ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Болезнь Альцгеймера (AD; от англ. “Alzheimer′s disease”) представляет собой нейродегенеративное расстройство центральной нервной системы и ведущей причиной прогрессирующей деменции у пожилого населения. Ее клиническими симптомами являются ухудшение памяти, познавательной способности, временной и пространственной ориентации, суждения и мышления, а также тяжелые эмоциональные нарушения. В настоящее время нет доступных способов лечения, которые могут предупреждать это заболевание или его прогрессирование, либо вызывать стабильную реверсию его клинических симптомов. AD стала значительной проблемой здравоохранения во всех обществах с высокой средней продолжительностью жизни, а также значительным экономическим бременем для их систем здравоохранения.
AD характеризуется двумя основными патологиями в центральной нервной системе (ЦНС), возникновением амилоидных бляшек и нейрофибриллярных клубков (Hardy et al., The amyloid hypothesis of Alzheimer′s disease: progress and problems on the road to therapeutics, Science. 2002 Jul 19; 297(5580): 353-6, Selkoe, Cell biology of the amyloid beta-protein precursor and the mechanism of Alzheimer′s disease, Annu Rev Cell Biol. 1994; 10: 373-403). Обе патологии также обычно наблюдают у пациентов с синдромом Дауна (трисомией 21), у которых также развиваются AD-подобные симптомы в раннем возрасте. Нейрофибриллярные клубки представляют собой внутриклеточные агрегаты ассоциированного с микротрубочками белка tau (МАРТ; от англ. “microtubule-associated protein tau”). Амилоидные бляшки возникают во внеклеточном пространстве, их основными компонентами являются Аβ-пептиды. Последние представляют собой группу протеолитических фрагментов, образованных от белка-предшественника β-амилоида (АРР; от англ. “β-amyloid precursor protein”) в результате серии стадий протеолитического расщепления. Идентифицировано несколько форм АРР, из которых наиболее распространенными являются белки длиной 695, 751 и 770 аминокислот. Все они имеют происхождение от одного гена посредством дифференциального сплайсинга. Аβ-пептиды образованы из одного и того же домена АРР, но отличаются их N- и С-концами, где основные виды молекул имеют длину 40 и 42 аминокислоты. Существует несколько научных доказательств, которые позволяют с большой вероятностью предположить, что Аβ-пептиды являются существенными молекулами в патогенезе AD: 1) амилоидные бляшки, образованные из Аβ-пептидов, составляют неизменную часть патологии AD; 2) Аβ-пептиды токсичны для нейронов; 3) при семейной болезни Альцгеймера (FAD; от англ. “Familial Alzheimer′s Disease”) мутации в генах этого заболевания АРР, PSN1, PSN2 приводят к повышенным уровням Аβ-пептидов и раннему амилоидозу головного мозга; 4) у трансгенных мышей, которые экспрессируют такие гены FAD, развивается патология, которая имеет много сходств с заболеванием у человека. Аβ-пептиды продуцируются из АРР посредством последовательного действия двух протеолитических ферментов, называемых β- и γ-секретазой. β-Секретаза сначала расщепляет во внеклеточном домене АРР примерно 28 аминокислот снаружи от трансмембранного домена (ТМ; от англ. “transmembrane domain”) с образованием С-концевого фрагмента АРР, содержащего ТМ и цитоплазматический домен (CTFβ). CTFβ является субстратом для γ-секретазы, которая расщепляет в нескольких соседних положениях внутри ТМ с образованием Аβ пептидов и цитоплазматического фрагмента. γ-Секретаза представляет собой комплекс, состоящий по меньшей мере 4 различных белков, и ее каталитической субъединицей весьма вероятно является белок пресенилин (PSEN1, PSEN2). β-Секретаза (ВАСЕ1, Asp2; ВАСЕ обозначает β-сайт АРР-расщепляющего фермента) представляет собой аспартилпротеазу, которая заякорена в мембране посредством трансмембранного домена (Vassar et al., Beta-secretase cleavage of Alzheimer′s amyloid precursor protein by the transmembrane aspartic protease BACE, Science. 1999 Oct 22; 286(5440): 735). Она экспрессируется во многих тканях организма человека, но ее уровень особенно высок в ЦНС. Генетическое разрушение гена ВАСЕ1 у мышей четко показало, что его активность существенна для процессинга АРР, который приводит к образованию Аβ-пептидов, в отсутствие ВАСЕ1 Аβ-пептиды не продуцируются (Luo et al., Mice deficient in BACE1, the Alzheimer′s beta-secretase, have normal phenotype and abolished beta-amyloid generation, Nat Neurosci. 2001 Mar; 4(3): 231-2, Roberds et al., BACE knockout mice are healthy despite lacking the primary beta-secretase activity in brain: implications for Alzheimer′s disease therapeutics, Hum Mol Genet. 2001 Jun 1; 10(12): 1317-24). Мыши, у которых генно-инженерным путем получена экспрессия гена АРР человека, и у которых образуются обширные амилоидные бляшки и патологии, подобные болезни Альцгеймера, при старении, этого не происходит, когда активность β-секретазы снижена в результате генетического разрушения одного из аллелей ВАСЕ1 (McConlogue et al., Partial reduction of BACE1 has dramatic effects on Alzheimer plaque and synaptic pathology in APP Transgenic Mice. J Biol Chem. 2007 Sep 7; 282(36): 26326). Следовательно, предполагают, что ингибиторы активности ВАСЕ1 могут быть полезными агентами для терапевтического вмешательства в болезнь Альцгеймера (AD).
Диабет типа 2 (T2D; от англ. “type 2 diabetes”) вызван устойчивостью к инсулину и неадекватной секрецией инсулина из β-клеток поджелудочной железы, приводящей к слабому контролю глюкозы в крови и гипергликемии (М Prentki & CJ Nolan, “Islet beta-cell failure in type 2 diabetes.” J. Clin. Investig. 2006, 116(7), 1802-1812). Пациенты, страдающие T2D, обладают повышенным риском микрососудистого и макрососудистого заболевания и ряда родственных осложнений, включающих диабетическую нефропатию, ретинопатию и сердечнососудистое заболевание. В 2000 г. по оценкам 171 млн. человек страдал этим состоянием, причем ожидают, что эта цифра удвоится к 2030 г. (S Wild, G Roglic, А Green, R. Sicree & Н King, “Global prevalence of diabetes”, Diabetes Care 2004, 27(5), 1047-1053), что делает это заболевание значительной проблемой здравоохранения. Рост распространенности T2D связан с все возрастающим сидячим образом жизни и приемом высококалорийных продуктов питания населением во всем мире (Р Zimmet, KGMM Alberti & J Shaw, “Global and societal implications of the diabetes epidemic” Nature 2001, 414, 782-787).
Недостаточность β-клеток и вследствие этого резкое снижение секреции инсулина и гипергликемия отмечает начало T2D. Большинство современных терапий не предотвращает потерю массы β-клеток, характеризующую выраженный T2D. Однако недавние разработки аналогов GLP-1, гастрина и других агентов показывают, что сохранения и пролиферации β-клеток можно достичь, что приведет к улучшенной толерантности к глюкозе и замедлению прогрессирования до выраженного T2D (LL Baggio & DJ Drucker, “Therapeutic approaches to preserve islet mass in type 2 diabetes”, Annu. Rev. Med. 2006, 57, 265-281).
Tmem27 идентифицирован как белок, стимулирующий пролиферацию бета-клеток (Р Akpinar, S Kuwajima, J Krützfeldt, M Stoffel, “Tmem27: A cleaved and shed plasma membrane protein that stimulates pancreatic β cell proliferation”, Cell Metab. 2005, 2, 385-397) и секрецию инсулина (К Fukui, Q Yang, Y Cao, N Takahashi et al., “The HNF-1 target Collectrin controls insulin exocytosis by SNARE complex formation”, Cell Metab. 2005, 2, 373-384). Tmem27 представляет собой мембранный гликопротеин, имеющий молекулярную массу 42 кДа, который конститутивно выделяется с поверхности β-клеток в результате расщепления полноразмерного клеточного Tmem27. Гиперэкспрессия Tmem27 у трансгенных мышей увеличивает массу β-клеток и улучшает толерантность к глюкозе в модели диабета, вызванного алиментарным ожирением DIO (от англ. “diet-induced obesity”). Кроме того, нокаут Tmem27 посредством siRNA в анализе пролиферации β-клеток грызунов (например, с использованием клеток INS1e) снижает скорость пролиферации, что указывает на роль Tmem27 в контроле массы β-клеток.
В таком же анализе пролиферации ингибиторы ВАСЕ2 также увеличивают пролиферацию. Однако ингибирование ВАСЕ2 в сочетании с нокаутом Tmem27 siRNA приводит в результате к низким скоростям пролиферации. Поэтому сделан вывод, что ВАСЕ2 является протеазой, ответственной за расщепление Tmem27. Кроме того, in vitro ВАСЕ2 расщепляет пептид, основанный на последовательности Tmem27. Близкородственная протеаза ВАСЕ1 не расщепляет этот пептид, и селективное ингибирование только ВАСЕ1 не усиливает пролиферацию β-клеток.
Близким гомологом ВАСЕ2 является мембраносвязанная аспартилпротеаза, и она совместно локализована с Tmem27 в β-клетках поджелудочной железы человека (G Finzi, F Franzi, С Placidi, F Acquati et al., “BACE2 is stored in secretory granules of mouse and rat pancreatic beta cells”, Ultrastruct Pathol. 2008, 32(6), 246-251 Также известно, что она способна к расщеплению АРР (I Hussain, D Powell, D Howlett, G Chapman et al., “ASP1 (BACE2) cleaves the amyloid precursor protein at the β-secretase site” Mol Cell Neurosci. 2000, 16, 609-619), IL-1R2 (P Kuhn, E Marjaux, A Imhof, В De Strooper et al., “Regulated intramembrane proteolysis of the interleukin-1 receptor II by alpha-, beta-, and gamma-secretase” J. Biol. Chem. 2007, 282(16), 11982-11995) и ACE2. Способность к расщеплению АСЕ2 указывает на возможную роль ВАСЕ2 в контроле гипертензии.
Ингибирование ВАСЕ2, следовательно, предложено в качестве терапии T2D с потенциалом сохранения и восстановления массы β-клеток и стимуляции секреции инсулина у преддиабетических и диабетических пациентов. Следовательно, цель настоящего изобретения состоит в разработке селективных ингибиторов ВАСЕ2. Такие соединения полезны в качестве терапевтически активных веществ, в частности, при лечении и/или предупреждении заболеваний, которые связаны с ингибированием ВАСЕ2.
Кроме того, образование или образование и отложение β-амилоидных пептидов в неврологической ткани или вокруг нее (например, в головном мозге) ингибируется настоящими соединениями, то есть ингибированием продуцирования Аβ из АРР или фрагмента АРР.
В настоящем изобретении предложены новые соединения, имеющие формулу I, их получение, лекарственные средства на основе соединений в соответствии с изобретением и их получение, а также применение соединений, имеющих формулу I, при контроле или предупреждении заболеваний, таких как болезнь Альцгеймера и диабет типа 2.
ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к N-[3-(5-амино-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-фенил]-амидам, обладающих ингибиторными свойствами в отношении ВАСЕ1 и/или ВАСЕ2, к их получению, к содержащим их фармацевтическим композициям и к их применению в качестве терапевтически активных веществ.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к соединениям, имеющим формулу I,
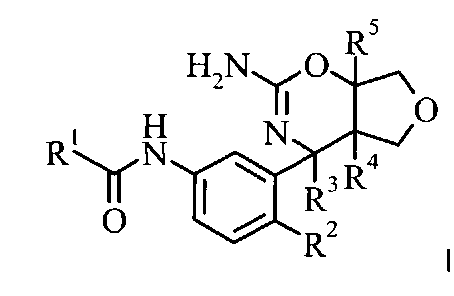 ,
,
в которых заместители и переменные являются такими, как описано ниже и в формуле изобретения, или к их фармацевтически приемлемым солям.
Настоящие соединения обладают ингибиторной активностью по отношению к Asp2 (β-секретазе, ВАСЕ1 или Мемапсин-2) и, следовательно, их можно применять при терапевтическом и/или профилактическом лечении заболеваний и расстройств, характеризующихся повышенными уровнями β-амилоида и/или олигомеров β-амилоида и/или β-амилоидных бляшек и других отложений, в частности, болезни Альцгеймера, и/или настоящие соединения обладают ингибиторной активностью по отношению к ВАСЕ2 и, следовательно, их можно применять при терапевтическом и/или профилактическом лечении заболеваний и расстройств, таких как диабет типа 2 и другие метаболические расстройства.
СВЕДЕНИЯ, ПОДТВЕРЖДАЮЩИЕ ВОЗМОЖНОСТЬ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
В настоящем изобретении предложено соединение, имеющее формулу I, и его фармацевтически приемлемые соли, получение вышеуказанных соединений, содержащие их лекарственные средства и их получение, а также применение вышеуказанных соединений при терапевтическом и/или профилактическом лечении заболеваний и расстройств, связанных с ингибированием активности ВАСЕ1 и/или ВАСЕ2, таких как болезнь Альцгеймера и диабет типа 2. Кроме того, настоящие соединения ингибируют образование или образование и отложение β-амилоидных бляшек в неврологической ткани (например, в головном мозге) или вокруг нее посредством ингибирования продуцирования Аβ из АРР или фрагмента АРР.
Приведенные ниже определения общих терминов, используемых в настоящем описании, применяют независимо от того, встречаются ли обсуждаемые термины отдельно или в сочетании с другими группами.
Если не указано иное, приведенные ниже термины, используемые в данной заявке, включая описание и формулу изобретения, имеют приведенные ниже определения. Необходимо отметить, что, как используют в описании и прилагаемой формуле изобретения, формы единственного числа включают множественные объекты ссылки, если контекстом явным образом не продиктовано иное.
Термин "C1-6-алкил", отдельно или в комбинации с другими группами, означает углеводородный радикал, который может быть нормальным или разветвленным, с одиночным или множественным разветвлением, где алкильная группа, как правило, включает от 1 до 6 атомов углерода, например, метил (Me), этил (Et), пропил, изопропил (i-пропил), н-бутил, i-бутил (изобутил), 2-бутил (втор-бутил), t-бутил (трет-бутил), изопентил, 2-этил-пропил, 1,2-диметил-пропил и тому подобное. Конкретный “C1-6-алкил” представляет собой группы, включающие от 1 до 5 атомов углерода. Конкретные группы представляют собой метил, этил и трет-бутил. Наиболее конкретная группа представляет собой метил.
Термин “циано-C1-6-алкил”, отдельно или в комбинации с другими группами, относится к C1-6-алкилу, как определено в данной работе, замещенному одной или множественными цианогруппами, в частности, 1-5 цианогруппами, более конкретно одной цианогруппой. Примерами являются циано-метил и тому подобное.
Термин “галстен-C1-6-алкил”, отдельно или в комбинации с другими группами, относится к C1-6-алкилу, как определено в данной работе, замещенному одним или множественными атомами галогена, предпочтительно 1-5 атомами галогена, более предпочтительно 1-3 атомами галогена, наиболее предпочтительно одним атомом галогена или 3 атомами галогена. Конкретный атом галогена представляет собой атом фтора. Примерами являются дифторметил, хлорметил, фторметил и тому подобное, в частности, -CH2CH2F, CH2CHF2 или -CF3. Конкретным примером является трифторметил.
Термин “C1-6-алкокси-C1-6-алкил”, отдельно или в комбинации с другими группами, относится к C1-6-алкилу, как определено в данной работе, замещенному одной или множественными C1-6-алкоксигруппами, как определено в данной работе. Примерами являются MeO-Me, 1MeO-Et, 2MeO-Et, 1МеО-2EtO-пропил и тому подобное.
Термин “циано”, отдельно или в комбинации с другими группами, относится к группе N≡C-(NC-).
Термин "атом галогена", отдельно или в комбинации с другими группами, означает атом хлора (Cl), йода (I), фтора (F) и брома (Br). Конкретный “атом галогена” представляет собой Cl и F. Конкретным является F.
Термин “арил", отдельно или в комбинации с другими группами, относится к ароматической карбоциклической группе, включающей от 6 до 14, предпочтительно от 6 до 10 атомов углерода и имеющей по меньшей мере одно ароматическое кольцо или множественные конденсированные кольца, в которых по меньшей мере одно кольцо является ароматическим. Примеры “арила” включают бензил, дифенил, инданил, нафтил, фенил (Ph) и тому подобное. Конкретный “арил” представляет собой фенил.
Термин "гетероарил", отдельно или в комбинации с другими группами, относится к ароматической карбоциклической группе, имеющей одно 4-8-членное кольцо или множественные конденсированные кольца, включающие от 6 до 14, более предпочтительно от 6 до 10, кольцевых атомов и включающие 1, 2 или 3 гетероатома, индивидуально выбранные из N, О и S, в частности, N и О, причем, в этой группе по меньшей мере одно гетероциклическое кольцо является ароматическим. Примеры "гетероарила" включают бензофурил, бензоимидазолил, 1Н-бензоимидазолил, бензоксазинил, бензоксазолил, бензотиазинил, бензотиазолил, бензотиенил, бензотриазолил, фурил, имидазолил, индазолил, 1H-индазолил, индолил, изохинолинил, изотиазолил, изоксазолил, оксазолил, пиразинил, пиразолил (пиразил), 1Н-пиразолил, пиразоло[1,5-а]пиридинил, пиридазинил, пиридинил, пиримидинил, пирролил, хинолинил, тетразолил, тиазолил, тиенил, триазолил, 6,7-дигидро-5Н-[1]пиримидинил и тому подобное. Конкретный "гетероарил" представляет собой пиридинил и пиразинил. Конкретными примерами являются пиридин-2-ил и пиразин-2-ил.
Термин "C1-6-алкокси", отдельно или в комбинации с другими группами, обозначает -О-C1-6-алкильный радикал, который может быть нормальным или разветвленным, с одиночным или множественным разветвлением, в котором алкильная группа, как правило, включает от 1 до 6 атомов углерода, например, метокси (ОМе, МеО), этокси (OEt), пропокси, изопропокси (i-пропокси), н-бутокси, i-бутокси (изо-бутокси), 2-бутокси (втор-бутокси), t-бутокси (трет-бутокси), изопентилокси (i-пентилокси) и тому подобное. Конкретными “C1-6-алкокси” являются группы, включающие от 1 до 4 атомов углерода. Конкретными примерами являются метокси, этокси и этилокси.
Термин “галоген-C1-6-алкокси”, отдельно или в комбинации с другими группами, относится к группе C1-6-алкокси, как определено в данной работе, замещенной одним или множественными атомами галогена, в частности, атомом фтора. Конкретная группа “галоген-C1-6-алкокси” представляет собой фтор-C1-6-алкокси. Конкретным примером является 2,2,2-трифтор-этокси-.
Термин “C2-6-алкинил-C1-6-алкокси”, отдельно или в комбинации с другими группами, относится к группе C1-6-алкокси, как определено в данной работе, замещенной одним или множественными C2-6-алкинилами, как определено в данной работе. Конкретная группа “C2-6-алкинил-C1-6-алкокси” представляет собой 5-бут-2-инилокси-пиразин-2-ил.
Термин “C2-6-алкинил”, отдельно или в комбинации с другими группами, означает одновалентную нормальную или разветвленную насыщенную углеводородную группу, состоящую из атомов углерода в количестве от 2 до 6, в частности, от 2 до 4 атомов углерода, и включающую одну, две или три тройные связи. Примеры “C2-6-алкинила” включают этинил, пропинил, проп-2-инил, изопропинил и н-бутинил. Конкретными примерами являются этинил и пропинил.
Термин "фармацевтически приемлемые соли" относится к солям, которые пригодны для применения в контакте с тканями людей и животных. Примерами подходящих солей с неорганическими и органическими кислотами являются, но не ограничены ими, соли со следующими кислотами: уксусной кислотой, лимонной кислотой, муравьиной кислотой, фумаровой кислотой, соляной кислотой, молочной кислотой, малеиновой кислотой, яблочной кислотой, метансульфоновой кислотой, азотной кислотой, фосфорной кислотой, пара-толуолсульфоновой кислотой, янтарной кислотой, серной кислотой, сернистой кислотой, винной кислотой, трифторуксусной кислотой и тому подобное. Предпочтительны соли с муравьиной кислотой, трифторуксусной кислотой и соляной кислотой.
