Настоящее изобретение относится к определенным замещенным тиазолилфенилбензолсульфонамидо соединениям, которые модулируют активность протеинкиназ. Таким образом, соединения по настоящему изобретению могут быть использованы для лечения заболеваний, вызываемых нарушением регуляции активности протеинкиназ. Также настоящее изобретение относится к способам получения этих соединений, фармацевтическим композициям, содержащим эти соединения, и к способам лечения заболеваний с использованием фармацевтических композиций, содержащих эти соединения.
Классический каскад Ras, Raf, MEK (активируемая митогеном протеинкиназа/киназа регулируемой внеклеточным сигналом киназы), ERK (регулируемая внеклеточным сигналом киназа) играет центральную роль в регуляции различных клеточных функций, в зависимости от клеточного контекста, включая пролиферацию, дифференцировку, выживание, иммортализацию и ангиогенез клеток (рассмотрено в Peyssonnaux and Eychene, Biology of the Cell, 2001, 93, 3-62). В этом каскаде представители семейства Raf привлекаются к плазматической мембране при связывании с Ras, нагруженной гуанозинтрифосфатом (GTP), что приводит к фосфорилированию и активации белков Raf. Затем активированные Raf фосфорилируют и активируют MEK, которые в свою очередь фосфорилируют и активируют ERK. При активации ERK транслоцируются из цитоплазмы в ядро, что приводит к фосфорилированию и регуляции активности факторов транскрипции, таких как Elk-I и Myc. Описано, что Ras/Raf/MEK/ERK вносит вклад в онкогенный фенотип путем индукции иммортализации, роста, независимого от факторов роста, нечувствительности к ингибирующим рост сигналам, способности к инвазии и метастазированию, путем стимуляции ангиогенеза и путем ингибирования апоптоза (рассмотрено в Kolch et al., Exp. Rev. Mol. Med., 2002, 25 April, http://www.expertreviews.org/02004386h.htm). Действительно, фосфорилирование ERK повышено приблизительно в 30% всех опухолей человека (Hoshino et al., Oncogene, 1999, 18, 813-822). Это может быть результатом сверхэкспрессии и/или мутации ключевых представителей каскада.
Описаны три изоформы серин/треониновой протеинкиназы Raf: Raf-1/C-Raf, B-Raf и A-Raf (рассмотрено в Mercer and Pritchard, Biochim. Biophys. Acta, 2003, 1653, 25-40), гены для которых, как полагают, появились в результате дупликации гена. Все три гена Raf экспрессируются в большинстве тканей, но с отличиями: C-Raf экспрессируется повсеместно на высоких уровнях, в то время как экспрессия B-Raf на высоком уровне встречается в нейрональной ткани, а экспрессия A-Raf встречается в тканях мочеполовых путей. Высоко гомологичные представители семейства Raf имеют перекрывающиеся, но отличающиеся биохимические виды активности и биологические функции (Hagemann and Rapp, Expt. Cell Res. 1999, 253, 34-46). Экспрессия всех трех генов Raf требуется для нормального развития мышей, однако как C-Raf, так и B-Raf, требуются для завершения гестации. Мыши B-Raf -/- погибают на сроке E12,5 вследствие сосудистой геморрагии, вызываемой повышенным апоптозом эндотелиальных клеток (Wojnowski et al. Nature Genet., 1997, 16, 293-297). Описано, что B-Raf является главной изоформой, вовлеченной в пролиферацию клеток, и является первичной мишенью онкогенной Ras. Активирующие соматические миссенс-мутации были идентифицированы исключительно для B-Raf, встречающихся с частотой 66% в злокачественных меланомах кожи (Davies et al., Nature, 2002, 417, 949-954) и также присутствующих в широком диапазоне злокачественных опухолей человека, включая, но не ограничиваясь ими, папиллярные опухоли щитовидной железы (Cohen et al., J. Natl. Cancer Inst., 2003, 95, 625-627), холангиокарциномы (Tannapfel et al., Gut, 2003, 52, 706-712), рак толстого кишечника и рак яичника (Davies et al., Nature, 2002, 417, 949-954). Наиболее частой мутацией в B-Raf (80%) является замещение глутаминовой кислотой валина в положении 600. Эти мутации увеличивают базальную активность киназы B-Raf, и полагают, что они разобщают передачу сигнала Raf/MEK/ERK от вышележащих механизмов включения пролиферации, включая активацию Ras и рецепторов факторов роста, что приводит к конститутивной активации ERK. Мутантные белки B-Raf являются трансформирующими в клетках NIH3T3 (Davies et al., Nature, 2002, 15 417, 949-954) и меланоцитах (Wellbrock et al., Cancer Res., 2004, 64, 2338-2342) и также было показано, что они необходимы для жизнеспособности и трансформации клеток меланомы (Hingorani et al., Cancer Res., 2003, 63, 5198-5202). В качестве ключевого запускающего фактора каскада передачи сигнала Raf/MEK/ERK, B-Raf представляет собой возможную точку вмешательства в опухоли, зависимые от этого каскада.
Замещенные производные тиазола для лечения опосредуемых протеинкиназами заболеваний, таких как злокачественная опухоль, описаны в WO 2009/137391 на имя SKB & Co, в WO 2011/059610 на имя Glaxosmithkline LLC и в WO 2011/161216 на имя Novartis AG.
Авторы настоящего изобретения открыли, что соединения формулы (I), описанные ниже, являются ингибиторами киназ и, таким образом, пригодны в терапии в качестве противоопухолевых средств.
Таким образом, первой задачей настоящего изобретения является предоставление замещенного тиазолилфенилбензолсульфонамидо соединения, соответствующего формуле (I)
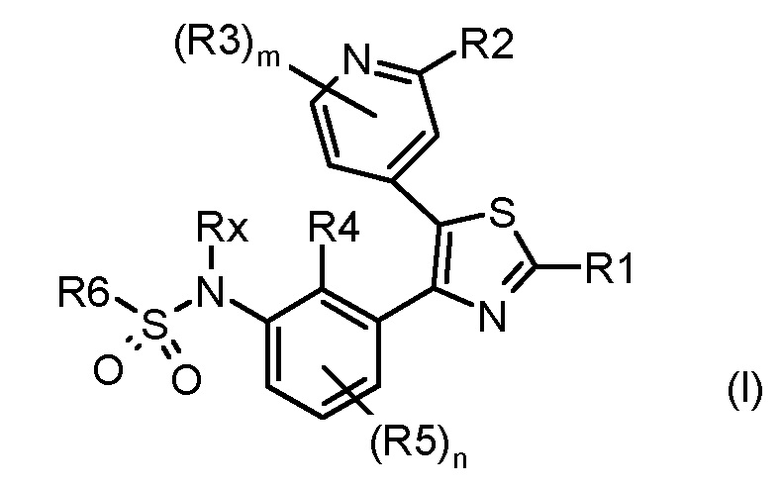
где:
каждый из n и m независимо равен 1 или 2;
R1 представляет собой водород, галоген, циано или необязательно замещенную группу, выбранную из прямого или разветвленного (C1-C8)алкила, (C2-C8)алкенила, (C2-C8)алкинила, (C3-C8)циклоалкила, (C3-C8)циклоалкенила, гетероциклила, арила и гетероарила; или R1 представляет собой NR7R8 или COR9,
где:
каждый из R7 и R8 независимо представляет собой водород или необязательно замещенную группу, выбранную из прямого или разветвленного (C1-C8)алкила, (C2-C8)алкенила, (C2-C8)алкинила, (C3-C8)циклоалкила, (C3-C8)циклоалкенила, гетероциклила, арила и гетероарила; или, взятые вместе с атомом азота, с которым они связаны, R7 и R8 могут образовывать необязательно замещенный 3-8-членный гетероциклил, необязательно содержащий один дополнительный гетероатом или гетероатомную группу, выбранные из S, O, N и NH; или
R7 представляет собой водород и R8 представляет собой COR10,
где:
R10 представляет собой OR11, NR12R13 или необязательно замещенную группу, выбранную из прямого или разветвленного (C1-C8)алкила, (C2-C8)алкенила, (C2-C8)алкинила, (C3-C8)циклоалкила, (C3-C8)циклоалкенила, гетероциклила, арила и гетероарила, где:
R11 представляет собой необязательно замещенную группу, выбранную из прямого или разветвленного (C1-C8)алкила, (C2-C8)алкенила, (C2-C8)алкинила, (C3-C8)циклоалкила, гетероциклила, арила и гетероарила;
каждый из R12 и R13 независимо представляет собой водород или необязательно замещенную группу, выбранную из прямого или разветвленного (C1-C8)алкила, (C2-C8)алкенила, (C2-C8)алкинила, (C3-C8)циклоалкила, гетероциклила, арила и гетероарила; или, взятые вместе с атомом азота, с которым они связаны, R12 и R13 могут образовывать необязательно замещенный 3-8-членный гетероциклил или гетероарил, необязательно содержащий один дополнительный гетероатом или гетероатомную группу, выбранные из S, O, N и NH;
R9 представляет собой OR14 или NR15R16, где:
R14 представляет собой водород или необязательно замещенную группу, выбранную из прямого или разветвленного (C1-C8)алкила, (C2-C8)алкенила, (C2-C8)алкинила, (C3-C8)циклоалкила, гетероциклила, арила и гетероарила;
каждый из R15 и R16 независимо представляет собой водород или необязательно замещенную группу, выбранную из прямого или разветвленного (C1-C8)алкила, (C2-C8)алкенила, (C2-C8)алкинила, (C3-C8)циклоалкила, гетероциклила, арила и гетероарила; или, взятые вместе с атомом азота, с которым они связаны, R15 и R16 могут образовывать необязательно замещенный 3-8-членный гетероциклил или гетероарил, необязательно содержащий один дополнительный гетероатом или гетероатомную группу, выбранные из S, O, N и NH;
каждый из R2 и R3 независимо представляет собой водород, галоген, циано или необязательно замещенную группу, выбранную из прямого или разветвленного (C1-C8)алкила, (C2-C8)алкенила, (C2-C8)алкинила, (C3-C8)циклоалкила, (C3-C8)циклоалкенила, гетероциклила, арила и гетероарила; или каждый из R2 и R3 независимо представляет собой NR17R18, CONR19R20, OR21, SR21 или SO2R21,
где:
R17 и R18 независимо представляют собой водород или необязательно замещенную группу, выбранную из прямого или разветвленного (C1-C8)алкила, (C2-C8)алкенила, (C2-C8)алкинила, (C3-C8)циклоалкила, гетероциклила, арила и гетероарила; или, взятые вместе с атомом азота, с которым они связаны, R17 и R18 могут образовывать необязательно замещенный 3-8-членный гетероциклил или гетероарил, необязательно содержащий один дополнительный гетероатом или гетероатомную группу, выбранные из S, O, N и NH; или
R17 представляет собой водород и R18 представляет собой COR22,
где:
R22 представляет собой OR23, NR24R25 или необязательно замещенную группу, выбранную из прямого или разветвленного (C1-C8)алкила, (C2-C8)алкенила, (C2-C8)алкинила, (C3-C8)циклоалкила, (C3-C8)циклоалкенила, гетероциклила, арила и гетероарила, где:
R23 представляет собой необязательно замещенную группу, выбранную из прямого или разветвленного (C1-C8)алкила, (C2-C8)алкенила, (C2-C8)алкинила, (C3-C8)циклоалкила, (C3-C8)циклоалкенила, гетероциклила, арила и гетероарила, и
каждый из R24 и R25 независимо представляет собой водород или необязательно замещенную группу, выбранную из прямого или разветвленного (C1-C8)алкила, (C2-C8)алкенила, (C2-C8)алкинила, (C3-C8)циклоалкила, гетероциклила, арила и гетероарила: или, взятые вместе с атомом азота, с которым они связаны, R24 и R25 могут образовывать необязательно замещенный 3-8-членный гетероциклил или гетероарил, необязательно содержащий один дополнительный гетероатом или гетероатомную группу, выбранные из S, O, N и NH;
каждый из R19 и R20 независимо представляет собой водород или необязательно замещенную группу, выбранную из прямого или разветвленного (C1-C8)алкила, (C2-C8)алкенила, (C2-C8)алкинила, (C3-C8)циклоалкила, гетероциклила, арила и гетероарила; или, взятые вместе с атомом азота, с которым они связаны, R19 и R20 могут образовывать необязательно замещенный 3-8-членный гетероциклил или гетероарил, необязательно содержащий один дополнительный гетероатом или гетероатомную группу, выбранные из S, O, N и NH;
R21 представляет собой водород или необязательно замещенную группу, выбранную из прямого или разветвленного (C1-C8)алкила, (C2-C8)алкенила, (C2-C8)алкинила, (C3-C8)циклоалкила, гетероциклила, арила и гетероарила;
или R2 и R3, взятые вместе, могут быть частью гетероциклила, арила или гетероарила, когда m равно 1, и R3 находится в положении 3 ядра пиридина;
каждый из R4 и R5 независимо представляет собой водород, галоген, трифторметил, трихлорметил, циано, OR26 или необязательно замещенную группу, выбранную из прямого или разветвленного (C1-C8)алкила, (C2-C8)алкенила, (C2-C8)алкинила и (C3-C8)циклоалкила, где:
R26 представляет собой водород или необязательно замещенную группу, выбранную из прямого или разветвленного (C1-C8)алкила, (C2-C8)алкенила, (C2-C8)алкинила и (C3-C8)циклоалкила;
Rx представляет собой водород, необязательно замещенный прямой или разветвленный (C1-C3)алкил, необязательно замещенную (C2-C6)ацильную группу или необязательно замещенную (C2-C6)алкоксикарбонильную группу;
R6 представляет собой необязательно замещенную группу, выбранную из прямого или разветвленного (C2-C8)алкенила, (C2-C8)алкинила, (C3-C8)циклоалкила, (C3-C8)циклоалкенила, гетероциклила, арила и гетероарила;
или его фармацевтически приемлемых солей.
Также настоящее изобретение относится к способам получения замещенных тиазолилфенилбензолсульфонамидо соединений, соответствующих формуле (I), получаемых с помощью процесса, состоящего из стандартных синтетических преобразований.
Также настоящее изобретение относится к способу лечения заболеваний, вызываемых и/или ассоциированных с нарушением регуляции активности протеинкиназы, в частности, семейства Raf, ABL, ACK1, AKT1, ALK, AUR1, AUR2, BRK, BUB1, CDC7/DBF4, CDK2/CYCA, CHK1, CK2, EEF2K, EGFR1, EphA2, EphB4, ERK2, FAK, FGFR1, FLT3, GSK3-бета, Haspin, IGFR1, IKK2, IR, JAK1, JAK2, JAK3, KIT, LCK, LYN, MAPKAPK2, MELK, MET, MNK2, MPS1, MST4, NEK6, NIM1, P38-альфа, PAK4, PDGFR, PDK1, PERK, PIM1, PIM2, PKA-альфа, PKC-бета, PLK1, RET, ROS1, SULU1, Syk, TLK2, TRKA, TYK, VEGFR2, VEGFR3, ZAP70, более конкретно семейства Raf, который включает введение млекопитающему, нуждающемуся в этом, эффективного количества замещенного тиазолилфенилбензолсульфонамидо соединения, соответствующего формуле (I), как определено выше.
Предпочтительным способом по настоящему изобретению является лечение заболевания, вызываемого и/или ассоциированного с нарушением регуляции активности протеинкиназ, выбранного из группы, состоящей из злокачественной опухоли, нарушений пролиферации клеток, вирусных инфекций, аутоиммунных и нейродегенеративных нарушений.
Другим предпочтительным способом по настоящему изобретению является лечение конкретных типов злокачественной опухоли, включая, но не ограничиваясь ими: карциному, такую как карцинома мочевого пузыря, молочной железы, толстого кишечника, почки, печени, легкого, включая мелкоклеточный рак легкого, карциному пищевода, желчного пузыря, яичника, поджелудочной железы, желудка, шейки матки, щитовидной железы, предстательной железы и кожи, включая плоскоклеточную карциному; гемопоэтические опухоли лимфоидного ростка, включая лейкоз, острый лимфоцитарный лейкоз, острый лимфобластный лейкоз, B-клеточную лимфому, T-клеточную лимфому, лимфому Ходжкина, неходжскинскую лимфому, волосатоклеточную лимфому и лимфому Беркитта; гемопоэтические опухоли миелоидного ростка, включая острые и хронические миелогенные лейкозы, миелодиспластический синдром и промиелоцитарный лейкоз; опухоли мезенхимного происхождения, включая фибросаркому и рабдомиосаркому; опухоли центральной и периферической нервной системы, включая астроцитому, нейробластому, глиому и шванномы; другие опухоли, включая меланому, семиному, тератокарциному, остеосаркому, пигментную ксеродерму, кератоксантому, фолликулярный рак щитовидной железы и саркому Капоши.
Другим предпочтительным способом по настоящему изобретению является лечение определенных клеточно-пролиферативных нарушений, например, таких как доброкачественная гиперплазия предстательной железы, семейный аденоматоз, полипоз, нейрофиброматоз, псориаз, пролиферация гладкомышечных клеток сосудов, ассоциированная с атеросклерозом, фиброзом легких, артритом, гломерулонефритом и послеоперационным стенозом и рестенозом.
Другим предпочтительным способом по настоящему изобретению является лечение вирусных инфекций, в частности, профилактика развития СПИД у ВИЧ-инфицированных индивидуумов.
Другим предпочтительным способом по настоящему изобретению является лечение ассоциированных с иммунными клетками заболеваний и нарушений, таких как воспалительные и аутоиммунные заболевания, например, рассеянный склероз, системная красная волчанка, воспалительные заболевания кишечника (IBD), болезнь Крона, синдром раздраженной кишки, панкреатит, язвенный колит, дивертикулез, миастения gravis, васкулит, псориаз, склеродермия, астма, аллергия, системная склеродермия, витилиго, артрит, такой как остеоартрит, ювенильный ревматоидный артрит, анкилозирующий спондилит.
Другим предпочтительным способом по настоящему изобретению является лечение нейродегенеративных нарушений, таких как болезнь Альцгеймера, болезнь Паркинсона и болезнь Гентингтона.
Кроме того, способ по настоящему изобретению также предусматривает ингибирование ангиогенеза в опухоли и метастазирования опухоли, а также лечение отторжения трансплантата органа и реакции "хозяин против трансплантата".
В следующем предпочтительном варианте осуществления способ по настоящему изобретению дополнительно включает воздействие на млекопитающего, нуждающегося в этом, режима лучевой терапии или химиотерапии в комбинации по меньшей мере с одним цитостатическим или цитотоксическим средством.
Более того, изобретение относится к способу ингибирования in vitro активности белков семейства Raf, который включает контактирование указанного белка с эффективным количеством соединения формулы (I).
Также настоящее изобретение относится к фармацевтической композиции, содержащей одно или несколько соединений формулы (I) или их фармацевтически приемлемые солей и фармацевтически приемлемый эксципиент, носитель или разбавитель.
Кроме того, настоящее изобретение относится к фармацевтической композиции, содержащей соединение формулы (I), в комбинации с известным способом лечения злокачественной опухоли, таким как режим лучевой терапии или химиотерапии в комбинации с цитостатическими или цитотоксическими средствами, средствами типа антибиотиков, алкилирующими средствами, антиметаболитными средствами, гормональными средствами, иммунологическими средствами, средствами типа интерферонов, ингибиторами циклооксигеназы (например, ингибиторы COX-2), ингибиторами матриксной металлопротеиназы, ингибиторами теломеразы, ингибиторами тирозинкиназы, средствами против рецепторов факторов роста, средствами против HER, средствами против EGFR, антиангиогенными средствами (например, ингибиторы ангиогенеза), ингибиторами фарнезилтрансферазы, ингибиторами каскада передачи сигнала RAS-RAF, ингибиторами клеточного цикла, другими ингибиторами Cdk, связывающими тубулин средствами, ингибиторами топоизомеразы I, ингибиторами топоизомеразы II и т.п. Кроме того, изобретение относится к продукту или набору, содержащим соединение формулы (I) или его фармацевтически приемлемую соль, как определено выше, или их фармацевтические композиции и одно или несколько химиотерапевтических средств, в качестве комбинированного препарата для одновременного, отдельного или последовательного применения в терапии против злокачественной опухоли.
В другом аспекте изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли, как определено выше, для применения в качестве лекарственного средства.
Более того, изобретение относится к применению соединения формулы (I) или его фармацевтически приемлемой соли, как определено выше, для изготовления лекарственного средства с противоопухолевой активностью.
Наконец, изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли, как определено выше, для применения в способе лечения злокачественной опухоли.
Если нет иных указаний, при указании на соединения формулы (I) непосредственно, а также на любую их фармацевтическую композицию или на любое медикаментозное лечение, включающее их, настоящее изобретение включает все изомеры, таутомеры, гидраты, сольваты, комплексы, метаболиты, пролекарства, носители, N-оксиды и фармацевтически приемлемые соли соединений по настоящему изобретению.
Метаболит соединения формулы (I) представляет собой любое соединение, в которое это соединение формулы (I) преобразуется in vivo, например, при введении млекопитающему, нуждающемуся в этом. Как правило, но в качестве неограничивающего примера, при введении соединения формулы (I) это производное может преобразовываться в различные соединения, например, включающие более растворимые производные, такие как гидроксилированные производные, которые легко экскретируются. Таким образом, в зависимости от метаболического каскада, протекающего таким путем, любое из этих гидроксилированных производных может быть воспринято как метаболит соединений формулы (I).
Пролекарства представляют собой любые ковалентно связанные соединения, которые высвобождают in vivo активное исходное лекарственное средство формулы (I).
N-оксиды представляют собой соединения формулы (I), где азот и кислород связаны через донорно-акцепторную связь.
Если в соединении по настоящему изобретению присутствует хиральный центр или другая форма изомерного центра, подразумевается, что все формы такого изомера или изомеров, включая энантиомеры и диастереомеры, охватываются настоящим изобретением. Соединения, содержащие хиральный центр, можно использовать в качестве рацемической смеси или энантиомерно обогащенной смеси, или рацемическую смесь можно разделять с использованием хорошо известных способов и отдельный энантиомер можно использовать отдельно. В случаях, когда соединения имеют ненасыщенные углерод-углеродные двойные связи, как цис(Z)-, так и транс(E)-изомеры входят в объем настоящего изобретения.
В случаях, когда соединения могут существовать в таутомерных формах, предусматривается, что каждая форма включена в настоящее изобретение, существуя либо в равновесии, либо с преобладанием одной формы.
В случаях, когда соединения могут существовать в других таутомерных формах, таких как кето-енольные таутомеры, предусматривается, что каждая таутомерная форма включена в настоящее изобретение, существуя либо в равновесии, либо с преобладанием одной формы.
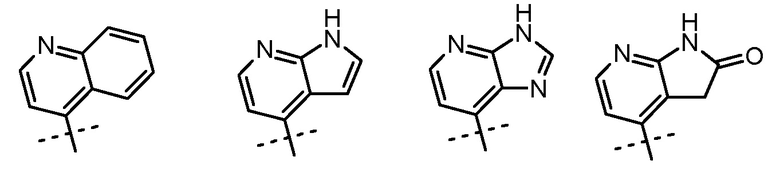
В случаях, когда m равно 1 и R3 находится в положении 3 ядра пиридина, R2 и R3, взятые вместе, могут быть частью гетероциклила, арила или гетероарила, так что в таком случае предусматривается группа, изображенная ниже:
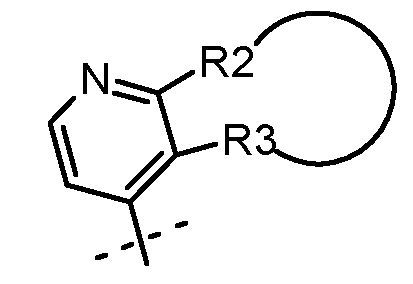
Неограничивающими примерами таких групп являются:
Под термином "прямой или разветвленный (C1-C8)алкил" понимают любую из групп, например, такую как метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, втор-бутил, н-пентил, н-гексил, н-гептил, н-октил и т.п.
Под термином "прямой или разветвленный (C1-C6)алкил" понимают любую из групп, например, такую как метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, втор-бутил, н-пентил, н-гексил и т.п.
Под термином "прямой или разветвленный (C1-C3)алкил", понимают любую из групп, например, такую как метил, этил, н-пропил, изопропил.
Под термином "(C3-C8)циклоалкил" понимают, если нет иных указаний, 3-8-членное полностью углеродное моноциклическое кольцо, которое может содержать одну или несколько двойных связей, но не имеет полностью сопряженной π-электронной системы. Примерами циклоалкильных групп являются, но не ограничиваясь ими, циклопропан, циклобутан, циклопентан, циклопентен, циклогексан, циклогексен и циклогексадиен.
Под термином "гетероциклил" понимают 3-8-членное насыщенное или частично ненасыщенное карбоциклическое кольцо, где один или несколько атомов углерода заменены гетероатомами, такими как азот, кислород и сера. Неограничивающими примерами гетероциклильных групп являются, например, пиран, пирролидин, пирролин, имидазолин, имидазолидин, пиразолидин, пиразолин, тиазолин, тиазолидин, дигидрофуран, тетрагидрофуран (THF), 1,3-диоксолан, пиперидин, пиперазин, морфолин и т.п.
Под термином "(C2-C8)алкенил" понимают алифатическую (C2-C8)углеводородную цепь, содержащую по меньшей мере одну углерод-углеродную двойную связь, и которая может быть прямой или разветвленной. Репрезентативные примеры включают, но не ограничиваются ими, этенил, 1-пропенил, 2-пропенил, 1- или 2-бутенил и т.п.
Под термином "(C2-C8)алкинил" понимают алифатическую (C2-C8)углеводородную цепь, содержащую по меньшей мере одну углерод-углеродную тройную связь, и которая может быть прямой или разветвленной. Репрезентативные примеры включают, но не ограничиваются ими, этинил, 1-пропинил, 2-пропинил, 1- или 2-бутинил и т.п.
Термин "арил" относится к моно-, би- или поли-карбоциклическому углеводороду с 1-4 кольцевыми системами, необязательно дополнительно конденсированными или связанными друг с другом одинарными связями, где по меньшей мере одно из карбоциклических колец является "ароматическим", где термин "ароматический" относится к системе с полностью сопряженной π-электронной связью. Неограничивающими примерами таких арильных групп являются фенильная, α- или β-нафтильная или бифенильная группы.
Термин "гетероарил" относится к ароматическим гетероциклическим кольцам, как правило, 5-8-членным гетероциклам с 1-3 гетероатомами, выбранными из N, O или S; гетероарильное кольцо может быть необязательно дополнительно конденсированным или связанным с ароматическими и неароматическими карбоциклическими и гетероциклическими кольцами. Неограничивающими примерами таких гетероарильных групп являются, например, пиридил, пиразинил, пиримидинил, пиридазинил, индолил, имидазолил, тиазолил, изотиазолил, пирролил, фенилпирролил, фурил, фенилфурил, оксазолил, изоксазолил, пиразолил, тиенил, бензотиенил, изоиндолинил, бензоимидазолил, хинолинил, изохинолинил, 1,2,3-триазолил, 1-фенил-1,2,3-триазолил, 2,3-дигидроиндолил, 2,3-дигидробензофуранил, 2,3-дигидробензотиофенил; бензопиранил, 2,3-дигидробензоксазинил, 2,3-дигидрохиноксалинил и т.п.
В соответствии с настоящим изобретением и если не предусмотрено иное, любая из указанных выше групп Rx, R1, R2, R3, R4, R5 и R6 может быть необязательно замещенной в любом из ее свободных положений одной или несколькими группами, например 1-6 группами, независимо выбранными из: галогена, нитро, оксогруппы (=O), циано, (C1-C8)алкила, полифторированного алкила, полифторированного алкокси, (C2-C8)алкенила, (C2-C8)алкинила, гидроксиалкила, арила, арилалкила, гетероарила, гетероарилалкила, гетероциклила, гетероциклилалкила, (C3-C8)циклоалкила, гидрокси, алкокси, арилокси, гетероциклилокси, метилендиокси, алкилкарбонилокси, арилкарбонилокси, циклоалкенилокси, гетероциклилкарбонилокси, алкилиденаминоокси, карбокси, алкоксикарбонила, арилоксикарбонила, циклоалкилоксикарбонила, гетероциклилалкилоксикарбонил-амино, уреидо, алкиламино, диалкиламино, ариламино, диариламино, гетероциклиламино, формиламино, алкилкарбониламино, арилкарбониламино, гетероциклилкарбониламино, аминокарбонила, алкиламинокарбонила, диалкиламинокарбонила, ариламинокарбонила, гетероциклиламинокарбонила, алкоксикарбониламино, гидроксиаминокарбонилалкоксиимино, алкилсульфониламино, арилсульфониламино, гетероциклилсульфониламино, формила, алкилкарбонила, арилкарбонила, циклоалкилкарбонила, гетероциклилкарбонила, алкилсульфонила, арилсульфонила, аминосульфонила, алкиламиносульфонила, диалкиламиносульфонила, ариламиносульфонила, гетероциклиламиносульфонила, арилтио, алкилтио, фосфоната и алкилфосфоната. В свою очередь, когда это является пригодным, каждый из описанных выше заместителей может быть дополнительно замещен одной или несколькими из упомянутых выше групп.
Под термином "галоген" понимают фтор, хлор, бром или иод.
Под термином "циано" понимают остаток -CN.
Под термином "нитро" понимают группу -NO2.
Под термином "полифторированный алкил" или "полифторированный алкокси" понимают любую из указанных выше прямых или разветвленных (C1-C8)алкильных или алкоксигрупп, которые замещены более чем одним атомом фтора, например, такую как трифторметил, трифторэтил, 1,1,1,3,3,3-гексафторпропил, трифторметокси и т.п.
Под термином "гидроксиалкил" понимают любой из указанных выше (C1-C8)алкилов, имеющий гидроксильную группу, например, такой как гидроксиметил, 2-гидроксиэтил, 3-гидроксипропил и т.п.
Из вышесказанного специалисту в данной области очевидно, что любая группа, название которой является составным названием, как например, ариламино, традиционно следует толковать по частям, из которых она происходит, например, как аминогруппа, которая дополнительно замещена арилом, где арил является таким, как определено выше.
Аналогично, любой из терминов, например, такой как алкилтио, алкиламино, диалкиламино, алкоксикарбонил, алкоксикарбониламино, гетероциклилкарбонил, гетероциклилкарбониламино, циклоалкилоксикарбонил и т.п., включает группы, где алкильная, алкокси, арильная, (C3-C8)циклоалкильная и гетероциклильная части являются такими, как определено выше.
Фармацевтически приемлемые соли соединений формулы (I) включают кислотно-аддитивные соли с неорганическими или органическими кислотами, например, азотной, хлористоводородной, бромистоводородной, серной, перхлорной, фосфорной, уксусной, трифторуксусной, пропионовой, гликолевой, молочной, щавелевой, фумаровой, малоновой, яблочной, малеиновой, виннокаменной, лимонной, бензойной, коричной, миндальной, метансульфоновой, изотионовой и салициловой кислотой.
Фармацевтически приемлемые соли соединений формулы (I) также включают соли с неорганическими или органическими основаниями, например, щелочными или щелочноземельными металлами, особенно гидроксиды, карбонаты или бикарбонаты натрия, калия, кальция, аммония или магния, ациклические или циклические амины, предпочтительно метиламин, этиламин, диэтиламин, триэтиламин, пиперидин и т.п.
Предпочтительным классом соединений формулы (I) являются соединения, где:
R1 представляет собой NR7R8 или необязательно замещенный гетероциклил, где R7 и R8 являются такими, как определено выше.
Более предпочтительным классом соединений формулы (I) являются соединения, где:
R2 представляет собой водород или группу NR17R18, где R17 и R18 являются такими, как определено выше.
Еще более предпочтительным классом соединений формулы (I) являются соединения, где:
R2 представляет собой группу NR17R18, где R17 представляет собой водород и R18 представляет собой COR22, где R22 является таким, как определено выше.
Наиболее предпочтительным классом соединений формулы (I) являются соединения, где:
R3 представляет собой водород, R4 представляет собой галоген и R5 представляет собой водород или галоген.
Следующим наиболее предпочтительным классом соединений формулы (I) являются соединения, где:
Rx представляет собой водород и R6 представляет собой необязательно замещенную фенильную группу.
Предпочтительные конкретные соединения (cmpd.) формулы (I) или их фармацевтически приемлемые соли представляют собой соединения, приведенные ниже:
1) N-{3-[2-амино-5-(пиридин-4-ил)-1,3-тиазол-4-ил]-2,4-дифторфенил}-2,5-дифторбензолсульфонамид;
2) N-{2,4-дифтор-3-[2-(метиламино)-5-(пиридин-4-ил)-1,3-тиазол-4-ил]фенил}-2,5-дифторбензолсульфонамид;
3) N-{3-[2-(диэтиламино)-5-(пиридин-4-ил)-1,3-тиазол-4-ил]-2,4-дифторфенил}-2,5-дифторбензолсульфонамид;
4) N-(2,4-дифтор-3-{2-[(2-метилпропил)амино]-5-(пиридин-4-ил)-1,3-тиазол-4-ил}фенил)-2,5-дифторбензолсульфонамид;
5) N-(2,4-дифтор-3-{2-[(2-метоксиэтил)амино]-5-(пиридин-4-ил)-1,3-тиазол-4-ил}фенил)-2,5-дифторбензолсульфонамид;
6) N-{2,4-дифтор-3-[2-(пиперидин-4-иламино)-5-(пиридин-4-ил)-1,3-тиазол-4-ил]фенил}-2,5-дифторбензолсульфонамид;
7) N-(3-{2-[циклогексил(метил)амино]-5-(пиридин-4-ил)-1,3-тиазол-4-ил}-2,4-дифторфенил)-2,5-дифторбензолсульфонамид;
8) N-{2,4-дифтор-3-[2-(4-метилпиперазин-1-ил)-5-(пиридин-4-ил)-1,3-тиазол-4-ил]фенил}-2,5-дифторбензолсульфонамид;
9) N-{2,4-дифтор-3-[2-(пиперидин-1-ил)-5-(пиридин-4-ил)-1,3-тиазол-4-ил]фенил}-2,5-дифторбензолсульфонамид;
10) N-(3-{2-[4-(диметиламино)пиперидин-1-ил]-5-(пиридин-4-ил)-1,3-тиазол-4-ил}-2,4-дифторфенил)-2,5-дифторбензолсульфонамид;
11) N-{3-[2-(1,4-диокса-8-азаспиро[4.5]дец-8-ил)-5-(пиридин-4-ил)-1,3-тиазол-4-ил]-2,4-дифторфенил}-2,5-дифторбензолсульфонамид;
12) N-{2,4-дифтор-3-[2-(4-оксопиперидин-1-ил)-5-(пиридин-4-ил)-1,3-тиазол-4-ил]фенил}-2,5-дифторбензолсульфонамид;
13) N-{2,4-дифтор-3-[2-(4-гидроксипиперидин-1-ил)-5-(пиридин-4-ил)-1,3-тиазол-4-ил]фенил}-2,5-дифторбензолсульфонамид;
14) N-{3-[2-(4,4-дифторпиперидин-1-ил)-5-(пиридин-4-ил)-1,3-тиазол-4-ил]-2,4-дифторфенил}-2,5-дифторбензолсульфонамид;
15) N-{2,4-дифтор-3-[2-(морфолин-4-ил)-5-(пиридин-4-ил)-1,3-тиазол-4-ил]фенил}-2,5-дифторбензолсульфонамид;
16) N-{3-[2-(диэтиламино)-5-(пиридин-4-ил)-1,3-тиазол-4-ил]-2-фторфенил}-2,5-дифторбензолсульфонамид;
17) N-{3-[2-(1,4-диокса-8-азаспиро[4.5]дец-8-ил)-5-(пиридин-4-ил)-1,3-тиазол-4-ил]-2-фторфенил}-2,5-дифторбензолсульфонамид;
18) N-{3-[2-(4,4-дифторпиперидин-1-ил)-5-(пиридин-4-ил)-1,3-тиазол-4-ил]-2-фторфенил}-2,5-дифторбензолсульфонамид;
19) 2,5-дифтор-N-{2-фтор-3-[2-(1-метилпиперидин-4-ил)-5-(пиридин-4-ил)-1,3-тиазол-4-ил]фенил}бензолсульфонамид;
20) 2,5-дифтор-N-{2-фтор-3-[5-(пиридин-4-ил)-2-(тетрагидро-2H-пиран-4-ил)-1,3-тиазол-4-ил]фенил}бензолсульфонамид;
21) N-{3-[2-(1-циклопропилпиперидин-4-ил)-5-(пиридин-4-ил)- 1,3-тиазол-4-ил]-2-фторфенил}-2,5-дифторбензолсульфонамид;
22) N-{3-[5-(2-аминопиридин-4-ил)-2-(1-циклопропилпиперидин-4-ил)-1,3-тиазол-4-ил]-2-фторфенил}-2,5-дифторбензолсульфонамид;
23) N-{4-[4-(3-{[(2,5-дифторфенил)сульфонил]амино}-2-фторфенил)-2-(1-метилпиперидин-4-ил)-1,3-тиазол-5-ил]пиридин-2-ил}ацетамид;
24) N-{3-[5-(2-аминопиридин-4-ил)-2-(тетрагидро-2H-пиран-4-ил)-1,3-тиазол-4-ил]-2-фторфенил}-2,5-дифторбензолсульфонамид;
25) 2,5-дифтор-N-(2-фтор-3-{5-[2-(метиламино)пиридин-4-ил]-2-(тетрагидро-2H-пиран-4-ил)-1,3-тиазол-4-ил}фенил)бензолсульфонамид;
26) 2,5-дифтор-N-{2-фтор-3-[2-(1-метилпиперидин-4-ил)-5-(2-метилпиридин-4-ил)-1,3-тиазол-4-ил]фенил}бензолсульфонамид;
27) 2,5-дифтор-N-{2-фтор-3-[5-(2-фторпиридин-4-ил)-2-(1-метилпиперидин-4-ил)-1,3-тиазол-4-ил]фенил}бензолсульфонамид;
28) 2,5-дифтор-N-{2-фтор-3-[5-(2-метилпиридин-4-ил)-2-(тетрагидро-2H-пиран-4-ил)-1,3-тиазол-4-ил]фенил}бензолсульфонамид;
29) N-{4-[4-(3-{[(2,5-дифторфенил)сульфонил]амино}-2-фторфенил)-2-(1-метилпиперидин-4-ил)-1,3-тиазол-5-ил]пиридин-2-ил}-2-метилпропанамид;
30) N-{4-[2-(1-циклопропилпиперидин-4-ил)-4-(3-{[(2,5-дифторфенил)сульфонил]амино}-2-фторфенил)-1,3-тиазол-5-ил]пиридин-2-ил}ацетамид;
31) N-{3-[2-(1-циклопропилпиперидин-4-ил)-5-(2-метилпиридин-4-ил)-1,3-тиазол-4-ил]-2-фторфенил}-2,5-дифторбензолсульфонамид;
32) N-{3-[2-(1-циклопропилпиперидин-4-ил)-5-(2-фторпиридин-4-ил)-1,3-тиазол-4-ил]-2-фторфенил}-2,5-дифторбензолсульфонамид;
33) N-[4-(3-{[(2,5-дифторфенил)сульфонил]амино}-2,6-дифторфенил)-5-(пиридин-4-ил)-1,3-тиазол-2-ил]ацетамид;
34) 2,5-дифтор-N-{2-фтор-3-[5-(3-фторпиридин-4-ил)-2-(тетрагидро-2H-пиран-4-ил)-1,3-тиазол-4-ил]фенил}бензолсульфонамид;
35) N-{4-[4-(3-{[(2,5-дифторфенил)сульфонил]амино}-2-фторфенил)-2-(тетрагидро-2H-пиран-4-ил)-1,3-тиазол-5-ил]пиридин-2-ил}ацетамид;
36) N-[2-({4-[4-(3-{[(2,5-дифторфенил)сульфонил]амино}-2-фторфенил)-2-(тетрагидро-2H-пиран-4-ил)-1,3-тиазол-5-ил]пиридин-2-ил}амино)этил]ацетамид;
37) N-(3-{2-(1-циклопропилпиперидин-4-ил)-5-[2-(метиламино)пиридин-4-ил]-1,3-тиазол-4-ил}-2-фторфенил)-2,5-дифторбензолсульфонамид;
38) 2,5-дифтор-N-(2-фтор-3-{5-[2-(метиламино)пиридин-4-ил]-2-(1-метилпиперидин-4-ил)-1,3-тиазол-4-ил}фенил)бензолсульфонамид;
39) N-{3-[5-(2-аминопиридин-4-ил)-2-(1-метилпиперидин-4-ил)-1,3-тиазол-4-ил]-2-фторфенил}-2,5-дифторбензолсульфонамид;
40) N-{3-[5-(2-{[2-(диметиламино)этил]амино}пиридин-4-ил)-2-(тетрагидро-2H-пиран-4-ил)-1,3-тиазол-4-ил]-2-фторфенил}-2,5-дифторбензолсульфонамид;
41) метил [(2S)-1-({4-[4-(3-{[(2,5-дифторфенил)сульфонил]амино}-2-фторфенил)-2-(тетрагидро-2H-пиран-4-ил)-1,3-тиазол-5-ил]пиридин-2-ил}амино)пропан-2-ил]карбамат;
42) N-{4-[2-трет-бутил-4-(3-{[(2,5-дифторфенил)сульфонил]амино}-2-фторфенил)-1,3-тиазол-5-ил]пиридин-2-ил}ацетамид;
43) N-{3-[2-трет-бутил-5-(пиридин-4-ил)-1,3-тиазол-4-ил]-2-фторфенил}-2,5-дифторбензолсульфонамид;
44) N-{4-[4-(3-{[(2,5-дифторфенил)сульфонил]амино}-2-фторфенил)-2-(пиперидин-4-ил)-1,3-тиазол-5-ил]пиридин-2-ил}ацетамид;
45) метил [(2S)-1-({4-[4-(3-{[(2,5-дифторфенил)сульфонил]амино}-2-фторфенил)-2-(1-метилпиперидин-4-ил)-1,3-тиазол-5-ил]пиридин-2-ил}амино)пропан-2-ил]карбамат;
46) 2,5-дифтор-N-{2-фтор-3-[5-(3-фторпиридин-4-ил)-2-(1-метилпиперидин-4-ил)-1,3-тиазол-4-ил]фенил}бензолсульфонамид;
47) N-{4-[4-(3-{[(2,5-дифторфенил)сульфонил]амино}-2-фторфенил)-2-(1-этилпиперидин-4-ил)-1,3-тиазол-5-ил]пиридин-2-ил}ацетамид;
48) метил [(2S)-1-({4-[2-(1-циклопропилпиперидин-4-ил)-4-(3-{[(2,5-дифторфенил)сульфонил]амино}-2-фторфенил)-1,3-тиазол-5-ил]пиридин-2-ил}амино)пропан-2-ил]карбамат;
49) N-{3-[2-(1-циклопропилпиперидин-4-ил)-5-(3-фторпиридин-4-ил)-1,3-тиазол-4-ил]-2-фторфенил}-2,5-дифторбензолсульфонамид;
50) N-{3-[5-(2-аминопиридин-4-ил)-2-трет-бутил-1,3-тиазол-4-ил]-2-фторфенил}-2,5-дифторбензолсульфонамид;
51) N-(3-{2-трет-бутил-5-[2-(метиламино)пиридин-4-ил]-1,3-тиазол-4-ил}-2-фторфенил)-2,5-дифторбензолсульфонамид и
52) метил [(2S)-1-({4-[2-трет-бутил-4-(3-{[(2,5-дифторфенил)сульфонил]амино}-2-фторфенил)-1,3-тиазол-5-ил]пиридин-2-ил}амино)пропан-2-ил]карбамат.
Также настоящее изобретение относится к способу получения соединения формулы (I), как определено выше, с использованием путей реакций и синтетических схем, описанных ниже, с использованием способов, доступных в данной области, и легко доступных исходных материалов. Получение определенных вариантов осуществления настоящего изобретения описано в примерах, которые следуют далее, однако специалисту в данной области будет понятно, что описанные способы получения можно легко адаптировать для получения других вариантов осуществления настоящего изобретения. Например, синтез не проиллюстрированных соединений согласно изобретению можно проводить путем модификаций, понятных специалистам в данной области, например, путем защиты надлежащим образом препятствующих синтезу групп, путем замены на другие подходящие реагенты, известные в данной области, или путем общепринятых модификаций условий реакции. Альтернативно другие реакции, указанные в настоящем описании или известные в данной области, могут быть признаны как адаптируемые для получения других соединений по изобретению.
Соединение формулы (I) можно получать в соответствии с общими процессами синтеза, описанными далее в способах A, B, C, D, E и F.
Всем специалистам в данной области будет понятно, что любое преобразование, проводимое в соответствии с указанными способами, может требовать стандартных модификаций, например, таких как защита препятствующих синтезу групп, замена на другие реагенты, известные в данной области, или проведение обычных модификаций условий реакции.
В общем процессе синтеза соединение формулы (I)A, (I)B, (I)U1 или (I)V1 получают способом A, представленным ниже.
Способ A
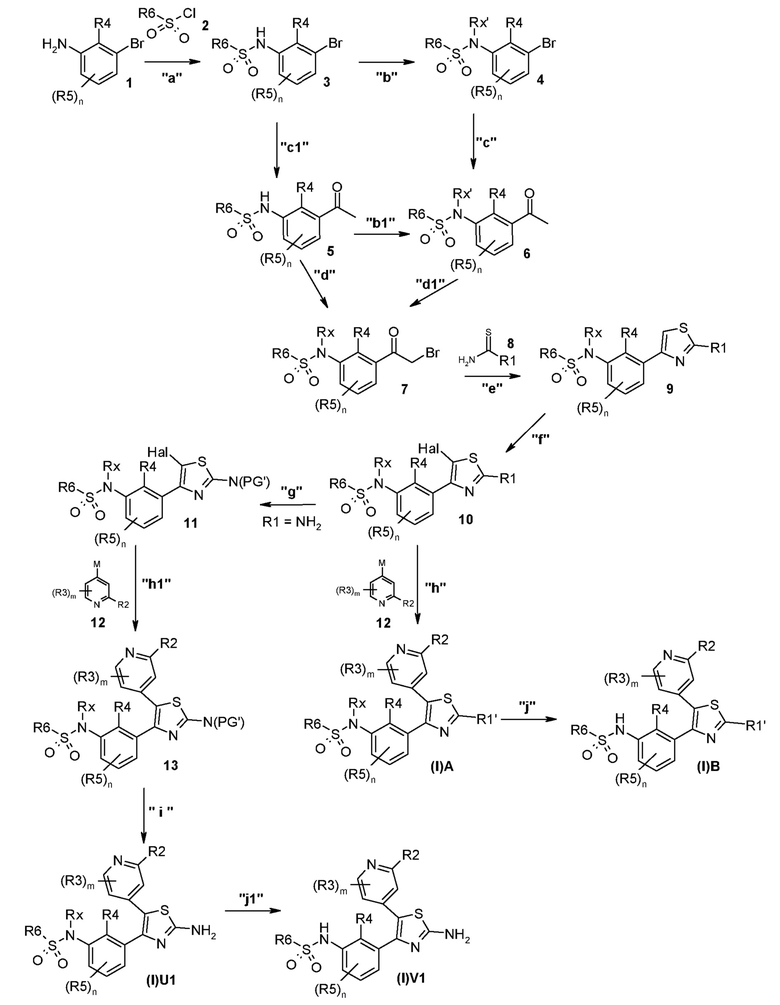
В описанной выше схеме m, n, R2, R3, R4, R5, R6, R7, R8 и Rx являются такими, как определено выше, Rx’ является таким же, как и Rx, за исключением водорода, R1’ является таким же, как и R1, за исключением NH2, PG’ представляет собой защитную группу, такую как ацетильная, бензоильная или диметиламиноиминогруппа, Hal представляет собой галоген, такой как иод или бром, и M представляет собой Li, B(OH)2, B(OAlk)2, Sn(Alk)3, Al(Alk)2, ZnHal, MgHal или ZrCp2Hal, где Alk обозначает алкильную группу и Cp обозначает циклопентадиенид.
