Способ относится к получению медицинских препаратов, а именно к синтезу радиофармпрепаратов (РФП) и может найти применение в радиофармацевтическом производстве в центрах позитронно-эмиссионной томографии (ПЭТ).
ПЭТ - одно из наиболее бурно развивающихся направлений современной ядерной медицины, позволяет выявлять злокачественные новообразования, очаги воспаления и другие патологические процессы, а также визуализировать метаболизм миокарда.
Использование ПЭТ требует применения радиофармпрепаратов (РФП), меченных ультракороткоживущими позитрон-излучающими радионуклидами, которые получают с использованием циклотронов. Периоды полураспада таких радионуклидов составляют минуты или десятки минут. Наиболее широко используемым радионуклидом для ПЭТ-томографии является фтор-18 (более 90% обследований пациентов проводят с РФП, меченными фтором-18).
Радионуклид фтор-18 производится по ядерной реакции 18O(p,n)18F при облучении протонами с энергией 12-18 МэВ кислорода, обогащенного изотопом 18O, в химической форме H2 18O. Радионуклид обладает периодом полураспада, равным 109 минутам.
Возможности метода ПЭТ в значительной степени определяются арсеналом доступных РФП. Именно выбор подходящего РФП позволяет изучать с помощью ПЭТ такие разные процессы, как метаболизм, транспорт веществ, лиганд-рецепторные взаимодействия, экспрессию генов и т.д. Широкий спектр различных классов РФП делает ПЭТ достаточно универсальным инструментом современной молекулярной биологии и клинической медицины. Поэтому радиохимический синтез новых РФП и разработка эффективных методик синтеза уже зарекомендовавших себя РФП в настоящее время становится одной из важнейших задач, решаемой для развития метода ПЭТ.
Наиболее распространенным РФП для ПЭТ является 2-[18F]фтор-2-дезокси-D-глюкоза (ФДГ), с помощью данного препарата проводится до 95% всех обследований. ФДГ позволяет оценить региональную скорость утилизации глюкозы. Замена гидроксильной группы в молекуле глюкозы атомом фтора приводит к тому, что ФДГ транспортируется в ткани аналогично глюкозе, однако в клетках метаболизм ФДГ останавливается на стадии фосфорилирования, что приводит к накоплению ФДГ в областях повышенного энергетического метаболизма глюкозы. Следовательно, распределение ФДГ в тканях организма описывается в рамках простой фармакокинетической модели, и данные ПЭТ исследования могут быть достаточно легко интерпретированы. Однако данный РФП имеет ряд ограничений в диагностике опухолей мозга, связанных с тем, что глюкоза является основным энергетическим субстратом для мозга и, кроме того, утилизируется в макрофагах, продуцируемых в абсцессах или очагах воспаления в послеоперационный период, в результате чего дифференциация очага воспаления и опухоли с помощью ФДГ представляет определенные трудности. В связи с этим в последние годы ведется интенсивный поиск РФП, механизм накопления которых является специфичным именно для опухолей. В данном аспекте широко изучаются аминокислоты, которые можно было бы использовать в ПЭТ.
В настоящее время в ряде ПЭТ-центров Европы, Японии и США интенсивно исследуются фторированные аналоги тирозина, содержащие фтор-18 в ароматическом кольце - O-(2′-[18F]фторэтил)-L-тирозин. Предполагается, что фторированные аналоги аминокислот (ФАА), в частности фторпроизводные тирозина, в гораздо меньшей степени накапливаются в очагах воспаления по сравнению с ФДГ, и именно они считаются наиболее перспективными агентами для дифференциации опухолевого и воспалительного процессов. Оптимальными характеристиками такого агента являются низкий индекс накопления в абсцессе при относительно высоком накоплении в злокачественных клетках.
В рутинной практике синтеза ФЭТ нашел применение предшественник трет-бутиловый эфир O-(2-тозилоксиэтил)-N-тритил-L-тирозина [K. Hamacher, Н.Н. Coenen / Applied Radiation and Isotopes 57 (2002) 853-856]. В качестве «уходящей» группы выступает тозильная группа, замещение которой на [18F]фторид протекает с высоким выходом. С данным предшественником разработан метод синтеза, который был адаптирован для модуля GE Healthcare TracerLab FX F-N [Bourdier Т., Greguric I., Roselt P., Jackson T, Faragalla J., Katsifis A. Fully automated one-pot radiosynthesis of O-(2-[18F]fluoroethyl)-L-tyrosine on the TracerLab FXFN module. // Nucl. Med. Biol. - 2011. - V. 38. - №5. - P. 645-651]. По такому методу радиохимический выход конечного продукта составляет 35% без поправки на распад при времени синтеза около 60 минут, энантиомерная чистота [18F]ФЭТ - более 99%.
