Изобретение относится к области химии биологически активных веществ, в частности к новому пептидному сополимеру, способу его получения и фармацевтической композиции на его основе.
Синтетические полипептиды являются важным классом биополимеров, представляющих большой интерес в качестве биоматериалов для инженерии тканей [X.Y.Wang, et al., Mater. Today 2006, 9, 44-53], катализаторов [Mart, R.J., et al., Soft Matter, 2006, 2, 822-835; S.Juliá, et al., Angew. Chem., Int. Ed. Engl. 1980, 19, 929. doi: 10.1002/anie.198009291], средств доставки лекарств [T.J.Deming, Adv/. Drug Delivery Rev., 2002, 54, 1145-1155], a также в качестве лекарственных средств [J.S.Wolinsky, Ann. Neurol., 2007, 61, 14-24; doi: 10.1002/ana.21079]. Примером такого лекарственного препарата служит Глатирамер ацетат, успешно используемый при лечении рассеянного склероза [J.S.Wolinsky, et al., Ann. Neur., 2007, 61, 14-24]. Он представляет собой статистический сополимер L-аланина, L-лизина, L-глутаминовой кислоты и L-тирозина. Средняя молекулярная масса Глатирамера ацетата лежит в интервале 4,7-11 кДа.
Хорошо известны сложности, возникающие при получении пептидных полимеров сополимеризацией N-карбоксиангидридов аминокислот, связанные с неустойчивостью процесса при масштабировании, а также трудным контролем степени полимеризации и свойств образующегося полимера. Для решения этой задачи могут быть использованы металлорганические инициаторы [Т.J.Deming, Nature, 1997, 390, 386-389; doi:10.1038/37084], a также использованы микрофлюдные системы, обеспечивающие большую степень контроля над реакцией полимеризации [T.Honda, et al., Lab Chip, 2005, 5, 812-818].
Известные способы получения Глатирамера ацетата основаны на совместной полимеризации N-карбоксиангидридов аминокислот, содержащих защитные группы, под действием основных индукторов, таких как первичные и вторичные амины, с последующим удалением этих защитных групп. N-Карбоксиангидриды аминокислот, являющиеся исходными веществами для стадии полимеризации, получают реакцией аминокислот или их производных с фосгеном, дифосгеном, трифосгеном или их синтетическими эквивалентами в среде безводных апротонных растворителей, таких как тетрагидрофуран (ТГФ). В усовершенствованном варианте синтез этих полупродуктов может проводиться в присутствии нейтральных акцепторов HCl, таких как пропиленоксид или алкены.

Конечный полимер очищают методами микрофильтрации его водных растворов на мембранах от 3 до 30 кДа либо диализом. Продукт в сухом состоянии получают методом лиофилизации его водных растворов.
В уровне технике описано применение различных защищенных N-карбоксиангидридов лизина для получения Глатирамера ацетата. Так, в патентах США US 3,849,550; 5,800,808; 5,981,589; 6,048,898; 6,054,430; 6,342,476; 6,362,161 был использован N-карбоксиангидрид Nε-трифторацетил-L-лизина (IV) при сополимеризации с N-карбоксиангидридами L-аланина (II), γ-O-бензил L-глутаминовой кислоты (III) и L-тирозина (V). Для удаления трифторацетильных групп использовали амины, предпочтительно моно- и диизопропиламин. Ацетат полимера получали действием 50% уксусной кислоты в течение 30 минут или проведением конечной очистки в уксуснокислой среде.
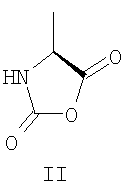
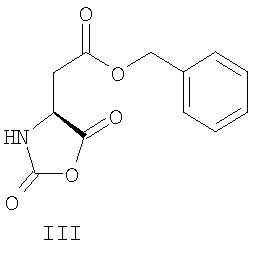

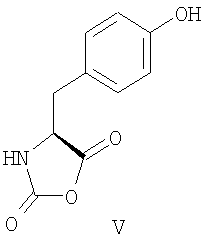
Описанный способ неудобен ввиду широкого использования метода центрифугирования для ускорения разделения фаз. Это усложняет технологию и увеличивает стоимость процесса при масштабировании синтеза до промышленных объемов.
Известен способ получения Глатирамера ацетата, который заключается в сополимеризации N-карбоксиангидрида Nε-Вос-L-лизина (VI) с N-карбоксиангидридами (II), (III), (V) с последующим удалением защитных групп раствором бромоводорода в уксусной кислоте или смесью концентрированной соляной и уксусной кислот [международная заявка WO 2006/05012 А1, патенты США US 2008/0021200 А1, US 2006/0154862 А1].
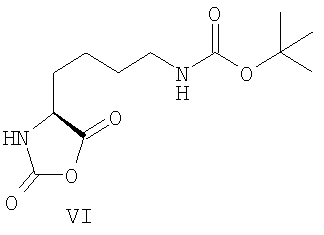
В связи с трудностью очистки исходного Nε-Boc-L-лизина и сильным влиянием посторонних примесей на выход соответствующего N-карбоксиангидрида и его дальнейшее поведение в реакции полимеризации применение исходного соединения (IV) в синтезе Глатирамер ацетата является малоэффективным.
