Родственные заявки
Настоящая заявка заявляет приоритет относительно временных заявок США №№ 61/156447, поданной 27 февраля 2009 года, 61/294083 поданной 11 января 2010 года, и 61/294490 поданной 13 января 2010 года. Раскрытия указанных выше заявок включены в настоящую заявку посредством ссылки во всей их полноте.
Область изобретения
В настоящей заявке представлены соединения, которые являются модуляторами JAK киназ, композиции, включающие такие соединения, и способы их применения. Представленные соединения являются полезными для лечения, профилактики или облегчения заболевания или расстройства, связанного с JAK, включая JAK2, JAK3 или TYK2 киназы, или одного или нескольких симптомов, связанных с такими заболеваниями или расстройствами. Кроме того, представлены способы лечения рака, включая гематологические и солидные опухоли.
Предпосылки изобретения
Семейство JAK киназ представляет собой семейство цитоплазматических протеинкиназ, включающее такие члены как JAK1, JAK2, JAK3 и TYK2. Рецепторы ростовых факторов или цитокинов для рекрутмента JAK киназ включают рецепторы интерферона, рецепторы интерлейкинов (рецепторы для цитокинов IL-2 - IL-7, IL-9 - IL-13, IL-15, IL-23), рецепторы различных гормонов (рецептор эритропоэтина (Epo), рецептор тробпопоэтина (Tpo), рецептор лептина, рецептор инсулина, рецептор пролактина (PRL), рецептор гранулоцитарного колониестимулирующего фактора (G-CSF) и рецептор гормона роста, рецептор тирозиновых протеинкиназ (такой как EGFR и PDGFR) и рецепторы для других ростовых факторов, таких как фактор ингибирования лейкоза (LIF), Онкостатин M (OSM), IFNα/β/γ, Гранулоцитарный-макрофагальный колониестимулирующий фактор (GM-CSF), Цилиарный нейротрофический фактор (CNTF), кардиотрофин-1 (CT-1)(См., Rane, S. G. and Reddy E.P., Oncogene 2000 19, 5662-5679).
Фосфорилированные рецепторы служат в качестве докинговых сайтов для другого SH-2 домена, содержащего сигнальные молекулы, которые взаимодействуют с JAK, такие как STAT семейство транскрипционных факторов, Src семейство киназ, MAP киназы, PI3 киназа и протеинтирозинфосфатазы (Rane S.G. and Reddy E.P., Oncogene 2000 19, 5662-5679). Семейство латентных цитоплазматических транскрипционных факторов, STAT, представляет собой наиболее хорошо охарактеризованные присутствующие по ходу этого пути субстраты для JAK. STAT белки связываются с фосфорилированными цитокиновыми рецепторами через их SH2 домены для осуществления их фосфорилирования посредством JAK, что приводит к их димеризации и высвобождению и, в конечном счете, транслокации к ядру, где они активируют генную транскрипцию. Различные члены STAT, которые идентифицированы к настоящему времени, представляют собой STAT1, STAT2, STAT3, STAT4, STAT5 (включая STAT5a и STAT5b) и STAT6.
Поскольку JAK киназы могут играть важную роль в передаче сигнала через такие рецепторы, нарушения жирового метаболизма, нарушения роста и расстройства иммунной системы - все являются потенциальными терапевтическими мишенями.
JAK киназы и JAK2 мутации вовлечены в миелопролиферативные расстройства, раковые заболевания, включая гематологические и солидные опухоли. Примеры расстройств включают хронический миелогенный лейкоз (CML), злокачественную полицитемию (PV), эссенциальную тромбоцитемию (ET), первичный миелофиброз (PMF), хронический эозинофильный лейкоз (CEL), хронический миеломоноцитарный лейкоз (CMML) и системный мастоцитоз (SM). Считается, что миелопролиферативные расстройства возникают либо из мутаций с приобретением функции JAK как таковой, либо из активации онкопротеином BCR-ABL, который специфическим образом активирует путь JAK2. Некоторые литературные источники описывают роль JAK2 мутаций в различных расстройствах. См., Samanta et al. Cancer Res 2006, 66(13), 6468-6472, Sawyers et al. Cell, 1992, 70, 901-910, Tefferi N. Eng J. Med. (2007) 356(5): 444-445) Baxter et al. Lancet (2005) 365: 1054-1056, Levine et al. Blood (2006, Jones et al. Blood (2005) 106:2162-2168) 107:4139-4141, Campbell et al. Blood (2006) 107(5): 2098-2100, Scott et al. N Eng J Med 2007 356(5): 459-468, Mercher et al. Blood (2006) 108(8): 2770-2778, Lacronique et al. Science (1997) 278: 1309-1312, Lacronique et al. Blood (2000) 95:2535-2540, Griesinger F. et al. Genes Chromosomes Cancer (2005) 44:329-333, Bousquet et al. Oncogene (2005) 24:7248-7252, Schwaller et al. Mol. Cell. 2000 6,693-704, Zhao et al. EMBO 2002 21(9), 2159-2167.
Литературные источники указывают, что JAK также может служить мишенью для рака предстательной железы, включая адроген-резистентный рак предстательной железы. См., Barton et al. Mol. Canc. Ther. 2004 3(1), 11-20, Blume-Jensen et al. Nature (2001) 411(6835):355-356 и Bromberg J Clin Invest. (2002) 109(9): 1139-1142, Rane Oncogene (2000) 19(49):5662-5679. JAK в качестве заметного медиатора пути передачи сигнала цитокинов считается терапевтической мишенью при воспалении и отторжении трансплантата. См., Borie et al., Transplantation (2005) 79(7):791-801, и Milici et al., Arthritis Research (2008) 10(R14): 1-9
Учитывая большое количество различных заболеваний, связанных с нарушением регуляции сигнальной активности JAK, в настоящее время на стадии разработки находится множество малых молекул-ингибиторов JAK. Примеры соединений, находящихся в предклинической стадии разработки, включают TG101209 (TargeGen), примеры соединений, находящихся в клинических стадиях исследования, включают INCBO 18424 Incyte), XL019 (Exelixis) и TG101348 (TargeGen). См., Pardanani et al. Leukemia 2007, 21: 1658-1668; и Pardanai, A. Leukemia 2008 22:23-20.
Однако все еще существует необходимость в обеспечении новых классов соединений, которые являются полезными в качестве ингибиторов ферментов в сигнальном пути JAK.
Краткое описание изобретения
В настоящей заявке представлены соединения формулы (I)
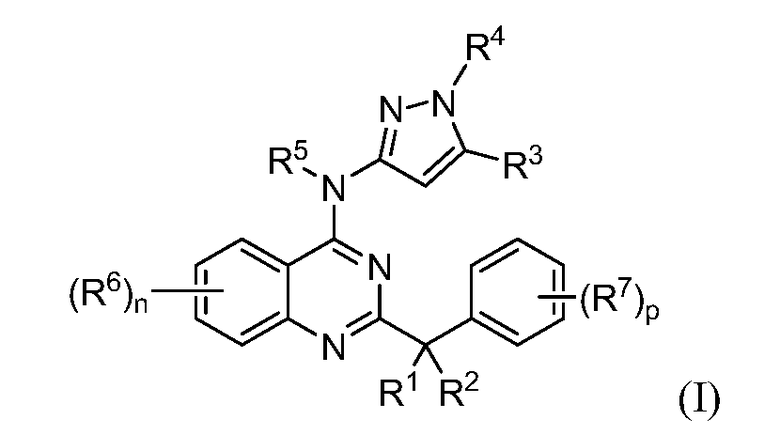
или их фармацевтически приемлемые соли, сольваты или гидраты, где
R1 и R2 выбраны из (i), (ii), (iii), (iv) и (v) и имеют следующие значения:
(i) R1 и R2 вместе образуют =О, =S, =NR9 или =CR10R11;
(ii) R1 и R2 оба представляют собой -OR8, или R1 и R2, вместе с атомом углерода, с которым они связаны, образуют диоксациклоалкил;
(iii) R1 представляет собой водород или галоген; и R2 представляет собой галоген;
(iv) R1 представляет собой алкил, алкенил, алкинил, циклоалкил или арил, где алкил, алкенил, алкинил, циклоалкил или арил необязательно замещен одним или несколькими, в одном варианте воплощения одним-четырьмя, в одном варианте воплощения одним-тремя, в одном варианте воплощения одним, двумя или тремя заместителями, выбранными из галогена, циано, алкила, -RXORW, -RxS(O)qRv, -RxNRyRz и -C(O)ORW; и R2 представляет собой водород, галоген или -OR8; и
(v) R1 представляет собой галоген, дейтеро, -OR12, -NR13R14 или -S(O)qR15; и R2 представляет собой водород, дейтеро, алкил, алкенил, алкинил, циклоалкил или арил, где алкил, алкенил, алкинил, циклоалкил или арил необязательно замещен одним или несколькими, в одном варианте воплощения одним-четырьмя, в одном варианте воплощения одним-тремя, в одном варианте воплощения одним, двумя или тремя заместителями, выбранными из галогена, циано, алкила, -RXORW, -RxS(O)qRv и -RxNRyRz;
R3 представляет собой водород, галоген, алкил, циано, галогеналкил, циклоалкил, циклоалкилалкил, гидрокси или алкокси;
R4 и R5, каждый независимо, представляет собой водород или алкил;
каждый R6 независимо выбран из галогена, алкила, алкенила, алкинила, галогеналкила, циклоалкила, -RXOR18, -RXNR19R20 и -RxS(O)qRv;
каждый R7 независимо представляет собой галоген, алкил, галогеналкил или -RXORW;
R8 представляет собой алкил, алкенил или алкинил;
R9 представляет собой водород, алкил, галогеналкил, гидрокси, алкокси или амино;
R10 представляет собой водород или алкил;
R11 представляет собой водород, алкил, галогеналкил или -C(O)OR8;
R12 выбран из водорода, алкила, алкенила, алкинила, циклоалкила, циклоалкилалкила, гетероциклила, гетероциклилалкила, арила, аралкила, гетероарила, гетероаралкила, -C(O)RV, -C(O)ORW и -C(О)NRyRz, где алкил, алкенил, алкинил, циклоалкил, циклоалкилалкил, гетероциклил, гетероциклилалкил, арил, аралкил, гетероарил и гетероаралкил, каждый, необязательно замещен одним или несколькими, в одном варианте воплощения одним-четырьмя, в одном варианте воплощения одним-тремя, в одном варианте воплощения одним, двумя или тремя заместителями, независимо выбранными из галогена, оксо, алкила, гидрокси, алкокси, амино и алкилтио;
R13 и R14 выбраны следующим образом:
(i) R13 представляет собой водород или алкил; и R14 выбран из водорода, алкила, алкенила, алкинила, циклоалкила, циклоалкилалкила, гетероциклила, гетероциклилалкила, арила, аралкила, гетероарила, гетероаралкила, алкокси, -C(O)RV, -C(O)ORW, -C(О)NRyRz и -S(O)qRv, где алкил, алкенил, алкинил, циклоалкил, циклоалкилалкил, гетероциклил, гетероциклилалкил, арил, аралкил, гетероарил или гетероаралкил, каждый, необязательно замещен одним или несколькими, в одном варианте воплощения одним-четырьмя, в одном варианте воплощения одним-тремя, в одном варианте воплощения одним, двумя или тремя заместителями, независимо выбранными из галогена, оксо, алкила, гидрокси, алкокси, амино и алкилтио; или
(ii) R13 и R14, вместе с атомом азота, с которым они связаны, образуют гетероциклил или гетероарил, где гетероциклил или гетероарил замещены одним или несколькими, в одном варианте воплощения одним-четырьмя, в одном варианте воплощения одним-тремя, в одном варианте воплощения одним, двумя или тремя заместителями, независимо выбранными из галогена, алкила, гидрокси, алкокси, амино и алкилтио, и, где гетероциклил необязательно замещен группой оксо;
R15 представляет собой алкил, алкенил, алкинил, циклоалкил, циклоалкилалкил, гетероциклил, гетероциклилалкил, арил, аралкил, гетероарил, гетероаралкил, -C(O)NRyRz или -NRyRz, где алкил, алкенил, алкинил, циклоалкил, циклоалкилалкил, гетероциклил, гетероциклилалкил, арил, аралкил, гетероарил или гетероаралкил, каждый, необязательно замещен одним или несколькими, в одном варианте воплощения одним-четырьмя, в одном варианте воплощения одним-тремя, в одном варианте воплощения одним, двумя или тремя заместителями, независимо выбранными из галогена, оксо, алкила, гидрокси, алкокси, амино и алкилтио;
R18 представляет собой водород, алкил, галогеналкил, гидроксиС2-6алкил, алкенил, алкинил, циклоалкил, циклоалкилалкил, гетероциклил, гетероциклилалкил, арил, аралкил, гетероарил или гетероарилалкил; где R18 необязательно замещен 1-3 группами Q1, каждый Q1 независимо выбран из алкила, гидроксила, галогена, галогеналкила, алкокси, арилокси, алкоксиалкила, алкоксикарбонила, алкоксисульфонила, карбоксила, циклоалкила, гетероциклила, арила, гетероарила, галогенарила и амино;
R19 и R20 выбраны следующим образом:
(i) R19 и R20, каждый независимо, представляет собой водород или алкил; или (ii) R19 и R20, вместе с атомом азота, с которым они связаны, образуют гетероциклил или гетероарил, который необязательно замещен 1-2 группами, каждая из которых независимо выбрана из галогена, алкила, галогеналкила, гидроксила и алкокси;
каждый Rx независимо представляет собой алкилен или простую связь;
Rv представляет собой водород, алкил, алкенил или алкинил;
Rw независимо представляет собой водород, алкил, алкенил, алкинил или галогеналкил;
Ry и Rz выбраны следующим образом:
(i) Ry и Rz, каждый независимо, представляет собой водород, алкил, алкенил, алкинил, циклоалкил или галогеналкил;
(ii) Ry и Rz, вместе с атомом азота, с которым они связаны, образуют гетероциклил или гетероарил, которые необязательно замещены 1-2 группами, каждая из которых независимо выбрана из галогена, алкила, галогеналкила, гидроксила и алкокси;
n имеет значение 0-4;
p имеет значение 0-5; и
каждый q, независимо, имеет значение 0, 1 или 2.
В некоторых вариантах воплощения соединения обладают активностью модуляторов JAK киназы, в том числе JAK2 киназы. Соединения являются полезными в медицинском лечении, фармацевтических композициях и способах модулирования активности JAK киназы, в том числе дикого типа и/или мутированных форм JAK киназы. В некоторых вариантах воплощения соединения, представленные в настоящем изобретении, обладают активностью модуляторов JAK2 киназы. В некоторых вариантах воплощения соединения являются ингибиторами JAK киназы, в том числе JAK2 киназы. В некоторых вариантах воплощения соединения являются ингибиторами JAK киназы, в том числе JAK2 и TYK2 киназ.
