Область, к которой относится изобретение
Настоящее изобретение относится к новым соединениям формулы (I), их таутомерным формам, их стереоизомерам, их фармацевтически приемлемым солям, содержащим их фармацевтическим композициям, способам их получения. Эти соединения демонстрируют активность против инфекции, вызванной Mycobacterium tuberculosis.
Предпосылки создания изобретения
Туберкулез (TB) является одной из основных причин смерти людей от инфекционных заболеваний в мире. Считается, что около 33% населения мира инфицированы Mycobacterium tuberculosis (Mtb), патогеном, вызывающим заболевание TB (WHO “Global Tuberculosis Report 2016”, World Health Organization, 2017). Современное лечение потенциально способно вылечить чувствительный к лекарственным средствам TB, однако лечение резистентного к лекарственным средствам или мультирезистентного к лекарственным средствам TB (MDR-TB) является сложной задачей и требует двухлетнего курса комбинированной химиотерапии (WHO “Multidrug-Resistant Tuberculosis (MDR-TB) 2016 Update”, World Health Organization, 2016). Резистентность к современным способам лечения для MDR-TB и экстенсивно резистентного TB (XDR-TB) подчеркивает потребность в новых лекарственных средствах с новым механизмом действия.
Линезолид представляет собой одобренный антибиотик для лечения грамположительных бактериальных инфекций. Он также показал антибактериальную активность против патогенов mycobacterium tuberculosis. Линезолид и другие препараты класса оксазолидинонов ингибируют синтез бактериального белка путем связывания с пептидилтрансферазным центром субъединицы рибосомы 50S и препятствуя размещению аминоацил-тРНК. Они не связываются с цитоплазматическими рибосомами млекопитающих, но связываются с митохондриальными рибосомами, которые ответственны за токсичность для костного мозга, связанную с Линезолидом и другими оксазолидинонами. Лечение TB долгое, поэтому Линезолид не подходит для его лечения. Следовательно, требуются новые оксазолидиноны с улучшенным профилем безопасности, которые можно использовать для лечения TB. В WO 2017015106 описаны замещенные фенилоксазолидиноны для лечения туберкулеза. В KR 101271224 описаны производные оксазолидинона, содержащие бициклическую группу, обладающую антибактериальной активностью против грамположительных бактерий, включая различные устойчивые штаммы. WO 2005054234 описывает новые замещенные производные пиперидинофенилоксазолидинона в качестве активных ингредиентов и способы лечения бактериальной инфекции. Также раскрыты другие документы, которые описывают класс ингибиторов оксазолидона, включая WO 9323384, WO 2002080841, WO 2001042242, WO 2003064415, WO 2009020616, WO 2017070024, WO 2005054234, WO 2002051819, WO 2017156519, WO 2013044865 и WO 2006059221.
Сущность изобретения
Настоящее изобретение раскрывает новые соединения формулы (I). Соединения по настоящему изобретению полезны для лечения человека или животного путем регулирования синтеза бактериального белка. Следовательно, соединения по настоящему изобретению подходят для лечения туберкулеза или инфекции Mycobacterium.
Варианты осуществления изобретения
Основной целью настоящего изобретения является обеспечение новых соединений общей формулы (I), их таутомерных форм, новых промежуточных соединений, участвующих в их синтезе, их фармацевтически приемлемых солей, их фармацевтически приемлемых сольватов и содержащих их фармацевтических композиций или их смесей, подходящих для лечения туберкулеза или инфекции Mycobacterium.
В другом варианте осуществления представлены фармацевтические композиции, содержащие соединения общей формулы (I), их таутомерные формы, их фармацевтически приемлемые соли, сольваты и их смеси, содержащие фармацевтически приемлемые носители, растворители, разбавители, эксципиенты и другие среды, обычно используемые при их изготовлении.
Еще в одном варианте осуществления представлено применение новых соединений по настоящему изобретению для лечения инфекций у млекопитающих, таких как туберкулез, путем введения млекопитающим терапевтически эффективного и нетоксичного количества соединения формулы (I) или его фармацевтически приемлемых композиций.
Еще в одном варианте осуществления представлен способ лечения инфекции у млекопитающих, такой как туберкулез, с использованием соединения формулы (I) или его фармацевтически приемлемых композиций для млекопитающих.
В последнем варианте осуществления представлена фармацевтическая композиция, содержащая соединение формулы (I) и второе терапевтическое средство, для лечения инфекций у млекопитающих, таких как туберкулез.
Описание изобретения
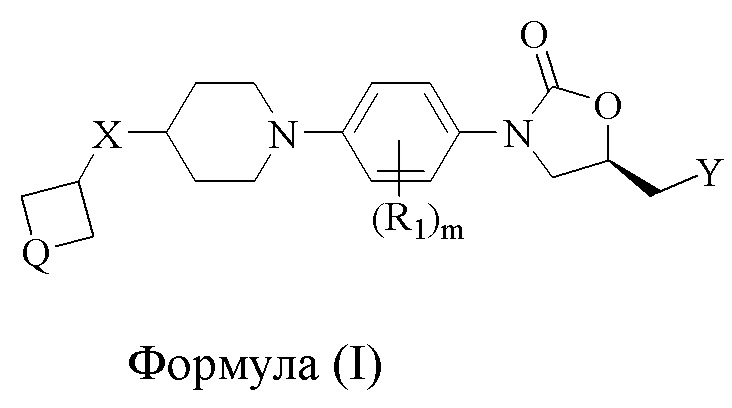
Соответственно, настоящее изобретение относится к соединениям формулы (I),
где
X либо отсутствует, либо является связью; в случаях, когда X отсутствует, четырехчленное кольцо непосредственно присоединено к 6-членному пиперидиновому кольцу, образуя спироциклическую систему;
Q либо представляет собой O, либо S(O)p; p = целое число в диапазоне 0-2;
Y представляет собой OH, NR2R3, NHC(O)R4;
R1 выбран из H, F, Cl, CH3, CN и OCH3; m = целое число в диапазоне 1-4;
R2 и R3 независимо выбраны из H, (C1-C6)алкила, (C3-C6)циклоалкила, арила, гетероциклила и гетероарила, каждый из которых может быть дополнительно необязательно замещен; R2 и R3, взятые вместе с азотом, к которому они присоединены, могут образовывать 4-8-членный гетероциклил или гетероарил с 1-3 дополнительными гетероатомами, выбранными из O, S или N, и могут быть дополнительно необязательно замещены;
R4 независимо выбран из (C1-C6)алкила, (C3-C6)циклоалкила, арила, гетероарила и (C1-C6)алкокси, каждый из которых может быть дополнительно необязательно замещен;
В варианте осуществления, когда любая из (C1-C6)алкильной, (C3-C6)циклоалкильной, арильной, гетероциклильной и гетероарильной групп необязательно замещена, группы выбраны из галогена, гидроксила, (C1-C6)алкила, (C1-C6)алкокси, (C1-C6)ацилокси, галогеналкила, NO2, CN и NH2;
В другом варианте осуществления группы, радикалы, описанные выше, могут быть выбраны из следующих групп:
- “алкильная” группа, используемая отдельно или в комбинации с другими радикалами, означает линейный или разветвленный радикал, содержащий от одного до шести атомов углерода, выбранный из метила, этила, н-пропила, изопропила, н-бутила, втор-бутила, трет-бутила, амила, трет-амила, н-пентила, н-гексила и т.д.;
- “алкокси” группа, используемая отдельно или в комбинации с другими радикалами, означает линейный или разветвленный радикал, содержащий от одного до шести атомов углерода, присоединенный к атому кислорода, выбранный из метокси, этокси, н-пропокси, изопропокси, н-бутокси, втор-бутокси, изобутокси, пентилокси, гексилокси и т.п.;
- “циклоалкильная” или “алициклическая” группа, используемая отдельно или в комбинации с другими радикалами, выбрана из циклического радикала, содержащего от трех до шести атомов углерода, более предпочтительно циклопропила, циклобутила, циклопентила, циклогексила и т.п.;
- “галогеналкильная” группа выбрана из алкильного радикала, как определено выше, подходящим образом замещенного одним или более галогенами; например, фторметильная, дифторметильная, трифторметильная, фторэтильная, дифторэтильная, трифторэтильная, моно- или полигалогензамещенная метильная, этильная, пропильная, бутильная, пентильная или гексильная группы;
- “арильная” или “ароматическая” группа, используемая отдельно или в комбинации с другими радикалами, выбрана из подходящей ароматической системы, содержащей одно, два или три кольца, где такие кольца могут быть соединены вместе по типу боковых подвешенных групп или могут быть конденсированы, более предпочтительно группы выбраны из фенила, нафтила, тетрагидронафтила, индана, бифенила и т.д.;
- “гетероциклильная” или “гетероциклическая” группа, используемая отдельно или в комбинации с другими радикалами, выбрана из подходящих насыщенных, частично насыщенных или ненасыщенных ароматических или неароматических моно-, би- или трициклических радикалов, содержащих один или более гетероатомов, выбранных из азота, серы и кислорода, более предпочтительно выбрана из азиридинила, азетидинила, пирролидинила, имидазолидинила, пиперидинила, пиперазинила, 2-оксопиперидинила, 4-оксопиперидинила, 2-оксопиперазинила, 3-оксопиперазинила, морфолинила, тиоморфолинила, 2-оксоморфолинила, азепинила, диазепинила, оксапинила, тиазепинила, оксазолидинила, тиазолидинила, дигидротиофена, дигидропирана, дигидрофурана, дигидротиазола, бензопиранила, бензопиранонила, бензодигидрофуранила, бензодигидротиенила, пиразолопиримидонила, азахиназолиноила, тиенопиримидонила, хиназолонила, пиримидонила, бензоксазинила, бензоксазинонила, бензотиазинила, бензотиазинонила, тиенопиперидинила и т.п.; В одном варианте осуществления гетероциклильная группа, где это применимо, может состоять из подходящего количества атомов углерода и включать от 1 до 4 гетероатомов, выбранных из группы, состоящей из N, O и S(O)p, p = 0-2;
- “гетероарильная” или “гетероароматическая” группа, используемая отдельно или в комбинации с другими радикалами, выбрана из подходящих одиночных или конденсированных моно-, би- или трициклических ароматических гетероциклических радикалов, содержащих один или более гетероатомов, выбранных из O, N или S, более предпочтительно группы выбраны из пиридила, тиенила, фурила, пирролила, оксазолила, тиазолила, изотиазолила, имидазолила, изоксазолила, оксадиазолила, тиадиазолила, триазолила, тетразолила, бензофуранила, бензотиенила, индолинила, индолила, азаиндолила, азаиндолинила, пиразолопиримидинила, азахиназолинила, пиридофуранила, пиридотиенила, тиенопиримидила, хинолинила, пиримидинила, пиразолила, хиназолинила, пиридазинила, триазинила, бензимидазолила, бензотриазолила, фталазинила, нафтилидинила, пуринила, карбазолила, фенотиазинила, феноксазинила, бензоксазолила, бензотиазолила и т.п.;
- “аралкильная” группа, используемая отдельно или в комбинации с другими радикалами, выбрана из групп, содержащих арильный радикал, как определено выше, присоединенный непосредственно к алкильному радикалу, как определено выше, более предпочтительно группы выбраны из бензила, фенэтила и т.д.;
- “гетероциклилалкильная” группа, используемая отдельно или в комбинации с другими радикалами, выбрана из групп, содержащих гетероциклильный радикал, как определено выше, присоединенный непосредственно к алкильному радикалу, как определено выше;
- “спироциклическая система” представляет собой группу, в которой одно циклическое кольцо присоединено к другому циклическому кольцу через один атом углерода, который является общим атомом углерода для обоих циклических колец.
Другие предпочтительные варианты осуществления раскрыты ниже.
Предпочтительная “(C1-C6)алкильная” группа R2, R3 и R4 выбрана из метила, этила, н-пропила, изопропила;
Предпочтительная “(C1-C6)алкокси” группа R2, R3 и R4 выбрана из метокси и этокси;
Предпочтительная “(C3-C6)циклоалкильная” группа R2, R3 и R4 выбрана из циклопропила и циклобутила;
Предпочтительная “гетероарильная” группа R2 и R3 выбрана из триазолила, изоксазолила, тиенила, фурила.
Подходящие группы и заместители в группах могут быть выбраны из тех, которые описаны где-либо в настоящем описании.