В общем способе синтеза для получения соединения формулы (I)A, (I)B, (I)A1 и (I)B1, который описан в способе A, на стадии "a" соединение формулы 1 подвергают реакции с соединением формулы 2 с получением сульфонамида формулы 3. На стадии "b" необязательно в сульфонамидогруппу вводят группу с получением соединения формулы 4. На стадии "c" получают метилкетон формулы 6 в двухстадийном процессе, вовлекающем реакцию по типу Хека с последующим кислотным гидролизом. На стадии "c1" получают метилкетон формулы 5 из соединения формулы 3 в двухстадийном процессе, вовлекающем реакцию типа Хека с последующим кислотным гидролизом. На стадии "b1" в сульфонамидогуппу соединения формулы 5 необязательно вводят группу с получением соединения формулы 6. На стадии "d" и "d1" кетон формулы 5 или 6, соответственно, преобразуют в соответствующий α-бромкетон формулы 7, с помощью подходящего способа бромирования. На стадии "e" проводят получение тиазольной системы путем конденсации производного тиомочевины или тиоамида формулы 8 с получением соединения формулы 9. На стадии "f" тиазольное кольцо галогенируют с получением соединения формулы 10. На стадии "g" соединение формулы 10, где R1 представляет собой NH2, защищают по аминогруппе с получением соединения формулы 11. На стадии "h" соединение формулы 10, где R1 отличается от NH2, подвергают реакции перекрестного сочетания, подходящей для образований углерод-углеродных связей, с получением соединения формулы (I)A; на стадии "h1" соединение формулы 11 подвергают реакции перекрестного сочетания, указанной выше, с получением соединения формулы 13. Указанные реакции, которые хорошо известны в данной области, подразумевают сочетание с подходящим металлоорганическим реагентом общей формулы 12, например, таким как борорганическое соединение, оловоорганическое соединение, цинкорганическое соединение, алюминийорганическое соединение или цирконийорганическое соединение и т.п. На стадии "i" соединение формулы 13 подвергают реакции с подходящим гидролизующим агентом с получением соединения формулы (I)U1. На стадии "j" соединение формулы (I)A, где Rx является таким, как определено выше, за исключением водорода, преобразуют в соответствующее соединение формулы (I)B. На стадии "j1" соединение формулы (I)U1, где Rx является таким, как определено выше, за исключением водорода, преобразуют в соответствующее соединение формулы (I)V1.
Согласно стадии "a" способа A, соединение формулы 1 подвергают реакции с сульфонилхлоридом формулы 2 в присутствии подходящего основания, например, такого как пиридин, N-метилморфолин, диизопропилэтиламин (DIPEA), триэтиламин (TEA), в подходящем растворителе, таком как пиридин, дихлорметан (DCM) или THF, при температуре в диапазоне от приблизительно 0°C до температуры кипения с обратным холодильником и в течение периода времени, варьирующего приблизительно от 1 до 8 ч.
Согласно стадии "b" способа A, соединение формулы 3 необязательно подвергают реакции с подходящим реагентом, таким как трет-бутоксикарбонилангидрид, п-метоксибензилхлорид, алкоксиалкилхлорид, такой как метоксиметилхлорид, ацилхлорид, такой как ацетилхлорид, алкоксикарбонилхлорид, такой как этилхлорформиат и т.п., необязательно в присутствии подходящего основания, такого как диметиламинопиридин, TEA, DIPEA, в подходящем растворителе, таком как DCM, ацетонитрил, пиридин или THF, при температуре в диапазоне от 0°C до к.т. и в течение периода времени, варьирующего от 1 до приблизительно 6 ч.
Согласно стадии "b1" способа A, необязательное преобразование соединения формулы 5 в соединение формулы 6 проводят, как описано для стадии "b" способа A.
Согласно стадии "c" способа A, соединение формулы 4 подвергают реакции с винил-н-алкилом, предпочтительно винил-н-бутиловым простым эфиром, в присутствии основания, такого как TEA, фосфинового лиганда, такого как 1,3-бис(дифенилфосфино)пропан (DPPP), и в присутствии подходящего катализатора, такого как ацетат палладия, в подходящем растворителе, таком как этиленгликоль, при температуре в диапазоне от 80 до 150°C в закрытой бутылке в атмосфере азота и в течение периода времени в диапазоне от 1 до приблизительно 12 ч. Промежуточное соединение, полученное таким образом, гидролизуют в кислых условиях, например, водным раствором хлористоводородной кислоты (HCl), в подходящем растворителе, таком как диоксан или THF при комнатной температуре и в течение периода времени в диапазоне от 1 до приблизительно 6 ч с получением соединения формулы 6.
Согласно стадии "c1" способа A, преобразование соединения формулы 3 в соединение формулы 5 проводят, как описано для стадии "c" способа A.
Согласно стадии "d" способа A, соединение формулы 5 подвергают реакции с подходящим бромирующим агентом, таким как пиридиния бромид пербромид или тетрабутиламмония пербромид, в подходящем растворителе, таком как THF или DCM, при температуре в диапазоне от 60 до 100°C в микроволновом устройстве или в классических температурных условиях и в течение периода времени в диапазоне от 15 мин до 3 ч. Альтернативно бромирование соединения формулы 5 проводят в двухстадийном процессе, вовлекающем сначала реакцию соединения 5 с триметилсилилтрифторметансульфонатом в присутствии основания, такого как TEA или DIPEA, в подходящем растворителе, таком как DCM или THF, при температуре в диапазоне от -10 до 0°C и в течение периода времени в диапазоне от 10 до 30 мин. Затем полученный триметилсилиленоловый простой эфир обрабатывают N-бромсукцинимидом в подходящем растворителе, таком как DCM или THF, при температуре в диапазоне от -10 до 0°C и в течение периода времени в диапазоне от 30 мин до 1 ч.
Согласно стадии "d1" способа A, преобразование соединения формулы 6 в соединение формулы 7 проводят, как описано для стадии "d" способа A.
Согласно стадии "e" способа A, соединение формулы 7 подвергают реакции с производным тиомочевины или тиоамида формулы 8, в подходящем растворителе, таком как этанол или метанол (MeOH), при температуре в диапазоне от 60°C до температуры кипения с обратным холодильником, в микроволновом устройстве или в классических температурных условиях и в течение периода времени в диапазоне от 15 мин до 3 ч.
Согласно стадии "f" способа A, соединение формулы 9 подвергают реакции с подходящим бромирующим агентом, таким как N-бромсукцинимид, в подходящем растворителе, таком как DCM или THF, или Br2, в подходящем растворителе, таком как уксусная кислота, необязательно в присутствии KOAc, при комнатной температуре и в течение периода времени в диапазоне от 1 до приблизительно 6 ч.
Согласно стадии "g" способа A, соединение формулы 10, где R1 представляет собой NH2, подвергают реакции с подходящим защитным агентом. Предпочтительной является реакция с диметилформамиддиметилацеталем в подходящем растворителе, таком как диметилформамид (DMF), при комнатной температуре и в течение периода времени в диапазоне от 1 до 16 ч.
Согласно стадии "h" способа A, соединение формулы 10, где R1 отличается от NH2, подвергают реакции перекрестного сочетания с подходящим металлоорганическим соединением общей формулы 12, например, таким как борорганическое соединение (реакция Сузуки), оловоорганическое соединение (реакция Стилла), цинкорганическое, алюминийорганическое или цирконийорганическое соединение (реакция Негиши) и т.п. с получением соединения формулы (I)A. Указанные реакции хорошо известны специалистам в данной области. Предпочтительной реакцией является реакция Сузуки, где используют производное бороната в присутствии катализатора на основе палладия, например, такого как комплекс дихлорид дифенилфосфиноферроцен палладия с DCM, и подходящего основания, такого как Cs2CO3, Na2CO3, K2CO3, Rb2CO3, NaOH, CsF и т.п. Указанные реакции можно проводить в растворителе, таком как DMF, диметилсульфоксид, вода, диметоксиэтан (DME), 1,4-диоксан, THF и т.п. и их смеси, при температуре в диапазоне от 100 до 120°C, в микроволновом устройстве или в классических температурных условиях, в течение периода времени в диапазоне от 1 до 16 ч.
Согласно стадии "h1" способа A, преобразование соединения формулы 11 в соединение формулы 13 проводят, как описано для стадии "h" способа A.
Согласно стадии "i" способа A, соединение формулы 13 подвергают реакции с подходящим гидролизующим агентом, в зависимости от природы защитной группы PG’, с получением соединения формулы (I)U1. Например, когда такая защитная группа представляет собой диметиламиноиминогруппу, удаления защитной группы достигают с использованием 7н раствора аммиака в MeOH, этилендиамина в этаноле, гидрата лития или гидрата натрия в смесях вода/THF или MeOH или этанол, при температуре в диапазоне от комнатной температуры до температуры кипения с обратным холодильником и в течение периода времени в диапазоне от 8 до 96 ч.
Согласно стадии "j" способа A, соединение формулы (I)A, где Rx представляет собой Rx’, преобразуют в соединение формулы (I)B с использованием условий, зависящих от природы такой группы Rx’. Например, когда такая группа представляет собой метоксиметильную, бензильную, трет-бутоксикарбонильную группу и т.п., удаление можно проводить с использованием сильных кислот, таких как трифторуксусная кислота (TFA) или HCl. Указанную реакцию можно проводить необязательно в присутствии подходящего сорастворителя, такого как вода, THF или 1,4-диоксан, при температуре в диапазоне от комнатной температуры до 90°C и в течение периода времени в диапазоне от 1 ч до приблизительно 8 ч. Когда такая группа представляет собой, например, ацетильную, этоксикарбонильную группу и т.п., удаление можно проводить с использованием основания, такого как TEA или DIPEA, в MeOH или этаноле, или с использованием водного раствора неорганического основания, такого как карбонат натрия или калия, гидроксид натрия или калия и т.п. Указанные реакции можно проводить при температурах в диапазоне от 0°C до температуры кипения с обратным холодильником и в течение периода времени в диапазоне от 30 мин до приблизительно 48 ч.
Согласно стадии "j1" способа A, соединение формулы (I)U1 преобразуют в соединение формулы (I)V1, как описано для стадии "j" выше.
Соединение формулы (I), полученное согласно способу A, описанному выше, можно далее преобразовывать в другое соединение формулы (I) согласно методикам, хорошо известным специалистам в данной области.
Например, когда R1 представляет собой группу NH2 (соединение формулы (I)U1), указанное соединение можно далее преобразовывать в другое соединение формулы (I)C, (I)D, (I)E, (I)F, (I)G, (I)H, (I)I, (I)J, (I)K, (I)L, (I)M, (I)N, (I)O, (I)P, (I)U или (I)V в соответствии со способом B, представленным ниже.
Способ B
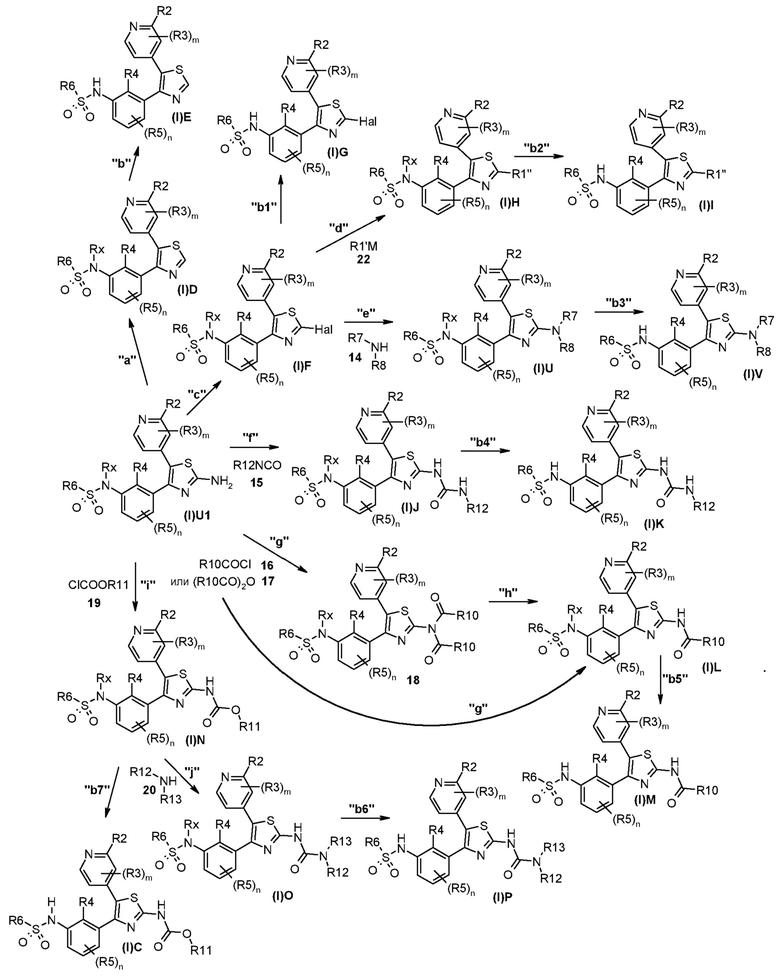
На представленной выше схеме m, n, R2, R3, R4, R5, R6, R7, R8, R10, R11, R12, R13, Rx и Hal являются такими, как описано выше, и R1’’ представляет собой необязательно замещенный (C2-C8)алкенил, (C3-C8)циклоалкенил, гетероциклил, арил или гетероарил.
В способе синтеза для получения соединений формулы (I)C, (I)D, (I)E, (I)F, (I)G, (I)H, (I)I, (I)J, (I)K, (I)L, (I)M, (I)N, (I)O, (I)P, (I)U или (I)V, который описан в способе B, на стадии "a" соединение формулы (I)U1, полученное, как описано для способа A, подвергают реакции по типу Сандмейера с последующим восстановлением промежуточной соли диазония с получением соединения общей формулы (I)D. На стадии "b" соединение формулы (I)D, где Rx является таким, как определено выше, за исключением водорода, преобразуют в соответствующее соединение формулы (I)E. На стадии "c" соединение формулы (I)U1 подвергают реакции по типу Сандмейера, а затем реакции с подходящим галогенирующим агентом с получением галогенпроизводного формулы (I)F. На стадии "b1" соединение формулы (I)F, где Rx является таким, как определено выше, за исключением водорода, преобразуют в соответствующее соединение формулы (I)G. На стадии "d" соединение формулы (I)F преобразуют в соединение формулы (I)H с использованием любой из реакций перекрестного сочетания, пригодных для образования углерод-углеродных связей. Указанные реакции, которые хорошо известны в данной области, предполагают реакцию сочетания с подходящим металлоорганическим реагентом формулы 22, например, таким как борорганическое соединение, оловоорганическое соединение, цинкорганическое соединение, алюминийорганическое соединение или цирконийорганическое соединение и т.п. На стадии "b2" соединение формулы (I)H, где Rx является таким, как определено выше, за исключением водорода, преобразуют в соответствующее соединение формулы (I)I. На стадии "e" соединение формулы (I)F преобразуют в соединение формулы (I)U путем реакции с амином формулы 14. На стадии "b3" соединение формулы (I)U, где Rx является таким, как определено выше, за исключением водорода, преобразуют в соответствующее соединение формулы (I)V. На стадии "f" соединение формулы (I)U1 конденсируют с изоцианатом формулы 15 с получением производного мочевины формулы (I)J. На стадии "b4" соединение формулы (I)J, где Rx является таким, как определено выше, за исключением водорода, преобразуют в соответствующее соединение формулы (I)K. На стадии "g" соединение формулы (I)U1 конденсируют с соединением формулы 16 или 17 с получением имидопроизводного формулы 18 или непосредственно соединения формулы (I)L. Последнее можно иным образом получать на стадии "h" из соединения формулы 18 путем селективного гидролиза одной из ацильных групп, который можно осуществлять в основных условиях. На стадии "b5" соединение формулы (I)L, где Rx является таким, как определено выше, за исключением водорода, преобразуют в соответствующее соединение формулы (I)M. На стадии "i" соединение формулы (I)U1 подвергают реакции с подходящим хлорформиатом формулы 19 с получением карбаматного производного формулы (I)N. На стадии "j" последнее преобразуют в производное мочевины формулы (I)O путем реакции с амином формулы 20. На стадии "b6" соединение формулы (I)O, где Rx является таким, как определено выше, за исключением водорода, преобразуют в соответствующее соединение формулы (I)P. На стадии "b7" соединение формулы (I)N, где Rx является таким, как определено выше, за исключением водорода, преобразуют в соответствующее соединение формулы (I)C.
Согласно стадии "a" способа B, синтез соединения формулы (I)D из соединения формулы (I)U1 проводят путем получения соли диазония, которое можно проводить с использованием нитрита натрия в воде или водных растворителях, в присутствии минеральной кислоты, такой как HCl, серная кислота и т.п., с последующим восстановлением указанной соли, которое можно проводить с использованием спирта, такого как этанол, или подходящего восстановителя, такого как гипофосфорная кислота. Альтернативно соль диазония можно получать с использованием изоамилнитрита в подходящем растворителе, таком как DCM, DME, THF и т.п. при температуре в диапазоне от 0°C до температуры кипения с обратным холодильником и в течение периода времени в диапазоне от 30 мин до приблизительно 24 ч.
Согласно стадии "b" способа B, преобразование соединения формулы (I)D в соответствующее соединение формулы (I)E проводят, как описано для стадии "j" способа A.
Согласно стадии "c" способа B, преобразование соединения формулы (I)U1 в соединение формулы (I)F проводят с использованием протокола Сандмейера. Это осуществляют путем получения соли диазония, которое можно проводить с использованием нитрита натрия в воде или водных растворителях, в присутствии минеральной кислоты, такой как HCl, серная кислота и т.п., с последующей обработкой солью галогенводородной кислоты. Предпочтительным является бромирование, и, соответственно, предпочтительной солью галогенводородной кислоты является CuBr. Альтернативно можно проводить иодирование, которое осуществляют с использованием KI, NaI, CsI, CuI, необязательно в присутствии иода. Альтернативно соль диазония можно получать с использованием изоамилнитрита или трет-бутилнитрита в подходящем растворителе, таком как ацетонитрил, DCM, DME, THF и т.п. при температуре в диапазоне от 0°C до температуры кипения с обратным холодильником и в течение периода времени в диапазоне от 30 мин до приблизительно 3 суток. Согласно стадии "b1" способа B, преобразование соединения формулы (I)F в соответствующее соединение формулы (I)G проводят, как описано для стадии "j" способа A.
Согласно стадии "d" способа B, соединение формулы (I)F подвергают реакции перекрестного сочетания с подходящим металлоорганическим соединением общей формулы 22, например, таким как борорганическое соединение (реакция Сузуки), оловоорганическое соединение (реакция Стилла), цинкорганическое соединение, алюминийорганическое соединение или цирконийорганическое соединение (реакция Негиши) и т.п. Указанные реакции хорошо известны среди специалистов в данной области. Предпочтительной реакцией является реакция Сузуки, где используют соответствующий арил- или гетероарилборонат в присутствии катализатора на основе палладия, например, такого как палладий-тетракис(трифенилфосфин)(Pd(PPh3)4), и подходящего основания, такого как Cs2CO3, Na2CO3, K2CO3, Rb2CO3, NaOH, CsF и т.п. Указанные реакции можно проводить в растворителе, таком как DMF, диметилсульфоксид, вода, DME, 1,4-диоксан, THF и т.п., и их смесь, при температуре в диапазоне от 20°C до температуры кипения с обратным холодильником и в течение периода времени в диапазоне от 30 мин до приблизительно 24 ч. Согласно стадии "b2" способа B, преобразование соединения формулы (I)H в соответствующее соединение формулы (I)I проводят, как описано для стадии "j" способа A.
Согласно стадии "e" способа B, реакцию соединения формулы (I)F с соединением формулы 14 можно проводить в подходящем растворителе, таком как диметилацетамид, DMF, ацетонитрил и т.п. необязательно в присутствии подходящего основания, такого как TEA или DIPEA. Реакцию можно проводить в микроволновом устройстве или в классических температурных условиях, при температуре в диапазоне от 80 до 120°C и в течение периода времени в диапазоне от 1 ч до 2 суток. Согласно стадии "b3" способа B, преобразование соединения формулы (I)U в соответствующее соединение формулы (I)V проводят, как описано для стадии "j" способа A.
Согласно стадии "f" способа B, реакцию соединения формулы (I)U1 с соединением формулы 15 можно проводить в подходящем растворителе, таком как 1,4-диоксан, THF, DME при температуре в диапазоне от комнатной температуры до температуры кипения с обратным холодильником и в течение периода времени в диапазоне от 5 ч до 2 суток. Согласно стадии "b4" способа B, преобразование соединения формулы (I)J в соответствующее соединение формулы (I)K проводят, как описано для стадии "j" способа A.
Согласно стадии "g" способа B, реакцию соединения формулы (I)U1 с соединением формулы 16 или 17 можно проводить в присутствии подходящего основания, такого как TEA или DIPEA или пиридин, в подходящем растворителе, таком как DCM, THF, 1,4-диоксан или ацетонитрил, при комнатной температуре и в течение периода времени в диапазоне от 1 до приблизительно 8 ч.
Согласно стадии "h" способа B, преобразование соединения формулы 18 в соединение формулы (I)L проводят путем селективного гидролиза с использованием основных условий гидролиза, таких как TEA в MeOH или разбавленный водный раствор NaOH или LiOH в подходящем растворителе, таком как этанол, MeOH или THF, при комнатной температуре и в течение периода времени в диапазоне от 1 до приблизительно 6 ч. Согласно стадии "b5" способа B, преобразование соединения формулы (I)L в соответствующее соединение формулы (I)M проводят, как описано для стадии "j" способа A.
Согласно стадии "i" способа B, преобразование соединения формулы (I)U1 в соединение формулы (I)N осуществляют путем реакции с хлорформиатом формулы 19 в подходящем растворителе, таком как THF, DMF, DCM, хлороформ, ацетонитрил, толуол или их смеси, при температуре в диапазоне от приблизительно -10°C до температуры кипения с обратным холодильником и в течение периода времени в диапазоне от приблизительно 30 мин до приблизительно 96 ч. Реакцию обычно проводят в присутствии подходящего акцептора протонов, такого как TEA, DIPEA или пиридин.
Согласно стадии "b7" способа B, преобразование соединения формулы (I)N в соответствующее соединение формулы (I)C проводят, как описано для стадии "j" способа A.
Согласно стадии "j" способа B, соединение формулы (I)O получают из соединения формулы (I)N путем реакции с соответствующим амином формулы 20. Указанную реакцию, как правило, поводят в подходящем растворителе, таком как диметилсульфоксид, THF, DMF, N,N-диметилацетамид, ацетонитрил, толуол или их смесь, необязательно в присутствии дополнительного основания, такого как TEA, DIPEA, диазабициклоундецен или металлоорганический реагент, такой как реагент Гриньяра или триметилалюминий, при температуре в диапазоне от приблизительно -10°C до температуры кипения с обратным холодильником и в течение периода времени в диапазоне от приблизительно 30 мин до приблизительно 96 ч. Согласно стадии "b6" способа B, преобразование соединения формулы (I)O в соответствующее соединение формулы (I)P проводят, как описано для стадии "j" способа A.
В другом общем способе синтеза, описанном в способе C, представленном ниже, описан синтез соединения формулы (I), где R1 представляет собой гетероциклильную группу; в частности, соединение формулы (I)H1 получают и далее преобразуют в соединение формулы (I)Q, (I)R, (I)R1 или (I)R2.
Способ C
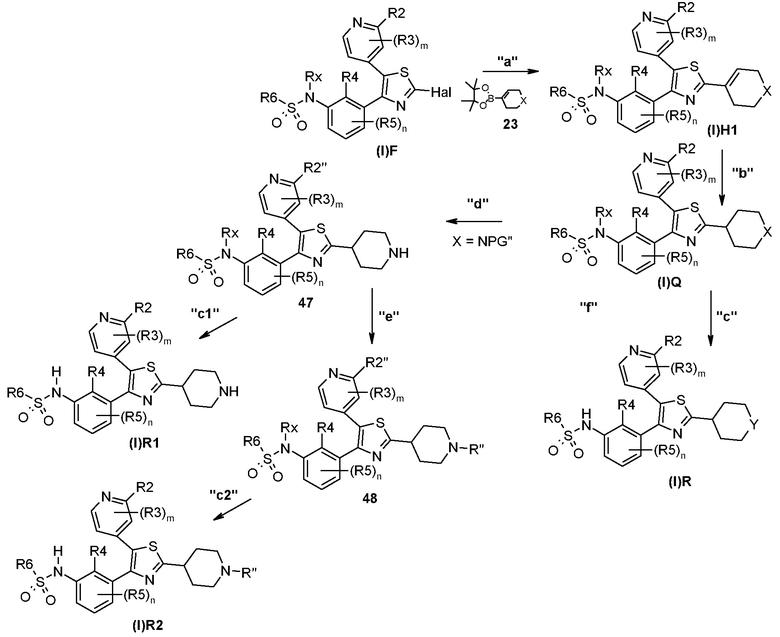
На представленной выше схеме m, n, R2, R3, R4, R5, R6, Rx и Hal являются такими, как определено выше; R2’’ является таким, как R2 или NR18COR22, где R18 и R22 являются такими, как определено выше.
X представляет собой S, O, NH или NR’, где R’ представляет собой прямой или разветвленный (C1-C8)алкил, (C3-C8)циклоалкил, гетероциклил, арил, гетероарил или группу PG", где PG" представляет собой подходящую защитную группу азота, например, такую как трет-бутоксикарбонил, бензил или бензилоксикарбонил;
R’’ представляет собой прямой или разветвленный (C1-C8)алкил или (C3-C8)циклоалкил;
Y представляет собой S, O, NH или группу NR’’’, где R’’’ представляет собой прямой или разветвленный (C1-C8)алкил, (C3-C8)циклоалкил, гетероциклил, арил или гетероарил.
В способе синтеза для получения соединения формулы (I)H1, (I)Q, (I)R, (I)R1 и (I)R2, которое описано в способе C, на стадии "a" соединение формулы (I)F подвергают реакции с соединением формулы 23 в соответствии с протоколом реакции Сузуки, с получением соединения формулы (I)H1. На стадии "b" соединение формулы (I)H1 подвергают каталитической гидрогенизации с получением соединения формулы (I)Q. На стадии "c", когда X представляет собой S, O, NH или NR’, получают соединение формулы (I)R. На стадии "d", когда X представляет собой группу NPG", получают соединение формулы 47 путем мягкого селективного удаления группы PG’’. На стадии "e" соединение формулы 47 подвергают восстановительному аминированию с получением соединения формулы 48. На стадии "c1" или "c2" получают соединение формулы (I)R1 или (I)R2, соответственно.
Согласно стадии "a" способа C, гетероциклоалкенильное производное формулы 23 подвергают реакции сочетания с соединением формулы (I)F согласно протоколу реакции Сузуки. Указанную реакцию можно проводить в присутствии подходящего основания, такого как карбонат цезия или карбонат или фосфат калия, и в присутствии подходящего катализатора, такого как комплекс дихлориддифенилфосфиноферроцена палладия с DCM, с использованием смесей DME/вода или 1,4-диоксан/вода, в качестве системы растворителей. Реакцию проводят в подходящем растворителе, таком как смеси DME/вода или 1,4-диоксан/вода, в микроволновом устройстве или в классических температурных условиях, при температуре в диапазоне от 90 до 120°C и в течение периода времени в диапазоне от 1 ч до приблизительно 16 ч.
Согласно стадии "b" способа C, восстановление соединения формулы (I)H1 с получением соединения формулы (I)Q проводят с использованием любых способов, хорошо известных в данной области, для восстановления углерод-углеродной двойной связи, например, с формиатом аммония в присутствии катализатора, такого как 10% палладий на угле или карбонат палладия на угле 10% в подходящем растворителе, таком как MeOH или этанол, при кипячении с обратным холодильником и в течение периода времени в диапазоне от 4 ч до приблизительно 16 ч. Альтернативно восстановление можно проводить путем гидрогенизации в устройстве Парра при давлении от 20 до 50 фунт./кв. дюйм (от 138 до 345 кПа) в присутствии катализатора, такого как 5 или 10% палладий на угле при комнатной температуре в течение периода времени в диапазоне от 2 до приблизительно 6 ч.
Согласно стадии "c" способа C, преобразование соединения формулы (I)Q в соответствующее соединение формулы (I)R проводят, как описано для стадии "j" способа A.
Согласно стадии "d" способа C, селективное удаление группы PG" из соединения формулы (I)Q с получением соединения формулы 47 можно проводить с использованием кислых или восстанавливающих условий. Например, реакцию проводят с использованием сильных кислот, таких как TFA, необязательно в присутствии подходящего сорастворителя, такого как DCM, при температурах в диапазоне от 20°C до температуры кипения с обратным холодильником и в течение периода времени в диапазоне от 30 мин до приблизительно 48 ч. Альтернативно, когда PG’’ представляет собой бензильную или бензилоксигруппу, указанную реакцию проводят с использованием восстанавливающих условий, таких как H2, в присутствии подходящего катализатора гидрогенизации. Катализатор гидрогенизации, как правило, представляет собой металл, наиболее часто палладий, который можно использовать в чистом виде или на подложке из угля, в подходящем растворителе, например, таком как THF, 1,4-диоксан, DMF, MeOH, этилацетат или их смесь.
Согласно стадии "c1" способа C, преобразование соединения формулы 47 в соответствующее соединение формулы (I)R1 проводят, как описано для стадии "j" способа A.
Согласно стадии "e" способа C, восстановительное аминирование соединения формулы 47 с получением соединения формулы 48 проводят путем реакции с подходящим карбонильным производном, таким как альдегид, кетон или их соответствующие ацетальные производные. Реакцию проводят в присутствии подходящего восстановителя, такого как цианоборгидрид натрия или триацетоксиборгидрид натрия, в подходящем растворителе, таком как MeOH или смесь уксусная кислота-MeOH, при температуре в диапазоне от комнатной температуры до температуры кипения с обратным холодильником и в течение периода времени в диапазоне от 1 до приблизительно 24 ч.
Согласно стадии "c2" способа C, преобразование соединения формулы 48 в соответствующее соединение формулы (I)R2 проводят, как описано для стадии "j" способа A.
В другом общем способе синтеза соединение общей формулы (I)S или (I)T получают согласно способу D, представленному ниже.
Способ D
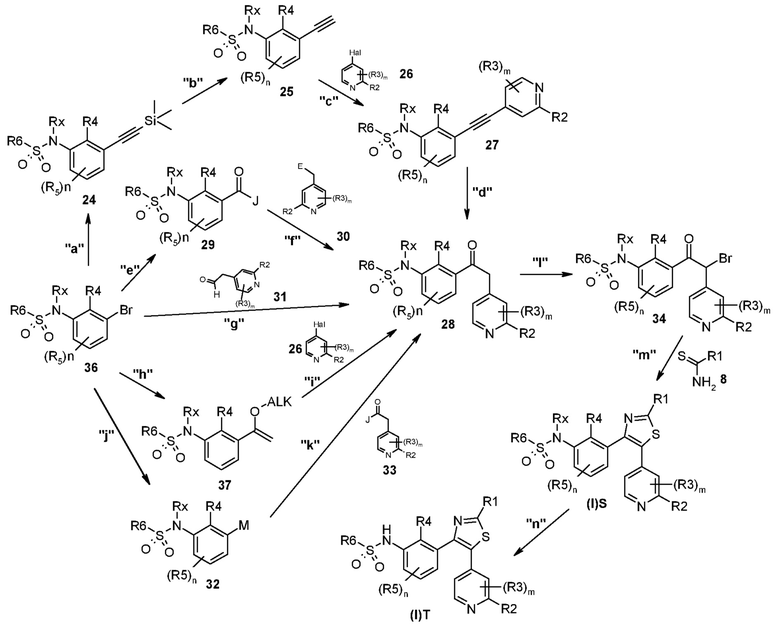
На представленной выше схеме m, n, R1, R2, R3, R4, R5, R6, Rx и Hal являются такими, как описано выше; J представляет собой галоген, такой как хлорид или бромид, O-алкильную группу или -N(CH3)O-алкильную группу, где алкил является таким, как определено выше; E представляет собой водород или алкоксикарбонильную группу;
В способе синтеза для получения соединения формулы (I)S и (I)T, который описан в способе D, на стадии "a" соединение формулы 36, которое, когда Rx представляет собой водород, соответствует соединению формулы 3, или, когда Rx представляет собой Rx’, соответствует соединению формулы 4, представленному на схеме A, подвергают реакции по типу Соногашира с триметилсилилацетиленом с образованием промежуточного соединения формулы 24. На стадии "b" десилилирование последнего дает другое промежуточное соединение формулы 25, которое, после второй реакции сочетания по типу Соногашира с соединением формулы 26 на стадии "c", дает производное ацетилена формулы 27. Гидратацию последнего проводят на стадии "d" с образованием ключевого кетонового промежуточного соединения формулы 28. Альтернативно на стадии "e" соединение формулы 36 преобразуют в соединение формулы 29 путем метилирования, например, с хлоридом изопропилмагния или бутиллитием, и гашения подходящим реагентом формулы Hal-CO-J. Альтернативно гашение с помощью CO2 с последующей активацией карбоновой кислоты или катализируемым палладием карбоксилированием может давать то же соединение формулы 29. На стадии "f" последнее подвергают реакции с соединением формулы 30, которое сначала обычно преобразуют в соответствующий анион металла с получением ключевого промежуточного соединения формулы 28. Альтернативно на стадии "g" соединение формулы 36 прямо преобразуют в соединение формулы 28 путем катализируемой палладием реакции перекрестного сочетания с подходящим альдегидом формулы 31. Альтернативно на стадии "h" соединение формулы 36 преобразуют в соединение формулы 37, которое на стадии "i" подвергают реакции по типу Хека с подходящим соединением формулы 26 с получением ключевого промежуточного соединения 28. Альтернативно на стадии "j" соединение формулы 36 преобразуют в металлоорганическое производное формулы 32, например, такое как литийорганическое соединение, борорганическое или магнийорганическое соединение, которое, в свою очередь, на стадии "k" подвергают реакции с подходящим электрофилом формулы 33 с получением соединения формулы 28. Такое преобразование можно проводить согласно множеству способов, в зависимости от природы M и J, как понятно любому специалисту в данной области. На стадии "l" галогенирование промежуточного соединения 28 дает соединение формулы 34, которое на стадии "m" подвергают реакции с подходящим производным тиомочевины или тиоамида формулы 8 с получением соединения тиазола формулы (I)S. На стадии "n" последнее преобразуют в соответствующее соединение формулы (I)T.
Согласно стадии "a" способа D, соединение формулы 36 подвергают реакции с триметилсилилацетиленом в присутствии подходящего палладиевого катализатора, такого как PdCl2(PPh3)2, Pd(PPh3)4 и т.п., и подходящего медного катализатора, такого как CuI. Указанную реакцию проводят в присутствии подходящего основания, такого как TEA, диэтиламин, диизопропиламин и т.п., необязательно в присутствии фосфинового лиганда, такого как трифенилфосфин. Реакцию обычно проводят при температурах в диапзаоне от -20°C до температуры кипения с обратным холодильником и в течение периода времени в диапазоне от 30 мин до приблизительно 48 ч. Согласно стадии "b" способа D, триметилсилильную группу удаляют с использованием основания, такого как KOH, NaOH, K2CO3, в растворителе, таком как MeOH, этанол и т.п., или с использованием подходящей фторидной соли, такой как KF, n-Bu4NF, в растворителях, таких как THF, DME, DMF и т.п. Согласно стадии "c" способа D, соединение формулы 25 преобразуют в соединение формулы 27 путем реакции с подходящим ароматическим галогенидом формулы 26 в соответствии с условиями, описанными для стадии "a" способа D. Согласно стадии "d" способа D, гидратацию алкина формулы 27 с получением соединения формулы 28 проводят с использованием, например, уксусной кислоты, TFA, трифторметансульфоновой кислоты, серной кислоты, Hg(OTf)2, HgSO4, NaHSO3 и т.п. в подходящем водном растворителе, таком как ацетонитрил, 1,4-диоксан, ацетон, этанол и т.п. при температуре в диапазоне от 0°C до температуры кипения с обратным холодильником и в течение периода времени в диапазоне от 1 ч до 72 ч.
Согласно стадии "e" способа D, преобразование соединения формулы 36 в соединение формулы 29 можно проводить рядом путей. Один такой путь вовлекает замещение металлом с помощью подходящего металлоорганического соединения, например, такого как бутиллитий или хлорид изопропилмагния, с последующим гашением реагентом формулы Hal-CO-J. Такие реакции обычно проводят в инертной атмосфере с использованием подходящего растворителя, например, такого как THF, 1,4-диоксан, DME или сходные с ними, при температуре в диапазоне от -80°C до комнатной температуры. Альтернативно гашение можно проводить с помощью сухого льда и полученную карбоновую кислоту можно активировать с получением соединения формулы 29 согласно способам, хорошо известным в данной области. Согласно стадии "f" способа D, соединение формулы 30 подвергают реакции с сильным основанием, таким как натрия гексаметилдисилазан (NaHMDS), лития гексаметилдисилазан (LiHMDS), лития диизопропиламид (LDA), реагент Гриньяра и т.п., с последующей конденсацией с соединением формулы 29. Указанную реакцию, как правило, проводят с использованием различных растворителей, таких как толуол, THF, 1,4-диоксан, DME и т.п., при температуре в диапазоне от 0°C до температуры кипения с обратным холодильником и в течение периода времени в диапазоне от 30 мин до приблизительно 24 ч.
Согласно стадии "g" способа D, преобразование соединения формулы 36 в соединении формулы 28 можно проводить путем реакции с подходящим альдегидом формулы 31. Такую реакцию обычно проводят в присутствии подходящего катализатора, такого как Pd(dba)2 или Pd(OAc)2, и основания, например, такого как пирролидин, необязательно в присутствии дополнительного лиганда, такого как DPPP, и молекулярных сит. Указанную реакцию обычно проводят в растворителях, например, таких как DMF, при температуре в диапазоне от 80°C до температуры кипения с обратным холодильником, в течение периода времени от 1 ч до 10 ч.
Согласно стадии "h" способа D, преобразование соединения формулы 36 в соединение формулы 37 можно проводить путем реакции с винил-н-алкиловым простым эфиром, предпочтительно винил-н-бутиловым простым эфиром, в присутствии основания, такого как TEA, фосфинового лиганда, такого как DPPP, и в присутствии подходящего катализатора, такого как ацетат палладия, в подходящем растворителе, таком как этиленгликоль, при температуре в диапазоне от 80 до 120°C в закрытой бутылке в атмосфере азота и в течение периода времени в диапазоне от 1 до приблизительно 6 ч.
Согласно стадии "i" способа D, преобразование соединения формулы 37 в соединение формулы 28 можно проводить с использованием протокола Мизороки-Хека, где олефиновое производное 37 подвергают реакции с арилгалогенидом формулы 26 в присутствии катализатора на основе палладия, например, такого как Pd(PPh3)4, и подходящего основания, такого как TEA и т.п. Такую реакцию обычно проводят в растворителях, таких как DMF, THF, 1,4-диоксан, DME и т.п., при температуре в диапазоне от 0°C до температуры кипения с обратным холодильником и в течение периода времени в диапазоне от 30 мин до приблизительно 24 ч.
Согласно стадии "j" способа D, соединение формулы 36 преобразуют в металлоорганическое производное формулы 32, такое как литийорганическое соединение, борорганическое соединение, магнийорганическое соединение и т.п. Такое соединение можно получать различными способами, в зависимости от природы самого металлоорганического соединения. Например, литийорганические соединения можно получать путем реакции соединения формулы 36 с бутиллитием или трет-бутиллитием. Борорганические соединения можно получать путем реакции соединения формулы 36 с подходящим соединением бора, таким как бис(пинаколято)дибор, пинаколборан и т.п., в присутствии подходящего палладиевого катализатора, такого как ацетат палладия, PdCl2(dppf) [(1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий(II)], и подходящего основания, такого как KOAc, TEA и т.п., в растворителях, таких как DMF, диметилсульфоксид, DME, 1,4-диоксан, THF и т.п., при температуре в диапазоне от 20°C до температуры кипения с обратным холодильником и в течение периода времени в диапазоне от 30 мин до приблизительно 24 ч. Магнийорганические соединения можно получать путем реакции соединения формулы 36 с подходящим производным магния, таким как хлорид изопропилмагния, или металлическим магнием в растворителях, таких как DME, 1,4-диоксан, THF и т.п., при температуре в диапазоне от -78°C до температуры кипения с обратным холодильником и в течение периода времени в диапазоне от 30 мин до приблизительно 24 ч.
Согласно стадии "k" способа D, соединение формулы 32 преобразуют в соединение формулы 28 путем реакции сочетания с подходящим реагентом формулы 33. Условия для такого сочетания зависят от природы металлоорганического соединения 32 и реагента формулы 33. Например, когда соединение формулы 32 представляет собой борорганическое соединение, реакцию сочетания можно осуществлять с соединением формулы 33. Такую реакцию обычно проводят в присутствии катализатора на основе палладия, например, такого как PdCl2 или Pd(OAc)2, и подходящего основания, такого как K3PO4, Cs2CO3, K2CO3, Rb2CO3, NaOH, CsF и т.п., в растворителе, таком как DME, 1,4-диоксан, THF и т.п., при температуре в диапазоне от 20°C до температуры кипения с обратным холодильником и в течение периода времени в диапазоне от 30 мин до приблизительно 24 ч. Когда соединение 32 представляет собой магнийорганическое соединение, реакцию сочетания можно удобным образом осуществлять со сложным эфиром или амидом Вейнреба формулы 33. Такие реакции обычно проводят в растворителе, таком как DME, 1,4-диоксан, THF и т.п., при температуре в диапазоне от -20°C до температуры кипения с обратным холодильником и в течение периода времени в диапазоне от 30 мин до приблизительно 24 ч.
Согласно стадии "l" способа D, соединение формулы 28 подвергают реакции с подходящим бромирующим агентом, таким как Br2, N-бромсукцинимид, пиридиния бромид пербромид или тетрабутиламмония пербромид, в подходящем растворителе, таком как THF или DCM, при температуре в диапазоне от 60 до 100°C в микроволновом устройстве или в классических температурных условиях и в течение периода времени в диапазоне от 15 мин до 3 ч. Альтернативно бромирование соединения формулы 28 осуществляют в двухстадийном процессе, вовлекающем сначала реакцию соединения формулы 28 с триметилсилилтрифторметансульфонатом в присутствии основания, такого как TEA или DIPEA, в подходящем растворителе, таком как DCM или THF, при температуре в диапазоне от -10 до 0°C и в течение периода времени в диапазоне от 10 до 30 мин. Затем полученный триметилсилиленоловый простой эфир обрабатывают N-бромсукцинимидом в подходящем растворителе, таком как DCM или THF, при температуре в диапазоне от -10 до 0°C и в течение периода времени в диапазоне от 30 мин до 1 ч.
Согласно стадии "m" способа D, соединение формулы 34 подвергают реакции с соединением формулы 8 в подходящем растворителе, таком как этанол или MeOH, при температуре в диапазоне от 60°C до температуры кипения с обратным холодильником, в микроволновом устройстве или в классических температурных условиях, в течение периода времени в диапазоне от 15 мин до 3 ч. Согласно стадии "n" способа D, преобразование соединения формулы (I)S в соответствующее соединение формулы (I)T проводят, как описано для стадии "j" способа A.
В другом общем способе синтеза соединение общей формулы (I)T получают согласно способу E, представленному ниже.
Способ E
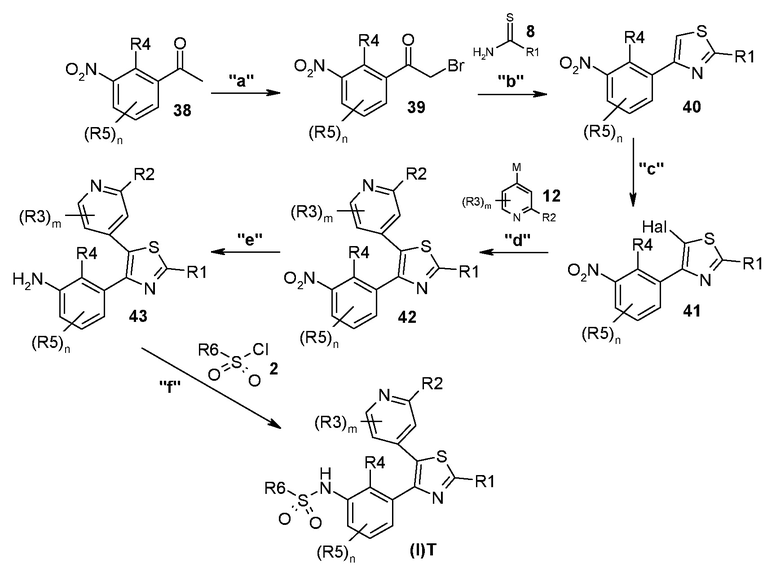
На представленной выше схеме m, n, R1, R2, R3, R4, R5, R6, M и Hal являются такими, как описано выше.
В способе синтеза для получения соединения формулы (I)T, который описан для способа E, на стадии "a" соединение формулы 38 преобразуют в соответствующий α-бромкетон формулы 39, с помощью подходящего способа бромирования. На стадии "b" проводят получение тиазольной системы путем конденсации с тиомочевиной или тиоамидным производным формулы 8 с получением соединения формулы 40. На стадии "c" тиазольное кольцо галогенируют с получением соединения формулы 41. На стадии "d" соединение формулы 41 подвергают реакции перекрестного сочетания, подходящей для образования углерод-углеродных связей, с получением соединения формулы 42. Указанные реакции, которые хорошо известны в данной области, предполагают реакцию сочетания с подходящим металлоорганическим реагентом общей формулы 12, например, таким как борорганическое, оловоорганическое, цинкорганическое соединение, алюминийорганическое соединение или цирконийорганическое соединение и т.п. На стадии "e" соединение формулы 42 обрабатывают подходящим восстановителем с получением аминопроизводного формулы 43, которое на стадии "f" подвергают реакции с соединением формулы 2 с получением соединения формулы (I)T.
Согласно стадии "a" способа E, синтез соединения общей формулы 39 из соединения общей формулы 38 проводят, как описано для стадии "d" способа A.
Согласно стадии "b" способа E, синтез тиазольного производного общей формулы 40 из соединения общей формулы 39 проводят, как описано для стадии "e" способа A.
Согласно стадии "c" способа E, галогенирование тиазольного производного общей формулы 40 с получением соединения общей формулы 41 проводят, как описано для стадии "f" способа A.
Согласно стадии "d" способа E, проводят реакцию перекрестного сочетания между производным общей формулы 41 и металлоорганическим соединением общей формулы 12 с получением соединения общей формулы 42, как описано для стадии "h" способа A.
Согласно стадии "e" способа E, нитрогруппу соединения формулы 42 восстанавливают в аминогруппу с получением соединения формулы 43. Реакцию можно проводить различными способами и в действующих условиях, которые широко известны в данной области, для восстановления нитрогруппы в аминогруппу. Предпочтительно, эту реакцию проводят в подходящем растворителе, например, таком как вода, MeOH, THF, 1,4-диоксан, DMF, этилацетат или их смесь, в присутствии подходящего восстановителя, например, такого как водород, и катализатора гидрогенизации, или путем обработки циклогексеном или циклогексадиеном и катализатором гидрогенизации, или путем обработки хлоридом олова(II), или путем обработки цинком или хлоридом цинка(II) и водным раствором HCl или уксусной кислотой или хлоридом аммония, при температуре в диапазоне от 0°C до температуры кипения с обратным холодильником и в течение периода времени в диапазоне от приблизительно 1 ч до приблизительно 96 ч. Катализатор гидрогенизации, как правило, представляет собой металл, наиболее часто палладий, который можно использовать в чистом виде или на подложке из угля.
Согласно стадии "f" способа E, синтез соединения общей формулы (I)T из соединения общей формулы 43 проводят, как описано для стадии "a" способа A.
Соединение формулы (I), полученное согласно способу A, способу B, способу C, способу D и способу E, выше, можно далее преобразовывать в другое соединение формулы (I) согласно методикам, хорошо известным специалистам в данной области.
Например, когда R2 представляет собой водород (соединение формулы (I)W) или когда R2 представляет собой галоген (соединение формулы (I)Y), указанные соединения можно далее преобразовывать в другие соединения формулы (I)X, (I)Z, (I)Z1 и (I)Z2. Кроме того, соединение формулы (I)Y можно далее преобразовывать в другое соединение формулы (I)AB и (I)AC в соответствии со способом F.
Способ F

На представленной выше схеме m, n, R1, R3, R4, R5, R6, R17, R18, Rx и Hal являются такими, как описано выше; R2’ представляет собой алкильную группу; R17’ является таким, как и R17, или представляет собой COR22, где R22 является таким, как определено выше.
