Область техники
[001] Настоящее изобретение относится к области фармацевтической биохимической инженерии, в частности, относится к способу получения промежуточного соединения для получения авибактама и, более конкретно, относится к способу получения (2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамида.
Уровень техники
[002] Авибактам, поскольку он является одним из ингибиторов диазабициклооктаноновых соединений не на основе β-лактама, может ингибировать β-лактамазы типа A (в том числе ESBL и KPC) и типа C. При введении в комбинации с различными типами антибиотиков, представляющих собой цефалоспорины и карбапенем, авибактам обладает активностью широкого спектра действия в отношении бактерий, в частности, обладает значительной активностью в отношении Escherichia coli и Klebsiella pneumoniae, содержащих расширенный спектр β-лактамаз, Escherichia coli, содержащей избыточное количество фермента AmpC, и Escherichia coli, содержащей как AmpC, так и расширенный спектр β-лактамаз. Авибактам (I), характеризующийся номером CAS № 1192491-61-4 и химическим названием [(1R,2S,5R)-2-(аминокарбонил)-7-оксо-1,6-диазабицикло[3.2.1]окт-6-ил]сульфат натрия, характеризуется структурной формулой, представленной на формуле I:
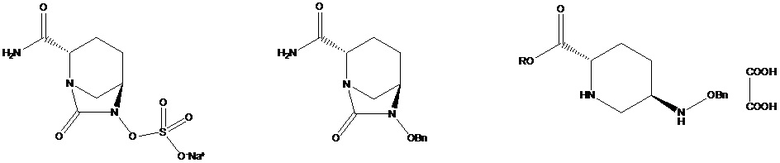
[003] В патентных литературных источниках CN 103649051 A, CN 105294690 A, CN 106866668 A, WO 2012086241, US 8148540, US 9284273 и US 9567335 авибактам (I) во всех случаях получали с применением (2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамида (II) в качестве промежуточного соединения. Осуществляли дебензилирование (2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамида (II) при катализе с помощью палладия на угле в присутствии различных восстановителей (таких как водород, триэтилсилан, формиат натрия и гидрат гидразина), затем осуществляли сульфатирование с помощью комплекса триоксида серы, и преобразовывали продукт в солевую форму четвертичного аммония с последующим ионным обменом с получением авибактама (I), как показано на схеме 1.
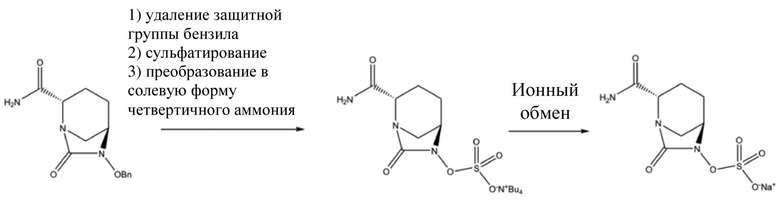
Схема 1
[004] Различные способы получения (2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамида (II) главным образом делятся на две схемы: амидирование с последующей циклизацией мочевины и циклизация мочевины с последующим амидированием, как показано на схеме 2.
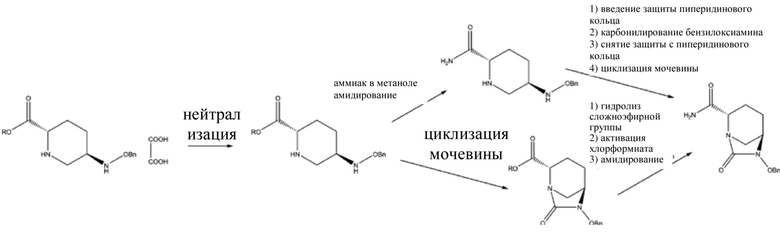
Схема 2
[005] В патентах CN 103649051 A и CN 105294690 A применяется схема амидирования с последующей циклизацией мочевины. 5R-[(бензилокси)амино]пиперидин-2S-карбоксилата оксалат (III) в качестве исходного вещества амидировали в метанольном растворе газообразного аммиака или в водном растворе аммиака в спирте, и реакционную смесь фильтровали для удаления оксалата аммония; осадок на фильтре, представляющий собой оксалат аммония, промывали метанолом, и полученный метанольный раствор концентрировали; продукт экстрагировали с помощью метилбензола и перекристаллизовывали с использованием подходящего растворителя с получением (2S,5R)-5-[(бензилокси)амино]пиперидин-2-карбоксамида (выход: 68-99%); затем проводили реакцию карбонилирования с участием карбонилдиимидазола и бензилосиламина в присутствии защитной группы при аминогруппе в пиперидиновом кольце полученного (2S, 5R)-5-[(бензилокси)амино]пиперидин-2-карбоксамида с помощью 9-фторенилметилхлорформиата (FMOC-CI), и после удаления защитной группы в пиперидиновом кольце с помощью диэтиламина проводили циклизацию мочевины с получением (2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамида (II) (выход: 90%, общий выход: 61,2-89,1%). В данном способе получения обработка после амидирования осложнена, и защитное средство, представляющее собой 9-фторенилметилхлорформиат, применяемое для циклизации мочевины, является дорогостоящим. Кроме того, 9-фторенилметилхлорформиат и карбонилдиимидазол обеспечивают только один карбонил, так что реакция характеризуется низкой атомной эффективностью, что не способствует защите окружающей среды и снижению затрат. Дополнительно, прямая циклизация мочевины в отношении (2S, 5R)-5-[(бензилокси)амино]пиперидин-2-карбоксамида с применением трифосгена и карбонилдиимидазола без введения защитной группы для аминогруппы в пиперидиновом кольце характеризовалась низким выходом (50-56%) с отсутствием промышленного значения.
