Изобретение относится к аналитической химии.
Глицирризиновая кислота (20β-карбокси-11-оксо-30-норолеан-12-ен-3β-ил-2-O-β-D-глюкопиранфуранозил-альфа-D-глюко-пиранозид-уроновая кислота) относится к стероидным сапонинам. Она состоит из двух молекул глюкуроновой кислоты, и ее строение сходно со строением молекул гормонов, вырабатываемых корой надпочечников (кортизон и др.). Это дает возможность применять глицирризиновую кислоту в заместительной терапии гормонами и существенно снижать их дозу (например, дозу кортизона).
Глицирризиновая кислота проявляет также антиаллергическое, противовоспалительное действие. Поэтому ее применяют при лечении различного рода аллергических заболеваний, а также артрита.
Глицирризиновая кислота стимулирует выработку интерферона, обладающего антивирусной активностью; подавляет рост клеток Герпеса. Глицирризин защищает клетки печени от химических и иммунологических агентов.
Количественное определение глицирризиновой кислоты является актуальным при оценке безопасности лекарственных препаратов.
Сведения по количественному определению микроколичеств глицирризиновой кислоты вольтамперометрическим методом отсутствуют.
Известны следующие физико-химические методы количественного определения глицирризиновой кислоты в лекарственном растительном сырье (ЛРС). Глицирризиновую кислоту определяют в основном в корнях солодки и полученных из них экстрактах, где она является основным действующим веществом.
1. Спектрофотометрическое определение глицирризиновой кислоты в корне солодки (Radix Glycyrrhizae). Метод основан на осаждении глицирризиновой кислоты из ацетонового извлечения 25% раствором аммиака с последующей спектрофотометрией. Определяют оптическую плотность полученного раствора на спектрофотометре при длине волны 258 нм в кювете с толщиной слоя 1 см, применяя в качестве контрольного раствора воду. Нормативная документация на сырье солодки регламентирует нижнюю границу содержания глицирризиновой кислоты на уровне 6%. Это может быть связано как с неполнотой извлечения, так и с недостаточной очисткой выделенной глицирризиновой кислоты [Государственная фармакопея X (ГФХ), ст. 573, ст. 260].
Количественному определению глицирризиновой кислоты данным методом предшествует сложная пробоподготовка с использованием токсичных реактивов, требующая значительных затрат времени.
2. Высокоэффективная жидкостная хроматография (ВЭЖХ).
a. В основе метода лежит определение глицирризиновой кислоты на колонке VarianPolarisRPC18-A длиной 250 мм. В качестве подвижной фазы применяли смесь раствора фосфорной кислоты (1%) и ацетонитрила (60:40), детектирование осуществляли при 254 нм. Линейность находилась в интервале 14 - 558 мкг/мл, открываемость составила 98,43% (Государственная фармакопея СССР. XI издание. - М.: Медицина, 1989. - Вып. II. Лекарственное растительное сырье. - 400 с.).
b. Отечественные исследователи для количественного определения глицирризиновой кислоты методом ВЭЖХ использовали систему растворителей: метанол - ледяная уксусная кислота - вода (60:5:35). Исследования проводили на жидкостном хроматографе Стайер фирмы Аквилон (Россия - США - Чехия), снабженном колонкой Luna С-18 4,6150 мм (Phenomenex, США), с содержанием углерода около 16%. В качестве подвижной фазы использовали смесь ацетонитрила и раствора муравьиной кислоты (20 г/л) в соотношении (40:60) при расходе элюента 1 мл/мин. Детектирование осуществляли при 257 нм, объем вводимой пробы составлял 20 мкл (Государственная фармакопея СССР. XI издание. - М.: Медицина, 1987. - Вып. I. Общие методы анализа. - 337 с.; Столярова О.В., Балтина Л.А., Михайлова Л.Р. и др. Оптимизация метода получения моноаммонийной соли глицирризиновой кислоты из корней солодки уральской (Glycyrrhiza uralensis Fisch.) сибирских популяций // Химия: в интересах устойчивого развития. 2008. Т. 16. №5. С. 571-576).
