24 часа, 48 часов, 72 часов или более, после введения соединения по настоящему изобретению (например, ингибитор Фактора XIa или калликреина). В некоторых вариантах осуществления дополнительное терапевтическое средство вводят (например, перорально) после выписки из медицинского учреждения (например, из больницы).
В некоторых вариантах осуществления соединение по изобретению (например, ингибитор Фактора XIa или калликреина) и дополнительное терапевтическое средство введены вместе в единую композицию или лекарственную форму. В некоторых вариантах осуществления соединение по изобретению (например, ингибитор Фактора XIa или калликреина) и дополнительное терапевтическое средство вводят раздельно. В некоторых вариантах осуществления соединение по изобретению (например, ингибитор Фактора XIa или калликреина) и дополнительное терапевтическое средство вводят последовательно. В некоторых вариантах осуществления соединение по изобретению (например, ингибитор Фактора XIa или калликреина) и дополнительное терапевтическое средство вводят раздельно последовательно. В целом, по меньшей мере, одно соединение по настоящему изобретению (например, ингибитор Фактора XIa или калликреина) и дополнительное терапевтическое средство вводят парентерально (например, интраназально, внутримышечно, буккально, путем ингаляции, имплантации, трансдермально, внутривенно (например, внутривенной инфузией, внутривенной болюсной инъекцией), подкожно, внутрикожно, интраназально, пульмонарно, чрескожно, внутрисуставно, внутриартериально, интрасиновиально, интрастернально, интратекально, внутрь пораженной ткани и внутричерепной инъекцией или другими способами инфузии); перорально; или ректально, например, внутримышечной инъекцией или внутривенно (например, внутривенной инфузией, внутривенной болюсной инъекцией)). В некоторых вариантах осуществления соединение по настоящему изобретению вводят парентерально (например, интраназально, буккально, внутривенно (например, внутривенная инфузия, внутривенная болюсная инъекция) или внутримышечно). В некоторых вариантах осуществления дополнительное лекарственное средство вводят перорально. В некоторых вариантах осуществления соединение по изобретению (например, ингибитор Фактора XIa или калликреина) вводят парентерально (например, интраназально, буккально, внутривенно (например, внутривенная инфузия, внутривенная болюсная инъекция) или внутримышечно) и дополнительное терапевтическое средство вводят перорально.
В некоторых вариантах осуществления соединение по изобретению (например, ингибитор Фактора XIa или калликреина) можно вводить один раз или несколько раз в день. Продолжительность лечения может составлять, например, один раз в день в течение периода приблизительно 1, 2, 3, 4, 5, 6, 7 и более дней. В некоторых вариантах осуществления лечение является длительным (например, всю жизнь). В некоторых вариантах осуществления лечение вводят перорально. В некоторых вариантах осуществления вводят либо разовую дозу в виде единичной дозы или нескольких более мелких доз, либо путем множественного введения разделенных доз через определенные промежутки времени. Например, лекарственная форма может быть введена от приблизительно 0 часов до примерно 1 часа, приблизительно от 1 часа до приблизительно 24 часов, приблизительно от 1 часа до приблизительно 72 часов, приблизительно от 1 часа до приблизительно 120 часов или от приблизительно 24 часов до по меньшей мере приблизительно 120 часов после травмы. В качестве альтернативы лекарственная форма может быть введена через примерно 0,5, 1, 1,5, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 30, 40, 48, 72, 96, 120 часов или более после травмы. Последующие лекарственные формы можно вводить в любое время после первоначального введения, так, чтобы достигался терапевтический эффект. В некоторых вариантах осуществления начальная доза вводится перорально. В некоторых вариантах осуществления дозы после первоначальной дозы вводят парентерально (например, интраназально, внутримышечно, буккально, путем ингаляции, имплантации, трансдермально, внутривенно (например, внутривенной инфузией, внутривенная болюсная инъекция), подкожно, внутрикожно, интраназально, пульмонарно, чрескожно, внутрисуставно, внутриартериально, интрасинавиально, интрастернально, интратекально, внутрь пораженных тканей и внутричерепной инъекцией или другими способами инфузии); перорально; или ректально.
В случае, когда у индивида, получающего лечение, имеется частичный ответ, или риск развития рецидива после завершения первого цикла лечения, последующие курсы терапии могут быть необходимы для достижения частичного или полного терапевтического ответа (например, длительное лечение, например, всю жизнь).
В некоторых вариантах осуществления соединение по изобретению (например, ингибитор Фактора XIa или калликреина) вводят внутривенно, например, в виде внутривенной инфузии или внутривенной болюсной инъекции в течение от приблизительно 5 минут до приблизительно 1 недели; от приблизительно 30 минут до приблизительно 24 часов, от приблизительно 1 часа до приблизительно 12 часов, от приблизительно 2 часов до приблизительно 12 часов, от приблизительно 4 часов до приблизительно 12 часов, от приблизительно 6 часов до приблизительно 12 часов, от приблизительно 6 часов до приблизительно 10 часов; от приблизительно 5 минут до приблизительно 1 часа, от приблизительно 5 минут до приблизительно 30 минут; от приблизительно 12 часов до приблизительно 1 недели, от приблизительно 24 часов до приблизительно 1 недели, от приблизительно 2 дней до приблизительно 5 дней или от приблизительно 3 дней до приблизительно 5 дней. В одном из вариантов осуществления соединение по настоящему изобретению (например, ингибитор Фактора XIa или калликреина) вводят в виде внутривенной инфузии в течение приблизительно 5, 10, 15, 30, 45 или 60 минут, или более; приблизительно 1, 2, 4, 6, 8, 10, 12, 16 или 24 часа, или более; приблизительно 1, 2, 3, 4, 5, 6, 7, 8, 9 или 10 дней, или более.
Дозы и режимы дозирования
Эффективное количество низкомолекулярного Фактора XIa или калликреина, вводимого в соответствии с настоящим изобретением, может быть определено средним специалистом в данной области. Конкретный уровень дозы и частота дозировки для любого конкретного индивида может изменяться и будет зависеть от множества факторов, в том числе от активности конкретного используемого соединения, метаболической стабильности и продолжительности действия этого соединения, вида, возраста, массы тела, общего состояния здоровья, пола и режима питания индивида, пути и времени введения, скорости экскреции, комбинации лекарственного средства и тяжести конкретного состояния.
При улучшении состояния пациента можно вводить, при необходимости, поддерживающую дозу соединения, композиции или комбинации по изобретению. Соответственно, доза или частота введения, или то и другое, могут быть уменьшены, в зависимости от симптомов, до уровня, при котором сохраняется улучшение состояния, когда симптомы облегчены до желаемого уровня. Однако, пациентам может требоваться периодическое лечение на длительной основе при любом рецидиве симптомов заболевания.
ПРИМЕРЫ
Соединения по настоящему изобретению получены как описано на Таблицах, Схемах, Получении промежуточных соединений и Примерах ниже, или получены способами, аналогичными этим способам, которые хорошо известны и доступны специалистам в области органического синтеза.
Синтез соединений по изобретению:
Температура дана в градусах Цельсия (°C). Если не указано иного, то действия проводили при комнатной температуре или температуре окружающей среды, то есть, при температуре от 18 до 25°C в инертной атмосфере с удалением влаги. Хроматография означает флэш-хроматографию на силикагеле, как описано в Still, W.C, Kahn, M.; Mitra, A. J. Org. Chem. 1978, 43, 2923.; тонкослойную хроматографию (ТСХ) проводили на силикагелевых планшетах. Данные ЯМР даны в частях на миллион (ppm) относительно лок-сигнала дейтерия, используемого дейтерированного растворителя. Используются обычные сокращения для формы сигнала. Для масс-спектров (MS), для молекул указывается самая низкая масса основного иона, где изотопное расщепление дает в результате несколько масс-спектральных пиков. Композиции смесей растворителя даны в процентах объема или объемных соотношениях. В тех случаях, когда спектры ЯМР являются комплексными, указываются только диагностические сигналы.
Аналитическая ВЭЖХ
Способ A: Agilent 1100 ВЭЖХ, Zorbax Eclipse XDB-C18 50×4,6 мм колонка, 1,5 мл/мин, Растворитель A - Вода (0,1% TFA), Растворитель B - Ацетонитрил (0,07% TFA), Градиент - 5 мин от 95%A до 90%B; 1 мин удержания; затем повторение цикла (до 95% A в течение 1 мин), УФ детекция @ 214 и 254 нм.
Способ B: Agilent 1100 ВЭЖХ, Zorbax Eclipse XDB-C18 50×4,6 мм колонка, 1,5 мл/мин, Растворитель A - Вода (0,1% TFA), Растворитель B - Ацетонитрил (0,07% TFA), Градиент - 5 мин от 95%A до 90%B; 1 мин удержание; затем повторение цикла (до 95% A в течение 1 мин), УФ детекция @ 210 и 254 нм.
Способ C: Agilent 1100 ВЭЖХ, Zorbax Eclipse XDB-C18 50×4,6 мм колонка, 1,5 мл/мин, Растворитель A - Вода (0,1% TFA), Растворитель B - Ацетонитрил (0,07% TFA), Градиент - 6 мин от 95%A до 95%B; 1 мин удержание; затем повторение цикла (до 95% A в течение 1 мин), УФ детекция @ 210 и 254 нм.
Термины и аббревиатура:
ACN = ацетонитрил,
BOC = Boc = трет-бутоксикарбонил,
ушир. = уширенный,
t-BuOH = трет-бутиловый спирт,
кат. = каталитический,
конц. = концентрированный,
д = дублет,
дд = дублет дублетов,
ддд = дублет дублета из дублетов,
дт = дублет триплетов,
DCM = дихлорметан,
Перйодинан Десс-Мартина = 1,1,1-трис(ацетилокси)1,1-дигидро-1,2-бензйодоксол-3(1H)-он,
DIAD = диизопропил азодикарбоксилат,
ДМФ = N,N-диметилформамид,
DMSO = диметил сульфоксид,
Et2O = диэтиловый эфир,
Et3N = триэтиламин,
EtOAc = этилацетат,
EtOH = этиловый спирт,
экв. = эквивалент(ы),
ч = час(ы),
H2O = вода,
HCl = хлористоводородная кислота
ВЭЖХ = высокоэффективная жидкостная хроматография,
HOAc = уксусная кислота,
IPA = изопропиловый спирт,
ISCO = силикагелевый картридж нормальной фазы, поставляемый Teledyne ISCO,
K2CO3 = карбонат калия,
LiBH4 = тетрагидроборат лития,
LiBr = бромид лития,
LiCl = хлорид лития,
LAH = тетрагидроалюминат лития,
м = мультиплет,
мин = минута(ы)
MgCl2 = хлорид магния
MeOH = метанол,
MsCl = метансульфонил хлорид,
MTBE = метил трет-бутиловый эфир,
NaHCO3 = бикарбонат натрия,
Na2SO4 = сульфат натрия,
NH4OH = гидроксид аммония,
NH4OAc = ацетат аммония,
NH4Cl = хлорид аммония,
ЯМР = ядерно-магнитный резонанс,
NMP = N-метилпирролидинон,
Pd-C = палладий на активированном угле,
п = пентет,
PMB = п-метоксибензил,
PMBCl = п-метоксибензил хлорид,
удерж. = удержание
к.т. = комнатная температура,
с = синглет,
насыщ. = насыщенный,
т = триплет,
TFA = трифторуксусная кислота,
TBDPS = трет-бутилдифенилсилил,
TBS = трет-бутилдиметилсилил,
THF = тетрагидрофуран,
THP = тетрагидропиран,
ТСХ = тонкослойная хроматография
Пример 1. Общие схемы синтеза и способы
В целом, в случаях, когда соединение по настоящему изобретению содержат основной заместитель (например, аминогруппу, пиридил), кислый заместитель (например, карбоновая кислота) или представляет собой цвиттерион (т.е. содержит как основной заместитель, так и кислотный заместитель), для облегчения выделения и обработки, предпочтительно, выделить соединение в виде соли. Эта форма соли облегчает характеристику и непосредственное использование в биологических анализах.
Схема 1. Синтез соединений со структурой A-8 и А-9, как показано на Диаграмме А.
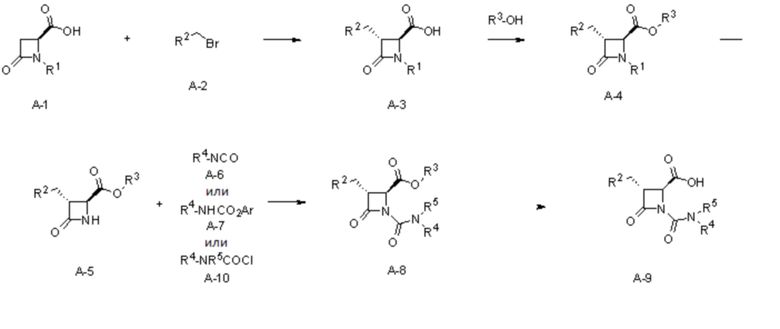
На Схеме 1 описан общий способ получения (2S, 3р) -транс-дизамещенной 4-оксоазетидин-2-карбоновой кислоты с общей структурой А-8 и А-9. Алкилирование дианиона N-защищенного азетидинона А-1 (например, R1 = TBS), с помощью коммерчески доступного или легко получаемого алкил галогенида с общей структурой А-2, приводит к получению желаемого промежуточного продукта, в виде преимущественно транс продукта и с сохранением стереохимии в положении 2 (Baldwin, JE, и др., Tetrahedron, 1990 46, 4733). Для соединений, содержащих аминопиридин, бромид, описанный для получения промежуточного продукта 1, является предпочтительным в качестве алкилирующего агента. Кроме того, последовательность синтеза, описанная для получения промежуточного соединения 1, является общей для получения ряда необходимых бромидов с общей структурой А-2, из коммерчески доступных сложных эфиров и спиртов. Этерификация карбоновой кислоты в стандартных условиях конденсации дает промежуточное соединение А-4, которое легко очищают с помощью хроматографии с нормальной фазой. Для временной защиты карбоновой кислоты особенно удобными являются бензиловый и 4-метоксибензиловый эфиры. Удаление N-защитной группы бета-лактама дает промежуточное соединение А-5, которое легко ацилируется с помощью коммерчески доступного или легко получаемого изоцианата А-6 (например, Tsai, M.H., Takaoka, L.R., Powell, N.A., Nowick, J.S. Org. Syn. 2002, 78, 220), с получением бета-лактамного промежуточного соединения А-8. Промежуточное соединение А-5 также может быть преобразовано в А-8 путем реакции с рядом других реагентов, которые, как известно, реагируют с аминами с получением мочевины, такими как фенилкарбаматы (А-7 где Ar = фенил, например: Thavonekham, B. Synthesis, 1997, 1189) и 4-нитрофенилкарбаматы (A-7, где Ar представляет собой 4-нитрофенил, например: Hutchins, S.M., Chapman, K.T. Tet. Lett. 1994, 35, 4055). Кроме того, реакция А-5 с карбамоилхлоридом (А-10, где R5 = Me, например: Holmes, D.L., Smith, E. M., Nowick, J. S. J. Am. Chem. Soc. 1997, 119 (33), 7665) приводит к получению тетра-замещенной мочевины А-8 (R5 = Me). Коммерчески доступные или легко получаемые амины, R4-NH2(недавний обзор по асимметричному синтезу аминов, представляющих особый интерес, см., Robak, M.T., Herbage, M.A., Ellman, J.A. Chem. Rev. 2010, 110, 3600) служат в качестве подходящих исходных материалов для получения изоцианатов А-6 и карбаматов А-7. Снятие временной защитной группы карбоксилата (либо путем гидрирования в тех случаях, когда R3 представляет собой бензил, либо путем ацидолиза вместе с TFA, когда R3 представляет собой 4-метоксибензил) и снятие любых других защитных групп, присутствующих в молекуле, приводит к получению примеров с общей структурой А-9. В тех примерах, когда сложный эфир СО2R3 сохраняется, снятие любых других защитных групп, присутствующих в молекулах, приводит к получению примеров с общей структурой А-8. Для некоторых структур А-9 карбоновая кислота также может быть этерифицирована с использованием способов, известных специалистам в этой области, с получением дополнительных примеров с общей структурой А-8. Примеры соединений по настоящему изобретению, синтезированных в соответствии со способом, описанным на Схеме 1, обобщены на Диаграмме А (ФИГ,1А-1М).
Схема 2. Синтез соединений со Структурой B-5, как показано на Диаграмме В.
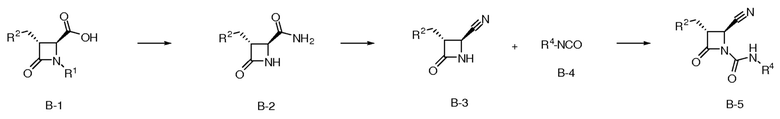
На Схеме 2 описан общий способ получения 2,3-дизамещенных 4-оксоазетидинов, содержащих 2-цианогруппу. Превращение карбоновой кислоты в промежуточное соединение B-1 (получен, как описано для промежуточного соединения А-3 на Схеме 1) в карбоксамид (например, под действием ди-трет-бутилдикарбоатана и бикарбоната аммония в присутствии пиридина, как описано Pozdnev В., Tet. Lett., 1995, 36, 7115) приводит к получению промежуточного соединения B-2 с одновременным удалением силильной защитной группы на N-1. Обезвоживание алифатического амида обеспечивает получение нитрильного промежуточного соединения B-3 (общий обзор см. Mowry, D., Chem. Rev. 1948, 42, 189). Реакция с изоцианатом B-4 и, в случае необходимости, снятие любых оставшихся защитных групп, приводит к получению примеров с общей формулой B-5. Примеры соединений по настоящему изобретению, синтезированных в соответствии со способом, описанным на Схеме 2, обобщены на Диаграмме B (ФИГ. 2).
Схема 3. Синтез соединений со Структурой С-8, как показано на Диаграмме C и D.
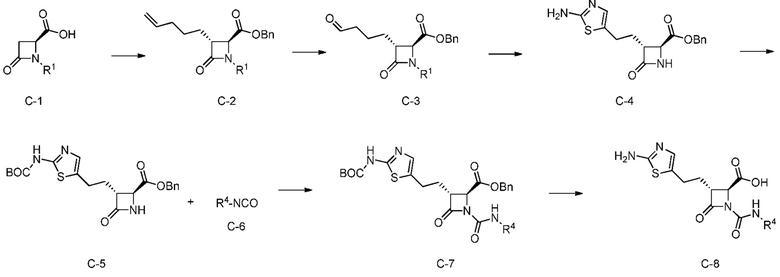
На Схеме 3 описан синтез 3-[2-(2-амино-1,3-тиазол-5-ил)этил]азетидин-2-онов с общей структурой С-8. Окисление алкена С-2 (может быть легко получен способами, описанными на Схеме 1) приводит к получению альдегида С-3, который последовательно обрабатывают бромидом тетрабутиламмония, а затем тиомочевиной с получением промежуточного соединения аминотиазола С-4 с одновременным снятием защитной группы на N-1. Защита аминотиазола с помощью подходящей защитной группы, такой как ВОС, дает промежуточное соединение С-5. Реакция с изоцианатом С-6, с последующим снятием любых защитных групп, приводит к получению примеров с общей формулой С-8. Примеры соединений по настоящему изобретению, синтезированных в соответствии со способом, описанным на Схема 1, обобщены в Диаграмме С и D (ФИГ. 3).
Схема 4. Синтез соединений со Структурой D-8, как показано на Диаграмме C и D.
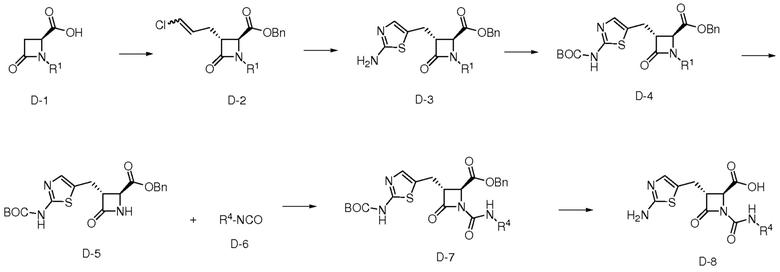
На Схеме 4 описан синтез 3-(2-аминотиазол-5-илметил) азетидин-2-онов с общей структурой D-8. Обработка винилгалогенида D-2 (может быть легко получен способами, описанными на Схеме 1) с помощью MCPBA, а затем конденсация промежуточного соединения хлорэпоксида с помощью тиомочевины, дает промежуточное соединение аминотиазола D-3. Защита аминотиазола подходящей группой, такой как BOC, дает промежуточное соединение D-4. Снятие защиты азота бета-лактама и реакция с изоцианатом D-6 дает промежуточное соединение D-7. Снятие любых оставшихся защитных групп приводит к получению примеров с общей формулой D-8. Примеры соединений по настоящему изобретению, синтезированных в соответствии со способом, описанным на Схеме 4, обобщены в Таблицах С и D (ФИГ. 3).
Схема 5. Синтез соединений со Структурой Е-6, как показано на Диаграмме E.

На Схеме 5 описан общий способ получения 2-карбоновой кислоты с общей структурой Е-6. Конденсация кислоты Е-1 (полученной, как описано для А-3 на Схеме 1) приводит к получению амида Е-2. Снятие защитной группы N-1, с последующей конденсацией изоцианата E-4, дает промежуточное соединение Е-5. Удаление амидной защитной группы и любых других защитных групп, присутствующих на молекуле, приводит к получению примеров с общей формулой Е-6. Примеры соединений по настоящему изобретению, синтезированных в соответствии со способом, описанным на Схеме 5, обобщены в Диаграмме E (ФИГ.4).
Схема 6. Синтез соединений со Структурой F-8, как показано на Диаграмме F.
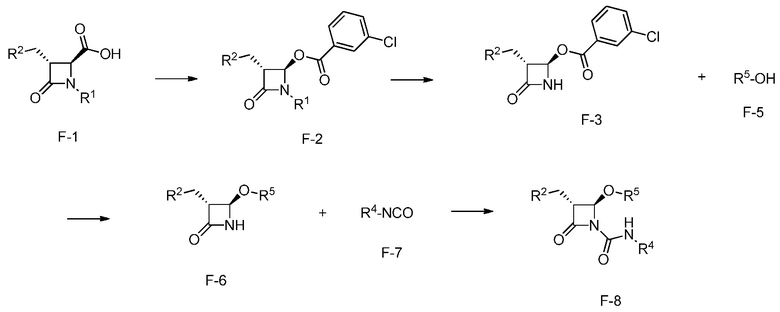
На Схеме 6 описан синтез (2R, 3R) -транс-дизамещенных эфиров с общей формулой F-8. Окислительное декарбоксилирование F-1 (получен, как описано для А-3 из Схемы 1) приводит к получению бензоата F-2 (например, см. Shiozaki, M., Synthesis, 1990, 691). Снятие защитной группы N-1, с последующим замещением бензоата на нуклеофильный кислород F-5, приводит к получению эфира F-6. Реакция с изоцианатом F-7 и снятие любых защитных групп, присутствующих в молекуле, приводит к получению примеров с общей формулой F-8. Примеры соединений по настоящему изобретению, синтезированных в соответствии со способом, описанным на Схеме 6, обобщены на Диаграмме F (ФИГ. 5).
Схема 7. Синтез соединений со Структурой G-4, как показано на Диаграмме G.

На Схеме 7 описан синтез сульфонов общей формулы G-4. Реакция бензоата G-1 (получен, как описано для F-3, Схема 6) с метансульфинатом натрия дает сульфон G-2 (Clauss, K., et. al. Liebigs Ann. Chem. 1974, 539). Реакция с изоцианатом G-3 и последующее снятие любых защитных групп, присутствующих в молекуле, дает примеры с общей формулой G-4. Примеры соединений по настоящему изобретению, синтезированных в соответствии со способом, описанным на Схеме 7, обобщены в Диаграмме G (ФИГ. 6).
Схема 8. Синтез соединений со Структурой H-9, как показано на Диаграмме H.
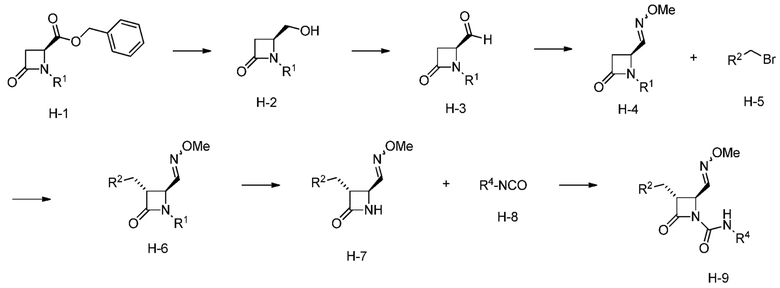
На Схеме 8 описан общий способ получения оксимов с общей структурой H-9. Восстановление N-защищенного азетидинона Н-1 (например, R1 = TBDPS) приводит к получению промежуточного соединения гидроксиметила Н-2, которое легко окисляется до альдегида Н-3. Конденсация с метоксиамином дает оксим Н-4 в виде смеси геометрических изомеров. Алкилирование аниона оксима Н-4, с коммерчески доступным или легко получаемым алкилгалогенидом с общей структурой Н-5, приводит к получению 2,3-транс продукта Н-6. Снятие N-защитной группы бета-лактама дает промежуточное соединение Н-7. Реакция с изоцианатом H-8 и снятие любых защитных групп, присутствующих в молекуле, дает примеры с общей формулой H-9. Примеры соединений по настоящему изобретению, синтезированных в соответствии со способом, описанным на Схеме 8, обобщены в Диаграмме H (ФИГ. 7).
Схема 9. Синтез соединений со Структурами I-4-I-7, как показано на Диаграмме I.
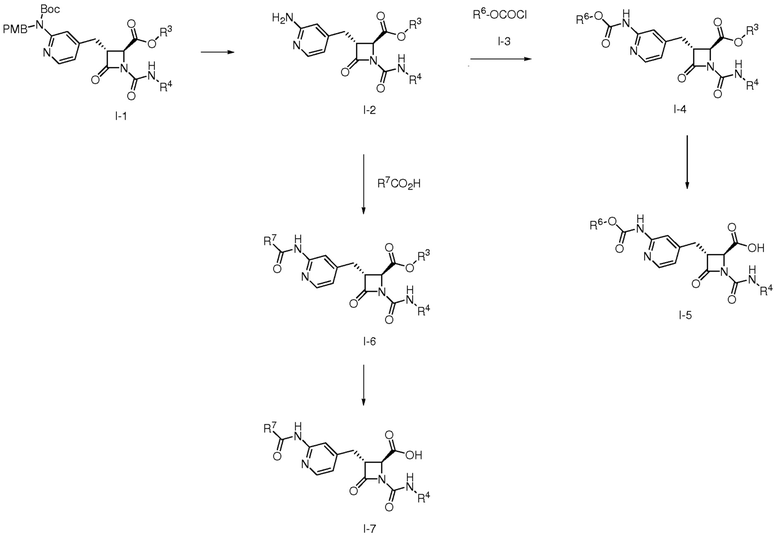
На Схеме 9 описан общий способ получения примеров с общей структурой I-4-I-7. Снятие Вос и PMB защитных групп I-1 (получен, как описано для А-8, Схема 1) приводит к получению аминопиридина I-2. Реакция с хлорформиатом или другим подходящим производным карбоновой кислоты приводит к получению карбамата с общей структурой I-4. Снятие группы карбоновой кислоты, при желании, дает соединения с общей структурой I-5. Кроме того, реакция I-2 с карбоновой кислотой R7СО2Н в присутствии подходящего агента конденсации, приводит к получению соединений с общей структурой I-6. Снятие защитной группой карбоновой кислоты и конденсация с другим спиртом R3ОН в присутствии подходящего агента конденсации приводит к получению R3 группы I-6. В тех случаях, когда желательно иметь свободную карбоновую кислоту, снятие защитной группы карбоновой кислоты и любых других защитных групп, присутствующих в молекуле, приводит к получению соединений с общей структурой I-7. Примеры соединений по настоящему изобретению, синтезированных в соответствии со способом, описанным на Схеме 9, обобщены на Диаграмме I (ФИГ. 8).
Схема 10. Синтез соединений со структурой J-3 и J-4, как показано на Диаграмме J.

На Схеме 10 описан общий способ получения примеров с общей структурой J-3 и J-4. Переходное силилирование соединений общей структуры J-1 (получены, как описано на Схеме 1), с последующей реакцией с нитрофенилкарбонатом J-2 (R5 = Н или Me), приводит к получению соединения с общей структурой J-3. Реакция J-3 со спиртом (R3-ОН) в присутствии подходящего агента конденсации или бромида (R3-Br), в присутствии подходящего основания, обеспечивает получение сложных эфиров с общей структурой J-4. Примеры соединений по настоящему изобретению, синтезированных в соответствии со способом, описанным на Схеме 10, обобщены на Диаграмме J (ФИГ,9А-9B).
Схема 11. Синтез соединений со Структурой K-9, как показано на Диаграмме K.
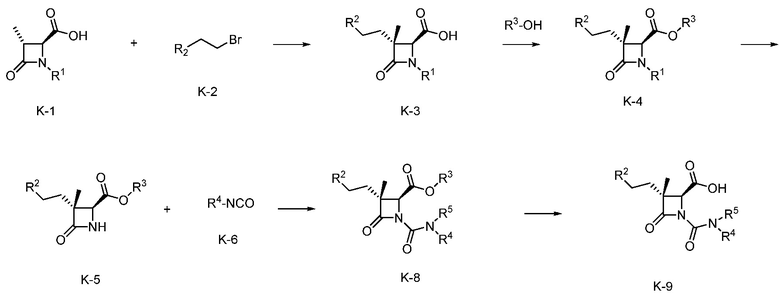
На Схеме 11 описан общий способ получения (2S, 3R) -тризамещенного 4-оксоазетидин-2-карбоксилата с общей структурой К-9. Химизм полностью аналогичен способу, описанному на Схеме 1, в котором используемым исходным материалом, является известный лактам К-1 (см Finke, P.E., et. al., J. Med. Chem. 1995, 38, 2449). Примеры соединений по настоящему изобретению, синтезированных в соответствии со способом, описанным на Схеме 11, обобщены в Диаграмме K (ФИГ.10A-10B).
Пример 2. Получение промежуточных соединений.
Промежуточный продукт 1: трет-Бутил [4-(бромметил)пиридин-2-ил](4-метоксибензил) карбамат
Стадия 1. Получение метил 2-[( трет бутоксикарбонил) амино] изоникотината
Раствор ди-трет-бутилдикарбоната (47,8 г, 219 ммоль) в т-BuOH (650 мл, 6,80 моль) нагревали при 33°С и обрабатывали вместе с метил-2-аминоизоникотинатом (30,4 г, 200 ммоль) порциями в течение 1 ч [~5 г добавляется каждые 10 мин]. Реакционную смесь перемешивали при 33°С в течение ночи (18 ч); ВЭЖХ/LC MS показала практически полное превращение. Твердые вещества собирали вакуумной фильтрацией [стеклянные нутч-фильтры (фритты) из спекшегося стекла] и промывали Et2O (~200 мл). Светло-коричневый твердый продукт сушили в высоком вакууме в течение 3,5 ч до достижения постоянной массы (39,65 г, 79%): MS (ESI+) для C12H16N2O4 m/z 253,2 [M+H]+, 275,2 [M+Na]+; Время удержания ВЭЖХ: 3,25 мин (Способ B).
Стадия 2. Получение трет бутил[4-(гидроксиметил)пиридин-2-ил] (4-метоксибензил) карбамата
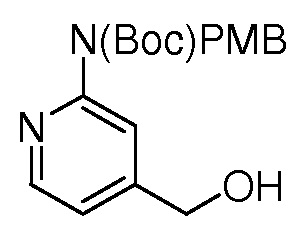
Суспензию метил 2-[(трет-бутоксикарбонил)амино]изоникотина (41,5 г, 164 ммоль) в ДМФ (610 мл, 7,90 моль) охлаждали до ~0°C (лед/NaCl) и обрабатывали 1,0 M раствором NaHMDS в THF (197 мл, 197 ммоль) по каплям в течение 2 часов с получением прозрачного коричневого раствора. Реакционную смесь перемешивали в течение 30 мин при 0°С и обрабатывали PMBCl (24,5 мл, 181 ммоль) в течение 11 мин. Реакционную смесь перемешивали в течение 10 мин с получением желтой/коричневой смеси, охлаждающую баню удаляли, и реакционную смесь перемешивали при комнатной температуре в течение 1 ч 40 мин; ВЭЖХ показала почти полное превращение. Реакционную смесь охлаждали до ~0°C (лед/NaCl), и реакционную смесь гасили добавлением насыщенного водного раствора NH4Cl (100 мл). Смесь оставляли нагреваться до комнатной температуры и экстрагировали Et2O (1×200 мл). Отделенный водный слой разбавляли водой (100 мл) и экстрагировали Et2O (2×100 мл). Объединенные органические слои промывали насыщенным водным раствором NaHCO3 (1×400 мл), и отделенный водный слой фильтровали [грубое спекшееся стекло] и экстрагировали Et2O (2×100 мл). Объединенные органические экстракты промывали 10% водным раствором LiCl (2×600 мл), сушили (Na2SO4) и концентрировали в вакууме с получением оранжевого масла. Сырое вещество затем сушили в высоком вакууме в течение ночи с получением твердого продукта светлого цвета/оранжевого масла (80,86 г), которое использовали без дополнительной обработки: MS (ESI+) для C20H24N2O5 m/z 373,2 [M+H]+; время удержания ВЭЖХ: 4,53 мин (Способ B); 1H ЯМР показал, что вещество было загрязнено гексаметилдисилазаном.
Вышеуказанный неочищенный метил 2-[(третбутоксикарбонил) (4-метоксибензил)амино]изоникотинат (80,86 г) разделяли на две равные части, и каждую часть обрабатывали следующим образом: Раствор сырого вещества в THF (400 мл) охлаждали до ~0°C (лед/NaCl) и обрабатывали LiBH4 (3,94 г, 181 ммоль) одной порцией. Реакционную смесь перемешивали в течение 5 мин, охлаждающую баню удаляли, и реакционную смесь оставляли нагреваться до комнатной температуры при перемешивании в течение 30 мин. Колбу переносили в предварительно нагретую масляную баню с 50°C, и реакционную смесь перемешивали при 40°С в течение 2,5 ч; ВЭЖХ показала полное превращение. Реакционную смесь охлаждали до ~0°С, и реакционную смесь осторожно гасили медленным добавлением по каплям насыщенного водного раствора NaHCO3 (100 мл). Смесь разбавляли водой (150 мл) и экстрагировали EtOAc (1×200 мл, 2×100 мл). Объединенные органические слои промывали насыщенным водным раствором NH4Cl (1×200 мл), водой (1×200 мл) и солевым раствором (1×200 мл), сушили (Na2SO4) и концентрировали в вакууме с получением указанного в заголовке соединения в виде вязкого масла желтого масла (55,6 г, 89%), которое использовали без дополнительной обработки: MS (ESI+) для C19SO24N2О4 m/z 345,3 [М+Н]; Время удержания ВЭЖХ: 3,19 мин (Способ В).
Стадия 3. Получение трет-бутил[4-(бромметил)пиридин-2-ил](4-метоксибензил)карбамата
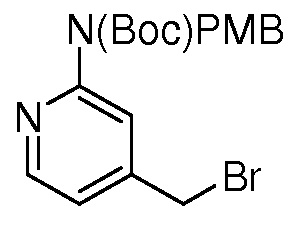
К перемешиваемому раствору трет-бутил [4-(гидроксиметил)пиридин-2-ил](4-метоксибензил)карбамата (23,14 г, 67,19 ммоль) в THF (300 мл), охлажденному на лед/вода баня, добавляли Et3N (13,6 мл, 97,8 ммоль). MsCl (7,20 мл, 93,1 ммоль) медленно добавляли в течение 15 мин, и реакционную смесь перемешивали в течение 1 ч при 0°C; ВЭЖХ показала практически полное превращение. LiBr (8,54 g, 98,3 ммоль) добавляли одной порцией, и холодную баню удаляли. Реакционную смесь перемешивали в течение ночи при комнатной температуре; ВЭЖХ показала полное превращение в желаемый продукт. Реакционную смесь разделяли между EtOAc (250 мл) и водой (250 мл). Отделенную водную фазу экстрагировали EtOAc (200 мл), и объединенные органические фазы промывали водой (300 мл), сушили (MgSO4) и упаривали в вакууме с получением вязкого оранжевого масла (12,05 г). Сырую смесь абсорбировали на силикагеле и очищали флэш-хроматографией на колонке (500 г силикагель, заполняли гексаном, элюировали 15% EtOAc/гексан) с получением соединения, указанного в заголовке, в виде светло-желтого масла (11,59 г, 42%): MS (ESI+) для C19H23BrN2O3 m/z 407,1, 409,1 [M+H]+; время удержания ВЭЖХ: 4,48 мин (Способ B); 1H ЯМР (CDCl3, 300 МГц) δ 8,42-8,30 (м, 1H), 7,73 (ушир.c, 1H), 7,26-7,19 (м, 2H), 7,07-6,9,9 (м, 1H), 6,85-6,77 (м, 2H), 5,14 (c, 2H), 4,38 (c, 2H), 3,78 (c, 3H), 1,44 (c, 9H).
Промежуточное соединение 2: 1-[(1R)-1-изоцианатоэтил]-3-(трифторметокси)бензол
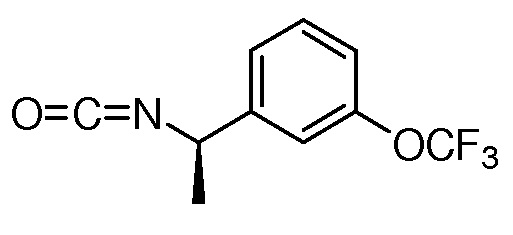
К охлажденному (0-5°C), энергично перемешанному раствору трихлорида (1R)-1-[3-(трифторметокси)фенил]этанамина (получен, как описано в стадии 1-3 в Примере 4 из 3-трифторметоксибензальдегид) (0,197 г, 0,815 ммоль) в метиленхлориде (4 мл) и насыщ. NaHCO3 (4 мл) добавляли трифосген (0,080 г, 0,269 ммоль) одной порцией. Через 30 мин смесь переносили в разделительную воронку, и органический слой собирали. Водный слой промывали DCM, и объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением соединения, указанного в заголовке, (155 мг, 82%) в виде вязкого масла, которое использовали незамедлительно без дополнительной очистки. 1H ЯМР (300 МГц, CDCl3) δ 1,63 (д, J=7 Гц, 3H), 4,82 (кв., J=7 Гц, 1H), 7,16-7,29 (м, 3H), 7,41 (т, J=8 Гц, 1H).
Промежуточное соединение 3. 4-метоксибензил (2S,3R)-3-({2-[(трет-бутоксикарбонил)(4-метоксибензил)амино]пиридин-4-ил}метил)-4-оксо-1-{[(1S)-2,2,2-трифтор-1-фенилэтил]карбамоил}азетидин-2-карбоксилат
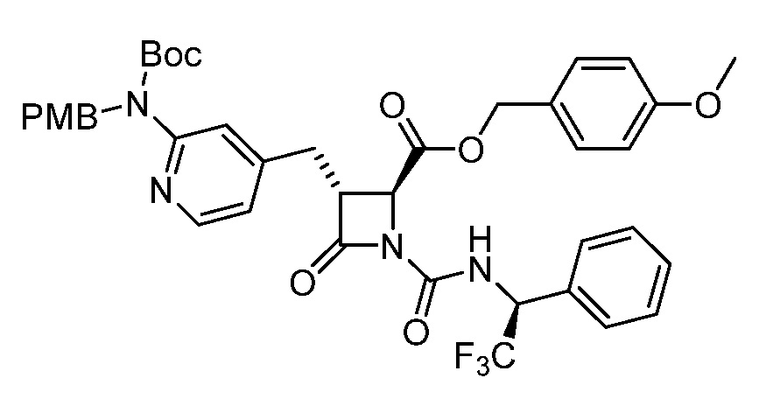
К раствору 4-нитрофенилхлорформиата (0,378 г, 2,22 ммоль) в тетрагидрофуране (20 мл) при температуре окружающей среды по каплям добавляли раствор (S)-2,2,2-трифтор-1-фенилэтиламина (0,390 г, 2,22 ммоль) и пиридин (0,180 мл, 2,22 ммоль) в THF (20 мл) в течение 45 мин. Через 15 мин смесь разбавляли этилацетатом и промывали 0,1 н водным HCl, насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением 4-нитрофенил [(1S)-2,2,2-трифтор-1-фенилэтил]карбамата, которое использовали без дополнительной очистки. MS (ESI+) для C15H11F3N2O4 m/z 341,3 (M+H)+; Время удержания ВЭЖХ: 4,4 мин (Способ C). К раствору карбамата, описанного выше в THF (5 мл) добавляли 4-метоксибензил (2S,3R)-3-({2-[(трет-бутоксикарбонил)(4-метоксибензил)амино]пиридин-4-ил}метил)-4-оксоазетидин-2-карбоксилат (185 мг, 0,329 ммоль), триэтиламин (0,080 мл, 0,57 ммоль) и 4-диметиламинопиридин (0,002 г, 0,016 ммоль) при температуре окружающей среды. Через 3 дня летучие вещества удаляли при пониженном давлении, и остаток очищали флэш-хроматографией, используя гексан и этилацетат (15-30%) в качестве элюента с получением соединения, указанного в заголовке, (220 мг, 74%) в виде белого твердого продукта, который содержал приблизительно 15% (S,S)-1,3-бис-(2,2,2-трифтор-1-фенил-этил)мочевины, которую сразу удаляли с помощью хроматографии после снятия защитной группы. 1H ЯМР (300 МГц, CDCl3) δ 1,43 (c, 9H), 3,02-3,24 (м, 2H), 3,50-3,56 (м, 1H), 3,78 (c, 3H), 3,80 (c, 3H), 4,29 (д, J=3 Гц, 1H), 5,50-5,62 (м, 1H), 6,79-6,85 (перекрываемый м, 5H), 7,10 (д, J=9 Гц, 2H), 7,21-7,23 (перекрываемый м, 3H), 7,39-7,45 (перекрываемый м, 6H), 7,64 (c, 1H), 8,27 (д, J=5 Гц, 1H); MS (ESI+) для C40H41F3N4O8 m/z 763,0 (M+H)+; Время удержания ВЭЖХ: 5,54 мин (Способ C).
Промежуточное соединение 4. трет-бутил [4-(2-бромэтил)пиридин-2-ил](4-метоксибензил)карбамат
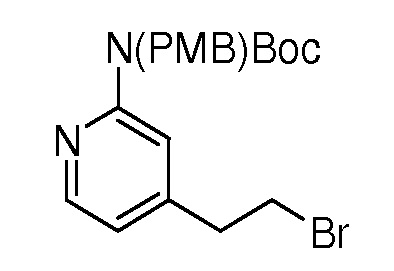
Стадия 1. Получение трет-бутил (4-метилпиридин-2-ил)карбамата
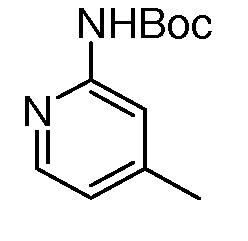
Раствор 2-пиридинамина, 4-метил- (0,60 г, 5,5 ммоль) в трет-BuOH (35 мл) обрабатывали ди-трет-бутилдикарбонатом (1,3 г, 6,1 ммоль). Реакционную смесь перемешивали при 30°C в течение 2 дней 17 часов; ВЭЖХ/ТСХ указали на превращение в продукт. Растворитель удаляли в вакууме с получением светло-коричневого кристаллического твердого вещества, которое перекристаллизовывали из горячего изопропанола с получением соединения, указанного в заголовке, в виде белого кристаллического твердого вещества (0,87 г, 75%): (последующая 1H ЯМР не показывает никаких примесей, что согласуется с LC МC хотя ВЭЖХ показывает два пика) MS (ESI+) для C11H16N2O2 m/z 153,2 [M-tBu+H]+; Время удержания ВЭЖХ: 2,24 мин (второй пик при 2,11 мин) (Способ B).
Стадия 2. Получение трет-бутил (4-метоксибензил)(4-метилпиридин-2-ил)карбамата

