Область техники
[001] Настоящее изобретение относится к области фармацевтической биохимической инженерии, и в частности относится к простому способу получения авибактама.
Уровень техники
[002] Являясь не-β-лактамным ингибитором и одним из диазабициклооктаноновых соединений, авибактам (I) может ингибировать β-лактамазы типа A (включая ESBL и KPC) и типа C. При введении в комбинации с различными типами цефалоспоринов и карбапенемовых антибиотиков авибактам обладает широким спектром активности в отношении бактерий, в частности обладает значительной активностью в отношении Escherichia coli и Klebsiella pneumoniae, содержащих расширенный спектр β-лактамаз, Escherichia coli, содержащей дополнительный фермент AmpC, и Escherichia coli, содержащей как AmpC, так и расширенный спектр β-лактамаз. Авибактам (I), c № CAS 1192491-61-4 и химическим названием [(1R,2S,5R)-2-(аминокарбонил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил]сульфат натрия, характеризуется структурной формулой, представленной формулой I:
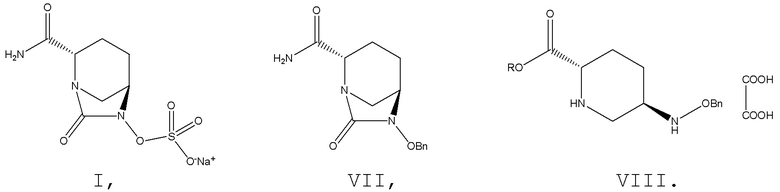
В существующие технологии синтеза авибактама главным образом вовлечены два промежуточных соединения, т. е. промежуточное соединение VII: (2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид; и промежуточное соединение VIII: 5R-[(бензилокси)амино]пиперидин-2S-карбоксилата оксалат.
[003] В предшествующем уровне техники в патентных литературных источниках CN103649051A, CN105294690A, CN106866668A, WO2012086241, US8148540, US9284273 и US9567335 авибактам (I) получали из (2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамида (VII) в качестве промежуточного соединения. Осуществляли дебензилирование соединения VII при катализе с помощью палладия-на-угле в присутствии различных восстановителей (таких как водород, триэтилсилан, формиат натрия и гидрат гидразина), затем осуществляли сульфатирование с помощью комплекса триоксида серы, и преобразовывали продукт в солевую форму четвертичного аммония с последующим ионным обменом с получением авибактама (I), как показано на схеме 1.
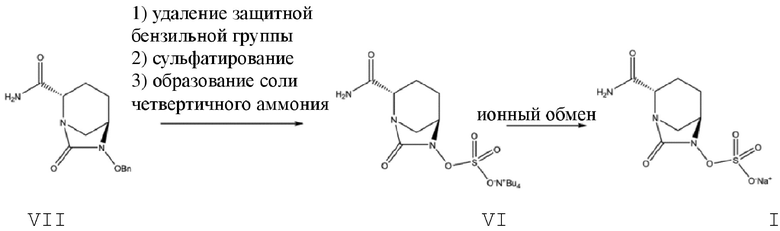
Схема 1
[004] Промежуточный продукт, представляющий собой (2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид, полученный путем дебензилирования при гидролизе в соответствии с данным способом, характеризуется низкой стабильностью и тенденцией вызывать отравление катализатора; кроме того, он требует использования значительного количества катализатора, представляющего собой палладий-на-угле (10% от концентрации субстрата), что не способствует снижению затрат и обуславливает низкую пригодность для промышленного использования.
I. Синтез промежуточного соединения, представляющего собой (2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (VII)
[005] В литературных источниках, указанных выше, различные способы получения промежуточного соединения, представляющего собой (2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (VII), главным образом подразделяются на две схемы: амидирование с последующей циклизацией мочевины и циклизация мочевины с последующим амидированием, как показано на схеме 2.
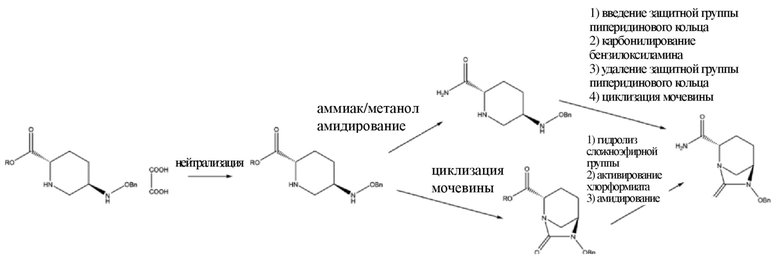
Схема 2
[006] В патентах CN103649051A и CN105294690A применяется схема амидирования с последующей циклизацией мочевины. 5R-[(бензилокси)амино]пиперидин-2S-карбоксилата оксалат (VIII) в качестве исходного вещества амидировали в метанольном растворе газообразного аммиака или спиртовом растворе аммиака, и реакционную смесь фильтровали для удаления оксалата аммония; осадок на фильтре, представляющий собой оксалат аммония, промывали метанолом, и полученный метанольный раствор концентрировали; продукт экстрагировали с помощью метилбензола и перекристаллизовывали с использованием подходящего растворителя с получением (2S,5R)-5-[(бензилокси)амино]пиперидин-2-карбоксамида (выход: 68-99%); затем осуществляли введение защитной группы для аминогруппы в пиперидиновом кольце полученного (2S, 5R)-5-[(бензилокси)амино]пиперидин-2-карбоксамида с помощью 9-флуоренилметилхлорформиата (FMOC-Cl) и осуществляли реакцию карбонилирования между карбонилдиимидазолом и бензилоксиламином; и после удаления защитной группы в пиперидиновом кольце с помощью диэтиламина проводили циклизацию мочевины с получением (2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамида (выход: 90%, общий выход: 61,2-89,1%). В данном способе получения обработка после амидирования осложнена, и защитное средство, представляющее собой 9-флуоренилметилхлорформиат, применяемое для циклизации мочевины являлось дорогостоящим. Кроме того, 9-флуоренилметилхлорформиат и карбонилдиимидазол обеспечивают только один карбонил, так что реакция характеризуется низкой атомной эффективностью, что не способствует защите окружающей среды и снижению затрат. Дополнительно, прямая циклизация мочевины в отношении (2S, 5R)-5-[(бензилокси)амино]пиперидин-2-карбоксамида с применением трифосгена и карбонилдиимидазола без введения защитной группы для аминогруппы в пиперидиновом кольце характеризовалась низким выходом (50-56%) с отсутствием промышленного значения.
[007] Помимо этого, все из патентов CN102834395A, CN103649051A, CN103328476A, CN106279163A, CN106565712A, US9284273 и US9567335 включают способ циклизации мочевины с последующим амидированием. Осуществляли циклизацию мочевины в отношении 5R-[(бензилокси)амино]пиперидин-2S-карбоксилата оксалата (VIII) в качестве исходного вещества с применением трифосген-органического основания, карбонилдиимидазола или других средств карбонилирования, затем гидролизовали в щелочных условиях, таких как водный гидроксид лития, с получением (2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты; затем карбоксил активировали до ангидрида с применением триметилацетилхлорида или других средств, и затем ангидрид амидировали с применением водного раствора аммиака с получением (2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамида (VII) с общим выходом 34,5-65,5%. (2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксилат, полученный циклизацией мочевины, характеризовался низкой реакционной способностью, который невозможно было непосредственно амидировать в метанольном растворе газообразного аммиака. Вместо этого, для эффективного амидирования сложноэфирную группу необходимо было гидролизовать до карбоксила, и затем карбоксил активировали до ангидрида. Данный способ характеризовался сложностью выполнения операций и низкой атомной эффективностью, что, таким образом, не способствовало защите окружающей среды и осуществлению промышленного производства.
II. Синтез промежуточного соединения, представляющего собой 5R-[(бензилокси)амино]пиперидин-2S-карбоксилата оксалат (VIII)
[008] В патентах США US2010197928 и US2013012712 раскрыт синтез 5R-[(бензилокси)амино]пиперидин-2S-карбоксилата оксалата (VIII) (см. схему 3). Вкратце, осуществляли реакцию раскрытия кольца L-пироглутамата, содержащего защитную группу для атома N, в качестве исходного вещества с йодидом триметилсульфоксида для удлинения углеродной цепи, при этом карбонил полученного соединения превращали в имин с помощью бензилоксиамина, и затем из промежуточного соединения удаляли защитную группу в кислых условиях, осуществляли циклизацию в щелочных условиях, и, в конечном итоге, осуществляли восстановление с помощью восстановителя и подвергали хиральному разделению с получением продукта, представляющего собой 5R-[(бензилокси)амино]пиперидин-2S-карбоксилата оксалат. Применяемые в данном способе исходные вещества, представляющие собой L-пироглутамат, содержащий защитную группу для атома N, йодид триметилсульфоксида, метансульфоновую кислоту, были дорогостоящими, причем получали значительное количество сточных вод при использовании диметилсульфоксида в качестве растворителя, вследствие чего это являлось неблагоприятным для окружающей среды; кроме того, общий выход был низким (59%).