Данный метод также включает использование токсичных реактивов, характеризуется длительностью осуществления и трудоемкостью. Он требует применения дорогостоящего оборудования и реактивов.
3. Метод капиллярного зонного электрофореза на немодифицированном кварцевом капилляре. Работу проводили с использованием системы капиллярного электрофореза Капель 103 Р (НПФ Люмэкс, Санкт-Петербург) с кварцевым капилляром диаметром 50 мкм, общей длиной 75 см и эффективной длиной 65 см. Детектирование осуществляли спектрофотометрически при 254 нм в катодной области капилляра. Работу проводили при комнатной температуре. В качестве ведущего электролита использовали раствор декагидрататетрабората натрия, применение которого для анализа слабых кислот можно считать оптимальным. Описано использование капиллярного электрофореза в боратном электролите с добавлением натрия додецилсульфата и тетрабутиламмония бромида. Первоначально анализировали раствор моноаммонийной соли глицирризиновой кислоты (глицирам) фармакопейной чистоты. В условиях капиллярного зонного электрофореза в боратном электролите (10 мг/мл) данное вещество фиксируется в виде одиночного неоднородного пика. Добавление натрия додецилсульфата характера пика не изменяет. Модификация ведущего электролита циклодекстрином в количестве 1 мМ существенно изменила профиль электрофореграммы, и, кроме основного, наблюдали еще два достаточно заметных пика. Это свидетельствует о том, что модификация электролита позволила отделить очень близкие по структуре вещества. Для вычисления содержания глицирризиновой кислоты в сырье был использован градуировочный график (Гаврилин М.В. Совершенствование способов оценки качества корней и сиропа солодки / М.В. Гаврилин, С.П. Сенченко, A.M. Тамирян, А.В. Печенова // Химия растительного сырья. 2009. №4. С. 147-150). Недостатком метода является низкая концентрационная чувствительность по сравнению с ВЭЖХ. Хотя метод более прост и быстр в исполнении и отличается экспрессностью и дешевизной оборудования по сравнению с ВЭЖХ.
Раскрытие изобретения
Целью изобретения является разработка способа определения глицирризиновой кислоты методом вольтамперометрии.
Разработанный вольтамперометрический способ количественного определения глицирризиновой кислоты в фармацевтических субстанциях характеризуется тем, что в течение 150 с проводят электрохимическое концентрирование глицирризиновой кислоты на поверхности ртутно-пленочного электрода при потенциале электролиза (-1,8) В на фоне 0,01 М раствора калия хлорида с последующей регистрацией вольтамперных (поляризационных) кривых при линейной скорости развертки потенциала 50 В/с, а концентрацию глицирризиновой кислоты определяют по высоте пика в диапазоне потенциалов (-0,2) до (-0,3) В относительно хлорид-серебряного электрода.
Установленные экспериментальные условия определения глицирризиновой кислоты методом вольтамперометрии позволяют с высокой чувствительностью (минимальная концентрация 1 пг/мл) и экспрессностью (время единичного анализа не превышает 10-15 мин) определить глицирризиновую кислоту в фармацевтических субстанциях.
Определение глицирризиновой кислоты осуществляется на электрохимическом анализаторе ТА-4. Использовали 0,01 М раствор калия хлорида с рН 6.0-7.0 без добавления буферных растворов.
Приготовление фоновых и стандартных растворов глицирризиновой кислоты в воде было общепринятым.
Все условия определения глицирризиновой кислоты были подобраны экспериментально.
