ОБЛАСТЬ ТЕХНИКИ
[0001] Настоящее изобретение относится к новым хинолиновым соединениям, фармацевтическим композициям, содержащим такие соединения, а также к способам их получения и применения. Эти соединения являются полезными в качестве ингибиторов S-нитрозоглутатион-редуктазы (GSNOR).
УРОВЕНЬ ТЕХНИКИ
[0002] Химическое соединение оксид азота является газом с химической формулой NO. NO является одной из немногих газообразных сигнальных молекул, известных в биологических системах, и он играет важную роль в контролировании различных биологических событий. Например, в эндотелии NO используется для сигнализации окружающих гладких мышц в стенках артериол о расслаблении, что приводит к вазодилатации и увеличенному кровотоку к гипоксическим тканям. NO также участвует в регуляции пролиферации гладких мышц, функции тромбоцитов и нейротрансмиссии, и играет роль в защитных силах организма. Хотя NO является высоко активным и имеет продолжительность существования в несколько секунд, он может легко диффундировать через мембраны и связываться со многими молекулярными мишенями. Это способствует тому, что NO является идеальной сигнальной молекулой, способной контролировать биологические события между соседними клетками и внутри клеток.
[0003] NO является свободнорадикальным газом, что делает его химически активным и неустойчивым, поэтому NO имеет короткую продолжительность существования in vivo, имея период полураспада 3-5 секунд в физиологических условиях. В присутствии кислорода, NO может объединяться с тиолами с образованием биологически важного класса устойчивых NO аддуктов, называемых S-нитрозотиолами (SNO). Постулировано, что эта устойчивая группа NO действует как источник биоактивного NO и, следовательно, является жизненно важной в здоровом и болезненном состоянии, учитывая центральную роль NO в клеточном гомеостазе (Stamler et al., Proc. Natl. Acad. Sci. USA, 89: 7674-7677 (1992)). Белковые SNO играют разнообразные роли в функционировании сердечнососудистой, дыхательной, метаболической, желудочно-кишечной, иммунной и центральной нервной системы (Foster et al., Trends in Molecular Medicine, 9(4): 160-168, (2003)). Одним из наиболее изученных SNO в биологических системах является S-нитрозоглутатион (GSNO) (Gaston et al., Proc. Natl. Acad. Sci. USA 90: 10957-10961 (1993)), выясняющийся ключевой регулятор NO передачи сигналов, поскольку он является эффективным транс-нитрозирующим агентом и поддерживает равновесие с другими S-нитрозированными белками (Liu et al., Nature, 410: 490-494 (2001)) в клетках. Учитывая это центральное положение в среде NO-SNO, GSNO представляет собой терапевтически перспективную мишень, которую следует рассматривать, когда фармацевтически оправдано модулирование NO.
[0004] В свете этого понимания GSNO как ключевого регулятора гомеостаза NO и клеточных уровней SNO, исследования были направлены на изучения эндогенной выработки белков GSNO и SNO, что происходит после выработки NO радикала ферментами синтетазы оксида азота (NOS). В последнее время было достигнуто углубленное понимание ферментативного катаболизма GSNO, который играет важную роль в управлении доступными концентрациями GSNO и, следовательно, доступными NO и SNO.
[0005] Центральным в этом понимании катаболизма GSNO является то, что исследователи недавно идентифицировали высококонсервативную S-нитрозоглутатион-редуктазу (GSNOR) (Jensen et al., Biochem J., 331: 659-668 (1998); Liu et al., (2001)). GSNOR известна также как глутатион-зависимая формальдегид-дегидрогеназа (GSH-FDH), алкоголь-дегидрогеназа 3 (ADH-3) (Uotila and Koivusalo, Coenzymes and Cofactors., D. Dolphin, ed. p.517-551 (New York, John Wiley & Sons, (1989)) и алкоголь-дегидрогеназа 5 (ADH-5). Важно, что GSNOR обладает более высокой активностью в отношении GSNO, чем другие субстраты (Jensen et al., (1998); Liu et al., (2001)) и опосредует важную денитрозирующую активность белков и пептидов у бактерий, растений и животных. GSNOR является основным GSNO-метаболизирующим ферментом у эукариотов (Liu et al., (2001)). Следовательно, GSNO может накапливаться в биологических отделах, где активность GSNOR является низкой или отсутствует (например, жидкость дыхательных путей) (Gaston et al., (1993)).
[0006] Недостаток дрожжей в GSNOR накапливает S-нитрозилированные белки, которые не являются субстратами этого фермента, что является мощным толчком к мысли, что GSNO существует в равновесии с SNO-белками (Liu et al., (2001)). Точный ферментативный контроль окружающих уровней GSNO и, следовательно, SNO-белков увеличивает вероятность того, что GSNO/GSNOR могут играть роли в физиологических и патологических функциях организма, включая защиту от нитрозативного стресса, где NO вырабатывается в избытке от физиологических потребностей. Действительно, конкретно GSNO участвует в физиологических процессах, варьирующихся от движения до дыхания (Lipton et al., Nature, 413: 171-174 (2001)), для регуляции трансмембранного регулятора фиброзно-кистозной дегенерации (Zaman et al., Biochem Biophys Res Commun, 284: 65-70 (2001)), для регуляции сосудистого тонуса, тромбоза и функции тромбоцитов (de Beider et al., Cardiovasc Res.; 28(5): 691-4 (1994)), Z. Kaposzta, et al., Circulation;, 106(24): 3057-3062, (2002)), а также в защитных силах организма (de Jesus-Berrios et al., Curr. Biol., 13: 1963-1968 (2003)). В других исследованиях было найдено, что GSNOR защищает дрожжевые клетки от нитрозативного стресса, как in vitro (Liu et al., (2001)), так и in vivo (de Jesus-Berries et al., (2003)).
[0007] Все вместе, эти данные позволяют предположить, что GSNO является первичным физиологическим лигандом для фермента S-нитрозоглутатион-редуктазы (GSNOR), который катаболизирует GSNO и, следовательно, снижает доступные SNO и NO в биологических системах (Liu et al., (2001)), (Liu et al., Cell, 116(4), 617-628 (2004)) и (Que et al., Science, 308, (5728): 1618-1621 (2005)). Поэтому этот фермент играет центральную роль в регуляции локального и системного биоактивного NO. Поскольку нарушения биодоступности NO связаны с патогенезом многих болезненных состояний, включая гипертонию, атеросклероз, тромбоз, астму, желудочно-кишечные расстройства, воспаление и рак, то средства, которые регулируют активность GSNOR, являются потенциальными терапевтическими средствами для лечения заболеваний, связанных с дисбалансом NO.
[0008] Оксид азота (NO), S-нитрозоглутатион (GSNO) и S-нитрозоглутатион-редуктаза (GSNOR) регулируют нормальную физиологию легких и участвуют в патофизиологии легких. При нормальных условиях, NO и GSNO поддерживают нормальную физиологию легких и действуют за счет своих противовоспалительных и бронхорасширяющих функций. Пониженные уровни этих медиаторов при легочных заболеваниях, таких как астма, хроническая обструктивная болезнь легких (ХОБЛ), могут возникать за счет повышенной регуляции активности фермента GSNOR. Эти пониженные уровни NO и GSNO, и, следовательно, пониженные противовоспалительные возможности, являются ключевыми событиями, которые способствуют легочным заболеваниям и которые могут быть потенциально реверсированы ингибированием GSNOR.
[0009] Было показано, что S-нитрозоглутатион (GSNO) промотирует восстановление и/или регенерацию органов млекопитающих, таких как сердце (Lima et al., 2010), кровеносные сосуды (Lima et al., 2010), кожа (Georgii et al., 2010), глаза или глазные структуры (Haq et al., 2007) и печень (Prince et al., 2010). S-нитрозоглутатион-редуктаза (GSNOR) является основным катаболическим ферментом GSNO. Предполагается, что ингибирование GSNOR увеличивает эндогенный GSNO.
[0010] Воспалительные заболевания кишечника (IBD), включая болезнь Крона и язвенный колит, являются хроническими воспалительными расстройствами желудочно-кишечного (ЖК) тракта, на которые могут оказывать влияние NO, GSNO и GSNOR. При нормальных условиях, NO и GSNO действуют для поддержания нормальной кишечной физиологии за счет противовоспалительного действия и сохранения кишечного эпителиального клеточного барьера. При IBD, пониженные уровни GSNO и NO очевидно и вероятно возникают за счет повышенной регуляции активности GSNOR. Пониженные уровни этих медиаторов способствуют патофизиологии IBD за счет разрушения эпителиального барьера путем дисрегуляции белков, участвующих в поддержании прочных эпителиальных связей. Эта дисфункция эпителиального барьера, с результирующим проникновением микроорганизмов из полости, а также общие пониженные противовоспалительные возможности в присутствии пониженных NO и GSNO, являются ключевыми событиями прогрессировании IBD, на которые можно потенциально влиять, нацеливаясь на GSNOR.
[0011] Гибель клеток является решающим событием, приводящим к клиническому проявлению гепатотоксичности лекарств, вирусов и спирта. Глутатион (GSH) является наиболее распространенной редокс-молекулой в клетках и, следовательно, наиболее важным определителем клеточного редокс-статуса. Тиолы в белках подвергаются широкому ряду обратимых редокс-модификаций при воздействии частиц активного кислорода и активных азотных частиц, что может ухудшить активность белка. Поддержание GSH печени является динамическим процессом, который достигается балансом между скоростями синтеза GSH, оттоком GSH и GSSG, реакциями GSH с активными частицами кислорода и активными азотными частицами, а также использованием GSH-пероксидазой. GSNO и GSNOR играют роли в регуляции белкового редокс-статуса за счет GSH.
[0012] Передозировки ацетаминофена являются основной причиной острой печеночной недостаточности (ALF) в Соединенных штатах, Великобритании и большинстве стран Европы. Ежегодно в этой стране с ацетаминофеном связано более 100000 звонков в токсикологический центр США, 56000 случаев экстренной медицинской помощи, 2600 госпитализаций, около 500 смертей. Около 60% выздоравливают без необходимости в трансплантации печени, 9% переносят пересадку печени, и 30% пациентов погибают из-за болезни. Уровень смертности, связанной с ацетаминофеном, превышает, по меньшей мере, в три раза количество всех остальных смертей из-за других идиосинкратических реакций на лекарства (Lee, Hepatol Res 2008; 38 (Suppl.1): S3-S8).
[0013] Пересадка печени стала основным лечением пациентов со скоротечной печеночной недостаточностью и конечной стадией хронической болезни печени, а также некоторых метаболических заболеваний печени. Поэтому потребность в трансплантации в настоящее время сильно превышает наличие донорских органов. По оценкам, в настоящее время в Службе обеспечения донорскими органами (UNOS) зарегистрировано более 18000 пациентов, и еще 9000 пациентов каждый год добавляются в список ожидания донорской печени, но для трансплантации доступно лишь менее 5000 трупных доноров.
[0014] В настоящее время в данной области существует огромная потребность в диагностировании, профилактике, улучшении и лечении медицинских состояний, связанных с увеличенным синтезом NO и/или увеличенной биоактивностью NO. Кроме того, существует значительная потребность в новых соединениях, композициях и способах предупреждения, улучшения или реверсирования других NO-связанных расстройств. В настоящем изобретении удовлетворяются эти потребности.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
[0015] В настоящем изобретении представлены новые хинолиновые соединения. Эти соединения являются полезными в качестве ингибиторов S-нитрозоглутатион-редуктазы («GSNOR»). Настоящее изобретение охватывает фармацевтически приемлемые соли, стереоизомеры, пролекарства, метаболиты и N-оксиды описанных соединений. Настоящим изобретением охватываются также фармацевтические композиции, включающие, по меньшей мере, одно соединение настоящего изобретения и, по меньшей мере, один фармацевтически приемлемый носитель.
[0016] Композиции настоящего изобретения могут быть получены в любой фармацевтически приемлемой лекарственной форме.
[0017] В настоящем изобретении представлен способ ингибирования GSNOR у пациента, нуждающегося в этом. Такой способ включает введение терапевтически эффективного количества фармацевтической композиции, включающей, по меньшей мере, один ингибитор GSNOR или его фармацевтически приемлемую соль, стереоизомер, пролекарство, метаболит или N-оксид, в комбинации, по меньшей мере, с одним фармацевтически приемлемым носителем. Ингибитор GSNOR может быть новым соединением в соответствии с настоящим изобретением, или он может быть известным соединением, которое ранее не было известно как ингибитор GSNOR.
[0018] В настоящем изобретении представлен также способ лечения расстройства, которое улучшается NO-донорной терапией у пациента, нуждающегося в этом. Такой способ включает введение терапевтически эффективного количества фармацевтической композиции, включающей, по меньшей мере, один ингибитор GSNOR или его фармацевтически приемлемую соль, стереоизомер, пролекарство, метаболит или N-оксид, в комбинации, по меньшей мере, с одним фармацевтически приемлемым носителем. Ингибитор GSNOR может быть новым соединением в соответствии с настоящим изобретением, или он может быть известным соединением, которое ранее не было известно как ингибитор GSNOR.
[0019] В настоящем изобретении представлен также способ лечения клеточного пролиферативного расстройства у пациента, нуждающегося в этом. Такой способ включает введение терапевтически эффективного количества фармацевтической композиции, включающей, по меньшей мере, один ингибитор GSNOR или его фармацевтически приемлемую соль, стереоизомер, пролекарство, метаболит или N-оксид, в комбинации, по меньшей мере, с одним фармацевтически приемлемым носителем. Ингибитор GSNOR может быть новым соединением в соответствии с настоящим изобретением, или он может быть известным соединением, которое ранее не было известно как ингибитор GSNOR.
[0020] Способы настоящего изобретения охватывают введение с одним или несколькими вторичными активными средствами. Такое введение может быть последовательным или к комплексной композиции.
[0021] Хотя в практическом осуществлении или испытании настоящего изобретения могут использоваться такие же или эквивалентные способы и материалы, как описано в настоящем документе, ниже описаны пригодные способы и материалы. Все открыто доступные публикации, патентные заявки, патенты и другие ссылки, упомянутые в настоящем документе, включены в настоящий документ путем ссылки в полном объеме. В случае противоречий, следует руководствоваться настоящим описанием, включая определения.
[0022] Как вышеизложенная сущность изобретения, так и последующее подробное описание являются иллюстративными и пояснительными, и они предназначены для предоставления дополнительных подробностей заявленных композиций и способов. Другие объекты, преимущества и новые характеристики являются легко понятными специалистам в данной области из следующего подробного описания.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
[0023] A. Обзор изобретения
[0024] До недавнего времени было известно, что S-нитрозоглутатион-редуктаза (GSNOR) окисляет формальдегид-глутатионовый аддукт, S-гидроксиметилглутатион. С тех пор GSNOR была идентифицирована в различных бактериях, дрожжах, растениях и животных, и она является очень консервативной. Белки из Е. coli, S. cerevisiae и мышиных макрофагов имеют свыше 60% идентичности аминокислотной последовательности. Активность GSNOR (то есть разложение GSNO в присутствии НАДФ в качестве необходимого кофактора) была обнаружена в Е. coli, в макрофагах мышей, в эндотелиальных клетках мышей, в клетках гладких мышц мышей, в дрожжах и в клетках HeLa, эпителия и моноцитарных клетках человека. Информация о нуклеотидной и аминокислотной последовательности GSNOR человека может быть получена из баз данных Национального центра биотехнологической информации (NCBI) под номерами доступа M29872, NM_000671. Информация о нуклеотидной и аминокислотной последовательности GSNOR мышей может быть получена из баз данных NCBI под номерами доступа NM_007410. В нуклеотидной последовательности подчеркнут сайт инициации и терминирующей сайт.CDS обозначает кодирующую последовательность. SNP обозначает однонуклеотидный полиморфизм. Другие родственные нуклеотидные и аминокислотные последовательности GSNOR, включая нуклеотидные и аминокислотные последовательности других видов, можно найти в заявке на патент США 2005/0014697.
[0025] В соответствии с настоящим изобретением показано, что GSNOR действует in vivo и in vitro для метаболизирования S-нитрозоглутатиона (GSNO) и белковых S-нитрозотиолов (SNO) для модулирования биоактивности NO за счет контролирования внутриклеточных уровней низкомолекулярных соединений-доноров NO и предотвращения достижения токсичных уровней нитрозилирования белка.
[0026] На основании этого следует, что ингибирование этого фермента усиливает биоактивность при заболеваниях, при которых показана NO-донорная терапия, ингибирует пролиферацию патологически разрастающихся клеток и увеличивает биоактивность NO при заболеваниях, где это является благотворным.
[0027] В настоящем изобретении представлены фармацевтические средства, которые являются потенциальными ингибиторами GSNOR. В частности, представлены замещенные хинолиновые аналоги, имеющие структуры, изображенные ниже (Формула I) или их фармацевтически приемлемые соли, стереоизомеры, пролекарства, метаболиты или N-оксиды.
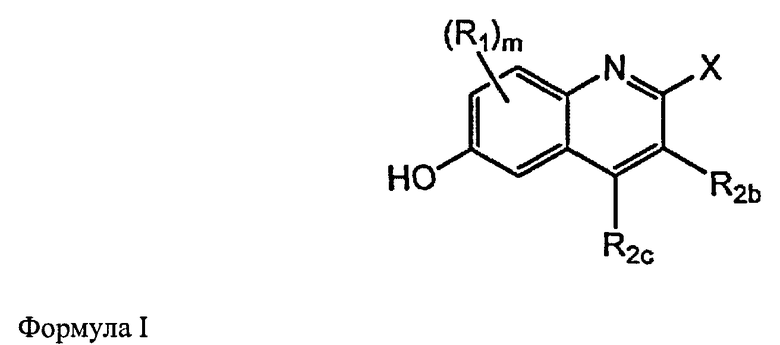
где
m выбран из группы, состоящей из 0, 1, 2, или 3;
R1 независимо выбран из группы, состоящей из хлора, фтора, брома, циано и метокси;
R2b и R2c независимо выбраны из группы, состоящей из водорода, галогена, C1-C3 алкила, фторированного C1-C3 алкила, циано, C1-C3 алкокси и N(CH3)2;
X выбран из группы, состоящей из
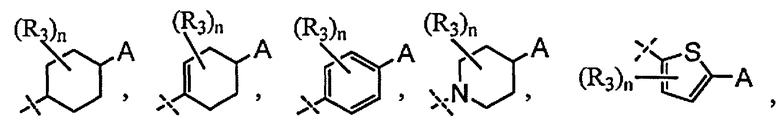
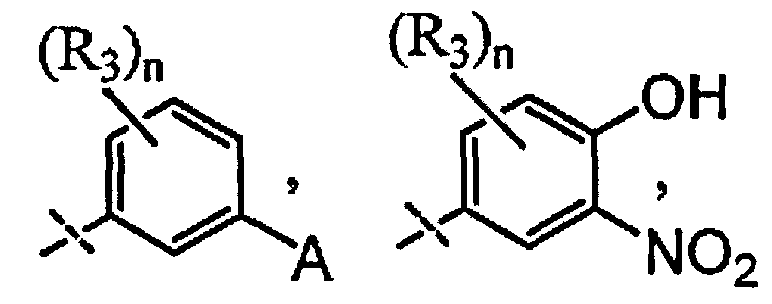 и
и 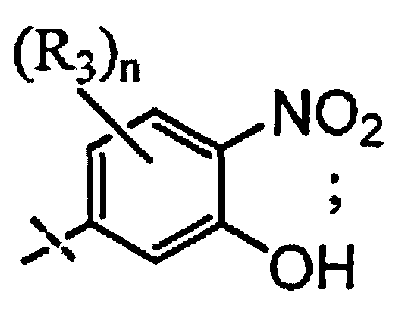
n выбран из группы, состоящей из 0, 1 и 2;
R3 независимо выбран из группы, состоящей из галогена, C1-C3 алкила, фторированного C1-C3 алкила, циано, гидрокси, C1-C3 алкокси и NR4R4′, где R4 и R4′ независимо выбраны из группы, состоящей из C1-C3 алкила, или R4, взятый вместе с R4', образует кольцо из 3-6 членов; и
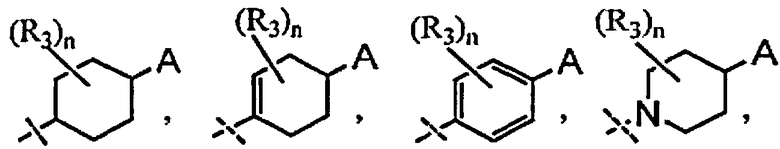
A выбран из группы, состоящей из
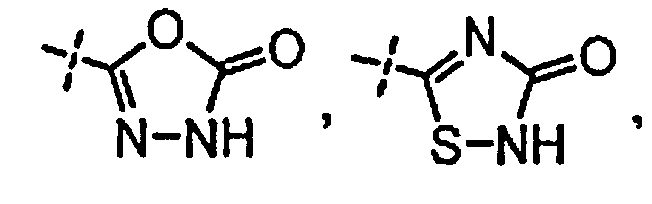 и
и 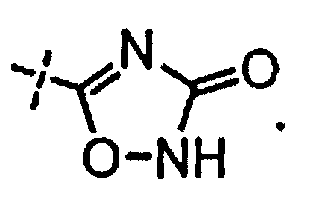
[0028] При использовании в настоящем контексте, термин «аналог» относится к соединению, имеющему аналогичную химическую структуру и функцию, что и соединения Формулы I, которое сохраняет хинолиновое кольцо.
[0029] Некоторые хинолиновые аналоги настоящего изобретения могут также существовать в различных изомерных формах, включая конфигурационные, геометрические и конформационные изомеры, а также существование в различных таутомерных формах, в частности, в тех, которые отличаются точкой присоединения атома водорода. При использовании в настоящем документе, термин «изомер» предназначен для охвата всех изомерных форм соединения, включая таутомерные формы этого соединения.
[0030] Иллюстративные соединения, имеющие асимметричные центры, могут существовать в различных энантиомерных и диастереомерных формах. Соединение может существовать в форме оптического изомера или диастереомера. Соответственно, настоящее изобретение охватывает соединения в формах их оптических изомеров, диастереомеров и их смесей, включая рацемические смеси.
[0031] Следует отметить, что если есть расхождение между изображенной структурой и названием, данным этой структуре, то следует руководствоваться изображенной структурой. Кроме того, если стереохимия структуры или части структуры не указана, например, жирными, клинообразными или пунктирными линиями, то эту структуру или часть структуры следует интерпретировать как охватывающую все стереоизомеры описанного соединения.
[0032] В. Ингибиторы S-нитрозоглутатион-редуктазы
[0033] 1. Соединения по изобретению
[0034] В одном из аспектов настоящего изобретения представлены соединения, имеющие структуру, показанную в Формуле I, или их фармацевтически приемлемые соли, стереоизомеры, пролекарства, метаболиты или N-оксиды:
где
m выбран из группы, состоящей из 0, 1, 2, или 3;
R1 независимо выбран из группы, состоящей из хлора, фтора, брома, циано и метокси;
R2b и R2c независимо выбраны из группы, состоящей из водорода, галогена, C1-C3 алкила, фторированного C1-C3 алкила, циано, C1-C3 алкокси и N(CH3)2;
X выбран из группы, состоящей из
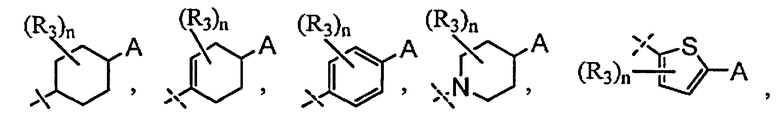
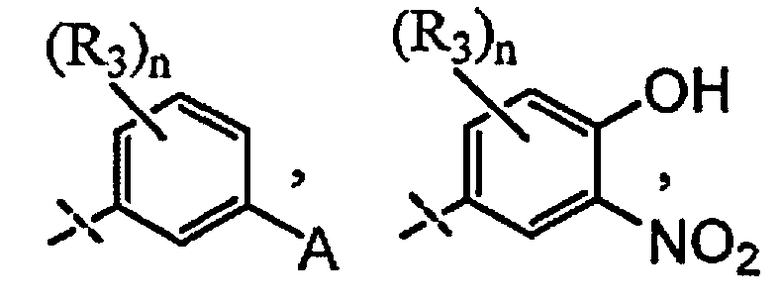 и
и 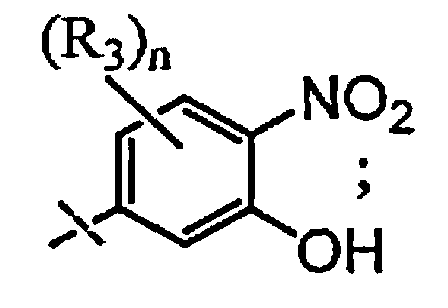
n выбран из группы, состоящей из 0, 1 и 2;
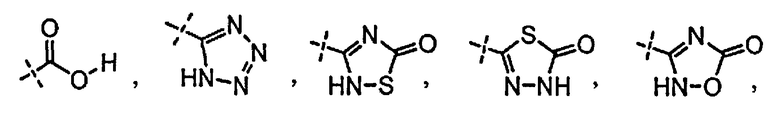
R3 независимо выбран из группы, состоящей из галогена, C1-C3 алкила, фторированного C1-C3 алкила, циано, гидрокси, C1-C3 алкокси и NR4R4′, где R4 и R4′ независимо выбраны из группы, состоящей из C1-C3 алкила, или R4, взятый вместе с R4′, образует кольцо из 3-6 членов; и
A выбран из группы, состоящей из
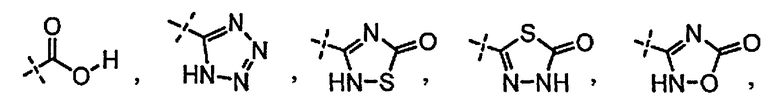
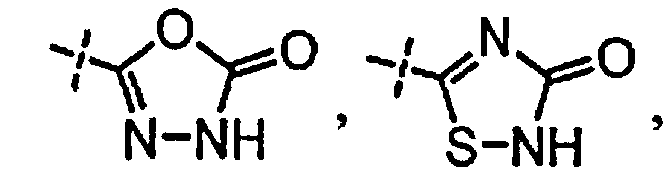 и
и 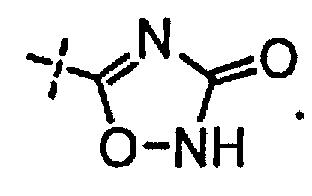
[0035] В следующем аспекте настоящего изобретения R1 независимо выбран из группы, состоящей из хлора, фтора и брома; R3 независимо выбран из группы, состоящей из галогена, C1-C3 алкила, фторированного C1-C3 алкила, циано, C1-C3 алкокси и NR4R4′, где R4 и R4′ независимо выбраны из группы, состоящей из C1-C3 алкила, или R4, взятый вместе с R4′, образует кольцо из 3-6 членов; и
X выбран из группы, состоящей из
 и
и 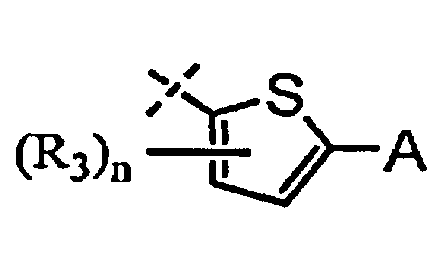 .
.
[0036] В следующем аспекте настоящего изобретения R3 независимо выбран из группы, состоящей из галогена, C1-C3 алкила, фторированного C1-C3 алкила, циано, C1-C3 алкокси и NR4R4′, где R4 и R4′ являются метилом, или, альтернативно, вместе с указанным N образуют кольцо азиридин-1-ил или морфолино.
[0037] В следующем аспекте настоящего изобретения m выбран из группы, состоящей из 0 и 1; R2b и R2c независимо выбраны из группы, состоящей из водорода, хлора, фтора, метила, трифторметила, циано, метокси и N(CH3)2; n выбран из группы, состоящей из 0 и 1; и R3 независимо выбран из группы, состоящей из фтора, хлора, брома, метила, трифторметила, циано, гидрокси, метокси и N(CH3)2.
[0038] В следующем аспекте настоящего изобретения X является 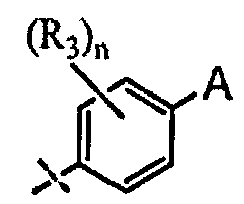 .
.
[0039] В следующем аспекте настоящего изобретения A является COOH.
[0040] В следующем аспекте настоящего изобретения применимые соединения Формулы I включают, но не ограничиваясь этим, следующие:
4-(6-гидрокси-3-метилхинолин-2-ил)бензойную кислоту;
2-(4-(1H-тетразол-5-ил)фенил)-3-метилхинолин-6-ол;
4-(6-гидроксихинолин-2-ил)бензойную кислоту;
2-(4-(1H-тетразол-5-ил)фенил)хинолин-6-ол;
1-(6-гидроксихинолин-2-ил)пиперидин-4-карбоновую кислоту;
(1r,4r)-4-(6-гидроксихинолин-2-ил)циклогексанкарбоновую кислоту;
(1s,4s)-4-(6-гидроксихинолин-2-ил)циклогексанкарбоновую кислоту;
3-хлор-4-(6-гидроксихинолин-2-ил)бензойную кислоту;
2-хлор-4-(6-гидроксихинолин-2-ил)бензойную кислоту;
2-фтор-4-(6-гидроксихинолин-2-ил)бензойную кислоту;
2-(4-(2H-тетразол-5-ил)фенил)-4-хлорхинолин-6-ол;
3-(4-(6-гидроксихинолин-2-ил)фенил)-1,2,4-оксадиазол-5-(2H)-он;
3-фтор-4-(6-гидроксихинолин-2-ил)бензойную кислоту;
4-(6-гидроксихинолин-2-ил)-3-метоксибензойную кислоту;
5-(6-гидроксихинолин-2-ил)тиофен-2-карбоновую кислоту;
4-(6-гидроксихинолин-2-ил)циклогекс-3-енкарбоновую кислоту;
4-(3-фтор-6-гидроксихинолин-2-ил)бензойную кислоту;
4-(4-хлор-3-фтор-6-гидроксихинолин-2-ил)бензойную кислоту;
4-(3-хлор-6-гидроксихинолин-2-ил)бензойную кислоту;
3-(2-фтор-4-(6-гидроксихинолин-2-ил)фенил)-1,2,4-оксадиазол-5(4H)-он;
3-(3-фтор-4-(6-гидроксихинолин-2-ил)фенил)-1,2,4-оксадиазол-5(4H)-он;
4-(4-хлор-6-гидроксихинолин-2-ил)бензойную кислоту;
2-(2-хлор-4-(2H-тетразол-5-ил)фенил)хинолин-6-ол;
5-(4-(6-гидроксихинолин-2-ил)фенил)-1,3,4-оксадиазол-2(3H)-он;
3-(диметиламино)-4-(6-гидроксихинолин-2-ил)бензойную кислоту;
4-(4-фтор-6-гидроксихинолин-2-ил)бензойную кислоту;
4-(6-гидроксихинолин-2-ил)-3-метилбензойную кислоту;
4-(3-хлор-6-гидроксихинолин-2-ил)-3-фторбензойную кислоту;
3-(4-(6-гидроксихинолин-2-ил)фенил)-1,2,4-тиадиазол-5(2H)-он;
4-(6-гидроксихинолин-2-ил)-3-(трифторметил)бензойную кислоту;
4-(6-гидрокси-3-(трифторметил)хинолин-2-ил)бензойную кислоту;
2-(4-карбоксифенил)-6-гидроксихинолин 1-оксид;
5-(4-(6-гидроксихинолин-2-ил)фенил)-1,3,4-тиадиазол-2(3H)-он;
5-(4-(6-гидроксихинолин-2-ил)фенил)-1,2,4-оксадиазол-3(2H)-он;
(1r,4r)-4-(3-хлор-6-гидроксихинолин-2-ил)циклогексанкарбоновую кислоту;
(1s,4s)-4-(3-хлор-6-гидроксихинолин-2-ил)циклогексанкарбоновую кислоту;
3-хлор-4-(4-фтор-6-гидроксихинолин-2-ил)бензойную кислоту;
2-(5-(2H-тетразол-5-ил)тиофен-2-ил)хинолин-6-ол;
5-(4-(6-гидроксихинолин-2-ил)фенил)-1,2,4-тиадиазол-3(2H)-он;
3-фтор-4-(4-фтор-6-гидроксихинолин-2-ил)бензойную кислоту;
1-(6-гидрокси-3-(трифторметил)хинолин-2-ил)пиперидин-4-карбоновую кислоту;
4-(5-хлор-6-гидроксихинолин-2-ил)бензойную кислоту;
(1r,4r)-4-(6-гидрокси-3-(трифторметил)хинолин-2-ил)циклогексанкарбоновую кислоту;
(1s,4s)-4-(6-гидрокси-3-(трифторметил)хинолин-2-ил)циклогексанкарбоновую кислоту;
4-(5-бром-6-гидроксихинолин-2-ил)бензойную кислоту;
3-бром-4-(6-гидроксихинолин-2-ил)бензойную кислоту;
4-(4-(диметиламино)-6-гидроксихинолин-2-ил)бензойную кислоту;
4-(4-фтор-6-гидроксихинолин-2-ил)-3-метоксибензойную кислоту;
3-циано-4-(6-гидроксихинолин-2-ил)бензойную кислоту;
2-(4-карбокси-2-хлорфенил)-6-гидроксихинолин 1-оксид;
4-(4-амино-6-гидроксихинолин-2-ил)бензойную кислоту;
4-(3-циано-6-гидроксихинолин-2-ил)бензойную кислоту;
4-(5-фтор-6-гидроксихинолин-2-ил)бензойную кислоту;
4-(8-фтор-6-гидроксихинолин-2-ил)бензойную кислоту;
3-гидрокси-4-(6-гидроксихинолин-2-ил)бензойную кислоту; и
3-фтор-4-(5-фтор-6-гидроксихинолин-2-ил)бензойную кислоту.
[0041] Если показано, что связь с заместителем пересекает связь, связывающую два атома в кольце, то такой заместитель может быть связан с любым атомом в этом кольце. Если заместитель перечислен без указания атома, через который такой заместитель связан с остальной частью соединения данной формулы, то такой заместитель может быть связан через любой атом в таком заместителе. Допустимы комбинации заместителей и/или переменных, но только если такие комбинации приводят к устойчивым соединениям.
[0042] Соединения, описанные в настоящем документе, могут иметь асимметричные центры. Соединения настоящего изобретения, содержащие асимметрично замещенный атом, могут быть выделены в оптически активных или рацемических формах. В данной области хорошо известно, как получить оптически активные формы, как, например, разделением рацемических форм или синтезом из оптически активных исходных материалов. Многие геометрические изомеры олефинов, двойных связей C=N и тому подобные также могут присутствовать в соединениях, описанных в настоящем документе, и все устойчивые изомеры таких соединений входят в настоящее изобретение. Описаны цис- и транс-геометрические изомеры соединений настоящего изобретения, и они могут быть выделены в виде смеси изомеров или как отдельные изомерные формы. Подразумеваются все хиральные, диастереомерные, рацемические и геометрические изомерные формы структур, если специально не указана определенная стереохимия или изомерная форма. Все таутомеры изображенных или описанных соединений также считаются частью настоящего изобретения.
[0043] Следует понимать, что изомеры, возникающие из такой асимметрии (например, все энантиомеры и диастереомеры) включены в рамки настоящего изобретения, если не указано иное. Такие изомеры могут быть получены, в основном, в чистой форме классическими способами разделения и путем стереохимически контролируемого синтеза. Более того, структуры и другие соединения и фрагменты, описанные в настоящей заявке, также включают все их таутомеры. Алкены могут включать как E-, так и Z-геометрию, где это уместно.
[0044] 2. Иллюстративные соединения
[0045] В Примерах 1-56 перечислены иллюстративные новые хинолиновые аналоги Формулы I. Синтетические способы, которые могут использоваться для получения каждого соединения, детализированы в Примерах 1-56, со ссылкой на схемы синтеза, изображенные перед этим в Примере 1, и со ссылкой на промежуточные соединения, описанные в Примере 57. Подтверждающие данные масс-спектрометрии и/или данные протонного ЯМР для каждого соединения также включены в Примеры 1-56. Активность ингибитора GSNOR была определена анализом, описанным в Примере 58, и были получены значения IC50. Соединения-ингибиторы GSNOR в Примерах 1-56 имеют IC50 около <10 мкМ. Соединения-ингибиторы GSNOR в Примерах 1-4, 6, 8, 10-14, 16-35, 37-43, 45-50 и 52-56 имеют IC50 около <0,5 мкМ. Соединения-ингибиторы GSNOR в Примерах 1-4, 8, 10-14, 17-28, 30, 31, 37, 40-41, 43, 46, 48-49 и 52-56 имеют IC50 около <0,1 мкМ.
[0046] C. Определения
[0047] При использовании в настоящем документе, термин «около» является понятным для специалистов в данной области и варьируется до некоторого предела в зависимости от контекста, в котором он используется. Если применение этого термина не понятно для специалиста в данной области в контексте, в котором он используется, то термин «около» обозначает среднее значение плюс или минус 10% от конкретного значения.
[0048] Термин «ацил» включает соединения и фрагменты, которые содержат ацетиловый радикал (CH3CO-) или карбонильную группу, к которой присоединен низший алкиловый остаток прямого или разветвленного строения.
[0049] Термин «алкил», используемый в настоящем документе, относится к насыщенному углеводороду прямого или разветвленного строения, имеющему указанное количество углеродных атомов. Например, (C1-C6) алкил включает, но не ограничиваясь этим, метил, этил, пропил, изопропил, бутил, втор-бутил, трет-бутил, пентил, изопентил, неопентил, гексил, изогексил и неогексил. Алкиловая группа может быть незамещенной или необязательно замещенной одним или несколькими заместителями, как описано в настоящем документе.
[0050] Термин «алкенил», используемый в настоящем документе, относится к ненасыщенному углеводороду прямого или разветвленного строения, имеющему указанное количество углеродных атомов и, по меньшей мере, одну двойную связь. Примеры (C2-C8) алкениловой группы включают, но не ограничиваясь этим, этилен, пропилен, 1-бутилен, 2-бутилен, изобутилен, втор-бутилен, 1-пентен, 2-пентен, изопентен, 1-гексен, 2-гексен, 3-гексен, изогексен, 1-гептен, 2-гептен, 3-гептен, изогептен, 1-октен, 2-октен, 3-октен, 4-октен и изооктен. Алкениловая группа может быть незамещенной или необязательно замещенной одним или несколькими заместителями, как описано в настоящем документе.
[0051] Термин «алкинил», используемый в настоящем документе, относится к ненасыщенному углеводороду прямого или разветвленного строения, имеющему указанное количество углеродных атомов и, по меньшей мере, одну тройную связь. Примеры (C2-C8) алкиниловой группы включают, но не ограничиваясь этим, ацетилен, пропин, 1-бутан, 2-бутин, 1-пентин, 2-пентин, 1-гексин, 2-гексин, 3-гексин, 1-гептан, 2-гептин, 3-гептан, 1-октан, 2-октин, 3-октан и 4-октин. Алкиниловая группа может быть незамещенной или необязательно замещенной одним или несколькими заместителями, как описано в настоящем документе.
[0052] Термин «алкокси», используемый в настоящем документе, относится к -O-алкиловой группе, имеющей указанное количество углеродных атомов. Например, (C1-C6) алкокси-группа включает -O-метил, -O-этил, -O-пропил, -O-изопропил, -O-бутил, -O-втор-бутил, -O-трет-бутил, -O-пентил, -O-изопентил, -O-неопентил, -O-гексил, -O-изогексил и -O-неогексил.
[0053] Термин «аминоалкил», используемый в настоящем документе, относится к алкиловой группе (обычно из одного-шести углеродных атомов), где один или несколько водородных атомов C1-C6 алкиловой группы замещены амином формулы -N(Rc)2, где Rc в каждом случае независимо является -H или (C1-C6)алкилом. Примеры аминоалкиловых групп включают, но не оганичиваясь этим, -CH2NH2, -CH2CH2NH2, -CH2CH2CH2NH2, -CH2CH2CH2CH2NH2, -CH2CH2CH2CH2CH2NH2, -CH2CH2CH2CH2CH2CH2NH2, -CH2CH2CH2N(CH3)2, трет-бутиламинометил, изопропиламинометил и тому подобные.
[0054] Термин «арил», используемый в настоящем документе, относится к 5-14-членной моноциклической, бициклической или трициклической ароматической кольцевой системе. Примеры ариловой группы включают фенил и нафтил. Ариловая группа может быть незамещенной или необязательно замещенной одним или несколькими заместителями, как описано ниже в настоящем документе. Примеры ариловых групп включают фенил или ариловые гетероциклы, такие как пиррол, фуран, тиофен, тиазол, изотиазол, имидазол, триазол, тетразол, пиразол, оксазол, изоксазол, пиридин, пиразин, пиридазин и пиримидин, и тому подобные.
[0055] При использовании в настоящем документе, термин «биоактивность» обозначает действие на один или несколько клеточных или внеклеточных процессов (например, за счет связывания, передачи сигнала и так далее), которое может влиять на физиологические или патофизиологические процессы.
[0056] Термин «карбонил» включает соединения и фрагменты, которые содержат углерод, связанный двойной связью с атомом кислорода. Примеры фрагментов, содержащих карбонил, включают, но не ограничиваясь этим, альдегиды, кетоны, карбоновые кислоты, амиды, сложные эфиры, ангидриды и так далее.
[0057] Термин «карбокси» или «карбоксил» означает группу -COOH или карбоновую кислоту.