В способе синтеза для получения соединения формулы (I)X, (I)Z, (I)Z1, (I)Z2, (I)AB и (I)AC, который описан для способа F, на стадии "a" азот пиридина в соединении формулы (I)W окисляют с образованием N-оксидного производного формулы 44. На стадии "b" и "b1" реакция последнего с подходящим электрофильным соединением, таким как тозилангидрид, в присутствии или после обработки подходящим нуклеофилом, таким как соединение общей формулы NHR17’R18 (45) или первичный амин общей формулы NH2R18 (46), дает соединение формулы 49 или (I)X, соответственно. На стадии "d" и "d1" последние соединения, где Rx является таким, как определено выше, за исключением водорода, преобразуют в соответствующее соединение формулы (I)Z или (I)Z1, соответственно. Необязательно на стадии "e" соединение формулы 49, где R17’ представляет собой трет-бутильную группу, бензильную группу, трет-бутоксикарбонильную группу и т.п., обрабатывают кислотой или в восстановительных условиях для удаления указанной группы с получением соединения формулы (I)X. Аналогично, на стадии "e1" соединение формулы (I)X, где R18 представляет собой трет-бутильную группу, бензильную группу, трет-бутоксикарбонильную группу и т.п., обрабатывают кислотой или в восстановительных условиях для удаления указанной группы с получением соединения формулы (I)Z2. На стадии "f" соединение формулы (I)X необязательно обрабатывают электрофилом общей формулы R17’Hal с получением соединения 49. На стадиях "c" и "c1" соединения общей формулы 49 и (I)X, соответственно, можно получать из соединения общей формулы (I)Y путем катализируемой палладием реакции перекрестного сочетания с подходящим соединением общей формулы NHR17’R18 (45) или NH2R18 (46), соответственно.
На стадии "g" соединение формулы (I)Y преобразуют в соединение общей формулы (I)AB путем катализируемой палладием реакции перекрестного сочетания с подходящим металлоорганическим производным, таким как алюминийорганическое соединение; на стадии "d2" последнее, где Rx является таким, как определено выше, за исключением водорода, преобразуют в соответствующее соединение формулы (I)AC.
Согласно стадии "a" способа F, окисление азота пиридина проводят с использованием окислителей, хорошо известных специалистам в данной области, например, таких как пероксид водорода, в растворителе, таком как уксусная кислота или м-хлорпербензойная кислота, в растворителях, таких как DCM, ацетон, THF и т.п. при температурах в диапазоне от 0°C до температуры кипения с обратным холодильником и в течение периода времени в диапазоне от 30 мин до приблизительно 48 ч.
Согласно стадии "b" способа F, преобразование соединения формулы 44 в соединение формулы 49 проводят путем активации N-оксида пиридина и реакции его с соединением общей формулы NHR17’R18 (45). Активацию обычно проводят с использованием подходящего электрофильного реагента, такого как оксалилхлорид, трифторметансульфонилхлорид, тозилхлорид, бензоилхлорид, уксусный ангидрид, тозилангидрид, PyBroP (бром-трис-пирролидинофосфоний-гексафторфосфат), BroP (бром-трис-(диметиламино)фосфоний-гексафторфосфат), PyBOP (бензотриазол-1-ил-окси-трис-пирролидино-фосфоний-гексафторфосфат), BOP (бензотриазол-1-ил-окси-трис-(диметиламино)-фосфоний-гексафторфосфат) и т.п., в растворителе, таком как DCM, THF, дихлорэтан, этилацетат, ацетонитрил, толуол, трифторметилбензол и т.п., в присутствии основания, такого как TEA, DIPEA, 2,6-лутидин и т.п. Предпочтительным является применение PyBroP и DIPEA в DCM. Реакцию обычно проводят в присутствии вторичного амина, и ее можно проводить при температурах в диапазоне от 20°C до температуры кипения с обратным холодильником и в течение периода времени в диапазоне от 30 мин до приблизительно 48 ч.
Согласно стадии "b1" способа F, преобразование соединения формулы 44 в соединение общей формулы (I)X проводят, как описано для стадии "b" способа F, но с использованием подходящего первичного амина общей формулы 46.
Согласно стадии "c" способа F, соединение формулы (I)Y подвергают реакции перекрестного сочетания с подходящим амином, амидом или карбамоильным соединением общей формулы 45 (реакция Бухвальда-Хартвига). Указанная реакция хорошо известна специалистам в данной области. Ее проводят с использованием катализатора на основе палладия, например, такого как хлорид палладия, ацетат палладия, Pd(dba)2, Pd2(dba)3 и т.п., в присутствии подходящего основания, такого как т-бутоксид натрия, Cs2CO3, Na2CO3, K2CO3, K3PO4, LiHMDS и т.п., необязательно в присутствии лиганда на основе фосфина, такого как P(o-tol)3, P(t-Bu)3, DPPP, DPPF, BINAP (2,2'-бис(дифенилфосфино)-1,1'-бинафтил), Xantphos (4,5-бис(дифенилфосфино)-9,9-диметилксантен) или монофосфинобифениловые лиганды. Указанные реакции можно проводить в растворителе, таком как DMF, диметилсульфоксид, вода, DME, 1,4-диоксан, THF и т.п., и их смесь, при температуре в диапазоне от 20°C до температуры кипения с обратным холодильником и в течение периода времени в диапазоне от 30 мин до приблизительно 24 ч.
Согласно стадии "c1" способа F, преобразование соединения формулы (I)Y в соединение общей формулы (I)X проводят, как описано для стадии "c" способа F, но с использованием подходящего амина, амида или карбамоильного соединения общей формулы 46.
Согласно стадии "d" способа F, преобразование соединения формулы 49 в соответствующее соединение формулы (I)Z проводят, как описано для стадии "j" способа A.
Согласно стадии "d1" способа F, преобразование соединения формулы (I)X в соответствующее соединение формулы (I)Z1 проводят, как описано для стадии "j" способа A.
Согласно стадии "e" способа F, соединение формулы 49, где R17’ представляет собой группу, такую как трет-бутил, бензил, трет-бутоксикарбонил и т.п., группу, такую как R17’, можно удалять с получением соединения общей формулы (I)X. Реакцию обычно проводят с использованием сильных кислот, таких как TFA, необязательно в присутствии подходящего сорастворителя, такого как DCM или вода, при температурах в диапазоне от 20°C до температуры кипения с обратным холодильником и в течение периода времени в диапазоне от 30 мин до приблизительно 48 ч. Альтернативно указанную реакцию проводят с использованием восстановительных условий, таких как H2, в присутствии подходящего катализатора гидрогенизации. Катализатор гидрогенизации обычно представляет собой металл, наиболее часто палладий, который можно использовать в чистом виде или на подложке из угля, в подходящем растворителе, например, таком как THF, 1,4-диоксан, DMF, MeOH, этилацетат или их смесь.
Согласно стадии "e1" способа F, преобразование соединения формулы (I)X в соединение формулы (I)Z2 проводят, как описано для стадии "e" способа F.
Согласно стадии "f" способа F, соединение формулы (I)X можно преобразовывать в другое соединение формулы 49 путем реакции с подходящим электрофилом общей формулы R17’Hal. Специалисту в данной области очевидно, что эту реакцию можно проводить различными путями и с помощью рабочих условий, которые широко известны в данной области для получения аминов, карбоксамидов или карбаматов. Такие реакции можно проводить в подходящем растворителе, например, таком как DCM, хлороформ, THF, диэтиловый эфир, 1,4-диоксан, ацетонитрил, толуол или DMF, в присутствии подходящего основания, такого как TEA, DIPEA, DBU и т.п., при температуре в диапазоне от приблизительно -10°C до температуры кипения с обратным холодильником и в течение подходящего периода времени, например, от приблизительно 30 мин до приблизительно 96 ч.
Согласно стадии "g" способа F, соединение формулы (I)Y можно преобразовывать в соединение формулы (I)AB, где R2’ представляет собой алкильную группу. Реакцию обычно проводят с помощью металлоорганического реагента, такого как R2’MgBr, R2’ZnCl, R2’2CuMgBr или R2’3Al и т.п., в присутствии координирующего агента, такого как Fe(acac)3, NiBr2, CuBr, ZnBr2, Ni(acac)2 и т.п., в растворителе, таком как THF или N-метилпирролидон и т.п., при температуре в диапазоне от 0°C до температуры кипения с обратным холодильником. Альтернативно реакцию сочетания металлоорганического соединения с субстратом может опосредовать палладиевый катализатор, такой как Pd(PPh3)4 и т.п., в растворителе, таком как диоксан или толуол, при температуре в диапазоне от комнатной температуры до температуры кипения с обратным холодильником. Предпочтительной является реакция с R2’3Al и Pd(PPh3)4 в диоксане.
Согласно стадии "d2" способа F, преобразование соединения формулы (I)AB в соответствующее соединение формулы (I)AC проводят, как описано для стадии "j" способа A.
При получении соединений формулы (I) согласно любому варианту процесса, все из которых включены в объем изобретения, необязательные функциональные группы в исходных материалах, реагенты или их промежуточные соединения, которые могут привести к нежелательным побочным реакциям, должны быть надлежащим образом защищены общепринятыми способами.
Исходные материалы способов, являющихся объектом настоящего изобретения, охватывающих любой возможный вариант, а также любые их реагенты являются известными и, если не являются коммерчески доступными непосредственно, могут быть получены хорошо известными способами.
ФАРМАКОЛОГИЯ
Анализы
Анализ пролиферации клеток in vitro
Экспоненциально растущие клетки меланомы человека A375 (с мутантной B-Raf) и клетки меланомы человека Mewo (с B-Raf дикого типа) высевали и инкубировали при 37°C в увлажненной атмосфере с 5% CO2. Через 24 ч в среду добавляли ступенчато возрастающие дозы соединения и клетки инкубировали в течение 72 ч. В конце обработки клетки промывали и подсчитывали. Количество клеток определяли с помощью системы мониторинга клеточного аденозинтрифосфата. Пролиферацию клеток сравнивали с контрольными клетками и вычисляли концентрацию, ингибирующую рост клеток на 50%.
Анализ ArrayScan для p-MAPK (T202/Y204)
Клетки меланомы человека A375, имеющие мутантную B-Raf, высевали в 384-луночные покрытые полилизином планшеты (Matrix) с плотностью 1000 клеток/лунка с подходящей средой, дополненной 10% FCS, и инкубировали в течение 16-24 ч. Клетки обрабатывали в течение 1,5 или 2 ч возрастающими дозами соединений (исходная доза 10 мкМ, фактор разведения 2,5). В конце обработки клетки фиксировали 3,7% п-формальдегидом в течение 15-30 мин, а затем промывали два раза D-PBS (80 мкл/лунка) и обеспечивали их проницаемость с помощью D-PBS, содержавшего 0,1% Triton X-100 и 1% BSA (Sigma-Aldrich) в течение 15 мин при комнатной температуре (исходный раствор). Добавляли моноклональное антитело против фосфо-MAPK (T202/Y204) E10 (Cell Signaling, каталожный номер № 9106), разбавленное 1:100, в растворе для окрашивания и инкубировали в течение 1 ч при 37°C. После удаления раствора первичного антитела добавляли конъюгированное с CyTM2 (Green) вторичное антитело против антител мыши (Amersham), разбавленное 1:500, в растворе для окрашивания, содержавшем 2 мкг/мл DAPI. Планшет инкубировали в течение 1 ч при 37°C, промывали два раза, а затем считывали на Cellomics’ ArrayScan VTI (4 поля/лунка, алгоритм CytoNucTrans).
Параметр "MEAN_RingAvgIntenCh2", который измеряет среднюю интенсивность флуоресценции цитоплазмы, ассоциированную с окрашиванием p-MAPK, сообщают в качестве конечного результата.
Мутации B-Raf, которые конститутивно активируют киназу, идентифицированы в большинстве меланом и в большей части карцином толстой и прямой кишки и папиллярных карцином щитовидной железы. Рост клеток с активированной B-Raf строго зависит от активности B-Raf. Учитывая описанные выше анализы, соединения формулы (I) обладают примечательной активностью ингибирования пролиферации клеток с величинами IC50 ниже 0,2 мкМ, причем она является более высокой в отношении клеточной линии с мутантной B-Raf (A375), чем в отношении клеточной линии с B-Raf (Mewo) дикого типа, как описано в следующей таблице.
В той же таблице также описаны данные, полученные для соединений формулы (I) в анализе ArrayScan, и они демонстрируют способность соединений формулы (I) ингибировать каскад передачи сигнала, контролируемый активацией B-Raf, в клеточной линии A375 с мутантной B-Raf. Величины IC50 всегда ниже 0,2 мкМ и согласуются с величинами IC50, полученными в анализе пролиферации на той же клеточной линии, что подтверждает, что антипролиферативная активность соединений является следствием ингибирования активности B-Raf.
IC50 (мкМ)
IC50 (мкМ)
Противоопухолевая эффективность на модели меланомы человека с ксенотрансплантатом
Самцов мышей Balb Nu/Nu от Harlan (Италия) содержали в клетках, накрытых бумажным фильтром, с кормом и стерилизованной подстилкой и подкисленной водой. Подкожно инъецировали 3×106 A375 клеток меланомы человека (от American Type Culture Collection). Соединения вводили пероральным путем в объеме 10 мл/кг в течение 10 последовательных дней. Рост опухоли и массу тела измеряли каждые 3 суток. Рост опухоли оценивали с помощью толщиномера. Массу опухоли вычисляли следующим образом: Масса опухоли (мг) = длина (мм) · ширина2 (мм2)/2 · d (мг/мм3), допуская, что плотность d = 1 мг/мм3 для опухолевой ткани. Ингибирование роста опухоли (TGI) определяли согласно уравнению % TGI = 100 − (средняя масса опухоли в обработанной группе/средняя масса опухоли в контрольной группе) × 100. Токсичность оценивали, исходя из снижения массы тела.
Исходя из указанного выше анализа, группа соединений по настоящему изобретению продемонстрировала TGI более 90% без снижения массы тела.
Биодоступность
В дополнение к способам in vitro, можно проводить способы in vivo, такие как фармакокинетические исследования, у ряда животных. Соединение формулы (I) по настоящему изобретению можно вводить животным, например, мыши или крысе, в различных дозировках и различными путями введения, предпочтительно перорально. Взятие образцов крови можно проводить в последовательные моменты времени и образцы анализировать на присутствие указанного соединения формулы (I).
Соединение формулы (I) по настоящему изобретению, составленное в 0,5% Methocel®, вводили перорально мышам (от 10 до 100 мг/кг) в фармакокинетических испытаниях, и мониторинг его концентрации в крови проводили с помощью анализа ВЭЖХ/MS через 15 и 30 мин, 1, 6 и 24 ч после дозирования. Взятие всех образцов крови проводили через подкожную вену.
Пероральную биодоступность (Fos) вычисляли в качестве процентного отношения средней величины пероральной AUC соединения к средней величине в/в AUC соединения после нормализации дозы соединения.
Исходя из указанного выше анализа, группа соединений по настоящему изобретению продемонстрировала пероральную биодоступность выше 20%.
Из всего вышеуказанного, новые соединения формулы (I) по изобретению оказались особенно преимущественными в терапии заболеваний, вызванных нарушением регуляции активности протеинкиназ, таких как злокачественная опухоль.
Соединения по настоящему изобретению можно вводить либо в качестве единичных средств, либо, альтернативно, в комбинации с известными способами лечения злокачественной опухоли, такими как лучевая терапия или режим химиотерапии, в комбинации, например, с антигормональными средствами, такими как антиэстрогены, антиандрогены и ингибиторы ароматазы, ингибиторами топоизомеразы I, ингибиторами топоизомеразы II, средствами, нацеленными на микротрубочки, средствами на основе платины, алкилирующими средствами, повреждающими ДНК или интеркалирующими средствами, антинеопластическими антиметаболитами, другими ингибиторами киназ, другими антиангиогенными средствами, ингибиторами кинезинов, терапевтическими моноклональными антителами, ингибиторами mTOR, ингибиторами деацетилазы гистонов, ингибиторами фарнезилтрансферазы и ингибиторами гипоксического ответа.
При составлении в качестве фиксированной дозы, в таких комбинированных продуктах используются соединения по настоящему изобретению в диапазоне дозировок, описанном ниже, и другое фармацевтически активное вещество в одобренном диапазоне дозировок.
Соединения формулы (I) можно использовать последовательно с известными средствами против злокачественной опухоли, когда комбинированный состав не пригоден.
Соединения формулы (I) по настоящему изобретению, пригодные для введения млекопитающему, например, человеку, можно вводить обычными путями и уровень дозировки зависит от возраста, массы тела и состояния пациента, и пути введения.
Например, подходящая дозировка соединения формулы (I) для перорального введения может находиться в диапазоне от приблизительно 10 мг до приблизительно 2 г на дозу, от 1 до 5 раз в сутки. Соединения по изобретению можно вводить в различных дозированных формах, например, перорально в форме таблеток, капсул, покрытых сахаром или пленкой таблеток, жидких растворов или суспензий; ректально в форме суппозиториев; парентерально, например, внутримышечно, или путем внутривенной, и/или интратекальной, и/или интраспинальной инъекции или инфузии.
Также настоящее изобретение относится к фармацевтическим композициям, содержащим соединение формулы (I) или его фармацевтически приемлемую соль, вместе с фармацевтически приемлемым эксципиентом, который может представлять собой носитель или разбавитель.
Фармацевтические композиции, содержащие соединения по изобретению, обычно получают общепринятыми способами и вводят в подходящей фармацевтической форме.
Например, твердые пероральные формы могут содержать вместе с активным соединением разбавители, например, лактозу, декстрозу, сахарозу, сахаробиозу, целлюлозу, кукурузный крахмал или картофельный крахмал; смазывающие вещества, например, диоксид кремния, тальк, стеариновую кислоту, стеарат магния или кальция, и/или полиэтиленгликоли; связующие вещества, например, крахмалы, гуммиарабик, желатин, метилцеллюлоза, карбоксиметилцеллюлоза или поливинилпирролидон; дезинтегрирующие вещества, например, крахмал, альгиновая кислота, альгинаты или натрия крахмал гликолят; шипучие смеси; красители; подсластители; смачивающие вещества, такие как лецитин, полисорбаты, лаурилсульфаты и, как правило, нетоксические и фармакологически неактивные вещества, используемые в фармацевтических составах. Эти фармацевтические препараты могут быть изготовлены известным способом, например, путем смешения, гранулирования, таблетирования, покрытия сахаром или нанесения пленочного покрытия.
Жидкие дисперсии для перорального введения могут представлять собой, например, сиропы, эмульсии и суспензии.
В качестве примера, сиропы могут содержать в качестве носителя сахарозу или сахарозу с глицерином и/или маннит и сорбит.
Суспензии и эмульсии могут содержать, в качестве примеров носителей, природную камедь, агар, альгинат натрия, пектин, метилцеллюлозу, карбоксиметилцеллюлозу или поливиниловый спирт.
Суспензия или растворы для внутримышечных инъекций могут содержать, вместе с активным соединением, фармацевтически приемлемый носитель, например, стерильную воду, оливковое масло, этилолеат, гликоли, например, пропиленгликоль и, если желательно, подходящее количество лидокаина гидрохлорида.
Растворы для внутривенных инъекций или инфузий могут содержать, в качестве носителя, стерильную воду или предпочтительно они могут быть в форме стерильных водных изотонических солевых растворов или они могут содержать пропиленгликоль в качестве носителя.
Суппозитории могут содержать, вместе с активным соединением, фармацевтически приемлемый носитель, например, масло какао, полиэтиленгликоль, поверхностно-активное вещество на основе сложного эфира полиоксиэтилена сорбитана и жирной кислоты или лецитин.
ЭКСПЕРИМЕНТАЛЬНЫЙ РАЗДЕЛ
Для отсылки к любому конкретному соединению формулы (I) по изобретению, необязательно в форме фармацевтически приемлемой соли, см. экспериментальный раздел и формулу изобретения. Ссылаясь на примеры, которые следуют далее, соединения по настоящему изобретению синтезировали с использованием способов, описанных в настоящем описании, или других способов, которые хорошо известны в данной области.
Короткие формы и сокращения, использованные в настоящем описании, имеют следующее значение:
В целях лучшей иллюстрации настоящего изобретения, без какого-либо его ограничения, далее приведены следующие примеры.
Как используют в рамках изобретения, символы и условные обозначения, используемые в способах, на схемах и в примерах, соответствуют символам и условным обозначениям, используемым в современной научной литературе, например, Journal of the American Chemical Society или Journal of Biological Chemistry.
Если нет иных указаний, все материалы приобретали от коммерческих поставщиков, они представляли собой материалы наилучшей категории и их использовали без дальнейшей очистки. Безводный растворитель, такой как DMF, THF, DCM и толуол, приобретали от Aldrich Chemical Company. Все реакции, вовлекающие чувствительные к воздуху или влаге соединения, проводили в атмосфере азота или аргона.
Общие способы очистки и аналитические способы
Флэш-хроматографию проводили на силикагеле (Merck grade 9395, 60A).
ВЭЖХ проводили на колонке Waters X Terra RP 18 (4,6×50 мм, 3,5 мкм) с использованием системы ВЭЖХ Waters 2790, оборудованной детектором 996 Waters PDA и одноквадрупольным масс-спектрометром Micromass mod. ZQ, оборудованным электрораспылительным (ESI) источником ионов. Подвижная фаза A представляла собой 5 мМ буфер на основе ацетата аммония (pH 5,5, с соотношением уксусная кислота-ацетонитрил 95:5), и подвижная фаза B представляла собой воду-ацетонитрил (5:95). Градиент от 10 до 90% B в течение 8 мин с удерживанием 90% B 2 мин. УФ-детекция при 220 нм и 254 нм. Скорость потока 1 мл/мин. Объем инжекции 10 мкл. Полное сканирование, диапазон масс от 100 до 800 а.е.м. Напряжение на капиллярах составляло 2,5 кВ; температура источника составляла 120°C; напряжение на конусе составляло 10 В. Время удерживания (ВЭЖХ Rt) приведено в минутах (мин) при 220 нм или при 254 нм. Масса приведена в качестве соотношения m/z.
Когда было необходимо, соединения очищали с помощью препаративной ВЭЖХ на колонке Waters Symmetry C18 (19×50 мм, 5 мкм) или на колонке Waters X Terra RP 18 (30×150 мм, 5 мкм) с использованием Waters preparative HPLC 600, оборудованного детектором 996 Waters PDA и одноквадрупольным масс-спектрометром Micromass mod. ZMD, с ионизацией электрораспылением, положительный режим. Подвижная фаза A представляла собой смесь вода-0,01% TFA, и подвижная фаза B представляла собой ацетонитрил. Градиент от 10 до 90% B в течение 8 мин, удерживание 90% B 2 мин. Скорость потока 20 мл/мин. Альтернативно, подвижная фаза A представляла собой смесь вода-0,1% NH3, и подвижная фаза B представляла собой ацетонитрил. Градиент от 10 до 100% B в течение 8 мин, удерживание 100% B 2 мин. Скорость потока 20 мл/мин.
Спектры 1H-NMR регистрировали при постоянной температуре 28°C на спектрометре Varian INOVA 400, работающем при 400,50 МГц и оборудованным 5 мм зондом с непрямой детекцией PFG с z-осью (1H{15N-31P}).
Химические сдвиги указывали относительно сигналов остаточного растворителя (DMSO-d6: 2,50 м.д. для 1H, в остальных случаях не указано). Представленные данные являются следующими: химический сдвиг (δ), мультиплетность (с = синглет, д = дублет, т = триплет, кв = квартет, ушир.c. = уширенный синглет, тд = триплет дублетов, дд = дублет дублетов, ддд = дублет дублетов дублетов, м = мультиплет, спт = септет), константа связи (J, Гц), и количество протонов.
Как описано ранее (M. Colombo, F.R. Sirtori, V. Rizzo, Rapid Commun Mass Spectrom 2004, 18(4), 511-517), спектры масс высокого разрешения (HRMS) ESI(+) получали на масс-спектрометре Q-Tof Ultima (Waters, Manchester, Великобритания), прямо соединенном с микро-ВЭЖХ системой Agilent 1100 (Palo Alto, США).
Получение N-(3-ацетил-2-фторфенил)-2,5-дифтор-N-(метоксиметил)бензолсульфонамид, соединение формулы 6 [n = 1; R4 = F; R5 = H; R6 = 2,5-дифторфенил; Rx’ = метоксиметил] (методика получения 1, способ A)
N-(3-Бром-2-фторфенил)-2,5-дифторбензолсульфонамид, соединение формулы 3 [n = 1; R4 = F; R5 = H; R6 = 2,5-дифторфенил]
Способ A, стадия a
3-Бром-2-фторанилин (10 г, 52,63 ммоль) растворяли в DCM (100 мл) в атмосфере азота. Добавляли сухой пиридин (6 мл, 73,68 ммоль), а затем 2,5-дифторбензолсульфонилхлорид (7,08 мл, 52,63 ммоль) и смесь перемешивали при к.т. в течение 2 ч. Затем ее разбавляли DCM и промывали водной 0,5н HCl (3×80 мл) и насыщенным солевым раствором. Органический слой сушили над Na2SO4 и упаривали до сухого состояния. Твердое вещество отбирали в диэтиловый эфир и перемешивали в течение 30 мин. Затем его фильтровали и сушили при 40°C при пониженном давлении с получением 17,8 г N-(3-бром-2-фторфенил)-2,5-дифторбензолсульфонамида в виде светло-желтого твердого вещества (92%).
ВЭЖХ: Rt: 6,28 мин
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 10,86 (с, 1H), 7,50-7,73 (м, 4H), 7,23-7,31 (м, 1H), 7,12 (дт, J=1,3, 8,1 Гц, 1H)
N-(3-Бром-2-фторфенил)-2,5-дифтор-N-(метоксиметил)бензолсульфонамид, соединение формулы 4 [n = 1; R4 = F; R5 = H; R6 = 2,5-дифторфенил; Rx’ = метоксиметил]
Способ A, стадия b
К раствору N-(3-бром-2-фторфенил)-2,5-дифторбензолсульфонамида (17,8 г, 48,61 ммоль) в безводном DCM (160 мл) при 0°C добавляли DIPEA (12,5 мл, 73 ммоль), а затем метоксиметилхлорид (5,7 мл, 73 ммоль). Реакционную смесь перемешивали при 0°C в течение 10 мин, а затем позволяли ей нагреться до к.т. Через 2 ч добавляли насыщенный раствор хлорида аммония и смесь перемешивали при к.т. в течение 10 мин. Затем ее разбавляли DCM и промывали водой и насыщенным солевым раствором, сушили над Na2SO4 и упаривали до сухого состояния. Осадок обрабатывали Hex и перемешивали в течение 30 мин. Твердое вещество отфильтровывали и сушили с получением 18,52 г (93%) указанного в заголовке соединения в виде порошка белого цвета.
ВЭЖХ: Rt: 6,88 мин
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 7,78 (ддд, J=1,7, 6,4, 8,1 Гц, 1H), 7,67-7,73 (м, 1H), 7,61 (дт, J=4,3, 9,6, 1H), 7,49-7,55 (м, 1H), 7,28-7,34 (м, 1H), 7,17-7,23 (м, 1H), 5,06 (с, 2H), 3,35 (с, 3H)
HRMS (ESI): вычислено для C14H11BrF3NO3SNa [M+Na]+ 431,9487, найдено 431,9487.
N-(3-Ацетил-2-фторфенил)-2,5-дифтор-N-(метоксиметил)бензолсульфонамид, соединение формулы 6 [n = 1; R4 = F; R5 = H; R6 = 2,5-дифторфенил; Rx’ = метоксиметил]
Способ A, стадия c
К раствору 6,15 г (15 ммоль) N-(3-бром-2-фторфенил)-2,5-дифтор-N-(метоксиметил)бензолсульфонамида в 35 мл этиленгликоля в колбе, оборудованной резиновой мембраной, через которую через иглу подают азот, последовательно добавляли 37,5 мг (0,15 ммоль) ацетата палладия, 129 мг (0,30 ммоль) DPPP, 5,4 мл (37,5 ммоль) TEA и 5,9 мл (45 ммоль) н-бутилвинилового простого эфира. Смесь нагревали при 120°C при перемешивании в течение 6 ч, а затем разбавляли DCM и промывали насыщенным солевым раствором. Органический слой сушили над Na2SO4 и упаривали с получением N-[3-(1-бутоксиэтенил)-2-фторфенил]-2,5-дифтор-N-(метоксиметил)бензолсульфонамида в виде масла коричневого цвета. Последнее без какой-либо дальнейшей очистки растворяли в 65 мл 1,4-диоксана и к полученному раствору добавляли 11 мл 1н HCl. После перемешивания в течение 1 ч при к.т. реакция завершалась. Затем растворитель удаляли, осадок перерастворяли в DCM и промывали водным раствором NaHCO3. Органический слой сушили над безводным Na2SO4 и упаривали. После растирания с диэтиловым эфиром 4,82 г (86%) указанное в заголовке соединение собирали фильтрацией.
ВЭЖХ: Rt: 6,28 мин
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 7,81-7,89 (м, 1H), 7,47-7,73 (м, 5H), 7,36 (т, J=7,8 Гц, 1H), 5,05-5,10 (м, 2H), 3,35-3,38 (м, 3H), 2,48-2,49 (м, 3H)
HRMS (ESI): вычислено для C16H14F3NO4SNa [M+Na]+ 396,0488, найдено 396,0488.
Получение 3-{[(2,5-дифторфенил)сульфонил](метоксиметил)-амино}-2-фтор-N-метокси-N-метилбензамида, соединение формулы 29 [n = 1; R4 = F; R5 = H; Rx = метоксиметил; R6 = 2,5-дифторфенил; J=NMe(OMe)] (методика получения 2, способ D)
3-{[(2,5-Дифторфенил)сульфонил](метоксиметил)амино}-2-фторбензойная кислота
N-(3-Бром-2-фторфенил)-2,5-дифтор-N-(метоксиметил)бензолсульфонамид (полученный, как описано в методике получения 1, 10,92 г, 26,62 ммоль) растворяли в безводном THF (53 мл) и охлаждали до 0°C. По каплям добавляли раствор хлорида изопропилмагния (2н в THF) (13,3 мл, 26,62 ммоль, 1 экв.). После завершения добавления реакционной смеси позволяли нагреться до к.т. и перемешивали в течение 2 ч. Затем раствор желтого цвета обратно охлаждали до 0°C, а затем через раствор барботировали газообразный угольный ангидрид (полученный из твердого угольного ангидрида и высушенный с помощью концентрированной серной кислоты) в течение 20 мин. Затем добавляли 0,5н раствор HCl (50 мл) и смесь экстрагировали диизопропиловый эфиром (3×130 мл). Затем желаемый продукт экстрагировали из органической фазы 1н раствором гидроксида натрия (3×100 мл). Затем при энергичном перемешивании добавляли 2н HCl (150 мл). Осадок собирали фильтрацией, промывали водой и Hex и сушили в печи с получением 9,07 г (91%) указанного в заголовке соединения в виде твердого вещества белого цвета.
ВЭЖХ: Rt: 3,67 мин
1H ЯМР (600 МГц, DMSO-d6) δ м.д. 7,88 (т, J=6,7 Гц, 1H), 7,71-7,64 (м, 1H), 7,61 (дт, J=4,1, 9,4 Гц, 1H), 7,53-7,44 (м, 2H), 7,32 (т, J=7,9 Гц, 1H), 5,06 (с, 2H).
HRMS (ESI): вычислено для C15H12NO5F3SNa [M+Na]+ 398,0280, найдено 398,0280.
3-{[(2,5-Дифторфенил)сульфонил](метоксиметил)амино}-2-фтор-N-метокси-N-метилбензамид, соединение формулы 29 [n = 1; R4 = F; R5 = H; Rx = метоксиметил; R6 = 2,5-дифторфенил; J=NMe(OMe)]
3-{[(2,5-Дифторфенил)сульфонил](метоксиметил)амино}-2-фторбензойную кислоту (8,86 г, 23,61 ммоль) растворяли в DCM (73 мл) в атмосфере азота. Добавляли сухой DMF (14 мл), а затем N-метокси-N-метиламина гидрохлорид (3,72 г, 38,13 ммоль, 1,6 экв.), N-метилморфолин (4,1 мл, 37,3 ммоль, 1,6 экв.) и DMAP (293 мг, 2,4 ммоль, 0,1 экв.). Затем реакционную смесь охлаждали до 0°C и порционно добавляли гидрохлорид EDC (5,38 г, 28,06 ммоль, 1,2 экв.). Смеси позволяли нагреться до к.т. и перемешивали в течение 4 ч. Затем ее охлаждали до 0°C и добавляли холодный 1н раствор HCl (100 мл). Смесь экстрагировали диизопропиловым эфиром и органическую фазу промывали насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над Na2SO4 и упаривали до сухого состояния. Неочищенный продукт очищали флэш-хроматографией на силикагеле (DCM/MeOH 98:2) с получением 8,97 г (91%) указанного в заголовке соединения в виде аморфного бесцветного твердого вещества.
ВЭЖХ: Rt: 5,25 мин
1H ЯМР (600 МГц, DMSO-d6) δ м.д. 7,70-7,64 (м, 1H), 7,61 (дт, J=3,9, 9,4 Гц, 1H), 7,54-7,44 (м, 2H), 7,42 (т, J=7,2 Гц, 1H), 7,31 (т, J=7,9 Гц, 1H), 5,07 (с, 2H), 3,36 (с, 3H), 3,29 (с, 3H), 3,21 (ушир.с, 1H).
HRMS (ESI): вычислено для C17H18N2O5F3S [M+H]+ 419,0883, найдено 419,0893.
Получение трет-бутил-4-карбамотиоилпиперидин-1-карбоксилата, соединение формулы 8 [R1 = 1-трет-бутоксикарбонилпиперидин-4-ил] (методика получения 3, способы A, D и E)
трет-Бутил 4-карбамоилпиперидин-1-карбоксилат
К раствору пиперидин-4-карбоксамида (2 г, 15,6 ммоль) в ацетонитриле (30 мл) добавляли трет-бутилдикарбонат (4,4 г, 20,2 ммоль, 1,3 экв.) и DMAP (190 мг, 1,56 ммоль, 0,1 экв.) и смесь перемешивали при к.т. в течение ночи. Затем ее концентрировали при пониженном давлении, отбирали в DCM и промывали насыщенным водным NaHCO3, насыщенным водным NH4Cl, насыщенным солевым раствором и водой. Органическую фазу сушили над Na2SO4 и упаривали до сухого состояния. Осадок растирали с диэтиловым эфиром и белое твердое вещество фильтровали и сушили с получением 2,65 г (74%) указанного в заголовке продукта.
ВЭЖХ: Rt: 4,23 мин
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 7,24 (ушир.с, 1H), 6,75 (ушир.с, 1H), 4,06-3,73 (м, 2H), 2,71 (ушир.с, 2H), 2,23 (тт, J=3,8, 11,5 Гц, 1H), 1,66 (дд, J=2,7, 13,2 Гц, 2H), 1,44-1,26 (м, 2H), 1,36 (с, 9H).
трет-Бутил-4-карбамотиоилпиперидин-1-карбоксилат, соединение формулы 8 [R1 = 1-трет-бутоксикарбонилпиперидин-4-ил]
К раствору трет-бутил-4-карбамоилпиперидин-1-карбоксилата (2,65 г, 11,62 ммоль) в смеси DME/DCM 2:1 (78 мл), добавляли реагент Лоуссона (2,35 г, 5,81 ммоль, 0,5 экв.) и смесь перемешивали при к.т. в течение ночи. Растворитель удаляли и осадок отбирали в этилацетат и промывали насыщенным водным K2CO3. Органический слой отделяли, сушили над Na2SO4 и концентрировали до сухого состояния. Осадок растирали с диэтиловым эфиром и сушили с получением 2,65 г (92%) указанного в заголовке соединения в виде белого твердого вещества.
ВЭЖХ: Rt: 5,06 мин
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 9,39 (ушир.с, 1H), 9,09 (ушир.с, 1H), 4,00 (д, J=12,6 Гц, 2H), 2,77-2,61 (м, 3H), 1,71-1,51 (м, 4H), 1,39 (ушир.с, 9H)
Получение бензил-4-карбамотиоилпиперидин-1-карбоксилата, соединение формулы 8 [R1 = 1-бензилоксикарбонилпиперидин-4-ил] (методика получения 4, способы A, D и E)
Бензил-4-карбамоилпиперидин-1-карбоксилат
К раствору пиперидин-4-карбоксамида (5 г, 39 ммоль) и бензилхлорформиата (5,54 мл, 39 ммоль) в смеси воды (30 мл) и ацетона (40 мл), по каплям добавляли 1н NaOH (39 мл, 39 ммоль), при поддержании pH от 6 до 8. Смесь перемешивали при к.т. в течение 3 ч; затем ацетон выпаривали и полученный осадок фильтровали и сушили при 70°C при пониженном давлении с получением 7,75 г бензил-4-карбамоилпиперидин-1-карбоксилата (76%).
ВЭЖХ: Rt: 4,54 мин
1H ЯМР (600 МГц, DMSO-d6) δ м.д. 7,41-7,29 (м, 5H), 7,25 (ушир.с, 1H), 6,76 (ушир.с, 1H), 5,07 (с, 2H), 4,06-3,81 (м, J=13,2 Гц, 2H), 2,92-2,72 (м, 2H), 2,27 (тт, J=3,7, 11,5 Гц, 1H), 1,75-1,64 (м, 2H), 1,40 (д, кв, J=4,3, 12,4 Гц, 2H)
HRMS (ESI) [M+H]+: вычислено для C14H18O3N2 263,1390, найдено 263,1390.
Бензил-4-карбамотиоилпиперидин-1-карбоксилат, соединение формулы 8 [R1 = 1-бензилоксикарбонилпиперидин-4-ил]
Бензил-4-карбамоилпиперидин-1-карбоксилат (7,5 г, 38,6 ммоль) растворяли в THF (160 мл) и добавляли реагент Лоуссона (6,9 г, 17,1 ммоль). Через 4 ч растворитель выпаривали и осадок очищали флэш-хроматографией на силикагеле (DCM-MeOH 95/5) с получением 1,8 г (23%) указанного в заголовке соединения, кристаллизованного из MeOH.
ВЭЖХ: Rt: 5,28 мин
1H ЯМР (600 МГц, DMSO-d6) δ м.д. 9,40 (ушир.с, 1H), 9,11 (ушир.с, 1H), 7,42-7,24 (м, 5H), 5,07 (с, 2H), 4,07 (д, J=13,2 Гц, 2H), 2,90-2,73 (м, 2H), 2,69 (тт, J=3,8, 11,6 Гц, 1H), 1,70-1,64 (м, 2H), 1,64-1,55 (м, 2H)
HRMS (ESI) [M+H]+: вычислено для C14H18O2N2S 279,1162, найдено 279,1163.
Получение 1-метилпиперидин-4-карботиоамида, соединение формулы 8 [R1 = 1-метилпиперидин-4-ил] (методика получения 5, способы A, D и E)
К суспензии трет-бутил-4-карбамотиоилпиперидин-1-карбоксилата (полученного, как описано в методике получения 3, 2,65 г, 10,86 ммоль) в сухом диоксане (40 мл) добавляли 4M раствор HCl в диоксане (16 мл, 64 ммоль, 5,9 экв.) и смесь перемешивали при к.т. Через 1 ч проводили дополнительное добавление HCl (10 мл) и перемешивание проводили в течение 2 дополнительных часов. Растворитель выпаривали до сухого состояния и осадок отбирали в толуол и упаривали до сухого состояния два раза.
Осадок растворяли в MeOH (64 мл) и добавляли водный раствор формальдегида (2 мл, 26,86 ммоль, 2,5 экв.), а затем уксусную кислоту (2,24 мл, 39,17 ммоль, 3,6 экв.) и цианоборгидрид натрия (2,08 г, 28,24 ммоль, 2,6 экв.) и смесь перемешивали при к.т. в течение 2 ч. Затем растворитель концентрировали при пониженном давлении и осадок отбирали в этилацетат, промывали насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над Na2SO4 и упаривали до сухого состояния с получением 2,2 г указанного в заголовке соединения в виде белого твердого вещества.
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 9,35 (ушир.с, 1H), 9,08 (ушир.с, 1H), 3,44-3,19 (м, 2H), 2,86 (ушир.с, 2H), 2,44 (т, J=10,2 Гц, 1H), 2,21 (ушир.с, 3H), 1,82-1,73 (м, 2H), 1,63 (д, J=11,9 Гц, 2H)
HRMS (ESI): вычислено для C7H15N2S [M+H]+ 159,0951, найдено 159,0944.
Получение тетрагидро-2H-пиран-4-карботиоамида, соединение формулы 8 [R1 = тетрагидропиран-4-ил] (методика получения 6, способы A, D и E)
Тетрагидро-2H-пиран-4-карбоксамид
Смесь метил тетрагидро-2H-пиран-4-карбоксилата (7 г, 48,6 ммоль) и 30% водного раствора аммиака (20 мл) перемешивали в закрытой бутылке при к.т. в течение 18 ч. Избыток аммиака удаляли при пониженном давлении и осадок кристаллизовали из этанола с получением 5,6 г (89%) тетрагидро-2H-пиран-4-карбоксамида.
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 7,21 (ушир.с, 1H), 6,73 (ушир.с, 1H), 3,90-3,80 (м, 2H), 3,30-3,23 (м, 2H), 2,36-2,24 (м, 1H), 1,66-1,47 (м, 4H)
Тетрагидро-2H-пиран-4-карботиоамид, соединение формулы 8 [R1 = тетрагидропиран-4-ил]
Тетрагидро-2H-пиран-4-карбоксамид (2 г, 15,5 ммоль) суспендировали в сухом THF (20 мл) и добавляли реагент Лоуссона (3,13 г, 7,75 ммоль). После кипячения с обратным холодильником в течение 4 ч смесь выливали в насыщенный водный раствор NaHCO3 (200 мл), а затем экстрагировали диэтиловым эфиром (4×100 мл). Органический слой сушили над Na2SO4 и упаривали до сухого состояния с получением 1,2 г (54%) указанного в заголовке соединения.
ВЭЖХ: Rt: мин 2,79
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 9,37 (ушир.с, 1H), 9,08 (ушир.с, 1H), 3,87 (дд, J=4,0, 11,0 Гц, 2H), 3,37-3,23 (м, 2H), 2,78-2,67 (м, 1H), 1,75 (д, кв, J=4,5, 12,5 Гц, 2H), 1,63-1,52 (м, 2H)
HRMS (ESI): вычислено для C16H11NOS [M+H]+ 146,0634, найдено 146,0634.
Получение 1-циклопропилпиперидин-4-карботиоамида, соединение формулы 8 [R1 = 1-циклопропилпиперидин-4-ил] (методика получения 7, способы A, D и E)
К раствору пиперидин-4-карбоксамида (1 г, 7,8 ммоль) в MeOH (80 мл) добавляли 1-этокси-1-триметилсилилоксициклопропан (2,35 мл, 11,7 ммоль, 1,5 экв.), а затем уксусную кислоту (1,34 мл, 23,4 ммоль, 3 экв.) и цианоборгидрид натрия (923 мг, 12,48 ммоль, 1,6 экв.) и смесь перемешивали при 60°C в течение ночи. Затем растворитель концентрировали при пониженном давлении и осадок очищали флэш-хроматографией на силикагеле (DCM/MeOH/7н NH3 в MeOH 90:9:1) с получением 1,6 г 1-циклопропилпиперидин-4-карбоксамида.
1-Циклопропилпиперидин-4-карбоксамид суспендировали в сухом THF (20 мл) и добавляли реагент Лоуссона (2,7 г, 6,67 ммоль). После кипячения с обратным холодильником в течение 6 ч растворитель концентрировали при пониженном давлении. Осадок растворяли в смеси этилацетат/MeOH и промывали насыщенным водным NaHCO3. Водную фазу обратно экстрагировали этилацетатом и упаривали до сухого состояния. Неочищенный продукт обрабатывали этанолом и фильтровали. Белое твердое вещество сушили в высоком вакууме с получением 2,7 г указанного в заголовке соединения.
1H ЯМР (600 МГц, DMSO-d6) δ м.д. 9,32 (ушир.с, 1H), 9,04 (ушир.с, 1H), 2,95 (ушир.с, 2H), 2,54-2,43 (м, 1H), 2,20-2,04 (м, 2H), 1,75-1,38 (м, 5H), 0,39 (ушир.с, 2H), 0,27 (ушир.с, 2H)
HRMS (ESI): вычислено для C9H17N2S [M+H]+ 185,1107, найдено 185,1104.
Получение трет-бутилметилкарбамата, соединение формулы 45 [R17’ = трет-бутоксикарбонил; R18 = метил] (методика получения 8, способ F)
2M Метиламин в THF (15 мл, 33 ммоль) и TEA (4,35 мл) добавляли к сухому DCM (25 мл), охлажденному до -20°C, при перемешивании. К реакционной смеси по каплям добавляли раствор ди-трет-бутилдикарбоната (7,2 г, 33 ммоль) в DCM (25 мл) при поддержании температуры между -20 и -10°C. Затем температуре позволяли подняться до к.т. в течение ночи. Затем смесь промывали насыщенным солевым раствором и органический слой сушили над Na2SO4 и упаривали до сухого состояния с получением 2,83 г (66%) указанного в заголовке соединения в виде бесцветного масла.
1H ЯМР (600 МГц, DMSO-d6) δ м.д. 6,63 (ушир.с, 1H), 2,49 (ушир.с, 3H), 1,37 (с, 9H)
Пример 1
Синтез N-{2,4-дифтор-3-[2-(4-метилпиперазин-1-ил)-5-(пиридин-4-ил)-1,3-тиазол-4-ил]фенил}-2,5-дифторбензолсульфонамида, соединение формулы (I)A (соединение 8) [m, n = 1; R2, R3, Rx = H; R4 = F; R5 = 4-F; R6 = 2,5-дифторфенил; R1’ = 4-метилпиперазин-1-ил]
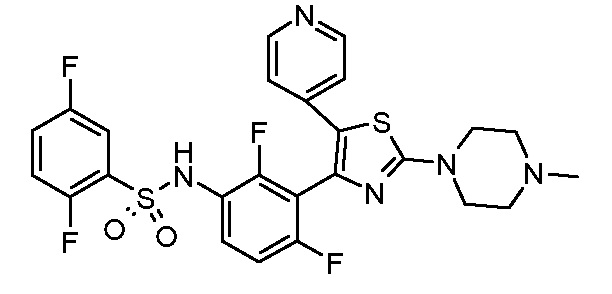
N-[3-(2-Бромацетил)-2,4-дифторфенил]-2,5-дифторбензолсульфонамид, соединение формулы 7 [n = 1; Rx = H; R4 = F; R5 = 4-F; R6 = 2,5-дифторфенил]
Способ A, стадия d
N-(3-Ацетил-2,4-дифторфенил)-2,5-дифторбензолсульфонамид (7,6 г, 21,88 ммоль), полученный, как описано в WO 2010/10154, растворяли в уксусной кислоте (30 мл). Затем добавляли раствор 1,12 мл (26,25 ммоль) брома в 2 мл уксусной кислоты тремя порциями. Затем добавляли 48% водный раствор бромистоводородной кислоты (0,248 мл, 2,19 ммоль), растворенный в 1 мл уксусной кислоты, и смесь перемешивали при к.т. в течение 21 ч. По каплям добавляли 5% водный раствор NaHCO3 (50 мл) и смесь экстрагировали метил-трет-бутиловым эфиром (2×30 мл). Объединенные органические слои сушили над Na2SO4 и упаривали до сухого состояния. Неочищенное вещество перекристаллизовывали из смеси этилацетат/толуол/циклогексан с получением 5,79 г (61%) указанного в заголовке соединения в виде светло-коричневого твердого вещества.
ВЭЖХ: Rt: 6,10 мин
1H-ЯМР (401 МГц, DMSO-d6) δ м.д. 10,75 (ушир.с, 1H), 7,68-7,39 (м, 4H), 7,29-7,17 (м, 1H), 4,34 (с, 2H).
N-{2,4-Дифтор-3-[2-(4-метилпиперазин-1-ил)тиазол-4-ил]-фенил}-2,5-дифторбензолсульфонамид, соединение формулы 9 [n = 1; Rx = H; R4 = F; R5 = 4-F; R6 = 2,5-дифторфенил; R1 = 4-метилпиперазин-1-ил]
Способ A, стадия e
50 мг (0,31 ммоль) 4-метилпиперазин-1-карботиоамида (полученного, как описано в Heterocycles, 1989, 29, 1601) растворяли в 15 мл этанола и к полученному раствору добавляли 134 мг (0,31 ммоль) N-[3-(бромацетил)-2,4-дифторфенил]-2,5-дифторбензолсульфонамида. Смесь перемешивали при 60°C в течение 20 мин в микроволновой печи. Затем растворитель упаривали и осадок растирали с диизопропиловым эфиром с получением 140 мг (97%) указанного в заголовке соединения.
ВЭЖХ: Rt: 5,51 мин
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 10,50 (с, 1H), 7,60-7,66 (м, 1H), 7,53-7,60 (м, 1H), 7,46-7,53 (м, 1H), 7,27 (тд, J=5,7, 8,7 Гц, 1H), 7,15 (тд, J=1,5, 9,2 Гц, 1H), 7,11 (с, 1H), 2,80 (ушир.с, 3H)
HRMS (ESI): вычислено для C20H19F4N4O2S2 [M+H]+ 487,0880, найдено 487,0857.