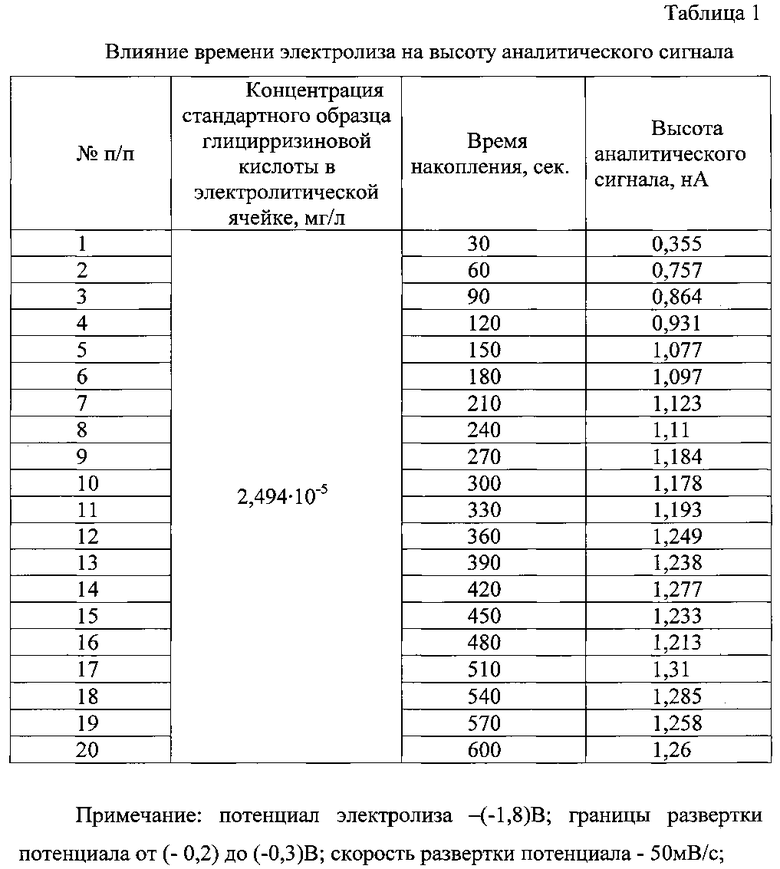
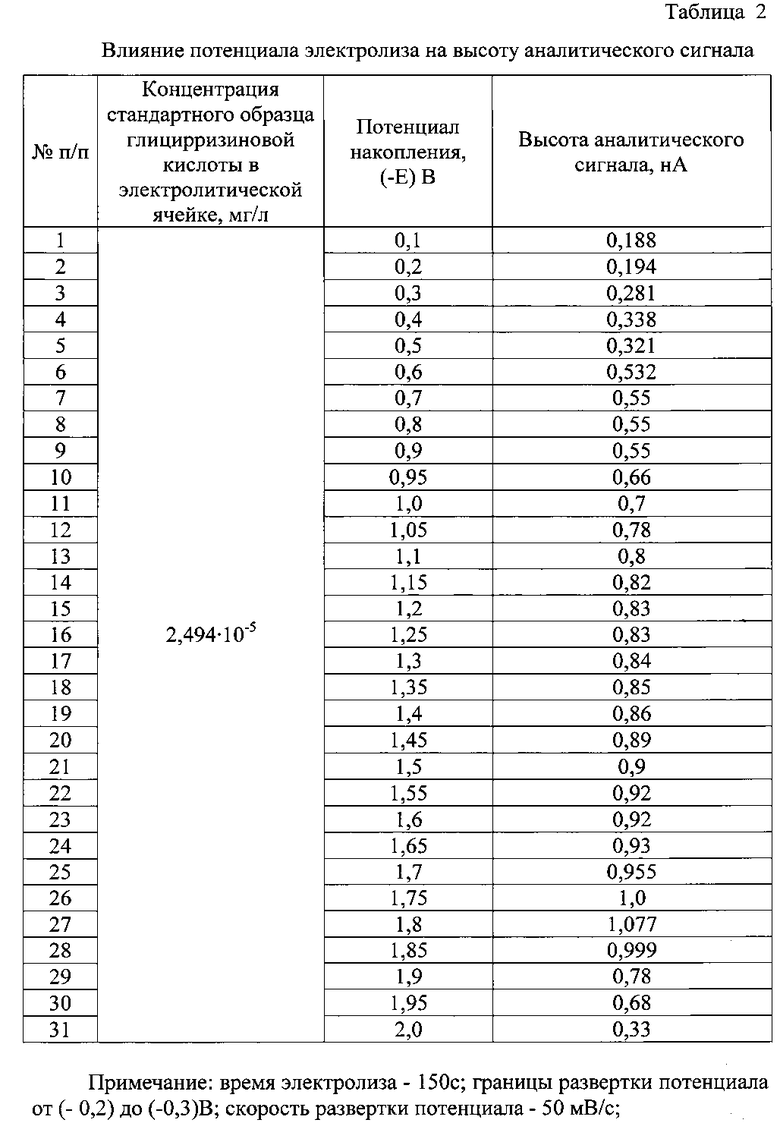
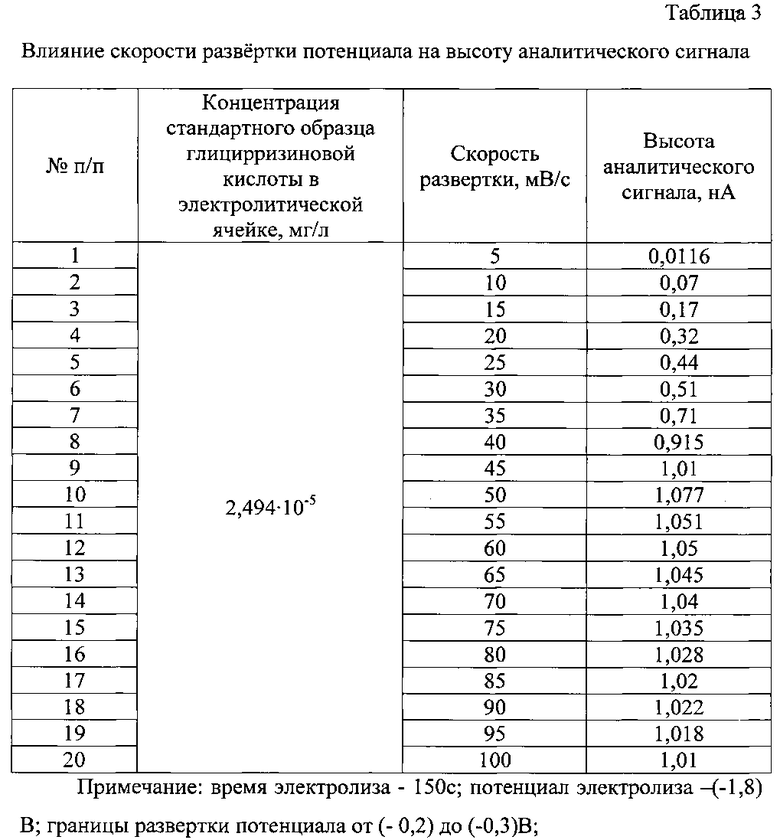
В процессе поиска оптимальных условий вольтамперометрического определения глицирризиновой кислоты было изучено влияние ряда факторов (индикаторный электрод, фоновый электролит, его концентрация и рН, время и потенциал электролиза, скорость развертки потенциала) на высоту аналитического сигнала (табл. 1-3).
В предлагаемом способе в качестве индикаторного (рабочего) электрода использовали ртутно-пленочный из-за хорошей воспроизводимости сигнала и низкого остаточного тока. Глицирризиновая кислота легко адсорбируется на рабочей поверхности электрода, что позволяет концентрировать ее на электроактивном электроде.
В качестве фоновых электролитов были исследованы растворы: Бриттона-Робинсона, хлоридов натрия, калия, лития, кальция, сульфатов и нитратов натрия, калия, аммония. Исходя из полученных результатов, в качестве фонового электролита был выбран раствор калия хлорида, так как только он обеспечивал широкую рабочую область, хорошую электропроводность и необходимую площадь для обработки сигнала, был прост в приготовлении и имел продолжительный срок годности.
Оптимальная концентрация раствора калия хлорида составила 0,01 моль/л. В более концентрированных растворах не наблюдали прироста концентрации глицирризиновой кислоты от ее добавления при наличии большого остаточного тока, тогда как более разбавленный раствор был неустойчив во времени.
Значение рН фонового электролита составляет 6,0-7,0. В сильнощелочной среде при подобранных условиях (потенциал электролиза, время накопления, скорость развертки) сигнал глицирризиновой кислоты был выражен слабо, а в кислой - резко сужалась рабочая область и возрастала величина остаточного тока, при этом невозможно было зафиксировать пик глицирризиновой кислоты. Результаты эксперимента показали, что оптимальным решением является электролиз без добавления дополнительных буферных растворов, что соответствует необходимым границам рН 6,0-7,0.
