Область техники, к которой относится изобретение
Настоящее изобретение относится к конденсированному производному аминодигидротиазина и его фармацевтическому применению. Более конкретно, настоящее изобретение относится к конденсированному производному аминодигидротиазина, которое обладает эффектом ингибирования продукции β-амилоидного белка (далее обозначенного как Aβ) или эффектом ингибирования фермента расщепления бета-сайта белка-предшественника амилоида-β типа 1 (фермента, далее обозначенного как BACE1 или бета-секретаза) и является эффективным при лечении нейродегенеративного заболевания, вызываемого Aβ белком, в частности деменции Альцгеймера, синдрома Дауна или тому подобное, и фармацевтической композиции, содержащей конденсированное производное аминодигидротиазина в качестве активного компонента.
Предпосылки создания изобретения
Болезнь Альцгеймера является заболеванием, характеризующимся дегенерацией и потерей нейронов, а также образованием старческих бляшек и нейрофибриллярной дегенерацией. В настоящее время болезнь Альцгеймера лечат только методами симптоматической терапии с использованием средств для уменьшения симптомов, типичным представителем которых является ингибитор ацетилхолинэстеразы, а фундаментальное лекарственное средство, ингибирующее прогрессирование болезни, все еще не создано. Необходимо разработать способ контролирования причины возникновения патологии, чтобы создать фундаментальное лекарственное средство для лечения болезни Альцгеймера.
Считается, что Aβ-белки как метаболиты амилоидных белков-предшественников (далее обозначенных как APP) сильно вовлечены в процессы дегенерации и потери нейронов и возникновения симптомов деменции (см., например, непатентные документы 3 и 4). Aβ-белки содержат в качестве основных компонентов Aβ40, состоящий из 40 аминокислот, и Aβ42, который дополнительно содержит 2 аминокислоты на С-конце. Известно, что Aβ40 и Aβ42 обладают высокой агрегационной способностью (см., например, непатентный документ 5) и являются основными составляющими старческих бляшек (см., например, непатентные документы 5, 6 и 7). Известно также, что Aβ40 и Aβ42 отличаются повышенной мутацией в генах APP и пресенилина, которая наблюдается при семейной форме болезни Альцгеймера (см., например, непатентные документы 8, 9 и 10). Поэтому ожидается, что соединение, понижающее продукцию Aβ40 и Aβ42, будет для болезни Альцгеймера ингибитором развития или профилактическим средством.
Aβ продуцируется в результате расщепления APP бета-секретазой (BACE1) и затем гамма-секретазой. Поэтому были предприняты попытки создания ингибиторов гамма-секретазы и бета-секретазы, чтобы ингибировать продукцию Aβ. Уже известные ингибиторы бета-секретазы описаны в указанных ниже патентных документах 1-13 и непатентных документах 1 и 2 и тому подобное. В частности, в патентном документе 1 описаны производное аминодигидротиазина и соединение, обладающее ингибирующей активностью в отношении BACE1.
Патентный документ 1: WO 2007/049532
Патентный документ 2: US 3235551
Патентный документ 3: US 3227713
Патентный документ 4: JP-A-09-067355
Патентный документ 5: WO 01/87293
Патентный документ 6: WO 04/014843
Патентный документ 7: JP-A-2004-149429
Патентный документ 8: WO 02/96897
Патентный документ 9: WO 04/043916
Патентный документ 10: WO 2005/058311
Патентный документ 11: WO 2005/097767
Патентный документ 12: WO 2006/041404
Патентный документ 13: WO 2006/041405
Непатентный документ 1: Journal of Heterocyclic Chemistry, vol. 14, p. 717-723 (1977).
Непатентный документ 2: Journal Organic Chemistry, vol. 33, p. 3126-3132 (1968).
Непатентный документ 3: Klein WL, and seven others, Alzheimer's disease-affected brain: Presence of oligomeric Aβ ligands (ADDLs) suggests molecular basis for reversible memory loss, Proceeding National Academy of Science USA 2003, Sep 2; 100 (18), p. 10417-10422.
Непатентный документ 4: Nitsch RM, and sixteen others, Antibodies against β-amyloid slow cognitive decline in Alzheimer's disease, Neuron, 2003, May 22; 38, p. 547-554.
Непатентный документ 5: Jarrett JT, and two others, The carboxy terminus of the β amyloid protein is critical for the seeding of amyloid formation: Implications for the pathogenesis of Alzheimer's disease, Biochemistry, 1993, 32 (18), p. 4693-4697.
Непатентный документ 6: Glenner GG, and one other, Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein, Biochemical and biophysical research communications, 1984, May 16, 120 (3), p. 885-890.
Непатентный документ 7: Masters CL, and five others, Amyloid plaque core protein in Alzheimer disease and Down syndrome, Proceding National Academy of Science USA, 1985, Jun, 82 (12), p. 4245-4249.
Непатентный документ 8: Gouras GK, and eleven others, Intraneuronal Aβ42 accumulation in human brain, American Journal of Pathology, 2000, Jan, 156 (1), p. 15-20.
Непатентный документ 9: Scheuner D, and twenty others, Secreted amyloid β-protein similar to that in the senile plaques of Alzheimer's disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's disease, Nature Medicine, 1996, Aug, 2 (8), p. 864-870.
Непатентный документ 10: Forman MS, and four others, Differential effects of the swedish mutant amyloid precursor protein on β-amyloid accumulation and secretion in neurons and nonneuronal cells, The Journal of Biological Chemistry, 1997, Dec 19, 272 (51), p. 32247-32253.
Раскрытие сущности изобретения
Проблемы, решаемые изобретением
Целью настоящего изобретения является создание конденсированного производного аминодигидротиазина, которое является соединением, отличающимся от производного аминодигидротиазина и соединения, обладающего ингибирующей BACE1 активностью, описанных в патентном документе 1, и которое ингибирует продукцию Aβ или ингибирует BACE1 и применимо в качестве профилактического или терапевтического средства от вызываемого белком Aβ нейродегенеративного заболевания, типичным примером которого является деменция Альцгеймера, и его фармацевтическое применение.
В соответствии с настоящим изобретением предлагаются:
[1] Соединение, представленное формулой (I):
[Формула 1]

или его фармацевтически приемлемая соль или сольват, где
кольцо A представляет собой C6-14 арильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, 5-6-членную гетероарильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, или 9-10-членную бензоконденсированную гетероциклическую группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α,
L представляет собой одинарную связь, атом кислорода, группу формулы -NReCO- (где Re представляет собой атом водорода или C1-6 алкильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α), группу формулы -NReSO2- (где Re представляет собой атом водорода или C1-6 алкильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α), группу формулы -NRe- (где Re представляет собой атом водорода или C1-6 алкильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α), C1-6 алкиленовую группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, C2-6 алкениленовую группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, или C2-6 алкиниленовую группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α,
кольцо B представляет собой C3-8 циклоалкильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, C6-14 арильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, или 5-10-членную гетероциклическую группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α,
X представляет собой одинарную связь или C1-3 алкиленовую группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α,
Y представляет собой одинарную связь, -NRY- (где RY представляет собой атом водорода, C1-6 алкильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, C1-6 алкилкарбонильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, C6-14 арилкарбонильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, C1-6 алкилсульфонильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, C6-14 арилсульфонильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, C6-14 арильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, или 5-10-членную гетероциклическую группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α), атом кислорода, атом серы, сульфоксид или сульфон,
Z представляет собой одинарную связь, C1-3 алкиленовую группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, или C2-3 алкениленовую группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α,
R1 и R2 каждый независимо представляет собой атом водорода, C1-6 алкильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, C1-6 алкилкарбонильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, C6-14 арилкарбонильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, C1-6 алкилсульфонильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, C6-14 арилсульфонильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, 3-10-членную карбоциклическую группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, или 5-10-членную гетероциклическую группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, и
R3, R4, R5 и R6 независимо представляют собой атом водорода, атом галогена, гидроксигруппу, C1-6 алкильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, C1-6 алкоксигруппу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, 3-10-членную карбоциклическую группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, или 5-10-членную гетероциклическую группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, или
R4 и R6 вместе могут образовывать кольцо, представленное формулой (II):
[Формула 2]

где Y, Z, R5 и R3 являются такими, как определено выше, и Q представляет собой атом кислорода, метиленовую группу или этиленовую группу
[группа заместителей α: атом водорода, атом галогена, гидроксигруппа, нитрогруппа, C1-6 алкилтиогруппа, C6-14 арильная группа, C6-14 арилоксикарбонильная группа, C6-14 арилкарбонильная группа, цианогруппа, C3-8 циклоалкоксигруппа, C3-8 циклоалкильная группа, C3-8 циклоалкилтиогруппа, сульфониламиногруппа (где сульфониламиногруппа может быть замещена C1-6 алкильной группой), C2-6 алкенильная группа, которая может содержать 1-3 заместителя, выбранных из группы заместителей β, C2-6 алкинильная группа, которая может содержать 1-3 заместителя, выбранных из группы заместителей β, карбамоильная группа, которая может быть замещена одной или двумя C1-6 алкильными группами, C1-6 алкоксигруппа, которая может содержать 1-3 заместителя, выбранных из группы заместителей β, C1-6 алкильная группа, которая может содержать 1-3 заместителя, выбранных из группы заместителей β, и 5-10-членная гетероциклическая группа, которая может содержать 1-3 заместителя, выбранных из группы заместителей β,
группа заместителей β: атом галогена, цианогруппа, гидроксигруппа, C1-6 алкоксигруппа, C1-6 алкильная группа, C3-8 циклоалкильная группа и оксогруппа];
[2] Соединение или его фармацевтически приемлемая соль или сольват по пункту [1] выше, где X представляет собой метилен, который может содержать 1-2 заместителя, выбранных из группы заместителей α;
[3] Соединение или его фармацевтически приемлемая соль или сольват по пункту [1] или [2] выше, где Y представляет собой одинарную связь и Z представляет собой C1-3 алкилен, который может содержать 1-3 заместителя, выбранных из группы заместителей α;
[4] Соединение или его фармацевтически приемлемая соль или сольват по пункту [1] или [2] выше, где Y представляет собой атом кислорода и Z представляет собой C1-3 алкилен, который может содержать 1-3 заместителя, выбранных из группы заместителей α;
[5] Соединение или его фармацевтически приемлемая соль или сольват по пункту [1] или [2] выше, где Y представляет собой атом кислорода и Z представляет собой одинарную связь;
[6] Соединение или его фармацевтически приемлемая соль или сольват по пункту [1] или [2] выше, где Y представляет собой -NRY- (где RY представляет собой C1-6 алкильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, C1-6 алкилкарбонильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, C6-14 арилкарбонильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, C6-14 арилсульфонильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, C6-14 арильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, или 5-10-членную гетероциклическую группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α), атом серы, сульфоксид или сульфон и Z представляет собой одинарную связь, C1-3 алкилен, который может содержать 1-3 заместителя, выбранных из группы заместителей α;
[7] Соединение или его фармацевтически приемлемая соль или сольват по любому из пунктов [1]-[6] выше, где L представляет собой одинарную связь, группу формулы -NReCO- (где Re представляет собой атом водорода или C1-6 алкильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α) или группу формулы -NReSO2- (где Re представляет собой атом водорода или C1-6 алкильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α);
[8] Соединение или его фармацевтически приемлемая соль или сольват по любому из пунктов [1]-[6] выше, где L представляет собой одинарную связь, атом кислорода, C1-6 алкиленовую группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, C2-6 алкениленовую группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, или C2-6 алкиниленовую группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α;
[9] Соединение или его фармацевтически приемлемая соль или сольват по любому из пунктов [1]-[6] выше, где L представляет собой группу формулы -NReCO- (где Re представляет собой атом водорода или C1-6 алкильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α);
[10] Соединение или его фармацевтически приемлемая соль или сольват по любому из пунктов 1-9, которое выбрано из следующих соединений:
1) (+)-N-{3-[(4aR*,8aS*)-2-амино-4,4a,5,6,7,8-гексагидробензо[d][1,3]тиазин-8a-ил]-4-фторфенил}-5-хлорпиридин-2-карбоксамид,
2) (+)-N-{3-[(4aR*,7aS*)-2-амино-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил]-4-фторфенил}-5-хлорпиридин-2-карбоксамид,
3) N-{3-[(4aR*,7aS*)-2-амино-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил]-4-фторфенил}пиридин-2-карбоксамид,
4) N-{3-[(4aR*,7aS*)-2-амино-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил]-4-фторфенил}-5-фторпиридин-2-карбоксамид,
5) N-[3-((4aR*,8aS*)-2-амино-4,4a,5,6,7,8-гексагидробензо[d][1,3]тиазин-8a-ил)-4-фторфенил]-5-цианопиридин-2-карбоксамид,
6) N-[3-((4aR*,7aS*)-2-амино-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]-5-дифторметоксипиразин-2-карбоксамид,
7) N-[3-((4aR*,7aS*)-2-амино-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]-5-фторметоксипиразин-2-карбоксамид,
8) N-[3-((4aR*,7aS*)-2-амино-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]-5-цианопиридин-2-карбоксамид,
9) N-[3-((4aR*,7aS*)-2-амино-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]-5-фторметоксипиридин-2-карбоксамид,
10) N-[3-((4aS*,7aS*)-2-амино-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил]-5-цианопиридин-2-карбоксамид,
11) N-[3-((4aS*,7aS*)-2-амино-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил]-5-дифторметоксипиразин-2-карбоксамид,
12) N-[3-((4aS*,7aS*)-2-амино-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил]-5-хлорпиридин-2-карбоксамид,
13) N-[3-((7S*,7aS*)-2-амино-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил]-5-фторметоксипиразин-2-карбоксамид,
14) N-[3-((4aS*,8aS*)-2-амино-4a,5,7,8-тетрагидро-4H-6-окса-3-тиа-1-азанафталин-8a-ил)-4-фторфенил]-5-цианопиридин-2-карбоксамид,
15) N-[3-((4aS*,8aS*)-2-амино-4a,5,7,8-тетрагидро-4H-6-окса-3-тиа-1-азанафталин-8a-ил)-4-фторфенил]-5-дифторметоксипиразин-2-карбоксамид,
16) N-[3-((4aS*,8aS*)-2-амино-4a,5,7,8-тетрагидро-4H-6-окса-3-тиа-1-азанафталин-8a-ил)-4-фторфенил]-5-хлорпиридин-2-карбоксамид,
17) (+)-N-[3-((4aR*,6S*,7aS*)-2-амино-6-метокси-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]-5-цианопиридин-2-карбоксамид,
18) (+)-N-[3-((4aR*,6R*,7aS*)-2-амино-6-метокси-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]-5-цианопиридин-2-карбоксамид,
19) (+)-N-[3-((4aR*,9aS*)-2-амино-4a,5,6,7,8,9-гексагидро-4H-циклогепта[d][1,3]тиазин-9a-ил)-4-фторфенил]-5-цианопиридин-2-карбоксамид,
20) N-[3-((4aR*,7aS*)-2-амино-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-метоксифенил]-5-хлорпиридин-2-карбоксамид,
21) N-[3-((4aS*,7aS*)-2-амино-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил]-5-дифторметилпиразин-2-карбоксамид,
22) (4aR*,7aS*)-7a-[3-(2-фторпиридин-3-ил)фенил]-6-фенил-4,4a,5,6,7,7a-гексагидропирроло[3,4-d][1,3]тиазин-2-иламин,
23) (4aR*,7aS*)-7a-[3-(2-фторпиридин-3-ил)фенил]-6-пиримидин-2-ил-4,4a,5,6,7,7a-гексагидропирроло[3,4-d][1,3]тиазин-2-иламин,
24) N-[3-((4aS*,5R*,7aS*)-2-амино-5-метил-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил]-5-цианопиридин-2-карбоксамид,
25) N-[3-((4aS*,5R*,7aS*)-2-амино-5-метил-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил]-5-дифторметилпиразин-2-карбоксамид,
26) N-[3-((4aS*,8aS*)-2-амино-4a,5,7,8-тетрагидро-4H-6-окса-3-тиа-1-азанафталин-8a-ил)-4-фторфенил]-5-фторметоксипиразин-2-карбоксамид,
27) N-[3-((4aS*,5R*,7aS*)-2-амино-5-этил-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил]-5-дифторметоксипиразин-2-карбоксамид,
28) N-[3-((4aS,5S,7aS)-2-амино-5-фторметил-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил]-5-дифторметилпиразин-2-карбоксамид,
29) N-[3-((4aS,5S,7aS)-2-амино-5-фторметил-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил]-5-фторметоксипиразин-2-карбоксамид,
30) N-[3-((4aS*,5S*,7aS*)-2-амино-5-фторметил-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил]-5-цианопиридин-2-карбоксамид,
31) N-[3-((4aS*,5S*,8aS*)-2-амино-5-фторметил-4a,5,7,8-тетрагидро-4H-6-окса-3-тиа-1-азанафталин-8a-ил)-4-фторфенил]-5-цианопиридин-2-карбоксамид,
32) N-[3-((4aS*,5S*,8aS*)-2-амино-5-фторметил-4a,5,7,8-тетрагидро-4H-6-окса-3-тиа-1-азанафталин-8a-ил)-4-фторфенил]-5-фторметоксипиразин-2-карбоксамид,
33) N-[3-((4aS*,5S*,8aS*)-2-амино-5-фторметил-4a,5,7,8-тетрагидро-4H-6-окса-3-тиа-1-азанафталин-8a-ил)-4-фторфенил]-5-хлорпиридин-2-карбоксамид,
34) N-[3-((4aS*,6S*,7aS*)-2-амино-6-метокси-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]-5-дифторметилпиразин-2-карбоксамид,
35) N-[3-((4aR*,6R*,7aS*)-2-амино-6-метокси-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]-5-дифторметилпиразин-2-карбоксамид и
36) N-[3-((4aR*,6S*,7aS*)-2-амино-6-фтор-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]-5-дифторметилпиразин-2-карбоксамид;
[11] Фармацевтическая композиция, содержащая соединение или его фармацевтически приемлемую соль или сольват по любому из пунктов [1]-[10] выше в качестве активного компонента;
[12] Фармацевтическая композиция по пункту [11] выше для ингибирования продукции β-амилоидного белка;
[13] Фармацевтическая композиция по пункту [11] выше для ингибирования фермента расщепления бета-сайта белка-предшественника β-амилоида типа 1 (BACE1);
[14] Фармацевтическая композиция по любому из пунктов [11]-[13] выше для лечения нейродегенеративного заболевания и
[15] Фармацевтическая композиция по пункту [14] выше, где указанным нейродегенеративным заболеванием является деменция Альцгеймера или синдром Дауна.
Далее поясняются значения символов, терминов и тому подобное, использованных в данном описании, и дается подробное описание настоящего изобретения.
В данном описании структурная формула соединения может подходящим образом представлять какой-либо изомер. Однако настоящее изобретение включает все изомеры и изомерные смеси, такие как геометрические изомеры, которые могут быть образованы в связи со структурой соединения, оптические изомеры, образование которых основано на наличии асимметрического атома углерода, стереоизомеры и таутомеры. Настоящее изобретение не ограничивается описанием выбранной из соображений удобства химической формулы и может включать любой из изомеров или их смеси. Таким образом, соединение по настоящему изобретению может иметь в молекуле асимметрический атом углерода и существовать в виде оптически активного соединения или рацемата, и настоящее изобретение включает без ограничений как оптически активное соединение, так и рацемат. Хотя могут существовать кристаллические полиморфы соединения, соединение точно также не ограничивается ими и может существовать в виде монокристалла или смеси монокристаллов. Соединение может быть ангидридом или гидратом. Любая из указанных форм включена в формулу изобретения данного описания.
"Атом галогена" в данном описании относится к атому фтора, атому хлора, атому брома, атому йода или тому подобное и предпочтительно представляет собой атом фтора или атом хлора.
"C1-6 алкильная группа" относится к алкильной группе, содержащей 1-6 атомов углерода. Предпочтительные примеры группы включают неразветвленные или разветвленные алкильные группы, такие как метильная группа, этильная группа, н-пропильная группа, изопропильная группа, н-бутильная группа, изобутильная группа, трет-бутильная группа, н-пентильная группа, изопентильная группа, неопентильная группа, н-гексильная группа, 1-метилпропильная группа, 1,2-диметилпропильная группа, 1-этилпропильная группа, 1-метил-2-этилпропильная группа, 1-этил-2-метилпропильная группа, 1,1,2-триметилпропильная группа, 1-метилбутильная группа, 2-метилбутильная группа, 1,1-диметилбутильная группа, 2,2-диметилбутильная группа, 2-этилбутильная группа, 1,3-диметилбутильная группа, 2-метилпентильная группа и 3-метилпентильная группа. Более предпочтительно данная группа представляет собой метильную группу, этильную группу или н-пропильную группу.
"C2-6 алкенильная группа" относится к алкенильной группе, содержащей 2-6 атомов углерода. Предпочтительные примеры группы включают неразветвленные или разветвленные алкенильные группы, такие как винильная группа, аллильная группа, 1-пропенильная группа, изопропенильная группа, 1-бутен-1-ильная группа, 1-бутен-2-ильная группа, 1-бутен-3-ильная группа, 2-бутен-1-ильная группа и 2-бутен-2-ильная группа.
"C2-6 алкинильная группа" относится к алкинильной группе, содержащей 2-6 атомов углерода. Предпочтительные примеры группы включают неразветвленные или разветвленные алкинильные группы, такие как этинильная группа, 1-пропинильная группа, 2-пропинильная группа, бутинильная группа, пентинильная группа и гексинильная группа.
"C1-6 алкоксигруппа" относится к алкильной группе, содержащей 1-6 атомов углерода, в которой один атом водорода заменен на атом кислорода. Примеры группы включают метоксигруппу, этоксигруппу, н-пропоксигруппу, изопропоксигруппу, н-бутоксигруппу, изобутоксигруппу, втор-бутоксигруппу, трет-бутоксигруппу, н-пентоксигруппу, изопентоксигруппу, втор-пентоксигруппу, трет-пентоксигруппу, н-гексоксигруппу, изогексоксигруппу, 1,2-диметилпропоксигруппу, 2-этилпропоксигруппу, 1-метил-2-этилпропоксигруппу, 1-этил-2-метилпропоксигруппу, 1,1,2-триметилпропоксигруппу, 1,1-диметилбутоксигруппу, 2,2-диметилбутоксигруппу, 2-этилбутоксигруппу, 1,3-диметилбутоксигруппу, 2-метилпентоксигруппу, 3-метилпентоксигруппу и гексилоксигруппу.
"C1-6 алкилтиогруппа" относится к алкильной группе, содержащей 1-6 атомов углерода, в которой один атом водорода заменен на атом серы. Примеры группы включают метилтиогруппу, этилтиогруппу, н-пропилтиогруппу, изопропилтиогруппу, н-бутилтиогруппу, изобутилтиогруппу, трет-бутилтиогруппу, н-пентилтиогруппу, изопентилтиогруппу, неопентилтиогруппу, н-гексилтиогруппу и 1-метилпропилтиогруппу.
"C1-6 алкилсульфонильная группа" относится к алкильной группе, содержащей 1-6 атомов углерода, в которой один атом водорода заменен на сульфонильную группу. Примеры группы включают метилсульфонильную группу, этилсульфонильную группу, н-пропилсульфонильную группу, изопропилсульфонильную группу, н-бутилсульфонильную группу, изобутилсульфонильную группу, трет-бутилсульфонильную группу, н-пентилсульфонильную группу, изопентилсульфонильную группу, неопентилсульфонильную группу, н-гексилсульфонильную группу и 1-метилпропилсульфонильную группу.
"C1-6 алкилкарбонильная группа" относится к алкильной группе, содержащей 1-6 атомов углерода, в которой один атом водорода заменен на карбонильную группу. Предпочтительные примеры группы включают ацетильную группу, пропионильную группу и бутирильную группу.
"C6-14 арильная группа" относится к ароматической углеводородной циклической группе, содержащей 6-14 атомов углерода. Примеры группы включают фенильную группу, нафтильную группу и антрильную группу. Фенильная группа является особенно предпочтительной.
"C7-12 аралкильная группа" относится к группе, содержащей 7-12 атомов углерода, в которой ароматический углеводородный цикл, такой как фенильная группа или нафтильная группа, замещен C1-6 алкильной группой. Примеры группы включают бензильную группу, фенетильную группу, фенилпропильную группу и нафтилметильную группу. Бензильная группа является особенно предпочтительной.
"C6-14 арилоксикарбонильная группа" относится к группе, в которой оксикарбонил связан с ароматической углеводородной циклической группой, содержащей 6-14 атомов углерода. Предпочтительные примеры группы включают фенилоксикарбонильную группу, нафтилоксикарбонильную группу и антрилоксикарбонильную группу. Фенилоксикарбонильная группа является более предпочтительной.
"C6-14 арилкарбонильная группа" относится к группе, в которой карбонильная группа связана с ароматической углеводородной циклической группой, содержащей 6-14 атомов углерода. Предпочтительные примеры группы включают бензоильную группу и нафтоильную группу. Бензоильная группа является более предпочтительной.
"C6-14 арилсульфонильная группа" относится к группе, в которой сульфонильная группа связана с ароматической углеводородной циклической группой, содержащей 6-14 атомов углерода. Предпочтительные примеры группы включают бензолсульфонильную группу и нафтилсульфонильную группу. Бензолсульфонильная группа является более предпочтительной.
"C3-8 циклоалкильная группа" относится к циклической алкильной группе, содержащей 3-8 атомов углерода. Предпочтительные примеры группы включают циклопропильную группу, циклобутильную группу, циклопентильную группу, циклогексильную группу, циклогептильную группу и циклооктильную группу.
"C3-8 циклоалкоксигруппа" относится к циклической алкильной группе, содержащей 3-8 атомов углерода, в которой один атом водорода заменен на атом кислорода. Примеры группы включают циклопропоксигруппу, циклобутоксигруппу, циклопентоксигруппу, циклогексоксигруппу, циклогептилоксигруппу и циклооктилоксигруппу.
"C3-8 циклоалкилтиогруппа" относится к циклической алкильной группе, содержащей 3-8 атомов углерода, в которой один атом водорода заменен на атом серы. Примеры группы включают циклопропилтиогруппу, циклобутилтиогруппу, циклопентилтиогруппу, циклогексилтиогруппу, циклогептилтиогруппу и циклооктилтиогруппу.
"5-10-Членная гетероциклическая группа" относится к циклической группе, содержащей гетероатом и имеющей суммарно 5-10 членов. Предпочтительные примеры группы включают пиперидинильную группу, пирролидинильную группу, азепинильную группу, азоканильную группу, пиперазинильную группу, 1,4-диазепанильную группу, морфолинильную группу, тиоморфолинильную группу, пирролильную группу, имидазолильную группу, пиразолильную группу, пиридинильную группу, пиридазинильную группу, пиримидинильную группу, пиразинильную группу, триазолильную группу, триазинильную группу, тетразолильную группу, изоксазолильную группу, оксазолильную группу, оксадиазолильную группу, изотиазолильную группу, тиазолильную группу, тиадиазолильную группу, фурильную группу, тиенильную группу, хинолинильную группу, изохинолинильную группу, бензофурильную группу, бензопиранильную группу, бензимидазолильную группу, бензотриазолильную группу, бензизотиазолильную группу, индолинильную группу, изоиндолинильную группу, хроманильную группу, изохроманильную группу, 1,3-диоксаинданильную группу и 1,4-диоксатетралинильную группу.
"5-6-Членная гетероарильная группа" относится к "5-10-членной гетероциклической группе", которая является ароматической циклической группой, содержащей гетероатом и имеющей суммарно 5-6 членов. Примеры группы включают пирролильную группу, имидазолильную группу, пиразолильную группу, пиридинильную группу, пиридазинильную группу, пиримидинильную группу, пиразинильную группу, триазолильную группу, триазинильную группу, тетразолильную группу, изоксазолильную группу, оксазолильную группу, оксадиазолильную группу, изотиазолильную группу, тиазолильную группу, тиадиазолильную группу, фурильную группу и тиенильную группу.
"9-10-Членная бензоконденсированная гетероциклическая группа" относится к "5-10-членной гетероциклической группе", которая является циклической группой, содержащей гетероатом и имеющей суммарно 9-10 членов, конденсированной с бензольным кольцом. Предпочтительные примеры группы включают индолинильную группу, изоиндолинильную группу, хроманильную группу, изохроманильную группу, 1,3-диоксаинданильную группу и 1,4-диоксатетралинильную группу.
"3-10-Членная карбоциклическая группа" относится к карбоциклической группе, имеющей суммарно 3-10 членов. Предпочтительные примеры группы включают циклопропильную группу, циклобутильную группу, циклопентильную группу, циклогексильную группу, циклогептильную группу, циклооктильную группу, спиро[3,4]октанильную группу, деканильную группу, инданильную группу, 1-аценафтенильную группу, циклопентациклооктенильную группу, бензоциклооктенильную группу, инденильную группу, тетрагидронафтильную группу, 6,7,8,9-тетрагидро-5H-бензоциклогептенильную группу и 1,4-дигидронафталенильную группу.
"C1-6 алкиленовая группа" относится к двухвалентной группе, полученной удалением любого одного атома водорода из "C1-6 алкильной группы", определенной выше. Примеры группы включают метиленовую группу, 1,2-этиленовую группу, 1,1-этиленовую группу, 1,3-пропиленовую группу, тетраметиленовую группу, пентаметиленовую группу и гексаметиленовую группу.
"C2-6 алкениленовая группа" относится к двухвалентной группе, полученной удалением любого одного атома водорода из "C2-6 алкенильной группы", определенной выше. Примеры группы включают 1,2-виниленовую группу (этениленовую группу), пропениленовую группу, бутениленовую группу, пентениленовую группу и гексениленовую группу.
"C2-6 алкиниленовая группа" относится к двухвалентной группе, полученной удалением любого одного атома водорода из "C2-6 алкинильной группы", определенной выше. Примеры группы включают этиниленовую группу, пропиниленовую группу, бутиниленовую группу, пентиниленовую группу и гексиниленовую группу.
Примеры "C1-3 алкиленовой группы" включают метиленовую группу, этиленовую группу и пропиленовую группу.
Примеры "C2-3 алкениленовой группы" включают 1,2-виниленовую группу (этениленовую группу) и пропениленовую группу.
Примеры "C2-3 алкиниленовой группы" включают этиниленовую группу и пропиниленовую группу.
Примеры сульфониламиногруппы, которая может быть замещена C1-6 алкильной группой в "сульфониламиногруппе (где сульфониламиногруппа может быть замещена C1-6 алкильной группой)", включают метилсульфонилметиламиногруппу, этилсульфонилметиламиногруппу и этилсульфонилэтиламиногруппу.
"Группа заместителей α" относится к атому водорода, атому галогена, гидроксигруппе, нитрогруппе, C1-6 алкилтиогруппе, C6-14 арильной группе, C6-14 арилоксикарбонильной группе, C6-14 арилкарбонильной группе, цианогруппе, C3-8 циклоалкоксигруппе, C3-8 циклоалкильной группе, C3-8 циклоалкилтиогруппе, сульфониламиногруппе (где сульфониламиногруппа может быть замещена C1-6 алкильной группой), C2-6 алкенильной группе, которая может содержать 1-3 заместителя, выбранных из группы заместителей β, C2-6 алкинильной группе, которая может содержать 1-3 заместителя, выбранных из группы заместителей β, карбамоильной группе, которая может быть замещена одной или двумя C1-6 алкильными группами, C1-6 алкоксигруппе, которая может содержать 1-3 заместителя, выбранных из группы заместителей β, C1-6 алкильной группе, которая может содержать 1-3 заместителя, выбранных из группы заместителей β, и 5-10-членной гетероциклической группе, которая может содержать 1-3 заместителя, выбранных из группы заместителей β.
"Группа заместителей β" относится к атому галогена, цианогруппе, гидроксигруппе, C1-6 алкоксигруппе, C1-6 алкильной группе, C3-8 циклоалкильной группе и оксогруппе.
Конденсированное производное аминодигидротиазина формулы (I) в соответствии с настоящим изобретением может представлять собой фармацевтически приемлемую соль. Конкретные примеры фармацевтически приемлемой соли включают соли неорганических кислот (такие как сульфаты, нитраты, перхлораты, фосфаты, карбонаты, бикарбонаты, гидрофториды, гидрохлориды, гидробромиды и гидройодиды), органические карбоксилаты (такие как ацетаты, оксалаты, малеаты, тартраты, фумараты и цитраты), органические сульфонаты (такие как метансульфонаты, трифторметансульфонаты, этансульфонаты, бензолсульфонаты, толуолсульфонаты и камфорсульфонаты), соли аминокислот (такие как аспартаты и глутаматы), соли четвертичных аминов, соли щелочных металлов (такие как натриевые соли и калиевые соли) и соли щелочноземельных металлов (такие как магниевые соли и кальциевые соли).
Конденсированное производное аминодигидротиазина формулы (I) или фармацевтически приемлемая соль по настоящему изобретению может представлять собой сольват. Примеры сольвата включают гидрат.
Соединение (I) не ограничивается конкретным изомером и включает все возможные изомеры (такие как кето-енольный изомер, имин-енаминовый изомер, диастереоизомер, оптический изомер и ротамер) и рацематы. Например, соединение (I), где R1 представляет собой водород, включает следующие таутомеры.
[Формула 3]

Конденсированное производное аминодигидротиазина формулы (I) по настоящему изобретению предпочтительно является соединением формулы (I), где X представляет собой метилен, который может содержать 1-2 заместителя, выбранных из группы заместителей α. Соединение формулы (I), где Y представляет собой одинарную связь и Z представляет собой C1-3 алкилен, который может содержать 1-3 заместителя, выбранных из группы заместителей α; где Y представляет собой атом кислорода и Z представляет собой C1-3 алкилен, который может содержать 1-3 заместителя, выбранных из группы заместителей α; или где Y представляет собой атом кислорода и Z представляет собой одинарную связь, является особенно предпочтительным.
Конденсированное производное аминодигидротиазина формулы (I) по настоящему изобретению предпочтительно является соединением формулы (I), где L представляет собой одинарную связь, группу формулы -NReCO- (где Re представляет собой атом водорода или C1-6 алкильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α) или группу формулы -NReSO2- (где Re представляет собой атом водорода или C1-6 алкильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α) или где L представляет собой одинарную связь, атом кислорода, C1-6 алкиленовую группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, C2-6 алкениленовую группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, или C2-6 алкиниленовую группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α. Соединение, где L представляет собой группу формулы -NReCO- (где Re представляет собой атом водорода или C1-6 алкильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α), является особенно предпочтительным.
Предпочтительные соединения по настоящему изобретению включают следующие соединения:
1) (+)-N-{3-[(4aR*,8aS*)-2-амино-4,4a,5,6,7,8-гексагидробензо[d][1,3]тиазин-8a-ил]-4-фторфенил}-5-хлорпиридин-2-карбоксамид,
2) (+)-N-{3-[(4aR*,7aS*)-2-амино-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил]-4-фторфенил}-5-хлорпиридин-2-карбоксамид,
3) N-{3-[(4aR*,7aS*)-2-амино-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил]-4-фторфенил}пиридин-2-карбоксамид,
4) N-{3-[(4aR*,7aS*)-2-амино-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил]-4-фторфенил}-5-фторпиридин-2-карбоксамид,
5) N-[3-((4aR*,8aS*)-2-амино-4,4a,5,6,7,8-гексагидробензо[d][1,3]тиазин-8a-ил)-4-фторфенил]-5-цианопиридин-2-карбоксамид,
6) N-[3-((4aR*,7aS*)-2-амино-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]-5-дифторметоксипиразин-2-карбоксамид,
7) N-[3-((4aR*,7aS*)-2-амино-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]-5-фторметоксипиразин-2-карбоксамид,
8) N-[3-((4aR*,7aS*)-2-амино-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]-5-цианопиридин-2-карбоксамид,
9) N-[3-((4aR*,7aS*)-2-амино-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]-5-фторметоксипиридин-2-карбоксамид,
10) N-[3-((4aS*,7aS*)-2-амино-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил]-5-цианопиридин-2-карбоксамид,
11) N-[3-((4aS*,7aS*)-2-амино-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил]-5-дифторметоксипиразин-2-карбоксамид,
12) N-[3-((4aS*,7aS*)-2-амино-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил]-5-хлорпиридин-2-карбоксамид,
13) N-[3-((7S*,7aS*)-2-амино-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил]-5-фторметоксипиразин-2-карбоксамид,
14) N-[3-((4aS*,8aS*)-2-амино-4a,5,7,8-тетрагидро-4H-6-окса-3-тиа-1-азанафталин-8a-ил)-4-фторфенил]-5-цианопиридин-2-карбоксамид,
15) N-[3-((4aS*,8aS*)-2-амино-4a,5,7,8-тетрагидро-4H-6-окса-3-тиа-1-азанафталин-8a-ил)-4-фторфенил]-5-дифторметоксипиразин-2-карбоксамид,
16) N-[3-((4aS*,8aS*)-2-амино-4a,5,7,8-тетрагидро-4H-6-окса-3-тиа-1-азанафталин-8a-ил)-4-фторфенил]-5-хлорпиридин-2-карбоксамид,
17) (+)-N-[3-((4aR*,6S*,7aS*)-2-амино-6-метокси-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]-5-цианопиридин-2-карбоксамид,
18) (+)-N-[3-((4aR*,6R*,7aS*)-2-амино-6-метокси-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]-5-цианопиридин-2-карбоксамид,
19) (+)-N-[3-((4aR*,9aS*)-2-амино-4a,5,6,7,8,9-гексагидро-4H-циклогепта[d][1,3]тиазин-9a-ил)-4-фторфенил]-5-цианопиридин-2-карбоксамид,
20) N-[3-((4aR*,7aS*)-2-амино-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-метоксифенил]-5-хлорпиридин-2-карбоксамид,
21) N-[3-((4aS*,7aS*)-2-амино-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил]-5-дифторметилпиразин-2-карбоксамид,
22) (4aR*,7aS*)-7a-[3-(2-фторпиридин-3-ил)фенил]-6-фенил-4,4a,5,6,7,7a-гексагидропирроло[3,4-d][1,3]тиазин-2-иламин,
23) (4aR*,7aS*)-7a-[3-(2-фторпиридин-3-ил)фенил]-6-пиримидин-2-ил-4,4a,5,6,7,7a-гексагидропирроло[3,4-d][1,3]тиазин-2-иламин,
24) N-[3-((4aS*,5R*,7aS*)-2-амино-5-этил-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил]-5-цианопиридин-2-карбоксамид,
25) N-[3-((4aS*,5R*,7aS*)-2-амино-5-этил-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил]-5-дифторметилпиразин-2-карбоксамид,
26) N-[3-((4aS*,8aS*)-2-амино-4a,5,7,8-тетрагидро-4H-6-окса-3-тиа-1-азанафталин-8a-ил)-4-фторфенил]-5-фторметоксипиразин-2-карбоксамид,
27) N-[3-((4aS*,5R*,7aS*)-2-амино-5-этил-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил]-5-дифторметоксипиразин-2-карбоксамид,
28) N-[3-((4aS,5S,7aS)-2-амино-5-фторметил-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил]-5-дифторметилпиразин-2-карбоксамид,
29) N-[3-((4aS,5S,7aS)-2-амино-5-фторметил-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил]-5-фторметоксипиразин-2-карбоксамид,
30) N-[3-((4aS*,5S*,7aS*)-2-амино-5-фторметил-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил]-5-цианопиридин-2-карбоксамид,
31) N-[3-((4aS*,5S*,8aS*)-2-амино-5-фторметил-4a,5,7,8-тетрагидро-4H-6-окса-3-тиа-1-азанафталин-8a-ил)-4-фторфенил]-5-цианопиридин-2-карбоксамид,
32) N-[3-((4aS*,5S*,8aS*)-2-амино-5-фторметил-4a,5,7,8-тетрагидро-4H-6-окса-3-тиа-1-азанафталин-8a-ил)-4-фторфенил]-5-фторметоксипиразин-2-карбоксамид,
33) N-[3-((4aS*,5S*,8aS*)-2-амино-5-фторметил-4a,5,7,8-тетрагидро-4H-6-окса-3-тиа-1-азанафталин-8a-ил)-4-фторфенил]-5-хлорпиридин-2-карбоксамид,
34) N-[3-((4aS*,6S*,7aS*)-2-амино-6-метокси-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]-5-дифторметилпиразин-2-карбоксамид,
35) N-[3-((4aR*,6R*,7aS*)-2-амино-6-метокси-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]-5-дифторметилпиразин-2-карбоксамид и
36) N-[3-((4aR*,6S*,7aS*)-2-амино-6-фтор-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]-5-дифторметилпиразин-2-карбоксамид.
Ниже описаны способы получения соединения формулы (I) [далее называемого соединением (I); соединение, представленное другой формулой, описывается аналогичным образом] или его фармацевтически приемлемой соли по настоящему изобретению.
Соединение, представленное формулой (I):
[Формула 4]
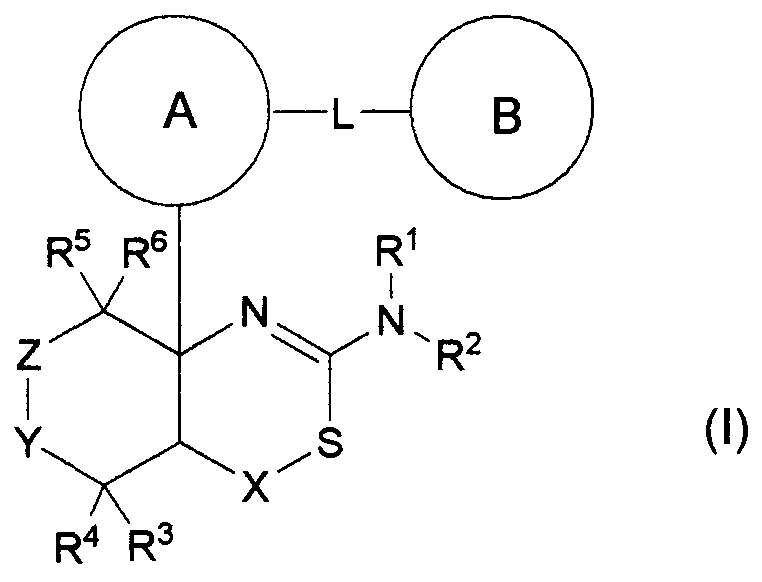
(где кольцо A, кольцо B, R1, R2, R3, R4, R5, R6, L, X, Y и Z являются такими, как определено выше), или его промежуточное соединение синтезируют, например, общими способами получения 1-15, описанными ниже.
"Уходящая группа" в исходном соединении, используемом для получения соединения (I) по настоящему изобретению, может представлять собой любую уходящую группу, используемую для реакции нуклеофильного замещения. Предпочтительные примеры уходящей группы включают атом галогена, C1-6 алкилсульфонилоксигруппу, которая может быть замещена указанной выше группой заместителей α, и арилсульфонилоксигруппу, которая может быть замещена указанной выше группой заместителей α. Конкретные примеры уходящей группы включают атом хлора, атом брома, атом йода, метансульфонилоксигруппу, трифторметансульфонилоксигруппу и п-толуолсульфонилоксигруппу.
1. Общий способ получения 1:
[Формула 5]

На данной схеме R7 представляет собой C1-6 алкильную группу, такую как метильная группа или этильная группа, C7-12 аралкильную группу, такую как бензильная группа, или тому подобное, LV является уходящей группой и представляет собой, например, атом галогена (такой как атом хлора, атом брома или атом йода) или, например, сульфонилоксигруппу, такую как метансульфонилоксигруппа, п-толуолсульфонилоксигруппа или трифторметансульфонилоксигруппа (представленная на схеме в виде TfO), и кольцо A, R3, R4, R5, R6, Y и Z являются такими, как определено выше.
Общий способ получения 1 представляет собой способ получения соединения (1-7), которое является промежуточным соединением в синтезе соединения (I) по настоящему изобретению, получаемым из соединения (1-1) в качестве исходного вещества путем проведения нескольких стадий от стадии 1-1 до стадии 1-6.
Соединение (1-1) может быть коммерчески доступным продуктом, используемым как он есть, может быть также получено из коммерчески доступного продукта способом, известным специалисту в данной области техники, и, кроме того, может быть получено способом, описанным в примерах получения в разделе "Примеры".
Стадия 1-1:
Данная стадия является стадией получения соединения (1-2) трифторметансульфонилированием соединения (1-1).
Реакция на данной стадии может быть осуществлена в таких же условиях, какие обычно используют в реакции трифторметансульфонилирования карбонильного соединения (такие как условия, описанные в J. Org. Chem., 57, 6972-6975 (1992), Tetrahedron Letters., 40, 8133-8136 (1999) and Tetrahedron., 61, 4128-4140 (2005)).
В частности, соединение (1-2) может быть получено действием основания на соединение (1-1), а затем взаимодействием соединения, например, с N-фенилтрифторметансульфонимидом или трифторметансульфоновым ангидридом. Данная реакция может быть осуществлена действием одного или нескольких эквивалентов основания на соединение (1-1) в органическом растворителе, таком как, например, простой эфир, тетрагидрофуран, 1,4-диоксан, 1,2-диметоксиэтан, дихлорметан, 1,2-дихлорэтан, бензол или толуол. Примеры используемого основания включают гидрид натрия, LDA (диизопропиламид лития), бис(триметилсилил)амид лития, диизопропилэтиламин, пиридин и 2,6-лутидин. Время реакции особо не ограничивают, и оно обычно составляет от 5 минут до 24 часов, предпочтительно от 5 минут до 12 часов. Температура реакции обычно составляет от -100°C до комнатной температуры, а более предпочтительно от -78°C до комнатной температуры.
Стадия 1-2:
Данная стадия является стадией получения соединения (1-3) по реакции сочетания соединения (1-2) с использованием переходного металла.
Данная реакция может быть осуществлена в условиях, обычно применяемых в реакции сочетания с использованием переходного металла (такой как реакция Сузуки-Мияуры или реакция Стилле).
Примеры реакции с использованием борорганического реагента в качестве металлоорганического соединения включают реакции, описанные в таких документах, как Tetrahedron: Asymmetry 16 (2005) 2, 528-539 and Org. Lett. 6 (2004) 2, 277-279. Примеры реакции с использованием оловоорганического реагента включают реакцию, описанную в таком документе, как Tetrahedron 61 (2005) 16, 4128-4140. Примеры реакции с использованием цинкорганического реагента в качестве металлоорганического соединения включают реакцию, описанную в таком документе, как Tetrahedron 61 (2005) 16, 4128-4140. Металлоорганический катализатор, используемый в данной реакции, особо не ограничивают. Предпочтительные примеры металлоорганического катализатора включают тетракис(трифенилфосфин)палладий(0), дихлорбис(трифенилфосфин)палладий(II), дихлорид [1,1'-бис(дифенилфосфино)ферроцен]палладия(II), бис(трет-бутилфосфин)палладий(0), ацетат палладия(II) и [1,3-бис(дифенилфосфино)пропан]никель(II). Количество используемого металлоорганического катализатора составляет примерно 0,001-0,1 эквивалента относительно исходного вещества. Металлоорганическое соединение особо не ограничивают. Предпочтительные примеры металлоорганического соединения включают оловоорганические реагенты, такие как арилтри-н-бутилолово, и борорганические реагенты, такие как арилбороновая кислота. Количество используемого металлоорганического соединения составляет один-пять эквивалентов относительно исходного вещества. Растворитель, используемый в данной реакции, особо не ограничивают, лишь бы он не замедлял реакцию. Предпочтительные примеры растворителя включают бензол, толуол, ксилол, N,N-диметилформамид, 1-метил-2-пирролидон, тетрагидрофуран, 1,4-диоксан, ацетонитрил и пропионитрил. Температуру реакции особо не ограничивают, и она обычно находится, например, в пределах от температуры охлаждения льдом до температуры кипения растворителя с обратным холодильником, а предпочтительно от комнатной температуры до температуры кипения растворителя с обратным холодильником. Время реакции особо не ограничивают, и оно составляет обычно от 0,5 до 48 часов, предпочтительно от 0,5 до 24 часов.
Более предпочтительный результат, такой как повышенный выход, может быть достигнут осуществлением данной реакции в присутствии основания. Такое основание особо не ограничивают. Предпочтительные примеры основания включают такие основания, как карбонат натрия, карбонат калия, карбонат цезия, фосфат калия и их растворы, и триэтиламин.
Стадия 1-3:
Данная стадия является стадией получения спиртового соединения (1-4) путем восстановления сложноэфирного соединения (1-3). Спиртовое соединение (1-4) может быть получено из сложноэфирного соединения (1-3) способом, известным специалисту в данной области техники.
Примеры восстановителя, используемого в данной реакции, включают алюмогидрид лития, боргидрид лития и диизобутилалюмогидрид. Температуру реакции особо не ограничивают, и она находится обычно в пределах от -78°C до температуры кипения растворителя с обратным холодильником, а предпочтительно от -78°C до комнатной температуры. Растворитель, используемый в данной реакции, особо не ограничивают, лишь бы он не замедлял реакцию и позволял исходному веществу растворяться в нем до определенной степени. Предпочтительные примеры растворителя включают тетрагидрофуран, диэтиловый эфир, толуол и дихлорметан.
Стадия 1-4:
Данная стадия является стадией получения соединения (1-5) путем преобразования гидроксильной группы соединения (1-4) в уходящую группу.
Примеры уходящей группы включают атомы галогенов (такие как атом хлора, атом брома и атом йода) и сульфонилоксигруппы, такие как метансульфонилоксигруппа, п-толуолсульфонилоксигруппа и трифторметансульфонилоксигруппа.
Реакция может быть осуществлена в таких же условиях, какие обычно используют в реакции преобразования гидроксильной группы в такую уходящую группу. Если уходящая группа представляет собой, например, атом галогена, то соединение (1-5) может быть получено путем взаимодействия соединения (1-4), например, с тионилхлоридом, тионилбромидом, трибромидом фосфора или тетрагалогенметантрифенилфосфином. Растворитель, используемый в данной реакции, особо не ограничивают, лишь бы он не замедлял реакцию и позволял исходному веществу растворяться в нем до определенной степени. Предпочтительные примеры растворителя включают бензол, толуол, ксилол, дихлорметан и хлороформ. Температура реакции находится обычно в пределах от -78°C до температуры кипения растворителя с обратным холодильником, а предпочтительно от температуры охлаждения льдом до температуры кипения растворителя с обратным холодильником. Время реакции особо не ограничивают, и оно составляет обычно от 5 минут до 48 часов, предпочтительно от 5 минут до 12 часов.
Если уходящей группой является сульфонилоксигруппа, то соединение (1-5) может быть получено путем взаимодействия соединения (1-4), например, с метансульфонилхлоридом, п-толуолсульфонилхлоридом или трифторметансульфоновым ангидридом.
Растворитель, используемый в данной реакции, особо не ограничивают, лишь бы он не замедлял реакцию и позволял исходному веществу растворяться в нем до определенной степени. Предпочтительные примеры растворителя включают тетрагидрофуран, толуол, ксилол, дихлорметан, хлороформ и N,N-диметилформамид. Температура реакции находится обычно в пределах от -78°C до температуры кипения растворителя с обратным холодильником, а предпочтительно от -78°C до комнатной температуры. Положительный результат, такой как повышенный выход, может быть достигнут добавлением основания. Используемое основание особо не ограничивают, лишь бы оно не замедляло реакцию. Предпочтительные примеры основания включают карбонат натрия, карбонат калия, триэтиламин, пиридин и диизопропилэтиламин.
Стадия 1-5:
Данная стадия является стадией получения соединения (1-6) из соединения (1-5). Производное тиомочевины (1-6) может быть получено из соединения (1-5) способом, известным специалисту в данной области техники.
В частности, соединение (1-6) может быть получено, например, путем взаимодействия соединения (1-5) с тиомочевиной в растворителе. Данная реакция может быть осуществлена действием одного или нескольких эквивалентов тиомочевины на соединение (1-5) в органическом растворителе, таком как, например, этанол, 1-пропанол, 2-пропанол, 1-бутанол, тетрагидрофуран, 1,4-диоксан или N,N-диметилформамид. Время реакции особо не ограничивают, и оно составляет обычно от 5 минут до 24 часов, предпочтительно от 5 минут до 12 часов. Температура реакции находится обычно в пределах от 0°C до 150°C, а более предпочтительно от комнатной температуры до 100°C.
Стадия 1-6:
Данная стадия представляет собой способ получения соединения (1-7) циклизацией соединения (1-6) кислотой.
Реакционный раствор особо не ограничивают, лишь бы он не замедлял реакцию и позволял исходному веществу растворяться в нем до определенной степени. Например, реакция может быть осуществлена действием подходящей кислоты в количестве от одного эквивалента до большого избытка на соединение (1-6) в присутствии или в отсутствие растворителя, такого как бензол, толуол или дихлорметан. Кроме того, кислота может быть также использована в качестве растворителя. Примеры используемой кислоты включают серную кислоту, трифторуксусную кислоту, метансульфоновую кислоту, трифторметансульфоновую кислоту и их смеси. Время реакции особо не ограничивают, и оно составляет обычно от 1 до 72 часов, предпочтительно от 1 до 48 часов. Температура реакции обычно находится в пределах от температуры охлаждения льдом до температуры кипения растворителя с обратным холодильником.
Аминогруппа в соединении (1-7) может быть преобразована в соответствующую группу -NR1R2 в формуле (I), в которой R1 и R2 являются замещенными, дополнительным взаимодействием соединения (1-7) с соответствующим галогенидом или тому подобное, таким как C1-6 алкилгалогенид, C1-6 алкилкарбонилгалогенид, C6-14 арилкарбонилгалогенид, C1-6 алкилсульфонилгалогенид, C6-14 арилсульфонилгалогенид, 3-10-членный карбоциклический галогенид или 5-10-членный гетероциклический галогенид.
2. Общий способ получения 2:
Способ 2A:
[Формула 6]

На данной схеме кольцо A, R3, R4, R5, R6, Y и Z являются такими, как определено выше.
Общий способ получения 2 состоит из указанного выше способа 2A и описанного ниже способа 2B. Способ 2A представляет собой способ получения соединения общей формулы (1-4), которое является промежуточным соединением в синтезе соединения (I) по настоящему изобретению, получаемым из соединения (2-1) в качестве исходного вещества путем проведения нескольких стадий от стадии 2A-1 до стадии 2A-3.
Соединение (2-1) может быть коммерчески доступным продуктом, используемым как он есть, может быть также получено из коммерчески доступного продукта способом, известным специалисту в данной области техники, и, кроме того, может быть получено способом, описанным в примерах получения в разделе "Примеры".
Стадия 2A-1:
Данная стадия является стадией получения соединения (2-2) из соединения (2-1). Данная реакция может быть осуществлена в таких же условиях, какие обычно используют в реакции синтеза соединения (2-2) из карбонильного соединения (такие как условия, описанные в J. Org. Chem., 47, 3597-3607 (1982)).
Стадия 2A-2:
Данная стадия является стадией синтезирования соединения (2-3) из соединения (2-2) в качестве исходного вещества с использованием способа получения, описанного выше (стадия 1-2).
Стадия 2A-3:
Данная стадия является стадией получения спиртового соединения (1-4) восстановлением альдегидного соединения (2-3).
Спиртовое соединение (1-4) может быть получено из альдегидного соединения (2-3) способом, известным специалисту в данной области техники. Примеры восстановителя, используемого в данной реакции, включают боргидрид натрия, цианоборгидрид натрия и триацетоксиборгидрид натрия. Температуру реакции особо не ограничивают, и она находится обычно в пределах от -78°C до температуры кипения растворителя с обратным холодильником, а предпочтительно от -20°C до комнатной температуры. Растворитель, используемый в данной реакции, особо не ограничивают, лишь бы он не замедлял реакцию и позволял исходному веществу растворяться в нем до определенной степени. Предпочтительные примеры растворителя включают метанол, этанол, тетрагидрофуран, простой эфир, толуол и дихлорметан.
Способ 2B:
[Формула 7]

На данной схеме кольцо A, R3, R4, R5, R6, R7, Y и Z являются такими, как определено выше.
Как описано выше в способе 2B, соединение (1-4) может быть также получено преобразованием соединения (1-3) в соединение (2-4) и восстановлением полученного соединения.
Соединение (1-3) может быть получено из коммерчески доступного продукта общим способом получения 1 и может быть также получено способом, описанным в примерах получения в разделе "Примеры".
Стадия 2B-1:
Данная стадия является стадией получения соединения (2-4) щелочным гидролизом соединения (1-3).
Реакция может быть осуществлена в таких же условиях реакции, какие описаны, например, в J. Med. Chem., 33 (9), 2621-2629 (1990).
В частности, соединение (2-4) может быть получено добавлением основания, такого как гидроксид натрия, к раствору соединения (1-3), перемешиванием смеси в течение от нескольких часов до одних суток и затем обработкой раствора кислотой, такой как, например, раствор уксусной кислоты.
Растворитель, используемый в данной реакции, особо не ограничивают, лишь бы он не замедлял реакцию и позволял исходному веществу растворяться в нем до определенной степени. Примеры растворителя включают метанол, этанол, 2-пропанол, тетрагидрофуран и 1,4-диоксан. Используемое основание особо не ограничивают, и предпочтительно оно представляет собой, например, гидроксид натрия, гидроксид калия или гидроксид лития. Количество используемого основания составляет от одного эквивалента до большого избытка и предпочтительно от 1 до 20 эквивалентов относительно соединения (1-3). Время реакции особо не ограничивают, и оно составляет обычно 1-24 часа, предпочтительно 1-6 часов. Температуру реакции особо не ограничивают, и она находится обычно в пределах от комнатной температуры до температуры кипения растворителя с обратным холодильником.
Стадия 2B-2:
Данная стадия является стадией получения соединения (1-4) восстановлением соединения (2-4).
Соединение (1-4) может быть получено преобразованием соединения (2-4) в смешанный ангидрид кислоты и затем взаимодействием смешанного ангидрида кислоты с боргидридом натрия. Смешанный ангидрид кислоты может быть синтезирован способом, известным специалисту в данной области техники. Синтез выполняют путем взаимодействия соединения (2-4) с хлорформиатом, таким как этилхлорформиат, в присутствии основания, такого как, например, триэтиламин. Используют один-два эквивалента хлорформиата и основания относительно соединения (2-4). Температура реакции составляет от -30°C до комнатной температуры, предпочтительно от -20°C до комнатной температуры.
Стадию взаимодействия смешанного ангидрида кислоты с восстановителем, таким как боргидрид натрия, выполняют, например, реакцией в растворителе, таком как тетрагидрофуран или 1,2-диметоксиэтан, или в смешанном растворе из растворителя и воды. Восстановитель, такой как боргидрид натрия, используют в количестве от одного эквивалента до большого избытка относительно смешанного ангидрида кислоты.
Время реакции особо не ограничивают, и оно составляет обычно от 0,5 до 48 часов, предпочтительно от 0,5 до 24 часов. Температуру реакции особо не ограничивают, и она находится обычно в пределах от -78°C до температуры кипения растворителя с обратным холодильником, а предпочтительно от -20°C до комнатной температуры. Растворитель, используемый в данной реакции, особо не ограничивают, лишь бы он не замедлял реакцию и позволял исходному веществу растворяться в нем до определенной степени. Предпочтительные примеры растворителя включают тетрагидрофуран и простой эфир.
3. Общий способ получения 3:
[Формула 8]

На данной схеме кольцо A, R1, R2, R3, R4, R5, R6, X, Y, Z и кольцо B являются такими, как определено выше.
Общий способ получения 3 представляет собой способ получения соединения общей формулы (I) по настоящему изобретению, где L представляет собой -NHCO- и R1 и R2 представляют собой атомы водорода, из соединения (3-1) в качестве исходного вещества путем проведения нескольких стадий от стадии 3-1 до стадии 3-4.
Соединение (3-1) может быть получено из коммерчески доступного продукта описанным выше общим способом получения 1 или комбинацией трех способов получения: общего способа получения 1, общего способа получения 2 и общего способа получения 4, и может быть также получено способом, описанным в примерах получения в разделе "Примеры". Каждое из соединений (3-4) и (3-5) может быть коммерчески доступным продуктом, используемым как он есть, может быть также получено из коммерчески доступного продукта способом, известным специалисту в данной области техники, и, кроме того, может быть получено способом, описанным в примерах получения в разделе "Примеры".
Стадия 3-1:
Данная стадия является стадией получения соединения (3-2) трет-бутоксикарбонилированием аминогруппы соединения (3-1), когда R1 и R2 оба представляют собой водород.
Реакция может быть осуществлена в таких же условиях, какие обычно используют при трет-бутоксикарбонилировании аминосоединения, такие как условия, описанные в таком документе, как T. W. Green and P. G. M. Wuts, "Protective Groups in Organic Chemistry, Second Edition", John Wiley & Sons (1991), P. 327-330. Соединение (3-2) может быть получено, например, путем взаимодействия соединения (3-1) с ди-трет-бутилдикарбонатом с использованием триэтиламина в качестве основания в растворителе, таком как тетрагидрофуран.
Стадия 3-2:
Данная стадия является стадией получения соединения (3-3) из соединения (3-2).
Соединение (3-3) синтезируют восстановлением нитросоединения (3-2) способом синтеза, известным специалисту в данной области техники. Примеры способа включают восстановление путем каталитического гидрирования с использованием катализатора на основе благородного металла, такого как никель Ренея, палладий, рутений, родий или платина. В данном случае предпочтительной является, например, реакция восстановления железом в нейтральных условиях с использованием хлорида аммония.
Стадия 3-3:
Данная стадия является стадией получения соединения (3-6) конденсацией соединения (3-3) с соединением (3-4) с использованием конденсирующего агента. Альтернативно, данная стадия является стадией получения соединения (3-6) конденсацией соединения (3-3) с соединением (3-5) путем проведения реакции ацилирования.
Реакция конденсации соединения (3-3) с соединением (3-4) с использованием конденсирующего агента может быть осуществлена в таких же условиях, как обычно используемые условия, описанные в следующих документах. Примеры известного способа включают примеры, описанные у Rosowsky, A.; Forsch, R. A.; Moran, R. G.; Freisheim, J. H.; J. Med. Chem., 34 (1), 227-234 (1991), Brzostwska, M.; Brossi, A.; Flippen-Anderson, J. L.; Heterocycles, 32 (10), 1968-1972 (1991), and Romero, D. L.; Morge, R. A.; Biles, C.; Berrios-Pena, N.; May, P. D.; Palmer, J. R.; Johnson, P. D.; Smith, H. W.; Busso, M.; Tan, C.-K.; Voorman, R. L.; Reusser, F.; Althaus, I. W.; Downey, K. M.; So, A. G.; Resnick, L.; Tarpley, W. G., Aristoff, P. A.; J. Med. Chem., 37 (7), 998-1014 (1994).
Соединение (3-3) может представлять собой свободную форму или соль.
Растворитель в данной реакции особо не ограничивают, лишь бы он не замедлял реакцию. Примеры растворителя включают тетрагидрофуран, 1,4-диоксан, этилацетат, метилацетат, дихлорметан, хлороформ, N,N-диметилформамид, толуол и ксилол. Примеры конденсирующего агента включают CDI (N,N'-карбонилдиимидазол), Bop (гексафторфосфат 1H-1,2,3-бензотриазол-1-илокси(три(диметиламино))фосфония), WSC (гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида), DCC (N,N-дициклогексилкарбодиимид), диэтилфосфорилцианид, PyBOP (гексафторфосфат бензотриазол-1-илокситрис(пирролидино)фосфония) и EDC·HCl (гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида). Соединение (3-4) используют в количестве от одного эквивалента до большого избытка относительно соединения (3-3). При необходимости может быть добавлено органическое основание, такое как триэтиламин, в количестве от одного эквивалента до большого избытка.
Время реакции особо не ограничивают, и оно составляет обычно от 0,5 до 48 часов, предпочтительно от 0,5 до 24 часов. Температура реакции изменяется в соответствии с используемым исходным веществом, растворителем и тому подобное, и ее особо не ограничивают. Предпочтительной является температура в пределах от температуры охлаждения льдом до температуры кипения растворителя с обратным холодильником.
Соединение формулы (I) по настоящему изобретению, где по меньшей мере один из R1 и R2 представляет собой С1-6 алкильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, C1-6 алкилкарбонильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, C6-14 арилкарбонильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, C1-6 алкилсульфонильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, C6-14 арилсульфонильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, 3-10-членную карбоциклическую группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, или 5-10-членную гетероциклическую группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, может быть получено дополнительным взаимодействием соединения (I-a), полученного общим способом получения 3, с соответствующим галогенидом, таким как C1-6 алкилгалогенид.
Альтернативно, группа -NHCO- группы L в соединении (I-a) по настоящему изобретению может быть преобразована в группу -NReCO- (где Re представляет собой С1-6 алкильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α) дополнительным взаимодействием соединения (I-a), полученного общим способом получения 3, с соответствующим галогенидом, таким как C1-6 алкилгалогенид.
Соединение формулы (I) по настоящему изобретению, где L представляет собой -NReSO2-, может быть получено с использованием соответствующего сульфонилгалогенида вместо соединения (3-4) или (3-5), используемого в общем способе получения 3.
В общем способе получения 3 соединение (3-6) может быть также получено из соединения (3-3) и соединения (3-4) способом, описанным в следующем альтернативном способе (1) или (2).
Альтернативный способ (1):
Соединение (3-6) может быть получено преобразованием соединения (3-4) в смешанный ангидрид кислоты и затем взаимодействием смешанного ангидрида кислоты с соединением (3-3). Смешанный ангидрид кислоты может быть синтезирован способами, известными специалисту в данной области техники. Синтез выполняют, например, путем взаимодействия соединения (3-4) с хлорформиатом, таким как этилхлорформиат, в присутствии основания, такого как триэтиламин. Используют один-два эквивалента хлорформиата и основания относительно соединения (3-4). Температура реакции составляет от -30°C до комнатной температуры, предпочтительно от -20°C до комнатной температуры.
Стадию конденсации смешанного ангидрида кислоты с соединением (3-3) выполняют, например, путем взаимодействия смешанного ангидрида кислоты с соединением (3-3) в растворителе, таком как дихлорметан, тетрагидрофуран или N,N-диметилформамид. Соединение (3-3) используют в количестве от одного эквивалента до большого избытка относительно смешанного ангидрида кислоты.
Время реакции особо не ограничивают, и оно составляет обычно от 0,5 до 48 часов, предпочтительно от 0,5 до 12 часов. Температура реакции составляет от -20°C до 50°C и предпочтительно от -20°C до комнатной температуры.
Альтернативный способ (2):
Соединение (3-6) может быть получено преобразованием соединения (3-4) в активированный сложный эфир и затем взаимодействием активированного сложного эфира с соединением (3-3). Стадию получения активированного сложного эфира выполняют, например, путем взаимодействия соединения (3-4) с активирующим реагентом для синтеза сложного эфира в растворителе, таком как 1,4-диоксан, тетрагидрофуран или N,N-диметилформамид, в присутствии конденсирующего агента, такого как DCC. Примеры активирующего реагента для синтеза сложного эфира включают N-гидроксисукцинимид. Используют от одного до 1,5 эквивалентов активирующего реагента для синтеза сложного эфира и конденсирующего агента относительно соединения (3-4). Время реакции особо не ограничивают, и оно составляет обычно от 0,5 до 48 часов, предпочтительно от 0,5 до 24 часов.
Температура реакции составляет от -20°C до 50°C, предпочтительно от -20°C до комнатной температуры.
Стадию конденсации активированного сложного эфира с соединением (3-3) выполняют, например, путем взаимодействия активированного сложного эфира с соединением (3-3) в растворителе, таком как дихлорметан, тетрагидрофуран или N,N-диметилформамид. Соединение (3-3) используют в количестве от одного эквивалента до большого избытка относительно активированного сложного эфира. Время реакции особо не ограничивают, и оно составляет обычно от 0,5 до 48 часов, предпочтительно от 0,5 до 24 часов. Температура реакции составляет от -20°C до 50°C, предпочтительно от -20°C до комнатной температуры.
В данной реакции ацилирования соединение (3-6) может быть получено из соединений (3-3) и (3-5) способом, известным специалисту в данной области техники.
Примеры основания, используемого в данной реакции, включают триэтиламин, пиридин, карбонат калия и диизопропилэтиламин. Температуру реакции особо не ограничивают, и она находится обычно в пределах от -78°C до температуры кипения растворителя с обратным холодильником, а предпочтительно от -20°C до комнатной температуры. Растворитель, используемый в данной реакции, особо не ограничивают, лишь бы он не замедлял реакцию и позволял исходному веществу растворяться в нем до определенной степени. Предпочтительные примеры растворителя включают тетрагидрофуран, простой эфир, толуол и дихлорметан.
Стадия 3-4:
Данная стадия является стадией получения соединения (I-a) проведением реакции снятия трет-бутоксикарбонильной защитной группы соединения (3-6).
Реакция может быть осуществлена в таких же условиях, какие обычно используют в реакции снятия трет-бутоксикарбонильной защитной группы, такие как условия, описанные в таком документе, как T. W. Green and P. G. M. Wuts, "Protective Groups in Organic Chemistry, Second Edition", John Wiley & Sons (1991), P. 327-330. Соединение (I-a) может быть получено, например, путем взаимодействия трифторуксусной кислоты с соединением (3-6) в растворителе, таком как дихлорметан.
4. Общий способ получения 4:
[Формула 9]

На данной схеме кольцо A, R1, R2, R3, R4, R5, R6, X, Y и Z являются такими, как определено выше.
Общий способ получения 4 представляет собой способ получения соединения общей формулы (3-1), которое является промежуточным соединением в синтезе соединения по настоящему изобретению и используется в общем способе получения 3, получаемым из соединения (4-1) в качестве исходного вещества проведением стадии 4-1.
Соединение (4-1) может быть получено из коммерчески доступного продукта общим способом получения 1, общим способом получения 5 или комбинацией общего способа получения 1 и общего способа получения 2 и может быть также получено способом, описанным в примерах получения в разделе "Примеры".
Стадия 4-1:
Данная стадия является стадией получения соединения (3-1) реакцией нитрования соединения (4-1). В данной реакции нитрования соединение (3-1) может быть получено из соединения (4-1) способом, известным специалисту в данной области техники. Примеры нитрующего агента, используемого в данной реакции, включают смеси нитрат калия/концентрированная серная кислота и дымящая азотная кислота/уксусный ангидрид. Температуру реакции особо не ограничивают, и она составляет обычно от -20°C до комнатной температуры.
5. Общий способ получения 5:
[Формула 10]

На данной схеме Prt представляет собой защитную группу, такую как бензоильная группа, ацетильная группа или 8-флуоренметилоксикарбонильная группа (Fmoc группа), и кольцо A, R3, R4, R5, R6, Y и Z являются такими, как определено выше.
Общий способ получения 5 представляет собой способ получения соединения (1-7), которое является промежуточным соединением в синтезе соединения (I) по настоящему изобретению, получаемым из соединения (5-1) в качестве исходного вещества проведением нескольких стадий от стадии 5-1 до стадии 5-7.
Соединение (5-1) может быть получено из коммерчески доступного продукта описанным ниже общим способом получения 6 или 7, может быть также получено из коммерчески доступного продукта способом, известным специалисту в данной области техники, и, кроме того, может быть получено способом, описанным в примерах получения в разделе "Примеры".
Стадия 5-1:
Данная стадия является стадией получения соединения (5-2) оксимированием соединения (5-1).
Реакция на данной стадии может быть осуществлена в таких же условиях, какие обычно используют в реакции оксимирования карбонильного соединения, такие как условия, описанные в Org. Lett. 9 (2007) 5, 753-756, Tetrahedron: Asymmetry 5 (1994) 6, 1018-1028 and Tetrahedron 54 (1998) 22, 5868-5882.
В частности, соединение (5-2) может быть получено, например, путем взаимодействия соединения (5-1) с гидроксиламином или гидроксиламиновой солью (такой как гидрохлорид гидроксиламина или сульфат гидроксиламина) в присутствии основания или в отсутствие основания.
Растворитель, используемый в данной реакции, особо не ограничивают, лишь бы он не замедлял реакцию. Предпочтительные примеры растворителя включают органические растворители, такие как этанол, метанол, тетрагидрофуран, 1,4-диоксан, 1,2-диметоксиэтан и дихлорметан, и смеси указанных растворителей и воды. Примеры используемого основания включают ацетат натрия, пиридин, гидроксид натрия, гидроксид цезия, гидроксид бария и 2,6-лутидин. Время реакции особо не ограничивают, и оно составляет обычно от 5 минут до 24 часов, предпочтительно от 5 минут до 12 часов. Температура реакции находится обычно в пределах от -20°C до температуры кипения растворителя с обратным холодильником, а более предпочтительно от 0°C до температуры кипения растворителя с обратным холодильником.
Стадия 5-2:
Данная стадия является стадией получения соединения (5-3) преобразованием соединения (5-2) в нитрилоксидное производное и выполнением реакции 1,3-диполярного циклоприсоединения с олефиновым фрагментом в той же самой молекуле.
Реакция на данной стадии может быть осуществлена в таких же условиях, какие обычно используют в реакции 1,3-диполярного циклоприсоединения, такие как условия, описанные в таком документе, как Org. Lett. 9 (2007) 5, 753-756, Tetrahedron: Asymmetry 5 (1994) 6, 1018-1028 and Tetrahedron 54 (1998) 22, 5868-5882. Примеры реагента для преобразования соединения оксима в нитрилоксид включают N-хлорсукцинимид и гипохлорит натрия. Растворитель, используемый в данной реакции, особо не ограничивают, лишь бы он не замедлял реакцию. Предпочтительные примеры растворителя включают дихлорметан, хлороформ, бензол, толуол, ксилол, N,N-диметилформамид, тетрагидрофуран и 1,4-диоксан. Температуру реакции особо не ограничивают, и она находится обычно в пределах от температуры охлаждения льдом до температуры кипения растворителя с обратным холодильником. Время реакции особо не ограничивают, и оно составляет обычно от 0,5 до 48 часов, предпочтительно от 0,5 до 24 часов.
Более предпочтительный результат, такой как повышенный выход, может быть достигнут осуществлением данной реакции в присутствии основания. Такое основание особо не ограничивают. Примеры основания включают такие основания, как карбонат натрия, карбонат калия, карбонат цезия, фосфат калия и их растворы, и триэтиламин и пиридин.
Стадия 5-3:
Данная стадия является стадией получения соединения (5-4) путем дополнительного взаимодействия ариллитиевого реагента (включая гетероциклический) или реактива Гриньяра (включая гетероциклический) с соединением (5-3).
Реакция на данной стадии может быть осуществлена, например, в таких же условиях, как описано в J. Am. Chem. Soc. 2005, 127, 5376-5383, Bull. Chem. Soc. Jpn., 66, 2730-2737 (1993) and SYNLETT. 2004, No. 8, pp 1408-1413.
Ариллитиевый реагент (включая гетероциклический) или реактив Гриньяра (включая гетероциклический) может быть получен способом, известным специалисту в данной области техники. В частности, соответствующий ариллитиевый реагент (включая гетероциклический) или арилмагниевый реагент (включая гетероциклический) может быть получен, например, обменом галоген-металл между арилгалогенидом и коммерчески доступным металлоорганическим реагентом, таким как алкиллитиевый реагент, такой как н-, втор- или трет-бутиллитий, или реактив Гриньяра, такой как изопропилмагнийбромид, или металлическим магнием.
Растворитель, используемый на данной стадии, изменяется в соответствии с используемыми исходным веществом и реагентом, и его особо не ограничивают, лишь бы он не замедлял реакцию, позволял исходному веществу растворяться в нем до определенной степени и всегда был инертным во время реакции. Предпочтительные примеры растворителя включают органические растворители, такие как диэтиловый эфир, тетрагидрофуран, 1,4-диоксан, 1,2-диметоксиэтан, бензол и толуол, и их смешанные растворители. Время реакции особо не ограничивают, и оно составляет обычно 0,1-48 часов, предпочтительно 0,1-12 часов. Температура реакции изменяется в соответствии с используемыми исходным веществом, реагентом и тому подобное, и ее предпочтительно поддерживают низкой, например, на уровне -78°C, чтобы минимизировать образование побочного продукта.
Положительные результаты, такие как повышенный выход и пониженное время реакции, могут быть достигнуты, например, добавлением TMEDA (тетраметилэтилендиамин), HMPA (гексаметилфосфорамид) или кислоты Льюиса, такой как комплекс трифторид бора-диэтиловый эфир (BF3 ·OEt2) в качестве добавки.
Стадия 5-4:
Данная стадия является стадией получения соединения (5-5) реакцией восстановительного расщепления N-O связи соединения (5-4).
Реакция восстановительного расщепления N-O связи может быть осуществлена в условиях с использованием, например, смеси цинк-уксусная кислота, металлического катализатора, такого как водород-оксид платины, или алюмогидрида лития.
Реакция с использованием цинка, такого как смесь цинк-уксусная кислота, может быть осуществлена, например, в таких же условиях, как описано в J. Org. Chem. 2003, 68, 1207-1215 and Org. Lett. 7 (2005) 25, 5741-5742. Примеры используемой кислоты включают уксусную кислоту, муравьиную кислоту и хлористо-водородную кислоту. Растворитель, используемый в данной реакции, особо не ограничивают, лишь бы он не замедлял реакцию и позволял исходному веществу растворяться в нем до определенной степени. Примеры растворителя включают метанол, этанол, 1,4-диоксан, ТГФ и воду. Указанная выше кислота может быть также использована в качестве растворителя. Температура реакции находится в пределах обычно от -20°C до температуры кипения растворителя с обратным холодильником, а предпочтительно от температуры охлаждения льдом до температуры кипения растворителя с обратным холодильником. Время реакции особо не ограничивают, и оно составляет обычно от 5 минут до 48 часов, предпочтительно от 5 минут до 24 часов.
Реакция с использованием металлического катализатора, такого как водород-оксид платины, может быть осуществлена, например, в таких же условиях, как описано в Tetrahedron: Asymmetry 5 (1994) 6, 1018-1028 and Tetrahedron, Vol. 53, No. 16, pp 5752-5746, 1997. Соединение (5-5) может быть получено, например, гидрированием соединения (5-4) с использованием оксида платины в качестве катализатора в растворителе, таком как метанол.
Реакция с использованием алюмогидрида лития может быть осуществлена, например, в таких же условиях, как описано в Bull. Chem. Soc. Jpn., 66, 2730-2737 (1993). Соединение (5-5) может быть получено, например, восстановлением соединения (5-4) с использованием алюмогидрида лития в растворителе, таком как простой эфир.
Стадия 5-5:
Данная стадия является стадией получения соединения (5-6) из соединения (5-5). Производное тиомочевины (5-6) может быть получено из соединения (5-5) способом, известным специалисту в данной области техники.
Если защитной группой является бензоильная группа, то соединение (5-6) может быть получено на данной стадии путем взаимодействия соединения (5-5) с бензоилизотиоцианатом в растворителе, таком как дихлорметан или толуол. Данная реакция может быть осуществлена, например, в таких же условиях, как описано в J. Org. Chem. 1994, 59, 1911-1917. Растворитель, используемый в данной реакции, особо не ограничивают, лишь бы он не замедлял реакцию и позволял исходному веществу растворяться в нем до определенной степени. Примеры растворителя включают дихлорметан, хлороформ, толуол, метанол, этанол, 1,4-диоксан и ТГФ. Температура реакции находится обычно в пределах от -20°C до температуры кипения растворителя с обратным холодильником, а предпочтительно от температуры охлаждения льдом до температуры кипения растворителя с обратным холодильником. Время реакции особо не ограничивают, и оно составляет обычно от 5 минут до 48 часов, предпочтительно от 5 минут до 24 часов.
Если защитной группой является 8-флуоренметилоксикарбонильная группа (Fmoc группа), то соединение (5-6) может быть получено на данной стадии путем взаимодействия соединения (5-5) с флуоренметилоксикарбонилизотиоцианатом в растворителе, таком как дихлорметан или толуол. Данная реакция может быть осуществлена, например, в таких же условиях, как описано в J. Org. Chem. 1998, 63, 196-200. Растворитель, используемый в данной реакции, особо не ограничивают, лишь бы он не замедлял реакцию и позволял исходному веществу растворяться в нем до определенной степени. Примеры растворителя включают дихлорметан, хлороформ, толуол, метанол, этанол, 1,4-диоксан и ТГФ. Температура реакции находится обычно в пределах от -20°C до температуры кипения растворителя с обратным холодильником, а предпочтительно от температуры охлаждения льдом до температуры кипения растворителя с обратным холодильником. Время реакции особо не ограничивают, и оно составляет обычно от 5 минут до 48 часов, предпочтительно от 5 минут до 24 часов.
Стадия 5-6:
Данная стадия является способом получения соединения (5-7) циклизацией соединения (5-6).
В данной реакции соединение (5-6) может быть подвергнуто циклизации в различных условиях с получением соединения (5-7) путем подбора защитной группы соединения (5-6).
Если защитной группой является, например, Fmoc группа или бензоильная группа, то соединение (5-7) может быть получено в данной реакции нагреванием соединения (5-6) в растворителе, таком как метанол, в присутствии кислоты, такой как, например, концентрированная хлористо-водородная кислота. Растворитель, используемый в данной реакции, особо не ограничивают, лишь бы он не замедлял реакцию и позволял исходному веществу растворяться в нем до определенной степени. Примеры растворителя включают такие растворители, как метанол, этанол, 1-пропанол и вода, их смешанные растворители и кислоты, используемые в качестве растворителя. Реакция может быть осуществлена действием подходящей кислоты в количестве от одного эквивалента до большого избытка в присутствии или в отсутствие такого растворителя. Примеры используемой кислоты включают концентрированную хлористо-водородную кислоту, бромисто-водородную кислоту, серную кислоту, трифторуксусную кислоту, метансульфоновую кислоту, трифторметансульфоновую кислоту и их смеси. Время реакции особо не ограничивают, и оно составляет обычно от 0,5 до 72 часов, предпочтительно от 0,5 до 24 часов. Температура реакции находится в пределах обычно от температуры охлаждения льдом до температуры кипения растворителя с обратным холодильником.
Если защитной группой является Fmoc группа или бензоильная группа, то соединение (5-7) может быть получено альтернативным способом 1 взаимодействия соединения (5-6) с трифторметансульфоновым ангидридом в растворителе, таком как дихлорметан, в присутствии основания, такого как пиридин. Данная реакция может быть осуществлена в таких же условиях, как, например, описано в Chem Bio Chem. 2005, 6, 186-191. Растворитель, используемый в данной реакции, особо не ограничивают, лишь бы он не замедлял реакцию и позволял исходному веществу растворяться в нем до определенной степени. Примеры растворителя включают такие растворители, как дихлорметан, 1,2-дихлорэтан, ТГФ, 1,2-диметоксиэтан и толуол, и их смешанные растворители. Реакция может быть осуществлена с использованием 1-20 эквивалентов подходящего основания в таком растворителе. Примеры используемого основания включают пиридин, 2,6-лутидин, карбонат натрия, карбонат калия и их смеси. Время реакции особо не ограничивают, и оно составляет обычно от 0,5 до 24 часов, предпочтительно от 0,5 до 12 часов. Температура реакции составляет обычно от -78°C до комнатной температуры.
Если защитной группой является бензоильная группа, то соединение (5-7) может быть получено альтернативным способом 2 взаимодействия соединения (5-6) с трифенилфосфином и тетрабромидом углерода (или бромом) в растворителе, таком как дихлорметан. Условия реакции являются такими же, как условия бромирования первичного спирта, которые известны специалисту в данной области техники.
Стадия 5-7:
Данная стадия представляет собой способ получения соединения (1-7) удалением защитной группы соединения (5-7). Соединение (1-7) может быть получено в условиях снятия защиты, известных специалисту в данной области техники.
Если защитной группой является, например, Fmoc группа, то соединение (1-7) может быть получено в таких же условиях, какие обычно используют при удалении защитной группы аминосоединения (такие как условия, описанные в таком документе, как T. W. Green and P. G. M. Wuts, "Protective Groups in Organic Chemistry, Third Edition", John Wiley & Sons, p. 506-507 и J. Org. Chem. 1998, 63, 196-200). В данной реакции соединение (1-7) может быть получено, например, путем взаимодействия соединения (5-7) с избытком амина, такого как пирролидин, в растворителе, таком как ацетонитрил. Растворитель, используемый в данной реакции, особо не ограничивают, лишь бы он не замедлял реакцию и позволял исходному веществу растворяться в нем до определенной степени. Примеры растворителя включают дихлорметан, ТГФ и ацетонитрил. Реакция может быть осуществлена действием подходящего основания в количестве от одного эквивалента до большого избытка в присутствии такого растворителя. Примеры используемого основания включают пиперидин, морфолин, пирролидин, TBAF и DBU. Время реакции особо не ограничивают, и оно составляет обычно от 0,5 до 72 часов, предпочтительно от 0,5 до 24 часов. Температура реакции находится обычно в пределах от температуры охлаждения льдом до температуры кипения растворителя с обратным холодильником.
Положительные результаты, такие как повышенный выход и пониженное время реакции, могут быть достигнуты, например, введением тиольного соединения, такого как 1-октантиол, в качестве добавки.
Если защитной группой является бензоильная группа, то соединение (1-7) может быть получено в данной реакции, например, нагреванием соединения (5-7) в растворителе, таком как метанол, в присутствии основания, такого как DBU. Данная реакция может быть осуществлена, например, в таких же условиях, как описано в Synth. Commun. 32 (2), 265-272 (2002). Растворитель, используемый в данной реакции, особо не ограничивают, лишь бы он не замедлял реакцию и позволял исходному веществу растворяться в нем до определенной степени. Примеры растворителя включают такие растворители, как метанол, этанол и 1-пропанол. Реакция может быть осуществлена с использованием 1-20 эквивалентов подходящего основания в таком растворителе. Примеры используемого основания включают DBU. Время реакции особо не ограничивают, и оно составляет обычно от 0,5 до 24 часов, предпочтительно от 0,5 до 12 часов. Температура реакции находится обычно в пределах от комнатной температуры до температуры кипения растворителя с обратным холодильником.
6. Общий способ получения 6:
[Формула 11]

На данной схеме Prt2 представляет собой первичную гидроксилзащитную группу, R8 представляет собой C1-6 алкильную группу и Z, R3, R4, R5, R6, R7 и LV являются такими, как определено выше.
Общий способ получения 6 представляет собой способ получения соединения (6-4), которое является соединением (5-1), служащим в качестве исходного вещества для общего способа получения 5, где Y представляет собой атом кислорода.
Каждое из соединений (6-1), (6-2), (6-5), (6-7) и (6-9) может быть коммерчески доступным продуктом, используемым как он есть, может быть также получено из коммерчески доступного продукта способом, известным специалисту в данной области техники, и, кроме того, может быть получено способом, описанным в примерах получения в разделе "Примеры".
Стадия 6-1:
Данная стадия является стадией получения соединения (6-3) путем взаимодействия соединения (6-1) с соединением (6-2).
Данная реакция может быть осуществлена в таких же условиях, какие обычно используют в реакции O-алкилирования спиртового соединения (такие как условия, описанные в Tetrahedron Lett. 46 (2005) 45, 7751-7755). В данной реакции соединение (6-3) может быть получено, например, добавлением основания, такого как гидрид натрия, к раствору соединения (6-1) в ТГФ с получением алкоксида и затем взаимодействием алкоксида с соединением (6-2). Растворитель, используемый в данной реакции, особо не ограничивают, лишь бы он не замедлял реакцию и позволял исходному веществу растворяться в нем до определенной степени. Примеры растворителя включают такие растворители, как ТГФ, ДМФА и диметилсульфоксид. Реакция может быть осуществлена действием 1-3 эквивалентов подходящего основания в присутствии такого растворителя. Примеры используемого основания включают гидрид натрия, гидрид калия и трет-бутоксикалий. Время реакции особо не ограничивают, и оно составляет обычно от 0,5 до 72 часов, предпочтительно от 0,5 до 12 часов. Температура реакции составляет обычно от -20°C до 50°C.
Более предпочтительный результат, такой как повышенный выход, может быть достигнут в данной реакции добавлением соли, такой как йодид тетрабутиламмония.
Стадия 6-2:
Данная стадия является стадией получения альдегидного соединения (6-4) путем реакции окисления спиртового соединения (6-3). Альдегидное соединение может быть получено из спиртового соединения способом, известным специалисту в данной области техники.
Примеры известного способа окисления, используемого в данной реакции, включают окисление по Сверну, окисление по Corey-Kim, окисление по Moffatt, PCC окисление, PDC окисление, окисление по Дессу-Мартину, окисление с использованием комплекса SO3-пиридин и TEMPO окисление.
Растворитель, используемый в данной реакции, особо не ограничивают, лишь бы он не замедлял реакцию и позволял исходному веществу растворяться в нем до определенной степени. Примеры растворителя включают диметилсульфоксид, тетрагидрофуран, толуол, дихлорметан и хлороформ.
Температуру реакции особо не ограничивают, и она находится обычно в пределах от -78°C до температуры кипения растворителя с обратным холодильником, а предпочтительно от -78°C до комнатной температуры. Время реакции особо не ограничивают, и оно составляет обычно от 0,5 до 48 часов, предпочтительно от 0,5 до 24 часов.
Стадия 6-3:
Данная стадия является стадией синтезирования соединения (6-6) из соединения (6-5) в качестве исходного вещества с использованием способа получения, описанного выше (стадия 6-1).
Стадия 6-4:
Данная стадия является стадией получения соединения (6-4) снятием защиты с ацетальной группы соединения (6-6).
Данная реакция может быть осуществлена в таких же условиях, какие обычно используют при снятии защиты с альдегидной группы, такие как условия, описанные в таком документе, как T. W. Green and P. G. M. Wuts, "Protective Groups in Organic Chemistry, Third Edition", John Wiley & Sons, P. 293-329.
Стадия 6-5:
Данная стадия является стадией синтезирования соединения (6-8) из соединения (6-7) в качестве исходного вещества с использованием способа получения, описанного выше (стадия 6-1).
Стадия 6-6:
Данная стадия является стадией получения соединения (6-3) удалением гидроксилзащитной группы соединения (6-8). Гидроксилзащитную группу, используемую на данной стадии, особо не ограничивают.
Данная реакция может быть осуществлена в таких же условиях, какие обычно используют при удалении защитной группы для спирта, такие как условия, описанные в таком документе, как T. W. Green and P. G. M. Wuts, "Protective Groups in Organic Chemistry, Third Edition", John Wiley & Sons, P. 17-245.
Стадия 6-7:
Данная стадия является стадией синтезирования соединения (6-3) из соединения (6-9) в качестве исходного вещества с использованием способа получения, описанного выше ((стадия 1-3) или (стадии 2B-1 и 2)).
7. Общий способ получения 7:
[Формула 12]
На данной схеме R9 представляет собой C1-6 алкильную группу или два R9 вместе могут образовывать кольцо, Prt3 представляет собой защитную группу, такую как 2,4-диметоксибензильная группа, и Z, R3, R4, R5, R6, Z и LV являются такими, как определено выше.
Общий способ получения 7 представляет собой способ получения соединения (7-5), которое является соединением (5-1), служащим в качестве исходного вещества для общего способа получения 5, где Y представляет собой атом азота.
Каждое из соединений (7-1) и (7-3) может быть коммерчески доступным продуктом, используемым как он есть, может быть также получено из коммерчески доступного продукта способом, известным специалисту в данной области техники, и, кроме того, может быть получено способом, описанным в примерах получения в разделе "Примеры".
Стадия 7-1:
Данная стадия является стадией получения соединения (7-2) защитой аминогруппы соединения (7-1).
Данная реакция может быть осуществлена в таких же условиях, какие обычно используют при защите аминогруппы, такие как условия, описанные в таком документе, как T. W. Green and P. G. M. Wuts, "Protective Groups in Organic Chemistry, Third Edition", John Wiley & Sons, P. 494-572 and J. Med. Chem. 2007, 50, 5493-5508.
Стадия 7-2:
Данная стадия является стадией получения соединения (7-4) по реакции N-алкилирования соединения (7-2) соединением (7-3).
Данная реакция может быть осуществлена в таких же условиях, какие обычно используют в реакции N-алкилирования соединения (7-2) (такие как условия, описанные в J. Med. Chem. 2007, 50, 5493-5508). В данной реакции соединение (7-4) может быть получено, например, добавлением основания, такого как порошкообразный гидроксид натрия, к раствору соединения (7-2) в толуоле, а затем взаимодействием полученной смеси с соединением (7-3). Растворитель, используемый в данной реакции, особо не ограничивают, лишь бы он не замедлял реакцию и позволял исходному веществу растворяться в нем до определенной степени. Примеры растворителя включают такие растворители, как толуол, ТГФ, ДМФА и диметилсульфоксид. Реакция может быть осуществлена действием 1-5 эквивалентов подходящего основания в присутствии такого растворителя. Примеры используемого основания включают гидроксид натрия, гидроксид калия, гидрид натрия, гидрид калия и трет-бутоксикалий. Время реакции особо не ограничивают, и оно составляет обычно от 0,5 до 72 часов, предпочтительно от 0,5 до 24 часов. Температура реакции составляет обычно от -20°C до 100°C.
Более предпочтительный результат, такой как повышенный выход, может быть достигнут в данной реакции добавлением соли, такой как йодид тетрабутиламмония.
Стадия 7-3:
Данная стадия является стадией получения соединения (7-5) снятием защиты с ацетальной группы соединения (7-4).
Данная реакция может быть осуществлена в таких же условиях, какие обычно используют при снятии защиты с альдегидной группы, такие как условия, описанные в таком документе, как T. W. Green and P. G. M. Wuts, "Protective Groups in Organic Chemistry, Third Edition", John Wiley & Sons, P. 293-329.
8. Общий способ получения 8:
[Формула 13]

На данной схеме Prt представляет собой защитную группу, такую как бензоильная группа, ацетильная группа или 8-флуоренметилоксикарбонильная группа (Fmoc группа), Prt3 представляет собой защитную группу, такую как 2,4-диметоксибензильная группа, и кольцо A, R3, R4, R5 и R6 являются такими, как определено выше.
Общий способ получения 8 представляет собой стадии способа получения соединений общих формул (8-7) и (8-8), которые являются промежуточными соединениями для синтеза соединения (I) по настоящему изобретению в общем способе получения 5, где Y представляет собой атом азота и Z представляет собой одинарную связь. Указанные соединения могут быть получены из соединения (8-1) в качестве исходного вещества проведением стадий, показанных выше.
Соединение (8-1) может быть коммерчески доступным продуктом, используемым как он есть, может быть также получено из коммерчески доступного продукта способом, известным специалисту в данной области техники, и, кроме того, может быть получено способом, описанным в примерах получения в разделе "Примеры". Соединение (8-2) может быть получено из коммерчески доступного продукта способом, известным специалисту в данной области техники, и, кроме того, может быть получено способом, описанным в примерах получения в разделе "Примеры".
Стадия 8-1:
Данная стадия является стадией получения соединения (8-3) путем взаимодействия соединения (8-1) с соединением (8-2). Данная реакция может быть осуществлена в таких же условиях, какие обычно используют в реакции N-алкилирования аминосоединения (такие как условия, описанные в J. Med. Chem. 2002, 45, 3794-3804 и J. Med. Chem. 2000, 43, 3808-3812). В данной реакции соединение (8-3) может быть получено, например, взаимодействием соединения (8-1) с соединением (8-2) в растворителе, таком как дихлорметан, в присутствии основания, такого как N,N-диизопропилэтиламин. Растворитель, используемый в данной реакции, особо не ограничивают, лишь бы он не замедлял реакцию и позволял исходному веществу растворяться в нем до определенной степени. Примеры растворителя включают дихлорметан, ТГФ, ацетонитрил и ДМФА. Реакция может быть осуществлена действием 1-10 эквивалентов подходящего основания в таком растворителе. Примеры используемого основания включают N,N-диизопропилэтиламин, триэтиламин, карбонат натрия и карбонат калия. Время реакции особо не ограничивают, и оно составляет обычно от 0,5 до 72 часов, предпочтительно от 0,5 до 12 часов. Температура реакции составляет обычно от температуры охлаждения льдом до 50°C.
Стадия 8-2:
Данная стадия является стадией получения соединения (8-4) оксимированием соединения (8-3).
Реакция на данной стадии может быть осуществлена в таких же условиях, какие обычно используют в реакции оксимирования карбонильного соединения, такие как условия, описанные в J. Med. Chem. 2002, 45, 3794-3804 и J. Med. Chem. 2000, 43, 3808-3812.
В частности, соединение (8-4) может быть получено, например, путем взаимодействия соединения (8-3) с гидроксиламином или гидроксиламиновой солью (такой как гидрохлорид гидроксиламина или сульфат гидроксиламина) в присутствии основания или в отсутствие основания. Растворитель, используемый в данной реакции, особо не ограничивают, лишь бы он не замедлял реакцию. Предпочтительные примеры растворителя включают органические растворители, такие как этанол, метанол, тетрагидрофуран, 1,4-диоксан, 1,2-диметоксиэтан и дихлорметан, и смеси указанных растворителей и воды. Примеры используемого основания включают карбонат натрия, карбонат калия, ацетат натрия, пиридин, гидроксид натрия, гидроксид цезия, гидроксид бария и 2,6-лутидин. Время реакции особо не ограничивают, и оно составляет обычно от 5 минут до 24 часов, предпочтительно от 5 минут до 12 часов. Температура реакции находится обычно в пределах от 0°C до температуры кипения растворителя с обратным холодильником, а более предпочтительно от комнатной температуры до температуры кипения растворителя с обратным холодильником.
Стадия 8-3:
Данная стадия является стадией получения соединения (8-5) путем введения оксимного соединения (8-4) в реакцию 1,3-диполярного циклоприсоединения.
Реакция на данной стадии может быть осуществлена в таких же условиях, какие обычно используют в реакции 1,3-диполярного циклоприсоединения, такие как условия, описанные в J. Org. Chem. 1993, 58, 4538-4546 и Tetrahedron Letters, Vol. 29, No. 41, pp 5312-5316.
В частности, соединение (8-5) может быть получено, например, кипячением с обратным холодильником раствора соединения (8-4) в толуоловом растворителе. Растворитель, используемый в данной реакции, особо не ограничивают, лишь бы он не замедлял реакцию. Предпочтительные примеры растворителя включают органические растворители, такие как толуол, ксилол и хлорбензол. Время реакции особо не ограничивают, и оно составляет обычно от 5 минут до 24 часов, предпочтительно от 5 минут до 12 часов. Температура реакции находится обычно в пределах от 0°C до температуры кипения растворителя с обратным холодильником, а более предпочтительно от комнатной температуры до температуры кипения растворителя с обратным холодильником.
Положительные результаты, такие как повышенный выход и пониженное время реакции, могут быть достигнуты добавлением кислоты Льюиса, такой как хлорид цинка, в виде добавки.
Положительные результаты, такие как уменьшенное время реакции и повышенный выход, могут быть получены проведением данной реакции с использованием микроволнового реактора.
Стадия 8-4:
Соединение (8-6) может быть синтезировано из соединения (8-5) с использованием серии способов, показанных в описанном выше способе получения от ((стадии 5-4) до (стадии 5-6)).
Стадия 8-5:
Данная стадия является стадией синтезирования соединения (8-7) из соединения (8-6) в качестве исходного вещества с использованием способа получения, описанного выше (стадия 5-7).
Стадия 8-6:
Данная стадия является стадией получения соединения (8-8) снятием защиты с аминогруппы соединения (8-6). Аминозащитную группу, используемую на данной стадии, особо не ограничивают. Если Prt3 представляет собой, например, 2,4-диметоксибензильную группу, то данная стадия может быть осуществлена в таких же условиях, какие обычно используют (такие как условия, описанные в таком документе, как Tetrahedron Vol. 47, No. 26, pp 4591-4602, 1991). Если на данной стадии Prt3 представляет собой 2,4-диметоксибензильную группу, то растворитель, используемый на данной стадии, особо не ограничивают, лишь бы он не замедлял реакцию и позволял исходному веществу растворяться в нем до определенной степени. Например, растворителем в реакции первой стадии может быть метиленхлорид или хлороформ, а растворителем в реакции второй стадии может быть метанол. Температура реакции на данной стадии обычно составляет от 0°C до комнатной температуры. Время реакции на данной стадии особо не ограничивают, и оно составляет обычно от 0,5 до 24 часов, предпочтительно от 0,5 до 12 часов.
9. Общий способ получения 9:
[Формула 14]

