Область техники
Настоящее изобретение относится к микробиологической промышленности и медицинской биотехнологии и представляет собой способ препаративного получения рекомбинантного белка SAV-RGD, специфически узнающего клетки меланомы, где: SAV обозначает мономер стрептавидина, имеющий аминокислотную последовательность SEQ NO 1; RGD - меланома-адресующий олигопептид, имеющий аминокислотную последовательность SEQ NO 2, с использованием штамма MG1655/pSAV-RGD Escherichia coli - продуцента данных слитых белков.
Предшествующий уровень техники
Известно, что RGD-содержащие пептиды способны оказывать токсический эффект на клетки, связываясь с интегриновыми рецепторами на поверхности опухолевых клеток, они запускают внутриклеточные сигнальные каскады, приводящие к остановке роста, снижению пролиферации и, как следствие, гибели клеток. Данный принцип положен в основу действия препарата Cilengitide производства Merck KGaA (Германия), представляющего собой циклический пептид RGDfV (Arg-Gly-Asp-(D-Phe)-Val). [Mas-Moruno С., Rechenmacher F., Kessler H. (2010) Anti-Cancer Agents in Medicinal Chemistry, 10, 753-768.] Тем не менее, использование малых концентраций таких пептидов имеет обратный эффект, обусловленный кластеризацией рецепторов интегрина и активацией другого сигнального пути. Это создает дополнительные сложности при использовании препаратов, в состав которых входят RGD-содержащие пептиды, не несущий токсической части. Для устранения вышеописанного эффекта такие пептиды конъюгируют с токсическими агентами либо располагают на поверхности нагруженных токсическим агентом наноструктур или липосом [Pattillo СВ. et al. (2005) Pharm. Res., 22, 1117-1120; Dubey P.K. Mishra V., Jain S., Manor S., Vyas S.P. (2004) J. Drug Target, 12, 257-264], что является достаточно трудоемкой процедурой.
Для эффективного связывания с интегриновыми рецепторами на поверхности меланомных клеток белок должен содержать трипептид Arg-Gly-Asp (RGD). [Pierschbacher M.D. and Ruoslahti E. (1984) Nature, 309, 30-33] Однако связывать RGD-мотив способны несколько интегриновых рецепторов (это группа интегринов, содержащих субчастицу αv, а также интегрины α5β1, α8β1 αIIbβ3). Для обеспечения селективности важно окружение RGD-мотива. Варьируя последовательность аминокислотных остатков, окружающих RGD-мотив, была создана панель RGD-содержащих олигопептидов с различной тканевой специфичностью, обусловленной селективным взаимодействием с различными интегриновыми рецепторами. В ряду прочих, были получены RGD-содержащие олигопептиды, обладающие способностью селективно связываться с рецепторами на поверхности меланомных клеток и клеток эндотелия опухолевых сосудов человека и мыши (RGD10, RGD13, RGD15). [Hölig P., Bach М, Völkel Т., Nahde Т., Hoffmann S., Müller R. and Kontermann R.E. (2004) Protein Eng. Des. Sel., 17,433-441.]
Следует отметить, что для эффективного связывания с рецептором трипептид RGD должен принимать изогнутую конформацию, которую стабилизируют либо посредством циклизации пептидной цепочки [Aumailey М., Gurrath М., Müller G., Calvete J., Timple R., Kessler H. (1991) FEBS Lett., 291, 50-54; Gurrath M., Müller G., Kessler H., Aumailey M. Timple R. (1992) Eur. J. Biochem., 210, 911-921], либо путем образования дисульфидной связи между фланкирующими RGD-мотив остатками цистеина [Pierschbacher M.D. and Ruoslahti Е. (1987) J. Biol. Chem., 262, 17924-17928; Hölig P., Bach M., Völkel Т., Nahde Т., Hoffmann S., Müller R. and Kontermann R.E. (2004) Protein Eng. Des. Sel., 17, 433-441].
Короткие пептиды, как правило, получают методом химического синтеза. Однако при реализации данного способа возможна рацемизация аминокислотных остатков. Замена L-формы аминокислот на D-форму может приводить к образованию нефункциональных пептидов, в данном случае - со сниженной аффинностью к рецептору. [Han Y., Albericio F., Barany G. (1997), J Org Chem, 62, 4307-4312; Palasek S.A., Cox Z.J., Collins J.M. (2007) J. Peptide Sci., 13, 143-148].
Короткие RGD-пептиды имеют и другие недостатки помимо вышеперечисленных.
Изогнутая конформация трипептида RGD, обусловленная кольцевой структурой химически синтезируемых пептидов, подобных Cilengitide, может также поддерживаться за счет дисульфидной связи, образуемой между фланкирующими RGD-мотив остатками цистеина. [Pierschbacher M.D. and Ruoslahti E. (1987) J. Biol. Chem., 262, 17924-17928; Hölig P., Bach M., Völkel Т., Nahde Т., Hoffmann S., Müller R. and Kontermann R.E. (2004) Protein Eng. Des. Sel., 17, 433-441]. Это предоставляет возможность для биотехнологического получения пептидов, исключающего саму возможность рацемизации. Тем не менее, основной проблемой, возникающей при наработке белков в клетках микроорганизмов, является деградация коротких молекул, осуществляемая протеолитическими ферментами в цитоплазме клетки. Небольшие пептиды, как правило, имеют малое время циркуляции в организме, что, наряду с пониженным сродством к рецептору, может вести к необходимости увеличения как количества применяемого препарата, так и частоты его введения. Эти процессы могут существенно снижать эффективность действия препарата.
Из уровня техники известна аминокислотная последовательность растворимого, экскретируемого в периплазму стрептавидина; нуклеотидная последовательность, кодирующая данный белок; создан вектор, в состав которого входит вышеупомянутая нуклеотидная последовательность, находящаяся под контролем сильного конститутивного промотора или промотора уридинфосфорилазы Е. coli (Pudp); штамм Е. coli, содержащий упомянутый вектор и являющийся продуцентом стрептавидина; а также способ получения данного полипептида в бактериальных клетках с использованием хроматографической очистки [патент РФ 2153535]. Прототипами таких экспрессионных систем могут служить векторы, описанные нами ранее в патенте [патент РФ 2153535] и статье [Syrkina MS и др. Preparation and functional evaluation of RGD-modified streptavidin targeting to integrin-expressing melanoma cells. Protein Eng Des Sel. 2013 Feb; 26(2): 143-50]».
Кроме того, известна аминокислотная последовательность меланома-адресующих олигопептидов RIO, R13 и R15 (упоминающихся в литературном источнике как RGD10, RGD13 и RGD15) [Hölig P., Bach М., Völkel Т., Nahde Т., Hoffmann S., Müller R. and Kontermann R.E. (2004) Protein Eng. Des. Sel., 17,433-441].
Наконец, подробно описан и охарактеризован RGD-содержащий циклический пептид RGDfV, выпускаемый под торговой маркой Cilengitide компанией Merck KgaA (Германия). [Mas-Moruno С., Rechenmacher F., Kessler H. (2010) Anti-Cancer Agents in Medicinal Chemistry, 10, 753-768].
Включение олигопептида в состав тетрамеризующегося белка-носителя, стрептавидина, приводит к тому, что рекомбинантный слитый белок будет содержать в своем составе четыре интегрин-адресующих олигопептида. В результате взаимодействия такой структуры с рецептором на поверхности клетки происходит увеличение локальной концентрации RGD-мотивов вблизи данного рецептора, что повышает эффективность связывания. Литературные данные подтверждают повышенную эффективность связывания таких поливалентных структур по сравнению с моновалентными [патент РФ 2153535].
Помимо этого, стрептавидин способен с высоким сродством связывать остаток d-биотина (Kd=10-15). Учитывая тот факт, что каждая субъединица стрептавидина связывает 1 молекулу биотина, тетрамер способен связать 4 молекулы биотина, что может приводить к амплификации сигнала при использовании биотинилированного диагностического агента или усилению токсического эффекта при использовании биотинилированного терапевтического агента [Xia N., Liu L., Harrington M.G., Zhou F.(2010), Anal. Chem., 82, 10151-10157].
Использование запатентованной экспрессионной системы, позволяющей в препаративных количествах нарабатывать рекомбинантный стрептавидин в периплазме Е. coli [патент РФ 2153535], обеспечивает локализацию химерного белка на основе стрептавидина в периплазматическом пространстве клетки, что позволяет не только ограничить его расщепление протеолитическими ферментами, присутствующими в цитоплазме, но и существенно упростить стадию выделения с использованием аффинной хроматографии.
Ранее авторы настоящего изобретения создали систему для экспрессии рекомбинантных слитых белков SAV-RGD, специфически узнающих меланомные клетки, в состав которых входит стрептавидин (S) и меланома-адресующий олигопептид (R), соединенный со стрептавидином посредством пептидного линкера, содержащего сайт расщепления эн-теропептидазой; конструировали рекомбинантные плазмидные ДНК, обеспечивающие гетерологичную экспрессию указанных слитых белков; а также получили штаммы бактерий, принадлежащих к роду Escherichia, - продуценты указанных слитых белков. Указанные плазмидные ДНК обеспечивают экспрессию пробелка, содержащего лидерный пептид, обеспечивающий его секрецию в периплазматическое пространство клеток бактерий, где происходит отщепление лидерного пептида, и указанный белок SAV-RGD локализуется в биологически активном растворимом состоянии [Syrkina MS и др. Preparation and functional evaluation of RGD-modified streptavidin targeting to integrin-expressing melanoma cells. Protein Eng Des Sel. 2013 Feb;26(2): 143-50].
Краткое описание настоящего изобретения
Целью настоящего изобретения является создание высокоэффективного и экономичного способа препаративного получения рекомбинантного белка SAV-RGD, специфически узнающего клетки меланомы, в качестве препарата для производства лекарственных препаратов для лечения меланомы человека.
Данная цель была достигнута за счет разработки и оптимизации способа препаративного получения рекомбинантного белка SAV-RGD с использованием штамма MG1655/pSAV-RGD Escherichia coli.
Поставленная задача решается тем, что разработан и оптимизирован способ препаративного получения рекомбинантного белка SAV-RGD с использованием штамма Escherichia coli MG1655/pSAV-RGD.
Бактериальный штамм Escherichia coli MG1655/pSAV-RGD получен в результате трансформации штамма Escherichia coli MG1655 рекомбинантной плазмидной ДНК pSAV-RGD на базе вектора pUC18, которая включает последовательность промотора уридинфосфорилазы E. coli (Pudp), а также последовательность, кодирующую слитой белок SAV-RGD. В свою очередь, рекомбинантный слитой белок SAV-RGD, специфически узнающий меланомные клетки, имеет формулу SAV-RGD, где:
SAV - мономер стрептавидина, имеющий аминокислотную последовательность SEQ NO 1;
RGD - меланома-адресующий олигопептид, имеющий аминокислотную последовательность SEQ NO 2, где
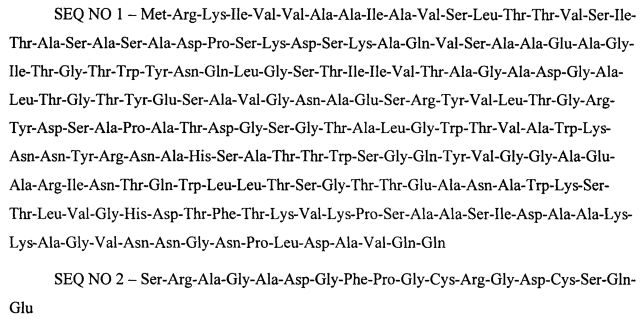
Вышеупомянутый способ препаративного получения рекомбинантного белка SAV-RGD включает следующие стадии:
- культивирование в питательной среде (LB, М9++глюкоза) штамма Escherichia coli MG1655/pSAV-RGD с рН-статированием в диапазоне рН 7,5-7,8 и сниженной аэрацией (концентрация растворенного кислорода не превышает 20-25%), включающее 2 этапа, где на первом этапе осуществляют подпитку источником глюкозы, а на втором этапе осуществляют подпитку источником аминокислот (пептоном и/или дрожжевой экстрактом);
- получение периплазматической фракции клеток бактериального штамма Escherichia coli MG1655/pSAV-RGD с использованием лизирующего буфера, в состав которого входит лизоцим в концентрации 1 г/л;
- очистку рекомбинантного белка по SAV-RGD методом ионообменной хроматографии с последующей ультрафильтрацией; и
- лиофилизацию очищенного рекомбинантного белка SAV-RGD с получением препарата на основе самособирающегося слитого белка, содержащего меланома-узнающие пептиды и адапторную часть для присоединения токсического агента.