[006] Помимо этого, все из патентов CN 102834395 A, CN 103649051 A, CN 103328476 A, CN 106279163 A, CN 106565712 A, US 9284273 и US 9567335 относятся к способу циклизации мочевины с последующим амидированием. Осуществляли циклизацию мочевины в отношении 5R-[(бензилокси)амино]пиперидин-2S-карбоксилата оксалата (III) в качестве исходного вещества с применением трифосген-органического основания, карбонилдиимидазола или других средств карбонилирования, затем гидролизовали в щелочной среде, такой как водный раствор гидроксида лития, с получением (2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты; затем карбоксил активировали до ангидрида с применением триметилацетилхлорида или других средств, и затем ангидрид амидировали с применением водного раствора аммиака с получением (2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамида (II) с общим выходом 34,5-65,5%. (2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-формиат, полученный циклизацией мочевины, характеризуется низкой реакционной способностью, и его невозможно непосредственно амидировать в метанольном растворе газообразного аммиака. Вместо этого, для эффективного амидирования сложноэфирную группу необходимо гидролизовать до карбоксила, и затем карбоксил активируют до ангидрида. Данный способ предусматривает трудоемкую рабочую процедуру и низкую атомную эффективность, что, таким образом, не способствует защите окружающей среды и осуществлению промышленного производства.
Краткое описание
[007] Для устранения недостатков предшествующего уровня техники, в настоящем изобретении предусмотрен способ получения промежуточного соединения для получения авибактама, а именно (2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамида (II). Исходный материал для настоящего изобретения является недорогим и легкодоступным; способ получения является простым и в высокой степени пригодным к применению без необходимости в специальном защитном средстве или реагенте для карбонилирования; кроме того, реакция характеризуется высокой атомной эффективностью и низкой стоимостью; способ получения является экологичным и безвредным для окружающей среды; полученный в результате продукт (II) характеризуется высоким показателем чистоты и высоким выходом; полученный в результате 2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (II) можно применять для получения авибактама (I).
[008] Определение терминов:
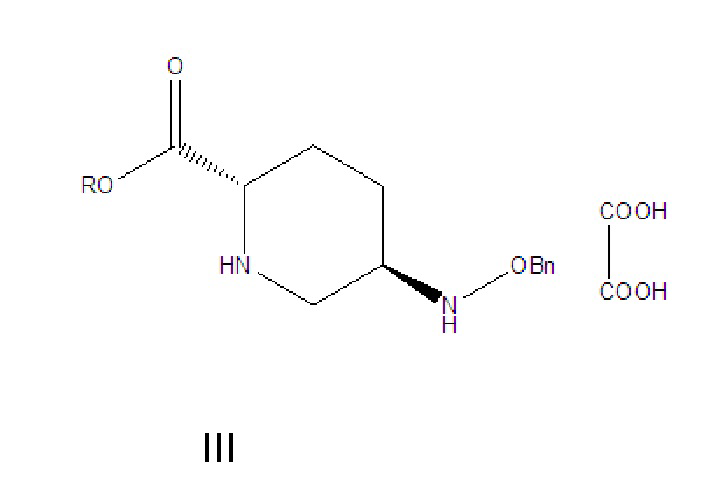
[009] соединение формулы III: 5R-[(бензилокси)амино]пиперидин-2S-карбоксилата оксалат, где -Bn означает бензил;
[0010] соединение формулы IV: 5R-[(бензилокси)амино]пиперидин-2S-карбоновая кислота, где -Bn означает бензил;
[0011] соединение формулы V: (2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-формилхлорид, где -Bn означает бензил.
[0012] Номера соединений в описании полностью соответствуют номерам их структурных формул, и они имеют одни и те же ссылки.
[0013] Техническое решение в соответствии с настоящим изобретением представлено ниже.
[0014] Способ получения промежуточного соединения для получения авибактама, предусматривающий следующие стадии, на которых:
[0015] (1) соединение формулы III растворяют в растворителе A, гидролизуют в присутствии основания A и затем подкисляют с получением соединения формулы IV;
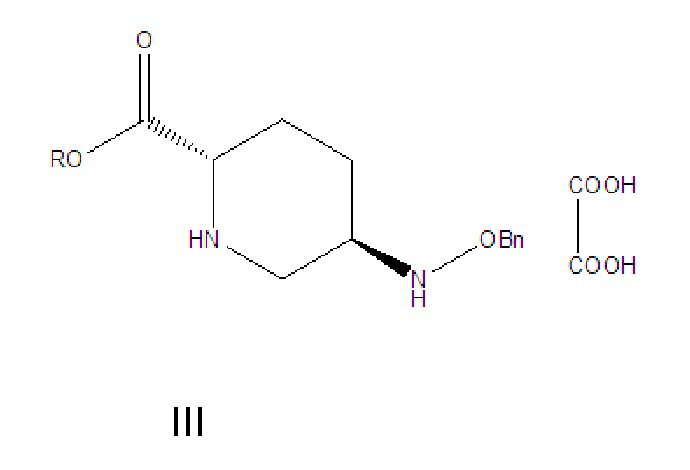
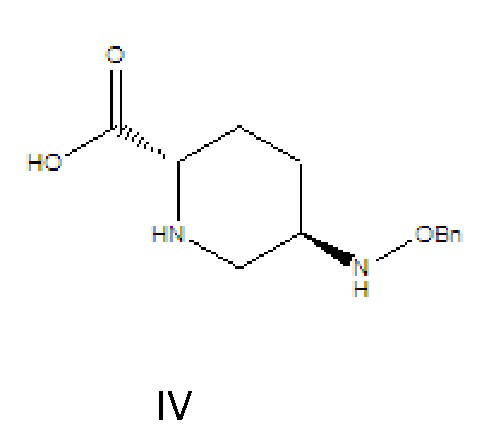
[0016] где R в соединении формулы III означает C1-6-алифатическую группу или -алкилзамещенный фенил; предпочтительно R выбран из группы, состоящей из метила, этила, н-пропила, изопропила, н-бутила, изобутила, трет-бутила, н-пентила, изопентила, трет-пентила, гексила, бензила, о-метилбензила и п-метилбензила;
[0017] 2) соединение формулы IV и твердый фосген или дифосген параллельно вводят в реакцию циклизации мочевины и реакции хлорформилирования в присутствии органического основания B и катализатора в растворителе B с получением соединения формулы V, которое применяли непосредственно на следующей стадии реакции без очистки;
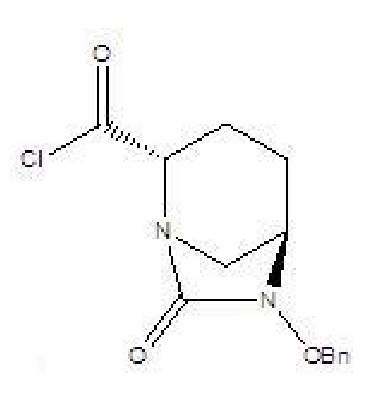
V
[0018] (3) проводят реакцию амидирования с участием соединения формулы V и аммиака с получением (2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамида (II).