На данной схеме L1 представляет собой одинарную связь или C1-6 алкиленовую группу в соединениях (9-3) и (9-4) и представляет собой одинарную связь или C1-4 алкиленовую группу в соединениях (9-5) и (9-6), L представляет собой одинарную связь, атом кислорода, C1-6 алкиленовую группу, C2-6 алкениленовую группу или C2-6 алкиниленовую группу, Alk представляет собой C1-6 алкильную группу и кольцо A, кольцо B, R3, R4, R5, R6, Y, Z и LV являются такими, как определено выше.
Общий способ получения 9 представляет собой способ получения соединения (I-b) общей формулы (I) по настоящему изобретению, где L представляет собой одинарную связь, атом кислорода, C1-6 алкиленовую группу, C2-6 алкениленовую группу или C2-6 алкиниленовую группу и R1 и R2 представляют собой атомы водорода, из соединения (9-1) в качестве исходного вещества проведением показанных выше стадий.
Соединение (9-1) может быть получено из коммерчески доступного продукта общим способом получения 1, общим способом получения 5 или комбинацией общего способа получения 1 и способа 2B общего способа получения 2 и может быть также получено способом, описанным в примерах получения в разделе "Примеры". Каждое из соединений (9-3), (9-4), (9-5), (9-6) и (9-7) может быть коммерчески доступным продуктом, используемым как он есть, может быть также получено из коммерчески доступного продукта способом, известным специалисту в данной области техники, и, кроме того, может быть получено способом, описанным в примерах получения в разделе "Примеры".
Стадия 9-1:
Данная стадия является стадией получения соединения (9-2) ди-трет-бутоксикарбонилированием соединения (9-1). Данная реакция может быть осуществлена в таких же условиях, какие обычно используют при трет-бутоксикарбонилировании амидного соединения, такие как условия, описанные в T. W. Green and P. G. M. Wuts, "Protective Groups in Organic Chemistry, Third Edition", John Wiley & Sons, P. 642-643 и J. Org. Chem. 2005, 70, 2445-2454. Соединение (9-2) может быть получено, например, путем взаимодействия соединения (9-1) с ди-трет-бутилдикарбонатом с использованием 4-диметиламинопиридина в качестве основания в растворителе, таком как ТГФ.
Растворитель, используемый в данной реакции, особо не ограничивают, лишь бы он не замедлял реакцию. Предпочтительные примеры растворителя включают органические растворители, такие как тетрагидрофуран, 1,4-диоксан, 1,2-диметоксиэтан, дихлорметан, ДМФА и ацетонитрил, и их смешанные растворители. Примеры используемого основания включают триэтиламин, 4-диметиламинопиридин, DBU и их смеси. Основание используют в количестве от каталитического количества до избытка, а более предпочтительно 0,1-5 эквивалентов относительно соединения (9-1). Ди-трет-бутилдикарбонат используют в количестве от двух эквивалентов до избытка, а более предпочтительно 2-10 эквивалентов относительно соединения (9-1). Время реакции особо не ограничивают, и оно составляет обычно от 5 минут до 24 часов, предпочтительно от 5 минут до 12 часов. Температура реакции находится обычно в пределах от -20°C до температуры кипения растворителя с обратным холодильником, а более предпочтительно от 0°C до температуры кипения растворителя с обратным холодильником.
Стадия 9-2:
Данная стадия является стадией получения соединения (9-8) реакцией сочетания соединения (9-2) с соединением (9-3), (9-4), (9-5), (9-6) или (9-7) с использованием переходного металла. Данная реакция может быть осуществлена в условиях, обычно используемых в реакции сочетания с использованием переходного металла (такой как реакция Сузуки-Мияуры, реакция Стилле, реакция Соногаширы, реакция Хека или реакция Буквальда и др. с использованием арилового простого эфира).
Примеры реакции Сузуки-Мияуры включают реакции, описанные в таких документах, как J. Org. Chem. 2007, 72, 7207-7213, J. Am. Chem. Soc. 2000, 122, 4020-4028 и J. Org. Chem. 2007, 72, 5960-5967. Примеры реакции сочетания по Стилле включают реакцию, описанную в таком документе, как J. Am. Chem. Soc. 1990, 112, 3093-3100. Примеры реакции Соногаширы включают реакции, описанные в таких документах, как J. Org. Chem. 2007, 72, 8547-8550 и J. Org. Chem. 2008, 73, 234-240. Примеры реакции Хека включают реакцию, описанную в таком документе, как J. Am. Chem. Soc. 2005, 127, 16900-16911. Примеры реакции Буквальда и др. с использованием арилового простого эфира включают реакцию, описанную в таком документе, как Buckwald, S. L. et al., J Am Chem Soc (1999) 121 (18), 4369-4378. Металлоорганический катализатор, используемый в данной реакции, особо не ограничивают. Предпочтительные примеры металлоорганического катализатора включают металлические катализаторы, такие как тетракис(трифенилфосфин)палладий(0), дихлорбис(трифенилфосфин)палладий(II), дихлорид [1,1'-бис(дифенилфосфино)ферроцен]палладия(II), бис(трет-бутилфосфин)палладий(0), ацетат палладия(II) и [1,3-бис(дифенилфосфино)пропан]никель(II), и смеси указанных металлических катализаторов. Количество используемого металлоорганического катализатора составляет примерно 0,001-0,5 эквивалента относительно исходного вещества. Количество используемого соединения (9-3), (9-4), (9-5), (9-6) или (9-7) особо не ограничивают, и оно составляет обычно 1-5 эквивалентов относительно соединения (9-2). Растворитель, используемый в данной реакции, особо не ограничивают, лишь бы он не замедлял реакцию. Предпочтительные примеры растворителя включают бензол, толуол, ксилол, N,N-диметилформамид, 1-метил-2-пирролидон, тетрагидрофуран, 1,2-диметоксиэтан, 1,4-диоксан, ацетонитрил и пропионитрил. Температуру реакции особо не ограничивают, и она находится обычно в пределах от температуры охлаждения льдом до температуры кипения растворителя с обратным холодильником, а предпочтительно, например, от комнатной температуры до температуры кипения растворителя с обратным холодильником. Время реакции особо не ограничивают, и оно составляет обычно от 0,5 до 48 часов, предпочтительно от 0,5 до 24 часов.
Более предпочтительный результат, такой как повышенный выход, может быть достигнут осуществлением данной реакции в присутствии основания или соли. Такое(ую) основание или соль особо не ограничивают. Предпочтительные примеры основания или соли включают такие основания или соли, как карбонат натрия, карбонат калия, гидроксид бария, карбонат цезия, фосфат калия, фторид калия и их растворы, и триэтиламин, N,N-диизопропилэтиламин, хлорид лития и йодид меди(I).
Стадия 9-3:
Данная стадия является стадией синтезирования соединения (I-b) из соединения (9-8) в качестве исходного вещества с использованием способа получения, описанного выше (стадия 3-4).
Соединение формулы (I) по настоящему изобретению, где по меньшей мере один из R1 и R2 представляет собой С1-6 алкильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, C1-6 алкилкарбонильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, C6-14 арилкарбонильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, C1-6 алкилсульфонильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, C6-14 арилсульфонильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, 3-10-членную карбоциклическую группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, или 5-10-членную гетероциклическую группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, может быть получено дополнительным взаимодействием соединения (I-b), полученного общим способом получения 9, с соответствующим галогенидом, таким как C1-6 алкилгалогенид.
10. Общий способ получения 10:
[Формула 15]

На данной схеме кольцо A, кольцо B, R3, R4, R5, R6, Z, Y, L1, L и LV являются такими, как определено выше.
Общий способ получения 10 представляет собой способ получения соединения (I-b) общей формулы (I) по настоящему изобретению, где L представляет собой одинарную связь и R1 и R2 представляют собой атомы водорода, из соединения (10-1).
Соединение (10-1) может быть получено из коммерчески доступного продукта общим способом получения 1, общим способом получения 5 или комбинацией общего способа получения 1 и способа 2B общего способа получения 2 и может быть также получено способом, описанным в примерах получения в разделе "Примеры".
Стадия 10-1:
Данная стадия является стадией получения соединения (10-2) бензилоксикарбонилированием соединения (10-1).
Реакция может быть осуществлена в таких же условиях, какие обычно используют при бензилоксикарбонилировании аминосоединения, такие как условия, описанные в таком документе, как T. W. Green and P. G. M. Wuts, "Protective Groups in Organic Chemistry, Third Edition", John Wiley & Sons, P. 531-537. Соединение (10-2) может быть получено, например, путем взаимодействия соединения (10-1) с бензилхлорформиатом в смешанном растворителе из 1,4-диоксана и насыщенного раствора бикарбоната натрия.
Стадия 10-2:
Данная стадия является стадией синтезирования соединения (I-b) из соединения (10-2) в качестве исходного вещества с использованием такого же способа, как реакция Сузуки-Мияуры, описанная в показанном выше способе получения (стадия 9-2).
Соединение формулы (I) по настоящему изобретению, где по меньшей мере один из R1 и R2 представляет собой С1-6 алкильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, C1-6 алкилкарбонильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, C6-14 арилкарбонильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, C1-6 алкилсульфонильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, C6-14 арилсульфонильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, 3-10-членную карбоциклическую группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, или 5-10-членную гетероциклическую группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, может быть получено дополнительным взаимодействием соединения (I-b), полученного общим способом получения 10, с соответствующим галогенидом, таким как C1-6 алкилгалогенид.
11. Общий способ получения 11:
[Формула 16]
На данной схеме кольцо A, кольцо B, R3, R4, R5, R6, L1, L, LV, Alk и Prt3 являются такими, как определено выше.
Общий способ получения 11 представляет собой общий способ получения 9 в случае, когда в общей формуле Y представляет собой атом азота и Z представляет собой одинарную связь. Данный способ является способом получения соединения (11-4), которое представляет собой промежуточное соединение в синтезе соединения (I) по настоящему изобретению, полученное из соединения (11-1).
Соединение (11-1) может быть получено из коммерчески доступного продукта общим способом получения 5 или общим способом получения 8 и может быть также получено способом, описанным в примерах получения в разделе "Примеры".
Стадия 11-1:
Данная стадия является стадией синтезирования соединения (11-2) из соединения (11-1) в качестве исходного вещества с использованием способа получения, описанного выше (стадия 9-1).
Стадия 11-2:
Данная стадия является стадией синтезирования соединения (11-3) из соединения (11-2) в качестве исходного вещества с использованием способа получения, описанного выше (стадия 9-2).
Стадия 11-3:
Данная стадия является стадией получения соединения (11-4) снятием защиты с аминогруппы соединения (11-3). Аминозащитную группу, используемую на данной стадии, особо не ограничивают. Если, например, Prt3 представляет собой 2,4-диметоксибензильную группу, то данная стадия может быть осуществлена в таких же условиях, какие обычно используют (такие как условия, описанные в таком документе, как Tetrahedron Vol. 47, No. 26, pp 4591-4602, 1991). На данной стадии, если Prt3 представляет собой 2,4-диметоксибензильную группу, то может быть снята одна защитная Boc группа одновременно со снятием 2,4-диметоксибензильной защитной группы. Если на данной стадии Prt3 представляет собой 2,4-диметоксибензильную группу, то растворитель, используемый на данной стадии, особо не ограничивают, лишь бы он не замедлял реакцию и позволял исходному веществу растворяться в нем до определенной степени. Например, растворителем в реакции первой стадии может быть метиленхлорид или хлороформ, а растворителем в реакции второй стадии может быть метанол. Температура реакции на данной стадии обычно составляет от 0°C до комнатной температуры. Время реакции на данной стадии особо не ограничивают, и оно составляет обычно от 0,5 до 24 часов, предпочтительно от 0,5 до 12 часов.
12. Общий способ получения 12:
[Формула 17]

На данной схеме кольцо A, кольцо B, R3, R4, R5, R6, Y, Z, L и LV являются такими, как определено выше.
Общий способ получения 12 представляет собой способ получения соединения (I-b) общей формулы (I) по настоящему изобретению, где L представляет собой одинарную связь и R1 и R2 представляют собой атомы водорода, из соединения (9-2).
Соединение (9-2) может быть получено из коммерчески доступного продукта общим способом получения 9 и может быть также получено способом, описанным в примерах получения в разделе "Примеры". Соединение (12-2) может быть коммерчески доступным продуктом, используемым как он есть, может быть также получено из коммерчески доступного продукта способом, известным специалисту в данной области техники, и, кроме того, может быть получено способом, описанным в примерах получения в разделе "Примеры".
Стадия 12-1:
Данная стадия является стадией получения соединения (12-1) реакцией сочетания соединения (9-2) с использованием переходного металла.
Реакция на данной стадии может быть осуществлена в таких же условиях, какие обычно используют в реакции сочетания с использованием переходного металла, такие как условия, описанные в Org. Lett. 2007, Vol. 9, No. 4, 558-562 и Bioorg. Med. Chem, 14 (2006) 4944-4957. В частности, соединение (12-1) может быть получено, например, путем взаимодействия соединения (9-2) с бис(пинаколато)дибораном в условиях нагревания в растворителе, таком как ДМФА, в присутствии катализатора, такого как ацетат калия или дихлорид [1,1'-бис(дифенилфосфино)ферроцен]палладия (II).
Металлоорганический катализатор, используемый в данной реакции, особо не ограничивают. Предпочтительные примеры металлоорганического катализатора включают металлические катализаторы, такие как дихлорбис(трифенилфосфин)палладий(II), дихлорид [1,1'-бис(дифенилфосфино)ферроцен]палладия(II), бис(трет-бутилфосфин)палладий(0), ацетат палладия(II) и [1,3-бис(дифенилфосфино)пропан]никель(II). Количество используемого металлоорганического катализатора составляет примерно 0,001-0,5 эквивалента относительно исходного вещества. Растворитель, используемый в данной реакции, особо не ограничивают, лишь бы он не замедлял реакцию. Предпочтительные примеры растворителя включают бензол, толуол, ксилол, N,N-диметилформамид, 1-метил-2-пирролидон, диметилсульфоксид, тетрагидрофуран, 1,2-диметоксиэтан, 1,4-диоксан, ацетонитрил и пропионитрил. Температуру реакции особо не ограничивают, и она, например, находится обычно в пределах от температуры охлаждения льдом до температуры кипения растворителя с обратным холодильником, а предпочтительно от комнатной температуры до температуры кипения растворителя с обратным холодильником. Время реакции особо не ограничивают, и оно составляет обычно от 0,5 до 72 часов, предпочтительно от 0,5 до 24 часов.
Более предпочтительный результат, такой как повышенный выход, может быть достигнут осуществлением данной реакции в присутствии основания. Такое основание особо не ограничивают. Предпочтительные примеры основания включают такие основания, как ацетат калия, ацетат натрия, карбонат натрия, карбонат калия, карбонат цезия, фосфат калия, фторид калия, триэтиламин и N,N-диизопропилэтиламин.
Стадия 12-2:
Данная стадия является стадией синтезирования соединения (12-3) из соединения (12-1) в качестве исходного вещества с использованием способа получения, описанного выше (стадия 9-2).
Стадия 12-3:
Данная стадия является стадией синтезирования соединения (I-b) из соединения (12-3) в качестве исходного вещества с использованием способа получения, описанного выше (стадия 3-4).
Соединение формулы (I) по настоящему изобретению, где по меньшей мере один из R1 и R2 представляет собой С1-6 алкильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, C1-6 алкилкарбонильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, C6-14 арилкарбонильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, C1-6 алкилсульфонильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, C6-14 арилсульфонильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, 3-10-членную карбоциклическую группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, или 5-10-членную гетероциклическую группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, может быть получено дополнительным взаимодействием соединения (I-b), полученного общим способом получения 12, с соответствующим галогенидом, таким как C1-6 алкилгалогенид.
13. Общий способ получения 13:
[Формула 18]
На данной схеме кольцо A, кольцо B, R3, R4, R5, R6, Y и Z являются такими, как определено выше.
Общий способ получения 13 представляет собой способ получения соединения (I-a) общей формулы (I) по настоящему изобретению, где L представляет собой -NHCO- и R1 и R2 представляют собой атомы водорода, из соединения (12-1).
Соединение (12-1) может быть получено из коммерчески доступного продукта общим способом получения 12 и может быть также получено способом, описанным в примерах получения в разделе "Примеры".
Стадия 13-1:
Данная стадия является стадией получения соединения (13-1) путем взаимодействия соединения (12-1) с азидом натрия в присутствии медного катализатора.
Реакция на данной стадии может быть осуществлена, например, в таких же условиях, как описано в Org. Lett. 2007, Vol. 9, No. 5, 761-764 и Tetrahedron Lett. 2007, 48, 3525-3529. В частности, соединение (13-1) может быть получено, например, взаимодействием соединения (12-1) с азидом натрия при комнатной температуре с использованием растворителя, такого как метанол, в присутствии катализатора, такого как ацетат меди(II).
Катализатор, используемый в данной реакции, особо не ограничивают. Предпочтительные примеры катализатора включают металлические катализаторы, такие как ацетат меди(II), сульфат меди(II), йодид меди(I) и хлорид меди(I). Количество используемого катализатора особо не ограничивают, и оно составляет обычно примерно 0,1-0,5 эквивалента относительно исходного вещества. Растворитель, используемый в данной реакции, особо не ограничивают, лишь бы он не замедлял реакцию и позволял исходному веществу растворяться в нем до определенной степени. Предпочтительные примеры растворителя включают метанол, N,N-диметилформамид, 1-метил-2-пирролидон, тетрагидрофуран, 1,2-диметоксиэтан, 1,4-диоксан, ацетонитрил, пропионитрил и дихлорметан. Температуру реакции особо не ограничивают, и она, например, находится обычно в пределах от температуры охлаждения льдом до температуры кипения растворителя с обратным холодильником, а предпочтительно от комнатной температуры до температуры кипения растворителя с обратным холодильником. Время реакции особо не ограничивают, и оно составляет обычно от 0,5 до 100 часов, предпочтительно от 1 до 72 часов.
Более предпочтительный результат, такой как повышенный выход, может быть достигнут осуществлением данной реакции в атмосфере кислорода.
Стадия 13-2:
Данная стадия является стадией получения соединения (13-2) восстановлением азидосоединения (13-1). Реакция на данной стадии может быть осуществлена, например, в таких же условиях, как описано в J. Org. Chem. 2003, 68, 4693-4699. В частности, соединение (13-2) может быть получено, например, растворением соединения (13-1) в растворителе, таком как метанол, и взаимодействием полученного раствора с боргидридом натрия.
Стадия 13-3:
Данная стадия является стадией синтезирования соединения (13-3) из соединения (13-2) в качестве исходного вещества с использованием способа получения, описанного выше (стадия 3-3).
Стадия 13-4:
Данная стадия является стадией синтезирования соединения (I-a) из соединения (13-3) в качестве исходного вещества с использованием способа получения, описанного выше (стадия 3-4).
Соединение формулы (I) по настоящему изобретению, где по меньшей мере один из R1 и R2 представляет собой С1-6 алкильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, C1-6 алкилкарбонильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, C6-14 арилкарбонильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, C1-6 алкилсульфонильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, C6-14 арилсульфонильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, 3-10-членную карбоциклическую группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, или 5-10-членную гетероциклическую группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, может быть получено дополнительным взаимодействием соединения (I-a), полученного общим способом получения 13, с соответствующим галогенидом, таким как C1-6 алкилгалогенид.
Альтернативно, -NHCO- группы L в соединении (I-a) по настоящему изобретению может быть превращена в группу -NReCO- (где Re представляет собой С1-6 алкильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α) дополнительным взаимодействием соединения (I-a), полученного общим способом получения 13, с соответствующим галогенидом, таким как C1-6 алкилгалогенид.
Соединение формулы (I) по настоящему изобретению, где L представляет собой -NReSO2-, может быть получено с использованием соответствующего производного сульфонилгалогенида вместо соединения (3-4) или (3-5), используемого в общем способе получения 13.
14. Общий способ получения 14:
[Формула 19-1]

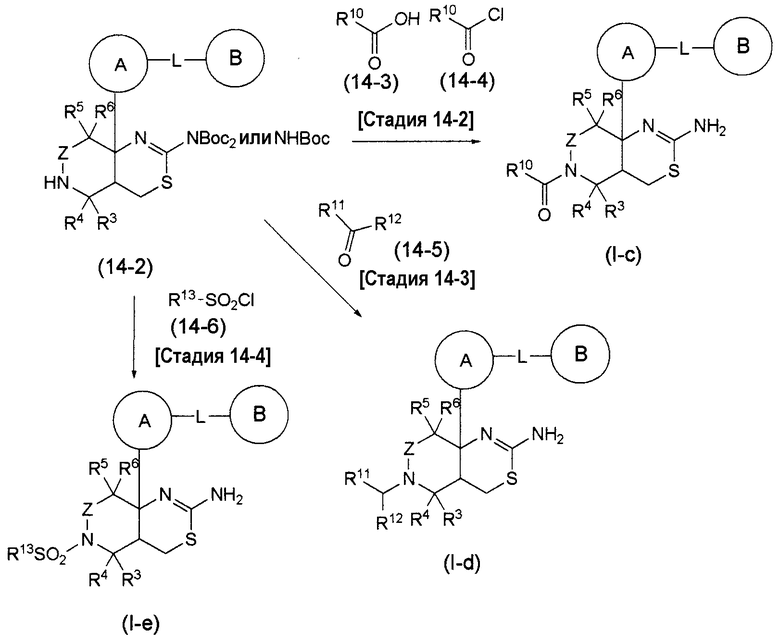
[Формула 19-2]
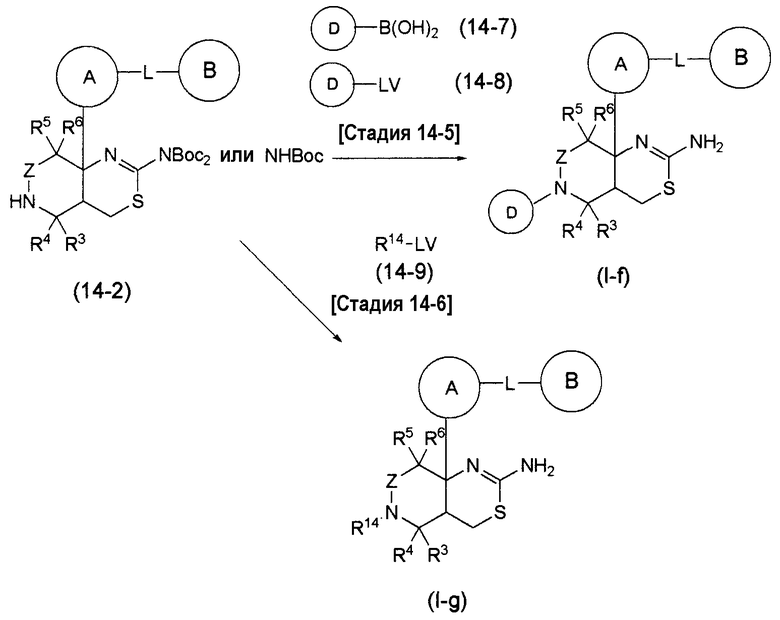
На данной схеме кольцо A, кольцо B, R3, R4, R5, R6, L, Z, Prt3 и LV являются такими, как определено выше; кольцо D представляет собой C6-14 арильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, или 5-6-членную гетероарильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α; R10 представляет собой C6-14 арильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, 5-10-членную гетероциклическую группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, C3-8 циклоалкильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, C1-6 алкильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, или 3-10-членную карбоциклическую группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α; R11 и R12 каждый независимо представляет собой атом водорода, C6-14 арильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, 5-10-членную гетероциклическую группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, C3-8 циклоалкильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, C1-6 алкильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, или 3-10-членную карбоциклическую группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, или R11 и R12 вместе могут образовывать кольцо; R13 представляет собой C6-14 арильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, 5-10-членную гетероциклическую группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, C3-8 циклоалкильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, C1-6 алкильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, или 3-10-членную карбоциклическую группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α; и R14 представляет собой C7-12 аралкильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α.
Общий способ получения 14 представляет собой способ получения соединений (I-c)-(I-g) общей формулы (I) по настоящему изобретению, где Y представляет собой атом азота и R1 и R2 представляют собой атомы водорода, из соединения (14-1).
Соединение (14-1) может быть получено из коммерчески доступного продукта общим способом получения 5, общим способом получения 8, общим способом получения 9, общим способом получения 10, общим способом получения 11, общим способом получения 12 или их комбинацией и может быть также получено способом, описанным в примерах получения в разделе "Примеры".
Каждое из соединений (14-3), (14-4), (14-5), (14-6), (14-7), (14-8) и (14-9) может быть коммерчески доступным продуктом, используемым как он есть, может быть также получено из коммерчески доступного продукта способом, известным специалисту в данной области техники, и, кроме того, может быть получено способом, описанным в примерах получения в разделе "Примеры".
Стадия 14-1:
Данная стадия является стадией получения соединения (14-2) снятием защиты с аминогруппы соединения (14-1).
Реакция может быть осуществлена в таких же условиях, какие обычно используют при удалении защитной группы аминосоединения, такие как условия, описанные в таком документе, как T. W. Green and P. G. M. Wuts, "Protective Groups in Organic Chemistry, Third Edition", John Wiley & Sons, P. 494-572.
Аминозащитную группу, используемую на данной стадии, особо не ограничивают. Если Prt3 представляет собой, например, 2,4-диметоксибензильную группу, то данная стадия может быть осуществлена в таких же условиях, как обычно используемые условия (такие как условия, описанные в таком документе, как Tetrahedron Vol. 47, No. 26, pp 4591-4602, 1991). Может быть снята одна защитная Boc группа одновременно со снятием 2,4-диметоксибензильной защитной группы. Растворитель, используемый на данной стадии, особо не ограничивают, лишь бы он не замедлял реакцию и позволял исходному веществу растворяться в нем до определенной степени. Например, растворителем в реакции первой стадии может быть метиленхлорид или хлороформ, а растворителем в реакции второй стадии может быть метанол. Температура реакции на данной стадии обычно составляет от 0°C до комнатной температуры. Время реакции на данной стадии особо не ограничивают, и оно составляет обычно от 0,5 до 24 часов, предпочтительно от 0,5 до 12 часов.
Если Prt3 представляет собой бензилоксикарбонильную группу, то соединение (14-2) может быть получено снятием защиты у соединения (14-1) путем гидрирования с использованием палладия на углероде в качестве катализатора в растворителе, таком как, например, спирт.
Стадия 14-2:
Данная стадия является стадией синтезирования соединения (I-c) из соединения (14-2) в качестве исходного вещества с использованием способа получения, описанного выше ((стадия 3-3) и (стадия 3-4)).
Стадия 14-3:
Данная стадия является стадией синтезирования соединения (I-d) с использованием способа получения, описанного выше (стадия 3-4), после реакции восстановительного аминирования соединения (14-2) соединением (14-5).
Реакция восстановительного аминирования может быть осуществлена в таких же условиях, какие обычно используют в реакции восстановительного аминирования между карбонильным соединением и аминосоединением. Реакцию восстановления на данной стадии особо не ограничивают. Примеры реакции восстановления включают реакцию восстановительного аминирования с использованием восстановителя, такого как борановое или боргидридное комплексное соединение. Примеры реакции восстановительного аминирования с использованием боргидридного комплексного соединения включают способ, описанный в таком документе, как J. Org. Chem. 1996, 61, 3849. Примеры боргидридного комплексного соединения, которое может быть использовано, включают боргидрид натрия, цианоборгидрид натрия и триацетоксиборгидрид натрия.
Если боргидридное комплексное соединение используют в качестве восстановителя, то растворитель особо не ограничивают, лишь бы он не замедлял реакцию и позволял исходному веществу растворяться в нем до определенной степени. Конкретные примеры растворителя, который может быть использован, включают метанол, этанол, тетрагидрофуран, N,N-диметилформамид, дихлорметан и 1,2-дихлорэтан. Более предпочтительный результат, такой как повышенный выход, может быть получен осуществлением данной реакции в присутствии кислоты. Такую кислоту особо не ограничивают. Предпочтительные примеры кислоты включают минеральные кислоты, такие как хлористо-водородная кислота, органические кислоты, такие как уксусная кислота, и кислоты Льюиса, такие как хлорид цинка, комплекс трифторид бора-диэтиловый эфир и тетраизопропоксид титана(IV).
Стадия 14-4:
Данная стадия является стадией синтезирования соединения (I-e) с использованием способа получения, описанного выше (стадия 3-4), после сульфонилирования аминогруппы соединения (14-2). Для сульфонилирования реакцией, известной специалисту в данной области техники, является реакция с использованием производного сульфонилхлорида.
Стадия 14-5:
Данная стадия является стадией синтезирования соединения (I-f) с использованием способа получения, описанного выше (стадия 3-4), после реакции сочетания между соединением (14-2) и соединением (14-7) или (14-8). На данной стадии используют реакцию, такую как сочетание с использованием комплекса переходного металла или тому подобное, или нуклеофильное ароматическое замещение (SNAr реакция).
Реакция сочетания на данной стадии может быть осуществлена, например, в таких же условиях, как описано в Org. Lett. 2007, Vol. 9, No. 5, 761-764 и Org. Lett. 2003, Vol. 5, No. 23, 4397-4400. В частности, реакция сочетания может быть осуществлена путем взаимодействия соединения (14-2) с соединением (14-7) при температуре в пределах от комнатной температуры до 50°C с использованием растворителя, такого как дихлорметан, в присутствии молекулярного сита 4A и катализатора, такого, например, как ацетат меди(II).
Катализатор, используемый в данной реакции, особо не ограничивают. Предпочтительные примеры катализатора включают металлические катализаторы, такие как ацетат меди(II), сульфат меди(II), йодид меди(I) и хлорид меди(I). Количество используемого катализатора особо не ограничивают, и оно составляет обычно примерно 0,1-0,5 эквивалента относительно исходного вещества. Растворитель, используемый в данной реакции, особо не ограничивают, лишь бы он не замедлял реакцию и позволял исходному веществу растворяться в нем до определенной степени. Предпочтительные примеры растворителя включают N,N-диметилформамид, 1-метил-2-пирролидон, тетрагидрофуран, 1,2-диметоксиэтан, 1,4-диоксан, ацетонитрил, пропионитрил и дихлорметан. Температуру реакции особо не ограничивают, и она находится обычно в пределах от температуры охлаждения льдом до температуры кипения растворителя с обратным холодильником, а предпочтительно, например, от комнатной температуры до температуры кипения растворителя с обратным холодильником. Время реакции особо не ограничивают, и оно составляет обычно от 0,5 до 100 часов, предпочтительно от 1 до 72 часов.
Более предпочтительный результат, такой как повышенный выход, может быть достигнут осуществлением данной реакции в атмосфере кислорода.
Когда данная стадия представляет собой реакцию сочетания с использованием комплекса переходного металла или тому подобное в качестве катализатора, реакция может быть осуществлена с использованием соединения (14-2) и соединения (14-8), которым является производное арилгалогенида, производное гетероарилгалогенида, производное арилокситрифторметансульфоната или производное гетероарилокситрифторметансульфоната, в таких же условиях, как обычно используемые условия (такие как условия, описанные в таком документе, как Org. Lett. 2002, Vol. 4, No. 4, 581). Производное арилгалогенида, производное гетероарилгалогенида, производное арилокситрифторметансульфоната или производное гетероарилокситрифторметансульфоната, используемое на данной стадии, может быть коммерчески доступным продуктом, используемым как он есть, и может быть также получено из коммерчески доступного продукта способом, известным специалисту в данной области техники. Примеры комплекса переходного металла, используемого на данной стадии, включают дихлорбис(трифенилфосфин)палладий(II), тетракис(трифенилфосфин)палладий(0), трис(дибензилиденацетон)палладий(0) и комплекс медь-диольный лиганд. В данной реакции может быть также добавлен фосфорный лиганд (такой как предпочтительно трифенилфосфин, три-о-толилфосфин, три-трет-бутилфосфин, 2,2'-бис(дифенилфосфино)-1,1'бинафтил или 1,1'-бис(дифенилфосфино)ферроцен), чтобы получить положительные результаты (такие как пониженная температура реакции, уменьшенное время реакции и повышенный выход). Если используемым комплексом переходного металла является палладиевый комплекс, то реакцию на данной стадии предпочтительно проводят в атмосфере азота или аргона. Растворитель, используемый на данной стадии, особо не ограничивают, лишь бы он не замедлял реакцию и позволял исходному веществу растворяться в нем до определенной степени. Например, если используемым комплексом переходного металла является палладиевый комплекс, то может быть использован N,N-диметилформамид, N-метил-2-пирролидон, 1,4-диоксан, толуол, ксилол или тому подобное. Если используемым комплексом переходного металла является медь-диольный комплекс, то может быть использован 2-пропанол или тому подобное. Температура реакции на данной стадии обычно находится в пределах от комнатной температуры до температуры кипения растворителя с обратным холодильником. Время реакции на данной стадии особо не ограничивают, и оно составляет обычно от 0,5 до 72 часов, предпочтительно от 0,5 до 24 часов.
Если данная стадия представляет собой нуклеофильное ароматическое замещение (SNAr реакция), то реакция может быть осуществлена с использованием соединения (14-2) и соединения (14-8), которым является производное арилгалогенида, производное гетероарилгалогенида, производное арилокситрифторметансульфоната или производное гетероарилокситрифторметансульфоната, в присутствии основания в таких же условиях, как обычно используемые условия. Производное арилгалогенида, производное гетероарилгалогенида, производное арилокситрифторметансульфоната или производное гетероарилокситрифторметансульфоната, используемое на данной стадии, может быть коммерчески доступным продуктом, используемым как он есть, и может быть также получено из коммерчески доступного продукта способом, известным специалисту в данной области техники. Нуклеофильное ароматическое замещение (SNAr реакция), используемое на данной стадии, может быть осуществлено в таких же условиях, как обычно используемые условия (такие как условия в соответствии со способами, описанными в таких документах, как Org. Prep. Proced. int. 39 (2007) 4, 399-402, Bioorg. Med. Chem. Lett. 15 (2005) 9, 2409-2413 и Bioorg. Med. Chem. Lett. 15 (2005) 3, 719-723). Растворитель, используемый на данной стадии, особо не ограничивают, лишь бы он не замедлял реакцию и позволял исходному веществу растворяться в нем до определенной степени. Примеры растворителя, который может быть использован, включают N,N-диметилформамид, N-метил-2-пирролидон, диметилсульфоксид и ацетонитрил. Используемое основание на данной стадии особо не ограничивают. Примеры основания включают карбонат калия, карбонат натрия, гидрид натрия и тетрабутиламмонийфторид. Предпочтительно используют карбонат калия, карбонат натрия и тетрабутиламмонийфторид. Температура реакции на данной стадии обычно находится в пределах от комнатной температуры до температуры кипения растворителя с обратным холодильником. Время реакции на данной стадии особо не ограничивают, и оно составляет обычно от 0,5 до 24 часов, предпочтительно от 0,5 до 12 часов.
Стадия 14-6:
Данная стадия является стадией синтезирования соединения (I-g) из соединения (14-2) в качестве исходного вещества с использованием способа получения, описанного выше ((стадия 8-1) и (стадия 3-4)).
Соединение формулы (I) по настоящему изобретению, где по меньшей мере один из R1 и R2 представляет собой С1-6 алкильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, C1-6 алкилкарбонильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, C6-14 арилкарбонильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, C1-6 алкилсульфонильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, C6-14 арилсульфонильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, 3-10-членную карбоциклическую группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, или 5-10-членную гетероциклическую группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, может быть получено дополнительным взаимодействием любого из соединений (I-c)-(I-g), полученного общим способом получения 14, с соответствующим галогенидом, таким как C1-6 алкилгалогенид.
Соединение формулы (I) по настоящему изобретению, полученное описанным выше образом, может быть при необходимости преобразовано в фармацевтически приемлемую соль традиционным способом. Соль может быть получена способом, в котором подходящим образом объединены способы, обычно используемые в области химии органического синтеза, и тому подобное. Конкретные примеры способа включают нейтрализационное титрование свободного раствора соединения по настоящему изобретению раствором кислоты. Соединение формулы (I) по настоящему изобретению можно, если необходимо, преобразовать в сольват, подвергая соединение известной самой по себе сольватобразующей реакции.
Конденсированное производное аминодигидротиазина или его фармацевтически приемлемая соль или сольват по настоящему изобретению обладает очень высоким ингибирующим эффектом в отношении продукции Aβ или эффектом ингибирования BACE1 и является очень полезным(ой) в качестве профилактического или терапевтического средства при нейродегенеративном заболевании, вызываемом Aβ, типичным примером которого является деменция Альцгеймера.
Конденсированное производное аминодигидротиазина или его фармацевтически приемлемая соль или сольват по настоящему изобретению могут быть получены в виде лекарственной формы традиционным способом. Предпочтительные примеры лекарственной формы включают таблетки, таблетки с покрытием, такие как таблетки с пленочным покрытием и таблетки с сахарным покрытием, микрогранулы, гранулы, порошки, капсулы, сиропы, пастилки, ингалянты, суппозитории, растворы для инъекций, мази, глазные капли, капли в нос, ушные капли, припарки и лосьоны.
Указанные твердые препараты, такие как таблетки, капсулы, гранулы и порошки, могут содержать обычно 0,01-100 масс.%, предпочтительно 0,1-100 масс.%, конденсированного производного аминодигидротиазина или его фармацевтически приемлемой соли или сольвата по настоящему изобретению в качестве активного компонента.
Для изготовления лекарственной формы активный компонент традиционным способом смешивают с компонентами, обычно используемыми в качестве материалов для фармацевтического препарата, и добавляют обычно используемые эксципиент, разрыхлитель, связывающее вещество, смазывающее вещество, красящее вещество и корригент, а также добавляют при необходимости, например, стабилизатор, эмульгатор, вещество, облегчающее абсорбцию, поверхностно-активное вещество, регулятор pH, консервант и антиоксидант. Примеры таких компонентов включают животные и растительные масла, такие как соевое масло, говяжий жир и синтетический глицерид; углеводороды, такие как жидкий парафин, сквалан и твердый парафин; эфирные масла, такие как октилдодецилмиристат и изопропилмиристат; высшие спирты, такие как цетостеариловый спирт и бегениловый спирт; силиконовую смолу; силиконовое масло; поверхностно-активные вещества, такие как сложный эфир полиоксиэтилена и жирной кислоты, сложный эфир сорбитана и жирной кислоты, сложный эфир глицерина и жирной кислоты, сложный эфир полиоксиэтиленсорбитана и жирной кислоты, полиоксиэтилен гидрогенизированное касторовое масло и блок-сополимер полиоксиэтилена и полиоксипропилена; водорастворимые полимеры, такие как гидроксиэтилцеллюлоза, полиакриловая кислота, карбоксивиниловый полимер, полиэтиленгликоль, поливинилпирролидон и метилцеллюлоза; низшие спирты, такие как этанол и изопропанол; многоатомные спирты, такие как глицерин, пропиленгликоль, дипропиленгликоль и сорбит; сахара, такие как глюкоза и сахароза; неорганические порошки, такие как кремниевый ангидрид, алюмосиликат магния и силикат алюминия, и дистиллированную воду. Примеры используемого эксципиента включают лактозу, кукурузный крахмал, сахарозу, глюкозу, маннит, сорбит, кристаллическую целлюлозу и диоксид кремния. Примеры используемого связывающего вещества включают поливиниловый спирт, простой поливиниловый эфир, метилцеллюлозу, этилцеллюлозу, аравийскую камедь, трагакант, желатин, шеллак, гидроксипропилметилцеллюлозу, гидроксипропилцеллюлозу, поливинилпирролидон, блок-сополимер полипропиленгликоля и полиоксиэтилена и меглумин. Примеры используемого разрыхлителя включают крахмал, агар, желатиновый порошок, кристаллическую целлюлозу, карбонат кальция, бикарбонат натрия, цитрат кальция, декстрин, пектин и кальций-карбоксиметилцеллюлозу. Примеры используемого смазывающего вещества включают стеарат магния, тальк, полиэтиленгликоль, диоксид кремния и гидрогенизированное растительное масло. Примеры используемого красящего вещества включают красящие вещества, приемлемые для фармацевтических препаратов. Примеры используемого корригента включают порошок какао, ментол, эмпазм (empasm), мятное масло, борнеол и порошок корицы. Очевидно, что указанные компоненты не ограничиваются указанными добавками.
Например, препарат для перорального применения получают добавлением конденсированного производного аминодигидротиазина или его фармацевтически приемлемой соли или сольвата по настоящему изобретению в качестве активного компонента, эксципиента и, если требуется, связывающего вещества, разрыхлителя, смазывающего вещества, красящего вещества, корригента и тому подобное и затем превращением смеси в порошок, микрогранулы, гранулы, таблетки, таблетки с покрытием, капсулы или тому подобное традиционным способом. Очевидно, что таблетки или гранулы при необходимости могут быть снабжены подходящим покрытием, например сахарным покрытием.
Например, сироп или препарат для инъекций получают добавлением регулятора рН, солюбилизатора, изотонизирующего средства и тому подобное и, если требуется, солюбилизирующего средства, стабилизатора и тому подобное традиционным способом. Препарат для инъекций может представлять собой предварительно приготовленный раствор или порошок как таковой, или порошок, содержащий подходящую добавку, который растворяют перед применением. Препарат для инъекций может содержать обычно 0,01-100 масс.%, предпочтительно 0,1-100 масс.% активного компонента. Кроме того, жидкий препарат для перорального применения, такой как суспензия или сироп, может содержать обычно 0,01-100 масс.%, предпочтительно 0,1-100 масс.% активного компонента.
Например, препарат для наружного применения может быть получен любым традиционным способом без особых ограничений. В качестве материала для основы может быть использован любой из многочисленных материалов, обычно применяемых для получения фармацевтического препарата, квазилекарственного средства, косметического средства или тому подобное. Примеры материала для основы включают такие материалы, как животные и растительные масла, минеральные масла, эфирные масла, воски, высшие спирты, жирные кислоты, силиконовые масла, поверхностно-активные вещества, фосфолипиды, спирты, многоатомные спирты, водорастворимые полимеры, глинистые минералы и дистиллированную воду. При необходимости могут быть добавлены регулятор рН, антиоксидант, хелатообразователь, консервант и фунгицид, красящее вещество, вкусовое вещество или тому подобное. Кроме того, при необходимости могут быть подмешаны компоненты, такие как компонент, обладающий эффектом индуцирования дифференциации, усилитель кровотока, бактерицид, противовоспалительное средство, активатор клеток, витамин, аминокислота, увлажнитель и кератолитическое средство.
Дозу конденсированного производного аминодигидротиазина или его фармацевтически приемлемой соли или сольвата по настоящему изобретению в качестве активного компонента изменяют, например, в соответствии со степенью проявления симптомов, возрастом, полом, массой тела, способом введения, типом соли и конкретным видом заболевания. Обычно активный компонент вводят перорально взрослому в количестве примерно от 30 мкг до 10 г, предпочтительно от 100 мкг до 5 г и более предпочтительно от 100 мкг до 1 г в сутки или вводят взрослому путем инъекции примерно от 30 мкг до 1 г, предпочтительно от 100 мкг до 500 мг и более предпочтительно от 100 мкг до 300 мг в сутки в виде разовой дозы или в виде разделенных доз, соответственно.
Наилучший вариант осуществления изобретения
Настоящее изобретение описано более подробно ниже со ссылками на примеры, примеры получения и примеры испытания. Однако настоящее изобретение не ограничивается этим. Сокращения, используемые в примерах, являются обычными сокращениями, известными специалисту в данной области техники. Некоторые сокращения показаны ниже:
ТГФ: тетрагидрофуран
ДМФА: N,N-диметилформамид
ТФУК: трифторуксусная кислота
EDC·HCl: гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида
пТСХ: препаративная тонкослойная хроматография
ЖХ-МС: жидкостная хроматография-масс-спектрометрия
PyBOP: гексафторфосфат бензотриазол-1-илокситрис(пирролидино)фосфония
Pd2DBA3: трис(дибензилиденацетон)дипалладий
Pd(t-Bu3P)2: бис(три-трет-бутилфосфин)палладий
Химические сдвиги в спектре протонного ядерного магнитного резонанса записаны в единицах δ (м.д.) относительно тетраметилсилана и константы связывания записаны в герцах (Гц). Виды сигналов в спектре обозначены как с: синглет, д: дублет, т: триплет, уш.: уширенный.
"Комнатная температура" в следующих примерах и примерах получения обычно относится к температуре в пределах от примерно 10°C до примерно 35°C. "%" означает масс.%, если не указано иное.
Пример получения 1
Синтез трет-бутил (±)-[(4aR*,8aS*)-8a-(5-амино-2-фторфенил)-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата
[Формула 20]

(1) Синтез этил 2-трифторметансульфонилоксициклогекс-1-енкарбоксилата
Диизопропилэтиламин (38,0 мл) добавляли к раствору этил 2-оксоциклогексанкарбоксилата (8,00 г) в дихлорметане (100 мл) в атмосфере азота при -78°C. После перемешивания при той же температуре в течение 10 минут добавляли трифторметансульфоновый ангидрид (8,80 мл). Смесь перемешивали в течение ночи при постепенном нагревании до комнатной температуры. Смесь промывали водой и затем дважды промывали 5% раствором лимонной кислоты (150 мл). Органический слой сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения в виде неочищенного продукта (15,5 г). Неочищенный продукт использовали для следующей реакции без дополнительной очистки.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,33 (т, J=7,2 Гц, 3H), 1,66 (м, 2H), 1,78 (м, 2H), 2,40 (м, 2H), 2,48 (м, 2H), 4,28 (кв, J=7,2 Гц, 2H).
(2) Синтез этил 2-(2-фторфенил)циклогекс-1-енкарбоксилата
Этанол (100 мл) добавляли к раствору этил 2-трифторметансульфонилоксициклогекс-1-енкарбоксилата, полученного в примере получения 1-(1) (17,0 г), в толуоле (200 мл). Добавляли 2-фторфенилбороновую кислоту (7,74 г) и тетракис(трифенилфосфин)палладий (1,60 г). Добавляли 1 н. раствор карбоната натрия (55,3 мл), после чего атмосферу реакции заменяли азотом. Реакционный раствор нагревали до 80°C и перемешивали в течение восьми часов. После доведения до комнатной температуры избыток этанола выпаривали при пониженном давлении и остаток экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (10,5 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 0,87 (т, J=7,2 Гц, 3H), 1,75 (м, 4H), 2,36 (м, 2H), 2,46 (м, 2H), 3,89 (кв, J=7,2 Гц, 2H), 6,99-7,08 (м, 3H), 7,22 (м, 1H).
(3) Синтез [2-(2-фторфенил)циклогекс-1-енил]метанола
Алюмогидрид лития (1,90 г) помещали в колбу для отгонки и добавляли к нему ТГФ (300 мл) на ледяной бане. Раствор этил 2-(2-фторфенил)циклогекс-1-енкарбоксилата, полученного в примере получения 1-(2) (10,3 г), в ТГФ (100 мл) добавляли по каплям к реакционному раствору при той же температуре и смесь перемешивали в течение одного часа. Последовательно добавляли к реакционному раствору воду (1,90 мл), 5 н. раствор гидроксида натрия (1,90 мл) и воду (5,70 мл). Дополнительно добавляли безводный сульфат магния с последующим экстрагированием этилацетатом. Нерастворимое вещество отделяли фильтрованием и фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (8,88 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,22 (дт, J=2,0, 5,2 Гц, 1H), 1,75 (уш., 4H), 2,25 (уш., 2H), 2,30 (уш., 2H), 3,86 (д, J=5,2 Гц, 2H), 7,04 (т, J=9,2 Гц, 1H), 7,08-7,13 (м, 2H), 7,22 (м, 1H).
(4) Синтез 1-(2-хлорметилциклогекс-1-енил)-2-фторбензола
N,N-Диизопропилэтиламин (14,7 мл) добавляли к раствору [2-(2-фторфенил)циклогекс-1-енил]метанола, полученного в примере получения 1-(3) (8,88 г), в дихлорметане (300 мл). Метансульфонилхлорид (4,00 мл) добавляли по каплям к реакционному раствору на ледяной бане. Реакционный раствор постепенно нагревали до комнатной температуры и перемешивали в течение ночи. К реакционному раствору добавляли воду с последующим экстрагированием хлороформом. Органический слой сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении. Полученный неочищенный продукт очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (5,88 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,75 (уш.с, 4H), 2,27 (м, 4H), 3,89 (уш.с, 2H), 7,03-7,29 (м, 4H).
(5) Синтез гидрохлорида 2-[2-(2-фторфенил)циклогекс-1-енилметил]изотиомочевины
Тиомочевину (2,09 г) добавляли к раствору 1-(2-хлорметилциклогекс-1-енил)-2-фторбензола, полученного в примере получения 1-(4) (5,88 г), в этаноле (200 мл). Реакционный раствор нагревали до 80°C и перемешивали в течение 270 минут. Тиомочевину (399 мг) добавляли к реакционному раствору с последующим перемешиванием при той же температуре в течение одного часа. После охлаждения до комнатной температуры растворитель выпаривали при пониженном давлении. К остаточному сиропу добавляли диэтиловый эфир и этилацетат. Белое твердое вещество осаждали ультразвуковой обработкой. После отстаивания при комнатной температуре в течение 30 минут удаляли супернатант. Затем твердое вещество промывали диэтиловым эфиром и снова удаляли супернатант. Полученное твердое вещество сушили при пониженном давлении с получением указанного в заголовке соединения (6,38 г).
1H-ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 1,69 (с, 4H), 2,20 (с, 2H), 2,21 (с, 2H), 3,60 (с, 2H), 7,17 (дт, J=2,0, 7,6 Гц, 1H), 7,24 (м, 2H), 7,38 (м, 1H), 9,04 (уш.с, 3H).
(6) Синтез (±)-(4aR*,8aS*)-8a-(2-фторфенил)-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-иламина
Гидрохлорид 2-[2-(2-фторфенил)циклогекс-1-енилметил]изотиомочевины, полученный в примере получения 1-(5) (6,38 г), растворяли в ТФУК (32,0 мл) при охлаждении льдом и трифторметансульфоновую кислоту (6,40 мл) добавляли по каплям при той же температуре. Реакционный раствор перемешивали в течение ночи при постепенном нагревании до комнатной температуры. Реакционный раствор выливали на лед, разбавляли диэтиловым эфиром и затем нейтрализовали бикарбонатом натрия. Полученную реакционную смесь экстрагировали этилацетатом и органический слой сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении с получением неочищенного продукта. Неочищенный продукт очищали хроматографией на колонке с NH-силикагелем с получением указанного в заголовке соединения (4,58 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,47-1,83 (м, 7H), 2,26 (м, 1H), 2,52 (дд, J=2,8, 12,0 Гц, 1H), 2,70 (ддд, J=4,0, 6,8, 12,0 Гц, 1H), 2,86 (дд, J=4,4, 12,4 Гц, 1H), 7,01 (ддд, J=1,2, 8,0, 12,8 Гц, 1H), 7,08 (дт, J=1,2, 7,6 Гц, 1H), 7,21 (м, 1H), 7,28 (дт, J=2,0, 8,0 Гц, 1H).
(7) Синтез трет-бутил (±)-[(4aR*,8aS*)-8a-(2-фтор-5-нитрофенил)-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата
Соединение, полученное в примере получения 1-(6) (3,50 г), добавляли к концентрированной серной кислоте (25,0 мл) на ледяной бане. К реакционному раствору по каплям добавляли дымящую азотную кислоту (удельный вес: 1,53, 800 мкл) с последующим перемешиванием при той же температуре в течение 30 минут. Реакционную смесь выливали на лед и нейтрализовали 5 н. раствором гидроксида натрия. Полученное твердое вещество собирали фильтрованием через стеклянный фильтр и промывали водой. Твердое вещество растворяли в смешанном растворителе из ТГФ и этилацетата и раствор сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат выпаривали при пониженном давлении при комнатной температуре или ниже с получением промежуточного соединения в виде неочищенного продукта реакции. К раствору неочищенного продукта в ТГФ (100 мл) добавляли триэтиламин (9,20 мл). К реакционному раствору добавляли ди-трет-бутилдикарбонат (8,64 г) с последующим перемешиванием в течение двух суток. Насыщенный раствор бикарбоната натрия добавляли к реакционному раствору с последующим экстрагированием этилацетатом. Органический слой сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (4,70 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,55 (с, 9H), 1,65-1,88 (м, 7H), 2,23 (м 1H), 2,55 (дд, J=2,8, 12,8 Гц, 1H), 2,83 (м, 2H), 7,23 (м, 1H), 8,20 (м, 2H).
(8) Синтез трет-бутил (±)-[(4aR*,8aS*)-8a-(5-амино-2-фторфенил)-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата
К раствору соединения, полученного в примере получения 1-(7) (58,0 мг), в метаноле (14,5 мл) добавляли 10% палладий на углероде (15,8 мг). Атмосферу реакционной системы заменяли водородом с последующим перемешиванием при комнатной температуре в течение двух часов. Реакционный раствор фильтровали через целит и фильтрат концентрировали при пониженном давлении с получением неочищенного продукта указанного в заголовке соединения (58,0 мг). Полученный неочищенный продукт использовали для следующей реакции без дополнительной очистки.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,53 (с, 9H), 1,64-1,87 (м, 7H), 2,35 (м 1H), 2,47 (дд, J=2,8, 12,8 Гц, 1H), 2,88 (м, 2H), 3,64 (с, 2H), 6,54 (м, 2H), 6,85 (м, 1H).
Пример получения 2
Синтез трет-бутил [(4aR*,8aS*)-8a-(5-амино-2-фторфенил)-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата
[Формула 21]

(1) Синтез трет-бутил (+)-[(4aR*,8aS*)-8a-(2-фтор-5-нитрофенил)-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата
Соединение, полученное в примере получения 1-(7) (80,0 мг), оптически разделяли, используя CHIRALPAK™ OJ-H производства Daicel Chemical Industries, Ltd. (2 см × 25 см, подвижная фаза: гексан:этанол = 8:2, скорость потока: 20 мл/мин). Компоненты, имеющие время удерживания от 9,38 до 18,3 минут, собирали с получением указанного в заголовке соединения. Эту же операцию повторяли с получением указанного в заголовке соединения (433 мг; >99% ee) из рацемата (1,00 г).
(2) Синтез трет-бутил [(4aR*,8aS*)-8a-(5-амино-2-фторфенил)-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата
Раствор дитионита натрия (923 мг) в воде (20,0 мл) добавляли по каплям к раствору соединения, полученного в примере получения 2-(1) (433 мг), в этаноле (100 мл) при комнатной температуре. Реакционный раствор перемешивали в течение 30 минут и затем этанол выпаривали при пониженном давлении при комнатной температуре или ниже. Остаток нейтрализовали раствором бикарбоната натрия с последующим экстрагированием этилацетатом. Органический слой сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении. Полученный неочищенный продукт очищали хроматографией на колонке с NH-силикагелем с получением указанного в заголовке соединения (111 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,53 (с, 9H), 1,57-2,05 (м, 7H), 2,36 (дт, J=4,4, 14,4 Гц, 1H), 2,47 (дд, J=2,8, 12,4 Гц, 1H), 2,84 (м, 1H), 2,90 (дд, J=4,0, 12,4 Гц, 1H), 3,64 (с, 2H), 6,55 (м, 2H), 6,85 (м, 1H).
Пример получения 3
Синтез трет-бутил (-)-[(4aR*,7aS*)-7a-(5-амино-2-фторфенил)-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]карбамата
[Формула 22]

(1) Синтез этил 2-трифторметансульфонилоксициклопент-1-енкарбоксилата
N,N-Диизопропилэтиламин (27,2 мл) добавляли к раствору этил 2-оксо-циклопентанкарбоксилата (5,00 г) в дихлорметане (100 мл) при -78°C в течение 10 минут. Трифторметансульфоновый ангидрид (5,92 мл) добавляли по каплям к реакционному раствору при той же температуре. Реакционный раствор перемешивали в течение ночи при постепенном нагревании до комнатной температуры. К реакционной смеси добавляли воду, после чего дважды промывали 5% раствором лимонной кислоты (150 мл). Органический слой сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и к фильтрату добавляли толуол (200 мл). Дихлорметан выпаривали при пониженном давлении при комнатной температуре или ниже с получением раствора указанного в заголовке соединения в толуоле. Соединение использовали для следующей реакции без дополнительной очистки.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,33 (т, J=6,0 Гц, 3H), 2,02 (м, 2H), 2,72 (м, 4H), 4,27 (кв, J=6,0 Гц, 2H).
(2) Синтез этил 2-(2-фторфенил)циклопент-1-енкарбоксилата
2-Фторбензолбороновую кислоту (4,48 г) и тетракис(трифенилфосфин)палладий (740 мг) добавляли к раствору этил 2-трифторметансульфонилоксициклопент-1-енкарбоксилата, полученного в примере получения 3-(1), в толуоле. Затем добавляли к реакционному раствору этанол (100 мл) и 1 н. раствор карбоната натрия (32 мл), после чего заменяли реакционную атмосферу азотом. Реакционный раствор нагревали до 85°C и перемешивали в течение ночи. Реакционный раствор охлаждали до комнатной температуры с последующим экстрагированием этилацетатом. Органический слой сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (7,60 г).
ESI-MS; m/z 235 [M+H].
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,06 (т, J=7,2 Гц, 3H), 2,02 (м, 2H), 2,83 (т, J=7,6 Гц, 4H), 4,05 (кв, J=7,2 Гц, 2H), 7,04 (м, 1H), 7,10 (дт, J=1,2, 7,2 Гц, 1H), 7,19-7,29 (м, 2H).
(3) Синтез [2-(2-фторфенил)циклопент-1-енил]метанола
Раствор этил 2-(2-фторфенил)циклопент-1-енкарбоксилата, полученного в примере получения 3-(2) (7,60 г), в ТГФ (100 мл) добавляли по каплям к суспензии алюмогидрида лития (1,34 г) в ТГФ (300 мл) на ледяной бане. Реакционный раствор перемешивали при той же температуре в течение одного часа. Затем последовательно добавляли по каплям воду (1,35 мл), 5 н. раствор гидроксида натрия (1,35 мл) и воду (4,05 мл) на ледяной бане. К полученной реакционной смеси добавляли безводный сульфат магния с последующим экстрагированием этилацетатом. Реакционную смесь фильтровали через целит и фильтрат концентрировали при пониженном давлении с получением остатка. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (6,50 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,34 (дт, J=1,6, 5,8 Гц, 1H), 2,00 (м, 2H), 2,65 (м, 2H), 2,75 (м, 2H), 4,15 (д, J=5,8 Гц, 2H), 7,03-7,27 (м, 4H).
(4) Синтез 1-(2-хлорметилциклопент-1-енил)-2-фторбензола
N,N-Диизопропилэтиламин (17,2 мл) добавляли к раствору [2-(2-фторфенил)циклопент-1-енил]метанола, полученного в примере получения 3-(3) (6,50 г), в дихлорметане (300 мл) на ледяной бане. Метансульфонилхлорид (2,88 мл) добавляли к реакционному раствору при той же температуре. Затем реакционный раствор нагревали до комнатной температуры и перемешивали в течение ночи. К реакционной смеси добавляли воду с последующим экстрагированием хлороформом. Органический слой сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (7,23 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,02 (м, 2H), 2,67 (м, 2H), 2,77 (м, 2H), 4,11 (с, 2H), 7,07 (м, 1H), 7,15 (м, 1H), 7,23-7,30 (м, 2H).
(5) Синтез (±)-(4aR*,7aS*)-7a-(2-фторфенил)-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-иламина
Тиомочевину (2,60 г) добавляли к раствору 1-(2-хлорметилциклопент-1-енил)-2-фторбензола, полученного в примере получения 3-(4) (7,20 г), в этаноле (100 мл) и смесь перемешивали при кипении с обратным холодильником в течение пяти часов. Реакционный раствор охлаждали до комнатной температуры и выпаривали при пониженном давлении растворитель. Остаточный сироп промывали гептаном, после чего сушили при пониженном давлении. Трифторуксусную кислоту (50,0 мл) и трифторметансульфоновую кислоту (10,0 мл) добавляли к остатку на ледяной бане. Затем реакционную смесь нагревали до комнатной температуры и перемешивали в течение четырех суток. Реакционный раствор выливали на лед, разбавляли простым эфиром и затем нейтрализовали бикарбонатом натрия с последующим экстрагированием этилацетатом. Органический слой сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении с получением остатка. Остаток очищали хроматографией на колонке с NH-силикагелем с получением указанного в заголовке соединения (4,98 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,69-1,95 (м, 5H), 2,62 (м, 1H), 2,74 (дд, J=4,0, 12,4 Гц, 1H), 2,77 (м, 1H), 2,94 (дд, J=3,2, 12,4 Гц, 1H), 7,00 (ддд, J=1,6, 8,4, 12,8 Гц, 1H), 7,09 (ддд, J=1,2, 7,2, 7,6 Гц, 1H), 7,20 (м, 1H), 7,33 (ддд, J=2,0, 8,4, 8,8 Гц, 1H).
(6) Синтез трет-бутил (±)-[(4aR*,7aS*)-7a-(2-фтор-5-нитрофенил)-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]карбамата
Соединение, полученное в примере получения 3-(5) (1,00 г), растворяли в серной кислоте (6,00 мл) на ледяной бане. Дымящую азотную кислоту (215 мкл, удельный вес: 1,53) добавляли по каплям к реакционному раствору при той же температуре с последующим перемешиванием в течение 30 минут. Реакционную смесь выливали на лед и нейтрализовали 5 н. раствором гидроксида натрия. Полученное твердое вещество собирали фильтрованием через стеклянный фильтр и затем растворяли в смешанном растворителе из ТГФ и этилацетата. Раствор сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат выпаривали при пониженном давлении с получением промежуточного соединения. Триэтиламин (2,77 мл) и ди-трет-бутилдикарбонат (2,47 г) добавляли к раствору промежуточного соединения в ТГФ (50 мл) с последующим перемешиванием в течение двух суток. Насыщенный раствор бикарбоната натрия добавляли к реакционной смеси с последующим экстрагированием этилацетатом. Органический слой сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении с получением остатка. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (1,00 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,53 (с, 9H), 1,80-2,16 (м, 5H), 2,52 (м, 1H), 2,76 (м, 1H), 2,97 (м, 2H), 7,22 (м, 1H), 8,20 (м, 1H), 8,25 (м, 1H).
(7) Синтез трет-бутил (±)-[(4aR*,7aS*)-7a-(5-амино-2-фторфенил)-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]карбамата
Насыщенный раствор хлорида аммония (2,10 мл) и железный порошок (905 мг) добавляли к раствору соединения, полученного в примере получения 3-(6) (800 мг), в этаноле (21,0 мл) и смесь перемешивали при кипении с обратным холодильником в течение 30 минут. Реакционный раствор охлаждали до комнатной температуры и затем выпаривали при пониженном давлении растворитель. Остаток очищали хроматографией на колонке с NH-силикагелем с получением указанного в заголовке соединения (545 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,51 (с, 9H), 1,87-2,08 (м, 5H), 2,62 (м, 1H), 2,70 (дд, J=4,4, 14,0 Гц, 1H), 3,02 (дд, J=3,4, 14,0 Гц, 1H), 3,03 (м, 1H), 3,63 (с, 2H), 6,55 (м, 1H), 6,59 (дд, J=2,6, 7,0 Гц, 1H), 6,85 (дд, J=8,4, 12,0 Гц, 1H).
(8) Синтез трет-бутил (-)-[(4aR*,7aS*)-7a-(5-амино-2-фторфенил)-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]карбамата
Соединение, полученное в примере получения 3-(7) (50 мг), оптически разделяли, используя CHIRALPAK™ AD-H производства Daicel Chemical Industries, Ltd. (2 см × 25 см, подвижная фаза: гексан:этанол = 9:1, скорость потока: 20 мл/мин), и компоненты, имеющие время удерживания от 17,1 до 22,8 минут, собирали. Данную операцию повторяли с получением указанного в заголовке соединения (200 мг; >99% ee) из 500 мг рацемата.
Пример получения 4
Синтез трет-бутил [(4aR*,8aS*)-8a-(3-аминофенил)-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата
[Формула 23]

(1) Синтез 2-(3-нитрофенил)циклогекс-1-енкарбальдегида
Этанол (11,1 мл) добавляли к раствору 2-бромциклогекс-1-енкарбальдегида (2,22 г) в толуоле (22,2 мл). К смеси добавляли 3-нитрофенилбороновую кислоту (2,34 г), тетракис(трифенилфосфин)палладий (270 мг) и 1 н. раствор карбоната натрия (14,0 мл). Атмосферу реакционной системы заменяли азотом. Затем реакционный раствор перемешивали при кипении с обратным холодильником в течение трех часов. Реакционный раствор охлаждали до комнатной температуры с последующим экстрагированием этилацетатом. Органический слой сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении с получением остатка. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (2,00 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,76 (м, 2H), 1,83 (м, 2H), 2,39 (м, 2H), 2,56 (м, 2H), 7,58 (м, 2H), 8,14 (м, 1H), 8,24 (м, 1H), 9,46 (с, 1H).
(2) Синтез [2-(3-нитрофенил)циклогекс-1-енил]метанола
Гептагидрат хлорида церия (1,22 г) добавляли к смешанному раствору из 2-(3-нитрофенил)циклогекс-1-енкарбальдегида, полученного в примере получения 4-(1) (630 мг), в метаноле (60,0 мл) и ТГФ (20,0 мл) на ледяной бане. Боргидрид натрия (130 мг) добавляли к реакционному раствору при той же температуре с последующим перемешиванием в течение 30 минут. Насыщенный раствор хлорида аммония добавляли к реакционной смеси с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным раствором хлорида натрия. Органический слой сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении с получением неочищенного продукта. Полученный неочищенный продукт очищали хроматографией на колонке с NH-силикагелем с получением указанного в заголовке соединения (610 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,28 (т, J=5,2 Гц, 1H), 1,77 (м, 4H), 2,30 (с, 4H), 3,93 (д, J=5,2 Гц, 2H), 7,50 (м, 2H), 8,04 (м, 1H), 8,11 (дт, J=2,0, 7,2 Гц, 1H).
(3) Синтез 1-(2-хлорметилциклогекс-1-енил)-3-нитробензола
N,N-Диизопропилэтиламин (3,64 мл) добавляли к раствору [2-(3-нитрофенил)циклогекс-1-енил]метанола, полученного в примере получения 4-(2) (1,67 г), в дихлорметане (109 мл) на ледяной бане. Затем по каплям добавляли метансульфонилхлорид (668 мкл). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение ночи. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (1,56 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,78 (м, 4H), 2,32 (с, 4H), 3,86 (с, 2H), 7,54 (т, J=7,6 Гц, 1H), 7,60 (м, 1H), 8,10 (м, 1H), 8,15 (м, 1H).
(4) Синтез гидрохлорида 2-[2-(3-нитрофенил)циклогекс-1-енилметил]изотиомочевины
Тиомочевину (495 мг) добавляли к раствору 1-(2-хлорметилциклогекс-1-енил)-3-нитробензола, полученного в примере получения 4-(3) (1,56 г), в этаноле (71,6 мл) и смесь перемешивали при кипении с обратным холодильником в течение четырех часов. Реакционный раствор охлаждали до комнатной температуры и затем выпаривали при пониженном давлении растворитель. Остаточное твердое вещество промывали эфиром с получением указанного в заголовке соединения (2,04 г).
1H-ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 1,70 (с, 4H), 2,22 (с, 2H), 2,30 (с, 2H), 3,68 (с, 2H), 7,65 (дт, J=1,2, 7,6 Гц, 1H), 7,71 (т, J=8,0 Гц, 1H), 7,99 (т, J=2,0 Гц, 1H), 8,19 (ддд, J=1,6, 2,4, 8,4 Гц, 1H), 9,02 (уш.с, 3H).
(5) Синтез (±)-(4aR*,8aS*)-8a-(3-нитрофенил)-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-иламина
Трифторметансульфоновую кислоту (1,00 мл) добавляли к раствору гидрохлорида 2-[2-(3-нитрофенил)циклогекс-1-енилметил]изотиомочевины, полученного в примере получения 4-(4) (2,04 г), в ТФУК (10,0 мл) на ледяной бане. Реакционный раствор нагревали до комнатной температуры с последующим перемешиванием в течение ночи. Трифторметансульфоновую кислоту (1,00 мл) дополнительно добавляли к реакционному раствору с последующим перемешиванием в течение двух суток. После подтверждения окончания реакции реакционную смесь осторожно вливали в смешанный раствор насыщенного раствора бикарбоната натрия и простого эфира на ледяной бане. Водный слой экстрагировали этилацетатом и органический слой промывали насыщенным раствором бикарбоната натрия и насыщенным раствором хлорида натрия. Органический слой сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении с получением неочищенного продукта. Полученный неочищенный продукт очищали хроматографией на колонке с NH-силикагелем с получением указанного в заголовке соединения (1,62 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,47-1,86 (м, 8H), 2,23 (ддд, J=4,0, 6,4, 11,6 Гц, 1H), 2,51 (дд, J=2,8, 12,0 Гц, 1H), 2,78 (дд, J=4,4, 12,0 Гц, 1H), 4,45 (с, 2H), 7,49 (т, J=8,0 Гц, 1H), 7,67 (ддд, J=1,2, 2,0, 8,0 Гц, 1H), 8,08 (ддд, J=1,2, 2,4, 8,0 Гц, 1H), 8,19 (т, J=2,0 Гц, 1H).
(6) Синтез трет-бутил (±)-[(4aR*,8aS*)-8a-(3-нитрофенил)-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата
Триэтиламин (3,08 мл) добавляли к раствору соединения, полученного в примере получения 4-(5) (1,62 г), в ТГФ (30,0 мл). Ди-трет-бутилдикарбонат (1,33 г) добавляли к реакционному раствору с последующим перемешиванием при комнатной температуре в течение трех суток. Реакционный раствор концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с NH-силикагелем с получением указанного в заголовке соединения (2,28 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,46-1,95 (м, 8H), 1,54 (с, 9H), 2,46 (м, 1H), 2,48 (дд, J=2,4, 13,2 Гц, 1H), 2,74 (дд, J=4,4, 12,8 Гц, 1H), 7,57 (т, J=8,0 Гц, 1H), 7,70 (д, J=8,0 Гц, 1H), 8,18 (м, 2H).
(7) Синтез трет-бутил (-)-[(4aR*,8aS*)-8a-(3-нитрофенил)-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата
Соединение, полученное в примере получения 4-(6) (70,0 мг), растворяли в этаноле (1,2 мл) и оптически разделяли, используя CHIRALPAK™ OJ-H производства Daicel Chemical Industries, Ltd. (2 см × 25 см, подвижная фаза: гексан:этанол = 8:2, скорость потока: 20 мл/мин). Компоненты, имеющие время удерживания от 12,0 до 21,51 минут, собирали с получением указанного в заголовке (-)-изомера. Данную операцию повторяли с получением указанного в заголовке (-)-изомера (144 мг) из 290 мг исходного вещества.
(8) Синтез трет-бутил [(4aR*,8aS*)-8a-(3-аминофенил)-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата
Насыщенный раствор дитионита натрия (879 мг) добавляли к раствору трет-бутил (-)-[(4aR*,8aS*)-8a-(3-нитрофенил)-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата (395 мг) в этаноле (20 мл) при комнатной температуре. После перемешивания при комнатной температуре в течение 10 минут дополнительно добавляли к реакционному раствору этанол (10 мл). После перемешивания при комнатной температуре в течение пяти минут дополнительно добавляли воду (10 мл). Реакционный раствор нагревали до 40°C и перемешивали в течение 30 минут. После подтверждения окончания реакции реакционный раствор охлаждали до комнатной температуры. Избыток этанола в реакционном растворе выпаривали при пониженном давлении и затем водный слой экстрагировали этилацетатом. Органический слой промывали насыщенным раствором хлорида натрия и сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и затем фильтрат концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с NH-силикагелем с получением указанного в заголовке соединения (110 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,53 (с, 9H), 1,61-1,99 (м, 8H), 2,38 (дд, J=2,4, 13,2 Гц, 1H), 2,39 (м, 1H), 2,89 (дд, J=4,8, 13,2 Гц, 1H), 3,74 (с, 2H), 6,60 (м, 2H), 6,66 (д, J=8,4 Гц, 1H), 7,15 (т, J=7,6 Гц, 1H).
Пример получения 5
Синтез трет-бутил (-)-[(4aR*,8aS*)-8a-(5-амино-2,3-дифторфенил)-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата
[Формула 24]

(1) Синтез этил 2-(2,3-дифторфенил)циклогекс-1-енкарбоксилата
Указанное в заголовке соединение (675 мг) получали из этил 2-трифторметансульфонилоксициклогекс-1-енкарбоксилата, полученного в примере получения 1-(1) (1,00 г), и 2,3-дифторфенилбороновой кислоты (627 мг) в соответствии со способом примера получения 1-(2).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 0,92 (т, J=7,2 Гц, 3H), 1,76 (м, 4H), 2,34 (м, 2H), 2,46 (м, 2H), 3,92 (кв, J=7,2 Гц, 2H), 6,82 (м, 1H), 7,02 (м, 2H).
(2) Синтез [2-(2,3-дифторфенил)циклогекс-1-енил]метанола
Указанное в заголовке соединение (490 мг) получали из этил 2-(2,3-дифторфенил)циклогекс-1-енкарбоксилата, полученного в примере получения 5-(1) (675 мг), в соответствии со способом примера получения 1-(3).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,19 (дт, J=1,6, 5,8 Гц, 1H), 1,75 (м, 4H), 2,24 (м, 2H), 2,30 (м, 2H), 3,86 (д, J=5,8 Гц, 2H), 6,87 (м, 1H), 7,04 (м, 2H).
(3) Синтез 1-(2-хлорметилциклогекс-1-енил)-2,3-дифторбензола
Указанное в заголовке соединение (588 мг) получали из [2-(2,3-дифторфенил)циклогекс-1-енил]метанола, полученного в примере получения 5-(2) (490 мг), в соответствии со способом примера получения 1-(4).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,77 (с, 4H), 2,29 (уш.с, 4H), 3,85 (с, 2H), 6,96 (м, 1H), 7,07 (м, 2H).
(4) Синтез гидрохлорида 2-[2-(2,3-дифторфенил)циклогекс-1-енилметил]изотиомочевины
Указанное в заголовке соединение (635 мг) получали из 1-(2-хлорметилциклогекс-1-енил)-2,3-дифторбензола, полученного в примере получения 5-(3) (588 мг), и тиомочевины (193 мг) в соответствии со способом примера получения 1-(5).
1H-ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 1,67 (с, 4H), 2,19 (с, 2H), 2,20 (с, 2H), 3,62 (с, 2H), 6,97 (м, 1H), 7,22 (м, 1H), 7,38 (м, 1H), 8,99 (с, 3H).
(5) Синтез (±)-(4aR*,8aS*)-8a-(2,3-дифторфенил)-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-иламина
Указанное в заголовке соединение (447 мг) получали из гидрохлорида 2-[2-(2,3-дифторфенил)циклогекс-1-енилметил]изотиомочевины, полученного в примере получения 5-(4) (635 мг), в соответствии со способом примера получения 1-(6).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,48-1,81 (м, 7H), 2,22 (м, 1H), 2,55 (дд, J=2,8, 12,0 Гц, 1H), 2,66 (м, 1H), 2,87 (дд, J=4,4, 12,0 Гц, 1H), 4,45 (с, 2H), 7,03 (м, 3H).
(6) Синтез трет-бутил (±)-[(4aR*,8aS*)-8a-(2,3-дифтор-5-нитрофенил)-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата
Указанное в заголовке соединение (530 мг) получали из (±)-(4aR*,8aS*)-8a-(2,3-дифторфенил)-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-иламина, полученного в примере получения 5-(5) (420 мг), в соответствии со способом примера получения 1-(7).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,54 (с, 9H), 1,54-1,90 (м, 7H), 2,19 (м, 1H), 2,58 (дд, J=2,8, 12,8 Гц, 1H), 2,79 (м, 1H), 2,82 (м, 1H), 8,05 (м, 2H).
(7) Синтез трет-бутил (±)-[(4aR*,8aS*)-8a-(5-амино-2,3-дифторфенил)-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата
Указанное в заголовке соединение (174 мг) получали из трет-бутил (±)-[(4aR*,8aS*)-8a-(2,3-дифтор-5-нитрофенил)-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата, полученного в примере получения 5-(6) (530 мг), в соответствии со способом примера получения 3-(7).
ESI-MS; m/z 398 [M+H].
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,54 (с, 9H), 1,63-1,92 (м, 7H), 2,31 (м, 1H), 2,51 (дд, J=2,4, 12,8 Гц, 1H), 2,81 (м, 1H), 2,92 (дд, J=4,0, 12,8 Гц, 1H), 3,72 (с, 2H), 6,29 (м, 1H), 6,42 (ддд, J=2,8, 6,0, 11,2 Гц, 1H).
(8) Синтез трет-бутил (-)-[(4aR*,8aS*)-8a-(5-амино-2,3-дифторфенил)-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата
трет-Бутил (±)-[(4aR*,8aS*)-8a-(5-амино-2,3-дифторфенил)-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамат, полученный в примере получения 5-(7) (150 мг), оптически разделяли, используя CHIRALPAK™ AD-H производства Daicel Chemical Industries, Ltd. (2 см × 25 см, подвижная фаза: гексан:этанол = 8:2, скорость потока: 20 мл/мин). Компоненты, имеющие время удерживания от 9,13 до 12,4 минут, собирали с получением указанного в заголовке соединения (42 мг).
Пример получения 6
Синтез трет-бутил (±)-[(4aR*,7aS*)-7a-(5-амино-2-метоксифенил)-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]карбамата
[Формула 25]

(1) Синтез трет-бутил (±)-[(4aR*,7aS*)-7a-(2-метокси-5-нитрофенил)-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]карбамата
К раствору соединения, полученного в примере получения 3-(6) (97 мг), в метаноле (2,0 мл) добавляли 28% раствор метоксида натрия в метаноле (100 мкл). Реакционный раствор перемешивали при комнатной температуре в течение одного часа и перемешивали при 45°C в течение пяти часов. Реакционный раствор концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (75,8 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,50 (с, 9H), 1,75-2,36 (м, 6H), 2,82-2,91 (м, 1H), 3,13-3,22 (м, 1H), 3,22-3,29 (м, 1H), 4,03 (с, 3H), 7,02 (д, J=9,2 Гц, 1H), 8,14 (д, J=2,8 Гц, 1H), 8,23 (дд, J=9,2, 2,8 Гц, 1H).
(2) Синтез трет-бутил (±)-[(4aR*,7aS*)-7a-(5-амино-2-метоксифенил)-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]карбамата
Железо (83,1 мг) и насыщенный раствор хлорида аммония (168 мкл) добавляли к раствору соединения, полученного в примере получения 6-(1) (75,8 мг), в этаноле (1,68 мл). Реакционный раствор кипятили с обратным холодильником при наружной температуре 100°C в течение 20 минут. После охлаждения до комнатной температуры добавляли к реакционному раствору этилацетат. Нерастворимое вещество отфильтровывали. К фильтрату добавляли воду с последующим экстрагированием этилацетатом. Органический слой концентрировали. Остаток очищали хроматографией на NH-силикагеле с получением указанного в заголовке соединения (50 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,50 (с, 9H), 1,78-2,15 (м, 5H), 2,49-2,61 (м, 1H), 2,67-2,74 (дд, J=13,1, 4,3 Гц, 1H), 3,09-3,16 (дд, J=13,1, 4,3 Гц, 1H), 3,16-3,25 (м, 1H), 3,42-3,54 (уш.с, 2H), 3,79 (с, 3H), 6,59-6,62 (м, 2H), 6,73-6,77 (м, 1H).
Пример получения 7
Синтез трет-бутил (-)-[(4aR*,9aS*)-9a-(5-амино-2-фторфенил)-4,4a,5,6,7,8,9,9a-октагидроциклогепта[d][1,3]тиазин-2-ил]карбамата
[Формула 26]
(1) Синтез метил 2-(2-фторфенил)циклогепт-1-енкарбоксилата
Указанное в заголовке соединение (10,2 г) получали из метил 2-трифторметансульфонилоксициклогепт-1-енкарбоксилата, полученного в соответствии с примером получения 1-(1) (16,0 г), и 2-фторфенилбороновой кислоты (4,50 г) в соответствии со способом примера получения 1-(2).
ESI-MS; m/z 249 [M+H].
(2) Синтез [2-(2-фторфенил)циклогепт-1-енил]метанола
Указанное в заголовке соединение (4,50 г) получали из метил 2-(2-фторфенил)циклогепт-1-енкарбоксилата, полученного в примере получения 7-(1) (10,2 г), в соответствии со способом примера получения 1-(3).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,21 (дт, J=2,0, 6,0 Гц, 1H), 1,56-1,67 (м, 4H), 1,84 (м, 2H), 2,46 (м, 4H), 3,89 (м, 2H), 7,04 (м, 1H), 7,09 (м, 2H), 7,21 (м, 1H).
(3) Синтез 1-хлорметил-2-(2-фторфенил)циклогептена
Указанное в заголовке соединение (1,56 г) получали из [2-(2-фторфенил)циклогепт-1-енил]метанола, полученного в примере получения 7-(2) (2,10 г), в соответствии со способом примера получения 1-(4).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,55-1,68 (м, 4H), 1,83 (м, 2H), 2,46 (м, 4H), 3,92 (д, J=3,2 Гц, 2H), 7,05 (ддд, J=1,6, 8,0, 9,6 Гц, 1H), 7,11 (дт, J=1,2, 7,2 Гц, 1H), 7,20 (дт, J=2,0, 7,2 Гц, 1H), 7,26 (м, 1H).
(4) Синтез гидрохлорида 2-[2-(2-фторфенил)циклогепт-1-енилметил]изотиомочевины
Указанное в заголовке соединение (2,01 г) получали из 1-хлорметил-2-(2-фторфенил)циклогептена, полученного в примере получения 7-(3) (1,56 г), и тиомочевины (507 мг) в соответствии со способом примера получения 1-(5).
1H-ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 1,54 (м, 4H), 1,77 (м, 2H), 2,37 (м, 4H), 3,65 (м, 2H), 7,11 (дт, J=2,0, 7,2 Гц, 1H), 7,19 (м, 1H), 7,23 (м, 1H), 7,35 (м, 1H), 8,99 (с, 3H).
(5) Синтез (±)-(4aR*,9aS*)-9a-(2-фторфенил)-4,4a,5,6,7,8,9,9a-октагидроциклогепта[d][1,3]тиазин-2-иламина
Указанное в заголовке соединение (1,35 г) получали из гидрохлорида 2-[2-(2-фторфенил)циклогепт-1-енилметил]изотиомочевины, полученного в примере получения 7-(4) (2,00 г), в соответствии со способом примера получения 1-(6).
ESI-MS; m/z 279 [M+H].
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,48-1,89 (м, 9H), 2,40 (м, 1H), 2,50 (дд, J=3,6, 12,0 Гц, 1H), 2,65 (м, 1H), 2,77 (дд, J=3,6, 12,0 Гц, 1H), 7,00 (ддд, J=1,6, 8,0, 12,8 Гц, 1H), 7,07 (дт, J= 1,6, 7,6 Гц, 1H), 7,17-7,24 (м, 2H).
(6) Синтез трет-бутил (±)-[(4aR*,9aS*)-9a-(2-фтор-5-нитрофенил)-4,4a,5,6,7,8,9,9a-октагидроциклогепта[d][1,3]тиазин-2-ил]карбамата
Указанное в заголовке соединение (1,83 г) получали из (±)-(4aR*,9aS*)-9a-(2-фторфенил)-4,4a,5,6,7,8,9,9a-октагидроциклогепта[d][1,3]тиазин-2-иламина, полученного в примере получения 7-(5) (1,35 г), в соответствии со способом примера получения 1-(7).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,50-1,98 (м, 9H), 1,54 (с, 9H), 2,38 (м, 1H), 2,53 (дд, J=3,2, 12,8 Гц, 1H), 2,70 (3,2, 9,6 Гц, 1H), 2,82 (м, 1H), 7,23 (м, 1H), 8,19 (м, 2H).
(7) Синтез трет-бутил (±)-[(4aR*,9aS*)-9a-(5-амино-2-фторфенил)-4,4a,5,6,7,8,9,9a-октагидроциклогепта[d][1,3]тиазин-2-ил]карбамата
Указанное в заголовке соединение (1,36 г) получали из трет-бутил (±)-[(4aR*,9aS*)-9a-(2-фтор-5-нитрофенил)-4,4a,5,6,7,8,9,9a-октагидроциклогепта[d][1,3]тиазин-2-ил]карбамата, полученного в примере получения 7-(6) (1,83 г), в соответствии со способом примера получения 3-(7).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,53 (с, 9H), 1,57-1,99 (м, 9H), 2,45 (м, 2H), 2,83 (м, 2H), 3,64 (с, 2H), 6,53 (м, 2H), 6,84 (м, 1H).
(8) Синтез трет-бутил (-)-[(4aR*,9aS*)-9a-(5-амино-2-фторфенил)-4,4a,5,6,7,8,9,9a-октагидроциклогепта[d][1,3]тиазин-2-ил]карбамата
трет-Бутил (±)-[(4aR*,9aS*)-9a-(5-амино-2-фторфенил)-4,4a,5,6,7,8,9,9a-октагидроциклогепта[d][1,3]тиазин-2-ил]карбамат, полученный в примере получения 7-(7) (140 мг), оптически разделяли, используя CHIRALPAK™ AD-H производства Daicel Chemical Industries, Ltd. (2 см × 25 см, подвижная фаза: гексан:этанол = 8:2, скорость потока: 10 мл/мин). Компоненты, имеющие время удерживания от 14,3 до 17,9 минут, собирали с получением указанного в заголовке соединения (60 мг).
Пример получения 8
Синтез трет-бутил (-)-[(4aS*,8aS*)-8a-(5-амино-2-фторфенил)-4a,7,8,8a-тетрагидро-4H,5H-6-окса-3-тиа-1-азанафталин-2-ил]карбамата
[Формула 27]

(1) Синтез оксима 3-аллилоксипропиональдегида
Раствор, содержащий оксалилхлорид (5,45 мл) в дихлорметане (130 мл), охлаждали до -78°C в атмосфере азота. Раствор, содержащий диметилсульфоксид (4,85 мл) в дихлорметане (5 мл), добавляли по каплям к реакционному раствору при той же температуре. После перемешивания при той же температуре в течение 10 минут к реакционному раствору добавляли по каплям раствор, содержащий 3-аллилоксипропан-1-ол (4,99 г) в дихлорметане (5 мл). После перемешивания при той же температуре в течение одного часа к реакционному раствору добавляли триэтиламин (20,4 мл). Охлаждающую баню удаляли. Реакционный раствор нагревали до комнатной температуры и перемешивали при комнатной температуре в течение одного часа. К реакционному раствору добавляли насыщенный водный раствор хлорида аммония и отделяли органический слой. Органический слой промывали насыщенным водным раствором хлорида натрия и затем сушили над безводным сульфатом магния. Нерастворимое вещество отделяли фильтрованием и фильтрат концентрировали при пониженном давлении. Остаток растворяли в этаноле (100 мл) и воде (10 мл). Ацетат натрия (12 г) и сульфат гидроксиламина (8,02 г) добавляли к реакционному раствору при комнатной температуре. Реакционный раствор перемешивали при комнатной температуре в течение 15 часов. Затем добавляли насыщенный раствор хлорида натрия и этилацетат и отделяли органический слой. Органический слой снова промывали насыщенным водным раствором хлорида натрия. Органический слой сушили над безводным сульфатом магния. Нерастворимое вещество отделяли фильтрованием и фильтрат концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (5,5 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,47-2,52 (м, 1H), 2,65-2,70 (м, 1H), 3,58-3,62 (м, 2H), 3,98-4,01 (м, 2H), 5,17-5,22 (м, 1H), 5,24-5,31 (м, 1H), 5,85-5,96 (м, 1H), 6,86 (т, J=5,2 Гц, 0,5H), 7,50 (т, J=5,6 Гц, 0,5H).
(2) Синтез (±)-3a,4,6,7-тетрагидро-3H-пирано[4,3-c]изоксазола
5% Раствор гипохлорита натрия (63,2 мл) добавляли к раствору, содержащему соединение, полученное в примере получения 8-(1) (5,5 г), в дихлорметане (200 мл) при комнатной температуре и смесь перемешивали при комнатной температуре в течение четырех часов. Воду и бисульфит натрия (10 г) добавляли к реакционному раствору с последующим перемешиванием при комнатной температуре в течение 10 минут. Органический слой отделяли и промывали насыщенным раствором хлорида натрия. Органический слой сушили над безводным сульфатом магния. Нерастворимое вещество отделяли фильтрованием и фильтрат концентрировали. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (3,33 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,47-2,56 (м, 1H), 2,73 (дд, J=2,8, 14,0 Гц, 1H), 3,23-3,47 (м, 3H), 3,73 (дд, J=2,4, 10,4 Гц, 1H), 4,26 (дд, J=2,4, 11,2 Гц, 1H), 4,34 (дд, J=6,8, 10,8 Гц, 1H), 4,47 (дд, J=8,0, 10,8 Гц, 1H).
(3) Синтез (±)-(3aS*,7aS*)-7a-(2-фторфенил)гексагидропирано[4,3-c]изоксазола
Раствор н-бутиллития в гексане (2,77 М; 18,9 мл) добавляли по каплям к раствору, содержащему 2-бромфторбензол (9,61 г) в смеси тетрагидрофуран/толуол (35 мл/115 мл), в атмосфере азота при -78°C. Реакционный раствор перемешивали при той же температуре в течение одного часа. Комплекс трифторид бора-диэтиловый эфир (6,6 мл) добавляли по каплям к раствору, содержащему соединение, полученное в примере получения 8-(2) (3,33 г), в толуоле (235 мл) в атмосфере азота при -78°C. Ранее полученный раствор 2-фторфениллития добавляли по каплям к реакционному раствору при той же температуре. После перемешивания при той же температуре в течение одного часа к реакционному раствору добавляли водный раствор хлорида аммония и реакционный раствор нагревали до комнатной температуры. К реакционному раствору добавляли воду и этилацетат и отделяли органический слой. Органический слой промывали насыщенным раствором хлорида натрия. Органический слой сушили над безводным сульфатом магния и нерастворимое вещество отделяли фильтрованием. Фильтрат концентрировали и остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (5,1 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,90-1,95 (м, 1H), 2,42-2,50 (м, 1H), 3,05-3,11 (м, 1H), 3,54-3,60 (м, 1H), 3,67-3,87 (м, 4H), 3,98 (дд, J=4,8, 12,0 Гц, 1H), 7,07 (дд, J=8,0, 12,0 Гц, 1H), 7,14-7,19 (м, 1H), 7,27-7,32 (м, 1H), 7,73-7,78 (м, 1H).
(4) Синтез (±)-[(3R*,4S*)-4-амино-4-(2-фторфенил)тетрагидропиран-3-ил]метанола
Цинковый порошок (19,1 г) добавляли к раствору, содержащему соединение, полученное в примере получения 8-(3) (5,1 г), в уксусной кислоте (130 мл) при комнатной температуре. Реакционный раствор перемешивали при комнатной температуре в течение 16 часов. Нерастворимое вещество отделяли фильтрованием через целит и фильтрат концентрировали. К остатку добавляли этилацетат и раствор бикарбоната натрия и отделяли органический слой. Органический слой промывали насыщенным водным раствором хлорида натрия. Органические слои дополнительно экстрагировали из водного слоя четыре раза этилацетатом. Органические слои объединяли и сушили над безводным сульфатом магния. Нерастворимое вещество отделяли фильтрованием и фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (5,1 г).
1H-ЯМР (CDCl3) δ (м.д.): 1,45 (д, J=14,0 Гц, 1H), 2,52-2,58 (м, 1H), 2,62-2,70 (м, 1H), 3,54 (д, J=4,0 Гц, 2H), 3,89-3,96 (м, 4H), 7,07 (дд, J=8,0, 12,0 Гц, 1H), 7,15-7,19 (м, 1H), 7,26-7,31 (м, 1H), 7,57-7,61 (м, 1H).
(5) Синтез (±)-9H-флуорен-9-илметил({[(3R*,4S*)-4-(2-фторфенил)-3-(гидроксиметил)тетрагидро-2H-пиран-4-ил]амино}карбонотиоил)карбамата
Флуоренилметилоксикарбонилизотиоцианат (7,02 г) добавляли к раствору, содержащему соединение, полученное в примере получения 8-(4) (5,1 г), в дихлорметане (100 мл) и смесь перемешивали при комнатной температуре в течение двух часов. Реакционный раствор концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (10,03 г).
1H-ЯМР (CDCl3) δ (м.д.): 1,64-1,66 (уш.м, 1H), 2,45-2,55 (уш.м, 1H), 2,59-2,67 (м, 1H), 3,49-3,52 (уш.м, 1H), 3,67-3,87 (м, 4H), 4,00-4,04 (м, 2H), 4,24-4,27 (м, 1H), 4,50-4,59 (м, 2H), 7,05 (ддд, J=1,2, 8,0, 12,8 Гц, 1H), 7,13-7,17 (м, 1H), 7,26-7,46 (м, 6H), 7,56-7,59 (м, 2H), 7,79 (д, J=8,0 Гц, 2H), 7,96 (уш.с, 1H), 10,6 (уш.с, 1H).
(6) Синтез (±)-(4aS*,8aS*)-8a-(2-фторфенил)-4a,7,8,8a-тетрагидро-4H,5H-6-окса-3-тиа-1-азанафталин-2-иламина
Концентрированную хлористо-водородную кислоту (5 мл) добавляли к раствору, содержащему соединение, полученное в примере получения 8-(5) (10 г), в метаноле (200 мл) и реакционный раствор кипятили с обратным холодильником в течение двух часов. Реакционный раствор охлаждали до комнатной температуры и концентрировали при пониженном давлении. К остатку добавляли этилацетат и насыщенный раствор бикарбоната натрия и отделяли органический слой. Органический слой концентрировали при пониженном давлении. Остаток растворяли в ацетонитриле (200 мл). К раствору добавляли пиперидин (20 мл) с последующим перемешиванием при комнатной температуре в течение двух часов. Реакционный раствор концентрировали и остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (3,17 г).
1H-ЯМР (CDCl3) δ (м.д.): 1,65 (д, J=13,2 Гц, 1H), 2,53 (дд, J=2,8, 12,8 Гц, 1H), 2,65-2,73 (м, 1H), 2,87 (дд, J=4,4, 12,4 Гц, 1H), 2,98-3,10 (м, 1H), 3,69-3,80 (м, 3H), 3,88 (дд, J=4,4, 10,8 Гц, 1H), 4,55 (уш.с, 2H), 7,04 (дд, J=8,0, 12,8 Гц, 1H), 7,09-7,13 (м, 1H), 7,21-7,32 (м, 2H).
(7) Синтез трет-бутил (±)-[(4aS*,8aS*)-8a-(2-фтор-5-нитрофенил)-4a,7,8,8a-тетрагидро-4H,5H-6-окса-3-тиа-1-азанафталин-2-ил]карбамата
Дымящую азотную кислоту (682 мкл) добавляли по каплям к раствору соединения, полученного в примере получения 8-(6) (2,08 г), в концентрированной серной кислоте (30 мл) при охлаждении льдом. После перемешивания реакционного раствора при той же температуре в течение 30 минут реакционный раствор вливали в ледяную воду. Реакционную смесь подщелачивали 5 н. раствором гидроксида натрия. Добавляли к смеси хлороформ и отделяли органический слой. Органический слой сушили над безводным сульфатом магния. Нерастворимое вещество отделяли фильтрованием и фильтрат концентрировали при пониженном давлении. Остаток растворяли в тетрагидрофуране (120 мл). Триэтиламин (6,6 мл) и ди-трет-бутилдикарбонат (6,5 г) добавляли к раствору и смесь перемешивали при комнатной температуре в течение 17 часов. К реакционному раствору добавляли насыщенный раствор хлорида натрия и этилацетат и отделяли органический слой. Органический слой сушили над безводным сульфатом магния. Нерастворимое вещество отделяли фильтрованием и фильтрат концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (4,7 г).
ESI-MS; m/z 412 [M++H].
1H-ЯМР (CDCl3) δ (м.д.): 1,53 (с, 9H), 1,64-1,70 (м, 1H), 2,52-2,62 (м, 2H), 2,79 (дд, J=3,6, 13,2 Гц, 1H), 3,05-3,15 (уш.м, 1H), 3,60-3,93 (м, 4H), 7,22-7,28 (м, 1H), 8,18-8,22 (м, 2H).
(8) Синтез трет-бутил (±)-[(4aS*,8aS*)-8a-(5-амино-2-фторфенил)-4a,7,8,8a-тетрагидро-4H,5H-6-окса-3-тиа-1-азанафталин-2-ил]карбамата
Железный порошок (5,28 г) и насыщенный водный раствор хлорида аммония (18,6 мл) добавляли к раствору соединения, полученного в примере получения 8-(7) (4,7 г), в этаноле (150 мл). Реакционный раствор кипятили с обратным холодильником в течение 30 минут и затем добавляли железный порошок (5,28 г). Реакционный раствор дополнительно кипятили с обратным холодильником в течение 30 минут и затем еще добавляли железный порошок (5,28 г). Реакционный раствор дополнительно кипятили с обратным холодильником в течение 30 минут и затем охлаждали до комнатной температуры. Реакционный раствор разбавляли этилацетатом и нерастворимое вещество отделяли фильтрованием через целит. Фильтрат концентрировали при пониженном давлении. К остатку добавляли этилацетат и насыщенный водный раствор хлорида натрия и отделяли органический слой. Органический слой сушили над безводным сульфатом магния и нерастворимое вещество отделяли фильтрованием. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (3,48 г).
ESI-MS; m/z 382 [M++H].
1H-ЯМР (CDCl3) δ (м.д.): 1,53 (с, 9H), 1,55-1,66 (м, 1H), 2,48 (дд, J=2,8, 9,2 Гц, 1H), 2,72-2,81 (м, 1H), 2,92 (дд, J=4,0, 13,2 Гц, 1H), 3,09-3,13 (м, 1H), 3,66 (с, 2H), 3,71-3,94 (м, 4H), 6,53-6,59 (м, 2H), 6,88 (дд, J=8,0, 12,0 Гц, 1H).
(9) Синтез трет-бутил (-)-[(4aS*,8aS*)-8a-(5-амино-2-фторфенил)-4a,7,8,8a-тетрагидро-4H,5H-6-окса-3-тиа-1-азанафталин-2-ил]карбамата
Соединение, полученное в примере получения 8-(8) (75 мг), оптически разделяли, используя CHIRALPAK™ AD-H производства Daicel Chemical Industries, Ltd. (2 см × 25 см, подвижная фаза: гексан:этанол = 7:3, скорость потока: 10 мл/мин), и компоненты, имеющие время удерживания от 31,8 до 38,3 минут, собирали. Данную операцию повторяли с получением указанного в заголовке соединения (444 мг; >99% ee) из 1 г рацемата.
ESI-MS; m/z 382 [M++H].
1H-ЯМР (CDCl3) δ (м.д.): 1,53 (с, 9H), 1,55-1,66 (м, 1H), 2,48 (дд, J=2,8, 9,2 Гц, 1H), 2,72-2,81 (м, 1H), 2,92 (дд, J=4,0, 13,2 Гц, 1H), 3,09-3,13 (м, 1H), 3,66 (с, 2H), 3,71-3,94 (м, 4H), 6,53-6,59 (м, 2H), 6,88 (дд, J=8,0, 12,0 Гц, 1H).
Пример получения 9
Синтез трет-бутил [(4aS*,7aS*)-7a-(5-амино-2-фторфенил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил]карбамата
[Формула 28]

