Область техники, к которой относится изобретение
Настоящее изобретение относится к высокочувствительному способу количественного определения глицирризина, его метаболитов или тому подобного, в биологических образцах.
Для настоящего изобретения испрашивается приоритет по заявке на патент Японии №2010-256187, поданной 16 ноября 2010, содержание которой включено в настоящее описании посредством ссылки.
Уровень техники
Глицирризин (далее сокращенно "GL") и его соли являются компонентами экстракта корня солодки, имеющими разнообразные физиологические свойства, такие как противоаллергенные свойства и противовоспалительные свойства, а также иммунорегуляторные свойства, гепатопротекторные свойства, свойство стимулирования роста гепатоцитов и свойство ингибирования/инактивации размножения вирусов. В Японии GL и его соли уже давно используются в качестве клинических лекарственных препаратов, например в китайской медицине лекарственных трав, и в настоящее время широко используется в лечении хронических заболеваний печени, мокнущей экземы, детского строфулюса, очаговой алопеции, при различных аллергиях, воспалительных заболеваниях и т.д. Что касается применения в областях, отличных от фармацевтики, GL часто используется в качестве подсластителя в широком диапазоне пищевых продуктов, таких как маринады и приправы, из-за его свойств солевого созревания и подслащивания.
На основе не-клинических исследований можно сделать вывод, что GL имеет тенденцию к накоплению в печени, быстро секретируется с желчью и выбрасывается, подвергаясь при этом метаболической и печеночно-кишечной рециркуляции. В частности, при пероральном введении GL, являющийся гликозидом и водорастворимым полярным веществом, абсорбируется слабо, и его концентрация в периферической крови оказывается крайне низкой из-за эффекта первого прохождения и метаболизма кишечных бактерий. Как следствие, ранее были опробованы различные способы для измерения кинетики GL в крови, но обнаружить GL в крови во время введения составов, содержащих GL или китайские травяные составы на основе солодки, весьма трудно, и его кинетика долго была неясна.
Например, существуют методы, в рамках которых осуществляют обработку метанолом для удаления белков из плазмы крови, собранной от здорового человека, которому был перорально введен GL, с последующим измерением количества GL в плазме крови путем проведения: иммуноферментного анализа (ИФА); или измерения с помощью высокоэффективной жидкостной хроматографии (ВЭЖХ) с использованием колонки, работающей в обращенно-фазовом режиме, такой как колонка с носителем типа химически связанной группы октадецила (ODS) (см., например, непатентные документы 1-4). Тем не менее, с такими способами измерения количественный предел обнаружения составляет самое большее около 0,1 мкг/мл, и GL не обнаруживается, даже притом, что глицирретиновая кислота (далее сокращенно "GA"), которая является основным метаболитом, может быть обнаружена. По этой причине миграция GL в крови была лишь косвенно подтверждена обнаружением в моче (см. непатентный документ 4). В том, что касается фармакологической эффективности составов, содержащих GL, вместо GL оценочным показателем стала количественно измеряемая GA.
Документы предшествующего уровня техники
Непатентный документ 1: Ishiwata и 7 соавторов. Biological and Pharmaceutical Bulletin, 2000, Vol.23, No.8, pp.904-905.
Непатентный документ 2: De Groot и один соавтор. Journal of Chromatography, 1988, Vol.456, pp.71-81.
Непатентный документ 3: Nakata, и 3 соавтора. Medical and Pharmaceutical Society for Wakan-Yaku, 1986, Vol.3, No. 3, pp.278-279.
Непатентный документ 4: Yamamura и 7 соавторов. Journal of Pharmaceutical Sciences, 1992, Vol.81, No.10, pp.1042-1046.
Раскрытие изобретения
Задачи, решаемые настоящим изобретением
Фармакокинетика неизмененного GL, представляющего собой активатор, является полезной для правильного использования с точки зрения эффективности и безопасности. Таким образом, желательно иметь способ количественного определения количества GL в биологических образцах, таких как плазма крови, не завязанный на изучение GA.
Настоящее изобретение было сделано в свете вышеуказанной проблемы, и задачей изобретения является получение способа обнаружения и количественного определения GL в биологических образцах, таких как плазма крови.
Средства решения указанных задач
В результате кропотливых исследований, нацеленных на решение вышеуказанной задачи, авторы настоящего изобретения обнаружили, что GL в биологических образцах можно обнаружить и количественно определить путем экстракции компонентов, включающих GL, из биологического образца посредством твердофазной экстракции с использованием твердой фазы, обладающей обращенно-фазовой распределительной функцией и функцией анионного обмена, с последующим направлением GL в виде экстракта на масс-спектрометрию. Так было разработано настоящее изобретение.
Таким образом, настоящее изобретение описывает:
(1) высокочувствительный способ определения количества компонентов, полученных из лекарственных трав, включающий:
стадию экстракции, на которой экстракт, содержащий компоненты, полученные из лекарственных трав, готовят следующим образом: смесь, содержащую биологический образец со щелочью или спиртом, вводят в твердую фазу, обладающую обращенно-фазовой распределительной функцией и функцией анионного обмена; твердую фазу промывают по крайней мере один раз с очищающей жидкостью; а затем осуществляют элюирование из твердой фазы с кислым спиртом, и
стадию количественного определения, на которой по меньшей мере один компонент, выбранный из группы, состоящей из глицирризина, глицирретиновой кислоты, метаболитов глицирризина и глицирретиновой кислоты, родственных глицирризину и глицирретиновой кислоте соединений, компонентов сапонина, содержащихся в лакричнике (солодке), и их фармакологически приемлемых солей, и содержащийся в экстракте, извлеченном на стадии экстракции, обнаруживают и определяют количественно методом масс-спектрометрии;
где очищающая жидкость представляет собой однокомпонентную жидкость или жидкую смесь по меньшей мере двух компонентов, выбранных из группы, включающей воду, щелочь, спирт и ацетонитрил;
(2) высокочувствительный способ определения количества компонентов, полученных из лекарственных трав, по пункту (1) выше, где на стадии экстракции твердую фазу промывают жидкой смесью щелочи, спирта и воды, с последующим промыванием спиртом, жидкой смесью спирта и воды или ацетонитрилом;
(3) высокочувствительный способ определения количества компонентов, полученных из лекарственных трав, по пункту (2) выше, где в жидкой смеси щелочи, спирта и воды 0,5-28 об.% водный раствор аммиака и метанол смешаны в соотношении от 99:1 до 1:3, а также
(4) высокочувствительный способ определения количества компонентов, полученных из лекарственных трав, по любому из пунктов (1)-(3) выше, в котором биологический образец представляет собой кровь, плазму крови или экстракт ткани.
Преимущества изобретения
В соответствии с высокочувствительным способом определения количества полученных из лекарственных трав компонентов по настоящему изобретению можно обнаружить и количественно определить очень маленькие количества GL в биологическом образце. Как следствие, при использовании высокочувствительного способа определения количества полученных из лекарственных трав компонентов по настоящему изобретению можно количественно определить GL в биологических образцах, взятых у людей, которыми были поглощены GL-содержащие составы или пищевые продукты или напитки, а также можно точно проанализировать его фармакокинетику.
Краткое описание чертежей
Фиг.1A представляет собой графическое изображение, соответствующее хроматограмме GL в контрольном образце плазмы крови по примеру 6.
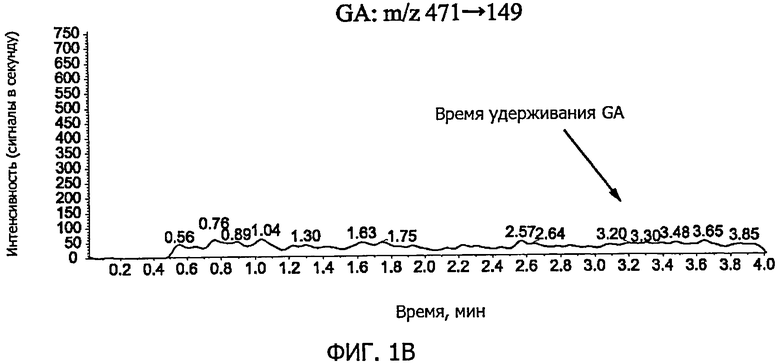
Фиг.1B представляет собой графическое изображение, соответствующее хроматограмме GA в контрольном образце плазмы крови по примеру 6.