В одном варианте воплощения соединения для использования в композициях и способах, представленных в настоящей заявке, представляют собой соединения формулы (I).
В одном варианте воплощения соединение, представленное в настоящем изобретении, представляет собой соединение формулы (I). В одном варианте воплощения соединение, представленное в настоящем изобретении, представляет собой фармацевтически приемлемую соль соединения формулы (I). В одном варианте воплощения соединение, представленное в настоящем изобретении, представляет собой сольват соединения формулы (I). В одном варианте воплощения соединение, представленное в настоящем изобретении, представляет собой гидрат соединения формулы (I).
Также представлены фармацевтические композиции, сформулированные для введения подходящим путем и способом, содержащие эффективные концентрации одного или нескольких соединений, представленных в настоящем изобретении, или их фармацевтически приемлемых солей, сольватов и гидратов, и необязательно включающие, по меньшей мере, один фармацевтический носитель.
Такие фармацевтические композиции доставляют количества, эффективные для лечения, профилактики или облегчения заболеваний или расстройств, которые включают, без ограничения, миелопролиферативные расстройства, такие как истинная полицитемия (PCV), эссенциальная тромбоцитемия (ET), первичный миелофиброз (PMF), хронический эозинофильный лейкоз (CEL), хронический миеломоноцитарный лейкоз (CMML), системный мастоцитоз (SM) и идиопатический миелофиброз (IMF); лейкоз, такой как миелоидный лейкоз, включая хронический миелоидный лейкоз (CML), иматиниб-резистентные формы CML, острый миелоидный лейкоз (AML) и подтип AML, острый мегакариобластный лейкоз (AMKL); лимфопролиферативные заболевания, такие как миелома; рак, такой как рак головы и шеи, рак предстательной железы, рак молочной железы, рак яичника, меланома, рак легкого, опухоли головного мозга, рак поджелудочной железы и рак почек; и воспалительные заболевания или расстройства, связанные с иммунной дисфункцией, иммунодефицитом, иммуномодуляцией, аутоиммунные заболевания, отторжение тканевого трансплантата, болезнь трансплантат-против-хозяина, заживление ран, заболевание почек, рассеянный склероз, тиреоидит, диабет типа 1, саркоидоз, псориаз, аллергический ринит, воспалительное заболевание кишечника, включая болезнь Крона и язвенный колит (UC), системная красная волчанка (SLE), артрит, остеоартрит, ревматоидный артрит, остеопороз, астма, хроническое обструктивное заболевание легких (COPD) и синдром сухих глаз (или сухой кератоконъюнктивит (KCS)). В одном варианте воплощения, такие заболевания или расстройства модулируются, или на них каким-либо иным образом влияют JAK киназы, включая JAK2, JAK3 или TYK2.
Также в настоящей заявке представлены комбинированные терапии с использованием одного или нескольких соединений или композиций, представленных в настоящем изобретении, или их фармацевтически приемлемых солей, сольватов или гидратов в сочетании с другими фармацевтически активными средствами для лечения заболеваний и расстройств, описанных в настоящей заявке.
В одном варианте воплощения такие дополнительные фармацевтические средства включают одно или несколько химиотерапевтических средств, антипролиферативных средств, противовоспалительных средств, иммуномодулирующих средств или иммуносупрессивных средств.
Соединения или композиции, представленные в настоящем изобретении, или их фармацевтически приемлемые соли, сольваты или гидраты можно вводить одновременно с введением, до или после введения одного или нескольких из указанных выше средств. Также обеспечиваются фармацевтические композиции, содержащие соединение, представленное в настоящем изобретении, и одно или несколько из указанных выше средств.
В некоторых вариантах воплощения в настоящей заявке представлены способы лечения, профилактики или облегчения заболевания или расстройства, которое модулируется JAK киназами или на которое иным образом влияют JAK киназы, включая JAK2 киназу, такую как дикого типа и/или мутантную JAK2 киназу, или одного или нескольких его симптомов или причин. В другом варианте воплощения, в настоящей заявке представлены способы лечения, профилактики или облегчения заболевания или расстройства путем модулирования JAK2 киназы селективным образом по сравнению с JAK3 киназой. В следующем варианте воплощения, в настоящей заявке представлены способы лечения, профилактики или облегчения заболевания или расстройства путем модулирования JAK3 киназы селективным образом по сравнению с JAK2 киназой. В другом варианте воплощения, в настоящей заявке представлены способы лечения, профилактики или облегчения заболевания или расстройства путем модулирования обоих киназ JAK2 и JAK3. В одном варианте воплощения представлены способы лечения рака, включая гематологические и солидные опухоли.
При осуществлении на практике способов, эффективные количества соединений или композиций, содержащих терапевтически эффективные концентрации соединений, которые сформулированы для системной доставки, включая парентеральную, пероральную или внутривенную доставку, или для локального или местного применения, вводят субъекту, у которого проявляются симптомы заболевания или расстройства, подлежащего лечению. Количества являются эффективными для облегчения или устранения одного или нескольких симптомов заболевания или расстройства.
Эти и другие аспекты изобретения, описанного в настоящей заявке, будут очевидны при обращении к представленному ниже подробному описанию.
Краткое описание рисунков
Фиг. 1 представляет in vivo данные, демонстрирующие ответную реакцию на введение дозы соединения формулы I в модели тип II коллаген-индуцированного артрита (CIA) у крыс.
Фиг. 2 представляет эффекты введения различных доз соединения формулы I и контроль массы тела в модели тип II коллаген-индуцированного артрита (CIA) у крысы.
Фиг. 3 представляет анализ выживания Каплана-Мейера для фирменного соединения Ambit, предназначенного только для внутреннего использования, TGEN101348 и контроля в мышиной модели TELJAK.
Фиг. 4 представляет анализ выживания Каплана-Мейера для фирменного соединения Ambit, предназначенного только для внутреннего использования, соединения формулы I и контроля в мышиной модели TELJAK.
Фиг. 5 представляет анализ выживания Каплана-Мейера для фирменного соединения Ambit, предназначенного только для внутреннего использования, соединения формулы I и контроля в HELV617F мышиной модели жидкой опухоли.
Фиг. 6 представляет анализ выживания Каплана-Мейера для фирменного соединения Ambit, предназначенного только для внутреннего использования, TGEN101348 и контроля в HELV617F мышиной модели жидкой опухоли.
Подробное описание изобретения
В настоящей заявке представлены соединения формулы (I), которые обладают активностью в качестве модуляторов JAK киназы, в том числе JAK2 киназы. Кроме того, представлены способы лечения, профилактики или облегчения заболеваний, которые модулируются JAK киназами, включая JAK2 киназу, и фармацевтические композиции и лекарственные формы, полезные для таких способов. Способы и композиции описаны подробно в представленных ниже разделах.
В некоторых вариантах воплощения соединения, представленные в настоящей заявке, являются селективными в отношении JAK2, т.е. соединения связываются или взаимодействуют с JAK2 при существенно более низких концентрациях по сравнению с их связыванием или взаимодействием с другими JAK рецепторами, включая JAK3 рецептор, при той же самой концентрации. В некоторых вариантах воплощения соединения связываются с JAK3 рецептором при константе связывания, по меньшей мере, примерно в 3 раза выше, примерно в 5 раз выше, примерно в 10 раз выше, примерно в 20 раз выше, примерно в 25 раз выше, примерно в 50 раз выше, примерно в 75 раз выше, примерно в 100 раз выше, примерно в 200 раз выше, примерно в 225 раз выше, примерно в 250 раз выше или примерно в 300 раз выше по сравнению с их связыванием с JAK2 рецептором.
В некоторых вариантах воплощения соединения, представленные в настоящей заявке, являются селективными в отношении JAK3, т.е. соединения связываются или взаимодействуют с JAK3 при существенно более низких концентрациях по сравнению с их связыванием или взаимодействием с другими JAK рецепторами, включая JAK2 рецептор, при той же самой концентрации. В некоторых вариантах воплощения соединения связываются с JAK2 рецептором при константе связывания, по меньшей мере, примерно в 3 раза выше, примерно в 5 раз выше, примерно в 10 раз выше, примерно в 20 раз выше, примерно в 25 раз выше, примерно в 50 раз выше, примерно в 75 раз выше, примерно в 100 раз выше, примерно в 200 раз выше, примерно в 225 раз выше, примерно в 250 раз выше или примерно в 300 раз выше по сравнению с их связыванием с JAK3 рецептором.
В некоторых вариантах воплощения соединения, представленные в настоящем изобретении, связываются или взаимодействуют с TYK2. В некоторых вариантах воплощения соединения, представленные в настоящем изобретении, имеют Kd меньше чем 20 нМ, меньше чем 40 нМ, меньше чем 50 нМ, меньше чем 75 нМ, меньше чем 80 нМ, меньше чем 90 нМ или меньше чем 100 нМ против TYK2. В некоторых вариантах воплощения соединения, представленные в настоящем изобретении, имеют Kd больше чем около 10 нМ, 20 нМ, 25 нМ, 40 нМ, 50 нМ или 70 нМ против киназы Aurora B. Способы определения Kd соединений против киназ, таких как TYK2 и Aurora B киназы, известны специалистам в данной области. Иллюстративные способы описаны во временной заявке США № 61/294413 и в Fabian et al., Nature Biotechnology 2005, 23,329-336.
A. Определения
Если не указано иное, все технические и научные термины, используемые в настоящей заявке, имеют обычные значения, которые известны специалистам в данной области. Все патенты, заявки, опубликованные заявки и другие публикации включены посредством ссылки во всей их полноте. В случае, когда в описании встречается несколько определений термина, преимущество имеют определения, указанные в данном разделе, если не указано иное.
"Алкил" относится к группе с линейной или разветвленной углеводородной цепью, состоящей исключительно из атомов углерода и водорода, не содержащей какой-либо ненасыщенности, содержащей от одного до десяти, от одного до восьми, от одного до шести или от одного до четырех атомов углерода, и которая присоединена к остальной части молекулы при помощи простой связи, например, такой как метил, этил, н-пропил, 1-метилэтил (изопропил), н-бутил, н-пентил, 1,1-диметилэтил (трет-бутил) и подобные.
"Алкенил" относится к группе с линейной или разветвленной углеводородной цепью, состоящей исключительно из атомов углерода и водорода, содержащей, по меньшей мере, одну двойную связь, в некоторых вариантах воплощения, содержащей от 2 до 10 атомов углерода, от 2 до 8 атомов углерода или от 2 до 6 атомов углерода, и которая присоединена к остальной части молекулы при помощи простой связи или двойной связи, например, этенил, проп-1-енил, бут-1-енил, пент-1-енил, пента-1,4-диенил и подобные.
"Алкинил" относится к группе с линейной или разветвленной углеводородной цепью, состоящей исключительно из атомов углерода и водорода, содержащей, по меньшей мере, одну тройную связь, содержащей от двух до десяти атомов углерода, и которая присоединена к остальной части молекулы при помощи простой связи или тройной связи, например, такой как этинил, проп-1-инил, бут-1-инил, пент-1-инил, пент-3-инил и подобные.
"Алкилен" и "алкиленовая цепь" относятся к линейной или разветвленной двухвалентной углеводородной цепи, состоящей исключительно из углерода и водорода, не содержащей какой-либо ненасыщенности и содержащей от одного до восьми атомов углерода, например, такой как метилен, этилен, пропилен, н-бутилен и подобные. Алкиленовая цепь может быть присоединена к остальной части молекулы через любые два атома углерода в цепи.
"Алкокси" относится к группе, имеющей формулу -OR, где R представляет собой алкил или галогеналкил, где алкил, необязательно, может быть замещен одним или несколькими заместителями, в одном варианте воплощения одним, двумя или тремя заместителями, независимо выбранными из группы, включающей нитро, галоген, гидроксил, алкокси, оксо, тиоксо, амино, карбонил, карбокси, азидо, циано, циклоалкил, гетероарил и гетероциклил.
"Алкоксиалкил" относится к группе, имеющей формулу -RhOR, где Rh представляет собой линейную или разветвленную алкиленовую цепь и OR представляет собой алкокси, определенный выше.
"Алкилтио" относится к группе, имеющей формулу -SR, где R представляет собой алкил или галогеналкил.
"Арилокси" относится к группе -OR, в которой R представляет собой арил, включая низший арил, такой как фенил.
"Амин" или "амино" относится к группе, имеющей формулу -NR'R", где R' и R'', каждый независимо, представляют собой водород, алкил, галогеналкил, гидроксиалкил или алкоксиалкил, или где R' и R" вместе с атомом азота, с которым они связаны, образуют гетероциклил, необязательно замещенный галогеном, оксо, гидрокси или алкокси.
"Аминоалкил" относится к группе, имеющей формулу -RhNR'R", где Rh представляет собой линейную или разветвленную алкиленовую цепь, и где NR'R" представляет собой амино, определенный выше.
"Аминокарбонил" относится к группе, имеющей формулу -C(O)NR'R'', где -NR'R" представляет собой амино, определенный выше.
"Арил" относится к группе, представляющей собой карбоциклическую кольцевую систему, включая моноциклические, бициклические, трициклические, тетрациклические C6-C18 кольцевые системы, где, по меньшей мере, одно из колец является ароматическим. Арил может быть полностью ароматическим, примерами которого являются фенил, нафтил, антраценил, аценафтиленил, азуленил, флуоренил, инденил и пиренил. Арил также может содержать ароматическое кольцо в сочетании с неароматическим кольцом, примерами которого являются аценафен, инден и флуорен. Термин включает как замещенные, так и незамещенные группы. Арильная группа может быть замещена любой описанной группой, включая, но не ограничиваясь этим, одну или несколько групп, выбранных из группы, включающей галоген (фтор, хлор, бром или иод), алкил, гидроксил, амино, алкокси, арилокси, нитро и циано.
"Карбоксил" относится к группе, имеющей формулу -C(O)OH.
"Циклоалкил" относится к подходящей одновалентной моноциклической или бициклической углеводородной группе, состоящей исключительно из атомов углерода и водорода, содержащей от трех до десяти атомов углерода, и которая является насыщенной и присоединена к остальной части молекулы при помощи простой связи, например, такой как циклопропил, циклобутил, циклопентил, циклогексил, декалинил, норборнан, норборнен, адамантил, бицикло[2,2,2]октан и подобные.
"Циклоалкилалкил" относится к группе формулы -RaRd, где Ra представляет собой алкильную группу, определенную выше, и Rd представляет собой циклоалкильную группу, определенную выше. Алкильная группа и циклоалкильная группа, необязательно, могут быть замещены, как определено в настоящей заявке.
"Дейтеро" или "дейтерий" относится к изотопу водорода дейтерию, имеющему химическое обозначение D или 2H.
"Галоген", "галоген" или "галогенид" относится к F, Cl, Br или I.
