Родственная заявка
Данная заявка испрашивает приоритет по предварительной заявки на патент Индии №409/MUM/2011, поданной 14 февраля 2011 года.
Область изобретения
Настоящее изобретение относится к аналитическим методам, таким как определение молекулярной массы полипептида, в частности, глатирамера ацетата. Настоящее изобретение также относится к улучшенному способу получения полипептидов или их фармацевтически приемлемых солей, в частности, глатирамера ацетата, также известного как сополимер-1. Настоящее изобретение также относится к определению характеристик глатирамера ацетата пептидным картированием.
Предпосылки создания изобретения
Одно из наиболее распространенных неврологических заболеваний взрослых людей представляет собой рассеянный склероз. Это состояние является хроническим, воспалительным заболеванием ЦНС, которое характеризуется демиелинизацией в головном мозге и спинном мозге. Глатирамера ацетат (ГА), известный также как сополимер-1, как было показано, является эффективным при лечении рассеянного склероза (PC). Ежедневные подкожные инъекции глатирамера ацетатом (20 мг/инъекцию) уменьшают патологические изменения, частоту рецидивов и прогрессирование нетрудоспособности (Johnson K.P. et al, Neurol., 1995, 45 (7): 1268-76). Глатирамера ацетат уменьшает долю новых поражений PC, прогрессирующих в «черные дыры» (Filippi М. et al, Neurol., 2001, 57: 731-733).
Глатирамера ацетат (Сополимер-1) обозначен торговой маркой КОПАКСОН®. Он предназначен для снижения частоты рецидивов у пациентов с рецидивирующе-ремиттирующим рассеянным склерозом (РРМС). Глатирамера ацетат состоит из ацетатных солей синтетических полипептидов, содержащих четыре встречающиеся в природе аминокислоты: L-глутаминовую кислоту, L-аланин, L-тирозин и L-лизин со средней мольной долей 0,141, 0,427, 0,095 и 0,338, соответственно. Он синтезируется путем химической полимеризации четырех аминокислот с получением продукта с желаемым диапазоном молекулярных масс. Средняя молекулярная масса глатирамера ацетата составляет 4,700-11,000 дальтон [маркировка США для лиофилизированного порошка] или 5000-9000 дальтон [маркировка США для предварительно заполненного шприца/общая характеристика продукта]. Копаксон содержит смесь полипептидов, имеющих различные молекулярные массы и последовательности.
Структурная формула Копаксона представляет собой
(Glu, Ala, Lys, Tyr)х. xCH3COOH
(C5H9NO4. C3H7NO2.C6H14N2O2.C9H11NO3)x. xC2H4O2
Способ получения глатирамера ацетата описан в Euro. J. Immun. 1, 242-248 (1971) [Tietelbaum et al] и US3849550 [Tietelbaum et al]. В US 3849550 раскрыт способ, в котором N-карбоксиангидриды тирозина, аланина, γ-бензил-глутамата и ε-N-трифторацетил-лизина полимеризуют при комнатной температуре в безводном диоксане с диэтиламином в качестве инициатора. Удаление защитной γ-карбоксильной группы глутаминовой кислоты осуществляется с помощью бромистого водорода в ледяной уксусной кислоте, за которым следует удаление трифторацетильных групп от остатков лизина при помощи 1М пиперидина.
Способ получения сополимера-1 также описан в US 5800808, IN 190759, US 5981589, US 6054430, US 6342476, US 6362161 и WO 00/05250. Эти документы подробно останавливаются на способе получения сополимера-1, включающем полимеризацию четырех N-карбоксиангидридов аминокислот, чтобы получить защищенный сополимер и снятие защиты с защищенного сополимера с использованием бромистого водорода в уксусной кислоте и пиперидине для получения сополимера с удаленной защитной группой, который затем подвергают диализу для очистки и солевого обмена.
В US 20060172942 раскрывается способ получения глатирамера ацетата, который включает полимеризацию N-карбоксиангидридов тирозина, аланина, γ-бензилглутамата и трифторацетил-лизина, чтобы получить защищенные полипептиды. Бензильная защитная группа из защищенных полипептидов удаляется путем каталитического гидрогенолиза с последующим удалением трифторацетильной защитной группы за счет взаимодействия полипептида с органическим основанием. Свободные трифторацетильные группы и низкомолекулярные примеси удаляют из полипептидной смеси путем ультрафильтрации с последующим контактированием полипептидной смеси с водным раствором уксусной кислоты с получением соли уксусной кислоты полипептида.
В US 7049399 описывается способ получения полипептида, содержащего полимеризацию смеси N-карбоксиангидридов L-аланина, L-тирозина, защищенного L-глутамата и защищенного L-лизина, чтобы получить защищенный сополимер или его соль; снятие защиты с защищенного сополимера (или его соли), чтобы получить полипептид или его фармацевтически приемлемую соль в одну стадию, разделение и очистку полипептида (или его фармацевтически приемлемой соли), с получением очищенного полипептида. Снятие защиты с защищенного сополимера осуществляют либо путем передачи каталитического гидрирования или каталитическим гидрированием под давлением водорода в пределах от 40-100 фунтов на квадратный дюйм (psi) и при температуре в диапазоне от 50°-80°C.
Недостатком по отношению к способу, описанному в этих документах, является то, что гидрогенолиз требует высоких давления и температуры, что в свою очередь, требует дополнительных мер предосторожности при работе в крупных масштабах, тем самым увеличивая стоимость производства. Так как Копаксон является стабильным при низкой температуре от 2°C до 8°C, высокая температура во время последней стадии может привести к разложению продукта, снижая тем самым качество и выход целевого продукта.
В WO 2006050122 описан двухстадийный способ получения глатирамера ацетата. Способ включает полимеризацию смеси N-карбоксиангидрида L-тирозина, N-карбоксиангидрида L-аланина, N-карбоксиангидрида защищенного L-глутамата и N-карбоксиангидрида N-защищенного L-лизина, в полярном апротонном растворителе в присутствии инициатора с образованием защищенного глатирамера, добавление кислоты и/или органического или неорганического основания к образованному защищенному глатирамеру, чтобы формировать глатирамер, и обработку полученного глатирамера с уксусной кислотой с образованием глатирамера ацетата. Снятие защиты с защищенного глатирамера осуществляется двумя различными способами. Первый способ для удаления защитных групп заключается в обработке кислотой и второй способ для снятия защиты осуществляют путем обработки гидроксидом щелочноземельного металла. Недостатком указанного способа является то, что использование гидроксида щелочноземельного металла для удаления защитных групп у пептидов приводит к медленным реакциям и образованию высоких уровней диастереомера в результате рацемизации/эпимеризации стереогенных центров (Ahmed F. Abdel-Magid et al, Tetrahedron Letters 39, 3391 (1998)).
В WO 2006029393 раскрыт способ получения смеси трифторацетил- полипептидов, у которых могут быть разные аминокислотные последовательности, где каждый полипептид состоит, по существу, из аланина, глутаминовой кислоты, тирозина и лизина, где смесь имеет желаемую среднюю молекулярную массу, содержащий удаление защитной группы у смеси полипептидов, каждый из которых состоит по существу из аланина, γ-бензилглутамата, тирозина и трифторацетил-лизина с помощью раствора бромистоводородной кислоты в уксусной кислоте, причем раствор содержит менее, чем 0,5% свободного брома и менее, чем 1000 частей на миллион (мд) примесей ионов металлов. В данном документе также раскрыто, что раствор бромистоводородной кислоты в уксусной кислоте, имеющей менее чем 0,5% свободного брома, может быть получен путем предварительной обработки раствора брома акцептором, предпочтительно фенолом, чтобы уменьшить уровень свободного брома. Также в этом документе раскрыто использование от 10% до 36% бромистоводородной кислоты в уксусной кислоте.
В US 20080021200 раскрыт способ получения полипептида, содержащего L-тирозин, L-аланин, L-глутамат и L-лизин или их фармацевтически приемлемую соль, где указанный способ включает: (а) полимеризацию смеси N-карбоксиангидрида L-тирозина, N-карбоксиангидрида L-аланина, N-карбоксиангидрида защищенного L-глутамата и N-карбоксиангидрида защищенного L-лизина, в полярном апротонном растворителе в присутствии инициатора, с образованием защищенного полипептида, (b) смешивание кислоты с защищенным полипептидом, полученным на стадии (а) и растворителем, с образованием продукта, и (с) смешивание вещества, выбранного из группы, состоящей из диизопропиламина, изопропиламина, аммиака и их смесей, с продуктом, полученным на стадии (b) и водой, или смеси растворителя и воды, чтобы сформировать полипептид с удаленной защитной группой или его фармацевтически приемлемую соль. Кроме того, раскрывается, что диизопропиламин и изопропиламин были единственными аминами, которые успешно удалили Nε-трифторацетильную группу из фрагмента лизина. Также в данном документе раскрыто, что, когда диизопропиламин или изопропиламин используются для удаления трифторацетильных групп, получают прозрачный раствор через 1 ч или примерно через 1,5 часа, соответственно.
В WO 2009016643 описывается способ получения глатирамера ацетата (Сополимер-1), где трифторацетил-сополимер-1, полученный после реакции дебензилирования, промывают органическим растворителем, чтобы удалить реакционный бензилбромид, вырабатываемый в качестве побочного продукта реакции. Высвобожденный бензилбромид является высоко реакционноспособным электрофилом и реагирует с нуклеофилами, подобно первичным и вторичным аминам, чтобы генерировать нежелательные N-алкилированные продукты. Также он высоко слезоточивый и обработка его в больших количествах в промышленных масштабах является вредной и небезопасной. В WO′643 также раскрывается реакция дебензилирования защищенного сополимера-1 в течение более короткого периода времени при высокой температуре и способ для удаления бензилбромида из реакционной смеси. Удаление бензильной защитной группы глутаминовой кислоты зависит от 33% бромистого водорода в уксусной кислоте при температуре 35-45°C в течение 1-5 часов, тем самым уменьшая время реакции, и для того, чтобы удалить бензилбромид, отфильтрованный продукт промывают органическим растворителем. Этот способ использует дополнительный органический растворитель, который является нежелательным в промышленном масштабе. Таким образом, способы получения глатирамера ацетата, как описано выше, являются промышленно неосуществимыми.
Авторы настоящего изобретения разработали простой и экономически эффективный, рентабельный и унифицированный способ получения сополимера-1 (глатирамера ацетата). Применение 33% HBr приводит к расщеплению пептидных связей в полимере приводя к получению низкомолекулярных соединений. Настоящее изобретение обеспечивает более рентабельный способ получения глатирамера ацетата с использованием примерно 7% до 20% HBr, предпочтительно, около 15% HBr, в уксусной кислоте для удаления бензильной защитной группы при комнатной температуре, предпочтительно приблизительно при 23°C до 30°C, более предпочтительно при около 25°C, наиболее предпочтительно при 25°C±0,2°C. Настоящее изобретение также относится к способу получения глатирамера ацетата, где влажное твердое вещество, полученное после первой стадии удаления защитных групп, не подвергают сушке и используют как таковое в последующей реакции, что экономит много времени и, следовательно, считается промышленно осуществимым способом. Настоящее изобретение также относится к способу удаления трифторацетильной защитной группы с использованием амина, предпочтительно вторичного амина, такого как диалкиламин, выбранного из диэтиламина, диметиламина или диизопропиламина, предпочтительно, диэтиламина.
Предшествующий уровень техники обеспечивает способы определения молекулярной массы полипептидов, в частности глатирамера ацетата. В WO 200018794 (WO′794) раскрывается использование множества маркеров молекулярной массы, чтобы установить связь между временем удерживания на хроматографической колонке и молекулярной массой для определения средней молекулярной массы партии глатирамера ацетата. Каждый из этих маркеров молекулярной массы, раскрытый в WO′794, представляет собой полипептид, и имеет предварительно определенную аминокислотную последовательность и определенную молекулярную массу. Настоящее изобретение относится к применению фракций полипептида (маркеров молекулярной массы), полученных, воздействуя на Копаксон/глатирамера ацетат/глатирамоиды эксклюзионной хроматографией (ЭХ), или гельпроникающей хроматографией (ГПХ), для определения молекулярной массы партий глатирамера ацетата. Полипептидные фракции (маркеры молекулярной массы), полученные способом по настоящему изобретению обладают определенной молекулярной массой. Однако аминокислотная последовательность указанной полипептидной фракции является случайной.
Другой аналитический метод, используемый для определения характеристик полипептида/белка представляет собой пептидное картирование. Пептидное картирование является сравнительной процедурой, в которой полученная информация, по сравнению с образцом для сравнения или стандартным веществом, обрабатываемым аналогичным образом, подтверждает первичную структуру полипептида/белка. Это помогает обнаружить, произошли ли изменения в структуре, и свидетельствует о постоянстве процесса и генетической стабильности. В WO 2010129851 предлагается способ, где гидролиз ферментами используют, чтобы расщепить стандартную сложную смесь полипептидов, такую как глатирамера ацетат, на несколько фрагментов пептида. Пептидные фрагменты анализируют с помощью масс-спектрометрии. Результаты масс-спектрометрии каждого образца используют как «отпечаток» для сравнения с другими образцами. Каждый фрагмент пептида, обнаруженный первым масс-анализатором, выбирают и подвергают второму масс-спектрометрическому анализу для расщепления ионов предшественника пептида на еще более мелкие фрагменты. Масс-спектры, полученные из тандемного масс спектрометрического анализа, анализируются с помощью программного обеспечения, такого как BioTools, чтобы получить последовательность каждого пептидного фрагмента.