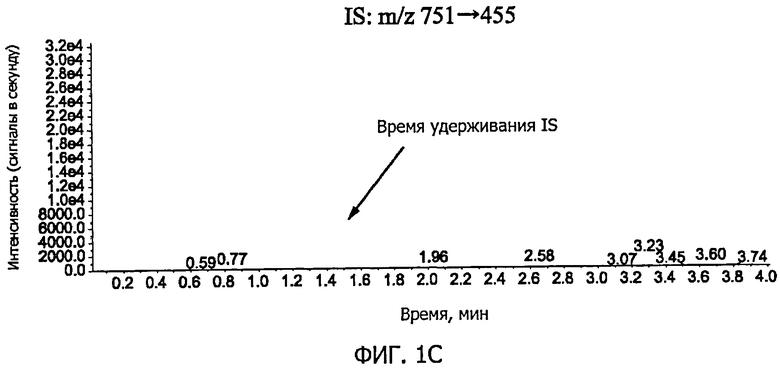
Фиг.1C представляет собой графическое изображение, соответствующее хроматограмме внутреннего стандарта (IS) в контрольном образце плазмы крови по примеру 6.
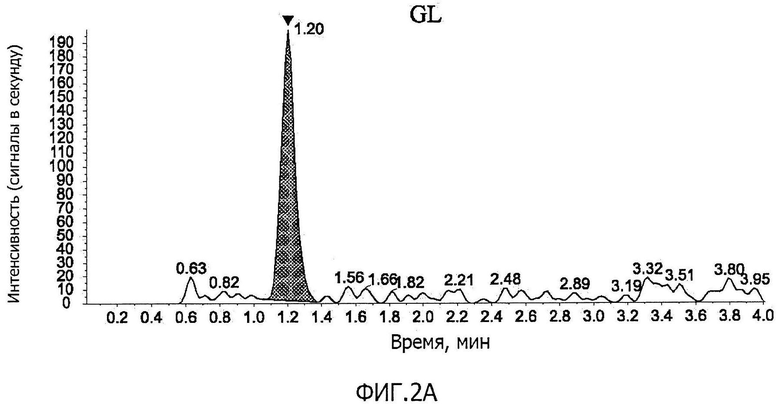
Фиг.2A представляет собой графическое изображение, соответствующее хроматограмме GL в образце плазмы крови, к которому добавляют стандартный раствор (G) (содержание GL в плазме крови: 0,5 нг/мл), по примеру 6.
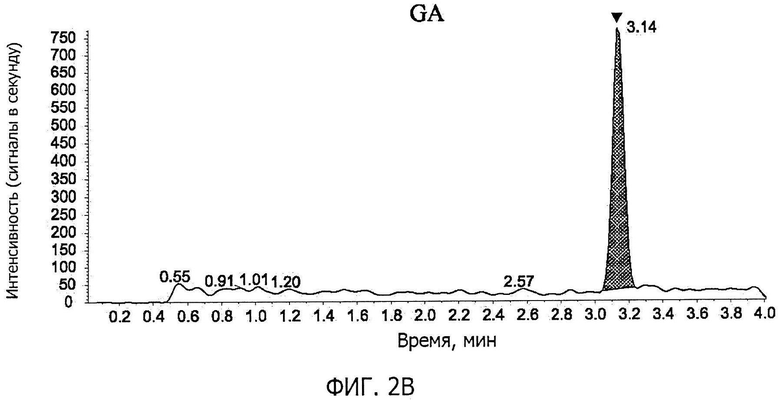
Фиг.2B представляет собой графическое изображение, соответствующее хроматограмме GA в образце плазмы крови, к которому добавляют стандартный раствор (G) (содержание GA в плазме крови: 2 нг/мл), по примеру 6.
Фиг.2C представляет собой графическое изображение, соответствующее хроматограмме внутреннего стандарта (IS) в образце плазмы крови, к которому добавляют стандартный раствор (G), по примеру 6.
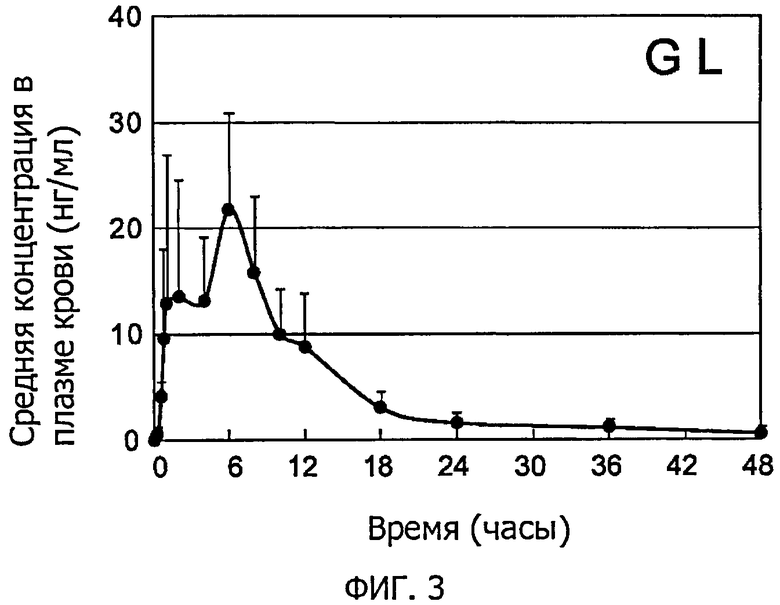
Фиг.3 представляет собой графическое изображение, которое иллюстрирует изменения средней концентрации GL в плазме крови в примере 7.
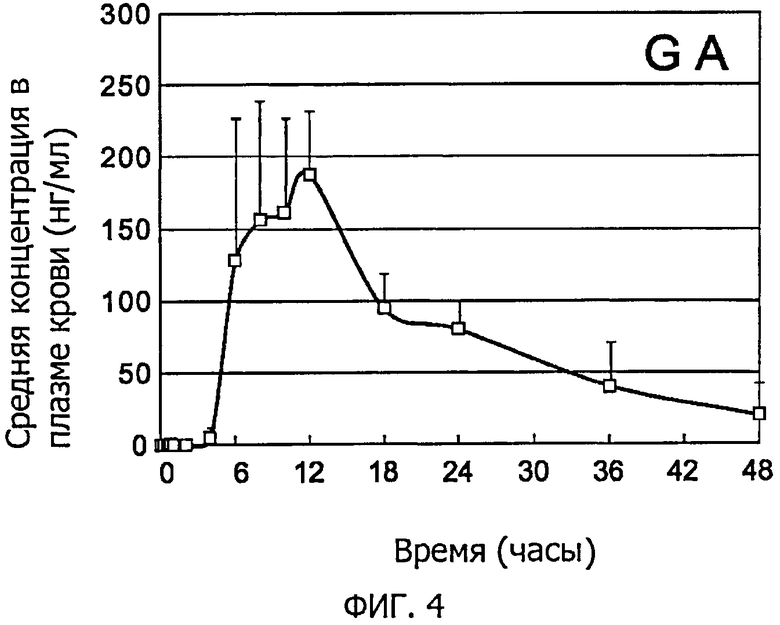
Фиг.4 представляет собой графическое изображение, которое иллюстрирует изменения средней концентрации GA в плазме крови в примере 7.
Осуществление изобретения
Высокочувствительный способ определения количества полученных из лекарственных трав компонентов по настоящему изобретению включает экстракцию GL из биологического образца посредством твердофазной экстракции с использованием твердой фазы, обладающей обращенно-фазовой распределительной функцией и анионообменной функцией, с последующим осуществлением количественного определения содержания GL в полученном экстракте методом масс-спектрометрии. Чувствительность обнаружения GL увеличивается при проведении количественного определения с использованием масс-спектрометрии вместо обычного метода ВЭЖХ и тому подобного. Кроме того, в отличие от твердофазной экстракции с использованием твердой фазы, обладающей только обращенно-фазовой распределительной функцией, в качестве обычной колонки ODS, способ по изобретению осуществляется с использованием твердой фазы, обладающий как обращенно-фазовой распределительной функцией, так и анионообменной функцией, что позволяет удалять из экстракта, отправляемого на масс-спектрометрию, множество чужеродных веществ, и в значительной мере повышать чувствительность масс-спектрометрии.