"Галогеналкил" относится к алкильной группе, в некоторых вариантах воплощения С1-6алкильной группе, в которой один или несколько атомов водорода замещены галогеном. Такие группы включают, но не ограничиваются этим, хлорметил, трифторметил, 1-хлор-2-фторэтил, 2,2-дифторэтил, 2-фторпропил, 2-фторпропан-2-ил, 2,2,2-трифторэтил, 1,1-дифторэтил, 1,3-дифтор-2-метилпропил, 2,2-дифторциклопропил, (трифторметил)циклопропил, 4,4-дифторциклогексил и 2,2,2-трифтор-1,1-диметилэтил.
"Гетероциклил" относится к подходящей 3-15-членной кольцевой группе, которая состоит из атомов углерода и от одного до пяти гетероатомов, выбранных из группы, включающей азот, кислород и серу. В одном варианте воплощения, группа, представляющая собой гетероциклическую кольцевую систему, может представлять собой моноциклическую, бициклическую или трициклическую или тетрациклическую кольцевую систему, которая может включать конденсированные или связанные мостиковой связью кольцевые системы; и атомы азота или серы в гетероциклической кольцевой системе, необязательно, могут быть окислены; атом азота, необязательно, может быть кватернизирован; и гетероциклильная группа может быть частично или полностью насыщенной или ароматической. Гетероциклическая кольцевая система может быть присоединена к основной структуре по любому гетероатому или атому углерода, который обеспечивает получение подходящего соединения. Примеры гетероциклических радикалов включают, азетидинил, бензопиранонил, бензопиранил, бензотетрагидрофуранил, бензотетрагидротиенил, хроманил, хромонил, кумаринил, декагидроизохинолинил, дибензофуранил, дигидробензизотиазинил, дигидробензизоксазинил, дигидрофурил, дигидропиранил, диоксоланил, дигидропиразинил, дигидропиридинил, дигидропиразолил, дигидропиримидинил, дигидропирролил, диоксоланил, 1,4-дитианил, изобензотетрагидрофуранил, изобензотетрагидротиенил, изохроманил, изокумаринил, бензо[1,3]диоксол-5-ил, бензодиоксолил, 1,3-диоксолан-2-ил, диоксоланил, морфолинил, октагидроиндолил, октагидроизоиндолил, тетрагидрофуран, оксазолидин-2-онил, оксазолидинонил, пиперидиноил, пиперазинил, пиранил, тетрагидрофурил, тетрагидрофуранил, тетрагидроизохинолинил, тетрагидропиранил, тетрагидротиенил, пирролидинонил, оксатиоланил и пирролидинил.
"Гетероарил" относится к гетероциклильной группе, определенной выше, которая является ароматической. Гетероарильная группа может быть присоединена к основной структуре по любому гетероатому или атому углерода, который обеспечивает получение подходящего соединения. Примеры таких гетероарильных групп включают, но не ограничиваются этим: акридинил, бензимидазолил, бензиндолил, бензизоксазинил, бензо[4,6]имидазо[1,2-α]пиридинил, бензофуранил, бензонафтофуранил, бензотиадиазолил, бензотиазолил, бензотиофенил, бензотриазолил, бензотиопиранил, бензоксазинил, бензоксазолил, бензотиазолил, β-карболинил, карбазолил, циннолинил, дибензофуранил, фуранил, имидазолил, имидазопиридинил, имидазотиазолил, индазолил, индолизинил, индолил, изобензотиенил, изоиндолинил, изохинолинил, изотиазолидинил, изотиазолил, нафтиридинил, октагидроиндолил, октагидроизоиндолил, оксазолидинонил, оксазолидинил, оксазолопиридинил, оксазолил, изоксазолил, оксиранил, перимидинил, фенантридинил, фенантролинил, фенарсазинил, феназинил, фенотиазинил, феноксазинил, фталазинил, птеридинил, пуринил, пиразинил, пиразолил, пиридазинил, пиридинил, пиридопиридинил, пиримидинил, пирролил, хиназолинил, хинолинил, хиноксалинил, тетразолил, тиадиазолил, тиазолил, тиенил, триазинил и триазолил.
"Аралкил" относится к группе формулы -RaRb, где Ra представляет собой алкильную группу, определенную выше, замещенную Rb, арильной группой, определенной выше, например, бензил. Как алкильная, так и арильная группа, необязательно, могут быть замещены, как определено в настоящей заявке.
"Гетероаралкил" относится к группе формулы -RaRf, где Ra представляет собой алкильную группу, определенную выше, и Rf представляет собой гетероарильную группу, определенную в настоящей заявке. Алкильная группа и гетероарильная группа, необязательно, могут быть замещены, как определено в настоящей заявке.
"Гетероциклилалкил" относится к группе формулы -RaRe, где Ra представляет собой алкильную группу, определенную выше, и Re представляет собой гетероциклильную группу, определенную в настоящей заявке, где алкильная группа Ra может быть присоединена по любому атому углерода или гетероатому гетероциклильной группы Re. Алкильная группа и гетероциклильная группа, необязательно, могут быть замещены, как определено в настоящей заявке.
"Алкоксикарбонил" относится к группе, имеющей формулу -C(O)OR, в которой R представляет собой алкил, включая низший алкил.
Термин "диоксациклоалкил", как он используется в настоящей заявке, означает гетероциклическую группу, содержащую два кольцевых атома кислорода и два или более кольцевых атомов углерода.
"Оксо" относится к группе =О, присоединенной к атому углерода.
"Тиоалкил" относится к группе, имеющей формулу -RhSRi, где Rh представляет собой линейную или разветвленную алкиленовую цепь, и Ri представляет собой алкил или галогеналкил.
"Тиоксо" относится к группе =S, присоединенной к атому углерода.
"ИК50" относится к количеству, концентрации или дозе конкретного испытываемого соединения, которые обеспечивают 50% ингибирование максимального ответа, такого как клеточный рост или пролиферация, измеренного при помощи любого in vitro или клеточного анализа, описанного в настоящей заявке.
Если не указано иное или конкретно не описано в настоящем описании, должно быть понятно, что замещение может иметь место по любому атому алкильной, алкенильной, алкинильной, циклоалкильной, гетероциклильной, арильной или гетероарильной группы.
Фармацевтически приемлемые соли включают, но не ограничиваются этим, соли минеральных кислот, такие как гидрохлориды; и соли органических кислот, такие как, но не ограничиваясь этим, мезилат, эзилат, тозилат, безилат, брозилат, камфорсульфонат, гидробромид, фосфат, сульфат, трифторацетат, ацетат, бензоат, фумарат, малат, малеат, оксалат, сукцинат и тартрат.
Как он используется в настоящей заявке, и если не указано иное, термин "гидрат" означает соединение, представленное в настоящем изобретении, или его соль, который, кроме того, включает стехиометрическое или нестехиометрическое количество воды, связанной нековалентными межмолекулярными связями.
Как он используется в настоящей заявке, и если не указано иное, термин "сольват" означает сольват, образованный в результате ассоциации одной или нескольких молекул растворителя с соединением, представленным в настоящем изобретении. Термин "сольват" включает гидраты (например, моногидрат, дигидрат, тригидрат, тетрагидрат и подобные).
Как это используется в настоящей заявке, "по существу чистый" означает вещество, на вид достаточно гомогенное и не содержащее примесей, которые легко можно определить стандартными методами анализа, такими как тонкослойная хроматография (ТСХ), гель-электрофорез, высоко-эффективная жидкостная хроматография (ВЭЖХ) и масс-спектрометрия (MS), используемыми специалистами в данной области для оценки такой чистоты, или достаточно чистый настолько, что дополнительная очистка не может определяемым образом изменить физические и химические свойства вещества, такие как ферментативная и биологическая активность. Способы очистки соединений для получения, по существу, химически чистых соединений известны специалистам в данной области. Однако, по существу, химически чистое соединение может представлять собой смесь стереоизомеров. В таких случаях дополнительная очистка может увеличить специфическую активность соединения.
Если конкретно не указано иное, когда соединение может принимать альтернативные таутомерные, региоизомерные и/или стереоизомерные формы, предполагается, что все альтернативные изомеры охватываются объемом заявленного изобретения. Например, когда соединение описано как имеющее одну из двух таутомерных форм, предполагается, что оба таутомера охватываются настоящим изобретением. Таким образом, соединения, представленные в настоящем изобретении, могут быть энантиомерно чистыми или могут представлять собой стереоизомерные или диастереомерные смеси.
Должно быть понятно, что соединения, представленные в настоящем изобретении, могут содержать хиральные центры. Такие хиральные центры могут быть либо в (R), либо в (S) конфигурации, или это может быть их смесь.
Оптически активные (+) и (-), (R)- и (S)- или (D)- и (L)-изомеры можно получить с использованием хирального синтеза или хиральных реагентов или путем разделения с использованием традиционных методов, таких как обращено-фазовая ВЭЖХ или кристаллизация.
Как он используется в настоящей заявке, термин "энантиомерно чистый" или "чистый энантиомер" означает, что соединение включает более чем 75% масс., более чем 80% масс., более чем 85% масс., более чем 90% масс., более чем 91% масс., более чем 92% масс., более чем 93% масс., более чем 94% масс., более чем 95% масс., более чем 96% масс., более чем 97% масс., более чем 98% масс., более чем 98,5% масс., более чем 99% масс., более чем 99,2% масс., более чем 99,5% масс., более чем 99,6% масс., более чем 99,7% масс., более чем 99,8% масс. или более чем 99,9% масс. желаемого энантиомера.
Когда количество любого присутствующего заместителя не указано (например, галогеналкил), может присутствовать один или несколько заместителей. Например, "галогеналкил" может включать один или несколько одинаковых или отличных друг от друга галогенов.
В представленном в настоящей заявке описании, если существует какое-либо расхождение между химическим названием и химической структурой, в этом случае определяющей, предпочтительно, является структура.
Как он используется в настоящей заявке, термин "изотопная композиция" относится к количеству каждого присутствующего изотопа для данного атома, и "природная изотопная композиция" относится к встречающейся в природе изотопной композиции или относительному изотопному составу для данного атома. Атомы, имеющие их природную изотопную композицию, также могут быть указаны как "необогащенные" атомы. Если не указано иное, атомы соединений, описанных в настоящей заявке, представляют любой стабильный изотоп этого атома. Например, если не указано иное, когда положение конкретно обозначено как "H" или "водород", имеется в виду, что это положение включает водород в его природной изотопной композиции.
Как он используется в настоящей заявке, термин "изотопно обогащенный" относится к атому, имеющему изотопную композицию, отличную от природной изотопной композиции этого атома. "Изотопно обогащенный" может также относиться к соединению, содержащему, по меньшей мере, один атом, имеющий изотопную композицию, отличную от природной изотопной композиции этого атома.
Как он используется в настоящей заявке, термин "изотопное обогащение" относится к процентному количеству включения определенного изотопа на данном атоме в молекуле вместо природного изотопного состава этого атома. Например, обогащение дейтерием 1% в указанном положении означает, что 1% молекул в данном образце содержат дейтерий в указанном положении. Поскольку естественное распределение дейтерия составляет около 0,0156%, обогащение дейтерием в любом положении в соединении, синтезированном с использованием необогащенных исходных веществ, составляет около 0,0156%. Изотопное обогащение соединений, представленных в настоящем изобретении, можно определить с использованием традиционных аналитических методов, известных любым специалистам в данной области, имеющим среднюю квалификацию, включая масс-спектрометрию и спектроскопию ядерного магнитного резонанса.
"Противораковые средства" относится к анти-метаболитам (например, 5-фтор-урацил, метотрексат, флударабин), средствам против образования микротрубочек (например, алкалоиды барвинка, такие как винкристин, винбластин; таксаны, такие как паклитаксел, доцетаксел), алкилирующим средствам (например, циклофосфамид, мелфалан, кармустин, средства на основе нитрозомочевины, такие как бисхлорэтилнитрозомочевина и гидроксимочевина), средствам на основе платины (например, цисплатин, карбоплатин, оксалиплатин, JM-216 или сатраплатин, CI-973), антрациклинам (например, доксрубицин, даунорубицин), противоопухолевым антибиотикам (например, митомицин, идарубицин, адриамицин, дауномицин), ингибиторам топоизомеразы (например, этопозид, камптотецины), средствам против ангиогенеза (например, Sutent® и Бевацизумаб) или любым другим цитотоксическим средствам, (эстрамустин фосфат, преднимустин), гормонам или агонистам, антагонистам, частичным агонистам или частичным антагонистам гормонов, ингибиторам киназы и лучевой терапии.
"Противовоспалительные средства" относится к ингибиторам металлопротеиназы матрикса, ингибиторам провоспалительных цитокинов (например, анти-TNF молекулы, растворимые рецепторы TNF и IL1), нестероидным противовоспалительным лекарственным средствам (NSAID), таким как ингибиторы простагландинсинтазы (например, холин магний салицилат, салицилсалициловая кислота), ингибиторы COX-1 или COX-2) или агонистам глюкокортикоидного рецептора, таким как кортикостероиды, метилпреднизон, преднизон или кортизон.
Как это используется в настоящей заявке, аббревиатуры для любых защитных групп, аминокислот и других соединений, если не указано иное, соответствуют их обычному употреблению или являются известными аббревиатурами, включая аббревиатуры, которые можно найти в J. Org. Chem. 2007 72(1): 23A-24A, или аббревиатуры, установленные IUPAC-IUB Commission on Biochemical Nomenclature (см. Biochem. 1972, 77:942-944).
