Область техники
Настоящее изобретение относится к новым пептидам, которые индуцируют аналгезию и ингибируют ASIC-каналы (протон-чувствительные ионные каналы), более конкретно, гомомерные ASIC1a-каналы, гомомерные ASIC1b-каналы и/или гетеромерные каналы, содержащие по меньшей мере одну субъединицу, выбранную из ASIC1a и ASIC1b, к полинуклеотидам, кодирующим указанные пептиды, а также к содержащим их фармацевтическим композициям, клеткам-хозяевам и векторам. В частности, указанные пептиды имеют минимально 56%-ную идентичность с аминокислотной последовательностью SEQ ID NO:1, представленной в прилагаемом списке последовательностей. В частности, они представляют собой два пептида с 57 аминокислотами, ASICалгин (π-Dp1) и ASICалгин (π-Dp2), выделенные из яда змеи Dendroaspis polylepis (черная мамба), с последовательностями соответственно представленными SEQ ID NO: 2 и SEQ ID NO: 3.
Настоящее изобретение относится также к применению названных пептидов для получения диагностического или лекарственного средства, в частности, анальгетического средства, а также для идентификации анальгетических молекул или молекул, ингибирующих ASIC-каналы.
Настоящее изобретение предлагает, в частности, применение пептидов для профилактики или лечения боли, в частности, боли, ассоциированной с активацией ASIC-каналов (например, воспалительных, нейропатических, раковых, послеоперационных, мышечно-скелетных, висцеральных и т.д. болей), неврологических заболеваний центральной нервной системы (например, посттравматический стресс, депрессия, тревожные состояния, нарушения мозгового кровообращения, эпилепсия, рассеянный склероз, воспаления головного мозга, нейродегенеративные заболевания и т.д.) и патологий, в которых предполагается участие ASIC-каналов (например, воспалительные процессы, раковые заболевания, фибромиалгии, синдром раздраженной толстой кишки и т.д.).
В нижеследующем описании ссылки, указанные в квадратных скобках, сведены в общий список ссылок, представленный в конце текста.
Уровень техники
Изучение и лечение боли являются существенными аспектами улучшения качества жизни пациентов. Боль поражает каждый год в Европе значительное количество людей, порядка 60 миллионов, что составляет затраты в 1 миллиард долларов в год на обезболивающие лекарственные средства для ее лечения. Затраты, расходуемые ежегодно в мире на обезболивающие препараты, оцениваются в сумме около 25 миллиардов долларов и должны будут составить 42 миллиарда в 2010 году. Боль разделяют на две категории: острая боль и хроническая боль. Острая боль соответствует боли внезапной и короткой, ограниченной во времени. Напротив, хроническая боль это непрекращающаяся боль, которая может быть связана, например, с гипералгезией, и является фактором, значительно отягощающим течение болезни, поражающим примерно 20% взрослого населения и 50% населения более старшего возраста.
Лечение боли основывается главным образом на назначении противовоспалительных препаратов, которые могут быть нестероидными (НПВП) или стероидными (кортикоидными) препаратами, и сильных или слабых опиатов. НПВП образуют наиболее назначаемый в мире класс терапевтических препаратов в связи с их высокой эффективностью как в отношении воспалительного процесса, так и в отношении самой боли. Их применяют при всех типах воспалительных болей, как острых, так и хронических [Bertin et Vergne-Salle, 2007] [1]. Когда НПВП и/или кортикоиды недостаточно эффективны для того, чтобы успокоить воспалительную боль, то врач добавляет к анальгетику, не являющемуся противовоспалительным средством, например, парацетамолу, слабые опиаты (кодеин, трамадол), а если боль не поддается лечению, то назначает сильные опиаты (морфин, оксикодон, фентанил) [Gutstein & Akil, 2006][2].
Хотя НПВП являются очень эффективными средствами, тем не менее, они остаются серьезным источником нежелательных побочных эффектов. Среди обычных нежелательных побочных эффектов чаще всего встречаются эффекты со стороны органов пищеварения, что ограничивает применение НПВП в многочисленных клинических ситуациях. Отмечают также нежелательные побочные эффекты со стороны почек, кожи, слизистой оболочки, аллергические и респираторные нежелательные эффекты и гематологические побочные эффекты, побочные эффекты со стороны печени и, наконец, нейросенсорные и психические побочные эффекты [Bertin et Vergne-Salle, 2007, см. выше] [1]. Кроме того, препараты НПВП не являются эффективными при всех видах боли. Опиоиды также играют важную роль в борьбе с болью, однако они могут провоцировать галлюцинаторные явления и кардиореспираторную депрессию. Анальгетические средства могут также стать источником возникновения зависимости, как, например, зависимость от морфина, метадона и т.д. Имеются также случаи привыкания к анальгетикам, т.е. случаи, когда доза, необходимая для достижения постоянного эффекта, должна быть увеличена. Это привыкание усиливается со временем и поэтому приводит к необходимости увеличивать дозировку, что может повлечь за собой потерю эффективности лекарственного средства. Так, доза, необходимая для снятия боли, может оказаться выше токсической дозы вышеназванного лекарственного средства. Наконец, лечение опиоидами может также ассоциироваться с нежелательными эффектами, такими как серьезные запоры или гипералгезия после окончания лечения (например, послеоперационная боль) [Gutstein & Akil, 2006, см. выше; Bannister & Dickenson, 2010] [2, 3].
Несмотря на разнообразный арсенал существующих терапевтических средств, многие виды боли остаются мало чувствительными к известным анальгетикам, такие как нейропатические боли, возникающие вследствие поражения нервной системы (50% больных не испытывают облегчения), висцеральные хронические боли, такие как при синдроме раздраженной толстой кишки или хронических воспалительных заболеваниях кишечника, фибромиалгии, боли, связанные с раковыми заболеваниями и с костными метастазами, и т.д. [Yennurajalingam et al., 2004; Mizoguchi et al., 2009] [4, 5].
В этом контексте открытие новых анальгетических или антальгетических средств, устраняющих указанные проблемы и недостатки, и/или открытие новых мишеней для анальгетиков было бы реальным прогрессом в данной области.
Исследования фармацевтической промышленности в области боли в последние годы привели лишь к нескольким ограниченным разработкам. Можно назвать, например, триптаны, используемые при мигренях, и некоторые новые лекарственные средства, применение которых остается пока ограниченным, такие как ассоциация тетрагидроканнабинола с каннабидиолом для борьбы с раковыми и нейропатическими болями. В самом деле, прогресс, отмеченный за последние два десятилетия, основывался главным образом на наилучшем использовании и адаптации дозировок имеющихся анальгетических препаратов. Однако ни одно из крупных семейств этих анальгетиков не имеет соотношения польза/риск, которое было бы оптимальным, поскольку они имеют ограниченную эффективность и/или серьезные побочные эффекты.
Среди молекулярных мишеней, идентифицированных в последние годы, ионные каналы занимают особенно важное место, потому что они непосредственно участвуют в детектировании и передаче болевых сигналов сенсорными и центральными нейронами. ASIC-каналы (протон-чувствительные ионные каналы) являются катионными каналами, активируемыми путем ацидификации внеклеточной среды (внеклеточный ацидоз) [Waldemann & Lazdunski, 1998; Wemmie et al., 2006; Lingueglia et al., 2007] [6, 7, 8]. К настоящему времени идентифицированы у млекопитающих четыре гена, кодирующие по меньшей мере семь субъединиц (ASIC1a, ASIC1b, ASIC1b2, ASIC2a, ASIC2b, ASIC3 и ASIC4). Функциональные ASIC-каналы появляются в результате ассоциации различных ASIC-субъединиц c образованием трехмерной структуры [Jasti et al., 2007] [9], приводящей к образованию гомомерных или гетеромерных каналов [Lingueglia et al., 1997; Benson et al., 2002; Hesselager et al., 2004] [10, 11, 12]. ASIC-каналы экспрессируются главным образом в ноцицептивных сенсорных нейронах периферической нервной системы и в нейронах центральной нервной системы [Waldmann et al., 1997a; Lingueglia et al., 2007, см. выше; Noel et al., 2010] [13, 8, 14]. Когда изоформы ASIC1a и ASIC2 находятся одновременно в центральной и периферической нервной системах, то экспрессия изоформ ASIC1b и ASIC3 ограничивается сенсорными нейронами [Waldmann et al., 1997b; Bassler et al., 2001; Chen et al., 1998] [15, 16, 17].
Известно, что ASIC-каналы, экспрессируемые сенсорными нейронами, в частности, ASIC3-канал, способны детектировать внеклеточную ацидификацию, которая может развиваться при ишемии, воспалительном процессе, гематоме, переломе, ране, при хирургическом вмешательстве (послеоперационная боль), или при развитии некоторых опухолей [Reeh et Steen, 1996] [18]. Однако в течение нескольких лет уже известно, что внеклеточный ацидоз генерирует боль [Steen et al., 1995a; Issbermer et al., 1996] [19, 20], и эксперименты, проведенные на здоровых волонтерах [Ugawa et al., 2002; Jones et al., 2004] [21, 22] показали участие ASIC-каналов в механизме возникновения кислотной кожной боли с использованием амилорида и некоторых НПВП препаратов, являющихся неспецифическими игибиторами ASIC-каналов [Waldmann et al., 1997a, см. выше; Voilley et al., 2001] [13, 23]. Была также доказана важная роль некоторых ASIC-каналов, экспрессирующихся в нейронах центральной нервной системы, в нейрональной активности (синоптическая пластичность гиппокампа, миндалины) и в нейромодуляции передачи болевой информации (ASIC1a-каналы) нейронами спинного мозга [Noel et al., 2010] [14].
До настоящего времени список активных лигандов, способных ингибировать ASIC-каналы, ограничивался в основном амилоридом, некоторыми НПВП и соединением А-317567 [Dubé et al., 2005] [24]. Однако ни одна из этих молекул не является абсолютно специфичной к ASIC-каналам или к конкретной ASIC-субъединице. С целью проведения идентификации специфических эффекторов ASIC-каналов, было отобрано большое число ядов: скорпиона, пчелы, паука, змеи или морской анемоны. Недавно были идентифицированы два пептидных токсина РсТх1 и АРЕТх2 животного происхождения, которые ингибируют, соответственно, гомомерные ASIC1a-каналы и каналы, содержащие субъединицу ASIC3 [Escoubas et al., 2000; Diochot et al., 2004] [25, 26]. Периферическая инъекция (подкожная) АРЕТх2 вызывает анальгетический эффект по отношению к воспалительной и кислотной боли у крысы [Deval et al., 2008] [27] и по отношению к послеоперационной боли у крысы после внутриоперационной аппликации АРЕТх2 [Deval et al., J.Neurosci., 31(16): 6059-6066, 2011] [36], а инъекция РсТх1 в центральную нервную систему вызывает у мыши мощный анальгетический эффект [Mazzuca et al., 2007] [28]. Анальгетические эффекты этих двух токсинов позволили продемонстрировать участие ASIC-каналов в восприятии и передаче болевой информации.