В частности, высокочувствительный способ определения количества полученных из лекарственных трав компонентов по настоящему изобретению содержит стадию экстракции, на которой готовят экстракт, содержащий компоненты, полученные из лекарственных трав, путем подачи смеси биологического образца со щелочью или спиртом в твердую фазу, обладающую обращенно-фазовой распределительной функцией и анионообменной функцией, и затем твердую фазу промывают один, два или более раза очищающей жидкостью, после чего осуществляют элюирование из твердой фазой с кислым спиртом; и стадию количественного определения, на которой осуществляют обнаружение и количественное определение методом масс-спектрометрии вещества по меньшей мере одного типа, выбранного из группы, состоящей из GL, GA, метаболитов GL и GA, родственных веществ GL и GA, компонентов сапонина, содержащихся в солодке, и их фармакологически приемлемых солей, в экстракте, полученном на стадии экстракции; при этом очищающая жидкость представляет собой однокомпонентную жидкость или жидкую смесь по крайней мере двух компонентов, выбранных из группы, состоящей из воды, щелочи, спирта и ацетонитрила.
Предпочтительно, чтобы экстракт, содержащий по меньшей мере два компонента, полученных из лекарственных трав, был подготовлен на стадии экстракции с последующим обнаружением и количественным определением по меньшей мере двух компонентов, полученных из лекарственного препарата низшего сорта, на стадии количественного определения в одно измерение (с использованием различных способов обнаружения).
Компоненты, полученные из лекарственных трав, которые могут быть количественно определены высокочувствительным способом определения количества полученных из лекарственных трав компонентов по настоящему изобретению, представляют собой по крайней мере один компонент, выбранный из группы, состоящей из GL, GA, метаболитов GL и GA, родственных соединений GL и GA, компонентов сапонина, содержащихся в солодке, а также их фармакологически приемлемых солей (далее сокращенно «GL и т.д.»).
Примеры метаболитов GL и GA включают 3-моноглюкуронил-глицирретиновую кислоту (20β-карбокси-11-оксо-30-норолеан-12-ен-3β-ил-β-D-глюкопиранозидуроновую кислоту), 30-моноглюкуронил-глицирретиновую кислоту (3β-гидрокси-11-оксоолеан-12-ен-30-оил-β-D-глюкопиранозидуроновую кислоту), 3-оксо-GA (3,11-диоксоолеан-12-ен-30-овую кислоту), 3α-GA (3α-гидрокси-11-оксоолеан-12-ен-30-овую кислоту), тройной сульфоконьюгат (3β-гидроксисульфонилокси-11-оксоолеан-12-ен-30-овую кислоту), 3β,22α-дигидрокси-11-оксоолеан-12-ен-30-овую кислоту, 3β,24-дигидрокси-11-оксоолеан-12-ен-30-овую кислоту и тому подобное.
Примеры родственных соединений GL и GA включают 20α-карбокси-11-оксо-29-норолеан-12-ен-3β-ил (β-D-глюкопиранозилуроновую кислоту)-(1→2)-β-D-глюкопиранозидуроновую кислоту, 20β-карбокси-24-гидрокси-11-оксо-30-норолеан-12-ен-3β-ил (β-D-глюкопиранозилуроновую кислоту)-(1→2)-β-D-глюкопиранозидуроновую кислоту), 18α-GL (20β-карбокси-11-оксо-(18αH)-30-норолеан-12-ен-3β-ил (β-D-глюкопиранозилуроновую кислоту)-(1→2)-β-D-глюкопиранозидуроновую кислоту), 18α-GA (3β-гидрокси-11-оксо-(18αH)-олеан-12-ен-30-овую кислоту) и тому подобное.
Примеры компонентов сапонина, содержащихся в солодке, включают солодка-сапонин C1 (20β-карбокси-11-оксо-30-норолеан-12-ен-3β-ил(β-D-глюкопиранозилуроновую кислоту)-(1→2)-β-D-глюкопиранозидуроновую кислоту) и тому подобное.
Никаких особых ограничений в отношении фармакологически приемлемых солей GL и т.д., используемых в настоящем изобретении, не накладывается, при условии, что они представляют собой соли, обладающие теми же самыми фармакологическими свойствами in vivo, что и GL и т.д. Конкретные примеры солей включают аммониевые соли, натриевые соли, калиевые соли и тому подобное.
В настоящем изобретении может количественно определяться один тип GL и т.д. или по крайней мере два типа GL и т.д. могут быть количественно определены за одну операцию. В особенно предпочтительном варианте осуществления настоящего изобретения одновременно количественно определяют GL и GA.
Хотя биологические образцы, используемыми в высокочувствительном способе определения количества полученных из лекарственных трав компонентов по настоящему изобретению, могут быть любыми образцами, взятыми из живого организма, в предпочтительном варианте биологические образцы представляют собой образцы, забранные у людей или животных, таких как мыши или крысы. Биологическими образцами могут быть кровь, плазма крови, сыворотка крови, моча, перитонеальная жидкость, плевральная жидкость, синовиальная жидкость, костный мозг, желчь, экстракты фрагментов ткани или тому подобного (тканевые экстракты), собранные из таких тканей, как печень, поджелудочная железа или почки, или тому подобное. Тканевые экстракты могут быть приготовлены путем гомогенизации тканевых фрагментов или т.п. с использованием обычных методов. В настоящем изобретении биологические образцы предпочтительно представляют собой кровь, плазму крови, мочу или тканевые экстракты, а более предпочтительно - плазму крови.
Ниже приведено описание каждой из стадий высокочувствительного способа определения количества полученных из лекарственных трав компонентов по настоящему изобретению.
На стадии экстракции сначала готовят смесь биологического образца со щелочью или спиртом. Нет никаких конкретных ограничений в отношении щелочи, которую смешивают с биологическим образцом, при условии, что pH полученной смеси может быть установлен в щелочной области. Примеры щелочей включают аммиак, аммиачную воду (водный раствор аммиака), раствор гидроксида натрия, раствор гидрокарбоната натрия и тому подобное. В качестве спирта предпочтительными являются низшие спирты с числом атомов углерода 1-6, Примеры спиртов включают метанол, этанол, изопропанол и тому подобное. Также могут быть использованы и спиртовые растворы, разбавленные водой. В настоящем изобретении предпочтительно используют аммиак, аммиачную воду, метанол или водный раствор метанола, и более предпочтительно в качестве щелочи или спирта, смешиваемых с биологическим образцом, использовать аммиак или аммиачную воду.
Никаких конкретных ограничений в отношении концентрации аммиачной воды, добавляемой к биологическому образцу, не налагается, и эта концентрация может быть соответствующим образом скорректирована с учетом таких факторов, как тип используемых биологического образца и твердой фазы и концентрация аммиака полученной смеси. В настоящем изобретении концентрация аммиака или аммиачной воды, добавляемых к биологическому образцу, предпочтительно составляет такую величину, чтобы концентрация аммиака в полученной смеси составляла 0,01-30 об.%, более предпочтительно - 0,05-25 об.%, и еще более предпочтительно - 0,05-20 об.%.
Затем полученную смесь вводят в твердую фазу, обладающую обращенно-фазовой распределительной функцией и анионообменной функцией, для адсорбции GL и т.д. из смеси в твердую фазу. Примеры твердых фаз, обладающих распределительной обращенно-фазовой функцией и анионообменной функцией, включают картридж для твердофазной экстракции смешанного типа обращенно-фазовой распределительной/анионообменной функциональности Oasis MAX (от Waters Corp.,) и тому подобное.
Твердую фазу, в которой адсорбируется GL и т.д., промывают один, два или более раз очищающей жидкостью для удаления неспецифически связанных компонентов. Очищающая жидкость представляет собой однокомпонентную жидкость или жидкую смесь по крайней мере двух компонентов, выбранных из группы, состоящей из воды, щелочи, спирта и ацетонитрила. В качестве щелочи или спирта, используемых в качестве очищающей жидкости, могут быть использованы точно такие же щелочь или спирт, которые добавляются в биологический образец. Промывание твердой фазы может быть осуществлено дважды или большее число раз с использованием одного типа очищающей жидкости или с последовательным использованием различных видов очищающих жидкостей.
