ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к биоразрушаемому материалу и способу получения биоразрушаемого материала.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
В целях остановки кровотечения после разреза пораженной зоны, блокирования поступления питательных веществ к опухоли, поддержания концентрации противоракового препарата в опухоли и подобных целей, в качестве полимерных частиц для эмболизации кровеносных сосудов и т.п. применялись сополимер поли(молочная кислота/гликолевая кислота) (патентный документ 1), блок-сополимер полиэтиленгликоля и полимолочной кислоты и т.д. (патентные документы 2-5) или мультиблок сополимер, полученный сополимеризацией молочной кислоты, полиэтиленгликоля, поликарбоновой кислоты и т.п. (патентный документ 6) и т.д.
Применение подобных полимерных частиц для эмболизации кровеносных сосудов и аналогичных целей было связано с такими проблемами, как невозможность добиться быстрого биоразрушения после выполнения целевых функций. Кроме того, поскольку эти полимерные частицы, которые применяются в форме сферических частиц с тем, чтобы плотно и надежно эмболизировать кровеносные сосуды и т.п., доставляются к целевому участку в кровеносном сосуде и т.п. через микрокатетер, имеющий небольшой диаметр, возникает такая проблема, как забивание катетера из-за недостаточной эластичности полимерных частиц или агрегации между ними, или же необратимая деформация частиц перед тем, как они достигнут целевого участка.
Для решения этих проблем предпринимались попытки регулировать эластичность полимерных частиц путем разработки частиц, сформированных из смеси полимеров нескольких типов (патентный документ 7) или путем разработки полимерных частиц, сшитых поперечными химическими связями (патентный документ 8). Кроме того, сообщалось о попытках покрытия поверхности полимерных частиц полиэтиленгликолем, чтобы предупредить агрегацию полимерных частиц и, тем самым, улучшить их способность проходить через катетер (патентный документ 9).
Кроме того, для предотвращения адгезии (образования спаек) между поврежденным участком поверхности органа, который появляется из-за хирургического вмешательства, и окружающими тканями, в качестве биоразрушаемого материала, например, антиадгезионного материала, перевязочного материала, гемостатического материала или материала, предупреждающего недержание мочи, применяется образующийся in situ гель, состоящий из такого сополимера, как, например, поли(этиленгликоль/полимолочная кислота) и полигликолевая кислота и т.п. (патентный документ 10); или гель, состоящий из декстрана и поли N-изопропилакриламида (патентный документ 11); или двухкомпонентный гель, состоящий из полиэтиленгликоля и т.п. и поликарбокси полисахарида (патентный документ 12); гель, состоящий из 2 типов полиэтиленгликолей и т.п. (патентный документ 13); или поперечно-сшитый ионный гель, например, карбоксиметилхитозан (патентный документ 14).
Сополимер поли(этиленгликоль/пропиленгликоль) (патентный документ 15), сополимер поли(молочная кислота/диоксанон) (патентный документ 16), сополимер поли(этиленгликоль/ модифицированная аминокислота/немодифицированная аминокислота) (патентный документ 17), сополимер поли(молочная кислота/депсипептид/этиленгликоль) (патентный документ 18), пористый лист, состоящий из сополимера поли(молочная кислота/этиленгликоль) (патентный документ 19) и т.д. также применялись, в качестве биоразрушаемого материала, например, антиадгезионного материала, перевязочного материала, кровоостанавливающего материала или материала для предупреждения недержания мочи; кроме того, предпринимались попытки регулировать способность материала к разрушению под действием биологической среды и его эластичность.
ССЫЛКИ НА ИСТОЧНИКИ ИЗВЕСТНОГО УРОВНЯ ТЕХНИКИ
Патентные документы
Патентный документ 1: JP 5-969 A
Патентный документ 2: JP 5-17245 B
Патентный документ 3: JP 2004-167229 A
Патентный документ 4: JP 2005-312623 A
Патентный документ 5: JP 2007-291323 A
Патентный документ 6: US 2009/0117033 A
Патентный документ 7: JP 2007-146146 A
Патентный документ 8: JP 4 655505 B
Патентный документ 9: JP 2007-145826 A
Патентный документ 10: JP 3107514 B
Патентный документ 11: JP 2003-252936 A
Патентный документ 12: JP 2003-531682 A
Патентный документ 13: JP 2002-541923 A
Патентный документ 14: JP 7-90041 B
Патентный документ 15: WO 96/21056
Патентный документ 16: JP 3483753 B
Патентный документ 17: JP 4735260 B
Патентный документ 18: JP 4734772 B
Патентный документ 19: JP 2008-36134 A
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Проблемы, которые предполагалось решить настоящим изобретением
Хотя усовершенствованные методики, такие как смешивание полимеров нескольких типов (патентный документ 7), применение химически сшитых полимерных частиц (патентный документ 8) и покрытие поверхности полимерных частиц (патентный документ 9), были способны улучшить регулирование эластичности полимерных частиц или их способности проходить через катетер, не удалось добиться существенного улучшения в отношении проблемы необратимой деформации полимерных частиц. Для достижения достаточного эффекта эмболизации кровеносных сосудов и т.п. необходимы дополнительные усовершенствования. Конкретно, имелась необходимость разработки материала для эмболизации кровеносных сосудов и т.п., например, полимерных частиц с высокой способностью восстанавливать свою исходную форму после прохождения через катетер (далее по тексту именуемой «степень восстановления формы частиц»).
Далее, хотя были достигнуты определенные успехи в улучшении биоразрушаемости или эластичности материалов, например, антиадгезионных материалов, перевязочных материалов, кровоостанавливающих материалов или материалов для предотвращения недержания мочи, биоразрушаемые материалы, состоящие, например, из двухкомпонентных гелей сталкиваются с той проблемой, что их физические свойства могут меняться в зависимости от факторов окружающей среды (например, температуры, влажности или pH) или соотношения компонентов смеси на целевом участке. Кроме того, поскольку поверхность органа или ткани, поврежденная при хирургическом вмешательстве, испытывает постоянное растяжение и сжатие, нанесенный на них биоразрушаемый материал может подвергаться необратимой деформации. В отношении этой проблемы, не удалось добиться существенного улучшения традиционных биоразрушаемых материалов, и существует потребность в разработке материала, например, антиадгезионного материала, перевязочного материала, кровоостанавливающего материала или материала для предотвращения недержания мочи, имеющего стабильные физические свойства и высокую степень восстановления формы.
Соответственно, цель настоящего изобретения заключалась в разработке биоразрушаемого материала с улучшенной степенью восстановления формы после деформации материала и улучшенной эластичностью.
Средства для решения указанных проблем
Настоящее изобретение конкретно относится к биоразрушаемому материалу и способу его получения, которые описаны ниже в пп. 1-15.
1. Биоразрушаемый материал, который представляет собой сшитый поперечными химическими связями продукт: мультивалентного соединения A, включающего 3 или более функциональные группы X, выбранные из группы, состоящей из гидроксильной группы, тиольной группы и аминогруппы; мультивалентного соединения B, включающего 3 или более функциональные группы Y, выбранные из группы, состоящей из карбоксильной группы, изоцианатной группы и тиоизоцианатной группы; а также соединения C, имеющего структуру, образованную из гидроксикарбоновой кислоты, гомополимер которой, полученный при гомополимеризации, имеет температуру стеклования -40°C или ниже.
2. Биоразрушаемый материал по п. 1, где массовая доля структуры, образовавшейся из соединения C, составляет от 18 до 70 мас.%.
3. Биоразрушаемый материал по пп. 1 или 2, где мультивалентное соединение A представляет собой одно из следующих соединений a)-e):
a) гомополимер или сополимер мономера(ов) водорастворимого полимера(ов), выбранного из группы, состоящей из полиэтиленгликоля, полипропиленгликоля, поливинилового спирта, полигидроксиэтилакрилата, полигидроксиэтилэтилметакрилата, карбоксиметилцеллюлозы, гидроксиметилцеллюлозы и гидроксиэтилцеллюлозы;
b) сополимер мономера водорастворимого полимера и мономера(ов) гидрофобного полимера(ов), выбранного из группы, состоящей из винилацетата и винилкапролактама;
c) сополимер мономера водорастворимого полимера и гидроксикарбоновой кислоты(кислот), гомополимер(ы) которых, образующийся при гомополимеризации, имеет/имеют температуру стеклования -39°C или выше;
d) разветвленный полимер, образованный связыванием всех гидроксильных групп полиола с гомополимером или сополимером мономера(ов) водорастворимого полимера(ов), выбранного из группы, состоящей из полиэтиленгликоля и полипропиленгликоля;
e) сополимер разветвленного полимера и гидроксикарбоновой кислоты(кислот), гомополимер(ы) которой, образующийся при гомополимеризации, имеет/имеют температуру стеклования -39°C или выше.
4. Биоразрушаемый материал по любому из пп. 1-3, где мультивалентное соединение B представляет собой одно из следующих соединений f)-i):
f) соединение, образованное при связывании гидроксильной группы(групп) гомополимера или сополимера мономера(ов) водорастворимого полимера(ов), выбранного из группы, состоящей из полиэтиленгликоля, полипропиленгликоля, поливинилового спирта, полигидроксиэтилакрилата, полигидроксиэтилметакрилата, карбоксиметилцеллюлозы, гидроксиметилцеллюлозы и гидроксиэтилцеллюлозы с поликарбоновой кислотой(ами);
g) соединение, образованное при связывании гидроксильной группы(групп) сополимера мономера водорастворимого полимера и гидроксикарбоновой кислоты(кислот), гомополимер(ы) которой, образующийся при гомополимеризации, имеет/имеют температуру стеклования -39°C или выше, с поликарбоновой кислотой(ами);
h) соединение, образованное при связывании гидроксильной группы(групп) разветвленного полимера, образованного при связывании всех гидроксильных групп полиола с гомополимером или сополимером мономера(ов) водорастворимого полимера(ов), выбранного из группы, состоящей из полиэтиленгликоля и полипропиленгликоля, с поликарбоновой кислотой(ами);
i) соединение, образованное связыванием гидроксильной группы(групп) сополимера разветвленного полимера и гидроксикарбоновой кислоты(кислот), гомополимер(ы) которой, образующийся при гомополимеризации, имеет/имеют температуру стеклования -39°C или выше, с поликарбоновой кислотой(ами).
5. Биоразрушаемый материал по любому из пп. 1-4, где соединение C представляет собой сополимер мономера(ов) водорастворимого полимера(ов), выбранного из группы, состоящей из полиэтиленгликоля, полипропиленгликоля, поливинилового спирта, полигидроксиэтилакрилата, полигидроксиэтилметакрилата, карбоксиметилцеллюлозы, гидроксиметилцеллюлозы и гидроксиэтилцеллюлозы, и гидроксикарбоновой кислоты(кислот), гомополимер(ы) которой, полученный при гомополимеризации, имеет/имеют температуру стеклования -40°C или ниже.
6. Биоразрушаемый материал по любому из пп. 1-5, где соединение C представляет собой 6-гидроксикапроновую кислоту (6-гидроксигексановую кислоту).
7. Биоразрушаемый материал по любому из пп. 3-6, где разветвленный полимер имеет степень разветвления от 3 до 16.
8. Биоразрушаемый материал по любому из пп. 3-7, где полиол выбран из группы, состоящей из глицерина, полиглицерина и пентаэритрита.
9. Биоразрушаемый материал по любому из пп. 3-8, где гидроксикарбоновая кислота(кислоты), гомополимер(ы) которой, образующийся при гомополимеризации, имеет/имеют температуру стеклования -39°C или выше, выбрана(ы) из группы, состоящей из гликолевой кислоты, молочной кислоты, глицериновой кислоты, гидроксимасляной кислоты, яблочной кислоты, винной кислоты, гидроксивалериановой кислоты и 3-гидроксигексановой кислоты.
10. Биоразрушаемый материал по любому из пп. 4-9, где поликарбоновая кислота(кислоты) выбрана/выбраны из группы, состоящей из щавелевой кислоты, малоновой кислоты, янтарной кислоты, глутаровой кислоты, адипиновой кислоты, пимелиновой кислоты, пробковой кислоты, азелаиновой кислоты, себациновой кислоты, яблочной кислоты, винной кислоты и фумаровой кислоты.
11. Материал для эмболизации сосудов, состоящий из биоразрушаемого материала по любому из пп. 1-10.
12. Антиадгезионный материал, состоящий из биоразрушаемого материала по любому из пп. 1-10.
13. Материал для перевязки ран, состоящий из биоразрушаемого материала по любому из пп. 1-10.