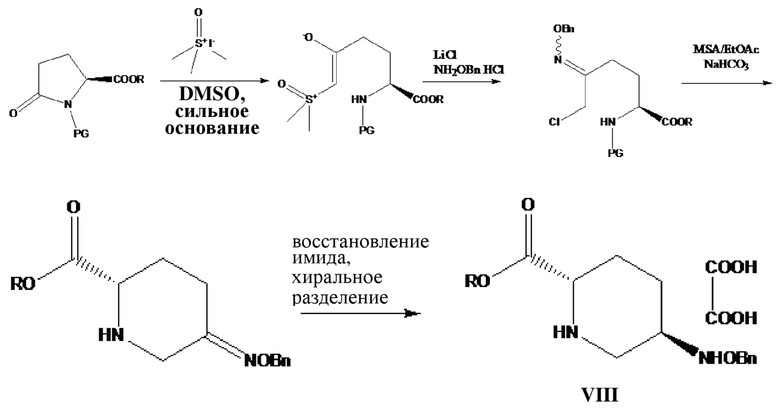
Схема 3
[009] В заявке на патент США № US20140275001 раскрыт другой способ синтеза 5R-[(бензилокси)амино]пиперидин-2S-карбоксилата (свободная форма VIII) (схема 4), где L-пироглутамат, содержащий защитную группу для атома N, также использовали в качестве исходного вещества и подвергали реакции раскрытия кольца с йодидом триметилсульфоксида для удлинения углеродной цепи. Разница заключается в том, что в патенте US20140275001 циклизацию сначала проводят с помощью иридиевого катализатора с получением спирта с S-конформацией посредством хирального восстановления карбонила; и затем инверсию SN2-конфигурации реализовывали посредством применения N-бензилокси-2-нитробензолсульфонамида, и гидроксил превращали в амино; 2-нитробензолсульфонилхлор-группу сначала удаляли под действием гидроксида лития и меркаптоуксусной кислоты, и затем защитную группу для атома N удаляли с помощью трифторуксусной кислоты с получением свободной формы продукта VIII. Способ характеризовался сложностью выполнения операций, и в нем применяли дорогостоящий иридиевый катализатор и меркаптоуксусную кислоту со специфическим запахом; кроме того, при этом получали значительное количество сточных вод, но общий выход составлял только 15%.
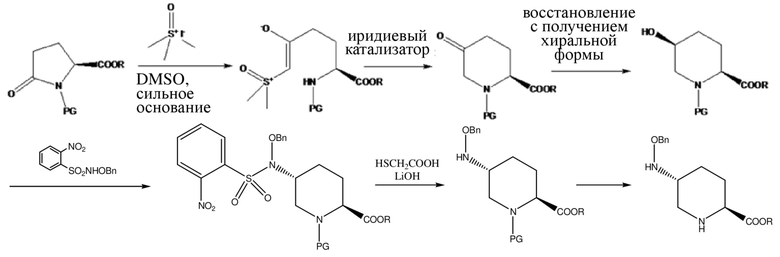
Схема 4
[0010] Ввиду вышеизложенного, схемы синтеза, включающие промежуточное соединение, представляющее собой 5R-[(бензилокси)амино]пиперидин-2S-карбоксилата оксалат (VIII), и промежуточное соединение, представляющее собой (2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (VII), для синтезирования авибактама являются продолжительными, и в них применяются дорогостоящие исходные вещества; они требуют большого количества дорогостоящего катализатора, представляющего собой палладий-на-угле; кроме того, они предусматривают сброс значительного количества сточных вод, отработанного газа и отработанных остатков, что является неблагоприятным для окружающей среды, и характеризуются низкой атомной эффективностью. Помимо этого, сложность выполнения операций не способствует промышленному производству.
Краткое описание
[0011] Для устранения недостатков предшествующего уровня техники, в настоящем изобретении предусмотрен простой способ получения авибактама. Настоящее изобретение характеризуется простыми стадиями получения, простыми схемами, простотой выполнения операций и недорогими исходными веществами; кроме того, в настоящем изобретении не требуется применение дорогостоящего катализатора, представляющего собой палладий-на-угле, ввиду чего оно обеспечивает снижение затрат; кроме того, оно предусматривает сброс меньшего количества сточных вод, отработанного газа и отработанных остатков, вследствие чего это является благоприятным для окружающей среды; при этом значения выхода его соответствующих стадий являются высокими, что способствует промышленному производству авибактама.
[0012] Определение терминов
[0013] Соединение формулы II: пиперидин-5-он-2S-карбоксилат.
[0014] Соединение формулы III: 5-замещенный оксииминопиперидин-2S-карбоксилат, где волнистая линия в структурной формуле представляет смесь двух хиральных структур.
[0015] Соединение формулы IV: 5R-замещенный оксиаминопиперидин-2S-карбоксилата оксалат.
[0016] Соединение формулы V: 5R-замещенный оксиаминопиперидин-2S-карбоновая кислота.
[0017] Соединение формулы VI: (2S,5R)-6-замещенный окси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид.
[0018] Соединение формулы VII: (2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил]окси}сульфонил-тетра-н-бутиламмониевая соль; при этом в данной структуре -Bu относится к н-бутилу.
[0019] Номера соединений в описании полностью соответствуют номерам их структурных формул, и они имеют одни и те же ссылки.
[0020] Техническое решение по настоящему изобретению представлено ниже.
[0021] Способ получения авибактама предусматривает стадии:
[0022] (1) осуществление реакции конденсации соединения формулы II и гидроксиламина гидрохлорида, содержащего защитную группу для атома O, в растворителе и при катализе с помощью основания a с получением соединения формулы III:

[0023] где R в соединении формулы II является идентичным R в соединении формулы III, который при этом выбран из группы, состоящей из метила, этила, изопропила, н-пропила, трет-бутила, н-бутила, изобутила или бензила; PG в соединении формулы III выбран из группы, состоящей из метоксиметила, бензилоксиметила, трет-бутилдиметилсилила, трет-бутилдифенилсилила, триэтилсилила или триизопропилсилила;
[0024] (2) осуществление восстановления соединения формулы III с помощью восстановителя в присутствии концентрированной серной кислоты и этилацетата и хирального разделения с получением соединения формулы IV:
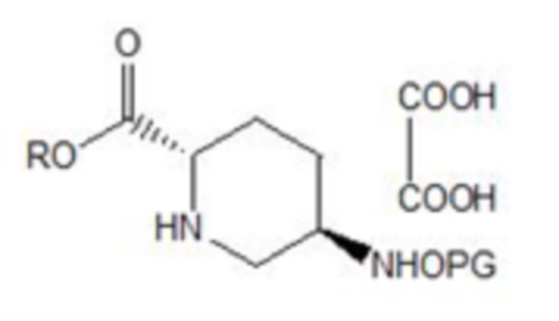

IV,
[0025] где R и PG в соединении формулы IV имеют такие же значения, что и R и PG в соединении формулы III;
[0026] (3) осуществление гидролиза соединения формулы IV в присутствии основания b и в растворителе b с получением соединения формулы V:
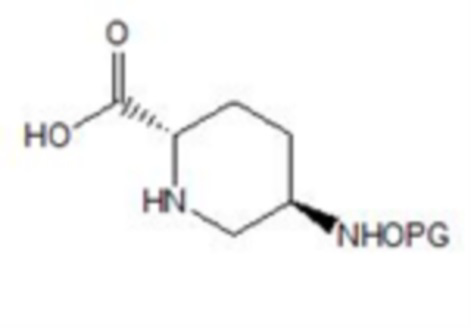

V,
[0027] где PG в соединении формулы V имеет такое же значение, что и PG в соединении формулы IV;
[0028] (4) осуществление в отношении соединения формулы V циклизации мочевины и ацилхлорирования с использованием фосгена, твердого фосгена или дифосгена в присутствии основания c и катализатора и в растворителе c, и затем осуществление амидирования с получением соединения формулы VI:
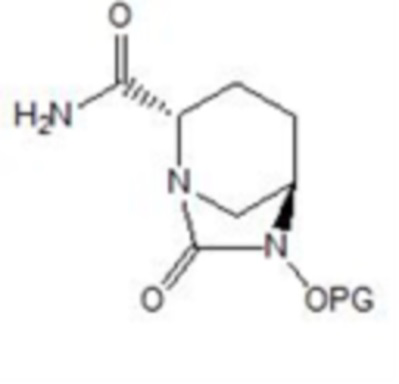

VI,
[0029] где PG в соединении формулы VI имеет такое же значение, что и PG в соединении формулы V;
[0030] (5) осуществление в отношении соединения формулы VI удаления защитной группы с использованием реагента для удаления защитной группы, сульфатирования и образования тетрабутиламмониевой соли при катализе с помощью основания d и в растворителе d с получением соединения формулы VII:
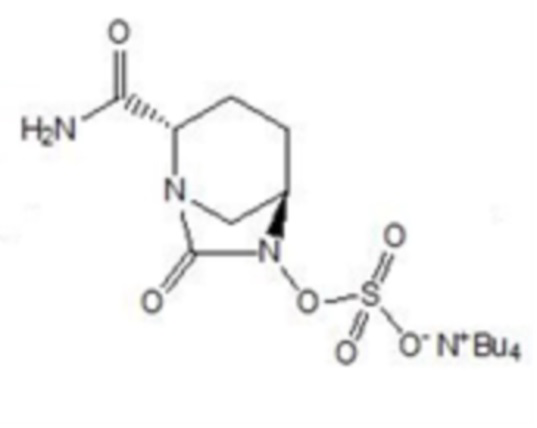

VII;
[0031] (6) осуществление в отношении соединения формулы VII ионного обмена с получением авибактама (I).
[0032] Предпочтительно, в соответствии с настоящим изобретением, используемый на стадии (1) гидроксиламина гидрохлорид, содержащий защитную группу для атома O, выбран из группы, состоящей из метоксиметилгидроксиламина гидрохлорида, бензилоксиметилгидроксиламина гидрохлорида, трет-бутилдиметилсилилгидроксиламина гидрохлорида, трет-бутилдифенилсилилгидроксиламина гидрохлорида, триэтилсилилгидроксиламина гидрохлорида, триизопропилсилилгидроксиламина гидрохлорида; при этом молярное соотношение гидроксиламина гидрохлорида, содержащего защитную группу для атома O, и соединения формулы II составляет 0,9-1,5:1.
[0033] Предпочтительно, в соответствии с настоящим изобретением, используемый на стадии (1) растворитель выбран из группы, состоящей из метанола, этанола, пропанола, бутанола, этилацетата, тетрагидрофурана, ацетонитрила, дихлорметана, хлороформа, 1,2-дихлорэтана, бензола и толуола или смеси двух или более из них; при этом массовое соотношение растворителя и соединения формулы II составляет 3-15:1, и предпочтительно массовое соотношение растворителя и соединения формулы II составляет 6-10:1.
[0034] Предпочтительно, в соответствии с настоящим изобретением, используемое на стадии (1) основание a представляет собой неорганическое основание или органическое основание; предпочтительно неорганическое основание выбрано из группы, состоящей из карбоната калия, карбоната натрия, карбоната кальция, гидрокарбоната калия, гидрокарбоната натрия, гидрокарбоната кальция, ацетата калия, ацетата натрия или ацетата кальция или комбинации двух или более из них, и органическое основание выбрано из группы, состоящей из триэтиламина или три-н-бутиламина или комбинации двух из них; при этом массовое соотношение основания a и соединения формулы II составляет 0,5-1,5:1.