Раствор трет-бутил (4-метилпиридин-2-ил)карбамата (81 мг, 0,39 ммоль) в ДМФ (1,2 мл) охлаждали до -10°C (лед/солевой раствор) и обрабатывали гидроксидом натрия (60% в минеральном масле, 22 мг, 0,55 ммоль). Реакционную смесь энергично перемешивали в течение 20 мин при -10°C с получением практически гомогенного раствора, который обрабатывали PMBCl (0,07 мл, 0,5 ммоль). Реакционную смесь перемешивали в течение 40 мин с получением розовой смеси, холодную баню удаляли, и смесь перемешивали при комнатной температуре в течение 17 часов с получением оранжевой смеси; ВЭЖХ/LC MS указала полное превращение в продукт. Реакцию гасили дополнительным количеством воды (1 мл) и разбавляли водой (5 мл) и Et2O (40 мл). Отделенный органический слой промывали водой (10 мл), 0,1 н водным раствором HCl (10 мл), насыщенным водным раствором NaHCO3 (10 мл) и насыщенным солевым раствором (10 мл), сушили (Na2SO4) и концентрировали в вакууме с получением небольшого количества оранжевого масла. Очистка колоночной хроматографией (2×8 см силикагеля; Гекс., 5%, 10%, 20% EtOAc/Гекс.) давала соединение, указанное в заголовке, в виде бесцветного прозрачного масла (91 мг, 71%); MS (ESI+) для C19H24N2O3 m/z 329,2 [M+H]+; Время удержания ВЭЖХ: 3,66 мин (Способ B).
Стадия 3. Получение этил {2-[(трет-бутоксикарбонил)(4-метоксибензил)амино]пиридин-4-ил}ацетата и трет-бутил 2-[(4-метоксибензил)амино]-4-метилникотината
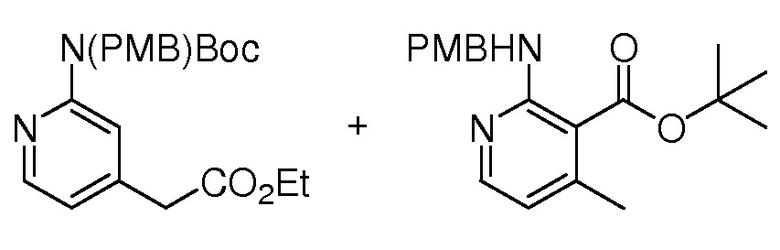
Раствор трет-бутил (4-метоксибензил)(4-метилпиридин-2-ил)карбамата (3,3 г, 10,0 ммоль) и диэтилкарбоната (6,1 мл, 50,0 ммоль) в THF (82 мл) охлаждали до -78°C (ацетон/CO2) и обрабатывали каплями 1,33 M раствора LDA в гексан /THF/этилбензол (9,1 мл, 12 ммоль) в течение 15 мин. Желто-оранжевую реакционную смесь перемешивали в течение 30 мин при -78°C; ВЭЖХ указала 1,1:1:0,5 смесь трет-бутил (4-метоксибензил)(4-метилпиридин-2-ил)карбамата, желаемого продукта этил {2-[(трет-бутоксикарбонил)(4-метоксибензил)амино]пиридин-4-ил}ацетата и побочного продукта трет-бутил 2-[(4-метоксибензил)амино]-4-метилникотинат. На 45 мин реакцию гасили дополнительным количеством уксусной кислоты (0,68 мл, 12 ммоль), и смесь разбавляли водой (75 мл) и EtOAc (36 мл). Смесь оставляли нагреваться до комнатной температуры, и отделенный водный слой экстрагировали EtOAc (2×36 мл). Объединенные органические слои сушили (Na2SO4) и концентрировали в вакууме с получением желтого масла. Очистка колоночной хроматографией (5×18 см силикагель; Гекс., 5%, 10%, 15%, 20% EtOAc/Гекс.) давала 70:30 смесь этил {2-[(трет-бутоксикарбонил)(4-метоксибензил)амино]пиридин-4-ил}ацетата и трет-бутил 2-[(4-метоксибензил)амино]-4-метилникотината в виде бесцветного прозрачного масла (1,77 г, 33%) и восстанавливала трет-бутил (4-метоксибензил)(4-метилпиридин-2-ил)карбамат в виде бесцветного прозрачного масла (1,78 г, 54%): 1H ЯМР указала 2,9:1 смесь этил {2-[(трет-бутоксикарбонил)(4-метоксибензил)амино]пиридин-4-ил}ацетата и трет-бутил 2-[(4-метоксибензил)амино]-4-метилникотината; MS (ESI+) для C22H28N2O5 m/z 401,3 [M+H]+, MS (ESI+) для C19H24N2O3 m/z 329,3 [M+H]+; ВЭЖХ время удержания для этил {2-[(трет-бутоксикарбонил)(4-метоксибензил)амино]пиридин-4-ил}ацетата: 4,22 мин (Способ B), Время удержания ВЭЖХ для трет-бутил 2-[(4-метоксибензил)амино]-4-метилникотината: 3,24 мин (Способ B).
Стадия 4. Получение трет-бутил [4-(2-гидроксиэтил)пиридин-2-ил](4-метоксибензил)карбамата
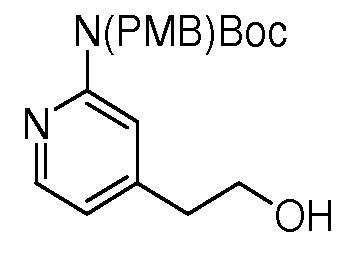
~2:1 Смесь этил {2-[(трет-бутоксикарбонил)(4-метоксибензил)амино]пиридин-4-ил}ацетата (1,77 г, 3,27 ммоль) и трет-бутил 2-[(4-метоксибензил)амино]-4-метилникотината в THF (16 мл) охлаждали до 0°C (лед/солевой раствор) и обрабатывали LiBH4 (0,21 г, 9,6 ммоль). Холодную баню удаляли, и реакционную смесь оставляли нагреваться до комнатной температуры в течение 20 мин, а затем нагревали при 50°C в течение 3 ч; ВЭЖХ/LC MS указала на полное потребление {2-[(трет-бутоксикарбонил)(4-метоксибензил)амино]пиридин-4-ил}ацетата. Реакционную смесь оставляли охлаждаться до комнатной температуры и реакцию аккуратно гасили дополнительным количеством насыщенного водного раствора NaHCO3 (30 мл). Смесь разбавляли водой (60 мл) и экстрагировали EtOAc (3×60 мл). Объединенные органические слои сушили (Na2SO4) и концентрировали в вакууме с получением ярко-желтого масла. Очистка колоночной хроматографией (4×14 см силикагель; Гекс., 10%, 30%, 50% EtOAc/Гекс.) давала соединение, указанное в заголовке, в виде бесцветного прозрачного масла (0,91 г, 78%): MS (ESI+) для C20H26N2O4 m/z 359,3 [M+H]+; ВЭЖХ время удержания: 3,17 мин (Способ B).
Стадия 5. Получение трет-бутил [4-(2-бромэтил)пиридин-2-ил](4-метоксибензил)карбамата
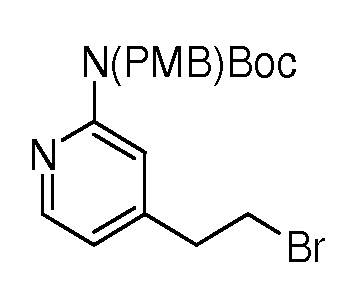
Смесь трет-бутил [4-(2-гидроксиэтил)пиридин-2-ил](4-метоксибензил)карбамата (0,91 г, 2,5 ммоль) и тетрабромида углерода (0,93 г, 2,8 ммоль) в CH2Cl2 (15 мл) охлаждали до 0°C (лед/вода). Одной порцией добавляли трифенилфосфин (0,73 г, 2,8 ммоль) и ярко-желтую реакционную смесь перемешивали в течение 30 мин при 0°C. Холодную баню удаляли, и реакционную смесь перемешивали при комнатной температуре в течение 1 ч; ВЭЖХ показала практически полное превращение в продукт. На 2 ч ВЭЖХ указала на отсутствие изменений. Реакционную смесь разбавляли CH2Cl2 (120 мл) и промывали насыщенным водным раствором NaHCO3 (70 мл). Отделенную водную фазу экстрагировали CH2Cl2 (2×70 мл), и объединенные органические слои сушили (Na2SO4) и концентрировали в вакууме с получением бесцветного прозрачного масла. Очистка колоночной хроматографией (4×16 см силикагель; Гекс., 10%, 20%, 30% EtOAc/Гекс.) давала соединение, указанное в заголовке, в виде бесцветного прозрачного масла (0,69 г, 64%): MS (ESI+) для C20H25BrN2O3 m/z 421,2 [M+H]+; ВЭЖХ время удержания: 4,29 мин (Способ B); 1H ЯМР (CDCl3, 300 МГц) δ 8,37-8,28 (м, 1H), 7,54 (ушир.c, 1H), 7,26-7,16 (м, 2H), 6,90-6,84 (м, 1H), 6,84-6,74 (м, 2H), 5,38-4,88 (м, 2H), 3,77 (c, 3H), 3,62-3,51 (м, 2H), 3,19-3,09 (м, 2H), 1,44 (c, 9H).
Промежуточное соединение 5: трет-бутил [4-(бромметил)пиридин-2-ил]метилкарбамат
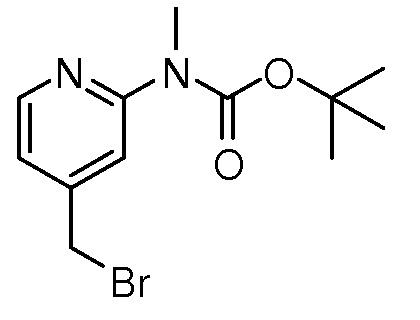
Стадия 1. Получение метил 2-[(трет-бутоксикарбонил)(метил)амино]изоникотината
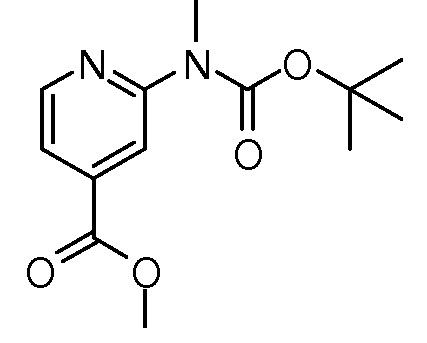
Взвесь метил 2-[(трет-бутоксикарбонил)амино]изоникотината (5,0 г, 2,0E1 ммоль) в ДМФ (75 мл) охлаждали до ~0°C (лед/NaCl) и обрабатывали 1,0 M раствором гексаметилдисилазина натрия в THF (24 мл, 24 ммоль) по каплям в течение 25 мин с получением прозрачного желтого/коричневого раствора. Реакционную смесь перемешивали в течение 30 мин при 0°C и обрабатывали метил йодидом (1,4 мл, 22 ммоль). Реакционную смесь перемешивали в течение 20 мин, холодную баню удаляли, и реакционную смесь перемешивали при комнатной температуре в течение 2 ч; ВЭЖХ/LC MS указала полное превращение в продукт. Реакционную смесь охлаждали до ~0°C, и реакцию гасили дополнительным количеством насыщенного водного раствора NH4Cl (25 мл). Смесь оставляли нагреваться до комнатной температуры и экстрагировали Et2O (3×50 мл). Объединенные органические слои промывали насыщенным водным раствором NaHCO3 (1×50 мл) и 10% водным раствором LiCl (2×50 мл), сушили (Na2SO4) и концентрировали в вакууме с получением ярко-желтого масла с осадком. Сырое вещество дополнительно сушили в высоком вакууме в течение ночи с получением сырого соединения, указанного в заголовке в виде твердого продукта светлого цвета/желтого масла (5,29 г). MS (ESI+) для C13H18N2O4LC m/z 267,1 (M+H)+; Время удержания ВЭЖХ: 3,78 мин (Способ B).
Стадия 2. Получение трет-бутил [4-(гидроксиметил)пиридин-2-ил]метилкарбамата
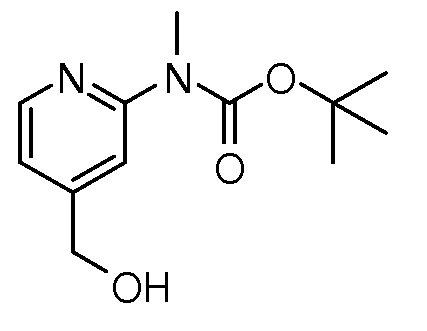
Раствор сырого метил 2-[(трет-бутоксикарбонил)(метил)амино]изоникотината (5,29 г) в THF (50 мл) охлаждали до ~0°C (лед/солевой раствор) и обрабатывали одной порцией тетрагидробората лития (0,47 г, 22 ммоль). Реакционную смесь перемешивали в течение 10 мин, холодную баню удаляли, и реакционную смесь оставляли нагреваться до комнатной температуры в течение 30 мин. Колбу помещали на заранее нагретую до 40°C масляную баню, и реакционную смесь перемешивали при 40°C в течение 3,5 ч; ВЭЖХ указала смесь продукта и исходного вещества. Температуру масляной бани повышали до 50°C в течение 50 минут; ВЭЖХ показала практически полное превращение в продукт. Реакционную смесь оставляли охлаждаться до комнатной температуры и хранили при 0-5°C в течение ночи. Реакционную смесь охлаждали до ~0°C, и реакцию аккуратно гасили медленно по каплям добавляя насыщенный водный раствор NaHCO3 (15 мл). Смесь разбавляли водой (20 мл) и экстрагировали EtOAc (3×20 мл). Объединенные органические слои промывали насыщенным водным раствором NH4Cl (1×20 мл), водой (1×20 мл) и насыщенным солевым раствором (1×30 мл), сушили (Na2SO4) и концентрировали в вакууме с получением сырого соединения, указанного в заголовке, в виде светло-коричневого масла (4,49 г). MS (ESI+) для C12H18N2O3LC m/z 239,2 (M+H)+; Время удержания ВЭЖХ: 2,10 мин (Способ B).
Стадия 3. Получение трет-бутил [4-(бромметил)пиридин-2-ил]метилкарбамата
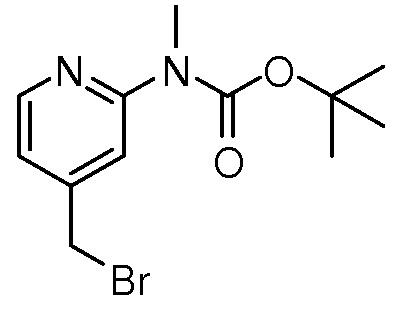
Смесь сырого трет-бутил [4-(гидроксиметил)пиридин-2-ил]метилкарбамата (4,49 г) и тетрабромида углерода (7,2 г, 22 ммоль) помещали в CH2Cl2 (110 мл) и охлаждали до ~0°C (ледяная вода). Одной порцией добавляли трифенилфосфин (5,7 г, 22 ммоль), и ярко-желтую реакционную смесь перемешивали в течение 20 мин, холодную баню удаляли, и оранжево-красную смесь перемешивали при комнатной температуре в течение 1 ч; ВЭЖХ/LC MS указала полное превращение в продукт. Реакционную смесь разбавляли CH2Cl2 (50 мл) и промывали насыщенным водным раствором NaHCO3 (1×50 мл). Отделенный водный слой экстрагировали CH2Cl2 (2×20 мл), и объединенные органические слои сушили (Na2SO4) и концентрировали в вакууме с получением красно-оранжевого масла, которое хранили при 0-5°C в течение ночи. Очистка колоночной хроматографией (5×14 см силикагель; Гекс., 10%, 20%, 30% EtOAc/Гекс.) давала соединение, указанное в заголовке, в виде прозрачного, практически бесцветного масла (4,74 г, 80%). MS (ESI+) для C12H17BrN2O2 m/z 301,1, 303,1 (M+H)+; Время удержания ВЭЖХ: 3,52 мин (Способ B); 1H ЯМР (300 МГц, CDCl3) δ 8,43-8,27 (м, 1H), 7,84-7,66 (м, 1H), 7,12-6,92 (м, 1H), 4,39 (ушир.c, 2H), 3,41 (c, 3H), 1,54 (c, 9H).
Пример 3, Схема 1: трифторацетат (2S,3R)-3-[(2-аминопиридин-4-ил)метил]-1-{[(1R)-1-циклогексилэтил]карбамоил}-4-оксоазетидин-2-карбоновой кислоты
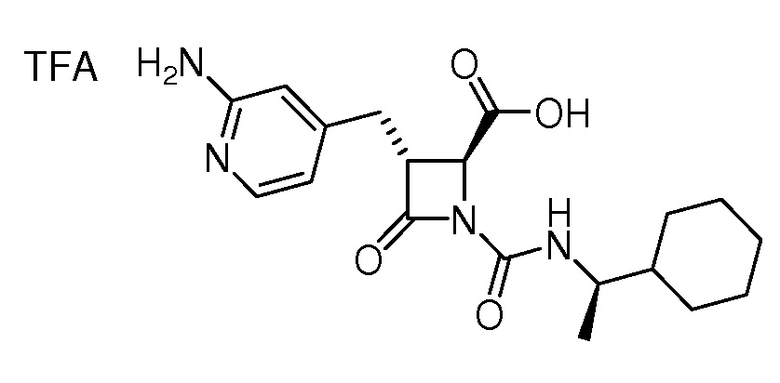
Стадия 1. Получение (2S,3R)-3-({2-[(трет-бутоксикарбонил)(4-метоксибензил)амино]пиридин-4-ил}метил)-1-[трет-бутил(диметил)силил]-4-оксоазетидин-2-карбоновой кислоты
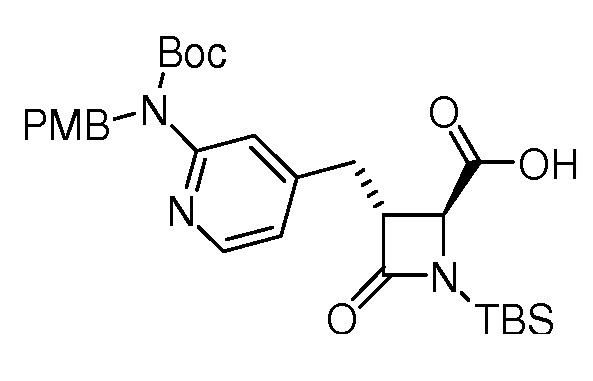
Высушенную в печи трехгорлую колбу охлаждали в азоте, наполняли (2S)-1-[трет-бутил(диметил)силил]-4-оксоазетидин-2-карбоновой кислотой (3,00 г, 13,1 ммоль) и сухим THF (110 мл), и смесь охлаждали до -70°C. По каплям добавляли диизопропиламид лития в THF [1,25 M (титрованный), 21,4 мл, 26,8 ммоль] в течение 8 мин, поддерживая внутреннюю температуру ниже -55°C, и полученную светло-коричневую смесь перемешивали при -78°C в течение 35 мин ,а затем помещали на баню лед-насыщенный солевой раствор-MeOH и перемешивали при температуре ниже -15°C еще 30 мин. Затем добавляли раствор трет-бутил [4-(бромметил)пиридин-2-ил](4-метоксибензил)карбамата (5,86 г, 14,4 ммоль) в сухом THF (21 мл), который охлаждали до 0°C в течение 5 мин, поддерживая внутреннюю температуру ниже -5°C, и полученную темную смесь перемешивали при этой температуре в течение 1 ч, гасили водным раствором лимонной кислоты (0,5 M, 120 мл), разбавляли водой (120 мл) и экстрагировали EtOAc (3×100 мл). Объединенную органическую фазу промывали насыщенным солевым раствором (60 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении с получением сырого продукта в виде прозрачного липкого осадка, который использовали как есть в следующей стадии. MS (ESI+) для C29H41N3O6Si m/z 556,5 (M+H)+.
Стадия 2. Получение бензил (2S,3R)-3-({2-[(трет-бутоксикарбонил)(4-метоксибензил)амино]пиридин-4-ил}метил)-1-[трет-бутил(диметил)силил]-4-оксоазетидин-2-карбоксилата
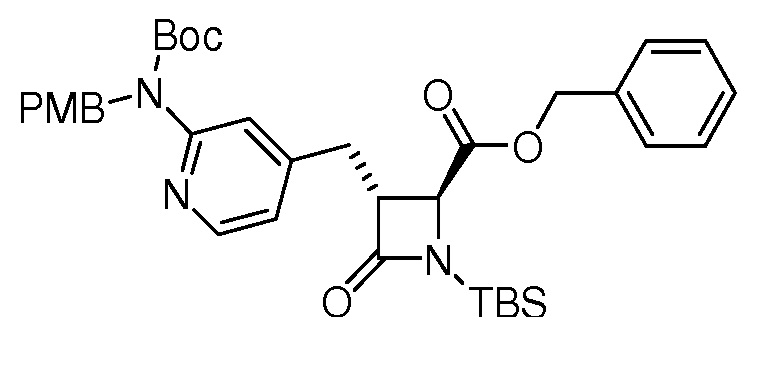
Перемешанный раствор сырой (2S,3R)-3-({2-[(трет-бутоксикарбонил)(4-метоксибензил)амино]пиридин-4-ил}метил)-1-[трет-бутил(диметил)силил]-4-оксоазетидин-2-карбоновой кислоты в DCM (60 мл) в азоте обрабатывали гидрохлоридом N-(3-диметиламинопропил)-N'-этилкарбодиимида (3,26 г, 17,0 ммоль), а затем бензиловым спиртом (1,62 мл, 15,7 ммоль) и 4-диметиламинопиридином (160 мг, 1,31 ммоль), и полученную коричневую смесь перемешивали при комнатной температуре в течение 2 ч, и в этот момент ВЭЖХ указала на завершение реакции. Смесь разбавляли водой (60 мл) и экстрагировали DCM (2×60 мл), и объединенную органическую фазу промывали водой (60 мл) и насыщенным солевым раствором (50 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Очистка хроматографией на силикагеле [120 г картридж; элюция градиентом 10-20% EtOAc/гексан] давала 4,16 г соединения, указанного в заголовке, (загрязненный ~15 масс.% бензилового спирта; 42% выход за две первые стадии) в виде светло-коричневого вязкого масла, которое использовали без дополнительной очистки. MS (ESI+) для C36H47N3O6Si m/z 646,7 (M+H)+.
Стадия 3. Получение бензил (2S,3R)-3-({2-[(трет-бутоксикарбонил)(4-метоксибензил)амино]пиридин-4-ил}метил)-4-оксоазетидин-2-карбоксилата
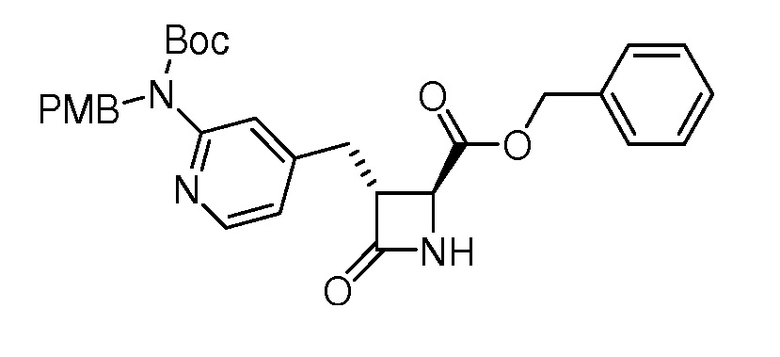
Перемешанный раствор бензил (2S,3R)-3-({2-[(трет-бутоксикарбонил)(4-метоксибензил)амино]пиридин-4-ил}метил)-1-[трет-бутил(диметил)силил]-4-оксоазетидин-2-карбоксилата (2,98 г, 3,92 ммоль) в MeOH (39 мл) в азоте обрабатывали уксусной кислотой (0,780 мл, 13,7 ммоль), а затем раствором фторида аммония в MeOH (0,5 M, 10,2 мл, 5,10 ммоль), и полученную гомогенную смесь перемешивали при комнатной температуре в течение 1 ч, и в этот момент ВЭЖХ указала на завершение реакции. Смесь концентрировали при пониженном давлении, и остаток помещали в DCM (120 мл), промывали насыщенным водным раствором NaHCO3 (80 мл), водой (60 мл) и насыщенным солевым раствором (40 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Очистка хроматографией на силикагеле [40 г картридж; элюция 20-50% EtOAc/гексан] давала 1,95 г (94%) соединения, указанного в заголовке, в виде белого твердого продукта. MS (ESI+) для C30H33N3O6 m/z 532,4 (M+H)+.
Стадия 4. Получение бензил (2S,3R)-3-({2-[(трет-бутоксикарбонил)(4-метоксибензил)амино]пиридин-4-ил}метил)-1-{[(1R)-1-циклогексилэтил]карбамоил}-4-оксоазетидин-2-карбоксилата

Перемешанный раствор бензил (2S,3R)-3-({2-[(трет-бутоксикарбонил)(4-метоксибензил)амино]пиридин-4-ил}метил)-4-оксоазетидин-2-карбоксилата (2,72 г, 5,12 ммоль) в DCM (51 мл) в азоте обрабатывали Et3N (2,50 мл, 17,9 ммоль), а затем раствором коммерческого [(1R)-1-изоцианатоэтил]циклогексана (1,02 г, 6,65 ммоль) в DCM (5 мл), и смесь перемешивали при комнатной температуре в течение ночи, и в этот момент ВЭЖХ указала, что исходный продукт был практический полностью использован. В 17 ч смесь концентрировали при пониженном давлении, и остаток очищали хроматографией на силикагеле [120 г; элюция 15-25% EtOAc/гексан] с получением 2,31 g (66%) соединения, указанного в заголовке, в виде белой пены. MS (ESI+) для C39H48N4O7 m/z 685,7 (M+H)+.
Стадия 5. Получение (2S,3R)-3-({2-[(трет-бутоксикарбонил)(4-метоксибензил)амино]пиридин-4-ил}метил)-1-{[(1R)-1-циклогексилэтил]карбамоил}-4-оксоазетидин-2-карбоновой кислоты
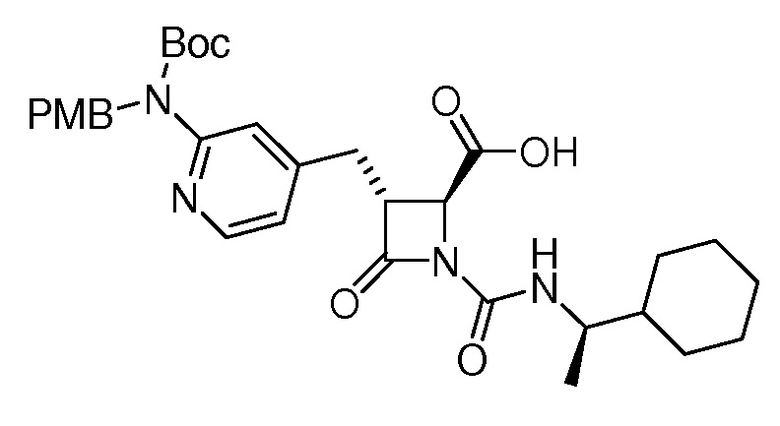
Смесь бензил (2S,3R)-3-({2-[(трет-бутоксикарбонил)(4-метоксибензил)амино]пиридин-4-ил}метил)-1-{[(1R)-1-циклогексилэтил]карбамоил}-4-оксоазетидин-2-карбоксилата (1,94 г, 2,83 ммоль) в 1:1 MeOH/EtOAc (56 мл) в азоте обрабатывали 10% палладием на угле (301 мг, 0,283 ммоль Pd), и смесь откачивали и наполняли два раза азотом, а затем перемешивали в атмосфере водорода (двухслойный баллон) в течение 70 мин, и в этот момент ВЭЖХ указала на завершение реакции. Катализатор отфильтровали через Solka-Floc®, промывали 1:1 MeOH/EtOAc, и фильтрат концентрировали при пониженном давлении с получением 1,67 г (99%) соединения, указанного в заголовке, в виде белого липкого твердого вещества. MS (ESI+) для C32H42N4O7 m/z 595,5 (M+H)+.
Стадия 6. Получение трифторацетата (2S,3R)-3-[(2-аминопиридин-4-ил)метил]-1-{[(1R)-1-циклогексилэтил]карбамоил}-4-оксоазетидин-2-карбоновой кислоты
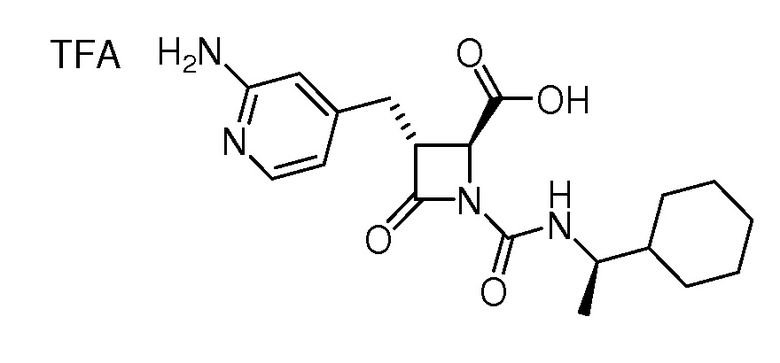
Перемешанный раствор (2S,3R)-3-({2-[(трет-бутоксикарбонил)(4-метоксибензил)амино]пиридин-4-ил}метил)-1-{[(1R)-1-циклогексилэтил]карбамоил}-4-оксоазетидин-2-карбоновой кислоты (78,0 мг, 0,131 ммоль) в DCM (2,1 мл) в азоте охлаждали на бане «лед-вода» и обрабатывали каплями TFA (0,7 мл, 9 ммоль). Полученную смесь перемешивали при 0-5°С в течение 30 мин, а затем нагревали до комнатной температуры и перемешивали в течение ночи, и в этот момент ВЭЖХ указала на завершение реакции. Смесь концентрировали при пониженном давлении, и остаток очищали препаративной ВЭЖХ на системе CombiFlash Rf [30 г C18 Gold колонка; элюция градиентом от 10% ацетонитрил (0,07% TFA)/вода (0,1% TFA) до 100% ацетонитрил (0,07% TFA)]. Лиофилизация фракций продукта давала 45 мг (70%) соединения, указанного в заголовке, в виде белого аморфного твердого продукта. 1H ЯМР (400 МГц, МeОD) δ 7,79 (д, J=6,4 Гц, 1H), 6,98 (c, 1H), 6,91 (д, J=6,8 Гц, 1H), 6,64 (ушир.д, J=9,2 Гц, 1H), 4,27 (д, J=2,8 Гц, 1H), 3,70 (м, 2H), 3,24 (м, 2H), 1,85-1,65 (м, 5H), 1,42 (м, 1H), 1,36-1,15 (м, 3H), 1,17 (д, J=6,8 Гц, 3H), 1,15-0,95 (м, 2H); MS (ESI+) для C19H26N4O4 (исходный) m/z 375,3 (M+H)+; Время удержания ВЭЖХ: 3,21 мин (Способ A).
Пример 4, Схема 1: трифторацетат (2S,3R)-3-[(2-аминопиридин-4-ил)метил]-1-{[(1R)-1-(2,2-дифтор-1,3-бензодиоксол-5-ил)этил]карбамоил}-4-оксоазетидин-2-карбоновой кислоты
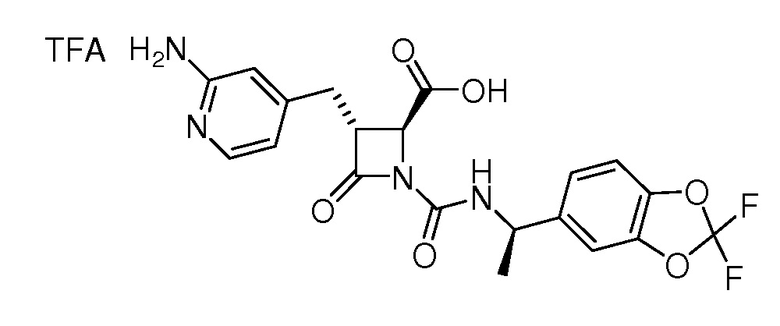
Стадия 1. Получение (S)-N-[(1E)-(2,2-дифтор-1,3-бензодиоксол-5-ил)метилene]-2-метилпропан-2-сульфинамида

К раствору амида (S)-2-метил-пропан-2-сульфиновой кислоты (0,207 г, 1,71 ммоль) в DCM (5 мл) добавляли сульфат меди (II) (0,8186 г, 5,129 ммоль), а затем раствор 2,2-дифтор-1,3-бензодиоксол-5-карбальдегида (0,350 г, 1,88 ммоль) в DCM (5 мл). Смесь перемешивали при температуре окружающей среды в течение 16 ч, фильтровали через целит и концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией (гексан и этилацетат (2-8%) в качестве элюента) с получением соединения, указанного в заголовке, (0,468 г, 95%) в виде масла. MS (ESI+) для C13H17F2NO3S m/z 290,1 (M+H)+; Время удержания ВЭЖХ: 4,73 мин (Способ C).
Стадия 2. Получение N-[(1R)-1-(2,2-дифтор-1,3-бензодиоксол-5-ил)этил]-2-метилпропан-2-сульфинамида

К охлажденному (-20°C) раствору (S)-N-[(1E)-(2,2-дифтор-1,3-бензодиоксол-5-ил)метилен]-2-метилпропан-2-сульфинамида (0,274 г, 0,947 ммоль) в THF (9 мл) добавляли 3 M метилмагния бромид (3 M в этиловой эфире, 3,157 мл, 9,471 ммоль) в течение 15 мин со скоростью, достаточно медленной для поддержания реакционной температуры ниже -15°C. Через 60 мин при -20°C, реакцию гасили дополнительным количеством насыщенного водного раствора NH4Cl. Смесь разбавляли этилацетатом и промывали насыщенным раствором NaHCO3, насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали флэш-хроматографией (гексан с этилацетатом 20-30% в качестве элюента) с получением продукта, указанного в заголовке (основной и медленно элюирирующий диастереомер, 0,234 г, 81%) в виде масла: MS (ESI+) для C13H17F2NO3S m/z 306,2 (M+H)+; Время удержания ВЭЖХ: 4,20 мин (Способ C).
Стадия 3. Получение гидрохлорида (1R)-1-(2,2-дифтор-1,3-бензодиоксол-5-ил)этанамина

К охлажденному (0-5°C) раствору N-[(1R)-1-(2,2-дифтор-1,3-бензодиоксол-5-ил)этил]-2-метилпропан-2-сульфинамида (0,234 г, 0,766 ммоль) в метаноле (4,6 мл) добавляли гидрохлорид в 1,4-диоксане (4 M, 0,958 мл, 3,83 ммоль). Через 5 мин удаляли ледяную баню, и реакционную смесь перемешивали при температуре окружающей среды в течение 1 ч. Летучие вещества удаляли в вакууме, и остаток концентрировали дважды из метанола, дважды из этилового эфира и сушили в вакууме с получением соединения, указанного в заголовке, (0,175 г, 96%) в виде белого твердого продукта: 1H ЯМР (300 МГц, CD3ОD) δ 1,67 (т, J=7 Гц, 3H), 4,55 (кв., J=7 Гц, 2H), 7,29-7,36 (м, 2H), 7,43-7,44 (м, 1H); Время удержания ВЭЖХ: 2,69 мин (Способ C).
Стадия 4. Получение фенил [(1R)-1-(2,2-дифтор-1,3-бензодиоксол-5-ил)этил]карбамата