Однако способ обладает значительными недостатками, а именно: использование метода ВЭЖХ (высокоэффективная жидкостная хроматография) очистки препарата делает систему громоздкой, а сам метод синтеза - длительным и трудоемким.
Очистка конечного продукта возможна более простым и удобным методом твердофазной экстракции (ТФЭ). Это существенно упрощает автоматизацию процесса, позволяет проводить синтез за более короткое время и использовать одноразовые недорогие компоненты (картриджи) [Mueller D., Klette I., Kalb F., Baum R.R Synthesis of O-(2-[18F]fluoroethyl)-L-tyrosine based on a cartridge purification method. // Nucl. Med. Biol. - 2011. - V. 38. - №5. - Р. 653-658].
Реакционную смесь, содержащую [18F]ФЭТ, пропускают через два одноразовых картриджа С18 Chromafix (Macherey Nagel, обращено фазовый сорбент), картриджи промывают последовательно раствором ацетата аммония и 5% раствором этанола в 0.9% NaCl, после чего [18F]ФЭТ элюируют раствором 0.9% хлорида натрия, но содержащим 10% этанола.
Время синтеза существенно ниже по отношению метода ВЭЖХ очистки ФЭТ и составляет 35 минут, радиохимический выход около 45%. Однако, несмотря на хорошие результаты, полученные отдельными группами авторов, синтез ФЭТ, даже в комбинации с очисткой методом ТФЭ, не получил широкого применения в рутинной практике ПЭТ, так как в данной методике синтеза ФЭТ в качестве алкилирующего агента используется фтор-18-этил-бромид. Очистка фтор-18-этил-бромида достигается методом дистилляции, что, с одной стороны, является преимуществом, поскольку нет необходимости в дополнительной очистке алкилирующего агента, но, с другой стороны, предъявляет огромные требования к герметичности автоматизированной системы синтеза. Этот метод часто приводит к потере радиоактивности именно на стадии дистилляции фтор-18-этил-бромида и может приводить к невоспроизводимым радиохимическим выходам. Из этого следует, что такой метод синтеза ФЭТ непригоден для его серийного производства.
Настоящее изобретение касается синтеза O-(2′-[18F]фторэтил)-L-тирозина с целью использования его для серийного производства данного препарата и широкого применения его в клинической практике.
Наиболее близким к предлагаемому является способ получения O-(2′-[18F]фторэтил)-L-тирозина (ФЭТ), описанный в [Bioorg. Med. Chem. 16 (2008) 4994-5003, вместе с тем сам синтез подробнее представлен в Дипломе, выполненном в той же организации, Институте Мозга Человека РАН (копия прилагается)].
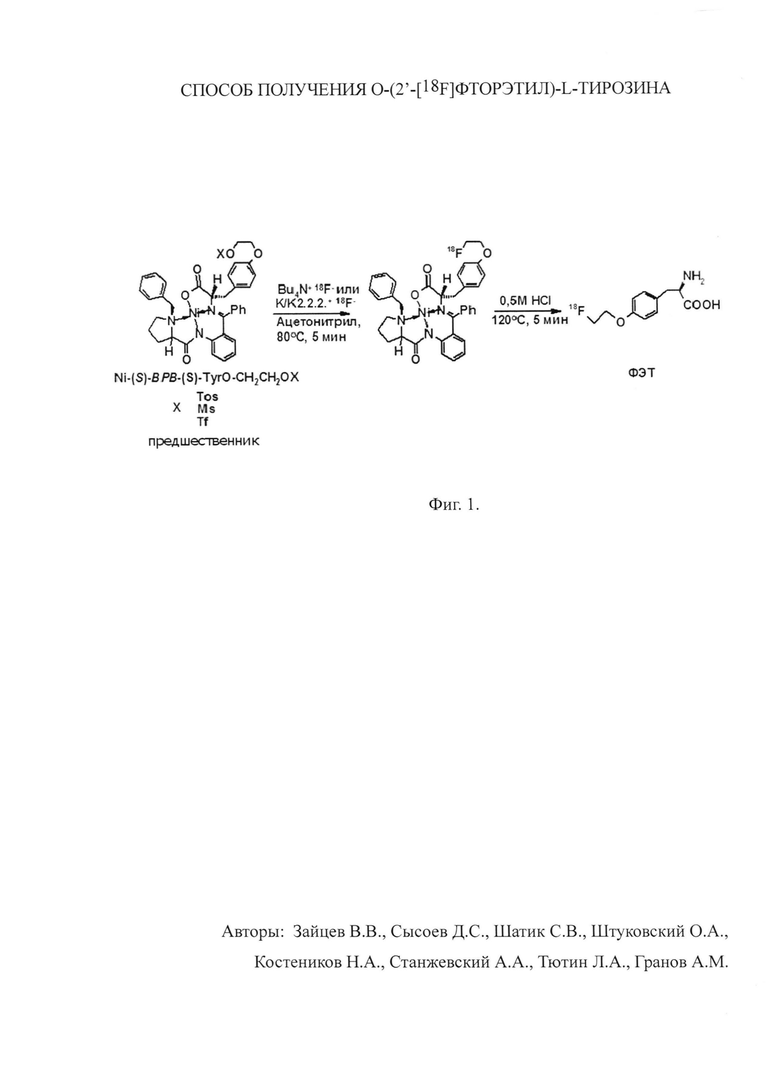
Способ заключается в нуклеофильном фторировании Ni-(S)-BPB-(S)-TyrO-CH2CH2OX (X=Ms, Ts, Tf; ВРВ=[N-2-(N′-бензил-пролил)амино]бензофенон) (предшественника) (фиг. 1), гидролизе фторированного предшественника и очистке конечного полупродукта в модуле синтеза с последующей очисткой самого модуля.