14. Кровоостанавливающий материал, состоящий из биоразрушаемого материала по любому из пп. 1-10.
15. Материал, предупреждающий недержание мочи, состоящий из биоразрушаемого материала по любому из пп. 1-10.
16. Способ получения биоразрушаемого материала, где указанный способ включает стадию поперечного сшивания, в которой мультивалентное соединение A, включающее 3 или более функциональные группы X, выбранные из группы, состоящей из гидроксильной группы, тиольной группы и аминогруппы, мультивалентное соединение B, включающее 3 или более функциональные группы Y, выбранные из группы, состоящей из карбоксильной группы, изоцианатной группы и тиоизоцианатной группы, а также соединение C, имеющее структуру, образованную из гидроксикарбоновой кислоты, гомополимер которого, полученный при гомополимеризации, имеет температуру стеклования -40°C или ниже, растворяют в растворителе, который позволяет проводить химическую реакцию поперечного сшивания, с получением биоразрушаемого материала.
ЭФФЕКТ ИЗОБРЕТЕНИЯ
Биоразрушаемый материал по настоящему изобретению имеет улучшенную биоразрушаемость и улучшенную степень восстановления формы после деформации материала, и может успешно применяться в качестве материала для эмболизации сосудов, который, например, можно легко доставить к целевому участку кровеносного сосуда или другого органа без забивания катетера, и который претерпевает быстрое разрушение в биологической среде и рассасывается после эффективной эмболизации целевого участка. Далее, поскольку биоразрушаемый материал по настоящему изобретению имеет улучшенную прочность на разрыв и прочность при сдвиге, и способен восстанавливать свою форму после деформации от растягивающего или сдвигающего усилия, его можно успешно применять в качестве такого материала, как антиадгезионный материал, перевязочный материал, кровоостанавливающий материал или материал, предупреждающий недержание мочи, например, наклеивая на орган или окружающую ткань, которая постоянно претерпевает растяжение и сжатие.
ПОДРОБНОЕ ОПИСАНИЕ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Используемые в заявке термины соответствуют данным ниже определениям, если не указано иное.
Биоразрушаемый материал по настоящему изобретению характеризуется тем, что он является сшитым поперечными химическими связями продуктом, состоящим из: мультивалентного соединения A, включающего 3 или более функциональные группы X, выбранные из группы, состоящей из гидроксильной группы, тиольной группы и аминогруппы; мультивалентного соединения B, включающего 3 или более функциональные группы Y, выбранные из группы, состоящей из карбоксильной группы, изоцианатной группы и тиоизоцианатной группы; а также соединения C, имеющего структуру, образованную из гидроксикарбоновой кислоты, гомополимер которой, полученный при гомополимеризации, имеет температуру стеклования -40°C или ниже.
Термин «биоразрушаемый» относится к способности биоразрушаемого материала разрушаться, растворяться, поглощаться или подвергаться метаболизму в живом организме, или выводиться из организма за его пределы. Примеры реакций разрушения материала включают гидролиз и разрушение под действием ферментов. Предпочтительным путем является гидролиз, т.к. он не зависит от ферментов.
Термин «сшитый поперечными химическими связями» относится к связыванию мультивалентного соединения A, мультивалентного соединения B и соединения C с использованием реагента, способствующего образованию поперечных связей (кросс-линкера). Примеры связей включают сложноэфирные связи, тиоэфирные связи, амидные связи и т.п. Предпочтительными являются сложноэфирные связи, поскольку они обеспечивают повышенную способность материала к биоразрушению. Кросс-линкер предпочтительно представляет собой реагент, осуществляющий дегидратацию-конденсацию. Наличие «сшивания поперечными химическими связями» можно подтвердить отсутствием изменения внешнего вида биоразрушаемого материала после погружения материала в воду на 1 час при температуре 25°C.
Массовая доля структуры, образовавшейся из упомянутого соединения C, в биоразрушаемом материале по настоящему изобретению предпочтительно составляет от 18 до 70 мас.%. Для улучшения эластичности и биоразрушаемости полученного биоразрушаемого материала, эта массовая доля, более предпочтительно, составляет от 20 до 65 мас.%.
Примеры «мультивалентного соединения A» включают:
i) гомополимер или сополимер, состоящий из мономера(ов) водорастворимого полимера(ов), выбранного из группы, состоящей из полиэтиленгликоля (далее по тексту именуемого «ПЭГ»), полипропиленгликоля (далее по тексту именуемого «ППГ»), поливинилового спирта (далее по тексту именуемого «PVA»), полигидроксиэтилакрилата, полигидроксиэтилэтилметакрилата, карбоксиметилцеллюлозы, гидроксиметил целлюлозы и гидроксиэтилцеллюлозы;
ii) сополимер мономера водорастворимого полимера и мономера(ов) гидрофобного полимера(ов), выбранного из группы, состоящей из винилацетата и винилкалролактама;
iii) сополимер мономера водорастворимого полимера и гидроксикарбоновой кислоты(кислот), гомополимер(ы) которой, образующийся при гомополимеризации, имеет/имеют температуру стеклования -39°C или выше;
iv) разветвленный полимер, образующийся при связывании всех гидроксильных групп полиола с гомополимером или сополимером мономера(ов) водорастворимого полимера(ов), выбранного из группы, состоящей из ПЭГ и ППГ; и
v) сополимер разветвленного полимера и гидроксикарбоновой кислоты(кислот), гомополимер(ы) которой, образующийся при гомополимеризации, имеет/имеют температуру стеклования -39°C или выше.
Мультивалентное соединение A содержит 3 или более функциональные группы X, выбранные из группы, состоящей из гидроксильной группы, тиольной группы и аминогруппы. Производные мультивалентного соединения A, например, галогенангидриды, сложные эфиры, ангидриды и гидрохлориды, также включены в число мультивалентных соединений А.
Для достижения устойчивого химического сшивания мультивалентного соединения A с мультивалентным соединением В и соединением С, и для улучшения биосовместимости полученного биоразрушаемого материала «водорастворимый полимер» предпочтительно является полиалкиленгликолем, например, ПЭГ или ППГ; полигидроксиалкил(мет)акрилатом, например, PVA, полигидроксиэтилметакрилатом или полигидроксиэтилакрилатом; или производным целлюлозы, например, карбоксиметилцеллюлозой, гидроксиметилцеллюлозой или гидроксиэтилцеллюлозой; более предпочтительно, полиалкиленгликолем.
Для увеличения плотности химического сшивания получаемого биоразрушаемого материала, мультивалентное соединение A предпочтительно является разветвленным соединением, например, разветвленным полимером (разветвленным полимером a1), образованным при связывании всех гидроксильных групп полиола с гомополимером или сополимером, состоящим из мономера(ов) водорастворимого полимера(ов), выбранного из группы, состоящей ПЭГ и ППГ, более предпочтительно, сополимера разветвленного полимера и гидроксикарбоновой кислоты(кислот) (гидроксикарбоновой кислоты a2), еще более предпочтительно, блок-сополимера, в котором гидроксикарбоновая кислота(кислоты) связана/связаны с концом(ами) разветвленного полимера. Полиол предпочтительно является глицерином, полиглицерином или моносахаридом, например, пентаэритритом.
Примеры «мультивалентного соединения B» включают:
i) соединение, образованное при связывании гидроксильной группы(групп) гомополимера или сополимера мономера(ов) водорастворимого полимера(ов), выбранного из группы, состоящей из ПЭГ, ППГ, PVA, полигидроксиэтилакрилата, полигидроксиэтилметакрилата, карбоксиметилцеллюлозы, гидроксиметилцеллюлозы и гидроксиэтилцеллюлозы с поликарбоновой кислотой(ами);
ii) соединение, образованное при связывании гидроксильной группы(групп) сополимера мономера водорастворимого полимера и гидроксикарбоновой кислоты(кислот), гомополимер(ы) которой, образующийся при гомополимеризации, имеет/имеют температуру стеклования -39°C или выше, с поликарбоновой кислотой(ами);
iii) соединение, образованное при связывании гидроксильной группы(групп) разветвленного полимера, образованного при связывании всех гидроксильных групп полиола с гомополимером или сополимером мономера(ов) водорастворимого полимера(ов), выбранного из группы, состоящей из ПЭГ и ППГ, с поликарбоновой кислотой(ами); и
vi) соединение, образованное при связывании гидроксильной группы(групп) сополимера разветвленного полимера и гидроксикарбоновой кислоты(кислот), гомополимер(ы) которой, образующийся при гомополимеризации, имеет/имеют температуру стеклования -39°C или выше, с поликарбоновой кислотой(ами).
Мультивалентное соединение B содержит 3 или более функциональные группы Y, выбранные из группы, состоящей из карбоксильной группы, изоцианатной группы и тиоизоцианатной группы. Производные мультивалентного соединения B, такие как галогенангидриды, сложные эфиры и ангидриды кислот также входят в число мультивалентных соединений В.
В качестве поликарбоновой кислоты, которая является одним из компонентов мультивалентного соединения B, предпочтительными с точки зрения доступности являются дикарбоновые кислоты, такие как щавелевая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, глутаровая кислота, адипиновая кислота, пимелиновая кислота, пробковая кислота, азелаиновая кислота, себациновая кислота, яблочная кислота, винная кислота или додекандиовая кислота; или лимонная кислота. Более предпочтительной является янтарная кислота, которая присутствует в живом организме и является весьма безопасной.
Мультивалентное соединение B предпочтительно является разветвленным соединением, например, соединением, образовавшимся при связывании гидроксильной группы(групп) разветвленного полимера (разветвленного полимера b1), образованного при связывании всех гидроксильных групп полиола с гомополимером или сополимером мономера(ов) водорастворимого полимера(ов), выбранного из группы, состоящей из ПЭГ и ППГ, с поликарбоновой кислотой(ами) (поликарбоновой кислотой b2). Полиол предпочтительно представляет собой глицерин, полиглицерин или моносахарид, например, пентаэритрит.
«Гидроксикарбоновая кислота», которая является одним из компонентов мультивалентного соединения A, мультивалентного соединения B, а также соединения C, включает циклические соединения, например, циклические димеры гидроксикарбоновых кислот. Производные гидроксикарбоновых кислот, такие как галогенангидриды, сложные эфиры и ангидриды кислот также включены в число гидроксикарбоновых кислот. Что касается гидроксикарбоновых кислот, имеющих оптические изомеры, например, яблочной кислоты и винной кислоты, термин «гидроксикарбоновая кислота» включает все их D-изомеры, L-изомеры, а также их смеси. Далее, гидроксикарбоновые кислоты включают сополимеры, образованные в результате сополимеризации этих гидроксикарбоновых кислот. Примеры гидроксикарбоновых кислот включают гликолевую кислоту, молочную кислоту, глицериновую кислоту, гидроксимасляную кислоту, яблочную кислоту, винную кислоту, гидроксивалериановую кислоту, 3-гидроксигексановую кислоту и 6-гидроксикапроновую кислоту. Примеры циклических соединений, состоящих из гидроксикарбоновых кислот, включают гликолид, который является циклическим димером гликолевой кислоты, лактид, который является циклическим димером молочной кислоты и s-капролактон, который соответствует 6-гидроксикапроновой кислоте. Примеры сополимеров, образующихся при сополимеризации гидроксикарбоновых кислот, включают сополимеры молочной кислоты и гликолевой кислоты, сополимеры молочной кислоты и терефталевой кислоты, сополимеры молочной кислоты и изофталевой кислоты, сополимеры 6-гидроксикапроновой кислоты и гликолевой кислоты, а также сополимеры 6-гидроксикапроновой кислоты и полибутилен сукцината (сополимера 1,4-бутандиола и янтарной кислоты). Гидроксикарбоновая кислота, гомополимер которой, образующийся при гомополимеризации, имеет температуру стеклования -39°C или выше, предпочтительно является молочной кислотой.
Примеры «гидроксикарбоновой кислоты, гомополимер которой, образующийся при гомополимеризации, имеет температуру стеклования -40°C или ниже», которая является компонентом «соединения C», включают 6-гидроксикапроновую кислоту, сополимеры 6-гидроксикапроновой кислоты и гликолевой кислоты, а также сополимеры 6-гидроксикапроновой кислоты и полибутилен сукцината (сополимера 1,4-бутандиола и янтарной кислоты). Предпочтительной является 6-гидроксикапроновая кислота. Термин «гомополимер» относится к полимеру, образующемуся при полимеризации одного типа мономера, например, полимолочной кислоте, образующейся при полимеризации чистой молочной кислоты. Однако понятие «гомополимер, образующийся при гомополимеризации» в настоящем изобретении охватывает также полимеры, образовавшиеся при полимеризации сополимеров одного типа, например, сополимера молочной кислоты и гликолевой кислоты.