Предпочтительные соединения в соответствии с настоящим изобретением включают, но не ограничиваются этим:
(R)-5-((1H-1,2,3-триазол-1-ил)метил)-3-(4-(2,2-диоксидо-2-тиа-7-азаспиро[3.5]нонан-7-ил)-3,5-дифторфенил)оксазолидин-2-он;
(S)-3-(4-(2,2-диоксидо-2-тиа-7-азаспиро[3.5]нонан-7-ил)-3,5-дифторфенил)-5-((изоксазол-3-иламино)метил)оксазолидин-2-он;
(S)-N-((3-(4-(2,2-диоксидо-2-тиа-7-азаспиро[3.5]нонан-7-ил)-3,5-дифторфенил)-2-оксооксазолидин-5-ил)метил)ацетамид;
(S)-метил((3-(4-(2,2-диоксидо-2-тиа-7-азаспиро[3.5]нонан-7-ил)-3,5-дифторфенил)-2-оксооксазолидин-5-ил)метил)карбамат;
(S)-N-((3-(4-(2,2-диоксидо-2-тиа-7-азаспиро[3.5]нонан-7-ил)-3-фторфенил)-2-оксооксазолидин-5-ил)метил)ацетамид;
(S)-N-((3-(3,5-дифтор-4-(2-окса-7-азаспиро[3.5]нонан-7-ил)фенил)-2-оксооксазолидин-5-ил)метил)ацетамид;
(S)-N-((3-(3-фтор-4-(2-окса-7-азаспиро[3.5]нонан-7-ил)фенил)-2-оксооксазолидин-5-ил)метил)ацетамид;
(S)-N-((3-(3,5-дифтор-4-(4-(оксетан-3-ил)пиперидин-1-ил)фенил)-2-оксооксазолидин-5-ил)метил)ацетамид;
(S)-N-((3-(4-(4-(1,1-диоксидотиетан-3-ил)пиперидин-1-ил)-3,5-дифторфенил)-2-оксооксазолидин-5-ил)метил)ацетамид;
(S)-N-((3-(4-(4-(1,1-диоксидотиетан-3-ил)пиперидин-1-ил)-3-фторфенил)-2-оксооксазолидин-5-ил)метил)ацетамид;
метил-(S)-((3-(4-(4-(1,1-диоксидотиетан-3-ил)пиперидин-1-ил)-3,5-дифторфенил)-2-оксооксазолидин-5-ил)метил)карбамат;
(S)-5-(аминометил)-3-(4-(4-(1,1-диоксидотиетан-3-ил)пиперидин-1-ил)-3,5-дифторфенил)оксазолидин-2-он;
(R)-3-(4-(4-(1,1-диоксидотиетан-3-ил)пиперидин-1-ил)-3,5-дифторфенил)-5-(гидроксиметил)оксазолидин-2-он;
(S)-5-(аминометил)-3-(3,5-дифтор-4-(4-(оксетан-3-ил)пиперидин-1-ил)фенил)оксазолидин-2-он;
(S)-5-(аминометил)-3-(3,5-дифтор-4-(2-окса-7-азаспиро[3.5]нонан-7-ил)фенил)оксазолидин-2-он;
(S)-5-(аминометил)-3-(4-(2,2-диоксидо-2-тиа-7-азаспиро[3.5]нонан-7-ил)-3,5-дифторфенил)оксазолидин-2-он;
(R)-3-(4-(2,2-диоксидо-2-тиа-7-азаспиро[3.5]нонан-7-ил)-3,5-дифторфенил)-5-(гидроксиметил)оксазолидин-2-он;
(S)-N-((3-(4-(2,2-диоксидо-2-тиа-7-азаспиро[3.5]нонан-7-ил)-3,5-дифторфенил)-2-оксооксазолидин-5-ил)метил)тиофен-2-карбоксамид;
(S)-N-((3-(4-(2,2-диоксидо-2-тиа-7-азаспиро[3.5]нонан-7-ил)-3-фторфенил)-2-оксооксазолидин-5-ил)метил)тиофен-2-карбоксамид;
(S)-N-((3-(4-(2,2-диоксидо-2-тиа-7-азаспиро[3.5]нонан-7-ил)-3,5-дифторфенил)-2-оксооксазолидин-5-ил)метил)фуран-2-карбоксамид;
(S)-N-((3-(4-(2,2-диоксидо-2-тиа-7-азаспиро[3.5]нонан-7-ил)-3-фторфенил)-2-оксооксазолидин-5-ил)метил)фуран-2-карбоксамид;
(S)-N-((3-(4-(4-(1,1-диоксидотиетан-3-ил)пиперидин-1-ил)-3,5-дифторфенил)-2-оксооксазолидин-5-ил)метил)тиофен-2-карбоксамид;
(S)-N-((3-(4-(4-(1,1-диоксидотиетан-3-ил)пиперидин-1-ил)-3-фторфенил)-2-оксооксазолидин-5-ил)метил)тиофен-2-карбоксамид;
(S)-N-((3-(4-(4-(1,1-диоксидотиетан-3-ил)пиперидин-1-ил)-3,5-дифторфенил)-2-оксооксазолидин-5-ил)метил)фуран-2-карбоксамид;
(S)-N-((3-(4-(4-(1,1-диоксидотиетан-3-ил)пиперидин-1-ил)-3-фторфенил)-2-оксооксазолидин-5-ил)метил)фуран-2-карбоксамид;
(S)-N-((3-(4-(2,2-диоксидо-2-тиа-7-азаспиро[3.5]нонан-7-ил)-3,5-дифторфенил)-2-оксооксазолидин-5-ил)метил)бутирамид;
(S)-N-((3-(4-(2,2-диоксидо-2-тиа-7-азаспиро[3.5]нонан-7-ил)-3-фторфенил)-2-оксооксазолидин-5-ил)метил)бутирамид;
(S)-N-((3-(4-(4-(1,1-диоксидотиетан-3-ил)пиперидин-1-ил)-3,5-дифторфенил)-2-оксооксазолидин-5-ил)метил)бутирамид;
(S)-N-((3-(4-(4-(1,1-диоксидотиетан-3-ил)пиперидин-1-ил)-3-фторфенил)-2-оксооксазолидин-5-ил)метил)бутирамид;
этил-(S)-((3-(4-(2,2-диоксидо-2-тиа-7-азаспиро[3.5]нонан-7-ил)-3,5-дифторфенил)-2-оксооксазолидин-5-ил)метил)карбамат;
этил-(S)-((3-(4-(2,2-диоксидо-2-тиа-7-азаспиро[3.5]нонан-7-ил)-3-фторфенил)-2-оксооксазолидин-5-ил)метил)карбамат;
этил-(S)-((3-(4-(4-(1,1-диоксидотиетан-3-ил)пиперидин-1-ил)-3,5-дифторфенил)-2-оксооксазолидин-5-ил)метил)карбамат;
этил-(S)-((3-(4-(4-(1,1-диоксидотиетан-3-ил)пиперидин-1-ил)-3-фторфенил)-2-оксооксазолидин-5-ил)метил)карбамат;
(S)-N-((3-(4-(2,2-диоксидо-2-тиа-7-азаспиро[3.5]нонан-7-ил)-3,5-дифторфенил)-2-оксооксазолидин-5-ил)метил)циклопропанкарбоксамид;
(S)-N-((3-(4-(2,2-диоксидо-2-тиа-7-азаспиро[3.5]нонан-7-ил)-3-фторфенил)-2-оксооксазолидин-5-ил)метил)циклопропанкарбоксамид;
(S)-N-((3-(4-(4-(1,1-диоксидотиетан-3-ил)пиперидин-1-ил)-3,5-дифторфенил)-2-оксооксазолидин-5-ил)метил)циклопропанкарбоксамид;
(S)-N-((3-(4-(4-(1,1-диоксидотиетан-3-ил)пиперидин-1-ил)-3-фторфенил)-2-оксооксазолидин-5-ил)метил)циклопропанкарбоксамид;
(S)-N-((3-(4-(2,2-диоксидо-2-тиа-7-азаспиро[3.5]нонан-7-ил)-3,5-дифторфенил)-2-оксооксазолидин-5-ил)метил)циклобутанкарбоксамид;
(S)-N-((3-(4-(2,2-диоксидо-2-тиа-7-азаспиро[3.5]нонан-7-ил)-3-фторфенил)-2-оксооксазолидин-5-ил)метил)циклобутанкарбоксамид;
(S)-N-((3-(4-(4-(1,1-диоксидотиетан-3-ил)пиперидин-1-ил)-3,5-дифторфенил)-2-оксооксазолидин-5-ил)метил)циклобутанкарбоксамид;
(S)-N-((3-(4-(4-(1,1-диоксидотиетан-3-ил)пиперидин-1-ил)-3-фторфенил)-2-оксооксазолидин-5-ил)метил)циклобутанкарбоксамид;
(R)-5-((1H-1,2,3-триазол-1-ил)метил)-3-(4-(2,2-диоксидо-2-тиа-7-азаспиро[3.5]нонан-7-ил)-3-фторфенил)оксазолидин-2-он;
(R)-5-((1H-1,2,3-триазол-1-ил)метил)-3-(4-(4-(1,1-диоксидотиетан-3-ил)пиперидин-1-ил)-3,5-дифторфенил)оксазолидин-2-он;
(R)-5-((1H-1,2,3-триазол-1-ил)метил)-3-(4-(4-(1,1-диоксидотиетан-3-ил)пиперидин-1-ил)-3-фторфенил)оксазолидин-2-он;
(R)-5-((1H-1,2,4-триазол-1-ил)метил)-3-(4-(2,2-диоксидо-2-тиа-7-азаспиро[3.5]нонан-7-ил)-3,5-дифторфенил)оксазолидин-2-он;
(R)-5-((1H-1,2,4-триазол-1-ил)метил)-3-(4-(2,2-диоксидо-2-тиа-7-азаспиро[3.5]нонан-7-ил)-3-фторфенил)оксазолидин-2-он;
(S)-3-(4-(2,2-диоксидо-2-тиа-7-азаспиро[3.5]нонан-7-ил)-3-фторфенил)-5-((изоксазол-3-иламино)метил)оксазолидин-2-он;
(S)-3-(4-(4-(1,1-диоксидотиетан-3-ил)пиперидин-1-ил)-3,5-дифторфенил)-5-((изоксазол-3-иламино)метил)оксазолидин-2-он;
метил-(S)-((3-(4-(4-(1,1-диоксидотиетан-3-ил)пиперидин-1-ил)-3-фторфенил)-2-оксооксазолидин-5-ил)метил)карбамат;
метил-(S)-((3-(4-(2,2-диоксидо-2-тиа-7-азаспиро[3.5]нонан-7-ил)-3-фторфенил)-2-оксооксазолидин-5-ил)метил)карбамат;
метил-(S)-((3-(3-фтор-4-(2-окса-7-азаспиро[3.5]нонан-7-ил)фенил)-2-оксооксазолидин-5-ил)метил)карбамат;
(R)-5-((1H-1,2,3-триазол-1-ил)метил)-3-(3-фтор-4-(2-окса-7-азаспиро[3.5]нонан-7-ил)фенил)оксазолидин-2-он;
метил-(S)-((3-(3,5-дифтор-4-(2-окса-7-азаспиро[3.5]нонан-7-ил)фенил)-2-оксооксазолидин-5-ил)метил)карбамат;
(R)-5-((1H-1,2,3-триазол-1-ил)метил)-3-(3,5-дифтор-4-(2-окса-7-азаспиро[3.5]нонан-7-ил)фенил)оксазолидин-2-он;
(R)-5-((1H-1,2,3-триазол-1-ил)метил)-3-(3,5-дифтор-4-(4-(оксетан-3-ил)пиперидин-1-ил)фенил)оксазолидин-2-он.
Новые соединения по настоящему изобретению можно получить с использованием реакций и процедур, показанных на схеме ниже и описанных в этом разделе. Реакции осуществляют в растворителях, подходящих для используемых реагентов и веществ и подходящих для соответствующих преобразований. Специалистам в данной области должно быть понятно, что природа и порядок представленных стадий синтеза могут быть изменены с целью оптимизации образования соединений по настоящему изобретению. Также хорошо известно, что одно или более участвующих в реакции веществ можно защитить и осуществить снятие защиты для облегчения синтеза методами, известными специалистам в данной области. Также должно быть понятно, что одно или более соединений по настоящему изобретению могут существовать в стереоизомерных и/или диастереомерных формах. Такие стереоизомеры и/или диастереоизомеры, а также их оптические антиподы должны рассматриваться как входящие в объем настоящего изобретения. Также должно быть понятно, что одно или более из этих соединений можно преобразовать в их соли и другие производные, на основе конкретных групп, присутствующих в соединениях, что может быть хорошо известно специалистам в данной области. Такие соли и/или другие производные, в зависимости от обстоятельств, также следует рассматривать как входящие в объем настоящего изобретения.
Схема 1: Синтез соединений общей формулы (I)
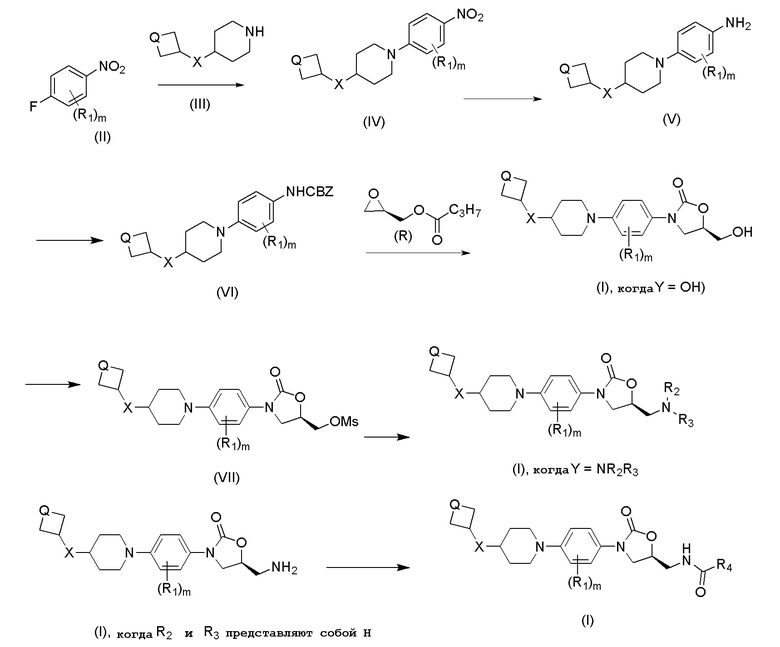
Соединение (IV) можно получить путем взаимодействия соединений формулы (II) с (III) в присутствии основания, такого как карбонат натрия, K2CO3, гидрид натрия и т.д., в растворителях, таких как THF, DMF, MeOH и т.д. Соединения общей формулы (V) можно получить путем восстановления с использованием дигидрата хлорида олова в этилацетате. Соединения общей формулы (VI) можно получить путем взаимодействия (V) с бензилоксикарбонилхлоридом, используя карбонат натрия в качестве основания, в растворителях, таких как вода, этилацетат, ацетонитрил или их смесь. Соединение формулы (I, когда Y=OH) можно получить путем обработки н-бутиллитием и R-глицидилбутиратом в THF. Затем его преобразовывали в мезилатные производные (VII) с использованием метансульфонилхлорида и TEA в растворителях, таких как THF, ACN и т.д., которые затем подвергали взаимодействию с соответствующим образом замещенными аминами с получением желаемых соединений (I, когда Y=NR2R3). Соединения (I, когда Y=NH2) далее подвергали взаимодействию с подходящим хлорангидридом или ангидридом в присутствии основания, такого как TEA или пиридин, с получением соединений формулы (I) с амидной или карбаматной связью.
Схема 2: Синтез соединений общей формулы (I)
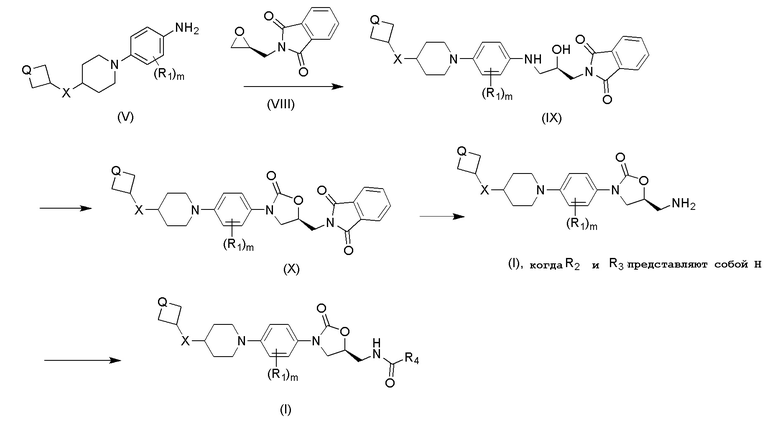
Альтернативно, можно осуществить взаимодействие соединения общей формулы (V) с соединениями формулы (VIII) (полученными в соответствии с процедурой, описанной в Tetrahedron: Asymmetry, 7(6), 1641-1648; 1996) в растворителях, таких как MeOH, EtOH и т.д., с получением соединений (IX). Соединения общей формулы (X) можно получить путем циклизации (X) с использованием CDI в растворителе, таком как DCM, CHCl3 и т.д. Соединения (I) (когда Y=NH2) получают снятием фталимидной защиты с использованием водн. метиламина в протонных растворителях, таких как MeOH, EtOH и т.д. Соединения (I, когда Y=NH2) дополнительно подвергают взаимодействию с соответствующим хлорангидридом или ангидридом кислоты в присутствии основания, такого как TEA или пиридин, с получением соединений формулы (I) с амидной или карбаматной связью.
Перечень аббревиатур
ACN: Ацетонитрил
CDI: 1,1'-Карбонилдиимидазол
CHCl3: Хлороформ
DCM: Дихлорметан
DMAP: 4-диметиламинопиридин
DMF: Диметилформамид
DMSO: Диметилсульфоксид
EtOAc: Этилацетат
EtOH: Этанол
K2CO3: Карбонат калия
LiAlH4: Литийалюминийгидрид
MeOH: Метанол
NaHCO3: Бикарбонат натрия
Na2SO4: Сульфат натрия
NaOH: Гидроксид натрия
Pd-C: палладий на угле
TEA: Триэтиламин
TFA: Трифторуксусная кислота
THF: Тетрагидрофуран
1H ЯМР: протонный ядерный магнитный резонанс
ч: час(часы)
мин: минута(минуты)
J: Константа взаимодействия в Гц
Гц: Герц
MABA: анализ в микропланшете с использованием красителя аламарового синего.
Получение соединений
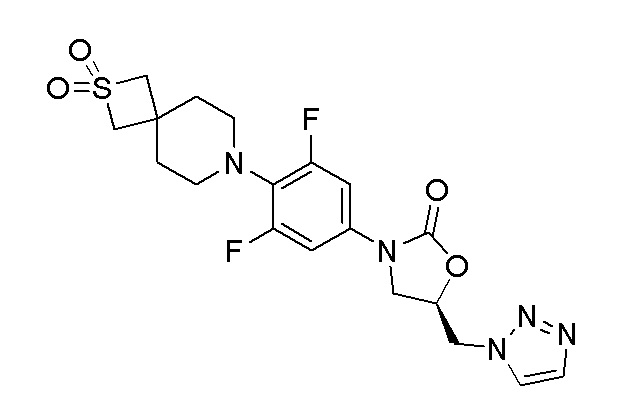
ПРИМЕР 1
Получение (R)-5-((1H-1,2,3-триазол-1-ил)метил)-3-(4-(2,2-диоксидо-2-тиа-7-азаспиро[3.5]нонан-7-ил)-3,5-дифторфенил)оксазолидин-2-она
Стадия 1: N-бензил-2-хлор-N-(2-хлорэтил)этан-1-амин
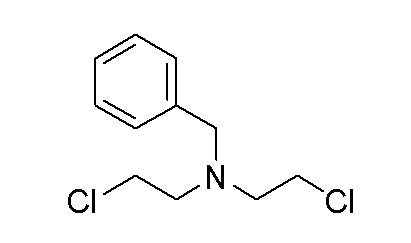
Бис(2-хлорэтил)амин (208 г, 1462 ммоль) добавляли к DCM (200 мл), затем добавляли 2M NaOH раствор и перемешивали в течение 30 мин. при 25-30°C. Отделенный DCM слой сушили над Na2SO4 и упаривали с получением масла (свободный амин). К этому добавляли ACN (500 мл), затем бензилбромид (69,5 мл, 585 ммоль) и K2CO3 (242 г, 1754 ммоль) при 25-30°C и перемешивали в течение 16 часов при 80°C. Реакционную смесь фильтровали и фильтрат упаривали с получением неочищенного масла, которое очищали колоночной хроматографией с использованием смеси гексан/EtOAc. ESI-MS (m/z): 232,05 [M+H]+.