В указанном способе условия культивирования оптимизированы для наиболее эффективного получения белка SAV-RGD, очистка целевого рекомбинантного белка SAV-RGD методом ионообменной хроматографии позволяет существенно снизить издержки процесса на стадии очистки по сравнению с использованием аффинной хроматографии на 2-иминобиотин-агарозе при сопоставимых выходах по целевому белку и степени его чистоты.
Настоящее изобретение более детально описано ниже.
Подробное описание настоящего изобретения.
Способом согласно настоящему изобретению является способ препаративного получения рекомбинантного белка SAV-RGD, где SAV - мономер стрептавидина, имеющий аминокислотную последовательность SEQ ID NO 1, RGD - меланома-адресующий олиго-пептид, имеющий аминокислотную последовательность SEQ ID NO 2, с использованием бактериального штамма Escherichia coli MG1655/pSAV-RGD.
Указанный способ включает следующие стадии:
- культивирование в питательной среде бактериального штамма Escherichia coli MG1655/pSAV-RGD - продуцента рекомбинантного белка SAV-RGD, при этом культивирование ведут в питательной среде с рН-статированием в диапазоне рН 7,5-7,8 и сниженной аэрацией (концентрация растворенного кислорода не превышает 20-25%), в 2 этапа, где на первом этапе осуществляют подпитку источником глюкозы, а на втором этапе осуществляют подпитку источником аминокислот (пептоном и/или дрожжевой экстрактом);
- получение периплазматической фракции клеток указанного штамма-продуцента рекомбинантного белка SAV-RGD;
- очистку рекомбинантного белка SAV-RGD методом ионообменной хроматографии с последующей ультрафильтрацией; и
- лиофилизацию очищенного рекомбинантного белка SAV-RGD с получением препарата на основе самособирающегося слитого белка, содержащего меланома-узнающие пептиды и адапторную часть для присоединения токсического агента.
Рекомбинантным белком SAV-RGD, получаемым способом согласно настоящему изобретению, является белок формулы SAV - мономер стрептавидина, имеющий аминокислотную последовательность SEQ ID NO 1, RGD - меланома-адресующий олигопептид, имеющий аминокислотную последовательность SEQ ID NO 2.
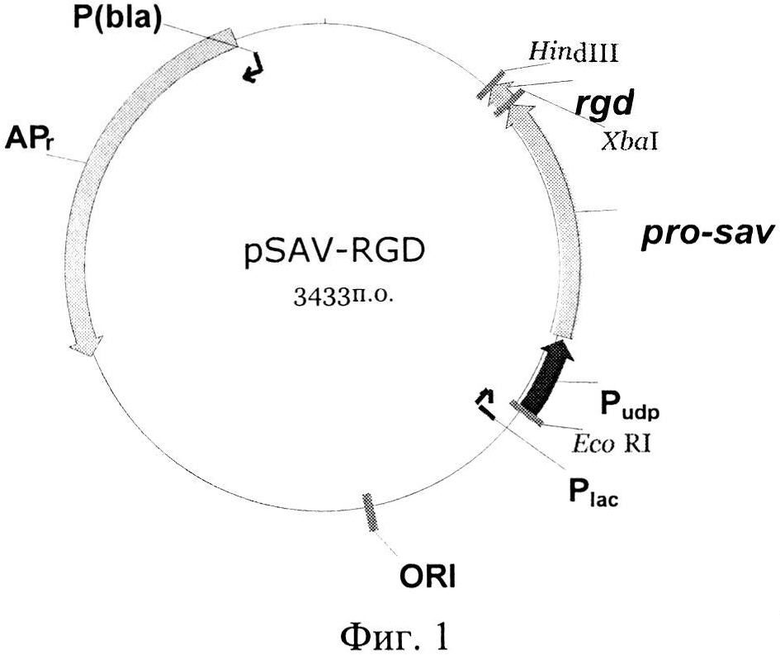
Примером рекомбинантной плазмиды, экспрессирующей указанный рекомбинант-ный белок SAV-RGD, содержащий лидерный пептид, обеспечивающий секрецию пробелка в периплазматическое пространство клеток Е. coli, где происходит отщепление лидерного пептида и стрептавидин локализуется в биологически активном растворимом состоянии, является плазмида pSAV-RGD на базе вектора pUC18, включающая последовательность, кодирующую слитой пробелок под контролем промотора уридинфосфорилазы Е. coli (Pudp). Схематическое изображение плазмиды pSAV-RGD представлено на Фиг. 1. Следует понимать, что круг плазмид, которые возможно использовать в настоящем способе, не ограничивается только указанной плазмидой.
Бактерией, используемой в способе согласно настоящему изобретению, является бактерия, принадлежащая к роду Escherichia - продуцент рекомбинантного белка SAV-RGD.
Термин «бактерия, принадлежащая к роду Escherichia, означает, что бактерия относится к роду Escherichia в соответствии с классификацией, известной специалисту в области микробиологии. Среди примеров бактерий, принадлежащих к роду Escherichia, но не ограничивающихся ими, может быть упомянута бактерия Escherichia coli (E. coli).
Круг бактерий, принадлежащих к роду Escherichia, которые могут быть использованы в настоящем изобретении, не ограничен каким-либо образом, однако, например, бактерии, описанные в книге Neidhardt F.C. et al. (Escherichia coli and Salmonella typhimurium, American Society for Microbiology, Washington D.C., 1208, Таблица 1), могут быть включены в число бактерий согласно настоящему изобретению.
Согласно настоящему изобретению термин «бактерия - продуцент рекомбинантного белка SAV-RGD» означает бактерию, способную производить и накапливать рекомбинантный белок SAV-RGD, предпочтительно в периплазме указанной бактерии, когда такая бактерия культивируется в питательной среде. Термин «бактерия - продуцент рекомбинантного белка SAV-RGD» означает, что микроорганизм способен производить и накапливать рекомбинантный белок SAV-RGD в количестве не меньше чем 0,05 г на 1 л культуральной жидкости и более предпочтительно не меньше чем 0,1 г на 1 л культуральной жидкости после выращивания в течение предпочтительно 18 часов.
Примером бактериального штамма, принадлежащего к роду Escherichia, - продуцента рекомбинантного белка SAV-RGD, является штамм Escherichia coli MG1655, трансформированный указанной выше плазмидой, например штамм Escherichia coli MG1655/pSAV-RGD, но не ограничивается им. Указанный штамм хранится в коллекции Биологического факультета МГУ.
Согласно настоящему изобретению выращивание бактерии - продуцента рекомбинантного белка SAV-RGD, осуществляют в подходящей питательной среде. Питательная среда для выращивания может быть как синтетической, так и натуральной, при условии, что она содержит источники углерода, азота, минеральные соединения и, если необходимо, питательные добавки в количестве, необходимом для роста бактерии. Источники углерода включают в себя различные углеводы, такие как глюкоза и сахароза, и различные органические кислоты. В зависимости от способа ассимиляции используемых бактерий, могут быть использованы спирты, такие как этанол и глицерин. В качестве источников азота используются аммиак, различные соли аммония, такие как сульфат аммония, другие соединения азота, такие как амины, природные источники азота, такие как пептон, гидролизат соевых бобов и микробный ферментолизат. В качестве минеральных соединений используются однозамещенный фосфат калия, хлорид натрия, хлорид кальция, соли магния, соли железа, соли марганца и подобные им. В качестве витаминов используются тиамин, дрожжевой экстракт и подобные им.
Выращивание проводят предпочтительно в аэробных условиях, таких как взбалтывание и аэрация с перемешиванием, при температуре от 20°С до 40°С, предпочтительно от 30°С до 38°С, наиболее предпочтительно при 37°С. Значение рН обычно варьирует в пределах от 5 до 9, предпочтительно в пределах от 7,0 до 8,0, наиболее предпочтительно при рН 7,5-7,8. рН среды может быть скорректирован аммиаком, карбонатом кальция, различными кислотами, основаниями и буферами. Обычно, выращивание в течение 18 часов приводит к накоплению биомассы бактерий, содержащих целевой белок в периплазматическом пространстве.
В ходе оптимизации условий культивирования штамма-продуцента рекомбинантного белка SAV-RGD установлено, что наиболее оптимальным является рН питательной среды 7.5.
Также в ходе оптимизации условий культивирования штамма-продуцента рекомбинантного белка SAV-RGD установлено, что снижение аэрации, за счет перемешивания со скоростью предпочтительно 450 об/мин, ведет к увеличению выхода целевого рекомбинантного белка SAV-RGD. Принудительную аэрацию проводят путем подачи 1 объема воздуха на 1 объем питательной среды.
Также в ходе оптимизации условий культивирования штамма-продуцента рекомбинантного белка SAV-RGD установлено, что наиболее оптимальным является двухступенчатое культивирование штамма. В первой части культивирования осуществляют наращивание популяции клеток штамма-продуцента путем подпитки источником углерода, в частности глюкозой, при этом предпочтительно контролировать скорость подпитки таким образом, чтобы конечная концентрация глюкозы в питательной среде не превышала 1-2 г/л. Во второй части культивирования осуществляют подпитку аминокислотами для эффективного биосинтеза рекомбинантного белка SAV-RGD. В качестве источников аминокислотами используют пептон и/или дрожжевой экстракт.
После выращивания получают периплазматическую фракцию указанных клеток. Для этого осаждают клетки центрифугированием, предпочтительно при 5000 об/мин при температуре 4°С в течение предпочтительно 10 минут. Культуральную жидкость удаляют, а полученную биомассу ресуспендируют в лизирующем буферном растворе, после чего центрифугированием при предпочтительно 12500 об/мин при 4°С в течение предпочтительно 20 минут получают супернатант, представляющий собой суммарную фракцию пе-риплазматических белков Е. coli. Варианты указанного процесса подробно описаны в Sambrook J., Fritsch E.F., Maniatis Т. (1989) [Cold Spring Harbor Laboratory Press].
В ходе оптимизации условий получения периплазматической фракции бактериальных клеток установлено, что экономически целесообразным является использование лизирующего буфера, содержащего лизоцим в концентрации 1 г/л, в соотношении объемов 1:20 к объему биомассы клеток.