В настоящем изобретении твердую фазу предпочтительно промывают жидкой смесью из щелочи, спирта и воды, жидкой смесью щелочи и воды, или водой с последующим промыванием спиртом, жидкой смесью спирта и воды, или ацетонитрилом, и более предпочтительно твердую фазу промывают жидкой смесью щелочи, спирта и воды, с последующим промыванием спиртом, жидкой смесью спирта и воды, или ацетонитрилом. Предпочтительно, чтобы жидкая смесь щелочи, спирта и воды была жидкой смесью аммиака, спирта и воды, и еще более предпочтительно - жидкой смесью аммиака, метанола и воды.
Никаких конкретных ограничений в отношении соотношения количеств соответствующих компонентов жидкой смеси аммиака, метанола и воды не накладывается, при условии, что поддерживается состояние, в котором GL и т.д. адсорбируется в твердую фазу. Например, предпочтительно, чтобы концентрация метанола в жидкой смеси была 1-75 об.%, а концентрация аммиака в жидкой фазе была 0,1-21 об.%, и еще более предпочтительно, чтобы концентрация метанола в жидкой фазе была 25-75 об.%, а концентрация аммиака в жидкой фазе была 0,1-21 об.%. Жидкая смесь с таким соотношением количеств компонентов может, например, быть приготовлена путем смешивания 0,5-28 об.% аммиачной воды с метанолом при соотношении от 99:1 до 1:3.
Предпочтительно, чтобы спирт, более предпочтительно метанол или этанол, и еще более предпочтительно метанол, использовался в качестве очищающей жидкости для второй промывки после осуществления первой промывки с помощью жидкой смеси щелочи, спирта и воды.
Затем путем элюирования из твердой фазы с кислым спиртом получают экстракт, содержащий компоненты, полученные из лекарственных трав. Примеры растворителей для подкисления включают муравьиную кислоту-спирт, соляную кислоту, трифторуксусную кислоту и тому подобное. В настоящем изобретении муравьиная кислота-спирт является предпочтительным вариантом. В качестве муравьиной кислоты-спирта, используемого для элюирования, предпочтительно используется сложный эфир муравьиной кислоты и низшего спирта с числом атомов углерода 1-6, более предпочтительно - муравьиная кислота-метанол или муравьиная кислота-этанол, и еще более предпочтительно - муравьиная кислота-метанол. Полученный элюированный продукт выпаривают досуха с целью последующей масс-спектрометрии.
Затем GL и т.д. в продукте элюирования, экстрагированном на стадии экстракции, обнаруживают и количественно определяют методом масс-спектрометрии на стадии количественного определения. В качестве метода масс-спектрометрии предпочтительно используют метод ЖХ-МС (жидкостной хроматографии/масс-спектрометрии) или метод ЖХ-МС/МС (жидкостной хроматографии/тандемной масс-спектрометрии). В частности, упаренный досуха продукт элюирования растворяют в подвижной фазе ЖХ с последующим осуществлением ЖХ и МС. ЖХ-МС и ЖХ-МС/МС могут быть осуществлены с использованием устройства, сочетающего ВЭЖХ с масс-спектрометром.
Как отмечено в непатентных документах 1-4, ЖХ предпочтительно осуществляют с использованием колонки, обладающей обращенно-фазовой распределительной функцией, такой как колонки с ODS. МС может быть осуществлена с помощью обычных методов. Например, образцы ионизируют методом ESI (ионизация электрораспылением) или методом APCI (химическая ионизация при атмосферном давлении) с последующим разделением и обнаружением соответствующих ионов с использованием устройства типа магнитного отклонения, квадрупольного типа, времяпролетного типа или тому подобного.
При проведении масс-спектрометрии методом ЖХ-МС количественно определяемый уровень GL и т.д. в крови, плазме крови или сыворотке крови может быть улучшен вплоть до 20 нг/мл. Аналогичным образом, при проведения масс-спектрометрии методом ЖХ-МС/МС количественно определяемый уровень GL и т.д. в крови и т.п. может быть улучшен до не более чем 10 нг/мл, например до примерно 0,5 нг/мл.
Ранее сообщалось, что концентрации GL в плазме крови составляют приблизительно 500 нг/мл в случае, когда коммерчески доступный реагент вводится перорально в высокой дозе (1600 мг в пересчете на GL) (Environmental Health Perspectives, 102 (9), 65-68, 1994). В результате пропорционального вычисления на основе вышеупомянутого наблюдения можно сделать вывод, что концентрация в крови при введении в дозе 75 мг (обычная клиническая доза), по оценкам, должна приблизительно равняться 23 нг/мл в случае, когда целью является лечение заболевания печени. Таким образом, высокочувствительный метод определения количества полученных из лекарственных трав компонентов по настоящему изобретению позволяет количественно определить уровень GL в крови, который не может быть обнаружен обычными способами. Хотя концентрации GL в крови после перорального приема составов, включающих GL, или тому подобного, характеризуются значительной изменчивостью, бывают случаи, когда методы измерений, имеющие недостаточные количественные границы обнаружения, могут привести к тому, что обнаружить GL в некоторых образцах крови будет невозможно или невозможно будет измерить долгосрочную фармакокинетику GL, что приведет к недостаточной точности измерений или недостаточной достоверности результатов измерений, и высоко чувствительный метод определения количества полученных из лекарственных трав компонентов по настоящему изобретению сделает возможным получение результатов измерений с более высокой надежностью, в частности путем масс-спектрометрии методом ЖХ-МС/МС.
Примеры
Следующие примеры представлены для того, чтобы описать настоящее изобретение более подробно, но при этом настоящее изобретение не ограничивается этими примерами.
Пример 1
Измерялись предельные обнаружимые количества GL и GA в плазме крови, и строили калибровочные кривые для случая, когда высокочувствительный способ определения количества полученных из лекарственных трав компонентов по настоящему изобретению был осуществлен с использованием ЖХ-МС масс-спектрометрии.
Получение стандартного раствора
Во-первых, точно отвешивали 10,0 мг GL (производства Tokiwa Phytochemical., Ltd.) и затем его растворяли в метаноле. После этого объем раствора точно устанавливали на уровень 100 мл, получая 100 мкг/мл стандартного исходного раствора GL. Аналогичным образом, точно отвешивали 10,0 мг GA (производства Alps Pharmaceutical Industries Co., Ltd.), и затем его растворяли в метаноле. После этого объем раствора точно устанавливали на уровень 100 мл, получая 100 мкг/мл стандартного исходного раствора GA.
Затем точно смешивали 2 мл стандартного исходного раствора GL и 8 мл стандартного исходного раствора GA. Суммарный объем доводили метанолом точно до 50 мл для получения стандартного раствора (А), в котором концентрация GL составляла 4000 нг/мл, а концентрация GA составляла 16000 нг/мл. Полученный стандартный раствор А последовательно разбавляли метанолом для получения стандартных растворов (B)-(F), приведенных в таблице 1. Исходные стандартные растворы и полученные стандартные растворы хранили при низких температурах (5±4°C) после приготовления. При приготовлении использовали стеклянные инструменты.
Получение образцов плазмы крови
50 мкл стандартного раствора (А) добавляли к 0,5 мл плазмы крови человека, и тщательно перемешивали для получения образца плазмы крови. Такую же процедуру проводили соответственно со стандартными растворами (B)-(F) для получения соответствующих образцов плазмы крови. Затем в соответствующие образцы плазмы крови добавляли 50 мкл раствора внутреннего стандарта (IS). В настоящем примере в качестве вещества внутреннего стандарта α-хедерин (α-Hederin) был использован. Контрольные образцы плазмы крови получали путем добавления 100 мкл метанола к 0,5 мл плазмы крови человека.
Стадия экстракции
К каждому образцу плазмы крови добавляли 1 мл аммиачной воды (0,56 об.%) и тщательно перемешивали (концентрация аммиака в смеси: 0,37 об.%) с последующим введением всей полученной смеси в Oasis МАХ (производства Waters Corp.) (далее сокращенно ″МАХ″). Перед введением образца плазмы крови МАХ был предварительно обработан путем введения в него раствора муравьиной кислоты-метанола (2 об.%) с последующим введением воды.
После того введения образца плазмы крови МАХ промывали раствором аммиачной воды/метанола с концентрацией 0,56 об.% (жидкая смесь с объемным соотношением 1:1) с последующим дополнительным промыванием метанолом.