Для осуществления синтеза в реакционный сосуд, содержащий комплекс [ТБА]+18F- или [К/2.2.2.]+18F-, добавляли раствор предшественника (4-5 мг Ni-(S)-BPB-(S)-TyrO-CH2CH2OX (X=Ms, Ts, Tf) в 0.5 мл ацетонитрила). Смесь выдерживали при 80°С в течение 5 мин без перемешивания, после чего добавляли в реакционный сосуд 0.5 мл водного раствора 0.5 М HCl и проводили гидролиз и снятие защиты при температуре 120°С в течение 5 мин.
Согласно прототипу, наилучшие выходы фторирования промежуточного комплекса достигались при использовании тозильной (Ts) уходящей группы и криптофикса 2.2.2 ([К/2.2.2.]+) в качестве межфазного катализатора, 57.3±8.5% (n=3).
Очистка от непрореагировавшего фтора и побочных неорганических и органических примесей и выделение полупродукта производились при помощи колонок с сорбентом С18 SepPak.
Для автоматизации синтеза, согласно прототипу, в ИМЧ РАН был сконструирован модуль синтеза с использованием стандартных коммерчески доступных комплектующих: клапанов фирмы Burkert Compromatic (Германия), терморегулятора ТРМ1А, регулятора потока газа, пневматического поршня SMC Pneumatics для поднятия и опускания игл в реакционный сосуд, 5 стандартных конических сосудов объемом 5 мл.
Транспортировка жидкостей осуществлялась током азота, а управление пневматическим поршнем для устройства поднятия иглы - сжатым воздухом. Для промывки линий модуля (тефлоновых капилляров 1/16″ с внутренним диаметром 0,75 мм) сосуды для реагентов заполняли растворителями, которые транспортировались током азота в различных направлениях. Модуль включал в себя 15 трехходовых и двухходовых клапанов, пять сосудов объемом 1-5 мл для добавления реагентов, основной сосуд для проведения реакций, герметично закрытый тефлоновой крышкой, снабженной четырьмя входами-трубками и одной иглой. Основной реакционный сосуд находился в нагревательном устройстве, которое обеспечивало диапазон нагрева до 200°С. Обдув реакционного сосуда сжатым воздухом давал возможность его быстрого охлаждения. Кроме того, модуль был снабжен устройством для фильтрования, что давало возможность очистки препарата методом твердофазной экстракции и стерильного фильтрования. Система для ВЭЖХ-очистки продукта не интегрирована в систему синтеза, а находится отдельно. Управление процессом проводилось дистанционно с помощью выключателей, установленных вне горячей зоны.
Как отметили авторы, время синтеза составило 55 минут, радиохимический выход 40-45% (с коррекцией на распад), энантиомерная чистота более 98%, радиохимическая чистота более 95%.
