Область техники настоящего изобретения
Настоящее изобретение относится к катализаторам. В частности, оно относится к способу получения предшественника кобальтсодержащего катализатора, который является предшественником катализатора синтеза углеводородов, к способу получения катализатора, представляющего собой катализатор синтеза углеводородов, а также к способу получения углеводородов, который включает применение указанного катализатора.
Уровень техники настоящего изобретения
Как известно, содержащие носители кобальтсодержащие катализаторы синтеза Фишера-Тропша (FTS) получают посредством пропитывания солью кобальта, такой как нитрат кобальта, которую наносят на поверхность и/или вводят в объем носителя катализатора, такого как носитель на основе диоксида кремния, с последующим прокаливанием полученного в результате пропитанного, т.е. модифицированного носителя катализатора и получением предшественника катализатора FTS. Предшественник катализатора затем восстанавливают с получением катализатора FTS, содержащего кобальтовые кристаллиты, диспергированные в носителе.
Условия прокаливания, используемые в процессе получения содержащих диоксид кремния носителей катализаторов, известны в технике и описаны, например, в публикациях WO 2016/135577 и WO2018/029548. В условиях прокаливания, которые упомянуты в указанных публикациях, предусмотрено нагревание при скорости, составляющей 1°С в минуту, до 250°С.
Неожиданно было обнаружено, что если модифицированный носитель катализатора прокаливают при значительно более высокой скорости нагревания, которую поддерживают в пределах заданного температурного диапазона согласно настоящему изобретению, то получается улучшенный катализатор.
В публикации WO 2012/153217 раскрыты прокаливание при повышенных значениях скорости нагревания (это также описано в сравнительных примерах публикации WO 2012/153218) и важность того, что периоды уменьшенной скорости нагревания включены в температурный диапазон, описанный в настоящем изобретении как диапазон, в пределах которого используется только высокая скорость нагревания. Однако согласно настоящему изобретению неожиданно было обнаружено, что никакие периоды низкой скорости нагревания не являются желательными в пределах этого температурного диапазона. Согласно настоящему изобретению неожиданно было обнаружено, что если высокая скорость нагревания не сохраняется в пределах этого температурного диапазона (то есть если периоды низкой скорости нагревания присутствуют в указанном диапазоне, что требуется в публикациях предшествующего уровня техники), то это отрицательно влияет, например, на активность катализатора.
Кроме того, неожиданно было обнаружено, что для модифицированных носителей катализаторов согласно настоящему изобретению отсутствие периодов низкой скорости нагревания в этом диапазоне не приводит к повышенной степени разрушения катализатора.
Краткое раскрытие настоящего изобретения
Предшественник кобальтсодержащего катализатора
Согласно первому аспекту настоящего изобретения предложен способ получения предшественника кобальтсодержащего катализатора, причем способ включает
прокаливание модифицированного носителя катализатора, представляющего собой носитель катализатора на основе диоксида кремния (SiO2), содержащий нитрат кобальта, причем в процессе прокаливания модифицированного носителя катализатора происходит превращение нитрата кобальта в оксид кобальта; и
прокаливание, представляющее собой нагревание модифицированного носителя катализатора при высокой скорости нагревания, которая составляет не менее чем 10°С в минуту, в пределах по меньшей мере температурного диапазона А, который составляет от минимальной температуры, при которой начинается прокаливание модифицированного носителя катализатора, до 165°С, и при этом газовый поток пропускают над модифицированным носителем катализатора в пределах по меньшей мере температурного диапазона А, и в результате этого получается предшественник кобальтсодержащего катализатора.
Предшественник катализатора может представлять собой предшественник катализатора синтеза углеводородов, который применяют для синтеза углеводородов и/или кислородсодержащих производных углеводородов, используя по меньшей мере водород и монооксид углерода. Предпочтительно в качестве предшественника катализатора присутствует предшественник катализатора синтеза Фишера-Тропша (FT), используемый для осуществления синтез Фишера-Тропша. Синтез FT можно осуществлять, используя реактор с неподвижным слоем, реактор с взвешенным слоем или реактор с неподвижным псевдоожиженным слоем. Предпочтительно способ синтеза FT представляет собой способ синтеза FT в трехфазной системе с взвешенным слоем. Модифицированный носитель катализатора
Носитель катализатора на основе диоксида кремния (SiO2) может представлять собой носитель на основе осажденного диоксида кремния. Предпочтительно он представляет собой носитель на основе аэрозольного диоксида кремния (который также может называться пирогенным) или носитель на основе геля диоксида кремния. Предпочтительно он представляет собой носитель на основе аморфного диоксида кремния, в частности, носитель на основе аморфного аэрозольного диоксида кремния или носитель на основе геля аморфного диоксида кремния.
Носитель катализатора на основе диоксида кремния (SiO2) может представлять собой пористый материал, который может иметь средний диаметр пор, составляющий по меньшей мере от 10 нм до 20 нм, предпочтительно более чем 20 нм, но менее чем 50 нм. Предпочтительно носитель на основе диоксида кремния имеет средний диаметр пор, составляющий более чем 22 нм, предпочтительно по меньшей мере 25 нм, предпочтительно менее чем 40 нм, предпочтительно от 25 до 35 нм, предпочтительно 30 нм. Средний диаметр пор был определен с применением анализа физической адсорбции азота методом Баррета-Джойнера-Халенды (BJH). Следует понимать, что средний диаметр пор представляет собой средний диаметр пор собственно носителя, который измеряют перед добавлением нитрата кобальта или какого-либо из других соединений, описанных ниже, в носитель катализатора на основе диоксида кремния.
Объем пор носителя может составлять от 0,1 до 1 мл на 1 г носителя катализатора, предпочтительно от 0,3 до 0,9 мл на 1 г носителя катализатора.
Носитель катализатора на основе диоксида кремния может быть изготовлен предварительно. Предварительно изготовленный носитель может представлять собой зернистый носитель, предпочтительно имеющий средний размер частиц, составляющий от 1 до 500 микрометров, предпочтительнее от 10 до 250 микрометров, еще предпочтительнее от 45 до 200 микрометров.
Нитрат кобальта может представлять собой Со(NO3)2⋅6H2O.
Модифицированный носитель катализатора также может содержать соль кобальта, которая не представляет собой нитрат кобальта. Предпочтительно модифицированный носитель катализатора содержит гидроксид кобальта (Со(ОН)2) в дополнение к нитрату кобальта. Массовое соотношение Со(ОН)2 и нитрата кобальта может составлять от 0,02 до 0,1, предпочтительно приблизительно 0,028 до 0,067. Предпочтительно Со(ОН)2 наносят на поверхность и/или вводят в объем носителя в жидкой среде, и добавляемое количество Со(ОН)2 является таким, что значение рН составляет от 4,5 до 5,5, предпочтительно приблизительно 5.
Нитрат кобальта может быть добавлен (предпочтительно путем пропитывания) посредством нанесения на поверхность и/или введения в объем носителя на более чем одной стадии добавления (предпочтительно путем пропитывания). Предпочтительно модифицированный носитель катализатора прокаливают после каждой стадии добавления (предпочтительно путем пропитывания). Следует понимать, что в таких случаях после первой стадии добавления (предпочтительно путем пропитывания) и последующего прокаливания в результате следующей стадии добавления (предпочтительно путем пропитывания) получается модифицированный носитель катализатора, одновременно содержащий при этом оксид кобальта и нитрат кобальта. Следует понимать, что согласно некоторым вариантам осуществления настоящего изобретения модифицированный носитель катализатора может содержать оксид кобальта и нитрат кобальта.
Добавление Со(ОН)2 может быть осуществлено вместе с добавлением нитрата кобальта в течение первой стадии добавления (предпочтительно путем пропитывания), и предпочтительно на следующей стадии добавления нитрат кобальта (предпочтительно путем пропитывания), как правило, отсутствует добавление Со(ОН)2.
Предшественник катализатора может содержать кобальт (Со) в количестве, составляющем от 5 до 70 г Со на 100 г носителя катализатора, предпочтительно от 20 до 40 г Со на 100 г носителя катализатора и предпочтительнее от 25 до 35 г Со на 100 г носителя катализатора.
Предпочтительно модифицированный носитель катализатора содержит соединение титана (Ti) на поверхности и/или в объеме носителя катализатора. Предпочтительно модифицированный носитель катализатора содержит соединение марганца (Mn) на поверхности и/или в объеме носителя катализатора. Предпочтительно модифицированный носитель катализатора одновременно содержит соединение титана на поверхности и/или в объеме носителя катализатора и соединение марганца на поверхности и/или в объеме носителя катализатора.
Предпочтительно, модифицированный носитель катализатора содержит более чем 1 мас. % и не более чем 10 мас. % Ti по отношению к массе носителя катализатора на основе диоксида кремния (SiO2), причем Ti присутствует в форме одного или нескольких соединений титана.
Предпочтительно модифицированный носитель катализатора содержит не более чем 5 мас. % Ti, предпочтительно не более чем 3,5 мас. % Ti. Предпочтительно титан в форме одного или нескольких соединений титана может присутствовать в объеме и/или на поверхности носителя катализатора в количестве, составляющем более чем 1,5 мас. %, предпочтительно по меньшей мере 2,0 мас. %, предпочтительнее по меньшей мере 2,4 мас. % Ti.
Предпочтительно титан в форме одного или нескольких соединений титана может присутствовать в объеме и/или на поверхности носителя катализатора в количестве, составляющем менее чем 3,5 мас. %, предпочтительно не более чем 3,0 мас. %, но предпочтительно более чем 2,0 мас. % Ti.
Предпочтительное количество титана в форме одного или нескольких соединений титана, присутствующего в объеме и/или на поверхности носителя катализатора, составляет 2,6 мас. % Ti. Титан предпочтительно присутствует в форме оксида титана.
Предпочтительно Ti присутствует в качестве модификатора носителя, то есть представляет собой соединение титана, которое было нанесено на поверхность и/или введено в объем носителя катализатора (и предпочтительно также подвергнуто прокаливанию) перед тем, как нитрат кобальта был нанесен на поверхность и/или введен в объем носителя катализатора. Предпочтительно соединение титана представляет собой оксид титана.
В качестве альтернативы, Ti может присутствовать в качестве промотора, то есть представляет собой соединение титана, которое было нанесено на поверхность и/или введено в объем носителя катализатора одновременно и/или после того, как нитрат кобальта был нанесен на поверхность и/или введен в объем носителя катализатора. Соединение титана может представлять собой соль титана. Соль титана может быть подвергнута прокаливанию с нитратом кобальта для превращения соли титана в оксид титана.
Предпочтительно модифицированный носитель катализатора содержит более чем 0,5 мас. % и менее чем 10 мас. % марганца по отношению к массе носителя катализатора на основе диоксида кремния (SiO2), причем Mn присутствует в форме одного или нескольких соединений марганца.
Предпочтительно модифицированный носитель катализатора содержит не более чем 7,5 мас. % Mn, предпочтительно не более чем 5 мас. % Mn. Предпочтительно марганец в форме одного или нескольких соединений марганца может присутствовать в объеме и/или на поверхности носителя катализатора в количестве, составляющем более чем 1 мас. %, предпочтительно по меньшей мере 1,5 мас. %, предпочтительнее по меньшей мере 1,6 мас. % Mn.
Предпочтительно марганец в форме одного или нескольких соединений марганца может присутствовать в объеме и/или на поверхности носителя катализатора в количестве, составляющем менее чем 3,2 мас. %, предпочтительно не более чем 2,3 мас. %, но предпочтительно более чем 1,5 мас. % Mn.
Предпочтительное количество марганца в форме одного или нескольких соединений марганца, которые присутствуют в объеме и/или на поверхности носителя катализатора, составляет 2,2 мас. % Mn. В качестве альтернативы, это количество может составлять 1,6 мас. % Mn.
Предпочтительно соединение марганца представляет собой неорганическое соединение марганца, такое как нитрат марганца. В качестве альтернативы, оно может представлять собой органическое соединение марганца. В настоящем описании органическое соединение марганца представляет собой соединение марганца, в котором с атомом марганца соединена по меньшей мере одна органическая группа посредством связи, например, посредством ковалентной связи, координационной связи между металлом и лигандом или ионного взаимодействия.
Предпочтительно Mn может присутствовать в качестве промотора, то есть представляет собой соединение марганца, которое было нанесено на поверхность и/или введено в объем носителя катализатора одновременно и/или после того, как соединение кобальта было нанесено на поверхность и/или введено в объем носителя катализатора. Соединение марганца может присутствовать в форме соли марганца, предпочтительно представляя собой нитрат марганца. Предпочтительно соль марганца подвергают прокаливанию вместе с нитратом кобальта в целях превращения соли марганца в оксид марганца.
В качестве альтернативы, Mn может присутствовать в качестве модификатора носителя, то есть представлять собой соединение марганца, которое был нанесено на поверхность и/или введено в объем носителя катализатора (и предпочтительно также подвергнуто прокаливания) перед тем, как соединение кобальта было нанесено на поверхность и/или введено в объем носителя катализатора. Марганец может присутствовать в форме оксида марганца.
Согласно предпочтительному варианту осуществления настоящего изобретения модифицированный носитель катализатора, который подвергают прокаливанию, представляет собой носитель катализатора на основе диоксида кремния, содержащий нитрат кобальта и гидроксид кобальта. Носитель на основе диоксида кремния предпочтительно содержит соединение титана, предпочтительно оксид титана; и носитель на основе диоксида кремния также содержит соединение марганца, предпочтительно соль марганца, предпочтительно нитрат марганца. В процессе прокаливания нитрат кобальта и гидроксид кобальта могут реагировать с кислородом и превращаться в оксид кобальта; и соединение марганца может превращаться в оксид марганца. Предпочтительно соединение титана представляет собой оксид титана, однако если оно не представляет собой оксид титана, оно превращается в оксид титана в процессе прокаливания.
Модифицированный носитель катализатора может быть получен таким способом, как описано в публикациях WO2016/135577 и WO2018/029548, которые включены в настоящий документ посредством ссылки.
Согласно одному варианту осуществления настоящего изобретения рений отсутствует или практически отсутствует в каталитической композиции. Предпочтительно, если какое-либо количество Re присутствует в каталитической композиции, массовое соотношение Re и Со составляет менее чем 0,001:1.