В Expert Opin. Pharmacother. (2009) 10 (4) раскрывается, что полипептидное картирование с помощью разделения капиллярным электрофорезом полипептидных фрагментов, полученных после расщепления трипсином и картирование на основе протеолитического гидролиза с помощью карбоксипептидазы P, с последующим разделением фрагментов обращенно-фазовой ВЭЖХ является методом определения различий между последовательностями ГА структуры и других структур глатирамоидов. Карбоксипептидаза P является дорогим и труднодоступным реагентом. Авторы настоящего изобретения испробовали различные комбинации протеолитических ферментов. Было обнаружено авторами настоящего изобретения, что смесь полипептидов, обработанная трипсином с последующей обработкой карбоксипептидазой B дает результаты, аналогичные результаты с карбоксипептидазой P.
Задача изобретения
Задачей настоящего изобретения является создание маркеров молекулярной массы и их использование при определении молекулярной массы партий глатирамера ацетата посредством ГПХ.
Другой целью настоящего изобретения является создание простого, экономически эффективного, рентабельного и унифицированного способа получения полипептидов, в частности, глатирамера ацетата.
Другой целью настоящего изобретения является создание способа получения трифторацетил-сополимера путем обработки защищенного сополимера примерно от 7% до 20% HBr для получения сополимера-1.
Еще одной целью настоящего изобретения является создание способа получения сополимера-1 (глатирамера ацетата) обработкой защищенного сополимера или трифторацетил-сополимера диалкиламином, выбранным из диэтиламина, диметиламина, или диизопропиламина, предпочтительно диэтиламина, чтобы получить сополимер с частично удаленной защитной группой или сополимер с полностью удаленной защитной группой.
Другой целью настоящего изобретения является создание способа получения глатирамера ацетата, где влажное твердое вещество, полученное после первой стадии удаления защитных групп, не подвергают сушке и используют как таковое в последующей реакции, что экономит много времени и, следовательно, считается промышленно применимым способом.
Другая цель настоящего изобретения относится к способу определения характеристик полипептидной смеси, в частности глатирамера ацетата, с использованием трипсина и карбоксипептидазы В.
Сущность изобретения
Настоящее изобретение относится к способу получения полипептидных фракций со случайной последовательностью аминокислот, аналогичной последовательности глатирамера ацетата, которые служат в качестве маркеров молекулярной массы для глатирамера ацетата, содержащему стадии,
а) воздействие на полипептидную смесь, которая подобна глатирамера ацетату по аминокислотному составу и молярному соотношению аминокислот, гель-проникающей хроматографией (ГПХ)/эксклюзионной хроматографией (ЭХ), чтобы получить полипептидные фракции, имеющие молекулярные массы в диапазоне от 2кДа до 30 кДа;
b) выбор полипептидных фракций, которые будут использоваться в качестве маркеров молекулярной массы из указанных полипептидных фракций, полученных на стадии а), таким образом, что одна из фракций представляет собой вершину пика молекулярной массы, а другие фракции распределены по обе стороны от вершины пика молекулярной массы, и
c) определение коэффициента асимметрии пика этих выбранных полипептидных фракций.
Предпочтительно, коэффициент асимметрии пика выбранных полипептидных фракций находится в диапазоне от 0,8 до 1.
Предпочтительно, полидисперсность указанных выбранных полипептидных фракций составляет меньше, чем или равна 1,20.
Предпочтительно, смесь полипептидов представляет собой глатирамера ацетат.
Еще один аспект настоящего изобретения относится к применению полипептидных фракций, полученных способом по настоящему изобретению, в качестве маркеров молекулярной массы для определения молекулярной массы партии глатирамера ацетата посредством способа, содержащего следующие стадии
a) калибровка ЭХ-ВЭЖХ колонки с использованием указанных выбранных полипептидных фракций;
b) установление взаимосвязи между временем удерживания и логарифмом (log) молекулярной массы указанных выбранных фракций с помощью калибровочной кривой;
c) проведение ЭХ-ВЭЖХ партии Глатирамера ацетата, молекулярная масса которого должна быть определена, чтобы получить время удерживания;
d) использование соотношения между временем удерживания и логарифмом молекулярной массы для определения молекулярной массы партии глатирамера ацетата.
Предпочтительно, ЭХ-ВЭЖХ колонку выбирают из Superose-12 или Superdex-75, подвижную фазу выбирают от 0,1 до 0,3 М ацетата аммония или формиата аммония, и скорость потока регулируют в диапазоне от 0,4 до 0,6 мл/мин.
Другой аспект настоящего изобретения обеспечивает способ проверки стабильности и воспроизводимости способа получения маркеров молекулярной массы, при этом способ содержит стадии,
а) получение множественного набора маркеров молекулярной массы, с использованием способа по настоящему изобретению, из различных партий глатирамера ацетата/Копаксона, каждый набор, содержащий маркеры молекулярной массы, имеет молекулярную массу в диапазоне от 2000 до 13000 дальтон;
b) сопоставление маркеров молекулярной массы из каждого набора, полученного на стадии а), с использованием статистического способа рандомизации, чтобы дополнительно выделить больший набор маркеров молекулярной массы, и
c) использование этих более крупных наборов, полученных на стадии b) в качестве маркеров молекулярной массы для определения молекулярной массы партии Копаксон/глатирамера ацетат посредством ГПХ.
Предпочтительно, маркеры молекулярной массы, используемые на стадии с) обеспечивают предсказуемые результаты, основанные на 95% доверительного интервала.
Предпочтительно, глатирамера ацетат, используемый для приготовления маркеров молекулярной массы получают с помощью способа, включающего следующие стадии
a) обработку защищенного сополимера с примерно 7% до 20% HBr в уксусной кислоте, при температуре около 25°C в течение около 21-23 часов, чтобы получить трифторацетил-сополимер;
b) обработку указанного трифторацетил-сополимера с диэтиламином, чтобы получить сополимер с полностью удаленной защитной группой;
c) превращение сополимера с полностью удаленной защитной группой в глатирамера ацетат.
Другой аспект настоящего изобретения обеспечивает способ получения глатирамера ацетата, содержащего стадии
a) обработку защищенного сополимера с примерно 7% до 20% HBr в уксусной кислоте при температуре около 25°C в течение примерно 21-23 часов, чтобы получить трифторацетил-сополимер;
b) обработку указанного трифторацетил-сополимера с основанием, выбранным из диэтиламина, диметиламина или диизопропиламина, чтобы получить сополимер с полностью удаленной защитной группой;
c) превращение сополимера с полностью удаленной защитной группой в глатирамера ацетат.
Предпочтительно, обработку на стадии а) осуществляют при температуре 25°C±0,2°C, HBr в уксусной кислоте, необязательно предварительно обрабатывают акцептором, выбранным из фенола, резорцина, тирозина, бисульфита натрия, тиосульфат натрия, нафталина, 1-нафтола или 2-нафтола; и основание выбирают из диэтиламина.
Другой аспект настоящего изобретения обеспечивает превращение сополимера с полностью удаленной защитной группой до глатирамера ацетата, содержащий стадии
а) концентрирование водного раствора сополимера с полностью удаленной защитной группой до минимального объема с использованием тангенциальной поточной фильтрации пропуская через кассету мембраны из полиэфирсульфона, имеющей номинальное отсечение по молекулярной массе 1 кДа;
b) промывка концентрированного раствора со стадии а), водой до достижения pH от 8 до 9, с последующей промывкой 0,3% раствором уксусной кислоты для солевого обмена, чтобы получить глатирамера ацетат; и
c) промывка глатирамера ацетата, полученного на стадии b), водой до достижения pH от 5,5 до 7, чтобы получить чистый глатирамера ацетат.
Предпочтительно, глатирамера ацетат, полученный способом по настоящему изобретению, имеет содержание бромированного тирозина не более, чем 0,15% и содержание диэтиламида не более, чем 5000 частей на миллион (мд). Предпочтительно, глатирамера ацетата, полученный способом по настоящему изобретению, имеет среднюю молекулярную массу в диапазоне 5000-9000 дальтон.
Предпочтительно, глатирамера ацетат, полученный способом по настоящему изобретению, используется в приготовлении фармацевтической композиции или в виде лиофилизированного твердого вещества или в виде водного раствора, содержащих глатирамера ацетат в диапазоне концентраций приблизительно 25-45 мг/мл.
Другой аспект настоящего изобретения относится к способу для определения характеристик глатирамера ацетата пептидным картированием с использованием трипсина и карбоксипептидазы B, содержащем стадии
a) ферментативной обработки глатирамера ацетата с использованием трипсина для получения пептидных короткоцепочечных фрагментов;
b) обработку полученных пептидных фрагментов карбоксипептидазой В, что приводит к дальнейшей фрагментации пептидной цепи;
c) отделение и анализ пептидных фрагментов, полученных на стадии b) с использованием ВЭЖХ-УФ, чтобы получить хроматограмму, и
d) сравнение, полученной таким образом, хроматограммы с хроматограммой образца для сравнения, чтобы определить структурное сходство.
Предпочтительно, образцом для сравнения, используемым для пептидного картирования, является Копаксон® (продаваемый состав).
Краткое описание фигур
Фиг.1: График, показывающий фракционирование Копаксона® партия №Р53289.
Фиг.2: Наложение профиля пика выбранных фракций (маркеров молекулярной массы).
Фиг.3: Калибровочная кривая фракционированных маркеров молекулярной массы.
Фиг.4: Определение молекулярной массы Копаксона® партия №53119 ГПХ с использованием фракционированных маркеров молекулярной массы.
Фиг.5: График, показывающий фракционирование глатирамера ацетата, полученного способом по настоящему изобретению.
Фиг.6: Калибровочная кривая фракционированного глатирамера ацетата, полученного способом по настоящему изобретению.
Фиг.7: Определение молекулярной массы глатирамера ацетата с использованием маркеров, приведенных в таблице 4.
Фиг.8: Наложение хроматограммы сравнения расщепления структуры Копаксона® и глатирамера ацетат, полученного способом по настоящему изобретению, при обработке ферментом трипсином.
Фиг.9: Наложение хроматограммы сравнения расщепления структуры Копаксона® и глатирамера ацетата, полученного способом по настоящему изобретению, при обработке трипсином с последующей обработкой ферментом карбоксипептидазой В.
Подробное описание изобретения
Настоящее изобретение относится к маркерам молекулярной массы для определения молекулярной массы полипептидов, в частности глатирамера ацетата (Сополимер-1).
Маркеры молекулярной массы, используемые в настоящем изобретении, являются полипептидными фракциями, состоящими из полипептидов со случайной последовательностью аминокислот подобной последовательности глатирамера ацетата. Предпочтительно, указанные маркеры молекулярной массы содержат аланин, глутаминовую кислоту, тирозин и лизин в том же мольном соотношении, которое присутствует в глатирамера ацетате.
Один из вариантов осуществления настоящего изобретения относится к полипептидным фракциям со случайной аминокислотной последовательностью подобной последовательности глатирамера ацетата, которые служат в качестве маркеров молекулярной массы для глатирамера ацетата, полученного по способу, содержащему следующие стадии
a) воздействие на полипептидную смесь, которая аналогична глатирамера ацетату по аминокислотному составу и молярному соотношению аминокислот, гель-проникающей хроматографией (ГПХ)/эксклюзионной хроматографией (ЭХ), чтобы получить полипептидные фракции, имеющие молекулярные массы в диапазоне от 2 кДа до 30 кДа;
b) выбор полипептидных фракций, которые будут использоваться в качестве маркеров молекулярной массы, из указанных полипептидных фракций, полученных на стадии а) таким образом, что одна из фракций представляет собой вершину пика молекулярной массы, а другие выбранные фракции распределены по обе стороны от вершины пика молекулярной массы, и
с) определение коэффициента асимметрии пика этих выбранных полипептидных фракций.
Предпочтительно, коэффициент асимметрии пика этих выбранных полипептидных фракций находится в диапазоне от 0,8 до 1. Пик симметрии, как измерено при помощи коэффициента асимметрии пика, имеет большое значение в хроматографическом анализе. Когда коэффициент асимметрии пика увеличивается, интеграция становится менее надежной.
Коэффициент асимметрии пика может быть рассчитан в соответствии с методом фармакопейного комитета США (USP).
Предпочтительно, полидисперсность этих отдельных полипептидных фракций составляет меньше, чем или равна 1,20.
В другом варианте осуществления, настоящее изобретение относится к способу получения полипептидных фракций со случайной аминокислотной последовательностью аналогичной последовательности глатирамера ацетата, которые служат в качестве маркеров молекулярной массы для глатирамера ацетата, содержащего стадии
a) воздействие на полипептидную смесь, которая аналогична глатирамера ацетату по аминокислотному составу и молярному соотношению аминокислот, гель-проникающей хроматографией (ГПХ)/эксклюзионной хроматографией (ЭХ), чтобы получить полипептидные фракции, имеющие молекулярные массы в диапазоне от 2 кДа до 30 кДа;
b) выбор полипептидных фракций, которые будут использоваться в качестве маркеров молекулярной массы, из указанных полипептидных фракций, полученных на стадии а) таким образом, что одна из фракций представляет собой вершину пика молекулярной массы, а другие выбранные фракции распределены по обе стороны от вершины пика молекулярной массы, и
c) определение коэффициента асимметрии пика этих выбранных полипептидных фракций.
Предпочтительно, коэффициент асимметрии пика этих выбранных полипептидных фракций находится в диапазоне от 0,8 до 1. Полидисперсность указанных выбранных полипептидных фракций, предпочтительно составляет меньше, чем или равна 1,20.
Предпочтительно, полипептидная смесь представляет собой Копаксон® или глатирамера ацетат, полученный способом по настоящему изобретению или любым другим известным способом.