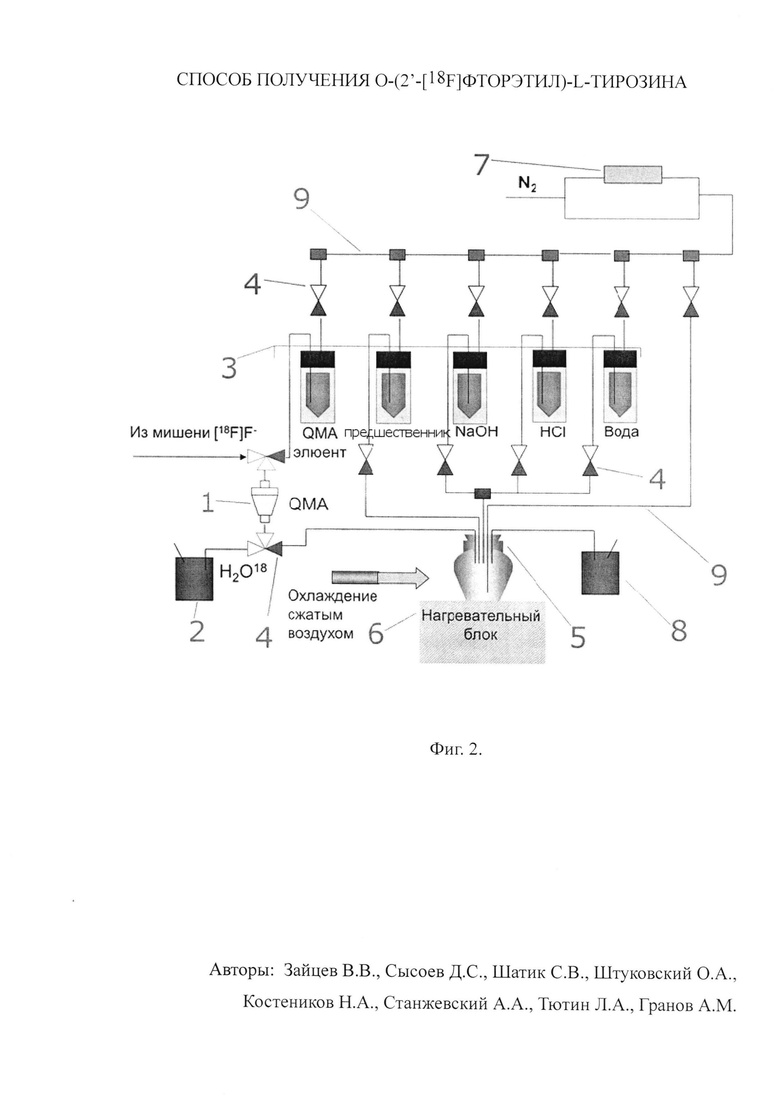
Для лучшего понимания приводим технологическую схему синтеза согласно прототипу (фиг.2), где 1 - колонка с анионообменным сорбентом, 2 - сосуд для регенерата, 3 - сосуды для реагентов, 4 - клапаны (двух- и трехходовые), 5 - реакционный сосуд, 6 - нагревательный блок, 7 - регулятор потоков, 8 - сосуд для жидких отходов, 9 - капилляры.
Такой способ, по данным авторов, позволяет получить ФЭТ с высоким радиохимическим выходом и высокой радиохимической и энантиомерной чистотой.
Однако неоднократное повторение нами данного синтеза по способу-прототипу показало, что радиохимический выход препарата нестабилен, а именно составляет 5-35% без коррекции на распад. Это, на наш взгляд, связано с тем, что, как отмечают авторы, ими не достигнута полная автоматизация синтеза (она находится еще в стадии разработки) и потому очистка модуля синтеза, в котором при многократных синтезах накапливаются различные примеси, проводилась частично вручную.
Кроме того, выпадение осадка при нейтрализации реакционной массы после гидролиза фторированного предшественника с большой вероятностью может вызвать закупоривание коммуникаций (стандартных капилляров с внутренним диаметром 0.75 мм) и привести к потере всего синтеза, что значительно снижает ценность способа-прототипа.
Технический результат настоящего изобретения состоит в усовершенствовании способа получения O-(2′-[18F]фторэтил)-L-тирозина за счет дополнительной очистки модуля синтеза и конечного полупродукта, что обеспечивает стабильный высокий выход препарата и полную автоматизацию процесса.