Аналогично получали следующее промежуточное соединение:
N-[3-(2-Амино-1,3-тиазол-4-ил)-2,4-дифторфенил]-2,5-дифторбензолсульфонамид, соединение формулы 9 [n = 1; Rx = H; R1 = NH2; R4 = F; R5 = 4-F; R6 = 2,5-дифторфенил]
ВЭЖХ: Rt: 5,50 мин
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 10,60 (с, 1H), 7,46-7,65 (м, 3H), 7,24 (тд, J=5,7, 8,8 Гц, 1H), 7,11 (тд, J=1,6, 9,3 Гц, 1H), 7,07 (ушир.с, 2H), 6,69 (с, 7H)
HRMS (ESI): вычислено для C15H10F4N3O2S2 [M+H]+ 404,0145, найдено 404,0142.
N-{3-[5-Бром-2-(4-метилпиперазин-1-ил)тиазол-4-ил]-2,4-дифторфенил}-2,5-дифторбензолсульфонамид, соединение формулы 10 [n = 1; Rx = H; R4 = F; R5 = 4-F; R6 = 2,5-дифторфенил; R1 = 4-метилпиперазин-1-ил; Hal = Br]
Способ A, стадия f
150 мг (0,31 ммоль) N-{2,4-Дифтор-3-[2-(4-метилпиперазин-1-ил)тиазол-4-ил]фенил}-2,5-дифторбензолсульфонамида суспендировали в 15 мл в сухом DCM и добавляли 103 мг (0,62 ммоль) N-бромсукцинимида. Реакцию поддерживали при к.т. в течение 1 ч, а затем разбавляли тем же растворителем и промывали водным раствором NaHCO3. Органический слой сушили над Na2SO4 и упаривали. Осадок растирали с петролейным эфиром и фильтровали с получением 128 мг (73%) N-{3-[5-бром-2-(4-метилпиперазин-1-ил)-1,3-тиазол-4-ил]-2,4-дифторфенил}-2,5-дифторбензолсульфонамида.
ВЭЖХ: Rt: 6,33 мин
HRMS (ESI): вычислено для C20H18BrF4N4O2S2 [M+H]+ 564,9985, найдено 564,9982.
Аналогично получали следующее промежуточное соединение:
N-[3-(2-Амино-5-бром-1,3-тиазол-4-ил)-2,4-дифторфенил]-2,5-дифторбензолсульфонамид, соединение формулы 10 [n = 1; Rx = H; R1 = NH2; R4 = F; R5 = 4-F; R6 = 2,5-дифторфенил; Hal = Br]
ВЭЖХ: Rt: 5,91 мин
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 10,67 (с, 1H), 7,43-7,65 (м, 3H), 7,36-7,42 (м, 1H), 7,35 (ушир.с, 2H), 7,17 (тд, J=1,5, 8,9 Гц, 1H).
HRMS (ESI): вычислено для C15H9BrF4N3O2S2 [M+H]+ 481,9250, найдено 481,9232.
N-{2,4-Дифтор-3-[2-(4-метилпиперазин-1-ил)-5-(пиридин-4-ил)-1,3-тиазол-4-ил]фенил}-2,5-дифторбензолсульфонамид, соединение формулы (I)A (соединение 8) [m, n = 1; R2, R3, Rx = H; R4 = F; R5 = 4-F; R6 = 2,5-дифторфенил; R1’ = 4-метилпиперазин-1-ил]
Способ A, стадия h
100 мг (0,18 ммоль) N-{3-[5-Бром-2-(4-метилпиперазин-1-ил)тиазол-4-ил]-2,4-дифторфенил}-2,5-дифторбензолсульфонамида растворяли в 9 мл DME и 1 мл воды. К смеси добавляли 170 мг (0,54 ммоль) Cs2CO3, 116 мг (0,35 ммоль) 4-пиридилборпинаколята и 42 мг (0,05 ммоль) PdCl2(dppf)2·CH2Cl2, а затем ее подвергали облучению микроволновым излучением при 110°C в течение 1 ч. Неочищенное вещество фильтровали через слой целита и фильтрат упаривали. Осадок отбирали в DCM и промывали 15% NH4OH. Органический слой сушили над Na2SO4 и упаривали. В конце продукт очищали флэш-хроматографией на предварительно покрытой колонке с силикагелем, элюируя DCM-MeOH 95/5 с получением 30 мг (30%) указанного в заголовке соединения.
ВЭЖХ: Rt: 5,44 мин
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 8,38-8,44 (м, 2H), 7,53-7,62 (м, 1H), 7,38-7,52 (м, 3H), 7,19 (тд, J=1,4, 8,8 Гц, 1H), 6,79-6,96 (м, 2H), 3,64 (ушир.с, 4H), 3,15 (ушир.с, 4H), 2,73 (ушир.с, 3H)
HRMS (ESI): вычислено для C25H21F4N5O2S2 [M+H]+ 523,0516, найдено 523,0508.
Аналогично получали следующие соединения:
N-{2,4-Дифтор-3-[2-(метиламино)-5-(пиридин-4-ил)-1,3-тиазол-4-ил]фенил}-2,5-дифторбензолсульфонамид, соединение формулы (I)A (соединение 2) [m, n = 1; R2, R3, Rx = H; R4 = F; R5 = 4-F; R6 = 2,5-дифторфенил; R1’ = метиламино]
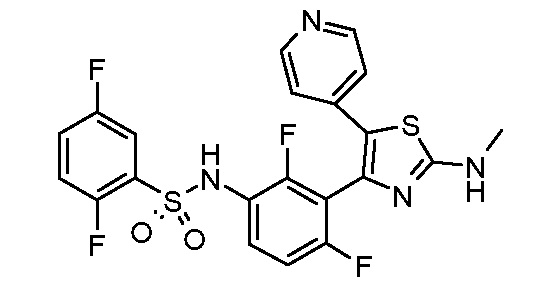
ВЭЖХ: Rt: 5,55 мин
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 10,67 (ушир.с, 1H), 8,29-8,44 (м, 2H), 8,14 (кв, J=4,8 Гц, 1H), 7,52-7,61 (м, 1H), 7,38-7,52 (м, 3H), 7,19 (тд, J=1,5, 8,8 Гц, 1H), 6,83-6,91 (м, 2H), 2,85 (д, J=4,8 Гц, 3H)
HRMS (ESI): вычислено для C21H14F4N4O2S2 [M+H]+ 495,0567, найдено 495,0548.
N-{3-[2-Амино-5-(пиридин-4-ил)-1,3-тиазол-4-ил]-2,4-дифторфенил}-2,5-дифторбензолсульфонамид, соединение формулы (I)U1 (соединение 1) [m, n = 1; R2, R3, Rx = H; R4 = F; R5 = 4-F; R6 = 2,5-дифторфенил]
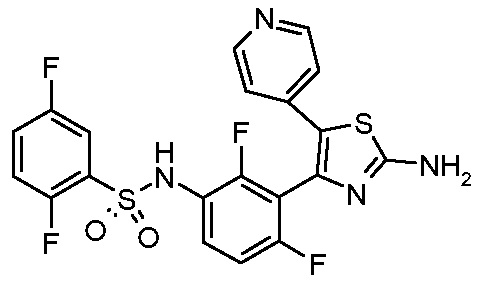
ВЭЖХ: Rt: 5,15 мин
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 10,67 (ушир.с, 1H), 8,33 (д, J=5,7 Гц, 2H), 7,59-7,51 (м, 3H), 7,50-7,35 (м, 3H), 7,20-7,11 (м, 1H), 6,82 (д, J=5,9 Гц, 2H)
HRMS (ESI): вычислено для C20H12F4N4O2S2 [M+H]+ 481,0411, найдено 481,0394.
Пример 2
Синтез N-[4-(3-{[(2,5-дифторфенил)сульфонил]амино}-2,6-дифторфенил)-5-(пиридин-4-ил)-1,3-тиазол-2-ил]ацетамида, соединение формулы (I)M (соединение 33) [m, n = 1; R2, R3 = H; R4 = F; R5 = 4-F; R6 = 2,5-дифторфенил; R10 = метил]
Способ B, стадии g и b5
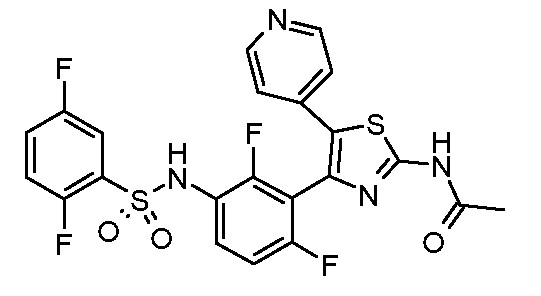
50 мг (0,104 ммоль) N-{3-[2-амино-5-(пиридин-4-ил)-1,3-тиазол-4-ил]-2,4-дифторфенил}-2,5-дифторбензолсульфонамида растворяли в 5 мл уксусного ангидрида и раствор перемешивали при к.т. в течение 3 суток. Смесь выливали в насыщенный водный раствор NaHCO3 и экстрагировали несколько раз DCM. Органический слой сушили над Na2SO4 и упаривали. Неочищенное вещество растворяли в 5 мл MeOH и добавляли 2 мл TEA. Раствор перемешивали в течение 3 суток при к.т. Затем растворитель удаляли, осадок отбирали в DCM и промывали водой. Органический слой сушили над Na2SO4 и упаривали с получением 30 мг (58%) указанного в заголовке соединения, растертого с диизопропиловым эфиром-петролейным эфиром.
ВЭЖХ: Rt: 5,23 мин
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 12,48 (с, 1H), 8,40-8,48 (м, 2H), 7,30-7,62 (м, 4H), 7,12 (ушир.с, 1H), 7,02-7,07 (м, 2H), 2,19 (с, 3H)
HRMS (ESI): вычислено для C22H14F4N4O3S2 [M+H]+ 523,0516, найдено 523,0497.
Пример 3
Синтез 2,5-дифтор-N-{2-фтор-3-[2-(1-метилпиперидин-4-ил)-5-(пиридин-4-ил)-1,3-тиазол-4-ил]фенил}бензолсульфонамида, соединение формулы (I)R2 (соединение 19) [m, n = 1; R2, R3, R5 = H; R4 = F, R6 = 2,5-дифторфенил; R’’ = метил]

N-[3-(Бромацетил)-2-фторфенил]-2,5-дифтор-N-(метоксиметил)бензолсульфонамид, соединение формулы 7 [n = 1; R5 = H; R4 = F; R6 = 2,5-дифторфенил; Rx = метоксиметил]
Способ A, стадия d1
Условия 1
100 мг (0,27 ммоль) N-(3-Ацетил-2-фторфенил)-2,5-дифтор-N-(метоксиметил)бензолсульфонамида растворяли в 10 мл сухого THF и добавляли 100 мг (0,3 ммоль) пиридиния бромида пербромида. Полученный раствор нагревали в микроволновом устройстве при 80°C в течение 15 мин. Затем растворитель выпаривали, осадок отбирали в DCM и промывали 0,5н HCl. Затем органический слой сушили над Na2SO4 и упаривали с получением указанного в заголовке соединения в виде масла.
Условия 2
100 мг (0,27 ммоль) N-(3-ацетил-2-фторфенил)-2,5-дифтор-N-(метоксиметил)бензолсульфонамида растворяли в 4 мл сухого DCM в атмосфере азота. Раствор охлаждали до 0°C и добавляли 94 мкл (0,65 ммоль) TEA и 106 мкл (0,54 ммоль) триметилсилил-трифторметансульфоната. Раствор перемешивали в течение 15 мин при той же температуре, а затем разбавляли DCM и быстро промывали насыщенным солевым раствором. Органический слой сушили над Na2SO4 и упаривали с получением 2,5-дифтор-N-(2-фтор-3-{1-[(триметилсилил)окси]этенил}фенил)-N-(метоксиметил)бензолсульфонамида в виде прозрачного масла. Последнее перерастворяли в 4 мл сухого DCM в атмосфере азота и добавляли 58 мг (0,32 ммоль) N-бромсукцинимида при 0°C. Через 30 мин реакция завершалась. Смесь разбавляли DCM и промывали водным раствором NaHCO3. Органический слой сушили над Na2SO4 и упаривали с получением указанного в заголовке соединения, которое использовали без какой-либо дальнейшей очистки.
ВЭЖХ: Rt: 6,77 мин.
ВЭЖХ/MS (ESI): 469-471 [M+NH4]+
N-[3-(2-Амино-1,3-тиазол-4-ил)-2-фторфенил]-2,5-дифтор-N-(метоксиметил)бензолсульфонамид, соединение формулы 9 [n = 1; R1 = NH2; R4 = F; R5 = H; R6 = 2,5-дифторфенил; Rx = метоксиметил]
Способ A, стадия e
К раствору 122 мг (0,27 ммоль) N-[3-(бромацетил)-2-фторфенил]-2,5-дифтор-N-(метоксиметил)бензолсульфонамида в 5 мл этанола добавляли 21 мг (0,27 ммоль) тиомочевины. Смесь нагревали при 60°C в течение 20 мин в микроволновом устройстве. Затем растворитель удаляли, осадок отбирали в DCM и промывали водой. Органический слой отделяли, сушили над Na2SO4 и упаривали. Неочищенное вещество очищали флэш-хроматографией, элюируя DCM-CH3OH 95/5, с получением 92 мг (80%) указанного в заголовке соединения.
ВЭЖХ: Rt: 6,26 мин.
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 7,97 (тд, J=1,8, 7,5 Гц, 1H), 7,57-7,76 (м, 2H), 7,40-7,54 (м, 1H), 7,21-7,30 (м, 1H), 7,13-7,19 (м, 1H), 7,11 (с, 2H), 6,82 (д, J=2,6 Гц, 1H), 5,09 (с, 2H), 3,37 (с, 3H).
HRMS (ESI): вычислено для C17H15F3N3O3S2 [M+H]+ 430,0505, найдено 430,0493.
N-[3-(2-Амино-5-бром-1,3-тиазол-4-ил)-2-фторфенил]-2,5-дифтор-N-(метоксиметил)бензолсульфонамид, соединение формулы 10 [n = 1; R1 = NH2; R4 = F; R5 = H; R6 = 2,5-дифторфенил; Rx = метоксиметил; Hal = Br]
Способ A, стадия f
1,26 г (2,94 ммоль) N-[3-(2-амино-1,3-тиазол-4-ил)-2-фторфенил]-2,5-дифтор-N-(метоксиметил)бензолсульфонамида растворяли в 30 мл сухого DCM и добавляли 523 мг (2,94 ммоль) N-бромсукцинимида. Полученный раствор перемешивали при к.т. в течение 30 мин. Смесь разбавляли тем же растворителем и промывали водным раствором NaHCO3. Органический слой сушили над Na2SO4 и упаривали. Осадок растирали с диизопропиловым эфиром и фильтровали с получением 1,4 г (93%) указанного в заголовке соединения.
ВЭЖХ: Rt: 6,52 мин.
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 7,57-7,71 (м, 2H), 7,44-7,55 (м, 2H), 7,31-7,42 (м, 2H), 7,24-7,32 (м, 1H), 4,96-5,21 (м, 2H), 3,36-3,39 (м, 3H).
N-[3-(5-Бром-2-{[(E)-(диметиламино)метилиден]амино}-1,3-тиазол-4-ил)-2-фторфенил]-2,5-дифтор-N-(метоксиметил)бензолсульфонамид, соединение формулы 11 [n = 1; R4 = F; R5 = H; R6 = 2,5-дифторфенил; Rx = метоксиметил; PG’ = диметиламинометилен; Hal = Br]
Способ A, стадия g
1,4 г (2,75 ммоль) N-[3-(2-Амино-5-бром-1,3-тиазол-4-ил)-2-фторфенил]-2,5-дифтор-N-(метоксиметил)бензолсульфонамида растворяли в 30 мл сухого DMF и к смеси добавляли 448 мкл (2,75 ммоль) диметилацеталя диметилформамида. Реакционную смесь перемешивали при к.т. в течение ночи, а затем растворитель удаляли при пониженном давлении. Осадок отбирали в DCM и промывали насыщенным солевым раствором. Затем органический слой сушили над Na2SO4 и упаривали. Наконец, неочищенное вещество очищали флэш-хроматографией, элюируя смесью циклогексан-этанол 9/1, с получением 0,8 г (52%) указанного в заголовке соединения.
ВЭЖХ: Rt: 7,18 мин.
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 8,25 (с, 1H), 7,72-7,57 (м, 2H), 7,57-7,52 (м, 1H), 7,52-7,45 (м, 1H), 7,45-7,37 (м, 1H), 7,34-7,28 (м, 1H), 5,09 (с, 2H), 3,38 (с, 3H), 3,11 (с, 3H), 2,97 (с, 3H).
HRMS (ESI): вычислено для C20H19BrF3N4O3S2 [M+H]+ 563,0029, найдено 563,0049.
Аналогично, но начиная с подходящих аминотиазольных производных, получали следующие соединения:
N-[3-(5-Бром-2-{[(E)-(диметиламино)метилиден]амино}-1,3-тиазол-4-ил)-2,4-дифторфенил]-2,5-дифтор-N-(метоксиметил)бензолсульфонамид, соединение формулы 11 [n = 1; R4 = F; R5 = 4-F; R6 = 2,5-дифторфенил; Rx = метоксиметил; PG’ = диметиламинометилен; Hal = Br]
ВЭЖХ: Rt: 7,00 мин.
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 8,25 (с, 1H), 7,57-7,71 (м, 2H), 7,45-7,56 (м, 2H), 7,25-7,34 (м, 1H), 5,07 (с, 2H), 3,37 (с, 3H), 3,11 (с, 3H), 2,97 (с, 3H).
HRMS (ESI): вычислено для C20H18BrF4N4O3S2 [M+H]+ 580,9935, найдено 580,9921.
N-[3-(5-Бром-2-{[(E)-(диметиламино)метилиден]амино}-1,3-тиазол-4-ил)-2,4-дифторфенил]-2,5-дифторбензолсульфонамид, соединение формулы 11 [n = 1; R4 = F; R5 = 4-F; R6 = 2,5-дифторфенил; Rx = H; PG’ = диметиламинометилен; Hal = Br]
ВЭЖХ: Rt: 6,47 мин.
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 10,70 (с, 1H), 8,24 (с, 1H), 7,65-7,59 (м, 1H), 7,59-7,52 (м, 1H), 7,51-7,46 (м, 1H), 7,42 (дт, J=5,9, 8,9 Гц, 1H), 7,24-7,17 (м, 1H), 3,11 (с, 3H), 2,97 (с, 3H)
HRMS (ESI): вычислено для C18H14BrF4N4O2S2 [M+H]+ 536,9672, найдено 536,9646.
N-{3-[2-{[(E)-(Диметиламино)метилиден]амино}-5-(пиридин-4-ил)-1,3-тиазол-4-ил]-2-фторфенил}-2,5-дифтор-N-(метоксиметил)бензолсульфонамид, соединение формулы 13 [m, n = 1; R2, R3, R5 = H; R4 = F; R6 = 2,5-дифторфенил; Rx = метоксиметил; PG’ = диметиламинометилен]
Способ A, стадия h1
К раствору 342 мг (0,61 ммоль) N-[3-(5-бром-2-{[(E)-(диметиламино)метилиден]амино}-1,3-тиазол-4-ил)-2-фторфенил]-2,5-дифтор-N-(метоксиметил)бензолсульфонамида в смеси 12 мл DME и 2 мл воды последовательно добавляли 596 мг (1,83 ммоль) Cs2CO3, 160 мг (0,2 ммоль) PdCl2(dppf)2·CH2Cl2 и 244 мг (1,22 ммоль) 4-пиридилборпинаколята. Смесь нагревали в микроволновой печи при 110°C в течение 1 ч, а затем фильтровали через слой целита. Фильтрат упаривали при пониженном давлении, неочищенное вещество отбирали в DCM и промывали насыщенным солевым раствором. Органический слой отделяли, сушили над Na2SO4 и растворитель удаляли. Продукт очищали флэш-хроматографией, элюируя DCM-MeOH 98/2 с получением 240 мг (70%) указанного в заголовке соединения.
ВЭЖХ: Rt: 6,30 мин.
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 8,39 (с, 1H), 8,40-8,37 (м, 2H), 7,68-7,59 (м, 1H), 7,59-7,52 (м, 2H), 7,50-7,45 (м, 1H), 7,38-7,25 (м, 2H), 7,03-6,90 (м, 2H), 4,98 (с, 2H), 3,27 (с, 3H), 3,14 (с, 3H), 3,01 (с, 3H).
HRMS (ESI): вычислено для C25H23F3N5O3S2 [M+H]+ 562,1189, найдено 562,1183.
Аналогично, но начиная с подходящего бромтиазольного производного, получали следующее соединение:
N-{3-[2-{[(E)-(Диметиламино)метилиден]амино}-5-(пиридин-4-ил)-1,3-тиазол-4-ил]-2,4-дифторфенил}-2,5-дифтор-N-(метоксиметил)бензолсульфонамид, соединение формулы 13 [m, n = 1; R2, R3, = H; R4 = F; R5 = 4-F; R6 = 2,5-дифторфенил; Rx = метоксиметил; PG’ = диметиламинометилен]
ВЭЖХ: Rt: 6,20 мин.
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 8,40-8,44 (м, 2H), 8,38 (с, 1H), 7,58-7,68 (м, 1H), 7,42-7,58 (м, 4H), 7,22-7,30 (м, 1H), 6,93-6,99 (м, 2H), 5,01-5,04 (м, 2H), 5,03 (с, 2H), 3,14 (с, 3H), 3,01 (с, 3H).
HRMS (ESI): вычислено для C25H22F4N5O3S2 [M+H]+ 580,1095, найдено 580,1072.
N-{3-[2-Амино-5-(пиридин-4-ил)-1,3-тиазол-4-ил]-2-фторфенил}-2,5-дифтор-N-(метоксиметил)бензолсульфонамид, соединение формулы (I)U1 [m, n = 1; R2, R3, R5 = H; R4 = F; R6 = 2,5-дифторфенил; Rx = метоксиметил]
Способ A, стадия i
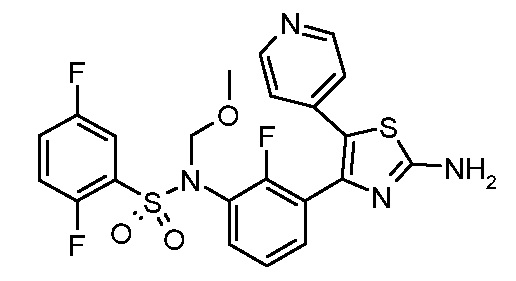
550 мг (0,98 ммоль) N-{3-[2-{[(E)-(диметиламино)метилиден]амино}-5-(пиридин-4-ил)-1,3-тиазол-4-ил]-2-фторфенил}-2,5-дифтор-N-(метоксиметил)бензолсульфонамида суспендировали в 15 мл этанола и добавляли 460 мкл (6,86 ммоль) этилендиамина. Смесь поддерживали при кипячении с обратным холодильником с получением прозрачного раствора. Через 8 ч растворитель удаляли при пониженном давлении и осадок перерастворяли в DCM и промывали насыщенным солевым раствором. Органическую фазу сушили над Na2SO4 и растворитель выпаривали. Неочищенное вещество растирали с диэтиловым эфиром с получением после фильтрации 450 мг (90%) указанного в заголовке соединения.
ВЭЖХ: Rt: 5,68 мин.
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 8,15-8,35 (м, 2H), 7,58-7,66 (м, 1H), 7,42-7,57 (м, 5H), 7,24-7,36 (м, 2H), 6,84-6,91 (м, 2H), 4,99 (с, 2H), 3,27 (с, 3H).
HRMS (ESI): вычислено для C22H18F3N4O3S2 [M+H]+ 507,0767, найдено 507,0769.
Аналогично, но начиная с подходящего иминопроизводного, получали следующее соединение:
N-{3-[2-Амино-5-(пиридин-4-ил)-1,3-тиазол-4-ил]-2,4-дифторфенил}-2,5-дифтор-N-(метоксиметил)бензолсульфонамид, соединение формулы (I)U1 [m, n = 1; R2, R3, = H; R4 = F; R5 = 4-F; R6 = 2,5-дифторфенил; Rx = метоксиметил]

ВЭЖХ: Rt: 5,62 мин.
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 8,34-8,38 (м, 2H), 7,59-7,67 (м, 2H), 7,49-7,59 (м, 5H), 7,41-7,49 (м, 1H), 7,20-7,27 (м, 1H), 6,85-6,90 (м, 2H), 5,02 (с, 2H)
HRMS (ESI): вычислено для C22H17F4N4O3S2 [M+H]+ 525,0673, найдено 525,0659.
N-{3-[2-Бром-5-(пиридин-4-ил)-1,3-тиазол-4-ил]-2-фторфенил}-2,5-дифтор-N-(метоксиметил)бензолсульфонамид, соединение формулы (I)F [m, n = 1; R2, R3, R5 = H; R4 = F; R6 = 2,5-дифторфенил; Rx = метоксиметил; Hal = Br]
Способ B, стадия c
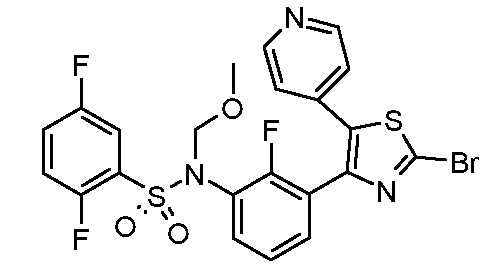
450 мг (0,89 ммоль) N-{3-[2-амино-5-(пиридин-4-ил)-1,3-тиазол-4-ил]-2-фторфенил}-2,5-дифтор-N-(метоксиметил)бензолсульфонамида суспендировали в 45 мл сухого ацетонитрила и добавляли 298 мг (1,34 ммоль) CuBr2 и 2 мл (16,8 ммоль) трет-бутилнитрита. Смесь перемешивали при 85°C в течение 8 ч. После этого реакционную смесь фильтровали через слой целита и фильтрат упаривали. Осадок отбирали в DCM и промывали водным раствором NaHCO3. Органический слой отделяли, сушили над Na2SO4 и упаривали. Неочищенное вещество очищали флэш-хроматографией, элюируя DCM-MeOH 98/2, с получением 350 мг (69%) указанного в заголовке соединения.
ВЭЖХ: Rt: 7,14 мин.
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 8,52 (д, J=5,2 Гц, 2H), 7,57-7,70 (м, 2H), 7,42-7,57 (м, 2H), 7,25-7,42 (м, 2H), 7,14 (д, J=6,0 Гц, 2H), 4,97 (с, 2H), 3,23 (с, 3H)
HRMS (ESI): вычислено для C22H16BrF3N3O3S2 [M+H]+ 569,9763, найдено 569,9789.
Аналогично, но начиная с соответствующего производного аминотиазола, получали следующее соединение:
N-{3-[2-Бром-5-(пиридин-4-ил)-1,3-тиазол-4-ил]-2,4-дифторфенил}-2,5-дифтор-N-(метоксиметил)бензолсульфонамид, соединение формулы (I)F [m, n = 1; R2, R3, = H; R4 = F; R5 = 4-F; R6 = 2,5-дифторфенил; Rx = метоксиметил; Hal = Br]
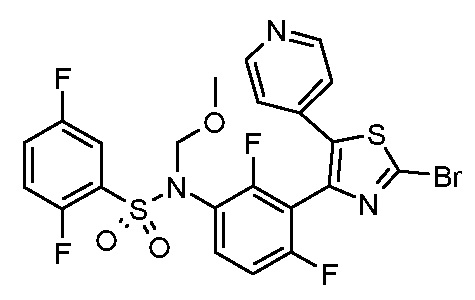
ВЭЖХ: Rt: 6,95 мин.
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 8,53-8,59 (м, 2H), 7,60-7,68 (м, 1H), 7,45-7,59 (м, 3H), 7,26-7,34 (м, 1H), 7,12-7,18 (м, 2H), 5,01 (с, 2H), 3,27 (с, 3H)
HRMS (ESI): вычислено для C22H15BrF4N3O3S2 [M+H]+ 587,9669, найдено 587,9645.
трет-Бутил-4-[4-(3-{[(2,5-дифторфенил)сульфонил](метоксиметил)амино}-2-фторфенил)-5-(пиридин-4-ил)-1,3-тиазол-2-ил]-3,6-дигидропиридин-1(2H)-карбоксилат, соединение формулы (I)H1 [m, n = 1; R2, R3, R5 = H; R4 = F; R6 = 2,5-дифторфенил; Rx = метоксиметил; X = NCOOt-Bu]
Способ C, стадия a
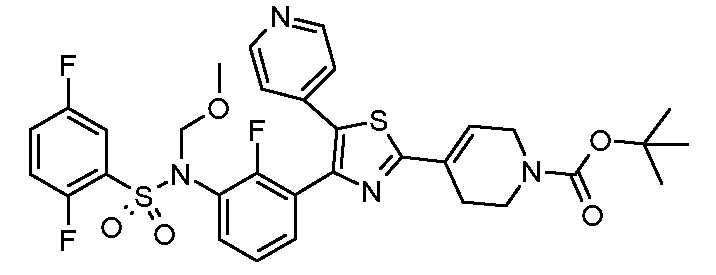
200 мг (0,35 ммоль) N-{3-[2-Бром-5-(пиридин-4-ил)-1,3-тиазол-4-ил]-2-фторфенил}-2,5-дифтор-N-(метоксиметил)бензолсульфонамида растворяли в смеси 7,8 мл DME и 1,2 мл воды. Добавляли 344 мг (1,05 ммоль) Cs2CO3, 216 мг (0,70 ммоль) трет-бутилового эфира 4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-3,6-дигидро-2H-пиридин-1-карбоновой кислоты и 96 мг (0,12 ммоль) PdCl2(dppf)2·CH2Cl2 и полученную смесь нагревали в микроволновой печи при 100°C в течение одного часа. Реакционную смесь фильтровали через слой целита и фильтрат упаривали. Затем осадок отбирали в DCM и промывали насыщенным солевым раствором. Органический слой сушили над Na2SO4 и упаривали. Наконец, неочищенное вещество очищали флэш-хроматографией, элюируя смесью циклогексан-этилацетат-этанол 4/1/0,5, с получением, после растирания с диэтиловым эфиром и фильтрации, 225 мг (85%) указанного в заголовке соединения.
ВЭЖХ: Rt: 7,91 мин.
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 8,28-8,65 (м, 2H), 7,49-7,73 (м, 4H), 7,46 (ддд, J=3,2, 5,1, 7,9 Гц, 1H), 7,24-7,40 (м, 2H), 7,03-7,19 (м, 2H), 4,97 (с, 2H), 3,96-4,08 (м, J=12,8 Гц, 2H), 3,21-3,24 (м, 3H), 2,92 (ушир.с, 2H), 2,08 (дд, J=3,4, 14,1 Гц, 2H), 1,60 (кв, д, J=4,3, 12,1 Гц, 2H), 1,41 (с, 9H)
HRMS (ESI): вычислено для C32H32F3N4O5S22 [M+H]+ 673,1761, найдено 673,1771.
Аналогично, но с использованием подходящего производного борпинаколята, получали следующее соединение:
N-{3-[2-(3,6-Дигидро-2H-пиран-4-ил)-5-(пиридин-4-ил)-1,3-тиазол-4-ил]-2-фторфенил}-2,5-дифтор-N-(метоксиметил)бензолсульфонамид, соединение формулы (I)H1 [m, n = 1; R2, R3, R5 = H; R4 = F; R6 = 2,5-дифторфенил; Rx = метоксиметил; X = O]
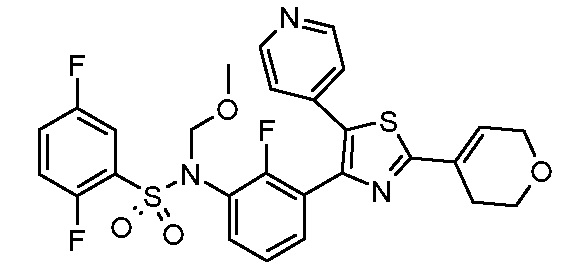
ВЭЖХ: Rt: 6,12 мин.
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 8,49 (д, J=6,1 Гц, 2H), 7,57-7,69 (м, 2H), 7,50-7,57 (м, 1H), 7,42-7,49 (м, 1H), 7,28-7,41 (м, 2H), 7,07-7,17 (м, 2H), 6,79 (с, 1H), 4,98 (с, 2H), 4,29 (д, J=2,8 Гц, 2H), 3,79-3,87 (м, 2H), 3,24 (с, 3H), 2,57 (ушир.с, 2H), 2,02-2,10 (м, 1H)
HRMS (ESI): вычислено для C27H23F3N3O4S2 [M+H]+ 574,1077, найдено 574,1086.
трет-Бутил-4-[4-(3-{[(2,5-дифторфенил)сульфонил](метоксиметил)амино}-2-фторфенил)-5-(пиридин-4-ил)-1,3-тиазол-2-ил]пиперидин-1-карбоксилат, соединение формулы (I)Q [m, n = 1; R2, R3, R5 = H; R4 = F; R6 = 2,5-дифторфенил; Rx = метоксиметил; X = NCOOt-Bu]
Способ C, стадия b
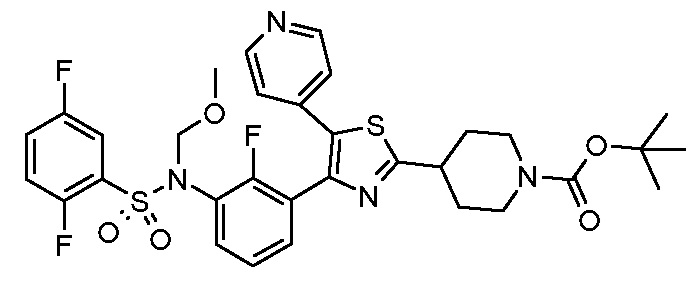
220 мг (0,33 ммоль) трет-бутил-4-[4-(3-{[(2,5-дифторфенил)сульфонил](метоксиметил)амино}-2-фторфенил)-5-(пиридин-4-ил)-1,3-тиазол-2-ил]-3,6-дигидропиридин-1(2H)-карбоксилата суспендировали в 30 мл MeOH и 880 мг (14 ммоль) формиата аммония и добавляли 50 мг Pd-C 10%. Реакционную смесь перемешивали при кипячении с обратным холодильником в течение 2 суток. После этого смесь фильтровали через слой целита и фильтрат упаривали. Неочищенное вещество отбирали в DCM и промывали насыщенным солевым раствором. Органический слой сушили над Na2SO4 и упаривали. Затем неочищенное вещество растирали со смесью диэтиловый эфир-петролейный эфир и собирали фильтрацией с получением 200 мг (91%) указанного в заголовке соединения.
ВЭЖХ: Rt: 7,72 мин.
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 8,28-8,65 (м, 2H), 7,49-7,73 (м, 4H), 7,46 (ддд, J=3,2, 5,1, 7,9 Гц, 1H), 7,24-7,40 (м, 2H), 7,03-7,19 (м, 2H), 4,97 (с, 2H), 3,96-4,08 (м, J=12,8 Гц, 2H), 3,21-3,24 (м, 3H), 2,92 (ушир.с, 2H), 2,08 (дд, J=3,4, 14,1 Гц, 2H), 1,60 (кв, д, J=4,3, 12,1 Гц, 2H), 1,41 (с, 9H)
HRMS (ESI): вычислено для C32H34F3N4O5S2 [M+H]+ 675,1917, найдено 675,1938.
Аналогично, получали следующее соединение из подходящего ненасыщенного производного:
2,5-Дифтор-N-{2-фтор-3-[5-(пиридин-4-ил)-2-(тетрагидро-2H-пиран-4-ил)-1,3-тиазол-4-ил]фенил}-N-(метоксиметил)бензолсульфонамид, соединение формулы (I)Q [m, n = 1; R2, R3, R5 = H; R4 = F; R6 = 2,5-дифторфенил; Rx = метоксиметил; X = O]
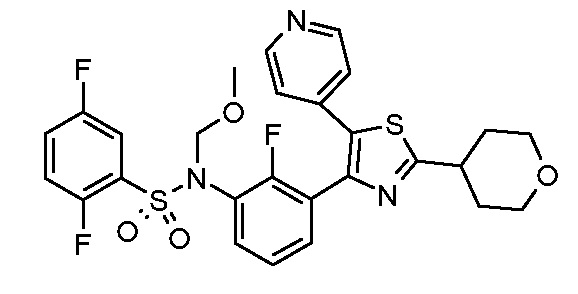
ВЭЖХ: Rt: 5,83 мин.
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 8,46-8,50 (м, J=4,9 Гц, 2H), 7,41-7,67 (м, 4H), 7,28-7,38 (м, 2H), 7,07-7,17 (м, 3H), 4,97 (с, 2H), 3,91-3,99 (м, 1H), 3,44-3,52 (м, 1H), 1,98-2,06 (м, 2H), 1,70-1,84 (м, 2H)
HRMS (ESI): вычислено для C27H25F3N3O4S2 [M+H]+ 576,1233, найдено 576,1252.
2,5-Дифтор-N-{2-фтор-3-[2-(пиперидин-4-ил)-5-(пиридин-4-ил)-1,3-тиазол-4-ил]фенил}-N-(метоксиметил)бензолсульфонамид, соединение формулы 47 [m, n = 1; R2’’, R3, R5 = H; R4 = F; R6 = 2,5-дифторфенил; Rx = метоксиметил], соответствующее соединению формулы (I), где m, n = 1; R1 = 4-пиперидинил; R2, R3, R5 = H; R4 = F; R6 = 2,5-дифторфенил; Rx = метоксиметил.
Способ C, стадия d
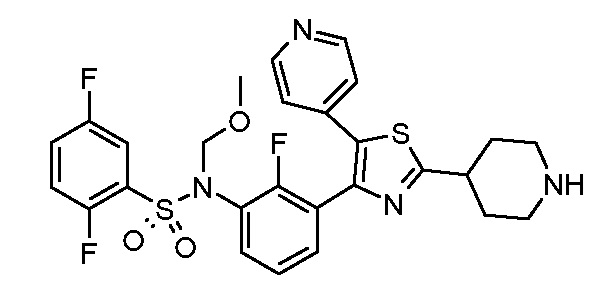
200 мг (0,3 ммоль) трет-бутил-4-[4-(3-{[(2,5-дифторфенил)сульфонил](метоксиметил)амино}-2-фторфенил)-5-(пиридин-4-ил)-1,3-тиазол-2-ил]пиперидин-1-карбоксилата растворяли в 20 мл сухого DCM и добавляли 2 мл TFA. После 3 ч при к.т. растворитель удаляли при 30°C. Осадок отбирали в DCM, промывали 15% NH4OH и экстрагировали несколько раз смесью DCM-MeOH 9/1. Органический слой отделяли, сушили над Na2SO4 и упаривали. Осадок растирали с диэтиловым эфиром с получением после фильтрации 155 мг (91%) указанного в заголовке соединения.
ВЭЖХ: Rt: 4,82 мин.
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 8,48 (д, J=6,1 Гц, 2H), 7,42-7,69 (м, 5H), 7,27-7,39 (м, 2H), 7,04-7,15 (м, 2H), 4,97 (с, 2H), 3,23 (с, 3H), 3,15 (тт, J=3,6, 11,6 Гц, 1H), 2,96-3,08 (м, J=8,9 Гц, 2H), 2,60-2,69 (м, 2H), 1,97-2,06 (м, J=11,1 Гц, 2H), 1,63 (кв, д, J=3,8, 12,1 Гц, 2H)
HRMS (ESI): вычислено для C27H26F3N4O3S2 [M+H]+ 575,1393, найдено 575,1418.
2,5-Дифтор-N-{2-фтор-3-[2-(1-метилпиперидин-4-ил)-5-(пиридин-4-ил)-1,3-тиазол-4-ил]фенил}-N-(метоксиметил)бензолсульфонамид, соединение формулы 48 [m, n = 1; R2’’, R3, R5 = H; R4 = F; R6 = 2,5-дифторфенил; Rx = метоксиметил; R’’ = метил], соответствующий соединению формулы (I), где m, n = 1; R1 = 1-метил-4-пиперидинил; R2, R3, R5 = H; R4 = F; R6 = 2,5-дифторфенил; Rx = метоксиметил.
Способ C, стадия e
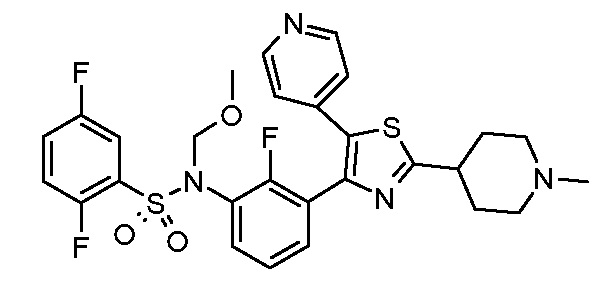
150 мг (0,26 ммоль) 2,5-дифтор-N-{2-фтор-3-[2-(пиперидин-4-ил)-5-(пиридин-4-ил)-1,3-тиазол-4-ил]фенил}-N-(метоксиметил)бензолсульфонамида растворяли в 15 мл MeOH и добавляли 45 мкл (0,78 ммоль) ледяной уксусной кислоты, 26 мг (0,52 ммоль) NaBH3CN и 20 мкл (0,39 ммоль) 37% формальдегида. Полученный раствор перемешивали при к.т. в течение 2 ч, а затем растворитель выпаривали. Осадок отбирали в DCM, промывали NH4OH 15% и экстрагировали несколько раз смесью DCM-MeOH 9/1. Органический слой сушили над Na2SO4 и упаривали, с получением 145 мг (94%) указанного в заголовке соединения.
ВЭЖХ: Rt: 4,91 мин
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 8,48 (д, J=6,0 Гц, 2H), 7,41-7,71 (м, 5H), 7,27-7,39 (м, 2H), 6,97-7,18 (м, 2H), 4,97 (с, 2H), 3,23 (с, 3H), 3,01 (тт, J=3,6, 11,5 Гц, 1H), 2,73-2,91 (м, J=11,5 Гц, 2H), 2,20 (с, 3H), 1,90-2,12 (м, 4H), 1,60-1,87 (м, 2H)
HRMS (ESI): вычислено для C28H28F3N4O3S2 [M+H]+ 589,1549, найдено 589,1551.
2,5-Дифтор-N-{2-фтор-3-[2-(1-метилпиперидин-4-ил)-5-(пиридин-4-ил)-1,3-тиазол-4-ил]фенил}бензолсульфонамид, соединение формулы (I)R2 (соединение 19) [m, n = 1; R2, R3, R5 = H; R4 = F; R6 = 2,5-дифторфенил; R’’ = метил]
Способ C, стадия c2
Раствор 140 мг (0,24 ммоль) 2,5-дифтор-N-{2-фтор-3-[2-(1-метилпиперидин-4-ил)-5-(пиридин-4-ил)-1,3-тиазол-4-ил]фенил}-N-(метоксиметил)бензолсульфонамида в 9 мл TFA и 1 мл воды нагревали при перемешивании при 80°C в течение 1,5 ч. Затем растворитель выпаривали, осадок перерастворяли в DCM, промывали 15% NH4OH и экстрагировали несколько раз смесью DCM-MeOH 9/1. Органический слой сушили над Na2SO4 и упаривали. Наконец, продукт очищали препаративной ОФ-ВЭЖХ, элюируя 0,05% NH4OH-CH3CN 95/5, с получением, после растирания со смесью диэтиловый эфир-диизопропиловый эфир, 70 мг (54%) указанного в заголовке соединения.
ВЭЖХ: Rt: 4,57 мин
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 8,46 (д, J=5,9 Гц, 2H), 7,35-7,43 (м, 2H), 7,21-7,34 (м, 2H), 7,07-7,16 (м, 2H), 7,00 (т, J=7,9 Гц, 1H), 6,80-6,95 (м, 1H), 2,97-3,17 (м, 4H), 2,42 (с, 4H), 2,10-2,21 (м, 2H), 1,68-1,96 (м, 2H)
HRMS (ESI): вычислено для C26H23F3N4O2S2 [M+H]+ 545,1287, найдено 545,1298.
Аналогично, но начиная с подходящего производного с защищенным сульфонамидом, получали следующее соединение:
2,5-Дифтор-N-{2-фтор-3-[5-(пиридин-4-ил)-2-(тетрагидро-2H-пиран-4-ил)-1,3-тиазол-4-ил]фенил}бензолсульфонамид, соединение формулы (I)R (соединение 20) [m, n = 1; R2, R3, R5 = H; R4 = F; R6 = 2,5-дифторфенил]
Способ C, стадия c
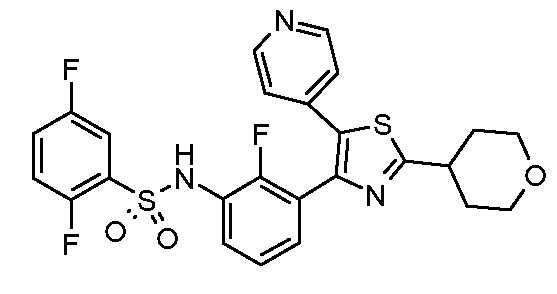
ВЭЖХ: Rt: 5,38 мин
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 10,66 (ушир.с, 1H), 8,46 (д, J=6,0 Гц, 2H), 7,51-7,61 (м, 1H), 7,27-7,49 (м, 4H), 7,17-7,26 (м, 1H), 7,07 (д, J=6,1 Гц, 2H), 3,94 (дт, J=2,3, 9,4 Гц, 2H), 3,47 (тд, J=1,9, 11,6 Гц, 2H), 1,94-2,05 (м, J=1,9, 12,8 Гц, 2H), 1,76 (кв, д, J=4,2, 12,2 Гц, 2H)
HRMS (ESI): вычислено для C25H20F3N3O3S2 [M+H]+ 532,0971, найдено 532,0991.
Пример 4
N-{3-[2-Бром-5-(пиридин-4-ил)-1,3-тиазол-4-ил]-2-фторфенил}-2,5-дифторбензолсульфонамид, соединение формулы (I)G [m, n = 1; R2, R3, R5 = H; R4 = F; R6 = 2,5-дифторфенил; Hal = Br]
Способ B, стадия b1
N-{3-[2-Бром-5-(пиридин-4-ил)-1,3-тиазол-4-ил]-2,4-дифторфенил}-2,5-дифтор-N-(метоксиметил)бензолсульфонамид (полученный, как описано в примере 3, 174 мг, 0,305 ммоль) растворяли в 6 мл TFA. Добавляли воду (0,5 мл) и смесь перемешивали при 80°C в течение 6 ч. Растворитель концентрировали при пониженном давлении и осадок отбирали в DCM и промывали насыщенным водным NaHCO3 и насыщенным солевым раствором. Органическую фазу сушили над Na2SO4 и упаривали до сухого состояния. Неочищенный продукт обрабатывали петролейным эфиром, фильтровали и сушили в высоком вакууме с получением 130 мг (80%) указанного в заголовке соединения в виде твердого вещества белого цвета.
ВЭЖХ: Rt: 5,81 мин
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 10,71 (с, 1H), 8,53 (д, J=4,8 Гц, 2H), 7,32-7,72 (м, 5H), 7,26 (кв, J=7,7 Гц, 1H), 7,11-7,19 (м, 2H)
HRMS (ESI): вычислено для C20H11BrF3N3O2S2 [M+H]+ 525,9501, найдено 525,9508.
Пример 5
Синтез N-{3-[2-(4,4-дифторпиперидин-1-ил)-5-(пиридин-4-ил)-1,3-тиазол-4-ил]-2-фторфенил}-2,5-дифторбензолсульфонамид, соединение формулы (I)U (соединение 18) [m, n = 1; R2, R3, R5, Rx = H; R4 = F; R6 = 2,5-дифторфенил; R7-R8 = -(CH2CH2)2CF2]
Способ B, стадия e
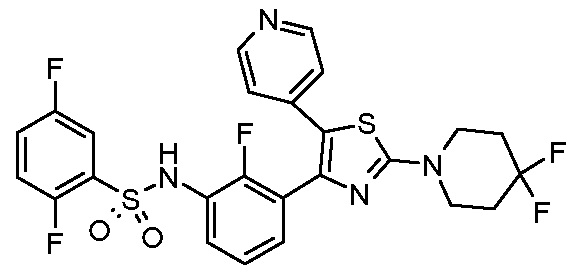
47 мг (0,09 ммоль) N-{3-[2-Бром-5-(пиридин-4-ил)-1,3-тиазол-4-ил]-2-фторфенил}-2,5-дифторбензолсульфонамида растворяли в 3 мл диметилацетамида и к смеси добавляли 100 мкл (1,39 ммоль) TEA и 100 мг (0,6 ммоль) 4,4-дифторпиперидина гидрохлорида. Раствор нагревали в микроволновой печи при 120°C в течение 3 ч. После этого растворитель удаляли при пониженном давлении, осадок отбирали в DCM и промывали водным раствором NaHCO3. Органическую фазу сушили над Na2SO4 и упаривали. Затем неочищенное вещество очищали препаративной ОФ-ВЭЖХ, элюируя 0,05% NH4OH-CH3CN 95/5, с получением 30 мг (59%) указанного в заголовке соединения.