B. Соединения
В настоящей заявке представлены соединения формулы (I) или их фармацевтически приемлемые соли, сольваты или гидраты, где
R1 и R2 выбраны из (i), (ii), (iii), (iv) и (v) и имеют следующие значения:
(i) R1 и R2 вместе образуют =О, =S, =NR9 или =CR10R11;
(ii) R1 и R2 оба представляют собой -OR8, или R1 и R2, вместе с атомом углерода, с которым они связаны, образуют диоксациклоалкил;
(iii) R1 представляет собой водород или галоген; и R2 представляет собой галоген;
(iv) R1 представляет собой алкил, алкенил, алкинил, циклоалкил или арил, где алкил, алкенил, алкинил, циклоалкил или арил необязательно замещен одним или несколькими заместителями, выбранными из галогена, алкила, -RXORW, -RxS(O)qRv, -RxNRyRz и -C(O)ORW; и R2 представляет собой водород, галоген или -OR8; и
(v) R1 представляет собой галоген, -OR12, -NR13R14 или -S(O)qR15; и R2 представляет собой водород, алкил, алкенил, алкинил, циклоалкил или арил, где алкил, алкенил, алкинил, циклоалкил или арил необязательно замещен одним или несколькими заместителями, выбранными из галогена, алкила, -RXORW, -RxS(O)qRv и -RxNRyRz;
R3 представляет собой водород, галоген, алкил, циано, галогеналкил, циклоалкил, циклоалкилалкил, гидрокси или алкокси;
R4 и R5, каждый независимо, представляет собой водород или алкил; каждый R6 независимо выбран из галогена, алкила, алкенила, алкинила, галогеналкила, циклоалкила, -RXOR18 и -RXNR19R20; каждый R7 независимо представляет собой галоген, алкил, галогеналкил или -RXORW;
R8 представляет собой алкил, алкенил или алкинил;
R9 представляет собой водород, алкил, галогеналкил, гидрокси, алкокси или амино;
R10 представляет собой водород или алкил;
R11 представляет собой водород, алкил, галогеналкил или -C(O)OR8;
R12 выбран из водорода, алкила, алкенила, алкинила, циклоалкила, циклоалкилалкила, гетероциклила, гетероциклилалкила, арила, аралкила, гетероарила, гетероаралкила, -C(O)RV, -C(O)ORW и -C(О)NRyRz, где алкил, алкенил, алкинил, циклоалкил, циклоалкилалкил, гетероциклил, гетероциклилалкил, арил, аралкил, гетероарил и гетероаралкил, каждый, необязательно замещен одним или несколькими заместителями, независимо выбранными из галогена, оксо, алкила, гидрокси, алкокси, амино и алкилтио;
R13 и R14 выбраны следующим образом:
(i) R13 представляет собой водород или алкил; и R14 выбран из водорода, алкила, алкенила, алкинила, циклоалкила, циклоалкилалкила, гетероциклила, гетероциклилалкила, арила, аралкила, гетероарила, гетероаралкила, алкокси, -C(O)RV, -C(O)ORW, -C(О)NRyRz и -S(O)qRv, где алкил, алкенил, алкинил, циклоалкил, циклоалкилалкил, гетероциклил, гетероциклилалкил, арил, аралкил, гетероарил или гетероаралкил, каждый, необязательно замещен одним или несколькими заместителями, независимо выбранными из галогена, оксо, алкила, гидрокси, алкокси, амино и алкилтио; или
(ii) R13 и R14, вместе с атомом азота, с которым они связаны, образуют гетероциклил или гетероарил, где гетероциклил или гетероарил необязательно замещен одним или несколькими заместителями, независимо выбранными из галогена, алкила, гидрокси, алкокси, амино и алкилтио, и где гетероциклил также необязательно замещен группой оксо;
R15 представляет собой алкил, алкенил, алкинил, циклоалкил, циклоалкилалкил, гетероциклил, гетероциклилалкил, арил, аралкил, гетероарил, гетероаралкил, -C(О)NRyRz или -NRyRz, где алкил, алкенил, алкинил, циклоалкил, циклоалкилалкил, гетероциклил, гетероциклилалкил, арил, аралкил, гетероарил или гетероаралкил, каждый, необязательно замещен одним или несколькими заместителями, независимо выбранными из галогена, оксо, алкила, гидрокси, алкокси, амино и алкилтио;
R18 представляет собой водород, алкил, галогеналкил, гидроксиС2-6алкил, алкенил, алкинил, циклоалкил, циклоалкилалкил, гетероциклил, гетероциклилалкил, арил, аралкил, гетероарил или гетероарилалкил; где R18 необязательно замещен 1-3 группами Q1, каждый Q1 независимо выбран из алкила, гидроксила, галогена, галогеналкила, алкокси, арилокси, алкоксиалкила, алкоксикарбонила, карбоксила, циклоалкила, гетероциклила, арила, гетероарила, галогенарила и амино;
R19 и R20 выбраны следующим образом:
(i) R19 и R20, каждый независимо, представляет собой водород или алкил; или
(ii) R19 и R20, вместе с атомом азота, с которым они связаны, образуют гетероциклил или гетероарил, который необязательно замещен 1-2 группами, каждая из которых независимо выбрана из галогена, алкила, галогеналкила, гидроксила и алкокси;
каждый Rx независимо представляет собой алкилен или простую связь;
Rv представляет собой алкил, алкенил или алкинил;
Rw независимо представляет собой водород, алкил, алкенил, алкинил или галогеналкил;
Ry и Rz выбраны следующим образом:
(i) Ry и Rz, каждый независимо, представляет собой водород, алкил, алкенил, алкинил, циклоалкил или галогеналкил;
(ii) Ry и Rz, вместе с атомом азота, с которым они связаны, образуют гетероциклил или гетероарил, который необязательно замещен 1-2 группами, каждая из которых независимо выбрана из галогена, алкила, галогеналкила, гидроксила и алкокси;
n имеет значение 0-4;
p имеет значение 0-5; и
каждый q, независимо, имеет значение 0, 1 или 2.
В некоторых вариантах воплощения в настоящей заявке представлены соединения формулы (II)
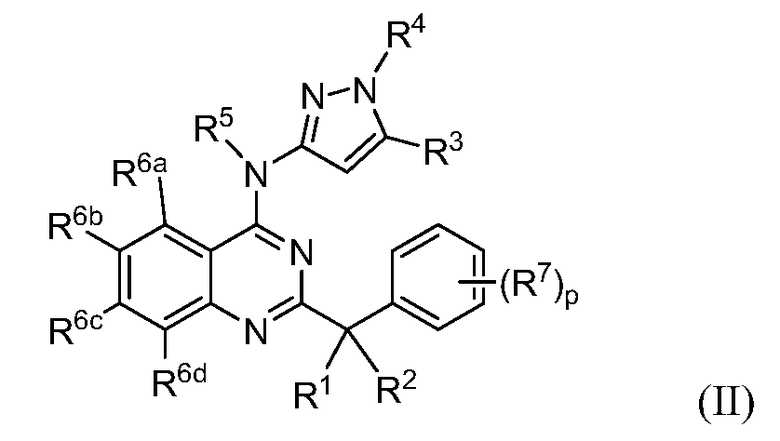
или их фармацевтически приемлемые соли, сольваты или гидраты, где R1 и R2 выбраны из (i), (ii), (iii), (iv) и (v) и имеют следующие значения:
(i) R1 и R2 вместе образуют =О, =S, =NR9 или =CR10R11;
(ii) R1 и R2 оба представляют собой -OR8, или R1 и R2, вместе с атомом углерода, с которым они связаны, образуют диоксациклоалкил;
(iii) R1 представляет собой водород или галоген, и R2 представляет собой галоген;
(iv) R1 представляет собой алкил, алкенил, алкинил, циклоалкил или арил, где алкил, алкенил, алкинил, циклоалкил или арил необязательно замещен одним или несколькими заместителями, выбранными из галогена, алкила, -RXORW, -RxS(O)qRv и -RxNRyRz, и R2 представляет собой водород, галоген и -OR8; и
(v) R1 представляет собой галоген, -OR12, -NR13R14, -S(O)qR15 или -R17C(O)OR12, и R2 представляет собой водород, алкил, алкенил, алкинил, циклоалкил или арил, где алкил, алкенил, алкинил, циклоалкил или арил необязательно замещен одним или несколькими заместителями, выбранными из галогена, алкила, -RXORW, -RxS(O)qRv и -RxNRyRz;
R3 представляет собой водород, алкил или циклоалкил,
R4 и R5, каждый независимо, представляет собой водород или алкил;
R6a, R6b, R6c и R6d, каждый независимо, выбраны из водорода, галогена, алкила, галогеналкила, RxS(O)qRv и -RXOR18; каждый R7 независимо представляет собой галоген, алкил, галогеналкил или -RXORW;
R8 представляет собой алкил, алкенил или алкинил;
R9 представляет собой водород, алкил, галогеналкил, гидрокси, алкокси или амино;
R10 представляет собой водород или алкил;
R11 представляет собой водород, алкил, галогеналкил или -C(O)OR8; каждый R12 независимо представляет собой водород, алкил, галогеналкил, гидроксиалкил, алкоксиалкил, аминоалкил, тиоалкил, гетероциклилалкил или -C(О)NRyRz;
R13 и R14 выбраны следующим образом:
(i) R13 представляет собой водород или алкил, и R14 выбран из водорода, алкила, галогеналкила, гидроксиалкила, алкоксиалкила, аминоалкила, тиоалкила, гетероциклилалкила, -C(O)RV, -C(O)ORW, -C(O)NRyRz и -S(O)qRv; или
(ii) R13 и R14, вместе с атомом азота, с которым они связаны, образуют гетероциклил, необязательно замещеннный одним или несколькими заместителями, независимо выбранными из галогена, оксо, алкила, гидрокси, алкокси, амино и алкилтио;
R15 выбран из водорода, алкила, галогеналкила, гидроксиалкила, алкоксиалкила, аминоалкила, тиоалкила, гетероциклилалкила, -C(О)NRyRz или -NRyRz;
R18 представляет собой водород, алкил, галогеналкил, гидроксиалкил, алкенил, алкинил, циклоалкил, циклоалкилалкил, гетероциклил, гетероциклилалкил, арил, аралкил, гетероарил или гетероарилалкил; где R18 необязательно замещен 1-3 группами Q1, каждый Q1 независимо выбран из алкила, гидроксила, галогена, галогеналкила, алкокси, арилокси, алкоксиалкила, алкоксикарбонила, карбоксила, циклоалкила, гетероциклила, арила, гетероарила, галогенарила и амино;
Rv представляет собой водород, алкил, алкенил или алкинил;
каждый Rx независимо представляет собой алкилен или простую связь;
Rw независимо представляет собой водород или алкил;
Ry и Rz выбраны следующим образом:
(i) Ry и Rz, каждый независимо, представляет собой водород, алкил, алкенил, алкинил, циклоалкил или галогеналкил;
(ii) Ry и Rz, вместе с атомом азота, с которым они связаны, образуют гетероциклил или гетероарил, который необязательно замещен 1-2 группами, каждая из которых независимо выбрана из галогена, алкила, галогеналкила, гидроксила и алкокси; и каждый q, независимо, имеет значение 0, 1 или 2.
В некоторых вариантах воплощения в настоящей заявке представлены соединения формулы (III) или (IIIa)
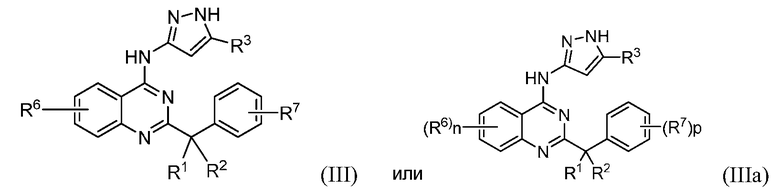
их фармацевтически приемлемые соли, сольваты или гидраты, где
R3 представляет собой водород, алкил, галогеналкил или циклоалкил;
каждый R6 независимо выбран из галогена, алкила, галогеналкила, -RxS(O)qRv и -RXOR18;
каждый R7 независимо представляет собой галоген, алкил, галогеналкил или -RXORW;
p имеет значение 1 или 2;
и другие переменные имеют значение, определенное в настоящей заявке.
В некоторых вариантах воплощения, в настоящей заявке представлены соединения формулы (III), (IIIa) или их фармацевтически приемлемые соли, сольваты или гидраты, где
R3 представляет собой водород, алкил, галогеналкил или циклоалкил;
каждый R6 независимо выбран из галогена, алкила, галогеналкила, -RxS(O)qRv и -RXOR18;
каждый R7 независимо представляет собой галоген, алкил, галогеналкил или -RXORW;
и другие переменные имеют значение, определенное в настоящей заявке.
В некоторых вариантах воплощения в настоящей заявке представлены соединения формулы (III), (IIIa) или их фармацевтически приемлемые соли, сольваты или гидраты, где
R3 представляет собой водород или алкил или циклоалкил;
каждый R6 независимо выбран из галогена, алкила, галогеналкила и -RXOR18;
каждый R7 независимо представляет собой галоген, алкил, галогеналкил или -RXORW;
и другие переменные имеют значение, определенное в настоящей заявке.
В некоторых вариантах воплощения в настоящей заявке представлены соединения формулы (III), (IIIa) или их фармацевтически приемлемые соли, сольваты или гидраты, где
R3 представляет собой водород или алкил;
каждый R6 независимо выбран из галогена, алкила, алкенила, алкинила, галогеналкила, циклоалкила, -RXOR18, -RxS(O)qRv и -RXNR19R20;
каждый R7 независимо представляет собой галоген, алкил, галогеналкил или -RXORW;
p имеет значение 1;
и другие переменные имеют значение, определенное в настоящей заявке.
В некоторых вариантах воплощения в настоящей заявке представлены соединения формулы (III), (IIIa) или их фармацевтически приемлемые соли, сольваты или гидраты, где
R3 представляет собой водород или алкил;
каждый R6 независимо выбран из галогена, алкила, галогеналкила и -RXOR18;
каждый R7 независимо представляет собой галоген, алкил, галогеналкил или -RXORW;
и другие переменные имеют значение, определенное в настоящей заявке.
В некоторых вариантах воплощения в настоящей заявке представлены соединения формулы (III), (IIIa) или их фармацевтически приемлемые соли, сольваты или гидраты, где
R3 представляет собой водород, алкил или галогеналкил;
каждый R6 независимо выбран из галогена, алкила, галогеналкила, -RxS(O)qRv и -RXOR18;
каждый R7 независимо представляет собой галоген, алкил, галогеналкил или -RXORW;
и другие переменные имеют значение, определенное в настоящей заявке.
В некоторых вариантах воплощения в настоящей заявке представлены соединения формулы (III), (IIIa) или их фармацевтически приемлемые соли, сольваты или гидраты, где
R3 представляет собой водород, алкил, циклоалкил, гидроксил или алкокси;
каждый R6 независимо выбран из галогена, алкила, галогеналкила и -RXOR18;
каждый R7 независимо представляет собой галоген, алкил, галогеналкил или -RXORW;
и другие переменные имеют значение, определенное в настоящей заявке.
В некоторых вариантах воплощения в настоящей заявке представлены соединения формулы (III), (IIIa) или их фармацевтически приемлемые соли, сольваты или гидраты, где
R3 представляет собой водород, алкил, циклоалкил, гидроксил или алкокси;
каждый R6 независимо выбран из галогена, алкила, алкенила, алкинила, галогеналкила, циклоалкила, -RXOR18 и -RXNR19R20;
каждый R7 независимо представляет собой галоген, алкил, галогеналкил или -RXORW;
и другие переменные имеют значение, определенное в настоящей заявке.
