Настоящее изобретение относится к катализатору (каталитическому нейтрализатору для) селективного каталитического восстановления оксидов азота в отработавших газах (ОГ) дизельных двигателей, обладающему улучшенной стойкостью к также присутствующим в ОГ дизельных двигателей углеводородам, а также к его изготовлению и применению для снижения токсичности ОГ дизельных двигателей прежде всего на автомобилях.
Отработавшие газы дизельных двигателей наряду с образующимися в результате неполного сгорания топлива вредными газами, к которым относятся монооксид углерода (СО) и углеводороды (НС), содержат твердые частицы в виде сажи и оксиды азота (NOx). Помимо этого ОГ дизельных двигателей содержат кислород в количестве, которое может достигать 15 об. %. Известно, что окисляемые вредные газы СО и НС можно путем их пропускания над приемлемым катализатором окисления превращать в безвредный диоксид углерода (СО2), а твердые частицы можно удалять из ОГ путем их пропускания через пригодный для этого сажевый фильтр. В уровне техники хорошо известны также технологии удаления оксидов азота в присутствии кислорода. Одним из таких "способов денитрификации" является способ селективного каталитического восстановления (сокращенно СКВ) оксидов азота аммиаком в качестве восстановителя на пригодном для этого катализаторе, сокращенно называемом СКВ-катализатором. Аммиак может при этом добавляться в поток ОГ как таковой или в виде соединения-предшественника, разлагающегося до аммиака в окружающих условиях, под которыми в данном случае подразумевается окружающая среда, в которой разлагающееся до аммиака соединение находится в потоке ОГ перед СКВ-катализатором. Для реализации СКВ-способа необходимы источник восстановителя, впрыскивающее устройство для дозирования восстановителя по мере необходимости в ОГ и расположенный на пути потока ОГ СКВ-катализатор. Всю такую совокупность из источника восстановителя, СКВ-катализатора и расположенного по ходу потока ОГ перед СКВ-катализатором впрыскивающего устройства называют также СКВ-системой.
С учетом вступающих в будущем в силу законодательно устанавливаемых норм на предельно допустимые показатели выброса вредных веществ с ОГ в принципе обязательным для всех вновь допускаемых к эксплуатации автомобилей с дизельными двигателями является снижение токсичности ОГ в целях удаления всех выбрасываемых двигателем вредных газов. Поэтому на сегодняшний день для снижения токсичности ОГ дизельных двигателей необходимо использовать в комбинации между собой каталитические нейтрализаторы окислительного типа для снижения токсичности ОГ дизельного двигателя (так называемые дизельные катализаторы окисления), фильтры для улавливания твердых частиц, присутствующих в ОГ дизельного двигателя (так называемые сажевые фильтры), и СКВ-системы, при этом использование таких агрегатов в комбинации между собой влечет за собой изменение рабочих условий прежде всего для СКВ-катализатора. В настоящее время на стадии испытаний находятся три подобные системы. В так называемой "SCRT®-системе" согласно ЕР 1054722 в направлении потока ОГ последовательно располагают дизельный катализатор окисления, сажевый фильтр и СКВ-систему. Альтернативно этому СКВ-система может располагаться под днищем кузова автомобиля между установленным вблизи двигателя дизельным катализатором окисления (ДКО) и сажевым фильтром (СФ) (система типа ДКО-СКВ-СФ) или перед узлом из дизельного катализатора окисления и сажевого фильтра (система типа СКВ-ДКО-СФ).
Использование в комбинации между собой сажевого фильтра и СКВ-системы в выпускном тракте приводит к тому, что СКВ-катализатор в определенных режимах работы длительно подвергается воздействию углеводородов (НС) в существенно больших концентрациях, чем это было ранее. Появление углеводородов в ОГ в повышенных концентрациях обусловлено несколькими следующими причинами.
Во-первых, калибровка процесса сгорания, происходящего внутри двигателя, в настоящее время не преследует более цель сэкономить затратоемкие ступени снижения токсичности ОГ, а выполняется в целях оптимизации мощности, при этом твердые частицы и углеводороды, а также оксиды азота допускаются на равных началах в качестве вредных выбросов. Этим фактором обусловлена определенная основная нагрузка, создаваемая углеводородами на систему снижения токсичности ОГ, в которых при этом углеводороды содержатся в уже явно бóльших концентрациях, чем в обычных до настоящего времени системах снижения токсичности ОГ, где использовались СКВ-системы. Во-вторых, сажевый фильтр необходимо с регулярной периодичностью подвергать регенерации, которая помимо прочего заключается в контролируемом выжигании твердых частиц, скопившихся на фильтре. С этой целью фильтр необходимо нагревать до температуры выше температуры воспламенения сажи. Такой нагрев обеспечивают путем дополнительного впрыскивания топлива в камеру сгорания в цилиндре на такте выпуска либо в выпускной тракт и путем последующего каталитического превращения несгоревших углеводородов на обладающем окислительным действием катализаторе (так называемом "нагревательном катализаторе"). В большинстве случае выполнение функции такого "нагревательного катализатора" берет на себя расположенный выше по ходу потока ОГ дизельный катализатор окисления. При его отсутствии нагревательные функции при определенных условиях может также выполнять СКВ-катализатор. В любом случае в процессе регенерации сажевого фильтра перед СКВ-катализатором углеводороды присутствуют в повышенных концентрациях, поскольку (дополнительно) впрыснутые после воспламенения рабочей смеси углеводороды не полностью каталитически сгорают в фазе "нагрева". В SCRT®-системе, в которой перед СКВ-катализатором расположены дизельный катализатор окисления и сажевый фильтр, по истечении определенного времени работы наступает, кроме того, длительное отравление СКВ-катализатора углеводородами, обусловленное постепенной утратой своих окислительных функций дизельным катализатором окисления и при определенных условиях снабженным каталитическим покрытием фильтром в результате гидротермального старения.
Независимо от регенерации сажевого фильтра нагрев путем дополнительного впрыскивания топлива может требоваться и в иных целях, например, для компенсации задержек при пуске холодного двигателя и его прогреве и может равным образом приводить к кратковременному резкому возрастанию концентраций НС перед СКВ-катализатором.
По причине указанных эффектов СКВ-катализатор работает в современных комбинированных системах снижения токсичности ОГ в изменившихся условиях, при этом содержание присутствующих в ОГ перед СКВ-катализатором углеводородов явно выше того, которое характерно для прежних систем снижения токсичности ОГ. У традиционных СКВ-катализаторов в подобных условиях в целом наблюдается явное падение их производительности по превращению оксидов азота в сравнении с их же активностью в не содержащих углеводороды ОГ. В последнее время в уровне технике были описаны также стойкие к углеводородам СКВ-катализаторы.
Так, в частности, в WO 2009/135588 описан способ снижения токсичности содержащих оксиды азота (NOx) и углеводороды (НС) отработавших газов дизельного двигателя, предусматривающий а) добавление аммиака (NH3) как такового или в виде соединения, которое позволяет образовывать из него аммиак в окружающих условиях, в содержащий оксиды азота и углеводороды поток отработавших газов из не относящегося к выпускному тракту источника и б) избирательное превращение NOx их взаимодействием с подаваемым в поток отработавших газов NH3 на СКВ-катализаторе, содержащем замещенный медью (Cu) и/или железом (Fe) цеолит, и отличающийся тем, что содержащиеся в отработавших газах углеводороды удерживают благодаря действию цеолита, подобному действию молекулярного сита, на расстоянии от активных центров в катализаторе, на которых происходят превращения. В качестве цеолитов при этом используют мелкопористые цеолиты, прежде всего ферриерит, шабазит и эрионит, в которые углеводороды не могут проникать из-за своих размеров.
Такие стойкие к углеводородам СКВ-катализаторы отличаются тем, что их активность по превращению оксидов азота в содержащих углеводороды ОГ снижается существенно меньше по сравнению с их активностью в не содержащих углеводороды ОГ. Однако степень превращения оксидов азота, достижимая при применении таких катализаторов даже в не содержащих углеводороды ОГ, в целом значительно хуже, чем у традиционных СКВ-катализаторов. Устойчивость таких стойких к НС катализаторов к гидротермальному старению часто оказывается также явно хуже, чем у традиционных СКВ-катализаторов.
Повышенное содержание углеводородов в нейтрализуемых ОГ большей частью отрицательно сказывается далее на долговременной стабильности СКВ-катализаторов. Сказанное относится прежде всего к традиционным СКВ-катализаторам, функциональность которых основана на СКВ-активности замещенных переходными металлами цеолитов с порами средних или увеличенных размеров, таких, например, как морденит, β-цеолит, USY, ZSM-5 или ZSM-20, поскольку такие углеводороды из ОГ могут накапливаться в цеолитном каркасе. Содержащиеся в ОГ углеводороды при меньших рабочих температурах накапливаются в цеолитном каркасе, конкурируя с аммиаком. В этом случае при воздействии на катализатор повышенных рабочих температур, которые превышают начальную температуру (так называемую минимальную рабочую температуру или температуру воспламенения), по достижении которой начинается каталитическое окисление углеводородов, накопленные углеводороды "сгорают" в цеолите. В этом случае под действием выделяющейся теплоты реакции в катализаторе возникает значительный экзотермический эффект, проявляющийся в соответствующем повышении температуры, которое может привести к существенному повреждению каталитически активных центров в цеолитном катализаторе.