Известен способ получения Глатирамера ацетата, который заключается в сополимеризации N-карбоксиангидридов (II), (III), (V) и Nε-бензил-L-лизина (VII) в N,N-диметилформамиде (ДМФА), смеси ДМФА с хлористым метиленом, диоксане или тетрагидрофуране (ТГФ) [патентная заявка Канады СА 2411786 А1, международная заявка WO 2004/043995 А2]. Снятие защитных групп осуществляют каталитическим гидрогенолизом. В качестве источника водорода используют водород, циклогексен или циклогексадиен.

Данный способ неудобен тем, что защищенный полимер малорастворим как в воде, так и большинстве органических растворителей, пригодных для гидрирования. Протекание реакции в подобной гетерофазной системе затрудняет масштабирование процесса, который в большей степени контролируется явлениями переноса вещества, чем кинетикой реакции [см., напр., Roberge, D.M. Org. Process Res. Devel. 2004, 8, 1049]. При этом возможно медленное и/или неполное деблокирование полимера, что изменяет характеристики конечного продукта. Это является крайне негативным фактором, затрудняющим переход к промышленным объемам производства. Кроме того, в процессе используют обогащенный палладием уголь, что приводит к существенному удорожанию процесса. Обогащенный палладием уголь также способен самовозгораться при контакте с горючими растворителями, что создает повышенные требования к технике безопасности на производстве [А.О.King, I.Shinkai. "Palladium on carbon" in "Encyclopedia of Reagents for Organic Synthesis", L.A. Paquette Ed.-in-Chief; John Wiley & Sons, Inc., Chichester, 2005 (electronic version)].
Описана фракция сополимера-1, представляющая собой смесь полипептидов, состоящих из остатков аланина, глутаминовой кислоты, лизина и тирозина, и имеющая среднюю молекулярную массу примерно от 4 до 8,6 кДа. На основе этой фракции известна фармацевтическая композиция для лечения рассеянного склероза [патент RU 2161489].
Ближайшим аналогом Глатирамера ацетата и сополимеров лизина является «усовершенствованный сополимер-1», представляющий собой статистический сополимер аланина, глутаминовой кислоты, лизина и тирозина в мольном соотношении 6:2:5:1 соответственно и имеющий среднюю молекулярную массу в интервале от 4 до 8,6 кДа. Его получают взаимодействием сополимера-1, имеющего бензильные и трифторацетильные защитные группы, с бромистоводородной кислотой с образованием сополимера-1, содержащего только трифторацетильные группы, который затем обрабатывают водным раствором пиперидина с последующей очисткой и выделением сополимера-1 с молекулярной массой примерно от 5 до 9 кДа [патент RU 2198900]. Этот метод не позволяет контролировать степень полимеризации N-карбоксиангидридов аминокислот, в частности получать полимер с более высокой молекулярной массой. Поскольку процесс протекает в гетерофазных условиях, он трудно поддается масштабированию.
Таким образом, задача настоящего изобретения состоит в разработке нового пептидного сополимера, полученного более эффективным способом, причем этот способ, в частности, может быть использован для получения лекарственного препарата «Глатирамера ацетата». При этом целевой продукт должен быть получен по масштабируемой технологии, совместимой с промышленным производством, обеспечивающей приемлемый контроль таких свойств продукта, как средняя молекулярная масса и аминокислотный состав.
Вышеуказанная задача решается новым сополимером, содержащим остатки лизина, аланина, тирозина и глутаминовой кислоты в соотношении 0,4-0,45:0,3-0,36:0,09-0,11:0,14-0,18, включающим его кислотно-аддитивную соль - ацетат и имеющим среднюю молекулярную массу, лежащую в интервале примерно 8,7-14,5 кДа.
Техническая задача достигается также и способом получения пептидного сополимера по п.1 путем совместной полимеризации N-карбоксиангидрида Nε-трифторацетил-L-лизина и/или N-карбоксиангидрида Nε-бензилоксикарбонил-L-лизина с N-карбоксиангидридами L-аланина, гамма-О-бензил-L-глутаминовой кислоты и L-тирозина с инициатором диэтиламином в среде апротонных растворителей диоксан или тетрагидрофуран или их смесях, с последующим удалением защитных групп бензильного типа с остатков лизина и глутаминовой кислоты раствором бромводорода в уксусной кислоте с каталитическим количеством воды, а для удаления защитных трифторацетильных групп с остатков лизина используют легколетучие амины диэтиламин или метиламин в виде водного раствора их.
Итак, в общем виде, поставленная задача также решается предложенным способом получения пептидного сополимера, содержащего остатки лизина, аланина, тирозина и глутаминовой кислоты в соотношении 0,4-0,45:0,3-0,36:0,09-0,11:0,14-0,18, включая его кислотно-аддитивную соль - ацетат, и имеющего среднюю молекулярную массу, лежащую в интервале примерно 8,7-14,5 кДа, - Глатирамера ацетата, путем совместной полимеризации N-карбоксиангидридов лизина, имеющих различные защитные группы, с N-карбоксиангидридами других аминокислот в среде апротонных растворителей или их смесях с последующим удалением защитных групп.