Выделение целевого рекомбинантного белка SAV-RGD из фракции периплазматических белков осуществляют методом ионообменной хроматографии. В ходе оптимизации условий очистки установлено, что в качестве носителя в методе ионообменной хроматографии экономически целесообразным является использование SP-сефарозы (гранулированная агароза, модифицированная сульфопропильными группами) вместо 2-иминобиотинагарозы. К тому же SP-сефароза в качестве носителя достаточно стабильна и выдерживает многократные циклы очистки белка. Выходы целевого белка и его чистота при использовании SP-сефарозы и 2-иминобиотинагарозы сопоставимы.
После проведения хроматографии целевые рекомбинантные белки подвергают очистке методом ультрафильтрации через мембрану, позволяющую фильтровать белки размером свыше предпочтительно 30 кДа, для концентрирования и перевода в раствор. Для ультрафильтрации предпочтительно использование мембраны YM-30 (Amicon, США). Предпочтительно использовать разведение 1:20 с не менее чем пятикратной сменой буфера. Ультрафильтрацию осуществляют под давлением инертного газа (аргон, гелий с давлением не менее 4,7 атм).
В результате вышеописанных процедур получают очищенные препараты рекомбинантных белков SAV-RGD. Структуры полученных гибридных белков подтверждают методом масс-спектрометрии (MALDI-TOF MS). Гомогенность рекомбинантных белков подтверждают электрофорезом в денатурирующем полиакриламидном геле. Чистота полученного препарата рекомбинантных белков SAV-RGD, полученного указанным способом, составляла не менее 97%.
Для получения препарата для приготовления лекарственного средства для лечения меланомы человека полученный раствор очищенного рекомбинантного белка SAV-RGD лиофилизуют. Лиофилизацию осуществляют, разливая раствор во флаконы, замораживают раствор при температуре предпочтительно -70°С и помещают в лиофильную сушку. Процесс лиофильного высушивания проводят в течение 10-16 часов в вакууме. По окончании высушивания флаконы запаковывают. Хранение лиофилизированного препарата осуществляют при температуре 4°С.
Краткое описание чертежей
На Фиг. 1 приведено схематическое изображение плазмиды pSAV - RGD, содержащей гибридный ген pro-sav-rgd. Pudp - промотор гена уридинфосфорилазы из Е. coli, рrо-sav - ген предшественника стрептавидина (про-стрептавидин) из Streptomyces avidinii, rgd - искусственный ген меланома-адресующего пептида, АРr - ген устойчивости к ампициллину (бета-лактамаза).
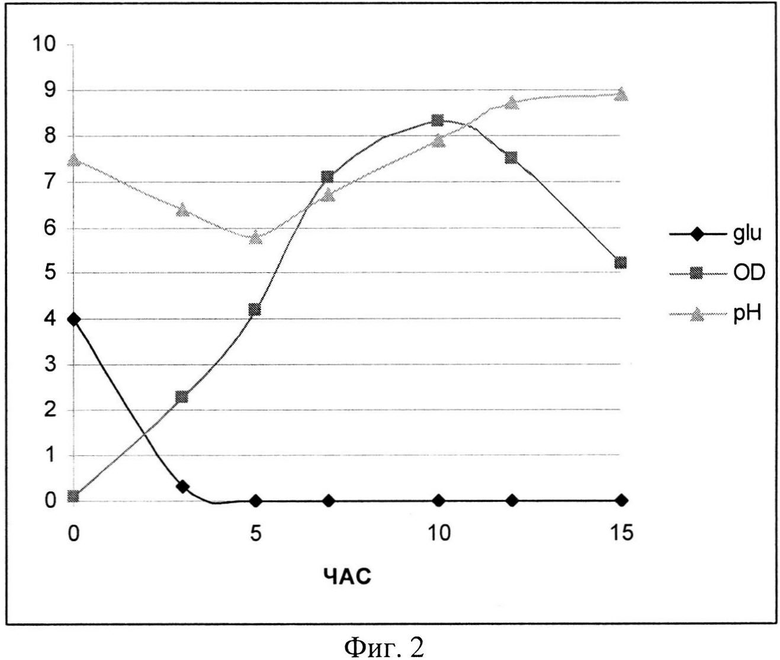
На Фиг. 2 показана динамика роста штамма-продуцента рекомбинантного белка SAV-RGD (контрольная ферментация). Glu - содержание глюкозы в среде культивирования (г/л); OD - оптическая плотность суспензии клеток при 600 нм; рН - значение рН среды культивирования.
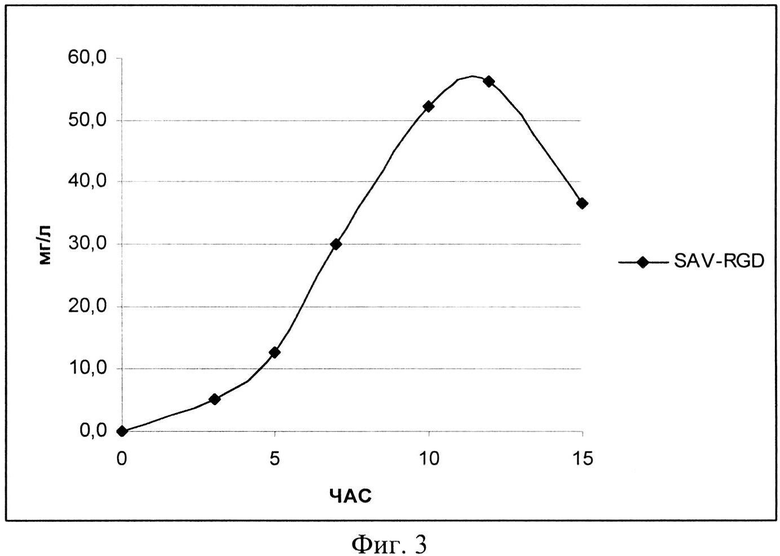
На Фиг. 3 показана кинетика накопления целевого продукта - рекомбинантного белка SAV-RGD (мг/л) в условиях контрольной ферментации.
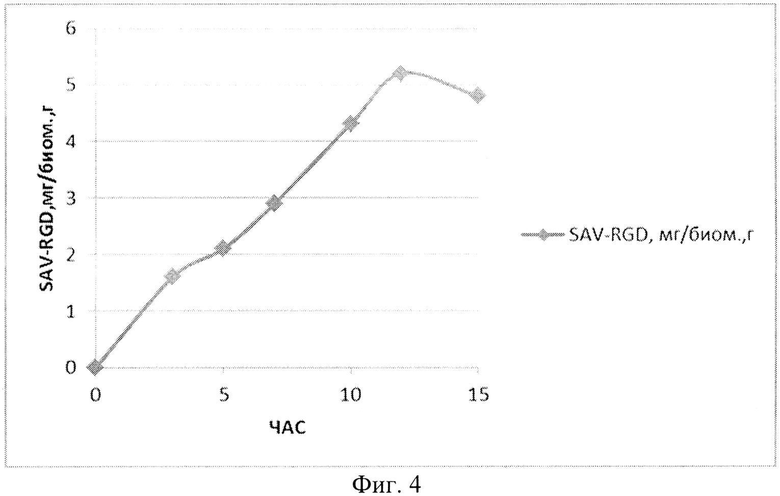
На Фиг. 4 показано содержание рекомбинантного белка SAV-RGD мг в пересчете на единицу биомассы в условиях контрольной ферментации.
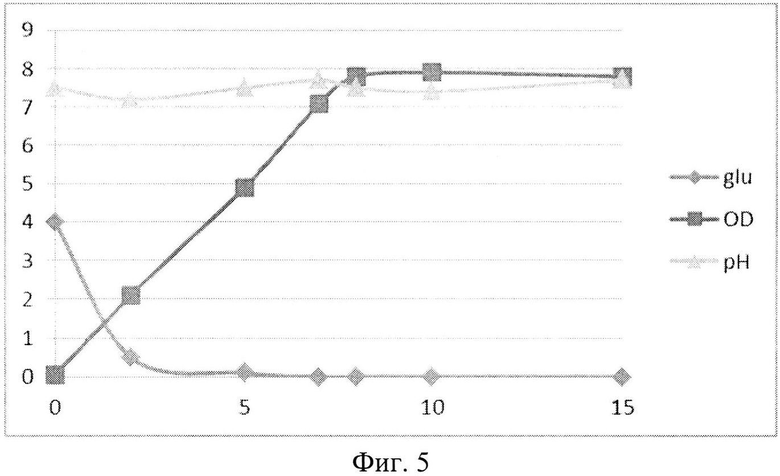
На Фиг. 5 показана динамика роста штамма-продуцента рекомбинантного белка SAV-RGD в 1,5-литровом ферментере при рН-статировании. Glu - содержание глюкозы в среде культивирования (г/л); OD - оптическая плотность суспензии клеток при 600 нм; рН - значение рН среды культивирования.
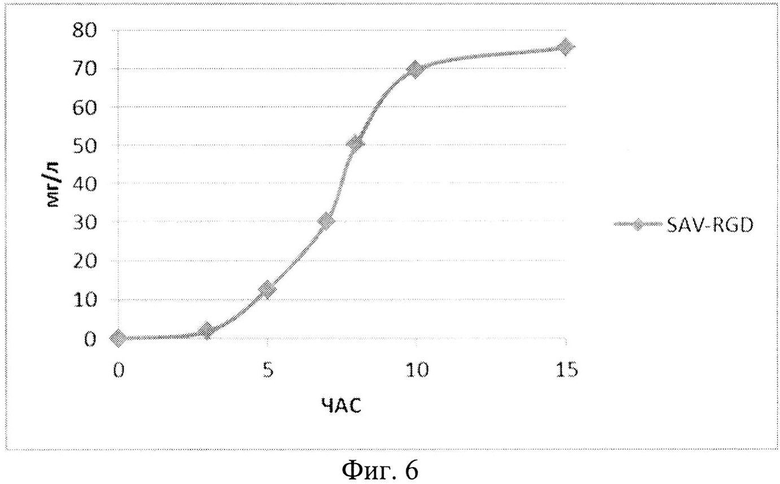
На Фиг. 6 показана кинетика накопления целевого продукта - рекомбинантного белка SAV-RGD (мг/л) в 1,5-литровом ферментере при рН-статировании.
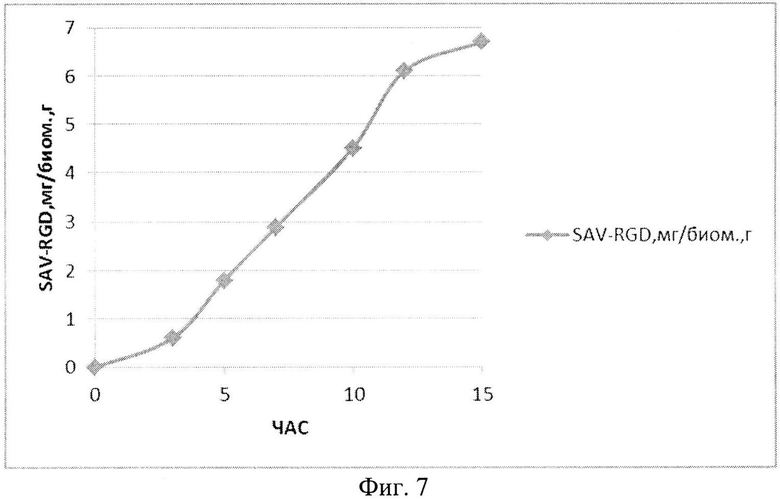
На Фиг. 7 показано содержание рекомбинантного белка SAV-RGD мг в пересчете на единицу биомассы в 1,5-литровом ферментере при рН-статировании.