Затем для элюирования адсорбированного GL и т.д. вводили раствор муравьиной кислоты/метанола с концентрацией 2 об.%. Элюат (экстракт) выпаривали досуха с использованием при нагревании при 40°C в газообразном азоте.
Стадия количественного определения
Упаренный досуха экстракт растворяли в 250 мкл следующей подвижной фазы: жидкость A/жидкость B (жидкая смесь с объемным соотношением 1:1). 20 мкл жидкой смеси вводили в аналитическую колонку и подвергали ЖХ в условиях, приведенных ниже.
Устройство: Alliance HT 2695 (произведено Waters Corp.)
Аналитическая колонка: CAPCELL РАК С 18 (UG120, 5 мкл, 1,5 мм × 150 мм, произведено Shiseido Co., Ltd.)
Подвижная фаза: жидкость A представляет собой 0,1 об.% муравьиную кислоту, жидкость B представляет собой 0,1 об.% муравьиную кислоту/ацетонитрил, градиентный режим (таблица 2)
Скорость потока: 0,30 мл/мин
Температура колонки: 40°C
Объем вводимого образца: 20 мкл
Затем в отношении фракции, содержащей GL и т.д. и полученной методом ЖХ, осуществляли МС в указанных ниже условиях.
Устройство: ZQ2000 (произведено Waters Corp.)
Способ ионизации: ESI (турбо-спрей)
Способ детектирования: GL (детектирование положительных и отрицательных ионов), GA и IS (детектирование положительных ионов), MRM (мониторинг множественных реакций)
Напряжение капилляра: 2,5 кВ
Экстрактор: 2 B
Радиочастотная линза: 0,2 B
Исходная температура: 110 C
Температура десольватации: 350°C
Поток десольватирующего газа: 350 л/час
Конусный газовый поток: 50 л/час
Результаты измерений ЖХ-МС увеличивались в зависимости от концентраций GL или GA в образцах плазмы крови, и калибровочные кривые, полученные из результатов измерений ЖХ-МС и концентраций GL или GA в образцах плазмы крови, оказались линейными. Таким образом, исходя из результатов концентрация GL в биологических образцах плазмы крови составляла 20-400 нг/мл, а концентрация GA составляла 80-1600 нг/мл, из чего ясно видно, что удалось осуществить высокочувствительное количественное определение GL и т.д. в биологических образцах с помощью высокочувствительного способа определения количества полученных из лекарственных трав компонентов по настоящему изобретению с использованием ЖХ-МС.
Изучение специфичности, линейности и воспроизводимости в течение дня
Далее было проведено исследование специфичности, линейности и воспроизводимости в течение дня. В результате высокочувствительный способ обнаружения количества полученных из лекарственных трав компонентов по настоящему изобретению с использованием ЖХ-МС продемонстрировал удовлетворительную линейность (коэффициент корреляции калибровочной кривой составил 0,9979) и воспроизводимость в пределах, соответственно, диапазонов концентраций 20-400 нг/мл и 80-1600 нг/мл. Что касается результатов дневной воспроизводимости, то, как показано в Таблице 4, правильность отрицательного режима определения GL (GL-) составила от -1,2 до 0,6%, а точность от 5,2 до 6,0%; правильность положительного режима определения GL (GL+) составила от -5,9 до +5,9%, а точность составила от 3,3 до 7,7%; правильность определения GA составила от -7,0 до +7,2%, а точность составила от 3,0 до 7,7%. Из указанных выше результатов был сделан вывод, что пределы определяемых концентраций GL и GA составили, соответственно, 20 нг/мл и 80 нг/мл.
Пример 2
Было проведено исследование влияния типа щелочи или спирта, которые смешивают с биологическим образцом, на количественную чувствительность высокочувствительного способа определения количества полученных из лекарственных трав компонентов по настоящему изобретению.
50 мкл стандартного раствора (Е) из примера 1 и 50 мкл IS добавляли к 0,5 мл плазмы крови человека, а затем тщательно перемешивали для получения образцов плазмы крови.
Количественно определяли GL и GA по той же методике, что и в примере 1, за исключением того, что 1 мл растворов, приведенных в таблице 5, добавляли к каждому образцу плазмы крови для приготовления смеси, для введения в МАХ.
Рассматривая каждый из результатов количественного определения с учетом того, что случай, когда раствором, смешиваемым с образцом плазмы крови, была 0,56 об.% аммиачная вода, берут за 100% степени извлечения, рассчитывали относительные степени извлечения GL, GA и IS для каждого образца плазмы крови. Результаты расчетов приведены в таблице 5. Цифры, указанные в скобках после "аммиачной воды" в таблице 5, показывают концентрацию аммиака (об.%) в смеси, полученной путем добавления аммиачной воды.
В результате, когда использовали щелочь или спирт, степени извлечения оказались лучше, чем когда использовали воду. И наоборот, когда добавляли кислотный раствор, такой как фосфорная кислота или хлорная кислота, GL обнаружить не удавалось, и извлечь GA практически не удалось. Среди спиртов, степени извлечения как для GL, так и для GA были лучше, когда использовали метанол, чем когда использовали этанол. Среди щелочей результаты были лучше, когда использовали аммиачную воду (водный раствор аммиака), чем когда использовали водный раствор гидроксида натрия или водный раствор гидрокарбоната натрия. Тем не менее, различия при разных концентрациях аммиачной воды едва наблюдались.
Пример 3
Было проведено исследование влияния типа очищающей жидкости для твердой фазы на количественную чувствительность высокочувствительного способа определения количества полученных из лекарственных трав компонентов по настоящему изобретению.
50 мкл стандартного раствора (E) и 50 мкл IS из примера 1 добавляли к 0,5 мл плазмы крови человека, а затем тщательно перемешивали для получения образцов плазмы крови.
Количественно определяли GL и GA таким же образом, как в примере 1, за исключением того, что после промывания МАХ растворами, представленными в таблице 6, после введения образца плазмы крови осуществляли дополнительное промывание с метанолом.
Рассматривая каждый из результатов количественного определения с учетом того, что случай, когда очищающая жидкость МАХ после введения образца плазмы крови состояла из 0,56 об.% аммиачной воды (водного раствора аммиака) и раствора метанола (жидкая смесь с объемным соотношением 1:1), принимали за 100% степени извлечения, рассчитывали относительные степени извлечения GL, GA и IS для каждого образца плазмы крови. Результаты расчетов приведены в таблице 6. Соотношения, приведенные в скобках после наименования раствора, соответствуют соотношению смеси соответствующего раствора.
В результате, хотя степень извлечения GA была в некоторой степени низкой только в случае, когда промывание проводили с 0,0025М водным раствором гидроксида натрия/раствором метанола (жидкая смесь с объемным соотношением 1:1), степени извлечения GL, GA и IS были удовлетворительными во всех случаях, когда в качестве очищающей жидкости использовали воду, аммиачную воду или смешанный раствор аммиачной воды и метанола.
Пример 4
Было проведено исследование влияния типа очищающей жидкости для твердой фазы на количественную чувствительность высокочувствительного способа определения количества полученных из лекарственных трав компонентов по настоящему изобретению.
50 мкл стандартного раствора (E) и 50 мкл IS из примера 1 добавляли к 0,5 мл плазмы крови человека, а затем тщательно перемешивали для получения образца плазмы крови.
Количество GL и GA определяли таким же образом, как в примере 1, за исключением того, что после промывки МАХ 0,56 об.% раствором аммиачной воды/метанолом (жидкая смесь с объемным соотношением 1:1), следующей за введением образца плазмы крови, осуществляли дополнительную промывку с этанолом, метанолом, 50 об.% водным раствором метанола или ацетонитрилом.
Рассматривая случай, когда второй очищающей жидкостью был метанол, за 100%, рассчитывали относительные степени извлечения GL, GA и IS для каждого образца плазмы крови. Результаты расчетов приведены в таблице 7. В результате, когда промывание осуществляли с любым из используемых растворителей, степень извлечения GL была удовлетворительной - 80% или более. Степень извлечения GA также была удовлетворительной - 80% или более, за исключением одного случая, когда использовали 50 об.% водный раствор метанола. В частности, использование метанола дало наиболее благоприятные результаты.
Пример 5
Было проведено исследование влияния типа элюата из твердой фазы на количественную чувствительность высокочувствительного способа определения количества полученных из лекарственных трав компонентов по настоящему изобретению.