[0035] Предпочтительно, в соответствии с настоящим изобретением, используемая на стадии (1) температура реакции конденсации находится в диапазоне от 30°C до 80°C; предпочтительно температура реакции конденсации находится в диапазоне от 30°C до 60°C. Продолжительность реакции конденсации находится в диапазоне от 2 часов до 5 часов.
[0036] Предпочтительно, в соответствии с настоящим изобретением, используемая на стадии (2) концентрированная серная кислота представляет собой серную кислоту с массовой долей, находящейся в диапазоне от 95% до 98%, и молярное соотношение концентрированной серной кислоты и соединения формулы III составляет (3,0-6,0):1; предпочтительно концентрированная серная кислота представляет собой серную кислоту с массовой долей, составляющей 98%. В настоящем изобретении предусмотрен способ объединения концентрированной серной кислоты и субстрата с образованием соли, повышая таким образом селективность реакции восстановления.
[0037] Предпочтительно, в соответствии с настоящим изобретением, используемое на стадии (2) массовое соотношение этилацетата и соединения формулы III составляет 5-20:1; еще более предпочтительно массовое соотношение этилацетата и соединения формулы III находится в диапазоне 10-14:1. Причиной применения этилацетата в настоящем изобретении является облегчение отделения от водной фазы в последующей обработке. Полученный продукт, т. е. соединение формулы IV, характеризуется более высокой растворимостью в этилацетате.
[0038] Предпочтительно, в соответствии с настоящим изобретением, используемый на стадии (2) восстановитель выбран из группы, состоящей из борогидрида натрия, трицианоборогидрида натрия, триацетоксиборогидрида натрия, трипропионилоксиборогидрида натрия, борогидрида калия, трицианоборогидрида калия, триацетоксиборогидрида калия или трипропионилоксиборогидрида калия; при этом молярное соотношение восстановителя и соединения формулы III составляет 2,0-4,0:1.
[0039] Предпочтительно, в соответствии с настоящим изобретением, используемая на стадии (2) температура реакции восстановления находится в диапазоне от (-30) - (-10)°C. Продолжительность реакции восстановления находится в диапазоне от 2 часов до 8 часов.
[0040] В соответствии с настоящим изобретением, применяемый на стадии (2) способ хирального разделения проводят в соответствии с предшествующим уровнем техники.
[0041] Предпочтительно, в соответствии с настоящим изобретением, используемый на стадии (3) растворитель b выбран из группы, состоящей из воды, метанола, этанола, пропанола, бутанола, этилацетата, дихлорметана, хлороформа, 1,2-дихлорэтана, бензола и толуола или комбинации двух или более из них; при этом массовое соотношение растворителя b и соединения формулы IV составляет 3-12:1; и массовое соотношение растворителя b и соединения формулы IV составляет 3-6:1.
[0042] Предпочтительно, в соответствии с настоящим изобретением, используемое на стадии (3) основание b выбрано из группы, состоящей из гидроксида натрия, гидроксида калия, гидроксида лития, карбоната калия, карбоната натрия, гидрокарбоната калия или гидрокарбоната натрия или смеси двух или более из них; при этом молярное соотношение основания b и соединения формулы IV составляет 1,5-4,0:1.
[0043] Предпочтительно, в соответствии с настоящим изобретением, используемую на стадии (3) реакцию гидролиза проводят при температуре, составляющей 10-100°C. Предпочтительно реакцию гидролиза проводят при температуре, составляющей 20-50°C. Продолжительность реакции гидролиза находится в диапазоне от 2 часов до 7 часов.
[0044] Предпочтительно, в соответствии с настоящим изобретением, используемый на стадии (4) растворитель c выбран из группы, состоящей из дихлорметана, 1,2-дихлорэтана, трихлорметана, тетрахлорметана, ацетонитрила, тетрагидрофурана, 2-метилтетрагидрофурана, метоксициклопентана и метилбензола или комбинации двух или более из них; при этом массовое соотношение растворителя c и соединения формулы V составляет 4-30:1, предпочтительно массовое соотношение растворителя c и соединения формулы V составляет 18-30:1.
[0045] Предпочтительно, в соответствии с настоящим изобретением, используемое на стадии (4) основание c выбрано из группы, состоящей из триметиламина, триэтиламина, три-н-бутиламина, диизопропилэтиламина, карбоната калия, карбоната натрия или карбоната кальция или комбинации двух или более из них; при этом молярное соотношение основания c и соединения формулы V составляет 3,0-8,0:1.
[0046] Предпочтительно, в соответствии с настоящим изобретением, катализатор, используемый на стадии (4), выбран из группы, состоящей из N,N-диметилформамида, пиридина или 4-диметиламинопиридина или комбинации двух или более из них; при этом масса катализатора составляет 0,1-5,0% от массы соединения формулы V.
[0047] Предпочтительно, в соответствии с настоящим изобретением, используемое на стадии (4) молярное соотношение твердого фосгена/дифосгена/фосгена и соединения формулы V составляет 0,6-5,0:1; предпочтительно молярное соотношение твердого фосгена и соединения формулы V составляет 1,2-2,0:1; при этом молярное соотношение дифосгена и соединения формулы V составляет 1,0-2,5:1, и молярное соотношение фосгена и соединения формулы V составляет 2,0-4,0:1.
[0048] Предпочтительно, в соответствии с настоящим изобретением, в качестве аммиака на стадии (4) применяется аммиак, выбранный из группы, состоящей из газообразного аммиака, спиртового раствора газообразного аммиака, раствора газообразного аммиака в тетрагидрофуране, раствора газообразного аммиака в ацетонитриле или гидроксида аммония; при этом массовая концентрация газообразного аммиака в спиртовом растворе газообразного аммиака, растворе газообразного аммиака в тетрагидрофуране, растворе газообразного аммиака в ацетонитриле или гидроксиде аммония составляет 5-20%.
[0049] Предпочтительно, в соответствии с настоящим изобретением, молярное соотношение аммиака и соединения формулы V на стадии (4) составляет 1,0-6,0:1.
[0050] Предпочтительно, в соответствии с настоящим изобретением, все значения температуры реакций циклизации мочевины, ацилхлорирования, амидирования находятся в диапазоне от -20°C до 60°C; предпочтительно все значения температуры реакций циклизации мочевины, ацилхлорирования, амидирования находятся в диапазоне от 10°C до 30°C. Все значения продолжительности циклизации мочевины, ацилхлорирования, амидирования находятся в диапазоне от 1 часа до 8 часов.
[0051] Предпочтительно, в соответствии с настоящим изобретением, используемый на стадии (5) растворитель d выбран из группы, состоящей из воды, изопропанола, изобутанола, этилацетата, дихлорметана, хлороформа, 1,2-дихлорэтана или изобутилметилкетона или комбинации двух или более из них; при этом массовое соотношение растворителя d и соединения формулы VI составляет 4-20:1; предпочтительно массовое соотношение растворителя d и соединения формулы VI составляет 4-8:1.
[0052] Предпочтительно, в соответствии с настоящим изобретением, используемое на стадии (5) основание d выбрано из группы, состоящей из триметиламина, триэтиламина, три-н-бутиламина и диизопропилэтиламина; при этом молярное соотношение основания d и соединения формулы VI составляет 0,2-0,7:1.
[0053] Предпочтительно, в соответствии с настоящим изобретением, если используемая на стадии (5) PG в соединении формулы VI представляет собой защитную группу, не содержащую кремний, то средство для удаления защитной группы выбрано из группы, состоящей из комплекса триоксида серы и триметиламина, комплекса триоксида серы и триэтиламина и комплекса триоксида серы и пиридина; если PG в соединении формулы VI представляет собой защитную группу, содержащую кремний, то средство для удаления защитной группы представляет собой фтортетрабутиламмоний; при этом молярное соотношение средства для удаления защитной группы и соединения формулы VI составляет 1,0-3,0:1.
[0054] Предпочтительно, в соответствии с настоящим изобретением, используемый на стадии (5) реагент, применяемый при сульфатировании, выбран из группы, состоящей из комплекса триоксида серы и триметиламина, комплекса триоксида серы и триэтиламина или комплекса триоксида серы и пиридина; при этом молярное соотношение реагента, применяемого для сульфатирования, и соединения формулы VI составляет 1,0-3,0:1.
[0055] Предпочтительно, в соответствии с настоящим изобретением, используемый на стадии (5) реагент для образования соли, применяемый в реакции образования тетрабутиламмониевой соли, представляет собой ацетат тетрабутиламмония или фтортетрабутиламмоний, и молярное соотношение реагента для образования соли, применяемого в реакции образования тетрабутиламмониевой соли, и соединения формулы VI составляет 0,5-2:1.
[0056] Предпочтительно, в соответствии с настоящим изобретением, на стадии (5) удаление защитной группы, сульфатирование и реакцию образования тетрабутиламмониевой соли проводят с помощью «однореакторного» способа; при этом температура реакции находится в диапазоне от 0°C до 60°C; предпочтительно температура реакции находится в диапазоне от 10°C до 30°C. Все значения продолжительности в отношении удаления защитной группы, сульфатирования и реакции образования тетрабутиламмониевой соли находятся в диапазоне от 1 часа до 8 часов.
[0057] Предпочтительно, в соответствии с настоящим изобретением, используемый на стадии (6) реагент, применяемый в ионном обмене, представляет собой изооктаноат натрия; при этом молярное соотношение реагента, применяемого в ионном обмене, и соединения формулы VII составляет 1,5-3,0:1.
[0058] Предпочтительно, в соответствии с настоящим изобретением, используемая на стадии (6) температура реакции ионного обмена находится в диапазоне от 0°C до 50°C; предпочтительно температура реакции ионного обмена находится в диапазоне от 10°C до 40°C. Продолжительность реакции ионного обмена находится в диапазоне от 1 часа до 5 часов.