(1) Синтез аллилоксиацетальдегидоксима
Раствор, содержащий оксалилхлорид (27,3 мл) в дихлорметане (600 мл), охлаждали до -78°C в атмосфере азота. Раствор, содержащий диметилсульфоксид (24,3 мл) в дихлорметане (50 мл), добавляли по каплям к реакционному раствору при той же температуре. После перемешивания при той же температуре в течение 10 минут к реакционному раствору добавляли по каплям раствор, содержащий 2-аллилоксиэтанол (25 г) в дихлорметане (50 мл), при той же температуре. После перемешивания при той же температуре в течение одного часа к реакционному раствору добавляли триэтиламин (102 мл). Охлаждающую баню удаляли. Реакционный раствор нагревали до комнатной температуры и перемешивали при комнатной температуре в течение одного часа. К реакционному раствору добавляли насыщенный водный раствор хлорида аммония. Органический слой отделяли и промывали насыщенным водным раствором хлорида натрия. Органический слой сушили над безводным сульфатом магния и нерастворимое вещество отделяли фильтрованием. Фильтрат концентрировали при пониженном давлении. Остаток растворяли в этаноле (500 мл) и воде (50 мл). Ацетат натрия (60,2 г) и сульфат гидроксиламина (40,2 г) добавляли к реакционному раствору при комнатной температуре. Реакционный раствор перемешивали при комнатной температуре в течение 15 часов. Затем добавляли воду и этилацетат и отделяли органический слой. Органический слой промывали насыщенным водным раствором хлорида натрия и сушили над безводным сульфатом магния. Нерастворимое вещество отделяли фильтрованием и фильтрат концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (13,2 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 4,00-4,04 (м, 2H), 4,09-4,11 (м, 1H), 4,35 (д, J=3,6 Гц, 1H), 5,21-5,25 (м, 1H), 5,27-5,35 (м, 1H), 5,85-5,95 (м, 1H), 6,92 (т, J=4,0 Гц, 0,5 H), 7,51 (т, J=5,6 Гц, 0,5H).
(2) Синтез (±)-3a,4-дигидро-3H,6H-фуро[3,4-c]изоксазола
5% Раствор гипохлорита натрия (170 мл) добавляли к раствору, содержащему соединение, полученное в примере получения 9-(1) (13,2 г), в дихлорметане (400 мл) при комнатной температуре и смесь перемешивали при комнатной температуре в течение шести часов. Воду и бисульфит натрия (7,95 г) добавляли к реакционному раствору с последующим перемешиванием при комнатной температуре в течение 10 минут. Затем отделяли органический слой. Органический слой сушили над безводным сульфатом магния. Нерастворимое вещество отделяли фильтрованием и фильтрат концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (4,8 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 3,65 (дд, J=9,2, 8,0 Гц, 1H), 4,00 (дд, J=12,0, 8,0 Гц, 1H), 4,17-4,29 (м, 2H), 4,40-4,49 (м, 2H), 4,59 (дд, J=9,2, 8,0 Гц, 1H).
(3) Синтез (±)-(3aS*,6aS*)-6a-(2-фторфенил)тетрагидрофуро[3,4-c]изоксазола
2,77 М раствор н-бутиллития в гексане (30,7 мл) добавляли по каплям к раствору, содержащему 2-бромфторбензол (15,6 г) в смеси тетрагидрофуран/толуол (50 мл/150 мл), в атмосфере азота при -78°C. Реакционный раствор перемешивали при той же температуре в течение одного часа. Комплекс трифторид бора-диэтиловый эфир (10,7 мл) добавляли по каплям к раствору, содержащему соединение, полученное в примере получения 9-(2) (4,8 г), в толуоле (350 мл) в атмосфере азота при -78°C. Ранее полученный 2-фторфениллитий добавляли по каплям к реакционному раствору при той же температуре. После перемешивания при той же температуре в течение одного часа к реакционному раствору добавляли водный раствор хлорида аммония и реакционный раствор нагревали до комнатной температуры. К реакционному раствору добавляли воду и этилацетат и отделяли органический слой. Органический слой промывали насыщенным раствором хлорида натрия. Органический слой сушили над безводным сульфатом магния и нерастворимое вещество отделяли фильтрованием. Фильтрат концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (5,6 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 3,39-3,45 (м, 1H), 3,52-3,62 (уш.м, 1H), 3,84-3,92 (уш.м, 2H), 3,98 (уш.д, J=9,2 Гц, 1H), 4,16 (ддд, J=2,4, 6,4, 11,2 Гц, 1H), 4,50-4,58 (уш.м, 1H), 5,11 (уш.с, 1H), 7,06 (ддд, J=1,2, 8,4, 11,6 Гц, 1H), 7,16 (ддд, J=1,2, 7,6, 7,6 Гц, 1H), 7,25-7,31 (м, 1H), 7,84-7,95 (м, 1H).
(4) Синтез (±)-[(3R*,4S*)-4-амино-4-(2-фторфенил)тетрагидрофуран-3-ил]метанола
Цинк (порошок: 21 г) добавляли к раствору, содержащему соединение, полученное в примере получения 9-(3) (5,6 г), в уксусной кислоте (140 мл) при комнатной температуре. Реакционный раствор перемешивали при комнатной температуре в течение 16 часов. Нерастворимое вещество отделяли фильтрованием через целит и фильтрат концентрировали при пониженном давлении. К остатку добавляли этилацетат и раствор бикарбоната натрия и отделяли органический слой. Органический слой промывали насыщенным водным раствором хлорида натрия. Органические слои дополнительно экстрагировали из водного слоя три раза этилацетатом. Органические слои объединяли и сушили над безводным сульфатом магния. Нерастворимое вещество отделяли фильтрованием и фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (5,46 г).
ESI-MS; m/z 212 [M++H].
1H-ЯМР (CDCl3) δ (м.д.): 2,81-2,88 (м, 1H), 3,83 (дд, J=6,8, 12,0 Гц, 1H), 3,92 (дд, J=3,2, 8,8 Гц, 1H), 3,94-4,00 (м, 2H), 4,07 (дд, J=8,4, 9,2 Гц, 1H), 4,14 (дд, J=1,2, 8,8 Гц, 1H), 7,09 (ддд, J=1,2, 8,0, 12,4 Гц, 1H), 7,16 (ддд, J=1,2, 7,6, 8,0 Гц, 1H), 7,26-7,32 (м, 1H), 7,53 (дт, J=2,0, 8,0 Гц, 1H).
(5) Синтез (±)-1-бензоил-3-[(3S*,4R*)-3-(2-фторфенил)-4-гидроксиметилтетрагидрофуран-3-ил]тиомочевины
Соединение, полученное в примере получения 9-(4) (2,5 г), добавляли к раствору бензоилизотиоцианата (2,13 г) в дихлорметане (75 мл) и смесь перемешивали при комнатной температуре в течение трех часов. Реакционный раствор концентрировали и остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (4,19 г).
ESI-MS; m/z 307 [M++Na].
1H-ЯМР (CDCl3) δ (м.д.): 2,83 (дд, J=4,4, 6,8 Гц, 1H), 3,15-3,22 (м, 1H), 3,81 (дд, J=2,8, 8,8 Гц, 1H), 3,89-3,95 (м, 1H), 4,01-4,07 (м, 1H), 4,13-4,17 (м, 1H), 4,43 (дд, J=2,8, 9,6 Гц, 1H), 4,69 (д, J=10,0 Гц, 1H), 7,04 (ддд, J=1,2, 8,0, 12,0 Гц, 1H), 7,18 (ддд, J=1,2, 8,0, 8,0 Гц, 1H), 7,24-7,33 (м, 1H), 7,52 (т, J=7,6 Гц, 2H), 7,64 (тд, J=1,2, 8,0 Гц, 1H), 7,71 (тд, J=1,6, 8,0 Гц, 1H), 7,86 (дд, J=1,6, 6,4 Гц, 1H), 8,90 (уш.с, 1H), 11,8 (уш.с, 1H).
(6) Синтез (±)-N-[(4aS*,7aS*)-7a-(2-фторфенил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил]бензамида
Трифенилфосфин (7,08 г) добавляли к раствору соединения, полученного в примере получения 9-(5) (3,89 г), и тетрабромида углерода (8,95 г) в дихлорметане (100 мл) при комнатной температуре. Реакционный раствор охлаждали до 0°C, перемешивали в течение 20 минут и затем нагревали до комнатной температуры. Реакционный раствор перемешивали при комнатной температуре в течение 15 часов. Затем к реакционному раствору добавляли воду и отделяли органический слой. Органический слой сушили над безводным сульфатом магния и нерастворимое вещество отделяли фильтрованием. Фильтрат концентрировали при пониженном давлении и остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (1,93 г).
1H-ЯМР (CDCl3) δ (м.д.): 2,90 (дд, J=4,8, 13,6 Гц, 1H), 3,23 (дд, J=4,0, 13,6 Гц, 1H), 3,39-3,46 (м, 1H), 4,06 (дд, J=2,8, 9,2 Гц, 1H), 4,22-4,25 (м, 2H), 4,44 (д, J=9,2 Гц, 1H), 7,13 (ддд, J=1,2, 8,4, 12,4 Гц, 1H), 7,21 (ддд, J=1,2, 7,6, 7,6 Гц, 1H), 7,33-7,52 (м, 5H), 8,15 (д, J=7,6 Гц, 2H).
(7) Синтез (±)-(4aS*,7aS*)-7a-(2-фторфенил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-иламина
Раствор соединения, полученного в примере получения 9-(6) (2,08 г), и 1,8-диазабицикло[5,4,0]ундец-7-ена (1,6 мл) в метаноле (20 мл) кипятили с обратным холодильником в течение пяти часов. После охлаждения реакционного раствора до комнатной температуры выпаривали при пониженном давлении растворитель. Остаток подвергали хроматографии на силикагеле. Полученный неочищенный продукт суспендировали в диэтиловом эфире. Полученное твердое вещество собирали фильтрованием с получением указанного в заголовке соединения (1,19 г).
ESI-MS; m/z 253 [M++H].
1H-ЯМР (CDCl3) δ (м.д.): 2,83 (дд, J=5,2, 12,4 Гц, 1H), 2,99-3,08 (м, 2H), 3,82 (дд, J=2,0, 8,4 Гц, 1H), 4,05-4,15 (м, 2H), 4,44 (уш.с, 2H), 4,49 (д, J=8,8 Гц, 1H), 7,05 (ддд, J=1,6, 8,0, 12,0 Гц, 1H), 7,13 (ддд, J=1,2, 7,2, 8,0 Гц, 1H), 7,22-7,30 (м, 1H), 7,46 (дт, J=1,6, 8,0 Гц, 1H).
(8) Синтез трет-бутил (±)-[(4aS*,7aS*)-7a-(2-фтор-5-нитрофенил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил]карбамата
Дымящую азотную кислоту (293 мкл) добавляли по каплям к раствору соединения, полученного в примере получения 9-(7) (1,19 г), в концентрированной серной кислоте (20 мл) при охлаждении льдом. Реакционный раствор перемешивали при той же температуре в течение 30 минут и затем вливали в ледяную воду. Реакционную смесь нейтрализовали 5 н. раствором гидроксида натрия. Добавляли к смеси хлороформ и отделяли органический слой. Органический слой сушили над безводным сульфатом магния и нерастворимое вещество отделяли фильтрованием. Фильтрат концентрировали при пониженном давлении и остаток растворяли в тетрагидрофуране (50 мл). Триэтиламин (2,62 мл) и ди-трет-бутилдикарбонат (2,58 г) добавляли к раствору и смесь перемешивали при комнатной температуре в течение 18 часов. К реакционному раствору добавляли воду и этилацетат и отделяли органический слой. Органический слой промывали насыщенным раствором хлорида натрия и сушили над безводным сульфатом магния. Нерастворимое вещество отделяли фильтрованием и фильтрат концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (1,68 г).
ESI-MS; m/z 398 [M++H].
1H-ЯМР (CDCl3) δ (м.д.): 1,51 (с, 9H), 2,74-2,99 (уш.м, 2H), 3,15-3,44 (уш.м, 1H), 3,72-3,85 (уш.м, 1H), 4,17-4,19 (м, 2H), 4,37 (дд, J=8,4, 1,6 Гц, 1H), 7,21-7,29 (м, 1H), 8,19-8,24 (м, 1H), 8,35 (дд, J=7,2, 2,8 Гц, 1H).
(9) Синтез трет-бутил (±)-[(4aS*,7aS*)-7a-(5-амино-2-фторфенил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил]карбамата
Железный порошок (1,89 г) и насыщенный водный раствор хлорида аммония (5 мл) добавляли к раствору соединения, полученного в примере получения 9-(8) (1,68 г), в этаноле (50 мл). Реакционный раствор кипятили с обратным холодильником в течение 30 минут и затем охлаждали до комнатной температуры. Реакционный раствор разбавляли этилацетатом и нерастворимое вещество отделяли фильтрованием через целит. Фильтрат концентрировали при пониженном давлении. К остатку добавляли этилацетат и насыщенный водный раствор хлорида натрия и отделяли органический слой. Органический слой сушили над безводным сульфатом магния и нерастворимое вещество отделяли фильтрованием. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (1,54 г).
ESI-MS; m/z 368 [M++H].
1H-ЯМР (CDCl3) δ (м.д.): 1,50 (с, 9H), 2,75 (дд, J=4,4, 13,2 Гц, 1H), 3,01 (дд, J=3,2, 13,2 Гц, 1H), 3,25-3,30 (м, 1H), 3,62 (уш.с, 2H), 3,84 (д, J=7,6 Гц, 1H), 4,14-4,17 (м, 2H), 4,41 (дд, J=0,8, 9,2 Гц, 1H), 6,55-6,59 (м, 1H), 6,65 (дд, J=3,2, 6,4 Гц, 1H), 6,87 (дд, J=8,4, 12,4 Гц, 1H).
(10) Синтез трет-бутил (-)-[(4aS*,7aS*)-7a-(5-амино-2-фторфенил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил]карбамата
Соединение, полученное в примере получения 9-(9) (50 мг), оптически разделяли, используя CHIRALPAK™ AD-H производства Daicel Chemical Industries, Ltd. (2 см × 25 см, подвижная фаза: гексан:этанол = 7:3, скорость потока: 10 мл/мин), и компоненты, имеющие время удерживания от 23,1 до 26,3 минут, собирали. Данную операцию повторяли с получением указанного в заголовке соединения (220 мг; >99% ee) из 600 мг рацемата.
ESI-MS; m/z 368 [M++H].
1H-ЯМР (CDCl3) δ (м.д.): 1,50 (с, 9H), 2,75 (дд, J=4,4, 13,2 Гц, 1H), 3,01 (дд, J=3,2, 13,2 Гц, 1H), 3,25-3,30 (м, 1H), 3,62 (уш.с, 2H), 3,84 (д, J=7,6 Гц, 1H), 4,14-4,17 (м, 2H), 4,41 (дд, J=0,8, 9,2 Гц, 1H), 6,55-6,59 (м, 1H), 6,65 (дд, J=3,2, 6,4 Гц, 1H), 6,87 (дд, J=8,4, 12,4 Гц, 1H).
Пример получения 10
Синтез (3aR*,5S*,6aS*)-6a-(2-фторфенил)-5-метоксигексагидроциклопента[c]изоксазола и (3aR*,5R*,6aS*)-6a-(2-фторфенил)-5-метоксигексагидроциклопента[c]изоксазола
[Формула 29]

(1) Синтез 3-метокси-5-гексенала
Диметилсульфоксид (0,612 мл) добавляли по каплям к раствору оксалилхлорида (0,652 мл) в дихлорметане (15 мл) при -55°C и смесь перемешивали при -70°C в течение 10 минут. Раствор 3-метокси-5-гексенола (Tetrahedron, 61, 3183-3194 (2005)) (660 мг) в дихлорметане (5 мл) добавляли по каплям к раствору при -60°C и смесь перемешивали при -60°C в течение 15 минут. Триэтиламин (4,95 мл) добавляли по каплям к раствору при -60°C и реакционный раствор перемешивали при температуре от -60°C до комнатной температуры в течение 30 минут. Реакционный раствор вливали в воду с последующим экстрагированием дихлорметаном. Экстракт промывали насыщенным раствором хлорида натрия и затем сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения, содержащего дихлорметан и триэтиламин (3,0 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,24-2,70 (м, 4H), 3,38 (с, 3H), 3,76-3,86 (м, 1H), 5,00-5,40 (м, 2H), 5,70-5,90 (м, 1H), 9,80 (т, J=1,6 Гц, 1H).
(2) Синтез 3-метокси-5-гексеналоксима
Смесь 3-метокси-5-гексенала (3,0 г, с примесью дихлорметана и триэтиламина), сульфата гидроксиламина (990 мг) и ацетата натрия (624 мг) в смеси этанола (6,5 мл) и воды (0,65 мл) перемешивали при комнатной температуре в течение 12 часов. Реакционный раствор вливали в ледяную воду с последующим экстрагированием этилацетатом. Экстракт промывали насыщенным раствором хлорида натрия и затем сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении. Полученный неочищенный продукт очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (500 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,22-2,48 (м, 3H), 2,52-2,68 (м, 1H), 3,37 и 3,38 (с, суммарн. 3H), 3,40-3,54 (м, 1H), 5,05-5,20 (м, 2H), 5,72-5,90 (м, 1H), 6,86 и 7,48 (т, J=5,6 Гц, суммарн. 1H), 7,80 и 8,22 (уш.с, суммарн. 1H).
(3) Синтез 5-метокси-3a,4,5,6-тетрагидро-3H-циклопента[c]изоксазола
Раствор гипохлорита натрия (5% активный хлор, 9,36 мл) добавляли по каплям к раствору 3-метокси-5-гексеналоксима (450 мг) в дихлорметане (20 мл) при 0°C и смесь перемешивали при температуре от 0°C до комнатной температуры в течение 1,5 часов. Реакционный раствор вливали в ледяную воду с последующим экстрагированием дихлорметаном. Экстракт промывали насыщенным раствором хлорида натрия и затем сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при нормальном давлении. Полученный неочищенный продукт очищали хроматографией на колонке с силикагелем с получением менее полярного указанного в заголовке соединения (230 мг) и более полярного указанного в заголовке соединения (150 мг).
Низкополярное указанное в заголовке соединение
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,54-1,68 (м, 1H), 2,20-2,30 (м, 1H), 2,46-2,56 (м, 1H), 2,72-2,84 (м, 1H), 3,30-3,34 (м, 3H), 3,72-3,80 (м, 1H), 3,92-4,06 (м, 1H), 4,26-4,32 (м, 1H), 4,54-4,61 (м, 1H).
Более полярное указанное в заголовке соединение
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,45-1,60 (м, 1H), 2,40-2,55 (м, 2H), 2,83 (дд, J=8,4, 18,0, 1H), 3,35 (с, 3H), 3,55-3,70 (м, 1H), 3,75-3,85 (м, 1H), 4,20-4,33 (м, 1H), 4,50-4,60 (м, 1H).
(4) Синтез (3aR*,5S*,6aS*)-6a-(2-фторфенил)-5-метоксигексагидроциклопента[c]изоксазола и (3aR*,5R*,6aS*)-6a-(2-фторфенил)-5-метоксигексагидроциклопента[c]изоксазола
[Формула 30]

н-Бутиллитий (2,77 M, 2,29 мл) добавляли по каплям к раствору 2-бромфторбензола (1,22 г) в смеси толуола (20 мл) и тетрагидрофурана (6 мл) при -78°C и смесь перемешивали при той же температуре в течение одного часа. Комплекс трифторид бора-диэтиловый эфир (0,797 мл) добавляли по каплям к раствору 5-метокси-3a,4,5,6-тетрагидро-3H-циклопента[c]изоксазола (427 мг, смесь более полярного и менее полярного соединений) в толуоле (30 мл) при -78°C. Ранее полученный раствор 2-фторфениллития добавляли по каплям к раствору при температуре от -78°C до -60°C. Реакционный раствор перемешивали при -78°C в течение одного часа. К реакционному раствору добавляли раствор хлорида аммония при температуре -78°C с последующим нагреванием до комнатной температуры в течение одного часа. Реакционный раствор экстрагировали этилацетатом. Экстракт промывали насыщенным раствором хлорида натрия и затем сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении. Полученный неочищенный продукт очищали хроматографией на колонке с силикагелем с получением менее полярного указанного в заголовке соединения (5S, 247 мг) и более полярного указанного в заголовке соединения (5R, 275 мг).
Менее полярное указанное в заголовке соединение (5S)
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,95-2,17 (м, 2H), 2,17-2,32 (м, 2H), 3,20-3,35 (м, 1H), 3,30 (с, 3H), 3,69 (т, J=8,0 Гц, 1H), 4,05-4,15 (м, 1H), 4,39 (т, J=8,0 Гц, 1H), 6,03 (с, 1H), 6,90-7,32 (м, 3H), 7,94 (т, J=8,0 Гц, 1H).
Более полярное указанное в заголовке соединение (5R)
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,00-2,26 (м, 3H), 2,34-2,44 (м, 1H), 3,26-3,38 (м, 1H), 3,34 (с, 3H), 3,69 (уш.с, 1H), 4,08-4,22 (м, 2H), 7,06 (дд, J=8,0, 12,0 Гц, 1H), 7,12 (т, J=8,0 Гц, 1H), 7,16-7,32 (м, 1H), 7,58-7,72 (м, 1H).
Пример получения 11
Синтез трет-бутил [(4aR*,6S*,7aS*)-7a-(5-амино-2-фторфенил)-6-метокси-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]карбамата ((+)-изомера и (-)-изомера)
[Формула 31]

(1) Синтез [(1R*,2S*,4S*)-2-амино-2-(2-фторфенил)-4-метоксициклопентил]метанола
Цинк (533 мг) добавляли к раствору (3aR*,5S*,6aS*)-6a-(2-фторфенил)-5-метоксигексагидроциклопента[c]изоксазола (247 мг) в уксусной кислоте (5 мл) и смесь перемешивали при комнатной температуре в течение 12 часов. Цинк (500 мг) добавляли к реакционному раствору с последующим перемешиванием при комнатной температуре в течение трех часов. Цинк удаляли фильтрованием и фильтрат вливали в насыщенный раствор бикарбоната натрия с последующим экстрагированием этилацетатом. Экстракт сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (245 мг).
ESI-MS; m/z 240 [M++H].
(2) Синтез 9H-флуорен-9-илметил ({[(1S*,2R*,4S*)-1-(2-фторфенил)-2-(гидроксиметил)-4-метоксициклопентил]амино}карбонотиоил)карбамата
Раствор [(1R*,2S*,4S*)-2-амино-2-(2-фторфенил)-4-метоксициклопентанил]метанола (225 мг) и флуоренилметилоксикарбонилизотиоцианата (316 мг) в дихлорметане (18 мл) перемешивали при комнатной температуре в течение трех суток. Реакционный раствор очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (330 мг).
ESI-MS; m/z 543[M++Na].
(3) Синтез [(4aR*,6S*,7aS*)-7a-(2-фторфенил)-6-метокси-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]амина
Раствор 9H-флуорен-9-илметил ({[(1S*,2R*,4S*)-1-(2-фторфенил)-2-(гидроксиметил)-4-метоксициклопентил]амино}карбонотиоил)карбамата (330 мг) в смеси метанола (20 мл) и концентрированной хлористо-водородной кислоты (1 мл) кипятили с обратным холодильником в течение трех часов. Реакционный раствор концентрировали при пониженном давлении. Ацетонитрил (10 мл) и пиперидин (1 мл) добавляли к остатку при комнатной температуре и смесь перемешивали при комнатной температуре в течение одного часа. Реакционный раствор концентрировали при пониженном давлении. Полученный неочищенный продукт очищали хроматографией на колонке с NH-силикагелем с получением указанного в заголовке соединения (170 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,98-2,10 (м, 2H), 2,14-2,24 (м, 1H), 2,58-2,68 (м, 1H), 2,77 (дд, J=4,8, 12,8 Гц, 1H), 2,91 (дд, J=3,2, 12,8 Гц, 1H), 2,98 (дд, J=8,4, 14,4 Гц, 1H), 3,33 (с, 3H), 4,10-4,22 (м, 1H), 7,01 (дд, J=8,0, 12,4 Гц, 1H), 7,09 (т, J=8,0 Гц, 1H), 7,17-7,35 (м, 2H).
(4) Синтез трет-бутил [(4aR*,6S*,7aS*)-7a-(2-фтор-5-нитрофенил)-6-метокси-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]карбамата
Дымящую азотную кислоту (0,02 мл) добавляли к раствору [(4aR*,6S*,7aS*)-7a-(2-фторфенил)-6-метокси-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]амина (151 мг) в концентрированной серной кислоте (2 мл) при 0°C и смесь перемешивали при 0°C в течение 30 минут. Реакционный раствор вливали в смесь 5 н. раствора гидроксида натрия и ледяной воды с последующим экстрагированием этилацетатом. Экстракт промывали насыщенным раствором хлорида натрия и затем сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении. Триэтиламин (0,301 мл) и ди-трет-бутилдикарбонат (176 мг) добавляли к раствору полученного неочищенного продукта в тетрагидрофуране (10 мл) при комнатной температуре и смесь перемешивали при комнатной температуре в течение 12 часов. Реакционный раствор вливали в насыщенный раствор бикарбоната натрия с последующим экстрагированием этилацетатом. Экстракт промывали насыщенным раствором хлорида натрия и затем сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении. Полученный неочищенный продукт очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (118 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,52 (с, 9H), 2,00-2,25 (м, 2H), 2,25-2,40 (м, 1H), 2,70-2,95 (м, 4H), 3,32 (с, 3H), 4,10-4,20 (м, 1H), 7,14-7,30 (м, 1H), 8,12-8,26 (м, H).
(5) Синтез трет-бутил [(4aR*,6S*,7aS*)-7a-(5-амино-2-фторфенил)-6-метокси-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]карбамата
Раствор трет-бутил [(4aR*,6S*,7aS*)-7a-(2-фтор-5-нитрофенил)-6-метокси-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]карбамата (118 мг) и железа (133 мг) в смеси этанола (8 мл) и насыщенного раствора хлорида аммония (0,303 мл) перемешивали при 87°C в течение 30 минут. Реакционный раствор охлаждали до комнатной температуры и затем вливали в смесь вода-этилацетат с последующим экстрагированием этилацетатом. Экстракт промывали насыщенным раствором хлорида натрия и затем сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении. Полученный неочищенный продукт очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (83 мг).
ESI-MS; m/z 396 [M++H].
(6) Синтез трет-бутил [(4aR*,6S*,7aS*)-7a-(5-амино-2-фторфенил)-6-метокси-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]карбамата ((+)-изомера и (-)-изомера)
[Формула 32]

трет-Бутил (±)-[(4aR*,6S*,7aS*)-7a-(5-амино-2-фторфенил)-6-метокси-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]карбамат (83 мг) оптически разделяли, используя CHIRALPAK™ ADH производства Daicel Chemical Industries, Ltd. (2 см × 25 см, подвижная фаза: гексан:этанол = 8:2, скорость потока: 10 мл/мин). Компоненты, имеющие время удерживания от 14 до 17,5 минут, собирали с получением указанного в заголовке (+)-изомера (44 мг; >99% ee). Компоненты, имеющие время удерживания от 17,5 до 23 минут, собирали с получением указанного в заголовке (-)-изомера (45 мг; 95% ee). Характеристические значения (-)-соединения показаны ниже.
Оптическое вращение (-)
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,51 (с, 9H), 1,92-2,08 (м, 1H), 2,13 (ддд, J=6,4, 6,4, 12,0 Гц, 1H), 2,25-2,35 (м, 1H), 2,69 (дд, J=3,6, 13,2 Гц, 1H), 2,80-3,05 (м, 3H), 3,29 (с, 3H), 3,63 (уш.с, 2H), 4,10-4,20 (м, 1H), 6,54 (ддд, J=3,2, 3,6, 8,4 Гц, 1H), 6,59 (дд, J=3,2, 7,2 Гц, 1H), 6,84 (дд, J=8,4, 12,0 Гц, 1H).
Пример получения 12
Синтез трет-бутил [(4aR*,6R*,7aS*)-7a-(5-амино-2-фторфенил)-6-метокси-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]карбамата ((+)-изомера и (-)-изомера)
[Формула 33]

(1) Синтез [(1R*,2S*,4R*)-2-амино-2-(2-фторфенил)-4-метоксициклопентил]метанола
Цинк (533 мг) добавляли к раствору (3aR*,5R*,6aS*)-6a-(2-фторфенил)-5-метоксигексагидроциклопента[c]изоксазола (275 мг) в уксусной кислоте (5,57 мл) и смесь перемешивали при комнатной температуре в течение 12 часов. Еще добавляли к реакционному раствору цинк (500 мг) с последующим перемешиванием при комнатной температуре в течение трех часов. Цинк удаляли фильтрованием и фильтрат вливали в насыщенный раствор бикарбоната натрия с последующим экстрагированием этилацетатом. Экстракт сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (270 мг).
ESI-MS; m/z 240 [M++H].
(2) Синтез 9H-флуорен-9-илметил ({[(1S*,2R*,4R*)-1-(2-фторфенил)-2-(гидроксиметил)-4-метоксициклопентил]амино}карбонотиоил)карбамата
Раствор [(1R*,2S*,4R*)-2-амино-2-(2-фторфенил)-4-метоксициклопентил]метанола (250 мг) и флуоренилметилоксикарбонилизотиоцианата (351 мг) в дихлорметане (20 мл) перемешивали при комнатной температуре в течение трех суток. Реакционный раствор очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (340 мг).
ESI-MS; m/z 543 [M++Na].
(3) Синтез [(4aR*,6S*,7aS*)-7a-(2-фторфенил)-6-метокси-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]амина
Раствор 9H-флуорен-9-илметил ({[(1S*,2R*,4R*)-1-(2-фторфенил)-2-(гидроксиметил)-4-метоксициклопентил]амино}карбонотиоил)карбамата (340 мг) в смеси метанола (20 мл) и концентрированной хлористо-водородной кислоты (1 мл) кипятили с обратным холодильником в течение трех часов. Реакционный раствор концентрировали при пониженном давлении. Ацетонитрил (10 мл) и пиперидин (2 мл) добавляли к остатку при комнатной температуре и смесь перемешивали при комнатной температуре в течение одного часа. Реакционный раствор концентрировали при пониженном давлении. Неочищенный продукт очищали хроматографией на колонке с NH-силикагелем с получением указанного в заголовке соединения (130 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,87 (ддд, J=3,6, 9,2, 13,2 Гц, 1H), 2,20-2,38 (м, 2H), 2,64 (дд, J=6,8, 12,8 Гц, 1H), 2,73 (дд, J=3,6, 12,8 Гц, 1H), 2,95 (дд, J=3,6, 12,8 Гц, 1H), 3,05-3,15 (м, 1H), 3,33 (с, 3H), 3,87-4,00 (м, 1H), 7,02 (дд, J=7,6, 12,0 Гц, 1H), 7,09 (т, J=7,6 Гц, 1H), 7,17-7,33 (м, 2H).
(4) Синтез трет-бутил [(4aR*,6R*,7aS*)-7a-(2-фтор-5-нитрофенил)-6-метокси-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]карбамата
Дымящую азотную кислоту (0,0193 мл) добавляли к раствору [(4aR*,6R*,7aS*)-7a-(2-фторфенил)-6-метокси-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]амина (146 мг) в концентрированной серной кислоте (2 мл) при 0°C и смесь перемешивали при 0°C в течение 30 минут. Реакционный раствор вливали в смесь 5 н. раствора гидроксида натрия и ледяной воды с последующим экстрагированием этилацетатом. Экстракт промывали насыщенным раствором хлорида натрия и затем сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении. Триэтиламин (0,291 мл) и ди-трет-бутилдикарбонат (170 мг) добавляли к раствору полученного неочищенного продукта в тетрагидрофуране (9,67 мл) при комнатной температуре и смесь перемешивали при комнатной температуре в течение 12 часов. Реакционный раствор вливали в насыщенный раствор бикарбоната натрия с последующим экстрагированием этилацетатом. Экстракт промывали насыщенным раствором хлорида натрия и затем сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении. Полученный неочищенный продукт очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (198 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,53 (с, 9H), 1,92-2,06 (м, 1H), 2,30-2,45 (м, 2H), 2,54 (дд, J=6,0, 12,8 Гц, 1H), 2,68-2,80 (м, 1H), 2,80-3,00 (м, 1H), 3,18-3,36 (м, 1H), 3,30 (с, 3H), 3,90-4,10 (м, 1H), 7,00-7,40 (м, 1H), 8,05-8,30 (м, 2H).
(5) Синтез трет-бутил [(4aR*,6R*,7aS*)-7a-(5-амино-2-фторфенил)-6-метокси-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]карбамата ((+)-изомера и (-)-изомера)
[Формула 34]
Раствор трет-бутил [(4aR*,6R*,7aS*)-7a-(2-фтор-5-нитрофенил)-6-метокси-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]карбамата (198 мг) и железа (223 мг) в смеси этанола (13,4 мл) и насыщенного раствора хлорида аммония (0,508 мл) перемешивали при 87°C в течение 30 минут. Реакционный раствор вливали в смесь этилацетат-вода при комнатной температуре с последующим экстрагированием этилацетатом. Экстракт промывали насыщенным раствором хлорида натрия и затем сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении. Полученный неочищенный продукт очищали хроматографией на колонке с силикагелем с получением указанной в заголовке (±)-смеси (118 мг).
Полученную смесь оптически разделяли, используя CHIRALPAK™ ADH производства Daicel Chemical Industries, Ltd. (2 см × 25 см, подвижная фаза: гексан:этанол = 5:5, скорость потока: 10 мл/мин). Компоненты, имеющие время удерживания от 8 до 11 минут, собирали с получением указанного в заголовке (+)-изомера (52 мг; >95% ee). Компоненты, имеющие время удерживания от 17,5 до 23 минут, собирали с получением указанного в заголовке (-)-изомера (52 мг; >95% ee). Характеристические значения (-)-изомера указанного в заголовке соединения показаны ниже. Оптическое вращение (-).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,52 (с, 9H), 1,90-2,02 (м, 1H), 2,26-2,44 (м, 2H), 2,65 (дд, J=6,0, 13,6 Гц, 1H), 2,70 (дд, J=3,6, 13,6 Гц, 1H), 3,03 (дд, J=3,6, 13,6 Гц, 1H), 3,20-3,36 (м, 1H), 3,32 (с, 3H), 3,62 (уш.с, 2H), 4,00-4,10 (м, 1H), 6,45-6,66 (м, 2H), 6,86 (дд, J=8,4, 12,0 Гц, 1H).
Пример получения 13
Синтез 5-цианопиридин-2-карбоновой кислоты
[Формула 35]

Синтез метил 5-цианопиридин-2-карбоксилата
Смесь метил 5-бромпиридин-2-карбоксилата (2,8 г) и цианида меди (3,6 г) в NMP (30 мл) нагревали при перемешивании при 170°C в течение 1,5 часов. К реакционному раствору добавляли воду при комнатной температуре и нерастворимое вещество удаляли фильтрованием. Фильтрат экстрагировали этилацетатом. Экстракт промывали насыщенным раствором хлорида натрия и затем сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении. Полученный неочищенный продукт очищали хроматографией на колонке с силикагелем (система этилацетат-гептан) с получением указанного в заголовке соединения (920 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 4,06 (с, 3H), 8,16 (дд, J=2,0, 8,0 Гц, 1H), 8,27 (д, J=8,0 Гц, 1H), 9,01 (д, J=2,0 Гц, 1H).
Синтез 5-цианопиридин-2-карбоновой кислоты
Раствор соединения из примера получения 13-(1) (920 мг) и 5 н. раствора гидроксида натрия (2,26 мл) в этаноле (30 мл) перемешивали при комнатной температуре в течение 10 минут. К реакционному раствору добавляли 5 н. хлористо-водородную кислоту (5,2 мл) при комнатной температуре с последующим экстрагированием этилацетатом. Экстракт сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (800 мг).
1H-ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 8,18 (д, J=8,0 Гц, 1H), 8,51 (дд, J=2,0, 8,0 Гц, 1H), 9,12-9,18 (м, 1H).
Пример получения 14
Синтез 5-дифторметоксипиразин-2-карбоновой кислоты
[Формула 36]

(1) Синтез метил 5-дифторметоксипиразин-2-карбоксилата
Карбонат калия (8,82 г) и хлордифторацетат натрия (6,53 г) добавляли к раствору соединения (CAS 13924-95-3) (3,3 г) в ДМФА (42,8 мл). Реакционный раствор перемешивали при 100°C в течение 30 минут и затем добавляли насыщенный водный раствор хлорида аммония с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным раствором бикарбоната натрия и насыщенным раствором хлорида натрия и затем сушили над сульфатом магния. Осушающий агент удаляли фильтрованием и затем выпаривали при пониженном давлении растворитель. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (928 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 4,04 (с, 3H), 7,49 (т, J=71,2 Гц, 1H), 8,47 (д, J=0,8 Гц, 1H), 8,92 (д, J=0,8 Гц, 1H).
(2) Синтез 5-дифторметоксипиразин-2-карбоновой кислоты
Воду (1,54 мл) и 5 н. раствор гидроксида натрия (492 мл) добавляли к раствору соединения, полученного в примере получения 14-(1) (250 мг), в ТГФ (4,60 мл). Реакционный раствор перемешивали при комнатной температуре в течение пяти минут и затем добавляли 2 н. раствор хлористо-водородной кислоты с последующим экстрагированием этилацетатом. Органические слои промывали насыщенным раствором хлорида натрия и затем сушили над сульфатом магния. Осушающий агент удаляли фильтрованием и затем концентрировали при пониженном давлении растворитель с получением указанного в заголовке соединения (200 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 7,51 (т, J=71,2 Гц, 1H), 8,39 (д, J=1,2 Гц, 1H), 9,04 (д, J=1,2 Гц, 1H).
Пример получения 15
Синтез 5-фторметоксипиразин-2-карбоновой кислоты
[Формула 37]

(1) Синтез метил 5-фторметоксипиразин-2-карбоксилата
Фторметилтолуол-4-сульфонат (Journal of Labelled Compounds & Radiopharmaceuticals, 46 (6), 555-566; 2003) (344 мг) и карбонат цезия (824 мг) добавляли к раствору метил 5-гидроксипиразин-2-карбоксилата (130 мг) в N,N-диметилформамиде (2,0 мл). Реакционный раствор перемешивали при 70°C в течение пяти часов и 30 минут и затем охлаждали до комнатной температуры. К реакционному раствору добавляли воду с последующим экстрагированием этилацетатом. Органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с NH-силикагелем с получением указанного в заголовке соединения (18,0 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 4,03 (с, 3H), 6,14 (д, J=51,2 Гц, 2H), 8,42 (д, J=1,2 Гц, 1H), 8,94 (д, J=1,2 Гц, 1H).
(2) Синтез 5-фторметоксипиразин-2-карбоновой кислоты
Триметилсиланолат калия (18,6 мг) добавляли к раствору метил 5-фторметоксипиразин-2-карбоксилата, полученного в примере получения 15-(1) (18,0 мг), в тетрагидрофуране (1,0 мл). Реакционный раствор перемешивали при комнатной температуре в течение одного часа. Добавляли к реакционному раствору воду и этилацетат и отделяли водный слой. Водный слой подкисляли 1 М хлористо-водородной кислотой с последующим экстрагированием этилацетатом. Органический слой сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении с получением неочищенного продукта, состоящего из указанного в заголовке соединения (10,2 мг). Соединение использовали для следующей реакции без дополнительной очистки.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 6,16 (д, J=50,8 Гц, 2H), 8,34 (д, J=1,4 Гц, 1H), 9,05 (д, J=1,4 Гц, 1H).
Пример получения 16
Синтез 5-фторметоксипиридин-2-карбоновой кислоты
[Формула 38]

(1) Синтез метил 5-фторметоксипиридин-2-карбоксилата
Раствор, содержащий фторметилтолуол-4-сульфонат (233 мг), в ДМФА добавляли к раствору, содержащему метил 5-гидроксипиридин-2-карбоксилат (100 мг) и карбонат цезия (532 мг), в ДМФА (5 мл). Реакционный раствор перемешивали при 70°C в течение трех часов. Реакционный раствор охлаждали до комнатной температуры. Добавляли к реакционному раствору этилацетат и насыщенный водный раствор хлорида аммония и отделяли органический слой. Органический слой сушили над безводным сульфатом магния и нерастворимое вещество отделяли фильтрованием. Фильтрат концентрировали и остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (51 мг).
1H-ЯМР (CDCl3) δ (м.д.): 4,00 (с, 3H), 5,80 (д, J=45,1 Гц, 2H), 7,51 (ддд, J=0,8, 2,8, 8,8 Гц, 1H), 8,16 (д, J=0,4, 8,8 Гц, 1H), 8,54 (д, J=2,8 Гц, 1H).
(2) Синтез 5-фторметоксипиридин-2-карбоновой кислоты
К раствору, содержащему метил 5-фторметоксипиридин-2-карбоксилат (50 мг) в смеси тетрагидрофуран/вода (2 мл, 3/1), добавляли 5 н. гидроксид натрия (81 мкл) и смесь перемешивали при комнатной температуре в течение 10 минут. К реакционному раствору добавляли воду (1 мл) с последующим перемешиванием в течение 20 минут. Реакционный раствор подкисляли 5 н. хлористо-водородной кислотой. К реакционному раствору добавляли этилацетат и насыщенный раствор хлорида натрия и отделяли органический слой. Органический слой сушили над безводным сульфатом магния и нерастворимое вещество отделяли фильтрованием. Фильтрат концентрировали с получением указанного в заголовке соединения (22,6 мг).
1H-ЯМР (CDCl3) δ (м.д.): 5,81 (д, J=53,2 Гц, 2H), 7,61 (ддд, J=0,8, 2,8, 8,8 Гц, 1H), 8,25 (д, J=0,8, 8,8 Гц, 1H), 8,42 (д, J=2,4 Гц, 1H).
Пример получения 17
Синтез 5-дифторметилпиразин-2-карбоновой кислоты
[Формула 39]

(1) Синтез трет-бутил 5-метилпиразин-2-карбоксилата
Комплекс трифторид бора-диэтиловый эфир (91,7 мкл) добавляли по каплям к суспензии 2-метилпиразин-5-карбоновой кислоты (1 г) и трет-бутил 2,2,2-трихлорацетимидата (4,75 г) в тетрагидрофуране (20 мл) при охлаждении льдом. Реакционный раствор нагревали до комнатной температуры с последующим перемешиванием в течение двух часов. К реакционному раствору добавляли насыщенный раствор хлорида натрия и этилацетат и отделяли органический слой. Органический слой сушили над безводным сульфатом магния и нерастворимое вещество отделяли фильтрованием. Фильтрат концентрировали и очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (1,4 г).
1H-ЯМР (CDCl3) δ (м.д.): 1,65 (с, 9H), 2,65 (с, 3H), 8,57 (д, J=1,2 Гц, 1H), 9,10 (д, J =1,6 Гц, 1H).
(2) Синтез трет-бутил 5-((E)-2-диметиламиновинил)пиразин-2-карбоксилата
Смесь трет-бутил 5-метилпиразин-2-карбоксилата (1,35 г), N,N-диметилформамида (25 мл) и диметилацеталя N,N-диметилформамида (25 мл) перемешивали при 130°C в течение пяти часов. Реакционный раствор охлаждали до комнатной температуры и разбавляли этилацетатом. Смесь промывали три раза насыщенным раствором хлорида натрия. Органический слой сушили над безводным сульфатом магния и нерастворимое вещество отделяли фильтрованием. Фильтрат концентрировали и остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (648 мг).
1H-ЯМР (CDCl3) δ (м.д.): 1,63 (с, 9H), 3,00 (с, 6H), 5,16 (д, J=12,8 Гц, 1H), 7,72 (д, J=12,8 Гц, 1H), 8,16 (д, J=1,2 Гц, 1H), 8,81 (д, J=1,6 Гц, 1H).
(3) Синтез трет-бутил 5-формилпиразин-2-карбоксилата
Периодат натрия (1,67 г) добавляли к раствору трет-бутил 5-((E)-2-диметиламиновинил)пиразин-2-карбоксилата (645 мг) в смеси 50% тетрагидрофуран-вода (26 мл) и смесь перемешивали при комнатной температуре в течение четырех часов. К реакционному раствору добавляли насыщенный раствор бикарбоната натрия и этилацетат и отделяли органический слой. Органический слой промывали насыщенным раствором хлорида натрия и сушили над безводным сульфатом магния. Нерастворимое вещество отделяли фильтрованием и фильтрат концентрировали. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (249 мг).
1H-ЯМР (CDCl3) δ (м.д.): 1,68 (с, 9H), 9,25 (д, J=1,2 Гц, 1H), 9,36 (д, J=1,6 Гц, 1H), 10,2 (с, 1H).
(4) Синтез трет-бутил 5-дифторметилпиразин-2-карбоксилата
Трифторид [бис(2-метоксиэтил)амино]серы (662 мкл) добавляли по каплям к раствору трет-бутил 5-формилпиразин-2-карбоксилата (249 мг) в дихлорметане (12 мл) в атмосфере азота при охлаждении льдом. Реакционный раствор перемешивали в течение двух часов, постепенно доводя его до комнатной температуры. К реакционному раствору добавляли насыщенный раствор бикарбоната натрия и этилацетат и отделяли органический слой. Органический слой промывали насыщенным раствором хлорида натрия и сушили над безводным сульфатом магния. Нерастворимое вещество отделяли фильтрованием и фильтрат концентрировали. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (175 мг).
1H-ЯМР (CDCl3) δ (м.д.): 1,67 (с, 9H), 6,75 (т, J=54,4 Гц, 1H), 9,02 (д, J=0,8 Гц, 1H), 9,25 (д, J=0,8 Гц, 1H).
(5) Синтез 5-дифторметилпиразин-2-карбоновой кислоты
Трифторуксусную кислоту (1 мл) добавляли к раствору трет-бутил 5-дифторметилпиразин-2-карбоксилата (175 мг) в дихлорметане (1 мл) и смесь перемешивали при комнатной температуре в течение пяти часов. Простой эфир и 5 н. гидроксид натрия добавляли к реакционному раствору. Водный слой отделяли и подкисляли 5 н. хлористо-водородной кислотой. Добавляли к водному слою этилацетат и отделяли органический слой. Органический слой сушили над безводным сульфатом магния и нерастворимое вещество отделяли фильтрованием. Фильтрат концентрировали с получением указанного в заголовке соединения (100 мг).
1H-ЯМР (CDCl3) δ (м.д.): 6,80 (т, J=54,4 Гц, 1H), 9,02 (с, 1H), 9,47 (с, 1H).
Пример получения 18
Синтез трет-бутил (±)-{(4aR*,7aS*)-7a-[3-(2-фторпиридин-3-ил)фенил]-4,4a,5,6,7,7a-гексагидропирроло[3,4-d][1,3]тиазин-2-ил}карбамата
[Формула 40]


(1) Синтез 2-[аллил(2,4-диметоксибензил)амино]-1-(3-бромфенил)этанона
3-Бромфенацилбромид (20,9 г) растворяли в дихлорметане (400 мл). После охлаждения раствора льдом растворяли N,N-диизопропилэтиламин (14,3 мл) и аллил(2,4-диметоксибензил)амин (18,7 г) в дихлорметане (50 мл) и добавляли по каплям. Реакционный раствор постепенно нагревали до комнатной температуры, погружая в ванну с ледяной водой, и перемешивали в течение примерно 20 часов. Хлороформ, насыщенный раствор бикарбоната натрия и насыщенный раствор хлорида натрия последовательно добавляли к реакционному раствору и отделяли органический слой. Органический слой сушили над безводным сульфатом магния (2 г). Осушающий агент удаляли фильтрованием и затем фильтрат концентрировали при пониженном давлении с получением неочищенного продукта. Продукт подвергали хроматографии на колонке с силикагелем с получением указанного в заголовке соединения (29,88 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 3,22-3,27 (м, 2H), 3,62 (с, 3H), 3,72 (с, 2H), 3,75 (с, 2H), 3,80 (с, 3H), 5,12-2,28 (м, 2H), 5,87-6,00 (м, 1H), 6,39 (д, J=2,4 Гц, 1H), 6,44 (дд, J=2,4, 8 Гц, 1H), 7,16 (д, J=8 Гц, 1H), 7,22-7,32 (м, 1H), 7,60-7,68 (м, 1H), 7,80-7,87 (м, 1H), 8,07 (т, J=1,6 Гц, 1H).
(2) Синтез (3aS*,6aR*)-6a-(3-бромфенил)-5-(2,4-диметоксибензил)гексагидропирроло[3,4-c]изоксазола
Соединение, полученное в примере получения 18-(1) (29,8 г), растворяли в этаноле (475 мл). Гидрохлорид гидроксиламина (10,3 г) и ацетат натрия (12,1 г) добавляли к раствору и смесь нагревали при перемешивании при 90°C. Через пять часов реакционный раствор охлаждали и затем фильтровали. Фильтрат концентрировали при пониженном давлении. К остатку добавляли этилацетат и смесь последовательно промывали насыщенным раствором бикарбоната натрия и насыщенным раствором хлорида натрия. Органический слой сушили над безводным сульфатом магния (2 г). Осушающий агент удаляли фильтрованием и затем фильтрат концентрировали при пониженном давлении с получением оксимного соединения (31,3 г). Оксимное соединение (31,3 г) растворяли в толуоле (600 мл). Раствор кипятили с обратным холодильником в атмосфере азота в течение восьми часов. Реакционный раствор доводили до комнатной температуры и затем концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем с получением указанного в заголовке соединения (19 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,44 (уш.с, 1H), 2,65 (уш.с, 1H), 2,91 (уш.с, 1H), 2,97-3,14 (м, 2H), 3,50-3,62 (м, 1H), 3,66 (с, 2H), 3,79 (с, 3H), 3,81 (с, 3H), 4,47 (уш.с, 1H), 5,42 (уш.с, 1H), 6,44-6,50 (м, 2H), 7,15-7,22 (м, 2H), 7,33-7,40 (м, 1H), 7,42-7,49 (м, 1H), 7,69-7,74 (м, 1H).
(3) Синтез (±)-[(3S*,4R*)-4-амино-4-(3-бромфенил)-1-(2,4-диметоксибензил)пирролидин-3-ил]метанола
Соединение, полученное в примере получения 18-(2) (19 г), растворяли в уксусной кислоте (230 мл). К раствору добавляли цинк (30 г) с последующим перемешиванием при комнатной температуре. Через 20 часов реакционный раствор фильтровали через целит. Фильтрат концентрировали при пониженном давлении. К остатку добавляли 5 н. раствор гидроксида натрия и хлороформ с последующим фильтрованием через целит. Фильтрат отделяли. Органический слой сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении с получением остатка. Остаток подвергали хроматографии на колонке с силикагелем с получением указанного в заголовке соединения (19,1 г).
ESI-MS; m/z 421 [M+H].
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,44-2,53 (м, 1H), 2,68-2,77 (м, 3H), 2,97 (д, J=9,2 Гц, 1H), 3,62-3,82 (м, 4H), 3,81 (с, 3H), 3,82 (с, 3H) 6,44 (м, 2H), 7,18-7,29 (м, 2H), 7,34-7,40 (м, 1H), 7,46-7,53 (м, 1H), 7,71 (т, J=2,0 Гц, 1H).
(4) Синтез (±)-1-бензоил-3-[(3R*,4S*)-3-(3-бромфенил)-1-(2,4-диметоксибензил)-4-гидроксилметилпирролидин-3-ил]тиомочевины
Соединение, полученное в примере получения 18-(3) (2,29 г), растворяли в дихлорметане (50 мл). Бензоилизотиоцианат (807 мкл) добавляли к раствору с последующим перемешиванием при комнатной температуре. Через 11 часов реакционный раствор концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем с получением указанного в заголовке соединения (1,96 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,55 (т, J=8,4 Гц, 1H), 2,81-2,90 (м, 1H), 2,98-3,06 (м, 1H), 3,20 (д, J=10,4 Гц, 1H), 3,63-3,83 (м, 3H), 3,81 (с, 3H), 3,83 (с, 3H), 3,87-3,99 (м, 2H), 6,44-6,52 (м, 2H), 7,18 (т, J=8,0 Гц, 1H), 7,24-7,32 (м, 1H), 7,33-7,38 (м, 1H), 7,40-7,46 (м, 1H), 7,49-7,57 (м, 2H), 7,61-7,70 (м, 2H), 7,84-7,91 (м, 2H), 8,91 (с, 1H), 11,7 (с, 1H).
(5) Синтез (±)-N-[(4aR*,7aS*)-7a-(3-бромфенил)-6-(2,4-диметоксибензил)-4,4a,5,6,7,7a-гексагидропирроло[3,4-d][1,3]тиазин-2-ил]бензамида
Раствор соединения, полученного в примере получения 18-(4) (1,43 г), и пиридина (810 мкл) в дихлорметане (55 мл) охлаждали до -50°C. Трифторметансульфоновый ангидрид (1,1 мл) добавляли по каплям к раствору и смесь постепенно нагревали до 0°C. Через один час и 30 минут реакционный раствор охлаждали до -20°C, разбавляли хлороформом и промывали насыщенным раствором бикарбоната натрия. Органический слой сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием. Фильтрат концентрировали при пониженном давлении с получением остатка. Остаток подвергали хроматографии на колонке с силикагелем с получением указанного в заголовке соединения (1,30 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,73 (дд, J=4,4, 13,6 Гц, 1H), 2,90 (д, J =10,4 Гц, 1H), 2,92-3,18 (м, 4H), 3,38 (д, J=10,4 Гц, 1H), 3,70 (с, 2H), 3,77 (с, 3H), 3,81 (с, 3H), 6,40 (дд, J=2,4, 8,4 Гц, 1H), 6,44 (д, J=2,4 Гц, 1H), 7,18-7,30 (м, 2H), 7,40-7,54 (м, 5H), 7,68-7,72 (м, 1H), 8,17-8,26 (м, 2H).
(6) Синтез (±)-(4aR*,7aS*)-7a-(3-бромфенил)-6-(2,4-диметоксибензил)-4,4a,5,6,7,7a-гексагидропирроло[3,4-d][1,3]тиазин-2-иламина
1,8-Диазабицикло[5,4,0]ундец-7-ен (684 мкл) добавляли к раствору соединения, полученного в примере получения 18-(5) (1,3 г), в метаноле (22 мл) и смесь кипятили с обратным холодильником. Через четыре часа и 30 минут реакционному раствору давали охладиться и затем концентрировали при пониженном давлении с получением остатка. Остаток суспендировали в трет-бутилметиловом эфире и собирали фильтрованием с получением указанного в заголовке соединения (837 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,52-2,68 (м, 3H), 2,72-2,80 (м, 1H), 2,92 (дд, J=4,0, 12,8 Гц, 1H), 3,06-3,14 (м, 1H), 3,39 (д, J=9,6 Гц, 1H), 3,64 (д, J=10 Гц, 2H), 3,81 (с, 3H), 3,83 (с, 3H), 4,48 (уш.с, 2H), 6,44-6,50 (м, 2H), 7,13-7,19 (м, 1H), 7,27-7,34 (м, 2H), 7,46-7,52 (м, 1H), 7,70-7,74 (м, 1H).
(7) Синтез (±)-ди-трет-бутил [(4aR*,7aS*)-7a-(3-бромфенил)-6-(2,4-диметоксибензил)-4,4a,5,6,7,7a-гексагидропирроло[3,4-d][1,3]тиазин-2-ил]имидодикарбоната
Соединение, полученное в примере получения 18-(6) (1,14 г), растворяли в ТГФ (45 мл) и ДМФА (20 мл). Ди-трет-бутилдикарбонат (3,23 г) и 4-диметиламинопиридин (2,11 г) последовательно добавляли к раствору и затем смесь перемешивали при комнатной температуре. Через примерно 17 часов реакционный раствор концентрировали при пониженном давлении. К остатку добавляли этилацетат и насыщенный раствор бикарбоната натрия и отделяли органический слой. Органический слой промывали насыщенным раствором хлорида натрия и сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении с получением остатка. Остаток подвергали хроматографии на силикагеле с получением указанного в заголовке соединения (1,33 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,56 (с, 18H), 2,54 (д, J=10 Гц, 1H), 2,60-2,80 (м, 3H), 2,92-3,00 (м, 1H), 3,10-3,18 (м, 1H), 3,53-3,70 (м, 3H), 3,81 (с, 3H), 3,82 (с, 3H), 6,42-6,54 (м, 2H), 7,16-7,22 (м, 1H), 7,26-7,38 (м, 2H), 7,46-7,53 (м, 1H), 7,64 (т, J=2,0 Гц, 1H).
(8) Синтез (±)-ди-трет-бутил {(4aR*,7aS*)-6-(2,4-диметоксибензил)-7a-[3-(2-фторпиридин-3-ил)фенил]-4,4a,5,6,7,7a-гексагидропирроло[3,4-d][1,3]тиазин-2-ил}имидодикарбоната
Соединение, полученное в примере получения 18-(7) (1,33 г), растворяли в ТГФ (25 мл). 2-Фторпиридин-3-бороновую кислоту (848 мг), фторид калия (495 мг), Pd2DBA3 (172 мг) и Pd(t-Bu3P)2 (195 мг) добавляли к раствору и смесь перемешивали в атмосфере азота при комнатной температуре. Через 3,5 часа реакционный раствор разбавляли этилацетатом и фильтровали через NH силикагель (80 мл). Полученный продукт дополнительно промывали смесью этилацетат:гептан = 1:1 (500 мл). Фильтрат концентрировали при пониженном давлении с получением остатка. Остаток подвергали хроматографии на силикагеле с получением указанного в заголовке соединения (1,15 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,52 (с, 18H), 2,63 (д, J=10,4 Гц, 1H), 2,68-2,87 (м, 3H), 3,00-3,07 (м, 1H), 3,13-3,20 (м, 1H), 3,60-3,69 (м, 3H), 3,78 (с, 3H), 3,81 (с, 3H), 6,40-6,52 (м, 2H), 7,22-7,29 (м, 1H), 7,29-7,36 (м, 1H), 7,38-7,50 (м, 2H), 7,53-7,59 (м, 1H), 7,72 (с, 1H), 7,87-7,94 (м, 1H), 8,16-8,22 (м, 1H).
(9) Синтез трет-бутил (±)-{(4aR*,7aS*)-7a-[3-(2-фторпиридин-3-ил)фенил]-4,4a,5,6,7,7a-гексагидропирроло[3,4-d][1,3]тиазин-2-ил}карбамата
Триэтиламин (387 мкл) добавляли к раствору соединения, полученного в примере получения 18-(8) (411,00 мг), в дихлорметане (20,00 мл). Смесь в достаточной мере охлаждали на ледяной бане в атмосфере азота и затем медленно добавляли трифторуксусный ангидрид (344 мкл). По окончании добавления смесь перемешивали в течение трех часов и 30 минут. Растворитель выпаривали из смеси при пониженном давлении и затем остаток растворяли в метаноле (30 мл). Добавляли к раствору 5 н. водный раствор гидроксида калия (1,6 мл) и смесь перемешивали при комнатной температуре в течение двух часов и 30 минут. Реакционную смесь разбавляли хлороформом и затем промывали насыщенным раствором хлорида натрия. Полученный органический слой сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (122 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,58 (с, 9H), 2,73-2,78 (м, 1H), 2,91-2,95 (м, 1H), 3,10-3,15 (м, 1H), 3,24 (д, J=12,0 Гц, 1H), 3,34-3,45 (м, 2H), 3,53 (д, J=12,0 Гц, 1H), 7,28-7,32 (м, 1H), 7,42-7,46 (м, 1H), 7,50-7,54 (м, 3H), 7,85-7,89 (м, 1H), 8,22-8,24 (м, 1H).
Пример получения 19
Синтез трет-бутил [(4aS,7R,8aS)-8a-(5-амино-2-фторфенил)-7-метоксиметил-4a,7,8,8a-тетрагидро-4H,5H-6-окса-3-тиа-1-азанафталин-2-ил]карбамата
[Формула 41]

(1) Синтез (R)-1-бензилокси-3-[1,3]дитиан-2-илпропан-2-ола
Раствор, содержащий 1,3-дитиан (11 г) в ТГФ (190 мл), охлаждали до -70°С. Добавляли к реакционному раствору н-бутиллитий (2,64 М раствор в гексане, 35 мл) и затем смесь нагревали до -30°С и перемешивали в течение одного часа. Реакционный раствор охлаждали до -70°С и добавляли по каплям раствор, содержащий бензил (R)-(-)-глицидиловый простой эфир (16,5 г) в ТГФ (18 мл). Охлаждающую баню удаляли и смесь постепенно нагревали до комнатной температуры. После перемешивания в течение ночи к реакционному раствору добавляли насыщенный раствор хлорида аммония, насыщенный раствор соли и трет-бутилметиловый эфир и отделяли органический слой. Органический слой промывали насыщенным раствором соли и сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и затем фильтрат концентрировали при пониженном давлении с получением остатка. Остаток подвергали хроматографии на колонке с силикагелем с получением указанного в заголовке соединения (22,1 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,78-2,00 (м, 3H), 2,08-2,16 (м, 1H), 2,42-2,46 (м, 1H), 2,79-2,96 (м, 4H), 3,38 (дд, J=6,8, 9,6 Гц, 1H), 3,52 (дд, J=3,4, 9,6 Гц, 1H), 4,10-4,18 (м, 1H), 4,24-4,30 (м, 1H), 4,56 (д, J=1,6 Гц, 2H), 7,27-7,39 (м, 5H).
(2) Синтез 2-((R)-2-аллилокси-3-бензилоксипропил)-[1,3]дитиана
Раствор (R)-1-бензилокси-3-[1,3]-дитиан-2-илпропан-2-ола (22,1 г) в ТГФ (300 мл) охлаждали до 0°C. Добавляли 60% гидрид натрия (4,35 г) с последующим перемешиванием. Через 12 минут добавляли аллилбромид (10 мл) и затем ледяную баню удаляли. Смесь дополнительно перемешивали при комнатной температуре. После перемешивания в течение ночи реакционный раствор добавляли к смеси льда и трет-бутилметилового эфира и отделяли органический слой. Органический слой промывали насыщенным раствором соли и затем сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении с получением остатка. Остаток подвергали хроматографии на силикагеле с получением указанного в заголовке соединения (24,7 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,80-2,16 (м, 4H), 2,76-2,92 (м, 4H), 3,51 (дд, J=2, 4,8 Гц, 2H), 3,78-3,88 (м, 1H), 4,04-4,11 (м, 1H), 4,12-4,22 (м, 2H), 4,55 (д, J=5,6 Гц, 2H), 5,12-5,19 (м, 1H), 5,23-5,32 (м, 1H), 5,88-6,00 (м, 1H), 7,27-7,39 (м, 5H).
(3) Синтез (R)-3-аллилокси-4-бензилоксибутиральдегидоксима
Карбонат калия (5,14 г) и метилиодид (4,90 мл) добавляли к смешанному раствору 2-((R)-2-аллилокси-3-бензилоксипропил)-[1,3]-дитиана (12 г) в ацетонитриле (27 мл) и воде (4,5 мл) и затем смесь перемешивали при 40°C. Через четыре часа воду (3 мл) и метилиодид (2 мл) добавляли к реакционному раствору. Еще через три часа добавляли к реакционному раствору метилиодид (1 мл). После перемешивания в течение восьми часов в сумме к реакционному раствору добавляли воду и трет-бутилметиловый эфир и отделяли органический слой. Органический слой промывали насыщенным раствором соли. Органический слой сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении с получением неочищенного (R)-3-аллилокси-4-бензилоксибутиральдегида (примерно 10 г). Неочищенный продукт использовали для следующей реакции без дополнительной очистки. Гидрохлорид гидроксиламина (4,29 г) и ацетат натрия (4,92 г) последовательно добавляли к раствору этанола (60 мл) и воды (15 мл). Указанный выше альдегид (10 г) добавляли к смеси с последующим перемешиванием в течение 20 часов. Добавляли к реакционному раствору насыщенный раствор бикарбоната натрия и этилацетат и отделяли органический слой. Органический слой последовательно промывали насыщенным раствором бикарбоната натрия и насыщенным раствором соли и сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и затем фильтрат концентрировали при пониженном давлении с получением остатка. Остаток подвергали хроматографии на колонке с силикагелем с получением указанного в заголовке соединения (5,74 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,41-2,55 (м, 1H), 2,59-2,74 (м, 1H), 3,47-3,60 (м, 2H), 3,68-3,82 (м, 1H), 4,02-4,19 (м, 2H), 4,52-4,58 (м, 2H), 5,14-5,20 (м, 1H), 5,23-5,32 (м, 1H), 5,84-5,98 (м, 1H), 6,87 (т, J=5,4 Гц, 0,5H) 7,24-7,40 (м, 5H), 7,48 (т, J=6,2 Гц, 0,5H).
(4) Синтез (3aS,6R)-6-бензилоксиметил-3a,4,6,7-тетрагидро-3H-пирано[4,3-c]изоксазола
Гипохлорит натрия (5% водный раствор, 42 мл) добавляли по каплям к раствору, содержащему оксим, синтезированный на предшествующей стадии (5,74 г), в дихлорметане (120 мл) при комнатной температуре с последующим перемешиванием в течение одного часа. К реакционному раствору добавляли раствор тиосульфата натрия и отделяли органический слой. Органический слой промывали насыщенным раствором соли и сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и затем фильтрат концентрировали при пониженном давлении с получением остатка. Остаток подвергали хроматографии на колонке с силикагелем с получением указанного в заголовке соединения (4,49 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,27-2,39 (м, 1H), 2,70-2,78 (м, 1H), 3,30-3,52 (м, 2H), 3,53-3,62 (м, 3H), 3,74 (дд, J=8,4, 10,4 Гц, 1H), 4,40 (дд, J=6,0, 10,0 Гц, 1H), 4,48 (дд, J=8,4, 10,0 Гц, 1H), 4,61 (с, 2H), 7,27-7,39 (м, 5H).
(5) Синтез (3aS,6R,7aS)-6-бензилоксиметил-7a-(2-фторфенил)гексагидропирано[4,3-c]изоксазола
ТГФ (10 мл) и толуол (90 мл) добавляли к 2-бромфторбензолу (4,14 мл) в атмосфере азота и смесь охлаждали до -78°C. К раствору медленно добавляли н-бутиллитий (2,64 M раствор в гексане, 13,8 мл). После перемешивания при той же температуре в течение 10 минут последовательно добавляли по каплям комплекс трифторид бора-диэтиловый эфир (4,57 мл) и раствор, содержащий изоксазол, синтезированный на предшествующей стадии (4,49 г), в толуоле (10 мл). Смесь дополнительно перемешивали при той же температуре в течение четырех часов. К реакционному раствору добавляли насыщенный раствор хлорида аммония с последующим нагреванием до комнатной температуры. Затем добавляли этилацетат и воду и отделяли органический слой. Органический слой промывали насыщенным раствором соли и затем сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем и хроматографии на колонке с NH-силикагелем с получением указанного в заголовке соединения (5,73 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,81 (дд, J=3,2, 15,2 Гц, 1H), 2,28 (дд, J=12,4, 15,2 Гц, 1H), 3,07-3,16 (м, 1H), 3,40-3,85 (м, 6H), 4,08-4,19 (м, 1H), 4,58 (д, J=5,2 Гц, 2H), 5,94 (с, 1H), 7,00-7,08 (м, 1H), 7,10-7,17 (м, 1H), 7,22-7,39 (м, 6H), 7,83-7,92 (м, 1H).
(6) Синтез [(3R,4S,6R)-4-амино-6-бензилоксиметил-4-(2-фторфенил)тетрагидропиран-3-ил]метанола
Изоксазол, синтезированный на предшествующей стадии (5,73 г), растворяли в уксусной кислоте (70 мл). К раствору добавляли цинк (11 г) с последующим перемешиванием при комнатной температуре. Через 15 часов реакционный раствор фильтровали через целит и промывали метанолом. Фильтрат концентрировали при пониженном давлении. К остатку добавляли 2 н. раствор гидроксида натрия и хлороформ и отделяли органический слой. Органический слой сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (6,08 г). Соединение использовали для следующей реакции без дополнительной очистки.
ESI-MS; m/z 346 [M++H].
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,44 (дд, J=2,4, 13,6 Гц, 1H), 2,37 (дд, J=11,6, 13,6 Гц, 1H), 2,49-2,58 (м, 1H), 3,41-3,59 (м, 4H), 3,99-4,16 (м, 3H), 4,59 (д, J=10,0 Гц, 2H), 7,01-7,09 (м, 1H), 7,14-7,20 (м, 1H), 7,24-7,36 (м, 6H), 7,57-7,63 (м, 1H).
(7) Синтез 1-бензоил-3-[(2R,4S,5R)-2-бензилоксиметил-4-(2-фторфенил)-5-гидроксиметилтетрагидропиран-4-ил]тиомочевины
Амин, синтезированный на предшествующей стадии (6,08 г), растворяли в дихлорметане (60 мл). Бензоилизотиоцианат (2,58 мл) добавляли к раствору с последующим перемешиванием при комнатной температуре. Через 15 часов реакционный раствор концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем с получением указанного в заголовке соединения (7,83 г).
ESI-MS; m/z 509 [M++H], 531 [M++Na].
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,25-2,50 (м, 1H), 3,50-3,90 (м, 5H), 3,90-4,05 (м, 1H), 4,10-4,30 (м, 2H), 4,61 (д, J=2,4 Гц, 2H), 7,02-7,10 (м, 1H), 7,13-7,20 (м, 1H), 7,24-7,44 (м, 7H), 7,50-7,57 (м, 2H), 7,61-7,68 (м, 1H), 7,85-7,91 (м, 2H), 8,90 (с, 1H), 11,7 (с, 1H).
(8) Синтез N-[(4aS,7R,8aS)-7-бензилоксиметил-8a-(2-фторфенил)-4a,7,8,8a-тетрагидро-4H,5H-6-окса-3-тиа-1-азанафталин-2-ил]бензамида
Тиомочевину, синтезированную на предшествующей стадии (7,83 г), растворяли в метаноле (100 мл) и концентрированной хлористо-водородной кислоте (3 мл). Раствор кипятили с обратным холодильником при 95°C. Через четыре часа реакционному раствору давали охладиться и затем концентрировали при пониженном давлении. Хлороформ, 5 н. раствор гидроксида натрия и насыщенный раствор соли добавляли к остатку и отделяли органический слой. Органический слой сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (10 г). Соединение использовали для следующей реакции без дополнительной очистки.
ESI-MS; m/z 491 [M++H].
(9) Синтез (4aS,7R,8aS)-7-бензилоксиметил-8a-(2-фторфенил)-4a,7,8,8a-тетрагидро-4H,5H-6-окса-3-тиа-1-азанафталин-2-иламина
Соединение, полученное в примере получения 19-(8) (10 г), растворяли в метаноле (60 мл). Добавляли к раствору DBU (5 мл) и смесь кипятили с обратным холодильником при 95°C. Через пять часов реакционному раствору давали охладиться и концентрировали его при пониженном давлении. Этилацетат, насыщенный раствор бикарбоната натрия и насыщенный раствор соли добавляли к остатку и отделяли органический слой. Органический слой сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении. Остаток подвергали хроматографии на силикагеле с получением указанного в заголовке соединения (6,42 г).
ESI-MS; m/z 387 [M++H].
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,67 (дд, J=2,0, 12,8 Гц, 1H), 2,36-2,46 (м, 1H), 2,52-2,60 (м, 1H), 2,87 (дд, J=4,4, 12,4 Гц, 1H), 2,94-3,02 (м, 1H), 3,42-3,58 (м, 2H), 3,79-3,99 (м, 3H), 4,40-4,70 (м, 4H), 6,99-7,07 (м, 1H), 7,07-7,13 (м, 1H), 7,20-7,36 (м, 7H).
(10) Синтез [(4aS,7R,8aS)-2-амино-8a-(2-фторфенил)-4a,7,8,8a-тетрагидро-4H,5H-6-окса-3-тиа-1-азанафталин-7-ил]метанола
Концентрированную хлористо-водородную кислоту (35 мл) добавляли к соединению, полученному в примере получения 19-(9) (6,42 г), и смесь кипятили с обратным холодильником при 125°C. Через два часа реакционному раствору давали охладиться. Добавляли трет-бутилметиловый эфир и отделяли органический слой. Хлороформ и 5 н. раствор гидроксида натрия добавляли к водному слою и отделяли органический слой. Органические слои сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (4,16 г).
ESI-MS; m/z 297 [M++H].
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,64 (дд, J=2,0, 12,8 Гц, 1H), 2,34-2,44 (м, 1H), 2,53-2,62 (м, 1H), 2,85-2,93 (м, 1H), 2,93-3,03 (м, 1H), 3,54-3,69 (м, 2H), 3,78-3,91 (м, 3H), 4,55 (уш.с, 2H), 6,99-7,07 (м, 1H), 7,08-7,15 (м, 1H), 7,20-7,32 (м, 2H).
(11) Синтез трет-бутил [(4aS,7R,8aS)-8a-(2-фторфенил)-7-гидроксиметил-4a,7,8,8a-тетрагидро-4H,5H-6-окса-3-тиа-1-азанафталин-2-ил]карбамата
ТГФ (100 мл), метанол (50 мл) и триэтиламин (2,80 мл) добавляли к соединению, полученному в примере получения 19-(10) (3,8 г). К реакционной смеси добавляли ди-трет-бутилдикарбонат (3,7 г) с последующим перемешиванием. После перемешивания в течение ночи реакционный раствор концентрировали при пониженном давлении с получением остатка. Остаток подвергали колоночной хроматографии и осаждали, используя простой эфир, с получением указанного в заголовке соединения (5,35 г).
ESI-MS; m/z 397 [M++H].
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,53 (с, 9H), 1,63 (дд, J=2,0, 14,0 Гц, 1H), 1,98 (уш.с, 1H), 2,46-2,58 (м, 2H), 2,86 (дд, J=4,4, 13,2 Гц, 1H), 3,04-3,16 (м, 1H), 3,54-3,74 (м, 2H), 3,76-3,89 (м, 1H), 3,96 (д, J=7,6 Гц, 2H), 7,04-7,12 (м, 1H), 7,14-7,21 (м, 1H), 7,21-7,36 (м, 2H).
(12) Синтез трет-бутил [(4aS,7R,8aS)-8a-(2-фторфенил)-7-метоксиметил-4a,7,8,8a-тетрагидро-4H,5H-6-окса-3-тиа-1-азанафталин-2-ил]карбамата
Раствор соединения, полученного в примере получения 19-(11) (613 мг), в дихлорметане (15 мл) охлаждали льдом. К раствору добавляли триэтиламин (432 мкл) и метансульфонилхлорид (144 мкл). Реакционный раствор перемешивали при той же температуре в течение одного часа. Затем дихлорметан и насыщенный раствор бикарбоната натрия добавляли к реакционному раствору и отделяли органический слой. Органический слой сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и затем фильтрат концентрировали при пониженном давлении с получением мезила. Мезил растворяли в метаноле (10 мл) с последующим охлаждением льдом. Метоксид натрия (28% раствор в метаноле, 1,7 мл) добавляли к раствору и смесь перемешивали при комнатной температуре. Через один час реакционный раствор нагревали до 70°C. Через примерно три часа добавляли дополнительно метоксид натрия (28% раствор в метаноле, 5 мл) и смесь перемешивали еще в течение четырех часов. Реакционному раствору давали охладиться. Затем добавляли хлороформ и насыщенный раствор бикарбоната натрия и отделяли органический слой. Органический слой промывали насыщенным раствором соли. Органический слой сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и затем фильтрат концентрировали при пониженном давлении с получением остатка. Остаток подвергали хроматографии на силикагеле с получением указанного в заголовке соединения (262 мг).
ESI-MS; m/z 41 [M++H].
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,53 (с, 9H), 1,67 (дд, J=2,0, 13,6 Гц, 1H), 1,98 (уш.с, 1H), 2,46-2,56 (м, 2H), 2,81-2,90 (м, 1H), 3,07-3,16 (м, 1H), 3,40 (с, 3H), 3,40-3,52 (м, 2H), 3,86-4,01 (м, 3H), 7,04-7,12 (м, 1H), 7,14-7,21 (м, 1H), 7,25-7,36 (м, 2H).
(13) Синтез трет-бутил [(4aS,7R,8aS)-8a-(2-фтор-5-нитрофенил)-7-метоксиметил-4a,7,8,8a-тетрагидро-4H,5H-6-окса-3-тиа-1-азанафталин-2-ил]карбамата
Соединение, полученное в примере получения 19-(12) (262 мг), растворяли в дихлорметане (3 мл) с последующим охлаждением льдом. К раствору добавляли ТФУК (1 мл) с последующим перемешиванием при комнатной температуре. Через четыре часа реакционный раствор концентрировали при пониженном давлении с получением остатка. К остатку добавляли ТФУК (1 мл) с последующим охлаждением льдом. К раствору добавляли концентрированную серную кислоту (0,5 мл). Затем добавляли дымящую азотную кислоту (37 мкл) и смесь перемешивали в течение одного часа. Реакционный раствор вливали в воду. Осторожно добавляли хлороформ и 5 н. раствор гидроксида натрия и отделяли органический слой. Органический слой сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении с получением остатка. Остаток (239 мг) растворяли в ТГФ (3 мл) и метаноле (1 мл), а затем добавляли триэтиламин (200 мкл). Ди-трет-бутилдикарбонат (215 мг) растворяли в ТГФ (2 мл) и раствор добавляли к указанному выше раствору. Через 18 часов реакционный раствор концентрировали при пониженном давлении с получением остатка. Остаток подвергали колоночной хроматографии с получением указанного в заголовке соединения (256 мг).
ESI-MS; m/z 456 [M++H].
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,54 (с, 9H), 1,66-1,73 (м, 1H), 2,26-2,42 (м, 1H), 2,56 (дд, J=2,8, 13,2 Гц, 1H), 2,74-2,84 (м, 1H), 3,00-3,12 (м, 1H), 3,39 (с, 3H), 3,36-3,50 (м, 2H), 3,72-4,02 (м, 3H), 7,18-7,32 (м, 1H), 8,12-8,24 (м, 2H).
(14) Синтез трет-бутил [(4aS,7R,8aS)-8a-(5-амино-2-фторфенил)-7-метоксиметил-4a,7,8,8a-тетрагидро-4H,5H-6-окса-3-тиа-1-азанафталин-2-ил]карбамата
Соединение, полученное в примере получения 19-(13) (255 мг), растворяли в этаноле (9 мл). К раствору добавляли насыщенный раствор хлорида аммония (0,9 мл) и железный порошок (440 мг) и смесь нагревали при 90°C в течение 40 минут. Реакционному раствору давали охладиться и фильтровали его через целит. К фильтрату добавляли этилацетат и раствор бикарбоната натрия и отделяли органический слой. Органический слой сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении с получением остатка. Остаток подвергали хроматографии на колонке с силикагелем и затем осаждали, используя трет-бутилметиловый эфир и гексан, с получением указанного в заголовке соединения (151 мг).
ESI-MS; m/z 426 [M++H].
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,53 (с, 9H), 1,63 (дд, J=2,0, 14,0 Гц, 1H), 2,44-2,55 (м, 2H), 2,92 (дд, J=4,0, 13,2 Гц, 1H), 3,04-3,13 (м, 1H), 3,39 (с, 3H), 3,38-3,49 (м, 2H), 3,64 (уш.с, 2H), 3,86-4,00 (м, 3H), 6,50-6,60 (м, 2H), 6,87 (дд, J=8,4, 12,0 Гц, 1H).
Пример получения 20
Синтез трет-бутил [(4aS,7R,8aS)-8a-(5-амино-2-фторфенил)-7-фторметил-4a,7,8,8a-тетрагидро-4H,5H-6-окса-3-тиа-1-азанафталин-2-ил]карбамата
[Формула 42]
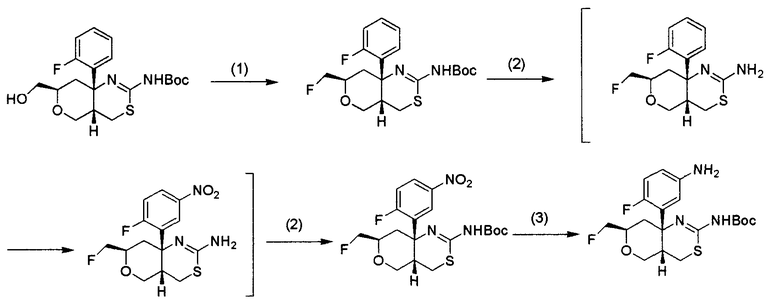
(1) Синтез трет-бутил [(4aS,7R,8aS)-7-фторметил-8a-(2-фторфенил)-4a,7,8,8a-тетрагидро-4H,5H-6-окса-3-тиа-1-азанафталин-2-ил]карбамата
Перфторбутансульфонилфторид (990 мкл), тригидрофторид триэтиламина (842 мкл) и триэтиламин (2,2 мл) последовательно добавляли к смеси соединения, полученного в примере получения 19-(11) (1 г), в ацетонитриле (15 мл) и ТГФ (4 мл) с последующим перемешиванием при комнатной температуре. Через примерно 22 часа добавляли к реакционному раствору насыщенный раствор бикарбоната натрия. Органический слой экстрагировали хлороформом и промывали насыщенным раствором соли. Органический слой сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и затем фильтрат концентрировали при пониженном давлении с получением остатка. Остаток подвергали хроматографии на силикагеле с получением указанного в заголовке соединения (307 мг).
ESI-MS; m/z 399 [M++H].
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,53 (с, 9H), 1,69 (дд, J=2,0, 13,6 Гц, 1H), 2,48-2,60 (м, 2H), 2,85 (дд, J=4,4, 13,2 Гц, 1H), 3,04-3,18 (м, 1H), 3,85-4,08 (м, 3H), 4,32-4,44 (м, 1H), 4,45-4,56 (м, 1H), 7,05-7,14 (м, 1H), 7,14-7,21 (м, 1H), 7,24-7,37 (м, 2H).
(2) Синтез трет-бутил [(4aS,7R,8aS)-7-фторметил-8a-(2-фтор-5-нитрофенил)-4a,7,8,8a-тетрагидро-4H,5H-6-окса-3-тиа-1-азанафталин-2-ил]карбамата
Соединение, полученное в примере получения 20-(1) (307 мг), растворяли в дихлорметане (3 мл) с последующим охлаждением льдом. К раствору добавляли ТФУК (1 мл) с последующим перемешиванием при комнатной температуре. Через четыре часа реакционный раствор концентрировали при пониженном давлении с получением остатка. К полученному остатку добавляли ТФУК (1 мл) с последующим охлаждением льдом. К раствору добавляли концентрированную серную кислоту (0,5 мл). Затем добавляли дымящую азотную кислоту (42 мкл) и смесь перемешивали в течение одного часа. Реакционный раствор вливали в воду. Осторожно добавляли хлороформ и 5 н. раствор гидроксида натрия и отделяли органический слой. Органический слой сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении с получением остатка (260 мг). Остаток (260 мг) растворяли в ТГФ (3 мл) и метаноле (1 мл), а затем добавляли триэтиламин (200 мкл). Ди-трет-бутилдикарбонат (285 мг) растворяли в ТГФ (2 мл) и раствор добавляли к раствору, полученному выше. Через 18 часов добавляли к реакционному раствору этилацетат и насыщенный раствор бикарбоната натрия и отделяли органический слой. Органический слой сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении с получением остатка. Остаток подвергали колоночной хроматографии с получением указанного в заголовке соединения (292 мг).
ESI-MS; m/z 444 [M++H].
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,54 (с, 9H), 1,66-1,74 (м, 1H), 2,27-2,48 (м, 1H), 2,57 (дд, J=2,8, 13,6 Гц, 1H), 2,72-2,88 (м, 1H), 2,98-3,16 (м, 1H), 3,76-4,04 (м, 3H), 4,30-4,56 (м, 2H), 4,45-4,56 (м, 1H), 7,18-7,29 (м, 1H), 8,12-8,25 (м, 2H).
(3) Синтез трет-бутил [(4aS,7R,8aS)-8a-(5-амино-2-фторфенил)-7-фторметил-4a,7,8,8a-тетрагидро-4H,5H-6-окса-3-тиа-1-азанафталин-2-ил]карбамата
Соединение, полученное в примере получения 20-(2) (290 мг), растворяли в этаноле (10 мл). К раствору добавляли насыщенный раствор хлорида аммония (1 мл) и железный порошок (490 мг) и смесь нагревали при 90°C в течение 40 минут. Реакционному раствору давали охладиться и фильтровали его через целит. К фильтрату добавляли этилацетат и раствор бикарбоната натрия и отделяли органический слой. Органический слой сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении с получением остатка. Остаток подвергали колоночной хроматографии с получением указанного в заголовке соединения (186 мг).
ESI-MS; m/z 414 [M++H].
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,53 (с, 9H), 1,58-1,72 (м, 1H), 2,47-2,60 (м, 2H), 2,93 (дд, J=4,0, 13,2 Гц, 1H), 3,03-3,12 (м, 1H), 3,65 (уш.с, 2H), 3,87-4,03 (м, 3H), 4,33-4,43 (м, 1H), 4,44-4,54 (м, 1H), 6,48-6,60 (м, 2H), 6,83-6,92 (м, 1H).
Пример получения 21
Синтез трет-бутил (-)-[(4aS*,5R*,7aS*)-7a-(5-амино-2-фторфенил)-5-этил-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил]карбамата
[Формула 43]

(1) Синтез (1-этилаллилокси)ацетальдегидоксима
Раствор 1-пентен-3-ола (15,0 мл) в N-метил-2-пирролидоне (292 мл) охлаждали до 0°C в атмосфере азота. Гидрид натрия (60%, 6,42 г) и диэтилацеталь бромацетальдегида (31,6 г) добавляли к реакционному раствору при той же температуре и смесь перемешивали при 80°C в течение 30 минут. К реакционному раствору при 0°C добавляли насыщенный раствор хлорида аммония с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным бикарбонатом натрия и насыщенным водным раствором хлорида натрия и затем сушили над безводным сульфатом магния. Нерастворимое вещество отделяли фильтрованием и фильтрат концентрировали при пониженном давлении. Остаток растворяли в гептане. Раствор фильтровали через силикагель, используя смесь 30% этилацетат/гептан, и концентрировали при пониженном давлении. К остатку добавляли муравьиную кислоту (100 мл), гидрохлорид гидроксиламина (15,2 г) и ацетат натрия (24,0 г) и смесь перемешивали при комнатной температуре в течение двух суток. К реакционному раствору добавляли этилацетат и насыщенный раствор хлорида натрия и отделяли органический слой. Органический слой промывали насыщенным водным раствором хлорида натрия и затем сушили над безводным сульфатом магния. Нерастворимое вещество отделяли фильтрованием и фильтрат концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (4,30 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 0,88-0,92 (м, 3H), 1,55-1,63 (м, 2H) 3,59-4,36 (м, 3H), 5,19-5,24 (м, 2H), 5,61-5,68 (м, 1H), 6,90-6,92 (м, 0,5H), 7,48-7,50 (м, 0,5H).
(2) Синтез (3aR*,4R*)-4-этил-3a,4-дигидро-3H,6H-фуро[3,4-c]изоксазола
5% Раствор гипохлорита натрия (53,6 г) добавляли к раствору, содержащему соединение, полученное в примере получения 21-(1) (4,30 г), в дихлорметане (95,7 мл) при 0°C и смесь перемешивали при 0°C в течение 30 минут. Раствор бисульфита натрия добавляли к реакционному раствору при той же температуре. Органический слой отделяли и сушили над безводным сульфатом магния. Нерастворимое вещество отделяли фильтрованием и фильтрат концентрировали. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (2,02 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 0,92 (т, J=7,2 Гц, 3H), 1,56-1,83 (м, 2H), 3,74-3,78 (м, 2H), 3,97-4,02 (м, 1H), 4,44-4,57 (м, 3H).
(3) Синтез (3aR*,4R*,6aS*)-4-этил-6a-(2-фторфенил)тетрагидрофуро[3,4-c]изоксазола
Раствор н-бутиллития в гексане (2,60 M; 12,2 мл) добавляли по каплям к раствору, содержащему 2-бромфторбензол (3,62 мл) в смеси тетрагидрофуран/толуол (5,80 мл/58,0 мл), в атмосфере азота при -78°C. Реакционный раствор перемешивали при той же температуре в течение 10 минут. К реакционному раствору последовательно добавляли по каплям комплекс трифторид бора-диэтиловый эфир (3,92 мл) и раствор, содержащий соединение, полученное в примере получения 21-(2) (2,02 г), в толуоле (20 мл) при той же температуре. После перемешивания при той же температуре в течение 40 минут к реакционному раствору добавляли водный раствор хлорида аммония с последующим нагреванием до комнатной температуры. К реакционному раствору добавляли воду и этилацетат и отделяли органический слой. Органический слой промывали насыщенным раствором хлорида натрия. Органический слой сушили над безводным сульфатом магния и нерастворимое вещество отделяли фильтрованием. Фильтрат концентрировали и остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (3,39 г).
ESI-MS; m/z 238 [M++H].
(4) Синтез 1-бензоил-3-[(3S*,4R*,5R*)-5-этил-3-(2-фторфенил)-4-гидроксиметилтетрагидрофуран-3-ил]тиомочевины
Цинковый порошок (9,35 г) добавляли к раствору, содержащему соединение, полученное в примере получения 21-(3) (3,39 г), в уксусной кислоте (59,8 мл) при комнатной температуре. Реакционный раствор перемешивали при комнатной температуре в течение 18 часов. Нерастворимое вещество отделяли фильтрованием через целит и фильтрат концентрировали. К остатку добавляли этилацетат и раствор бикарбоната натрия и отделяли органический слой. Органический слой промывали насыщенным водным раствором хлорида натрия и сушили над безводным сульфатом магния. Нерастворимое вещество отделяли фильтрованием и фильтрат концентрировали при пониженном давлении. Бензоилизотиоцианат (2,12 мл) добавляли к раствору, содержащему остаток, в дихлорметане (43,2 мл) и смесь перемешивали при комнатной температуре в течение 10 минут. Реакционный раствор концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (3,83 г).
ESI-MS; m/z 402 [M++H].
(5) Синтез N-[(4aS*,5R*,7aS*)-5-этил-7a-(2-фторфенил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил]бензамида
Пиридин (2,15 мл) и трифторметансульфоновый ангидрид (3,20 мл) добавляли к раствору соединения, полученного в примере получения 21-(4) (4,11 г), в дихлорметане (18,0 мл) при 0°C и смесь перемешивали при той же температуре в течение 10 минут. Насыщенный раствор бикарбоната натрия добавляли к реакционному раствору с последующим экстрагированием этилацетатом. Органический слой сушили над безводным сульфатом магния и нерастворимое вещество отделяли фильтрованием. Фильтрат концентрировали при пониженном давлении и остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (3,53 г).
1H-ЯМР(400МГц, CDC13) δ (м.д.): 1,02(т, J=8,0 Гц, 3H), 1,57-1,61(м, 2H), 2,72-2,74(м, 1H), 2,95-2,97(м, 1H), 3,83-3,99(м, 2H), 4,40-4,43(м, 1H), 4,60-4,63(м, 1H), 7,02-7,04(м, 1H), 7,16-7,19(м, 1H), 7,26-7,28 (м, 1H), 7,50-7,54(м, 2H), 7,62-7,64(м, 1H), 7,73-7,74(м, 1H), 7,85-7,88(м, 2H), 8,87(уш., 1H).
(6) Синтез (4aS*,5R*,7aS*)-5-этил-7a-(2-фторфенил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-иламина
Раствор соединения, полученного в примере получения 21-(5) (3,53 г), и метоксида натрия (28% раствор в метаноле; 3,67 мл) в метаноле (23,4 мл) кипятили с обратным холодильником в течение 2,5 часов. После охлаждения реакционного раствора до комнатной температуры растворитель выпаривали при пониженном давлении. Остаток очищали хроматографией на силикагеле с получением указанного в заголовке соединения (1,21 г).
ESI-MS; m/z 281 [M++H].
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,06(т, J=7,6 Гц, 3H), 1,62-1,73(м, 2H), 2,62-2,75(м, 2Н), 3,05-3,09(м, 1H), 3,79-3,81(м, 1H), 4,10-4,19(м, 1H), 4,57-4,59(м, 1H), 7,01-7,24(м, 3H), 7,40-7,44(м, 1H).
(7) Синтез (4aS*,5R*,7aS*)-5-этил-7a-(2-фтор-5-нитрофенил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-иламина
Дымящую азотную кислоту (215 мкл) добавляли по каплям к раствору соединения, полученного в примере получения 21-(6) (1,21 г), в концентрированной серной кислоте (21,6 мл) при охлаждении льдом. Реакционный раствор перемешивали при той же температуре в течение 30 минут и затем вливали в ледяную воду. Реакционную смесь нейтрализовали 5 н. раствором гидроксида натрия. Смесь дважды экстрагировали этилацетатом. Органические слои сушили над безводным сульфатом магния и нерастворимое вещество отделяли фильтрованием. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (1,41 г).
ESI-MS; m/z 326 [M++H].
(8) Синтез трет-бутил [(4aS*,5R*,7aS*)-5-этил-7a-(2-фтор-5-нитрофенил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил]карбамата
Соединение, полученное в примере получения 21-(7) (1,34 г), растворяли в дихлорметане (20,4 мл). Триэтиламин (2,41 мл) и ди-трет-бутилдикарбонат (1,88 г) добавляли к раствору и смесь перемешивали при комнатной температуре в течение 10 минут. Реакционный раствор концентрировали при пониженном давлении и остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (1,41 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,06 (т, J=7,6 Гц, 3H), 1,52 (с, 9H), 1,52-1,65 (м, 2H), 2,66-2,70 (м, 2H), 2,91-2,93 (м, 1H), 3,78-3,79 (м, 1H), 4,25-4,26 (м, 1H), 4,46-4,48 (м, 1H), 7,20-7,22 (м, 1H), 8,21-8,29 (м, 2H).
(9) Синтез трет-бутил [(4aS*,5R*,7aS*)-7a-(5-амино-2-фторфенил)-5-этил-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил]карбамата
Насыщенный раствор хлорида аммония (3,1 мл) и железный порошок (1,48 г) добавляли к раствору соединения, полученного в примере получения 21-(8) (1,41 г), в этаноле (33,1 мл). Реакционный раствор кипятили с обратным холодильником в течение 30 минут и затем охлаждали до комнатной температуры. Реакционный раствор разбавляли этилацетатом и нерастворимое вещество отделяли фильтрованием через целит. К фильтрату добавляли этилацетат и воду и отделяли органический слой. Органический слой сушили над безводным сульфатом магния и нерастворимое вещество отделяли фильтрованием. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (920 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,06 (т, J=7,6 Гц, 3H), 1,46-1,71 (м, 2H), 1,49 (с, 9H), 2,63-2,67 (м, 1H), 2,84-2,85 (м, 1H), 3,07-3,09 (м, 1H), 3,62 (уш., 2H), 3,81-3,83 (м, 1H), 4,27-4,28 (м, 1H), 4,49-4,51 (м, 1H), 6,55-6,63 (м, 2H), 6,84-6,89 (м, 1H).
(10) Синтез трет-бутил (-)-[(4aS*,5R*,7aS*)-7a-(5-амино-2-фторфенил)-5-этил-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил]карбамата
Соединение, полученное в примере получения 21-(9) (50 мг), оптически разделяли, используя CHIRALPAK™ OJ-H производства Daicel Chemical Industries, Ltd. (2 см × 25 см, подвижная фаза: гексан:этанол = 1:1, скорость потока: 10 мл/мин), и компоненты, имеющие время удерживания от 19 до 25 минут, собирали. Данную операцию повторяли с получением указанного в заголовке соединения (365 мг; >99% ee) из 920 мг рацемата.
Пример получения 22
Синтез трет-бутил (-)-[(4aS*,5R*,7aS*)-7a-(5-амино-2-фторфенил)-5-метил-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил]карбамата
[Формула 44]