Модифицированный носитель катализатора также может содержать модифицирующий элемент, предпочтительно модифицирующий элемент, способный повышать восстанавливаемость нитрата кобальта после его прокаливания. Модифицирующий элемент может присутствовать в форме соединения модифицирующего элемента, которое представляет собой соединение металла, выбранного из группы, которую составляют палладий (Pd), платина (Pt), рутений (Ru), рений (Re) и смесь двух или большего числа этих металлов. Массовое соотношение металла, представляющего собой модифицирующий элемент, в частности, соотношение палладия или платины в качестве модифицирующего металла и кобальта в пересчете на металл может составлять от 1:300 до 1:3000.
Модифицированный носитель катализатора также может содержать кислоту, предпочтительно органическую кислоту, предпочтительно малеиновую кислоту.
Прокаливание
В процессе прокаливания модифицированного носителя катализатора нитрат кобальта превращается в оксид кобальта, предпочтительно, оксид кобальта, выбранный из СоО, СоО(ОН), CO3O4, CO2O3 или смеси, содержащей один или несколько указанных веществ. Предпочтительно оксид кобальта представляет собой CO3O4.
Высокая скорость нагревания в пределах температурного диапазона А не становится ниже 10°С в минуту, предпочтительно высокая скорость нагревания не становится ниже 15°С в минуту в пределах температурного диапазона А, и предпочтительно высокая скорость нагревания не становится ниже 20°С в минуту в пределах температурного диапазона А. Предпочтительно высокая скорость нагревания не становится ниже 48°С в минуту в пределах температурного диапазона А, предпочтительно высокая скорость нагревания не становится ниже 55°С в минуту в пределах температурного диапазона А, и предпочтительно высокая скорость нагревания не становится ниже 66°С в минуту в пределах температурного диапазона А. Высокая скорость нагревания не может становиться ниже 144°С в минуту в пределах температурного диапазона А, и она не может даже становиться ниже 221°С в минуту в пределах температурного диапазона А. Предпочтительно высокая скорость нагревания в пределах температурного диапазона А не становится выше 300°С в минуту, предпочтительно не становится выше 233°С в минуту и предпочтительно не становится выше 221°С в минуту.
Согласно предпочтительному варианту осуществления настоящего изобретения прокаливание представляет собой нагревание модифицированного носителя катализатора до температуры выше температурного диапазона А, предпочтительно до температуры, составляющей по меньшей мере 170°С, предпочтительно до температуры, составляющей по меньшей мере 180°С, предпочтительно до температуры, составляющей по меньшей мере 200°С, предпочтительно до температуры, составляющей по меньшей мере 220°С, предпочтительно до температуры, составляющей по меньшей мере 250°С, и предпочтительно до температуры не выше 400°С.
Предпочтительно прокаливание также представляет собой нагревание модифицированного носителя катализатора до температуры выше температурного диапазона А при высокой скорости нагревания, описанной выше, предпочтительно до температуры, составляющей по меньшей мере 170°С, предпочтительно по меньшей мере 180°С, предпочтительно по меньшей мере 200°С, предпочтительно по меньшей мере 220°С, предпочтительно до температуры, составляющей по меньшей мере 250°С и предпочтительно до температуры, составляющей не выше 350°С, предпочтительно не выше 300°С.
Если прокаливание при высокой скорости нагревания прекращается при температуре ниже 250°С, то прокаливание может происходить до температуры, составляющей по меньшей мере 250°С, при скорости нагревания, которая является ниже, чем высокая скорость нагревания.
Предпочтительно высокая скорость нагревания в пределах температурного диапазона А является такой же, как высокая скорость нагревания выше температурного диапазона А.
Предпочтительно прокаливание представляет собой нагревание модифицированного носителя катализатора при высокой скорости нагревания, которая составляет не менее чем 10°С в минуту, в пределах по меньшей мере температурного диапазона, который составляет от 100°С до 170°С, предпочтительно от 100°С до 180°С, предпочтительно от 100°С до 200°С, предпочтительно от 100°С до 220°С, предпочтительно от 100°С до 250°С. Предпочтительно указанные температурные диапазоны начинаются при 90°С, предпочтительно при 85°С, а не при 100°С. Высокая скорость нагревания может быть выше, чем высокие скорости нагревания, которые описаны выше.
Минимальная температура, при которой начинается прокаливание модифицированного носителя катализатора, представляет собой минимальную температуру, при которой нитрат кобальта начинает превращаться в оксид кобальта. Эта температура представляет собой температуру, при которой нитрат кобальта начинает разлагаться с высвобождением газообразного NO2 (температура высвобождения NO2) в количестве, превышающем 1500 объемных частей на миллион, или 0,15 об. %. Температура, при которой начинается высвобождение газообразного NO2 в количестве, превышающем 1500 объемных частей на миллион, измеряется методом инфракрасной спектроскопии с преобразованием Фурье с анализом газовой фазы при скорости нагревания, составляющей 0,5°С в минуту, в газовой смеси на основе Не, содержащей 12% О2, при скорости газового потока, составляющей 0,5 мл/с.
Температура, при которой начинается высвобождение газообразного NO2 в количестве, превышающем 1500 объемных частей на миллион, может зависеть от композиции модифицированного носителя катализатора. Как известно, присутствие Со(ОН)2 и/или малеиновой кислоты влияет на эту температуру. Согласно одному варианту осуществления настоящего изобретения температура, при которой начинается высвобождение газообразного NO2 в количестве, превышающем 1500 объемных частей на миллион, составляет 125°С; в качестве альтернативы, эта температура составляет 115°С; в качестве альтернативы, она составляет 100°С.
Согласно предпочтительному варианту осуществления настоящего изобретения нагревание модифицированного носителя катализатора при высокой скорости нагревания может начинаться при температуре, составляющей 120°С, предпочтительно 110°С, предпочтительно 100°С, предпочтительно 90°С или даже 85°С, и это нагревание может быть осуществлено до температуры, составляющей 165°С, при которой завершается температурный диапазон А. Следует понимать, что высокая скорость нагревания может начинаться при температуре ниже минимальной температуры, при которой начинается прокаливание модифицированного носителя катализатора.
При прокаливании может происходить реакция нитрата кобальта с источником кислорода, и в результате этого превращение нитрата кобальта в оксид кобальта. Источник кислорода может представлять собой любой подходящий источник, и предпочтительно он представляет собой кислородсодержащий газ. Кислородсодержащий газ может представлять собой кислород, и предпочтительно кислородсодержащий газ представляет собой воздух.
Газовый поток, который пропускают над модифицированным носителем катализатора в пределах температурного диапазона А, может иметь часовую объемную скорость газа (GHSV), составляющую по меньшей мере 5 Нм3 на 1 кг нитрата кобальта в час; предпочтительно она составляет по меньшей мере 9 Нм3 на 1 кг нитрата кобальта в час или даже по меньшей мере 14 Нм3 на 1 кг нитрата кобальта в час. В тех случаях, когда модифицированный носитель катализатора поступает в непрерывном режиме или в форме множества порций, количество нитрата кобальта представляет собой количество нитрата кобальта, поступающего в течение часа. Таким образом, если газовый поток составляет 20 Нм3 в час, и 2 кг нитрата кобальта поступают в течение часа, то газовый поток, составляющий 20 Нм3 в час, делят на 2 кг нитрата кобальта и получают значение GHSV, составляющее 10 Нм3 на 1 кг нитрата кобальта в час.
Газ, используемый в процессе прокаливания, может представлять собой любой подходящий газ, такой как инертный газ или кислородсодержащий газ. Инертный газ может представлять собой азот. Кислородсодержащий газ может представлять собой кислород, и предпочтительно кислородсодержащий газ представлять собой воздух.
Предпочтительно, когда прокаливание представляет собой нагревание модифицированного носителя катализатора ниже и/или выше температурного диапазона А, при прокаливании также пропускают газовый поток над модифицированным носителем катализатора в процессе этого нагревания. Газовый поток в процессе этого нагревания может иметь такое же значение GHSV, которое присутствует в процессе нагревания в пределах температурного диапазона А.
Прокаливание может быть осуществлено в любом подходящем прокалочном устройстве (прокалочной печи). Такое прокалочное устройство может представлять собой прокалочную печь с псевдоожиженным слоем. Следует понимать, что скорость нагревания означает скорость, при которой модифицированный носитель катализатора, введенный в прокалочное устройство (описанное ниже) нагревается до достижения температуры реактора. Таким образом, скорость нагревания представляет собой результат деления разности между температурой реактора (конечной температурой) и температурой (начальной температурой) модифицированного носителя катализатора, который поступает в реактор, на время, которое требуется модифицированному носителю катализатора для достижения конечной температуры от начальной температуры.
Модифицированный носитель катализатора может быть введен в прокалочное устройство (прокалочную печь) любым подходящим способом. Предпочтительно модифицированный носитель катализатора может поступать в непрерывном режиме. В качестве альтернативы, он может поступать в периодическом режиме, предпочтительно в форме множества порций.
Модифицированный носитель катализатора поступает в прокалочную печь таким образом, что скорость нагревания в пределах температурного диапазона А не становится ниже высокой скорости нагревания.
Модифицированный носитель катализатора может быть превращен в немодифицированный материал в одной или нескольких порциях, выходящих из прокалочной печи. Множество порций, поступающих в прокалочную печь, можно превратить в немодифицированный материал в одной порции.
Модифицированный носитель катализатора может быть подвергнут прокаливанию таким образом, что содержание азота в прокаленном модифицированном носителе катализатора может составлять не более чем 0,4 мас. %, предпочтительно не более чем 0,3 мас. %, что определяется посредством анализа элементов CHNS.
В способе также может быть предусмотрено высушивание модифицированного носителя катализатора перед прокаливанием модифицированного носителя катализатора при высокой скорости нагревания в пределах температурного диапазона А. Предпочтительно высушивание не происходит при температуре выше 100°С. Высушивание может быть осуществлено при давлении, составляющим менее чем атмосферное давление. Предпочтительно по меньшей мере две из шести молекул кристаллизационной воды, составляющие 33,3% кристаллизационной воды, необходимо удалить из внедренного посредством пропитывания Со(NO3)2⋅6H2O в течение процесса высушивания. Предпочтительно не более чем 50% (три из шести молекул) кристаллизационной воды необходимо удалить из Со(NO3)2⋅6H2O в течение процесса высушивания. Следует понимать, что если процентная доля удаляемой в процессе высушивания кристаллизационной воды составляет от 33,3% до 50%, это означает удаление первых двух молекул кристаллизационной воды и некоторой части третьих молекул кристаллизационной воды.
Процентная доля удаляемой кристаллизационной воды может быть вычислена на основании изменения массы, измеряемой в процессе потери массы при прокаливании (LOI) при температуре 400°С.
Активация
Согласно второму аспекту настоящего изобретения предложен способ получения кобальтсодержащего катализатора, причем способ включает получение предшественника кобальтсодержащего катализатора, как указано выше; и восстановление предшественника катализатора, и в результате этого происходит активация предшественника катализатора.
Таким образом, может быть получен активный кобальтсодержащий катализатор, содержащий носитель катализатора, в котором содержится кобальт.
При восстановлении предшественника катализатора может происходить превращение оксида кобальта в кобальт в нулевой степени окисления.
При восстановлении предшественника катализатора предпочтительно осуществляется его обработка газообразным восстановителем в целях активации. Предпочтительно газообразный восстановитель представляет собой водород или водородсодержащий газ. Водородсодержащий газ может содержать водород и один или несколько инертных газов, которые являются инертными по отношению к активному катализатору. Водородсодержащий газ предпочтительно содержит по меньшей мере 90 об. % водорода.
Газообразный восстановитель может быть введен в контакт с предшественником катализатора любым подходящим способом. Предпочтительно предшественник катализатора присутствует в форме слоя частиц, и при этом поток газообразного восстановителя пропускают через слой частиц. Слой частиц может представлять собой неподвижный слой, но предпочтительно он представляет собой псевдоожиженный слой, и при этом газообразный восстановитель предпочтительно выступает в качестве псевдоожижающей среды для слоя частиц предшественника катализатора.
Восстановление может быть осуществлено при абсолютном давлении, составляющем от 0,6 до 1,5 бар, предпочтительно от 0,8 до 1,3 бар. В качестве альтернативы, абсолютное давление может составлять от 1,5 бар до 20 бар. Однако давление предпочтительно находится приблизительно на уровне атмосферного давления.
Восстановление предпочтительно осуществляется при температуре выше 25°С, при которой предшественник катализатора будет восстанавливаться до активной формы. Предпочтительно восстановление осуществляется при температуре выше 150°С и предпочтительно ниже 600°С. Предпочтительно восстановление осуществляется при температуре ниже 500°С, предпочтительнее ниже 450°С.
В процессе восстановления температура может изменяться, и предпочтительно она увеличивается до максимальной температуры, при которой восстановление осуществляется, как указано выше.
Поток газообразного восстановителя через слой частиц предпочтительно регулируют для обеспечения того, чтобы содержание загрязняющих веществ, которые образуются в процессе восстановления, оставалось на достаточно низком уровне. Газообразный восстановитель может рециркулировать, и при этом рециркулирующий газообразный восстановитель предпочтительно обрабатывают для удаления одного или нескольких загрязняющих веществ, которые образуются в процессе восстановления. Загрязняющие вещества могут содержать одно или несколько веществ, представляющих собой воду и аммиак.
Восстановление может быть осуществлено в течение двух или большего числа стадий, в процессе которых варьируют один или оба параметра, представляющих собой скорость нагревания и объемную скорость газообразного восстановителя.
Согласно одному варианту осуществления настоящего изобретения на активный катализатор может быть нанесено покрытие предпочтительно посредством введения смеси частиц активного катализатора и среды для нанесения покрытия в форме расплавленного органического вещества, которое присутствует при температуре T1, и которое затвердевает или застывает при менее высокой температуре Т2, таким образом, что Т2<T1, по меньшей мере в одной форме; и при этом форму по меньшей мере частично погружают в охлаждающую жидкость, в результате чего органическое вещество охлаждается температуры Т3, где Т3≤Т2
В процессе восстановления, парциальное давление водяного пара предпочтительно поддерживается на минимально возможном уровне, предпочтительнее ниже 0,1 атмосферы. Объемная скорость водорода может составлять от 2 до 4 литров в час на грамм катализатора.