[0059] В соответствии с настоящим изобретением, применяемый на стадии (6) способ ионного обмена осуществляют в соответствии с предшествующим уровнем техники.
[0060] В настоящем изобретении пиперидин-5-он-2S-карбоксилат II в качестве исходного вещества подвергают реакции конденсации с гидроксиламина гидрохлоридом, содержащим защитную группу для атома O, в присутствии основного реагента с получением соединения формулы III, представляющего собой 5-замещенный оксииминопиперидин-2S-карбоксилат; соединение формулы III подвергают восстановлению и хиральному разделению с получением соединения формулы IV, представляющего собой 5R-замещенный оксиаминопиперидин-2S-карбоксилата оксалат; соединение формулы IV гидролизуют в щелочных условиях с получением соединения формулы V, представляющего собой 5R-замещенную оксиаминопиперидин-2S-карбоновую кислоту; в присутствии растворителя, основания и катализатора соединение формулы V при использовании фосгена, твердого фосгена или дифосгена подвергают циклизации мочевины, ацилхлорированию и амидированию с помощью «однореакторного» способа с получением соединения формулы VI: (2S,5R)-6-замещенного окси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамида; соединение формулы VI подвергают удалению защитной группы, сульфатированию и реакции образования тетрабутиламмониевой соли с получением соединения формулы VII, представляющего собой (2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил]окси}сульфонил-тетра-н-бутиламмониевую соль, и соединение формулы VII подвергают ионному обмену с получением авибактама (I). Схема представлена ниже.
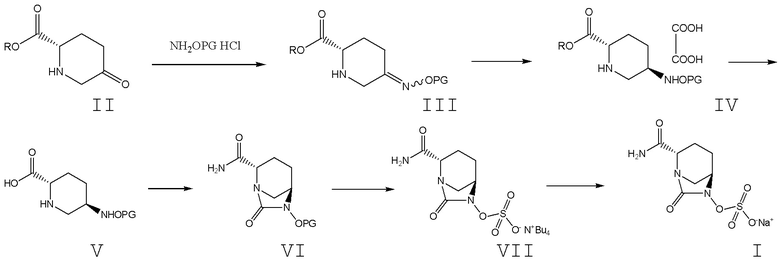
Схема 5
Технические характеристики и преимущественные эффекты настоящей заявки
[0061] 1. В настоящем изобретении применяется небензильная защитная группа для атома O гидроксиламина гидрохлорида, и полученное промежуточное соединение, представляющее собой (2S,5R)-6-замещенный окси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид, подвергают удалению защитной группы, сульфатированию, образованию тетрабутиламмониевой соли в кислой среде с помощью «однореакторного» способа, вследствие чего стадии являются простыми; кроме того, в качестве реагента для введения защитной группы и реагента для сульфатирования может применяться один и тот же реагент, что дополнительно снижает затраты; при этом в настоящем изобретении применяется простой способ удаления небензильной защитной группы без необходимости применения дорогостоящих йодида триметилсульфоксида, 9-флуоренилметилхлорформиата (FMOC-Cl), карбонилдиимидазола и 10% палладия на угле, применяемых в способах из предшествующего уровня техники, что, таким образом, снижает остатки тяжелых металлов, улучшает качество продукта и дополнительно снижает затраты.
[0062] 2. В способе получения согласно настоящему изобретения на стадии (2) концентрированную серную кислоту с определенной концентрацией объединяют с субстратом с образованием соли, что способствует повышению селективности реакции восстановления. В способе получения согласно настоящему изобретению стадию (4) завершают с помощью разработанного «однореакторного» способа, т. е. циклизация мочевины - ацилхлорирование - амидирование в «одном реакторе», вследствие чего стадия является простой, что позволяет избежать проблем, присущих традиционным способам, таких как сложная обработка после амидирования, применение дорогостоящего реагента для введения защитной группы при циклизации мочевины и низкая атомная эффективность реакций.
[0063] 3. В сравнении с традиционными способами получения авибактама способ получения авибактама согласно настоящему изобретению характеризуется простыми стадиями получения, простыми схемами, простотой выполнения операций и недорогими исходными веществами; кроме того, в настоящем изобретении не требуется использование дорогостоящего катализатора, представляющего собой палладий-на-угле, вследствие чего он характеризуется низкими затратами; кроме того, он предусматривает сброс меньшего количества сточных вод, отработанного газа и отработанных остатков, вследствие чего он является благоприятным для окружающей среды; при этом значения выхода его соответствующих стадий являются высокими, что способствует промышленному производству авибактама.
Краткое описание графических материалов
[0064] На фиг. 1 показаны результаты 1H-ЯМР-спектроскопии метил-5R-метоксиметилоксиаминопиперидин-2S-карбоксилата оксалата (IV1), полученного на стадии (2) примера 1.
[0065] На фиг. 2 показаны результаты 1H-ЯМР-спектроскопии 5R-метоксиметилоксиаминопиперидин-2S-карбоновой кислоты (V1), полученной на стадии (3) примера 1.
[0066] На фиг 3 представлены результаты 1H-ЯМР-спектроскопии (2S,5R)-6-метоксиметилокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамида (VI1), полученного на стадии (4) примера 1.
[0067] На фиг. 4 представлены результаты 1H-ЯМР-спектроскопии {[(2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил]окси}сульфонил-тетра-н-бутиламмониевой соли (VII), полученной на стадии (5) примера 1.
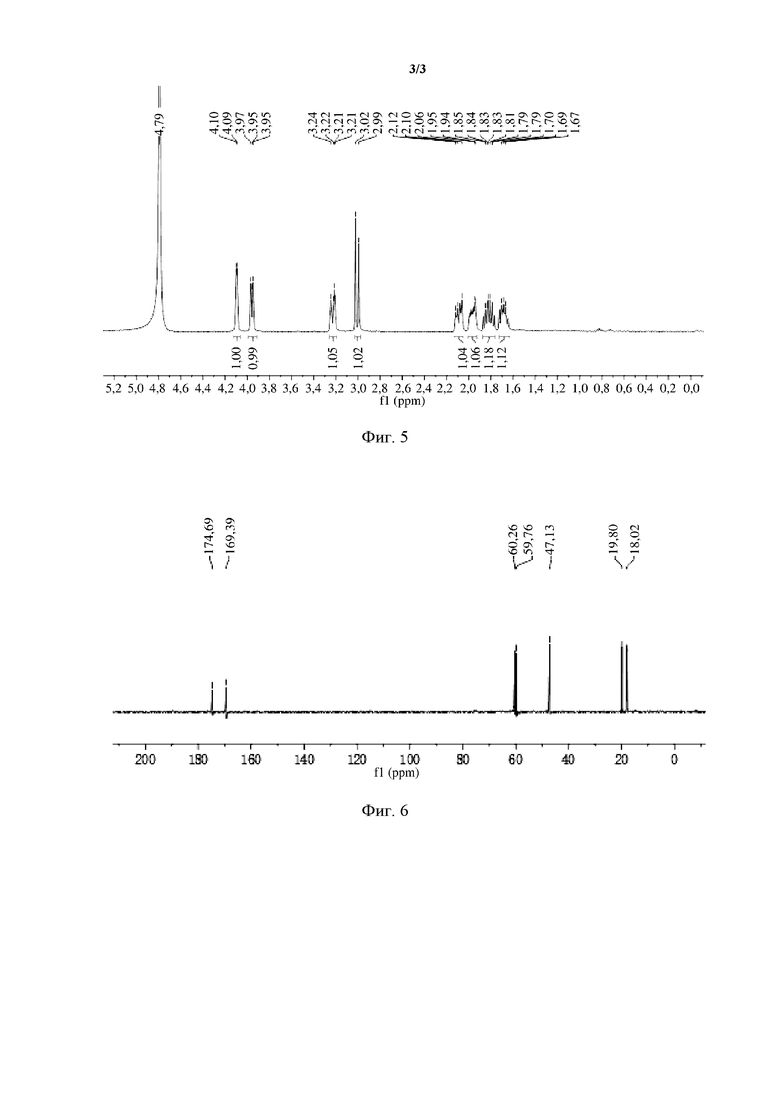
[0068] На фиг. 5 представлены результаты 1H-ЯМР-спектроскопии авибактама (I), полученного на стадии (6) примера 1.
[0069] На фиг. 6 представлены результаты 13C-ЯМР-спектроскопии авибактама (I), полученного на стадии (6) примера 1.
Подробное описание вариантов осуществления
[0070] Далее в данном документе настоящее изобретение будет описано подробно со ссылкой на примеры, хотя настоящее изобретение ими не ограничивается.
[0071] Если не указано иное, все значения процентного содержания (%) в примерах означают массовые процентные доли.
[0072] Исходные вещества, представляющие собой пиперидин-5-он-2S-карбоксилат, метоксиметилгидроксиламина гидрохлорид, трет-бутилдиметилсилилгидроксиламина гидрохлорид, являются коммерчески доступными (реализуются фармацевтической компанией Jinan Qinsi).
[0073] Процесс протекания реакции и чистоту продукта контролировали с помощью газового хроматографа или жидкостного хроматографа. Жидкостный хроматограф, оснащенный хиральной колонкой (ES-OVS, 150 мм x 4,6 мм, Agilent), применяют для определения оптической чистоты (% отношение значений площади) и расчета выхода, а также % значения энантиомерной чистоты.
Пример 1. Получение авибактама (I)
Стадия (1). Получение 5-метоксиметилоксииминопиперидин-2S-карбоксилата (III1)
[0074] В 4-горлую колбу объемом 500 мл, оснащенную мешалкой, термометром и конденсатором с обратным холодильником, добавляли по отдельности 200 г 1,2-дихлорметана, 23,5 г (0,15 моль) метилпиперидин-5-он-2S-карбоксилата, 20,5 г (0,18 моль) метоксиметилгидроксиламина гидрохлорида и 25 г триэтиламина, затем перемешивали для осуществления реакции при температуре, составляющей от 40°C до 45°C, в течение 4 часов. После охлаждения смеси до 20°C - 25°C добавляли 100 г воды. Затем раствор разделяли и водный слой экстрагировали два раза с помощью 1,2-дихлорэтана (по 50 г каждый раз). Органические фазы объединяли и промывали дважды с помощью насыщенного солевого раствора (по 25 г каждый раз). Органическую фазу перегоняли с извлечением растворителя и затем перегоняли при пониженном давлении с получением 31,3 г метил-5-метоксиметилоксииминопиперидин-2S-карбоксилата в виде желтоватой жидкости с показателем чистоты согласно GC, составляющим 99,8%, и выходом, составляющим 96,5%.