К охлажденному (0-5°C) раствору гидрохлорида (1R)-1-(2,2-дифтор-1,3-бензодиоксол-5-ил)этанамина (100 мг, 0,421 ммоль) в метиленхлориде (2 мл) добавляли триэтиламин (0,132 мл, 0,947 ммоль), а затем по каплям фенил хлорформиат (0,054 мл, 0,433 ммоль). Через 15 мин ледяную баню удаляли, и реакционную смесь перемешивали при температуре окружающей среды в течение 3 ч. Смесь разбавляли этилацетатом и промывали 0,1 н HCl, насыщенным раствором NaHCO3, насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали флэш-хроматографией (гексан с этилацетатом 5% в качестве элюента) с получением соединения, указанного в заголовке, (140 мг, 93%) в виде белого твердого продукта: MS (ESI+) для C16H13F2NO4 m/z 322,2 (M+H)+; Время удержания ВЭЖХ: 4,75 мин (Способ C).
Стадия 5. Получение 4-метоксибензил (2S,3R)-3-({2-[(трет-бутоксикарбонил)(4-метоксибензил)амино]пиридин-4-ил}метил)-1-{[(1R)-1-(2,2-дифтор-1,3-бензодиоксол-5-ил)этил]карбамоил}-4-оксоазетидин-2-карбоксилата
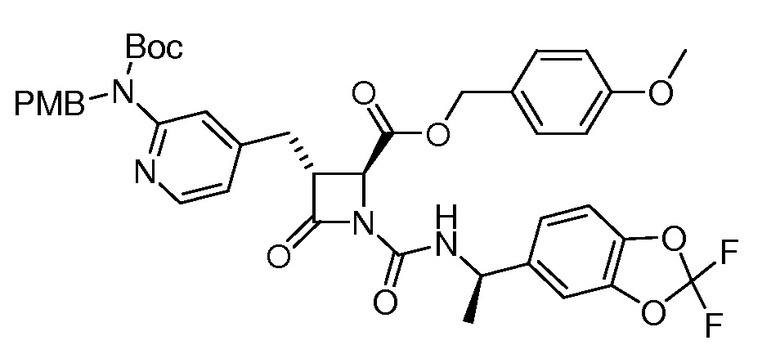
К раствору 4-метоксибензил (2S,3R)-3-({2-[(трет-бутоксикарбонил)(4-метоксибензил)амино]пиридин-4-ил}метил)-4-оксоазетидин-2-карбоксилата (получен, как описано для Примера 3, заменяя 4-метоксибензиловым спиртом, вместо бензилового спирта на стадии 2, 0,161 г, 0,287 ммоль) в диметилсульфоксиде (2 мл) добавляли фенил [(1R)-1-(2,2-дифтор-1,3-бензодиоксол-5-ил)этил]карбамат (0,120 г, 0,374 ммоль). Через 16 ч смесь разбавляли этилацетатом и промывали 0,1 н HCl, насыщенным раствором NaHCO3, насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали флэш-хроматографией (гексан с этилацетатом 5-20% в качестве элюента) с получением соединения, указанного в заголовке, (156 мг, 69%) в виде белого твердого продукта: MS (ESI+) для C41H42F2N4O10 m/z 789,5 (M+H)+; Время удержания ВЭЖХ: 5,71 мин (Способ C).
Стадия 6. Получение трифторацетата (2S,3R)-3-[(2-аминопиридин-4-ил)метил]-1-{[(1R)-1-(2,2-дифтор-1,3-бензодиоксол-5-ил)этил]карбамоил}-4-оксоазетидин-2-карбоновой кислоты
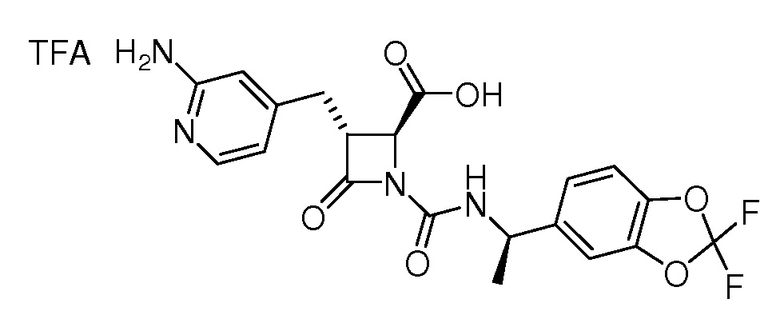
К охлажденному (0-5°C) раствору 4-метоксибензил (2S,3R)-3-({2-[(трет-бутоксикарбонил)(4-метоксибензил)амино]пиридин-4-ил}метил)-1-{[(1R)-1-(2,2-дифтор-1,3-бензодиоксол-5-ил)этил]карбамоил}-4-оксоазетидин-2-карбоксилата (0,160 г, 0,203 ммоль) в метиленхлориде (6 мл) добавляли триэтилсилан (1 мл), затем трифторуксусную кислоту (3 мл). Через 2 ч ледяную баню удаляли, и реакционную смесь перемешивали при температуре окружающей среды в течение 16 ч. Летучие вещества удаляли, и остаток очищали хроматографией CombiFlash [30 г RediSep C-18 gold силикагелевый картридж, градиент растворителя: от 10% ацетонитрил (0,07% TFA)/вода (0,1% TFA) до 100% ацетонитрил (0,07% TFA)] и лиофилизовали с получением соединения, указанного в заголовке, (80 мг, 70%) в виде белого твердого продукта: 1H ЯМР (300 МГц, CDCD3ОD) δ 1,54 (д, J=7Гц, 3H), 3,20-3,29 (м, 2H), 3,69-3,75 (м, 1H), 4,30 (д, J=3 Гц, 1H), 4,94-5,00 (м, 1H), 6,90-6,98 (м, 2H), 7,13-7,26 (м, 3H); MS (ESI+) для C20H18F2N4O6 m/z 449,2 (M+H)+; Время удержания ВЭЖХ: 3,39 мин (Способ C).
Пример 5, Схема 1: трифторацетат (2S,3R)-3-[(2-аминопиридин-4-ил)метил]-1-{[(1S)-1-циклогексил-2,2,2-трифторэтил]карбамоил}-4-оксоазетидин-2-карбоновой кислоты
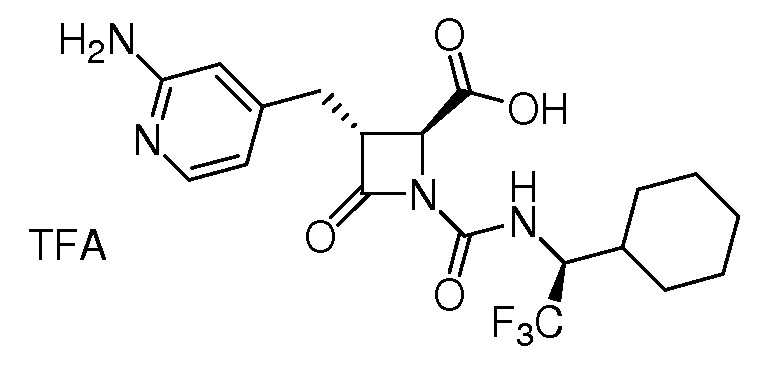
Стадия 1. Получение бензил (2S,3R)-3-({2-[(трет-бутоксикарбонил)(4-метоксибензил)амино]пиридин-4-ил}метил)-1-{[(1S)-1-циклогексил-2,2,2-трифторэтил]карбамоил}-4-оксоазетидин-2-карбоксилата
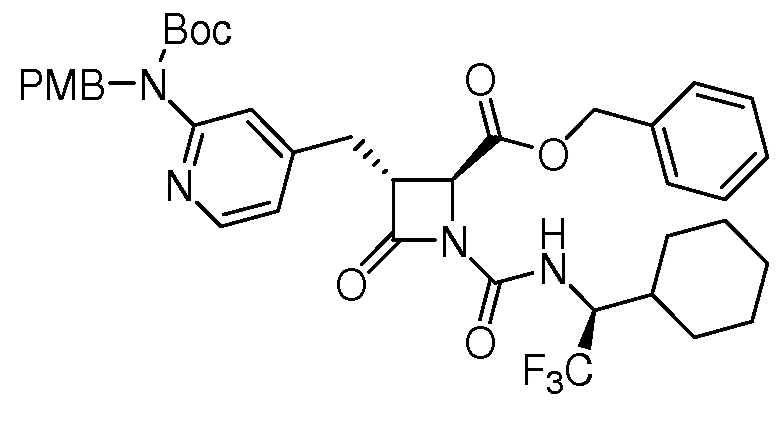
К раствору бензил (2S,3R)-3-({2-[(трет-бутоксикарбонил)(4-метоксибензил)амино]пиридин-4-ил}метил)-4-оксоазетидин-2-карбоксилата (100 мг, 0,19 ммоль, получен общими способами, описанными на Схеме 1) в диметил сульфоксиде (0,58 мл) добавляли фенил [(1S)-1-циклогексил-2,2,2-трифторэтил]карбамат (74 мг, 0,24 ммоль), а затем тритиламин (28 мкл, 0,21 моль). Смесь перемешивали при температуре окружающей среды в течение ночи, разбавляли этилацетатом, промывали водой, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Осадок очищали флэш-хроматографией, используя гексан/этилацетат (5-15%) в качестве элюента с получением соединения, указанного в заголовке (70 мг, 50%) в виде светло-коричневого твердого вещества: 1H ЯМР (CDCl3) δ 0,97-1,33 (м, 5 H), 1,42 (с, 9 H), 1,59-1,86 (м, 6 H), 3,05-3,25 (м, 2H), 3,53-3,59 (м, 1Н), 3,78 (с, 3Н), 4,31-4,39 (перекрываемый м, 2Н), 5,09-5,25 (перекрываемый м, 4Н), 6,64 (d, J=10 Гц, 1Н), 6,78-6,86 (перекрываемый м, 3Н), 7,19-7,23 (м, 4Н), 7,32-7,36 (м, 3Н), 7,65 (с, 1Н), 8,27 (d, J=5 Гц, 1 H); МС (EСI+) для C39H45F3N4O7 м/z 739,3 (М+H)+, ВЭЖХ время удержания: 5,99 мин (Способ C).
Стадия 2. Получение трифторацетата (2S,3R)-3-[(2-аминопиридин-4-ил)метил]-1-{[(1S)-1-циклогексил-2,2,2-трифторэтил]карбомоил}-4-оксоазетидин-2-карбоновой кислоты
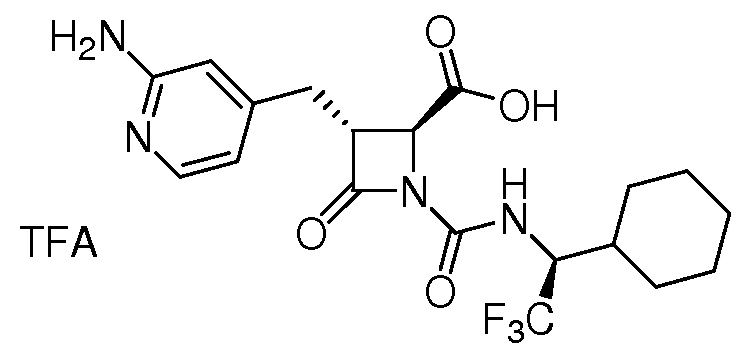
В колбу, содержащую Pd/C (10%, 10 мг) добавляли раствор бензил (2S,3R)-3-({2-[(трет-бутоксикарбонил)(4-метоксибензил)амино]пиридин-4-ил}метил)-1-{[(1S)-1-циклогексил-l-2,2,2-трифторэтил]карбамоил}-4-оксоазетидин-2-карбоксилат (70 мг, 0,095 ммоль) в этаноле (40 мл). Смесь перемешивали в 1 атмосфере H2 в течение 3 ч, фильтровали через пад из Solka-floc и концентрировали при пониженном давлении. К охлажденному (0-5°C) раствору остатка, полученной выше в метиленхлориде (3 мл) добавляли триэтилсилан (0,5 мл), а затем трифторуксусную кислоту (1,5 mL). Через 2 ч ледяную баню удаляли, и реакционную смесь перемешивали при температуре окружающей среды в течение 16 ч. Летучие вещества удаляли, и остаток очищали хроматографией CombiFlash [30 г RediSep C-18 gold силикагелевый картридж, градиент растворителя: от 10% ацетонитрил (0,07% TFA)/вода (0,1% TFA) до 100% ацетонитрил (0,07% TFA)] и лиофилизовали с получением соединения, указанного в заголовке, (80 мг, 70%) в виде белого твердого продукта: 1H ЯМР (300 МГц, CD3OD) δ 1,54 (д, J=7 Гц, 3Н), 3,20-3,29 (м, 2Н), 3,69-3,75 (м, 1Н), 4,30 (д, J=3 Гц, 1Н), 4,94-5,00 (м, 1Н), 6,90-6,98 (м, 2Н), 7,13-7,26 (м, 3Н); MS (ESI+) для C20H18F2N4O6 m/z 449,2 (M+H)+; Время удержания ВЭЖХ: 3,39 мин (Способ C).
Пример 6, Схема 1: трифторацетат (2S,3R)-1-{[(1S)-1-циклогексил-2,2,2-трифторэтил]карбамоил}-4-оксо-3-(пиридин-4-илметил)азетидин-2-карбоновой кислоты

К охлажденному (0-5°C) раствору 4-метоксибензил (2S,3R)-3-[(2-хлорпиридин-4-ил)метил]-1-{[(1S)-1-циклогексил-2,2,2-трифторэтил]карбамоил}-4-оксоазетидин-2-карбоксилата (0,190 г, 0,33 ммоль, полученного общими способами, описанными на Схеме 1) в CH2Cl2 (6 мл) добавляли трифторуксусную кислоту (3 мл). Через 5 мин ледяную баню удаляли, и реакционную смесь перемешивали при температуре окружающей среды еще 30 мин. Летучие вещества удаляли при пониженном давлении, и остаток концентрировали дважды из эфира. Остаток растворяли в этаноле (20 мл) и добавляли к колбе, содержащей PdPd/C (10%, 20 мг). Добавляли триэтиламин (0,102 мл, 0,74 ммоль), и смесь перемешивали при 1 атмосфере Н2 в течение 30 мин. Смесь фильтровали через целит и концентрировали при пониженном давлении. Остаток очищали CombiFlash хроматографией [30 г RediSep C-18 gold силикагелевый картридж, градиент растворителя: от 10% ацетонитрил (0,07% TFA)/вода (0,1% TFA) до 100% ацетонитрил (0,07% TFA)] и лиофилизовали с получением соединения, указанного в заголовке, (39 мг, 22%) в виде белого твердого продукта. 1H ЯМР (CD3OD) δ 1,07-1,39 (м, 5Н), 1,68-1,93 (м, 6Н), 3,46-3,61 (м, 2Н), 3,89-3,95 (м, 1Н), 4,36-4,44 (перекрываемый м, 2Н), 8,02 (д, J=7 Гц, 2Н), 8,76 (д, J=7 Гц, 2Н); MS (ESI+) для C19H22F3N3O4 m/z 414,0 (M+H)+; Время удержания ВЭЖХ: 3,45 мин (Способ C).
Пример 7, Схема 2: трифторацетат (2S,3R)-3-[(2-аминопиридин-4-ил)метил]-2-циано-4-оксо-N-[(1R)-1-фенилэтил]азетидин-1-карбоксамида

Стадия 1. Получение (2S,3R)-3-({2-[(трет-бутоксикарбонил)(4-метоксибензил)амино]пиридин-4-ил}метил)-1-[трет-бутил(диметил)силил]-4-оксоазетидин-2-карбоновой кислоты
Соединение, указанное в заголовке, получали из бензил (2S,3R)-3-({2-[(трет-бутоксикарбонил)(4-метоксибензил)амино]пиридин-4-ил}метил)-1-[трет-бутил(диметил)силил]-4-оксоазетидин-2-карбоксилата в соответствии с общей процедурой, описанной в стадии 5 Примера 3 с выходом 98% в виде белого липкого твердого вещества. MS (ESI+) для C29H41N3O6Si m/z 556,5 (M+H)+.
Стадия 2. Получение трет-бутил (4-{[(2S,3R)-2-карбамоил-4-оксоазетидин-3-ил]метил}пиридин-2-ил)(4-метоксибензил)карбамата

Перемешанный раствор (2S,3R)-3-({2-[(трет-бутоксикарбонил)(4-метоксибензил)амино]пиридин-4-ил}метил)-1-[трет-бутил(диметил)силил]-4-оксоазетидин-2-карбоновой кислоты (Стадия 1, 417 мг, 0,750 ммоль) в сухом ДМФ (3,5 мл) в азоте обрабатывали последовательно пиридином (38 мкл, 0,47 ммоль), ди-трет-бутилдикарбонатом (205 мг, 0,938 ммоль) и бикарбонатом аммония (71,2 мг, 0,900 ммоль), и полученную светло-коричневую смесь перемешивали при комнатной температуре в течение ночи. ВЭЖХ в 24 ч указала на завершение реакции, таким образом, смесь разбавляли водой (30 мл) и экстрагировали DCM (3×30 мл). Объединенную органическую фазу промывали насыщенным солевым раствором (15 мл), сушили над Na2SO4 и концентрировали при пониженном давлении. Очистка радиальной хроматографией [2000 микрон силикагелевый ротор; 5-10% MeOH/DCM элюент] давала 225 мг (68%) соединения, указанного в заголовке, в виде стекловидного твердого продукта. MS (ESI+) для C23H28N4O5 m/z 441,4 (M+H)+.
Стадия 3. Получение трет-бутил (4-{[(2S,3R)-2-циано-4-оксоазетидин-3-ил]метил}пиридин-2-ил)(4-метоксибензил)карбамата
Перемешанный раствор трет-бутил (4-{[(2S,3R)-2-карбамоил-4-оксоазетидин-3-ил]метил}пиридин-2-ил)(4-метоксибензил)карбамата (Стадия 2, 75,0 мг, 0,170 ммоль) и пиридина (33,0 мкл, 0,409 ммоль) в сухом THF (2 мл) в азоте охлаждали на бане «лед-насыщенный солевой раствор-MeOH» при -10°C и обрабатывали, медленно по каплям добавляя трифторуксусный ангидрид (28,8 мкл, 0,204 ммоль) ~3 мин. Полученную гомогенную смесь оставляли медленно нагреваться до 0°C в течение 25 мин, и в этот момент ВЭЖХ указала на практически полное завершение реакции. Смесь разбавляли водой (15 мл) и экстрагировали EtOAc (3×15 мл), и объединенную органическую фазу промывали насыщенным солевым раствором (10 мл), сушили над MgSO4 и концентрировали при пониженном давлении с получением 75 мг прозрачной пленки. Этот сырой продукт объединяли с другими 72 мг сырого продукта из дополнительной реакционной смеси и очищали радиальной хроматографией [2000 микрон силикагелевый ротор; 5% MeOH/DCM элюент] с получением 119 мг (86% для объединенных реакционных смесей) соединения, указанного в заголовке, в виде белого твердого продукта. MS (ESI+) для C23H26N4O4 m/z 423,4 (M+H)+.
Стадия 4. Получение трет-бутил (4-{[(2S,3R)-2-циано-4-оксо-1-{[(1R)-1-фенилэтил]карбамоил}азетидин-3-ил]метил}пиридин-2-ил)(4-метоксибензил)карбамата
Соединение, указанное в заголовке, получали из трет-бутил (4-{[(2S,3R)-2-циано-4-оксоазетидин-3-ил]метил}пиридин-2-ил)(4-метоксибензил)карбамата и [(1R)-1-изоцианатоэтил]бензола в соответствии с общей процедурой Стадии 4 Примера 3 в 52% в виде стекловидного твердого вещества. MS (ESI+) для C32H35N5O5 m/z 570,5 (M+H)+.
Стадия 5. Получение трифторацетата (2S,3R)-3-[(2-аминопиридин-4-ил)метил]-2-циано-4-оксо-N-[(1R)-1-фенилэтил]азетидин-1-карбоксамида
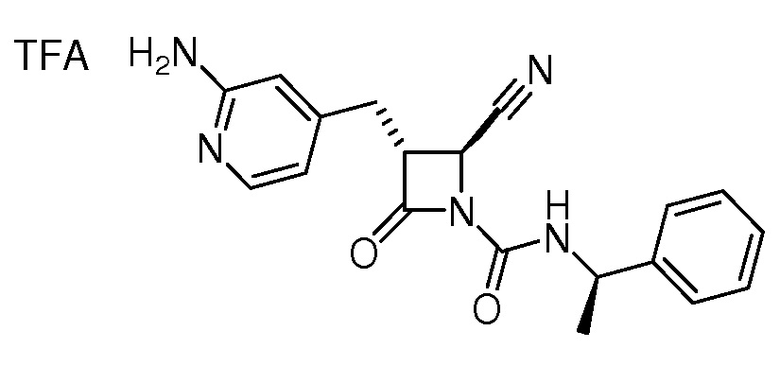
Соединение, указанное в заголовке, получали из трет-бутил (4-{[(2S,3R)-2-циано-4-оксо-1-{[(1R)-1-фенилэтил]карбамоил}азетидин-3-ил]метил}пиридин-2-ил)(4-метоксибензил)карбамата в соответствии с общей процедурой, описанной в Стадии 6 Примера 3 в 51 % в виде белого твердого продукта. 1H ЯМР (400 МГц, МeOD) δ 7,82 (д, J=6,8 Гц, 1H), 7,38 (м, 4H), 7,29 (м, 1Н), 7,09 (ушир.д, J=7,6 Гц, 1H), 6,97 (с, 1Н), 6,91 (дд, J=6,8, 1,6 Гц, 1H), 5,00 (м, 1H), 4,70 (д, J=3,2 Гц, 1H), 4,17 (ттд, J=8,0, 3,2 Гц, 1H), 3,27 (д, J=8,0 Гц, 2H), 1,55 (д, J=6,8 Гц, 3H); MS (ESI+) для C19H19N5O2 (исход.) m/z 350,3 (M+H)+; Время удержания ВЭЖХ: 2,85 мин (Способ A).
Пример 8, Схема 2: трифторацетат (2S,3R)-3-[(2-Аминопиридин-4-ил)метил]-2-циано-N-(дифенилметил)-4-оксоазетидин-1-карбоксамида
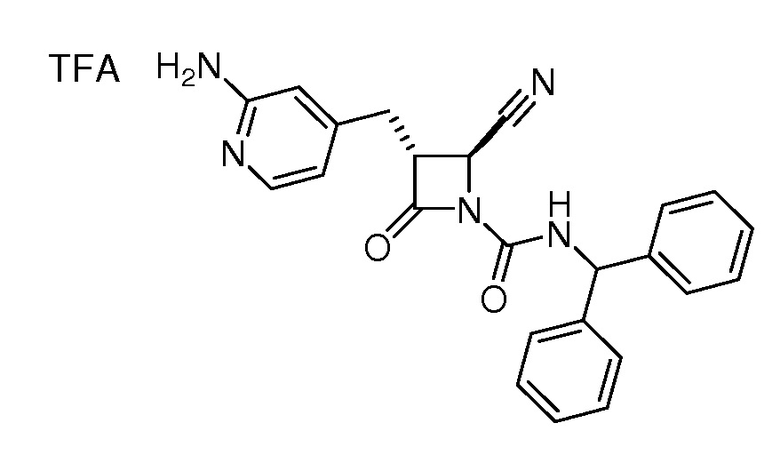
Стадия 1. Получение трет-бутил [4-({(2S,3R)-2-циано-1-[(дифенилметил)карбамоил]-4-оксоазетидин-3-ил}метил)пиридин-2-ил](4-метоксибензил)карбамата

Соединение, указанное в заголовке, получали из трет-бутил (4-{[(2S,3R)-2-циано-4-оксоазетидин-3-ил]метил}пиридин-2-ил)(4-метоксибензил)карбамат и 1,1'-(изоцианатометилен)дибензол в соответствии с общей процедурой стадии 4 Примера 3, используя 1,5 ч времени реакции в 84% в виде стекловидного твердого вещества. MS (ESI+) для C37H37N5O5 m/z 632,7 (M+H)+.
Стадия 2. Получение (2S,3R)-3-[(2-аминопиридин-4-ил)метил]-2-циано-N-(дифенилметил)-4-оксоазетидин-1-карбоксамид трифторацетат
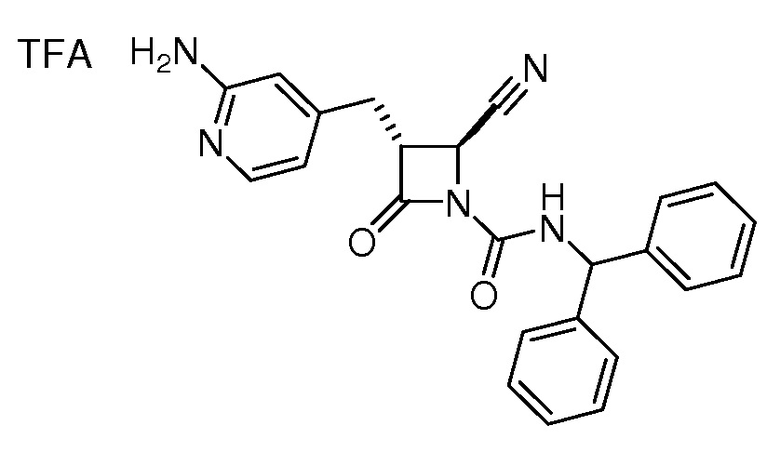
Соединение, указанное в заголовке, получали из трет-бутил [4-({(2S,3R)-2-циано-1-[(дифенилметил)карбамоил]-4-оксоазетидин-3-ил}метил)пиридин-2-ил](4-метоксибензил)карбамата в соответствии с общей процедурой стадии 6 Примера 3 в 54% в виде белого твердого продукта. 1H ЯМР (400 МГц, МeOD) δ 7,81 (д, J=6,8 Гц, 1H), 7,44 (ушир.д, J=7,6 Гц, 1H), 7,28-7,40 (м, 10Н), 6,96 (м, 1Н), 6,91 (дд, J=6,8, 1,6 Гц, 1H), 6,15 (д, J=8,0 Гц, 1H), 4,73 (д, J=3,2 Гц, 1H), 4,22 (ттд, J=8,0, 3,2 Гц, 1H), 3,27 (д, J=8,0 Гц, 2H); MS (ESI+) для C24H21N5O2 (исход.) m/z 412,4 (M+H)+; Время удержания ВЭЖХ: 3,36 мин (Способ A).
Пример 9, Схема 3: трифторацетат (2S,3R)-3-[2-(2-Амино-1,3-тиазол-5-ил)этил]-1-[(дифенилметил)карбамоил]-4-оксоазетидин-2-карбоновой кислоты
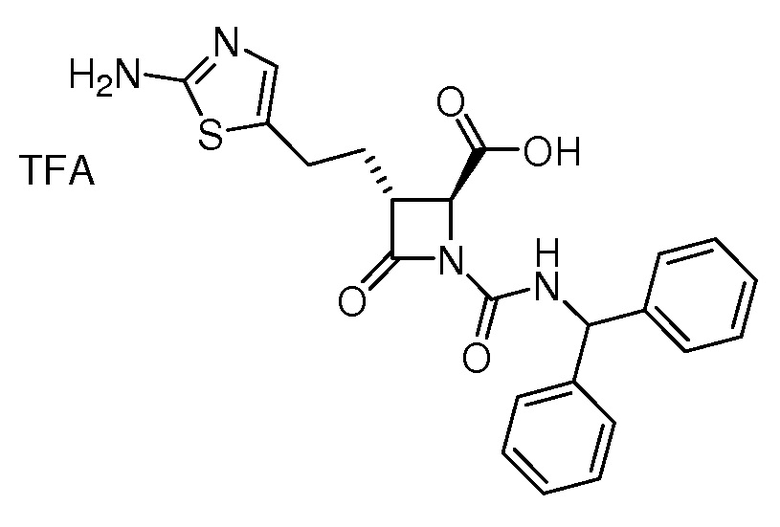
Стадия 1. Получение бензил (2S,3R)-1-[трет-бутил(диметил)силил]-4-оксо-3-пент-4-ен-1-илазетидин-2-карбоксилатаа
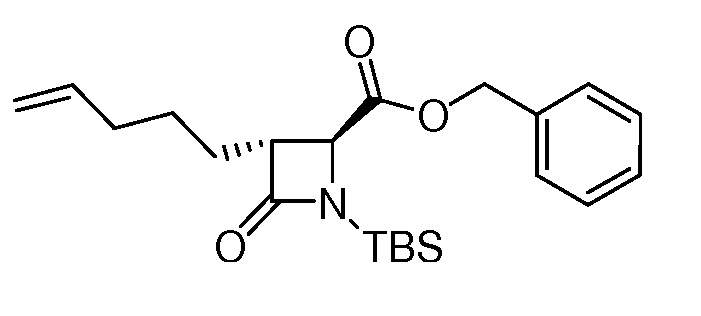
Соединение, указанное в заголовке, получали следуя общей процедуре стадий 1 и 2 Примера 3, но используя 5-бромпент-1-ен в стадии 1 и позволяя нагреваться при алкилировании до 0°C. Соединение, указанное в заголовке, получали как 29% в виде вязкого масла. MS (ESI+) для C22H33NO3Si m/z 388,3 (M+H)+.
Стадия 2. Получение бензил (2S,3R)-1-[трет-бутил(диметил)силил]-4-оксо-3-(4-оксобутил)азетидин-2-карбоксилата
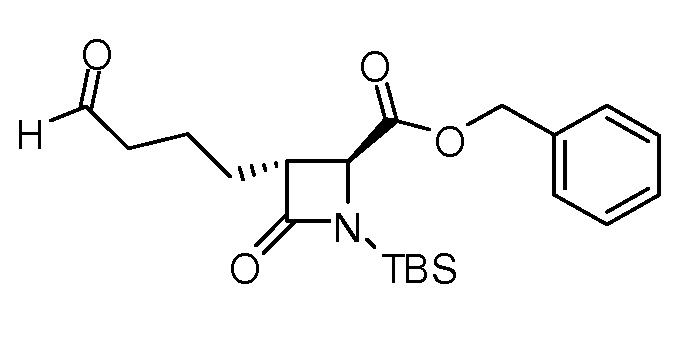
Перемешанную смесь бензил (2S,3R)-1-[трет-бутил(диметил)силил]-4-оксо-3-пент-4-ен-1-илазетидин-2-карбоксилата (412 мг, 1,06 ммоль) в 1,4-диоксане (12 мл)/вода (3 мл) обрабатывали N-метилморфолин N-оксидом (156 мг, 1,33 ммоль), а затем тетроксидом осмия (2,5 масс.% в 2-метил-2-пропаноле, 670 мкл, 0,053 ммоль), и полученную смесь перемешивали при комнатной температуре в азоте в течение 2,5 ч, и в этот момент превращение в бензил (2S,3R)-1-[трет-бутил(диметил)силил]-3-(4,5-дигидроксипентил)-4-оксоазетидин-2-карбоксилатное промежуточное соединение [MS (ESI+) для C22H35NO5Si m/z 422,2 (M+H)+] оказалось завершенным по данным ВЭЖХ. Добавляли метаперийодат натрия (284 мг, 1,33 ммоль), и полученную гетерогенную белую смесь перемешивали при комнатной температуре в течение 2,5 ч, и в этот момент ВЭЖХ указала на расход промежуточного вещества. Смесь гасили полунасыщенным водным раствором тиосульфата натрия (15 мл), разбавляли водой (15 мл) и экстрагировали EtOAc (2×40 мл). Объединенную органическую фазу промывали насыщенным солевым раствором (15 мл), сушили над MgSO4, фильтровали, концентрировали и сушили при пониженном давлении с получением 18 мг (100%) соединения, указанного в заголовке, в виде вязкого масла, которое использовали без дополнительной очистки. MS (ESI+) для C21H31NO4Si m/z 390,3 (M+H)+.
Стадия 3. Получение бензил (2S,3R)-3-[2-(2-амино-1,3-тиазол-5-ил)этил]-4-оксоазетидин-2-карбоксилата
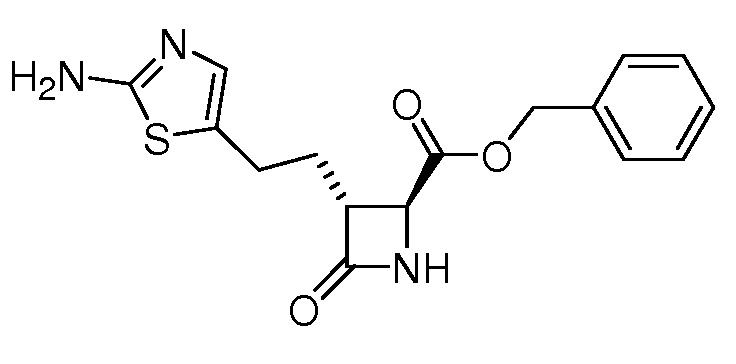
Перемешанный раствор бензил (2S,3R)-1-[трет-бутил(диметил)силил]-4-оксо-3-(4-оксобутил)азетидин-2-карбоксилата (414 мг, 1,06 ммоль) в ацетонитриле (11 мл) в азоте обрабатывали трибромидом тетрабутиламмония 512 мг, 1,06 ммоль) в одной порции, и полученную темно-желтую гомогенную смесь перемешивали при комнатной температуре в течение 75 мин, и в этот момент ВЭЖХ указала на расход исходного вещества. Смесь разбавляли водой (40 мл) и экстрагировали EtOAc (2×40 мл), и объединенную органическую фазу промывали водой (2×40 мл) и насыщенным солевым раствором (20 мл), сушили над MgSO4, фильтровали и концентрировали при пониженном давлении с получением 302 мг сырого промежуточного соединения бензил (2S,3R)-3-(3-бром-4-оксобутил)-1-[трет-бутил(диметил)силил]-4-оксоазетидин-2-карбоксилата [MS (ESI+) для C21H30BrNO4Si m/z 468 (M+H)+] в виде светло-коричневого вязкого масла. Раствор этого промежуточного соединения в EtOH (11 мл) обрабатывали тиомочевиной (89,0 мг, 1,17 ммоль), и смесь нагревали при осторожном перемешивании в течение 2 ч, охлаждали до комн. темп., разбавляли DCM (45 мл), промывали насыщенным водным раствором NaHCO3 (30 мл), водой (30 мл) и насыщенным солевым раствором (20 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Очистка хроматографией на силикагеле [40 г, 2,5-7,5% MeOH/DCM элюент] давала 60 мг (17%) соединения, указанного в заголовке, в виде светло-коричневого твердого вещества. MS (ESI+) для C16H17N3O3S m/z 332,2 (M+H)+. TBS-защищенный побочный продукт, бензил (2S,3R)-3-[2-(2-амино-1,3-тиазол-5-ил)этил]-1-[трет-бутил(диметил)силил]-4-оксоазетидин-2-карбоксилат, также выделяли (145 мг, 24%, чистота 80%). MS (ESI+) для C22H31N3O3SSi m/z 446,2 (M+H)+.
Стадия 4. Получение бензил (2S,3R)-3-(2-{2-[(трет-бутоксикарбонил)амино]-1,3-тиазол-5-ил}этил)-4-оксоазетидин-2-карбоксилата
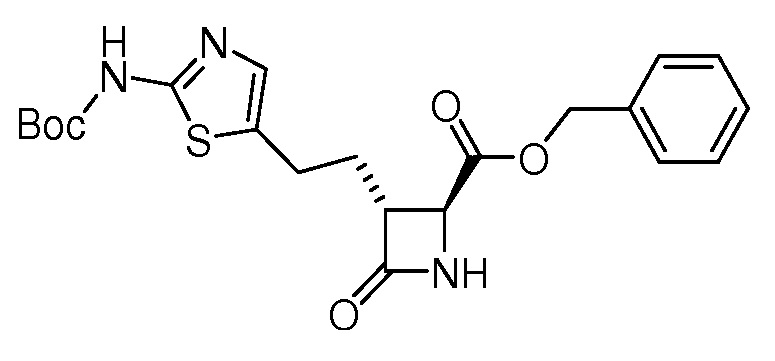
Перемешанный раствор бензил (2S,3R)-3-[2-(2-амино-1,3-тиазол-5-ил)этил]-4-оксоазетидин-2-карбоксилата (57,0 мг, 0,172 ммоль) в ацетонитриле (5 мл) в азоте обрабатывали ди-трет-бутилдикарбонатом (48,8 мг, 0,224 ммоль) и перемешивали при комнатной температуре в течение приблизительно 2 дней, в течение которых одной порцией добавляли дополнительное количество ди-трет-бутилдикарбоната (13 мг, 0,060 ммоль). Смесь концентрировали при пониженном давлении и очищали радиальной хроматографией [2000 микрон силикагелевый ротор, 2,5-10% MeOH/DCM элюент] с получением 74 мг (81%) соединения, указанного в заголовке, в виде стекловидной пленки (загрязнена бис-BOC побочным продуктом бензил (2S,3R)-3-(2-{2-[бис(трет-бутоксикарбонил)амино]-1,3-тиазол-5-ил}этил)-4-оксоазетидин-2-карбоксилат). MS (ESI+) для C21H25N3O5S m/z 432,2 (M+H)+.
Стадия 5. Получение бензил (2S,3R)-3-(2-{2-[(трет-бутоксикарбонил)амино]-1,3-тиазол-5-ил}этил)-1-[(дифенилметил)карбамоил]-4-оксоазетидин-2-карбоксилата