[0019] В соответствии с настоящим изобретением, предпочтительно после гидролиза соединения формулы III на стадии (1) в щелочной среде и его подкисления получают экстракт, содержащий соединение формулы IV, с применением экстрагирующего средства; экстракт, содержащий соединение формулы IV, подвергают перегонке с удалением экстрагирующего средства с получением соединения формулы IV, или экстракт, содержащий соединение формулы IV, непосредственно применяют на следующей стадии реакции без перегонки.
[0020] Предпочтительно, в соответствии с настоящим изобретением, растворитель A на стадии (1) выбран из группы, состоящей из дихлорметана, 1,2-дихлорэтана, трихлорметана, тетрахлорметана, тетрагидрофурана, 2-метилтетрагидрофурана, метоксициклопентана, метилбензола и комбинаций из двух или более из них.
[0021] Предпочтительно, в соответствии с настоящим изобретением, на стадии (1) массовое соотношение растворителя A и соединения формулы III составляет 3-4,5:1.
[0022] Предпочтительно, в соответствии с настоящим изобретением, используемое на стадии (1) основание A выбрано из группы, состоящей из гидроксида натрия, гидроксида калия, гидроксида лития, карбоната калия, карбоната натрия, гидрокарбоната калия, гидрокарбоната натрия и смеси двух или более из них; предпочтительно основание A представляет собой водный раствор, содержащий основание при концентрации 5-15% по массе.
[0023] Предпочтительно, в соответствии с настоящим изобретением, на стадии (1) молярное соотношение основания A и соединения формулы III составляет 2,0-5,0:1.
[0024] Предпочтительно, в соответствии с настоящим изобретением, на стадии (1) реакцию гидролиза осуществляют при температуре 0-80°C; на стадии (1) реакцию гидролиза предпочтительно осуществляют при температуре 10-40°C. Длительность реакции составляет 2-5 часов.
[0025] Предпочтительно на стадии (1) подкисление осуществляют при температуре 20-25°C в течение 1-2 часов.
[0026] Предпочтительно на стадии (1) подкисление предусматривает регулирование значения pH системы до 2-3 с применением подкисляющего средства, при этом подкисляющее средство представляет собой водный раствор хлористоводородной кислоты, серной кислоты или азотной кислоты при концентрации 10-40% по массе.
[0027] Предпочтительно на стадии (2) растворитель B выбран из группы, состоящей из дихлорметана, 1,2-дихлорэтана, трихлорметана, тетрахлорметана, тетрагидрофурана, 2-метилтетрагидрофурана, метоксициклопентана, метилбензола и комбинаций из двух или более из них.
[0028] Предпочтительно, в соответствии с настоящим изобретением, на стадии (2) массовое соотношение растворителя B и соединения формулы IV составляет 4-20:1.
[0029] Предпочтительно, в соответствии с настоящим изобретением, на стадии (2) органическое основание B выбрано из группы, состоящей из триэтиламина, три-н-бутиламина, диизопропилэтиламина и комбинаций из двух или более из них.
[0030] Предпочтительно, в соответствии с настоящим изобретением, на стадии (2) молярное соотношение органического основания B и соединения формулы IV составляет 3,0-8,0:1.
[0031] Предпочтительно, в соответствии с настоящим изобретением, на стадии (2) катализатор выбран из группы, состоящей из N, N-диметилформамида, пиридина, 4-диметиламинопиридина и комбинаций из двух или более из них.
[0032] Предпочтительно, в соответствии с настоящим изобретением, на стадии (2) катализатор присутствует в количестве 0,1-5,0% по массе соединения формулы IV.
[0033] Предпочтительно, в соответствии с настоящим изобретением, на стадии (2) молярное соотношение твердого фосгена или дифосгена и соединения формулы IV составляет 0,6-2,5:1.
[0034] Предпочтительно, в соответствии с настоящим изобретением, молярное соотношение твердого фосгена и соединения формулы IV составляет 0,6-2,0:1.
[0035] Предпочтительно, в соответствии с настоящим изобретением, молярное соотношение дифосгена и соединения формулы IV составляет 1,0-2,5:1.
[0036] Предпочтительно, в соответствии с настоящим изобретением, на стадии (2) как реакцию циклизации мочевины, так и реакцию хлорформилирования предпочтительно осуществляют при температуре −20-60°C; на стадии (2) как реакцию циклизации мочевины, так и реакцию хлорформилирования осуществляют при температуре 0-40°C. Длительность реакции составляет 1-8 часов.
[0037] Предпочтительно, в соответствии с настоящим изобретением, на стадии (3) соединение аммония выбрано из группы, состоящей из аммиака, раствора газообразного аммиака в метаноле или водного раствора аммиака.
[0038] Предпочтительно, в соответствии с настоящим изобретением, на стадии (3) молярное соотношение аммиака и соединения формулы IV составляет 1,0-5,0:1.
[0039] Предпочтительно, в соответствии с настоящим изобретением, на стадии (3) реакцию амидирования осуществляют при температуре −20-80°C; на стадии (3) реакцию амидирования предпочтительно осуществляют при температуре 10-50°C. Длительность реакции составляет 1-6 часов.
[0040] В соответствии с настоящим изобретением, 5R-[(бензилокси)амино]пиперидин-2S-карбоксилата оксалат (III) в качестве исходного материала гидролизуют в щелочной среде, затем окисляют с получением 5R-[(бензилокси)амино]пиперидин-2S-карбоновой кислоты (IV) и полученное соединение формулы IV и твердый фосген или дифосген одновременно вводят в реакцию циклизации мочевины и хлорформилирования в присутствии органического основания и катализатора с получением (2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-формилхлорида (V), который затем амидируют с получением (2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамида (II), при этом реакции циклизации мочевины, хлорформилирования и амидирования осуществляют посредством “однореакторного” способа, и промежуточные продукты не нужно разделять и очищать. Схема (схема 3) представлена ниже.
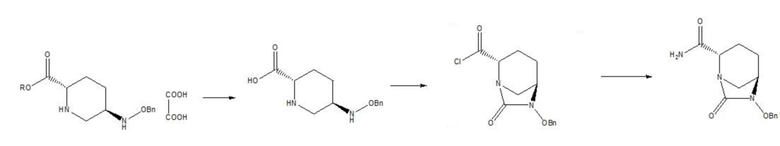
Схема 3
[0041] В настоящем изобретении предусмотрены следующие преимущественные эффекты.