В качестве апротонного растворителя используют диоксан и/или тетрагидрофуран (ТГФ), а в качестве одного из защищенных N-карбоксиангидридов лизина используют N-карбоксиангидрид Nε-трифторацетил-L-лизина или N-карбоксиангидрид Nε-бензилоксикарбонил-L-лизина.
На стадии удаления защитных групп бензильного типа - Nε-бензилоксикарбонильной на остатках лизина и бензилового эфира в боковой цепи остатков глутаминовой кислоты - используют раствор бромоводорода в уксусной кислоте в сочетании с каталитическим количеством воды, а на стадии удаления защитных Nε-трифторацетильных групп с остатков лизина используют легколетучие амины, такие как диэтиламин или метиламин, в виде водного раствора их.
Поставленная задача также решается предложенной фармацевтической композицией для получения лекарственного средства при лечении рассеянного склероза, которая содержит терапевтически эффективное количество указанного выше пептидного сополимера и фармацевтически приемлемый носитель. В качестве фармацевтически приемлемого носителя композиция содержит, например, воду.
В предложенном способе последующее дебензилирование может быть проведено при комнатной температуре в течение 17-24 часа. Удаление трифторацетильных защитных групп с фрагментов лизина может быть осуществлено при комнатной температуре в течение 24-48 часов. Раствор конечного полимера может быть очищен одним из известных методов, например дробной микрофильтрацией водного раствора на мембранах с пределом отсечения по молекулярной массе 30-100 кДа, с последующим концентрированием на мембранах с пределом отсечения по молекулярной массе 2-8 кДа, и выделением конечного полимера лиофилизацией.
В ходе изучения совместной полимеризации N-карбоксиангидридов (II), (III), (V) и (VIII) после снятия защитных групп был получен полимер средней молекулярной массой 18-20 кДа, что выше, чем средняя молекулярная масса целевого полимера. При этом использование метода микрофильтрации для отсечения тяжелых фракций резко понижало выход целевого продукта. Исследование полученного полимера методом обращенно-фазной высокоэффективной хроматографии на колонке С-18 показало большее время удерживания, чем для Глатирамера ацетата, в тех же условиях.
В другом эксперименте после проведения полимеризации N-карбоксиангидридов (II), (III), (IV) и (V) и удаления защитных групп с остатков лизина конечный полимер имел среднюю молекулярную массу 5-7 кДа по данным гель-хроматографии, что ниже, чем у полимера (I). Исследование полученного полимера методом обращенно-фазной высокоэффективной жидкостной хроматографии на колонке С-18 показало меньшее время удерживания, чем для Глатирамера ацетата, в тех же условиях.
При проведении совместной полимеризации Nε-трифторацетил-L-лизина (IV) и Nε-бензилоксикарбонил-L-лизина (VIII) с N-карбоксиангидридами (II), (III) и (V) наблюдалось изменение свойств получаемых после удаления защитных групп с полимеров в зависимости от соотношения N-карбоксиангидридов (IV) и (VIII). При этом увеличение доли N-карбоксиангидрида Nε-бензилоксикарбонил-L-лизина (VIII) приводило к увеличению средней молекулярной массы и времени удерживания на ВЭЖХ. Таким образом, предлагаемый в данном изобретении метод сочетания различных защищенных N-карбоксиангидридов лизина в совместной полимеризации с N-карбоксиангидридами других аминокислот позволяет точно регулировать как среднюю молекулярную массу конечного полимера, так и время его удерживания на ВЭЖХ, которое, в частности, связано с такой характеристикой пептидного сополимера, как липофильность.
Для получения Глатирамера ацетата способом по изобретению было использовано следующее соотношение N-карбоксиангидридов аминокислот.
После стадии полимеризации было проведено одновременное удаление бензильных групп с фрагментов глутаминовой кислоты и бензилоксикарбонильных групп с фрагментов лизина действием 32-33% раствора бромоводорода в уксусной кислоте при комнатной температуре с добавлением каталитического количества воды. Удаление трифторацетильных защитных групп с фрагментов лизина было осуществлено действием водного раствора этиламина при комнатной температуре. Раствор конечного полимера очищали методом дробной микрофильтрации водного раствора на мембранах с пределом отсечения по молекулярной массе 30-100 кДа, и дополнительно очищается и концентрируется на мембранах с пределом отсечения по молекулярной массе 2-8 кДа. Продукт выделяли методом лиофилизации водного раствора. В обоих случаях полимер, полученный методом по изобретению, был идентичен с ацетатом Глатирамера. Это свидетельствует о том, что предложенный способ получения пептидного сополимера позволяет синтезировать полимеры с заданными свойствами, такими как средняя молекулярная масса и липофильность. Аминокислотный состав полимера, полученного способом по изобретению, также совпадал с аминокислотным составом Глатирамера ацетата.