Для увеличения плотности сшивания поперечными химическими связями соединения C с мультивалентным соединением А и мультивалентным соединением В, гидроксикарбоновая кислота, гомополимер которой, образующийся при гомополимеризации, имеет температуру стеклования -40°C или ниже, предпочтительно является сополимером мономера(ов) водорастворимого полимера(ов), выбранного из группы, состоящей из ПЭГ, ППГ, PVA, полигидроксиэтилакрилата, полигидроксиэтилметакрилата, карбоксиметилцеллюлозы, гидроксиметилцеллюлозы и гидроксиэтилцеллюлозы и гидроксикарбоновой кислоты(кислот), гомополимер(ы) которой, образующийся при гомополимеризации, имеет/имеют температуру стеклования -40°C или ниже; более предпочтительно, сополимером разветвленного полимера, образующегося при связывании всех гидроксильных групп полиола с гомополимером или сополимером мономера(ов) водорастворимого полимера(ов), выбранного из группы, состоящей из ПЭГ и ППГ, и гидроксикарбоновой кислоты(кислот), гомополимер(ы) которой, образующийся при гомополимеризации, имеет/имеют температуру стеклования -40°C или ниже.
В случаях, когда мультивалентное соединение A, мультивалентное соединение B и соединение C являются разветвленными соединениями, они предпочтительно имеют степень разветвления от 3 до 16, более предпочтительно, от 4 до 12. В то время, как слишком низкая степень разветвления приводит к невозможности увеличить плотность сшивания химическими связями и обеспечить достаточную прочность биоразрушаемого материала, слишком высокая степень разветвления может помешать реакции образования поперечных химических связей из-за пространственных препятствий.
В случаях, когда мультивалентное соединение A, мультивалентное соединение B и соединение C являются сополимерами, они могут быть полимером любого из следующих типов: статистическим сополимером, блок-сополимером или чередующимся сополимером. Однако предпочтительно, они являются блок-сополимерами, поскольку механические и другие свойства получаемого биоразрушаемого материала можно легко регулировать и можно улучшить его эластичность и биоразрушаемость. Термин «сополимер» в данном описании относится к высокомолекулярному соединению, образовавшемуся при сополимеризации двух или нескольких типов мономеров. Термин «блок-сополимер» относится к сополимеру, в котором ковалентно связаны как минимум два или несколько типов полимеров, состоящих из различных повторяющихся звеньев, с образованием молекулярной структуры, напоминающей длинную цепь, где термин «блок» относится к каждому из «как минимум двух или нескольких типов полимеров, состоящих из различных повторяющихся звеньев», составляющих блок-сополимер.
Массовая доля структуры, образовавшейся из гидроксикарбоновой кислоты(кислот), гомополимер(ы) которой, образующийся при гомополимеризации, имеет/имеют температуру стеклования -39°C или выше в упомянутом выше мультивалентном соединении А предпочтительно составляет от 10 до 300 мас.%. Для достижения достаточной эластичности и биоразрушаемости получаемого биоразрушаемого материала, это массовое соотношение предпочтительно составляет от 30% до 250 мас.%, еще более предпочтительно от 40 до 200 мас.%.
Массовая доля структуры, образовавшейся из гидроксикарбоновой кислоты(кислот), гомополимер(ы) которой, образующийся при гомополимеризации, имеет/имеют температуру стеклования -40°C или ниже в упомянутом выше соединении С предпочтительно составляет от 10 до 300 мас.%. Для достижения достаточной эластичности и биоразрушаемости получаемого биоразрушаемого материала, это массовое соотношение предпочтительно составляет от 30% до 250 мас.%, еще более предпочтительно от 40 до 200 мас.%.
Если среднемассовые молекулярные массы мультивалентного соединения A, мультивалентного соединения B и соединения C будут слишком низкими, скорость биологического разрушения биоразрушаемого материала увеличится слишком сильно, и не удастся получить необходимый эффект эмболизации, например, при эмболизации сосудов. С другой стороны, если среднемассовые молекулярные массы мультивалентного соединения A, мультивалентного соединения B и соединения C будут слишком высокими, понизится способность биоразрушаемого материала к разрушению. Поэтому среднемассовые молекулярные массы упомянутых мультивалентного соединения A, мультивалентного соединения B и соединения C предпочтительно находятся в пределах от 1000 до 50000, более предпочтительно от 3000 до 30000. Среднемассовые молекулярные массы упомянутых мультивалентного соединения A, мультивалентного соединения B и соединения C можно измерить с помощью гель-проникающей хроматографии (далее по тексту именуемой «методика GPC») в следующих условиях:
Условия измерения:
Аппарат (колонка): TSKgel GMHHR-M (производства Tosoh Corporation; внутренний диаметр: 7,8 мм; длина: 30 см; две колонки расположены линейно);
Элюент: хлороформ;
Температура колонки: 35°C;
Скорость потока: 1,0 мл/мин;
Способ детектирования: показатель преломления;
Калибровочная кривая: строили с использованием стандартных образцов полистирола.
Предпочтительно, чтобы величина NB/(NA+NC) имела значение от 1,2 до 4,0, где МВ≥МАС и величина (NA+NC)/NB имела значение от 1,2 до 4,0, где МВ<МАС, более предпочтительно, чтобы значения указанных величин находились в пределах от 1,3 до 3,0, еще более предпочтительно от 1,4 до 2,5, где NA означает число молей функциональных групп X, содержащихся в мультивалентном соединении A; NB означает число молей функциональных групп Y, содержащихся в мультивалентном соединении В; и NC означает число молей функциональных групп соединения C, выбранных из группы, состоящей из гидроксильной группы, тиольной группы, аминогруппы, карбоксильной группы, изоцианатной группы и тиоизоцианатной группы; и где переменные величины определяются указанным ниже образом:
MA: среднемассовая молекулярная масса мультивалентного соединения A;
MB: среднемассовая молекулярная масса мультивалентного соединения B;
MC: среднемассовая молекулярная масса соединения C;
MAC: среднемассовая молекулярная масса смеси мультивалентного соединения A и соединения C.
Согласно знаниям известного уровня техники (JP 2007-145826 A), равенство значений NB и (NA+NC) увеличит до максимума количество сформировавшихся связей, т.е. максимально повысит плотность поперечного сшивания, в результате чего теоретически не останется не вступивших в реакцию функциональных групп. Однако в настоящем изобретении предпочтительно, чтобы одна из величин NB и (NA+NC) превышала другую, находясь в оптимальном диапазоне относительно нее.
Массовые доли и прочие параметры конкретных структур в каждом компоненте из числа мультивалентного соединения A, мультивалентного соединения B и соединения C можно вычислить на основании результатов измерений, полученных с применением методики ядерного магнитного резонанса (далее по тексту именуемой «1H-ЯМР») в указанных ниже условиях. Например, в случае, если гидроксикарбоновая кислота является молочной кислотой, для определения указанных параметров может использоваться каждый атом водорода метиновой группы в α-положении (величина химического сдвига: примерно 5,2 м.д.). В случае, если гидроксикарбоновая кислота представляет собой 6-гидроксикарбоновую кислоту, для определения указанных параметров может использоваться атом водорода метиленовой группы в соположении (величина химического сдвига: примерно 2,3 м.д.). В случае, если гидроксикарбоновая кислота представляет собой гликолевую кислоту, для определения указанных параметров может использоваться атом водорода метиленовой группы в α-положении (величина химического сдвига: примерно 4,8 м.д.). С другой стороны, что касается ПЭГ, для определения указанных параметров могут использоваться 4 атома водорода этиленовых групп (величина химического сдвига: примерно 3,5 м.д.). Каждую массовую долю можно вычислить исходя из интегральных значений сигнала, появляющегося при каждом из указанных характеристических химических сдвигов атомов водорода.
Условия регистрации спектров ЯМР
Спектрометр: JNM-EX270 (производства JEOL Ltd., 270 МГц);
Растворитель: дейтерированный хлороформ (содержащий 0,05 об.% TMS в качестве внутреннего стандарта);
Температура регистрации спектра: 20°C.
В случаях, если биоразрушаемый материал по настоящему изобретению получают в виде ацетонитрил-содержащей пленки, комплексный модуль упругости этого материала предпочтительно составляет от 40 до 400 кПа. Комплексный модуль упругости можно вычислить, исходя из результатов измерений, полученных на устройстве для измерения вязкоупругих свойств (далее по тесту именуемого «реометром») при указанных ниже условиях.
Конкретно, указанные количества мультивалентного соединения A, мультивалентного соединения B и соединения C (все в виде 0,3 г/мл растворов в ацетонитриле), а также катализатор (0,1 г/мл раствор в ацетонитриле) и концентрированный раствор конденсирующего агента быстро смешивали с получением смешанного раствора. Затем 500 мкл смешанного раствора капали на пластину устройства, помещая смешанный раствор между неподвижной и подвижной пластинами устройства, и осуществляли тест на динамическую вязкоупругость в течение 105 с после смешивания.
Условия проведения измерений
Наименование теста: тест на динамическую вязкоупругость;
Прибор (реометр): MCR301 (производства Anton Parr Ltd.);
Неподвижная пластина: CP40-1 (диаметр: 39,958 мм; угол: 1°);
Зазор: 0,081 мм (расстояние между неподвижной пластиной и пластиной устройства, между которыми помещают образец);
Деформация: 0,1% (постоянная);
Угловая скорость: 10 рад/с (постоянная);
Температура измерения: 25°C;
Время измерения: 18000 с.
Термин «ацетонитрил-содержащая пленка» относится к пленке, сформированной поперечным сшиванием химически связями мультивалентного соединения A, мультивалентного соединения B и соединения C, в которой по-прежнему содержится ацетонитрил, причем указанная пленка получена после измерения реометром.
Термин «комплексный модуль упругости» означает величину, отображающую эластичность биоразрушаемого материала и относящуюся к значению модуля Е* (кПа), вычисленному по приведенному ниже уравнению 1, причем эта величина описывает как упругие так и вязкие свойства измеряемого образца, который является вязкоупругим телом.
Конкретно, в случаях, когда биоразрушаемый материал по настоящему изобретению применяется в качестве материала для эмболизации сосудов, слишком низкое значение комплексного модуля упругости приводит к невозможности сохранения формы биоразрушаемого материала и неспособности обеспечить желаемый эффект эмболизации; в то же время слишком высокое значение комплексного модуля упругости увеличивает сопротивление биоразрушаемого материала при прохождении через катетер или аналогичные устройства. В случае, когда биоразрушаемый материал по настоящему изобретению применяется в качестве антиадгезионного материала, перевязочного материала, кровоостанавливающего материала, материала, предотвращающего недержание мочи и т.п., слишком низкое значение комплексного модуля упругости приводит к неспособности сохранять форму биоразрушаемого материала и обеспечивать желаемое антиадгезионное действие на орган или окружающую ткань; тогда как слишком высокое значение комплексного модуля упругости слишком сильно затрудняет колебательные движения органа или окружающей ткани.
Конкретно, что касается биоразрушаемого материала по настоящему изобретению, комплексный модуль упругости ацетонитрил-содержащей пленки при постоянной деформации 0,1% и постоянной угловой скорости 10 рад/с предпочтительно составляет от 40 до 400 кПа, более предпочтительно от 100 до 300 кПа.

Е′: динамический модуль упругости (кПа);
Е′′: модуль потерь (кПа);
i: мнимая единица.
Термин «динамический модуль упругости» в настоящей заявке относится к компоненту комплексного модуля упругости, совпадающему по фазе с приложенным усилием (к реальной части комплексного модуля упругости), измеренному при бесконечно малых деформациях вязкоупругого тела при постоянном усилии и постоянной угловой частоте, и представляет собой величину, отражающую упругие свойства измеряемого образца. Что касается биоразрушаемого материала по настоящему изобретению, динамический модуль упругости ацетонитрил-содержащей пленки при постоянной деформации 0,1% и постоянной угловой частоте 10 рад/с предпочтительно составляет от 40 до 400 кПа, более предпочтительно, от 100 до 300 кПа. С другой стороны, термин «модуль потерь» относится к компоненту, отличающемуся по фазе от приложенного усилия на π/2 (мнимая часть комплексного модуля упругости), и представляет собой величину, отражающую вязкие свойства измеряемого образца.