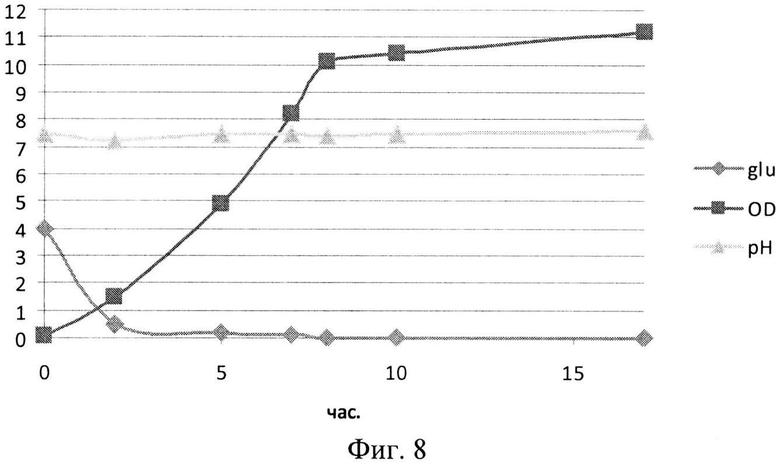
На Фиг. 8 показана динамика роста штамма-продуцента рекомбинантного белка SAV-RGD в 1,5-литровом ферментере при сниженном уровне аэрации. Glu - содержание глюкозы в среде культивирования (г/л); OD - оптическая плотность суспензии клеток при 600 нм; рН - значение рН среды культивирования.
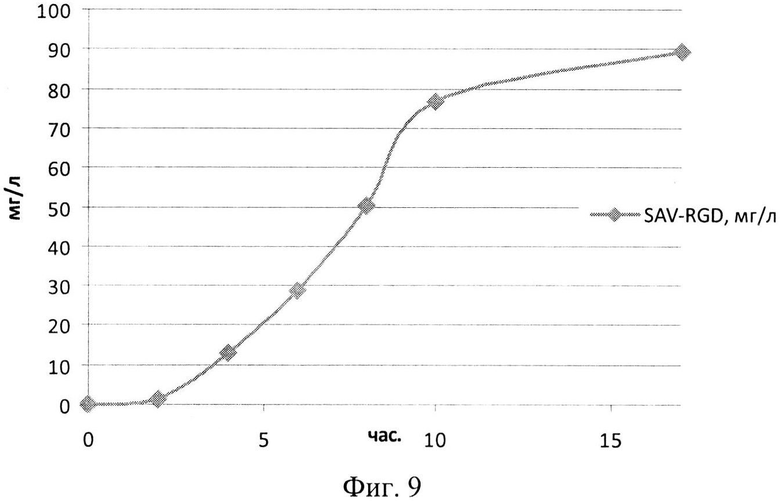
На Фиг. 9 показана кинетика накопления целевого продукта - рекомбинантного белка SAV-RGD (мг/л) в 1,5-литровом ферментере при сниженном уровне аэрации.
На Фиг. 10 показано содержание рекомбинантного белка SAV-RGD мг в пересчете на единицу биомассы в 1,5-литровом ферментере при сниженном уровне аэрации.
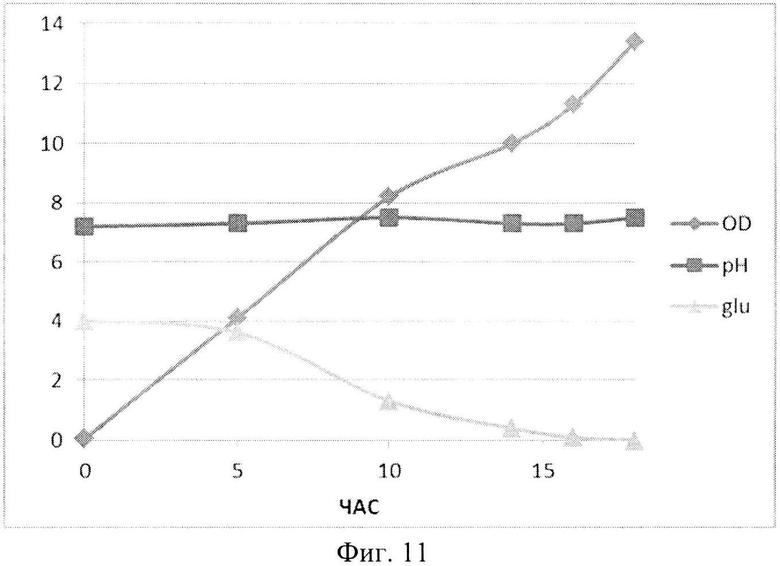
На Фиг. 11 показана динамика роста штамма-продуцента рекомбинантного белка SAV-RGD в 1,5-литровом ферментере в условиях подпитки источниками углерода и аминокислот. Glu - содержание глюкозы в среде культивирования (г/л); OD - оптическая плотность суспензии клеток при 600 нм; рН - значение рН среды культивирования.
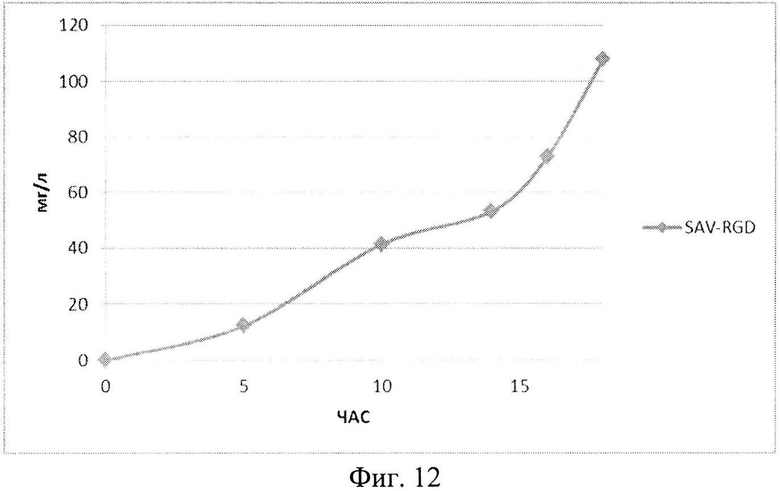
На Фиг. 12 показана кинетика накопления целевого продукта - рекомбинантного белка SAV-RGD (мг/л) в 1,5-литровом ферментере в условиях подпитки источниками углерода и аминокислот.
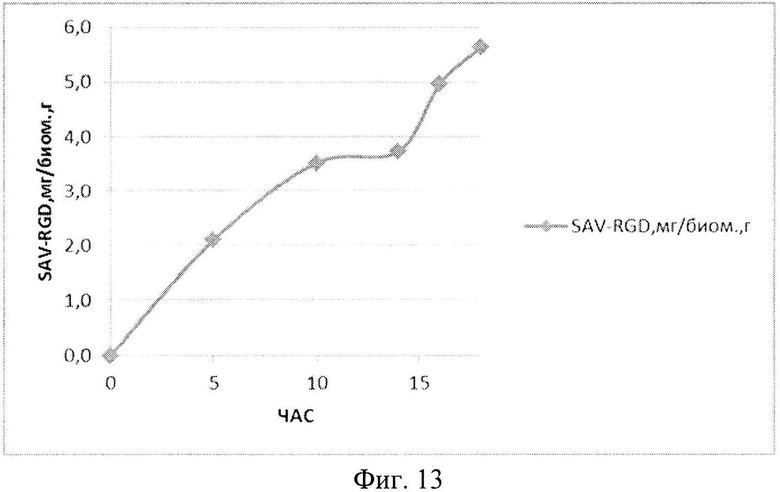
На Фиг. 13 показано содержание рекомбинантного белка SAV-RGD мг в пересчете на единицу биомассы в 1,5-литровом ферментере в условиях подпитки источниками углерода и аминокислот.
На Фиг. 14 показана оценка чистоты рекомбинантного белка SAV-RGD, очищенного методом аффинной хроматографии (А) и ионообменной хроматографии (Б), с помощью ВЭЖХ.
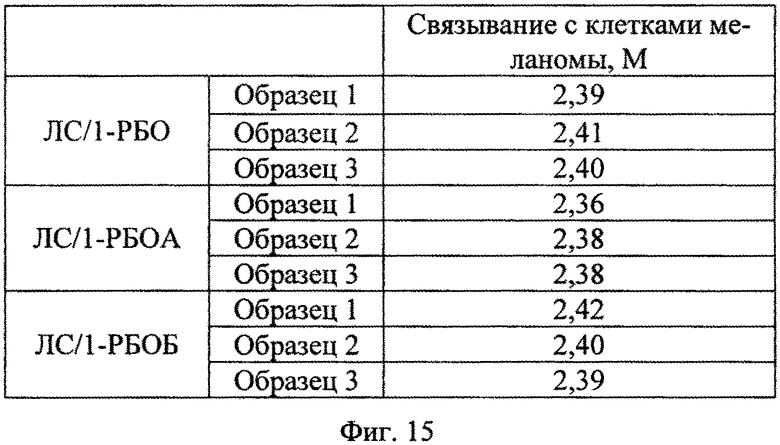
На Фиг. 15 показаны результаты эксперимента по оценке эффективности связывания рекомбинантного белка SAV-RGD клетками меланомы человека линии Me Wo.
Рекомбинантная плазмида pSAV-RGD, представленная на фиг. 1, состоит из следующих фрагментов ДНК:
1) последовательность с 1 нуклеотида по 404 нуклеотид (н.) включает фрагмент ДНК размером 404 п. о., содержащий фрагмент ДНК плазмиды pUC18 с 1н. по 404 н.
2) последовательность с 405 н. по 455 н. включает фрагмент ДНК размером 51 п. о., содержащий ген RGD.
3) последовательность с 456 н. по 461 н. включает синтетический фрагмент ДНК размером 6 п. о., содержащий синтетический полилинкер;
4) последовательность с 462 н. по 1010 н. включает фрагмент ДНК размером 549 п. о., содержащий ген прострептавидина, SAV (последовательность, кодирующая стоп-кодон, отсутствует);
5) последовательность с 1011 н. по 1017 н. включает фрагмент ДНК размером 6 п. о., содержащий синтетический полилинкер;
6) последовательность с 1018 н. по 1196 н. включает фрагмент ДНК размером 180 п. о., содержащий последовательность промотора уридинфосфорилазы Е. coli, Pudp;
7) последовательность с 1197 н. по 3433 н. включает фрагмент ДНК плазмиды pUC18 с 450 н. по 2686 н. размером 2237 п. о., содержащий промотор гена β-лактамазы (P(BLA)), ген устойчивости к ампициллину (АРr), последовательность, ответственную за репликацию плазмиды (ori) и lac промотер (P(LAC)).
Настоящее изобретение будет более подробно описано ниже со ссылкой на следующие никоим образом не ограничивающие настоящее изобретение Примеры.
ПРИМЕРЫ
Пример 1. Конструирование гена SAV-RGD и создание плазмид pSAV-RGD.