Обеспечение характеристик продукта статистической полимеризации, таких как средняя молярная масса и липофильность, достигается путем варьирования соотношения N-карбоксиангидридов лизина с различными защитными группами. Изобретение включает применение данного метода контролируемой полимеризации в синтезе лекарственного препарата «Глатирамера ацетат» (I).
Изобретение иллюстрируется следующими примерами, которые не ограничивают его объем.
Пример 1. N-карбоксиангидрид L-аланина
Суспензию 100 г (1,12 моль) L-аланина в 1,5 л абсолютного ТГФ кипятили 1 час в атмосфере сухого аргона при перемешивании механической мешалкой. Раствор 170 г (0,6 моль) трифосгена в 500 мл абсолютного THF прибавляли по каплям в течение 30-40 минут. Реакционную смесь кипятили при перемешивании 2-3 часа до растворения основного количества аминокислоты, охладили до комнатной температуры, декантировали с незначительного осадка и разбавили 6 л сухого петролейного эфира. Полученный раствор выдерживали в морозильной камере при -20°С сутки. Образовавшийся кристаллический осадок в холодном виде быстро отфильтровывали, промыли на фильтре сухим петролейным эфиром и сушили в вакууме масляного насоса. Получили 65-75 г продукта в виде белых кристаллов, чувствительных к влаге воздуха. Выход 50-60%.
Пример 2. N-карбоксиан гидрид γOBz-L-глутаминовой кислоты
Суспензию 200 г (0,84 моль) γOBz-L-глутаминовой кислоты в 1,5 л абсолютного ТГФ кипятили 1 час в атмосфере сухого аргона при перемешивании механической мешалкой. Раствор 90 г (0,3 моль) трифосгена в 0,5 л абсолютного ТГФ прибавляли по каплям в течение 30-40 минут. Реакционную смесь кипятили 2-3 часа при перемешивании до растворения аминокислоты, охладили до комнатной температуры, декантировали с незначительного осадка и разбавили 6 л сухого петролейного эфира. Полученный раствор с осадком выдерживали в морозильной камере при -20°С сутки. Образовавшийся кристаллический осадок в холодном виде быстро отфильтровывали, промывали на фильтре сухим петролейным эфиром и сушили в вакууме масляного насоса. Получили 205-210 г продукта в виде белых кристаллов, чувствительных к влаге воздуха. Выход 90-93%.
Пример 3. N-карбоксиангидрид Nε-трифторацетил-L-лизина
Суспензию 200 г (0,71 моль) перекристаллизованного из воды и тщательно высушенного Nε-TFA-L-лизина в 1,5 л абсолютного ТГФ кипятили 1 час в атмосфере сухого аргона при перемешивании механической мешалкой. Раствор 100 г (0,33 моль) трифосгена в 0,5 л абсолютного THF прибавляли по каплям в течение 30-40 минут. Реакционную смесь кипятили 3 часа при перемешивании, охладили, декантировали с незначительного осадка и разбавили 6 л сухого петролейного эфира. Полученный раствор с осадком выдерживали в морозильной камере при -20°С сутки. Образовавшийся кристаллический осадок в холодном виде быстро отфильтровывали и промыли на фильтре сухим петролейным эфиром и сушили в вакууме масляного насоса. Получили 200-205 г продукта в виде белых кристаллов, чувствительных к влаге воздуха. Выход 88-92%.
Пример 4. N-карбоксиангидрид Nε-бензилоксикарбонил-L-лизина
Суспензию 200 г (0,65 моль) перекристаллизованного из воды и тщательно высушенного Nε-бензилоксикарбонил-L-лизина в 1,5 л абсолютного ТГФ кипятили 1 час в атмосфере сухого аргона при перемешивании механической мешалкой. Раствор 75 г (0,25 моль) трифосгена в 0,5 л абсолютного ТГФ прибавляли по каплям в течение 30-40 минут. Реакционную смесь кипятили 3 часа при перемешивании, охладили, декантировали с незначительного осадка и разбавили 6 л сухого петролейного эфира. Полученный раствор с осадком выдерживали в морозильной камере при -20°С сутки. Образовавшийся кристаллический осадок в холодном виде быстро отфильтровывали, промыли на фильтре сухим петролейным эфиром и сушили в вакууме масляного насоса. Получили 190-195 г продукта в виде белых кристаллов, чувствительных к влаге воздуха. Выход 82-84%.
Пример 5. N-карбоксиангидрид-L-тирозина
Суспензию 200 г (1,1 моль) L-тирозина в 1500 мл абсолютного ТГФ кипятили 1 час в атмосфере сухого аргона при перемешивании механической мешалкой. Раствор 120 г (0,4 моль) трифосгена в 500 мл абсолютного ТГФ прибавляли по каплям в течение 30-40 минут. Реакционную смесь умеренно кипятили 3 часа при перемешивании, охладили, декантировали с незначительного осадка и разбавили 6 л сухого петролейного эфира. Полученный раствор с осадком выдерживали в морозильной камере при -20°С сутки. Образовавшийся кристаллический осадок в холодном виде быстро отфильтровывали и промыли на фильтре сухим петролейным эфиром. Продукт сушили в вакууме масляного насоса. Выход 180-205 г. Белое кристаллическое вещество, чувствительное к влаге воздуха, устойчивое при хранении в морозильной камере -20°С. Выход 80-92%.