Оптимальное время накопления составило 150 с - при этом достигалось максимальное значение величины тока растворения накопленных осадков глицирризиновой кислоты с поверхности ртутно-пленочного электрода и хорошая воспроизводимость результатов количественного определения исследуемого вещества. При увеличении времени накопления более 150 с происходит насыщение осадка на электроде, аналитический сигнал глицирризиновой кислоты искажается и затрудняется обработка вольтамперограмм. При времени накопления менее 150 с величина тока растворения не достигает максимального значения, что снижает чувствительность определения глицирризиновой кислоты (табл. 1).
Оптимальный потенциал электролиза составил (-1,8) В. Смещение потенциала накопления в более положительную или в более отрицательную область приводило к уменьшению величины регистрируемого тока (табл. 2).
Важным для определения глицирризиновой кислоты методом вольтамперометрии является выбор скорости развертки потенциала. Оптимальной экспериментально установленной скоростью является 50 мВ/с. Изменение скорости развертки потенциала в сторону увеличения или уменьшения заметно снижало высоту аналитического сигнала, что затрудняло обработку вольтамперных кривых (табл. 3).
Минимальная определяемая концентрация глицирризиновой кислоты составила 1 пг/л.
Пример. Определение глицирризиновой кислоты методом вольтамперометрии.
В кварцевый стаканчик емкостью 20 мл наливают 10 мл 0.01 М раствора калия хлорида. При потенциале (-1,8) В проводят электролиз в течение 150 с. Фиксируют вольтамперограмму при линейной скорости развертки потенциала 50 мВ/с. Отсутствие пиков свидетельствует о чистоте фона.
Затем добавляют 0,01 мл стандартного раствора глицирризиновой кислоты определенной концентрации (использовали стандартные растворы разной концентрации, см. таблицу 4), перемешивают раствор и проводят электрохимическое концентрирование вещества при потенциале (-1,8) В в течение 150 с. Фиксируют вольтамперограмму при линейной скорости развертки потенциала 50 мВ/с. Аналитический сигнал для указанной концентрации глициррицизиновой кислоты регистрируют в диапазоне потенциалов от (-0,2) до (-0,3) В.
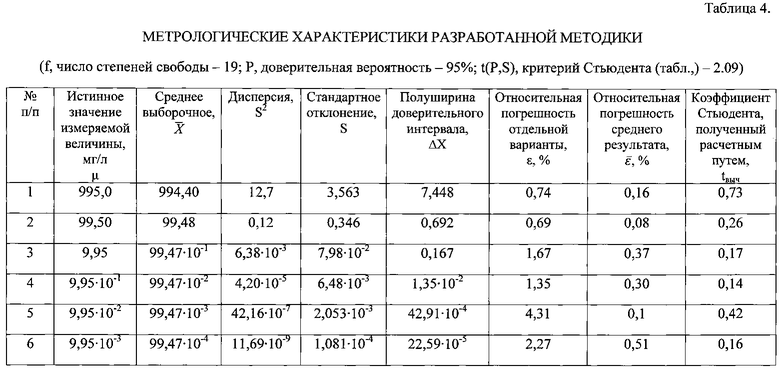
Полученные результаты подвергли статистической обработке (таблица 4). Как следует из таблицы, погрешность методики составляет не более 5%, что соответствует погрешности вольтамперометрического метода, причем коэффициент Стьюдента, полученный расчетным путем, не превышает табличный.
Время единичного анализа не превышает 10-15 мин.
Установленные условия впервые позволили количественно определить глицирризиновую кислоту путем регистрации вольтамперных кривых при потенциале (-1,8) В на фоне 0,01 моль/л калия хлорида.
| название | год | авторы | номер документа |
|---|---|---|---|
| ВОЛЬТАМПЕРОМЕТРИЧЕСКИЙ СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ L-ТИРОКСИНА | 2010 |
|
RU2428690C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ КАРВЕДИЛОЛА МЕТОДОМ ИНВЕРСИОННОЙ ВОЛЬТАМПЕРОМЕТРИИ | 2007 |
|
RU2334510C1 |
| ВОЛЬТАМПЕРОМЕТРИЧЕСКИЙ СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ ЛИДОКАИНА ГИДРОХЛОРИДА | 2007 |
|
RU2348925C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ПЛАТИНЫ В ВОДНЫХ РАСТВОРАХ МЕТОДОМ ИНВЕРСИОННОЙ ВОЛЬТАМПЕРОМЕТРИИ ПО ПИКУ СЕЛЕКТИВНОГО ЭЛЕКТРООКИСЛЕНИЯ PtPb | 2012 |
|
RU2491539C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ СПИРАПРИЛА ГИДРОХЛОРИДА МЕТОДОМ ИНВЕРСИОННОЙ ВОЛЬТАМПЕРОМЕТРИИ | 2004 |
|
RU2280860C2 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ФОЗИНОПРИЛА НАТРИЯ МЕТОДОМ ИНВЕРСИОННОЙ ВОЛЬТАМПЕРОМЕТРИИ | 2005 |
|
RU2288469C1 |
| ВОЛЬТАМПЕРОМЕТРИЧЕСКИЙ СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ БЕНЗОЙНОЙ КИСЛОТЫ | 2013 |
|
RU2537168C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ АМИОДАРОНА (КОРДАРОНА) МЕТОДОМ ИНВЕРСИОННОЙ ВОЛЬТАМПЕРОМЕТРИИ | 2003 |
|
RU2246722C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ТАЛЛИЯ В ВОДНЫХ РАСТВОРАХ И ТЕХНОЛОГИЧЕСКИХ СЛИВАХ МЕТОДОМ ИНВЕРСИОННОЙ ВОЛЬТАМПЕРОМЕТРИИ | 2011 |
|
RU2494386C2 |
| СПОСОБ ОПРЕДЕЛЕНИЯ БЕНАЗЕПРИЛА ГИДРОХЛОРИДА (ЛОТЕНЗИНА) МЕТОДОМ ИНВЕРСИОННОЙ ВОЛЬТАМПЕРОМЕТРИИ | 2004 |
|
RU2280861C2 |
Изобретение относится к аналитической химии. Способ заключается в том, что в течение 150 с проводят электрохимическое концентрирование глицирризиновой кислоты на поверхности ртутно-пленочного электрода при потенциале электролиза (-1,8) В на фоне 0,01 М калия хлорида с последующей регистрацией вольтамперных кривых при линейной скорости развертки потенциала 50 В/с, а концентрацию глицирризиновой кислоты определяют по высоте пика в диапазоне потенциалов (-0,2) до (-0,3) В относительно хлорид-серебряного электрода. Способ характеризуется высокой чувствительностью (1 пг/мл) и экспрессностью (время единичного анализа не превышает 10-15 мин). 1 пр., 4 табл.
Вольтамперометрический способ количественного определения глицирризиновой кислоты в фармацевтических субстанциях, характеризующийся тем, что в течение 150 с проводят электрохимическое концентрирование глицирризиновой кислоты на поверхности ртутно-пленочного электрода при потенциале электролиза (-1,8) В на фоне 0,01 М калия хлорида с последующей регистрацией вольтамперных кривых при линейной скорости развертки потенциала 50В/с, а концентрацию глицирризиновой кислоты определяют по высоте пика в диапазоне потенциалов (-0,2) до (-0,3) В относительно хлорид-серебряного электрода.
| 0 |
|
SU153140A1 | |
| ВЫСОКОЧУВСТВИТЕЛЬНЫЙ СПОСОБ ОПРЕДЕЛЕНИЯ КОЛИЧЕСТВА КОМПОНЕНТОВ, ПОЛУЧЕННЫХ ИЗ ЛЕКАРСТВЕННЫХ ТРАВ | 2011 |
|
RU2558042C2 |
| СN104880430А, 02.09.2015 | |||
| CN103529151A, 22.01.2014. | |||