ВЭЖХ: Rt: 6,1 мин
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 10,66 (ушир.с, 1H), 8,35 (д, J=5,7 Гц, 2H), 7,55 (ушир.с, 1H), 7,39-7,49 (м, 2H), 7,32-7,37 (м, 1H), 7,10-7,30 (м, 2H), 6,73-6,96 (м, 2H), 3,59-3,73 (м, 4H), 2,03-2,19 (м, 4H)
HRMS (ESI): вычислено для C25H19F5N4O2S2 [M+H]+ 567,0943, найдено 567,0963.
Аналогично, но с использованием надлежащих коммерческих аминопроизводных, получали следующие соединения:
N-{3-[2-(1,4-Диокса-8-азаспиро[4.5]дец-8-ил)-5-(пиридин-4-ил)-1,3-тиазол-4-ил]-2-фторфенил}-2,5-дифторбензолсульфонамид, соединение формулы (I)U (соединение 17) [m, n = 1; R2, R3, R5, Rx = H; R4 = F; R6 = 2,5-дифторфенил; R7-R8 = -(CH2CH2)2C(OCH2CH2O)]
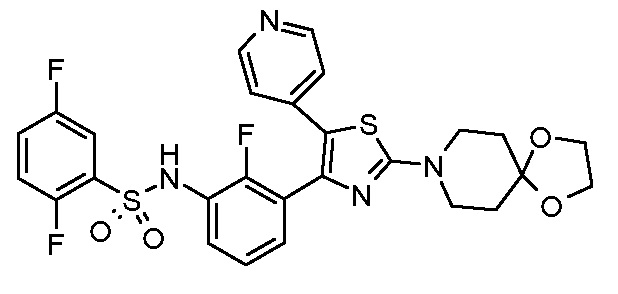
ВЭЖХ: Rt: 5,76 мин
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 10,66 (ушир.с, 1H), 8,18-8,40 (м, 2H), 7,52-7,61 (м, 1H), 7,40-7,48 (м, 2H), 7,36 (тд, J=1,8, 7,6 Гц, 1H), 7,25-7,31 (м, 1H), 7,17-7,24 (м, 1H), 6,83-6,90 (м, 2H), 3,93 (с, 4H), 3,54-3,60 (м, 4H), 1,70-1,78 (м, 4H)
HRMS (ESI): вычислено для C27H23F3N4O4S2 [M+H]+ 589,1186, найдено 589,1193.
N-{3-[2-(Диэтиламино)-5-(пиридин-4-ил)-1,3-тиазол-4-ил]-2-фторфенил}-2,5-дифторбензолсульфонамид, соединение формулы (I)U (соединение 16) [m, n = 1; R2, R3, R5, Rx = H; R4 = F; R6 = 2,5-дифторфенил; R7, R8 = этил]

ВЭЖХ: Rt: 6,17 мин
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 10,66 (ушир.с, 1H), 8,31 (д, J=6,1 Гц, 2H), 7,51-7,59 (м, 1H), 7,40-7,48 (м, 1H), 7,32-7,39 (м, J=1,8 Гц, 1H), 7,26-7,31 (м, 1H), 7,27 (д, J=5,6 Гц, 1H), 7,16-7,25 (м, 1H), 6,82-6,87 (м, 2H), 3,47 (кв, J=7,0 Гц, 4H), 1,14-1,23 (м, 6H)
HRMS (ESI): вычислено для C24H21F3N4O2S2 [M+H]+ 519,1131, найдено 519,1134.
Пример 6
Синтез N-{3-[2-(циклогексиламино)-5-(пиридин-4-ил)-1,3-тиазол-4-ил]-2,4-дифторфенил}-2,5-дифторбензолсульфонамида, соединение формулы (I)V [m, n = 1; R2, R3, R7 = H; R4 = F; R5 = 4-F; R6 = 2,5-дифторфенил; R8 = циклогексил]
Способ B, стадии e и b3
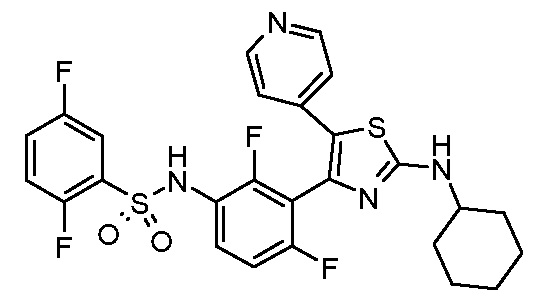
N-{3-[2-Бром-5-(пиридин-4-ил)-1,3-тиазол-4-ил]-2,4-дифторфенил}-2,5-дифтор-N-(метоксиметил)бензолсульфонамид (полученный, как описано в примере 3, 50 мг, 0,09 ммоль) растворяли в 3 мл диметилацетамида и добавляли 74 мкл (0,85 ммоль) циклогексиламина. Смесь перемешивали при 110°C в течение 16 ч. Растворитель выпаривали при пониженном давлении и осадок отбирали в DCM и промывали насыщенным солевым раствором. Органический слой сушили над Na2SO4 и упаривали с получением N-{3-[2-(циклогексиламино)-5-(пиридин-4-ил)-1,3-тиазол-4-ил]-2,4-дифторфенил}-2,5-дифтор-N-(метоксиметил)бензолсульфонамида. Последний без какой-либо дальнейшей очистки обрабатывали 4 мл TFA и 1 мл воды и перемешивали при 75°C в течение 8 ч. Затем растворитель удаляли, осадок отбирали в DCM и промывали водном раствором NaHCO3. Органический слой сушили над Na2SO4 и упаривали. Неочищенное вещество очищали флэш-хроматографией, элюируя смесью циклогексан-этанол 9/1, с получением 20 мг (42%) указанного в заголовке соединения.
ВЭЖХ: Rt: 7,08 мин
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 10,67 (ушир.с, 1H), 8,33 (д, J=5,6 Гц, 2H), 8,12 (д, J=7,6 Гц, 1H), 7,54 (тд, J=4,1, 8,1 Гц, 1H), 7,37-7,48 (м, 3H), 7,17 (т, J=9,1 Гц, 1H), 6,77-6,83 (м, 2H), 3,40-3,52 (м, J=7,8 Гц, 1H), 1,84-1,97 (м, J=3,1, 9,6 Гц, 2H), 1,65-1,78 (м, J=4,6 Гц, 2H), 1,48-1,60 (м, J=3,3 Гц, 1H), 1,09-1,37 (м, J=11,8 Гц, 6H)
HRMS (ESI): вычислено для C26H22F4N4O2S2 [M+H]+ 563,1193, найдено 563,1194.
Аналогично получали следующие соединения:
N-(2,4-Дифтор-3-{2-[(2-метоксиэтил)амино]-5-(пиридин-4-ил)-1,3-тиазол-4-ил}фенил)-2,5-дифторбензолсульфонамид, соединение формулы (I)V (соединение 5) [m, n = 1; R2, R3, R7 = H; R4 = F; R5 = 4-F; R6 = 2,5-дифторфенил; R8 = 2-метоксиэтил]
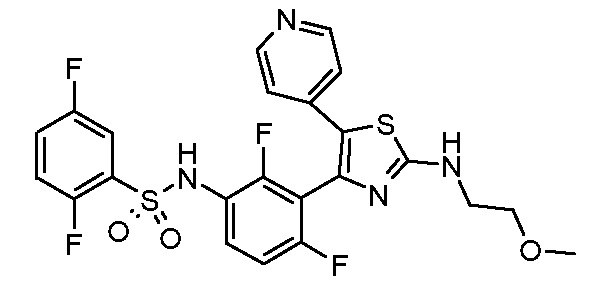
ВЭЖХ: Rt: 6,01 мин
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 10,67 (ушир.с, 1H), 8,34 (д, J=5,9 Гц, 2H), 8,18-8,28 (м, 1H), 7,51-7,61 (м, 1H), 7,36-7,51 (м, 3H), 7,17 (т, J=8,7 Гц, 1H), 6,82 (д, J=6,1 Гц, 2H), 3,45-3,51 (м, 2H), 3,39-3,44 (м, 2H), 3,27 (с, 3H)
HRMS (ESI): вычислено для C23H18F4N4O3S2 [M+H]+ 539,0829, найдено 539,0845.
N-(2,4-Дифтор-3-{5-(пиридин-4-ил)-2-[(пиридин-3-илметил)амино]-1,3-тиазол-4-ил}фенил)-2,5-дифторбензолсульфонамид, соединение формулы (I)V [m, n = 1; R2, R3, R7 = H; R4 = F; R5 = 4-F; R6 = 2,5-дифторфенил; R8 = 3-пиридилметил]
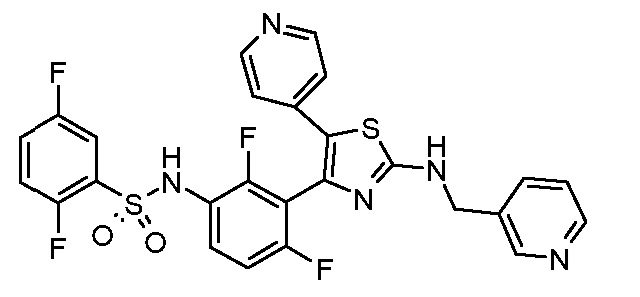
ВЭЖХ: Rt: 6,11 мин
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 8,66 (т, J=5,7 Гц, 1H), 8,57 (д, J=1,6 Гц, 1H), 8,50 (д, J=1,3 Гц, 1H), 8,35 (д, J=6,1 Гц, 2H), 7,70-7,85 (м, 1H), 7,50-7,57 (м, J=6,5 Гц, 1H), 7,32-7,48 (м, 4H), 7,16 (т, J=9,9 Гц, 1H), 6,79-6,86 (м, 2H), 4,48-4,54 (м, 2H)
HRMS (ESI): вычислено для C26H17F4N5O2S2 [M+H]+ 572,0833, найдено 572,0829.
N-(2,4-Дифтор-3-{2-[(2-метилпропил)амино]-5-(пиридин-4-ил)-1,3-тиазол-4-ил}фенил)-2,5-дифторбензолсульфонамид, соединение формулы (I)V (соединение 4) [m, n = 1; R2, R3, R7 = H; R4 = F; R5 = 4-F; R6 = 2,5-дифторфенил; R8 = изобутил]
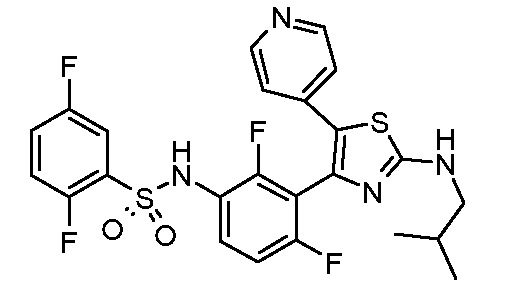
ВЭЖХ: Rt: 6,75 мин
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 10,67 (ушир.с, 1H), 8,33 (д, J=6,1 Гц, 2H), 8,20 (т, J=5,6 Гц, 1H), 7,33-7,60 (м, 5H), 7,15 (ушир.с, 1H), 6,78-6,85 (м, 2H), 3,05 (дд, J=5,9, 6,7 Гц, 2H), 1,80-1,94 (м, 1H), 0,87-0,94 (м, 6H)
HRMS (ESI): вычислено для C24H20F4N4O2S2 [M+H]+ 537,1037, найдено 537,1043.
N-(3-{2-[Циклогексил(метил)амино]-5-(пиридин-4-ил)-1,3-тиазол-4-ил}-2,4-дифторфенил)-2,5-дифторбензолсульфонамид, соединение формулы (I)V (соединение 7) [m, n = 1; R2, R3 = H; R4 = F; R5 = 4-F, R6 = 2,5-дифторфенил; R7 = метил; R8 = циклогексил]

ВЭЖХ: Rt: 7,64 мин
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 10,68 (ушир.с, 1H), 8,34 (д, J=6,0 Гц, 2H), 7,35-7,61 (м, 4H), 7,11-7,20 (м, 1H), 6,79-6,87 (м, 2H), 3,70-3,84 (м, 1H), 2,95 (с, 3H), 1,77 (ушир.с, 4H), 1,57 (ушир.с, 3H), 1,35 (ушир.с, 2H), 1,06-1,23 (м, 1H)
HRMS (ESI): вычислено для C27H24F4N4O2S2 [M+H]+ 577,135, найдено 577,1358.
N-{2,4-Дифтор-3-[2-(4-оксопиперидин-1-ил)-5-(пиридин-4-ил)-1,3-тиазол-4-ил]фенил}-2,5-дифторбензолсульфонамид соединение формулы (I)V (соединение 12) [m, n = 1; R2, R3, R4 = F; R5 = 4-F; R6 = 2,5-дифторфенил; R7-R8 = -(CH2CH2)2CO]
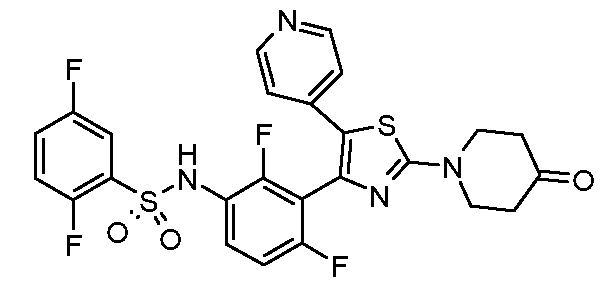
ВЭЖХ: Rt: 6,14 мин
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 8,25-8,44 (м, 2H), 7,49 (с, 4H), 7,05 (т, J=7,6 Гц, 1H), 6,91 (д, J=6,2 Гц, 2H), 3,84 (т, J=6,3 Гц, 4H), 2,56 (т, J=6,3 Гц, 4H)
HRMS (ESI): вычислено для C25H18F4N4O3S2 [M+H]+ 563,0829, найдено 563,083.
N-{3-[2-(Диэтиламино)-5-(пиридин-4-ил)-1,3-тиазол-4-ил]-2,4-дифторфенил}-2,5-дифторбензолсульфонамид, соединение формулы (I)V (соединение 3) [m, n = 1; R2, R3 = H; R4 = F; R5 = 4-F; R6 = 2,5-дифторфенил; R7, R8 = этил]
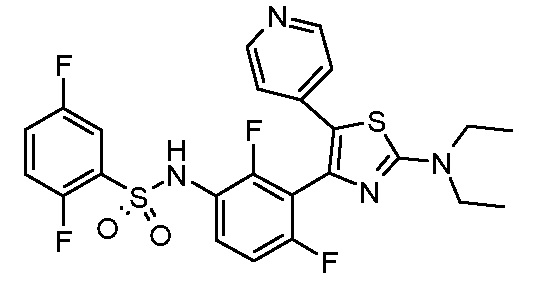
ВЭЖХ: Rt: 6,99 мин
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 10,67 (ушир.с, 1H), 8,35 (д, J=5,6 Гц, 2H), 7,36-7,61 (м, 4H), 7,18 (с, 1H), 6,80-6,87 (м, 2H), 3,47 (кв, J=7,2 Гц, 4H), 1,18 (т, J=7,0 Гц, 6H)
HRMS (ESI): вычислено для C24H20F4N4O2S2 [M+H]+ 537,1037, найдено 537,1025.
N-(3-{2-[4-(Диметиламино)пиперидин-1-ил]-5-(пиридин-4-ил)-1,3-тиазол-4-ил}-2,4-дифторфенил)-2,5-дифторбензолсульфонамида трифторацетат, соединение формулы (I)V (соединение 10) [m, n = 1; R2, R3 = H; R4 = F; R5 = 4-F; R6 = 2,5-дифторфенил; R7-R8 = -(CH2CH2)2CHN(Me2)]
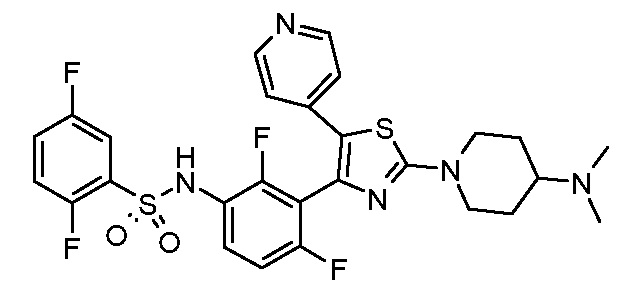
ВЭЖХ: Rt: 4,88 мин
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 10,67 (ушир.с, 1H), 9,60 (ушир.с, 1H), 8,35-8,44 (м, 2H), 7,53-7,62 (м, 1H), 7,37-7,50 (м, 3H), 7,16-7,23 (м, 1H), 6,85-6,89 (м, 2H), 4,04 (д, J=13,7 Гц, 2H), 3,38-3,47 (м, 1H), 3,15 (т, J=11,7 Гц, 3H), 2,78 (с, 6H), 2,07-2,14 (м, 2H), 1,62-1,78 (м, 2H)
HRMS (ESI): вычислено для C27H25F4N5O2S2.C2HF3O2 [M+H]+ 592,1459, найдено 592,1483.
N-{2,4-Дифтор-3-[2-(пиперидин-1-ил)-5-(пиридин-4-ил)-1,3-тиазол-4-ил]фенил}-2,5-дифторбензолсульфонамид, соединение формулы (I)V (соединение 9) [m, n = 1; R2, R3 = H; R4 = F; R5 = 4-F; R6 = 2,5-дифторфенил; R7-R8 = -(CH2CH2)2CH2]
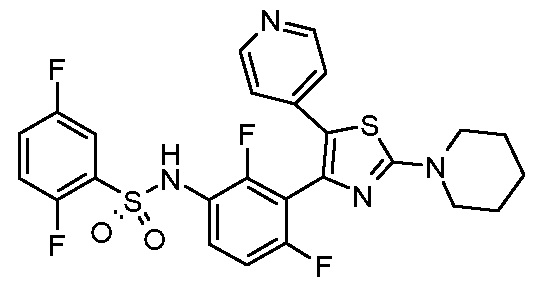
ВЭЖХ: Rt: 6,84 мин
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 10,67 (ушир.с, 1H), 8,33-8,45 (м, 2H), 7,52-7,61 (м, 1H), 7,38-7,50 (м, 3H), 7,18 (т, J=8,4 Гц, 1H), 6,81-6,91 (м, 2H), 3,46 (ушир.с, 4H), 1,61 (ушир.с, 6H)
HRMS (ESI): вычислено для C25H20F4N4O2S2 [M+H]+ 549,1037, найдено 549,1046.
N-{2,4-Дифтор-3-[2-(морфолин-4-ил)-5-(пиридин-4-ил)-1,3-тиазол-4-ил]фенил}-2,5-дифторбензолсульфонамид, соединение формулы (I)V (соединение 15) [m, n = 1; R2, R3 = H; R4 = F; R5 = 4-F; R6 = 2,5-дифторфенил; R7-R8 = -(CH2CH2)2O]
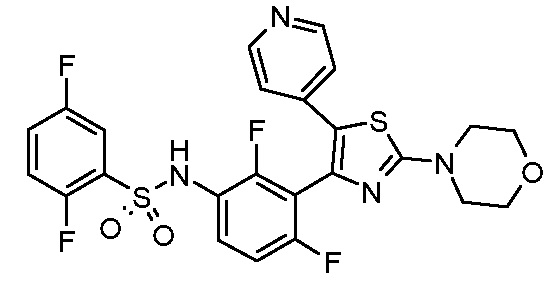
ВЭЖХ: Rt: 5,95 мин
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 10,67 (ушир.с, 1H), 8,34-8,47 (м, 2H), 7,51-7,63 (м, 1H), 7,33-7,51 (м, 3H), 7,18 (т, J=8,2 Гц, 1H), 6,78-7,00 (м, 2H), 3,66-3,79 (м, 4H), 3,39-3,49 (м, 4H)
HRMS (ESI): вычислено для C24H18F4N4O3S2 [M+H]+ 551,0829, найдено 551,0855.
1-[4-(3-{[(2,5-Дифторфенил)сульфонил]амино}-2,6-дифторфенил)-5-(пиридин-4-ил)-1,3-тиазол-2-ил]пиперидин-4-карбоксамид, соединение формулы (I)V [m, n = 1; R2, R3 = H; R4 = F; R5 = 4-F; R6 = 2,5-дифторфенил; R7-R8 = -(CH2CH2)2CHCONH2]
ВЭЖХ: Rt: 5,25 мин
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 10,67 (ушир.с, 1H), 8,32-8,41 (м, 1H), 7,52-7,63 (м, J=7,8 Гц, 1H), 7,37-7,51 (м, 3H), 7,32 (с, 1H), 7,18 (т, J=8,2 Гц, 1H), 6,84-6,87 (м, J=1,5 Гц, 2H), 6,82 (ушир.с, 1H), 3,89 (д, J=12,6 Гц, 2H), 3,03-3,19 (м, 2H), 1,82 (д, J=10,1 Гц, 2H), 1,49-1,69 (м, 2H)
HRMS (ESI): вычислено для C26H21F4N5O3S2 [M+H]+ 592,1095, найдено 592,11.
N-{2,4-Дифтор-3-[2-(4-гидроксипиперидин-1-ил)-5-(пиридин-4-ил)-1,3-тиазол-4-ил]фенил}-2,5-дифторбензолсульфонамид, соединение формулы (I)V (соединение 13) [m, n = 1; R2, R3 = H; R4 = F; R5 = 4-F; R6 = 2,5-дифторфенил; R7-R8 = -(CH2CH2)2CHOH]
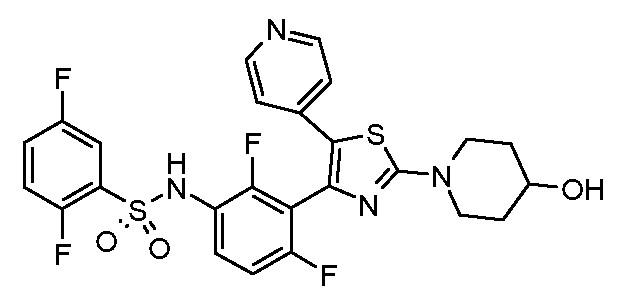
ВЭЖХ: Rt: 5,5 мин
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 10,67 (ушир.с, 1H), 8,36 (д, J=6,1 Гц, 2H), 7,50-7,59 (м, 1H), 7,36-7,50 (м, 5H), 7,17 (т, J=8,5 Гц, 1H), 6,78-6,88 (м, 2H), 4,82 (д, J=4,2 Гц, 1H), 3,66-3,85 (м, 3H), 1,77-1,91 (м, J=4,3, 8,5 Гц, 1H), 1,39-1,56 (м, 2H)
HRMS (ESI): вычислено для C25H20F4N4O3S2 [M+H]+ 565,0986, найдено 565,0993.
N-{3-[2-(4,4-Дифторпиперидин-1-ил)-5-(пиридин-4-ил)-1,3-тиазол-4-ил]-2,4-дифторфенил}-2,5-дифторбензолсульфонамид, соединение формулы (I)V (соединение 14) [m, n = 1; R2, R3 = H; R4 = F; R5 = 4-F; R6 = 2,5-дифторфенил; R7-R8 = -(CH2CH2)2CF2]
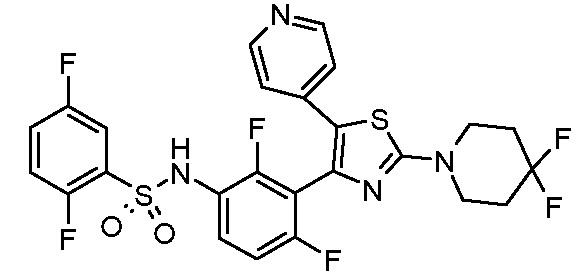
ВЭЖХ: Rt: 6,64 мин
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 10,67 (ушир.с, 1H), 8,39 (д, J=6,1 Гц, 2H), 7,50-7,61 (м, 1H), 7,34-7,50 (м, 3H), 7,18 (т, J=8,5 Гц, 1H), 6,84-6,95 (м, 2H), 3,54-3,69 (м, 4H), 2,00-2,22 (м, 4H)
HRMS (ESI): вычислено для C25H18F6N4O2S2 [M+H]+ 585,0848, найдено 585,0858.
N-{2,4-Дифтор-3-[2-{[2-(морфолин-4-ил)этил]амино}-5-(пиридин-4-ил)-1,3-тиазол-4-ил]фенил}-2,5-дифторбензолсульфонамид, соединение формулы (I)V [m, n = 1; R2, R3, R7 = H; R4 = F; R5 = 4-F; R6 = 2,5-дифторфенил; R8 = 2-(морфолин-4-ил)этил]
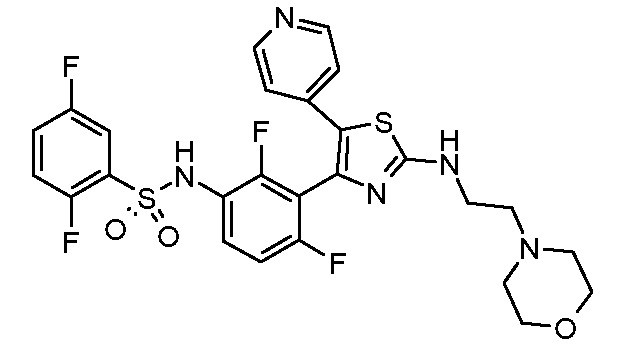
ВЭЖХ: Rt: 5,2 мин
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 10,70 (с, 1H), 8,28-8,38 (м, 2H), 8,10 (т, J=5,4 Гц, 1H), 7,50-7,59 (м, 1H), 7,35-7,50 (м, 3H), 7,10-7,20 (м, 1H), 6,76-6,86 (м, 2H), 3,50-3,60 (м, 4H), 3,37 (ушир.с, 2H)
HRMS (ESI): вычислено для C26H23F4N5O3S2 [M+H]+ 594,1251, найдено 594,127.
N-(2,4-Дифтор-3-{2-[(1-метилпиперидин-4-ил)амино]-5-(пиридин-4-ил)-1,3-тиазол-4-ил}фенил)-2,5-дифторбензолсульфонамид, соединение формулы (I)V [m, n = 1; R2, R3, R7 = H; R4 = F; R5 = 4-F; R6 = 2,5-дифторфенил; R8 = 1-метилпиперидин-4-ил]
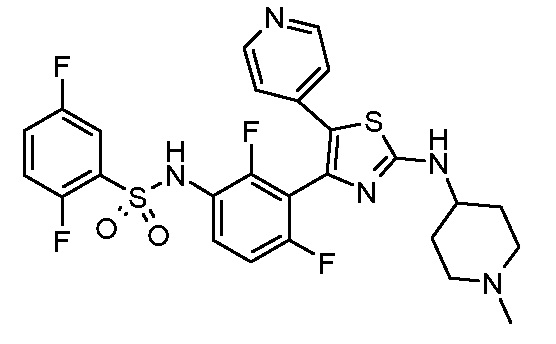
ВЭЖХ: Rt: 4,65 мин
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 8,28-8,41 (м, 2H), 8,18 (д, J=7,1 Гц, 1H), 7,22-7,49 (м, 4H), 6,91-7,00 (м, 1H), 6,85-6,91 (м, 2H), 3,70 (ушир.с, 1H), 2,01-2,13 (м, 2H), 1,54-1,66 (м, 2H)
HRMS (ESI): вычислено для C26H23F4N5O2S2 [M+H]+ 578,1302, найдено 578,1301.
Пример 7
Синтез N-(3-{2-[(этилкарбамоил)амино]-5-(пиридин-4-ил)-1,3-тиазол-4-ил}-2,4-дифторфенил)-2,5-дифторбензолсульфонамида, соединение формулы (I)K [m, n = 1; R2, R3 = H; R4 = F; R5 = 4-F; R6 = 2,5-дифторфенил; R12 = этил]
Способ B, стадии f и b4
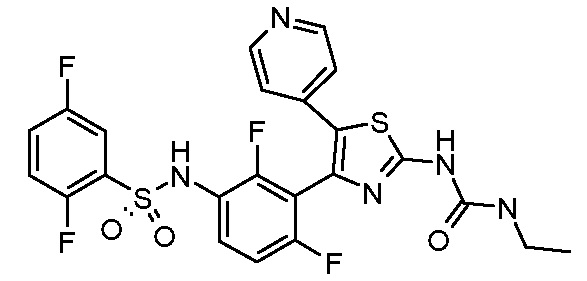
N-{3-[2-Амино-5-(пиридин-4-ил)-1,3-тиазол-4-ил]-2,4-дифторфенил}-2,5-дифтор-N-(метоксиметил)бензолсульфонамид (полученный, как описано в примере 3, 50 мг, 0,1 ммоль) растворяли в 2 мл сухого 1,4-диоксана и порционно добавляли общее количество 320 мкл (4 ммоль) этилизоцианата до тех пор, пока реакция не завершится. Раствор нагревали при 80°C при перемешивании в течение 18 ч. Затем растворитель удаляли и осадок отбирали в DCM и промывали водой. Органическую фазу сушили над Na2SO4 и упаривали до сухого состояния с получением N-(3-{2-[(этилкарбамоил)амино]-5-(пиридин-4-ил)-1,3-тиазол-4-ил}-2,4-дифторфенил)-2,5-дифтор-N-(метоксиметил)бензолсульфонамида. Последней без какой-либо дальнейшей очистки растворяли в 9 мл TFA и 1 мл воды и полученную смесь поддерживали при 60°C при перемешивании в течение 8 ч. Растворитель выпаривали и осадок отбирали в DCM и промывали водным раствором NaHCO3. Затем органический слой сушили над Na2SO4 и вновь упаривали. Продукт очищали флэш-хроматографией на предварительно покрытой колонке с силикагелем, элюируя DCM-MeOH 95/5, с получением после растирания со смесью диэтиловый эфир-диизопропиловый эфир, 17 мг (31%) указанного в заголовке соединения.
ВЭЖХ: Rt: 5,59 мин
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 10,82 (с, 1H), 10,68 (ушир.с, 1H), 8,42 (д, J=5,4 Гц, 2H), 7,56 (д, J=11,4 Гц, 1H), 7,36-7,51 (м, 4H), 7,17 (т, J=8,6 Гц, 1H), 6,93-7,02 (м, 2H), 6,59 (ушир.с, 1H), 3,11-3,23 (м, 2H), 1,05-1,11 (м, 3H)
HRMS (ESI): вычислено для C23H17F4N5O3S2 [M+H]+ 552,0782, найдено 552,078.
Пример 8
Синтез N-[4-(3-{[(2,5-дифторфенил)сульфонил]амино}-2,6-дифторфенил)-5-(пиридин-4-ил)-1,3-тиазол-2-ил]циклогексанкарбоксамида, соединение формулы (I)M [m, n = 1; R2, R3 = H; R4 = F; R5 = 4-F; R6 = 2,5-дифторфенил; R10 = циклогексил]
Способ B, стадии g, h и b5
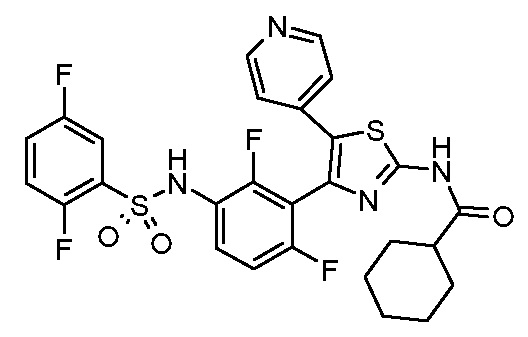
50 мг (0,39 ммоль) циклогексанкарбоновой кислоты растворяли в 3 мл сухого DCM и к смеси добавляли 56 мкл (0,39 ммоль) TEA. Затем по каплям добавляли раствор 33 мкл оксалилхлорида в 2 мл DCM при к.т. Через 3 ч растворитель удаляли при пониженном давлении, несколько раз добавляли толуол и вновь повторно выпаривали. Осадок растворяли в 2 мл сухого DCM и по каплям добавляли к раствору 50 мг (0,095 ммоль) N-{3-[2-амино-5-(пиридин-4-ил)-1,3-тиазол-4-ил]-2,4-дифторфенил}-2,5-дифтор-N-(метоксиметил)бензолсульфонамида в 3 мл того же растворителя, содержавшего 56 мкл TEA. Реакционную смесь поддерживали при к.т. в течение ночи при перемешивании. Затем раствор разбавляли DCM и промывали водным раствором NaHCO3. Органический слой сушили над Na2SO4 и упаривали с получением N-(циклогексилкарбонил)-N-[4-(3-{[(2,5-дифторфенил)сульфонил]-(метоксиметил)амино}-2,6-дифторфенил)-5-(пиридин-4-ил)-1,3-тиазол-2-ил]циклогексанкарбоксамида в виде масла. Последнее отбирали в 5 мл MeOH и к полученному раствору добавляли 1 мл TEA. Реакционную смесь поддерживали при к.т. в течение ночи. Затем растворитель выпаривали, осадок перерастворяли в DCM и промывали насыщенным солевым раствором. Органическую фазу сушили над Na2SO4 и вновь упаривали с получением N-[4-(3-{[(2,5-дифторфенил)сульфонил](метоксиметил)амино}-2,6-дифторфенил)-5-(пиридин-4-ил)-1,3-тиазол-2-ил]циклогексанкарбоксамида. Наконец, последний растворяли в 9 мл TFA и 1 мл воды и нагревали при 75°C в течение 6 ч. Растворитель удаляли при пониженном давлении, осадок перерастворяли в DCM и промывали водным раствором NaHCO3. Органический слой сушили над Na2SO4 и упаривали. Наконец, неочищенное вещество очищали флэш-хроматографией на предварительно покрытой колонке с силикагелем, элюируя DCM-MeOH 98/2, с получением 35 мг (общий выход 63%) указанного в заголовке соединения.
ВЭЖХ: Rt: 6,75 мин
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 12,43 (с, 1H), 10,69 (ушир.с, 1H), 8,45 (д, J=6,0 Гц, 2H), 7,55 (ушир.с, 2H), 7,34-7,49 (м, 4H), 7,17 (ушир.с, 1H), 7,00-7,05 (м, 2H)
HRMS (ESI): вычислено для C27H22F4N4O3S2 [M+H]+ 591,1142, найдено 591,1124.
Аналогично получали следующие соединения:
N-[4-(3-{[(2,5-Дифторфенил)сульфонил]амино}-2,6-дифторфенил)-5-(пиридин-4-ил)-1,3-тиазол-2-ил]-2-метилпропанамид, соединение формулы (I)M [m, n = 1; R2, R3 = H; R4 = F; R5 = 4-F; R6 = 2,5-дифторфенил; R10 = изопропил]
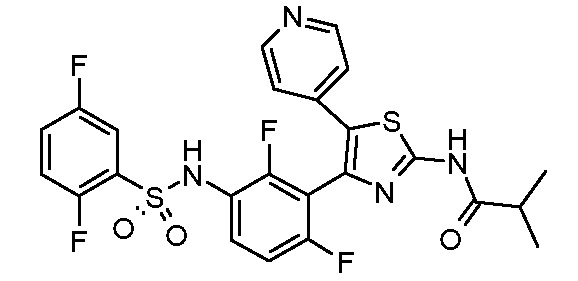
ВЭЖХ: Rt: 6,11 мин
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 12,48 (с, 1H), 10,69 (д, J=0,5 Гц, 1H), 8,43-8,47 (м, 2H), 7,51-7,60 (м, 1H), 7,36-7,52 (м, 4H), 7,18 (ушир.с, 1H), 7,00-7,06 (м, 2H), 2,76 (квин, J=6,8 Гц, 1H), 1,15 (д, J=6,8 Гц, 6H)
HRMS (ESI): вычислено для C24H18F4N4O3S2 [M+H]+ 551,0829, найдено 551,0809.
N-[4-(3-{[(2,5-Дифторфенил)сульфонил]амино}-2,6-дифторфенил)-5-(пиридин-4-ил)-1,3-тиазол-2-ил]бензамид, соединение формулы (I)M [m, n = 1; R2, R3 = H; R4 = F; R5 = 4-F; R6 = 2,5-дифторфенил; R10 = фенил]
Способ B, стадии g и b5
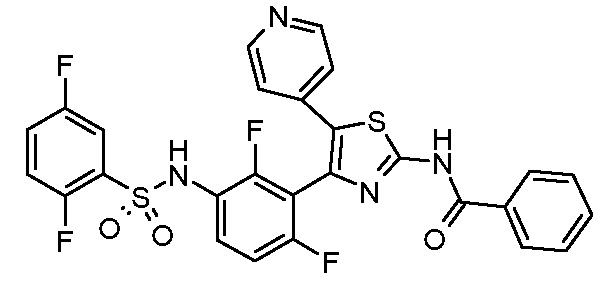
ВЭЖХ: Rt: 6,46 мин
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 13,07 (с, 1H), 10,72 (с, 1H), 8,44-8,50 (м, 2H), 8,07-8,19 (м, 2H), 7,63-7,73 (м, 1H), 7,54-7,62 (м, 3H), 7,40-7,50 (м, 4H), 7,18-7,26 (м, 1H), 7,01-7,12 (м, 2H)
HRMS (ESI): вычислено для C27H16F4N4O3S2 [M+H]+ 585,0673, найдено 585,0679.
Пример 9
Синтез N-{3-[5-(2-хлорпиридин-4-ил)-2-(тетрагидро-2H-пиран-4-ил)-1,3-тиазол-4-ил]-2-фторфенил}-2,5-дифторбензолсульфонамида, соединение формулы (I)T [m, n = 1; R1 = тетрагидропиран-4-ил; R2 = Cl; R3, R5 = H; R4 = F; R6 = 2,5-дифторфенил]
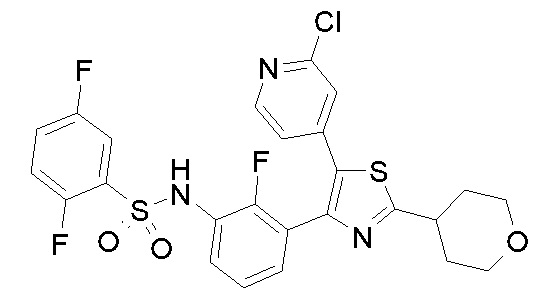
2,5-Дифтор-N-{2-фтор-3-[(2-хлорпиридин-4-ил)ацетил]фенил}-N-(метоксиметил)бензолсульфонамид, соединение формулы 28 [m, n = 1; R2 = Cl; R3, R5 = H; R4 = F; Rx = метоксиметил; R6 = 2,5-дифторфенил]
Способ D, стадия f
Сухой диизопропиламин (1,2 мл, 8,604 ммоль, 1,2 экв.) растворяли в сухом THF (16 мл) в атмосфере аргона и охлаждали до -78°C. Затем добавляли 2,5M бутиллитий в гексане (3,44 мл, 8,604 ммоль, 1,2 экв.), а затем через 5 мин раствор 2-Cl-4-метилпиридина (0,628 мл, 7,17 ммоль, 1 экв.) в THF (10 мл). Смесь перемешивали при -78°C в течение 1 ч, а затем по каплям добавляли раствор 3-{[(2,5-дифторфенил)-сульфонил](метоксиметил)амино}-2-фтор-N-метокси-N-метилбензамида (полученный, как описано в методике получения 2, 3,0 г, 7,17 ммоль, 1 экв.) в THF (20 мл). После 10 мин при -78°C темно-желтую смесь нагревали до 0°C и перемешивали в течение 1 ч. Затем ее гасили насыщенным водным раствором хлорида аммония и экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над Na2SO4 и концентрировали при пониженном давлении. Неочищенный продукт очищали флэш-хроматографией на силикагеле (Hex/этилацетат 1:1) с получением 2,37 г (68%) указанного в заголовке соединения в виде желтого масла.
ВЭЖХ: Rt: 6,04 мин
1H ЯМР (600 МГц, DMSO-d6) δ м.д. 8,35 (д, J=5,1 Гц, 1H), 7,97-7,93 (м, 1H), 7,73-7,66 (м, 1H), 7,65-7,56 (м, 2H), 7,52 (ддд, J=3,3, 5,0, 7,8 Гц, 1H), 7,42 (с, 1H), 7,40 (т, J=7,9 Гц, 1H), 7,29 (дд, J=5,1, 1,1, 1H), 5,09 (с, 2H), 4,39 (с, 2H), 3,37 (с, 3H).
HRMS (ESI): вычислено для C21H17N2O4F3SCl [M+H]+ 485,0544, найдено 485,0539.
Аналогичным путем, но с использованием подходящего производного пиридина, также получали следующие промежуточные соединения:
2,5-Дифтор-N-{2-фтор-3-[(2-фторпиридин-4-ил)ацетил]фенил}-N-(метоксиметил)бензолсульфонамид, соединение формулы 28 [m, n = 1; R2 = F; R3, R5 = H; R4 = F; Rx = метоксиметил; R6 = 2,5-дифторфенил]
ВЭЖХ: Rt: 5,97 мин
1H ЯМР (600 МГц, DMSO-d6) δ м.д. 8,18 (д, J=5,1 Гц, 1H), 7,95 (т, J=6,5 Гц, 1H), 7,71-7,65 (м, 1H), 7,64-7,55 (м, 2H), 7,54-7,49 (м, 1H), 7,40 (т, J=7,9 Гц, 1H), 7,22 (д, J=5,1 Гц, 1H), 7,07 (с, 1H), 5,09 (с, 2H), 4,42 (с, 2H), 3,37 (с, 3H).
HRMS (ESI): вычислено для C21H17N2O4F4S [M+H]+ 469,0840, найдено 469,0828.
2,5-Дифтор-N-{2-фтор-3-[(пиридин-4-ил)ацетил]фенил}-N-(метоксиметил)бензолсульфонамид, соединение формулы 28 [m, n = 1; R2, R3, R5 = H; R4 = F; Rx = метоксиметил; R6 = 2,5-дифторфенил]
ВЭЖХ: Rt: 5,31 мин
HRMS (ESI): вычислено для C21H18N2O4F3S [M+H]+ 451,0934, найдено 451,0922.
трет-Бутил-{4-[2-(3-{[(2,5-дифторфенил)сульфонил](метоксиметил)амино}-2-фторфенил)-2-оксоэтил]пиридин-2-ил}карбамат, соединение формулы 28 [m, n = 1; R2 = трет-бутоксикарбониламино; R3, R5 = H; R4 = F; Rx = метоксиметил; R6 = 2,5-дифторфенил]
ВЭЖХ: Rt: 6,49 мин
1H ЯМР (600 МГц, DMSO-d6) δ м.д. 9,67 (с, 1H), 8,15 (д, J=4,9 Гц, 1H), 7,93 (т, J=6,9 Гц, 1H), 7,70-7,65 (м, 2H), 7,61-7,54 (м, 2H), 7,53-7,46 (м, 1H), 7,38 (т, J=7,8 Гц, 1H), 6,84 (д, J=4,8 Гц, 1H), 5,08 (с, 2H), 4,27 (с, 2H), 3,31 (с, 3H), 1,46 (с, 9H)
HRMS (ESI): вычислено для C26H27N3O6F3S [M+H]+ 566,1567, найдено 566,1579.
N-{3-[5-(2-Хлорпиридин-4-ил)-2-(тетрагидро-2H-пиран-4-ил)-1,3-тиазол-4-ил]-2-фторфенил}-2,5-дифтор-N-(метоксиметил)бензолсульфонамид, соединение формулы (I)S [m, n = 1; R1 = тетрагидропиран-4-ил; R2 = Cl; R3, R5 = H; R4 = F; Rx = метоксиметил; R6 = 2,5-дифторфенил]
Способ D, стадии l и m
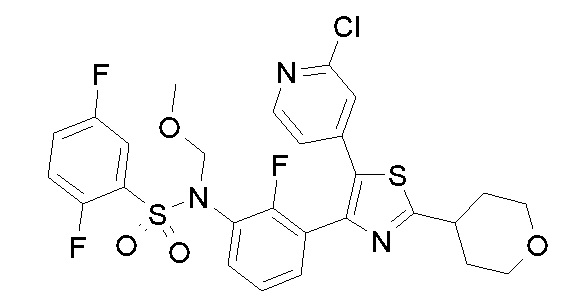
2,5-Дифтор-N-{2-фтор-3-[(2-хлорпиридин-4-ил)ацетил]фенил}-N-(метоксиметил)бензолсульфонамид (937 мг, 1,932 ммоль) растворяли в сухом DMF (17 мл) в атмосфере аргона. Добавляли пиридиния бромид пербромид (556 мг, 1,739 ммоль, 0,9 экв.) и смесь перемешивали при к.т. Через 50 мин добавляли тетрагидро-2H-пиран-4-карботиоамид (полученный, как описано в методике получения 5, 305 мг, 2,1 ммоль, 1,09 экв.) и реакционную смесь нагревали до 60°C и перемешивали в течение 2 ч. Смесь концентрировали при пониженном давлении и отбирали в этилацетат и промывали насыщенным водным раствором NaHCO3. Водную фазу обратно экстрагировали этилацетатом. Объединенные органические слои промывали водой и насыщенным солевым раствором, сушили над Na2SO4 и концентрировали при пониженном давлении. Неочищенный продукт очищали флэш-хроматографией на силикагеле (DCM/MeOH 98:2) с получением 996 мг (72%) указанного в заголовке продукта в виде масла.
ВЭЖХ: Rt: 7,14 мин
1H ЯМР (600 МГц, DMSO-d6) δ м.д. 8,33 (дд, J=0,5, 5,1 Гц, 1H), 7,68-7,60 (м, 2H), 7,55 (дт, J=3,9, 9,4 Гц, 1H), 7,47-7,44 (м, 1H), 7,35-7,32 (м, 2H), 7,24 (дд, J=0,6, 1,6 Гц, 1H), 7,16 (дд, J=1,6, 5,1 Гц, 1H), 4,98 (с, 2H), 3,98-3,93 (м, 2H), 3,52-3,46 (м, 2H), 3,39-3,31 (м, 1H), 3,24 (с, 3H), 2,06-2,01 (м, J=2,9, 13,7 Гц, 2H), 1,82-1,74 (м, 2H)
HRMS (ESI): вычислено для C27H24N3O4F3S2Cl [M+H]+ 610,0844, найдено 610,0860.
Аналогичным путем, но с использованием подходящего тиоамида, также получали следующие промежуточные соединения:
N-{3-[5-(2-Хлорпиридин-4-ил)-2-(1-циклопропилпиперидин-4-ил)-1,3-тиазол-4-ил]-2-фторфенил}-2,5-дифтор-N-(метоксиметил)бензолсульфонамид, соединение формулы (I)S [m, n = 1; R1 = 1-циклопропилпиперидин-4-ил; R2 = Cl; R3, R5 = H; R4 = F; Rx = метоксиметил; R6 = 2,5-дифторфенил]
Способ D, стадии l и m
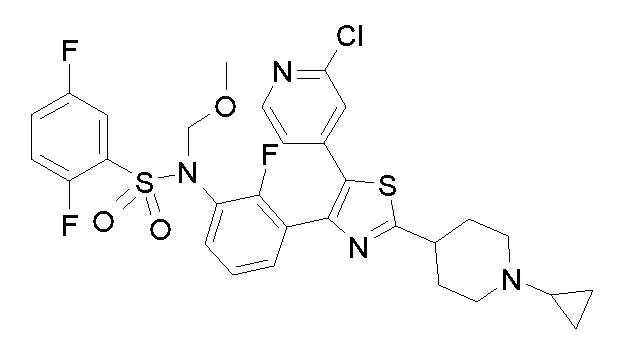
ВЭЖХ: Rt: 7,74 мин
1H ЯМР (600 МГц, DMSO-d6) δ м.д. 8,32 (д, J=5,2 Гц, 1H), 7,70-7,60 (м, 2H), 7,56 (дт, J=4,0, 9,3 Гц, 1H), 7,46 (ддд, J=3,2, 5,1, 7,8 Гц, 1H), 7,37-7,27 (м, 2H), 7,26-7,20 (м, 1H), 7,15 (дд, J=1,4, 5,3 Гц, 1H), 4,98 (с, 2H), 3,24 (с, 3H), 3,14-2,94 (м, 3H), 2,32 (т, J=11,1 Гц, 2H), 2,06 (д, J=11,9 Гц, 2H), 1,73-1,56 (м, 3H), 0,50-0,35 (м, J=5,0 Гц, 2H), 0,31 (ушир.с, 2H).
HRMS (ESI): вычислено для C30H29N4O3F3S2Cl [M+H]+ 649,1316, найдено 649,1313.