Пример 6. Полимеризация на основе Nε-трифторацетил-L-лизина
Смесь N-карбоксиангидридов L-аланина (2) 72,0 г, γ-O-бензил L-глутаминовой кислоты (3) 40,0 г, Nε-трифторацетил-L-лизина (4) 90,0 г, L-тирозина (5) 24,7 г и Nε-бензилоксикарбонил-L-лизина (8) 10,0 г растворяли в 4 л безводного диоксана при комнатной температуре. Реакцию полимеризации индуцировали добавлением 4 мл (3 моль %) диэтиламина. Реакционную смесь перемешивали при комнатной температуре (20°С) без доступа влаги 24 часа. Реакцию останавливали, выливая реакционную смесь в избыток воды (20-25 л) при интенсивном перемешивании. Полученный защищенный полимер тщательно промывали водой на фильтре, отжимали, сушили на воздухе и в сушильном шкафу при 45°С. Выход защищенного полимера 144 г.
Пример 7. Снятие защитных бензильных и трифторацетильных групп с полимера, полученного в примере 6.
К 200 г защищенного полимера добавляли 2 л 33% HBr в уксусной кислоте и 2 мл дистиллированной воды. Реакционную смесь перемешивали без доступа влаги 19 часов при комнатной температуре, полимер осаждали добавлением 4 л сухого диэтилового эфира. Выпавший осадок дважды промывали на стеклянном фильтре сухим диэтиловым эфиром, а после два раза дистиллированной водой. К полученному осадку добавляли 3 л воды и 442 мл 70% водного раствора этиламина, реакционную смесь перемешивали в течение 24 часов, затем при охлаждении в бане с водой разбавляли раствором уксусной кислоты в деминерализованной воде (100 мл/л) до слабокислой реакции, фильтровали на стеклянном фильтре от механических примесей и очищали на установке микрофильтрации. Пропускали через мембрану с пределом отсечения по молекулярному весу 100 кДа и концентрировали на мембране с пределом отсечения по молекулярному весу 3 кДа. Полученный прозрачный бесцветный раствор подвергали лиофилизации. Конечный продукт рыхлый пористый упругий полимер. Выход полимера (I) 70 г. Средние молекулярные массы и времена выхода для полученного полимера и препарата «Глатирамера ацетат» в условиях ВЭЖХ совпадают в пределах допустимых отклонений. Данные аминокислотного состава Глатирамера ацетата и полимера (I):
Примером фармацевтической композиции является композиция, в которой фармацевтическим приемлемым носителем является вода.
Биологическая активность глатиромера ацетата подтверждена в следующем эксперименте.
Тест блокирования развития экспериментального аутоиммунного энцефаломиелита (ЭАЭ) у мышей.
ЭАЭ представляет собой острое аутоиммунное заболевание центральной нервной системы, вызываемое у животных иммунизацией материалом, полученным из центральной нервной системы.
Блокада ЭАЭ определяется как уменьшение клинических симптомов, типичных для данного экспериментального заболевания. Биологическая активность глатирамера ацетата определяется по его способности блокировать развитие ЭАЭ у мышей. Энцефалитогенным антигеном, используемым для индукции ЭАЭ, является гомогенат спинного мозга мышей (ГСММ).
Около 0,200 г (точная навеска) Глатирамера ацетата вносят в стерильную одноразовую пластиковую пробирку объемом 15 мл (Falcon, Greiner bio-one, cat. 188261 или аналогичную), растворяют в 10 мл воды для инъекций (ФС 42-2620-97 или аналогичного качества). 2,5 мл Полученного раствора вносят в стерильную одноразовую пластиковую пробирку объемом 15 мл (Falcon, Greiner bio-one, cat. 188261 или аналогичную) и добавляют воду для инъекций (ФС 42-2620-97 или аналогичного качества) до метки 10 мл. Полученный таким образом раствор с концентрацией 5 мг/мл используют для приготовления эмульсии для подкожного введения.
Приготовление эмульсии.
В стерильной пластиковой одноразовой пробирке вместимостью 50 мл (Falcon, Greiner bio-one, cat. 210261 или аналогичной) взвешивают 0,072 г гомогената спинного мозга мышей (ГСММ). Добавляют 1,2 мл раствора испытуемого образца Глатирамера ацетата с концентрацией 5 мг/мл. Инкубируют в течение 1 ч при (37±0,1)°С. Добавляют 1,2 мл ПАФ, предварительно подогретого до (37±0,1)°С. Эмульгируют суспензиию на ультразвуковой бане в течение 5-10 мин. Стабильная эмульсия соответствует необходимому качеству, если капля эмульсии плавает, не диспергируясь, на поверхности воды, эмульгируют вместе с ГСММ с полным адьювантом Фрейнда (ПАФ) и вводят мышам линий, чувствительных к ЭАЭ.