(1) Синтез (1-метилаллилокси)ацетальдегидоксима
Раствор 3-бутен-2-ола (5,00 г) в N-метил-2-пирролидоне (70,0 мл) охлаждали до 0°C в атмосфере азота. Гидрид натрия (60%, 3,33 г) и диэтилацеталь бромацетальдегида (15,0 г) добавляли к реакционному раствору при той же температуре и смесь перемешивали при 60°C в течение 30 минут. К реакционному раствору добавляли насыщенный раствор хлорида аммония при 0°C с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным раствором бикарбоната натрия и насыщенным водным раствором хлорида натрия и затем сушили над безводным сульфатом магния. Нерастворимое вещество отделяли фильтрованием и фильтрат концентрировали при пониженном давлении. Остаток растворяли в гептане. Раствор фильтровали через силикагель и концентрировали при пониженном давлении. К остатку добавляли муравьиную кислоту (30,0 мл) и воду (10,0 мл) и смесь перемешивали при комнатной температуре в течение 30 минут. Гидрохлорид гидроксиламина (5,78 г) и ацетат натрия (11,4 г) добавляли к реакционному раствору при той же температуре и смесь перемешивали при комнатной температуре в течение пяти суток. К реакционному раствору добавляли этилацетат и насыщенный раствор хлорида натрия и отделяли органический слой. Органический слой промывали насыщенным водным раствором хлорида натрия и затем сушили над безводным сульфатом магния. Нерастворимое вещество отделяли фильтрованием и фильтрат концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (2,29 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,26-1,29 (м, 3H), 3,85-4,36 (м, 3H), 5,17-5,24 (м, 2H), 5,68-5,77 (м, 1H), 6,90 (т, J=3,6 Гц, 0,5H), 7,49 (т, J=6,0 Гц, 0,5H).
(2) Синтез (3aR*,4R*)-4-метил-3a,4-дигидро-3H,6H-фуро[3,4-c]изоксазола
5% Раствор гипохлорита натрия (87,1 г) добавляли по каплям к раствору, содержащему соединение, полученное в примере получения 22-(1) (7,55 г), в дихлорметане (168 мл) при 0°C и смесь перемешивали при 0°C в течение 30 минут. Раствор бисульфита натрия добавляли к реакционному раствору при той же температуре. Органический слой отделяли и сушили над безводным сульфатом магния. Нерастворимое вещество отделяли фильтрованием и фильтрат концентрировали. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (3,95 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,37 (д, J=5,6 Гц, 3H), 3,75-3,80 (м, 2H), 3,90-4,00 (м, 1H), 4,40-4,57 (м, 3H).
(3) Синтез (3aR*,4R*,6aS*)-6a-(2-фторфенил)-4-метилтетрагидрофуро[3,4-c]изоксазола
Раствор н-бутиллития в гексане (2,60 M; 6,05 мл) добавляли по каплям к раствору, содержащему 2-бромфторбензол (1,79 мл) в смеси тетрагидрофуран/толуол (2,88 мл/28,8 мл), в атмосфере азота при -78°C. Реакционный раствор перемешивали при той же температуре в течение 10 минут. Комплекс трифторид бора-диэтиловый эфир (1,94 мл) и раствор, содержащий соединение, полученное в примере получения 22-(2) (1,00 г), в толуоле (10 мл) последовательно добавляли по каплям к реакционному раствору при той же температуре. После перемешивания при той же температуре в течение одного часа к реакционному раствору добавляли водный раствор хлорида аммония и реакционный раствор нагревали до комнатной температуры. К реакционному раствору добавляли воду и этилацетат и отделяли органический слой. Органический слой промывали насыщенным раствором хлорида натрия. Органический слой сушили над безводным сульфатом магния и нерастворимое вещество отделяли фильтрованием. Фильтрат концентрировали и остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (1,60 г).
ESI-MS; m/z 224 [M++H].
(4) Синтез [(2R*,3R*,4S*)-4-амино-4-(2-фторфенил)-2-метилтетрагидрофуран-3-ил]метанола
Цинковый порошок (4,69 г) добавляли к раствору, содержащему соединение, полученное в примере получения 22-(3) (1,60 г), в уксусной кислоте (30,0 мл) при комнатной температуре. Реакционный раствор перемешивали при комнатной температуре в течение 17 часов. Нерастворимое вещество отделяли фильтрованием через целит и фильтрат концентрировали. К остатку добавляли этилацетат и раствор бикарбоната натрия и отделяли органический слой. Органический слой промывали насыщенным водным раствором хлорида натрия и сушили над безводным сульфатом магния. Нерастворимое вещество отделяли фильтрованием и фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (1,57 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,32 (д, J=6,4 Гц, 1H), 2,20-2,22 (м, 1H), 3,76-3,86 (м, 2H), 4,04-4,10 (м, 1H), 4,31-4,38 (м, 2H), 7,06-7,26 (м, 3H), 7,45-7,49 (м, 1H).
(5) Синтез 1-бензоил-3-[(3S*,4R*,5R*)-3-(2-фторфенил)-4-гидроксиметил)-5-метилтетрагидрофуран-3-ил]тиомочевины
Бензоилизотиоцианат (1,25 г) добавляли к раствору, содержащему соединение, полученное в примере получения 22-(4) (1,57 г), в дихлорметане (21,0 мл) и смесь перемешивали при комнатной температуре в течение 10 минут. Реакционный раствор концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (1,43 г).
ESI-MS; m/z 389 [M++H].
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,37 (д, J=6,0 Гц, 1H), 2,61-2,62 (м, 2H), 2,85-2,86 (м, 1H), 3,95-4,07 (м, 2H), 4,41-4,44 (м, 1H), 4,68 (д, J=10 Гц, 1H), 7,00-7,30 (м, 3H), 7,52 (т, J=8,0 Гц, 2H), 7,62-7,72 (м, 2H), 7,85-7,87 (м, 2H), 8,88 (уш., 1H).
(6) Синтез N-[(4aS*,5R*,7aS*)-7a-(2-фторфенил)-5-метил-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил]бензамида
Трифторметансульфоновый ангидрид (1,55 мл) добавляли к раствору соединения, полученного в примере получения 22-(5) (1,43 г), в пиридине (7,0 мл) при 0°C и смесь перемешивали при той же температуре в течение 10 минут. К реакционному раствору добавляли насыщенный раствор бикарбоната натрия с последующим экстрагированием этилацетатом. Органический слой сушили над безводным сульфатом магния и нерастворимое вещество отделяли фильтрованием. Фильтрат концентрировали при пониженном давлении и остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (780 мг).
ESI-MS; m/z 371 [M++H].
(7) Синтез (4aS*,5R*,7aS*)-7a-(2-фторфенил)-5-метил-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-иламина
Раствор соединения, полученного в примере получения 22-(6) (3,02 г), и метоксида натрия (28% раствор в метаноле; 3,14 мл) в метаноле (20 мл) кипятили с обратным холодильником в течение 2,5 часов. После охлаждения реакционного раствора до комнатной температуры выпаривали при пониженном давлении растворитель. Остаток очищали хроматографией на силикагеле с получением указанного в заголовке соединения (980 мг).
ESI-MS; m/z 267 [M++H].
(8) Синтез (4aS*,5R*,7aS*)-7a-(2-фтор-5-нитрофенил)-5-метил-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-иламина
Дымящую азотную кислоту (199 мкл) добавляли по каплям к раствору соединения, полученного в примере получения 22-(7) (980 мг), в концентрированной серной кислоте (36,6 мл) при охлаждении льдом. Реакционный раствор перемешивали при той же температуре в течение 30 минут и затем вливали в ледяную воду. Реакционную смесь нейтрализовали 5 н. раствором гидроксида натрия. Смесь дважды экстрагировали этилацетатом. Органические слои сушили над безводным сульфатом магния и нерастворимое вещество отделяли фильтрованием. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (1,02 г).
ESI-MS; m/z 312 [M++H].
(9) Синтез трет-бутил [(4aS*,5R*,7aS*)-7a-(2-фтор-5-нитрофенил)-5-метил-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил]карбамата
Соединение, полученное в примере получения 22-(8) (1,02 г), растворяли в дихлорметане (15,5 мл). Триэтиламин (1,83 мл) и ди-трет-бутилдикарбонат (1,43 г) добавляли к раствору и смесь перемешивали при комнатной температуре в течение 14 часов. Реакционный раствор концентрировали при пониженном давлении и остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (1,68 г).
ESI-MS; m/z 412 [M++H].
(10) Синтез трет-бутил [(4aS*,5R*,7aS*)-7a-(5-амино-2-фторфенил)-5-метил-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил]карбамата
Насыщенный раствор хлорида аммония (2,7 мл) и железный порошок (1,25 г) добавляли к раствору соединения, полученного в примере получения 22-(9) (1,15 г), в этаноле (27 мл). Реакционный раствор кипятили с обратным холодильником в течение 30 минут и затем охлаждали до комнатной температуры. Реакционный раствор разбавляли этилацетатом и нерастворимое вещество отделяли фильтрованием через целит. К фильтрату добавляли этилацетат и воду и отделяли органический слой. Органический слой сушили над безводным сульфатом магния и нерастворимое вещество отделяли фильтрованием. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (1,01 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,39-1,50 (м, 3H), 1,50 (с, 9H), 2,64-2,74 (м, 2H), 3,08-3,12 (м, 1H), 3,61 (уш., 2H), 3,82-3,84 (м, 1H), 4,43-4,56 (м, 2H), 6,58-6,60 (м, 2H), 6,84-6,89 (м, 1H).
(11) Синтез трет-бутил (-)-[(4aS*,5R*,7aS*)-7a-(5-амино-2-фторфенил)-5-метил-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил]карбамата
Соединение, полученное в примере получения 22-(10) (50 мг), оптически разделяли, используя CHIRALPAK™ OJ-H производства Daicel Chemical Industries, Ltd. (2 см × 25 см, подвижная фаза: гексан:этанол = 9:1, скорость потока: 10 мл/мин), и компоненты, имеющие время удерживания от 19 до 30 минут, собирали. Данную операцию повторяли с получением указанного в заголовке соединения (363 мг; >99% ee) из 1,01 г рацемата.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,38 (д, J=6,0 Гц, 3H), 2,64-2,68 (м, 1H), 2,73-2,75 (м, 1Н), 3,08-3,12 (м, 1H), 3,62 (уш., 2H), 3,82-3,85 (м, 1H), 4,42-4,45 (м, 1H), 4,54-4,56 (м, 1H), 6,55-6,62 (м, 2H), 6,84-6,89 (м, 1H).
Пример получения 23
[Формула 45]

(1) Синтез 4-(трет-бутилдиметилсиланилокси)бутан-1-ола
Имидазол (6,77 г) добавляли к раствору, содержащему 1,4-бутандиол (58,8 мл), в ДМФА (60,0 мл) при комнатной температуре. Добавляли по каплям раствор трет-бутилдиметилсилилхлорида (10,0 г) в дихлорметане (5,0 мл) и смесь перемешивали при той же температуре в течение трех часов. К реакционному раствору добавляли диэтиловый эфир и воду. Органический слой отделяли, промывали водой и насыщенным раствором хлорида натрия и затем сушили над безводным сульфатом магния. Нерастворимое вещество отделяли фильтрованием и фильтрат концентрировали с получением указанного в заголовке соединения (13,4 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 0,07 (с, 6H), 0,90 (с, 9H), 1,62-1,68 (м, 4H), 3,64-3,69 (м, 4H).
(2) Синтез 4-(трет-бутилдиметилсиланилокси)бутиральдегида
Диметилсульфоксид (23,1 мл), N,N-диизопропилэтиламин (45,4 мл) и комплекс триоксид серы-пиридин (36,3 г) добавляли к раствору, содержащему соединение, полученное в примере получения 23-(1) (13,3 г), в дихлорметане (162 мл) при 0°C и смесь перемешивали при той же температуре в течение 20 минут. Насыщенный раствор бикарбоната натрия добавляли к реакционному раствору при той же температуре. Органический слой отделяли и сушили над безводным сульфатом магния. Нерастворимое вещество отделяли фильтрованием и фильтрат концентрировали. К остатку добавляли диэтиловый эфир и 2 н. раствор хлористо-водородной кислоты. Органический слой отделяли, промывали насыщенным раствором бикарбоната натрия и затем сушили над безводным сульфатом магния. Нерастворимое вещество отделяли фильтрованием через силикагель и фильтрат концентрировали с получением указанного в заголовке соединения (12,0 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 0,05 (с, 6H), 0,89 (с, 9H), 1,83-1,89 (м, 2H), 2,49-2,62 (м, 2H), 3,64-3,67 (м, 2H), 9,79 (т, J=1,6 Гц, 1H).
(3) Синтез 6-(трет-бутилдиметилсиланилокси)гекс-1-ен-3-ола
Раствор винилхлорида магния в ТГФ (1,38 M, 51,6 мл) добавляли по каплям к раствору, содержащему соединение, полученное в примере получения 23-(2) (12,0 г), в ТГФ (138 мл) при -78°C и смесь перемешивали при 0°C в течение 10 минут. К реакционному раствору добавляли насыщенный раствор хлорида аммония при той же температуре с последующим добавлением этилацетата и 2 н. раствора хлористо-водородной кислоты. Органический слой отделяли, промывали насыщенным раствором бикарбоната натрия и затем сушили над безводным сульфатом магния. Нерастворимое вещество отделяли фильтрованием через силикагель и фильтрат концентрировали с получением указанного в заголовке соединения (13,5 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 0,07 (с, 6H), 0,90 (с, 9H), 1,63-1,66 (м, 4H), 3,65-3,68 (м, 2H), 4,15-4,17 (м, 1H), 5,09-5,11 (м, 1H), 5,22-5,27 (м, 1H), 5,85-5,87 (м, 1H).
(4) Синтез 4-метоксигекс-5-ен-1-ола
Метилиодид (10,9 мл) и гидрид натрия (60%, 2,34 г) добавляли к раствору, содержащему соединение, полученное в примере получения 23-(3) (13,5 г), в N-метил-2-пирролидоне (146 мл) при 0°C и смесь перемешивали при 60°C в течение 30 минут. Насыщенный водный раствор хлорида аммония и диэтиловый эфир добавляли к реакционному раствору при 0°C. Органический слой отделяли, промывали насыщенным раствором бикарбоната натрия и насыщенным раствором хлорида натрия и затем сушили над безводным сульфатом магния. Нерастворимое вещество отделяли фильтрованием через силикагель и фильтрат концентрировали с получением остатка. Ацетилхлорид (15 мл) добавляли по каплям к метанолу (135 мл) при 0°C и смесь добавляли к полученному выше остатку. Реакционный раствор концентрировали и полученный остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (5,52 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,64-1,68 (м, 4H), 3,29 (с, 3H), 3,55-3,65 (м, 3H), 5,18-5,23 (м, 2H), 5,63-5,72 (м, 1H).
(5) Синтез 4-метоксигекс-5-еналя
Диметилсульфоксид (12,0 мл), N,N-диизопропилэтиламин (29,5 мл) и комплекс триоксид серы-пиридин (20,2 г) добавляли к раствору, содержащему соединение, полученное в примере получения 23-(4) (5,52 г), в дихлорметане (84,8 мл) при 0°C и смесь перемешивали при той же температуре в течение 20 минут. Насыщенный раствор бикарбоната натрия добавляли к реакционному раствору при той же температуре. Органический слой отделяли и сушили над безводным сульфатом магния. Нерастворимое вещество отделяли фильтрованием и фильтрат концентрировали. К остатку добавляли диэтиловый эфир и 2 н. раствор хлористо-водородной кислоты. Органический слой отделяли, промывали насыщенным раствором бикарбоната натрия и затем сушили над безводным сульфатом магния. Нерастворимое вещество отделяли фильтрованием и фильтрат концентрировали с получением указанного в заголовке соединения (3,85 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,84-1,91 (м, 2H), 2,50-2,53 (м, 2H), 3,26 (с, 3H), 3,54-3,57 (м, 1H), 5,20-5,25 (м, 2H), 5,61-5,70 (м, 1H), 9,76 (т, J=1,6 Гц, 1H).
(6) Синтез 4-метоксигекс-5-енальоксима
Ацетат натрия (4,92 г) и гидрохлорид гидроксиламина (3,13 г) добавляли к раствору, содержащему соединение, полученное в примере получения 23-(5) (3,85 г), в метаноле (60,0 мл) при комнатной температуре и смесь перемешивали при той же температуре в течение трех часов. Этилацетат и насыщенный раствор бикарбоната натрия добавляли к реакционному раствору. Органический слой отделяли, промывали насыщенным раствором хлорида натрия и затем сушили над безводным сульфатом магния. Нерастворимое вещество отделяли фильтрованием и фильтрат концентрировали с получением указанного в заголовке соединения (3,40 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,75-1,82 (м, 2H), 2,26-2,31 (м, 1H), 2,42-2,48 (м, 1H), 3,27-3,29 (м, 3H), 3,52-3,58 (м, 1H), 5,20-5,25 (м, 2H), 5,61-5,71 (м, 1H), 6,75 (т, J=5,6 Гц, 0,5Н), 7,43 (т, J=5,2 Гц, 0,5H).
(7) Синтез 4-метокси-3a,4,5,6-тетрагидро-3H-циклопента[c]изоксазола
Указанное в заголовке соединение (1,30 г) получали из соединения, полученного в примере получения 23-(6) (3,40 г), в соответствии с примером получения 22-(2).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,18-2,70 (м, 5H), 3,26-3,28 (м, 3H), 3,42-3,79 (м, 1H), 3,96-4,20 (м, 1H), 4,35-4,68 (м, 1H).
(8) Синтез [2-амино-2-(2-фторфенил)-5-метоксициклопентил]метанола
Раствор н-бутиллития в гексане (2,60 M; 7,07 мл) добавляли по каплям к раствору, содержащему 2-бромфторбензол (2,09 мл) в смеси тетрагидрофуран/толуол (3,36 мл/33,6 мл), в атмосфере азота при -78°C. Реакционный раствор перемешивали при той же температуре в течение 10 минут. Комплекс трифторид бора-диэтиловый эфир (2,27 мл) и раствор, содержащий соединение, полученное в примере получения 23-(7) (1,30 г), в толуоле (10 мл) добавляли по каплям к реакционному раствору при той же температуре. После перемешивания при той же температуре в течение 40 минут к реакционному раствору добавляли водный раствор хлорида аммония с последующим нагреванием до комнатной температуры. К реакционному раствору добавляли воду и этилацетат и отделяли органический слой. Органический слой промывали насыщенным раствором хлорида натрия. Органический слой сушили над безводным сульфатом магния и нерастворимое вещество отделяли фильтрованием. Фильтрат концентрировали и остаток растворяли в смеси этилацетат-гептан. Раствор фильтровали через силикагель и концентрировали. Уксусную кислоту (31,0 мл) и цинковый порошок (5,02 г) добавляли при комнатной температуре. Реакционный раствор перемешивали при комнатной температуре в течение четырех часов. Нерастворимое вещество отделяли фильтрованием через целит и фильтрат концентрировали. К остатку добавляли этилацетат и раствор бикарбоната натрия и отделяли органический слой. Органический слой промывали насыщенным водным раствором хлорида натрия и сушили над безводным сульфатом магния. Нерастворимое вещество отделяли фильтрованием и фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (1,41 г).
ESI-MS; m/z 240 [M++H].
(9) Синтез N-[7a-(2-фторфенил)-5-метокси-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]бензамида
Бензоилизотиоцианат (0,871 мл) добавляли к раствору, содержащему соединение, полученное в примере получения 23-(8) (1,41 г), в дихлорметане (19,6 мл) и смесь перемешивали при комнатной температуре в течение 10 минут. Реакционный раствор концентрировали при пониженном давлении и остаток растворяли в смеси этилацетат-гептан. Раствор фильтровали через силикагель и концентрировали. Дихлорметан (24,0 мл), пиридин (1,16 мл) и трифторметансульфоновый ангидрид (2,20 мл) добавляли при -78°C и смесь перемешивали при той же температуре в течение 30 минут. Насыщенный раствор бикарбоната натрия добавляли к реакционному раствору с последующим экстрагированием дихлорметаном. Органический слой сушили над безводным сульфатом магния и нерастворимое вещество отделяли фильтрованием. Фильтрат концентрировали при пониженном давлении и остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (1,71 г).
ESI-MS; m/z 385 [M++H].
(10) Синтез 7a-(2-фторфенил)-5-метокси-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-иламина
Указанное в заголовке соединение (681 мг) получали из соединения, полученного в примере получения 23-(9) (1,71 г), в соответствии со способом примера получения 22-(7).
ESI-MS; m/z 281 [M++H].
(11) Синтез 7a-(2-фтор-5-нитрофенил)-5-метокси-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-иламина
Указанное в заголовке соединение (506 мг) получали из соединения, полученного в примере получения 23-(10) (681 мг), в соответствии со способом примера получения 22-(8).
ESI-MS; m/z 326 [M++H].
(12) Синтез трет-бутил [(4aS*,5S*,7aS*)-7a-(2-фтор-5-нитрофенил)-5-метокси-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]карбамата и трет-бутил [(4aS*,5R*,7aS*)-7a-(2-фтор-5-нитрофенил)-5-метокси-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]карбамата
Более полярное указанное в заголовке соединение (283 мг) и менее полярное указанное в заголовке соединение (135 мг) получали из соединения, полученного в примере получения 23-(11) (506 мг), в соответствии со способом примера получения 22-(9).
Более полярное указанное в заголовке соединение (трет-бутил [(4aS*,5S*,7aS*)-7a-(2-фтор-5-нитрофенил)-5-метокси-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]карбамат)
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,53 (с, 9H), 1,87-2,10 (м, 5H), 2,66-2,90 (м, 2H), 3,41 (с, 3H), 4,10-4,15 (м, 1H), 7,20-7,26 (м, 1H), 8,18-8,21 (м, 2H).
Менее полярное указанное в заголовке соединение (трет-бутил [(4aS*,5R*,7aS*)-7a-(2-фтор-5-нитрофенил)-5-метокси-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]карбамат)
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,50 (с, 9H), 1,81-2,45 (м, 5H), 2,88-3,01 (м, 2H), 3,41 (с, 3H), 4,11-4,13 (м, 1H), 7,20-7,26 (м, 1H), 8,19-8,28 (м, 2H).
(13) Синтез трет-бутил [(4aS*,5S*,7aS*)-7a-(5-амино-2-фторфенил)-5-метокси-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]карбамата
Указанное в заголовке соединение (244 мг) получали из более полярного соединения, полученного в примере получения 23-(12) (283 мг), в соответствии со способом примера получения 22-(10).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,51 (с, 9H), 1,52-2,15 (м, 5H), 2,75-3,00 (м, 2H), 3,40 (с, 3H), 4,10-4,15 (м, 1H), 6,54-6,57 (м, 2H), 6,83-6,86 (м, 1H).
(14) Синтез трет-бутил [(4aS*,5R*,7aS*)-7a-(5-амино-2-фторфенил)-5-метокси-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]карбамата
Указанное в заголовке соединение (79,0 мг) получали из менее полярного соединения, полученного в примере получения 23-(12) (100 мг), в соответствии со способом примера получения 22-(10).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,49 (с, 9H), 1,57-2,20 (м, 4H), 2,45-2,49 (м, 1H), 2,86-3,03 (м, 3H), 3,32 (с, 3H), 3,98-4,00 (м, 1H), 6,54-6,55 (м, 2H), 6,83-6,88 (м, 1H).
(15) Синтез трет-бутил [(4aS*,5S*,7aS*)-7a-(5-амино-2-фторфенил)-5-метокси-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]карбамата
Соединение, полученное в примере получения 23-(13) (50 мг), оптически разделяли, используя CHIRALPAK™ OJ-H производства Daicel Chemical Industries, Ltd. (2 см × 25 см, подвижная фаза: гексан:этанол = 1:1 - 0:1 (градиент, 30 мин), скорость потока: 10 мл/мин), и компоненты, имеющие время удерживания от 14 до 20 минут, собирали. Данную операцию повторяли с получением указанного в заголовке соединения (89 мг; >99% ee) из 244 мг рацемата.
Пример получения 24
трет-Бутил [(4aS,5S,7aS)-7a-(5-амино-2-фторфенил)-5-метоксиметил-4,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил]карбамат
[Формула 46]
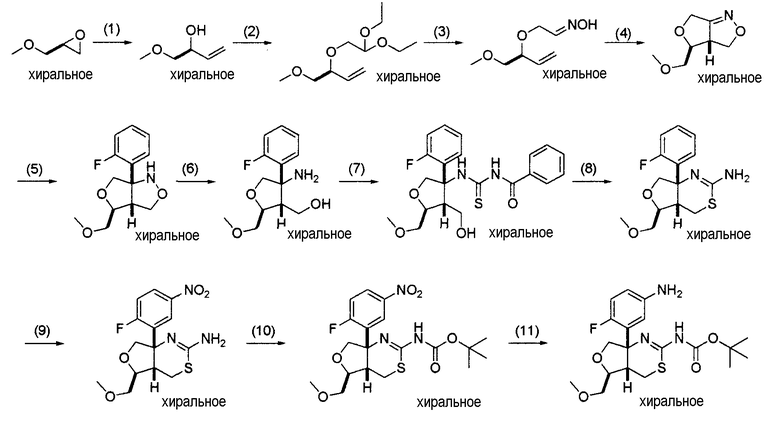
(1) Синтез (S)-1-метоксибут-3-ен-2-ола
Раствор йодида триметилсульфония (50,0 г) в толуоле (200 мл) кипятили с обратным холодильником, используя ловушку Дина-Старка, в течение одного часа и концентрировали. Остаток растворяли в ТГФ (444 мл) и раствор н-BuLi в гексане (2,6 M; 94,0 мл) добавляли по каплям при -15°C. После перемешивания при той же температуре в течение 30 минут добавляли по каплям (S)-глицидилметиловый простой эфир (7,31 мл) и смесь перемешивали при комнатной температуре в течение 14 часов. К реакционному раствору добавляли воду при 0°C с последующим экстрагированием диэтиловым эфиром. Органический слой сушили над безводным сульфатом магния и нерастворимое вещество отделяли фильтрованием. Фильтрат концентрировали при пониженном давлении. Остаток очищали хроматографией на силикагеле с получением указанного в заголовке соединения (3,30 г).
(2) Синтез (S)-3-(2,2-диэтоксиэтокси)-4-метоксибут-1-ена
Раствор соединения, полученного в примере получения 24-(1) (5,00 г), в N-метил-2-пирролидоне (98,0 мл) охлаждали до 0°C. Гидрид натрия (60%, 2,16 г) и диэтилацеталь бромацетальдегида (8,86 г) добавляли к реакционному раствору при той же температуре и смесь перемешивали при 100°C в течение двух часов. К реакционному раствору добавляли насыщенный раствор хлорида аммония при 0°C с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным раствором бикарбоната натрия и насыщенным водным раствором хлорида натрия и затем сушили над безводным сульфатом магния. Нерастворимое вещество отделяли фильтрованием и фильтрат концентрировали при пониженном давлении. Остаток очищали хроматографией на силикагеле с получением указанного в заголовке соединения (5,82 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,91-1,26 (м, 6H), 3,37 (с, 3H), 3,38-4,00 (м, 5H), 4,63-4,67 (м, 1H), 5,25-5,35 (м, 2H), 5,70-5,74 (м, 1H).
(3) Синтез ((S)-1-метоксиметилаллилокси)ацетальдегидоксима
Муравьиную кислоту (35,1 мл), гидрохлорид гидроксиламина (2,83 г) и ацетат натрия (4,46 г) добавляли к соединению, полученному в примере получения 24-(2) (5,82 г), и смесь перемешивали при комнатной температуре в течение 30 минут. К реакционному раствору добавляли этилацетат и насыщенный раствор хлорида натрия и отделяли органический слой. Органический слой промывали насыщенным водным раствором хлорида натрия и затем сушили над безводным сульфатом магния. Нерастворимое вещество отделяли фильтрованием и фильтрат концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (2,00 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 3,34-3,51 (м, 4H), 3,93-4,23 (м, 3H), 4,33-4,46 (м, 1H), 5,31-5,38 (м, 2H), 5,69-5,78 (м, 1H), 6,98 (т, J=3,6 Гц, 0,5H), 7,53 (т, J=4,8 Гц, 0,5H).
(4) Синтез (3aR,4S)-4-метоксиметил-3a,4-дигидро-3H,6H-фуро[3,4-c]изоксазола
Указанное в заголовке соединение (870 мг) получали из соединения, полученного в примере получения 24-(3) (2,00 г), в соответствии с примером получения 22-(2).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 3,38-3,55 (м, 4H), 3,41 (с, 3H), 3,97-4,13 (м, 2H), 4,48-4,58 (м, 2H).
(5) Синтез (3aR,4S,6aS)-6a-(2-фторфенил)-4-метоксиметилтетрагидрофуро[3,4-c]изоксазола
Указанное в заголовке соединение (940 мг) получали из соединения, полученного в примере получения 24-(4) (870 мг), в соответствии с примером получения 22-(3).
ESI-MS; m/z 254 [M++H].
(6) Синтез [(2S,3R,4S)-4-амино-4-(2-фторфенил)-2-метоксиметилтетрагидрофуран-3-ил]метанола
Указанное в заголовке соединение (850 мг) получали из соединения, полученного в примере получения 24-(5) (940 мг), в соответствии с примером получения 22-(4).
ESI-MS; m/z 256 [M++H].
(7) Синтез 1-бензоил-3-[(3S,4R,5S)-3-(2-фторфенил)-4-гидроксиметил-5-метоксиметилтетрагидрофуран-3-ил]тиомочевины
Указанное в заголовке соединение (1,22 г) получали из соединения, полученного в примере получения 24-(6) (850 мг), в соответствии с примером получения 22-(5).
ESI-MS; m/z 441 [M++Na].
(8) Синтез (4aS,5S,7aS)-7a-(2-фторфенил)-5-метоксиметил-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-иламина
Пиридин (0,66 мл) и трифторметансульфоновый ангидрид (0,983 мл) добавляли к раствору соединения, полученного в примере получения 24-(7) (1,22 г), в дихлорметане (5,80 мл) при -78°C и смесь перемешивали при 0°C в течение 10 минут. Насыщенный раствор бикарбоната натрия добавляли к реакционному раствору с последующим экстрагированием дихлорметаном. Органический слой сушили над безводным сульфатом магния и нерастворимое вещество отделяли фильтрованием. Фильтрат концентрировали при пониженном давлении. Остаток фильтровали через силикагель, используя этилацетат и гептан, и концентрировали при пониженном давлении. Раствор метоксида натрия (28% раствор в метаноле; 1,08 мл) в метаноле (7,00 мл) добавляли к остатку с последующим кипячением с обратным холодильником в течение 2,5 часов. После охлаждения реакционного раствора до комнатной температуры выпаривали при пониженном давлении растворитель. Остаток очищали хроматографией на силикагеле с получением указанного в заголовке соединения (231 мг).
ESI-MS; m/z 297 [M++H].
(9) Синтез (4aS,5S,7aS)-7a-(2-фтор-5-нитрофенил)-5-метоксиметил-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-иламина
Указанное в заголовке соединение (211 мг) получали из соединения, полученного в примере получения 24-(8) (231 мг), в соответствии с примером получения 22-(8).
ESI-MS; m/z 342 [M++H].
(10) Синтез трет-бутил [(4aS,5S,7aS)-7a-(2-фтор-5-нитрофенил)-5-метоксиметил-4,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил]карбамата
Указанное в заголовке соединение (144 мг) получали из соединения, полученного в примере получения 24-(9) (211 мг), в соответствии с примером получения 22-(9).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,52 (с, 9H), 2,68-2,77 (м, 1H), 2,88-2,93 (м, 2H), 3,39 (с, 3H), 3,44-3,62 (м, 2H), 3,97-3,99 (м, 1H), 4,46-4,52 (м, 2H), 7,14-7,17 (м, 1H), 8,21-8,30 (м, 2H).
(11) трет-Бутил [(4aS,5S,7aS)-7a-(5-амино-2-фторфенил)-5-метоксиметил-4,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил]карбамат
Указанное в заголовке соединение (86 мг) получали из соединения, полученного в примере получения 24-(10) (114 мг), в соответствии с примером получения 22-(10).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,50 (с, 9H), 2,72-2,76 (м, 1H), 3,12-3,13 (м, 2H), 3,56 (с, 3H), 3,58-3,65 (м, 2H), 3,81-3,82 (м, 1H), 4,49-4,51 (м, 2H), 6,56-6,63 (м, 2H), 6,84-6,89 (м, 1H).
Пример получения 25
Синтез трет-бутил [(4aS,5S,7aS)-7a-(5-амино-2-фторфенил)-5-фторметил-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил]карбамата
[Формула 47]

(1) Синтез (S)-1-тритилоксибут-3-ен-2-ола
Раствор н-BuLi в гексане (2,6 M; 182 мл) добавляли по каплям к раствору йодида триметилсульфония (96,8 г) в ТГФ (800 мл) при -30°C. После перемешивания при -20°C в течение 20 минут добавляли (S)-тритилглицидиловый простой эфир (50,0 г) при той же температуре и смесь перемешивали при комнатной температуре в течение 30 минут. К реакционному раствору добавляли воду с последующим экстрагированием диэтиловым эфиром. Органический слой сушили над безводным сульфатом магния. Нерастворимое вещество отделяли фильтрованием и фильтрат концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (52,0 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 3,09-3,13 (м, 1H), 3,20-3,23 (м, 1H), 4,26-4,29 (м, 1H), 5,14-5,32 (м, 2H), 5,76-5,84 (м, 1H), 7,23-7,45 (м, 15H).
(2) Синтез этил ((S)-1-тритилоксиметилаллилокси)ацетата
Гидрид натрия (60%, 6,18 г) и бромэтилацетат (17,1 мл) добавляли к раствору, содержащему соединение, полученное в примере получения 25-(1) (25,5 г), в N-метил-2-пирролидоне (210 мл) при 0°C. Смесь перемешивали при 50°C в течение 18 часов и перемешивали при 100°C в течение одного часа. К реакционному раствору добавляли насыщенный раствор хлорида аммония при 0°C с последующим экстрагированием диэтиловым эфиром. Органический слой сушили над безводным сульфатом магния. Нерастворимое вещество отделяли фильтрованием и фильтрат концентрировали. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (15,5 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,32 (т, J=7,2 Гц, 3H), 3,13-3,17 (м, 1H), 3,31-3,35 (м, 1H), 3,98-4,27 (м, 5H), 5,28-5,33 (м, 2H), 5,74-5,76 (м, 1H), 7,20-7,47 (м, 15H).
(3) Синтез ((S)-1-тритилоксиметилаллилокси)ацетальдегидоксима
Раствор диизобутилалюминийгидрида в толуоле (1,0 M; 55,2 мл) добавляли по каплям к раствору, содержащему соединение, полученное в примере получения 25-(2) (15,5 г), в дихлорметане (74,0 мл) при -78°C. Смесь перемешивали при той же температуре в течение 30 минут. К реакционному раствору добавляли 2 н. раствор хлористо-водородной кислоты с последующим экстрагированием дихлорметаном. Органический слой промывали насыщенным раствором бикарбоната натрия и сушили над безводным сульфатом магния. Нерастворимое вещество отделяли фильтрованием и фильтрат концентрировали. Метанол (70,0 мл), ацетат натрия (6,04 г) и гидрохлорид гидроксиламина (3,84 г) добавляли к остатку при комнатной температуре и смесь перемешивали при той же температуре в течение 15 минут. Этилацетат и воду добавляли к реакционному раствору и органический слой отделяли и сушили над безводным сульфатом магния. Нерастворимое вещество отделяли фильтрованием и фильтрат концентрировали. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (11,3 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 3,08-3,12 (м, 1H), 3,24-3,26 (м, 1H), 3,81-4,42 (м, 4H), 5,23-5,30 (м, 2H), 5,70-5,72 (м, 1H), 6,95-6,96 (м, 0,5H), 7,21-7,47 (м, 15H), 7,52-7,53 (м, 0,5H).
(4) Синтез (3aR,4S)-4-тритилоксиметил-3a,4-дигидро-3H,6H-фуро[3,4-c]изоксазола
5% Раствор гипохлорита натрия (52,2 мл) добавляли по каплям к раствору, содержащему соединение, полученное в примере получения 25-(3) (11,3 г), в дихлорметане (100 мл) при 0°C и смесь перемешивали при 0°C в течение 30 минут. Раствор бисульфита натрия добавляли к реакционному раствору при той же температуре. Органический слой отделяли и сушили над безводным сульфатом магния. Нерастворимое вещество отделяли фильтрованием и фильтрат концентрировали. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (5,20 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 3,21-3,35 (м, 1H), 3,37-3,40 (м, 1H), 3,93-4,07 (м, 3H), 4,47-4,57 (м, 3Н), 7,23-7,42 (м, 15H).
(5) Синтез (3aR,4S,6aS)-6a-(2-фторфенил)-4-тритилоксиметилтетрагидрофуро[3,4-c]изоксазола
Раствор н-бутиллития в гексане (2,60 M; 10,4 мл) добавляли по каплям к раствору, содержащему 2-бромфторбензол (2,93 мл) в смеси тетрагидрофуран/толуол (10,8 мл/108 мл), в атмосфере азота при -78°C. Реакционный раствор перемешивали при той же температуре в течение 10 минут. Комплекс трифторид бора-диэтиловый эфир (3,33 мл) и раствор, содержащий соединение, полученное в примере получения 25-(4) (5,20 г), в толуоле (50 мл) последовательно добавляли по каплям к реакционному раствору при той же температуре. После перемешивания при той же температуре в течение 40 минут к реакционному раствору добавляли водный раствор хлорида аммония с последующим нагреванием до комнатной температуры. К реакционному раствору добавляли воду и этилацетат и отделяли органический слой. Органический слой промывали насыщенным раствором хлорида натрия. Органический слой сушили над безводным сульфатом магния и нерастворимое вещество отделяли фильтрованием. Фильтрат концентрировали и остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (6,23 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 3,24 (м, 3H), 3,91-3,98 (м, 2H), 4,07-4,35 (м, 3H), 7,00-7,62 (м, 19H).
(6) Синтез [(2S,3R,4S)-4-амино-4-(2-фторфенил)-2-тритилоксиметилтетрагидрофуран-3-ил]метанола
Цинковый порошок (8,44 г) добавляли к раствору, содержащему соединение, полученное в примере получения 25-(5) (6,22 г), в уксусной кислоте (50,0 мл) при комнатной температуре. Реакционный раствор перемешивали при комнатной температуре в течение 18 часов. Нерастворимое вещество отделяли фильтрованием через целит и фильтрат концентрировали. К остатку добавляли этилацетат и раствор бикарбоната натрия и отделяли органический слой. Органический слой промывали насыщенным водным раствором хлорида натрия и сушили над безводным сульфатом магния. Нерастворимое вещество отделяли фильтрованием и фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с получением указанного в заголовке соединения (4,10 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,63-2,65 (м, 1H), 3,24-3,31 (м, 2H), 3,61-3,65 (м, 1H), 3,92-3,97 (м, 2H), 4,15-4,26 (м, 1H), 4,37-4,41 (м, 1H), 7,00-7,52 (м, 19H).
(7) Синтез 1-бензоил-3-[(3S,4R,5S)-3-(2-фторфенил)-4-гидроксиметил-5-тритилоксиметилтетрагидрофуран-3-ил]тиомочевины
Бензоилизотиоцианат (1,37 мл) добавляли к раствору, содержащему соединение, полученное в примере получения 25-(6) (4,10 г), в дихлорметане (16,0 мл) и смесь перемешивали при комнатной температуре в течение 10 минут. Реакционный раствор концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (4,32 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 3,19-3,36 (м, 3H), 3,79-4,05 (м, 3H), 4,57-4,58 (м, 2H), 7,03-7,89 (м, 24H), 8,89 (уш., 1H).
(8) Синтез N-[(4aS,5S,7aS)-7a-(2-фторфенил)-5-тритилокси-4,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил]бензамида
Пиридин (2,01 мл) и трифторметансульфоновый ангидрид (2,25 мл) добавляли к раствору соединения, полученного в примере получения 25-(7) (4,32 г), в дихлорметане (27,7 мл) при 0°C и смесь перемешивали при той же температуре в течение 20 минут. Насыщенный раствор бикарбоната натрия добавляли к реакционному раствору с последующим экстрагированием дихлорметаном. Органический слой сушили над безводным сульфатом магния и нерастворимое вещество отделяли фильтрованием. Фильтрат концентрировали при пониженном давлении и остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (3,52 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,65-2,69 (м, 1H), 3,15-3,19 (м, 1H), 3,32-3,47 (м, 3H), 4,08-4,10 (м, 1H), 4,55-4,58 (м, 2H), 7,11-7,52 (м, 22H), 8,15-8,17 (м, 2H).
(9) Синтез N-[(4aS,5S,7aS)-7a-(2-фторфенил)-5-гидроксиметил-4,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил]бензамида
Муравьиную кислоту (15,0 мл) и диэтиловый эфир (15,0 мл) добавляли к соединению, полученному в примере получения 25-(8) (3,52 г), при комнатной температуре и смесь перемешивали при той же температуре в течение восьми часов. Реакционный раствор концентрировали при пониженном давлении. Муравьиную кислоту (20 мл) добавляли при комнатной температуре и смесь перемешивали при той же температуре в течение 15 часов. Реакционный раствор концентрировали при пониженном давлении. Раствор триэтиламина в метаноле (10%; 20,0 мл) добавляли при комнатной температуре с последующим кипячением с обратным холодильником в течение 30 минут. Реакционный раствор концентрировали. Добавляли этилацетат и насыщенный раствор соли и органический слой отделяли и сушили над безводным сульфатом магния. Нерастворимое вещество отделяли фильтрованием. Фильтрат концентрировали при пониженном давлении и остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (1,72 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,80-2,84 (м, 1H), 3,25-3,29 (м, 1H), 3,40-3,44 (м, 1H), 3,73-3,77 (м, 1H), 3,94-3,98 (м, 1H), 4,08-4,11 (м, 1H), 4,13-4,57 (м, 2H), 7,12-7,53 (м, 7H), 8,14-8,16 (м, 2H).
(10) Синтез N-[(4aS,5S,7aS)-5-фторметил-7a-(2-фторфенил)-4,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил]бензамида
Триэтиламин (3,52 мл), тригидрофторид триэтиламина (1,37 мл) и перфторбутансульфонилфторид (1,45 мл) добавляли к раствору соединения, полученного в примере получения 25-(9) (1,62 г), в ацетонитриле (16,2 мл) при 0°C и смесь перемешивали при комнатной температуре в течение 20 минут. Реакционный раствор очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (920 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,81-2,85 (м, 1H), 3,28-3,31 (м, 1H), 3,41-3,43 (м, 1H), 4,05-4,07 (м, 1H), 4,55-4,74 (м, 4H), 7,12-7,54 (м, 7H), 8,12-8,14 (м, 2H).
(11) Синтез (4aS,5S,7aS)-5-фторметил-7a-(2-фторфенил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-иламина
Раствор соединения, полученного в примере получения 25-(10) (970 мг), и метоксид натрия (28% раствор в метаноле; 0,965 мл) в метаноле (6,43 мл) кипятили с обратным холодильником в течение 14 часов. После охлаждения реакционного раствора до комнатной температуры выпаривали при пониженном давлении растворитель. Остаток очищали хроматографией на силикагеле с получением указанного в заголовке соединения (310 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,76-2,80 (м, 1H), 3,00-3,04 (м, 1H), 3,09-3,13 (м, 1H), 3,85-3,88 (м, 1H), 4,47-4,63 (м, 4H), 7,00-7,16 (м, 2H), 7,27-7,44 (м, 2H).
(12) Синтез трет-бутил [(4aS,5S,7aS)-5-фторметил-7a-(2-фтор-5-нитрофенил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил]карбамата
Дымящую азотную кислоту (55 мкл) добавляли по каплям к раствору соединения, полученного в примере получения 25-(11) (310 мг), в концентрированной серной кислоте (5,53 мл) при охлаждении льдом. Реакционный раствор перемешивали при той же температуре в течение 10 минут и затем вливали в ледяную воду. Реакционную смесь нейтрализовали 5 н. раствором гидроксида натрия. Смесь дважды экстрагировали этилацетатом. Органические слои сушили над безводным сульфатом магния и нерастворимое вещество отделяли фильтрованием. Фильтрат концентрировали при пониженном давлении и остаток растворяли в дихлорметане (5,40 мл). Триэтиламин (0,602 мл) и ди-трет-бутилдикарбонат (471 мг) добавляли к раствору. Реакционный раствор концентрировали при пониженном давлении и остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (419 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,56 (с, 9H), 2,70-2,74 (м, 1H), 2,96-2,99 (м, 1H), 3,18-3,19 (м, 1H), 3,84-3,85 (м, 1H), 4,47-4,70 (м, 4H), 7,22-7,24 (м, 1H), 8,20-8,31 (м, 2H).
(13) Синтез трет-бутил [(4aS,5S,7aS)-7a-(5-амино-2-фторфенил)-5-фторметил-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил]карбамата
Насыщенный раствор хлорида аммония (1,0 мл) и железный порошок (436 мг) добавляли к раствору соединения, полученного в примере получения 25-(12) (419 мг), в этаноле (10 мл). Реакционный раствор кипятили с обратным холодильником в течение 30 минут и затем охлаждали до комнатной температуры. Реакционный раствор разбавляли этилацетатом и нерастворимое вещество отделяли фильтрованием через целит. К фильтрату добавляли этилацетат и воду и отделяли органический слой. Органический слой сушили над безводным сульфатом магния и нерастворимое вещество отделяли фильтрованием. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (291 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,51 (с, 9H), 3,10-3,21 (м, 2H), 3,62 (уш., 1H), 3,85-3,86 (м, 1H), 4,15-4,66 (м, 4H), 6,56-6,62 (м, 2H), 6,85-6,92 (м, 1H).
Пример получения 26
Синтез 3-этокси-5-гексен-1-ола
[Формула 48]
(1) Синтез трет-бутил-(3-этокси-5-гексенилокси)дифенилсилана
Раствор 1-(трет-бутил-дифенилсиланилокси)-5-гексен-3-ола (Tetrahedron, 57, 4023-4034 (2001)) (5,0 г) и этилиодида (2,03 мл) в ТГФ (10 мл) добавляли по каплям к суспензии гидрида натрия (60%, 1,85 г) в ТГФ (40 мл) при 50°C в течение 10 минут. Реакционный раствор перемешивали при 50°C в течение одного часа. Этилиодид (6,0 мл) добавляли к раствору при 50°C и смесь перемешивали при 60°C в течение 1,5 часов. Реакционный раствор доводили до комнатной температуры и вливали в ледяную воду с последующим экстрагированием этилацетатом. Экстракт промывали насыщенным раствором хлорида натрия и затем сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении. Полученный неочищенный продукт очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (4,1 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,05 (с, 9H), 1,14 (т, J=7,2 Гц, 3H) 1,66-1,80 (м, 2H), 2,22-2,30 (м, 2H), 3,36-3,86 (м, 5H), 5,00-5,10 (м, 2H), 5,74-5,88 (м, 1H), 7,30-7,46 (м, 6H), 7,60-7,74 (м, 4H).
(2) Синтез 3-этокси-5-гексен-1-ола
Тетрабутиламмонийфторид (1 M раствор в ТГФ, 13 мл) добавляли к раствору трет-бутил-(3-этокси-5-гексенилокси)дифенилсилана (4,1 г), в ТГФ (40 мл) при комнатной температуре с последующим перемешиванием в течение одного часа. Реакционный раствор концентрировали. Полученный неочищенный продукт очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (1,1 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,20 (т, J=7,2 Гц, 3H) 1,68-1,82 (м, 2H), 2,22-2,44 (м, 2H), 2,70 (с, 1H), 3,36-3,88 (м, 5H), 5,00-5,16 (м, 2H), 5,70-5,90 (м, 1H).
Пример получения 27
Синтез (3aR*,5S*,6aS*)-5-этокси-6a-(2-фторфенил)гексагидроциклопента[c]изоксазола и (3aR*,5R*,6aS*)-5-этокси-6a-(2-фторфенил)гексагидроциклопента[c]изоксазола
[Формула 49]

Менее полярное указанное в заголовке соединение и более полярное указанное в заголовке соединение получали обработкой 3-этокси-5-гексен-1-ола в соответствии со способом примера получения 10-(1)-(4).
Менее полярное указанное в заголовке соединение ((3aR*,5S*,6aS*)-5-этокси-6a-(2-фторфенил)гексагидроциклопента[c]изоксазол)
ESI-MS; m/z 252 [M++H].
Более полярное указанное в заголовке соединение ((3aR*,5R*,6aS*)-5-этокси-6a-(2-фторфенил)гексагидроциклопента[c]изоксазол)
ESI-MS; m/z 252 [M++H].
Пример получения 28
Синтез трет-бутил [(4aR*,6R*,7aS*)-7a-(5-амино-2-фторфенил)-6-этокси-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]карбамата ((+)-изомера и (-)-изомера)
[Формула 50]

(1) Синтез [(1R*,2S*,4R*)-2-амино-4-этокси-2-(2-фторфенил)циклопентил]метанола
Цинк (3,68 г) добавляли к раствору (3aR*,5R*,6aS*)-5-этокси-6a-(2-фторфенил)гексагидроциклопента[c]изоксазола (540 мг) в уксусной кислоте (15 мл) и смесь перемешивали при комнатной температуре в течение двух часов. Добавляли к реакционному раствору насыщенный раствор бикарбоната натрия и этилацетат и цинк удаляли фильтрованием. Фильтрат экстрагировали этилацетатом. Экстракт сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (540 мг).
ESI-MS; m/z 254 [M++H].
(2) Синтез N-({[(1S*,2R*,4R*)-4-этокси-1-(2-фторфенил)-2-(гидроксиметил)циклопентил]амино}карбонотиоил)бензамида
Бензоилизотиоцианат (0,345 мл) добавляли к раствору амина, синтезированного на предшествующей стадии (540 мг), в дихлорметане (9 мл) и смесь перемешивали при комнатной температуре в течение 12 часов. Реакционный раствор концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (180 мг).
ESI-MS; m/z 439 [M++Na].
(3) Синтез (4aR*,6S*,7aS*)-6-этокси-7a-(2-фторфенил)-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]амина
Раствор тиомочевины, полученной на предшествующей стадии (180 мг), в смеси метанола (20 мл) и концентрированной хлористо-водородной кислоты (0,7 мл) кипятили с обратным холодильником в течение трех часов. Реакционный раствор доводили до комнатной температуры и затем вливали в охлажденный раствор бикарбоната натрия с последующим экстрагированием этилацетатом. Экстракт сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении. Полученный неочищенный продукт очищали хроматографией на колонке с силикагелем с получением циклизованного соединения (94 мг). DBU (0,2 мл) добавляли к раствору циклизованного соединения в метаноле (10 мл) с последующим кипячением с обратным холодильником в течение четырех часов. Реакционный раствор концентрировали при пониженном давлении. Неочищенный продукт очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (69 мг).
ESI-MS; m/z 295 [M++H].
(4)-(6) Синтез трет-бутил [(4aR*,6R*,7aS*)-7a-(5-амино-2-фторфенил)-6-этокси-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]карбамата ((+)-изомера и (-)-изомера)
Указанное в заголовке (-)-соединение получали обработкой циклизованного соединения, полученного на предшествующей стадии, в соответствии со способом стадий (4)-(5) примера получения 12. Оптическое разделение осуществляли, используя CHIRALPAK™ ADH производства Daicel Chemical Industries, Ltd. (2 см × 25 см, подвижная фаза: гексан:этанол = 8:2, скорость потока: 10 мл/мин). Компоненты, имеющие время удерживания от 7 до 10 минут, собирали с получением указанного в заголовке (+)-изомера. Компоненты, имеющие время удерживания от 16 до 19 минут, собирали с получением указанного в заголовке (-)-изомера. (-)-Изомер использовали для синтеза хирального соединения в данном примере.
Характеристические значения (-)-изомера даны ниже.
оптическое вращение (-)
ESI-MS; m/z 410 [M++H].
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,21 (т, J=7,2 Гц, 3H), 1,52 (с, 9H), 1,90-2,05 (м, 1H), 2,25-2,45 (м, 2H), 2,60-2,75 (м, 2H), 3,03 (дд, J=3,6, 13,6 Гц, 1H), 3,25-3,35 (м, 1H), 3,46 (кв, J=7,2 Гц, 2H), 3,62 (уш.с, 2H), 4,10-4,20 (м, 1H), 6,51-6,58 (м, 2H), 6,86 (дд, J=8,0, 12,0 Гц, 1H).
Пример получения 29
Синтез трет-бутил (±)-[(4aR*,6S*,7aS*)-7a-(5-амино-2-фторфенил)-6-этокси-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]карбамата
[Формула 51]

Указанное в заголовке соединение получали обработкой (3aR*,5S*,6aS*)-5-этокси-6a-(2-фторфенил)гексагидроциклопента[c]изоксазола, полученного в примере получения 27, в соответствии со способом примера получения 28-(1)-(3) и примера получения 11-(4)-(5).
ESI-MS; m/z 410 [M++H].
Пример получения 30
Синтез трет-бутил (-)-[(4aR*,6R*,7aS*)-7a-(5-амино-2-фторфенил)-6-бутокси-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]карбамата
[Формула 52]

Указанное в заголовке соединение получали обработкой 1-(трет-бутил-дифенилсиланилокси)-5-гексен-3-ола и йодбутана в соответствии со способом примеров получения 26-29.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 0,92 (т, J=7,2 Гц, 3H), 1,32-1,42 (м, 2H), 1,52 (с, 9H), 1,48-1,60 (м, 2H), 1,85-2,05 (м, 1H), 2,20-2,45 (м, 2H), 2,55-2,75 (м, 2H), 3,03 (д, J=13,2 Гц, 1H), 3,20-3,45 (м, 3H), 3,62 (с, 2H), 4,05-4,20 (м, 1H), 6,50-6,60 (м, 2H), 6,80-6,95 (м, 1Н).
ESI-MS m/z 438 [M++H]
Пример получения 31
Синтез трет-бутил (±)-[(4aR*,6S*,7aS*)-7a-(5-амино-2-фторфенил)-6-бутокси-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]карбамата
[Формула 53]

Указанное в заголовке соединение получали обработкой 1-(трет-бутил-дифенилсиланилокси)-5-гексен-3-ола и йодбутана в соответствии со способом примеров получения 26-29.
ESI-MS m/z 438 [M++H]
Пример получения 32
Синтез (3aR*,5S*,6aS*)-6a-(2-фторфенил)-5-(3,4-дифторбензил)оксигексагидроциклопента[c]изоксазола и (3aR*,5R*,6aS*)-6a-(2-фторфенил)-5-(3,4-дифторбензил)оксигексагидроциклопента[c]изоксазола
[Формула 54]

Указанное в заголовке соединение получали обработкой 1-(трет-бутилдифенилсиланилокси)-5-гексен-3-ола и 3,4-дифторбензилбромида в соответствии со способом примера получения 26-(1)-(2) и примера получения 10-(1)-(4).
Менее полярное указанное в заголовке соединение ((3aR*,5S*,6aS*)-6a-(2-фторфенил)-5-(3,4-дифторбензил)оксигексагидроциклопента[c]изоксазол);
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,00-2,20 (м, 2H), 2,20-2,40 (м, 2H), 3,02 (уш.с, 1H), 3,31 (кв, J=7,6 Гц, 1H), 3,73 (т, J=7,6 Гц, 1H), 4,30 (с, 1H), 4,30-4,55 (м, 2H), 4,39 (т, J=8,0 Гц, 1H), 6,03 (с, 1H), 6,90-8,00 (м, 7H).
Более полярное указанное в заголовке соединение ((3aR*,5R*,6aS*)-6a-(2-фторфенил)-5-(3,4-дифторбензил)оксигексагидроциклопента[c]изоксазол);
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,00-2,28 (м, 2H), 2,32 (дд, J=8,8, 13,6 Гц, 1H), 2,41 (дд, J=5,6, 13,6 Гц, 1H), 3,30-3,45 (м, 1H), 3,73 (уш.с, 1H), 4,13 (уш.с, 1H), 4,25-4,40 (м, 1H), 4,44 (с, 2H), 6,94-7,32 (м, 6H), 7,56-7,66 (м, 1H).
Пример получения 33
Синтез трет-бутил [(4aR*,6S*,7aS*)-7a-(2-фторфенил)-6-гидрокси-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]карбамата
[Формула 55]
(1) Синтез [(1R*,2S*,4S*)-2-амино-2-(2-фторфенил)-4-(3,4-дифторбензилокси)циклопентил]метанола
Цинк (3,0 г) добавляли к раствору (3aR*,5S*,6aS*)-6a-(2-фторфенил)-5-(3,4-дифторбензилокси)гексагидроциклопента[c]изоксазола (620 мг) в уксусной кислоте (25 мл) и смесь перемешивали при комнатной температуре в течение двух часов. К реакционному раствору добавляли насыщенный раствор бикарбоната натрия и этилацетат и цинк удаляли фильтрованием. Фильтрат экстрагировали этилацетатом. Экстракт сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (630 мг).
ESI-MS; m/z 352 [M++H].
(2) Синтез N-({[(1S*,2R*,4S*)-1-(2-фторфенил)-2-(гидроксиметил)-4-(3,4-дифторбензилокси)циклопентил]амино}карбонотиоил)бензамида
Бензоилизотиоцианат (0,362 мл) добавляли к раствору амина, синтезированного на предшествующей стадии (630 мг), в дихлорметане (20 мл) и смесь перемешивали при комнатной температуре в течение 12 часов. Реакционный раствор очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (550 мг).
ESI-MS; m/z 537 [M++Na].
(3) Синтез N-((4aR*,6S*,7aS*)-7a-(2-фторфенил)-6-(3,4-дифторбензилокси)-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил)бензамида
Концентрированную хлористо-водородную кислоту (1,7 мл) добавляли к раствору тиомочевины, синтезированной на предшествующей стадии (550 мг), в метаноле (50 мл) и смесь кипятили с обратным холодильником в течение трех часов. Реакционному раствору давали охладиться и затем его вливали в охлажденный льдом раствор бикарбоната натрия с последующим экстрагированием этилацетатом. Экстракт промывали насыщенным раствором соли и сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (480 мг).
ESI-MS; m/z 497 [M++H].
(4) Синтез трет-бутил [(4aR*,6S*,7aS*)-7a-(2-фторфенил)-6-гидрокси-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]карбамата
Раствор амидного соединения, полученного на предшествующей стадии (135 мг), в концентрированной хлористо-водородной кислоте (20 мл) кипятили с обратным холодильником в течение четырех часов. Реакционный раствор доводили до комнатной температуры и вливали в охлажденный льдом раствор бикарбоната натрия с последующим экстрагированием этилацетатом. Экстракт промывали насыщенным раствором соли и сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении. Остаток растворяли в ТГФ (3,3 мл). Добавляли триэтиламин (0,0525 мл) и ди-трет-бутилдикарбонат (49,3 мг) и реакционный раствор перемешивали при комнатной температуре в течение двух часов. Реакционный раствор вливали в насыщенный раствор бикарбоната натрия с последующим экстрагированием этилацетатом. Экстракт промывали насыщенным раствором соли и сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (64 мг).
ESI-MS; m/z 367 [M++H].
Пример получения 34
Синтез трет-бутил [(4aR*,6R*,7aS*)-7a-(5-амино-2-фторфенил)-6-фтор-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]карбамата ((+)-изомера и (-)-изомера)
[Формула 56]

(1) Синтез трет-бутил [(4aR*,6R*,7aS*)-7a-(2-фторфенил)-6-фтор-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]карбамата
Трифторид [бис(2-метоксиэтил)амино]серы (0,0564 мл) добавляли к раствору трет-бутил [(4aR*,6S*,7aS*)-7a-(2-фторфенил)-6-гидрокси-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]карбамата (51 мг) в дихлорметане (2 мл) при 0°C и смесь перемешивали при 0°C в течение 1,5 часов. К реакционному раствору добавляли воду с последующим экстрагированием этилацетатом. Экстракт промывали насыщенным раствором соли и затем сушили над безводным сульфатом магния. Указанное в заголовке соединение (51 мг) получали удалением осушающего агента и концентрированием при пониженном давлении.
ESI-MS; m/z 369 [M++H].
(2) Синтез трет-бутил [(4aR*,6R*,7aS*)-7a-(2-фтор-5-нитрофенил)-6-фтор-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]карбамата
ТФУК (1,0 мл) добавляли к раствору трет-бутилового сложного эфира, полученного на предшествующей стадии (51 мг), в дихлорметане (2,0 мл) при комнатной температуре и реакционный раствор перемешивали при комнатной температуре в течение одного часа. Реакционный раствор вливали в охлажденный раствор бикарбоната натрия с последующим экстрагированием этилацетатом. Экстракт промывали насыщенным раствором соли и затем сушили над безводным сульфатом магния. Осушающий агент удаляли с последующим концентрированием при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением аминосоединения (34 мг). Дымящую азотную кислоту (4,48 мкл) добавляли к раствору аминосоединения (34 мг) в концентрированной серной кислоте (1,5 мл) при 0°C и смесь перемешивали при 0°C в течение 30 минут. Реакционный раствор вливали в смесь 5 н. раствора гидроксида натрия и ледяной воды с последующим экстрагированием этилацетатом. Экстракт промывали насыщенным раствором хлорида натрия и затем сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении. Триэтиламин (44,2 мкл) и ди-трет-бутилдикарбонат (41,6 мг) добавляли к раствору полученного неочищенного продукта в ТГФ (5 мл) при комнатной температуре и смесь перемешивали при комнатной температуре в течение 12 часов. Реакционный раствор вливали в насыщенный раствор бикарбоната натрия с последующим экстрагированием этилацетатом. Экстракт промывали насыщенным раствором хлорида натрия и затем сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении. Полученный неочищенный продукт очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (27 мг).
ESI-MS; m/z 414 [M++H].
(3) Синтез трет-бутил [(4aR*,6R*,7aS*)-7a-(5-амино-2-фторфенил)-6-фтор-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]карбамата
Раствор нитросоединения, полученного на предшествующей стадии (27 мг), и железа (60 мг) в этаноле (1,25 мл) и насыщенный раствор хлорида аммония (0,1 мл) перемешивали при 87°C в течение 30 минут. После доведения реакционного раствора до комнатной температуры железо удаляли фильтрованием. Фильтрат вливали в смесь вода-этилацетат с последующим экстрагированием этилацетатом. Экстракт промывали насыщенным раствором хлорида натрия и затем сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и затем фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (23 мг).
ESI-MS; m/z 384 [M++H].
(4) Синтез трет-бутил [(4aR*,6R*,7aS*)-7a-(5-амино-2-фторфенил)-6-фтор-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]карбамата ((+)-изомера и (-)-изомера)
трет-Бутил (±)-[(4aR*,6R*,7aS*)-7a-(5-амино-2-фторфенил)-6-метокси-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]карбамат (46 мг) оптически разделяли, используя CHIRALPAK™ ADH производства Daicel Chemical Industries, Ltd. (2 см × 25 см, подвижная фаза: гексан:этанол = 8:2, скорость потока: 10 мл/мин). Получали 15 мг компонентов, имеющих время удерживания от 15 до 20 минут. Получали также 11 мг компонентов, имеющих время удерживания от 28 до 33 минут.
Соединение, полученное из компонентов, имеющих время удерживания от 28 до 33 минут, использовали для синтеза хирального соединения в данном примере.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,51 (с, 9H), 2,10-2,30 (м, 1H), 2,30-2,55 (м, 2H), 2,73 (д, J=13,2 Гц, 1H), 2,92 (дд, J=14,4, 32,4 Гц, 1H), 3,05 (д, J=13,2 Гц, 1H), 3,33 (уш.с, 1H), 3,63 (уш.с, 2H), 5,02-5,45 (м, 1H), 6,45-6,60 (м, 2H), 6,80-6,95 (м, 1H).
Пример получения 35
Синтез трет-бутил [(4aR*,6R*,7aS*)-7a-(2-фторфенил)-6-гидрокси-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]карбамата
[Формула 57]

Указанное в заголовке соединение получали обработкой (3aR*,5R*,6aS*)-6a-(2-фторфенил)-5-(3,4-дифторбензилокси)гексагидроциклопента[c]изоксазола в соответствии со способом примера получения 33.
ESI-MS; m/z 367 [M++H].
Пример получения 36
Синтез трет-бутил (-)-[(4aR*,6S*,7aS*)-7a-(5-амино-2-фторфенил)-6-фтор-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]карбамата
[Формула 58]

Указанное в заголовке соединение получали обработкой трет-бутил [(4aR*,6R*,7aS*)-7a-(2-фторфенил)-6-гидрокси-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]карбамата в соответствии со способом примера получения 34. Оптическое разделение осуществляли, используя CHIRALPAK™ ADH производства Daicel Chemical Industries, Ltd. (2 см × 25 см, подвижная фаза: гексан:этанол = 7:3, скорость потока: 10 мл/мин). Компоненты, имеющие время удерживания от 18 до 23 минут, собирали с получением указанного в заголовке (-)-изомера.
Характеристические значения (-)-изомера даны ниже.
оптическое вращение (-)
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,51 (с, 9H), 2,10-2,60 (м, 3H), 2,70 (д, J=12,0 Гц, 1H), 2,75-3,10 (м, 3H), 3,64 (уш.с, 2H), 5,20-5,50 (м, 1H), 6,45-6,65 (м, 2H), 6,75-6,90 (м, 1H).
Пример получения 37
Синтез трет-бутил [(4aR*,7aS*)-7a-(5-амино-2-фторфенил)-6,6-дифтор-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]карбамата
[Формула 59]

(1) Синтез трет-бутил [(4aR*,7aS*)-7a-(2-фторфенил)-4a,5,7,7a-тетрагидро-4H-циклопента[d][1,3]тиазин-6-он-2-ил]карбамата
Диметилсульфоксид (0,014 мл) добавляли по каплям к раствору оксалилхлорида (0,0169 мл) в дихлорметане (5 мл) при -55°C и смесь перемешивали при -70°C в течение 10 минут. Раствор спирта, полученного в примере получения 33 (48 мг), в дихлорметане (5 мл) добавляли по каплям к раствору при -60°C и смесь перемешивали при -60°C в течение 15 минут. К раствору при -60°C добавляли по каплям триэтиламин (0,128 мл) и реакционный раствор перемешивали при температуре от -60°C до комнатной температуры в течение 30 минут. Реакционный раствор вливали в воду с последующим экстрагированием этилацетатом. Экстракт промывали насыщенным раствором хлорида натрия и затем сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (28 мг).
ESI-MS; m/z 365 [M++H].
(2) Синтез трет-бутил [(4aR*,7aS*)-7a-(2-фторфенил)-6,6-дифтор-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]карбамата
Трифторид [бис(2-метоксиэтил)амино]серы (1,42 мл) добавляли к раствору производного кетона, полученного на предшествующей стадии (281 мг), в дихлорметане (3 мл) при комнатной температуре и смесь перемешивали при комнатной температуре в течение 12 часов. К реакционному раствору добавляли ледяную воду с последующим экстрагированием этилацетатом. Экстракт промывали насыщенным раствором соли и затем сушили над безводным сульфатом магния. Осушающий агент удаляли с последующим концентрированием при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (50 мг).
ESI-MS; m/z 387 [M++H].
(3)-(5) Синтез трет-бутил [(4aR*,7aS*)-7a-(5-амино-2-фторфенил)-6,6-дифтор-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]карбамата
Указанное в заголовке соединение получали обработкой дифторсоединения, полученного в примере получения 37-(2), в соответствии со способом примера получения 34-(2)-(4). Оптическое разделение осуществляли, используя CHIRALPAK™ ADH производства Daicel Chemical Industries, Ltd. (2 см × 25 см, подвижная фаза: гексан:этанол = 7:3, скорость потока: 10 мл/мин).
Соединение, полученное из компонентов, имеющих время удерживания от 18,5 до 21 минуты, использовали для синтеза хирального соединения в данном примере.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,52 (с, 9H), 2,30-2,76 (м, 4H), 2,98 (д, J=13,2 Гц, 1H), 3,10-3,30 (м, 2H), 3,63 (уш.с, 2H), 6,50 (дд, J=2,8, 7,2 Гц, 1H), 6,53-6,63 (м, 1H), 6,87 (дд, J=8,4, 12,0 Гц, 1H).
Пример получения 38
Синтез ди-трет-бутил [(4aR*,7aS*)-7a-(5-амино-2-фторфенил)-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]имидодикарбоната
[Формула 60]

(1) Синтез ди-трет-бутил [(4aR*,7aS*)-7a-(2-фтор-5-нитрофенил)-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]имидодикарбоната
N,N-Диметиламинопиридин (721 мг) и ди-трет-бутилдикарбонат (1,03 г) добавляли к раствору [(4aR*,7aS*)-7a-(2-фтор-5-нитрофенил)-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]амина, полученного в примере получения 3-(6) (350 мг), в дихлорметане (10 мл) и смесь перемешивали при комнатной температуре в течение 12 часов. Реакционный раствор вливали в воду с последующим экстрагированием этилацетатом. Экстракт промывали насыщенным раствором соли и затем сушили над безводным сульфатом магния. Осушающий агент удаляли с последующим концентрированием при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (580 мг).
ESI-MS; m/z 496 [M++H].
(2) Синтез ди-трет-бутил [(4aR*,7aS*)-7a-(5-амино-2-фторфенил)-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]имидодикарбоната
Раствор диимидного соединения, полученного на предшествующей стадии (630 мг), и железа (945 мг) в этаноле (10 мл) и насыщенный раствор хлорида аммония (1 мл) перемешивали при 87°C в течение 30 минут. После охлаждения реакционного раствора до комнатной температуры реакционный раствор вливали в этилацетат и нерастворимое вещество удаляли фильтрованием. Фильтрат концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (380 мг).
ESI-MS; m/z 466 [M++H].
Пример получения 39
Синтез 5-циклопропилэтинилпиридин-2-карбоновой кислоты
[Формула 61]
(1) Синтез этил 5-циклопропилэтинилпиридин-2-карбоксилата
Циклопропилацетилен (73,7 мкл), йодид меди (3,16 мг) и тетракис(трифенилфосфин)палладий (9,59 мг) добавляли к раствору этил 5-бромпиридин-2-карбоксилата (95,5 мг) в диизопропиламине (2 мл) при комнатной температуре и смесь перемешивали при комнатной температуре в течение 17 часов и пяти минут. Реакционный раствор концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с NH-силикагелем с получением указанного в заголовке соединения (19,4 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 0,86-0,90 (м, 2H), 0,91-0,97 (м, 2H), 1,44 (т, J=7,2 Гц, 3H), 1,47-1,54 (м, 1H), 4,47 (кв, J=7,2 Гц, 2H), 7,77 (дд, J=8,0, 2,0 Гц, 1H), 8,04 (дд, J=8,0, 0,8 Гц, 1H), 8,70 (дд, J=2,0, 0,8 Гц, 1H).
(2) Синтез 5-циклопропилэтинилпиридин-2-карбоновой кислоты
5 M раствор гидроксида натрия (36,6 мкл) добавляли к раствору соединения, полученного на предшествующей стадии (19,4 мг), в этаноле (500 мкл) и смесь перемешивали при комнатной температуре в течение 40 минут. 5 M хлористо-водородную кислоту (36,6 мкл) добавляли к реакционному раствору с последующим экстрагированием этилацетатом. Органический слой сушили над безводным сульфатом магния и осушающий агент отфильтровывали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (18,7 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 0,87-0,92 (м, 2H), 0,94-1,00 (м, 2H), 1,48-1,55 (м, 1H), 7,86 (дд, J=8,0, 2,0 Гц, 1H), 8,13 (дд, J=8,0, 1,1 Гц, 1H), 8,56 (дд, J=2,0, 1,1 Гц, 1H).
Пример получения 40
Синтез 5-тиазол-2-илпиридин-2-карбоновой кислоты
[Формула 62]

(1) Синтез этил 5-тиазол-2-илпиридин-2-карбоксилата
2-Трибутилстаннилтиазол (325 мг) и бис(три-трет-бутилфосфин)палладий(0) (25 мг) добавляли к раствору этил 5-бромпиридин-2-карбоксилата (100 мг) в 1,4-диоксане (2 мл). После замены азотом смесь перемешивали при 100°C в течение восьми часов. Реакционный раствор доводили до комнатной температуры и выпаривали при пониженном давлении растворитель с получением указанного в заголовке соединения (10 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,47 (т, J=7,2 Гц, 3H), 4,52 (кв, J=7,2, 2H), 7,50 (д, J=3,2 Гц, 1H), 7,99 (д, J=3,2 Гц, 1H), 8,22 (дд, J=0,8, 8,4 Гц, 1H), 8,39 (дд, J=2,2, 8,4 Гц, 1H), 9,31 (дд, J=0,8, 2,2 Гц, 1H).
(2) Синтез 5-тиазол-2-илпиридин-2-карбоновой кислоты
5 M раствор гидроксида натрия (17,1 мкл) добавляли к раствору соединения, полученного на предшествующей стадии (10 мг), в этаноле (250 мкл) при комнатной температуре с последующим перемешиванием в течение 35 минут. 5 M хлористо-водородную кислоту (17,1 мкл) добавляли к реакционному раствору с последующим экстрагированием этилацетатом. Органический слой сушили над безводным сульфатом магния и осушающий агент отфильтровывали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (8,6 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 7,54 (д, J=3,2 Гц, 1H), 8,02 (д, J=3,2 Гц, 1H), 8,30-8,34 (м, 1H), 8,46-8,51 (м, 1H), 9,22-9,24 (м, 1H).
Пример получения 41
Синтез 5-циклопропилпиридин-2-карбоновой кислоты
[Формула 63]
(1) Синтез трет-бутил 5-циклопропилпиридин-2-карбоксилата
Циклопропилбороновую кислоту (43,2 мг), трициклогексилфосфин (10,9 мг), ацетат палладия (4,34 мг) и фосфат калия (288 мг) добавляли к смешанному раствору трет-бутил 5-бромпиридин-2-карбоксилата (100 мг) в толуоле (2 мл) и воде (100 мкл) и смесь перемешивали при 100°C в течение девяти часов и 30 минут. После доведения до комнатной температуры к реакционному раствору добавляли воду. После экстрагирования этилацетатом органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (11,6 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 0,76-0,82 (м, 2H), 1,08-1,14 (м, 2H), 1,63 (с, 9H), 1,91-2,00 (м, 1H), 7,36 (дд, J=8,4, 2,4 Гц, 1H), 7,93 (д, J=8,4 Гц, 1H), 8,51 (д, J=2,4 Гц, 1H).
(2) Синтез 5-циклопропилпиридин-2-карбоновой кислоты
Смешанному раствору соединения, полученного на предшествующей стадии (11,6 мг), в трифторуксусной кислоте (333 мкл) и дихлорметане (666 мкл) давали отстояться при комнатной температуре в течение двух часов и 20 минут. Реакционный раствор концентрировали при пониженном давлении с получением указанного в заголовке соединения (16,6 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 0,95-1,03 (м, 2H), 1,31-1,41 (м, 2H), 2,08-2,20 (м, 1H), 7,94 (уш.д, J=8,0 Гц, 1H), 8,33 (уш.д, J=8,0 Гц, 1H), 8,65 (уш.с, 1H).
Пример получения 42
Синтез 5-метилсульфанилпиразин-2-карбоновой кислоты
[Формула 64]

(1) Синтез метил 5-метилсульфанилпиразин-2-карбоксилата
Метантиолат натрия (44,6 мг) добавляли к раствору метил 5-хлорпиразин-2-карбоксилата (87 мг) в гексаметилфосфортриамиде (1 мл) при комнатной температуре с последующим перемешиванием в течение 13 часов и 30 минут. К реакционному раствору добавляли воду с последующим экстрагированием этилацетатом. Органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с NH-силикагелем с получением указанного в заголовке соединения (6,8 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,63 (с, 3H), 4,02 (с, 3H), 8,53 (д, J=1,4 Гц, 1H), 9,07 (д, J=1,4 Гц, 1H).
(2) Синтез 5-метилсульфанилпиразин-2-карбоновой кислоты
Триметилсиланолат калия (6,15 мг) добавляли к раствору соединения, полученного на предшествующей стадии (6,8 мг), в тетрагидрофуране (500 мкл) при комнатной температуре с последующим перемешиванием в течение одного часа. К реакционному раствору добавляли воду и этилацетат и отделяли водный слой. К водному слою добавляли 1 M хлористо-водородную кислоту с последующим экстрагированием этилацетатом. Органический слой сушили над безводным сульфатом магния и осушающий агент отфильтровывали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (6,6 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,67 (с, 3H), 8,49 (уш.с, 1H), 9,17 (уш.с, 1H).
Пример получения 43
Синтез 5-(3-метоксипропин-1-ил)пиридин-2-карбоновой кислоты
[Формула 65]

(1) Синтез метил 5-(3-метоксипропин-1-ил)пиридин-2-карбоксилата
Хлорид бис(трифенилфосфин)палладия(II) (82,4 мг), йодид меди (22,3 мг), метилпропаргиловый простой эфир (828 мкл) и триэтиламин (1,9 мл) добавляли к раствору метил 5-бром-пиридин-2-карбоксилата (423 мг) в тетрагидрофуране (10,6 мл) и смесь перемешивали при комнатной температуре в течение 19 часов и 50 минут. Реакционный раствор концентрировали при пониженном давлении и к остатку добавляли воду. После экстрагирования этилацетатом органический слой промывали насыщенным раствором соли и выпаривали при пониженном давлении растворитель. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (88,1 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 3,47 (с, 3H), 4,02 (с, 3H), 4,36 (с, 2H), 7,88 (дд, J=8,0, 2,0 Гц, 1H), 8,10 (дд, J=8,0, 0,8 Гц, 1H), 8,78 (дд, J=2,0, 0,8 Гц, 1H).
(2) Синтез 5-(3-метоксипропин-1-ил)пиридин-2-карбоновой кислоты
Триметилсилилсиланолат калия (26,9 мг) добавляли к раствору соединения, полученного на предшествующей стадии (33 мг), в тетрагидрофуране (1 мл) и смесь перемешивали при комнатной температуре в течение одного часа и 20 минут. Реакционный раствор концентрировали при пониженном давлении. К остатку добавляли воду и диэтиловый эфир и отделяли водный слой. К водному слою добавляли 1 M хлористо-водородную кислоту с последующим экстрагированием этилацетатом. Органический слой сушили над безводным сульфатом магния и осушающий агент отфильтровывали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (25,4 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 3,48 (с, 3H), 4,37 (с, 2H), 7,97 (дд, J=8,0, 1,8 Гц, 1H), 8,19 (дд, J=8,0, 0,8 Гц, 1H), 8,66 (дд, J=1,8, 0,8 Гц, 1H).
Пример получения 44
Синтез трет-бутил (-)-[(4aS*,5S*,8aS*)-8a-(5-амино-2-фторфенил)-5-фторметил-4a,7,8,8a-тетрагидро-4H,5H-6-окса-3-тиа-1-азанафталин-2-ил]карбамата
[Формула 66]

(1) Синтез 1-бензилокси-3-бутен-2-ола
2,64 M раствор н-бутиллития в гексане (35,7 мл) добавляли к раствору йодида триметилсульфония (19,9 г) в тетрагидрофуране (300 мл) при -25°C. Смесь перемешивали при той же температуре в течение 30 минут. Бензилглицидиловый эфир (5,00 мл) добавляли к реакционному раствору при той же температуре и затем смесь нагревали до комнатной температуры на протяжении двух часов и 50 минут. К реакционному раствору добавляли воду с последующим экстрагированием диэтиловым эфиром. Органический слой сушили над безводным сульфатом магния. Осушающий агент отфильтровывали и фильтрат концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (6,98 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,44 (д, J=3,6 Гц, 1H), 3,38 (дд, J=9,6, 8,0 Гц, 1H), 3,55 (дд, J=9,6, 3,0 Гц, 1H), 4,32-4,40 (м, 1H), 4,58 (с, 2H), 5,18-5,23 (м, 1H), 5,33-5,40 (м, 1H), 5,79-5,89 (м, 1H), 7,28-7,40 (м, 5H).
В данном примере получения 44-(2)-(8) синтез осуществляли в соответствии с примером получения 22-(1)-(5). Но вместо диэтилацеталя бромацетальдегида использовали диметилацеталь 3-бромпропиональдегида.
В данном примере получения 44-(9)-(11) синтез осуществляли в соответствии с примером получения 19-(8)-(10).
(12) Синтез [(4aS*,5S*,8aS*)-2-амино-8a-(2-фтор-5-нитрофенил)-4a,7,8,8a-тетрагидро-4H,5H-6-окса-3-тиа-1-азанафталин-5-ил]метанола
Дымящую азотную кислоту (121 мкл) добавляли к смешанному раствору соединения, полученного на предшествующей стадии (720 мг), в трифторуксусной кислоте (12 мл) и серной кислоте (6 мл) при 0°C с последующим перемешиванием в течение одного часа. Реакционный раствор выливали на лед и 2 M раствор гидроксида натрия добавляли при 0°C. После экстрагирования этилацетатом органический слой сушили над безводным сульфатом магния. Осушающий агент отфильтровывали и фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (908 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,68-1,77 (м, 1H), 2,56-2,67 (м, 1H), 2,73-2,79 (м, 2H), 2,90-3,00 (м, 1H), 3,67-3,75 (м, 1H), 3,77-3,92 (м, 3H), 3,93-4,00 (м, 1H), 7,19-7,26 (м, 1H), 8,16-8,22 (м, 1H), 8,24-8,28 (м, 1H).
(13) Синтез трет-бутил [(4aS*,5S*,8aS*)-8a-(2-фтор-5-нитрофенил)-5-гидроксиметил-4a,7,8,8a-тетрагидро-4H,5H-6-окса-3-тиа-1-азанафталин-2-ил]карбамата
Ди-трет-бутилдикарбонат (1,16 г) и триэтиламин (1,48 мл) добавляли к раствору соединения, полученного на предшествующей стадии (908 мг), в тетрагидрофуране (30 мл) и смесь перемешивали при комнатной температуре в течение пяти часов и 30 минут. Реакционный раствор концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (454 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,54 (с, 9H), 1,63-1,71 (м, 1H), 2,55-2,77 (м, 3H), 2,97-3,08 (м, 1H), 3,67-4,02 (м, 5H), 7,21-7,27 (м, 1H), 8,14-8,24 (м, 2H).
(14) Синтез трет-бутил [(4aS*,5S*,8aS*)-5-фторметил-8a-(2-фтор-5-нитрофенил)-4a,7,8,8a-тетрагидро-4H,5H-6-окса-3-тиа-1-азанафталин-2-ил]карбамата
Трифторид [бис(2-метоксиэтил)амино]серы (500 мкл) добавляли к раствору соединения, полученного на предшествующей стадии (398 мг), в дихлорметане (20 мл) при -78°C и смесь перемешивали при той же температуре в течение 30 минут. После этого реакционный раствор перемешивали при 0°C в течение 30 минут и при комнатной температуре в течение четырех часов. К реакционному раствору добавляли насыщенный раствор бикарбоната натрия с последующим экстрагированием хлороформом. Органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (414 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,55 (с, 9H), 1,62-1,71 (м, 1H), 2,56-2,80 (м, 3H), 3,05-3,16 (м, 1H), 3,64-4,03 (м, 3H), 4,55-4,75 (м, 2H), 7,22-7,28 (м, 1H), 8,15-8,24 (м, 2H).
В данном примере получения 44-(15) синтез осуществляли в соответствии с примером получения 20-(3).
(16) Синтез трет-бутил (-)-[(4aS*,5S*,8aS*)-8a-(5-амино-2-фторфенил)-5-фторметил-4a,7,8,8a-тетрагидро-4H,5H-6-окса-3-тиа-1-азанафталин-2-ил]карбамата
Соединение, полученное на предшествующей стадии (33 мг), оптически разделяли, используя CHIRALPAK™ AD-H производства Daicel Chemical Industries, Ltd. (2 см × 25 см, подвижная фаза: гексан:этанол = 7:3, скорость потока: 8 мл/мин), и компонент, имеющий время удерживания от 20 до 27 минут, собирали. Данную операцию повторяли с получением указанного в заголовке соединения (174 мг; >99% ee) из 364 мг рацемата.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,53 (с, 9H), 1,58-1,66 (м, 1H), 2,62-2,69 (м, 1H), 2,75-2,86 (м, 1H), 2,89-2,96 (м, 1H), 3,08-3,15 (м, 1H), 3,66 (уш.с, 2H), 3,78-4,04 (м, 3H), 4,65 (дд, J=47,6, 2,8 Гц, 2H), 6,52-6,61 (м, 2H), 6,85-6,93 (м, 1H).
Пример получения 45
Синтез трет-бутил [(4aS*,8aS*)-8a-(5-амино-2-трифторметоксифенил)-4a,7,8,8a-тетрагидро-4H,5H-6-окса-3-тиа-1-азанафталин-2-ил]карбамата
[Формула 67]

В данном примере получения 45 в качестве исходного вещества использовали соединение, полученное в примере получения 8-(2).
В данном примере получения 45-(1)-(3) синтез осуществляли в соответствии с примером получения 22-(3)-(5). Но вместо 2-бромфторбензола использовали 1-бром-2-трифторметоксибензол.
(4) Синтез N-[(4aS*,8aS*)-8a-(2-трифторметоксифенил)-4a,7,8,8a-тетрагидро-4H,5H-6-окса-3-тиа-1-азанафталин-2-ил]бензамида
Тетрабромид углерода (542 мг) и трифенилфосфин (429 мг) добавляли к раствору соединения, полученного на предшествующей стадии (286 мг), в дихлорметане (6,41 мл) при комнатной температуре с последующим перемешиванием в течение четырех часов. Реакционный раствор концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (52,4 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,65-1,75 (м, 1H), 2,55-2,64 (м, 1H), 2,77-2,95 (м, 2H), 3,25-3,35 (м, 1H), 3,80-4,03 (м, 4H), 7,27-7,55 (м, 7H), 8,22-8,27 (м, 2H).
(5)-(8) Синтез трет-бутил [(4aS*,8aS*)-8a-(5-амино-2-трифторметоксифенил)-4a,7,8,8a-тетрагидро-4H,5H-6-окса-3-тиа-1-азанафталин-2-ил]карбамата
Указанное в заголовке соединение получали синтезом в данном примере получения 45-(5) в соответствии с примером получения 19-(9) и синтезом в данном примере получения 45-(6), (7) и (8) в соответствии с примером получения 22-(8), (9) и (10).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,54 (с, 9H), 1,54-1,62 (м, 1H), 2,45-2,53 (м, 1H), 2,67-2,80 (м, 1H), 2,83-2,92 (м, 1H), 3,09-3,18 (м, 1H), 3,69-3,96 (м, 6H), 6,58-6,64 (м, 2H), 7,09-7,14(м, 1H).
Пример получения 46
Синтез трет-бутил (-)-[(6S*,7S*,7aS*)-7a-(5-амино-2-фторфенил)-7-метил-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил]карбамата
[Формула 68]

(1) Синтез 1,1-диметоксипропан-2-ола
Боргидрид натрия добавляли к смешанному раствору диметилацеталя пировиноградного альдегида (10 мл) в метаноле (50 мл) и тетрагидрофуране (50 мл) при 0°C с последующим перемешиванием в течение 10 минут. Реакционный раствор нагревали до комнатной температуры и перемешивали в течение одного часа и 20 минут. К реакционному раствору добавляли насыщенный раствор хлорида аммония. После экстрагирования диэтиловым эфиром органический слой сушили над безводным сульфатом магния. Осушающий агент отфильтровывали и фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (9,86 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,20 (д, J=6,4 Гц, 3H), 2,17 (уш.д, J=3,2 Гц, 1H), 3,44 (с, 3H), 3,46 (с, 3H), 3,72-3,80 (м, 1H), 4,08 (д, J=6,4 Гц, 1H).
(2) Синтез 3-(2,2-диметокси-1-метилэтокси)пропена
60% Гидрид натрия (992 мг) добавляли к раствору 1,1-диметоксипропан-2-ола (2,49 г) в диметилформамиде (50 мл) при 0°C с последующим перемешиванием в течение 15 минут. Аллилбромид (1,96 мл) добавляли при той же температуре с последующим перемешиванием в течение 15 минут. К реакционному раствору добавляли лед с последующим экстрагированием этилацетатом. Органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (3,46 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,16 (д, J=6,5 Гц, 3H), 3,43 (с, 3H), 3,45 (с, 3H), 3,48-3,54 (м, 1H), 4,03-4,14 (м, 2H), 4,18 (д, J=5,2 Гц, 1H), 5,14-5,19 (м, 1H), 5,25-5,32 (м, 1H), 5,87-5,98 (м, 1H).
В данном примере получения 46-(3) и (4) синтез осуществляли в соответствии с примером получения 24-(3).
В данном примере получения 46-(5)-(9) синтез осуществляли в соответствии с примером получения 22-(2)-(6).
В данном примере получения 46-(10) синтез осуществляли в соответствии с примером получения 19-(9).
В данном примере получения 46-(11), (12) и (13) синтез осуществляли в соответствии с примером получения 22-(8), (9) и (10).
(14) Синтез трет-бутил (-)-[(6S*,7S*,7aS*)-7a-(5-амино-2-фторфенил)-7-метил-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил]карбамата
Соединение, полученное на предшествующей стадии (12 мг), оптически разделяли, используя CHIRALPAK™ AD-H производства Daicel Chemical Industries, Ltd. (2 см × 25 см, подвижная фаза: гексан:этанол = 8:2, скорость потока: 10 мл/мин), и компонент, имеющий время удерживания от 16 до 21 минут, собирали. Данную операцию повторяли с получением указанного в заголовке соединения (112 мг; >99% ee) из 240 мг рацемата.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 0,96 (д, J=6,0 Гц, 3H), 1,49 (с, 9H), 2,81-2,90 (м, 1H), 3,30-3,40 (м, 1H), 3,43-3,52 (м, 1H), 3,61 (уш.с, 2H), 4,10-4,19 (м, 1H), 4,20-4,38 (м, 2H), 6,56-6,64 (м, 2H), 6,87-6,94 (м, 1H).
Пример получения 47
Синтез трет-бутил (-)-[(6R*,7S*,7aS*)-7a-(5-амино-2-фторфенил)-7-метил-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил]карбамата
[Формула 69]
(1)-(8) Синтез трет-бутил (-)-[(6R*,7S*,7aS*)-7a-(5-амино-2-фторфенил)-7-метил-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил]карбамата
В данном примере получения в качестве исходного вещества использовали (3S*,3aS*,5R*)-6a-(2-фторфенил)-6-метилтетрагидрофуро[3,4-c]изоксазол, полученный в примере получения 46-(6).
Указанное в заголовке соединение получали синтезом в данном примере получения 47-(1)-(8) в соответствии с примером получения 46-(7)-(14).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,14 (уш.д, J=6,4 Гц, 3H), 1,52 (с, 9H), 2,62-2,70 (м, 1H), 3,03-3,15 (м, 1H), 3,46-3,60 (м, 1H), 3,64 (уш.с, 2H), 4,10-4,23 (м, 2H), 4,56-4,65 (м, 1H), 6,55-6,62 (м, 1H), 6,63-6,67 (м, 1H), 6,83-6,90 (м, 1H).
Пример получения 48
Синтез трет-бутил (-)-[(4aS*,5R*,8aS*)-8a-(5-амино-2-фторфенил)-5-метил-4a,7,8,8a-тетрагидро-4H,5H-6-окса-3-тиа-1-азанафталин-2-ил]карбамата
[Формула 70]
В данном примере получения 48-(1)-(7) синтез осуществляли в соответствии с примером получения 22-(1)-(5). Но вместо диэтилацеталя бромацетальдегида использовали диметилацеталь 3-бромпропиональдегида.
В данном примере получения 48-(8) и (9) синтез осуществляли в соответствии с примером получения 19-(8) и (9). В данном примере получения 48-(10), (11) и (12) синтез осуществляли в соответствии с примером получения 22-(8), (9) и (10).
(13) Синтез трет-бутил (-)-[(4aS*,5R*,8aS*)-8a-(5-амино-2-фторфенил)-5-метил-4a,7,8,8a-тетрагидро-4H,5H-6-окса-3-тиа-1-азанафталин-2-ил]карбамата
Соединение, полученное на предшествующей стадии (44 мг), оптически разделяли, используя CHIRALPAK™ OJ-H производства Daicel Chemical Industries, Ltd. (2 см × 25 см, подвижная фаза: гексан:этанол = 2:8, скорость потока: 10 мл/мин), и компонент, имеющий время удерживания от 14 до 28 минут, собирали. Данную операцию повторяли с получением указанного в заголовке соединения (223 мг; >99% ee) из 700 мг рацемата.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,32 (д, J=5,2 Гц, 3H), 1,53 (с, 9H), 1,56-1,65 (м, 1H), 2,56-2,70 (м, 2H), 2,73-2,91 (м, 2H), 3,66 (уш.с, 2H), 3,75-3,97 (м, 3H), 6,53-6,59 (м, 2H), 6,84-6,91 (м, 1H).
Пример получения 49
Синтез 5-дифторметоксипиридин-2-карбоновой кислоты
[Формула 71]
(1) Синтез метил 5-дифторметоксипиридин-2-карбоксилата
Карбонат цезия (7,45 г) и 2-хлор-2,2-дифторацетофенон (5,75 г) добавляли к раствору метил 5-гидроксипиридин-2-карбоксилата (2,5 г) в ДМФА и смесь перемешивали при 100°C в течение трех часов. Реакционный раствор доводили до комнатной температуры. Добавляли водный хлорид аммония и этилацетат и отделяли органический слой. Органический слой промывали насыщенным водным раствором хлорида натрия и сушили над безводным сульфатом магния. Органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (760 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 4,02 (с, 3H), 6,64 (т, J=72,0 Гц, 1H), 7,62 (дд, J=2,8, 8,8 Гц, 1H), 8,18 (д, J=9,2 Гц, 1H), 8,58 (д, J=2,8 Гц, 1H).
(2) Синтез 5-дифторметоксипиридин-2-карбоновой кислоты
К раствору метил 5-дифторметоксипиридин-2-карбоксилата, полученного в примере получения 49-(1) (760 мг), в метаноле (15 мл) добавляли 2 н. раствор гидроксида натрия (3,74 мл) и смесь перемешивали при комнатной температуре в течение 30 минут. Реакционный раствор подкисляли хлористо-водородной кислотой. К реакционному раствору добавляли насыщенный водный раствор хлорида натрия и этилацетат и отделяли органический слой. Органический слой сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении с получением указанного в заголовке соединения (482 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 6,67 (т, J=71,6 Гц, 1H), 7,72 (дд, J=2,0, 8,4 Гц, 1H), 8,28 (д, J=8,8 Гц, 1H), 8,50 (д, J=2,0 Гц, 1H).
Пример получения 50
Синтез 5-(2,2,2-трифторэтокси)пиразин-2-карбоновой кислоты
[Формула 72]

(1) Синтез метил 5-(2,2,2-трифторэтокси)пиразин-2-карбоксилата
Карбонат цезия (2,96 г) и 2,2,2-трифторэтилтрифторметансульфонат (1,57 г) добавляли к раствору метил 5-гидроксипиразин-2-карбоксилата (700 мг) в ДМФА (20 мл) и смесь перемешивали при комнатной температуре в течение 20 часов. К реакционному раствору добавляли водный хлорид аммония и этилацетат и отделяли органический слой. Органический слой промывали насыщенным водным раствором хлорида натрия и сушили над безводным сульфатом магния. Органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на силикагеле с получением указанного в заголовке соединения (197 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 4,02 (с, 3H), 4,85 (кв, J=8,0 Гц, 2H), 8,44 (д, J=1,2 Гц, 1H), 8,88 (д, J=1,2 Гц, 1H).
(2) Синтез 5-(2,2,2-трифторэтокси)пиразин-2-карбоновой кислоты
К раствору метил 5-(2,2,2-трифторэтокси)пиразин-2-карбоксилата, полученного в примере получения 50-(1) (197 мг), в ТГФ (5 мл) добавляли 5 н. раствор гидроксида натрия (3 мл) и этанол (3 мл) и смесь кипятили с обратным холодильником в течение 15 минут. После доведения до комнатной температуры добавляли воду и этилацетат и водный слой отделяли. Водный слой доводили до pH 1 хлористо-водородной кислотой и добавляли к водному слою этилацетат. Органический слой отделяли и сушили над безводным сульфатом магния. Органический слой концентрировали при пониженном давлении с получением указанного в заголовке соединения (87 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 4,88 (кв, J=8,4 Гц, 2H), 8,37 (д, J=1,2 Гц, 1H), 8,99 (д, J=1,2 Гц, 1H).
Пример получения 51
Синтез 5-(2,2-дифторэтокси)пиразин-2-карбоновой кислоты
[Формула 73]

(1) Синтез метил 5-(2,2-дифторэтокси)пиразин-2-карбоксилата
Карбонат цезия (2,12 г) и 2-бром-1,1-дифторэтан (939 мг) добавляли к раствору метил 5-гидроксипиразин-2-карбоксилата (500 мг) в ДМФА (20 мл) и смесь перемешивали при 80°C в течение четырех часов. Реакционный раствор доводили до комнатной температуры. Добавляли насыщенный водный раствор хлорида натрия и этилацетат и отделяли органический слой. Органический слой сушили над безводным сульфатом магния. Органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (145 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 4,02 (с, 3H), 4,64 (дт, J=4,0, 13,2 Гц, 2H), 6,15 (тт, J=4,0, 54,8 Гц, 1H), 8,39 (д, J=1,2 Гц, 1H), 8,88 (д, J=1,2 Гц, 1H).
(2) Синтез 5-(2,2-дифторэтокси)пиразин-2-карбоновой кислоты
К раствору метил 5-(2,2-дифторэтокси)пиразин-2-карбоксилата, полученного в примере получения 51-(1) (145 мг), в этаноле (4 мл) добавляли 5 н. раствор гидроксида натрия (266 мкл) и смесь перемешивали при комнатной температуре в течение одного часа. К реакционному раствору добавляли 5 н. хлористо-водородную кислоту для подкисления раствора. К реакционному раствору добавляли этилацетат и насыщенный раствор соли и отделяли органический слой. Органический слой сушили над безводным сульфатом магния. Органический слой концентрировали при пониженном давлении с получением указанного в заголовке соединения (92 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 4,68 (дт, J=4,0, 13,2 Гц, 2H), 6,16 (тт, J=4,0, 54,8 Гц, 1H), 8,34 (с, 1H), 8,99 (д, J=0,8 Гц, 1H).
Пример получения 52
Синтез 5-(2,2-дифторэтокси)пиридин-2-карбоновой кислоты
[Формула 74]

Карбонат цезия (423 мг) и 2-бром-1,1-дифторэтан (189 мг) добавляли к раствору метил 5-гидроксипиридин-2-карбоксилата (100 мг) в ДМФА (4 мл) и смесь перемешивали при комнатной температуре в течение 20 часов. К реакционному раствору добавляли насыщенный водный раствор хлорида натрия и этилацетат и отделяли органический слой. Органический слой сушили над безводным сульфатом магния. Органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке промежуточного соединения. К раствору полученного промежуточного соединения в этаноле (5 мл) добавляли 5 н. раствор гидроксида натрия (262 мкл). Реакционный раствор перемешивали при комнатной температуре в течение 30 минут. Реакционный раствор подкисляли 5 н. хлористо-водородной кислотой (1 мл). К реакционному раствору добавляли насыщенный водный раствор хлорида натрия и этилацетат и отделяли органический слой. Органический слой сушили над безводным сульфатом магния. Органический слой концентрировали при пониженном давлении с получением указанного в заголовке соединения (22,4 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 4,33 (дт, J=4,0, 12,8 Гц, 2H), 6,15 (тт, J=4,0, 54,8 Гц, 1H), 7,42 (дд, J=2,8, 8,4 Гц, 1H), 8,22 (д, J=8,8 Гц, 1H), 8,34 (с, 1H).
Пример получения 53
Синтез 5-(2-фторэтокси)пиразин-2-карбоновой кислоты
[Формула 75]
(1) Синтез метил 5-(2-фторэтокси)пиразин-2-карбоксилата
Карбонат цезия (6,34 г) и 1-йод-2-фторэтан (2,26 г) добавляли к раствору метил 5-гидроксипиразин-2-карбоксилата (1 г) в ДМФА (30 мл) и смесь перемешивали при комнатной температуре в течение 20 часов. К реакционному раствору добавляли водный хлорид аммония и этилацетат и отделяли органический слой. Органический слой промывали насыщенным водным раствором хлорида натрия и затем сушили над безводным сульфатом магния. Органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (200 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 4,00 (с, 3H), 4,63-4,86 (м, 4H), 8,36 (д, J=1,2 Гц, 1H), 8,87 (д, J=1,6 Гц, 1H).
(2) Синтез 5-(2-фторэтокси)пиразин-2-карбоновой кислоты
К раствору метил 5-(2-фторэтокси)пиразин-2-карбоксилата, полученного в примере получения 53-(1) (200 мг), в этаноле (4 мл) добавляли 5 н. раствор гидроксида натрия (400 мкл). Добавляли воду до тех пор, пока реакционный раствор не становился завершенным раствором, с последующим перемешиванием при комнатной температуре в течение 10 минут. Реакционный раствор подкисляли 5 н. хлористо-водородной кислотой. К реакционному раствору добавляли насыщенный водный раствор хлорида натрия и этилацетат и отделяли органический слой. Органический слой сушили над безводным сульфатом магния. Органический слой концентрировали при пониженном давлении с получением указанного в заголовке соединения (150 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 4,67-4,87 (м, 4H), 8,27 (д, J=1,2 Гц, 1H), 8,97 (д, J=1,2 Гц, 1H).
Пример получения 54
Синтез (±)-(4aR*,7aS*)-7a-(5-бром-2-фторфенил)-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-иламина
[Формула 76]

(1)-(4) Синтез (±)-(4aR*,7aS*)-7a-(5-бром-2-фторфенил)-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-иламина
Указанное в заголовке соединение (4,03 г) получали из соединения, полученного в примере получения 3-(1) (10 г), в соответствии с примером получения 3, используя 5-бром-2-фторфенилбороновую кислоту на указанной выше стадии (2) и используя боргидрид лития при кипячении с обратным холодильником вместо алюмогидрида лития на стадии (3).
ESI-MS; m/z 331 [M++H].
Пример получения 55
Синтез (±)-ди-трет-бутил [(4aR*,7aS*)-7a-(3-амино-5-фторфенил)-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]имидодикарбоната
[Формула 77]
(1)-(2) Синтез этил 2-(3-бром-5-фторфенил)циклопент-1-енкарбоксилата
Указанное в заголовке соединение (12,9 г) получали из этил 2-оксоциклопентанкарбоксилата (6,8 г) в соответствии с примером получения 3.
ESI-MS; m/z 313 [M++H].
(3) Синтез 2-(3-бром-5-фторфенил)циклопент-1-енкарбоновой кислоты
К раствору соединения, полученного в примере получения 55-(2) (12,9 г), в этаноле (130 мл) добавляли 5 н. раствор гидроксида натрия (16,5 мл) и смесь перемешивали при комнатной температуре в течение 16 часов. Этанол выпаривали при пониженном давлении. К остатку добавляли воду (100 мл) и эфир (150 мл) и отделяли водный слой. Водный слой подкисляли 5 н. хлористо-водородной кислотой и добавляли к водному слою этилацетат. Органический слой отделяли и сушили над безводным сульфатом магния. Органический слой концентрировали при пониженном давлении. Полученное твердое вещество промывали гептаном (150 мл) с получением указанного в заголовке соединения (10,58 г).
1H-ЯМР (CDCl3) δ (м.д.): 1,96-2,05 (м, 2H), 2,80-2,85 (м, 4H), 6,96-7,00 (м, 1H), 7,15-7,18 (м, 1H), 7,22-7,24 (м, 1H).
(4) Синтез [2-(3-бром-5-фторфенил)циклопент-1-енил]метанола
Изобутилхлорформиат (5,08 мл) добавляли по каплям к раствору соединения, полученного в примере получения 55-(3) (10,6 г), и триэтиламина (5,41 мл) в тетрагидрофуране (230 мл) в атмосфере азота при -20°C. Реакционный раствор перемешивали при той же температуре в течение 30 минут и затем полученное нерастворимое вещество отделяли фильтрованием через целит. Фильтрат добавляли по каплям к раствору боргидрида натрия в воде (2,81 г/162 мл) при температуре от 0 до -10°C. Смесь перемешивали при той же температуре в течение двух часов и затем нагревали до комнатной температуры. Реакционный раствор перемешивали при комнатной температуре в течение 3 часов. К реакционному раствору добавляли этилацетат и насыщенный водный раствор хлорида натрия и отделяли органический слой. Органический слой сушили над безводным сульфатом магния. Органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (9,3 г).
1H-ЯМР (CDCl3) δ (м.д.): 1,93-2,05 (м, 2H), 2,66-2,76 (м, 4H), 4,30 (д, J=2,8 Гц, 2H), 6,89-6,92 (м, 1H), 7,11-7,19 (м, 2H).
(5)-(6) Синтез (±)-(4aR*,7aS*)-7a-(3-бром-5-фторфенил)-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-иламина
Указанное в заголовке соединение (12,2 г) получали из соединения, полученного в примере получения 55-(4) (9,3 г), в соответствии с примером получения 3.
ESI-MS; m/z 331 [M++H].
(7) Синтез трет-бутил (±)-[(4aR*,7aS*)-7a-(3-бром-5-фторфенил)-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]карбамата
Триэтиламин (1,11 мл) и ди-трет-бутилдикарбонат (1,16 г) добавляли к раствору соединения, полученного в примере получения 55-(6) (1 г), в тетрагидрофуране (30 мл) и смесь перемешивали при комнатной температуре в течение 14 часов. К реакционному раствору добавляли воду и этилацетат и отделяли органический слой. Органический слой промывали насыщенным водным раствором хлорида натрия и сушили над безводным сульфатом магния. Органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (1,03 г).
ESI-MS; m/z 431 [M++H].
(8) Синтез (±)-ди-трет-бутил [(4aR*,7aS*)-7a-(3-бром-5-фторфенил)-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]имидодикарбоната
4-Диметиламинопиридин (880 мг) и ди-трет-бутилдикарбонат (1,05 г) добавляли к раствору соединения, полученного в примере получения 55-(7) (1,03 г), в ацетонитриле (20 мл). Реакционный раствор перемешивали при комнатной температуре в течение двух часов. К реакционному раствору добавляли этилацетат и насыщенный водный раствор хлорида натрия и отделяли органический слой. Органический слой снова промывали насыщенным раствором соли и сушили над безводным сульфатом магния. Органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (1,12 г).
ESI-MS; m/z 531 [M++H].
(9) Синтез (±)-ди-трет-бутил {(4aR*,7aS*)-7a-[3-фтор-5-(4,4,5,5-тетраметил-1,3,2-диоксаборан-2-ил)фенил]-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил}имидодикарбоната
Раствор соединения, полученного в примере получения 55-(8) (635 мг), бис(пинаколато)диборана (3,04 г), ацетата калия (471 мг) и 1,1'-бис(дифенилфосфино)ферроцендихлорпалладия(II) (87,5 мг) в ДМФА (12 мл) перемешивали в атмосфере азота при 80°C в течение пяти часов. Реакционный раствор доводили до комнатной температуры. К реакционному раствору добавляли этилацетат и насыщенный водный раствор хлорида натрия и отделяли органический слой. Органический слой снова промывали насыщенным водным раствором хлорида натрия. Органический слой сушили над безводным сульфатом магния. Органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (692 мг).
ESI-MS; m/z 577 [M++H].
(10) Синтез (±)-ди-трет-бутил [(4aR*,7aS*)-7a-(3-азидо-5-фторфенил)-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]имидодикарбоната
Раствор соединения, полученного в примере получения 55-(9) (695 мг), азида натрия (118 мг) и ацетата меди(II) (44 мг) в метаноле (15 мл) перемешивали при комнатной температуре в течение 72 часов. К реакционному раствору добавляли воду и этилацетат и отделяли органический слой. Органический слой дважды промывали насыщенным водным раствором хлорида натрия. Органический слой сушили над безводным сульфатом магния. Органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (370 мг).
ESI-MS; m/z 492 [M++H].
(11) Синтез (±)-ди-трет-бутил [(4aR*,7aS*)-7a-(3-амино-5-фторфенил)-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]имидодикарбоната
Воду (2 мл) и трифенилфосфин (257 мг) добавляли к раствору соединения, полученного в примере получения 55-(10) (370 мг), в тетрагидрофуране (8 мл) и смесь перемешивали при 60°C в течение трех часов. Реакционный раствор далее кипятили с обратным холодильником в течение 40 часов. Реакционный раствор доводили до комнатной температуры. К реакционному раствору добавляли этилацетат и насыщенный водный раствор хлорида натрия и отделяли органический слой. Органический слой сушили над безводным сульфатом магния. Органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (103 мг).
ESI-MS; m/z 466 [M++H].
Пример получения 56
Синтез трет-бутил (-)-[(4aS*,8aR*)-8a-(5-амино-2-фторфенил)-4,4a,7,8,8a-тетрагидро-4H,5H-6-окса-3-тиа-1-азанафталин-2-ил]карбамата
[Формула 78]

(1) Синтез 4-аллилокси-2-(2-фторфенил)бутиронитрила
трет-Бутоксид калия (9,93 г) добавляли к раствору 2-фторфенилацетонитрила (10 г), 2-аллилоксиэтилового эфира толуол-4-сульфоновой кислоты (19 г) и 18-краун-6 (3,91 г) в тетрагидрофуране (400 мл) при охлаждении льдом. Реакционный раствор перемешивали при той же температуре в течение 10 минут. Реакционный раствор нагревали до комнатной температуры и дополнительно перемешивали в течение четырех часов. К реакционному раствору добавляли водный хлорид аммония и этилацетат и отделяли органический слой. Органический слой промывали насыщенным водным раствором хлорида натрия. Органический слой сушили над безводным сульфатом магния. Органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (10,9 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,18 (кв, J=6,4 Гц, 2H), 3,46-3,52 (м, 1H), 3,60-3,66 (м, 1H), 3,99 (д, J=6,4 Гц, 2H), 4,35 (т, J=7,2 Гц, 1H), 5,21-5,87 (м, 2H), 5,86-5,96 (м, 1H), 7,07-7,12 (м, 1H), 7,16-7,20 (м, 1H), 7,30-7,35 (м, 1H), 7,42-7,47 (м, 1H).
(2) Синтез 4-(2,3-дигидроксипропокси)-2-(2-фторфенил)бутиронитрила
Тетроксид осмия (2,5 масс.% раствор в трет-бутиловом спирте, 15,6 мл) добавляли к раствору соединения, полученного в примере получения 56-(1) (10,9 г), и 4-метилморфолин-4-оксида (8,75 г) в смеси ацетон/вода (2/1, 390 мл) при охлаждении льдом. Реакционный раствор нагревали до комнатной температуры и перемешивали в течение 15 часов. Бисульфит натрия (5,18 г) добавляли к реакционному раствору и смесь перемешивали при той же температуре в течение 20 минут. К реакционному раствору добавляли этилацетат и насыщенный водный раствор хлорида натрия и отделяли органический слой. Органический слой сушили над безводным сульфатом магния. Органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (10,5 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,99-2,02 (м, 1H), 2,17-2,22 (м, 2H), 2,53-2,57 (м, 1H), 3,50-3,59 (м, 3H), 3,61-3,76 (м, 3H), 3,87-3,91 (м, 1H), 4,32 (т, J=6,4 Гц, 1H), 7,08-7,13 (м, 1H), 7,18-7,23 (м, 1H), 7,32-7,38 (м, 1H), 7,45-7,50 (м, 1H).
(3) Синтез 4-[3-(трет-бутилдифенилсиланилокси)-2-гидроксипропокси]-2-(2-фторфенил)бутиронитрила
Имидазол (7,05 г) и трет-бутилдифенилхлорсилан (12,3 мл) добавляли к раствору соединения, полученного в примере получения 56-(2) (10,5 г), в ДМФА (125 мл) при охлаждении льдом. Реакционный раствор нагревали до комнатной температуры и перемешивали при комнатной температуре в течение 18 часов. К реакционному раствору добавляли насыщенный водный раствор хлорида натрия и этилацетат и отделяли органический слой. Органический слой снова промывали насыщенным водным раствором хлорида натрия. Органический слой сушили над безводным сульфатом магния и органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (16,4 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,07 (с, 9H), 2,11-2,18 (м, 2H), 2,47-2,49 (м, 1H), 3,45-3,55 (м, 3H), 3,59-3,64 (м, 1H), 3,69-3,71 (м, 2H), 3,88-3,91 (м, 1H), 4,21-4,26 (м, 1H), 7,05-7,10 (м, 1H), 7,14-7,19 (м, 1H), 7,29-7,35 (м, 1H), 7,36-7,45 (м, 7H), 7,64-7,68 (м, 4H).
(4) Синтез 1-(трет-бутилдифенилсиланилоксиметил)-2-[3-циано-3-(2-фторфенил)пропокси]этилтрифторметансульфоната
Раствор соединения, полученного в примере получения 56-(3) (16,4 г), и N,N-диизодиизопропилэтиламина (17,4 мл) в дихлорметане (330 мл) охлаждали до -78°C в атмосфере азота. Трифторметансульфоновый ангидрид (8,28 мл) добавляли по каплям к реакционному раствору при той же температуре. Реакционный раствор перемешивали в течение шести часов, постепенно нагревая до комнатной температуры. К реакционному раствору добавляли водный хлорид аммония и отделяли органический слой. Органический слой промывали насыщенным водным раствором хлорида натрия и сушили над безводным сульфатом магния. Органический слой концентрировали и остаток очищали колоночной хроматографией с получением указанного в заголовке соединения (15,1 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,07 (с, 9H), 2,12-2,17 (м, 2H), 3,48-3,53 (м, 1H), 3,62-3,80 (м, 3H), 3,85-3,87 (м, 2H), 4,23-4,28 (м, 1H), 5,01-5,04 (м, 1H), 7,06-7,11 (м, 1H), 7,15-7,19 (м, 1H), 7,29-7,34 (м, 1H), 7,37-7,48 (м, 6H), 7,52-7,67 (м, 4H), 7,67-7,73 (м, 1H).
(5) Синтез 3-(трет-бутилдифенилсиланилоксиметил)-4-(2-фторфенил)тетрагидропиран-4-карбонитрила
Трет-бутоксид калия (2,98 г) добавляли к раствору соединения, полученного в примере получения 56-(4) (15,1 г), и 18-краун-6 (1,28 г) в тетрагидрофуране (250 мл) при охлаждении льдом. Реакционный раствор перемешивали при той же температуре в течение одного часа. К реакционному раствору добавляли водный хлорид аммония и этилацетат и отделяли органический слой. Органический слой промывали насыщенным водным раствором хлорида натрия и сушили над безводным сульфатом магния. Органический слой концентрировали при пониженном давлении и остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (11,1 г).
ESI-MS; m/z 496 [M++Na].
(6) Синтез 4-(2-фторфенил)-3-гидроксиметилтетрагидропиран-4-карбонитрила
Тетрабутиламмонийфторид (1 M раствор в тетрагидрофуране, 46,9 мл) добавляли по каплям к раствору соединения, полученного в примере получения 56-(5) (11,1 г), в тетрагидрофуране (240 мл). Реакционный раствор перемешивали при комнатной температуре в течение 12 часов. Реакционный раствор концентрировали и к остатку добавляли этилацетат и насыщенный водный раствор хлорида натрия. Органический слой отделяли и сушили над безводным сульфатом магния. Органический слой концентрировали при пониженном давлении и остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (4,0 г).
ESI-MS; m/z 236 [M++H].
(7) Синтез (±)-(3R*,4S*)-4-(2-фторфенил)-3-метоксиметоксиметилтетрагидропиран-4-карбонитрила
N,N-Диизопропилэтиламин (14,8 мл) и хлорметилметиловый эфир (3,87 мл) добавляли к соединению, полученному в примере получения 56-(6) (4 г), в дихлорметане (100 мл). Реакционный раствор перемешивали при комнатной температуре в течение 16 часов. К реакционному раствору добавляли воду и хлороформ и отделяли органический слой. Органический слой сушили над безводным сульфатом магния. Органический слой концентрировали и остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (1,65 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,00 (д, J=11,6 Гц, 1H), 2,38-2,46 (м, 1H), 2,72 (д, J=8,8 Гц, 1H), 2,93 (дд, J=4,0, 9,6 Гц, 1H), 3,19 (с, 3H), 3,62 (т, J=9,6 Гц, 1H), 3,99-4,06 (м, 2H), 4,17 (д, J=12,0 Гц, 2H), 4,37-4,43 (м, 2H), 7,14-7,21 (м, 3H), 7,36-7,41 (м, 1H).
(8) Синтез (±)-(3R*,4S*)-4-(2-фторфенил)-3-метоксиметоксиметилтетрагидропиран-4-карбоксамида
Гидроксид калия (1,33 г) добавляли к раствору соединения, полученного в примере получения 56-(7) (1,65 г), в трет-бутиловым спирте (35 мл). Реакционный раствор кипятили с обратным холодильником в течение шести часов. Реакционный раствор охлаждали до комнатной температуры. Этилацетат и насыщенный водный раствор хлорида натрия добавляли к реакционному раствору и отделяли органический слой. Органический слой снова промывали насыщенным водным раствором хлорида натрия. Органический слой сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (1,37 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,18-2,32 (м, 2H), 2,86-2,90 (м, 1H), 3,02 (дд, J=3,2, 9,6 Гц, 1H), 3,23 (с, 3H), 3,58-3,72 (м, 2H), 3,99-4,15 (м, 3H), 4,44-4,48 (м, 2H), 5,30-5,55 (м, 2H), 7,04-7,10 (м, 1H), 7,16-7,20 (м, 1H), 7,29-7,35 (м, 2H).
(9) Синтез (±)-(3R*,4S*)-4-(2-фторфенил)-3-метоксиметоксиметилтетрагидропиран-4-иламина
[Бис(трифторацетокси)йод]бензол (2,36 г) добавляли к раствору соединения, полученного в примере получения 56-(8) (1,37 г), в смеси ацетонитрил/вода (35 мл/15 мл). Реакционный раствор перемешивали при комнатной температуре в течение 18 часов. Реакционный раствор концентрировали при пониженном давлении. К остатку добавляли 5 н. гидроксид натрия и хлороформ и отделяли органический слой. Органический слой сушили над безводным сульфатом магния. Органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (619 мг).
ESI-MS; m/z 270 [M++H].
(10) Синтез (±)-1-бензоил-3-[(3R*,4S*)-4-(2-фторфенил)-3-метоксиметоксиметилтетрагидропиран-4-ил]тиомочевины
Бензоилизоцианат (412 мг) добавляли к раствору соединения, полученного в примере получения 56-(9) (619 мг), в дихлорметане (15 мл). Реакционный раствор перемешивали при комнатной температуре в течение 15 часов. Реакционный раствор концентрировали и остаток очищали хроматографией на колонке с NH-силикагелем с получением указанного в заголовке соединения (956 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,24-2,33 (м, 1H), 2,58 (д, J=9,2 Гц, 1H), 2,99-3,03 (м, 1H), 3,21 (с, 3H), 3,55 (дд, J=1,6, 14,0 Гц, 1H), 3,71-3,84 (м, 2H), 3,98-4,08 (м, 2H), 4,19 (д, J=12,4 Гц, 1H), 4,41-4,45 (м, 2H), 6,99-7,06 (м, 1H), 7,14-7,18 (м, 1H), 7,26-7,38 (м, 2H), 7,52 (т, J=7,2 Гц, 2H), 7,63 (тт, J=2,0, 7,2 Гц, 1H), 7,84-7,87 (м, 2H), 8,76 (с, 1H), 11,7 (с, 1H).
(11) Синтез (±)-1-бензоил-3-[(3S*,4S*)-4-(2-фторфенил)-3-гидроксиметилтетрагидропиран-4-ил]тиомочевины
Концентрированную хлористо-водородную кислоту (1 мл) добавляли к раствору соединения, полученного в примере получения 56-(10) (956 мг), в метаноле (20 мл). Реакционный раствор кипятили с обратным холодильником в течение четырех часов. Реакционный раствор концентрировали при пониженном давлении. К остатку добавляли этилацетат и водный бикарбонат натрия и отделяли органический слой. Органический слой промывали насыщенным водным раствором хлорида натрия. Органический слой сушили над безводным сульфатом магния. Органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (465 мг).
ESI-MS; m/z 411 [M++Na].
(12)-(15) Синтез трет-бутил (±)-[(4aR*,8aS*)-8a-(5-амино-2-фторфенил)-4a,7,8,8a-тетрагидро-4H,5H-6-окса-3-тиа-1-азанафталин-2-ил]карбамата
Указанное в заголовке соединение (58 мг) получали из соединения, полученного в примере получения 56-(11) (465 мг), в соответствии с примером получения 9.
ESI-MS; m/z 382 [M++H].
(16) Синтез трет-бутил (-)-[(4aR*,8aS*)-8a-(5-амино-2-фторфенил)-4a,7,8,8a-тетрагидро-4H,5H-6-окса-3-тиа-1-азанафталин-2-ил]карбамата
Соединение, полученное в примере получения 56-(15) (19 мг), оптически разделяли, используя CHIRALPAK™ AD-H производства Daicel Chemical Industries, Ltd. (2 см × 25 см, подвижная фаза: гексан:этанол = 7:3, скорость потока: 10 мл/мин), и компонент, имеющий время удерживания от 15,3 до 18,3 минут, собирали. Данную операцию повторяли с получением указанного в заголовке соединения (23 мг; >99% ee) из 58 мг рацемата.
ESI-MS; m/z 382 [M++H].
Пример получения 57
Синтез трет-бутил (-)-[(4aR*,7S*,8aS*)-8a-(5-амино-2-фторфенил)-7-метокси-4a,5,6,7,8,8a-гидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата
[Формула 79]

(1) Синтез 1,1-диэтоксигепт-6-ен-3-ола
Раствор оксалилхлорида (4,07 мл, удельный вес: 1,455 г/см3) в дихлорметане (200 мл) охлаждали до -78°C в атмосфере азота. Затем медленно добавляли раствор ДМСО (6,62 мл, удельный вес: 1,101 г/см3) в дихлорметане (50 мл), так чтобы внутренняя температура не превышала -60°C. После перемешивания в течение 15 минут медленно добавляли раствор 3,3-диэтокси-1-пропанола в дихлорметане (50 мл), так чтобы внутренняя температура не превышала -65°C. После перемешивания еще в течение одного часа и 45 минут медленно добавляли TEA (25,9 мл). Смесь перемешивали после указанного добавления еще в течение 30 минут. После нагревания до комнатной температуры добавляли насыщенный раствор хлорида аммония с последующим перемешиванием. Водный слой отделяли и затем органический слой промывали насыщенным раствором хлорида аммония. Полученный органический слой сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении. Полученный остаток суспендировали в диэтиловом эфире и твердое вещество удаляли фильтрованием. Полученный фильтрат концентрировали при пониженном давлении. К полученному остатку добавляли ТГФ (60 мл) и смесь в достаточной мере охлаждали на ледяной бане в атмосфере азота. Раствор 3-бутенилмагнийбромида в ТГФ (0,5 M, 100 мл) добавляли к смеси, так чтобы внутренняя температура не превышала 10°C. По окончании добавления смесь перемешивали в течение 13 часов и 30 минут, постепенно нагревая до комнатной температуры. Медленно добавляли к реакционной системе воду с последующим перемешиванием в течение некоторого времени. Затем добавляли этилацетат и насыщенный раствор хлорида аммония с последующим перемешиванием. Водный слой отделяли, после чего полученный органический слой последовательно промывали насыщенным раствором хлорида аммония, водой и насыщенным раствором соли. Полученный органический слой сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Затем остаток очищали хроматографией на колонке с NH-силикагелем с получением указанного в заголовке соединения (3,71 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,19-1,25 (м, 6H), 1,50-1,64 (м, 2H), 1,77-1,79 (м, 2H), 2,10-2,24 (м, 2H), 3,18 (с, 1H), 3,49-3,58 (м, 2H), 3,62-3,78 (м, 2H), 3,83 (уш.с, 1H), 4,69-4,71 (м, 1H), 4,95-5,06 (м, 2H), 5,79-5,89 (м, 1H).
(2) Синтез 7,7-диэтокси-5-метоксигепт-1-ена
ДМФА (30 мл) добавляли к 1,1-диэтоксигепт-6-ен-3-олу, полученному в примере получения 57-(1) (3,07 г), и смесь охлаждали на ледяной бане в атмосфере азота. Затем добавляли гидрид натрия (60%, 699 мг) с последующим перемешиванием в течение 10 минут. Добавляли метилиодид (1,8 мл, удельный вес: 2,28 г/см3) с последующим перемешиванием в течение одного часа и 50 минут. Затем смесь нагревали до комнатной температуры и дополнительно перемешивали в течение одного часа и 30 минут. Дополнительно добавляли гидрид натрия (60%, 300 мг) и метилиодид (0,9 мл, удельный вес: 2,28 г/см3) с последующим перемешиванием в течение двух часов и 30 минут. Затем медленно добавляли воду и насыщенный раствор хлорида аммония с последующим перемешиванием в течение некоторого времени. После экстрагирования этилацетатом органический слой последовательно промывали насыщенным раствором хлорида аммония, водой и насыщенным раствором соли. Полученный органический слой сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и затем фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (2,98 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,19-1,23 (м, 6H), 1,56-1,63 (м, 2H), 1,70-1,84 (м, 2H), 2,08-2,14 (м, 2H), 3,31-3,37 (м, 4H), 3,46-3,56 (м, 2H), 3,62-3,71 (м, 2H), 4,66 (дд, J=4,4, 8,0 Гц, 1H), 4,94-5,06 (м, 2H), 5,76-5,88 (м, 1H).
(3) Синтез 3-метоксигепт-6-енальоксима
80% Раствор муравьиной кислоты (30 мл) добавляли к 7,7-диэтокси-5-метоксигепт-1-ену, полученному в примере получения 57-(2) (2,98 г), и смесь перемешивали при комнатной температуре в течение 10 минут. Добавив далее 75% раствор этанола (64 мл), добавляли ацетат натрия (3,75 г) и гидрохлорид гидроксиламина (1,92 г) с последующим дополнительным перемешиванием в течение одного часа и 20 минут. Растворитель концентрировали до примерно 40 мл при пониженном давлении с последующим экстрагированием этилацетатом. Полученный органический слой последовательно промывали насыщенным раствором бикарбоната натрия (три раза), водой и насыщенным раствором соли. Полученный органический слой сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и затем фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (1,95 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,51-1,74 (м, 2H), 2,08-2,16 (м, 2H), 2,35-2,68 (м, 2H), 3,36 (с, 3H), 3,38-3,46 (м, 1H), 4,96-5,08 (м, 2H), 5,76-5,86 (м, 1H), 6,84-6,86 и 7,47-7,49 (м, суммарн. 1H), 7,56 и 7,97 (уш., суммарн. 1H).
(4) Синтез (±)-(3aR*,6S*)-6-метокси-3,3a,4,5,6,7-гексагидробенз[c]изоксазола
Раствор гипохлорита натрия (5%, 18,5 мл) добавляли к раствору 3-метоксигепт-6-енальоксима, полученного в примере получения 57-(3) (1,95 г), в дихлорметане и смесь перемешивали при комнатной температуре в течение одного часа и 10 минут. Избыток гипохлорита натрия разлагали тиосульфатом натрия с последующим экстрагированием три раза хлороформом. Полученные органические слои сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и затем фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (796 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,54 (ддт, J=2,0, 3,6, 14,0, 1H), 1,68-1,79 (м, 1H), 1,90-1,97 (м, 1H), 2,05-2,13 (м, 1H), 2,20 (ддд, J=1,6, 7,6, 11,2 Гц, 1H), 3,04 (тд, J=2,4, 14,8 Гц, 1H), 3,13-3,21 (м, 1H), 3,32 (с, 3H), 3,78 (т, J=2,4 Гц, 1H), 3,79 (дд, J=8,0, 11,2 Гц, 1H), 4,53 (дд, J=7,6, 10,0 Гц, 1H).
(5) Синтез (±)-(3aR*,6S*,7aS*)-7a-(2-фторфенил)-6-метоксиоктагидробенз[c]изоксазола
ТГФ (3 мл) и толуол (20 мл) добавляли к 2-бромфторбензолу (1,23 мл, удельный вес: 1,614 г/см3) в атмосфере азота и смесь охлаждали до -78°C. Раствор н-бутиллития в гексане (3,9 мл, 2,63 M) медленно добавляли, так чтобы внутренняя температура находилась на уровне -60°C или ниже. По окончании добавления смесь перемешивали в течение 10 минут. После медленного добавления комплекса трифторид бора-диэтиловый эфир (1,29 мл) добавляли раствор (±)-(3aR*,6S*)-6-метокси-3,3a,4,5,6,7-гексагидробенз[c]изоксазола, полученного в примере получения 57-(4) (796 мг), в толуоле (10 мл) медленно, так чтобы внутренняя температура находилась на уровне -60°C или ниже. По окончании добавления смесь перемешивали в течение одного часа и 50 минут. Добавляли насыщенный раствор хлорида аммония с последующим нагреванием до комнатной температуры. Добавляли этилацетат и воду с последующим дополнительным перемешиванием. Отделяли водный слой, после чего органический слой последовательно промывали насыщенным раствором хлорида аммония, водой и насыщенным раствором соли. Полученный органический слой сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и затем фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (711 мг).
ESI-MS; m/z 252 [M+H]
(6) Синтез (±)-1-бензоил-3-[(1S*,2R*,5S*)-1-(2-фторфенил)-2-гидроксиметил-5-метоксициклогексил]тиомочевины
(±)-(3aR*,6S*,7aS*)-7a-(2-фторфенил)-6-метоксиоктагидробенз[c]изоксазол, полученный в примере получения 57-(5) (872 мг), растворяли в уксусной кислоте (20 мл). Затем добавляли цинковый порошок (2,27 г) и смесь перемешивали при комнатной температуре в течение 14 часов и 10 минут. Твердое вещество удаляли фильтрованием через целит и затем целит промывали этилацетатом. Полученный фильтрат концентрировали при пониженном давлении. Остаток растворяли в этилацетате. Затем добавляли насыщенный раствор бикарбоната натрия с последующим интенсивным перемешиванием. Органический слой отделяли и затем водный слой снова экстрагировали дважды этилацетатом. Объединенные органические слои сушили над безводным сульфатом магния. Твердое вещество удаляли фильтрованием и фильтрат концентрировали при пониженном давлении. Полученный остаток растворяли в дихлорметане (7 мл). Затем добавляли бензоилизотиоцианат (524 мкл, удельный вес: 1,21 г/см3) и смесь перемешивали при комнатной температуре в течение 16 часов и 30 минут. Реакционный раствор концентрировали при пониженном давлении и затем очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (453 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,67-1,76 (м, 2H), 2,05 (уш., 3H), 2,26 (уш., 2H), 2,63 (уш., 1H), 3,39 (с, 3H); 3,57 (уш., 1H), 3,67 (уш., 1H), 3,78 (уш., 1H), 6,99-7,04 (м, 1H), 7,13 (с, 1H), 7,51-7,62 (м, 4H), 7,87-7,88 (м, 2H), 8,83 (с, 1H), 11,60 (с, 1H).
(7) Синтез (±)-N-[(4aR*,7S*,8aS*)-8a-(2-фторфенил)-7-метокси-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]бензамида
Дихлорметан (20 мл) и пиридин (264 мкл, удельный вес: 0,978 г/см3) добавляли к (±)-1-бензоил-3-[(1S*,2R*,5S*)-1-(2-фторфенил)-2-гидроксиметил-5-метоксициклогексил]тиомочевине, полученной в примере получения 57-(6) (453 мг), в соответствии со способом примера получения 18-(5). Смесь охлаждали до -78°C в атмосфере азота и перемешивали в течение 15 минут. Трифторметансульфоновый ангидрид (358 мкл, удельный вес: 1,72 г/см3) медленно добавляли к реакционному раствору. По окончании добавления смесь перемешивали в течение 15 минут и затем перемешивали еще в течение одного часа, нагревая до 0°C. После добавления этилацетата добавляли насыщенный раствор бикарбоната натрия с последующим экстрагированием этилацетатом. Органический слой последовательно промывали водой и насыщенным раствором соли и сушили над безводным сульфатом магния. Твердое вещество удаляли фильтрованием. После концентрирования при пониженном давлении остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (268 мг).
ESI-MS; m/z 399 [M+H].
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,50-1,52 (м, 1H), 1,72 (тт, J=3,2, 13,6 Гц, 1H), 2,18-2,30 (м, 3H), 2,43 (дд, J=3,6, 15,2 Гц, 1H), 2,61 (дд, J=2,8, 12,8 Гц, 1H), 2,90 (дд, J=4,0, 12,8 Гц, 1H), 2,95-3,01 (м, 1H), 3,35 (с, 3H), 3,63 (т, J=2,8 Гц, 1H), 7,07 (ддд, J=1,2, 8,0, 12,8 Гц, 1H), 7,15 (дт, J=1,2, 8,0 Гц, 1H), 7,28-7,33 (м, 1H), 7,38-7,43 (м, 3H), 7,45-7,49 (м, 1H), 8,26 (дд, J=1,6, 8,4 Гц, 2H).
(8) Синтез (±)-(4aR*,7S*,8aS*)-8a-(2-фторфенил)-7-метокси-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-иламина
(±)-N-[(4aR*,7S*,8aS*)-8a-(2-Фторфенил)-7-метокси-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]бензамид, полученный в примере получения 57-(7) (268 мг), растворяли в метаноле (8 мл). Затем добавляли DBU (202 мкл, удельный вес: 1,018 г/см3) и смесь перемешивали при кипении с обратным холодильником в течение четырех часов и 15 минут. Затем реакционный раствор перемешивали при 64°C в течение 13 часов и 30 минут. После этого реакционный раствор перемешивали при кипении с обратным холодильником в течение девяти часов и 30 минут. Реакционному раствору давали охладиться до комнатной температуры и затем концентрировали его при пониженном давлении. Остаток очищали хроматографией на колонке с NH-силикагелем с получением указанного в заголовке соединения (150 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,37-1,40 (м, 1H), 1,66-1,73 (м, 1H), 2,05-2,19 (м, 3H), 2,35-2,39 (м, 1H), 2,58-2,60 (м, 1H), 2,72-2,75 (м, 1H), 2,81-2,90 (м, 1H), 3,34 (с, 3H), 3,61 (уш., 1H), 6,98-7,03 (м, 1H), 7,09-7,12 (м, 1H), 7,20-7,23 (м, 1H), 7,38 (уш., 1H).
(9) Синтез трет-бутил (±)-[(4aR*,7S*,8aS*)-8a-(2-фтор-5-нитрофенил)-7-метокси-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата
ТФУК (1 мл) и концентрированную серную кислоту (0,5 мл) добавляли к (±)-(4aR*,7S*,8aS*)-8a-(2-фторфенил)-7-метокси-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-иламину, полученному в примере получения 57-(8) (150 мг). Смесь в достаточной мере охлаждали на ледяной бане и затем медленно добавляли дымящую азотную кислоту (27,3 мкл). По окончании добавления смесь перемешивали в течение 15 минут. Реакционный раствор разбавляли дихлорметаном и затем медленно выливали на раздробленный лед. Добавляли 5 н. раствор гидроксида натрия до тех пор, пока реакционный раствор не становился щелочным, после чего его три раза экстрагировали дихлорметаном. Полученные органические слои сушили над безводным сульфатом магния и твердое вещество удаляли фильтрованием. Фильтрат концентрировали при пониженном давлении. Затем ТГФ (2,5 мл), воду (2,5 мл) и ди-трет-бутилдикарбонат (170 мг) добавляли к остатку при комнатной температуре и смесь перемешивали при комнатной температуре в течение двух часов. Добавляли этилацетат и воду и затем отделяли водный слой. Органический слой последовательно промывали насыщенным раствором хлорида аммония, водой и насыщенным раствором соли. Полученный органический слой сушили над безводным сульфатом магния и твердое вещество удаляли фильтрованием. Фильтрат концентрировали при пониженном давлении и затем очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (138 мг).
ESI-MS; m/z 440 [M+H].
(10) Синтез трет-бутил (±)-[(4aR*,7S*,8aS*)-8a-(5-амино-2-фторфенил)-7-метокси-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата
Этанол (5 мл), насыщенный раствор хлорида аммония (0,5 мл) и железный порошок (175 мг) добавляли к трет-бутил (±)-[(4aR*,7S*,8aS*)-8a-(2-фтор-5-нитрофенил)-7-метокси-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамату, полученному в примере получения 57-(9) (138 мг), и смесь перемешивали при кипении с обратным холодильником в течение 30 минут. Реакционный раствор охлаждали до комнатной температуры и затем твердое вещество удаляли фильтрованием через целит. Фильтрат концентрировали при пониженном давлении. Затем остаток суспендировали в дихлорметане и твердое вещество удаляли фильтрованием. Фильтрат концентрировали при пониженном давлении и затем очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (98 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,42-1,49 (м, 1H), 1,53 (с, 9H), 1,66-1,70 (м, 1H), 2,06-2,10 (м, 1H), 2,14-2,19 (м, 2H), 2,38 (дд, J=3,6, 15,2 Гц, 1H), 2,49-2,54 (м, 1H), 2,84-2,90 (м, 2H), 3,31 (с, 3H), 3,58-3,60 (м, 1H), 3,66 (уш., 2H), 6,52-6,56 (м, 1H), 6,62 (дд, J=2,8, 6,8 Гц, 1H), 6,84 (дд, J=8,4, 12,4 Гц, 1H).
(11) Синтез трет-бутил (-)-[(4aR*,7S*,8aS*)-8a-(5-амино-2-фторфенил)-7-метокси-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата
трет-Бутил (±)-[(4aR*,7S*,8aS*)-8a-(5-амино-2-фторфенил)-7-метокси-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамат, полученный в примере получения 57-(10), оптически разделяли, используя CHIRALPAK™ AD-H производства Daicel Chemical Industries, Ltd. (2 см × 25 см, подвижная фаза: гексан:этанол = 85:15, скорость потока: 20 мл/мин, загружаемый раствором примерно 10 мг в 0,5 мл этанола для одного цикла). Компонент, имеющий время удерживания от 18,1 до 21,2 минут, собирали с получением указанного в заголовке соединения (41 мг, >99% ee, оптическое вращение (-)).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,43-1,46 (м, 1H), 1,53 (с, 9H), 1,67-1,70 (м, 1H), 2,06-2,18 (м, 3H), 2,36-2,39 (м, 1H), 2,50-2,53 (м, 1H), 2,86-2,89 (м, 2H), 3,31 (с, 3H), 3,58 (уш., 1H), 3,67 (уш., 2H), 6,53-6,55 (м, 1H), 6,60-6,62 (м, 1H), 6,81-6,86 (м, 1H).
Пример получения 58
Синтез трет-бутил (-)-[(4aR*,7R*,8aS*)-8a-(5-амино-2-фторфенил)-7-метокси-4a,5,6,7,8,8a-гидро-4H-бензо[d][1,3]тиазин-2-ил]карбамат
[Формула 80]
(1) Синтез [1-(2,2-диэтоксиэтил)пент-4-енилоксиметил]бензола
ДМФА (40 мл) и бензилбромид (2,4 мл, удельный вес: 1,44 г/см3) добавляли к 1,1-диэтоксигепт-6-ен-3-олу, полученному в примере получения 57-(1) (3,71 г), и смесь охлаждали на ледяной бане в атмосфере азота. Затем добавляли гидрид натрия (60%, 883 мг) с последующим перемешиванием в течение 60 минут. Добавляли дополнительно бензилбромид (1,09 мл) с последующим перемешиванием в течение одного часа. Добавляли дополнительно гидрид натрия (60%, 116 мг) и смесь перемешивали в течение одного часа и 50 минут, постепенно нагревая до комнатной температуры. Далее добавляли тетрабутиламмонийиодид (680 мг) с последующим перемешиванием в течение одного часа и 10 минут. Медленно добавляли воду и насыщенный раствор хлорида аммония. После перемешивания в течение некоторого времени отделяли водный слой. Органический слой последовательно промывали насыщенным раствором хлорида аммония, водой и насыщенным раствором соли. Полученный органический слой сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и затем фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (4,06 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,19 (дт, J=4,4, 7,2 Гц, 6H), 1,63-1,70 (м, 2H), 1,76-1,93 (м, 2H), 2,11-2,17 (м, 2H), 3,39-3,54 (м, 2H), 3,56-3,69 (м, 3H), 4,46-4,56 (м, 2H), 4,68 (дд, J=4,0, 7,2 Гц, 1H), 4,95-4,97 (м, 1H), 4,99-5,05 (м, 1H), 5,82 (тдд, J=6,8, 10,0, 16,8 Гц, 1H), 7,25-7,32 (м, 1H), 7,32-7,36 (м, 4H).
(2) Синтез 3-бензилоксигепт-6-енальоксима
Указанное в заголовке соединение (3,53 г) получали из [1-(2,2-диэтоксиэтил)пент-4-енилоксиметил]бензола, полученного в примере получения 58-(1) (3,71 г), в соответствии со способом примера получения 57-(3) без очистки хроматографией на колонке с силикагелем.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,59-1,81 (м, 2H), 2,11-2,23 (м, 2H), 2,42-2,51 (м, 1H), 2,65-2,68 (м, 1H), 3,64 (тд, J=6,0, 18,4 Гц, 1H), 4,50-4,58 (м, 2H), 4,95-5,04 (м, 2H), 5,73-5,85 (м, 1H), 6,86-6,89 и 7,48-7,51 (м, суммарн. 1H), 7,17 и 7,53 (уш., суммарн. 1H), 7,29 (уш., 1H), 7,34 (с, 4H).
(3) Синтез 6-бензилокси-3,3a,4,5,6,7-гексагидробенз[c]изоксазола
Указанное в заголовке соединение (2,91 г) получали из 3-бензилоксигепт-6-енальоксима, полученного в примере получения 58-(2) (3,53 г), в соответствии со способом примера получения 57-(4).
ESI-MS; m/z 232 [M+H].
(4) Синтез (±)-(3aR*,6R*,7aS*)-6-бензилокси-7a-(2-фторфенил)октагидробенз[c]изоксазола
Указанное в заголовке соединение (1,69 г) получали из 6-бензилокси-3,3a,4,5,6,7-гексагидробенз[c]изоксазола, полученного в примере получения 58-(3) (2,91 г), в соответствии со способом примера получения 57-(5).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,38-1,49 (м, 1H), 1,59-1,69 (м, 1H), 1,98-2,05 (м, 1H), 2,12-2,17 (м, 1H), 2,22-2,35 (м, 2H), 2,90-2,96 (м, 1H), 3,50-3,58 (м, 2H), 3,67 (д, J=6,8 Гц, 1H), 4,52 (с, 2H), 5,77 (уш., 1H), 7,03 (ддд, J=1,6, 8,0, 12,4 Гц, 1H), 7,13 (дт, J=1,2, 7,6 Гц, 1H), 7,22-7,35 (м, 6H), 7,82 (дт, J=1,6, 8,0 Гц, 1H).
(5) Синтез (±)-1-бензоил-3-[(1S*,2R*,5R*)-5-бензилокси-1-(2-фторфенил)-2-гидроксиметилциклогексил]тиомочевины
Указанное в заголовке соединение (2,26 г) получали из (±)-(3aR*,6R*,7aS*)-6-бензилокси-7a-(2-фторфенил)октагидробенз[c]изоксазола, полученного в примере получения 58-(4) (1,69 г), в соответствии со способом примера получения 57-(6).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,70-1,76 (м, 1H), 1,83-1,87 (м, 1H), 2,14-2,25 (м, 3H), 2,42-2,45 (м, 2H), 3,66 (уш., 2H), 3,84 (уш., 1H), 4,49-4,64 (м, 2H), 6,94-7,14 (м, 5H), 7,22-7,28 (м, 1H), 7,29-7,32 (м, 2H), 7,40-7,49 (м, 3H), 7,55-7,60 (м, 3H), 8,57 (уш., 1H), 11,57 (уш., 1H).
(6) Синтез (±)-N-[(4aR*,7R*,8aS*)-7-бензилокси-8a-(2-фторфенил)-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]бензамида
Указанное в заголовке соединение (1,81 г) получали из (±)-1-бензоил-3-[(1S*,2R*,5R*)-5-бензилокси-1-(2-фторфенил)-2-гидроксиметилциклогексил]тиомочевины, полученной в примере получения 58-(5) (2,26 г), в соответствии со способом примера получения 57-(7).
ESI-MS; m/z 475 [M+H].
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,59-1,69 (м, 1H), 1,78-1,85 (м, 1H), 1,97-2,10 (м, 1H), 2,27-2,32 (м, 2H), 2,41-2,47 (м, 1H), 2,62 (дд, J=2,8, 12,8 Гц, 1H), 2,90-3,00 (м, 2H), 3,78-3,86 (м, 1H), 4,58 (с, 2H), 7,07-7,13 (м, 1H), 7,14-7,18 (м, 1H), 7,24-7,28 (м, 1H), 7,30-7,36 (м, 6H), 7,42-7,46 (м, 2H), 7,49-7,54 (м, 1H) 8,24-8,27 (м, 2H).
(7) Синтез (±)-(4aR*,7R*,8aS*)-7-бензилокси-8a-(2-фторфенил)-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-иламина
(±)-N-[(4aR*,7R*,8aS*)-7-Бензилокси-8a-(2-фторфенил)-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]бензамид, полученный в примере получения 58-(6) (1,81 г), растворяли в метаноле (60 мл). Затем добавляли DBU (1,14 мл, удельный вес: 1,018 г/см3) и смесь перемешивали при кипении с обратным холодильником в течение трех часов. Затем реакционный раствор перемешивали при 64°C в течение 14 часов. Реакционному раствору давали охладиться до комнатной температуры и затем концентрировали его при пониженном давлении. Остаток очищали хроматографией на колонке с NH-силикагелем с получением указанного в заголовке соединения (1,20 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,53-1,63 (м, 2H), 1,76-1,88 (м, 1H), 2,18-2,22 (м, 1H), 2,26-2,30 (м, 2H), 2,58 (дд, J=2,8, 12,0 Гц, 1H), 2,68-2,74 (м, 1H), 2,85 (дд, J=4,0, 12,0 Гц, 1H), 3,63-3,71 (м, 1H), 4,42 (уш., 2H), 4,52-4,59 (м, 2H), 7,00-7,05 (м, 1H), 7,10 (дт, J=1,2, 7,6 Гц, 1H), 7,20-7,27 (м, 3H), 7,29-7,34 (м, 4H).
(8) Синтез трет-бутил (±)-[(4aR*,7R*,8aS*)-8a-(2-фторфенил)-7-гидрокси-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата
(±)-(4aR*,7R*,8aS*)-7-Бензилокси-8a-(2-фторфенил)-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-иламин, полученный в примере получения 58-(7) (1,2 г), перемешивали в концентрированной хлористо-водородной кислоте (120 мл) при кипении с обратным холодильником в течение трех часов и 10 минут. Реакционный раствор охлаждали до комнатной температуры и растворитель концентрировали при пониженном давлении. Затем к остатку добавляли 1 н. раствор гидроксида натрия (16,2 мл), ТГФ (16 мл) и ди-трет-бутилдикарбонат (1,06 г) и смесь перемешивали при комнатной температуре. Через один час и 30 минут добавляли дополнительно ди-трет-бутилдикарбонат (15 г) с последующим перемешиванием в течение 12 часов и 30 минут. К реакционному раствору добавляли этилацетат и воду с последующим дополнительным перемешиванием. Затем отделяли водный слой. Органический слой последовательно промывали насыщенным раствором хлорида аммония, водой и насыщенным раствором соли. Полученный органический слой сушили над безводным сульфатом магния и твердое вещество удаляли фильтрованием. Фильтрат концентрировали при пониженном давлении и затем очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (1,31 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,53 (с, 9H), 1,55-1,65 (м, 1H), 1,71-1,76 (м, 1H), 1,96-2,20 (м, 3H), 2,32-2,38 (м, 1H), 2,52-2,54 (м, 1H), 2,79-2,87 (м, 2H), 3,95-4,00 (м, 1H), 7,08 (ддд, J=1,2, 8,0, 12,8 Гц, 1H), 7,16-7,20 (м, 1H), 7,24-7,27 (м, 1H), 7,28-7,34 (м, 1H).
(9) Синтез трет-бутил (±)-[(4aR*,7R*,8aS*)-8a-(2-фторфенил)-7-гидрокси-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]-(4-метоксибензил)карбамата
трет-Бутил (±)-[(4aR*,7R*,8aS*)-8a-(2-фторфенил)-7-гидрокси-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамат, полученный в примере получения 58-(8) (500 мг), растворяли в ДМФА (10 мл) и смесь охлаждали на ледяной бане в атмосфере азота. К смеси добавляли п-метоксибензилхлорид (161 мкл, удельный вес: 1,154 г/см3) и карбонат калия (247 мг) с последующим перемешиванием в течение одного часа. После этого реакционный раствор нагревали до комнатной температуры и перемешивали в течение 19 часов. К реакционному раствору добавляли этилацетат и воду с последующим дополнительным перемешиванием. Затем отделяли водный слой. Органический слой последовательно промывали насыщенным раствором хлорида аммония, водой и насыщенным раствором соли. Полученный органический слой сушили над безводным сульфатом магния и твердое вещество удаляли фильтрованием. Фильтрат концентрировали при пониженном давлении и затем очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (541 мг).
ESI-MS; m/z 501 [M+H].
(10) Синтез трет-бутил (±)-[(4aR*,7R*,8aS*)-8a-(2-фторфенил)-7-метокси-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]-(4-метоксибензил)карбамата
трет-Бутил (±)-[(4aR*,7R*,8aS*)-8a-(2-фторфенил)-7-гидрокси-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]-(4-метоксибензил)карбамат, полученный в примере получения 58-(9) (541 мг), растворяли в ДМФА (5 мл). Добавляли метилиодид (113 мкл, удельный вес: 2,28 г/см3) и затем смесь охлаждали на ледяной бане в атмосфере азота. Добавляли гидрид натрия (60%, 55 мг) с последующим перемешиванием в течение одного часа и 45 минут. После этого добавляли метилиодид (113 мкл, удельный вес: 2,28 г/см3) и гидрид натрия (60%, 55 мг) и смесь нагревали до комнатной температуры и перемешивали в течение одного часа и 45 минут. Затем дополнительно добавляли метилиодид (113 мкл, удельный вес: 2,28 г/см3) с последующим перемешиванием в течение 13 часов. К реакционному раствору добавляли этилацетат и воду с последующим перемешиванием. Затем отделяли водный слой. Органический слой последовательно промывали насыщенным раствором хлорида аммония, водой и насыщенным раствором соли. Полученный органический слой сушили над безводным сульфатом магния и твердое вещество удаляли фильтрованием. Фильтрат концентрировали при пониженном давлении и затем очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (409 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,32-1,42 (м, 1H) 1,47-1,52 (м, 2H), 1,53 (с, 9H), 1,95-1,99 (м, 1H), 2,10-2,16 (м, 1H), 2,26-2,31 (м, 1H), 2,47 (дд, J=2,8, 12,4 Гц, 1H), 2,59-2,64 (м, 1H), 2,77 (дд, J=4,0, 12,4 Гц, 1H), 2,96-3,04 (м, 1H), 3,26 (с, 3H), 3,80 (с, 3H), 4,92-5,04 (м, 2H), 6,85-6,88 (м, 2H), 6,96-7,05 (м, 3H), 7,18-7,23 (м, 1H), 7,31-7,34 (м, 2H).
(11) Синтез трет-бутил (±)-[(4aR*,7R*,8aS*)-8a-(2-фтор-5-нитрофенил)-7-метокси-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата
Указанное в заголовке соединение (283 мг) получали из трет-бутил (±)-[(4aR*,7R*,8aS*)-8a-(2-фторфенил)-7-метокси-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]-(4-метоксибензил)карбамата, полученного в примере получения 58-(10) (409 мг), в соответствии со способом примера получения 57-(9), используя 82,1 мкл дымящей азотной кислоты (удельный вес: 1,52 г/см3, 2,6 эквивалента относительно исходного вещества).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,38-1,52 (м, 2H), 1,54 (с, 9H), 1,70-1,74 (м, 1H), 1,89-1,92 (м, 1H), 2,08-2,24 (м, 3H), 2,56-2,60 (м, 1H), 2,74-2,81 (м, 2H), 3,35-3,39 (м, 4H), 7,20-7,25 (м, 1H), 8,12-8,21 (м, 2H).
(12) Синтез трет-бутил (±)-[(4aR*,7R*,8aS*)-8a-(5-амино-2-фторфенил)-7-метокси-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата
Указанное в заголовке соединение (221 мг) получали из трет-бутил (±)-[(4aR*,7R*,8aS*)-8a-(2-фтор-5-нитрофенил)-7-метокси-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата, полученного в примере получения 58-(11) (283 мг), в соответствии со способом примера получения 57-(10), где очистку осуществляли хроматографией на колонке с NH-силикагелем.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,30-1,49 (м, 1H), 1,53 (с, 9H), 1,70-1,77 (м, 1H), 1,89-1,99 (м, 1H), 2,09-2,12 (м, 1H), 2,23-2,29 (м, 2H), 2,52 (дд, J=2,4, 12,8 Гц, 1H), 2,81-2,91 (м, 2H), 3,36 (с, 3H), 3,42-3,47 (м, 1H), 3,65 (уш., 2H), 6,49-6,57 (м, 2H), 6,83-6,88 (м, 1H).
(13) Синтез трет-бутил (-)-[(4aR*,7R*,8aS*)-8a-(5-амино-2-фторфенил)-7-метокси-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата
трет-Бутил (±)-[(4aR*,7R*,8aS*)-8a-(5-амино-2-фторфенил)-7-метокси-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамат, полученный в примере получения 58-(12) (221 мг), оптически разделяли, используя CHIRALPAK™ AD-H производства Daicel Chemical Industries, Ltd. (2 см × 25 см, подвижная фаза: гексан:этанол = 70:30, скорость потока: 10 мл/мин, загружаемый раствором примерно 35 мг в 1 мл этанола для одного цикла). Компонент, имеющий время удерживания от 17,8 до 23,7 минут, собирали с получением указанного в заголовке соединения (93 мг, >99% ee, оптическое вращение (-)).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,46-1,49 (м, 1H), 1,54 (с, 9H), 1,71-1,75 (м, 1H), 1,89-1,99 (м, 1H), 2,09-2,12 (м, 1H), 2,23-2,29 (м, 2H), 2,50-2,53 (м, 1H), 2,81-2,91 (м, 2H), 3,36 (с, 3H), 3,44-3,49 (м, 1H), 3,65 (уш., 2H), 6,49-6,52 (м, 1H), 6,53-6,57 (м, 1H), 6,83-6,88 (м, 1H).
Пример получения 59
Синтез (±)-N-[(4aR*,6S*,8aS*)-8a-(2-фторфенил)-6-метокси-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]бензамида и (±)-N-[(4aR*,6R*,8aS*)-8a-(2-фторфенил)-6-метокси-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]бензамида
[Формула 81]

(1) Синтез 7,7-диметоксигепт-1-ен-4-ола
4,4-Диметоксибутиральдегид (Org. Biomol. Chem. 4 (2006) 2158) (5,47 г) растворяли в ТГФ (55 мл) и раствор охлаждали на ледяной бане в атмосфере азота. Затем медленно добавляли раствор аллилмагнийхлорида в ТГФ (62,1 мл, 1 M). По окончании добавления смесь перемешивали в течение трех часов. После медленного добавления воды добавляли этилацетат и насыщенный раствор хлорида аммония с последующим дополнительным перемешиванием. Водный слой отделяли и затем органический слой последовательно промывали насыщенным раствором хлорида аммония, водой и насыщенным раствором соли. Полученный органический слой сушили над безводным сульфатом магния и твердое вещество удаляли фильтрованием. Фильтрат концентрировали при пониженном давлении и затем очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (5,55 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,44-1,62 (м, 2H), 1,67-1,86 (м, 2H), 2,09 (д, J=3,6, 1H), 2,14-2,23 (м, 1H), 2,24-2,32 (м, 1H), 3,34 (с, 6H), 3,63-3,69 (м, 1H), 4,38-4,41 (м, 1H), 5,11 (д, J=1,2 Гц, 1H), 5,13-5,16 (м, 1H), 5,78-5,89 (м, 1H).
(2) Синтез 4,7,7-триметоксигепт-1-ена
7,7-Диметоксигепт-1-ен-4-ол, полученный в примере получения 59-(1) (6,16 г), растворяли в 1-метил-2-пирролидиноне (60 мл) и раствор охлаждали на ледяной бане в атмосфере азота. Затем добавляли гидрид натрия (60%, 2,12 г) с последующим перемешиванием в течение 10 минут. Далее добавляли метилиодид (6,61 г, 2,28 г/см3) и смесь дополнительно перемешивали в течение двух часов и 10 минут. После медленного добавления воды добавляли этилацетат с последующим дополнительным перемешиванием. Водный слой отделяли и затем органический слой последовательно промывали насыщенным раствором хлорида аммония, водой и насыщенным раствором соли. Полученный органический слой сушили над безводным сульфатом магния и твердое вещество удаляли фильтрованием. Фильтрат концентрировали при пониженном давлении и затем очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (5,83 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,47-1,76 (м, 4H), 2,20-2,33 (м, 2H), 3,20-3,26 (м, 1H), 3,32 (с, 6H), 3,34 (с, 3H), 4,36 (т, J=5,6 Гц, 1H), 5,05-5,10 (м, 2H), 5,76-5,86 (м, 1H).
(3) Синтез 4-метоксигепт-6-енальоксима
Указанное в заголовке соединение (4,61 г) получали из 4,7,7-триметоксигепт-1-ена, полученного в примере получения 59-(2) (5,83 г), в соответствии со способом примера получения 57-(3) без очистки хроматографией на колонке с силикагелем.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,62-1,73 (м, 2H), 2,21-2,36 (м, 3H), 2,46 (дт, J=5,6, 8,0 Гц, 1H), 3,21-3,28 (м, 1H), 3,36 (д, J=3,6 Гц, 3H), 5,06-5,13 (м, 2H), 5,70-5,86 (м, 1H), 6,74-6,77 и 7,43-7,46 (м, суммарн. 1H), 7,44 и 7,82 (уш., суммарн. 1H).
(4) Синтез 5-метокси-3,3a,4,5,6,7-гексагидробенз[c]изоксазола
Указанное в заголовке соединение (3,95 г) получали из 4-метоксигепт-6-енальоксима, полученного в примере получения 59-(3) (4,61 г), в соответствии со способом примера получения 57-(4).
ESI-MS; m/z 156 [M+H].
(5) Синтез (±)-(3aR*,7aS*)-7a-(2-фторфенил)-5-метоксиоктагидробенз[c]изоксазола
Указанное в заголовке соединение (5,60 г) получали из 5-метокси-3,3a,4,5,6,7-гексагидробенз[c]изоксазола, полученного в примере получения 59-(4) (3,95 г), в соответствии со способом примера получения 57-(5).
ESI-MS; m/z 252 [M+H].
(6) Синтез (±)-[(1R*,2S*)-2-амино-2-(2-фторфенил)-5-метоксициклогексил]метанола
(±)-(3aR*,7aS*)-7a-(2-Фторфенил)-5-метоксиоктагидробенз[c]изоксазол, полученный в примере получения 59-(5) (5,60 г), растворяли в уксусной кислоте (128 мл). Затем добавляли цинковый порошок (14,1 г) и смесь перемешивали при комнатной температуре в течение восьми часов. Твердое вещество удаляли фильтрованием через целит и затем целит промывали этилацетатом. Полученный фильтрат концентрировали при пониженном давлении. Остаток растворяли в этилацетате. Затем добавляли насыщенный раствор бикарбоната натрия с последующим интенсивным перемешиванием. Органический слой отделяли и затем водный слой снова экстрагировали дважды этилацетатом. Объединенные органические слои сушили над безводным сульфатом магния. Твердое вещество удаляли фильтрованием и фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (5,49 г).
ESI-MS; m/z 254 [M+H].
(7) Синтез (±)-1-бензоил-3-[(1S*,2R*)-1-(2-фторфенил)-2-гидроксиметил-4-метоксициклогексил]тиомочевины
(±)-[(1R*,2S*)-2-Амино-2-(2-фторфенил)-5-метоксициклогексил]метанол, полученный в примере получения 59-(6) (5,49 г), растворяли в дихлорметане (22 мл). Затем добавляли бензоилизотиоцианат (3,04 мл, удельный вес: 1,21 г/см3) и смесь перемешивали при комнатной температуре в течение 16 часов и 30 минут. Реакционный раствор концентрировали при пониженном давлении и затем очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (7,16 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,51-1,70 (м, 3H), 1,84-2,19 (м, 2H), 2,27-2,30 (м, 1H), 2,57 (уш., 1H), 3,39-3,42 (м, 3H), 3,56 (уш., 2H), 3,67 (уш., 1H), 7,04-7,15 (м, 2H), 7,43-7,63 (м, 5H), 7,88 (с, 2H), 8,90 (уш., 1H), 11,53 (уш., 1H).
(8) Синтез (±)-N-[(4aR*,6S*,8aS*)-8a-(2-фторфенил)-6-метокси-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]бензамида и (±)-N-[(4aR*,6R*,8aS*)-8a-(2-фторфенил)-6-метокси-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]бензамида
Указанные в заголовке соединения (±)-N-[(4aR*,6S*,8aS*)-8a-(2-фторфенил)-6-метокси-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]бензамид (3,11 г) и (±)-N-[(4aR*,6R*,8aS*)-8a-(2-фторфенил)-6-метокси-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]бензамид (1,65 г) получали из (±)-1-бензоил-3-[(1S*,2R*)-1-(2-фторфенил)-2-гидроксиметил-4-метоксициклогексил]тиомочевины, полученной в примере получения 59-(7) (7,16 г), в соответствии со способом примера получения 57-(7).
(±)-N-[(4aR*,6S*,8aS*)-8a-(2-Фторфенил)-6-метокси-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]бензамид
ESI-MS; m/z 399 [M++H].
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,64 (д, J=14,8 Гц, 1H), 1,77-1,85 (м, 1H), 1,89-1,94 (м, 1H), 1,99-2,08 (м, 2H), 2,54 (дд, J=2,8, 12,8 Гц, 1H), 2,75 (дт, J=4,0, 13,6 Гц, 1H), 2,97 (дд, J=4,0, 12,8 Гц, 1H), 3,30-3,38 (м, 1H), 3,40 (с, 3H), 3,68 (т, J=2,4 Гц, 1H), 7,09 (ддд, J=1,2, 8,0, 12,4 Гц, 1H), 7,14 (дт, J=1,6, 7,6 Гц, 1H), 7,28-7,35 (м, 2H), 7,41-7,45 (м, 2H), 7,48-7,52 (м, 1H), 8,24-8,27 (м, 2H).
(±)-N-[(4aR*,6R*,8aS*)-8a-(2-Фторфенил)-6-метокси-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]бензамид
ESI-MS; m/z 399 [M++H].
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,63-1,74 (м, 1H), 1,89-1,95 (м, 2H), 2,05-2,13 (м, 2H), 2,46 (дт, J=3,6, 14,4 Гц, 1H), 2,60 (дд, J=2,8, 12,8 Гц, 1H), 2,93 (дд, J=4,0, 12,8 Гц, 1H), 2,98-3,04 (м, 1H), 3,40 (с, 3H), 3,47-3,54 (м, 1H), 7,09 (ддд, J=1,2, 8,0, 12,8 Гц, 1H), 7,16 (дт, J=1,6, 7,6 Гц, 1H), 7,29-7,38 (м, 2H), 7,41-7,45 (м, 2H), 7,48-7,52 (м, 1H), 8,22-8,25 (м, 2H).
Пример получения 60
Синтез трет-бутил (-)-[(4aR*,6S*,8aS*)-8a-(5-амино-2-фторфенил)-6-метокси-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата
[Формула 82]

(1) Синтез (±)-(4aR*,6S*,8aS*)-8a-(2-фторфенил)-6-метокси-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-иламина
(±)-N-[(4aR*,6S*,8aS*)-8a-(2-Фторфенил)-6-метокси-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]бензамид, полученный в примере получения 59-(8) (3,11 г), растворяли в метаноле (100 мл). Затем добавляли DBU (2,34 мл, удельный вес: 1,018 г/см3) и смесь перемешивали при кипении с обратным холодильником в течение двух часов и 15 минут. Затем реакционный раствор перемешивали при 64°C в течение 13 часов и 30 минут. Реакционному раствору давали охладиться до комнатной температуры и затем концентрировали его при пониженном давлении. Остаток очищали хроматографией на колонке с NH-силикагелем с получением указанного в заголовке соединения (1,96 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,53 (тд, J=3,6, 13,2 Гц, 1H), 1,66-1,75 (м, 2H), 1,81-1,91 (м, 2H), 2,49 (дд, J=2,8, 12,4 Гц, 1H), 2,61 (дт, J=4,0, 13,2 Гц, 1H), 2,90 (дд, J=4,4, 12,0 Гц, 1H), 3,09 (квд, J=3,6, 12,4 Гц, 1H), 3,39 (с, 3H), 3,60 (т, J=2,8 Гц, 1H), 7,02 (ддд, J=1,6, 8,0, 12,8 Гц, 1H), 7,06-7,10 (м, 1H), 7,18-7,24 (м, 1H), 7,25-7,29 (м, 1H).
(2) Синтез трет-бутил (±)-[(4aR*,6S*,8aS*)-8a-(2-фтор-5-нитрофенил)-6-метокси-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата
Указанное в заголовке соединение (1,37 г) получали из (±)-(4aR*,6S*,8aS*)-8a-(2-фторфенил)-6-метокси-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-иламина, полученного в примере получения 60-(1) (1 г), в соответствии со способом примера получения 57-(9).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,42-1,55 (м, 1H), 1,54 (с, 9H), 1,66-1,73 (м, 1H), 1,85-2,00 (м, 3H), 2,49-2,61 (м, 2H), 2,83-2,87 (м, 1H), 3,21-3,25 (м, 1H), 3,39 (с, 3H), 3,66 (уш., 1H), 7,21-7,25 (м, 1H), 8,19-8,21 (м, 2H).
(3) Синтез трет-бутил (±)-[(4aR*,6S*,8aS*)-8a-(5-амино-2-фторфенил)-6-метокси-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата
Указанное в заголовке соединение (1,21 г) получали из трет-бутил (±)-[(4aR*,6S*,8aS*)-8a-(2-фтор-5-нитрофенил)-6-метокси-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата, полученного в примере получения 60-(2) (2,56 г), в соответствии со способом примера получения 57-(10).
ESI-MS; m/z 410 [M+H].
(4) Синтез трет-бутил (-)-[(4aR*,6S*,8aS*)-8a-(5-амино-2-фторфенил)-6-метокси-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата
трет-Бутил (±)-[(4aR*,6S*,8aS*)-8a-(5-амино-2-фторфенил)-6-метокси-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамат, полученный в примере получения 60-(3), оптически разделяли, используя CHIRALPAK™ AD-H производства Daicel Chemical Industries, Ltd. (2 см × 25 см, подвижная фаза: гексан:этанол = 70:30, скорость потока: 10 мл/мин, загружаемый раствором примерно 50 мг в 2 мл этанола для одного цикла). Компонент, имеющий время удерживания от 15,7 до 20,5 минут, собирали с получением указанного в заголовке соединения (502 мг, >99% ee, оптическое вращение (-)).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,48-1,54 (м, 2H), 1,53 (с, 9H), 1,66-1,75 (м, 1H), 1,82-1,88 (м, 1H), 1,91-2,00 (м, 2H), 2,41-2,45 (м, 1H), 2,66-2,74 (м, 1H), 2,94 (дд, J=4,0, 12,0 Гц, 1H), 3,19-3,25 (м, 1H), 3,38 (с, 3H), 3,64-3,65 (м, 2H), 6,52-6,57 (м, 2H), 6,83-6,88 (м, 1H).
Пример получения 61
Синтез трет-бутил (-)-[(4aR*,6R*,8aS*)-8a-(5-амино-2-фторфенил)-6-метокси-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата
[Формула 83]

(1) Синтез (±)-(4aR*,6R*,8aS*)-8a-(2-фторфенил)-6-метокси-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-иламина
(±)-N-[(4aR*,6R*,8aS*)-8a-(2-Фторфенил)-6-метокси-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]бензамид, полученный в примере получения 59-(8) (1,65 г), растворяли в метаноле (60 мл). Затем добавляли DBU (1,24 мл, удельный вес: 1,018 г/см3) и смесь перемешивали при кипении с обратным холодильником в течение 13 часов и 30 минут. Реакционному раствору давали охладиться до комнатной температуры и затем концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с NH-силикагелем с получением указанного в заголовке соединения (1,16 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,52-1,74 (м, 2H), 1,80-1,90 (м, 2H), 1,94-2,01 (м, 1H), 2,27-2,35 (м, 1H), 2,56 (дд, J=2,8, 12,0 Гц, 1H), 2,71-2,77 (м, 1H), 2,87 (дд, J=4,0, 12,0 Гц, 1H), 3,36-3,49 (м, 4H), 4,46 (уш., 2H), 7,01 (ддд, J=1,2, 8,0, 12,8 Гц, 1H), 7,10 (ддд, J=1,2, 7,2, 7,6 Гц, 1H), 7,19-7,25 (м, 1H), 7,26-7,31 (м, 1H).
(2) Синтез трет-бутил (±)-[(4aR*,6R*,8aS*)-8a-(2-фтор-5-нитрофенил)-6-метокси-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата
Указанное в заголовке соединение (1,27 г) получали из (±)-(4aR*,6R*,8aS*)-8a-(2-фторфенил)-6-метокси-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-иламина, полученного в примере получения 61-(1) (1,16 г), в соответствии со способом примера получения 57-(9).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,46-1,55 (м, 1H), 1,54 (с, 9H), 1,77-1,87 (м, 2H), 1,99-2,08 (м, 2H), 2,21-2,27 (м, 1H), 2,54-2,58 (м, 1H), 2,76-2,80 (м, 1H), 2,86-2,90 (м, 1H), 3,39-3,49 (м, 4H), 7,20-7,28 (м, 1H), 8,18-8,21 (м, 2H).
(3) Синтез трет-бутил (±)-[(4aR*,6R*,8aS*)-8a-(5-амино-2-фторфенил)-6-метокси-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата
Указанное в заголовке соединение (838 мг) получали из трет-бутил (±)-[(4aR*,6R*,8aS*)-8a-(2-фтор-5-нитрофенил)-6-метокси-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата, полученного в примере получения 61-(2) (1,27 г), в соответствии со способом примера получения 57-(10).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,46-1,54 (м, 1H), 1,53 (с, 9H), 1,77-1,86 (м, 2H), 1,98-2,07 (м, 2H), 2,41 (дт, J=4,0, 6,4 Гц, 1H), 2,47-2,52 (м, 1H), 2,87-2,93 (м, 2H), 3,38-3,49 (м, 4H), 3,65 (уш., 2H), 6,53-6,57 (м, 2H), 6,82-6,87 (м, 1H).
ВЭЖХ (CHIRALPAK™ AD-H производства Daicel Chemical Industries, Ltd., 2 см × 25 см, подвижная фаза: гексан:этанол = 70:30, скорость потока: 1 мл/мин): 3,1 минуты (оптическое вращение (+)), 4,3 минуты (оптическое вращение (-))
(4) Синтез трет-бутил (-)-[(4aR*,6R*,8aS*)-8a-(5-амино-2-фторфенил)-6-метокси-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата
трет-Бутил (±)-[(4aR*,6R*,8aS*)-8a-(5-амино-2-фторфенил)-6-метокси-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамат, полученный в примере получения 58-(12) (838 мг), оптически разделяли, используя CHIRALPAK™ AD-H производства Daicel Chemical Industries, Ltd. (2 см × 25 см, подвижная фаза: гексан:этанол = 50:50, скорость потока: 10 мл/мин, загружаемый раствором примерно 16 мг в 2 мл этанола для одного цикла). Компонент, имеющий из двух основных компонентов более длинное время удерживания, собирали с получением указанного в заголовке соединения (335 мг, >99% ee, оптическое вращение (-)).
ВЭЖХ (CHIRALPAK™ AD-H производства Daicel Chemical Industries, Ltd., 2 см × 25 см, подвижная фаза: гексан:этанол = 70:30, скорость потока: 1 мл/мин): 4,3 минуты
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,44-1,54 (м, 1H), 1,53 (с, 9H), 1,80-1,86 (м, 2H), 1,98-2,07 (м, 2H), 2,37-2,45 (м, 1H), 2,48-2,51 (м, 1H), 2,87-2,92 (м, 2H), 3,41 (с, 3H), 3,45-3,49 (м, 1H), 3,65 (уш., 2H), 6,53-6,55 (м, 2H), 6,82-6,87 (м, 1H).
Пример получения 62
Синтез (±)-(3aR*,5S*,7aS*)-5-бензилокси-7a-(2-фторфенил)октагидробенз[c]изоксазола и (±)-(3aR*,5R*,7aS*)-5-бензилокси-7a-(2-фторфенил)октагидробенз[c]изоксазола
[Формула 84]

(1) Синтез [1-(3,3-диметоксипропил)-бут-3-енилоксиметил]бензола
7,7-Диметоксигепт-1-ен-4-ол, полученный в примере получения 59-(1) (5,47 г), растворяли в 1-метил-2-пирролидиноне (35 мл). Добавляли бензилбромид (3,12 мл, удельный вес: 1,44 г/см3) и смесь в достаточной мере охлаждали на ледяной бане в атмосфере азота. Добавляли гидрид натрия (60%, 2,12 г) с последующим перемешиванием в течение одного часа и 30 минут. Медленно добавляли воду и насыщенный хлорид аммония и затем добавляли этилацетат с последующим дополнительным перемешиванием. Водный слой отделяли и затем органический слой последовательно промывали насыщенным раствором хлорида аммония, водой и насыщенным раствором соли. Полученный органический слой сушили над безводным сульфатом магния и твердое вещество удаляли фильтрованием. Фильтрат концентрировали при пониженном давлении и затем очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (4,14 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,55-1,68 (м, 3H), 1,73-1,81 (м, 1H), 2,27-2,41 (м, 2H), 3,30 (д, J=2,0 Гц, 6H), 3,44-3,50 (м, 1H), 4,33-4,37 (м, 1H), 4,47-4,59 (м, 2H), 5,05-5,12 (м, 2H), 5,79-5,90 (м, 1H), 7,28-7,34 (м, 5H).
(2) Синтез 4-бензилоксигепт-6-енальоксима
Указанное в заголовке соединение (6,42 г) получали из [1-(3,3-диметоксипропил)-бут-3-енилоксиметил]бензола, полученного в примере получения 62-(1) (7,78 г), в соответствии со способом примера получения 57-(3) без очистки хроматографией на колонке с силикагелем.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,70-1,75 (м, 2H), 2,22-2,43 (м, 3H), 2,46-2,51 (м, 1H), 3,45-3,52 (м, 1H), 4,46-4,50 (м, 1H), 4,58-4,61 (м, 1H), 5,06-5,14 (м, 2H), 5,78-5,89 (м, 1H), 6,71-6,74 и 7,41-7,44 (м, суммарн. 1H), 7,16 и 7,53 (уш., суммарн. 1H), 7,26-7,32 (м, 1H), 7,34-7,35 (м, 4H).
(3) Синтез 5-бензилокси-3,3a,4,5,6,7-гексагидробенз[c]изоксазола
Указанное в заголовке соединение (5,66 г) получали из 4-бензилоксигепт-6-енальоксима, полученного в примере получения 62-(2) (6,42 г), в соответствии со способом примера получения 57-(4).
ESI-MS; m/z 232 [M+H].
(4) Синтез (±)-(3aR*,5S*,7aS*)-5-бензилокси-7a-(2-фторфенил)октагидробенз[c]изоксазола и (±)-(3aR*,5R*,7aS*)-5-бензилокси-7a-(2-фторфенил)октагидробенз[c]изоксазола
Указанные в заголовке соединения (±)-(3aR*,5S*,7aS*)-5-бензилокси-7a-(2-фторфенил)октагидробенз[c]изоксазол (5,04 г) и (±)-(3aR*,5R*,7aS*)-5-бензилокси-7a-(2-фторфенил)октагидробенз[c]изоксазол (2,49 г) получали из 5-бензилокси-3,3a,4,5,6,7-гексагидробенз[c]изоксазола, полученного в примере получения 62-(3) (5,66 г), в соответствии со способом примера получения 57-(5).
(±)-(3aR*,5S*,7aS*)-5-Бензилокси-7a-(2-фторфенил)октагидробенз[c]изоксазол
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,63 (уш., 2H), 1,76-1,80 (м, 1H), 1,87 (уш., 1H), 2,10-2,15 (м, 1H), 2,58-2,64 (м, 1H), 3,21 (уш., 1H), 3,66 (уш., 2H), 3,81 (уш., 1H), 4,56-4,63 (м, 2H), 5,95 (уш., 1H), 7,02-7,07 (м, 1H), 7,11-7,15 (м, 1H), 7,23-7,31 (м, 2H), 7,35-7,43 (м, 4H), 7,82 (уш., 1H).
(±)-(3aR*,5R*,7aS*)-5-Бензилокси-7a-(2-фторфенил)октагидробенз[c]изоксазол
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,40-1,54 (м, 2H), 1,99-2,05 (м, 1H), 2,07-2,13 (м, 1H), 2,18-2,25 (м, 1H), 2,28-2,36 (м, 1H), 2,99-3,05 (м, 1H), 3,53-3,56 (м, 1H), 3,64-3,74 (м, 2H), 4,61 (д, J=1,6 Гц, 2H), 6,00 (уш., 1H), 7,01 (ддд, J=1,2, 8,4, 12,4 Гц, 1H), 7,13 (дт, J=1,2, 7,6 Гц, 1H), 7,21-7,27 (м, 1H), 7,28-7,33 (м, 1H), 7,36-7,37 (м, 4H), 7,87 (дт, J=1,6, 8,0 Гц, 1H).
Пример получения 63
Синтез трет-бутил (-)-[(4aR*,6R*,8aS*)-8a-(5-амино-2-фторфенил)-6-фтор-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата
[Формула 85]
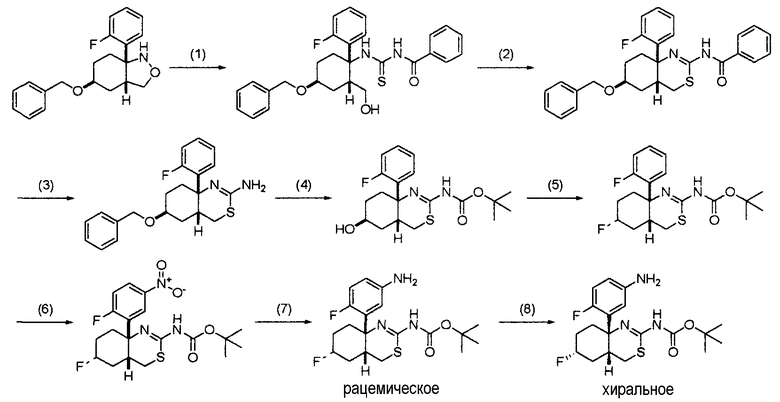
(1) Синтез (±)-1-бензоил-3-[(1S*,2R*,4S*)-4-бензилокси-1-(2-фторфенил)-2-гидроксиметилциклогексил]тиомочевины
Указанное в заголовке соединение (6,24 г) получали из (±)-(3aR*,5S*,7aS*)-5-бензилокси-7a-(2-фторфенил)октагидробенз[c]изоксазола, полученного в примере получения 62-(4) (5,04 г), в соответствии со способом примера получения 57-(6).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,40-1,80 (м, 3H), 1,99-2,02 (м, 1H), 2,13-2,19 (м, 3H), 3,54-3,73 (м, 3H), 3,90 (с, 1H), 4,54-4,65 (м, 2H), 7,00-7,08 (м, 1H), 7,16 (уш., 1H), 7,26-7,30 (м, 2H), 7,35-7,41 (м, 4H), 7,44-7,47 (м, 1H), 7,50-7,54 (м, 2H), 7,61-7,64 (м, 1H), 7,88 (д, J=3,6 Гц, 2H), 8,88 (уш., 1H), 11,52 (уш., 1H).
(2) Синтез (±)-N-[(4aR*,6S*,8aS*)-6-бензилокси-8a-(2-фторфенил)-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]бензамида
Указанное в заголовке соединение (4,57 г) получали из (±)-1-бензоил-3-[(1S*,2R*,4S*)-4-бензилокси-1-(2-фторфенил)-2-гидроксиметилциклогексил]тиомочевины, полученной в примере получения 63-(1) (6,24 г), в соответствии со способом примера получения 57-(7).
ESI-MS; m/z 475 [M++H].
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,68 (д, J=14,4 Гц, 1H), 1,81-1,89 (м, 1H), 1,93-1,97 (м, 1H), 2,03-2,10 (м, 2H), 2,52 (дд, J=3,2, 12,8 Гц, 1H), 2,85-2,93 (м, 1H), 2,97 (дд, J=4,0, 12,8 Гц, 1H), 3,43-3,49 (м, 1H), 3,92 (с, 1H), 4,54-4,66 (м, 2H), 7,09-7,17 (м, 2H), 7,28-7,45 (м, 9H), 7,48-7,52 (м, 1H), 8,24-8,27 (м, 2H), 12,32 (уш., 1H).
(3) Синтез (±)-(4aR*,6S*,8aS*)-6-бензилокси-8a-(2-фторфенил)-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-иламина
Указанное в заголовке соединение (3,96 г, чистота согласно 1H-ЯМР: примерно 90%) получали из (±)-N-[(4aR*,6S*,8aS*)-6-бензилокси-8a-(2-фторфенил)-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]бензамида, полученного в примере получения 63-(2) (4,58 г), в соответствии со способом примера получения 61-(1).
ESI-MS; m/z 371 [M++H].
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,54-1,72 (м, 1H), 1,74-1,83 (м, 2H), 1,84-1,88 (м, 1H), 1,90-1,97 (м, 1H), 2,48 (дд, J=2,8, 12,0 Гц, 1H), 2,76 (дт, J=3,6, 13,2 Гц, 1H), 2,87-2,91 (м, 1H), 3,18-3,24 (м, 1H), 3,84 (т, J=2,8 Гц, 1H), 4,54 (уш., 2H), 4,51-4,66 (м, 2H), 7,04 (ддд, J=1,2, 8,0, 12,4 Гц, 1H), 7,09 (дт, J=1,6, 7,6 Гц, 1H), 7,19-7,25 (м, 1H), 7,26-7,31 (м, 2H), 7,35-7,39 (м, 2H), 7,43-7,45 (м, 2H).
(4) Синтез трет-бутил (±)-[(4aR*,6S*,8aS*)-8a-(2-фторфенил)-6-гидрокси-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата
Указанное в заголовке соединение (3,58 г) получали из (±)-(4aR*,6S*,8aS*)-6-бензилокси-8a-(2-фторфенил)-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-иламина, полученного в примере получения 63-(3) (3,96 г, чистота согласно 1H-ЯМР: примерно 90%), в соответствии со способом примера получения 58-(8).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,54-1,68 (м, 1H), 1,71-1,79 (м, 2H), 1,87-1,96 (м, 1H), 2,07-2,15 (м, 1H), 2,45 (дд, J=2,8, 10,0 Гц, 1H), 2,82-2,91 (м, 2H), 3,31-3,45 (м, 2H), 4,29-4,33 (м, 1H), 7,06-7,11 (м, 1H), 7,15-7,19 (м, 1H), 7,29-7,34 (м, 2H).
(5) Синтез трет-бутил (±)-[(4aR*,6R*,8aS*)-6-фтор-8a-(2-фторфенил)-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата
трет-Бутил (±)-[(4aR*,6S*,8aS*)-8a-(2-фторфенил)-6-гидрокси-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамат, полученный в примере получения 63-(4) (1,5 г), растворяли в ТГФ. Затем перфторбутансульфонилфторид (1,36 мл, удельный вес: 1,682 г/см3), комплекс триэтиламин-трифтороводородная кислота (1,23 мл, удельный вес: 0,989 г/см3) и триэтиламин (3,15 мл, удельный вес: 0,73 г/см3) последовательно добавляли при комнатной температуре. По окончании добавления смесь перемешивали в течение 16 часов. Смесь нагревали до 50°C и перемешивали еще в течение 24 часов. Перфторбутансульфонилфторид (0,68 мл), комплекс триэтиламин-трифтороводородная кислота (0,62 мл) и триэтиламин (1,58 мл) еще последовательно добавляли к смеси с последующим дополнительным перемешиванием при кипении с обратным холодильником в течение восьми часов. Реакционный раствор охлаждали до комнатной температуры и концентрировали при пониженном давлении. Затем остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (106 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,53 (с, 9H), 1,84-1,89 (м, 2H), 2,06-2,13 (м, 3H), 2,39-2,47 (м, 1H), 2,52 (дд, J=3,2, 12,8 Гц, 1H), 2,80-2,85 (м, 1H), 2,89-2,97 (м, 1H), 4,69-4,90 (м, 1H), 7,04-7,11 (м, 1H), 7,15-7,20 (м, 1H), 7,27-7,34 (м, 2H).
(6) Синтез трет-бутил (±)-[(4aR*,6R*,8aS*)-6-фтор-8a-(2-фтор-5-нитрофенил)-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата
Указанное в заголовке соединение (406 мг) получали из трет-бутил (±)-[(4aR*,6R*,8aS*)-6-фтор-8a-(2-фторфенил)-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата, полученного в примере получения 63-(5) (402 мг), в соответствии со способом примера получения 57-(9), используя дымящую азотную кислоту (87,1 мкл, два эквивалента относительно исходного вещества).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,55 (с, 9H), 1,80-1,88 (м, 2H), 2,06 (уш., 3H), 2,18-2,27 (м, 1H), 2,56 (дд, J=2,8, 12,8 Гц, 1H), 2,75-2,80 (м, 1H), 2,87 (уш., 1H), 4,66-4,86 (м, 1H), 7,19-7,25 (м, 1H), 8,14-8,22 (м, 2H).
(7) Синтез трет-бутил (±)-[(4aR*,6R*,8aS*)-8a-(5-амино-2-фторфенил)-6-фтор-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата
Указанное в заголовке соединение (330 мг) получали из трет-бутил (±)-[(4aR*,6R*,8aS*)-6-фтор-8a-(2-фтор-5-нитрофенил)-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата, полученного в примере получения 63-(6) (406 мг), в соответствии со способом примера получения 57-(10).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,54 (с, 9H), 1,80-1,88 (м, 2H), 2,04-2,11 (м, 3H), 2,38-2,47 (м, 1H), 2,48-2,53 (м, 1H), 2,89-2,95 (м, 2H), 3,65 (с, 2H), 4,68-4,88 (м, 1H), 6,51-6,57 (м, 2H), 6,83-6,88 (м, 1H).
ВЭЖХ (CHIRALPAK™ AD-H производства Daicel Chemical Industries, Ltd., 2 см × 25 см, подвижная фаза: гексан:этанол = 80:20, скорость потока: 1 мл/мин): 4,4 минуты (оптическое вращение (+)), 6,8 минут (оптическое вращение (-))
(8) Синтез трет-бутил (-)-[(4aR*,6R*,8aS*)-8a-(5-амино-2-фторфенил)-6-фтор-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата
трет-Бутил (±)-[(4aR*,6R*,8aS*)-8a-(5-амино-2-фторфенил)-6-фтор-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамат, полученный в примере получения 58-(12) (330 мг), оптически разделяли, используя CHIRALPAK™ AD-H производства Daicel Chemical Industries, Ltd. (2 см × 25 см, подвижная фаза: гексан:этанол = 50:50, скорость потока: 10 мл/мин, загружаемый раствором примерно 40 мг в 1 мл этанола для одного цикла). Из двух основных компонентов компонент, имеющий время удерживания от 12,2 до 15,5 минут, собирали с получением указанного в заголовке соединения (155 мг, >99% ee, оптическое вращение (-)).
ВЭЖХ (CHIRALPAK™ AD-H производства Daicel Chemical Industries, Ltd., 2 см × 25 см, подвижная фаза: гексан:этанол = 80:20, скорость потока: 1 мл/мин): 7,0 минут.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,54 (с, 9H), 1,80-1,88 (м, 2H), 2,04-2,12 (м, 3H), 2,38-2,46 (м, 1H), 2,48-2,53 (м, 1H), 2,89-2,94 (м, 2H), 3,66 (уш., 2H), 4,68-4,88 (м, 1H), 6,51-6,57 (м, 2H), 6,83-6,88 (м, 1H).
Пример получения 64
Синтез трет-бутил (-)-[(4aR*,6S*,8aS*)-8a-(5-амино-2-фторфенил)-6-фтор-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата
[Формула 86]
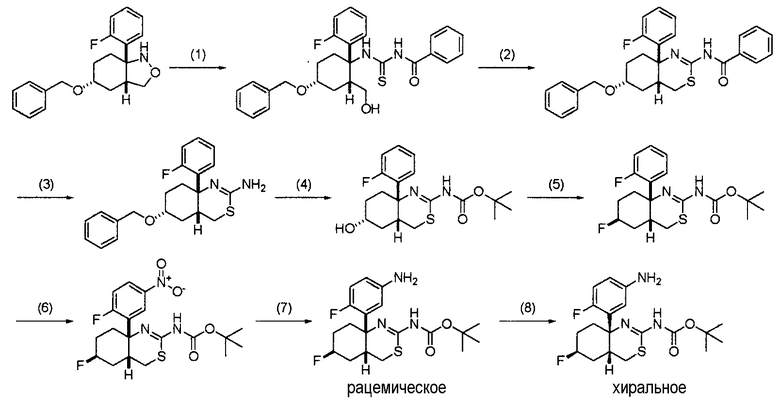
(1) Синтез (±)-1-бензоил-3-[(1S*,2R*,4R*)-4-бензилокси-1-(2-фторфенил)-2-гидроксиметилциклогексил]тиомочевины
Указанное в заголовке соединение (3,13 г) получали из (±)-(3aR*,5R*,7aS*)-5-бензилокси-7a-(2-фторфенил)октагидробенз[c]изоксазола, полученного в примере получения 62-(4) (2,48 г), в соответствии со способом примера получения 57-(6).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,59-1,69 (м, 2H), 1,95-2,10 (м, 2H), 2,31-2,34 (м, 2H), 3,58-3,72 (м, 3H), 3,85 (уш., 1H), 4,64 (с, 2H), 7,02-7,08 (м, 1H), 7,12-7,16 (м, 1H), 7,27-7,30 (м, 2H), 7,35-7,46 (м, 5H), 7,51-7,55 (м, 2H), 7,61-7,65 (м, 1H), 7,86-7,89 (м, 2H), 8,89 (уш., 1H), 11,56 (уш., 1H).
(2) Синтез (±)-N-[(4aR*,6R*,8aS*)-6-бензилокси-8a-(2-фторфенил)-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]бензамида
Указанное в заголовке соединение (2,03 г) получали из (±)-1-бензоил-3-[(1S*,2R*,4R*)-4-бензилокси-1-(2-фторфенил)-2-гидроксиметилциклогексил]тиомочевины, полученной в примере получения 64-(1) (3,13 г), в соответствии со способом примера получения 57-(7).
ESI-MS; m/z 475 [M++H].
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,80 (кв, J=12,8 Гц, 1H), 1,93 (д, J=14,8 Гц, 1H), 2,01-2,17 (м, 3H), 2,42-2,49 (м, 1H), 2,59 (д, J=12,4 Гц, 1H), 2,93 (д, J=12,8 Гц, 1H), 3,00 (д, J=10,8, 1H), 3,68-3,73 (м, 1H), 4,62 (с, 2H), 7,05-7,11 (м, 1H), 7,14-7,18 (м, 1H), 7,29-7,36 (м, 7H), 7,41-7,45 (м, 2H), 7,48-7,52 (м, 1H), 8,24 (д, J=7,2 Гц, 2H), 12,30 (уш., 1H).
(3) Синтез (±)-(4aR*,6S*,8aS*)-6-бензилокси-8a-(2-фторфенил)-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-иламина
Указанное в заголовке соединение (1,50 г) получали из (±)-N-[(4aR*,6R*,8aS*)-6-бензилокси-8a-(2-фторфенил)-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]бензамида, полученного в примере получения 64-(2) (2,03 г), в соответствии со способом примера получения 58-(7).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,64-1,77 (м, 1H), 1,80-1,86 (м, 2H), 1,88-1,94 (м, 1H), 2,00-2,06 (м, 1H), 2,26-2,33 (м, 1H), 2,53-2,57 (м, 1H), 2,71-2,76 (м, 1H), 2,84-2,88 (м, 1H), 3,61-3,69 (м, 1H), 4,43 (уш., 2H), 4,62 (д, J=4,4 Гц, 2H), 7,00 (ддд, J=1,2, 8,0, 12,8 Гц, 1H), 7,07-7,11 (м, 1H), 7,19-7,24 (м, 1H), 7,28-7,31 (м, 2H), 7,33-7,39 (м, 4H).
(4) Синтез трет-бутил (±)-[(4aR*,6R*,8aS*)-8a-(2-фторфенил)-6-гидрокси-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата
Указанное в заголовке соединение (1,59 г) получали из (±)-(4aR*,6R*,8aS*)-6-бензилокси-8a-(2-фторфенил)-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-иламина, полученного в примере получения 64-(3) (1,50 г), в соответствии со способом примера получения 58-(8).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,53 (с, 9H), 1,62-1,73 (м, 1H), 1,80-2,02 (м, 4H), 2,42-2,54 (м, 2H), 2,80-2,84 (м, 1H), 2,93-2,99 (м, 1H), 3,90-3,98 (м, 1H), 7,04-7,09 (м, 1H), 7,15-7,20 (м, 1H), 7,28-7,34 (м, 2H).
(5) Синтез трет-бутил (±)-[(4aR*,6S*,8aS*)-6-фтор-8a-(2-фторфенил)-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата
трет-Бутил (±)-[(4aR*,6R*,8aS*)-8a-(2-фторфенил)-6-гидрокси-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамат, полученный в примере получения 64-(4) (500 мг), растворяли в ТГФ. Затем перфторбутансульфонилфторид (0,43 мл, удельный вес: 1,682 г/см3), комплекс триэтиламин-трифтороводородная кислота (0,39 мл, удельный вес: 0,989 г/см3) и триэтиламин (0,99 мл, удельный вес: 0,73 г/см3) последовательно добавляли при комнатной температуре. По окончании добавления смесь перемешивали в течение 15 часов. Реакционный раствор концентрировали при пониженном давлении и затем остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (351 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,53 (с, 9H), 1,63-1,67 (м, 1H), 1,76-2,19 (м, 4H), 2,46-2,49 (м, 1H), 2,76-2,83 (м, 1H), 2,88-2,92 (м, 1H), 3,29-3,32 (м, 1H), 5,04 (д, J=48,0 Гц, 1H), 7,07-7,12 (м, 1H), 7,16-7,20 (м, 1H), 7,30-7,32 (м, 2H).
(6) Синтез трет-бутил (±)-[(4aR*,6S*,8aS*)-6-фтор-8a-(2-фтор-5-нитрофенил)-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата
Указанное в заголовке соединение (266 мг) получали из трет-бутил (±)-[(4aR*,6S*,8aS*)-6-фтор-8a-(2-фторфенил)-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата, полученного в примере получения 64-(5) (351 мг), в соответствии со способом примера получения 57-(9) добавлением ди-трет-бутилдикарбоната (401 мг), перемешиванием при комнатной температуре в течение 14 часов и затем снова добавлением ди-трет-бутилдикарбоната (200 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,54 (с, 9H), 1,65-1,68 (м, 1H), 1,77-1,91 (м, 1H), 1,98-2,14 (м, 3H), 2,54 (дд, J=2,8, 12,8 Гц, 1H), 2,61-2,67 (м, 1H), 2,86 (дд, J=3,6, 12,8 Гц, 1H), 3,27 (уш., 1H), 5,03 (д, J=48,4 Гц, 1H), 7,23-7,28 (м, 1H), 8,18-8,23 (м, 2H).
(7) Синтез трет-бутил (±)-[(4aR*,6S*,8aS*)-8a-(5-амино-2-фторфенил)-6-фтор-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата
Указанное в заголовке соединение (191 мг) получали из трет-бутил (±)-[(4aR*,6S*,8aS*)-6-фтор-8a-(2-фтор-5-нитрофенил)-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата, полученного в примере получения 64-(6) (266 мг), в соответствии со способом примера получения 57-(10).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,53 (с, 9H), 1,56-1,68 (м, 1H), 1,75-1,93 (м, 1H), 1,99-2,16 (м, 3H), 2,47 (д, J=13,2 Гц, 1H), 2,78 (т, J=13,2 Гц, 1H), 2,98 (д, J=12,0 Гц, 1H), 3,26-3,28 (м, 1H), 3,65 (уш., 2H), 5,03 (д, J=48,0 Гц, 1H), 6,53-6,58 (м, 2H), 6,85-6,90 (м, 1H).
ВЭЖХ (CHIRALPAK™ AD-H производства Daicel Chemical Industries, Ltd., 2 см × 25 см, подвижная фаза: гексан:этанол = 80:20, скорость потока: 1 мл/мин): 3,3 минуты (оптическое вращение (+)), 5,9 минут (оптическое вращение (-))
(8) Синтез трет-бутил (-)-[(4aR*,6S*,8aS*)-8a-(5-амино-2-фторфенил)-6-фтор-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата
трет-Бутил (±)-[(4aR*,6S*,8aS*)-8a-(5-амино-2-фторфенил)-6-фтор-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамат, полученный в примере получения 58-(12) (191 мг), оптически разделяли, используя CHIRALPAK™ AD-H производства Daicel Chemical Industries, Ltd. (2 см × 25 см, подвижная фаза: гексан:этанол = 50:50, скорость потока: 10 мл/мин, загружаемый раствором примерно 6 мг в 2 мл этанола для одного цикла). Из двух основных компонентов компонент, имеющий время удерживания от 10,9 до 12,6 минут, собирали с получением указанного в заголовке соединения (72 мг, >99% ee, оптическое вращение (-)).
ВЭЖХ (CHIRALPAK™ AD-H производства Daicel Chemical Industries, Ltd., 2 см × 25 см, подвижная фаза: гексан:этанол = 80:20, скорость потока: 1 мл/мин): 5,9 минут.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,52 (с, 9H), 1,60-1,68 (м, 1H), 1,74-1,93 (м, 1H), 1,98-2,15 (м, 3H), 2,47 (д, J=12,8 Гц, 1H), 2,77 (т, J=13,2 Гц, 1H), 2,97 (д, J=12,4 Гц, 1H), 3,26 (д, J=10,0 Гц, 1H), 3,72 (уш., 2H), 5,02 (д, J=48,0 Гц, 1H), 6,53-6,58 (м, 2H), 6,85-6,90 (м, 1H).
Пример получения 65
Синтез (±)-N,N-бис(трет-бутоксикарбонил)[(4aR*,7aS*)-7a-(3-бромфенил)-6-пиразин-2-ил-4,4a,5,6,7,7a-гексагидропирроло[3,4-d][1,3]тиазин-2-ил]амина
[Формула 87]
(1) Синтез (±)-N-[(4aR*,7aS*)-7a-(3-бромфенил)-4,4a,5,6,7,7a-гексагидропирроло[3,4-d][1,3]тиазин-2-ил]бензамида
Указанное в заголовке соединение (7,52 г) получали из (±)-N-[(4aR*,7aS*)-7a-(3-бромфенил)-6-(2,4-диметоксибензил)-4,4a,5,6,7,7a-гексагидропирроло[3,4-d][1,3]тиазин-2-ил]бензамида, полученного в примере получения 18-(5) (14,5 г), в соответствии со способом примера получения 18-(9).
ESI-MS; m/z 416 [M+H].
(2) Синтез (±)-N-(4aR*,7aS*)-7a-(3-бромфенил)-6-пиразин-2-ил-4,4a,5,6,7,7a-гексагидропирроло[3,4-d][1,3]тиазин-2-иламина
2-Хлорпиразин (2,14 мл, удельный вес: 1,284 г/см3) добавляли к (±)-N-[(4aR*,7aS*)-7a-(3-бромфенил)-4,4a,5,6,7,7a-гексагидропирроло[3,4-d][1,3]тиазин-2-ил]бензамиду, полученному в примере получения 65-(1) (2 г), и смесь нагревали до 90°C и перемешивали в атмосфере азота. Через 13 часов реакционный раствор охлаждали до комнатной температуры. Добавляли хлороформ и насыщенный раствор бикарбоната натрия с последующим экстрагированием три раза хлороформом. Полученные органические слои сушили над безводным сульфатом магния и твердое вещество удаляли фильтрованием. Полученный фильтрат концентрировали при пониженном давлении и затем очищали хроматографией на колонке с NH-силикагелем. К смеси, содержащей полученное N-арильное соединение, добавляли метанол (9,6 мл). Затем добавляли раствор метоксида натрия в метаноле (373 мкл, 25%, удельный вес: 0,945 г/см3) и смесь перемешивали при кипении с обратным холодильником. Через три часа и 50 минут реакционный раствор охлаждали до комнатной температуры. Добавляли хлороформ, насыщенный раствор бикарбоната натрия и насыщенный раствор соли с последующим экстрагированием три раза хлороформом. Полученные органические слои сушили над безводным сульфатом магния и твердое вещество удаляли фильтрованием. Полученный фильтрат концентрировали при пониженном давлении и затем очищали хроматографией на колонке с NH-силикагелем с получением указанного в заголовке соединения (138 мг).
ESI-MS; m/z 390 [M+H].
(3) Синтез (±)-N,N-бис(трет-бутоксикарбонил)[(4aR*,7aS*)-7a-(3-бромфенил)-6-пиразин-2-ил-4,4a,5,6,7,7a-гексагидропирроло[3,4-d][1,3]тиазин-2-ил]амина
(±)-N-(4aR*,7aS*)-7a-(3-Бромфенил)-6-пиразин-2-ил-4,4a,5,6,7,7a-гексагидропирроло[3,4-d][1,3]тиазин-2-иламин, полученный в примере получения 65-(2) (138 мг), растворяли в ТГФ (10 мл). Затем добавляли ди-трет-бутилдикарбонат (232 мг) и 4-диметиламинопиридин (151 мг) и смесь перемешивали при комнатной температуре. Через 11 часов и 30 минут реакционный раствор разбавляли этилацетатом. Затем добавляли насыщенный раствор хлорида аммония с последующим экстрагированием этилацетатом. Полученный органический слой последовательно промывали водой и насыщенным раствором соли и сушили над безводным сульфатом магния. Твердое вещество удаляли фильтрованием. Фильтрат концентрировали при пониженном давлении и затем остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (154 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,41 (м, 18H), 2,88-3,12 (м, 3H), 3,17-3,21 (м, 1H), 3,80-3,96 (м, 3H), 7,40-7,47 (м, 3H), 7,65-7,66 (м, 1H), 7,85 (д, J=2,0 Гц, 1H), 7,88 (д, J=1,6 Гц, 1H), 8,04-8,05 (м, 1H).
Следующие соединения синтезировали из этил 2-трифторметансульфонилоксициклогекс-1-енкарбоксилата и соответствующих бороновых кислот в соответствии с примером получения 1.
Пример получения 66
Синтез трет-бутил (±)-[(4aR*,8aS*)-8a-(5-амино-2,4-дифторфенил)-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата
[Формула 88]
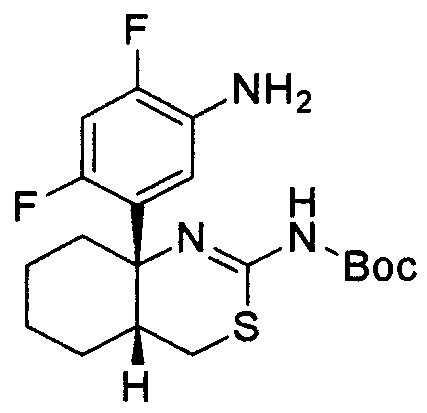
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,53 (с, 9H), 1,46-1,73 (м, 6H), 1,83-1,89 (м, 1H), 2,28-2,34 (м, 1H), 2,48 (дд, J=3,2, 12,4 Гц, 1H), 2,77-2,81 (м, 1H), 2,87 (дд, J=4,0, 12,4 Гц, 1H), 3,68 (с, 2H), 6,66 (дд, J=8,0, 9,6 Гц, 1H), 6,78 (дд, J=10,4, 11,6 Гц, 1H).
Пример получения 67
Синтез трет-бутил (±)-[(4aR*,8aS*)-8a-(5-амино-2,6-дифторфенил)-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата
[Формула 89]

1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,51 (с, 9H), 1,43-2,05 (м, 7H), 2,16-2,24 (м, 1H), 2,54-2,58 (м, 1H), 2,97-3,01 (м, 1H), 3,18 (дд, J=4,4, 12,8 Гц, 1H), 3,63 (с, 2H), 6,64-6,73 (м, 2H).
Пример получения 68
Синтез 5-(1-этоксивинил)пиридин-2-карбоновой кислоты
[Формула 90]
(1) Синтез этил 5-(1-этоксивинил)пиридин-2-карбоксилата
1-Этоксивинилтри-н-бутилолово (880 мкл) и тетракис(трифенилфосфин)палладий (125 мг) добавляли к раствору 5-бромпиридин-2-карбоновой кислоты (500 мг) в ДМФА (10 мл). После замены азотом смесь нагревали до 85°C и перемешивали в течение ночи. После охлаждения до комнатной температуры к реакционной смеси добавляли этилацетат. Органический слой промывали водой и насыщенным раствором соли и сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (342 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,42-1,47 (м, 6H), 3,96 (кв, J=6,4 Гц, 2H), 4,40 (д, J=3,4 Гц, 1H), 4,49 (кв, J=6,8 Гц, 2H), 4,81 (д, J=3,4 Гц, 1H), 8,03 (дд, J=2,2, 8,4 Гц, 1H), 8,10 (дд, J=0,8, 8,4 Гц, 1H), 8,98 (дд, J=0,8, 2,2 Гц, 1H).
(2) Синтез 5-(1-этоксивинил)пиридин-2-карбоновой кислоты
К раствору этил 5-(1-этоксивинил)пиридин-2-карбоксилата (100 мг) в этаноле (5 мл) добавляли 5 н. раствор гидроксида натрия (2 мл). Смесь перемешивали при комнатной температуре в течение двух часов. После подтверждения окончания реакции реакционный раствор нейтрализовали 1 н. хлористо-водородной кислотой. Водный слой экстрагировали этилацетатом и органический слой сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении с получением указанного в заголовке соединения (92 мг).
1H-ЯМР (400 МГц, CD3OD) δ (м.д.): 1,44 (т, J=6,8 Гц, 3H), 3,99 (кв, J=6,8 Гц, 2H), 4,52 (д, J=3,4 Гц, 1H), 4,96 (д, J=3,4 Гц, 1H), 8,13 (дд, J=0,8, 8,4 Гц, 1H), 8,21 (дд, J=2,4, 8,4 Гц, 1H), 8,90 (дд, J=0,8, 2,4 Гц, 1H).
Пример получения 69
Синтез 3-метоксипиридин-2-карбоновой кислоты
[Формула 91]
(1) Синтез метил 3-метоксипиридин-2-карбоксилата
Гидрид натрия (60%, 253 мг) добавляли к раствору 3-гидроксипиколиновой кислоты (440 мг) в ДМФА (4 мл) на ледяной бане. После перемешивания при комнатной температуре в течение 30 минут добавляли йодметан (393 мкл) и смесь перемешивали при комнатной температуре в течение ночи. К реакционной смеси добавляли лед, чтобы остановить реакцию. Водный слой экстрагировали этилацетатом и органический слой промывали водой и насыщенным раствором соли. Органический слой сушили над безводным сульфатом магния и затем выпаривали при пониженном давлении растворитель. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (112 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 3,85 (с, 3H), 3,90 (с, 3H), 7,30 (дд, J=1,2, 8,6 Гц, 1H), 7,35 (дд, J=4,4, 8,6 Гц, 1H), 8,20 (дд, J=1,2, 4,4 Гц, 1H).
(2) Синтез 3-метоксипиридин-2-карбоновой кислоты
К раствору метил 3-метоксипиридин-2-карбоксилата (112 мг) в метаноле (2 мл) добавляли 5 н. раствор гидроксида натрия (147 мкл) и смесь перемешивали при комнатной температуре в течение ночи. Избыток метанола выпаривали при пониженном давлении и остаток разбавляли водой. Водный слой промывали эфиром и затем слабо подкисляли 5 н. хлористо-водородной кислотой. Водный слой экстрагировали смешанным растворителем ТГФ-этилацетат и затем органический слой сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении с получением указанного в заголовке соединения (19 мг).
1H-ЯМР (400 МГц, CD3OD) δ (м.д.): 3,95 (с, 3H), 7,96 (дд, J=4,4, 8,2 Гц, 1H), 7,71 (д, J=8,2 Гц, 1H), 8,18 (д, J=4,4 Гц, 1H).
Пример получения 70
Синтез 5-(1-метил-1H-пиразол-4-ил)пиридин-2-карбоновой кислоты
[Формула 92]

(1) Синтез трет-бутил 5-(1-метил-1H-пиразол-4-ил)пиридин-2-карбоксилата
1-Метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол (80,7 мг), бис(три-трет-бутилфосфин)палладий(0) (9,91 мг) и 1 н. раствор фосфата калия (388 мкл) добавляли к раствору трет-бутил 5-бромпиридин-2-карбоксилата (50 мг) в 1,4-диоксане (2 мл). После замены азотом смесь нагревали до 100°C и перемешивали в течение восьми часов. Реакционный раствор охлаждали до комнатной температуры и выпаривали при пониженном давлении растворитель. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения в виде смеси (81 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,65 (с, 9H), 3,99 (с, 3H), 7,74 (с, 1H), 7,82-7,85 (м, 2H), 8,04 (дд, J=1,2, 8,4 Гц, 1H), 8,85 (дд, J=0,8, 2,4 Гц, 1H).
ESI-MS; m/z 260 [M++H].
(2) Синтез 5-(1-метил-1H-пиразол-4-ил)пиридин-2-карбоновой кислоты
ТФУК (1 мл) добавляли к раствору трет-бутил 5-(1-метил-1H-пиразол-4-ил)пиридин-2-карбоксилата (81 мг) в дихлорметане (4 мл) и смесь перемешивали при комнатной температуре в течение ночи. Растворитель выпаривали при пониженном давлении с получением неочищенного продукта, состоящего из указанного в заголовке соединения (170 мг).
1H-ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 3,90 (с, 3H), 8,03 (дд, J=0,8, 8,2 Гц, 1H), 8,09 (д, J=0,8 Гц, 1H), 8,14 (дд, J=2,0, 8,2 Гц, 1H), 8,41 (с, 1H), 8,95 (дд, J=0,8, 2,0 Гц, 1H).
Пример получения 71
Синтез (±)-N,N-бис(трет-бутоксикарбонил)[(4aR*,7aS*)-7a-(3-амино-5-хлорфенил)-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]амина
[Формула 93]

(1) Синтез (±)-(4aR*,7aS*)-7a-(3-бром-5-хлорфенил)-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-иламина
Указанное в заголовке соединение синтезировали из гекс-5-енальоксима (JOC, 41(5), 863-9; 1976) в соответствии со способом примера получения 8, используя 1,3-дибром-5-хлорбензол вместо 2-фторбромбензола.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,66-2,00 (м, 5H), 2,17-2,24 (м, 1H), 2,33-2,39 (м, 1H), 2,73 (дд, J=4,0, 12,6 Гц, 1H), 2,97 (дд, J=3,2, 12,6 Гц, 1H), 4,36 (уш., 2H), 7,27 (т, J=2,0 Гц, 1H), 7,36 (т, J=2,0 Гц, 1H), 7,37 (т, J=1,6 Гц, 1H).
(2) Синтез (±)-N,N-бис(трет-бутоксикарбонил)[(4aR*,7aS*)-7a-(3-бром-5-хлорфенил)-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]амина
Ди-трет-бутилдикарбонат (1,05 г) и 4-диметиламинопиридин (393 мг) добавляли к раствору (±)-(4aR*,7aS*)-7a-(3-бром-5-хлорфенил)-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-иламина (555 мг) в дихлорметане (30 мл). Смесь перемешивали при комнатной температуре в течение ночи и затем выпаривали при пониженном давлении растворитель при комнатной температуре или ниже. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (140 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,53 (с, 18Н), 1,77-2,14 (м, 5H), 2,22-2,30 (м, 1H), 2,48-2,54 (м, 1H), 2,82 (дд, J=3,2, 13,0 Гц, 1H), 3,06 (дд, J=3,6, 13,0 Гц, 1H), 7,34 (т, J=2,0 Гц, 1H), 7,38 (т, J=1,6 Гц, 1H), 7,44 (т, J=1,6 Гц, 1H).
ESI-MS; m/z 547 [M++H].
(±)-N-(трет-Бутоксикарбонил)-N-(метоксикарбонил)[(4aR*,7aS*)-7a-(3-бром-5-хлорфенил)-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]амин, образующийся во время очистки (575 мг), получали в качестве побочного продукта.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,55 (с, 9H), 1,78-2,04 (м, 4H), 2,12 (ддд, J=3,2, 7,2, 13,2 Гц, 1H), 2,26-2,33 (м, 1H), 2,44-2,50 (м, 1H), 2,83 (дд, J=3,6, 12,8 Гц, 1H), 3,06 (дд, J=3,2, 12,8 Гц, 1H), 3,92 (с, 3H), 7,32 (т, J=2,0 Гц, 1H), 7,40 (т, J=2,0 Гц, 1H), 7,42 (т, J=1,6 Гц, 1H).
ESI-MS; m/z 505 [M++H].
(3) Синтез (±)-N,N-бис(трет-бутоксикарбонил){(4aR*,7aS*)-7a-[3-хлор-5-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)фенил]-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил}амина
1,1'-Бис(дифенилфосфино)ферроцендихлорпалладий(II) (40,2 мг), бис(пинаколато)дибор (1,40 г) и ацетат калия (216 мг) добавляли к раствору (±)-N,N-бис(трет-бутоксикарбонил)[(4aR*,7aS*)-7a-(3-бром-5-хлорфенил)-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]амина (300 мг) в ДМФА (6 мл). После замены азотом смесь перемешивали при 80°C в течение шести часов. Реакционную смесь охлаждали до комнатной температуры и разбавляли этилацетатом. Органический слой промывали два раза водой и один раз насыщенным раствором соли и сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (1,16 г) в виде смеси.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,34 (с, 12H), 1,52 (с, 18H), 1,76-2,14 (м, 5H), 2,22-2,30 (м, 1H), 2,48-2,54 (м, 1H), 2,80 (дд, J=3,6, 13,2 Гц, 1H), 3,08 (дд, J=3,6, 13,2 Гц, 1H), 7,50 (т, J=2,0 Гц, 1H), 7,65 (м, 2H).
ESI-MS; m/z 593 [M++H].
(4) Синтез (±)-N,N-бис(трет-бутоксикарбонил)[(4aR*,7aS*)-7a-(3-азидо-5-хлорфенил)-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]амина
Азид натрия (53 мг) и ацетат меди(II) (19,9 мг) добавляли к раствору (±)-N,N-бис(трет-бутоксикарбонил){(4aR*,7aS*)-7a-[3-хлор-5-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)фенил]-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил}амина (1,16 г) в метаноле (20 мл). Смесь перемешивали при комнатной температуре в течение шести суток. К реакционной смеси добавляли насыщенный раствор соли с последующим экстрагированием этилацетатом. Органический слой сушили над безводным сульфатом магния и выпаривали при пониженном давлении растворитель. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (58,0 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,53 (с, 18H), 1,78-2,15 (м, 5H), 2,25-2,32 (м, 1H), 2,46-2,51 (м, 1H), 2,81 (дд, J=3,2, 12,8 Гц, 1H), 3,05 (дд, J=3,2, 12,8 Гц, 1H), 6,89 (т, J=2,0 Гц, 1H), 7,03 Гц (т, J=2,0 Гц, 1H), 7,16 (т, J=1,6 Гц, 1H).
ESI-MS; m/z 508 [M++H].
(5) Синтез (±)-N,N-бис(трет-бутоксикарбонил)[(4aR*,7aS*)-7a-(3-амино-5-хлорфенил)-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]амина
Формиат аммония (12,4 мг) и цинк (3,86 мг) добавляли к раствору (±)-N,N-бис(трет-бутоксикарбонил)[(4aR*,7aS*)-7a-(3-азидо-5-хлорфенил)-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]амина (20,0 мг) в метаноле (2 мл). Смесь перемешивали при комнатной температуре в течение ночи и затем избыток метанола выпаривали при пониженном давлении. Остаток разбавляли этилацетатом и органический слой промывали раствором хлорида аммония. Растворитель выпаривали при пониженном давлении и остаток очищали пТСХ с получением указанного в заголовке соединения (13,0 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,52 (с, 18H), 1,75-2,11 (м, 5H), 2,25-2,33 (м, 1H), 2,44-2,50 (м, 1H), 2,81 (дд, J=3,6, 12,8 Гц, 1H), 3,12 (дд, J=3,6, 12,8 Гц, 1H), 3,72 (уш.с, 2H), 6,53 (т, J=2,0 Гц, 1H), 6,65 Гц (т, J=1,6 Гц, 1H), 6,74 (т, J=1,6 Гц, 1H).
ESI-MS; m/z 482 [M++H].
Пример получения 72
Синтез (±)-N,N-бис(трет-бутоксикарбонил)[(4aR*,7aS*)-7a-(5-бром-2-фторфенил)-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]амина
[Формула 94]

4-Диметиламинопиридин (1,62 г) и ди-трет-бутилдикарбонат (2,31 г) добавляли к раствору соединения, полученного в примере получения 54-(4) (1,00 г), в дихлорметане (25 мл). После перемешивания при комнатной температуре в течение 14 часов добавляли к реакционной смеси воду, чтобы остановить реакцию. Водный слой экстрагировали дихлорметаном и органический слой сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (1,13 г).
ESI-MS; m/z 531 [M++H].
Пример получения 73
Синтез (±)-N,N-бис(трет-бутилоксикарбонил)[(4aR*,6R*,7aS*)-7a-(5-бром-2-фторфенил)-6-метокси-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]амина и (±)-N,N-бис(трет-бутилоксикарбонил)[(4aR*,6R*,7aS*)-7a-(5-бром-2-фторфенил)-6-метокси-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]амина
[Формула 95]

(1) Синтез (±)-(3aR*,5S*,6aS*)-6a-(5-бром-2-фторфенил)-5-метоксигексагидроциклопента[c]изоксазола и (±)-(3aR*,5R*,6aS*)-6a-(5-бром-2-фторфенил)-5-метоксигексагидроциклопента[c]изоксазола
Менее полярное указанное в заголовке соединение (5S; 62 мг) и более полярное указанное в заголовке соединение (5R; 170 мг) получали из соединения примера получения 10-(3) (смесь более полярного и менее полярного соединений; 450 мг) и 2,4-дибром-1-фторбензола (1,78 г) в соответствии с примером получения 21-(3).
Менее полярное указанное в заголовке соединение
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,08-2,26 (м, 3H), 3,21-3,29 (м, 2H), 3,30 (с, 3H),3,68 (т, J=8,0 Гц, 1H), 4,08 (т, J=5,2 Гц, 1H), 4,40 (т, J=8,4 Гц, 1H), 6,88 (дд, J=8,4, 11,2 Гц, 1H), 7,31 (ддд, J=2,8, 4,4, 8,4 Гц, 1H), 8,09 (дд, J=2,8, 7,2 Гц, 1H).
Более полярное указанное в заголовке соединение
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,03-2,20 (м, 3H), 2,33 (дд, J=5,8, 13,4 Гц, 1H), 3,23-3,29 (м, 1H), 3,35 (с, 3H), 3,59 (уш.с, 1H), 4,10-4,16 (м, 1H), 4,25-4,28 (м, 1H), 6,94 (дд, J=9,0, 10,8 Гц, 1H), 7,35 (ддд, J=2,8, 4,8, 8,8 Гц, 1H), 7,85-7,87 (м, 1H).
(2) Синтез (±)-(4aR*,6S*,7aS*)-7a-(5-бром-2-фторфенил)-6-метокси-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-иламина
Указанное в заголовке соединение получали из (±)-(3aR*,5S*,6aS*)-6a-(5-бром-2-фторфенил)-5-метоксигексагидроциклопента[c]изоксазола в соответствии со способом примера получения 28.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,98-2,10 (м, 2H), 2,16-2,22 (м, 1H), 2,57-2,64 (м, 1H), 2,76 (дд, J=4,4, 13,2 Гц, 1H), 2,87-2,93 (м, 2H), 3,33 (с, 3H), 4,10-4,17 (м, 1H), 6,90 (дд, J=4,4, 11,6 Гц, 1H), 7,32 (ддд, J=2,4, 4,0, 8,4 Гц, 1H), 7,43 (дд, J=2,4, 7,6 Гц, 1H).
ESI-MS; m/z 359 [M++H].
(3) Синтез (±)-N,N-бис(трет-бутилоксикарбонил)[(4aR*,6S*,7aS*)-7a-(5-бром-2-фторфенил)-6-метокси-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]амина
Указанное в заголовке соединение (22 мг) получали из (±)-(4aR*,6S*,7aS*)-7a-(5-бром-2-фторфенил)-6-метокси-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-иламина (15 мг) в соответствии с примером получения 72.
ESI-MS; m/z 559 [M++H].
(4) Синтез (±)-(4aR*,6R*,7aS*)-7a-(5-бром-2-фторфенил)-6-метокси-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-иламина
Указанное в заголовке соединение получали из (±)-(3aR*,5R*,6aS*)-6a-(5-бром-2-фторфенил)-5-метоксигексагидроциклопента[c]изоксазола в соответствии со способом примера получения 19.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,87 (ддд, J=4,0, 9,6, 13,6 Гц, 1H), 2,25-2,30 (м, 2H), 2,56 (дд, J=7,0, 13,0 Гц, 1H), 2,72 (дд, 3,6, 12,8 Гц, 1H), 2,94 (дд, J=3,2, 12,8 Гц, 1H), 3,06-3,12 (м, 1H), 3,32 (с, 3H), 3,87-3,94 (м, 1H), 4,41 (уш., 2H), 6,91 (дд, J=8,8, 12,0 Гц, 1H), 7,33 (ддд, J=2,4, 4,4, 8,8 Гц, 1H), 7,40 (дд, J=2,4, 7,2 Гц, 1H).
ESI-MS; m/z 361 [M++H].
(5) Синтез (±)-N,N-бис(трет-бутилоксикарбонил)[(4aR*,6R*,7aS*)-7a-(5-бром-2-фторфенил)-6-метокси-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]амина
Указанное в заголовке соединение (92 мг) получали из (±)-(4aR*,6R*,7aS*)-7a-(5-бром-2-фторфенил)-6-метокси-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-иламина (65 мг) в соответствии с примером получения 72.
ESI-MS; m/z 559 [M++H].
Пример получения 74
Синтез бензил (±)-[(4aR*,8aS*)-8a-(5-бром-2,4-дифторфенил)-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата
[Формула 96]

(1) Синтез (±)-(4aR*,8aS*)-8a-(2,4-дифторфенил)-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-иламина
Указанное в заголовке соединение синтезировали из этил 2-трифторметансульфонилоксициклогекс-1-енкарбоксилата и 2,4-дифторфенилбороновой кислоты в соответствии с примером получения 1.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,43-1,80 (м, 7H), 2,16-2,24 (м, 1H), 2,48 (дд, J=3,2, 12,0 Гц, 1H), 2,60-2,66 (м, 1H), 2,85 (дд, J=4,0, 12,0 Гц, 1H), 4,47 (с, 2H), 6,74-6,84 (м, 1H), 7,21-7,28 (м, 1H).
(2) Синтез (±)-(4aR*,8aS*)-8a-(5-бром-2,4-дифторфенил)-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-иламина
(±)-(4aR*,8aS*)-8a-(2,4-Дифторфенил)-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-иламин (270 мг) растворяли в концентрированной серной кислоте (3 мл). N-Бромсукцинимид (170 мг) добавляли на ледяной бане и смесь перемешивали при той же температуре в течение двух часов. Добавляли лед, чтобы остановить реакцию, с последующим разбавлением эфиром. Реакционный раствор нейтрализовали раствором бикарбоната натрия и водный слой экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом магния и выпаривали при пониженном давлении растворитель. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (330 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,43-1,80 (м, 7H), 2,09-2,16 (м, 1H), 2,56 (дд, J=2,8, 12,0 Гц, 1H), 2,59-2,63 (м, 1H), 2,85 (дд, J=4,0, 12,0 Гц, 1H), 6,86 (дд, J=8,0, 11,6 Гц, 1H), 7,45 (т, J=8,4, 1H).
ESI-MS; m/z 361 [M++H].
(3) Синтез бензил (±)-[(4aR*,8aS*)-8a-(5-бром-2,4-дифторфенил)-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата
(±)-(4aR*,8aS*)-8a-(5-Бром-2,4-дифторфенил)-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-иламин (330 мг) суспендировали в 1,4-диоксане (10 мл) и насыщенном растворе карбоната натрия (10 мл).
Добавляли бензилхлорформиат (156 мкл) и смесь перемешивали при комнатной температуре в течение ночи. Водный слой экстрагировали этилацетатом и органический слой сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (501 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,47-1,90 (м, 7H), 2,25-2,30 (м, 1H), 2,55 (дд, J=2,4, 12,8 Гц, 1H), 2,81-2,87 (м, 2H), 4,71 (с, 1H), 5,18 (с, 2H), 6,93 (дд, J=8,0, 12,0 Гц, 1H), 7,26-7,45 (м, 6H).
Пример получения 75
Синтез трет-бутил (±)-[(3aS*,7aR*)-7a-(3-аминофенил)-3a,6,7,7a-тетрагидро-4H-пирано[4,3-d]тиазол-2-ил]карбамата
[Формула 97]

(1) Синтез 4-(3-бромфенил)-3,6-дигидро-2H-пирана
н-Бутиллитий (2,64 M, 37,9 мл) добавляли по каплям к раствору 1,3-дибромбензола (23,5 г) в тетрагидрофуране (469 мл) при -78°C. Смесь перемешивали при той же температуре в течение 30 минут и затем по каплям добавляли тетрагидро-4H-пиран-4-он (10,0 г). Смесь перемешивали при той же температуре в течение 30 минут и затем нагревали до комнатной температуры. После перемешивания при комнатной температуре в течение четырех часов к реакционной смеси добавляли раствор хлорида аммония, чтобы остановить реакцию. Водный слой экстрагировали этилацетатом и органический слой промывали водой и насыщенным раствором соли. Органический слой сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и полученный остаток растворяли в толуоле (400 мл). Добавляли моногидрат п-толуолсульфоновой кислоты (2 г) и смесь перемешивали при кипении с обратным холодильником в течение двух часов. Реакционную смесь охлаждали до комнатной температуры и очищали хроматографией на колонке с NH-силикагелем с получением указанного в заголовке соединения (21,4 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,46-2,50 (м, 2H), 3,94 (т, J=5,6 Гц, 2H), 4,32 (дд, J=2,4, 5,6 Гц, 2H), 6,13-6,15 (м, 1H), 7,20 (т, J=8,0 Гц, 1H), 7,30-7,32 (м, 1H), 7,38 (ддд, J=1,2, 1,6, 8,0 Гц, 1H), 7,52 (т, J=1,6 Гц, 1H).
(2) Синтез (±)-6-(3-бромфенил)-3,7-диоксабицикло[4,1,0]гептана
3-Хлорпербензойную кислоту (чистота: 80%, 25,1 г) добавляли к раствору 4-(3-бромфенил)-3,6-дигидро-2H-пирана (21,4 г) в дихлорметане (400 мл) на ледяной бане. Смесь перемешивали при той же температуре в течение трех часов и затем нагревали до комнатной температуры и перемешивали в течение пяти часов. К реакционной смеси добавляли лед и насыщенный раствор бикарбоната натрия и отделяли органический слой. Органический слой последовательно промывали насыщенным раствором бикарбоната натрия, раствором тиосульфата натрия и насыщенным раствором соли. Водные слои объединяли, экстрагировали этилацетатом и органический слой последовательно промывали раствором бикарбоната натрия, раствором тиосульфата натрия и насыщенным раствором соли. Органические слои объединяли, сушили над безводным сульфатом магния и выпаривали при пониженном давлении растворитель. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (17,5 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,09 (дт, J=3,6, 14,8 Гц, 1H), 2,51 (ддд, 6,0, 8,0, 14,8 Гц, 1H), 3,17 (д, J=3,2 Гц, 1H), 3,65-3,68 (м, 2H), 4,01 (д, J=13,6 Гц, 1H), 4,13 (дд, J=3,2, 13,2 Гц, 1H), 7,24 (т, J=8,0 Гц, 1H), 7,33 (ддд, J=1,2, 1,6, 8,0 Гц, 1H), 7,44 (ддд, 1,2, 2,0, 8,0 Гц, 1H), 7,55 (т, J=2,0 Гц, 1H).
(3) Синтез (±)-(3R*,4R*)-4-азидо-4-(3-бромфенил)тетрагидропиран-3-ола
Воду (88 мл) и хлорид аммония (11,7 г) добавляли к раствору (±)-6-(3-бромфенил)-3,7-диоксабицикло[4,1,0]гептана (17,5 г) в метаноле (700 мл) с последующим перемешиванием при комнатной температуре. Затем добавляли азид натрия (32,1 г) с последующим перемешиванием при 80°C в течение восьми часов. После доведения до комнатной температуры избыток метанола выпаривали при пониженном давлении. Остаточный водный слой три раза экстрагировали хлороформом и органические слои сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (22,1 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,94 (ддд, J=1,6, 3,6, 14,4 Гц, 1H), 2,09 (д, J=7,2 Гц, 1H), 2,61 (ддд, J=5,2, 12,4, 14,4 Гц, 1H), 3,71 (дд, J=1,6, 7,2 Гц, 1H), 3,84 (д, J=1,6 Гц, 1H), 3,88 (т, J=1,6 Гц, 1H), 3,97-4,00 (м, 1H), 4,03 (дд, J=1,2, 12,4 Гц, 1H), 7,34 (т, J=8,0 Гц, 1H), 7,42 (ддд, J=1,2, 2,0, 8,0 Гц, 1H), 7,52 (ддд, J=1,2, 2,0, 8,0 Гц, 1H), 7,62 (т, J=2,0 Гц, 1H).
(4) Синтез (±)-(3R*,4R*)-4-амино-4-(3-бромфенил)тетрагидропиран-3-ола
Формиат аммония (16,9 г) и цинк (5,25 г) добавляли к раствору (±)-(3R*,4R*)-4-азидо-4-(3-бромфенил)тетрагидропиран-3-ола (16,0 г) в метаноле (250 мл). Смесь перемешивали при комнатной температуре в течение ночи и затем избыток метанола выпаривали при пониженном давлении. Остаток разбавляли раствором хлорида аммония и водный слой экстрагировали хлороформом. Органический слой промывали раствором хлорида аммония и насыщенным раствором соли и сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (10,9 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,62 (дд, J=2,0, 14,0 Гц, 1H), 2,58 (дт, 8,8, 14,0 Гц, 1H), 3,68 (д, 1,6 Гц, 1H), 3,85 (дд, J=2,0, 12,0 Гц, 1H), 3,92 (д, J=2,0 Гц, 1H), 3,95 (д, J=2,0 Гц, 1H), 4,14 (дд, J=1,6, 12,0 Гц, 1H), 7,27 (дд, J=8,0 Гц, 10,0 Гц, 1H), 7,40-7,43 (м, 2H), 7,63 (т, J=2,0 Гц, 1H).
(5) Синтез (±)-[(3R*,4R*)-4-(3-бромфенил)-3-гидрокситетрагидропиран-4-ил]тиомочевины
Бензоилизотиоцианат (7,20 г) добавляли к суспензии (±)-(3R*,4R*)-4-амино-4-(3-бромфенил)тетрагидропиран-3-ола (10,9 г) в толуоле (200 мл). Смесь перемешивали при комнатной температуре в течение ночи и затем разбавляли тетрагидрофураном с последующим добавлением силикагеля. Растворитель выпаривали при пониженном давлении и остаток очищали хроматографией на колонке с силикагелем с получением промежуточного соединения в виде неочищенного продукта. Полученное промежуточное соединение суспендировали в метаноле (300 мл) и добавляли карбонат калия (20,0 г). Смесь перемешивали при комнатной температуре в течение ночи. Затем нерастворимое вещество удаляли фильтрованием через целит и выпаривали при пониженном давлении растворитель. Остаток разбавляли этилацетатом и полученное твердое вещество удаляли фильтрованием через целит. Растворитель выпаривали при пониженном давлении и остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (8,63 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,12 (д, J=13,6 Гц, 1H), 2,34 (д, J=6,4 Гц, 1H), 2,58 (ддд, J=4,4, 10,8, 14,0 Гц, 1H), 3,77-3,83 (м, 2H), 3,88 (дт, J=4,0, 12,0 Гц, 1H), 3,95 (дд, J=2,8, 12,8 Гц, 1H), 4,03 (дд, J=2,0, 12,8 Гц, 1H), 6,60 (с, 1H), 7,35 (т, J=8,0 Гц, 1H), 7,50 (ддд, J=1,2, 2,0, 8,0 Гц, 1H), 7,54 (ддд, J=1,2, 2,0, 8,0 Гц, 1H), 7,71 (т, J=2,0 Гц, 1H).
ESI-MS; m/z 333 [M++H].
(6) Синтез (±)-(3aS*,7aR*)-7a-(3-бромфенил)-3a,6,7,7a-тетрагидро-4H-пирано[4,3-d]тиазол-2-иламина
Диэтилазодикарбонат (13,0 мл) добавляли по каплям к раствору трифенилфосфина (7,51 г) в тетрагидрофуране (200 мл) на ледяной бане. Смесь нагревали до комнатной температуры и перемешивали в течение 30 минут. Реакционный раствор охлаждали до 0°C и добавляли по каплям раствор (±)-[(3R*,4R*)-4-(3-бромфенил)-3-гидрокситетрагидропиран-4-ил]тиомочевины (6,33 г) в тетрагидрофуране (44 мл). Смесь перемешивали в течение ночи, постепенно нагревая до комнатной температуры. К реакционной смеси добавляли воду и водный слой экстрагировали этилацетатом. Растворитель выпаривали при пониженном давлении и остаток очищали хроматографией на колонке с силикагелем с получением неочищенного продукта. Полученный неочищенный продукт снова очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (3,55 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,13-2,23 (м, 2H), 3,36 (дд, J=10,8, 11,6 Гц, 1H), 3,71 (дт, J=3,4 Гц, 1H), 3,89-3,96 (м, 2H), 4,08 (дд, J=6,4, 12,0 Гц, 1H), 7,21 (т, J=8,0 Гц, 1H), 7,37-7,40 (м, 2H), 7,60 (т, J=2,0 Гц, 1H).
(7) Синтез (±)-N,N-бис(трет-бутоксикарбонил)[(3aS*,7aR*)-7a-(3-бромфенил)-3a,6,7,7a-тетрагидро-4H-пирано[4,3-d]тиазол-2-ил]амина
4-Диметиламинопиридин (2,77 г) и ди-трет-н-бутилдикарбонат (12,3 г) добавляли к раствору (±)-(3aS*,7aR*)-7a-(3-бромфенил)-3a,6,7,7a-тетрагидро-4H-пирано[4,3-d]тиазол-2-иламина (3,55 г) в тетрагидрофуране (500 мл). Смесь перемешивали при комнатной температуре в течение четырех часов и затем выпаривали при пониженном давлении растворитель при комнатной температуре или ниже. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (4,49 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,54 (с, 18H), 2,11 (ддд, J=5,2, 12,4, 14,4 Гц, 1H), 2,31 (дт, J=2,0, 14,4 Гц, 1H), 3,41 (дд, J=10,4, 12,0 Гц, 1H), 3,60-3,67 (м, 1H), 3,83-3,94 (м, 2H), 4,10 (дд, J=6,2, 11,8 Гц, 1H), 7,20 (т, J=8,0 Гц, 1H), 7,37-7,40 (м, 2H), 7,59 (д, J=2,0 Гц, 1H).
(8) Синтез трет-бутил (±)-{(3aS*,7aR*)-7a-[3-(бензгидрилиденамино)фенил]-3a,6,7,7a-тетрагидро-4H-пирано[4,3-d]тиазол-2-ил}карбамата
Бензофенонимин (244 мкл), BINAP (60,6 мг), трис(дибензилиденацетон)дипалладий(0) (44,6 мг) и трет-бутоксид натрия (233 мг) добавляли к раствору (±)-N,N-бис(трет-бутоксикарбонил)[(3aS*,7aR*)-7a-(3-бромфенил)-3a,6,7,7a-тетрагидро-4H-пирано[4,3-d]тиазол-2-ил]амина (250 мг) в толуоле (20 мл). После замены азотом смесь перемешивали при 85°C в течение четырех часов. Реакционный раствор доводили до комнатной температуры и добавляли к реакционной смеси воду. Водный слой экстрагировали этилацетатом и органический слой промывали насыщенным раствором соли. Органический слой сушили над безводным сульфатом магния и выпаривали при пониженном давлении растворитель. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (165 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,38 (с, 9H), 1,98-2,06 (уш., 2H), 3,24 (дд, J=6,0, 10,8 Гц, 1H), 3,35 (т, J=10,8 Гц, 1H), 3,61 (т, J=12,6 Гц, 1H), 3,80-3,87 (м, 1H), 3,96 (дд, J=6,0, 11,6 Гц, 1H), 6,57 (уш.с, 1H), 6,82 (д, J=7,6 Гц, 1H), 6,95-6,97 (м, 1H), 7,06-7,08 (м, 2H), 7,17 (т, J=8,0 Гц, 1H), 7,25-7,28 (м, 4H), 7,39-7,44 (м, 2H), 7,47-7,51 (м, 1H), 7,75-7,77 (м, 2Н).
ESI-MS; m/z 536 [M++Na].
(9) Синтез трет-бутил (±)-[(3aS*,7aR*)-7a-(3-аминофенил)-3a,6,7,7a-тетрагидро-4H-пирано[4,3-d]тиазол-2-ил]карбамата
К раствору трет-бутил (±)-{(3aS*,7aR*)-7a-[3-(бензгидрилиденамино)фенил]-3a,6,7,7a-тетрагидро-4H-пирано[4,3-d]тиазол-2-ил}карбамата (165 мг) в простом эфире (1 мл) добавляли 1 н. хлористо-водородную кислоту (1 мл) и смесь перемешивали при комнатной температуре в течение одного часа. Реакционный раствор нейтрализовали раствором бикарбоната натрия, органический слой сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и остаток очищали хроматографией на колонке с NH-силикагелем с получением указанного в заголовке соединения (77 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,41 (с, 9H), 2,04-2,28 (м, 2H), 3,38 (т, J=11,2 Гц, 1H), 3,65-4,12 (м, 4H), 6,56-6,60 (м, 1H), 6,69-6,85 (м, 2H), 7,10-7,15 (м, 1H).
ESI-MS; m/z 350 [M++H].
Пример получения 76
Синтез (±)-N-трет-бутоксикарбонил-N-[(4aS*,8aS*)-8a-(4-бромтиофен-2-ил)-4a,7,8,8a-тетрагидро-4H,5H-6-окса-3-тиа-1-азанафталин-2-ил]бензамида
[Формула 98]

(1) Синтез (±)-(3aS*,7aS*)-7a-(4-бромтиофен-2-ил)гексагидропирано[4,3-c]изоксазола
Указанное в заголовке соединение (166 мг) получали из соединения, полученного в примере получения 8-(2) (100 мг), и 2,4-дибромтиофена (400 мг) в соответствии со способом примера получения 21-(3).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,05 (уш., 1H), 2,26 (уш., 1H), 2,80 (уш., 1H), 3,44-4,23 (м, 6H), 7,03 (с, 1H), 7,21 (с, 1H).
(2) Синтез (±)-[(3R*,4S*)-4-амино-4-(4-бромтиофен-2-ил)-тетрагидропиран-3-ил]метанола
Цинк (374 мг) добавляли к раствору (±)-(3aS*,7aS*)-7a-(4-бромтиофен-2-ил)гексагидропирано[4,3-c]изоксазола (166 мг) в уксусной кислоте (5 мл) и смесь перемешивали при комнатной температуре в течение ночи. Нерастворимое вещество удаляли фильтрованием через целит и выпаривали при пониженном давлении растворитель. К остатку добавляли лед с последующей нейтрализацией 2 н. раствором гидроксида натрия. Водный слой экстрагировали этилацетатом и органический слой сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (147 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,65 (дт, J=2,8, 14,0 Гц, 1H), 2,00-2,06 (м, 1H), 2,25 (ддд, J=5,2, 10,8, 14,0 Гц, 1H), 3,62 (дд, J=4,0, 11,6 Гц, 1H), 3,70 (дд, J=4,4, 11,6 Гц, 1H), 3,81-3,95 (м, 4H), 6,94 (д, J=1,4 Гц, 1H), 7,14 (д, J=1,4 Гц, 1H).
(3) Синтез (±)-1-бензоил-3-[(3R*,4S*)-4-(4-бромтиофен-2-ил)-3-гидроксиметилтетрагидропиран-4-ил]тиомочевины
Бензоилизотиоцианат (66,5 мкл) добавляли к раствору (±)-[(3R*,4S*)-4-амино-4-(4-бромтиофен-2-ил)тетрагидропиран-3-ил]метанола (144 мг) в дихлорметане (5 мл). Смесь перемешивали при комнатной температуре в течение ночи и затем выпаривали при пониженном давлении растворитель. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (233 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,65 (дд, 4,4, 6,0 Гц, 1H), 2,19 (ддд, J=3,6, 10,8, 14,4 Гц, 1H), 2,27 (уш.с, 1H), 3,74-4,06 (м, 6H), 6,94 (д, J=1,6 Гц, 1H), 7,18 (д, J=1,6 Гц, 1H), 7,52-7,56 (м, 2H), 7,63-7,67 (м, 1H), 7,86-7,88 (м, 2H), 8,88 (с, 1H), 11,64 (с, 1H).
(4) Синтез (±)-N-[(4aS*,8aS*)-8a-(4-бромтиофен-2-ил)-4a,7,8,8a-тетрагидро-4H,5H-6-окса-3-тиа-1-азанафталин-2-ил]бензамида
(±)-1-Бензоил-3-[(3R*,4S*)-4-(4-бромтиофен-2-ил)-3-гидроксиметилтетрагидропиран-4-ил]тиомочевину (233 мг) растворяли в метаноле (5 мл). Добавляли несколько капель концентрированной хлористо-водородной кислоты с последующим перемешиванием в течение двух часов. Реакционный раствор доводили до комнатной температуры и выпаривали при пониженном давлении растворитель с получением указанного в заголовке соединения.
ESI-MS; m/z 439 [M++H].
(5) Синтез (±)-N-трет-бутоксикарбонил-N-[(4aS*,8aS*)-8a-(4-бромтиофен-2-ил)-4a,7,8,8a-тетрагидро-4H,5H-6-окса-3-тиа-1-азанафталин-2-ил]бензамида
4-Диметиламинопиридин (40,3 мг) и ди-трет-бутилдикарбонат (48 мг) добавляли к раствору (±)-N-[(4aS*,8aS*)-8a-(4-бромтиофен-2-ил)-4a,7,8,8a-тетрагидро-4H,5H-6-окса-3-тиа-1-азанафталин-2-ил]бензамида (48 мг) в тетрагидрофуране (2 мл). Смесь перемешивали при комнатной температуре в течение двух часов и затем выпаривали при пониженном давлении растворитель. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (60,0 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,36 (с, 9H), 2,06 (д, J=12,8 Гц, 1H), 2,26 (дт, J=4,4, 12,8 Гц, 1H), 2,37-2,43 (м, 1H), 2,65 (дд, J=2,8, 13,2 Гц, 1H), 3,25 (дд, J=4,0, 13,2 Гц, 1H), 3,36 (дт, J=2,0 Гц, 12,4 Гц, 1H), 3,78-3,83 (м, 3H), 7,01 (д, J=1,8 Гц, 1H), 7,15 (д, J=1,8 Гц, 1H), 7,44-7,48 (м, 2H), 7,54-7,58 (м, 1H), 7,74-7,76 (м, 2H).
Пример получения 77
Синтез (±)-6-{(E)-2-[3-((4aR*,7aS*)-2-амино-4a,5,6,7,-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]винил}никотинонитрила
[Формула 99]
Синтез (±)-N,N-бис(трет-бутоксикарбонил)-6-{(E)-2-[3-((4aR*,7aS*)-2-амино-4a,5,6,7,7a-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]винил}никотинонитрила
Соединение, полученное в примере получения 72 (100 мг), смешивали с транс-1,2-бис(три-н-бутилстаннилом) (114 мг), 2-бром-5-цианопиридином (35 мг), три(о-толил)фосфином (9,2 мг) и бис(ацетонитрил)дихлорпалладием(II) (2,8 мг) в толуоле (3 мл) и смесь перемешивали в атмосфере азота при 80°C в течение ночи. Реакционную суспензию концентрировали и затем остаток очищали препаративной ВЭЖХ. Полученный продукт снова очищали пТСХ с получением указанного в заголовке соединения (17 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,57 (с, 18H), 1,80-2,20 (м, 5H), 2,68-2,88 (м, 3H), 3,07 (дд, J=3,4, 13,3 Гц, 1H), 7,07 (дд, J=8,5, 12,2 Гц, 1H), 7,32 (д, J=15,8 Гц, 1H), 7,41 (м, 2H), 7,81 (д, J=12,2 Гц, 1H), 7,87 (дд, (дд, J=2,1, 8,2 Гц, 2H), 8,81 (д, J=2,1 Гц, 1H).
Пример получения 78
Синтез (±)-(4aR*,7aS*)-7a-(5-этинил-2-фторфенил)-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-амина
[Формула 100]

(1) Синтез (±)-(4aR*,7aS*)-7a-{2-фтор-5-[(триметилсилил)этинил]фенил}-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-амина
Соединение, полученное в примере получения 54-(4) (100 мг), смешивали с дихлоридом бис(трифенилфосфин)палладия(II) (60 мг), йодидом меди(I) (2,3 мг) и этинилтриметилсиланом (60 мг) в триэтиламине (1,75 мл) и тетрагидрофуране (0,25 мл) и смесь перемешивали в атмосфере азота при 90°C в течение ночи. Реакционную суспензию фильтровали и концентрировали. Затем полученный остаток очищали препаративной ВЭЖХ с получением указанного в заголовке соединения (31 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 0,26 (с, 9H), 2,00 (м, 4H), 2,30 (м, 1H), 2,55 (м, 1H), 2,88 (дд, J=3,5, 12,5 Гц, 1H), 3,00-3,14 (м, 2H), 7,00 (дд, J=8,4, 12,3 Гц, 1H), 7,34 (дд, J=2,0, 7,9 Гц, 1H), 7,40 (м, 1H), 9,31 (уш.с, NH2).
(2) Синтез (±)-(4aR*,7aS*)-7a-(5-этинил-2-фторфенил)-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-амина
Тетрабутиламмонийфторид (2,9 мл) добавляли к раствору соединения, полученного в примере получения 78-(1) (500 мг), в тетрагидрофуране (30 мл) и смесь перемешивали при комнатной температуре в течение 30 минут. К реакционной смеси добавляли насыщенный раствор соли с последующим экстрагированием этилацетатом. Органический слой сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении с получением остатка. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (59 мг).
ESI-MS; m/z 275 [M++H].
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,74-2,00 (м, 5H), 2,58 (м, 1H), 2,81 (м, 2H), 2,97 (дд, J=3,1, 12,5 Гц, 1 H), 3,05 (с, 1H), 6,99 (дд, J=8,3, 12,2 Гц, 1H), 7,37 (м, 1H), 7,48 (дд, J=2,2, 8,00 Гц, 1H).
Пример 1
Синтез трет-бутил (±)-((4aR*,8aS*)-8a-{5-[(5-хлорпиридин-2-карбонил)амино]-2-фторфенил}-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил)карбамата
[Формула 101]
5-Хлорпиридин-2-карбоновую кислоту (30,4 мг) и триэтиламин (89,0 мкл) добавляли к раствору соединения примера получения 1-(8) (58,0 мг) в ДМФА (5,63 мл). 1-Гидроксибензотриазол (26,0 мг) и EDC·HCl (61,4 мг) добавляли к реакционному раствору на ледяной бане. Реакционный раствор нагревали до комнатной температуры с последующим перемешиванием в течение ночи. К реакционной смеси добавляли лед с последующим экстрагированием этилацетатом. Органический слой промывали водой и насыщенным раствором хлорида натрия и сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении. Полученный неочищенный продукт очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (23,0 мг).
ESI-MS; m/z 519 [M+H].
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,57 (с, 9H), 1,64-1,89 (м, 7H), 2,37 (м, 1H), 2,53 (м, 1H), 2,88 (уш.с, 1H), 2,91 (д, J=2,4 Гц, 1H), 7,12 (дд, J=8,8, 12,0 Гц, 1H), 7,18 (м, 1H), 7,88 (дд, J=2,8, 8,4 Гц, 1H), 8,22 (д, J=8,4 Гц, 1H), 8,23 (м, 1H), 8,59 (д, J=2,0 Гц, 1H), 9,84 (с, 1H).
Пример 2
Синтез (±)-N-[3-((4aR*,8aS*)-2-амино-4,4a,5,6,7,8-гексагидробензо[d][1,3]тиазин-8a-ил)-4-фторфенил]-5-хлорпиридин-2-карбоксамида
[Формула 102]

Трифторуксусную кислоту (200 мкл) добавляли к раствору соединения из примера 1 (23,0 мг) в дихлорметане (1,00 мл) и смесь перемешивали при комнатной температуре в течение четырех часов. Реакционный раствор разбавляли диэтиловым эфиром и нейтрализовали раствором бикарбоната натрия. Реакционную смесь экстрагировали этилацетатом и органический слой промывали насыщенным раствором хлорида натрия. Органический слой сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении с получением остатка. Остаток очищали хроматографией на колонке с NH-силикагелем с получением указанного в заголовке соединения (18,0 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,37-1,90 (м, 7H), 2,20 (м, 1H), 2,56 (дд, J=2,8, 12,2 Гц, 1H), 2,74 (м, 1H), 2,94 (дд, J=4,0, 12,2 Гц, 1H), 7,06 (дд, J=8,8, 12,0 Гц, 1H), 7,27 (м, 1H), 7,87 (дд, J=2,8, 8,4 Гц, 1H), 7,98 (м, 1H), 8,24 (дд, J=0,8, 8,4 Гц, 1H), 8,56 (дд, J=0,8, 2,4 Гц, 1H), 9,77 (с, 1H).
Пример 3
Синтез (+)-N-[3-((4aR*,8aS*)-2-амино-4,4a,5,6,7,8-гексагидробензо[d][1,3]тиазин-8a-ил)-4-фторфенил]-5-хлорпиридин-2-карбоксамида
[Формула 103]

(±)-2-[(4aR*,8aS*)-2-Амино-4,4a,5,6,7,8-гексагидробензо[d][1,3]тиазин-8a-ил]-4-(5-хлорпиридин-2-карбониламино)фторбензол, полученный в примере 2 (10 мг), оптически разделяли, используя CHIRALPAK™ OD-H производства Daicel Chemical Industries, Ltd. (2 см × 25 см, подвижная фаза: гексан:этанол = 8:2, скорость потока: 20 мл/мин). Компонент, имеющий время удерживания шесть минут, собирали и снова очищали последовательно NH-пТСХ, ЖХ-МС и хроматографией на колонке с NH-силикагелем с получением указанного в заголовке (+)-изомера (2,0 мг; >99% ee).
ESI-MS; m/z 419 [M+H].
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,37-1,90 (м, 7H), 2,20 (м, 1H), 2,56 (дд, J=2,8, 12,2 Гц, 1H), 2,74 (м, 1H), 2,94 (дд, J=4,0, 12,2 Гц, 1H), 7,06 (дд, J=8,8, 12,0 Гц, 1H), 7,27 (м, 1H), 7,87 (дд, J=2,8, 8,4 Гц, 1H), 7,98 (м, 1H), 8,24 (дд, J=0,8, 8,4 Гц, 1H), 8,56 (дд, J=0,8, 2,4 Гц, 1H), 9,77 (с, 1H).
Пример 4 (способ, альтернативный примеру 3)
Синтез (+)-N-[3-((4aR*,8aS*)-2-амино-4,4a,5,6,7,8-гексагидробензо[d][1,3]тиазин-8a-ил)-4-фторфенил]-5-хлорпиридин-2-карбоксамида
[Формула 104]

N,N-Диметилформамид (одна капля) и тионилхлорид (1 мл) добавляли к суспензии 5-хлорпиридинкарбоновой кислоты (55,2 мг) в толуоле (5 мл). Смесь кипятили с обратным холодильником в течение одного часа. Реакционный раствор охлаждали до комнатной температуры и затем выпаривали при пониженном давлении растворитель с получением хлорида 5-хлорпиридинкарбоновой кислоты. Раствор хлорида 5-хлорпиридинкарбоновой кислоты в ТГФ (5 мл) и пиридине (115 мкл) последовательно добавляли к раствору соединения примера получения 2-(2) (111 мг) в ТГФ (10 мл) при охлаждении льдом. Смесь нагревали до комнатной температуры и перемешивали в течение 30 минут. После подтверждения окончания реакции к смеси добавляли насыщенный раствор бикарбоната натрия с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным раствором хлорида натрия и сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении с получением амида синтетического промежуточного соединения. Полученный амид растворяли в дихлорметане (10 мл). Добавляли трифторуксусную кислоту (2 мл) и смесь перемешивали при комнатной температуре в течение трех часов. Реакционный раствор разбавляли диэтиловым эфиром. Затем реакционную смесь нейтрализовали раствором бикарбоната натрия с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным раствором хлорида натрия и затем сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении. Неочищенный продукт очищали хроматографией на колонке с NH-силикагелем с получением указанного в заголовке соединения (115 мг; >99% ee).
Пример 5
Синтез (+)-N-[3-((4aR*,7aS*)-2-амино-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]-5-хлорпиридин-2-карбоксамида
[Формула 105]

Указанное в заголовке соединение (39,5 мг; >99% ee) получали из соединения, полученного в примере получения 3-(8) (45,0 мг), и 5-хлорпиридин-2-карбоновой кислоты (23,3 мг) в соответствии с примером 4.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,73-2,00 (м, 5H), 2,61 (м, 1H), 2,75 (дд, J=4,0, 12,6 Гц, 1H), 2,83 (м, 1H), 2,99 (дд, J=3,2, 12,6 Гц, 1H), 7,05 (дд, J=8,8, 12,0 Гц, 1H), 7,40 (дд, J=2,8, 7,2 Гц, 1H), 7,87 (дд, J=2,4, 8,4 Гц, 1H), 7,94 (м, 1H), 8,23 (д, J=8,4 Гц, 1H), 8,55 (дд, J=0,8, 2,4 Гц, 1H), 9,79 (с, 1H).
Пример 6
Синтез N-[3-((4aR*,7aS*)-2-амино-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]пиридин-2-карбоксамида
[Формула 106]

Указанное в заголовке соединение (4,20 мг) получали из соединения, полученного в примере получения 3-(8) (15,0 мг), и гидрохлорида пиколиноилхлорида (8,76 мг) в соответствии со способом примера 5.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,76-2,02 (м, 5H), 2,62 (м, 1H), 2,76 (дд, J=4,0, 12,8 Гц, 1H), 2,84 (м, 1H), 3,01 (дд, J=3,2, 12,8 Гц, 1H), 7,06 (дд, J=8,8, 12,0 Гц, 1H), 7,39 (дд, 2,4, 6,8 Гц, 1H), 7,49 (дд, J=4,4, 7,6 Гц, 1H), 7,91 (м, 1H), 8,00 (м, 1H), 8,29 (дд, J=1,2, 7,6 Гц, 1H), 8,62 (ддд, J=0,8, 1,2, 4,8 Гц, 1H), 9,99 (с, 1H).
Пример 7
Синтез N-[3-((4aR*,7aS*)-2-амино-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]никотинамида
[Формула 107]
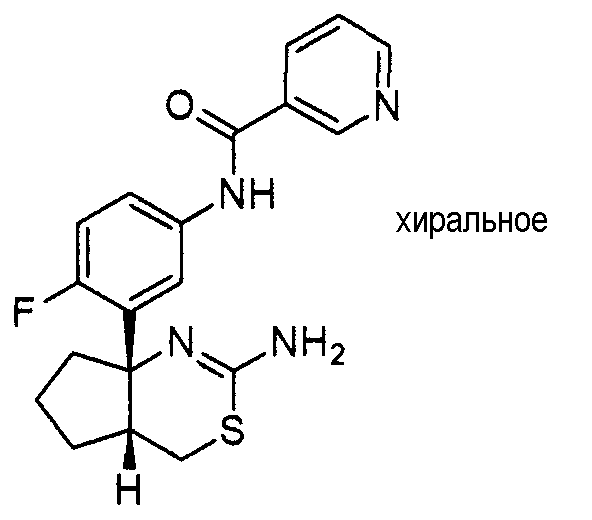
Триэтиламин (30,4 мкл), 1-гидроксибензотриазол (12,2 мг) и EDC·HCl (21,0 мг) добавляли к раствору соединения, полученного в примере получения 3-(8) (10,0 мг), и никотиновой кислоты (10,1 мг) в ДМФА (2,00 мл). Реакционный раствор перемешивали при комнатной температуре в течение четырех суток. Затем к реакционной смеси добавляли ледяную воду с последующим экстрагированием этилацетатом. Органический слой промывали водой и насыщенным раствором хлорида натрия и затем сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и затем выпаривали при пониженном давлении растворитель. Полученный неочищенный продукт очищали пТСХ с получением соответствующего амида. ТФУК (2,00 мл) добавляли к раствору полученного амида в дихлорметане (5,00 мл) и смесь перемешивали при комнатной температуре в течение трех часов. После подтверждения окончания реакции реакционный раствор разбавляли толуолом и выпаривали при пониженном давлении растворитель при комнатной температуре или ниже. Остаток очищали NH-пТСХ с получением указанного в заголовке соединения (3,80 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,75-1,94 (м, 5H), 2,36 (уш., 2H), 2,60 (м, 1H), 2,76 (дд, J=4,0, 12,8 Гц, 1H), 2,85 (м, 1H), 2,97 (дд, 3,6, 12,8 Гц, 1H), 7,06 (дд, J=8,8, 12,0 Гц, 1H), 7,26 (м, 1H), 7,43 (дд, J=4,8, 7,2 Гц, 1H), 7,89 (м, 1H), 8,20 (д, J=8,0 Гц, 1H), 8,77 (дд, J=1,6, 4,8 Гц, 1H), 9,09 (д, J=1,6 Гц, 1H).
Пример 8
Синтез N-[3-((4aR*,7aS*)-2-амино-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]изоникотинамида
[Формула 108]
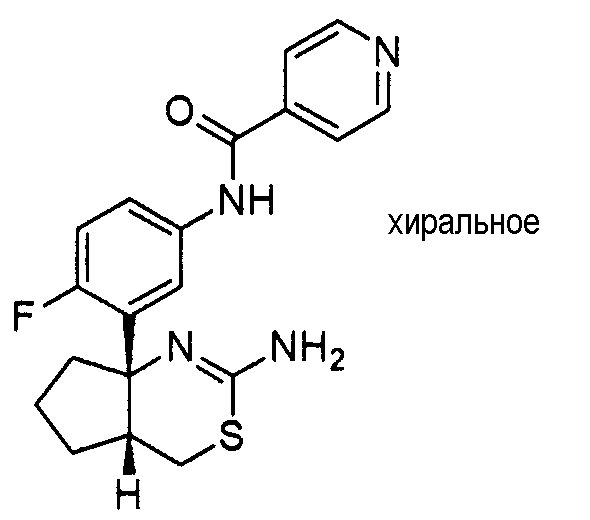
Указанное в заголовке соединение (3,20 мг) получали из соединения, полученного в примере получения 3-(8) (10,0 мг), и изоникотиновой кислоты (10,1 мг) в соответствии со способом примера 7.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,75-1,97 (м, 5H), 2,59 (м, 1H), 2,76 (дд, J=4,0, 12,8 Гц, 1H), 2,84 (м, 1H), 2,96 (дд, J=3,2, 12,8 Гц, 1H), 7,07 (дд, J=8,8, 12,4 Гц, 1H), 7,27 (м, 1H), 7,69 (дд, J=1,6, 4,4 Гц, 2H), 7,88 (м, 1H), 8,79 (дд, J=1,2, 4,4 Гц, 2H).
Пример 9
Синтез N-[3-((4aR*,7aS*)-2-амино-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]-5-фторпиридин-2-карбоксамида
[Формула 109]

Указанное в заголовке соединение (7,20 мг) получали из соединения, полученного в примере получения 3-(8) (12,0 мг), и 5-фторпиридин-2-карбоновой кислоты (10,5 мг) в соответствии со способом примера 4.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,63-2,00 (м, 5H), 2,61 (м, 1H), 2,76 (дд, J=4,0, 12,8 Гц, 1H), 2,82 (м, 1H), 3,00 (дд, J=3,2, 12,8 Гц, 1H), 4,43 (уш., 2H), 7,05 (дд, J=8,8, 12,4 Гц, 1H), 7,38 (дд, J=2,8, 7,2 Гц, 1H), 7,59 (м, 1H), 7,95 (м, 1H), 8,33 (дд, J=4,8, 8,8 Гц, 1H), 8,45 (д, J=2,8 Гц, 1H), 9,77 (с, 1H).
Пример 10
Синтез N-[3-((4aR*,7aS*)-2-амино-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]-6-хлорпиридин-2-карбоксамида
[Формула 110]

Указанное в заголовке соединение (10,8 мг) получали из соединения, полученного в примере получения 3-(8) (10,0 мг), и 6-хлорпиридин-2-карбоновой кислоты (7,75 мг) в соответствии со способом примера 4.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,70-2,07 (м, 5H), 2,60 (м, 1H), 2,76 (дд, J=4,0, 12,8 Гц, 1H), 2,85 (м, 1H), 3,01 (дд, J=3,2, 12,8 Гц, 1H), 7,05 (дд, J=8,8, 12,0 Гц, 1H), 7,41 (дд, J=2,8, 7,2 Гц, 1H), 7,51 (дд, J=1,2, 8,0 Гц, 1H), 7,87 (т, J=8,0 Гц, 1H), 7,95 (м, 1H), 8,22 (дд, J=0,8, 7,6 Гц, 1H), 9,66 (с, 1H).
Пример 11
Синтез N-[3-((4aR*,7aS*)-2-амино-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]-4-хлорпиридин-2-карбоксамида
[Формула 111]

Указанное в заголовке соединение (9,60 мг) получали из соединения, полученного в примере получения 3-(8) (10,0 мг), и 4-хлорпиридин-2-карбоновой кислоты (7,75 мг) в соответствии со способом примера 4.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,69-1,98 (м, 5H), 2,61 (м, 1H), 2,76 (дд, J=4,0, 12,8 Гц, 1H), 2,83 (м, 1H), 2,99 (дд, 3,2, 12,8 Гц, 1H), 7,06 (дд, J=8,8, 12,0 Гц, 1H), 7,39 (дд, J=2,8, 7,2 Гц, 1H), 7,48 (дд, J=2,0, 5,2 Гц, 1H), 7,96 (м, 1H), 8,29 (дд, J=0,4, 2,0 Гц, 1H), 8,51 (дд, J=0,4, 5,2 Гц, 1H), 9,87 (с, 1H).
Пример 12
Синтез N-[3-((4aR*,7aS*)-2-амино-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]-3-хлорпиридин-2-карбоксамида
[Формула 112]
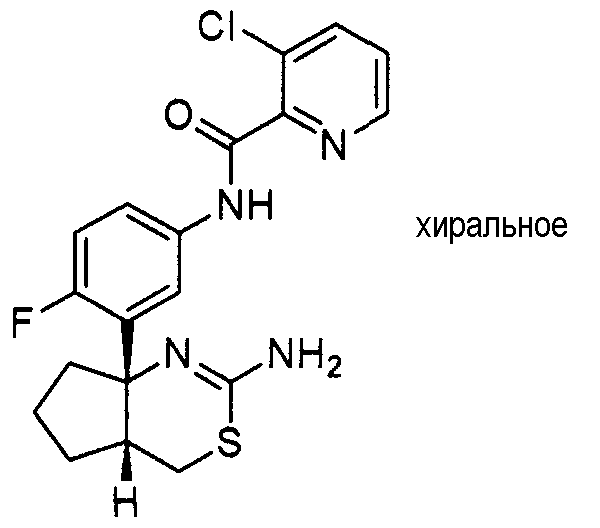
Указанное в заголовке соединение (4,20 мг) получали из соединения, полученного в примере получения 3-(8) (11,0 мг), и 3-хлорпиридин-2-карбоновой кислоты (7,11 мг) в соответствии со способом примера 4.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,69-1,97 (м, 5H), 2,60 (м, 1H), 2,76 (дд, J=4,0, 12,6 Гц, 1H), 2,84 (м, 1H), 9,40 (дд, J=3,2, 12,6, 1H), 7,05 (дд, J=8,8, 12,0 Гц, 1H), 7,25 (м, 1H), 7,43 (дд, J=4,4, 8,0 Гц, 1H), 7,88 (дд, 1,2, 8,0 Гц, 1H), 8,08 (м, 1H), 8,53 (дд, J=1,2, 4,8 Гц, 1H), 9,86 (с, 1H).
Пример 13
Синтез (+)-N-[3-((4aR*,8aS*)-2-амино-4,4a,5,6,7,8-гексагидробензо[d][1,3]тиазин-8a-ил)фенил]5-хлорпиридин-2-карбоксамида
[Формула 113]
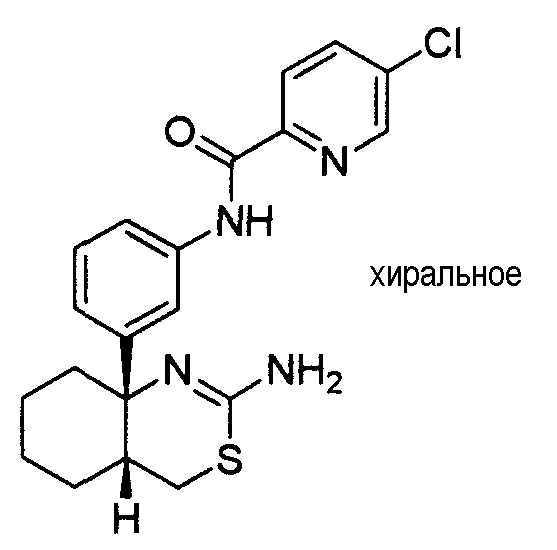
Указанное в заголовке соединение (66,1 мг; >99% ee) получали из трет-бутил [(4aR*,8aS*)-8a-(3-аминофенил)-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата, полученного в примере получения 4-(8) (110 мг), и 5-хлорпиридин-2-карбоновой кислоты (62,3 мг) в соответствии со способом примера 4.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,49-1,89 (м, 8H), 2,26 (м, 1H), 2,47 (дд, J=2,8, 12,0 Гц, 1H), 2,93 (дд, J=4,4, 12,0 Гц, 1H), 7,12 (дт, J=1,6, 8,0 Гц, 1H), 7,35 (т, J=8,0 Гц, 1H), 7,67 (м, 2H), 7,88 (дд, J=2,4, 8,4 Гц, 1H), 8,25 (д, J=8,4 Гц, 1H), 8,58 (д, J=0,8 Гц, 1H), 9,83 (с, 1H).
Пример 14
Синтез (+)-N-[3-((4aR*,8aS*)-2-амино-4,4a,5,6,7,8-гексагидробензо[d][1,3]тиазин-8a-ил)-4,5-дифторфенил]-5-хлорпиридин-2-карбоксамида
[Формула 114]

5-Хлорпиридинкарбоновую кислоту (4,16 мг), N,N-диизопропилэтиламин (8,98 мкл) и PyBOP (22,9 мг) добавляли к раствору соединения, полученного в примере получения 5-(8) (7,00 мг), в дихлорметане (4,00 мл). Реакционный раствор перемешивали при комнатной температуре в течение трех суток. Затем реакционную смесь очищали хроматографией на колонке с NH-силикагелем с получением амида. Полученный амид растворяли в дихлорметане (4,00 мл) и добавляли трифторуксусную кислоту (1,00 мл). Реакционный раствор перемешивали при комнатной температуре в течение трех часов. Затем добавляли насыщенный раствор бикарбоната натрия с последующим экстрагированием этилацетатом. Органический слой сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и затем выпаривали при пониженном давлении растворитель. Остаток очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (5,80 мг; >99% ee).
ESI-MS; m/z 437 [M+H].
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,51-1,95 (м, 7H), 2,20 (м, 1H), 2,62 (дд, J=2,8, 12,2 Гц, 1H), 2,79 (м, 1H), 2,98 (дд, J=4,0, 12,2 Гц, 1H), 6,95 (м, 1H), 7,89 (дд, J=2,4, 8,4 Гц, 1H), 8,12 (м, 1H), 8,22 (д, J=8,4 Гц, 1H), 8,58 (д, J=2,0 Гц, 1H), 9,84 (с, 1H).
Пример 15
Синтез N-[3-((4aR*,8aS*)-2-амино-4,4a,5,6,7,8-гексагидробензо[d][1,3]тиазин-8a-ил)-4,5-дифторфенил]-пиридин-2-карбоксамида
[Формула 115]

Указанное в заголовке соединение (6,30 мг) получали из соединения, полученного в примере получения 5-(8) (7,00 мг), и пиколиновой кислоты (4,16 мг) в соответствии со способом примера 14.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,52-1,84 (м, 7H), 2,21 (м, 1H), 2,59 (дд, J=2,8, 12,0 Гц, 1H), 2,73 (м, 1H), 2,96 (дд, J=4,0, 12,0 Гц, 1H), 6,96 (м, 1H), 7,50 (ддд, J=1,2, 4,8, 8,0 Гц, 1H), 7,91 (дт, J=1,6, 8,0 Гц, 1H), 8,12 (ддд, J=2,8, 6,8, 11,6 Гц, 1H), 8,27 (дд, J=1,2, 8,0 Гц, 1H), 8,61 (ддд, J=0,8, 1,6, 4,8 Гц, 1H), 9,99 (с, 1H).
Пример 16
Синтез N-[3-((4aR*,8aS*)-2-амино-4,4a,5,6,7,8-гексагидробензо[d][1,3]тиазин-8a-ил)-4,5-дифторфенил]-5-фторпиридин-2-карбоксамида
[Формула 116]

Указанное в заголовке соединение (2,30 мг) получали из соединения, полученного в примере получения 5-(8) (7,00 мг), и 5-фторпиридин-2-карбоновой кислоты (7,45 мг) в соответствии со способом примера 14.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,53-1,83 (м, 7H), 2,21 (м, 1H), 2,60 (д, J=12,0 Гц, 1H), 2,73 (м, 1H), 2,96 (дд, J=3,2, 12,0 Гц, 1H), 6,95 (м, 1H), 7,60 (дт, J=2,8, 8,4 Гц, 1H), 8,08 (ддд, J=2,0, 6,8, 11,6 Гц, 1H), 8,31 (дд, J=4,8, 8,8 Гц, 1H), 8,45 (д, J=2,8 Гц, 1H), 9,78 (с, 1H).
Пример 17
Синтез N-[3-((4aR*,8aS*)-2-амино-4,4a,5,6,7,8-гексагидробензо[d][1,3]тиазин-8a-ил)-4,5-дифторфенил]-3-хлорпиридин-2-карбоксамида
[Формула 117]

Указанное в заголовке соединение (6,20 мг) получали из соединения, полученного в примере получения 5-(8) (7,00 мг), и 3-хлорпиколиновой кислоты (4,16 мг) в соответствии со способом примера 14.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,52-1,82 (м, 7H), 2,19 (м, 1H), 2,59 (дд, J=2,8, 12,4 Гц, 1H), 2,72 (м, 1H), 2,96 (дд, J=4,0, 12,4 Гц, 1H), 6,82 (м, 1H), 7,44 (дд, J=4,8, 8,0 Гц, 1H), 7,89 (дд, J=1,2, 8,0 Гц, 1H), 8,21 (ддд, J=2,8, 6,8, 11,6 Гц, 1H), 8,52 (дд, J=1,2, 4,8 Гц, 1H), 9,89 (с, 1H).
Пример 18
Синтез N-[3-((4aR*,8aS*)-2-амино-4,4a,5,6,7,8-гексагидробензо[d][1,3]тиазин-8a-ил)-4,5-дифторфенил]-3,5-дифторпиридин-2-карбоксамида
[Формула 118]

Указанное в заголовке соединение (5,20 мг) получали из соединения, полученного в примере получения 5-(8) (7,00 мг), и 3,5-дифторпиколиновой кислоты (4,16 мг) в соответствии со способом примера 14.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,49-1,82 (м, 7H), 2,19 (м, 1H), 2,59 (дд, J=2,8, 12,2 Гц, 1H), 2,72 (м, 1H), 2,96 (дд, J=4,0, 12,2 Гц, 1H), 6,87 (м, 1H), 7,39 (ддд, J=2,4, 8,0, 10,4 Гц, 1H), 8,12 (ддд, J=2,8, 6,8, 11,6 Гц, 1H), 8,35 (д, J=2,4 Гц, 1H), 9,57 (с, 1H).
Пример 19
Синтез N-[3-((4aR*,8aS*)-2-амино-4,4a,5,6,7,8-гексагидробензо[d][1,3]тиазин-8a-ил)-4-фторфенил]-5-цианопиридин-2-карбоксамида
[Формула 119]

(1) Синтез трет-бутил ((4aR*,8aS*)-8a-{5-[(5-цианопиридин-2-карбонил)амино]-2-фторфенил}-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил)карбамата
PyBOP (219 мг) добавляли к раствору соединения примера получения 13-(2) (46 мг), N,N-диизопропилэтиламина (0,11 мл) и соединения примера получения 2-(2) (40 мг) в дихлорметане (4 мл). Смесь перемешивали при комнатной температуре в течение 1,5 часов. Реакционный раствор вливали в раствор бикарбоната натрия с последующим экстрагированием этилацетатом. Экстракт промывали насыщенным раствором хлорида натрия и сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении. Неочищенный продукт очищали хроматографией на колонке с силикагелем (система этилацетат-гептан) с получением указанного в заголовке соединения (47 мг).
ESI-MS; m/z 510 [M+H].
(2) Синтез N-[3-((4aR*,8aS*)-2-амино-4,4a,5,6,7,8-гексагидробензо[d][1,3]тиазин-8a-ил)-4-фторфенил]-5-цианопиридин-2-карбоксамида
Трифторуксусную кислоту (1,0 мл) добавляли к раствору соединения, полученного в примере 19-(1) (47 мг), в дихлорметане (2 мл), и реакционный раствор перемешивали при комнатной температуре в течение 1,5 часов. Реакционный раствор вливали в водный раствор бикарбоната натрия с последующим экстрагированием этилацетатом. Экстракт промывали насыщенным раствором хлорида натрия и сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении. Неочищенный продукт очищали хроматографией на колонке с NH-силикагелем (система этилацетат-гептан) с получением указанного в заголовке соединения (28 мг).
ESI-MS; m/z 410 [M+H].
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,40-1,86 (м, 7H), 2,16-2,30 (м, 1H), 2,50-2,60 (м, 1H), 2,66-2,78 (м, 1H), 2,92 (дд, J=4,0, 12,0 Гц, 1H), 7,01 (дд, J=8,8, 12,0 Гц, 1H), 7,24-7,36 (м, 1H), 7,86-7,98 (м, 1H), 8,18 (д, J=8,0 Гц, 1H), 8,39 (д, J=8,0 Гц, 1H), 8,84 (с, 1H), 9,73 (с, 1H).
Пример 20
Синтез N-[3-((4aR*,7aS*)-2-амино-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил-4-фторфенил]-5-дифторметоксипиразин-2-карбоксамида
[Формула 120]

Указанное в заголовке соединение (275 мг) получали из соединения, полученного в примере получения 3-(8) (230 мг), и 5-дифторметоксипиразин-2-карбоновой кислоты, полученной в примере получения 14-(2) (180 мг), в соответствии со способом примера 14.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,78-1,99 (м, 6H), 2,60-3,00 (м, 3H), 7,00-7,10 (м, 1H), 7,28-7,35 (м, 1H), 7,51 (т, J=71,6 Гц, 1H), 7,94-7,97 (м, 1H), 8,34 (с, 1H), 9,08 (с, 1H), 9,45 (с, 1H).
Пример 21
Синтез N-[3-((4aR*,7aS*)-2-амино-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]-5-фторметоксипиразин-2-карбоксамида
[Формула 121]

5-Фторметоксипиразин-2-карбоновую кислоту, полученную в примере получения 15-(2) (10,2 мг), N,N-диизопропилэтиламин (24,7 мкл) и PyBOP (61,5 мг) добавляли к раствору соединения, полученного в примере получения 3-(8) (17,3 мг), в дихлорметане (1,0 мл). Реакционный раствор перемешивали при комнатной температуре в течение одного часа. Затем реакционную смесь очищали хроматографией на колонке с силикагелем с получением амида. Полученный амид растворяли в дихлорметане (750 мкл) и добавляли трифторуксусную кислоту (250 мкл). Реакционному раствору давали отстояться при комнатной температуре в течение 55 минут и затем выпаривали при пониженном давлении растворитель. К остатку добавляли насыщенный раствор бикарбоната натрия с последующим экстрагированием этилацетатом. Органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с NH-силикагелем с получением указанного в заголовке соединения (12,1 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,69-2,02 (м, 5H), 2,55-2,66 (м, 1H), 2,76 (дд, J=12,6, 4,9 Гц, 1H), 2,79-2,88 (м, 1H), 3,00 (дд, J=12,6, 4,0 Гц, 1H), 6,07-6,11 (м, 1H), 6,19-6,23 (м, 1H), 7,06 (дд, J=12,0, 8,6 Гц, 1H), 7,36 (дд, J=7,2, 3,0 Гц, 1H), 7,92-7,98 (м, 1H), 8,29 (д, J=1,2 Гц, 1H), 9,08 (д, J=1,2 Гц, 1H), 9,47 (уш.с, 1H).
Пример 22
Синтез N-[3-((4aR*,7aS*)-2-амино-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]-5-цианопиридин-2-карбоксамида
[Формула 122]

5 н. Раствор гидроксида натрия (50,1 мкл) добавляли к раствору соединения, полученного в примере получения 13-(1), метил 5-цианопиридин-2-карбоксилата (30 мг) в этаноле и смесь перемешивали при комнатной температуре в течение 15 минут. Реакционный раствор подкисляли 5 н. хлористо-водородной кислотой. К реакционному раствору добавляли этилацетат и насыщенный раствор соли и отделяли органический слой. Органический слой сушили над безводным сульфатом магния и выпаривали при пониженном давлении растворитель. Указанное в заголовке соединение (28 мг) получали из полученной 5-цианопиридин-2-карбоновой кислоты и соединения, полученного в примере получения 3-(8) (35 мг), в соответствии со способом примера 14.
ESI-MS; m/z 396 [M++H].
1H-ЯМР (CDCl3) δ (м.д.): 1,74-2,00 (м, 5H), 2,57-2,64 (м, 1H), 2,78 (дд, J=4,0, 12,8 Гц, 1H), 2,85-2,91 (м, 1H), 3,00 (дд, J=3,2, 12,8 Гц, 1H), 7,08 (дд, J=8,8, 12,4 Гц, 1H), 7,40 (дд, J=2,8, 7,2 Гц, 1H), 7,95-7,99 (м, 1H), 8,20 (дд, J=2,0, 8,0 Гц, 1H), 8,42 (дд, J=0,8, 8,0 Гц, 1H), 8,90 (дд, J=0,8, 2,0 Гц, 1H), 9,84 (уш.с, 1H).
Пример 23
Синтез N-[3-((4aR*,7aS*)-2-амино-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]-5-фторметоксипиридин-2-карбоксамида
[Формула 123]

Указанное в заголовке соединение (31 мг) получали из 5-фторметоксипиридин-2-карбоновой кислоты, полученной в примере получения 16-(2) (22,4 мг), и соединения, полученного в примере получения 3-(8) (35 мг), в соответствии со способом примера 14.
ESI-MS; m/z 419 [M++H].
1H-ЯМР (CDCl3) δ (м.д.): 1,60-2,00 (м, 5H), 2,57-2,64 (м, 1H), 2,76 (дд, J=4,0, 12,4 Гц, 1H), 2,80-2,85 (м, 1H), 2,99 (дд, J=3,2, 12,4 Гц, 1H), 5,80 (д, J=53,2 Гц, 2H), 7,05 (дд, J=8,8, 12,4 Гц, 1H), 7,37 (дд, J=2,8, 7,2 Гц, 1H), 7,57 (дд, J=2,8, 8,8 Гц, 1H), 7,94-7,98 (м, 1H), 8,29 (д, J=8,8 Гц, 1H), 8,41 (д, J=2,8 Гц, 1H), 9,80 (уш.с, 1H).
Пример 24
Синтез N-[3-((4aS*,7aS*)-2-амино-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил]-5-цианопиридин-2-карбоксамида
[Формула 124]
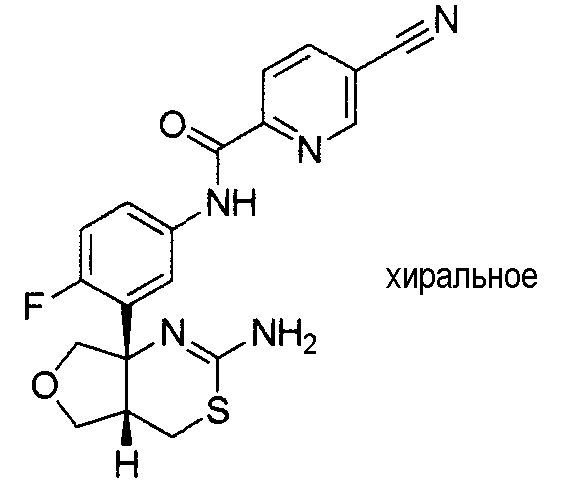
Указанное в заголовке соединение (30 мг) получали из 5-цианопиридин-2-карбоновой кислоты, полученной в примере получения 13-(2) (21,2 мг), и соединения, полученного в примере получения 9-(10) (35 мг), в соответствии со способом примера 14.
ESI-MS; m/z 398 [M++H].
1H-ЯМР (CDCl3) δ (м.д.): 2,82-2,88 (м, 1H), 3,07-3,10 (м, 2H), 3,84 (дд, J=2,4, 8,8 Гц, 1H), 4,08-4,19 (м, 2H), 4,46 (дд, J=2,4, 8,8 Гц, 1H), 7,11 (дд, J=8,4, 12,0 Гц, 1H), 7,62 (дд, J=2,8, 7,2 Гц, 1H), 7,94-7,98 (м, 1H), 8,21 (дд, J=1,6, 8,0 Гц, 1H), 8,43 (д, J=8,0 Гц, 1H), 8,90 (д, J=1,6 Гц, 1H), 9,86 (с, 1H).
Пример 25
Синтез N-[3-((4aS*,7aS*)-2-амино-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил]-5-дифторметоксипиразин-2-карбоксамида
[Формула 125]

Указанное в заголовке соединение (27 мг) получали из 5-дифторметоксипиразин-2-карбоновой кислоты, полученной в примере получения 14-(2) (21,7 мг), и соединения, полученного в примере получения 9-(10) (35 мг), в соответствии со способом примера 14.
ESI-MS; m/z 440 [M++H].
1H-ЯМР (CDCl3) δ (м.д.): 2,82-2,87 (м, 1H), 3,06-3,13 (м, 2H), 3,83 (дд, J=2,4, 9,2 Гц, 1H), 4,07-4,18 (м, 2H), 4,46 (дд, J=1,2, 8,4 Гц, 1H), 7,09 (дд, J=8,8, 11,6 Гц, 1H), 7,51 (т, J=71,6 Гц, 1H), 7,59 (дд, J=2,8, 7,2 Гц, 1H), 7,91-7,95 (м, 1H), 8,34 (д, J=0,8 Гц, 1H), 9,07 (д, J=1,2 Гц, 1H), 9,45 (с, 1H).
Пример 26
Синтез N-[3-((4aS*,7aS*)-2-амино-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил]-5-хлорпиридин-2-карбоксамида
[Формула 126]
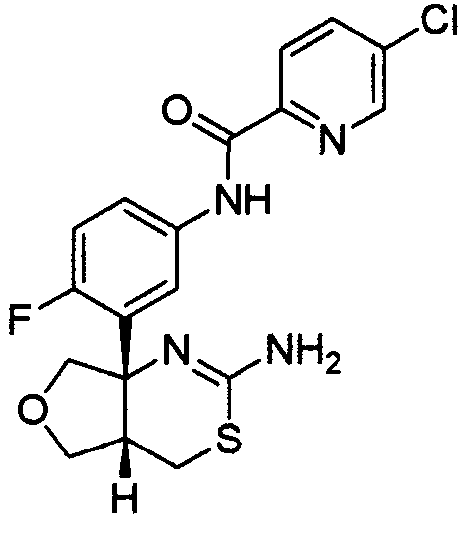
Указанное в заголовке соединение (30 мг) получали из 5-хлорпиридин-2-карбоновой кислоты (21,7 мг) и соединения, полученного в примере получения 9-(10) (35 мг), в соответствии со способом примера 14.
ESI-MS; m/z 407 [M++H].
1H-ЯМР (CDCl3) δ (м.д.): 2,81-2,86 (м, 1H), 3,07-3,13 (м, 2H), 3,83 (дд, J=2,4, 8,8 Гц, 1H), 4,08-4,18 (м, 2H), 4,46 (дд, J=1,6, 8,4 Гц, 1H), 7,09 (дд, J=8,8, 11,6 Гц, 1H), 7,61 (дд, J=2,4, 7,2 Гц, 1H), 7,88 (дд, J=2,4, 8,4 Гц, 1H), 7,91-7,95 (м, 1H), 8,24 (д, J=8,4 Гц, 1H), 8,56 (дд, J=0,8, 2,4 Гц, 1H), 9,83 (с, 1H).
Пример 27
Синтез N-[3-((7S*,7aS*)-2-амино-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил]-5-фторметоксипиразин-2-карбоксамида
[Формула 127]

Соединение, полученное в примере получения 15-(2) (18,4 мг), N,N-диизопропилэтиламин (39,8 мкл) и PyBOP (99,1 мг) добавляли к раствору соединения, полученного в примере получения 9-(10) (28,0 мг), в дихлорметане (800 мкл). Реакционный раствор перемешивали при комнатной температуре в течение 16 часов и 30 минут. Затем реакционную смесь очищали хроматографией на колонке с силикагелем с получением амида. Полученный амид растворяли в дихлорметане (600 мкл) и добавляли трифторуксусную кислоту (200 мкл). Реакционному раствору давали отстояться при комнатной температуре в течение одного часа и затем выпаривали при пониженном давлении растворитель. К остатку добавляли насыщенный раствор бикарбоната натрия с последующим экстрагированием этилацетатом. Органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с NH-силикагелем с получением указанного в заголовке соединения (14,5 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,80-2,88 (м, 1H), 3,05-3,14 (м, 2H), 3,81-3,85 (м, 1H), 4,06-4,19 (м, 2H), 4,44-4,49 (м, 1H), 6,08-6,10 (м, 1H), 6,21-6,23 (м, 1H), 7,10 (дд, J=11,6, 8,8 Гц, 1H), 7,58 (дд, J=7,0, 3,0 Гц, 1H), 7,93-7,97 (м, 1H), 8,29 (д, J=1,2 Гц, 1H), 9,08 (д, J=1,2 Гц, 1H), 9,50 (уш.с, 1H).
Пример 28
Синтез N-[3-((4aS*,8aS*)-2-амино-4a,5,7,8-тетрагидро-4H-6-окса-3-тиа-1-азанафталин-8a-ил)-4-фторфенил]-5-цианопиридин-2-карбоксамида
[Формула 128]

Указанное в заголовке соединение (135 мг) получали из 5-цианопиридин-2-карбоновой кислоты, полученной в примере получения 13-(2) (113 мг), и соединения, полученного в примере получения 8-(9) (195 мг), в соответствии со способом примера 14.
ESI-MS; m/z 412 [M++H].
1H-ЯМР (CDCl3) δ (м.д.): 1,68 (д, J=13,6 Гц, 1H), 2,58 (дд, J=2,4, 13,2 Гц, 1H), 2,63-2,71 (м, 1H), 2,95 (дд, J=4,4, 12,4 Гц, 1H), 3,03-3,06 (м, 1H), 3,69-3,82 (м, 3H), 3,90 (дд, J=4,0, 11,2 Гц, 1H), 7,10 (дд, J=9,2, 12,0 Гц, 1H), 7,42 (дд, J=2,8, 6,8 Гц, 1H), 7,91-7,95 (м, 1H), 8,20 (дд, J=2,0, 8,4 Гц, 1H), 8,42 (д, J=8,0 Гц, 1H), 8,89 (д, J=2,4 Гц, 1H), 9,82 (с, 1H).
Пример 29
Синтез N-[3-((4aS*,8aS*)-2-амино-4a,5,7,8-тетрагидро-4H-6-окса-3-тиа-1-азанафталин-8a-ил)-4-фторфенил]-5-дифторметоксипиразин-2-карбоксамида
[Формула 129]

Указанное в заголовке соединение (24 мг) получали из 5-дифторметоксипиразин-2-карбоновой кислоты, полученной в примере получения 14-(2) (26,1 мг), и соединения, полученного в примере получения 8-(9) (35 мг), в соответствии со способом примера 14.
ESI-MS; m/z 454 [M++H].
1H-ЯМР (CDCl3) δ (м.д.): 1,68 (д, J=12,8 Гц, 1H), 2,58 (дд, J=2,4, 12,8 Гц, 1H), 2,63-2,71 (м, 1H), 2,95 (дд, J=4,4, 12,4 Гц, 1H), 3,03-3,07 (м, 1H), 3,69-3,82 (м, 3H), 3,89 (дд, J=4,4, 11,6 Гц, 1H), 7,10 (дд, J=9,2, 12,4 Гц, 1H), 7,36 (дд, J=2,8, 6,8 Гц, 1H), 7,90-7,94 (м, 1H), 8,34 (д, J=1,2 Гц, 1H), 9,07 (д, J=1,2 Гц, 1H), 9,44 (с, 1H).
Пример 30
Синтез N-[3-((4aS*,8aS*)-2-амино-4a,5,7,8-тетрагидро-4H-6-окса-3-тиа-1-азанафталин-8a-ил)-4-фторфенил]-5-хлорпиридин-2-карбоксамида
[Формула 130]

Указанное в заголовке соединение (21 мг) получали из 5-хлорпиридин-2-карбоновой кислоты (21,7 мг) и соединения, полученного в примере получения 8-(9) (35 мг), в соответствии со способом примера 14.
ESI-MS; m/z 421 [M++H].
1H-ЯМР (CDCl3) δ (м.д.): 1,70-1,83 (м, 1H), 2,62 (дд, J=3,2, 12,8 Гц, 1H), 2,68 (дд, J=4,4, 12,8 Гц, 1H), 2,99 (дд, J=4,0, 12,4 Гц, 1H), 3,08-3,15 (м, 1H), 3,70-3,77 (м, 2H), 3,83 (дд, J=4,4, 11,6 Гц, 1H), 3,92 (дд, J=4,0, 11,2 Гц, 1H), 7,10 (дд, J=8,8, 12,0 Гц, 1H), 7,41 (дд, J=2,4, 7,2 Гц, 1H), 7,88 (дд, J=2,4, 8,4 Гц, 1H), 7,92-7,96 (м, 1H), 8,24 (д, J=8,4 Гц, 1H), 8,58 (д, J=2,4 Гц, 1H), 9,82 (с, 1H).
Пример 31
Синтез (+)-N-[3-((4aR*,6S*,7aS*)-2-амино-6-метокси-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]-5-цианопиридин-2-карбоксамида
[Формула 131]

(1) Синтез трет-бутил ((4aR*,6S*,7aS*)-7a-{5-[(5-цианопиридин-2-карбонил)амино]-2-фторфенил}-6-метокси-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил)карбамата
PyBOP (86,9 мг) добавляли к раствору трет-бутил (-)-[(4aR*,6S*,7aS*)-7a-(5-амино-2-фторфенил)-6-метокси-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]карбамата (22 мг), соединения примера получения 26-(2) (11,6 мг) и N,N-диизопропилэтиламина (0,11 мл) в дихлорметане (2,2 мл). Смесь перемешивали при комнатной температуре в течение одного часа. Реакционный раствор вливали в насыщенный раствор бикарбоната натрия с последующим экстрагированием этилацетатом. Экстракт промывали насыщенным раствором хлорида натрия и сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении. Неочищенный продукт очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (29 мг).
ESI-MS; m/z 526 [M++H].
(2) Синтез (+)-N-[3-((4aR*,6S*,7aS*)-2-амино-6-метокси-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]-5-цианопиридин-2-карбоксамида
Трифторуксусную кислоту (1,0 мл) добавляли к раствору трет-бутил (4aR*,6S*,7aS*)-7a-{5-[(5-цианопиридин-2-карбонил)амино]-2-фторфенил}-6-метокси-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]карбамата (29 мг) в дихлорметане (2 мл), и реакционный раствор перемешивали при комнатной температуре в течение одного часа. Реакционный раствор вливали в насыщенный раствор бикарбоната натрия с последующим экстрагированием этилацетатом. Экстракт промывали насыщенным раствором хлорида натрия и сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении. Неочищенный продукт очищали хроматографией на колонке с NH-силикагелем с получением указанного в заголовке соединения (12 мг).
Оптическое вращение (+)
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,98-2,14 (м, 2H), 2,22 (ддд, J=6,8, 6,8, 13,2 Гц, 1H), 2,64-2,74 (м, 1H), 2,78 (дд, J=4,4, 13,2 Гц, 1H), 2,88-3,02 (м, 2H), 3,34 (с, ЗН), 4,08-4,24 (м, 1Н), 7,07 (дд, J=8,8, 12,0 Гц, 1H), 7,38 (дд, J=2,8, 7,2 Гц, 1H), 7,90-8,02 (м, 1H), 8,20 (дд, J=2,0, 8,0 Гц, 1H), 8,42 (д, J=8,0 Гц, 1H), 8,90 (с, 1H), 9,82 (с, 1H).
Пример 32
Синтез (+)-N-[3-((4aR*,6R*,7aS*)-2-амино-6-метокси-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]-5-цианопиридин-2-карбоксамида
[Формула 132]
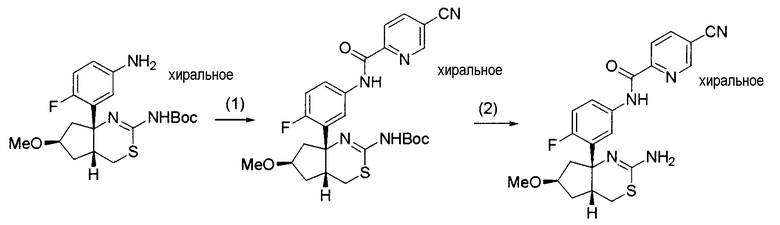
(1) Синтез трет-бутил ((4aR*,6R*,7aS*)-7a-{5-[(5-цианопиридин-2-карбонил)амино]-2-фторфенил}-6-метокси-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил)карбамата
PyBOP (86,9 мг) добавляли к раствору трет-бутил (-)-[(4aR*,6R*,7aS*)-7a-(5-амино-2-фторфенил)-6-метокси-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]карбамата (22 мг), соединения примера получения 26-(2) (11,6 мг) и N,N-диизопропилэтиламина (0,11 мл) в дихлорметане (2,2 мл). Смесь перемешивали при комнатной температуре в течение одного часа. Реакционный раствор вливали в насыщенный раствор бикарбоната натрия с последующим экстрагированием этилацетатом. Экстракт промывали насыщенным раствором хлорида натрия и сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении. Неочищенный продукт очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (29 мг).
ESI-MS; m/z 526 [M++H].
(2) Синтез (+)-N-[3-((4aR*,6R*,7aS*)-2-амино-6-метокси-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]-5-цианопиридин-2-карбоксамида
Трифторуксусную кислоту (1,0 мл) добавляли к раствору трет-бутил ((4aR*,6R*,7aS*)-7a-{5-[(5-цианопиридин-2-карбонил)амино]-2-фторфенил}-6-метокси-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил)карбамата (29 мг) в дихлорметане (2 мл), и реакционный раствор перемешивали при комнатной температуре в течение одного часа. Реакционный раствор вливали в насыщенный раствор бикарбоната натрия с последующим экстрагированием этилацетатом. Экстракт промывали насыщенным раствором хлорида натрия и сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении. Неочищенный продукт очищали хроматографией на колонке с NH-силикагелем с получением указанного в заголовке соединения (12 мг).
Оптическое вращение (+)
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,91 (ддд, J=3,6, 9,6, 13,2 Гц, 1H), 2,22-2,40 (м, 2H), 2,62 (дд, J=6,8, 12,8 Гц, 1H), 2,76 (дд, J=3,2, 12,8 Гц, 1H), 2,99 (дд, J=3,2, 12,8 Гц, 1H), 3,10-3,22 (м, 1H), 3,34 (с, 3H), 3,88-4,00 (м, 1H), 7,08 (дд, J=8,8, 12,0 Гц, 1H), 7,34-7,46 (м, 1H), 7,86-7,98 (м, 1H), 8,20 (дд, J=1,2, 8,0 Гц, 1H), 8,42 (д, J=8,0 Гц, 1H), 8,89 (с, 1H), 9,82 (с, 1H).
Пример 33
Синтез (+)-N-[3-((4aR*,9aS*)-2-амино-4a,5,6,7,8,9-гексагидро-4H-циклогепта[d][1,3]тиазин-9a-ил)-4-фторфенил]-5-цианопиридин-2-карбоксамида
[Формула 133]

Указанное в заголовке соединение (41 мг) получали из трет-бутил (-)-[(4aR*,9aS*)-9a-(5-амино-2-фторфенил)-4,4a,5,6,7,8,9,9a-октагидроциклогепта[d][1,3]тиазин-2-ил]карбамата, полученного в примере получения 7-(8) (60,0 мг), и 5-цианопиридин-2-карбоновой кислоты, полученной в примере получения 13-(2) (97,8 мг), в соответствии со способом примера 14.
ESI-MS; m/z 424 [M+H].
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,50-1,91 (м, 9H), 2,39 (м, 1H), 2,56 (дд, J=3,2, 12,0 Гц, 1H), 2,71 (м, 1H), 2,86 (дд, J=3,2, 12,0 Гц, 1H), 7,07 (дд, J=8,8, 12,0 Гц, 1H), 7,27 (м, 1H), 7,97 (ддд, J=2,4, 4,8, 8,8 Гц, 1H), 8,19 (дд, J=2,0, 8,0 Гц, 1H), 8,42 (дд, J=1,2, 8,0 Гц, 1H), 8,88 (дд, J=1,2, 2,4 Гц, 1H), 9,79 (с, 1H).
Пример 34
Синтез (±)-N-[3-((4aR*,7aS*)-2-амино-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-метоксифенил]-5-хлорпиридин-2-карбоксамида
[Формула 134]
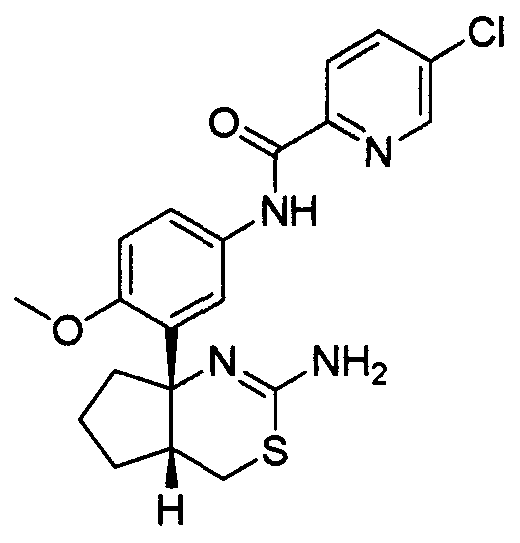
5-Хлорпиридин-2-карбоновую кислоту (6,26 мг), N,N-диизопропилэтиламин (13,8 мкл) и PyBOP (34,5 мг) добавляли к раствору соединения, полученного в примере получения 6-(2) (10,0 мг), в дихлорметане (1,0 мл). Реакционный раствор перемешивали при комнатной температуре в течение одного часа и 50 минут и затем добавляли трифторуксусную кислоту (250 мкл). Реакционному раствору давали отстояться при комнатной температуре в течение 40 минут и затем выпаривали при пониженном давлении растворитель. К остатку добавляли насыщенный раствор бикарбоната натрия с последующим экстрагированием этилацетатом. Органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с NH-силикагелем с получением указанного в заголовке соединения (1,0 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,72-1,98 (м, 5H) 2,72 (дд, J=12,8, 4,7 Гц, 1H), 2,74-2,82 (м, 1H), 2,97 (дд, J=12,8, 4,0 Гц, 1H), 3,05-3,12 (м, 1H), 3,86 (с, 3H), 6,93 (д, J=8,8 Гц, 1H), 7,31 (уш.д, J=3,5 Гц, 1H), 7,86 (дд, J=8,4, 3,5 Гц, 1H), 7,99 (дд, J=8,8, 3,5 Гц, 1H), 8,24 (д, J=8,4 Гц, 1H), 8,56 (д, J=3,5 Гц, 1H), 9,73 (уш.с, 1H).
Пример 35
Синтез N-[3-((4aS*,7aS*)-2-амино-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил]-5-дифторметилпиразин-2-карбоксамида
[Формула 135]