Следовательно, существует реальная необходимость в идентификации других специфических эффекторов ASIC-каналов или конкретной ASIC-субъединицы, способных производить анальгетический эффект, устраняя при этом все проблемы, недостатки и препятствия, связанные с анальгетическими средствами известного уровня техники.
Описание изобретения
Авторами изобретения были открыты и идентифицированы, на основе яда змеи Dendroaspis polylepis (черная мамба), новые пептиды, которые вызывают обезболивание и ингибируют ASIC-каналы (протон-чувствительные ионные каналы), более конкретно, гомомерные ASIC1a-каналы, гомомерные ASIC1b-каналы, гетеромерные ASIC1a+2a-каналы и гетеромерные ASIC1a+1b-каналы.
В частности, они представляют собой пептиды ASICалгин-1 (π-Dp1) и ASICалгин-2 (π-Dp2), которые после инъекции в центральную нервную систему (интратекальной и интрацеребровентрикулярной) in vivo мышам, оказывают мощный анальгетический эффект в отношении различных видов боли (химической, термической, воспалительной), сравнимый по силе с эффектом морфина, но в большой степени независим от активации опиоидных рецепторов. Кроме того, эти пептиды не оказывают нейротоксического действия, когда их инъецируют в центральную нервную систему. При периферической подкожной инъекции эти пептиды также производят анальгетический эффект с реверсией воспалительной гипералгезии. Эти два пептида являются первыми пептидами, экстрагированными из яда Dendroaspis polylepis (черная мамба), имеющими анальгетический эффект.
Если не ограничиваться этим объяснением, то наиболее вероятный механизм действия этих пептидов вытекает из их способности ингибировать ионные ASIC-каналы, которые играют главную роль в восприятии, передаче и модулировании болевой информации. Действительно, эти пептиды эффективно ингибируют гомомерные ASIC1a-каналы, гомомерные ASIC1b-каналы и/или гетеромерные каналы, содержащие субъединицу ASIC1a и/или ASIC1b, крысы и человека. Эти два пептида являются, в частности, первыми известными ингибиторами гомомерных ASIC1b и ASIC1a-каналов и гетеромерных ASIC1a+ASIC1b и ASIC1a+ASIC2a-каналов.
Пептиды ASICалгин-1 (π-Dp1) и ASICалгин-2 (π-Dp2) ингибируют также нативные ASIC-токи в сенсорных и в центральных нейронах. Они не оказывают никакого влияния на TRPV1 ток, активируемый капсаином и нагреванием, который сам тоже задействован в восприятии боли. Они не изменяют электрические свойства нейронов в исходном состоянии, но уменьшают нейрональную возбудимость в ответ на внеклеточную ацидификацию, способную активировать ASIC-каналы.
Поэтому пептиды ASICалгин-1 (π-Dp1) и ASICалгин-2 (π-Dp2) грызунов обладают анальгетическими свойствами, сравнимыми по эффективности с морфином, и эти свойства продолжают наблюдаться даже в случае, когда опиоидные рецепторы блокированы, что можно объяснить их способностью специфически ингибировать некоторые ASIC-каналы грызунов и человека. Инъекция их в центральную нервную систему не вызывает никакой токсичности (нейротоксичность, конвульсии….), и не оказывает никакого влияния на моторную активность (тест ускоряющегося вращающегося стержня).
Следовательно, пептиды ASICалгин-1 (π-Dp1) и ASICалгин-2 (π-Dp2) могут рассматриваться в качестве новых молекул, обладающих терапевтическим потенциалом в борьбе с болью (например, с раковой, нейропатической, послеоперационной… и т.д. болью) у человека.
Таким образом, настоящее изобретение относится к пептиду, включающему:
(i) аминокислотную последовательность:
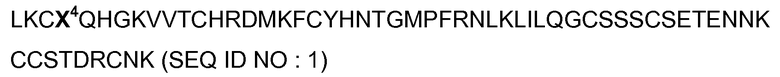
где Х4 обозначает любую аминокислоту;
или
(ii) природную или синтезированную последовательность, имеющую идентичность по меньшей мере 56% с последовательностью SEQ ID NO: 1, предпочтительно, по меньшей мере 70%, более предпочтительно по меньшей мере 80%, особенно предпочтительно по меньшей мере 98%, и сохраняющую биологические свойства пептидов, включающих последовательность SEQ ID NO: 1, такие, как описаны выше, а именно, индуцировать аналгезию и ингибировать по меньшей мере один ASIC-канал, содержащий по меньшей мере одну субъединицу, выбранную из группы, состоящей из субъединиц ASIC1a и ASIC1b.
Вышеназванные пептиды согласно изобретению функционируют, в частности, как блокаторы гомомерных ASIC1a-каналов, гомомерных ASIC1b-каналов и/или гетеромерных ASIC-каналов, содержащих по меньшей мере одну субъединицу, выбранную из группы, состоящей из субъединиц ASIC1a и ASIC1b, в частности, гетеромерных ASIC1a+ASIC1b-каналов и/или гетеромерных ASIC1a+ASIC2a-каналов.
Под «блокатором» подразумевают согласно изобретению пептид, способный ингибировать концентрационно-зависимым способом ток, продуцируемый вышеупомянутыми каналами. Например, он может представлять собой пептид, который, как и РсТх1 [Escoubas et al., 2000, см. выше] [25], способен ингибировать при концентрации 1 нМ 50% тока, продуцируемого гомомерными ASIC1a-каналами, или который, как и АРЕТх2 [Diochot et al., 2004, см. выше] [26], способен ингибировать при концентрации 2 мкМ 50% тока, продуцируемого гетеромерными ASIC1a+ASIC3-каналами крысы.
Предпочтительно, вышеназванные пептиды согласно изобретению экстрагируют из яда змеи Dendroaspis polylepis. Например, их выделяют из яда путем последовательного фракционирования с помощью высокоэффективной жидкостной хроматографии при полярности обращенной фазы (ОФ-ВЭЖХ). Вышеназванные пептиды могут быть также получены методами рекомбинации ДНК или химического синтеза.
Предпочтительно, вышеназванные пептиды согласно изобретению включают пептид ASICалгин-1 (π-Dp1) или пептид ASICалгин-2 (π-Dp2), имеющие последовательность SEQ ID NO: 1, где Х4 обозначает Y или F, соответственно (SEQ ID NO: 2 и 3, соответственно).
Настоящее изобретение относится также к полинуклеотиду, включающему нуклеотидную последовательность, кодирующую пептид согласно изобретению.
Предпочтительно, вышеназванный полинуклеотид согласно изобретению включает нуклеотидную последовательность:

или

где 73txx75 и 10txx12 обозначают tac, tat или ttt, ttc.
Предпочтительно, вышеназванный полинуклеотид согласно изобретению включает нуклеотидную последовательность, которая может гибридизироваться в условиях связывания с нуклеотидной последовательностью SEQ ID NO: 4 или SEQ ID NO: 22 или с комплементарной ей последовательностью. Например, это может быть полинуклеотид, который включает природную или искусственно синтезированную последовательность, которая имеет идентичность по меньшей мере 76% с последовательностью SEQ ID NO: 4 или с последовательностью SEQ ID NO: 22, предпочтительно по меньшей мере 80%, особенно предпочтительно по меньшей мере 98%.
Настоящее изобретение относится также к вектору, содержащему полинуклеотид согласно изобретению.
Предпочтительно, вышеназванный вектор согласно изобретению является вектором экспрессии.
Настоящее изобретение относится также к клетке-хозяину, содержащей один или несколько пептидов согласно изобретению, полинуклеотидов согласно изобретению или векторов согласно изобретению.
Настоящее изобретение относится также к фармацевтической композиции, содержащей один или несколько пептидов согласно изобретению, полинуклеотидов согласно изобретению, векторов согласно изобретению или клеток-хозяев согласно изобретению. Фармацевтическая композиция согласно изобретению может также содержать один или несколько фармацевтически приемлемых носителей (карбонат кальция, крахмал, тальк, лактозу, стеарат магния, смолу акации и т.д.) и может быть получена в форме раствора, суспензии, мази, желатиновой капсулы, таблетки, облатки, порошка, гранулы, лиофилизата, системы с контролируемым высвобождением, микрочастицы, микро- или наносферы, липосомы и т.д. Фармацевтическая композиция согласно изобретению может вводиться пероральным, внутримышечным, внутривенным, подкожным, топическим путем, через легкие, интраназальным, буккальным, ректальным, подъязычным, внутрикожным, внутрибрюшинным, интратекальным и т.д. путем. Эффективное количество действующего вещества (пептид, полинуклеотид, вектор или клетка-хозяин) в фармацевтической композиции согласно изобретению подбирается таким образом, чтобы была достигнута эффективная доза и чтобы при введении млекопитающему достигался анальгетический эффект. Доза, вводимая конкретному млекопитающему, зависит от многих факторов: способа введения, продолжительности лечения, массы тела и физического состояния млекопитающего, уровня активности действующего вещества и от реакции млекопитающего на это действующее вещество. Например, анальгетически эффективное количество действующего вещества, вводимого интратекальным путем, находится обычно в интервале примерно от 5 нг/кг до 500 мкг/кг массы тела млекопитающего, предпочтительно, примерно от 50 нг/кг до 50 мкг/кг массы тела млекопитающего, особенно предпочтительно примерно от 500 нг/кг до 5 мкг/кг массы тела млекопитающего. Эффективные дозы действующего вещества могут варьировать при использовании других способов введения. Анальгетически эффективное количество может быть установлено путем тестирования действующего вещества в одном или нескольких тестах на боль, описанных ниже, при дозе, которую можно варьировать в зависимости от одного или нескольких описанных выше критериев для определения эффективного количества действующего вещества, вводимого млекопитающему.
Настоящее изобретение относится также к веществу, выбранному из пептида согласно изобретению, полинуклеотида согласно изобретению, вектора согласно изобретению, клетки-хозяина согласно изобретению или фармацевтической композиции согласно изобретению, предназначенному для применения его в качестве лекарственного средства.
Предпочтительно, вышеназванное лекарственное средство является анальгетическим средством, например, предназначенным для профилактики или лечения боли, в которой участвует активация ASIC-каналов, в частности ASIC-каналов, содержащих по меньшей мере одну субъединицу, выбранную из группы, составленной из субъединиц ASIC1a и ASIC1b, особенно предпочтительно, гомомерных ASIC1a-каналов, гомомерных ASIC1b-каналов, гетеромерных ASIC1a+ASIC1b-каналов и/или гетеромерных ASIC1a+ASIC2a-каналов. Например, боль может (i) появиться в результате активации центральных или периферических ASIC-каналов или (ii) индуцировать их активацию. Например, она может представлять собой, в частности, такие боли, которые ассоциированы с активацией ASIC-каналов, и выбраны из группы, включающей воспалительные, нейропатические, раковые, послеоперационные, мышечно-скелетные, висцеральные и т.д. боли.