В тесте на динамическую вязкоупругость можно также оценить время гелеобразования, которое представляет собой время, необходимое биоразрушаемому материалу для превращения в гель. Термин «время гелеобразования» относится ко времени (c), необходимому для того, чтобы динамический модуль упругости и модуль потерь достигли одинаковых значений, т.е. времени достижения тангенсом угла потерь значения 1 (tanδ=1). Время гелеобразования ацетонитрил-содержащей пленки при постоянной деформации 0,1% и постоянной угловой частоте 10 рад/с предпочтительно составляет от 100 до 1000 с, более предпочтительно, от 200 до 800 с. «Тангенс угла потерь» в настоящей заявке относится к величине, отражающей эластичность биоразрушаемого материала и способность деформированной ацетонитрил-содержащей пленки восстанавливать исходную форму, и соответствует величине tanδ, рассчитанной по приведенному ниже уравнению 2. Tanδ является безразмерной величиной, которая отражает способность ацетонитрил-содержащей пленки поглощать приложенную энергию при деформации и превращать эту энергию в тепло.

В случаях, когда биоразрушаемый материал по настоящему изобретению получают в виде биоразрушаемой пленки, усилие сжатия пленки на 50% в состоянии насыщения водой представляет собой величину, отражающую эластичность биоразрушаемого материала. Термин «биоразрушаемая пленка» в настоящей заявке относится к пленке, полученной при растворении мультивалентного соединения A, мультивалентного соединения B и соединения C в растворителе с последующим проведением химической реакции поперечного сшивания при удалении растворителя.
Термин «состояние насыщения водой» относится к состоянию, когда примерно 20 мг биоразрушаемой пленки погружают в 10 мл фосфатного буферного солевого раствора при 37°C (при вращении тестовой пробирки на ротаторе со скоростью 0,5 оборотов/секунду для перемешивания содержимого), и содержание воды в биоразрушаемой пленке стало постоянным. Выражение «содержание воды является постоянным» относится к состоянию, когда скорость изменения массы биоразрушаемой пленки, погруженной в фосфатный буферный солевой раствор при 37°C, при измерении каждую минуту со временем становится равной 10% или менее. Скорость изменения массы со временем представляет собой величину Rw(%), рассчитанную по приведенному ниже уравнению 3.
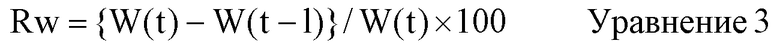
W(t): масса (г) биоразрушаемой пленки после погружения в воду на t минут;
W(t-1): масса (г) биоразрушаемой пленки после погружения в воду на (t-1) минут.
Термин «содержание воды» относится к величине Wr(%), вычисленной по приведенному ниже уравнению 4. «Биоразрушаемая пленка в сухом состоянии» в настоящей заявке относится к биоразрушаемой пленке, которую погрузили в деионизированную воду при 25°C на 3 часа и затем высушили в вакууме при 25°C в течение 12 часов. «Биоразрушаемая пленка в состоянии насыщения водой» относится к биоразрушаемой пленке, которую для удаления фосфатного буферного солевого раствора подвергали центрифугированию (25°C, 1000 g × 5 минут) после того, как содержание воды в образце становилось постоянным. Содержание воды в биоразрушаемой пленке повышается за счет впитывания воды в пленку. Чем выше плотность химического сшивания биоразрушаемого материала, тем более ограниченным становится впитывание в биоразрушаемую пленку. Конкретно, поскольку существует корреляция между содержанием воды и плотностью поперечного сшивания химическими связями в биоразрушаемом материале по настоящему изобретению, содержание воды в состоянии насыщения водой может использоваться в качестве показателя степени поперечного сшивания химическими связями.
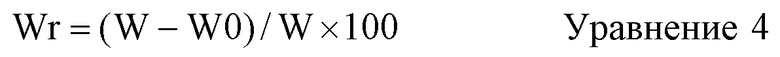
W: масса биоразрушаемой пленки в состоянии насыщения водой;
W0: масса биоразрушаемой пленки в сухом состоянии (стандартно примерно 20 мг).
«Усилие сжатия на 50%» представляет собой величину, отображающую эластичность биоразрушаемого материала, и соответствует усилию, необходимому для сжатия одного слоя биоразрушаемой пленки до 50% от ее исходной толщины. Слишком низкое значение усилия сжатия пленки на 50% приводит к потере формы биоразрушаемого материала, а слишком высокое значение усилия сжатия пленки на 50% вызывает проблемы, связанные с сопротивлением при прохождении материала через катетер. Поэтому, что касается биоразрушаемого материала по настоящему изобретению, усилие сжатия биоразрушаемой пленки на 50% в состоянии насыщения водой предпочтительно составляет от 10 до 100 мН, более предпочтительно, от 20 до 80 мН.
Усилие сжатия биоразрушаемой пленки на 50% в состоянии насыщения водой можно измерить с помощью устройства для измерения микроусилий, используя следующие условия. Конкретно, усилие (изменение усилия) прилагают к каждому образцу биоразрушаемой пленки, описанному выше, и измеряют усилие, необходимое для сжатия пленки до 50% от ее исходной толщины.
Условия проведения измерения
Наименование теста: тест на сжатие;
Прибор: Micro Auto Model MST-I (производства Shimadzu Corporation);
Методика измерения: методика с поперечной головкой;
Условия окружающей среды: комнатная температура, на открытом воздухе;
Форма образца: 5 мм × 5 мм;
Толщина образца: 1 мм;
Предварительная обработка образца: погружение в дистиллированную воду до состояния насыщения водой;
Скорость тестирования: 0,1 мм/мин;
Верхний нажимной элемент: диаметр 0,7 мм;
Термин «степень восстановления» относится к способности биоразрушаемого материала после снятия давления восстанавливать исходную форму перед следующим сжатием, например, после прохождения через катетер с небольшим внутренним диаметром. Конкретно, степень восстановления представляет собой показатель степени восстановления исходной формы. Степень восстановления биоразрушаемой пленки в состоянии насыщения водой после приложения усилия сжатия на 50% предпочтительно составляет 70% или, более предпочтительно, 75% или более, поскольку слишком низкая степень восстановления приводит к тому, что биоразрушаемый материал проходит сквозь целевой участок в кровеносном сосуде, который предполагается, например, эмболизировать, и проходит с кровотоком дальше.
Степень восстановления биоразрушаемой пленки в состоянии насыщения водой после приложения усилия сжатия на 50% измеряют с помощью того же прибора для измерения микроусилий, который используется в тесте на сжатие, при следующих условиях, и полученный результат соответствует значению Rr(%), вычисленному согласно приведенным ниже уравнениям 5-7. Конкретно, усилие (изменение усилия) прилагают к биоразрушаемой пленке вплоть до достижения усилия сжатия на 50% (т.е. чтобы максимальное усилие в тесте равнялось усилию сжатия на 50%), определенного в тесте на сжатие, и затем убирают (ослабляют) усилие до минимального усилия в тесте.
Условия проведения измерения
Наименование теста: тест приложение/снятие усилия; Прибор: Micro Auto Model MST-I (производства Shimadzu Corporation);
Методика измерения: методика с поперечной головкой;
Условия окружающей среды: комнатная температура, на открытом воздухе;
Форма образца: 5 мм × 5 мм;
Толщина образца: 1 мм;
Предварительная обработка образца: погружение в дистиллированную воду до состояния насыщения водой;
Скорость тестирования: 0,1 мм/мин;
Верхний нажимной элемент: диаметр 0,7 мм;
Максимальное усилие в тесте: усилие сжатия на 50% для каждого образца пленки, полученное в тесте на сжатие;
Минимальное усилие в тесте: 0,0001 Н;
Конечное усилие после снятия нагрузки: 0,001 Н;
Скорость измерения нагрузки:
Время выдерживания под нагрузкой:
 ;
;
L1a: диаметр частиц (мкм) при приложении минимального усилия в тесте;
L1b: диаметр частиц (мкм) при приложении максимального усилия в тесте;
 ;
;
L2b: изменение размера частиц (мкм) при приложения максимального усилия в тесте с последующим уменьшением нагрузки до минимального усилия в тесте;
 ;
;
Термин «степень сжатия» относится к отношению толщины пленки биоразрушаемого материала после сжатия к исходной толщине пленки, и степень сжатия соответствует величине Cr(%), которая вычисляется по приведенному ниже уравнению 8. В настоящем изобретении, степень восстановления соответствует степени восстановления после приложения усилия (изменения усилия) до усилия сжатия на 50%, поэтому Cr=50%.
 ;
;
d: средняя толщина биоразрушаемой пленки (мм);
Биоразрушаемый материал по настоящему изобретению хорошо подходит для применения в качестве материала для эмболизации сосудов. Кроме того, он подходит для применения в качестве антиадгезионного материала, материала для перевязки ран, кровоостанавливающего материала, материала для предотвращения недержания мочи или подобных материалов.
В случаях, когда биоразрушаемый материал по настоящему изобретению применяется в качестве материала для эмболизации сосудов, могут применяться биоразрушаемые частицы сами по себе, либо в виде жидкой дисперсии в подходящей контрастной среде или в дисперсионной среде. Примеры контрастной среды включают водорастворимую контрастную среду, например, среду для инъекций йопамидол, среду для инъекций йоксагаловая кислота и среду для инъекций йогексол; а также масляную контрастную среду, например, йодированное маковое масло. Предпочтительными являются водорастворимые контрастные среды. Примеры дисперсионной среды включают водные растворы для инъекций и растительные масла, такие как кунжутное масло и кукурузное масло, содержащие диспергирующее средство, например, сложный эфир полиоксисорбитана и жирной кислоты, консервант, например, метилпарабен или изотонический агент, например, хлорид натрия. Упомянутый материал для эмболизации сосудов может дополнительно включать антисептик, стабилизатор, солюбилизатор, эксципиент и/или действующий компонент, например, противоопухолевый агент.
Dr(%) означает массовую долю остатка биоразрушаемого материала по настоящему изобретению после погружения пленки в фосфатный солевой буфер при 37°C на определенный период времени, и значение Dr вычисляют по приведенному ниже уравнению 9. Массовая доля остатка после погружения материала на 30 дней предпочтительно составляет 5% или более, более предпочтительно 50% или более, еще более предпочтительно 60% или более.

Dt: масса биоразрушаемой пленки после погружения на определенный период времени;
D0: масса биоразрушаемой пленки перед погружением.
Способ получения биоразрушаемого материала по настоящему изобретению включает стадию поперечного сшивания, во время которой мультивалентное соединение A, включающее 3 или более функциональные группы X, выбранные из группы, состоящей из гидроксильной группы, тиольной группы и аминогруппы, мультивалентное соединение B, включающее 3 или более функциональные группы Y, выбранные из группы, состоящей из карбоксильной группы, изоцианатной группы и тиоизоцианатной группы, а также соединение C, имеющее структуру производного гидроксикарбоновой кислоты, гомополимер которой, полученный при гомополимеризации, имеет температуру стеклования -40°C или ниже, растворяют в растворителе для проведения реакции химического сшивания, с получением биоразрушаемого материала.
Примеры мультивалентного соединения A включают блок-сополимер разветвленного полимера a1, образованного при связывании всех гидроксильных групп полиола с ПЭГ и ППГ, и гидроксикарбоновой кислоты a2 (далее по тексту именуемой "гидроксикарбоновой кислотой a2») гомополимер которой, образующийся при гомополимеризации, имеет температуру стеклования -39°C или выше. Примеры разветвленного полимера a1 включают 4-разветвленный ПЭГ (серия РТЕ; производства NiGK Corporation) и 8-разветвленный ПЭГ (серия HGEO; производства NiGK Corporation).
В случаях, когда гидроксикарбоновая кислота a2 является молочной кислотой, гликолевой кислотой и т.п., конденсационная полимеризация является предпочтительным способом получения мультивалентного соединения A, которое является блок-сополимером разветвленного полимера a1 и гидроксикарбоновой кислоты a2. В случаях, когда гидроксикарбоновая кислота a2 является циклическим соединением, например, лактидом или гликолидом, предпочтительной является полимеризация с раскрытием цикла.
В качестве растворителя для реакции конденсационной полимеризации применяют растворитель, который хорошо растворяет разветвленный полимер a1, например 4-разветвленный ПЭГ или 8-разветвленный ПЭГ, а также гидроксикарбоновую кислоту a2 (хороший растворитель). Примеры таких растворителей включают дихлорметан, хлороформ, ацетонитрил и тетрагидрофуран, а также их смеси. Предпочтительно выбирают такую температуру проведения реакции, чтобы применяемый растворитель кипел. Давление, применяемое для проведения реакции, может являться пониженным давлением, но предпочтительным является нормальное давление, поскольку это облегчает осуществление реакции на практике. Время проведения реакции предпочтительно составляет от 2 до 48 часов, более предпочтительно, от 4 до 24 часов для надлежащего регулирования молекулярной массы образующегося мультивалентного соединения A.