Соединение, указанное в заголовке, получали из (2S,3R)-3-(2-{2-[(трет-бутоксикарбонил)амино]-1,3-тиазол-5-ил}этил)-4-оксоазетидин-2-карбоксилата и 1,1'-(изоцианатометилен)дибензола, следуя общей процедуре стадии 4 Примера 3 и используя 5 ч времени реакции, в количестве 82% в виде стекловидного твердого вещества. MS (ESI+) для C35H36N4O6S m/z 641,3 (M+H)+.
Стадия 6. Получение (2S,3R)-3-(2-{2-[(трет-бутоксикарбонил)амино]-1,3-тиазол-5-ил}этил)-1-[(дифенилметил)карбамоил]-4-оксоазетидин-2-карбоновой кислоты
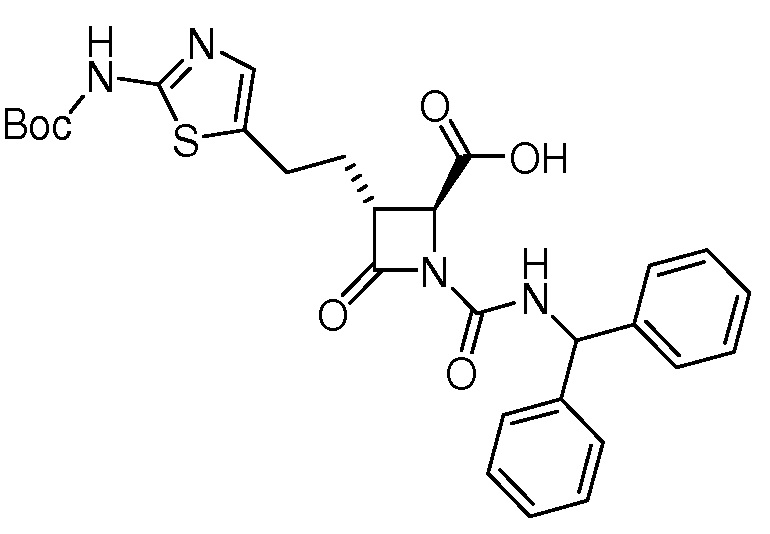
Смесь бензил (2S,3R)-3-(2-{2-[(трет-бутоксикарбонил)амино]-1,3-тиазол-5-ил}этил)-1-[(дифенилметил)карбамоил]-4-оксоазетидин-2-карбоксилата (85,0 мг, 0,133 ммоль) в 1:1 MeOH/EtOAc (6 мл) в азоте обрабатывали 10% палладием на угле (35 мг, 0,013 ммоль Pd), и смесь откачивали и наполняли два раза азотом, затем перемешивали в атмосфере водорода (двухслойный баллон) в течение приблизительно 6 ч, в течение которых одной порцией добавляли дополнительное количество 10% палладия на угле (35 мг, 0,013 ммоль Pd). Катализатор отфильтровали через Solka-Floc®, промывая 1:1 MeOH/EtOAc, и фильтрат концентрировали при пониженном давлении с получением 71 мг (97%) соединения, указанного в заголовке, в виде не совсем белой пены, которую использовали без дополнительной очистки. MS (ESI+) для C28H30N4O6S m/z 551,2 (M+H)+.
Стадия 7. Получение (2S,3R)-3-[2-(2-амино-1,3-тиазол-5-ил)этил]-1-[(дифенилметил)карбамоил]-4-оксоазетидин-2-карбоновой кислоты трифторацетат
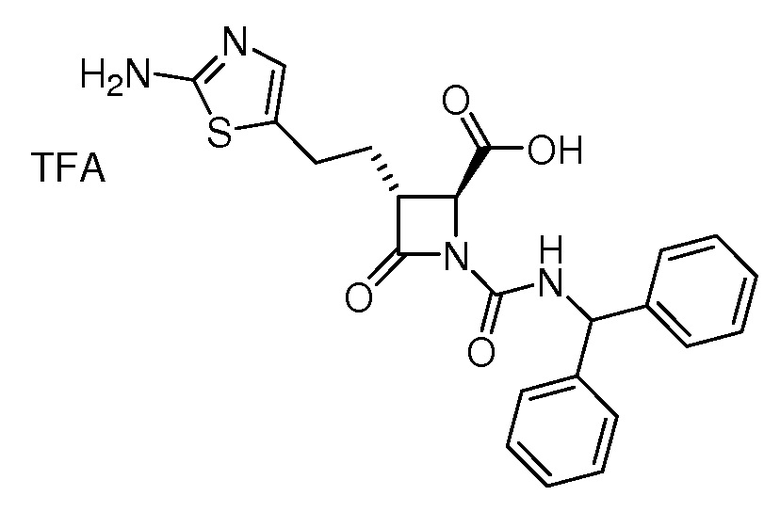
Соединение, указанное в заголовке, получали из (2S,3R)-3-(2-{2-[(трет-бутоксикарбонил)амино]-1,3-тиазол-5-ил}этил)-1-[(дифенилметил)карбамоил]-4-оксоазетидин-2-карбоновой кислоты, следуя общей процедуре стадии 6 Примера 3 и используя 5 ч на время реакции, в количестве 47% в виде белого твердого продукта. 1H ЯМР (400 МГц, МeOD) δ 7,26-7,39 (м, 10H), 6,96 (ушир.с, 1H), 6,11 (ушир.с, 1H), 4,31 (ушир.с, 1H), 3,40 (м, 1H), 2,88 (м, 2H), 2,16 (м, 2H); MS (ESI+) для C23H22N4O4S (исход.) m/z 451,0 (M+H)+; Время удержания ВЭЖХ: 3,25 мин (Способ A).
Пример 10, Схема 3: трифторацетат (2S,3R)-3-[2-(2-амино-1,3-тиазол-5-ил)этил]-4-оксо-1-{[(1R)-1-фенилэтил]карбамоил}азетидин-2-карбоновой кислоты
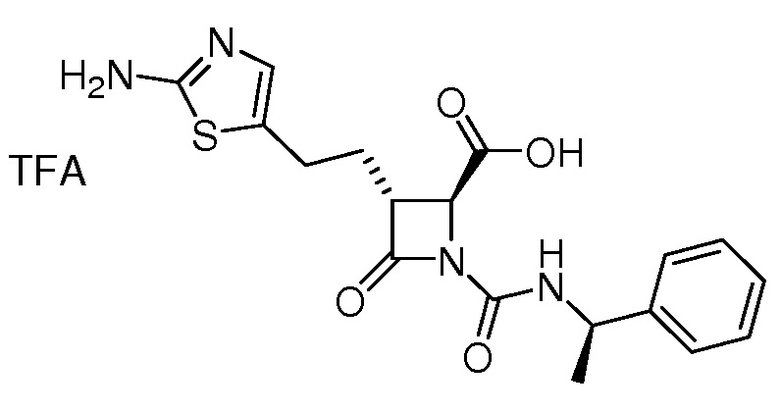
Стадия 1. Получение бензил (2S,3R)-3-(2-{2-[(трет-бутоксикарбонил)амино]-1,3-тиазол-5-ил}этил)-4-оксо-1-{[(1R)-1-фенилэтил]карбамоил}азетидин-2-карбоксилата
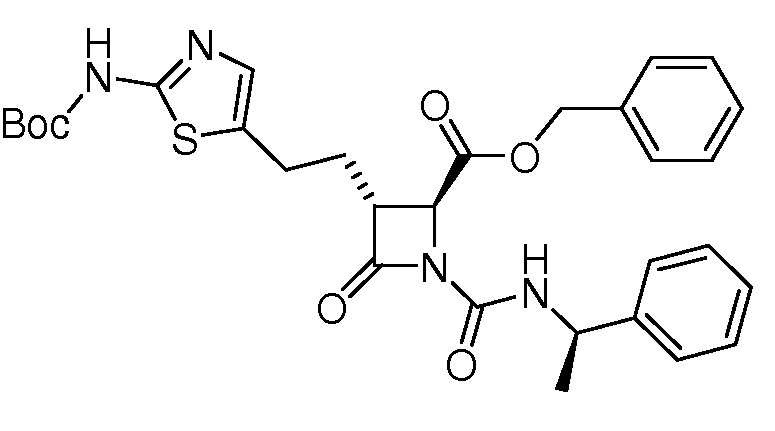
Соединение, указанное в заголовке, получали из бензил (2S,3R)-3-(2-{2-[(трет-бутоксикарбонил)амино]-1,3-тиазол-5-ил}этил)-4-оксоазетидин-2-карбоксилата и [(1R)-1-изоцианатоэтил]бензола, следуя общей процедуре стадии 4 Примера 3, в количестве 66 % в виде стекловидного твердого вещества. MS (ESI+) для C30H34N4O6S m/z 579,3 (M+H)+.
Стадия 2. Получение (2S,3R)-3-(2-{2-[(трет-бутоксикарбонил)амино]-1,3-тиазол-5-ил}этил)-4-оксо-1-{[(1R)-1-фенилэтил]карбамоил}азетидин-2-карбоновой кислоты
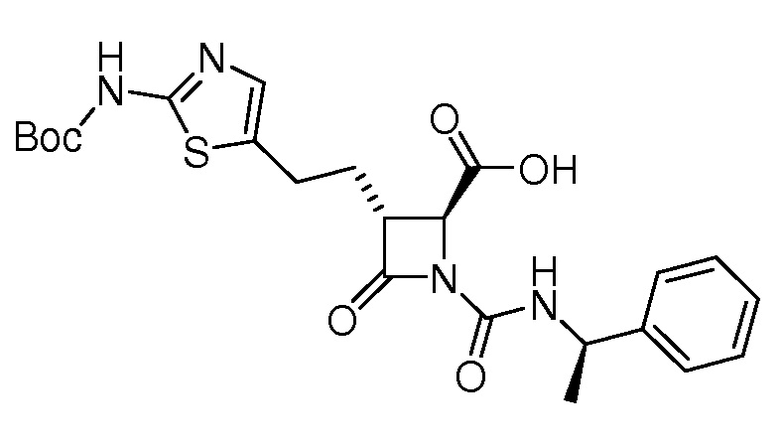
Соединение, указанное в заголовке, получали из бензил (2S,3R)-3-(2-{2-[(трет-бутоксикарбонил)амино]-1,3-тиазол-5-ил}этил)-4-оксо-1-{[(1R)-1-фенилэтил]карбамоил}азетидин-2-карбоксилата, следуя общей процедуре стадии 6 Примера 9, в количестве 82% в виде белого твердого продукта. MS (ESI+) для C23H28N4O6S m/z 489,1 (M+H)+.
Стадия 3. Получение трифторацетат (2S,3R)-3-[2-(2-амино-1,3-тиазол-5-ил)этил]-4-оксо-1-{[(1R)-1-фенилэтил]карбамоил}азетидин-2-карбоновой кислоты
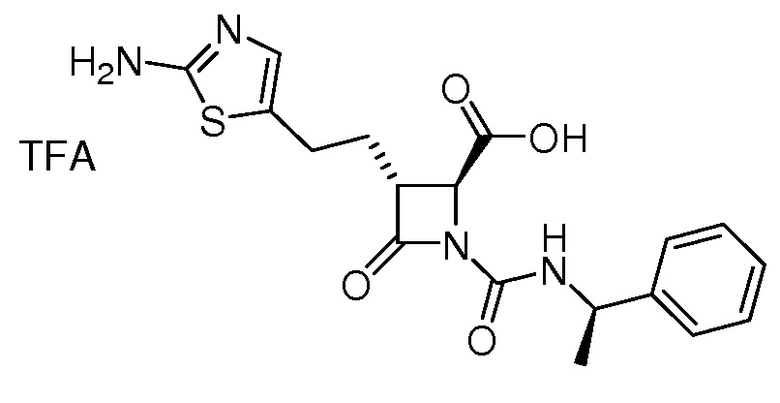
Соединение, указанное в заголовке, получали из (2S,3R)-3-(2-{2-[(трет-бутоксикарбонил)амино]-1,3-тиазол-5-ил}этил)-4-оксо-1-{[(1R)-1-фенилэтил]карбамоил}азетидин-2-карбоновой кислоты, следуя общей процедуре стадии 6 Примера 3 и используя 5 ч на время реакции, в количестве 66% в виде белого твердого продукта. 1H ЯМР (400 МГц, МeOD) δ 7,36 (м, 4H), 7,27 (м, 1H), 6,96 (ушир.с, 1H), 4,98 (q, J=6,8 Гц, 1H), 4,26 (ушир.с, 1H), 3,35 (м, 1H), 2,89 (т, J=7,2 Гц, 2H), 2,16 (м, 2H), 1,53 (д, J=6,8 Гц, 3Н); MS (ESI+) для C18H20N4O4S (исход.) m/z 389,1 (M+H)+; Время удержания ВЭЖХ: 2,71 мин (Способ A).
Пример 11, Схема 4: трифторацетат (2S,3R)-3-[(2-амино-1,3-тиазол-5-ил)метил]-1-[(дифенилметил)карбамоил]-4-оксоазетидин-2-карбоновой кислоты
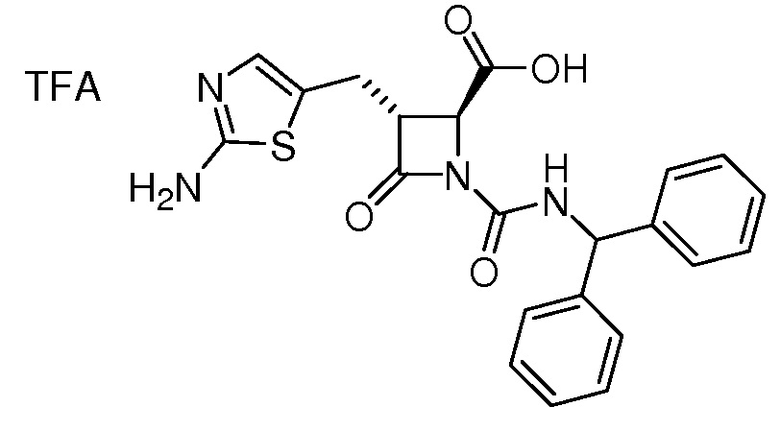
Стадия 1. Получение бензил (2S,3R)-1-[трет-бутил(диметил)силил]-3-[(2E)-3-хлорпроп-2-ен-1-ил]-4-оксоазетидин-2-карбоксилата
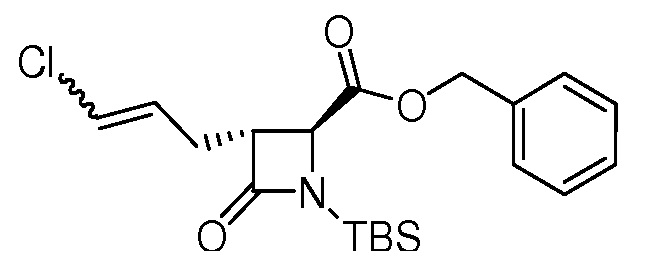
Соединение, указанное в заголовке, получали, следуя общей процедуре стадий 1-2 Примера 3, но используя (1E)-1,3-дихлорпроп-1-ен вместо трет-бутил [4-(бромметил)пиридин-2-ил](4-метоксибензил)карбамата в стадии 1 и позволяя температуре реакции подняться до 0°C. Соединение, указанное в заголовке, получали в количестве 39% (смесь E/Z изомеров) в виде вязкого масла. MS (ESI+) для C20H28ClNO3Si m/z 394,2 (M+H)+.
Стадия 2. Получение бензил (2S,3R)-3-[(2-амино-1,3-тиазол-5-ил)метил]-1-[трет-бутил(диметил)силил]-4-оксоазетидин-2-карбоксилата
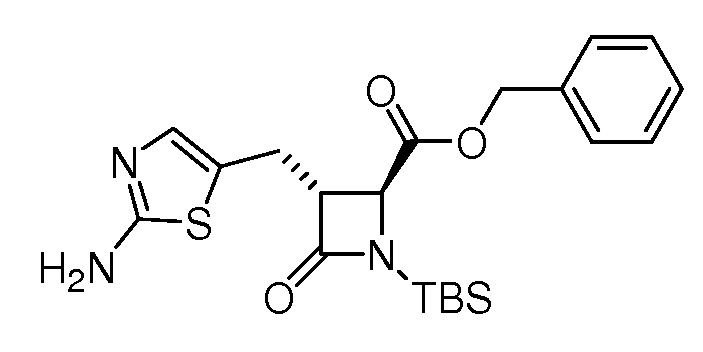
Перемешанный раствор бензил (2S,3R)-1-[трет-бутил(диметил)силил]-3-[(2E)-3-хлорprop-2-en-1-ил]-4-оксоазетидин-2-карбоксилата (220 мг, 0,558 ммоль) в сухом 1,2-дихлорэтане (2,8 мл) в сосуде с завинчивающейся крышкой, обрабатывали м-хлорпербензойной кислотой (289 мг, 1,68 ммоль, промывали и сушили, чистота >90%) и 2,6-ди-трет-бутил-4-метилфенол (7,0 мг, 0,032 ммоль), быстро нагревали до 60°C и перемешивали при этой температуре в течение 8 ч. Смесь охлаждали до комн. темп., разбавляли EtOAc (20 мл), промывали насыщенным водным раствором NaHCO3 (2×20 мл), полунасыщенным раствором водного раствора тиосульфата натрия (2×15 мл), водой (20 мл) и насыщенным солевым раствором (10 мл), сушили над MgSO4 и концентрировали при пониженном давлении с получением сырого промежуточного продукта бензил (2S,3R)-1-[трет-бутил(диметил)силил]-3-[(3-хлороксиран-2-ил)метил]-4-оксоазетидин-2-карбоксилата [MS (ESI+) для C20H28ClNO4Si m/z 410,2 (M+H)+] в виде вязкого масла, которое использовали без дополнительной очистки. Перемешанную смесь этого промежуточного вещества в сухом 1,2-диметоксиэтане (5,5 мл) в азоте обрабатывали тиомочевиной (53,1 мг, 0,698 ммоль), нагревали до 60°C и перемешивали при этой температуре в течение 2,5 ч, и в этот момент ВЭЖХ и LC-MS указала на расход промежуточного соединения. Смесь охлаждали до комнатной температуры, разбавляли EtOAc (30 мл), водой (8 мл) и насыщенным водным раствором NaHCO3 (8 мл), и слои разделяли. Органическую фазу промывали насыщенным водным раствором NaHCO3 (15 мл), водой (15 мл) и насыщенным солевым раствором (10 мл), сушили над MgSO4 и концентрировали при пониженном давлении с получением 240 мг сырого продукта, который объединяли с 100 мг сырого продукта из предыдущей реакции. Очистка радиальной хроматографией [2000 микрон силикагелевый ротор; 5-10% MeOH/DCM элюент] давала 172 мг (49% для объединенных реакций) соединения, указанного в заголовке, в виде вязкого масла. MS (ESI+) для C21H29N3O3SSi m/z 432,3 (M+H)+.
Стадия 3. Получение бензил (2S,3R)-3-({2-[(трет-бутоксикарбонил)амино]-1,3-тиазол-5-ил}метил)-1-[трет-бутил(диметил)силил]-4-оксоазетидин-2-карбоксилатаа
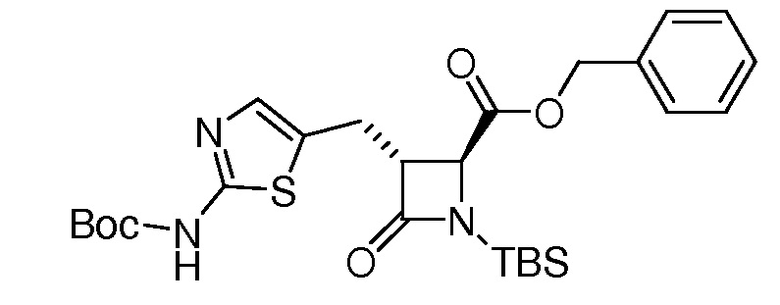
Перемешанный раствор бензил (2S,3R)-3-[(2-амино-1,3-тиазол-5-ил)метил]-1-[трет-бутил(диметил)силил]-4-оксоазетидин-2-карбоксилата (168 мг, 0,389 ммоль) в ацетонитриле (6 мл) в азоте обрабатывали ди-трет-бутилдикарбонатом (170 мг, 0,778 ммоль), и полученную смесь перемешивали при комнатной температуре в течение приблизит. 24 ч, и в этот момент ВЭЖХ указала на завершение реакции. Смесь концентрировали при пониженном давлении и очищал радиальной хроматографией [2000 микрон силикагелевый ротор; 30-50% EtOAc/гексан элюент] с получением 242 мг (100%) соединения, указанного в заголовке, в виде масляной пленки (загрязнена бис-BOC побочным продуктом бензил (2S,3R)-3-({2-[бис(трет-бутоксикарбонил)амино]-1,3-тиазол-5-ил}метил)-1-[трет-бутил(диметил)силил]-4-оксоазетидин-2-карбоксилат). MS (ESI+) для C26H37N3O5SSi m/z 532,3 (M+H)+.
Стадия 4. Получение бензил (2S,3R)-3-({2-[(трет-бутоксикарбонил)амино]-1,3-тиазол-5-ил}метил)-4-оксоазетидин-2-карбоксилата
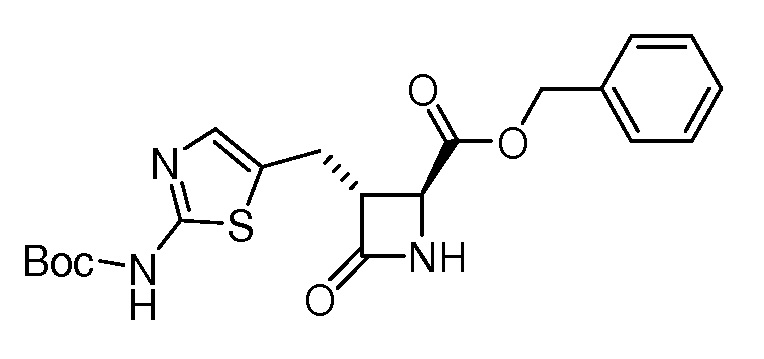
Соединение, указанное в заголовке, получали из бензил (2S,3R)-3-({2-[(трет-бутоксикарбонил)амино]-1,3-тиазол-5-ил}метил)-1-[трет-бутил(диметил)силил]-4-оксоазетидин-2-карбоксилата, следуя общей процедуре стадии 3 Примера 3, в количестве 99% (загрязнен бис-BOC побочным продуктом бензил (2S,3R)-3-({2-[бис(трет-бутоксикарбонил)амино]-1,3-тиазол-5-ил}метил)-4-оксоазетидин-2-карбоксилат). MS (ESI+) для C20H23N3O5S m/z 418,2 (M+H)+.
Стадия 5. Получение бензил (2S,3R)-3-({2-[(трет-бутоксикарбонил)амино]-1,3-тиазол-5-ил}метил)-1-[(дифенилметил)карбамоил]-4-оксоазетидин-2-карбоксилата
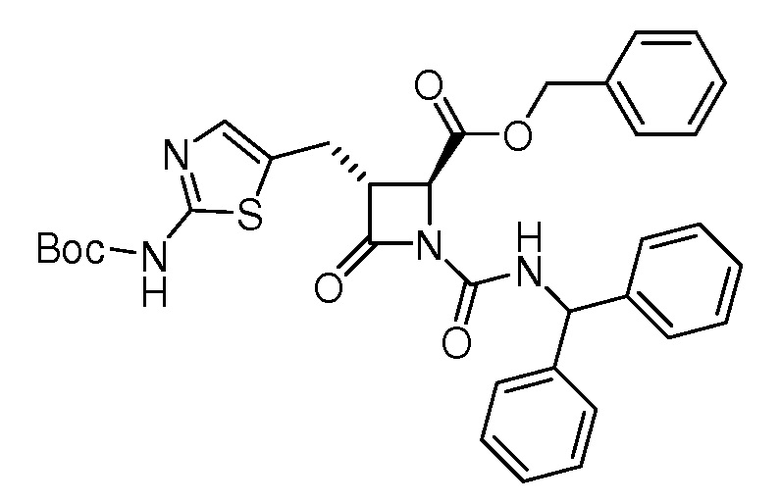
Соединение, указанное в заголовке, получали из бензил (2S,3R)-3-({2-[(трет-бутоксикарбонил)амино]-1,3-тиазол-5-ил}метил)-4-оксоазетидин-2-карбоксилата и 1,1'-(изоцианатометилен)дибензола, следуя общей процедуре стадии 4 Примера 3, используя 3 ч на время реакции в количестве 88% в виде стекловидного твердого вещества. MS (ESI+) для C34H34N4O6S m/z 627,4 (M+H)+.
Стадия 6. Получение (2S,3R)-3-({2-[(трет-бутоксикарбонил)амино]-1,3-тиазол-5-ил}метил)-1-[(дифенилметил)карбамоил]-4-оксоазетидин-2-карбоновой кислоты
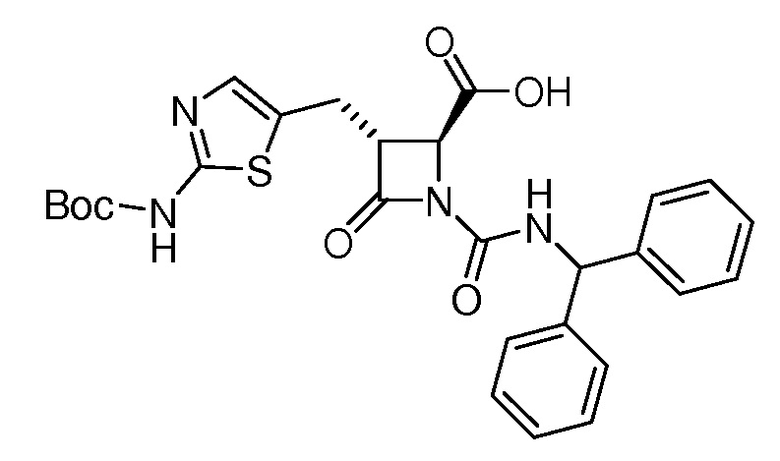
Соединение, указанное в заголовке, получали из бензил (2S,3R)-3-({2-[(трет-бутоксикарбонил)амино]-1,3-тиазол-5-ил}метил)-1-[(дифенилметил)карбамоил]-4-оксоазетидин-2-карбоксилата, следуя общей процедуре стадии 6 Примера 9, с количественным выходом в виде белого твердого продукта. MS (ESI+) для C27H28N4O6S m/z 537,3 (M+H)+.
Стадия 7. Получение трифторацетата (2S,3R)-3-[(2-амино-1,3-тиазол-5-ил)метил]-1-[(дифенилметил)карбамоил]-4-оксоазетидин-2-карбоновой кислоты
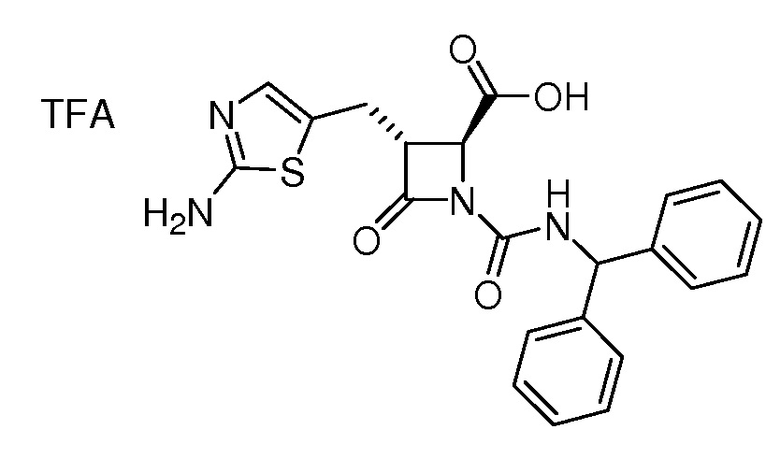
Соединение, указанное в заголовке, получали из (2S,3R)-3-({2-[(трет-бутоксикарбонил)амино]-1,3-тиазол-5-ил}метил)-1-[(дифенилметил)карбамоил]-4-оксоазетидин-2-карбоновой кислоты, следуя общей процедуре стадии 6 Примера 3 и используя 3 ч на время реакции в количестве 53% в виде белого твердого продукта. 1H ЯМР (400 МГц, МeOD) δ 7,49 (д, J=8,4 Гц, 1H), 7,40-7,27 (м, 10Н), 7,14 (с, 1Н), 6,12 (м, 1Н), 4,34 (д, J=2,8 Гц, 1H), 3,69 (ттд, J=7,2, 2,8 Гц, 1H), 3,25 (м, 2Н); MS (ESI+) для C22H20N4O4S (исход.) m/z 437,1 (M+H)+; Время удержания ВЭЖХ: 3,11 мин (Способ A).
Пример 12, Схема 4: трифторацетат (2S,3R)-3-[(2-амино-1,3-тиазол-5-ил)метил]-4-оксо-1-{[(1R)-1-фенилэтил]карбамоил}азетидин-2-карбоновой кислоты
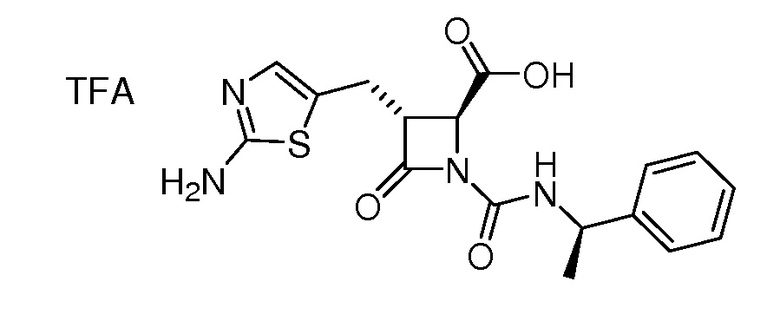
Стадия 1. Получение бензил (2S,3R)-3-({2-[(трет-бутоксикарбонил)амино]-1,3-тиазол-5-ил}метил)-4-оксо-1-{[(1R)-1-фенилэтил]карбамоил}азетидин-2-карбоксилата
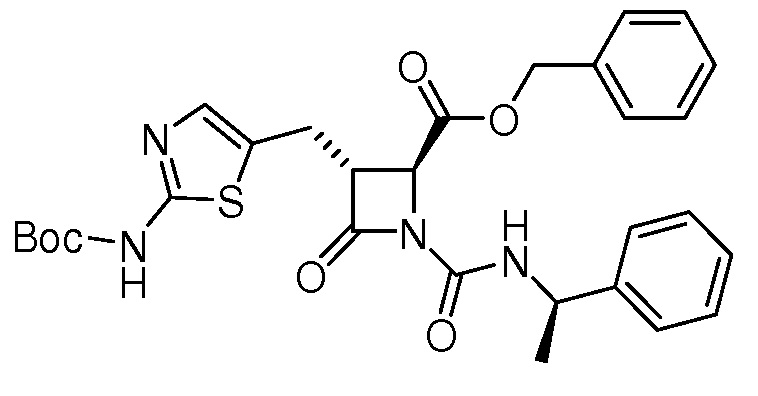
Соединение, указанное в заголовке, получали из бензил (2S,3R)-3-({2-[(трет-бутоксикарбонил)амино]-1,3-тиазол-5-ил}метил)-4-оксоазетидин-2-карбоксилата и [(1R)-1-изоцианатоэтил]бензола, следуя общей процедуре стадии 4 Примера 3, в количестве приблизительно 95% в виде стекловидного твердого вещества. MS (ESI+) для C29H32N4O6S m/z 565,2 (M+H)+.
Стадия 2. Получение (2S,3R)-3-({2-[(трет-бутоксикарбонил)амино]-1,3-тиазол-5-ил}метил)-4-оксо-1-{[(1R)-1-фенилэтил]карбамоил}азетидин-2-карбоновой кислоты
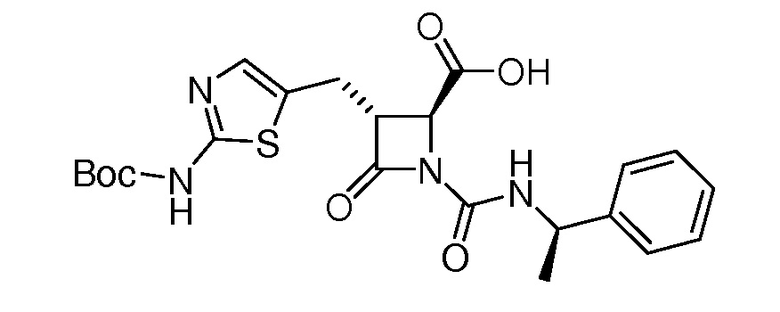
Соединение, указанное в заголовке, получали из бензил (2S,3R)-3-({2-[(трет-бутоксикарбонил)амино]-1,3-тиазол-5-ил}метил)-4-оксо-1-{[(1R)-1-фенилэтил]карбамоил}азетидин-2-карбоксилата, следуя общей процедуре стадии 6 Примера 9, в количестве 75% в виде белого твердого продукта. MS (ESI+) для C22H26N4O6S m/z 475,2 (M+H)+.
Стадия 3. Получение (2S,3R)-3-[(2-амино-1,3-тиазол-5-ил)метил]-4-оксо-1-{[(1R)-1-фенилэтил]карбамоил}азетидин-2-карбоновой кислоты трифторацетат
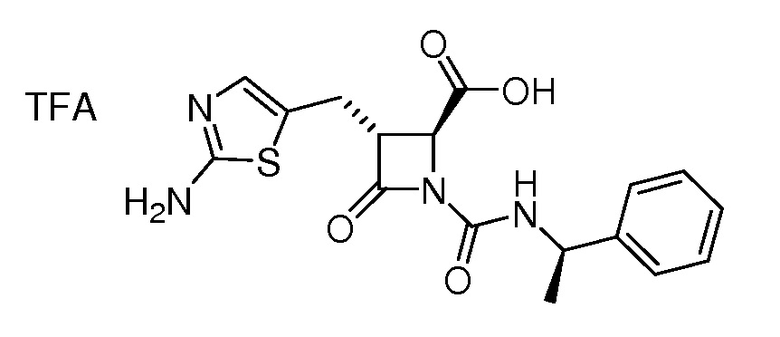
Соединение, указанное в заголовке, получали из (2S,3R)-3-({2-[(трет-бутоксикарбонил)амино]-1,3-тиазол-5-ил}метил)-4-оксо-1-{[(1R)-1-фенилэтил]карбамоил}азетидин-2-карбоновой кислоты, следуя общей процедуре стадии 6 Примера 3, используя 5 ч на время реакции, в количестве 54% в виде белого твердого продукта. 1H ЯМР (400 МГц, МeOD) δ 7,37 (м, 4H), 7,28 (м, 1H), 7,16 (с, 1H), 4,98 (q, J=6,8 Гц, 1H), 4,32 (д, J=2,8 Гц, 1H) 3,65 (тд, J=7,2, 2,8 Гц, 1H), 3,25 (м, 2H), 1,54 (д, J=7,2Н, 3H); MS (ESI+) для C17H18N4O4S (исход.) m/z 375,3 (M+H)+; Время удержания ВЭЖХ: 2,58 мин (Способ A).
Пример 13, Схема 5: трифторацетат (2S,3R)-3-[(2-аминопиридин-4-ил)метил]-4-оксо-N-1-[(1R)-1-фенилэтил]азетидин-1,2-дикарбоксамида
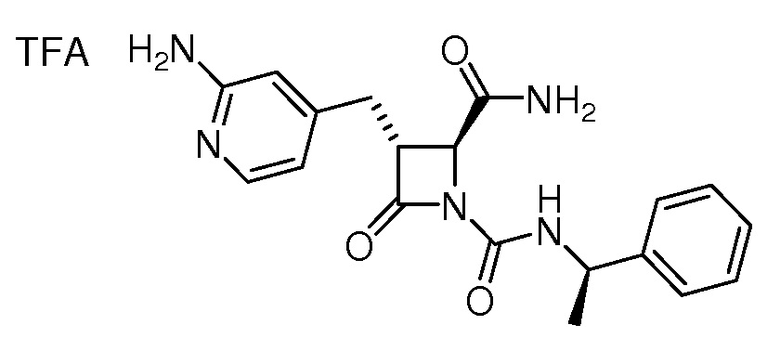
Стадия 1. Получение ди-трет-бутил [4-({(3R,4S)-1-[трет-бутил(диметил)силил]-2-оксо-4-[(2,4,6-триметоксибензил)карбамоил]азетидин-3-ил}метил)пиридин-2-ил]имидодикарбоната
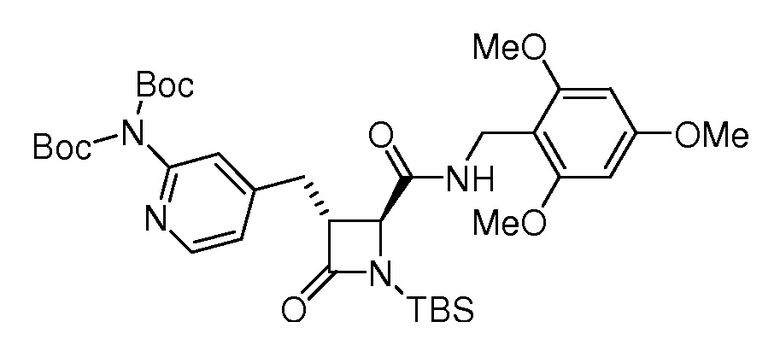
Раствор(2S,3R)-3-({2-[бис(трет-бутоксикарбонил)амино]пиридин-4-ил}метил)-1-[трет-бутил(диметил)силил]-4-оксоазетидин-2-карбоновой кислоты (170 мг, 0,32 ммоль) в N,N-диметилформамиде (2,0 мл) обрабатывали гидрохлоридом (2,4,6-триетоксифенил)метанамина (81,6 мг, 0,349 ммоль) и гексафторфосфатом N,N,N',N'-тетраметил-O-(7-азабензотриазол-1-ил)урония (145 мг, 0,381 ммоль), а затем по каплям добавляли N,N-диизопропилэтиламин (182 мкл, 1,05 ммоль), и реакционную смесь перемешивали при 0°C. Анализ ВЭЖХ через 15 мин указал на расход исходного вещества. Реакционную смесь разбавляли 25 мл Н2O, экстрагировали двумя порциями по 25 мл CH2Cl2, и объединенную органическую фазу промывали порциями по 20 мл Н2O и насыщенным солевым раствором. Органическую фазу сушили над Na2SO4, фильтровали и концентрировали с выходом слегка окрашенной в светло-коричневый цвет жидкости, которую помещали в высокий вакуум для удаления ДМФ. Сырое вещество очищали флэш-хроматографией (60 г силикагель, 15-50% EtOAc/CH2Cl2) с выходом соединения, указанного в заголовке, (130 мг, 60%) в виде слегка окрашенного в желтый цвет стекла: Время удержания ВЭЖХ: 5,05 мин (Способ A); MS (ESI+) для C36H54N4O9Si m/z 715,5 (M+H)+.
Стадия 2. Получение ди-трет-бутил [4-({(3R,4S)-2-оксо-4-[(2,4,6-триметоксибензил)карбамоил]азетидин-3-ил}метил)пиридин-2-ил]имидодикарбоната
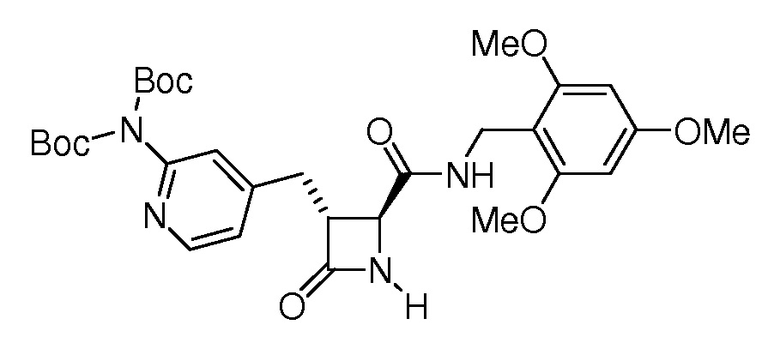
Раствор ди-трет-бутил [4-({(3R,4S)-1-[трет-бутил(диметил)силил]-2-оксо-4-[(2,4,6-триметоксибензил)карбамоил]азетидин-3-ил}метил)пиридин-2-ил]имидоикарбоната (137 мг, 0,192 ммоль) в метаноле (3,3 мл) обрабатывали каплями уксусной кислоты (38 мкл, 0,67 ммоль), а затем 0,5M фторида аммония в метаноле (0,46 мл, 0,23 ммоль), и реакционную смесь перемешивали при комнатной температуре. ВЭЖХ через 1,5 часов указала на завершение реакции. Реакционную смесь концентрировали, остаток разбавляли 7 мл толуола и концентрировали. Остаток помещали в 20 мл CH2Cl2 и промывали порциями по 10 мл Н2O и насыщенным раствором NaHCO3. Органическую фазу сушили над Na2SO4, фильтровали и концентрировали с получением соединения, указанного в заголовке, (70 мг, 95%) в виде светло-желтой крепкой пены: Время удержания ВЭЖХ, 3,84 мин (Способ A); MS (ESI+) для C30H40N4O9 m/z 601,3 (M+H)+.
Стадия 3. Получение ди-трет-бутил [4-({(3R,4S)-2-оксо-1-{[(1R)-1-фенилэтил]карбамоил}-4-[(2,4,6-триметоксибензил)карбамоил]азетидин-3-ил}метил)пиридин-2-ил]имидодикарбоната

Раствор ди-трет-бутил [4-({(3R,4S)-2-оксо-4-[(2,4,6-триметоксибензил)карбамоил]азетидин-3-ил}метил)пиридин-2-ил]имидодикарбоната (94 мг, 0,16 ммоль) в метиленхлориде (2,5 мл) обрабатывали каплями триэтиламина (87 мкл, 0,63 ммоль), а затем [(1R)-1-изоцианатоэтил]бензолом (29 мкл, 0,20 ммоль), и реакционную смесь перемешивали при комнатной температуре. Через 4 часа ВЭЖХ указала на завершение реакции. Реакционную смесь концентрировали, и остаток помещали в 5 мл CH2Cl2 и концентрировали до слегка окрашенного в желтый цвет стекла. Сырое вещество очищали флэш-хроматографией (30 г силикагель, 40-70% EtOAc/гекс.) с выходом соединения, указанного в заголовке, (94 мг, 80%) в виде бесцветного стекла: Время удержания ВЭЖХ, 4,88 мин (Способ A); MS (ESI+) для C39H49N5O10 m/z 748,9 (M+H)+; MS (ESI-) для C39H49N5O10 m/z 746,5 (M-H)-.
Стадия 4. Получение трифторацетата (2S,3R)-3-[(2-аминопиридин-4-ил)метил]-4-оксо-N-1-[(1R)-1-фенилэтил]азетидин-1,2-дикарбоксамида

Раствор ди-трет-бутил [4-({(3R,4S)-2-оксо-1-{[(1R)-1-фенилэтил]карбамоил}-4-[(2,4,6-триметоксибензил)карбамоил]азетидин-3-ил}метил)пиридин-2-ил]имидодикарбоната (94 мг, 0,12 ммоль) в метиленхлориде (4,0 мл) охлаждали при 0°C, обрабатывали каплями трифторуксусной кислоты (1,0 мл, 13 ммоль), и смесь перемешивали при 0°С в течение 60 минут, в течение которых реакцию оставляли нагреваться до комнатной температуры. После перемешивания в течение 24 ч было обнаружено, что реакция была завершена по данным ВЭЖХ. Реакционную смесь концентрировали, и сырое вещество очищали CombiFlash хроматографией [30 г RediSep C-18 gold силикагелевый картридж, градиент растворителя: от 10% ацетонитрил (0,07% TFA)/вода (0,1% TFA) до 100% ацетонитрил (0,07% TFA)] с выходом соединения, указанного в заголовке, (31 мг, 51%) в виде белого твердого продукта после лиофилизации: Время удержания ВЭЖХ: 2,58 мин (Способ A); MS (ESI+) для C19H21N5O3 m/z 368,1 (M+H)+; 1H ЯМР (400 МГц, МeOD) δ 7,96 (1Н, ушир.с.) 7,77 (1Н, д, J=6,6 Гц) 7,40 (1Н, ушир.с.) 7,35 (4Н, м) 7,26 (1Н, м) 7,09 (1Н, д, J=7,8 Гц) 6,96 (1Н, с) 6,89 (1Н, дд, J=6,7, 1,6 Гц) 4,96 (1Н, м) 4,28 (1Н, д, J=2,8 Гц) 3,67 (1Н, дт, J=8,0, 2,8 Гц) 3,23 (2Н, м) 1,52 (3Н, д, J=7,1 Гц).
Пример 14, Схема 6: трифторацетат 4-{[(2R,3R)-3-[(2-аминопиридин-4-ил)метил]-4-оксо-1-{[(1R)-1-фенилэтил]карбамоил}азетидин-2-ил]окси}бензойной кислоты

Стадия 1. Получение (2R,3R)-3-({2-[бис(трет-бутоксикарбонил)амино]пиридин-4-ил}метил)-1-[трет-бутил(диметил)силил]-4-оксоазетидин-2-ил 3-хлорбензоата
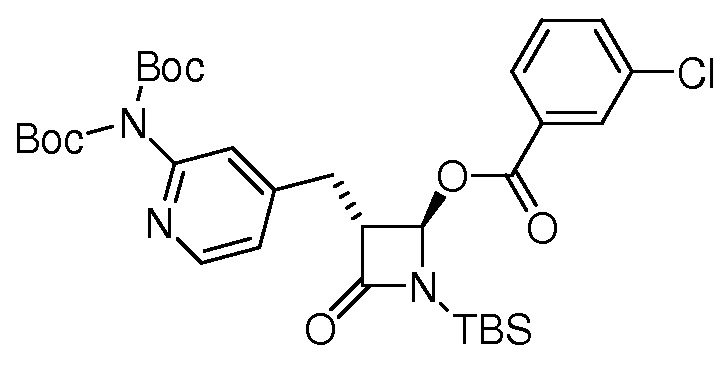
Раствор(2S,3R)-3-({2-[бис(трет-бутоксикарбонил)амино]пиридин-4-ил}метил)-1-[трет-бутил(диметил)силил]-4-оксоазетидин-2-карбоновой кислоты (201 мг, 0,375 ммоль) в метиленхлориде (7,0 мл) охлаждали при 0°C на ледяной бане и обрабатывали м-хлорпербензойной кислотой (79 мг, 0,41 ммоль), а затем N,N'-дициклогексилкарбодиимидом (85 мг, 0,41 ммоль), и реакционную смесь перемешивали при 0°С в течение 25 мин, и в этот момент ВЭЖХ указала на расход исходного вещества. Реакционную смесь фильтровали, твердое вещество промывали 20 мл 1/1 Et2O/CH2Cl2, и фильтрат промывали 15 мл Н2O и насыщенным солевым раствором. Органическую фазу сушили над Na2SO4, фильтровали и концентрировали с выходом светло-желтого стекла. Сырое вещество очищали флэш-хроматографией (45 г силикагель, 10-30% EtOAc/гекс.) с выходом соединения, указанного в заголовке, (131 мг, 54%) в виде бесцветного стекла: Время удержания ВЭЖХ, 5,95 мин (Способ A); MS (ESI+) для C32H44ClN3O7Si m/z 646,3/648,3 (M+H)+.
Стадия 2. Получение 3-хлорбензота (2R,3R)-3-({2-[бис(трет-бутоксикарбонил)амино]пиридин-4-ил}метил)-4-оксоазетидин-2-ила