[0058] «Кислотный фрагмент», используемый в настоящем документе, обозначает карбоновую кислоту или биоизостер карбоновой кислоты. Биоизостеры являются заместителями или группами с аналогичными физическими или химическими свойствами, которые обеспечивают, в основном, аналогичные биологические свойства химического соединения. Обзор биоизостеров представлен в публикации J. Med. Chem, 2011, 54, 2529-2591. Примеры «кислотного фрагмента» включают, но не ограничиваясь этим,
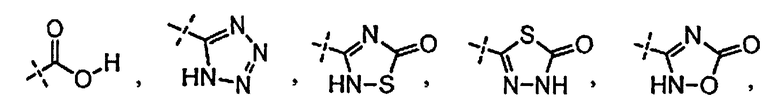
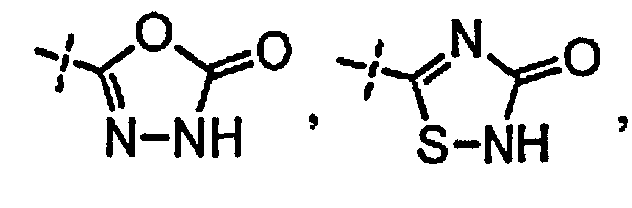 и
и 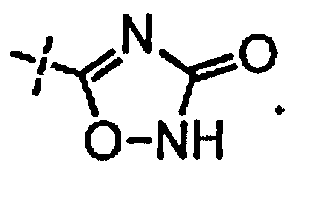
[0059] «Фармакофор» определяется как «набор структурных особенностей молекулы, который распознается на рецепторном сайте и отвечает за биологическую активность этой молекулы» (Gund, Prog. Mol. Subcell. Biol., 5: pp 117-143 (1977)).
[0060] Термин «Cm-Cn» означает количество от «m» углеродных атомов до «n» углеродных атомов. Например, термин «C1-C6» означает от одного до шести углеродных атомов (C1, C2, C3, C4, C5 или C6). Термин «C2-C6» включает от двух до шести углеродных атомов (C2, C3, C4, C5 или C6) Термин «C3-C6» включает от трех до шести углеродных атомов (C3, C4, C5 или C6)
[0061] Термин «циклоалкил», используемый в настоящем документе, относится к 3-14-членной насыщенной или ненасыщенной неароматической моноциклической, бициклической или трициклической углеводородной кольцевой системе. В этот класс включены циклоалкиловые группы, которые являются конденсированными с бензольным кольцом. Иллюстративные циклоалкиловые группы включают, но не ограничиваясь этим, циклопропил, циклобутил, циклобутенил, циклопентил, циклопентенил, циклопентадиенил, циклогексил, циклогексенил, 1,3-циклогексадиенил, циклогептил, циклогептенил, 1,3-циклогептадиенил, 1,4-циклогептадиенил, 1,3,5-циклогептатриенил, циклооктил, циклооктенил, 1,3-циклооктадиенил, 1,4-циклооктадиенил, 1,3,5-циклооктатриенил, декагидронафталин, октагидронафталин, гексагидронафталин, октагидроинден, гексагидроинден, тетрагидроинден, декагидробензоциклогептен, октагидробензоциклогептен, гексагидробензоциклогептен, тетрагидробензоциклогептен, додекагидрогептален, декагидрогептален, октагидрогептален, гексагидрогептален, тетрагидрогептален, (1s,3s)-бицикло[1.1.0]бутан, бицикло[1.1.1]пентан, бицикло[2.1.1]гексан, бицикло[2.2.1]гептан, бицикло[2.2.2]октан, бицикло[3.1.1]гептан, бицикло[3.2.1]октан, бицикло[3.3.1]нонан, бицикло[3.3.2]декан, бицикло[3.3.]ундекан, бицикло[4.2.2]декан и бицикло[4.3.1]декан. Циклоалкиловая группа может быть незамещенной или необязательно замещенной одним или несколькими заместителями, как описано ниже в настоящем документе.
[0062] Термин «галоген» включает фтор, бром, хлор, йод и так далее.
[0063] Термин «галоалкил», используемый в настоящем документе, относится к C1-C6 алкиловой группе, где один или несколько атомов водорода C1-C6 алкиловой группы замещены атомами галогена, которые могут быть одинаковыми или различными. Примеры галоалкиловых групп включают, но не ограничиваясь этим, трифторметил, 2,2,2-трифторэтил, 4-хлорбутил, 3-бромпропил, пентахлорэтил и 1,1,1-трифтор-2-бром-2-хлорэтил.
[0064] Термин «гетероалкил», самостоятельно или в комбинации с другим термином, означает, если не указано иное, устойчивый алкил прямого или разветвленного строения, или их комбинацию, состоящую из атомов углерода и от одного до трех гетероатомов, выбранных из группы, состоящей из O, N и S, и где атомы азота и серы могут быть необязательно окислены, а атом азота может быть необязательно кватернизован. Гетероатом(-ы) O, N и S могут находиться в любом положении гетероалкиловой группы. Примеры включают -CH2-CH2-O-CH3, -CH2-CH2-NH-CH3, -CH2-CH2-N(CH3)-CH3, CH2-S-CH2-CH3, -CH2-CH2-S(O)-CH3, -CH2-CH2-S(O)2-CH3 и -CH2-CH=N-OCH3. Последовательно могут располагаться до двух гетероатомов, например, -CH2-NH-OCH3. При использовании приставки, такой как (C2-C8), для обозначения гетероалкиловой группы, количество атомов углерода (в этом примере от 2 до 8) включает также гетероатомы. Например, C2-гетероалкиловая группа включает, например, -CH2OH (один атом углерода и один гетероатом, заменяющий атом углерода) и -CH2SH.
[0065] Для дополнительной иллюстрации определения гетероалкиловой группы, где гетероатомом является кислород, гетероалкиловой группой может быть оксиалкиловая группа. Например, (C2-C5) оксиалкил включает, например, -CH2-O-CH3 (C3-оксиалкиловая группа с двумя атомами углерода и одним кислородом, заменяющим атом углерода), -CH2CH2CH2CH2OH, -OCH2CH2OCH2CH2OH, -OCH2CH(OH)CH2OH и тому подобные.
[0066] Термин «гетероарил», используемый в настоящем документе, относится к ароматическому гетероциклическому кольцу из 5-14 членов, имеющему, по меньшей мере, один гетероатом, выбранный из азота, кислорода и серы, и содержащему, по меньшей мере, 1 атом углерода, включая моноциклические, бициклические и трициклические кольцевые системы. Иллюстративными гетероарилами являются триазолил, тетразолил, оксадиазолил, пиридил, фурил, бензофуранил, тиенил, бензотиенил, хинолинил, пирролил, индолил, оксазолил, бензоксазолил, имидазолил, бензимидазолил, тиазолил, бензотиазолил, изоксазолил, пиразолил, изотиазолил, пиридазинил, пиримидинил, пиразинил, триазинил, циннолинил, фталазинил, хиназолинил, пиримидил, азепинил, оксепинил, хиноксалинил и оксазолил. Гетероариловая группа может быть незамещенной или необязательно замещенной одним или несколькими заместителями, как описано ниже в настоящем документе.
[0067] При использовании в настоящем документе, термин «гетероатом» включает кислород (O), азот (N) и серу (S).
[0068] При использовании в настоящем документе, термин «гетероцикл» относится к 3-14-членным кольцевым системам, которые являются насыщенными, ненасыщенными или ароматическими, и которые содержат от 1 до 4 гетероатомов, независимо выбранных из азота, кислорода и серы, и где гетероатомы азота и серы могут быть необязательно окислены, а гетероатом азота может быть необязательно кватернизован, включая моноциклические, бициклические и трициклические кольцевые системы. Бициклические и трициклические кольцевые системы могут охватывать гетероциклы или гетероарилы, конденсированные с бензольным кольцом. Гетероцикл может быть присоединен через любой гетероатом или атом углерода, где это химически приемлемо. Гетероциклы включают гетероарилы, как определено выше. Иллюстративные примеры гетероциклов включают, но не ограничиваясь этим, азиридинил, оксиранил, тииранил, триазолил, тетразолил, азиринил, диазиридинил, диазиринил, оксазиридинил, азетидинил, азетидинонил, оксетанил, тиетанил, пиперидинил, пиперазинил, морфолинил, пирролил, оксазинил, тиазинил, диазинил, диоксанил, триазинил, тетразинил, имидазолил, тетразолил, пирролидинил, изоксазолил, фуранил, фуразанил, пиридинил, оксазолил, бензоксазолил, бензизоксазолил, тиазолил, бентиазолил, тиенил, пиразолил, триазолил, пиримидинил, бензимидазолил, изоиндолил, индазолил, бензодиазолил, бензотриазолил, бензоксазолил, бензизоксазолил, пуринил, индолил, изохинолинил, хинолинил и хиназолинил. Гетероциклическая группа может быть незамещенной или необязательно замещенной одним или несколькими заместителями, как описано ниже в настоящем документе.
[0069] Термин «гетероциклоалкил», самостоятельно или в комбинации с другими терминами, представляет, если не указано иное, циклические версии «гетероалкила». Кроме того, гетероатом может занимать положение, в котором этот гетероцикл присоединен к остальной части молекулы. Примеры гетероциклоалкила включают 1-(1,2,5,6-тетрагидропиридил), 1-пиперидинил, 2-пиперидинил, 3-пиперидинил, 4-морфолинил, 3-морфолинил, тетрагидрофуран-2-ил, тетрагидрофуран-3-ил, тетрагидротиен-2-ил, тетрагидротиен-3-ил, 1-пиперазинил, 2-пиперазинил и тому подобные.
[0070] Термин «гидроксиалкил», используемый в настоящем документе, относится к алкиловой группе, имеющей указанное количество атомов углерода, где один или несколько атомов водорода в алкиловой группе замещено группой -OH. Примеры гидроксиалкиловых групп включают, но не ограничиваясь этим, -CH2OH, -CH2CH2OH, CH2CH2CH2OH, CH2CH2CH2CH2OH, -CH2CH2CH2CH2CH2OH, -CH2CH2CH2CH2CH2CH2OH и их разветвленные варианты.
[0071] Термин «гидрокси» или «гидроксил» включает группы с OH или -O-.
[0072] При использовании в настоящем документе, N-оксид или аминоксид, относится к соединению, полученному из третичного амина присоединением одного атома кислорода к атому азота, R3N+-O-. Путем расширения, этот термин включает аналоги, полученные из первичных и вторичных аминов.
[0073] При использовании в настоящем документе и если не указано иное, термин «стереоизомер» обозначает один стереоизомер соединения, который является, в основном, чистым от других стереоизомеров этого соединения. Например, стереоизомерно чистое соединение, имеющее один хиральный центр, является, в основном, чистым от противоположного энантиомера этого соединения. Стереоизомерно чистое соединение, имеющее два хиральных центра, являются, в основном, чистым от других диастереомеров этого соединения. В некоторых вариантах воплощения стереоизомерно чистое соединение включает более чем около 80% по весу одного стереоизомера этого соединения и менее чем около 20% по весу других стереоизомеров этого соединения, например, более чем около 90% по весу одного стереоизомера этого соединения и менее чем около 10% по весу других стереоизомеров этого соединения, или более чем около 95% по весу одного стереоизомера этого соединения и менее чем около 5% по весу других стереоизомеров этого соединения, или более чем около 97% по весу одного стереоизомера этого соединения и менее чем около 3% по весу других стереоизомеров этого соединения.
[0074] При использовании в настоящем документе, «белок» используется как синоним с «пептидом», «полипептидом» или «пептидным фрагментом». «Очищенный» полипептид, белок, пептид или пептидный фрагмент является, в основном, чистым от клеточного материала или других загрязняющих белков из клеток, тканей или бесклеточного источника, из которого получена данная аминокислотная последовательность, или, в основном, чистым от химических предшественников или других химических веществ при химическом синтезировании.
[0075] При использовании в настоящем документе, термин «модулирует» относится к увеличению или снижению уровней пептида или полипептида, или к увеличению или снижению устойчивости или активности пептида или полипептида. Термин «ингибирует» относится к снижению уровней пептида или полипептида, или к снижению устойчивости или активности пептида или полипептида. В предпочтительных вариантах пептидом, который модулируется или ингибируется, является S-нитрозоглутатион (GSNO) или белковые S-нитрозотиолы (SNO).
[0076] При использовании в настоящем документе, термины «оксид азота» и «NO» охватывают незаряженный оксид азота и заряженные частицы оксида азота, в частности, включая ион нитрозония (NO+) и нитроксильный ион (NO-). Химически активная форма оксида азота может предоставляться газообразным оксидом азота. Соединения, имеющие структуру X-NOy, где X является фрагментом, высвобождающим, доставляющим или переносящим оксид азота, включают любые и все такие соединения, которые предоставляют оксид азота в заданном месте его действия в форме, активной для заданной цели, a Y равен 1 или 2.
[0077] «Заживление» обозначает восстановление структурной целостности и нормальной физиологической функции. В качестве примера, эпителий ротовой полости и верхних дыхательных путей может заживляться после травмы, причиненной термическим повреждением или вирусной инфекцией.
[0078] «Регенерация» означает способность органа осуществлять не злокачественный клеточный, сосудистый и стромальный рост для восстановления функциональных тканей органа.
В качестве примера, заживление раны включает регенерацию тканей и органов (например, кожи, слизистой оболочки желудка и кишечника), также как происходит с костями после перелома и печенью после частичного хирургического удаления и воздействия инфекционного или токсического инсульта.
[0079] При использовании в настоящем документе, термин «фармацевтически приемлемый» означает одобренный регулирующим органом федерального или государственного руководства, или перечисленный в Фармакопее США или других общепринятых фармакопеях для применения для животных, и, более предпочтительно, для людей. Термин «носитель» относится к разбавителю, адъюванту, формообразующему средству или среде, с которыми вводится терапевтическое средство, и включает, но не ограничиваясь этим, стерильные жидкости, такие как вода и масла.
[0080] «Фармацевтически приемлемая соль» или «соль» соединения настоящего изобретения является продуктом описанного соединения, который содержит ионную связь и обычно получается по реакции описанного соединения с кислотой или основанием, и который пригоден для введения пациенту. Фармацевтически приемлемая соль может включать, но не ограничиваясь этим, соли присоединения кислот, включая гидрохлориды, гидробромиды, фосфаты, сульфаты, гидросульфаты, алкилсульфонаты, арилсульфонаты, арилалкилсульфонаты, ацетаты, бензоаты, цитраты, малеаты, фумараты, сукцинаты, лактаты и тартраты; катионы щелочных металлов, таких как Li, Na и K, соли щелочноземельных металлов, таких как Mg или Ca, или соли органических аминов.
[0081] «Фармацевтическая композиция» является композицией, включающей описанные соединения в форме, пригодной для введения пациенту. Фармацевтическая композиция настоящего изобретения предпочтительно составляется так, чтобы быть совместимой с заданным способом введения. Примеры способов введения включают, но не ограничиваясь этим, пероральное и парентеральное, например, внутривенное, интрадермальное, подкожное, ингаляционное, локальное, трансдермальное, трансмукозальное и ректальное введение.
[0082] Термин «замещенный», при использовании в настоящем документе, обозначает, что любой один или несколько атомов водорода у обозначенного атома замещены выбором из указанной группы, при условии, что не превышается нормальная валентность обозначенного атома, и что такое замещение приводит к устойчивому соединению. Если заместителем является кето (то есть =O), то замещаются 2 атома водорода у указанного атома. Кольцевые двойные связи, при использовании в настоящем документе, являются двойными связями, которые образованы между двумя соседними кольцевыми атомами (например, C=C, C=N или N=N).
[0083] Заместители для групп, которые упоминаются как алкил, гетероалкил, алкилен, алкенил, алкинил, циклоалкил, гетероциклоалкил, циклоалкенил и гетероциклоалкенил, могут быть выбраны из различных групп, включая -ORd′, =O, =NRd′, =N-ORd′, -NRd′Rd″, -SRd′, -гало, SiRd′Rd″Rd′″, -OC(O)Rd′, -C(O)Rd′, -CO2Rd′, -CONRd′Rd″, -OC(O)NRd′Rd″, -NRd″C(O)Rd′, -NRd′″C(O)NRd′Rd″, -NRd′″SO2NRd′Rd″, -NRd″CO2Rd′, -NHC(NH2)=NH, -NRa′C(NH2)=NH, -NHC(NH2)=NRd′, -S(O)Rd′,-SO2Rd′, -SO2NRd′Rd″, -NRd″SO2Rd′, -CN и -NO2, в количестве, варьирующемся от нуля до трех, где эти иллюстративные группы, например, имеют ноль, один или два заместителя.
[0084] Rd′, Rd″ и Rd′″, каждый независимо, относятся к водороду, незамещенному (C1-C8)алкилу, незамещенному гетеро(C1-C8)алкилу, незамещенному арилу и арилу, замещенному одним-тремя заместителями, выбранными из галогена, незамещенного алкила, незамещенного алкокси, незамещенного тиоалкокси и незамещенного арил(C1-C4)алкила. Если Rd′ и Rd″ присоединены к одному атому азота, они могут объединяться с этим атомом азота с образованием 5-, 6- или 7-членного кольца. Например, NRd′Rd″ может представлять 1-пирролидинил или 4-морфолинил.
[0085] Как правило, алкиловая или гетероалкиловая группа имеет от нуля до трех заместителей, где эти группы, имеющие два или менее заместителей, являются иллюстративными для настоящего изобретения. Алкиловый или гетероалкиловый радикал может быть незамещенным или монозамещенным.
В некоторых вариантах алкиловый или гетероалкиловый радикал является незамещенным.
[0086] Иллюстративные заместители для алкиловых и гетероалкиловых радикалов включают, но не ограничиваясь этим -ORd′, =O, =NRd′, =N-ORd′, -NRd′Rd″, -SRd′, -гало, SiRd′Rd″Rd′″, -OC(O)Rd′, -C(O)Rd′, -CO2Rd′, -CONRd′Rd″, -OC(O)NRd′Rd″, -NRd″C(O)Rd′, -NRd′″C(O)NRd′Rd″, -NRd′″SO2NRd′Rd″, -NRd″CO2Rd′, -NHC(NH2)=NH, -NRa′C(NH2)=NH, -NHC(NH2)=NRd′, -S(O)Rd′, -SO2Rd′, -SO2NRd′Rd″, -NRd″SO2Rd′, -CN и -NO2, где Rd′, Rd″ и Rd′″ являются такими, как определено выше. Типичные заместители могут быть выбраны из: -ORd′, =O, -NRd′Rd″, -галогена, -OC(O)Rd′, -CO2Rd′, C(O)NRd′Rd″, -OC(O)NRd′Rd″, -NRd″C(O)Rd′, NRd″CO2Rd′, -NRd′″SO2NRd′Rd″, -SO2Rd′, -SO2NRd′Rd″, -NRd″SO2Rd′, -CN и -NO2.
[0087] Точно так же, заместителя для ариловых и гетероариловых групп варьируются и выбираются из: -галогена, -ORe′, -OC(O)Re′, -NRe′Re″, -SRe′, -Re′, -CN, -NO2, -CO2Re′, -C(O)NRe′Re″, -C(O)Re′, -OC(O)NRe′Re″, -NRe″C(O)Re′, -NRe″CO2Re′, -NRe′″C(O)NRe′Re″, -NRe′″SO2NRe′Re″, -NHC(NH2)=NH, NRe′C(NH2)=NH, -NH-C(NH2)NRe′, -S(O)Re′, SO2Re′, -SO2NRe′Re″, -NRe″SO2Re′, -N3, -CH(Ph)2, перфторалкокси и перфтор(C1-C4)алкила, в количестве, варьирующемся от нуля до общего количества открытых валентностей ароматической кольцевой системы.
[0088] Re′, Re″ и Re′″ независимо выбраны из водорода, незамещенного (C1-C8)алкила, незамещенного гетеро(C1-C8)алкила, незамещенного арила, незамещенного гетероарила, незамещенного арил(C1-C4)алкила и незамещенного арилокси(C1-C4)алкила. Как правило, ариловая или гетероариловая группа имеет от нуля до трех заместителей, где эти группы, имеющие два или менее заместителей, являются иллюстративными для настоящего изобретения. В одном варианте настоящего изобретения ариловая или гетероариловая группа является незамещенной или монозамещенной. В другом варианте ариловая или гетероариловая группа является незамещенной.
[0089] Два заместителя у соседних атомов арилового или гетероарилового кольца в гетероариловой группе, как описано в настоящем документе, могут быть необязательно замещены заместителем формулы -T-C(O)-(CH2)q-U-, где T и U независимо являются -NH-, -O-, -CH2- или одинарной связью, a q является целым числом от 0 до 2. Альтернативно, два заместителя у соседних атомов арилового или гетероарилового кольца могут быть необязательно замещены заместителем формулы -J-(CH2)r-K-, где J и K независимо являются -CH2-, -O-, -NH-, -S-, -S(O)-, -S(O)2-, -S(O)2NRf′- или одинарной связью, а r является целым числом от 1 до 3. Одна из одинарных связей образованного таким образом нового кольца может быть необязательно заменена двойной связью. Альтернативно, два заместителя у соседних атомов арилового или гетероарилового кольца могут быть необязательно замещены заместителем формулы -(CH2)s-X-(CH2)t-, где s и t независимо являются целыми числами от 0 до 3, и X является -O-, -NRf′-, -S-, -S(O)-, -S(O)2- или -S(O)2NRa′-. Заместитель Rf′ в -NRf′- и -S(O)2NRf′- выбран из водорода или незамещенного (C1-C6)алкила.
[0090] «Устойчивое соединение» и «устойчивая структура» обозначают соединение, которое является достаточно прочным, чтобы выдерживать его выделение из реакционной смеси до полезной степени чистоты и составление в композицию эффективного терапевтического агента.
[0091] При использовании в настоящем документе, термин «терапевтически эффективное количество», в общем, обозначает количество, необходимое для улучшения, по меньшей мере, одного симптома расстройства, подлежащего предупреждению, снижению или лечению, как описано в настоящем документе. Выражение «терапевтически эффективное количество» в отношении ингибиторов GSNOR настоящего изобретения означает дозу ингибитора GSNOR, которая обеспечивает определенную фармакологическую реакцию, для которой вводится ингибитор GSNOR, у значимого количества пациентов, нуждающихся в таком лечении. Подчеркивается, что терапевтически эффективное количество ингибитора GSNOR, которое вводится определенному пациенту в конкретном случае, не всегда является эффективным для лечения состояний/заболеваний, описанных в настоящем документе, даже если такая доза считается специалистом в данной области терапевтически эффективным количеством.
[0092] Термин «биологический образец» включает, но не ограничиваясь этим, образцы крови (например, сыворотку, плазму или цельную кровь), мочу, слюну, пот, грудное молоко, влагалищные секреты, сперму, волосяные фолликулы, кожу, зубы, кости, ногти или другие секреты, жидкости, ткани или клетки организма. В соответствии с настоящим изобретением, уровни GSNOR в биологическом образце могут быть определены по способам, описанным в публикации заявки на патент США 2005/0014697.
[0093] D. Фармацевтические композиции
[0094] Настоящим изобретением охватываются фармацевтические композиции, включающие, по меньшей мере, одно соединение настоящего изобретения, описанное в настоящем документе, и, по меньшей мере, один фармацевтически приемлемый носитель. Соответствующие носители описаны в книге "Remington: The Science and Practice", двенадцатое издание, опубликованной издательством Lippincott Williams & Wilkins, которая включена в настоящий документ путем ссылки. Фармацевтические композиции по настоящему изобретению могут также включать один или несколько активных агентов, не являющихся соединениями настоящего изобретения.
[0095] Фармацевтические композиции настоящего изобретения могут включать новые соединения, описанные в настоящем документе, фармацевтические композиции могут включать известные соединения, для которых ранее не была известна активность ингибитора GSNOR, или их комбинацию.
[0096] Соединения настоящего изобретения могут использоваться в любой фармацевтически приемлемой лекарственной форме, включая, но не ограничиваясь этим, лекарственные формы для инъекций, жидкие дисперсии, гели, аэрозоли, мази, кремы, лиофилизированные композиции, сухие порошки, таблетки, капсулы, композиции с контролируемым высвобождении, быстроплавкие композиции, композиции с замедленным высвобождением, композиции с пролонгированным высвобождением, композиции с пульсирующим высвобождением, смешанные композиции с быстрым и контролируемым высвобождением и так далее. В частности, соединения настоящего изобретения, описанные в настоящем документе, могут составляться в композиции: (а) для введения, выбранного из группы, состоящей из перорального, пульмонального, внутривенного, внутриартериального, интратекального, внутрисуставного, ректального, введения в ободочную кишку, парентерального, ушного, интрацистернального, интравагинального, внутрибрюшинного, локального, буккального, назального и местного введения; (б) в лекарственную форму, выбранную из группы, состоящей из жидких дисперсий, гелей, аэрозолей, мазей, кремов, таблеток, саше и капсул; (в) в лекарственную форму, выбранную из группы, состоящей из лиофилизированных композиций, сухих порошков, быстроплавких композиций, композиций с контролируемым высвобождением, композиций с замедленным высвобождением, композиций с пролонгированным высвобождением, композиций с пульсирующим высвобождением и смешанных композиций с быстрым и контролируемым высвобождением; или (г) любые их комбинации.
[0097] Для респираторных инфекций могут использоваться ингаляционные композиции для достижения высоких локальных концентраций. Композиции, пригодные для ингаляции, включают сухие порошки или выпаренные растворы, дисперсии или суспензии, которые могут распыляться ингалятором или распылителем в эндобронхиальной или носовой полости инфицированного пациента для лечения бактериальных инфекций верхних и нижних дыхательных путей.
[0098] Растворы или суспензии, используемые для парентерального, интрадермального или подкожного введения, могут включать один или несколько из следующих компонентов: (1) стерильный разбавитель, такой как вода для инъекций, солевой раствор, нелетучие масла, полиэтиленгликоли, глицерин, пропиленгликоль или другие синтетические растворители; (2) антибактериальные средства, такие как бензиловый спирт или метилпарабены; (3) антиоксиданты, такие как аскорбиновая кислота или бисульфит натрия; (4) хелатирующие агенты, такие как этилендиаминтетрауксусная кислота; (5) буферы, такие как ацетаты, цитраты или фосфаты; и (5) средства для регулирования тоничности, такие как хлорид натрия или декстроза. pH может регулироваться кислотами или основаниями, такими как хлороводородная кислота или гидроксид натрия. Композиция для парентерального введения может быть заключена в ампулы, одноразовые шприцы или емкости для многократных доз, сделанные из стекла или пластика.
[0099] Фармацевтические композиции, пригодные для инъекционного применения, могут включать стерильные водные растворы (в случае водорастворимых композиций) или дисперсии и стерильные порошки для приготовления стерильных инъекционных растворов или дисперсий для немедленного приема. Для внутривенного введения пригодные носители включают физиологический солевой раствор, бактериостатическую воду, Cremophor EL (BASF, Парсиппани, штат Нью-Джерси) или фосфатно-солевой буферный раствор (PBS). Во всех случаях композиция должна быть стерильной и должна быть жидкой настолько, чтобы обеспечивать простое введение через шприц. Фармацевтическая композиция должна быть устойчивой в условиях производства и хранения и должна быть защищена от заражающего действия микроорганизмов, таких как бактерии и грибки.
[00100] Носителем может быть растворитель или дисперсионная среда, включающая, например, воду, этанол, полиол (например, глицерин, пропиленгликоль и жидкий полиэтиленгликоль, и так далее) и их соответствующие смеси. Необходимая текучесть может поддерживаться, например, за счет использования покрытий, таких как лецитин, путем сохранения нужного размера частиц в случае дисперсий, а также при помощи поверхностно-активных веществ. Предотвращение заражения микроорганизмами может быть достигнуто различными антибактериальными и противогрибковыми средствами, например, парабенами, хлорбутанолом, фенолом, аскорбиновой кислотой, тимеросалом и тому подобными. Во многих случаях предпочтительно включать в композицию изотонические агенты, например, сахара, многоатомные спирты, такие как маннит или сорбит, и неорганические соли, такие как хлорид натрия. Пролонгированное поглощение инъецируемых композиций может быть осуществлено за счет включения в композиции агентов, замедляющих абсорбцию, например, моностеарата алюминия и желатина.
[00101] Стерильные инъецируемые растворы могут быть получены введением активного агента в необходимом количестве в соответствующий растворитель, при необходимости, с одним компонентом, перечисленным выше, или их композицией, с последующей стерилизацией фильтрованием. В общем, дисперсии получаются введением, по меньшей мере, одного соединения настоящего изобретения, в стерильный жидкий носитель, который содержит основную дисперсионную среду и любые другие необходимые ингредиенты. В случае стерильных порошков для приготовления стерильных инъецируемых растворов, иллюстративные способы получения включают вакуумную сушку и вымораживание, в результате которых получают порошок соединения настоящего изобретения, плюс любой дополнительный заданный компонент из его предварительно стерилизованного фильтрацией раствора.
[00102] Пероральные композиции, в общем, включают инертный разбавитель или съедобный носитель. Они могут быть заключены, например, в желатиновые капсулы или спрессованы в таблетки. Для перорального терапевтического введения, соединения настоящего изобретения могут быть смешаны с формообразующими средствами и использоваться в форме таблеток, пастилок или капсул. Пероральные композиции также могут быть получены с использованием жидкого носителя для применения в виде жидкостей для полоскания рта, где соединение настоящего изобретения в жидком носителе применяется перорально, полощется и отплевывается или проглатывается. Фармацевтически совместимые связующие агенты и/или вспомогательные материалы могут вводиться как часть композиции.
[00103] Для введения ингаляцией, соединения настоящего изобретения доставляются в форме аэрозольного спрея из емкости под давлением или распылителя, содержащего соответствующий газ-вытеснитель, например, газ, такой как диоксид углерода, распыленную жидкость или сухой порошок из соответствующего устройства. Для трансмукозального или трансдермального введения в композиции используются пенетранты, соответствующие барьеру, через которое должно пройти лекарство. Такие пенетранты обычно известны в данной области и включают, например, для трансмукозального введения, детергенты, соли желчной кислоты и производные фусидовой кислоты. Трансмукозальное введение может осуществляться путем применения назальных спреев или суппозиториев. Для трансдермального введения, активные агенты составляются в композиции притирок, мазей, гелей или кремов, как, в общем, известно в данной области. Эти агенты могут быть также получены в форме суппозиториев (например, с обычными основами для суппозиториев, такими как какао-масло и другие глицериды) или удерживающих клизм для ректальной доставки.
[00104] В одном варианте воплощения соединения настоящего изобретения приготавливаются с носителями, которые защищают от быстрого вывода из организма. Например, может использоваться композиция с контролируемым высвобождением, включая имплантаты и микроинкапсулированные системы доставки. Могут использоваться биоразлагаемые, биосовместимые полимеры, такие как этиленвинилацетат, полиангидриды, полигликолевые кислоты, коллаген, полиортоэфиры и полимолочная кислота. Способы получения таких композиций являются очевидными для специалистов в данной области.
[00105] В качестве фармацевтически приемлемых носителей могут использоваться также липосомные суспензии (включая липосомы, направленные на инфицированные клетки с моноклональными антителами для вирусных антигенов). Их можно получить по способам, известным специалистам в данной области, например, как описано в патенте США №4522811.
[00106] Кроме того, суспензии соединений настоящего изобретения могут быть приготовлены в виде соответствующих масляных инъекционных суспензий. Подходящие липофильные растворители или среды включают жирные масла, такие как кунжутное масло, или сложные эфиры синтетических жирных кислот, такие как этилолеат, триглицериды или липосомы. Для доставки могут также использоваться нежирные поликатионные аминополимеры. Суспензия необязательно может включать также подходящие стабилизаторы или агенты для увеличения растворимости соединений настоящего изобретения и для обеспечения возможности приготовления высококонцентрированных растворов.
[00107] Особенно выгодно составлять пероральные или парентеральные композиции в единичной дозированной форме для простоты введения и равномерности дозирования. Единичная дозированная форма, при использовании в настоящем документе, относится к физически дискретным единицам, пригодным в качестве однократных доз для пациента, подлежащего лечению, где каждая единица содержит определенное количество соединения настоящего изобретения, рассчитанное для обеспечения заданного терапевтического эффекта, в сочетании с заданным фармацевтическим носителем. Характеристики единичных дозированных форм настоящего изобретения продиктованы и напрямую зависят от уникальных характеристик соединения настоящего изобретения и конкретного ожидаемого терапевтического эффекта, а также от ограничений, существующих в области составления композиций, таких как активный агент для лечения пациентов.
[00108] Фармацевтические композиции по настоящему изобретению, включающие, по меньшей мере, одно соединение настоящего изобретения, могут включать одно или несколько фармацевтических формообразующих средств. Примеры таких формообразующих средств включают, но не ограничиваясь этим, связующие агенты, наполнители, смазывающие агенты, суспендирующие агенты, подсластители, ароматизаторы, консерванты, буферы, увлажняющие агенты, средства для улучшения распадаемости таблеток, шипучие агенты и другие формообразующие средства. Такие формообразующие средства известны в данной области. Примеры формообразующих средств включают: (1) связующие агенты, которые включают различные целлюлозы и поперечно-сшитый поливинилпирролидон, микрокристаллическую целлюлозу, такую как Avicel® PH101 и Avicel® PH102, силикатированную микрокристаллическую целлюлозу (ProSolv SMCC™), трагакантовую камедь и желатин; (2) наполнители, такие как различные крахмалы, лактоза, моногидрат лактозы и безводная лактоза; (3) средства для улучшения распадаемости таблеток, такие как альгиновая кислота, Primogel, кукурузный крахмал, слабо сшитый поливинилпирролидон, картофельный крахмал, маисовый крахмал и модифицированные крахмалы, кроскармеллоза натрия, кросс-повидон, натрия крахмалгликолат и их смеси; (4) смазывающие вещества, включая агенты, которые действуют на текучесть прессуемого порошка, включая стеарат магния, коллоидный диоксид кремния, такой как Aerosil® 200, тальк, стеариновая кислота, стеарат кальция и силикагель; (5) глиданты, такие как коллоидный диоксид кремния; (6) консерванты, такие как сорбат калия, метилпарабен, пропилпарабен, бензойная кислота и ее соли, другие сложные эфиры пара-гидроксибензойной кислоты, такие как бутилпарабен, спирты, такие как этиловый или бензиловый спирт, фенольные соединения, такие как фенол, или четвертичные соединения, такие как хлорид бензалкония; (7) разбавители, такие как фармацевтически приемлемые инертные наполнители, такие как микрокристаллическая целлюлоза, лактоза, двухосновной фосфат кальция, сахариды и/или смеси любых из вышеперечисленных; примеры разбавителей включают микрокристаллическую целлюлозу, такую как Avicel® PH101 и Avicel® PH102; лактозу, такую как моногидрат лактозы, безводная лактоза и Pharmatose® DCL21; двухосновной фосфат кальция, такой как Emcompress®; маннит; крахмал; сорбит; сахарозу; и глюкозу; (8) подсластители, включая любые природные или искусственные подсластители, такие как сахароза, сахарин-сахароза, ксилит, сахарин натрия, цикламат, аспартам и ацесульфам; (9) ароматизаторы, такие как мята перечная, метилсалицилат, апельсиновый ароматизатор, Magnasweet® (торговая марка MAFCO), ароматизатор жевательной резинки, фруктовые ароматизаторы и тому подобные; и (10) шипучие агенты, включая шипучие пары, такие как органическая кислота и карбонат или бикарбонат. Соответствующие органические кислоты включают, например, лимонную, винную, яблочную, фумаровую, адипиновую, янтарную и альгиновую кислоты, ангидриды и соли кислот. Соответствующие карбонаты и бикарбонаты включают, например, карбонат натрия, бикарбонат натрия, карбонат калия, бикарбонат калия, карбонат магния, натрия глицинкарбонат, L-лизинкарбонат и аргининкарбонат. Альтернативно, из шипучей пары может присутствовать только компонент бикарбонат натрия.
[00109] E. Наборы, включающие композиции настоящего изобретения
[00110] Настоящим изобретением охватываются также наборы, включающие композиции настоящего изобретения. Такие наборы могут включать, например, (1) по меньшей мере, одно соединение настоящего изобретения; и (2) по меньшей мере, один фармацевтически приемлемый носитель, такой как растворитель или раствор. Дополнительные компоненты набора могут необязательно включать, например: (1) любые фармацевтически приемлемые формообразующие средства, указанные в настоящем документе, такие как стабилизаторы, буферы и так далее, (2) по меньшей мере, один контейнер, емкость или аналогичное приспособление для хранения и/или смешивания компонентов набора; и (3) устройство доставки, такое как ингалятор, распылитель, шприц и так далее.
[00111] F. Способы получения соединений настоящего изобретения
[00112] Соединения настоящего изобретения могут быть легко синтезированы с использованием известных синтетических методик или путем модификации известных синтетических методик. Как легко понятно специалисту в данной области, способы, описанные в ниже, позволяют синтезировать хинолины, имеющие различные заместители. Иллюстративные способы синтеза описаны ниже в разделе Примеров.
[00113] При необходимости, дополнительная очистка и разделение энантиомеров и диастереомеров могут быть достигнуты обычными способами, известными в данной области. Так, например, разделение энантиомеров соединения может быть достигнуто использованием хиральной ВЭЖХ и родственных хроматографических способов. Таким же образом могут быть разделены диастереомеры. Однако в некоторых случаях диастереомеры могут быть просто разделены физически, как, например, контролируемым осаждением или кристаллизацией.
[00114] Способы настоящего изобретения, будучи осуществлены так, как описано в настоящем документе, могут быть легко осуществлены при температурах, которые обычно доступны в данной области. В одном варианте, способ выполняется при температуре в диапазоне от около 25°C до около 110°C. В другом варианте, температура находится в диапазоне от около 40°C до около 100°C. в другом варианте, температура находится в диапазоне от около 50°C до около 95°C.
[00115] Этапы синтеза, для которых необходимо основание, выполняются с использованием обычных органических или неорганических оснований. Как правило, основание не является нуклеофильным. Так, в одном варианте основание выбрано из карбонатов, фосфатов, гидроксидов, алкоксидов, солей дисилазанов и третичных аминов.
[00116] Способы настоящего изобретения, которые выполняются так, как описано в настоящем документе, могут быть, по существу, завершены через время от нескольких минут до нескольких часов, в зависимости от природы и количества реагентов, а также от температуры реакции. Определение времени, когда реакция, по существу, завершена, может быть просто оценено обычными способами, известными в данной области, такими как, например, ВЭЖХ, ЖХМС, ТСХ и 1H ЯМР.
[00117] G. Способы лечения
[00118] Настоящим изобретением охватываются способы предупреждения или лечения (например, облегчения одного или нескольких симптомов) медицинских состояний путем использования одного или нескольких описанных соединений. Эти способы включают введение нуждающемуся пациенту терапевтически эффективного количества соединения настоящего изобретения. Композиции настоящего изобретения могут использоваться также для профилактической терапии.
[00119] Соединения настоящего изобретения, используемые в способах лечения по настоящему изобретению, могут быть: (1) новым соединением, описанным в настоящем документе, или его фармацевтически приемлемой солью, его стереоизомером, его пролекарством, его метаболитом или его N-оксидом; (2) соединением, которое было известно до настоящего изобретения, но не было известно, что это соединение является ингибитором GSNOR, или его фармацевтически приемлемой солью, его стереоизомером, его пролекарством, его метаболитом или его N-оксидом; или (3) соединением, которое было известно до настоящего изобретения, и было известно, что это соединение является ингибитором GSNOR, но не было известно, что это соединение является полезным для способов лечения, описанных в настоящем документе, или его фармацевтически приемлемой солью, стереоизомером, пролекарством, метаболитом или N-оксидом.
[00120] Пациентом может быть любое животное, домашнее, сельскохозяйственное или дикое, включая, но не ограничиваясь этим, котов, собак, лошадей, свиней и крупный рогатый скот, и, предпочтительно, пациентом является человек. При использовании в настоящем документе, термины пациент и субъект могут использоваться взаимозаменяемо.
[00121] При использовании в настоящем документе, «лечение» описывает контроль и обслуживание пациента в целях борьбы с заболеванием, состоянием или расстройством, и включает введение соединения настоящего изобретения для предупреждения возникновения симптомов или осложнений, облегчения симптомов или осложнений или устранения заболевания, состояния или расстройства. Более конкретно, «лечение» включает реверсирование, ослабление, облегчение, минимизацию, подавление или исключение, по меньшей мере, одного опасного симптома или результата состояния болезни (расстройства), прогрессирования заболевания, причинного фактора заболевания (например, бактерий или вирусов) или другого патологического состояния. Лечение продолжается до тех пор, пока не улучшатся симптомы и/или патология.
[00122] В общем, доза, то есть терапевтически эффективное количество, находится в диапазоне от 1 мкг/кг до 10 г/кг, и зачастую находится в диапазоне от 10 мкг/кг до 1 г/кг или от 10 мкг/кг до 100 мг/кг веса тела пациента, подлежащего лечению, в день.
[00123] H. Применение GSNOR
[00124] У пациентов с разрушительно высокими уровнями GSNOR или активности GSNOR, модулирование может быть достигнуто, например, введением одного или нескольких из описанных соединений, которые разрушают или понижающее регулируют действие GSNOR, или снижают уровни GSNOR. Эти соединения могут вводиться с другими средствами-ингибиторами GSNOR, такими как анти-GSNOR антитела или фрагменты антител, антисмысловая GSNOR, мРНК или низкомолекулярные соединения, или с другими ингибиторами, отдельно или в комбинации с другими средствами, как подробно описано в настоящем документе.
[00125] В настоящем изобретении представлен способ лечения пациента, пораженного расстройством, которое улучшается NO-донорной терапией. Такой способ включает введение пациенту терапевтически эффективного количества ингибитора GSNOR.
[00126] Расстройства могут включать легочные расстройства, связанные с гипоксемией и/или сокращением гладких мышц в легких и дыхательных путях, и/или легочной инфекцией, и/или воспалением легких, и/или травмой легких (например, легочная гипертензия, респираторный дистресс-синдром у взрослых, астма, пневмония, фиброз легких/интерстициальный легочный процесс, фиброзно-кистозная дегенерация, хроническое обструктивное заболевание легких); сердечнососудистые заболевания и болезни сердца (например, гипертония, ишемический коронарный синдром, атеросклероз, сердечная недостаточность, глаукома); заболевания, характеризующиеся ангиогенезом (например, коронарная болезнь сердца); расстройства, при которых существует риск возникновения тромбоза; расстройства, при которых существует риск возникновения рестеноза; воспалительные заболевания (например, деменция, связанная со СПИДом, воспалительная болезнь кишечника (IBD), болезнь Крона, колит и псориаз); функциональные расстройства кишечника (например, синдром раздраженного кишечника (IBS)); расстройства, при которых существует риск возникновения апоптоза (например, сердечная недостаточность, атеросклероз, дегенеративные неврологические расстройства, артрит и поражение печени (например, вызванное лекарствами, ишемическое или алкогольное)); импотенцию, приступы апноэ во сне; заживление диабетических ран; кожные инфекции; лечение псориаза; ожирение, обусловленное питанием в ответ на желание пищи; приступ; реперфузионные повреждения (например, травматическое повреждение мышц сердца или легких, или размозжение); и расстройства, при которых полезна предварительная адаптация сердца или мозга для NO-защиты от последующих ишемических событий; расстройства центральной нервной системы (ЦНС) (например, боязнь, депрессия, психоз и шизофрения); и инфекции, обусловленные бактериями (например, среди прочих, туберкулез, инфекции С. difficile).