В одном варианте воплощения, R1 и R2 выбраны из (i), (ii), (iii), (iv) и (v) и имеют следующие значения:
(i) R1 и R2 вместе образуют =О, =S, =NR9 или =CR10R11;
(ii) R1 и R2 оба представляют собой алкокси, или R1 и R2, вместе с атомом углерода, с которым они связаны, образуют диоксациклоалкил;
(iii) R1 представляет собой водород или галоген; и R2 представляет собой галоген; и
(iv) R1 представляет собой алкил, алкенил, алкинил или циклоалкил, где алкил, алкенил, алкинил или циклоалкил замещен одним или несколькими, в одном варианте воплощения одним-четырьмя, в одном варианте воплощения одним-тремя, в одном варианте воплощения одним, двумя или тремя заместителями, выбранными из галогена, циано, алкила, -RXORW, -RxS(O)qRv, -RxNRyRz и -C(O)ORW; и R2 представляет собой водород, галоген или гидрокси; и
(v) R1 представляет собой галоген, дейтеро, гидрокси или амино; и R2 представляет собой водород, дейтеро, алкил, алкенил, алкинил или циклоалкил, где алкил, алкенил, алкинил или циклоалкил, необязательно замещен одним или несколькими, в одном варианте воплощения одним-четырьмя, в одном варианте воплощения одним-тремя, в одном варианте воплощения одним, двумя или тремя заместителями, выбранными из галогена, циано, алкила, -RXORW, -RxS(O)qRv и -RxNRyRz;
и другие переменные имеют значение, определенное в настоящей заявке.
В другом варианте воплощения, R1 и R2 выбраны из (i), (ii), (iii), (iv) и (v) и имеют следующие значения:
(i) R1 и R2 вместе образуют =О;
(ii) R1 и R2 оба представляют собой алкокси, или R1 и R2, вместе с атомом углерода, с которым они связаны, образуют диоксациклоалкил;
(iii) R1 представляет собой водород или галоген; и R2 представляет собой галоген; и
(iv) R1 представляет собой алкил, алкенил, алкинил или циклоалкил, где алкил, алкенил, алкинил или циклоалкил замещен одним или несколькими, в одном варианте воплощения одним-четырьмя, в одном варианте воплощения одним-тремя, в одном варианте воплощения одним, двумя или тремя заместителями, выбранными из галогена, циано, алкила, -RXORW, -RxS(O)qRv, -RxNRyRz и -C(O)ORW; и R2 представляет собой водород, галоген или гидрокси; и
(v) R1 представляет собой галоген, дейтеро, гидрокси или амино; и R2 представляет собой водород, дейтеро, алкил, алкенил, алкинил или циклоалкил, где алкил, алкенил, алкинил или циклоалкил, необязательно замещен одним или несколькими, в одном варианте воплощения одним-четырьмя, в одном варианте воплощения одним-тремя, в одном варианте воплощения одним, двумя или тремя заместителями, выбранными из галогена, циано, алкила, -RXORW, -RxS(O)qRv и -RxNRyRz;
и другие переменные имеют значение, определенное в настоящей заявке.
В одном варианте воплощения, R1 и R2 выбраны из (i), (ii), (iii), (iv) и (v) и имеют следующие значения:
(i) R1 и R2 вместе образуют =О, =S, =NR9 или =CR10R11;
(ii) R1 и R2 оба представляют собой алкокси, или R1 и R2, вместе с атомом углерода, с которым они связаны, образуют диоксациклоалкил;
(iii) R1 представляет собой водород или галоген и R2 представляет собой галоген;
(iv) R1 представляет собой алкил, алкенил, алкинил, циклоалкил или арил и R2 представляет собой водород, галоген гидрокси и алкокси; и
(v) R1 представляет собой дейтеро, гидроксил, алкокси, амино, алкоксикарбониламино или -NHC(O)H и R2 представляет собой водород, дейтеро, алкил, арил или галогенарил.
В одном варианте воплощения, R1 и R2 выбраны из (i), (ii), (iii), (iv) и (v) и имеют следующие значения:
(i) R1 и R2 вместе образуют =О, =S, =NR9 или =CR10R11;
(ii) R1 и R2 оба представляют собой алкокси, или R1 и R2, вместе с атомом углерода, с которым они связаны, образуют диоксациклоалкил;
(iii) R1 представляет собой водород или галоген, и R2 представляет собой галоген;
(iv) R1 представляет собой алкил, алкенил, алкинил, циклоалкил или арил, и R2 представляет собой водород, галоген гидрокси и алкокси; и
(v) R1 представляет собой гидроксил, алкокси, амино или алкоксикарбониламино, и R2 представляет собой водород, алкил, арил или галогенарил.
В одном варианте воплощения, R1 и R2 выбраны из (i), (ii), (iii), (iv) и (v) и имеют следующие значения:
(i) R1 и R2 вместе образуют =О, =S, =NR9 или =CR10R11;
(ii) R1 и R2 оба представляют собой алкокси, или R1 и R2, вместе с атомом углерода, с которым они связаны, образуют диоксациклоалкил;
(iii) R1 представляет собой водород или галоген, и R2 представляет собой галоген;
(iv) R1 представляет собой алкил, цианоалкил, алкенил, алкинил, циклоалкил или арил, и R2 представляет собой водород, галоген гидрокси и алкокси; и
(v) R1 представляет собой гидроксил, алкокси, амино или алкоксикарбониламино, и R2 представляет собой водород, алкил, арил или галогенарил.
В одном варианте воплощения, R1 и R2 выбраны из (i), (ii), (iii) и (iv) и имеют следующие значения:
(i) R1 и R2 вместе образуют =О;
(ii) R1 и R2 оба представляют собой алкокси, или R1 и R2 вместе с атомом углерода, с которым они связаны, образуют диоксациклоалкил;
(iii) R1 представляет собой водород или галоген, и R2 представляет собой галоген; и
(iv) R1 представляет собой гидроксил, алкокси, амино или алкоксикарбониламино, и R2 представляет собой водород, алкил, арил или галогенарил.
В одном варианте воплощения, R1 и R2 вместе образуют =О.
В одном варианте воплощения, R1 представляет собой водород, галоген или дейтеро, и R2 представляет собой галоген или дейтеро.
В одном варианте воплощения, R1 и R2 выбраны из (i), (ii), (iii) и (iv) и имеют следующие значения:
(i) R1 и R2 вместе образуют =О;
(ii) R1 и R2, вместе с атомом углерода, с которым они связаны, образуют диоксациклоалкил;
(iii) R1 представляет собой водород или галоген; и R2 представляет собой галоген; и
(iv) R1 представляет собой галоген, дейтеро или гидроксил, и R2 представляет собой водород или дейтеро;
где другие переменные имеют значение, определенное в настоящей заявке.
В одном варианте воплощения, R1 и R2 выбраны из (i) и (ii) и имеют следующие значения:
(i) R1 и R2 оба представляют собой алкокси, или R1 и R2, вместе образуют =О; и
(ii) R1 представляет собой гидроксил, -OR12 или -NR13R14; и R2 представляет собой водород, алкил, арил или галогенарил;
R12 выбран из водорода, алкила, алкенила, алкинила, циклоалкила, циклоалкилалкила, гетероциклила, гетероциклилалкила, арила, аралкила, гетероарила, гетероаралкила, -C(O)RV, -C(O)ORW и -C(O)NRyRz, где алкил, алкенил, алкинил, циклоалкил, циклоалкилалкил, гетероциклил, гетероциклилалкил, арил, аралкил, гетероарил и гетероаралкил, каждый, необязательно замещен одним или несколькими заместителями, независимо выбранными из галогена, оксо, алкила, гидрокси, алкокси, амино и алкилтио;
R13 и R14 выбраны следующим образом:
(i) R13 представляет собой водород или алкил, и R14 выбран из водорода, алкила, алкенила, алкинила, циклоалкила, циклоалкилалкила, гетероциклила, гетероциклилалкила, арила, аралкила, гетероарила, гетероаралкила, алкокси, -C(O)RV, -C(O)ORW, -C(O)NRyRz и -S(O)qRv, где алкил, алкенил, алкинил, циклоалкил, циклоалкилалкил, гетероциклил, гетероциклилалкил, арил, аралкил, гетероарил или гетероаралкил, каждый, необязательно замещен одним или несколькими заместителями, независимо выбранными из галогена, оксо, алкила, гидрокси, алкокси, амино и алкилтио; или
(ii) R13 и R14, вместе с атомом азота, с которым они связаны, образуют гетероциклил или гетероарил, где гетероциклил или гетероарил необязательно замещен одним или несколькими заместителями, независимо выбранными из галогена, алкила, гидрокси, алкокси, амино и алкилтио, и где гетероциклил также необязательно замещен группой оксо;
Rv представляет собой водород, алкил, алкенил или алкинил;
Rw независимо представляет собой водород, алкил, алкенил, алкинил или галогеналкил;
Ry и Rz выбраны следующим образом:
(i) Ry и Rz, каждый независимо, представляет собой водород, алкил, алкенил, алкинил, циклоалкил или галогеналкил;
(ii) Ry и Rz, вместе с атомом азота, с которым они связаны, образуют гетероциклил или гетероарил, который необязательно замещен 1-2 группами, каждая из которых независимо выбрана из галогена, алкила, галогеналкила, гидроксила и алкокси; и каждый q, независимо, имеет значение 0, 1 или 2.
В одном варианте воплощения, R1 и R2 выбраны из (i) и (ii) и имеют следующие значения:
(i) R1 и R2 оба представляют собой алкокси, или R1 и R2, вместе образуют =О; и
(ii) R1 представляет собой гидроксил, -OR12 или -NR13R14; и R2 представляет собой водород, алкил, арил или галогенарил;
R12 выбран из водорода, алкила, алкенила, алкинила, циклоалкила, циклоалкилалкила, гетероциклила, гетероциклилалкила, арила, аралкила, гетероарила, гетероаралкила, -C(O)RV, -C(O)ORW и -C(O)NRyRz, где алкил, алкенил, алкинил, циклоалкил, циклоалкилалкил, гетероциклил, гетероциклилалкил, арил, аралкил, гетероарил или гетероаралкил, каждый, необязательно замещен одним или несколькими заместителями, независимо выбранными из галогена, оксо, алкила, гидрокси, алкокси, амино и алкилтио;
R13 и R14 выбраны следующим образом:
(i) R13 представляет собой водород или алкил, и R14 выбран из водорода, алкила, алкенила, алкинила, циклоалкила, циклоалкилалкила, гетероциклила, гетероциклилалкила, арила, аралкила, гетероарила, гетероаралкила, алкокси, -C(O)RV, -C(O)ORW, -C(O)NRyRz и -S(O)qRv, где алкил, алкенил, алкинил, циклоалкил, циклоалкилалкил, гетероциклил, гетероциклилалкил, арил, аралкил, гетероарил или гетероаралкил, каждый, необязательно замещен одним или несколькими заместителями, независимо выбранными из галогена, оксо, алкила, гидрокси, алкокси, амино и алкилтио; или
(ii) R13 и R14, вместе с атомом азота, с которым они связаны, образуют гетероциклил или гетероарил, где гетероциклил или гетероарил необязательно замещен одним или несколькими заместителями, независимо выбранными из галогена, алкила, гидрокси, алкокси, амино и алкилтио, и где гетероциклил также необязательно замещен группой оксо;
Rv представляет собой алкил, алкенил или алкинил;
Rw независимо представляет собой водород, алкил, алкенил, алкинил или галогеналкил;
Ry и Rz выбраны следующим образом:
(i) Ry и Rz, каждый независимо, представляет собой водород, алкил, алкенил, алкинил, циклоалкил или галогеналкил;
(ii) Ry и Rz, вместе с атомом азота, с которым они связаны, образуют гетероциклил или гетероарил, который необязательно замещен 1-2 группами, каждая из которых независимо выбрана из галогена, алкила, галогеналкила, гидроксила и алкокси; и каждый q, независимо, имеет значение 0, 1 или 2.
В другом варианте воплощения, R12 представляет собой водород или алкил; R13 представляет собой водород или алкил, и R14 представляет собой алкил, циклоалкил, -C(O)Rv или -C(O)ORW, где Rv и Rw, каждый независимо, представляет собой водород или алкил.
В другом варианте воплощения, R12 представляет собой водород или алкил; R13 представляет собой водород или алкил, и R14 представляет собой алкил, циклоалкил или -C(O)ORW, где Rv и Rw, каждый независимо, представляет собой водород или алкил.
В одном варианте воплощения, R1 и R2 выбраны из (i), (ii) и (iii) и имеют следующие значения:
(i) R1 и R2 вместе образуют =О;
(ii) R1 и R2 оба представляют собой алкокси; и
(iii) R1 представляет собой гидрокси или алкокси, и R2 представляет собой водород.
В одном варианте воплощения, R1 и R2 выбраны из (i) и (ii) и имеют следующие значения:
(i) R1 и R2 вместе образуют =О; и
(ii) R1 представляет собой гидрокси или алкокси, и R2 представляет собой водород.
В одном варианте воплощения, R1 представляет собой -OR12 или -NR13R14, и R2 представляет собой водород, где R12 выбран из водорода, алкила, алкенила, алкинила, циклоалкила, циклоалкилалкила, гетероциклила, гетероциклилалкила, арила, аралкила, гетероарила, гетероаралкила, -C(O)RV, -C(O)ORW и -C(O)NRyRz, где алкил, алкенил, алкинил, циклоалкил, циклоалкилалкил, гетероциклил, гетероциклилалкил, арил, аралкил, гетероарил и гетероаралкил, каждый, необязательно замещен одним или несколькими заместителями, независимо выбранными из галогена, оксо, алкила, гидрокси, алкокси, амино и алкилтио;
R13 и R14 выбраны следующим образом:
(i) R13 представляет собой водород или алкил, и R14 выбран из водорода, алкила, алкенила, алкинила, циклоалкила, циклоалкилалкила, гетероциклила, гетероциклилалкила, арила, аралкила, гетероарила, гетероаралкила, алкокси, -C(O)RV, -C(O)ORW, -C(O)NRyRz и -S(O)qRv, где алкил, алкенил, алкинил, циклоалкил, циклоалкилалкил, гетероциклил, гетероциклилалкил, арил, аралкил, гетероарил или гетероаралкил, каждый, необязательно замещен одним или несколькими заместителями, независимо выбранными из галогена, оксо, алкила, гидрокси, алкокси, амино и алкилтио; или
(ii) R13 и R14, вместе с атомом азота, с которым они связаны, образуют гетероциклил или гетероарил, где гетероциклил и гетероарил необязательно замещен одним или несколькими заместителями, независимо выбранными из галогена, алкила, гидрокси, алкокси, амино и алкилтио, и где гетероциклил также необязательно замещен группой оксо;
Rv представляет собой алкил, алкенил или алкинил;
Rw независимо представляет собой водород, алкил, алкенил, алкинил или галогеналкил;
Ry и Rz выбраны следующим образом:
(i) Ry и Rz, каждый независимо, представляет собой водород, алкил, алкенил, алкинил, циклоалкил или галогеналкил;
(ii) Ry и Rz, вместе с атомом азота, с которым они связаны, образуют гетероциклил или гетероарил, который необязательно замещен 1-2 группами, каждая из которых независимо выбрана из галогена, алкила, галогеналкила, гидроксила и алкокси; и каждый q, независимо, имеет значение 0, 1 или 2.