В основу настоящего изобретения была положена задача разработать СКВ-катализаторы, которые отличались бы улучшенной по сравнению с традиционными СКВ-катализаторами на основе цеолитов стойкостью к углеводородам (НС-стойкостью), но при этом одновременно обладали бы более высокой СКВ-активностью до и после гидротермального старения, чем стойкие к углеводородам СКВ-катализаторы, известные из уровня техники.
Указанная задача решается с помощью катализатора селективного каталитического восстановления оксидов азота аммиаком в содержащих углеводороды отработавших газах (ОГ), имеющего
- носитель,
- первое, нанесенное непосредственно на носитель каталитически активное покрытие, содержащее замещенный одним или несколькими переходными металлами цеолит и/или замещенное одним или несколькими переходными металлами цеолитоподобное соединение, и
- второе покрытие, которое перекрывает первое покрытие с обращенной к ОГ стороны и обладает такими свойствами, что оно препятствует контакту присутствующих в отработавших газах углеводородов, содержащих по меньшей мере 3 атома углерода, с нижерасположенным первым покрытием, но при этом не блокирует прохождение к нему оксидов азота и аммиака, и которое содержит один или несколько мелкопористых цеолитов и/или одно или несколько мелкопористых цеолитоподобных соединений, выбранных из группы, включающей SAPO-34, СНА, FER, ERI, OFF, ALPO-34 и их смеси, или один или несколько оксидов, выбранных из группы, включающей диоксид кремния, диоксид германия, оксид алюминия, диоксид титана, оксид олова, оксид церия, диоксид циркония и их смешанные оксиды.
Под "цеолитоподобными соединениями" согласно настоящему изобретению подразумеваются соединения, имеющие типичную цеолитную структуру ("цеолитный каркас"), но образованные не алюмосиликатом, соответственно не исключительно им. К таковым относятся прежде всего алюмофосфаты кремния (SAPO) и алюмофосфаты (ALPO).
Первое, нанесенное непосредственно на носитель каталитически активное покрытие в предлагаемом в изобретении катализаторе катализирует взаимодействие оксидов азота с аммиаком. В предпочтительном варианте это первое покрытие содержит замещенный переходным металлом цеолит и/или замещенное переходным металлом цеолитоподобное соединение, выбранный/выбранное из группы, включающей β-цеолит, ZSM-5, ZSM-20, USY, MOR и их смеси. Особенно предпочтительны при этом β-цеолит, USY и MOR. Содержащийся в цеолите, соответственно в цеолитоподобном соединении переходный металл в предпочтительном варианте выбран из группы, включающей церий, марганец, железо, медь, серебро, золото, платину, палладий и их смеси. Наиболее предпочтительны при этом церий, железо и медь.
Второе покрытие в предпочтительном варианте полностью перекрывает первое покрытие с обращенной к ОГ стороны. Однако соответствующий изобретению эффект все еще проявляется и в том случае, когда второе покрытие почти полностью перекрывает первое покрытие. Тем самым степень перекрытия первого покрытия вторым покрытием прежде всего составляет от 90 до 100%, особенно предпочтительно от 95 до 100%, в каждом случае в пересчете на площадь первого покрытия.
Предпочтительными мелкопористыми цеолитами, которые могут содержаться во втором покрытии, являются SAPO-34, СНА и FER.
Мелкопористые цеолиты, которые могут содержаться во втором покрытии, в предпочтительном варианте замещены одним или несколькими переходными металлами, выбранными из группы, включающей церий, марганец, железо, медь, серебро, золото, платину, палладий и их смеси. Особенно предпочтительны при этом церий, железо и медь.
В одном из вариантов осуществления настоящего изобретения второе покрытие образовано одним или несколькими мелкопористыми цеолитами и/или одним или несколькими мелкопористыми цеолитоподобными соединениями, выбранными из группы, включающей SAPO-34, СНА, FER, ERI, OFF, ALPO-34 и их смеси.
Второе покрытие в том случае, когда оно содержит мелкопористые цеолиты, соответственно образовано ими, самó может обладать СКВ-каталитической активностью.
К предпочтительным оксидам, которые могут содержаться во втором покрытии, относятся диоксид кремния, диоксид титана, оксид алюминия и оксид церия, среди которых наиболее предпочтителен диоксид кремния. С целью обеспечить достаточную пористость этого покрытия и тем самым достаточную его проходимость для реагентов - аммиака и оксидов азота - при одновременно достаточном барьерном действии в отношении содержащихся в ОГ углеводородов значение d50 в распределении частиц оксидов во втором покрытии по размерам предпочтительно должно составлять не более 100 нм, особенно предпочтительно не более 70 нм. Под значением d50 при этом подразумевается такое распределение частиц оксидов по размерам, при котором 50% от всего объема оксидов приходится только на те частицы, диаметр которых не превышает указанного в виде d50 значения.
В одном из вариантов осуществления настоящего изобретения второе покрытие образовано одним или несколькими оксидами, выбранными из группы, включающей диоксид кремния, диоксид германия, оксид алюминия, диоксид титана, оксид олова, оксид церия, диоксид циркония и их смешанные оксиды.
Второе покрытие в том случае, когда оно содержит оксиды, соответственно образовано ими, можно получать способом, заключающимся в том, что приемлемый оксид суспендируют в воде, распределение частиц оксидов в суспензии по размерам при необходимости доводят путем размола до вышеуказанного предпочтительного и затем из этой суспензии на уже снабженный первым покрытием носитель наносят покрытие традиционным, известным специалисту методом погружения, просасывания или прокачивания.
Согласно изобретению предпочтительны оксиды, которые не имеют дальнего порядка, т.е. представлены в аморфном виде.
В соответствии с этим второе покрытие предлагаемого в изобретении катализатора в предпочтительном варианте получают способом, предусматривающим пропитку уже снабженного первым покрытием носителя раствором, содержащим один или несколько алкоголятов формулы (I)
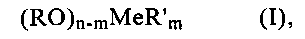
в которой
n обозначает число 3 или 4, причем m<n,
Me обозначает кремний, германий, алюминий, титан, олово или цирконий,
R обозначает С1-С4алкил или фенил, а
R′ обозначает С1-С8алкил, амино-С1-С4алкил, амино-С1-С4алкил, аминогруппа которого замещена амино-С1-С4алкилом, или С1-С4алкиловый эфир метакриловой кислоты,
и последующую сушку.
Образование оксидов происходит при этом в результате гидролиза алкоголятов и конденсации продуктов гидролиза с образованием Ме-О-цепей и сетчатых Ме-О-структур, из которых в конечном итоге формируются оксиды.
В предпочтительном варианте после сушки проводят прокаливание.
Указанные выше алкильные группы могут иметь прямую или разветвленную цепь и представлять собой, например, метил, этил, н-пропил, изопропил, н-бутил или изобутил. Помимо этого С1-С8алкил может также представлять собой, например, пентил, гексил, гептил или октил.
R в предпочтительном варианте обозначает метил, этил, изопропил, бутил или фенил.
R′ в предпочтительном варианте обозначает амино-С1-С4алкил, прежде всего аминометил или аминоэтил, N-(2-аминоэтил)-3-аминопропил, изопропил, изобутил, фенил, октил или С1-С4алкиловый эфир метакриловой кислоты, прежде всего метиловый эфир метакриловой кислоты, этиловый эфир метакриловой кислоты или пропиловый эфир метакриловой кислоты.
В особенно предпочтительном варианте Me обозначает кремний. В качестве алкоголятов формулы (I) в этом случае используют прежде всего тетраэтоксисилан, диизобутилдиметоксисилан, N-(2-аминоэтил)-3-аминопропилтриметоксисилан, фенилтриэтоксисилан, метакрилоксипропилтриметоксисилан и триэтоксиоктилсилан.
Предлагаемый в изобретении катализатор в различных вариантах его выполнения особо пригоден для применения на автомобилях и помимо этого экономичен в изготовлении, в том числе и кратко рассмотренными выше способами, когда второе покрытие образовано диоксидом кремния.