[00127] В одном варианте воплощения расстройством является повреждение печени. Повреждение печени может включать, например, острую печеночную токсичность. Острая печеночная токсичность может приводить к острой печеночной недостаточности. Острая печеночная недостаточность (ALF) является редким, но потенциально летальным связанным с лекарствами неблагоприятным эффектом, который зачастую приводит к трансплантации печение (LT) или смерти. Ацетаминофен является наиболее распространенной причиной острой печеночной токсичности и острой печеночной недостаточности, хотя острая печеночная токсичность может быть и результатом других агентов, таких как алкоголь и другие лекарства. Независимо от причины возникновения, в результате однократной передозировки или после многократного приема в дозах, превышающих терапевтические дозы, прогрессирование отравления ацетаминофеном можно разделить по категориям на четыре этапа: доклиническое токсическое воздействие (нормальная концентрация аланин-аминотрансферазы в сыворотке), повреждение печени (повышенная концентрация аланин-аминотрансферазы), печеночная недостаточность (повреждение печени с печеночной энцефалопатией) и восстановление. Пока присутствет достаточное количество глутатиона, печень защищена по повреждения. Передозировки ацетаминофена (как в результате однократного приема большого количества, так и в результате многократного приема в дозах, превышающих терапевтические дозы) могут истощить печеночные запасы глутатиона и привести к возникновению повреждения печени. Соединения настоящего изобретения способны лечить и/или предупреждать повреждение печени и/или острую печеночную токсичность. В этом варианте, соответствующие количества соединений настоящего изобретения являются количествами, достаточными для лечения и/или предупреждения повреждения печени, и могут быть определены без излишних экспериментов в доклинических и/или клинических испытаниях. В одном варианте воплощениия, количество для лечения составляет, по меньшей мере, 0,001 мг/кг, по меньшей мере, 0,002 мг/кг, по меньшей мере, 0,003 мг/кг, по меньшей мере, 0,004 мг/кг, по меньшей мере, 0,005 мг/кг, по меньшей мере, 0,006 мг/кг, по меньшей мере, 0,007 мг/кг, по меньшей мере, 0,008 мг/кг, по меньшей мере, 0,009 мг/кг, по меньшей мере, 0,01 мг/кг, по меньшей мере, 0,02 мг/кг, по меньшей мере, 0,03 мг/кг, по меньшей мере, 0,04 мг/кг, по меньшей мере, 0,05 мг/кг, по меньшей мере, 0,06 мг/кг, по меньшей мере, 0,07 мг/кг, по меньшей мере, 0,08 мг/кг, по меньшей мере, 0,09 мг/кг, по меньшей мере, 0,1 мг/кг, по меньшей мере, 0,2 мг/кг, по меньшей мере, 0,3 мг/кг, по меньшей мере, 0,4 мг/кг, по меньшей мере, 0,5 мг/кг, по меньшей мере, 0,6 мг/кг, по меньшей мере, 0,7 мг/кг, по меньшей мере, 0,8 мг/кг, по меньшей мере, 0,9 мг/кг, по меньшей мере, 1 мг/кг, по меньшей мере, 1,5 мг/кг, по меньшей мере, 2 мг/кг, по меньшей мере, 2,5 мг/кг, по меньшей мере, 3 мг/кг, по меньшей мере, 3,5 мг/кг, по меньшей мере, 4 мг/кг, по меньшей мере, 4,5 мг/кг, по меньшей мере, 5 мг/кг, по меньшей мере, 6 мг/кг, по меньшей мере, 7 мг/кг, по меньшей мере, 8 мг/кг, по меньшей мере, 9 мг/кг, по меньшей мере, 10 мг/кг, по меньшей мере, 15 мг/кг, по меньшей мере, 20 мг/кг, по меньшей мере, 30 мг/кг, по меньшей мере, 40 мг/кг, по меньшей мере, 50 мг/кг, по меньшей мере, 60 мг/кг, по меньшей мере, 70 мг/кг, по меньшей мере, 80 мг/кг, по меньшей мере, 90 мг/кг, по меньшей мере, 100 мг/кг. Дозирование может быть ежечасным, четыре раза, дважды или один раз в день, или четыре раза, дважды или один раз в неделю, или еженедельно, или каждую вторую неделю, каждую третью неделю или ежемесячно.
[00128] В одном варианте воплощения расстройством является травма (включая хирургическую и термальную), инфекционное, токсическое, возрастное и ишемическое повреждение органов известной регенеративной способности, таких как кожа, слизистая оболочка желудка, эпителий дыхательных путей и хрящевые структуры, печень, нейронные структуры, такие как спинной мозг, костный мозг и кости. Авторы настоящего изобретения показали, что ингибирование GSNOR при помощи высокоспецифичных низкомолекулярных соединений ингибирует, восстанавливает и ускоряет регенерацию тканей млекопитающих. Например, низкомолекулярные ингибиторы являются эффективными для лечения и ускорения восстановления и регенерации легочной ткани млекопитающих, поврежденной инстилляцией химического агента, который, как известно, вызывает сильное повреждение легких (свиная панкреатическая эластаза) (Blonder et al., реферативная ссылка ATS 2011). В этом варианте воплощения, соответствующие количества соединений настоящего изобретения являются количествами, достаточными для регенерации ткани/органов, и могут быть определены без излишних экспериментов в доклинических и/или клинических испытаниях.
[00129] В одном варианте воплощения расстройством является травма (включая хирургическую и термальную), инфекционное, токсическое, возрастное и ишемическое повреждение органов, для которых обычно не известна регенеративная способность. Примеры включают регенерацию: сердца, легких, почек, центральной нервной системы, периферической нервной системы, периферических сосудистых тканей, печени, поджелудочной железы, надпочечников, щитовидной железы, яичек, яичников, сетчатки, языка, костей, мочевого пузыря, пищевода, гортани, тимуса, селезенки, хрящевых структур головы и хрящевых структур суставов. В этом варианте воплощения, соответствующие количества соединений настоящего изобретения являются количествами, достаточными для регенерации ткани/органов, и могут быть определены без излишних экспериментов в доклинических и/или клинических испытаниях.
[00130] В одном варианте воплощения ex и in vivo имплантация и регенерация органов и структур, включая стволовые клетки. В этом варианте воплощения, соответствующие количества соединений настоящего изобретения являются количествами, достаточными для регенерации ткани/органов, и могут быть определены без излишних экспериментов в доклинических и/или клинических испытаниях.
[00131] Водном варианте воплощения соединения настоящего изобретения или их фармацевтически приемлемые соли или пролекарства, стереоизомеры, метаболиты или N-оксиды могут вводиться в комбинации с донором NO. Донор NO предоставляет оксид азота или родственные редокс-частицы и, более типично, обеспечивает биоактивность оксида азота, то есть активность, которая определяется оксидом азота, например, вазорелаксацию или стимуляцию или ингибирование рецепторного белка, например, белка ras, адренергического рецептора, NFκB. Доноры NO, включая S-нитрозо, O-нитрозо, C-нитрозо и N-нитрозосоединения и их нитропроизводные, а также металлические комплексы NO, но не исключая другие соединения, создающие биоактивность NO, которые являются применимыми в настоящем документе, описаны в публикации "Methods in Nitric Oxide Research," Feelisch et al. eds., страницы 71-115 (J.S., John Wiley & Sons, Нью-Йорк, 1996), которая включена в настоящий документ путем ссылки. Доноры NO, которые являются C-нитрозосоединениями, где нитрозо-группа присоединена к третичному углероду, и которые являются применимыми в настоящем документе, включают соединения, описанные в патенте США №6359182 и WO 02/34705. Примеры S-нитрозосоединений, включая S-нитрозотиолы, применимые в настоящем документе, включают, например, S-нитрозоглутатион, S-нитозо-N-ацетилпеницилламин, S-нитрозо-цистеин и его этиловый эфир, S-нитрозо-цистеин глицин, S-нитрозо-гамма-метил-L-гомоцистеин, S-нитрозо-L-гомоцистеин, S-нитрозо-гамма-тио-L-лейцин, S-нитрозо-дельта-тио-L-лейцин и S-нитрозоальбумин. Примерами других доноров NO, применимых в настоящем документе, являются нитропроссид натрия (ниприд), этилнитрит, изосорбид, нитроглицерин, SIN 1, который является молсидомином, фуроксамины, N-гидрокси (N-нитрозамин) и перфторуглероды, которые насыщены NO или гидрофобные доноры NO.
[00132] Комбинация ингибитора GSNOR с R(+) энантиомером амлодипина, известным NO-высвобождающим средством (Zhang at al., J. Cardiovasc. Pharm. 39: 208-214 (2002)), также является вариантом настоящего изобретения.
[00133] В настоящем изобретении представлен также способ лечения пациента, пораженного патологически разрастающимися клетками, где этот способ включает введение указанному пациенту терапевтически эффективного количества ингибитора GSNOR. Ингибиторами GSNOR являются соединения, как определено выше, или их фармацевтически приемлемые соли, или их стереоизомеры, пролекарства, метаболиты или N-оксиды, в комбинации с фармацевтически приемлемым носителем. Лечение продолжается до тех пор, пока не улучшатся симптомы и/или патология.
[00134] В другом варианте воплощения патологически разрастающимися клетками могут быть патологически разрастающиеся микробы. Микробами, которых это касается, могут быть микробы, в которых экспрессируется GSNOR для защиты микроба от нитрозативного стресса, или в которых инфицированная микробом клетка хозяина экспрессирует фермент, защищая, таким образом, микроб от нитрозативного стресса. Термин «патологически разрастающиеся микробы» используется в настоящем документе для обозначения патологических микроорганизмов, включая, но не ограничиваясь этим, патологические бактерии, патологические вирусы, патологические хламидии, патологические простейшие, патологические риккетсии, патологические грибки и патологические микоплазмы. Более подробное описание применимых микробов изложено далее в колонках 11 и 12 патента США №6057367. Термин «клетки хозяина, инфицированные патологическими микробами» включает не только клетки млекопитающих, инфицированные патологическими вирусами, но и клетки млекопитающих, содержащие внутриклеточные бактерии или простейшие, например, макрофаги, содержащие Mycobacterium tuberculosis, Mycobacterium leper (проказа) или Salmonella typhi (брюшной тиф).
[00135] В другом варианте воплощения патологически разрастающимися клетками могут быть патологические гельминты. Термин «патологические гельминты» используется в настоящем документе для обозначения патологических нематод, патологических трематод и патологических цестод. Более подробная информация о применимых гельминтах представлена далее в колонке 12 патента США №6057367.
[00136] В другом варианте патологически разрастающимися клетками могут быть патологически разрастающиеся клетки млекопитающих. Термин «патологически разрастающиеся клетки млекопитающих», при использовании в настоящем документе, обозначает клетки млекопитающего, которые так вырастают в организме указанного млекопитающего по размеру или количеству, что вызывают губительное действие на млекопитающее или его органы. Этот термин включает, например, патологически разрастающиеся или увеличивающиеся клетки, вызывающие рестеноз, патологически разрастающиеся или увеличивающиеся клетки, вызывающие доброкачественную гипертрофию простаты, патологически разрастающиеся клетки, вызывающие миокартиальную гипертрофию, и разрастающиеся клетки в воспалительных очагах, такие как синовиальные клетки при артрите или клетки, связанные с нарушением клеточной пролиферации.
[00137] При использовании в настоящем документе, термин «нарушение клеточной пролиферации» относится к состояниям, при которых нерегулируемый и/или патологический рост клеток может приводить к развитию нежелательного состояния или заболевания, которое может быть раковым или нераковым, например, псориатическое состояние. При использовании в настоящем документе, термин «псориатическое состояние» относится к расстройствам, включающим гиперпролиферацию кератиноцитов, инфильтрацию клеток воспаления и альтерацию цитокинов. Расстройство клеточной пролиферации может быть предраковым состоянием или раком. Рак может быть первичным раком или метастатическим раком, или ими обоими.
[00138] При использовании в настоящем документе, термин «рак» включает солидные опухоли, такие как рак легких, молочной железы, ободочной кишки, яичников, поджелудочной железы, простаты, аденокарцинома, сквамозная карцинома, саркома, злокачественная глиома, лейомиосаркома, гепатома, рак головы и шеи, злокачественная меланома, немеланомный рак кожи, а также гематологические опухоли и/или злокачественные образования, такие как лейкоз, детский лейкоз и лимфома, множественная миелома, болезнь Ходжкина, лимфома лимфоцитарного и кожного происхождения, острый и хронический лейкоз, такой как острый лимфобластный, острый миелоцитарный или хронический миелоцитарный лейкоз, неоплазма клеток плазмы, лимфоидная неоплазма и рак, связанный со СПИДом.
[00139] Помимо псориатических состояний, типами пролиферативных заболеваний, которые можно лечить при помощи композиций настоящего изобретения, являются эпидермальные и дермоидные кисты, липомы, аденомы, капиллярные и кожные гемангиомы, лимфангиомы, повреждение невусов, тератомы, нефромы, миофиброматозы, остеопластические опухоли и другие диспластические массы, и тому подобные. В одном варианте воплощения, пролиферативные заболевания включают дисплазию и подобные расстройства.
[00140] В одном варианте воплощения, лечение рака включает уменьшение объема опухоли, снижение количества опухолей, отсрочку роста опухоли, снижение метастатических повреждений в других тканях и органах, удаленных от первичного очага опухоли, улучшение выживаемости пациентов или улучшение качества жизни пациента, или, по меньшей мере, двух из вышеперечисленных.
[00141] В другом варианте воплощения, лечение нарушения клеточной пролиферации включает снижение скорости клеточной пролиферации, снижение доли разрастающихся клеток, уменьшение размера области или зоны клеточной пролиферации или снижение количества или доли клеток, имеющих патологический внешний вид или морфологию, или, по меньшей мере, двух из вышеперечисленных.
[00142] В другом варианте воплощения, соединения настоящего изобретения или их фармацевтически приемлемые соли, их стереоизомеры, их пролекарства, их метаболиты или их N-оксиды могут вводиться в комбинации со вторым химиотерапевтическим средством. В следующем варианте воплощения, второе химиотерапевтическое средство выбрано из группы, состоящей из тамоксифена, ралоксифена, анастрозола, экземестана, летрозола, цисплатина, карбоплатина, паклитаксела, циклофосфамида, ловастатина, минозина, гемцитабина, araC, 5-фторурацила, метотрексата, доцетаксела, гозерелина, винкристина, винбластина, нокодазола, тенипозида, этопозида, эпотилона, навелбина, камтотецина, даунонибицина, дактиномицина, митоксантрона, амсакрина, доксорубицина, эпирубицина, идарубицина, иматиниба, гефитиниба, эрлотиниба, сорафениба, сунитиниб малата, трастузумаба, ритуксимаба, цетуксимаба и бевацизумаба.
[00143] В одном варианте воплощения, соединения настоящего изобретения или их фармацевтически приемлемые соли, их стереоизомеры, их пролекарства, их метаболиты или их N-оксиды могут вводиться в комбинации со средством, которое налагает нитрозативный или окислительный стресс. Средства для селективного наложения нитрозативного стресса для ингибирования пролиферации патологически разрастающихся клеток в комплексной терапии с ингибиторами GSNOR настоящего документа, а также дозы и способы их введения включают те, которые описаны в патенте США №6057367, который включен в настоящий документ. Дополнительные средства для наложения окислительного стресса (то есть средства, которые увеличивают соотношение GSSG (окисленного глутатиона) к GSH (глутатиону) или отношение НАД(Ф) к НАД(Ф)Н или увеличивают производные тиобарбитуровой кислоты) в комплексной терапии с ингибиторами GSNOR настоящего документа включают, например, L-бутионин-3-сульфоксимин (BSO), ингибиторы глутатион-редуктазы (например, BCNU), ингибиторы или разобщающие агенты митохондриального дыхания, а также лекарства, которые увеличивают активные формы кислорода (ROS), например, адриамицин, в стандартных дозах при стандартных способах введения.
[00144] Ингибиторы GSNOR могут также вводиться совместно с ингибитором фосфодиэстеразы (например, ролипрам, циломиласт, рофлумиласт, Viagra® (силденифил цитрат), Cialis® (тадалафил), Levitra® (варденифил) и так далее), β-агонистами, стероидами, антимускариновыми средствами или агонистом лейкотриена (LTD-4). Специалисты в данной области могут легко определить соответствующее терапевтически эффективное количество в зависимости от расстройства, подлежащего улучшению.
[00145] Ингибиторы GSNOR могут использоваться к средства для улучшения β-адренергического сигналинга. В частности, ингибиторы GSNOR, самостоятельно или в комбинации с β-агонистами, могут использоваться для лечения или защиты от сердечной недостаточности или других сосудистых расстройств, таких как гипертония и астма. Ингибиторы GSNOR могут также использоваться для модулирования рецепторов, сопряженных с G-белком (GPCR), за счет усиления Gs G-белка, приводящего к релаксации гладких мышц (например, дыхательных путей и кровеносных сосудов) и за счет ослабления Gq G-белка, предотвращая, таким образом, сокращение гладких мышц (например, в дыхательных путях и кровеносных сосудах).
[00146] Терапевтически эффективное количество для лечения пациента, пораженного расстройством, которое облегчается NO-донорной терапией, является количеством, ингибирующим GSNOR in vivo, которое вызывает улучшение расстройства, подлежащего лечению, или защищает от риска, связанного с этим расстройством. Например, для астмы, терапевтически эффективным количеством является бронходилатирующее эффективное количество; для фиброзно-кистозной дегенерации, терапевтически эффективным количеством является эффективное количество для улучшения обструкции дыхательных путей; для респираторного дистресс-синдрома взрослых (ARDS), терапевтически эффективным количеством является эффективное количество для улучшения гипоксемии; для болезней сердца, терапевтически эффективным количеством является эффективное количество, вызывающее облегчение стенокардии или ангиогенез; для гипертонии, терапевтически эффективным количеством является эффективное количество для понижения кровяного давления; для ишемических коронарных расстройств, терапевтическим количеством является эффективное количество для увеличения кровотока; для атеросклероза, терапевтически эффективными количеством является эффективное количество для реверсирования эндотелиальной дисфункции; для глаукомы, терапевтически эффективным количеством является эффективное количество для снижения внутриглазного давления; для заболеваний, характеризующихся ангиогенезом, терапевтически эффективным количеством является эффективное количество для ингибирования ангиогенеза; для расстройств, при которых существует риск возникновения тромбоза, терапевтически эффективным количеством является эффективное количество для предотвращения тромбоза; для расстройств, при которых существует риск возникновения рестеноза, терапевтически эффективным количеством является эффективное количество для ингибирования рестеноза; для хронических воспалительных заболеваний, терапевтически эффективным количеством является эффективное количество для уменьшения воспаления; для расстройств, при которых существует риск возникновения апоптоза, терапевтически эффективным количеством является эффективное количество для предотвращения апоптоза; для импотенции, терапевтически эффективным количеством является эффективное количество для достижения или оддержания потенции; для ожирения, терапевтически эффективным количеством является эффективное количество для вызывания насыщения; для удара, терапевтически эффективным количеством является эффективное количество для увеличения кровотока или защиты от транзиторной ишемической атаки (TIA); для реперфузионного повреждения, терапевтически эффективным количеством является эффективное количество для повышения функции; для предварительной адаптации сердца и мозга, терапевтически эффективным количеством является эффективное количество для защиты клеток, например, по измерениям тропонина или креатинфосфокиназы (CPK).
[00147] Терапевтически эффективное количество для лечения пациента, пораженного патологически разрастающимися клетками, означает количество, ингибирующее GSNOR in vivo, которое является антипролиферативным эффективным количеством. Такое антипролиферативное эффективное количество, при использовании в настоящем документе, означает количество, вызывающее снижение скорости пролиферации, по меньшей мере, примерно на 20%, по меньшей мере, примерно на 10%, по меньшей мере, примерно на 5% или, по меньшей мере, примерно на 1%.
[00148] I. Применение в приборах
[00149] Соединения настоящего изобретения или их фармацевтически приемлемые соли, их стереоизомеры, пролекарства, метаболиты или N-оксиды могут применяться в различных приборах при обстоятельствах, когда присутствие таких соединений является благотворным. Такими приборами могут быть любые устройства или контейнеры, например, имплантируемые устройства, в которых соединение настоящего изобретения может использоваться для покрытия хирургической сетки или сердечнососудистого стента перед имплантацией пациенту. Соединения настоящего изобретения могут также наноситься на различные приборы для анализов in vitro или для культивируемых клеток.
[00150] Соединения настоящего изобретения или их фармацевтически приемлемые соли, их стереоизомеры, пролекарства, метаболиты или N-оксиды могут применяться в качестве средств для создания, выделения или очистки участников, связывающихся соединениями настоящего изобретения, таких как антитела, природные лиганды и тому подобные. Специалисты в данной области могут легко определить родственные области применения соединений настоящего изобретения.
ПРИМЕРЫ
[00151] Следующие примеры представлены для иллюстрации настоящего изобретения. Однако следует понимать, что настоящее изобретение не ограничивается конкретными условиями или деталями, описанными в этих примерах. В настоящем описании, любые и все ссылки на общедоступные документы, включая патенты США, отдельно включены путем ссылки.
[00152] В Примерах 1-56 перечислены иллюстративные новые хинолиновые аналоги Формулы I, применимые в качестве ингибиторов GSNOR настоящего изобретения. На представленных ниже иллюстративных схемах изображены некоторые общие способы получения хинолиновых аналогов Формулы I. Синтетические способы, которые могут использоваться для получения каждого соединения, описаны в Примерах 1-56. Подтверждающие данные масс-спектрометрии и/или данные протонного ЯМР для каждого соединения также включены в Примеры 1-56. Подробности синтеза соответствующих Промежуточных соединений детализированы в Примере 57.
[00153] На Схемах 1-5 ниже представлены общие способы получения хинолиновых аналогов.
Схема 1
[00154] Подробный пример Общей Схемы 1 представлен в Примере 1, Соединение 1.
Схема 2
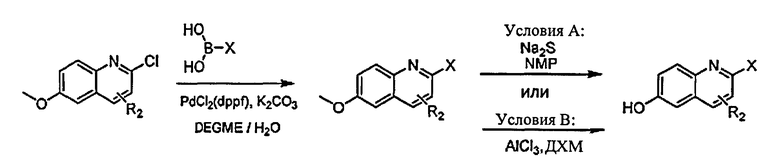
[00155] Подробный пример Схемы 2, условия A, представлен в Примере 2, Соединение 2.
[00156] Подробный пример Схемы 2, условия B, представлен в Примере 8, Соединение 8.
Схема 3
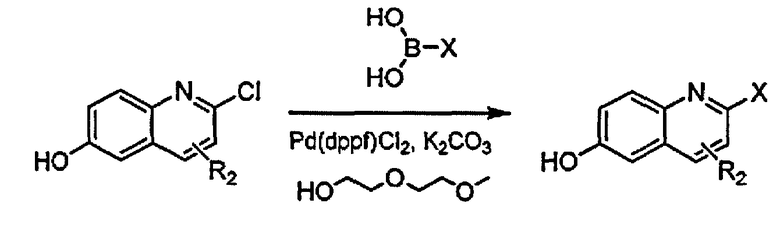
[00157] Подробный пример Схемы 3 представлен в  , Соединение 9.
, Соединение 9.
Схема 4
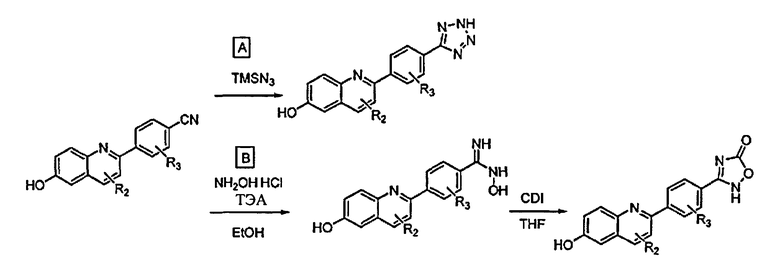
[00158] Подробный пример Схемы 4, путь A, представлен в Примере 11, Соединение 11.
[00159] Подробный пример Схемы 4, путь B, представлен в Примере 12, Соединение 12.
Схема 5
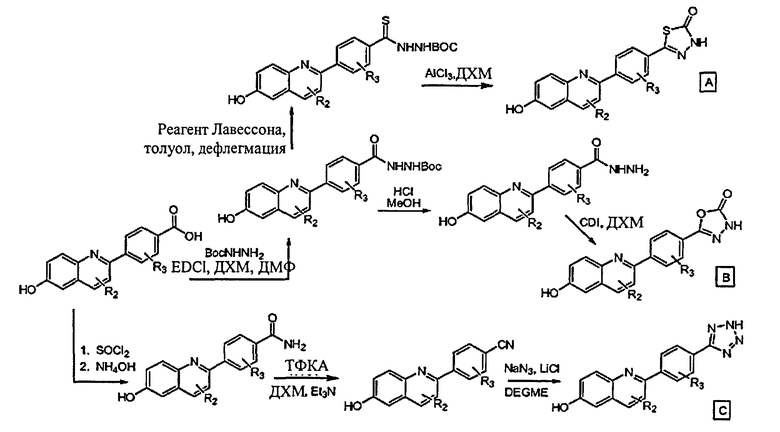
[00160] Подробный пример Схемы 5, соединение A, представлен в Примере 33, Соединение 33.
[00161] Подробный пример Схемы 5, соединение B, представлен в Примере 24, Соединение 24.
[00162] Подробный пример Схемы 5, соединение C, представлен в Примере 23, Соединение 23.
[00163] Пример 1: Соединение 1: 4-(6-гидрокси-3-метилхинолин-2-ил)бензойная кислота
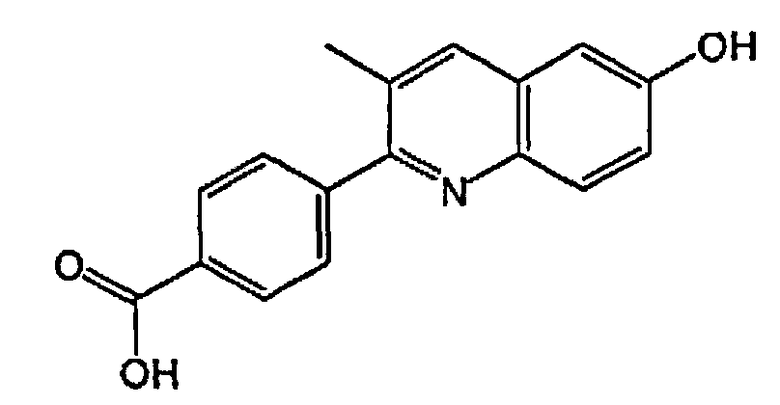
[00164] Следовали Схеме 1.
[00165] Этап 1: Синтез метил 4-(6-метокси-3-метилхинолин-2-ил)бензоата:
[00166] К смеси 2-хлор-6-метокси-3-метилхинолина (100 мг, 0,482 ммоль), 4-(метоксикарбонил)фенилбороновой кислоты (184 мг, 1,02 ммоль), ТЭА (0,35 мл, 2,41 ммоль) и PdCl2(dppf) (35 мг, 0,048 ммоль) добавили 2 мл ДМФ под аргоном. Затем смесь перемешивали в течение 2,5 часов при 120°C в микроволновом реакторе. Затем неочищенную реакционную смесь разбавили водой (25 мл) и экстрагировали EtOAc (25 мл × 2). Органические слои промыли насыщенным солевым раствором (50 мл), высушили над сульфатом натрия и концентрировали для получения 250 мг неочищенного материала. Неочищенный материал очистили колоночной хроматографией с градиентом от 5% EtOAc в гексанах до 80% EtOAc в гексанах для получения 37 мг (выход 25%) метил 4-(6-метокси-3-метилхинолин-2-ил)бензоата.
[00167] Этап 2: Синтез 4-(6-гидрокси-3-метилхинолин-2-ил)бензойной кислоты.
[00168] Метил 4-(6-метокси-3-метилхинолин-2-ил)бензоат (37 мг, 0,121 ммоль) растворили в 2 мл ДХМ и добавили BBr3 (150 мкл). Смесь перемешивали при комнатной температуре в течение 1 дня, а затем добавили 10 мл H2O. Раствор энергично перемешивали в течение 1 часа, а затем отфильтровали твердые вещества. Твердые вещества промыли H2O и высушили in vacuo для получения 9,5 мг (выход 28%) Соединения 1. 1H ЯМР (ДМСО-d6, 400 MHz): δ 8.37 (s, 1H), 8.10 (d, 2H), 7.94 (d, 1H), 7.78 (d, 2H), 7.42-7.39 (dd, 1H), 7.24-7.23 (d, 1H), 2.43 (s, 3H). MS (ESI): m/z 280.10 [M+H]+.
[00169] Пример 2: Соединение 2: 2-(4-(1H-тетразол-5-ил)фенил)-3-метилхинолин-6-ол
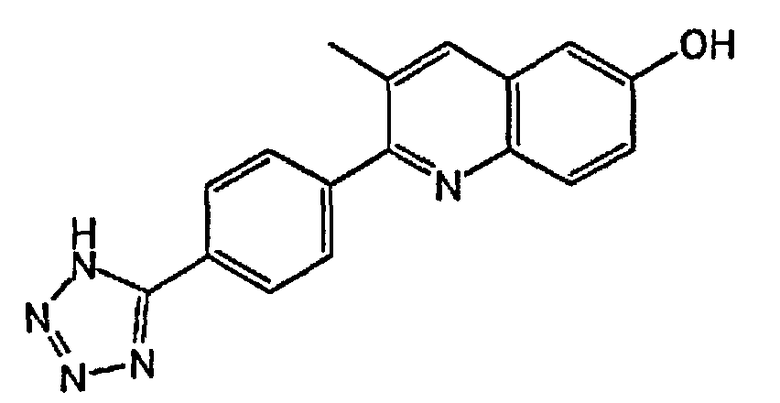
[00170] Следовали Схеме 2: условия A.
[00171] Этап 1: Синтез 2-(4-(1H-тетразол-5-ил)фенил)-6-метокси-3-метилхинолина.
[00172] К смеси 2-хлор-6-метокси-3-метилхинолина (100 мг, 0,482 ммоль), 4-(1H-тетразол-5-ил)фенилбороновой кислоты (91,2 мг, 0,482 ммоль), K2CO3 (199 мг, 1,45 ммоль) и PdCl2(dppf) (17,6 мг, 0,024 ммоль) добавили 7 мл DEGME и 3 мл H2O под аргоном. Смесь перемешивали при 150°C в микроволновом реакторе в течение 1,5 часов. Неочищенную смесь разбавили 1 н. NaOH (10 мл) и медленно подкислили до pH 4,0 при помощи концентрированной HCl. Твердые вещества отфильтровали для получения 128 мг (выход 84%) заданного продукта.
[00173] Этап 2: Синтез (2-(4-(1H-тетразол-5-ил)фенил)-3-метилхинолин-6-ола).
[00174] 2-(4-(1H-тетразол-5-ил)фенил)-6-метокси-3-метилхинолин (128 мг, 0,40 ммоль) растворили в 5 мл NMP, и к нему добавили Na2S (47 мг, 0,60 ммоль). Затем смесь перемешивали в течение 4 часов при 140°C в микроволновом реакторе. После концентрирования in vacuo неочищенный продукт растворили в 5 мл 1 н. NaOH и медленно подкислили при помощи 1 н. HCl до pH 4. Твердые вещества отфильтровали и высушили in vacuo для получения 46,2 мг (выход 38%) Соединения 2. 1H ЯМР (ДМСО-d6, 400 MHz): δ 10.10-10.00 (bs, 1H), 8.18-8.15 (d, 2H), 8.08 (s, 1H), 7.87-7.84 (m, 3H), 7.30-7.26 (d, 1H), 7.13 (s, 1H), 2.45 (s, 3H). MS (ESI): m/z 304.11 [M+H]+.
[00175] Пример 3: Соединение 3: 4-(6-гидроксихинолин-2-ил)бензойная кислота
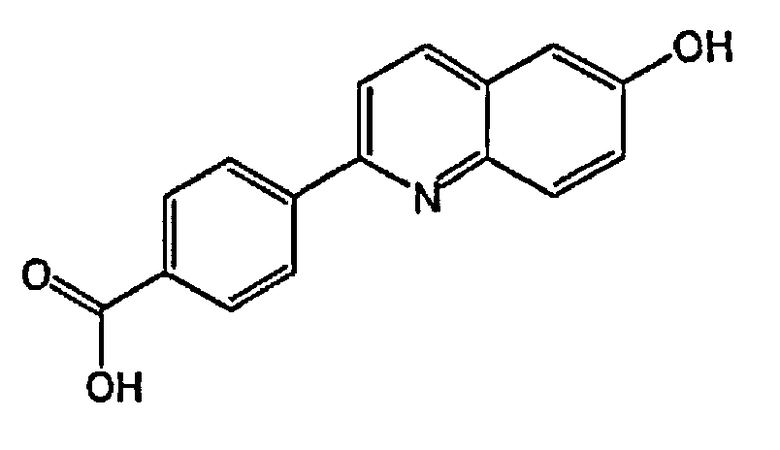
[00176] Следовали Схеме 2, условия A: исходные материалы: 2-хлор-6-метоксихинолин (Промежуточное соединение 1) (100 мг, 0,52 ммоль) и (4-(метоксикарбонил)фенил)бороновая кислота. 1H ЯМР (ДМСО-d6, 400 MHz): δ 13.06-12.90 (bs, 1H), 10.14 (s, 1H), 8.36-8.33 (d, 2H), 8.30-8.27 (d, 1H), 8.11-8.06 (m, 3H), 7.97-7.94 (d, 1H), 7.38-7.34 (dd, 1H), 7.20 (s, 1H). MS (ESI): m/z 266.08 [M+H]+.
[00177] Пример 4: Соединение 4: 2-(4-(1H-тетразол-5-ил)фенил)хинолин-6-ол
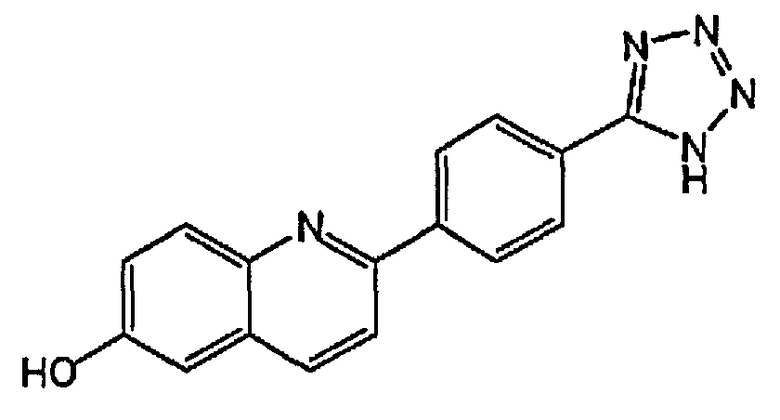
[00178] Следовали Схеме 2, условия A: исходные материалы: 2-хлор-6-метоксихинолин (Промежуточное соединение 1) и (4-(1H-тетразол-5-ил)фенил)бороновая кислота. 1H ЯМР (ДМСО-d6, 400 MHz): δ 10.15 (s, 1H), 8.45 (d, 2H), 8.31-8.28 (d, 1H), 8.22-8.12 (m, 3H), 7.98-7.95 (d, 1H), 7.39-7.35 (dd, 1H), 7.21 (s, 1H). MS (ESI): m/z 290.08 [M+H]+.
[00179] Пример 5: Соединение 5: 1-(6-гидроксихинолин-2-ил)пиперидин-4-карбоновая кислота
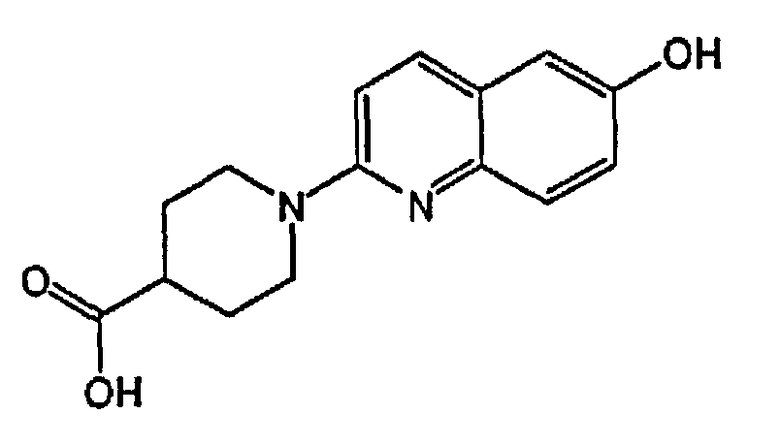
[00180] Этап 1: Синтез этил 4-(6-метоксихинолин-2-ил)циклогексан карбоксилата: Этил пиперидин-4-карбоксилат (100 мг) обрабатывали 2-хлор-6-метоксихинолином (Промежуточное соединение 1) (90 мг) в MeCN (0,8 мл) и ТЭА (100 мг) в закрытой пробирке при 180°C в течение 6 часов в микроволновом реакторе. После выделения продукта при помощи воды и EtOAc и колоночной очистки, элюируя EtOAc/гексаном, получили этил 4-(6-метоксихинолин-2-ил)циклогексанкарбоксилат (90 мг) в виде твердого вещества.
[00181] Этап 2: Синтез Соединения 5: Этил 4-(6-метоксихинолин-2-ил)циклогексанкарбоксилат (90 мг) обрабатывали этантиолатом натрия (150 мг) в NMP (2 мл) при 100°C в течение 48 часов. Заданный продукт (50 мг) очистили при помощи катионообменной колонки Dowex 50W X8, элюируя водой и 2 н. раствором NH4OH. 1H ЯМР (ДМСО-d6, 300 MHz): δ 7.82 (1H, d, J=9 Hz), 7.42 (1H, d, J=9 Hz), 7.13 (1H, d, J=9 Hz), 7.08 (1H, dd, J=3,9 Hz), 6.93 (1H, d, J=3 Hz), 4.2-4.4 (2H, m), 2.88-2.97 (2H, m), 2.15-2.21 (1H, m), 1.80-1.86 (2H, m), 1.45-1.60 (1H, m) ppm. MS (ESI): m/z 273.0 [M+1]+.
[00182] Пример 6: Соединение 6 (1r,4r)-4-(6-гидроксихинолин-2-ил)циклогексанкарбоновая кислота
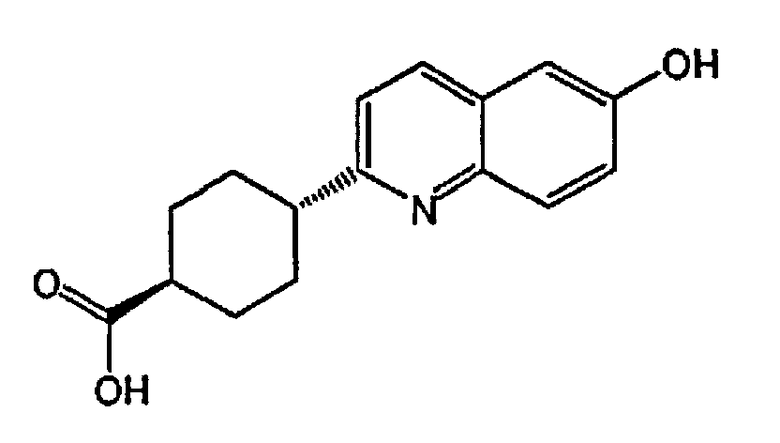
[00183] Этап 1: Синтез (1r,4r)-метил 4-(6-метоксихинолин-2-ил)циклогексанкарбоксилата и (1s,4s)-метил 4-(6-метоксихинолин-2-ил)циклогексанкарбоксилата: Метил 4-(6-гидроксихинолин-2-ил)циклогекс-3-енкарбоксилат (261 мг) (Соединение 16) растворили в EtOAc (10 мл) и смешали с 10% Pd/C (38,5 мг). Систему быстро вакуумировали и заполнили водородом. Эту процедуру повторили три раза. Реакционную смесь перемешивали под водородом в течение 3 часов. После фильтрации для удаления катализатора и концентрирования под пониженным давлением, полученную смесь разделили силикагелевой флэш-хроматографией, элюируя EtOAc/гексаном для получения (1s,4s)-метил 4-(6-метоксихинолин-2-ил)циклогексанкарбоксилата (153 мг) и (1r,4r)-метил 4-(6-метоксихинолин-2-ил)циклогексанкарбоксилата (72 мг) по отдельности.
[00184] Этап 2: Синтез Соединения 6: Следовали способу, описанному на Этапе 2 Примера 5, начиная с (1r,4r)-метил 4-(6-метоксихинолин-2-ил)циклогексанкарбоксилата (72 мг, продукт, полученный выше), для получения заданного Соединения 6. 1H ЯМР (ДМСО-d6, 300 MHz): δ 8.03 (1H, d, J=9 Hz), 7.75 (1H, d, J=9 Hz), 7.33 (1H, d, J=9 Hz), 7.24 (1H, dd, J=3,9 Hz), 7.08 (1H, d, J=3 Hz), 3.30 (1H, m), 2.76 (1H, m), 2.25-1.90 (4H, m) 1.75-1.50 (4Н, m) ppm. MS (ESI): m/z 272.0 [M+1]+.