Предпочтительно, вышеназванное лекарственное средство предназначено для профилактики или лечения патологии, в которой участвует активация ASIC-каналов, в частности, ASIC-каналов, содержащих по меньшей мере одну субъединицу, выбранную из группы, состоящей из субъединиц ASIC1a и ASIC1b, особенно предпочтительно, гомомерных ASIC1a-каналов, гомомерных ASIC1b- каналов, гетеромерных ASIC1a+ASIC1b-каналов и/или гетеромерных ASIC1a+ASIC2a-каналов. Например, они могут представлять собой патологии, выбранные из группы, включающей воспалительные заболевания, раковые заболевания, фибромиалгии, синдром раздраженной толстой кишки и т.д.
Предпочтительно, вышеназванное лекарственное средство предназначено для профилактики или лечения неврологического заболевания центральной нервной системы, выбранного, например, из группы, включающей посттравматический стресс, депрессию, тревожное состояние, нарушения мозгового кровообращения, эпилепсию, воспалительные заболевания центральной нервной системы, рассеянный склероз, нейродегенеративные заболевания и т.д.
Предпочтительно, вышеназванное лекарственное средство вводят парентеральным путем, а именно местнорегиональным или центральным путем (интраперитональным, перидуральным, интратекальным, интрацеребровентрикулярным, интрадермальным и т.д.) и общепринятым путем (внутримышечно, внутривенно, подкожно и т.д.), пероральным путем, местным путем (чрескожно и т.д.) или через дыхательные пути (ингаляция, инстилляция и т.д.). Особенно предпочтительно, вышеназванное лекарственное средство вводят интратекальным, перидуральным, интрацеребровентрикулярным, внутрибрюшинным или подкожным путем.
Настоящее изобретение относится также к веществу, выбранному из пептида согласно изобретению, полинуклеотида согласно изобретению, вектора согласно изобретению, клетки-хозяина согласно изобретению или фармацевтической композиции согласно изобретению, предназначенному для применения его в качестве диагностического средства.
Настоящее изобретение относится также к способу идентификации соединения, имитирующего анальгетическую активность пептида согласно изобретению, включающему следующие стадии:
а) определение анальгетической активности пептида согласно изобретению;
b) определение анальгетической активности соединения-кандидата;
c) сравнение уровней анальгетической активности, выявленных на стадиях а) и b);
d) отбор соединения-кандидата, обладающего анальгетической активностью, эквивалентной или превышающей анальгетическую активность пептида согласно изобретению.
Настоящее изобретение относится также к способу идентификации соединения, имитирующего анальгетическую активность пептида согласно изобретению, включающему следующие стадии:
а) введение в контакт пептида согласно изобретению с образцом и измерение связи вышеназванного пептида с вышеназванным образцом;
b) добавление соединения-кандидата и оценка эффекта вышеназванного соединения на связь вышеназванного пептида с вышеназванным образцом;
c) отбор соединения-кандидата, способного модулировать связь вышеназванного пептида с вышеназванным образцом.
Под «соединением, имитирующим анальгетическую активность пептида согласно изобретению» подразумевают согласно изобретению соединение-кандидат, способное вызвать обезболивание и связаться с ASIC-каналами, с которыми связывается пептид согласно изобретению, или же действовать физиологически идентично или схожим образом с действием пептидов согласно изобретению, а именно, модулировать (ингибировать или стимулировать) обратимо и в зависимости от концентрации ток, продуцируемый по меньшей мере одним каналом, содержащим по меньшей мере одну субъединицу, выбранную в группе, состоящей из субъединиц ASIC1a и ASIC1b, в частности, гомомерными ASIC1a-каналами, гомомерными ASIC1b- каналами, гетеромерными ASIC1a+ASIC1b-каналами и/или гетеромерными ASIC1a+ASIC2a-каналами. Например, он может представлять собой пептид, который, как и РсТх1 [Escoubas et al., 2000, см. выше] [25], способен ингибировать при концентрации 1 нМ 50% тока, продуцируемого гомомерными ASIC1a-каналами, или который, как и АРЕТх2 [Diochot et al., 2004, см. выше] [26], способен ингибировать при концентрации 2 мкМ 50% тока, продуцируемого гетеромерными ASIC1a+ASIC3-каналами крысы.
Под «образцом» подразумевают согласно изобретению клетки или ткани, предварительно выделенные из целого организма или из иммортализованных линий клеток насекомых или млекопитающих.
Термин «анальгетическая активность» используется как ссылка на способность пептида согласно изобретению лечить или облегчать боль у млекопитающих, как это показано на одной или нескольких обычных лабораторных моделях для тестирования боли или оценки обезболивания, так как это описано ниже. Так, анальгетическая активность пептида согласно изобретению может быть определена, например, с помощью одного или нескольких следующих тестов in vivo: i) определение латентного периода отдергивания хвоста/лап (измерение термической ноцицепции) [Abott et al., 1982; Cridland et Henry, 1992] [29, 30], ii) определение болевого порога на горячей/холодной пластине (измерение термической ноцицепции) [Woolfe et Macdonald, 1944; Ankier, 1974] [31, 32], iii) определение болевого порога в тесте с волосками фон Фрея или в тесте Рэндалла-Селитто или в инструментальном тесте с зажимом (измерение механической ноцицепции) [Kim et al., 1993] [33], iv) тест с динамическим распределением массы тела (измерение ноцицептивной активности, связанной с положением тела), v) измерение ноцицептивного спонтанного поведения, и/или с помощью одного или нескольких тестов in vitro: например, отбор соединений-кандидатов путем конкурентного связывания, в котором меченый пептид согласно изобретению (например, с помощью радиометки, такой как С14, Н3, I125, фермента, такого как пероксидаза, щелочная или кислая фосфатаза, флуоресцентного маркера, такого как FITC, родамин, с помощью антитела, антигена, биотина, парамагнитного иона, частицы латекса и т.д.) вводят в контакт с образцом в условиях, обеспечивающих связывание пептида с вышеназванным образцом, и измеряют связывание вышеназванного меченого пептида с образцом, при котором соединения, имитирующие активность вышеназванного меченого пептида, вступают в конкуренцию с вышеназванным пептидом за сайты связывания на рецепторе (ASIC-каналы, содержащие по меньшей мере одну субъединицу, выбранную в группе, состоящей из субъединиц ASIC1a и ASIC1b). Таким образом, когда тестируемые соединения имитируют активность вышеназванного пептида, связываясь с рецептором, то измеряется более низкое количество детектируемых меток, чем в случае, когда тестируемые соединения не имитируют активность вышеназванного пептида и не связываются с рецептором, или связываются с меньшей аффинностью. Альтернативно, отбор путем конкурентного связывания может быть осуществлен с мечением соединения-кандидата вместо мечения пептида согласно изобретению. Таким образом, когда тестируемые соединения имитируют активность вышеназванного пептида, связываясь с рецептором, измеряется более высокое количество детектируемых меток, чем в случае, когда тестируемые соединения не имитируют активность вышеназванного пептида и не связываются с рецептором, или связываются с меньшей аффинностью.
Примеры способа идентификации соединений, имитирующих анальгетическую активность пептидов, описаны (не ограничиваясь настоящим описанием), например, в патенте US 5877026 [34].
Другие преимущества могут быть раскрыты для специалиста при чтении нижеследующих примеров, приведенных в качестве иллюстрации, которые иллюстрируются прилагаемыми фигурами.
Краткое описание фигур
- Фигура 1 иллюстрирует: (А) аминокислотную последовательность пептидов ASIСалгина-1 (π-Dp1) и ASICалгина-2 (π-Dp2), выделенных из яда змеи Dendroaspis polylepis (SEQ ID NO: 2 и 3, соответственно), (В) нуклеотидную последовательность комплементарной ДНК (ADNc), кодирующую ASIСалгин-1 (SEQ ID NO: 5). Выделены сигнальная последовательность (курсив), конечный пептид (жирный шрифт) (SEQ ID NO: 6) и стоп-кодон (*). Некодирующая 5'-концевая последовательность не представлена (соответствующая последовательности SEQ ID NO: 9).
- Фигура 2 иллюстрирует анальгетический эффект пептидов ASIСалгина-1 (π-Dp1) и ASICалгина-2 (π-Dp2) in vivo после интратекальной инъекции мыши. Химическая боль (формол): (А) кинетика спонтанного болезненного поведения, измеряемая с интервалом в 5 минут, (В) общая продолжительность фазы I (0-10 минут) острой боли и фазы II (10-45 минут) воспалительной боли. Острая термическая боль: (С) кинетика латентного периода отдергивания хвоста, (D) средние значения площадей под кривой (AUC), вычисленные для каждого животного. Воспалительная гипералгезия: (Е) кинетика латентного периода отдергивания лапы. Значимые величины гипералгезии по отношению к контрольной величине: #, р<0,05; ##, р<0,01; ###, р<0,001; тест ANOVA с последующим множественным сравнением по Newman Keuls. (F) средние значения площадей под кривой (AUC), вычисленные для каждого животного, начиная с уровня гипералгезии в момент Т-7 минут. Приведены средние значения ± sem (средняя величина стандартной ошибки), а количество (n) тестируемых животных приведено в обозначениях к схеме. Значимые величины по отношению к инъекции носителя ± наксолон (NAL) или же как указано чертой на фигуре: незначимая величина ns, р>0,05; *, р<0,05; **, р<0,01; ***, р<0,001; тест ANOVA с последующим множественным сравнением по Newman Keuls.