Предлагаемые в изобретении катализаторы, у которых их второе покрытие образовано диоксидом кремния, альтернативно описанному выше способу можно также изготавливать путем нанесения покрытия из водной суспензии пирогенной кремниевой кислоты на уже снабженный первым, цеолитным покрытием носитель. При этом следует выбирать пирогенную кремниевую кислоту, у которой ее первичные частицы имеют в их распределении по размерам значение d50 не более 100 нм, предпочтительно не более 70 нм, особенно предпочтительно не более 50 нм. Под значением d50 при этом подразумевается такое распределение частиц по размерам, при котором 50% от всего объема пирогенной кремниевой кислоты приходится только на те частицы, диаметр которых не превышает указанного в виде d50 значения. Пирогенная кремниевая кислота, применение которой предпочтительно при изготовлении предлагаемых в изобретении катализаторов этим способом, имеет модифицированную галогенидом и/или гидроксидом поверхность, благодаря функциональности которой в процессе или при определенных условиях после нанесения покрытия из водной суспензии кремниевой кислоты в результате гидролиза и/или конденсации происходит сшивание кремнекислотных частиц в результирующем покрытии.
Предлагаемый в изобретении катализатор во всех описанных выше и других возможных вариантах его выполнения пригоден для снижения содержания оксидов азота, включая монооксид азота и диоксид азота, в содержащих углеводороды ОГ дизельных двигателей. С этой целью нейтрализуемые ОГ, содержащие оксиды азота и углеводороды, после добавления аммиака или разлагающегося до него соединения-предшественника в качестве восстановителя пропускают через предлагаемый в изобретении СКВ-катализатор. В предпочтительном варианте нейтрализуемые ОГ, содержащие оксиды азота и углеводороды, перед добавлением аммиака или разлагающегося до него соединения-предшественника пропускают через катализатор окисления, который обладает активностью по превращению по меньшей мере части содержащегося в ОГ монооксида азота в диоксид азота. Благодаря этому в идеальном случае на долю диоксида азота приходится от 30 до 70% от всего количества оксидов азота в нейтрализуемых ОГ перед их входом в СКВ-катализатор. При этом такой катализатор окисления может быть представлен в виде каталитически активного покрытия на монолитном проточном сотовом элементе и/или на подложке фильтра с проницаемыми стенками каналов.
Ниже изобретение более подробно рассмотрено на некоторых примерах со ссылкой на прилагаемые к описанию графические материалы, на которых показано:
на фиг. 1 - температура ОГ перед известным из уровня техники СКВ-катализатором (VK1) и за ним, который перед проведением измерений насыщали углеводородами, при этом повышение температуры в реакторе, соответственно температуры перед катализатором до 400°С приводит к явному, обусловленному выгоранием углеводородов экзотермическому эффекту,
на фиг. 2 - разность ΔТ между температурой перед катализатором и температурой за ним, когда его перед проведением измерений насыщали углеводородами, после повышения температуры в реакторе, соответственно температуры перед катализатором до 400°С, при этом между собой сравниваются обусловленный выгоранием углеводородов экзотермический эффект в предлагаемом в изобретении катализаторе (K1), с одной стороны, и обусловленный выгоранием углеводородов экзотермический эффект в известном из уровня техники катализаторе (VK1), с другой стороны,
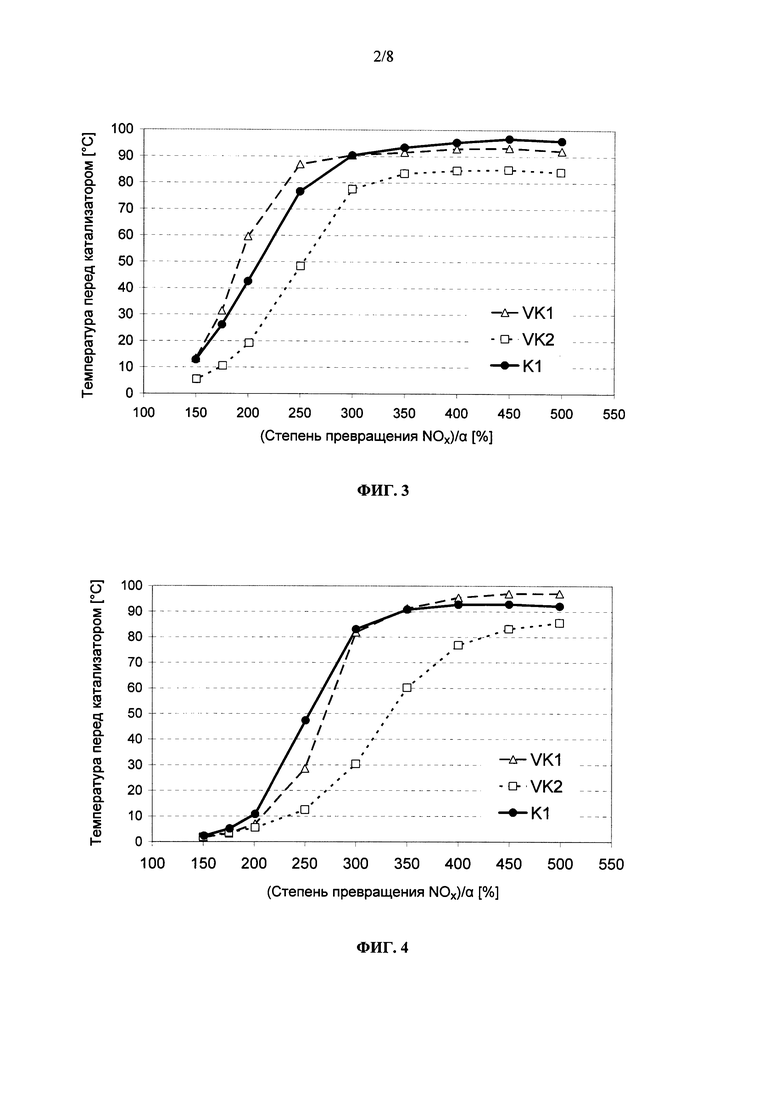
на фиг. 3 - нормированная на величину α степень превращения NOx в свежеизготовленных катализаторах VK1, VK2 (оба согласно уровню техники) и K1 (согласно изобретению) при пропускании через них не содержащих углеводороды ОГ,