[00185] Пример 7: Соединение 7: (1s,4s)-4-(6-гидроксихинолин-2-ил)циклогексанкарбоновая кислота
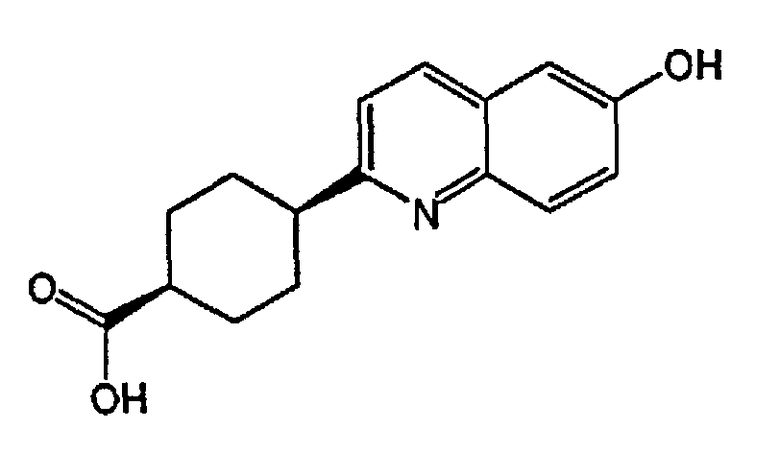
[00186] Следовали способу, описанному на Этапе 2 Примера 5, начиная с (1s,4s)-метил 4-(6-метоксихинолин-2-ил)циклогексанкарбоксилата (72 мг, смотри Пример 6. Этап 1, где описан синтез), для получения заданного Соединения 7. 1H ЯМР (ДМСО-d6, 300 MHz): δ 7.97 (1H, d, J=9 Hz), 7.74 (1H, d, J=9 Hz), 7.26 (1H, dd, J=3,9 Hz), 7.24 (1H, d, J=9 Hz), 7.08 (1H, d, J=3 Hz), 3.34 (1H, m), 2.75 (1H, m), 2.25-1.50 (8H, m) ppm. MS (ESI): m/z 272.0 [M+1]+.
[00187] Пример 8: Соединение 8: 3-хлор-4-(6-гидроксихинолин-2-ил)бензойная кислота
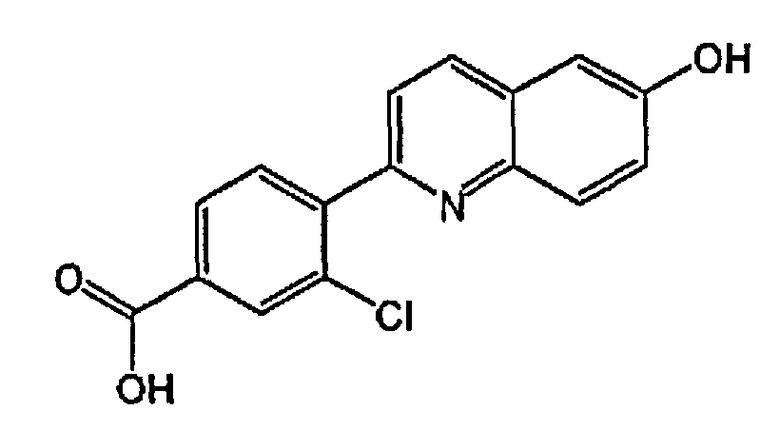
[00188] Следовали Схеме 2, условия B:
[00189] Этап 1: Синтез 3-хлор-4-(6-метоксихинолин-2-ил)бензойной кислоты:
[00190] Смесь 2-хлор-6-метоксихинолина (Промежуточное соединение 1) (200 мг, 1,04 ммоль), 4-карбокси-2-хлорфенилбороновой кислоты (247 мг, 1,24 ммоль) и K2CO3 (369 мг, 2,70 ммоль) в DEGME/H2O (7,0 мл/2,0 мл) три раза дегазировали под атмосферой N2. Затем добавили PdCl2(dppf) (75 мг, 0,104 ммоль) и нагревали смесь до 110°C в течение 3 часов под атмосферой N2. Реакционную смесь разбавили EtOAc (100 мл) и отфильтровали. Фильтрат промыли насыщенным солевым раствором (20 мл), высушили над Na2SO4, отфильтровали и концентрировали для получения 3-хлор-4-(6-метоксихинолин-2-ил)бензойной кислоты (150 мг, выход 46%) в виде твердого желтого вещества, которое использовали на следующем этапе без дополнительной очистки.
[00191] Этап 2: Синтез Соединения 8: К суспензии 3-хлор-4-(6-метоксихинолин-2-ил)бензойной кислоты (150 мг, 0,479 ммоль) в безводном CH2Cl2 (5 мл) добавили Al2Cl3 (320 мг, 2,40 ммоль). Реакционную смесь дефлегмировали в течение ночи. Смесь погасили насыщенным раствором NH4Cl (10 мл), а водный слой экстрагировали CH2Cl2/MeOH (об./об. = 10:1, 30 мл × 3). Объединенный органический слой промыли насыщенным солевым раствором, высушили над Na2SO4, отфильтровали и концентрировали для получения неочищенного продукта, который очистили препаративной ВЭЖХ (с добавлением 0,1% ТФК) для получения 3-хлор-4-(6-гидроксихинолин-2-ил)бензойной кислоты (25 мг, выход 18%). 1H ЯМР (ДМСО, 400 MHz): δ 10.20 (brs, 1H), 8.30 (d, J=8.4 Hz, 1H), 8.10-8.00 (m, 2H), 7.95 (d, J=9.2 Hz, 1H), 7.80 (d, J=8.0 Hz, 1H), 7.72 (d, J=8.8 Hz, 1H), 7.38 (dd, J=6.4, 2.8 Hz, 1H), 7.22 (d, J=2.4 Hz, 1H), MS (ESI): m/z 299.9 [M+H]+.
[00192] Пример 9: Соединение 9: 2-хлор-4-(6-гидроксихинолин-2-ил)бензойная кислота
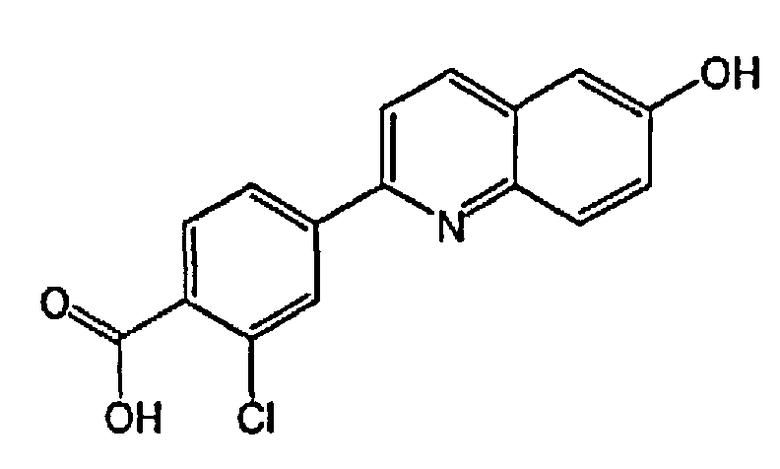
[00193] Следовали Схеме 3:
[00194] Смесь 2-хлорхинолин-6-ола (Промежуточное соединение 2) (50 мг, 0,270 ммоль), 4-бороно-2-хлорбензойной кислоты (55 мг, 0,270 ммоль), K2CO3 (75 мг, 0,540 ммоль) и Pd(dppf)Cl2 (25 мг, 0,0306 ммоль) в 2-(2-метоксиэтокси)этаноле (1,5 мл) и воде (0,4 мл) перемешивали под атмосферой N2 при 130°C в течение 3 часов. Полученную смесь охладили до комнатной температуры и отфильтровали. Фильтрат концентрировали под пониженным давлением, а остаток очистили препаративной ВЭЖХ (с добавлением 0,1% ТФК) для получения Соединения 9 (33 мг, выход 40%) в виде твердого желтого вещества. 1H ЯМР (CD3OD, 400 MHz): δ 8.39 (d, J=8.4 Hz, 1H), 8.29 (d, J=1.6 Hz, 1H), 8.16-8.02 (m, 4H), 7.47 (dd, J=9.2, 2.8 Hz, 1H), 7.25 (d, 7=2.4 Hz, 1H). MS (ESI): m/z 298.0 [M-1]-.
[00195] Пример 10: Соединение 10: 2-фтор-4-(6-гидроксихинолин-2-ил)бензойная кислота
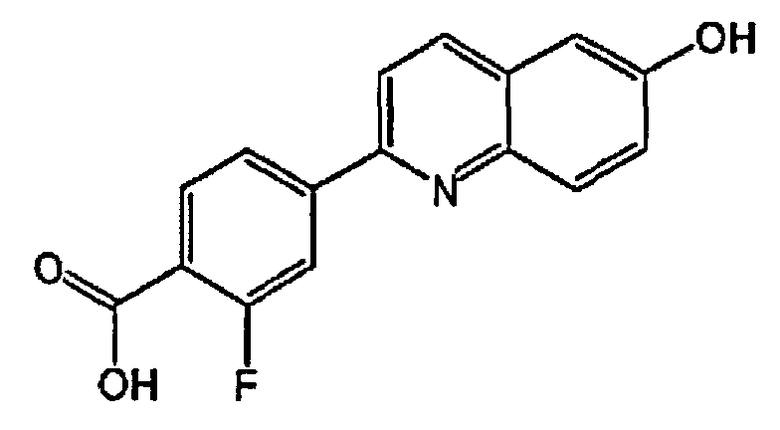
[00196] Следовали Схеме 3: Исходные материалы: 2-хлорхинолин-6-ол (Промежуточное соединение 2) и 4-бороно-2-фторбензойная кислота 1H ЯМР (CD3OD, 400 MHz): δ 8.54 (d, J=8.4 Hz, 1H), 8.20-8.06 (m, 3H), 8.06-7.96 (m, 2H), 7.55 (dd, J=9.2, 2.4 Hz, 1H), 7.32 (d, J=2.8 Hz, 1H). MS (ESI): m/z 283.6 [M+1]+.
[00197] Пример 11: Соединение 11: 2-(4-(2H-тетразол-5-ил)фенил)-4-хлорхинолин-6-ол
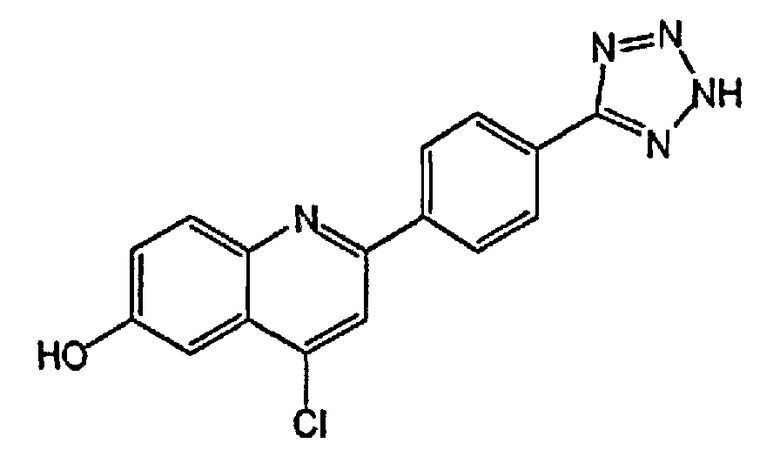
[00198] Этап 1: Синтез 4-(4-хлор-6-метоксихинолин-2-ил)бензонитрила: Следовали Схеме 2, Этап 1, начиная с 4-цианофенилбороновой кислоты и 2,4-дихлор-6-метоксихинолина, где используемым растворителем был ДМФ, а используемым катализатором был Pd(PPh3)4. Смесь нагревали до 100°C в течение 3 часов, оставили остывать, а затем вылили в ледяную воду. Полученное твердое вещество отделили фильтрацией, промыли водой и высушили с последующей перекристаллизацией из метанола для получения заданного продукта.
[00199] Этап 2: Синтез 4-(4-хлор-6-гидроксихинолин-2-ил)бензонитрила: Следовали способу Схемы 1, этап 2, с выделением при помощи этилацетата/воды. В результате очистки препаративной TCX получили заданный продукт.
[00200] Этап 3: Синтез 2-(4-(2H-тетразол-5-ил)фенил)-4-хлорхинолин-6-ола: Следовали Схеме 4, путь A. К раствору 4-(4-хлор-6-гидроксихинолин-2-ил)бензонитрила (65 мг, 0,23 ммоль) в толуоле (2 мл) добавили TMSN3 (455 мг, 4,18 ммоль) и Bu2SnO (15 мг, 0,069 ммоль) при комнатной температуре. Смесь нагревали с дефлегматором в течение ночи. Летучие вещества удалили под пониженным давлением. Остаток очистили препаративной ВЭЖХ для получения Соединения 11 (10 мг, 13,5%). 1H ЯМР (MeOD-d4, 500 MHz): δ 8.34 (d, J=8.5 Hz, 2H), 8.21 (s, 1H), 8.19 (s, 2H), 8.07 (d, J=9.0 Hz, 1H), 7.51 (d, J=2.5 Hz, 1H), 7.47 (dd, J=2.5 Hz, J=9.5 Hz, 1H). MS (ESI): m/z 324.0 [M+1]+.
[00201] Пример 12: Соединение 12: 3-(4-(6-гидроксихинолин-2-ил)фенил)-1,2,4-оксадиазол-5(2H)-он
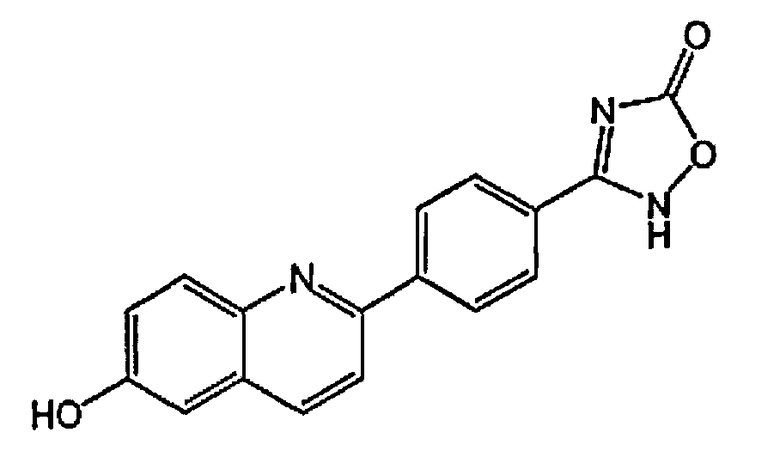
[00202] Этап 1; Синтез 4-(6-гидроксихинолин-2-ил)бензонитрила: Следовали Схеме 3, начиная с 2-хлорхинолин-6-ола (Промежуточное соединение 2) и 4-цианофенилбороновой кислоты, и используя раствор 1,4-диоксан : H2O. Реакция протекала при 100°C в микроволновом реакторе в течение 1 часа. После выделения при помощи этилацетата, выполнили колоночную хроматографию (градиент от 5% до 50% EtOAc в гексанах).
[00203] Этап 2: Синтез N-гидрокси-4-(6-гидроксихинолин-2-ил)бензимидамида. Смотри Схему 4, путь B. 4-(6-гидроксихинолин-2-ил)бензонитрил (900 мг, 3,68 ммоль) растворили в 25 мл EtOH, и к нему добавили NH2OH·HCl (500 мг, 7,36 ммоль) и ТЭА (1,5 мл). Смесь перемешивали при 80°C в течение 2 часов, а затем концентрировали in vacuo. Неочищенные твердые вещества затем суспендировали в 25 мл H2O и перемешивали в течение 1 часа. В результате фильтрации и высушивания твердых веществ получили заданный продукт (800 мг, выход 78%).
[00204] Этап 3: Синтез (3-(4-(6-гидроксихинолин-2-ил)фенил)-1,2,4-оксадиазол-5(2H)-она): N-гидрокси-4-(6-гидроксихинолин-2-ил)бензимидамид (800 мг, 2,86 ммоль) растворили в 25 мл ТГФ и добавили CDI (557 мг, 3,44 ммоль) и ТЭА (0,2 мл). Смесь перемешивали при 65°C в течение 2 часов, а затем концентрировали in vacuo. Неочищенный материал растворили в 10 мл 1 н. NaOH и отфильтровали через целит. Затем смесь подкислили 1 н. HCl до pH 4,5, а твердые вещества отфильтровали и высушили. Твердые вещества суспендировали в 10 мл EtOAc в течение ночи при 50°C, а затем отфильтровали для получения Соединения 12 (315 мг, выход 36%). 1H ЯМР (ДМСО-d6, 400 MHz): δ 10.19 (s, 1H), 8.42 (d, 2H), 8.28 (d, 1H), 8.14-8.11 (d, 1H), 8.00-7.94 (m, 3H), 7.39-7.35 (dd, 1H), 7.21 (s, 1H). MS (ESI): m/z 306.44 [M+H]+.
[00205] Пример 13: Соединение 13: 3-фтор-4-(6-гидроксихинолин-2-ил)бензойная кислота
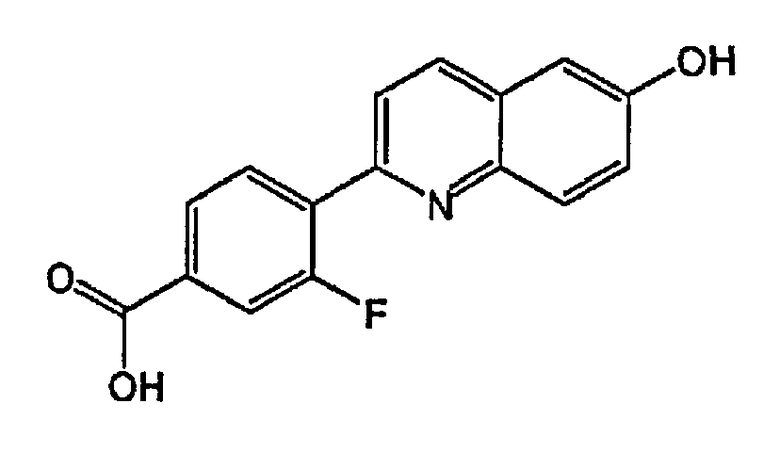
[00206] Следовали Схеме 3: Исходные материалы: 2-хлорхинолин-6-ол и 4-бороно-3-фторбензойная кислота. 1H ЯМР (CD3OD, 400 MHz): δ 8.31 (d, J=8.4 Hz, 1H), 7.94-7.86 (m, 3H), 7.81-7.74 (m, 2H), 7.35 (dd, J=9.2, 2.8 Hz, 1H), 7.15 (d, J=2.8 Hz, 1H). MS (ESI): m/z 283.6 [M+H]+.
[00207] Пример 14: Соединение 14: 4-(6-гидроксихинолин-2-ил)-3-метоксибензойная кислота
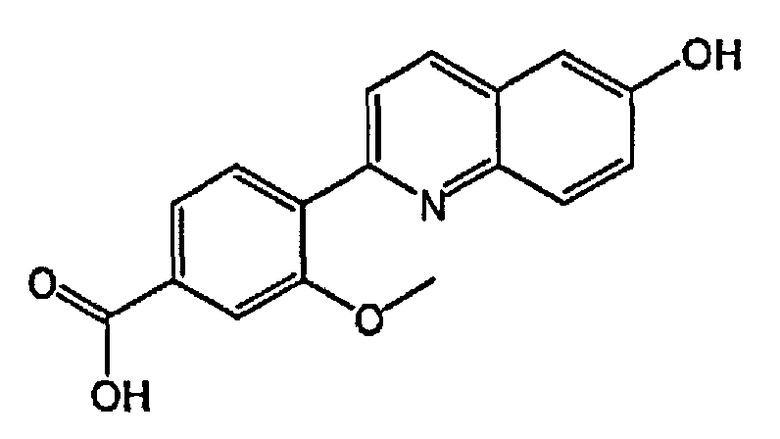
[00208] Следовали Схеме 3: Исходные материалы: 2-хлорхинолин-6-ол и 4-бороно-3-метоксибензойная кислота. 1H ЯМР (CD3OD, 400 MHz): δ 8.81 (d, J=8.4 Hz, 1H), 8.14 (d, J=9.2 Hz, 1H), 8.10 (d, 7=8.4 Hz, 1H), 7.89 (dd, J=6.8, 1.6 Hz, 2H), 7.85 (d, J=8.4 Hz, 1H), 7.69 (dd, J=9.2, 2.8 Hz, 1H), 7.48 (d, J=2.8 Hz, 1H). MS (ESI): m/z 295.7 [M+H]+.
[00209] Пример 15: Соединение 15: 5-(6-гидроксихинолин-2-ил)тиофен-2-карбоновая кислота
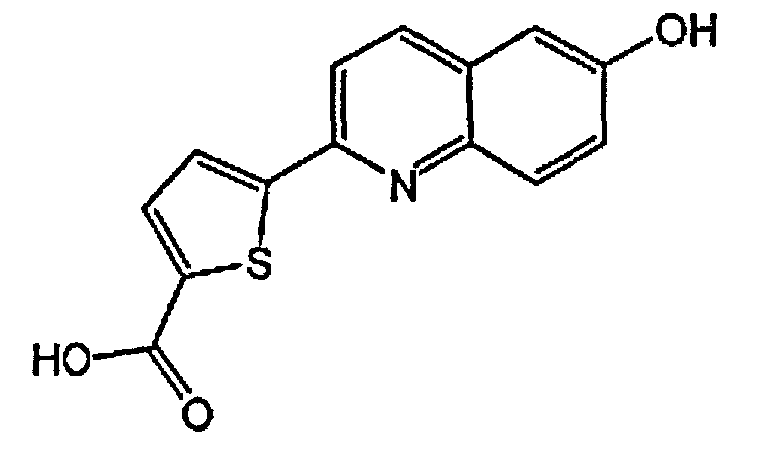
[00210] Следовали Схеме 3: Исходные материалы: 2-хлорхинолин-6-ол и 5-боронотиофен-2-карбоновая кислота. 1H ЯМР (CD3OD, 400 MHz): δ 8.25 (d, J=8.8 Hz, 1H), 7.97 (dd, J=9.2, 3.2 Hz, 2H), 7.88-7.82 (m, 2H), 7.41 (dd, J=9.2, 2.8 Hz, 1H), 7.19 (d, J=2.4 Hz, 1H). MS (ESI): m/z 271.6 [M+H]+.
[00211] Пример 16: Соединение 16: 4-(6-гидроксихинолин-2-ил)циклогекс-3-енкарбоновая кислота
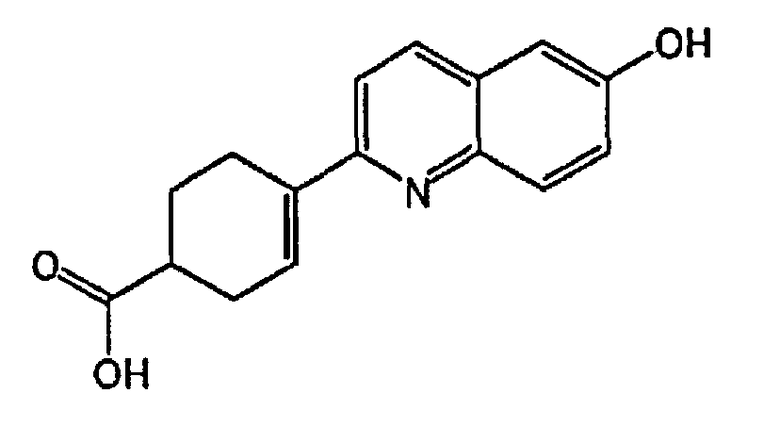
[00212] Этап 1: Синтез метил 4-(6-гидроксихинолин-2-ил)циклогекс-3-енкарбоксилата: Следовали Схеме 3: исходные материалы: 2-хлорхинолин-6-ол и метил 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)циклогекс-3-енкарбоксилат.
[00213] Этап 2: Синтез 4-(6-гидроксихинолин-2-ил)циклогекс-3-енкарбоновой кислоты: Основные условия гидролиза с LiOH дают конечный заданный продукт. 1H ЯМР (ДМСО-d6, 300 MHz): δ 12.24 (1H, s), 9.92 (1H, s), 8.04 (1H, d, J=9 Hz), 7.75 (1H, d, J=9 Hz), 7.66 (1H, d, J=9 Hz), 7.24 (1H, dd, J=3,9 Hz), 7.08 (1H, d, J=3 Hz), 6.74 (1H, s), 3.30 (1H, m), 2.84 (1H, m), 2.50 (4H, m), 2.08 (1H, m), 1.71 (1H, m) ppm. MS (ESI): m/z 270.0 [M+1]+.
[00214] Пример 17: Соединение 17: 4-(3-фтор-6-гидроксихинолин-2-ил)бензойная кислота
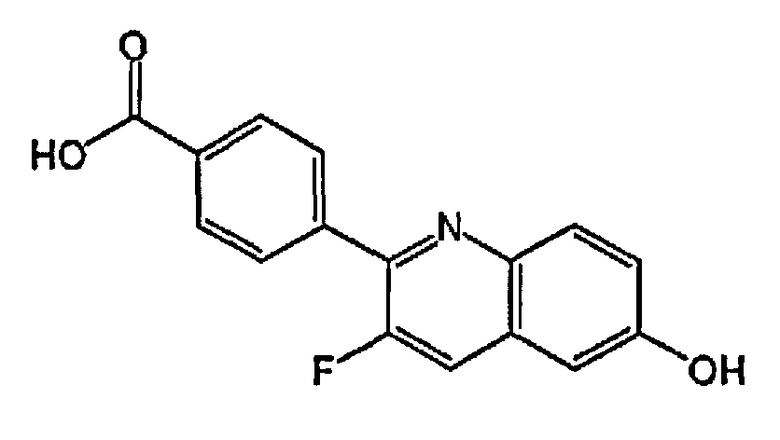
[00215] Этап 1: Синтез 4-(4-хлор-3-фтор-6-метоксихинолин-2-ил)бензойной кислоты: Следовали Схеме 3, где исходными материалами были 2,4-дихлор-3-фтор-6-метоксихинолин (Промежуточное соединение 3) и 4-боронобензойная кислота, где неочищенный продукт очистили силикагелевой колонкой (PE/EtOAc=1/1) для получения смеси соединения 4-(4-хлор-3-фтор-6-метоксихинолин-2-ил)бензойной кислоты и4-(3-фтор-6-метоксихинолин-2-ил)бензойной кислоты, которую использовали на следующем этапе без дополнительной очистки.
[00216] Этап 2: Синтез 4-(3-фтор-6-метоксихинолин-2-ил)бензойной кислоты: К раствору смеси из Этапа 1 в абсолютном MeOH (5 мл) добавили Pd/C (10% Pd, 100 мг). Смесь перемешивали при 25°C в течение 1 часа под атмосферой H2. Твердые вещества отфильтровали, а фильтрат концентрировали для получения продукта (60 мг, выход за два этапа 66%).
[00217] Этап 3: Синтез 4-(3-фтор-6-гидроксихинолин-2-ил)бензойной кислоты: Следовали Схеме 2, этап 2, условия B. 1H ЯМР (ДМСО, 400 MHz): δ 13.35 (brs, 1H), 10.40 (brs, 1H), 8.25 (d, J=12.8 Hz, 1H), 8.27 (s, 4H), 8.01 (d, J=9.2 Hz, 1H), 7.40 (dd, J=8.8, 2.4 Hz, 1H), 7.25 (d, J=2.8 Hz, 1H).
[00218] Пример 18: Соединение 18: 4-(4-хлор-3-фтор-6-гидроксихинолин-2-ил)бензойная кислота
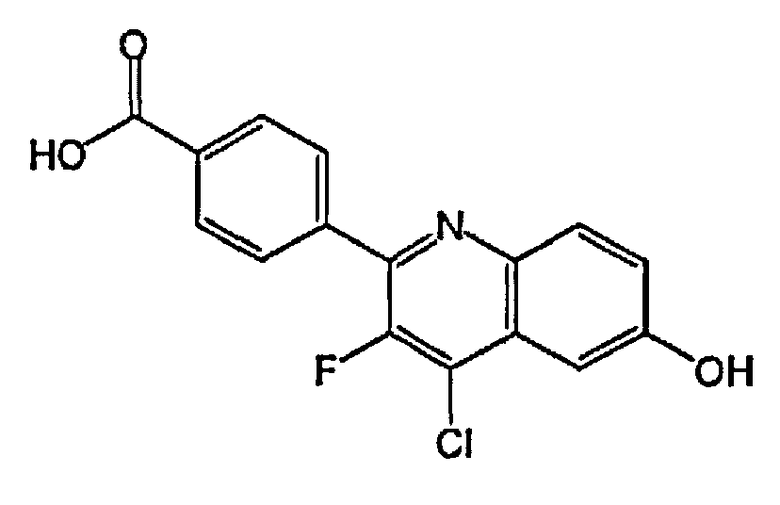
[00219] Этап 1: Синтез 4-(4-хлор-3-фтор-6-метоксихинолин-2-ил)бензойной кислоты: Синтез описан на Этапе 1 Примера 17.
[00220] Этап 2: Синтез 4-(4-хлор-3-фтор-6-гидроксихинолин-2-ил)бензойной кислоты: Следовали Схеме 2, Этап 2, условия B. 1H ЯМР (ДМСО, 400 MHz): δ 13.25 (brs, 1H), 10.75 (brs, 1H), 8.10 (s, 4H), 8.05 (d, J=9.2 Hz, 1H), 7.41 (dd, J=9.2, 2.8 Hz, 1H), 7.35 (d, J=2.4 Hz, 1H).
[00221] Пример 19: Соединение 19: 4-(3-хлор-6-гидроксихинолин-2-ил)бензойная кислота
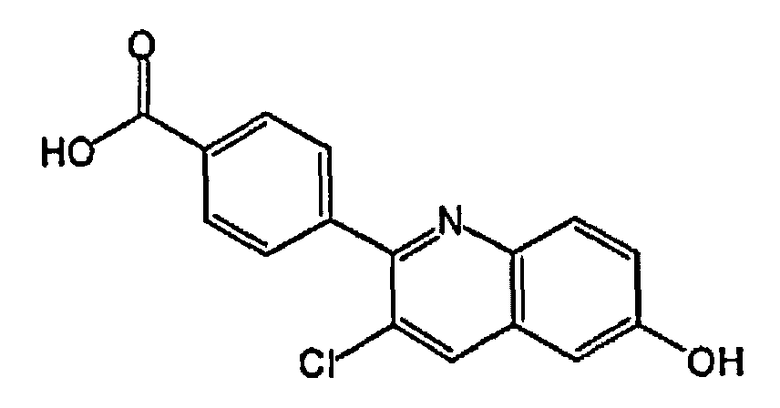
[00222] Этап 1: Синтез 4-(3-хлор-6-метоксихинолин-2-ил)бензойной кислоты: Следовали Схеме 3, где исходными материалами были 2,3-дихлор-6-метоксихинолин (Промежуточное соединение 4) и 4-боронобензойная кислота.
[00223] Этап 2: Синтез 4-(4-хлор-3-фтор-6-гидроксихинолин-2-ил)бензойной кислоте: Следовали Схеме 2, Этап 2, условия B. 1H ЯМР (CD3OD, 400 MHz): δ 8.35 (s, 1H), 8.17 (d, J=8.0 Hz, 2H), 7.95 (d, J=9.2 Hz, 1H), 7.80 (d, J=8.0 Hz, 2H), 7.40 (dd, J=9.2, 2.4 Hz, 1H), 7.15 (d, J=2.8 Hz, 1H). MS (ESI): m/z 299.8 [M+H]+.
[00224] Пример 20: Соединение 20: 3-(2-фтор-4-(6-гидроксихинолин-2-ил)фенил)-1,2,4-оксадиазол-5(4H)-он
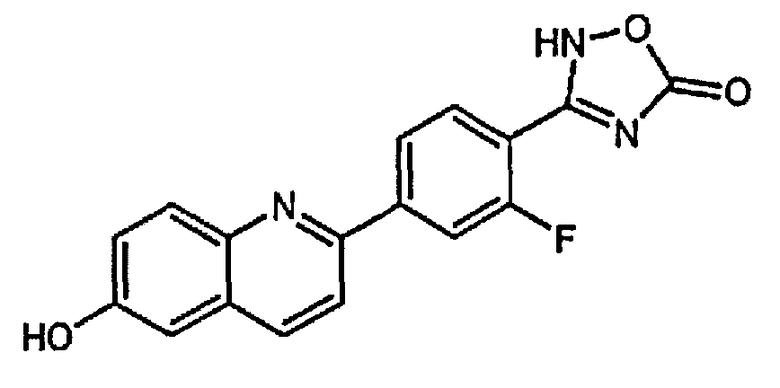
[00225] Этап 1: Синтез 2-фтор-4-(6-гидроксихинолин-2-ил)бензонитрила: Следовали Схеме 3, начиная с 2-хлорхинолин-6-ола (Промежуточное соединение 2) и 4-циано-3-фторфенилбороновой кислоты, где неочищенный продукт очистили силикагелевой колоночной хроматографией.
[00226] Этап 2 и 3: Синтез 3-(2-фтор-4-(6-гидроксихинолин-2-ил)фенил)-1,2,4-оксадиазол-5(4H)-она. Следовали Схеме 4, путь B, этап 1: После удаления растворителя in vacuo, неочищенный 2-фтор-N-гидрокси-4-(6-гидроксихинолин-2-ил)бензимидамид использовали без очистки. Следовали Схеме 4, путь B, этап 2: В результате очистки на силикагелевой колонке, элюируя 10% МеОН в ДХМ, получили заданный продукт. 1H ЯМР (ДМСО-d6, 300 MHz): δ 10.22 (1H, s), 8.27-8.33 (2H, m), 8.16 (1H, d, J=9 Hz), 7.91-7.99 (2H, m), 7.66 (1H, d, J=9 Hz), 7.39 (1H, dd, J=3,9 Hz), 7.21 (1H, d, J=3 Hz) ppm. MS (ESI): m/z 324.0 [M+1]+.
[00227] Пример 21: Соединение 21: 3-(3-фтор-4-(6-гидроксихинолин-2-ил)фенил)-1,2,4-оксадиазол-5(4H)-он
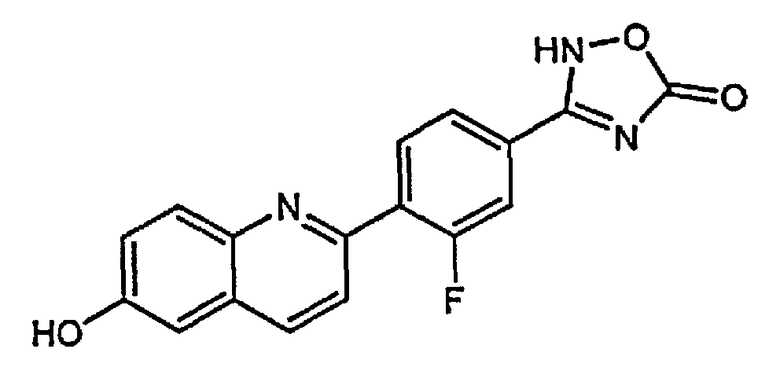
[00228] Следовали способу, описанному для Соединения 20 в Примере 20, начиная с 2-хлорхинолин-6-ола (Промежуточное соединение 2) и 4-циано-2-фторфенилбороновой кислоты. 1H ЯМР (ДМСО-d6, 300 MHz): δ 10.22 (1H, s), 8.27-8.33 (2H, m), 7.97 (1H, d, J=9 Hz), 7.77-7.82 (2H, m), 7.39 (1H, dd, J=3,9 Hz), 7.21 (1H, d, J=3 Hz) ppm. MS (ESI): m/z 324.0 [M+1]+.
[00229] Пример 22: Соединение 22: 4-(4-хлор-6-гидроксихинолин-2-ил)бензойная кислота
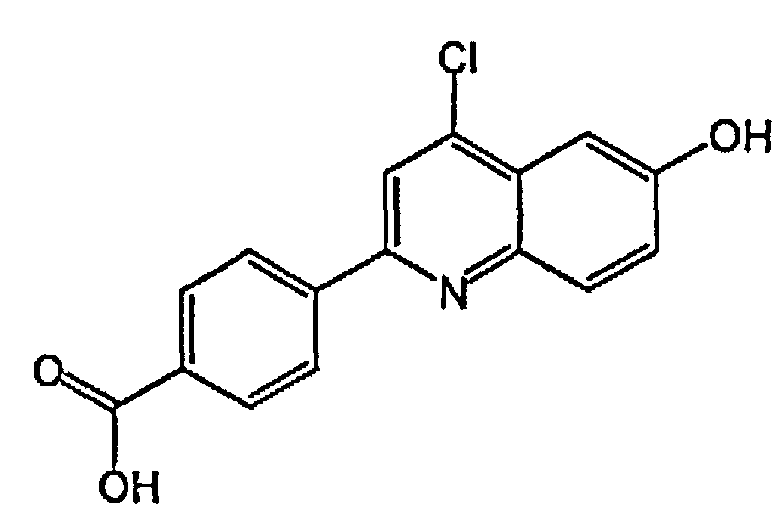
[00230] Этап 1: Синтез 4-(4-хлор-6-гидроксихинолин-2-ил)бензонитрила: Следовали Этапу 2 Схемы 1, начиная с 4-(4-хлор-6-метоксихинолин-2-ил)бензонитрила (смотри этап 1 Примера 11, где описан синтез).
[00231] Этап 2: Синтез 4-(4-хлор-6-гидроксихинолин-2-ил)бензойной кислоты: Раствор 4-(4-хлор-6-гидроксихинолин-2-ил)бензонитрила (40 мг, 0,14 ммоль), концентрированной HCl (1 мл) и диоксана (1 мл) нагревали при 90°C в течение ночи. Смесь экстрагировали этилацетатом. Органический слой концентрировали и очистили препаративной ВЭЖХ для получения Соединения 22 (10 мг, выход 23%). 1H ЯМР (MeOD-d4, 500 MHz): 8.16 (d, J=8.0 Hz, 2H), 8.10 (d, J=8.5 Hz, 2H), 8.07 (s, 1H), 7.96 (d, J=9.0 Hz, 1H), 7.41 (d, J=2.5 Hz, 1H), 7.35 (dd, J=3.0 Hz, J=9.0 Hz, 1H). MS 5 (ESI): m/z 300 [M+1]+.
[00232] Пример 23: Соединение 23: 2-(2-хлор-4-(2H-тетразол-5-ил)фенил)хинолин-6-ол
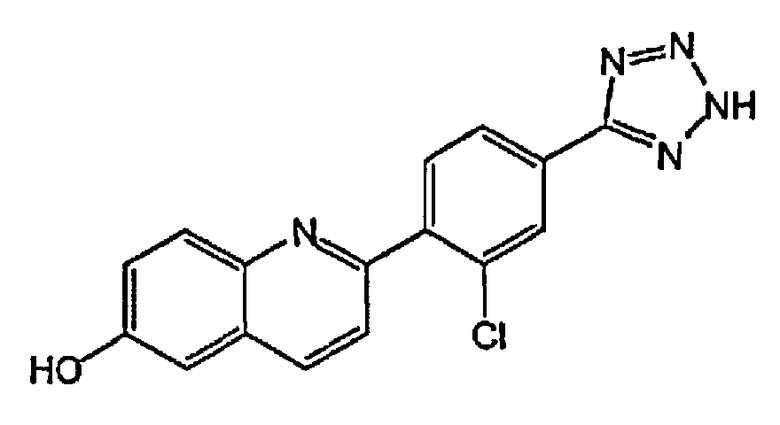
[00233] Иллюстративный способ для Схемы 5, Соединение C.
[00234] Этап 1: Синтез 3-хлор-4-(6-гидроксихинолин-2-ил)бензамида: Смесь 3-хлор-4-(6-гидроксихинолин-2-ил)бензойной кислоты (Соединение 8, Пример 8) (400 мг, 1,33 ммоль) и SOCl2 (10 мл) дефлегмировали в течение 1 часа, затем концентрировали под пониженным давлением для получения неочищенного продукта (400 мг) в виде грязновато-белого твердого вещества. К этому твердому веществу добавили NH4OH (10 мл) и перемешивали реакционную смесь при 30°C в течение 1 часа. Полученную смесь подкислили водным раствором HCl (2 М) до pH=6 и экстрагировали EtOAc (50 мл × 3), высушили над Na2SO4 и концентрировали для получения неочищенного продукта (360 мг) в виде твердого вещества.
[00235] Этап 2: Синтез 3-хлор-4-(6-гидроксихинолин-2-ил)бензонитрила: Смесь неочищенного 3-хлор-4-(6-гидроксихинолин-2-ил)бензамида (360 мг), ангидрида ТФКА (505 мг, 2,40 ммоль) и Et3N (364 мг, 3,62 ммоль) в ДХМ (20 мл) перемешивали при 30°C в течение ночи. Полученную смесь суспендировали в воде (50 мл) и экстрагировали EtOAc (50 мл × 3). Объединенные органические слои высушили над Na2SO4 и концентрировали под пониженным давлением. Остаток очистили колоночной хроматографией на силикагеле (PE/EtOAc=10/1) для получения прдоукта (250 мг, выход за 3 этапа 67%) в виде твердого вещества.
[00236] Этап 3: Синтез 2-(2-хлор-4-(2H-тетразол-5-ил)фенил)хинолин-6-ола: Смесь 3-хлор-4-(6-гидроксихинолин-2-ил)бензонитрила (230 мг, 0,819 ммоль), NaN3 (55 мг, 0,820 ммоль) и LiCl (70 мг, 1,64 ммоль) в монометиловом эфире диэтиленгликоля (5 мл) дефлегмировали в течение 4 часов. Полученную смесь охладили, отфильтровали через слой силикагеля и очистили препаративной ВЭЖХ (с добавлением 0,1% ТФК) для получения Соединения 23 (32 мг, выход 12%). 1H ЯМР (CD3OD, 400 MHz): δ 8.52 (d, J=8.4 Hz, 1H), 8.34 (s, 1H), 8.21 (d, J=8.0 Hz, 1H), 8.04 (d, J=9.2 Hz, 1H), 7.92-7.82 (m, 2H), 7.54 (dd, J=9.2, 2.0 Hz, 1H), 7.34 (d, J=2.0 Hz, 1H). MS (ESI): m/z 323.6 [M+H]+.