Колонка, используемая в хроматографической методике выбирается из Superose-12 или Superdex-75, предпочтительно Superose-12. Подвижная фаза выбрана из ацетата аммония или формиата аммония. Ацетат аммония или формиат аммония используют в диапазоне концентрации от 0,1 до 0,3 М, предпочтительно 0,2 М. pH подвижной фазы составляет приблизительно от 4 до 6, предпочтительно от 5. Скорость потока и количество Копаксон®/глатирамера ацетата раствора, которое загружают в колонку зависит от радиуса и длины колонки. Детекторы, используемые для хроматографического метода, выбирают из УФ-детектора, рефрактометрического детектора, с рассеиванием лазерного излучения с кратными углами, детектора рассеивания лазерного излучения с кратными углами (РЛИКУ), детектора флуоресценции или их комбинации.
В предпочтительном варианте осуществления настоящего изобретения, Копаксон®/глатирамера ацетата приблизительно от 0,5 до 4 мг, предпочтительно 2 мг, подвергают ГПХ (ЭХ) на колонке Superose-12 или Superdex-75 с использованием 0,2 М формиата аммония или ацетата аммония, pH 5,0, в качестве подвижной фазы. 22 отдельные фракции собирают в интервальном промежутке в течение 1 минуты, как на фиг.1. Все фракции лиофилизируют отдельно. Из 22 фракций, пять фракций выбирают таким образом, чтобы выбрать две от каждой стороны вершины пика и фракцию вершины пика. Определяют коэффициент асимметрии пика этих полипептидных фракций. Коэффициент асимметрии пика предпочтительно находится в диапазоне от 0,8 до 1. Эти выбранные фракции суспендируют в 0,2 М ацетате аммония или формиате аммония при pH 5,0, и конечная концентрация каждой из этих фракций составляет 1-10 мг/мл, предпочтительно 6 мг/мл. Молекулярную массу этих выбранных фракций определяют с помощью ЭХ-РЛИКУ. Определяют Mp (вершину пика молекулярной массы), Mw (среднемассовую молекулярную массу), Mn (среднечисленную молекулярную массу) и полидисперсность каждой выбранной фракции. Почти одинаковые значения Mp, Mw и Mn предполагают, что эти фракции являются почти монодисперсными. Полидисперсность этих фракций составляет менее 1,05. Указанные охарактеризованные фракции используют в качестве маркеров молекулярной массы для определения Mp, Mw и Mn партий глатирамера ацетата. Каждая из этих полипептидных фракций состоит из полипептида со случайной аминокислотной последовательностью. Калибровочная кривая отображается с использованием логарифма (log) молекулярной массы и времени удерживания этих фракций, как показано на фиг.3. Коэффициент корреляции (R2) для калибровочной кривой составляет 0,996.
Условия ЭХ-ВЭЖХ, используемые для фракционирования Копаксон® являются такими, как приведены в таблице 1 ниже.
Mp (Вершина пика молекулярной массы), Mw (среднемассовая молекулярная масса) и Mn (среднечисленная молекулярная масса) для выбранной фракции приведены в таблице 2, ниже
Индекс полидисперсности (ИПД) является мерой распределения молекулярной массы в данном образце полимера. Он рассчитывается путем деления среднемассовой молекулярной массы на среднечисленную молекулярную массу. Почти одинаковые значения Mp, Mw и Mn предполагают, что эти фракции являются почти монодисперсными.
В другом предпочтительном варианте осуществления настоящего изобретения, около 40 мг глатирамера ацетата подвергают ГПХ на Superose-12 (10/300) GL системы с использованием 0,2 М ацетата аммония, pH 5,0, в качестве подвижной фазы. Глатирамер начинает элюироваться через 20 минут. Около 30 фракций собирают с интервалом 0,5 мин. Выбирают от 12 до 15 фракций. Эти фракции анализируют на коэффициент асимметрии пика, молекулярную массу и поли дисперсность. Фракции выбраны таким образом, что одна из этих фракций представляет вершину пика молекулярной массы, а другие фракции распределены по обе стороны от вершины пика молекулярной массы. Фракции с полидисперсностью меньше, чем 1,2 отбирают и лиофилизируют. Фракции с полидисперсностью более чем 1,2 подвергают дальнейшей очистке с целью получения фракций, имеющих полидисперсность меньше, чем 1,2. Предпочтительно, выбирают фракции с молекулярной массой 3 кДа, 5 кДа, 7 кДа, 10 кДа и 12 кДа, чтобы использовать в качестве маркеров молекулярной массы.
Условия ГПХ, используемые для фракционирования Глатирамера ацетата, полученного способом по настоящему изобретению, представлены в таблице 3 ниже
Молекулярную массу указанных выбранных фракций определяют, используя ЭХ-РЛИКУ. Mp и полидисперность указанных выбранных фракций приведены в таблице 4, ниже
Любое количество фракций может быть выбрано для использования в качестве маркеров. Критерием для отбора является то, что одна из фракций должна представлять вершину пика молекулярной массы, а другие фракции, которые выбраны, должны быть распределены по обе стороны от вершины пика молекулярной массы.
Предпочтительно, коэффициент асимметрии пика этих отобранных фракций находится в диапазоне от 0,8 до 1.
Эти выбранные полипептидные фракции, которые будут использоваться в качестве маркеров молекулярной массы, далее характеризованы посредством ЯМР и соотношения аминокислот. Способ получения маркеров молекулярной массы согласно настоящему изобретению является стабильным и воспроизводимым. Маркеры молекулярной массы, полученные способом по настоящему изобретению, имитируют глатирамера ацетата по показателям гидродинамического объема.
В другом варианте осуществления, настоящее изобретение относится к применению выбранных полипептидных фракций, которые служат в качестве маркеров молекулярной массы для определения молекулярной массы партии глатирамера ацетата, при помощи способа, содержащему следующие стадии
a) калибровка колонки ЭХ/ГПХ с использованием выбранных полипептидных фракций;
b) установление взаимосвязи между временем удерживания и логарифмом молекулярной массы этих выбранных фракций посредством калибровочной кривой;
c) проведение ЭХ/ГПХ партии глатирамера ацетата, молекулярную массу которого определяют, чтобы получить время удерживания;
d) использование соотношения между временем удерживания и логарифмом молекулярной массой для определения молекулярной массы партии глатирамера ацетата.
Другой вариант осуществления настоящего изобретения относится к способу для определения молекулярной массы партии глатирамера ацетата с использованием выбранных полипептидных фракций, которые служат в качестве маркеров молекулярной массы, содержащему стадии
a) калибров колонки ЭХ/ГПХ с использованием выбранных полипептидных фракций;
b) установление взаимосвязи между временем удерживания и логарифмом молекулярной массы этих полипептидных фракций с помощью калибровочной кривой;
c) проведение ЭХ/ГПХ партии глатирамера ацетата, молекулярная которого масса должна быть определена, чтобы получить время удерживания;
d) использование соотношения между временем удерживания и логарифмом молекулярной массы для определения молекулярной массы партии глатирамера ацетата.
Предпочтительно, определяют вершину пика молекулярной массой глатирамера ацетата.
В предпочтительном варианте изобретения, вершина пика молекулярной массы глатирамера ацетат определяется посредством ЭХ-ВЭЖХ с использованием маркеров молекулярной массы, указанных здесь. Колонку ЭХ-ВЭЖХ калибруют с использованием выбранных полипептидных фракций, которые служат в качестве маркеров молекулярной массы, как указано в настоящем документе. Соотношение между временем удерживания и логарифмом молекулярной массы этих маркеров молекулярной массы устанавливают путем построения калибровочной кривой, как показано на фиг.3. Копаксон®/глатирамера ацетат подвергают ЭХ-ВЭЖХ на Superose-12 или Superdex-75 колонке, предпочтительно Superose-12 (10×300 мм) с использованием 0,2 М формиата аммония или ацетата аммония, pH 5,0, предпочтительно, ацетата аммония, в качестве подвижной фазы. Вводимый объем, составляющий 50 мкл образца, имеющего концентрацию около 2 мг/мл, наносят на колонку. Скорость потока поддерживается при 0,5 мл/мин и общее время работы составляет 60 мин. Вершину пика молекулярной массы определяют с помощью УФ-детектора при длине волны 275 нм. С помощью стандартной кривой, две партии Копаксон® и глатирамера ацетата анализируют, как показано на фиг.4, с использованием маркеров молекулярной массы, указанных в таблице 2 выше, и результаты показаны в таблице 5 ниже:
Результаты исследования для ГПХ КОПАКСОН® с использованием маркеров молекулярной массы, указанных в таблице 2 выше, приведены в таблице 6, ниже:
Две партии Копаксон® и глатирамера ацетата анализируют, как показано на фиг.7 с использованием маркеров молекулярной массы, указанных выше в таблице 4, а результаты показаны в Таблице 7 ниже.
Партии Копаксона/глатирамера ацетата, проанализированные указанным способом имеют среднюю молекулярную массу между 5000-9000 Дальтон.
Партия глатирамера ацетата, чья молекулярная масса определяется с помощью указанного выше способа, может быть использована для получения маркеров молекулярной массы, как указано выше.
Другой вариант осуществления настоящего изобретения обеспечивает способ для анализа стабильности и воспроизводимости способа получения маркеров молекулярной массы с использованием статистических методов.
Способ включает следующие стадии:
a) получение множественного набора маркеров молекулярной массы, с использованием способа по настоящему изобретению из различных партий глатирамера ацетата/Копаксона, каждый набор содержит маркеры молекулярной массы, имеющие молекулярную массу в диапазоне от 2000 до 13000 дальтон;
b) распределение маркеров молекулярной массы из каждого набора, полученного на стадии а), с использованием статистического способа рандомизации для дальнейшего выделения большего набора маркеров молекулярной массы, и
c) использование этих более крупных наборов, полученных на стадии b) в качестве маркеров молекулярной массы для определения молекулярной массы Копаксона/глатирамера ацетата партии при помощи ГПХ.
В предпочтительном варианте осуществления, пять наборов маркеров молекулярной массы выделяют с использованием разных партий глатирамера ацетата. Каждый набор содержит семь маркеров молекулярной массы, имеющих разные молекулярные массы в диапазоне от 2000 до 13000 дальтон, как показано ниже в таблице 8.
Статистический способ рандомизации применяют к указанным пяти наборам маркеров молекулярной массы для получения большего набора маркеров молекулярной массы, каждый набор содержит семь маркеров молекулярной массы, как в таблице 9 ниже
Эти более крупные наборы маркеров затем используются для определения вершины пика молекулярной массы партии глатирамера ацетата, и результаты показаны ниже в таблице 10,
Это исследование рандомизации показывает, что любое изменение комбинации маркеров молекулярной массы, взятых из различных наборов обеспечит предсказуемые результаты на основе 95% доверительного интервала. Это показывает, что способ по настоящему изобретению для получения маркеров молекулярной массы является стабильным, воспроизводимым и не зависит от изменения партии к партии исходного материала. Маркеры молекулярной массы, выделенные с использованием способа по настоящему изобретению из различных партий глатирамера ацетата, обеспечивают предсказуемые результаты на основе 95% доверительного интервала.
В альтернативном варианте осуществления настоящего изобретения, молекулярные массы глатирамера ацетата могут быть определены с помощью глатирамоидов в качестве маркеров молекулярной массы. Глатирамоиды являются гетерогенной полипептидной смесью, содержащей четыре аминокислоты, L-глутаминовую кислоту, L-аланин, L-лизин и L-тирозин в том же мольном соотношении, что и глатирамер. Глатирамоиды с различными молекулярными массами получают гашением реакционной смеси в различные промежутки времени во время стадии снятия защиты HBr - уксусной кислотой, с последующим снятием защиты с использованием аминов, таких как диэтиламин, диметиламин, диизопропиламин или пиперидин, предпочтительно, диэтиламин.
В другом варианте осуществления, настоящее изобретение обеспечивает улучшенный способ получения полипептидов, в частности, глатирамера ацетата, также известного как сополимер-1, со средней молекулярной массой 5000-9000 дальтон, из аминокислот, а именно, L-тирозина, L-аланина, L-глутаминовой кислоты и L-лизина или их фармацевтически приемлемой соли.
В соответствии с одним из вариантов осуществления настоящего изобретения, предлагается способ получения сополимера-1 (глатирамера ацетата) со средней молекулярной массой 5000-9000 дальтон, содержащий стадии:
a) обработку защищенного сополимера с примерно 7% до 20% HBr в уксусной кислоте при температуре около 25°C в течение примерно 21-23 часов, чтобы получить трифторацетил-сополимер;
b) обработку указанного трифторацетил-сополимера основанием с получением сополимера с полностью удаленной защитной группой;
c) превращение сополимера с полностью удаленной защитной группой в сополимер-1.
Защищенный сополимер получают полимеризацией смеси N-карбоксиангидрида L-аланина, N-карбоксиангидрида γ-защищенного L-глутамата, N-карбоксиангидрида ε-N-защищенного L-лизина и N-карбоксиангидрида L-тирозина в присутствии инициатора, чтобы получить защищенный сополимер. Защитная группа (ЗГ) для свободной γ-карбоксильной группы L-глутаминовой кислоты выбирается из замещенного или незамещенного алкила, арила или аралкильной группы, предпочтительно, бензильной группы или замещенной бензильной группы, и защитную группу (ЗГ) для свободной ε-аминогруппы L-лизина выбирают из ацила или замещенного ацила, где заместители выбраны из алкила, арила или галогена, предпочтительно, трифторацетильной группы.