на фиг. 4 - нормированная на величину α степень превращения NOx в подвергнутых гидротермальному старению катализаторах VK1′, VK2′ (оба согласно уровню техники) и К1′ (согласно изобретению) при пропускании через них не содержащих углеводороды ОГ,
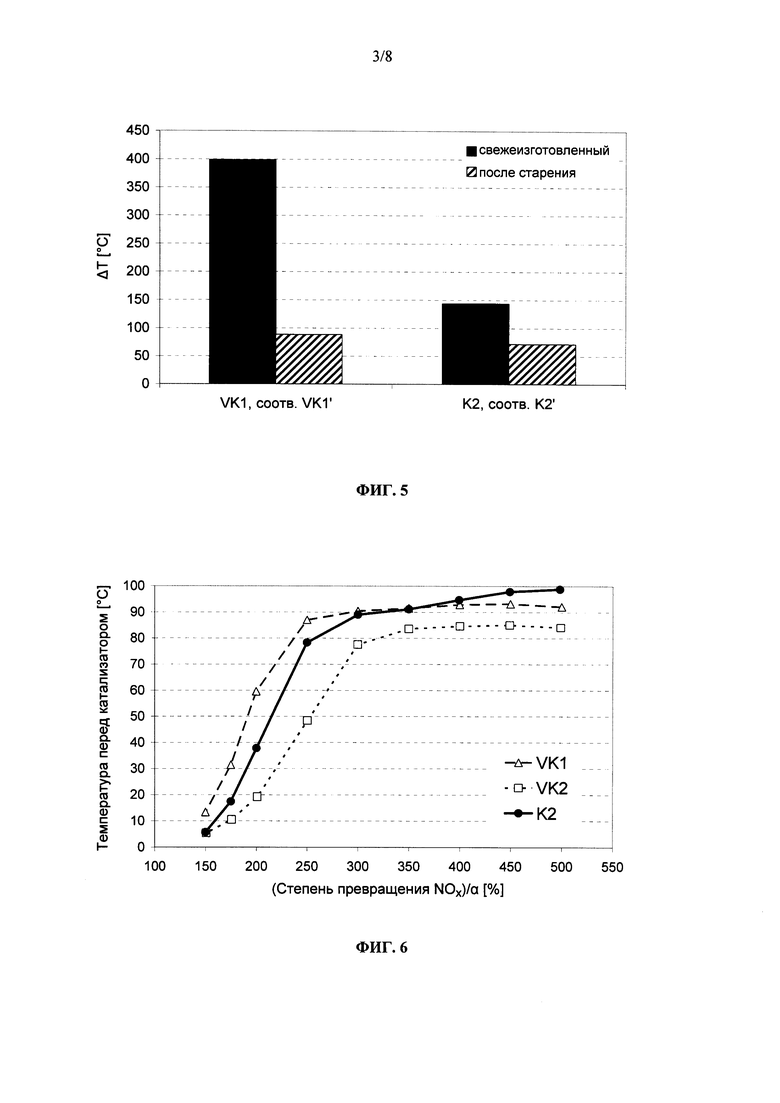
на фиг. 5 - разность ΔТ между температурой перед катализатором и температурой за ним, когда его перед проведением измерений насыщали углеводородами, после повышения температуры в реакторе, соответственно температуры перед катализатором до 400°С, при этом между собой сравниваются обусловленный выгоранием углеводородов экзотермический эффект в предлагаемом в изобретении катализаторе K2 (свежеизготовленном), соответственно K2′ (подвергнутом гидротермальному старению), с одной стороны, и обусловленный выгоранием углеводородов экзотермический эффект в известном из уровня техники катализаторе (VK1, соответственно VK1′), с другой стороны,
на фиг. 6 - нормированная на величину α степень превращения NOx в свежеизготовленных катализаторах VK1, VK2 (оба согласно уровню техники) и K2 (согласно изобретению) при пропускании через них не содержащих углеводороды ОГ,
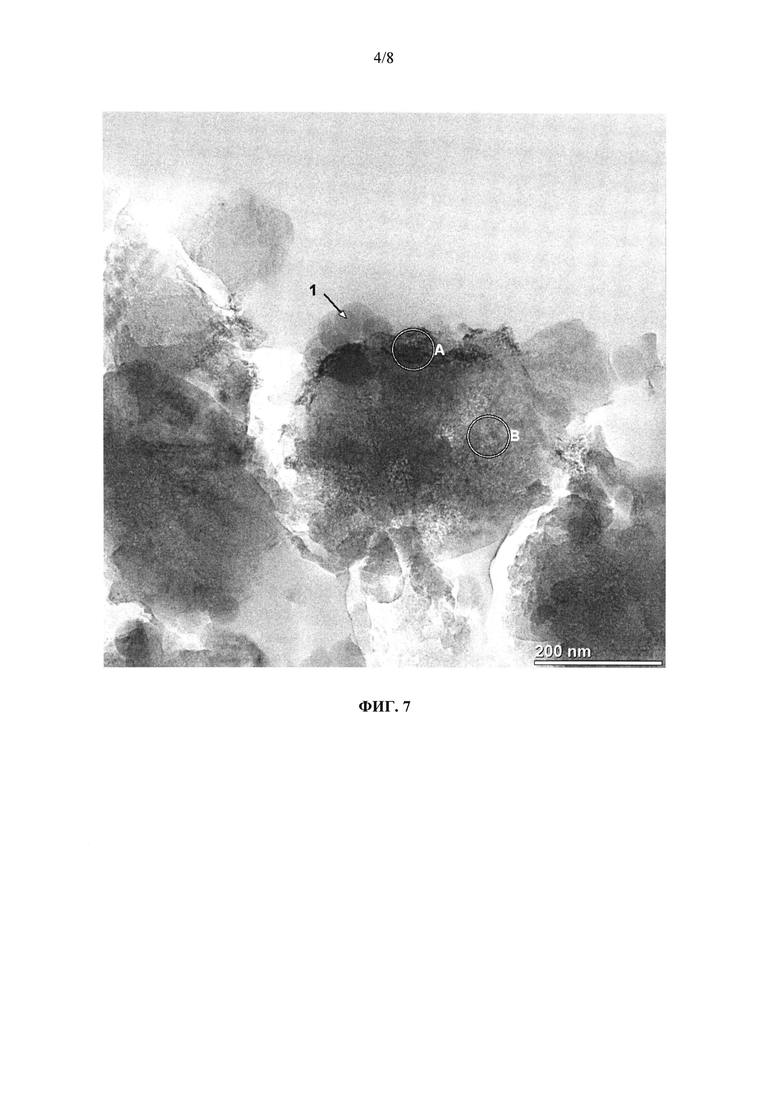
на фиг. 7 - полученный с помощью просвечивающего электронного микроскопа снимок образца, взятого из предлагаемого в изобретении катализатора K4, на каковом снимке можно увидеть пространственное расположение компонентов каталитически активного материала друг относительно друга, при этом цифрой 1 обозначены шаровидные частицы SiO2, буквой А обозначен замещенный железом цеолит, а буквой В обозначен оксид железа из содержащегося в этом материале избыточного железа,
на фиг. 8 - полученный с помощью просвечивающего электронного микроскопа снимок образца каталитически активного материала в предлагаемом в изобретении катализаторе K4, при этом в ограниченной окружностью зоне вокруг окружности А можно увидеть SiO2, который отличается тем, что он не имеет дальнего порядка (плоскости кристаллической решетки) и тем самым является аморфным,
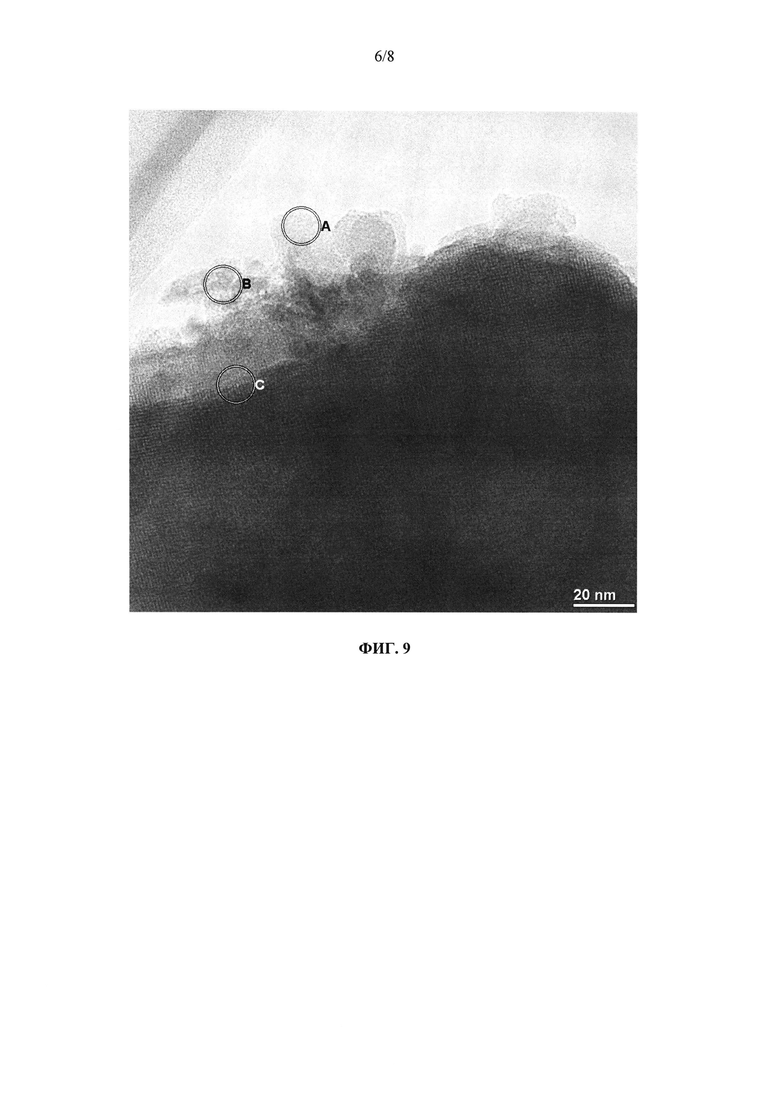
на фиг. 9 - полученный с помощью просвечивающего электронного микроскопа снимок образца каталитически активного материала в предлагаемом в изобретении катализаторе K4, при этом в зоне вокруг окружности С можно увидеть замещенный железом цеолит, имеющий плоскости кристаллической решетки с межплоскостным расстоянием примерно 1,1 нм,
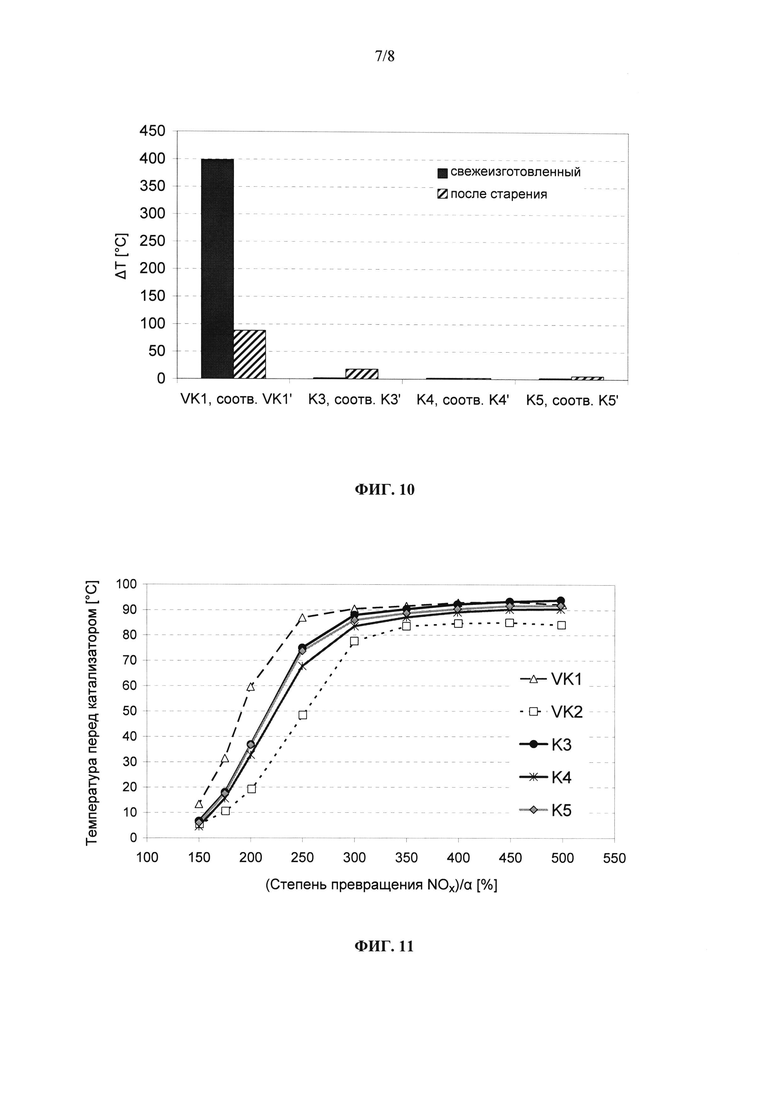
на фиг. 10 - разность ΔT между температурой перед катализатором и температурой за ним, когда его перед проведением измерений насыщали углеводородами, после повышения температуры в реакторе, соответственно температуры перед катализатором до 400°С, при этом между собой сравниваются обусловленный выгоранием углеводородов экзотермический эффект в предлагаемых в изобретении катализаторах K3-K5 (свежеизготовленных), соответственно K3′-K5′ (подвергнутых гидротермальному старению), с одной стороны, и обусловленный выгоранием углеводородов экзотермический эффект в известном из уровня техники катализаторе (VK1, соответственно VK1′), с другой стороны,
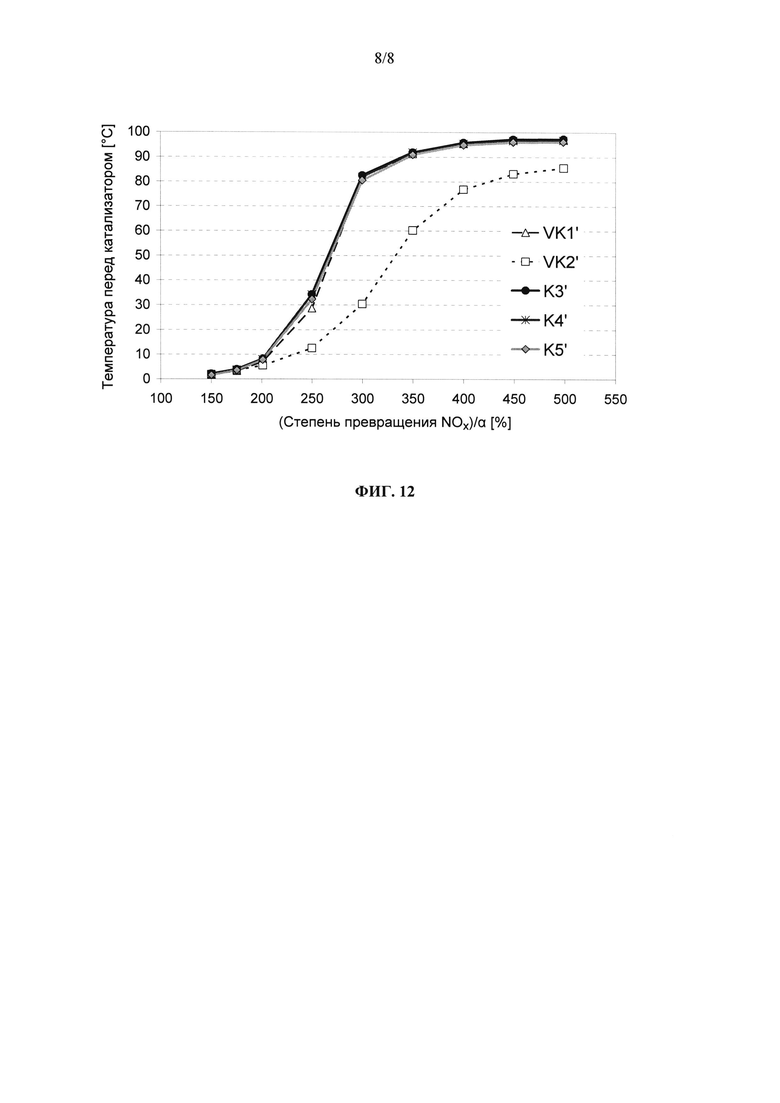
на фиг. 11 - нормированная на величину α степень превращения NOx в свежеизготовленных катализаторах VK1, VK2 (оба согласно уровню техники) и K3-K5 (согласно изобретению) при пропускании через них не содержащих углеводороды ОГ и
на фиг. 12 - нормированная на величину α степень превращения NOx в подвергнутых гидротермальному старению катализаторах VK1′, VK2′ (оба согласно уровню техники) и K3′-K5′ (согласно изобретению) при пропускании через них не содержащих углеводороды ОГ.