В другом варианте воплощения, R12 представляет собой водород или алкил; R13 и R14 выбраны следующим образом:
(i) R13 представляет собой водород или алкил, и R14 представляет собой алкил, циклоалкил или -C(O)ORW; или
(ii) R13 и R14, вместе с атомом азота, с которым они связаны, образуют гетероциклил.
В другом варианте воплощения, R12 представляет собой водород или алкил; R13 представляет собой водород или алкил, и R14 представляет собой алкил, циклоалкил, -C(O)Rv или -C(O)ORW, где Rv и Rw, каждый независимо, представляет собой водород или алкил.
В другом варианте воплощения, R12 представляет собой водород или алкил; R13 представляет собой водород или алкил, и R14 представляет собой алкил, циклоалкил или -C(O)ORW. В одном варианте воплощения, R1 и R2, вместе с атомом углерода, с которым они связаны, образуют диоксациклоалкил.
В одном варианте воплощения, R1 представляет собой водород или галоген, и R2 представляет собой галоген. В одном варианте воплощения, R1 представляет собой водород или фтор, и R2 представляет собой фтор. В одном варианте воплощения, R1 представляет собой фтор, и R2 представляет собой фтор.
В одном варианте воплощения, R1 представляет собой гидроксил, алкокси, амино или алкоксикарбониламино, и R2 представляет собой водород, алкил, арил или галогенарил. В одном варианте воплощения, R1 представляет собой гидроксил или алкокси, и R2 представляет собой водород. В одном варианте воплощения, R1 представляет собой гидроксил, и R2 представляет собой водород. В одном варианте воплощения, R1 представляет собой алкокси, и R2 представляет собой водород. В одном варианте воплощения, R1 представляет собой гидроксил, метокси, амино или метоксикарбониламино, и R2 представляет собой водород, фенил или фторфенил.
В одном варианте воплощения, R3 представляет собой водород, алкил, циклоалкил или алкокси. В другом варианте воплощения, R3 представляет собой водород, алкил или циклоалкил. В одном варианте воплощения, R3 представляет собой водород, алкил или алкокси. В следующем варианте воплощения, R3 представляет собой водород или алкил. В другом варианте воплощения, R3 представляет собой водород или метил. В одном варианте воплощения, R3 представляет собой водород, метил или циклопропил.
В одном варианте воплощения, R3 представляет собой алкил, циклоалкил или циано. В одном варианте воплощения, R3 представляет собой метил, циклопропил или циано. В одном варианте воплощения, R3 представляет собой алкил или циклоалкил. В одном варианте воплощения, R3 представляет собой метил или циклопропил.
В одном варианте воплощения, каждый R6 независимо выбран из галогена, алкила, алкенила, алкинила, галогеналкила, циклоалкила и -OR18, где R18 представляет собой водород, алкил, галогеналкил, гидроксиалкил, алкенил, алкинил, циклоалкил, циклоалкилалкил, гетероциклил или гетероциклилалкил; где R18 необязательно замещен 1-3 группами Q1, каждый Q1 независимо выбран из алкила, гидроксила, циано, галогена, галогеналкила, алкокси, арилокси, алкоксиалкила, алкоксикарбонила, карбоксила, циклоалкила, гетероциклила, арила, гетероарила, галогенарила и амино.
В одном варианте воплощения, каждый R6 независимо выбран из галогена, алкила, алкенила, алкинила, галогеналкила, гидроксиалкила; циклоалкила, -RxS(O)qRv и -OR18, где Rx представляет собой простую связь или алкилен; Rv представляет собой водород или алкил; q имеет значение 1 или 2; R18 представляет собой водород, алкил, галогеналкил, гидроксиалкил, алкенил, алкинил, циклоалкил, циклоалкилалкил, гетероциклил или гетероциклилалкил; где R18 необязательно замещен 1-3 группами Q1, каждый Q1 независимо выбран из алкила, гидроксила, галогена, галогеналкила, алкокси, арилокси, алкоксиалкила, алкоксикарбонила, карбоксила, циклоалкила, гетероциклила, арила, гетероарила, галогенарила и амино.
В одном варианте воплощения, каждый R6 независимо выбран из галогена, алкила, галогеналкила и -OR18; где R18 представляет собой водород, алкил, гидроксиалкил, алкоксиалкил, гетероциклилалкил или гетероциклил, где R18 необязательно замещен 1-3 группами Q1, каждый Q1 независимо выбран из алкила, гидроксила, галогена, галогеналкила, алкокси, арилокси, алкоксиалкила, алкоксикарбонила, карбоксила, циклоалкила, гетероциклила, арила, гетероарила, галогенарила и амино. В одном варианте воплощения, R18 представляет собой водород, алкил, галогеналкил, гидроксиС2-6алкил, алкенил, алкинил, циклоалкил, циклоалкилалкил, гетероциклил, гетероциклилалкил, арил, аралкил, гетероарил или гетероарилалкил; где R18 необязательно замещен 1-3 группами Q1, каждый Q1 независимо выбран из алкила, гидроксила, галогена, галогеналкила, алкокси, арилокси, алкоксиалкила, алкоксикарбонила, карбоксила, циклоалкила, гетероциклила, арила, гетероарила, галогенарила и амино.
В одном варианте воплощения, каждый R6 независимо выбран из галогена, алкила, галогеналкил и -RXOR18; где R18 представляет собой водород, алкил, галогеналкил, гидроксиалкил или гетероциклил; где R18 необязательно замещен группой Q1, где Q1 выбран из гидроксила, циано, алкокси, алкоксикарбонила, карбоксила, гетероциклила и амино. В одном варианте воплощения, R18 представляет собой водород или алкил. В другом варианте воплощения, R18 представляет собой водород или метил.
В одном варианте воплощения, каждый R6 независимо выбран из водорода, алкила, галогена, гидрокси или алкокси. В одном варианте воплощения, каждый R6 независимо выбран из фтора, иода, метила, трифторметила и -OR18; где R18 представляет собой водород, метил, гидроксиэтил, гидроксипропил, морфолиноэтил, метоксиэтил, трет-бутилоксикарбонилметил, карбоксиметил или пиперидинил.
В одном варианте воплощения, R6a представляет собой водород или галоген. В одном варианте воплощения, R6b представляет собой водород или алкокси. В одном варианте воплощения, R6c представляет собой водород, галоген, алкил, галогеналкил, -RxOR18, -RxS(O)qRv, где Rx представляет собой простую связь или алкилен; Rv представляет собой водород или алкил; q имеет значение 1 или 2; R18 представляет собой водород, алкил, галогеналкил, гидроксиалкил или гетероциклил; где R18 необязательно замещен группой Q1, где Q1 выбран из гидрокси, алкокси, алкоксикарбонила, карбоксила, гетероциклила и амино. В одном варианте воплощения, R6c представляет собой водород, галоген, алкил, гидрокси или алкокси. В одном варианте воплощения, R6d представляет собой водород или галоген.
В одном варианте воплощения, R6a представляет собой водород или галоген. В одном варианте воплощения, R6b представляет собой водород или алкокси. В одном варианте воплощения, R6c представляет собой водород, галоген, алкил, галогеналкил, -RxOR18; где R18 представляет собой водород, алкил, галогеналкил, гидроксиалкил или гетероциклил; где R18 необязательно замещен группой Q1, где Q1 выбран из гидрокси, алкокси, алкоксикарбонила, карбоксила, гетероциклила и амино. В одном варианте воплощения, R6c представляет собой водород, галоген, алкил, гидрокси или алкокси. В одном варианте воплощения, R6d представляет собой водород или галоген.
В одном варианте воплощения, R6a представляет собой водород или галоген. В одном варианте воплощения, R6a представляет собой водород или фтор. В одном варианте воплощения, R6b представляет собой водород или метокси. В одном варианте воплощения, R6c представляет собой водород, фтор, иод, метил, трифторметил или -OR18; где R18 представляет собой водород, метил, гидроксиэтил, гидроксипропил, морфолиноэтил, метоксиэтил, трет-бутилоксикарбонилметил, карбоксиметил или пиперидинил. В одном варианте воплощения, R6d представляет собой водород или фтор.
В одном варианте воплощения, каждый R7 независимо представляет собой галоген, алкил, галогеналкил или -RXORW, где Rw представляет собой водород или алкил. В одном варианте воплощения, каждый R7 независимо представляет собой фтор или метокси. В одном варианте воплощения, R7 представляет собой галоген. В одном варианте воплощения, R7 представляет собой фтор.
В одном варианте воплощения, Rx представляет собой простую связь. В одном варианте воплощения, n имеет значение 0-4. В одном варианте воплощения, n имеет значение 0, 1, 2 или 3. В одном варианте воплощения, n имеет значение 1. В одном варианте воплощения, n имеет значение 0. В одном варианте воплощения, n имеет значение 2. В одном варианте воплощения, p имеет значение 0, 1 или 2. В одном варианте воплощения, p имеет значение 1 или 2. В одном варианте воплощения, p имеет значение 1.
В некоторых вариантах воплощения в настоящей заявке представлены соединения формулы (III) или (IIIa), где
R1 и R2 выбраны из (i), (ii), (iii) и (iv) и имеют следующие значения:
(i) R1 и R2 вместе образуют =О;
(ii) R1 и R2, оба представляют собой -OR8, или R1 и R2, вместе с атомом углерода, с которым они связаны, образуют диоксациклоалкил;
(iii) R1 представляет собой водород или галоген, и R2 представляет собой галоген; и
(iv) R1 представляет собой гидроксил, алкокси, цианоалкил, амино, алкоксикарбониламино или -NHC(O)H, и R2 представляет собой водород, алкил, арил или галогенарил;
R3 представляет собой водород или алкил; каждый R6 независимо выбран из галогена, алкила, галогеналкила и -RxOR18; где R18 представляет собой водород, алкил, галогеналкил, гидроксиалкил или гетероциклил; где R18 необязательно замещен группой Q1, где Q1 выбран из гидроксила, алкокси, алкоксикарбонила, карбоксила, гетероциклила и амино; каждый R7 независимо представляет собой галоген, алкил, галогеналкил, гидрокси или алкокси; и
R8 представляет собой алкил, алкенил или алкинил.
В некоторых вариантах воплощения в настоящей заявке представлены соединения формулы (III) или (IIIa), где
R1 и R2 выбраны из (i), (ii), (iii) и (iv) и имеют следующие значения:
(i) R1 и R2 вместе образуют =О;
(ii) R1 и R2, оба представляют собой -OR8, или R1 и R2, вместе с атомом углерода, с которым они связаны, образуют диоксациклоалкил;
(iii) R1 представляет собой водород или галоген, и R2 представляет собой галоген; и
(iv) R1 представляет собой гидроксил, алкокси, амино или алкоксикарбониламино, и R2 представляет собой водород, алкил, арил или галогенарил; R3 представляет собой водород или алкил; каждый R6 независимо выбран из галогена, алкила, галогеналкила и -RxOR18; где R18 представляет собой водород, алкил, галогеналкил, гидроксиалкил или гетероциклил; где R18 необязательно замещен группой Q1, Q1 выбран из гидроксила, алкокси, алкоксикарбонила, карбоксила, гетероциклила и амино; каждый R7 независимо представляет собой галоген, алкил, галогеналкил, гидрокси или алкокси; и R8 представляет собой алкил, алкенил или алкинил.
В некоторых вариантах воплощения в настоящей заявке представлены соединения формулы (III) или (IIIa), где
R1 и R2 выбраны из (i), (ii) и (iii) и имеют следующие значения:
(i) R1 и R2 вместе образуют =О;
(ii) R1 представляет собой водород или галоген, и R2 представляет собой галоген; и
(iii) R1 представляет собой гидроксил, алкокси, амино, -NHCH(O) или алкоксикарбониламино, и R2 представляет собой водород или алкил; R3 представляет собой водород или алкил; каждый R6 независимо выбран из галогена, алкила, галогеналкила, -RXOR18 и -RxS(O)qRv, где Rx представляет собой простую связь или алкилен; Rv представляет собой водород или алкил; q имеет значение 2; R18 представляет собой водород, алкил, галогеналкил, гидроксиалкил или гетероциклил; где R18 необязательно замещен группой Q1, Q1 выбран из гидроксила, алкокси, алкоксикарбонила, карбоксила, гетероциклила и амино; R7 представляет собой галоген; и p имеет значение 1.
В некоторых вариантах воплощения в настоящей заявке представлены соединения формулы (III) или (IIIa), где
R1 и R2 выбраны из (i), (ii) и (iii) и имеют следующие значения:
(i) R1 и R2 вместе образуют =О;
(ii) R1 представляет собой водород или галоген, и R2 представляет собой галоген; и
(iii) R1 представляет собой гидроксил, алкокси, амино, -NHCH(O) или алкоксикарбониламино, и R2 представляет собой водород или алкил;
R3 представляет собой водород или алкил;
каждый R6 независимо выбран из галогена, алкила, галогеналкила, -RXOR18 и -RxS(O)qRv, где Rx представляет собой простую связь или алкилен; Rv представляет собой алкил; q имеет значение 2; R18 представляет собой водород, алкил, галогеналкил, гидроксиалкил или гетероциклил; где R18 необязательно замещен группой Q1, где Q1 выбран из гидроксила, алкокси, алкоксикарбонила, карбоксила, гетероциклила и амино;
R7 представляет собой галоген; и
p имеет значение 1.
В некоторых вариантах воплощения в настоящей заявке представлены соединения формулы (IV) или (IVa)
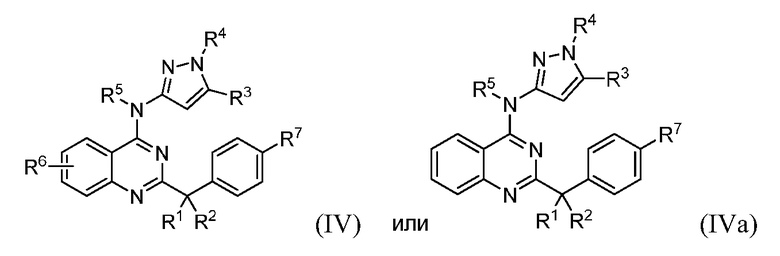
или их фармацевтически приемлемые соли, сольваты или гидраты, где переменные имеют значения, определенные в настоящей заявке. В одном варианте воплощения, R7 представляет собой галоген. В одном варианте воплощения, R7 представляет собой фтор.