В предпочтительном варианте осуществления настоящего изобретения, смесь N-карбоксиангидрида L-аланина, N-карбоксиангидрида из γ-бензил-L-глутамата, N-карбоксиангидрида Nε-трифторацетил-L-лизина и N-карбоксиангидрида L-тирозина, растворяют в подходящем растворителе, предпочтительно безводном диоксане в течение 1 часа, предпочтительно от 30 мин, более предпочтительно 15 мин. К этой смеси добавляют 1% диэтиламина в безводном диоксане в один прием. Реакционную смесь перемешивают при комнатной температуре в течение 24 часов при 200-215 оборотов в минуту. После завершения реакции, деионизированную воду добавляют к реакционной смеси, чтобы получить защищенный сополимер виде белого твердого вещества. Полученное твердое вещество отфильтровывают под вакуумом, промывают деионизированной водой и сушат до достижения потери при сушке (LOD) NMT 10%. Защищенный сополимер обрабатывают примерно 7% до 20% бромистого водорода (HBr), предпочтительно от примерно 15% HBr, в уксусной кислоте и смесь перемешивают при около 25°C, предпочтительно при 25°C±0,2°C, в течение приблизительно 21-23 часов при 200-215 оборотов в минуту до получения трифторацетил-сополимера. Затем реакционную смесь выливают в ледяную воду, чтобы получить белое твердое вещество, которое затем нейтрализуют путем обработки раствором аммиака до достижения pH 6-7 и перемешивают в течение 24 часов. Твердое вещество отфильтровывают и промывают водой. Эта обработка полученной суспензии твердого вещества с раствором аммиака также разлагает бензилбромид, который выделяется во время реакции.
Сополимер с частично удаленной защитной группой, трифторацетил-сополимер, полученный таким образом, преобразуют в глатирамера ацетат способом, описанным в нашей заявке, по которой принято решение о выдаче патента, индийской патентной заявке 1082/MUM/2009 от 23 апреля 2009, или любым другим известным способом.
Предшествующий уровень техники описывает удаление бензильной защитной группы глутаминовой кислоты с использованием 33% бромистого водорода в уксусной кислоте. Использование 33% HBr-уксусной кислоты (жесткие условия для пептидов) может привести к нестабильному и случайному расщеплению пептидных связей в полимере, которое при дальнейшем снятии защиты приводит к получению глатирамера ацетата, который имеет нестабильную молекулярную массу. Способ в соответствии с настоящим изобретением применяет использование примерно 7% до 20% HBr, предпочтительно от около 15%, до около 25°C в течение примерно 21-23 часов для удаления бензильной защитной группы, которые при дальнейшем снятии защиты приводит к глатирамера ацетату, имеющему молекулярные массы, которые являются стабильными и воспроизводимыми.
Согласно еще одному варианту осуществления настоящего изобретения, около 7% до 20% HBr, предпочтительно, около 15% HBr, в уксусной кислоте подают в реактор с регулируемой температурой при комнатной температуре и перемешивают при 200-215 оборотов в минуту в течение 20-60 минут, предпочтительно, 30 минут, до достижения приблизительно 25°C. Акцептор добавляют к реакционной смеси и перемешивают в течение 1-3 часов, предпочтительно, в течение 2 часов, при комнатной температуре, предпочтительно при температуре около 25°C, при 200-215 оборотов в минуту с последующим добавлением защищенного сополимера в реактор и перемешивают при комнатной температуре, предпочтительно при около 25°C, более предпочтительно при температуре 25°C±0,2°C, при 200-215 оборотов в минуту в течение 20-25 часов, предпочтительно 21-23 часов. Реакционную смесь охлаждают в холодной воде с получением белого твердого вещества с последующим добавлением раствора аммиака до достижения pH 6-7 и перемешивают в течение 24 часов. Полученное твердое вещество, трифторацетил-сополимер фильтруют, промывают водой и сушат на воздухе. Акцептор, использованный для снижения содержания свободного брома, выбирают из фенола, резорцина, тирозина, бисульфита натрия, тиосульфата натрия, нафталина, 1-нафтола и 2-нафтола, предпочтительно, фенола или резорцина.
В другом варианте осуществления настоящее изобретение относится к способу получения глатирамера ацетата, который включает воздействие на защищенный сополимер для уменьшения размера частиц, таким образом, чтобы получить однородные по размеру частицы перед снятием защиты. Это обеспечивает стабильность и воспроизводимость в молекулярных массах партий глатирамера ацетата. В предпочтительном варианте осуществления, защищенный сополимер, пропускают через сито 30 меш таким образом, чтобы получить однородный размер частиц.
В другом варианте осуществления настоящее изобретение относится к способу получения сополимера-1 (глатирамера ацетата), который включает обработку защищенного сополимера или трифторацетил-сополимера с диалкиламином, таким как диэтиламин, диметиламин, диизопропиламин, предпочтительно, диэтиламин, чтобы получить сополимер с частично удаленной защитной группой или сополимер с полностью удаленной защитной группой и превращение полученного сополимера с частично удаленной защитной группой или сополимера с полностью удаленной защитной группой в сополимер-1 (глатирамера ацетат).
В предпочтительном варианте осуществления настоящего изобретения, сополимер с частично удаленной защитной группой, трифторацетил-сополимер, суспендируют в подходящем растворителе, предпочтительно в воде и обрабатывают диэтиламином с последующим перемешиванием в течение 20-30 часов, предпочтительно 24 часов, при комнатной температуре. Реакционную смесь пропускают через фильтр 0,2µ, подвергают тангенциальной поточной фильтрации через кассету, имеющую номинальное отсечение по молекулярной массе 1 кДа, диафильтрации с деионизированной водой до достижения pH 8-9, затем с 0,3% водного раствора уксусной кислоты с последующим промывкой деионизированной водой до достижения pH 5,5-7,0. Полученный раствор затем лиофилизируют с получением сополимера-1 (глатирамера ацетат) с желаемым диапазоном молекулярной массы. Полученное твердое вещество проявляет среднюю молекулярную массу в диапазоне от 5000 до 9000 дальтон, определенную посредством ГПХ и РЛИКУ. Альтернативно, указанный раствор может быть принят как таковой для состава:
В способе согласно настоящему изобретению, диэтиламин используется в качестве инициатора, а также агента для снятия защиты. Диэтиламин, при использовании в меньших количествах в диапазоне от 0,5% до 5% по отношению к NCAs L-аминокислот действует как инициатор и при использовании в больших количествах в диапазоне от 1 до 5 раз больше массы защищенного сополимера действует как агент для снятия защиты для удаления Nε-трифторацетильных групп.
Схема реакции показана на Схеме II, ниже:
В другом предпочтительном варианте осуществления настоящего изобретения, способ получения сополимера-1 (глатирамера ацетата) содержит стадии
a) полимеризация смеси N-карбоксиангидрида L-аланина, N-карбоксиангидрид γ-защищенного L-глутамата, N-карбоксиангидрида ε-N-защищенного L-лизина и N-карбоксиангидрида L-тирозина в присутствии инициатора, чтобы получить защищенный сополимера;
b) необязательная обработка защищенного сополимера, чтобы получить однородные по размеру частицы;
c) обработка защищенного сополимера со стадии а) или Ь) примерно 7% до 20% HBr, предпочтительно около 15% HBr, в уксусной кислоте при комнатной температуре, предпочтительно при около 25°C, более предпочтительно при 25°C±0,2°C, в течение приблизительно 21-23 часов, чтобы получить сополимер с частично удаленной защитной группой, трифторацетил-сополимер;
d) обработка полученного сополимера с частично удаленной защитной группой, трифторацетил-сополимера, с диэтиламином, чтобы получить полимер с полностью удаленной защитной группой;
e) очистка сополимера с полностью удаленной защитной группой на очистку и подвергание его солевому обмену.
Предпочтительно, защищенный сополимер, однородный по размеру частиц, может быть получен путем пропускания защищенного сополимера через сито с желаемым размером ячеек, предпочтительно 30 меш.
Альтернативный вариант осуществления настоящего изобретения относится к способу получения глатирамера ацетата, где последовательность снятия защиты является обратной, содержащей первую обработку защищенного сополимера с диэтиламином, чтобы удалить трифторацетильную группу, а затем с HBr/уксусной кислотой для удаления бензильной группы.
Другой вариант осуществления настоящего изобретения относится к способу получения глатирамера ацетата, где влажное твердое вещество, полученное после первой стадии удаления защитной группы, не подвергают сушке и используют как таковое в последующей реакции, что экономит много времени и, следовательно, считается промышленно осуществимым способом. Схема реакции показана на схеме III ниже 
В другом варианте осуществления, настоящее изобретение относится к способу очистки и солевого обмена сополимера с полностью удаленной защитной группой (глатирамера) с помощью ультрафильтрации, содержащий стадии
a) концентрирование водного раствора сополимера с полностью удаленной защитной группой (глатирамера) до минимального объема с использованием тангенциальной поточной фильтрации пропусканием через кассету мембраны из полиэфирсульфона, имеющей номинальное отсечение по молекулярной массе 1 кДа;
b) промывка концентрированного раствора со стадии а) водой до достижения pH от 8 до 9 с последующей промывкой 0,3% раствором уксусной кислоты для солевого обмена, чтобы получить глатирамера ацетат;
c) промывка полученного глатирамера ацетата водой до достижения pH 5,5 до 7, чтобы получить чистый глатирамера ацетат.
Вода, используемая для описанного выше способа, предпочтительно, представляет собой воду RO (обратный осмос) или WFI (вода для инъекций).
Нежелательные низкомолекулярные полипептиды, образующиеся в ходе процесса, удаляют фильтрованием. Методы фильтрования, используемые, чтобы удалить полипептиды с нежелательный молекулярной массой, могут быть выбраны из диализа, ультрафильтрации или тангенциальной поточной фильтрации.
Глатирамера ацетат, полученный способом по настоящему изобретению, имеет содержание бромированного тирозина не более, чем примерно 0,15% при анализе методом обращенно-фазовой ВЭЖХ и содержание диэтиламида не более, чем приблизительно 5000 частей на миллион (мд), при анализе методом ионной хроматографии.
N-карбоксиангидрид (NCA) L-аминокислоты, такой как L-тирозин, L-аланин, защищенная L-глутаминовая кислота и защищенный L-лизин, может быть получен, как описано в нашей индийской заявке, по которой вынесено решение о выдаче патента 1082/MUM/2009 от 23 апреля 2009 или любым другим известным способом.
Сополимер-1 (глатирамера ацетат), полученный в соответствии с настоящим изобретением, охарактеризованный посредством МАЛДИ, ИК, ЯМР и ГПХ, CD-, УФ и аминокислотным анализом (AAA), оказывается эквивалентным КОПАКСОНу®.
Глатирамера ацетат, полученный в соответствии с настоящим изобретением, имеет среднюю молекулярную массу между 5000-9000 Дальтон и, по существу, свободен от продуктов с высокой молекулярной массой, имеющих молекулярную массу, равную или больше, чем приблизительно 35 кДа. Молекулярная масса глатирамера ацетата, полученного в соответствии с настоящим изобретением, может быть определена с использованием маркеров, описанных здесь.
Глатирамера ацетат представляет собой смесь полипептидов, состоящую из смеси аланина, глутаминовой кислоты, лизина и тирозина в молярном соотношении приблизительно 4,6:1,5:3,6:1, соответственно. Соответствующие мольные доли составляют примерно 0,427 для аланина, 0,141 для глутаминовой кислоты, 0,338 для лизина и 0,095 для тирозин, которые могут варьироваться посредством примерно ±10%. Мольные доли аминокислоты определяются сверхпроизводительной жидкостной хроматографии (СВЭЖХ), с использованием ультраколонки AccQ. Tag (2.1×100 мм, 1,7 мкм) и УФ-детектора. Систему градиента используют в качестве подвижной фазы.
Молярная фракция, измеренная для инновационного образца (КОПАКСОН®) и сополимера-1 (глатирамера ацетата), полученного в соответствии с настоящим изобретением, обнаружена в пределах, указанных в таблице 11.
Еще один способ для определения характеристик полипептидов представляет собой пептидное картирование. Способ включает химическую или ферментативную обработку полипептида, приводящую образованию пептидных фрагментов с последующим разделением и идентификацией фрагментов воспроизводимым образом. Пептидное картирование является сравнительной процедурой, так как полученная информация, сравниваемая со стандартным образцом, обработанным аналогично, подтверждает первичную структуру полипептида. Пептидная карта может рассматриваться как обзорная пептидная хроматограмма белка/полипептида.
В другом варианте осуществления, настоящее изобретение относится к аналитическому методу для определения характеристик полипептидов, в частности, глатирамера ацетата, с использованием трипсина и карбоксипептидазы В, содержащий стадии
a) ферментативной обработки полипептидов, в частности, глатирамера ацетата, с использованием трипсина для получения пептидных фрагментов с короткими длинами цепи;
b) обработки пептидных фрагментов, полученных на стадии а), с карбоксипептидазой В, приводящей к дальнейшей фрагментации пептидной цепи;
c) отделение и анализ пептидных фрагментов, полученных на стадии b) с помощью метода ВЭЖХ, чтобы получить хроматограмму, и
d) сравнение хроматограммы, полученной таким образом, с хроматограммой образца для сравнения, чтобы определить структурное сходство.
Предпочтительно, образец для сравнения представляет собой Копаксон® (продаваемый состав).