[0042] 1. По сравнению с предшествующим уровнем техники, в настоящем изобретении отсутствуют недостатки способа амидирования с последующей циклизацией мочевины, для которого необходимы защита пиперидинового кольца и применение специфического реагента для карбонилирования, и дополнительно отсутствуют трудоемкие операции циклизации мочевины с последующим амидированием, для чего необходимы гидролиз сложноэфирной группы, активация с образованием ангидрида и амидирование. В настоящем изобретении применяется «однореакторный» способ реакций циклизации мочевины, хлорформилирования и амидирования, и промежуточные продукты не нужно подвергать последующим видам обработки, таким как разделение и очистка; способ предусматривает простые стадии, экологичные и безвредные для окружающей среды процедуры и низкую стоимость.
[0043] 2. В настоящем изобретении применяется недорогой и легкодоступный исходный материал, и типы применяемых реакций являются типичными реакциями; условия реакции являются легко контролируемыми; операции являются простыми; пригодность к применению является высокой; способ является простым. В способе циклизации мочевины не требуется специальная защита или реагент для карбонилирования; реакция характеризуется высокой атомной эффективностью; способ получения является экологичным и безвредным для окружающей среды; продукт, получаемый в результате циклизации мочевины, характеризуется соответствующей активностью, при этом он может быть амидирован с помощью газообразного аммиака, водного раствора аммиака и т. д., так что стадии являются простыми, и стоимость является низкой. Кроме того, (2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (II), полученный в соответствии со способом по настоящему изобретению, характеризуется высокими показателями чистоты и выхода, что способствует снижению стоимости и экологичному получению авибактама (I).
Примеры
[0044] Далее в данном документе настоящее изобретение будет подробно проиллюстрировано со ссылкой на примеры; однако настоящее изобретение не ограничено ими.
[0045] Если не указано иное, все значения процентного содержания в примерах означают массовые процентные доли.
[0046] Исходный материал, представляющий собой 5R-[(бензилокси)амино]пиперидин-2S-карбоксилата оксалат (III), является коммерчески доступным (от фармацевтической компании Jinan Qinsi), при этом он представляет собой белый порошок с оптической чистотой, составляющей 99,6%.
[0047] Протекание реакции и чистоту продукта контролируют с помощью газового хроматографа или жидкостного хроматографа. Жидкостный хроматограф, оснащенный хиральной колонкой (ES-OVS, 150 мм x 4,6 мм, Agilent), применяют для определения оптической чистоты (отношение значений площади %) и расчета выхода, а также % значения энантиомерной чистоты.
Пример 1. Получение 5R-[(бензилокси)амино]пиперидин-2S-карбоновой кислоты (IV)
[0048] В 4-горлую колбу объемом 500 мл, оснащенную мешалкой и термометром, добавляли 150 г дихлорметана, 150 г 10% (по массе) водного раствора гидроксида натрия, 43,0 г (0,1 моль) бензил-5R-[(бензилокси)амино]пиперидин-2S-карбоксилата оксалата (III) и затем реакционную смесь перемешивали при 20-30°C в течение 3 часов. Затем реакционную смесь подкисляли до значения pH 2,5-3,0 с помощью 30% (по массе) водного раствора хлористоводородной кислоты и перемешивали при комнатной температуре в течение 1-2 часов. Смешанный раствор разделяли и затем водную фазу трижды экстрагировали дихлорметаном (каждый раз по 50 г). Органические фазы объединяли и один раз промывали насыщенным раствором хлорида натрия (каждый раз по 20 г). После извлечения растворителя из полученной органической фазы получали 24,5 г 5R-[(бензилокси)амино]пиперидин-2S-карбоновой кислоты с выходом 98,0% и чистотой 99,9%, определенными с помощью HPLC.
[0049] Данные ЯМР (ядерного магнитного резонанса) полученного продукта приведены ниже: 1H-ЯМР (400 МГц, DMSO-d6), δ: 1,10 (1H, q), 1,27 (1H, q), 1,82 (2H, d), 2,23 (1H, t), 2,76 (1H, m), 2,90 (1H, d), 3,13 (1H, d), 4,70 (2H, s), 6,54 (1H, d), 7,35 (5H, m), 13,51 (1H, br).
Пример 2. Получение 5R-[(бензилокси)амино]пиперидин-2S-карбоновой кислоты (IV)
[0050] В 4-горлую колбу объемом 500 мл, оснащенную мешалкой и термометром, добавляли 150 г 1,2-дихлорэтана, 80 г 10% (по массе) водного раствора гидроксида лития, 43,0 г (0,1 моль) бензил-5R-[(бензилокси)амино]пиперидин-2S-карбоксилата оксалата (III) и затем реакционную смесь перемешивали при 20-25°C в течение 4 часов. Затем реакционную смесь подкисляли до значения pH 2,5-3,0 с помощью 30% (по массе) водного раствора хлористоводородной кислоты и перемешивали при комнатной температуре в течение 1-2 часов. Смешанный раствор разделяли и затем водную фазу трижды экстрагировали 1,2-дихлорэтаном (каждый раз по 50 г). Органические фазы объединяли и один раз промывали насыщенным раствором хлорида натрия (каждый раз по 20 г). После извлечения растворителя из полученной органической фазы получали 24,6 г 5R-[(бензилокси)амино]пиперидин-2S-карбоновой кислоты с выходом 98,5% и чистотой 99,9%, определенными с помощью HPLC.
Пример 3. Получение 5R-[(бензилокси)амино]пиперидин-2S-карбоновой кислоты (IV)
[0051] В 4-горлую колбу объемом 500 мл, оснащенную мешалкой и термометром, добавляли 150 г дихлорметана, 120 г 10% (по массе) водного раствора гидроксида натрия, 37,0 г (0,1 моль) этил-5R-[(бензилокси)амино]пиперидин-2S-карбоксилата оксалата (III) и затем реакционную смесь перемешивали при 20-25°C в течение 4 часов. Затем реакционную смесь подкисляли до значения pH 2,5-3,0 с помощью 30% (по массе) водного раствора хлористоводородной кислоты и перемешивали при комнатной температуре в течение 1-2 часов. Смешанный раствор разделяли и затем водную фазу трижды экстрагировали дихлорметаном (каждый раз по 50 г). Органические фазы объединяли и один раз промывали насыщенным раствором хлорида натрия (каждый раз по 20 г). После извлечения растворителя из полученной органической фазы получали 24,1 г 5R-[(бензилокси)амино]пиперидин-2S-карбоновой кислоты с выходом 96,4% и чистотой 99,9%, определенными с помощью HPLC.