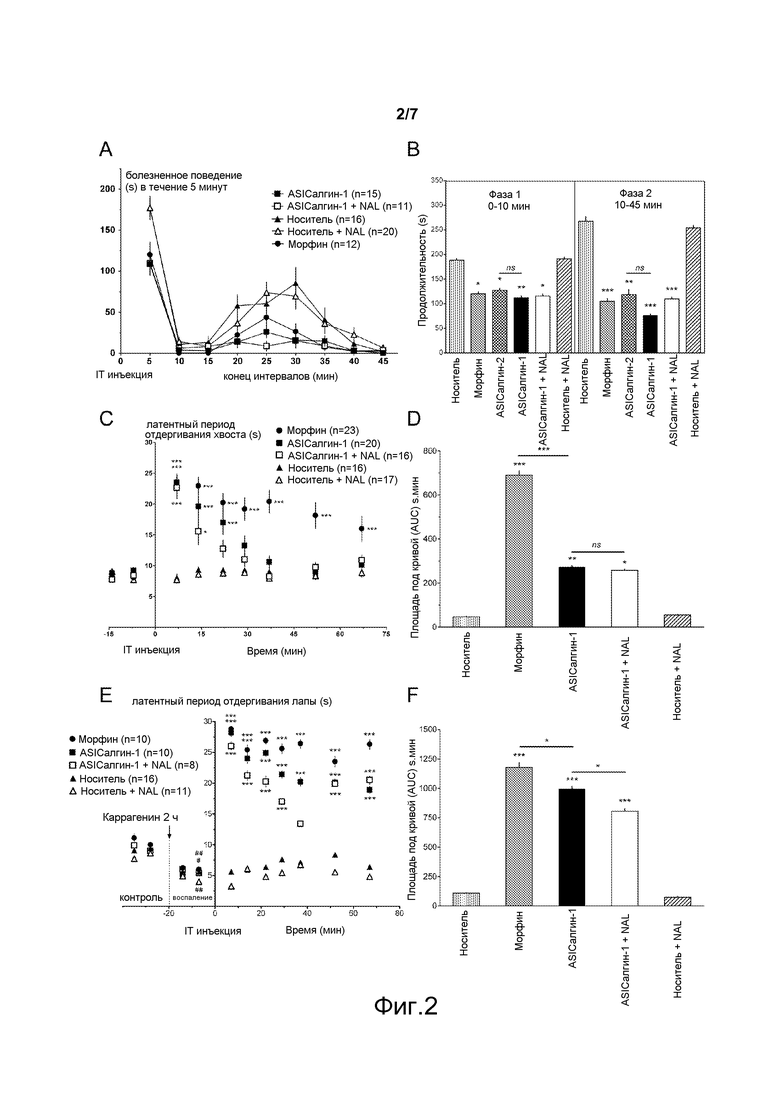
- Фигура 3 иллюстрирует анальгетический эффект пептида ASIСалгина-1 (π-Dp1) in vivo после интрацеребровентрикулярной инъекции мыши в тесте острой термической боли: (А) кинетика латентного периода отдергивания хвоста, (В) средние значения площадей под кривой (AUC), вычисленные для каждого животного. Приведены средние значения ± sem (средняя величина стандартной ошибки). Количество (n) тестируемых животных приведено в обозначениях к схеме. Значимые величины по отношению к инъекции носителя или же как указано чертой на фигуре: незначимая величина ns, р>0,05; *, р<0,05; **, р<0,01; ***, р<0,001; тест ANOVA с последующим множественным сравнением по Newman Keuls.
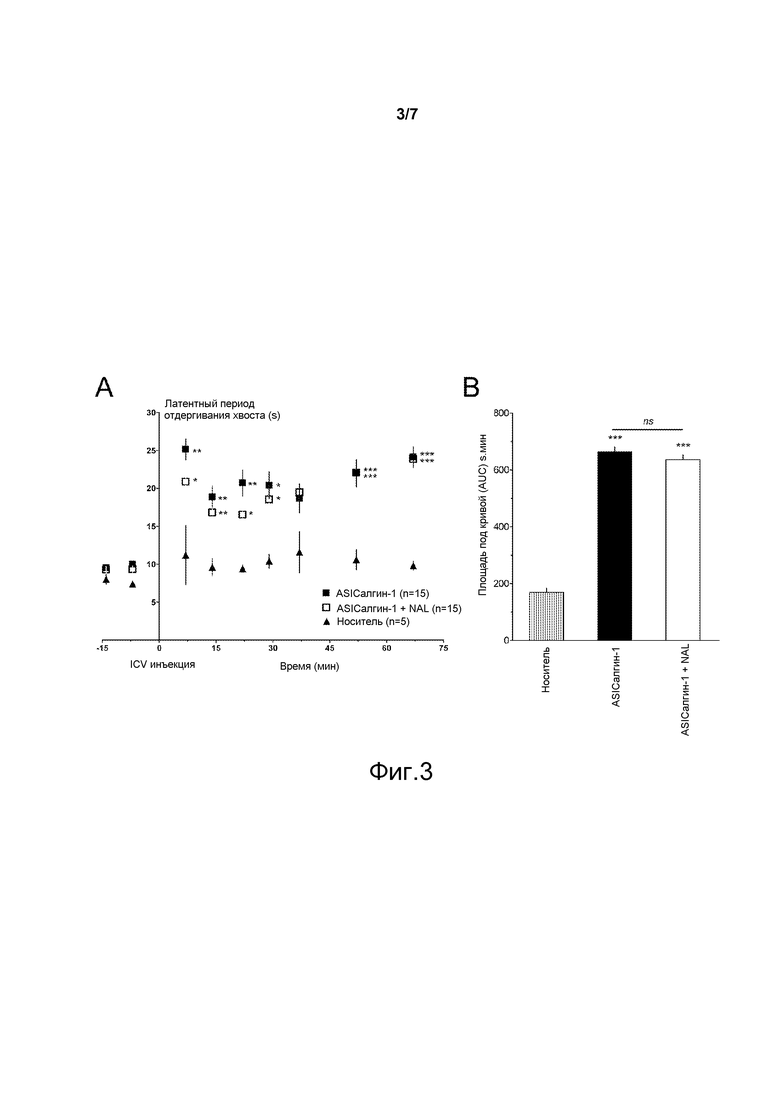
- Фигура 4 иллюстрирует анальгетический эффект пептида ASIСалгина-1 (π-Dp1) in vivo после подкожной инъекции в тесте воспалительной гипералгезии: (А) кинетика латентного периода отдергивания лапы. Пептид ASIСалгин-1 введен подкожным путем в заднюю левую лапу одновременно с каррагенином (□), затем снова введен спустя два часа (время 0 или Т0, □). Осуществляли аналогичную процедуру, но с инъекцией только одного пептида ASIСалгина-1 в момент Т0, т.е. после проявления воспалительной гипералгезии (■). (В) средние значения площадей под кривой (AUC), вычисленные для каждого животного, начиная со значения в момент Т-7 минут. Приведены средние значения ± sem. Количество (n) тестируемых животных приведено в обозначениях к схеме. Значимые величины по отношению к инъекции носителя: *, р<0,05; **, р<0,01; ***, р<0,001; тест ANOVA с последующим множественным сравнением по Newman Keuls. Значимые величины по сравнению с контрольной величиной перед инъекцией каррагенина: #, р<0,05; ##, р<0,01; ###, р<0,001; Т-тест для спаренных выборок.
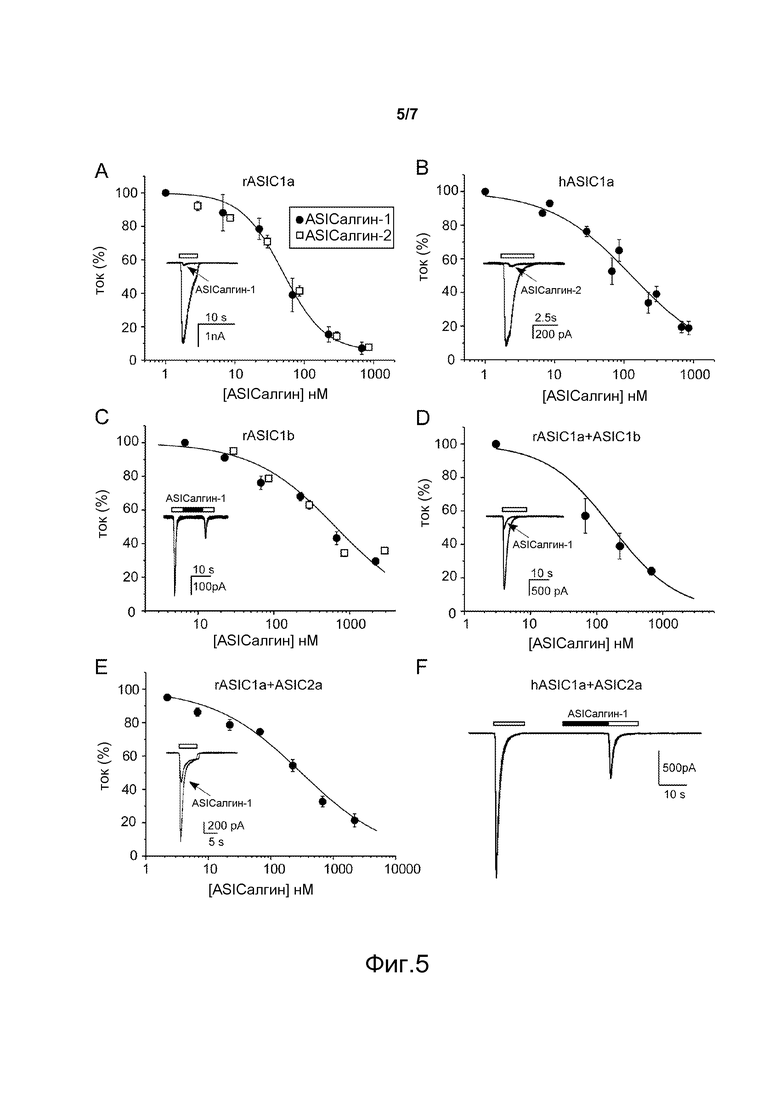
- Фигура 5 показывает ингибирование пептидами ASIСалгин-1 (π-Dp1) и ASIСалгин-2 (π-Dp2) токов, генерируемых ASIC-каналами, гетерологично экспрессирующимися в клетках COS. Кривая доза-ответ ингибирования пептидом ASIСалгин-1 (π-Dp1) тока гомомерного ASIC1a-канала крысы, CI50=49 нМ (А), тока гомомерного ASIC1a-канала человека, CI50=127 нМ (В), тока гомомерного ASIC1b-канала крысы, CI50=650 нМ (С), тока гетеромерного ASIC1a+ASIC1b-канала крысы (соотношение 1:1), CI50=223 нМ (D), тока гетеромерного ASIC1a+ASICa-канала крысы (соотношение 2:1), CI50=308 нМ (Е). Приведены средние значения ± sem, где n обозначает от 4 до 15 экспериментов. Первоначальные кривые токов, ингибируемых ASIСалгином-1 (π-Dp1, 674 нМ) и ASIСалгином-2 (π-Dp2, 852 нМ), изображены в миниатюре. (F) Первоначальные кривые токов, показывающие ингибирование тока гетеромерного ASIC1a+ASIC2a-канала человека (соотношение 2:1) с помощью ASIСалгина-1 (π-Dp1, 674 нМ). Потенциал покоя: -60 мВ, белые бруски: скачок рН от 7,4 до 5,5, черные бруски: применение пептида.