Согласно одному варианту осуществления настоящего изобретения способ получения кобальтсодержащего катализатора может включать:
- на стадии образования карбидов обработку активированного катализатора, содержащего носитель катализатора, в котором содержится кобальт в нулевой степени окисления, содержащим СО газом (предпочтительно при температуре T1, где T1 составляет от 200°С до 280°С) в целях превращения кобальта в карбид кобальта и последующего получения содержащего карбид кобальта предшественника катализатора; и
- на последующей стадии активации обработку содержащего карбид кобальта предшественника катализатора водородсодержащий газом (предпочтительно при температуре Т2, где Т2 составляет по меньшей мере 300°С) в целях превращения карбида кобальта в металлический кобальт и последующей активации содержащего карбид кобальта предшественника катализатора и получения кобальтсодержащего катализатора синтеза углеводородов.
Катализатор может представлять собой катализатор синтеза углеводородов, используемый в целях синтеза углеводородов и/или кислородсодержащих производных углеводородов с применением по меньшей мере водорода и монооксида углерода. Предпочтительно катализатор представляет собой катализатор синтеза Фишера-Тропша (FT), используемый для осуществления синтез Фишера-Тропша. Синтез FT может быть осуществлен в реакторе с неподвижным слоем, в реакторе с взвешенным слоем или в реакторе с неподвижным псевдоожиженным слоем. Предпочтительно синтез FT представляет собой способ синтеза FT в трехфазной системе с взвешенным слоем.
Синтез углеводородов
Согласно третьему аспекту настоящего изобретения предложен способ синтеза углеводородов для получения углеводородов и необязательно кислородсодержащих производных углеводородов, причем способ включает введение кобальтсодержащего катализатора, который описан выше, в контакт с водородом и монооксидом углерода при температуре выше 100°С и при давлении, составляющем по меньшей мере 10 бар, для получения углеводородов и необязательно кислородсодержащих производных углеводородов.
Согласно четвертому аспекту настоящего изобретения предложен способ синтеза углеводородов для получения углеводородов и необязательно кислородсодержащих производных углеводородов, причем способ включает введение синтетического газа, содержащего водород, монооксид углерода и азотсодержащие загрязняющие вещества, выбранный из группы, которую составляют HCN, NH3, NO, RxNH3-x, где R представляет собой органическую группу, и х составляет 1, 2 или 3, причем группы R являются одинаковыми или различными, когда х составляет 2 или 3, R1-CN, где R1 представляет собой органическую группу, и гетероциклические соединения, содержащие по меньшей мере один атом азота в составе цикла, представляющего собой гетероциклическое кольцо гетероциклического соединения, причем азотсодержащие загрязняющие вещества составляют в сумме по меньшей мере 100 объемных частей на миллиард, но менее чем 1000000 объемных частей на миллиард по отношению к объему синтетического газа, при температуре, составляющей по меньшей мере 180°С, и абсолютном давлении, составляющим по меньшей мере 10 бар (1000 кПа), в контакт с катализатором, который описан выше, в целях получения углеводородов и необязательно кислородсодержащих производных углеводородов посредством синтеза Фишера-Тропша в процессе реакции водорода с монооксидом углерода.
Синтетический газ (синтез-газ) может содержать в сумме по меньшей мере 200 объемных частей на миллиард азотсодержащих загрязняющих веществ. Предпочтительно синтетический газ содержит по меньшей мере 250 объемных частей на миллиард азотсодержащих загрязняющих веществ. Предпочтительнее синтетический газ содержит по меньшей мере 500 объемных частей на миллиард азотсодержащих загрязняющих веществ. Как правило, синтетический газ содержит по меньшей мере 1000 объемных частей на миллиард азотсодержащих загрязняющих веществ. Предпочтительно синтетический газ содержит не более чем 100000 объемных частей на миллиард азотсодержащих загрязняющих веществ. Предпочтительнее синтетический газ содержит не более чем 20000 объемных частей на миллиард азотсодержащих загрязняющих веществ. Как правило, синтетический газ может содержать не более чем 10000 объемных частей на миллиард азотсодержащих загрязняющих веществ. Например, согласно одному варианту осуществления настоящего изобретения синтетический газ может содержать приблизительно 2000 объемных частей на миллиард азотсодержащих загрязняющих веществ. Однако согласно другому варианту осуществления синтетический газ может содержать приблизительно 5000 объемных частей на миллиард азотсодержащих загрязняющих веществ. Как правило, когда синтетический газ получен посредством ожижения газа, он содержит HCN и NH3 в качестве азотсодержащих загрязняющих веществ; когда синтетический газ получен посредством ожижения угля, он содержит NH3 и NO в качестве азотсодержащих загрязняющих веществ.
Предпочтительно, R в RxNH3-x представляет собой гидрокарбильную группу и/или содержащую кислород гидрокарбильную группу. Предпочтительнее R в RxNH3-x представляет собой алкильную группу и/или спирт. Предпочтительно х составляет 1 или 2. Согласно предпочтительному варианту осуществления настоящего изобретения RxNH3-x представляет собой дипропиламин (CH3CH2CH2)2NH. В качестве альтернативы, RxNH3-x может представлять собой диэтаноламин или метилдиэтаноламин.
Предпочтительно, R1 в R1-CN представляет собой гидрокарбильную группу. Предпочтительнее R1 в R1-CN представляет собой алкильную группу. Согласно одному предпочтительному варианту осуществления настоящего изобретения, R1 представляет собой метальную группу.
Гетероциклические соединения могут содержать кислородсодержащие группы. Примеры таких кислородсодержащих соединений и бескислородных соединений представляют собой 4-пиперидиноацетофенон (гетероциклическое кислородсодержащее соединение), 1,4-бипиперидин (гетероциклическое бескислородное соединение), 1-пиперидинпропионитрил (моноциклическое соединение) и 3-пиперидино-1,2-пропандиол (моноциклическое кислородсодержащее соединение).
В качестве альтернативы, способ синтеза углеводородов может быть таким, как описано выше, за исключением того, что синтетический газ не содержит или содержит менее чем 100 объемных частей на миллиард азотсодержащих загрязняющих веществ, которые описаны выше.
Согласно одному предпочтительному варианту осуществления настоящего изобретения катализатор может быть получен способом, который включает:
- на стадии образования карбидов обработку активированный катализатор, содержащий носитель катализатора, в котором содержится кобальт в нулевой степени окисления, с содержащим СО газом (предпочтительно при температуре T1, где T1 составляет от 200°С до 280°С) в целях превращения кобальта в карбид кобальта и последующего получения содержащего карбид кобальта предшественника катализатора; и
- на последующей стадии активации обработку содержащего карбид кобальта предшественника катализатора водородсодержащий газом (предпочтительно при температуре Т2, где Т2 составляет по меньшей мере 300°С) в целях превращения карбида кобальта в металлический кобальт и последующей активации содержащего карбид кобальта предшественника катализатора и получения кобальтсодержащего катализатора синтеза углеводородов.
Предпочтительно способ синтеза углеводородов представляет собой процесс Фишера-Тропша, предпочтительно трехфазный процесс Фишера-Тропша, предпочтительнее процесс Фишера-Тропша во взвешенном слое для получения воскового продукта.
Абсолютное парциальное давление водяного пара во взвешенном слое может достигать по меньшей мере 5 бар, предпочтительно по меньшей мере 8 бар. Полное молярное соотношение исходных H2 и СО может составлять от 1,4 до 2, предпочтительно приблизительно 1,5, в качестве альтернативы, приблизительно 1,8. Согласно альтернативному варианту осуществления абсолютное парциальное давление водяного пара во взвешенном слое может составлять ниже 5 бар. Полное молярное соотношение исходных H2 и СО может составлять от 1,4 до 2, предпочтительно приблизительно 1,6.
Способ синтеза углеводородов также может включать стадию гидропереработки для превращения углеводородов и необязательно соответствующих кислородсодержащих производных в жидкие горючие материалы и/или другие химические продукты.
Согласно пятому аспекту настоящего изобретения предложены продукты, получаемые способом синтеза углеводородов, который описан выше.
Краткое описание фигур
Далее настоящее изобретение будет описано более подробно, исключительно в качестве примера, со ссылкой на следующие сопровождающие фигуры, где:
на фиг. 1 представлен график температуры прокаливания псевдоожиженного слоя при низкой скорости нагревания, высоты слоя, перепада давления в слое и концентрации NO2 по отношению к примеру 1;
на фиг. 2 представлен график температуры прокаливания псевдоожиженного слоя при высокой скорости нагревания, высоты слоя, перепада давления в слое и концентрации NO2 по отношению к примеру 4;
на фиг. 3 представлен увеличенный профиль прокаливания в течение одночасового периода, выбранного из фиг.2;
на фиг. 4 представлен график процентного отличия скорости FTS по отношению к примерам 1, 7, 10, 11, 12, 13, 15 и 30 по сравнению с примером 4;
на фиг. 5 представлен график мутности в зависимости от температуры реактора по отношению к примерам 15-19 и 21;
на фиг. 6 представлен график процентного отличия скорости FTS в примере 20 в зависимости от температуры реактора по отношению к примерам 16 до 22;
на фиг. 7 представлен профиль окисления с программированием температуры (ТРО) по отношению к примеру 28; и
на фиг. 8 представлен профиль ТРО по отношению к примеру 29.
Представленные выше и другие задачи, признаки и преимущества настоящего изобретения становятся более понятными из следующего описание определенных вариантов осуществления настоящего изобретения посредством приведенных ниже неограничительных примеров.
Примеры
Определения, соответствующие примерам
Полунепрерывное введение модифицированного носителя катализатора. -Модифицированный носитель катализатора в непрерывном режиме или в составе нескольких порций поступает в прокалочную печь, где исходный материал объединяется с образованием слоя и находится в течение периода выдерживания. Объединенный исходный материал затем превращается в немодифицированный материал в периодическом режиме (предпочтительно в составе единственной порции) после периода выдерживания в прокалочной печи.
Часовая объемная скорость газа (GHSV). - Для типичного прокаливания модифицированного носителя катализатора, содержащего Со(NO3)2⋅6H2O, значение GHSV (часовая объемная скорость газа) определено как Нм3 воздуха в час на 1 кг Со(NO3)2⋅6H2O и вычислено в результате деления скорости потока воздуха (Нм3/ч) (или вместо воздуха может присутствовать другой подходящий газ) на полное количество Со(NO3)2⋅6H2O, модифицирующего определенное количество или порцию катализатора. Однако в течение полунепрерывного введения поток (кг/ч) модифицированного носителя катализатора осуществляется вместо загрузки полной порции до начала прокаливания. В таком случае количество Со(NO3)2⋅6H2O, используемое в выражении GHSV, представляет собой количество Со(NO3)2⋅6H2O, которое поступает в прокалочную печь в течение одного часа. Таким образом, если газовый поток составляет 20 Нм3 в час, и 2 кг нитрата кобальта поступает в час, то газовый поток 20 Нм3 в час следует разделить на 2 кг нитрата кобальта, чтобы получить значение GHSV, составляющее 10 Нм3 на 1 кг нитрата кобальта в час.
Скорость нагревания предшественника. - Скорость нагревания модифицированного носителя катализатора означает скорость, при которой модифицированный носитель катализатора, поступающий в прокалочное устройство (прокалочную печь), нагревается до достижения температуры реактора. Таким образом, скорость нагревания представляет собой результат деления разности между температурой реактора (конечной температурой) и температурой модифицированного носителя катализатора при его поступлении в реактор (начальной температурой) на время, которое требуется модифицированному носителю катализатора для достижения конечной температуры от начальной температуры.
Мутность. Сверхтонкие частицы, определяемые как частицы мельче 5 мкм, не всегда можно точно измерить методом распределения частиц по размеру (PSD) с применением прибора Saturn DigiSizer или посредством просеивания. Измерения мутности используются вместо измерения сверхтонких частиц катализатора с применением ультразвуковой обработки. При ультразвуковом воздействии на катализатор от основной частицы отделяются тонкие частицы, которые рассеивают или поглощают свет, придавая исследуемой среде облачный внешний вид. Измеритель мутности определяет интенсивность рассеянного света. Чем выше интенсивность рассеянного света, тем более высоким будет наблюдаемое показание мутности.
Мутность измеряли, помещая в четыре лабораторных стакана объемом 100 мл по 2,00 г образца катализатора и 38 мл очищенной от хлопьев воды, имеющей мутность ниже 0,5 нефелометрических единиц мутности (NTU), и помещая стаканы в ультразвуковую ванну, содержащую 700 мл воды при температуре 25°С. Образцы обрабатывали ультразвуком в течение четырех минут при частоте 40 кГц и мощности ультразвука 70 Вт, измеряя мутность в единицах NTU.
Прокаливание при высокой скорости нагревания. Прокаливание осуществляли при скорости нагревания, составляющей более чем 10°С в минуту.
Примеры
Пример 1 (сравнительный). - Прокаливание псевдоожиженного слоя, содержащего 30 г Со/0,075 г Pt/3,1 г Mn/100 г Ti-SiO2 (С5121, рН=2,3), при низкой скорости нагревания, составляющей 1°С в минуту
Модифицированный носитель на основе диоксида кремния. - Изопропоксид титана(IV) (17,2 г) добавляли в сухой этанол (130 г) и осуществляли перемешивание в течение 10 минут. Предварительно изготовленный аморфный гель диоксида кремния CARiACT Q-30 (100 г, средний диаметр пор 30 нм), полученный от компании Fuji Silysia Chemical Ltd., добавляли в полученный в результате раствор и осуществляли перемешивание в течение следующих 10 минут. Этанол удаляли при пониженном давлении с применением профиль высушивания, представленного в таблице 1, и получали сыпучий порошок. Полученный таким способом материал модифицированного носителя катализатора Ti-SiO2 прокаливали в муфельной печи при температуре 550°С и скорости нагревания 5°С в минуту, и продолжительность заключительного выдерживания составляла 5 часов. Полученный в результате модифицированный носитель содержал 2,6 г Ti на 100 г SiO2.