Стадия 2: диэтил-1-бензилпиперидин-4,4-дикарбоксилат
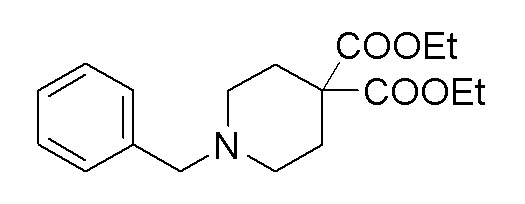
К перемешиваемому раствору продукта стадии 1 (17 г, 73,2 ммоль) в DMF добавляли K2CO3 (30,4 г, 220 ммоль), диэтилмалонат (11,17 мл, 73,2 ммоль) и тетрабутиламмонийбромид (2,361 г, 7,32 ммоль) при 25-30°C. Реакционную смесь перемешивали в течение 16 часов при 90°C. После завершения реакции смесь разбавляли EtOAc и промывали водой. EtOAc слой сушили над Na2SO4 и упаривали с получением неочищенного продукта, который очищали колоночной хроматографией с использованием смеси гексан/EtOAc. ESI-MS (m/z): 320,22 [M+H]+.
Стадия 3: (1-бензилпиперидин-4,4-диил)диметанол
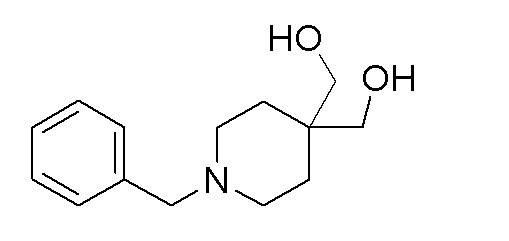
К перемешиваемому раствору продукта стадии 2 (26 г, 81 ммоль) в THF добавляли LiAlH4 (9,27 г, 244 ммоль) при 0°C и смесь перемешивали в течение 16 часов при 25-30°C. После завершения реакции смесь охлаждали до 0°C и разбавляли 10 мл воды, затем 10 мл 10% водного раствора NaOH и фильтровали через слой Hyflow. Остаток промывали при помощи 10% MeOH в EtOAc. Фильтрат выпаривали с получением указанного в заголовке продукта. ESI-MS (m/z): 236,15 [M+H]+.
Стадия 4: пиперидин-4,4-диилдиметанол
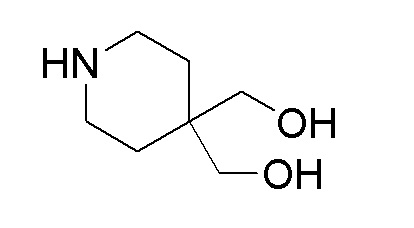
К перемешиваемому раствору продукта стадии 3 (4,50 г, 19,12 ммоль) в MeOH добавляли Pd-C (0,407 г, 3,82 ммоль), затем формиат аммония (3,62 г, 57,4 ммоль). Реакционную смесь кипятили с обратным холодильником в течение 6 часов. После завершения реакции смесь пропускали через слой hyflow и промывали при помощи 20% MeOH в EtOAc. Органический слой отгоняли дистилляцией с получением указанного в заголовке продукта. ESI-MS (m/z): 146,10 [M+H]+.
Стадия 5: трет-бутил-4,4-бис(гидроксиметил)пиперидин-1-карбоксилат
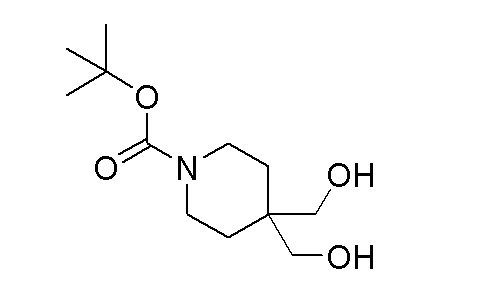
К перемешиваемому раствору продукта стадии 4 (9,4 г, 64,7 ммоль) в DMF добавляли TEA (13,54 мл, 97 ммоль) и ди-трет-бутилдикарбонат (15,03 мл, 64,7 ммоль). Реакционную смесь перемешивали в течение 3 часов при 25-30°C. После завершения реакции реакционную смесь разбавляли EtOAc (150 мл) и промывали водой (30 мл). Органический слой отгоняли дистилляцией с получением указанного в заголовке продукта.
Стадия 6: трет-бутил-4,4-бис(((метилсульфонил)окси)метил)пиперидин-1-карбоксилат
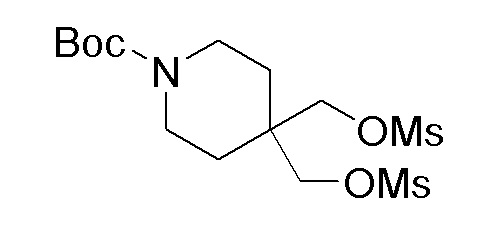
К перемешиваемому раствору продукта стадии 5 (10 г, 40,8 ммоль) в DCM добавляли TEA (22,92 мл, 163 ммоль), затем добавляли метансульфонилхлорид (9,46 мл, 122 ммоль) при 0-5°C и реакционную смесь перемешивали в течение 2 часов при 25-30°C. После завершения реакции смесь разбавляли DCM (50 мл) и водой (25 мл). Органический слой отделяли, сушили над Na2SO4 и концентрировали при пониженном давлении с получением указанного в заголовке продукта.
Стадия 7: трет-бутил-2-тиа-7-азаспиро[3.5]нонан-7-карбоксилат
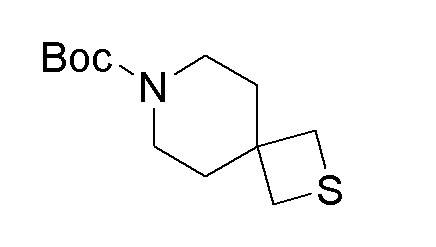
К перемешиваемому раствору продукта стадии 6 (4,90 г, 12,20 ммоль) в EtOH добавляли нонагидрат сульфида натрия (3,52 г, 14,65 ммоль) и реакционную смесь кипятили с обратным холодильником в течение 16 часов. После завершения реакции смесь разбавляли при помощи EtOAc (100 мл). Органический слой отделяли, сушили над Na2SO4 и упаривали с получением неочищенного продукта, который очищали колоночной хроматографией с использованием смеси гексан/EtOAc с получением указанного в заголовке продукта.
Стадия 8: трет-бутил-2-тиа-7-азаспиро[3.5]нонан-7-карбоксилат 2,2-диоксид
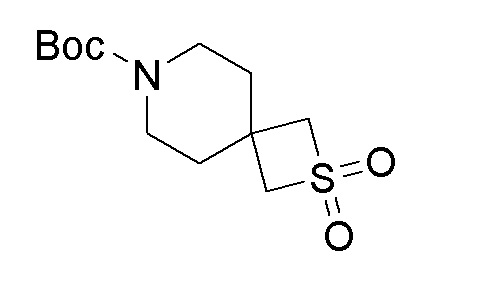
В круглодонную колбу добавляли оксон (9,60 г, 15,61 ммоль) в воде (45 мл) при 0-5°C. Затем по каплям добавляли продукт стадии 7 (1,90 г, 7,81 ммоль), растворенный в MeOH при 0-5°C. Реакционную смесь перемешивали при 15-20°C в течение 4 часов. Реакционную смесь фильтровали и промывали при помощи MeOH (30 мл). MeOH выпаривали и остаток экстрагировали при помощи DCM. Органический слой сушили над Na2SO4 и упаривали при пониженном давлении с получением указанного в заголовке продукта в виде твердого вещества.
Стадия 9: 7-(2,6-дифтор-4-нитрофенил)-2-тиа-7-азаспиро[3.5]нонан 2,2-диоксид
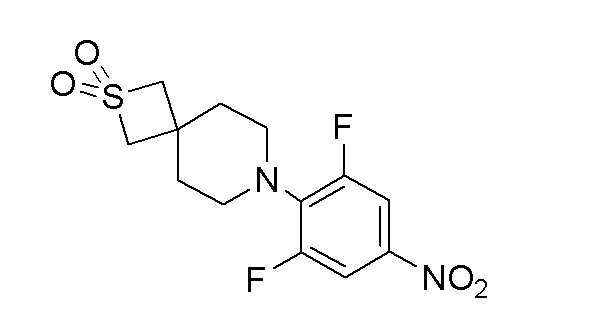
К перемешиваемому раствору продукта стадии 8 (0,650 г, 2,361 ммоль) в DCM добавляли TFA (0,903 мл, 11,80 ммоль) при 0-5°C и перемешивали в течение 3 часов при 25-30°C. После завершения реакции DCM выпаривали при пониженном давлении с получением неочищенного продукта, который разбавляли DMF (10 мл). К этому добавляли K2CO3 (0,946 г, 6,85 ммоль) и 1,2,3-трифтор-5-нитробензол (0,404 г, 2,283 ммоль) и перемешивали в течение 4 часов при 80°C. Реакционную смесь затем разбавляли водой, полученное твердое вещество фильтровали с получением указанного в заголовке продукта. ESI-MS (m/z): 333,14 [M+H]+.
Стадия 10: 7-(4-амино-2,6-дифторфенил)-2-тиа-7-азаспиро[3.5]нонан 2,2-диоксид
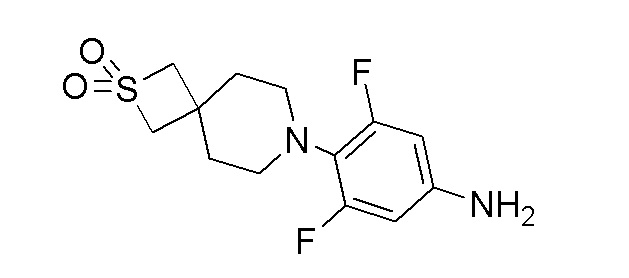
К перемешиваемому раствору продукта стадии 9 (0,690 г, 2,076 ммоль) в EtOAc добавляли дигидрат хлорида олова(II) (2,342 г, 10,38 ммоль) и перемешивали в течение 1,5 часов при 80°C. После завершения реакции смесь охлаждали до 25°C и подщелачивали водным раствором аммиака. EtOAc декантировали и остаток снова экстрагировали при помощи EtOAc. Объединенный EtOAc слой сушили над Na2SO4 и концентрировали с получением указанного в заголовке продукта. ESI-MS (m/z): 303,15 [M+H]+.
Стадия 11: бензил(4-(2,2-диоксидо-2-тиа-7-азаспиро[3.5]нонан-7-ил)-3,5-дифторфенил)карбамат
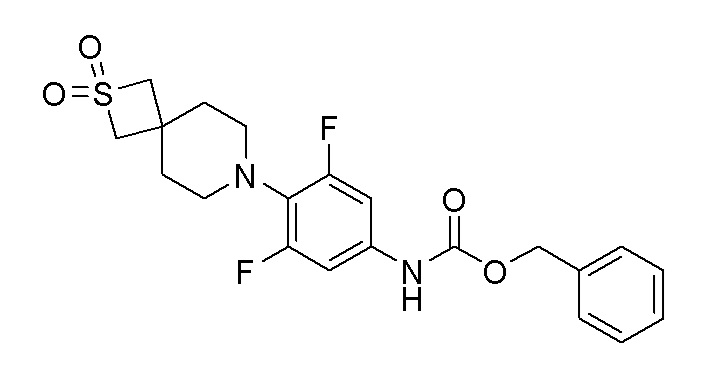
К перемешиваемому раствору продукта стадии 10 (0,535 г, 1,770 ммоль) в THF добавляли NaHCO3 (0,223 г, 2,65 ммоль) и бензилхлорформиат (0,785 г, 2,300 ммоль) при 0-5°C. Реакционную смесь перемешивали при 25-30°C в течение 7 часов. После завершения реакции смесь разбавляли EtOAC и водой. Органический слой отделяли, сушили и упаривали при пониженном давлении с получением неочищенного продукта, который очищали колоночной хроматографией с использованием смеси гексан/EtOAc с получением чистого продукта. ESI-MS (m/z): 437,16 [M+H]+.
Стадия 12: (R)-3-(4-(2,2-диоксидо-2-тиа-7-азаспиро[3.5]нонан-7-ил)-3,5-дифторфенил)-5-(гидроксиметил)оксазолидин-2-он
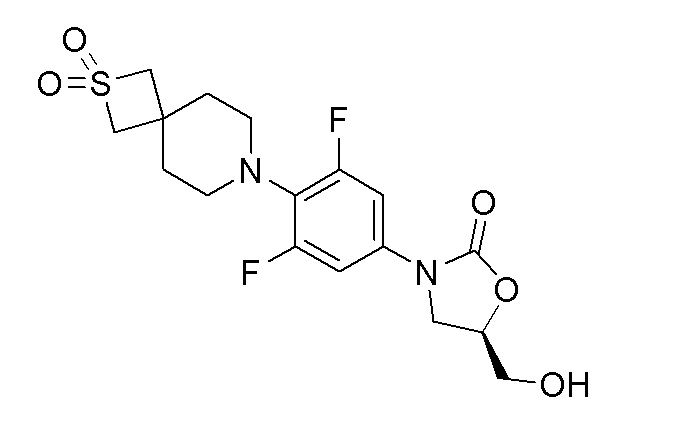
К перемешиваемому раствору продукта стадии 11 (830 мг, 1,902 ммоль) в безводном THF добавляли н-бутиллитий (2,282 мл (2,5 M), 5,70 ммоль) при -78°C. Полученный светло-желтый раствор перемешивали при -78°C в течение 1 часа и затем по каплям добавляли (R)-глицидилбутират (685 мг, 4,75 ммоль) при -70-75°C. Реакционную смесь перемешивали еще 1 час при -78°C. Реакционной смеси давали нагреться до комнатной температуры и перемешивали в течение 16 часов. Реакционную смесь разбавляли водой и EtOAc. Органический слой отделяли, сушили над Na2SO4 и концентрировали с получением неочищенного продукта в виде масла. Неочищенный продукт очищали колоночной хроматографией с получением указанного в заголовке продукта. ESI-MS (m/z): 403,14 [M+H]+.
Стадия 13: (R)-(3-(4-(2,2-диоксидо-2-тиа-7-азаспиро[3.5]нонан-7-ил)-3,5-дифторфенил)-2-оксооксазолидин-5-ил)метилметансульфонат
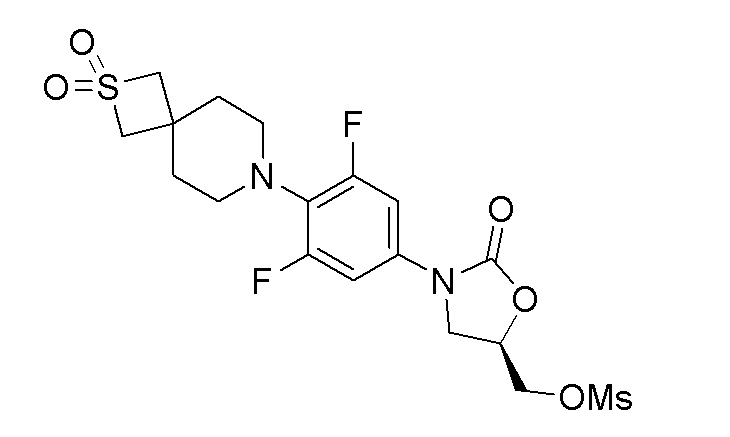
К перемешиваемому раствору продукта стадии 12 (220 мг, 0,547 ммоль) в DCM добавляли TEA (0,133 мл, 0,957 ммоль) и метансульфонилхлорид (0,047 мл, 0,601 ммоль) при 0-5°C. Реакционную смесь перемешивали в течение 2 часов при 25-30°C. После завершения реакции смесь разбавляли DCM и промывали водой. DCM слой отделяли, сушили над Na2SO4 и упаривали с получением указанного в заголовке продукта. ESI-MS (m/z): 481,16 [M+H]+.
Стадия 14: (R)-5-((1H-1,2,3-триазол-1-ил)метил)-3-(4-(2,2-диоксидо-2-тиа-7-азаспиро[3.5]нонан-7-ил)-3,5-дифторфенил)оксазолидин-2-он
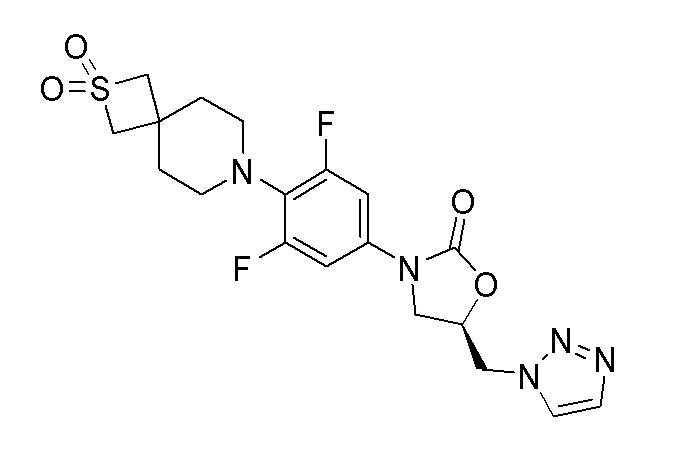
К перемешиваемому раствору продукта стадии 13 (100 мг, 0,208 ммоль) в DMF добавляли 1H-1,2,3-триазол (0,024 мл, 0,416 ммоль) и K2CO3 (57,5 мг, 0,416 ммоль). Реакционную смесь нагревали до 80°C. После завершения реакции смесь разбавляли EtOAc и водой. EtOAc слой отделяли, промывали водой, сушили над Na2SO4 и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали колоночной хроматографией с получением указанного в заголовке продукта. 1H ЯМР (DMSO-d6): 8,16 (с, 1H), 7,77 (с, 1H), 7,22 (д, J=8,4 Гц, 2H), 5,23 (м, 1H), 5,17-5,11 (м, 2H), 4,83-4,81 (м, 1H), 4,21-4,17 (м, 1H), 4,03 (с, 4H), 3,87-3,84 (м, 1H), 3,01 (с, 4H), 1,95 (с, 4H). ESI-MS (m/z): 454,12 [M+H]+.
ПРИМЕР 2
Получение (S)-3-(4-(2,2-диоксидо-2-тиа-7-азаспиро[3.5]нонан-7-ил)-3,5-дифторфенил)-5-((изоксазол-3-иламино)метил)оксазолидин-2-она
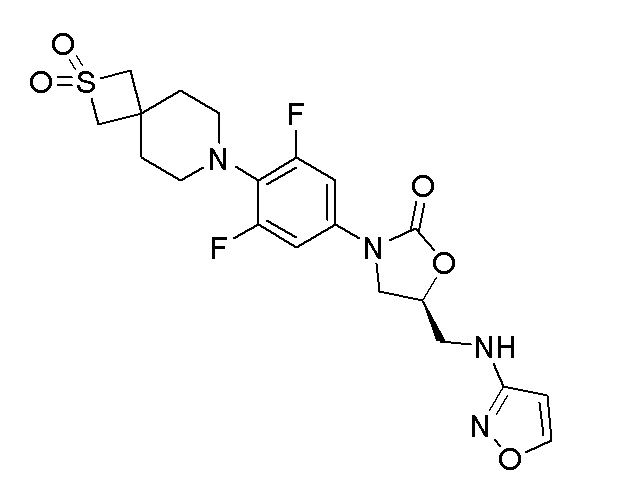
Стадия 1: (R)-трет-бутил((3-(4-(2,2-диоксидо-2-тиа-7-азаспиро[3.5]нонан-7-ил)-3,5-дифторфенил)-2-оксооксазолидин-5-ил)метил)(изоксазол-3-ил)карбамат
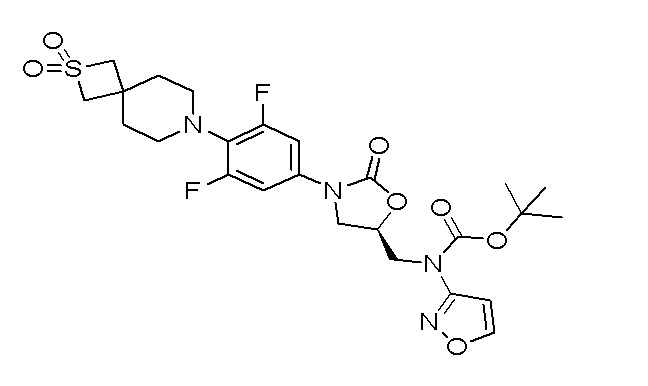
К перемешиваемому раствору (R)-(3-(4-(2,2-диоксидо-2-тиа-7-азаспиро[3.5]нонан-7-ил)-3,5-дифторфенил)-2-оксооксазолидин-5-ил)метилметансульфоната (100 мг, 0,208 ммоль) в DMF добавляли трет-бутилизоксазол-3-илкарбамат (57,5 мг, 0,312 ммоль) и K2CO3 (57,5 мг, 0,416 ммоль). Реакционную смесь перемешивали при 80°C в течение 3 часов. После завершения реакции смесь разбавляли EtOAc и водой. EtOAc слой отделяли, сушили над Na2SO4 и концентрировали с получением неочищенного продукта, который очищали колоночной хроматографией с использованием смеси EtOAc:гексан с получением указанного в заголовке продукта. ESI-MS (m/z): 569,16 [M+H]+.
Стадия 2: (S)-3-(4-(2,2-диоксидо-2-тиа-7-азаспиро[3.5]нонан-7-ил)-3,5-дифторфенил)-5-((изоксазол-3-иламино)метил)оксазолидин-2-он
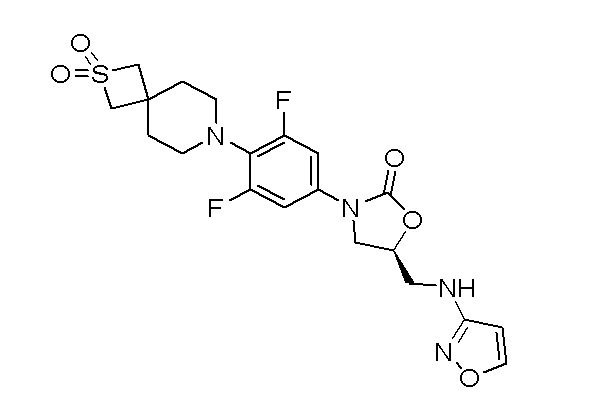
К перемешиваемому раствору стадии 1 (70 мг, 0,123 ммоль) в DCM (10 мл) добавляли TFA (0,047 мл, 0,616 ммоль) при 0-5°C и перемешивали в течение 2 часов. После завершения реакции смесь разбавляли при помощи EtOAc и промывали насыщенным раствором NaHCO3. Органический слой отделяли, сушили над Na2SO4 и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали колоночной хроматографией с использованием смеси EtOAc:гексан с получением указанного в заголовке продукта. 1H ЯМР (DMSO): 8,39 (д, J=1,6 Гц, 1H), 7,25 (д, J=11,6 Гц, 2H), 6,52 (т, 1H), 5,99 (д, J=2,0 Гц, 1H), 5,89 (м, 1H), 4,11 (м, 1H), 4,02 (с, 4H), 3,79-3,77 (с, 1H), 3,44-3,41 (м, 2H), 3,03-3,00 (м, 4H), 1,90-1,88 (м, 4H). ESI-MS (m/z): 469,12 [M+H]+.
ПРИМЕР 3
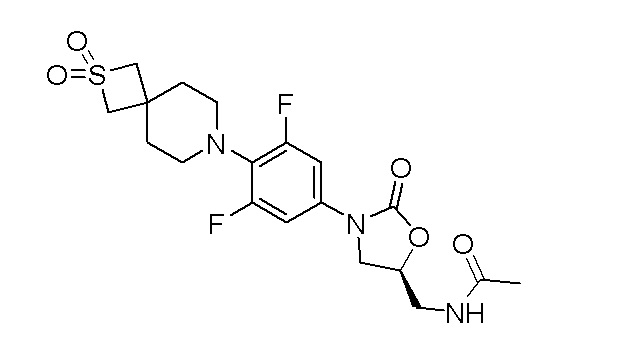
Получение (S)-N-((3-(4-(2,2-диоксидо-2-тиа-7-азаспиро[3.5]нонан-7-ил)-3,5-дифторфенил)-2-оксооксазолидин-5-ил)метил)ацетамида
Стадия 1: (R)-5-(азидометил)-3-(4-(2,2-диоксидо-2-тиа-7-азаспиро[3.5]нонан-7-ил)-3,5-дифторфенил)оксазолидин-2-он
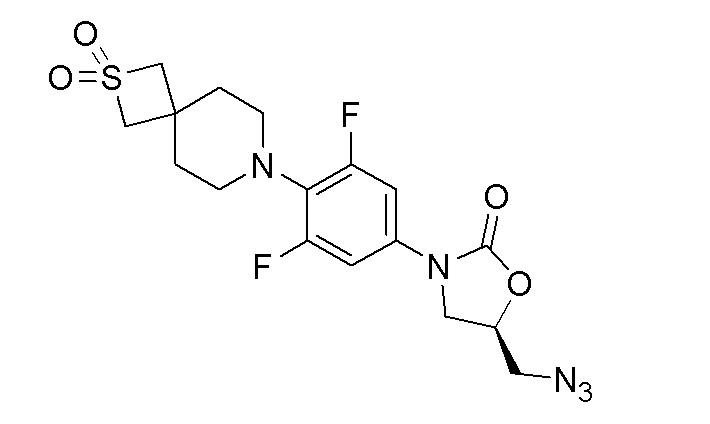
К перемешиваемому раствору (R)-(3-(4-(2,2-диоксидо-2-тиа-7-азаспиро[3.5]нонан-7-ил)-3,5-дифторфенил)-2-оксооксазолидин-5-ил)метилметансульфоната (380 мг, 0,791 ммоль) в DMF добавляли азид натрия (257 мг, 3,95 ммоль) и перемешивали в течение 16 часов при 60-65°C. После завершения реакции смесь разбавляли EtOAc и водой. Органический слой отделяли, сушили над Na2SO4 и концентрировали с получением указанного в заголовке продукта. ESI-MS (m/z): 428,05 [M+H]+.
Стадия 2: (S)-5-(аминометил)-3-(4-(2,2-диоксидо-2-тиа-7-азаспиро[3.5]нонан-7-ил)-3,5-дифторфенил)оксазолидин-2-он
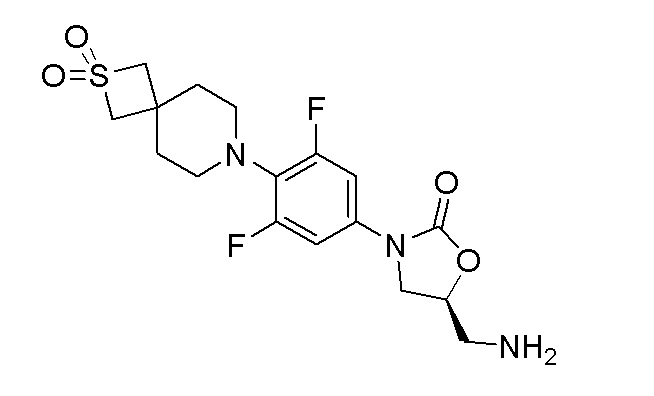
К перемешиваемому раствору продукта стадии 1 (0,330 г, 0,772 ммоль) в MeOH (5 мл) добавляли Pd-C (0,016 г, 0,154 ммоль) при 0°C. Затем порциями добавляли боргидрид натрия (0,088 г, 2,316 ммоль) и перемешивали в течение 3 часов при 25-30°C. После завершения реакции смесь пропускали через слой hyflow и промывали при помощи EtOAc (50 мл). Органический слой сушили над Na2SO4 и концентрировали с получением указанного в заголовке продукта. ESI-MS (m/z): 402,09 [M+H]+.
Стадия 3: (S)-N-((3-(4-(2,2-диоксидо-2-тиа-7-азаспиро[3.5]нонан-7-ил)-3,5-дифторфенил)-2-оксооксазолидин-5-ил)метил)ацетамид
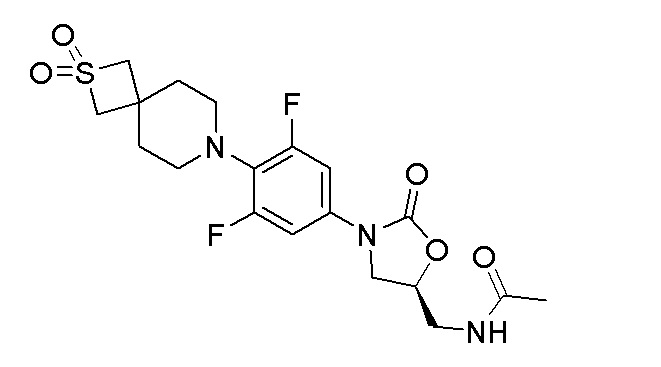
К перемешиваемому раствору продукта стадии 2 (150 мг, 0,374 ммоль) в DCM добавляли пиридин (0,030 мл, 0,374 ммоль) и уксусный ангидрид (0,035 мл, 0,374 ммоль) и перемешивали в течение 3 часов при 25-30°C. После завершения реакции смесь разбавляли водой и продукт экстрагировали при помощи DCM. Органический слой сушили над Na2SO4 и концентрировали с получением неочищенного продукта, который очищали колоночной хроматографией с получением указанного в заголовке продукта. 1H ЯМР (DMSO-d6): 8,24 (т, J=5,8 Гц, 1H), 7,23 (дд, J=5,2 и 16,8 Гц, 2H), 4,74-4,70 (м, 1H), 4,10-4,05 (м, 1H), 4,03 (с, 4H), 3,70-3,68 (м, 1H), 3,41-3,38 (м, 2H), 3,01-3,00 (м, 4H), 1,91-1,87 (м, 4H), 1,83 (с, 4H). ESI-MS (m/z): 444,20 [M+H]+.
ПРИМЕР 4
Получение (S)-метил((3-(4-(2,2-диоксидо-2-тиа-7-азаспиро[3.5]нонан-7-ил)-3,5-дифторфенил)-2-оксооксазолидин-5-ил)метил)карбамата
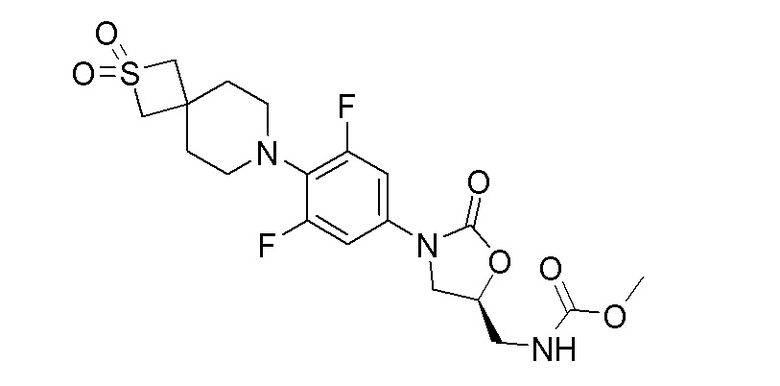
К перемешиваемому раствору (S)-5-(аминометил)-3-(4-(2,2-диоксидо-2-тиа-7-азаспиро[3.5]нонан-7-ил)-3,5-дифторфенил)оксазолидин-2-она (100 мг, 0,249 ммоль) в DCM добавляли пиридин (0,030 мл, 0,374 ммоль), затем метилхлорформиат (0,023 мл, 0,299 ммоль) и перемешивали в течение 3 часов при 25-30°C. После завершения реакции смесь разбавляли водой и продукт экстрагировали при помощи DCM (20 мл). Органический слой сушили над Na2SO4 и концентрировали с получением неочищенного продукта, который очищали колоночной хроматографией с получением указанного в заголовке продукта. 1H ЯМР (DMSO-d6): 7,53 (уш.с, 1H), 7,24 (д, J=12 Гц, 2H), 4,73 (с, 1H), 4,10-4,06 (м, 2H), 4,02 (с, 4H), 3,741-3,61 (м, 2H), 3,40 (с, 3H), 3,19-3,13 (м, 4H), 1,89 (м, 4H). ESI-MS (m/z): 460,17 (M+H)+.
ПРИМЕР 5
Получение (S)-N-((3-(4-(2,2-диоксидо-2-тиа-7-азаспиро[3.5]нонан-7-ил)-3-фторфенил)-2-оксооксазолидин-5-ил)метил)ацетамида
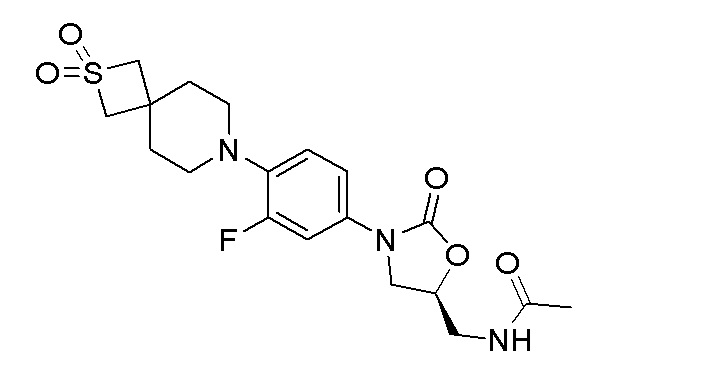
Получали с использованием способа, аналогичного описанному в примере 3. 1H ЯМР (DMSO-d6): 8,24 (т, 1H), 7,49-7,45 (м, 1H), 7,18-7,16 (м, 1H), 7,07-7,03 (м, 1H), 4,72 (м, 1H), 4,10-4,05 (м, 1H), 4,02 (с, 4H), 2,94-2,91 (м, 4H), 1,95-1,92 (м, 4H), 1,24 (с, 3H). ESI-MS (m/z): 426,14 [M+H]+.
ПРИМЕР 6
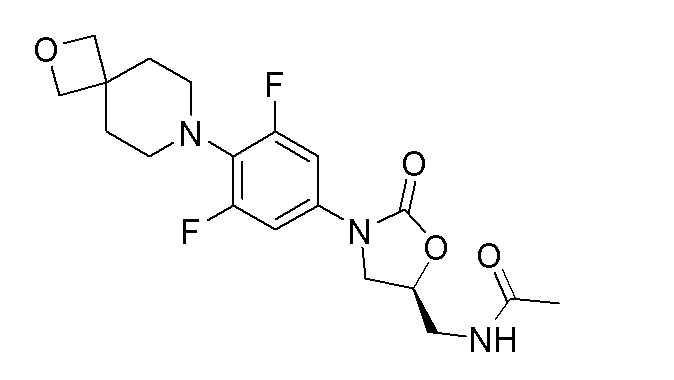
Получение (S)-N-((3-(3,5-дифтор-4-(2-окса-7-азаспиро[3.5]нонан-7-ил)фенил)-2-оксооксазолидин-5-ил)метил)ацетамида
Стадия 1: трет-бутил-2-окса-7-азаспиро[3.5]нонан-7-карбоксилат
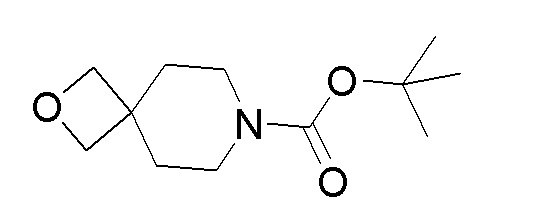
К перемешиваемому раствору трет-бутил-4,4-бис(гидроксиметил)пиперидин-1-карбоксилата (880 мг, 3,59 ммоль) в THF по каплям добавляли н-бутиллитий (1,435 мл, 3,59 ммоль) при 0-5°C. Реакционную смесь перемешивали при 0-5°C в течение 30 мин. К смеси добавляли раствор п-толуолсульфонилхлорида (684 мг, 3,59 ммоль) в THF по каплям при 0-5°C. Реакционную смесь перемешивали при 0-5°C в течение 30 мин. К этому добавляли н-бутиллитий (1,435 мл, 3,59 ммоль) при 0-5°C. Реакционную смесь нагревали при 60°C в течение 2 часов. Реакционную смесь выливали в воду и продукт экстрагировали при помощи EtOAC. Органический слой отгоняли дистилляцией с получением неочищенного продукта, который очищали колоночной хроматографией с использованием смеси гексан/EtOAc с получением чистого продукта.
Стадия 2: 7-(2,6-дифтор-4-нитрофенил)-2-окса-7-азаспиро[3.5]нонан
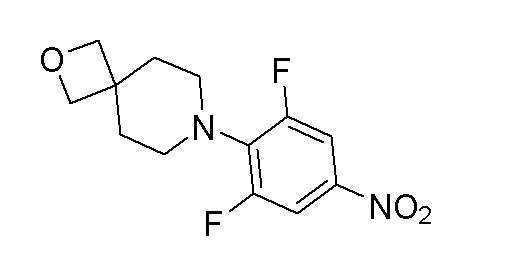
К перемешиваемому раствору продукта стадии 1 (840 мг, 3,70 ммоль) в DCM добавляли TFA (1,4 мл, 18,48 ммоль) при 0-5°C и перемешивали в течение 3 часов при 25-30°C. После завершения реакции DCM выпаривали при пониженном давлении с получением неочищенного продукта, который разбавляли при помощи DMF. К этому добавляли K2CO3 (1,17 г, 8,47 ммоль) и 1,2,3-трифтор-5-нитробензол (600 мг, 3,39ммоль) и перемешивали в течение 4 часов при 80°C. Реакционную смесь разбавляли водой с получением твердого продукта. ESI-MS (m/z): 285,08 [M+H]+.
Стадия 3: 3,5-дифтор-4-(2-окса-7-азаспиро[3.5]нонан-7-ил)анилин
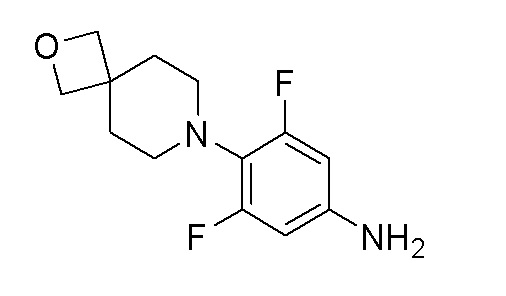
К перемешиваемому раствору продукта стадии 2 (720 мг, 2,53 ммоль) в THF (10 мл) добавляли Pd-C (27 мг) и перемешивали под давлением водорода в течение 16 часов при 25°C. После завершения реакции смесь пропускали через слой hyflow и растворитель выпаривали с получением указанного в заголовке продукта. ESI-MS (m/z): 255,10 [M+H]+.
Стадия 4: бензил(3,5-дифтор-4-(2-окса-7-азаспиро[3.5]нонан-7-ил)фенил)карбамат
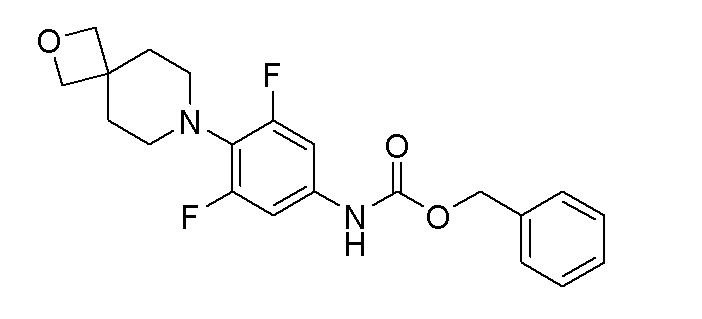
К перемешиваемому раствору продукта стадии 3 (700 мг, 2,75 ммоль) в THF (10 мл) добавляли NaHCO3 (925 мг, 11,01 ммоль) и бензилхлорформиат (0,865 мл, 3,03 ммоль) при 0°C. Реакционную смесь перемешивали при 25-30°C в течение 16 часов. После завершения реакции смесь разбавляли EtOAc и водой. Органический слой выпаривали при пониженном давлении с получением неочищенного продукта. Неочищенный продукт очищали колоночной хроматографией с использованием смеси гексан/EtOAc с получением указанного в заголовке продукта. ESI-MS (m/z): 389,16 [M+H]+.
Стадия 5: (R)-3-(3,5-дифтор-4-(2-окса-7-азаспиро[3.5]нонан-7-ил)фенил)-5-(гидроксиметил)оксазолидин-2-он
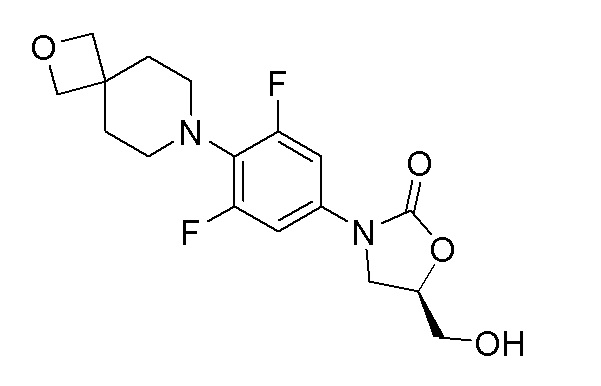
К перемешиваемому раствору продукта стадии 4 (540 мг, 1,390 ммоль) в безводном THF добавляли н-бутиллитий (0,612 мл, 1,529 ммоль) при -78°C. Полученный светло-желтый раствор перемешивали при -78°C в течение 1 часа и затем добавляли (R)-глицидилбутират (220 мг, 1,529 ммоль) при -70°C по каплям. Реакционную смесь перемешивали еще 1 час при -78°C. Реакционной смеси давали нагреться до комнатной температуры и перемешивали в течение 16 часов. После завершения реакции смесь выливали в водный раствор хлорида аммония, и неочищенный продукт экстрагировали при помощи EtOAc, затем очищали колоночной хроматографией с получением указанного в заголовке продукта. ESI-MS (m/z): 355,14 [M+H]+.
Стадия 6: (R)-(3-(3,5-дифтор-4-(2-окса-7-азаспиро[3.5]нонан-7-ил)фенил)-2-оксооксазолидин-5-ил)метилметансульфонат
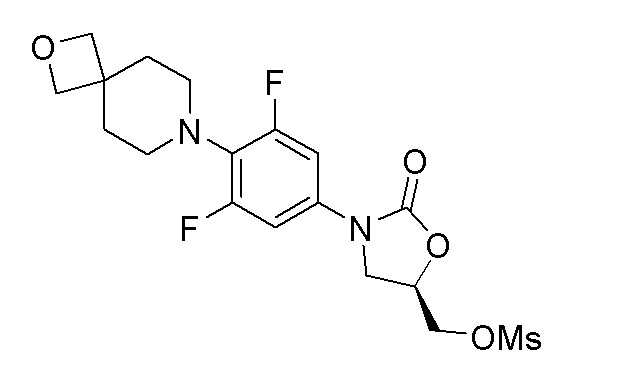
К перемешиваемому раствору продукта стадии 5 (316 мг, 0,892 ммоль) в DCM добавляли TEA (0,162 мл, 1,159 ммоль) и метансульфонилхлорид (0,07 мл, 0,936 ммоль) при 0°C. Реакционную смесь перемешивали в течение 2 часов при 25-30°C. После завершения реакции смесь разбавляли DCM (10 мл) и промывали водой. DCM слой сушили над Na2SO4 и отгоняли дистилляцией с получением указанного в заголовке продукта. ESI-MS (m/z): 433,12 [M+H]+.
Стадия 7: (R)-5-(азидометил)-3-(3,5-дифтор-4-(2-окса-7-азаспиро[3.5]нонан-7-ил)фенил)оксазолидин-2-он
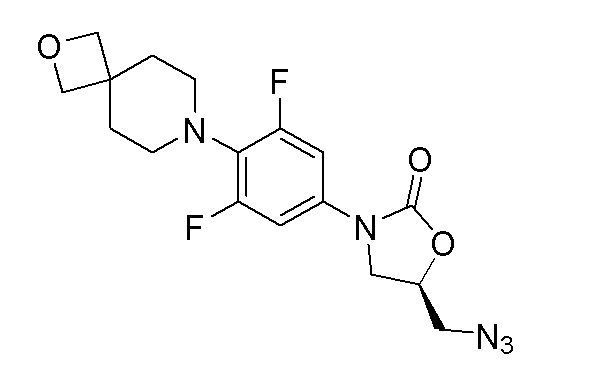
К перемешиваемому раствору продукта стадии 6 (397 мг, 0,918 ммоль) в DMF добавляли азид натрия (298 мг, 4,59 ммоль) и перемешивали в течение 3 часов при 60-65°C. После завершения реакции смесь разбавляли водой и фильтровали с получением указанного в заголовке продукта. ESI-MS (m/z): 380,14 [M+H]+.
Стадия 8: (S)-5-(аминометил)-3-(3,5-дифтор-4-(2-окса-7-азаспиро[3.5]нонан-7-ил)фенил)оксазолидин-2-он
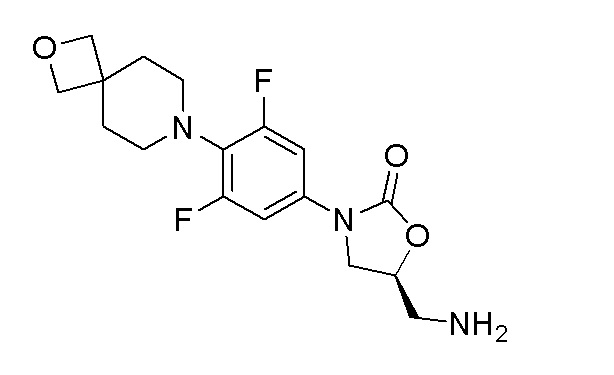
К перемешиваемому раствору продукта стадии 7 (242 мг, 0,638 ммоль) в MeOH добавляли Pd-C при 0-5°C. Затем порциями добавляли боргидрид натрия (72,4 мг, 1,914 ммоль) и перемешивали в течение 3 часов при 25-30°C. После завершения реакции смесь пропускали через слой hyflow и промывали при помощи EtOAc (20 мл). Органический слой сушили над Na2SO4 и концентрировали с получением указанного в заголовке продукта. ESI-MS (m/z): 354,15 [M+H]+.
Стадия 9: (S)-N-((3-(3,5-дифтор-4-(2-окса-7-азаспиро[3.5]нонан-7-ил)фенил)-2-оксооксазолидин-5-ил)метил)ацетамид
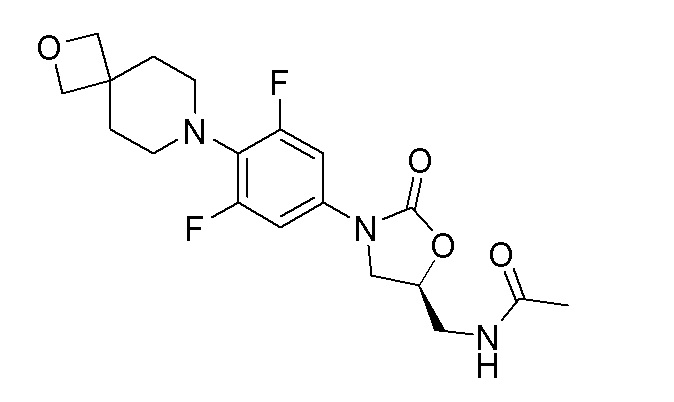
К перемешиваемому раствору продукта стадии 8 (100 мг, 0,283 ммоль) в DCM добавляли пиридин (0,034 мл, 0,424 ммоль) и уксусный ангидрид (0,037 мл, 0,396 ммоль). Реакционную смесь перемешивали в течение 1 часа при 25-30°C. После завершения реакции смесь разбавляли DCM и промывали водой. Органический слой сушили над Na2SO4 и концентрировали с получением указанного в заголовке продукта. 1H ЯМР (DMSO-d6): 8,22 (т, J=5,8 Гц, 1H), 7,23 (д, J=11,6 Гц, 2H), 4,73-4,70 (м, 1H), 4,34 (с, 4H), 4,09-4,05 (м, 1H), 3,70-3,66 (м, 1H), 3,40-3,37 (м, 2H), 2,96-2,93 (м, 4H), 1,99-1,86 (м, 4H), 1,83 (с, 3H). ESI-MS (m/z): 396,16 (M+H)+.
ПРИМЕР 7
Получение (S)-N-((3-(3-фтор-4-(2-окса-7-азаспиро[3.5]нонан-7-ил)фенил)-2-оксооксазолидин-5-ил)метил)ацетамида
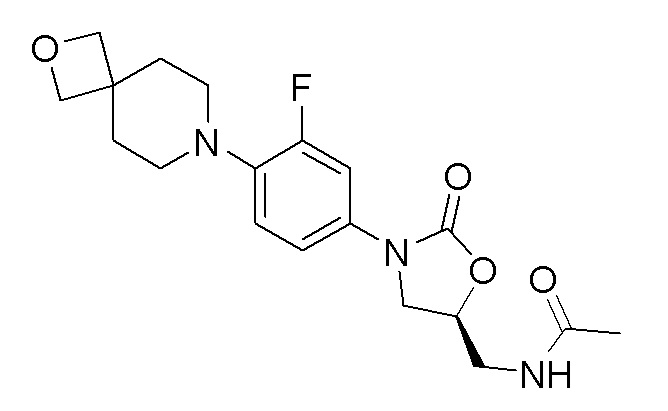
Получали в соответствии с процедурой, аналогичной описанной в примере 6, с использованием соответствующих модификаций. 1H ЯМР (CDCl3): 7,41 (дд, J=2,4 и 14,0 Гц, 1H), 7,08-7,06 (м, 1H), 6,95-6,90 (м, 1H), 5,99 (м, 1H), 4,78-4,77 (м, 1H), 4,50 (с, 4H), 4,05-4,01 (м, 1H), 3,77-3,75 (м, 2H), 3,73-3,70 (м, 1H), 2,96-2,93 (м, 4H), 2,07-2,04 (м, 7H). ESI-MS (m/z): 378,15 (M+H)+
ПРИМЕР 8
Получение (S)-N-((3-(3,5-дифтор-4-(4-(оксетан-3-ил)пиперидин-1-ил)фенил)-2-оксооксазолидин-5-ил)метил)ацетамида
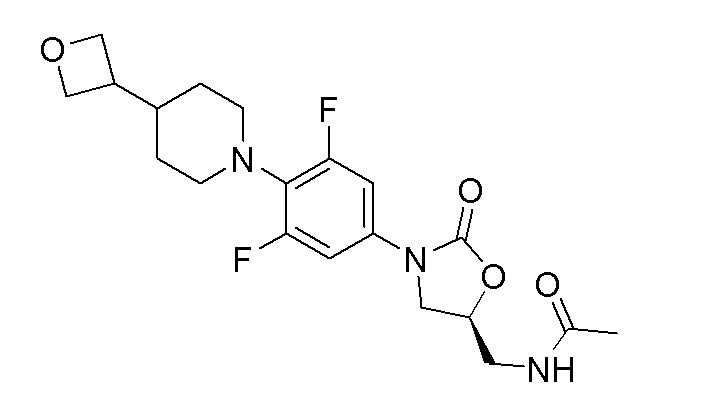
Стадия 1: (R)-2-(3-((3,5-дифтор-4-(4-(оксетан-3-ил)пиперидин-1-ил)фенил)амино)-2-гидроксипропил)изоиндолин-1,3-дион
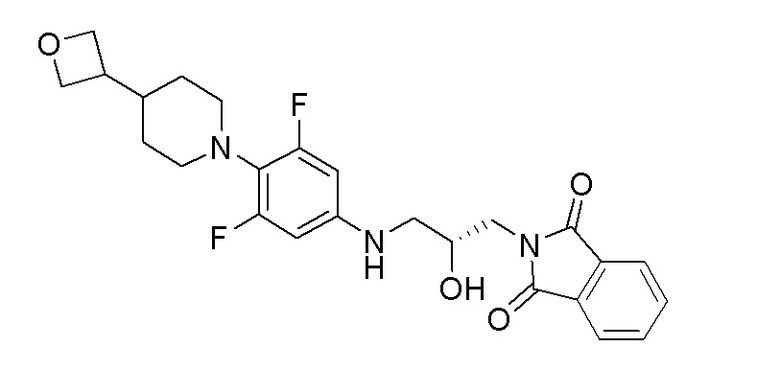
Перемешиваемый раствор (S)-2-(оксиран-2-илметил)изоиндолин-1,3-диона (1,988 г, 9,78 ммоль) и 3,5-дифтор-4-(4-(оксетан-3-ил)пиперидин-1-ил)анилина (1,50 г, 5,59 ммоль) в водн. EtOH нагревали при 100°C в течение 4 часов. При 80°C добавляли еще одну порцию (S)-2-(оксиран-2-илметил)изоиндолин-1,3-диона (1,988 г, 9,78 ммоль) и перемешивание продолжали в течение 16 часов. Реакционную смесь фильтровали с получением указанного в заголовке продукта. ESI-MS (m/z): 472,16 [M+H]+.
Стадия 2: (S)-2-((3-(3,5-дифтор-4-(4-(оксетан-3-ил)пиперидин-1-ил)фенил)-2-оксооксазолидин-5-ил)метил)изоиндолин-1,3-дион
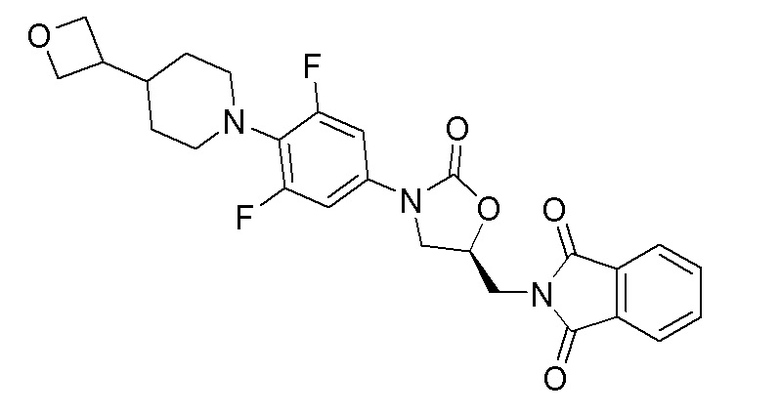
К перемешиваемому раствору продукта стадии 1 (2,0 г, 4,24 ммоль) в CHCl3 добавляли CDI (2,75 г, 16,97 ммоль) при 25-30°C. Реакционную смесь перемешивали при 70°C в течение 5 часов. Растворитель отгоняли дистилляцией и реакционную смесь подкисляли 35% раствором хлористоводородной кислоты до pH=2. Продукт экстрагировали при помощи DCM. Органический слой отделяли, сушили над Na2SO4 и отгоняли дистилляцией с получением указанного в заголовке продукта. ESI-MS (m/z): 498,14 [M+H]+.
Стадия 3: (S)-5-(аминометил)-3-(3,5-дифтор-4-(4-(оксетан-3-ил)пиперидин-1-ил)фенил)оксазолидин-2-он
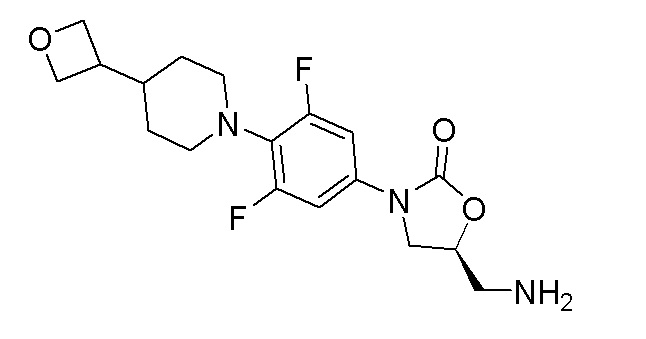
К перемешиваемому раствору продукта стадии 2 (1,90 г, 3,82 ммоль) в EtOH добавляли водный раствор метиламина (2,97 г, 38,2 ммоль) при 25-30°C. Реакционную смесь перемешивали при 80°C в течение 3 часов. После полной конверсии исходного вещества реакционную смесь разбавляли DCM (100 мл) и промывали холодной водой. Органический слой сушили над Na2SO4 и отгоняли дистилляцией с получением указанного в заголовке продукта в виде твердого вещества. ESI-MS (m/z): 368,15 [M+H]+.
Стадия 4: (S)-N-((3-(3,5-дифтор-4-(4-(оксетан-3-ил)пиперидин-1-ил)фенил)-2-оксооксазолидин-5-ил)метил)ацетамид
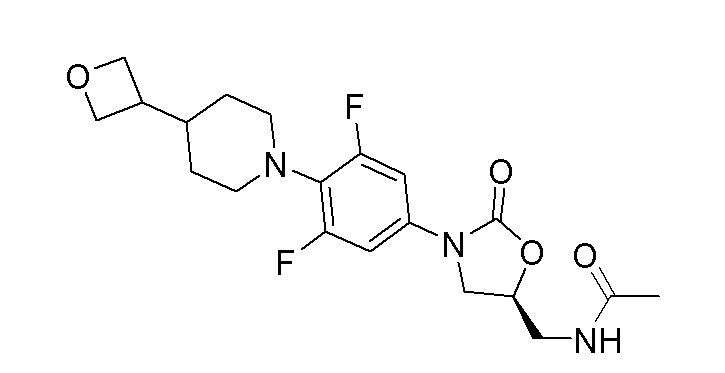
К перемешиваемому раствору продукта стадии 3 (1,00 г, 2,72 ммоль) в DCM добавляли пиридин (0,330 мл, 4,08 ммоль) и уксусный ангидрид (0,360 мл, 3,81 ммоль) и перемешивали в течение 2 часов при 25-30°C. После полной конверсии исходного вещества реакционную смесь разбавляли водой и экстрагировали при помощи DCM. Органический слой отделяли, сушили над Na2SO4 и отгоняли дистилляцией с получением неочищенного продукта, который очищали препаративной ВЭЖХ с получением указанного в заголовке продукта в виде белого твердого вещества. 1H ЯМР (DMSO): 8,23 (т, 1H), 7,28-7,21 (м, 2H), 4,75-4,69 (м, 1H), 4,63-4,60 (м, 2H), 4,38-4,35 (м, 2H), 4,10-4,05 (м, 1H), 3,70-3,66 (м, 1H), 3,43-3,38 (м, 1H), 3,11-2,97 (м, 4H), 2,78-2,72 (м, 1H), 1,83 (с, 3H), 1,76-1,71 (м, 1H), 1,68-1,60 (м, 2H), 1,19-1,06 (м, 2H). ESI-MS (m/z): 410,16 [M+H]+.
ПРИМЕР 9
Получение (S)-N-((3-(4-(4-(1,1-диоксидотиетан-3-ил)пиперидин-1-ил)-3,5-дифторфенил)-2-оксооксазолидин-5-ил)метил)ацетамида
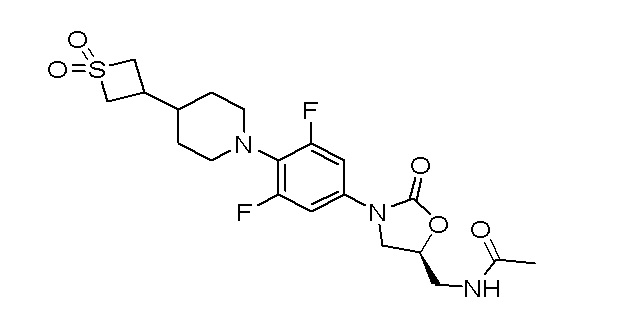
Стадия 1: трет-бутил-4-оксопиперидин-1-карбоксилат
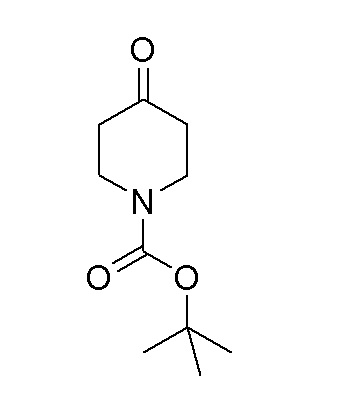
К перемешиваемой суспензии гидрохлорида пиперидин-4-она (30 г, 221 ммоль) в безводном DCM добавляли TEA (83 мл, 597 ммоль), затем добавляли по каплям ди-трет-бутилдикарбонат (64,2 мл, 277 ммоль) в течение 15 минут при 10-15°C. Полученную смесь перемешивали при 25-30°C в течение 16 часов. Реакционную смесь разбавляли DCM и промывали водой. Органический слой сушили над Na2SO4 и упаривали при пониженном давлении с получением указанного в заголовке продукта.
Стадия 2: диэтил-2-(1-(трет-бутоксикарбонил)пиперидин-4-илиден)малонат
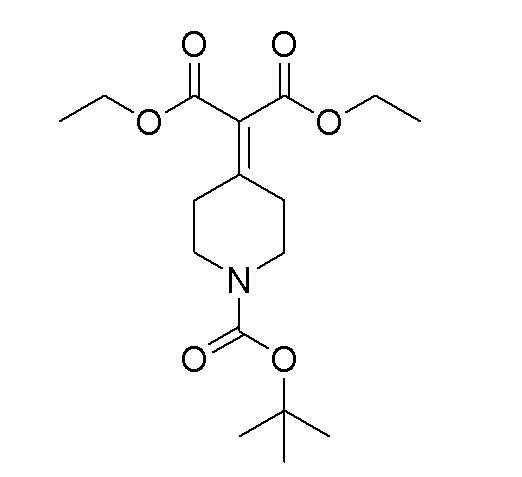
Перемешиваемый раствор TiCl4 (52,0 мл, 472 ммоль) в тетрахлориде углерода (40 мл) добавляли к THF (150 мл) при 0°C. Затем добавляли раствор продукта стадии 1 (37,3 г, 187 ммоль) и диэтилмалонат (25,7 мл, 168 ммоль) в THF (150 мл) при 0°C и перемешивали в течение 15 мин. К этому добавляли пиридин (98 мл, 1217 ммоль) и перемешивали в течение 16 часов при 25-30°C. Реакционную смесь выливали в 10% раствор лимонной кислоты и продукт экстрагировали при помощи EtOAc. Органический слой отделяли, сушили над Na2SO4 и упаривали при пониженном давлении с получением указанного в заголовке продукта.
Стадия 3: диэтил-2-(1-(трет-бутоксикарбонил)пиперидин-4-ил)малонат
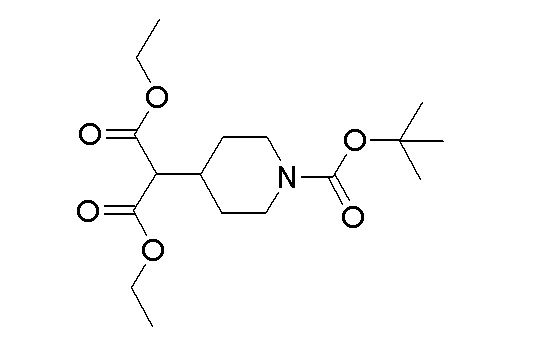
К перемешиваемому раствору продукта стадии 2 (63 г, 185 ммоль) в EtOH добавляли боргидрид натрия (6,98 г, 185 ммоль) и перемешивали в течение 2 часов при 25-30°C. После полной конверсии исходного вещества реакционную смесь выливали в ледяную воду и экстрагировали при помощи DCM. Органический слой отделяли, сушили над Na2SO4 и упаривали при пониженном давлении с получением указанного в заголовке продукта.
Стадия 4: трет-бутил-4-(1,3-дигидроксипропан-2-ил)пиперидин-1-карбоксилат
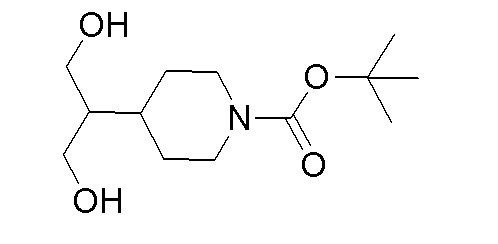
К перемешиваемому раствору продукта стадии 3 (63 г, 183 ммоль) в EtOH (315 мл) добавляли боргидрид натрия (69,4 г, 1834 ммоль) при 25-30°C и полученную смесь перемешивали при 70°C в течение 3 часов. После полной конверсии исходного вещества реакционную смесь разбавляли холодной водой и экстрагировали при помощи EtOAc. Органический слой отделяли, сушили над Na2SO4 и упаривали при пониженном давлении с получением указанного в заголовке продукта.
Стадия 5: трет-бутил-4-(1,3-бис(тозилокси)пропан-2-ил)пиперидин-1-карбоксилат
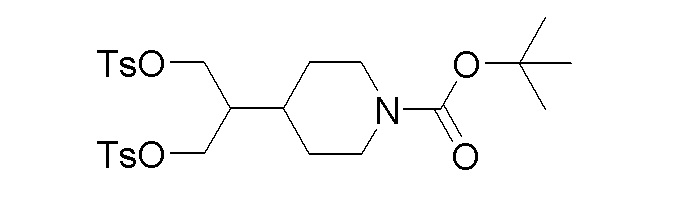
К перемешиваемому раствору продукта стадии 4 (10 г, 38,6 ммоль) в DCM добавляли TEA (21,50 мл, 154 ммоль) и DMAP (9,42 г, 77 ммоль). Затем добавляли порциями п-толуолсульфонилхлорид (22,05 г, 116 ммоль) при 0-5°C. Реакционную смесь перемешивали при 25-30°C в течение 3 часов. После завершения реакции реакционную смесь разбавляли DCM и промывали водой и насыщенным солевым раствором, сушили над Na2SO4 и упаривали при пониженном давлении с получением указанного в заголовке продукта.
Стадия 6: трет-бутил-4-(тиетан-3-ил)пиперидин-1-карбоксилат
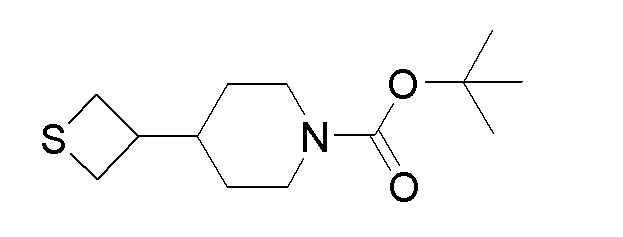
К перемешиваемому раствору продукта стадии 5 (12,1 г, 21,31 ммоль) в DMF добавляли нонагидрат сульфида натрия (6,14 г, 25,6 ммоль) и реакционную смесь нагревали при 80-85°C в течение 16 часов. После завершения реакции смесь разбавляли EtOAc и водой. Органический слой отделяли, сушили над Na2SO4 и концентрировали с получением маслянистого продукта. Неочищенный продукт очищали колоночной хроматографией с использованием смеси гексан/EtOAc с получением указанного в заголовке продукта.
Стадия 7: трет-бутил-4-(1,1-диоксидотиетан-3-ил)пиперидин-1-карбоксилат
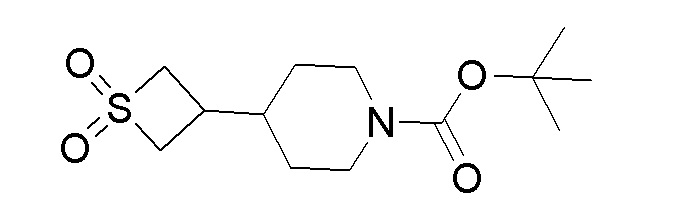
К перемешиваемому раствору продукта стадии 6 (5,5 г, 21,371 ммоль) в DCM добавляли мета-хлорпербензойную кислоту (18,44 г, 107 ммоль) и перемешивали в течение 4 часов при 25-30°C. После завершения реакции смесь разбавляли DCM и промывали водным раствором NaHCO3. DCM слой отделяли, сушили над Na2SO4 и концентрировали с получением указанного в заголовке продукта. 1H ЯМР (CDCl3): 4,19-3,99 (м, 4H), 3,88-3,85 (м, 2H), 2,70-2,60 (м, 2H), 2,45-2,41 (м, 1H), 2,35-2,22 (м, 1H), 1,87-1,86 (м, 2H), 1,55 (с, 9H), 1,21-1,1 (м, 2H).
Стадия 8: 3-(1-(2,6-дифтор-4-нитрофенил)пиперидин-4-ил)тиетан-1,1-диоксид
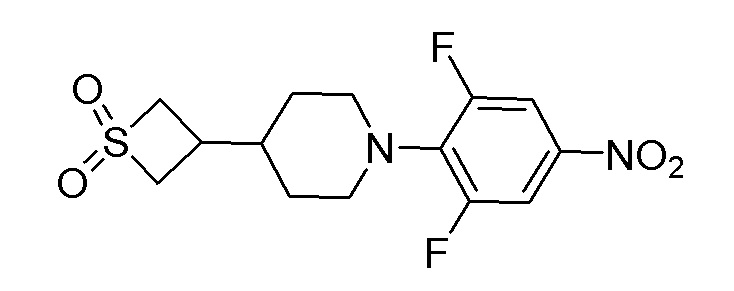
К перемешиваемому раствору продукта стадии 7 (5,5 г, 19,01 ммоль) в DCM добавляли TFA (2,93 мл, 38,0 ммоль) при 0°C и перемешивали в течение 3 часов 25-30°C. После завершения реакции DCM выпаривали с получением неочищенного продукта, который разбавляли при помощи DMF. К этому добавляли K2CO3 (5,39 г, 39,0 ммоль) и 1,2,3-трифтор-5-нитробензол (2,3 г, 12,99 ммоль) и перемешивали в течение 4 часов при 80°C. Реакционную смесь разбавляли водой с получением продукта в виде твердого вещества. ESI-MS (m/z): 347,07 [M+H]+.
Стадия 9: 3-(1-(4-амино-2,6-дифторфенил)пиперидин-4-ил)тиетан-1,1-диоксид

К перемешиваемому раствору продукта стадии 8 (2,3 г, 6,64 ммоль) в THF добавляли Pd-C (71 мг) и перемешивали под давлением водорода в течение 3 часов при 25-30°C. После завершения реакции смесь пропускали через слой hyflow и растворитель выпаривали с получением указанного в заголовке продукта. ESI-MS (m/z): 317,10 [M+H]+.
Стадия 10: бензил(4-(4-(1,1-диоксидотиетан-3-ил)пиперидин-1-ил)-3,5-дифторфенил)карбамат
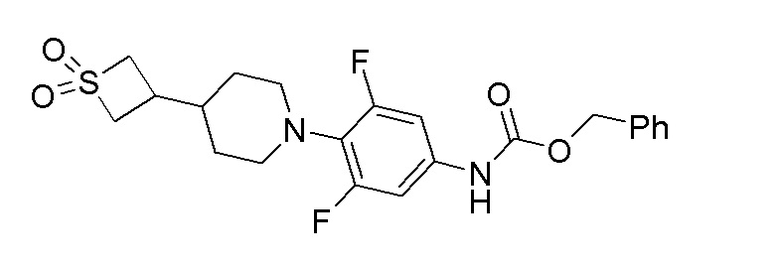
К перемешиваемому раствору продукта стадии 9 (2,14 г, 6,76 ммоль) в THF добавляли NaHCO3 (2,27 г, 27,1 ммоль) и бензилхлорформиат (2,124 мл, 7,44 ммоль) при 0-5°C. Реакционную смесь перемешивали при 25-30°C в течение 7 часов. После завершения реакции смесь разбавляли EtOAc и водой. Органический слой отделяли и упаривали при пониженном давлении с получением неочищенного продукта. Неочищенный продукт очищали колоночной хроматографией с использованием смеси гексан/EtOAc с получением указанного в заголовке продукта. ESI-MS (m/z): 451,10 [M+H]+.
Стадия 11: (R)-3-(4-(4-(1,1-диоксидотиетан-3-ил)пиперидин-1-ил)-3,5-дифторфенил)-5-(гидроксиметил)оксазолидин-2-он
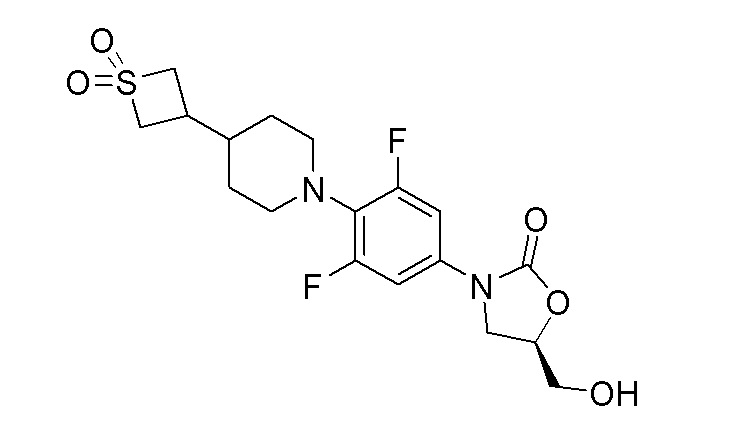
К перемешиваемому раствору продукта стадии 10 (2,47 г, 5,48 ммоль) в безводном THF добавляли н-бутиллитий (2,41 мл, 6,03 ммоль) при -78°C. Полученный светло-желтый раствор перемешивали при -78°C в течение 1 часа и затем добавляли по каплям (R)-глицидилбутират (0,869 г, 6,03 ммоль) при -70°C. Реакционную смесь перемешивали еще 1 час при -78°C. Реакционной смеси давали нагреться до комнатной температуры и перемешивали в течение 16 часов. После завершения реакции смесь выливали в водный раствор хлорида аммония и неочищенный продукт экстрагировали при помощи EtOAc, затем очищали колоночной хроматографией с получением указанного в заголовке продукта. ESI-MS (m/z): 439,10 [M+Na]+.
Стадия 12: (R)-(3-(4-(4-(1,1-диоксидотиетан-3-ил)пиперидин-1-ил)-3,5-дифторфенил)-2-оксооксазолидин-5-ил)метилметансульфонат
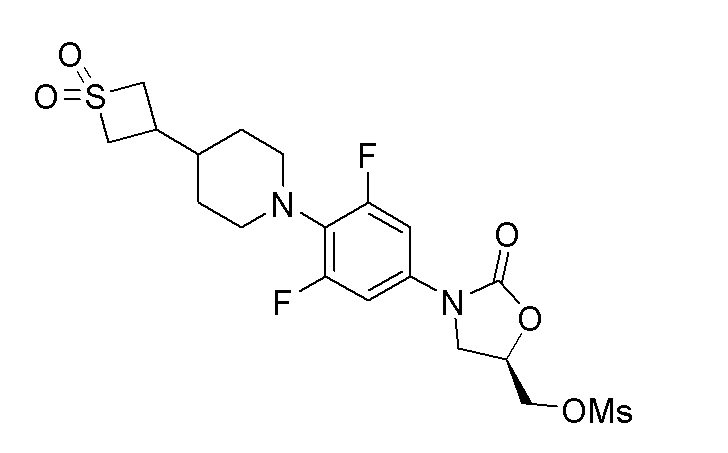
К перемешиваемому раствору продукта стадии 11 (590 мг, 1,417 ммоль) в DCM добавляли TEA (0,257 мл, 1,842 ммоль) и метансульфонилхлорид (0,116 мл, 1,488 ммоль) при 0-5°C. Реакционную смесь перемешивали в течение 2 часов при 25-30°C. После завершения реакции смесь разбавляли DCM и промывали водой. DCM слой сушили над Na2SO4 и отгоняли дистилляцией с получением указанного в заголовке продукта. ESI-MS (m/z): 495,10 [M+H]+.
Стадия 13: (R)-5-(азидометил)-3-(4-(4-(1,1-диоксидотиетан-3-ил)пиперидин-1-ил)-3,5-дифторфенил)оксазолидин-2-он
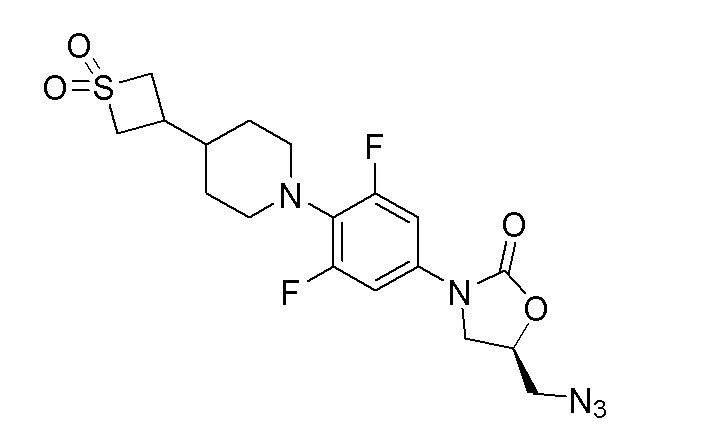
К перемешиваемому раствору продукта стадии 12 (650 мг, 1,314 ммоль) в DMF добавляли азид натрия (427 мг, 6,57 ммоль) и перемешивали в течение 3 часов при 60-65°C. После завершения реакции смесь разбавляли водой и фильтровали с получением указанного в заголовке продукта. ESI-MS (m/z): 442,17 [M+H]+.
Стадия 14: (S)-5-(аминометил)-3-(4-(4-(1,1-диоксидотиетан-3-ил)пиперидин-1-ил)-3,5-дифторфенил)оксазолидин-2-он
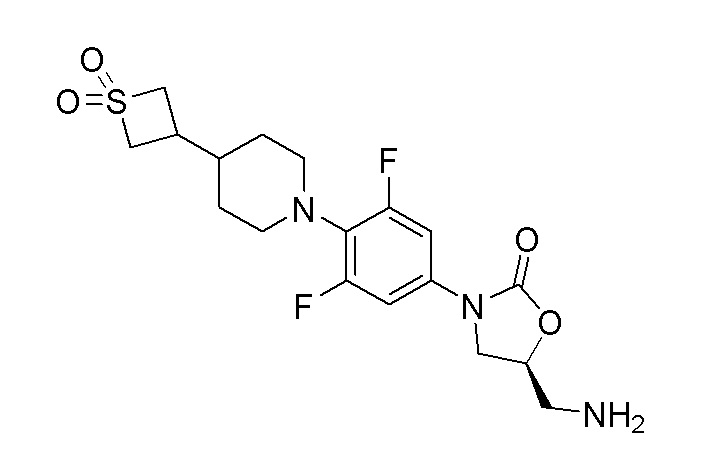
К перемешиваемому раствору продукта стадии 13 (500 мг, 1,133 ммоль) в MeOH добавляли Pd-C (12 мг) при 0-5°C. Затем порциями добавляли боргидрид натрия (129 мг, 3,40 ммоль) и перемешивали в течение 3 часов при 25-30°C. После завершения реакции смесь пропускали через слой hyflow и промывали при помощи EtOAc (50 мл). Органический слой сушили над Na2SO4 и концентрировали с получением указанного в заголовке продукта. ESI-MS (m/z): 416,15 [M+H]+.
Стадия 15: (S)-N-((3-(4-(4-(1,1-диоксидотиетан-3-ил)пиперидин-1-ил)-3,5-дифторфенил)-2-оксооксазолидин-5-ил)метил)ацетамид
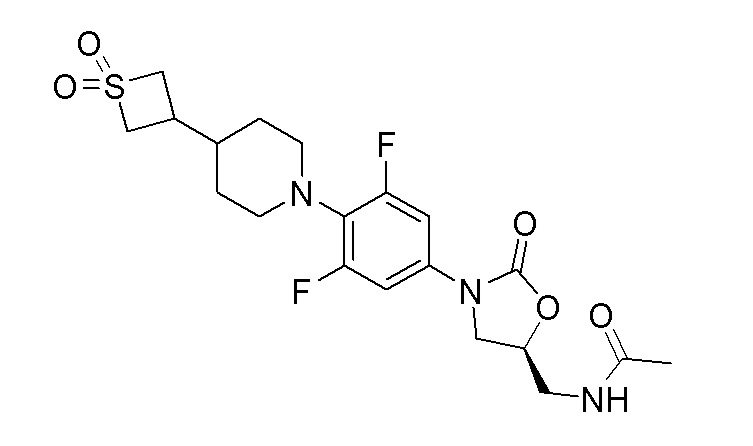
К перемешиваемому раствору продукта стадии 14 (200 мг, 0,481 ммоль) в DCM (5 мл) добавляли пиридин (0,058 мл, 0,722 ммоль) и уксусный ангидрид (0,064 мл, 0,674 ммоль). Реакционную смесь перемешивали в течение 1 часа при 25-30°C. После завершения реакции смесь разбавляли DCM и промывали водой. Органический слой сушили над Na2SO4 и концентрировали с получением указанного в заголовке продукта. 1H ЯМР (DMSO-d6): 8,23 (т, J=5,8 Гц, 1H), 7,28-7,23 (м, 2H), 4,74-4,70 (м, 1H), 4,19-4,15 (м, 2H), 4,13-1,05 (м, 1H), 4,01-3,96 (м, 2H), 3,70-3,66 (м, 1H), 3,41-3,39 (м, 2H), 3,12-3,10 (м, 2H), 2,99-2,94 (м, 2H), 2,30-2,27 (м, 1H), 1,83 (с, 3H), 1,71-1,68 (м, 2H), 1,57-1,54 (м, 1H), 1,27-1,21 (м, 2H). ESI-MS (m/z): 458,18 (M+H)+
ПРИМЕР 10
Получение (S)-N-((3-(4-(4-(1,1-диоксидотиетан-3-ил)пиперидин-1-ил)-3-фторфенил)-2-оксооксазолидин-5-ил)метил)ацетамида
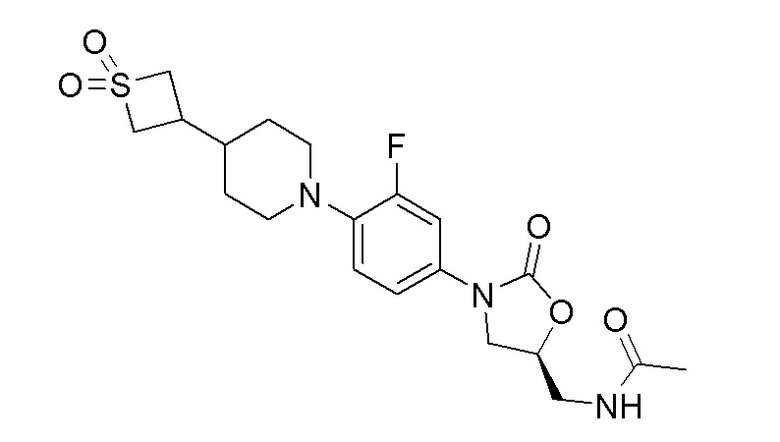
Получали в соответствии с процедурой, аналогичной описанной в примере 9, с использованием соответствующих модификаций. 1H ЯМР (DMSO-d6): 8,24 (т, 1H), 7,44 (дд, J=2,4 и 14,8 Гц, 1H), 7,16-7,14 (м, 1H), 7,09-7,06 (м, 1H), 4,72 (м, 1H), 4,20-4,10 (м, 2H), 4,07-4,02 (м, 1H), 4,01-3,97 (м, 2H), 3,71-3,67 (м, 1H), 3,40 (т, 2H), 3,29 (уш.с, 2H), 2,62-2,59 (м, 2H), 2,35-2,28 (м, 1H), 1,83 (с, 3H), 1,70-1,82 (м, 2H), 1,51-1,62 (м, 1H), 1,23-1,34 (м, 2H). ESI-MS (m/z): 440,12 (M+H)+.
ПРИМЕР 11
Получение метил-(S)-((3-(4-(4-(1,1-диоксидотиетан-3-ил)пиперидин-1-ил)-3,5-дифторфенил)-2-оксооксазолидин-5-ил)метил)карбамата
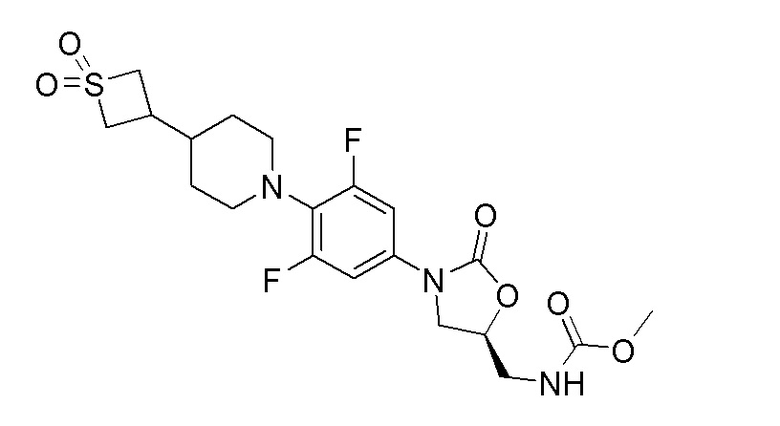
К перемешиваемому раствору (S)-5-(аминометил)-3-(4-(4-(1,1-диоксидотиетан-3-ил)пиперидин-1-ил)-3,5-дифторфенил)оксазолидин-2-она (100 мг, 0,241 ммоль) в DCM добавляли пиридин (0,029 мл, 0,361 ммоль) и метилхлорформиат (0,026 мл, 0,337 ммоль). Реакционную смесь перемешивали в течение 1 часа при 25-30°C. После завершения реакции смесь разбавляли при помощи DCM. Органический слой отделяли, сушили над Na2SO4 и концентрировали с получением указанного в заголовке продукта. 1H ЯМР (CDCl3): 7,53 (т, 1H), 7,50 (д, J=5,6 Гц, 2H), 4,73-4,70 (м, 1H), 4,19-4,15 (м, 2H), 4,13-4,10 (м, 1H), 4,08-3,96 (м, 2H), 3,74-3,70 (м, 1H), 3,54 (с, 3H), 3,12-3,00 (м, 2H), 2,97-2,94 (м, 2H), 2,30-2,26 (м, 1H), 1,71-1,68 (м, 2H), 1,57-1,55 (м, 1H), 1,24-1,21 (м, 4H). ESI-MS (m/z): 474,151 (M+H)+.
С использованием подходящих исходных веществ и подходящих модификаций способа, описанного в приведенных выше примерах, включая подходящее добавление и/или исключение стадий, при необходимости, что находится в пределах компетенции специалиста в данной области, следующие соединения были получены аналогичным способом.
Биологическая оценка:
Определение минимальной ингибирующей концентрации (MIC)
Для оценки ингибирующего эффекта соединений по изобретению на рост бактерий осуществляли MABA анализ с использованием культуры Mycobacterium tuberculosis H37Rv в фазе логарифмического роста. Бактериальную культуру с оптической плотностью 0,6-0,8 разводили до конечной плотности 0,02 в среде 7Н9. 100 мкл разведенной Mycobacterium tuberculosis H37Rv инкубировали с испытываемыми соединениями в микротитровальном планшете. Также подготавливали не содержащий лекарственного средства контроль, содержащий DMSO. После инкубации соединения с культурой в течение 7 дней при 37°C в каждую лунку добавляли 20 мкл 0,02% раствора аламарового синего (свежеприготовленного). Цвету давали проявиться в течение 16 часов при 37°C. Синий цвет в лунке указывал на отсутствие роста, тогда как розовый цвет означал рост в лунке. Значение MIC определяли как самую низкую концентрацию, которая предотвращает изменение цвета с синего на розовый. Флуоресценцию измеряли при возбуждении при 530 нм и эмиссии при 590 нм. Значения MIC выбранных соединений приведены в Таблице 2.
Новые соединения по настоящему изобретению могут быть сформулированы в подходящие фармацевтически приемлемые композиции путем объединения с подходящими эксципиентами с использованием хорошо известных процедур, способов и концентраций. Фармацевтическая композиция, содержащая соединения по настоящему изобретению, может содержать подходящее связующее, подходящий наполнитель и/или разбавитель и любые другие подходящие агенты, которые могут быть необходимы.
Новые соединения по настоящему изобретению можно использовать для лечения инфекций, таких как туберкулез, у млекопитающих путем введения терапевтически активного и нетоксичного количества соединений формулы (I) или их фармацевтически приемлемых композиций.
Способ лечения от туберкулезной инфекции у субъекта, включающий введение пациенту, нуждающемуся в этом, эффективного количества соединения формулы (I) или его подходящей фармацевтической композиции.
Фармацевтические композиции по настоящему изобретению могут существовать в различных формах. В некоторых вариантах осуществления фармацевтическая композиция предназначена для местного, перорального или парентерального введения.
Одно или более дополнительных фармацевтических средств или способов лечения можно использовать в комбинации с соединениями по настоящему изобретению для лечения или профилактики инфекции млекопитающих. Терапевтические средства можно комбинировать с соединениями по настоящему изобретению в одной лекарственной форме, или средства можно вводить одновременно или последовательно в раздельных лекарственных формах. Соединение формулы (I) можно вводить в комбинации с другими терапевтическими средствами, такими как изониазид, рифампин, рифапентин, рифабутин, этамбутол, пиразинамид, стрептомицин, амикацин, левофлоксацин, офлоксацин, пара-аминосалициловая кислота.
Хотя настоящее изобретение описано при помощи его конкретных вариантов осуществления, определенные модификации и эквиваленты будут очевидны специалистам в данной области техники и предназначены для включения в объем настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ОКСАЗОЛИДИНОНОВЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ В КАЧЕСТВЕ ПРОТИВОБАКТЕРИАЛЬНЫХ СРЕДСТВ | 2016 |
|
RU2794494C2 |
| ПРОИЗВОДНЫЕ ЦИКЛИЧЕСКОГО ЦИАНОЕНОНА КАК МОДУЛЯТОРЫ KEAP1 | 2021 |
|
RU2822828C1 |
| ИНГИБИТОРЫ ВЗАИМОДЕЙСТВИЯ МЕНИН-MLL | 2017 |
|
RU2799820C2 |
| 1,1,1-ТРИФТОР-3-ГИДРОКСИПРОПАН-2-ИЛКАРБАМАТНЫЕ ПРОИЗВОДНЫЕ КАК ИНГИБИТОРЫ MAGL | 2018 |
|
RU2720203C1 |
| ИНГИБИТОРЫ ИОННОГО КАНАЛА, ФАРМАЦЕВТИЧЕСКИЕ СОСТАВЫ И ПРИМЕНЕНИЕ | 2016 |
|
RU2746188C2 |
| ПРОИЗВОДНЫЕ ТЕТРАГИДРОИМИДАЗОПИРИДИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2696270C1 |
| СПИРОЦИКЛИЧЕСКИЕ НИТРИЛЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕАЗЫ | 2012 |
|
RU2621695C2 |
| СПИРОЦИКЛИЧЕСКИЕ НИТРИЛЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕАЗЫ | 2007 |
|
RU2478620C2 |
| БИЦИКЛИЧЕСКИЕ НИТРОИМИДАЗОЛЫ, КОВАЛЕНТНО СОЕДИНЕННЫЕ С ЗАМЕЩЕННЫМИ ФЕНИЛОКСАЗОЛИДИНОНАМИ | 2009 |
|
RU2504547C2 |
| ПРОИЗВОДНЫЕ 5-ОКСА-2-АЗАСПИРО[3.4]ОКТАНА В КАЧЕСТВЕ АГОНИСТОВ M4 | 2020 |
|
RU2820477C1 |
Группа изобретений относится к фармацевтической химии и включает соединение формулы (I), в которой X либо отсутствует, либо является связью; в случаях, когда X отсутствует, четырехчленное кольцо непосредственно присоединено к 6-членному пиперидиновому кольцу, образуя спироциклическую систему; Q либо представляет собой O, либо S(O)p; p = целое число в диапазоне 0-2; Y выбран из NR2R3, NHC(O)R4; R1 выбран из H, F; m = целое число в диапазоне 1-4; один из R2 и R3 означает H, а другой изоксазолил; или R2 и R3, взятые вместе с азотом, к которому они присоединены, могут образовывать 5-членный гетероарил с 2 дополнительными гетероатомами, представляющими собой N; R4 независимо выбран из (C1-C6)алкила, (C3-C6)циклоалкила, гетероарила, представляющего собой тиенил или фурил, и (C1-C6)алкокси. Также группа изобретений включает конкретные промежуточные соединения для получения соединения формулы (I), представляющие собой (S)-5-(аминометил)-3-(4-(4-(1,1-диоксидотиетан-3-ил)пиперидин-1-ил)-3,5-дифторфенил)оксазолидин-2-он; (R)-3-(4-(4-(1,1-диоксидотиетан-3-ил)пиперидин-1-ил)-3,5-дифторфенил)-5-(гидроксиметил)оксазолидин-2-он; (S)-5-(аминометил)-3-(3,5-дифтор-4-(4-(оксетан-3-ил)пиперидин-1-ил)фенил)оксазолидин-2-он; (S)-5-(аминометил)-3-(3,5-дифтор-4-(2-окса-7-азаспиро[3.5]нонан-7-ил)фенил)оксазолидин-2-он; (S)-5-(аминометил)-3-(4-(2,2-диоксидо-2-тиа-7-азаспиро[3.5]нонан-7-ил)-3,5-дифторфенил)оксазолидин-2-он; (R)-3-(4-(2,2-диоксидо-2-тиа-7-азаспиро[3.5]нонан-7-ил)-3,5-дифторфенил)-5-(гидроксиметил)оксазолидин-2-он. Технический результат – соединение формулы (I), обладающее активностью против Mycobacterium tuberculosis. 2 н. и 2 з.п. ф-лы, 2 табл., 11 пр.
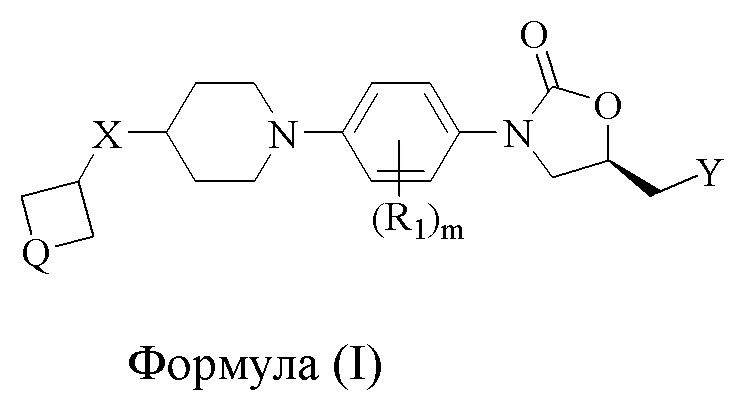
1. Соединения общей формулы (I),
формула (I)
где
X либо отсутствует, либо является связью; в случаях, когда X отсутствует, четырехчленное кольцо непосредственно присоединено к 6-членному пиперидиновому кольцу, образуя спироциклическую систему;
Q либо представляет собой O, либо S(O)p; p = целое число в диапазоне 0-2;
Y выбран из NR2R3, NHC(O)R4;
R1 выбран из H, F; m = целое число в диапазоне 1-4;
один из R2 и R3 означает H, а другой изоксазолил; или R2 и R3, взятые вместе с азотом, к которому они присоединены, могут образовывать 5-членный гетероарил с 2 дополнительными гетероатомами, представляющими собой N;
R4 независимо выбран из (C1-C6)алкила, (C3-C6)циклоалкила, гетероарила, представляющего собой тиенил или фурил, и (C1-C6)алкокси.
2. Соединения формулы (I) по п. 1, в котором R4 выбран из метила или метокси.
3. Соединение формулы (I) по п. 1, которое выбрано из:
(R)-5-((1H-1,2,3-триазол-1-ил)метил)-3-(4-(2,2-диоксидо-2-тиа-7-азаспиро[3.5]нонан-7-ил)-3,5-дифторфенил)оксазолидин-2-она;
(S)-3-(4-(2,2-диоксидо-2-тиа-7-азаспиро[3.5]нонан-7-ил)-3,5-дифторфенил)-5-((изоксазол-3-иламино)метил)оксазолидин-2-она;
(S)-N-((3-(4-(2,2-диоксидо-2-тиа-7-азаспиро[3.5]нонан-7-ил)-3,5-дифторфенил)-2-оксооксазолидин-5-ил)метил)ацетамида;
(S)-метил((3-(4-(2,2-диоксидо-2-тиа-7-азаспиро[3.5]нонан-7-ил)-3,5-дифторфенил)-2-оксооксазолидин-5-ил)метил)карбамата;
(S)-N-((3-(4-(2,2-диоксидо-2-тиа-7-азаспиро[3.5]нонан-7-ил)-3-фторфенил)-2-оксооксазолидин-5-ил)метил)ацетамида;
(S)-N-((3-(3,5-дифтор-4-(2-окса-7-азаспиро[3.5]нонан-7-ил)фенил)-2-оксооксазолидин-5-ил)метил)ацетамида;
(S)-N-((3-(3-фтор-4-(2-окса-7-азаспиро[3.5]нонан-7-ил)фенил)-2-оксооксазолидин-5-ил)метил)ацетамида;
(S)-N-((3-(3,5-дифтор-4-(4-(оксетан-3-ил)пиперидин-1-ил)фенил)-2-оксооксазолидин-5-ил)метил)ацетамида;
(S)-N-((3-(4-(4-(1,1-диоксидотиетан-3-ил)пиперидин-1-ил)-3,5-дифторфенил)-2-оксооксазолидин-5-ил)метил)ацетамида;
(S)-N-((3-(4-(4-(1,1-диоксидотиетан-3-ил)пиперидин-1-ил)-3-фторфенил)-2-оксооксазолидин-5-ил)метил)ацетамида;
метил-(S)-((3-(4-(4-(1,1-диоксидотиетан-3-ил)пиперидин-1-ил)-3,5-дифторфенил)-2-оксооксазолидин-5-ил)метил)карбамата;
(R)-5-((1H-1,2,3-триазол-1-ил)метил)-3-(3,5-дифтор-4-(4-(оксетан-3-ил)пиперидин-1-ил)фенил)оксазолидин-2-она.
4. Промежуточное соединение для получения соединения по п. 1, выбранное из:
(S)-5-(аминометил)-3-(4-(4-(1,1-диоксидотиетан-3-ил)пиперидин-1-ил)-3,5-дифторфенил)оксазолидин-2-она;
(R)-3-(4-(4-(1,1-диоксидотиетан-3-ил)пиперидин-1-ил)-3,5-дифторфенил)-5-(гидроксиметил)оксазолидин-2-она;
(S)-5-(аминометил)-3-(3,5-дифтор-4-(4-(оксетан-3-ил)пиперидин-1-ил)фенил)оксазолидин-2-она;
(S)-5-(аминометил)-3-(3,5-дифтор-4-(2-окса-7-азаспиро[3.5]нонан-7-ил)фенил)оксазолидин-2-она;
(S)-5-(аминометил)-3-(4-(2,2-диоксидо-2-тиа-7-азаспиро[3.5]нонан-7-ил)-3,5-дифторфенил)оксазолидин-2-она;
(R)-3-(4-(2,2-диоксидо-2-тиа-7-азаспиро[3.5]нонан-7-ил)-3,5-дифторфенил)-5-(гидроксиметил)оксазолидин-2-она.
| WO 2007023507 А2, 01.03.2007 | |||
| WO 9525106 А1, 21.09.1995 | |||
| WO 2017015106 A1, 26.01.2017 | |||
| ЗАМЕЩЕННЫЕ ЦИКЛОПРОПИЛЬНОЙ ГРУППОЙ ОКСАЗОЛИДИНОНОВЫЕ АНТИБИОТИКИ И ИХ ПРОИЗВОДНЫЕ | 2004 |
|
RU2348628C2 |
| RU 2011136537 A, 10.03.2013. | |||