Для получения слитых генов необходимо получение гена стрептавидина (SAV) и генов RGD-coдержащего олигопептида (RGD). Нуклеотидная последовательность кодирующей цепи ДНК гена RGD: GCGGGTGCAGATGGCTTCCCAGGTTGCCGTGGTGATTGCAGTCAGGAATGA
Ген RGD-содержащего олигопептида получали путем отжига и PCR-достройки синтетических олигонуклеотидов RGD_F (5'-GCGATCTAGAGCGGGTGCAGATGGCTTCCCAGGTTGCCGTGGTG-3') и RGD_R (5'-TGACAAGCTTTCATTCCTGACTGCAATCACCACGGCAACCTGGGAAG-3')
Реакционная смесь имела следующий состав: 10х буфер для ПЦР (конечная концентрация в смеси: 75mM Tris-HCl (рН 8.8 at 25°С), 20 mМ (NH4)2S04, 0.01% (v/v) Tween 20) - 2 мкл, 25 mМ MgC12 (конечная концентрация - 3 mМ) - 3 мкл, 1.25 mМ dNTP (конечная концентрация - 0.125 mМ) - 2 мкл, праймеры - по 10 pmol на реакцию каждого, Taq-полимераза - 5 ед. акт. на реакцию, mQ Н20 - до 20 мкл. Амплификация включала: предварительную денатурацию при 94°С в течение 2 мин, 5 циклов (денатурация 94°С - 10 сек., отжиг праймеров 65°С - 10 сек, элонгация 72°С - 20 секунд), заключительная элонгация 72°С - 2 минуты. Последовательности нуклеотидов, полученные таким образом, содержали сайты расщепления эндонуклеазами рестрикции Xbal и Hindlll для лигирования с вектором.
После проведения ПЦР полученные фрагменты ДНК осаждали 3,5 объемами 96% этанола с добавлением 1/10 по объему 3 М ацетата натрия (рН 5,0) в течение 30 минут на -70°С, после чего центрифугировали при 13400 об/мин в течение 10 минут, удаляли супернатант, осадок промывали 70% этанолом, затем центрифугировали еще 5 минут при 13400 об/мин и высушивали на вакуумной сушке.
Для получения слитых генов в составе вектора pUC18 под контролем промотора уридинфосфорилазы E. coli (Pudp) использовали вектор pLSAVdS(H), аналогичной плазмиде pflSAV19 [Гулько Л.Б., Дьяков Н.А., Окорокова Н.А., Вейко В.П., Дебабов В.Г. (1999) Биотехнология, 4, с. 3-8]. Данный вектор содержит ген стрептавидина (последовательность, кодирующая стоп-кодон, отсутствует) под контролем промотора уридинфосфорилазы Е. coli (Pudp).
Затем проводили совместное расщепление вектора и фрагмента эндонуклеазами рестрикции Xbal и Hindlll. Реакционная смесь рестрикции имела следующий состав: плаз-мида - 6 мкл (900 нг); фрагмент - 5 мкл (100 нг); 10х буфер Trango Yellow+(Fermentas (Литва) (конечная концентрация в смеси: 33 mM Tris-acetate, рН 7,9, 10 mМ Mg-acetate, 66 mМ K-acetate, 0,1 mg/ml BSA) - 2 мкл; рестриктаза XbaI - 1 мкл (10 ед.акт.); рестриктаза Hindlll - 1 мкл (10 ед.акт.); mQ Н2O - 5 мкл. Молярное соотношение вектор: фрагмент в смеси составило 1:5. Рестрикцию проводили при 37°С в течение 1 часа.
После проведения расщепления ДНК осаждали 3,5 объемами 96% этанола в присутствии 0,3М ацетата натрия (рН 5,0) в течение 30 минут при -70°С, после чего центрифугировали при 13400 об/мин в течение 10 минут, удаляли супернатант, промывали осадок 70% этанолом и высушивали.
Полученную смесь расщепленных гена и плазмиды лигировали в течение 1 часа при 22 °С. Реакционная смесь лигирования имела следующий состав: 10х буфер для лигирования (Fermentas (Литва); конечная концентрация в смеси: 40 mM Tris-HCl, 10 mМ MgC12, 10 mМ DTT, 2,5 mМ АТР (рН 7.8 при 2°С)) - 2 мкл, расщепленные по сайтам Xbal и Hindlll вектор pLSAVdS(H) и ген RGD в молярном соотношении 1:5-15 мкл, Т4-лигаза - 2 мкл (10 ед.акт.), mQ Н2O - 1 мкл. После проведения лигирования ДНК в составе лигазной смеси осаждали 3,5 объемами 96% этанола в присутствии 0,3 М ацетата натрия (рН 5,0) в течение 30 минут при -70°С, после чего центрифугировали при 13400 об/мин в течение 10 минут, удаляли супернатант, осадок промывали 70% этанолом, осаждали центрифугированием и высушивали.
Далее лигазной смесью трансформировали компетентные клетки штамма E. coli JM110 и рассевали на чашке Петри с агаризованной средой LB, содержащей 100-150 мг/л ампициллина. Переосажденную лигазную смесь добавляли к компетентным клеткам. Количество ДНК, использовавшейся для трансформации, составляло около 300 нг в пересчете на плазмидную ДНК), 40 минут инкубировали на ледяной бане, далее 2 минуты при 42°С, потом добавляли по 1 мл среды LB, 1 час инкубировали при 32-37°С и рассевали клетки по чашке. Чашки инкубировали при 32-37°С в течение ночи.
Скрининг выросших клонов осуществляли с помощью ПЦР с использованием праймеров M13F (5-TAACTAGTACGCAAGTTCACG-3') и Seq2SAV (5'-AGACCGCTTCTGCGTTCTG-3'). Реакционная смесь имела следующий состав: 10х буфер для ПЦР (конечная концентрация в смеси: 75 mM Tris-HCl (рН 8.8 при 25°С), 20 mМ (NH4)2S04, 0.01% (v/v) Tween 20) - 2 мкл, 25 mМ MgC12 (конечная концентрация - 3 mМ) - 2,4 мкл, 1.25 mМ dNTP (конечная концентрация - 0.125 mМ) - 2 мкл, праймеры - по 5 pmol на реакцию каждого, клетки с колонии, суспендированные в стерильной воде, - 1 мкл на реакцию, Taq-полимераза - 5 ед. акт. на реакцию, mQ Н2O - до 20 мкл. Амплификация включала: предварительную денатурацию при 94°С в течение 2 мин, 25 циклов` (денатурация 94°С - 10 сек, отжиг праймеров 60°С - 10 сек, элонгация 72°С - 10 секунд), заключительная элонгация 72°С - 2 минуты.
После проведения ПЦР реакционную смесь подвергали электрофоретическому разделению в 3% агарозном геле, содержащем этидий бромид. Наличие слитого гена в ДНК бактериальных клеток идентифицировали по наличию светящихся продуктов ПЦР-амплификации требуемого размера (около 150 п. о.).
Колонии, продемонстрировавшие положительный ответ при ПЦР-скрининге, высевали в жидкую среду LB, содержащую 100-150 мг/л ампициллина, и культивировали при 32-37°С в течение ночи при покачивании.
На следующий день клетки осаждали центрифугированием (3-5 минут при 4500-6000 об/мин) и выделяли плазмидные ДНК при помощи набора GeneJETTM Plasmid Miniprep Kit (Fermentas, Литва). Схематическое изображение плазмиды pSR представлено на фиг. 1.
Пример 2. Получение штаммов E. coli MG1655/pSAV-RGD - продуцента рекомбинантного слитого белка SAV-RGD
Полученную на предыдущем этапе плазмиду pSAV-RGD секвенировали на участке, содержащем ген RGD. Реакцию секвенирования проводили на амплификаторе Eppendorf (Германия) по следующей программе: предварительная денатурация 94°С - 2 мин, 25 циклов (денатурация 94°С - 10 сек, отжиг праймеров 60°С - 10 сек, элонгация 72°С - 20 секунд), заключительная элонгация 72°С - 5 минут. Секвенирование проводили с помощью следующих праймеров: Seq2SAV и M13F. Реакционная смесь имела следующий состав: 10х буфер для секвенирования - 1 мкл, праймер - по 10 pmol на реакцию, плазмида -200 нг на реакцию, радиоактивная метка (α32P-dATP, активность 250 mkKu) - 1 мкл, ди-дезоксирибонуклеозидтрифосфаты-терминаторы - 2 мкл (свой в каждую реакцию), Taq-полимераза - 1 мкл (10 ед.акт.), mQ Н2O - до 8 мкл. Остановка реакции осуществлялась добавлением реагента StopSolution. Далее продукты реакции секвенирования прогревали до 95°С и наносили на 5% полиакриламидный гель с мочевиной. Электорофорез проводили в течение 2-3 часов. После этого гель вымачивали 20 минут в 10% уксусной кислоте, переносили на лист 3 мм ватмана и высушивали на гель-драйере. Далее гель закладывали в кассету с рентгеновской пленкой и оставляли на ночь в полной темноте. На следующий день пленку проявляли, последовательность нуклеотидов читали вручную, оценку проводили в программе VectorNTI 8.0 (Invitrogen, США). Плазмидой с выверенной нуклеотидной последовательностью трансформировали штамм E. coli MG1655. Трансформацию проводили химическим методом: плазмиду в количестве 20 нг добавляли в компетентные клетки, смесь 40 минут инкубировали сначала на ледяной бане, затем 2 минуты при 42°С, потом добавляли по 1 мл среды LB, 1 час инкубировали при 32-37°С и рассевали клетки на чашке Петри со средой LB с агаром, содержащей 100-150 мг/л ампициллина. Чашки инкубировали при 37°С в течение ночи.
В результате вышеописанных процедур получали штамм E. coli MG1655/p SAV-RGD - продуцент гибридного белка SAV-RGD.
Пример 3. Подготовка стерильных питательных сред.
Стерильная среда LB
Среду готовят в мерном стакане емкостью 1000 мл, в который наливают 900 мл дистиллированной воды, добавляют навеску бакто-триптона (10 г), бакто-дрожжевого экстракта (5 г), натрий хлористого (10 г), перемешивают. рН среды доводят 10 N водным раствором NaOH до значения 7,5. Приготовленный раствор переносят в колбу емкостью 2000 мл, закрывают ватно-марлевой пробкой и стерилизуют в течение 20 мин при 121°С в автоклаве (Sanyo MLS 2420 U, Япония). После стерилизации добавляют в асептических условиях стерильный ампициллин (Россия) до концентрации 150 мкг/мл. Среду хранят при комнатной температуре не более 3 месяцев.
Стерильная среда М9++глюкоза
Стерильная среда имеет следующий состав (г/л):
Среду готовят в мерном стакане емкостью 1000 мл, в который наливают 990 мл дистиллированной воды, добавляют навеску бакто-дрожжевого экстракта (2 г), аммоний сернокислый (4 г), натрий хлористого (0,6 г), калий фосфорнокислый 2-замещенный (2 г), магний сернокислый (0,4 г), железо сернокислое (0,02 г), марганец сернокислый (0,02 г), перемешивают. рН среды доводят 10 н. водным раствором NaOH до значения 7,0-7,2. Приготовленный раствор переносят в колбу емкостью 2000 мл, закрывают ватно-марлевой пробкой и стерилизуют в течение 20 мин при 121°С в автоклаве (Sanyo MLS 2420 U, Япония). После стерилизации добавляют в асептических условиях стерильный ампициллин (Россия) до концентрации 150 мкг/мл и стерильный тиамин до концентрации 0,04 г/л. Среду хранят при комнатной температуре не более 3 месяцев.
Для приготовления 40% раствора глюкозы в 100 мл дистиллированной воды растворяют 40 г глюкозы и стерилизуют в течение 15 мин при 115°С в автоклаве (Sanyo MLS 2420 U, Япония). Раствор хранят при комнатной температуре не более 6 месяцев.
Перед употреблением к стерильной среде добавляют в асептических условиях 10 мл стерильного 40% раствора глюкозы. Полученная среда с глюкозой не подлежит хранению.
Пример 4. Оптимизация процесса культивирования штамма-продуцента путем рН-статирования среды культивирования.