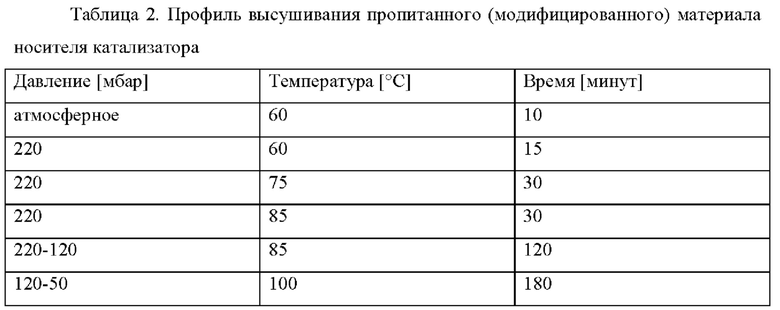
Предшественник катализатора. - На первой стадия введения металла Со(NO3)2⋅6H2O (7,56 кг), Mn(NO3)2⋅4H2O (1,44 кг) и (NH4)3Pt(NO3)2 (5,06 г) растворяли в воде (12,24 кг). Значение рН раствора устанавливали на уровне 2,3 после добавления Со(NO3)2⋅6H2O с применением разбавленной азотной кислоты. Описанный выше материал модифицированного носителя катализатора Ti-SiO2 (10,2 кг) добавляли в раствор и перемешивали в течение 10 минут. Избыточную воду удаляли при пониженном давлении в конусной сушилке до желательного удаления 33,3% кристаллизационной воды Со(NO3)2⋅6H2O посредством применения профиля высушивания, представленного в таблице 2.
Процентную долю удаленной кристаллизационной воды вычисляли по изменению массы, измеряемой в процессе потери массы при прокаливании (LOI) при температуре 400°С.
Полученный таким способом сыпучий порошок высушенного пропитанного материала носителя прокаливали в прокалочной печи с псевдоожиженным слоем при скорости нагревания 1°С в минуту до 250°С и продолжительностью выдерживания 6 часов с применением значения GHSV, составляющего 2,5 Нм3 воздуха на 1 кг Со(NO3)2⋅6H2O в час. Полученный в результате предшественник катализатора содержал 15 г Со/0,0255 г Pt/3,1 г Mn/100 г Ti-SiO2.
На второй стадии введения металла Со(NO3)2⋅6H2O (6,62 кг) и (NH4)3Pt(NO3)2 (8,87 г) растворяли в воде (13,47 кг). Значение рН раствора устанавливали на уровне 2,3 с применением разбавленной азотной кислоты. Прокаленный материал после первой стадии пропитывания (11,2 кг) добавляли в раствор и перемешивали в течение 10 минут. Избыточную воду удаляли при пониженном давлении в конусной сушилке до желательного удаления 33,3% кристаллизационной воды Со(NO3)2⋅6H2O посредством применения профиля высушивания, представленного в таблице 2. Прокаливание осуществляли в таких же условиях псевдоожижения, которые были применены после первой стадии пропитывания. Полученный в результате предшественник катализатора содержал 30 г Со/0,075 г Pt/3,1 г Mn/100 г Ti-SiO2.
Распределение по размеру частиц предшественника катализатора анализировали с применением имеющегося в продаже прибора Saturn DigiSizer™ 5200, и процентную долю тонкодисперсного материала мельче 45 микрон определяли, чтобы установить разрушение катализатора.
Пример 2 (сравнительный). - Прокаливание псевдоожиженного слоя, содержащего 15 г Со/0,025 г Pt/3,1 г Mn/100 г Ti-SiO2, при низкой скорости нагревания, составляющей 5°С в минуту (С5459, рН=2,3)
Предшественник катализатора был получен, как описано в примере 1, но без второй стадии введения металла, и высушенный пропитанный сыпучий порошок материала носителя, полученный после первой стадии пропитывания, прокаливали в прокалочной печи с псевдоожиженным слоем при скорости нагревания 5°С в минуту в отличие от 1°С в минуту, как описано в примере 1. Полученный в результате предшественник катализатора содержал 15 г Со/0,025 г Pt/3,1 г Mn/100 г Ti-SiO2.
Пример 3 (сравнительный). - Прокаливание псевдоожиженного слоя, содержащего 15 г Со/0,0255 г Pt/2,5 г МАс/100 г модифицированного Ti/Mn SiO2 (2,6 г Ti/ 3,1 г Mn на 100 г SiO2), при высоком значении GHSV, составляющем 8,2 Нм3 воздуха на 1 кг Со(NO3)2⋅6H2O в час (С2170, рН=2,3)
Модифицированный носитель. - Изопропоксид титана(IV) (2,57 кг) добавляли в сухой этанол (15,9 кг) и осуществляли перемешивание в течение 10 минут. В этот раствор добавляли предварительно изготовленный аморфный гель диоксида кремния (15 кг), CARiACT Q-30, полученный от компании Fuji Silysia, и осуществляли перемешивание в течение следующих 10 минут. Этанол удаляли при пониженном давлении с применением профиля высушивания, представленного в таблице 3, и получали сыпучий порошок.


Тетрагидрат ацетата марганца(II) (2,07 кг) растворяли в воде (22,5 кг) и осуществляли перемешивание в течение 10 минут. В этот раствор добавляли сыпучий порошок, полученный из модифицированного изопропоксидом титана(IV) диоксида кремния (17,6 кг), и осуществляли перемешивание в течение следующих 10 минут. Воду удаляли при пониженном давлении с применением профиля высушивания, представленного в таблице 3, и получали сыпучий порошок. Материал носителя прокаливали в муфельной печи при температуре 550°С, скорости нагревания 5°С в минуту и продолжительности заключительного выдерживания, составляющей 5 часов. Полученный в результате модифицированный носитель катализатора TiMn-SiO2 содержал 3,1 г Mn/2,6 г Ti на 100 г SiO2.
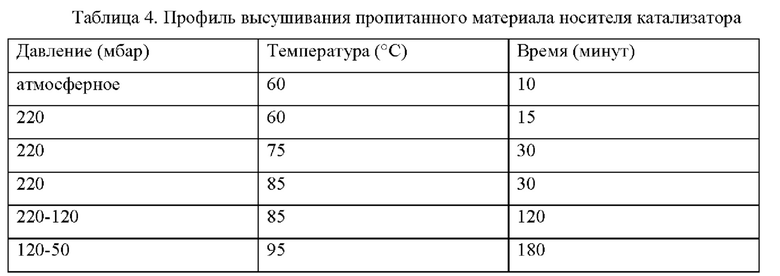
Предшественник катализатора. На первой стадии пропитывания 7,9 кг Со(NO3)2⋅6H2O растворяли в воде (10,31 кг), и значение рН раствора устанавливали на уровне 2,3 с применением разбавленной азотной кислоты. После этого в раствор добавляли малеиновую кислоту (МАс) (250 г) и (NH4)3Pt(NO3)2 (5,06 г). В смесь добавляли модифицированный носитель катализатора MnTi-SiO2 (10 кг), избыточную воду удаляли при пониженном давлении с применением профиля высушивания, представленного в таблице 4, и получали сыпучий порошок.
Сыпучий порошок прокаливали в прокалочной печи с псевдоожиженным слоем при скорости нагревания 0,5°С в минуту до 250°С и продолжительности выдерживания, составляющей 6 часов, с применением значения GHSV, составляющего 8,2 Нм3 воздуха на 1 кг Со(NO3)2⋅6H2O в час. Полученный в результате предшественник катализатора содержал 15 г Со/0,0255 г Pt/2,5 г МАс/100 г модифицированного Ti/Mn SiO2 (2,6 г Ti/3,1 г Mn/100 г SiO2) после первого пропитывания.
Пример 4 (согласно настоящему изобретению). - Прокаливание псевдоожиженного слоя, содержащего 30 г Со/0,075 г Pt/3,1 г Mn/100 г Ti-SiO2, при высокой скорости нагревания 67°С в минуту и температуре 210°С (С2173≡С5134, рН=2,3)
Высушенный пропитанный металлом материал носителя катализатора был получен, как описано в примере 1; однако модифицированный материал носителя поступал в полунепрерывном режиме при высокой скорости нагревания в прокалочную печь с псевдоожиженным слоем в отличие от прокаливания в условиях псевдоожижения, которое описано в примере 1.
Прокаливание при высокой скорости нагревания осуществляли посредством введения множества небольших порций пропитанного материала носителя катализатора через врезную трубную секцию в предварительно нагретый шестидюймовый реактор с псевдоожиженным слоем или в непрерывном режиме через вращающийся клапан в течение промышленного производства с регулированием постоянного высвобождения NO2. Прокалочную печь предварительно нагревали в потоке воздуха до температуры реактора, составляющей 210°С. Пропитанный материал носителя катализатора (75-100 г) модифицировали, используя испаряющийся материал, вводимый в реактор в токе азота через врезную трубную секцию. Процедуру повторяли через каждые 4-10 минут в течение периода, составляющего от 10 до 12 часов, для порций, составляющих приблизительно 10 кг. При размере загрузки 75 г и потоке воздуха 10 кг/ч вычисленная максимальная концентрация NO2 в отходящем газе составляла приблизительно 15000 частей на миллион. Когда весь материал был модифицирован в прокалочной печи с псевдоожиженным слоем, температуру реактора увеличивали до 250°С для заключительного выдерживания, которое осуществляли в течение периода полного высвобождения оставшегося NO2. Условия прокаливания при высокой скорости нагревания и заключительного выдерживания кратко представлены в таблице 5. Полученный в результате предшественник катализатора содержал 15 г Со/0,0255 г Pt/3,1 г Mn/100 г Ti-SiQ2.
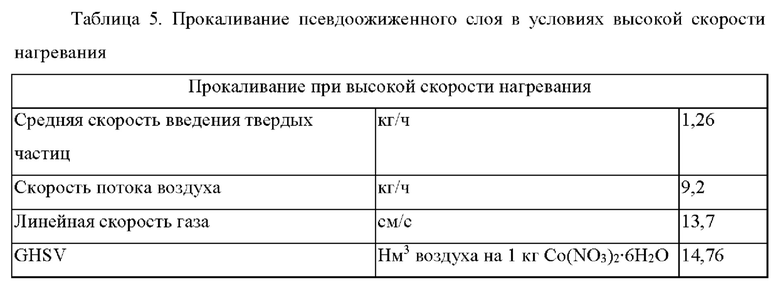
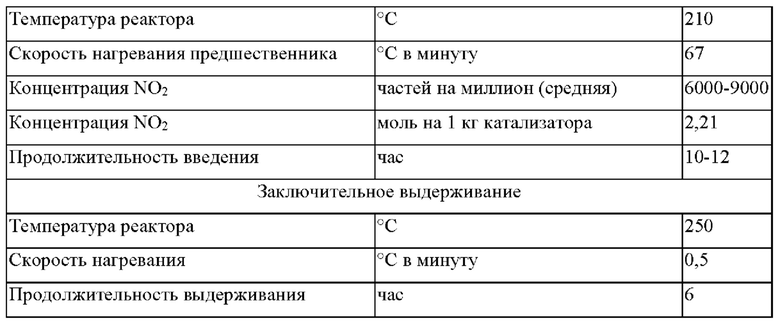
Вторая стадия введения активного металла была осуществлена, как описано в примере 1, и прокаливание было выполнено при высокой скорости нагревания, как описано выше в таблице 5, с получением каталитической композиции, содержащей 30 г Со/0,075 г Pt/3,1 г Mn/100 г Ti-SiO2.
Пример 5. - Активация катализатора
Прокаленные предшественники катализаторов, которые описаны выше в примере 1 и в примере 4, восстанавливали перед синтезом Фишера-Тропша (FTS) в потоке водорода при значении GHSV, составляющем 2,0 Нм3 в час на 1 кг прокаленного катализатора, и скорости нагревания 1°С в минуту до 390°С и выдерживали в течение 7 часов. Восстановленный катализатор охлаждали, погружали в расплавленный воск и модифицировали в реакторе, представляющем собой резервуар с непрерывным перемешиванием (CSTR), в атмосфере инертного газа.
Пример 6. - Синтез Фишера-Тропша (FTS)
Эксплуатационные характеристики FTS активированных и защищенных воском катализаторов, которые описаны в примере 5, оценивали, используя лабораторный суспензионный микрореактор CSTR при температуре реактора 230°С, давлении реактора, составляющем приблизительно 15 бар, и полном молярное соотношение исходных H2 и СО, составляющем приблизительно 1,5/1. В реакторе использовали электрическое нагревательное устройство и достаточно высокую скорость перемешивания для устранения любых ограничений в отношении массопереноса между газом и жидкостью. Объемную скорость исходного газа изменяли таким образом, что степень превращения синтетического газа составляла приблизительно от 75 до 78%. Парциальное давление водяного пара составляло приблизительно 5,5 бар.
Обсуждение
Неудовлетворительная устойчивость псевдоожижения, представляющая собой неустойчивость перепада давления или профиля высвобождения NO2 или распределения температуры в слое, наблюдалась для псевдоожиженных прокаленных предшественников катализаторов, которые описаны в примере 1 (см фиг. 1), в то время как псевдоожиженный предшественник катализатора, прокаленный при высокой скорости нагревания, который описан в примере 4, продемонстрировал хорошую устойчивость псевдоожижения слоя при температуре реактора 210°С (см. фиг. 2) без неустойчивости перепада давления и распределения температуры. Увеличенный профиль одночасового периода прокаливания, представленного на фиг. 2, который проиллюстрирован на фиг. 3, демонстрирует регулируемые многочисленные одиночные пики NO2 в процессе прокаливания при высокой скорости нагревания.
Размер кристаллитов Co3O4 прокаленного при высокой скорости нагревания предшественника катализатора (пример 4) оказался меньше, чем в случае прокаленного в псевдоожиженном слое предшественника катализатора в примере 1 и примере 2 (см. таблицу 6), и в результате этого получается катализатор FTS с более высокой активностью по сравнению с псевдоожиженном слое, прокаленном при низкой скорости нагревания, как описано в примере 1 (см. фиг. 4). Повышение скорости нагревания в процессе прокаливания псевдоожиженного слоя до 5°С в минуту в примере 2 не решило проблемы неудовлетворительного псевдоожижения, о чем свидетельствуют большие размеры наблюдаемых кристаллитов CO3O4, которые могут быть обусловлены миграцией кобальта (см. таблицу 6).
Даже несмотря на то, что никакая неустойчивость перепада давления в слое не наблюдалась при высоком значении GHSV, составляющем 8 Нм3 воздуха на 1 кг Со(NO3)2⋅6H2O в час в примере 3, все же наблюдался профиль высвобождения NO2, который, как правило, приводит к неудовлетворительному качеству катализатора.
Аналогично низкому процентному содержанию тонкодисперсного материала с размером частиц мельче 45 микрон, составляющему 1,7% в случае предшественника катализатора в примере 1, который прокаливали при низкой скорости нагревания, процентное содержание тонкодисперсного материала с размером частиц мельче 45 микрон в случае предшественника катализатора в примере 4 составляет лишь 1,3% (см. таблицу 6) и является показателем очень низкой степени разрушения предшественника катализатора, прокаленного при высокой скорости нагревания.