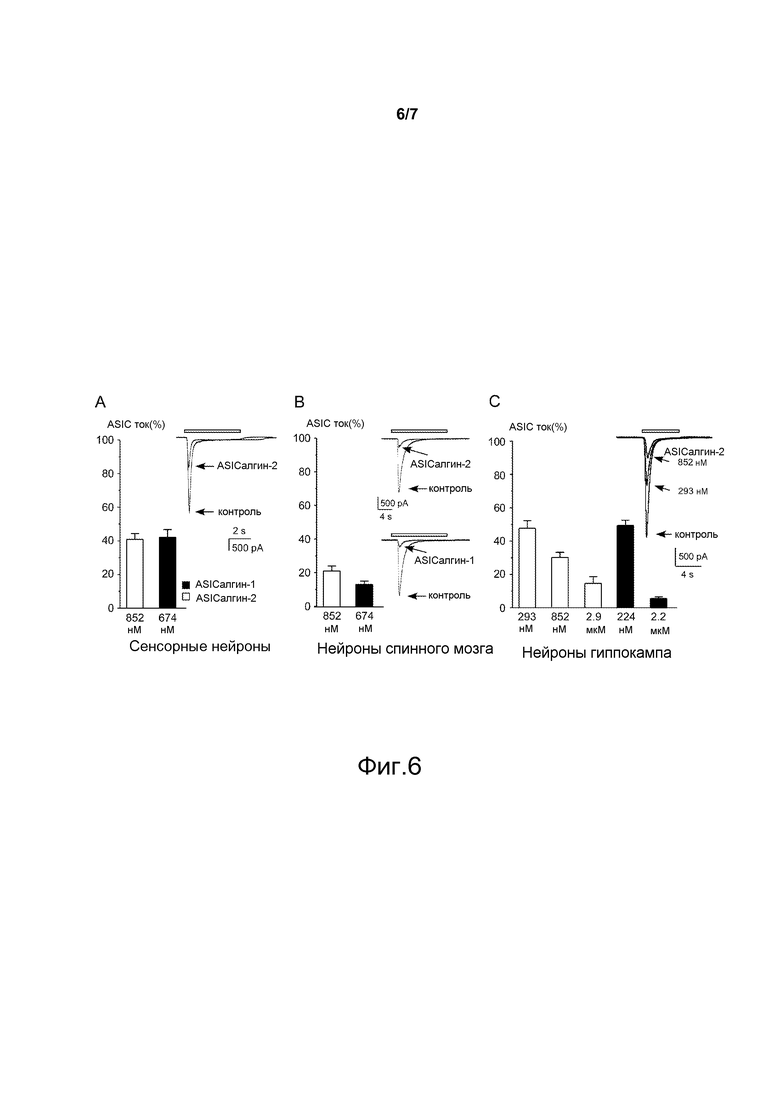
- Фигура 6 показывает ингибирование с помощью пептидов ASIСалгина-1 (π-Dp1) и ASIСалгина-2 (π-Dp2) токов нативных ASIC-каналов в культуре сенсорных и центральных мышиных нейронов. (А) Среднее значение ингибирования ASIC-тока в сенсорных нейронах крысы (n=34) с помощью ASIСалгина-2 (π-Dp2, 852 нМ), □) и ASIСалгина-1 (π-Dp1, 674 нМ, ■). Справа примеры первоначальных токов, зарегистрированных при потенциале покоя -50 мВ и активированных путем скачкообразного изменения рН от 7,4 до 6 (белый брусок). В сенсорных нейронах суммарный ASIC-ток является результатом смешения тока гомомерного ASIC1a-канала, тока типа ASIC1b и тока типа ASIC3. (В) Среднее значение ингибирования ASIC-тока в нейронах спинного мозга мыши (n=18) с помощью ASIСалгина-2 (π-Dp2, 852 нМ), □) и ASIСалгина-1 (π-Dp1, 674 нМ, ■). Справа пример первоначальных токов, зарегистрированных при потенциале покоя -50 мВ и активированных путем скачкообразного изменения рН от 7,4 до 6 (белый брусок). (С) Среднее значение ингибирования ASIC-тока в нейронах гиппокампа мыши (n=26) с помощью ASIСалгина-2 (π-Dp2, 293 нМ, 852 нМ и 2,9 мкМ, □) и ASIСалгина-1 (π-Dp1, 224 нМ и 2,2 мкМ, ■). Справа пример первоначальных токов, зарегистрированных при потенциале покоя -50 мВ и активированных путем скачкообразного изменения рН от 7,4 до 5,5 (белый брусок). В центральных нейронах (спинной мозг и гиппокамп мыши) суммарный ASIC-ток является результатом смешения тока гомомерного ASIC1a-канала и тока гетеромерного ASIC1a+ASIC2a-канала. Приведены средние значения ± sem.
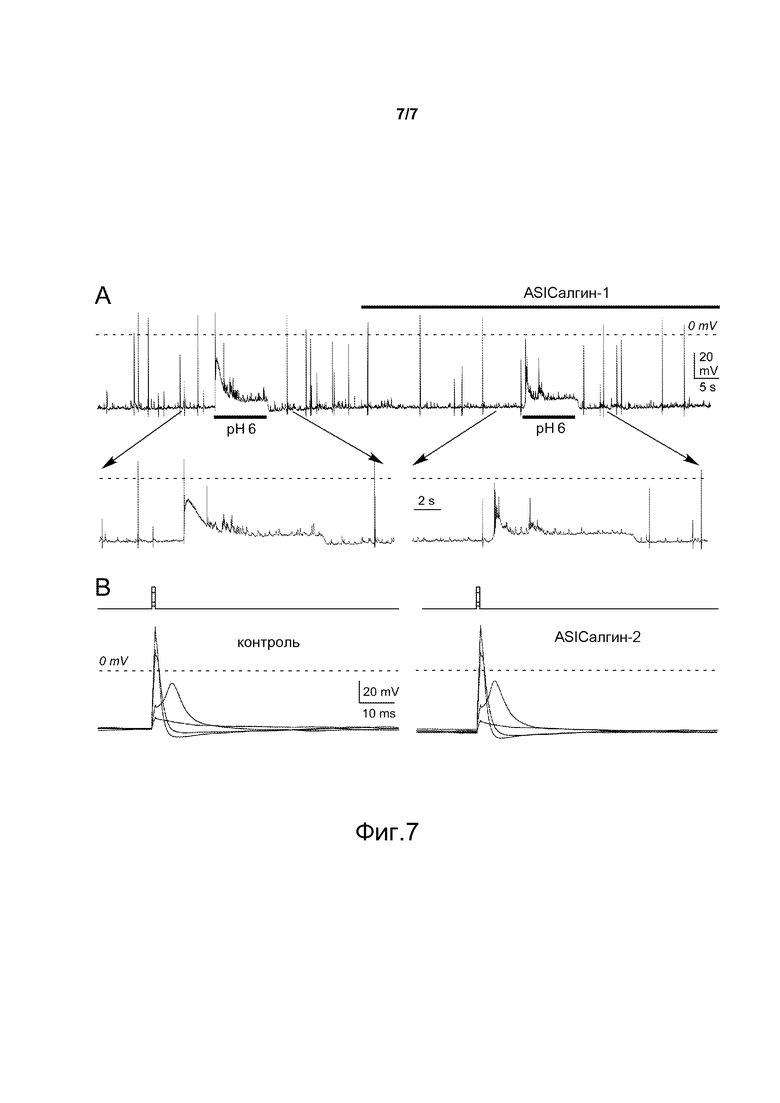
- Фигура 7 показывает действие пептидов ASIСалгина-1 (π-Dp1) и ASIСалгина-2 (π-Dp2) на мембранный потенциал нейронов спинного мозга мыши. (А) Мембранный потенциал нейрона, при котором спонтанные потенциалы действия отражают синаптическую активность (пунктирная линия: 0 мВ). Активация ASIC-тока путем скачкообразного изменения рН от 7,4 до 6 индуцирует деполяризацию, пик которой запускает потенциал действия (развитие). Применение пептида ASIСалгина-1 (π-Dp1, 674 нМ) не изменяет ни потенциал покоя нейрона, ни спонтанную активность. Напротив, деполяризация, индуцированная скачкообразным изменением кислого рН, снижается и более не индуцирует потенциал действия. (В) Потенциал действия, возникший под воздействием электростимуляции (зубцы кривой тока, изображенные выше). Четыре ответа, наложенные друг на друга (изображение слева): два ответа, приведших к допороговой деполяризации и два ответа, приведших к запуску потенциала действия (пунктирная линия: 0 мВ). Применение ASIСалгина-2 (π-Dp2, 852 нМ, изображение справа) не изменяет возбудимость нейрона.
ПРИМЕРЫ
ПРИМЕР 1: ВЫДЕЛЕНИЕ ДВУХ ПЕПТИДОВ, ASICАЛГИНА-1 (π-Dp1) И ASICАЛГИНА-2 (π-Dp2), ИЗ ЯДА ЗМЕИ DENDROASPIS POLYLEPIS
Яд и очистка пептидных токсинов
Лиофилизированный яд приобретен у фирмы Latoxan (Валенсия, Франция). Яд разбавляли уксусной кислотой (1%) и фракционировали пропусканием через колонку, содержащую смолу Сефадекс G50. Пептидную фракцию собирали и затем фракционировали на системе жидкостной хроматографии высокого давления (HPLC): 1) Пептидную фракцию загружали на катионобменную колонку и элюировали в градиенте рН растворителей (1% уксусная кислота, рН3,0 и 1М ацетат аммония, рН6,8). Фракцию, содержащую ASICалгины, элюировали начиная с 60% ацетата аммония. 2) Эту пептидную фракцию загружали на колонку с обращенной фазой С18 и ее компоненты элюировали градиентом гидрофобности между растворителями вода-ТФУ 0,1% и ацетонитрил-ТФУ 0,1%. Чистые пептиды ASICалгин-1 и ASICалгин-2 элюировали при 26 и 27%-ной концентрации ацетонитрила, соответственно.
Характеристика пептидов
Анализ аминокислотного состава
ASIСалгин-1 и ASIСалгин-2 являются двумя основными изопептидами, имеющими последовательность из 57 аминокислот, содержащих 8 цистеинов, образующих 4 дисульфидных мостика (фигура 1А, последовательности SEQ ID NO: 2 и 3, соответственно), с предшествующей сигнальной последовательностью из 21 аминокислоты (фигура 1В, см. последовательность SEQ ID NO: 6). Они отличаются друг от друга только аминокислотой в 4 положении последовательностей SEQ ID NO: 1, 2 или 3 (= положение 25 последовательности SEQ ID NO: 6).
Определение аминокислотной последовательности методом клонирования
Полная последовательность (фигура 1В) ASIСалгина-1 была определена методом клонирования кДНК с помощью ПЦР на основе матричных РНК (мРНК), присутствующих в следовых количествах в неочищенном яде. 25 мг яда змеи Dendroaspis polylepsis (Sigma) ресуспендировали в буфере для лизиса: 500 мМ LiCl, 10 мМ EDTA, 1% (масс./об.) LiDS и 5 мМ дитиотреитол в 100 мМ буфере Tris-HCl, рН 7,5. Матричные РНК были захвачены магнитными шариками с привитыми олигонуклеотидами dT25 (Dynal, Великобритания). Эти олигонуклеотиды были использованы непосредственно в качестве затравок для проведения обратной транскрипции мРНК в кДНК с использованием обратной транскриптазы PrimeScriptTM (TaKaRa Bio Inc., Japan). Полученная таким путем кДНК-библиотека на твердой фазе использовалась для осуществления ПЦР-реакции с использованием вырожденных смысловых (TGITTYCARCAYGGIAARGT, SEQ ID NO: 7) и антисмысловых (YTTIARRTTICGRAAIGGCAT, SEQ ID NO: 8) олигонуклеотидов, характеризуемых на основе частичной белковой последовательности токсина, определяемой прямым N-терминальным секвенированием. Для исключения какой-либо ошибки, связанной с полимеразой, были проведены три независимые ПЦР-реакции, которые привели к получению фрагментов желаемой длины (89 пар оснований), затем фрагменты субклонировали в векторе pGEMTeasy (Promega) и секвенировали. Благодаря этим последовательностям был синтезирован специфический смысловой олигонуклеотид с добавленным к нему сайтом рестрикции EcoRI, (ACAC(GAATTC)GCTATCATAACACTGGCATG, SEQ ID NO: 9), а также неспецифический антисмысловой олигонуклеотид поли-dT30, также с добавленным к нему сайтом EcoRI (ACAC(GAATTC)dT30, SEQ ID NO: 10). Использование этих двух олигонуклеотидов позволяет амплифицировать с помощью ПЦР любую 3'-концевую кодирующую и 3'-концевую некодирующую последовательность с целью идентификации. Три независимые ПЦР-реакции генерировали последовательность длиной примерно 400 пар оснований и ее субклонирование в векторе pGETeasy с сайтом рестрикции ECoRI позволило секвенировать ее, исключив любую ошибку, вызванную полимеразой.