[00237] Пример 24: Соединение 24: 5-(4-(6-гидроксихинолин-2-ил)фенил)-1,3,4-оксадиазол-2(3H)-он
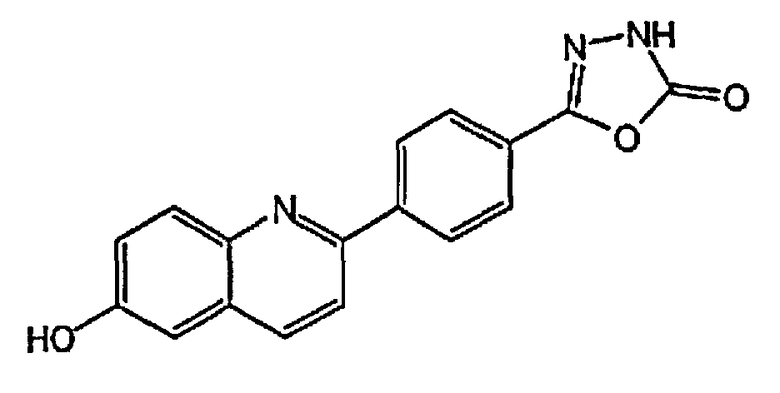
[00238] Иллюстративный способ для Схемы 5, Соединение B.
[00239] Этап 1: Синтез трет-бутил 2-(4-(6-гидроксихинолин-2-ил)бензоил)гидразинкарбоксилата: Смесь 4-(6-гидроксихинолин-2-ил)бензойной кислоты (Соединение 3, Пример 3) (200 мг, 0,754 ммоль), EDCI (145 мг, 0,754 ммоль) и BocNHNH2 (100 мг, 0,754 ммоль) в ДХМ (10 мл) и ДМФ (10 мл) перемешивали при 25°C в течение ночи, а затем выделили из воды/EtOAc. Неочищенный продукт очистили колоночной хроматографией на силикагеле (PE/EtOAc=1/1) для получения продукта (200 мг, выход 70%) в виде твердого желтого вещества.
[00240] Этап 2: Синтез 4-(6-гидроксихинолин-2-ил)бензогидразида: Смесь трет-бутил 2-(4-(6-гидроксихинолин-2-ил)бензоил)гидразинкарбоксилата (240 мг, 0,633 ммоль) и HCl/MeOH (20 мл, 4 M) перемешивали при 25°C в течение ночи. Полученную смесь концентрировали под пониженным давлением до сухости для получения продукта (120 мг, выход 68%).
[00241] Этап 3: Синтез 5-(4-(6-гидроксихинолин-2-ил)фенил)-1,3,4-оксадиазол-2(3H)-она: Смесь 4-(6-гидроксихинолин-2-ил)бензогидразида (90 мг, 0,322 ммоль) и CDI (454 мг, 3,22 ммоль) в ДХМ (20 мл) дефлегмировали в течение ночи. Полученную смесь охладили до комнатной температуры, а затем выделили из воды/EtOAc. Неочищенный продукт очистили препаративной ВЭЖХ (с добавлением 0,1% ТФК) для получения Соединения 24 (18 мг, выход 11%). 1H ЯМР (CD3OD, 400 MHz): δ 8.64-8.48 (m, 1H), 8.38-7.96 (m, 6H), 7.66-7.24 (m, 2H). MS (ESI): m/z 305.7 [M+H]+.
[00242] Пример 25: Соединение 25: 3-(диметиламино)-4-(6-гидроксихинолин-2-ил)бензойная кислота
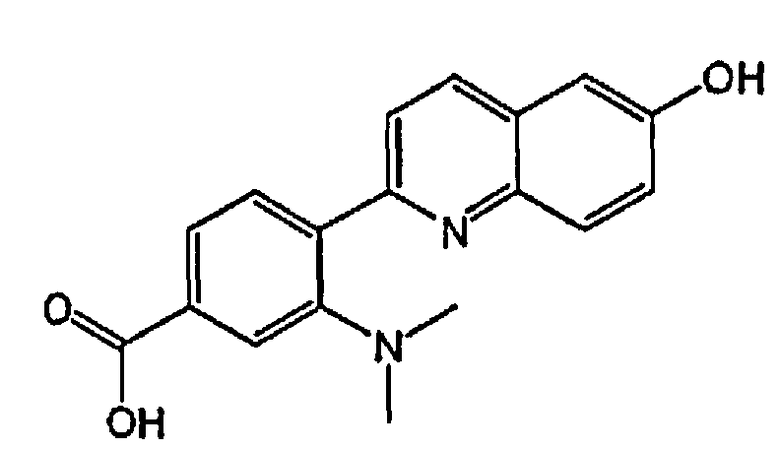
[00243] Этап 1: Синтез метил 3-амино-4-(6-метоксихинолин-2-ил)бензоата: Следовали Схеме 2, этап 1, условия B, начиная с 2-амино-4-(метоксикарбонил)фенилбороновой кислоты и 2-хлор-6-метоксихинолана, с очисткой колоночной хроматографией на силикагеле (PE/EtOAc=8/1) для получения продукта.
[00244] Этап 2: Синтез метил 3-(диметиламино)-4-(6-метоксихинолин-2-ил)бензоата: К раствору полученного выше соединения (400 мг, 1,30 ммоль) в MeOH/CH2Cl2 (10 мл/20 мл) добавили водный HCHO (37% в воде, 0,4 мл), а затем NaBH3CN (327 мг, 5,19 ммоль) и ZnCl2 (348 мг, 2,60 ммоль) при 0°C. Смесь перемешивали при 30°C в течение 6 часов. Смесь погасили ледяной водой, водный слой экстрагировали CH2Cl2 (30 мл × 3), объединенный органический слой промыли насыщенным солевым раствором (30 мл), высушили над безводным Na2SO4 и концентрировали in vacuo для получения продукта (400 мг, выход 92%).
[00245] Этап 3: Синтез метил 3-(диметиламино)-4-(6-гидроксихинолин-2-ил)бензоата: Следовали Схеме 2, этап 2, условия B, где неочищенный продукт очистили колоночной хроматографией на силикагеле (РЕ/EtOAc=5/1) для получения заданного продукта.
[00246] Этап 4: Синтез 3-(диметиламино)-4-(6-гидроксихинолин-2-ил)бензойной кислоты: К раствору полученного выше соединения (290 мг, 0,901 ммоль) в ТГФ/MeOH (8 мл/4 мл) добавили водный раствор LiOH (1 M, 4 мл). Смесь перемешивали при 30°C в течение 3 часов. Смесь разбавили H2O (10 мл), водный слой нейтрализовали 2 M HCl до pH 7. После выделения продукта при помощи воды/CH2Cl2 выполнили колоночную хроматографию на силикагеле (CH2Cl2/MeOH=10/1) для получения Соединения 25 (80 мг, выход 29%). 1H ЯМР (MeOD-d4, 400 MHz): δ 8.15 (d, J=8.8 Hz, 1H), 7.95 (d, J=9.2 Hz, 1H), 7.87-7.81 (m, 2H), 7.76 (d, J=8.0 Hz, 1H), 7.60 (d, J=8.0 Hz, 1H), 7.38 (dd, J=8.8, 2.4 Hz, 1H), 7.20 (d, J=2.4 Hz, 1H), 2.65 (s, 6H). MS (ESI): m/z 308.9 [M+H]+.
[00247] Пример 26: Соединение 26: 4-(4-фтор-6-гидроксихинолин-2-ил)бензойная кислота
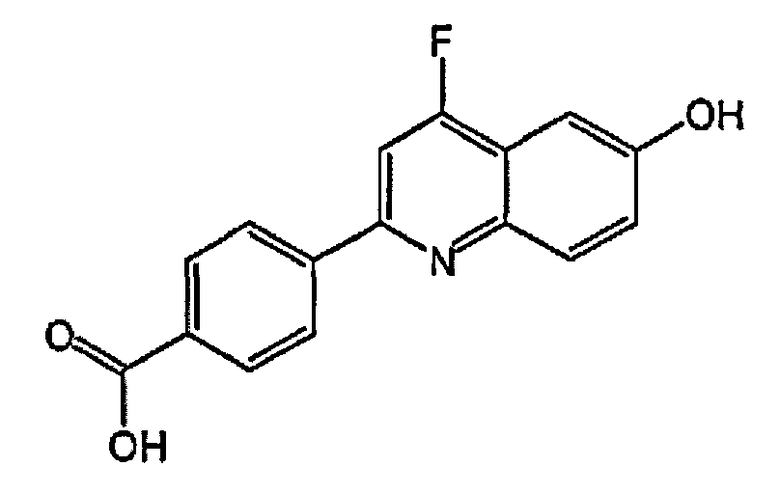
[00248] Этап 1: Синтез 4-(4-фтор-6-метоксихинолин-2-ил)бензонитрила: Смесь 4-(4-хлор-6-метоксихинолин-2-ил)бензонитрила (смотри Пример 11, этап 1) (1 г, 3,4 ммоль), CsF (5,2 г, 34 ммоль) и n-Bu4NBr (109 мг, 0,34 ммоль) в ДМСО (10 мл) нагревали до 150°C в течение 2 часов. После выделения продукта при помощи воды/EtOAc выполнили очистку колоночной хроматографией (PE/EA=10/1) для получения заданного продукта (300 мг, 31,7%).
[00249] Этап 2: Синтез 4-(4-фтор-6-гидроксихинолин-2-ил)бензонитрила: Следовали Этапу 2 Схемы 1, где после выделения продукта при помощи воды/EtOAc выполнили колоночную хроматографию (PE:EA=4:1) для получения заданного продукта.
[00250] Этап 3: Синтез 4-(4-фтор-6-гидроксихинолин-2-ил)бензойной кислоты: Смесь полученного выше соединения (100 мг, 0,38 ммоль) и NaOH (152 мг, 3,8 ммоль) в 1,4-диоксане/H2O (1 мл /1 мл) нагревали с дефлегматором в течение ночи. Летучие вещества удалили под пониженным давлением. Остаток очистили препаративной ВЭЖХ, а затем препаративной TCX (EtOAc) для получения Соединения 26 (10 мг, 9,3%). 1H ЯМР (MeOD-d4, 500 MHz): 8.22-8.17 (m, 4H), 8.04 (d, J=9.5 Hz, 1H), 7.79 (d, J=12.0 Hz, 1H). 7.43 (dd, J=2.5 Hz, J=9.0 Hz, 1H), 7.31 (d, J=2.0 Hz, 1H). MS (ESI): m/z 284.0 [M+1]+.
[00251] Пример 27: Соединение 27: 4-(6-гидроксихинолин-2-ил)-3-метилбензойная кислота
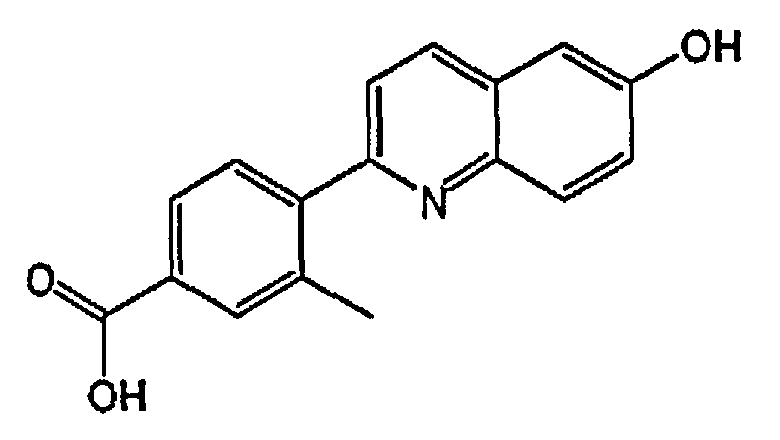
[00252] Следовали Схеме 3: Исходные материалы: 2-хлорхинолин-6-ол и метил 3-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензоат. 1H ЯМР (ДМСО-d6, 400 MHz): δ 10.45 (brs, 1H), 8.61-8.46 (m, 1H), 8.03 (d, J=8.8 Hz, 1H), 7.96 (s, 1H), 7.92 (d, J=8.0 Hz, 1H), 7.78 (d, J=8.8 Hz, 1H), 7.66 (d, J=8.0 Hz, 1H), 7.50 (d, J=8.0 Hz, 1H), 7.35 (s, 1H), 2.42 (s, 3H). MS (ESI): m/z 279.9 [M+H]+.
[00253] Пример 28: Соединение 28: 4-(3-хлор-6-гидроксихинолин-2-ил)-3-фторбензойная кислота
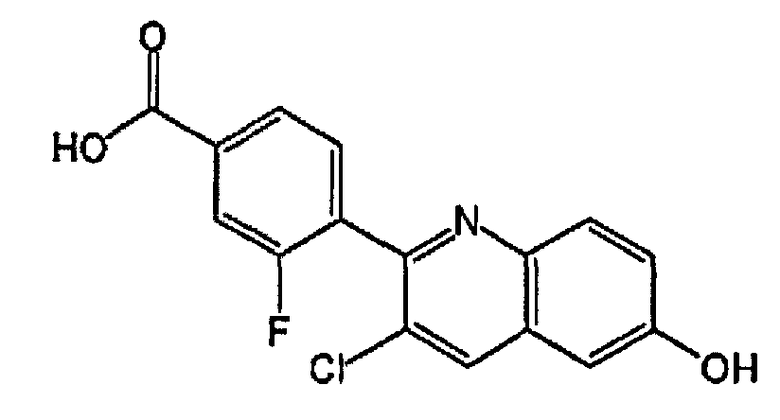
[00254] Этап 1: Синтез 4-(3-хлор-6-метоксихинолин-2-ил)-3-фторбензойиой кислоты: Схема 3: Начиная с 2,3-дихлор-6-метоксихинолина (Промежуточное соединение 4) и 4-бороно-3-фторбензойной кислоты. Выполнили выделение продукта при помощи воды/EtOAc с последующей очисткой на силикагелевой колонке (PE/EtOAc=1/1) для получения заданного продукта.
[00255] Этап 2: Синтез 4-(3-хлор-6-гидроксихинолин-2-ил)-3-фторбензойной кислоты: Схема 1, этап 2: где реакционную смесь дефлегмировали в течение 18 часов, а затем выделили продукт из воды/EtOAc с 10% MeOH. В результате очистки препаративной ВЭЖХ получили Соединение 28. 1H ЯМР (ДМСО-d6, 400 MHz): δ 13.45 (brs, 1H), 10.39 (brs, 1H), 8.52 (s, 1H), 7.94-7.88 (m, 2H), 7.80 (d, J=10.4 Hz, 1H), 7.67 (t, J=7.6 Hz, 1H), 7.38 (dd, J=6.8, 2.4 Hz, 1H), 7.21 (d, J=2.8 Hz, 1H). MS (ESI): m/z 317.8 [M+H]+.
[00256] Пример 29: Соединение 29: 3-(4-(6-гидроксихинолин-2-ил)фенил)-1,2,4-тиадиазол-5(2H)-он
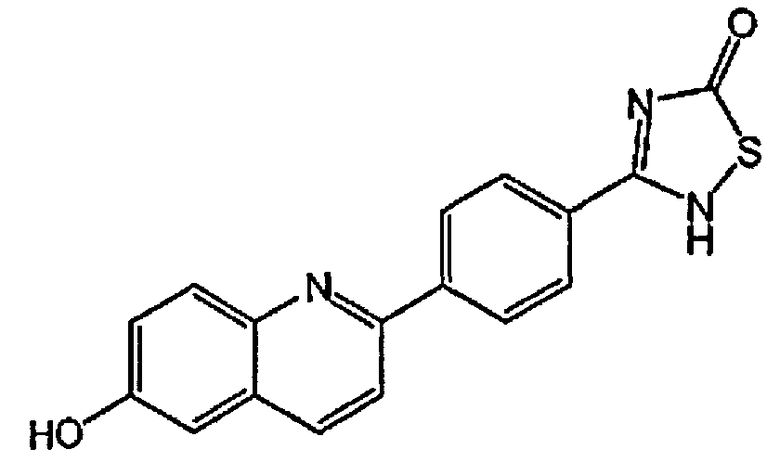
[00257] Этап 1: Синтез N-гидрокси-4-(6-метоксихинолин-2-ил)бензимидамида: Схема 4, путь B, этап 1: 4-(6-метоксихинолин-2-ил)бензонитрил (полученный по Схеме 3) (780 мг, 3,00 ммоль) суспендировали в метаноле (10 мл) и добавили гидроксиламина гидрохлорид (639 мг, 10,7 ммоль), K2CO3 (414 мг, 3,00 ммоль). Реакционную смесь дефлегмировали в течение 12 часов. Добавили воду (15 мл), а выпавшее в осадок твердое вещество собрали фильтрацией, промыли этанолом (5 мл) и высушили под высоким вакуумом для получения продукта (600 мг).
[00258] Этап 2: Синтез 3-(4-(6-метоксихинолин-2-ил)фенил)-1,2,4-тиадиазол-5(2H)-она: Схема 4, путь B, этап 2: К раствору полученного выше продукта (450 мг) в ТГФ (10 мл) добавили TCDI (410 мг, 2,30 ммоль) и перемешивали смесь при 30°C в течение 3 часов. После завершения реакции растворитель удалили для получения продукта (300 мг).
[00259] Этап 3: Синтез 3-(4-(6-гидроксихинолин-2-ил)фенил)-1,2,4-тиадиазол-5(2H)-она: Схема 1, этап 2: Реакционную смесь дефлегмировали в течение ночи, а затем выделили продукт из воды/EtOAc и очистили препаративной ВЭЖХ (с добавлением 0,1% ТФК) для получения соединения 29. 1H ЯМР (ДМСО-d6, 300 MHz): δ 13.50 (brs, 1H), 8.40-8.26 (m, 3H), 8.18-8.04 (m, 3H), 7.95 (d, J=9.0 Hz, 1H), 7.36 (dd, J=9.0, 2.4 Hz, 1H), 7.20 (d, J=2.7 Hz, 1H). MS (ESI): m/z 322.0 [M+H]+.
[00260] Пример 30: Соединение 30: 4-(6-гидроксихинолин-2-ил)-3-(трифторметил)бензойная кислота
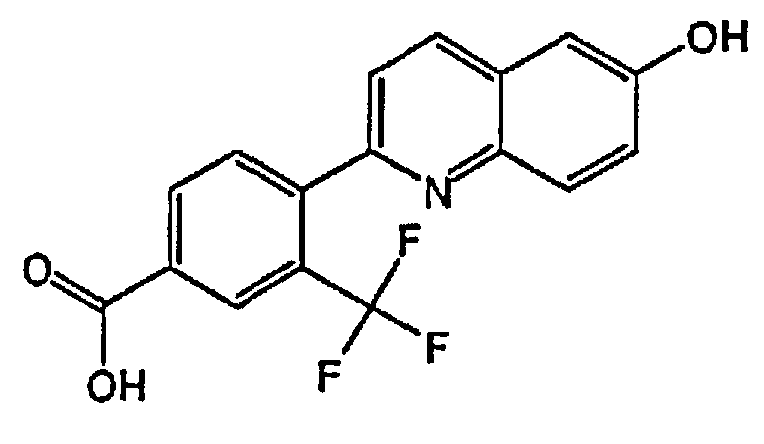
[00261] Следовали Схеме 3: начиная с 2-хлорхинолин-6-ола и метил 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3-(трифторметил)бензоата (Промежуточное соединение 5). 1H ЯМР (ДМСО-d6, 400 MHz): δ 10.21 (brs, 1H), 8.35-8.25 (m, 3H), 7.89 (d, J=9.2 Hz, 1H), 7.79 (d, J=8.0 Hz, 1H), 7.57 (d, J=8.4 Hz, 1H), 7.38 (dd, J=9.2, 2.8 Hz, 1H), 7.23 (d, J=2.4 Hz, 1H). MS (ESI): m/z 333.7 [M+H]+.
[00262] Пример 31: Соединение 31: 4-(6-гидрокси-3-(трифторметил)хинолин-2-ил)бензойная кислота
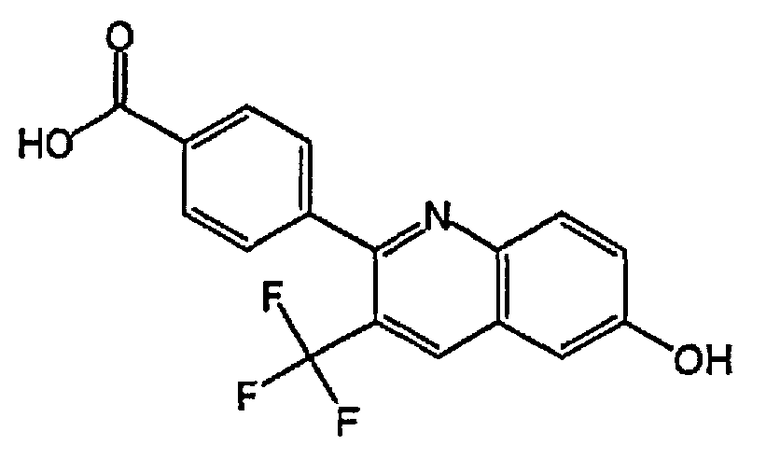
[00263] Следовали Схеме 3: начиная с 4-карбоксифенилбороновой кислоты и 2-хлор-3-(трифторметил)хинолин-6-ола (Промежуточное соединение 6). 1H ЯМР (ДМСО, 400 MHz): δ 13.10 (brs, 1H), 10.45 (brs, 1H), 8.84 (s, 1H), 8.02 (d, J=8.4 Hz, 2H), 7.97 (d, J=8.8 Hz, 1H), 7.60 (d, J=8.0 Hz, 2H), 7.50 (dd, J=9.2, 2.8 Hz, 1H), 7.40 (d, J=2.8 Hz, 1H). MS (ESI): m/z 333.9 [M+H]+.
[00264] Пример 32: Соединение 32: 2-(4-карбоксифенил)-6-гидроксихинолин 1-оксид
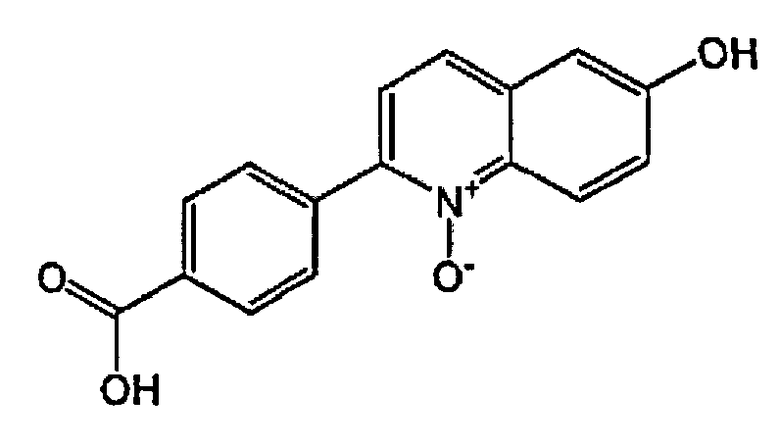
[00265] Этап 1: Синтез метил 4-(6-гидроксихинолин-2-ил)бензоата: Схема 1, этап 1, начиная с 4-(метоксикарбонил)фенилбороновой кислоты и 2-хлорхинолин-6-ола, используя Na2CO3 вместо ТЭА.
[00266] Этап 2: Синтез 6-гидрокси-2-(4-(метоксикарбонил)фенил)хинолин 1-оксида: К раствору полученного выше соединения (200 мг, 0,72 ммоль) в 1,4-диоксане (5 мл) добавили mCPBA (372 мг, 2,16 ммоль). Смесь перемешивали при комнатной температуре в течение трех дней. Летучие вещества удалили под пониженным давлением. Остаток разделили между насыщенным раствором Na2CO3 (10 мл) и EtOAc (20 мл). Органическую фазу промыли насыщенным солевым раствором (10 мл), высушили над Na2SO4, концентрировали и очистили колоночной хроматографией для получения продукта (40 мг, 18,9%).
[00267] Этап 3: Синтез 2-(4-карбоксифенил)-6-гидроксихинолин 1 оксида: Раствор полученного выше соединения (40 мг, 0,14 ммоль) и NaOH (16 мг, 0,41 ммоль) в 1,4-диоксане/воде (1,5 мл/0,5 мл) нагревали при 80°C в течение 2 часов. Летучие вещества удалили in vacuo. Остаток разделили между водой (8 мл) и EtOAc (10 мл). Водную фазу отделили, подкислили при помощи 1 н. HCl до pH=5. Полученный осадок отфильтровали, промыли водой и EtOH и высушили in vacuo для получения Соединения 32 (10 мг, 25,6%). 1H ЯМР (MeOD-d4, 500 MHz): 8.61 (d, J=9.5 Hz, 1H), 8.20 (d, J=8.0 Hz, 2H), 8.04-7.99 (m, 3H), 7.65 (d, J=8.5 Hz, 1H), 7.48 (dd, J=2.0 Hz, J=9.0 Hz, 1H), 7.30 (d, J=2.0 Hz, 1H); MS (ESI): m/z 282.0 [M+1]+.
[00268] Пример 33: Соединение 33: 5-(4-(6-гидроксихинолин-2-ил)фенил)-1,3,4-тиадиазол-2(3H)-он
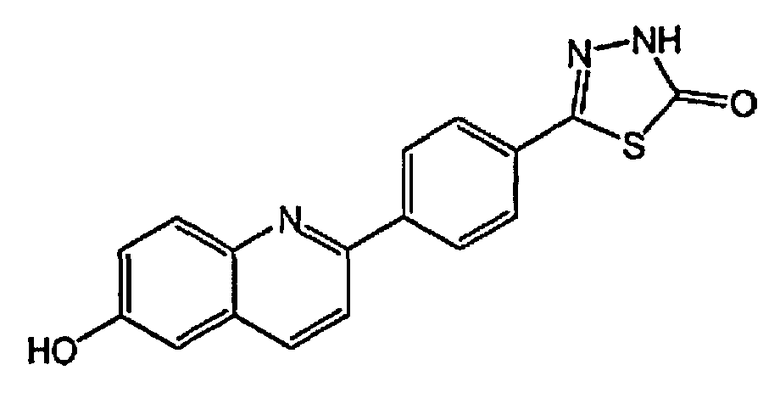
[00269] Иллюстративный способ для Схемы 5, Соединение A:
[00270] Этап 1: Синтез трет-бутил 2-(4-(6-метоксихинолин-2-ил)бензоил)гидразинкарбоксилата: Схема 5, этап 1 пути A (смотри Пример 24, этап 1), начиная с 4-(6-метоксихинолин-2-ил)бензойной кислоты (полученной по Схеме 3).
[00271] Этап 2: Синтез трет-бутил 2-(4-(6-метоксихинолин-2-ил)фенилкарбонотиоил)гидразинкарбоксилата: Смесь полученного выше соединения (2,30 г, 5,85 ммоль) и реагента Лавессона (2,30 г, 5,69 ммоль) в безводном толуоле (150 мл) дефлегмировали в течение ночи. Полученную смесь концентрировали in vacuo. Остаток промыли MeOH, а твердое вещество отделили. Фильтрат концентрировали in vacuo, а остаток очистили на силикагелевой колонке (PE/EtOAc=3/1) для получения неочищенного трет-бутил 2-(4-(6-метоксихинолин-2-ил)фенилкарбонотиоил)гидразинкарбоксилата (2,00 г).
[00272] Этап 3: Синтез 5-(4-(6-гидроксихинолин-2-ил)фенил)-1,3,4-тиадиазол-2(3H)-она: Смесь трет-бутил 2-(4-(6-метоксихинолин-2-ил)фенилкарбонотиоил)гидразинкарбоксилата (500 мг, 1,49 ммоль) и AlCl3 (600 мг, 4,50 ммоль) в безводном ДХМ (50 мл) дефлегмировали в течение ночи. Реакционную смесь охладили, а затем выделили продукт из воды/EtOAc. Остаток очистили препаративной ВЭЖХ (с добавлением 0,1% ТФК) для получения Соединения 33 (35 мг, выход 7%) в виде твердого вещества. 1H ЯМР (ДМСО-d6, 400 MHz): δ 13.22 (brs, 1H), 10.22 (brs, 1H), 8.40-8.28 (m, 3H), 8.11 (d, J=8.8 Hz, 1H), 7.98 (d, J=8.8 Hz, 1H), 7.86 (d, J=8.4 Hz, 2H), 7.38 (dd, J=9.2, 2.8 Hz, 1H), 7.22 (d, J=2.8 Hz, 1H). MS (ESI): m/z 321.7 [M+H]+.
[00273] Пример 34: Соединение 34: 5-(4-(6-гидроксихинолин-2-ил)фенил)-1,2,4-оксадиазол-3(2H)-он
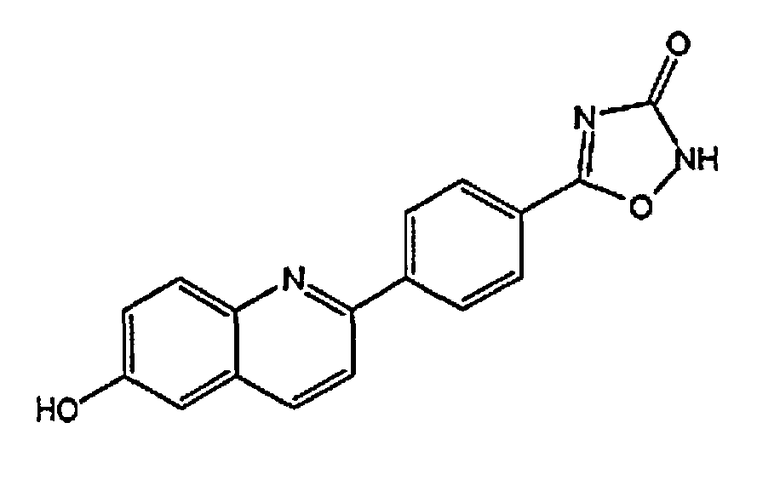
[00274] Этап 1: Синтез 4-(6-метоксихинолин-2-ил)бензамида: К суспензии 4-(6-метоксихинолин-2-ил)бензонитрила (5,00 г, 15,4 ммоль, полученного по Схеме 2, Этап 1, начиная с Промежуточного соединения 1 и 4-цианофенилбороновой кислоты) в ДМСО (40 мл) добавили водный раствор NaOH (1 M, 10 мл). Смесь охладили до 0°C. По каплям добавили водный раствор H2O2 (30%, 30 мл). После добавления смесь перемешивали при 0°C в течение 30 минут. Смесь погасили насыщенным раствором Na2SO3 (100 мл) и отфильтровали. Осадок промыли H2O (50 мл) и MeOH (50 мл). Осадок на фильтре высушили под высоким вакуумом для получения заданного продукта (5,50 г, выход 99+%) в виде твердого вещества.
[00275] Этап 2: Синтез 5-(4-(6-метоксихинолин-2-ил)фенил)-1,2,4-оксадиазол-3-ола: К суспензии 4-(6-метоксихинолин-2-ил)бензамида (2,00 г, 7,19 ммоль) в ДХЭ (20 мл) быстро добавили оксалилхлорид (1,10 г, 8,99 ммоль) при 30°C. Смесь нагревали до 70°C в течение 16 часов. Растворитель удалили in vacuo для получения твердого желтого вещества (2,00 г), которое использовали напрямую, без дополнительной очистки. Суспензию этого материала (2,00 г) в TMSN3 (30 мл) нагревали до 90°C в течение двух дней. Избыток TMSN3 удалили in vacuo, а остаток разбавили EtOH (200 мл). Смесь отфильтровали, а фильтрат концентрировали для получения 5-(4-(6-метоксихинолин-2-ил)фенил)-1,2,4-оксадиазол-3-ола (700 мг, выход за два этапа: 30%) в виде твердого вещества.
[00276] Этап 3: Синтез 5-(4-(6-гидроксихинолин-2-ил)фенил)-1,2,4-оксадиазол-3-ола: Следовали Схеме 2, Этап 2, условия B, для получения Соединения 34 (60 мг, выход: 18%) в виде твердого вещества. 1H ЯМР (ДМСО 400 MHz): δ 12.90 (brs, 1H), 10.15 (brs, 1H), 8.45 (d, J=8.4 Hz, 2H), 8.30 (d, J=8.4 Hz, 1H), 8.15-8.10 (m, 3H), 7.92 (d, J=9.2 Hz, 1H), 7.5 (dd, J=8.8, 2.4 Hz, 1H), 7.20 (d, J=2.8 Hz, 1H). MS (ESI): m/z 305.9 [M+H]+.
[00277] Пример 35: Соединение 35: (1r,4r)-4-(3-хлор-6-гидроксихинолин-2-ил)циклогексанкарбоновая кислота
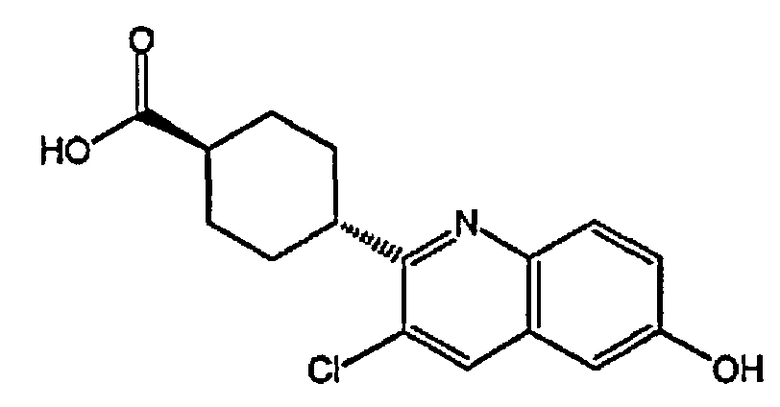
[00278] Этап 1: Синтез этил 4-(3-хлор-6-метоксихинолин-2-ил)циклогекс-3-енкарбоксилата: Следовали Схеме 2, Этап 1 (смотри Пример 8, Этап 1), начиная с этил 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)циклогекс-3-енкарбоксилата и 2,3-дихлор-6-метоксихинолина (Промежуточное соединение 4).
[00279] Этап 2: Синтез этил 4-(3-хлор-6-метоксихинолин-2-ил)циклогексанкарбоксилата: К этил 4-(3-хлор-6-метоксихинолин-2-ил)циклогекс-3-енкарбоксилату (900 мг, 2,60 ммоль) в EtOH (40 мл) добавили Rh(PPh3)3Cl (90,0 мг, 0,0900 ммоль). Смесь перемешивали при 30°C в течение 4 дней под H2 (15 psi). Смесь отфильтровали, а фильтрат концентрировали для получения неочищенного продукта, который очистили на силикагелевой колонке (PE/EtOAc=10/1) для получения заданного продукта (234 мг, выход 26%).
[00280] Этап 3: Синтез (1r,4r)-4-(3-хлор-6-гидроксихинолин-2-ил)циклогексанкарбоновой кислоты: К смеси этил 4-(3-хлор-6-метоксихинолин-2-ил)циклогексанкарбоксилата (200 мг, 0,580 ммоль) в безводном CH2Cl2 (10 мл) по каплям добавили BBr3 (0,3 мл, 2,9 ммоль) при 0°C. Смесь перемешивали при 0°C в течение 2 часов. Добавили воду (10 мл) и экстрагировали смесь EtOAc (30 мл × 3). Объединенные органические слои промыли насыщенным солевым раствором (20 мл × 2), высушили над Na2SO4, отфильтровали и концентрировали для получения неочищенного продукта, который очистили препаративной ВЭЖХ (с добавлением 0,1% ТФК) для получения (1r,4r)-4-(3-хлор-6-гидроксихинолин-2-ил)циклогексанкарбоновой кислоты (Соединение 35, 65 мг, выход 37%) в виде твердого вещества, и (1s,4s)-4-(3-хлор-6-гидроксихинолин-2-ил)циклогексанкарбоновой кислоты
(Соединение 36, 26 мг, выход 15%) в виде твердого вещества. Структуру подтвердили при помощи ядерного эффекта Оверхаузера (NOE). Данные для Соединения 35: 1H ЯМР (ДМСО 400 MHz): δ 10.08 (brs, 1H), 8.27 (s, 1H), 7.79 (d, J=8.8 Hz, 1H), 7.26 (dd, J=9.2, 2.8 Hz, 1H), 7.08 (d, J=2.8 Hz, 1H), 3.18 (tt, Jaa=12.0 Hz, Jea=3.2 Hz, 1H), 2.28 (tt, Jaa=12.0 Hz, Jea=3.2 Hz, 1H), 2.03 (d, J=10.4 Hz, 2H), 1.91 (d, J=10.8 Hz, 2H), 1.72-1.60 (m, 2H), 1.55-1.40 (m, 2H). MS (ESI): m/z 306.0 [M+H]+.
[00281] Пример 36: Соединение 36: (1s,4s)-4-(3-хлор-6-гидроксихинолин-2-ил)циклогексанкарбоновая кислота
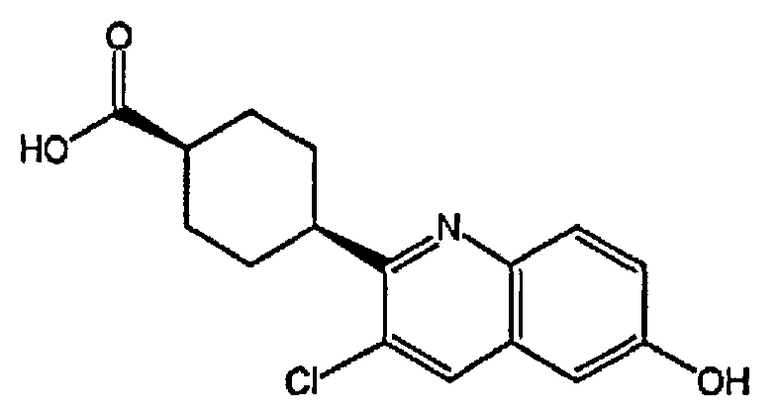
[00282] Синтез представлен в Примере 35. 1H ЯМР (ДМСО 400 MHz): δ 10.11 (brs, 1H), 8.28 (s, 1H), 7.82 (d, J=9.2 Hz, 1H), 7.27 (dd, J=9.2, 2.8 Hz, 1H), 7.09 (d, J=2.8 Hz, 1H), 3.32-3.19 (m, 1H), 2.70-2.60 (m, 1H), 2.25-2.14 (m, 2H), 1.85-1.70 (m, 4H), 1.70-1.58 (m, 2H). MS (ESI): m/z 306.0 [M+H]+.
[00283] Пример 37: Соединение 37: 3-хлор-4-(4-фтор-6-гидроксихинолин-2-ил)бензойная кислота
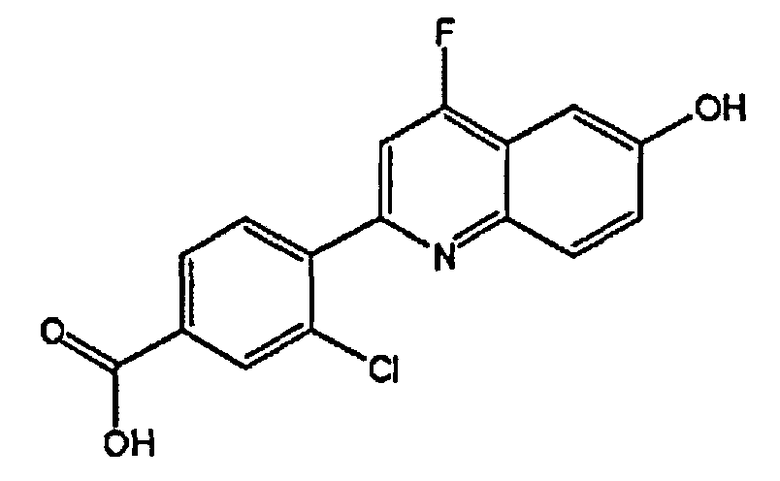
[00284] Следовали Схеме 2, условия B, начиная с 2-хлор-4-фтор-6-метоксихинолина (Промежуточное соединение 7) и 4-карбокси-2-хлорфенилбороновой кислоты. 1H ЯМР (MeOD 400 MHz): δ 8.20 (d, J=1.2 Hz, 1H), 8.10 (dd, J=8.0, 1.6 Hz, 1H), 8.02 (d, J=9.2, 1.2 Hz, 1H),7.75 (d, J=8.0 Hz, 1H), 5 7.52-7.45 (m, 2H), 7.47 (d, J=2.4 Hz, 1H). MS (ESI): m/z 317.9 [M+H]+.
[00285] Пример 38: Соединение 38: 2-(5-(2H-тетразол-5-ил)тиофен-2-ил)хинолин-6-ол
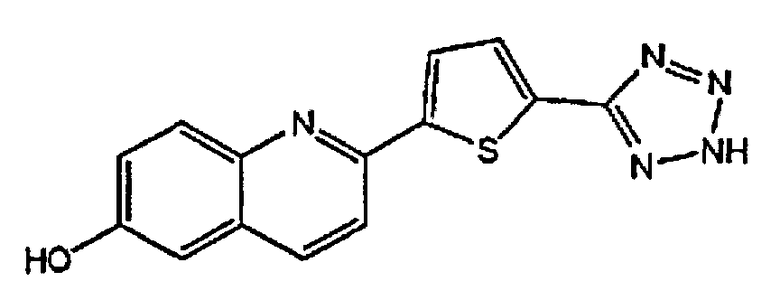
[00286] Этап 1 и 2: Синтез 5-(6-гидроксихинолин-2-ил)тиофен-2-карбонитрила: Следовали двум этапам синтеза, показанным на Схеме 1, начиная с 2-хлор-6-метоксихинолина и 5-цианотиофен-2-илбороновой кислоты, с небольшими изменениями: основанием, используемым на этапе 1, был карбонат натрия, а растворителем был DME (диметоксиэтан)/вода. На этапе 2, после погашения ледяной водой, выполнили стандартную экстракцию этилацетатом с последующей очисткой препаративной TCX (PE:EA=1:1).