Термины “фармацевтически приемлемый носитель” и “фармацевтически приемлемое вспомогательное вещество” относятся к носителям и вспомогательным веществам, таким как разбавители или наполнители, которые совместимы с другими ингредиентами препарата.
Термин "фармацевтическая композиция" включает препарат, содержащий указанные ингредиенты в предопределенных количествах или долях, а также любой препарат, который является результатом, прямо или косвенно, объединения указанных ингредиентов в указанных количествах. Предпочтительно он включает препарат, содержащий один или более активных ингредиентов, и необязательный носитель, включающий инертные ингредиенты, а также любой препарат, который является результатом, прямо или косвенно, объединения, комплексообразования или агрегации любых двух или более ингредиентов, либо диссоциации одного или более ингредиентов, либо других типов реакций или взаимодействий одного или более ингредиентов.
Термин “ингибитор” означает соединение, конкурирующее за связывание конкретного лиганда с конкретным рецептором, либо уменьшающее или предотвращающее это связывание, либо соединение, либо уменьшающее или предотвращающее ингибирование функции конкретного белка.
Термин “половинная максимальная ингибиторная концентрация” (IC50) означает концентрацию конкретного соединения, необходимую для получения 50% ингибирования биологического процесса in vitro. Значения IC50 можно логарифмически преобразовать в значения pIC50 (-log IC50), в которых более высокие значения указывают на экспоненциально более высокую эффективность. Значение IC50 не является абсолютной величиной, а зависит от экспериментальных условий, например, от используемых концентраций. Значение IC50 можно преобразовать в абсолютную константу ингибирования (Ki), используя уравнение Ченга-Прусоффа (Biochem. Pharmacol. (1973) 22: 3099). Термин “константа ингибирования” (Ki) означает абсолютное сродство связывания конкретного ингибитора с рецептором. Ее измеряют, используя анализы конкурентного связывания, и она равна концентрации, при которой конкретный ингибитор занимал бы 50% рецепторов, если отсутствует конкурирующий лиганд (например, радиолиганд). Значения Ki можно логарифмически преобразовать в значения pKi (-log Ki), в которых более высокие значения указывают на экспоненциально более высокую эффективность.
“Терапевтически эффективное количество” означает количество соединения, которое при введении субъекту для лечения болезненного состояния, является достаточным, чтобы осуществить такое лечение болезненного состояния. “Терапевтически эффективное количество” варьирует в зависимости от соединения, болезненного состояния, подлежащего лечению, тяжести заболевания, подлежащего лечению, возраста и относительного состояния здоровья субъекта, пути и формы введения, мнения лечащего врача или ветеринара и от других факторов.
Термин “как определено в данной работе” и “как описано в данной работе” при отнесении к переменной включает посредством ссылки как широкое определение этой переменной, так и предпочтительные, более предпочтительные и наиболее предпочтительные определения, если они есть.
Термины “обработка”, “приведение в контакт” и “взаимодействие” при отнесении к химической реакции означают добавление или смешивание двух или более реагентов в соответствующих условиях с получением указанного и/или желаемого продукта. Понятно, что реакция, которая дает указанный и/или желаемый продукт, может необязательно являться результатом объединения двух реагентов, которые были первоначально добавлены, то есть может существовать одно или более промежуточных соединений, которые образуются в смеси, что, в конце концов, приводит к образованию указанного и/или желаемого продукта.
Термин “ароматический” означает традиционное понятие ароматичности, определенное в литературе, в частности, в руководстве IUPAC - Compendium of Chemical Terminology, 2nd, A.D. McNaught & A. Wilkinson (Eds). Blackwell Scientific Publications, Oxford (1997).
Термин “фармацевтически приемлемый наполнитель” означает любой ингредиент, не обладающий терапевтической активностью, и не являющийся токсичным, такой как, например, разрыхлители, связующие вещества, объемообразующие агенты, растворители, буферы, тонические агенты, стабилизаторы, антиоксиданты, сурфактанты или смазывающие вещества, применяемые при приготовлении фармацевтических препаратов.
В любом случае, когда в химической структуре присутствует хиральный атом углерода, подразумевают, что все стереоизомеры, связанные с этим хиральным атомом углерода, включены в структуру.
В изобретении также предложены фармацевтические композиции, способы применения и способы получения вышеупомянутых соединений.
Все отдельные формы осуществления можно объединять.
Одна форма осуществления изобретения представляет собой соединение, имеющее формулу I:
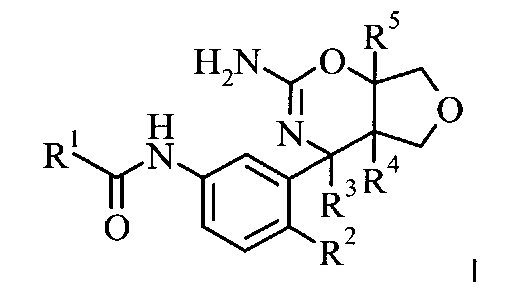
в котором
R1 выбран из группы, состоящей из
i) арила,
ii) арила, замещенного 1-4 заместителями, индивидуально выбранными из циано, циано-C1-6-алкила, атома галогена, галоген-C1-6-алкокси, галоген-C1-6-алкила, C1-6-алкокси, C1-6-алкокси-C1-6-алкила, C2-6-алкинил-C1-6-алкокси, C2-6-алкинила и C1-6-алкила,
iii) гетероарила и
iv) гетероарила, замещенного 1-4 заместителями, индивидуально выбранными из циано, циано-C1-6-алкила, атома галогена, галоген-C1-6-алкокси, галоген-C1-6-алкила, C1-6-алкокси, C1-6-алкокси-C1-6-алкила, C2-6-алкинил-C1-6-алкокси, C2-6-алкинила и C1-6-алкила;
R2 выбран из группы, состоящей из
i) атома водорода,
ii) C1-6-алкила и
iii) атома галогена;
R3 выбран из группы, состоящей из
i) C1-6-алкила и
ii) галоген-С1-6-алкила,
R4 выбран из группы, состоящей из
iii) атома водорода и
iv) C1-6-алкила, и
R5 выбран из группы, состоящей из
i) атома водорода,
ii) галоген-С1-6-алкила и
iii) C1-6-алкила;
или его фармацевтически приемлемые соли.
Конкретная форма осуществления изобретения представляет собой соединение, имеющее формулу I:
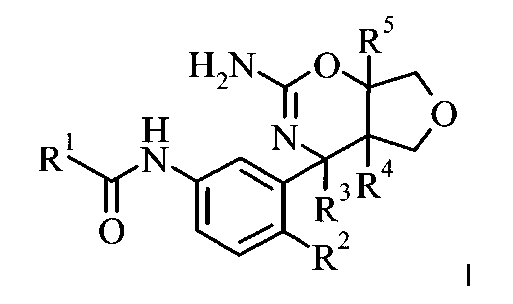
в котором
R1 выбран из группы, состоящей из
i) арила,
ii) арила, замещенного 1-4 заместителями, индивидуально выбранными из циано, циано-C1-6-алкила, атома галогена, галоген-C1-6-алкокси, галоген-C1-6-алкила, C1-6-алкокси, C1-6-алкокси-C1-6-алкила, C2-6-алкинил-C1-6-алкокси, C2-6-алкинила и C1-6-алкила,
iii) гетероарила и
iv) гетероарила, замещенного 1-4 заместителями, индивидуально выбранными из циано, циано-C1-6-алкила, атома галогена, галоген-C1-6-алкокси, галоген-C1-6-алкила, C1-6-алкокси, C1-6-алкокси-C1-6-алкила, C2-6-алкинил-C1-6-алкокси, C2-6-алкинила и C1-6-алкила;
R2 выбран из группы, состоящей из
i) атома водорода,
ii) C1-6-алкила и
iii) атома галогена;
R3 представляет собой C1-6-алкил,
R4 выбран из группы, состоящей из
i) атома водорода и
ii) C1-6-алкила, и
R5 выбран из группы, состоящей из
i) атома водорода и
ii) C1-6-алкила;
или его фармацевтически приемлемые соли.
Конкретная форма осуществления изобретения относится к соединению, имеющему формулу Ia:
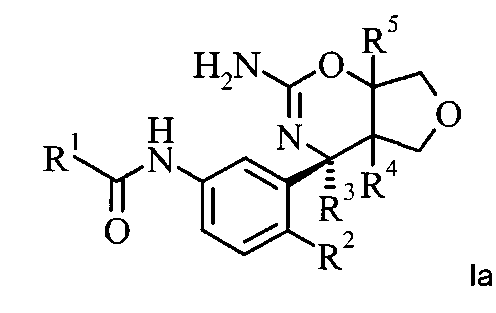 ,
,
в котором R1, R2, R3, R4 и R5 являются такими, как определено в данной работе; или к его фармацевтически приемлемым солям.
Конкретная форма осуществления изобретения относится к соединению, имеющему формулу Ia:
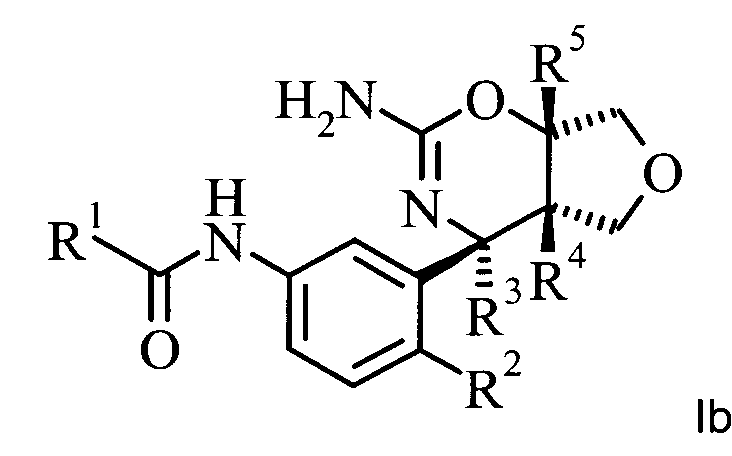 ,
,
в котором R1, R2, R3, R4 и R5 являются такими, как определено в данной работе; или к его фармацевтически приемлемым солям.
Конкретная форма осуществления изобретения относится к соединению, как определено в данной работе, в котором R2 представляет собой атом галогена.
Конкретная форма осуществления изобретения относится к соединению, как определено в данной работе, в котором R2 представляет собой F.
Конкретная форма осуществления изобретения относится к соединению, как определено в данной работе, в котором R3 представляет собой C1-6-алкил.
Конкретная форма осуществления изобретения относится к соединению, как определено в данной работе, в котором R3 представляет собой метил.
Конкретная форма осуществления изобретения относится к соединению, как определено в данной работе, в котором R3 представляет собой галоген-C1-6-алкил.
Конкретная форма осуществления изобретения относится к соединению, как определено в данной работе, в котором R3 представляет собой -CH2CH2F или -CH2CHF2.
Конкретная форма осуществления изобретения относится к соединению, как определено в данной работе, в котором R3 представляет собой -CH2CH2F.
Конкретная форма осуществления изобретения относится к соединению, как определено в данной работе, в котором R3 представляет собой -CH2CHF2.
Конкретная форма осуществления изобретения относится к соединению, как определено в данной работе, в котором R3 представляет собой метил, -CH2CH2F или -CH2CHF2.
Конкретная форма осуществления изобретения относится к соединению, как определено в данной работе, в котором R4 представляет собой C1-6-алкил.
Конкретная форма осуществления изобретения относится к соединению, как определено в данной работе, в котором R4 представляет собой атом водорода.
Конкретная форма осуществления изобретения относится к соединению, как определено в данной работе, в котором R5 представляет собой атом водорода.
Конкретная форма осуществления изобретения относится к соединению, как определено в данной работе, в котором R5 представляет собой C1-6-алкил.
Конкретная форма осуществления изобретения относится к соединению, как определено в данной работе, в котором R5 представляет собой галоген-C1-6-алкил.
Конкретная форма осуществления изобретения относится к соединению, как определено в данной работе, в котором R5 представляет собой -CF3.
Конкретная форма осуществления изобретения относится к соединению, как определено в данной работе, в котором R1 представляет собой гетероарил или гетероарил, замещенный 1-2 заместителями, индивидуально выбранными из циано, атома галогена, галоген-C1-6-алкокси и C2-6-алкинил-C1-6-алкокси.
Конкретная форма осуществления изобретения относится к соединению, как определено в данной работе, в котором R1 представляет собой гетероарил.
Конкретная форма осуществления изобретения относится к соединению, как определено в данной работе, в котором R1 представляет собой пиридинил.
Конкретная форма осуществления изобретения относится к соединению, как определено в данной работе, в котором R1 представляет собой пиридин-2-ил.
Конкретная форма осуществления изобретения относится к соединению, как определено в данной работе, в котором R1 представляет собой гетероарил, замещенный 1-2 заместителями, индивидуально выбранными из циано, атома галогена, галоген-C1-6-алкокси и C2-6-алкинил-C1-6-алкокси.
Конкретная форма осуществления изобретения относится к соединению, как определено в данной работе, в котором R1 представляет собой пиридинил, замещенный 1-2 заместителями, индивидуально выбранными из циано, атома галогена и галоген-C1-6-алкокси.
Конкретная форма осуществления изобретения относится к соединению, как определено в данной работе, в котором R1 представляет собой 5-хлор-пиридин-2-ил.
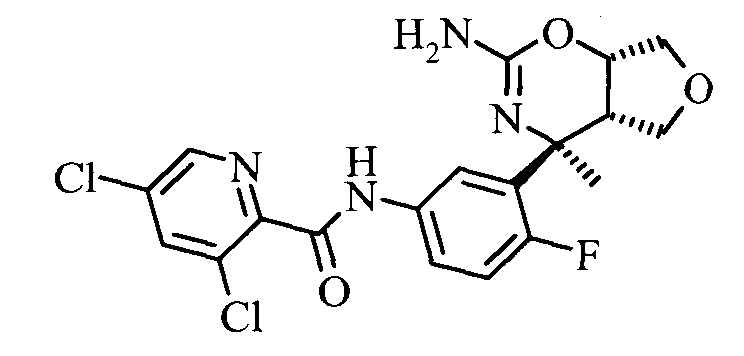
Конкретная форма осуществления изобретения относится к соединению, как определено в данной работе, в котором R1 представляет собой 3,5-дихлор-пиридин-2-ил.
Конкретная форма осуществления изобретения относится к соединению, как определено в данной работе, в котором R1 представляет собой 5-(2,2,2-трифтор-этокси)-пиридин-2-ил.
Конкретная форма осуществления изобретения относится к соединению, как определено в данной работе, в котором R1 представляет собой 5-циано-пиридин-2-ил.
Конкретная форма осуществления изобретения относится к соединению, как определено в данной работе, в котором R1 представляет собой 5-фтор-пиридин-2-ил.
Конкретная форма осуществления изобретения относится к соединению, как определено в данной работе, в котором R1 представляет собой пиразинил, замещенный галоген-C1-6-алкокси или C2-6-алкинил-C1-6-алкокси.
Конкретная форма осуществления изобретения относится к соединению, как определено в данной работе, в котором R1 представляет собой 5-(2,2,2-трифтор-этокси)-пиразин-2-ил.
Конкретная форма осуществления изобретения относится к соединению, как определено в данной работе, в котором R1 представляет собой 5-бут-2-инилокси-пиразин-2-ил.
Конкретная форма осуществления изобретения относится к соединению, как определено в данной работе, в котором R1 представляет собой пиридинил, пиридинил, замещенный 1-2 заместителями, индивидуально выбранными из циано, атома галогена и галоген-C1-6-алкокси, или пиразинил, замещенный галоген-C1-6-алкокси или C2-6-алкинил-C1-6-алкокси.
Конкретная форма осуществления изобретения относится к соединению, как определено в данной работе, в котором R1 представляет собой 5-хлор-пиридин-2-ил, 3,5-дихлор-пиридин-2-ил, 5-(2,2,2-трифтор-этокси)-пиразин-2-ил, 5-(2,2,2-трифтор-этокси)-пиридин-2-ил, 5-бут-2-инилокси-пиразин-2-ил, 5-циано-пиридин-2-ил, 5-фтор-пиридин-2-ил или пиридин-2-ил.
Конкретная форма осуществления изобретения относится к соединению, как определено в данной работе, в котором R1 представляет собой 5-циано-пиридин-2-ил или 5-бут-2-инилокси-пиразин-2-ил.
Конкретная форма осуществления изобретения относится к соединению, как определено в данной работе, в котором R1 представляет собой арил.
Конкретная форма осуществления изобретения относится к соединению, как определено в данной работе, в котором R1 представляет собой арил, замещенный 1-4 заместителями, индивидуально выбранными из циано, циано-C1-6-алкила, атома галогена, галоген-C1-6-алкокси, галоген-C1-6-алкила, C1-6-алкокси, C1-6-алкокси-C1-6-алкила, C2-6-алкинил-C1-6-алкокси, C2-6-алкинила и C1-6-алкила.