В некоторых вариантах воплощения в настоящей заявке представлены соединения формулы (V) или (Va)
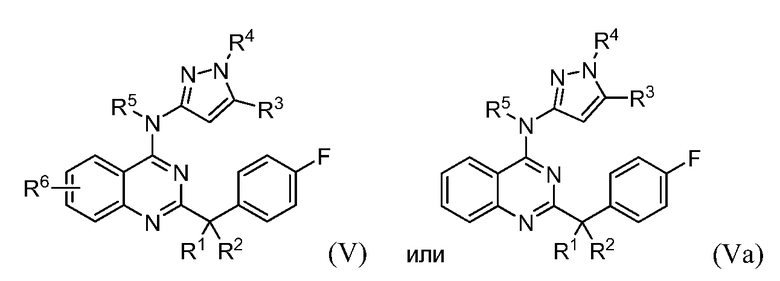
или их фармацевтически приемлемые соли, сольваты или гидраты, где переменные имеют значения, определенные в настоящей заявке. В некоторых вариантах воплощения в настоящей заявке представлены соединения формулы (V) или (Va), где
R1 и R2 выбраны из (i), (ii) и (iii) и имеют следующие значения:
(i) R1 и R2 вместе образуют =О;
(ii) R1 представляет собой водород или галоген, и R2 представляет собой галоген; и
(iii) R1 представляет собой гидроксил, алкокси, амино, -NHCH(O) или алкоксикарбониламино, и R2 представляет собой водород или алкил;
R3 представляет собой водород или алкил; и
R4 представляет собой водород;
R5 представляет собой водород;
R6 выбран из галогена, алкила, галогеналкила, -RXOR18 и -RxS(O)qRv, где Rx представляет собой простую связь или алкилен; Rv представляет собой алкил; q имеет значение 2; R18 представляет собой водород, алкил, галогеналкил, гидроксиалкил или гетероциклил; где R18 необязательно замещен группой Q1, где Q1 выбран из гидроксила, алкокси, алкоксикарбонила, карбоксила, гетероциклила и амино.
В некоторых вариантах воплощения в настоящей заявке представлены соединения формулы (VI)
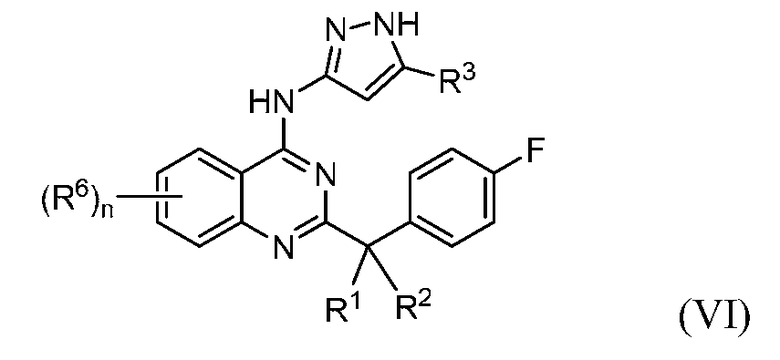
или их фармацевтически приемлемые соли, сольваты или гидраты, где переменные имеют значения, определенные в настоящей заявке. В одном варианте воплощения, R1 представляет собой гидроксил, амино, алкокси или алкоксикарбониламино; R2 представляет собой водород, галоген или галогенарил; каждый R6 независимо выбран из галогена, алкила, галогеналкила, -RxS(O)qRv и -RXOR18; где Rx представляет собой простую связь или алкилен; Rv представляет собой водород или алкил; q имеет значение 1 или 2; R18 представляет собой водород, алкил, галогеналкил, гидроксиалкил или гетероциклил; R18 необязательно замещен группой Q1, выбранной из гидроксила, алкокси, алкоксикарбонила, карбоксила, гетероциклила и амино; n имеет значение 0 или 1; и R3 представляет собой водород или алкил. В одном варианте воплощения, R1 представляет собой гидроксил; и R2 представляет собой водород; n имеет значение 0, и R3 представляет собой алкил.
В некоторых вариантах воплощения в настоящей заявке представлены соединения формулы (VII)
или их фармацевтически приемлемые соли, сольваты или гидраты, где переменные имеют значения, определенные в настоящей заявке. В одном варианте воплощения, каждый R6 независимо выбран из галогена, алкила, галогеналкила, -RxS(O)qRv и -RXOR18; где Rx представляет собой простую связь или алкилен; Rv представляет собой водород или алкил; q имеет значение 1 или 2; где R18 представляет собой водород, алкил, галогеналкил, гидроксиалкил или гетероциклил; R18 необязательно замещен группой Q1, выбранной из гидроксила, алкокси, алкоксикарбонила, карбоксила, гетероциклила и амино; n имеет значение 0 или 1; каждый R7 независимо представляет собой галоген, алкил, галогеналкил, гидрокси или алкокси; p имеет значение 1; и R3 представляет собой водород, алкил или алкокси.
В некоторых вариантах воплощения в настоящей заявке представлены соединения формулы (VII) или их фармацевтически приемлемые соли, сольваты или гидраты, где n имеет значение 0, и другие переменные имеют значение, определенное в настоящей заявке.
В некоторых вариантах воплощения в настоящей заявке представлены соединения формулы (VIII)
или их фармацевтически приемлемые соли, сольваты или гидраты, где переменные имеют значения, определенные в настоящей заявке. В одном варианте воплощения, каждый R6 независимо выбран из галогена, алкила, галогеналкила, -RxS(O)qRv и -RXOR18; где Rx представляет собой простую связь или алкилен; Rv представляет собой водород или алкил; q имеет значение 1 или 2; где R18 представляет собой водород, алкил, галогеналкил, гидроксиалкил или гетероциклил; R18 необязательно замещен группой Q1, выбранной из гидроксила, алкокси, алкоксикарбонила, карбоксила, гетероциклила и амино; n имеет значение 0 или 1; каждый R7 независимо представляет собой галоген, алкил, галогеналкил, гидрокси или алкокси; p имеет значение 1; и R3 представляет собой водород, алкил или циклоалкил.
В некоторых вариантах воплощения в настоящей заявке представлены соединения формулы (IX)
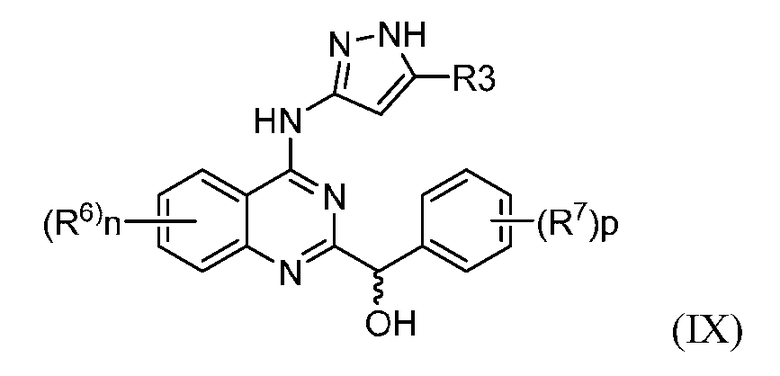
или их фармацевтически приемлемые соли, сольваты или гидраты, где переменные имеют значения, определенные в настоящей заявке. В одном варианте воплощения, каждый R6 независимо выбран из галогена, алкила, галогеналкила, -RxS(O)qRv и -RXOR18; где Rx представляет собой простую связь или алкилен; Rv представляет собой водород или алкил; q имеет значение 1 или 2; где R18 представляет собой водород, алкил, галогеналкил, гидроксиалкил или гетероциклил; R18 необязательно замещен группой Q1, выбранной из гидроксила, алкокси, алкоксикарбонила, карбоксила, гетероциклила и амино; n имеет значение 0 или 1; каждый R7 независимо представляет собой галоген, алкил, галогеналкил, гидрокси или алкокси; p имеет значение 1; и R3 представляет собой водород или алкил. В одном варианте воплощения, в настоящей заявке представлены соединения формулы (IX) или их фармацевтически приемлемые соли, сольваты или гидраты, где R3 представляет собой алкил; R7 представляет собой галоген; n имеет значение 0, и p имеет значение 1.
В некоторых вариантах воплощения в настоящей заявке представлены соединения формулы (X)
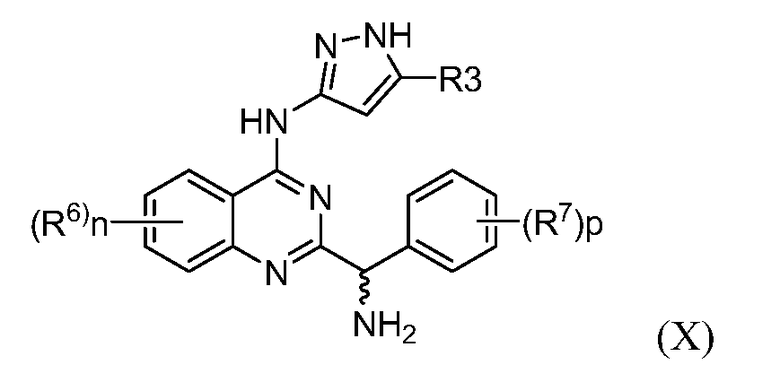
или их фармацевтически приемлемые соли, сольваты или гидраты, где переменные имеют значения, определенные в настоящей заявке. В одном варианте воплощения, каждый R6 независимо выбран из галогена, алкила, галогеналкила, -RxS(O)qRv и -RXOR18; где Rx представляет собой простую связь или алкилен; Rv представляет собой водород или алкил; q имеет значение 1 или 2; где R18 представляет собой водород, алкил, галогеналкил, гидроксиалкил или гетероциклил; R18 необязательно замещен группой Q1, выбранной из из гидроксила, алкокси, алкоксикарбонила, карбоксила, гетероциклила и амино; n имеет значение 0 или 1; каждый R7 независимо представляет собой галоген, алкил, галогеналкил, гидрокси или алкокси; p имеет значение 1; и R3 представляет собой водород или алкил.
В некоторых вариантах воплощения в настоящей заявке представлены соединения формулы (XI)
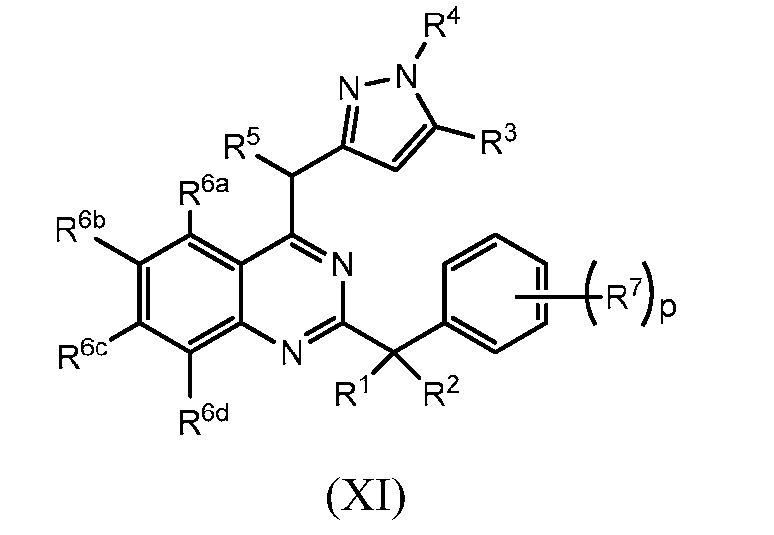
или их фармацевтически приемлемые соли, сольваты или гидраты, где R1 и R2 выбраны из (i), (ii), (iii), (iv) и (v) и имеют следующие значения:
(i) R1 и R2 вместе образуют =О, =S, =NR9 или =CR10R11;
(ii) R1 и R2 оба представляют собой -OR8, или R1 и R2, вместе с атомом углерода, с которым они связаны, образуют диоксациклоалкил;
(iii) R1 представляет собой водород или галоген; и R2 представляет собой галоген; и
(iv) R1 представляет собой алкил, алкенил, алкинил, циклоалкил или арил, где алкил, алкенил, алкинил, циклоалкил или арил необязательно замещен одним или несколькими, в одном варианте воплощения одним-четырьмя, в одном варианте воплощения одним-тремя, в одном варианте воплощения одним, двумя или тремя заместителями, выбранными из галогена, циано, алкила, -RXORW, -RxS(O)qRv, -RxNRyRz и -C(O)ORW; и R2 представляет собой водород, галоген или -OR8; и
(v) R1 представляет собой галоген, дейтеро, -OR12, -NR13R14 или -S(O)qR15; и R2 представляет собой водород, дейтеро, алкил, алкенил, алкинил или циклоалкил, где алкил, алкенил, алкинил или циклоалкил необязательно замещен одним или несколькими, в одном варианте воплощения одним-четырьмя, в одном варианте воплощения одним-тремя, в одном варианте воплощения одним, двумя или тремя заместителями, выбранными из галогена, циано, алкила, -RXORW, -RxS(O)qRv и -RxNRyRz;
R3 представляет собой галоген, алкил, галогеналкил, гидрокси или алкокси;
R4 и R5, каждый независимо, представляет собой водород или алкил;
R6a, R6b, R6c и R6d, каждый независимо, выбраны из водорода, галогена, алкила, алкенила, алкинила, галогеналкила, циклоалкила, -RXOR18, -RXNR19R20 и -RxS(O)qRv; каждый R7 независимо представляет собой галоген, алкил, галогеналкил или -RXORW;
R8 представляет собой алкил, алкенил или алкинил;
R9 представляет собой водород, алкил, галогеналкил, гидрокси, алкокси или амино;
R10 представляет собой водород или алкил;
R11 представляет собой водород, алкил, галогеналкил или -C(O)OR8;
R12 выбран из водорода, алкила, алкенила, алкинила, циклоалкила, циклоалкилалкила, гетероциклила, гетероциклилалкила, арила, аралкила, гетероарила, гетероаралкила, -C(O)RV, -C(O)ORW и -C(О)NRyRz, где алкил, алкенил, алкинил, циклоалкил, циклоалкилалкил, гетероциклил, гетероциклилалкил, арил, аралкил, гетероарил и гетероаралкил, каждый, необязательно замещен одним или несколькими, в одном варианте воплощения одним-четырьмя, в одном варианте воплощения одним-тремя, в одном варианте воплощения одним, двумя или тремя заместителями, независимо выбранными из галогена, оксо, алкила, гидрокси, алкокси, амино и алкилтио;
R13 и R14 выбраны следующим образом:
(i) R13 представляет собой водород или алкил; и R14 выбран из водорода, алкила, алкенила, алкинила, циклоалкила, циклоалкилалкила, гетероциклила, гетероциклилалкила, арила, аралкила, гетероарила, гетероаралкила, алкокси, -C(O)RV, -C(O)ORW, -C(O)NRyRz и -S(O)qRv, где алкил, алкенил, алкинил, циклоалкил, циклоалкилалкил, гетероциклил, гетероциклилалкил, арил, аралкил, гетероарил и гетероаралкил, каждый, необязательно замещен одним или несколькими, в одном варианте воплощения одним-четырьмя, в одном варианте воплощения одним-тремя, в одном варианте воплощения одним, двумя или тремя заместителями, независимо выбранными из галогена, оксо, алкила, гидрокси, алкокси, амино и алкилтио; или
(ii) R13 и R14, вместе с атомом азота, с которым они связаны, образуют гетероциклил или гетероарил, где гетероциклил или гетероарил необязательно замещен одним или несколькими, в одном варианте воплощения одним-четырьмя, в одном варианте воплощения одним-тремя, в одном варианте воплощения одним, двумя или тремя заместителями, независимо выбранными из галогена, алкила, гидрокси, алкокси, амино и алкилтио, и где гетероциклил также необязательно замещен группой оксо;
R15 представляет собой алкил, алкенил, алкинил, циклоалкил, циклоалкилалкил, гетероциклил, гетероциклилалкил, арил, аралкил, гетероарил, гетероаралкил, -C(O)NRyRz или -NRyRz, где алкил, алкенил, алкинил, циклоалкил, циклоалкилалкил, гетероциклил, гетероциклилалкил, арил, аралкил, гетероарил или гетероаралкил, каждый, необязательно замещен одним или несколькими, в одном варианте воплощения одним-четырьмя, в одном варианте воплощения одним-тремя, в одном варианте воплощения одним, двумя или тремя заместителями, независимо выбранными из галогена, оксо, алкила, гидрокси, алкокси, амино и алкилтио;
R18 представляет собой водород, алкил, галогеналкил, гидроксиС2-6алкил, алкенил, алкинил, циклоалкил, циклоалкилалкил, гетероциклил, гетероциклилалкил, арил, аралкил, гетероарил или гетероарилалкил; где R18 необязательно замещен 1-3 группами Q1, каждый Q1 независимо выбран из алкила, гидроксила, галогена, галогеналкила, алкокси, арилокси, алкоксиалкила, алкоксикарбонила, алкоксисульфонила, карбоксила, циклоалкила, гетероциклила, арила, гетероарила, галогенарила и амино;
R19 и R20 выбраны следующим образом:
(i) R19 и R20, каждый независимо, представляет собой водород или алкил; или
(ii) R19 и R20, вместе с атомом азота, с которым они связаны, образуют гетероциклил или гетероарил, который необязательно замещен 1-2 группами, каждая из которых независимо выбрана из галогена, алкила, галогеналкила, гидроксила и алкокси;
каждый Rx независимо представляет собой алкилен или простую связь;
Rv представляет собой водород, алкил, алкенил или алкинил;
Rw независимо представляет собой водород, алкил, алкенил, алкинил или галогеналкил;
Ry и Rz выбраны следующим образом:
(i) Ry и Rz, каждый независимо, представляет собой водород, алкил, алкенил, алкинил, циклоалкил или галогеналкил;
(ii) Ry и Rz, вместе с атомом азота, с которым они связаны, образуют гетероциклил или гетероарил, который необязательно замещен 1-2 группами, каждая из которых независимо выбрана из галогена, алкила, галогеналкила, гидроксила и алкокси;
p имеет значение 0-5; и
каждый q, независимо, имеет значение 0, 1 или 2; при условии, что, когда R1 и R2 вместе образуют =О, тогда R6a и R6d представляют собой водород, R6b выбран из водорода, алкила, алкенила, алкинила, галогеналкила, циклоалкила, -RXOR18, -RXNR19R20 и -RxS(O)qRv, R6c выбран из водорода, алкила, алкенила, алкинила, циклоалкила, гидрокси, -RXNR19R20 и -RxS(O)qRv, и другие переменные имеют значение, определенное в настоящей заявке.