Исходя из 3'- и 5'-некодирующих последовательностей были синтезированы смысловой (ACAC(GAATTC)TCCAGAGAAGATCGCAAGATG, SEQ ID NO: 11) и антисмысловой (ACAC(GAATTC)ATTTAGCCACTCGTAGAGCTA, SEQ ID NO: 12) олигонуклеотиды, образующие кодирующую последовательность. Четыре независимые ПЦР-реакции были осуществлены на основе кДНК-библиотеки на твердой фазе и ПЦР-продукты были снова субклонированы в векторе pGEMTeasy для получения полной нуклеотидной последовательности предшественника токсина ASICалгина-1.
Пептидное секвенирование
Была подтверждена последовательность вплоть до аминокислоты Asp40 прямым N-терминальным секвенированием восстановленных и алкилированных пептидов на автоматическом секвенаторе (LC491, Applied Biosystems, USA).
Ферментативное расщепление
Для подтверждения последовательностей пептиды подвергали следующим ферментативным обработкам: (а) протеазой V8 с разделением переваренных пептидов на колонке ВЭЖХ с обращенной фазой С18. Расщепление в положении D15 и Е43 и разделение на ВЭЖХ колонке 2 пептидов: с молекулярной массой 1786,9 Да (пептид 1-15), полностью ресеквенированный, и 3194,5 Да (пептид 16-43), частичная последовательность, позволяет подтвердить N-терминальную последовательность; (b) перевариванию трипсином с использованием прямого масс-спектрометрического анализа. Трипсиновые фрагменты также подтверждают N-терминальную последовательность и другие части пептидной последовательности ASIC-алгинов. Трипсиновые пептиды, детектированные и секвенированные с помощью анализа МС/МС, представлены в следующей таблице 1.

Масс-спектрометрия (MALDI-TOF)
Во всех случаях масс-спектрометрический анализ осуществляли на спектрометре 4800 MALDI-TOF-TOF (Applied Biosysteme, USA).
Нативные белки анализировались в линейном режиме, детектируя положительные ионы и используя синапиновую кислоту в качестве матрицы (Sigma Aldrich, USA), внутреннюю калибровку проводили с использованием смеси известных белков с точным значением молекулярной массы.
Измеренная молекулярная масса составляла 6554,8 Да для ASICалгина-1 и 6538,4 Да для ASICалгина-2. Она полностью согласуется с молекулярными массами 6554,58 Да и 6538,58 Да, соответственно, в расчете на последовательность, полученными с помощью программного обеспечения GPMAW protein analysis. Эти результаты указывают на свободную карбоксильную группу на С- конце. Молекулярное поглощение на длине волны 280 нм составило ε=3040 (для ASICалгина-1) и ε=1760 (для ASICалгина-2).
Поиск гомологов в базах данных
ASICалгин-1 и ASICалгин-2 не фигурируют в базах данных и не имеют никакой идентичности полной последовательности с другими известными пептидами или токсинами.
Выравнивание последовательностей по программе BLAST показывает, что ASICалгин-1 и ASICалгин-2 имеют, тем не менее, 47-55%-ную идентичность последовательностей (63-66%-ная гомология последовательностей) с очищенными пептидами СМ-1b, CM-3 и CM-2a, выделенными из ядов разных видов кобр (Naja et Ringhals), а также с нейротоксином-α ОН-26, присутствующим в яде королевской кобры Ophiophagus hannah. Однако при описании этих СМ- и ОН-26-нейротоксинов не говорится о том, что они воздействуют на ASIC-каналы или обладают анальгетической активностью in vivo, а говорится как о продуктах, обладающих цитотоксическим эффектом. Поэтому может казаться, что при идентичности последовательностей ниже 55% с ASICалгином-1 и ASICалгином-2, биологические свойства, проявляемые пептидами согласно изобретению, исчезнут. Однако, наоборот, из уровня техники известно, что изоформы и/или ортологи токсинов сохраняют имеющуюся активность, несмотря на некоторые различия в их белковых последовательностях, что является результатом идентичности последовательностей на уровне порядка 70%. Поэтому представляется, что изоформы и/или ортологи токсинов, имеющие идентичность последовательностей на уровне по меньшей мере 56-70% с ASICалгином-1 и ASICалгином-2, будут сохранять свойства вышеназванных пептидов согласно изобретению.
ПРИМЕР 2: АНАЛЬГЕТИЧЕСКИЙ ЭФФЕКТ ПЕПТИДОВ ASICАЛГИНА-1 (π-Dp1) и ASICАЛГИНА-2 (π-Dp-2) В ТЕСТЕ IN VIVO ПОСЛЕ ИНТРАТЕКАЛЬНОЙ ИНЪЕКЦИИ
Болезненное поведение мышей (C57B16J, самцы в возрасте 7-11 недель) оценивалось после интратекальной инъекции (IT) пептидов между L5 и L6 позвонками согласно протоколу Гильдена и Уилкоксона [1980] [35]. Эффекты IT инъекции в объеме 10 мкл раствора, содержащего 34 мкМ ASICалгина-1 (0,34 нмоль) или 20 мкМ ASICалгина-2 (0,2 нмоль), сравнивали с эффектами IT инъекции в объеме 10 мкл раствора, содержащего 3,1 мМ морфина (31 нмоль), или 10 мкл солевого раствора в качестве носителя (145 мМ NaCl, 5мМ KCl, 2 мМ CaCl2, 10 мМ HEPES, рН 7,4, 0,05% бычьего сывороточного альбумина). В некоторых сериях экспериментов вводили подкожно наксолон в дозе 2 мг/кг в 50 мкл NaCl 0,9% в качестве ингибитора опиоидных рецепторов (особенно типа мю) за десять минут до введения IT инъекции пептида ASICалгина.
Токсичность
Интратекальная инъекция пептидов ASICалгина-1 и ASICалгина-2 не вызывала никакого изменения в поведении животного типа моторных нарушений, нарушений равновесия, апатии, паралича или конвульсий, и не привела ни к одному смертельному исходу у мышей.
Острая химическая и воспалительная боль
Подкожная инъекция формола (15 мкл при концентрации 2% в растворе NaCl 0,9%) в свод стопы задней лапы мыши индуцировала боль, развивавшуюся в две фазы: фаза I острой химической боли в течение 10 минут и фаза II воспалительной боли в течение 15-45 минут (фигура 2А).
Боль измеряли, наблюдая за спонтанным болезненным поведением животного, т.е. сравнивали время, в течение которого мышь поднимала инъецированную лапу, облизывала ее, покусывала и встряхивала ею (интегрированное произвольное поведение).
IT инъекция ASICалгина-1 значительно снижала, на 40%, боль в фазе I и на 70% в фазе II. Этот анальгетический эффект близок к эффекту морфина (фигура 2В).
IT инъекция ASICалгина-2 производила эффект, идентичный эффекту ASICалгина-1.
Предварительная обработка наксолоном, ингибитором опиоидных рецепторов, не оказала никакого действия на анальгетический эффект ASICалгина-1, ни в одной, ни в другой болезненной фазе I и II (фигура 2В).
Пептиды ASICалгин-1 и ASICалгин-2 являются первыми пептидами, обладающими анальгетическим эффектом, экстрагированными из яда змеи Dendroaspis polylepis.
Острая термическая боль
Термическая боль исследовалась на мышах путем измерения латентного периода отдергивания хвоста, погруженного в воду с температурой 46°С (рефлекторное движение, животное зафиксировано в неподвижном состоянии) в течение времени, ограниченного 30 секундами.
Пептид ASICалгин-1 индуцировал анальгетический эффект такого же уровня, что и эффект морфина (7 минут после IT инъекции), но был более продолжительного действия при используемой концентрации (фигура 2С).
Анальгетический эффект ASICалгина-1 не был заметно ингибирован предварительной обработкой наксолоном (фигура 2D).
Воспалительная гипералгезия
Подкожная инъекция 2%-ного каррагенина в свод стопы задней лапы мыши вызвала развитие воспалительного процесса в ткани и гипералгезии, измеренное путем определения латентного периода отдергивания задней лапы, погруженной в воду с температурой 46°С (интегрированное произвольное движение, животное зафиксировано в неподвижном состоянии) в течение времени, ограниченного 30 секундами спустя два часа после инъекции каррагенина (фигура 2Е, #).
IT инъекция ASICалгина-1 вызвала быстрое обезболивание, сравнимое с обезболиванием морфином, и продолжалось в течение всего эксперимента (67 минут, фигура 2Е).
Анальгетический эффект ASICалгина-1 был лишь слегка ингибирован (на 20%) предварительной обработкой наксолоном (фигура 2F).
Моторная активность
Эффекты пептида ASICалгина-1 на моторную активность оценивали с помощью теста ускоряющегося вращающегося стержня. Мышей помещали на стержень, вращающийся со скоростью 4 оборота в минуту (об/мин), и сообщали стержню постоянное ускорение от 5 оборотов в минуту до 40 оборотов в минуту. Измеряли время удерживания животного до падения его со стержня с максимальным временем вращения 300 секунд.
IT инъекция ASICалгина-1 не производила никакого значимого эффекта (тест ANOVA) на проявления мышей со стороны их моторной активности по сравнению с инъекцией раствора-носителя.
ПРИМЕР 3: АНАЛЬГЕТИЧЕСКИЙ ЭФФЕКТ ПЕПТИДА ASICАЛГИНА-1 (π-Dp1) и В ТЕСТЕ IN VIVO ПОСЛЕ ИНТРАЦЕРЕБРОВЕНТРИКУЛЯРНОЙ ИНЪЕКЦИИ
Интрацеребровентрикулярная инъекция (ICV) пептида ASICалгина-1 была введена в третий желудочек мозга мышей (C57B16J, самцы в возрасте от 7 до 11 недель), анестезированных изофлураном (1,5%), по следующим стереотаксическим координатам: -2,4 мм под кортикальной поверхностью, -0,5 мм по передне-задней оси и +1,6 мм по медио-латеральной оси по отношению к темени. Эффекты ICV инъекции в объеме 5 мкл раствора, содержащего 34 мкМ ASICалгина-1 (0,34 нмоль), сравнивали с эффектами от введения 5 мкл солевого раствора-носителя (145 мМ NaCl, 5 мМ KCl, 2 мМ CaCl2, 10 мМ HEPES, рН 7,4, 0,05% бычьего сывороточного альбумина). Наксолон, как ингибитор опиоидных рецепторов (особенно мю класса), был инъецирован подкожно в дозе 2 мг/кг в 50 мкл NaCl 0,9%, за десять минут до ICV инъекции пептида ASICалгина-1.