[00287] Этап 3; Синтез 2-(5-(2H-тетразол-5-ил)тиофен-2-ил)хинолин-6-ола: Следовали Схеме 4, путь A. 1H-ЯМР (ДМСО-d6 500 MHz): 10.15 (s, 1H), 8.23 (d, J=9.0 Hz, 1H), 8.05 (d, J=8.5 Hz, 1H), 7.99 (d, J=4.0 Hz, 1H), 7.86 (d, J=9.0 Hz, 1H), 7.78 (d, J=3.5 Hz, 1H), 7.34 (dd, J=2.5, 9.0 Hz, 1H), 7.17 (d, J=2.0 Hz, 1H). MS (ESI): m/z 296.0 [M+1]+.
[00288] Пример 39: Соединение 39: 5-(4-(6-гидроксихинолин-2-ил)фенил)-1,2,4-тиадиазол-3(2H)-он
[00289] Этап 1: Синтез 4-(6-метоксихинолин-2-ил)бензотиоамида: К суспензии 4-(6-метоксихинолин-2-ил)бензамида (смотри Пример 34, этап 1, где описан синтез, 3,00 г, 10,8 ммоль) в безводном толуоле (50 мл) добавили реагент Лавессона (2,60 г, 6,44 ммоль). Смесь дефлегмировали в течение 3 часов. Смесь разбавили MeOH (50 мл) и отфильтровали. Фильтрат концентрировали под пониженным давлением для получения неочищенного продукта, который очистили на силикагелевой колонке (PE/EtOAc=1/1) для получения продукта (1,30 г, выход 41%) в виде твердого вещества.
[00290] Этап 2 и 3: Синтез 5-(4-(6-метоксихинолин-2-ил)фенил)-1,2,4-тиадиазол-3(2H)-она: К раствору 4-(6-метоксихинолин-2-ил)бензотиоамида (500 мг, 1,07 ммоль) в ДХЭ (10 мл) по каплям добавили оксалилхлорид (0,3 мл, 3,9 ммоль) при 0°C. Смесь нагревали до 90°C в течение 2 часов. По каплям добавили TMSN3 (0,8 мл, 5,7 ммоль). Смесь перемешивали при 100°C в течение 2 часов. К смеси добавили воду (10 мл). Смесь отфильтровали, а осадок на фильтре промыли IPA (20 мл) для получения продукта (280 мг, выход 49%).
[00291] Этап 4: Синтез 5-(4-(6-гидроксихинолин-2-ил)фенил)-1,2,4-тиадиазол-3(2H)-она: Следовали Схеме 2, Этап 2, условия B, для деметилирования. 1H ЯМР (ДМСО 400 MHz): δ 12.76 (brs, 1H), 10.16 (brs, 1H), 8.41 (d, J=8.4 Hz, 2H), 8.29 (d, J=8.8 Hz, 1H), 8.15-8.02 (m, 3H), 7.95 (d, J=9.2 Hz, 1H), 7.36 (dd, J=9.2, 2.8 Hz, 1H), 7.19 (d, J=2.4 Hz, 1H), MS (ESI): m/z 321.9 [M+H]+.
[00292] Пример 40: Соединение 40: 3-фтор-4-(4-фтор-6-гидроксихинолин-2-ил)бензойная кислота
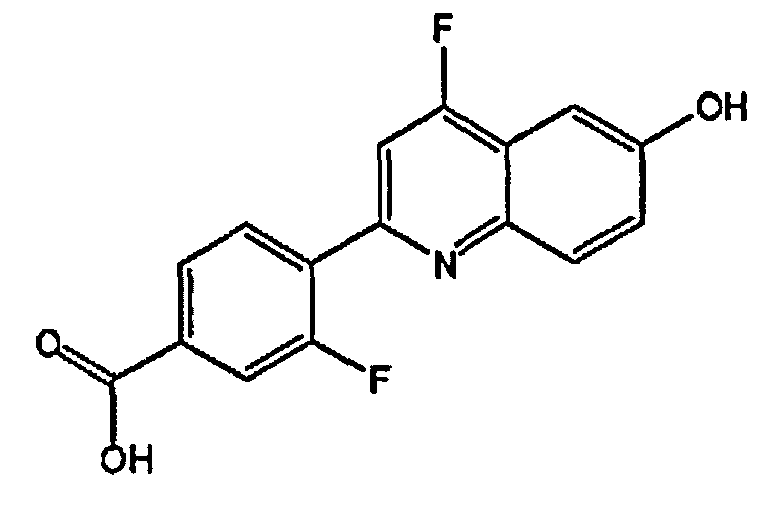
[00293] Соединение получили по Схеме 2, условия В, начиная с 2-хлор-4-фтор-6-метоксихинолина (Промежуточное соединение 7) и 4-карбокси-2-фторфенилбороновой кислоты. Примечание: на этапе 2, после очистки препаративной ВЭЖХ, собранные фракции сразу нейтрализовали насыщенным раствором NaHCO3 до pH 6 с последующей экстракцией EtOAc/MeOH (об./об.=10/1, 50 мл × 3). Объединенные органические слои промыли насыщенным солевым раствором (50 мл), высушили над Na2SO4 и концентрировали in vacuo для получения заданного продукта. 1H ЯМР (MeOD 400 MHz): δ 8.10-8.00 (m, 2H), 7.98 (dd, J=8.0, 1.2 Hz, 1H), 7.85 (dd, J=11.6, 1.2 Hz, 1H), 7.62 (dd, J=11.6, 2.0 Hz, 1H), 7.45 (dd, J=9.2, 2.8 Hz, 1H), 7.31 (d, J=2.8 Hz, 1H). MS (ESI): m/z 301.8 [M+H]+.
[00294] Пример 41: Соединение 41: 1-(6-гидрокси-3-(трифторметил)хинолин-2-ил)пиперидин-4-карбоновая кислота
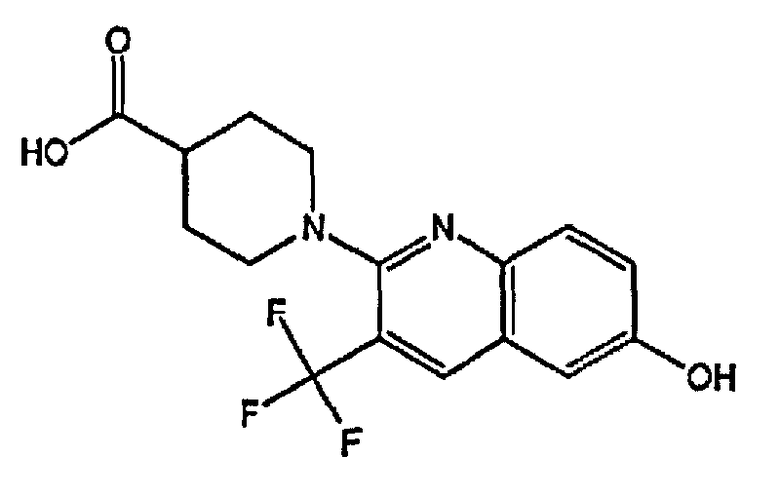
[00295] Этап 1: Синтез метил 1-(6-метокси-3-(трифторметил)хинолин-2-ил)пиперидин-4-карбоксилата: К раствору 2-хлор-6-метокси-3-(трифторметил)хинолина (1,00 г, 3,85 ммоль) (смотри Промежуточное соединение 6, Этап 8, где описан синтез) в IPA (5 мл) добавили метил-4-пиперидинкарбоксилат (5,50 г, 38,5 ммоль) и Et3N (1,17 г, 11,6 ммоль). Смесь нагревали с дефлегматором в течение 48 часов. Смесь разбавили водой (100 мл) и экстрагировали ДХМ (100 мл × 3), объединенные органические слои промыли насыщенным солевым раствором (200 мл), высушили над безводным Na2SO4, отфильтровали и концентрировали под пониженным давлением для получения неочищенного продукта, который очистили колоночной хроматографией на силикагеле (PE/EtOAc=15/1) для получения продукта (600 мг, выход 43%) в виде твердого вещества.
[00296] Этап 2: Синтез 1-(6-гидрокси-3-(трифторметил)хинолин-2-ил)пиперидин-4-карбоновой кислоты: К раствору метил 1-(6-метокси-3-(трифторметил)хинолин-2-ил)пиперидин-4-карбоксилата (300 мг, 0,815 ммоль) в ДХМ (5 мл) по каплям добавили BBr3 (0,4 мл, 4,08 ммоль) при 0°C. Смесь перемешивали при 0°C в течение 20 часов. К смеси по каплям добавили воду (10 мл) при 0°C. Смесь концентрировали in vacuo, затем разбавили EtOAc (150 мл), отфильтровали и концентрировали in vacuo. Очистили препаративной ВЭЖХ (с добавлением 0,1% ТФК) для получения Соединения 41 (26 мг, выход 9,4%) в виде твердого вещества. 1H ЯМР (MeOD 400 MHz): δ 8.51 (s, 1H), 7.82 (d, J=8.8 Hz, 1H), 7.41 (dd, J=8.8, 2.8 Hz, 1H), 7.21 (d, J=2.8 Hz, 1H), 3.55-3.49 (m, 2H), 3.13-3.02 (m, 2H), 2.58-2.49 (m, 1H), 2.09-2.00 (m, 2H), 1.93-1.82 (m, 2H). MS (ESI): m/z 340.7 [M+H]+.
[00297] Пример 42: Соединение 42: 4-(5-хлор-6-гидроксихинолин-2-ил)бензойная кислота
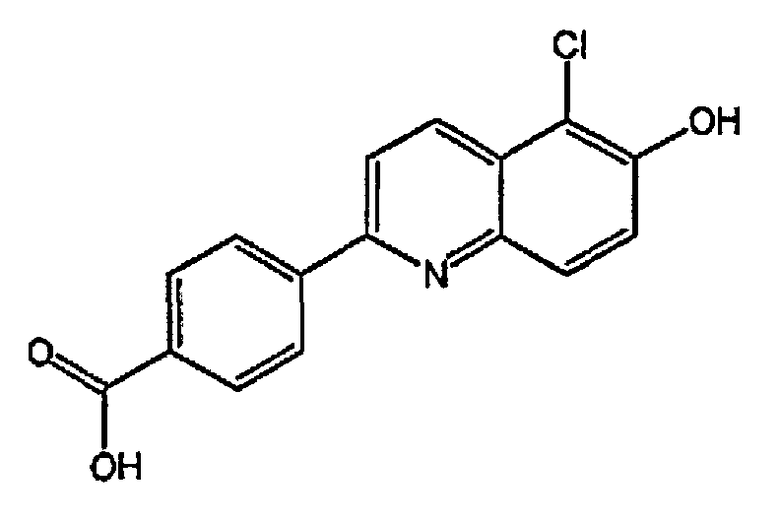
[00298] Этап 1: Синтез метил 4-(6-метоксихинолин-2-ил)бензоата: Смесь 2-хлор-6-метоксихинолина (Промежуточное соединение 1, 200 мг, 1,0 ммоль), 4-(метоксикарбонил)фенилбороновой кислоты (205 мг, 1,1 ммоль), Pd(dppf)Cl2 (366 мг, 0,5 ммоль) и карбоната натрия (212 мг, 2,0 ммоль) в 1,4-диоксане/воде (3 мл/0,6 мл) нагревали до 120°C в микроволновом реакторе в течение 1 часа. Осадок отфильтровали; промыли по отдельности EA (10 мл), ацетоном (10 мл) и водой (10 мл); высушили для получения продукта (120 мг, 40,9%).
[00299] Этап 2: Синтез метил 4-(5-хлор-6-метоксихинолин-2-ил)бензоата: К раствору полученного выше соединения (100 мг, 0,34 ммоль) в AcOH (2 мл) добавили SO2Cl2 (55 мг, 0,41 ммоль). Реакционную смесь нагревали до 60°C в течение 3 часов. Затем смесь перемешивали при комнатной температуре в течение ночи. Осадок отфильтровали, промыли AcOH (10 мл × 3) и высушили для получения продукта в виде порошка (100 мг, 90,1%).
[00300] Этап 3: Синтез 4-(5-хлор-6-гидроксихинолин-2-ил)бензойной кислоты: Следовали Схеме 1, этап 2, с очисткой препаративной ВЭЖХ для получения продукта. 1H-ЯМР (ДМСО-d6 500 MHz): 8.50 (d, J=9.0 Hz, 1H), 8.32 (d, J=8.0 Hz, 2H), 8.25 (d, J=8.5 Hz, 1H), 8.10 (d, J=8.0 Hz, 2H), 7.96 (d, J=9.5 Hz, 1H), 7.62 (d, J=9.0 Hz, 1H); MS (ESI): m/z 300.0 [M+1]+.
[00301] Пример 43: Соединение 43: (1r,4r)-4-(6-гидрокси-3-(трифторметил)хинолин-2-ил)циклогексанкарбоновая кислота
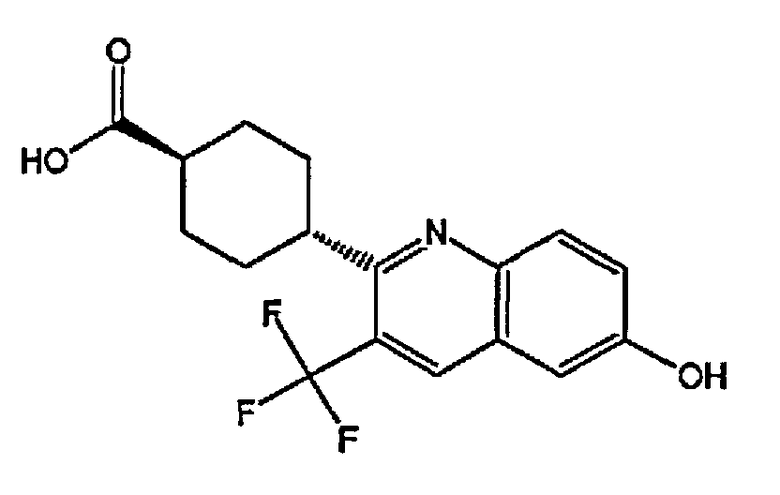
[00302] Этап 1: Синтез этил 4-(6-метокси-3-(трифторметил)хинолин-2-ил)циклогекс-3-енкарбоксилата: Следовали Схеме 2, Этап 1 (смотри Пример 8, Этап 1), начиная с этил 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)циклогекс-3-енкарбоксилата и 2-хлор-6-метокси-3-(трифторметил)хинолина (смотри Промежуточное соединение 6, Этап 8, где описан синтез), с очисткой колоночной хроматографией (PE/EtOAc=10/1).
[00303] Этап 2: Синтез этил 4-(6-метокси-3-(трифторметил)хинолин-2-ил)циклогексанкарбоксилата: Следовали этапу 2 Соединения 35 с 0,15 экв. катализатора при 20°C в течение 4 дней под H2 (15 psi).
[00304] Этап 3: Синтез (1r,4r)-4-(6-гидрокси-3-(трифторметил)хинолин-2-ил)циклогексанкарбоновой кислоты: Следовали этапу 3 Соединения 35, где реакционную смесь перемешивали при 10°C в течение 20 часов, а затем погасили водой. Очистили неочищенный продукт препаративной ВЭЖХ для получения этил 4-(6-метокси-3-(трифторметил)хинолин-2-ил)циклогексанкарбоксилата (Соединение 43, 109 мг, выход 29%) в виде твердого вещества, и (1s,4s)-4-(6-гидрокси-3-(трифторметил)хинолин-2-ил)циклогексанкарбоновой кислоты (Соединение 44,25 мг, выход 7%) в виде твердого вещества. Данные для Соединения 43: 1H ЯМР (ДМСО 400 MHz): δ 10.27 (brs, 1H), 8.61 (s, 1H), 7.89 (d, J=9.2 Hz, 1H), 7.44 (dd, J=9.2, 2.8 Hz, 1H), 7.32 (d, J=2.8 Hz, 1H), 3.01-2.91 (m, 1H), 2.40-2.30 (m, 1H), 2.00-1.90 (m, 2H), 1.95-1.78 (m, 4H), 1.53-1.38 (m, 2H). MS (ESI): m/z 339.9 [M+H]+.
[00305] Пример 44: Соединение 44: (1s,4s)-4-(6-гидрдокси-3-(трифторметил)хинолин-2-ил)циклогексанкарбоновая кислота
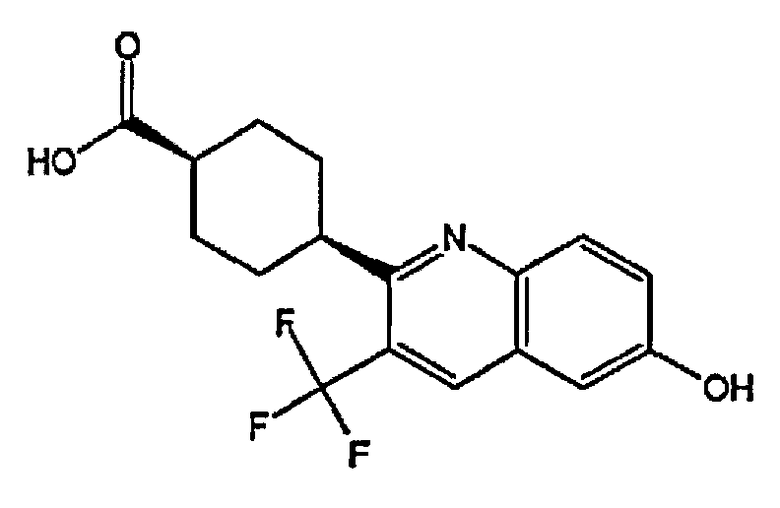
[00306] Смотри Пример 43, где описан синтез. 1H ЯМР (ДМСО 400 MHz): δ 10.26 (brs, 1H), 8.59 (s, 1H), 7.89 (d, J=9.2 Hz, 1H), 7.43 (dd, J=9.2, 2.8 Hz, 1H), 7.31 (d, J=2.0 Hz, 1H), 3.09-2.95 (m, 1H), 2.75-2.62 (m, 1H), 2.28-2.16 (m, 2H), 1.98-1.80 (m, 2H), 1.60-1.55 (m, 4H). MS (ESI): m/z 339.9 [M+H]+.
[00307] Пример 45: Соединение 45: 4-(5-бром-6-гидроксихинолин-2-ил)бензойная кислота
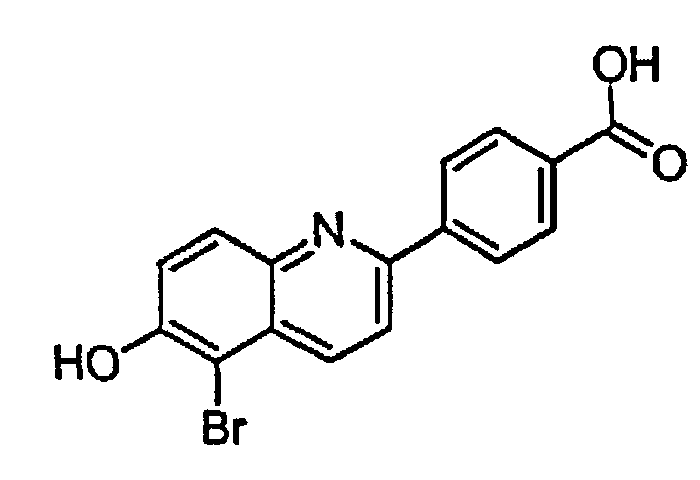
[00308] Этап 1: Синтез метил 4-(6-метоксихинолин-2-ил)бензоата: Смесь 2-хлор-6-метоксихинолина (смотри публикацию US 61/391225, где описан синтез) (200 мг, 1,0 ммоль), 4-(метоксикарбонил)фенилбороновой кислоты (205 мг, 1,1 ммоль), Pd(dppf)Cl2 (366 мг, 0,5 ммоль) и карбоната натрия (212 мг, 2,0 ммоль) в 1,4-диоксане/воде (3 мл/0,6 мл) нагревали до 120°C в микроволновом реакторе в течение 1 часа. Осадок отфильтровали; промыли по отдельности EtOAc (10 мл), ацетоном (10 мл) и водой (10 мл); высушили для получения продукта в виде твердого черного вещества (120 мг, 40,9%).
[00309] Этап 2: синтез метил 4-(5-бром-6-метоксихинолин-2-ил)бензоата: К раствору метил 4-(6-метоксихинолин-2-ил)бензоата (630 мг, 2,15 ммоль) в ДХМ (9 мл) добавили Br2 (0,3 мл, 6,45 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Затем смесь разделили при помощи насыщенного солевого раствора и ДХМ. Осадок отфильтровали и высушили для получения продукта в виде твердого вещества (800 мг, 100%). MS (ESI): m/z=373.0 [M+1]+.
[00310] Этап 3: Синтез Соединения 45: К раствору полученного выше продукта (150 мг, 0,40 ммоль) в ДХМ (5 мл) добавили BBr3 (0,38 мл, 4,0 ммоль) и перемешивали при комнатной температуре в течение ночи. Осторожно добавили воду (20 мл), смесь экстрагировали EtOAc (20 мл × 3), концентрировали и очистили препаративной ВЭЖХ для получения Соединения 45 в виде серого порошка (40 мг, 29,2%). MS (ESI): m/z=346.0 [M+1]+. 1H-ЯМР (ДМСО-d6 500 MHz): 8.48 (d, J=8.5 Hz, 1H), 8.35 (d, J=8.0 Hz, 2H), 8.25 (d, J=9.0 Hz, 1H), 8.10 (d, J=7.5 Hz, 2H), 8.01 (d, J=8.5 Hz, 1H), 7.60 (d, J=9.0 Hz, 1H) ppm.
[00311] Пример 46: Соединение 46: 3-бром-4-(6-гидроксихинолин-2-ил)бензойная кислота
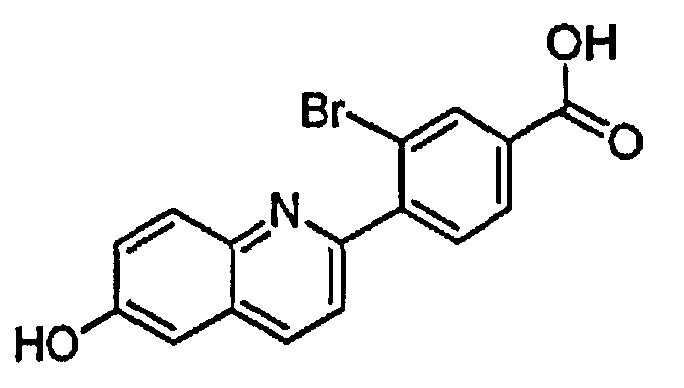
[00312] Этап 1: Синтез метил 3-амино-4-(6-метоксихинолин-2-ил)бензоата: К смеси соединения 2-хлор-6-метоксихинолина (1,70 г, 8,78 ммоль), 2-амино-4-(метоксикарбонил)фенилбороновой кислоты (2,05 г, 10,5 ммоль) и K2CO3 (2,43 г, 17,6 ммоль) в монометиловом эфире этиленгликоля/H2O (35 мл/5 мл) добавили Pd(dppf)Cl2 (158 мг, 0,193 ммоль) под атмосферой N2. Затем смесь нагревали до 80°C в течение 3 часов. После выделения продукта при помощи воды с экстракцией EtOAc, полученный неочищенный продукт очистили на силикагелевой колонке (PE/EtAOc = от 15/1 до 3/1) для получения продукта (500 мг, выход 19%) в виде твердого желтого вещества.
[00313] Этап 2: Синтез метил 3-бром-4-(6-метоксихинолин-2-ил)бензоата: К смеси полученного выше продукта (200 мг, 0,649 ммоль) в HBr (40%)/H2O (5 мл/5 мл) по каплям добавили NaNO2 (44,8 мг, 0,649 ммоль) в H2O (3 мл) при 0°C, и перемешивали реакционную смесь при 0°C в течение 30 минут. Добавили CuBr (186 мг, 1,30 ммоль) и перемешивали смесь при 25°C в течение 2 часов. Реакционную смесь подщелочили водным раствором NaOH (2 М) до pH=7-8, экстрагировали EtOAc (30 мл × 3). Объединенные органические слои промыли насыщенным солевым раствором (30 мл), высушили над безводным Na2SO4, отфильтровали и концентрировали под пониженным давлением для получения продукта (205 мг, выход 85%) в виде твердого желтого вещества.
[00314] Этап 3: Синтез Соединения 46: К раствору полученного выше продукта (205 мг, 0,550 ммоль) в безводном ДХМ (6 мл) по каплям добавили BBr3 (0,26 мл, 2,8 ммоль, d=2,64 г/мл) при 0°C. Полученную смесь перемешивали при 25°C в течение 48 часов. Реакционную смесь погасили H2O (30 мл) и отфильтровали, осадок на фильтре промыли EtOAc (10 мл) для получения соединения 46 (90 мг, выход 48%) в виде твердого желтого вещества. 1H ЯМР (ДМСО-d6 400 MHz): δ 10.27 (brs, 1H), 8.36 (d, J=8.8 Hz, 1H), 8.24 (d,J=1.6 Hz, 1H), 8.06 (dd, J=8.0, 1.6 Hz, 1H), 7.95 (d, J=9.2 Hz, 1H), 7.76 (d, J=8.0 Hz, 1H), 7.71 (d, J=8.8 Hz, 1H), 7.41 (dd, J=9.2, 2.4 Hz, 1H), 7.26 (d, J=2.4 Hz, 1H). Чистота по ЖХ-МС: 94,8%. MS (ESI): m/z 343.9 [M+H]+.
[00315] Пример 47: Соединение 47: 4-(4-(диметиламино)-6-гидроксихинолин-2-ил)бензойная кислота
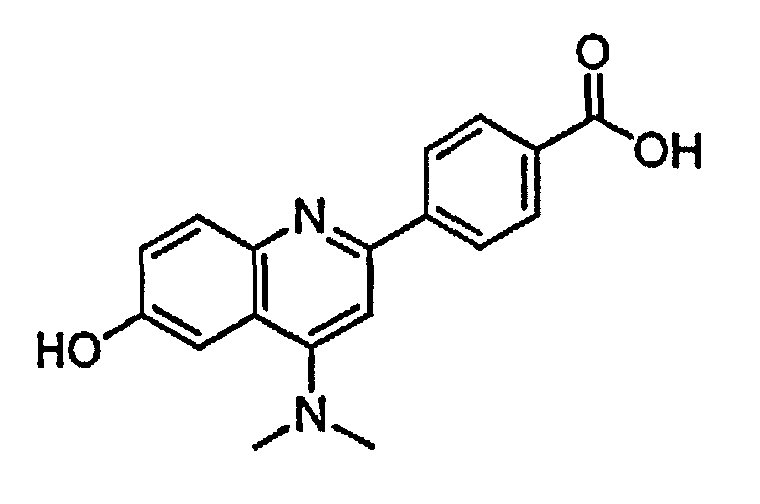
[00316] 30 мг 4-(4-фтор-6-гидроксихинолин-2-ил)бензойной кислоты (Соединение 26) смешали с 1 ммоль Me2NH·HCl и 0,5 мл DIEA в 3 мл ДМФ и нагревали до 160°C в течение 30 минут. Растворитель выпарили до сухости и добавили воду. Твердое вещество отфильтровали и промыли водой, и высушили. Неочищенный продукт растерли с ацетоном для получения 10,5 мг Соединения 47. 1H ЯМР (ДМСО-d6 300 MHz TMS): δ 13.2 (b, 1H), 10.28 (s, 1Н), 8.22 (m, 2H), 8.11 (d, 1H), 7.99 (d, 1H), 7.50 (s, 1H), 7.39 (d, 1H), 7.26 (s, 1H), 3.24 (s, 6H), MS (ESI): m/z=309.30 [M+1]+.
[00317] Пример 48: Соединение 48: 4-(4-фтор-6-гидроксихинолин-2-ил)-3-метоксибензойная кислота
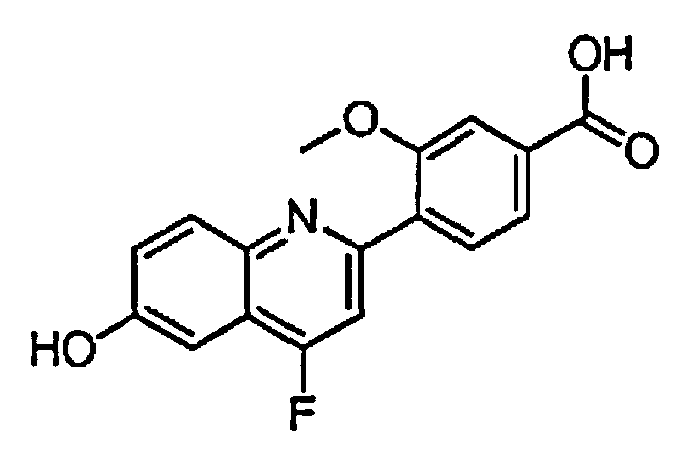
[00318] Этап 1: Синтез 2-хлор-4-фторхинолин-6-ола: Смесевой раствор 2-хлор-4-фтор-6-метоксихинолина (смотри публикацию US 61/391225, где описан синтез) (280 мг, 1,32 ммоль) и BBr3 (0,3 мл, 3,2 ммоль, 2,64 г/мл) в ДХМ (5 мл) перемешивали при 20°C в течение 12 часов. В результате выделения продукта при помощи воды с экстракцией ДХМ получили неочищенный продукт, который очистили на силикагелевой колонке (PE/EtOAc=50/1) для получения продукта (177 мг, выход 69%) в виде твердого белого вещества.
[00319] Этап 2: Синтез Соединения 48: Смесь полученного выше продукта (133 мг, 0,670 ммоль), 4-(дигидроксиборил)-3-метоксибензойной кислоты (156 мг, 0,801 ммоль), K2CO3 (278 мг, 2,01 ммоль), Pd(dppf)Cl2 (30 мг, 0,026 ммоль) в ДМФ (3 мл) и H2O (0,6 мл) перемешивали под атмосферой N2 при 130°C в течение 2,5 часов. Смесь охладили до комнатной температуры, подкислили водным раствором HCl (1M) до pH=6 и экстрагировали EtOAc (30 мл × 3). Объединенный органический слой промыли насыщенным солевым раствором (50 мл), высушили над безводным Na2SO4 и концентрировали in vacuo. Остаток очистили на силикагелевой колонке (ДХМ/MeOH=15/1) для получения Соединения 48 (10,5 мг, выход 5%) в виде грязновато-белого твердого вещества. 1H ЯМР (CD3OD 400 MHz TMS): δ 8.01 (dd, J=8.8, 1.2 Hz, 1H), 7.88-7.76 (m, 3H), 7.63 (d, J=11.2 Hz, 1H), 7.43 (dd, J=9.2, 2.8 Hz, 1H), 7.33 (d, J=2.8 Hz, 1H), 3.98 (s, 3H). MS (ESI): m/z 313.8 [M+H]+.
[00320] Пример 49: Соединение 49: 3-циано-4-(6-гидроксихинолин-2-ил)бензойная кислота
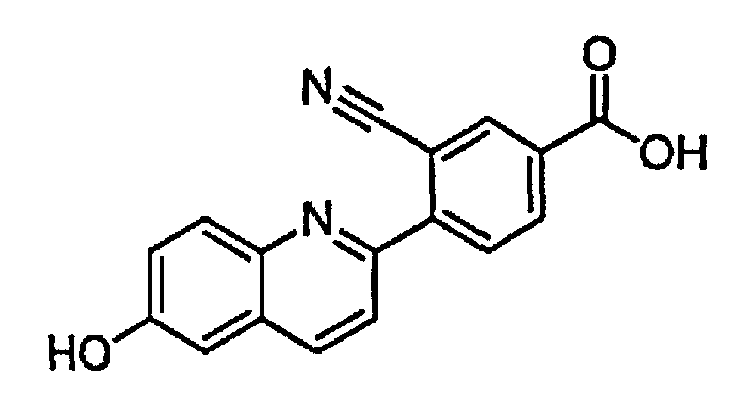
[00321] Этап 1: Синтез 2-метоксиэтил 3-амино-4-(6-метоксихинолин-2-ил)бензоата: Следовали способу связывания, описанному на этапе 1 Соединения 46, начиная с 2-хлор-6-метоксихинолина (1,70 г, 8,78 ммоль) и 2-амино-4-(метоксикарбонил)фенилбороновой кислоты (2,05 г, 10,5 ммоль). Примечание: между заданным соединением и растворителем произошел сложноэфирный обмен. Получили продукт (1,10 г, выход 35%) в виде твердого желтого вещества.
[00322] Этап 2: Синтез 2-метоксиэтил 3-бром-4-(6-метоксихинолин-2-ил)бензоата: К смеси полученного выше продукта (500 мг, 1,42 ммоль) в HBr (40%) / H2O (10 мл/10 мл) по каплям добавили NaNO2 (97,9 мг, 1,42 ммоль) в H2O (5 мл) при 0°C, и перемешивали реакционную смесь при 0°C в течение 30 минут. Добавили CuBr (407 мг, 2,84 ммоль) и перемешивали смесь при 25°C в течение 2 часов, затем подщелочили водным раствором NaOH (2 М) до pH=7-8 и экстрагировали EtOAc (20 мл × 3). Объединенные органические слои промыли насыщенным солевым раствором (20 мл), высушили над безводным Na2SO4, отфильтровали и концентрировали in vacuo для получения продукта (580 мг, выход 98%) в виде твердого желтого вещества.
[00323] Этап 3: Синтез 2-метоксиэтил 3-циано-4-(6-метоксихинолин-2-ил)бензоата: К раствору полученного выше продукта (580 мг, 1,40 ммоль) в ДМФ (15 мл) добавили Zn(CN)2 (329 мг, 2,80 ммоль) и Pd(PPh3)4 (162 мг, 0,140 ммоль). Полученную смесь перемешивали при 120°C под атмосферой N2 в течение 16 часов. После охлаждения до комнатной температуры смесь отфильтровали, а фильтрат разбавили EtOAc (60 мл), промыли H2O (20 мл × 3) и насыщенным солевым раствором (20 мл), высушили над безводным Na2SO4, отфильтровали и концентрировали под пониженным давлением для получения неочищенного продукта. Неочищенный продукт очистили на силикагелевой колонке (PE/EtOAc = от 50/1 до 10/1) для получения продукта (180 мг, выход 36%) в виде твердого желтого вещества.
[00324] Этап 4: Синтез 2-гидроксиэтил 3-циано-4-(6-гидроксихинолин-2-ил)бензоата: К раствору полученного выше продукта (180 мг, 0,497 ммоль) в безводном ДХМ (10 мл) по каплям добавили BBr3 (0,24 мл, 2,5 ммоль, d=2,64 г/мл) при 0°C. Полученную смесь перемешивали при 25°C в течение 2 часов. Добавили воду (20 мл), затем подщелочили водным раствором NaOH (2М) до pH=7~8 и экстрагировали ДХМ (30 мл × 3). Объединенные органические слои промыли насыщенным солевым раствором (30 мл), высушили над безводным Na2SO4, отфильтровали и концентрировали in vacuo для получения продукта (100 мг, выход 60%) в виде грязновато-белого твердого вещества.
[00325] Этап 5: Синтез Соединения 49: К раствору полученного выше продукта (100 мг, 0,299 ммоль) в MeOH (5 мл) и ТГФ (5 мл) добавили LiOH·H2O (25,1 мг, 0,598 ммоль). Смесь перемешивали при 25°C в течение 16 часов. Реакционную смесь подкислили при помощи 1 н. HCl до pH=5~6, а полученную смесь экстрагировали EtOAc (20 мл × 3). Объединенные органические слои промыли насыщенным солевым раствором (20 мл), высушили над безводным Na2SO4, отфильтровали и концентрировали для получения неочищенного продукта. Неочищенный продукт промыли EtOAc (10 мл) для получения Соединения 49 (35 мг, выход 45%) в виде твердого желтого вещества. 1H ЯМР (ДМСО-d6 400 MHz): δ 13.66 (brs, 1H), 10.32 (brs, 1H), 8.40 (d, J=1.6 Hz, 1H), 8.37 (d,J=8.4 Hz, 1H), 8.32 (dd, J=8.0, 1.6 Hz, 1H), 8.16 (d, J=8.0 Hz, 1H), 8.02-7.90 (m, 2H), 7.42 (dd, J=9.2, 2.8 Hz, 1H), 7.25 (d, J=2.8 Hz, 1H). MS (ESI): m/z 290.6 [M+H]+.
[00326] Пример 50: Соединение 50: 2-(4-карбокси-2-хлорфенил)-6-гидроксихинолин 1-оксид
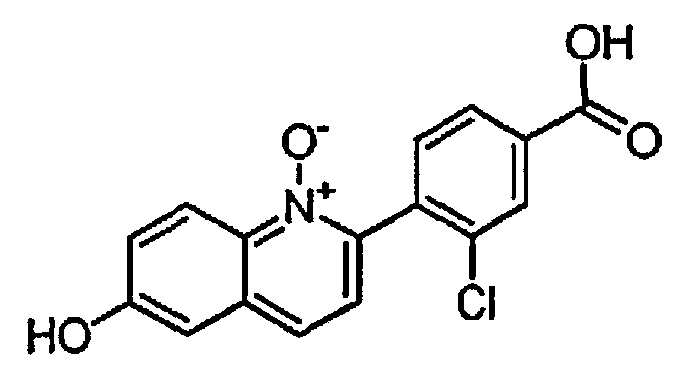
[00327] К раствору 3-хлор-4-(6-гидроксихинолин-2-ил)бензойной кислоты (Соединение 8) (620 мг) в HOAc (8 мл) добавили 3-хлорпероксибензойную кислоту (чистота 77%, 1,19 г). Полученную смесь нагревали при 90°C в течение 3 часов. После удаления HOAc под пониженным давлением, полученную смесь растерли с ДХМ и дважды перекристаллизовали из EtOH/воды для получения заданного продукта (245 мг) в виде бесцветного твердого вещества. 1H ЯМР (ДМСО-d6 300 MHz TMS): δ 10.49 (1H, s), 8.43 (1H, d, J=9 Hz), 8.06 (1H, d, J=3 Hz), 8.01 (1H, dd, J=9 и 3 Hz), 7.82 (1H, d, J=6 Hz), 7.69 (1H, d, J=6 Hz), 7.47 (1H, d, J=9 Hz), 7.37 (1H, dd, J=9 и 3 Hz), 7.31 (1H, d, J=3 Hz) ppm; MS (ESI): m/z 316, [M+H]+.
[00328] Пример 51: Соединение 51: 4-(4-амино-6-гидроксихинолин-2-ил)бензойная кислота
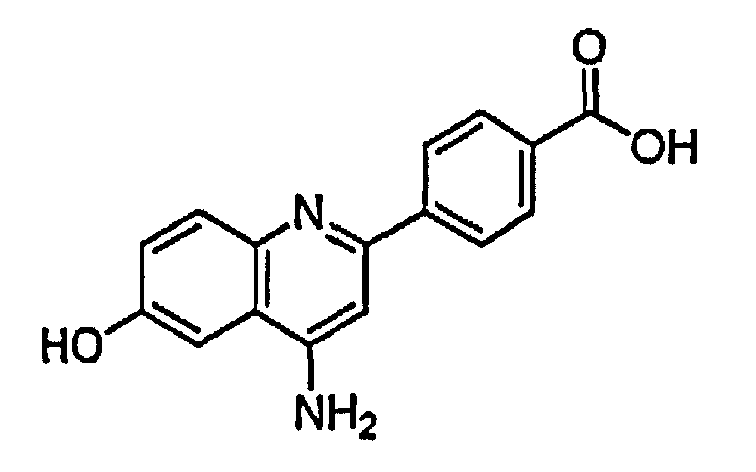
[00329] Этап 1: Синтез метил 4-(4-амино-6-метоксихинолин-2-ил)бензоата: Смесевой раствор Промежуточного соединения 8 (4,70 г, 22,5 ммоль), 4-метоксикарбонилфенилбороновой кислоты (4,01 г, 22,5 ммоль), K2CO3 (6,53 г, 47,2 ммоль), Pd(dppf)Cl2 (470 мг, 0,407 ммоль) в ДМФ (20 мл) и H2O (4 мл) перемешивали под атмосферой N2 при 130°C в течение 5 часов. Смесь охладили, подкислили водным раствором HCl (1М) до pH=6 и экстрагировали EtOAc (80 мл × 3). Объединенные органические слои промыли насыщенным солевым раствором (150 мл), высушили над безводным Na2SO4 и концентрировали in vacuo. Остаток промыли EtOAc (50 мл), смесь отфильтровали, затем концентрировали для получения продукта (3,20 г, выход 46%) в виде грязновато-белого твердого вещества.
[00330] Этап 2: Синтез Соединения 51: Смесь полученного выше продукта (300 мг, 0,97 ммоль) и BBr3 (1 мл, 10,5 ммоль, 2,64 г/мл) в ДХМ (10 мл) перемешивали при 20°C в течение 72 часов. Добавили воду (20 мл) и экстрагировали ДХМ (20 мл × 3), высушили над безводным Na2SO4 и концентрировали in vacuo для получения неочищенного продукта. В результате растирания с MeOH (20 мл) получили Соединение 51 (29,0 мг, выход 11%) в виде твердого желтого вещества. 1H ЯМР (ДМСО-d6, 400 MHz TMS): δ 13.52 (brs, 1H), 10.53 (brs, 1H), 8.74 (brs, 2H), 8.20 (d, J=8.4 Hz, 2H), 8.01-7.96 (m, 3H), 7.65 (d, J=1.6 Hz, 1H), 7.56 (dd, J=9.2, 2.4 Hz, 1H), 6.98 (s, 1H). MS (ESI): m/z 280.9 [M+H]+.