В предпочтительном варианте осуществления, глатирамера ацетат, полученный по настоящему изобретению, подвергают ферментативной обработке с использованием трипсина (обработанный тозил-L-фенилаланин-хлорметилкетоном) при температуре около 37°C и pH около 8. Такую обработку продолжают в течение приблизительно 4 часов для получения пептидных фрагментов с короткими длинами цепи. Эта смесь пептидных фрагментов, полученная после обработки трипсином, подвергается обработке карбоксипептидазой B при 37°C в течение 18 часов, что приводит к дальнейшей фрагментации пептидной цепи. Эти фрагменты разделяют и анализируют на С18, 250×4,6 мм колонке ВЭЖХ, получая хроматограмму. Хроматограмма, полученная таким образом, сравнивается с хроматограммой, полученной при обработке Копаксон® тем же способом, который указан выше.
Наложение хроматограммы Копаксона® партии 53378 и партий глатирамера ацетата, полученных способом по настоящему изобретению, обработанные только одним трипсином, показано на фиг.8. Наложение хроматограммы Копаксона® партии 53378 и партий глатирамера ацетата, полученных способом по настоящему изобретению, обработанных трипсином с последующей карбоксипептидазой B, показано на фиг.9. Эти профили хроматограммы показывают структурное сходство между Копаксоном и партиями глатирамера ацетата, полученными способом по настоящему изобретению.
На фиг.8 и фиг.9, (1) представляет собой холостой раствор, (2) представляет собой образец фрагментации стандартного образца для сравнения, т.е. Копаксон® партии 53378; и (3) и (4) представляют образец фрагментации для двух партий Глатирамера ацетата, полученного способом настоящего изобретения.
В предшествующем уровне техники раскрывается использование трипсина и карбоксипептидазы P для пептидного картирования глатирамера ацетата. Авторами настоящего изобретения было установлено, что глатирамера ацетат, обработанный трипсином с последующей обработкой карбоксипептидазой B, обеспечивает расщепление структуры подобно расщеплению карбоксипептидазой P. Карбоксипептидаза B предпочтительнее карбоксипептидазы P, из-за ее доступности и экономичности.
Согласно другому варианту осуществления настоящее изобретение относится к фармацевтической композиции в форме инъекцируемой лекарственной формы, содержащей эффективное количество глатирамера ацетата и фармацевтически приемлемое вещество, регулирующее тоничность.
Согласно предпочтительному варианту осуществления, изобретение обеспечивает фармацевтическую композицию в форме для подкожной инъекции, содержащей разовую дозу водного раствора в 1 мл, содержащую:
a) 20 мг глатиромера ацетат и
b) 40 мг маннита.
Согласно другому варианту осуществления, настоящее изобретение относится к способу получения фармацевтической композиции в форме подкожной инъекции, содержащему:
a) получение концентрированного раствора глатирамера ацетата с концентрацией (согласно анализу) в пределах от 25 мг/мл до 45 мг/мл;
b) растворение вещества, регулирующего тоничность, в части от общего количества воды для инъекций с образованием раствора;
c) добавление необходимого количества концентрированного раствора глатирамера ацетата к раствору стадии (b);
d) доведение объема раствора до конечного используя воду для инъекций;
e) фильтрацию раствора со стадии (d) через стерилизующие фильтры с размером пор 0,2µ для получения отфильтрованного раствора;
f) заполнение ампул указанным отфильтрованным раствором.
В предпочтительном варианте осуществления, настоящее изобретение относится к способу получения фармацевтической композиции в форме подкожной инъекции, содержащему:
a) получение концентрированного раствора глатирамера ацетата с концентрацией (анализ) в пределах от 30 мг/мл до 40 мг/мл;
b) растворение маннита в части от общего количества воды для инъекций;
c) добавление необходимого количества концентрированного раствора глатирамера ацетата к раствору со стадии b);
d) доведение объема раствора до конечного используя воду для инъекций;
e) фильтрацию раствора со стадии d) через стерилизующие фильтры с размером пор 0,2µ для получения отфильтрованного раствора;
f) заполнение ампул указанным отфильтрованным раствором.
Согласно другому варианту осуществления, настоящее изобретение относится к способу получения фармацевтической композиции в форме подкожной инъекции, содержащему:
a) обеспечение глатирамера ацетата;
b) растворение вещества, регулирующего тоничность, в части от общего количества воды для инъекций с образованием раствора;
c) растворение глатирамера ацетата в растворе стадии b);
d) доведение объема раствора до конечного используя воду для инъекций;
e) фильтрацию раствора со стадии d) через стерилизующие фильтры с размером пор 0,2µ для получения отфильтрованного раствора;
f) заполнение ампул указанным отфильтрованным раствором.
Согласно предпочтительному варианту осуществления, изобретение относится к способу получения фармацевтической композиции в форме подкожной инъекции, содержащему:
a) обеспечение глатирамера ацетата;
b) растворение маннита в части от общего количества воды для инъекций с образованием раствора;
c) растворение глатирамера ацетата в растворе стадии (b);
d) доведение объема раствора до конечного используя воду для инъекций;
e) фильтрацию раствора со стадии (d) через стерилизующие фильтры с размером пор 0,2µ для получения отфильтрованного раствора;
f) заполнение ампул отфильтрованным раствором.
Глатирамера ацетат используется в приготовлении фармацевтической композиции или в виде лиофилизированного твердого вещества или в виде водного раствора, содержащих глатирамера ацетата в концентрации от около 25-45 мг/мл.
Глатирамера ацетат может быть использован в диапазоне приблизительно от 1,0% до 4,0% от массы всей композиции. Вещества, регулирующие тоничность, могут быть использованы в диапазоне от примерно 0,9% до 9,75% от массы всей композиции.
При осуществлении настоящего изобретения, вещество, регулирующее тоничность, которое может быть использовано, включает, но не ограничивается, хлорид натрия, декстрозу, маннит, сахарозу, лактозу, галактозу, трегалозу, глицин или их смесь.
При осуществлении настоящего изобретения, композиция по настоящему изобретению может быть стерилизована с использованием мембранных фильтров с размером пор около 0,2 микрон. Подходящие мембранные фильтры, которые могут быть использованы, включают мембраны из поливинилиденфторида (ПВДФ), полиэфирсульфона (ПЭС) и ацетата целлюлозы.
Если не указано иное, следующие определения приведены для иллюстрации и определения значения и объема различных терминов, используемых в описании настоящего изобретения.
Термин "комнатная температура" следует понимать, как температура в диапазоне от приблизительно 20°C до примерно 40°C, предпочтительно от 25°C до 35°C.
Термин глатирамера ацетат будет включать Копаксон®, глатирамера ацетат, полученный способом по настоящему изобретению или полученный любым другим способом.
Термин "сополимер-1" и "глатирамера ацетат", используемый здесь, являются синонимами.
Термин "по существу свободный", как используется здесь, означает, глатирамера ацетат, содержащий менее, чем приблизительно 0,2% продуктов с высокой молекулярной массой, имеющих молекулярную массу, равную или больше, чем примерно 40 кДа, предпочтительно, менее чем около 0,2% продуктов с 35 кДа, более предпочтительно, менее примерно от 0,2% продуктов с 35 кДа и не обнаруживаемые количества продуктов с 40 кДа.
Инициаторы могут быть выбраны из оснований, нуклеофилов или их комбинации. В частности, инициатор может включать один или несколько аминов, спирты, воду или их комбинации.
Используемые амины могут быть первичными, вторичными или третичными аминами. Подходящие амины включают, но не ограничиваются, диметиламин, диэтиламин, ди-n-пропиламин, диизопропиламин, ди-вторбутиламин, N-этилметиламин, ди-n-бутиламин, ди-изобутиламин, ди-трет-бутиламин, диамиламин, ди-n-октиламин, ди(2-этилгексил)амин, ди-изонониламин, диаллиламин, N-метиланилин, дифениламин, или их комбинации, предпочтительным является диэтиламин. Другие инициаторы, которые могут быть использованы для полимеризации включают K-tOBu, NaH, КН, триэтиламин, тетраметилпиперидин, дициклогексиламин, дициклогексилундекан, диизопропиламин лития, трет-BuLi или их комбинация. Инициатор используют в количестве от примерно 0,2% до 4%, предпочтительно от 0,5 до 1,5%.
Подходящий используемый растворитель выбирают из группы, состоящей из воды, спирта, полярного апротонного растворителя, хлорированного углеводорода или углеводорода. Предпочтительный растворитель, используемый для удаления защитных групп представляет собой воду. Спирт может быть выбран из линейного или разветвленного спирта. Полярный апротонный растворитель выбирают из метилацетата, этилацетата, диметилфурана, диметилформамида, 1,4-диоксана, тетрагидрофурана или их смеси. Хлорированный углеводород может быть выбран из метилендихлорида, хлороформа или дихлорэтана. Углеводород может быть выбран из гексана, пентана, циклогексана и тому подобного.
Другие кислоты, которые могут быть использованы вместо смеси HBr - уксусная кислота, могут быть выбраны из HCl, H2SO4, HNO3, фосфористой кислоты, фосфорной кислоты, фторсульфоновой кислоты, хлорсульфоновой кислоты, уксусной кислоты, муравьиной кислоты, пропионовой кислоты, бензолсульфоновой кислоты, метансульфоновой кислоты, п-толуолсульфоновой кислоты, п-(н-додецил)бензолсульфоновой кислоты или их комбинации.
Хотя настоящее изобретение было описано в терминах его конкретных вариантов осуществления, для специалистов в данной области техники, очевидно, что возможны некоторые модификации и эквиваленты. Которые также включены в объем настоящего изобретения. Примеры приведены для иллюстрации определенных аспектов раскрытия, и не ограничивают объем настоящего изобретения.
Примеры:
Пример 1(A):
Определение молекулярной массы партий глатирамера ацетата
а) Фракционирование Копаксона при помощи ЭХ-ВЭЖХ:
- ЭХ-ВЭЖХ выполняли на колонке Superose 12 (10/300 GL) с использованием 0,2 М ацетата аммония pH 5,0, в качестве подвижной фазы, со скоростью потока 0,5 мл/мин.
- 100 мкл Копаксона (B.No. Р53289, 20 мг/мл) вводили в колонку в соответствии с оптимизированным методом, доводя конечную, вводимую концентрацию до 2 мг.
- пик Копаксона начал элюирование около 17 минут и завершил на отметке около 40 мин.
- 22 отдельных фракций были собраны в интервале разрыва 1 минута (фиг.1), начиная с 17-ой минуты, и заканчивая на 39-ой минуте с помощью воды для ВЭЖХ автосборника фракций (Autofraction collection).
- Все 22 фракций, собранные отдельно, полностью лиофилизировали.
b) Анализ фракционированного сополимера (Копаксона) при помощи ЭХ-РЛИКУ:
- Две фракции, каждая по обе стороны от вершины пика, и фракция, у вершины пика (Всего: 5 фракций, фиг.2) были взяты для ЭХ - РЛИКУ. Фракции повторно суспендировали в 0,2 М ацетата аммония pH 5,0, доводя конечную концентрацию каждой фракции до 6 мг/мл.
- Анализ проводили для определения Mp, Mw, Mn и полидисперсности каждой фракции.
- одинаковые значения Mp, Mw и Mn предполагают, что фракционированные образцы были почти монодисперсными. Полидисперсность фракционированных образцов составляла меньше, чем 1,05.
с) Использование охарактеризованного, фракционированного Сополимера в качестве маркеров молекулярной массы для определения Mp, Mw и Mn Глатирамера при помощи эксклюзионной хроматографии (ЭХ):
- Фракции 5, 7, 11, 13 и 17 элюировали при времени удерживания 21,317, 23,100, 26,833, 28,750 и 32,417 минут, соответственно.
- Калибровочная кривая было построена с помощью программного обеспечения Waters Empower. Соответствующая методика и установка 1-го порядка были использованы для построения кривой (фиг.3).
- R2 для калибровочной кривой фракций Копаксона равно 0,996.
- Используя такую стандартную кривую, две партии каждого из Копаксона и глатирамера ацетата, полученных способом по настоящему изобретению, были проанализированы (фиг.4), и результаты были следующими:
Пример 1 (В):
Определение молекулярной массы партий Глатирамера ацетата
а) Фракционирование глатирамера ацетата, полученного способом настоящего изобретения:
- около 40 мг глатирамера ацетата подвергали ГПХ на Superose 12 (10/300 GL), Система: Acta очиститель, с использованием 0,2 М ацетата аммония pH 5,0, в качестве подвижной фазы, со скоростью потока 0,5 мл/мин.
- Пик глатирамера начал элюирование примерно через 20 минут и закончил примерно через 40 мин.
- 30 отдельных фракций собирали в промежутке интервала 0,5 мин (фиг.5), начиная с 21ой минуты и заканчивая примерно на 41ой мин.
- около 15 фракций были проанализированы на полидисперсность и молекулярную массу.
- Фракции были выбраны таким образом, что одна из этих фракций представляет вершину пика молекулярной массы, а другие фракции были распределены по обе стороны вершины пика молекулярной массы.
- Фракции, имеющие полидисперсность меньше, чем 1,2, были отобраны и лиофилизированы.
- Фракции, имеющие полидисперсность более, чем 1,2, подвергали дальнейшей очистке с целью получения фракции, имеющей полидисперсность меньше, чем 1,2.
- фракции с молекулярной массой 3 кДа, 5 кДа, 7 кДа, 10 кДа и 12 кДа были выбраны для использования в качестве маркеров молекулярной массы.
- Молекулярная масса этих выбранных фракций была определена с использованием ЭХ-РЛИКУ [Условия такие же, как в примере 1 (А)]. Mp и полидисперсность этих выбранных фракций показаны в таблице ниже
Охарактеризованные фракции, использованные в качестве маркеров молекулярной массы
- Все указанные выбранные фракции были лиофилизированы и охарактеризованы.
b) применение охарактеризованных фракций в качестве маркеров молекулярной массы для определение Mp, Mw и Mn Глатирамера при помощи эксклюзионной хроматографии (ЭХ)/гель-проникающей хроматографии (ГПХ):
- Фракции 16, 18, 22, 24 и 26 элюировали при времени удерживания 24,60, 25,88, 27,41, 28,95 и 31,31 минут, соответственно.