Общая концентрация разветвленного полимера a1 и гидроксикарбоновой кислоты a2 в реакционной смеси при конденсационной полимеризации меняется в зависимости от типов применяемых компонентов a1 и a2 и т.п. и предпочтительно составляет от 10 до 100 мас.%, более предпочтительно от 50 до 100 мас.%. Концентрация катализатора в реакционном растворителе предпочтительно составляет от 0,01 до 0,5 мас.%, более предпочтительно от 0,1 до 0,3 мас.%, поскольку слишком высокая концентрация осложняет удаление катализатора после проведения реакции, тогда как слишком низкая концентрация мешает проведению реакции.
В качестве растворителя для проведения реакции полимеризации с раскрытием цикла может применяться тот же растворитель, который применяется для реакции конденсационной полимеризации. Однако для увеличения реакционной способности предпочтительно проводить реакцию без растворителя при температуре от 90°C до 150°C, более предпочтительно, от 100°C до 130°C. Давление, применяемое для проведения реакции, может являться пониженным давлением, но предпочтительным является нормальное давление, поскольку это облегчает осуществление реакции на практике. Время проведения реакции предпочтительно составляет от 2 до 48 часов, более предпочтительно, от 4 до 24 часов для надлежащего регулирования молекулярной массы образующегося мультивалентного соединения A.
Примеры катализаторов включают металлсодержащие катализаторы. Примеры металлсодержащих катализаторов включают алкоксиды металлов, галогениды металлов, соли органических карбоновых кислот, соли угольной кислоты, соли серной кислоты и оксиды олова, титана, свинца, цинка, кобальта, железа, лития или редкоземельных металлов. С точки зрения увеличения скорости реакции предпочтительными являются соединения олова. Примеры соединений олова включают порошок олова, хлорид олова (II), хлорид олова (IV), бромид олова (II), бромид олова (IV), этоксиолово (II), т-бутоксиолово (IV), изопропоксиолово (IV), ацетат олова (II), ацетат олова (IV), октилат олова (II), лаурат олова (II), миристат олова (II), пальмитат олова (II), стеарат олова (II), олеат олова (II), линолеат олова (II), ацетилацетонат олва (II), оксалат олова (II), лактат олова (II), тартрат олова (II), пирофосфат олова (II), п-фенолсульфонат олова (II), бис(метансульфонат) олова (II), сульфат олова (II), оксид олова (II), оксид олова (IV), сульфид олова (II), сульфид олова (IV), оксид диметилолова (IV), оксид метилфенилолова (IV), оксид дибутилолова (IV), оксид диоктилолова (IV), оксид дифенилолова (IV), оксид трибутилолова, гидроксид трибутилолова (IV), гидроксид трифенилолова (IV), гидрид трибутилолова, оксид монобутилолова (IV), тетраметилолово (IV), тетраэтилолово (IV), тетрабутилолово (IV), дибутилдифенилолово (IV), тетрафенилолово (IV), ацетат трибутилолова (IV), ацетат триизобутилолова (IV), ацетат трифенилолова (IV), диацетат дибутилолова, диоктоат дибутилолова, дилаурат дибутилолова (IV), малеат дибутилолова (IV):, бис (ацетилацетонат) дибутилолова, хлорид трибутилолова (IV), дихлорид дибутилолова, трихлорид монобутилолова, дихлорид диоктилолова, хлорид трифенилолова (IV), сульфид трибутилолова, сульфат трибутилолова, метансульфонат олова (II), этансульфонат олова (II), трифторметансульфонат олова (II), гексахлорстаннат аммония (IV), сульфид дибутилолова, сульфид дифенилолова, сульфат триэтилолова и фталоцианин олова (II). Предпочтительно, катализатором конденсационной полимеризации является оксид олова (II), и катализатором полимеризации с раскрытием цикла предпочтительно является октилат олова (II).
Примеры мультивалентного соединения B включают разветвленное соединение, образующееся при связывании разветвленного полимера b1, полученного при связывании всех гидроксильных групп полиола с ПЭГ или ППГ, с поликарбоновой кислотой b2. Примеры разветвленного полимера b1 включают 4-разветвленный ПЭГ и 8-разветвленный ПЭГ.
В качестве способа получения мультивалентного соединения B, образующегося при связывании разветвленного полимера b1 с поликарбоновой кислотой b2, предпочтительна реакция конденсации, в которой применяется реагент, осуществляющий дегидратацию-конденсацию. В качестве альтернативы, поликарбоновую кислоту a2 можно на первой стадии ввести во взаимодействие с электрофильным галогенирующим агентом, например, тионилхлоридом или оксалилхлоридом, с превращением в такое производное/ как галогенангидрид, ангидрид или сложный эфир кислоты, которое затем можно ввести в реакцию конденсации с получением мультивалентного соединения B.
Примеры реагента для дегидратации-конденсации включают карбодиимидные соединения, такие как N,N′-дициклогексилкарбодиимид, N,N′-диизопропилкарбодиимид, 1-этил-3-(3-диметиламинопропил)карбодиимид, гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида (далее по тексту именуемый «EDC»), 1,3-бис(2,2-диметил-1,3-диоксолан-4-илметил)карбодиимид, N-{3-(диметиламино)пропил}-N′-этилкарбодиимид, N-{3-(диметиламино)пропил}-N′-этилкарбодиимида метйодид, N-трет-бутил-N′-этилкарбодиимид, мезо-п-толуолсульфонат N-циклогексил-N′-(2-морфолиноэтил)карбодиимида, N,N′-дитрет-бутил карбодиимид и N,N′-ди-п-трикарбодиимид. Предпочтительным реагентом является EDC по причине легкости удаления побочного продукта реакции.
Реагент для дегидратации-конденсации может применяться совместно с ускорителем дегидратации-конденсации. Примеры ускорителей дегидратации-конденсации включают пиридин, 4-диметиламинопиридин (далее по тексту именуемый «DMAP»), триэтиламин, изопропиламин, 1-гидроксибензотриазол и имид N-гидроксиянтарной кислоты.
В качестве растворителя для проведения реакции конденсации разветвленного полимера b1 и поликарбоновой кислоты b2, используется растворитель, который хорошо растворяет соединения b1 и b2. Примеры таких растворителей включают дихлорметан, хлороформ, ацетонитрил и тетрагидрофуран, а также их смеси. Для проведения реакции обычно выбирают такую температуру, при которой кипит применяемый хороший растворитель. Давление, применяемое для проведения реакции, может являться пониженным давлением, но предпочтительным является нормальное давление, поскольку это облегчает осуществление реакции на практике. Время проведения реакции предпочтительно составляет от 2 до 48 часов, более предпочтительно, от 4 до 2 4 часов для надлежащего регулирования молекулярной массы образующегося мультивалентного соединения B.
Общая концентрация разветвленного полимера b1 и поликарбоновой кислоты b2′′ в реакционной смеси реакции конденсации меняется в зависимости от типов применяемых компонентов b1 и b2 и т.п. и предпочтительно составляет от 10 до 100 мас.%, более предпочтительно от 20 до 80 мас.%. Концентрация катализатора в реакционном растворителе предпочтительно составляет от 0,01 до 0,5 мас.%, более предпочтительно от 0,1 до 0,3 мас.%, поскольку слишком высокая концентрация осложняет удаление катализатора после проведения реакции, тогда как слишком низкая концентрация мешает проведению реакции.
Примеры катализатора включают пиридин, DMAP, триэтиламин и изопропиламин. Предпочтительным является пиридин, по причине легкости удаления.
В качестве соединения C еще более предпочтителен, например, разветвленный блок-сополимер (далее по тексту именуемый «блок-сополимер С») полимера c1, образованного при связывании всех гидроксильных групп полиола с ПЭГ и ППГ, и гидроксикарбоновой кислоты c2 (далее по тексту именуемой «гидроксикарбоновая кислота c2»), гомополимер которой, образовавшийся при гомополимеризации, имеет температуру стеклования -40°C или ниже.
В случаях, когда гидроксикарбоновая кислота c2 представляет собой 6-гидроксикапроновую кислоту и т.п., конденсационная полимеризация является предпочтительным способом получения блок-сополимера С, которые образуется при сополимеризации разветвленного полимера c1 и гидроксикарбоновой кислоты c2. В случаях, когда гидроксикарбоновая кислота c2 является циклическим соединением, например, ε-капролактоном и т.п., предпочтительной является полимеризация с раскрытием цикла.
В качестве растворителя для проведения реакции конденсационной полимеризации, используется растворитель, в котором хорошо растворим разветвленный полимер c1, например, 4-разветвленный ПЭГ или 8-разветвленный ПЭГ, и гидроксикарбоновая кислота c2. Примеры таких растворителей включают дихлорметан, хлороформ, ацетонитрил и тетрагидрофуран, а также их смеси. Для проведения реакции предпочтительно выбирают такую температуру, при которой кипит используемый растворитель. Давление, применяемое для проведения реакции, может являться пониженным давлением, но предпочтительным является нормальное давление, поскольку это облегчает осуществление реакции на практике. Время проведения реакции предпочтительно составляет от 2 до 48 часов, более предпочтительно, от 4 до 24 часов для надлежащего регулирования молекулярной массы образующегося соединения C.
Общая концентрация разветвленного полимера c1 и гидроксикарбоновой кислоты c2 в реакционной смеси реакции конденсационной полимеризации меняется в зависимости от типов применяемых компонентов c1 и c2 и т.п. Предпочтительно эта концентрация составляет от 10 до 100 мас.%, более предпочтительно от 50 до 100 мас.%. Концентрация катализатора в реакционном растворителе предпочтительно составляет от 0,01 до 0,5 мас.%, более предпочтительно от 0,1 до 0,3 мас.%, поскольку слишком высокая концентрация осложняет удаление катализатора после проведения реакции, тогда как слишком низкая концентрация мешает проведению реакции.
В качестве растворителя для проведения реакции полимеризации с раскрытием цикла, может применяться тот же самый растворитель, который применяется для реакции конденсационной полимеризации. Однако для увеличения скорости реакции предпочтительно проводить реакцию без растворителя при температуре реакционной смеси от 90°C до 150°C, более предпочтительно от 100°C до 130°C. Давление, применяемое для проведения реакции, может являться пониженным давлением, но предпочтительным является нормальное давление, поскольку это облегчает осуществление реакции на практике. Время проведения реакции предпочтительно составляет от 2 до 48 часов, более предпочтительно, от 4 до 24 часов для надлежащего регулирования молекулярной массы образующегося соединения C.
Примеры катализатора включают те же самые металлосодержащие катализаторы, которые применяются для получения мультивалентного соединения A.
Хотя полученные мультивалентное соединение A, мультивалентное соединение B и соединение C можно использовать на стадии поперечного сшивания химическими связями без очистки, их можно подвергнуть очистке для удаления не вступивших в реакцию веществ, растворителя и катализатора. Примеры подходящих способов очистки включают фракционное осаждение.
Фракционное осаждение представляет собой способ, в котором полученное мультивалентное соединение A, мультивалентное соединение B или соединение C растворяют в хорошем растворителе, и полученный раствор по каплям при перемешивании добавляют к плохому растворителю, чтобы получить очищенные мультивалентное соединение A, мультивалентное соединение B или соединение C в виде осадка. Термин «хороший растворитель» в настоящей заявке относится к органическому - растворителю, в котором можно растворить упомянутые мультивалентное соединение A, мультивалентное соединение B или соединение C, тогда как термин «плохой растворитель» относится к органическому растворителю, в котором нельзя растворить упомянутые мультивалентное соединение A, мультивалентное соединение B или соединение C.