Раствор 3-хлорбензоата (2R,3R)-3-({2-[бис(трет-бутоксикарбонил)амино]пиридин-4-ил}метил)-1-[трет-бутил(диметил)силил]-4-оксоазетидин-2-ила (130,0 мг, 0,201 ммоль) в метаноле (3,0 мл, 74 ммоль) обрабатывали уксусной кислотой (40 мкл, 0,70 ммоль), а затем 0,5 M фторидом аммония в метаноле (0,48 мл, 0,24 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 1 ч, и в этот момент ВЭЖХ указала на расход исходного продукта. Реакционную смесь концентрировали в вакууме, остаток помещали в 5 мл толуол, и раствор концентрировали. Процесс еще раз повторяли, остаток помещали в 25 мл CH2Cl2, и раствор промывали 15 мл Н2O и насыщенным водным раствором NaHCO3. Органическую фазу сушили над Na2SO4, фильтровали и концентрировали с выходом соединения, указанного в заголовке, (106 мг, 96%) в виде слегка окрашенного в желтый цвет стекла: Время удержания ВЭЖХ: 4,43 мин (Способ A); MS (ESI+) для C26H30ClN3O7 m/z 532,1/534,2 (M+H)+.
Стадия 3. Получение бензил 4-{[(2R,3R)-3-({2-[бис(трет-бутоксикарбонил)амино]пиридин-4-ил}метил)-4-оксоазетидин-2-ил]окси}бензоат
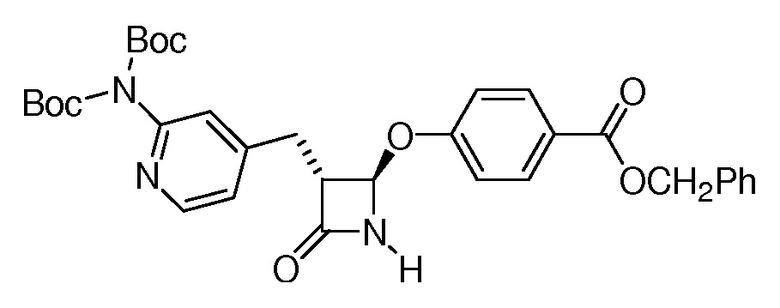
Раствор 3-хлорбензоат (2R,3R)-3-({2-[бис(трет-бутоксикарбонил)амино]пиридин-4-ил}метил)-4-оксоазетидин-2-ила (105 мг, 0,197 ммоль) в ацетонитриле (13 мл) и воде (1 мл) обрабатывали бензил п-гидроксибензоатом (45 мг, 0,20 ммоль), а затем карбонатом цезия (96 мг, 0,30 ммоль), и смесь перемешивали при комнатной температуре в течение 70 мин, и в этот момент ВЭЖХ указала на расход исходного продукта. Реакционную смесь разбавляли 25 мл Н2O и экстрагировали тремя порциями по 25 мл EtOAc. Органическую фазу промывали 25 мл насыщенного солевого раствора и сушили над Na2SO4. Раствор фильтровали и концентрировали с выходом бесцветного стекла, которое очищали флэш-хроматографией (30 г силикагель, 50-60% EtOAc/гекс.) с выходом соединения, указанного в заголовке, (85 мг, 71 %) в виде бесцветного стекла: Время удержания ВЭЖХ, 4,71 мин (Способ A); MS (ESI+) для C33H37N3O8 m/z 604,3 (M+H)+; MS (ESI-) для C33H37N3O8 m/z 604,6 (M-H)-.
Стадия 4. Получение бензил 4-{[(2R,3R)-3-({2-[бис(трет-бутоксикарбонил)амино]пиридин-4-ил}метил)-4-оксо-1-{[(1R)-1-фенилэтил]карбамоил}азетидин-2-ил]окси}бензоата

Раствор бензил 4-{[(2R,3R)-3-({2-[бис(трет-бутоксикарбонил)амино]пиридин-4-ил}метил)-4-оксоазетидин-2-ил]окси}бензоата (82 мг, 0,14 ммоль) в метиленхлориде (2,2 мл) обрабатывали каплями триэтиламина (76 мкл, 0,54 ммоль), а затем [(1R)-1-изоцианатоэтил]бензола (25 мкл, 0,18 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 4 ч, и в этот момент ВЭЖХ указала на практически полный расход исходного продукта. Добавляли дополнительные 10 мкл изоцианата, и реакцию продолжали еще час. Реакционную смесь концентрировали, остаток помещали в 5 мл CH2Cl2, и раствор концентрировали с выходом белой крепкой пены. Сырое вещество очищали флэш-хроматографией (25 г силикагеля 25-45 EtOAc/гекс.) с выходом соединения, указанного в заголовке, (87 мг, 85%) в виде бесцветного стекла: Время удержания ВЭЖХ: 5,54 мин (Способ A); MS (ESI+) для C42H46N4O m/z 751,4 (M+H)+.
Стадия 5. Получение 4-{[(2R,3R)-3-({2-[бис(трет-бутоксикарбонил)амино]пиридин-4-ил}метил)-4-оксо-1-{[(1R)-1-фенилэтил]карбамоил}азетидин-2-ил]окси}бензойной кислоты
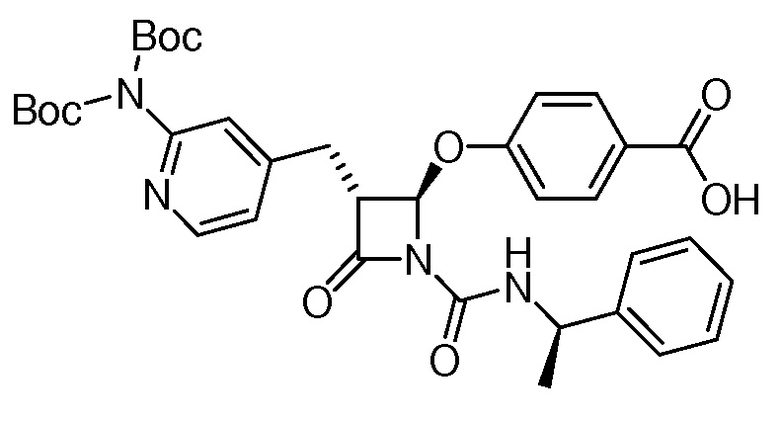
Раствор бензил 4-{[(2R,3R)-3-({2-[бис(трет-бутоксикарбонил)амино]пиридин-4-ил}метил)-4-оксо-1-{[(1R)-1-фенилэтил]карбамоил}азетидин-2-ил]окси}бензоата (87 мг, 0,12 ммоль) в метаноле (3,0 мл) и этилацетате (3,0 мл) аккуратно обрабатывали 10% палладием на угле (12 мг). Реакционную колбу откачивали и наполняли газообразным водородом три раза, и смесь перемешивали в атмосфере водорода в течение 4,5 ч, и в этот момент ТСХ указала на расход исходного продукта. Реакционную смесь фильтровали через пад из Solka-Floc®, и пад промывали 40 мл 1/1 EtOAc/MeOH. Фильтрат концентрировали с выходом соединения, указанного в заголовке, (77 мг, 100%) в виде бесцветного стекла: Время удержания ВЭЖХ, 4,56 мин (Способ A); MS (ESI+) для C35H40N4O9 m/z 661,3 (M+H)+; MS (ESI-) для C35H40N4O9 m/z 559,8 (M-H)-.
Стадия 6. Получение трифторацетата 4-{[(2R,3R)-3-[(2-аминопиридин-4-ил)метил]-4-оксо-1-{[(1R)-1-фенилэтил]карбамоил}азетидин-2-ил]окси}бензойной кислоты
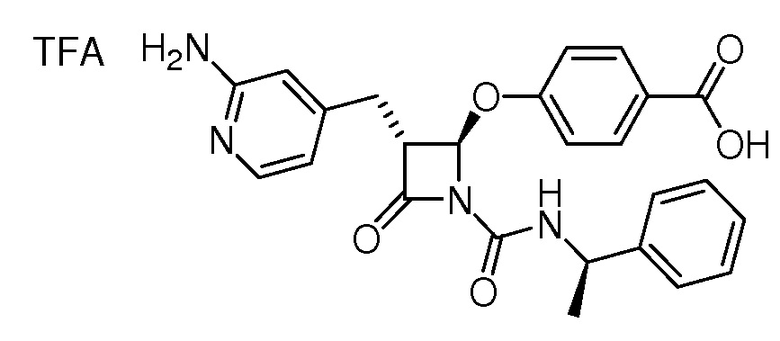
Раствор 4-{[(2R,3R)-3-({2-[бис(трет-бутоксикарбонил)амино]пиридин-4-ил}метил)-4-оксо-1-{[(1R)-1-фенилэтил]карбамоил}азетидин-2-ил]окси}бензойной кислоты (76 мг, 0,12 ммоль) в метиленхлориде (3,3 мл) охлаждали при 0°C и обрабатывали каплями трифторуксусной кислоты (0,66 мл, 8,6 ммоль). Реакционную смесь перемешивали при 0°С в течение 1 часа, после чего ледяную баню оставляли медленно нагреваться. Через 7 ч ВЭЖХ указала на расход исходного продукта. Реакционную смесь концентрировали в вакууме. Остаток помещали в 5 мл CH2Cl2 и концентрировали с выходом сырого продукта в виде светлого розового стекла. Вещество очищали CombiFlash хроматографией [30 г RediSep C-18 gold силикагелевый картридж, градиент растворителя: от 10% ацетонитрил (0,07% TFA)/вода (0,1% TFA) до 100% ацетонитрил (0,07% TFA)] с выходом соединения, указанного в заголовке, (32 мг, 48%) в виде белого порошка: Время удержания ВЭЖХ: 3,13 мин (Способ A); MS (ESI+) для C25H24N4O5 m/z 461,1 (M+H)+; 1H ЯМР (400 МГц, МeOD) δ 7,94 (2Н, м) 7,72 (1Н, д, J=6,6 Гц) 7,34 (4Н, м) 7,27 (2Н, м) 7,13 (2Н, м) 6,91 (1Н, с) 6,86 (1Н, дд, J=6,8, 1,5 Гц) 6,08 (1Н, д, J=1,5 Гц) 4,96 (1Н, м) 3,79 (1Н, дт, J=8,2, 1,5 Гц) 3,24 (2Н, д, J=8,1 Гц) 1,53 (3Н, д, J=7,1 Гц).
Пример 15, Схема 7: трифторацетат (2R,3S)-3-[(2-аминопиридин-4-ил)метил]-2-(метилсульфонил)-4-оксо-N-[(1R)-1-фенилэтил]азетидин-1-карбоксамида
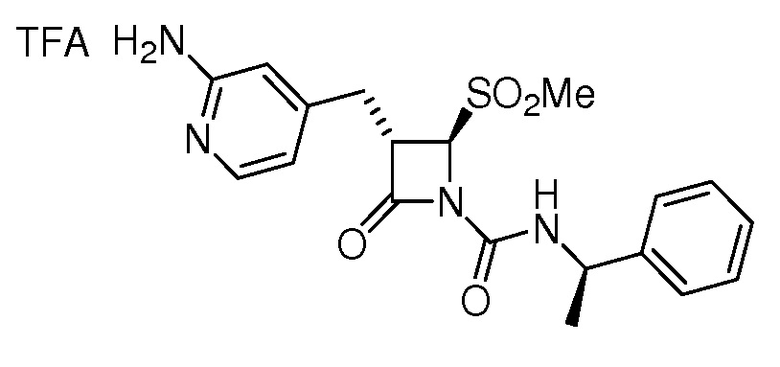
Стадия 1. Получение трет-бутил (4-метоксибензил)(4-{[(2R,3S)-2-(метилсульфонил)-4-оксоазетидин-3-ил]метил}пиридин-2-ил)карбамата
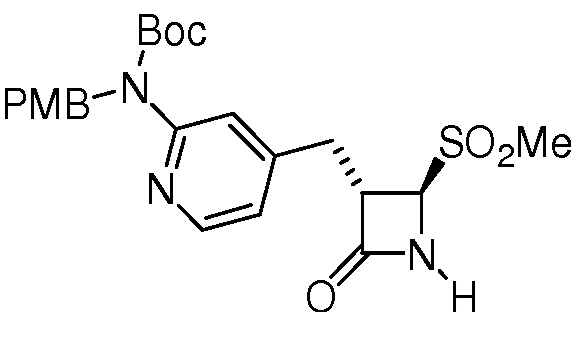
Раствор 3-хлорбензоат (2R,3R)-3-({2-[(трет-бутоксикарбонил)(4-метоксибензил)амино]пиридин-4-ил}метил)-4-оксоазетидин-2-ила (100 мг, 0,181 ммоль) в ацетонитриле (4,0 мл) и воде (1,1 мл) обрабатывали метансульфинатом натрия (87 мг, 0,72 ммоль). Смесь перемешивали при комнатной температуре в течение 4 ч, в это время ВЭЖХ указала на расход исходного продукта. Реакционную смесь концентрировали для удаления большей части CH3CN и разбавляли 10 мл Н2O. Водную фазу экстрагировали три раза порциями по 10 мл EtOAc. Объединенную органическую фазу сушили над MgSO4, фильтровали и концентрировали с выходом бесцветного стекла. Сырое вещество очищали препаративной ТСХ (20см × 10см × 1,0мм препарат. ТСХ планшет, 65% EtOAc/гекс.) с выходом соединения, указанного в заголовке, (52 мг, 60%) в виде бесцветного стекла: Время удержания ВЭЖХ, 3,50 мин (Способ A); MS (ESI+) для C23H29N3O6S m/z 476,4 (M+H)+; MS (ESI-) для C23H29N3O6S m/z 474,3 (M-H)-.
Стадия 2. Получение трет-бутил (4-метоксибензил)(4-{[(2R,3S)-2-(метилсульфонил)-4-оксо-1-{[(1R)-1-фенилэтил]карбамоил}азетидин-3-ил]метил}пиридин-2-ил)карбамата
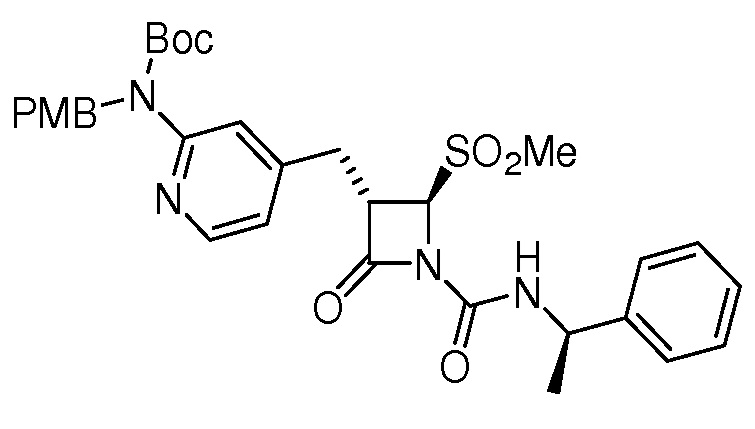
Раствор трет-бутил (4-метоксибензил)(4-{[(2R,3S)-2-(метилсульфонил)-4-оксоазетидин-3-ил]метил}пиридин-2-ил)карбамата (52,0 мг, 0,109 ммоль) в метиленхлориде (2,4 мл) обрабатывали каплями триэтиламина (61 мкл, 0,44 ммоль), а затем [(1R)-1-изоцианатоэтил]бензола (20 мкл, 0,14 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 2 ч, и в этот момент ВЭЖХ указала расход исходного вещества. Реакционную смесь концентрировали до бесцветного масла. Вещество помещали в 5 мл CH2Cl2 и концентрировали, и полученный сырой продукт очищали препарат. ТСХ (20см × 20см × 1,0мм препарат. ТСХ планшет, 50% EtOAc/гекс.) с выходом соединения, указанного в заголовке, (63 мг, 91 %) в виде бесцветной крепкой пены: Время удержания ВЭЖХ: 5,02 мин (Способ A); MS (ESI+) для C32H38N4O7S m/z 623,5 (M+H)+; MS (ESI+) для C32H38N4O7S m/z 621,5 (M+H)+.
Стадия 3. Получение трифторацетата (2R,3S)-3-[(2-аминопиридин-4-ил)метил]-2-(метилсульфонил)-4-оксо-N-[(1R)-1-фенилэтил]азетидин-1-карбоксамида
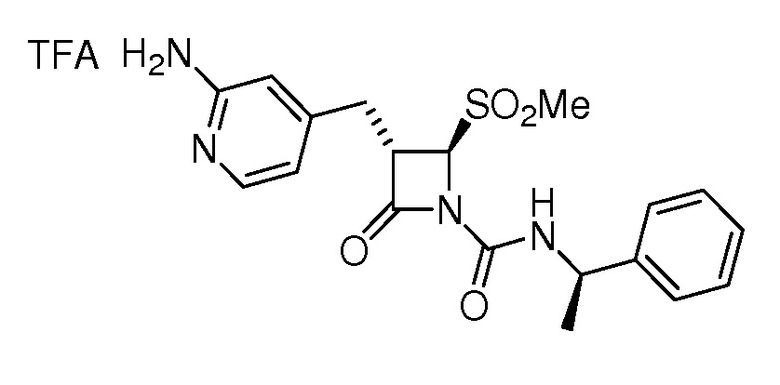
Раствор трет-бутил (4-метоксибензил)(4-{[(2R,3S)-2-(метилсульфонил)-4-оксо-1-{[(1R)-1-фенилэтил]карбамоил}азетидин-3-ил]метил}пиридин-2-ил)карбамата (62 мг, 0,10 ммоль) в метиленхлориде (3 мл) охлаждали при 0°C и обрабатывали трифторуксусной кислотой (1,0 мл, 13 ммоль). Раствор перемешивали при 0°С в течение 30 минут, затем оставляли нагреваться до комнатной температуры. ВЭЖХ реакционной смеси через 23 ч указала на расход исходного продукта. Сырую реакционную смесь концентрировали. Остаток помещали в 5 мл CH2Cl2 и концентрировали с выходом сырого продукта в виде фиолетового стекла. Сырое вещество очищали CombiFlash хроматографией [30 г RediSep C-18 gold силикагелевый картридж, градиент растворителя: от 10% ацетонитрил (0,07% TFA)/вода (0,1% TFA) до 100% ацетонитрил (0,07% TFA)] с выходом соединения, указанного в заголовке, (31 мг, 60%) в виде белого твердого продукта после лиофилизации: Время удержания ВЭЖХ: 2,99 мин (Способ A); MS (ESI+) для C19H22N4O4S m/z 403,2 (M+H)+; 1H ЯМР (400 МГц, МeOD) δ 7,77 (1Н, д, J=6,8 Гц) 7,35 (4Н, д, J=4,3 Гц) 7,27 (1Н, м) 7,19 (1Н, д, J=7,6 Гц) 6,98 (1Н, с) 6,91 (1Н, дд, J=6,8, 1,5 Гц) 5,30 (1Н, д, J=2,8 Гц) 4,97 (1Н, м) 4,02 (1Н, м) 3,34 (1Н, м) 3,21 (1Н, м) 3,17 (3Н, с) 1,54 (3Н, д, J=7,1 Гц).
Пример 16, Схема 8: трифторацетат (2S,3R)-3-[(2-аминопиридин-4-ил)метил]-2-[(метоксииминo)метил]-4-оксо-N-[(1R)-1-фенилэтил]азетидин-1-карбоксамида
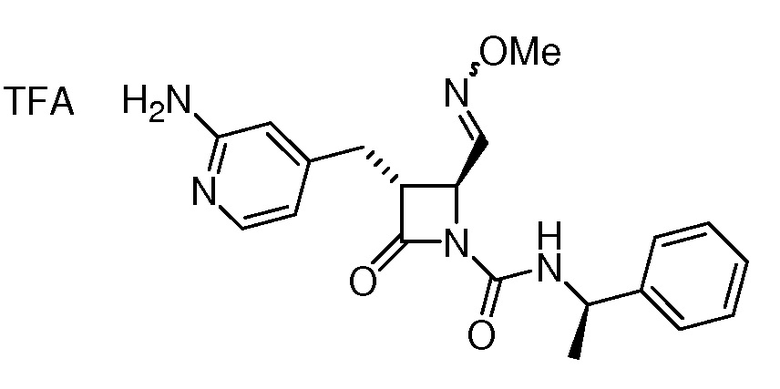
Стадия 1. Получение (4S)-1-[трет-бутил(дифенил)силил]-4-(гидроксиметил)азетидин-2-она
Раствор бензил (2S)-1-[трет-бутил(дифенил)силил]-4-оксоазетидин-2-карбоксилата (2,20 г, 4,96 ммоль) в метаноле (20 мл) охлаждали до 0°C (ледяная вода) и обрабатывали одной порцией тетрагидробората натрия (0,55 г, 15 ммоль). Реакционную смесь перемешивали и медленно нагревали до комнатной температуры. Через 18 ч ТСХ указала на расход исходного продукта. Реакционную смесь гасили 10 мл насыщенным раствором NaHCO3, и смесь концентрировали для удаления растворителя. Полученную молочную суспензию разбавляли 20 мл Н2O и экстрагировали тремя порциями 25 мл MTBE. Органическую фазу промывали 20 мл насыщенного солевого раствора и сушили над MgSO4. Органическую фазу фильтровали и концентрировали с выходом бесцветного вязкого масла. Сырое вещество очищали флэш-хроматографией (110 г, 15-50% EtOAc/гекс.) с выходом соединения, указанного в заголовке, (0,82 г, 48%) в виде белого твердого продукта: Время удержания ВЭЖХ: 4,47 мин (Способ A).
Стадия 2. Получение (2S)-1-[трет-бутил(дифенил)силил]-4-оксоазетидин-2-карбальдегида

Раствор (4S)-1-[трет-бутил(дифенил)силил]-4-(гидроксиметил)азетидин-2-она (530 мг, 1,56 ммоль) в метиленхлориде (39 мл) обрабатывали Десс-Мартина перйодинаном (1,32 г, 3,12 ммоль), и реакционную смесь перемешивали при комнатной температуре, получая прозрачный раствор. Через 1,5 ч при комнатной температуре, реакционную смесь разбавляли 40 мл Et2O и концентрировали в вакууме с получением прозрачного масла, которое помещали в 50 мл Et2O и добавляли смесь 1/1 40 мл 10% водного раствора Na2S2O3 и 40 мл насыщенного раствора NaHCO3. Смесь интенсивно перемешивали до тех пор, пока все твердые вещества не растворялись. Отделенный водный слой экстрагировали двумя порциями по 20 мл Et2O, и объединенную органическую фазу сушили над Na2SO4, фильтровали и концентрировали с получением соединения, указанного в заголовке (520 мг 99%), в виде бесцветного вязкого масла, которое использовали как есть в следующей стадии: 1H ЯМР (300 МГц, CDCl3) δ 9,12 (1Н, д, J=4,1 Гц) 7,68 (2Н, м) 7,59 (2Н, м) 7,45 (6Н, м) 3,76 (1Н, м) 3,39 (1Н, дд, J=15,8, 6,3 Гц) 3,02 (1Н, дд, J=15,8, 3,1 Гц) 1,25 (9Н, с).
Стадия 3. Получение (2S)-1-[трет-бутил(дифенил)силил]-4-оксоазетидин-2-карбальдегид O-метилоксима
(2S)-1-[Трет-бутил(дифенил)силил]-4-оксоазетидин-2-карбальдегид (520 мг, 1,54 ммоль) помещали в этанол (14 мл) и обрабатывали пиридином (177 мкл, 2,19 ммоль), а затем одной порцией гидроксихлорида метоксиамина (154 мг, 1,87 ммоль). Раствор перемешивали при комнатной температуре в течение 19 ч, и в этот момент ТСХ (30% EA/гекс.) указала на завершение реакции. Реакционную смесь концентрировали для удаления большей части растворителя, и полученную бесцветную жидкость разбавляли 20 мл Н2O и экстрагировали три раза порциями по 20 мл MTBE. Органическую фазу промывали 20 мл Н2O, сушили над Na2SO4, фильтровали и концентрировали с выходом бесцветного вязкого масла. Сырое вещество очищали флэш-хроматографией (50 г, 10-20% EtOAc/гекс.) с выходом соединения, указанного в заголовке, (469 мг, 82%) в виде бесцветного вязкого масла, которое было смесью E/Z геометрических изомеров: Время удержания ВЭЖХ: 4,93 мин (Способ A); MS (ESI+) для C21H26N2O2Si m/z 367,4 (M+H)+, m/z 389,3 (M+Na)+.
Стадия 4. Получение трет-бутил [4-({(2S,3R)-1-[трет-бутил(дифенил)силил]-2-[(метоксииминo)метил]-4-оксоазетидин-3-ил}метил)пиридин-2-ил](4-метоксибензил)карбамат
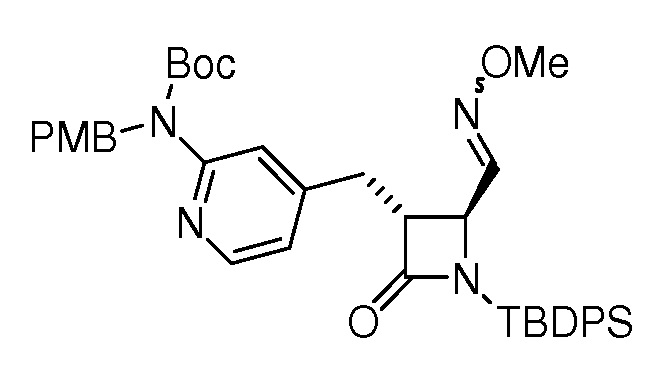
Раствор (2S)-1-[трет-бутил(дифенил)силил]-4-оксоазетидин-2-карбальдегида O-метилоксима (469 мг, 1,28 ммоль) в тетрагидрофуране (9,6 мл) охлаждали до -78°C и обрабатывали каплями 1,2 M раствора диизопропиламида лития в гексан/THF/этилбензол (1,2 мл, 1,4 ммоль). Светло-желтую реакционную смесь перемешивали в течение 15 мин и переносили посредством канюли в заранее охлажденный (-78°C) раствор трет-бутил [4-(бромметил)пиридин-2-ил](4-метоксибензил)карбамата (570 мг, 1,4 ммоль) в тетрагидрофуране (9,6 мл) по каплям в течение 5 мин, с получением желто-коричневого раствора. Реакционную смесь перемешивали в течение 90 мин при -78°C, в это время ВЭЖХ указала на расход исходного продукта. Реакцию гасили дополнительным количеством 10 мл насыщенного водного раствора NH4Cl. Охлаждающую баню удаляли, и смесь перемешивали в течение 5 мин. Реакционную смесь разбавляли 20 мл Н2O и экстрагировали два раза порциями по 40 мл EtOAc. Объединенную органическую фазу промывали порциями по 20 мл Н2O и насыщенным солевым раствором и сушили над MgSO4. Раствор фильтровали и концентрировали с выходом темно-бордового вязкого масла. Сырое вещество очищали флэш-хроматографией (80 г силикагель, 10-30% EtOAc/гекс.) с выходом соединения, указанного в заголовке, (553 мг, 62 %) в виде светло-желтой крепкой пены, которая была смесью E/Z геометрических изомеров: Время удержания ВЭЖХ: 5,77/5,82 мин (Способ A); MS (ESI+) для C40H48N4O5Si m/z 693,6 (M+H)+.
Стадия 5. Получение трет-бутил (4-метоксибензил)[4-({(2S,3R)-2-[(метоксиимино)метил]-4-оксоазетидин-3-ил}метил)пиридин-2-ил]карбамата
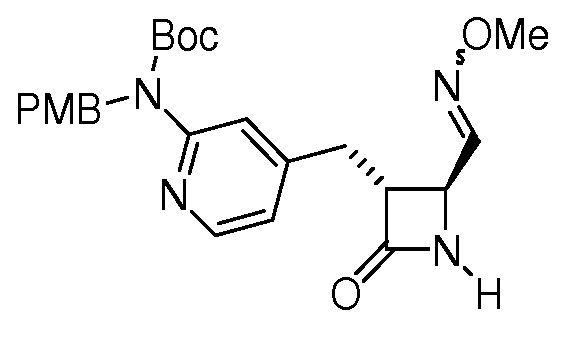
Раствор трет-бутил [4-({(2S,3R)-1-[трет-бутил(дифенил)силил]-2-[(метоксиимино)метил]-4-оксоазетидин-3-ил}метил)пиридин-2-ил](4-метоксибензил)карбамата (553 мг, 0,678 ммоль) в метаноле (10 мл) обрабатывали каплями уксусной кислоты (130 мкл, 2,4 ммоль), а затем 0,5 M фторидом аммония в метаноле (1,6 мл, 0,81 ммоль). Раствор перемешивали при комнатной температуре в течение 1 ч, в это время ВЭЖХ указала на расход исходного продукта. Реакционную смесь концентрировали для удаления MeOH, и полученное масло помещали в 50 мл CH2Cl2. Органическую фазу промывали порциями по 25 мл насыщенного раствора NaHCO3 иН2O, сушили над Na2SO4, фильтровали и концентрировали до светло-желтого масла. Сырое вещество очищали флэш-хроматографией (60 г силикагель, 40-80% EtOAc/гекс.) с выходом соединения, указанного в заголовке, (250 мг, 81%) в виде слегка окрашенного в желтый цвет стекла, которое было смесью E/Z изомеров: Время удержания ВЭЖХ: 3,59/3,66 мин (Способ A); MS (ESI+) для C24H30N4O5 m/z 455,3 (M+H)+; MS (ESI-) для C24H30N4O5 m/z 453,3 (M-H)-.
Стадия 6. Получение трет-бутил (4-метоксибензил)(4-{[(2S,3R)-2-[(метоксиимино)метил]-4-оксо-1-{[(1R)-1-фенилэтил]карбамоил}азетидин-3-ил]метил}пиридин-2-ил)карбамата