N-{3-[2-трет-Бутил-5-(2-хлорпиридин-4-ил)-1,3-тиазол-4-ил]-2-фторфенил}-2,5-дифтор-N-(метоксиметил)бензолсульфонамид, соединение формулы (I)S [m, n = 1; R1 = трет-бутил; R2 = Cl; R3, R5 = H; R4 = F; Rx = метоксиметил; R6 = 2,5-дифторфенил]
Способ D, стадии l и m
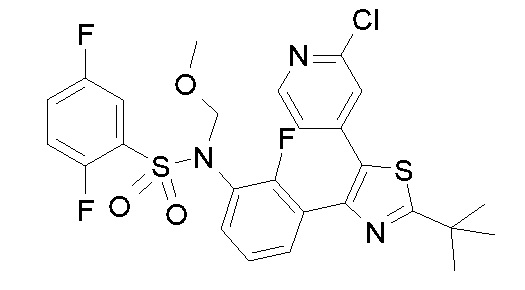
ВЭЖХ: Rt: 7,29 мин
1H ЯМР (600 МГц, DMSO-d6) δ м.д. 8,32 (д, J=5,1 Гц, 1H), 7,67-7,61 (м, 2H), 7,55 (дт, J=4,0, 9,4 Гц, 1H), 7,48-7,43 (м, 1H), 7,36-7,30 (м, 2H), 7,25 (д, J=0,9 Гц, 1H), 7,16 (дд, J=1,6, 5,2 Гц, 1H), 4,98 (с, 2H), 3,24 (с, 3H), 1,47-1,43 (м, 9H)
HRMS (ESI): вычислено для C26H24N3O3F3S2Cl [M+H]+ 582,0894, найдено 582,0913.
Бензил-4-[5-(2-хлорпиридин-4-ил)-4-(3-{[(2,5-дифторфенил)сульфонил](метоксиметил)амино}-2-фторфенил)-1,3-тиазол-2-ил]пиперидин-1-карбоксилат, соединение формулы (I)S [m, n = 1; R1 = 1-бензилоксикарбонилпиперидин-4-ил; R2 = Cl; R3, R5 = H; R4 = F; Rx = метоксиметил; R6 = 2,5-дифторфенил]
Способ D, стадии l и m

ВЭЖХ: Rt: 7,93 мин
1H ЯМР (600 МГц, DMSO-d6) δ м.д. 8,32 (д, J=5,1 Гц, 1H), 7,68-7,59 (м, 2H), 7,55 (дт, J=4,1, 9,4 Гц, 1H), 7,48-7,42 (м, 1H), 7,40-7,35 (м, 4H), 7,35-7,30 (м, 3H), 7,24 (д, J=0,9 Гц, 1H), 7,15 (дд, J=1,6, 5,2 Гц, 1H), 5,10 (с, 2H), 4,98 (с, 2H), 4,15-3,98 (м, 2H), 3,38-3,33 (м, 1H), 3,24 (с, 3H), 3,09-2,93 (ушир.с, 2H), 2,15-2,09 (м, 2H), 1,65 (д, кв, J=4,2, 12,2 Гц, 2H).
HRMS (ESI): вычислено для C35H31N4O5F3S2Cl [M+H]+ 743,1371, найдено 743,1379.
N-{3-[5-(2-Хлорпиридин-4-ил)-2-(тетрагидро-2H-пиран-4-ил)-1,3-тиазол-4-ил]-2-фторфенил}-2,5-дифторбензолсульфонамид, соединение формулы (I)T [m, n = 1; R1 = тетрагидропиран-4-ил; R2 = Cl; R3, R5 = H; R4 = F; R6 = 2,5-дифторфенил]
Способ D, стадия n
N-{3-[5-(2-Хлорпиридин-4-ил)-2-(тетрагидро-2H-пиран-4-ил)-1,3-тиазол-4-ил]-2-фторфенил}-2,5-дифтор-N-(метоксиметил)бензолсульфонамид (50 мг, 0,082 ммоль) растворяли в смеси TFA/вода 9:1 (1 мл) и перемешивали при 60°C в течение 5 ч. Смесь упаривали до сухого состояния, отбирали в DCM и промывали насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над Na2SO4 и концентрировали при пониженном давлении. Неочищенный продукт очищали флэш-хроматографией на силикагеле (толуол/этилацетат 6:4) с получением 38 мг (82%) продукта, который растирали со смесью диэтиловый эфир/этилацетат, фильтровали и сушили в высоком вакууме. Было получено 34 мг указанного в заголовке соединения в виде белого твердого вещества.
ВЭЖХ: Rt: 6,94 мин
1H ЯМР (600 МГц, DMSO-d6) δ м.д. 10,69 (с, 1H), 8,29 (д, J=5,1 Гц, 1H), 7,63-7,52 (м, 1H), 7,50-7,44 (м, 1H), 7,44-7,39 (м, 1H), 7,39-7,32 (м, 2H), 7,29-7,23 (м, 1H), 7,22 (д, J=0,9 Гц, 1H), 7,08 (дд, J=1,6, 5,2 Гц, 1H), 3,97-3,91 (м, 2H), 3,48 (дт, J=1,9, 11,6 Гц, 2H), 3,39-3,34 (м, 1H), 2,05-1,94 (м, 2H), 1,84-1,57 (м, 2H)
HRMS (ESI): вычислено для C25H20N3O3F3S2Cl [M+H]+ 566,0581, найдено 566,0588.
Аналогичным путем, но с использованием подходящего исходного материала и тиоамида, также получали следующие соединения:
2,5-Дифтор-N-{2-фтор-3-[5-(2-фторпиридин-4-ил)-2-(1-метилпиперидин-4-ил)-1,3-тиазол-4-ил]фенил}бензолсульфонамид, соединение формулы (I)T (соединение 27) [m, n = 1; R1 = 1-метилпиперидин-4-ил; R2 = F; R3, R5 = H; R4 = F; R6 = 2,5-дифторфенил]
Способ D, стадия n

ВЭЖХ: Rt: 4,19 мин
1H ЯМР (600 МГц, DMSO-d6) δ м.д. 8,14 (д, J=5,3 Гц, 1H), 7,45-7,35 (м, 2H), 7,35-7,24 (м, 2H), 7,15-6,96 (м, 3H), 6,89 (с, 1H), 3,41-3,36 (м, 1H), 3,23-3,09 (м, 2H), 2,60-2,40 (м, 3H), 2,18 (д, J=12,6 Гц, 2H), 1,93-1,77 (м, 2H).
HRMS (ESI): вычислено для C26H23N4O2F4S2 [M+H]+ 563,1193, найдено 563,1202.
N-{3-[2-трет-Бутил-5-(пиридин-4-ил)-1,3-тиазол-4-ил]-2-фторфенил}-2,5-дифторбензолсульфонамид, соединение формулы (I)T (соединение 43) [m, n = 1; R1 = трет-бутил; R2, R3, R5 = H; R4 = F; R6 = 2,5-дифторфенил]
Способ D, стадия n
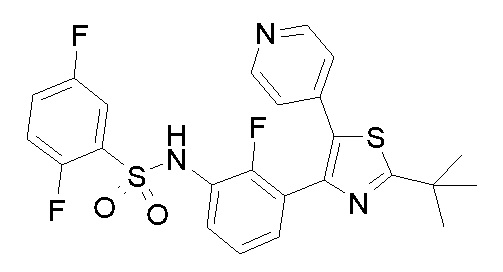
ВЭЖХ: Rt: 7,16 мин
1H ЯМР (600 МГц, DMSO-d6) δ м.д. 10,65 (ушир.с, 1H), 8,49-8,40 (м, 2H), 7,63-7,52 (м, 1H), 7,51-7,43 (м, 1H), 7,44-7,37 (м, 1H), 7,37-7,30 (м, 2H), 7,27-7,18 (м, 1H), 7,12-6,90 (м, 2H), 1,43 (с, 9H)
HRMS (ESI): вычислено для C24H21N3O2F3S2 [M+H]+ 504,1022, найдено 504,1031.
N-{3-[2-(1-Циклопропилпиперидин-4-ил)-5-(пиридин-4-ил)-1,3-тиазол-4-ил]-2-фторфенил}-2,5-дифторбензолсульфонамид, соединение формулы (I)T (соединение 21) [m, n = 1; R1 = 1-циклопропилпиперидин-4-ил; R2, R3, R5 = H; R4 = F; R6 = 2,5-дифторфенил]
Способ D, стадия n
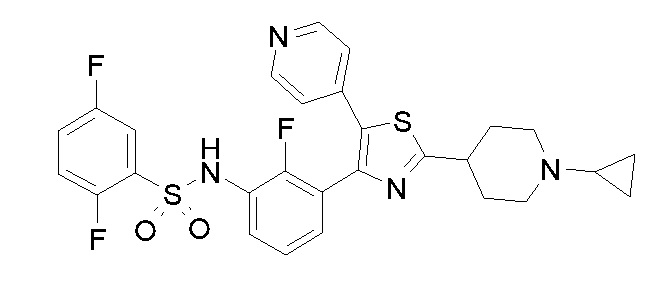
ВЭЖХ: Rt: 4,70 мин
1H ЯМР (600 МГц, DMSO-d6) δ м.д. 10,53 (ушир.с, 1H), 8,47-8,44 (м, 2H), 7,57-7,51 (м, 1H), 7,46-7,43 (м, 1H), 7,40 (ддд, J=3,2, 5,1, 7,8 Гц, 1H), 7,36-7,32 (м, 1H), 7,28 (м, 1H), 7,22-7,18 (м, 1H), 7,08-7,05 (м, 2H), 3,12-2,98 (м, 3H), 2,35-2,42 (м, 2H), 2,06 (д, J=9,9 Гц, 2H), 1,73 (м, 1H), 1,71-1,64 (м, 2H), 0,45 (д, J=4,8 Гц, 2H), 0,35 (ушир.с, 2H)
HRMS (ESI): вычислено для C28H26N4O2F3S2 [M+H]+ 571,1444, найдено 571,1463.
Пример 10
N-{3-[5-(2-Аминопиридин-4-ил)-2-(1-циклопропилпиперидин-4-ил)-1,3-тиазол-4-ил]-2-фторфенил}-2,5-дифторбензолсульфонамид, соединение формулы (I)T (соединение 22) [m, n = 1; R1 = 1-циклопропилпиперидин-4-ил; R2 = NH2; R3, R5 = H; R4 = F; R6 = 2,5-дифторфенил]
Способ D, стадии l, m, n
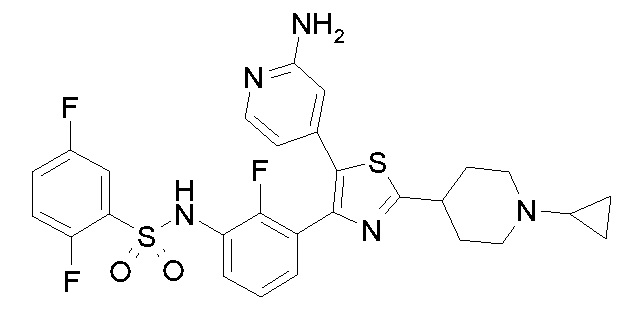
трет-Бутил-{4-[2-(3-{[(2,5-дифторфенил)сульфонил](метоксиметил)амино}-2-фторфенил)-2-оксоэтил]пиридин-2-ил}карбамат (полученный, как описано в примере 9, 374 мг, 0,662 ммоль) растворяли в сухом DMF (5 мл) в атмосфере аргона. Добавляли пиридина бромид пербромид (190 мг, 0,596 ммоль, 0,9 экв.) и реакционную смесь перемешивали при к.т. в течение 50 мин. Затем добавляли 1-циклопропилпиперидин-4-карботиоамид (146 мг, 0,794 ммоль, 1,2 экв.) и смесь нагревали до 70°C. Через 1 ч реакции позволяли остыть до к.т. в течение ночи. Растворитель концентрировали при пониженном давлении. Осадок отбирали в этилацетат и промывали насыщенным водным NaHCO3 и насыщенным солевым раствором. Органическую фазу сушили над Na2SO4 и упаривали до сухого состояния. Неочищенный продукт очищали флэш-хроматографией на силикагеле (DCM/MeOH 98:2) с получением 290 мг (60%) трет-бутил {4-[2-(1-циклопропилпиперидин-4-ил)-4-(3-{[(2,5-дифторфенил)сульфонил](метоксиметил)амино}-2-фторфенил)-1,3-тиазол-5-ил]пиридин-2-ил}карбамата в виде аморфного твердого вещества.
ВЭЖХ/MS (ESI): 730 [M+H]+, 728 [M-H]-
Это промежуточное соединение растворяли в смеси 9:1 TFA/H2O (4 мл) и перемешивали при 70°C в течение 2 ч. Затем смесь концентрировали при пониженном давлении, отбирали в DCM и промывали насыщенным водным NaHCO3 и насыщенным солевым раствором. Органическую фазу сушили над Na2SO4 и упаривали до сухого состояния. Неочищенный продукт очищали флэш-хроматографией на силикагеле (DCM/EtOH 95:5) и обрабатывали смесью изопропиловый эфир/этилацетат, фильтровали и сушили в высоком вакууме с получением 70 мг (30%) указанного в заголовке соединения в виде светло-оранжевого твердого вещества.
ВЭЖХ: Rt: 5,49 мин
1H ЯМР (600 МГц, DMSO-d6) δ м.д. 10,99-10,01 (ушир.с, 1H), 7,70 (д, J=5,3 Гц, 1H), 7,57-7,51 (м, 1H), 7,48-7,40 (м, 2H), 7,31 (м, 1H), 7,21 (ушир.с, 1H), 7,18-7,11 (м, 1H), 6,33 (с, 1H), 6,01 (с, 2H), 5,98 (д, J=5,3 Гц, 1H), 3,09-2,95 (м, 3H), 2,40-2,28 (м, 2H), 2,10-1,98 (м, 2H), 1,75-1,54 (м, 3H), 0,50-0,41 (м, 2H), 0,37-0,29 (м, 2H)
HRMS (ESI): вычислено для C28H27N5O2F3S2 [M+H]+ 586,1553, найдено 586,1556.
Пример 11
Синтез N-{4-[2-(1-циклопропилпиперидин-4-ил)-4-(3-{[(2,5-дифторфенил)сульфонил]амино}-2-фторфенил)-1,3-тиазол-5-ил]пиридин-2-ил}ацетамида, соединение формулы (I)T (соединение 30) [m, n = 1; R1 = 1-циклопропилпиперидин-4-ил; R2 = ацетиламино; R3, R5 = H; R4 = F; R6 = 2,5-дифторфенил]
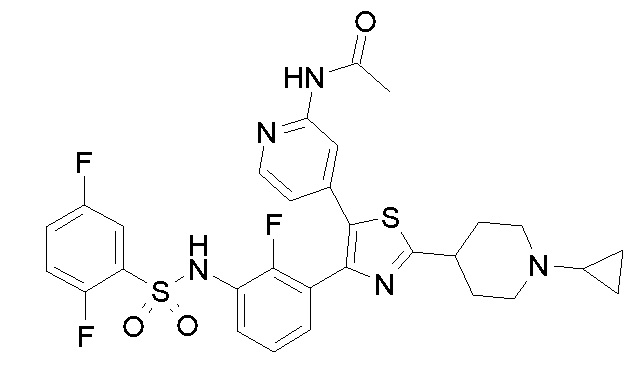
К раствору N-{3-[5-(2-аминопиридин-4-ил)-2-(1-циклопропилпиперидин-4-ил)-1,3-тиазол-4-ил]-2-фторфенил}-2,5-дифторбензолсульфонамида (полученного, как описано в примере 10, 66 мг, 0,113 ммоль) в сухом DCM (1 мл), добавляли TEA (0,063 мл, 0,452 ммоль, 4 экв.) и ацетилхлорид (0,024 мл, 0,338 ммоль, 3 экв.) и смесь перемешивали при к.т. в течение 16 ч. Затем ее разбавляли DCM и промывали насыщенным водным NaHCO3 и насыщенным солевым раствором. Органическую фазу сушили над Na2SO4 и упаривали до сухого состояния с получением смеси 1:1 диацетилированных и триацетилированных продуктов.
К раствору этой смеси в MeOH (2 мл) добавляли 1н гидроксид натрия (1 мл) и раствор перемешивали при к.т. в течение 1 ч. Растворитель выпаривали при пониженном давлении и осадок отбирали в этилацетат и промывали насыщенным водным NaHCO3 и насыщенным солевым раствором. Органическую фазу сушили над Na2SO4 и упаривали до сухого состояния. Неочищенный продукт очищали флэш-хроматографией на силикагеле (DCM/EtOH 95:5) с получением 47 мг (66%) указанного в заголовке соединения в виде белого твердого вещества.
ВЭЖХ: Rt: 5,57 мин
1H ЯМР (600 МГц, DMSO-d6) δ м.д. 10,54 (с, 1H), 8,12 (д, J=5,3 Гц, 1H), 8,04 (с, 1H), 7,53 (т, J=8,3 Гц, 1H), 7,47-7,37 (м, 2H), 7,33-7,23 (м, 2H), 7,21-7,12 (м, 1H), 6,62 (дд, J=1,6, 5,3 Гц, 1H), 3,14-2,96 (м, 3H), 2,38 (дд, J=1,8, 3,7 Гц, 2H), 2,12-2,01 (м, 2H), 2,06 (с, 3H), 1,79-1,56 (м, 3H), 0,45 (д, J=4,8 Гц, 2H), 0,35 (ушир.с, 2H)
HRMS (ESI): вычислено для C30H29N5O3F3S2 [M+H]+ 628,1659, найдено 628,1659.
Пример 12
Синтез N-{3-[5-(2-аминопиридин-4-ил)-2-(тетрагидро-2H-пиран-4-ил)-1,3-тиазол-4-ил]-2-фторфенил}-2,5-дифторбензолсульфонамида, соединение формулы (I)Z2 (соединение 24) [m, n = 1; R1 = тетрагидропиран-4-ил; R3, R5 = H; R4 = F; R6 = 2,5-дифторфенил]
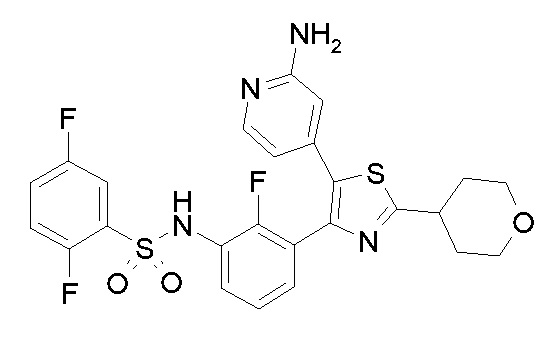
трет-Бутил-{4-[4-(3-{[(2,5-дифторфенил)сульфонил](метоксиметил)амино}-2-фторфенил)-2-(тетрагидро-2H-пиран-4-ил)-1,3-тиазол-5-ил]пиридин-2-ил}карбамат, соединение формулы (I)X [m, n = 1; R1 = тетрагидропиран-4-ил; R3, R5 = H; R4 = F; R6 = 2,5-дифторфенил; R18 = трет-бутоксикарбонил]
Способ F, стадия c1
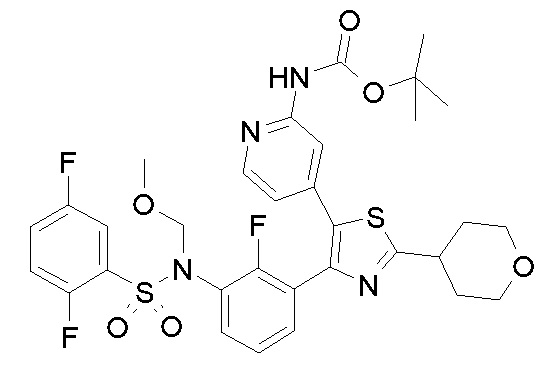
В пробирке для обработки микроволновым излучением N-{3-[5-(2-хлорпиридин-4-ил)-2-(тетрагидро-2H-пиран-4-ил)-1,3-тиазол-4-ил]-2-фторфенил}-2,5-дифтор-N-(метоксиметил)бензолсульфонамид (полученный, как описано в примере 9, 200 мг, 0,328 ммоль, 1 экв.) растворяли в безводном THF (3 мл) и раствор дегазировали барботированием аргона в течение 5 мин. Затем добавляли трет-бутилкарбамат (152 мг, 1,311 ммоль, 4 экв.), а затем карбонат цезия (212 мг, 0,656 ммоль, 2 экв.), ацетат палладия (8 мг, 0,033 ммоль, 0,1 экв.) и Xantphos (40 мг, 0,66 ммоль, 0,2 экв.) и смесь облучали в микроволновой печи при 120°C в течение 30 мин. Смесь фильтровали через слой целита и целит промывали этилацетатом. Фильтрат промывали насыщенным водным NaHCO3 и насыщенным солевым раствором, сушили над Na2SO4 и упаривали до сухого состояния. Неочищенный продукт очищали флэш-хроматографией на силикагеле (этилацетат/Hex 6:4) с получением 160 мг указанного в заголовке соединения в виде светло-желтого твердого вещества.
ВЭЖХ: Rt: 7,57 мин
1H ЯМР (600 МГц, DMSO-d6) δ м.д. 9,83 (с, 1H), 8,08 (д, J=5,1 Гц, 1H), 7,76 (с, 1H), 7,65-7,56 (м, 2H), 7,56-7,50 (м, 1H), 7,48-7,42 (м, 1H), 7,36-7,25 (м, 2H), 6,60 (дд, J=1,6, 5,2 Гц, 1H), 3,95 (тд, J=1,9, 9,6 Гц, 2H), 3,48 (дт, J=1,8, 11,6 Гц, 2H), 3,36-3,32 (м, 1H), 3,23 (с, 3H), 2,11-1,99 (м, 2H), 1,82-1,73 (м, 2H), 1,44 (с, 9H)
HRMS (ESI): вычислено для C32H34N4O6F3S2 [M+H]+ 691,1867, найдено 691,1866.
Аналогичным путем, но с использованием надлежащего исходного материала, также получали следующее промежуточное соединение:
Бензил-4-[5-{2-[(трет-бутоксикарбонил)амино]пиридин-4-ил}-4-(3-{[(2,5-дифторфенил)сульфонил](метоксиметил)амино}-2-фторфенил)-1,3-тиазол-2-ил]пиперидин-1-карбоксилат, соединение формулы (I)X [m, n = 1; R1 = 1-бензилоксикарбонилпиперидин-4-ил; R3, R5 = H; R4 = F; R6 = 2,5-дифторфенил; R18 = трет-бутоксикарбонил]
Способ F, стадия c1
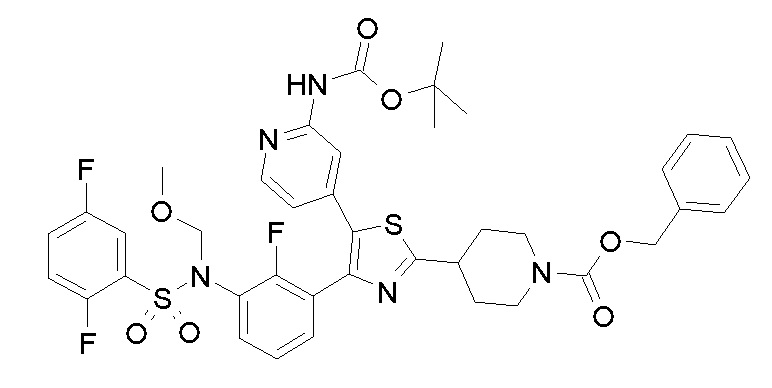
ВЭЖХ: Rt: 8,22 мин
1H ЯМР (600 МГц, DMSO-d6) δ м.д. 9,83 (с, 1H), 8,08 (д, J=5,5 Гц, 1H), 7,76 (с, 1H), 6,60 (дд, J=1,6, 5,2 Гц, 1H), 5,10 (с, 2H), 4,96 (с, 2H), 4,16-4,07 (м, J=13,4 Гц, 2H), 3,25-3,22 (м, 3H), 3,02 (ушир.с, 2H), 2,11 (д, J=11,0 Гц, 2H), 1,71-1,61 (м, 2H), 1,44 (с, 9H)
HRMS (ESI) [M+H]+: вычислено для C40H40O7N5S2F3 824,2394, найдено 824,2386.
N-{3-[5-(2-Аминопиридин-4-ил)-2-(тетрагидро-2H-пиран-4-ил)-1,3-тиазол-4-ил]-2-фторфенил}-2,5-дифторбензолсульфонамид, соединение формулы (I)Z2 (соединение 24) [m, n = 1; R1 = тетрагидропиран-4-ил; R3, R5 = H; R4 = F; R6 = 2,5-дифторфенил]
Способ F, стадия e1
трет-Бутил-{4-[4-(3-{[(2,5-дифторфенил)сульфонил](метоксиметил)амино}-2-фторфенил)-2-(тетрагидро-2H-пиран-4-ил)-1,3-тиазол-5-ил]пиридин-2-ил}карбамат (58 мг, 0,084 ммоль) растворяли в смеси 9:1 TFA/вода и перемешивали при 70°C в течение 1 ч. Смесь упаривали до сухого состояния, отбирали в DCM и промывали насыщенным водным NaHCO3 и насыщенным солевым раствором, сушили над Na2SO4 и концентрировали при пониженном давлении. Неочищенный продукт очищали флэш-хроматографией на силикагеле (чистый этилацетат) с получением 32 мг (70%) продукта, который растирали с диэтиловым эфиром, фильтровали и сушили в высоком вакууме. 30 мг указанного в заголовке соединения получали в виде белого твердого вещества.
ВЭЖХ: Rt: 5,86 мин
1H ЯМР (600 МГц, DMSO-d6) δ м.д. 10,69 (ушир.с, 1H), 7,70 (д, J=5,3 Гц, 1H), 7,61-7,52 (м, J=4,8, 7,7 Гц, 1H), 7,51-7,42 (м, 2H), 7,33 (т, J=7,0 Гц, 1H), 7,30-7,23 (м, 1H), 7,22-7,15 (м, 1H), 6,34 (с, 1H), 6,04 (ушир.с, 2H), 5,98 (д, J=5,1 Гц, 1H), 3,99-3,78 (м, 2H), 3,46 (дт, J=1,8, 11,6 Гц, 2H), 3,34-3,26 (м, 1H), 2,05-1,86 (м, 2H), 1,80-1,65 (м, 2H)
HRMS (ESI): вычислено для C25H22N4O3F3S2 [M+H]+ 547,1080, найдено 547,1092.
Аналогичным путем, но с использованием подходящего исходного материала, получали следующие соединения:
N-{3-[5-(2-Аминопиридин-4-ил)-2-(пиперидин-4-ил)-1,3-тиазол-4-ил]-2-фторфенил}-2,5-дифторбензолсульфонамид, соединение формулы (I)Z2 [m, n = 1; R1 = пиперидин-4-ил; R3, R5 = H; R4 = F; R6 = 2,5-дифторфенил]
Способ F, стадия e1
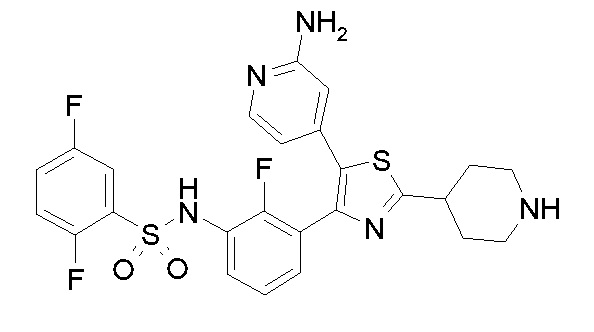
ВЭЖХ: Rt: 4,46 мин
1H ЯМР (600 МГц, DMSO-d6) (отдельные сигналы) δ м.д. 8,47 (с, 1H), 7,73 (д, J=5,3 Гц, 1H), 7,43 (ушир.с, 1H), 7,40-7,24 (м, 2H), 7,21 (ушир.с, 1H), 6,36 (с, 1H), 6,13 (ушир.с, 1H), 5,99 (с, 2H), 2,27-2,19 (м, 2H), 1,98-1,86 (м, 2H)
HRMS (ESI) [M+H]+: вычислено для C25H22O2N5S2F3 546,1240, найдено 546,1251.
Пример 13
Синтез N-{3-[5-(2-аминопиридин-4-ил)-2-(1-метилпиперидин-4-ил)-1,3-тиазол-4-ил]-2-фторфенил}-2,5-дифторбензолсульфонамида, соединение формулы (I)R2 (соединение 39) [m, n = 1; R2 = NH2; R3, R5 = H; R4 = F; R6 = 2,5-дифторфенил; R" = метил]
Способ C, стадии d, e и c2
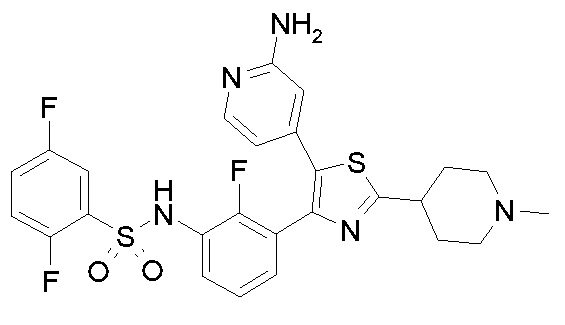
Бензил-4-[5-{2-[(трет-бутоксикарбонил)амино]пиридин-4-ил}-4-(3-{[(2,5-дифторфенил)сульфонил]-(метоксиметил)амино}-2-фторфенил)-1,3-тиазол-2-ил]пиперидин-1-карбоксилат (полученный, как описано в примере 10, 110 мг, 0,13 ммоль) растворяли в абсолютном этаноле (10 мл) в атмосфере азота. Затем добавляли формиат аммония (440 мг, 7 ммоль) и 5% палладий на угле (150 мг порционно). Смесь кипятили с обратным холодильником в течение 2 суток, затем фильтровали через слой целита и упаривали до сухого состояния. Осадок отбирали в DCM и промывали водой. Органический слой сушили над Na2SO4 и упаривали с получением 75 мг (84%) трет-бутил-{4-[4-(3-{[(2,5-дифторфенил)сульфонил]-(метоксиметил)амино}-2-фторфенил)-2-(пиперидин-4-ил)-1,3-тиазол-5-ил]пиридин-2-ил}карбамата.
ВЭЖХ/MS (ESI): 690 [M+H]+, 688 [M-H]-
Это промежуточное соединение (75 мг, 0,11 ммоль) растворяли в смеси MeOH (9 мл) и ледяной уксусной кислоты (28 мкл, 0,48 ммоль). Добавляли 37% водный раствор формальдегида (12 мкл, 0,24 ммоль) и цианоборгидрид натрия (16 мг, 0,32 ммоль). Раствор перемешивали при к.т. в течение 1 ч, а затем растворитель удаляли при пониженном давлении. Осадок отбирали в DCM и промывали разбавленным водным раствором аммиака. Наконец, органический слой сушили над Na2SO4 и упаривали до сухого состояния. Неочищенное вещество очищали флэш-хроматографией на силикагеле (циклогексан-этилацетат-этанол 4/2/1 для элюирования примесей, затем DCM-MeOH-NH4OH 20/5/0,5 для элюирования продукта) с получением 55 мг (71%) трет-бутил-{4-[4-(3-{[(2,5-дифторфенил)сульфонил](метоксиметил)амино}-2-фторфенил)-2-(1-метилпиперидин-4-ил)-1,3-тиазол-5-ил]пиридин-2-ил}карбамата.
ВЭЖХ/MS (ESI): 704 [M+H]+, 702 [M-H]-
Последнее промежуточное соединение (55 мг, 0,08 ммоль) растворяли в смеси TFA-H2O 9/1 (10 мл) и перемешивали при 70°C в течение 2 ч. Растворитель удаляли и осадок отбирали в DCM и промывали насыщенным водным раствором NaHCO3. Органический слой сушили над Na2SO4 и упаривали. Затем продукт очищали флэш-хроматографией на силикагеле (7н DCM-NH3 в MeOH для элюирования примесей, затем DCM-MeOH-30% NH4OH 20-5-0,5 для элюирования продукта) и растирали с диэтиловым эфиром с получением 30 мг (67%) указанного в заголовке соединения.
ВЭЖХ: Rt: 3,65 мин
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 7,72 (д, J=5,2 Гц, 1H), 7,47-7,37 (м, 2H), 7,38-7,29 (м, 1H), 7,25 (дт, J=1,8, 7,9 Гц, 1H), 7,04-6,96 (м, 1H), 6,92 (ушир.с, 1H), 6,35 (д, J=0,9 Гц, 1H), 6,07 (дд, J=1,3, 5,2 Гц, 1H), 5,98 (с, 2H), 3,18-2,91 (м, 4H), 2,43 (ушир.с, 5H), 2,18-2,10 (м, J=12,5 Гц, 2H), 1,89-1,76 (м, 2H)
HRMS (ESI) [M+H]+: вычислено для C26H24O2N5S2F3 560,1396, найдено 560,1403.
Пример 14
Синтез 2,5-дифтор-N-(2-фтор-3-{5-[2-(метиламино)пиридин-4-ил]-2-(тетрагидро-2H-пиран-4-ил)-1,3-тиазол-4-ил}фенил)бензолсульфонамида, соединение формулы (I)Z (соединение 25) [m, n = 1; R1 = тетрагидропиран-4-ил; R3, R5, R18 = H; R4 = F; R6 = 2,5-дифторфенил; R17 = метил]
Способ F, стадии f и d
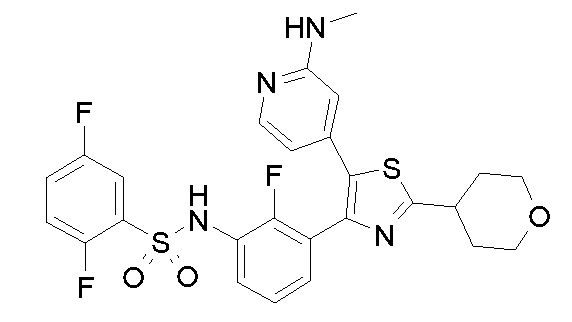
трет-Бутил-{4-[4-(3-{[(2,5-дифторфенил)сульфонил](метоксиметил)амино}-2-фторфенил)-2-(тетрагидро-2H-пиран-4-ил)-1,3-тиазол-5-ил]пиридин-2-ил}карбамат (полученный, как описано в примере 12, 97 мг, 0,140 ммоль) растворяли в сухом THF (1,5 мл) в атмосфере Ar. Раствор охлаждали до 0°C и добавляли метилиодид (0,02 мл, 0,321 ммоль, 2,3 экв.), а затем гидрид натрия (60% в минеральном масле) (20 мг, 0,353 ммоль, 2,5 экв.) и смесь перемешивали при к.т. в течение ночи. Затем смесь разбавляли водой и этилацетатом. Две фазы разделяли и водную фазу три раза экстрагировали этилацетатом. Объединенные органические слои промывали насыщенным солевым раствором, сушили над Na2SO4 и упаривали до сухого состояния. Осадок обрабатывали смесью TFA/вода 9:1 (2 мл) и перемешивали при 70°C в течение 1 ч. Смесь упаривали до сухого состояния, отбирали в DCM и промывали насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над Na2SO4 и концентрировали при пониженном давлении. Неочищенный продукт очищали флэш-хроматографией на силикагеле (этилацетат/Hex 3:1) с получением 59 мг продукта, который растирали с диэтиловым эфиром, фильтровали и сушили в высоком вакууме. Получили 36 мг (46%) указанного в заголовке соединения в виде белого твердого вещества.
ВЭЖХ: Rt: 6,24 мин
1H ЯМР (600 МГц, DMSO-d6) δ м.д. 10,68 (ушир.с, 1H), 7,80 (д, J=5,3 Гц, 1H), 7,56 (т, J=8,6 Гц, 1H), 7,52-7,40 (м, 2H), 7,36-7,31 (м, 1H), 7,30-7,23 (м, J=6,0 Гц, 1H), 7,22-7,16 (м, 1H), 6,54 (ушир.с, 1H), 6,24 (с, 1H), 6,03 (д, J=4,4 Гц, 1H), 3,96-3,89 (м, 2H), 3,46 (дт, J=1,9, 11,6 Гц, 2H), 3,32-3,26 (м, 1H), 2,66 (д, J=4,8 Гц, 3H), 2,04-1,94 (м, 2H), 1,79-1,68 (м, 2H)
HRMS (ESI): вычислено для C26H24N4O3F3S2 [M+H]+ 561,1237, найдено 561,1223.
Аналогичным путем, но с использованием подходящего исходного материала, также получали следующее соединение:
N-(3-{2-(1-Циклопропилпиперидин-4-ил)-5-[2-(метиламино)пиридин-4-ил]-1,3-тиазол-4-ил}-2-фторфенил)-2,5-дифторбензолсульфонамид, соединение формулы (I)Z (соединение 37) [m, n = 1; R1 = 1-циклопропилпиперидин-4-ил; R3, R5, R18 = H; R4 = F; R6 = 2,5-дифторфенил; R17 = метил]
Способ F, стадии f и d
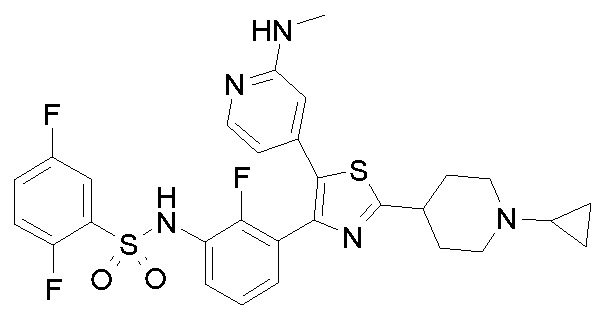
ВЭЖХ: Rt: 5,94 мин
1H ЯМР (600 МГц, DMSO-d6) δ м.д. 10,56 (ушир.с, 1H), 7,80 (д, J=5,5 Гц, 1H), 7,58-7,51 (м, 1H), 7,47-7,39 (м, 2H), 7,35-7,29 (м, 1H), 7,26-7,18 (м, 1H), 7,18-7,09 (м, 1H), 6,52 (кв, J=4,6 Гц, 1H), 6,23 (с, 1H), 6,04 (д, J=5,3 Гц, 1H), 3,03 (м, 3H), 2,66 (д, J=4,6 Гц, 3H), 2,38-2,30 (м, 2H), 2,09-2,01 (м, 2H), 1,76-1,60 (м, 3H), 0,49-0,42 (ушир.д, J =4,8 Гц, 2H), 0,34 (ушир.с, 2H)
HRMS (ESI): вычислено для C29H29N5O2F3S2 [M+H]+ 600,1709, найдено 600,1710.
Пример 15
N-{4-[4-(3-{[(2,5-дифторфенил)сульфонил]амино}-2-фторфенил)-2-(тетрагидро-2H-пиран-4-ил)-1,3-тиазол-5-ил]пиридин-2-ил}ацетамид, соединение формулы (I)Z1 (соединение 35) [m, n = 1; R1 = тетрагидропиран-4-ил; R3, R5 = H; R4 = F; R6 = 2,5-дифторфенил; R18 = ацетил]
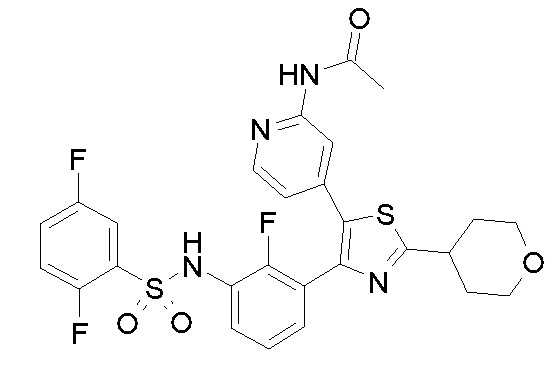
N-{4-[4-(3-{[(2,5-Дифторфенил)сульфонил](метоксиметил)амино}-2-фторфенил)-2-(тетрагидро-2H-пиран-4-ил)-1,3-тиазол-5-ил]пиридин-2-ил}ацетамид, соединение формулы (I)X [m, n = 1; R1 = тетрагидропиран-4-ил; R3, R5 = H; R4 = F; R6 = 2,5-дифторфенил; R18 = ацетил; Rx = метоксиметил]
Способ F, стадия c1
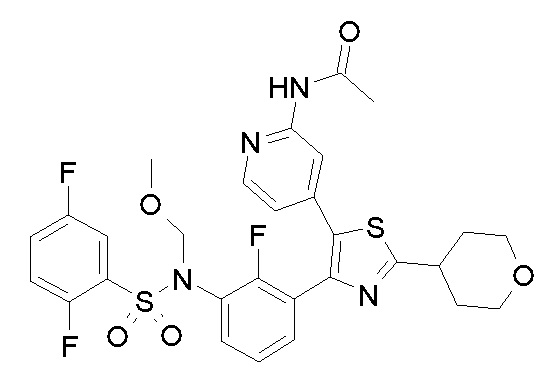
N-{3-[5-(2-Хлорпиридин-4-ил)-2-(тетрагидро-2H-пиран-4-ил)-1,3-тиазол-4-ил]-2-фторфенил}-2,5-дифтор-N-(метоксиметил)бензолсульфонамид (полученный, как описано в примере 9, 100 мг, 0,164 ммоль) растворяли в сухом THF (2 мл), и раствор дегазировали путем барботирования Ar в течение 5 мин. Затем добавляли ацетамид (20 мг, 0,339 ммоль, 2,1 экв.), а затем карбонат цезия (107 мг, 0,328 ммоль, 2 экв.), ацетат палладия (2 мг, 0,008 ммоль, 0,05 экв.) и Xantphos (10 мг, 0,016 ммоль, 0,1 экв.) и смесь облучали в микроволновой печи при 100°C в течение 30 мин. Смесь фильтровали через слой целита и целит промывали этилацетатом. Фильтрат промывали насыщенным водным раствором NaHCO3, водой и насыщенным солевым раствором, сушили над Na2SO4 и упаривали до сухого состояния. Неочищенный продукт очищали флэш-хроматографией на силикагеле (DCM/EtOH 98:2) с получением 50 мг указанного в заголовке соединения.
MS (ESI) [M+H]+ 633, [M-H]- 631
Аналогичным путем, но с использованием подходящего исходного материала, также получали следующие промежуточные соединения:
N-{4-[2-(1-Циклопропилпиперидин-4-ил)-4-(3-{[(2,5-дифторфенил)сульфонил](метоксиметил)амино}-2-фторфенил)-1,3-тиазол-5-ил]пиридин-2-ил}ацетамид, соединение формулы (I)X [m, n = 1; R1 = 1-циклопропилпиперидин-4-ил; R3, R5 = H; R4 = F; R6 = 2,5-дифторфенил; R18 = ацетил; Rx = метоксиметил]
Способ F, стадия c1
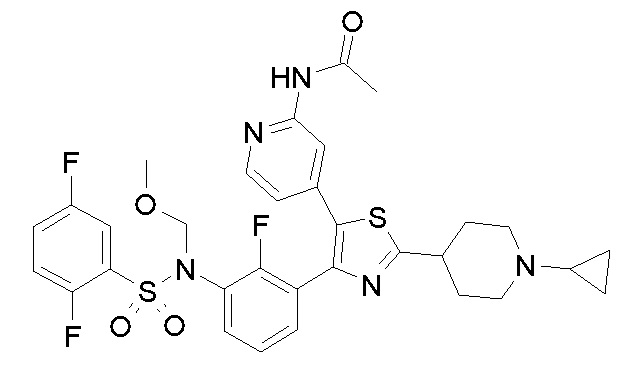
ВЭЖХ: Rt: 5,01 мин
1H ЯМР (600 МГц, DMSO-d6) δ м.д. 10,52 (с, 1H), 8,16 (д, J=5,4 Гц, 1H), 8,01 (с, 1H), 7,67-7,50 (м, 3H), 7,46-7,40 (м, 1H), 7,33-7,22 (м, 2H), 6,73 (дд, J=1,3, 5,1 Гц, 1H), 4,95 (с, 2H), 3,23 (с, 3H), 3,11-2,95 (м, 3H), 2,40-2,26 (м, 2H), 2,12-2,06 (м, J=1,7, 4,3 Гц, 2H), 2,04 (с, 3H), 1,76-1,55 (м, 3H), 0,49-0,38 (м, J=4,8 Гц, 2H), 0,31 (ушир.с, 2H)
HRMS (ESI): вычислено для C32H33N5O4F3S2 [M+H]+ 672,1921, найдено 672,1926.
N-{4-[2-трет-Бутил-4-(3-{[(2,5-дифторфенил)сульфонил](метоксиметил)амино}-2-фторфенил)-1,3-тиазол-5-ил]пиридин-2-ил}ацетамид, соединение формулы (I)X [m, n = 1; R1 = трет-бутил; R3, R5 = H; R4 = F; R6 = 2,5-дифторфенил; R18 = ацетил; Rx = метоксиметил]
Способ F, стадия c1
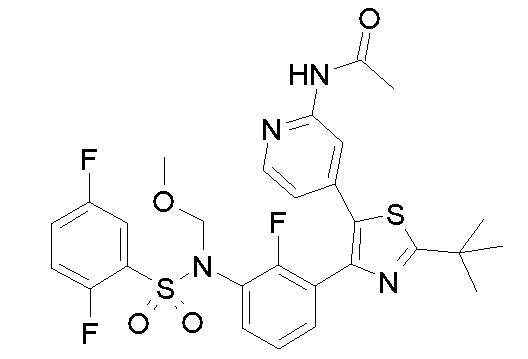
ВЭЖХ: Rt: 7,23 мин
MS (ESI) [M+H]+ 605, [M-H]- 603
Бензил-4-{5-[2-(ацетиламино)пиридин-4-ил]-4-(3-{[(2,5-дифторфенил)сульфонил](метоксиметил)амино}-2-фторфенил)-1,3-тиазол-2-ил}пиперидин-1-карбоксилат, соединение формулы (I)X [m, n = 1; R1 = 1-бензилоксикарбонилпиперидин-4-ил; R3, R5 = H; R4 = F; R6 = 2,5-дифторфенил; R18 = ацетил; Rx = метоксиметил]
Способ F, стадия c1
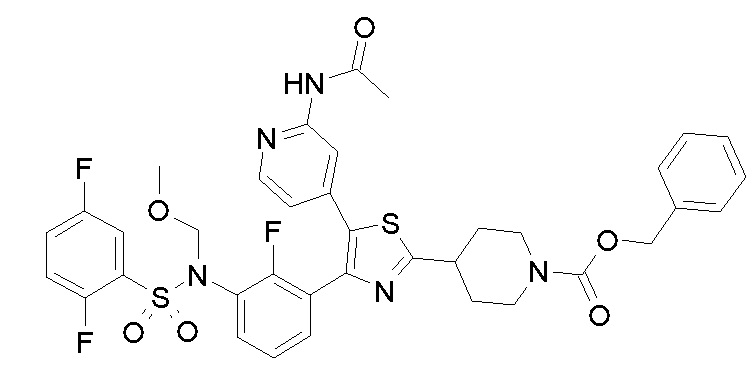
ВЭЖХ: Rt: 7,23 мин
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 10,52 (с, 1H), 8,17 (дд, J=0,7, 5,2 Гц, 1H), 8,02 (с, 1H), 7,66-7,48 (м, 3H), 7,46-7,41 (м, 1H), 7,39-7,35 (м, 3H), 7,34-7,26 (м, 3H), 6,73 (дд, J=1,6, 5,2 Гц, 1H), 5,10 (с, 2H), 4,96 (с, 2H), 4,10 (тд, J=3,9, 13,7 Гц, 1H), 3,23 (с, 3H), 3,05 (ушир.с, 2H), 2,11 (д, J=10,5 Гц, 2H), 2,04 (с, 3H), 1,76-1,56 (м, 2H)
HRMS (ESI) [M+H]+: вычислено для C37H34O6N5S2F3 766,1976, найдено 766,1995.
N-{4-[4-(3-{[(2,5-Дифторфенил)сульфонил]амино}-2-фторфенил)-2-(тетрагидро-2H-пиран-4-ил)-1,3-тиазол-5-ил]пиридин-2-ил}ацетамид, соединение формулы (I)Z1 (соединение 35) [m, n = 1; R1 = тетрагидропиран-4-ил; R3, R5 = H; R4 = F; R6 = 2,5-дифторфенил; R18 = ацетил]
Способ F, стадия d1
Неочищенный N-{4-[2-(тетрагидропиран-4-ил)-4-(3-{[(2,5-дифторфенил)сульфонил](метоксиметил)амино}-2-фторфенил)-1,3-тиазол-5-ил]пиридин-2-ил}ацетамид (50 мг) обрабатывали смесью TFA/вода 9:1 (1 мл) и перемешивали при 70°C в течение 1,5 ч. Смесь упаривали до сухого состояния, отбирали в DCM и промывали насыщенным водным NaHCO3 и насыщенным солевым раствором, сушили над Na2SO4 и концентрировали при пониженном давлении. Неочищенный продукт очищали флэш-хроматографией на силикагеле (DCM/MeOH 95:5) с получением 35 мг продукта, который растирали с этиловым эфиром, фильтровали и сушили в высоком вакууме. Получили 28 мг (60%) указанного в заголовке соединения в виде не совсем белого твердого вещества.