Стадия (2). Получение метил-5R-метоксиметилоксиаминопиперидин-2S-карбоксилата оксалата (IV1)
[0075] В 4-горлую колбу объемом 500 мл, оснащенную мешалкой и термометром, добавляли 200 г этилацетата, 17,3 г (0,08 моль) метил-5-метоксиметилоксииминопиперидин-2S-карбоксилата, полученного на стадии (1), и затем охлаждали. Затем добавляли по каплям 40,3 г (0,4 моль) концентрированной серной кислоты (массовая концентрация 98%) при -20°C, и затем перемешивали в течение 1 часа. Добавляли 38,0 г (0,18 моль) триацетоксиборогидрида при -20°C, затем перемешивали для осуществления реакции при температуре, составляющей от -20°C до -15°C, в течение 5 часов. Смесь поддерживали при температуре ниже 10°C, и затем реакционную смесь медленно добавляли в 200 г 10% водного раствора аммиака; затем раствор разделяли и органическую фазу промывали два раза с помощью насыщенного солевого раствора (по 25 г каждый раз). Органическую фазу концентрировали для извлечения растворителя, затем добавляли к остатку 80 г этилацетата, 40 г метанола и 11,5 г (0,09 моль) дигидрата щавелевой кислоты, и нагревали до 45°C, перемешивали в течение 1 часа, и затем охлаждали и фильтровали. Полученный осадок на фильтре сначала промывали с помощью 60 г смешанной жидкости, содержащей этилацетат/метанол (2:1), и затем промывали с помощью 50 г этилацетата. После высушивания под вакуумом получали 15,8 г оптического изомера метил-5R-метоксиметилоксиаминопиперидин-2S-карбоксилата оксалата с показателем чистоты согласно хиральной HPLC, составляющим 99,7%, и выходом, составляющим 64,0%.
[0076] Данные ЯМР продукта представлены ниже. 1H-ЯМР (400 MГц, DMSO-d6) δ: 1,39 (m, 1H), 1,64 (m, 1H), 1,85 (m, 1H), 2,12 (m, 1H), 2,62 (t, 1H), 3,06 (m, 1H), 3,36 (d, 1H), 3,74 (s, 3H), 3,93 (q, 1H), 4,58 (s, 2H), 7,26-7,38 (m, 5H).
[0077] Результаты 1H-ЯМР-спектроскопии продукта показаны на фиг. 1.
Стадия (3). Получение 5R-метоксиметилоксиаминопиперидин-2S-карбоновой кислоты (V1)
[0078] В 4-горлую колбу объемом 500 мл, оснащенную мешалкой и термометром, добавляли 30,8 г (0,1 моль) метил-5R-метоксиметилоксиаминопиперидин-2S-карбоксилата оксалат, полученный на стадии (2), 50 г воды, 100 г метанола и 70 г (0,35 моль) водного раствора гидроксида натрия (20 вес. %), и перемешивали для осуществления реакции при температуре, составляющей от 30°C до 35°C, в течение 3 часов. После завершения реакции гидролиза раствор охлаждали до температуры, составляющей от 0°C до 5°C, и затем подкисляли с помощью уксусной кислоты для регулирования значения pH до 3,5-3,0. Реакционную смесь фильтровали и высушивали с получением 18,6 г 5R-метоксиметилоксиаминопиперидин-2S-карбоновой кислоты с показателем чистоты согласно HPLC, составляющим 99,8%, и выходом, составляющим 91,2%.
[0079] Данные ЯМР продукта представлены ниже. 1H-ЯМР (400 MГц, DMSO-d6) δ: 1,25 (m, 1H), 1,44 (m, 1H), 1,79 (m, 1H), 2,10 (m, 1H), 3,02 (m, 1H), 3,07 (br, 1H), 3,21 (d, 1H), 4,57 (s, 2H), 6,75 (s, 1H), 7,29-7,34 (m, 5H).
[0080] Результаты 1H-ЯМР-спектроскопии продукта показаны на фиг. 2.
Стадия (4). Получение (2S,5R)-6-метоксиметилокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамида (VI1)
[0081] В 4-горлую колбу объемом 500 мл, оснащенную мешалкой и термометром, добавляли 200 г тетрагидрофурана, 10,2 г (0,05 моль) 5-метоксиметилоксиаминопиперидин-2S-карбоновой кислоты, полученной на стадии (3), 50 г диизопропилэтиламина и 0,1 г N,N-диметилформамида, и затем охлаждали; добавляли по каплям смешанный раствор 23,8 г (0,08 моль) твердого фосгена и 80 г тетрагидрофурана при температуре, составляющей от -10°C до 0°C, и после завершения добавления по каплям реакционную смесь перемешивали для осуществления реакции при температуре, составляющей от 10°C до 20°C, в течение 4 часов. Вводили 4,0-4,5 г газообразного аммиака при температуре, составляющей от 10°C до 20°C, и перемешивали для осуществления реакции при температуре, составляющей от 15°C до 20°C, в течение 3 часов. Реакционную жидкость выливали в 300 г смеси воды и льда и разделяли, а затем водную фазу дважды экстрагировали с помощью дихлорметана (по 50 г каждый раз). Органические фазы объединяли и промывали дважды с помощью насыщенного раствора хлорида натрия (по 20 г каждый раз). После извлечения растворителя из полученной органической фазы получали 10,7 г (2S,5R)-6-метоксиметилокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамида с показателем чистоты согласно HPLC, составляющим 99,8%, и выходом, составляющим 93,5%. Точка плавления: 169,1°C-170,0°C. Удельное вращение: [α]20 D= -26,2° (c=0,5, MeOH).
[0082] Данные ЯМР продукта представлены ниже. 1H-ЯМР (400 MГц, DMSO-d6) δ: 1,63 (m, 2H), 1,84 (m, 1H), 2,06 (m, 1H), 2,90 (s, 2H), 3,62 (br, 1H), 3,68 (d, 1H), 4,94 (q, 1H), 4,58 (s, 2H), 7,28-7,46 (m, 5H).
[0083] Результаты 1H-ЯМР-спектроскопии продукта показаны на фиг. 3.
Стадия (5). Получение {(2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил]окси}сульфонил-тетра-н-бутиламмониевой соли (VII)
[0084] В 4-горлую колбу объемом 500 мл, оснащенную мешалкой и термометром, добавляли 100 г изопропанола, 2,0 г воды, 23,0 г (0,1 моль) (2S,5R)-6-метоксиметилокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамида, полученного на стадии (4), 5,0 г триэтиламина и 36,0 г (0,12 моль) ацетата тетрабутиламмония и добавляли 46,5 г (0,25 моль) комплекса триоксида серы и триметиламина при температуре, составляющей от 10°C до 15°C; раствор смеси перемешивали для осуществления реакции при температуре, составляющей от 15°C до 20°C, в течение 4 часов. Реакционную жидкость выливали в 150 г дихлорметана и 150 г смеси льда и воды. Затем добавляли уксусную кислоту для регулирования значения pH системы до 3,5-2,5. Смешанный раствор разделяли, и затем водный слой экстрагировали два раза с помощью 1,2-дихлорметана (по 50 г каждый раз). Органические фазы объединяли и промывали дважды с помощью насыщенного раствора хлорида натрия (по 20 г каждый раз). После извлечения растворителя из полученной органической фазы остатки перекристаллизовывали с помощью 50 г дихлорметана-метилизобутилкетона (объемное соотношение 1:3) с получением 46,3 г {(2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил]окси}сульфонил-тетра-н-бутиламмониевой соли с показателем чистоты согласно HPLC, составляющим 99,9%, и выходом, составляющим 91,5%. Удельное вращение: [α]20 D= -29,4(c=0,5, H2O).
[0085] Данные ЯМР продукта представлены ниже. 1H-ЯМР (400 MГц, DMSO-d6) δ: 0,97 (t, 12H), 1,42 (m, 8H), 1,64 (m, 9H), 1,84 (m, 1H), 2,12 (m, 1H), 2,35 (m, 1H), 2,83 (d, 1H), 3,27 (m, 9H), 3,89 (d, 1H), 4,30 (s, 1H), 5,83 (s, 1H), 6,66 (s, 1H).
[0086] Результаты 1H-ЯМР-спектроскопии продукта показаны на фиг. 4.
Стадия (6). Получение авибактама (I)
[0087] В колбу объемом 500 мл, оснащенную мешалкой и термометром, добавляли 260 г этилового спирта (2 вес. % воды), 56,0 г (0,1 моль) {[(2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил]окси}сульфонил-тетра-н-бутиламмониевой соли, перемешивали и растворяли при температуре, составляющей от 20°C до 25°C.
[0088] 33,2 г (0,2 моль) изооктаноата натрия растворяли предварительно в 280,0 г этилового спирта для получения раствора. Раствор добавляли по каплям в систему при температуре, составляющей от 20°C до 25°C; белое твердое вещество выпадало в осадок; после завершения добавления по каплям раствор перемешивали при температуре, составляющей от 20°C до 25°C, в течение 3 часов. Смешанный раствор фильтровали и промывали с помощью 100,0 г этилового спирта с получением 26,2 г авибактама (I) с показателем чистоты согласно HPLC, составляющим 99,9%, и выходом, составляющим 91,5%. Удельное вращение: [α]20 D= -54,4°(c=0,5, H2O).
[0089] Данные ЯМР продукта представлены ниже. 1H-ЯМР (400 MГц, D2O) δ: 1,69 (m, 1H), 1,83 (m, 1H), 1,96 (m, 1H), 2,10 (m, 1H), 3,00 (d, 1H), 3,22 (d, 1H), 3,96 (d, 1H), 4,09 (q, 1H).