Раствор трет-бутил (4-метоксибензил)[4-({(2S,3R)-2-[(метоксиимино)метил]-4-оксоазетидин-3-ил}метил)пиридин-2-ил]карбамата (131 мг, 0,288 ммоль) в метиленхлориде (4,6 мл) обрабатывали каплями триэтиламина (160 мкл, 1,2 ммоль), а затем [(1R)-1-изоцианатоэтил]бензола (53 мкл, 0,37 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 21 ч, и в этот момент ВЭЖХ указала на расход исходного продукта. Реакцию концентрировали, остаток помещали в 5 мл CH2Cl2 и концентрировали до светло-коричневого остатка. Вещество помещали в 1/1 Et2O/гекс., и суспензию фильтровали через тонкую фритту. Твердые вещество промывали дополнительным количеством 1/1 Et2O/гекс., и фильтрат концентрировали до светло-коричневой крепкой пены. Сырое вещество очищали флэш-хроматографией (25 г силикагель, 25-40% EtOAc/гекс.) с выходом соединения, указанного в заголовке, (145 мг, 83%) в виде бесцветного стекла, которое было смесью E/Z изомеров: Время удержания ВЭЖХ: 4,84 мин (Способ A); MS (ESI+) для C33H39N5O6 m/z 602,5 (M+H)+; MS (ESI-) для C33H39N5O6 m/z 600,4 (M-H)-.
Стадия 7. Получение трифторацетата (2S,3R)-3-[(2-аминопиридин-4-ил)метил]-2-[(метоксиимино)метил]-4-оксо-N-[(1R)-1-фенилэтил]азетидин-1-карбоксамида
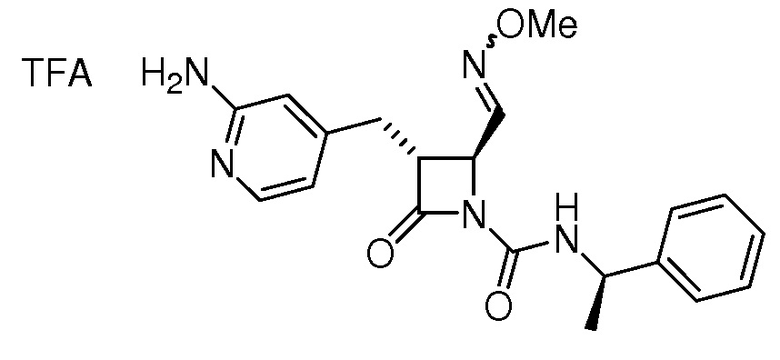
Раствор трет-бутил (4-метоксибензил)(4-{[(2S,3R)-2-[(метоксиимино)метил]-4-оксо-1-{[(1R)-1-фенилэтил]карбамоил}азетидин-3-ил]метил}пиридин-2-ил)карбамата (145 мг, 0,241 ммоль) в метиленхлориде (4,5 мл) охлаждали при 0°C и обрабатывали трифторуксусной кислотой (1,5 мл, 20,0 ммоль). Раствор перемешивали при 0°С в течение 30 минут, затем оставляли нагреваться до комнатной температуры. Реакционную смесь перемешивали в течение 24 ч, в это время ВЭЖХ указала на расход исходного продукта. Реакционную смесь концентрировали. Остаток помещали в 10 мл CH2Cl2 и концентрировали с выходом светло-коричневого стекла. Сырое вещество очищали CombiFlash хроматографией [30 г RediSep C-18 gold силикагелевый картридж, градиент растворителя: от 10% ацетонитрил (0,07% TFA)/вода (0,1% TFA) до 100% ацетонитрил (0,07% TFA)] с выходом соединения, указанного в заголовке, (71 мг, 59%) в виде белого твердого продукта после лиофилизации (данные приведены для смеси E/Z) Время удержания ВЭЖХ: 2,88/2,93 мин (Способ A); MS (ESI+) для C20H23N5O3 m/z 382,3 (M+H)+; 1H ЯМР (400 МГц, МeOD) δ 7,78 (1Н, м) 7,48 (0,6Н, д, J=6,6 Гц) 7,34 (4Н, м) 7,26 (1Н, м) 6,96 (0,4Н, д, J=4,8 Гц) 6,94 (1Н, с) 6,88 (1Н, м) 4,94 (1Н, м) 4,73 (0,4Н, дд, J=4,9, 3,2 Гц) 4,43 (0,6Н, дд, J=6,4, 2,9 Гц) 3,79 (1,2Н, с) 3,78 (1,8Н, с) 3,73 (0,6Н, дт, J=8,0, 3,0 Гц) 3,58 (0,4Н, дт, J=7,6, 3,2 Гц) 3,21 (2Н, м) 1,51 (3Н, д, J=7,1 Гц)
Пример 17, Схема 9: этил (2S,3R)-3-[(2-{[(гексилокси)карбонил]амино}пиридин-4-ил)метил]-4-оксо-1-{[(1R)-1-фенилэтил]карбамоил}азетидин-2-карбоксилат
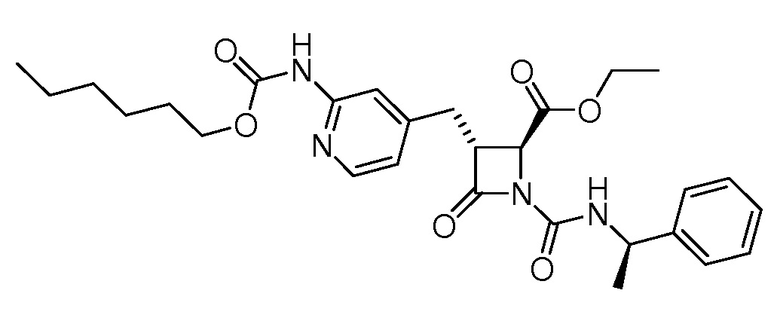
Стадия 1. Получение этил (2S,3R)-3-[(2-аминопиридин-4-ил)метил]-4-оксо-1-{[(1R)-1-фенилэтил]карбамоил}азетидин-2-карбоксилата
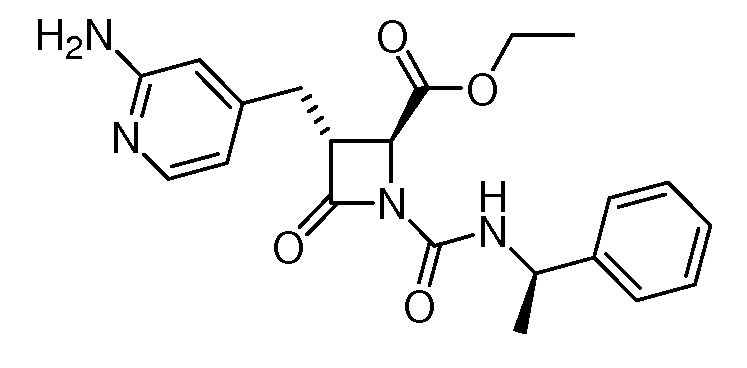
Раствор этил (2S,3R)-3-({2-[(трет-бутоксикарбонил)(4-метоксибензил)амино]-пиридин-4-ил}метил)-4-оксо-1-{[(1R)-1-фенилэтил]карбамоил}азетидин-2-карбоксилата (474 мг, 0,715 ммоль) в метиленхлориде (9,0 мл) охлаждали при 0°C и обрабатывали каплями трифторуксусной кислоты (3,0 мл). Раствор перемешивали при 0°С в течение 60 мин., а затем нагревали до комнатной температуры. Через 24 ч ВЭЖХ указала на расход исходного продукта. Фиолетовое масло помещали в 15 мл CH2Cl2 и концентрировали. Остаток растворяли в 11 мл CH2Cl2, обрабатывали 10 мл насыщенным раствором NaHCO3 и перемешивали при комнатной температуре, до тех пор, пока не прекращалось выделение пузырьков. Смесь выливали в делительную воронку, и фазы разделяли. Водную фазу промывали 20 мл CH2Cl2, и объединенные органические фазы сушили над Na2SO4. Раствор фильтровали и концентрировали с выходом крепкой пены. Сырое вещество очищали флэш-хроматографией (35 г силикагеля, 6-8% MeOH/CH2Cl2) с выходом соединения, указанного в заголовке, (254 мг, 89%) в виде бесцветной крепкой пены: Время удержания ВЭЖХ: 3,01 мин (Способ A); MS (ESI+) для C21H24N4O4 m/z 397,3 (M+H)+; MS (ESI-) для C21H24N4O4 m/z 395,3 (M-H)-.
Стадия 2. Получение этил (2S,3R)-3-[(2-{[(гексилокси)карбонил]амино}пиридин-4-ил)метил]-4-оксо-1-{[(1R)-1-фенилэтил]карбамоил}азетидин-2-карбоксилата
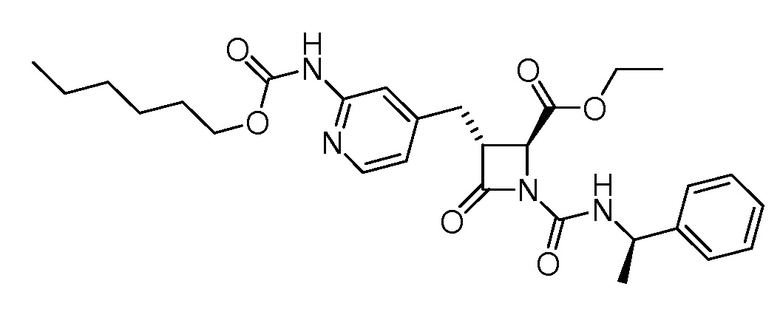
Перемешанный раствор этил (2S,3R)-3-[(2-аминопиридин-4-ил)метил]-4-оксо-1-{[(1R)-1-фенилэтил]карбамоил}азетидин-2-карбоксилата (50,0 мг, 0,126 ммоль) в сухом метиленхлориде (0,50 мл) в азоте охлаждали на бане «лед-вода» и обрабатывали пиридином (22 мкл, 0,28 ммоль), а затем каплями гексил хлорформиата (23 мкл, 0,14 ммоль). Реакционную смесь перемешивали при 0-5С в течение 1 ч, а затем медленно нагревали (в течение 2,5 ч) до комнатной температуры. Реакционную смесь разбавляли 10 мл Н2O и экстрагировали двумя порциями по 15 мл CH2Cl2. Органическую фазу промывали порциями по 15 мл Н2O и насыщенным солевым раствором и сушили над Na2SO4. Раствор концентрировали до практически бесцветного вязкого масла. Сырое вещество очищали флэш-хроматографией (25 г силикагель, 30-50% EtOAc/гекс.) с выходом соединения, указанного в заголовке, (43 мг, 66%) в виде бесцветного стекла: Время удержания ВЭЖХ: 4,27 мин (Способ A); MS (ESI+) для C28H36N4O6 m/z 525,3 (M+H)+; MS (ESI-) для C28H36N4O6 m/z 523,4 (M-H)-; 1H ЯМР (400 МГц, CDCl3) δ 8,21 (1Н, д, J=5,1 Гц) 7,93 (1Н, с) 7,73 (1Н, ушир.с.) 7,35 (4Н, м) 7,28 (1Н, м) 6,89 (1Н, дд, J=5,1, 1,5 Гц) 6,67 (1Н, д, J=8,1 Гц) 5,03 (1Н, м) 4,16 (5Н, м) 3,54 (1Н, ддд, J=8,3, 6,8, 2,7 Гц) 3,20 (1Н, м) 3,09 (1Н, м) 1,70 (2Н, м) 1,55 (3Н, д, J=7,1 Гц) 1,37 (6Н, м) 1,16 (3Н, т, J=7,2 Гц) 0,91 (3Н, м).
Пример 18, Схема 9: (2S,3R)-3-[(2-{[(гексилокси)карбонил]амино}пиридин-4-ил)метил]-4-оксо-1-{[(1R)-1-фенилэтил]карбамоил}азетидин-2-карбоновая кислота
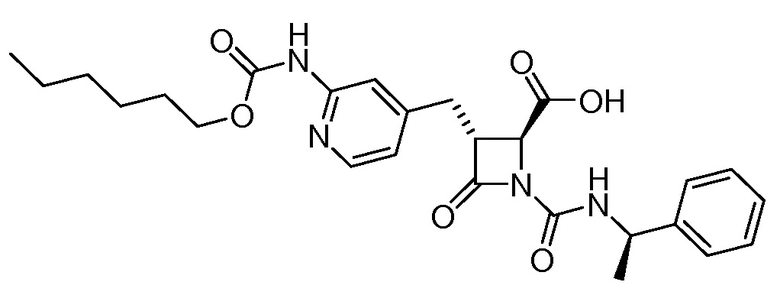
Стадия 1. Получение (2S,3R)-3-[(2-аминопиридин-4-ил)метил]-4-оксо-1-{[(1R)-1-фенилэтил]карбамоил}азетидин-2-карбоксилата
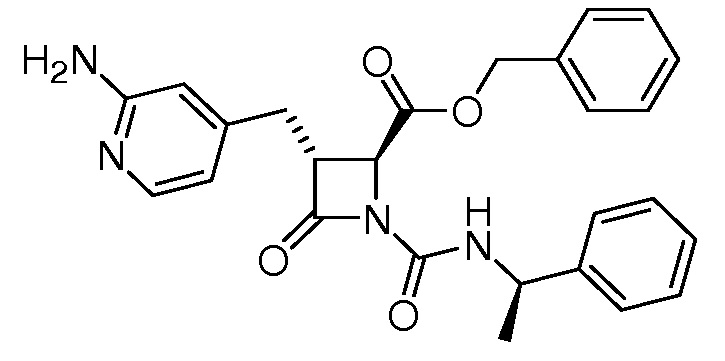
Раствор бензил (2S,3R)-3-({2-[(трет-бутоксикарбонил)(4-метоксибензил)амино]пиридин-4-ил}метил)-4-оксо-1-{[(1R)-1-фенилэтил]карбамоил}азетидин-2-карбоксилата (344 мг, 0,507 ммоль) в метиленхлориде (6,4 мл) охлаждали при 0°C и обрабатывали каплями трифторуксусной кислоты (2,1 мл, 28 ммоль). Раствор перемешивали при 0°С в течение 60 мин., а затем нагревали до комнатной температуры. Через 24 ч ВЭЖХ указала на завершение реакции. Фиолетовое масло помещали в 15 мл CH2Cl2 и концентрировали. Остаток растворяли в 20 мл CH2Cl2, обрабатывали 10 мл насыщенным раствором NaHCO3 и перемешивали при комнатной температуре до тех пор, пока не прекращалось выделение пузырьков. Смесь выливали в делительную воронку и фазы разделяли. Водную фазу промывали 10 мл CH2Cl2, и объединенные органические фазы сушили над Na2SO4. Раствор фильтровали и концентрировали с выходом крепкой пены. Сырое вещество очищали флэш-хроматографией (30 г силикагель gel, (4-6% MeOH/CH2Cl2) с выходом соединения, указанного в заголовке, (206 мг, 89%) в виде бесцветной крепкой пены: Время удержания ВЭЖХ: 3,48 мин (Способ A); MS (ESI+) для C26H26N4O4 m/z 459,3 (M+H)+; MS (ESI-) для C26H26N4O4 m/z 457,2 (M-H)-.
Стадия 2. Получение бензил (2S,3R)-3-[(2-{[(гексилокси)карбонил]амино}пиридин-4-ил)метил]-4-оксо-1-{[(1R)-1-фенилэтил]карбамоил}азетидин-2-карбоксилата
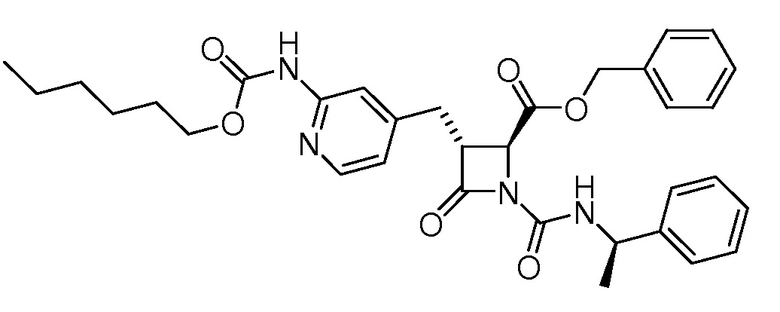
Перемешанный раствор бензил (2S,3R)-3-[(2-аминопиридин-4-ил)метил]-4-оксо-1-{[(1R)-1-фенилэтил]карбамоил}азетидин-2-карбоксилата (206 мг, 0,449 ммоль) в сухом метиленхлориде (3,0 мл) в азоте охлаждали на бане «лед-вода» и обрабатывали каплями пиридина (95 мкл, 1,2 ммоль), а затем гексил хлорформиата (110 мкл, 0,67 ммоль). Реакционную смесь перемешивали при 0-5°С в течение 1 ч, и в этот момент охлаждающую баню оставляли медленно нагреваться до комнатной температуры в течение 2,5 ч. В это время ВЭЖХ указала на расход исходного продукта. Реакционную смесь разбавляли 20 мл Н2O и экстрагировали двум порциями по 20 мл CH2Cl2. Органическую фазу промывали 15 мл Н2O насыщенным солевым раствором и сушили над Na2SO4. Раствор концентрировали до светло-желтого стекла. Сырое вещество очищали флэш-хроматографией (30 г силикагель, 30-50% EtOAc/гекс.) с выходом соединения, указанного в заголовке, (242 мг, 92%) в виде бесцветного стекла: Время удержания ВЭЖХ: 4,51 мин (Способ A); MS (ESI+) для C33H38N4O6 m/z 587,4 (M+H)+.
Стадия 3. Получение (2S,3R)-3-[(2-{[(гексилокси)карбонил]амино}пиридин-4-ил)метил]-4-оксо-1-{[(1R)-1-фенилэтил]карбамоил}азетидин-2-карбоновой кислоты
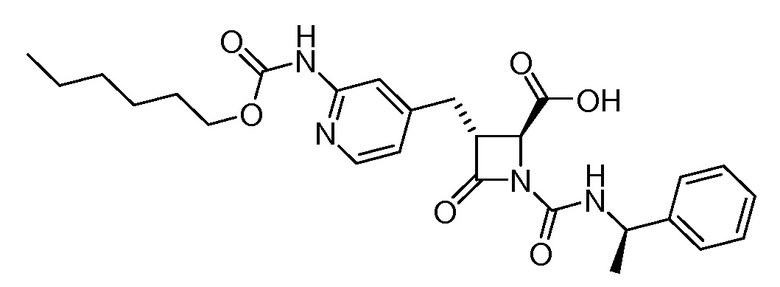
Раствор бензил (2S,3R)-3-[(2-{[(гексилокси)карбонил]амино}пиридин-4-ил)метил]-4-оксо-1-{[(1R)-1-фенилэтил]карбамоил}азетидин-2-карбоксилата (242 мг, 0,412 ммоль) в метаноле (4,0 мл) и этилацетате (4,0 мл) аккуратно обрабатывали 10% Pd-C катализатором (44 мг). Реакционную колбу откачивали и наполняли газообразным водородом три раза, и реакционную смесь перемешивали в атмосфере водорода в течение 1,5 ч, и в этот момент ВЭЖХ указала на расход исходного продукта. Реакционную смесь фильтровали через пад из Solka-Floc®, и пад промывали 40 мл 1/1 EtOAc/MeOH. Фильтрат концентрировали с выходом соединения, указанного в заголовке, (197 мг, 96%) в виде бесцветного твердого вещества: Время удержания ВЭЖХ: 3,77 мин (Способ A); MS (ESI+) для C26H32N4O6 m/z 497,3 (M+H)+; MS (ESI-) для C26H32N4O6 m/z 495,2 (M-H)-; ¹H ЯМР (400 МГц, CDCl3) δ 9,34 (1Н, ушир.с.) 8,04 (2Н, с) 7,33 (4Н, с) 7,28 (1Н, м) 6,95 (1Н, д, J=4,5 Гц) 6,78 (1Н, д, J=7,8 Гц) 5,05 (1Н, м) 4,31 (1Н, ушир.с.) 4,15 (2Н, т, J=6,7 Гц) 3,63 (1Н, ддд, J=8,8, 6,1, 2,8 Гц) 3,22 (1Н, м) 3,09 (1Н, м) 1,67 (2Н, м) 1,56 (3Н, д, J=6,8 Гц) 1,33 (6Н, м) 0,88 (3Н, м).
Пример 19, Схема 9: этил (2S,3R)-3-{[2-(L-аланиламино)пиридин-4-ил]метил}-1-{[(1R)-1-(2,2-дифтор-1,3-бензодиоксол-5-ил)этил]карбамоил}-4-оксоазетидин-2-карбоксилат трифторацетат
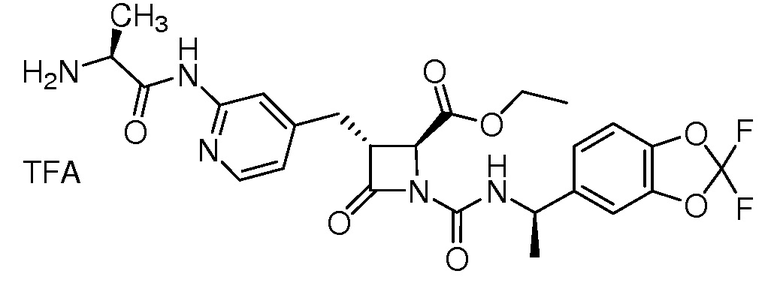
К раствору бензил (2S,3R)-3-[(2-аминопиридин-4-ил)метил]-1-{[(1R)-1-(2,2-дифтор-1,3-бензодиоксол-5-ил)этил]карбамоил}-4-оксоазетидин-2-карбоксилата (150 мг, 0,28 ммоль) и N-(трет-бутоксикарбонил)-L-аланина (79 мг, 0,42 ммоль) в N,N-диметилформамиде (1,7 мл) добавляли N,N-диизопропилэтиламин (0,194 мл, 1,11 ммоль), а затем гексафторфосфат N,N,N',N'-тетраметил-O-(7-азабензотриазол-1-ил)урония (159 мг, 0,42 ммоль). Через 72 ч реакцию разбавляли этилацетатом и промывали насыщенным раствором NaHCO3, насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток частично очищали флэш-хроматографией, используя гексан/этилацетат (30-40%) в качестве элюента с получением бензил (2S,3R)-3-[(2-{[N-(трет-бутоксикарбонил)-L-аланил]амино}пиридин-4-ил)метил]-1-{[(1R)-1-(2,2-дифтор-1,3-бензодиоксол-5-ил)этил]карбамоил}-4-оксоазетидин-2-карбоксилата (80 мг, приблизительно 80% чистота, 40%) в виде светло-коричневого твердого вещества, которое использовали без дополнительной очистки. MS (ESI+) для C35H37F2N5O9 m/z 710,2 (M+H)+.
В колбу, содержащую PdPd/C (10%, 13 мг) добавляли раствор вышеуказанного промежуточного вещества (130 мг, комбинация двух партий сходной чистоты) в этаноле (5 мл). Смесь перемешивали в 1 атмосфере Н2 в течение 5 ч. Добавляли еще PdPd/C (10%, 5 мг) в атмосфере азота, и смесь перемешивали еще 3 ч в 1 атмосфере Н2. Смесь фильтровали через целит и концентрировали при пониженном давлении с получением (2S,3R)-3-[(2-{[N-(трет-бутоксикарбонил)-L-аланил]амино}пиридин-4-ил)метил]-1-{[(1R)-1-(2,2-дифтор-1,3-бензодиоксол-5-ил)этил]карбамоил}-4-оксоазетидин-2-карбоновой кислоты (100 мг), в виде не совсем белого твердого вещества, которое использовали без дополнительной очистки. MS (ESI+) для C28H31F2N5O9 m/z 620,2 (M+H)+.
К раствору полученной (2S,3R)-3-[(2-{[N-(трет-бутоксикарбонил)-L-аланил]амино}пиридин-4-ил)метил]-1-{[(1R)-1-(2,2-дифтор-1,3-бензодиоксол-5-ил)этил]карбамоил}-4-оксоазетидин-2-карбоновой кислоты (100 мг, 0,16 ммоль) в CH2Cl2 (1 мл) добавляли этанол (0,28 мл, 4,84 ммоль), а затем гидрохлорид N-(3-диметиламинопропил)-N'-этилкарбодиимида (0,120 г, 0,62 ммоль) и DMAP (1 мг). Через 16 ч смесь разбавляли этилацетатом и промывали 0,1 водным раствором НCl, насыщенным раствором NaHCO3, насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток растворяли в CH2Cl2 (4 мл) и охлаждали до 0-5°C. К охлажденному раствору добавляли трифторуксусную кислоту (2 мл). Реакционную смесь перемешивали при 0-5°С в течение 30 мин, затем при температуре окружающей среды в течение 1 ч. Летучие вещества удаляли при пониженном давлении, и остаток очищали CombiFlash хроматографией [30 г RediSep C-18 gold силикагелевый картридж, градиент растворителя: от 10% ацетонитрил (0,07% TFA)/вода (0,1% TFA) до 100% ацетонитрил (0,07% TFA)] и лиофилизовали с получением соединения, указанного в заголовке, (24 мг, 22%) в виде белого твердого продукта. 1H ЯМР (CD3OD) δ 1,09 (т, J=7 Гц, 3Н), 1,54 (д, J=7 Гц, 3Н), 1,62 (д, J=7 Гц, 3Н), 3,15-3,29 (м, 2Н), 3,69-3,74 (м, 1Н), 4,05-4,18 (м, 3Н), 4,28 (д, J=3 Гц, 1Н), 4,95-4,98 (м, 1Н), 7,14-7,24 (перекрываемый м, 4Н), 8,07 (ушир.с, 1Н), 8,28 (д, J=5 Гц, 1Н); MS (ESI+) для C25H27F2N5O7 m/z 548,1 (M+H)+; Время удержания ВЭЖХ: 3,58 мин (Способ C).
Пример 20, Схема 10: этил (2S,3R)-1-{[(1R)-1-(2,2-дифтор-1,3-бензодиоксол-5-ил)этил]карбамоил}-3-{[2-({[(изобутирилокси)метокси]карбонил}амино)пиридин-4-ил]метил}-4-оксоазетидин-2-карбоксилат
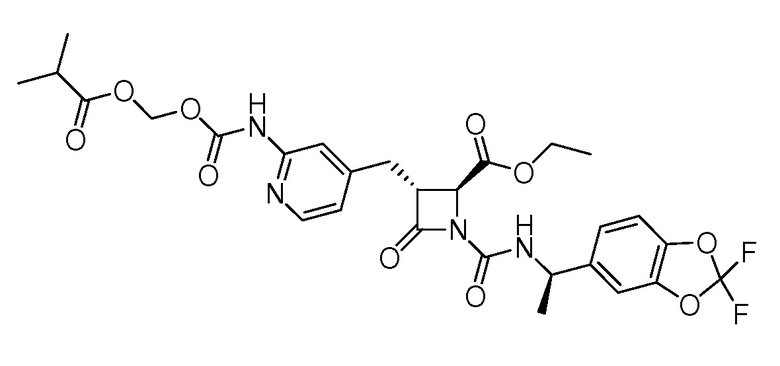
Стадия 1. Получение хлорметилового эфира 4-нитро-фенилового эфира карбоновой кислоты
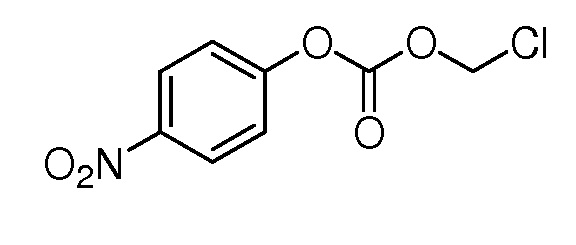
К раствору 4-нитрофенола (3,81 г, 0,027 моль) в THF (50 мл) добавляли хлорметил хлорформиат (4,00 г, 0,030 моль), а затем N,N-диизопропилэтиламин (5,29 мл, 0,030 моль). Смесь перемешивали в течение 2 ч, разбавляли этилацетатом и промывали насыщенным раствором NaHCO3, насыщенным солевым раствором, сушили безводным MgSO4, фильтровали и концентрировали с получением соединения, указанного в заголовке, (6,10 г, 96%) в виде желтого твердого вещества, которое использовали без дополнительной очитки. 1H ЯМР (CDCl3) δ 5,88 (с, 2Н), 7,44 (д, J=9 Гц, 2H), 8,32 (д, J=9 Гц, 2H).
Стадия 2. Получение йодметилового эфира 4-нитро-фенилового эфира карбоновой кислоты
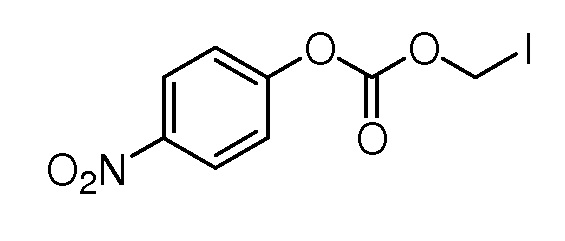
К раствору хлорметилового эфира 4-нитро-фенилового эфира карбоновой кислоты (3,00 г, 0,013 моль) в ацетоне (60 мл) добавляли йодид натрия (5,82 г, 0,039 моль) и 4 молекулярное сито (3,00 г). Смесь нагревали при 40°C до того, как считали реакцию завершенной путем исследования аликвоты реакционной смеси с помощью 1H ЯМР (приблизительно 6 ч). Смесь охлаждали до температуры окружающей среды и пропускали через пад из целита. Летучие вещества удаляли при пониженном давлении, и остаток растворяли в CH2Cl2, промывали насыщенным раствором NaHCO3, водой, насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением соединения, указанного в заголовке, (3,82 г, 91%) в виде твердого вещества, которое использовали без дополнительной очистки. 1H ЯМР (CDCl3) δ 6,08 (с, 2Н), 7,44 (д, J=9 Гц, 2Н), 8,32 (д, J=9 Гц, 2Н).
молекулярное сито (3,00 г). Смесь нагревали при 40°C до того, как считали реакцию завершенной путем исследования аликвоты реакционной смеси с помощью 1H ЯМР (приблизительно 6 ч). Смесь охлаждали до температуры окружающей среды и пропускали через пад из целита. Летучие вещества удаляли при пониженном давлении, и остаток растворяли в CH2Cl2, промывали насыщенным раствором NaHCO3, водой, насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением соединения, указанного в заголовке, (3,82 г, 91%) в виде твердого вещества, которое использовали без дополнительной очистки. 1H ЯМР (CDCl3) δ 6,08 (с, 2Н), 7,44 (д, J=9 Гц, 2Н), 8,32 (д, J=9 Гц, 2Н).
Стадия 3. Получение 2-метилпропионата серебра
К раствору 2-метилпропионовой кислоты (2,65 г, 0,030 моль) в ацетонитриле (100 мл) добавляли оксид серебра (I) (4,12 г, 0,018 моль). Колбу защищали от света и нагревали при 70°С в течение 90 мин. Смесь охлаждали до температуры окружающей среды и фильтровали через пад из целита. Летучие вещества удаляли в вакууме с получением соединения, указанного в заголовке, (5,65 г, 96%) в виде светло-коричневого твердого вещества, которое использовали без дополнительной очистки или характеристики.
Стадия 4. Получение 4-нитрофеноксикарбонилоксиметилового эфира 2-метилпропионовой кислоты
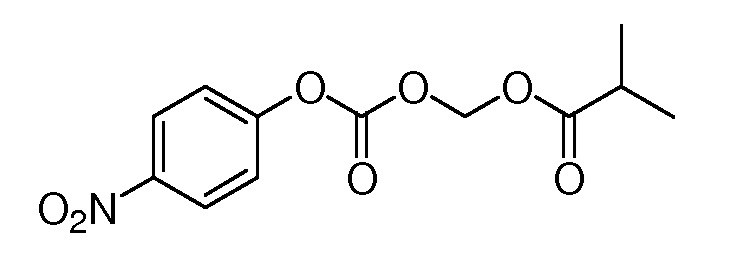
К раствору йодометилового эфира 4-нитрофенилового эфира карбоновой кислоты (2,10 г, 6,50 ммоль) в толуоле (30 мл) добавляли 2-метилпропаноат (2,53 г, 13,0 ммоль). Смесь нагревали при 55°C до того, как считали реакцию завершенной, путем исследования аликвоты реакционной смеси с помощью 1H ЯМР (приблизительно 5 h). Смесь охлаждали до температуры окружающей среды, фильтровали через пад из целита и промывали 10% водным раствором K2CO3, водой, насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали флэш-хроматографией, используя гексан и этилацетат (5 %) в качестве элюента с получением соединения, указанного в заголовке, (1,51 г, 82%) в виде масла: 1H ЯМР (CDCl3) δ 1,25 (д, J=7 Гц, 6Н), 2,68 (гептет, J=7 Гц, 1Н), 5,91 (с, 2Н), 7,42 (д, J=9 Гц, 2Н), 8,31 (д, J=9 Гц, 2Н).
Стадия 5. Получение (2S,3R)-1-{[(1R)-1-(2,2-дифтор-1,3-бензодиоксол-5-ил)этил]карбамоил}-3-{[2-({[(изобутирилокси)метокси]карбонил}амино)пиридин-4-ил]метил}-4-оксоазетидин-2-карбоновой кислоты
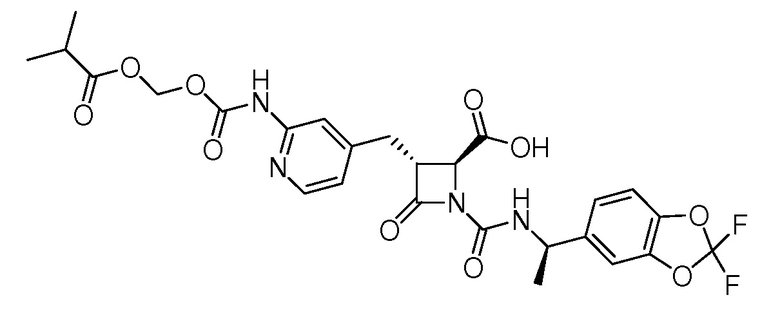
К перемешанной смеси трифторацетата (2S,3R)-3-[(2-аминопиридин-4-ил)метил]-1-{[(1R)-1-(2,2-дифтор-1,3-бензодиоксол-5-ил)этил]карбамоил}-4-оксоазетидин-2-карбоновой кислоты (0,200 г, 0,36 ммоль) в CH2Cl2 (0,5 мл) добавляли хлортриметилсилан (0,180 мл, 1,42 ммоль) и N,N-диизопропилэтиламин (0,217 мл, 1,24 ммоль). Смесь нагревали при 40°С в течение 1 ч, затем охлаждали до температуры окружающей среды. Добавляли раствор 4-нитрофеноксикарбонилоксиметилового эфира 2-метилпропионовой кислоты (0,201 г, 0,72 ммоль) в CH2Cl2 (0,36 мл), а затем N,N-диизопропилэтиламин (0,124 мл, 0,72 ммоль). Смесь нагревали при 40°С в течение 2 д, затем охлаждали до температуры окружающей среды. Реакцию гасили дополнительным количеством 0,1 водного раствора НCL (pH приблизительно 3), перемешивали в течение 15 мин, затем экстрагировали этилацетатом. Объединенные органические слои промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали флэш-хроматографией, используя гексан и этилацетат (20 %), а затем CH2Cl2/метаноле (1-2%) в качестве элюента с получением соединения, указанного в заголовке, (0,080 г, 38%) в виде стекловидного белого твердого продукта: 1H ЯМР (CDCl3) δ 1,19 (д, J=6 Гц, 6Н), 1,56 (д, J=8 Гц, 3Н), 2,61 (гептет, J=7 Гц, 1Н), 3,11-3,31 (м, 2Н), 3,68-3,74 (м, 1Н), 4,32 (д, J=2 Гц, 1Н), 5,01 (пентет, J=7 Гц, 1Н), 5,83-5,88 (м, 2Н), 6,75 (д, J=8 Гц, 1Н), 7,04-7,08 (м, 4Н), 8,06-8,12 (перекрываемый м, 2Н), 9,79 (очень ушир.с, 1Н); MS (ESI+) для C26H26F2N4O10 m/z 593,2 (M+H)+; Время удержания ВЭЖХ: 3,98 мин (Способ C).
Стадия 6. Получение этил (2S,3R)-1-{[(1R)-1-(2,2-дифтор-1,3-бензодиоксол-5-ил)этил]карбамоил}-3-{[2-({[(изобутирилокси)метокси]карбонил}амино)пиридин-4-ил]метил}-4-оксоазетидин-2-карбоксилата
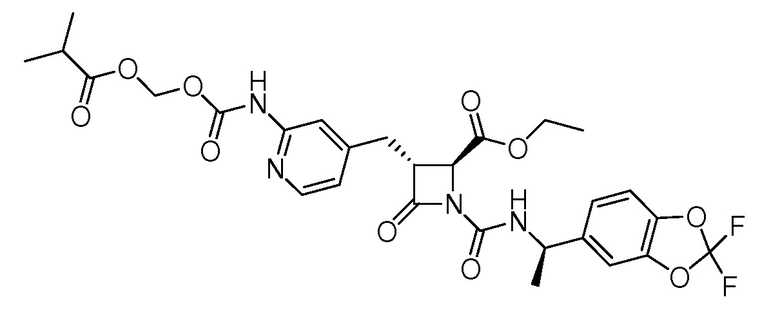
К раствору (2S,3R)-1-{[(1R)-1-(2,2-дифтор-1,3-бензодиоксол-5-ил)этил]карбамоил}-3-{[2-({[(изобутирилокси)метокси]карбонил}амино)пиридин-4-ил]метил}-4-оксоазетидин-2-карбоновой кислоты (60 мг, 0,10 ммоль) в CH2Cl2 (1 мл) добавляли этанол (0,118 мл, 2,02 ммоль), а затем гидрохлорид N-(3-диметиламинопропил)-N'-этилкарбодиимида (23 мг, 0,12 ммоль) и 4-диметиламинопиридина (0,6 мг, 0,006 ммоль). Смесь перемешивали при температуре окружающей среды в течение ночи, разбавляли этилацетатом и промывали 0,25 н водным раствором НCl, водой, насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали флэш-хроматографией, используя гексан/этилацетат (20-40%) в качестве элюента с получением соединения, указанного в заголовке, (35 мг, 60%) в виде белого твердого продукта: 1H ЯМР (CDCl3) δ 1,14-1,22 (перекрываемый триплет, 9Н), 1,54 (д, J=7 Гц, 3Н), 2,63 (гептет, J=7 Гц, 1Н), 3,10-3,27 (м, 2Н), 3,54-3,60 (м, 1Н), 4,13-4,24 (перекрываемый м, 3Н), 4,99 (пентет, J=6 Гц, 1Н), 5,89 (с, 2Н), 6,65 (д, J=7 Гц, 1Н), 6,98-7,07 (перекрываемый м, 4Н), 8,02 (с, 1Н), 8,31 (д, J=5 Гц, 1Н), 9,66 (с, 1Н); MS (ESI+) для C28H30F2N4O10 m/z 621,1 (M+H)+; Время удержания ВЭЖХ: 5,01 мин (Способ C).
Пример 21, Схема 11: (2S,3R)-3-[2-(4-метоксифенил)этил]-3-метил-4-оксо-1-{[(1R)-1-фенилэтил]карбамоил}азетидин-2-карбоновая кислота
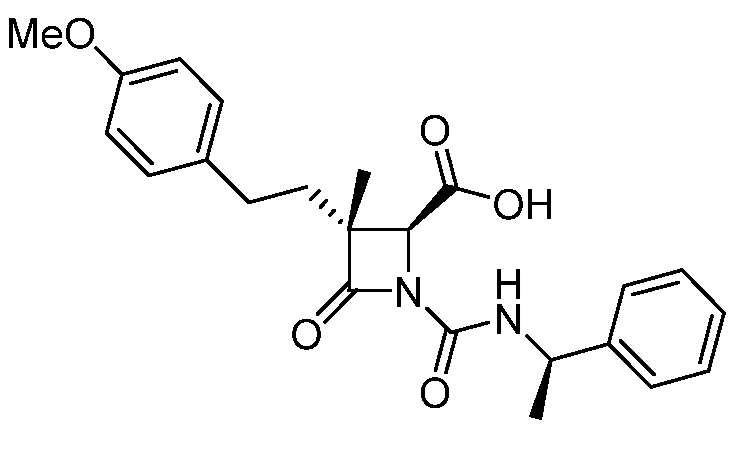
Стадия 1: Получение бензил-(2S,3R)-1-[трет-бутил(диметил)силил]-3-[2-(4-метоксифенил)этил]-3-метил-4-оксоазетидин-2-карбоксилата
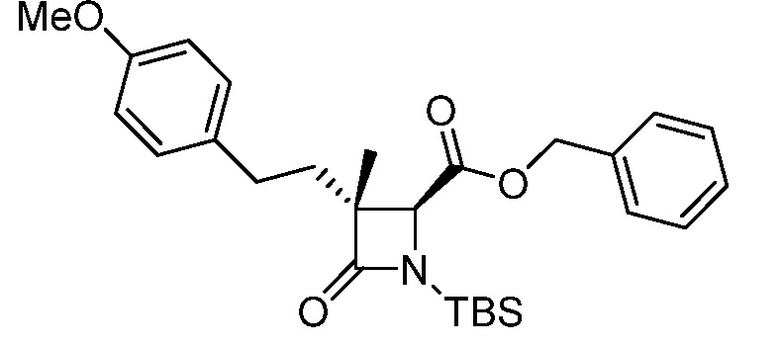
50 мл 2-горлую круглодонную колбу наполняли (2S,3R)-1-[трет-бутил(диметил)силил]-3-метил-4-оксоазетидин-2-карбоновой кислотой (450,0 мг, 1,849 ммоль), полученной способом Finke, P. E., et. al., J. Med. Chem. 1995, 38, 2449. Колбу откачивали и наполняли азотом три раза. Добавляли тетрагидрофуран (4,2 мл), и раствор охлаждали при 0°C на ледяной бане. По каплям добавляли 1,45M раствор LDA в гептан/THF/этилбензол (2,8 мл, 1,76 ммоль), и раствор перемешивали в течение 55 мин при 0°C. По каплям добавляли раствор 1-бром-2-(4-метоксифенил)этана (0,54 мл, 3,5 ммоль) в тетрагидрофуране (2 мл), и перемешивание продолжали при 0°С в течение 2 ч, а затем нагревали реакционную смесь до комн. темп. Через 6 ч при комнатной температуре реакционную смесь разбавляли 25 мл этилацетатом и выливали в 14 мл ледяного раствора 0,5M KHSO4, который находился в делительной воронке. Смесь экстрагировали, и фазы разделяли. Водную фазу промывали два раза порциями по 20 мл этилацетата, и объединенную органическую фазу промывали 20 мл насыщенным солевым раствором. Органическую фазу сушили над Na2SO4, фильтровали и концентрировали с выходом светло-коричневого масла. Сырой продукт помещали в метиленхлорид (11 мл) и обрабатывали гидрохлоридом N-(3-диметиламинопропил)-N'-этилкарбодиимида (390 мг, 2,03 ммоль), а затем бензиловым спиртом (210 мкл, 2,03 ммоль) и 4-диметиламинопиридином (11 мг, 0,094 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 7 ч. Реакционную смесь разбавляли 35 мл CH2Cl2 и промывали двумя порциями по 25 мл Н2O и 20 мл насыщенным солевым раствором. Органическую фазу сушили над Na2SO4, фильтровали и концентрировали с выходом светло-коричневого масла. Сырое вещество очищали флэш-хроматографией (75 г силикагель; 5-20% этилацетат/гекс.) с выходом 168 мг соединения, указанного в заголовке, загрязнен неидентифицированными примесями в виде желтого масла. Чистота по данным ВЭЖХ была 57-62%; Время удержания ВЭЖХ 5,77 мин (Способ A).
Стадия 2: Получение бензил (2S,3R)-3-[2-(4-метоксифенил)этил]-3-метил-4-оксоазетидин-2-карбоксилата
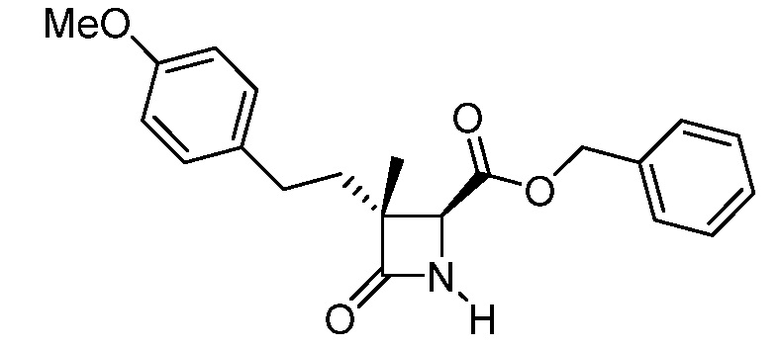
Раствор бензил (2S,3R)-3-[2-(4-метоксифенил)этил]-3-метил-4-оксоазетидин-2-карбоксилата (260 мг, 0,35 ммоль) в метаноле (5,3 мл) обрабатывали уксусной кислотой (72 мкл, 1,3 ммоль), а затем 0,5 M NH4F в метаноле (1,0 мл, 0,52 ммоль) по каплям, и смесь перемешивали при комнатной температуре в течение 20 ч, в это время ВЭЖХ указала на то, что исходное вещество все еще присутствовало. Реакционную смесь обрабатывали раствором 16 мкл НOAc и 190 мкл 0,5M NH4F и оставляли перемешиваться при комнатной температуре еще 20 ч, и в этот момент ВЭЖХ указала на завершение реакции. Реакционную смесь концентрировали, и остаток помещали в 10 мл толуола и концентрировали. Процесс еще раз повторяли, и остаток помещали в 25 мл CH2Cl2, и промывали порциями по 20 мл раствораН2O и NaHCO3. Органическую фазу сушили над Na2SO4, фильтровали и концентрировали с выходом светло-желтого вязкого масла. Сырой продукт очищали флэш-хроматографией (25 г силикагель; 20-40% этилацетат/гекс.) с выходом соединения, указанного в заголовке, (80 мг) в виде бесцветного стекла: Время удержания ВЭЖХ 3,81 мин (Способ A); ¹H ЯМР (400 МГц, CDCl3) δ 7,39 (м, 5Н), 7,11 (д, J=8,59 Гц, 2Н), 6,83 (м, 2Н), 6,22 (ушир.с., 1Н), 5,23 (м, 2Н), 4,08 (с, 1Н), 3,79 (с, 3Н), 2,68 (м, 2Н), 2,00 (м, 2Н), 1,19 (с, 3Н).
Стадия 3: Получение бензил (2S,3R)-3-[2-(4-метоксифенил)этил]-3-метил-4-оксо-1-{[(1R)-1-фенилэтил]карбамоил}азетидин-2-карбоксилата
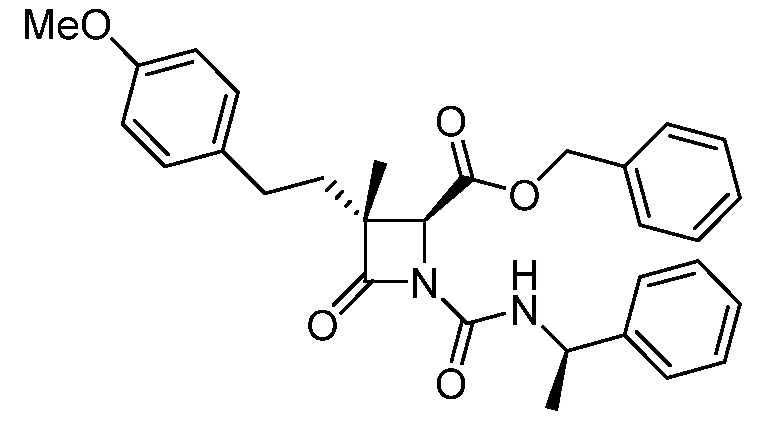
Раствор бензил (2S,3R)-3-[2-(4-метоксифенил)этил]-3-метил-4-оксоазетидин-2-карбоксилата (60,0 мг, 0,170 ммоль) в метиленхлориде (1,8 мл) обрабатывали триэтиламином (95 мкл, 0,679 ммоль), а затем [(1R)-1-изоцианатоэтил]бензолом (31 мкл, 0,221 ммоль), по каплям, и смесь перемешивали при комнатной температуре в течение 18 ч, и в этот момент ВЭЖХ указала на расход исходного продукта. Реакционную смесь концентрировали, остаток помещали в 10 мл CH2Cl2 и концентрировали до бесцветного вязкого масла/твердого вещества. Сырой продукт очищали препарат ТСХ (20см × 20см × 1,0мм преп. ТСХ планшет; 30% этилацетат/гекс.) с выходом соединения, указанного в заголовке, (72 мг) в виде бесцветного стекла: Время удержания ВЭЖХ 4,84 мин (Способ A); ¹H ЯМР (400 МГц, CDCl3) δ 7,35 (м, 9Н), 7,29 (м, 1Н), 7,10 (д, J=8,59 Гц, 2Н), 6,84 (м, 2Н), 6,71 (д, J=7,83 Гц, 1Н), 5,27 (м, 1Н), 5,18 (м, 1Н), 5,09 (м, 1Н), 4,43 (с, 1Н), 3,80 (с, 3Н), 2,72 (м, 1Н), 2,62 (м, 1Н), 2,04 (м, 2Н), 1,57 (д, J=6,82 Гц, 3Н), 1,17 (с, 3Н).
Стадия 4: Получение (2S,3R)-3-[2-(4-метоксифенил)этил]-3-метил-4-оксо-1-{[(1R)-1-фенилэтил]карбамоил}азетидин-2-карбоновой кислоты
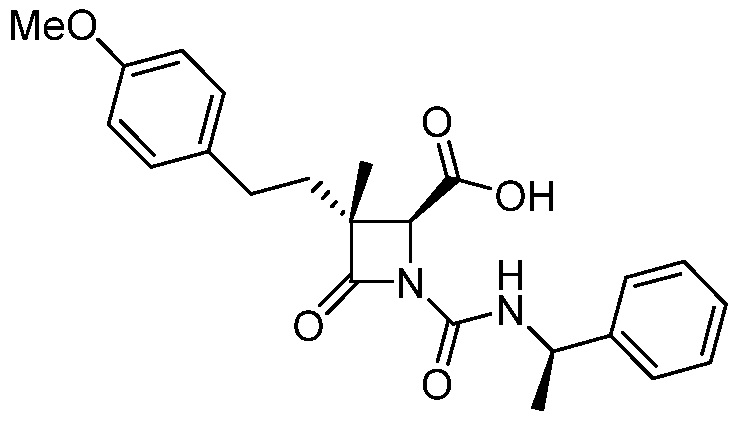
Раствор бензил (2S,3R)-3-[2-(4-метоксифенил)этил]-3-метил-4-оксо-1-{[(1R)-1-фенилэтил]карбамоил}азетидин-2-карбоксилата (72 мг, 0,14 ммоль) в метаноле (2,3 мл) и этилацетате (2,3 мл) аккуратно обрабатывали 10% палладием на угле (13 мг). Реакционную колбу откачивали и наполняли водородом три раза, и реакционную смесь перемешивали при комнатной температуре в атмосфере водорода в течение 3,5 ч, и в этот момент ТСХ (25% этилацетат/гекс.) указала на расход SM. Реакционную смесь фильтровали через пад из Solka-Floc®, и пад промывали 30 мл 1/1 этилацетат/MeOH. Фильтрат концентрировали до бесцветного стекла, которое помещали в водный раствор метанола и лиофилизовали с выходом соединения, указанного в заголовке, (56 мг) в виде белого твердого продукта: Время удержания ВЭЖХ 4,19 мин (Способ A); MS (ESI+) для C23H26N2O5 m/z 411,2 (M+H)+; MS (ESI-) для C23H26N2O5 m/z 409,2 (M-H)-; 1H ЯМР (400 МГц, МeOD) δ 7,35 (м, 4Н), 7,26 (м, 1Н), 7,15 (м, 2Н), 7,11 (д, J=7,83 Гц, 1Н), 6,84 (м, 2Н), 4,97 (м, 1Н), 4,43 (с, 1Н), 3,76 (с, 3Н), 2,77 (м, 1Н), 2,65 (м, 1Н), 2,05 (м, 2Н), 1,52 (д, J=7,07 Гц, 3Н), 1,31 (с, 3Н).
Пример 22. Анализ доза-ответ для ингибиторов протеазы
Вещества:
Аналитический буфер : 20 мMНepes, pH 7,4; 150 мM NaCl; 0,02% Tween 20
Соединения : 10 мM исходный раствор в DMSO
Субстрат : 20 мM Glp-Pro-Arg-AMC, 25 мг/2,3 мл Н2O (хранение при +4°C) [Фактор XIa, тромбин и трипсин]
20 мM Pro-Phe-Arg-AMC (Bachem I-1295), 25 мг/2,2 мл Н2O (хранение при +4°C) [Фактор Xa]
Фермент : Фактор XIa; 0,25 мкM в 50% глицерине (20 мкг/мл)
Трипсин; 0,2 мкМ в 50% глицирине (4,8 мкг/мл)
Тромбин; 0,2 мкM в 50% глицирине (7,34 мкг/мл)
Фактор Xa; 0,2 мкM в 50% глицирине (9,2 мкг/мл)
Из этих исходных растворов брали аликвоты (~100 мкл/аликвота) и хранили при-20°C
Способы:
1. Разбавляют субстрат до 100 мкМ в аналитическом буфере (30 мкл/6 мл). Фермент разбавляют до 0,5 нМ непосредственно перед использованием (12 мкл/6 мл для Фактор XIa; 15 мкл/6 мл для всех остальных).
2. Берут пипеткой 50 мкл субстрата в каждую лунку 96 луночного (12×8) микротитровального планшета. (Колонка 1 используется как контроль 100% активности, и в нее не помещают соединение, а колонка 12 является пустой и в нее не помещают фермент.) Добавляют дополнительные 46 мкл в колонку 2.
3. Берут пипеткой 4 мкл каждого соединения в соответствующую лунку в колонке 2 планшета (неизвестные анализируют в трех повторах, стандартные анализируют в двух повторах). Конечная концентрация соединения составляет 1/50 исходного раствора.
4. Серийно двукратно разбавляют соединение путем смешивания образца в колонке 2, перенося 50 мкл в следующую лунку (колонка 3), смешать и переместить в колонку 4, и так далее, до колонки 11. После смешивания колонки 11, отбирают 50 мкл и выбрасывают.
5. Берут пипеткой 50 мкл буфера в колонку 12. Инициируют реакцию, добавляя 50 мкл раствора фермента в каждую лунку колонок 1-11 так быстро, как возможно.
6. Считывают планшет на спектрофотометре (SpectraMax) при 30°C, где каждую лунку измеряют каждые 60 с в течение 30 мин. Для анализа Фактора Xa каждую лунку измеряют каждую 1 мин в течение общей сложности 60 мин.
7. Для анализа Фактора XIa соединения анализируют в повторе при 3 различных начальных концентрациях, 20, 2 и 0,2 мкм; 1:10, 1:100, 1:1000 разведение 10 мМ исходного раствора. Все наборы данных объединяют для построения графика и выравнивания данных.
8. Данные могут быть использованы как для оценки IC50 и для оценки Kon.
Пример 23. Стабильность β-лактама в плазме крысы
Исходные реагенты:
Нормальная плазма крови крыс, хранили при -80°C
Протокол:
1. Помещали 6 мкл каждого соединения в 0,5 мл пробирки для микроцентрифугирования или в 96-луночный полипропиленовый микротитровальный планшет с дном в форме U.
2. Помещали 40 мкл ацетонитрила (AcN) в 0,5 мл пробирки для микроцентрифугирования, обозначенные 1-9.
3. Добавляли 114 мкл плазмы к каждому соединению.
4. Отбирали образец 10 мкл соединение/плазма в AcN на 2, 10, 20, 30, 60, 90, 120, 240 и 480 мин инкубации при комнатной температуре.
5. После добавления образца каждой временной точки проводили смешивание и помещали на лед.
6. Центрифугировали образцы временных точек (12000 об/мин, 3 мин), удаляли 15 мкл супернатанта и смешивали с 15 мкл 0,1% TFA в микротитровальном планшете с V-образным дном.
7. Помещали микротитровальный планшет в автоматический пробоотборник ВЭЖХ и анализировали на колонке Restek Pinnacle C18 (2,1×100 мм).
8. Получали найденную ионную хроматограмму для родительского соединения и родительского соединения +18 (H2O). Интегрировали найденную ионную хроматограмму (EIC) и графически изображали % всей области пика для родительского соединения и продукта против времени. Подгоняли к модели одиночной фазы экспоненциального распада [Y=A0×ехр(-k×х)+C]. T½ равно ln(2)/k.
Пример 24. Анализы ингибирования фермента Фактора XIa
Способность соединений по настоящему изобретению ингибировать Фактор XIa оценивали путем определения концентрации ингибитора, что дает 50% уменьшение ферментной активности (IC50), используя очищенный фермент. Потенциальные ингибиторы фактора XIa оценивали, используя следующий анализ.
S-2366, pyroGlu-Pro-Arg-7-метиламинокурин (AMC), доступный от CPC Scientific, Inc., основан на субстрате pyro-Glu-Pro-Arg-pNA, доступный от Diapharma Group, Inc. (Columbus, Ohio), где п-нитроаналиновая группа заменена 7-метиламинокумарином (AMC).
Конечная концентрация субстрата в анализе была 50 мкM, и конечная концентрация фермента была 0,25 нМ. Ингибиторы протестировали путем серийного растворения в соответствующем диапазоне с получением кривой доза-ответ для определения значения IC50 ингибиторов. Аналитическую смесь считывали каждую минуту в течение 30 минут для получения прогрессивных кривых. Планшеты считывали на ридере для планшетов Spectramax m5 Multimode Plate Reader (Molecular Devices LLC, Sunnyvale, CA). Кривые доза-ответ подгоняли к уравнению 1, ниже в котором А является максимальным ингибированием, B является минимальным ингибированием, C является IC50, а D является коэффициентом Хилла.
[(А-В)/(1+(X/C)D)]+В (уравнение 1)
Желательные соединения имеют значение IC50 для ингибирования Фактора XIa менее чем 1 мкмоль, 100 наномоль, 10 наномоль или 1 наномоль (в порядке возрастания предпочтения).
Активность, селективность и стабильность соединений примеров
IC50 (нM)
Для Таблицы 2: FXIa и hFXIa относятся к фактору XIa и фактору XIa человека, соответственно.
Активность: “A” означает <10 нM, “B” означает 10-100 нM, “C” означает 100-1000 нM, “D” означает >1000 нM и “E” означает, что данные не доступны или не были определены.
Селективность: «F» означает <1, "G" означает 1-500, «H» означает 500-1000, «I» означает >1000 и «J» означает, что данные не доступны или не были определены.
Стабильность в плазме крысы: «K» означает 0-100 мин; "L" означает >100 мин; «М» означает, что данные не доступны или не были определены.
Пример 25. Анализы на растворимость
Следующая процедура была использована для определения водной растворимости тестируемого соединения в фосфатно-солевом буфере (PBS - NaCl 137 мМ, KCl 2,7 мМ, Na2HPO4 8,1 мМ, KН2PO4 1,5 мМ, рН 7,4) в формате 96-луночного планшета с помощью анализа ВЭЖХ-УФ/VIS. Тестируемое соединение получали при 200 мкМ в PBS из 10 мМ исходного раствора DMSO. Конечная концентрация DMSO была 2%. Образцы PBS буфера тщательно смешивали, а затем инкубировали при комнатной температуре в течение 24 ч. По окончании инкубирования образцы PBS центрифугировали и супернатанты анализировали с помощью ВЭЖХ. Водную растворимость (мкМ) тестируемого соединения в PBS определяли путем сравнения площади пика основного пика в калибровочном стандарте (200 мкМ) с площадью пика соответствующего пика в каждом образце PBS. Диапазон анализа был приблизительно от 0,5 мкМ до 200 мкМ. Эталонными соединениями, используемыми в каждом анализе, были метопролол, рифампицин, кетоконазол, фенитоин, галоперидол, симвастатин, диэтилстильбестрол и тамоксифен, расположенные от полностью растворимого (200 мкм) до труднорастворимого (<1 мкм).
Пример 26. Анализ метаболической стабильности
Следующая методика была использована для определения стабильности тестируемого соединения в объединенных микросомах печени человека (смешанного пола) в формате 96-луночного планшета. Исследуемое соединение количественно оценивали в пяти временных точках с помощью анализа ВЭЖХ-MS/MS. Конечная концентрация микросомального белка в анализе составляла 0,1 мг/мл. Каждое соединение тестировали при 0,1 мкМ вместе с 0,01% DMSO, 0,25% ацетонитрилом и 0,25% метанолом. Тестируемое соединение предварительно инкубировали с микросомами печени человека в фосфатном буфере (рН 7,4) в течение 5 мин при 37°C , встряхивая на водяной бане. Реакционную смесь инициировали, добавляя NADPH-генерирующую систему, и инкубировали в течение 0, 15, 30, 45, и 60 минут. Реакцию останавливают, перенося инкубационную смесь в смесь ацетонитрил/метанол. Затем образцы смешивали и центрифугировали, и супернатанты использовали для анализа ВЭЖХ-МС/МС. Площади пиков, соответствующие тестируемому соединению, записывали. Оставшееся соединение рассчитывали путем сравнения площади пика в каждый момент времени с нулевым моментом времени. В каждом анализе тестировали четыре ссылочных соединения; пропранолол и имипрамин относительно стабильны, в то время как верапамил и терфенадин легко метаболизируются в микросомах печени человека.
Пример 27. Анализы связывания белков плазмы
Следующую методику использовали для определения связывания белков плазмы испытуемым соединением в объединенной плазме человека (смешанного пола) посредством равновесного диализа в формате 96-луночного планшета. В диализный отсек наливали фосфатный буфер (рН 7,4), а в часть образца помещали с плазму, обогащенную испытуемым соединением, при концентрации 10 мкМ. После загрузки образцов накрывали и инкубировали в течение 4 часов при 37°С. После инкубации из каждого отсека брали образец, разбавляли ацетонитрил/буфер и центрифугировали. Супернатанты анализировали с помощью ВЭЖХ-МС/МС. Количество измеренного в отсеке с плазмой включает как свободное, так и связанное лекарственные средства, в то время как на стороне с буфером присутствует только свободное лекарственное средство; различия использовали для вычисления процента связанного белка плазмы. В каждом анализе тестировали три ссылочных соединения; ацебутолол, хинидин и варфарин. Эти соединения дают значения связывания с белком, соответствующие низкому, среднему и высокому значению связывания с белками плазмы человека, соответственно.
Связывание с белками плазмы, растворимость и метаболическая стабильность соединений примеров
Плазма человека: % связывания; 10 мкM: 4 ч инкубирования при 37°C
(мкM в PBS); 10 мM: 4 ч инкубирования при 37°C
Микросомы человека: полужизнь (мин)
0,1 мкM: точки времени: 0, 15, 30, 45, 60 мин при 37°C
R означает >100 мкМ; S означает 10-100 мкМ; T означает 1-10 мкМ; U означает <1 мкM
V означает >60 минут; W означает 30-60 минут; Х означает <30 минут; Y означает, что данные не доступны, не обнаруживаются или не были определены
| название | год | авторы | номер документа |
|---|---|---|---|
| ТЕРАПЕВТИЧЕСКИЕ СОЕДИНЕНИЯ И КОМПОЗИЦИИ | 2019 |
|
RU2817800C2 |
| ИНГИБИТОРЫ БЕТА-ЛАКТАМАЗ | 2013 |
|
RU2654692C2 |
| ЗАМЕЩЕННЫЕ ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И СПОСОБ ПРИМЕНЕНИЯ | 2016 |
|
RU2744766C2 |
| ПРОЛЕКАРСТВА МЕТИЛГИДРОФУМАРАТА, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ С НИМИ И СПОСОБЫ ПРИМЕНЕНИЯ | 2009 |
|
RU2554347C2 |
| ИНГИБИРОВАНИЕ CREB-СВЯЗЫВАЮЩЕГО БЕЛКА (CBP) | 2019 |
|
RU2803290C2 |
| ИНГИБИТОРЫ БЕТА-ЛАКТАМАЗ И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2815314C2 |
| ЗАМЕЩЕННЫЕ ДИАМИНОКАРБОКСАМИДНЫЕ И ДИАМИНОКАРБОНИТРИЛЬНЫЕ ПРОИЗВОДНЫЕ ПИРИМИДИНОВ, ИХ КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ С ИХ ПОМОЩЬЮ | 2012 |
|
RU2697712C2 |
| ЗАМЕЩЕННЫЕ ХРОМАНЫ | 2015 |
|
RU2718060C2 |
| ЗАМЕЩЕННЫЕ ПУРИНОВЫЕ И 7-ДЕАЗАПУРИНОВЫЕ СОЕДИНЕНИЯ | 2011 |
|
RU2606514C2 |
| 3-КАРБОНИЛАМИНО-5-ЦИКЛОПЕНТИЛ-1H-ПИРАЗОЛЬНЫЕ СОЕДИНЕНИЯ, ОБЛАДАЮЩИЕ АКТИВНОСТЬЮ ИНГИБИТОРОВ CDK2 | 2020 |
|
RU2797889C2 |
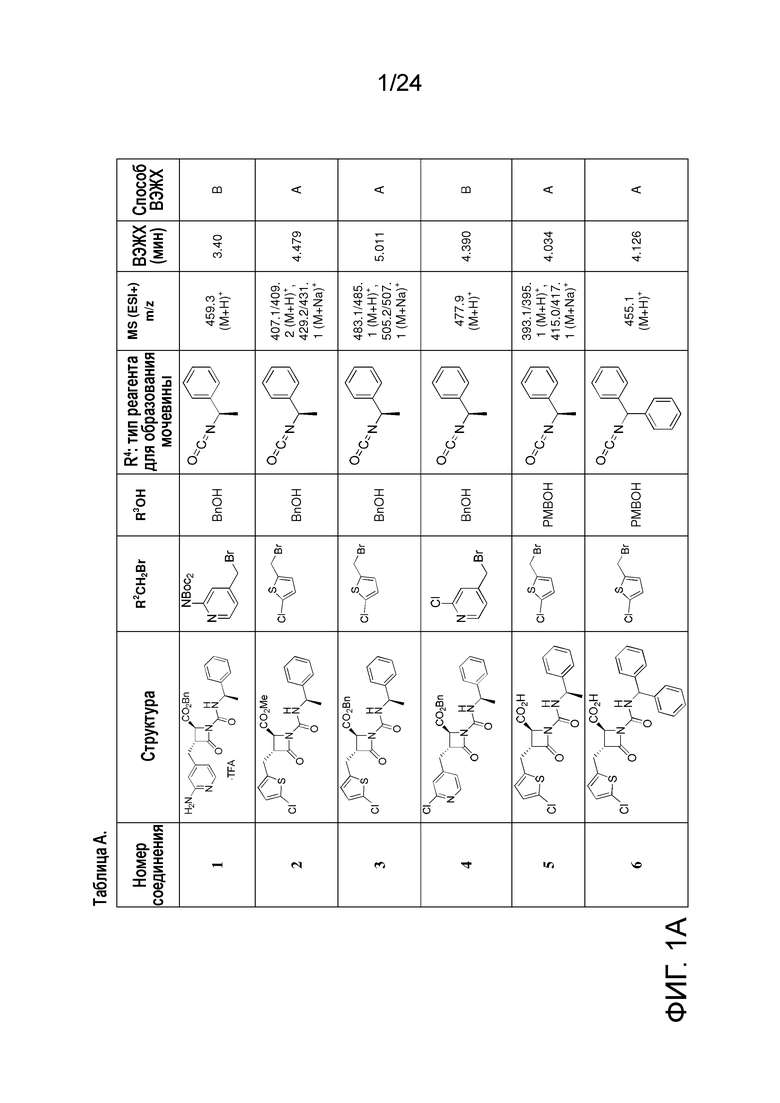