- Калибровочную кривую составляли с помощью программного обеспечения Waters Empower. Соответствующая методика и установка 1-го порядка были использованы для построения кривой (фиг.6).
- R2 для калибровочной кривой, охарактеризованных фракций, составило 0,99.
- Используя данную стандартную кривую, две порции каждого из Копаксона и глатирамера ацетата, полученных способом по настоящему изобретению, были проанализированы (фиг.7), и результаты были следующими:
Результаты с калибровочной кривой фракций Копаксона:
Пример 2
200 г (0,84 моль) γ-бензил-L-глутамата и 10 г активированного угля суспендировали в 2,4 л тетрагидрофурана (ТГФ). 124,6 г (0,42 моль) трифосгена добавляли к полученной смеси и перемешивали в течение 0,5 часа при 50-55°C. После завершения реакции, реакционную смесь охлаждали до комнатной температуры, отфильтровывали через слой хайфло (hyflo) и пропускают газообразный азот через полученный фильтрат в течение 2 часов ниже 35°C. Фильтрат концентрировали в вакууме, чтобы получить масло. Полученное масло трижды десорбировали с 1 л гексана, чтобы получить твердое вещество. Полученное твердое вещество смешивали с 2 л гексана, отфильтровывали, промывали 2 л гексана и сушили в вакууме при температуре ниже 45°C. Высушенное вещество пропускают через сито 30 меш и снова повторно сушат. Повторно высушенное твердое вещество хранили при -20°C в герметичном контейнере.
Выход: 200 г (100% мас./мас. относительно аминокислоты), т.пл: 92°-94°C
Пример 3
110 г (0,552 моль) L-тирозина и активированного угля (5,5 г) суспендировали в 1,32 л тетрагидрофурана (ТГФ). 89,76 г (0,302 моль) трифосгена добавляли к смеси. Реакционную смесь перемешивали в течение 2 часов при 50-55°C.После завершения реакции, реакционную смесь охлаждали до комнатной температуры, отфильтровывали через слой хайфло (hyfllo). Газообразный азот продували через полученный фильтрат в течение 2 часов ниже 35°C. Реакционную смесь затем концентрировали в вакууме, чтобы получить твердую массу. Полученную твердую массу трижды десорбировали 0,55 л гексана и затем перемешивали с 1,1 л гексана, фильтровали, промывали 1,1 л гексана и сушили в вакууме ниже 45°C. Высушенное вещество пропускали через сито 30 меш и снова повторно сушили. Повторно высушенное твердое вещество хранили при -20°C в герметичном контейнере.
Выход: 80 г (73% мас./мас. относительно аминокислоты). Соединение разлагается при 250°C до 270°C
Пример 4
400 г (1,65 моль) Nε-трифорацетил-L-лизина и активированного угля (20 г) суспендировали в 4,8 л тетрагидрофурана (ТГФ). 243,3 г (0,82 моля) трифосгена добавляли к смеси. Реакционную смесь перемешивали в течение 0,5 часов при 50-55°C. После завершения реакции, реакционную смесь охлаждали до комнатной температуры, отфильтровывали через слой хайфло (hyfllo). Газообразный азот продували через полученный фильтрат в течение 2 часов ниже 35°C. Реакционную смесь затем концентрировали в вакууме, чтобы получить масло. Полученное масло трижды десорбировали 2 л гексана, чтобы получить твердое вещество. Полученное твердое вещество смешивали с 4 л гексана, отфильтровывали, промывали 4 л гексана и сушили в вакууме ниже 45°C. Высушенное вещество пропускали через сито 30 меш и снова повторно сушили. Повторно высушенное твердое вещество хранили при -20°C в герметичном контейнере.
Выход: 415 г (104% мас./мас. относительно аминокислоты), т.пл: 83°C-86°C
Пример 5
200 г (2,24 моль) L-аланина суспендировали в 4 л тетрагидрофурана (ТГФ) и смесь нагревали до 50°-55°C в течение 1,5 часов. 10 г активированного угля и 333 г (1,12 моль) трифосгена, растворенного в 1 л ТГФ, добавляли к нагретой суспензии в течение примерно 10 мин при 50-55°C. Реакционную смесь перемешивали в течение 1,5 ч при той же самой температуре и вторую партию из 333 г (1,12 моль) трифосгена, растворенного в 1 л ТГФ, добавляли к смеси в течение примерно 10 мин с последующим перемешиванием смеси в течение 1,5 ч при 50-55°C. После завершения реакции смесь фильтровали, и пропускали газообразный азот через полученный фильтрат в течение 2 ч ниже 35°C.Фильтрат концентрировали под вакуумом ниже 35°C, чтобы получить масло. Добавляли 1,6 л этилацетата к полученному маслу, перемешивали в течение 10 мин и отфильтровывали через слой хайфло (hyflo). 13 л гексана добавляли к полученному фильтрату, чтобы получить твердое вещество, которое отфильтровывали, промывали 4 л гексана и сушили в вакууме при температуре ниже 45°C. Высушенное вещество пропускают через сито 30 меш и снова повторно сушили. Повторно высушенное твердое вещество хранили при -20°C в герметичном контейнере.
Выход: 180 г (90% мас./мас. относительно аминокислоты), т.пл: 91°C-92°C
Пример 6
Смесь NCAs L-аминокислоты [50 г N-карбоксиангидрида L-аланина, 35 г N-карбоксиангидрида γ-бензил-L-глутамата, 83 г N-карбоксиангидрида Nε-трифторацетил-L-лизина, 18 г из N-карбоксиангидрида L-тирозина] растворяли в 3,5 л безводного диоксана и перемешивали в течение 30 мин. Раствор фильтровали, если замечали любую нерастворимую взвешенную частицу. Раствор 1% диэтиламина в безводном диоксане (266 мл) добавляли к реакционной массе в одной партии. Полученную смесь перемешивали при комнатной температуре в течение 24 часов при 200-215 оборотов в минуту. 10 л деионизированной воды добавляли к реакционной смеси, чтобы получить белое твердое вещество. Полученное твердое вещество отфильтровывали под вакуумом, промывали 10 л деионизированной воды и сушили на воздухе до достижения LOD NMT 10%. Полученное твердое вещество пропускали через сито 30 меш.
Выход: 120 г (64,5% мас./мас.)
Пример 7
В реакторе подавали 960 мл ~15% HBr в уксусной кислоте при комнатной температуре и перемешивали при 200-215 оборотов в минуту в течение 30 мин до достижения приблизительно 25°C. 115 г защищенного сополимера (из примера 6) добавляли в реактор и перемешивали при температуре около 25°C при 200-215 оборотов в минуту в течение 23 часов. Реакционную смесь гасят в холодной воде, чтобы получить белое твердое вещество. Раствор аммиака добавляли к смеси до достижения pH 6-7, и перемешивали при комнатной температуре в течение примерно 20 часов. Твердое вещество отфильтровывали, промывали водой и сушили на воздухе. Выход: 75 г (65,2% мас./мас.)
Пример 8
75 г трифторацетил-сополимера (из примера 7) суспендировали в 3,75 л воды. 435 мл пиперидина добавляли к суспензию в один прием и перемешивали в течение 24 ч при комнатной температуре. Реакционную смесь пропускали через 0,2µ фильтр, пропускали через кассету для тангенциальной поточной фильтрации с номинальным отсечением по молекулярной массе 1 кДа, диафильтровали с деионизированной водой, затем 0,3% водной уксусной кислотой и снова деионизированной водой до достижения pH 5,5-7. Полученный раствор затем лиофилизировали до сухости с получением белого рассыпчатого твердого вещества.
Выход: 50,5 г (67,3% мас./мас.), вершина пика молекулярной массы (Mp)=7980 Да.
Кроме того, полученный раствор может быть принят как таковой для препарата.
Пример 9
75 г трифторацетил-сополимера (из примера 7) суспендировали в 3,75 л воды. 300 мл диэтиламина добавляли к суспензии одной порцией и перемешивали в течение 24 ч при комнатной температуре. Реакционную смесь пропускали через фильтр 0,2µ, пропускали через кассету для тангенциальной поточной фильтрации с номинальным отсечением по молекулярной массе 1 кДа, диафильтровали деионизированной водой, затем 0,3% водной уксусной кислотой и снова деионизированной водой до достижения pH 5,5-7. Полученный раствор затем лиофилизировали до сухости с получением белого рассыпчатого твердого вещества.
Выход: 52,86 г (70,4% мас./мас.), вершина пика молекулярной массы (Mp)=7730 Да. Кроме того, полученный раствор может быть принят как таковой для препарата.
Пример 10
В реактор подавали 960 мл ~15% HBr в уксусной кислоте при комнатной температуре и перемешивали при 200-215 оборотов в минуту в течение 30 мин до достижения 25°C. 115 г защищенного сополимера (пример 6) добавляли в реактор и перемешивали при температуре около 25°C при 200-215 оборотов в минуту в течение 23 часов. Реакционную смесь гасили в холодной воде, чтобы получить белое твердое вещество. Раствор аммиака добавляли к смеси до достижения pH 6-7, и перемешивали при комнатной температуре в течение примерно 20 часов. Твердое вещество центрифугировали, промывали водой и повторно центрифугировали, чтобы получить влажное твердое вещество. Влажное твердое вещество суспендировали в 4,6 л воды. 517 мл пиперидина добавляли к суспензии одной порцией и перемешивали в течение 24 ч при комнатной температуре. Реакционную смесь пропускали через фильтр 0,2µ, пропускали через кассету для тангенциальной поточной фильтрации потока с номинальным отсечением по молекулярной массе) 1 кДа, диафильтровали с деионизированной водой, затем 0,3% водной уксусной кислотой и снова деионизированной водой до достижения pH 5,5-7. Полученный раствор затем лиофилизировали до сухости с получением белого рассыпчатого твердого вещества.
Выход: 54,5 г (47,39% мас./мас.), вершина пика молекулярной массы (Mp)=6960 Да.
Кроме того, полученный раствор может быть принят как таковой для препарата.
Пример 11
В реактор подавали 960 мл ~15% HBr в уксусной кислоте при комнатной температуре и перемешивали при 200-215 оборотов в минуту, в течение 30 мин до достижения 25°C. 115 г защищенного сополимера (пример 6) добавляли в реактор и перемешивали при температуре около 25°C при 200-215 оборотов в минуту в течение 23 часов. Реакционную смесь гасили в холодной воде, чтобы получить белое твердое вещество. Раствор аммиака добавляли к смеси до достижения pH 6-7, и перемешивали при комнатной температуре в течение примерно 20 часов. Твердое вещество центрифугировали, промывали водой и повторно центрифугировали, чтобы получить влажное твердое вещество. Влажное твердое вещество суспендировали в 4,6 л воды. 287 мл диэтиламина добавляли к суспензии одной порцией и перемешивали в течение 24 ч при комнатной температуре. Реакционную смесь пропускали через фильтр 0,2µ, пропускали через кассету для тангенциальной поточной фильтрации с номинальным отсечением по молекулярной массе 1 кДа, диафильтровали деионизированной водой, затем 0,3% водной уксусной кислотой и снова деионизированной водой до достижения pH 5,5-7. Полученный раствор затем лиофилизировали до сухости с получением белого рассыпчатого твердого вещества. Выход: 55,86 г (48,57% мас./мас.), вершина пика молекулярной массы (Mp)=7291 Да. Кроме того, полученный раствор может быть принят как таковой для препарата.
Пример 12
480 мл приблиз. 15% HBr в уксусной кислоте подавали в реактор при комнатной температуре и перемешивали при 200-215 оборотов в минуту в течение 30 мин до достижения приблизительно 25°C. 2,4 г резорцина добавляли к смеси и перемешивали в течение 2 ч при 25°C при 200-215 оборотов в минуту. 60 г защищенного сополимера (пример 6) добавляли в реактор и перемешивали при температуре около 25°C при 200-215 оборотов в минуту в течение 23 часов. Реакционную смесь гасили в холодной воде, чтобы получить белое твердое вещество. Раствор аммиака у добавляли к смеси до достижения pH 6-7, и смесь перемешивали при комнатной температуре в течение примерно 20 часов. Твердое вещество отфильтровывали, промывали водой и сушили на воздухе. Выход: 36 г (60% мас./мас.).
Тот же пример был повторен с использованием другого акцептора брома, такого как тирозин, бисульфит натрия, тиосульфат натрия, нафталин, 1-нафтол и 2-нафтол.
Пример 13
36 г трифторацетил-сополимера (из примера 12) суспендировали в 1,8 л воды. 209 мл пиперидина добавляли в суспензию одной порцией и перемешивали в течение 24 ч при комнатной температуре. Реакционную смесь пропускали через фильтр 0,2µ, пропускали через кассету для тангенциальной поточной фильтрации с номинальным отсечением по молекулярной массе 1 кДа, диафильтровали с деионизированной водой, затем 0,3% водной уксусной кислотой и снова деионизированной водой до достижения pH 5,5-7. Полученный раствор затем лиофилизировали до сухости с получением белого рассыпчатого твердого вещества.
Выход: 24,48 г (68% мас/мас)
Кроме того, полученный раствор может быть принят как таковой для препарата.