Этот результат достигается тем, что в известном способе получения О-(2′-[18F]фторэтил)-L-тирозина в модуле синтеза с сосудами для реагентов, реакционным сосудом, капиллярами и колонкой с сорбентом С18 осуществляют нуклеофильное фторирование Ni-(S)-BPB-(S)-TyrO-СН2СН2ОХ (X=Ms, Ts, Tf) (предшественника), в присутствии межфазного катализатора в реакционном сосуде, гидролиз фторированного предшественника и очистку конечного полупродукта посредством колонки с сорбентом С18 с получением препарата и последующей очисткой самого модуля, при этом согласно изобретению конечный полупродукт дополнительно очищают от осадка, образующегося при нейтрализации реакционной массы после гидролиза фторированного предшественника, посредством фильтров грубой и тонкой очистки, установленных между реакционным сосудом и колонкой с сорбентом С18, а очистку модуля синтеза выполняют в две стадии с удалением образующихся при синтезе никелевых соединений и органических примесей с использованием деионизованной воды, ацетонитрила и одномолярной соляной кислоты - на первой стадии и следовых количеств воды и соляной кислоты посредством ацетонитрила, деионизованной воды и этилового спирта - на второй, причем эти растворы располагают в модуле в восьми сосудах для реагентов.
Целесообразно для удаления никелевых соединений и органических примесей из модуля синтеза деионизованную воду помещать в первый и четвертый сосуды для реагентов, ацетонитрил - во второй, третий и пятый, одномолярную соляную кислоту - в шестой.
Целесообразно также после первой стадии очистки модуля для удаления следовых количеств воды и кислоты в нем ацетонитрил помещать в первый, второй, третий, четвертый и пятый сосуды для реагентов, деионизованную воду в шестой, этанол - в седьмой и восьмой.
Использование для очистки коммуникаций и реакционного сосуда двухступенчатой очистки позволяет полностью удалить образующиеся при синтезе никелевые соединения и органические примеси, что обеспечивает достижение стабильно высокого выхода препарата при серийном производстве.
Использование фильтров грубой и тонкой очистки в капилляре между реакционным сосудом и колонкой с сорбентом С18 при синтезе препарата обеспечивает очистку полупродукта от осадка, выпадающего в реакционном сосуде при нейтрализации реакционной смеси щелочью после гидролиза. Это препятствует засорению пор колонки с сорбентом С18 образующимся осадком и обеспечивает безопасное и надежное прохождение реагентов по капиллярам модуля синтеза, позволяя проводить синтез в автоматизированном режиме.
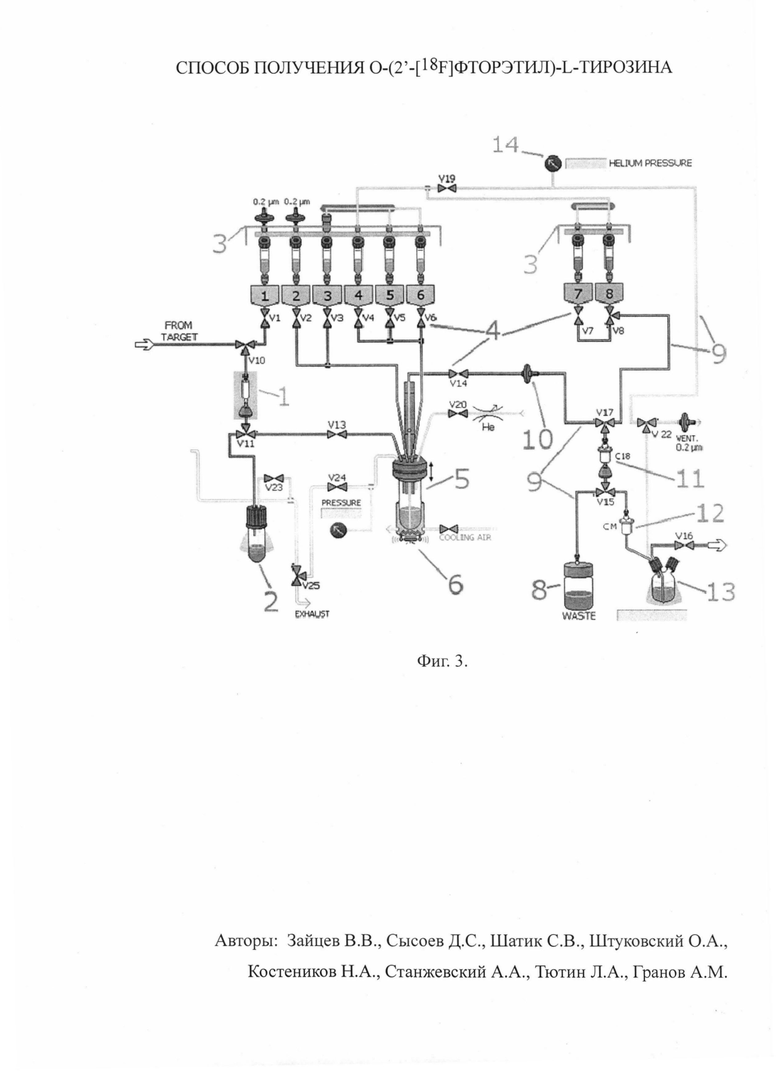
Для лучшего понимания приводим схему модуля синтеза согласно изобретению (фиг. 3), где 1 - колонка с анионообменной смолой, 2 - сосуд для регенерата, 3 - сосуды для реагентов, 4 - клапаны (двух- и трехходовые), 5 - реакционный сосуд, 6 - нагревательный блок, 8 - сосуд для жидких отходов, 9 - капилляры, 10 - фильтры грубой и тонкой очистки, 11 - колонка с сорбентом С18, 12 - колонка с катионообменным сорбентом, 13 - сосуд для продукта, 14 - датчик давления.