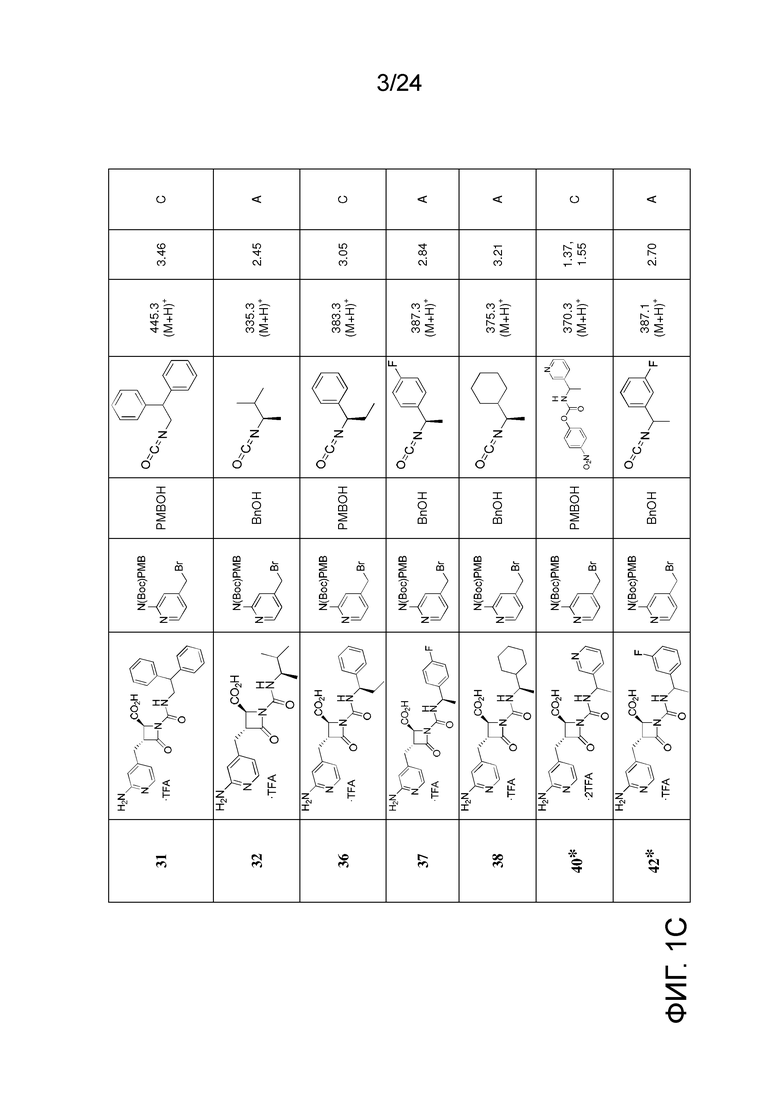
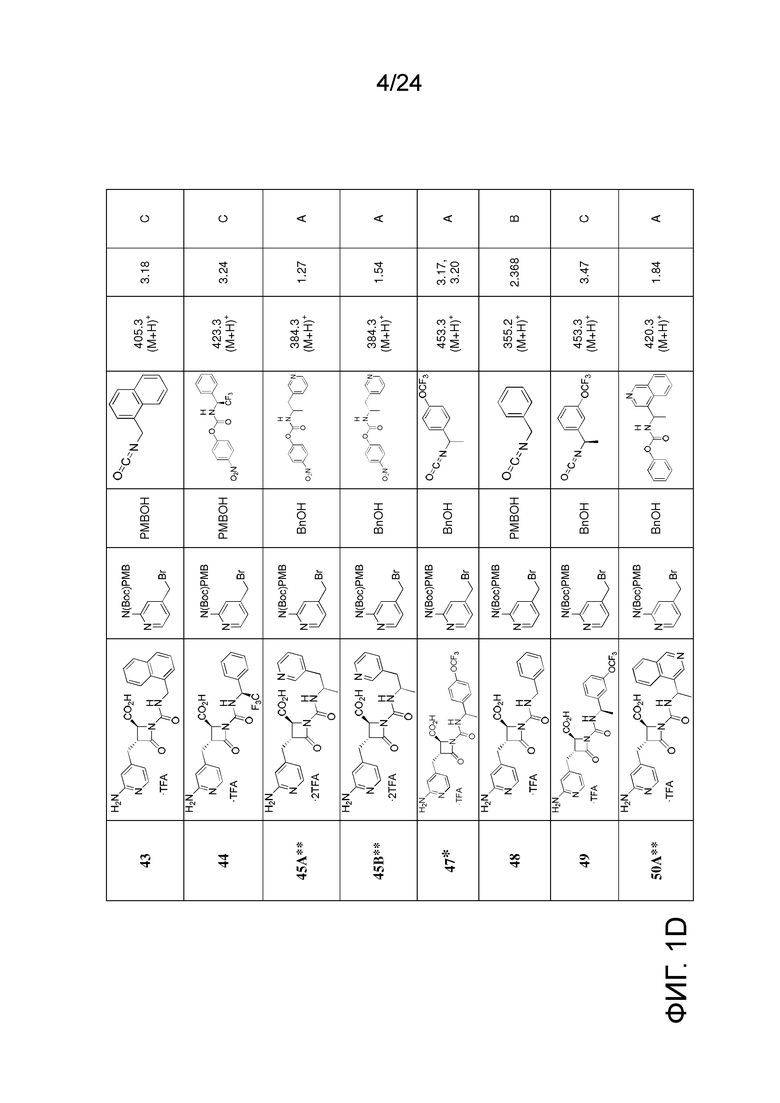

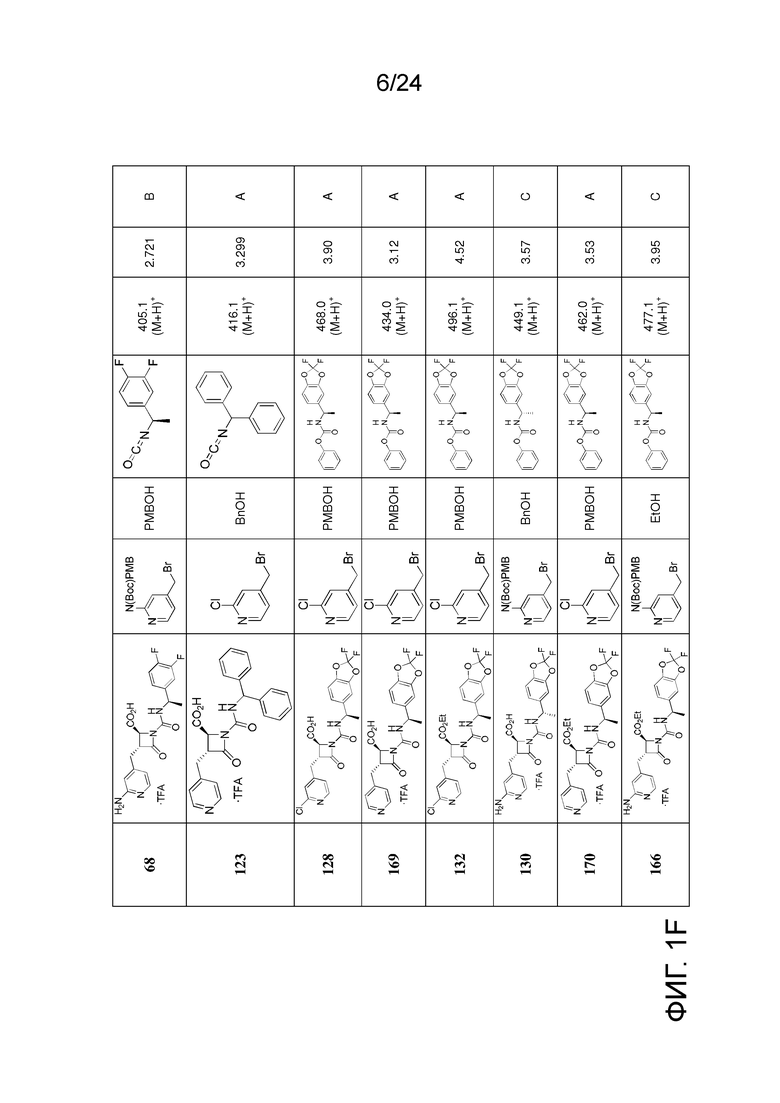
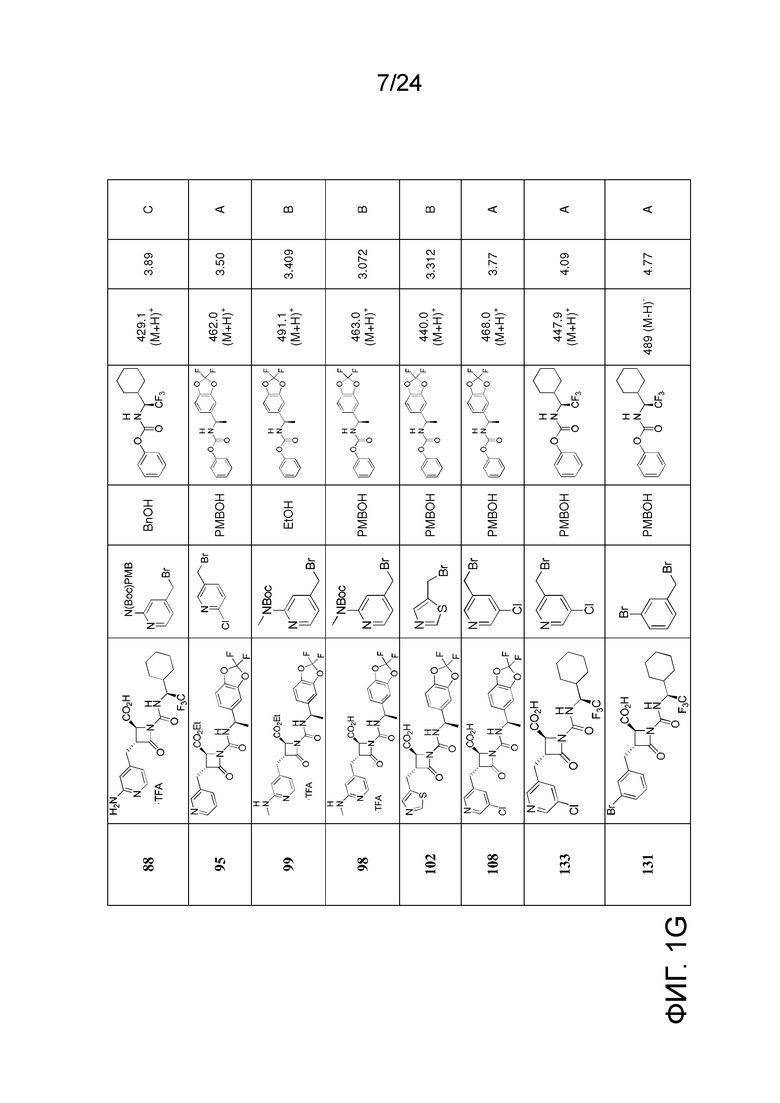
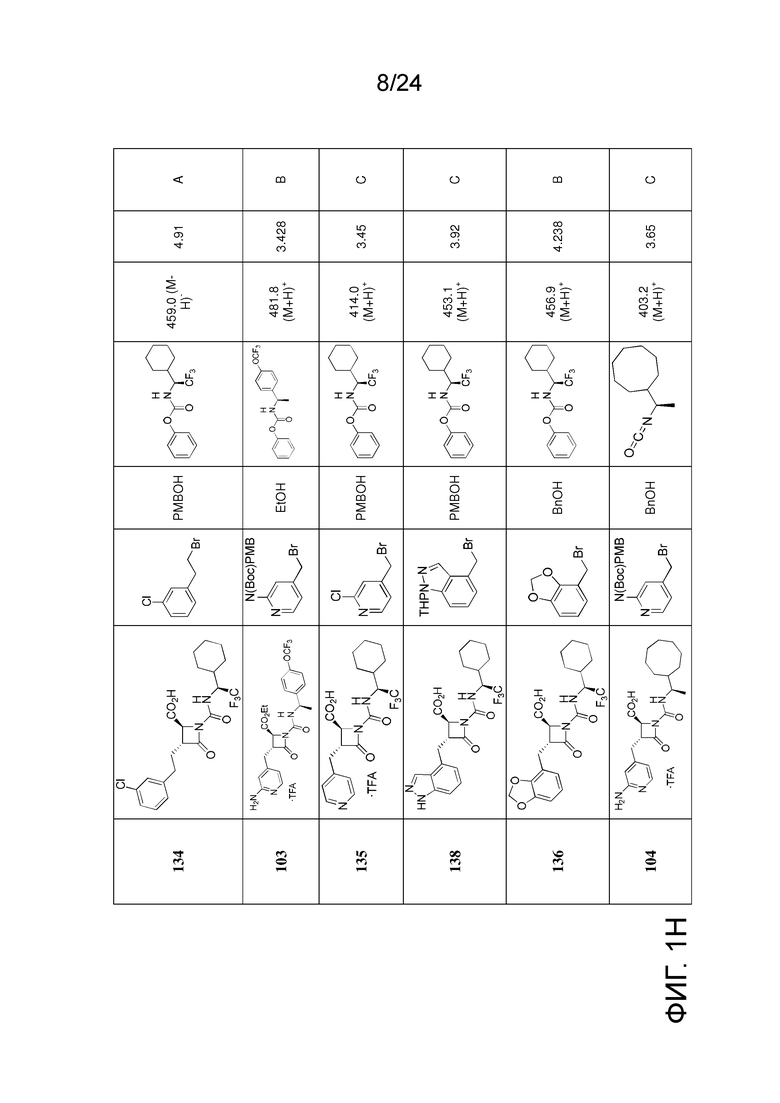
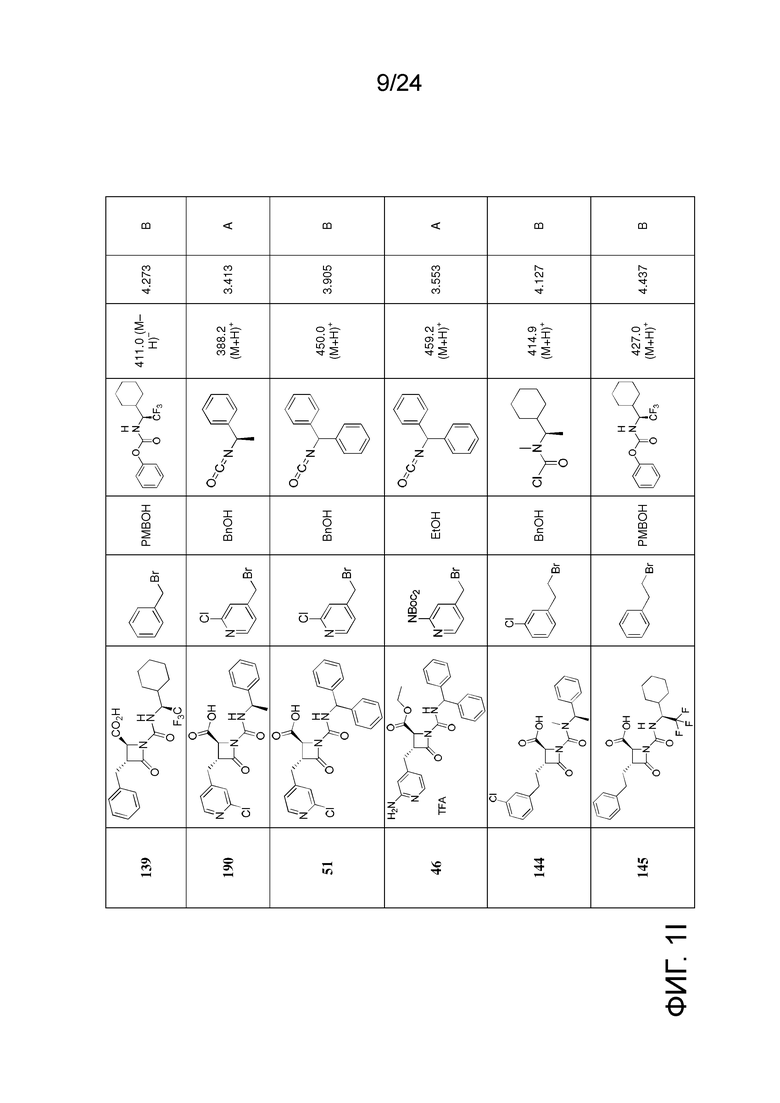
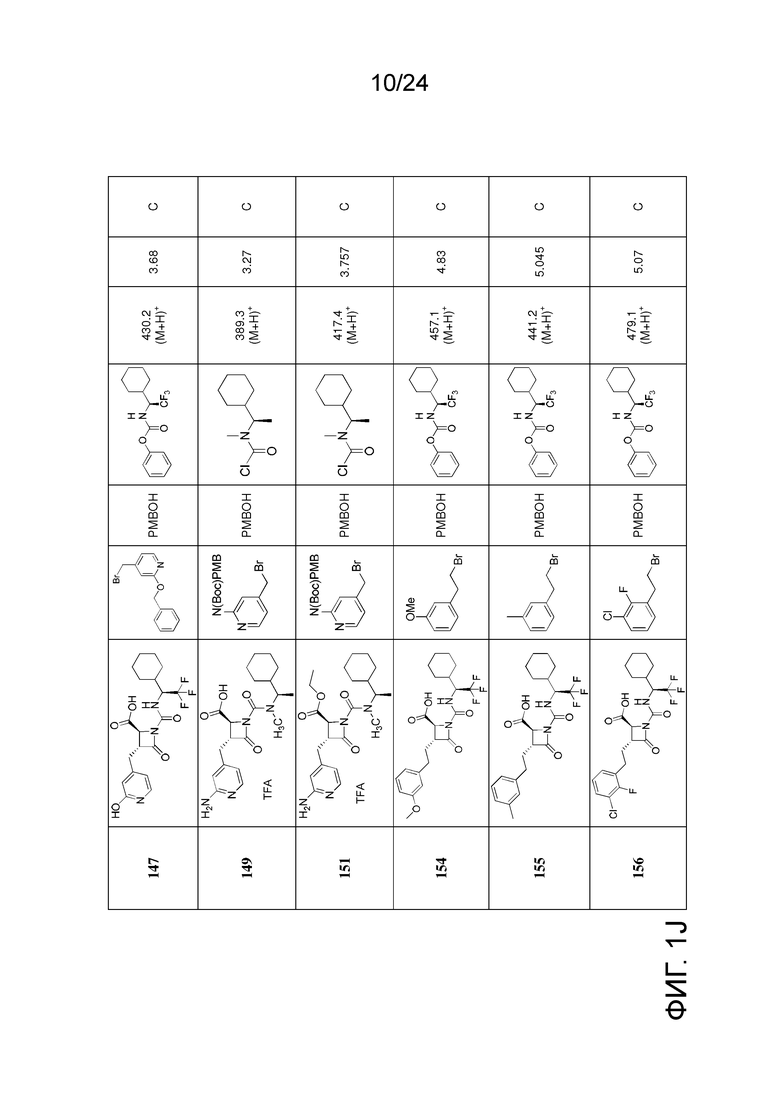
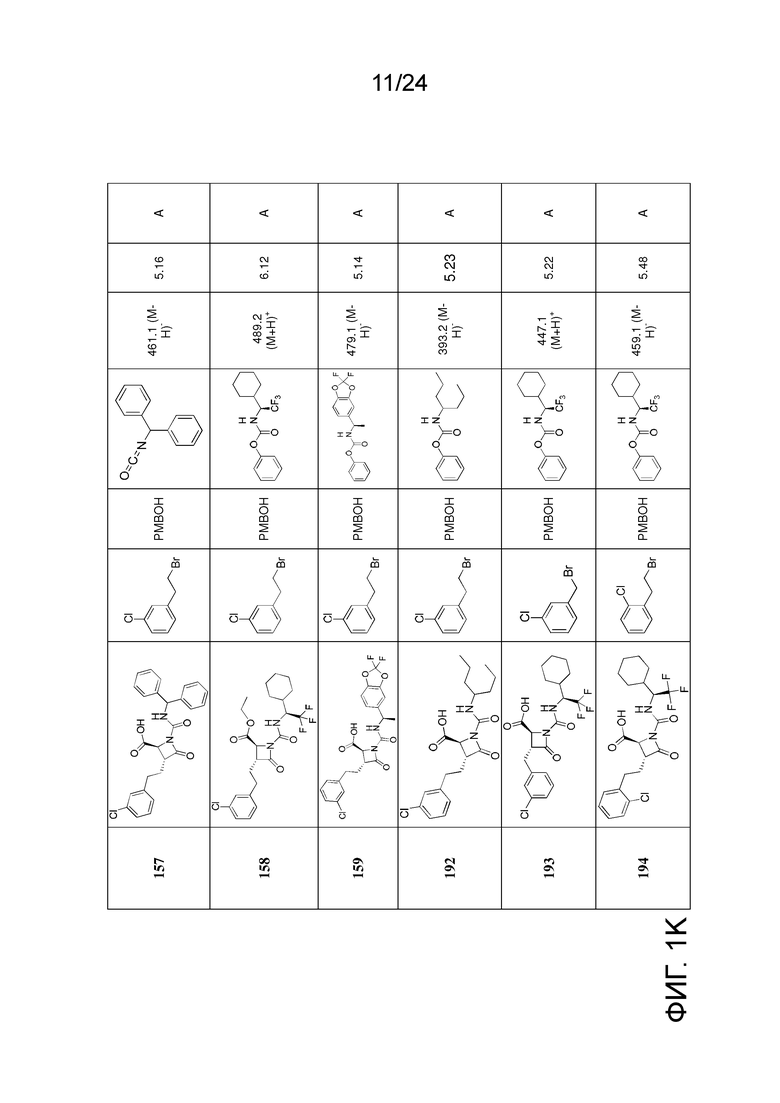
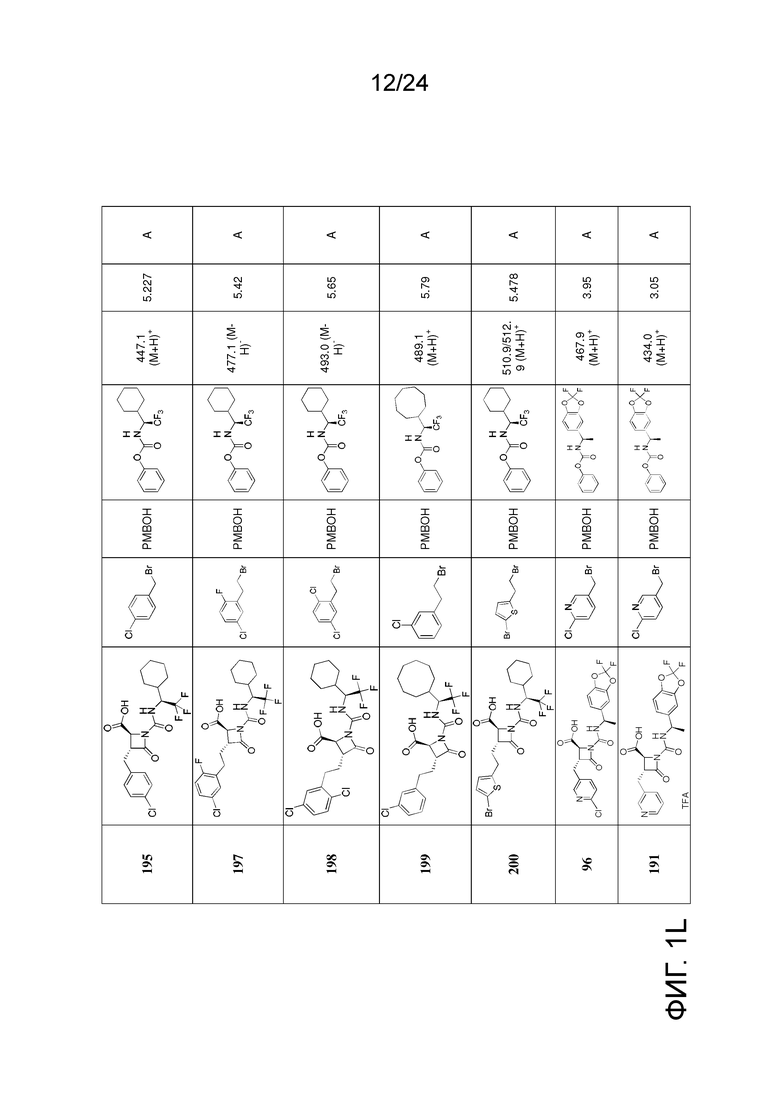
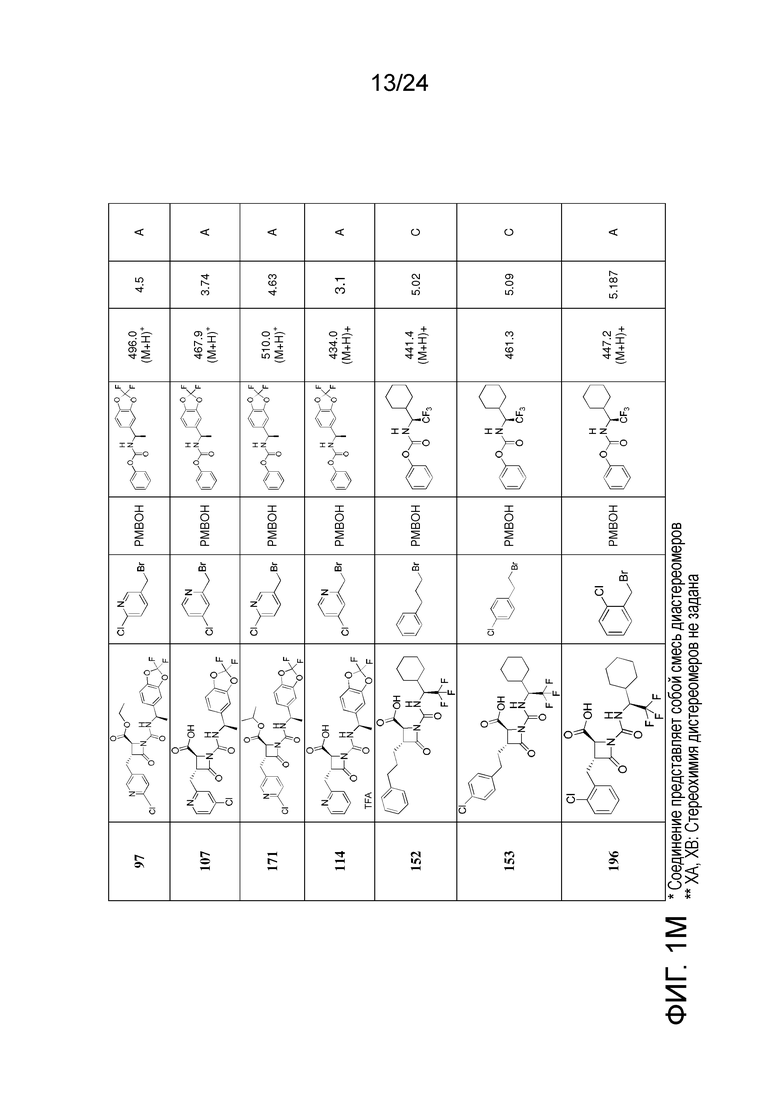