Конкретная форма осуществления изобретения относится к соединению, как описано в данной работе, выбранному из группы, состоящей из следующих соединений:
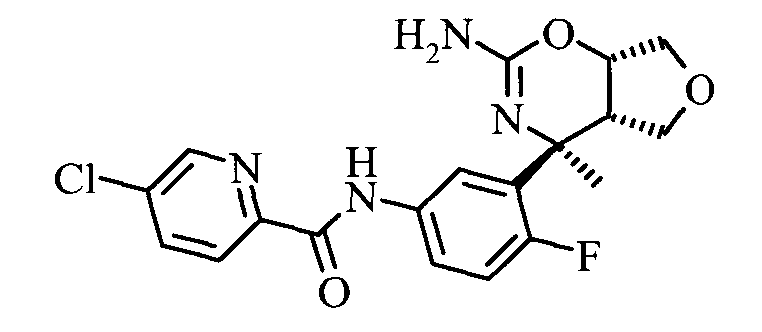
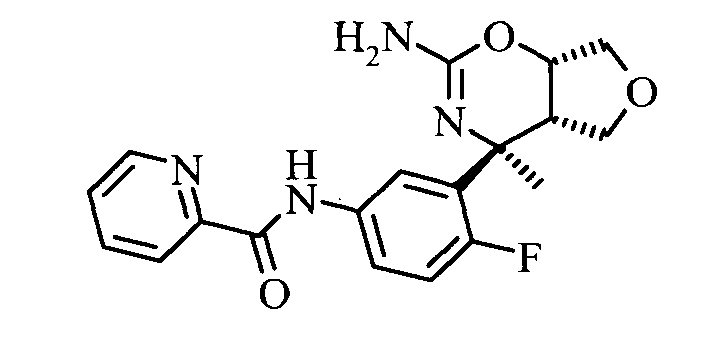
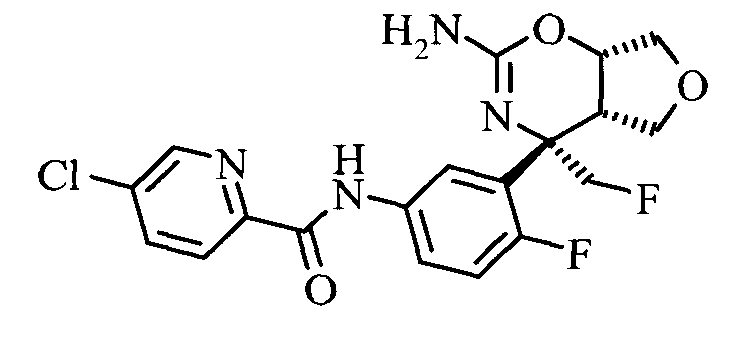
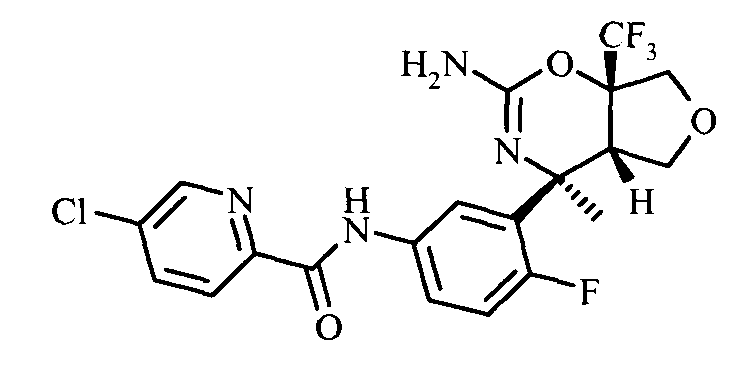
5-Хлор-пиридин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-MeTHn-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амида,
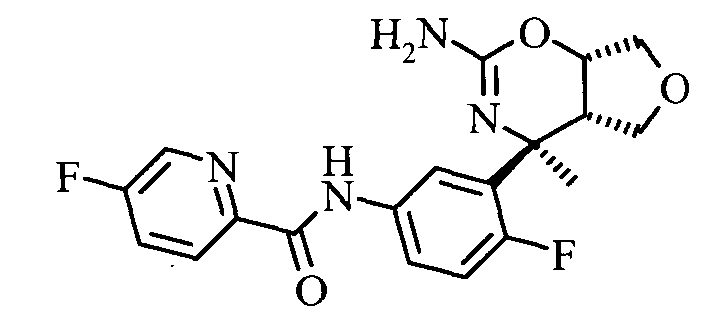
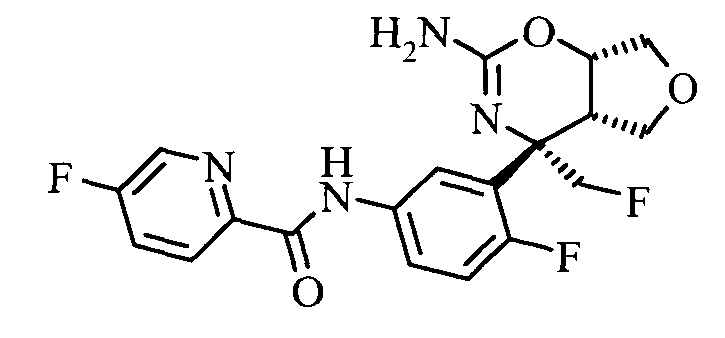
5-Фтор-пиридин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амида,
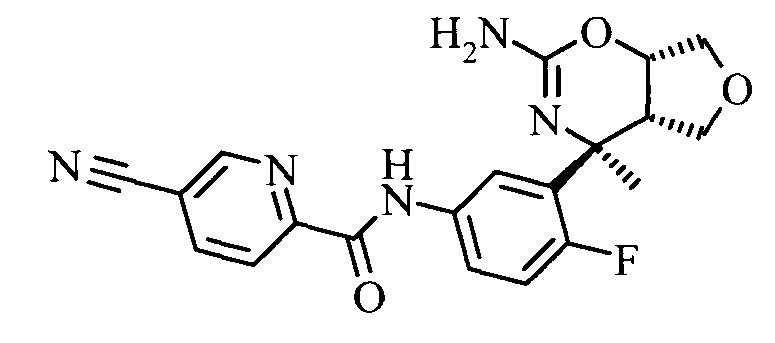
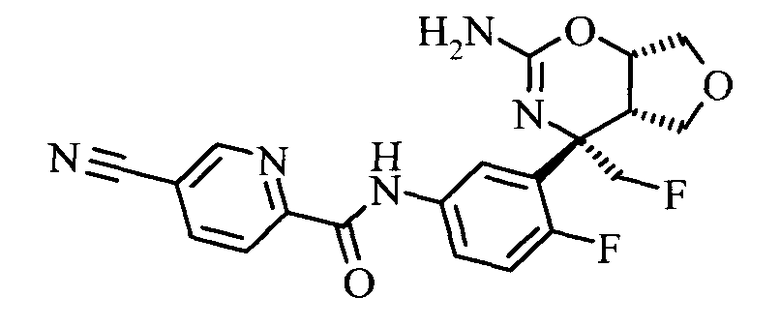
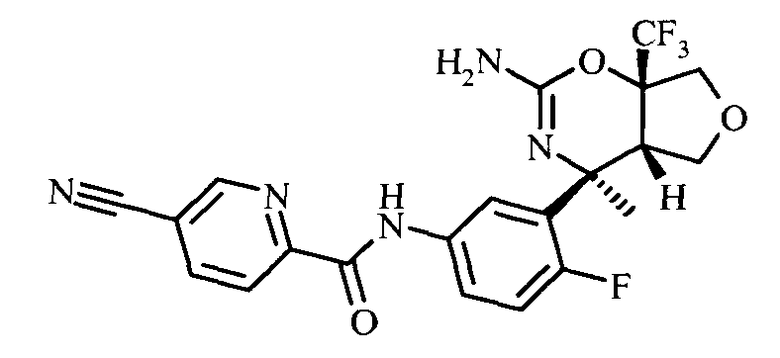
5-Циано-пиридин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амида,
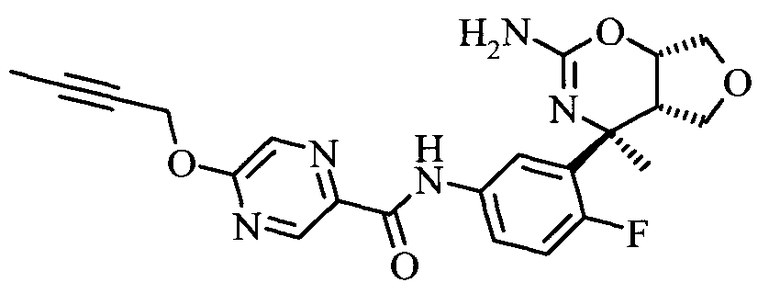
5-Бут-2-инилокси-пиразин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амида,
5-Циклопропилметокси-пиридин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амида,
5-Бут-2-инилокси-пиридин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амида,
5-Циано-пиридин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-фторметил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амида,
5-Хлор-пиридин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-фторметил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амида,
5-Фтор-пиридин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-фторметил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амида,
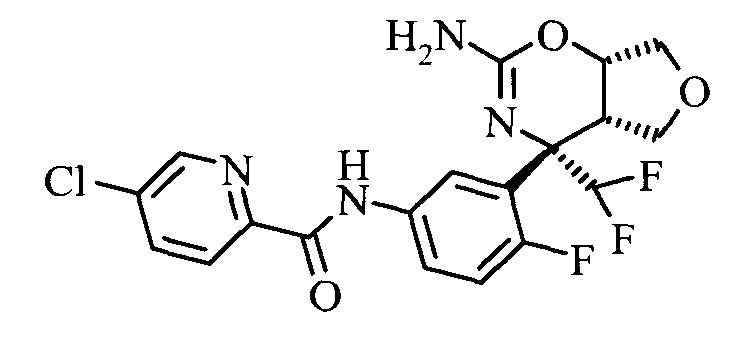
5-Хлор-пиридин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-дифторметил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амида,
5-Циано-пиридин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-дифторметил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амида,
5-Бут-2-инилокси-пиразин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-дифторметил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амида,
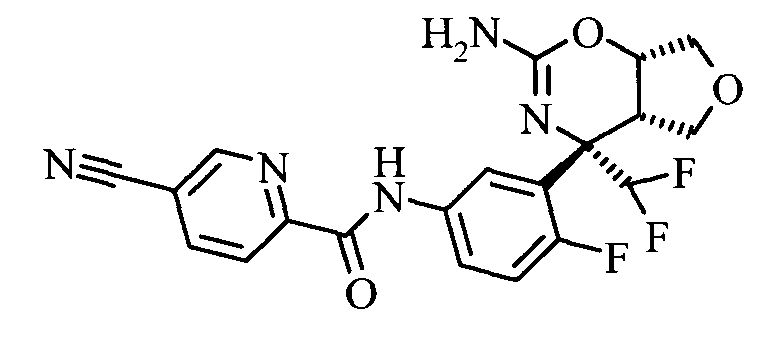
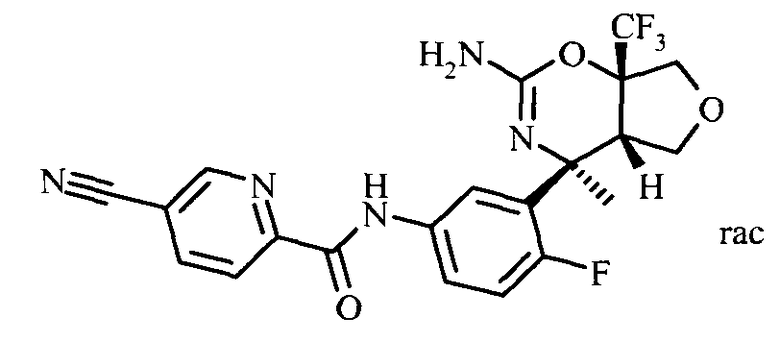
N-(3-(rel-(4SR,4aSR,7aSR)-2-амино-4-метил-7а-(трифторметил)-4а,5,7,7а-тетрагидро-4Н-фуро[3,4-е][1,3]оксазин-4-ил)-4-фторфенил)-5-цианопиколинамида,
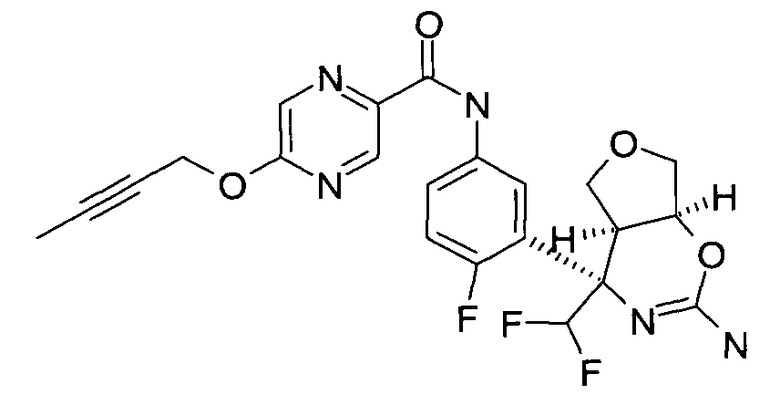
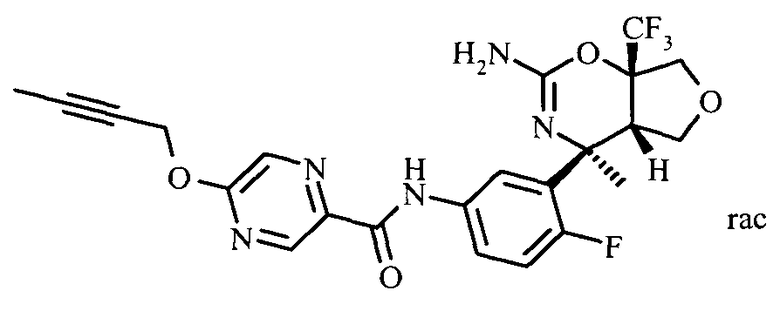
N-(3-(rel-(4SR,4aSR,7aSR)-2-амино-4-метил-7а-(трифторметил)-4а,5,7,7а-тетрагидро-4Н-фуро[3,4-е][1,3]оксазин-4-ил)-4-фторфенил)-5-(бут-2-инилокси)пиразин-2-карбоксамида,
N-(3-((4SR,4aSR,7aSR)-2-амино-4-метил-7а-(трифторметил)-4а,5,7,7а-тетрагидро-4Н-фуро[3,4-е][1,3]оксазин-4-ил)-4-фторфенил)-5-хлорпиколинамида,
N-(3-((4SR,4aSR,7aSR)-2-амино-4-метил-7а-(трифторметил)-4а,5,7,7а-тетрагидро-4Н-фуро[3,4-е][1,3]оксазин-4-ил)-4-фторфенил)-5-цианопиколинамида,
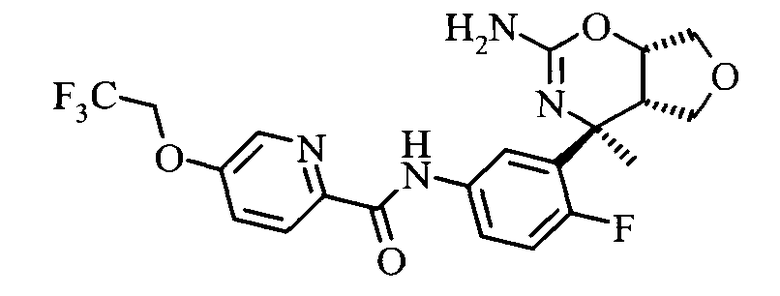
5-(2,2,2-Трифтор-этокси)-пиридин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амида,
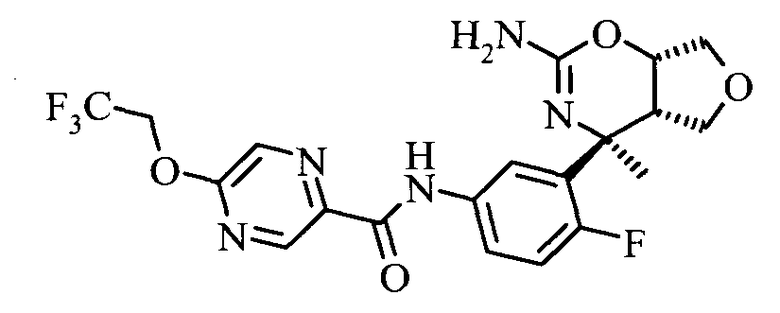
5-(2,2,2-Трифтор-этокси)-пиразин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амида,
5-(2,2,3,3-Тетрафтор-пропокси)-пиридин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амида,
N-(3-((4S,4aS,7aS)-2-амино-4-метил-4а,5,7,7а-тетрагидро-4Н-фуро[3,4-е][1,3]оксазин-4-ил)-4-фторфенил)-3-хлор-5-цианопиколинамида,
Пиридин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амида, и
3,5-Дихлор-пиридин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амида,
или их фармацевтически приемлемой соли.
Конкретная форма осуществления изобретения относится к соединению, как описано в данной работе, выбранному из группы, состоящей из следующих соединений:
5-Хлор-пиридин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амида,
3,5-Дихлор-пиридин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амида,
5-(2,2,2-Трифтор-этокси)-пиразин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амида,
5-(2,2,2-Трифтор-этокси)-пиридин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амида,
5-Бут-2-инилокси-пиразин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амида,
5-Циано-пиридин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амида,
5-Фтор-пиридин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амида, и
Пиридин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амида,
или их фармацевтически приемлемой соли.
Конкретная форма осуществления изобретения относится к способу синтеза соединения, имеющего формулу I, как описано в данной работе, включающему взаимодействие соединения, имеющего формулу XI, с соединением, имеющим формулу XII, с получением соединения, имеющего формулу I:
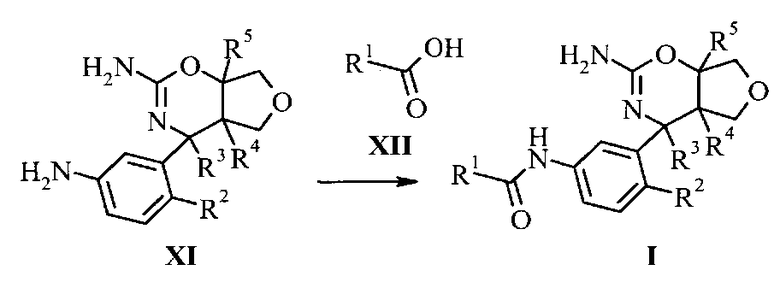
в котором R1, R2, R3, R4, R5 являются такими, как определено в данной работе.
Конкретная форма осуществления изобретения относится к соединению, имеющему формулу I, как описано в данной работе, всегда полученному способом, как определено выше.
Конкретная форма осуществления изобретения относится к соединению, имеющему формулу I, как описано в данной работе, применяемому в качестве терапевтически активного вещества.
Конкретная форма осуществления изобретения относится к соединению, имеющему формулу I, как описано в данной работе, применяемому в качестве ингибитора активности ВАСЕ1 и/или ВАСЕ2.
Конкретная форма осуществления изобретения относится к соединению, имеющему формулу I, как описано в данной работе, применяемому в качестве ингибитора активности ВАСЕ1.
Конкретная форма осуществления изобретения относится к соединению, имеющему формулу I, как описано в данной работе, применяемому в качестве ингибитора активности ВАСЕ2.
Конкретная форма осуществления изобретения относится к соединению, имеющему формулу I, как описано в данной работе, применяемому в качестве ингибитора активности ВАСЕ1 и ВАСЕ2.
Конкретная форма осуществления изобретения относится к соединению, имеющему формулу I, как описано в данной работе, применяемому в качестве терапевтически активного вещества для терапевтического и/или профилактического лечения заболеваний и расстройств, характеризующихся повышенными уровнями β-амилоида и/или олигомеров β-амилоида и/или β-амилоидных бляшек и дополнительных отложений, в частности, болезни Альцгеймера.
Конкретная форма осуществления изобретения относится к соединению, имеющему формулу I, как описано в данной работе, применяемому в качестве терапевтически активного вещества для терапевтического и/или профилактического лечения болезни Альцгеймера.
Конкретная форма осуществления изобретения относится к соединению, имеющему формулу I, как описано в данной работе, применяемому в качестве терапевтически активного вещества для терапевтического и/или профилактического лечения диабета, в частности, диабета типа 2.
Конкретная форма осуществления изобретения относится к соединению, имеющему формулу I, как описано в данной работе, применяемому в качестве терапевтически активного вещества для терапевтического и/или профилактического лечения диабета.
Конкретная форма осуществления изобретения относится к фармацевтической композиции, содержащей соединение, имеющее формулу I, как описано в данной работе, и фармацевтически приемлемый носитель и/или фармацевтически приемлемое вспомогательное вещество.
Конкретная форма осуществления изобретения относится к применению соединения, имеющего формулу I, как описано в данной работе, для получения лекарственного средства, применяемого при ингибировании активности ВАСЕ1 и/или ВАСЕ2.
Конкретная форма осуществления изобретения относится к применению соединения, имеющего формулу I, как описано в данной работе, для получения лекарственного средства, применяемого при ингибировании активности ВАСЕ1.
Конкретная форма осуществления изобретения относится к применению соединения, имеющего формулу I, как описано в данной работе, для получения лекарственного средства, применяемого при ингибировании активности ВАСЕ2.
Конкретная форма осуществления изобретения относится к применению соединения, имеющего формулу I, как описано в данной работе, для получения лекарственного средства, применяемого при ингибировании активности ВАСЕ1 и ВАСЕ2.
Конкретная форма осуществления изобретения относится к применению соединения, имеющего формулу I, как описано в данной работе, для получения лекарственного средства для терапевтического и/или профилактического лечения заболеваний и расстройств, характеризующихся повышенными уровнями β-амилоида и/или олигомеров β-амилоида и/или β-амилоидных бляшек и дополнительных отложений, в частности, болезни Альцгеймера.
Конкретная форма осуществления изобретения относится к применению соединения, имеющего формулу I, как описано в данной работе, для получения лекарственного средства для терапевтического и/или профилактического лечения болезни Альцгеймера.
Конкретная форма осуществления изобретения относится к применению соединения, имеющего формулу I, как описано в данной работе, для получения лекарственного средства для терапевтического и/или профилактического лечения диабета, в частности, диабета типа 2.
Конкретная форма осуществления изобретения относится к применению соединения, имеющего формулу I, как описано в данной работе, для получения лекарственного средства для терапевтического и/или профилактического лечения диабета.
Конкретная форма осуществления изобретения относится к соединению, имеющему формулу I, как описано в данной работе, применяемому при ингибировании активности ВАСЕ1 и/или ВАСЕ2.
Конкретная форма осуществления изобретения относится к соединению, имеющему формулу I, как описано в данной работе, применяемому при ингибировании активности ВАСЕ1.
Конкретная форма осуществления изобретения относится к соединению, имеющему формулу I, как описано в данной работе, применяемому при ингибировании активности ВАСЕ2.
Конкретная форма осуществления изобретения относится к соединению, имеющему формулу I, как описано в данной работе, применяемому при ингибировании активности ВАСЕ1 и ВАСЕ2.