Пример осуществления синтеза.
1. Получение радионуклида (РН) 18F.
Для получения РН проводят облучение протонами обогащенной по изотопу 18O воды в мишени ускорителя (циклотрона). Целевой РН 18F получают по ядерной реакции 18O(p,n)18F.
2. Выделение РН.
Облученный материал по капилляру транспортируют в модуль синтеза на колонку с анионообменной смолой (1), при этом обогащенная вода, из которой выделен целевой РН, собирается в сосуд для регенерата (2).
РН, сорбированный на колонке, элюируют в реакционный сосуд (5). В качестве элюента используют водный раствор карбоната калия, поступающий из сосуда для реагентов (сосуд 1).
Транспортировку жидкостей по капиллярам (9) осуществляют током инертного газа, причем распределение потоков производят с помощью клапанов (4). Давление инертного газа контролируют датчиком давления (14).
3. Образование комплекса РН с МФК и его азеотропная осушка.
Из сосуда для реагентов (сосуд 2) в реакционный сосуд (5) добавляют МФК (межфазный катализатор) в ацетонитриле. Для азеотропной осушки полученный комплекс РН с МФК нагревают до температуры 85°С в токе инертного газа в течение 10 минут с помощью нагревательного блока (6).
4. Фторирование предшественника.
В реакционный сосуд (5) из сосуда для реагентов (сосуд 3) добавляют раствор предшественника (коммерчески доступного) в ацетонитриле. Полученную смесь нагревают до температуры 85°С в течение 5 минут с помощью нагревательного блока (6).
5. Гидролиз и нейтрализация реакционной массы.
К реакционной массе из сосуда для реагентов (сосуд 4) добавляют 0.6 мл 0.5 М водного раствора соляной кислоты, нагревают полученную смесь до 125°С 5 минут. Затем реакционную массу охлаждают до температуры 40°С с помощью нагревательного блока (6).
К реакционной массе из сосуда для реагентов (сосуд 5) добавляют 10 мл деионизованной воды, затем из сосуда для реагентов (сосуд 6) добавляют 4 мл 0.1 М водного раствора гидроксида натрия. Полученную смесь перемешивают барботированием инертным газом в течение 5 секунд.
6. Очистка реакционной массы от осадка.
Реакционную массу пропускают через фильтры грубой и тонкой очистки (10) для удаления осадка, образовавшегося на стадии нейтрализации реакционной массы раствором гидроксида натрия.
7. Очистка конечного полупродукта.
Очищенный от осадка полупродукт поступает на колонку с сорбентом С18 (11), при этом целевой продукт задерживается на сорбенте, а отходы поступают в сосуд для жидких отходов (8). Колонку с сорбентом С18 (11) промывают 3 мл деионизованной воды из сосуда для реагентов (сосуд 7).
Конечный полупродукт элюируют с колонки с сорбентом С18 (11) 5 мМ раствором ацетата натрия с 5% этанолом (рН=4), из сосуда для реагентов (сосуд 8).