При проведении культивирования (контрольное, моделирующее процесс на лабораторном уровне «колба-ферментер») использовали среду следующего состава: М9++глюкоза, выбранную ранее как оптимальную для лабораторных экспериментов, которая содержала 2 г/л дрожжевого экстракта, 4 г/л глюкозы и 200 мкг/мл ампициллина. При внесении посевного материала контролировали оптическую плотность до достижения значения OD600, равного 0,09. Скорость мешалки поддерживали равной 750 об/мин. Принудительную аэрацию проводили путем подачи 1 V воздуха на V среды (1,5 л/мин). Следует отметить, что при таком режиме по показаниям датчика содержание растворенного кислорода снижалось в течение первых 5 часов (до мин. значения 15%), затем наблюдался подъем концентрации растворенного кислорода до 30 - 40%. рН-статирование отключали для оценки динамики рН при переходе к «крайним» условиям ферментации. Концентрацию глюкозы определяли на анализаторе глюкозы Biosen C-line (EKF Diagnostic). Результаты ферментаций приведены на Фиг. 2-4.
При переходе к культивированию в условиях рН-статирования использовали стандартную процедуру - титрование среды культивирования растворами 10% H2SO4 и 10% NH4OH, а рН поддерживали на уровне 7,5. При проведении ферментации с рН статированием использовали среду следующего состава: М9++глюкоза, выбранную ранее как оптимальную для лабораторных экспериментов, которая содержала 2 г/л дрожжевого экстракта, 4 г/л глюкозы и 200 мкг/мл ампициллина. При внесении посевного материала контролировали оптическую плотность до достижения значения OD600, равного 0,05. Скорость мешалки поддерживали равной 750 об/мин. Принудительную аэрацию проводили путем подачи 1 V воздуха на V среды (1,5 л/мин). Следует отметить, что при таком режиме по показаниям ДО содержание растворенного кислорода снижалось в течение первых 5 часов (до мин. значения 17%), затем наблюдался подъем концентрации растворенного кислорода до 35-40%. Концентрацию глюкозы определяли на анализаторе глюкозы Biosen C-line (EKF Diagnostic). Результаты культивирования приведены на Фиг. 5-7.
Полученные данные показывают, что рН-статирование замедляет процесс автолиза клеток штамма-продуцента и поддерживает накопление биомассы на постоянном уровне (Фиг. 5). Сравнительный анализ динамики роста культуры (OD600) и биосинтеза рекомбинантного белка SAV-RGD показывает, что скорость прироста содержания целевого продукта в клетках существенно выше скорости накопления биомассы: плотность культуры между 2 и 15 часом культивирования увеличилась в 3,7 раза, а количество синтезируемого целевого белка возросло в 10,8 раза, Фиг. 6 и 7). Соотнесение данных параметров, приведенное на Фиг. 8, показывает, что клетки штамма-продуцента не только сохраняют свой биосинтетический потенциал, но и увеличивают его.
Таким образом, рН-статирование является одним из ключевых факторов данного процесса культивирования, что позволяет сделать вывод о необходимости поддержания рН 7,5 в оптимизированном процессе культивирования.
Пример 5. Изучение влияния аэрации на уровень продукции целевого белка.
Как уже отмечалось выше, при культивировании клеток Е. coli в присутствии глюкозы как источника углерода наблюдается активная наработка кислого продукта. Анализ научно-технической литературы показывает, что таким кислым продуктом может ацетат и/или формиат [Тао, Н., С. Bausch, С. Richmond, F. R. Blattner, and Т. Conway. 1999. Functional genomics: expression analysis of Escherichia coli growing on minimal and rich media. J. Bacteriol. V. 181P. 6425-6440].
На данный процесс во многом влияет аэрация (степень насыщения среды культивирования кислородом). В связи с этим следующим этапом оптимизации было исследование влияния аэрации (скорости вращения мешалки лабораторного ферментера) на процесс культивирования клеток штамма-продуцента в лабораторном ферментере.
При проведении культивирования использовали среду следующего состава: М9++глюкоза, выбранную ранее как оптимальную для лабораторных экспериментов, которая содержала 2 г/л дрожжевого экстракта, 4 г/л глюкозы и 200 мкг/мл ампициллина. Для рН-статирования использовали стандартную процедуру, описанную выше, а рН поддерживали на уровне 7,5. При внесении посевного материала контролировали оптическую плотность до достижения значения OD600, равного 0,06. Скорость мешалки поддерживали равной 450 об/мин, уменьшая, таким образом, уровень аэрации в объеме культуральной среды. Принудительную аэрацию проводили путем подачи 1 V воздуха на V среды (1,5 л/мин). Следует отметить, что при таком режиме по показаниям ДО содержание растворенного кислорода снижалось в течение первых 5 часов (до минимального значения 10%), затем наблюдался подъем концентрации растворенного кислорода до 20 - 25%. Концентрацию глюкозы определяли на анализаторе глюкозы Biosen C-line (EKF Diagnostic).
Результаты культивирования при снижении уровня аэрации - снижении скорости вращения мешалки лабораторного ферментера - приведены на Фиг. 8-10. Из полученных экспериментальных данных следует, что снижение уровня аэрации до 450 оборотов мешалки в минуту, а следовательно, и снижение насыщения питательной среды кислородом, приводило к увеличению биомассы и количества целевого белка (Фиг. 8 и 9). Следует отметить, что содержание рекомбинантного белка SAV-RGD возрастало до 90 - 95 мг/л.
Однако выход целевого белка на единицу биомассы был ниже, чем в экспериментах при 750 оборотах/мин, и составлял 5,6 мг/г биомассы (Фиг. 10).
Таким образом, полученные данные показывают, что снижение оборотов мешалки ферментера (насыщение кислородом среды культивирования), наряду с дополнительным автоматическим режимом рН-статирования, оказывают существенное влияние на биосинтетический потенциал клеток штамма-продуцента, улучшая этот показатель в среднем на 15-18% по сравнению с ферментациями без рН-статирования и при высоких оборотах мешалки лабораторного ферментера.
Пример 6. Изучение влияния подпитки глюкозой и аминокислотами на уровень продукции целевого белка.
Известно, что одним из параметров, лимитирующих стадию биосинтеза, является быстрое истощение основных компонентов - источников углерода и азота - в питательной среде. Этот процесс особенно активно протекает в условиях культивирования клеток штамма-продуцента в ферментере (Фиг. 3, 7, 11). В связи с вышеизложенным была проведена экспериментальная ферментация на среде с дополнительным внесением глюкозы и источника аминокислот.
При выборе скорости подачи глюкозы учитывали особенности промотор-операторной области, под контролем которой находится искусственный ген sav-rgd. Согласно литературным данным активация этой промотор-операторной области осуществляется комплексом CRP-cAMP (Hammer - Jespersen К. // Metabolism of Nucleotides, Nucleosides and Nucleobases in Microorganisms / Ed. Munch - Petersen A. London, New York, San Francisco: Acad. Press - 1983. - P. 203 - 258. Kolb A., Busby S., Buc H., Garges S., Adhya S. (1993) Transcriptional regulation by cAMP and its receptor protein. Annu. Rev. Biochem. 62, 749-795). В самом общем виде: этот промотор является катаболит-чувствительным и репрессируется в условиях присутствия высоких концентраций глюкозы репрессором CytR. В связи с этим скорость подачи глюкозы регулировалась таким образом, чтобы конечная концентрация в питательной среде была не выше 1 - 2 г/л.
При проведении ферментации использовали среду следующего состава: М9++глюкоза, выбранную ранее как оптимальную для лабораторных экспериментов, которая содержала 2 г/л дрожжевого экстракта, 4 г/л глюкозы и 200 мкг/мл ампициллина. Для рН-статирования использовали стандартную процедуру, описанную выше, а рН поддерживали на уровне 7,5. При внесении посевного материала контролировали оптическую плотность до достижения значения OD600, равного 0,06. Скорость мешалки поддерживали равной 450 об/мин. Принудительную аэрацию проводили путем подачи 1 V воздуха на V среды (1,5 л/мин). Следует отметить, что при таком режиме по показаниям ДО содержание растворенного кислорода снижалось в течение первых 5 часов (до мин. значения 10%), затем наблюдался подъем концентрации растворенного кислорода до 20 - 25%. Концентрацию глюкозы определяли на анализаторе глюкозы Biosen C-line (EKF Diagnostic). Исходный объем среды в ферментере составлял 1,1 л. Инокулировали посевным материалом и в ферментер сразу принудительно подавали 40%-ный раствор глюкозы при скорости подачи 25 мл/час и культивирование клеток штамма-продуцента в этом режиме проводили в течение 10 часов. По окончании введения глюкозы дробно добавляли стерильный раствор пептона (10 г/л), дрожжевого экстракта (5 г/л) и ампициллина (100 мг/л) в течение еще 8 часов. Скорость подачи раствора составляла 25 мл/час. Общий объем культуральной суспензии составлял на момент окончания ферментации примерно 1,35 л. Суммарно процесс культивирования проводили в течение 18 часов.
Результаты ферментаций при условии подпиткой источниками углерода и аминокислот приведены на Фиг. 11-13.
Анализ приведенных данных (Фиг. 11-13) позволяет утверждать, что в период культивирования с 10 до 14 часов происходит исчерпание основного источника углерода - глюкозы (культура растет при лимитации источника углерода). В этот же период культура метаболитически перестраивается к потреблению аминокислот из пептона и дрожжевого экстракта, что выражается в понижении скорости роста культуры (несмотря на начало дробного введения источников аминокислот) и, одновременно, продукции целевого белка. После этого наблюдается резкое возрастание как плотности культуральной суспензии, так и уровня накопления рекомбинантного белка SAV-RGD (Фиг. 11-13). Эти данные позволяют предположить, что в условиях культивирования в лабораторном ферментере происходит начальная интенсивная утилизация глюкозы, что приводит к наработке популяции клеток штамма-продуцента, способного к активному продуцированию целевого рекомбинантного белка, а последующая дробная подпитка аминокислотными компонентами служит своеобразным «индуктором» активирующим биосинтез рекомбинантного белка SAV-RGD.
Одновременно следует признать, что избранный подход дробной подачи глюкозы является оптимальным, т.к. в питательной среде не создается высокая концентрация этого источника углерода, приводящая к слишком интенсивному формированию ацетата, угнетающее действие которого на рост культуры рекомбинантного штамма Е. coli-продуцента целевого белка в ферментере было обнаружено ранее.
В связи с этим можно было предложить разнести процесс культивирования клеток штамма-продуцента на два этапа: наращивание общей биомассы и этап активного биосинтеза рекомбинантного белка. При этом стадия «индукции» аминокислотными добавками может быть проведена не дробной подачей, а единовременным внесением аминокислотных компонентов.
Таким образом, оптимальными условиями культивирования рекомбинантного штамма Е. coli-продуцента целевого белка в ферментере являются: Среда М9++глюкоза следующего состава (г/л):
рН-статирование - рН 7,5.
Количество вносимого посевного материала - по OD600 0,05-0,09 культуральной среды.
Скорость мешалки - 450 об/мин.
Принудительная аэрация - 1,5 л воздуха в минуту.
Подпитка - 40%-ным раствором глюкозы (скорость подачи 25 мл/час, поддержание концентрации глюкозы на уровне 1 г/л) в течение 10 час.
Дробная подпитка - раствор пептона (10 г/л), дрожжевого экстракта (5 г/л) и ампициллина (100 мг/л) в течение последующих 8 часов со скоростью подачи 25 мл/час.
Время ферментации - 18 часов.
Пример 7. Приготовление растворов для хроматографии, лизиса клеток, гель-фильтрации и ультрафильтрации.
Для приготовления буфера «А» в мерный стакан объемом 1000 мл вносят 950 мл дистиллированной воды и добавляют навески NaCl (29 г), и Na2СО3 (5,3 г) и перемешивают на магнитной мешалке до полного растворения. Раствор титруют до достижения рН 10,8. После чего доводят объем до 1000 мл и хранят при температуре +4°С.
Для приготовления буфера «Б» в мерный стакан объемом 1000 мл вносят 950 мл дистиллированной воды и добавляют навески NaCl (29 г) и перемешивают на магнитной мешалке до полного растворения. После чего доводят объем до 1000 мл и хранят при температуре +4°С.
Для приготовления буфера «В» в мерный стакан объемом 1000 мл вносят 950 мл дистиллированной воды и добавляют навески СН3СООNa (4,1 г) и NaCl (29 г), и перемешивают на магнитной мешалке до полного растворения. Раствор титруют до достижения рН 4,0. После чего доводят объем до 1000 мл и хранят при температуре +4°С.
Для приготовления буфера для гель-фильтрации в мерный стакан объемом 1000 мл вносят 950 мл дистиллированной воды и добавляют навески Na2HPO4 (1,42 г) и NaCl (8,7 г), и перемешивают на магнитной мешалке до полного растворения. Раствор титруют до достижения рН 7,4. После чего доводят объем до 1000 мл и хранят при температуре +4°С.
Для приготовления буфера ТЕ-буфера в мерный стакан объемом 1000 мл вносят 950 мл дистиллированной воды и добавляют навески Трис-С1 (3,94 г) и ЭДТА (1,47 г), и перемешивают на магнитной мешалке до полного растворения. Раствор титруют до достижения рН 8,0. После чего доводят объем до 1000 мл и хранят при температуре +4°С.
Для приготовления буфера лизирующего буфера в мерный стакан объемом 1000 мл вносят 750 мл дистиллированной воды и добавляют навески Трис-С1 (1,57 г), ЭДТА (2,92 г), лизоцим (1 г) и сахарозу (200 г), перемешивают на магнитной мешалке до полного растворения. Раствор титруют до достижения рН 8,0. После чего доводят объем до 1000 мл и хранят при температуре +4°С.
Пример 8. Оптимизация процесса получения периплазматической фракции клеток штамма-продуцента рекомбинантного белка SAV-RGD.
Полученную биомассу промывают двукратно 100 мл ТЕ-буфером (10 мМ Трис-С1, рН 8,0; 5 мМ ЭДТА) с отделением ее центрифугированием при тех же условиях. К полученной биомассе добавляют лизирующий буфер, содержащий 20% сахарозы, 10 мМ ЭДТА, 1 мг/мл лизоцима, 25 мМ Трис-С1, рН 8,0, и инкубируют 15-20 минут при +4°С (ледяная баня) при постоянном перемешивании. Смесь центрифугируют 10 мин при 10000 об/мин (+4°С). Супернатант, содержащий фракцию периплазматических белков, собирают и используют для очистки целевого белка.
Процесс оптимизации включал подбор соотношения суспензии биомассы и объема лизирующей смеси с варьированием данного соотношения - 1:1, 1:5, 1:10, 1:15, 1:20 и 1:25. Контрольным параметром являлось количество выделенного целевого белка. Содержание целевого белка в периплазматической фракции определяли с помощью ВЭЖХ. Результаты оптимизации суммированы в Таблице 1.
Как видно из приведенных данных соотношение лизирующего буфера и биомассы можно значительно уменьшить, доведя его до величины 1:20 (1 объем лизирующего буфера: 20 объемов суспензии клеток после ферментации). При этом практически весь белок SAV-RGD локализовался в надосадочной жидкости (фракции периплазматических белков штамма-продуцента). Дальнейшее увеличение количество биомассы на один объем лизирующего приводило к увеличению вязкости получаемого раствора белков, а также к потерям при отделении раствора периплазматических белков от осадка после центрифугирования. В связи с этим в дальнейшем использовали соотношение лизирующего буфера к объему суспензии клеток 1:20.
Пример 9. Очистка рекомбинантного белка SAV-RGD методом ионообменной хроматографии.
Ранее авторы настоящего изобретения показали, что использование метода аффинной хроматографии на 2-иминобиотин-агарозе позволяет достичь степени очистки целевого продукта в 17,5 раз, а чистоты - 95-97%. Для увеличения степени чистоты целевого белка и удешевления стоимости данной стадии технологического процесса были проведены эксперименты по сравнению с процессами выделения рекомбинантного белка SAV-RGD из периплазматической фракции на колонках с носителями 2-иминобиотин-агароза и SP-сефароза. Объем носителя увеличивали в соответствии с коэффициентом масштабирования, что соответствовало увеличению получаемой культуральной жидкости на стадии 1 масштабирования - колбы, стадии 2 масштабирования - 1,5-литровый ферментер и стадии 3 масштабирования - 7-литровый ферментер.
Очистку рекомбинантного белка SAV-RGD методом ионообменной хроматографии проводили следующим образом. Периплазматическую фракцию вносили при постоянном перемешивании в буферный раствор 0,01 М ацетата натрия, рН 4,0 для удаления части балластных белков. Полученный раствор титровали до рН 4,0 ледяной уксусной кислотой и инкубировали в течение 10 минут при постоянном перемешивании. Затем раствор центрифугировали 30 мин (5000xg, +4°С). Супернатант осторожно сливали и наносили на колонку с SP-сефарозой fast flow (GE, США) с помощью перистальтического насоса (скорость протока 2,5 мл/мин). Колонку промывали последовательно 100 мл 0,01 М ацетата натрия, рН 4,0; 250 мл 0,15 М NaCl в 0,01 М ацетате натрия, рН 4,0. Белок элюировали 0,25 М NaCl в 0,01 М ацетате натрия, рН 4,0. Выход белка контролировали с помощью ультрафиолетового детектора. Полученный раствор белка собирали и определяли концентрацию белка с помощью метода ВСА (BCA-kit, Sigma), чистоту белка контролировали ВЭЖХ на колонке Symmetry 300С4 (4,6×150 мм) в градиенте 0,1% ТФУ - ацетонитрил, а также электрофорезом в 14%-ном полиакриламидном геле. Элюат концентрировали с помощью ячейки Amicon (мембрана РМ-30) до концентрации белка 10-20 мг/мл и диализовали против 3л 0,01 М фосфата натрия, рН 7,4 (+4°С, 18 ч). Диализат фильтруют через мембранный фильтр с размерами пор 0,22 мкм, аликвоты фильтрата переносят в стерильные пенициллиновые флаконы емкостью 10 мл, флаконы накрывают стерильной тканью Петрянова, выдерживают при - 70°С (как минимум 2 ч) и лиофилизуют 20 ч. Флаконы с высушенным препаратом укупоривают стерильными резиновыми пробками и зажимают алюминиевыми колпачками. Хранят при (+4°С).
Количество полученного рекомбинантного белка SAV-RGD определяли методом ВЭЖХ, концентрацию белка - с помощью метода ВСА (BCA-kit с протоколом методики фирмы-производителя Sigma, кат. №ВСА1-1КТ, США). Полученные данные суммированы в Таблице 2.
Как видно из полученных данных, как выход по целевому белку, так и степень его чистоты была сопоставима или выше при использовании ионообменной хроматографии. Чистоту полученных двумя методами целевых белков оценивали методом ВЭЖХ. Данные представлены на Фиг. 14. Результаты хроматографического анализа свидетельствуют, что чистота рекомбинантного белка SAV-RGD, выделенного методом аффинной хроматографии, и чистота рекомбинантного белка SAV-RGD, выделенного методом ионообменной хроматографии, сопоставима. Однако при сопоставимых выходах и чистоты целевого белка экономические показатели двух методов выделения различаются значительно. При максимальном масштабировании затраты на сорбент при выделении 1 мг целевого белка SAV-RGD составляют: при применении аффинной хроматографии 28,3 евро/мг, а при использовании ионообменной хроматографии - 0,14 евро/мг. То есть метод аффинной хроматографии дороже более чем в 200 раз. Следовательно, замена носителя для проведения выделения и очистки рекомбинантного белка SAV-RGD позволяет удешевить данную стадию технологического процесса. Кроме того, емкость 2-имнобиотин-агарозы и его невысокая стабильность (сшитая агароза) не позволяют рассчитывать на большое число циклов очистки белка на основе стрептавидина, необходимое при масштабировании процесса. В то же время SP-сефароза в качестве носителя достаточно стабильна и выдерживает многократные циклы очистки белка.
Таким образом, полученные результаты показали перспективность использования ионообменной хроматографии для выделения и очистки рекомбинантного белка SAV-RGD.
Пример 10. Проверка биологической активности полученного белка SAV-RGD.
Эффективность связывания целевого белка с клетками меланомы человека линии Me Wo определяли следующим образом:
Подготовка клеток
Клеточную культуру метастазирующей беспигментной меланомы человека MeWo (malignant melanoma (derived from metastatic site, lymph node by Y. Kodera and M. Bean, 1974), АТСС® HTB-65™) высевали на 10-сантиметровые культуральные чашки (Greiner, Австрия) и растили в течение 72 часов в модифицированной Дульбекко среде Игла (DMEM, Dulbecco's modified Eagles medium) (ПанЭко, Россия) с добавлением 10% фетальной сыворотки теленка (Hyclone, США), 100 ед./мл пенициллина и 100 мкг/мл стрептомицина (либо 200 мкг/мл канамицина). Культивирование вели при 37°С во влажной атмосфере, содержащей 5% углекислоты. Плотность культуры оценивали визуально. В экспериментах использовали клетки в экспоненциальной стадии роста, прошедшие 3-4 пассажа после размораживания. К моменту постановки эксперимента клетки занимали приблизительно 30-50% площади поверхности дна чашки.
Непосредственно перед проведением эксперимента по оценке эффективности связывания белка с клетками из культуральных чашек среду удаляли, клетки промывали 3 раза раствором DPBS (ПанЭко, Россия), после чего вносили 4 мл раствора Версена (ПанЭко, Россия) и инкубировали в течение 5 минут при комнатной температуре. Раствор Версена удаляли, клетки смывали с чашки при помощи 3 мл среды DMEM без сыворотки, ресуспендировали и инкубировали в течение 30 минут. Количество клеток определяли методом подсчета в камере Горяева.
Исходя из данных подсчета, клетки разводили средой DMEM без сыворотки до достижения плотности 200-250 тысяч клеток/1 мл суспензии.
Получение FITC-меченого рекомбинантного белка SAV-RGD.
Лиофилизированный рекомбинантный белок SAV-RGD растворяли в PBS до концентрации 1 мг/мл. FITC-конъюгированный биотин (5-((N-(5-(N-(6-(biotinoyl)amino)hexanoyl)amino)pentyl)thioureidyl)fluorescein (fluorescein biotin), В1370, Molecular Probes, Inc.) растворяли в DMSO до концентрации 25 мг/мл. Раствор FITC-биотина добавляли к раствору рекомбинантного белка SAV-RGD, перемешивали и инкубировали в темноте при комнатной температуре в течение 60 минут. Поскольку белок SAV-RGD содержит 4 сайта связывания биотина, молярное соотношение SAV-RGD:FITC-биотин в растворе составляло 1:4. Таким образом, для мечения 1 мг рекомбинантного белка использовали 44,5 мкг FITC-биотина.