Сравнительный пример 1
В данном примере для изготовления имеющегося в продаже СКВ-катализатора приготавливали суспензию для нанесения покрытия на основе замещенного железом β-цеолита. С этой целью в воде суспендировали имеющееся в продаже SiO2-связующее, имеющееся в продаже бемитное связующее (в качестве вспомогательных веществ для нанесения покрытия), нонагидрат нитрата железа(Ш) и имеющийся в продаже β-цеолит с молярным соотношением SiO2/Al2O3 (SAR), равным 25, и затем из полученной суспензии обычным методом погружения наносили покрытие на керамический проточный сотовый элемент с плотностью расположения каналов 62 канала на кв. см и с толщиной разделяющих каналы стенок 0,17 мм. Снабженную покрытием деталь прокаливали сначала в течение 15 мин при 350°С, а затем в течение 2 ч при 500°С. Покрытие в полученном таким путем (сравнительном) катализаторе VK1 состояло на 90% из β-цеолита и содержало железо в количестве 4,5 мас. %, рассчитанном в виде Fe2O3.
Сравнительный пример 2
В данном примере изготавливали стойкий к углеводородам СКВ-катализатор согласно WO 2009/135588. С этой целью имеющийся в продаже мелкопористый цеолит ферриеритного типа с молярным соотношением SiO2/Al2O3 (SAR), равным 20, взмучивали в воде. К полученной суспензии добавляли нонагидрат нитрата железа(Ш). После размола эту суспензию использовали для нанесения из нее покрытия на монолитный проточный сотовый элемент с плотностью расположения каналов 62 канала на кв. см и с толщиной разделяющих каналы стенок 0,17 мм обычным методом погружения. Снабженную покрытием деталь прокаливали сначала в течение 15 мин при 350°С, а затем в течение 2 ч при 500°С. Покрытие в полученном таким путем (сравнительном) катализаторе VK2 состояло из ферриерита с содержанием железа 4,5%, рассчитанным в виде Fe2O3.
Пример 1
На еще один керамический проточный сотовый элемент с плотностью расположения каналов 62 канала на кв. см и с толщиной разделяющих каналы стенок 0,17 мм наносили покрытие согласно сравнительному примеру 1 из имеющегося в продаже замещенного железом β-цеолита.
Затем приготавливали еще одну суспензию для нанесения покрытия, которая содержала имеющийся в продаже ферриерит FER с молярным соотношением SiO2/Al2O3 (SAR), равным 20, в воде и из которой затем на уже однократно снабженный покрытием проточный сотовый элемент вновь наносили покрытие обычным методом погружения, в результате чего второе покрытие из FER почти полностью перекрывало первое покрытие из β-цеолита с обращенной к ОГ стороны. После сушки и прокаливания при 350°С в течение 10-15 мин, а затем при 500°С в течение 2 ч получили (предлагаемый в изобретении) катализатор K1.
Из катализаторов из сравнительных примеров и из примера 1 вырезали по два цилиндрических керна диаметром 25,4 мм и длиной 76,2 мм. По одному керну из каждой пары кернов, вырезанных из одного катализатора, исследовали на каталитическую активность в свежеизготовленном состоянии (→ VK1, соответственно VK2, соответственно K1). Другой керн каждой пары кернов подвергали перед исследованием каталитической активности искусственному старению в печи в течение 16 ч при температуре 750°С в атмосфере азота с добавлением к нему водяного пара в количестве 10 об. % и кислорода в количестве 10 об. % (→ VK1′, VK2′ и К1′).
Исследование каталитической активности
В первом испытании в качестве меры НС-стойкости катализаторов исследовали, насколько высок экзотермический эффект, создаваемый насыщенным углеводородами катализатором при термической нагрузке. При этом обычный, известный из уровня техники СКВ-катализатор VK1 сравнивали с предлагаемыми в изобретении катализаторами.