Примеры хороших растворителей, применяемых при фракционном осаждении, включают дихлорметан, хлороформ, ацетонитрил и тетрагидрофуран, а также их смеси. Применяемое количество хорошего растворителя меняется в зависимости от состава и т.п. мультивалентного соединения A, мультивалентного соединения B или соединения C, и концентрация растворенного мультивалентного соединения A, мультивалентного соединения B или соединения C предпочтительно составляет от 10 до 80 мас.%, более предпочтительно от 20 до 70 мас.%. Примеры плохих растворителей, применяемых при фракционном осаждении, включают спиртовые органические растворители, например, метанол и этанол; простые эфиры, например, диметиловый эфир, этилметиловый эфир и диэтиловый эфир; углеводородные органические растворители, например, пентан, гексан, гептан и октан; а также смеси указанных растворителей. Применяемое количество плохого растворителя также изменяется в зависимости от состава и т.п. полученного мультивалентного соединения A, мультивалентного соединения B или соединения C. Предпочтительно, количество плохого растворителя составляет от 50 до 1000 мас.%, более предпочтительно от 100 до 500 мас.% от массы хорошего растворителя. С точки зрения регулирования распределения молекулярных масс, предпочтителен способ, в котором мультивалентное соединение A, мультивалентное соединение B или соединение C растворяют в дихлорметане, и полученный раствор по каплям при перемешивании добавляют к диэтиловому эфиру. Кроме того, для повышения чистоты очищенного продукта, полученный очищенный продукт предпочтительно промывают плохим растворителем, более предпочтительно, промывают от 2 до 5 раз.
На стадии поперечного сшивания химическими связями, когда мультивалентное соединение A, мультивалентное соединение B и соединение C растворяют в растворителе и дают пройти реакции поперечного сшивания с получением биоразрушаемого материала по настоящему изобретению, применение протонного растворителя, например, воды или спирта, не является предпочтительным, поскольку протонный растворитель сам может принять участие в химической реакции поперечного сшивания и плотность сшивания в полученном биоразрушаемом материале может существенно понизиться. В качестве растворителя на стадии химического сшивания поперечными связями предпочтительным является апротонный полярный органический растворитель с диэлектрической постоянной от 35 до 50.
В качестве апротонного полярного растворителя с диэлектрической постоянной от 35 до 50 предпочтителен N,N-диметилформамид (далее по тексту именуемый «ДМФА»), N,N-диметилацетамид, ацетонитрил или диметилсульфоксид (далее по тексту именуемый «ДМСО»). Более предпочтительным является ацетонитрил, т.к. его можно легко удалить выпариванием при пониженном давлении.
На стадии поперечного сшивания химическими связями может применяться реагент для дегидратации-конденсации. Примеры реагента, осуществляющего дегидратацию-конденсацию, применимого на стадии химического сшивания включают карбодиимиды, например, N,N′-дициклогексилкарбодиимид, N,N′-диизопропилкарбодиимид, EDC, N-{3-(диметиламино)пропил}-N′-этилкарбодиимид, N-{3-(диметиламино)пропил}-N′-этилкарбодиимида метйодид, N-трет-бутил-N′-этилкарбодиимид, мезо-п-толуолсульфонат N-циклогексил-N′-(2-морфолиноэтил)карбодиимида, N,N′-дитрет-бутил карбодиимид и N,N′-ди-п-трикарбодиимид. Предпочтительным реагентом является EDC по причине легкости удаления побочного продукта реакции.
Реагент для дегидратации-конденсации может применяться совместно с ускорителем дегидратации-конденсации. Примеры ускорителей дегидратации-конденсации включают пиридин, DMAP, триэтиламин, изопропиламин, 1-гидроксибензотриазол, имид N-гидроксиянтарной кислоты и т.п. Предпочтительным является DMAP, вследствие высокой реакционной способности и легкости удаления по окончании реакции.
Примеры способа получения биоразрушаемого материала по настоящему изобретению в форме биоразрушаемой пленки включают способ, в котором мультивалентное соединение A, мультивалентное соединение B и соединение C, растворенные в апротонном полярном органическом растворителе с диэлектрической постоянной от 35 до 50, вводят в плохой растворитель и затем дают пройти химической реакции сшивания при удалении апротонного полярного органического растворителя.
Предпочтительные примеры плохого растворителя, применяемого для получения биоразрушаемой" пленки, включают масла, например, синтетические масла и натуральные масла. Более предпочтительными являются натуральные масла.
Примеры синтетических масел включают силиконовые масла. Примеры натуральных масел включают масло хлопчатника, кукурузное масло, кокосовое масло, оливковое масло, пальмовое масло, масло рапса, сафлоровое масло, кунжутное масло, соевое масло, подсолнечное масло, терпентиновое масло (скипидар), миндальное масло, масло авокадо, масло бергамота, касторовое масло, кедровое масло, хлорофилловое масло, гвоздичное масло, кротоновое масло, эвкалиптовое масло, масло фенхеля, сивушное масло, масло виноградных косточек, масло жожоба, масло из плодов свечного дерева, лавандовое масло, лимонное масло, льняное масло, масло австралийского ореха, масло пенника лугового, апельсиновое масло, масло душицы, персиковое масло и масло плодов шиповника. Масло хлопчатника, кукурузное масло, оливковое масло, рапсовое масло, сафлоровое масло, кунжутное масло, соевое масло или подсолнечное масло являются предпочтительными с точки зрения высокой биологической безопасности и постоянной доступности.
ПРИМЕРЫ
Ниже по тексту настоящее изобретение будет подробно описано с привлечением примеров и сравнительных примеров, но следует понимать, что изобретение не ограничено этими примерами.
Пример 1
В колбу, имеющую форму баклажана, помещали 10,0 г 8-разветвленного ПЭГ (SUNBRIGHT (зарегистрированная торговая марка) HGEO5000 производства NiGK Corporation), взятого в качестве разветвленного полимера a1, и 22,0 г лактида (PURASORB L; производства Purac Biomaterials), взятого в качестве гидроксикарбоновой кислоты a2. Смесь перемешивали в расплаве в атмосфере аргона при 120°C и затем к полученной смеси в качестве катализатора добавляли 0,94 мл раствора октилата олова (II) (октилат олова (II) (производства Sigma-Aldrich Со.,), растворенный в толуоле (производства Wako Pure Chemical Industries, Ltd.) и доведенный до концентрации 0,16 г/мл), после чего осуществляли реакцию сополимеризации в течение 20 часов при нормальном давлении, получая неочищенное мультивалентное соединение A1.
Полученное неочищенное мультивалентное соединение A1 по каплям добавляли к 100 мл диэтилового эфира и собирали полученный осадок и жидкий компонент, отделяющийся от диэтилового эфира. Затем их три раза промывали 50 мл диэтилового эфира, получая очищенное мультивалентное соединение A1. Среднемассовая молекулярная масса очищенного мультивалентного соединения A1 по данным методики GPC составляла 15400.
В колбу формы «баклажан» помещали 10,0 г 8-разветвленного ПЭГ (SUNBRIGHT (зарегистрированная торговая марка) HGEO5000 производства NiGK Corporation), взятого в качестве разветвленного полимера b1, и 3,2 г безводной янтарной кислоты (производства Wako Pure Chemical Industries, Ltd.) в качестве поликарбоновой кислоты b2. В колбу добавляли 1 мл дегидратированного пиридина (производства Wako Pure Chemical Industries, Ltd.) в качестве катализатора, и 40 мл дегидратированного хлороформа (производства Wako Pure Chemical Industries, Ltd.) в качестве растворителя, и нагревали полученную смесь до 80°C, проводя реакцию при нормальном давлении в течение 24 часов и получая неочищенное мультивалентное соединение B1.
Полученное мультивалентное соединение B1 по каплям добавляли к 100 мл диэтилового эфира и собирали полученный осадок и жидкий компонент, отделяющийся от диэтилового эфира. Затем их три раза промывали 50 мл диэтилового эфира, получая очищенное мультивалентное соединение B1. Среднемассовая молекулярная масса очищенного мультивалентного соединения B1 по данным методики GPC составляла 5800.
В колбу формы «баклажан» помещали 10,0 г 8-разветвленного ПЭГ (SUNBRIGHT (R) HGEO5000 производства NiGK Corporation), взятого в качестве разветвленного полимера c1, и 20,0 г ε-капролактона (производства Wako Pure Chemical Industries, Ltd.) в качестве гидроксикарбоновой кислоты c2. Смесь перемешивали в расплаве в атмосфере аргона при 120°C и затем к полученной смеси в качестве катализатора добавляли 0,94 мл раствора октилата олова (II) (октилат олова (II) (производства Sigma-Aldrich Со.,) растворенный в толуоле (производства Wako Pure Chemical Industries, Ltd.) и доведенный до концентрации 0,16 г/мл), после чего осуществляли реакцию сополимеризации в течение 20 часов при нормальном давлении, получая неочищенное соединение C3. Среднемассовая молекулярная масса очищенного соединения C1 по данным методики GPC составляла 13600.
Полученное соединение C1 по каплям добавляли к 100 мл диэтилового эфира и собирали полученный осадок и жидкий компонент, отделяющийся от диэтилового эфира. Затем их три раза промывали 50 мл диэтилового эфира, получая очищенное соединение C3. Среднемассовая молекулярная масса очищенного мультивалентного соединения C3 по данным методики GPC составляла 13600.
Полученные очищенное мультивалентное соединение A1, очищенное мультивалентное соединение B1 и очищенное соединение C1 высушивали при пониженном давлении и каждое из указанных соединений растворяли в дегидратированном ацетонитриле (производства Wako Pure Chemical Industries, Ltd.) до концентрации 0,3 г/мл, получая растворы 1, 2 и 3, соответственно. В форму, состоящую из стеклянных пластин 1 мм толщины, наливали 0,295 мл раствора 1, 0,4 44 мл раствора 2, 0,261 мл раствора 3, 0, 022 мл раствора DMAP в ацетонитриле (0,1 г/мл) в качестве катализатора и 0,039 мл концентрированного раствора EDC в качестве конденсирующего агента, и удаляли ацетонитрил, погружая форму в масло хлопчатника, нагретое до 55°C, и получая биоразрушаемую пленку 1.
Проводили тест на биоразрушаемость, вычисляя массовую долю остатка полученной биоразрушаемой пленки 1. Также для пленки измеряли усилие сжатия и степень восстановления. Результаты показаны в таблице 1.
Затем раствор 1, раствор 2, раствор 3, раствор DMAP в ацетонитриле и концентрированный раствор EDC, имевшие те же концентрации, как и при получении описанной выше биоразрушаемой пленки 1, смешивали в тех же объемных пропорциях, получая ацетонитрил-содержащую пленку 1.
Осуществляли тест на динамическую вязкоупругость, измеряя комплексный модуль упругости полученной ацетонитрил-содержащей пленки 1. Результаты показаны в таблице 1.
Как видно из данных таблицы 1, биоразрушаемая пленка 1 имела высокую массовую долю остатка, высокое усилие сжатия и высокую степень восстановления. Ацетонитрил-содержащая пленка 1 обладала высоким комплексным модулем упругости.
Пример 2
Осуществляли те же действия, что и в примере 1, за исключением того, что количество раствора 1 изменяли до 0,199 мл, количество раствора 2 изменяли до 0,450 мл, количество раствора 3 изменяли до 0,351 мл, количество раствора DMAP изменяли до 0,023 мл и количество раствора EDC изменяли до 0,040 мл, получая биоразрушаемую пленку 2 и ацетонитрил-содержащую пленку 2.
Свойства биоразрушаемой пленки 2 и ацетонитрил-содержащей пленки 2 определяли аналогично Примеру 1. Результаты показаны в таблице 1.
Как видно из данных таблицы 1, биоразрушаемая пленка 2 имела высокую массовую долю остатка, высокое усилие сжатия и высокую степень восстановления. Ацетонитрил-содержащая пленка 2 обладала высоким комплексным модулем упругости.
Пример 3
При получении раствора 4 выполняли те же действия, что и в примере 1, за исключением того, что вместо разветвленного полимера Al применяли 8-разветвленный ПЭГ (SUNBRIGHT (зарегистрированная торговая марка) HGEO5000 производства NiGK Corporation). Среднемассовая молекулярная масса 8-разветвленного ПЭГ (SUNBRIGHT (зарегистрированная торговая марка) HGEO5000 производства NiGK Corporation) по данным методики GPC составляла 5000.
При получении биоразрушаемой пленки 3 и ацетонитрил-содержащей пленки 3, осуществляли те же действия, что и в примере 1, за исключением того, что 0,295 мл раствора 1 заменяли на 0,120 мл раствора 4, количество раствора 2 изменяли до 0,555 мл, количество раствора 3 изменяли до 0,325 мл, количество раствора DMAP изменяли до 0,028 мл и количество раствора EDC изменяли до 0,04 9 мл.
Свойства биоразрушаемой пленки 3 и ацетонитрил-содержащей пленки 3 определяли аналогично Примеру 1. Результаты показаны в таблице 1.
Как видно из данных таблицы 1, биоразрушаемая пленка 3 имела высокую массовую долю остатка, высокое усилие сжатия и высокую степень восстановления. Ацетонитрил-содержащая пленка 3 обладала высоким комплексным модулем упругости.