Пример 7 (согласно настоящему изобретению). - Прокаливание псевдоожиженного слоя, содержащего 30 г Со/0,075 г Pt/3,1 г Mn/100 г Ti-SiO2, при высокой скорости нагревания 106°С в минуту и температуре 210°С (С5123≡С2173 мелкий масштаб, рН=2,3)
Предшественник катализатора был получен, как описано в примере 4, но в меньшем масштабе, то есть использовали 50 г материала носителя в отличие от 10 кг или 15 кг материала носителя. Количества всех других исходных материалов, используемых в процессе получения предшественника катализатора, уменьшены в соответствующем масштабе. Условия прокаливания при высокой скорости нагревания и заключительного выдерживания кратко представлены в таблице 7. Осуществляли непрерывное введение предшественника катализатора при скорости 0,7 г/мин.
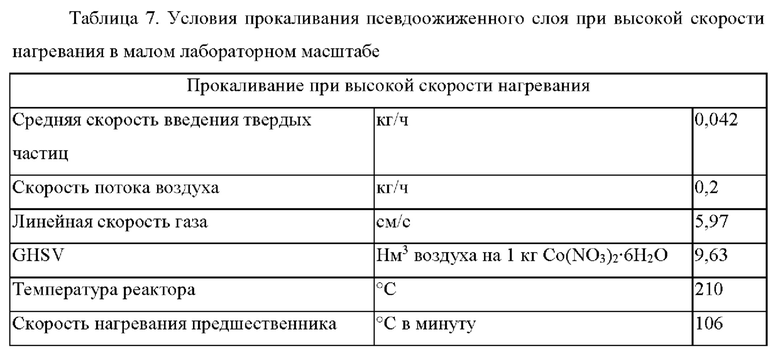

Предшественник катализатора был активирован, как описано в примере 5, и полученный в результате катализатор после этого был исследован в отношении эксплуатационных характеристик FTS своей суспензионной фазы в суспензионном микрореакторе CSTR в условиях, которые описаны в примере 6.
Пример 8 (сравнительный). - Прокаливание псевдоожиженного слоя, содержащего 15 г Со/0,0255 г Pt/3,1 г Mn/100 г Ti-SiO2, при высокой скорости нагревания 15°С в минуту и температуре 120°С (С5128, рН=2,3)
Предшественник катализатора был получен, как описано в примере 7, но без второй стадии введения металла, и температура прокаливания слоя, применяемая для данного слоя, составляла 120°С в отличие от температуры 210°С, которая описана в примере 7. Продолжительность заключительного выдерживания была такой же, как в примере 7 (таблица 7).
Пример 9 (согласно настоящему изобретению). - Прокаливание псевдоожиженного слоя, содержащего 30 г Со/0,075 г Pt/3,1 г Mn/100 г Ti-SiO2, при высокой скорости нагревания 106°С в минуту и температуре 210°С с заключительным выдерживанием при 340°С (С5143, рН=2,3)
Предшественник катализатора, имеющий состав 30 г Со/0,075 г Pt/3,1 г Mn/100 г Ti-SiO2, был получен, как описано в примере 7; однако температура заключительного выдерживания слоя составляла 340°С в отличие от 250°С.
Обсуждение
Из пропитанного носителя катализатора, который прокаливали при высокой скорости нагревания, составляющей 15°С в минуту, но только до 120°С (пример 8) (а не до температуры, составляющей по меньшей мере 165°С), был получен предшественник катализатора, имеющий большие размеры кристаллитов CO3O4 по сравнению с примером 7 и примером 9. Кристаллиты CO3O4, имеющие меньшие размеры, были получены для предшественников катализатора, которые прокаливали при 210°С; таким образом, прокаливание осуществляли в полном температурном диапазоне А (см. пример 7 и пример 9).
Кристаллитов CO3O4, имеющие аналогичные размеры, были получены при температуре заключительного выдерживания 340°С (пример 9) по сравнению с температурой заключительного выдерживания 250°С (пример 7).
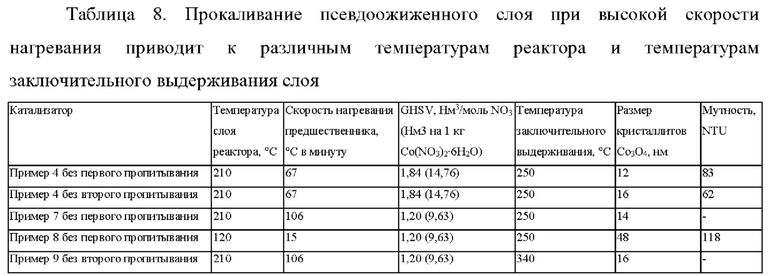
Без первого пропитывания = 15 г Со/0,0255 г Pt/3,1 г Mn/100 г носителя (2,6 г Ti/100 г SiO2)
Без второго пропитывания = 30 г Со/0,075 г Pt/3,1 г Mn/100 г носителя (2,6 г Ti/100 г SiO2)
Пример 10 (согласно настоящему изобретению). - Прокаливание псевдоожиженного слоя, содержащего 30 г Со/0,075 г Pt/2,2 г Mn/100 г Ti-SiO2, при высокой скорости нагревания 106°С в минуту при 210°С (С5166, рН=2,3)
Предшественник катализатора был получен, как описано в примере 7; однако содержание Mn(NO3)2⋅4H2O в течение первой стадии пропитывания было уменьшено с получением предшественника катализатора, имеющего состав 30 г Со/0,075 г Pt/2,2 г Mn/100 г Ti-SiO2. Продолжительность заключительного выдерживания была такой же, как в примере 7 (таблица 7).
Предшественник катализатора был активирован, как описано в примере 5, и полученный в результате катализатор после этого был исследован в отношении эксплуатационных характеристик FTS своей суспензионной фазы в суспензионном микрореакторе CSTR в условиях, которые описаны в примере 6.
Пример 11 (согласно настоящему изобретению). - Прокаливание псевдоожиженного слоя, содержащего 30 г Со/0,075 г Pt/2,2 г Mn/100 г Ti-SiO2, при высокой скорости нагревания 106°С в минуту при 210°С (С5167≡С2178, рН=5)
Предшественник катализатора, имеющий состав 30 г Со/0,075 г Pt/2,2 г Mn/100 г Ti-носитель на основе диоксида кремния (2,6 г Ti/100 г SiO2) был получен, как описано в примере 10. Однако первое пропитывание, за которым следовало 1 г гидроксида кобальта в дополнение к нитрату кобальта, изменило значение рН раствора до 5. Значение рН раствора для второго пропитывания снова устанавливали на уровне 2,3 без добавления гидроксида кобальта.
Предшественник был получен следующим образом. Материал модифицированного носителя на основе диоксида кремния, который описан в примере 1, использовали в качестве материала носителя. Со(NO3)2⋅6H2O (34,57 г) и (NH4)3Pt⋅ (NO3)2 (0,025 г) добавляли в 60 г воды и перемешивали в течение 10 минут при температуре 85°С, чтобы обеспечить растворение нитрата кобальта и соли платины. Значение рН устанавливали на уровне 2,3 с применением разбавленной азотной кислоты. В раствор добавляли Со(ОН)2 (0,79 г) и Mn(NO3)2⋅4H2O (5,02 г). Значение рН мутного раствора превышало 4,5. Никакое дополнительное регулирование рН не осуществляли, и перемешивание раствора продолжали в течение 10 минут при температуре 85°С. В раствор добавляли 50 г модифицированного титаном носителя. Избыточную воду удаляли при пониженном давлении с применением профиль высушивания, который описан в таблице 9, и получали сыпучий порошок. Модифицированный носитель вводили в непрерывном режиме при высокой скорости нагревания в прокалочную печь с псевдоожиженным слоем в условиях, которые описаны в примере 7 (таблица 7). Предшественник катализатора содержал 15 г Со/0,025 г Pt/2,2 г Mn/100 г Ti-SiO2 и 1 г Со из Со(ОН)2.
На второй стадии пропитывания Co(NO3)2⋅6H2O (30,75 г) и (NH4)3Pt(NO3)2 (0,049 г) растворяли в воде (60 г), и значение рН раствора устанавливали на уровне 2,3 с применением разбавленной азотной кислоты. Прокаленный материал после первой стадии пропитывания (50 г) добавляли в раствор и перемешивали в течение 10 минут. Избыточную воду удаляли при пониженном давлении с применением профиля высушивания, который описан в таблице 9, и получали сыпучий порошок, причем потеря массы при прокаливании при 400°С составляла 23,8% (удаление 35,2% кристаллизационной воды из Со(NO3)2⋅6H2O.

Предшественник катализатора был активирован, как описано в примере 5, и полученный в результате катализатор после этого был исследован в отношении эксплуатационных характеристик FTS своей суспензионной фазы в суспензионном микрореакторе CSTR в условиях, которые описаны в примере 6.
Пример 12 (согласно настоящему изобретению). - Прокаливание псевдоожиженного слоя, содержащего 30 г Со/0,075 г Pt/1,6 г Mn/100 г Ti-SiO2, при высокой скорости нагревания 106°С в минуту при 210°С (С5190, рН=5)
Предшественник катализатора был получен, как описано в примере 11; однако содержание Mn(NO3)2⋅4H2O в течение первой стадии пропитывания было уменьшено с получением предшественника катализатора, имеющего состав 30 г Со/0,075 г Pt/1,6 г Mn/100 г Ti-SiO2. Продолжительность заключительного выдерживания была такой же, как в примере 7 (таблица 7).
Предшественник катализатора был активирован, как описано в примере 5, и полученный в результате катализатор после этого был исследован в отношении эксплуатационных характеристик FTS своей суспензионной фазы в суспензионном микрореакторе CSTR в условиях, которые описаны в примере 6.
Пример 13 (сравнительный). - Прокаливание псевдоожиженного слоя, содержащего 30 г Со/0,075 г Pt/2,2 г Mn/100 г Ti-SiO2 (С5301, рН=5), при низкой скорости нагревания, составляющей 1°С в минуту
Предшественник катализатора был получен, как описано в примере 11; однако пропитанный носитель прокаливали, как описано в примере 1; таким образом, прокаливание псевдоожиженного слоя осуществляли при скорости нагревания 1°С в минуту до 250°С с продолжительностью выдерживания, составляющей 6 часов, при этом значение GHSV составляло 2,5 Нм3 воздуха на 1 кг Со(NO3)2⋅6Н2О в час.
Предшественник катализатора был активирован, как описано в примере 5, и полученный в результате катализатор после этого исследован в отношении эксплуатационных характеристик FTS своей суспензионной фазы в суспензионном микрореакторе CSTR в условиях, которые описаны в примере 6.
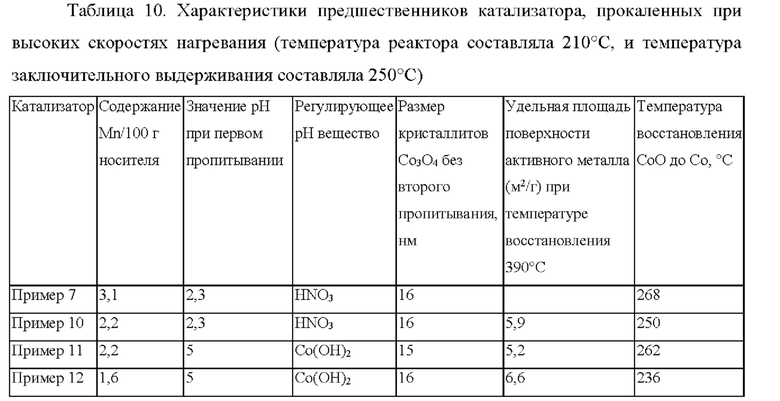
Обсуждение
На основании таблицы 10 площадь поверхности активного металла (MSA) в примере 12 (содержание 1,6 г Mn/100 г Ti-SiO2) является максимальной по сравнению с примером 10 и примером 11, и при этом соответствующая температура восстановления является минимальной с примером 7, примером 10 и примером 11. Активность FTS в примере 10 с применением 2,2 г Mn/100 г носителя оказывается выше по сравнению с применением 3,1 г Mn/100 г носителя в примере 7 (см. фиг. 4). Более высокое значение рН, получаемое при добавлении Со(ОН)2 в течение первой стадии пропитывания (пример 11 и пример 12), снова привело к еще более высокой активности FTS по сравнению с менее высоким значением рН раствора для пропитывания в примере 10 (см. фиг. 4).
Аналогично сравнению прокаливания псевдоожиженного слоя при низкой скорости нагревания в примере 1 с прокаливанием при высокой скорости нагревания в примере 4, размер кристаллитов Со3С4 в случае прокаливании псевдоожиженного слоя предшественника катализатора в примере 13 оказался больше, чем в случае прокаливания псевдоожиженного слоя предшественника катализатора при высокой скорости нагревания в примере 11 (см. таблицу 11), и в результате был получен катализатор FTS с менее высокой активностью (см. фиг. 4) по сравнению с прокаливанием при высокой скорости нагревания.
Даже несмотря на то, что снижение содержания Mn и повышение уровня рН в течение первой стадии пропитывания улучшали эксплуатационные характеристики FTS в примере 13 по сравнению с примером 1, неустойчивость псевдоожижения все же наблюдалась в процессе прокаливания псевдоожиженного слоя при низкой скорости нагревания.
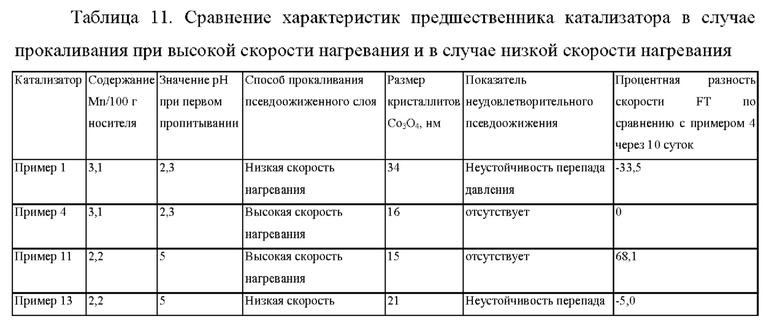

Пример 14 (согласно настоящему изобретению). - Прокаливание псевдоожиженного слоя, содержащего 30 г Со/0,075 г Pt/2,2 г Mn/100 г Ti-SiO2, при высокой скорости нагревания 138°С в минуту и температуре 250°С без периода заключительного выдерживания, но с выдерживанием в течение 20 минут в результате загрузки материала (С5315, рН=5)
Предшественник катализатора был получен, как описано в примере 11; однако температура прокаливания слоя, применяемого к данному слою, составляла 250°С. Предшественник катализатора удаляли из прокалочной печи немедленно после полной модификации всего материала.