Одновременно ЭАЭ воспроизводят у контрольной группы мышей, состоящей из того же количества животных той же линии.
Мышам контрольной группы вводят эмульсию ГСММ, не содержащую Глатирамера ацетат.
Для тестирования используются здоровые, свободные от патогенной микробной флоры (СПФ) мыши-самки в возрасте 6-9 недель линии SJL/J.
Каждая группа состоит из 10 мышей.
Животных рандомизируют на две группы. Первая является контрольной и служит для контроля индукции ЭАЭ, вторая группа предназначена для каждой серии Глатирамера ацетата, которая проходит тестирование. Каждая группа мышей помещается в клетки, имеющие специальную маркировку и идентификационные карты. Производят маркировку и идентификацию животных. Стерильным инсулиновым шприцем вводят животным по 0,1 мл раствора коклюшного токсина в хвостовую вену.
Перед введением эмульсию ГСММ тщательно перемешивают на ультразвуковой бане в течение 5-10 мин. Переносят эмульсию ГСММ в стерильный инсулиновый шприц (1,25 мл) и вводят ее подкожно в подошвы двух задних лап в объеме 0,1 мл (около 0,5 мкл в каждую подошву). 0,1 мл ГСММ содержит 0,25 мг Глатирамера ацетата.
Через 48 ч раствор коклюшного токсина и эмульсию ГСММ вводят повторно аналогичным образом.
Наблюдения за мышами проводят ежедневно в течение 20 дней, начиная с десятого дня после индукции ЭАЭ. Производят подсчет количества клинических симптомов в баллах во всех группах мышей.
Регистрацию клинических симптомов осуществляют, используя карту наблюдений, графы которой представлены в Таблице. Все мыши, имеющие балл 1 и выше, признаются заболевшими. Мыши, у которых степень заболевания достигает 4 баллов, немедленно забиваются, и им присваивают балл 4.
Таблица
Определение клинических признаков экспериментального аутоиммунного энцефаломиелита (ЭАЭ)
Баллы
Признаки
Описание (примечание)
0 Нормальное поведение
Нет неврологических синдромов
1 Слабость хвоста
Хвост мыши повис и при его поднятии падает
2 Слабость лап
Паралич конечностей, нарушения походки, неустойчивость
3 Паралич лап (частичный)
Мыши не могут двигать лапами, лапы волочатся при движении
4 Полный паралич
Мыши полностью неподвижны, выглядят изнуренными и похудевшими
5 Гибель животных
Подсчитывают количество больных животных в процентах для каждой группы. Рассчитывают ЭАЭ-блокирующую активность глатирамера ацетата (АЭАЭ) в процентах по следующей формуле:

Суммируют максимальные баллы для каждой мыши в группе и вычисляют средний максимальный балл в группе (СМБ) по формуле
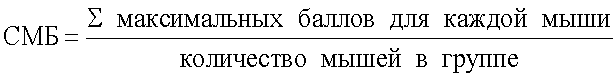
Вычисляют отношение СМБ следующим образом:
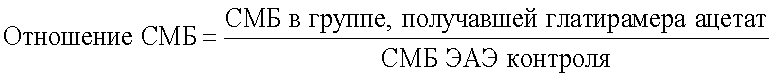
Результаты испытания приемлемы, если Глатирамера ацетат проявляет активность не менее чем в 80% случаев и отношение СМБ≤0,2.
Примечания. 1. Приготовление полного адьюванта Фрейнда. Для индукции ЭАЭ используют коммерческий полный адьювант Фрейнда (ПАФ) (фирмы «Calbiochem» кат. №344289 или аналогичного качества) без дополнительной подготовки.
2. Приготовление гомогената спинного мозга мышей (ГСММ). Мышей (нелинейных) обоих полов забивают с помощью эфира либо CO2. Выделяют позвоночник (с обоих концов) и удаляют спинной мозг с помощью давления воздуха. Ткань полученного спинного мозга хранят в морозильной камере (при -20°С) до гомогенизации и лиофилизации. Смешивают ткани спинного мозга, полученные из различных особей (300-500 мышей).
Взвешивают смешанные ткани спинного мозга, добавляют 1 часть РФБ (раствор фосфатного буфера) на 4 части ткани спинного мозга (W/W). Гомогенизируют ткани с помощью гомогенизатора в течение 3-5 мин при охлаждении льдом. Немедленно замораживают гомогенат жидким азотом и лиофилизируют в течение 48 ч. Сухой ГСММ хранят в плотно закрытых флаконах при температуре от -10°С до -20°С. Материал стабилен в течение 1 года.