[0090] 13C-ЯМР (400 MГц, D2O) δ: 174,64, 169,39, 60,26, 59,76, 47,13, 19,80, 18,02.
[0091] Результаты 1H-ЯМР-спектроскопии продукта показаны на фиг. 5, и результаты 13C-ЯМР-спектроскопии продукта показаны на фиг. 6.
Пример 2. Получение авибактама (I)
Стадия (1). Получение метил-5-трет-бутилдиметилсилилоксииминопиперидин-2S-карбоксилата (III2)
[0092] В 4-горлую колбу объемом 500 мл, оснащенную мешалкой, термометром и конденсатором с обратным холодильником, добавляли по отдельности 200 г дихлорметана, 23,5 г (0,15 моль) метилпиперидин-5-он-2S-карбоксилата, 36,5 г (0,2 моль) трет-бутилдиметилсилилгидроксиламина гидрохлорида и 25 г триэтиламина, затем перемешивали для осуществления реакции при температуре, составляющей от 38°C до 40°C, в течение 5 часов. После охлаждения смеси до 20°C - 25°C добавляли 100 г воды. Затем раствор разделяли и водный слой экстрагировали два раза с помощью дихлорметана (по 50 г каждый раз). Органические фазы объединяли и промывали дважды с помощью насыщенного солевого раствора (по 25 г каждый раз). Органическую фазу перегоняли с извлечением растворителя и затем перегоняли при пониженном давлении с получением 41,0 г метил-5-трет-бутилдиметилсилилоксииминопиперидин-2S-карбоксилата в виде желтоватой жидкости с показателем чистоты согласно GC, составляющим 99,9%, и выходом, составляющим 95,6%.
Стадия (2). Получение метил-5R-трет-бутилдиметилсилилоксиаминопиперидин-2S- карбоксилата оксалата (IV2)
[0093] В 4-горлую колбу объемом 500 мл, оснащенную мешалкой и термометром, добавляли 250 г этилацетата, 22,9 г (0,08 моль) метил-5-трет-бутилдиметилсилилоксииминопиперидин-2S-карбоксилата, полученного на стадии (1), и затем охлаждали. Затем добавляли по каплям 40,3 г (0,4 моль) концентрированной серной кислоты (массовая концентрация 98%) при -20°C, и затем перемешивали в течение 1 часа после завершения добавления по каплям. Добавляли 38,0 г (0,18 моль) триацетоксиборогидрида натрия при -20°C, и затем перемешивали для осуществления реакции при температуре, составляющей от -20°C до -15°C, в течение 5 часов. Смесь поддерживали при температуре ниже 10°C, и затем реакционную смесь медленно добавляли в 200 г 10% водного раствора аммиака; затем раствор разделяли и органическую фазу промывали два раза с помощью насыщенного солевого раствора (по 25 г каждый раз). Органическую фазу концентрировали для извлечения растворителя, затем добавляли к остатку 80 г этилацетата, 40 г метанола и 11,5 г (0,09 моль) дигидрата щавелевой кислоты, и нагревали до 45°C, перемешивали в течение 1 часа, и затем охлаждали и фильтровали. Полученный осадок на фильтре сначала промывали с помощью 60 г смешанной жидкости, содержащей этилацетат/метанол (2:1), и затем промывали с помощью 50 г этилацетата. После высушивания под вакуумом получали 19,7 г оптического изомера метил-5-трет-бутилдиметилсилилоксиаминопиперидин-2S-карбоксилата оксалата с показателем чистоты согласно хиральной HPLC, составляющим 99,8%, и выходом, составляющим 65,3%.
Стадия (3). Получение 5R-трет-бутилдиметилсилилоксиаминопиперидин-2S-карбоновой кислоты (V1)
[0094] В 4-горлую колбу объемом 500 мл, оснащенную мешалкой и термометром, добавляли 37,8 г (0,1 моль) метил-трет-бутилдиметилсилилоксиаминопиперидин-2S-карбоксилата оксалата, полученного на стадии (2), 50 г воды, 100 г этилового спирта и 70 г (0,35 моль) водного раствора гидроксида натрия (20 вес. %) и перемешивали для осуществления реакции при температуре, составляющей от 20°C до 25°C, в течение 4 часов. После завершения реакции гидролиза раствор охлаждали до температуры, составляющей от 0°C до 5°C, и затем подкисляли с помощью уксусной кислоты для регулирования значения pH до 3,5-3,0. Реакционную смесь фильтровали и высушивали с получением 25,3 г 5R-трет-бутилдиметилсилилоксиаминопиперидин-2S-карбоновой кислоты с показателем чистоты согласно HPLC, составляющим 99,9%, и выходом, составляющим 92,3%.
Стадия (4). Получение (2S,5R)-6-трет-бутилдиметилсилилокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамида (VI2)
[0095] В 4-горлую колбу объемом 500 мл, оснащенную мешалкой и термометром, добавляли 200 г ацетонитрила, 13,7 г (0,05 моль) 5-трет-бутилдиметилсилилоксиаминопиперидин-2S-карбоновой кислоты, полученной на стадии (3), 45 г диизопропилэтиламина и 0,1 г N,N-диметилформамида, и затем охлаждали; добавляли по каплям смешанный раствор 23,8 г (0,12 моль) дифосгена и 80 г ацетонитрила при температуре, составляющей от -10°C до 0°C, и после завершения добавления по каплям реакционную смесь перемешивали для осуществления реакции при температуре, составляющей от 10°C до 20°C, в течение 4 часов. 40 г (10 вес. %) раствора аммиака-ацетонитрила добавляли по каплям при температуре, составляющей от 10°C до 20°C, и перемешивали при температуре, составляющей от 15°C до 20°C, для осуществления реакции в течение 4 часов; реакционную жидкость выливали в 300 г смеси льда и воды. Смешанный раствор разделяли, и водный слой экстрагировали два раза с помощью дихлорметана (по 50 г каждый раз). Органические фазы объединяли и промывали дважды с помощью насыщенного раствора хлорида натрия (по 20 г каждый раз). После извлечения растворителя из полученной органической фазы получали 13,8 г (2S,5R)-6-трет-бутилдиметилсилилокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамида с показателем чистоты согласно HPLC, составляющим 99,9%, и выходом, составляющим 92,1%.
Стадия (5). Получение {[(2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил]окси}сульфонил-тетра-н-бутиламмониевой соли (VII)
[0096] В 4-горлую колбу объемом 500 мл, оснащенную мешалкой и термометром, добавляли 120 г изопропанола, 2,0 г воды, 30,0 г (0,1 моль) (2S,5R)-6-трет-бутилдиметилсилилокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамида, полученного на стадии (4), 5,5 г триэтиламина и 39,0 г (0,15 моль) фтортетрабутиламмония и добавляли 22,5 г (0,12 моль) комплекса триоксида серы и триметиламина при температуре, составляющей от 10°C до 15°C; раствор смеси перемешивали для осуществления реакции при температуре, составляющей от 15°C до 20°C, в течение 5 часов. Реакционную жидкость выливали в 150 г дихлорметана и 100 г смеси льда и воды. Затем добавляли уксусную кислоту для регулирования значения pH системы до 3,5-2,5. Смешанный раствор разделяли, и затем водный слой экстрагировали два раза с помощью дихлорметана (по 50 г каждый раз). Органические фазы объединяли и промывали дважды с помощью насыщенного раствора хлорида натрия (по 20 г каждый раз). После извлечения растворителя из полученной органической фазы остатки перекристаллизовывали с помощью 50 г дихлорметана-метилизобутилкетона (объемное соотношение 1:3) с получением 47,1 г {(2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил]окси}сульфонил-тетра-н-бутиламмониевой соли с показателем чистоты согласно HPLC, составляющим 99,9%, и выходом, составляющим 93,1%.
Стадия (6). Получение авибактама (I)
[0097] В колбу объемом 500 мл, оснащенную мешалкой и термометром, добавляли 260 г 98% этилового спирта (2 вес. % воды), 50.6 г (0,1 моль) {[(2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил]окси}сульфонил-тетра-н-бутиламмониевой соли, и смесь перемешивали и растворяли при температуре, составляющей от 20°C до 30°C.
[0098] Дополнительно, 33,2 г (0,2 моль) изооктаноата натрия растворяли в 280,0 г этилового спирта для получения раствора. Раствор добавляли по каплям в систему при температуре, составляющей от 20°C до 30°C; белое твердое вещество выпадало в осадок; после завершения добавления по каплям раствор перемешивали при температуре, составляющей от 20°C до 30°C, в течение 3 часов. Смешанный раствор фильтровали и промывали с помощью 100,0 г этилового спирта с получением 25,9 г авибактама (I) с показателем чистоты согласно HPLC, составляющим 99,8%, и выходом, составляющим 90,2%.
Сравнительный пример 1
[0099] Способ получения авибактама предусматривает следующие стадии.
[00100] Стадия (1). Получение метил-5-метоксиметилоксииминопиперидин-2S-карбоксилата (III1)
[00101] Данная стадия отличается от стадии (1) примера 1 тем, что:
[00102] условием реакции конденсации на данной стадии являлось перемешивание для осуществления реакции при температуре, составляющей от 20°C до 25°C, в течение 8 часов.
[00103] Остальные условия соответствовали примеру 1.
[00104] Получали 24,5 г метил-5-метоксиметилоксииминопиперидин-2S-карбоксилата в виде желтоватой жидкости с показателем чистоты согласно GC, составляющим 99,2%, и выходом, составляющим 75,5%.
[00105] Очевидно, что температура реакции конденсации оказывает значительное влияние на выход целевого продукта.
[00106] Стадия (2). Получение метил-5R-метоксиметилоксиаминопиперидин-2S-карбоксилата оксалата (IV1)
[00107] Данная стадия отличается от стадии (2) примера 1 тем, что:
[00108] количество концентрированной серной кислоты (массовая концентрация 98%) составляло 20,0 г (0,2 моль).
[00109] Добавляли 38,0 г (0,18 моль) триацетоксиборогидрида натрия при -20°C, и затем смесь перемешивали для осуществления реакции при температуре, составляющей от -20°C до -15°C, в течение 5 часов. Смесь поддерживали при температуре ниже 10°C, и затем реакционную смесь медленно добавляли в 100 г 10% водного раствора аммиака.