Оценка эффективности связывания FITC-меченого рекомбинантного белка SAV-RGD с клетками линии MeWo.
Подготовка экспериментального образца (Э). 250 мкл подготовленной клеточной суспензии, содержащей 50-60 тыс. клеток, помещали в 1,5 мл пробирку и добавляли 25 мкл раствора исследуемого белка SAV-RGD, меченного FITC. Таким образом, конечная концентрация белка в растворе составляла 1,25*10-6 моль/л. Пробирки с клеточной суспензией помещали в термостат и инкубировали в течение 1 часа при 37°С в атмосфере 5% СO2, встряхивая каждые 10-15 минут. По прошествии 1 часа клетки осаждали центрифугированием в настольной центрифуге (3 мин при 3000 об/мин (800g)) и удаляли супернатант. Осадок промывали 400 мкл раствора DPBS, осаждали центрифугированием, а затем ресуспендировали в 250 мкл раствора DPBS.
Подготовка контрольного образца (К)
250 мкл подготовленной клеточной суспензии, содержащей 50-60 тыс.клеток, помещали в пробирку и добавляли 25 мкл раствора DPBS. Пробирки с клеточной суспензией помещали в термостат и инкубировали в течение 1 часа при 37°С в атмосфере 5% СО2, периодически встряхивая (каждые 10-15 минут). По прошествии 1 часа клетки осаждали центрифугированием (3 мин при 3000 об/мин (800g)) и удаляли супернатант. Осадок промывали 400 мкл раствора DPBS, осаждали центрифугированием, а затем ресуспендировали в 250 мкл раствора DPBS.
Интенсивность флуоресценции клеток из экспериментального (Э) и контрольного (К) образцов анализировали при помощи проточного цитофлуориметра (FACS Aria SORP, Beckton Dickinson, либо аналога), определяя медиану распределения интенсивности флуоресценции клеток экспериментального (Мэ) и контрольного (Мк) образцов соответственно.
Вывод об эффективности белкового препарата делали на основании значения М, которое рассчитывается по формуле М=(Мэ/Мк*5). Если полученное значение больше либо равно 1, то анализируемый препарат считается эффективным, если меньше 1 - неэффективным.
Результаты эксперимента по оценке эффективности связывания рекомбинантного белка SAV-RGD клетками меланомы человека линии MeWo представлены на Фиг. 15.
Хотя указанное изобретение описано в деталях выше, для специалиста в указанной области техники очевидно, что могут быть внесены различные изменения и произведены эквивалентные замены, и такие изменения и замены не выходят за рамки настоящего изобретения. Все цитируемые документы являются частью описания настоящего изобретения и целиком включены в настоящее описание посредством ссылки.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ РЕКОМБИНАНТНОГО БЕЛКА SAV-RGD, СПЕЦИФИЧЕСКИ УЗНАЮЩЕГО КЛЕТКИ МЕЛАНОМЫ | 2013 |
|
RU2563540C2 |
| СЛИТНЫЙ БЕЛОК, СПЕЦИФИЧЕСКИ УЗНАЮЩИЙ МЕЛАНОМНЫЕ КЛЕТКИ | 2012 |
|
RU2535878C2 |
| РЕКОМБИНАНТНАЯ ПЛАЗМИДНАЯ ДНК PSAV27, КОДИРУЮЩАЯ СИНТЕЗ РАСТВОРИМОГО СТРЕПТАВИДИНА ИЗ STREPTOMYCES AVIDINII, И БАКТЕРИАЛЬНЫЙ ШТАММ ESCHERICHIA COLI - ПРОДУЦЕНТ РАСТВОРИМОГО СТРЕПТАВИДИНА ИЗ STREPTOMYCES AVIDINII | 1999 |
|
RU2153535C1 |
| Рекомбинантная плазмидная ДНК pET19b-SAV, обеспечивающая синтез полноразмерного белка стрептавидина Streptomyces avidinii, штамм бактерий Escherichia coli - продуцент растворимого полноразмерного белка стрептавидина Streptomyces avidinii | 2019 |
|
RU2728652C1 |
| Штамм Escherichia coli - продуцент L-треонина | 2023 |
|
RU2817252C1 |
| РЕКОМБИНАНТНАЯ ПЛАЗМИДНАЯ ДНК, СОДЕРЖАЩАЯ ПОСЛЕДОВАТЕЛЬНОСТЬ ЗРЕЛОЙ СТАФИЛОКИНАЗЫ Staphylococcus aureus С ЛИДЕРНЫМ ПЕПТИДОМ ИЗ ШЕСТИ ГИСТИДИНОВЫХ ОСТАТКОВ, ШТАММ Escherichia coli MZ08 И СПОСОБ ПОЛУЧЕНИЯ РЕКОМБИНАНТНОГО БЕЛКА, СОДЕРЖАЩЕГО ПОСЛЕДОВАТЕЛЬНОСТЬ ГЕНА ЗРЕЛОЙ СТАФИЛОКИНАЗЫ С ЛИДЕРНЫМ ПЕПТИДОМ ИЗ ШЕСТИ ГИСТИДИНОВЫХ ОСТАТКОВ | 2009 |
|
RU2422522C2 |
| Рекомбинантный штамм бактерии Escherichia coli - продуцент L-треонина | 2018 |
|
RU2697219C1 |
| РЕКОМБИНАНТНАЯ ПЛАЗМИДНАЯ ДНК pACYC-LANS(KM), ШТАММ Escherichia coli BL21(DE3), ТРАНСФОРМИРОВАННЫЙ РЕКОМБИНАНТНОЙ ДНК pACYC-LANS(KM), И СПОСОБ ПОЛУЧЕНИЯ РЕКОМБИНАНТНОЙ L-АСПАРАГИНАЗЫ Erwinia carotovora | 2010 |
|
RU2441916C1 |
| РЕКОМБИНАНТНАЯ ПЛАЗМИДНАЯ ДНК, СОДЕРЖАЩАЯ ПОСЛЕДОВАТЕЛЬНОСТЬ ЗРЕЛОЙ СТАФИЛОКИНАЗЫ STAPHYLOCOCCUS AUREUS С ЗАМЕНАМИ КОДОНОВ K74, E75 И R77 НА ТРИПЛЕТЫ, КОДИРУЮЩИЕ Ala, ШТАММ ESCHERICHIA COLI MZ09 И СПОСОБ ПОЛУЧЕНИЯ РЕКОМБИНАНТНОГО БЕЛКА, СОДЕРЖАЩЕГО ПОСЛЕДОВАТЕЛЬНОСТЬ ГЕНА ЗРЕЛОЙ СТАФИЛОКИНАЗЫ С ЗАМЕНАМИ КОДОНОВ K74, E75 И R77 НА ТРИПЛЕТЫ, КОДИРУЮЩИЕ Ala | 2010 |
|
RU2422153C1 |
| СПОСОБ МИКРОБИОЛОГИЧЕСКОГО СИНТЕЗА ГИБРИДНОГО БЕЛКА Е7-HSP70 (ВАРИАНТЫ) | 2013 |
|
RU2546917C1 |


Изобретение касается способа получения рекомбинантного белка SAV-RGD, где SAV - мономер стрептавидина, RGD - меланома-адресующий олигопептид, имеющий аминокислотную последовательность Ser-Arg-Ala-Gly-Ala-Asp-Gly-Phe-Pro-Gly-Cys-Arg-Gly-Asp-Cys-Ser-Gln-Glu. Представленный способ включает культивирование в питательной среде бактериального штамма E. coli MG1655/pSAV-RGD, полученного трансформацией штамма E. coli MG1655 плазмидой pSAV-RGD, в свою очередь полученной на базе вектора pUC18 и включающей последовательность, кодирующую слитый пробелок SAV-RGD (pro-sav-rgd) под контролем промотора уридинфосфорилазы (Pudp) Е. coli. Получение фракции клеток бактериального штамма, очистку полученного рекомбинантного белка. Причем культивирование проводят в два этапа. На первом этапе осуществляют подпитку источником глюкозы, а на втором этапе осуществляют подпитку пептоном и дрожжевым экстрактом. Изобретение может быть использовано для производства лекарственных средств для лечения меланомы человека. 4 з.п. ф-лы, 15 ил., 2 табл., 10 пр.
1. Способ получения рекомбинантного белка SAV-RGD, где SAV - мономер стрептавидина, RGD - меланома-адресующий олигопептид, имеющий аминокислотную последовательность Ser-Arg-Ala-Gly-Ala-Asp-Gly-Phe-Pro-Gly-Cys-Arg-Gly-Asp-Cys-Ser-Gln-Glu, включающий:
- культивирование в питательной среде штамма бактерий MG1655/pSAV-RGD, полученного трансформацией штамма Escherichia coli MG1655 плазмидой pSAV-RGD, в свою очередь полученной на базе вектора pUC18 и включающей последовательность, кодирующую слитый пробелок SAV-RGD (pro-sav-rgd) под контролем промотора уридинфосфорилазы (Pudp) Е. coli, при этом культивирование осуществляют с рН-статированием в диапазоне рН 7,5-7,8, и концентрацией растворенного кислорода, не превышающей 20-25%, в два этапа, где на первом этапе, продолжающемся в течение 8-12 часов, осуществляют подпитку источником глюкозы, а на втором этапе, продолжающемся в течение 6-10 часов, осуществляют подпитку источником аминокислот, в качестве которого используют пептон и дрожжевой экстракт;
- получение периплазматической фракции клеток бактериального штамма с использованием лизирующего буфера, в состав которого входит лизоцим в концентрации 1 г/л;
- очистку рекомбинантного белка методом ионообменной хроматографии с последующей ультрафильтрацией; и
- лиофилизацию очищенного рекомбинантного белка SAV-RGD.
2. Способ по п. 1, характеризующийся тем, что в качестве питательной среды используют М9++глюкоза.
3. Способ по п. 1, характеризующийся тем, что на первом этапе культивирования контролируют скорость подпитки глюкозой таким образом, чтобы конечная концентрация глюкозы в питательной среде не превышала 1-2 г/л.
4. Способ по п. 1, характеризующийся тем, что при получении периплазматической фракции клеток штамма-продуцента рекомбинантного белка SAV-RGD используют соотношение объемов лизирующего буфера и биомассы клеток 1:20.
5. Способ по п. 1, характеризующийся тем, что в качестве носителя в методе ионообменной хроматографии используют SP-сефарозу, а ультрафильтрацию проводят с использованием мембраны с размером пор 30 кДа.
| РЕКОМБИНАНТНАЯ ПЛАЗМИДНАЯ ДНК PSAV27, КОДИРУЮЩАЯ СИНТЕЗ РАСТВОРИМОГО СТРЕПТАВИДИНА ИЗ STREPTOMYCES AVIDINII, И БАКТЕРИАЛЬНЫЙ ШТАММ ESCHERICHIA COLI - ПРОДУЦЕНТ РАСТВОРИМОГО СТРЕПТАВИДИНА ИЗ STREPTOMYCES AVIDINII | 1999 |
|
RU2153535C1 |
| MARINA S.SYRKINA et al., Preparation and functional evaluation of RGD-modified streptavidin targeting to integrin-expressing melanoma cells, Protein Engineering, Design & Selection, Published online November 16, 2012, vol | |||
| Прибор для получения стереоскопических впечатлений от двух изображений различного масштаба | 1917 |
|
SU26A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Крутильная машина для веревок и проч. | 1922 |
|
SU143A1 |
| WO 2003072014 A2, 04.09.2003. | |||