Обсуждение
Небольшой пик NO2 на профиле NO2 в случае нагревания слоя при температуре от 210°С до 250°С на фиг. 2 показывает, что прокаливание при высокой скорости нагревания в условиях псевдоожижения не приводит к полному удалению следов NO2. На основании анализа элементов CHNS и содержания фазы CO3O4 методом рентгеновской дифракции показано, что продолжительность заключительного выдерживания более 20 минут требуется для полного удаления указанных остаточных соединений азота (см. таблицу 12).
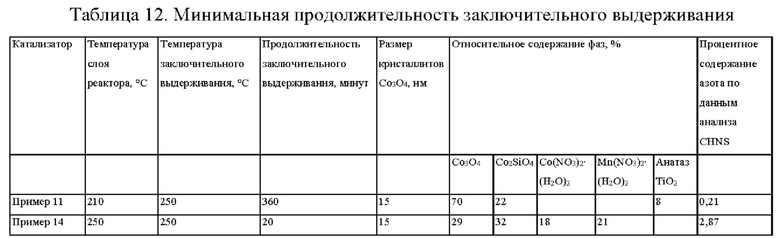
Пример 15 (согласно настоящему изобретению). - Прокаливание псевдоожиженного слоя, содержащего 30 г Со/0,075 г Pt/2,2 г Mn/100 г Ti-SiO2, при высокой скорости нагревания 68°С в минуту и температуре 210°С (С2179, рН=5)
Предшественник катализатора был получен, как описано в примере 11, но в большем масштабе, то есть использовали 15 кг материала модифицированного титаном носителя катализатора, который описан в примере 1, в отличие от 50 г материала носителя. Количества всех других исходных материалов, используемых в процессе получения предшественник катализатора, были увеличены в соответствующем масштабе. Избыточную воду удаляли при пониженном давлении в конусной сушилке с применением профиля высушивания, который описан в таблице 2, и получали сыпучий порошок. Удаляли 44% кристаллизационной воды Со(NO3)2⋅6Н2О. Немодифицированный высушенный пропитанный материал носителя разделяли на небольшие порции для прокаливания на экспериментальной установке при температуре 210°С, как описано в примере 4 (таблица 5). Продолжительность заключительного выдерживания была такой же, как в примере 4 (таблица 5).
Пример 16 (согласно настоящему изобретению). - Прокаливание псевдоожиженного слоя, содержащего 15 г Со/0,025 г Pt/2,2 г Mn/100 г Ti-SiO2, при высокой скорости нагревания 48°С в минуту и температуре 180°С (С2179, рН=5)
Предшественник катализатора был получен, как описано в примере 15, но без второй стадии введения металла, и температура прокаливания реактора, примененная для данного слоя после первой стадии пропитывания, составляла 180°С в отличие от температуры 210°С, которая описана в примере 15.
Пример 17 (согласно настоящему изобретению). - Прокаливание псевдоожиженного слоя, содержащего 15 г Со/0,025 г Pt/2,2 г Mn/100 г Ti-SiO2, при высокой скорости нагревания 144°С в минуту и температуре 250°С (С2179, рН=5)
Предшественник катализатора был получен, как описано в примере 15, но без второй стадии введения металла, и температура прокаливания реактора, примененная для данного слоя после первой стадии пропитывания, составляла 250°С в отличие от температуры 210°С, которая описана в примере 15.
Пример 18 (сравнительный). - Прокаливание псевдоожиженного слоя, содержащего 15 г Со/0,025 г Pt/2,2 г Mn/100 г Ti-SiO2, при высокой скорости нагревания 30°С в минуту и температуре 130°С (С5517 мелкий масштаб, эксперимент DI092, рН=5)
Предшественник катализатора был получен, как описано в примере 11, но без второй стадии введения металла, и температура прокаливания реактора, примененная для данного слоя после первой стадии пропитывания, составляла 130°С перед периодом заключительного выдерживания в отличие от температуры 210°С, которая описана в примере 11. Продолжительность заключительного выдерживания была такой же, как в примере 7 (таблица 7).
Пример 19 (сравнительный). - Прокаливание псевдоожиженного слоя, содержащего 15 г Со/0,025 г Pt/2,2 г Mn/100 г Ti-SiO2, при высокой скорости нагревания 38°С в минуту и температуре 140°С (С5446 мелкий масштаб, эксперимент DG093, рН=5)
Предшественник катализатора был получен, как описано в примере 11, но без второй стадии введения металла, и температура прокаливания реактора, примененная для данного слоя после первой стадии пропитывания, составляла 140°С перед периодом заключительного выдерживания в отличие от температуры 210°С, которая описана в примере 11. Продолжительность заключительного выдерживания была такой же, как в примере 7 (таблица 7).
Пример 20 (сравнительный). - Прокаливание псевдоожиженного слоя, содержащего 15 г Со/0,025 г Pt/2,2 г Mn/100 г Ti-SiO2, при высокой скорости нагревания 48°С в минуту и температуре 150°С (С5491 мелкий масштаб, эксперимент DD079, рН=5)
Предшественник катализатора был получен, как описано в примере 11, но без второй стадии введения металла, и температура прокаливания реактора, примененная для данного слоя после первой стадии пропитывания, составляла 150°С перед периодом заключительного выдерживания в отличие от температуры 210°С, которая описана в примере 11. Продолжительность заключительного выдерживания была такой же, как в примере 7 (таблица 7).
Пример 21 (сравнительный). - Прокаливание псевдоожиженного слоя, содержащего 15 г Со/0,025 г Pt/2,2 г Mn/100 г Ti-SiO2, при высокой скорости нагревания 53°С в минуту и температуре 160°С (С5506 мелкий масштаб, эксперимент DI091, рН=5)
Предшественник катализатора был получен, как описано в примере 11, но без второй стадии введения металла, и температура прокаливания реактора, примененная для данного слоя после первой стадии пропитывания, составляла 160°С перед периодом заключительного выдерживания в отличие от температуры 210°С, которая описана в примере 11. Продолжительность заключительного выдерживания была такой же, как в примере 7 (таблица 7).
Пример 22 (согласно настоящему изобретению). - Прокаливание псевдоожиженного слоя, содержащего 15 г Со/0,025 г Pt/2,2 г Mn/100 г Ti-SiO2, при высокой скорости нагревания 66°С в минуту и температуре 170°С (С5503 мелкий масштаб, эксперимент DC088, рН=5)
Предшественник катализатора был получен, как описано в примере 11, но без второй стадии введения металла, и температура прокаливания реактора, примененная для данного слоя после первой стадии пропитывания, составляла 170°С перед периодом заключительного выдерживания в отличие от температуры 210°С, которая описана в примере 11. Продолжительность заключительного выдерживания была такой же, как в примере 7 (таблица 7).
Пример 23А. - Активация катализатора и синтез Фишера-Тропша (FTS)
Образцы прокаленных предшественников катализаторов в примерах 16-22 были восстановлены после первой стадии пропитывания в потоке водорода при значении GHSV, составляющем 2,0 Нм3 в час на 1 кг прокаленного катализатора и при скорости нагревания 1°С в минуту до 370°С с последующим выдерживанием в течение 7 часов. Восстановленный катализатор охлаждали, погружали в расплавленный воск и подвергали модификации в реакторе, представляющем собой резервуар с непрерывным перемешиванием (CSTR), в атмосфере инертного газа для оценки соответствующих эксплуатационных характеристики в синтезе Фишера-Тропша.
Эксплуатационные характеристики FTS активированных и защищенных воском катализаторов, которые описаны выше, оценивали, используя лабораторный суспензионный микрореактор CSTR при температуре реактора 230°С, давлении реактора, составляющем приблизительно 20 бар, и полном молярном соотношении исходных H2 и СО, составляющем приблизительно 2/1. В реакторе использовали электрическое нагревательное устройство и достаточно высокую скорость перемешивания для устранения любых ограничений в отношении массопереноса между газом и жидкостью. Объемную скорость исходного газа изменяли таким образом, что степень превращения синтетического газа составляла приблизительно 80%. Парциальное давление водяного пара составляло приблизительно 8 бар.
Обсуждение
Результаты определения мутности показывают, что меньше сверхтонких частиц образуется при увеличении температура прокаливания реактора, причем минимальные показания мутности наблюдаются при температурах реактора прокаливания, составляющих 210°С (пример 15) и 250°С (пример 17) на фиг. 5.
Из пропитанного носителя катализатора в примере 18, который прокаливали при высокой скорости нагревания до температуры 130°С, были получены предшественники катализаторов, имеющие очень большие размеры кристаллитов Co3O4 (см. таблицу 13) по сравнению со случаем прокаливания при более высокой температуре в примере 22. Активность FTS также оказалась значительно ниже для катализатора в примере 18 (см. фиг. 6). Наиболее высокие значения активности FTS были получены для катализаторов, которые прокаливали при высоких скоростях нагревания до температур, превышающих 160°С (см. фиг. 6).
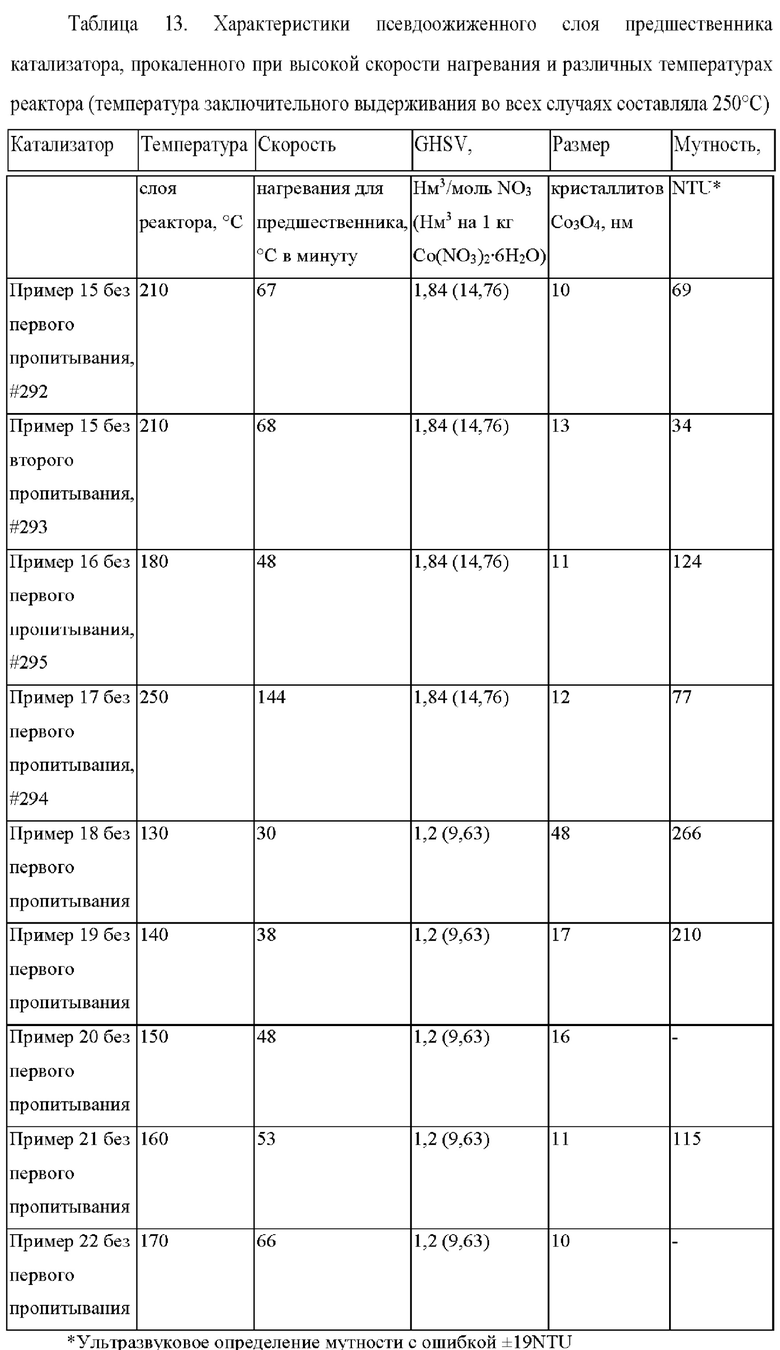
Пример 23 (согласно настоящему изобретению). - Прокаливание при низкой скорости нагревания в условиях псевдоожижения 15 г Со/0,025 г Pt/2,2 г Mn/100 г Ti-SiO2 (С5372, рН=5) до 80°С при скорости нагревания 1°С в минуту и последующее прокаливание при высокой скорости нагревания 138°С в минуту и температуре 250°С
Сыпучий порошок высушенного пропитанного носителя, который был получен согласно описанию в примере 11, прокаливали при низкой скорости нагревания, составляющей 1°С в минуту до температуры 80°С с применением значения GHSV, составляющего 2,5 Нм3 воздуха на 1 кг Со(NO3)2⋅6Н2О в час, с последующим прокаливанием е при высокой скорости нагревания и температуре 250°С с применением значения GHSV, которое представлено в таблице 7.
Пример 24 (согласно настоящему изобретению). - Прокаливание при низкой скорости нагревания в условиях псевдоожижения 15 г Со/0,025 г Pt/2,2 г Mn/100 г Ti-SiO2 (С5377, рН=5) до 90°С при скорости нагревания 1°С в минуту и последующее прокаливание при высокой скорости нагревания 138°С в минуту и температуре 250°С
Сыпучий порошок высушенного пропитанного носителя, который был получен согласно описанию в примере 11, прокаливали при низкой скорости нагревания, составляющей 1°С в минуту до температуры 90°С с применением значения GHSV, составляющего 2,5 Нм3 воздуха на 1 кг Со(NO3)2⋅6Н2О в час, с последующим прокаливанием е при высокой скорости нагревания и температуре 250°С с применением значения GHSV, которое представлено в таблице 7.