С этой целью в керны, вырезанные из свежеизготовленных катализаторов, на моторном стенде при 100°С в течение 60 мин подавали углеводороды. Затем керны предварительно кондиционировали в течение 10 мин в системе выпуска модельных ОГ (10% О2, 10% СО2, 5% Н2О, остальное N2, общий расход 4 м3/ч при нормальных условиях) при температуре в реакторе 100°С. Затем температуру в реакторе повышали в течение 30 с до 400°С при том же составе газовой смеси. Для оценки возникающего экзотермического эффекта регистрировали и анализировали температуру ОГ на расстоянии 5 мм перед входом в керн и на расстоянии 76,2 мм после выхода из него.
На фиг. 1 в качестве примера в графическом виде представлена температура ОГ на расстоянии 5 мм перед входом в сравнительный катализатор VK1 и температура ОГ, измеренная на расстоянии 76,2 мм за сравнительным катализатором VK1. Вскоре после повышения температуры в реакторе начиная с момента t=600 с можно наблюдать явный экзотермический эффект, проявляющийся в повышении температуры ОГ за катализатором до уровня, превышающего 800°С.
Для возможности более наглядного сравнения между собой создаваемых катализаторами экзотермических эффектов неопределенности в температурном режиме учитывали, вычисляя разность ΔT между температурой перед катализатором и температурой за ним. При этом температура Т(перед_кат) соответствовала температуре ОГ на расстоянии 5 мм перед керном, а температура Т(за_кат) соответствовала температуре ОГ на расстоянии 76,2 мм за керном. Температуры регистрировали в момент tперед_кат=tmax, перед_кат, соответственно tза_кат=tmax, за_кат, когда температура перед катализатором, соответственно за ним была максимальной.
На фиг. 2 в виде диаграммы представлена разность температур, измеренных на свежеизготовленных катализаторах VK1 и K1. В отличие от сравнительного катализатора VK1, для которого было выявлено повышение температуры на 400°С, обусловленное тем, что накопленные цеолитами в катализаторе углеводороды по достижении температуры воспламенения, необходимой для начала процесса их окисления, каталитически выгорают, у предлагаемого в изобретении катализатора K1 можно наблюдать повышение температуры на величину, которая явно меньше 50°С. Данный факт подтверждает, что предлагаемый в изобретении катализатор K1 накапливает углеводороды в существенно меньших количествах, чем традиционный СКВ-катализатор VK1, и тем самым обладает более высокой НС-стойкостью. Согласно предположению авторов изобретения данный эффект обусловлен тем, что имеющееся в предлагаемом в изобретении катализаторе верхнее покрытие из FER исключает возможность накопления углеводородов в расположенном под ним β-цеолите.
В ходе еще одного испытания исследовали превращение оксидов азота при их восстановлении аммиаком в предлагаемых в изобретении катализаторах и сравнительных катализаторах VK1 и VK2 в свежеизготовленном состоянии и после гидротермального старения.
Такое исследование проводили путем стационарного испытания с использованием системы выпуска модельных ОГ, которые имели указанный ниже состав.

Молярное соотношение между аммиаком и оксидами азота при исследованиях СКВ-активности обычно обозначают через α:
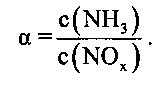
С учетом приведенных выше данных о концентрации компонентов ОГ значение α в данном случае равно 0,85. Среднечасовая скорость подачи газа в проводившихся испытаниях с модельными ОГ составляла 30000 ч-1.
Измеренную степень превращения NOx в настоящем описании указывают в нормированном на значение α виде, т.е. в виде значения, равного отношению степени превращения NOx к значению α [(степень превращения NOx)/α].
На фиг. 3 в графическом виде представлена нормированная на значение α степень превращения NOx в свежеизготовленных катализаторах VK1, VK2 и K1. Из приведенных на фиг. 3 графиков следует, что у предлагаемого в изобретении катализатора K1 в низкотемпературной области, прежде всего при рабочих температурах в интервале от 200 до 300°С, наблюдается слегка сниженная по сравнению с традиционным СКВ-катализатором VK1 производительность по превращению оксидов азота. Однако при температурах выше 300°С производительность по превращению оксидов азота у предлагаемого в изобретении катализатора полностью сопоставима с таковой у традиционного катализатора. По сравнению же с НС-стойким СКВ-катализатором VK2, известным из уровня техники, предлагаемый в изобретении катализатор K1 проявляет во всем интервале температур значительно лучшую производительность по превращению оксидов азота.
Сопоставление между собой результатов испытаний позволяет констатировать, что у предлагаемого в изобретении катализатора K1 в свежеизготовленном состоянии его SCR-активность существенно выше, чем у известных из уровня техники НС-стойких СКВ-катализаторов, при этом одновременно НС-стойкость предлагаемого в изобретении катализатора явно выше, чем у традиционных, известных из уровня техники СКВ-катализаторов на основе цеолитов (VK1).
На фиг. 4 в графическом виде представлена нормированная на значение α степень превращения оксидов азота в подвергнутых гидротермальному старению катализаторах VK1′, VK2′ и K′. Предлагаемый в изобретении катализатор проявляет сравнимую с традиционным СКВ-катализатором VK1 степень превращения оксидов азота. По сравнению с НС-стойким СКВ-катализатором VK2, известным из уровня техники, стойкость предлагаемого в изобретении катализатора K1 к гидротермальному старению оказывается явно выше, что приводит к существенно более высокой производительности по превращению оксидов азота после гидротермального старения.
Пример 2
На еще один керамический проточный сотовый элемент с плотностью расположения каналов 62 канала на кв. см и с толщиной разделяющих каналы стенок 0,17 мм наносили покрытие согласно сравнительному примеру 1 из имеющегося в продаже, замещенного железом β-цеолита.
На последующих стадиях уже однократно снабженный покрытием сотовый элемент методом погружения пропитывали раствором тетраэтоксисилана в этаноле до тех пор, пока силан не впитался в количестве 4,3 г/л SiO2 в пересчете на объем сотового элемента. После сушки воздуходувкой Leister при 50°С и прокаливания при 350°С в течение 10-15 мин и при 500°С в течение 2 ч получили катализатор K2. Из этого катализатора вырезали два керна диаметром 25,4 мм и длиной 76,2 мм, один из которых подвергали искусственному старению путем выдержки в печи в течение 16 ч при температуре 750°С в атмосфере азота с добавлением к нему водяного пара в количестве 10 об. % и кислорода в количестве 10 об. % (→ катализатор K2′). Затем керны K2 (свежеизготовленный) и K2′ (подвергнутый гидротермальному старению) подвергали описанным выше исследованиям каталитической активности.
На фиг. 5 в графическом виде представлен экзотермический эффект, возникающий в предлагаемом в изобретении катализаторе из примера 2 в свежеизготовленном состоянии (K2) и в состаренном состоянии (K2′) при разогреве после насыщения углеводородами, в сравнении с экзотермическим эффектом, возникающим в традиционном СКВ-катализаторе VK1 (свежеизготовленном), соответственно VK1′ (подвергнутом гидротермальному старению). В свежеизготовленном состоянии у предлагаемого в изобретении катализатора K2 наблюдается экзотермический эффект, который в численном выражении более чем на 250°С ниже, чем у сравнительного катализатора. После гидротермального старения экзотермический эффект, создаваемый в предлагаемом в изобретении катализаторе, также оказывается ниже, в данном случае на величину более чем 15°С. Тот факт, что и известный из уровня техники СКВ-катализатор (VK1) характеризуется после гидротермального старения существенно меньшим экзотермическим эффектом, обусловлен тем, что в результате старения произошло повреждение каталитически активных центров, которое отрицательно влияет также на способность обеспечивать каталитическое сгорание углеводородов. Тем самым предлагаемый в изобретении катализатор K2 в целом проявляет себя как обладающий явно более высокой НС-стойкостью, чем традиционный СКВ-катализатор.
На фиг. 6 в графическом виде представлена нормированная на значение α степень превращения NOx в свежеизготовленных катализаторах VK1, VK2 и K2. Из приведенных на данном чертеже графиков со всей очевидностью следует, что предлагаемый в изобретении катализатор K2 обладает во всем интервале температур существенно лучшей СКВ-активностью по сравнению с НС-стойким катализатором VK2, известным из уровня техники. По сравнению с традиционным СКВ-катализатором катализатор K2 проявляет в испытании с не содержащими углеводороды ОГ слегка меньшую активность в низкотемпературной области при одновременно лучшей степени превращения в высокотемпературной области начиная с 400°С.
Пример 3
На еще один керамический проточный сотовый элемент с плотностью расположения каналов 62 канала на кв. см и с толщиной разделяющих каналы стенок 0,17 мм наносили покрытие согласно сравнительному примеру 1 из имеющегося в продаже, замещенного железом β-цеолита.