[00110] Остальные условия соответствовали примеру 1.
[00111] Получали 8,8 г оптического изомера метил-5R-метоксиметилоксиаминопиперидин-2S-карбоксилата оксалата с показателем чистоты согласно хиральной HPLC, составляющим 98,3%, и выходом, составляющим 35,5%.
[00112] Очевидно, что количество концентрированной серной кислоты влияет на селективность реакции восстановления, что оказывает достаточно значительное влияние на выход и чистоту продукта.
[00113] Стадия (3). Получение 5R-метоксиметилоксиаминопиперидин-2S-карбоновой кислоты (V1)
[00114] Данная стадия отличается от стадии (3) примера 1 тем, что:
[00115] условием реакции гидролиза являлось перемешивание для осуществления реакции при температуре, составляющей от 70°C до 75°C, в течение 3 часов.
[00116] Остальные условия соответствовали примеру 1.
[00117] Получали 17,4 г 5R-метоксиметилоксиаминопиперидин-2S-карбоновой кислоты в виде желтоватого порошка с показателем чистоты согласно HPLC, составляющим 98,6%, и выходом, составляющим 85,5%.
[00118] Стадия (4). Получение (2S,5R)-6-метоксиметилокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамида (VI1)
[00119] Данная стадия отличается от стадии (4) примера 1 тем, что:
[00120] количество твердого фосгена, добавляемого по каплям, составляло 14,9 г (0,05 моль).
[00121] Остальные условия соответствовали примеру 1.
[00122] Получали 9,8 г (2S,5R)-6-метоксиметилокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамида с показателем чистоты согласно HPLC, составляющим 99,0%, и выходом, составляющим 85,6%.
[00123] Стадия (5). Получение {(2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил]окси}сульфонил-тетра-н-бутиламмониевой соли (VII)
[00124] Данная стадия отличается от стадии (5) примера 1 тем, что:
[00125] добавляли 28,0 г (0,15 моль) комплекса триоксида серы и триметиламина при температуре, составляющей от 32°C до 35°C, и смесь перемешивали для осуществления реакции при температуре, составляющей от 32°C до 35°C, в течение 4 часов.
[00126] Остальные условия соответствовали примеру 1.
[00127] Получали 40,3 г {[(2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил]окси}сульфонил-тетра-н-бутиламмониевой соли с показателем чистоты согласно HPLC, составляющим 99,9%, и выходом, составляющим 79,6%.
[00128] Очевидно, что температура реакции удаления защитной группы, сульфатирования, образования тетрабутиламмониевой соли оказывает значительное влияние на выход продукта.
[00129] Стадия (6). Получение авибактама (I)
[00130] Данная стадия отличается от стадии (6) примера 1 тем, что:
[00131] условием реакции ионного обмена являлось перемешивание для осуществления реакции при температуре, составляющей от 40°C до 45°C, в течение 3 часов.
[00132] Остальные условия соответствовали примеру 1.
[00133] Получали 26,0 г авибактама (I) с показателем чистоты согласно HPLC, составляющим 99,1%, и выходом, составляющим 90,8%.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ ДЛЯ ПОЛУЧЕНИЯ АВИБАКТАМА | 2018 |
|
RU2722932C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ ДЛЯ ПОЛУЧЕНИЯ АВИБАКТАМА | 2018 |
|
RU2722625C1 |
| СПОСОБ ПОЛУЧЕНИЯ 5R-[(БЕНЗИЛОКСИ)АМИНО]ПИПЕРИДИН-2S-КАРБОКСИЛАТА И ЕГО ОКСАЛАТОВ | 2018 |
|
RU2713400C1 |
| ОПТИЧЕСКИ АКТИВНОЕ ПРОИЗВОДНОЕ ДИАЗАБИЦИКЛООКТАНА И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2011 |
|
RU2591701C2 |
| НОВЫЙ ИНГИБИТОР бета-ЛАКТАМАЗЫ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2013 |
|
RU2693898C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ПРОИЗВОДНОГО ДИАЗАБИЦИКЛООКТАНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2014 |
|
RU2801220C2 |
| НОВЫЙ ИНГИБИТОР β-ЛАКТАМАЗЫ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2019 |
|
RU2800050C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ПРОИЗВОДНОГО ДИАЗАБИЦИКЛООКТАНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2014 |
|
RU2695219C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ПРОИЗВОДНОГО ДИАЗАБИЦИКЛООКТАНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2023 |
|
RU2838718C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ПРОИЗВОДНОГО ДИАЗАБИЦИКЛООКТАНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2023 |
|
RU2840014C2 |
Изобретение относится к простому способу получения авибактама. Пиперидин-5-он-2S-карбоксилат II в качестве исходного вещества подвергают реакции конденсации с гидроксиламина гидрохлоридом, содержащим защитную группу для атома O; полученное соединение подвергают восстановлению и хиральному разделению с получением 5R-замещенной оксиаминопиперидин-2S-карбоновой кислоты V в щелочных условиях; затем соединение формулы V подвергают циклизации мочевины, ацилхлорированию и амидированию с использованием фосгена, твердого фосгена или дифосгена с помощью «однореакторного» способа, и затем подвергают удалению защитной группы, сульфатированию и реакции образования тетрабутиламмониевой соли с получением (2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил]окси}сульфонил-тетра-н-бутиламмониевой соли VII, и, наконец, соединение формулы VII подвергают ионному обмену с получением авибактама I. Настоящее изобретение характеризуется простой схемой получения, простотой выполнения операций, недорогими исходными веществами и низкими затратами; предусматривается сброс меньшего количества сточных вод, отработанного газа и отработанных остатков. 9 з.п. ф-лы, 6 ил., 2 пр.
1. Способ получения авибактама, предусматривающий стадии:
(1) осуществление реакции конденсации соединения формулы II и гидроксиламина гидрохлорида, содержащего защитную группу для атома O, в растворителе a и при катализе с помощью основания a с получением соединения формулы III:
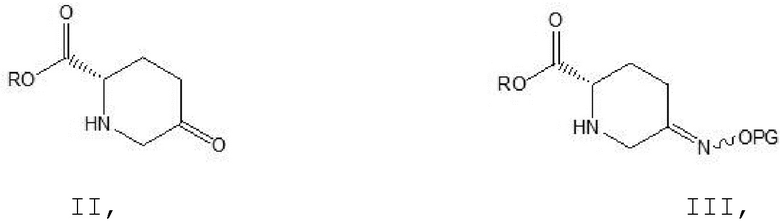
где R в соединении формулы II является идентичным R в соединении формулы III, который при этом выбран из группы, состоящей из метила, этила, изопропила, н-пропила, трет-бутила, н-бутила, изобутила или бензила; PG в соединении формулы III выбран из группы, состоящей из метоксиметила, бензилоксиметила, трет-бутилдиметилсилила, трет-бутилдифенилсилила, триэтилсилила или триизопропилсилила;
(2) осуществление восстановления соединения формулы III с помощью восстановителя в присутствии концентрированной серной кислоты и этилацетата и хирального разделения с получением соединения формулы IV:
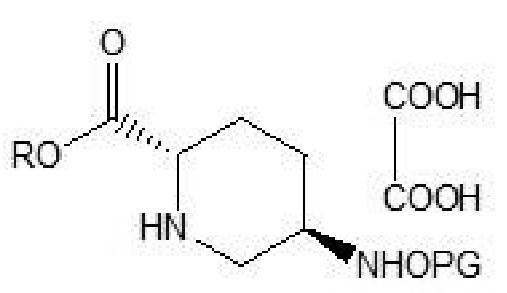
IV,
где R и PG в соединении формулы IV имеют такие же значения, что и R и PG в соединении формулы III;
(3) осуществление гидролиза соединения формулы IV в присутствии основания b и в растворителе b с получением соединения формулы V:
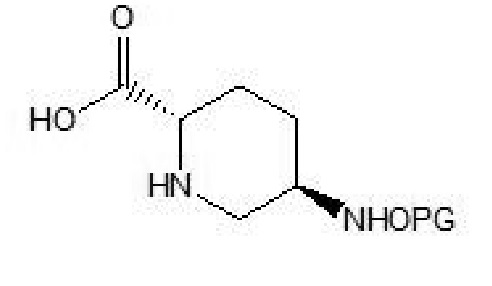
V,
где PG в соединении формулы V имеет такое же значение, что и PG в соединении формулы IV;
(4) осуществление в отношении соединения формулы V циклизации мочевины и ацилхлорирования с использованием фосгена, твердого фосгена или дифосгена в присутствии основания c и катализатора в растворителе c, и затем осуществление амидирования с получением соединения формулы VI:
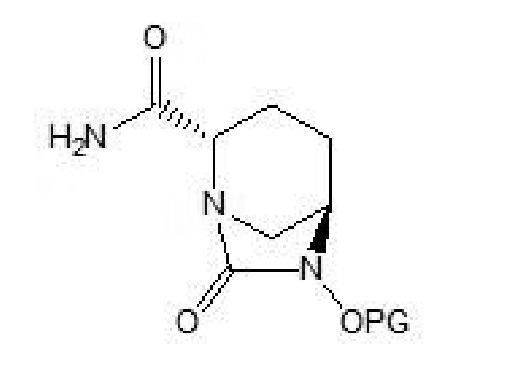
VI,
где PG в соединении формулы VI имеет такое же значение, что и PG в соединении формулы V;
(5) осуществление в отношении соединения формулы VI удаления защитной группы с использованием реагента для удаления защитной группы, сульфатирования и образования тетрабутиламмониевой соли при катализе с помощью основания d и в растворителе d с получением соединения формулы VII:
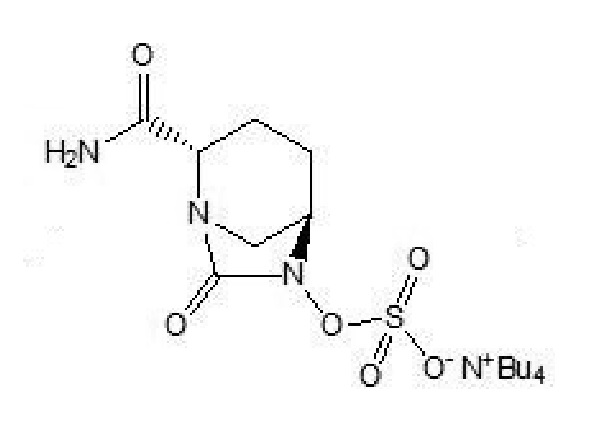
VII;
(6) осуществление в отношении соединения формулы VII ионного обмена с получением авибактама (I).