50 мкл стандартного раствора (E) и 50 мкл IS из примера 1 добавляли к 0,5 мл плазмы крови человека, а затем тщательно перемешивали для получения образца плазмы крови.
Количество GL и GA определяли таким же образом, как в примере 1, за исключением того, что элюирование из МАХ осуществляли с помощью растворов, приведенных в таблице 8.
Рассматривая случай, когда элюат представлял собой муравьиную кислоту-метанол, за 100%, вычисляли относительные степени извлечения GL, GA и IS для каждого образца плазмы крови. Результаты вычислений представлены в таблице 8. В результате, степени извлечения были самыми высокими в случае использования муравьиной кислоты-метанола. GL и GA также были извлечены с удовлетворительным результатом при использовании муравьиной кислоты-этанола, хотя и не до такой степени, как в случае муравьиной кислоты-метанола. Напротив, в случае, когда элюат представлял собой муравьиную кислоту-ацетонитрил или фосфорную кислоту-метанол, GL обнаружить не удалось.
Сравнительный пример 1
Количественное определение GL и GA в плазме крови проводили с использованием вместо МАХ твердой фазы, обладающей только обращенно-фазовой распределительной функцией.
50 мкл стандартного раствора (E) и 50 мкл IS из примера 1 добавляли в 0,5 мл плазмы крови человека, и тщательно перемешивали для получения образцов плазмы крови.
В целом, проводили ту же самую процедуру, что и в примере 1, за исключением следующего: вместо МАХ был использован Sep-Pak (3 см3, 60 мг, 30 мкм, производимый Waters Corp.), представляющий собой колонку ODS; требуемые условия в колонке устанавливали сначала с помощью метанола, а затем воды; 0,56 об.% аммиачную воду или 0,1 N соляную кислоту использовали в качестве раствора (разбавителя) для получения смеси для подачи в колонку; воду или смесь ацетонитрил/вода (объемное соотношение = 1:3, содержит 1 М бромида тетрабутиламмония) использовали в качестве очищающей жидкости для твердой фазы; и в качестве элюата для твердой фазы использовали 2 об.% муравьиную кислоту-метанол, 2 об.% муравьиную кислоту-ацетонитрил/воду (объемное соотношение 1:1) или 2 об.% муравьиную кислоту-метанол/воду (объемное соотношение 1:1).
Рассматривая случай, когда раствором, перемешиваемым с образцом плазмы крови в примере 2, являлась 0,56 об.% аммиачная вода, за 100% степени извлечения, рассчитывали относительные степени извлечения GL, GA, и IS для каждого образца плазмы крови. Результаты расчетов приведены в таблице 9. В таблице 9 "1-1" означает 0,56 об.% аммиачную воду (водный раствор аммиака), "1-2" означает 0,1 N соляную кислоту, "2-1" означает воду, "2-2" означает ацетонитрил/воду (объемное соотношение = 1:3, содержит 1 М бромида тетрабутиламмония), "3-1" означает 2 об.% муравьиную кислоту-метанол, "3-2" означает 2 об.% муравьиную кислоту-ацетонитрил/воду (объемное соотношение = 1:1), и "3-3" означает 2 об.% муравьиную кислоту-метанол/воду (объемное соотношение 1:1).
В результате, степень извлечения GL составила самое большее 50% даже в случае самой большой степени извлечения среди комбинаций разбавителя и т.д., из чего четко видно, что чувствительность обнаружения в отношении GL и т.д. была достаточно низкой, по сравнению со случаями, когда использовали МАХ.
Сравнительный пример 2
Количественное определение уровней GL и GA в плазме крови проводили с использованием вместо МАХ твердой фазы, обладающей обращенно-фазовой распределительной функцией и функцией полярного взаимодействия.
50 мкл стандартного раствора (E) и 50 мкл IS из примера 1 добавляли в 0,5 мл плазмы крови человека, и тщательно перемешивали для получения образцов плазмы крови.
В целом, проводили ту же самую процедуру, что и в примере 1, за исключением следующего: вместо МАХ использовали Oasis HLB (3 см3, 60 мг, 30 мкм, производимый Waters Corp.), представляющий собой пористый полимер; требуемые условия в колонке устанавливали сначала с помощью метанола, а затем воды; в качестве раствора (разбавителя) для получения смеси для подачи в колонку использовали 0,56 об.% аммиачную воду или 0,1 N соляную кислоту; 5 об.% водный раствор метанола использовали в качестве очищающей жидкости для твердой фазы.
Рассматривая случай, когда раствором, перемешиваемым с образцом плазмы крови в примере 2, являлась 0,56 об.% аммиачная вода (водный раствор аммиака), как 100% степени извлечения, рассчитывали относительные степени извлечения GL, GA, и IS для каждого образца плазмы крови. Результаты расчетов приведены в таблице 10.
В результате, в случае, когда в качестве разбавителя использовали 0,1 N соляную кислоту, GL обнаружить не удалось, а степень извлечения GA была очень низкой. С другой стороны, даже в том случае, когда использовали 0,56 об.% водный раствор аммиака, степень извлечения GL не превышала 55%, из чего однозначно следует, что чувствительность по отношению к GL и т.д. была намного ниже, чем в тех случаях, когда использовали МАХ.
Сравнительный пример 3
Количественное определение GL и GA в плазме крови проводили с использованием вместо MAX твердой фазы, обладающей обращенно-фазовой распределительной функцией и функцией положительного ионнообмена.
50 мкл стандартного раствора (E) и 50 мкл IS из примера 1 добавляли в 0,5 мл плазмы крови человека, и тщательно перемешивали для получения образцов плазмы крови.
В целом, проводили ту же самую процедуру, что и в примере 1, за исключением следующего: вместо МАХ использовали Oasis МСХ (3 см3, 60 мг, 30 мкм, производимый Waters Corp.,); требуемые условия в колонке создавали сначала с использованием метанола, а затем воды; в качестве раствора (разбавителя) для получения смеси для подачи в колонку использовали 0,1 N соляную кислоту; в качестве очищающей жидкости для твердой фазы использовали 0,1 N соляную кислоту; и в качестве элюата для твердой фазы использовали 2 об.% раствор аммиачной воды/метанол.
Рассматривая случай, когда раствором, перемешиваемым с образцом плазмы крови в примере 2, являлся 0,56 об.% водный раствор аммиака, в качестве 100% степени извлечения, рассчитывали относительные степени извлечения GL, GA, и IS для каждого образца плазмы крови. Результаты расчета для 5 независимых испытаний и их средние значения представлены в таблице 11.
В результате, степени извлечения GL и GA не достигали и 50%, что четко указывает на то, что чувствительность обнаружения по отношению к GL и т.д. была гораздо хуже, чем в тех случаях, когда был использован МАХ.
Пример 6
Измеряли предельные определяемые количества GL и GA в плазме крови и строили калибровочные кривые для случая, когда высокочувствительный способ определения количества полученных из лекарственных трав компонентов по настоящему изобретению осуществляли с использованием ЖХ-МС/МС метода в качестве метода масс-спектрометрии. В дополнение к этому подтверждали точность и надежность способа по изобретению.
Получение стандартного раствора
Стандартные исходные растворы, используемые в примере 1, разбавляли метанолом по той же самой процедуре, что и примере 1, для получения стандартных растворов (B)-(G), приведенных в таблице 12.
Приготовление образцов плазмы крови и стадия экстракции
Образцы плазмы человеческой крови, к которым добавляли, соответственно, стандартные растворы (B)-(G), а также контрольные (без добавок) образцы плазмы крови человека получали по той же методике, что и в примере 1. Образцы плазмы крови были, соответственно, введены в МАХ, чтобы добиться адсорбции на нем GL и т.д., и затем осуществляли промывку 0,56 об.% раствором аммиачной воды/метанолом, а затем чистым метанолом, после чего проводили элюирование 2 об.% раствором муравьиной кислоты/метанолом и полученную смесь упаривали досуха.
Стадия количественного определения
Выпаренный досуха экстракт растворяли в 250 мкл нижеприведенной подвижной фазы: растворы A/растворы B (жидкие смеси с объемным соотношением 1:1). 1 мкл каждого продукта вводили в аналитическую колонку и проводили ЖХ-МС/МС в условиях, приведенных ниже.
ЖХ-МС/МС система: API 4000 система (произведено Applied Biosystems Corp.)