Конкретная форма осуществления изобретения относится к соединению, имеющему формулу I, как описано в данной работе, применяемому при терапевтическом и/или профилактическом лечении заболеваний и расстройств, характеризующихся повышенными уровнями β-амилоида и/или олигомеров β-амилоида и/или β-амилоидных бляшек и дополнительных отложений, в частности, болезни Альцгеймера.
Конкретная форма осуществления изобретения относится к соединению, имеющему формулу I, как описано в данной работе, применяемому при терапевтическом и/или профилактическом лечении болезни Альцгеймера.
Конкретная форма осуществления изобретения относится к соединению, имеющему формулу I, как описано в данной работе, применяемому при терапевтическом и/или профилактическом лечении диабета, в частности, диабета типа 2.
Конкретная форма осуществления изобретения относится к соединению, имеющему формулу I, как описано в данной работе, применяемому при терапевтическом и/или профилактическом лечении диабета.
Конкретная форма осуществления изобретения относится к способу применения при ингибировании активности ВАСЕ1 и/или ВАСЕ2, в частности, для терапевтического и/или профилактического лечения заболеваний и расстройств, характеризующихся повышенными уровнями β-амилоида и/или олигомеров β-амилоида и/или β-амилоидных бляшек и дополнительных отложений, болезни Альцгеймера, диабета или диабета типа 2, включающему введение соединения, имеющего формулу I, как описано в данной работе, человеку или животному.
Конкретная форма осуществления изобретения относится к способу применения при терапевтическом и/или профилактическом лечении болезни Альцгеймера, диабета или диабета типа 2, включающему введение соединения, имеющего формулу I, как описано в данной работе, человеку или животному.
Кроме того, изобретение включает все оптические изомеры, то есть диастереоизомеры, диастереомерные смеси, рацемические смеси, все их соответствующие энантиомеры и/или таутомеры, а также их сольваты, соединений, имеющих формулу I.
Специалисту в данной области техники понятно, что соединения, имеющие формулу I, могут существовать в таутомерных формах, например, в приведенной ниже таутомерной форме.
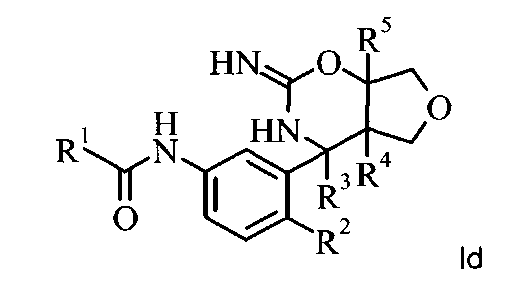
Все таутомерные формы включены в настоящее изобретение.
Соединения, имеющие формулу I, могут содержать один или более чем один асимметрический центр, и, следовательно, могут существовать в виде рацематов, рацемических смесей, отдельных энантиомеров, диастереомерных смесей и индивидуальных диастереомеров. Дополнительные асимметрические центры могут присутствовать в зависимости от природы различных заместителей на молекуле. Каждый такой асимметрический центр независимо образует два оптических изомера, и подразумевают, что все возможные оптические изомеры и диастереомеры в виде смесей и в виде чистых или частично очищенных соединений включены в данное изобретение. Подразумевают, что настоящее изобретение включает все такие изомерные формы этих соединений. Независимые синтезы этих диастереомеров или их хроматографические разделения могут быть достигнуты, как известно в данной области техники, путем соответствующей модификации методологии, описанной в настоящей работе. Их абсолютная стереохимия может быть определена с помощью рентгенокристаллографии кристаллических продуктов или кристаллических промежуточных соединений, которые являются дериватизированными при необходимости реагентом, содержащим асимметрический центр, имеющий известную абсолютную конфигурацию. При желании рацемические смеси соединений могут быть разделены таким образом, чтобы выделить индивидуальные энантиомеры. Это разделение можно осуществить способами, хорошо известными в данной области техники, такими как сочетание рацемической смеси соединений с энантиомерно чистым соединением с образованием диастереомерной смеси с последующим разделением индивидуальных диастереомеров стандартными способами, такими как фракционная кристаллизация или хроматография. Предпочтительными примерами изомеров соединения, имеющего формулу I, являются соединения, имеющие формулу Ib или соединения, имеющие формулу Ic, в которых остатки имеют значение, описанное в любой из форм осуществления.
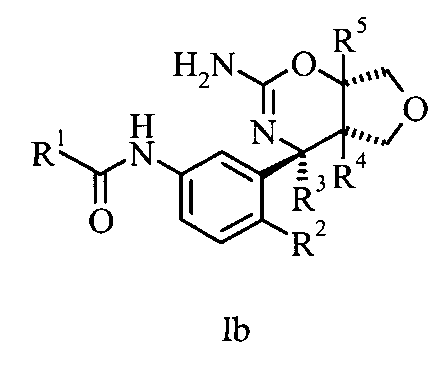
В тех формах осуществления, где предложены оптически чистые энантиомеры, оптически чистый энантиомер означает, что соединение содержит более 90% желаемого изомера по массе, предпочтительно более 95% желаемого изомера по массе или более предпочтительно более 99% желаемого изомера по массе, причем, процент по массе основан на суммарной массе изомера (изомеров) соединения. Хирально чистые или хирально обогащенные соединения могут быть получены путем хирально-селективного синтеза или путем разделения энантиомеров. Разделение энантиомеров можно осуществить на конечном продукте или альтернативно на подходящем промежуточном соединении.
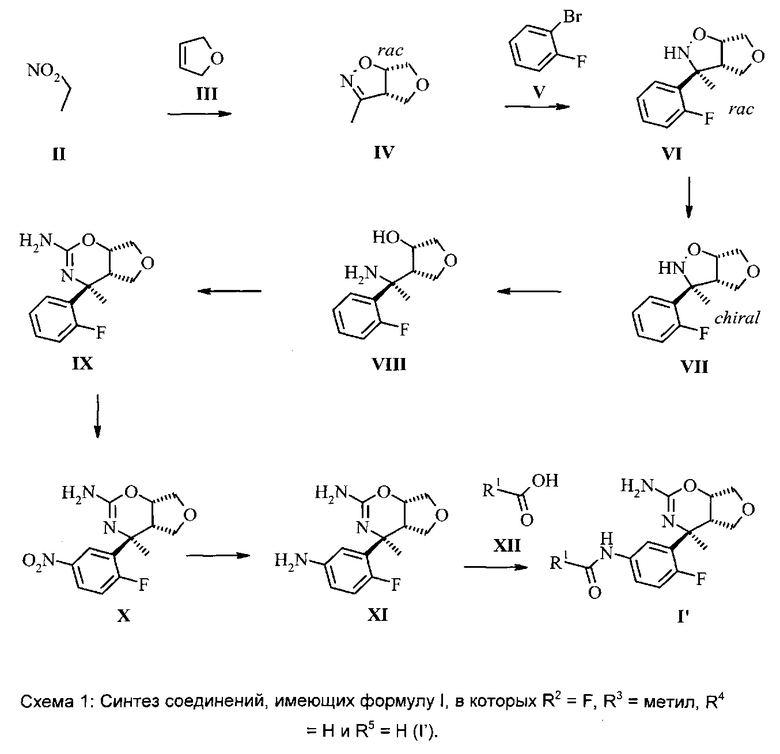
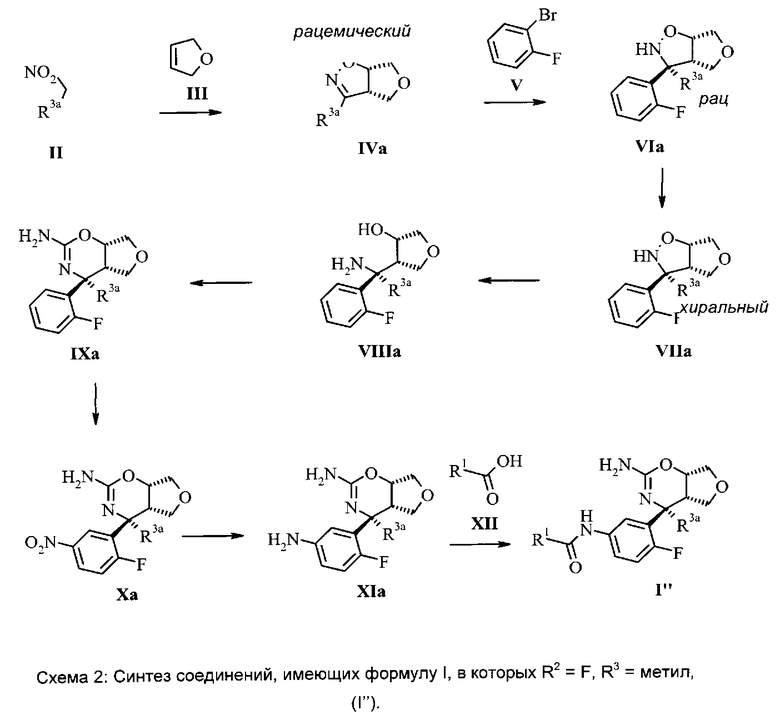
Соединения формулы I могут быть получены в соответствии с приведенной ниже схемой. Исходное вещество имеется в продаже или может быть получено в соответствии с известными способами. Любые определенные выше остатки и переменные продолжают иметь определенное выше значение, если не указано иное.
Нитросоединение (II) подвергают взаимодействию с олефином (III) в присутствии активирующего реагента, такого как, например, изоцианат, в частности, фенилизоцианат, и каталитического количества основания, в частности, алкиламина, более конкретно NEt3, в растворителе, таком как бензол или толуол, в частности, бензол, или алкиловый эфир, в частности, диэтиловый эфир, с получением дигидроизоксазола IV.
Арилирование дигидроизоксазола (IV) арилбромидом (V) с получением изоксазолидина (VI) выполняют путем взаимодействия арилгалогенида, в частности, арилбромида, с алкиллитиевым реагентом, в частности, с n-BuLi (н-бутиллитием), с получением ариллитиевых соединений, которые можно подвергать взаимодействию с дигидроизоксазолом (IV) в присутствии основания Льюиса, предпочтительно эфирата трифторида бора, в смеси растворителей, состоящей из эфира, в частности, ТГФ (тетрагидрофурана), и толуола, при температуре, составляющей от -100°C до -20°C, в частности, -78°C.
Разделение рацемического изоксазолидина (VI) с получением хирального изоксазолидина (VII) может быть выполнено с помощью хиральной высокоэффективной жидкостной хроматографии (ВЭЖХ), используя колонку Chiralpack AD, в смеси н-гептана и этанола.
Гидрогенолиз хирального изоксазолидина (VII) до аминоспирта (VIII) может быть лучше всего выполнен путем гидрогенолиза с переносом водорода, используя Pd-катализатор, в частности, Pd на углероде, и источник водорода, например, соль муравьиной кислоты, в частности, формиат аммония, в протонном растворителе, таком как спирт, в частности, этанол.
Оксазин (IX) может быть получен путем взаимодействия аминоспирта (VIII) с цианогенбромидом в растворителе, таком как спирт, в частности, этанол, при повышенной температуре. Альтернативно это взаимодействие может быть выполнено в последовательности двух стадий, используя цианобромид и буфер, такой как, например, ацетат натрия, в присутствии растворителя, такого как, например, CH3CN, с последующей циклизацией промежуточного соединения в присутствии минеральной кислоты, в частности, соляной кислоты, в растворителе, таком как эфир, в частности, 1,4-диоксан.
Нитрование оксазина (IX) с получением нитрооксазина (X) выполняют стандартным методом, включающим чистую серную кислоту и дымящуюся азотную кислоту без использования растворителя.
Восстановление нитрогруппы в промежуточном соединении (X) с получением анилина (XI) может быть выполнено путем гидрогенизации, используя катализатор, такой как Pd/C, в протонных растворителях, таких как спирты, в частности, этанол или метанол.
Селективное амидное сочетание анилина (XI) и карбоновой кислоты (XII) с получением амида (I) может быть выполнено с использованием гидрата 4-(4,6-диметокси[1.3.5]триазин-2-ил)-4-метилморфолиния хлорида (DMTMM) в растворителе, таком как спирт, в частности, метанол.
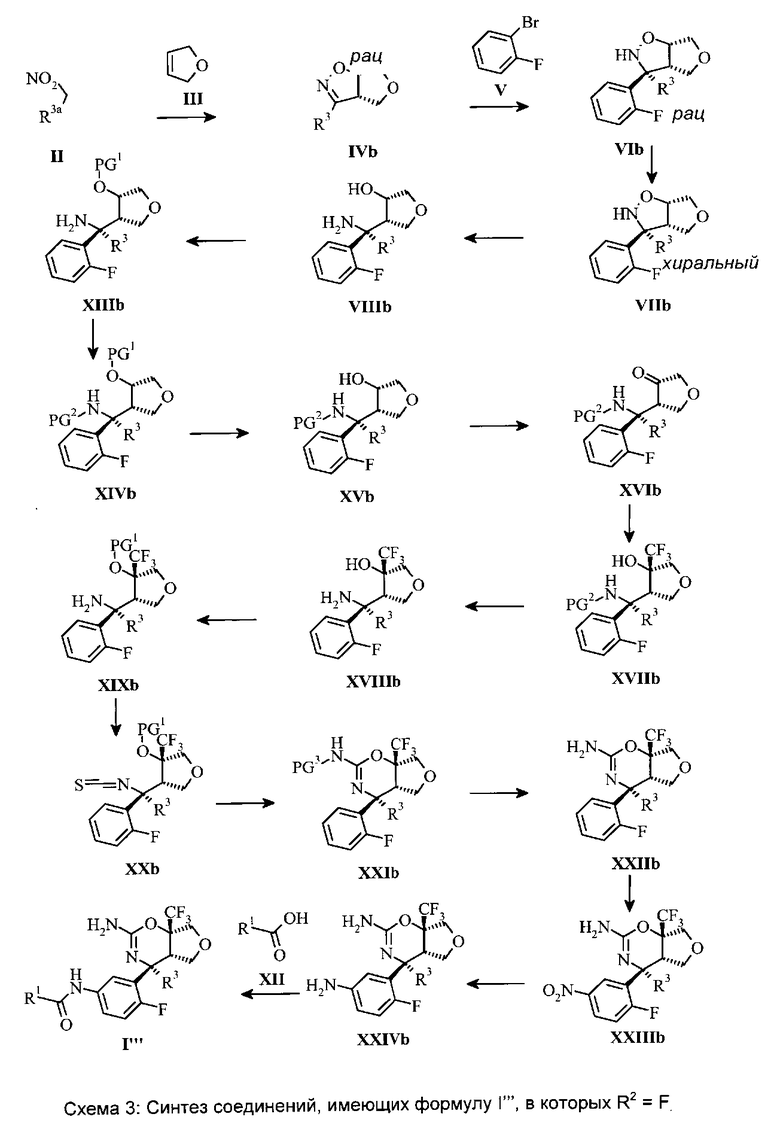
Соединения, имеющие общую формулу I′′′, могут быть получены, как изображено на схеме 3. Уже описанный выше аминоспирт VIIIb может быть селективно защищен на атоме кислорода путем O-силилирования с получением O-силилированного аминоспирта XIIIb хлорсиланом, в частности, трет-бутилхлордиметилсиланом (PG1=t-BuMe2Si), в хлорированном растворителе, таком как дихлорметан, в присутствии основания, представляющего собой триалкиламин, в частности, триэтиламин, и катализатора, представляющего собой пиридин, в частности, 4-диметиламинопиридин, при температуре, составляющей от 0°C до 23°C.
O-силилированный аминоспирт XIIIb может быть ацилирован с получением O-силилированного N-сульфинилированного аминоспирта XIVb сульфинилхлоридом, в частности, трет-бутилсульфинилхлоридом (PG2=t-BuSO), в хлорированном растворителе, в частности, в дихлорметане, в присутствии аминного основания, такого как триэтиламин или диизопропилэтиламин, при температуре, составляющей от 0°C до 60°C, предпочтительно 23°C.
O-силилированный N-сульфинилированный аминоспирт XIVb может быть десилилирован с получением N-сульфинилированного аминоспирта XVb путем взаимодействия с источником фторида, в частности, с тетрабутиламмония фторидом (TBAF; от англ. “tetrabutylammonium fluoride”), в растворителе, таком как ТГФ, при температуре, составляющей от 0°C до 50°C, предпочтительно 23°C.
N-сульфинилированный аминоспирт XVb может быть окислен с получением N-сульфинилированного аминокетона XVIb с помощью комбинации реагентов, такой как оксалилхлорид, диметилсульфоксид и аминное основание, такое как триэтиламин или диизопропилэтиламин, в хлорированном растворителе, в частности, в дихлорметане, при температурах, составляющих от -78°C до температуры окружающей среды.
N-сульфинилированный аминокетон XVIb может быть преобразован в N-сульфинилированный амино-α-трифторметиловый спирт XVIIb путем взаимодействия с трифторметилирующим реагентом, таким как (трифторметил)триметилсилан (реагент Рупперта), в присутствии источника фторида, в частности, тетрабутиламмония фторида (TBAF), в растворителе, таком как ТГФ, при температуре, составляющей от 0°C до 50°C, предпочтительно от 0 до 23°C.
Защитная группа N-сульфинилированного амино-α-трифторметилового спирта XVIIb может быть удалена с получением амино-α-трифторметилового спирта XVIIIb путем взаимодействия с сильной водной минеральной кислотой, в частности, с соляной кислотой, в растворителях, таких как ТГФ, этилацетат, метанол или этанол, при температурах, составляющих от 0 до 23°C.
Амино-α-трифторметиловый спирт XVIIIb может быть селективно защищен на атоме кислорода путем O-силилирования с получением O-силилированного амино-α-трифторметилового спирта XIXb сильным силилирующим реагентом, в частности, трет-бутилдиметилсилилтрифторметансульфонатом (PG1=t-BuMe2Si), в эфирном растворителе, таком как ТГФ, в присутствии сильного основания, в частности, гидрида натрия, при температурах, составляющих от 0 до 23°C.
O-силилированный амино-α-трифторметиловый спирт XIXb может быть преобразован в O-силилированного α-трифторметилового спирта изоцианат XXb путем обработки тиофосгеном или эквивалентным реагентом, таким как 1,1′-тиокарбонилдиимидазол, в присутствии слабого основания, такого как бикарбонат натрия, в хлорированном растворителе, в частности, в дихлорметане, при температурах, составляющих от 0 до 23°C, предпочтительно 23°C.
O-силилированного α-трифторметилового спирта изоцианат XXb может быть преобразован в N-бензилированный оксазин XXIb путем трехступенчатого метода, выполняемого в одном реакционном сосуде без выделения промежуточных соединений, как описано ниже: 1) взаимодействие с амином, в частности, с пара-метоксибензиламином (PG3=РМВ) или 2,4-диметоксибензиламином (PG3=DMB), с получением соответствующей O-силилированной тиомочевины в растворителе, в частности, в ацетонитриле, при температуре, составляющей от 0°C до 100°C, предпочтительно 80°C. 2) O-силилированная тиомочевина может быть десилилирована с получением α-трифторметилового спирта тиомочевины путем взаимодействия с источником фторида, в частности, с тригидратом тетрабутиламмония фторида (TBAF·3H2O), в растворителе, таком как ацетонитрил, при температуре, составляющей от 0°C до 80°C, предпочтительно 23°C. 3) α-трифторметилового спирта тиомочевину можно подвергать циклизации с получением N-бензилированного оксазина XXIb путем обработки карбодиимидом, таким как дициклогексилкарбодиимид, диизопропилкарбодиимид или 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид, в растворителе, таком как ацетонитрил, при температуре, составляющей от 23 до 100°C, предпочтительно 80°C.
N-бензилированный оксазин XXIb дебензилируют с получением оксазина XXIIb с помощью чистого взаимодействия с сильной органической кислотой, в частности, с трифторуксусной кислотой, при температурах, составляющих от 0°C до 50°C, предпочтительно 23°C.
Оксазин XXIIb может быть преобразован через нитрооксазин XXIIIb и анилин XXIVb в соединения, имеющие общую формулу I′′′, как уже описано для оксазинов IX и IXa.
Если их получение не описано в примерах, соединения, имеющие формулу I, а также все промежуточные продукты могут быть получены аналогичными способами или способами, описанными в данной работе. Исходные вещества имеются в продаже, известны в данной области техники или могут быть получены способами, известными в данной области техники, или по аналогии с ними.
Понятно, что соединения, имеющие общую формулу I, по данному изобретению могут быть дериватизированы по функциональным группам с получением производных, способных к обратному преобразованию в исходное соединение in vivo.
Фармакологические тесты
Соединения, имеющие формулу I, и их фармацевтически приемлемые соли обладают ценными фармакологическими свойствами. Обнаружено, что соединения по настоящему изобретению связаны с ингибированием активности ВАСЕ1 и/или ВАСЕ2. Эти соединения были исследованы в соответствии с тестом, приведенным в данной работе ниже.
Клеточный анализ снижения Аβ
Клетки НЕК293 человека, стабильно трансфицированные вектором, экспрессирующим кДНК гена АРР wt (дикого типа) (АРР695), использовали для оценки эффективности соединений в клеточном анализе. Клетки высевали в 96-луночные микротитрационные планшеты в среде для клеточных культур (Iscove, с добавлением 10% (об/об) фетальной бычьей сыворотки, глутамина, пенициллина/стрептомицина) примерно до 80% конфлюентности, и соединения добавляли в 10х концентрации в 1/10 объема среды без ФБС, содержащей 8% ДМСО (конечную концентрацию ДМСО поддерживали при 0,8% об/об). После 18-20 ч инкубации при 37°C и 5% CO2 в увлажненном термостате супернатант культуры собирали для определения концентраций Аβ40. 96-луночные планшеты для ELISA (твердофазного иммуноферментного анализа; от англ. “enzyme linked immunosorbent analysis”) (например, Nunc MaxiSorb) покрывали моноклональным антителом, которое специфично распознает С-конец Аβ40 (Brockhaus et al., NeuroReport 9, 1481-1486; 1998). После блокирования сайтов неспецифического связывания, например, 1% БСА, и отмывки супернатанты культуры добавляли в соответствующих разведениях вместе с идентифицирующим антителом Аβ, сшитым с пероксидазой хрена (например, антителом 4G8, Senetek, Maryand Heights, МО) и инкубировали в течение 5-7 ч. Затем лунки микротитрационного планшета интенсивно промывали физиологическим раствором, забуференным Трисом, содержащим 0,05% Твин 20, и анализ проявляли тетраметилбензидином/H2O2 в лимонно-кислотном буфере. После остановки реакции одним объемом 1 н. H2SO4 реакцию измеряли в считывающем устройстве для планшетов ELISA при длине волны 450 нм. Концентрации Аβ в супернатантах культуры вычисляли на основании стандартной кривой, полученной с известными количествами чистого пептида Аβ.
Анализ ингибирования ВАСЕ путем измерения расщепления клеточного ТМЕМ27
В данном анализе используют принцип ингибирования расщепления ТМЕМ27 человека эндогенным клеточным ВАСЕ2 в крысиной клеточной линии Ins1e и шеддинга с клеточной поверхности в культуральной среде с последующим обнаружением в анализе ELISA. Ингибирование ВАСЕ2 дозозависимо предотвращает это расщепление и шеддинг.
Стабильная клеточная линия “INS-TMEM27” представляет собой клеточную линию, имеющую происхождение от INS1e, с индуцибельной доксициклин-зависимой экспрессией (используя систему TetOn) полноразмерного hTMEM27. На протяжении всего эксперимента клетки культивируют в среде RPMI1640 с добавлением пенициллина/стрептомицина Glutamax (Invitrogen), 10% фетальной бычьей сыворотки, 100 мМ пирувата, 5 мМ бета-меркаптоэтанола, 100 мкг/мл G418 и 100 мкг/мл гигромицина и выращивают в адгезивной культуре при 37°C в стандартном CO2-инкубаторе для клеточных культур.
Клетки INS-TMEM27 высевают в 96-луночные планшеты. После 2 суток в культуре добавляют ингибитор ВАСЕ2 в диапазоне концентраций, который требуется для анализа, и еще через два часа добавляют доксициклин до конечной концентрации 500 нг/мл. Клетки инкубируют еще в течение 46 часов, и супернатант собирают для обнаружения шеддинга ТМЕМ27.
Анализ ELISA (с использованием пары антител мышь против ТМЕМ27 человека, вызванных против внеклеточного домена ТМЕМ27) используют для обнаружения ТМЕМ27 в культуральной среде. ЕС50 для ингибирования ВАСЕ2 вычисляют, используя результат ELISA для каждой концентрации ингибитора с помощью программы соответствия стандартной кривой, такой как XLFit для программы электронных таблиц Excel.
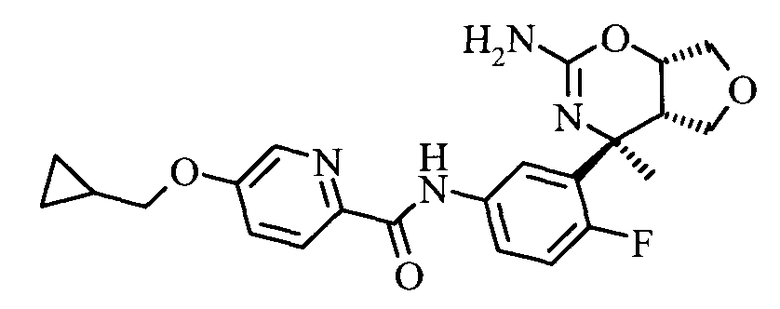
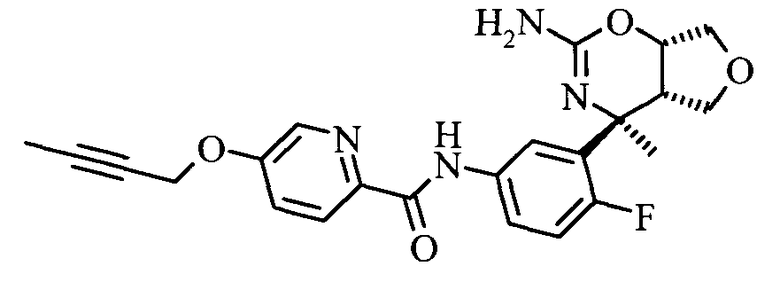
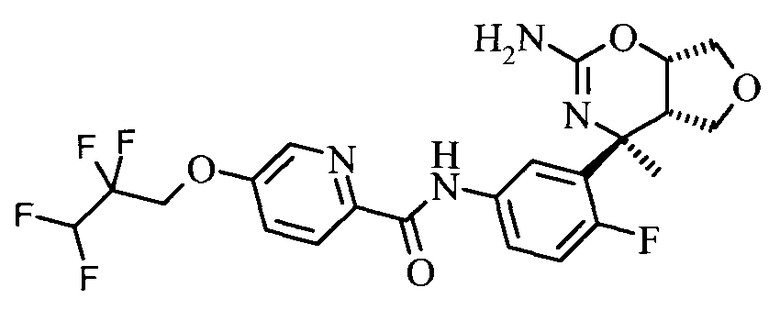
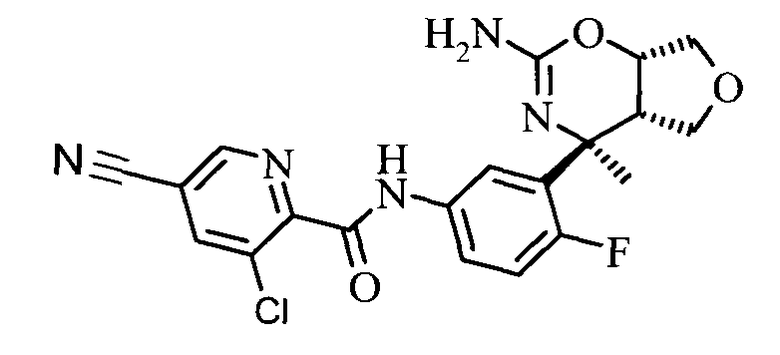
Фармацевтические композиции
Соединения, имеющие формулу I, и их фармацевтически приемлемые соли можно применять в качестве терапевтически активных веществ, например, в форме фармацевтических препаратов. Фармацевтические препараты можно вводить перорально, например, в форме таблеток, таблеток с покрытием, драже, твердых и мягких желатиновых капсул, растворов, эмульсий или суспензий. Введение можно, однако, также осуществлять ректально, например, в форме суппозиториев, или парентерально, например, в форме инъекционных растворов.
Соединения, имеющие формулу I, и их фармацевтически приемлемые соли можно обрабатывать фармацевтически инертными, неорганическими или органическими носителями, предназначенными для получения фармацевтических препаратов. Лактозу, кукурузный крахмал или его производные, тальк, стеариновые кислоты или их соли и тому подобное можно использовать, например, в качестве таких носителей для таблеток, таблеток с покрытием, драже и твердых желатиновых капсул. Подходящими носителями для мягких желатиновых капсул являются, например, растительные масла, воски, жиры, полутвердые и жидкие полиолы и тому подобное. Тем не менее, в зависимости от природы активного вещества в случае мягких желатиновых капсул носители обычно не требуются. Подходящими носителями для получения растворов и сиропов являются, например, вода, полиолы, глицерин, растительное масло и тому подобное. Подходящими носителями для суппозиториев являются, например, натуральные или отвержденные масла, воски, жиры, полужидкие или жидкие полиолы и тому подобное.
Фармацевтические препараты могут, кроме того, содержать фармацевтически приемлемые вспомогательные вещества, такие как консерванты, солюбилизаторы, стабилизаторы, увлажняющие агенты, эмульгаторы, подсластители, красители, корригенты, соли для варьирования осмотического давления, буферы, маскирующие агенты или антиоксиданты. Они могут также содержать другие терапевтически ценные вещества.
Лекарственные средства, содержащие соединение, имеющее формулу I, или его фармацевтически приемлемую соль и терапевтически инертный носитель, также являются объектом настоящего изобретения, как и способ их получения, который включает приведение одного или более соединений формулы I и/или их фармацевтически приемлемых солей и, если желательно, одного или более других терапевтически ценных веществ в форму галенова препарата для введения вместе с одним или более терапевтически инертных носителей.
Дозировка может варьировать в широких пределах и, конечно, ее следует регулировать по индивидуальным потребностям в каждом конкретном случае. В случае перорального введения доза для взрослых может варьировать от приблизительно 0,01 мг до приблизительно 1000 мг в сутки соединения, имеющего общую формулу I, или соответствующего количества его фармацевтически приемлемой соли. Суточную дозу можно вводить в виде однократной дозы или в разделенных дозах и, кроме того, верхний предел может быть также превышен, когда находят, что это показано.
Приведенные ниже примеры иллюстрируют настоящее изобретение без его ограничения, но служат исключительно как репрезентативные. Фармацевтические препараты обычно содержат примерно 1-500 мг, предпочтительно 1-100 мг соединения, имеющего формулу I. Примерами композиций в соответствии с изобретением являются следующие композиции:
Пример А
Таблетки приведенной ниже композиции готовят обычным способом:
Методика получения
1. Смешивают ингредиенты 1, 2, 3 и 4 и гранулируют с дистиллированной водой.
2. Высушивают гранулы при 50°C.
3. Пропускают гранулы через подходящее оборудование для измельчения.
4. Добавляют ингредиент 5 и смешивают в течение трех минут; прессуют на подходящем прессе.
Пример В-1
Готовят мягкие желатиновые капсулы приведенной ниже композиции:
Методика получения
1. Смешивают ингредиенты 1, 2 и 3 в подходящем смесителе в течение 30 минут.
2. Добавляют ингредиенты 4 и 5 и смешивают в течение 3 минут.
3. Заполняют в подходящую капсулу.
Соединение, имеющее формулу I, лактозу и кукурузный крахмал сначала смешивают в смесителе, а затем в измельчающем аппарате. Смесь возвращают в смеситель; добавляют к ней тальк и тщательно смешивают. Смесь заполняют с помощью аппарата в подходящие капсулы, например, в твердые желатиновые капсулы.
Пример В-2
Готовят капсулы приведенной ниже композиции:
Возможная композиция ингредиентов мягкой желатиновой капсулы
Методика получения
Соединение, имеющее формулу I, растворяют в теплом расплаве других ингредиентов, и смесь заполняют в мягкие желатиновые капсулы подходящего размера. Заполненные мягкие желатиновые капсулы обрабатывают в соответствии с обычными методами.
Пример С
Готовят суппозитории приведенной ниже композиции:
Методика получения
Суппозиторную массу плавят в стеклянном или стальном сосуде, тщательно смешивают и охлаждают до 45°C. После этого к ней добавляют тонкоизмельченное соединение, имеющее формулу I, и перемешивают до тех пор, пока оно полностью не диспергируется. Смесь заливают в формы для суппозиториев подходящего размера, оставляют до охлаждения; затем суппозитории извлекают из форм и упаковывают индивидуально в вощеную бумагу или металлическую фольгу.
Пример D
Готовят инъекционные растворы приведенной ниже композиции:
Методика получения
Соединение, имеющее формулу I, растворяют в смеси полиэтиленгликоля 400 и воды для инъекций (части). Доводят pH до 5,0 уксусной кислотой. Объем доводят до 1,0 мл добавлением остального количества воды. Раствор фильтруют, заполняют во флаконы, используя подходящий допустимый избыток, и стерилизуют.
Пример Е
Готовят пакеты-саше приведенной ниже композиции:
Методика получения
Соединение, имеющее формулу I, смешивают с лактозой, микрокристаллической целлюлозой и натриевой солью карбоксиметилцеллюлозы и гранулируют со смесью поливинилпирролидона в воде. Гранулят смешивают со стеаратом магния, добавляют добавки корригентов и заполняют в пакеты-саше.
Экспериментальный раздел
Приведенные ниже примеры приведены для иллюстрации изобретения. Их не следует рассматривать как ограничивающие объем изобретения, но исключительно как репрезентативные.
Общий метод А: Синтез промежуточного дигидроизоксазола IV
К перемешанному раствору нитросоединения II (72,8 ммоль) и олефина III (71,3 ммоль) в бензоле (105 мл) добавляли триэтиламин (NEt3) (10 капель) с последующим добавлением раствора фенилизоцианата (146 ммоль) в бензоле (15 мл), и перемешивание продолжали при 22°C в течение 1 ч, и при температуре образования флегмы в течение 1 ч. Альтернативно диэтиловый эфир можно использовать в качестве растворителя, и реакционную смесь перемешивали при 22°C в течение 3 суток. Суспензию фильтровали, и фильтрат подвергали хроматографии на силикагеле, используя смесь циклогексана и этилацетата (AcOEt), с получением чистого дигидрооксазола IV.
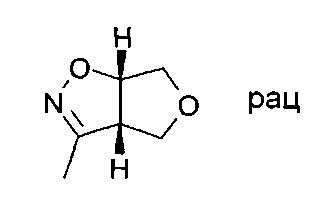
Промежуточное соединение IV-1: Начиная с нитроэтана и 2,5-дигидро-фурана, продукт (3aS,6aS)-rel-3-метил-3а,4,6,6а-тетрагидро-фуро[3,4-d]изоксазол был получен в виде бледно-желтого твердого вещества.
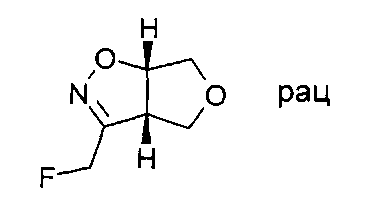
Промежуточное соединение IV-2: Раствор (3aS,6aS)-rel-1-(3а,4,6,6а-тетрагидро-фуро[3,4-d]изоксазол-3-ил)-метанола (14,0 ммоль) в дихлорметане (40 мл) обрабатывали добавлением по каплям раствора трифторида (диэтиламино)серы (15,4 ммоль) в дихлорметане (5 мл) при -70°C. Бесцветный мутный раствор перемешивали при -70°C в течение 30 минут, а затем давали подогреться до комнатной температуры, при этом его цвет становился коричневым. После перемешивания в течение одного часа темно-коричневый раствор охлаждали в ледяной бане и гасили насыщенным раствором гидрокарбоната натрия (50 мл). Водный слой отделяли и дважды экстрагировали дихлорметаном. Объединенные органические слои высушивали над сульфатом натрия, затем выпаривали. Сырой продукт очищали хроматографией на силикагеле, используя градиент гептана и этилацетата = 100:0-80:20 в качестве элюента. (3aS,6aS)-rel-3-Фторметил-3а,4,6,6а-тетрагидро-фуро[3,4-d]изоксазол (814 мг, выход 40%) был получен в виде светло-желтого твердого вещества. МС (масс-спектрометрия): m/z=146,2 [М+Н]+.
(3aS,6aS)-rel-1-(3а,4,6,6а-Тетрагидро-фуро[3,4-d]изоксазол-3-ил)-метанол был получен, как описано ниже:
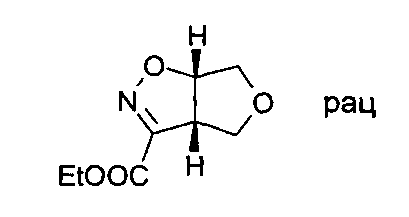
Начиная с имеющегося в продаже (Z)-этил-2-хлор-2-(гидроксиимино)ацетатнитроэтана и 2,5-дигидро-фурана, следуя общему методу А, продукт, представляющий собой (3aS,6aS)-rel-3а,4,6,6а-тетрагидро-фуро[3,4-d]изоксазол-3-карбоновой кислоты этиловый эфир, был получен в виде желтой жидкости.
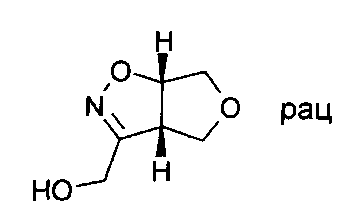
Раствор (3aS,6aS)-rel-3а,4,6,6а-тетрагидро-фуро[3,4-d]изоксазол-3-карбоновой кислоты этилового эфира (18,9 ммоль) (промежуточного соединения IV-2) в этаноле (60 мл) охлаждали до 5°C. Добавляли порциями боргидрид натрия (37,8 ммоль) за период, составляющий 15 минут. Во время выделения газа температуру поддерживали между 5 и 10°C. Затем реакционную смесь перемешивали при температуре, составляющей от 25 до 28°C, в течение 1 часа. Для обработки реакционную смесь охлаждали в ледяной бане и добавляли по каплям 3М соляную кислоту (12 мл). Смеси давали подогреться до комнатной температуры, а затем обрабатывали раствором карбоната натрия (2М; 10 мл), суспензию концентрировали при пониженном давлении, полученное в результате твердое вещество перемешивали в дихлорметане, затем фильтровали, и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали хроматографией на силикагеле, используя градиент гептана и этилацетата = 100:0-0:100 в качестве элюента. (3aS,6aS)-rel-1-(3а,4,6,6а-Тетрагидро-фуро[3,4-d]изоксазол-3-ил)-метанол (2,06 г, выход 76%) был получен в виде светло-желтого масла. МС: m/z=144,0 [М+Н]+.
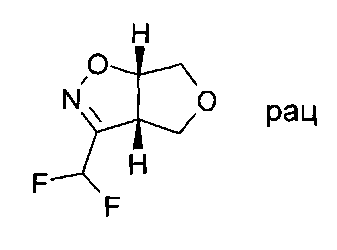
Промежуточное соединение IV-3: Раствор (3aS,6aS)-rel-3a,4,6,6a-тетрагидро-фуро[3,4-d]изоксазол-3-карбальдегида (35,4 ммоль) в дихлорметане (20 мл) обрабатывали добавлением по каплям трифторида (диэтиламино)серы (42,5 ммоль) при -2°C. Реакционную смесь перемешивали при 0°C в течение 2 часов. Для обработки реакционную смесь осторожно гасили добавлением по каплям насыщенного раствора гидрокарбоната натрия (25 мл), затем добавляли раствор карбоната натрия (10%) для доведения до щелочного pH. Смесь экстрагировали три раза дихлорметаном, органические слои объединяли, высушивали над сульфатом натрия и выпаривали. Для очистки сырой продукт перегоняли при пониженном давлении. (3aS,6aS)-rel-3-Дифторметил-3а,4,6,6а-тетрагидро-фуро[3,4-d]изоксазол (2,9 г, выход 50%) был получен в виде светло-желтого масла.
(3aS,6aS)-rel-3а,4,6,6а-Тетрагидро-фуро[3,4-d]изоксазол-3-карбальдегид был получен, как описано ниже:
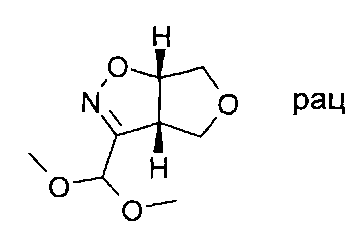
Начиная с имеющегося в продаже 1,1-диметокси-2-нитроэтана (CAS 69425-53-2) и 2,5-дигидро-фурана, следуя общему методу А, продукт, представляющий собой (3aS,6aS)-rel-3-диметоксиметил-3а,4,6,6а-тетрагидро-фуро[3,4-d]изоксазол, был получен в виде светло-коричневого масла.
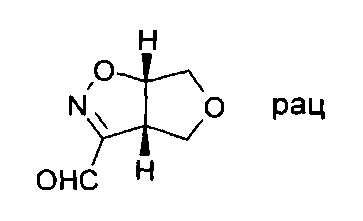
Раствор (3aS,6aS)-rel-3-диметоксиметил-3а,4,6,6а-тетрагидро-фуро[3,4-d]изоксазола (35,3 ммоль) в трифторуксусной кислоте (30 мл) обрабатывали водой (1,3 мл). Смесь перемешивали при комнатной температуре в течение 30 минут. Для обработки реакционную смесь разбавляли водой (100 мл) и экстрагировали три раза дихлорметаном. Объединенные органические слои высушивали над сульфатом натрия и выпаривали. Сырой продукт очищали хроматографией на силикагеле, используя смесь 5:1 дихлорметана и гептана в качестве элюента. (3aS,6aS)-rel-3а,4,6,6а-Тетрагидро-фуро[3,4-d]изоксазол-3-карбальдегид (4,79 г, выход 96%) был получен в виде желтого масла. МС: m/z=144,0 [М+Н]+.
Общий метод В: Синтез промежуточного изоксазолидина VI и VII
К перемешанному раствору арилбромида V (8,26 ммоль) в ТГФ (5 мл) и толуоле (15 мл) при -78°C добавляли n-BuLi (1,6 М в гексане, 4,9 мл) в течение 10 мин, и перемешивание продолжали при -78°C в течение 1 ч.
К раствору дигидроизоксазола IV (3,9 ммоль) в толуоле (35 мл) при -78°C добавляли эфират трифторида бора (BF3.Et2O) (7,9 ммоль) с последующим добавлением фениллитиевого реагента, полученного выше, с использованием теплоизоляционной канюли в течение 10 мин, поддерживая температуру ниже -70°C. Смесь перемешивали при -78°C в течение 1 ч, гасили насыщенным водным раствором хлорида аммония (NH4Cl) и экстрагировали AcOEt. Органический слой промывали рассолом, высушивали, выпаривали, и остаток подвергали хроматографии на силикагеле, используя смесь циклогексана и AcOEt, с получением чистого изоксазолидина VI.
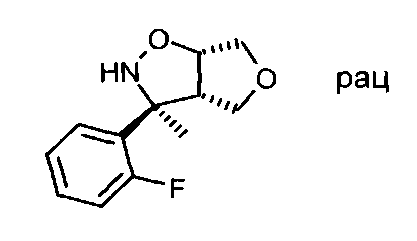
Промежуточное соединение VI-1: Начиная с (3aS,6aS)-rel-3-метил-3a,4,6,6а-тетрагидро-фуро[3,4-d]изоксазола, продукт, представляющий собой (3S,3aS,6aS)-rel-3-(2-фторфенил)-3-метилгексагидрофуро[3,4-d]изоксазол, был получен в виде беловатого твердого вещества. МС: m/z=224,2 [М+Н]+.
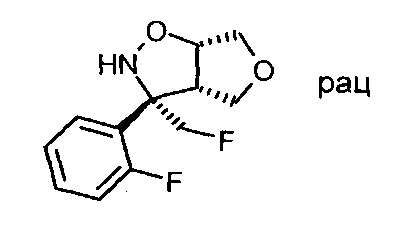
Промежуточное соединение VI-2: Начиная с (3aS,6aS)-rel-3-фторметил-3а,4,6,6а-тетрагидро-фуро[3,4-d]изоксазола и 1-бром-2-фторбензола, продукт, представляющий собой (3S,3aS,6aS)-rel-3-фторметил-3-(2-фтор-фенил)-гексагидро-фуро[3,4-d]изоксазол, был получен в виде светло-желтого масла. МС: m/z=242,1 [М+Н]+.
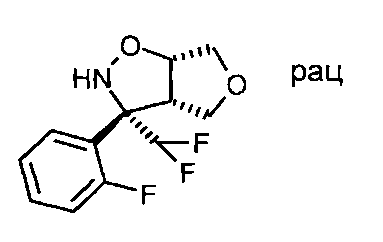
Промежуточное соединение Vl-3: Начиная с (3aS,6aS)-rel-3-дифторметил-3а,4,6,6а-тетрагидро-фуро[3,4-d]изоксазола и 1-бром-2-фторбензола, продукт, представляющий собой (3S,3aS,6aS)-rel-3-дифторметил-3-(2-фторфенил)-гексагидро-фуро[3,4-d]изоксазол, был получен в виде оранжевого масла. МС: m/z=260,2 [М+Н]+.
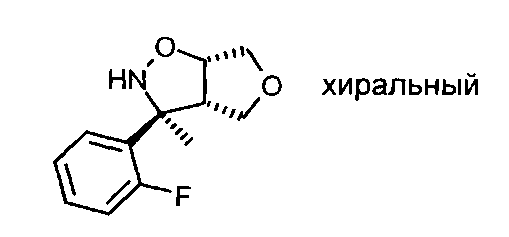
Промежуточное соединение VII-1: Рацемат (3S,3aS,6aS)-3-(2-фторфенил)-3-метилгексагидрофуро[3,4-d]изоксазола разделяли на хиральной колонке для высокоэффективной жидкостной хроматографии (ВЭЖХ) (Chiralpack AD), используя н-гептан/этанол (85:15), с получением желаемого (3S,3aS,6aS)-3-(2-фторфенил)-3-метилгексагидрофуро[3,4-d]изоксазола в виде энантиомера, элюирующего быстрее, и (3R,3aR,6aR)-3-(2-фтор-фенил)-3-метил-гексагидро-фуро[3,4-d]изоксазола в виде энантиомера, элюирующего медленнее.
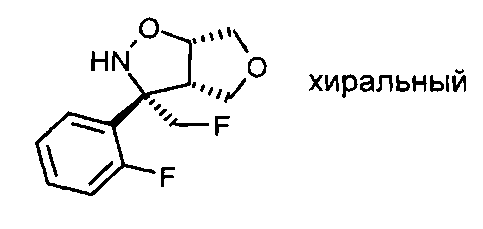
Промежуточное соединение VII-2: Рацемат (3S,3aS,6aS)-3-фторметил-3-(2-фторфенил)-гексагидро-фуро[3,4-d]изоксазола разделяли на хиральной колонке для высокоэффективной жидкостной хроматографии (ВЭЖХ) (Chiralpack AD), используя н-гептан/этанол (85:15), с получением желаемого (3S,3aS,6aS)-3-фторметил-3-(2-фтор-фенил)-гексагидро-фуро[3,4-d]изоксазола в виде энантиомера, элюирующего быстрее, и (3R,3aR,6aR-3-фторметил-3-(2-фтор-фенил)-гексагидро-фуро[3,4-d]изоксазола в виде энантиомера, элюирующего медленнее.
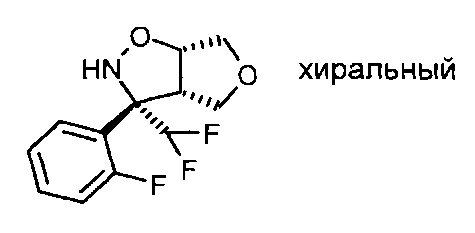
Промежуточное соединение VII-3: Рацемат (3S,3aS,6aS)-3-дифторметил-3-(2-фторфенил)-гексагидро-фуро[3,4-d]изоксазола разделяли на хиральной колонке для высокоэффективной жидкостной хроматографии (ВЭЖХ) (Chiralpack AD), используя н-гептан/изопропанол, с получением желаемого (3S,3aS,6aS)-3-дифторметил-3-(2-фтор-фенил)-гексагидро-фуро[3,4-d]изоксазола в виде энантиомера, элюирующего вторым, и (3R,3aR,6aR-3-дифторметил-3-(2-фтор-фенил)-гексагидро-фуро[3,4-d]изоксазола в виде энантиомера, элюирующего первым.
Общий метод С: Синтез промежуточного аминоспирта VIII
К раствору изоксазолидина VII (6,4 ммоль) в EtOH (40 мл) добавляли Pd/C (10%, 288 мг) и формиат аммония (3,2 г), и перемешивание смеси продолжали при 22°C в течение 3 ч. Суспензию фильтровали, фильтрат выпаривали, и остаток распределяли между AcOEt и насыщенным водным раствором гидрокарбоната натрия (NaHCO3). Органический слой высушивали, выпаривали, и остаток подвергали хроматографии на колонке Si-NH2, используя смесь циклогексана и AcOEt, с получением чистого аминоспирта VIII.
Промежуточное соединение VIII-1: Начиная с (3S,3aS,6aS)-3-(2-фторфенил)-3-метилгексагидрофуро[3,4-d]изоксазола, продукт, представляющий собой (3S,4S)-4-[(S)-1-амино-1-(2-фтор-фенил)-этил]-тетрагидро-фуран-3-ол, был получен в виде бесцветного масла. МС: m/z=226,2 [М+Н]+.
Промежуточное соединение VIII-2: Начиная с (3S,3aS,6aS)-3-фторметил-3-(2-фтор-фенил)-гексагидро-фуро[3,4-d]изоксазола, продукт, представляющий собой (3S,4S)-4-[(S)-1-амино-2-фтор-1-(2-фтор-фенил)-этил]-тетрагидро-фуран-3-ол, был получен в виде белого твердого вещества. МС: m/z=244,2 [М+Н]+.
Промежуточное соединение VIII-3: Начиная с (3S,3aS,6aS)-3-дифторметил-3-(2-фтор-фенил)-гексагидро-фуро[3,4-d]изоксазола, продукт, представляющий собой (3S,4S)-4-[(S)-1-амино-2,2-дифтор-1-(2-фтор-фенил)-этил]-тетрагидро-фуран-3-ол, был получен в виде бесцветного твердого вещества. МС: m/z=262,2 [М+Н]+.
Общий метод D: Синтез промежуточного оксазина IX
К раствору аминоспирта VIII (5,9 ммоль) в ТГФ (130 мл) последовательно добавляли ацетат натрия (6,8 ммоль) и раствор цианобромида (Br-CN) (5 М в ацетонитриле (CH3CN), 6,8 ммоль), и смесь перемешивали при температуре образования флегмы в течение 16 ч. Смесь разбавляли соляной кислотой (HCl) в 1,4-диоксане (4 М, 7,1 мл), и перемешивание продолжали при 22°C в течение 1 ч. Смесь распределяли между AcOEt и насыщенным водным раствором карбоната аммония (Na2CO3), органический слой высушивали, выпаривали, и остаток подвергали хроматографии на силикагеле, используя смесь AcOEt и метанола (МеОН) (9:1), с получением чистого оксазина IX.
Промежуточное соединение IX-1: Начиная с (3S,4S)-4-[(S)-1-амино-1-(2-фтор-фенил)-этил]-тетрагидро-фуран-3-ола, продукт, представляющий собой (3aS,7S,7aS)-7-(2-фтор-фенил)-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-5-иламин, был получен в виде бледно-желтого аморфного твердого вещества. МС: m/z=251,1 [М+Н]+.
Промежуточное соединение IX-2: Начиная с (3S,4S)-4-[(S)-1-амино-2-фтор-1-(2-фтор-фенил)-этил]-тетрагидро-фуран-3-ола, продукт, представляющий собой (3aS,7S,7aS)-7-фторметил-7-(2-фтор-фенил)-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-5-иламин, был получен в виде белой пены. МС: m/z=269,1 [М+Н]+.
Промежуточное соединение IX-3: Начиная с (3S,4S)-4-[(S)-1-амино-2,2-дифтор-1-(2-фтор-фенил)-этил]-тетрагидро-фуран-3-ола, продукт, представляющий собой (3aS,7S,7aS)-7-дифторметил-7-(2-фтор-фенил)-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-5-иламин, был получен в виде бесцветного твердого вещества. МС: m/z=287,2 [М+Н]+.
Общий метод Е: Синтез промежуточного нитро-оксазина X
К концентрированной серной кислоте (13 мл) добавляли порциями оксазине IX (3,0 ммоль) при 22°C, полученный раствор охлаждали до 0°C и обрабатывали красной дымящейся азотной кислотой (HNO3) (0,19 мл) в течение 20 мин, и перемешивание продолжали при 0°C в течение 1 ч. Реакционную смесь медленно добавляли к колотому льду (60 г), pH доводили до 10, используя гидроксид натрия (NaOH), водный слой экстрагировали AcOEt, органический слой высушивали, выпаривали, и остаток подвергали хроматографии на силикагеле, используя смесь AcOEt/MeOH (9:1), с получением чистого нитро-оксазина X.
Промежуточное соединение Х-1: Начиная с (3aS,7S,7aS)-7-(2-фтор-фенил)-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-5-иламина, продукт, представляющий собой (3aS,7S,7aS)-7-(2-фтор-5-нитро-фенил)-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-5-иламин, был получен в виде бледно-желтого аморфного твердого вещества. МС: m/z=296,2 [М+Н]+.
Промежуточное соединение Х-2: Начиная с (3aS,7S,7aS)-7-фторметил-7-(2-фтор-фенил)-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-5-иламина, продукт, представляющий собой (3aS,7S,7aS)-7-фторметил-7-(2-фтор-5-нитро-фенил)-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-5-иламин, был получен в виде белого твердого вещества. МС: m/z=313,9 [М+Н]+.
Промежуточное соединение Х-3: Начиная с (3aS,7S,7aS)-7-дифторметил-7-(2-фтор-фенил)-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-5-иламина, продукт, представляющий собой (3aS,7S,7aS)-7-дифторметил-7-(2-фтор-5-нитро-фенил)-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-5-иламин, был получен в виде светло-желтого твердого вещества. МС: m/z=332,2 [М+Н]+.
Общий метод F: Синтез промежуточного анилина XI
Суспензию нитро-оксазина X (2,6 ммоль) в EtOH (40 мл) и NEt3 (0,2 мл) обрабатывали Pd/C (10%, 80 мг), и смесь подвергали гидрогенизации при атмосферном давлении и 22°C в течение 2 ч. Смесь фильтровали, фильтрат выпаривали, и остаток подвергали хроматографии на колонке Si-NH2, используя смесь AcOEt/MeOH (9:1), с получением чистого анилина XI.
Промежуточное соединение XI-1: Начиная с (3aS,7S,7aS)-7-(2-фтор-фенил)-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-5-иламина, (3aS,7S,7aS)-7-(5-амино-2-фтор-фенил)-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-5-иламин был получен в виде белого аморфного твердого вещества. МС: m/z=266,1 [М+Н]+.
Промежуточное соединение XI-2: Начиная с (3aS,7S,7aS)-7-фторметил-7-(2-фтор-5-нитро-фенил)-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-5-иламина, (3aS,7S,7aS)-7-фторметил-7-(5-амино-2-фтор-5-фенил)-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-5-иламин был получен в виде белой пены. МС: m/z=284,1 [М+Н]+.
Промежуточное соединение XI-3: Начиная с (3aS,7S,7aS)-7-дифторметил-7-(2-фтор-5-нитро-фенил)-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-5-иламина, (3aS,7S,7aS)-7-дифторметил-7-(5-амино-2-фтор-5-фенил)-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-5-иламин был получен в виде бесцветной пены. МС: m/z=302,3 [М+Н]+.
Общий метод G: Синтез промежуточного O-защищенного аминоспирта XIIIb
Раствор аминоспирта VIIIb (13,9 ммоль) в дихлорметане (91 мл) обрабатывали при 0°C триэтиламином (4,48 г, 44,3 ммоль) и 4-(диметиламино)-пиридином (846 мг, 6,93 ммоль), а затем трет-бутилдиметилхлорсиланом (4,18 г, 27,7 ммоль). Реакционную смесь оставляли подогреться до комнатной температуры и перемешивали в течение ночи. В результате разбавления дихлорметаном и экстракции реакционной смеси насыщенным раствором гидрокарбоната натрия NaHCO3, отделения органического слоя, высушивания над сульфатом натрия и выпаривания получили сырой продукт, который очищали хроматографией на силикагеле, используя градиент гептана и этилацетата = 100:0-0:100 в качестве элюента.
Промежуточное соединение XIIIb-1: Начиная с rel-(3S,4S)-4-((S)-1-амино-1-(2-фторфенил)этил)тетрагидрофуран-3-ол), rel-(S)-1-((3S,4S)-4-(трет-бутилдиметилсилилокси)тетрагидрофуран-3-ил)-1-(2-фторфенил)этанамин был получен в виде бесцветного масла.
Промежуточное соединение XIIIb-2: Начиная с (3S,4S)-4-((S)-1-амино-1-(2-фторфенил)этил)тетрагидрофуран-3-ол), (S)-1-((3S,4S)-4-(трет-бутилдиметилсилилокси)тетрагидрофуран-3-ил)-1-(2-фторфенил)этанамин был получен в виде бесцветного масла. МС: m/z=340,3 [М+Н]+.
Общий метод Н: Синтез промежуточного дважды защищенного аминоспирта XIVb
Раствор защищенного аминоспирта XIIIb (19,1 ммоль) и триэтиламина (2,9 г, 28,6 ммоль) в дихлорметане (34 мл) обрабатывали добавлением по каплям при 0°C трет-бутилсульфинилхлорида (2,95 г, 21,0 ммоль). Реакционную смесь перемешивали при 0°C в течение 3 часов. С целью завершения реакции снова добавляли трет-бутилсульфинилхлорид (0,5 мл, 4 ммоль), и реакционную смесь перемешивали в течение 20 минут при 0°C. Для обработки реакционную смесь разбавляли дихлорметаном и экстрагировали насыщенным раствором гидрокарбоната натрия NaHCO3. Органический слой отделяли и высушивали над сульфатом натрия. В результате выпаривания получили сырой продукт, который очищали хроматографией на силикагеле, используя градиент гептана и этилацетата=100:0-0:100 в качестве элюента.
Промежуточное соединение XIVb-1: Начиная с rel-(S)-1-((3S,4S)-4-(трет-бутилдиметилсилилокси)тетрагидрофуран-3-ил)-1-(2-фторфенил)этанамина, оба соединения, представляющие собой (R)-N-rel-((S)-1-((3S,4S)-4-(трет-бутилдиметилсилилокси)тетрагидрофуран-3-ил)-1-(2-фторфенил)этил)-2-метилпропан-2-сульфинамид и (S)-N-rel-((S)-1-((3S,4S)-4-(трет-бутилдиметилсилилокси)тетрагидрофуран-3-ил)-1-(2-фторфенил)этил)-2-метилпропан-2-сульфинамид, были получены в виде бесцветных масел.
Промежуточное соединение XIVb-2: Начиная с (S)-1-((3S,4S)-4-(трет-бутилдиметилсилилокси)тетрагидрофуран-3-ил)-1-(2-фторфенил)этанамина, оба соединения, представляющие собой (R)-N-((S)-1-((3S,4S)-4-(трет-бутилдиметилсилилокси)тетрагидрофуран-3-ил)-1-(2-фторфенил)этил)-2-метилпропан-2-сульфинамид и (S)-N-((S)-1-((3S,4S)-4-(трет-бутилдиметилсилилокси)тетрагидрофуран-3-ил)-1-(2-фторфенил)этил)-2-метилпропан-2-сульфинамид, были получены в виде бесцветных масел.
Общий метод I: Синтез промежуточного N-защищенного аминоспирта XVb
Раствор дважды защищенного аминоспирта XIVb (5,43 ммоль) в тетрагидрофуране (7 мл) обрабатывали при 23°C тетрабутиламмония фторидом (1 М в тетрагидрофуране) (7,0 мл, 7,00 ммоль) и перемешивали в течение 45 минут. Для обработки реакционную смесь наливали в насыщенный раствор хлорида аммония, затем экстрагировали этилацетатом. Органический слой отделяли и промывали рассолом. Объединенные водные слои повторно экстрагировали этилацетатом, и объединенные органические слои высушивали над сульфатом натрия и выпаривали при пониженном давлении. Сырой продукт очищали хроматографией на силикагеле, используя градиент гептана и этилацетата = 100:0 до 0:100 в качестве элюента.
Промежуточное соединение XVb-1: Начиная с (S or R)-N-rel-((S)-1-((3S,4S)-4-(трет-бутилдиметилсилилокси)тетрагидрофуран-3-ил)-1-(2-фторфенил)этил)-2-метилпропан-2-сульфинамида, (S или R)-N-rel-((SR)-1-(2-фторфенил)-1-((3S,4S)-4-гидрокситетрагидрофуран-3-ил)этил)-2-метилпропан-2-сульфинамид был получен в виде белого твердого вещества.
Промежуточное соединение XVb-2: Начиная с (S или R)-N-((S)-1-((3S,4S)-4-(трет-бутилдиметилсилилокси)тетрагидрофуран-3-ил)-1-(2-фторфенил)этил)-2-метилпропан-2-сульфинамида, (S или R)-N-((S)-1-(2-фторфенил)-1-((3S,4S)-4-гидрокситетрагидрофуран-3-ил)этил)-2-метилпропан-2-сульфинамид был получен в виде беловатого твердого вещества. МС: m/z=330,1 [М+Н]+.
Общий метод J: Синтез промежуточного кетона XVIb
В сухой атмосфере аргона оксалилхлорид (896 мг, 7,06 ммоль) растворяли в сухом дихлорметане (16,5 мл). При -78°C добавляли сухой диметилсульфоксид (1,00 мл, 14,1 ммоль) по каплям через шприц. Полученную в результате смесь перемешивали в течение 15 минут, затем добавляли через шприц тонкоизмельченную суспензию N-защищенного аминоспирта XVb (4,71 ммоль) в сухом дихлорметане (19 мл). Смесь перемешивали при -78°C в течение 30 минут. Затем добавляли триэтиламин (3,28 мл, 23,5 ммоль), и полученную в результате смесь перемешивали при -78°C в течение 10 минут, затем при температуре, составляющей от -78°C до 10°C. Для обработки реакционную смесь гасили раствором хлорида аммония (20 мл), разбавляли дихлорметаном, экстрагировали лимонной кислотой (pH=2-3), промывали раствором гидрокарбоната натрия и рассолом. Органический слой высушивали над сульфатом натрия и выпаривали. Сырой продукт очищали хроматографией на силикагеле, используя градиент гептана и этилацетата = 100:0-20:80 в качестве элюента.
Промежуточное соединение XVIb-1: Начиная с (S или R)-N-rel-((S)-1-(2-фторфенил)-1-((3S,4S)-4-гидрокситетрагидрофуран-3-ил)этил)-2-метилпропан-2-сульфинамида, (S или R)-N-rel-((S)-1-(2-фторфенил)-1-((S)-4-оксотетрагидрофуран-3-ил)этил)-2-метилпропан-2-сульфинамид был получен в виде светло-желтого масла. МС: m/z=328,2 [М+Н]+.
Промежуточное соединение XVIb-2: Начиная с (S или R)-N-((S)-1-(2-фторфенил)-1-((3S,4S)-4-гидрокситетрагидрофуран-3-ил)этил)-2-метилпропан-2-сульфинамида, (S или R)-N-((S)-1-(2-фторфенил)-1-((S)-4-оксотетрагидрофуран-3-ил)этил)-2-метилпропан-2-сульфинамид был получен в виде светло-желтого масла. МС: m/z=328,2 [М+Н]+.
Общий метод К: Синтез промежуточного трифторметил-производного XVIIb
Раствор промежуточного кетона XVIb (2,72 ммоль) в тетрагидрофуране (15 мл) обрабатывали добавлением по каплям при 0°C (трифторметил)триметилсилана (580 мг, 603 мкл, 4,08 ммоль). Затем при 0°C добавляли тетрабутиламмония фторид (1 M в тетрагидрофуране) (136 мкл, 136 мкмоль). Коричневый раствор оставляли подогреться до комнатной температуры и перемешивали в течение 3 часов. С целью завершения реакции добавляли тетрабутиламмония фторид (1 M в тетрагидрофуране) (4,08 мл, 4,08 ммоль), и перемешивание продолжали в течение 1 часа. Для обработки реакционную смесь гасили водой, водный слой экстрагировали этилацетатом, органический слой промывали рассолом и высушивали над сульфатом натрия. После выпаривания при пониженном давлении остаток очищали хроматографией на силикагеле, используя градиент гептана и этилацетата = 100:0-0:100 в качестве элюента.
Промежуточное соединение XVIIb-1: Начиная с (S или R)-N-rel-((S)-1-(2-фторфенил)-1-((S)-4-оксотетрагидрофуран-3-ил)этил)-2-метилпропан-2-сульфинамида, (S или R)-N-rel-((S)-1-(2-фторфенил)-1-((3S,4S)-4-гидрокси-4-(трифторметил)тетрагидрофуран-3-ил)этил)-2-метилпропан-2-сульфинамид был получен в виде светло-коричневого твердого вещества. МС: m/z=398,1 [М+Н]+.
Промежуточное соединение XVIIb-2: Начиная с (S или R)-N-((S)-1-(2-фторфенил)-1-((S)-4-оксотетрагидрофуран-3-ил)этил)-2-метилпропан-2-сульфинамида, (S или R)-N-((S)-1-(2-фторфенил)-1-((3S,4S)-4-гидрокси-4-(трифторметил)тетрагидрофуран-3-ил)этил)-2-метилпропан-2-сульфинамид был получен в виде светло-коричневой пены. МС: m/z=398,1 [М+Н]+.
Общий метод L: Синтез промежуточного аминоспирта XVIIIb
Раствор промежуточного трифторметил-производного XVIIb (1,4 ммоль) в тетрагидрофуране (15 мл) обрабатывали при 23°C соляной кислотой (37% в воде) (573 мкл, 6,98 ммоль). Желтый раствор перемешивали при 23°C в течение 2 часов, затем наливали в раствор карбоната натрия (1 М). Смесь дважды экстрагировали этилацетатом, объединенные органические слои высушивали над сульфатом натрия и выпаривали при пониженном давлении. Объединенные органические слои высушивали над Na2SO4, фильтровали и выпаривали. Сырой продукт очищали хроматографией на силикагеле, используя градиент гептана и этилацетата = 100:0-50:50 в качестве элюента.
Промежуточное соединение XVIIIb-1: Начиная с (S или R)-N-rel-((S)-1-(2-фторфенил)-1-((3S,4S)-4-гидрокси-4-(трифторметил)тетрагидрофуран-3-ил)этил)-2-метилпропан-2-сульфинамида, rel-(3S,4S)-4-((S)-1-амино-1-(2-фторфенил)этил)-3-(трифторметил)тетрагидрофуран-3-ол был получен в виде светло-желтого масла. МС: m/z=294,1 [М+Н]+.
Промежуточное соединение XVIIIb-2: Начиная с (S или R)-N-((S)-1-(2-фторфенил)-1-((3S,4S)-4-гидрокси-4-(трифторметил)тетрагидрофуран-3-ил)этил)-2-метилпропан-2-сульфинамида, (3S,4S)-4-((S)-1-амино-1-(2-фторфенил)этил)-3-(трифторметил)тетрагидрофуран-3-ол был получен в виде коричневого масла. МС: m/z=294,2 [М+Н]+.
Общий метод М: Синтез промежуточного O-защищенного аминоспирта XIXb
Гидрид натрия (55% дисперсию в масле) (116 мг, 2,67 ммоль) добавляли при 0°C к раствору аминоспирта XVIIIb (1,48 ммоль, экв.: 1,00) в N,N-диметилформамиде (6 мл). После перемешивания в течение 30 минут при 23°C реакционную смесь обрабатывали при 0°C трет-бутилдиметилсилилтрифторметансульфонатом (728 мг, 632 мкл, 2,67 ммоль). После перемешивания в течение 16 часов реакционную смесь экстрагировали смесью дихлорметана и насыщенного раствора гидрокарбоната натрия. Органический слой отделяли, высушивали над сульфатом натрия и выпаривали. Остаток очищали хроматографией на силикагеле, используя градиент гептана и этилацетата = 100:0-50:50 в качестве элюента.
Промежуточное соединение XIXb-1: Начиная с rel-(3S,4S)-4-((S)-1-амино-1-(2-фторфенил)этил)-3-(трифторметил)тетрагидрофуран-3-ола, rel-(S)-1-((3S,4S)-4-(трет-бутилдиметилсилилокси)-4-(трифторметил)тетрагидрофуран-3-ил)-1-(2-фторфенил)этанамин был получен в виде бесцветного масла.
Промежуточное соединение XIXb-2: Начиная с (3S,4S)-4-((S)-1-амино-1-(2-фторфенил)этил)-3-(трифторметил)тетрагидрофуран-3-ола, (S)-1-((3S,4S)-4-(трет-бутилдиметилсилилокси)-4-(трифторметил)тетрагидрофуран-3-ил)-1-(2-фторфенил)этанамин был получен в виде светло-желтого масла. МС: m/z=408,3 [М+Н]+.
Общий метод N: Синтез промежуточного изотиоцианата XXb
Смесь O-защищенного аминоспирта XIXb (908 мкмоль) и бикарбоната натрия (381 мг, 4,54 ммоль) в дихлорметане (6 мл) обрабатывали при 0°C тиофосгеном (129 мг, 86,1 мкл, 1,09 ммоль). Реакционную смесь перемешивали при 23°C в течение 4 часов. Для обработки реакционную смесь экстрагировали смесью дихлорметана и воды. Органический слой отделяли, высушивали над сульфатом натрия и выпаривали. Сырой продукт вводили в следующую стадию без дополнительной очистки.
Промежуточное соединение XXb-1: Начиная с rel-(S)-1-((3S,4S)-4-(трет-бутилдиметилсилилокси)-4-(трифторметил)тетрагидрофуран-3-ил)-1-(2-фторфенил)этанамина, трет-бутил-rel-((3S,4S)-4-((S)-1-(2-фторфенил)-1-изотиоцианатоэтил)-3-(трифторметил)тетрагидрофуран-3-илокси)диметилсилан был получен в виде бесцветного масла.
Промежуточное соединение XXb-2: Начиная с (S)-1-((3S,4S)-4-(трет-бутилдиметилсилилокси)-4-(трифторметил)тетрагидрофуран-3-ил)-1-(2-фторфенил)этанамина, трет-бутил-((3S,4S)-4-((S)-1-(2-фторфенил)-1-изотиоцианатоэтил)-3-(трифторметил)тетрагидрофуран-3-илокси)диметилсилан был получен в виде коричневого масла.
Общий метод О: Синтез промежуточного N-бензилированного оксазина XXIb
Раствор изотиоцианата XXb (1,09 ммоль) в ацетонитриле (11 мл) обрабатывали при комнатной температуре 2,4-диметоксибензиламином (273 мг, 246 мкл, 1,63 ммоль). Бесцветный раствор перемешивали при 70°C в течение 16 часов. Добавляли тригидрат тетрабутиламмония фторида (378 мг, 1,2 ммоль) при 23°C, и перемешивание продолжали в течение 2 часов. Затем добавляли 1-этил-3-(3-диметиламинопропил)карбодиимид (418 мг, 2,18 ммоль), и перемешивание продолжали при 80°C в течение 16 часов. Для обработки реакционную смесь экстрагировали смесью этилацетата и насыщенного раствора гидрокарбоната натрия. Органический слой отделяли, высушивали над сульфатом натрия и выпаривали. Сырой продукт очищали хроматографией на силикагеле, используя градиент гептана и этилацетата = 100:0-65:35 в качестве элюента.
Промежуточное соединение XXIb-1: Начиная с трет-бутил-rel-((3S,4S)-4-((S)-1-(2-фторфенил)-1-изотиоцианатоэтил)-3-(трифторметил)тетрагидрофуран-3-илокси)диметилсилана, rel-(4S,4aS,7aS)-N-(2,4-диметоксибензил)-4-(2-фторфенил)-4-метил-7а-(трифторметил)-4а,5,7,7а-тетрагидро-4Н-фуро[3,4-е][1,3]оксазин-2-амин был получен в виде белого твердого вещества. МС: m/z=469,23 [М+Н]+.
Промежуточное соединение XXIb-2: Начиная с трет-бутил((3S,4S)-4-((S)-1-(2-фторфенил)-1-изотиоцианатоэтил)-3-(трифторметил)тетрагидрофуран-3-илокси)диметилсилана, (4S,4aS,7aS)-N-(2,4-диметоксибензил)-4-(2-фторфенил)-4-метил-7а-(трифторметил)-4а,5,7,7а-тетрагидро-4Н-фуро[3,4-е][1,3]оксазин-2-амин был получен в виде бесцветного масла.
Общий метод Р: Синтез промежуточного оксазина XXIIb
Раствор N-бензилированного оксазина XXIb (758 мкмоль) в трифторуксусной кислоте (4,32 г, 2,92 мл, 37,9 ммоль) перемешивали в течение 5 часов. Добавляли трифторметансульфоновую кислоту (341 мг, 202 мкл, 2,27 ммоль), и перемешивание продолжали еще в течение 2 часов. Темно-красный раствор наливали в раствор карбоната натрия (1 М) и дважды экстрагировали дихлорметаном. Объединенные органические слои высушивали над сульфатом натрия и выпаривали. Сырой продукт очищали хроматографией на силикагеле, используя смесь 19:1 гептана и метанола в качестве элюента.
Промежуточное соединение XXIIb-1: Начиная с rel-(4S,4aS,7aS)-N-(2,4-диметоксибензил)-4-(2-фторфенил)-4-метил-7а-(трифторметил)-4а,5,7,7а-тетрагидро-4Н-фуро[3,4-е][1,3]оксазин-2-амина, rel-(4S,4aS,7aS)-4-(2-фторфенил)-4-метил-7а-(трифторметил)-4а,5,7,7а-тетрагидро-4Н-фуро[3,4-е][1,3]оксазин-2-амин был получен в виде белого твердого вещества. МС: m/z=319,0 [М+Н]+.
Промежуточное соединение XXIIb-2: Начиная с (4S,4aS,7aS)-N-(2,4-диметоксибензил)-4-(2-фторфенил)-4-метил-7а-(трифторметил)-4а,5,7,7а-тетрагидро-4Н-фуро[3,4-е][1,3]оксазин-2-амина, (4S,4aS,7aS)-4-(2-фторфенил)-4-метил-7а-(трифторметил)-4а,5,7,7а-тетрагидро-4Н-фуро[3,4-е][1,3]оксазин-2-амин был получен в виде белой смолы. МС: m/z=319,0 [М+Н]+.
Синтез промежуточного нитрооксазина XXIIIb
Промежуточное соединение XXIIIb-1: Следуя общему методу Е, в результате нитрования rel-(4S,4aS,7aS)-4-(2-фторфенил)-4-метил-7а-(трифторметил)-4а,5,7,7а-тетрагидро-4Н-фуро[3,4-е][1,3]оксазин-2-амина получили rel-(4S,4aS,7aS)-4-(2-фтор-5-нитрофенил)-4-метил-7а-(трифторметил)-4а,5,7,7а-тетрагидро-4Н-фуро[3,4-е][1,3]оксазин-2-амин в виде светло-коричневого твердого вещества. МС: m/z=364,0 [М+Н]+.
Промежуточное соединение XXIIIb-2: Следуя общему методу Е, в результате нитрования (4S,4aS,7aS)-4-(2-фторфенил)-4-метил-7а-(трифторметил)-4а,5,7,7а-тетрагидро-4Н-фуро[3,4-е][1,3]оксазин-2-амина получили (4S,4aS,7aS)-4-(2-фтор-5-нитрофенил)-4-метил-7а-(трифторметил)-4а,5,7,7а-тетрагидро-4Н-фуро[3,4-е][1,3]оксазин-2-амин в виде светло-коричневой пены. МС: m/z=364,0 [М+Н]+.
Синтез промежуточного анилина XXIVb
Промежуточное соединение XXIVb-1: Следуя общему методу F, в результате восстановления rel-(4S,4aS,7aS)-4-(2-фтор-5-нитрофенил)-4-метил-7а-(трифторметил)-4а,5,7,7а-тетрагидро-4Н-фуро[3,4-е][1,3]оксазин-2-амина получили rel-(4S,4aS,7aS)-4-(5-амино-2-фторфенил)-4-метил-7а-(трифторметил)-4а,5,7,7а-тетрагидро-4Н-фуро[3,4-е][1,3]оксазин-2-амин в виде беловатого твердого вещества. МС: m/z=334,2 [М+Н]+.
Промежуточное соединение XXIVb-2: Следуя общему методу F, в результате восстановления (4S,4aS,7aS)-4-(2-фтор-5-нитрофенил)-4-метил-7а-(трифторметил)-4а,5,7,7а-тетрагидро-4Н-фуро[3,4-е][1,3]оксазин-2-амина получили (4S,4aS,7aS)-4-(5-амино-2-фторфенил)-4-метил-7а-(трифторметил)-4а,5,7,7а-тетрагидро-4Н-фуро[3,4-е][1,3]оксазин-2-амин в виде светло-коричневой пены. МС: m/z=334,2 [М+Н]+.
Общий метод Q синтеза конечных примеров
К раствору кислоты XII (0,16 ммоль) в МеОН (1 мл) добавляли при 22°C 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метил-морфолиния хлорид (0,19 ммоль), и перемешивание продолжали при 0°C в течение 30 мин. К смеси добавляли раствор анилина XI (0,15 ммоль) в МеОН (2 мл), и перемешивание продолжали при 0°C в течение 4 ч. Смесь выпаривали, и остаток распределяли между насыщенным водным раствором Na2CO3 и этилацетатом. Органический слой высушивали, выпаривали, и остаток очищали на колонке RP18 препаративной ВЭЖХ, используя градиент вода/NEt3 (99,9:0,1)→CH3CN. Альтернативно сырое вещество можно очистить хроматографией на колонке Si-NH2, используя AcOEt, с последующим растиранием с диэтиловым эфиром с получением конечных примеров соединений, имеющих формулу I.
Пример 1
5-Хлор-пиридин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амид
В результате сочетания (3aS,7S,7aS)-7-(5-амино-2-фтор-фенил)-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-5-иламина и 5-хлор-пиридин-2-карбоновой кислоты, следуя методу G, получили соединение, указанное в заголовке, в виде белого твердого вещества. МС: m/z=405,3 и 407.2 [М+Н]+.
Пример 2
3,5-Дихлор-пиридин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амид
В результате сочетания (3aS,7S,7aS)-7-(5-амино-2-фтор-фенил)-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-5-иламина и 3,5-дихлор-пиридин-2-карбоновой кислоты, следуя методу G, получили соединение, указанное в заголовке, в виде белого твердого вещества. МС: m/z=439,1 и 441,1 [М+Н]+.
Пример 3
5-Фтор-пиридин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амид
В результате сочетания (3aS,7S,7aS)-7-(5-амино-2-фтор-фенил)-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-5-иламина и 5-фтор-пиридин-2-карбоновой кислоты, следуя методу G, получили соединение, указанное в заголовке, в виде белого твердого вещества. МС: m/z=389,2 [М+Н]+.
Пример 4
5-Циано-пиридин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амид
В результате сочетания (3aS,7S,7aS)-7-(5-амино-2-фтор-фенил)-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-5-иламина и 5-циано-пиридин-2-карбоновой кислоты, следуя методу G, получили соединение, указанное в заголовке, в виде белого твердого вещества. МС: m/z=396,2 [М+Н]+.