Пример 4. Получение 5R-[(бензилокси)амино]пиперидин-2S-карбоновой кислоты (IV)
[0052] В 4-горлую колбу объемом 500 мл, оснащенную мешалкой и термометром, добавляли 150 г дихлорметана, 150 г 10% (по массе) водного раствора гидроксида натрия, 39,5 г (0,1 моль) трет-бутил-5R-[(бензилокси)амино]пиперидин-2S-карбоксилата оксалата (III) и затем реакционную смесь перемешивали при 20-30°C в течение 3 часов. Затем реакционную смесь подкисляли до значения pH 2,5-3,0 с помощью 30% (по массе) водного раствора хлористоводородной кислоты и перемешивали при комнатной температуре в течение 1-2 часов. Смешанный раствор разделяли и затем водную фазу трижды экстрагировали дихлорметаном (каждый раз по 50 г). Органические фазы объединяли и один раз промывали насыщенным раствором хлорида натрия (каждый раз по 20 г). После извлечения растворителя из полученной органической фазы получали 24,3 г 5R-[(бензилокси)амино]пиперидин-2S-карбоновой кислоты с выходом 97,2% и чистотой 99,9%, определенными с помощью HPLC.
Пример 5. Получение (2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамида (II)
[0053] В 4-горлую колбу объемом 500 мл, оснащенную мешалкой и термометром, добавляли 200 г тетрагидрофурана, 12,5 г (0,05 моль) 5R-[(бензилокси)амино]пиперидин-2S-карбоновой кислоты, полученной в примере 2, 50 г три-н-бутиламина и 0,1 г N,N-диметилформамида. После охлаждения добавляли по каплям раствор 23,8 г (0,08 моль) твердого фосгена в 80 г тетрагидрофурана при −10-0°C. После завершения добавления реакционную смесь перемешивали при 10-20°C в течение 4 часов. Вводили 3,0-3,5 г газообразного аммиака при 10-20°C. Затем реакционную смесь перемешивали при 15-20°C в течение 3 часов. Реакционную жидкость выливали в 300 г смеси воды и льда и разделяли и затем водную фазу дважды экстрагировали дихлорметаном (каждый раз по 50 г). Органические фазы объединяли и дважды промывали насыщенным раствором хлорида натрия (каждый раз по 20 г). После извлечения растворителя из полученной органической фазы добавляли 10,0 г холодного хлорбутана; затем смесь растирали, промывали и фильтровали с получением 12,6 г (2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло [3.2.1] октан-2-карбоксамида с выходом 91,6% и чистотой 99,9%, определенными с помощью HPLC.
[0054] Данные ЯМР (ядерного магнитного резонанса) полученного продукта приведены ниже: 1H-ЯМР (400 МГц, DMSO-d6), δ: 1,65 (2H, m), 1,84 (1H, br), 2,06(1H, m), 2,90 (2H, s), 3,62 (1H, s), 3,68 (1H, d), 4,93 (2H, dd), 7,30-7,46 (7H, m).
Пример 6. Получение (2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамида (II)
[0055] В 4-горлую колбу объемом 500 мл, оснащенную мешалкой и термометром, добавляли 200 г дихлорметана, 12,5 г (0,05 моль) 5R-[(бензилокси)амино]пиперидин-2S-карбоновой кислоты, полученной в примере 2, 60 г диизопропилэтиламина и 0,1 г N,N-диметилформамида. После охлаждения добавляли по каплям раствор 23,8 г (0,08 моль) твердого фосгена в 80 г дихлорметана при −10-0°C. После завершения добавления реакционную смесь перемешивали при 10-20°C в течение 4 часов. Добавляли 25 г 10% (по массе) раствора газообразного аммиака в метаноле при 10-20°C. Затем реакционную смесь перемешивали при 15-20°C в течение 3 часов. Реакционную жидкость выливали в 300 г смеси воды и льда и разделяли и затем водную фазу дважды экстрагировали дихлорметаном (каждый раз по 50 г). Органические фазы объединяли и дважды промывали насыщенным раствором хлорида натрия (каждый раз по 20 г). После извлечения растворителя из полученной органической фазы добавляли 10,0 г холодного хлорбутана; затем смесь растирали, промывали и фильтровали с получением 12,7 г (2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло [3.2.1] октан-2-карбоксамида с выходом 92,5% и чистотой 99,9%, определенными с помощью HPLC.
Пример 7. Получение (2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамида (II)
В 4-горлую колбу объемом 500 мл, оснащенную мешалкой и термометром, добавляли 200 г дихлорметана, 12,5 г (0,05 моль) 5R-[(бензилокси)амино]пиперидин-2S-карбоновой кислоты, полученной в примере 2, 60 г диизопропилэтиламина и 0,1 г N,N-диметилформамида. После охлаждения добавляли по каплям раствор 24,0 г (0,12 моль) дифосгена в 60 г дихлорметана при −10-0°C. После завершения добавления реакционную смесь перемешивали при 20-25°C в течение 3 часов. Добавляли 25 г 10% (по массе) раствора газообразного аммиака в метаноле при 20-25°C. Затем реакционную смесь перемешивали при 20-25°C в течение 3 часов. Реакционную жидкость выливали в 300 г смеси воды и льда и разделяли и затем водную фазу дважды экстрагировали дихлорметаном (каждый раз по 50 г). Органические фазы объединяли и дважды промывали насыщенным раствором хлорида натрия (каждый раз по 20 г). После извлечения растворителя из полученной органической фазы добавляли 10,0 г холодного хлорбутана; затем смесь растирали, промывали и фильтровали с получением 12,5 г (2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло [3.2.1] октан-2-карбоксамида с выходом 91,0% и чистотой 99,9%, определенными с помощью HPLC.