Токсичность
ICV инъекция пептида ASICалгина-1 не внесла никаких изменений в поведение животного, таких как моторные нарушения, нарушения равновесия, апатия, паралич или конвульсии, и не привела мышей к каком-либо летальному исходу, даже спустя 3 дня.
Острая термическая боль
Термическая боль изучалась на мышах путем измерения латентного периода отдергивания хвоста, погруженного в воду с температурой 46°С (рефлекторное движение, животное зафиксировано в неподвижном состоянии) в течение времени, ограниченного 30 секундами.
ICV инъекция пептида ASICалгина-1 вызвала мощный анальгетический эффект, который сохранялся в течение всего времени эксперимента (67 минут, фигура 3), этот эффект не был модифицирован предварительной обработкой животного наксолоном. Анальгетический эффект продолжался в течение нескольких часов до тех пор, пока величина латентного периода отдергивания хвоста не вернулась в своей контрольной величине.
ПРИМЕР 4: АНАЛЬГЕТИЧЕСКИЙ ЭФФЕКТ ПЕПТИДА ASIC-АЛГИНА-1 (π-Dp1) В ТЕСТЕ IN VIVO ПОСЛЕ ПОДКОЖНОЙ ИНЪЕКЦИИ
Эффект подкожной инъекции (SC) пептида ASICалгина-1 был исследован на модели воспалительной гипералгезии, вызванной каррагенином.
SC инъекция 2%-ного каррагенина в свод стопы задней лапы мыши (C57B16J, самцы в возрасте 7-11 недель) вызвала развитие тканевого воспаления и гипералгезии, измеренное с помощью определения латентного периода отдергивания задней лапы, погруженной в воду с температурой 46°С (интегрированное произвольное движение, животное зафиксировано в неподвижном состоянии) в течение времени, ограниченного 30 секундами, спустя два часа после инъекции каррагенина.
Когда пептид ASICалгин-1 (10 мкл раствора, содержащего 34 мкМ ASICалгина-1 (0,34 нмоль) в NaCl 0,9%, 0,05% бычьего сывороточного альбумина) был введен в виде инъекции вместе с каррагенином, не было отмечено никакой термической воспалительной гипералгезии (фигура 4, □). В этих условиях вторая SC инъекция пептида ASICалгина-1, сделанная спустя два часа, не произвела никакого значимого дополнительного эффекта (фигура 4, □).
Когда пептид ASICалгин-1 не был инъецирован совместно с каррагенином, а SC-инъекция этого пептида производилась два часа спустя после инъекции одного караггенина, т.е. когда воспалительный процесс уже распространился, то наблюдалась реверсия воспалительной гипералгезии и, кроме того, происходило значительное обезболивание по сравнению с контрольным экспериментом (фигура 4, ■ и #).
ПРИМЕР 5: АКТИВНОСТЬ ПЕПТИДОВ ASICАЛГИНА-1 (π-Dp1) и ASICАЛГИНА-2 (π-Dp2) В ОТНОШЕНИИ ASIC-ТОКОВ
Пептиды ASICалгин-1 и ASICалгин-2 ингибируют токи, генерируемые ASIC-каналами, гетерологично экспрессируемыми в клетках линии COS, и зарегистрированные по методу пэч-камп в конфигурации «целой клетки» (режим “фиксация потенциала”).
Пептиды вводили при рН 7,4 за 30 секунд перед внеклеточным скачком рН от 7,4 до 6 или 5,5.
Пептиды ASIC-алгин-1 и ASIC-алгин-2 имели одинаковый уровень эффективности и ингибировали ток, генерируемый крысиными гомомерными ASIC1a-каналами, при концентрации, производившей 50%-ное ингибирование (CI50), равной 49 нМ (фигура 5А) и ток, генерируемый гомомерными ASIC1a-каналами человека, при концентрации CI50, равной 127 нМ (фигура 5В). Ток, генерируемый гомомерными ASIC1b-каналами крысы, был ингибирован при концентрации CI50, равной 650 нМ (фигура 5С), генерируемый гетеромерными ASIC1a+ASIC2a-каналами крысы и человека при концентрации CI50, равной 308 нМ (фигура 5Е) и генерируемый гетеромерными ASIC1a+ASIC1b-каналами крысы при концентрации CI50, равной 223 нМ (фигура 5D) (канал ASIC1b человека не описан).
Пептиды ASIC-алгин-1 и ASIC-алгин-2 не ингибировали ни гомомерные ASIC2a- и ASIC3-каналы крысы и человека, ни гетеромерные ASIC1a+ASIC3 и ASIC1b+ASIC3-каналы крысы.
Эффекты применения пептидов ASICалгина-1 и ASICалгина-2 были обратимыми при прерывании введения пептидов (фигура 5).
Пептиды ASICалгин-1 и ASICалгин-2 являются единственными пептидами, описанными на сегодняшний день, способными ингибировать гомомерные ASIC1b-каналы и гетеромерные ASIC1a+ ASIC1b и ASIC1a+ASIC2a-каналы.
Напротив, пептид ASICалгин-1 (2 мкМ) не ингибирует ток, продуцируемый TRPV1-каналами крысы, активированными капсаином (1 мкМ), каналами, задействованными также в передаче боли сенсорными нервными окончаниями.
Не ограничиваясь этим объяснением, предполагается, что анальгетические эффекты пептидов ASICалгина-1 и ASICалгина-2 действуют как ингибиторы гомомерных ASIC1a-каналов и гетеромерных ASIC1a+ASIC2a-каналов в нейронах центральной нервной системы (IT и ICV инъекции) и ингибиторы гомомерных ASIC1a и ASIC1b-каналов и гетеромерных ASIC1a+ASIC1b-каналов в сенсорных нейронах (подкожная инъекция, IT инъекция при условии, что гомомерные ASIC1a и ASIC1b-каналы и гетеромерные ASIC1a+ASIC1b-каналы находятся в центральных окончаниях сенсорных нейронов на уровне дорсального рога спинного мозга).
ПРИМЕР 6: АКТИВНОСТЬ ПЕПТИДОВ ASICАЛГИНА-1 (π-Dp1) и ASICАЛГИНА-2 (π-Dp2) В ОТНОШЕНИИ НАТИВНЫХ ASIC-ТОКОВ В ЦЕНТРАЛЬНЫХ НЕЙРОНАХ И В СЕНСОРНЫХ НЕЙРОНАХ
Пептиды ASICалгин-1 и ASICалгин-2 обратимо ингибируют ASIC-токи, зарегистрированные по методу пэч-камп в конфигурации «целой клетки» (режим “фиксация потенциала”) в первичной культуре нейронов, выделенных из спинномозговых ганглиев взрослой крысы (фигура 6А), дорсального спинного мозга эмбриона мыши (фигура 6В) и гиппокампа новорожденных мышей (2 дня) (фигура 6С).
Частичный ингибирующий эффект пептидов ASICалгина-1 и ASICалгина-2 на суммарный ток ASIC-канала в сенсорных нейронах объясняется присутствием большой доли тока типа ASIC3 (не ингибируемого пептидами ASICалгин-1 и ASICалгин-2) в сенсорных нейронах, в то время как токи центральных ASIC-каналов (гиппокампа и спинного мозга) являются суммой тока гомомерного ASIC1a-канала и тока гетеромерного ASIC1a+ASIC2a-канала (оба тока ингибируются пептидами ASICалгин-1 и ASICалгин-2).
ПРИМЕР 7: АКТИВНОСТЬ ПЕПТИДОВ ASICАЛГИН-1 (π-Dp1) И ASICАЛГИН-2 (π-Dp2) В ОТНОШЕНИИ НЕЙРОНОВ
Пептиды ASICалгин-1 и ASICалгин-2, используемые в культуре спинальных нейронов, не производят никаких изменений ни на уровне базового тока, ни на уровне вариаций потенциала покоя (фигура 7А), зарегистрированных по методу пэч-камп в конфигурации «целой клетки».
Применение пептидов ASICалгина-1 и ASICалгина-2 не изменяет спонтанную синаптическую электрическую активность в культуре спинальных нейронов, ни форму или порог потенциалов действия, появившихся в результате электрического стимула (режим «фиксация тока» метода пэч-кламп) (фигура 7В).
Результаты показывают, что пептиды ASICалгин-1 и ASICалгин-2 не оказывали эффекта на свойства характеристической возбудимости нейронов, на синаптическую функцию или на потенциал-зависимые токи, участвующие в генезе (каналы Na+) или в реполяризации (каналы К+) потенциалов действия.
Напротив, специфическое ингибирование ASIC-каналов с помощью пептидов ASICалгин-1 и ASIСалгин-2 снижает возбудимость нейронов в ответ на скачок кислого рН (от 7,4 до 6,0), причем деполяризация, происходящая под действием ASIC-тока, является в этом случае недостаточной, чтобы запустить потенциал действия (фигура 7А).
Перечень ссылок
1. Bertin et Vergne-Salle, “Traitements médicamenteux de l'inflammation et des douleurs inflammatoires” Institut UPSA de la douleur - A Editorial Paris, р.113-132, 2007.
2. Gutstein et Akil, “Opioid analgesics”, in “The Pharmacological Basis of Therapeutics” Brunton et al., Eds. Mc Graw-Hill, 2006.
3. Bannister & Dickenson, Curr. Opin. Support Palliat. Care, 4: 1-5, 2-10.
4. Yennurajalingam et al., Support Cancer Ther., 1: 97-110, 2004.
5. Mizoguchi et al., Int. Rev. Neurobiol., 85: 249-260, 2009.
6. Waldmann et Lazdunski, Curr. Opin. Neurobiol., 3: 418-424, 1998.
7. Wemmie et al., Trends Neurosci., 29: 578-586, 2006.
8. Lingueglia et al., J. Biol. Chem., 282: 17325-17329, 2007
9. Jasti et al., Nature, 449: 316-323, 2007
10. Lingueglia et al., J. Biol. Chem., 272: 29778-29783, 1997.
11. Benson et al., Proc. Natl. Acad. Sci. U.S.A. 95: 10240-10245, 2002.
12. Hesselager et al., J. Biol. Chem., 279: 11006-11015, 2004.
13. Waldmann et al., Nature, 386: 173-177, 1997a.
14. Noel et al., «Current perspectives on acid-sensing ion channels: new advances and therapeuthic implications» Expert Rev. Clin. Pharmacol. «Clinical Pharmacology of Ion Channels», 3: 331-346, 2010.