ВЭЖХ: Rt: 5,84 мин
1H ЯМР (600 МГц, DMSO-d6) δ м.д. 10,63 (с, 1H), 10,54 (с, 1H), 8,12 (д, J=5,3 Гц, 1H), 8,04 (с, 1H), 7,59-7,51 (м, 1H), 7,47-7,42 (м, 1H), 7,42-7,38 (м, 1H), 7,37-7,28 (м, 2H), 7,25-7,17 (м, 1H), 6,62 (дд, J=1,6, 5,3 Гц, 1H), 3,93 (тд, J=2,2, 9,4 Гц, 2H), 3,47 (дт, J=2,2, 11,6 Гц, 2H), 3,39-3,31 (м, 1H), 2,07 (с, 3H), 2,03-1,97 (м, 2H), 1,81-1,69 (м, 2H)
HRMS (ESI): вычислено для C27H24N4O4F3S2 [M+H]+ 589,1186, найдено 589,1187.
Аналогичным путем, но с использованием подходящего исходного материала, также получали следующие соединения:
N-{4-[2-(1-Циклопропилпиперидин-4-ил)-4-(3-{[(2,5-дифторфенил)сульфонил]амино}-2-фторфенил)-1,3-тиазол-5-ил]пиридин-2-ил}ацетамид, соединение формулы (I)Z1 (соединение 30) [m, n = 1; R1 = 1-циклопропилпиперидин-4-ил; R3, R5 = H; R4 = F; R6 = 2,5-дифторфенил; R18 = ацетил]
Способ F, стадия d1
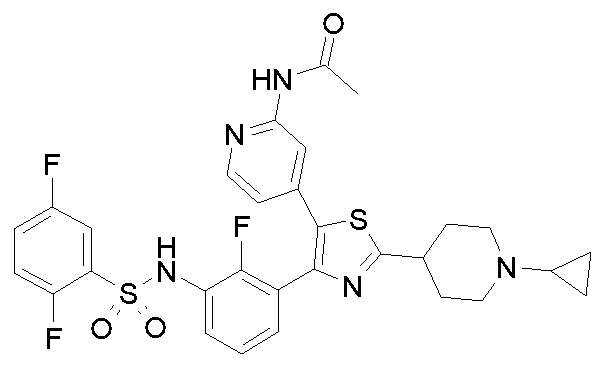
ВЭЖХ: Rt: 5,57 мин
1H ЯМР (600 МГц, DMSO-d6) δ м.д. 10,54 (с, 1H), 8,12 (д, J=5,3 Гц, 1H), 8,04 (с, 1H), 7,53 (т, J=8,3 Гц, 1H), 7,47-7,37 (м, 2H), 7,33-7,23 (м, 2H), 7,21-7,12 (м, 1H), 6,62 (дд, J=1,6, 5,3 Гц, 1H), 3,14-2,96 (м, 3H), 2,38 (дд, J=1,8, 3,7 Гц, 2H), 2,12-2,01 (м, 2H), 2,06 (с, 3H), 1,79-1,56 (м, 3H), 0,45 (д, J=4,8 Гц, 2H), 0,35 (ушир.с, 2H)
HRMS (ESI): вычислено для C30H29N5O3F3S2 [M+H]+ 628,1659, найдено 628,1659.
N-{4-[2-трет-Бутил-4-(3-{[(2,5-дифторфенил)сульфонил]амино}-2-фторфенил)-1,3-тиазол-5-ил]пиридин-2-ил}ацетамид, соединение формулы (I)Z1 (соединение 42) [m, n = 1; R1 = трет-бутил; R3, R5 = H; R4 = F; R6 = 2,5-дифторфенил; R18 = ацетил]
Способ F, стадия d1
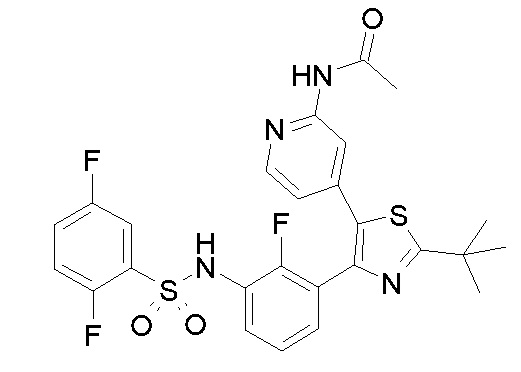
ВЭЖХ: Rt: 6,11 мин
1H ЯМР (600 МГц, DMSO-d6) δ м.д. 10,63 (с, 1H), 10,54 (с, 1H), 8,12 (д, J=5,3 Гц, 1H), 8,05 (с, 1H), 7,59-7,51 (ддд, J=4,2, 4,2, 8,4 Гц, 1H), 7,45 (дт, J=3,9, 9,1 Гц, 1H), 7,43-7,38 (м, 1H), 7,37-7,29 (м, 2H), 7,25-7,16 (м, 1H), 6,61 (дд, J=1,6, 5,3 Гц, 1H), 2,07 (с, 3H), 1,43 (с, 9H)
HRMS (ESI): вычислено для C26H24N4O3F3S2 [M+H]+ 561,1237, найдено 561,1257.
N-{4-[4-(3-{[(2,5-Дифторфенил)сульфонил]амино}-2-фторфенил)-2-(пиперидин-4-ил)-1,3-тиазол-5-ил]пиридин-2-ил}ацетамид; соединение формулы (I)Z1 (соединение 44) [m, n = 1; R1 = пиперидин-4-ил; R3, R5 = H; R4 = F; R6 = 2,5-дифторфенил; R18 = ацетил]
Способ F, стадия d1
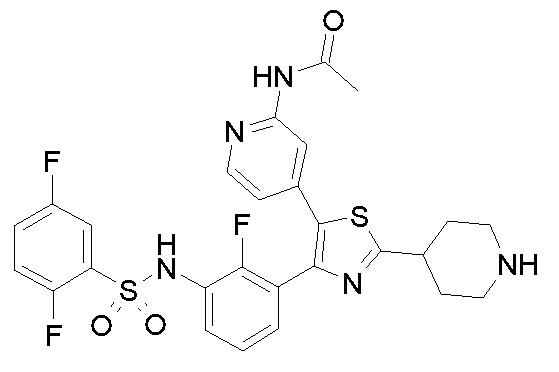
ВЭЖХ: Rt: 4,70 мин
1H ЯМР (401 МГц, DMSO-d6) (отдельные сигналы) δ м.д. 10,56 (с, 1H), 8,12 (д, J=4,8 Гц, 1H), 7,43-7,17 (м, 4H), 6,92 (ушир.с, 1H), 6,72-6,67 (м, 1H), 3,04 (дт, J=2,9, 12,5 Гц, 1H), 2,30-2,19 (м, J=2,8, 14,2 Гц, 2H), 2,07 (с, 3H), 1,99-1,85 (м, 2H)
HRMS (ESI) [M+H]+: вычислено для C27H24O3N5S2F3 588,1346, найдено 588,1349.
Пример 16
Синтез N-{4-[4-(3-{[(2,5-дифторфенил)сульфонил]амино}-2-фторфенил)-2-(1-метилпиперидин-4-ил)-1,3-тиазол-5-ил]пиридин-2-ил}ацетамида, соединение формулы (I)Z1 (соединение 23) [m, n = 1; R1 = 1-метилпиперидин-4-ил; R3, R5 = H; R4 = F; R6 = 2,5-дифторфенил; R18 = ацетил]
Способ F (аналогично способу C, стадия e)
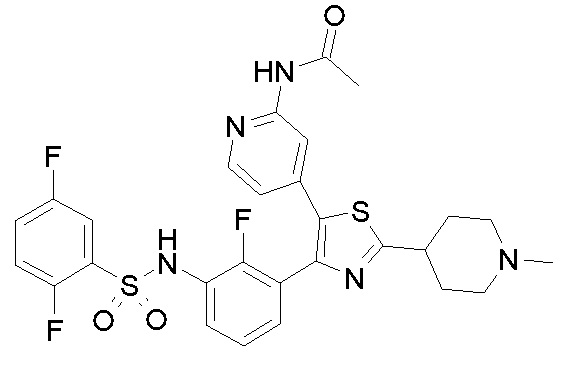
N-{4-[4-(3-{[(2,5-дифторфенил)сульфонил](метоксиметил)амино}-2-фторфенил)-2-(пиперидин-4-ил)-1,3-тиазол-5-ил]пиридин-2-ил}ацетамид (полученный, как описано в примере 15, 50 мг, 0,085 ммоль) растворяли в MeOH (5 мл) и ледяной уксусной кислоте (15 мкл, 0,26 ммоль). Затем в смесь добавляли 37% водный раствор формальдегида (6,5 мкл, 0,13 ммоль) и цианоборгидрид натрия (8,5 мг, 0,17 ммоль), и ее перемешивали при к.т. в течение 1 ч. Растворитель удаляли при пониженном давлении и осадок распределяли между этилацетатом и водным раствором NaHCO3. Органическую фазу сушили над Na2SO4 и упаривали до сухого состояния. Продукт очищали флэш-хроматографией на силикагеле (DCM-MeOH 4/1) с получением 30 мг (59%) указанного в заголовке соединения после растирания с петролейным эфиром.
ВЭЖХ: Rt: 3,72 мин
1H ЯМР (600 МГц, DMSO-d6) (отдельные сигналы) δ м.д. 10,55 (с, 1H), 8,11 (д, J=5,3 Гц, 1H), 8,09 (с, 1H), 7,44-7,36 (м, J=5,3 Гц, 2H), 7,35-7,28 (м, 1H), 7,26 (т, J=7,5 Гц, 1H), 7,02 (ушир.с, 2H), 6,66 (дд, J=1,4, 5,2 Гц, 1H), 3,18-2,97 (м, 3H), 2,44 (ушир.с, 3H), 2,20-2,12 (м, J=14,1 Гц, 2H), 2,07 (с, 3H), 1,91-1,78 (м, 2H)
HRMS (ESI) [M+H]+: вычислено для C28H26O3N5S2F3 602,1502, найдено 602,1503.
Пример 17
Синтез 2,5-дифтор-N-(2-фтор-3-{5-[2-(метиламино)пиридин-4-ил]-2-(1-метилпиперидин-4-ил)-1,3-тиазол-4-ил}фенил)бензолсульфонамида, соединение формулы (I)Z (соединение 38) [m, n = 1; R1 = 1-метилпиперидин-4-ил; R3, R5, R17 = H; R4 = F; R6 = 2,5-дифторфенил; R18 = метил]
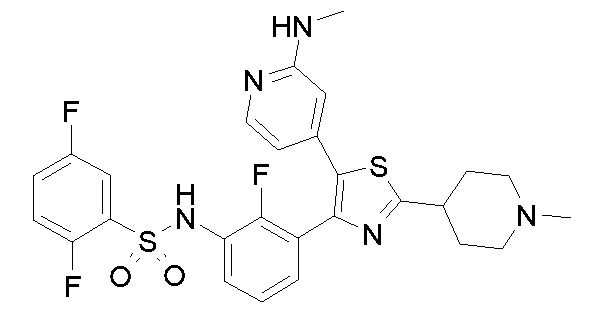
Бензил-4-[5-{2-[(трет-бутоксикарбонил)(метил)амино]пиридин-4-ил}-4-(3-{[(2,5-дифторфенил)сульфонил](метоксиметил)амино}-2-фторфенил)-1,3-тиазол-2-ил]пиперидин-1-карбоксилат, соединение формулы 49 [m, n = 1; R1 = 1-бензилоксикарбонилпиперидин-4-ил; R3, R5 = H; R4 = F; R6 = 2,5-дифторфенил; R17’ = трет-бутоксикарбонил; R18 = метил; Rx = метоксиметил]
Способ F, стадия c
Бензил-4-[5-(2-хлорпиридин-4-ил)-4-(3-{[(2,5-дифторфенил)сульфонил](метоксиметил)амино}-2-фторфенил)-1,3-тиазол-2-ил]пиперидин-1-карбоксилат (полученный, как описано в примере 9, 100 мг, 0,14 ммоль) растворяли в сухом THF (6 мл) и последовательно добавляли Cs2CO3 (90 мг, 0,28 ммоль), Xantphos (17 мг, 0,03 ммоль) трет-бутилметилкарбамат (полученный, как описано в методике получения 8, 74 мг, 0,56 ммоль) и Pd(AcO)2 (12 мг, 0,03 ммоль). Смесь подвергали микроволновому облучению в закрытой колбе при 120°C в течение 2 ч три раза. Затем суспензию фильтровали через слой целита и растворитель выпаривали при пониженном давлении. Затем суспензию фильтровали через слой целита и растворитель выпаривали при пониженном давлении. Затем неочищенное вещество очищали флэш-хроматографией (циклогексан-этилацетат-этанол 4/0,5/0,5) с получением 73 мг (62%) бензил-4-[5-{2-[(трет-бутоксикарбонил)(метил)амино]пиридин-4-ил}-4-(3-{[(2,5-дифторфенил)сульфонил](метоксиметил)амино}-2-фторфенил)-1,3-тиазол-2-ил]пиперидин-1-карбоксилата.
ВЭЖХ: Rt: 7,66 мин
1H ЯМР (600 МГц, DMSO-d6) δ м.д. 8,24 (д, J=5,3 Гц, 1H), 7,65-7,57 (м, 2H), 7,53 (дт, J=3,9, 9,4 Гц, 1H), 7,43 (ддд, J=3,1, 5,0, 7,6 Гц, 1H), 7,39-7,35 (м, 3H), 7,34-7,29 (м, 2H), 6,75 (дд, J=1,6, 5,2 Гц, 1H), 5,10 (с, 2H), 4,96 (с, 2H), 4,17-4,06 (м, J=7,9 Гц, 2H), 3,28-3,27 (м, 3H), 3,24-3,19 (м, 3H), 3,02 (ушир.с, 2H), 2,12 (д, J=4,6 Гц, 1H), 1,71-1,62 (м, 2H), 1,41 (с, 9H)
HRMS (ESI) [M+H]+: вычислено для C41H42O7N5S2F3 838,2551, найдено 838,2573.
трет-Бутил {4-[4-(3-{[(2,5-дифторфенил)сульфонил](метоксиметил)амино}-2-фторфенил)-2-(пиперидин-4-ил)-1,3-тиазол-5-ил]пиридин-2-ил}метилкарбамат, соединение формулы 47 [m, n = 1; R2’’ = N-метил-N-трет-бутоксикарбониламин; R3, R5 = H; R4 = F; R6 = 2,5-дифторфенил; Rx = метоксиметил]
Способ C, стадия d
Бензил-4-[5-{2-[(трет-бутоксикарбонил)(метил)амино]пиридин-4-ил}-4-(3-{[(2,5-дифторфенил)сульфонил](метоксиметил)амино}-2-фторфенил)-1,3-тиазол-2-ил]пиперидин-1-карбоксилат (177 мг, 0,21 ммоль) растворяли в абсолютном этаноле (7 мл) в атмосфере азота. Затем добавляли формиат аммония (53 мг, 0,84 ммоль) и 5% палладий на угле (150 мг порционно). Смесь кипятили с обратным холодильником в течение 2 суток, затем фильтровали через слой целита и упаривали до сухого состояния. Осадок отбирали в DCM и промывали водой. Органический слой сушили над Na2SO4 и упаривали. Затем неочищенное вещество очищали флэш-хроматографией на силикагеле (DCM-MeOH для элюирования примесей, затем DCM-MeOH-30% NH4OH 20-5-0,5 для элюирования продукта) с получением 65 мг (44%) указанного в заголовке соединения.
ВЭЖХ: Rt: 5,60 мин
1H ЯМР (500 МГц, DMSO-d6) δ м.д. 8,24 (д, J=5,2 Гц, 1H), 7,70-7,52 (м, 5H), 7,44 (ушир.с, 1H), 6,76 (д, J=5,2 Гц, 1H), 4,96 (с, 2H)
HRMS (ESI) [M+H]+: вычислено для C33H36O5N5S2F3 704,2183, найдено 704,2191.
трет-Бутил {4-[4-(3-{[(2,5-дифторфенил)сульфонил](метоксиметил)амино}-2-фторфенил)-2-(1-метилпиперидин-4-ил)-1,3-тиазол-5-ил]пиридин-2-ил}метилкарбамат, соединение формулы 48 [m, n = 1; R’’ = метил; R2’’ = N-метил-N-трет-бутоксикарбониламин; R3, R5 = H; R4 = F; R6 = 2,5-дифторфенил; Rx = метоксиметил]
Способ C, стадия e
трет-Бутил-{4-[4-(3-{[(2,5-дифторфенил)сульфонил](метоксиметил)амино}-2-фторфенил)-2-(пиперидин-4-ил)-1,3-тиазол-5-ил]пиридин-2-ил}метилкарбамат (65 мг, 0,09 ммоль) растворяли в MeOH (5 мл) и ледяной уксусной кислоте (15 мкл, 0,26 ммоль). Затем к смеси добавляли 37% водный раствор формальдегида (6,5 мкл, 0,13 ммоль) и цианоборгидрид натрия (8,5 мг, 0,17 ммоль), и ее перемешивали при к.т. в течение 1 ч. Растворитель удаляли при пониженном давлении и осадок распределяли между этилацетатом и 15% водным раствором аммиака. Органическую фазу сушили над Na2SO4 и упаривали до сухого состояния с получением 34 мг трет-бутил-{4-[4-(3-{[(2,5-дифторфенил)сульфонил](метоксиметил)амино}-2-фторфенил)-2-(1-метилпиперидин-4-ил)-1,3-тиазол-5-ил]пиридин-2-ил}метилкарбамата, который использовали на следующей стадии без какой-либо дополнительной очистки.
ВЭЖХ/MS (ESI): 718 [M+H]+, 716 [M-H]-
2,5-Дифтор-N-(2-фтор-3-{5-[2-(метиламино)пиридин-4-ил]-2-(1-метилпиперидин-4-ил)-1,3-тиазол-4-ил}фенил)бензолсульфонамид, соединение формулы (I)Z (соединение 38) [m, n = 1; R1 = 1-метилпиперидин-4-ил; R3, R5, R17 = H; R4 = F; R6 = 2,5-дифторфенил; R18 = метил]
Способ F, стадия d
трет-Бутил-{4-[4-(3-{[(2,5-дифторфенил)сульфонил](метоксиметил)амино}-2-фторфенил)-2-(1-метилпиперидин-4-ил)-1,3-тиазол-5-ил]пиридин-2-ил}метилкарбамат (34 мг, 0,05 ммоль) растворяли в смеси TFA-H2O 9/1 (5 мл) и перемешивали при 70°C в течение 2 ч. Растворитель удаляли при пониженном давлении и осадок распределяли между DCM и 15% водным раствором NH4OH. Затем водную фазу экстрагировали два раза этилацетатом и растворитель упаривали до сухого состояния. Неочищенное вещество очищали ОФ-ВЭЖХ (подвижная фаза A: 0,05% NH3 в воде/ацетонитрил (95:5), подвижная фаза B: ацетонитрил/H2O (95:5); градиент от 0 до 60% B в течение 15 мин, затем возрастание до 100% B в течение 0,1 мин) с получением 15 мг (37%) указанного в заголовке соединения в виде соли трифторуксусной кислоты.
ВЭЖХ: Rt: 3,85 мин
1H ЯМР (600 МГц, DMSO-d6) δ м.д. 10,70 (ушир.с, 1H), 9,46 (ушир.с, 1H), 7,83 (д, J=6,0 Гц, 1H), 7,64-7,54 (м, 1H), 7,52-7,41 (м, 2H), 7,39-7,30 (м, 1H), 7,27-7,21 (м, 1H), 6,65-6,39 (м, 1H), 6,27 (ушир.с, 1H), 3,56 (ушир.с, 2H), 3,14-3,04 (м, 2H), 2,82 (д, J=3,5 Гц, 2H), 2,78 (ушир.с, 2H), 2,35-2,28 (м, J=14,3 Гц, 2H), 1,99-1,86 (м, 2H)
HRMS (ESI) [M+H]+: вычислено для C27H26O2N5S2F3 574,1553, найдено 574,1564.
Аналогичным путем, но с использованием подходящего альдегида на стадии восстановительного аминирования, также получали следующее соединение:
N-(3-{2-(1-Этилпиперидин-4-ил)-5-[2-(метиламино)пиридин-4-ил]-1,3-тиазол-4-ил}-2-фторфенил)-2,5-дифторбензолсульфонамид, соединение формулы (I)Z [m, n = 1; R1 = 1-этилпиперидин-4-ил; R3, R5, R17 = H; R4 = F; R6 = 2,5-дифторфенил; R18 = метил]
Способ F, стадия d
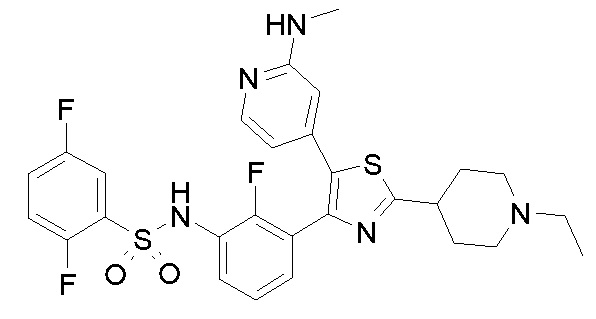
ВЭЖХ: Rt: 4,90 мин
1H ЯМР (600 МГц, DMSO-d6) δ м.д. 10,70 (ушир.с, 1H), 9,19 (ушир.с, 1H), 7,82 (д, J=5,9 Гц, 1H), 7,58 (т, J=8,4 Гц, 1H), 7,52-7,41 (м, 3H), 7,37-7,29 (м, 2H), 7,27-7,20 (м, 1H), 6,48 (ушир.с, 1H), 6,24 (ушир.с, 1H), 3,64-3,56 (м, J=12,3 Гц, 2H), 3,19-3,12 (м, 2H), 3,09-3,00 (м, 2H), 2,76 (ушир.с, 3H), 2,33 (д, J=13,6 Гц, 2H), 2,00-1,86 (м, 2H), 1,27-1,22 (м, 3H)
HRMS (ESI) [M+H]+: вычислено для C28H28O2N5S2F3 588,1709, найдено 588,1729.
Пример 18
Синтез N-{3-[2-(1-циклопропилпиперидин-4-ил)-5-(2-метилпиридин-4-ил)-1,3-тиазол-4-ил]-2-фторфенил}-2,5-дифторбензолсульфонамида, соединение формулы (I)AC (соединение 31) [m, n = 1; R1 = 1-циклопропилпиперидин-4-ил; R3, R5 = H; R4 = F; R6 = 2,5-дифторфенил; R2’ = метил]
Способ F, стадии g и d2

N-{3-[5-(2-Хлорпиридин-4-ил)-2-(1-циклопропилпиперидин-4-ил)-1,3-тиазол-4-ил]-2-фторфенил}-2,5-дифтор-N-(метоксиметил)бензолсульфонамид (полученный, как описано в примере 9, 100 мг, 0,154 ммоль) растворяли в сухом диоксане (1 мл). Через раствор барботировали аргон в течение 5 мин и добавляли 2M раствор AlMe3 в Hex (0,154 мл, 0,308 ммоль, 2 экв.), а затем Pd(PPh3)4 (4 мг, 0,004 ммоль, 0,02 экв.). Смесь нагревали до 105°C в закрытой пробирке в течение 2 ч. Затем ее разбавляли этилацетатом и насыщенным водным NaHCO3. Две фазы разделяли и водную фазу экстрагировали три раза этилацетатом. Объединенные органические слои промывали насыщенным солевым раствором, сушили над Na2SO4 и концентрировали при пониженном давлении. Неочищенный N-{3-[2-(1-циклопропилпиперидин-4-ил)-5-(2-метилпиридин-4-ил)-1,3-тиазол-4-ил]-2-фторфенил}-2,5-дифтор-N-(метоксиметил)бензолсульфонамид обрабатывали смесью TFA/вода 9:1 при 70°C в течение 1,5 ч. Смесь упаривали до сухого состояния, отбирали в DCM и промывали насыщенным водным NaHCO3 и насыщенным солевым раствором, сушили над Na2SO4 и концентрировали при пониженном давлении. Неочищенный продукт очищали флэш-хроматографией на силикагеле (DCM/MeOH 95:5) с получением 72 мг (80%) указанного в заголовке соединения в виде светло-желтого твердого вещества.
ВЭЖХ: Rt: 6,00 мин
1H ЯМР (600 МГц, DMSO-d6) δ м.д. 10,56 (ушир.с, 1H), 8,29 (д, J=5,3 Гц, 1H), 7,51-7,59 (м, 1H), 7,44 (дт, J=4,0, 9,2 Гц, 1H), 7,40-7,36 (м, 1H), 7,33 (дт, J=1,5, 7,7 Гц, 1H), 7,27 (м, 1H), 7,22-7,15 (м, 1H), 7,02 (с, 1H), 6,79 (дд, J=1,4, 5,3 Гц, 1H), 3,07-3,00 (м, 3H), 2,38 (с, 3H), 2,42-2,33 (м, 2H), 2,06 (д, J=12,6 Гц, 2H), 1,76-1,60 (м, 3H), 0,45 (ушир.д, J=4,6 Гц, 2H), 0,35 (ушир.с, 2H)
HRMS (ESI): вычислено для C29H28N4O2F3S2 [M+H]+ 585,1601, найдено 585,1599.
Аналогичным путем, но с использованием подходящего исходного материала, получали следующие соединения:
2,5-Дифтор-N-{2-фтор-3-[5-(2-метилпиридин-4-ил)-2-(тетрагидро-2H-пиран-4-ил)-1,3-тиазол-4-ил]фенил}бензолсульфонамид, соединение формулы (I)AC (соединение 28) [m, n = 1; R1 = тетрагидропиран-4-ил; R3, R5 = H; R4 = F; R6 = 2,5-дифторфенил; R2’ = метил]
Способ F, стадии g и d2
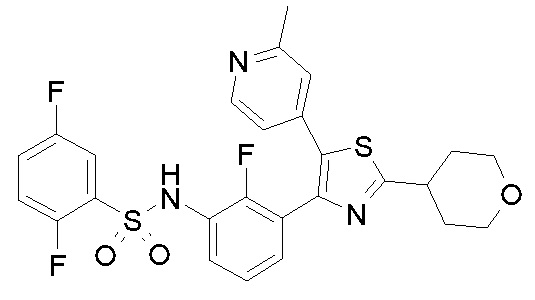
ВЭЖХ: Rt: 6,23 мин
1H ЯМР (600 МГц, DMSO-d6) (отдельные сигналы) δ м.д. 10,67 (с, 1H), 8,29 (д, J=5,2 Гц, 1H), 7,63-7,53 (м, 1H), 7,46 (дт, J=3,9, 9,2 Гц, 1H), 7,43-7,38 (м, 1H), 7,37-7,29 (м, 2H), 7,25-7,17 (м, 1H), 7,03 (с, 1H), 6,79 (дд, J=1,2, 5,2 Гц, 1H), 3,93 (тд, J=2,1, 9,6 Гц, 2H), 3,53-3,42 (м, 2H), 2,39 (с, 3H), 2,01 (м, 2H), 1,81-1,63 (м, 2H)
HRMS (ESI): вычислено для C26H23N3O3F3S2 [M+H]+ 546,1128, найдено 546,1127.
2,5-Дифтор-N-{2-фтор-3-[5-(2-метилпиридин-4-ил)-2-(пиперидин-4-ил)-1,3-тиазол-4-ил]фенил}бензолсульфонамид, соединение формулы (I)AC [m, n = 1; R1 = пиперидин-4-ил; R3, R5 = H; R4 = F; R6 = 2,5-дифторфенил; R2’ = метил]
Способ F, стадии g и d2
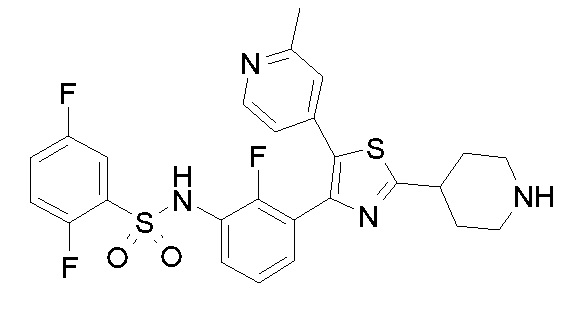
ВЭЖХ: Rt: 4,79 мин
1H ЯМР (600 МГц, DMSO-d6) δ м.д. 8,33 (д, J=5,3 Гц, 1H), 7,42-7,35 (м, 1H), 7,30-7,14 (м, 3H), 7,08 (ушир.с, 1H), 6,91 (д, J=5,3 Гц, 1H), 6,88-6,78 (м, 1H), 6,60 (ушир.с, 1H), 3,49-3,27 (м, 3H), 3,02 (дт, J=2,4, 12,4 Гц, 2H), 2,37 (с, 3H), 2,23 (дд, J=2,4, 14,0 Гц, 2H), 1,96-1,83 (м, 2H)
HRMS (ESI): вычислено для C26H24N4O2F3S2 [M+H]+ 545,1288, найдено 545,1304.
Пример 19
Синтез 2,5-дифтор-N-{2-фтор-3-[2-(1-метилпиперидин-4-ил)-5-(2-метилпиридин-4-ил)-1,3-тиазол-4-ил]фенил}бензолсульфонамида, соединение формулы (I)AC (соединение 26) [m, n = 1; R1 = 1-метилпиперидин-4-ил; R3, R5 = H; R4 = F; R6 = 2,5-дифторфенил; R2’ = метил]
Способ F (аналогично способу C, стадия e)
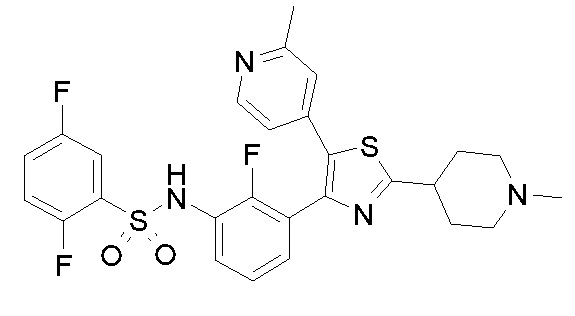
2,5-Дифтор-N-{2-фтор-3-[5-(2-метилпиридин-4-ил)-2-(пиперидин-4-ил)-1,3-тиазол-4-ил]фенил}бензолсульфонамид (полученный, как описано в примере 18, 100 мг, 0,184 ммоль) растворяли в MeOH (2 мл). Добавляли водный раствор формальдегида (37%, 0,021 мл, 0,276 ммоль, 1,5 экв.), а затем уксусную кислоту (0,032 мл, 0,552 ммоль, 3 экв.) и цианоборгидрид натрия (22 г, 0,294 ммоль, 1,6 экв.) и смесь перемешивали при к.т. в течение 1 ч. Затем смесь разбавляли этилацетатом, промывали насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над Na2SO4 и упаривали до сухого состояния. Неочищенный продукт очищали флэш-хроматографией на силикагеле (DCM/MeOH/7н NH3 в MeOH 90:9:1), обрабатывали диэтиловым эфиром, фильтровали и сушили в высоком вакууме с получением 68 мг (66%) указанного в заголовке соединения в виде светло-желтого твердого вещества.
ВЭЖХ: Rt: 4,87 мин
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 8,29 (д, J=5,1 Гц, 1H), 7,41-7,37 (м, 1H), 7,37-7,33 (м, 1H), 7,29 (дт, J=4,0, 8,9 Гц, 1H), 7,27-7,21 (м, 1H), 7,06 (ушир.с, 1H), 6,97 (т, J=7,4 Гц, 1H), 6,86 (дд, J=1,1, 5,1 Гц, 1H), 6,86-6,82 (м, 1H), 3,13-3,06 (м, 1H), 3,06-2,98 (м, 1H), 2,44-2,27 (м, 5H), 2,15-2,10 (м, 2H), 1,87-1,76 (м, 2H)
HRMS (ESI): вычислено для C27H26N4O2F3S2 [M+H]+ 559,1444, найдено 559,1464.
Аналогичным путем, но с использованием подходящего альдегида, также получали следующее соединение:
2,5-Дифтор-N-{2-фтор-3-[2-(1-этилпиперидин-4-ил)-5-(2-метилпиридин-4-ил)-1,3-тиазол-4-ил]фенил}бензолсульфонамид, соединение формулы (I)AC [m, n = 1; R1 = 1-этилпиперидин-4-ил; R3, R5 = H; R4 = F; R6 = 2,5-дифторфенил; R2’ = метил]
Способ F (аналогично способу C, стадия e)

ВЭЖХ: Rt: 5,00 мин
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 8,30 (д, J=5,3 Гц, 1H), 7,45-7,36 (м, 2H), 7,36-7,30 (м, 2H), 7,27 (м, 1H), 7,05 (ушир.с, 1H), 7,04-6,89 (м, 2H), 6,85 (ушир.д, J=5,3 Гц, 1H), 2,87-2,54 (м, 3H), 2,42-2,28 (м, 5H), 2,27-2,10 (м, 2H), 2,09-2,09 (м, 2H), 1,92-1,73 (м, 2H), 1,13 (т, J=7,1 Гц, 3H)
HRMS (ESI): вычислено для C28H28N4O2F3S2 [M+H]+ 573,1601, найдено 573,1600.
Пример 20
Синтез 2,5-дифтор-N-{2-фтор-3-[5-(3-фторпиридин-4-ил)-2-(тетрагидро-2H-пиран-4-ил)-1,3-тиазол-4-ил]фенил}бензолсульфонамида, соединение формулы (I)B (соединение 34) [m, n = 1; R2, R5 = H; R3 = 3-F; R4 = F; R6 = 2,5-дифторфенил; R1’ = тетрагидропиран-4-ил]
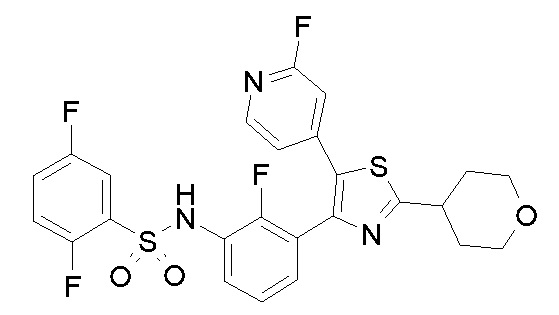
2,5-Дифтор-N-{2-фтор-3-[2-(тетрагидро-2H-пиран-4-ил)-1,3-тиазол-4-ил]фенил}-N-(метоксиметил)бензолсульфонамид, соединение формулы 9 [n = 1; R1 = тетрагидропиран-4-ил; R4 = F; R5 = H; R6 = 2,5-дифторфенил; Rx = метоксиметил]
Способ A, стадии d1 и e
К раствору N-(3-ацетил-2-фторфенил)-2,5-дифтор-N-(метоксиметил)бензолсульфонамида (полученный, как описано в методике получения 1, 2,55 г, 6,830 ммоль) в сухом THF (34 мл) добавляли пиридиния бромид пербромид (2,18 г, 6,83 ммоль, 1 экв.) и смесь кипятили с обратным холодильником в течение 1 ч. Затем смесь концентрировали при пониженном давлении, отбирали в этилацетат и промывали 0,25M HCl, водой и насыщенным солевым раствором. Органическую фазу сушили над Na2SO4 и упаривали до сухого состояния с получением 4,1 г неочищенного N-[3-(бромацетил)-2-фторфенил]-2,5-дифтор-N-(метоксиметил)бензолсульфонамида.
Половину этого материала (3,40 ммоль) растворяли в абсолютном этаноле (34 мл). Добавляли тетрагидро-2H-пиран-4-карботиоамид (полученный, как описано в методике получения 6, 595 мг, 1,2 экв.) и смесь перемешивали при 60°C в течение 1 ч. Затем смесь концентрировали при пониженном давлении, отбирали в этилацетат и промывали насыщенным водным NaHCO3, водой и насыщенным солевым раствором. Органическую фазу сушили над Na2SO4 и упаривали до сухого состояния. Неочищенный продукт очищали флэш-хроматографией на силикагеле (Hex/этилацетат 6:4) с получением 1,5 г (88%) указанного в заголовке соединения в виде аморфного твердого вещества.
ВЭЖХ: Rt: 7,07 мин
1H ЯМР (600 МГц, DMSO-d6) δ м.д. 8,17-8,05 (м, 1H), 7,78 (д, J=2,4 Гц, 1H), 7,71-7,64 (м, 1H), 7,61 (дт, J=4,0, 9,3 Гц, 1H), 7,54-7,46 (м, 1H), 7,34-7,29 (м, 1H), 7,29-7,21 (м, 1H), 5,10 (с, 2H), 3,98-3,90 (м, J=2,1, 2,1, 9,6 Гц, 2H), 3,48 (дт, J=1,9, 11,6 Гц, 2H), 3,38 (с, 3H), 3,37-3,31 (м, 1H), 2,04-1,99 (м, J=1,8, 12,8 Гц, 2H), 1,81-1,66 (м, 2H)
HRMS (ESI): вычислено для C22H22N2O4F3S2 [M+H]+ 499,0968, найдено 499,0974.
Аналогичным путем, но с использованием надлежащего тиоамида, также получали следующее промежуточное соединение:
Бензил 4-[4-(3-{[(2,5-дифторфенил)сульфонил](метоксиметил)амино}-2-фторфенил)-1,3-тиазол-2-ил]пиперидин-1-карбоксилат, соединение формулы 9 [n = 1; R1 = 1-бензилоксикарбонилпиперидин-4-ил; R4 = F; R5 = H; R6 = 2,5-дифторфенил; Rx = метоксиметил]
Способ A, стадии d1 и e
ВЭЖХ: Rt: 7,90 мин
1H ЯМР (600 МГц, DMSO-d6) δ м.д. 8,15-8,07 (м, 1H), 7,77 (д, J=2,4 Гц, 1H), 7,70-7,64 (м, 1H), 7,63-7,57 (м, 1H), 7,53-7,46 (м, 1H), 7,40-7,34 (м, 4H), 7,34-7,28 (м, 2H), 7,28-7,24 (м, 1H), 5,10 (с, 2H), 5,09 (с, 2H), 4,14-4,05 (м, 2H), 3,12-2,93 (м, 2H), 3,37 (с, 3H), 3,37-3,31 (м, 1H), 2,13-2,03 (м, J=11,5 Гц, 2H), 1,63 (д, кв, J=4,2, 12,2 Гц, 2H)
HRMS (ESI): вычислено для C30H29N3O5F3S2 [M+H]+ 632,1495, найдено 632,1501.
N-{3-[5-Бром-2-(тетрагидро-2H-пиран-4-ил)-1,3-тиазол-4-ил]- 2-фторфенил}-2,5-дифтор-N-(метоксиметил)бензолсульфонамид, соединение формулы 10 [n = 1; R1 = тетрагидропиран-4-ил; R4 = F; R5 = H; R6 = 2,5-дифторфенил; Rx = метоксиметил; Hal = Br]
Способ A, стадия f
2,5-дифтор-N-{2-фтор-3-[2-(тетрагидро-2H-пиран-4-ил)-1,3-тиазол-4-ил]фенил}-N-(метоксиметил)бензолсульфонамид (1,4 г, 2,808 ммоль) растворяли в уксусной кислоте (28 мл), добавляли ацетат натрия (461 мг, 5,616 ммоль, 2 экв.) и смесь перемешивали до полного растворения. Затем по каплям добавляли бром (0,2 мл, 3,9 ммоль, 1,4 экв.) в течение 45 мин и перемешивали при к.т. в течение ночи. Затем реакционную смесь по каплям добавляли к охлажденному 1н раствору гидроксида натрия (300 мл) и смесь экстрагировали этилацетатом. Органическую фазу промывали 0,5н гидроксидом натрия, 5% NaHSO3 и насыщенным солевым раствором, сушили над Na2SO4 и упаривали до сухого состояния с получением 1,26 г (78%) указанного в заголовке продукта в виде бесцветного масла.
ВЭЖХ: Rt: 7,24 мин
1H ЯМР (600 МГц, DMSO-d6) δ м.д. 7,69-7,64 (м, 1H), 7,63-7,56 (м, 2H), 7,53-7,48 (м, 1H), 7,47-7,42 (м, 1H), 7,35 (т, J=7,9, 1H), 5,10 (с, 2H), 3,95-3,85 (м, 2H), 3,44 (дт, J=1,9, 11,6 Гц, 2H), 3,38 (с, 3H), 3,31-3,23 (м, 1H), 1,98-1,92 (м, 2H), 1,75-1,65 (м, 2H)
HRMS (ESI): вычислено для C22H21N2O4F3S2Br [M+H]+ 577,0073, найдено 577,0075.
Аналогичным путем также получали следующее промежуточное соединение:
Бензил 4-[5-бром-4-(3-{[(2,5-дифторфенил)сульфонил](метоксиметил)амино}-2-фторфенил)-1,3-тиазол-2-ил]пиперидин-1-карбоксилат, соединение формулы 10 [n = 1; R1 = 1-бензилоксикарбонилпиперидин-4-ил; R4 = F; R5 = H; R6 = 2,5-дифторфенил; Rx = метоксиметил; Hal = Br]
Способ A, стадия f
ВЭЖХ: Rt: 8,03 мин
1H ЯМР (600 МГц, DMSO-d6) δ м.д. 7,67-7,62 (м, 1H), 7,62-7,55 (м, 2H), 7,53-7,47 (м, 1H), 7,47-7,42 (м, 1H), 7,40-7,33 (м, 5H), 7,33-7,29 (м, 1H), 5,09 (ушир.с, 2H), 5,08 (ушир.с, 2H), 4,10-4,04 (м, J=13,2 Гц, 2H), 3,38 (с, 3H), 3,29-3,22 (м, 1H), 3,05-2,93 (м, J=11,0 Гц, 2H), 2,05-1,99 (м, J=11,5 Гц, 2H), 1,63-1,54 (м, 2H)
HRMS (ESI): вычислено для C30H28N3O5F3S2Br [M+H]+ 710,0601, найдено 710,0618.
2,5-Дифтор-N-{2-фтор-3-[5-(3-фторпиридин-4-ил)-2-(тетрагидро-2H-пиран-4-ил)-1,3-тиазол-4-ил]фенил}бензолсульфонамид, соединение формулы (I)B (соединение 34) [m, n = 1; R2, R5 = H; R3 = 3-F; R4 = F; R6 = 2,5-дифторфенил; R1’ = тетрагидропиран-4-ил]
Способ A, стадии h и j
N-{3-[5-Бром-2-(тетрагидро-2H-пиран-4-ил)-1,3-тиазол-4-ил]-2-фторфенил}-2,5-дифтор-N-(метоксиметил)бензолсульфонамид (105 мг, 0,182 ммоль) растворяли в микроволновой пробирке в смеси DME/H2O 9:1 (2,2 мл) и через раствор барботировали аргон в течение 5 мин. Добавляли карбонат цезия (148 мг, 0,454 ммоль, 2,5 экв.), а затем пинаколовый эфир 3-F-4-пиридинбороновой кислоты (82 мг, 0,368 ммоль, 2 экв.) и Pd(dppf)Cl2·DCM (15 мг, 0,018 ммоль, 0,1 экв.) и смесь облучали в микроволновой печи при 100°C в течение 30 мин. Добавляли Pd(dppf)Cl2·DCM (15 мг, 0,018 ммоль, 0,1 экв.) и смесь подвергали второму циклу микроволнового облучения. Смесь фильтровали через слой целита и целит тщательно промывали этилацетатом. Фильтрат промывали насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над Na2SO4 и упаривали до сухого состояния. Неочищенный продукт очищали флэш-хроматографией на силикагеле с получением 76 мг 2,5-дифтор-N-{2-фтор-3-[5-(3-фторпиридин-4-ил)-2-(тетрагидро-2H-пиран-4-ил)-1,3-тиазол-4-ил]фенил}-N-(метоксиметил)бензолсульфонамида с примесями димерного побочного продукта. Этот продукт обрабатывали смесью TFA/вода 9:1 (1 мл) и перемешивали при 60°C в течение 1 ч. Смесь упаривали до сухого состояния, отбирали в DCM и промывали насыщенным водным NaHCO3 и насыщенным солевым раствором, сушили над Na2SO4 и концентрировали при пониженном давлении. Неочищенный продукт очищали флэш-хроматографией на силикагеле (Hex/ацетон 65:35) с получением 24 мг указанного в заголовке соединения в виде не совсем белого твердого вещества.
ВЭЖХ: Rt: 6,27 мин
1H ЯМР (401 МГц, DMSO-d6) (отдельные сигналы) δ м.д. 10,64 (с, 1H), 8,56 (д, J=1,6 Гц, 1H), 8,33 (д, J=4,9 Гц, 1H), 7,61-7,54 (м, 1H), 7,49-7,43 (м, 1H), 7,39-7,35 (м, 1H), 7,34-7,28 (м, 2H), 7,22-7,16 (м, 2H), 4,00-3,88 (м, 2H), 3,48 (дт, J=2,0, 11,6 Гц, 2H), 2,02 (дд, J=2,0, 12,8 Гц, 2H), 1,82-1,71 (м, 2H)
HRMS (ESI): вычислено для C25H20N3O3F4S2 [M+H]+ 550,0877, найдено 550,0894.
Пример 21
Синтез N-[2-({4-[4-(3-{[(2,5-дифторфенил)сульфонил]амино}-2-фторфенил)-2-(тетрагидро-2H-пиран-4-ил)-1,3-тиазол-5-ил]пиридин-2-ил}амино)этил]ацетамида, соединение формулы (I)Z1 (соединение 36) [m, n = 1; R1 = тетрагидропиран-4-ил; R3, R5 = H; R4 = F; R6 = 2,5-дифторфенил; R18 = 2-ацетиламиноэтил]
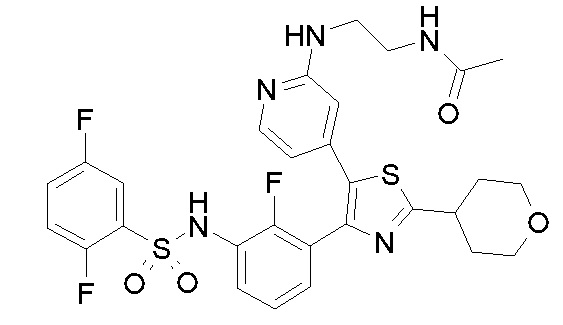
2,5-Дифтор-N-{2-фтор-3-[5-(пиридин-4-ил)-2-(тетрагидро-2H-пиран-4-ил)-1,3-тиазол-4-ил]фенил}-N-(метоксиметил)бензолсульфонамид, соединение формулы (I)A [m, n = 1; R2, R3, R5 = H; R4 = F; R6 = 2,5-дифторфенил; R1’ = тетрагидропиран-4-ил; Rx = метоксиметил] (соответствует соединению формулы (I)W способа F, где m, n = 1; R1 = тетрагидропиран-4-ил; R3, R5 = H; R4 = F; R6 = 2,5-дифторфенил; Rx = метоксиметил)
Способ A, стадия h
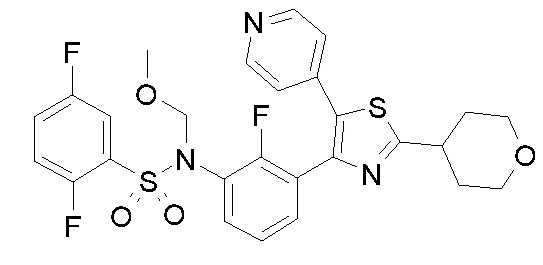
N-{3-[5-Бром-2-(тетрагидро-2H-пиран-4-ил)-1,3-тиазол-4-ил]-2-фторфенил}-2,5-дифтор-N-(метоксиметил)бензолсульфонамид (1,26 г, 2,182 ммоль) растворяли в смеси диоксан/H2O 9:1 (22 мл) и через раствор барботировали аргон в течение 5 мин. Добавляли карбонат цезия (1,78 мг, 5,455 ммоль, 2,5 экв.), а затем пинаколовый эфир 4-пиридинбороновой кислоты (895 мг, 4,364 ммоль, 2 экв.) и Pd(dppf)Cl2·DCM (178 мг, 0,218 ммоль, 0,1 экв.) и смесь перемешивали при 100°C в течение 1,5 ч. Смесь фильтровали через слой целита и целит тщательно промывали этилацетатом. Фильтрат концентрировали при пониженном давлении, отбирали в этилацетат и промывали насыщенным водным NaHCO3 и насыщенным солевым раствором, сушили над Na2SO4 и упаривали до сухого состояния. Неочищенный продукт очищали флэш-хроматографией на силикагеле (DCM/MeOH 97:3) с получением 1,09 г (87%) указанного в заголовке соединения в виде аморфного твердого вещества.
ВЭЖХ: Rt: 5,83 мин.
1H ЯМР (401 МГц, DMSO-d6) δ м.д. 8,46-8,50 (м, J=4,9 Гц, 2H), 7,41-7,67 (м, 4H), 7,28-7,38 (м, 2H), 7,07-7,17 (м, 3H), 4,97 (с, 2H), 3,91-3,99 (м, 1H), 3,44-3,52 (м, 1H), 1,98-2,06 (м, 2H), 1,70-1,84 (м, 2H)
HRMS (ESI): вычислено для C27H25F3N3O4S2 [M+H]+ 576,1233, найдено 576,1252.