Пример 5
Пиридин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амид
В результате сочетания (3aS,7S,7aS)-7-(5-амино-2-фтор-фенил)-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-5-иламина и пиридин-2-карбоновой кислоты, следуя методу G, получили соединение, указанное в заголовке, в виде бесцветного твердого вещества. МС: m/z=371,2 [М+Н]+.
Пример 6
5-(2,2,2-Трифтор-этокси)-пиридин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амид
В результате сочетания (3aS,7S,7aS)-7-(5-амино-2-фтор-фенил)-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-5-иламина и 5-(2,2,2-трифтор-этокси)-пиридин-2-карбоновой кислоты (полученной, как описано в документе Banner D. et al., WO 2010/128058), следуя методу G, получили соединение, указанное в заголовке, в виде бесцветного твердого вещества. МС: m/z=469,2 [М+Н]+.
Пример 7
5-Бут-2-инилокси-пиразин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амид
В результате сочетания (3aS,7S,7aS)-7-(5-амино-2-фтор-фенил)-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-5-иламина и 5-бут-2-инилокси-пиридин-2-карбоновой кислоты (полученной, как описано в документе Tamura Y. et al., WO 2010/113848), следуя методу G, получили соединение, указанное в заголовке, в виде бесцветного твердого вещества. МС: m/z=440,4 [М+Н]+.
Пример 8
5-(2,2,2-Трифтор-этокси)-пиразин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амид
В результате сочетания (3aS,7S,7aS)-7-(5-амино-2-фтор-фенил)-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-5-иламина и 5-(2,2,2-трифтор-этокси)-пиразин-2-карбоновой кислоты (полученной, как описано в документе Suzuki Y. et al., WO 2009/091016), следуя методу G, получили соединение, указанное в заголовке, в виде бесцветного твердого вещества. МС: m/z=470,2 [М+Н]+.
Пример 9
5-Циклопропилметокси-пиридин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амид
В результате сочетания (3aS,7S,7aS)-7-(5-амино-2-фтор-фенил)-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-5-иламина и 5-циклопропилметокси-пиридин-2-карбоновой кислоты (полученной, как описано в документе Scott, J. et al., WO 2011/044181), следуя методу G, получили соединение, указанное в заголовке, в виде бесцветного твердого вещества. МС: m/z=441,3 [М+Н]+.
Пример 10
5-Бут-2-инилокси-пиридин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амид
5-Бут-2-инилокси-пиридин-2-карбоновой кислоты метиловый эфир
Смесь 5-гидрокси-пиридин-2-карбоновой кислоты метилового эфира (271 мг) и 1-бром-2-бутина (283 мг) в ДМФ (5 мл) обрабатывали карбонатом калия (367 мг) и нагревали до 100°C в течение 16 ч. Смесь распределяли между водой и этилацетатом, органический слой высушивали, выпаривали, и остаток очищали хроматографией, используя гептан/этилацетат (градиент от 0 до 80% этилацетата), с получением продукта, указанного в заголовке (266 мг), в виде бледно-желтого твердого вещества. МС: m/z=206,1 [М+Н]+.
5-Бут-2-инилокси-пиридин-2-карбоновая кислота
К раствору 5-бут-2-инилокси-пиридин-2-карбоновой кислоты метилового эфира (234 мг) в ТГФ (20 мл) и воде (15 мл) добавляли водный раствор LiOH (1 М, 2,3 мл), и смесь перемешивали при 22°C в течение 1 ч. Смесь обрабатывали водной HCl (1 М, 2,3 мл), медленно выпаривали, полученную суспензию фильтровали, остаток промывали водой и высушивали с получением продукта, указанного в заголовке (165 мг), в виде белого твердого вещества. МС: m/z=192,1 [М+Н]+.
В результате сочетания (3aS,7S,7aS)-7-(5-амино-2-фтор-фенил)-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-5-иламина и 5-бут-2-инилокси-пиридин-2-карбоновой кислоты, следуя методу G, получили соединение, указанное в заголовке, в виде белого твердого вещества. МС: m/z=439,2 [М+Н]+.
Пример 11
5-(2,2,3,3-Тетрафтор-пропокси)-пиридин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амид
В результате сочетания (3aS,7S,7aS)-7-(5-амино-2-фтор-фенил)-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-5-иламина и 5-(2,2,3,3-тетрафтор-пропокси)-пиридин-2-карбоновой кислоты (полученной, как описано в документе Banner, D. et al., WO 2011/069934), следуя методу G, получили соединение, указанное в заголовке, в виде белого твердого вещества. МС: m/z=501,1 [М+Н]+.
Пример 12
N-(3-((4S,4aS,7aS)-2-амино-4-метил-4а,5,7,7а-тетрагидро-4Н-фуро[3,4-е][1,3]оксазин-4-ил)-4-фторфенил)-3-хлор-5-цианопиколинамид
В результате сочетания (3aS,7S,7aS)-7-(5-амино-2-фтор-фенил)-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-5-иламина и 3-хлор-5-циано-пиридин-2-карбоновой кислоты, следуя методу G, получили соединение, указанное в заголовке, в виде бледно-желтого твердого вещества. МС: m/z=430,2 [М+Н]+.
Пример 13
5-Циано-пиридин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-фторметил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амид
В результате сочетания (3aS,7S,7aS)-7-(5-амино-2-фтор-фенил)-7-фторметил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-5-иламина и 5-циано-пиридин-2-карбоновой кислоты, следуя методу G, получили соединение, указанное в заголовке, в виде белого твердого вещества. МС: m/z=414,1 [М+Н]+.
Пример 14
5-Хлор-пиридин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-фторметил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амид
В результате сочетания (3aS,7S,7aS)-7-(5-амино-2-фтор-фенил)-7-фторметил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-5-иламина и 5-хлор-пиридин-2-карбоновой кислоты, следуя методу G, получили соединение, указанное в заголовке, в виде белого твердого вещества. МС: m/z=423,0 [М+Н]+.
Пример 15
5-Фтор-пиридин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-фторметил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амид
В результате сочетания (3aS,7S,7aS)-7-(5-амино-2-фтор-фенил)-7-фторметил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-5-иламина и 5-фтор-пиридин-2-карбоновой кислоты, следуя методу G, получили соединение, указанное в заголовке, в виде белого твердого вещества. МС: m/z=407,3 [М+Н]+.
Пример 16
5-Хлор-пиридин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-дифторметил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амид
В результате сочетания (3aS,7S,7aS)-7-(5-амино-2-фтор-фенил)-7-дифторметил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-5-иламина и 5-хлор-пиридин-2-карбоновой кислоты, следуя методу G, получили соединение, указанное в заголовке, в виде бесцветного твердого вещества. МС: m/z=441,2 [М+Н]+.
Пример 17
5-Циано-пиридин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-дифторметил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амид
В результате сочетания (3aS,7S,7aS)-7-(5-амино-2-фтор-фенил)-7-дифторметил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-5-иламина и 5-циано-пиридин-2-карбоновой кислоты, следуя методу G, получили соединение, указанное в заголовке, в виде бесцветного твердого вещества. МС: m/z=432,3 [М+Н]+.
Пример 18
5-Бут-2-инилокси-пиразин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-дифторметил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амид
В результате сочетания (3aS,7S,7aS)-7-(5-амино-2-фтор-фенил)-7-дифторметил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-5-иламина и 5-бут-2-инилокси-пиридин-2-карбоновой кислоты (полученной, как описано в документе Tamura Y. et al., WO 2010/113848), следуя методу G, получили соединение, указанное в заголовке, в виде бесцветного твердого вещества. МС: m/z=476,2 [М+Н]+.
Пример 19
N-(3-(rel-(4SR,4aSR,7aSR)-2-амино-4-метил-7а-(трифторметил)-4а,5,7,7а-тетрагидро-4Н-фуро[3,4-е][1,3]оксазин-4-ил)-4-фторфенил)-5-цианопиколинамид
В результате сочетания rel-(4S,4aS,7aS)-4-(5-амино-2-фторфенил)-4-метил-7а-(трифторметил)-4а,5,7,7а-тетрагидро-4Н-фуро[3,4-е][1,3]оксазин-2-амина (промежуточного соединения XXIVb-1) и 5-циано-пиридин-2-карбоновой кислоты, следуя методу G, получили соединение, указанное в заголовке, в виде беловатой пены. МС: m/z=464,1 [М+Н]+.
Пример 20
N-(3-(rel-(4SR,4aSR,7aSR)-2-амино-4-метил-7а-(трифторметил)-4а,5,7,7а-тетрагидро-4Н-фуро[3,4-е][1,3]оксазин-4-ил)-4-фторфенил)-5-(бут-2-инилокси)пиразин-2-карбоксамид
В результате сочетания rel-(4S,4aS,7aS)-4-(5-амино-2-фторфенил)-4-метил-7а-(трифторметил)-4а,5,7,7а-тетрагидро-4Н-фуро[3,4-е][1,3]оксазин-2-амина (промежуточного соединения XXIVb-1) и 5-(бут-2-инилокси)пиразин-2-карбоновой кислоты, следуя методу G, получили соединение, указанное в заголовке, в виде белой пены. МС: m/z=508,2 [М+Н]+.
Пример 21
N-(3-((4SR,4aSR,7aSR)-2-амино-4-метил-7а-(трифторметил)-4а,5,7,7а-тетрагидро-4Н-фуро[3,4-е][1,3]оксазин-4-ил)-4-фторфенил)-5-хлорпиколинамид
В результате сочетания (4S,4aS,7aS)-4-(5-амино-2-фторфенил)-4-метил-7а-(трифторметил)-4а,5,7,7а-тетрагидро-4Н-фуро[3,4-е][1,3]оксазин-2-амина (промежуточного соединения XXIVb-2) и 5-хлор-пиридин-2-карбоновой кислоты, следуя методу G, получили соединение, указанное в заголовке, в виде белого твердого вещества. МС: m/z=473,1 [М+Н]+.
Пример 22
N-(3-((4SR,4aSR,7aSR)-2-амино-4-метил-7а-(трифторметил)-4а,5,7,7а-тетрагидро-4Н-фуро[3,4-е][1,3]оксазин-4-ил)-4-фторфенил)-5-цианопиколинамид
В результате сочетания (4S,4aS,7aS)-4-(5-амино-2-фторфенил)-4-метил-7а-(трифторметил)-4а,5,7,7а-тетрагидро-4Н-фуро[3,4-е][1,3]оксазин-2-амина (промежуточного соединения XXIVb-2) и 5-циано-пиридин-2-карбоновой кислоты, следуя методу G, получили соединение, указанное в заголовке, в виде белого твердого вещества. МС: m/z=464,1 [М+Н]+.
| название | год | авторы | номер документа |
|---|---|---|---|
| 1,4 ТИАЗЕПИНЫ/СУЛЬФОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ВАСЕ1 И(ИЛИ) ВАСЕ2 | 2012 |
|
RU2600931C2 |
| 2,5,6,7-ТЕТРАГИДРО-[1,4]ОКСАЗЕПИН-3-ИЛАМИНЫ ИЛИ 2,3,6,7-ТЕТРАГИДРО-[1,4]ОКСАЗЕПИН-5-ИЛАМИНЫ | 2011 |
|
RU2570796C2 |
| АМИДЫ КОНДЕНСИРОВАННОГО ПИПЕРИДИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ ИОННЫХ КАНАЛОВ | 2014 |
|
RU2741810C2 |
| ОКСАЗОЛЬНЫЕ ПРОИЗВОДНЫЕ ТЕТРАЦИКЛИНОВ | 2004 |
|
RU2415844C2 |
| МОДУЛЯТОРЫ SHIP1 И ОТНОСЯЩИЕСЯ К НИМ СПОСОБЫ | 2014 |
|
RU2679805C2 |
| КОНДЕНСИРОВАННОЕ ПРОИЗВОДНОЕ АМИНОДИГИДРОТИАЗИНА | 2009 |
|
RU2476431C2 |
| ПРОИЗВОДНЫЕ 2,6-ХИНОЛИНИЛА И 2,6-НАФТИЛА, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ИХ ОСНОВЕ, ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ VLA-4 И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 2003 |
|
RU2315041C2 |
| НОВЫЕ ЛИГАНДЫ КАННАБИОИДНЫХ РЕЦЕПТОРОВ, СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2006 |
|
RU2420518C2 |
| СЕЛЕКТИВНЫЕ АНТАГОНИСТЫ Н4-ГИСТАМИНОВЫХ РЕЦЕПТОРОВ ДЛЯ ЛЕЧЕНИЯ ВЕСТИБУЛЯРНЫХ НАРУШЕНИЙ | 2009 |
|
RU2589846C2 |
| НОВЫЕ 7-(ЗАМЕЩЕННЫЕ)-8-(ЗАМЕЩЕННЫЕ)-9-[(ЗАМЕЩЕННЫЕ ГЛИЦИЛ)АМИДО]-6-ДЕМЕТИЛ- 6-ДЕЗОКСИТЕТРАЦИКЛИНЫ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ ИЛИ КОМПЛЕКСЫ С МЕТАЛЛАМИ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ АНТИБИОТИЧЕСКОЙ АКТИВНОСТЬЮ IN VIVO | 1993 |
|
RU2125985C1 |
Изобретение относится к соединению, имеющему формулу Ia, где R1 представляет собой пиридинил или пиридинил, замещенный 1-2 заместителями, индивидуально выбранными из циано, атома галогена, галоген-C1-6-алкокси, или пиразинил, замещенный галоген-C1-6-алкокси или С2-6-алкинил-C1-6-алкокси; R2 представляет собой F; R3 выбран из группы, состоящей из i) метила, ii) -CH2CH2F, iii) -CH2CHF2; R4 представляет собой водород; и R5 выбран из группы, состоящей из i) атома водорода, ii) -CF3; или его фармацевтически приемлемые соли. Также изобретение относится к конкретным соединениям N-[3-(5-амино-3,3А,7,7А-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)фенил]амида, приведенным в формуле изобретения. Соединение по изобретению предназначено для применения в качестве терапевтически активного вещества, обладающего ингибирующей активностью в отношении ВАСЕ1 и/или ВАСЕ2, для изготовления фармацевтической композиции или лекарственного средства. Технический результат - N-[3-(5-амино-3,3А,7,7А-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)фенил]амиды, обладающие ингибирующими свойствами в отношении ВАСЕ1 и/или ВАСЕ2. 4 н. и 8 з.п. ф-лы, 8 табл., 22 пр.
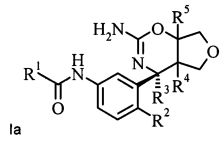
1. Соединение, имеющее формулу Ia:
в котором
R1 представляет собой пиридинил или пиридинил, замещенный 1-2 заместителями, индивидуально выбранными из циано, атома галогена, галоген-C1-6-алкокси, или пиразинил, замещенный галоген-C1-6-алкокси или С2-6-алкинил-C1-6-алкокси;
R2 представляет собой F;
R3 выбран из группы, состоящей из
i) метила,
ii) -CH2CH2F,
iii) -CH2CHF2;
R4 представляет собой водород; и
R5 выбран из группы, состоящей из
i) атома водорода,
ii) -CF3;
или его фармацевтически приемлемые соли.
2. Соединение по п. 1, в котором R3 представляет собой метил.
3. Соединение по п. 1, в котором R3 представляет собой -CH2CH2F или -CH2CHF2.
4. Соединение по п. 1, в котором R5 представляет собой атом водорода.
5. Соединение по п. 1, в котором R5 представляет собой -CF3.
6. Соединение по п. 1, в котором R1 представляет собой 5-хлор-пиридин-2-ил, 3,5-дихлор-пиридин-2-ил, 5-(2,2,2-трифтор-этокси)-пиразин-2-ил, 5-(2,2,2-трифтор-этокси)-пиридин-2-ил, 5-бут-2-инилокси-пиразин-2-ил, 5-циано-пиридин-2-ил, 5-фтор-пиридин-2-ил или пиридин-2-ил.
7. Соединение по п. 1, в котором R1 представляет собой 5-циано-пиридин-2-ил или 5-бут-2-инилокси-пиразин-2-ил.
8. Соединение по п. 1, выбранное из группы, состоящей из следующих соединений:
5-Хлор-пиридин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амида,
3,5-Дихлор-пиридин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амида,
5-(2,2,2-Трифтор-этокси)-пиразин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амида,
5-(2,2,2-Трифтор-этокси)-пиридин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амида,
5-Бут-2-инилокси-пиразин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амида,
5-Циано-пиридин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амида,
5-Фтор-пиридин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амида и
Пиридин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-метил-3,3a,7,7a-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амида,
или их фармацевтически приемлемой соли.
9. Соединение, выбранное из группы, состоящей из следующих соединений:
5-Хлор-пиридин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амида,
5-Фтор-пиридин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амида,
5-Циано-пиридин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амида,
5-Бут-2-инилокси-пиразин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амида,
5-Циклопропилметокси-пиридин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амида,
5-Бут-2-инилокси-пиридин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амида,
5-Циано-пиридин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-фторметил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амида,
5-Хлор-пиридин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-фторметил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амида,
5-Фтор-пиридин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-фторметил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амида,
5-Хлор-пиридин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-дифторметил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амида,
5-Циано-пиридин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-дифторметил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амида,
5-Бут-2-инилокси-пиразин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-дифторметил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амида,
N-(3-(rel-(4SR,4aSR,7aSR)-2-амино-4-метил-7а-(трифторметил)-4а,5,7,7а-тетрагидро-4Н-фуро[3,4-е][1,3]оксазин-4-ил)-4-фторфенил)-5-цианопиколинамида,
N-(3-(rel-(4SR,4aSR,7aSR)-2-амино-4-метил-7а-(трифторметил)-4а,5,7,7а-тетрагидро-4Н-фуро[3,4-е][1,3]оксазин-4-ил)-4-фторфенил)-5-(бут-2-инилокси)пиразин-2-карбоксамида,
N-(3-((4SR,4aSR,7aSR)-2-амино-4-метил-7a-(трифторметил)-4a,5,7,7a-тетрагидро-4Н-фуро[3,4-е][1,3]оксазин-4-ил)-4-фторфенил)-5-хлорпиколинамида,
N-(3-((4SR,4aSR,7aSR)-2-амино-4-метил-7a-(трифторметил)-4a,5,7,7a-тетрагидро-4Н-фуро[3,4-е][1,3]оксазин-4-ил)-4-фторфенил)-5-цианопиколинамида,
5-(2,2,2-Трифтор-этокси)-пиридин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амида,
5-(2,2,2-Трифтор-этокси)-пиразин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амида,
5-(2,2,3,3-Тетрафтор-пропокси)-пиридин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амида,
N-(3-((4S,4aS,7aS)-2-амино-4-метил-4а,5,7,7а-тетрагидро-4Н-фуро[3,4-е][1,3]оксазин-4-ил)-4-фторфенил)-3-хлор-5-цианопиколинамида,
Пиридин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-метил-3,3a,7,7a-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амида, и
3,5-Дихлор-пиридин-2-карбоновой кислоты [3-((3aS,7S,7aS)-5-амино-7-метил-3,3а,7,7а-тетрагидро-1Н-2,4-диокса-6-аза-инден-7-ил)-4-фтор-фенил]-амида,
или их фармацевтически приемлемой соли.
10. Соединение по любому из пп. 1-9, применяемое в качестве терапевтически активного вещества, обладающего ингибирующей активностью в отношении ВАСЕ1 и/или ВАСЕ2.
11. Фармацевтическая композиция, обладающая ингибирующей активностью в отношении ВАСЕ1 и/или ВАСЕ2, содержащая терапевтически эффективное количество соединения по любому из пп. 1-9 и фармацевтически приемлемый носитель и/или фармацевтически приемлемое вспомогательное вещество.
12. Применение соединения по любому из пп. 1-9 для получения лекарственного средства, обладающего ингибирующей активностью в отношении ВАСЕ1 и/или ВАСЕ2.
| RU 2012129168 А, даты приоритета 11.12.2009, 22.10.2010 | |||
| Колосоуборка | 1923 |
|
SU2009A1 |
| RU 2008120601 А, 10.12.2009. | |||