(ЖХ)
Устройство: LC-20A (произведено Shimadzu, Ltd.)
Аналитическая колонка: Inertsil ODS-3 (2,1 мм × 150 мм, 5µ размер зерна, произведено GL Sciences, Inc.)
Скорость потока: 0,50 мл/мин.
Температура колонки: 40°C
Температура автоматического дозатора: 5°C
Количество вводимого образца: 1 мкл
Подвижная фаза: раствор A - 0,1 об.% муравьиная кислота; раствор В 0,1 об.% муравьиная кислота-ацетонитрил; линейный градиент (Таблица 13)
(МС/МС)
Устройство: API 4000 (Applied Biosystems) Способ ионизации: ESI (турбо-спрей)
Способ детектирования: детектирование положительных ионов MRM (мониторинг множественных реакций)
Напряжение ионного спрея: 5500 B (положительное)
Температура источника ионов (температура): 400°C
Защищающий поток газа: 10 (азот)
Газообразный источник ионов 1: 70 (азот)
Газообразный источник ионов 2: 50 (азот)
Газ для соударений: 8 (азот)
Фиг.1A-фиг.1C представляют собой хроматограммы контрольных образцов плазмы крови. Фиг.1A показывает положение пика GL (время удерживания GL; M/z 823→453), фиг.1B показывает положение пика GA (время удерживания GA; м/z 471→149), и фиг.1C показывает положение пика IS (время удерживания IS; м /z 751→455). Фиг.2A-фиг.2C представляют собой хроматограммы образцов плазмы крови, к которым добавляли стандартный раствор (G) (концентрация GL в плазме крови: 0,5 нг/мл, концентрация GA: 2 нг/мл). Фиг.2A показывает пик GL (показано стрелкой на фигуре), фиг.2B показывает пик GA (показано стрелкой на фигуре), и фиг.2C показывает пик IS (показано стрелкой на фигуре). Время удерживания GL, GA и IS составляло приблизительно 1,2 минуты, 3,1 минуты и 1,4 минуты, соответственно; никаких пиков, препятствующих количественному определению, в хроматограммах исходных образцов плазмы крови не наблюдали, и формы пиков GL, GA и IS были удовлетворительными.
Таблица 15 показывает результаты количественного определения. Относительные погрешности в фактических добавленных концентрациях и измеренных значениях (количественные значения в таблице) также показаны в таблице 15. В результате, относительные погрешности как GL, так и GA были в пределах 15%. Таким образом, результаты показывают, что высокочувствительный способ определения количества полученных из лекарственных трав компонентов по настоящему изобретению с использованием ЖХ-МС/МС позволяет количественно определить GL и т.д. в биологических образцах с высокой степенью точности в случае, когда концентрация GL составляет 0,5-200 нг/мл, а концентрация GA составляет 2-800 нг/мл для таких биологических образцов, как плазма крови.
Исследование специфичности, линейности и воспроизводимости в течение дня/в разные дни
Затем проводили исследование специфичности, линейности и воспроизводимости в течение дня/в разные дни в соответствии "Guidance for Industry, Bioanalytical Method Validation," US Department of Health and Human Services, Food and Drug Administration, May 2001. В дополнение проводили также исследование стабильности (сохранение стабильности в биологических образцах при комнатной температуре в течение 4 ч и при низкотемпературном хранения в течение трех месяцев при температуре -20°C и -80°C, стабильность при замораживании и оттаивании, стабильность в измерении реальных образцов и стандартная стабильность раствора). Результаты показаны в таблице 16. В результате, высокочувствительный способ определения количества полученных из лекарственных трав компонентов по настоящему изобретению с использованием ЖХ-МС/МС показал удовлетворительную линейность и воспроизводимость в диапазонах 0,5-200 нг/мл и 2-800 нг/мл. Что касается результатов воспроизводимости в течение дня, то правильность результатов для GL составила от -12,8 до +4,8%, а точность составила от 4,0 до 9,5%; правильность для GA составила от -13,4 до +4,4%, а точность составила от 2,2 до 9,0%. С другой стороны, в том, что касается воспроизводимости в разные дни, правильность GL составила от -9,4 до +2,0%, а точность составила 2,1 до 5,5%, правильность для GA составила от -10,9 до +8,1%, а точность составила от 1,3 до 9,1%. Результаты воспроизводимости подтвердили, что пределы определяемых концентраций GL и GA составляют, соответственно, 0,5 нг/мл и 2 нг/мл.
Во всех тестах стабильности (сохранение стабильности в биологических образцах, стабильность при замораживании и оттаивании, стабильность в измерении реальных образцов и стабильность стандартного раствора), результаты находились в пределах ±15% от начального значения. Из приведенных результатов был сделан вывод, что в соответствии с «Guidance for Industry, Bioanalytical Method Validation» US Food and Drug Administration высокочувствительный способ определения количества полученных из лекарственных трав компонентов по настоящему изобретению является способом количественного определения с доказанной надежностью.
Пример 7
Детектировали GL в плазме крови после перорального введения GL-содержащих составов и измеряли его фармакокинетику.
Данный эксперимент был осуществлен в соответствии с этическими принципами, основанными на Хельсинской Декларации, согласно статье 14, параграфу 3 и статье 80(2) Закона о фармацевтике, а также в соответствии с "Ministerial Ordinance on Good Clinical Practice (GCP)" (Ministry of Health and Welfare Ordinance No. 28, 1997). В частности, пероральное введение GL-содержащего состава и сбор образцов плазмы крови проводился по просьбе авторов в Клинике EPS Corporation и Yamaguchi под руководством главного исследователя после получения одобрения независимого комитета. Измерение GL и GA в полученных образцах плазмы крови проводили при помощи Ecotechno Division (в настоящее время Pharmaceutical, Perfumery and Cosmetics Analysis Division) компании Japan Clinical Laboratories, Inc.
Что касается испытуемых субъектов, то после предоставления полного объяснения целей и содержания проводимых исследований, а также политики защиты личной информации и тому подобного, были отобраны шесть здоровых мужчин-японцев (возраст: от 20 до 35, ИМТ (индекс массы тела) во время предварительного осмотра: от 18,5 или более до 25,0 или менее), которые дали письменное согласие на участие в исследовании в качестве участника.
Покрытые сахаром таблетки с наименованием "таблетки GLYCYRON (товарный знак)", содержащие глицирризинат моноаммония, глицин и DL-метионин, использовались в качестве GL-содержащих составов. Таблетки GLYCYRON вводили перорально однократно в обычной дозе из трех таблеток с достаточным количеством воды. Таблица 17 показывает расписание исследования, включая время введения и время забора крови. 1 мл или более плазмы крови извлекали, подвергая собранную кровь центробежной обработке для разделения (4°C, 3000 об./мин, 10 мин) в течение 30 минут от момента сбора крови, и хранили в замороженном состоянии при -20°C или ниже до момента измерения концентрации.
После добавления 1 мл 0,56 об.% водного раствора аммиака к 0,5 мл собранному образцу плазмы крови и последующего тщательного перемешивания осуществляли ту же процедуру, что и в примере 1, в ходе которой полученная смесь наносилась на МАХ для адсорбции на нем GL и т.д. и промывалась 0,56 об.% водным раствором аммиака/метанолом, а затем метанолом, после чего проводили элюирование 2 об.% раствором муравьиной кислоты/метанолом с последующим выпариванием досуха.
Затем осуществляли анализ методом ЖХ-МС/МС, как это описано в примере 6, в отношении выпаренного досуха экстракта и измеряли концентрацию GL и концентрацию GA в каждом образце плазмы крови. Фиг.3 иллюстрирует изменения средней концентрации GL в плазме крови, а фиг.4 иллюстрирует изменения средней концентрации GA в плазме крови. Фиг.3 и фиг.4 показывают средние значения и стандартные отклонения для 6 образцов. В результате, было подтверждено, что высокочувствительным способом определения количества полученных из лекарственных трав компонентов по настоящему изобретению можно непосредственно измерить концентрацию в плазме крови не только GA, которая является основным метаболитом, ранее используемом в качестве альтернативного индикатора, но также и непосредственно GL, который является немодифицированным активным веществом. То есть настоящее изобретение впервые позволяет обнаруживать небольшие количества GL в плазме крови и проясняет кинетику крови GL, которая долгое время была неясна.