Аналогичным путем также получали следующее соединение:
Бензил-4-[4-(3-{[(2,5-дифторфенил)сульфонил](метоксиметил)амино}-2-фторфенил)-5-(пиридин-4-ил)-1,3-тиазол-2-ил]пиперидин-1-карбоксилат, соединение формулы (I)A [m, n = 1; R2, R3, R5 = H; R4 = F; R6 = 2,5-дифторфенил; R1’ = 1-бензилоксикарбонилпиперидин-4-ил; Rx = метоксиметил]
Способ A, стадия h
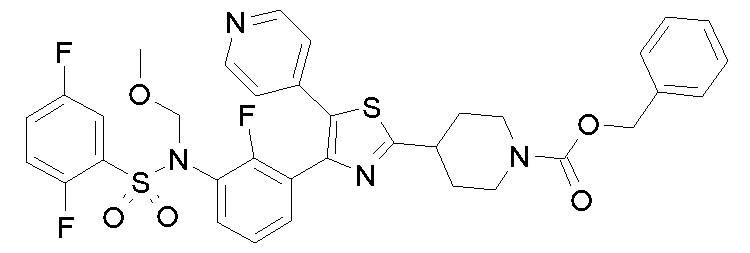
ВЭЖХ: Rt: 7,37 мин.
1H ЯМР (600 МГц, DMSO-d6) δ м.д. 8,54-8,43 (м, 2H), 7,65-7,56 (м, 2H), 7,52 (дт, J=4,0, 9,4 Гц, 1H), 7,47-7,42 (м, 1H), 7,40-7,35 (м, 4H), 7,35-7,28 (м, 3H), 7,14-7,03 (м, 2H), 5,09 (с, 2H), 4,96 (с, 2H), 4,16-4,01 (м, 2H), 3,35-3,29 (м, 1H), 3,23 (с, 3H), 3,03 (дд, J=7,1, 11,6 Гц, 2H), 2,13-2,07 (м, J=13,0 Гц, 2H), 1,65 (д, кв, J=4,2, 12,2 Гц, 2H)
HRMS (ESI): вычислено для C35H31N4O5F3S2 [M+H]+ 709,1761, найдено 709,1763.
2,5-Дифтор-N-{2-фтор-3-[5-(1-оксидопиридин-4-ил)-2-(тетрагидро-2H-пиран-4-ил)-1,3-тиазол-4-ил]фенил}-N-(метоксиметил)бензолсульфонамид, соединение формулы 44 [m, n = 1; R1 = тетрагидропиран-4-ил; R3, R5 = H; R4 = F; R6 = 2,5-дифторфенил; Rx = метоксиметил]
Способ F, стадия a
2,5-Дифтор-N-{2-фтор-3-[5-(пиридин-4-ил)-2-(тетрагидро-2H-пиран-4-ил)-1,3-тиазол-4-ил]фенил}-N-(метоксиметил)бензолсульфонамид (500 мг, 0,869 ммоль) растворяли в DCM (9 мл) и добавляли 70% м-хлорпербензойную кислоту (215 мг, 0,869 ммоль, 1 экв.). После перемешивания в течение 1 ч добавляли м-хлорпербензойную кислоту (215 мг, 0,869 ммоль, 1 экв.), а затем еще через 1 час проводили второе добавление 190 мг. Всего через 5 ч смесь разбавляли DCM и промывали насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над Na2SO4 и упаривали до сухого состояния. Неочищенный продукт очищали флэш-хроматографией на силикагеле (DCM/MeOH 95:5) с получением 420 мг (82%) указанного в заголовке соединения в виде аморфного твердого вещества.
ВЭЖХ: Rt: 5,40 мин.
1H ЯМР (600 МГц, DMSO-d6) δ м.д. 8,20-8,02 (м, 2H), 7,68-7,61 (м, 2H), 7,58-7,53 (м, 1H), 7,53-7,49 (м, 1H), 7,36-7,23 (м, 2H), 7,19-7,09 (м, 2H), 4,99 (с, 2H), 3,98-3,87 (м, 2H), 3,48 (дт, J=2,0, 11,6 Гц, 2H), 3,39-3,31 (м, 1H), 3,26 (с, 3H), 2,05-1,94 (м, 2H), 1,81-1,71 (м, 2H)
HRMS (ESI): вычислено для C27H25N3O5F3S2 [M+H]+ 592,1182, найдено 592,1188.
Аналогичным путем также получали следующее промежуточное соединение:
Бензил 4-[4-(3-{[(2,5-дифторфенил)сульфонил](метоксиметил)амино}-2-фторфенил)-5-(1-оксидопиридин-4-ил)-1,3-тиазол-2-ил]пиперидин-1-карбоксилат, соединение формулы 44 [m, n = 1; R1 = 1-бензилоксикарбонилпиперидин-4-ил; R3, R5 = H; R4 = F; R6 = 2,5-дифторфенил; Rx = метоксиметил]
Способ F, стадия a
ВЭЖХ: Rt: 6,52 мин
1H ЯМР (600 МГц, DMSO-d6) δ м.д. 8,16-8,02 (м, 2H), 7,66-7,61 (м, 2H), 7,56 (дд, J=3,9, 9,4 Гц, 1H), 7,52-7,48 (м, 1H), 7,39-7,36 (м, 4H), 7,34-7,29 (м, 3H), 7,19-7,11 (м, 2H), 5,10 (с, 2H), 4,99 (с, 2H), 4,15-3,97 (м, J=13,6 Гц, 2H), 3,34-3,29 (м, 1H), 3,26 (с, 3H), 3,08-2,96 (м, 2H), 2,13-2,07 (м, 2H), 1,69-1,59 (м, J=3,6, 12,2 Гц, 2H)
HRMS (ESI): вычислено для C35H32N4O6F3S2 [M+H]+ 725,1710, найдено 725,1710.
N-[2-({4-[4-(3-{[(2,5-Дифторфенил)сульфонил](метоксиметил)амино}-2-фторфенил)-2-(тетрагидро-2H-пиран-4-ил)-1,3-тиазол-5-ил]пиридин-2-ил}амино)этил]ацетамид, соединение формулы (I)X [m, n = 1; R1 = тетрагидропиран-4-ил; R3, R5 = H; R4 = F; R6 = 2,5-дифторфенил; Rx = метоксиметил; R18 = 2-ацетиламиноэтил]
Способ F, стадия b
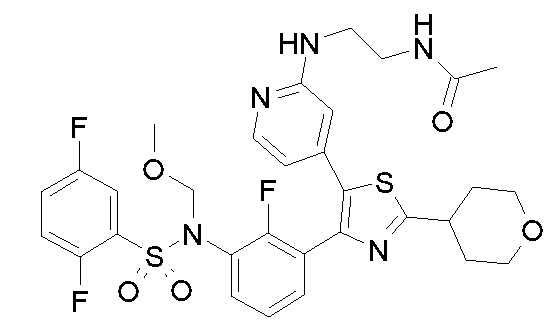
2,5-Дифтор-N-{2-фтор-3-[5-(1-оксидопиридин-4-ил)-2-(тетрагидро-2H-пиран-4-ил)-1,3-тиазол-4-ил]фенил}-N-(метоксиметил)бензолсульфонамид (104 мг, 0,175 ммоль) растворяли в DCM (1,5 мл). Добавляли DIPEA (0,112 мл, 0,656 ммоль, 3,75 экв.), а затем PyBroP (106 мг, 0,228 ммоль, 1,3 экв.) и N-ацетилэтилендиамин (0,021 мл, 0,219 ммоль, 1,25 экв.) и смесь перемешивали при к.т. в течение ночи. Проводили дополнительное добавление как ацетилэтилендиамина (0,01 мл), так и PyBroP (20 мг) и после перемешивания в течение дополнительных 2 ч смесь разбавляли DCM и промывали водой и насыщенным солевым раствором, сушили над Na2SO4 и упаривали до сухого состояния. Неочищенный продукт очищали флэш-хроматографией на силикагеле (DCM/MeOH 96:4) с получением 110 мг (93%) указанного в заголовке соединения.
ВЭЖХ: Rt: 5,78 мин.
1H ЯМР (600 МГц, DMSO-d6) δ м.д. 7,88 (т, J=5,4 Гц, 1H), 7,80 (д, J=5,1 Гц, 1H), 7,67-7,59 (м, 1H), 7,58-7,51 (м, 2H), 7,49-7,43 (м, 1H), 7,36-7,31 (м, 1H), 7,29 (т, J=7,9 Гц, 1H), 6,63 (т, J=5,6 Гц, 1H), 6,33 (с, 1H), 6,06 (дд, J=1,4, 5,2 Гц, 1H), 4,98 (с, 2H), 3,94 (ддд, J=1,9, 1,9, 9,7 Гц, 2H), 3,47 (дт, J=2,0, 11,6 Гц, 2H), 3,42-3,35 (м, 1H), 3,32-3,28 (м, 1H), 3,26 (с, 3H), 3,24-3,19 (м, 1H), 3,18-3,09 (м, 2H), 2,01 (дд, J=2,1, 12,7 Гц, 2H), 1,78 (с, 3H), 1,30-1,21 (м, 2H)
HRMS (ESI): вычислено для C31H33N5O5F3S2 [M+H]+ 676,1870, найдено 676,1880.
N-[2-({4-[4-(3-{[(2,5-Дифторфенил)сульфонил]амино}-2-фторфенил)-2-(тетрагидро-2H-пиран-4-ил)-1,3-тиазол-5-ил]пиридин-2-ил}амино)этил]ацетамид, соединение формулы (I)Z1 (соединение 36) [m, n = 1; R1 = тетрагидропиран-4-ил; R3, R5 = H; R4 = F; R6 = 2,5-дифторфенил; R18 = 2-ацетиламиноэтил]
Способ F, стадия d1
N-[2-({4-[4-(3-{[(2,5-Дифторфенил)сульфонил](метоксиметил)амино}-2-фторфенил)-2-(тетрагидро-2H-пиран-4-ил)-1,3-тиазол-5-ил]пиридин-2-ил}амино)этил]ацетамид (197 мг, 0,158 ммоль) обрабатывали смесью TFA/вода 9:1 (2 мл) и перемешивали при 60°C в течение 2 ч. Смесь упаривали до сухого состояния, отбирали в DCM и промывали насыщенным водным NaHCO3 и насыщенным солевым раствором, сушили над Na2SO4 и концентрировали при пониженном давлении. Неочищенный продукт очищали флэш-хроматографией на силикагеле (DCM/MeOH 95:5) с получением 55 мг указанного в заголовке соединения в виде не совсем белого твердого вещества.
ВЭЖХ: Rt: 5,47 мин.
1H ЯМР (600 МГц, DMSO-d6) δ м.д. 10,68 (ушир.с, 1H), 7,91 (т, J=5,4 Гц, 1H), 7,77 (д, J=5,5 Гц, 1H), 7,60-7,53 (м, 1H), 7,48-7,42 (м, 2H), 7,36-7,26 (м, 2H), 7,24-7,16 (м, 1H), 6,83-6,59 (м, 1H), 6,38 (ушир.с, 1H), 6,02 (ушир.с, 1H), 3,93 (тд, J=2,1, 9,5 Гц, 2H), 3,46 (дт, J=1,8, 11,6 Гц, 2H), 3,32-3,27 (м, 1H), 3,26-3,21 (м, J=5,3 Гц, 2H), 3,16 (кв, J=5,9 Гц, 2H), 2,03-1,96 (м, J=1,9, 12,9 Гц, 2H), 1,80 (с, 3H), 1,77-1,68 (м, 2H)
HRMS (ESI): вычислено для C29H29N5O4F3S2 [M+H]+ 632,1608, найдено 632,1625.
Также выделяли следующий побочный продукт:
N-(3-{5-[2-(3-Ацетилимидазолидин-1-ил)пиридин-4-ил]-2-(тетрагидро-2H-пиран-4-ил)-1,3-тиазол-4-ил}-2-фторфенил)-2,5-дифторбензолсульфонамид, соединение формулы (I)Z [m, n = 1; R3, R5 = H; R4 = F; R6 = 2,5-дифторфенил; R1 = тетрагидропиран-4-ил; R17-R18 = 3-ацетил-имидазолидин-1-ил]
Способ F, стадия d1
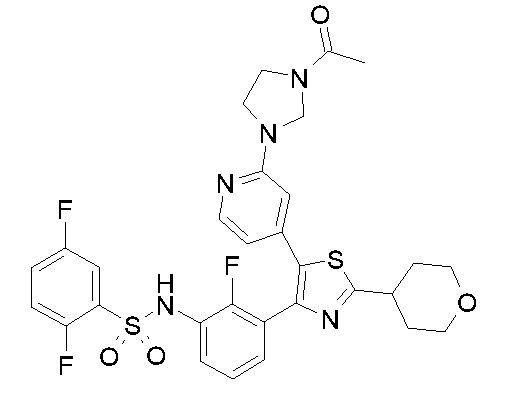
ВЭЖХ: Rt: 5,74 мин.
1H ЯМР (600 МГц, DMSO-d6) δ м.д. 10,67 (ушир.с, 1H), 7,99 (т, J=5,9 Гц, 1H), 7,55 (ушир.с, 1H), 7,45 (ушир.с, 1H), 7,41-7,37 (м, 1H), 7,33 (т, J=6,9 Гц, 2H), 7,23 (ушир.с, 1H), 6,34 (дд, J=4,9, 19,6 Гц, 1H), 6,28 (д, J=7,7 Гц, 1H), 4,78 и 4,66 (с, 2H, 2 ротамера), 3,98-3,87 (м, J=2,4, 9,0 Гц, 2H), 3,81-3,75 (м, 1H), 3,63 (т, J=6,8 Гц, 1H), 3,54-3,50 (м, 1H), 3,47 (т, J=11,0 Гц, 2H), 3,44-3,40 (м, 3H), 3,32-3,27 (м, 1H), 2,04-1,97 (м, 3H), 1,79-1,74 (м, 2H)
HRMS (ESI): вычислено для C30H29N5O4F3S2 [M+H]+ 644,1608, найдено 644,1623.
Аналогичным путем, но с использованием надлежащего амина, также получали следующие соединения:
N-{3-[5-(2-{[2-(Диметиламино)этил]амино}пиридин-4-ил)-2-(тетрагидро-2H-пиран-4-ил)-1,3-тиазол-4-ил]-2-фторфенил}-2,5-дифторбензолсульфонамид, соединение формулы (I)Z1 (соединение 40) [m, n = 1; R3, R5 = H; R4 = F; R6 = 2,5-дифторфенил; R1 = тетрагидропиран-4-ил; R18 = 2-диметиламиноэтил]
Способ F, стадия d1
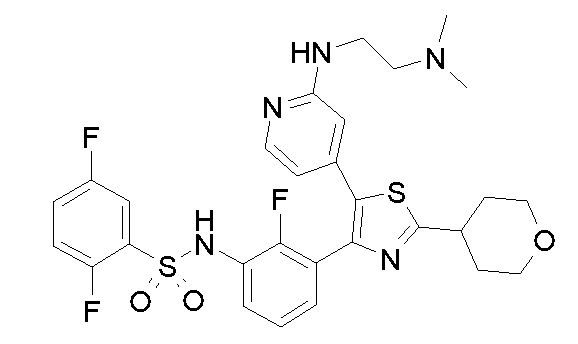
ВЭЖХ: Rt: 5,33 мин.
1H ЯМР (600 МГц, DMSO-d6) δ м.д. 7,85 (д, J=5,3 Гц, 1H), 7,42 (ддд, J=3,3, 5,1, 8,1 Гц, 1H), 7,39-7,34 (м, 1H), 7,31 (дт, J=4,2, 9,0 Гц, 1H), 7,23 (дт, J=1,4, 7,9 Гц, 1H), 6,96 (т, J=7,9 Гц, 1H), 6,86 (ушир.с, 1H), 6,66 (т, J=5,2 Гц, 1H), 6,40 (с, 1H), 6,22 (дд, J=1,2, 5,2 Гц, 1H), 3,93 (тд, J=2,0, 9,6 Гц, 2H), 3,47 (дт, J=1,8, 11,6 Гц, 2H), 3,41-3,25 (м, 3H), 2,86 (ушир.с, 2H), 2,56 (с, 6H), 2,07-1,93 (м, J=2,0, 12,8 Гц, 2H), 1,79-1,68 (м, 2H)
HRMS (ESI): вычислено для C29H31N5O3F3S2 [M+H]+ 618,1815, найдено 618,1822.
Метил [(2S)-1-({4-[4-(3-{[(2,5-дифторфенил)сульфонил]амино}-2-фторфенил)-2-(тетрагидро-2H-пиран-4-ил)-1,3-тиазол-5-ил]пиридин-2-ил}амино)пропан-2-ил]карбамат, соединение формулы (I)Z1 (соединение 41) [m, n = 1; R1 = тетрагидропиран-4-ил; R3, R5 = H; R4 = F; R6 = 2,5-дифторфенил; R18 = 2-метоксикарбонил-2-метиламиноэтил]
Способ F, стадия d1
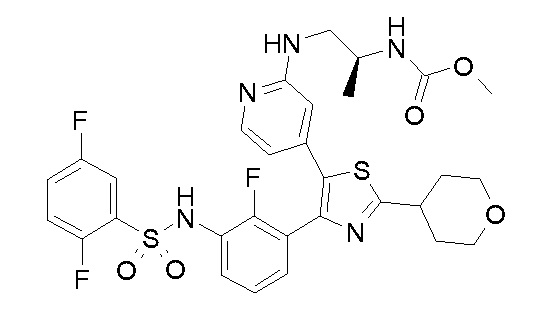
ВЭЖХ: Rt: 6,11 мин.
1H ЯМР (600 МГц, DMSO-d6) δ м.д. 10,66 (ушир.с, 1H), 7,75 (д, J=5,5 Гц, 1H), 7,60-7-54 (м, 1H), 7,51-7,42 (м, 2H), 7,33 (т, J=6,9 Гц, 1H), 7,27 (ушир.с, 1H), 7,22-7,16 (м, 1H), 7,01 (д, J=7,3 Гц, 1H), 6,63 (ушир.с, 1H), 6,40 (с, 1H), 5,96 (д, J=4,9 Гц, 1H), 3,93 (тд, J=2,0, 9,5 Гц, 2H), 3,64 (тд, J=6,8, 14,0 Гц, 1H), 3,50 (с, 3H), 3,47 (дт, J=1,9, 11,6 Гц, 2H), 3,34-3,26 (м, 1H), 3,21-3,15 (м, 2H), 2,00 (дд, J=1,9, 12,9 Гц, 2H), 1,80-1,65 (м, 2H), 1,03 (д, J=6,6 Гц, 3H)
HRMS (ESI): вычислено для C30H31N5O5F3S2 [M+H]+ 662,1713, найдено 662,1714.
Также выделяли следующий побочный продукт:
2,5-Дифтор-N-{2-фтор-3-[5-{2-[(4S)-3-(метоксиметил)-4-метил-2-оксоимидазолидин-1-ил]пиридин-4-ил}-2-(тетрагидро-2H-пиран-4-ил)-1,3-тиазол-4-ил]фенил}бензолсульфонамид, соединение формулы (I)Z [m, n = 1; R1 = тетрагидропиран-4-ил; R3, R5 = H; R4 = F; R6 = 2,5-дифторфенил; R17-R18 = 3-метоксиметил-4-метил-2-оксоимидазолидин-1-ил]
Способ F, стадия d1
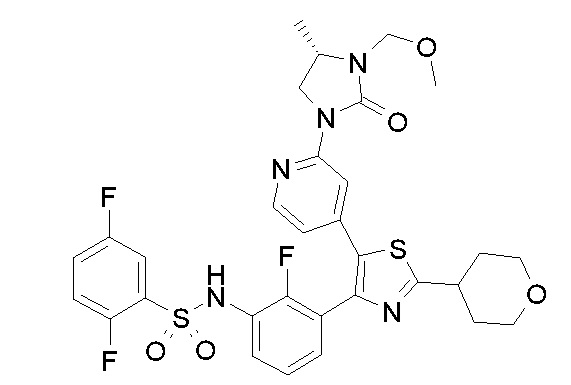
ВЭЖХ: Rt: 6,66 мин.
1H ЯМР (600 МГц, DMSO-d6) δ м.д. 10,67 (с, 1H), 7,98 (д, J=5,3 Гц, 1H), 7,55 (д, J=7,5 Гц, 1H), 7,46 (дт, J=3,8, 9,0 Гц, 1H), 7,42-7,38 (м, 1H), 7,37-7,29 (м, 2H), 7,26-7,14 (м, 1H), 6,33 (д, J=4,9 Гц, 1H), 6,25 (с, 1H), 4,67 (с, 2H), 4,19 (д, квин, J=2,8, 6,6 Гц, 1H), 3,98-3,87 (м, 2H), 3,65 (с, 3H), 3,51-3,42 (м, 3H), 3,29-3,14 (м, 2H), 2,05-1,97 (м, J=1,9, 12,7 Гц, 2H), 1,80-1,67 (м, 2H), 1,21 (д, J=6,4 Гц, 3H)
HRMS (ESI): вычислено для C31H31N5O5F3S2 [M+H]+ 674,1713, найдено 674,1726.
Пример 22
Синтез 2,5-дифтор-N-{3-[5-(пиридин-4-ил)-2-(тетрагидро-2H-пиран-4-ил)-1,3-тиазол-4-ил]фенил}бензолсульфонамид, соединение формулы (I)T [m, n = 1; R1 = тетрагидропиран-4-ил; R2, R3, R4, R5 = H; R6 = 2,5-дифторфенил]
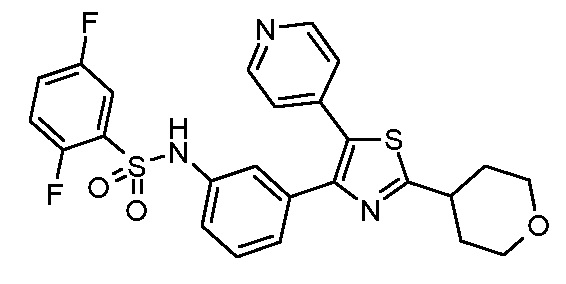
4-(3-Нитрофенил)-2-(тетрагидро-2H-пиран-4-ил)-1,3-тиазол, соединение формулы 40 [n = 1; R1 = тетрагидропиран-4-ил; R4, R5 = H]
Способ E, стадия b
Смесь 2-бром-1-(3-нитрофенил)этанона (245 мг, 1 ммоль) и тетрагидро-2H-пиран-4-карботиоамида (144 мг, 1 ммоль) суспендируют в абсолютном этаноле (20 мл) и нагревают до температуры кипения с обратным холодильником в течение 2 ч. Затем раствор охлаждают и упаривают до сухого состояния. Неочищенное вещество растворяют в DCM и два раза промывают раствором насыщенного бикарбоната натрия и один раз водой. Органический слой сушат сульфатом натрия, фильтруют и упаривают до сухого состояния с получением 285 мг указанного в заголовке соединения (98%).
ВЭЖХ: Rt: 5,66 мин
1H ЯМР (600 МГц, DMSO-d6) δ м.д. 8,75 (т, J=1,4 Гц, 1H), 8,40 (дт, J=1,4, 8,0 Гц, 1H), 8,33 (с, 1H), 8,19 (дд, J=1,4, 8,0 Гц, 1H), 7,75 (т, J=8,0 Гц, 1H), 3,95 (дт, J=1,8, 11,7 Гц, 2H), 3,50 (дт, J=1,8, 11,7 Гц, 2H), 3,40-3,33 (м, 1H), 2,06-1,99 (м, 2H), 1,84-1,71 (м, 2H).
HRMS (ESI): вычислено для C14H15N2O3S [M+H]+ 291,0798, найдено 291,0798.
5-Бром-4-(3-нитрофенил)-2-(тетрагидро-2H-пиран-4-ил)-1,3-тиазол, соединение формулы 41 [n = 1; R1 = тетрагидропиран-4-ил; R4, R5 = H; Hal = Br]
Способ E, стадия c
К раствору 4-(3-нитрофенил)-2-(тетрагидро-2H-пиран-4-ил)- 1,3-тиазола (113 мг, 0,390 ммоль) в уксусной кислоте (10,5 мл), добавляли ацетат натрия (71 мг, 0,866 ммоль, 2,2 экв.), и смесь перемешивали в течение 30 мин. К раствору, полученному таким образом, по каплям добавляли бром и за ходом реакции наблюдали с помощью ВЭЖХ вплоть до завершения (всего Br2: 44 мкл, 137 мг, 0,86 ммоль, 2,2 экв.). Раствор выливали в 100 мл 1 M гидроксида натрия и экстрагировали три раза этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили сульфатом натрия и упаривали до сухого состояния с получением 5-бром-4-(3-нитрофенил)-2-(тетрагидро-2H-пиран-4-ил)-1,3-тиазола (141 мг, 100%).
ВЭЖХ: Rt: 7,29 мин
1H ЯМР (600 МГц, DMSO-d6) δ м.д. 8,72 (т, J=2,2 Гц, 1H), 8,39 (ддд, J=1,2, 2,2, 8,0 Гц, 1H), 8,29 (ддд, J=1,2, 2,2, 8,0 Гц, 1H), 7,82 (т, J=8,0 Гц, 1H), 3,93 (дт, J=2,1, 2,1, 11,6 Гц, 2H), 3,47 (дт, J=2,1, 11,6 Гц, 2H), 2,03-1,98 (м, 2H), 1,81-1,69 (м, 2H).
HRMS (ESI): вычислено для C14H14N2O3SBr [M+H]+ 368,9903, найдено 368,9894.
4-[4-(3-Нитрофенил)-2-(тетрагидро-2H-пиран-4-ил)-1,3-тиазол-5-ил]пиридин, соединение формулы 42 [m, n = 1; R1 = тетрагидропиран-4-ил; R2, R3, R4, R5 = H]
Способ E, стадия d
5-Бром-4-(3-нитрофенил)-2-(тетрагидро-2H-пиран-4-ил)-1,3-тиазол (140 мг, 0,379 ммоль) растворяли в смеси дегазированного диоксана (8 мл) и воды (1,6 мл). К раствору последовательно добавляли 2-(4-пиридил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (78 мг, 0,379 ммоль, 1 экв.), карбонат цезия (370 мг, 1,137 ммоль, 3 экв.) и PdCl2(dppf)2·CH2Cl2 (31 мг, 0,0379 ммоль, 0,1 экв.). Смесь, полученную таким образом, перемешивали и нагревали до температуры кипения с обратным холодильником в атмосфере аргона в течение 5 ч. После охлаждения суспензию фильтровали через слой целита, и упаривали до сухого состояния. Неочищенное вещество перерастворяли в этилацетате и два раза промывали насыщенным солевым раствором. Органический слой сушили над сульфатом натрия и упаривали до сухого состояния. Затем твердый материал, полученный таким образом, очищали колоночной хроматографией на силикагеле, элюируя смесью этилацетат/Hex (6/4) с получением указанного в заголовке соединения (98 мг, 70%).
ВЭЖХ: Rt: 6,01 мин
1H ЯМР (600 МГц, DMSO-d6) δ м.д. 8,65-8,51 (м, 2H), 8,29 (т, J=2,1 Гц, 1H), 8,21 (ддд, J=1,2, 2,1, 8,1 Гц, 1H), 7,82 (тд, J=1,2, 8,1 Гц, 1H), 7,65 (т, J=8,1 Гц, 1H), 7,42-7,29 (м, 2H), 4,02-3,91 (м, 2H), 3,50 (дт, J=1,8, 11,6 Гц, 2H), 2,10-2,02 (м, J=2,6, 12,3 Гц, 2H), 1,85-1,73 (м, J=4,3, 13,1 Гц, 2H).
HRMS (ESI): вычислено для C19H18N3O3S [M+H]+ 368,1064, найдено 368,1062.
3-[5-(Пиридин-4-ил)-2-(тетрагидро-2H-пиран-4-ил)-1,3-тиазол-4-ил]анилин, соединение формулы 43 [m, n = 1; R1 = тетрагидропиран-4-ил; R2, R3, R4, R5 = H]
Способ E, стадия e
К раствору 4-[4-(3-нитрофенил)-2-(тетрагидро-2H-пиран-4-ил)-1,3-тиазол-5-ил]пиридина в абсолютном этаноле (5 мл) и уксусной кислоте (1 мл), добавляли цинк (300 мг). Смесь перемешивали и нагревали до температуры кипения с обратным холодильником в течение 3 ч. Смесь высушивали и обрабатывали небольшим количеством 1н HCl. После перемешивания в течение 1 ч раствор доводили до pH 14 с использованием гидроксида натрия и экстрагировали DCM. Органический слой сушили сульфатом натрия и упаривали до сухого состояния. Твердый материал, полученный таким образом, очищали колоночной хроматографией на силикагеле, элюируя смесью этилацетат/hex/гидроксид аммония 8/2/0,1 с получением указанного в заголовке соединения (52 мг, 59%).
ВЭЖХ: Rt: 4,97 мин
1H ЯМР (600 МГц, DMSO-d6) δ м.д. 8,70-8,30 (м, 2H), 7,34-7,18 (м, 2H), 6,96 (т, J=7,8 Гц, 1H), 6,74 (т, J=1,8 Гц, 1H), 6,58-6,50 (м, 1H), 6,48-6,31 (м, 1H), 5,13 (с, 2H), 3,95 (дт, J=2,1, 11,6 Гц, 2H), 3,48 (дт, J=2,1, 11,6 Гц, 3H), 3,33-3,26 (м, 2H), 2,02 (дд, J=1,8, 13,0 Гц, 2H), 1,82-1,71 (м, 2H).
HRMS (ESI): вычислено для C19H20N3OS [M+H]+ 338,1322, найдено 338,1331.
2,5-Дифтор-N-{3-[5-(пиридин-4-ил)-2-(тетрагидро-2H-пиран-4-ил)-1,3-тиазол-4-ил]фенил}бензолсульфонамид, соединение формулы (I)T [m, n = 1; R1 = тетрагидропиран-4-ил; R2, R3, R4, R5 = H; R6 = 2,5-дифторфенил]
Способ E, стадия f
3-[5-(Пиридин-4-ил)-2-(тетрагидро-2H-пиран-4-ил)-1,3-тиазол-4-ил]анилин (28 мг, 0,083 ммоль) растворяли в DCM (2 мл) в атмосфере азота. Добавляли сухой пиридин (10 мкл), а затем 2,5-дифторбензолсульфонилхлорид (20 мкл). Смесь перемешивали при к.т. в течение 2 ч, затем разбавляли DCM и промывали насыщенным раствором бикарбоната натрия. Органический слой высушивали сульфатом натрия и упаривали до сухого состояния. Неочищенное вещество очищали колоночной хроматографией на силикагеле, элюируя смесью DCM/MeOH/гидроксид аммония 100/2/0,2 с получением указанного в заголовке соединения (23 мг, 53%).
ВЭЖХ: Rt: 6,11 мин
1H ЯМР (600 МГц, DMSO-d6) δ м.д. 10,83 (ушир.с, 1H), 8,75-8,24 (м, 2H), 7,63-7,57 (м, 1H), 7,54-7,40 (м, 2H), 7,26-7,22 (м, 2H), 7,21-7,18 (м, 2H), 7,14-7,04 (м, 2H), 3,95 (тд, J=2,1, 11,7 Гц, 2H), 3,49 (дт, J=2,0, 11,6 Гц, 2H), 2,06-1,96 (м, 2H), 1,82-1,67 (м, 2H)
HRMS (ESI): вычислено для C25H22N3O3F2S2 [M+H]+ 514,1065, найдено 514,1075.
Аналогично, но с использованием надлежащего сульфонилхлорида, получали следующее соединение:
2,6-Дифтор-N-{3-[5-(пиридин-4-ил)-2-(тетрагидро-2H-пиран-4-ил)-1,3-тиазол-4-ил]фенил}бензолсульфонамид, соединение формулы (I)T [m, n = 1; R1 = тетрагидропиран-4-ил; R2, R3, R4, R5 = H; R6 = 2,6-дифторфенил]

ВЭЖХ: Rt: 5,89 мин
1H ЯМР (600 МГц, DMSO-d6) δ м.д. 10,96 (с, 1H), 8,60-8,38 (м, 2H), 7,80-7,62 (м, 1H), 7,30 (с, 1H), 7,28-7,21 (м, 3H), 7,20-7,17 (м, 2H), 7,13 (дд, J=1,2, 8,2 Гц, 1H), 7,06 (д, J=7,5 Гц, 1H), 3,99-3,91 (м, J=2,0, 2,0, 9,5 Гц, 2H), 3,49 (дт, J=1,9, 11,6 Гц, 2H), 3,36-3,32 (м, 1H), 2,05-1,98 (м, J=2,0, 12,8 Гц, 2H), 1,82-1,70 (м, 2H)
HRMS (ESI): вычислено для C25H22N3O3F2S2 [M+H]+ 514,1065, найдено 514,1074.
| название | год | авторы | номер документа |
|---|---|---|---|
| АЗАСПИРОПРОИЗВОДНЫЕ В КАЧЕСТВЕ АНТАГОНИСТОВ TRPM8 | 2015 |
|
RU2683309C2 |
| АМИДЫ КОНДЕНСИРОВАННОГО ПИПЕРИДИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ ИОННЫХ КАНАЛОВ | 2014 |
|
RU2741810C2 |
| ИНГИБИТОРЫ ПРОТЕИНКИНАЗ ДЛЯ УСИЛЕНИЯ РЕГЕНЕРАЦИИ ПЕЧЕНИ ИЛИ СНИЖЕНИЯ ИЛИ ПРЕДОТВРАЩЕНИЯ ГИБЕЛИ ГЕПАТОЦИТОВ | 2019 |
|
RU2822216C2 |
| НОВЫЕ СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ ИНФЕКЦИЙ МЛЕКОПИТАЮЩИХ | 2019 |
|
RU2798336C2 |
| (АЗА)ИНДОЛ-, БЕНЗОТИОФЕН- И БЕНЗОФУРАН-3-СУЛЬФОНАМИДЫ | 2017 |
|
RU2767904C2 |
| МОДУЛИРУЮЩИЕ JAK КИНАЗУ ХИНАЗОЛИНОВЫЕ ПРОИЗВОДНЫЕ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2010 |
|
RU2529019C2 |
| N-((ГЕТ)АРИЛМЕТИЛ)-ГЕТЕРОАРИЛ-КАРБОКСАМИДНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ ПЛАЗМЕННОГО КАЛЛИКРЕИНА | 2015 |
|
RU2707870C2 |
| N-((ГЕТ)АРИЛМЕТИЛ)-ГЕТЕРОАРИЛ-КАРБОКСАМИДНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ ПЛАЗМЕННОГО КАЛЛИКРЕИНА | 2015 |
|
RU2821520C2 |
| ПРОИЗВОДНЫЕ КОНДЕНСИРОВАННОГО 1,4-ДИГИДРОДИОКСИНА В КАЧЕСТВЕ ИНГИБИТОРОВ ФАКТОРА ТРАНСКРИПЦИИ 1 БЕЛКОВ ТЕПЛОВОГО ШОКА | 2014 |
|
RU2671979C2 |
| ИНГИБИТОРЫ ПРОТЕИНКИНАЗЫ MKK4 ДЛЯ СТИМУЛЯЦИИ РЕГЕНЕРАЦИИ ПЕЧЕНИ ИЛИ УМЕНЬШЕНИЯ ИЛИ ПРЕДОТВРАЩЕНИЯ ГИБЕЛИ ГЕПАТОЦИТОВ | 2019 |
|
RU2788000C2 |
Изобретение относится к соединению формулы (I), где каждый из n и m независимо равен 1; R1 представляет собой галоген или группу, выбранную из прямого или разветвленного (C1-C8)алкила и гетероциклила, выбранного из пиперидинила и тетрагидропиранила, где пиперидинил необязательно замещен (C1-C6)алкилом и (С3-С6)циклоалкилом; или R1 представляет собой NR7R8, где каждый из R7 и R8 независимо представляет собой водород или группу, выбранную из прямого или разветвленного (C1-C8)алкила, гетероциклила, выбранного из пиперидинила и необязательно замещенного метилом; или, взятые вместе с атомом азота, с которым они связаны, R7 и R8 могут образовывать необязательно замещенный 3-8-членный гетероциклил, необязательно содержащий один дополнительный гетероатом или гетероатомную группу, выбранные из О, N и NH, необязательно замещенный галогеном; каждый из R2 и R3 независимо представляет собой водород, галоген или необязательно замещенную группу, выбранную из прямого или разветвленного (C1-C8)алкила и гетероциклила, выбранного из имидазолидина, замещенного -СОСН3, -С=O, -СН2-ОСН3, -СН3; или каждый из R2 и R3 независимо представляет собой NR17R18, где R17 и R18 независимо представляют собой водород или необязательно замещенную группу, выбранную из прямого или разветвленного (С1-С8)алкила, необязательно замещенного -NH-СОСН3, -NHCOOCH3, -N(CH3)2; или R17 представляет собой водород и R18 представляет собой COR22, где R22 представляет собой OR23 или необязательно замещенную группу, выбранную из прямого или разветвленного (C1-C8)алкила, где R23 представляет прямой или разветвленный (С1-С8)алкил, каждый из R4 и R5 независимо представляет собой водород или галоген; Rx представляет собой водород; R6 представляет собой фенил, необязательно замещенный галогеном; или его фармацевтически приемлемой соли. Изобретение относится к способу ингибирования in vitro активности семейства Raf, который включает контактирование указанного рецептора с эффективным количеством соединения, формулы (I). Соединение формулы (I) или его фармацевтически приемлемая соль предназначены для применения в качестве лекарственного средства или в составе фармацевтической композиции, с активностью, направленной против злокачественной опухоли. Технический результат – тиазолилфенилбензолсульфонамидопроизводные в качестве ингибиторов киназ семейства Raf. 5 н. и 5 з.п. ф-лы, 1 табл., 22 пр.
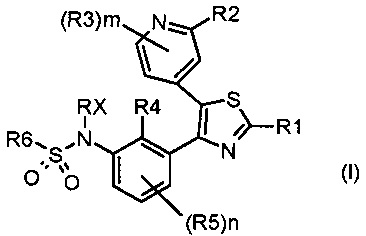
1. Соединение формулы (I):
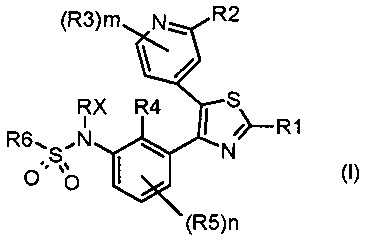
где:
каждый из n и m независимо равен 1;
R1 представляет собой галоген или группу, выбранную из прямого или разветвленного (C1-C8)алкила и гетероциклила, выбранного из пиперидинила и тетрагидропиранила, где пиперидинил необязательно замещен (C1-C6)алкилом и (С3-С6)циклоалкилом; или R1 представляет собой NR7R8,
где:
каждый из R7 и R8 независимо представляет собой водород или группу, выбранную из прямого или разветвленного (C1-C8)алкила, гетероциклила, выбранного из пиперидинила и необязательно замещенного метилом; или, взятые вместе с атомом азота, с которым они связаны, R7 и R8 могут образовывать необязательно замещенный 3-8-членный гетероциклил, необязательно содержащий один дополнительный гетероатом или гетероатомную группу, выбранные из О, N и NH, необязательно замещенный галогеном;
каждый из R2 и R3 независимо представляет собой водород, галоген или необязательно замещенную группу, выбранную из прямого или разветвленного (C1-C8)алкила и гетероциклила, выбранного из имидазолидина, замещенного -СОСН3, -С=O, -СН2-ОСН3, -СН3; или каждый из R2 и R3 независимо представляет собой NR17R18,
где:
R17 и R18 независимо представляют собой водород или необязательно замещенную группу, выбранную из прямого или разветвленного (С1-С8)алкила, необязательно замещенного -NH-СОСН3, -NHCOOCH3, -N(CH3)2; или
R17 представляет собой водород и R18 представляет собой COR22,
где:
R22 представляет собой OR23 или необязательно замещенную группу, выбранную из прямого или разветвленного (C1-C8)алкила, где:
R23 представляет прямой или разветвленный (С1-С8)алкил,
каждый из R4 и R5 независимо представляет собой водород или галоген;
Rx представляет собой водород;
R6 представляет собой фенил, необязательно замещенный галогеном;
или его фармацевтически приемлемые соли.
2. Соединение формулы (I) по п.1, где:
R1 представляет собой NR7R8 или гетероциклил, такой, как определено в п. 1.
3. Соединение формулы (I) по п.1 или 2, где:
R2 представляет собой водород или группу NR17R18, как определено в п. 1.
4. Соединение формулы (I) по п. 1, где:
R2 представляет собой группу NR17R18, где R17 представляет собой водород и R18 представляет собой COR22, где R22 является таким, как определено в п. 1.
5. Соединение формулы (I) по п. 1, где:
R3 представляет собой водород и R4 представляет собой галоген.
6. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 1, которые выбраны из группы, состоящей из:
N-{3-[2-амино-5-(пиридин-4-ил)-1,3-тиазол-4-ил]-2,4-дифторфенил}-2,5-дифторбензолсульфонамид;
N-{2,4-дифтор-3-[2-(метиламино)-5-(пиридин-4-ил)-1,3-тиазол-4-ил]фенил}-2,5-дифторбензолсульфонамид;
N-{3-[2-(диэтиламино)-5-(пиридин-4-ил)-1,3-тиазол-4-ил]-2,4-дифторфенил}-2,5-дифторбензолсульфонамид;
N-(2,4-дифтор-3-{2-[(2-метилпропил)амино]-5-(пиридин-4-ил)-1,3-тиазол-4-ил}фенил)-2,5-дифторбензолсульфонамид;
N-{2,4-дифтор-3-[2-(пиперидин-1-ил)-5-(пиридин-4-ил)-1,3-тиазол-4-ил]фенил}-2,5-дифторбензолсульфонамид;
N-{3-[2-(4,4-дифторпиперидин-1-ил)-5-(пиридин-4-ил)-1,3-тиазол-4-ил]-2,4-дифторфенил}-2,5-дифторбензолсульфонамид;
N-{2,4-дифтор-3-[2-(морфолин-4-ил)-5-(пиридин-4-ил)-1,3-тиазол-4-ил]фенил}-2,5-дифторбензолсульфонамид;
N-{3-[2-(диэтиламино)-5-(пиридин-4-ил)-1,3-тиазол-4-ил]-2-фторфенил}-2,5-дифторбензолсульфонамид;
N-{3-[2-(4,4-дифторпиперидин-1-ил)-5-(пиридин-4-ил)-1,3-тиазол-4-ил]-2-фторфенил}-2,5-дифторбензолсульфонамид;
2,5-дифтор-N-{2-фтор-3-[2-(1-метилпиперидин-4-ил)-5-(пиридин-4-ил)-1,3-тиазол-4-ил]фенил}бензолсульфонамид;
2,5-дифтор-N-{2-фтор-3-[5-(пиридин-4-ил)-2-(тетрагидро-2Н-пиран-4-ил)-1,3-тиазол-4-ил]фенил}бензолсульфонамид;
N-{3-[2-(1-циклопропилпиперидин-4-ил)-5-(пиридин-4-ил)-1,3-тиазол-4-ил]-2-фторфенил}-2,5-дифторбензолсульфонамид;
N-{3-[5-(2-аминопиридин-4-ил)-2-(1-циклопропилпиперидин-4-ил)-1,3-тиазол-4-ил]-2-фторфенил}-2,5-дифторбензолсульфонамид;
N-{4-[4-(3-{[(2,5-дифторфенил)сульфонил]амино}-2-фторфенил)-2-(1-метилпиперидин-4-ил)-1,3-тиазол-5-ил]пиридин-2-ил}ацетамид;
N-{3-[5-(2-аминопиридин-4-ил)-2-(тетрагидро-2Н-пиран-4-ил)-1,3-тиазол-4-ил]-2-фторфенил}-2,5-дифторбензолсульфонамид;
2,5-дифтор-N-(2-фтор-3-{5-[2-(метиламино)пиридин-4-ил]-2-(тетрагидро-2Н-пиран-4-ил)-1,3-тиазол-4-ил}фенил)бензолсульфонамид;
2,5-дифтор-N-{2-фтор-3-[2-(1-метилпиперидин-4-ил)-5-(2-метилпиридин-4-ил)-1,3-тиазол-4-ил]фенил}бензолсульфонамид;
2,5-дифтор-N-{2-фтор-3-[5-(2-фторпиридин-4-ил)-2-(1-метилпиперидин-4-ил)-1,3-тиазол-4-ил]фенил}бензолсульфонамид;
2,5-дифтор-N-{2-фтор-3-[5-(2-метилпиридин-4-ил)-2-(тетрагидро-2Н-пиран-4-ил)-1,3-тиазол-4-ил]фенил}бензолсульфонамид;
N-{4-[2-(1-циклопропилпиперидин-4-ил)-4-(3-{[(2,5-дифторфенил)сульфонил]амино}-2-фторфенил)-1,3-тиазол-5-ил]пиридин-2-ил}ацетамид;
N-{3-[2-(1-циклопропилпиперидин-4-ил)-5-(2-метилпиридин-4-ил)-1,3-тиазол-4-ил]-2-фторфенил}-2,5-дифторбензолсульфонамид;
2,5-дифтор-N-{2-фтор-3-[5-(3-фторпиридин-4-ил)-2-(тетрагидро-2Н-пиран-4-ил)-1,3-тиазол-4-ил]фенил}бензолсульфонамид;
N-{4-[4-(3-{[(2,5-дифторфенил)сульфонил]амино}-2-фторфенил)-2-(тетрагидро-2Н-пиран-4-ил)-1,3-тиазол-5-ил]пиридин-2-ил}ацетамид;
N-[2-({4-[4-(3-{[(2,5-дифторфенил)сульфонил]амино}-2-фторфенил)-2-(тетрагидро-2Н-пиран-4-ил)-1,3-тиазол-5-ил]пиридин-2-ил}амино)этил]ацетамид;
N-(3-{2-(1-циклопропилпиперидин-4-ил)-5-[2-(метиламино)пиридин-4-ил]-1,3-тиазол-4-ил}-2-фторфенил)-2,5-дифторбензолсульфонамид;
2,5-дифтор-N-(2-фтор-3-{5-[2-(метиламино)пиридин-4-ил]-2-(1-метилпиперидин-4-ил)-1,3-тиазол-4-ил}фенил)бензолсульфонамид;
N-{3-[5-(2-аминопиридин-4-ил)-2-(1-метилпиперидин-4-ил)-1,3-тиазол-4-ил]-2-фторфенил}-2,5-дифторбензолсульфонамид;
N-{3-[5-(2-{[2-(диметиламино)этил]амино}пиридин-4-ил)-2-(тетрагидро-2Н-пиран-4-ил)-1,3-тиазол-4-ил]-2-фторфенил}-2,5-дифторбензолсульфонамид;
метил[(2S)-1-({4-[4-(3-{[(2,5-дифторфенил)сульфонил]амино}-2-фторфенил)-2-(тетрагидро-2Н-пиран-4-ил)-1,3-тиазол-5-ил]пиридин-2-ил}амино)пропан-2-ил]карбамат;
N-{4-[2-трет-бутил-4-(3-{[(2,5-дифторфенил)сульфонил]амино}-2-фторфенил)-1,3-тиазол-5-ил]пиридин-2-ил}ацетамид;
N-{3-[2-трет-бутил-5-(пиридин-4-ил)-1,3-тиазол-4-ил]-2-фторфенил}-2,5-дифторбензолсульфонамид; и
N-{4-[4-(3-{[(2,5-дифторфенил)сульфонил]амино}-2-фторфенил)-2-(пиперидин-4-ил)-1,3-тиазол-5-ил]пиридин-2-ил}ацетамид.
7. Способ ингибирования in vitro активности семейства Raf, который включает контактирование указанного рецептора с эффективным количеством соединения, как определено в п. 1.
8. Фармацевтическая композиция с активностью, направленной против злокачественной опухоли, содержащая терапевтически эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли, как определено в п.1, и по меньшей мере один фармацевтически приемлемый эксципиент, носитель и/или разбавитель.
9. Соединение формулы (I) или его фармацевтически приемлемая соль, как определено в п. 1, для применения в качестве лекарственного средства, с активностью, направленной против злокачественной опухоли.
10. Применение соединения формулы (I) или его фармацевтически приемлемой соли, как определено в п.1, для изготовления лекарственного средства с активностью, направленной против злокачественной опухоли.
| Колосоуборка | 1923 |
|
SU2009A1 |
| Колосоуборка | 1923 |
|
SU2009A1 |
| J.TSAI et al.: "From the cover: Discover of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity", PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES, 2008, vol.105, no.8, p.3041-3046 | |||
| Деревянный щит, склеиваемый из отдельных досок | 1928 |
|
SU13249A1 |
| Способ приготовления лака | 1924 |
|
SU2011A1 |
| Способ приготовления лака | 1924 |
|
SU2011A1 |