Пример 8. Получение (2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамида (II)
[0056] В 4-горлую колбу объемом 500 мл, оснащенную мешалкой и термометром, добавляли 200 г дихлорметана, 12,5 г (0,05 моль) 5R-[(бензилокси)амино]пиперидин-2S-карбоновой кислоты, полученной в примере 2, 40 г триэтиламина и 0,1 г N,N-диметилформамида. После охлаждения добавляли по каплям раствор 23,8 г (0,08 моль) твердого фосгена в 80 г дихлорметана при −10-0°C. После завершения добавления реакционную смесь перемешивали в течение 4 часов при температуре 10-20°C. Добавляли 25 г 10% (по массе) водного раствора аммиака при 10-20°C. Затем реакционную смесь перемешивали при 15-20°C в течение 3 часов. Реакционную жидкость выливали в 200 г смеси воды и льда и разделяли и затем водную фазу дважды экстрагировали дихлорметаном (каждый раз по 50 г). Органические фазы объединяли и дважды промывали насыщенным раствором хлорида натрия (каждый раз по 20 г). После извлечения растворителя из полученной органической фазы добавляли 10,0 г холодного хлорбутана; затем смесь растирали, промывали и фильтровали с получением 12,1 г (2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло [3.2.1] октан-2-карбоксамида с выходом 88,0% и чистотой 99,8%, определенными с помощью HPLC.
Пример 9. Получение (2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамида (II)
[0057] При получении 5R-[(бензилокси)амино]пиперидин-2S-карбоновой кислоты (IV) смешанную жидкость полученной органической фазы непосредственно подвергали следующим стадиям.
[0058] В 4-горлую колбу объемом 500 мл, оснащенную мешалкой и термометром, добавляли 150 г дихлорметана, 120 г 10% (по массе) водного раствора гидроксида натрия, 37,0 г (0,1 моль) этил-5R-[(бензилокси)амино]пиперидин-2S-карбоксилата оксалата (III) и затем реакционную смесь перемешивали при 20-25°C в течение 4 часов. Затем реакционную смесь подкисляли до значения pH 2,5-3,0 с помощью 30% (по массе) водного раствора хлористоводородной кислоты и перемешивали при комнатной температуре в течение 1-2 часов. Смешанный раствор разделяли и затем водную фазу трижды экстрагировали дихлорметаном (каждый раз по 50 г). Органические фазы объединяли с получением смешанного раствора органических фаз.
[0059] Полученный смешанный раствор органической фазы переносили в другую 4-горлую колбу объемом 500 мл, в которую загружали 60 г диизопропилэтиламина и 0,1 г N,N-диметилформамида. После охлаждения добавляли по каплям раствор 26,7 г (0,09 моль) твердого фосгена в 80 г дихлорметана при −10-0°C. После завершения добавления реакционную смесь перемешивали при 10-20°C в течение 4 часов. Добавляли 25 г 10% (по массе) раствора газообразного аммиака в метаноле при 10-20°C. Затем реакционную смесь перемешивали при 15-20°C в течение 3 часов. Реакционную жидкость выливали в 300 г смеси воды и льда и разделяли и затем водную фазу дважды экстрагировали дихлорметаном (каждый раз по 50 г). Органические фазы объединяли и дважды промывали насыщенным раствором хлорида натрия (каждый раз по 20 г). После извлечения растворителя из полученной органической фазы добавляли 10,0 г холодного хлорбутана; затем смесь растирали, промывали и фильтровали с получением 24,9 г (2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло [3.2.1] октан-2-карбоксамида с общим выходом 90,5% и чистотой 99,9%, определенными с помощью HPLC.
Сравнительный пример 1. Получение 5R-[(бензилокси)амино]пиперидин-2S-карбоновой кислоты (IV)
[0060] В 4-горлую колбу объемом 500 мл, оснащенную мешалкой и термометром, добавляли 150 г дихлорметана, 70 г 10% (по массе) водного раствора гидроксида натрия, 43,0 г (0,1 моль) бензил-5R-[(бензилокси)амино]пиперидин-2S-карбоксилата оксалата (III) и затем реакционную смесь перемешивали при 20-30°C в течение 3 часов. Затем реакционную смесь подкисляли до значения pH 2,5-3,0 с помощью 30% (по массе) водного раствора хлористоводородной кислоты и перемешивали при комнатной температуре в течение 1-2 часов. Смешанный раствор разделяли и затем водную фазу трижды экстрагировали дихлорметаном (каждый раз по 50 г). Органические фазы объединяли и один раз промывали насыщенным раствором хлорида натрия (каждый раз по 20 г). После извлечения растворителя из полученной органической фазы получали 9,5 г 5R-[(бензилокси)амино]пиперидин-2S-карбоновой кислоты с выходом 38,1% и чистотой 98,1%, определенными с помощью HPLC.
[0061] В данном сравнительном примере показано, что если во время процедуры получения 5R-[(бензилокси)амино]пиперидин-2S-карбоновой кислоты количество основания является слишком низким, гидролиз будет неполным, что приводит к значительному снижению выхода 5R-[(бензилокси)амино]пиперидин-2S-карбоновой кислоты.
Сравнительный пример 2. Получение 5R-[(бензилокси)амино]пиперидин-2S-карбоновой кислоты (IV)
[0062] В 4-горлую колбу объемом 500 мл, оснащенную мешалкой и термометром, добавляли 150 г дихлорметана, 150 г 10% (по массе) водного раствора гидроксида натрия, 43,0 г (0,1 моль) бензил-5R-[(бензилокси)амино]пиперидин-2S-карбоксилата оксалата (III) и затем реакционную смесь перемешивали при 20-30°C в течение 3 часов. Затем реакционную смесь подкисляли до значения pH 1,5-1,9 с помощью 30% (по массе) водного раствора хлористоводородной кислоты и перемешивали при комнатной температуре в течение 1-2 часов. Смешанный раствор разделяли и затем водную фазу трижды экстрагировали дихлорметаном (каждый раз по 50 г). Органические фазы объединяли и один раз промывали насыщенным раствором хлорида натрия (каждый раз по 20 г). После извлечения растворителя из полученной органической фазы получали 18,2 г 5R-[(бензилокси)амино]пиперидин-2S-карбоновой кислоты с выходом 72,8% и чистотой 99,7%, определенными с помощью HPLC.