Пример 4
При получении очищенного мультивалентного соединения A2 осуществляли те же действия, что и в примере 1, за исключением того, что 10,0 г разветвленного полимера a1 заменяли на 8-разветвленный ПЭГ (SUNBRIGHT (зарегистрированная торговая марка) HGEO10000 производства NiGK Corporation). Среднемассовая молекулярная масса очищенного мультивалентного соединения A2 по данным методики GPC составляла 18600.
При получении очищенного мультивалентного соединения B2 осуществляли те же действия, что и в примере 1, за исключением того, что 10,0 г разветвленного полимера b1 заменяли на 8-разветвленный ПЭГ (SUNBRIGHT (зарегистрированная торговая марка) HGEO10000 производства NiGK Corporation). Среднемассовая молекулярная масса очищенного мультивалентного соединения B2 по данным методики GPC составляла 10800.
При получении очищенного соединения C2 осуществляли те же действия, что и в примере 1, за исключением того, что 10,0 г разветвленного полимера c1 заменяли на 8-разветвленный ПЭГ (SUNBRIGHT (зарегистрированная торговая марка) HGEO10000 производства NiGK Corporation). Среднемассовая молекулярная масса очищенного соединения C2 по данным методики GPC составляла 21700.
При получении раствора 5 осуществляли те же действия, что и в примере 1, за исключением того, что очищенное мультивалентное соединение A1 заменяли на очищенное мультивалентное соединение A2. При получении раствора 6 осуществления те же действия, что и в примере 1, за исключением того, что очищенное мультивалентное соединение B1 заменяли на очищенное мультивалентное соединение B2. При получении раствора 7 осуществления те же действия, что и в примере 1, за исключением того, что очищенное соединение C1 заменяли на очищенное соединение C2.
При получении биоразрушаемой пленки 4 и ацетонитрил-содержащей пленки 4 осуществляли те же действия, что и в примере 1, за исключением того, что 0,295 мл раствора 1 заменяли на 0,418 мл раствора 5, 0,444 мл раствора 2 заменяли на 0,534 мл раствора 6, 0,261 мл раствора 3 заменяли на 0,049 мл раствора 7, количество раствора DMAP изменяли до 0,014 мл и количество EDC изменяли до 0,025 мл.
Свойства биоразрушаемой пленки 4 и ацетонитрил-содержащей пленки 4 определяли аналогично Примеру 1. Результаты показаны в таблице 1.
Как видно из данных таблицы 1, биоразрушаемая пленка 4 имела высокую массовую долю остатка, высокое усилие сжатия и высокую степень восстановления. Ацетонитрил-содержащая пленка 4 обладала высоким комплексным модулем упругости.
Пример 5
При получении биоразрушаемой пленки 5 и ацетонитрил-содержащей пленки 5 осуществляли те же действия, что и в примере 1, за исключением того, что 0,295 мл раствора 1 заменяли на 0,223 мл раствора 5, 0,444 мл раствора 2 заменяли на 0,517 мл раствора 6, 0,261 мл раствора 3 заменяли на 0,260 мл раствора 7, количество раствора DMAP изменяли до 0,014 мл и количество EDC изменяли до 0,024 мл.
Свойства биоразрушаемой пленки 5 и ацетонитрил-содержащей пленки 5 определяли аналогично Примеру 1. Результаты показаны в таблице 1.
Как видно из данных таблицы 1, биоразрушаемая пленка 5 имела высокую массовую долю остатка, высокое усилие сжатия и высокую степень восстановления. Ацетонитрил-содержащая пленка 5 обладала высоким комплексным модулем упругости.
Пример 6
При получении биоразрушаемой пленки 6 и ацетонитрил-содержащей пленки 6 осуществляли те же действия, что и в примере 1, за исключением того, что 0,295 мл раствора 1 заменяли на 0,147 мл раствора 5, 0,444 мл раствора 2 заменяли на 0,511 мл раствора 6, 0,261 мл раствора 3 заменяли на 0,342 мл раствора 7, количество раствора DMAP изменяли до 0,014 мл и количество EDC изменяли до 0,024 мл.
Свойства биоразрушаемой пленки 6 и ацетонитрил-содержащей пленки 6 определяли аналогично Примеру 1. Результаты показаны в таблице 1.
Как видно из данных таблицы 1, биоразрушаемая пленка 6 имела высокую массовую долю остатка, высокое усилие сжатия и высокую степень восстановления. Ацетонитрил-содержащая пленка 6 обладала высоким комплексным модулем упругости.
Пример 7
При получении биоразрушаемой пленки 7 и ацетонитрил-содержащей пленки 7 осуществляли те же действия, что и в примере 1, за исключением того, что 0,295 мл раствора 1 заменяли на 0, 063 мл раствора 5, 0,444 мл раствора 2 заменяли на 0,201 мл раствора 6, 0,261 мл раствора 3 заменяли на 0,736 мл раствора 7, количество раствора DMAP изменяли до 0,005 мл и количество EDC изменяли до 0,010 мл.
Свойства биоразрушаемой пленки 7 и ацетонитрил-содержащей пленки 7 определяли аналогично Примеру 1. Результаты показаны в таблице 1.
Как видно из данных таблицы 1, биоразрушаемая пленка 7 имела высокую массовую долю остатка, высокое усилие сжатия и высокую степень восстановления. Ацетонитрил-содержащая пленка 7 обладала высоким комплексным модулем упругости.
Пример 8
При получении очищенного мультивалентного соединения A3 осуществляли те же действия, что и в примере 1, за исключением того, что 10,0 г разветвленного полимера a1 заменяли на 8-разветвленный ПЭГ (SUNBRIGHT (зарегистрированная торговая марка) HGEO20000 производства NiGK Corporation). Среднемассовая молекулярная масса очищенного мультивалентного соединения A3 по данным методики GPC составляла 32000.
При получении очищенного мультивалентного соединения B3 осуществляли те же действия, что и в примере 1, за исключением того, что 10,0 г разветвленного полимера b1 заменяли на 8-разветвленный ПЭГ (SUNBRIGHT (зарегистрированная торговая марка) HGEO20000 производства NiGK Corporation). Среднемассовая молекулярная масса очищенного мультивалентного соединения B3 по данным методики GPC составляла 20800.
При получении очищенного соединения C3 осуществляли те же действия, что и в примере 1, за исключением того, что 10,0 г разветвленного полимера c1 заменяли на 8-разветвленный ПЭГ (SUNBRIGHT (зарегистрированная торговая марка) HGEO20000 производства NiGK Corporation). Среднемассовая молекулярная масса очищенного соединения C3 по данным методики GPC составляла 33000.
При получении раствора 8 осуществляли те же действия, что и в примере 1, за исключением того, что очищенное мультивалентное соединение A1 заменяли на очищенное мультивалентное соединение A3. При получении раствора 9 осуществления те же действия, что и в примере 1, за исключением того, что очищенное мультивалентное соединение B1 заменяли на очищенное мультивалентное соединение B3. При получении раствора 10 осуществления те же действия, что и в примере 1, за исключением того, что очищенное соединение C1 заменяли на очищенное соединение C3.
При получении биоразрушаемой пленки 8 и ацетонитрил-содержащей пленки 8 осуществляли те же действия, что и в примере 1, за исключением того, что 0,295 мл раствора 1 заменяли на 0,216 мл раствора 8, 0,444 мл раствора 2 заменяли на 0,561 мл раствора 9, 0,261 мл раствора 3 заменяли на 0,223 мл раствора 10, количество раствора DMAP изменяли до 0,008 мл и количество EDC изменяли до 0,014 мл.
Свойства биоразрушаемой пленки 8 и ацетонитрил-содержащей пленки 8 определяли аналогично Примеру 1. Результаты показаны в таблице 1.
Как видно из данных таблицы 1, биоразрушаемая пленка 8 имела высокую массовую долю остатка, высокое усилие сжатия и высокую степень восстановления. Ацетонитрил-содержащая пленка 8 обладала высоким комплексным модулем упругости.
Пример 9
При получении очищенного мультивалентного соединения A4 осуществляли те же действия, что и в примере 1, за исключением того, что 10,0 г разветвленного полимера a1 заменяли на 4-разветвленный ПЭГ (SUNBRIGHT (зарегистрированная торговая марка) РТЕ20000; производства NiGK Corporation). Среднемассовая молекулярная масса очищенного мультивалентного соединения A4 по данным методики GPC составляла 34200.
При получении очищенного мультивалентного соединения B4 осуществляли те же действия, что и в примере 1, за исключением того, что 10,0 г разветвленного полимера b1 заменяли на 4-разветвленный ПЭГ (SUNBRIGHT (зарегистрированная торговая марка) РТЕ20000 производства; NiGK Corporation). Среднемассовая молекулярная масса очищенного мультивалентного соединения B4 по данным методики GPC составляла 20400.
При получении очищенного соединения C4 осуществляли те же действия, что и в примере 1, за исключением того, что 10,0 г разветвленного полимера c1 заменяли на 8-разветвленный ПЭГ (SUNBRIGHT (зарегистрированная торговая марка) HGEO20000 производства NiGK Corporation). Среднемассовая молекулярная масса очищенного соединения C4 по данным методики GPC составляла 34700.
При получении раствора 11 осуществляли те же действия, что и в примере 1, за исключением того, что очищенное мультивалентное соединение A1 заменяли на очищенное мультивалентное соединение A4. При получении раствора 12 осуществления те же действия, что и в примере 1, за исключением того, что очищенное мультивалентное соединение B1 заменяли на очищенное мультивалентное соединение B4. При получении раствора 13 осуществления те же действия, что и в примере 1, за исключением того, что очищенное соединение C1 заменяли на очищенное соединение C4.
При получении биоразрушаемой пленки 9 и ацетонитрил-содержащей пленки 9 осуществляли те же действия, что и в примере 1, за исключением того, что 0,295 мл раствора 1 заменяли на 0, 227 мл раствора 11, 0,444 мл раствора 2 заменяли на 0,542 мл раствора 12, 0,261 мл раствора 3 заменяли на 0,231 мл раствора 13, количество раствора DMAP изменяли до 0,008 мл и количество EDC изменяли до 0,014 мл.
Свойства биоразрушаемой пленки 9 и ацетонитрил-содержащей пленки 9 определяли аналогично Примеру 1. Результаты показаны в таблице 1.
Как видно из данных таблицы 1, биоразрушаемая пленка 9 имела высокую массовую долю остатка, высокое усилие сжатия и высокую степень восстановления. Ацетонитрил-содержащая пленка 9 обладала высоким комплексным модулем упругости.
Пример 10
При получении мультивалентного соединения A5 осуществляли те же действия, что и в примере 1, за исключением того, что 22,0 г лактида заменяли на 30,0 г гликолида (PURASORB G; производства PURAC BIOMATERIALS) и количество раствора октилата олова изменяли до 1,28 мл. Среднемассовая молекулярная масса мультивалентного соединения A5 по данным методики GPC составляла 14100. Для получения раствора 14 осуществляли те же действия, что и в примере 1, за исключением того, что очищенное мультивалентное соединение A1 заменяли на очищенное мультивалентное соединение A5.
При получении биоразрушаемой пленки 10 и ацетонитрил-содержащей пленки 10 осуществляли те же действия, что и в примере 1, за исключением того, что 0,295 мл раствора 1 заменяли на 0,277 мл раствора 14, количество раствора 2 изменяли до 0,456 мл, количество раствора 3 изменяли до 0,267 мл, количество раствора DMAP изменяли до 0,023 мл и количество EDC изменяли до 0,040 мл.
Свойства биоразрушаемой пленки 10 и ацетонитрил-содержащей пленки 10 определяли аналогично Примеру 1. Результаты показаны в таблице 1.
Как видно из данных таблицы 1, биоразрушаемая пленка 10 имела высокую массовую долю остатка, высокое усилие сжатия и высокую степень восстановления. Ацетонитрил-содержащая пленка 10 обладала высоким комплексным модулем упругости.
Пример 11
При получении мультивалентного соединения B5 осуществляли те же действия, что и в примере 1, за исключением того, что вместо поликарбоновой кислоты b2 использовали безводную малеиновую кислоту (производства Wako Pure Chemical Industries, Ltd.). Среднемассовая молекулярная масса мультивалентного соединения B14 по данным методики GPC составляла 5800. Для получения раствора 15 осуществляли те же действия, что и в примере 1, за исключением того, что очищенное мультивалентное соединение B2 заменяли на очищенное мультивалентное соединение B5.
При получении биоразрушаемой пленки 11 и ацетонитрил-содержащей пленки 11 осуществляли те же действия, что и в примере 1, за исключением того, что 0,444 мл раствора 2 заменяли на 0,444 мл раствора 15.