Затем сотовый элемент с нанесенным на него таким путем покрытием после погружения в разбавленную азотную кислоту пропитывали раствором, содержащим 15 мас. % тетраэтоксисилана, 5 мас. % воды и 80 мас. % этанола, сушили и прокаливали при 350°С в течение 10-15 мин и при 500°С в течение 2 ч. При этом получили катализатор K3.
Пример 4
На еще один керамический проточный сотовый элемент с плотностью расположения каналов 62 канала на кв. см и с толщиной разделяющих каналы стенок 0,17 мм наносили покрытие согласно сравнительному примеру 1 из имеющегося в продаже, замещенного железом β-цеолита.
Затем сотовый элемент с нанесенным на него таким путем покрытием пропитывали раствором, содержащим 15 мас. % тетраэтоксисилана, 5 мас. % воды и 80 мас. % этанола, сушили и прокаливали при 350°С в течение 10-15 мин и при 500°С в течение 2 ч. При этом получили катализатор K4.
Наряду с кернами для исследования НС-стойкости и каталитической активности от катализатора K4 отбирали образцы для определения характеристик каталитически активного материала. Способами, хорошо известными специалисту в области оптических методов анализа, от исходно изготовленного нанесенного катализатора K4 отбирали образец и целиком заделывали его в светопроницаемую смолу. Отделенный от нее тонкослойный образец исследовали в просвечивающем электронном микроскопе высокого разрешения со встроенным спектрометром для рентгеноспектрального электронно-зондового микроанализа (РСМА). На фиг. 7 показан репрезентативный фрагмент такого образца. На снимке можно видеть поверхностные шарики из диоксида кремния (обозначены цифрой 1; РСМА: 51,84 ат. % О, 48,16 ат. % Si), осевшие на частицы замещенного железом цеолита (обозначены буквой А; РСМА: 57,84 ат. % О, 2,59 ат. % Al, 39,18 ат. % Si, 0,4 ат. % Fe). Дополнительно в материале образца наряду с железозамещенным цеолитом содержится оксид железа (обозначен буквой В; РСМА: 55,78 ат. % О, 0,93 ат. % Na, 8,48 ат. % Al, 24,05 ат. % Si, 0,51 ат. % Cl, 10,26 ат. % Fe), присутствие которого обусловлено избыточными соединениями железа, добавленными для ионного обмена в процессе приготовления использовавшегося, имеющегося в продаже цеолита.
Для дальнейшего определения характеристик каталитически активного материала его отбирали в небольшом количестве путем механического отделения от носителя. Полученный порошок также исследовали путем просвечивающей электронной микроскопии и рентгеноспектрального электронно-зондового микроанализа. На фиг. 8 и 9 приведены детальные изображения фрагментов материала образца. На фиг. 8 в обозначенной окружностью зоне виден диоксид кремния, который отличается тем, что у него невозможно обнаружить никакого дальнего порядка (плоскости кристаллической решетки). Данный факт подтверждает, что присутствующий в предлагаемом в изобретении катализаторе SiO2 является аморфным и тем самым отличается от традиционных кремнеземных материалов, которые, например, можно приобрести, подвергнуть размолу и нанести обычным методом погружения на носитель. На фиг. 9 показан другой фрагмент образца. В обозначенной буквой "А" зоне присутствует замещенный железом цеолит. Он имеет явный дальний порядок с межплоскостными расстояниями примерно 1,1 нм.
Пример 5
На еще один керамический проточный сотовый элемент с плотностью расположения каналов 62 канала на кв. см и с толщиной разделяющих каналы стенок 0,17 мм наносили покрытие согласно сравнительному примеру 1 из имеющегося в продаже, замещенного железом β-цеолита.
Затем сотовый элемент с нанесенным на него таким путем покрытием пропитывали раствором, содержащим 15 мас. % тетраэтоксисилана, 35 мас. % воды и 50 мас. % этанола, до тех пор, пока количество нанесенного силана не составило 6,6 г/л SiO2. После сушки и прокаливания при 350°С в течение 10-15 мин и при 500°С в течение 2 ч получили катализатор K5.
Из катализаторов из примеров 3-5 вырезали по 2 керна диаметром 25,4 мм и длиной 76,2 мм, по одному из которых подвергали искусственному старению путем выдержки в печи в течение 16 ч при температуре 750°С в атмосфере азота с добавлением к нему водяного пара в количестве 10 об. % и кислорода в количестве 10 об. %. Затем керны K3, K4 и K5 (свежеизготовленные), а также K3′, K4′ и K5′ (подвергнутые старению) подвергали описанным выше исследованиям каталитической активности.
На фиг. 10 в графическом виде представлено сравнение наблюдаемых экзотермических эффектов при повышении температуры в насыщенных углеводородами катализаторах из примеров 3-5 с экзотермическим эффектом в традиционном СКВ-катализаторе из сравнительного примера 1 в свежеизготовленном и в подвергнутом старению состояниях. У всех предлагаемых в изобретении катализаторов и в их свежеприготовленном состоянии, и в их подвергнутом гидротермальному старению состоянии наблюдаются существенно более низкие, обусловленные выгоранием углеводородов экзотермические эффекты, соответственно наблюдается даже полное (!) их отсутствие. Следовательно, такие катализаторы отличаются также явно лучшей НС-стойкостью.
На фиг. 11 в графическом виде представлена нормированная на значение α степень превращения NOx в предлагаемых в изобретении катализаторах K3-K5 в сравнении с НС-стойким СКВ-катализатором VK2, известном из уровня техники, в свежеизготовленном состоянии. Все предлагаемые в изобретении катализаторы отличаются от НС-стойкого катализатора, известного из уровня техники, явно лучшей степенью превращения оксидов азота во всем интервале температур.
На фиг. 12 в графическом виде представлена нормированная на значение α степень превращения NOx в предлагаемых в изобретении катализаторах K3′-K5′ в сравнении с СКВ-катализаторами VK1′ и VK2′, известными из уровня техники, в подвергнутом гидротермальному старению состоянии. Все предлагаемые в изобретении катализаторы отличаются от НС-стойкого катализатора VK2′, известного из уровня техники, явно более высокой степенью превращения оксидов азота во всем интервале температур и тем самым значительно лучшей стойкостью к старению. Стойкость предлагаемых в изобретении катализаторов к старению соответствует таковой традиционного, не обладающего НС-стойкостью СКВ-катализатора VK1.
Полностью аналогично примеру 5 можно также изготавливать катализаторы с хорошей НС-стойкостью при хорошей каталитической активности, используя для пропитки сотовых элементов с покрытием из железозамещенного β-цеолита растворы описанного ниже в примерах 6-10 состава.

Пример 11
На еще один керамический проточный сотовый элемент с плотностью расположения каналов 62 канала на кв. см и с толщиной разделяющих каналы стенок 0,17 мм наносили покрытие согласно сравнительному примеру 1 из имеющегося в продаже, замещенного железом β-цеолита.
Затем приготавливали еще одну суспензию для нанесения покрытия, которая содержала имеющуюся в продаже пирогенную кремниевую кислоту, у которой ее первичные частицы имели значение d50 в их распределении по размерам, равное 12 нм, в воде, при этом под значением d50 подразумевается такое распределение частиц пирогенной кремниевой кислоты по размерам, при котором 50% от всего объема пирогенной кремниевой кислоты приходится только на те частицы, диаметр которых не превышает указанного в виде d50 значения. Затем из этой суспензии на уже однократно снабженный покрытием проточный сотовый элемент вновь наносили покрытие обычным методом погружения. После сушки и прокаливания при 350°С в течение 10-15 мин и при 500°С в течение 2 ч получили катализатор, у которого второе покрытие из SiO2 почти полностью перекрывало первое покрытие из β-цеолита с обращенной к ОГ стороны.
Полностью аналогично примеру 11 можно также изготавливать катализаторы с хорошей НС-стойкостью при хорошей каталитической активности, используя для нанесения второго покрытия из SiO2, почти полностью перекрывающего первое покрытие из β-цеолита с обращенной к ОГ стороны, суспензии пирогенной кремниевой кислоты с другим распределением частиц по размерам:
Пример 12: пирогенная кремниевая кислота с d50=40 нм
Пример 13: пирогенная кремниевая кислота с d50=20 нм
Пример 14: пирогенная кремниевая кислота с d50=16 нм
Пример 15: пирогенная кремниевая кислота с d50=14 нм
Пример 16: пирогенная кремниевая кислота с d50=7 нм
Пример 17
В данном примере приготавливали суспензию для нанесения покрытия из имеющегося в продаже цеолита ZSM-5. С этой целью в воде суспендировали нонагидрат нитрата железа(Ш) и имеющийся в продаже цеолит ZSM-5 с молярным соотношением SiO2/Al2O3 (SAR), равным 25, и затем из полученной суспензии обычным методом погружения наносили покрытие на керамический проточный сотовый элемент с плотностью расположения каналов 62 канала на кв. см и с толщиной разделяющих каналы стенок 0,17 мм. Снабженную покрытием деталь прокаливали сначала в течение 15 мин при 350°С, а затем в течение 2 ч при 500°С. Покрытие в полученном таким путем катализаторе содержало железо в количестве 4,5 мас. %, рассчитанном в виде Fe2O3.