[0063] В данном сравнительном примере показано, что если во время процедуры получения 5R-[(бензилокси)амино]пиперидин-2S-карбоновой кислоты значение pH при подкислении является слишком низким, часть продукта превратится в гидрохлорид, который растворяется в воде, что приводит к снижению выхода 5R-[(бензилокси)амино]пиперидин-2S-карбоновой кислоты.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОСТОЙ СПОСОБ ПОЛУЧЕНИЯ АВИБАКТАМА | 2018 |
|
RU2711358C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ ДЛЯ ПОЛУЧЕНИЯ АВИБАКТАМА | 2018 |
|
RU2722625C1 |
| СПОСОБ ПОЛУЧЕНИЯ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ, ВКЛЮЧАЯ ТРАНС-7-ОКСО-6-(СУЛЬФОКСИ)-1,6-ДИАЗАБИЦИКЛО[3.2.1]ОКТАН-2-КАРБОКСАМИД И ЕГО СОЛИ | 2012 |
|
RU2769076C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО ДИАЗАБИЦИКЛООКТАНА И ЕГО ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ | 2014 |
|
RU2719480C2 |
| НОВЫЙ ИНГИБИТОР бета-ЛАКТАМАЗЫ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2013 |
|
RU2693898C2 |
| (2S,5R)-5-[(бензилокси)амино]пиперидин-2-карбоксамид | 2012 |
|
RU2610091C2 |
| НОВЫЙ ИНГИБИТОР β-ЛАКТАМАЗЫ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2019 |
|
RU2800050C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ПРОИЗВОДНОГО ДИАЗАБИЦИКЛООКТАНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2014 |
|
RU2801220C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ПРОИЗВОДНОГО ДИАЗАБИЦИКЛООКТАНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2014 |
|
RU2695219C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ПРОИЗВОДНОГО ДИАЗАБИЦИКЛООКТАНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2023 |
|
RU2838718C2 |
Изобретение относится к области органической химии, а именно к способу получения промежуточного соединения для получения авибактама, предусматривающему следующие стадии, на которых: (1) соединение формулы III растворяют в растворителе A, гидролизуют в присутствии основания A и затем подкисляют с получением соединения формулы IV:
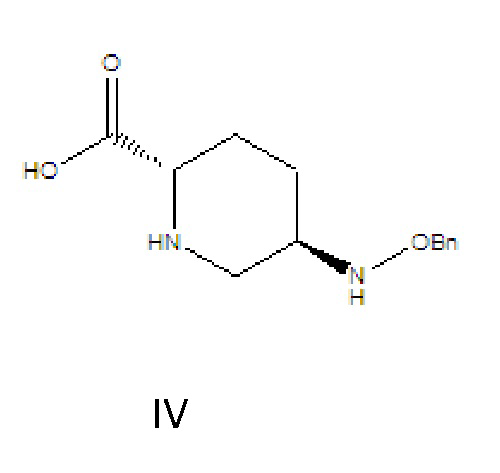
где R в соединении формулы III означает C1-6-алифатическую группу или -алкилзамещенный фенил; предпочтительно R выбран из группы, состоящей из метила, этила, н-пропила, изопропила, н-бутила, изобутила, трет-бутила, н-пентила, изопентила, трет-пентила, гексила, бензила, о-метилбензила и п-метилбензила; (2) соединение формулы IV и твердый фосген или дифосген параллельно вводят в реакцию циклизации мочевины и реакции хлорформилирования в присутствии органического основания B и катализатора в растворителе B с получением соединения формулы V, которое применяли непосредственно на следующей стадии реакции без очистки;
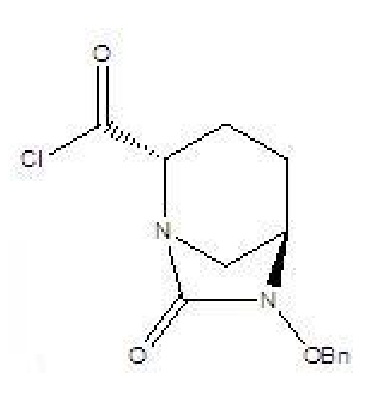
V,
(3) проводят реакцию амидирования с участием соединения формулы V и аммиака с получением (2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамида. Технический результат: предложен способ получения, который является простым и пригодным к применению без необходимости в специальном защитном средстве или реагенте для карбонилирования, а также является экологичным и безвредным для окружающей среды. 8 з.п. ф-лы, 9 пр., 3 ил.
1. Способ получения промежуточного соединения для получения авибактама, предусматривающий следующие стадии, на которых:
(1) соединение формулы III растворяют в растворителе A, гидролизуют в присутствии основания A и затем подкисляют с получением соединения формулы IV:
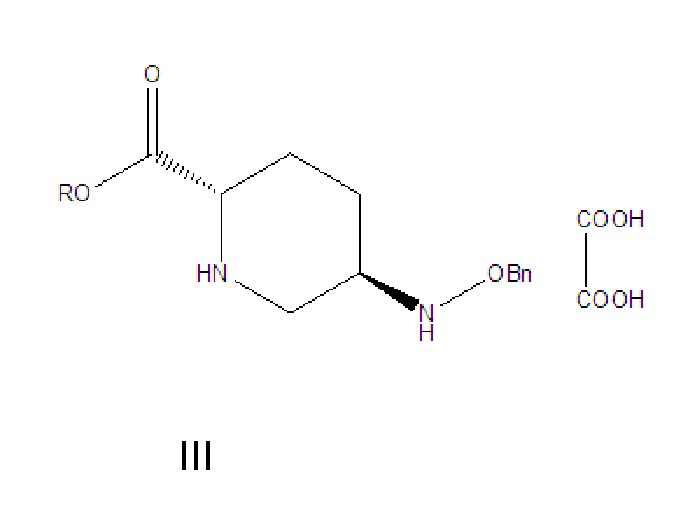
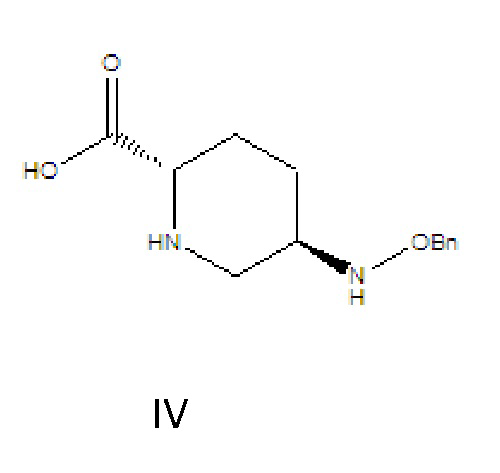
где R в соединении формулы III означает C1-6-алифатическую группу или -алкилзамещенный фенил; предпочтительно R выбран из группы, состоящей из метила, этила, н-пропила, изопропила, н-бутила, изобутила, трет-бутила, н-пентила, изопентила, трет-пентила, гексила, бензила, о-метилбензила и п-метилбензила;
2) соединение формулы IV и твердый фосген или дифосген параллельно вводят в реакцию циклизации мочевины и реакции хлорформилирования в присутствии органического основания B и катализатора в растворителе B с получением соединения формулы V, которое применяли непосредственно на следующей стадии реакции без очистки;
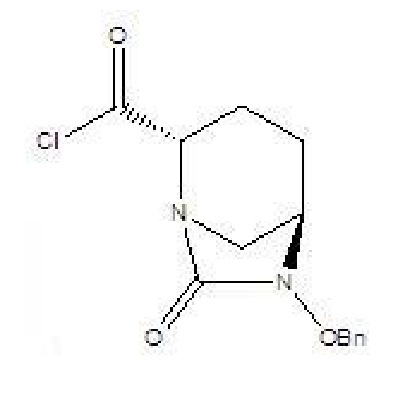
V,
(3) проводят реакцию амидирования с участием соединения формулы V и аммиака с получением (2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамида;