Свойства биоразрушаемой пленки 11 и ацетонитрил-содержащей пленки 11 определяли аналогично Примеру 1. Результаты показаны в таблице 1.
Как видно из данных таблицы 1, биоразрушаемая пленка 11 имела высокую массовую долю остатка, высокое усилие сжатия и высокую степень восстановления. Ацетонитрил-содержащая пленка 11 обладала высоким комплексным модулем упругости.
Сравнительный пример 1
При получении биоразрушаемой пленки 12 и ацетонитрил-содержащей пленки 12 осуществляли те же действия, что и в примере 1, за исключением того, что раствор 3 не использовали, и что 0,295 мл раствора 1 заменяли на 0,463 мл раствора 5, 0,444 мл раствора 2 заменяли на 0,537 мл раствора 6, количество раствора DMAP изменяли до 0,015 мл и количество EDC изменяли до 0,025 мл.
Свойства биоразрушаемой пленки 12 и ацетонитрил-содержащей пленки 12 определяли аналогично Примеру 1. Результаты показаны в таблице 1.
Как видно из данных таблицы 1, биоразрушаемая пленка 12 имела высокое усилие сжатия и высокую степень восстановления, но массовая доля остатка была низкой. Ацетонитрил-содержащая пленка 12 обладала высоким комплексным модулем упругости.
Сравнительный пример 2
При получении биоразрушаемой пленки 13 и ацетонитрил-содержащей пленки 13 осуществляли те же действия, что и в примере 1, за исключением того, что раствор 3 не использовали, и что 0,295 мл раствора 1 заменяли на 0,435 мл раствора 8, 0,444 мл раствора 2 заменяли на 0,565 мл раствора 9, количество раствора DMAP изменяли до 0,008 мл и количество EDC изменяли до 0,014 мл.
Свойства биоразрушаемой пленки 13 и ацетонитрил-содержащей пленки 13 определяли аналогично Примеру 1. Результаты показаны в таблице 1.
Как видно из данных таблицы 1, биоразрушаемая пленка 13 имела высокое усилие сжатия и высокую степень восстановления, но массовая доля остатка была низкой. Ацетонитрил-содержащая пленка 13 обладала высоким комплексным модулем упругости.
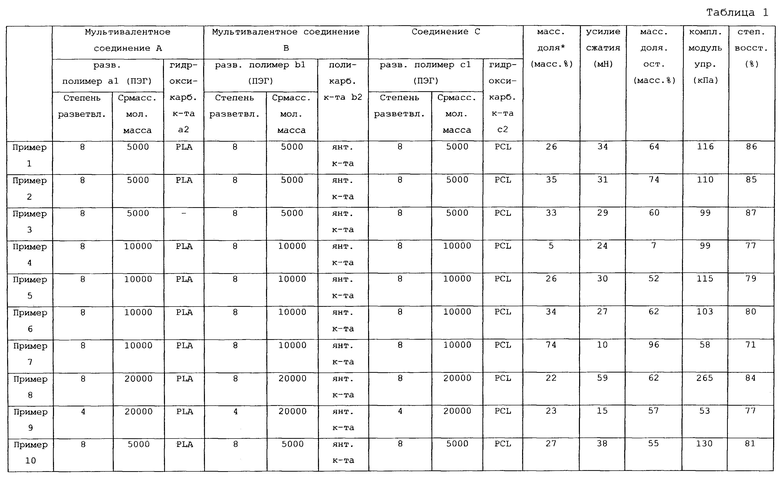
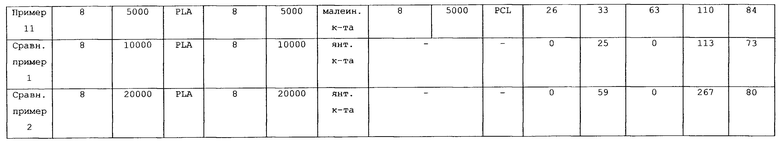
Промышленная применимость
Биоразрушаемый материал по настоящему изобретению может применяться в области медицины для эмболизации сосудов, предотвращения адгезии (образования спаек), перевязки ран, остановки кровотечения, предотвращения недержания мочи и т.п.
Изобретение направлено на разработку биоразрушаемого материала с улучшенной способностью к разрушению в биологической среде, повышенной способностью к восстановлению формы после деформации материала и улучшенной эластичностью. Биоразрушаемый материал, который представляет собой сшитый поперечными химическими связями продукт мультивалентного соединения A, включающего 3 или более функциональные группы X, например гидроксильные группы, мультивалентного соединения B, включающего 3 или более функциональные группы Y, например карбоксильные группы, а также соединения C, имеющего структуру производного гидроксикарбоновой кислоты, гомополимер которой, полученный при гомополимеризации, имеет температуру стеклования -40°C или ниже. Также изобретение относится к материалам для эмболизации сосудов, перевязки ран, к материалу, предупреждающему недержание мочи, антиадгезионному материалу, которые состоят из указанного биоразрушаемого материала. 7 н. и 6 з.п. ф-лы, 1 табл.
1. Биоразрушаемый материал, который представляет собой сшитый поперечными химическими связями продукт:
мультивалентного соединения A, включающего 3 или более функциональные группы X, выбранные из группы, состоящей из гидроксильной группы, тиольной группы и аминогруппы;
мультивалентного соединения B, включающего 3 или более функциональные группы Y, выбранные из группы, состоящей из карбоксильной группы, изоцианатной группы и тиоизоцианатной группы; а также
соединения C, имеющего структуру, образованную из гидроксикарбоновой кислоты, гомополимер которой, полученный при гомополимеризации, имеет температуру стеклования -40°C или ниже.
2. Биоразрушаемый материал по п. 1, где массовая доля структуры, образовавшейся из указанного соединения C, составляет от 18 до 70 масс.%.
3. Биоразрушаемый материал по п. 1 или 2, где указанное мультивалентное соединение A представляет собой одно из следующих соединений a)-e):
a) гомополимер или сополимер мономера(ов) водорастворимого полимера(ов), выбранного из группы, состоящей из полиэтиленгликоля, полипропиленгликоля, поливинилового спирта, полигидроксиэтилакрилата, полигидроксиэтилэтилметакрилата, карбоксиметилцеллюлозы, гидроксиметилцеллюлозы и гидроксиэтилцеллюлозы;
b) сополимер мономера указанного водорастворимого полимера и
мономера(ов) гидрофобного полимера(ов), выбранного из группы, состоящей из винилацетата и винилкапролактама;
c) сополимер мономера указанного водорастворимого полимера и гидроксикарбоновой кислоты(кислот), гомополимер(ы) которой, образующийся при гомополимеризации, имеет/имеют температуру стеклования -39°C или выше;
d) разветвленный полимер, образованный связыванием всех гидроксильных групп полиола с гомополимером или сополимером мономера(ов) водорастворимого полимера(ов), выбранного из группы, состоящей из полиэтиленгликоля и полипропиленгликоля;
e) сополимер разветвленного полимера и гидроксикарбоновой кислоты(кислот), гомополимер(ы) которой, образующийся при гомополимеризации, имеет/имеют температуру стеклования -39°C или выше.
4. Биоразрушаемый материал по п. 1 или 2, где указанное мультивалентное соединение B представляет собой одно из следующих соединений f)-i):
f) соединение, образованное при связывании гидроксильной группы(групп) гомополимера или сополимера мономера(ов) водорастворимого полимера(ов), выбранного из группы, состоящей из полиэтиленгликоля, полипропиленгликоля, поливинилового спирта, полигидроксиэтилакрилата, полигидроксиэтилметакрилата, карбоксиметилцеллюлозы, гидроксиметилцеллюлозы и гидроксиэтилцеллюлозы, с поликарбоновой кислотой(ами);
g) соединение, образованное при связывании гидроксильной группы(групп) сополимера мономера водорастворимого полимера и гидроксикарбоновой кислоты(кислот), гомополимер(ы) которой,
образующийся при гомополимеризации, имеет/имеют температуру стеклования -39°C или выше, с поликарбоновой кислотой(ами);
h) соединение, образованное при связывании гидроксильной группы(групп) разветвленного полимера, образованного при связывании всех гидроксильных групп полиола с гомополимером или сополимером мономера(ов) водорастворимого полимера(ов), выбранного из группы, состоящей из полиэтиленгликоля и полипропиленгликоля, с поликарбоновой кислотой(ами);
i) соединение, образованное при связывании гидроксильной группы(групп) сополимера разветвленного полимера и гидроксикарбоновой кислоты(кислот), гомополимер(ы) которой, образующийся при гомополимеризации, имеет/имеют температуру стеклования -39°C или выше, с поликарбоновой кислотой(кислотами).
5. Биоразрушаемый материал по п. 1 или 2, где указанное соединение C представляет собой сополимер мономера(ов) водорастворимого полимера(ов), выбранного из группы, состоящей из полиэтиленгликоля, полипропиленгликоля, поливинилового спирта, полигидроксиэтилакрилата, полигидроксиэтилметакрилата, карбоксиметилцеллюлозы, гидроксиметилцеллюлозы и гидроксиэтилцеллюлозы, и гидроксикарбоновой кислоты(кислот), гомополимер(ы) которой, полученный при гомополимеризации, имеет/имеют температуру стеклования -40°C или ниже.
6. Биоразрушаемый материал по п. 1 или 2, где указанное соединение C представляет собой 6-гидроксикапроновую кислоту.
7. Биоразрушаемый материал по п. 3, где указанный разветвленный полимер имеет степень разветвления от 3 до 16.
8. Биоразрушаемый материал по п. 3, где указанный полиол выбран из группы, состоящей из глицерина, полиглицерина и пентаэритрита.
9. Биоразрушаемый материал по п. 3, где указанная гидроксикарбоновая кислота(кислоты), гомополимер(ы) которой, образующийся при гомополимеризации, имеет/имеют температуру стеклования -39°C или выше, выбрана/выбраны из группы, состоящей из гликолевой кислоты, молочной кислоты, глицериновой кислоты, гидроксимасляной кислоты, яблочной кислоты, винной кислоты, гидроксивалериановой кислоты и 3-гидроксигексановой кислоты.
10. Биоразрушаемый материал по п. 4, где указанная поликарбоновая кислота(кислоты) выбрана/выбраны из группы, состоящей из щавелевой кислоты, малоновой кислоты, янтарной кислоты, глутаровой кислоты, адипиновой кислоты, пимелиновой кислоты, пробковой кислоты, азелаиновой кислоты, себациновой кислоты, яблочной кислоты, винной кислоты и фумаровой кислоты.
11. Материал для эмболизации сосудов, состоящий из указанного биоразрушаемого материала по п. 1 или 2.
12. Антиадгезионный материал, состоящий из указанного биоразрушаемого материала по п. 1 или 2.
13. Материал для перевязки ран, состоящий из указанного биоразрушаемого материала по п. 1 или 2.
14. Кровоостанавливающий материал, состоящий из указанного биоразрушаемого материала по п. 1 или 2.
15. Материал, предупреждающий недержание мочи, состоящий из указанного биоразрушаемого материала по п. 1 или 2.
16. Способ получения биоразрушаемого материала, где
указанный способ включает стадию поперечного сшивания химическими связями, в которой мультивалентное соединение A, включающее 3 или более функциональные группы X, выбранные из группы, состоящей из гидроксильной группы, тиольной группы и аминогруппы, мультивалентное соединение B, включающее 3 или более функциональные группы Y, выбранные из группы, состоящей из карбоксильной группы, изоцианатной группы и тиоизоцианатной группы, а также соединение C, имеющее структуру, образованную из гидроксикарбоновой кислоты, гомополимер которой, полученный при гомополимеризации, имеет температуру стеклования -40°C или ниже, растворяют в растворителе, и дают пройти химической реакции поперечного сшивания, с получением указанного биоразрушаемого материала.
| Устройство для охлаждения водою паров жидкостей, кипящих выше воды, в применении к разделению смесей жидкостей при перегонке с дефлегматором | 1915 |
|
SU59A1 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| ЩИТОВОЙ ДЛЯ ВОДОЕМОВ ЗАТВОР | 1922 |
|
SU2000A1 |
| СПОСОБ ПОЛУЧЕНИЯ ГЕЛЯ, КОМПОЗИЦИЯ ВОДНОГО ГЕЛЯ И СПОСОБ ИЗМЕНЕНИЯ ЕГО ФИЗИЧЕСКИХ ХАРАКТЕРИСТИК | 2002 |
|
RU2300541C2 |