Пример 25 (согласно настоящему изобретению). - Прокаливание при низкой скорости нагревания в условиях псевдоожижения 15 г Со/0,025 г Pt/2,2 г Mn/100 г Ti-SiO2 (С5378, рН=5) до 100°С при скорости нагревания 1°С в минуту и последующее прокаливание при высокой скорости нагревания 138°С в минуту и температуре 250°С
Сыпучий порошок высушенного пропитанного носителя, который был получен согласно описанию в примере 11, прокаливали при низкой скорости нагревания, составляющей 1°С в минуту до температуры 100°С с применением значения GHSV, составляющего 2,5 Нм3 воздуха на 1 кг Со(NO3)2⋅6Н2О в час, с последующим прокаливанием е при высокой скорости нагревания и температуре 250°С.
Пример 26 (согласно настоящему изобретению). - Прокаливание псевдоожиженного слоя, содержащего 15 г Со/0,025 г Pt/2,2 г Mn/100 г Ti-SiO2, при высокой скорости нагревания 138°С в минуту и температуре 250°С (С5390, рН=5)
Предшественник катализатора был получен, как описано в примере 17, но в меньшем масштабе, то есть использовали 50 г материала носителя в отличие от 10 кг материала носителя. Количества всех других исходных материалов, используемых в процессе получения предшественника катализатора, были уменьшены в соответствующем масштабе.
Обсуждение
Некоторые отрицательные эффекты начинали возникать при прокаливании пропитанного носителя катализатора при низкой скорость нагревания и низкой температуре, составляющей 90°С (пример 24). Они представляли собой ухудшение распределения Со, которое может быть вызвано миграцией Со к периферии частицы (см. таблицу 14). В случае предшественника катализатора в примере 25, который прокаливали при низкой скорости нагревания, составляющей 1°С в минуту до температуры 100°С в псевдоожиженном слое, также образовывались в большем количестве сверхтонкие частицы в течение ультразвуковой обработки, в результате чего показатели мутности повысились по сравнению с примером 26 (см. таблицу 14).
Количественная энергодисперсионная спектроскопия (ЭДС) была применена для определения нормированного массового процентного содержания в различных областях частицы в целях представления и сопоставления распределения и миграции кобальта в примере 23 и примере 26 в численной форме. После этого было определено массовое процентное соотношение Со и Si на периферии частицы (5 микрон от поверхности частицы) и в центре частицы.
Поскольку на периферии частицы в примере 24 и примере 25 было обнаружено более высокое нормированное массовое процентное соотношение Со и Si, чем в центре частицы, это количественно показывает, что присутствует неравномерное распределение по сравнению с примером 23 и примером 26. Равномерное распределение кобальта было получено в примере 23 и примере 26, где нормированные массовые процентные соотношения Со и Si являются одинаковыми на периферии частицы и в центре частицы (см. таблицу 14).

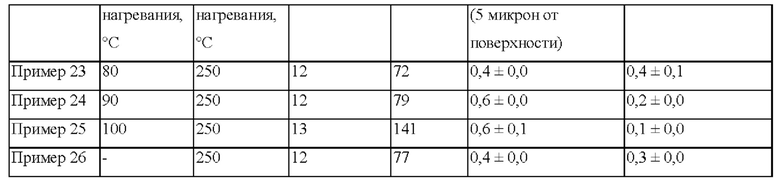
Пример 27. - Прокаливание псевдоожиженного слоя, содержащего 30 г Со/0,075 г Pt/2,2 г Mn/100 г Ti-SiO2, при высокой скорости нагревания 68°С в минуту и температуре 210°С (С2177, рН=5) с удалением лишь 16% кристаллизационной воды Со(NO3)2⋅6Н2О
Предшественник катализатора был получен, как описано в примере 15; однако избыточную воду удаляли при пониженном давлении в сушилке Lodige, а не в конусной сушилке, с применение профиля высушивания, представленного в таблице 15, и получали сыпучий порошок. Было удалено лишь 16% кристаллизационной воды Со(NO3)2⋅6Н2О, и конечная потеря массы при прокаливании при 400°С (LOL400) составляла 25,9%.
Предшественник катализатора был активирован, как описано в примере 5, и после этого исследован в отношении эксплуатационных характеристик FTS своей суспензионной фазы в суспензионном микрореакторе CSTR в условиях, которые описаны в примере 6.
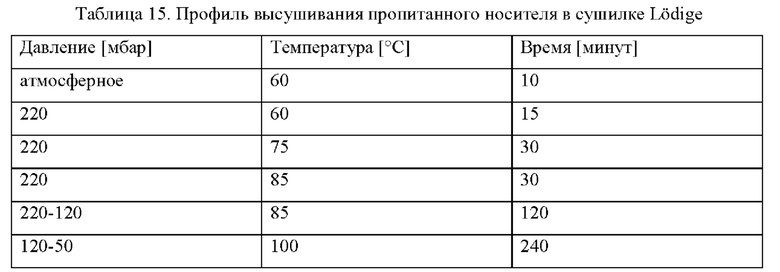
Обсуждение
Размер кристаллитов Co3O4 предшественника катализатора, который был получен в примере 27 с конечным значением LOI 25,9%, был больше, чем размер кристаллитов, полученных в примере 15 с конечным значением LOI 22,8%. Следовательно, активность в синтезе FT также была ниже по сравнению с примером 15 (см. таблицу 16).

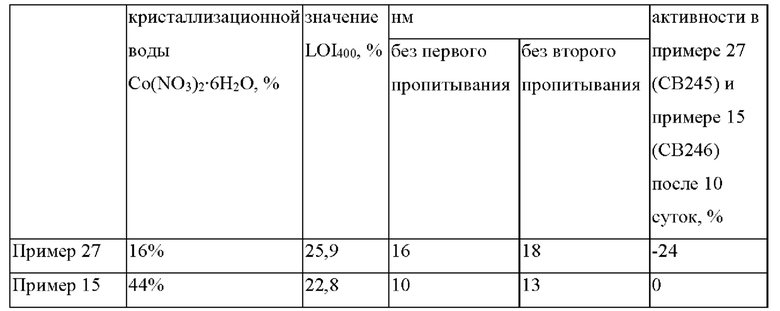
Пример 28 (сравнительный). - Прокаливание неподвижного слоя, содержащего 30 г Со/0,075 г Pt/2,2 г Mn/100 г Ti-SiO2 (С5253)
Предшественник катализатора был получен, как описано в примере 11; однако пропитанный носитель катализатора прокаливали в неподвижном слой при скорости нагревания 0,5°С в минуту в потоке (0,5 мл/с) гелия, содержащего 12% кислорода, до температуры 150°С, 175°С и 250°С.
Продукты разложения Со(NO3)2⋅6Н2О были исследованы методом инфракрасной спектроскопии с преобразованием Фурье с анализом газовой фазы.
Пример 29 (сравнительный). - Прокаливание неподвижного слоя, содержащего 30 г Со/0,075 г Pt/2,5 г МАс/100 г модифицированного Ti/Mn SiO2 (2,6 г Ti/3,1 г Mn/100 г SiO2) (С5252)
Предшественник катализатора был получен, как описано в примере 3; однако пропитанный носитель катализатора прокаливали в неподвижном слой при скорости нагревания 0,5°С в минуту в потоке (0,5 мл/с) гелия, содержащего 12% кислорода, до температуры 130°С, 175°С и 250°С.
Продукты разложения Со(NO3)2⋅6Н2О были исследованы методом инфракрасной спектроскопии с преобразованием Фурье с анализом газовой фазы.
Обсуждение
Профили ТРО Н2О, NO2 и HNO3 (газ) в течение линейного нагревание при скорости 0,5°С в минуту в примере 28 и примере 29 представлены на фиг. 7 и фиг. 8. На фиг. 7 видно, что вода высвобождается при 70°С и 105°С, в то время как NO2 и HNO3 (газ) высвобождаются при 140°С и 175°С. Температура высвобождения NO2 при содержании NO2 1500 объемных частей на миллион (0,15 об. %) составляет 115°С (см. фиг. 7). В отличие от примера 28, добавление органического модификатора в примере 29 привело к сдвигу указанной температуры высвобождения NO2 к 125°С с пиком NO2 при 155°С.
Пример 30. - Прокаливание псевдоожиженного слоя, содержащего 30 г Со/0,075 г Pt/2,2 г Mn/100 г Ti-SiO2, при высокой скорости нагревания 233°С в минуту и температуре 350°С (С5389)
Предшественник катализатора был получен, как описано в примере 11; однако температура прокаливания реактора, примененная к слою после первой и второй стадий пропитывания, составляла 350°С.
Предшественник катализатора был активирован, как описано в примере 5, и полученный в результате катализатор после этого был исследован в отношении эксплуатационных характеристик FTS своей суспензионной фазы в суспензионном микрореакторе CSTR в условиях, которые описаны в примере 6.
Обсуждение
Катализатор, который прокаливали при высокой скорости нагревания и температуре 350°С, проявил ухудшенные эксплуатационные характеристики FTS (см. фиг. 4). Кроме того, наблюдалось повышенное относительное процентное содержание фазы неактивного Co2SiO4 (см. таблицу 17).

| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ КОБАЛЬТСОДЕРЖАЩЕГО КАТАЛИЗАТОРА СИНТЕЗА ФИШЕРА-ТРОПША | 2012 |
|
RU2602803C2 |
| КАТАЛИЗАТОРЫ | 2012 |
|
RU2592271C2 |
| КАТАЛИЗАТОРЫ | 2012 |
|
RU2591702C2 |
| СПОСОБ ПОЛУЧЕНИЯ КАТАЛИЗАТОРА СИНТЕЗА ФИШЕРА-ТРОПША | 2009 |
|
RU2481156C2 |
| КАТАЛИЗАТОРЫ | 2011 |
|
RU2551433C1 |
| КАТАЛИЗАТОРЫ | 2012 |
|
RU2603136C2 |
| КАТАЛИЗАТОРЫ | 2012 |
|
RU2584915C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРЕДШЕСТВЕННИКА КАТАЛИЗАТОРА И КАТАЛИЗАТОРА ФИШЕРА-ТРОПША НА ОСНОВЕ КОБАЛЬТА | 2001 |
|
RU2298434C2 |
| КОБАЛЬТОВЫЕ КАТАЛИЗАТОРЫ | 2000 |
|
RU2252072C2 |
| КАТАЛИЗАТОРЫ НА ОСНОВЕ КОБАЛЬТА | 2001 |
|
RU2261143C2 |
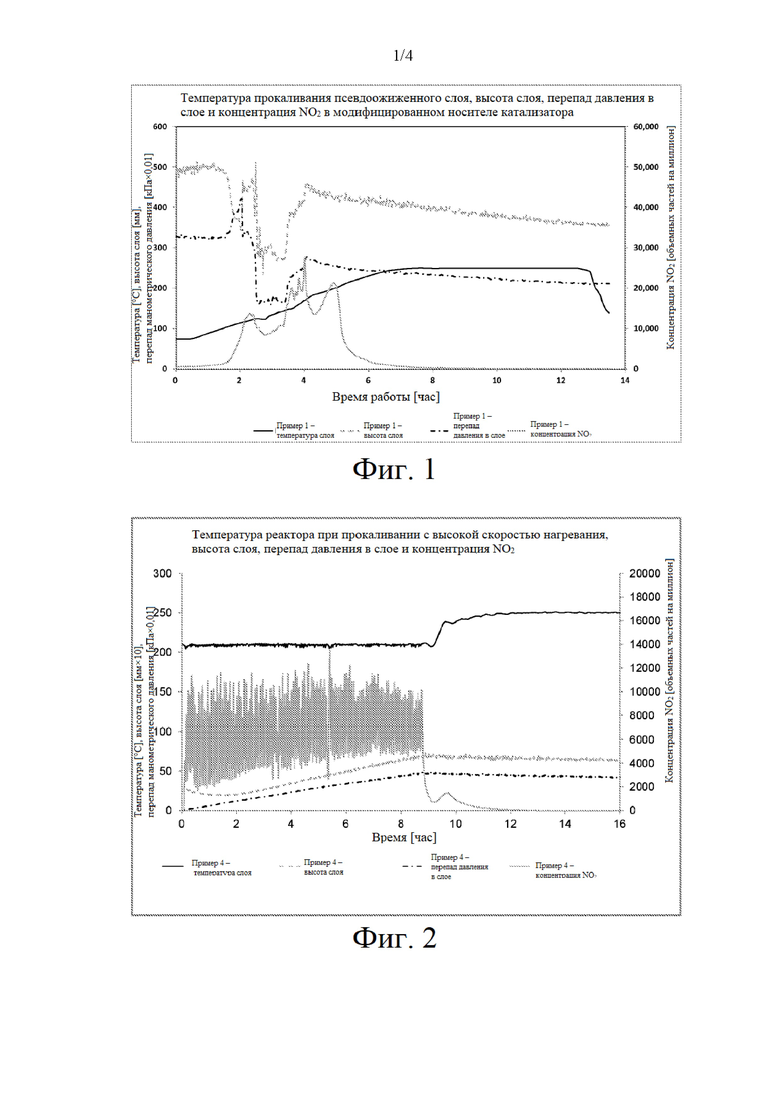
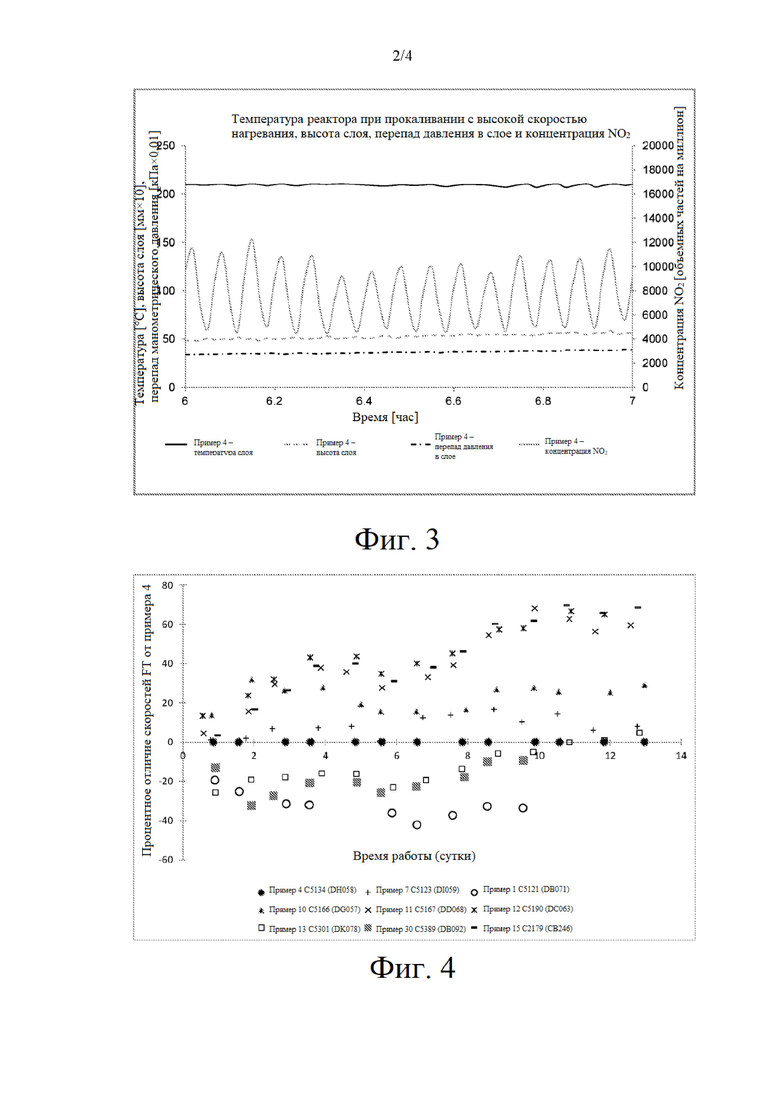
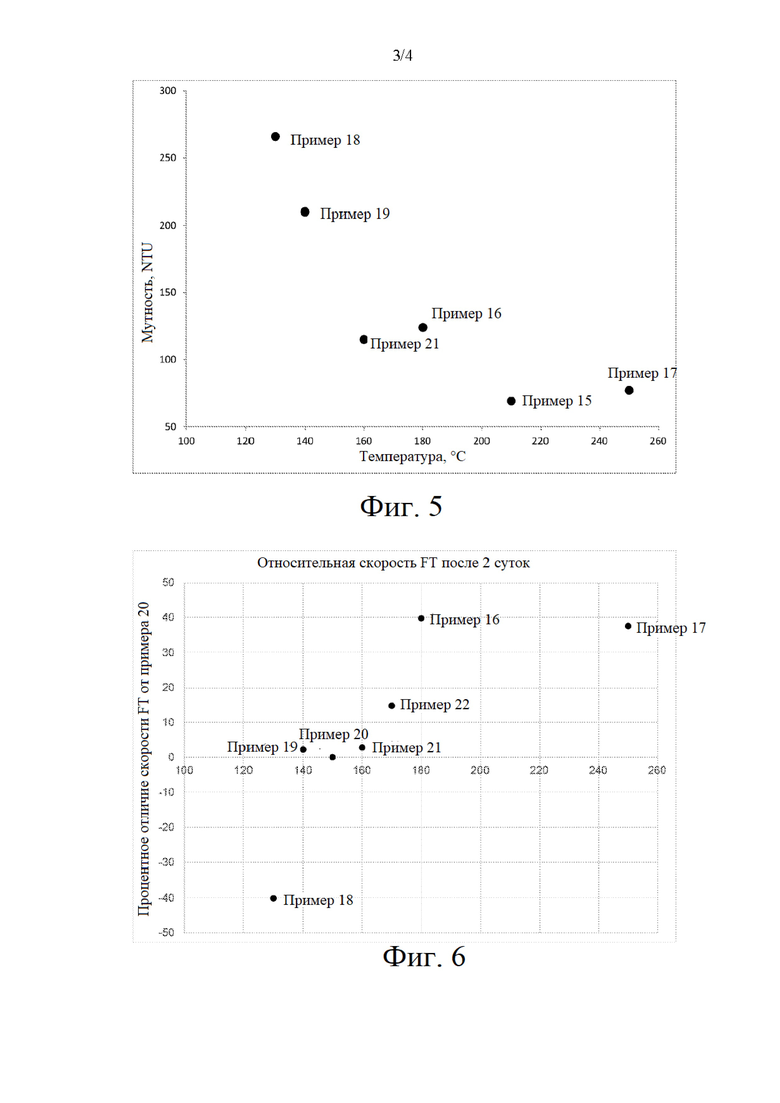
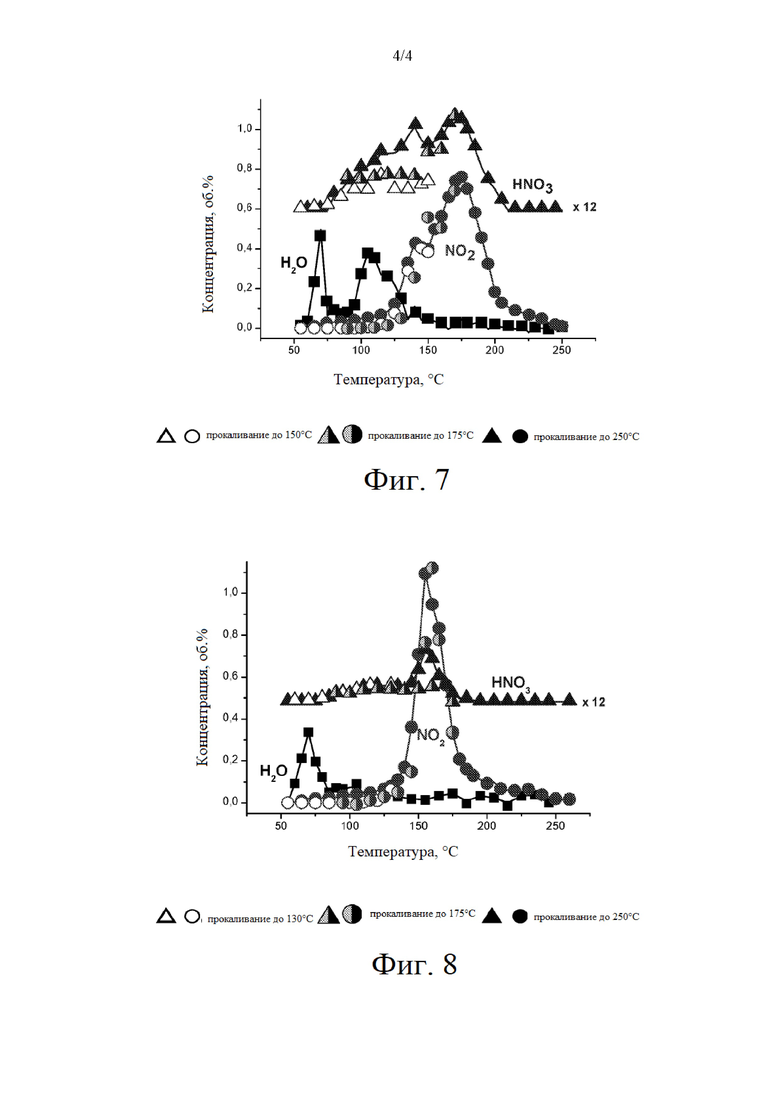