[00331] Пример 52: Соединение 52: 4-(3-циано-6-гидроксихинолин-2-ил)бензойная кислота
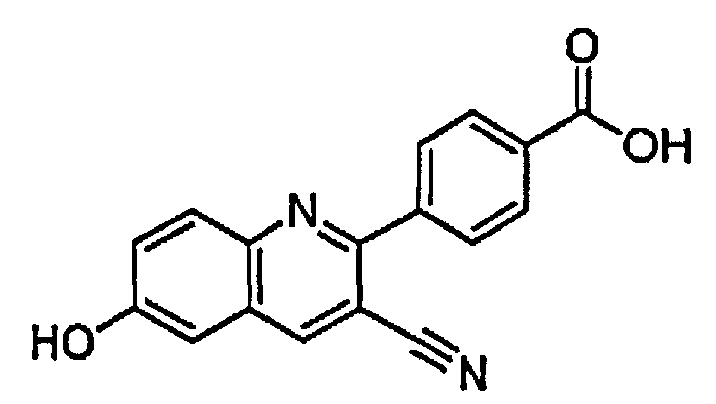
[00332] Этап 1: Синтез 2-хлор-6-метоксихинолин-3-карбонитрила: К смеси 2-хлорхинолин-3-карбоксальдегида (1,00 г, 4,50 ммоль) в ТГФ (30 мл) добавили NH3·H2O (30 мл, 25%) и I2 (1,26 г, 4,90 ммоль), смесь перемешивали при 20°C в течение 8 часов. В результате выделения продукта при помощи воды с экстракцией EtOAc получили неочищенный продукт. В результате очистки на силикагелевой колонке (PE/EtOAc = от 10/1 до 2/1) получили продукт (370 мг, выход 38%) в виде твердого желтого вещества.
[00333] Этап 2: Синтез 4-(3-циано-6-метоксихинолин-2-ил)бензойной кислоты: К смеси полученного выше продукта (75,0 мг, 0,350 ммоль) в ДМФ/H2O (5 мл/1 мл) добавили 4-карбоксифенилбороновую кислоту (57,0 мг, 0,350 ммоль), K2CO3 (73,0 мг, 0,525 ммоль) и PdCl2(dppf) (20,0 мг, 2,73×10-3 ммоль), реакционную смесь дегазировали (3 раза) и нагревали до 100°C в течение 3 часов. Реакционную смесь подкислили водным раствором HCl (1 М) до pH=6, экстрагировали EtOAc (5 мл × 3); органические слои промыли насыщенным солевым раствором (5 мл), высушили над безводным Na2S04, отфильтровали и концентрировали in vacuo. В результате очистки на силикагелевой колонке (PE/EtOAc = от 4/1 до 0/1) получили продукт (45,0 мг, выход 42%) в виде твердого желтого вещества.
[00334] Этап 3: Синтез Соединения 52: К раствору полученного выше продукта (80,0 мг, 0,263 ммоль) в ДХМ (2,5 мл) по каплям добавили BBr3 (0,4 мл). Полученную смесь перемешивали при 30°C под атмосферой N2 в течение 24 часов. В результате выделения продукта при помощи воды с экстракцией EtOAc получили неочищенный продукт. В результате препаративной ВЭЖХ (с добавлением 0,1% ТФК) получили Соединение 52 (6,0 мг, выход 8%) в виде твердого белого вещества. 1H ЯМР (MeOD-d6 400 MHz): δ 8.78 (s, 1H), 8.22 (d, J=8.0 Hz, 2H), 8.05-8.01 (m, 3H), 7.56 (dd, J=9.2, 2.8 Hz, 1H), 7.28 (d, J=2.4 Hz, 1H). MS (ESI): m/z 290.8 [M+H]+.
[00335] Пример 53: Соединение 53: 4-(5-фтор-6-гидроксихинолин-2-ил)бензойная кислота
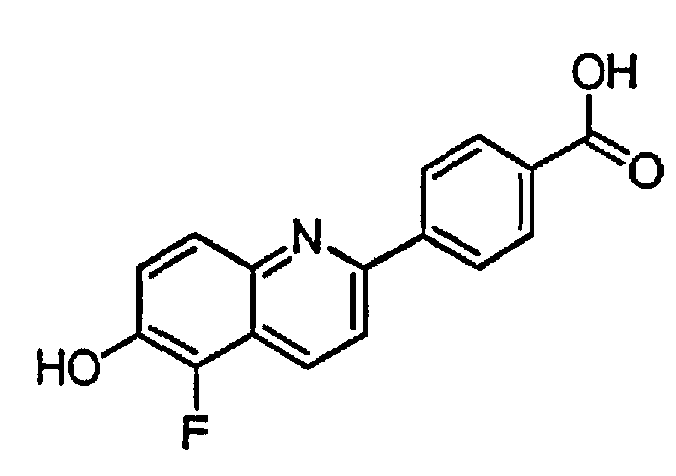
[00336] Этап 1: Синтез метил 4-(5-фтор-6-метоксихинолин-2-ил)бензоата: К раствору метил 4-(6-метоксихинолин-2-ил)бензоата (смотри Соединение 45, этап 1, где описан синтез) (250 мг, 0,85 ммоль) в MeCN (5 мл) добавили Selectfluoro (453 мг, 1,28 ммоль). Реакционную смесь нагревали при 50°C в течение ночи. Растворитель удалили под пониженным давлением, а затем выделили продукт из воды с экстракцией ДХМ для получения продукта в виде твердого коричневого вещества (300 мг, 113%).
[00337] Этап 2: Синтез Соединения 53: К раствору полученного выше продукта (300 мг, 0,96 ммоль) в ДХМ (2 мл) добавили BBr3 (2,4 г, 9,6 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Осторожно добавили воду (40 мл). Осадок собрали и очистили препаративной ВЭЖХ для получения Соединения 53 в виде коричневого порошка (76,8 мг, 25,9%). 2H-ЯМР (ДМСО-d6 500 MHz TMS): 8.45 (d, J=9.0 Hz, 1H), 8.35 (d, J=8.5 Hz, 2H), 8.22 (d, J=9.0 Hz, 1H), 8.10 (d, J=8.5 Hz, 2H), 7.84 (d, J=9.5 Hz, 1H), 7.56 (t, J=9.5 Hz, 1H) ppm. MS (ESI): m/z=284.1 [M+1]+.
[00338] Пример 54: Соединение 54: 4-(8-фтор-6-гидроксихинолин-2-ил)бензойная кислота
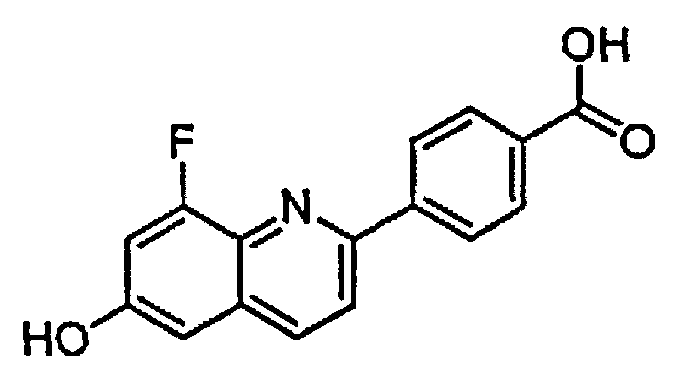
[00339] Этап 1: Синтез метил 4-(8-фтор-6-гидроксихинолин-2-ил)бензоата: Следовали общим условиям сочетания Сузуки, 2-хлор-8-фторхинолин-6-ил ацетат (Промежуточное соединение 9) (89,3 мг) обрабатывали (4-(метоксикарбонил)фенил)бороновой кислотой (74 мг), Pd(dppf)Cl2 (катализатор) и бикарбонатом натрия (69 мг) в диоксане (2 мл) и воде (0,4 мл) при 100°C при микроволновом нагревании в течение 2 часов. После выделения продукта при помощи воды, очистки колоночной флэш-хроматографией на силикагеле получили смесь метил 4-(8-фтор-6-гидроксихинолин-2-ил)бензоата и метил 4-(6-ацетокси-8-фторхинолин-2-ил)бензоата (120 мг) в виде светло-коричневых твердых веществ.
[00340] Этап 2: Синтез Соединения 54: Смесь метил 4-(8-фтор-6-гидроксихинолин-2-ил)бензоата и метил 4-(6-ацетокси-8-фторхинолин-2-ил)бензоата (120 мг) гидролизовали при помощи 2 н. NaOH (4 мл) в MeOH (4 мл). Заданный продукт, 4-(8-фтор-6-гидроксихинолин-2-ил)бензойную кислоту (65 мг) собрали фильтрацией после подкисления при помощи 12 н. HCl. 1H ЯМР (ДМСО-d6 300 MHz TMS): δ 8.36-8.33 (3H, m), 8.18 (1H, d, J=9 Hz), 8.09 (2H, d, J=9 Hz), 7.24 (1H, dd, J=12 и 3 Hz), 7.09 (1H, d, J=3 Hz) ppm, MS (ESI): m/z 284, [M+H]+.
[00341] Пример 55: Соединение 55: 3-гидрокси-4-(6-гидроксихинолин-2-ил)бензойная кислота
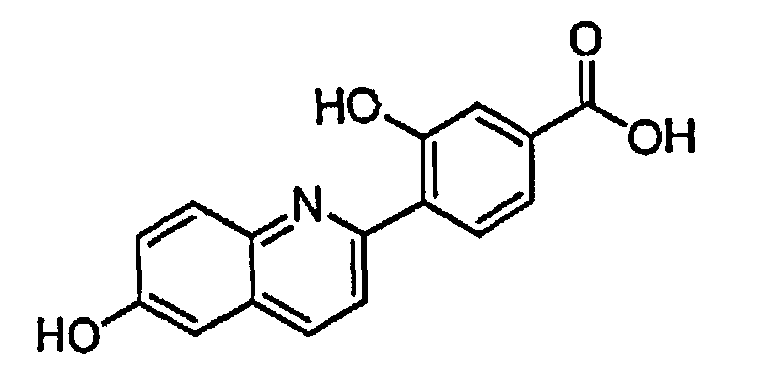
[00342] 70 мг 4-(6-гидроксихинолин-2-ил)-3-метоксибензойной кислоты (Соединение 48) суспендировали в 10 мл ДХМ и добавили 0,25 мл BBr3. Смесь перемешивали при комнатной температуре в течение 2 дней. Добавили 20 мл воды, чтобы погасить реакцию. ДХМ удалили выпариванием, а осадок отфильтровали и промыли водой, и высушили для получения 29 мг 3-гидрокси-4-(6-гидроксихинолин-2-ил)бензойной кислоты. 1H ЯМР (ДМСО-d6 300 MHz TMS): δ 10.34 (s, 1H), 8.45 (d, 1H), 8.30 (d, 1H), 8.24 (d, 1H), 7.97 (d, 1H), 7.46 (m, 3H), 7.26 (d, 1H), MS (ESI): m/z=282.30 [M+1]+.
[00343] Пример 56: Соединение 56: 3-фтор-4-(5-фтор-6-гидроксихинолин-2-ил)бензойная кислота
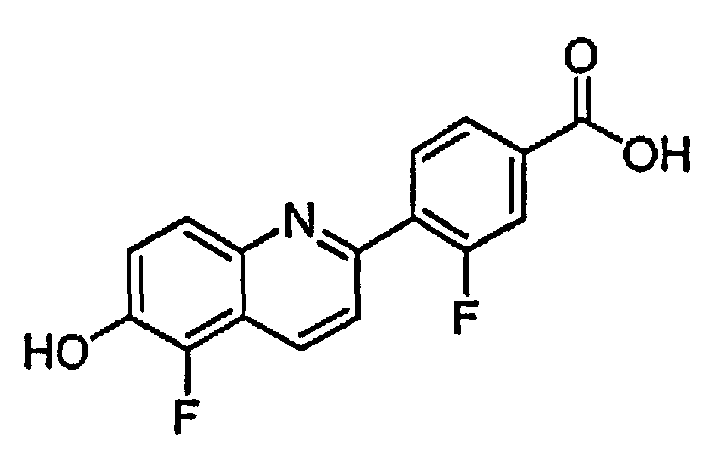
[00344] Этап 1: Синтез метил 3-фтор-4-(6-метоксихинолин-2-ил)бензоата: Смесь 2-хлор-6-метоксихинолина (Промежуточное соединение 1) (300 мг, 1,55 ммоль), метил 3-фтор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензоата (Промежуточное соединение 10) (415 мг, 1,48 ммоль), Pd(dppf)Cl2 (110 мг, 0,15 ммоль) и карбоната натрия (320 мг, 3,0 ммоль) в 1,4-диоксане/воде (6 мл/1 мл) нагревали до 120°C под микроволновым излучением в течение 4 часов. Осадок отфильтровали и промыли EA. Объединенный фильтрат промыли насыщенным солевым раствором, высушили над Na2SO4, отфильтровали и концентрировали in vacuo. Остаток очистили силикагелевой хроматографией (PE/EA=30/1) для получения продукта (220 мг, 46%) в виде твердого белого вещества. MS (ESI): m/z 312.2 [M+1]+.
[00345] Этап 2: Синтез метил 3-фтор-4-(5-фтор-6-метоксихинолин-2-ил)бензоата: К раствору полученного выше продукта (260 мг, 0,835 ммоль) в CH3CN (30 мл) добавили Selectfluor (296 мг, 0,835 ммоль). Реакционную смесь нагревали при 50°C в течение 3 часов и концентрировали. Остаток разделили между водой (20 мл) и ДХМ (20 мл). Водную фазу отделили и экстрагировали ДХМ (20 мл × 2). Объединенные органические слои промыли насыщенным солевым раствором (50 мл), высушили над Na2SO4, отфильтровали и концентрировали. Остаток очистили силикагелевой хроматографией (PE/EA=30/1) для получения продукта (190 мг, неочищенный). MS (ESI): m/z=330.1 [M+1]+.
[00346] Этап 3: Синтез Соединения 56: К ледяному раствор полученного выше продукта (190 мг, 0,577 ммоль) в ДХМ (2 мл) добавили BBr3 (1,45 мг, 5,77 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре и медленно погасили водой (40 мл). Осадок собрали и очистили препаративной ВЭЖХ для получения Соединения 56 в виде коричневого порошка (140 мг, 81%). 1H-ЯМР (ДМСО-d6, 500 MHz, TMS): δ 8.47 (d, J=8.5 Hz, 1H), 8.15 (t, J=8.0 Hz, 1H), 7.97 (dd, J=2.5 Hz, 1H), 7.93 (dd, J=1.5 Hz, 1H), 7.82-7.86 (m, 2H), 7.57 (t, J=9.0 Hz, 1H) ppm; MS (ESI): m/z=302.1 [M+1]+.
[00347] Пример 57: Синтез Промежуточных соединений
[00348] Промежуточное соединение 1: Синтез 2-хлор-6-метоксихинолина
[00349] Этап 1: Синтез 6-метоксихинолин 1-оксида: К раствору 6-метоксихинолина (2,00 г, 12,6 ммоль) в AcOH (10 мл) добавили H2O2 (30% в воде, 1,9 мл, 18,9 ммоль), смесь нагревали до 70°C в течение 21 часа. Смесь подщелочили при помощи 2M NaOH до рН 8-9 и экстрагировали CH2Cl2 (200 мл), объединенный органический слой промыли насыщенным солевым раствором (50 мл), высушили над Na2SO4, отфильтровали и концентрировали для получения неочищенного продукта, который очистили на силикагелевой колонке (EtOAc/MeOH=10/1) для получения 6-метоксихинолин 1-оксида (1,20 г, 55%) в виде твердого вещества.
[00350] Этап 2: Синтез 6-метоксихинолин-2-ола: Раствор 6-метоксихинолин 1-оксида (300 мг, 1,71 ммоль) в Ac2O (5,0 мл) дефлегмировали в течение 2 часов. Растворитель удалили под пониженным давлением. Остаток растворили в EtOAc (50 мл), органический слой промыли насыщенным солевым раствором (10 мл), высушили над Na2SO4, отфильтровали и концентрировали для получения неочищенного продукта, который очистили на силикагелевой колонке (PE/EtOAc=1/2) для получения 6-метоксихинолин-2-ола (200 мг, выход 67%).
[00351] Этап 3: Синтез 2-хлор-6-метоксихинолина: Раствор 6-метоксихинолин-2-ола (400 мг, 2,29 ммоль) в POCl3 (5,0 мл) дефлегмировали в течение 2 часов. Растворитель удалили под пониженным давлением, а остаток растворили в EtOAc (50 мл), органический слой промыли насыщенным раствором NaHCO3 (30 мл × 2), высушили над Na2SO4, отфильтровали и концентрировали для получения неочищенного продукта, который очистили на силикагелевой колонке (PE/EtOAc=10/1) для получения 2-хлор-6-метоксихинолина (380 мг, выход 86%) в виде твердого вещества. 1H ЯМР (CDCl3 400 MHz): δ 7.92 (d, J=8.4 Hz, 1H), 7.85 (d, J=9.2 Hz, 1H), 7.32-7.25 (m, 2H), 7.00 (d, J=2.4 Hz, 1H), 3.86 (s, 3H).
[00352] Промежуточное соединение 2: Синтез 2-хлорхинолин-6-ола
[00353] К раствору 2-хлор-6-метоксихинолина (Промежуточное соединение 1) (2,00 г, 10,4 ммоль) в безводном ДХМ (100 мл) по каплям добавили BBr3 (6 мл, 62,2 ммоль) при 0°C. Реакционную смесь перемешивали при 25°C в течение 2 часов, затем погасили насыщенным водным раствором NH4Cl (50 мл) и отфильтровали. Фильтрат экстрагировали CH2Cl2/MeOH (об./об.=10/1, 30 мл × 2), а объединенные органические слои промыли насыщенным солевым раствором (30 мл), высушили над Na2SO4, отфильтровали и концентрировали под пониженным давлением для получения 2-хлорхинолин-6-ола (1,30 г, выход 70%) в виде твердого желтого вещества. 1H ЯМР (CDCl3 300 MHz): δ 7.95 (t, J=8.1 Hz, 2H), 7.35 (dd, J=6.0, 3.3 Hz, 2H), 7.13 (d, J=2.7 Hz, 1H).
[00354] Промежуточное соединение 3: Синтез 2,4-дихлор-3-фтор-6-метоксихинолина
[00355] Этап 1: Синтез 2-фтормалоновой кислоты: К раствору диэтил 2-фтормалоната (5,00 г, 28,1 ммоль) в EtOH (100 мл) добавили LiOH·H2O (2,70 г, 64,3 ммоль) при 25°C. Смесь нагревали до 50°C в течение 16 часов. Смесь отфильтровали и собрали твердое вещество. Фильтрат концентрировали до сухости с получением маслянистого вещества. Маслянистое и твердое вещество растворили в H2O (30 мл) и МТБЭ (100 мл), смесь подкислили добавлением концентрированной HCl до pH 1, водный слой экстрагировали МТБЭ (30 мл × 2), объединенные органические слои высушили над Na2SO4, отфильтровали и концентрировали для получения 2-фтормалоновой кислоты (3,00 г, выход 88%) в виде твердого вещества.
[00356] Этап 2: Синтез 2,4-дихлор-3-фтор-6-метоксихинолина: Суспензию фтормалоновой кислоты (1,00 г, 8,13 ммоль) в POCl3 (10 мл) нагревали до 85°C для растворения твердого вещества. Смесь охладили до 60°C и частями добавили п-анизидин (900 мг, 7,32 ммоль) за 1 час. После добавления реакционную смесь дефлегмировали в течение 2 часов. Растворитель удалили in vacuo. Смесь разбавили ледяной водой и подщелочили добавлением NH3·H2O до pH 9. Водный слой экстрагировали EtOAc (30 мл × 3), объединенные органические слои промыли насыщенным солевым раствором (20 мл), высушили над Na2SO4, отфильтровали и концентрировали для получения неочищенного продукта, который очистили на силикагелевой колонке (PE/EtOAc=10/1) для получения 2,4-дихлор-3-фтор-6-метоксихинолина (650 мг, выход 36%). 1H ЯМР (CDCl3 300 MHz): δ 7.92 (d, J=9.0 Hz, 1H), 7.41-7.31 (m, 2H), 4.00 (s, 3H).
[00357] Промежуточное соединение 4: Синтез 2,3-дихлор-6-метоксихинолина
[00358] Этап 1: Синтез 2-хлор-N-(4-метоксифенил)ацетамида: Смесь 4-метоксианилина (5,00 г, 40,6 ммоль), хлоруксусной кислоты (8,6 г, 91,5 ммоль), EDCI (12,0 г, 61,2 ммоль), HOBT (8,4 г, 61,3 ммоль) и NMM (13 мл, 122 ммоль) в безводном CH2Cl2 (50 мл) перемешивали при 30°C в течение 3 часов. Смесь погасили ледяной водой, а затем экстрагировали CH2Cl2 (30 мл × 2). Объединенные органические слои промыли насыщенным солевым раствором (30 мл), высушили над Na2SO4, отфильтровали и концентрировали для получения неочищенного продукта, который очистили на силикагелевой колонке (PE/EtOAc=5/1) для получения продукта (1,60 г, выход 20%).
[00359] Этап 2: Синтез 2,3-дихлор-6-метоксихинолина: POCl3 (1,6 мл, 17,5 ммоль) по каплям добавили к ДМФ (0,29 мл, 3,80 ммоль) при 0°C. После добавления частями добавили 2-хлор-N-(4-метоксифенил)ацетамид (500 мг, 2,50 ммоль). Смесь перемешивали при 25°C в течение 15 минут и нагревали до 75°C в течение 3 часов. Реакционную смесь погасили ледяной водой и нейтрализовали при помощи 2 M NaOH до pH 7. После выделения продукта при помощи воды при помощи EtOAc выполнили очистку на силикагелевой колонке (PE/EtOAc=20/1) для получения Промежуточного соединения 4 (50 мг, выход 9%). 1H ЯМР (CDCl3 400 MHz): δ 8.06 (s, 1H), 7.81 (d, J=9.6 Hz, 1H), 7.30 (dd, J=9.2, 2.8 Hz, 1H), 6.92 (d, J=2.8 Hz, 1H), 3.87 (s, 3H).
[00360] Промежуточное соединение 5: Синтез метил 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3-(трифторметил)бензоата
[00361] К смеси метил 4-бром-3-(трифторметил)бензоата (1,00 г, 3,53 ммоль), бис(пинаколато)дибора (1,79 г, 7,06 ммоль) и KOAc (1,04 г, 10,6 ммоль) в ДМСО (15 мл) добавили Pd(PPh3)4 (816 мг, 0,706 ммоль) под атмосферой N2. Затем смесь нагревали до 120°C в течение 3 часов. Реакционную смесь охладили, а затем выделили продукт из воды/EtOAc для получения неочищенного продукта (2,3 г) в виде желтого маслянистого вещества.
[00362] Промежуточное соединение 6: Синтез 2-хлор-3-(трифторметил)хинолин-6-ола
[00363] Этап 1: Синтез (5-метокси-2-нитрофенил)метанола: К раствору 5-метокси-2-нитробензойной кислоты (20,0 г, 0,102 моль) в безводном ТГФ (200 мл) добавили SOCl2 (20 мл), смесь дефлегмировали в течение 4 часов. Растворитель удалили под пониженным давлением и растворили остаток в безводном ТГФ (100 мл). Раствор по каплям добавили к суспензии NaBH4 (7,70 г, 0,202 моль) в безводном ТГФ (100 мл) и ДМФ (140 мл) при 0°C за период 30 минут. Смесь перемешивали при 30°C в течение 3 часов, затем погасили ледяной водой (100 мл) и подщелочили при помощи 2М NaOH до pH 8. После выделения продукта при помощи EtOAc удалили растворитель in vacuo и выполнили очистку колоночной хроматографией (PE/EtOAc=1/1) для получения продукта (12,0 г, выход 66%).
[00364] Этап 2: Синтез трет-бутил(5-метокси-2-нитробензилокси)диметилсилана: К раствору (5-метокси-2-нитрофенил)метанола (12,0 г, 65,6 ммоль) в безводном ТГФ (200 мл) и ДМФ (20 мл) добавили имидазол (9,80 г, 144 ммоль). Затем частями добавили TBSC1 (11,8 г, 78,6 ммоль) при 0°C и перемешивали смесь при 30°C в течение 2 часов. Смесь погасили ледяной водой (100 мл). После выделения продукта при помощи EtOAc удалили растворитель in vacuo и выполнили очистку колоночной хроматографией (РЕ/EtOAc=10/1) для получения продукта (16,0 г, выход 84%) в виде желтого маслянистого вещества.
[003651 Этап 3: Синтез 2-((трет-бутилдиметилсилилокси)метил)-4-метоксианилина: К раствору трет-бутил(5-метокси-2-нитробензилокси)диметилсилана (14,0 г, 47,1 ммоль) в EtOH (200 мл) добавили 10% Pd/C (1,40 г), смесь перемешивали при 30°C в течение 2 часов под H2 (40 psi). Твердые вещества отфильтровали, а фильтрат концентрировали под пониженным давлением для получения продукта (14,0 г) в виде желтого маслянистого вещества. 1H ЯМР (ДМСО 400 MHz): δ 6.75 (d, J=2.4 Hz, 1H), 6.60-6.55 (m, 2H), 4.55 (s, 2H), 4.40 (brs, 2H), 3.61 (s, 3H), 0.90 (s, 9H), 0.09 (s, 6H).
[00366] Этап 4: Синтез N-(2-((трет-бутилдиметилсилилокси)метил)-4-метоксифенил)-3,3,3-трифторпропанамида: Смесь 2-((трет-бутилдиметилсилилокси)метил)-4-метоксианилина (14,0 г), 3,3,3-трифтор-пропионовой кислоты (7,30 г, 57,0 ммоль), EDCI (15,0 г, 76,5 ммоль), HOBT (11,0 г, 80,2 ммоль) и NMM (22 мл, 157 ммоль) в безводном CH2Cl2 (200 мл) перемешивали при 20°C в течение 2 часов. Смесь разбавили CH2Cl2 (100 мл), органический слой промыли 1M HCl (100 мл), H2O (100 мл) и насыщенным солевым раствором (100 мл), высушили над Na2SO4 и концентрировали для получения продукта.
[00367] Этап 5: Синтез 3,3,3-трифтор-N-(2-(гидроксиметил)-4-метоксифенил)пропанамида: К раствору N-(2-((трет-бутилдиметилсилилокси)метил)-4-метоксифенил)-3,3,3-трифторпропанамида (20,0 г) в безводном ТГФ (200 мл) добавили раствор TBAF (16,6 г, 63,6 ммоль) в безводном ТГФ (60 мл). Смесь перемешивали при 35°C в течение 30 минут. Смесь погасили ледяной водой. После выделения продукта при помощи воды/EtOAc удалили летучие вещества in vacuo. Неочищенный продукт очистили колоночной хроматографией на силикагеле (PE/EtOAc=1/2) для получения заданного продукта. 1H ЯМР (CDCl3 400 MHz): δ 8.60 (brs, 1H), 7.82 (d, J=8.8 Hz, 1H), 6.85 (dd, J=9.2, 3.2 Hz, 1H), 6.75 (d, J=2.8 Hz, 1H), 4.65 (s, 2H), 3.80 (s, 3H), 3.23 (q, J=10.4 Hz, 2H).
[00368] Этап 6; Синтез 3,3,3-трифтор-N-(2-формил-4-метоксифенил)пропанамида: К раствору 3,3,3-трифтор-N-(2-(гидроксиметил)-4-метоксифенил)пропанамида (6,30 г, 23,9 ммоль) в безводном CH2Cl2 (200 мл) добавили MnO2 (20,0 г, 229 ммоль). Смесь дефлегмировали в течение 16 часов. Смесь отфильтровали, а фильтрат концентрировали для получения заданного продукта (5,30 г, выход 84%).
[00369] Этап 7: Синтез 6-метокси-3-(трифторметил)хинолин-2(1H)-она: К раствору 3,3,3-трифтор-N-(2-формил-4-метоксифенил)пропанамида (5,30 г, 20,3 ммоль) в ДМФ (100 мл) добавили K2CO3 (14,0 г, 101 ммоль). Смесь нагревали до 60°C в течение 1,5 часов. Смесь разбавили EtOAc (200 мл) и отфильтровали. Фильтрат промыли H2O (100 мл) и насыщенным солевым раствором (100 мл), высушили над Na2SO4 и концентрировали in vacuo для получения заданного продукта (4,60 г, выход 94%). 1H ЯМР (CDCl3 300 MHz): δ 12.25 (brs, 1H), 8.18 (s, 1H), 7.40 (d, J=9.0 Hz, 1H), 7.25 (dd, J=9.0, 2.7 Hz, 1H), 7.02 (d, J=2.7 Hz, 1H), 3.87 (s, 3H).
[00370] Этап 8: Синтез 2-хлор-6-метокси-3-(трифторметил)хинолина: Раствор 6-метокси-3-(трифторметил)хинолин-2(1H)-она (4,60 г, 18,9 ммоль) в POCl3 (30 мл) дефлегмировали в течение 2,5 часов. Растворитель удалили под пониженным давлением, а остаток нейтрализовали при помощи 2М NaOH до рН 7. После выделения продукта при помощи EtOAc, удалили летучие вещества под пониженным давлением. Остаток очистили колоночной хроматографией на силикагеле (PE/EtOAc=15/1) для получения заданного продукта (4,60 г, выход 94%).
[00371] Этап 9: Синтез 2-хлор-3-(трифторметил)хинолин-6-ола: К раствору полученного выше продукта (1,00 г, 3,83 ммоль) в безводном CH2Cl2 (20 мл) добавили BBr3 (3,0 мл, 31,8 ммоль) при 0°C. Смесь перемешивали при 0°C в течение 2 часов. Смесь погасили ледяной водой (10 мл) и экстрагировали CH2Cl2 (30 мл × 3). Объединенный органический слой промыли насыщенным солевым раствором (50 мл), высушили над Na2SO4 и концентрировали под пониженным давлением для получения Промежуточного соединения 6 (600 мг, выход 63%) в виде твердого вещества. 1H ЯМР (CDCl3 400 MHz): δ 8.37 (s, 1H), 8.00 (d, J=9.2 Hz, 1H), 7.48 (dd, J=8.8, 2.4 Hz, 1H), 7.22 (d, J=2.8 Hz, 1H).
[00372] Промежуточное соединение 7: Синтез 2-хлор-4-фтор-6-метоксихинолина
[00373] Этап 1: Синтез 2,4-дихлор-6-метоксихинолина: Смесь п-анизидина (100 г, 0,813 моль) и малоновой кислоты (85,0 г, 0,817 моль) в POCl3 (500 мл) дефлегмировали в течение 6 часов. Избыток POCl3 удалили in vacuo, а остаток нейтрализовали при помощи 8М NaOH до pH 7. Водный слой экстрагировали CH2Cl2 (300 мл × 3), промыли насыщенным солевым раствором (500 мл), высушили над Na2SO4 и концентрировали in vacuo. В результате очистки колоночной хроматографией на силикагеле (PE/EtOAc=15/1) получили заданный продукт (35,0 г, выход: 19%).
[00374] Этап 2: Синтез 2-хлор-6-метоксихинолин-4-амина: Суспензию 2,4-дихлор-6-метоксихинолина (5,00 г, 22,0 ммоль) и NH3 (газ)/MeOH (насыщенный, 40 мл) нагревали до 150°C в течение 16 часов в закрытой пробирке. Растворитель удалили, а остаток разбавили MeOH (20 мл). Смесь отфильтровали, а фильтрат концентрировали для получения неочищенного продукта. В результате очистки колоночной хроматографией на силикагеле (PE/EtOAc=2/1) получили продукт (7,50 г, выход: 55%) в виде твердого вещества.
[00375] Этап 3: Синтез 2-хлор-4-фтор-6-метоксихинолина: Раствор 2-хлор-6-метоксихинолин-4-амина (4,50 г, 21,6 ммоль) в HF-пиридине (45 мл) охладили до -10-0°C. Затем частями добавили NaNO2 (1,80 г, 26,1 ммоль) и перемешивали смесь при 0°C в течение 1 часа и при 30°C в течение 1 часа. Затем смесь нагревали до 65°C в течение 1,5 часов. Смесь погасили ледяной водой (100 мл); водный слой нейтрализовали при помощи 2М NaOH до pH 7. Смесь отфильтровали, а фильтрат экстрагировали EtOAc (50 мл × 3), органический слой промыли насыщенным солевым раствором (100 мл), высушили над Na2SO4 и концентрировали in vacuo. В результате очистки колоночной хроматографией на силикагеле (PE/EtOAc=15/1) получили Промежуточное соединение 7 (3,60 г, выход: 40%). 1H ЯМР (CDCl3 400 MHz): δ 7.92 (dd, J=9.2, 1.6 Hz, 1H), 7.40 (dd, J=9.2, 2.8 Hz, 1H), 7.25 (s, 1H), 7.10 (d, J=9.2 Hz, 1H), 3.97 (s, 3H).
[00376] Промежуточное соединение 8: Синтез 2-хлор-6-метоксихинолин-4-амина
[00377] Этап 1: Синтез 2,4-дихлор-6-метоксихинолина: Смесь п-анизидина (100 г, 0,813 моль) и малоновой кислоты (85,0 г, 0,817 моль) в POCl3 (500 мл) дефлегмировали в течение 6 часов. Избыток POCl3 удалили in vacuo, а остаток нейтрализовали при помощи 8М NaOH до pH 7. Водный слой экстрагировали CH2Cl2 (300 мл × 3), объединенный органический слой промыли насыщенным солевым раствором (500 мл), высушили над Na2SO4 и концентрировали in vacuo для получения неочищенного продукта, который очистили колоночной хроматографией на силикагеле (PE/EtOAc=15/1) для получения 2,4-дихлор-6-метоксихинолина (35,0 г, выход: 19%) в виде твердого белого вещества.
[00378] Этап 2: Синтез Промежуточного соединения 8: Суспензию 2,4-дихлор-6-метоксихинолина (5,00 г, 22,0 ммоль) в NH3 (газ)/MeOH (насыщенный, 40 мл) нагревали до 150°C в течение 16 часов в закрытой пробирке. Растворитель удалили in vacuo, а остаток разбавили MeOH (20 мл). Смесь отфильтровали, а фильтрат концентрировали для получения неочищенного продукта, который очистили колоночной хроматографией на силикагеле (PE/EtOAc=2/1) для получения 2-хлор-6-метоксихинолин-4-амина (7,50 г, выход: 55%) в виде твердого желтого вещества.
[00379] Промежуточное соединение 9: Синтез 2-хлор-8-фторхинолин-6-ил ацетата
[00380] Этап 1: Синтез 3-хлор-N-(2-фтор-4-гидроксифенил)пропанамида: 4-Амино-3-фторфенол (3,4 г) смешали с 3-хлорпропаноил хлоридом (3,56 г) в ацетоне (60 мл) и нагревали с дефлегматором в течение 3 часов. После водного выделения продукта при помощи EtOAc/воды, отделенные органические слои высушили над Na2SO4 и концентрировали in vacuo. Неочищенный продукт очистили силикагелевой хроматографией для получения продукта (2,95 г) в виде светло-коричневого твердого вещества.
[00381] Этап 2: Синтез 8-фтор-6-гидрокси-3,4-дигидрохинолин-2(1Н)-она: 3-Хлор-N-(2-фтор-4-гидроксифенил)пропанамид (2,1 г) смешали с безводным Al2Cl3 (7 г) и нагревали при 160°C в течение ночи. Полученную смесь обработали 1 н. HCl и экстрагировали EtOAc. После отделения органического слоя и удаления растворителей под пониженным давлением, заданный неочищенный продукт (1,8 г) собрали в виде светло-коричневого твердого вещества.
[00382] Этап 3. Синтез 8-фтор-2-оксо-1,2,3,4-тетрагидрохинолин-6-ил ацетата: Неочищенный 8-фтор-6-гидрокси-3,4-дигидрохинолин-2(1H)-он (0,574 г) обрабатывали ацетилхлоридом (330 мг) и ТЭА (0,68 мл) в ДХМ (8 мл) в течение 3 часов. После водного выделения продукта при помощи EtOAc/воды, неочищенный продукт очистили колоночной флэш-хроматографией для получения заданного продукта (382 мг) в виде бесцветного твердого вещества.
[00383] Этап 4: Синтез 8-фтор-2-гидроксихинолин-6-ил ацетата: К раствору 8-фтор-2-оксо-1,2,3,4-тетрагидрохинолин-6-ил ацетата (718 мг) в толуоле (8 мл) добавили DDQ (1,2 г). Полученный раствор нагревали при 70°C в течение 48 часов. После водного выделения продукта при помощи EtOAc, неочищенный продукт очистили колоночной флэш-хроматографией на диоксиде кремния для получения чистого продукта (550 мг) в виде бесцветного твердого вещества.
[00384] Этап 5: Синтез Промежуточного соединения 9: К раствору 8-фтор-2-гидроксихинолин-6-ил ацетата (550 мг) в ДМФ (6 мл) добавили POCl3 (0,6 мл). Затем смесь нагревали при 80°C пару часов. После водного выделения продукта, заданный продукт, 2-хлор-8-фторхинолин-6-ил ацетат (380 мг) получили колоночной флэш-хроматографией на диоксиде кремния.
[00385] Промежуточное соединение 10: Синтез метил 3-фтор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензоата:
[00386] Этап 1: Синтез метил 4-бром-2-фторбензоата: К раствору 4-бром-2-фторбензойной кислоты (4 г, 18 ммоль) в метаноле (10 мл) по каплям добавили оксалилдихлорид (4,6 г, 36 ммоль). Реакционную смесь нагревали до 60°C в течение ночи и медленно добавили в ледяную воду, и экстрагировали ДХМ (50 мл × 2). Объединенные экстракты промыли насыщенным солевым раствором, высушили над Na2SO4 и концентрировали для получения заданного продукта (3,7 г, 88%) в виде твердого коричневого вещества.
[00387] Этап 2: Синтез Промежуточного соединения 10: К раствору метил 4-бром-2-фторбензоата (1 г, 4,29 ммоль) в 1,4-диоксане (20 мл) добавили Pin2B2 (1,31 г, 5,15 ммоль), ацетат калия (1,26 г, 12,87 ммоль) и Pd(dppf)Cl2 (106,2 мг, 0,128 ммоль). Систему вакуумировали и повторно заполнили N2. Реакционную смесь нагревали при 100°C в течение 17 часов. Смесь концентрировали, а остаток очистили силикагелевой хроматографией (PE/EA=15/1) для получения Промежуточного соединения 10 (950 мг, 79%) в виде твердого желтого вещества. 1H-ЯМР (CDCl3, 500 MHz, TMS): δ 7.80 (t, J=1.5 Hz, 2H), 7.67 (d, J=10 Hz, 1H), 3.92 (s, 3H), 1.37 (s, 12H) ppm.
[00388] Пример 58: Анализы GSNOR
[00389] Различные соединения испытали in vitro на их способность ингибировать активность GSNOR. Соединения-ингибиторы GSNOR в Примерах 1-56 имеют значения IC50 около <10 мкМ. Соединения-ингибиторы GSNOR в Примерах 1-4, 6, 8, 10-14, 16-35, 37-43, 45-50 и 52-56 имеют значения IC50 около <0,5 мкМ. Соединения-ингибиторы GSNOR в Примерах 1-4, 8, 10-14, 17-28, 30, 31, 37, 40-41, 43, 46, 48-49 и 52-56 имеют значение IC50 около <0,1 мкМ.
[00390] Экспрессия и очистка GSNOR описана в публикации Biochemistry 2000, 39, 10720-10729.
[00391] Ферментация GSNOR: Прекультуры выращивали из палочек глицериновой исходной культуры GSNOR в среде 2XYT, содержащей 100 мкг/мл ампициллина после инкубирования в течение ночи при 37°C. Затем клетки добавили к свежей среде 2XYT (4 л), содержащей ампициллин, и выращивали до оптической плотности (OD) (поглощение при 600 нм, A600) 0,6-0,9 при 37°C перед индуцированном. Экспрессию GSNOR индуцировали при помощи 0,1% арабинозы, инкубируя в течение ночи при 20°C.
[00392] Очистка GSNOR: Клеточную суспензию Е. coli лизировали порообразованием под действием азота, а осветленный лизат очистили Ni-аффинной хроматографией на жидкостном экспресс-хроматографе белков АКТА (Amersham Pharmacia). Колонку элюировали в 20 мМ Tris с pH 8,0/250 мМ NaCl с имидазоловым градиентом 0-500 мМ. Элюированные фракции GSNOR, содержащие слияние Smt-GSNOR, расщепляли в течение ночи при помощи Ulp-1 при 4°C для удаления метки аффинности, затем повторно пропустили через Ni-колонку при тех же условиях. GSNOR выделили в проточной фракции, и для кристаллографии дополнительно очистили Q-сефарозной и гепариновой проточной хроматографией в 20 мМ Tris с pH 8,0,1 мМ DTT, 10 мкМ ZnSO4.
[00393] Анализ GSNOR: Свежие растворы GSNO и фермента/НАДФ готовили каждый день. Эти растворы отфильтровали и оставили нагреваться до комнатной температуры. Раствор GSNO: 100 мМ NaPO4 (pH 7,4), 0,480 мМ GSNO. В кювету добавили 396 мкл раствора GSNO, а затем 8 мкл исследуемого соединения в ДМСО (или только ДМСО для полного контроля реакции) и перемешали кончиком пипетки. Исследуемые соединения приготовили в исходной концентрации 10 мМ в 100% ДМСО. 2-кратные серийные разбавления выполнили в 100% ДМСО. К образцу добавили 8 мкл каждого разбавления, так чтобы конечная концентрация ДМСО в образце составляла 1%. Концентрации исследуемых соединений находились в диапазоне от 100 до 0,003 мкМ. Раствор фермента/НАДФ: 100 мМ NaPO4 (pH 7,4), 0,600 мМ НАДФ, 1,0 мкг/мл GNSO-редуктазы. В кювету добавили 396 мкл раствора фермента/НАДФ, чтобы началась реакция. Кювету поместили в спектрофотометр УФ/видимого диапазона Cary 3E и в течение 3 минут записывали изменение поглощения при 340 нм в минуту при 25°C. Анализы выполнили в трех экземплярах для каждой концентрации соединения. IC50 для каждого соединения рассчитали, используя анализ калибровочной кривой в модуле Enzyme Kinetics программы SigmaPlot.
[00394] Конечные условия анализа: 100 мМ NaPO4, pH 7,4, 0,240 мМ GSNO, 0,300 мМ НАДФ, 0,5 мкг/мл GSNO-редуктазы и 1% ДМСО. Конечный объем: 800 мкл/кювету.
[00395] Пример 59: Эффективность ингибиторов GSNORi при экспериментальной астме
[00396] Экспериментальная модель астмы:
[00397] Для скрининга ингибиторов GSNOR на эффективность против метахолин (MCh)-индуцированной бронхоконстрикции/гиперчувствительности дыхательных путей, использовали модель овальбумин (OVA)-индуцированной астмы на мышах.