2. Способ получения авибактама по п. 1, где стадия (1) предусматривает одно или более условий, представленных ниже:
A) гидроксиламина гидрохлорид, содержащий защитную группу для атома O, выбран из группы, состоящей из метоксиметилгидроксиламина гидрохлорида, бензилоксиметилгидроксиламина гидрохлорида, трет-бутилдиметилсилилгидроксиламина гидрохлорида, трет-бутилдифенилсилилгидроксиламина гидрохлорида, триэтилсилилгидроксиламина гидрохлорида, триизопропилсилилгидроксиламина гидрохлорида; при этом молярное соотношение гидроксиламина гидрохлорида, содержащего защитную группу для атома O, и соединения формулы II составляет 0,9-1,5:1;
B) растворитель a выбран из группы, состоящей из метанола, этанола, пропанола, бутанола, этилацетата, тетрагидрофурана, ацетонитрила, дихлорметана, хлороформа, 1,2-дихлорэтана, бензола и толуола или смеси двух или более из них; при этом массовое соотношение растворителя a и соединения формулы II составляет 3-15:1, и предпочтительно массовое соотношение растворителя a и соединения формулы II составляет 6-10:1;
C) основание a представляет собой неорганическое основание или органическое основание; предпочтительно неорганическое основание выбрано из группы, состоящей из карбоната калия, карбоната натрия, карбоната кальция, гидрокарбоната калия, гидрокарбоната натрия, гидрокарбоната кальция, ацетата калия, ацетата натрия или ацетата кальция или комбинации двух или более из них, и органическое основание выбрано из группы, состоящей из триэтиламина или три-н-бутиламина или комбинации двух из них; при этом массовое соотношение основания a и соединения формулы II составляет 0,5-1,5:1.
3. Способ получения авибактама по п. 1, где на стадии (1) температура реакции конденсации находится в диапазоне от 30°C до 80°C; предпочтительно температура реакции конденсации находится в диапазоне от 30°C до 60°C.
4. Способ получения авибактама по п. 1, где на стадии (2) концентрированная серная кислота представляет собой серную кислоту с массовой долей, находящейся в диапазоне от 95% до 98%, и молярное соотношение концентрированной серной кислоты и соединения формулы III составляет (3,0-6,0):1; предпочтительно концентрированная серная кислота представляет собой серную кислоту с массовой долей, составляющей 98%.
5. Способ получения авибактама по п. 1, где стадия (2) предусматривает одно или более условий, представленных ниже:
A) массовое соотношение этилацетата и соединения формулы III составляет 5-20:1; еще более предпочтительно массовое соотношение этилацетата и соединения формулы III находится в диапазоне 10-14:1;
B) восстановитель выбран из группы, состоящей из борогидрида натрия, трицианоборогидрида натрия, триацетоксиборогидрида натрия, трипропионилоксиборогидрида натрия, борогидрида калия, трицианоборогидрида калия, триацетоксиборогидрида калия или трипропионилоксиборогидрида калия; при этом молярное соотношение восстановителя и соединения формулы III составляет 2,0-4,0:1;
C) температура реакции восстановления находится в диапазоне от (-30)°C до (-10)°C.
6. Способ получения авибактама по п. 1, где стадия (3) предусматривает одно или более условий, представленных ниже:
A) растворитель b выбран из группы, состоящей из воды, метанола, этанола, пропанола, бутанола, этилацетата, дихлорметана, хлороформа, 1,2-дихлорэтана, бензола и толуола или комбинации двух или более из них; при этом массовое соотношение растворителя b и соединения формулы IV составляет 3-12:1, и массовое соотношение растворителя b и соединения формулы IV составляет 3-6:1;
B) основание b выбрано из группы, состоящей из гидроксида натрия, гидроксида калия, гидроксида лития, карбоната калия, карбоната натрия, карбоната кальция, гидрокарбоната калия или гидрокарбоната натрия или смеси двух или более из них; при этом молярное соотношение основания b и соединения формулы IV составляет 1,5-4,0:1;
C) температура реакции гидролиза составляет от 10°C до 100°C; предпочтительно температура реакции гидролиза составляет от 20°C до 50°C.
7. Способ получения авибактама по п. 1, где стадия (4) предусматривает одно или более условий, представленных ниже:
A) растворитель c выбран из группы, состоящей из дихлорметана, 1,2-дихлорэтана, трихлорметана, тетрахлорметана, ацетонитрила, тетрагидрофурана, 2-метилтетрагидрофурана или метоксициклопентана, метилбензола или комбинации двух или более из них; при этом массовое соотношение растворителя c и соединения формулы V составляет 4-30:1; предпочтительно массовое соотношение растворителя c и соединения формулы V составляет 18-30:1;
B) основание c выбрано из группы, состоящей из триметиламина, триэтиламина, три-н-бутиламина, диизопропилэтиламина, карбоната калия, карбоната натрия или карбоната кальция или комбинации двух или более из них; при этом молярное соотношение основания c и соединения формулы V составляет 3,0-8,0:1;
C) катализатор выбран из группы, состоящей из N,N-диметилформамида, пиридина или 4-диметиламинопиридина или комбинации двух или более из них; при этом масса катализатора составляет 0,1-5,0% массы соединения формулы V;
D) молярное соотношение твердого фосгена/дифосгена/фосгена и соединения формулы V составляет 0,6-5,0:1; предпочтительно молярное соотношение твердого фосгена и соединения формулы V составляет 1,2-2,0:1; предпочтительно молярное соотношение дифосгена и соединения формулы V составляет 1,0-2,5:1; предпочтительно молярное соотношение фосгена и соединения формулы V составляет 2,0-4,0:1;
E) аммиак выбран из группы, состоящей из газообразного аммиака, спиртового раствора газообразного аммиака, раствора газообразного аммиака в тетрагидрофуране, раствора газообразного аммиака в ацетонитриле или гидроксида аммония; при этом массовая концентрация газообразного аммиака в спиртовом растворе газообразного аммиака, растворе газообразного аммиака в тетрагидрофуране, растворе газообразного аммиака в ацетонитриле или гидроксиде аммония составляет 5-20%;
F) молярное соотношение аммиака и соединения формулы V составляет 1,0-6,0:1;
G) все значения температуры реакций циклизации мочевины, ацилхлорирования, амидирования находятся в диапазоне от -20°C до 60°C; предпочтительно все значения температуры реакций циклизации мочевины, ацилхлорирования, амидирования находятся в диапазоне от 10°C до 30°C.
8. Способ получения авибактама по п. 1, где стадия (5) предусматривает одно или более условий, представленных ниже:
A) растворитель d выбран из группы, состоящей из воды, изопропанола, изобутанола, этилацетата, дихлорметана, хлороформа, 1,2-дихлорэтана или изобутилметилкетона или комбинации двух или более из них; при этом массовое соотношение растворителя d и соединения формулы VI составляет 4-20:1; предпочтительно массовое соотношение растворителя d и соединения формулы VI составляет 4-8:1;
B) основание d выбрано из группы, состоящей из триметиламина, триэтиламина, три-н-бутиламина и диизопропилэтиламина; при этом молярное соотношение основания d и соединения формулы VI составляет 0,2-0,7:1;
C) если PG в соединении формулы VI представляет собой защитную группу, не содержащую кремний, то средство для удаления защитной группы выбрано из группы, состоящей из комплекса триоксида серы и триметиламина, комплекса триоксида серы и триэтиламина и комплекса триоксида серы и пиридина; если PG в соединении формулы VI представляет собой защитную группу, содержащую кремний, то средство для удаления защитной группы представляет собой фтортетрабутиламмоний; при этом молярное соотношение средства для удаления защитной группы и соединения формулы VI составляет 1,0-3,0:1;
D) реагент, применяемый при сульфатировании, выбран из группы, состоящей из комплекса триоксида серы и триметиламина, комплекса триоксида серы и триэтиламина или комплекса триоксида серы и пиридина; при этом молярное соотношение реагента, применяемого для сульфатирования, и соединения формулы VI составляет 1,0-3,0:1;
E) реагент для образования соли, применяемый в реакции образования тетрабутиламмониевой соли, представляет собой ацетат тетрабутиламмония или фтортетрабутиламмоний, и молярное соотношение реагента для образования соли, применяемого в реакции образования тетрабутиламмониевой соли, и соединения формулы VI составляет 0,5-2:1.
9. Способ получения авибактама по п. 1, где на стадии (5) удаление защитной группы, сульфатирование и реакцию образования тетрабутиламмониевой соли проводят с помощью «однореакторного» способа; при этом температура реакции находится в диапазоне от 0°C до 60°C; предпочтительно температура реакции находится в диапазоне от 10°C до 30°C.
10. Способ получения авибактама по п. 1, где стадия (6) предусматривает одно или более условий, представленных ниже:
A) реагент, применяемый в ионном обмене, представляет собой изооктаноат натрия; при этом молярное соотношение реагента, применяемого в ионном обмене, и соединения формулы VII составляет 1,5-3,0:1;
B) температура реакции ионного обмена находится в диапазоне от 0°C до 50°C; предпочтительно температура реакции ионного обмена находится в диапазоне от 10°C до 40°C.
| (2S,5R)-5-[(бензилокси)амино]пиперидин-2-карбоксамид | 2012 |
|
RU2610091C2 |
| CN 106749242 A, 31.05.2017 | |||
| US 8148540 B2, 03.04.2012 | |||
| Устройство для подачи пара в тепловые аккумуляторы типа аккумуляторов Рутса | 1926 |
|
SU4920A1 |
| WO 2009091856 A2, 23.07.2009. | |||