Анализ фармакокинетики
Максимальная концентрация в плазме крови (Сmax) и время ее достижения (Тmax) вычисляли на основе данных измерений GL и GA. Площадь под зависимостью (AUC) концентрации плазмы крови от времени вплоть до 48 ч после введения (AUC 0→48 ч) вычисляли методом трапеций, а константу скорости выведения (Кэл) и период полувыведения (t1/2) вычисляли методом наименьших квадратов по фазе элиминации. Кроме того, вычисляли площадь под зависимостью концентрации плазмы крови от времени вплоть до бесконечного времени после введения (AUC 0-∞) и среднее время удержания (MRT).
В результате, в настоящем примере Cmax GL находилась в диапазоне приблизительно 10-40 нг/мл, а ее средняя величина составила 24,8 нг/мл. Так как величина была почти идентична величине, предсказанной на основе вышеупомянутого эксперимента по пероральному введению больших доз коммерчески доступного реагента (приблизительно 23 нг/мл), то была проиллюстрирована корреляция между дозировкой GL и концентрацией в плазме крови. Tmax составило в среднем 4,5 ч, но также наблюдали испытуемых, у которых наблюдалось множество пиков концентрации до (1-2 ч) или после (12 ч) этого момента. Так как последний пик концентрации проявлялся после приема пищи, было предположено, что потребление пищи может влиять на поглощение GL или что имеет место энтерогепатическая циркуляция.
С другой стороны, в отношении GA, которая является основным метаболитом GL, было подтверждено, что имела место 4-часовая задержка до того момента, когда ее присутствие в плазме кроме было подтверждено, а также то, что ее концентрация в плазме крови после 6 ч была приблизительно в десять раз (приблизительно в 20 раз в пересчете на моли) больше, чем концентрация GL. В результате in vitro инкубации GL в оптимальных условиях с использованием человеческих микросом печени, несмотря на превращение в 3-моноглюкуронил-глицирретиновую кислоту, которая является промежуточным метаболитом, был подтверждено и превращение в небольшое количество GA (Biochemical Pharmacology, 42 (6/7), 1025-1029, 1991). В результате перорального введения GL стерильным крысам присутствие GA в плазме крови или кале подтвердить не удалось, и поэтому был сделан вывод, что выработка GA in vivo регулируется кишечными бактериями (ссылка: J. Pharm. Pharmacol. 46, 135-137, 1994). Поэтому предполагается, что задержка абсорбции соответствует времени, необходимому для контактирования с кишечной флорой, и считается, что прием пищи (или условия голодания) негативно влияют на «заметность» GA путем воздействия на продвижение лекарственного препарата внутри желудочно-кишечного тракта, на метаболическую способность кишечной флоры, и на всасывание в пищеварительном тракте. Во время наблюдения у испытуемого кинетики GA после абсорбции, было обнаружено, что появление метаболитов была менее выраженным у испытуемых с высокой концентрацией неизменного вещества и наоборот, появление метаболитов было более выраженным у испытуемых с низкой концентрацией неизменного вещества. Таким образом, было предположено, что наряду с GA всасывание GL, который является неизменным веществом, в желудочно-кишечном тракте также связано с метаболической способностью кишечной флоры, которая приводит к соответствующим индивидуальным различиям (данные не опубликованы).
Промышленная применимость
Высокочувствительный способ определения количества полученных из лекарственных трав компонентов по настоящему изобретению позволяет определить уровень GL в крови после перорального поступления клинической дозы GL-содержащего состава, продукта питания или тому подобного, и, следовательно, этот способ может быть использован для контроля использования фармацевтических препаратов или Китайской медицины на лекарственных травах, при испытаниях на безопасность, фармакокинетических анализах, разработке новых GL-составов и тому подобном.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ количественного определения дексаметазона в биологических средах с помощью ВЭЖХ с ультрафиолетовым детектированием | 2022 |
|
RU2792274C1 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ 8-(ТРИФТОРМЕТИЛ)БЕНЗО[F][1,2,3,4,5]ПЕНТАТИЕПИН-6-АМИНА ГИДРОХЛОРИДА В БИОЛОГИЧЕСКИХ СРЕДАХ | 2018 |
|
RU2676487C1 |
| КОМПОЗИЦИИ, СОДЕРЖАЩИЕ РАСЩЕПЛЯЕМЫЕ ФЕРМЕНТАМИ ОПИОИДНЫЕ ПРОЛЕКАРСТВА С МОДИФИЦИРОВАННЫМ КЕТОНОМ И ИХ ДОПОЛНИТЕЛЬНЫЕ ИНГИБИТОРЫ | 2010 |
|
RU2600736C2 |
| Способ определения дабигатрана в сыворотке крови человека | 2018 |
|
RU2683032C1 |
| КОМПОЗИЦИИ, СОДЕРЖАЩИЕ РАСЩЕПЛЯЕМОЕ ФЕРМЕНТАМИ ПРОЛЕКАРСТВО ОКСИКОДОНА | 2012 |
|
RU2609412C2 |
| Способ количественного определения дисульфирама в биологических средах | 2019 |
|
RU2701524C1 |
| Способ определения лозартана, его основного метаболита лозартан карбоновой кислоты и глибенкламида в сыворотке крови и моче человека | 2020 |
|
RU2749567C1 |
| Способ определения амиодарона и его основного метаболита дезэтиламиодарона в сыворотке крови человека | 2020 |
|
RU2749566C1 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ РИВАРОКСАБАНА В КРОВИ МЕТОДОМ ВЫСОКОЭФФЕКТИВНОЙ ЖИДКОСТНОЙ ХРОМАТОГРАФИИ | 2025 |
|
RU2839935C1 |
| Способ количественного определения антигипертензивных лекарственных веществ в плазме крови | 2022 |
|
RU2803887C1 |

Изобретение относится к высокочувствительному способу определения количества глицирризина, глицирретиновой кислоты и их фармакологически приемлемых солей, присутствующих в плазме крови человека. Высокочувствительный способ определения количества глицирризина, глицирретиновой кислоты и их фармакологически приемлемых солей характеризуется тем, что смесь плазмы крови человека с метанолом или раствором аммиачной воды с определенной концентрацией вводят в твердую фазу, обладающую обращенно-фазовой распределительной функцией и функцией анионного обмена, затем промывают твердую фазу очищающей жидкостью, представляющей собой однокомпонентную жидкость или жидкую смесь, по меньшей мере, двух компонентов, выбранных из группы, включающей воду, щелочь, спирт и ацетонитрил. Далее проводят элюирование из твердой фазы кислым спиртом, выбранным из муравьиной кислоты-метанола или муравьиной кислоты-этанола, после чего проводят стадию количественного определения глицирризина, глицирретиновой кислоты и их фармакологически приемлемых солей методом ЖХ-МС или ЖХ-МС/МС. Высокочувствительный способ позволяет обнаружить и количественно определить глицирризин, глицирретиновую кислоту и их фармакологически приемлемые соли в плазме крови человека. 4 ил., 17 табл., 7 пр.
Высокочувствительный способ определения количества глицирризина, глицирретиновой кислоты и их фармакологически приемлемых солей, присутствующих в плазме крови человека, характеризующийся тем, что данный способ включает:
введение смеси плазмы крови человека с метанолом или раствором аммиачной воды с такой концентрацией, чтобы концентрация аммиака в полученной смеси составляла 0,05-20 об. %, в твердую фазу, обладающую обращенно-фазовой распределительной функцией и функцией анионного обмена;
промывание твердой фазы очищающей жидкостью, представляющей собой однокомпонентную жидкость или жидкую смесь по меньшей мере двух компонентов, выбранных из группы, включающей воду, щелочь, спирт и ацетонитрил; и
последующее элюирование из твердой фазы кислым спиртом, выбранным из муравьиной кислоты-метанола или муравьиной кислоты-этанола; и
стадию количественного определения глицирризина, глицирретиновой кислоты и их фармакологически приемлемых солей методом ЖХ-МС или ЖХ-МС/МС.
| Sakamaki N | |||
| Et al | |||
| simultaneous determination of stevioside, rebaudioside A and glycyrrhizic acid in foods by HPLC | |||
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| О.Б | |||
| РУДАКОВ и др | |||
| Физико-химические системы сорбат-сорбент-элюент в жидкостной хроматографии | |||
| - | |||