Она является широко применяемой и хорошо охарактеризованной моделью, которая представляет фенотип острой, аллергической астмы, схожей с астмой человека. Эффективность ингибиторов GSNOR была оценена с использованием протокола, в котором ингибиторы GSNOR вводились после OVA-сенсибилизации и затрудненности дыхательных путей, и перед испытанием с MCh. Бронхоконстрикция в ответ на испытание повышенными дозами MCh была оценена с использованием плетизмографии всего организма (Penh; Buxco). Также определили количество эозинофильного инфильтрата в жидкости бронхоальвеолярного лаважа (BALF) как меру воспаления легких. Влияние ингибиторов GSNO R сравнили с носителями и с комбивентом (ингаляция; IH) в качестве положительного контроля.
[00398] Материалы и способы
[00399] Аллергенная сенсибилизация и протокол испытания
[00400] OVA (500 мкг/мл) в PBS смешали с равными объемами 10% (вес./об.) алюминия-калия сульфата в дистиллированной воде и инкубировали в течение 60 минут при комнатной температуре после доведения pH до 6,5 при помощи 10 н. NaOH. После центрифугирования при 750×g в течение 5 минут, гранулу OVA/квасцов повторно суспендировали до исходного объема в дистиллированной воде. Мышам внутрибрюшинной (IP) инъекцией ввели 100 мкг OVA (0,2 мл раствора 500 мкг/мл в изотоническом растворе), вместе с квасцами в 0 день. Мышей анестезировали IP инъекцией 0,2 мл смеси кетамина и ксилазина (0,44 и 6,3 мг/мл, соответственно) в изотоническом растворе, и поместили на доску в положении лежа на спине. Двести пятьдесят микрограмм (100 мкл раствора 2,5 мг/мл) OVA (на 8 день) и 125 мкг (50 мкл раствора 2,5 мг/мл) OVA (на 15, 18 и 21 день) поместили на спинку языка каждого животного.
[00401] Испытание легочной функции (Penh)
[00402] In vivo восприимчивость к метахолину измерили через 24 часа после последнего испытания OVA у находящихся в сознании, свободно передвигающихся, самопроизвольно дышащих мышей при помощи плетизмографии всего тела с использованием камеры Buxco (уилмингтон, штат Северная Каролина). Мышей испытывали аэрозолем солевого раствора или увеличивающихся доз метахолина (5, 20 и 50 мг/мл), создаваемых ультразвуковым распылителем, в течение 2 минут. Степень бронхоконстрикции выразили как индекс PenH (Penh), расчетная безразмерная величина, которая коррелирует с измерением бронхиального сопротивления, импенданса и внутриплеврального давления у той же мыши. Значения Penh были взяты и усреднены в течение 4 минут после каждого испытания введением аэрозоля. Penh рассчитали следующим образом: Penh[(Te/Tr-1)×(PEF/PIF)], где Te является временем выдоха, Tr является временем релаксации, PEF является пиком выдоха, и PIF является пиком вдоха, x коэффициент 0,67. Время, за которое давление в камере изменяется с максимального до определенного пользователем процента от максимального значения, представляет время релаксации. Измерение Tr начинается в момент максимального давления в камере и заканчивается при 40%.
[00403] Эозинофильный инфильтрат в BALF
[00404] После измерения гиперчувствительности дыхательных путей, мышей обескровили пункцией сердца, а затем BALF собрали из обоих легких или из правого легкого после перерезания левого легкого у главного бронха. Общее количество клеток BALF подсчитали в аликвоте 0,05 мл, а остальную жидкость центрифугировали при 200×g в течение 10 минут при 4°C. Гранулы клеток повторно суспендировали в солевом растворе, содержащем 10% BSA, сделав мазки на предметном стекле. Эозинофилы окрашивали в течение 5 минут при помощи 0,05% водного эозина и 5% ацетона в дистиллированной воде, промыли дистиллированной водой и выполнили контрастное окрашивание при помощи 0,07% метиленового синего. Альтернативно, эозинофилы и другие лейкоциты окрасили при помощи DiffQuik.
[00405] Ингибиторы GSNOR и контрольные образцы
[00406] Ингибиторы GSNOR восстановили в фосфатно-солевом буферном растворе (PBS), pH 7,4, или в 0,5% вес./об. карбоксиметилцеллюлозе при концентрациях в диапазоне от 0,00005 до 3 мг/мл. Ингибиторы GSNOR ввели мышам (10 мл/кг) однократной дозой или многократными дозами, внутривенно (IV) или перорально через зонд. Дозирование выполнили за 30 минут - 72 часа до испытания MCh. Влияние ингибиторов GSNOR сравнили с носителем, дозированным таким же образом.
[00407] Во всех испытаниях в качестве положительного контроля использовали комбивент. Комбивент (Boehringer Ingelheim) вводили в легкие, используя ингалятор, подающий продукт, но адаптированный для введения мышам, используя кончик пипетки. Комбивент вводили за 48 часов, 24 часа и 1 час до испытания MCh. Каждое нажатие (или доза) комбивента обеспечивала дозу 18 мкг ипатропия бромида (IpBr) и 103 мкг альбутерол сульфата, или около 0,9 мг/кг IpBr и 5 мг/кг альбутерола.
[00408] Статистические анализы
[00409] Значения площади под кривой для Penh для базовой линии, солевого раствора и увеличивающихся доз при испытании MCh рассчитали при помощи GraphPad Prism 5.0 (Сан-Диего, штат Калифорния) и выразили в процентах от соответствующего (IV или перорально введенного) контроля с носителем. Статистические разницы между группами обработки и соответствующей контрольной группой в каждом исследовании рассчитали с использованием однофакторного дисперсионного анализа (ANOVA), критерия полученных результатов Дуннета или Бонферрони, или t-критерия (JMP 8.0, SAS Institute, Кэрри, штат Северная Каролина, или Microsoft Excel). Значение p<0,05 в группах обработки и соответствующей контрольной группе с носителем считали достоверно различным.
[00410] Результаты
[00411] В OVA модели астмы. Соединение 8 (Пример 8) существенно (p<0,05) снижает эозинофильную инфильтрацию в BAL на 37% от контроля с носителем, при введении тремя пероральными дозами по 10 мг/кг за 48 часов, 24 часа и 1 час до оценки.
[00412] В OVA модели астмы. Соединение 3 (Пример 3) существенно (p<0,05) снижает эозинофильную инфильтрацию в BAL на 42% от контроля с носителем, при введении тремя пероральными дозами по 10 мг/кг за 48 часов, 24 часа и 1 час до оценки.
[00413] В OVA модели астмы, Соединение 27 (Пример 27) существенно (p<0,05) снижает эозинофильную инфильтрацию в BAL на 23% от контроля с носителем, при введении тремя пероральными дозами по 10 мг/кг за 48 часов, 24 часа и 1 час до оценки.
[00414] В OVA модели астмы, Соединение 4 (Пример 4) существенно (p<0,05) снижает MCh-индуцированную гипервосприимчивость дыхательных путей (AHR) на 19% от контроля с носителем, и снижает эозинофильную инфильтрацию в BALF на 20% от контроля с носителем, при введении однократной IV дозой 10 мг/кг за 24 часа до оценки.
[00415] Пример 60: Фармакокинетическое (ФК) исследование на мышах
[00416] Экспериментальная модель
[00417] Для определения фармакокинетики соединений настоящего изобретения использовали мышей. Этот вид широко используется для оценки биодоступности соединений при введении пероральных (PO) и внутривенных (IV) опытных образцов. Эффективность соединений настоящего изобретения сравнили оценкой воздействия на плазму у самцов мышей BALB/c при IV или PO введении в моменты максимальной активности.
[00418] Материалы и способы
[00419] IV введение соединений настоящего изобретения
[00420] Соединения настоящего изобретения восстановили в фосфатно-солевом буферном растворе (PBS)/10% прозрачном растворе Solutol (HS 15), получив концентрацию 0,2 мг/мл, и ввели мышам (2 мг/кг) однократной IV дозой. Дозы вводили животным через боковую хвостовую вену. Образцы крови собрали в заданные временные точки (0,083, 0,25, 0,5, 1, 2, 4, 8, 16, 24 часа) пункцией сердца под изофлурановой анестезией (до 1 мл крови на одно животное). Кровь собрали в пробирки, содержащие Li-гепарин. Образцы крови хранили на льду до центрифугирования, в течение около 30 минут после сбора. Плазму перенесли в меченые полипропиленовые пробирки и заморозили при -70°C до анализа по ЖХ/МС/МС.
[00421] PO введение соединений настоящего изобретения
[00422] Соединения настоящего изобретения восстановили в 40% пропиленгликоле/40% пропиленкарбонате/20% растворе 5% прозрачного раствора сахарозы, получив концентрацию 2 мг/мл, и ввели мышам (10 мг/кг) однократной пероральной дозой через зонд. Образцы крови собрали через 0,25, 0,5, 1, 2, 4, 8, 12, 16, 20 и 24 часа после введения дозы, пункцией сердца под изофлурановой анестезией. Кровь собрали в пробирки, содержащие Li-гепарин. Образцы крови хранили на льду до центрифугирования, в течение около 30 минут после сбора. Плазму перенесли в меченые полипропиленовые пробирки и заморозили при -70°C до анализа по ЖХ/МС/МС.
[00423] Анализ ЖХ/МС/МС
[00424] Образцы плазмы в каждой временной точке анализировали с использованием ЖХ-МС/МС с нижним пределом количественного обнаружения (LLOQ) 1 нг/мл. Плазму анализировали для определения количества соединения настоящего изобретения в каждом образце, и построили кривые регрессии для каждого соединения настоящего изобретения в соответствующих матрицах.
[00425] Для расчета ФК параметров при IV и PO введении использовали анализ WinNonlin:
ФК параметры для IV части - AUClast; AUCINF; T1/2; Cl; Vss; Cmax; MRT
ФК параметры для PO части - AUClast; AUCINF; T1/2; Cmax; Cl; MRT.
[00426] Кроме перечисленных выше ФК параметров, рассчитали биодоступность (%F).
[00427] Исследовали соединения в Примерах 3, 4, 8, 13, 19, 27 и 28, и все они имеют значение пероральной доступности более 9%. Соединения в Примерах 3, 8, 13 и 27 имеют значение пероральной биодоступности более 45%.
[00428] Пример 61: Эффективность ингибиторов GSNOR в экспериментальной воспалительной болезни кишечника (IBD)
[00429] Обзор моделей:
[00430] Острые и хронические модели IBD, индуцированной декстран-сульфатом натрия (DSS) у мышей использовали для изучения эффективности ингибиторов GSNORi против этого заболевания. Острая и хроническая DSS-индуцированная IBD являются широко используемыми и хорошо охарактеризованными моделями, которые вызывают патологические изменения в ободочной кишке, такие же, как наблюдаются при заболевании человека. В этих моделях, а также при заболеваниях человека, эпителиальные клетки в криптах ободочной кишки разрушаются, приводя к дисфункции эпителиального барьера и результирующему воспалению ткани, отеку и образованию язвы. Терапия ингибиторами GSNORi может быть полезной при лечении IBD за счет восстановления уровней S-нитрозоглутатиона (GSNO) и, за счет этого, предупреждения или реверсирования дисфункции эпителиального барьера.
[00431] Модель профилактики острого заболевания:
[00432] Экспериментальную IBD индуцировали введением DSS в питьевую воду самцов мышей C57B1/6 (N = от 8 до 10 мышей на группу) в течение 6 последовательных дней. Ингибиторы GSNORi вводили перорально в дозах от 0,1 до 10 мг/кг/день в течение 10 дней, начиная за два дня до, и продолжая два дня после воздействия DSS. Через два дня после воздействия DSS, влияние ингибиторов GSNORi оценили слепым способом эндоскопией и гистопатологией, используя пятиточечную шкалу с оценками от = 0 (нормальная ткань) до оценки = 4 (язвенное поражение ткани и заметные патологические изменения). Оценили также уровни циркулирующих цитокинов, участвующих в воспалительных путях. Влияние ингибиторов GSNORi сравнили с контрольными образцами, обработанными носителем. В этом исследовании в качестве положительного контроля использовали кортикостероид преднизолон, его вводили ежедневно в дозировке 3 мг/кг/день путем перорального дозирования. Незараженных мышей (N=5) также оценили как контрольный образец нормальных тканей.
[00433] Модель лечения хронического заболевания:
[00434] Экспериментальную IBD индуцировали введением DSS в питьевую воду самцов мышей C57B1/6 (N = от 10 до 12 мышей на группу) в течение 6 последовательных дней. Ингибиторы GSNORi дозировали перорально в дозах 10 мг/кг/день в течение 14 дней, начиная на следующий день после прекращения воздействия DSS. Эффективность ингибиторов GSNORi оценили слепым способом эндоскопией через 7 дней и 14 дней дозирования ингибиторов GSNORi, а также гистопатологией через 14 дней дозирования ингибиторов GSNORi, используя пятиточечную шкалу, с оценками от = 0 (нормальная ткань) до оценки = 4 (язвенное поражение ткани и заметные патологические изменения). Оценили также уровни циркулирующих цитокинов, участвующих в воспалительных путях. Влияние ингибиторов GSNORi сравнили с контрольными образцами, обработанными носителем. В этом исследовании в качестве положительного контроля использовали кортикостероид преднизолон, его вводили ежедневно в дозировке 3 мг/кг/день путем перорального дозирования. Незараженных мышей (N=5) также оценили как контрольный образец нормальных тканей.
[00435] Результаты:
[00436] Соединение 3 (Пример 3) облегчает поражение ободочной кишки и понижает уровни цитокинов, участвующих в воспалительных реакциях в мышиной модели острой DSS-индуцированной IBD. Процент мышей, демонстрирующих тяжелые оценки поражения ободочной кишки, по оценкам эндоскопии и гистопатологии, значительно (p<0,05), снизился на 38%-88% от контроля с носителем, после пероральной обработки Соединением 3 при 0,1, 1 или 10 мг/кг/день в течение 10 последовательных дней, с использованием режима профилактического дозирования. Соединение 3 также восстанавливает циркулирующие воспалительные цитокины до уровней, наблюдаемых у необработанных, незараженных мышей. Эти действия Соединения 3 являются сравнимыми или превосходят результаты, наблюдаемые для преднизолона.
[00437] Соединение 8 (Пример 8) облегчает повреждение ободочной кишки в мышиной модели острой DSS-индуцированной IBD. Процент мышей, демонстрирующих тяжелые оценки поражения ободочной кишки, по оценкам эндоскопии или гистопатологии, снизился на 44% или 26%, соответственно, от контроля с носителем, после пероральной обработки Соединением 8 при 10 мг/кг/день в течение 10 последовательных дней, с использованием режима профилактического дозирования.
[00438] Соединение 19 (Пример 19) облегчает повреждение ободочной кишки в мышиной модели острой DSS-индуцированной IBD. Процент мышей, демонстрирующих тяжелые оценки поражения ободочной кишки, по оценкам эндоскопии, снизился на 31% от контроля с носителем, после пероральной обработки Соединением 19 при 10 мг/кг/день в течение 10 последовательных дней, с использованием режима профилактического дозирования.
[00439] Соединение 13 (Пример 13) облегчает повреждение ободочной кишки в мышиной модели хронической DSS-индуцированной IBD. Процент мышей, демонстрирующих тяжелые оценки поражения ободочной кишки, по оценкам эндоскопии или гистопатологии, существенно (p<0,05) снизился на 52% или 53%, соответственно, от контроля с носителем, после пероральной обработки Соединением 13 при 10 мг/кг/день в течение 14 последовательных дней, с использованием режима лечебного дозирования.
[00440] Пример 62: Эффективность ингибиторов GSNOR в экспериментальной хронической обструктивной болезни легких (COPD).
[00441] Модели COPD при кратковременном курении сигарет
[00442] Эффективность ингибиторов GSNOR была оценена в мышиной модели хронической обструктивной болезни легких (COPD), индуцированной краткосрочным (4 дня или 11 дней) воздействием сигаретного дыма. Для оценки воздействия ингибиторов GSNOR на некоторые ранние события, связанные с инициацией и прогрессированием COPD, измерили инфильтрацию воспалительных клеток в жидкость бронхоальвеолярного лаважа (BALF) и уровни в BALF хемокинов, участвующих в воспалении и обмене/восстановлении тканей.
[00443] Обзор моделей:
[00444] Эффективность ингибиторов GSNOR против COPD исследовали с использованием модели острой (4 дня) и субхронической (11 дней) моделей COPD, индуцированной сигаретным дымом, у мышей. Воздействие на животных сигаретного дыма дает модель COPD, в которой поражение вызывается таким же причинным фактором, что и в заболевании человека, и в котором это поражение демонстрирует сходства с заболеванием человека, включая обструкцию дыхательных путей, расширение легочной ткани и включение воспалительных реакций в эти патологии. В моделях на животных, изменения в легочной патологии являются очевидными лишь после продолжительного (несколько месяцев) воздействия сигаретного дыма, что препятствует использованию хронических моделей в качестве эффективных аналитических инструментов. Совсем недавно, в качестве средств для проверки эффективности и механизмов действия новых терапевтических средств против COPD, использовались модели изучения реакций после кратковременного (2 недели или менее) воздействия дыма на мышей. Ключевая роль воспаления при инициации и прогрессировании COPD делает эти краткосрочные модели релевантыми для первоначальных испытаний эффективности новых терапевтических средств.
[00445] Модель острого (4 дня) воздействия дыма: На самок мышей C57B1/6 (N=8 на группу) воздействовали сигаретным дымом, используя камеру для воздействия на весь организм. Мышей в течение 4 последовательных дней ежедневно обрабатывали 4 циклами дыма от 6 последовательных сигарет (Kentucky 3R4F без фильтра) с интервалами между циклами, без дыма, по 30 минут. Ингибиторы GSNOR вводили ежедневно пероральным дозированием по 10 мг/кг/день в течение 7 дней, начиная за 2 дня до обработки дымом, и продолжая еще 1 день после обработки. Влияние ингибиторов GSNOR оценили подсчетом общего количества клеток, лейкоцитов и разности лейкоцитов в BALF при помощи световой микроскопии, а также уровней хемокинов в BALF при помощи иммуноферментного твердофазного анализа (ELISA) примерно через 24 часа после последней обработки дымом. Влияние ингибиторов GSNOR сравнили с контрольными образцами, обработанными носителем. Ингибитор PDE4, рофлумиласт, использовали в качестве положительного контроля для этого испытания. На группу незараженных мышей (N=8) воздействовали воздухом, и ее использовали в качестве отрицательного контроля испытания.
[00446] Модель субхронического (11 дней) воздействия дыма: На самок мышей C57B1/6 (N=10 на группу) воздействовали сигаретным дымом, полученным от сигарет Marlboro 100 без фильтра. Время воздействия составляло 25 минут на 1 день испытания, 35 минут на 2 день испытания и 45 минут в 3-11 дни испытания. Ингибиторы GSNOR вводили каждый день, за один час до обработки дымом. Ингибиторы GSNOR дозировали перорально при 1-10 мг/кг/день в течение 11 дней. Влияние ингибиторов GSNOR оценили подсчетом общего количества клеток и разности лейкоцитов в BALF при помощи световой микроскопии через 24 часа после последней обработки дымом. Влияние ингибиторов GSNOR сравнили с контрольными образцами, обработанными носителем, и выразили в процентах ингибирования увеличения количества клеток в BALF, вызванного сигаретным дымом. Для этого исследования в качестве положительного контроля использовали рофлумиласт, и его дозировали по 5 мг/кг/день. На группу незараженных мышей (N=10) воздействовали воздухом и вводили носитель, и их использовали в качестве отрицательного контроля испытания.
[00447] Результаты:
[00448] Соединение 3 (Пример 3) облегчает вызванные дымом изменения в клеточном инфильтрате BALF и воспалительных хемокинах BALF. Соединение 3 существенно (p<0,05) снижает общее количество клеток, лейкоцитов, макрофагов, нейтрофилов и эозинофилов в BALF на 66%, 80%, 75%, 84% и 95%, соответственно, по сравнению с контрольными образцами, обработанными носителем, при пероральном дозировании по 10 мг/кг/день в течение 7 дней в модели острого воздействия дыма в течение 4 дней. Эти действия Соединения 3 являются сравнимыми или превосходят результаты, наблюдаемые для рофлумиласта. Соединение 3 также возвращает содержание хемокинов в BALF к уровням, наблюдаемым у незараженных мышей. В субхронической 11-дневной модели, Соединение 3 ингибирует вызванное дымом увеличение общего количества клеток (p<0,05), макрофагов, нейтрофилов (p<0,05), эозинофилов и лимфоцитов (p<0,05) в BALF на 25%, 24%, 41%, 70% и 49%, соответственно, при пероральном дозировании по 10 мг/кг/день в течение 11 дней. При пероральном дозировании по 5 мг/кг/день, Соединение 3 ингибирует общее количество клеток, макрофагов, нейтрофилов (p<0,05) и лимфоцитов (p<0,05) в BALF на 22%, 23%, 29% и 46%, соответственно.
[00449] Соединение 8 (Пример 8) существенно (p<0,05) ингибирует вызванное дымом увеличение общего количества клеток, макрофагов, нейтрофилов и лимфоцитов в BAL на 35%-48%, 24%-43%, 41%-70% и 49-65%, соответственно, при пероральном дозировании по 1-10 мг/кг/день в течение 11 дней в субхронической 11-дневной модели. Не наблюдалось дозозависимого эффекта при 1, 5 или 10 мг/кг/день. Влияние Соединения 8 является сравнимым с результатами, наблюдаемыми для рофлумиласта.
[00450] Соединение 13 (Пример 13) существенно (p<0,05) ингибирует вызванное дымом увеличение общего количества клеток, макрофагов, нейтрофилов и лимфоцитов в BAL на 56%, 53%, 67% и 60%, соответственно, при пероральном дозировании по 1 мг/кг/день в течение 11 дней в субхронической 11-дневной модели. Влияние Соединения 13 является сравнимым с результатами, наблюдаемыми для рофлумиласта.
[00451] Соединение 27 (Пример 27) существенно (p<0,05) ингибирует вызванное дымом увеличение общего количества клеток, макрофагов, нейтрофилов и лимфоцитов в BAL на 44%, 41%, 64% и 46%, соответственно, при пероральном дозировании по 1 мг/кг/день в течение 11 дней в субхронической 11-дневной модели. Влияние Соединения 27 является сравнимым с результатами, наблюдаемыми для рофлумиласта.
[00452] Пример 63: Исследовательское изучение токсичности ацетаминофена на мышах
[00453] Ранее авторами настоящего изобретения было показано, что ингибирование S-нитрозоглутатион-редуктазы (GSNOR) улучшает негативные проявления желудочно-кишечного поражения в моделях на животных. В продолжение этих наблюдений, можно оценить влияние S-нитрозоглутатиона (GSNO) или ингибиторов GSNOR (GSNORi) на ацетаминофен (ACAP)-индуцированную печеночную токсичность в модели поражения печени на мышах. Образцы крови собираются для анализов печеночной функции, а образцы тканей собираются в конце исследования для гистопатологического исследования.
[00454] Материалы и способы
[00455] Ингибиторы GSNORi, GSNO, ацетаминофен (ACAP, Sigma), носители (1/2 см3 шприцы для дозирования), изофлуран, 181 см3 шприцов с иглами 26 калибра для сбора крови, 90 пробирок для отделения сыворотки для проведения клинических химических анализов.
[00456] Общая схема испытания: Животные (5 на группу) прошли акклиматизацию, по меньшей мере, в течение 3 дней до начала дозирования. В 1 день испытания, обработку мышей ацетаминофеном (300 мг/кг, перорально) выполнили натощак во время = 0. Через 2 часа ингибиторы GSNORi (10 мг/кг/доза) или GSNO (5 мг/кг/доза) ввели внутривенно в группы обработки. GSNORi или GSNO вводили в группы обработки через 24 и 48 часов после их первого введения. Мышей наблюдали на появление признаков клинической токсичности, а образцы крови собрали через 6, 24 и 72 часа после введения ACAP для испытаний печеночной функции: щелочная фосфатаза (ALK); аланин-аминотрансфераза (ALT); аспартат-аминотрансфераза (AST); гамма-глутамилтрансфераза (GGT) и общий билирубин (TBILI). Через 72 часа собрали печень для гистопатологического исследования.
[00457] Схема испытания
[00458] Календарь испытания:
[00459] Подготовка носителя. GSNO и ингибиторов GSNORi
[00460] Контрольным образцом носителя является фосфатно-солевой буферный раствор (PBS) (не содержащий кальция, калия или магния) с pH 7,4. Компоненты носителя взвешиваются в контейнере с противовесом тары и доводятся до нужного объема очищенной водой (вес./об.). 10x маточный раствор перемешивается при помощи магнитной мешалки, при необходимости. Затем 10x маточный раствор разбавляется деионизированной водой в отношении 1:9 (об./об.). Перед приготовлением растворов GSNO нагревается до комнатной температуры. Перед использованием раствор PBS продувается азотом. Растворы GSNO 1 мг/мл хранят на холоде (то есть хранят на ледяной бане) в защищенном от света месте, и используют в течение 4 часов после приготовления. Приготовление ингибиторов GSNORi, концентрацию 1 мг/мл восстанавливают в фосфатно-солевом буферном растворе (PBS), pH 7,4. Ингибиторы GSNORi вводят мышам (10 мл/кг) однократной (IV) ежедневной дозой. Дозирование выполняется через 2 часа после введения ACAP, а затем спустя 26 и 50 часов. Влияние GSNO или ингибиторов GSNORi таким же образом сравнивается с введением ACAP и солевого раствора-носителя.
[00461] Расчеты: Средний вес тела, средний вес печеночного органа и конечные точки клинической патологии (+/- стандартное отклонение) сравниваются с контрольной группой, обработанной носителем, при помощи T-критерия и дисперсионного анализа (ANOVA) (альфа = 0,05) Данные клинической патологии получают как средние значения, если эти данные не имеют нормального распределения, и в этом случае средние значения могут быть представлены диапазоном от минимального до максимального значения.
[00462] Пример 64: Исследовательское изучение для оценки анти-NASH фибротической активности ингибиторов GSNORi у мышей STAM
[00463] Ранее авторами настоящего изобретения было показано, что ингибирование S-нитрозоглутатион-редуктазы (GSNOR) улучшает негативные проявления желудочно-кишечного поражения и поражения ACAP в моделях на мышах. В продолжение этих наблюдений, оценивается действие способности ингибиторов GSNOR (GSNORi) реверсировать фибротическую активность в печеночном заболевании, индуцированном неалкагольным стеатогепатитом (NASH), на мышах STAM (молекулах адаптера сигнальной трансдукции). У этих мышей в течение двух недель видны последовательные изменения от стеатоза печени до фиброза, и они очень похожи на гистопатологию NASH человека.
[00464] Материалы и способы
[00465] Ингибиторы GSNORi, Telmisartan, носители (1/2 см3 шприцы для дозирования), изофлуран, 18 1 см3 шприцов с иглами 26 калибра для сбора крови, 90 пробирок для отделения сыворотки для проведения клинических химических анализов.
[00466] Общая схема испытания: Животные (6 на группу) прошли акклиматизацию перед началом испытания. В возрасте 4 недель животных поместили на диету: группа 1 (нормальные мыши) получали обычное питание, тогда как животным в группах 2-4 (мыши STAM) получали питание с высоким содержанием жиров на протяжении всего испытания. На 7 неделю испытания мышам начали ежедневно перорально вводить ингибиторы GSNORi, и их умертвили на 9 неделю испытания. Мышей наблюдали на появление признаков клинической токсичности, а образцы крови/тканей собрали для анализов печени: триглицериды в плазме (TG), аланин-аминотрансфераза (ALT); аспартат-аминотрансфераза (AST); генная экспрессия: Timp-1, α-SMA, коллаген 3, TNF-α и MCP-1, а также гистопатологическое исследование с использованием гематоксилин-эозинового (HE) окрашивания для оценки активности неалкогольной жировой инфильтрации печени (NAFLD), а также окрашивания Сириусом красным (фиброзная область).
[00467] Схема испытания
[00468] Расчеты: Средний вес тела, средний вес печеночного органа и конечные точки клинической патологии (+/- стандартное отклонение) сравниваются с контрольной группой, обработанной носителем, при помощи T-критерия и дисперсионного анализа (ANOVA) (альфа = 0,05) Данные клинической патологии получают как средние значения, если эти данные не имеют нормального распределения, и в этом случае средние значения представлены диапазоном от минимального до максимального значения.
[00469] Специалистам в данной области понятно, что в способах и композициях настоящего изобретения могут быть сделаны различные модификации и изменения, без отклонения от идеи или рамок настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| БИ-АРИЛ-МЕТА-ПИРИМИДИНОВЫЕ ИНГИБИТОРЫ КИНАЗ | 2006 |
|
RU2597364C2 |
| БИ-АРИЛ-МЕТА-ПИРИМИДИНОВЫЕ ИНГИБИТОРЫ КИНАЗ | 2006 |
|
RU2589878C2 |
| ИНГИБИТОРЫ GCN2 И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2811408C2 |
| ПИРАЗОЛ-4-ИЛ-ГЕТЕРОЦИКЛИЛ-КАРБОКСАМИДНЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ПРИМЕНЕНИЯ | 2012 |
|
RU2638552C2 |
| {3-[(7H-ПИРРОЛО[2,3-d]ПИРИМИДИН-4-ИЛ)АЗОЛИЛ]АЗЕТИДИН-3-ИЛ}АЦЕТОНИТРИЛЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ЯНУС КИНАЗ | 2015 |
|
RU2601410C1 |
| НОВЫЕ ИНГИБИТОРЫ S-НИТРОЗОГЛУТАТИОНРЕДУКТАЗЫ | 2011 |
|
RU2585763C2 |
| ПРОИЗВОДНЫЕ ПИПЕРИДИНА В КАЧЕСТВЕ АГОНИСТОВ GPR119 | 2013 |
|
RU2603346C2 |
| ИНГИБИТОРЫ GCN2 И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2811403C2 |
| ПРОТИВОИНФЕКЦИОННЫЕ СОЕДИНЕНИЯ | 2014 |
|
RU2734760C2 |
| Производные бензимидазол-2-пиперазина, полезные в качестве ингибитора поли(АДФ-рибоза)-полимеразы (PARP) | 2014 |
|
RU2649002C2 |
Изобретение относится к области органической химии, а именно к новым производным хинолина общей формулы I или к их фармацевтически приемлемым солям, стереоизомерам или N-оксидам, где m = 0 и 1; R1 независимо выбран из группы, состоящей из хлора, фтора и брома; R2b и R2c независимо выбраны из группы, состоящей из водорода, галогена, C1-C3 алкила, фторированного C1-С3алкила, циано и N(CH3)2; X выбран из группы, состоящей из 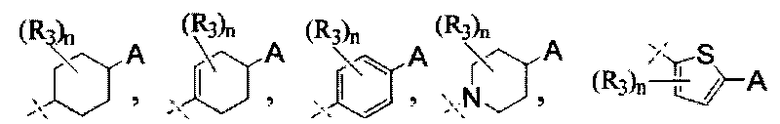 n выбран из группы, состоящей из 0 и 1; R3 независимо выбран из группы, состоящей из галогена, C1-С3 алкила, фторированного C1-С3 алкила, циано, C1-С3алкокси и NR4R4′, где R4 и R4′ независимо выбраны из группы, состоящей из C1-С3 алкила; и А выбран из группы, состоящей из
n выбран из группы, состоящей из 0 и 1; R3 независимо выбран из группы, состоящей из галогена, C1-С3 алкила, фторированного C1-С3 алкила, циано, C1-С3алкокси и NR4R4′, где R4 и R4′ независимо выбраны из группы, состоящей из C1-С3 алкила; и А выбран из группы, состоящей из 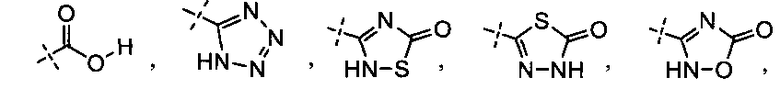
 . Также изобретение относится к применению соединения формулы I, фармацевтической композиции на основе соединения формулы I, способу лечения легочных расстройств и воспалительных заболеваний, основанному на использовании соединения формулы I, и способу получения фармацевтической композиции на основе соединения формулы I. Технический результат: получены новые производные хинолина, полезные в качестве ингибитора S-нитрозоглутатион-редуктазы (GSNOR). 6 н. и 11 з.п. ф-лы, 64 пр.
. Также изобретение относится к применению соединения формулы I, фармацевтической композиции на основе соединения формулы I, способу лечения легочных расстройств и воспалительных заболеваний, основанному на использовании соединения формулы I, и способу получения фармацевтической композиции на основе соединения формулы I. Технический результат: получены новые производные хинолина, полезные в качестве ингибитора S-нитрозоглутатион-редуктазы (GSNOR). 6 н. и 11 з.п. ф-лы, 64 пр.
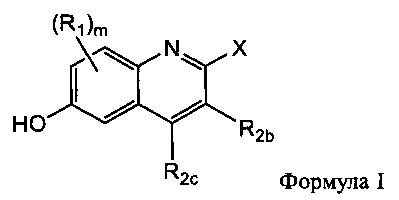
1. Соединение формулы I.
где
m выбран из группы, состоящей из 0 и 1;
R1 независимо выбран из группы, состоящей из хлора, фтора и брома;
R2b и R2c независимо выбраны из группы, состоящей из водорода, галогена, C1-C3 алкила, фторированного C1-С3алкила, циано и N(CH3)2;
X выбран из группы, состоящей из
n выбран из группы, состоящей из 0 и 1;
R3 независимо выбран из группы, состоящей из галогена, C1-С3 алкила, фторированного C1-С3 алкила, циано, C1-С3алкокси и NR4R4′, где R4 и R4′ независимо выбраны из группы, состоящей из C1-С3 алкила; и
А выбран из группы, состоящей из
или его фармацевтически приемлемая соль, стереоизомер или N-оксид.
2. Соединение по п.1, отличающееся тем, что
R2b и R2c независимо выбраны из группы, состоящей из водорода, хлора, фтора, метила, трифторметила, циано и N(CH3)2; и R3 независимо выбран из группы, состоящей из фтора, хлора, брома, метила, трифторметила, циано, метокси и N(CH3)2.
3. Соединение по п.2, отличающееся тем, что X является 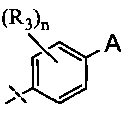 .
.
4. Соединение по п.3, отличающееся тем, что А является СООН.
5. Соединение по п.1, выбранное из группы, состоящей из:
4-(6-гидрокси-3-метилхинолин-2-ил)бензойной кислоты;
2-(4-(1Н-тетразол-5-ил)фенил)-3-метилхинолин-6-ола;
4-(6-(гидроксихинолин-2-ил)бензойной кислоты;
2-(4-(1Н-тетразол-5-ил)фенил)хинолин-6-ола;
1-(6-гидроксихинолин-2-ил)пиперидин-4-карбоновой кислоты;
(1r,4r)-4-(6-гидроксихинолин-2-ил)циклогексанкарбоновой кислоты;
(1s,4s)-4-(6-гидроксихинолин-2-ил)циклогексанкарбоновой кислоты;
3-хлор-4-(6-гидроксихинолин-2-ил)бензойной кислоты;
2-хлор-4-(6-гидроксихинолин-2-ил)бензойной кислоты;
2-фтор-4-(6-гидроксихинолин-2-ил)бензойной кислоты;
2-(4-(2Н-тетразол-5-ил)фенил)-4-хлорхинолин-6-ола;
3-(4-(6-гидроксихинолин-2-ил)фенил)-1,2,4-оксадиазол-5-(2Н)-она;
3-фтор-4-(6-гидроксихинолин-2-ил)бензойной кислоты;
4-(6-гидроксихинолин-2-ил)-3-метоксибензойной кислоты;
5-(6-гидроксихинолин-2-ил)тиофен-2-карбоновой кислоты;
4-(6-гидроксихинолин-2-ил)циклогекс-3-енкарбоновой кислоты;
4-(3-фтор-6-гидроксихинолин-2-ил)бензойной кислоты;
4-(4-хлор-3-фтор-6-гидроксихинолин-2-ил)бензойной кислоты;
4-(3-хлор-6-гидроксихинолин-2-ил)бензойной кислоты;
3-(2-фтор-4-(6-гидроксихинолин-2-ил)фенил)-1,2,4-оксадиазол-5(4Н)-она;
3-(3-фтор-4-(6-гидроксихинолин-2-ил)фенил)-1,2,4-оксадиазол-5(4Н)-она;
4-(4-хлор-6-гидроксихинолин-2-ил)бензойной кислоты;
2-(2-хлор-4-(2Н-тетразол-5-ил)фенил)хинолин-6-ола;
5-(4-(6-гидроксихинолин-2-ил)фенил)-1,3,4-оксадиазол-2(3Н)-она;
3-(диметиламино)-4-(6-гидроксихинолин-2-ил)бензойной кислоты;
4-(4-фтор-6-гидроксихинолин-2-ил)бензойной кислоты;
4-(6-гидроксихинолин-2-ил)-3-метилбензойной кислоты;
4-(3-хлор-6-гидроксихинолин-2-ил)-3-фторбензойной кислоты;
3-(4-(6-гидроксихинолин-2-ил)фенил)-1,2,4-тиадиазол-5(2Н)-она;
4-(6-гидроксихинолин-2-ил)-3-(трифторметил)бензойной кислоты;
4-(6-гидрокси-3-(трифторметил)хинолин-2-ил)бензойной кислоты;
2-(4-карбоксифенил)-6-гидроксихинолин 1-оксида;
5-(4-(6-гидроксихинолин-2-ил)фенил)-1,3,4-тиадиазол-2(3Н)-она;
5-(4-(6-гидроксихинолин-2-ил)фенил)-1,2,4-оксадиазол-3(2Н)-она;
(1r,4r)-4-(3-хлор-6-гидроксихинолин-2-ил)циклогексанкарбоновой кислоты;
(1s,4s)-4-(3-хлор-6-гидроксихинолин-2-ил)циклогексанкарбоновой кислоты;
3-хлор-4-(4-фтор-6-гидроксихинолин-2-ил)бензойной кислоты;
2-(5-(2Н-тетразол-5-ил)тиофен-2-ил)хинолин-6-ола;
5-(4-(6-гидроксихинолин-2-ил)фенил)-1,2,4-тиадиазол-3(2Н)-она;
3-фтор-4-(4-фтор-6-гидроксихинолин-2-ил)бензойной кислоты;
1-(6-гидрокси-3-(трифторметил)хинолин-2-ил)пиперидин-4-карбоновой кислоты;
4-(5-хлор-6-гидроксихинолин-2-ил)бензойной кислоты;
(1r,4r)-4-(6-гидрокси-3-(трифторметил)хинолин-2-ил)циклогексанкарбоновой кислоты;
(1s,4s)-4-(6-гидрокси-3-(трифторметил)хинолин-2-ил)циклогексанкарбоновой кислоты;
4-(5-бром-6-гидроксихинолин-2-ил)бензойной кислоты;
3-бром-4-(6-гидроксихинолин-2-ил)бензойной кислоты;
4-(4-(диметиламино)-6-гидроксихинолин-2-ил)бензойной кислоты;
4-(4-фтор-6-гидроксихинолин-2-ил)-3-метоксибензойной кислоты;
3-циано-4-(6-гидроксихинолин-2-ил)бензойной кислоты;
2-(4-карбокси-2-хлорфенил)-6-гидроксихинолин 1-оксида;
4-(3-циано-6-гидроксихинолин-2-ил)бензойной кислоты;
4-(5-фтор-6-гидроксихинолин-2-ил)бензойной кислоты;
4-(8-фтор-6-гидроксихинолин-2-ил)бензойной кислоты;
3-фтор-4-(5-фтор-6-гидроксихинолин-2-ил)бензойной кислоты.
6. Соединение по п.5, отличающееся тем, что указанное соединение представляет собой 3-хлор-4-(6-гидроксихинолин-2-ил)бензойную кислоту.
7. Соединение по п.5, отличающееся тем, что указанное соединение представляет собой 3-фтор-4-(6-гидроксихинолин-2-ил)бензойную кислоту.
8. Соединение по п.5, отличающееся тем, что указанное соединение представляет собой 4-(6-гидроксихинолин-2-ил)-3-метилбензойную кислоту.
9. Применение соединения формулы I по п.1 или его фармацевтически приемлемой соли в качестве ингибитора S-нитрозоглутатион-редуктазы (GSNOR).
10. Применение соединений по п.5 в качестве ингибиторов GSNOR.
11. Фармацевтическая композиция для лечения легочных расстройств или воспалительных заболеваний, содержащая терапевтически эффективное количество соединения по п.1 совместно с фармацевтически приемлемым носителем или формообразующим средством.
12. Фармацевтическая композиция по п.11, отличающаяся тем, что указанные легочные расстройства выбраны из группы, состоящей из астмы, хронической обструктивной болезни легких (ХОБЛ) и кистозного фиброза.
13. Фармацевтическая композиция по п.11, отличающаяся тем, что указанное воспалительное заболевание представляет собой воспалительное заболевание кишечника (ВЗК).
14. Способ лечения заболевания или состояния, включающий введение терапевтически эффективного количества соединения формулы I по п.1 пациенту, нуждающемуся в этом, при этом указанное заболевание или состояние выбрано из группы, состоящей из легочных расстройств и воспалительных заболеваний.
15. Способ лечения по п.14, отличающийся тем, что указанные легочные расстройства выбраны из астмы, хронической обструктивной болезни легких (ХОБЛ) и кистозного фиброза.
16. Способ лечения по п.14, отличающийся тем, что указанное воспалительное заболевание представляет собой воспалительное заболевание кишечника (ВЗК).
17. Способ получения фармацевтической композиции по п.11, включающий объединение соединения формулы I по п.1 с фармацевтически приемлемым носителем или формообразующим средством.
| WO 2010019909 A1, 18.02.2010 | |||
| WO 2006034491 A2, 30.03.2006 | |||
| ТЕТРАГИДРОХИНОЛИНОВЫЕ ПРОИЗВОДНЫЕ | 2002 |
|
RU2347570C2 |