Пример 14
36 г трифторацетил-сополимера (из примера 12) суспендировали в 1,8 л воды. 144 мл диэтиламина добавляли к суспензии одной порцией и перемешивали в течение 24 ч при комнатной температуре. Реакционную смесь фильтровали через фильтр 0,2µ, пропускали через кассету для тангенциальной поточной фильтрации с номинальным отсечением по молекулярной массе 1 кДа, диафильтровали с деионизированной водой, затем 0,3% водной уксусной кислоты и снова деионизированной водой до достижения pH 5,5-7. Полученный раствор затем лиофилизировали до сухости с получением белого рассыпчатого твердого вещества.
Выход: 26,8 г (74,4% мас./мас.)
Кроме того, полученный раствор может быть принят как таковой для препарата.
Пример 15
В реактор подавали 460 мл ~15% HBr в уксусной кислоте и 460 мл уксусной кислоты при комнатной температуре и перемешивали при 200-215 оборотов в минуту в течение 30 мин до достижения 25°C. 4,6 г фенола добавляли к смеси и перемешивали в течение 2 ч при температуре около 25°C при 200-215 оборотов в минуту. 115 г защищенного сополимера (пример 6) добавляли в реактор и перемешивали при температуре около 25°C при 200-215 оборотов в минуту в течение 21-23 часов. Реакционную смесь гасили в холодной воде, чтобы получить белое твердое вещество. Раствор аммиака добавляли к смеси до достижения pH 6-7, и перемешивали при комнатной температуре в течение примерно 20 часов. Твердое вещество центрифугировали, промывали водой и повторно центрифугировали в течение приблизительно 3 часов для получения влажного твердого вещества. Влажное твердое вещество суспендировали в 4,6 л воды. 287 мл диэтиламина добавляли к суспензии одной порцией и перемешивали в течение 24 ч при комнатной температуре. Реакционную смесь пропускали через фильтр 0,2µ, пропускали через кассету для тангенциальной поточной фильтрации с номинальным отсечением по молекулярной массе (MWCO) 1 кДа, диафильтровали с деионизированной водой, затем 0,3% водной уксусной кислотой и снова деионизированной водой до достижения pH 5,5-7. Полученный концентрированный раствор фильтровали в стерильных условиях и хранили при 2°C до 8°C в стерильной ПЭТ бутылке, которая была покрыта черным полиэтиленовым чехлом.
Выход: 1670 л (36,1 мг/мл), вершина пика молекулярной массы (Mp)=6974 Да. Полученный концентрированный раствор, имеющий концентрацию (анализ) глатирамера ацетата приблизительно 30-40 мг/мл может быть принят как таковой для препарата.
Пример 16
В реактор подавали 460 мл ~33% HBr в уксусной кислоте и 460 мл уксусной кислоты при комнатной температуре и перемешивали при 200-215 оборотов в минуту в течение 30 мин до достижения 25°C. 4,6 г фенола добавляли к смеси и перемешивали в течение 2 ч при температуре 25°C при 200-215 оборотов в минуту. 115 г защищенного сополимера (пример 6) добавляли в реактор и перемешивали при 25°C при 200-215 оборотов в минуту в течение 21 часа. Реакционную смесь гасили в холодной воде, чтобы получить белое твердое вещество. Раствор аммиака добавляли к смеси до достижения pH 6-7, и перемешивали при комнатной температуре в течение примерно 20 часов. Твердое вещество центрифугировали, промывали водой и повторно центрифугировали в течение приблизительно 3 часов. Влажное твердое вещество суспендировали в 4,6 л воды. 287 мл диэтиламина добавляли к суспензии одной порцией и перемешивали в течение 24 ч при комнатной температуре. Реакционную смесь пропускали через фильтр 0,2µ, пропускали через кассету для тангенциальной поточной фильтрации с номинальным отсечением по молекулярной массе (MWCO) 1 кДа, диафильтровали с деионизированной водой, затем 0,3% водной уксусной кислотой и снова деионизированной водой до достижения pH 5,5-7. Полученный концентрированный раствор фильтровали в стерильных условиях и хранили при 2°C до 8°C в стерильной ПЭТ G бутылке, которая была покрыта черным полиэтиленовым чехлом.
Выход: 1690 л (35 мг/мл), вершина пика молекулярной массы (Mp)=7324 Да. Полученный концентрированный раствор, имеющий концентрацию (анализ) глатирамера ацетата приблизительно 30-40 мг/мл может быть принят как таковой для препарата.
Пример 17
Анализ аминокислоты при помощи СЭЖХ (UPLC):
Условия хроматографии:
Колонка: AccQ.Tag Ultra колонка 2,1×100 мм, 1,7 мкм
Подвижная фаза A: элюент A1 (5% AccQ.Tag подвижная фаза, разбавленная водой)
Подвижная фаза B: элюент B (как таковой)
Длина волны: 260 нм
Частота отбора пробы: 10 точек/сек
Темп. колонки: 55°C
Темп. образца: 20°C
Скорость потока: 0,7 мл/мин
Программирование: программа градиента
Дериватизация: Дериватизация при помощи AccQ. Tag агента дериватизации
Результаты исследования молярных фракций аминокислот сведены в таблице 12:
Пример 18
Пептидное картирование с использованием фермента трипсина
0,25 мг глатирамера ацетата, расщепляли с использованием 5 мкг трипсина (обработанного тозил-L-фенилаланинхлорметилкетоном) в 0,05 М Трис буфера, pH 7,5, при температуре около 37°C. Данную обработку продолжали в течение 4 часов для получения пептидных фрагментов с короткими длинами цепи. Эти фрагменты отделяли и анализировали с помощью ВЭЖХ-УФ на С18, 250×4,6 мм колонка ВЭЖХ, получая хроматограмму. Хроматограмму, полученную таким образом, сравнивали с хроматограммой, полученной при воздействии на Копаксон такой же обработкой, которая приведена выше.
Пример 19
Пептидное картирование с использованием трипсина и карбоксипептидазы B
3,5 мг глатирамера ацетата, расщепляли с использованием 7 мкг трипсина (обработанного тозил-L-фенилаланинхлорметилкетоном) в 0,05 М Трис буфера, pH 7,5, при температуре около 37°C. Данную обработку продолжали в течение 4 часов для получения пептидных фрагментов с короткими длинами цепи. Эту смесь пептидных фрагментов, полученных после обработки трипсином, подвергали обработке 20 мкг карбоксипептидазой B при 37°C в течение 18 часов, что приводит к дальнейшей фрагментации пептидной цепи. Эти фрагменты отделяли и анализировали с помощью ВЭЖХ-УФ на С18, 250×4,6 мм колонка ВЭЖХ, получая хроматограмму. Хроматограмму, полученную таким образом, сравнивали с хроматограммой, полученной при воздействии на Копаксон такой же обработкой, как упоминалось выше.
Пример 20
(В) Получение:
Маннит растворяли в части количества воды для инъекций путем перемешивания. Концентрированный раствор глатирамера ацетата API добавляли в данный раствор маннита и перемешивали. Конечный объем составляли с оставшимся количеством воды для инъекций. Проверяли pH раствора (предел pH: от 5,5 до 7,0). Конечный раствор фильтровали через стерилизующий фильтр из полиэфирсульфона с размером пор 0,2µ. Отфильтрованный раствор разливали в ампулы и флаконы герметично упаковывали.
Пример 21
Статистический метод оценки стабильности и воспроизводимости способа получения маркеров молекулярной массы.
Пять наборов маркеров молекулярной массы были получены с использованием разных партий глатирамера ацетата. Каждая группа содержала семь маркеров молекулярной массы, имеющих разные молекулярные массы в диапазоне от 2000 до 13000 дальтон, как показано в таблице ниже
Статистический способ рандомизации был применен для этих пяти наборов маркеров молекулярной массы для получения большего набора маркеров молекулярной массы, каждый набор содержит семь маркеров молекулярной массы, как в таблице ниже
Приведенные большие наборы маркеров использовали затем, чтобы определить вершину пика молекулярной массы глатирамера ацетата и результаты приведены в таблице ниже.
Это исследование рандомизации показало, что любой перестановка- комбинация маркеров молекулярной массы, взятых из различных наборов обеспечит предсказуемые результаты на основе 95% доверительного интервала. Это показывает, что способ по настоящему изобретению для получения маркеров молекулярной массы был стабильным, воспроизводимым и не зависел от изменения от партии к партии исходного материала. Маркеры молекулярной массы вырабатывали используя способ настоящего изобретения из различных партий глатирамера ацетата при условии предсказуемых результатов на основе 95% доверительного интервала.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ СМЕСЕЙ ПОЛИПЕПТИДОВ С ИСПОЛЬЗОВАНИЕМ ОЧИЩЕННОЙ БРОМИСТОВОДОРОДНОЙ КИСЛОТЫ | 2005 |
|
RU2388764C2 |
| ПЕПТИДНЫЙ СОПОЛИМЕР, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ | 2009 |
|
RU2402576C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПОЛИПЕПТИДНЫХ СМЕСЕЙ С ИСПОЛЬЗОВАНИЕМ ГИДРОГЕНОЛИЗА | 2006 |
|
RU2419638C2 |
| УСОВЕРШЕНСТВОВАННЫЙ СОПОЛИМЕР-1 В СОПОЛИМЕРНЫХ КОМПОЗИЦИЯХ | 1995 |
|
RU2161489C2 |
| УСОВЕРШЕНСТВОВАННЫЙ СОПОЛИМЕР-1 И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1995 |
|
RU2198900C2 |
| ВАКЦИНА И СПОСОБ ЛЕЧЕНИЯ БОЛЕЗНЕЙ ДВИГАТЕЛЬНЫХ НЕЙРОНОВ | 2002 |
|
RU2303996C2 |
| СПОСОБ ПРОИЗВОДСТВА ПРОТЕОЛИТИЧЕСКИ ПРОЦЕССИРОВАННОГО ПОЛИПЕПТИДА | 2012 |
|
RU2673910C2 |
| СПОСОБЫ ПРОИЗВОДСТВА ПРОТЕОЛИТИЧЕСКИ ПРОЦЕССИРОВАННЫХ ПОЛИПЕПТИДОВ | 2019 |
|
RU2727402C1 |