Элюат пропускают через колонку с катионообменным сорбентом (12) и собирают в сосуде для продукта (13), содержащем 2 мл 0,85 М раствора ацетата натрия для придания изотоничности. При перемешивании барботированием очищенного полупродукта и раствора ацетата натрия получается продукт (препарат).
Время синтеза составляет 35 минут, радиохимический выход 45-50% (с коррекцией на распад), энантиомерная чистота более 98%, радиохимическая чистота более 95%.
После синтеза проводят очистку модуля синтеза, которая состоит из двух стадий. На первой стадии сосуды с реагентов (3) заправляют следующим образом: сосуд 1 - деионизованная вода 3 мл, сосуд 2 - ацетонитрил 3 мл, сосуд 3 - ацетонитрил 2 мл, сосуд 4 - деионизованная вода 1 мл, сосуд 5 - ацетонитрил 15 мл, сосуд 6 - 1М соляная кислота 14 мл. Реагенты последовательно транспортируют из сосудов для реагентов (3) в реакционный сосуд (5) и затем перемещают в сосуд для жидких отходов (8).
На второй стадии сосуды с реагентов (3) заправляют следующим образом: сосуд 1 - ацетонитрил 3 мл, сосуд 2 - ацетонитрил 3 мл, сосуд 3 - ацетонитрил 1 мл, сосуд 4 - ацетонитрил 1 мл, сосуд 5 - ацетонитрил 15 мл, сосуд 6 - деионизованная вода 14 мл, сосуд 7 - этанол 5 мл, сосуд 8 - этанол 5 мл. Реагенты последовательно транспортируют из сосудов для реагентов (3) в реакционный сосуд (5) и затем перемещают в сосуд для жидких отходов (8).
После такой очистки модуль готов к следующему синтезу.
Все параметры технологии синтеза получены опытным путем и позволили добиться хороших результатов.
В настоящее время препарат, получаемый описанным способом, проходит доклинические испытания в РНЦРХТ.
Предлагаемый способ по сравнению с известными имеет ряд существенных преимуществ:
1) полная автоматизация синтеза, что не может достичь прототип;
2) достижение стабильно высокого технологического выхода конечного продукта, что не позволяет прототип;
3) по сравнению с другими известными способами отличается простотой синтеза с более высоким технологическим выходом препарата, не уступающего им по энантиомерной и радиохимической чистоте.
Способ разработан в РНЦРХТ и прошел апробацию при 50-ти синтезах с положительным результатом.
| название | год | авторы | номер документа |
|---|---|---|---|
| Молекула общей структуры Y-Nic-F, способы получения, предшественники для её получения, а также применение в качестве действующего вещества в составе потенциального радиофармацевтического лекарственного препарата | 2021 |
|
RU2811181C2 |
| РЕАГЕНТЫ И СПОСОБЫ ВВЕДЕНИЯ РАДИОАКТИВНОЙ МЕТКИ | 2010 |
|
RU2524284C2 |
| Способ синтеза F-меченых биомолекул | 2012 |
|
RU2620598C2 |
| Устройство для производства радиофармпрепаратов | 2024 |
|
RU2836906C1 |
| МЕЧЕННЫЕ РАДИОАКТИВНЫМ ИЗОТОПОМ КОНЪЮГАТЫ RGD-СОДЕРЖАЩИХ ПЕПТИДОВ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ С ПОМОЩЬЮ CLICK-ХИМИИ | 2005 |
|
RU2419627C2 |
| СПОСОБЫ И УСТРОЙСТВО ДЛЯ СИНТЕЗИРОВАНИЯ РАДИОФАРМАЦЕВТИЧЕСКИХ ПРЕПАРАТОВ И ИХ ПРОМЕЖУТОЧНЫХ ПРОДУКТОВ | 2011 |
|
RU2631500C2 |
| ХИМИЧЕСКИЙ ПРОИЗВОДСТВЕННЫЙ МОДУЛЬ И КАРТА СИНТЕЗА ДОЗ ДЛЯ СИСТЕМЫ ПРОИЗВОДСТВА БИОМАРКЕРОВ ПЭТ | 2010 |
|
RU2541254C2 |
| ЛИОФИЛИЗАТ ДЛЯ ПРИГОТОВЛЕНИЯ РАДИОФАРМАЦЕВТИЧЕСКОГО ПРЕПАРАТА | 2017 |
|
RU2702238C2 |
| [F-18] МЕЧЕННАЯ L-ГЛЮТАМИНОВАЯ КИСЛОТА, [F-18] МЕЧЕННЫЙ L-ГЛЮТАМИН, ИХ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ, А ТАКЖЕ СПОСОБ ИХ ПОЛУЧЕНИЯ | 2006 |
|
RU2395489C2 |
| Способ радиоактивного мечения | 2014 |
|
RU2675371C2 |