Настоящее изобретение относится к катализаторам, в частности к способу получения предшественника кобальтсодержащего катализатора, который является предшественником катализатора синтеза углеводородов. Раскрывается способ получения предшественника кобальтсодержащего катализатора, включающий прокаливание модифицированного носителя катализатора на основе диоксида кремния, содержащего нитрат кобальта. При прокаливании модифицированного носителя катализатора происходит превращение нитрата кобальта в оксид кобальта. Прокаливание представляет собой нагревание модифицированного носителя катализатора при высокой скорости нагревания, которая составляет не менее чем 10°C в минуту, в пределах по меньшей мере температурного диапазона A, который составляет от минимальной температуры, при которой начинается прокаливание модифицированного носителя катализатора, до 165°C. Газовый поток пропускают над модифицированным носителем катализатора в пределах по меньшей мере температурного диапазона A при часовой объемной скорости газа (GHSV), составляющей по меньшей мере 5 Нм3 на 1 кг нитрата кобальта в час. При этом минимальная температура, при которой начинается прокаливание модифицированного носителя катализатора, представляет собой температуру, при которой нитрат кобальта начинает разлагаться с высвобождением газообразного NO2 в количестве, превышающем 1500 объемных частей на миллион. Измерение осуществляется методом инфракрасной спектроскопии с преобразованием Фурье с анализом газовой фазы при скорости нагревания, составляющей 0,5°C в минуту, в газовой смеси на основе He, содержащей 12% O2, при скорости газового потока, составляющей 0,5 мл/с. Также раскрывается способ получения катализатора, а также способы синтеза углеводородов, которые включают применение указанного катализатора. Технический результат заявленного изобретения состоит в повышении каталитической активности и снижении степени разрушения катализатора. 4 н. и 10 з.п. ф-лы, 8 ил., 17 табл., 30 пр.
1. Способ получения предшественника кобальтсодержащего катализатора, причем способ включает прокаливание модифицированного носителя катализатора, представляющего собой носитель катализатора на основе диоксида кремния (SiO2), содержащий нитрат кобальта, причем при прокаливании модифицированного носителя катализатора происходит превращение нитрата кобальта в оксид кобальта; и при этом
прокаливание представляет собой нагревание модифицированного носителя катализатора при высокой скорости нагревания, которая составляет не менее чем 10°C в минуту, в пределах по меньшей мере температурного диапазона A, который составляет от минимальной температуры, при которой начинается прокаливание модифицированного носителя катализатора, до 165°C, и при этом газовый поток пропускают над модифицированным носителем катализатора в пределах по меньшей мере температурного диапазона A при часовой объемной скорости газа (GHSV), составляющей по меньшей мере 5 Нм3 на 1 кг нитрата кобальта в час, причем минимальная температура, при которой начинается прокаливание модифицированного носителя катализатора, представляет собой температуру, при которой нитрат кобальта начинает разлагаться с высвобождением газообразного NO2 в количестве, превышающем 1500 объемных частей на миллион, при измерении методом инфракрасной спектроскопии с преобразованием Фурье с анализом газовой фазы при скорости нагревания, составляющей 0,5°C в минуту, в газовой смеси на основе He, содержащей 12% O2, при скорости газового потока, составляющей 0,5 мл/с, в результате чего получается предшественник кобальтсодержащего катализатора.
2. Способ по п. 1, в котором носитель катализатора на основе диоксида кремния (SiO2) является пористым и имеет средний диаметр пор, составляющий более чем 20 нм, но менее чем 50 нм, причем средний диаметр пор определяется посредством анализа физической адсорбции азота методом Баррета-Джойнера-Халенды (BJH).
3. Способ по п. 1, в котором модифицированный носитель катализатора одновременно содержит соединение титана на поверхности и/или в объеме носителя катализатора и соединение марганца на поверхности и/или в объеме носителя катализатора.
4. Способ по п. 1, в котором модифицированный носитель катализатора содержит гидроксид кобальта (Co(OH)2) в дополнение к нитрату кобальта.
5. Способ по п. 1, в котором модифицированный носитель катализатора содержит модифицирующий элемент, способный повышать восстанавливаемость нитрата кобальта после его прокаливания, причем модифицирующий элемент присутствует в форме соединения модифицирующего элемента, которое представляет собой соединение металла, выбранного из группы, которую составляют палладий (Pd), платина (Pt), рутений (Ru), рений (Re) и смесь двух или большего числа этих металлов.
6. Способ по п. 1, в котором прокаливание представляет собой нагревание модифицированного носителя катализатора до температуры выше температурного диапазона A.
7. Способ по п. 1, в котором прокаливание представляет собой нагревание модифицированного носителя катализатора при высокой скорости нагревания, которая составляет не менее чем 10°C в минуту, в пределах по меньшей мере температурного диапазона, который составляет от 100 до 170°C.
8. Способ по п. 7, в котором прокаливание представляет собой нагревание модифицированного носителя катализатора при высокой скорости нагревания, которая составляет не менее чем 10°C в минуту, в пределах по меньшей мере температурного диапазона, который составляет от 100 до 220°C.
9. Способ по п. 1, в котором газовый поток, который пропускают над модифицированным носителем катализатора в пределах температурного диапазона A, характеризуется часовой объемной скоростью газа (GHSV), составляющей по меньшей мере 9 Нм3 на 1 кг нитрата кобальта в час.
10. Способ по п. 1, в котором прокаливание осуществляется в прокалочной печи с псевдоожиженным слоем.
11. Способ по п. 1, который включает высушивание модифицированного носителя катализатора перед прокаливанием модифицированного носителя катализатора при высокой скорости нагревания в пределах температурного диапазона A.
12. Способ получения кобальтсодержащего катализатора, причем способ включает получение предшественника кобальтсодержащего катализатора по п. 1; и восстановление предшественника катализатора, в результате чего активируется предшественник катализатора, и получается катализатор.
13. Способ синтеза углеводородов для получения углеводородов и необязательно кислородсодержащих производных углеводородов, причем способ включает получение кобальтсодержащего катализатора способом по п. 12; и при этом способ также включает введение указанного катализатора в контакт с водородом и монооксидом углерода при температуре выше 100°C и при давлении, составляющем по меньшей мере 10 бар, для получения углеводородов и необязательно кислородсодержащих производных углеводородов.
14. Способ синтеза углеводородов, который включает получение кобальтсодержащего катализатора способом по п. 12; и при этом способ также включает введение указанного катализатора в контакт с водородом и монооксидом углерода при температуре выше 100°C и при давлении, составляющем по меньшей мере 10 бар, для получения углеводородов и необязательно кислородсодержащих производных углеводородов; и способ также включает стадию гидропереработки для превращения углеводородов и необязательно соответствующих кислородсодержащих производных в жидкие горючие материалы и/или другие химические продукты.
| КАТАЛИЗАТОРЫ | 2012 |
|
RU2592271C2 |
| СПОСОБ ПРОИЗВОДСТВА КОНСЕРВИРОВАННОГО НАПИТКА "АРОМАТНЫЙ" | 2004 |
|
RU2276563C2 |
| WO 2018029548 A1, 15.02.2018 | |||
| КОБАЛЬТОВЫЕ КАТАЛИЗАТОРЫ | 2000 |
|
RU2252072C2 |
| КАТАЛИЗАТОРЫ | 2012 |
|
RU2591702C2 |
| US 20180280939 A1, 04.10.2018 | |||
| Van De Loosdrecht et.al | |||
| "Calcination of Co-based Fischer-Tropsch synthesis catalysts", Topics in Catalysis, vol | |||
| Прибор для получения стереоскопических впечатлений от двух изображений различного масштаба | 1917 |
|
SU26A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |