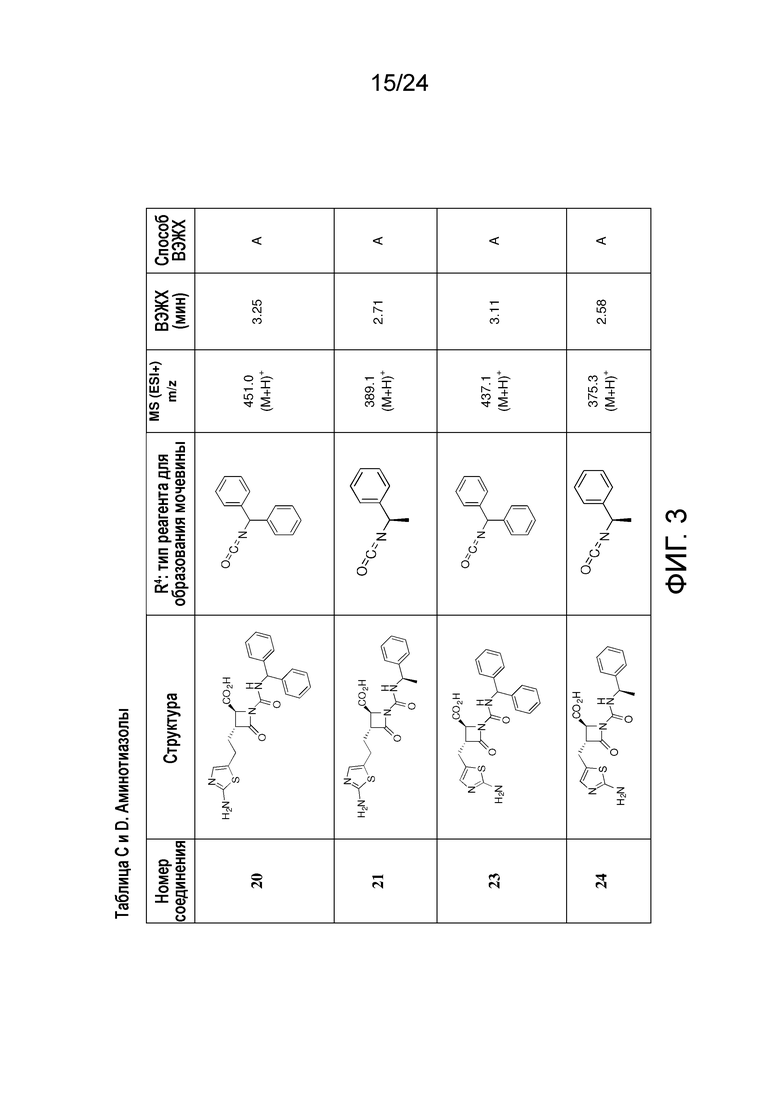
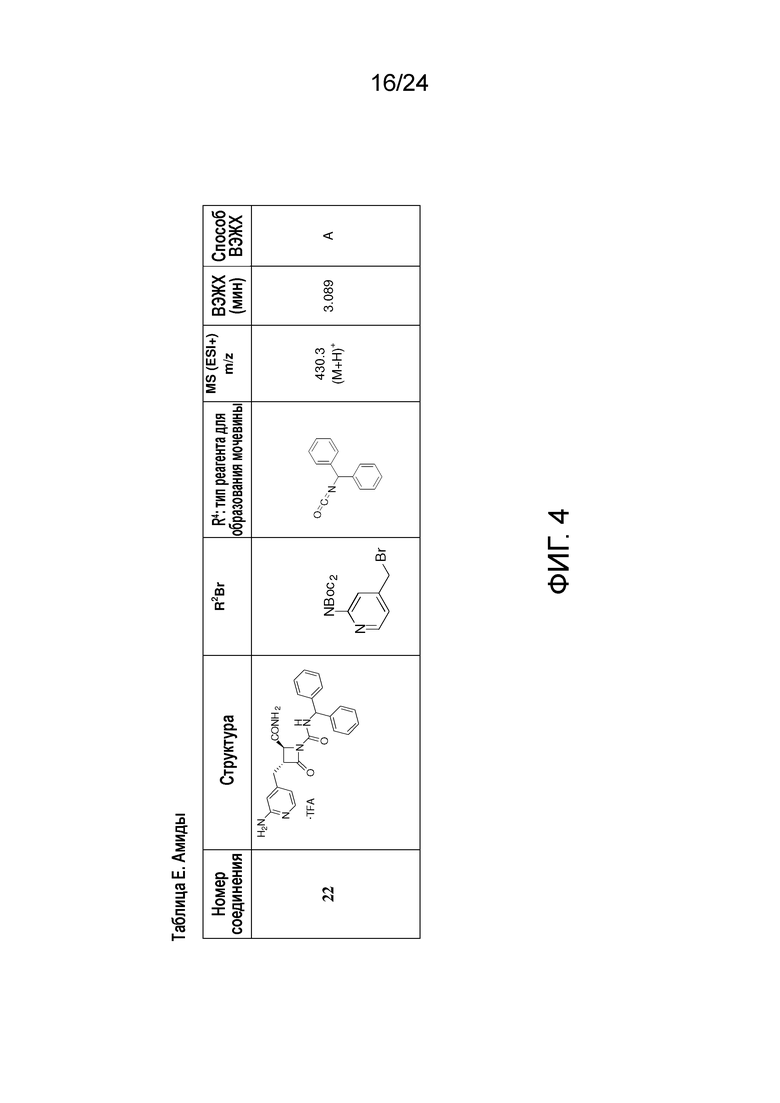
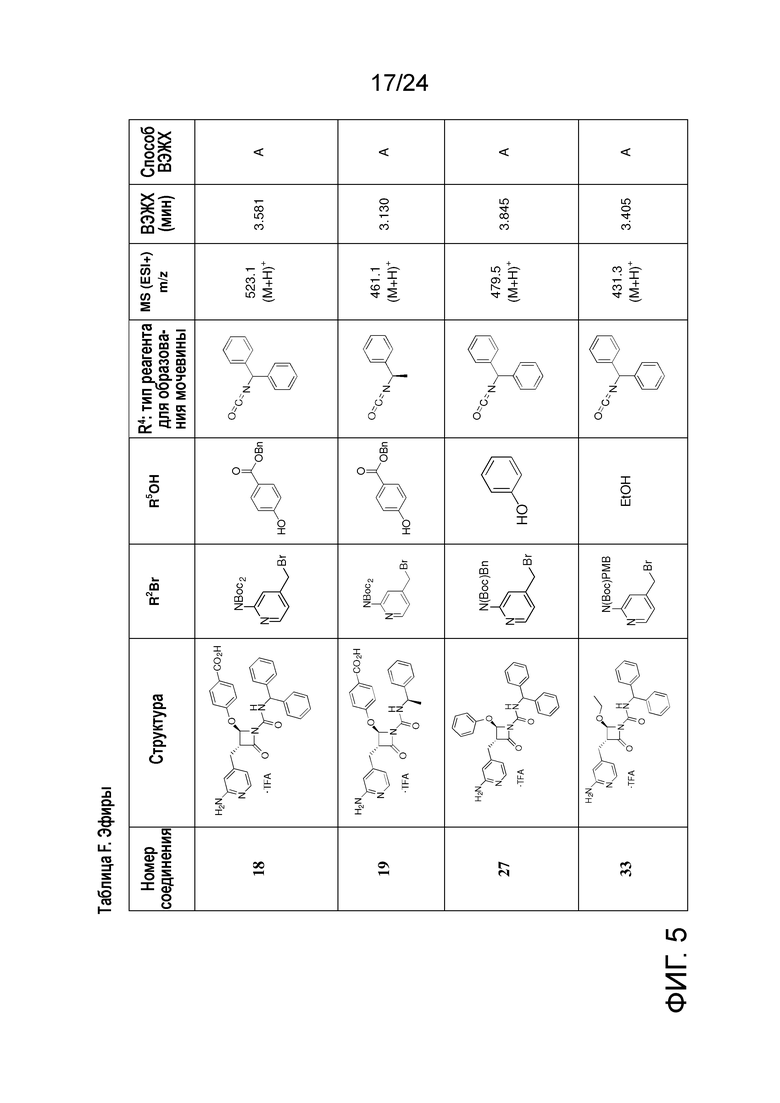
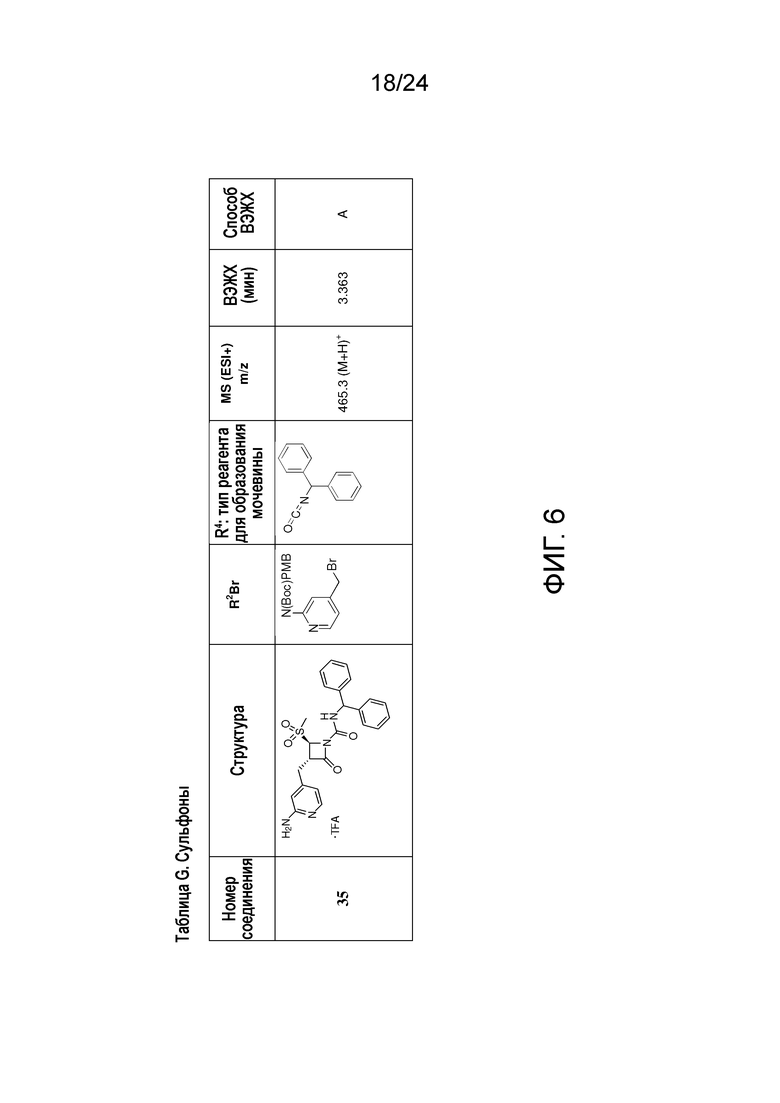
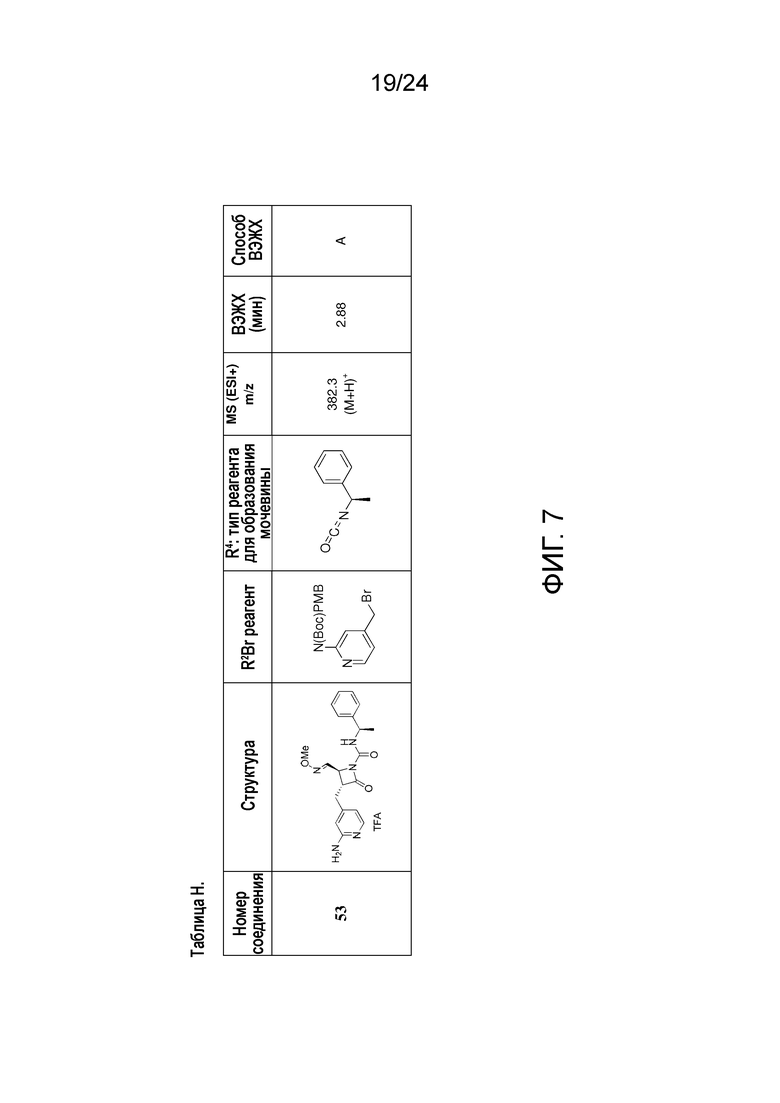
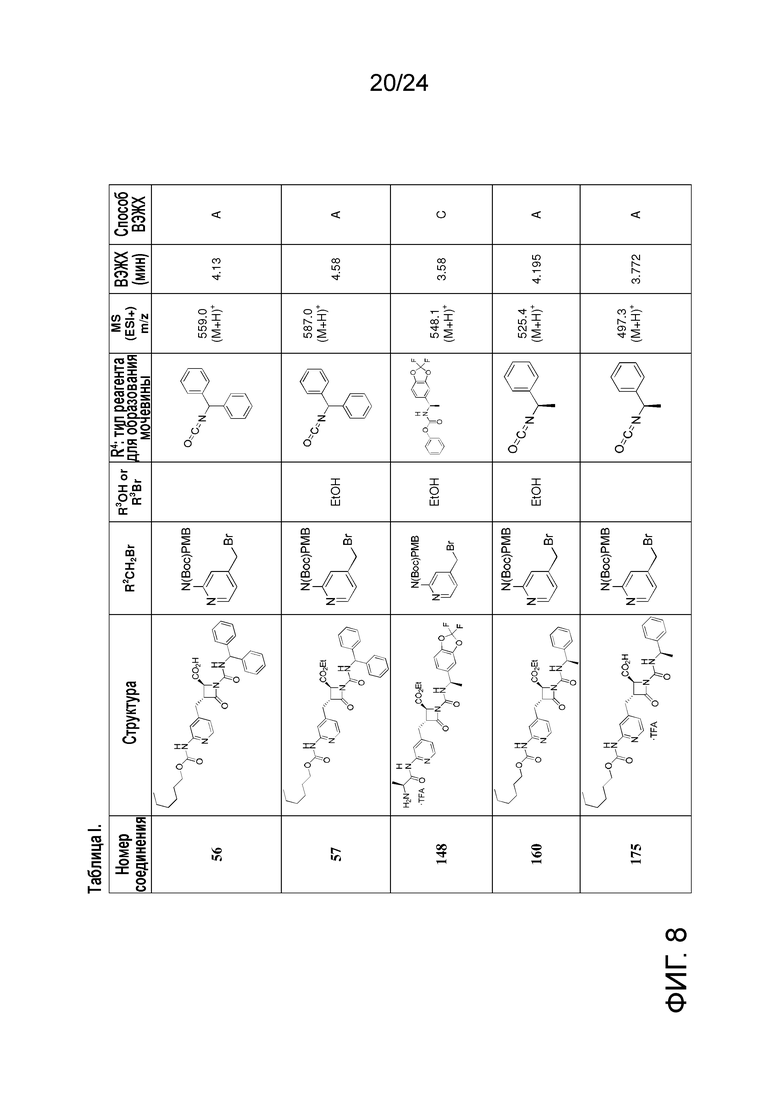
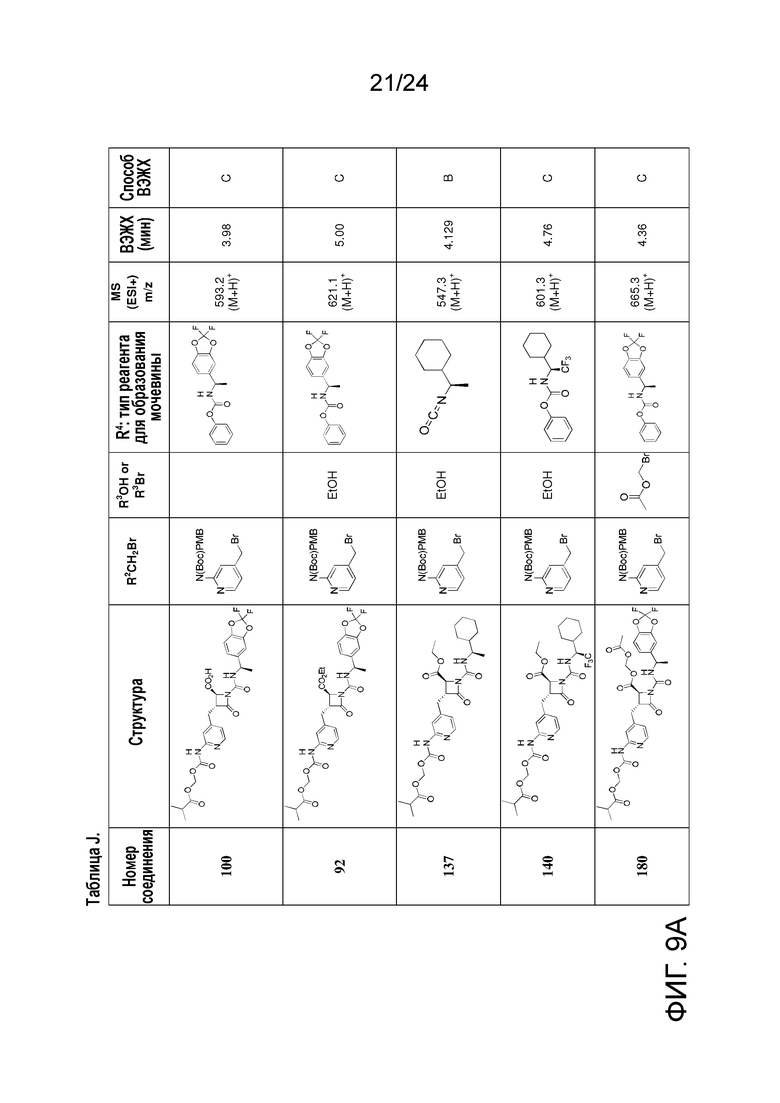
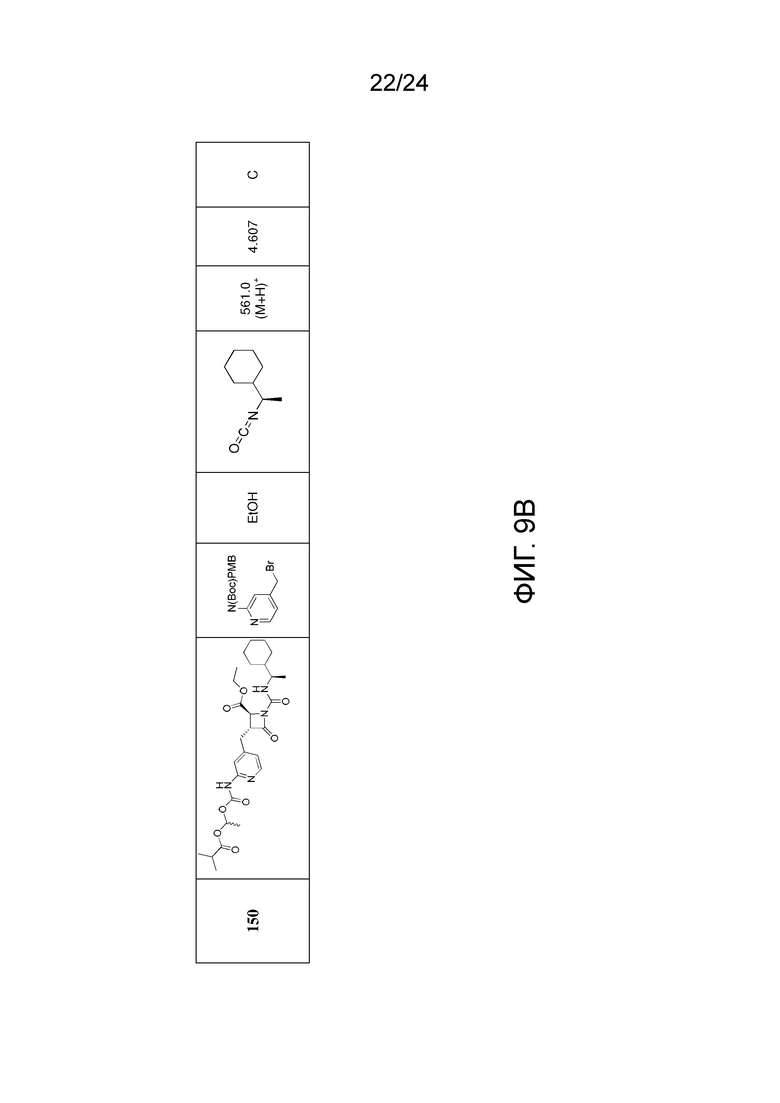

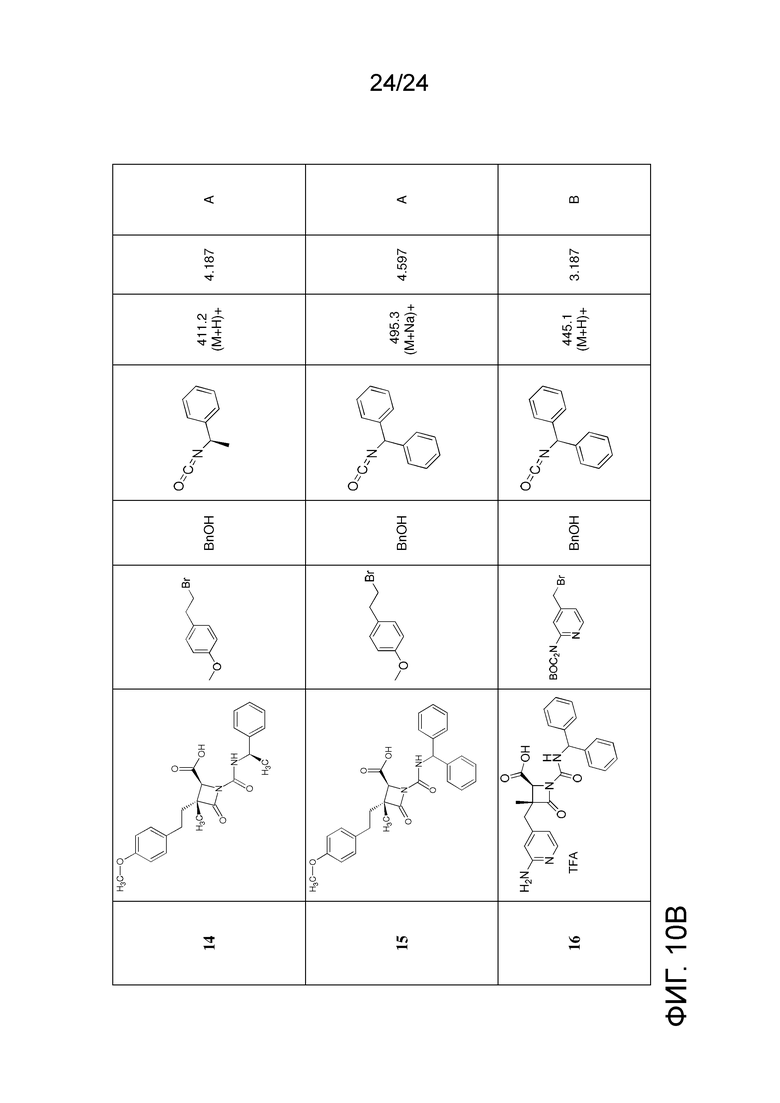
Настоящее изобретение относится к соединениям формулы (II) и его фармацевтически приемлемым солям, где значения R1-R7, A, X, Y указаны в формуле изобретения. Также предложены фармацевтические композиции, включающие соединения формулы (II), которые ингибируют Фактор XIa или калликреин, и способы применения этих соединений и композиций. 16 н. и 22 з.п. ф-лы, 10 ил., 3 таб., 27 пр.

1. Соединение формулы (II):
 (II),
(II),
или его фармацевтически приемлемая соль, где
R1 представляет собой H или -C1-6 алкил;
R2 представляет собой –CO2R5;
R3 представляет собой H или -C1-6 алкил;
A представляет собой C1-6 алкилен;
R4 представляет собой пиридил, замещенный 0-3 NH2 или R6;
каждый R5 независимо представляет собой H или -C1-6 алкил;
X представляет собой–C(O)N(R5)–;
Y представляет собой 5-8-членный циклоалкил, замещенный 0-3 –NH2 или R6;
R7 представляет собой –C1-6 алкил, фенил, пиридил или пиразинил, каждый из которых независимо замещен 0-3 –NH2 или R6;
где, если R6 представляет собой заместитель для любого R4 или R7, то каждый R6 независимо представляет собой галоген, гидрокси, C1-6 алкил, C1-6 алкокси или -NHR10;
если R6 является заместителем Y, то каждый R6 независимо представляет собой галоген, галогеналкокси;
R10 независимо представляет собой –C1-6 алкил; и
n равно целому числу от 0 до 2.
2. Соединение по п.1, где R1 представляет собой H.
3. Фармацевтически приемлемая соль соединения по п.1.
4. Соединение по п.1, где соединение формулы (II) выбрано из соединения формулы (IIa):
 (IIa),
(IIa),
где
R1, R2, R3, R4, R7 и Y такие, как описано для формулы (II), и
m равно целому числу от 1 до 6.
5. Соединение по п.4, где соединение формулы (II) выбрано из соединения формулы (IIb):
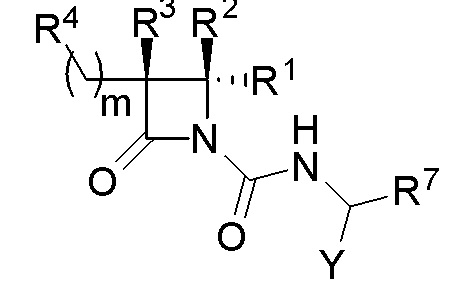 (Ib),
(Ib),
где
R1, R2, R3, R4, R7, Y и m такие, как описано для формулы (IIa).
6. Соединение по п.1, где соединение формулы (II) представляет собой:
 ,
,  ,
,  ,
,  ,
, 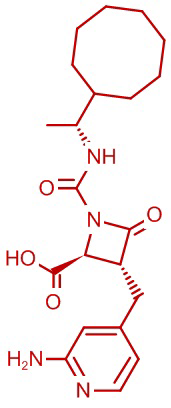 ,
, 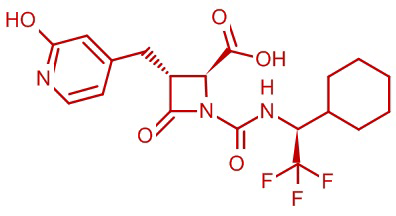 ,
, 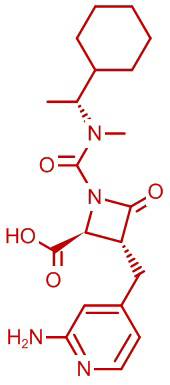 или
или 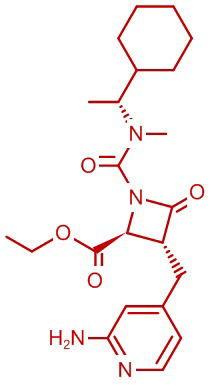 .
.
7. Соединение по п.5, где соединение формулы (IIb) выбрано из соединения формулы (IIc):
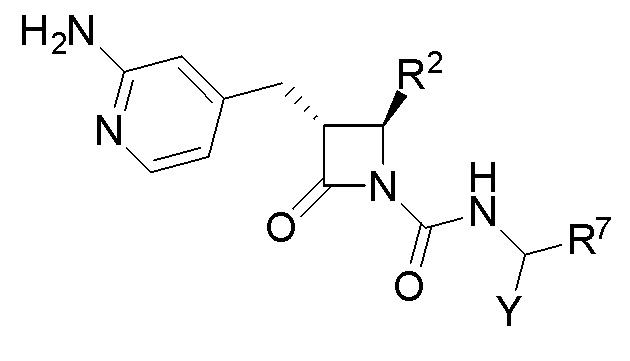 .
.
8. Фармацевтическая композиция, обладающая свойствами ингибирования Фактора XIa или калликреина, содержащая эффективное количество соединения формулы (II):
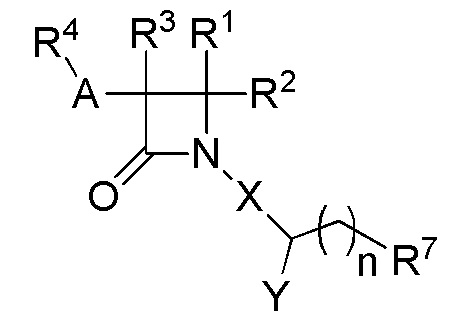 (II),
(II),
или его фармацевтически приемлемая соль, где
R1 представляет собой H или -C1-6 алкил;
R2 представляет собой–CO2R5;
R3 представляет собой H или -C1-6 алкил;
A представляет собой C1-6 алкилен;
R4 представляет собой пиридил, замещеный 0-3 –NH2 или R6;
каждый R5 независимо представляет собой H или -C1-6 алкил;
X представляет собой –C(O)N(R5)–;
Y представляет собой 5-8-членный циклоалкил, замещенный 0-3 –NH2 или R6; R7 представляет собой–C1-6 алкил, фенил, пиридил или пиразинил, каждый из которых замещен 0-3 –NH2 или R6;
где если R6 представляет собой заместителя для любого R4 или R7, то каждый R6 независимо представляет собой галоген, гидрокси, C1-6 алкил, C1-6 алкокси или -NHR10;
если R6 является заместителем для Y, то каждый R6 независимо представляет собой галоген, галогеналкокси; или
R10 независимо представляет собой –C1-6 алкил;
n равно целому числу от 0 до 2;
и один или несколько фармацевтически приемлемых носителей.
9.Соединение по п.1, где Y представляет собой циклоалкил.
10. Соединение по п.9, где Y представляет собой циклогексил.
11. Соединение по п.1, где R7 представляет собой –C1-6 алкил, замещенный 0-3 –NH2 или R6.
12. Соединение по п. 11, где R7 представляет собой метил или –CF3.
13. Соединение формулы
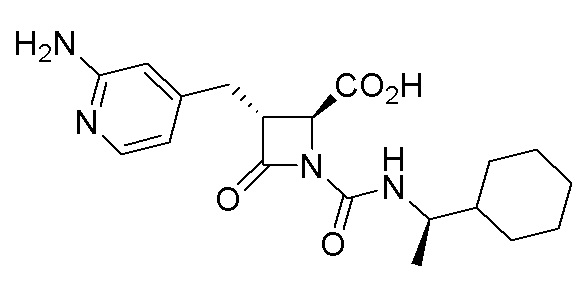
или его фармацевтически приемлемая соль.
14. Соединение формулы
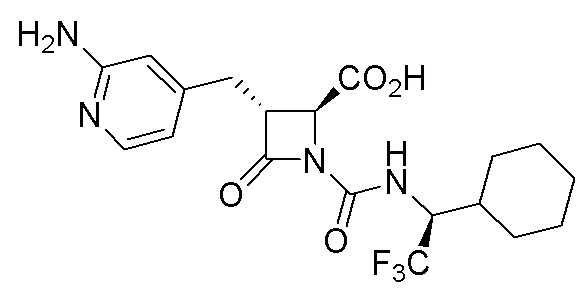
или его фармацевтически приемлемая соль.
15. Соединение формулы
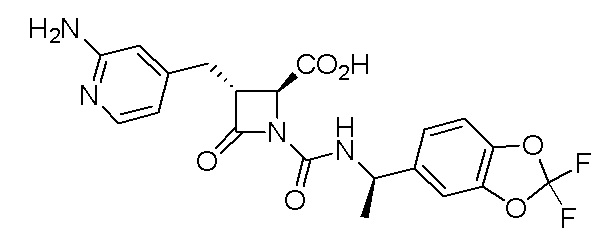
или его фармацевтически приемлемая соль.
16. Соединение формулы

или его фармацевтически приемлемая соль.
17. Фармацевтически приемлемая соль соединения формулы
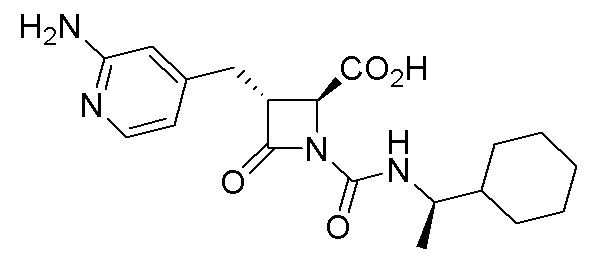 .
.
18. Фармацевтически приемлемая соль соединения формулы
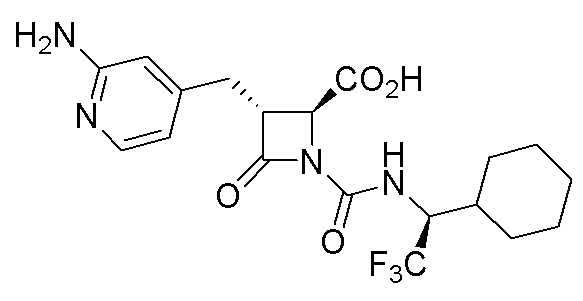 .
.
19. Фармацевтически приемлемая соль соединения формулы
 .
.
20. Фармацевтически приемлемая соль соединения формулы
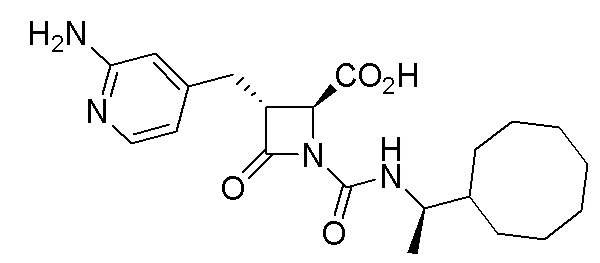 .
.
21. Фармацевтически приемлемая соль по п.17, где фармацевтически приемлемая соль представляет собой гидрохлорид.
22. Фармацевтически приемлемая соль по п.18, где фармацевтически приемлемая соль представляет собой гидрохлорид.
23. Фармацевтически приемлемая соль по п.19, где фармацевтически приемлемая соль представляет собой гидрохлорид.
24. Фармацевтически приемлемая соль по п.20, где фармацевтически приемлемая соль представляет собой гидрохлорид.
25. Способ снижения риска глубокого тромбоза вен у индивида, у которого был ишемический приступ, включающий введение индивиду эффективного количества соединения или его фармацевтически приемлемой соли по любому из пп.1-7 или 9-24, или фармацевтической композиции по п.8.
26. Способ ингибирования Фактора XIa у индивида, который страдает ишемией, включающий введение индивиду эффективного количества соединения или его фармацевтически примлемой соли по любому из пп.1-7 или 9-24, или фармацевтической композиции по п.8.
27. Способ по п.25 или 26, где индивидом является млекопитающее.
28. Способ по п.25 или 26, где индивидом является человек.
29. Способ по п.25 или 26, где индивиду было проведено хирургическое вмешательство.
30. Способ по п.25 или 26, где индивид подвергается хирургической замене коленного сустава или тазобедренного сустава.
31. Способ по п.25 или 26, где индивидом является индивид с «неклапанной» фибрилляцией предсердий.
32. Способ по п.25 или 26, где индивид имеет гипертонию.
33. Способ лечения индивида для поддержания экстракорпорального кровообращения, включающий контактирование крови индивида с эффективным количеством соединения или его фармацевтически приемлемой соли по любому из пп.1-7 или 9-24, или фармацевтической композиции по п.8.
34. Способ снижения риска тромбоза в результате сердечно-легочного шунтирования у субъекта, нуждающегося в этом, включающий контактирование крови индивида с эффективным количеством соединения или его фармацевтически приемлемой соли по любому из пп.1-7 или 9-24, или фармацевтической композиции по п.8.
35. Способ профилактики тромбоза в результате сердечно-легочного шунтирования у индивида, нуждающегося в этом, включающий контактирование крови индивида с эффективным количеством соединения или его фармацевтически приемлемой соли по любому из пп.1-7 или 9-24, или фармацевтической композиции по п.8.
36. Способ по любому из пп.33-35, где способ ингибирует коагуляцию крови.
37. Способ по любому из пп.33-35, где индивидом является млекопитающее.
38. Способ по любому из пп.33-35, где индивидом является человек.
| WO 2006108039 A2, 12.10.2006 | |||
| EP 1099690 A1, 16.05.2001 | |||
| RU 2001102266 A, 27.03.2003. |