15. Waldmann et al., J. Biol. Chem., 272: 20975-20978, 1997b.
16. Bassler et al., J. Biol. Chem., 276: 33782-33787, 2001.
17. Chen et al., Proc. Natl. Acad. Sci. U.S.A., 95: 10240-10245, 1998.
18. Reeh et Steen, Prog. Brain Res., 113: 143-151, 1996.
19. Steen et al., Neurosci. Lett., 199: 29-32, 1995a.
20. Issberner et al., Neurosci. Lett., 208: 191-194, 1996.
21. Ugawa et al., J. Clin. Invest., 10: 1185-1191, 2002.
22. Jones et al., J. Neurosci. 24: 10974-10979, 2004.
23. Voilley et al., J. Neurosci. 21: 8026-8033, 2001.
24. Dubé et al., Pain, 117: 88-96, 2005.
25. Escoubas et al., J. Biol. Chem., 275: 25116-25121, 2000.
26. Diochot et al., EMBO J., 23: 1516-1525, 2004.
27. Deval et al., EMBO J., 27: 3047-3055, 2008.
28. Mazzuca et al., Nature Neuroscience, 10: 943-945, 2007.
29. Abott et al., Pharmacol. Biochem. Behav., 17(6): 1213-1219, 1982.
30. Cridland et Henry, Brain Res., 584(1-2): 163-168, 1992.
31. Woolfe et Macdonald, J. Pharmacol. Exp. Ther., 80: 300-307, 1944.
32. Ankier, Eur. J. Pharmacol., 27(1): 1-4, 1974.
33. Kim et al., Pain, 55: 85-92, 1993.
34. Патент США 5877026.
35. Hylden et Wilcox, Eur. J. Pharmacol., 67: 313-316, 1980.
36. Deval et al., J. Neurosci., 31(16): 6059-6066, 2011.
| название | год | авторы | номер документа |
|---|---|---|---|
| АНАЛЬГЕТИЧЕСКИЙ ПЕПТИД ИЗ МОРСКОЙ АНЕМОНЫ URTICINA GREBELNYI | 2013 |
|
RU2521657C1 |
| ПОЛИПЕПТИД ИЗ МОРСКОЙ АНЕМОНЫ HETERACtis crispa, ОБЛАДАЮЩИЙ АНАЛЬГЕТИЧЕСКИМ ДЕЙСТВИЕМ | 2012 |
|
RU2475497C1 |
| ЛИГНАН, ОБЛАДАЮЩИЙ АНАЛЬГЕТИЧЕСКИМ ДЕЙСТВИЕМ | 2012 |
|
RU2491950C1 |
| АНАЛЬГЕТИЧЕСКИЕ СОЕДИНЕНИЯ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2020 |
|
RU2772045C2 |
| ПЕПТИДЫ ND2 И СПОСОБЫ ЛЕЧЕНИЯ НЕВРОЛОГИЧЕСКОГО ЗАБОЛЕВАНИЯ | 2011 |
|
RU2607033C2 |
| АНАЛЬГЕТИЧЕСКОЕ СРЕДСТВО НА ОСНОВЕ ПЛАЗМИДНОЙ ДНК, КОДИРУЮЩЕЙ HNP-1, ЛИБО HNP-2, ЛИБО HNP-3 (ВАРИАНТЫ) | 2014 |
|
RU2597789C2 |
| ПРИМЕНЕНИЕ РАМ | 2004 |
|
RU2345785C2 |
| Аналог альфа-конотоксина RgIA для лечения боли | 2016 |
|
RU2731217C2 |
| ПЕПТИДНЫЙ МОДУЛЯТОР ПУРИНЕРГИЧЕСКИХ РЕЦЕПТОРОВ | 2016 |
|
RU2650780C1 |
| СПОСОБ ПОЛИПЕПТИДНОЙ МОДИФИКАЦИИ ДЛЯ ОЧИСТКИ ПОЛИПЕПТИДНЫХ МУЛЬТИМЕРОВ | 2010 |
|
RU2606264C2 |









Настоящее изобретение относится к области биотехнологии, конкретно к получению новых выделенных пептидов, которые вызывают аналгезию и ингибируют ASIC-каналы (протон-чувствительные ионные каналы), к полинуклеотидам, кодирующим указанные пептиды, а также векторам и их применению, и может быть использовано в медицине. Указанные пептиды выделены из яда змеи Dendroaspis polylepis и имеют аминокислотную последовательность LKCX4QHGKVVTCHRDMKFCYHNTGMPFRNLKLILQGCSSSCSETENNKCCSTDRCNK, где X4 обозначает любую аминокислоту; или последовательность, имеющую идентичность по меньшей мере 56% с указанной последовательностью и сохраняющую аналгезирующую активность. Изобретение позволяет получить пептид, ингибирующий ASIC-каналы, содержащие по меньшей мере одну субъединицу, выбранную из группы, состоящей из субъединиц ASIC1a и ASIC1b. 4 н. и 10 з.п. ф-лы, 7 ил., 1 табл., 7 пр.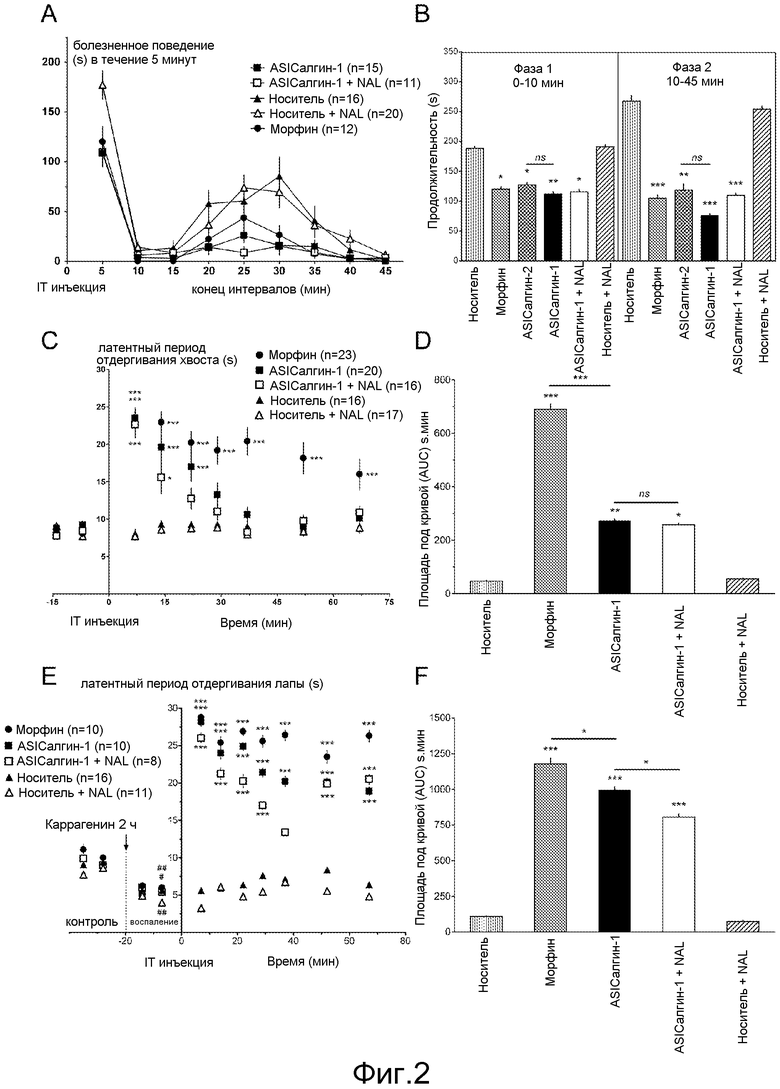
1. Пептид, демонстрирующий аналгезирующую активность, имеющий аминокислотную последовательность LKCX4QHGKVVTCHRDMKFCYHNTGMPFRNLKLILQGCSSSCSETENNKCCSTDRCNK (SEQ ID NO: 1),
где X4 обозначает любую аминокислоту; или
последовательность, имеющую идентичность по меньшей мере 56% с последовательностью SEQ ID NO: 1 и сохраняющую аналгезирующую активность указанного пептида, имеющего последовательность SEQ ID NO: 1.
2. Пептид по п. 1, в котором указанный пептид является блокатором по меньшей мере одного ASIC-канала.
3. Пептид по п. 2, в котором указанный пептид является блокатором по меньшей мере одного ASIC-канала, содержащего по меньшей мере одну субъединицу, выбранную из группы, состоящей из субъединиц ASICla и ASIClb.
4. Пептид по п. 3, в котором указанный пептид является блокатором гомомерного ASICla-канала, гомомерного ASIClb-канала, гетеромерного ASICla+ASIClb-канала и/или гетеромерного ASIC1a+ASIC2a-канала.
5. Пептид по любому из пп. 1-4, в котором X4 обозначает Y или F в последовательности SEQ ID NO: 1.
6. Полинуклеотид, имеющий нуклеотидную последовательность, кодирующую пептид, который определен в любом из пп. 1-5.
7. Полинуклеотид по п. 6, имеющий нуклеотидную последовательность:

где 73txx75 и 10txx12 обозначает tac, tat, ttt или ttc.
8. Полинуклеотид по п. 6, где указанный полинуклеотид гибридизируется в строгих условиях с нуклеотидной последовательностью SEQ ID NO: 4 или SEQ ID NO: 22 или с комплементарной ей последовательностью.
9. Полинуклеотид по п. 6 или 8, где указанный полинуклеотид включает природную или синтезированную последовательность, имеющую идентичность по меньшей мере 76% с последовательностью SEQ ID NO: 4 или с последовательностью SEQ ID NO: 22.
10. Вектор экспрессии, включающий полинуклеотид, который определен в любом из пп. 6-9.
11. Применение:
- пептида по любому из пп. 1-5;
- полинуклеотида по любому из пп. 6-9; или
- вектора по п. 10;
в качестве лекарственного средства для профилактики или лечения боли или патологии, в которой участвует активация ASIC-каналов.
12. Применение по п. 11, где боль и патология, в которых участвует активация ASIC-каналов, выбрана из группы, включающей воспалительные, нейропатические, онкологические, послеоперационные, мышечно-скелетные и висцеральные боли, воспалительные состояния, раковые заболевания, фибромиалгии и синдром раздраженной толстой кишки.
13. Применение по п. 11, где лекарственное средство предназначено для профилактики или лечения неврологического заболевания центральной нервной системы, выбранного из группы, включающей депрессию, тревожное состояние, геморрагический инсульт, эпилепсию, воспалительные заболевания центральной нервной системы и нейродегенеративные заболевания.
14. Применение по любому из пп. 11-13, где указанное лекарственное средство вводят в центральную нервную систему или подкожно, чрескожно, системным путем, перорально или через дыхательные пути.
| YING-YING HE et al., Cloning and purification of alpha-neurotoxins from king cobra (Ophiophagus hannah), Toxicon, 2004, v.44, is.3, p.295-303 | |||
| VEGA R | |||
| et al., Acid-sensing ionic-channel functional expression in the vestibular endorgans, Neurosci Lett., 2009, v.463, is.3, p.199-202 | |||
| СИДОРОВ А.В., Регуляция и модуляция нейронных функций при |