Затем сотовый элемент с нанесенным на него таким путем покрытием пропитывали раствором, содержащим 15 мас. % тетраэтоксисилана, 5 мас. % воды и 80 мас. % этанола, сушили и прокаливали при 350°С в течение 10-15 мин и при 500°С в течение 2 ч.
Полностью аналогично примеру 17 можно также получать катализаторы с хорошей НС-стойкостью при хорошей каталитической активности, когда первое, нанесенное непосредственно на носитель каталитически активное покрытие содержит указанные в следующей таблице, замещенные переходными металлами (ПМ) цеолиты.
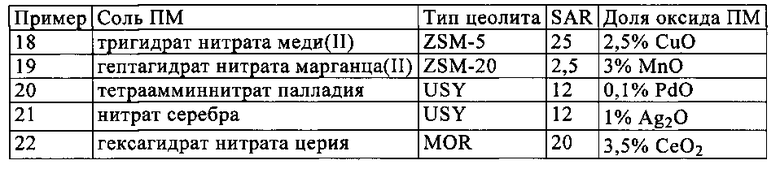
Полученные во всех примерах результаты свидетельствуют о том, что согласно изобретению удалось получить СКВ-катализаторы, которые по сравнению с традиционными цеолитными СКВ-катализаторами обладают явно лучшей НС-стойкостью, при этом с использованием таких катализаторов одновременно удается добиться существенно более высокой производительности по превращению оксидов азота, чем с использованием известных в настоящее время из уровня техники НС-стойких СКВ-катализаторов.

Изобретение относится к катализатору селективного каталитического восстановления оксидов азота аммиаком в содержащих углеводороды отработавших газах (ОГ). Катализатор имеет носитель, а также первое, нанесенное непосредственно на носитель каталитически активное покрытие, содержащее замещенный одним или несколькими переходными металлами цеолит и/или замещенное одним или несколькими переходными металлами цеолитоподобное соединение, и второе покрытие, которое перекрывает первое покрытие с обращенной к ОГ стороны и обладает такими свойствами, что оно препятствует контакту присутствующих в отработавших газах углеводородов, содержащих по меньшей мере 3 атома углерода, с нижерасположенным первым покрытием, но при этом не блокирует прохождение к нему оксидов азота и аммиака, и которое содержит один или несколько оксидов, выбранных из группы, включающей диоксид кремния, диоксид германия, оксид алюминия, диоксид титана, оксид олова, оксид церия, диоксид циркония и их смешанные оксиды. Изобретение также относится к способу снижения содержания оксидов азота, включая монооксид азота и диоксид азота, в содержащих углеводороды отработавших газах (ОГ) дизельных двигателей. 2 н. и 11 з.п. ф-лы, 12 ил., 22 пр.
1. Катализатор селективного каталитического восстановления оксидов азота аммиаком в содержащих углеводороды отработавших газах (ОГ), имеющий
- носитель,
- первое, нанесенное непосредственно на носитель каталитически активное покрытие, содержащее замещенный одним или несколькими переходными металлами цеолит и/или замещенное одним или несколькими переходными металлами цеолитоподобное соединение, и
- второе покрытие, которое перекрывает первое покрытие с обращенной к ОГ стороны и обладает такими свойствами, что оно препятствует контакту присутствующих в отработавших газах углеводородов, содержащих по меньшей мере 3 атома углерода, с нижерасположенным первым покрытием, но при этом не блокирует прохождение к нему оксидов азота и аммиака, и которое содержит один или несколько оксидов, выбранных из группы, включающей диоксид кремния, диоксид германия, оксид алюминия, диоксид титана, оксид олова, оксид церия, диоксид циркония и их смешанные оксиды.
2. Катализатор по п. 1, отличающийся тем, что замещенный переходным металлом цеолит, содержащийся в первом, нанесенном непосредственно на носитель каталитически активном покрытии, и/или замещенное переходным металлом цеолитоподобное соединение, содержащееся в первом, нанесенном непосредственно на носитель каталитически активном покрытии, выбран/выбрано из группы, включающей β-цеолит, ZSM-5, ZSM-20, USY, MOR и их смеси, при этом переходный металл выбран из группы, включающей церий, марганец, железо, медь, серебро, золото, платину, палладий и их смеси.
3. Катализатор по п. 1 или 2, отличающийся тем, что второе покрытие образовано одним или несколькими оксидами, выбранными из группы, включающей диоксид кремния, диоксид германия, оксид алюминия, диоксид титана, оксид олова, оксид церия, диоксид циркония и их смешанные оксиды.
4. Катализатор по п. 1, отличающийся тем, что значение d50 в распределении частиц оксидов во втором покрытии по размерам составляет не более 100 нм, при этом под значением d50 подразумевается такое распределение частиц оксидов по размерам, при котором 50% от всего объема оксидов приходится только на те частицы, диаметр которых не превышает указанного в виде d50 значения.
5. Катализатор по п. 1, отличающийся тем, что второе покрытие содержит один или несколько оксидов, выбранных из группы, включающей диоксид кремния, диоксид германия, оксид алюминия, диоксид титана, оксид олова, оксид церия, диоксид циркония и их смешанные оксиды, и получено путем пропитки уже снабженного первым покрытием носителя раствором, содержащим один или несколько алкоголятов формулы (I)
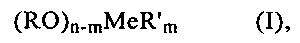
в которой
n обозначает число 3 или 4, причем m<n,
Me обозначает кремний, германий, алюминий, титан, олово или цирконий,
R обозначает С1-С4алкил или фенил, а
R′ обозначает С1-С8алкил, амино-С1-С4алкил, амино-С1-С4алкил, аминогруппа которого замещена амино-С1-С4алкилом, или С1-С4алкиловый эфир метакриловой кислоты,
и последующей сушки.
6. Катализатор по п. 5, отличающийся тем, что R обозначает метил, этил, изопропил, бутил или фенил.
7. Катализатор по п. 5, отличающийся тем, что R′ обозначает амино-С1-С4алкил, N-(2-аминоэтил)-3-аминопропил, изопропил, изобутил, фенил, октил или С1-С4алкиловый эфир метакриловой кислоты.
8. Катализатор по п. 1, 2, 4, 5, 6 или 7, отличающийся тем, что второе покрытие образовано диоксидом кремния.
9. Катализатор по п. 8, отличающийся тем, что второе покрытие из диоксида кремния получено путем его нанесения из водной суспензии пирогенной кремниевой кислоты, у которой ее первичные частицы имеют в их распределении по размерам значение d50 не более 100 нм, на уже снабженный первым покрытием носитель, при этом под значением d50 подразумевается такое распределение частиц пирогенной кремниевой кислоты по размерам, при котором 50% от всего ее объема приходится только на те частицы, диаметр которых не превышает указанного в виде d50 значения.
10. Катализатор по п. 9, отличающийся тем, что пирогенная кремниевая кислота имеет модифицированную галогенидом и/или гидроксидом поверхность, благодаря чему кремнекислотные частицы сшиты в результирующем покрытии в результате гидролиза и/или конденсации.
11. Способ снижения содержания оксидов азота, включая монооксид азота и диоксид азота, в содержащих углеводороды отработавших газах (ОГ) дизельных двигателей, заключающийся в том, что в нейтрализуемые ОГ, содержащие оксиды азота и углеводороды, добавляют аммиак или разлагающееся до него соединение-предшественник в качестве восстановителя и образовавшуюся смесь из ОГ и восстановителя пропускают через катализатор по одному из пп. 1-10.
12. Способ по п. 11, отличающийся тем, что ОГ перед добавлением в них аммиака или разлагающегося до него соединения-предшественника пропускают через катализатор окисления, который обладает активностью по превращению по меньшей мере части содержащегося в ОГ монооксида азота в диоксид азота.
13. Способ по п. 12, отличающийся тем, что катализатор окисления представлен в виде каталитически активного покрытия на монолитном проточном сотовом элементе и/или на подложке фильтра с проницаемыми стенками каналов.
| US 2010077738 A1, 01.04.2010 | |||
| US 20100290963 A1, 18.11.2010 | |||
| WO 2009135588 A1, 12.11.2009 | |||
| УСТРОЙСТВО И СПОСОБ ДЛЯ ОБРАБОТКИ ОТРАБОТАВШИХ ГАЗОВ, ОБРАЗУЮЩИХСЯ ПРИ РАБОТЕ ДВИГАТЕЛЯ НА БЕДНЫХ СМЕСЯХ, СЕЛЕКТИВНЫМ КАТАЛИТИЧЕСКИМ ВОССТАНОВЛЕНИЕМ ОКИСЛОВ АЗОТА | 2001 |
|
RU2278281C2 |