Указанное в заголовке соединение (27 мг) получали из 5-дифторметилпиразин-2-карбоновой кислоты, полученной в примере получения 17-(5) (17,0 мг), и соединения, полученного в примере получения 9-(10) (30 мг), в соответствии со способом примера 14.
ESI-MS; m/z 424 [M++H].
1H-ЯМР (CDCl3) δ (м.д.): 2,82-2,87 (м, 1H), 3,07-3,13 (м, 2H), 3,83 (дд, J=2,4, 8,8 Гц, 1H), 4,08-4,18 (м, 2H), 4,47 (дд, J=1,6, 8,4 Гц, 1H), 6,80 (т, J=54,8 Гц, 1H), 7,13 (дд, J=8,8, 12,0 Гц, 1H), 7,62 (дд, J=2,8, 6,8 Гц, 1H), 7,94-7,98 (м, 1H), 8,93 (с, 1H), 9,53 (с, 1H), 9,65 (с, 1H).
Пример 36
Синтез (±)-(4aR,7aS)-7a-[3-(2-фторпиридин-3-ил)фенил]-6-фенил-4,4a,5,6,7,7a-гексагидропирроло[3,4-d][1,3]тиазин-2-иламина
[Формула 136]
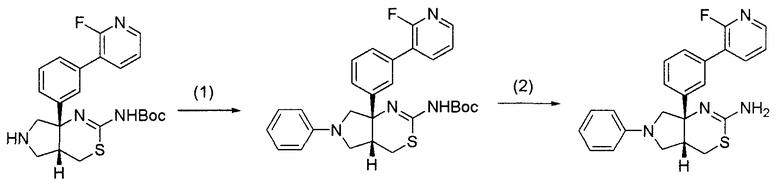
(1) Синтез трет-бутил (±)-{(4aR*,7aS*)-7a-[3-(2-фторпиридин-3-ил)фенил]-6-фенил-4,4a,5,6,7,7a-гексагидропирроло[3,4-d][1,3]тиадиазин-2-ил}карбамата
Соединение, полученное в примере получения 18-(9) (50,00 мг), смешивали с фенилбороновой кислотой (23,9 мг), ацетатом меди(II) (3,56 мг), триэтиламином (54,3 мкл) и молекулярными ситами 4A (порошок) (40,00 мг) в ТГФ, и смесь перемешивали в атмосфере азота при комнатной температуре в течение 23 часов. Реакционную суспензию очищали хроматографией на колонке с NH-силикагелем. Полученный продукт снова очищали NH-пТСХ с получением указанного в заголовке соединения (8 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,49 (с, 9H), 2,91 (дд, J=4,8, 13,2 Гц, 1H), 3,01-3,08 (м, 2H), 3,60-3,64 (м, 1H), 3,69 (д, J=10,4 Гц, 1H), 3,75-3,79 (м, 1H), 4,00 (д, J=10,0 Гц, 1H), 6,58 (д, J=8,0 Гц, 2H), 6,75 (т, J=7,2 Гц, 1H) 7,21-7,32 (м, 3H), 7,41-7,44 (м, 1H), 7,46-7,55 (м, 3H), 7,85 (ддд, J=2,0, 3,6, 9,6 Гц, 1H), 8,21-8,23 (м, 1H).
(2) Синтез (±)-(4aR*,7aS*)-7a-[3-(2-фторпиридин-3-ил)фенил]-6-фенил-4,4a,5,6,7,7a-гексагидропирроло[3,4-d][1,3]тиазин-2-иламина
ТФУК (0,50 мл) добавляли к раствору соединения, полученного в примере 36-(1) (8,00 мг), в хлороформе (0,50 мл). После перемешивания при комнатной температуре в течение 2,5 часов реакционный раствор разбавляли хлороформом и вливали в смесь насыщенного раствора бикарбоната натрия и насыщенного раствора хлорида натрия с последующим интенсивным встряхиванием. Водный слой отделяли и затем органический слой сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и затем выпаривали при пониженном давлении растворитель. К раствору остатка в хлороформе (0,50 мл) добавляли ТФУК (0,50 мл). После перемешивания при комнатной температуре в течение 23 часов реакционный раствор разбавляли хлороформом и вливали в смесь насыщенного раствора бикарбоната натрия и насыщенного раствора хлорида натрия с последующим интенсивным встряхиванием. Водный слой отделяли и затем органический слой сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и затем выпаривали при пониженном давлении растворитель. Остаток очищали хроматографией на колонке с NH-силикагелем. Полученный продукт растворяли в хлороформе и метаноле и растворитель выпаривали в токе азота. К остатку добавляли диэтиловый эфир и в достаточной мере осаждали твердое вещество. Затем растворитель выпаривали в токе азота. Остаток сушили при пониженном давлении с получением указанного в заголовке соединения (6,50 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,75-2,78 (м, 1H), 2,96-3,08 (м, 2H), 3,60 (д, J=7,6 Гц, 2H), 3,67 (д, J=9,6 Гц, 1H), 4,02 (д, J=10,0 Гц, 1H), 6,59 (д, J=8,4 Гц, 2H), 6,72 (т, J=7,2 Гц, 1H), 7,24-7,28 (м, 3H), 7,41-7,46 (м, 3H), 7,54 (с, 1H), 7,82-7,86 (м, 1H), 8,19 (д, J=4,4 Гц, 1H).
Пример 37
Синтез (±)-(4aR*,7aS*)-7a-[3-(2-фторпиридин-3-ил)фенил]-6-пиримидин-2-ил-4,4a,5,6,7,7a-гексагидропирроло[3,4-d][1,3]тиазин-2-иламина
[Формула 137]

2-Бромпиримидин (93,00 мг), трис(дибензилиденацетон)дипалладий(0) (10,70 мг), рац-2,2-бис(дифенилфосфино)-1,1-бинафтил (10,90 мг) и трет-бутоксид натрия (45,00 мг) добавляли к раствору соединения, полученного в примере получения 18-(9) (50,00 мг), в толуоле (1,00 мл). Смесь нагревали при перемешивании в атмосфере азота при 70°C в течение пяти часов и 45 минут. Реакционную смесь охлаждали до комнатной температуры и разбавляли этилацетатом. Затем реакционный раствор фильтровали через NH-силикагель при пониженном давлении и промывали этилацетатом. Полученный фильтрат концентрировали при пониженном давлении и затем очищали хроматографией на колонке с NH-силикагелем с получением Boc-защищенного соединения в качестве синтетического промежуточного соединения. ТФУК (0,50 мл) добавляли к раствору полученного продукта в хлороформе (0,50 мл). После перемешивания при комнатной температуре в течение шести часов реакционный раствор разбавляли хлороформом и вливали в смесь насыщенного раствора бикарбоната натрия и насыщенного раствора хлорида натрия с последующим интенсивным встряхиванием. Водный слой отделяли и затем органический слой сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и затем выпаривали при пониженном давлении растворитель. Остаток очищали хроматографией на колонке с NH-силикагелем. Полученный продукт растворяли в этилацетате и выпаривали растворитель в токе азота. К остатку добавляли диэтиловый эфир и в достаточной мере осаждали твердое вещество. Затем выпаривали в токе азота растворитель. Остаток сушили при пониженном давлении с получением указанного в заголовке соединения (1,60 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,82-2,88 (м, 1H), 2,98 (дд, J=5,2, 13,2 Гц, 1H), 3,13 (дд, J=4,0, 5,2 Гц, 1H), 3,87 (дд, J=2,8, 4,0 Гц, 2H), 4,07 (д, J=11,6 Гц, 1H), 4,23 (д, J=11,6 Гц, 1H), 6,53 (т, J=4,8 Гц, 1H), 7,24-7,29 (м, 1H), 7,44-7,48 (м, 3H), 7,56-7,57 (м, 1H), 7,86 (ддд, J=2,0, 7,6, 9,6 Гц, 1H), 8,19-8,21 (м, 1H), 8,34 (д, J=4,8 Гц, 2H).
Пример 38
Синтез N-[3-((4aS,7R,8aS)-2-амино-7-метоксиметил-4a,5,7,8-тетрагидро-4H-6-окса-3-тиа-1-азанафталин-8a-ил)-4-фторфенил]-5-фторметоксипиразин-2-карбоксамида
[Формула 138]

Соединение, полученное в примере получения 19-(14) (45 мг), растворяли в дихлорметане (2 мл). К раствору добавляли 5-фторметоксипиразин-2-карбоновую кислоту (28 мг), N,N-диизопропилэтиламин (48 мкл) и PyBOP (113 мг) с последующим перемешиванием при комнатной температуре. Через три часа реакционный раствор концентрировали и остаток подвергали хроматографии на колонке с силикагелем с получением амида (66 мг). Амид (66 мг) растворяли в дихлорметане (2 мл). Добавляли ТФУК (1 мл) и смесь перемешивали при комнатной температуре в течение 1,5 часов. Реакционный раствор концентрировали. К остатку добавляли хлороформ, насыщенный раствор бикарбоната натрия и 1 н. раствор гидроксида натрия и отделяли органический слой. Органический слой сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении с получением остатка. Остаток осаждали, используя трет-бутилметиловый эфир, этилацетат и гексан. Твердое вещество собирали фильтрованием с получением указанного в заголовке соединения (36 мг).
ESI-MS; m/z 480 [M++H].
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,69 (дд, J=1,6, 12,8 Гц, 1H), 2,38 (т, J=12,0 Гц, 1H), 2,56-2,64 (м, 1H), 2,96 (дд, J=4,4, 12,8 Гц, 1H), 2,98-3,06 (м, 1H), 3,40 (с, 3H), 3,37-3,51 (м, 2H), 3,79-3,95 (м, 3H), 6,09 (дд, J=2,0, 3,6 Гц, 1H), 6,22 (дд, J=2,0, 3,6 Гц, 1H), 7,08 (дд, J=8,8, 12,0 Гц, 1H), 7,30-7,40 (м, 1H), 7,85-7,95 (м, 1H), 8,29 (д, J=1,6 Гц, 1H), 9,08 (д, J=1,2 Гц, 1H) 9,45 (уш.с, 1H).
Примеры 39-40
Соединения примеров 39-40, показанные ниже в таблице 1, синтезировали в соответствии с примером 38, используя соответствующие карбоновые кислоты.
[Таблица 1]

Пример 41
Синтез N-[3-((4aS,7R,8aS)-2-амино-7-фторметил-4a,5,7,8-тетрагидро-4H-6-окса-3-тиа-1-азанафталин-8a-ил)-4-фторфенил]-5-фторметоксипиразин-2-карбоксамида
[Формула 139]
Соединение, полученное в примере получения 20-(3) (45 мг), растворяли в дихлорметане (2 мл). К раствору добавляли 5-фторметоксипиразин-2-карбоновую кислоту (28 мг), N,N-диизопропилэтиламин (48 мкл) и PyBOP (113 мг) с последующим перемешиванием при комнатной температуре. Через три часа реакционный раствор концентрировали и остаток подвергали хроматографии на колонке с силикагелем с получением амида (67 мг). Амид (67 мг) растворяли в дихлорметане (2 мл). Затем добавляли ТФУК (1 мл) и смесь перемешивали при комнатной температуре в течение 1,5 часов. Реакционный раствор концентрировали при пониженном давлении. Затем к остатку добавляли хлороформ, насыщенный раствор бикарбоната натрия и 1 н. раствор гидроксида натрия и отделяли органический слой. Органический слой сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении с получением остатка. Остаток отверждали добавлением трет-бутилметилового эфира и гексана. Твердое вещество собирали фильтрованием с получением указанного в заголовке соединения (41 мг).
ESI-MS; m/z 468 [M++H].
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,50-1,90 (м, 1H), 2,36-2,48 (м, 1H), 2,58-2,66 (м, 1H), 2,92-3,10 (м, 2H), 3,80-4,04 (м, 3H), 4,32-4,43 (м, 1H), 4,44-4,54 (м, 1H), 6,09 (дд, J=2,0, 4,0 Гц, 1H), 6,22 (дд, J=2,0, 4,0 Гц, 1H), 7,10 (дд, J=8,8, 12,0 Гц, 1H), 7,35-7,45 (м, 1H), 7,85-7,95 (м, 1H), 8,29 (д, J=1,6 Гц, 1H), 9,08 (д, J=1,6 Гц, 1H) 9,46 (уш.с, 1H).
Примеры 42-45
Соединения примеров 42-45, показанные ниже в таблице 2, синтезировали в соответствии с примером 41, используя соответствующие карбоновые кислоты.
[Таблица 2]




Пример 46
Синтез (±)-(4aR*,7aS*)-7a-[3-(2-фторпиридин-3-ил)фенил]-6-(2,2,2-трифторэтил)-4,4a,5,6,7,7a-гексагидропирроло[3,4-d][1,3]тиазин-2-иламина
[Формула 140]
Соединение, полученное в примере получения 18-(9) (50 мг), и N,N-диизопропилэтиламин (45 мкл) растворяли в ацетонитриле (1 мл). Затем добавляли 2,2,2-трифторэтилтрифторметансульфонат (24 мкл) с последующим перемешиванием при комнатной температуре. Через 14 часов добавляли к реакционному раствору воду и этилацетат и отделяли органический слой. Органический слой последовательно промывали насыщенным раствором соли и насыщенным раствором бикарбоната натрия. Органический слой сушили над сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонке с NH-силикагелем с получением N-алкильного соединения (38 мг) в виде неочищенного продукта. N-алкильное соединение (38 мг) растворяли в дихлорметане (2 мл). Затем добавляли ТФУК (0,5 мл) с последующим перемешиванием в течение двух часов. Реакционный раствор концентрировали при пониженном давлении. К остатку добавляли хлороформ и 2 н. гидроксид натрия и отделяли органический слой. Органический слой сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием. Фильтрат концентрировали при пониженном давлении с получением остатка. Остаток подвергали хроматографии на колонке с NH-силикагелем с получением указанного в заголовке соединения (18 мг).
ESI-MS; m/z 411 [M++H].
Пример 47
Синтез (±)-1-{(4aR*,7aS*)-2-амино-7a-[3-(2-фторпиридин-3-ил)фенил]-4a,5,7,7a-тетрагидро-4H-пирроло[3,4-d][1,3]тиазин-6-ил}-3,3,3-трифторпропан-1-она
[Формула 141]

Соединение, полученное в примере получения 18-(9) (45 мг), 3,3,3-трифторпропионовую кислоту (10 мкл), N,N-диизопропилэтиламин (17 мкл) растворяли в тетрагидрофуране (1 мл). Затем добавляли PyBOP (51 мг) с последующим перемешиванием при комнатной температуре. Через 14 часов добавляли к реакционному раствору насыщенный раствор бикарбоната натрия и этилацетат и отделяли органический слой. Органический слой последовательно промывали насыщенным раствором соли и насыщенным раствором хлорида аммония. Органический слой сушили над сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении. Остаток подвергали NH-пТСХ с получением амида (26 мг) в виде неочищенного продукта. Амид (26 мг) растворяли в дихлорметане (2 мл). Затем добавляли ТФУК (0,5 мл) с последующим перемешиванием в течение двух часов. Реакционный раствор концентрировали при пониженном давлении. К остатку добавляли хлороформ и 2 н. гидроксид натрия и отделяли органический слой. Органический слой сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении с получением остатка. Остаток подвергали хроматографии на колонке с силикагелем с получением указанного в заголовке соединения (15 мг).
ESI-MS; m/z 439 [M++H].
Пример 48
Соединение примера 48, показанное ниже в таблице 3, синтезировали в соответствии с примером 47, используя соответствующие карбоновые кислоты.
[Таблица 3]
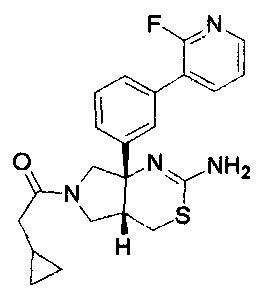
Пример 49
Синтез N-[3-((4aS*,5R*,7aS*)-2-амино-5-этил-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил]-5-цианопиридин-2-карбоксамида
[Формула 142]

Указанное в заголовке соединение (28,0 мг) получали из соединения, полученного в примере получения 21-(10) (30,0 мг), и 5-цианопиридин-2-карбоновой кислоты, полученной в примере получения 13-(2) (22,5 мг), в соответствии со способом примера 14.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,04-1,07 (м, 3H), 1,65-1,72 (м, 2H), 2,68-2,77 (м, 2H), 3,08-3,12 (м, 1H), 3,80-3,82 (м, 1H), 4,17-4,22 (м, 1H), 4,54-4,57 (м, 1H), 7,07-7,13 (м, 1H), 7,52-7,54 (м, 1H), 7,96-8,00 (м, 1H), 8,19-8,21 (м, 1H), 8,42-8,44 (м, 1H), 8,90 (с, 1H), 9,85 (уш., 1H).
Пример 50
Синтез N-[3-((4aS*,5R*,7aS*)-2-амино-5-этил-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил]-5-дифторметоксипиразин-2-карбоксамида
[Формула 143]
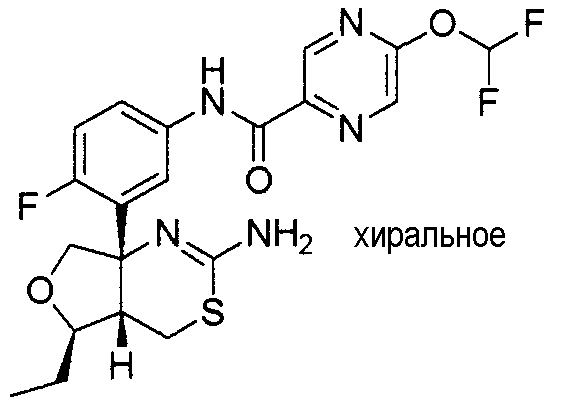
Указанное в заголовке соединение (22,0 мг) получали из соединения, полученного в примере получения 21-(10) (28,0 мг), и 5-дифторметоксипиразин-2-карбоновой кислоты, полученной в примере получения 14-(2) (20,2 мг), в соответствии со способом примера 14.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,04-1,07 (м, 3H), 1,67-1,70 (м, 2H), 2,70-2,76 (м, 2H), 3,08-3,11 (м, 1H), 3,80-3,83 (м, 1H), 4,17-4,20 (м, 1H), 4,54-4,56 (м, 1H), 7,06-7,11 (м, 1H), 7,51 (т, J=72 Гц, 1H), 7,93-7,95 (м, 1H), 8,33 (с, 1H), 9,06 (с, 1H), 9,48 (уш., 1H).
Пример 51
Синтез N-[3-((4aS*,5R*,7aS*)-2-амино-5-этил-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил]-5-хлорпиридин-2-карбоксамида
[Формула 144]
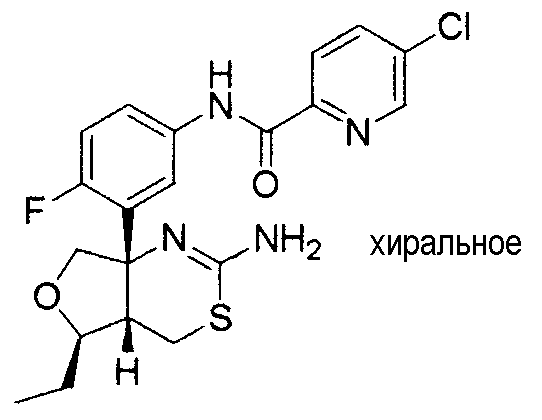
Указанное в заголовке соединение (26,0 мг) получали из соединения, полученного в примере получения 21-(10) (28,0 мг), и 5-хлорпиридин-2-карбоновой кислоты (23,2 мг) в соответствии со способом примера 14.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,05 (т, J=7,2 Гц, 3H), 1,65-1,72 (м, 2H), 2,68-2,76 (м, 2H), 3,08-3,13 (м, 1H), 3,81-3,83 (м, 1H), 4,17-4,22 (м, 1H), 4,54-4,56 (м, 1H), 7,05-7,10 (м, 1H), 7,51-7,53 (м, 1H), 7,87-7,98 (м, 2H), 8,23-8,25 (м, 1H), 8,56-8,56 (м, 1H), 9,83 (уш., 1H).
Пример 52
Синтез N-[3-((4aS*,5R*,7aS*)-2-амино-5-этил-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил]-5-фторметоксипиразин-2-карбоксамида
[Формула 145]

Указанное в заголовке соединение (23,0 мг) получали из соединения, полученного в примере получения 21-(10) (30,0 мг), и 5-фторметоксипиразин-2-карбоновой кислоты, полученной в примере получения 15-(2) (18,8 мг), в соответствии со способом примера 14.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,04-1,07 (м, 3H), 1,65-1,70 (м, 2H), 2,71-2,76 (м, 2H), 3,09-3,13 (м, 1H), 3,80-3,83 (м, 1H), 4,15-4,20 (м, 1H), 4,54-4,56 (м, 1H), 6,15 (д, J=51 Гц, 1H), 7,06-7,11 (м, 1H), 7,48-7,49 (м, 1H), 7,96-7,97 (м, 1H), 8,29 (с, 1H), 9,08 (с, 1H), 9,50 (уш., 1H).
Пример 53
Синтез N-[3-((4aS*,5R*,7aS*)-2-амино-5-этил-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил]-5-дифторметилпиразин-2-карбоксамида
[Формула 146]

Указанное в заголовке соединение (30,0 мг) получали из соединения, полученного в примере получения 21-(10) (28,0 мг), и 5-дифторметилпиразин-2-карбоновой кислоты, полученной в примере получения 17-(5) (18,5 мг), в соответствии со способом примера 14.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,04-1,08 (м, 3H), 1,66-1,73 (м, 2H), 2,69-2,78 (м, 2H), 3,09-3,13 (м, 1H), 3,81-3,83 (м, 1H), 4,18-4,23 (м, 1H), 4,55-4,57 (м, 1H), 6,80 (т, J= 55 Гц, 1H), 7,09-7,12 (м, 1H), 7,53-7,56 (м, 1H), 7,95-7,99 (м, 1H), 8,93 (с, 1H), 9,53 (с, 1H), 9,65 (уш., 1H).
Пример 54
Синтез N-[3-((4aS*,5R*,7aS*)-2-амино-5-метил-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил]-5-цианопиридин-2-карбоксамида
[Формула 147]

Указанное в заголовке соединение (18,0 мг) получали из соединения, полученного в примере получения 22-(11) (30,0 мг), и 5-цианопиридин-2-карбоновой кислоты, полученной в примере получения 13-(2) (23,3 мг), в соответствии со способом примера 14.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,37-1,39 (м, 3H), 2,57-2,58 (м, 1H), 2,73-2,76 (м, 1H), 3,10-3,13 (м, 1H), 3,81-3,83 (м, 1H), 4,35-4,38 (м, 1H), 4,59-4,61 (м, 1H), 7,08-7,13 (м, 1H), 7,52-7,53 (м, 1H), 7,96-7,98 (м, 1H), 8,20 (д, J=8,0 Гц, 1H), 8,43 (д, J=8,0 Гц, 1H), 8,90 (с, 1H), 9,85 (уш., 1H).
Пример 55
Синтез N-[3-((4aS*,5R*,7aS*)-2-амино-5-метил-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил]-5-дифторметоксипиразин-2-карбоксамида
[Формула 148]

Указанное в заголовке соединение (22,0 мг) получали из соединения, полученного в примере получения 22-(11) (30,0 мг), и 5-дифторметоксипиразин-2-карбоновой кислоты, полученной в примере получения 14-(2) (22,5 мг), в соответствии со способом примера 14.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,37-1,39 (м, 3H), 2,56-2,60 (м, 1H), 2,72-2,76 (м, 1H), 3,10-3,14 (м, 1H), 3,81-3,84 (м, 1H), 4,35-4,38 (м, 1H), 4,59-4,61 (м, 1H), 7,07-7,12 (м, 1H), 7,49-7,51 (м, 1H), 7,51 (т, J=71,6 Гц, 1H), 7,93-7,96 (м, 1H), 8,34 (д, J=1,6 Гц, 1H), 9,07 (д, J=1,6 Гц, 1H), 9,47 (уш., 1H).
Пример 56
Синтез N-[3-((4aS*,5R*,7aS*)-2-амино-5-метил-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил]-5-хлорпиридин-2-карбоксамида
[Формула 149]

Указанное в заголовке соединение (17,0 мг) получали из соединения, полученного в примере получения 22-(11) (28,0 мг), и 5-хлорпиридин-2-карбоновой кислоты (23,2 мг) в соответствии со способом примера 14.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,37-1,39 (м, 3H), 2,57-2,61 (м, 1H), 2,72-2,76 (м, 1H), 3,11-3,15 (м, 1H), 3,82-3,85 (м, 1H), 4,35-4,38 (м, 1H), 4,59-4,62 (м, 1H), 7,06-7,11 (м, 1H), 7,50-7,52 (м, 1H), 7,88-7,98 (м, 2H), 8,24 (д, J=8,4 Гц, 1H), 8,57 (с, 1H), 9,82 (уш., 1H).
Пример 57
Синтез N-[3-((4aS*,5R*,7aS*)-2-амино-5-метил-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил]-5-фторметоксипиразин-2-карбоксамида
[Формула 150]

Указанное в заголовке соединение (22,0 мг) получали из соединения, полученного в примере получения 22-(11) (28,0 мг), и 5-фторметоксипиразин-2-карбоновой кислоты, полученной в примере получения 15-(2) (19,0 мг), в соответствии со способом примера 14.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,37-1,39 (м, 3H), 2,59-2,61 (м, 1H), 2,73-2,77 (м, 1H), 3,10-3,15 (м, 1H), 3,82-3,85 (м, 1H), 4,35-4,37 (м, 1H), 4,59-4,61 (м, 1H), 6,08-6,22 (м, 1H), 7,07-7,12 (м, 1H), 7,49-7,51 (м, 1H), 7,93-7,97 (м, 1H), 8,29 (с, 1H), 9,08 (с, 1H), 9,50 (уш., 1H).
Пример 58
Синтез N-[3-((4aS*,5R*,7aS*)-2-амино-5-метил-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил]-5-дифторметилпиразин-2-карбоксамида
[Формула 151]
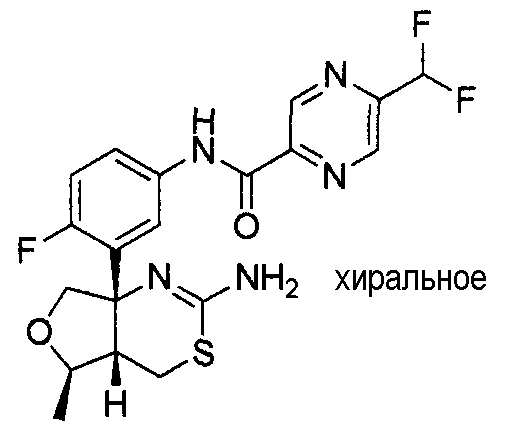
Указанное в заголовке соединение (21,0 мг) получали из соединения, полученного в примере получения 22-(11) (25,0 мг), и 5-дифторметилпиразин-2-карбоновой кислоты, полученной в примере получения 17-(5) (17,0 мг), в соответствии со способом примера 14.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,37-1,39 (м, 3H), 2,57-2,61 (м, 1H), 2,73-2,77 (м, 1H), 3,10-3,14 (м, 1H), 3,81-3,84 (м, 1H), 4,35-4,39 (м, 1H), 4,60-4,62 (м, 1H), 6,66-6,93 (м, 1H), 7,09-7,14 (м, 1H), 7,53-7,55 (м, 1H), 7,93-7,97 (м, 1H), 8,92 (д, J=8,0 Гц, 1H), 9,53 (с, 1H), 9,64 (уш., 1H).
Пример 59
Синтез N-[3-((4aS*,5R*,7aS*)-2-амино-5-метил-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил]-5-дифторметоксипиридин-2-карбоксамида
[Формула 152]

Указанное в заголовке соединение (28,0 мг) получали из соединения, полученного в примере получения 22-(11) (30,0 мг), и 5-дифторметоксипиридин-2-карбоновой кислоты (22,3 мг) в соответствии со способом примера 14.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,37-1,39 (м, 3H), 2,58-2,61 (м, 1H), 2,72-2,76 (м, 1H), 3,11-3,15 (м, 1H), 3,82-3,84 (м, 1H), 4,34-4,40 (м, 1H), 4,60-4,62 (м, 1H), 6,65 (т, J=72 Гц, 1H), 7,08-7,11 (м, 1H), 7,51-7,53 (м, 1H), 7,65-7,67 (м, 1H), 7,93-7,97 (м, 1H), 8,31-8,33 (м, 1H), 8,46 (с, 1H), 9,83 (уш., 1H).
Пример 60
Синтез N-[3-((4aS*,5R*,7aS*)-2-амино-5-метил-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил]пиримидин-4-карбоксамида
[Формула 153]

Указанное в заголовке соединение (16,0 мг) получали из соединения, полученного в примере получения 22-(11) (20,0 мг), и пиримидин-4-карбоновой кислоты (15,0 мг) в соответствии со способом примера 14.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,37-1,39 (м, 3H), 2,59-2,61 (м, 1H), 2,73-2,77 (м, 1H), 3,10-3,14 (м, 1H), 3,83-3,86 (м, 1H), 4,35-4,39 (м, 1H), 4,59-4,62 (м, 1H), 7,08-7,14 (м, 1H), 7,53-7,56 (м, 1H), 7,94-7,96 (м, 1H), 8,21-8,23 (м, 1H), 9,04-9,05 (м, 1H), 9,32-9,32 (м, 1H), 9,87 (уш., 1H).
Пример 61
Синтез N-[3-((4aS*,5R*,7aS*)-2-амино-5-метил-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил]пиридин-2-карбоксамида
[Формула 154]

Указанное в заголовке соединение (18,0 мг) получали из соединения, полученного в примере получения 22-(11) (20,0 мг), и пиколиновой кислоты (12,9 мг) в соответствии со способом примера 14.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,37-1,39 (м, 3H), 2,58-2,62 (м, 1H), 2,72-2,77 (м, 1H), 3,12-3,16 (м, 1H), 3,84-3,87 (м, 1H), 4,35-4,39 (м, 1H), 4,60-4,62 (м, 1H), 7,06-7,11 (м, 1H), 7,48-7,54 (м, 2H), 7,89-8,01 (м, 2H), 8,28-8,30 (м, 1H), 8,62-8,63 (м, 1H).
Пример 62
Синтез N-[3-((4aS*,5R*,7aS*)-2-амино-5-метил-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил]-5-фторпиридин-2-карбоксамида
[Формула 155]

Указанное в заголовке соединение (13,0 мг) получали из соединения, полученного в примере получения 22-(11) (20,0 мг), и 5-фторпиридин-2-карбоновой кислоты (15,0 мг) в соответствии со способом примера 14.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,37-1,39 (м, 3H), 2,59-2,61 (м, 1H), 2,72-2,77 (м, 1H), 3,11-3,15 (м, 1H), 3,83-3,86 (м, 1H), 4,35-4,39 (м, 1H), 4,59-4,62 (м, 1H), 7,06-7,12 (м, 1H), 7,49-7,62 (м, 2H), 7,95-7,97 (м, 1H), 8,31-8,47 (м, 2H), 9,80 (уш., 1H).
Пример 63
Синтез N-[3-((4aS*,5S*,7aS*)-2-амино-5-метокси-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]-5-цианопиридин-2-карбоксамида
[Формула 156]

Указанное в заголовке соединение (7,0 мг) получали из соединения, полученного в примере получения 23-(15) (20,0 мг), и 5-цианопиридин-2-карбоновой кислоты, полученной в примере получения 13-(2) (15,0 мг), в соответствии со способом примера 14.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,84-2,09 (м, 3H), 2,73-2,85 (м, 2H), 2,96-2,97 (м, 2H), 3,41 (с, 3H), 4,01-4,06 (м, 1H), 7,06-7,11 (м, 1H), 7,39-7,40 (м, 1H), 7,93-7,97 (м, 1H), 8,19-8,20 (м, 1H), 8,42-8,44 (м, 1H), 8,90 (с, 1H), 9,82 (уш., 1H).
Пример 64
Синтез N-[3-((4aS*,5S*,7aS*)-2-амино-5-метокси-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]-5-дифторметоксипиразин-2-карбоксамида
[Формула 157]

Указанное в заголовке соединение (10,0 мг) получали из соединения, полученного в примере получения 23-(15) (18,0 мг), и 5-дифторметоксипиразин-2-карбоновой кислоты, полученной в примере получения 14-(2) (13,0 мг), в соответствии со способом примера 14.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,85-2,06 (м, 3H), 2,75-2,84 (м, 2H), 2,96-2,97 (м, 2H), 3,41 (с, 3H), 4,02-4,04 (м, 1H), 7,05-7,10 (м, 1H), 7,35-7,38 (м, 1H), 7,51 (т, J=72 Гц, 1H), 7,90-7,94 (м, 1H), 8,33 (с, 1H), 9,07 (с, 1H), 9,45 (уш., 1H).
Пример 65
Синтез N-[3-((4aS*,5R*,7aS*)-2-амино-5-метокси-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]-5-цианопиридин-2-карбоксамида
[Формула 158]

Указанное в заголовке соединение (11,0 мг) получали из соединения, полученного в примере получения 23-(14) (20,0 мг), и 5-цианопиридин-2-карбоновой кислоты, полученной в примере получения 13-(2) (15,0 мг), в соответствии со способом примера 14.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,77-2,26 (м, 3H), 2,53-2,58 (м, 1H), 2,83-3,06 (м, 3H), 3,32 (с, 3H), 3,94-3,96 (м, 1H), 7,05-7,10 (м, 1H), 7,67-7,68 (м, 1H), 7,76-7,78 (м, 1H), 8,20-8,22 (м, 1H), 8,42-8,44 (м, 1H), 8,91 (с, 1H), 9,84 (уш., 1H).
Пример 66
Синтез N-[3-((4aS,5S,7aS)-2-амино-5-метоксиметил-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил]-5-цианопиридин-2-карбоксамида
[Формула 159]

Следующее соединение синтезировали в соответствии с примером 14, используя соединение, полученное в примере получения 24-(11), и соответствующую карбоновую кислоту.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,83-2,86 (м, 1H), 2,96-3,00 (м, 1H), 3,09-3,13 (м, 1H), 3,44 (с, 3H), 3,53-3,57 (м, 1H), 3,63-3,67 (м, 1H), 3,86-3,88 (м, 1H), 4,44-4,48 (м, 1H), 4,55-4,57 (м, 1H), 7,08-7,13 (м, 1H), 7,53-7,54 (м, 1H), 7,97-8,00 (м, 1H), 8,20-8,22 (м, 1H), 8,42-8,44 (м, 1H), 8,90 (с, 1H), 9,86 (уш., 1H).
Пример 67
Синтез N-[3-((4aS,5S,7aS)-2-амино-5-метоксиметил-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил]-5-дифторметоксипиразин-2-карбоксамида
[Формула 160]

Следующее соединение синтезировали в соответствии с примером 14, используя соединение, полученное в примере получения 24-(11), и соответствующую карбоновую кислоту.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,82-2,85 (м, 1H), 2,96-2,98 (м, 1H), 3,10-3,13 (м, 1H), 3,43 (с, 3H), 3,53-3,56 (м, 1H), 3,64-3,66 (м, 1H), 3,85-3,87 (м, 1H), 4,45-4,46 (м, 1H), 4,54-4,57 (м, 1H), 7,07-7,12 (м, 1H), 7,49-7,51 (м, 1H), 7,51 (т, J=72 Гц, 1H), 7,95-7,97 (м, 1H), 8,34 (с, 1H), 9,07 (с, 1H), 9,49 (уш., 1H).
Пример 68
Синтез N-[3-((4aS,5S,7aS)-2-амино-5-метоксиметил-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил]-5-дифторметилпиразин-2-карбоксамида
[Формула 161]

Следующее соединение синтезировали в соответствии с примером 14, используя соединение, полученное в примере получения 24-(11), и соответствующую карбоновую кислоту.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,82-2,86 (м, 1H), 2,95-2,99 (м, 1H), 3,09-3,14 (м, 1H), 3,44 (с, 3H), 3,54-3,57 (м, 1H), 3,65-3,67 (м, 1H), 3,85-3,87 (м, 1H), 4,44-4,48 (м, 1H), 4,55-4,57 (м, 1H), 6,80 (т, J=55 Гц, 1H), 7,08-7,14 (м, 1H), 7,55-7,56 (м, 1H), 7,95-7,99 (м, 1H), 8,92 (с, 1H), 9,52 (с, 1H), 9,65 (уш., 1H).
Пример 69
Синтез N-[3-((4aS,5S,7aS)-2-амино-5-метоксиметил-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил]-5-фторметоксипиразин-2-карбоксамида
[Формула 162]

Следующее соединение синтезировали в соответствии с примером 14, используя соединение, полученное в примере получения 24-(11), и соответствующую карбоновую кислоту.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,83-2,86 (м, 1H), 2,97-2,99 (м, 1H), 3,10-3,43 (м, 1H), 3,44 (с, 3H), 3,54-3,56 (м, 1H), 3,64-3,67 (м, 1H), 3,86-3,89 (м, 1H), 4,45-4,47 (м, 1H), 4,54-4,57 (м, 1H), 6,09-6,22 (м, 1H), 7,07-7,12 (м, 1H), 7,47-7,49 (м, 1H), 7,97-7,99 (м, 1H), 8,30 (с, 1H), 9,08 (с, 1H), 9,52 (уш., 1H).
Пример 70
Синтез N-[3-((4aS,5S,7aS)-2-амино-5-фторметил-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил]-5-трифторметилпиридин-2-карбоксамида
[Формула 163]
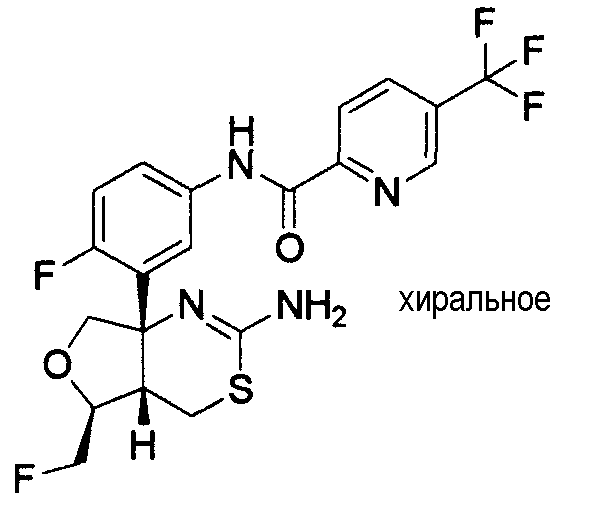
Указанное в заголовке соединение (29,0 мг) получали из соединения, полученного в примере получения 25-(13) (25,0 мг), и 5-трифторметилпиридин-2-карбоновой кислоты (17,9 мг) в соответствии со способом примера 14.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,79-2,83 (м, 1H), 3,08-3,19 (м, 2H), 3,88-3,91 (м, 1H), 4,51-4,66 (м, 4H), 7,09-7,14 (м, 1H), 7,58-7,61 (м, 1H), 7,96-8,00 (м, 1H), 8,16-8,18 (м, 1H), 8,42-8,44 (м, 1H), 8,90 (с, 1H).
Пример 71
Синтез N-[3-((4aS,5S,7aS)-2-амино-5-фторметил-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил]-5-хлорпиридин-2-карбоксамида
[Формула 164]

Указанное в заголовке соединение (16,0 мг) получали из соединения, полученного в примере получения 25-(13) (25,0 мг), и 5-хлорпиридин-2-карбоновой кислоты (14,8 мг) в соответствии со способом примера 14.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,79-2,83 (м, 1H), 3,08-3,19 (м, 2H), 3,88-3,91 (м, 1H), 4,51-4,66 (м, 4H), 7,07-7,12 (м, 1H), 7,54-7,56 (м, 1H), 7,87-8,00 (м, 2H), 8,23-8,25 (м, 1H), 8,57 (м, 1H), 9,83 (уш., 1H).
Пример 72
Синтез N-[3-((4aS,5,7aS)-2-амино-5-фторметил-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил]-5-дифторметоксипиридин-2-карбоксамида
[Формула 165]

Указанное в заголовке соединение (27,0 мг) получали из соединения, полученного в примере получения 25-(13) (25,0 мг), и 5-дифторметоксипиридин-2-карбоновой кислоты (17,8 мг) в соответствии со способом примера 14.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,79-2,83 (м, 1H), 3,08-3,19 (м, 2H), 3,88-3,91 (м, 1H), 4,51-4,66 (м, 4H), 6,65 (т, J=72,0 Гц, 1H), 7,08-7,13 (м, 1H), 7,55-7,57 (м, 1H), 7,66-7,69 (м, 1H), 7,95-7,99 (м, 1H), 8,31-8,34 (м, 1H), 8,47-8,48 (м, 1H), 9,84 (уш., 1H).
Пример 73
Синтез N-[3-((4aS,5S,7aS)-2-амино-5-фторметил-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил]-5-дифторметилпиразин-2-карбоксамида
[Формула 166]

Указанное в заголовке соединение (16,0 мг) получали из соединения, полученного в примере получения 25-(13) (25,0 мг), и 5-дифторметилпиразин-2-карбоновой кислоты, полученной в примере получения 17-(5) (16,3 мг), в соответствии со способом примера 14.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,80-2,84 (м, 1H), 3,11-3,19 (м, 2H), 3,91-3,94 (м, 1H), 4,51-4,66 (м, 4H), 6,80 (т, J=54,4 Гц, 1H), 7,10-7,16 (м, 1H), 7,56-7,59 (м, 1H), 7,96-8,00 (м, 1H), 8,94 (с, 1H), 9,53 (с, 1H), 9,66 (уш., 1H).
Пример 74
Синтез N-[3-((4aS,5S,7aS)-2-амино-5-фторметил-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил]-5-дифторметоксипиразин-2-карбоксамида
[Формула 167]

Указанное в заголовке соединение (26,0 мг) получали из соединения, полученного в примере получения 25-(13) (25,0 мг), и 5-дифторметоксипиразин-2-карбоновой кислоты, полученной в примере получения 14-(2) (17,9 мг), в соответствии со способом примера 14.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,79-2,83 (м, 1H), 3,09-3,18 (м, 2H), 3,89-3,92 (м, 1H), 4,51-4,65 (м, 4H), 7,09-7,14 (м, 1H), 7,52 (т, J=71,6 Гц, 1H), 7,52-7,55 (м, 1H), 7,94-7,97 (м, 1H), 8,35 (с, 1H), 9,07 (с, 1H), 9,49 (уш., 1H).
Пример 75
Синтез N-[3-((4aS,5,7aS)-2-амино-5-фторметил-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил]-5-фторметоксипиразин-2-карбоксамида
[Формула 168]

Указанное в заголовке соединение (28,0 мг) получали из соединения, полученного в примере получения 25-(13) (25,0 мг), и 5-фторметоксипиразин-2-карбоновой кислоты, полученной в примере получения 15-(2) (16,2 мг), в соответствии со способом примера 14.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,80-2,84 (м, 1H), 3,11-3,19 (м, 2H), 3,92-3,95 (м, 1H), 4,50-4,66 (м, 4H), 6,09-6,10 (м, 1H), 6,22-6,23 (м, 1H), 7,08-7,13 (м, 1H), 7,51-7,53 (м, 1H), 7,95-7,99 (м, 1H), 8,30 (с, 1H), 9,09 (с, 1H), 9,51 (уш., 1H).
Пример 76
Синтез N-[3-((4aS,5S,7aS)-2-амино-5-фторметил-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил]пиримидин-4-карбоксамида
[Формула 169]
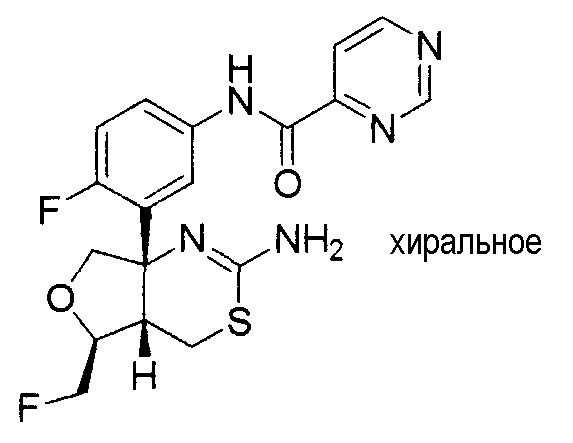
Указанное в заголовке соединение (15,0 мг) получали из соединения, полученного в примере получения 25-(13) (20,0 мг), и пиримидин-4-карбоновой кислоты (12,4 мг) в соответствии со способом примера 14.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,79-2,83 (м, 1H), 3,09-3,18 (м, 2H), 3,87-3,90 (м, 1H), 4,52-4,66 (м, 4H), 7,09-7,14 (м, 1H), 7,58-7,61 (м, 1H), 7,95-7,99 (м, 1H), 8,21-8,23 (м, 1H), 9,05 (д, J=5,6 Гц, 1H), 9,32 (д, J=5,6 Гц, 1H), 9,88 (уш., 1H).
Пример 77
Синтез N-[3-((4aS,5,7aS)-2-амино-5-фторметил-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил]пиридин-2-карбоксамида
[Формула 170]
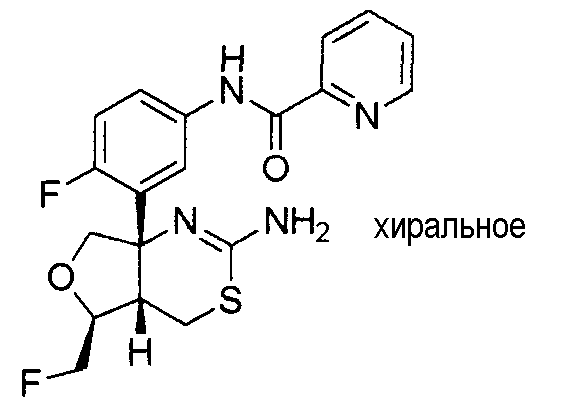
Указанное в заголовке соединение (18,0 мг) получали из соединения, полученного в примере получения 25-(13) (20,0 мг), и пиколиновой кислоты (12,9 мг) в соответствии со способом примера 14.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,79-2,83 (м, 1H), 3,08-3,20 (м, 2H), 3,90-3,92 (м, 1H), 4,51-4,66 (м, 4H), 7,07-7,20 (м, 2H), 7,48-7,58 (м, 2H), 7,90-8,01 (м, 1H), 8,28-8,30 (м, 1H), 8,63-8,65 (м, 1H).
Пример 78
Синтез N-[3-((4aS,5S,7aS)-2-амино-5-фторметил-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил]-5-фторпиридин-2-карбоксамида
[Формула 171]
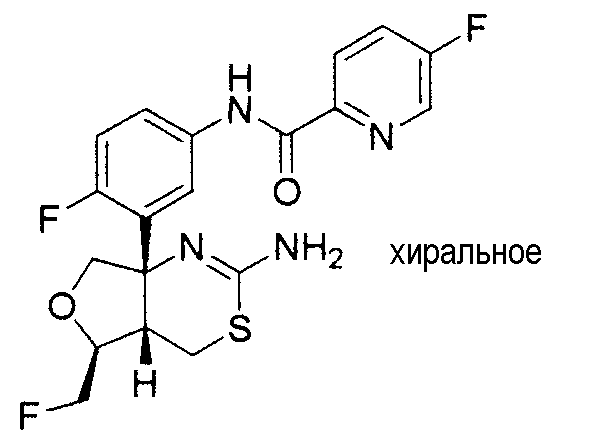
Указанное в заголовке соединение (13,0 мг) получали из соединения, полученного в примере получения 25-(13) (20,0 мг), и пиколиновой кислоты (15,0 мг) в соответствии со способом примера 14.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,78-2,82 (м, 1H), 3,06-3,19 (м, 2H), 3,88-3,90 (м, 1H), 4,51-4,67 (м, 4H), 7,07-7,12 (м, 1H), 7,55-7,62 (м, 1H), 7,93-7,97 (м, 1H), 8,31-8,45 (м, 2H).
Пример 79
Синтез N-[3-((4aS*,8aS*)-2-амино-4a,5,7,8-тетрагидро-4H-6-окса-3-тиа-1-азанафталин-8a-ил)-4-фторфенил]-5-дифторметилпиразин-2-карбоксамида
[Формула 172]
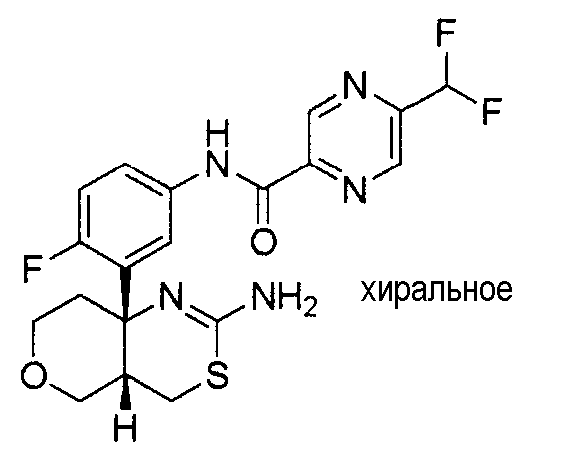
Указанное в заголовке соединение (29,0 мг) получали из соединения, полученного в примере получения 8 (30,0 мг), и 5-дифторметилпиразин-2-карбоновой кислоты, полученной в примере получения 17-(5) (22,3 мг), в соответствии со способом примера 14.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,70-1,73 (м, 1H), 2,60-2,70 (м, 2H), 2,93-3,05 (м, 2H), 3,70-3,91 (м, 4H), 4,59 (уш., 1H), 6,79 (т, J=54 Гц, 1H), 7,04-7,09 (м, 1H), 7,45-7,46 (м, 1H), 7,87-7,89 (м, 1H), 8,89 (с, 1H), 9,49 (с, 1H), 9,59 (уш., 1H).
Пример 80
Синтез N-[3-((4aS*,8aS*)-2-амино-4a,5,7,8-тетрагидро-4H-6-окса-3-тиа-1-азанафталин-8a-ил)-4-фторфенил]-5-фторметоксипиразин-2-карбоксамида
[Формула 173]
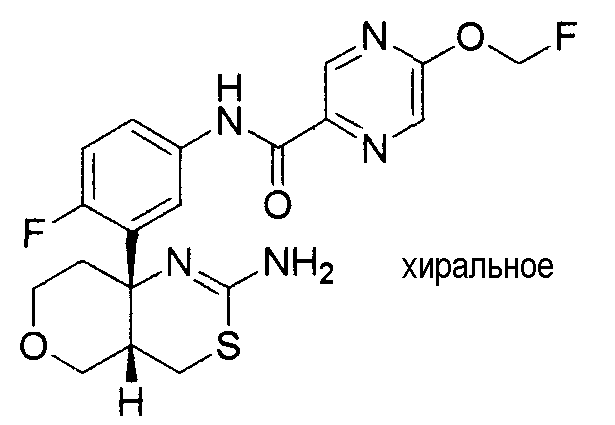
Указанное в заголовке соединение (24,0 мг) получали из соединения, полученного в примере получения 8 (30,0 мг), и 5-фторметоксипиразин-2-карбоновой кислоты, полученной в примере получения 15-(2) (20,3 мг), в соответствии со способом примера 14.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,68-1,71 (м, 1H), 2,57-2,69 (м, 2H), 2,93-3,06 (м, 2H), 3,70-3,91 (м, 4H), 6,08-6,21 (м, 1H), 7,05-7,10 (м, 1H), 7,37-7,38 (м, 1H), 7,89-7,93 (м, 1H), 8,27 (с, 1H), 9,07 (с, 1H), 9,45 (уш., 1H).
Пример 81
Синтез N-[3-((4aS*,8aS*)-2-амино-4a,5,7,8-тетрагидро-4H-6-окса-3-тиа-1-азанафталин-8a-ил)-4-фторфенил]-5-дифторметоксипиридин-2-карбоксамида
[Формула 174]

Указанное в заголовке соединение (31,0 мг) получали из соединения, полученного в примере получения 8 (30,0 мг), и 5-дифторметоксипиридин-2-карбоновой кислоты (21,5 мг) в соответствии со способом примера 14.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,69-1,72 (м, 1H), 2,56-2,71 (м, 2H), 2,94-3,05 (м, 2H), 3,70-3,91 (м, 4H), 6,65 (т, J=72 Гц, 1H), 7,03-7,09 (м, 1H), 7,40-7,41 (м, 1H), 7,64-7,66 (м, 1H), 7,89-7,93 (м, 1H), 8,28-8,30 (м, 1H), 8,43-8,43 (м, 1H), 9,77 (уш., 1H).
Примеры 82-86
Соединения примеров 82-86, показанные ниже в таблице 4, синтезировали в соответствии с примером 47.
[Таблица 4]





Примеры 87-88
Соединения примеров 87-88, показанные ниже в таблице 5, синтезировали в соответствии с примером 37.
[Таблица 5]


Пример 89
Синтез (+)-N-[3-((4aR*,6S*,7aS*)-2-амино-6-этокси-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]-5-цианопиридин-2-карбоксамида
[Формула 175]
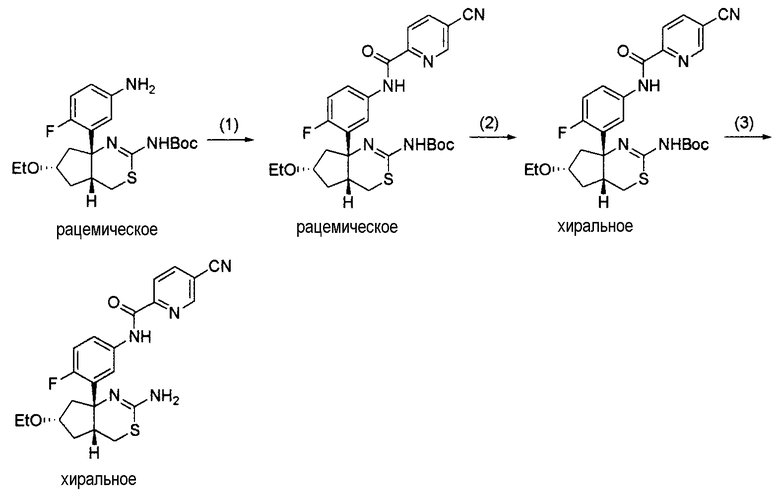
(1) Синтез трет-бутил ((4aR*,6S*,7aS*)-7a-{5-[(5-цианопиридин-2-карбонил)амино]-2-фторфенил}-6-этокси-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил)карбамата
PyBOP (205 мг) добавляли к раствору трет-бутил (±)-[(4aR*,6S*,7aS*)-7a-(5-амино-2-фторфенил)-6-этокси-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]карбамата (52 мг), соединения примера получения 26-(2) (27,3 мг) и N,N-диизопропилэтиламина (0,26 мл) в дихлорметане (5,2 мл). Смесь перемешивали при комнатной температуре в течение одного часа. Реакционный раствор вливали в насыщенный раствор бикарбоната натрия с последующим экстрагированием этилацетатом. Экстракт промывали насыщенным раствором хлорида натрия и сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении. Неочищенный продукт очищали хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (45 мг).
ESI-MS; m/z 540 [M++H].
(2) Синтез трет-бутил (-)-((4aR*,6S*,7aS*)-7a-{5-[(5-цианопиридин-2-карбонил)амино]-2-фторфенил}-6-этокси-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил)карбамата
Сложный трет-бутиловый эфир, полученный в (1) (45 мг), оптически разделяли, используя CHIRALPAK™ IB производства Daicel Chemical Industries, Ltd. (2 см × 25 см, подвижная фаза: гексан:этанол = 7:3, скорость потока: 10 мл/мин). Компоненты, имеющие время удерживания 21-28 минут, собирали с получением указанного в заголовке (-)-изомера (17 мг).
Оптическое вращение (-)
ESI-MS; m/z 540 [M++H].
(3) Синтез (+)-N-[3-((4aR*,6S*,7aS*)-2-амино-6-этокси-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]-5-цианопиридин-2-карбоксамида
Трифторуксусную кислоту (1,0 мл) добавляли к раствору трет-бутил (-)-карбамата, полученного в (2) (17 мг), в дихлорметане (1,0 мл), и реакционный раствор перемешивали при комнатной температуре в течение одного часа. Реакционный раствор вливали в насыщенный раствор бикарбоната натрия с последующим экстрагированием этилацетатом. Экстракт промывали насыщенным раствором хлорида натрия и сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении. Неочищенный продукт очищали хроматографией на колонке с NH-силикагелем с получением указанного в заголовке соединения (12 мг).
Оптическое вращение (+)
ESI-MS m/z 440 [M++H]
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,22 (т, J=6,8 Гц, 3H), 1,95-2,25 (м, 3H), 2,60-2,70 (м, 1H), 2,78 (дд, J=4,4, 13,2 Гц, 1H), 2,90-3,00 (м, 2H), 3,51 (кв, J=6,8 Гц, 2H), 4,20-4,35 (м, 1H), 7,07 (дд, J=8,8, 12,0 Гц, 1H), 7,38 (дд, J=2,8, 7,2 Гц, 1H), 7,90-8,00 (м, 1H), 8,20 (дд, J=2,0, 8,0 Гц, 1H), 8,42 (д, J=8,0 Гц, 1H), 8,86-8,92 (м, 1H), 9,82 (с, 1H).
Соединения примеров 90-103 синтезировали в соответствии с примером 19, используя соответствующие карбоновые кислоты и соответствующие анилиновые промежуточные соединения в примерах получения.
Пример 90
Синтез (+)-N-[3-((4aR*,6R*,7aS*)-2-амино-6-этокси-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]-5-цианопиридин-2-карбоксамида
[Формула 176]
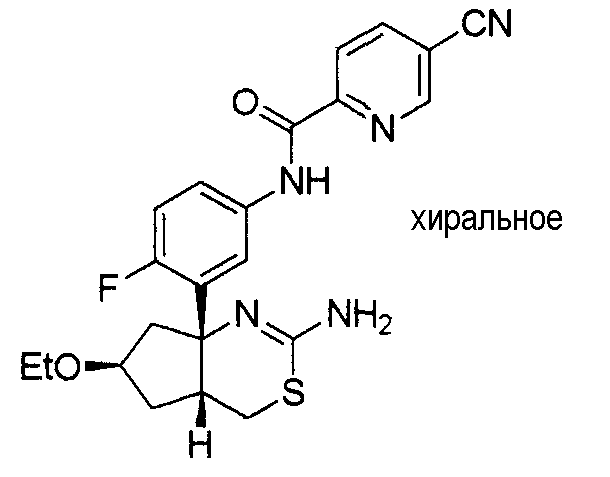
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,23 (т, J=7,2 Гц, 3H), 1,85-1,95 (м, 1H), 2,25-2,35 (м, 2H), 2,63 (дд, J=7,2, 12,8 Гц, 1H), 2,75 (дд, J=3,6, 12,8 Гц, 1H), 2,99 (дд, J=3,6, 12,8 Гц, 1H), 3,10-3,23 (м, 1H), 3,40-3,55 (м, 2H), 3,95-4,05 (м, 1H), 7,08 (дд, J=8,8, 12,0 Гц, 1H), 7,39 (дд, J=2,8, 7,2 Гц, 1H), 7,88-7,98 (м, 1H), 8,19 (дд, J=2,0, 8,0 Гц, 1H), 8,42 (д, J=8,0 Гц, 1H), 8,84-8,94 (м, 1H), 9,82 (с, 1H).
ESI-MS m/z 440 [M++H].
Пример 91
Синтез (+)-N-[3-((4aR*,6R*,7aS*)-2-амино-6-фтор-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]-5-цианопиридин-2-карбоксамида
[Формула 177]

1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,95-2,60 (м, 3H), 2,75-3,10 (м, 3H), 3,15-3,28 (м, 1H), 5,15-5,38 (м, 1H), 7,11 (дд, J=8,8, 12,0 Гц, 1H), 7,42 (дд, J=2,8, 7,2 Гц, 1H), 7,86-7,96 (м, 1H), 8,20 (дд, J=2,0, 8,0 Гц, 1H), 8,42 (д, J=8,0 Гц, 1H), 8,86-8,92 (м, 1H), 9,83 (с, 1H).
ESI-MS m/z 414 [M++H].
Пример 92
Синтез (+)-N-[3-((4aR*,6S*,7aS*)-2-амино-6-метокси-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]-5-дифторметилпиразин-2-карбоксамида
[Формула 178]

1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,00-2,14 (м, 2H), 2,18-2,28 (м, 1H), 2,64-2,74 (м, 1H), 2,78 (дд, J=4,0, 13,2 Гц, 1H), 2,90-3,00 (м, 2H), 3,34 (с, 3H), 4,08-4,24 (м, 1H), 6,79 (т, J=54,4 Гц, 1H), 7,00-7,13 (м, 1H), 7,39 (дд, J=2,8, 7,2 Гц, 1H), 7,90-8,00 (м, 1H), 8,91 (с, 1H), 9,51 (с, 1H), 9,61 (с, 1H).
ESI-MS m/z 452 [M++H].
Пример 93
Синтез (+)-N-[3-((4aR*,6R*,7aS*)-2-амино-6-метокси-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]-5-дифторметилпиразин-2-карбоксамида
[Формула 179]

1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,85-1,94 (м, 1H), 2,24-2,38 (м, 2H), 2,63 (дд, J=6,8, 12,8 Гц, 1H), 2,76 (дд, J=3,6, 12,8 Гц, 1H), 2,99 (дд, J=3,6, 12,8 Гц, 1H), 3,12-3,22 (м, 1H), 3,34 (с, 3H), 3,90-4,00 (м, 1H), 6,79 (т, J=54,8 Гц, 1H), 7,08 (дд, J=8,8, 12,0 Гц, 1H), 7,43 (дд, J=2,8, 7,2 Гц, 1H), 7,85-7,95 (м, 1H), 8,90 (д, J=0,8 Гц, 1H), 9,51 (д, J=0,8 Гц, 1H), 9,62 (с, 1H).
ESI-MS m/z 452 [M++H].
Пример 94
Синтез (+)-N-[3-((4aR*,7aS*)-2-амино-6,6-дифтор-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]-5-цианопиридин-2-карбоксамида
[Формула 180]
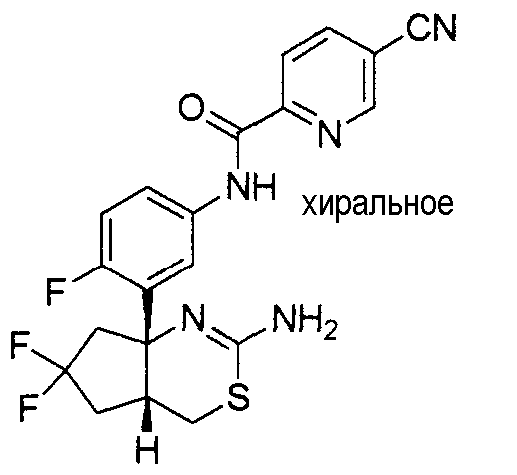
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,26-2,44 (м, 1H), 2,46-2,70 (м, 2H), 2,72-2,83 (м, 1H), 3,00 (д, J=13,2 Гц, 1H), 3,10-3,28 (м, 2H), 7,11 (дд, J=8,8, 11,6 Гц, 1H), 7,36-7,46 (м, 1H), 7,90-8,00 (м, 1H), 8,21 (дд, J=2,0, 8,0 Гц, 1H), 8,43 (д, J=8,0 Гц, 1H), 8,90 (д, J=0,8 Гц, 1H), 9,83 (с, 1H).
ESI-MS m/z 432 [M++H].
Пример 95
Синтез (+)-N-[3-((4aR*,6R*,7aS*)-2-амино-6-метокси-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]-5-дифторметилпиразин-2-карбоксамида
[Формула 181]

1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,80-1,95 (м, 1H), 2,25-2,40 (м, 2H), 2,62 (дд, J=6,8, 12,8 Гц, 1H), 2,76 (дд, J=3,6, 12,8 Гц, 1H), 2,99 (дд, J=3,6, 12,8 Гц, 1H), 3,10-3,20 (м, 1H), 3,34 (с, 3H), 3,85-4,00 (м, 1H), 7,07 (дд, J=8,8, 12,0 Гц, 1H), 7,38 (дд, J=2,4, 7,2 Гц, 1H), 7,51 (т, J=71,2 Гц, 1H), 7,84-7,94 (м, 1H), 8,32 (д, J=1,2 Гц, 1H), 9,06 (д, J=1,2 Гц, 1H), 9,45 (с, 1H).
ESI-MS m/z 468 [M++H].
Пример 96
Синтез (+)-N-[3-((4aR*,6R*,7aS*)-2-амино-6-метокси-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]-5-фторметоксипиразин-2-карбоксамида
[Формула 182]

1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,84-1,94 (м, 1H), 2,24-2,36 (м, 2H), 2,62 (дд, J=7,2, 12,8 Гц, 1H), 2,72-2,80 (м, 1H), 2,96-3,04 (м, 1H), 3,12-3,20 (м, 1H), 3,34 (с, 3H), 3,88-3,98 (м, 1H), 6,05-6,25 (м, 2H), 7,07 (дд, J=4,8, 12,0 Гц, 1H), 7,37 (дд, J=2,8, 7,2 Гц, 1H), 7,85-7,95 (м, 1H), 8,27 (д, J=1,2 Гц, 1H), 9,07 (д, J=1,2 Гц, 1H), 9,46 (с, 1H).
ESI-MS m/z 450 [M++H].
Пример 97
Синтез (+)-N-[3-((4aR*,6S*,7aS*)-2-амино-6-фтор-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]-5-дифторметилпиразин-2-карбоксамида
[Формула 183]
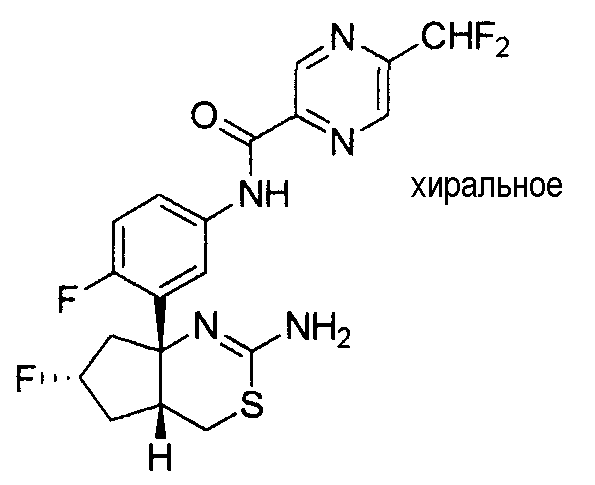
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,20-2,50 (м, 3H), 2,73-3,05 (м, 4H), 5,25-5,50 (м, 1H), 6,80 (т, J=54,8 Гц, 1H), 7,00-7,10 (м, 1H), 7,41 (дд, J=2,8, 7,2 Гц, 1H), 7,90-8,02 (м, 1H), 8,90 (с, 1H), 9,51 (с, 1H), 9,60 (с, 1H).
ESI-MS m/z 440 [M++H].
Пример 98
Синтез (+)-N-[3-((4aR*,6R*,7aS*)-2-амино-6-фтор-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]-5-дифторметоксипиразин-2-карбоксамида
[Формула 184]
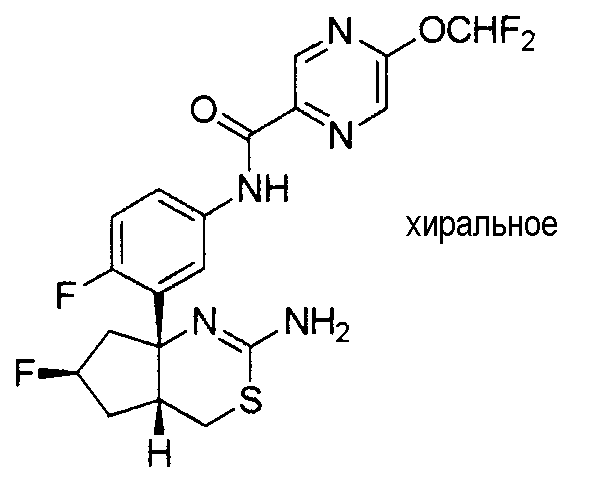
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,00-2,20 (м, 1H), 2,25-2,60 (м, 2H), 2,70-3,10 (м, 3H), 3,10-3,25 (м, 1H), 5,10-5,35 (м, 1H), 7,10 (дд, J=8,8, 12,0 Гц, 1H), 7,38 (дд, J=2,8, 7,2 Гц, 1H), 7,51 (т, J=71,6 Гц, 1H), 7,86-7,92 (м, 1H), 8,33 (д, J=1,2 Гц, 1H), 9,07 (д, J=1,2 Гц, 1H), 9,45 (с, 1H).
ESI-MS m/z 456 [M++H].
Пример 99
Синтез (+)-N-[3-((4aR*,6R*,7aS*)-2-амино-6-метокси-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]-5-хлорпиридин-2-карбоксамида
[Формула 185]
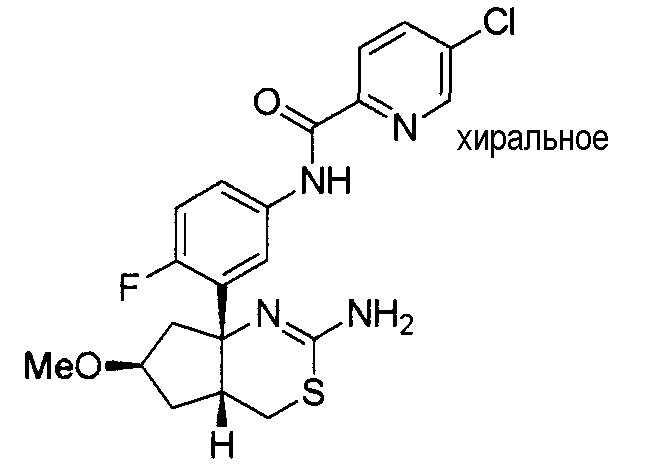
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,84-1,94 (м, 1H), 2,24-2,36 (м, 2H), 2,62 (дд, J=7,2, 12,8 Гц, 1H), 2,75 (дд, J=3,6, 12,8 Гц, 1H), 3,00 (дд, J=3,6, 12,8 Гц, 1H), 3,12-3,20 (м, 1H), 3,34 (с, 3H), 3,88-3,98 (м, 1H), 7,07 (дд, J=8,8, 12,0 Гц, 1H), 7,36 (дд, J=2,8, 7,2 Гц, 1H), 7,87 (дд, J=2,8, 8,4 Гц, 1H), 7,90-8,00 (м, 1H), 8,24 (д, J=8,4 Гц, 1H), 8,55 (д, J=2,4 Гц, 1H), 9,78 (с, 1H).
ESI-MS m/z 435 [M++H].
Пример 100
Синтез (+)-N-[3-((4aR*,6R*,7aS*)-2-амино-6-метокси-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]-5-дифторметоксипиридин-2-карбоксамида
[Формула 186]

1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,84-1,96 (м, 1H), 2,22-2,40 (м, 2H), 2,62 (дд, J=6,8, 12,8 Гц, 1H), 2,77 (дд, J=3,6, 12,8 Гц, 1H), 3,01 (дд, J=3,6, 12,8 Гц, 1H), 3,12-3,24 (м, 1H), 3,33 (с, 3H), 3,88-3,98 (м, 1H), 6,64 (т, J=72,0 Гц, 1H), 7,07 (дд, J=8,8, 12,4 Гц, 1H), 7,40 (дд, J=2,8, 7,2 Гц, 1H), 7,65 (дд, J=2,8, 8,8 Гц, 1H), 7,85-7,95 (м, 1H), 8,30 (д, J=8,8 Гц, 1H), 8,45 (д, J=2,0 Гц, 1H), 9,80 (с, 1H).
ESI-MS m/z 467 [M++H].
Пример 101
Синтез (±)-N-[3-((4aR*,6S*,7aS*)-2-амино-6-метокси-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]-5-фторпиридин-2-карбоксамида
[Формула 187]

1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,98-2,14 (м, 2H), 2,16-2,26 (м, 1H), 2,62-2,72 (м, 1H), 2,78 (дд, J=4,4, 12,8 Гц, 1H), 2,90-3,02 (м, 2H), 3,40 (с, 3H), 4,08-4,24 (м, 1H), 7,05 (дд, J=8,8, 12,0 Гц, 1H), 7,35 (дд, J=2,4, 7,2 Гц, 1H), 7,60 (дт, J=2,4, 8,8 Гц, 1H), 7,90-8,02 (м, 1H), 8,33 (дд, J=4,4, 8,8 Гц, 1H), 8,46 (д, J=2,4 Гц, 1H), 9,77 (с, 1H).
ESI-MS m/z 419 [M++H].
Пример 102
Синтез (±)-N-[3-((4aR*,6S*,7aS*)-2-амино-6-метокси-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]пиридин-2-карбоксамида
[Формула 188]

1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,96-2,12 (м, 2H), 2,16-2,26 (м, 1H), 2,62-2,74 (м, 1H), 2,78 (дд, J=4,8, 12,8 Гц, 1H), 2,90-3,02 (м, 2H), 3,34 (с, 3H), 4,08-4,24 (м, 1H), 7,05 (дд, J=8,8, 12,0 Гц, 1H), 7,38 (дд, J=2,8, 7,6 Гц, 1H), 7,45-7,55 (м, 1H), 7,85-7,95 (м, 1H), 7,95-8,05 (м, 1H), 8,25-8,35 (м, 1H), 8,60-8,70 (м, 1H), 9,99 (с, 1H).
ESI-MS m/z 401 [M++H].
Пример 103
Синтез (±)-N-[3-((4aR*,6S*,7aS*)-2-амино-6-метокси-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]пиримидин-4-карбоксамида
[Формула 189]

1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,00-2,14 (м, 2H), 2,16-2,28 (м, 1H), 2,64-2,74 (м, 1H), 2,78 (дд, J=4,4, 12,8 Гц, 1H), 2,90-3,00 (м, 2H), 3,34 (с, 3H), 4,10-4,24 (м, 1H), 7,07 (дд, J=8,8, 12,0 Гц, 1H), 7,40 (дд, J=2,8, 7,2 Гц, 1H), 7,90-8,00 (м, 1H), 8,21 (дд, J=1,6, 4,8 Гц, 1H), 9,04 (д, J=4,8 Гц, 1H), 9,31 (д, J=1,6 Гц, 1H), 9,85 (с, 1H).
ESI-MS m/z 402 [M++H].
Примеры 104-107
Соединения примеров 104-107, показанные ниже в таблице 6, синтезировали в соответствии с примером 19, используя соответствующие карбоновые кислоты и соответствующие анилиновые промежуточные соединения в примерах получения.
[Таблица 6]
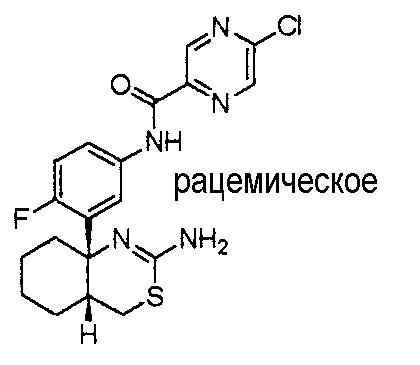



Примеры 108-110
Соединения примеров 108-110, показанные ниже в таблице 7, синтезировали в соответствии с примером 19 или 89, используя соответствующие карбоновые кислоты и соответствующие анилиновые соединения в примерах получения.
[Таблица 7]



Примеры 111-125
Соединения примеров 111-125, показанные ниже в таблице 8, синтезировали в соответствии с примером 1 или 19, используя соответствующие карбоновые кислоты или сульфонилхлориды и соединение примера получения 1-(8).
[Таблица 8]












Примеры 126-129
Соединения примеров 126-129, показанные ниже в таблице 9, синтезировали в соответствии с примером 1 или 19, используя соответствующие карбоновые кислоты и соединение примеров получения 3-(8).
[Таблица 9]




Пример 130
Синтез (±)-(4aR*,7aS*)-7a-{5-[(5-хлорпиридин-2-ил)амино]-2-фторфенил}-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-амина
[Формула 190]

(1) Синтез (±)-ди-трет-бутил [(4aR*,7aS*)-7a-{5-[(5-хлорпиридин-2-ил)амино]-2-фторфенил}-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]имидодикарбоната
2-Бром-5-хлорпиридин (10 мг), (R)-(+)-2,2'-бис(дифенилфосфино)-1,1'-бинафтил (4,86 мг), трис(дибензилиденацетон)дипалладий(0) (2,38 мг) и трет-бутоксинатрий (6,5 мг) добавляли к раствору (±)-ди-трет-бутил [(4aR*,7aS*)-7a-(5-амино-2-фторфенил)-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]имидодикарбоната (29,1 мг) в толуоле (10 мл). Смесь нагревали при перемешивании в атмосфере азота при 100°C в течение пяти часов. Реакционный раствор доводили до комнатной температуры и вливали в воду с последующим экстрагированием этилацетатом. Экстракт промывали насыщенным раствором соли и сушили над безводным сульфатом магния. Указанное в заголовке соединение (60 мг) получали удалением осушающего агента и концентрированием при пониженном давлении.
ESI-MS; m/z 577 [M+].
(2) (±)-(4aR*,7aS*)-7a-{5-[(5-Хлорпиридин-2-ил)амино]-2-фторфенил}-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-амин
ТФУК (0,5 мл) добавляли к раствору конденсата, полученного на предшествующей стадии (60 мг), в дихлорметане (3 мл), и смесь перемешивали при комнатной температуре в течение двух часов. Реакционный раствор вливали в насыщенный раствор бикарбоната натрия с последующим экстрагированием этилацетатом. Экстракт промывали насыщенным раствором соли и сушили над безводным сульфатом магния. Осушающий агент удаляли с последующим концентрированием при пониженном давлении. Остаток очищали колоночной хроматографией с получением указанного в заголовке соединения (2,4 мг).
ESI-MS; m/z 377 [M++H].
Пример 131
Соединение примера 131, показанное ниже в таблице 10, получали в соответствии со способом примера 130.
[Таблица 10]

Примеры 132-142
Соединения примеров 132-142, показанные ниже в таблице 11, синтезировали в соответствии с примером 14, используя соответствующие карбоновые кислоты и соединение, полученное в примере получения 3-(8).
[Таблица 11]




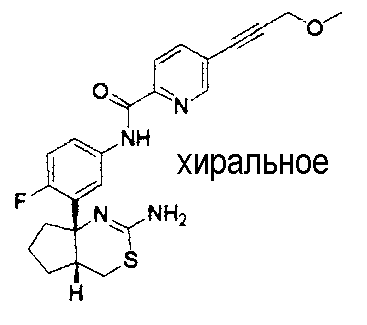

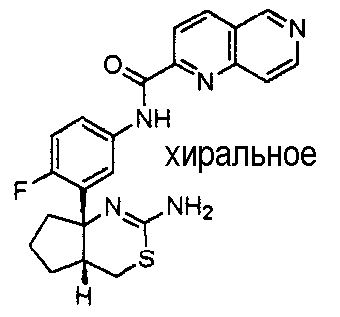
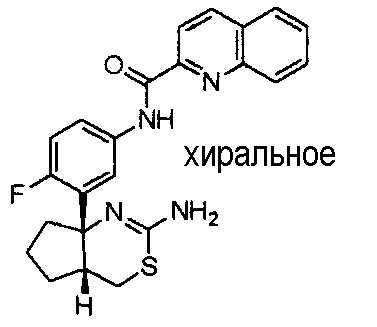



Соединения примеров 143-148, синтезировали в соответствии с примером 14, используя соответствующие карбоновые кислоты и соединение, полученное в примере получения 44-(16).
Пример 143
Синтез N-[3-((4aS*,5S*,8aS*)-2-амино-5-фторметил-4a,5,7,8-тетрагидро-4H-6-окса-3-тиа-1-азанафталин-8a-ил)-4-фторфенил]-5-цианопиридин-2-карбоксамида
[Формула 191]

1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,64-1,72 (м, 1H), 2,66-2,73 (м, 1H), 2,73-2,81 (м, 1H), 2,89-2,97 (м, 1H), 2,99-3,06 (м, 1H), 3,76-4,02 (м, 3H), 4,64 (дд, J=48,0, 3,8 Гц, 2H), 7,11 (дд, J=11,6, 8,8 Гц, 1H), 7,46 (дд, J=6,8, 2,8 Гц, 1H), 7,88-7,94 (м, 1H), 8,20 (дд, J=8,0, 2,5 Гц, 1H), 8,41-8,45 (м, 1H), 8,88-8,92 (м, 1H), 9,82 (уш.с, 1H).
ESI-MS m/z 444 [M++H].
Пример 144
Синтез N-[3-((4aS*,5S*,8aS*)-2-амино-5-фторметил-4a,5,7,8-тетрагидро-4H-6-окса-3-тиа-1-азанафталин-8a-ил)-4-фторфенил]-5-фторметоксипиразин-2-карбоксамида
[Формула 192]
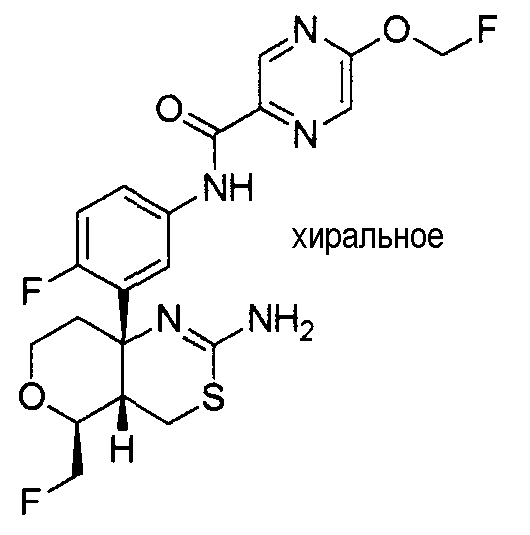
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,66-1,72 (м, 1H), 2,66-2,82 (м, 2H), 2,89-2,98 (м, 1H), 2,98-3,06 (м, 1H), 3,77-4,02 (м, 3H), 4,64 (уш.д, J=47,6 Гц, 2H), 6,16 (д, J=51,2 Гц, 2H), 7,05-7,16 (м, 1H), 7,37-7,46 (м, 1H), 7,84-7,93 (м, 1H), 8,29 (д, J=1,4 Гц, 1H), 9,08 (д, J=1,4 Гц, 1H), 9,47 (уш.с, 1H).
ESI-MS m/z 468 [M++H].
Пример 145
Синтез N-[3-((4aS*,5S*,8aS*)-2-амино-5-фторметил-4a,5,7,8-тетрагидро-4H-6-окса-3-тиа-1-азанафталин-8a-ил)-4-фторфенил]-5-дифторметоксипиразин-2-карбоксамида
[Формула 193]

1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,63-1,73 (м, 1H), 2,65-2,82 (м, 2H), 2,89-2,98 (м, 1H), 2,98-3,06 (м, 1H), 3,77-4,03 (м, 3H), 4,64 (дд, J=47,6, 3,5 Гц, 2H), 7,06-7,15 (м, 1H), 7,41-7,47 (м, 1H), 7,51 (т, J=71,4 Гц, 1H), 7,84-7,92 (м, 1H), 8,32-8,36 (м, 1H), 9,05-9,10 (м, 1H), 9,45 (уш.с, 1H).
ESI-MS m/z 486 [M++H].
Пример 146
Синтез N-[3-((4aS*,5S*,8aS*)-2-амино-5-фторметил-4a,5,7,8-тетрагидро-4H-6-окса-3-тиа-1-азанафталин-8a-ил)-4-фторфенил]-5-дифторметилпиразин-2-карбоксамида
[Формула 194]
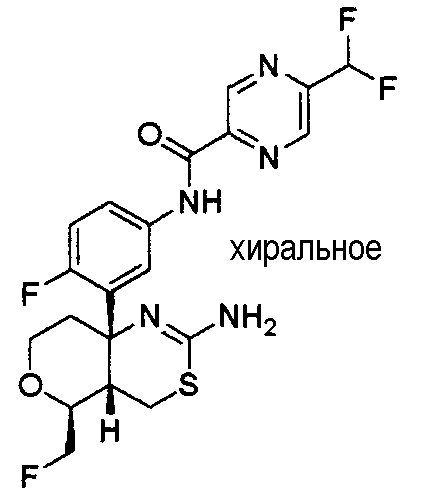
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,63-1,74 (м, 1H), 2,66-2,82 (м, 2H), 2,89-2,98 (м, 1H), 2,98-3,08 (м, 1H), 3,76-4,03 (м, 3H), 4,64 (дд, J=48,0, 2,8 Гц, 2H), 6,80 (т, J=54,4 Гц, 1H), 7,07-7,17 (м, 1H), 7,45-7,52 (м, 1H), 7,85-7,94 (м, 1H), 8,93 (с, 1H), 9,53 (с, 1H), 9,62 (уш.с, 1H).
ESI-MS m/z 470 [M++H].
Пример 147
Синтез N-[3-((4aS*,5S*,8aS*)-2-амино-5-фторметил-4a,5,7,8-тетрагидро-4H-6-окса-3-тиа-1-азанафталин-8a-ил)-4-фторфенил]-5-дифторметоксипиридин-2-карбоксамида
[Формула 195]

1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,63-1,72 (м, 1H), 2,67-2,81 (м, 2H), 2,88-2,98 (м, 1H), 2,98-3,07 (м, 1H), 3,76-4,02 (м, 3H), 4,64 (д, J=48,0 Гц, 2H), 6,65 (т, J=72,0 Гц, 1H), 7,03-7,15 (м, 1H), 7,38-7,47 (м, 1H), 7,62-7,71 (м, 1H), 7,84-7,95 (м, 1H), 8,27-8,36 (м, 1H), 8,42-8,50 (м, 1H), 9,80 (уш.с, 1H).
ESI-MS m/z 485 [M++H].
Пример 148
Синтез N-[3-((4aS*,5S*,8aS*)-2-амино-5-фторметил-4a,5,7,8-тетрагидро-4H-6-окса-3-тиа-1-азанафталин-8a-ил)-4-фторфенил]-5-хлорпиридин-2-карбоксамида
[Формула 196]

1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,63-1,72 (м, 1H), 2,67-2,80 (м, 2H), 2,90-2,98 (м, 1H), 2,98-3,06 (м, 1H), 3,77-4,01 (м, 3H), 4,64 (дд, J=47,6, 2,8 Гц, 2H), 7,06-7,14 (м, 1H), 7,41-7,47 (м, 1H), 7,86-7,93 (м, 2H), 8,22-8,28 (м, 1H), 8,55-8,60 (м, 1H), 9,80 (уш.с, 1H).
ESI-MS m/z 453 [M++H].
Примеры 149-151
Соединения примеров 149-151, показанные ниже в таблице 12, синтезировали в соответствии с примером 14, используя соответствующие карбоновые кислоты и соответствующие анилиновые промежуточные соединения в примерах получения.
[Таблица 12]



Соединения примеров 152-157 синтезировали в соответствии с примером 14, используя соответствующие карбоновые кислоты и соединение, полученное в примере получения 48-(13).
Пример 152
Синтез N-[3-((4aS*,5R*,8aS*)-2-амино-5-метил-4a,5,7,8-тетрагидро-4H-6-окса-3-тиа-1-азанафталин-8a-ил)-4-фторфенил]-5-хлорпиридин-2-карбоксамида
[Формула 197]

1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,34 (д, J=6,4 Гц, 3H), 1,72-1,82 (м, 1H), 2,64-2,82 (м, 3H), 2,92-3,00 (м, 1H), 3,75-3,86 (м, 2H), 3,92-3,97 (м, 1H), 7,08-7,16 (м, 1H), 7,40-7,46 (м, 1H), 7,87-7,96 (м, 2H), 8,22-8,26 (м, 1H), 8,57-8,60 (м, 1H), 9,84 (уш.с, 1H).
ESI-MS m/z 435 [M++H].
Пример 153
Синтез N-[3-((4aS*,5R*,8aS*)-2-амино-5-метил-4a,5,7,8-тетрагидро-4H-6-окса-3-тиа-1-азанафталин-8a-ил)-4-фторфенил]-5-фторметоксипиразин-2-карбоксамида
[Формула 198]
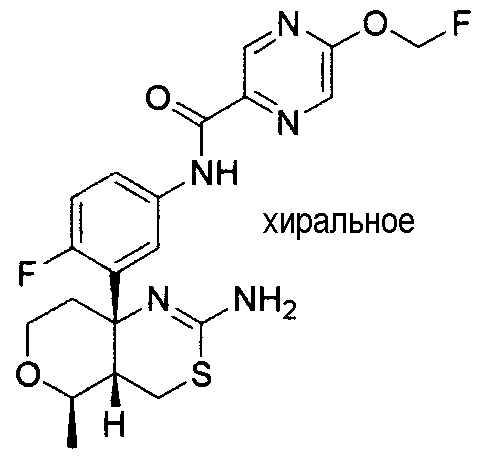
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,33 (д, J=6,0 Гц, 3H), 1,75-1,84 (м, 1H), 2,62-2,80 (м, 3H), 2,89-2,97 (м, 1H), 3,74-3,85 (м, 2H), 3,89-3,97 (м, 1H), 6,06-6,12 (м, 1H), 6,19-6,25 (м, 1H), 7,06-7,15 (м, 1H), 7,35-7,41 (м, 1H), 7,88-7,96 (м, 1H), 8,30 (д, J=1,2 Гц, 1H), 9,07 (д, J=1,2 Гц, 1H), 9,50 (уш.с, 1H).
ESI-MS m/z 450 [M++H].
Пример 154
Синтез N-[3-((4aS*,5R*,8aS*)-2-амино-5-метил-4a,5,7,8-тетрагидро-4H-6-окса-3-тиа-1-азанафталин-8a-ил)-4-фторфенил]-5-дифторметоксипиразин-2-карбоксамида
[Формула 199]
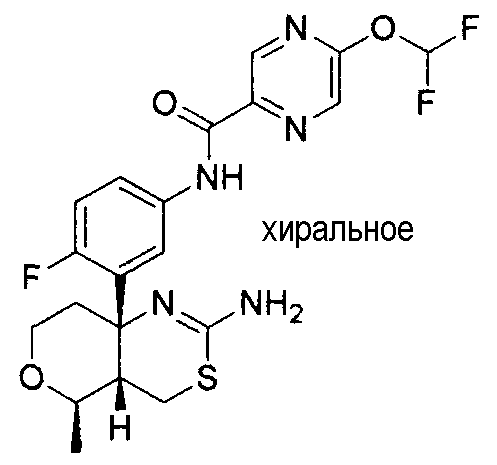
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,32 (д, J=6,0 Гц, 3H), 1,68-1,77 (м, 1H), 2,59-2,77 (м, 3H), 2,86-2,94 (м, 1H), 3,73-3,86 (м, 2H), 3,87-3,95 (м, 1H), 7,07-7,14 (м, 1H), 7,36-7,40 (м, 1H), 7,51 (т, J=71,4 Гц, 1H), 7,88-7,94 (м, 1H), 8,35 (д, J=1,2 Гц, 1H), 9,07 (д, J=1,2 Гц, 1H), 9,47 (уш.с, 1H).
ESI-MS m/z 468 [M++H].
Пример 155
Синтез N-[3-((4aS*,5R*,8aS*)-2-амино-5-метил-4a,5,7,8-тетрагидро-4H-6-окса-3-тиа-1-азанафталин-8a-ил)-4-фторфенил]-5-дифторметоксипиридин-2-карбоксамида
[Формула 200]

1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,33 (д, J=6,0 Гц, 3H), 1,74-1,83 (м, 1H), 2,62-2,79 (м, 3H), 2,89-2,97 (м, 1H), 3,74-3,86 (м, 2H), 3,88-3,96 (м, 1H), 6,65 (т, J=72,0 Гц, 1H), 7,05-7,14 (м, 1H), 7,37-7,42 (м, 1H), 7,64-7,69 (м, 1H), 7,90-7,96 (м, 1H), 8,30-8,34 (м, 1H), 8,46-8,50 (м, 1H), 9,82 (уш.с, 1H).
ESI-MS m/z 467 [M++H].
Пример 156
Синтез N-[3-((4aS*,5R*,8aS*)-2-амино-5-метил-4a,5,7,8-тетрагидро-4H-6-окса-3-тиа-1-азанафталин-8a-ил)-4-фторфенил]-5-дифторметилпиразин-2-карбоксамида
[Формула 201]

1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,33 (д, J=6,4 Гц, 3H), 1,72-1,81 (м, 1H), 2,62-2,79 (м, 3H), 2,88-2,95 (м, 1H), 3,74-3,86 (м, 2H), 3,89-3,96 (м, 1H), 6,79 (т, J=54,4 Гц, 1H), 7,08-7,16 (м, 1H), 7,41-7,46 (м, 1H), 7,88-7,96 (м, 1H), 8,92-8,95 (м, 1H), 9,51-9,54 (м, 1H), 9,64 (уш.с, 1H).
ESI-MS m/z 452 [M++H].
Пример 157
Синтез N-[3-((4aS*,5R*,8aS*)-2-амино-5-метил-4a,5,7,8-тетрагидро-4H-6-окса-3-тиа-1-азанафталин-8a-ил)-4-фторфенил]-5-цианопиридин-2-карбоксамида
[Формула 202]

1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,32 (д, J=6,4 Гц, 3H), 1,62-1,69 (м, 1H), 2,54-2,62 (м, 1H), 2,65-2,75 (м, 2H), 2,84-2,92 (м, 1H), 3,73-3,86 (м, 2H), 3,86-3,93 (м, 1H), 7,06-7,14 (м, 1H), 7,36-7,42 (м, 1H), 7,92-7,98 (м, 1H), 8,20 (дд, J=8,2, 1,8 Гц, 1H), 8,43 (дд, J=8,2, 1,0 Гц, 1H), 8,90 (дд, J=1,8, 1,0 Гц, 1H), 9,81 (уш.с, 1H).
ESI-MS m/z 426 [M++H].
Пример 158
Синтез (±)-N-[3-((4aR*,7aS*)-2-амино-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-5-фторфенил]-5-цианопиридин-2-карбоксамида
[Формула 203]

Указанное в заголовке соединение (52 мг) получали из 5-цианопиридин-2-карбоновой кислоты, полученной в примере получения 13 (49,3 мг), и соединения, полученного в примере получения 55-(11) (85 мг), в соответствии со способом примера 14.
ESI-MS; m/z 396 [M++H].
Пример 159
Синтез N-[3-((4aR*,8aS*)-2-амино-4a,5,7,8-тетрагидро-4H-6-окса-3-тиа-1-азанафталин-8a-ил)-4-фторфенил]-5-цианопиридин-2-карбоксамида
[Формула 204]

Указанное в заголовке соединение (15 мг) получали из 5-цианопиридин-2-карбоновой кислоты (12,9 мг) и соединения, полученного в примере получения 56-(16) (23 мг), в соответствии со способом примера 14.
ESI-MS; m/z 412 [M++H].
Пример 160
Синтез (±)-(4aR*,7aS*)-7a-[2-фтор-5-(2-фторпиридин-3-ил)фенил]-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-иламина
[Формула 205]

(1) Синтез бензил (±)-[(4aR*,7aS*)-7a-(5-бром-2-фторфенил)-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]карбамата
Бензилхлорформиат (1,16 мл) добавляли к раствору соединения, полученного в примере получения 54-(4) (2,24 г), в 1,4-диоксане и насыщенном растворе бикарбоната натрия (60 мл/60 мл). Реакционный раствор перемешивали при комнатной температуре в течение трех часов. К реакционному раствору добавляли этилацетат и насыщенный водный раствор хлорида натрия и отделяли органический слой. Органический слой снова промывали насыщенным водным раствором хлорида натрия. Органический слой сушили над безводным сульфатом магния. Органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на силикагеле с получением указанного в заголовке соединения (2,35 г).
ESI-MS; m/z 463 [M++H].
(2) Синтез бензил (±)-{(4aR*,7aS*)-7a-[2-фтор-5-(2-фторпиридин-3-ил)фенил]-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил}карбамата
1 M водный бикарбонат натрия (432 мкл), 2-фторпиридин-3-бороновую кислоту (67,1 мг) и тетракис(трифенилфосфин)палладий (0) (25,1 мг) добавляли к раствору соединения, полученного в примере 160-(1) (200 мг), в смеси толуол (4 мл)/этанол (2 мл), и смесь перемешивали при 85°C в течение 16 часов. Реакционный раствор доводили до комнатной температуры. Добавляли к реакционному раствору насыщенный водный раствор хлорида натрия и этилацетат. Органический слой отделяли и сушили над безводным сульфатом магния. Органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением грубо очищенного продукта, содержащего указанное в заголовке соединение (156 мг).
ESI-MS; m/z 480 [M++H].
(3) Синтез (±)-(4aR*,7aS*)-7a-[2-фтор-5-(2-фторпиридин-3-ил)фенил]-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-иламина
Йодтриметилсилан (116 мкл) добавляли к раствору соединения, полученного в примере 160-(2) (78 мг), в хлороформе (4 мл), и смесь перемешивали при комнатной температуре в течение ночи. Снова добавляли к реакционному раствору йодтриметилсилан (116 мкл) и смесь кипятили с обратным холодильником в течение 20 часов. Реакционный раствор доводили до комнатной температуры. Реакционный раствор подщелачивали 5 н. гидроксидом натрия и добавляли хлороформ. Органический слой отделяли и промывали насыщенным водным раствором хлорида натрия. Органический слой сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с NH-силикагелем с получением указанного в заголовке соединения (11 мг).
ESI-MS; m/z 346 [M++H].
Пример 161
Синтез (±)-(4aR*,7aS*)-7a-(4-фтор-3'-метоксибифенил-3-ил)-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-иламина
[Формула 206]

Синтез (±)-(4aR*,7aS*)-7a-(4-фтор-3'-метоксибифенил-3-ил)-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-иламина
Указанное в заголовке соединение (1,3 мг) получали из соединения, полученного в примере 160-(1) (100 мг), в соответствии с примером 160, используя соответствующую бороновую кислоту.
ESI-MS; m/z 357 [M++H].
Пример 162
Синтез (+)-(4aR*,7aS*)-7a-(3',5'-дихлор-4-фторбифенил-3-ил)-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-иламина
[Формула 207]

(1) Синтез (±)-(4aR*,7aS*)-7a-(3',5'-дихлор-4-фторбифенил-3-ил)-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-иламина
1 M водный бикарбонат натрия (486 мкл), 3,5-дихлорфенилбороновую кислоту (80,4 мг) и тетракис(трифенилфосфин)палладий(0) (18,8 мг) добавляли к раствору соединения, полученного в примере 160-(1) (120 мг), в ДМФА (3 мл), и смесь перемешивали при 110°C в течение восьми часов. Реакционный раствор доводили до комнатной температуры. К реакционному раствору добавляли насыщенный водный раствор хлорида натрия и этилацетат. Органический слой отделяли и сушили над безводным сульфатом магния. Органический слой концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем с получением грубо очищенного продукта, содержащего указанное в заголовке соединение (69 мг).
ESI-MS; m/z 395 [M++H].
(2) Синтез (+)-(4aR*,7aS*)-7a-(3',5'-дихлор-4-фторбифенил-3-ил)-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-иламина
Соединение, полученное в примере 162-(1) (69 г), промывали смешанным растворителем из этилацетата (0,5 мл) и гептана (3 мл). Полученное твердое вещество разбавляли 8 мл этанола и соединение оптически разделяли, используя CHIRALPAK™ IA производства Daicel Chemical Industries, Ltd. (2 см × 25 см, подвижная фаза: гексан:этанол = 7:3, скорость потока: 5 мл/мин). Компонент, имеющий время удерживания от 24 до 28 минут, собирали с получением указанного в заголовке соединения (11 мг).
ESI-MS; m/z 395 [M++H].
Пример 163
Синтез (+)-(4aR*,7aS*)-7a-[2-фтор-5-(5-метоксипиридин-3-ил)фенил]-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-иламина
[Формула 208]

(1) Синтез (±)-(4aR*,7aS*)-7a-[2-фтор-5-(5-метоксипиридин-3-ил)фенил]-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-иламина
Грубо очищенный продукт, содержащий указанное в заголовке соединение (150 мг), получали из соединения, полученного в примере 160-(1) (200 мг), в соответствии с примером 162, используя соответствующую бороновую кислоту.
ESI-MS; m/z 358 [M++H].
(2) Синтез (+)-(4aR*,7aS*)-7a-[2-фтор-5-(5-метоксипиридин-3-ил)фенил]-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-иламина
Соединение, полученное в примере 163-(1) (150 мг), снова очищали хроматографией на силикагеле и полученное твердое вещество разбавляли 12 мл этанола. Соединение оптически разделяли, используя CHIRALPAK™ IA производства Daicel Chemical Industries, Ltd. (2 см × 25 см, подвижная фаза: гексан:этанол = 1:1, скорость потока: 5 мл/мин). Компонент, имеющий время удерживания от 27 до 30 минут, собирали с получением указанного в заголовке соединения (18 мг).
ESI-MS; m/z 358 [M++H].
Соединения примеров 164-167 синтезировали в соответствии с примером 14, используя соответствующие карбоновые кислоты и соответствующие анилиновые промежуточные соединения в примерах получения.
Пример 164
Синтез N-[3-((4aS,5S,7aS)-2-амино-5-фторметил-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил]-5-цианопиридин-2-карбоксамида
[Формула 209]

1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,80 (дд, J=4,0, 13,2 Гц, 1H), 3,05-3,10 (м, 1H), 3,15 (дд, J=3,6, 13,2 Гц, 1H), 3,87 (дд, J=2,8, 8,8 Гц, 1H), 4,51-4,66 (м, 4H), 7,11 (дд, J=8,8, 12,0H, 1H), 7,58 (дд, J=2,8, 7,2 Гц, 1H), 7,93-7,97 (м, 1H), 8,21 (дд, J=2,4, 8,4 Гц, 1H), 8,42 (дд, J=0,8, 8,0 Гц, 1H), 8,90 (дд, J=0,8, 2,0 Гц, 1H), 9,86 (с, 1H).
ESI-MS m/z 430 [M++H].
Пример 165
Синтез N-[3-((4aS*,7aS*)-2-амино-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил]-5-трифторметилпиридин-2-карбоксамида
[Формула 210]

1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,84 (дд, J=5,6, 14,0 Гц, 1H), 3,08-3,13 (м, 2H), 3,83 (дд, J=2,0, 8,4 Гц, 1H), 4,08-4,15 (м, 2H), 4,47 (дд, J=1,2, 8,4 Гц, 1H), 7,10 (дд, J=8,8, 12,0 Гц, 1H), 7,65 (дд, J=2,8, 6,8 Гц, 1H), 7,93-7,97 (м, 1H), 8,17 (дд, J=2,4, 8,0 Гц, 1H), 8,43 (д, J=8,0 Гц, 1H), 8,90 (с, 1H), 9,94 (с, 1H).
ESI-MS m/z 441 [M++H].
Пример 166
Синтез N-[3-((4aS*,7aS*)-2-амино-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил]-5-бромпиридин-2-карбоксамида
[Формула 211]