| СПОСОБЫ ПРОИЗВОДСТВА ПРОТЕОЛИТИЧЕСКИ ПРОЦЕССИРОВАННЫХ ПОЛИПЕПТИДОВ | 2018 |
|
RU2719164C1 |
| КОВАЛЕНТНОЕ МОДИФИЦИРОВАНИЕ ПОЛИПЕПТИДОВ ПОЛИЭТИЛЕНГЛИКОЛЯМИ | 1992 |
|
RU2148586C1 |
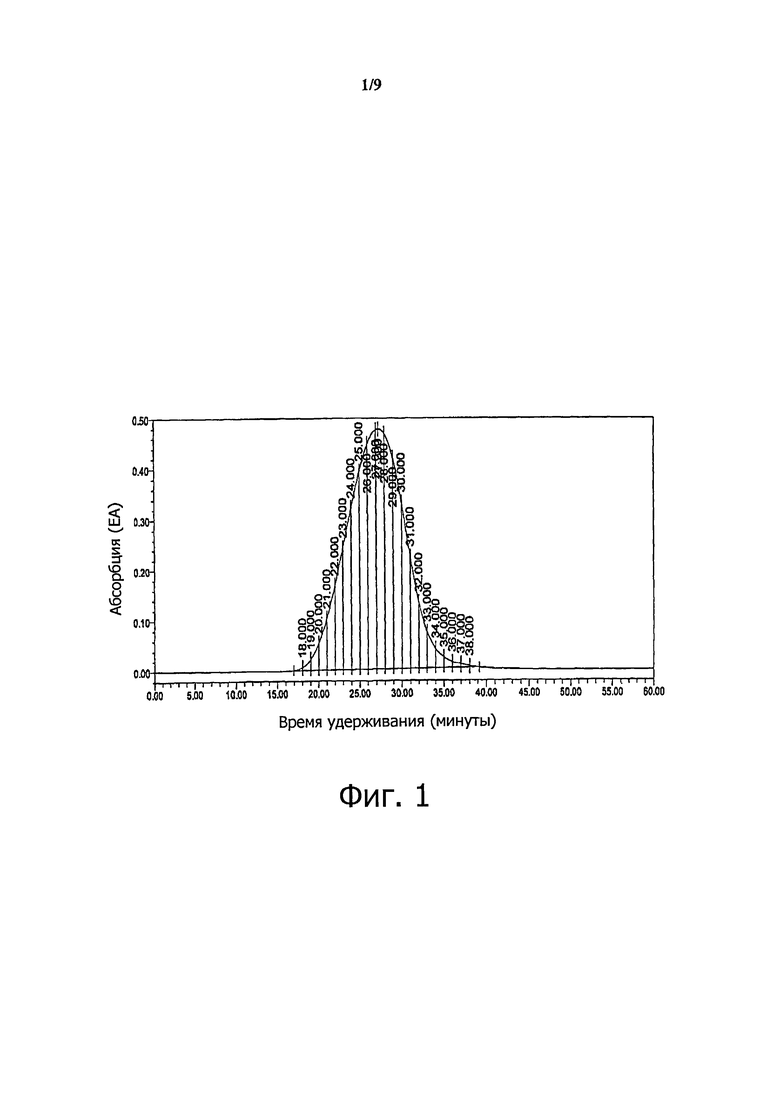



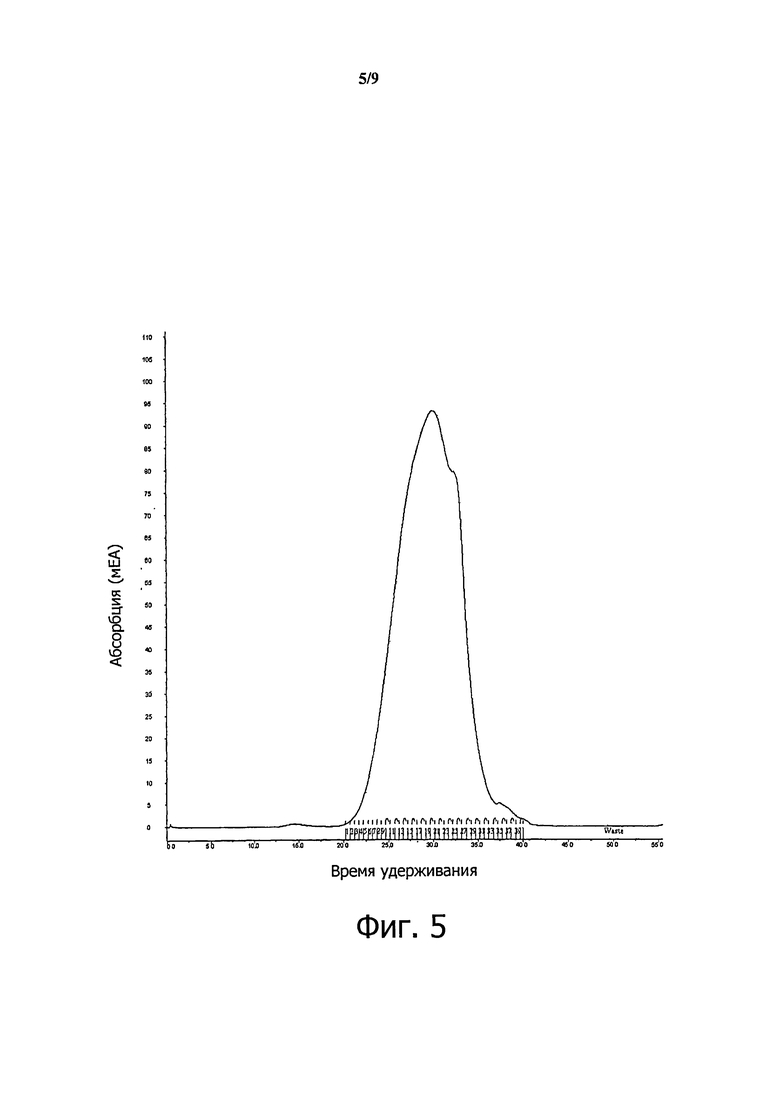
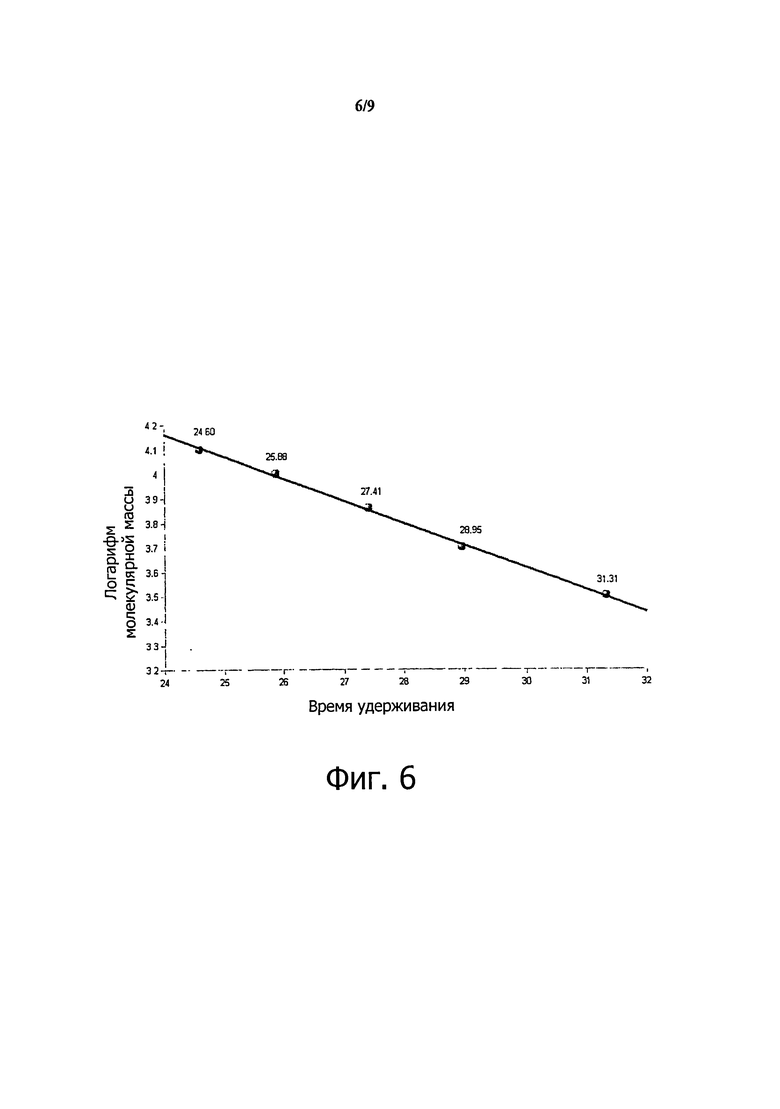



Изобретение относится к способу получения маркеров молекулярной массы для глатирамера ацетата со случайной аминокислотной последовательностью, аналогичной последовательности глатирамера ацетата, содержащему стадии: a) воздействие на полипептидную смесь, которая является смесью полипептидов, содержащих аланин, глутаминовую кислоту, тирозин и лизин в том же молярном соотношении, что у глатирамера ацетата, гель-проникающей хроматографией (ГПХ)/эксклюзионной хроматографией (ЭХ) для получения полипептидных фракций с молекулярными массами в диапазоне от 2 кДа до 30 кДа; b) выбор полипептидных фракций, которые будут использованы в качестве маркеров молекулярной массы, из указанных полипептидных фракций, полученных на стадии а), таким образом, что одна из фракций представляет вершину пика молекулярной массы, а другие фракции распределены по обе стороны от вершины пика молекулярной массы; и c) определение полидисперсности указанных выбранных полипептидных фракций, где полидисперсность указанных выбранных полипептидных фракций должна составлять меньше, чем или быть равной 1,20. 18 з.п. ф-лы, 9 ил., 21 табл., 21 пр.
1. Способ получения маркеров молекулярной массы для глатирамера ацетата, которые состоят из полипептидных фракций со случайной аминокислотной последовательностью, аналогичной последовательности глатирамера ацетата, содержащий стадии:
a) воздействие на полипептидную смесь, которая является смесью полипептидов, содержащих аланин, глутаминовую кислоту, тирозин и лизин в том же молярном соотношении, что у глатирамера ацетата, гель-проникающей хроматографией (ГПХ)/эксклюзионной хроматографией (ЭХ) для получения полипептидных фракций с молекулярными массами в диапазоне от 2 кДа до 30 кДа;
b) выбор полипептидных фракций, которые будут использованы в качестве маркеров молекулярной массы, из указанных полипептидных фракций, полученных на стадии а), таким образом, что одна из фракций представляет вершину пика молекулярной массы, а другие фракции распределены по обе стороны от вершины пика молекулярной массы; и
c) определение полидисперсности указанных выбранных полипептидных фракций, где полидисперсность указанных выбранных полипептидных фракций должна составлять меньше чем или быть равной 1,20.
2. Способ по п. 1, в котором коэффициент асимметрии пика выбранных полипептидных фракций находится в диапазоне от 0,8 до 1.
3. Способ по п. 1, в котором указанная полипептидная смесь представляет собой глатирамера ацетат.
4. Способ по п. 1, в котором указанную ГПХ/ЭХ проводят на колонке Superose-12 или Superdex-75, с использованием подвижной фазы, выбранной из ацетата аммония или формиата аммония с диапазоном концентрации от 0,1 до 0,3 М.
5. Способ по п. 4, в котором колонка представляет собой Superose-12 (10×300 GL); загрузка колонки составляет не более чем 50 мг; и скорость потока регулируют в пределах от 0,4 до 0,6 мл/мин.
6. Способ по п. 1, в котором указанные полипептидные фракции, полученные на стадии а), лиофилизируют и в котором Мр указанных выбранных фракций определяют с использованием ЭХ-РЛИКУ.
7. Способ по п. 1, в котором указанные выбранные полипептидные фракции используют в качестве маркеров молекулярной массы для определения молекулярной массы партии глатирамера ацетата способом, содержащим стадии
a) калибровка колонки для ЭХ/ГПХ с использованием указанных выбранных полипептидных фракций;
b) установление взаимосвязи между временем удерживания и логарифмом молекулярной массы этих выбранных полипептидных фракций посредством калибровочной кривой;
c) проведение ЭХ/ГПХ для партии глатирамера ацетата, чья молекулярная масса должна быть определена, чтобы получить время удерживания; и
d) использование соотношения между временем удерживания и логарифмом молекулярной массы для определения молекулярной массы партии глатирамера ацетата.
8. Способ по п. 7, в котором определяют вершину пика молекулярной массы партии глатирамера ацетата и в котором указанная колонка для ЭХ/ГПХ выбрана из Superose-12 или Superdex-75 и подвижная фаза выбрана из 0,1 до 0,3 М ацетата аммония или формиата аммония.
9. Способ по п. 1, где стабильность и воспроизводимость указанного способа тестируется посредством способа, который содержит стадии:
a) получение множественного набора маркеров молекулярной массы из различных партий глатирамера ацетата/Копаксона с помощью способа по п. 1, каждый набор содержит маркеры молекулярной массы, имеющие молекулярную массу в диапазоне от 2000 до 13000 дальтон;
b) распределение маркеров молекулярной массы из каждого набора, полученного на стадии а), с использованием статистического способа рандомизации для дальнейшего выделения большего набора маркеров молекулярной массы, и
c) использование этих более крупных наборов, полученных на стадии b), в качестве маркеров молекулярной массы для определения молекулярной массы Копаксона/партии глатирамера ацетата при помощи ГПХ.
10. Способ по п. 9, в котором указанные маркеры молекулярной массы обеспечивают предсказуемые результаты на основании 95% доверительного интервала, подтверждая стабильность и воспроизводимость способа получения маркеров молекулярной массы.
11. Способ по п. 3, в котором указанный глатирамера ацетат получен способом, содержащим стадии:
а) обработка защищенного сополимера с примерно 7% до 20% HBr в уксусной кислоте при температуре около 25°С, в течение 21-23 часов, чтобы получить трифторацетил-сополимер;
b) обработка указанного трифторацетил-сополимера основанием, чтобы получить сополимер с полностью удаленной защитной группой;
c) превращение сополимера с полностью удаленной защитной группой в глатирамера ацетат.
12. Способ по п. 11, в котором указанную обработку на стадии а) проводят при 25°С±0,2°С; указанный HBr в уксусной кислоте необязательно предварительно обрабатывают акцептором, выбранным из фенола, резорцина, тирозина, бисульфата натрия, тиосульфата натрия, нафталина, нафтола-1 или нафтола-2, и указанное основание выбирают из диэтиламина, диметиламина или диизопропиламина.
13. Способ по п. 11, где указанным основанием является диэтиламин.
14. Способ по п. 11, в котором указанное превращение сополимера с полностью удаленной защитной группой в глатирамера ацетат содержит стадии:
a) концентрирование водного раствора сополимера с полностью удаленной защитной группой до минимального объема, с использованием тангенциальной поточной фильтрации, пропусканием через кассету мембраны из полиэфирсульфона, имеющей номинальное отсечение по молекулярной массе 1 кДа;
b) промывание концентрированного раствора со стадии а) водой до достижения рН от 8 до 9, с последующим промыванием 0,3% раствора уксусной кислоты для солевого обмена, чтобы получить глатирамера ацетат; и
c) промывание глатирамера ацетата, полученного на стадии b), водой до достижения рН от 5,5 до 7, чтобы получить чистый глатирамера ацетат.
15. Способ по п. 14, в котором молекулярную массу указанного глатирамера ацетата определяют способом по п. 7 и в котором указанный глатирамера ацетат имеет содержание бромированного тирозина не более чем около 0,15%, при исследовании при помощи ВЭЖХ с обращенной фазой, и содержание диэтиламида не более чем около 5000 частей на миллион (мд), при исследовании методом ионной хроматографии
16. Способ по п. 14, в котором указанный глатирамера ацетат имеет среднюю молекулярную массу в диапазоне 5000-9000 дальтон.
17. Способ по п. 11, в котором глатирамера ацетат дополнительно охарактеризован пептидным картированием с использованием трипсина и карбоксипептидазы В, включающем следующие стадии:
a) ферментативную обработку глатирамера ацетата с использованием трипсина, чтобы получить пептидные фрагменты с короткой длиной цепи;
b) обработку пептидных фрагментов, полученных на стадии а) карбоксипептидазой В, приводящей к дополнительной фрагментации пептидной цепи;
c) отделение и анализ пептидных фрагментов, полученных на стадии b), с использованием методики ВЭЖХ, чтобы получить хроматограмму;
d) сравнение полученной таким образом хроматограммы с хроматограммой образца для сравнения, чтобы определить структурное сходство.
18. Способ по п. 17, в котором указанный образец для сравнения представляет собой Копаксон (продаваемый состав).
19. Способ по п. 7, в котором указанный глатирамера ацетат используют для приготовления фармацевтической композиции либо в виде лиофилизированного твердого вещества или в виде водного раствора, содержащих глатирамера ацетата в диапазоне концентраций около 25-45 мг/мл.
| US 2007059798 A1, 15.03.2007;RU 2388764 C2, 10.05.2010 | |||
| US 20050256046 A1, 17.11.2005. |