3. Определение активности ГСММ. До использования проверяют способность каждой серии ГСММ вызывать ЭАЭ у мышей-самок линии SJL/J. Активность серии ГСММ может быть тестирована с использованием 3-4 групп мышей по 10 животных в каждой с использованием доз, составляющих 2-6 мг/мышь. Пример титрования ГСММ в дозах 3, 4, 5 и 6 мг/мышь: взвешивают 72,0, 96,0, 120,0 и 144,0 мг (точные навески) лиофилизированного ГСММ. Суспендируют каждую навеску в 1,2 мл воды для инъекций для получения суспензии, содержащей 60, 80 100 или 120 мг/мл ГСММ соответственно. Инкубируют полученные образцы в течение 1 ч при (37±0,1)°С. Добавляют 1,2 мл ПАФ, предварительно подогретого до (37±0,1)°С. Эмульгируют суспензии на ультразвуковой бане (в течение 5-10 мин). Стабильная эмульсия соответствует необходимому качеству, если капля эмульсии плавает, не диспергируясь на поверхности воды. Индукция ЭАЭ производится по методике проведения ЭАЭ-блокирующего теста. Серия ГСММ считается пригодной, если она вызывает ЭАЭ, по крайней мере, у 70% мышей, которым была введена и вызвала развитие заболевания с выраженностью 2,0-4,0 балла. Если все четыре (3, 4, 5 и 6 мг/мышь) группы животных отвечают условиям приемлемости теста, в последующих экспериментах используют дозы ГСММ, дающие результаты среднего уровня. Если нет развития болезни у 70% мышей, проводят повторное титрование с использованием различных концентраций КГ.
4. Растворение коклюшного токсина. Используя одноразовый стерильный шприц объемом 2 мл, добавляют 1 мл стерильного раствора натрия хлорида изотонического 0,9% к содержимому флакона с коклюшным токсином (50 мкг) (Calbiochem, cat. №516560). Тщательно перемешивают, используя вортекс. Полученный раствор помещают в стерильную одноразовую пробирку вместимостью 15 мл (Falcon, Greiner bio-one, cat. 188261 или аналогичную). Во флакон, где находился коклюшный токсин, добавляют 1 мл стерильного раствора натрия хлорида изотонического 0,9%, тщательно перемешивают, используя вортекс, содержимое помещают в пробирку с раствором коклюшного токсина. Доводят объем полученного раствора до 10 мл с использованием стерильного раствора натрия хлорида изотонического 0,9%. Полученный раствор коклюшного токсина разливают в две стерильных одноразовых пластиковых пробирки по 5 мл в каждую, закрывают стерильными крышками. Содержимое одной пробирки используют для инъекции немедленно, вторую хранят при +4°С для повторной инъекции через 48 часов. Не замораживать!
5. Приготовление эмульсии гомогената спинного мозга мышей (ГСММ). Количество ГСММ, необходимое для каждого теста, определяют предварительным титрованием в соответствии с п.3. Около 72 мг (точная навеска) лиофилизированного ГСММ суспендируют в 1,2 мл раствора Глатирамера ацетата или воды для инъекций (контроль ЭАЭ), получая суспензию с концентрацией 60 мг/мл. Инкубируют суспензию в течение 1 ч при (37±0,1)°С, прибавляют к ней 1,2 мл ПАФ, предварительно подогретого до 37°С.
Эмульгируют суспензию на ультразвуковой бане в течение 5-10 мин. Стабильная эмульсия соответствует необходимому качеству, если капля эмульсии плавает, не диспергируясь, на поверхности воды.
Эмульсию используют свежеприготовленной, для повторного введения эмульсию хранят при +4°С в течение 48 часов.
6. Условия приемлемости теста. Тестирование может проводиться при наличии 10 животных в каждой группе. Количество мышей, наблюдение за которыми осуществляется в период проведения исследования, не может быть менее 9 в группе (в случае, если их гибель наступила от причин, не связанных с заболеванием).
Контрольная группа с ЭАЭ приемлема, если: по меньшей мере, у 70% животных регистрируется заболевание, СМБ в группе составляет 2,0-4,0.
В случае отсутствия указанных параметров тест должен быть повторен.
Пептидный сополимер, полученный вышеуказанным методом, по своим физико-химическим свойствам совпадал с препаратом сравнения «Глатирамера ацетат» в пределах допустимых отклонений. Он также показывал аналогичный профиль физиологической активности на модели рассеянного склероза. Сополимер был получен методом, позволяющим синтезировать аминокислотные сополимеры на основе лизина с заданными характеристиками, такими как средняя молекулярная масса, за счет варьирования соотношения N-карбоксиангидридов лизина IV и VIII в реакции сополимеризации с N-карбоксиангидридами других аминокислот.