1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,84 (дд, J=6,0, 14,0 Гц, 1H), 3,08-3,13 (м, 2H), 3,84 (дд, J=2,0, 8,8 Гц, 1H), 4,08-4,15 (м, 2H), 4,46 (дд, J=1,2, 8,4 Гц, 1H), 7,09 (дд, J=8,8, 11,6 Гц, 1H), 7,59 (дд, J=2,8, 6,8 Гц, 1H), 7,93-7,97 (м, 1H), 8,04 (дд, J=2,4, 8,0 Гц, 1H), 8,18 (д, J=8,4 Гц, 1H), 8,67 (д, J=1,6 Гц, 1H), 9,83 (с, 1H).
ESI-MS m/z 451 [M++H].
Пример 167
Синтез N-[3-((4aS*,7aS*)-2-амино-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил]-5-дифторметоксипиридин-2-карбоксамида
[Формула 212]

1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,84 (дд, J=6,4, 14,0 Гц, 1H), 3,07-3,12 (м, 2H), 3,83 (дд, J=2,0, 8,8 Гц, 1H), 4,08-4,18 (м, 2H), 4,46 (дд, J=1,2, 8,4 Гц, 1H), 6,65 (т, J=72,0 Гц, 1H), 7,09 (дд, J=8,8, 12,0 Гц, 1H), 7,59 (дд, J=2,8, 7,2 Гц, 1H), 7,67 (дд, J=2,8, 8,8 Гц, 1H), 7,94-7,98 (м, 1H), 8,32 (д, J=8,8 Гц, 1H), 8,47 (д, J=2,4 Гц, 1H), 9,83 (с, 1H).
ESI-MS m/z 439 [M++H].
Примеры 168-191
Соединения примеров 168-191, показанные ниже в таблице 13, синтезировали в соответствии с примером 14, используя соответствующие карбоновые кислоты и соответствующие анилиновые промежуточные соединения в примерах получения.
[Таблица 13]

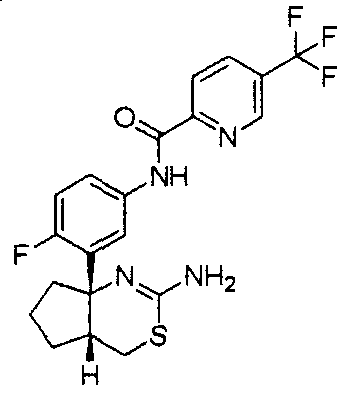
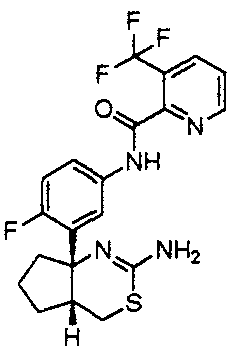
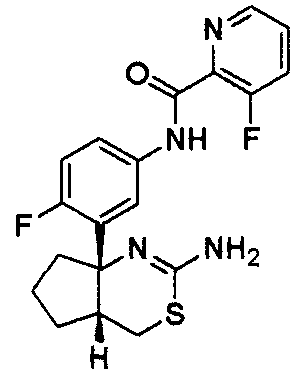
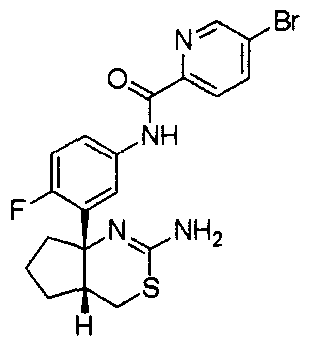

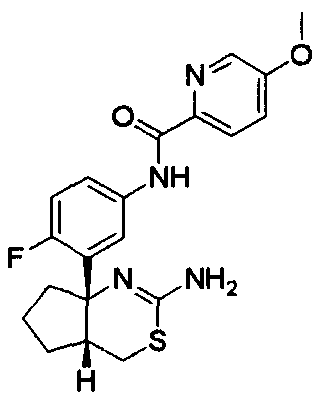




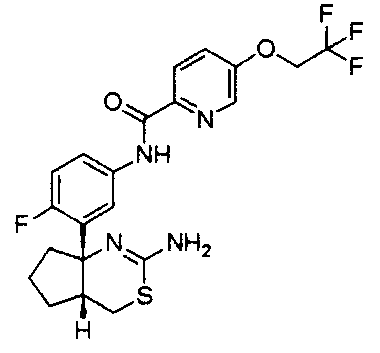









Примеры 192-200
Соединения примеров 192-200, показанные ниже в таблице 14, синтезировали в соответствии с примером 14, используя соответствующие карбоновые кислоты и соответствующие анилиновые промежуточные соединения в примерах получения.
[Таблица 14]

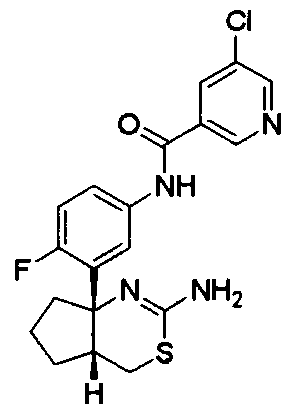






Пример 201
Синтез N-[3-((4aR*,7S*,8aS*)-2-амино-7-метокси-4,4a,5,6,7,8-гексагидробензо[d][1,3]тиазин-8a-ил)-4-фторфенил]-5-цианопиридин-2-карбоксамида
[Формула 213]
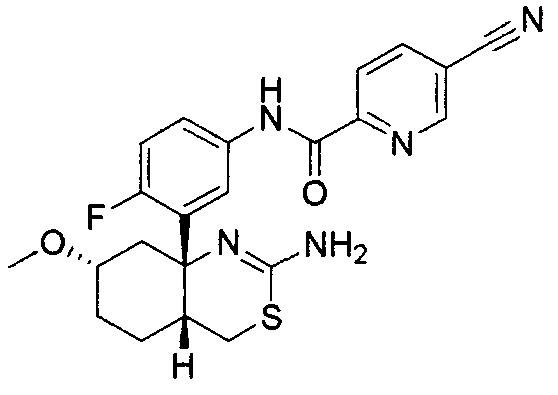
трет-Бутил (-)-[(4aR*,7S*,8aS*)-8a-(5-амино-2-фторфенил)-7-метокси-4a,5,6,7,8,8a-гидро-4H-бензо[d][1,3]тиазин-2-ил]карбамат, полученный в примере получения 57-(11) (41 мг), смешивали с 5-фторметоксипиразин-2-карбоновой кислотой (18 мг), N,N-диизопропилэтиламином (87 мкл, удельный вес: 0,742 г/см3) и PyBOP (104 мг) в дихлорметане (2 мл), и смесь перемешивали в атмосфере азота при комнатной температуре. После перемешивания в течение пяти часов реакционный раствор сразу же очищали хроматографией на колонке с силикагелем. К полученному амиду добавляли хлороформ (0,5 мл) и ТФУК (0,5 мл) и смесь перемешивали при комнатной температуре в течение двух часов и 30 минут. Реакционный раствор медленно вливали в насыщенный раствор бикарбоната натрия с последующим экстрагированием три раза хлороформом. Полученные органические слои сушили над безводным сульфатом магния и твердое вещество удаляли фильтрованием. Фильтрат концентрировали при пониженном давлении и затем очищали хроматографией на колонке с NH-силикагелем с получением указанного в заголовке соединения (34 мг).
ESI-MS; m/z 440 [M++H].
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,40-1,43 (м, 1H), 1,69-1,75 (м, 1H), 2,08-2,28 (м, 3H), 2,32-2,35 (м, 1H), 2,61-2,64 (м, 1H), 2,76-2,79 (м, 1H), 2,88-2,91 (м, 1H), 3,36 (с, 3H), 3,63 (уш., 1H), 7,05-7,10 (м, 1H), 7,37-7,38 (м, 1H), 8,04-8,07 (м, 1H), 8,19-8,20 (м, 1H), 8,41-8,43 (м, 1H), 8,90 (с, 1H), 9,84 (с, 1H).
Пример 202
Синтез N-[3-((4aR*,7R*,8aS*)-2-амино-7-метокси-4,4a,5,6,7,8-гексагидробензо[d][1,3]тиазин-8a-ил)-4-фторфенил]-5-цианопиридин-2-карбоксамида
[Формула 214]

Указанное в заголовке соединение (39 мг) получали из трет-бутил (-)-[(4aR*,7R*,8aS*)-8a-(5-амино-2-фторфенил)-7-метокси-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата, полученного в примере получения 58-(13) (50 мг), в соответствии со способом примера 201.
ESI-MS; m/z 440 [M++H].
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,42-1,53 (м, 1H), 1,61-1,65 (м, 1H), 1,77-1,89 (м, 1H), 2,09-2,24 (м, 3H), 2,61-2,64 (м, 1H), 2,71-2,74 (м, 1H), 2,92 (дд, J=4,0, 12,0 Гц, 1H), 3,36 (с, 3H), 3,39-3,45 (м, 1H), 7,08 (дд, J=8,8, 12,0 Гц, 1H), 7,32-7,34 (м, 1H), 7,92-7,95 (м, 1H), 8,18-8,21 (м, 1H), 8,42 (дд, J=0,8, 8,4 Гц, 1H), 8,88-8,89 (м, 1H), 9,80 (с, 1H).
Пример 203
Соединение примера 203, показанное ниже в таблице 15, синтезировали в соответствии с примером 202, используя соответствующую карбоновую кислоту.
[Таблица 15]

Пример 204
Синтез N-[3-((4aR*,6S*,8aS*)-2-амино-6-метокси-4,4a,5,6,7,8-гексагидробензо[d][1,3]тиазин-8a-ил)-4-фторфенил]-5-цианопиридин-2-карбоксамида
[Формула 215]
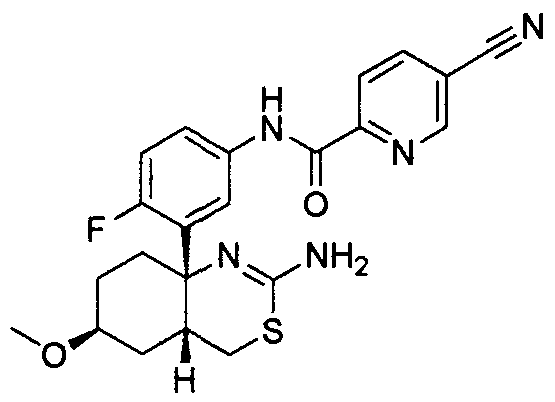
Указанное в заголовке соединение (37 мг) получали из трет-бутил (-)-[(4aR*,6S*,8aS*)-8a-(5-амино-2-фторфенил)-6-метокси-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата, полученного в примере получения 60-(4) (50 мг), в соответствии со способом примера 201.
ESI-MS; m/z 440 [M++H].
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,52-1,57 (м, 1H), 1,66-1,76 (м, 1H), 1,82-1,92 (м, 3H), 2,52-2,62 (м, 2H), 2,95-2,99 (м, 1H), 3,10-3,15 (м, 1H), 3,39 (с, 3H), 3,60-3,62 (м, 1H), 7,07 (дд, J=8,8, 11,6 Гц, 1H), 7,34 (дд, J=2,8, 7,2 Гц, 1H), 7,93 (ддд, J=2,8, 4,0, 8,8 Гц, 1H), 8,18-8,21 (м, 1H), 8,42 (дд, J=0,8, 4,4 Гц, 1H), 8,89 (дд, J=0,8, 2,0 Гц, 1H), 9,79 (с, 1H).
Соединение примера 205, показанное ниже в таблице 16, синтезировали в соответствии с примером 204, используя соответствующую карбоновую кислоту.
[Таблица 16]
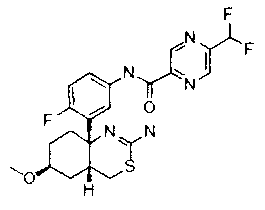
Пример 206
Синтез N-[3-((4aR*,6R*,8aS*)-2-амино-6-метокси-4,4a,5,6,7,8-гексагидробензо[d][1,3]тиазин-8a-ил)-4-фторфенил]-5-цианопиридин-2-карбоксамида
[Формула 216]
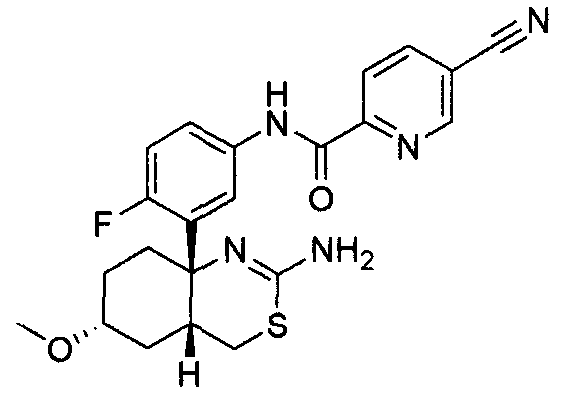
Указанное в заголовке соединение (33 мг) получали из трет-бутил (-)-[(4aR*,6R*,8aS*)-8a-(5-амино-2-фторфенил)-6-метокси-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата, полученного в примере получения 61-(4) (50 мг), в соответствии со способом примера 201.
ESI-MS; m/z 440 [M++H].
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,60-1,74 (м, 2H), 1,82-1,91 (м, 2H), 1,97-2,00 (м, 1H), 2,23-2,30 (м, 1H), 2,58-2,61 (м, 1H), 2,77-2,80 (м, 1H), 2,92-2,95 (м, 1H), 3,41 (с, 3H), 3,41-3,49 (м, 1H), 7,05-7,10 (м, 1H), 7,30-7,32 (м, 1H), 7,97-8,00 (м, 1H), 8,19 (д, J=8,4 Гц, 1H), 8,42 (д, J=8,0 Гц, 1H), 8,90 (с, 1H), 9,80 (с, 1H).
Примеры 207-211
Соединения примеров 207-211, показанные ниже в таблице 17, синтезировали в соответствии с примером 206, используя соответствующие карбоновые кислоты.
[Таблица 17]




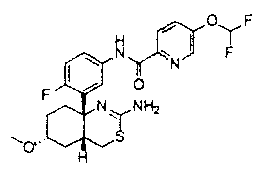
Пример 212
Синтез N-[3-((4aR*,6R*,8aS*)-2-амино-6-фтор-4,4a,5,6,7,8-гексагидробензо[d][1,3]тиазин-8a-ил)-4-фторфенил]-5-цианопиридин-2-карбоксамида
[Формула 217]
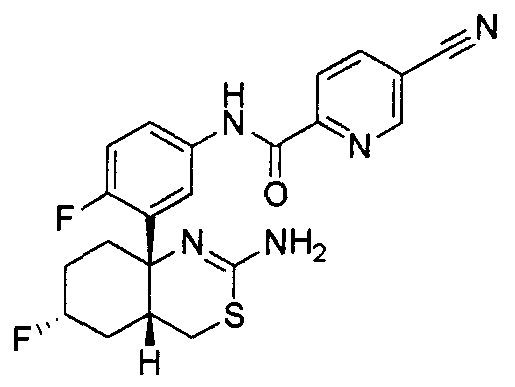
Указанное в заголовке соединение (37 мг) получали из трет-бутил (-)-[(4aR*,6R*,8aS*)-8a-(5-амино-2-фторфенил)-6-фтор-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата, полученного в примере получения 63-(8) (52 мг), в соответствии со способом примера 201.
ESI-MS; m/z 428 [M++H].
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,79-1,91 (м, 2H), 1,92-2,07 (м, 3H), 2,24-2,32 (м, 1H), 2,61 (дд, J=2,8, 12,0 Гц, 1H), 2,78-2,82 (м, 1H), 2,94 (ддд, J=3,2, 4,0, 12,0 Гц, 1H), 4,58 (уш., 2H), 4,68-4,88 (м, 1H), 7,07 (дд, J=8,8, 12,0 Гц, 1H), 7,34 (дд, J=2,8, 7,2 Гц, 1H), 7,97 (ддд, J=2,8, 4,4, 8,8 Гц, 1H), 8,20 (дд, J=2,4, 8,4 Гц, 1H), 8,42 (дд, J=1,2, 4,4 Гц, 1H), 8,89 (дд, J=1,2, 2,4 Гц, 1H), 9,80 (уш., 1H).
Примеры 213-214
Соединения примеров 213-214, показанные ниже в таблице 18, синтезировали в соответствии с примером 210, используя соответствующие карбоновые кислоты.
[Таблица 18]

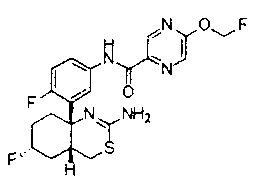
Пример 215
Синтез N-[3-((4aR*,6S*,8aS*)-2-амино-6-фтор-4,4a,5,6,7,8-гексагидробензо[d][1,3]тиазин-8a-ил)-4-фторфенил]-5-цианопиридин-2-карбоксамида
[Формула 218]

Указанное в заголовке соединение (22 мг) получали из трет-бутил (-)-[(4aR*,6S*,8aS*)-8a-(5-амино-2-фторфенил)-6-фтор-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата, полученного в примере получения 64-(8) (36 мг), в соответствии со способом примера 201.
ESI-MS; m/z 428 [M++H].
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,51-1,66 (м, 1H), 1,72-1,83 (м, 1H), 1,87-2,03 (м, 3H), 2,57 (дд, J=2,8, 12,4 Гц, 1H), 2,62-2,70 (м, 1H), 3,01 (дд, J=4,0, 12,0 Гц, 1H), 3,16-3,34 (м, 1H), 4,99 (д, J=48,8 Гц, 1H) 7,10 (дд, J=8,8, 11,6 Гц, 1H), 7,36-7,38 (м, 1H), 7,91-7,95 (м, 1H), 8,19-8,21 (м, 1H), 8,42-8,44 (м, 1H), 8,89-8,90 (м, 1H), 9,80 (уш., 1H).
Примеры 216-219
Соединения примеров 216-219, показанные ниже в таблице 19, синтезировали в соответствии с примером 202, используя соответствующие карбоновые кислоты.
[Таблица 19]

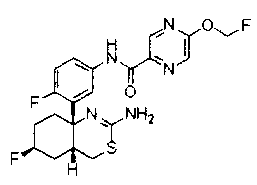

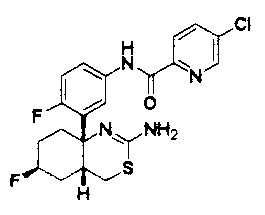
Пример 220
Синтез (±)-N-[3-((4aR*,8aS*)-2-амино-4,4a,5,6,7,8-гексагидробензо[d][1,3]тиазин-8a-ил)-4-фторфенил]бензамида
[Формула 219]

трет-Бутил (±)-[(4aR*,8aS*)-8a-(5-амино-2-фторфенил)-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамат, полученный в примере получения 1-(8) (100 мг), растворяли в ТГФ (5 мл). Затем добавляли пиридин (107 мкл, удельный вес 0,978 г/см3) и смесь охлаждали на ледяной бане в атмосфере азота. После достаточного охлаждения добавляли бензилхлорид (46 мкл, удельный вес 1,211 г/см3) с последующим перемешиванием в течение одного часа и 30 минут. После разбавления этилацетатом добавляли насыщенный раствор хлорида аммония с последующим экстрагированием этилацетатом. Полученный органический слой последовательно промывали насыщенным раствором хлорида аммония, водой и насыщенным раствором соли. Органический слой сушили над безводным сульфатом магния и твердое вещество удаляли фильтрованием. Фильтрат концентрировали при пониженном давлении и очищали пТСХ с получением амида (93 мг). Амид растворяли в смешанном растворителе из этилацетата (1 мл) и хлороформа (2 мл). Затем добавляли раствор хлороводорода в этилацетате (4 н., 1 мл) и смесь перемешивали при комнатной температуре. Через пять часов еще добавляли ТФУК (2 мл) с последующим перемешиванием. Через 17 часов концентрировали при пониженном давлении растворитель. К полученному остатку добавляли ТФУК (3 мл) с последующим перемешиванием. Через 23 часа и 30 минут реакционный раствор концентрировали при пониженном давлении. К полученному остатку добавляли этилацетат и насыщенный раствор бикарбоната натрия с последующим экстрагированием три раза этилацетатом. Полученные органические слои промывали насыщенным раствором соли и сушили над безводным сульфатом магния. Твердое вещество удаляли фильтрованием. После концентрирования при пониженном давлении остаток очищали NH-пТСХ с получением указанного в заголовке соединения (40 мг).
ESI-MS; m/z 384 [M++H].
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,47-1,81 (м, 7H), 2,20-2,27 (м, 1H), 2,55 (дд, J=2,8, 12,4 Гц, 1Н), 2,70-2,75 (м, 1H), 2,94 (дд, J=4,0, 12,0 Гц, 1H), 7,30-7,09 (м, 2H), 7,47-7,52 (м, 2H), 7,53-7,58 (м, 1H), 7,84-7,86 (м, 3H), 7,93-7,97 (м, 1H).
Пример 221
Синтез (±)-N-[3-((4aR*,8aS*)-2-амино-4,4a,5,6,7,8-гексагидробензо[d][1,3]тиазин-8a-ил)-4-фторфенил]фуран-2-карбоксамида
[Формула 220]
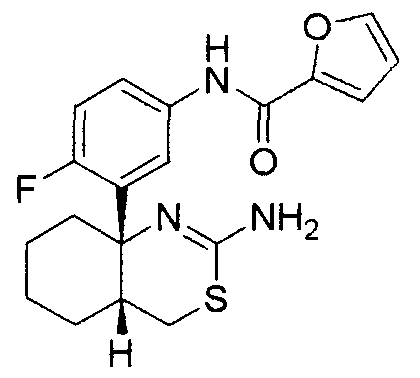
трет-Бутил (±)-[(4aR*,8aS*)-8a-(5-амино-2-фторфенил)-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамат, полученный в примере получения 1-(8) (100 мг), растворяли в ТГФ (5 мл). Затем добавляли пиридин (107 мкл, удельный вес 0,978 г/см3) и смесь охлаждали на ледяной бане в атмосфере азота. После достаточного охлаждения добавляли фуран-2-карбонилхлорид (39 мкл, удельный вес 1,324 г/см3) с последующим перемешиванием в течение одного часа и 30 минут. После разбавления этилацетатом добавляли насыщенный раствор хлорида аммония с последующим экстрагированием этилацетатом. Полученный органический слой последовательно промывали насыщенным раствором хлорида аммония, водой и насыщенным раствором соли. Органический слой сушили над безводным сульфатом магния и твердое вещество удаляли фильтрованием. Фильтрат концентрировали при пониженном давлении и очищали пТСХ с получением амида (59 мг). Амид растворяли в смешанном растворителе из этилацетата (2 мл) и хлороформа (2 мл). Затем добавляли раствор хлороводорода в этилацетате (4 н., 2 мл) и смесь перемешивали при комнатной температуре. Через пять часов еще добавляли ТФУК (2 мл) с последующим перемешиванием. Через 16 часов и 30 минут концентрировали при пониженном давлении растворитель. К полученному остатку добавляли этилацетат и насыщенный раствор бикарбоната натрия с последующим экстрагированием три раза этилацетатом. Полученные органические слои промывали насыщенным раствором соли и сушили над безводным сульфатом магния. Твердое вещество удаляли фильтрованием. После концентрирования при пониженном давлении остаток очищали NH-пТСХ с получением указанного в заголовке соединения (20 мг).
ESI-MS; m/z 374 [M++H].
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,50-1,81 (м, 7H), 2,19-2,26 (м, 1H), 2,56 (дд, J=2,4, 12,0 Гц, 1H), 2,71-2,74 (м, 1H), 2,94 (дд, J=4,0, 12,0 Гц, 1H), 6,56 (дд, J=1,6, 3,6 Гц, 1H), 7,05 (дд, J=8,8, 12,0 Гц, 1H), 7,13 (дд, J=2,4, 6,8 Гц, 1H), 7,23-7,25 (м, 1H), 7,52 (д, J=1,2 Гц, 1H), 7,89-7,93 (м, 1H), 8,07 (уш.с, 1H).
Примеры 222-225
Соединения примеров 222-225, показанные ниже в таблице 20, синтезировали в соответствии с примером 221, используя соответствующие карбоновые кислоты.
[Таблица 20]




Пример 226
Синтез (±)-(4aR*,7aS*)-6-(4-фторфенил)-7a-[3-(2-фторпиридин-3-ил)фенил]-4,4a,5,6,7,7a-гексагидропирроло[3,4-d][1,3]тиазин-2-иламина
[Формула 221]

(1) Синтез трет-бутил (±)-{(4aR*,7aS*)-6-(4-фторфенил)-7a-[3-(2-фторпиридин-3-ил)фенил]-4,4a,5,6,7,7a-гексагидропирроло[3,4-d][1,3]тиазин-2-ил}карбамата
трет-Бутил (±)-{(4aR*,7aS*)-7a-[3-(2-фторпиридин-3-ил)фенил]-4,4a,5,6,7,7a-гексагидропирроло[3,4-d][1,3]тиазин-2-ил}карбамат, полученный в примере получения 18-(9) (72 мг), смешивали с 4-фторбензолбороновой кислотой (24,7 мг), ацетатом меди(II) (6,1 мг), триэтиламином (93,2 мкл, удельный вес 0,73 г/см3) и молекулярными ситами 4A (порошок) (57,6 мг) в дихлорметане (3 мл) и смесь перемешивали в атмосфере азота при комнатной температуре в течение 11 часов и 30 минут. Дополнительно добавляли 4-фторбензолбороновую кислоту (23,5 мг) и ацетат меди(II) (12 мг). Атмосферу заменяли на открытую систему с последующим перемешиванием. Через 23 часа и 45 минут реакционную суспензию очищали хроматографией на колонке с NH- силикагелем. Полученный продукт снова очищали пТСХ с получением указанного в заголовке соединения (15 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,49 (с, 9H), 2,28-2,93 (м, 1H), 3,00-3,08 (м, 2H), 3,55-3,59 (м, 1H), 3,64 (д, J=10,0 Гц, 1H), 3,73-3,77 (м, 1H), 3,95 (д, J=10,0 Гц, 1H), 6,47-6,50 (м, 2H), 6,95-7,00 (м, 2H) 7,27-7,31 (м, 1H), 7,41-7,44 (м, 1H), 7,48-7,55 (м, 3H), 7,83-7,88 (м, 1H), 8,22 (тд, J=1,6, 4,8 Гц, 1H).
(2) Синтез (±)-(4aR*,7aS*)-6-(4-фторфенил)-7a-[3-(2-фторпиридин-3-ил)фенил]-4,4a,5,6,7,7a-гексагидропирроло[3,4-d][1,3]тиазин-2-иламина
Указанное в заголовке соединение (7,8 мг) получали из трет-бутил (±)-{(4aR*,7aS*)-6-(4-фторфенил)-7a-[3-(2-фторпиридин-3-ил)фенил]-4,4a,5,6,7,7a-гексагидропирроло[3,4-d][1,3]тиазин-2-ил}карбамата, полученного в примере 226-(1) (15 мг), в соответствии с примером 36-(2).
ESI-MS; m/z 423 [M++H].
Пример 227
Синтез (±)-(4aR*,7aS*)-7a-[3-(2-фторпиридин-3-ил)фенил]-6-o-толил-4,4a,5,6,7,7a-гексагидропирроло[3,4-d][1,3]тиазин-2-иламина
[Формула 222]

трет-Бутил (±)-{(4aR*,7aS*)-7a-[3-(2-фторпиридин-3-ил)фенил]-4,4a,5,6,7,7a-гексагидропирроло[3,4-d][1,3]тиазин-2-ил}карбамат, полученный в примере получения 18-(9) (50 мг), смешивали с o-толилбороновой кислотой (19,1 мг), ацетатом меди(II) (4,3 мг), триэтиламином (64,9 мкл, удельный вес 0,73 г/см3) и молекулярными ситами 4A (порошок) (40 мг) в дихлорметане (2 мл) с последующим перемешиванием при комнатной температуре. Через 20 часов добавляли о-толилбороновую кислоту (12,7 мг), ацетат меди(II) (4,3 мг) и триэтиламин (64,9 мкл) и смесь дополнительно перемешивали в атмосфере кислорода. Через двое суток еще добавляли о-толилбороновую кислоту (31,8 мг) с последующим перемешиванием. Через сутки добавляли дополнительно о-толилбороновую кислоту (63,6 мг), триэтиламин (130 мкл) и дихлорметан (1 мл) с последующим перемешиванием. Через трое суток реакционную суспензию очищали хроматографией на колонке с NH-силикагелем. Полученный продукт снова очищали пТСХ с получением N-арильного соединения. Его растворяли в хлороформе (1 мл) и затем добавляли ТФУК при комнатной температуре с последующим перемешиванием. Через 12 часов реакционный раствор разбавляли хлороформом и затем избыток ТФУК нейтрализовали насыщенным бикарбонатом натрия. Смесь экстрагировали три раза хлороформом. Полученные органические слои сушили над безводным сульфатом магния и твердое вещество удаляли фильтрованием. Фильтрат концентрировали при пониженном давлении и затем очищали хроматографией на колонке с NH-силикагелем с получением указанного в заголовке соединения (6,4 мг).
ESI-MS; m/z 419 [M++H]
Пример 228
Соединение примера 228, показанное ниже в таблице 21, синтезировали в соответствии с примером 227, используя соответствующую карбоновую кислоту.
[Таблица 21]

Пример 229
Синтез (±)-3'-((4aR*,7aS*)-2-амино-6-пиразин-2-ил-4a,5,6,7-тетрагидро-4H-пирроло[3,4-d][1,3]тиазин-7a-ил)бифенил-3-карбонитрила
[Формула 223]

(1) Синтез (±)-3'-{(4aR*,7aS*)-2-[N,N-бис(трет-бутоксикарбонил)амино]-6-пиразин-2-ил-4a,5,6,7-тетрагидро-4H-пирроло[3,4-d][1,3]тиазин-7a-ил}бифенил-3-карбонитрила
Указанное в заголовке соединение (23 мг) получали из (±)-N,N-бис(трет-бутоксикарбонил)[(4aR*,7aS*)-7a-(3-бромфенил)-6-пиразин-2-ил-4,4a,5,6,7,7a-гексагидропирроло[3,4-d][1,3]тиазин-2-ил]амина, полученного в примере получения 65-(3) (51 мг), в соответствии с примером получения 18-(8).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,41 (с, 18H), 2,99-3,07 (м, 2H), 3,20-3,23 (м, 1H), 3,87-3,98 (м, 2H), 4,07-4,21 (м, 2H), 7,40-7,55 (м, 4H), 7,63-7,66 (м, 1H), 7,83-7,84 (м, 2H), 7,88-7,93 (м, 2H), 7,96 (уш., 1H), 8,06 (уш., 1H).
(2) Синтез (±)-3'-((4aR*,7aS*)-2-амино-6-пиразин-2-ил-4a,5,6,7-тетрагидро-4H-пирроло[3,4-d][1,3]тиазин-7a-ил)бифенил-3-карбонитрила
Указанное в заголовке соединение (9,8 мг) получали из (±)-3'-{(4aR*,7aS*)-2-[N,N-бис(трет-бутоксикарбонил)амино]-6-пиразин-2-ил-4a,5,6,7-тетрагидро-4H-пирроло[3,4-d][1,3]тиазин-7a-ил}бифенил-3-карбонитрила, полученного в примере 229-(1) (23 мг), в соответствии с примером 36-(2).
ESI-MS; m/z 413 [M++H].
Пример 230
Соединение примера 230, показанное ниже в таблице 22, синтезировали в соответствии с примером 229, используя соответствующую карбоновую кислоту.
[Таблица 22]

Пример 231
Соединение примера 231, показанное ниже в таблице 23, синтезировали из соединения примера получения 66 и соответствующей карбоновой кислоты в соответствии с примером 14.
[Таблица 23]

Пример 232
Соединение примера 232, показанное ниже в таблице 24, синтезировали из соединения примера получения 67 и соответствующей карбоновой кислоты в соответствии с примером 14.
[Таблица 24]

Примеры 233-239
Соединения примеров 233-239, показанные ниже в таблице 25, синтезировали в соответствии с примером 14, используя соединение примера получения 3-(8) и соответствующие карбоновые кислоты.
[Таблица 25]
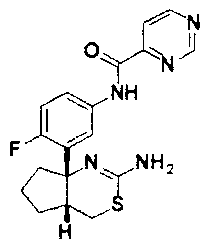






Пример 240
Синтез (±)-N-[3-((4aR*,7aS*)-2-амино-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-5-хлорфенил]-5-цианопиридин-2-карбоксамида
[Формула 224]

Указанное в заголовке соединение (1 мг) получали из соединения, полученного в примере получения 71-(5) (13 мг), и 5-цианопиридин-2-карбоновой кислоты (4,80 мг) в соответствии со способом примера 14.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,70-2,03 (м, 5H), 2,25-2,33 (м, 1H), 2,41-2,48 (м, 1H), 2,76 (дд, J=3,6, 12,8 Гц, 1H), 3,02 (дд, J=3,6, 12,8 Гц, 1H), 6,53 (дд, J=0,4, 3,6 Гц, 1H), 7,51 (дд, J=2,0, 3,6 Гц, 1H), 7,86 (т, J=2,0 Гц, 1H), 8,22 (дд, J=2,0, 8,0 Гц, 1H), 8,43 (дд, J=1,2, 8,0 Гц, 1H), 8,90 (дд, J=1,2, 3,2 Гц, 1H), 9,87 (с, 1H).
Пример 241
Синтез (±)-(4aR*,7aS*)-7a-[3-хлор-5-(2-фторпиридин-3-ил)фенил]-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-иламина
[Формула 225]

2-Фторпиридин-3-бороновую кислоту (55,8 мг), тетракис(трифенилфосфин)палладий(0) (22,9 мг) и 1 н. раствор карбоната натрия (396 мкл) добавляли к раствору побочного продукта (±)-N-(трет-бутоксикарбонил)-N-(метоксикарбонил)[(4aR*,7aS*)-7a-(3-бром-5-хлорфенил)-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]амина, полученного в примере получения 71-(2) (100 мг), в ДМФА (5 мл). После замены азотом смесь перемешивали при 85°C в течение трех часов. Реакционный раствор доводили до комнатной температуры и выпаривали при пониженном давлении растворитель. Остаток очищали хроматографией на колонке с NH-силикагелем с получением неочищенного продукта. Неочищенный продукт дополнительно очищали последовательно NH-пТСХ и хроматографией на колонке с силикагелем с получением указанного в заголовке соединения (25,6 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,74-2,06 (м, 5H), 2,26-2,34 (м, 1H), 2,43-2,48 (м, 1H), 2,78 (дд, J=3,6, 12,8 Гц, 1H), 3,04 (дд, J=3,6, 12,8 Гц, 1H), 7,28-7,31 (м, 1H), 7,39 (т, J=2,0 Гц, 1H), 7,41-7,42 (м, 2H), 7,87 (ддд, J=2,0, 7,2, 9,6 Гц, 1H), 8,22 (дт, J=1,6, 4,4 Гц, 1H).
ESI-MS; m/z 362 [M++H].
Пример 242
Синтез (±)-5-[3-((4aR*,7aS*)-2-амино-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]никотинонитрила
[Формула 226]
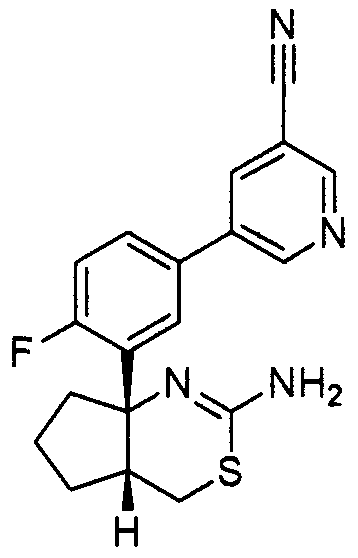
5-Циано-3-пиридинилбороновую кислоту (37,9 мг), тетракис(трифенилфосфин)палладий и 1 н. раствор карбоната натрия (256 мкл) добавляли к раствору (±)-N,N-бис(трет-бутоксикарбонил)[(4aR*,7aS*)-7a-(5-бром-2-фторфенил)-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]амина (70,0 мг) в ДМФА (5 мл). После замены азотом смесь перемешивали при 80°C в течение двух часов. После охлаждения до комнатной температуры добавляли к реакционной смеси воду. Водный слой экстрагировали этилацетатом и органический слой промывали водой и насыщенным раствором соли. Органический слой сушили над безводным сульфатом магния и выпаривали при пониженном давлении растворитель. Остаток растворяли в дихлорметане (3 мл). Добавляли трифторуксусную кислоту (1 мл) и смесь перемешивали при комнатной температуре в течение двух часов. Реакционную смесь разбавляли водой с последующей нейтрализацией насыщенным раствором бикарбоната натрия. Водный слой экстрагировали этилацетатом и органический слой сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и остаток очищали хроматографией на колонке с NH-силикагелем с получением указанного в заголовке соединения.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,74-2,01 (м, 5H), 2,59-2,66 (м, 1H), 2,78 (дд, J=4,0, 12,4 Гц, 1H), 2,81-2,87 (м, 1H), 2,96 (дд, J=3,2, 12,4 Гц, 1H), 7,17 (дд, J=8,4, 12,0 Гц, 1H), 7,41 (ддд, J=2,8, 4,4, 8,4 Гц, 1H), 7,56 (дд, J=2,8, 7,6 Гц, 1H), 8,09 (т, J=2,0 Гц, 1H), 8,83 (д, J=2,0 Гц, 1H), 8,99 (д, J=2,0 Гц, 1H).
Пример 243
Синтез (±)-(4aR*,6S*,7aS*)-7a-[2-фтор-5-(2-фторпиридин-3-ил)фенил-6-метокси-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-иламина
[Формула 227]
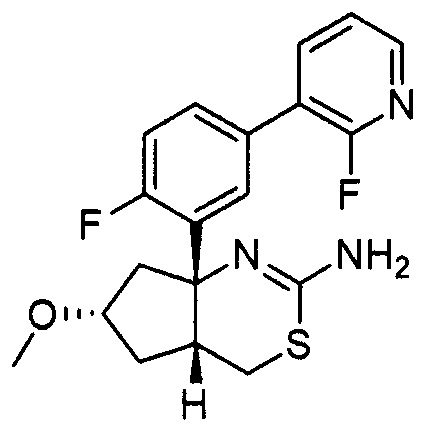
Указанное в заголовке соединение (2,1 мг) получали из (±)-N,N-бис(трет-бутилоксикарбонил)[(4aR*,6S*,7aS*)-7a-(5-бром-2-фторфенил)-6-метокси-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]амина (22,0 мг) и 2-фторпиридин-3-бороновой кислоты (11,0 мг) в соответствии с примером 242.
ESI-MS; m/z 376 [M++H].
Пример 244
Синтез (4aR,6R,7aS)-7a-[2-фтор-5-(2-фторпиридин-3-ил)фенил-6-метокси-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-иламина
[Формула 228]

Рацемат указанного в заголовке соединения (30,0 мг) получали из (±)-N,N-бис(трет-бутилоксикарбонил)[(4aR*,6R*,7aS*)-7a-(5-бром-2-фторфенил)-6-метокси-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-ил]амина (92,0 мг) и 2-фторпиридин-3-бороновой кислоты (46,2 мг) в соответствии с примером 242. Полученный рацемат (10,0 мг) оптически разделяли, используя CHIRALPAK™ AD-H производства Daicel Chemical Industries, Ltd. (2 см × 25 см, подвижная фаза: гексан:этанол = 8:2, скорость потока 10 мл/мин), и компонент, имеющий время удерживания от 33,8 до 38,1 минуты, собирали. Данную операцию повторяли с получением указанного в заголовке соединения (9,9 мг) из рацемата (26 мг).
ESI-MS; m/z 376 [M++H].
Пример 245
Синтез (±)-(4aR*,8aS*)-8a-[2,4-дифтор-5-(2-фторпиридин-3-ил)фенил]-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-иламина
[Формула 229]

Изопропиловый спирт (1 мл), 2-фтор-3-пиридинбороновую кислоту (49,8 мг), 1 н. раствор карбоната натрия (354 мкл) и бис(три-трет-бутилфосфин)палладий(0) (3,61 мг) добавляли к раствору бензил (±)-[(4aR*,8aS)-8a-(5-бром-2,4-дифторфенил)-4a,5,6,7,8,8a-гексагидро-4H-бензо[d][1,3]тиазин-2-ил]карбамата (35 мг) в толуоле (2 мл). После замены азотом смесь перемешивали при 85°C в течение 9,5 часов. Реакционный раствор охлаждали до комнатной температуры и выпаривали при пониженном давлении растворитель. Остаток очищали хроматографией на колонке с NH-силикагелем с получением промежуточного соединения. Полученное промежуточное соединение растворяли в хлороформе (2 мл). Добавляли йодтриметилсилан (30 мкл) с последующим перемешиванием в течение 14 часов. Реакционный раствор доводили до комнатной температуры и добавляли к реакционной смеси раствор бикарбоната натрия. Смесь разбавляли этилацетатом и добавляли тиосульфат натрия с последующим перемешиванием в течение 30 минут. Реакционную смесь, когда она становилась прозрачной, экстрагировали этилацетатом. Органический слой промывали насыщенным раствором соли. Органический слой сушили над безводным сульфатом магния и выпаривали при пониженном давлении растворитель. Остаток последовательно очищали NH-пТСХ и пТСХ с получением указанного в заголовке соединения (2,0 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,43-1,81 (м, 7H), 2,18-2,25 (м, 1H), 2,58 (дд, J=3,2, 12,4 Гц, 1H), 2,65-2,69 (м, 1H), 2,85 (дд, J=4,0, 12,4 Гц, 1H), 6,92 (дд, J=9,6, 12,0 Гц, 1H), 7,18-7,33 (м, 2H), 7,77-7,81 (м, 1H), 8,24 (д, J=4,8 Гц, 1H).
ESI-MS; m/z 378 [M++H].
Пример 246
Синтез (±)-N-[3-((3aS*,7aR*)-2-амино-3a,6,7,7a-тетрагидро-4H-пирано[4,3-d]тиазол-7a-ил)фенил]-5-хлорпиридин-2-карбоксамида
[Формула 230]

Указанное в заголовке соединение (39,5 мг) получали из соединения примера получения 75-(9) (99,0 мг) в соответствии со способом примера 14.
ESI-MS; m/z 389 [M++H].
Пример 247
Синтез (±)-(4aS*,8aS*)-8a-[4-(2-фторпиридин-3-ил)тиофен-2-ил]-4a,7,8,8a-тетрагидро-4H,5H-6-окса-3-тиа-1-азанафталин-2-иламина
[Формула 231]

2-Фторпиридин-3-бороновую кислоту (15,7 мг), тетракис(трифенилфосфин)палладий (4,3 мг) и 1 н. раствор карбоната натрия (112 мкл) добавляли к раствору (±)-N-трет-бутоксикарбонил-N-[(4aS*,8aS*)-8a-(4-бромтиофен-2-ил)-4a,7,8,8a-тетрагидро-4H,5H-6-окса-3-тиа-1-азанафталин-2-ил]бензамида (20,0 мг) в ДМФА (1 мл). После замены азотом смесь перемешивали при 90°C в течение семи часов. После охлаждения до комнатной температуры добавляли к реакционной смеси этилацетат. Органический слой промывали водой и насыщенным раствором соли и сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и остаток очищали хроматографией на колонке с NH-силикагелем с получением указанного в заголовке соединения (4,7 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,98-2,02 (м, 1H), 2,27 (ддд, J=4,4, 12,8, 13,6 Гц, 1H), 2,44-2,50 (м, 1H), 2,59 (дд, J=3,0, 12,8 Гц, 1H), 3,24 (дд, J=4,4, 12,8 Гц, 1H), 3,66-3,92 (м, 4H), 7,18 (т, J=1,4 Гц, 7,22-7,26 (м, 2H), 7,56 (т, J=1,6 Гц, 1H), 7,95 (ддд, J=1,6, 7,2, 9,6 Гц, 1H), 8,12 (дт, J=1,6, 4,8 Гц, 1H).
ESI-MS; m/z 350 [M++H].
Примеры 248-253
Соединения примеров 248-253, показанные ниже в таблице 26, синтезировали в соответствии с примером 33, используя соединение примера получения 7-(8) и соответствующие карбоновые кислоты.
[Таблица 26]


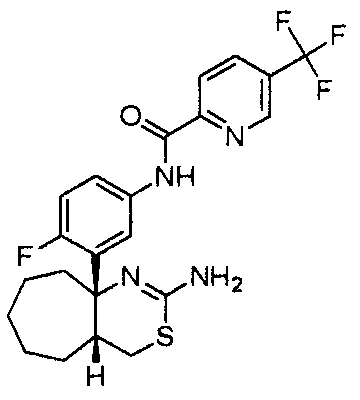



Пример 254
Соединение, синтезированное из соединения примера получения 1-(8) и 5-бромпиридин-2-карбоновой кислоты в соответствии с примером 14, оптически разделяли, используя CHIRALPAK™ OD-H производства Daicel Chemical Industries, Ltd. (2,5 см × 25 см, подвижная фаза гексан:этанол = 8:2, скорость потока 20 мл/мин), и компонент, имеющий время удерживания от 4,9 до 5,7 минут, собирали с получением соединения примера 254, показанного ниже в таблице 27.
[Таблица 27]
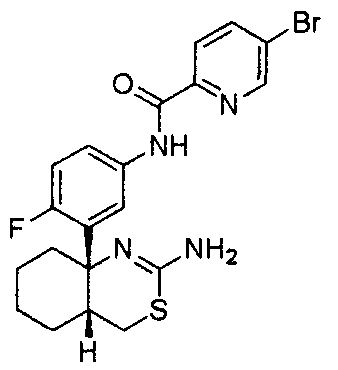
Пример 255
Соединение, синтезированное из соединения примера получения 1-(8) и пиридин-2-карбоновой кислоты в соответствии с примером 14, оптически разделяли, используя CHIRALPAK™ OD-H производства Daicel Chemical Industries, Ltd. (2,5 см × 25 см, подвижная фаза гексан:этанол = 8:2, скорость потока 20 мл/мин), и компонент, имеющий время удерживания от 5,1 до 6,4 минут, собирали с получением соединения примера 255, показанного ниже в таблице 28.
[Таблица 28]

Примеры 256-261
Соединения примеров 256-261, показанные ниже в таблице 29, синтезировали в соответствии с примером 14, используя соединение примера получения 2-(2) и соответствующие карбоновые кислоты.
[Таблица 29]

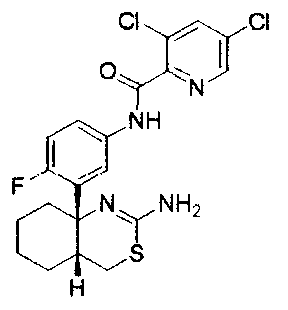


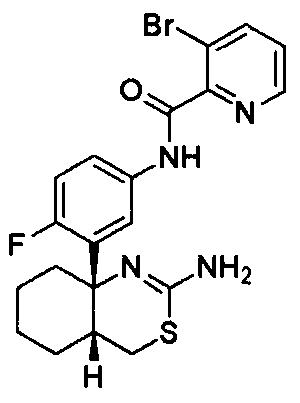

Пример 262
Синтез N-[3-((4aR*,7aS*)-2-амино-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]-2-хлороксазол-4-карбоксамида
[Формула 232]

Указанное в заголовке соединение (12,4 мг) получали из соединения, полученного в примере получения 3-(8) (20,0 мг), и 2-хлороксазол-4-карбоновой кислоты (12,1 мг) в соответствии с примером 14.
ESI-MS; m/z 393, 395 [M++H].
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,70-2,00 (м, 5H), 2,55-2,65 (м, 1H), 2,71-2,84 (м, 2H), 2,97 (дд, J=3,1, 12,5 Гц, 1H), 7,03 (дд, J=8,8, 12,0 Гц, 1H), 7,30 (дд, J=2,8, 7,2 Гц, 1H), 7,84 (ддд, J=2,4, 3,9, 8,8 Гц, 1H), 8,24 (с, 1H), 8,50 (с, 1H).
Примеры 263-267
Соединения примеров 263-267, показанные ниже в таблице 30, синтезировали в соответствии с примером 14, используя соответствующие карбоновые кислоты.
[Таблица 30]

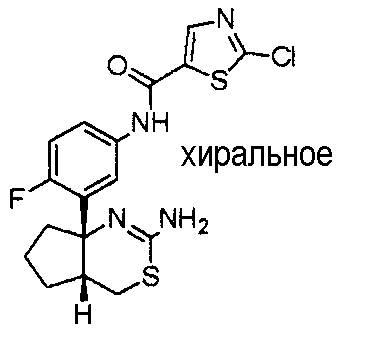
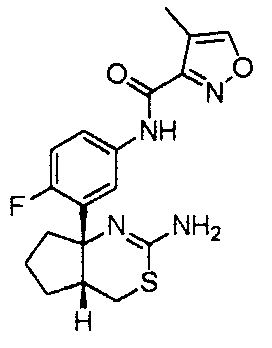
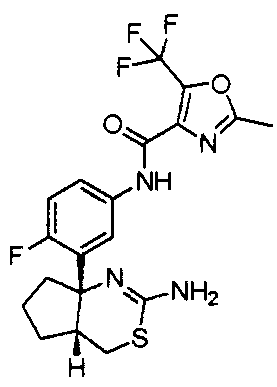

Пример 268
Синтез N-[3-((4aR*,7aS*)-2-амино-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]циклопентанкарбоксамида
[Формула 233]

Указанное в заголовке соединение (12,4 мг) получали из соединения, полученного в примере получения 3-(7) (20,0 мг), и 2-хлороксазол-4-карбоновой кислоты (12,1 мг) в соответствии с примером 14.
ESI-MS; m/z 334 [M++H].
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 0,79-0,86 (м, 2H), 1,03-1,07 (м, 2H), 1,53-1,62 (м, 1H), 1,76-2,04 (м, 5H), 2,60 (дт, J=13,4, 8,5 Гц, 1H), 2,79 (дд, J=3,7, 12,7 Гц, 1H), 2,91-3,06 (м, 1H), 6,98 (дд, J=8,8, 12,1 Гц, 1H), 7,09 (дд, J=2,2, 6,7 Гц, 1H), 7,88 (дд, J=3,3, 4,7 Гц, 1H), 8,39 (уш.с, 1H).
Пример 269
Синтез (+)-N-[3-((4aR*,7aS*)-2-амино-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]-3-бромтиазол-4-карбоксамида
[Формула 234]
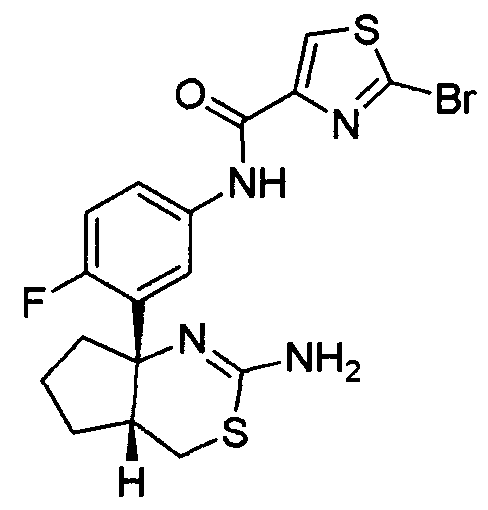
Указанное в заголовке соединение (12,5 мг) получали из соединения, полученного в примере получения 3-(8) (21,0 мг), и 2-бромтиазол-4-карбоновой кислоты (18,0 мг) в соответствии с примером 14.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,67-2,02 (м, 5H), 2,51-2,68 (м, 1H), 2,76 (дд, J=8,72, 12,0 Гц, 1H), 2,78-2,86 (м, 1H), 2,99 (дд, J=3,22, 12,57 Гц, 1H), 7,04 (дд, J=8,72, 12,00 Гц, 1H), 7,30 (дд, J=2,84, 7,14 Гц, 1H), 7,87 (ддд, J=2,78, 11,9, 8,78 Гц, 1H), 8,15 (с, 1H), 8,98 (с, 1H).
Пример 270
Синтез (+)-N-[3-((4aR*,7aS*)-2-амино-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]-1-метил-1H-имидазол-4-карбоксамида
[Формула 235]

Указанное в заголовке соединение (7 мг) получали из соединения, полученного в примере получения 3-(8) (20,0 мг), и 1-метил-1H-имидазол-4-карбоновой кислоты (10,0 мг) в соответствии с примером 14.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,74-2,01 (м, 5H), 2,61 (дт, J=12,00, 8,40 Гц, 1H), 2,74 (дд, J=4,04, 12,13 Гц, 1H), 2,77-2,83 (м, 1H), 2,99 (дд, J=3,03, 12,63 Гц, 1H), 3,77 (с, 3H), 7,01 (дд, J=8,84, 11,87 Гц, 1H), 7,30 (дд, J=2,53, 12,63 Гц, 1H), 7,42 (д, 1,52 Гц, 1H), 7,61 (д, 1,52 Гц, 1H), 7,92 (ддд, J=2,78, 4,04, 8,84, 1H), 8,89 (с, 1H).
Пример 271
Синтез (+)-N-[3-((4aR*,7aS*)-2-амино-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]-3-метил-3H-имидазол-4-карбоксамида
[Формула 236]

Указанное в заголовке соединение (10,5 мг) получали из соединения, полученного в примере получения 3-(8) (15,0 мг), и 3-метил-3H-имидазол-4-карбоновой кислоты (10,0 мг) в соответствии с примером 14.
ESI-MS; m/z 374 [M++H].
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,70-2,01 (м, 5H), 2,53-2,66 (м, 1H), 2,75 (дд, J=4,04, 12,76 Гц, 1H), 2,79-2,88 (м, 1H), 2,97 (дд, J=3,35, 12,69 Гц, 1H), 3,94 (с, 3H), 7,03 (дд, J=8,84, 12,00 Гц, 1H), 7,21 (дд, J=2,78, 7,07 Гц, 1H), 7,53 (с, 1H), 7,61 (д, J=0,76 Гц, 1H),7,79 (ддд, J=2,78, 4,01, 8,75 Гц, 1H).
Пример 272
Синтез (+)-N-[3-((4aR*,7aS*)-2-амино-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]-1-метил-1H-пиразол-4-карбоксамида
[Формула 237]

Указанное в заголовке соединение (14,7 мг) получали из соединения, полученного в примере получения 3-(8) (25,0 мг), и 1-метил-1H-пиразол-4-карбоновой кислоты (17,0 мг) в соответствии с примером 14.
ESI-MS; m/z 374 [M++H].
1H-ЯМР (400 МГц, CDCl3 + несколько капель MeOD) δ (м.д.): 1,64-2,01 (м, 5H), 2,51-2,65 (м, 1H), 2,73 (дд, J=3,79, 12,63 Гц, 1H), 2,84-2,92 (м, 1H), 2,96 (дд, J=3,41, 12,63 Гц, 1H), 3,93 (с, 3H), 6,95-7,04 (м, 2H), 7,97 (м, 3H).
Пример 273
Синтез (+)-N-[3-((4aR*,7aS*)-2-амино-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]-4-хлор-1-метил-1H-пиразол-3-карбоксамида
[Формула 238]

Указанное в заголовке соединение (8,5 мг) получали из соединения, полученного в примере получения 3-(8) (24,0 мг), и 4-хлор-1-метил-1H-пиразол-3-карбоновой кислоты (21,0 мг) в соответствии с примером 14.
ESI-MS; m/z 408 [M++H].
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,66-2,02 (м, 5H), 2,50-2,66 (м, 1H), 2,76 (дд, J=4,04, 12,76 Гц, 1H), 2,79-2,89 (м, 1H), 2,99 (дд, J=3,28, 12,63 Гц, 1H), 3,93 (с, 3H), 7,02 (дд, J=8,78, 12,06 Гц, 1H), 7,20 (дд, J=2,78, 7,20 Гц, 1H), 7,45 (с, 1H), 7,99 (ддд, J=2,78, 4,11, 8,78 Гц, 1H), 8,58 (с, 1H).
Пример 274
Синтез (±)-6-{(E)-2-[3-((4aR*,7aS*)-2-амино-4a,5,6,7,7a-тетрагидро-4H-циклопента[d][1,3]тиазин-7a-ил)-4-фторфенил]винил}никотинонитрила
[Формула 239]

Трифторуксусную кислоту (0,3 мл) добавляли к раствору соединения, полученного в примере получения 77 (17 мг), в дихлорметане (2 мл), и смесь перемешивали при комнатной температуре в течение 30 минут. Реакционный раствор нейтрализовали раствором карбоната натрия. Реакционную смесь экстрагировали дихлорметаном. Органический слой сушили над безводным сульфатом магния. Осушающий агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении с получением остатка. Остаток очищали препаративной ВЭЖХ с получением указанного в заголовке соединения (5,1 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,00 (м, 4H), 2,31 (м, 1H), 2,55 (м, 1H), 2,87 (дд, J=4,0, 12,9 Гц, 1H), 3,07 (дд, J=4,0, 12,9 Гц, 1H), 3,14 (м, 1H), 7,09 (дд, J=8,3, 12,2 Гц, 1H), 7,14 (д, J=16,2 Гц, 1H), 7,49 (дд, J=0,7, 8,3 Гц, 1H), 7,53 (м, 1H), 7,59 (дд, J=2,2, 8,0 Гц, 1H), 7,78 (д, J=16,2 Гц, 1H), 7,91 (дд, J=2,2, 8,2 Гц, 1H), 8,84 (дд, J=0,5, 2,1 Гц, 1H).
Пример 275
Синтез (±)-(4aR*,7aS*)-7a-(5((E)-2-(5-хлорпиридин-2-ил)винил)-2-фторфенокси)-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-амина
[Формула 240]

Указанное в заголовке соединение (13,4 мг) получали из соединения, полученного в примере получения 72 (200 мг), и 2-бром-5-хлорпиридина (73 мг) в соответствии с примером 274.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,70-2,00 (м, 4H), 2,19 (м, 1H), 2,40 (м, 1H), 2,76 (дд, J=3,8, 13,0 Гц, 1H), 2,96 (дд, J=3,8, 13,0 Гц, 1H), 3,06 (м, 1H), 6,90 (м, 2H), 7,18 (д, J=8,5 Гц, 1H), 7,28 (дд, J=2,1, 8,0 Гц, 1H), 7,34 (м, 1H), 7,39 (д, J=16,1 Гц, 1H), 7,49 (дд, J=2,5,8,4 Гц, 1H), 8,36 (д, J=2,4 Гц, 1H).
Пример 276
Синтез (±)-6-{[3-(2-амино-4a,5,6,7,-тетрагидро-4H-циклопента[d][1,3]тиазин-7a(4H)-ил)-4-фторфенил]этинил}никотинонитрила
[Формула 241]

Соединение, полученное в примере получения 78-(2) (135 мг), смешивали с дихлоридом бис(трифенилфосфин)палладия(II) (35 мг), йодидом меди(I) (9,4 мг) и 2-бром-5-цианопиридином (180 мг) в триэтиламине (3,5 мл) и тетрагидрофуране (0,45 мл), и смесь перемешивали в атмосфере азота при 90°C в течение пяти часов. Реакционную суспензию фильтровали и концентрировали. Затем полученный остаток очищали препаративной ЖХ-МС с получением указанного в заголовке соединения (113 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,60-1,90 (м, 5H), 2,43 (м, 1H), 2,72 (м, 1H), 2,84 (м, 2H), 5,77 (уш.с, 2H), 7,32 (дд, J=8,5, 12,3 Гц, 1H), 7,62 (м, 2H), 7,88 (д, J=8,2 Гц, 1H), 8,39 (дд, J=1,8, 8,1 Гц, 1H), 9,06 (с, 1H).
ESI-MS; m/z 377 [M++H].
Пример 277
Синтез (±)-(4aR*,7aS*)-7a-[2-фтор-5-(5-фторпиридин-2-ил)этинил]фенил}-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-амина
[Формула 242]

Указанное в заголовке соединение (59 мг) получали из соединения, полученного в примере получения 78-(2) (200 мг), и 3-бром-2-фторпиридина (257 мг) в соответствии с примером 276.
ESI-MS; m/z 370 [M++H].
Пример 278
Синтез (±)-(4aR*,7aS*)-7a-{2-фтор-5-[2-(2-фторпиридин-3-ил)этил]фенил]-4,4a,5,6,7,7a-гексагидроциклопента[d][1,3]тиазин-2-иламина
[Формула 243]
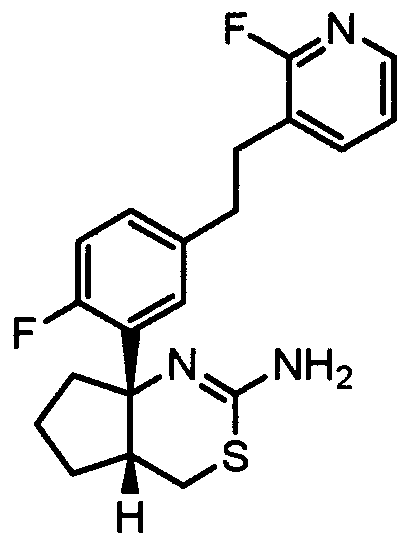
Катализатор в виде 10% гидроксида палладия добавляли к смешанному раствору соединения, полученного в примере 277 (20 мг), в метаноле (5 мл) и этилацетате (5 мл), и смесь перемешивали в атмосфере водорода при комнатной температуре в течение ночи. Реакционную суспензию фильтровали через целит и концентрировали. Затем полученный остаток очищали препаративной ЖХ-МС с получением указанного в заголовке соединения (18 мг).
Пример 279
Соединение примера 279, показанное ниже в таблице 31, синтезировали в соответствии с примером 278, используя соответствующее алкиновое соединение.
[Таблица 31]

Пример 280
Синтез N-[3-((4aS,5S,7aS)-2-амино-5-дифторметил-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил]-5-дигидропиридин-2-карбоксамида
[Формула 244]
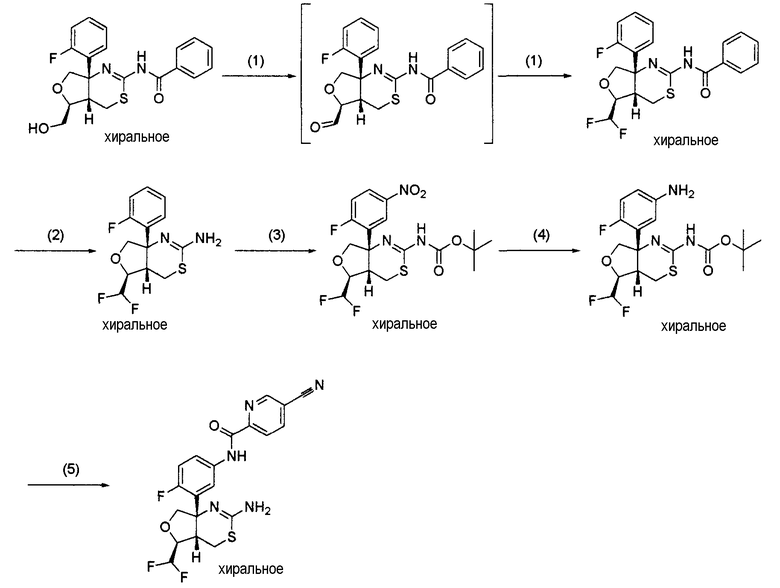
(1) Синтез N-[(4aS,5S,7aS)-5-дифторметил-7a-(2-фторфенил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил]бензамида
Раствор ДМСО (165 мкл) в дихлорметане (0,5 мл) добавляли по каплям к раствору оксалилхлорида (166 мкл) в дихлорметане (12 мл) в атмосфере азота при -78°C. После перемешивания смеси в течение 10 минут при той же температуре добавляли по каплям раствор соединения, полученного в примере получения 25-(9) (500 мг), в дихлорметане (2,5 мл). Смесь перемешивали в течение 45 минут при той же температуре. Диизопропиламин (1,12 мл) добавляли к реакционному раствору при той же температуре и нагревали реакционный раствор до комнатной температуры. Реакционный раствор перемешивали в течение одного часа при комнатной температуре. Добавляли водный раствор хлорида аммония и этилацетат и органический слой отделяли. Органический слой промывали насыщенным водным раствором хлорида натрия и сушили над безводным сульфатом магния. Органический слой концентрировали при пониженном давлении с получением неочищенного альдегидного сухого остатка. К неочищенному альдегидному сухому остатку добавляли дихлорметан (10 мл) и смесь охлаждали льдом. К смеси добавляли по каплям трифторид диэтиламиносеры (676 мкл) и смесь перемешивали в течение 30 минут с последующим нагреванием до комнатной температуры. Реакционный раствор дополнительно перемешивали в течение двух часов. К реакционному раствору добавляли насыщенный водный раствор бикарбоната натрия и хлороформ и отделяли органический слой. Органический слой сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток очищали хроматографией на силикагеле с получением указанного в заголовке соединения (170 мг).
Реакции, как описано выше, проводили при тех же параметрах, используя трифторид [бис(2-метоксиэтил)амино]серы (0,95 мл) вместо трифторида диэтиламиносеры, с получением указанного в заголовке соединения (140 мг).
ESI-MS; m/z 407 [M++H].
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,85-2,91 (м, 1H), 3,14-3,29 (м, 1H), 3,51-3,60 (м, 1H), 4,00-4,09 (м, 1H), 4,53 (д, J=9,2 Гц, 1H), 4,60-4,70 (м, 1H), 5,86-6,15 (м, 1H), 7,12-7,27 (м, 2H), 7,35-7,53 (м, 5H), 8,00-8,18 (м, 2H).
(2) Синтез (4aS,5S,7aS)-5-дифтор-7a-(2-фторфенил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-иламина
Гидразингидрат (738 мкл) добавляли к раствору соединения, полученного в примере 280-(1) (310 мг), в этаноле (8 мл) и смесь перемешивали в течение трех часов при комнатной температуре. К реакционному раствору добавляли насыщенный водный раствор хлорида натрия и этилацетат и отделяли органический слой. Органический слой сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток очищали хроматографией на силикагеле с получением указанного в заголовке соединения (185 мг).
ESI-MS; m/z 303 [M++H].
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,84 (дд, J=3,6, 13,2 Гц, 1H), 3,10 (дд, J=3,6, 13,2 Гц, 1H), 3,23-3,27 (м, 1H), 3,85-3,88 (м, 1H), 4,45-4,58 (м, 4H), 5,77-6,07 (м, 1H), 7,06 (ддд, J=1,2, 8,4, 12,8 Гц, 1Н), 7,13-7,18 (м, 1H), 7,28-7,31 (м, 1H), 7,41-7,45 (м, 1H).
(3) Синтез трет-бутил [(4aS,5S,7aS)-дифторметил-7a-(2-фтор-5-нитрофенил)-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил]карбамата
Белую дымящую азотную кислоту (39,7 мкл) добавляли по каплям к раствору соединения, полученного в примере 280-(2) (185 мг), при охлаждении льдом. Реакционный раствор перемешивали в течение 30 минут при комнатной температуре и затем выливали на лед. Полученную смесь подщелачивали 5 н. гидроксидом натрия при охлаждении льдом. К реакционному раствору добавляли хлороформ и отделяли органический слой. Органический слой сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток растворяли в ТГФ (801 мл), добавляли триэтиламин (0,85 мл) и ди-трет-бутилдикарбонат (801 мг) и полученную смесь перемешивали в течение двенадцати часов. К реакционному раствору снова добавляли триэтиламин (0,85 мл) и ди-трет-бутилдикарбонат (801 мг) и смесь перемешивали в течение пяти часов. К реакционному раствору добавляли насыщенный водный раствор хлорида натрия и этилацетат и отделяли органический слой. Органический слой сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток очищали хроматографией на силикагеле с получением указанного в заголовке соединения (213 мг).
ESI-MS; m/z 448 [M++H].
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,53 (с, 9H), 2,78 (дд, J=3,6, 14,0 Гц, 1H), 2,91-2,98 (м, 1H), 3,37-3,44 (м, 1H), 3,79-3,84 (м, 1H), 4,46 (д, J=8,0 Гц, 1H), 4,58-4,64 (м, 1H), 5,83-6,13 (м, 1H), 7,27-7,33 (м, 1H), 8,21-8,25 (м, 1H), 8,31 (дд, J=2,8, 6,8 Гц, 1H).
(4) Синтез трет-бутил [(4aS,5S,7aS)-7a-(5-амино-2-фторфенил)-5-дифторметил-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил]карбамата
Насыщенный водный раствор хлорида аммония (2 мл) и железный порошок (276 мг) добавляли к раствору соединения, полученного в примере 280-(3) (210 мг), в этаноле (10 мл) и кипятили с обратным холодильником в течение 30 минут. Температуру реакционного раствора доводили до комнатной температуры и разбавляли реакционный раствор этилацетатом. Вещества, нерастворимые в реакционном растворе, удаляли фильтрованием через целит. К фильтрату добавляли насыщенный водный раствор хлорида натрия и этилацетат и отделяли органический слой. Органический слой сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток очищали хроматографией на NH-силикагеле с получением указанного в заголовке соединения (144 мг).
ESI-MS; m/z 418 [M++H].
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,51 (с, 9H), 2,76 (дд, J=3,6, 14,0 Гц, 1H), 3,04-3,12 (м, 1H), 3,33-3,42 (м, 1H), 3,62 (уш.с, 2H), 3,80-3,85 (м, 1H), 4,50 (д, J=8,4 Гц, 1H), 4,49-4,59 (м, 1H), 5,94 (дт, J=3,6, 55,6 Гц, 1H), 6,55-6,62 (м, 2H), 6,87 (дд, J=8,4, 12,4 Гц, 1H).
(5) Синтез N-[3-((4aS,5S,7aS)-2-амино-5-дифторметил-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)-4-фторфенил]-5-цианопиридин-2-карбоксамида
Указанное в заголовке соединение (15 мг) получали в соответствии со способами примера 14 из соединения, полученного в примере 280-(4) (28 мг), и соединения, полученного в примере получения 13-(2) (19 мг).
ESI-MS; m/z 448 [M++H].
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 3,05 (дд, J=3,6, 14,0 Гц, 1H), 3,40 (дд, J=3,6, 13,6 Гц, 1H), 3,70-3,76 (м, 1H), 4,27-4,33 (м, 1H), 4,47-4,57 (м, 2H), 6,05 (дт, J=3,2, 54,8 Гц, 1H), 7,19 (дд, J=9,2, 12,0 Гц, 1H), 7,70 (дд, J=2,8, 7,2 Гц, 1H), 7,94-7,98 (м, 1H), 8,22 (дд, J=2,0, 8,0 Гц, 1H), 8,41 (дд, J=0,8, 8,0 Гц, 1H), 8,92 (дд, J=0,8, 2,0 Гц, 1H), 9,96 (с, 1H).
Примеры 281-284
Соединения примеров 281-284, показанные ниже в таблице 32, синтезировали в соответствии со способами примера 280, используя соответствующие карбоновые кислоты.
[Таблица 32]
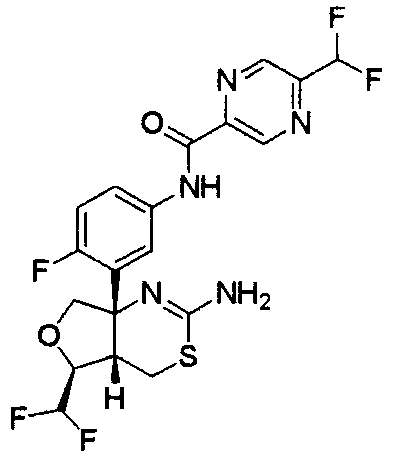
ESI-MS; m/z 474 [M++H]
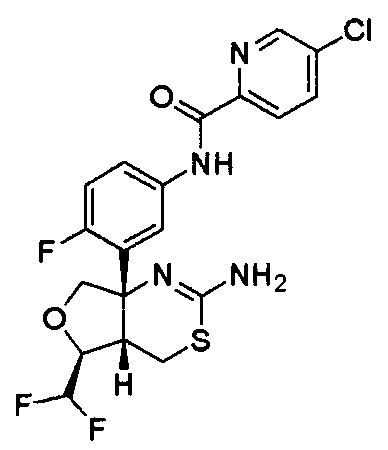
ESI-MS; m/z 457 [M++H].
ESI-MS; m/z 491 [M++H]
ESI-MS; m/z 489 [M++H]
Пример 285
Синтез (±)-N-[5-((4aS*,5R*,7aR*)-2-амино-5-метил-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)тиофен-3-ил]-5-цианопиридин-2-карбоксамида
[Формула 245]

(1) Синтез (±)-(3aR*,4R*,6aR*)-6a-(4-бромтиофен-2-ил)-4-метилтетрагидро-фуро[3,4-c]изоксазола
Указанное в заголовке соединение (956 мг) получали в соответствии со способами примера получения 76 из соединения, полученного в примере получения 22-(2)(410 мг), и 2,4-дибромтиофена (1,64 г).
ESI-NS; m/z 290 [M++H].
(2) Синтез (±)-(4aS*,5R*,7aR*)-7a-(4-бром-2-тиенил)-5-метил-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-иламина
Указанное в заголовке соединение (270 мг) получали в соответствии со способами примера получения 22 из (±)-(3aR*,4R*,6aR*)-6a-(4-бромтиофен-2-ил)-4-метилтетрагидро-flo[3,4-c]изоксазола. Однако реакцию дебензоилирования, соответствующую примеру получения 22-(7), проводили в соответствии со способами примера получения 19-(9).
ESI-MS; m/z 335 [M++H].
(3) Синтез (±)-ди-трет-бутил[(4aS*,5R*,7aR*)-7a-(4-азидо-2-тиенил)-5-метил-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил]имидодикарбоната
Указанное в заголовке соединение (75 мг) получали в соответствии со способами примера получения 71 из (±)-(4aS*,5R*,7aR*)-7a-(4-бром-2-тиенил)-5-метил-4a,5,7,7a-тетрагидро-4H-flo[3,4-d][1,3]тиазин-2-иламина.
ESI-MS; m/z 496 [M++H].
(4) Синтез (±)-трет-бутил [(4aS*,5R*,7aR*)-7a-(4-аминотиофен-2-ил)-5-метил-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил]карбамата
Цинк (19,7 мг) и формиат аммония добавляли к раствору (±)-ди-трет-бутил[(4aS*,5R*,7aR*)-7a-(4-азидо-2-тиенил)-5-метил-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-2-ил]имидодикарбоната (75 мг) в метаноле (5 мл). Смесь перемешивали в течение трех суток при комнатной температуре. Далее добавляли к реакционной смеси метанол (50 мл), цинк (197 мг) и формиат аммония (476 мг) и затем смесь перемешивали в течение трех часов. Избыток этанола выпаривали при пониженном давлении. К остатку добавляли воду и этилацетат, чтобы растворить нерастворимые вещества. Водный слой экстрагировали этилацетатом и органический слой промывали водой. Органический слой сушили над безводным сульфатом магния и выпаривали при пониженном давлении растворитель. Остаток очищали хроматографией на NH-силикагеле с получением указанного в заголовке соединения (35 мг).
ESI-MS; m/z 370 [M++H].
(5) Синтез (±)-N-[5-((4aS*,5R*,7aR*)-2-амино-5-метил-4a,5-дигидро-4H-фуро[3,4-d][1,3]тиазин-7a-ил)тиофен-3-ил]-5-цианопиридин-2-карбоксамида
Указанное в заголовке соединение (27 мг) получали в соответствии со способами примера 14 из соединения, полученного в примере 285-(4), и соединения, полученного в примере получения 13-(2).
ESI-MS; m/z 400 [M++H].
Пример 286
Синтез (±)-N-[5-((4aS*,8aR*)-2-амино-4a,5,7,8-тетрагидро-4H-6-окса-3-тиа-1-азанафталин-8a-ил)тиофен-3-ил]-5-цианопиридин-2-карбоксамида
[Формула 246]

(1) Синтез трет-бутил (±)-[(4aS*,8aR*)-8a-(4-аминотиофен-2-ил)-4a,7,8,8a-тетрагидро-4H,5H-6-окса-3-тиа-1-азанафталин-2-ил]карбамата
Указанное в заголовке соединение (89 мг) получали в соответствии со способами примера 285 из соединения, полученного в примере получения 8-(2), и 2,4-дибромтиофена.
ESI-MS; m/z 370 [M++H].
(2) Синтез (±)-N-[5-((4aS*,8aR*)-2-амино-4a,5,7,8-тетрагидро-4H-6-окса-3-тиа-1-азанафталин-8a-ил)тиофен-3-ил]-5-цианопиридин-2-карбоксамида
Указанное в заголовке соединение (23 мг) получали в соответствии со способами примера 14 из соединения, полученного в примере 286-(1), и соединения, полученного в примере получения 13-(2).
ESI-MS; m/z 400 [M++H].
Пример испытания 1
Количественное определение Aβ пептида в культуре нейронов из головного мозга плода крысы
(1) Первичная культура нервных клеток крысы
Первичные культуры нейронов получали из коры головного мозга 18-дневных эмбрионов крыс Wistar (Charles River Japan, Yokohama, Japan). В частности, эмбрионы были асептически извлечены у беременных крыс под эфирной анестезией. Мозг извлекали из эмбриона и погружали в охлаждаемую льдом среду L-15 (такую как Invitrogen Corp. Cat #11415-064, Carlsbad, CA, USA, или SIGMA L1518). Кору головного мозга выделяли из извлеченного мозга под стереоскопическим микроскопом. Собранные фрагменты коры головного мозга ферментативно обрабатывали в ферментном растворе, содержащем 0,25% трипсин (Invitrogen Corp. Cat #15050-065, Carlsbad, CA, USA) и 0,01% DNase (Sigma D5025, St. Louis, MO, USA), при 37°C в течение 30 минут для диспергирования клеток. После этого ферментативную реакцию останавливали добавлением инактивированной лошадиной сыворотки к раствору. Ферментативно обработанный раствор центрифугировали при 1500 об/мин в течение пяти минут для удаления супернатанта. К полученной клеточной массе добавляли 5-10 мл среды. В качестве среды (Neurobasal/B27/2-ME) использовали нейробазальную среду (Invitrogen Corp. Cat #21103-049, Carlsbad, CA, USA), дополненную 2% B27 добавкой (Invitrogen Corp. Cat #17504-044, Carlsbad, CA, USA), 25 мкМ 2-меркаптоэтанолом (2-ME, WAKO Cat #139-06861, Osaka, Japan), 0,5 мМ L-глутамином (Invitrogen Corp. Cat #25030-081, Carlsbad, CA, USA) и антибиотиками с антимикотиками (Invitrogen Corp. Cat #15240-062, Carlsbad, CA, USA). Однако для анализа использовали указанную выше нейробазальную среду, не дополненную 2-ME (Neurobasal/B27). Клетки снова диспергировали умеренной обработкой с помощью пипетки клеточной массы, к которой добавляли указанную среду. Клеточную дисперсию фильтровали через 40-мкм найлоновую сетку (Cell Strainer, Cat #35-2340, Becton Dickinson Labware, Franklin Lakes, NJ, USA) для удаления оставшейся клеточной массы и в результате получали суспензию нервных клеток. Суспензию нервных клеток разбавляли описанной средой и затем высевали в объеме 100 мкл/лунку при начальной плотности клеток 5×105 клеток/см2 на 96-луночный полистироловый культуральный планшет, предварительно покрытый поли-L или D-лизином (Falcon Cat #35-3075, Becton Dickinson Labware, Franklin Lakes, NJ, USA, покрытый поли-L-лизином с использованием способа, описанного ниже, или BIOCOAT™ cell environments Poly-D-lysine cell ware 96-well plate, Cat #35-6461, Becton Dickinson Labware, Franklin Lakes, NJ, USA). Покрытие поли-L-лизином осуществляли следующим образом. Асептически приготавливали, используя 0,15 M боратный буфер (pH 8,5), 100 мкг/мл раствора поли-L-лизина (SIGMA P2636, St. Louis, MO, USA). По 100 мкг/лунку полученного раствора вносили в лунки 96-луночного полистиролового культурального планшета и инкубировали при комнатной температуре в течение одного или более часов или при 4°C в течение ночи или более. После этого снабженный покрытием 96-луночный полистироловый культуральный планшет промывали стерильной водой четыре раза или более и затем сушили или прополаскивали, например, стерильной PBS или средой и использовали для посева клеток. Посеянные клетки выращивали на культуральном планшете при 37°C в атмосфере 5% CO2-95% воздуха в течение одних суток. Затем всю среду заменяли свежей средой Neurobasal™/B27/2-ME, после чего клетки культивировали еще трое суток.
(2) Внесение соединений
На день 4 культивирования в лунки планшета вносили лекарственное вещество следующим образом. Всю среду удаляли из лунок и вносили в них по 180 мкл/лунку нейробазальной среды, не содержащей 2-ME и содержащей 2% B-27 (Neurobasal/B27). Раствор испытуемого соединения в диметилсульфоксиде (далее называемом сокращенно ДМСО) разбавляли средой Neurobasal/B27 до концентрации, в 10 раз превышающей конечную концентрацию. В находящуюся в лунках среду добавляли по 20 мкл/лунку полученного разбавленного раствора и в достаточной мере смешивали его со средой. Конечная концентрация ДМСО составляла 1% или менее. В контрольную группу добавляли только ДМСО.
(3) Взятие образца
После добавления соединения клетки культивировали в течение трех суток и всю среду собирали. Полученную среду использовали в качестве образца для ELISA. Образец не разводили для измерения Aβx-42 методом ELISA и разводили до 5-кратного разведения разбавителем, поставляемым с набором ELISA, для ELISA измерения Aβx-40.
(4) Оценка выживаемости клеток
Выживаемость клеток оценивали MTT анализом по следующей методике. После сбора среды в лунки вносили по 100 мкл/лунку предварительно нагретой среды. Затем в лунки добавляли по 8 мкл/лунку раствора 8 мг/мл MTT (SIGMA M2128, St. Louis, MO, USA) в D-PBS(-) (Dulbecco's phosphate buffered Saline, SIGMA D8537, St. Louis, MO, USA). 96-Луночный полистироловый культуральный планшет инкубировали в инкубаторе при 37°C в атмосфере 5% CO2-95% воздуха в течение 20 минут. Добавляли по 100 мкл/лунку буфера для MTT лизиса и кристаллы MTT формазана в достаточной мере растворяли в буфере в инкубаторе при 37°C в атмосфере 5% CO2-95% воздуха. Затем измеряли оптическую плотность при 550 нм в каждой лунке. Буфер для MTT лизиса получали следующим образом. 100 г SDS (додецилсульфат натрия (лаурилсульфат натрия), WAKO 191-07145, Osaka, Japan) растворяли в смешанном растворе 250 мл N,N'-диметилформамида (WAKO 045-02916, Osaka, Japan) с 250 мл дистиллированной воды. Затем к раствору добавляли по 350 мкл концентрированной хлористо-водородной кислоты и уксусной кислоты для обеспечения конечного pH примерно 4,7.
При измерении лунки, не содержащие посеянных клеток и содержащие только среду и раствор MTT, считали фоном (bkg). Измеренные значения соответственно применяли в следующей формуле, вычитая из них bkg значения. Так, для сравнения и оценки активности в отношении выживаемости клеток вычисляли процентное отношение к контрольной группе (группа, не обработанная лекарственным веществом, CTRL) (% CTRL).
% CTRL=(A550_sample-A550_bkg)/(A550_CTRL-bkg)×100
(A550_sample: оптическая плотность при 550 нм лунки с образцом, A550_bkg: оптическая плотность при 550 нм фоновой лунки, A550_CTRL: оптическая плотность при 550 нм лунки контрольной группы).
(5) Aβ ELISA
Для определения Aβ методом ELISA использовали набор Human/Rat β Amyloid (42) ELISA Kit Wako (#290-62601) и набор Human/Rat β Amyloid (40) ELISA Kit Wako (#294-62501) от Wako Pure Chemical Industries, Ltd. Анализ Aβ ELISA проводили в соответствии с инструкциями, рекомендованными производителями (методики, описанные в прилагаемых документах). Однако Aβ калибрационную кривую получали, используя бета-амилоидный пептид 1-42, крысиный и бета-амилоидный пептид 1-40, крысиный (Calbiochem, #171596 [Aβ42], #171593 [Aβ40]). Результаты показаны ниже в таблицах 33, 34 и 35 в виде процентного отношения к концентрации Aβ в среде контрольной группы (% CTRL).
[Таблица 33]
IC50 (мкМ)
IC50 (мкМ)
IC50 (мкМ)
IC50 (мкМ)
IC50 (мкМ)
IC50 (мкМ)
Из результатов, представленных в таблицах, ясно, что соединение по настоящему изобретению обладает эффектом уменьшения продукции Aβ42.
Промышленная применимость
Соединение общей формулы (I) или его фармацевтически приемлемая соль или сольват по настоящему изобретению обладает эффектом уменьшения продукции Aβ42. Следовательно, настоящее изобретение может, в частности, обеспечить терапевтическое или профилактическое средство против нейродегенеративного заболевания, вызываемого Aβ, такого как деменция Альцгеймера или синдром Дауна.
| название | год | авторы | номер документа |
|---|---|---|---|
| КОНДЕНСИРОВАННЫЕ ПРОИЗВОДНЫЕ АМИНОДИГИДРОТИАЗИНА, ПРИГОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ВАСЕ | 2012 |
|
RU2593756C2 |
| ЦИННАМИДНОЕ СОЕДИНЕНИЕ | 2005 |
|
RU2361872C2 |
| НОВЫЕ ПИРИДИНОВОЕ ПРОИЗВОДНОЕ И ПИРИМИДИНОВОЕ ПРОИЗВОДНОЕ (1) | 2005 |
|
RU2330021C2 |
| ИНГИБИТОРЫ ПРОТЕИНКИНАЗ ДЛЯ УСИЛЕНИЯ РЕГЕНЕРАЦИИ ПЕЧЕНИ ИЛИ СНИЖЕНИЯ ИЛИ ПРЕДОТВРАЩЕНИЯ ГИБЕЛИ ГЕПАТОЦИТОВ | 2019 |
|
RU2822216C2 |
| ПРОИЗВОДНОЕ 1,2,4-ТРИАЗОЛОНА | 2011 |
|
RU2566754C2 |
| ПРОИЗВОДНОЕ ПИРАЗОЛА ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ | 2016 |
|
RU2687245C1 |
| АМИДЫ КОНДЕНСИРОВАННОГО ПИПЕРИДИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ ИОННЫХ КАНАЛОВ | 2014 |
|
RU2741810C2 |
| НОВОЕ ПРОИЗВОДНОЕ ПИРИДИНА И ПРОИЗВОДНОЕ ПИРИМИДИНА (3) | 2006 |
|
RU2362771C1 |
| НОВОЕ ПРОИЗВОДНОЕ ОКТАГИДРОТИЕНОХИНОЛИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ПРОИЗВОДНОЕ, И ИХ ПРИМЕНЕНИЕ | 2012 |
|
RU2573399C2 |
| НОВОЕ ПРОИЗВОДНОЕ ТЕТРАГИДРОИЗОХИНОЛИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ, ПРИМЕНЕНИЕ ЕГО И СПОСОБ ЛЕЧЕНИЯ И/ИЛИ ПРЕДОТВРАЩЕНИЯ ЗАБОЛЕВАНИЯ | 2009 |
|
RU2474575C2 |
Изобретение относится к соединению общей формулы:

или его фармацевтически приемлемой соли, где кольцо А представляет собой фенильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, или тиенильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, L представляет собой одинарную связь или группу формулы -NReСО- (где Re представляет собой атом водорода), кольцо В представляет собой С6-14 арильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, или 5-10-членную гетероциклическую группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, значения радикалов X, Y, Z, R1 и R2, R3, R4, R5 и R6 представлены в п.1 формулы изобретения, которое обладает эффектом ингибирования продукции белка Аβ или эффектом ингибирования ВАСЕ1 и применимо в качестве профилактического или терапевтического средства от нейродегенеративного заболевания, вызываемого Аβ. 2 н. и 11 з.п. ф-лы, 35 табл., 286 пр.
1. Соединение, представленное формулой (I)
[Формула 1]

или его фармацевтически приемлемая соль,
где кольцо А представляет собой фенильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, или тиенильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α,
L представляет собой одинарную связь или группу формулы -NReСО- (где Re представляет собой атом водорода),
кольцо В представляет собой С6-14 арильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, или 5-10-членную гетероциклическую группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α,
X представляет собой метиленовую группу, которая может содержать 1-2 заместителя, выбранных из группы заместителей α,
Y представляет собой одинарную связь, -NRY- (где RY представляет собой атом водорода, С6-14 арильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, или 5-10-членную гетероциклическую группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α), или атом кислорода,
Z представляет собой одинарную связь или С1-3 алкиленовую группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α,
R1 и R2 представляют собой атомы водорода и
R3, R4, R5 и R6 независимо представляют собой атом водорода, атом галогена, гидроксигруппу или С1-6 алкильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α,
[группа заместителей α: атом галогена, С1-6алкилтиогруппа, цианогруппа, С3-8циклоалкильная группа, С2-6 алкинильная группа, которая может содержать 1-3 заместителя, выбранных из группы заместителей β, С1-6алкоксигруппа, которая может содержать 1-3 заместителя, выбранных из группы заместителей β, C1-6 алкильная группа, которая может содержать 1-3 заместителя, выбранных из группы заместителей β, и 5-10-членная гетероциклическая группа, которая может содержать 1-3 заместителя, выбранных из группы заместителей β,
группа заместителей β: атом галогена, цианогруппа, гидроксигруппа, C1-6алкоксигруппа, C1-6 алкильная группа, С3-8 циклоалкильная группа и оксогруппа].
2. Соединение или его фармацевтически приемлемая соль по п.1, где Y представляет собой одинарную связь и Z представляет собой С1-3алкилен, который может содержать 1-3 заместителя, выбранных из группы заместителей α.
3. Соединение или его фармацевтически приемлемая соль по п.1, где Y представляет собой атом кислорода и Z представляет собой С1-3алкилен, который может содержать 1-3 заместителя, выбранных из группы заместителей α.
4. Соединение или его фармацевтически приемлемая соль по п.1, где Y представляет собой атом кислорода и Z представляет собой одинарную связь.
5. Соединение или его фармацевтически приемлемая соль по п.1, где Y представляет собой -NRY- (где RY представляет собой С6-14 арильную группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α, или 5-10-членную гетероциклическую группу, которая может содержать 1-3 заместителя, выбранных из группы заместителей α), и Z представляет собой одинарную связь.
6. Соединение или его фармацевтически приемлемая соль по п.1, где L представляет собой группу формулы -NReCO- (где Re представляет собой атом водорода).
7. Соединение или его фармацевтически приемлемая соль по п.1, где L представляет собой одинарную связь.
8. Соединение или его фармацевтически приемлемая соль по п.1, которое выбрано из следующих соединений:
1) (+)-N-{3-[(4aR*,8aS*)-2-амино-4,4a,5,6,7,8-гексагидробензо[d][1,3]тиазин-8а-ил]-4-фторфенил}-5-хлорпиридин-2-карбоксамид,
2) (+)-N-{3-[(4aR*,7aS*)-2-амино-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7а-ил]-4-фторфенил}-5-хлорпиридин-2-карбоксамид,
3) N-{3-[(4aR*,7aS*)-2-амино-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7а-ил]-4-фторфенил}пиридин-2-карбоксамид,
4) N-{3-[(4aR*,7aS*)-2-амино-4a,5,6,7-тетрагидро-4H-циклопента[(1][1,3]тиазин-7а-ил]-4-фторфенил}-5-фторпиридин-2-карбоксамид,
5) N-[3-((4aR*,8aS*)-2-амино-4,4a,5,6,7,8-гексагидробензо[d][1,3]тиазин-8а-ил)-4-фторфенил]-5-цианопиридин-2-карбоксамид,
6) N-[3-((4аR*,7аS*)-2-амино-4а,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7а-ил)-4-фторфенил]-5-дифторметоксипиразин-2-карбоксамид,
7) N-[3-((4aR*,7aS*)-2-амино-4a,5,6,7-тетрагидро-4H-циклопента[(1][1,3]тиазин-7а-ил)-4-фторфенил]-5-фторметоксипиразин-2-карбоксамид,
8) N-[3-((4aR*,7aS*)-2-амино-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7а-ил)-4-фторфенил]-5-цианопиридин-2-карбоксамид,
9) N-[3-((4aR*,7aS*)-2-амино-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7а-ил)-4-фторфенил]-5-фторметоксипиридин-2-карбоксамид,
10) N-[3-((4аS*,7aS*)-2-амино-4а,5,7,7а-тетрагидро-4Н-фуро[3,4-d][1,3]тиазин-7а-ил)-4-фторфенил]-5-цианопиридин-2-карбоксамид,
11) N-[3-((4аS*,7аS*)-2-амино-4а,5,7,7а-тетрагидро-4Н-фуро[3,4-d][1,3]тиазин-7а-ил)-4-фторфенил]-5-дифторметоксипиразин-2-карбоксамид,
12) N-[3-((4аS*,7аS*)-2-амино-4а,5,7,7а-тетрагидро-4Н-фуро[3,4-d][1,3]тиазин-7а-ил)-4-фторфенил]-5-хлорпиридин-2-карбоксамид,
13) N-[3-((7S*,7аS*)-2-амино-4а,5,7,7а-тетрагидро-4Н-фуро[3,4-d][1,3]тиазин-7а-ил)-4-фторфенил]-5-фторметоксипиразин-2-карбоксамид,
14) N-[3-((4аS*,8аS*)-2-амино-4а,5,7,8-тетрагидро-4Н-6-окса-3-тиа-1-азанафталин-8а-ил)-4-фторфенил]-5-цианопиридин-2-карбоксамид,
15) N-[3-((4аS*,8аS*)-2-амино-4а,5,7,8-тетрагидро-4Н-6-окса-3-тиа-1-азанафталин-8а-ил)-4-фторфенил]-5-дифторметоксипиразин-2-карбоксамид,
16) N-[3-((4аS*,8аS*)-2-амино-4а,5,7,8-тетрагидро-4Н-6-окса-3-тиа-1-азанафталин-8а-ил)-4-фторфенил]-5-хлорпиридин-2-карбоксамид,
17) (+)-N-[3-((4aR*,6S*,7aS*)-2-амино-6-метокси-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7а-ил)-4-фторфенил]-5-цианопиридин-2-карбоксамид,
18) (+)-N-[3-((4аR*,6R*,7аS*)-2-амино-6-метокси-4а,5,6,7-тетрагидро-4Н-циклопента[(1][1,3]тиазин-7а-ил)-4-фторфенил]-5-цианопиридин-2 карбоксамид,
19) (+)-N-[3-((4aR*,9aS*)-2-амино-4a,5,6,7,8,9-гексагидро-4H-циклогепта[(1][1,3]тиазин-9а-ил)-4-фторфенил]-5-цианопиридин-2-карбоксамид,
20) N-[3-((4aR*,7aS*)-2-амино-4a,5,6,7-тетрагидро-4H-циклопента[d][1,3]тиазин-7а-ил)-4-метоксифенил]-5-хлорпиридин-2-карбоксамид,
21) N-[3-((4аS*,7аS*)-2-амино-4а,5,7,7а-тетрагидро-4Н-фуро[3,4-а][1,3]тиазин-7а-ил)-4-фторфенил]-5-дифторметилпиразин-2-карбоксамид,
22) (4aR,7aS)-7a-[3-(2-фторпиридин-3-ил)фенил]-6-фенил-4,4a,5,6,7,7a-гeкcaгидpoпиppoлo[3,4-d][1,3]тиaзин-2-илaмин,
23) (4aR*,7aS*)-7a-[3-(2-фтopпиpидин-3-ил)фeнил]-6-пиpимидин-2-ил-4,4а,5,6,7,7а-гексагидропирроло[3,4-d][1,3]тиазин-2-иламин,
24) N-[3-((4aS*,5R*,7aS*)-2-aминo-5-мeтил-4a,5,7,7a-тeтpaгидpo-4H-фypo[3,4-d][1,3]тиaзин-7a-ил)-4-фтopфeнил]-5-циaнoпиpидин-2-карбоксамид,
25) N-[3-((4aS*,5R*,7aS*)-2-амино-5-метил-4a,5,7,7a-тетрагидро-4H-фypo[3,4-d][1,3]тиaзин-7a-ил)-4-фтopфeнил]-5-дифтopмeтилпиpaзин-2-карбоксамид,
26) Н-[3-((4аS*,8аS*)-2-амино-4а,5,7,8-тетрагидро-4Н-6-окса-3-тиа-1-азанафталин-8а-ил)-4-фторфенил]-5-фторметоксипиразин-2-карбоксамид,
27) N-[3-((4aS*,5R*,7aS*)-2-aминo-5-этил-4a,5,7,7a-тeтpaгидpo-4H-фypo[3,4-d][1,3]тиaзин-7a-ил)-4-фтopфeнил]-5-дифтopмeтoкcипиpaзин-2-карбоксамид,
28) N-[3-((4аS,5S,7аS)-2-амино-5-фторметил-4а,5,7,7а-тетрагидро-4Н-фypo[3,4-d][1,3]тиaзин-7a-ил)-4-фтopфeнил]-5-дифтopмeтилпиpaзин-2-карбоксамид,
29) N-[3-((4аS,5S,7аS)-2-амино-5-фторметил-4а,5,7,7а-тетрагидро-4Н-фуро[3,4-d][1,3]тиазин-7а-ил)-4-фторфенил]-5-фторметоксипиразин-2-карбоксамид,
30) N-[3-((4aS*,5S*,7aS*)-2-амино-5-фторметил-4a,5,7,7a-тетрагидро-4H-фуро[3,4-d][1,3]тиазин-7а-ил)-4-фторфенил]-5-цианопиридин-2-карбоксамид,
31) N-[3-((4аS*,5S*,8аS*)-2-амино-5-фторметил-4а,5,7,8-тетрагидро-4Н-6-окса-3-тиа-1-азанафталин-8а-ил)-4-фторфенил]-5-цианопиридин-2-карбоксамид,
32) N-[3-((4aS*,5S*,8aS*)-2-aминo-5-фтopмeтил-4a,5,7,8-тeтpaгидpo-4H-6-окса-3-тиа-1-азанафталин-8а-ил)-4-фторфенил]-5-фторметоксипиразин-2-карбоксамид,
33) N-[3-((4аS*,5S*,8аS*)-2-амино-5-фторметил-4а,5,7,8-тетрагидро-4Н-6-окса-3-тиа-1-азанафталин-8а-ил)-4-фторфенил]-5-хлорпиридин-2-карбоксамид,
34) N-[3-((4аS*,6S*,7аS*)-2-амино-6-метокси-4а,5,6,7-тетрагидро-4Н-циклопента[е][1,3]тиазин-7а-ил)-4-фторфенил]-5-дифторметилпиразин-2-карбоксамид,
35) N-[3-((4аR*,6R*,7аS*)-2-амино-6-метокси-4а,5,6,7-тетрагидро-4Н-циклопента[d][1,3]тиазин-7а-ил)-4-фторфенил]-5-дифторметилпиразин-2-карбоксамид и
36) N-[3-((4aR*,6S*,7aS*)-2-амино-6-фтор-4a,5,6,7-тетрагидро-4H-циклопента[(1][1,3]тиазин-7а-ил)-4-фторфенил]-5-дифторметилпиразин-2-карбоксамид.
9. Фармацевтическая композиция, обладающая эффектом ингибирования продукции β-амилоидного белка, содержащая соединение или его фармацевтически приемлемую соль по любому из пп.1-8 в качестве активного компонента.
10. Фармацевтическая композиция по п.9 для ингибирования продукции β-амилоидного белка.
11. Фармацевтическая композиция по п.9 для ингибирования фермента расщепления бета-сайта белка-предшественника β-амилоида типа 1 (ВАСЕ1).
12. Фармацевтическая композиция по любому из пп.9-11 для лечения нейродегенеративного заболевания.
13. Фармацевтическая композиция по п.12, где указанным нейродегенеративным заболеванием является деменция Альцгеймера или синдром Дауна.
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Производные 2-аминометил-4,4-диметил-4 @ -1,3-бензтиазина в свободном виде или в виде кислотно-аддитивной соли,обладающие антидепрессивной активностью | 1979 |
|
SU770029A1 |
| Бобров А.И | |||
| и др | |||
| Взаимодействие оксидов хинонов с тиомочевиной // Известия высших учебных заведений, Издательство Химия и химическая технология, №33(10), 1990, стр.15-18. | |||