Изобретение относится к способу получения O-(2′-[18F]фторэтил)-L-тирозина, который может найти применение в синтезе радиофармпрепаратов для позитронно-эмиссионной томографии. Предлагаемый способ осуществляют в модуле синтеза с сосудами для реагентов, реакционным сосудом, капиллярами и колонкой с сорбентом С18. Способ включает нуклеофильное фторирование предшественника Ni-(S)-BPB-(S)-TyrO-CH2CH2OX (X=Ms, Ts, Tf; ВРВ=[N-2-(N′-бензил-пролил)амино]бензофенон) в присутствии межфазного катализатора в реакционном сосуде, гидролиз фторированного предшественника и очистку конечного полупродукта посредством колонки с сорбентом С18 с получением препарата и последующей очисткой самого модуля. Способ характеризуется тем, что конечный полупродукт дополнительно очищают от осадка, образующегося при нейтрализации реакционной массы после гидролиза фторированного предшественника, посредством фильтров грубой и тонкой очистки, установленных между реакционным сосудом и колонкой с сорбентом С18. Очистку модуля синтеза выполняют в две стадии с удалением образующихся при синтезе никелевых соединений и органических примесей с использованием деионизованной воды, ацетонитрила и одномолярной соляной кислоты на первой стадии и следовых количеств воды и соляной кислоты посредством ацетонитрила, деионизованной воды и этилового спирта - на второй. При этом данные растворы располагают в модуле в восьми сосудах для реагентов. Способ позволяет получать целевой препарат со стабильно высоким радиохимическим выходом и высокой радиохимической и энантиомерной чистотой, а также обеспечивает полную автоматизацию синтеза. 2 з.п. ф-лы, 3 ил., 1 пр.
1. Способ получения O-(2′-[18F]фторэтил)-L-тирозина в модуле синтеза с сосудами для реагентов, реакционным сосудом, капиллярами и колонкой с сорбентом С18, включающий нуклеофильное фторирование предшественника Ni-(S)-BPB-(S)-TyrO-CH2CH2OX (X=Ms, Ts, Tf; ВРВ=[N-2-(N′-бензил-пролил)амино]бензофенон) в присутствии межфазного катализатора в реакционном сосуде, гидролиз фторированного предшественника и очистку конечного полупродукта посредством колонки с сорбентом С18 с получением препарата и последующей очисткой самого модуля, отличающийся тем, что конечный полупродукт дополнительно очищают от осадка, образующегося при нейтрализации реакционной массы после гидролиза фторированного предшественника, посредством фильтров грубой и тонкой очистки, установленных между реакционным сосудом и колонкой с сорбентом С18, а очистку модуля синтеза выполняют в две стадии с удалением образующихся при синтезе никелевых соединений и органических примесей с использованием деионизованной воды, ацетонитрила и одномолярной соляной кислоты на первой стадии и следовых количеств воды и соляной кислоты посредством ацетонитрила, деионизованной воды и этилового спирта - на второй, причем эти растворы располагают в модуле в восьми сосудах для реагентов.
2. Способ по п.1, отличающийся тем, что для удаления никелевых соединений и органических примесей из модуля синтеза деионизованную воду помещают в первый и четвертый сосуды для реагентов, ацетонитрил - во второй, третий и пятый, одномолярную соляную кислоту - в шестой.
3. Способ по п.1 или 2, отличающийся тем, что после первой стадии очистки модуля для удаления следовых количеств воды и кислоты в нем ацетонитрил помещают в первый, второй, третий, четвертый и пятый сосуды для реагентов, деионизованную воду - в шестой, этанол - в седьмой и восьмой.
| СТЕПАНОВА М.А., Оптимизация синтеза О-(2'-[F]фторэтил)-L-тирозина, диагностического агента для позитронной эмиссионной томографии, Дипломная работа, Государственное образовательное учреждение высшего профессионального образования Санкт-Петербургский государственный технологический институт, Санкт-Петербург, 2009 | |||
| R | |||
| N | |||
| KRASIKOVA et |