где растворитель A на стадии (1) выбран из группы, состоящей из дихлорметана, 1,2-дихлорэтана, трихлорметана, тетрахлорметана, тетрагидрофурана, 2-метилтетрагидрофурана, метоксициклопентана, метилбензола и комбинации двух или более из них;
при этом на стадии (1) массовое соотношение растворителя A и соединения формулы III составляет 3-4,5:1;
при этом на стадии (1) основание A выбрано из группы, состоящей из гидроксида натрия, гидроксида калия, гидроксида лития, карбоната калия, карбоната натрия, гидрокарбоната калия, или гидрокарбоната натрия, или смеси из двух или более из них; и
при этом на стадии (1) молярное соотношение основания A и соединения формулы III составляет 2,0-5,0:1;
при этом на стадии (1) реакцию гидролиза осуществляют при температуре 0-80°C;
при этом на стадии (1) подкисление осуществляют при температуре 20-25°C;
при этом на стадии (2) растворитель B выбран из группы, состоящей из дихлорметана, 1,2-дихлорэтана, трихлорметана, тетрахлорметана, тетрагидрофурана, 2-метилтетрагидрофурана, метоксициклопентана, метилбензола и комбинации двух или более из них;
при этом на стадии (2) массовое соотношение растворителя B и соединения формулы IV составляет 4-20:1;
при этом на стадии (2) органическое основание B выбрано из группы, состоящей из триэтиламина, три-н-бутиламина, диизопропилэтиламина и комбинации двух или более из них;
при этом на стадии (2) молярное соотношение органического основания B и соединения формулы IV составляет 3,0-8,0:1;
при этом на стадии (2) катализатор присутствует в количестве 0,1-5,0% по массе соединения формулы IV;
при этом на стадии (2) молярное соотношение твердого фосгена или дифосгена и соединения формулы IV составляет 0,6-2,5:1;
при этом на стадии (2) как реакцию циклизации мочевины, так и реакцию хлорформилирования осуществляют при температуре −20-60°C;
при этом на стадии (3) реакцию амидирования осуществляют при температуре −20-80°C.
2. Способ по п. 1, где после гидролиза соединения формулы III на стадии (1) в щелочной среде и его подкисления получают экстракт, содержащий соединение формулы IV, с применением экстрагирующего средства; экстракт, содержащий соединение формулы IV, подвергают перегонке с удалением экстрагирующего средства с получением соединения формулы IV, или экстракт, содержащий соединение формулы IV, непосредственно применяют на следующей стадии реакции без перегонки.
3. Способ по п. 1, где на стадии (1) реакцию гидролиза осуществляют при температуре 10-40°C.
4. Способ по п. 1, где на стадии (1) подкисление предусматривает регулирование значения pH системы до 2-3 с применением подкисляющего средства;
при этом подкисляющее средство представляет собой водный раствор хлористоводородной кислоты, серной кислоты или азотной кислоты при концентрации 10-40% по массе.
5. Способ по п. 1, где на стадии (2) катализатор выбран из группы, состоящей из N,N-диметилформамида, пиридина, 4-диметиламинопиридина и комбинации двух или более из них.
6. Способ по п. 1, где молярное соотношение твердого фосгена и соединения формулы IV составляет 0,6-2,0:1; или
молярное соотношение дифосгена и соединения формулы IV составляет 1,0-2,5:1.
7. Способ по п. 1, где на стадии (2) как реакцию циклизации мочевины, так и реакцию хлорформилирования осуществляют при температуре 0-40°C.
8. Способ по п. 1, где на стадии (3) соединение аммиака выбрано из группы, состоящей из газообразного аммиака, раствора газообразного аммиака в метаноле или водного раствора аммиака;
при этом на стадии (3) молярное соотношение аммиака и соединения формулы IV составляет 1,0-5,0:1.
9. Способ по п. 1, где на стадии (3) реакцию амидирования осуществляют при температуре 10-50°C.
| BALL M | |||
| et al | |||
| Development of a Manufacturing Route to Avibactam, a B-Lactamase Inhibitor, Org | |||
| Process Res | |||
| Dev., vol.20, no 10, p | |||
| Деревянный понтон с фанерной обшивкой | 1925 |
|
SU1799A1 |
| CN 106749242 A, 31.05.2017 | |||
| СПОСОБ ОЧИСТКИ РЕГЕНЕРИРОВАННОГО ПОЛИЭТИЛЕНА | 2017 |
|
RU2721005C1 |
| CN 106279163 A, 04.10.2017 | |||
| СПОСОБ ПОЛУЧЕНИЯ НАТРИЕВОЙ СОЛИ (2S,5R)-2-КАРБОКСАМИДО-7-ОКСО-6-СУЛЬФООКСИ-1,6-ДИАЗАБИЦИКЛО[3.2.1]ОКТАНА | 2013 |
|
RU2632192C2 |
| WO 2010126820 A2, 04.11.2010. | |||