Применение легколетучего амина (такого как диэтиламин) на стадии деблокирования защитной трифторацетильной группы с фрагментов лизина позволяет легко удалять следы амина во время сушки готового полимера.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОПОЛИМЕР-1, СПОСОБЫ ЕГО ПОЛУЧЕНИЯ И АНАЛИТИЧЕСКИЕ МЕТОДЫ | 2012 |
|
RU2604521C2 |
| СПОСОБ ПОЛУЧЕНИЯ СМЕСЕЙ ПОЛИПЕПТИДОВ С ИСПОЛЬЗОВАНИЕМ ОЧИЩЕННОЙ БРОМИСТОВОДОРОДНОЙ КИСЛОТЫ | 2005 |
|
RU2388764C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПОЛИПЕПТИДНЫХ СМЕСЕЙ С ИСПОЛЬЗОВАНИЕМ ГИДРОГЕНОЛИЗА | 2006 |
|
RU2419638C2 |
| УСОВЕРШЕНСТВОВАННЫЙ СОПОЛИМЕР-1 В СОПОЛИМЕРНЫХ КОМПОЗИЦИЯХ | 1995 |
|
RU2161489C2 |
| УСОВЕРШЕНСТВОВАННЫЙ СОПОЛИМЕР-1 И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1995 |
|
RU2198900C2 |
| БИОДЕГРАДИРУЕМЫЙ ПОЛИМЕРНЫЙ НОСИТЕЛЬ ДЛЯ ДОСТАВКИ ПРОТИВООПУХОЛЕВОГО ЛЕКАРСТВЕННОГО СРЕДСТВА | 2012 |
|
RU2500428C1 |
| ВАКЦИНА И СПОСОБ ЛЕЧЕНИЯ БОЛЕЗНЕЙ ДВИГАТЕЛЬНЫХ НЕЙРОНОВ | 2002 |
|
RU2303996C2 |
| СПОСОБЫ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ С ПОМОЩЬЮ СТАТИСТИЧЕСКИХ СОПОЛИМЕРОВ | 2005 |
|
RU2410115C2 |
| СОР1 ДЛЯ ЛЕЧЕНИЯ ВОСПАЛИТЕЛЬНЫХ ЗАБОЛЕВАНИЙ КИШЕЧНИКА | 2004 |
|
RU2339397C2 |
| Конъюгат дексаметазона с синтетическим статистическим полипептидом | 2020 |
|
RU2792146C2 |
Изобретение относится к пептидному сополимеру, содержащему остатки лизина, аланина, тирозина и глутаминовой кислоты в соотношении 0,4-0,45:0,3-0,36:0,09-0,11:0,14-0,18, включая его кислотно-аддитивную соль - ацетат, и имеющему среднюю молекулярную массу, лежащую в интервале примерно 8,7-14,5 кДа, который может быть использован в составе фармацевтической композиции для получения лекарственного средства против рассеянного склероза, в частности лекарственного препарата «Глатирамера ацетат». Пептидный сополимер получают путем совместной полимеризации N-карбоксиангидрида Nε-трифторацетил-L-лизина и/или N-карбоксиангидрида Nε-бензилоксикарбонил-L-лизина с N-карбоксиангидридами L-аланина, гамма-O-бензил-L-глутаминовой кислоты и L-тирозина с инициатором диэтиламином в среде апротонных растворителей диоксана или тетрагидрофурана или их смесях. Удаление защитных групп бензильного типа с остатков лизина и глутаминовой кислоты осуществляют раствором бромводорода в уксусной кислоте с каталитическим количеством воды, а для удаления защитных трифторацетильных групп с остатков лизина используют легколетучие амины диэтиламин или метиламин в виде их водного раствора. Способ позволяет контролировать такие свойства продукта, как средняя молекулярная масса и аминокислотный состав. 3 н.п. ф-лы.
1. Пептидный сополимер, содержащий остатки лизина, аланина, тирозина и глутаминовой кислоты в соотношении 0,4-0,45:0,3-0,36:0,09-0,11:0,14-0,18, включая его кислотно-аддитивную соль - ацетат, и имеющий среднюю молекулярную массу, лежащую в интервале примерно 8,7-14,5 кДа.
2. Способ получения пептидного сополимера по п.1 путем совместной полимеризации N-карбоксиангидрида Nε-трифторацетил-L-лизина и/или N-карбоксиангидрида Nε-бензилоксикарбонил-L-лизина с N-карбоксиангидридами L-аланина, гамма-О-бензил-L-глутаминовой кислоты и L-тирозина с инициатором диэтиламином в среде апротонных растворителей диоксана или тетрагидрофурана или их смесях с последующим удалением защитных групп бензильного типа с остатков лизина и глутаминовой кислоты раствором бромводорода в уксусной кислоте с каталитическим количеством воды, а для удаления защитных трифторацетильных групп с остатков лизина используют легколетучие амины диэтиламин или метиламин в виде их водного раствора.
3. Фармацевтическая композиция для получения лекарственного средства против рассеянного склероза, содержащая терапевтически эффективное количество пептидного сополимера по п.1 и фармацевтически приемлемый носитель.
| УСОВЕРШЕНСТВОВАННЫЙ СОПОЛИМЕР-1 И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1995 |
|
RU2198900C2 |
| УСОВЕРШЕНСТВОВАННЫЙ СОПОЛИМЕР-1 В СОПОЛИМЕРНЫХ КОМПОЗИЦИЯХ | 1995 |
|
RU2161489C2 |
| Самовар | 1916 |
|
SU2285A1 |
| СПОСОБ ВЫРАБОТКИ КОМПОТА ИЗ АБРИКОСОВ | 2009 |
|
RU2411786C1 |
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| US 3849550 A, 19.11.1974. | |||