В другом варианте воплощения, когда R1 и R2 вместе образуют =О, тогда R6a и R6 представляют собой водород, R6 и R6c, каждый независимо, выбраны из водорода, алкила, алкенила, алкинила, циклоалкила, гидрокси, -RXNR19R20 и -RxS(O)qRv, и другие переменные имеют значение, определенное в настоящей заявке.
В другом варианте воплощения, когда R1 и R2 вместе образуют =О, тогда R6a, R6b и R6d представляют собой водород, и R6c выбран из водорода, алкила, алкенила, алкинила, циклоалкила, гидрокси, -RXNR19R20 и -RxS(O)qRv, и другие переменные имеют значение, определенное в настоящей заявке.
В другом варианте воплощения, когда R1 и R2 вместе образуют =О, тогда R6a, R6b и R6d представляют собой водород, и R6c выбран из водорода, алкила, циклоалкила и гидрокси.
В другом варианте воплощения, R6a представляет собой водород, и R6b, R6c и R6d, каждый независимо, выбраны из водорода, галогена, алкила, алкенила, алкинила, галогеналкила, циклоалкила, -RXOR18, -RXNR19R20 и -RxS(O)qRv, при условии, что, когда R1 и R2 вместе образуют =О, тогда R6b и R6d представляют собой водород, и R6c выбран из водорода, алкила, алкенила, алкинила, циклоалкила, -RXOR18, -RXNR19R20 и -RxS(O)qRv, и другие переменные имеют значение, определенное в настоящей заявке.
В другом варианте воплощения, R6a, R6b и R6d, каждый, представляют собой водород, и R6c независимо выбран из водорода, алкила, алкенила, алкинила, циклоалкила, RXOR18, -RXNR19R20 и -RxS(O)qRv, и другие переменные имеют значение, определенное в настоящей заявке. В следующем варианте воплощения, R6a, R6b и R6d, каждый, представляют собой водород, и R6c представляет собой водород, алкил, циклоалкил или -RxOR18, и другие переменные имеют значение, определенное в настоящей заявке. В другом варианте воплощения, R6a, R6b, R6c и R6d представляют собой водород. В другом варианте воплощения, p имеет значение 2, и каждый R7 независимо выбран из галогена, гидрокси и алкокси. В следующем варианте воплощения, p имеет значение 1, и R7 представляет собой галоген.
В одном варианте воплощения, R6a и R6d представляют собой водород, и R6b и R6c, каждый независимо, выбраны из водорода, галогена, алкила, алкенила, алкинила, галогеналкила, циклоалкила, -RXOR18, -RXNR19R20 и -RxS(O)qRv, при условии, что, когда R1 и R2 вместе образуют =О, R6c выбран из водорода, алкила, алкенила, алкинила, циклоалкила, -RXOR18, -RXNR19R20 и -RxS(O)qRv, где другие переменные имеют значение, определенное в настоящей заявке.
В некоторых вариантах воплощения в настоящей заявке представлены соединения формулы (XII)
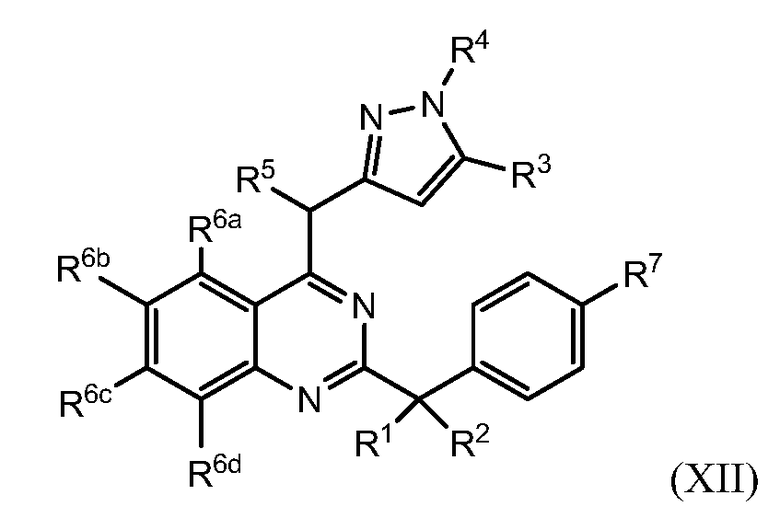
или их фармацевтически приемлемые соли, сольваты или гидраты, где другие переменные имеют значение, определенное в настоящей заявке. В одном варианте воплощения, R7 представляет собой галоген.
В некоторых вариантах воплощения в настоящей заявке представлены соединения формулы (XIII)
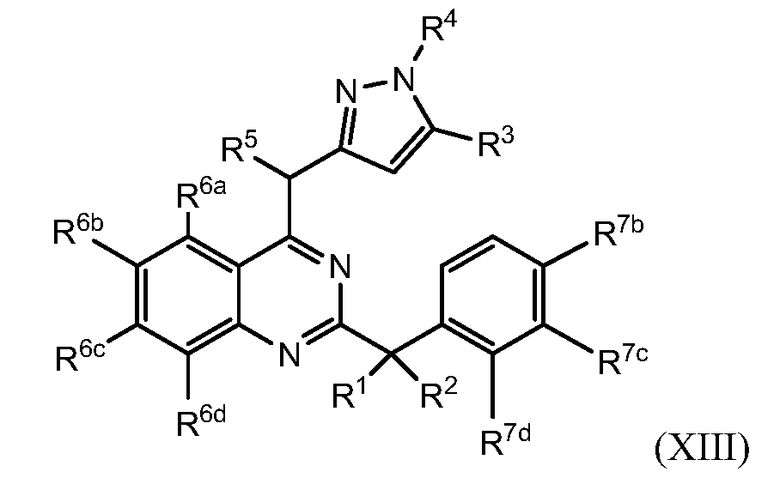
или их фармацевтически приемлемые соли, сольваты или гидраты, где R1 и R2 выбраны из (i), (ii), (iii), (iv) и (v) и имеют следующие значения:
(i) R1 и R2 вместе образуют =О, =S, =NR9 или =CR10R11;
(ii) R1 и R2 оба представляют собой -OR8, или R1 и R2, вместе с атомом углерода, с которым они связаны, образуют диоксациклоалкил;
(iii) R1 представляет собой водород или галоген; и R2 представляет собой галоген; и
(iv) R1 представляет собой алкил, алкенил, алкинил или циклоалкил, где алкил, алкенил, алкинил или циклоалкил необязательно замещен одним или несколькими, в одном варианте воплощения одним-четырьмя, в одном варианте воплощения одним-тремя, в одном варианте воплощения одним, двумя или тремя заместителями, выбранными из галогена, циано, алкила, -RXORW, -RxS(O)qRv, -RxNRyRz и -C(O)ORW; и R2 представляет собой водород, галоген или -OR8; и
(v) R1 представляет собой галоген, дейтеро, -OR12, -NR13R14 или -S(O)qR15; и R2 представляет собой водород, дейтеро, алкил, алкенил, алкинил или циклоалкил, где алкил, алкенил, алкинил или циклоалкил, необязательно замещен одним или несколькими, в одном варианте воплощения одним-четырьмя, в одном варианте воплощения одним-тремя, в одном варианте воплощения одним, двумя или тремя заместителями, выбранными из галогена, циано, алкила, -RXORW, -RxS(O)qRv и -RxNRyRz;
R3 представляет собой галоген, алкил, галогеналкил, циклоалкил, циклоалкилалкил, гидрокси или алкокси;
R4 и R5, каждый независимо, представляет собой водород или алкил;
R6a, R6b, R6c и R6d, каждый независимо, выбраны из водорода, галогена, алкила, алкенила, алкинила, галогеналкила, циклоалкила, -RXOR18, -RXNR19R20 и -RxS(O)qRv;
R7, R7c и R7, каждый независимо, выбраны из водорода, галогена, алкила, галогеналкил и -RXORW;
R8 представляет собой алкил, алкенил или алкинил;
R9 представляет собой водород, алкил, галогеналкил, гидрокси, алкокси или амино;
R10 представляет собой водород или алкил;
R11 представляет собой водород, алкил, галогеналкил или -C(O)OR8;
R12 выбран из водорода, алкила, алкенила, алкинила, циклоалкила, циклоалкилалкила, гетероциклила, гетероциклилалкила, арила, аралкила, гетероарила, гетероаралкила, -C(O)RV, -C(O)ORW и -C(О)NRyRz, где алкил, алкенил, алкинил, циклоалкил, циклоалкилалкил, гетероциклил, гетероциклилалкил, арил, аралкил, гетероарил или гетероаралкил, каждый, необязательно замещен одним или несколькими, в одном варианте воплощения одним-четырьмя, в одном варианте воплощения одним-тремя, в одном варианте воплощения одним, двумя или тремя заместителями, независимо выбранными из галогена, оксо, алкила, гидрокси, алкокси, амино и алкилтио;
R13 и R14 выбраны следующим образом:
(i) R13 представляет собой водород или алкил; и R14 выбран из водорода, алкила, алкенила, алкинила, циклоалкила, циклоалкилалкила, гетероциклила, гетероциклилалкила, арила, аралкила, гетероарила, гетероаралкила, алкокси, -C(O)RV, -C(O)ORW, -C(О)NRyRz и -S(O)qRv, где алкил, алкенил, алкинил, циклоалкил, циклоалкилалкил, гетероциклил, гетероциклилалкил, арил, аралкил, гетероарил или гетероаралкил, каждый, необязательно замещен одним или несколькими, в одном варианте воплощения одним-четырьмя, в одном варианте воплощения одним-тремя, в одном варианте воплощения одним, двумя или тремя заместителями, независимо выбранными из галогена, оксо, алкила, гидрокси, алкокси, амино и алкилтио; или
(ii) R13 и R14, вместе с атомом азота, с которым они связаны, образуют гетероциклил или гетероарил, где гетероциклил или гетероарил необязательно замещен одним или несколькими, в одном варианте воплощения одним-четырьмя, в одном варианте воплощения одним-тремя, в одном варианте воплощения одним, двумя или тремя заместителями, независимо выбранными из галогена, алкила, гидрокси, алкокси, амино и алкилтио, и где гетероциклил также необязательно замещен группой оксо;
R15 представляет собой алкил, алкенил, алкинил, циклоалкил, циклоалкилалкил, гетероциклил, гетероциклилалкил, арил, аралкил, гетероарил, гетероаралкил, -C(О)NRyRz или -NRyRz, где алкил, алкенил, алкинил, циклоалкил, циклоалкилалкил, гетероциклил, гетероциклилалкил, арил, аралкил, гетероарил или гетероаралкил, каждый, необязательно замещен одним или несколькими, в одном варианте воплощения одним-четырьмя, в одном варианте воплощения одним-тремя, в одном варианте воплощения одним, двумя или тремя заместителями, независимо выбранными из галогена, оксо, алкила, гидрокси, алкокси, амино и алкилтио;
R18 представляет собой водород, алкил, галогеналкил, гидроксиС2-6алкил, алкенил, алкинил, циклоалкил, циклоалкилалкил, гетероциклил, гетероциклилалкил, арил, аралкил, гетероарил или гетероарилалкил; где R18 необязательно замещен 1-3 группами Q1, каждый Q1 независимо выбран из алкила, гидроксила, галогена, галогеналкила, алкокси, арилокси, алкоксиалкила, алкоксикарбонила, алкоксисульфонила, карбоксила, циклоалкила, гетероциклила, арила, гетероарила, галогенарила и амино;
R19 и R20 выбраны следующим образом:
(i) R19 и R20, каждый независимо, представляет собой водород или алкил; или
(ii) R19 и R20, вместе с атомом азота, с которым они связаны, образуют гетероциклил или гетероарил, который необязательно замещен 1-2 группами, каждая из которых независимо выбрана из галогена, алкила, галогеналкила, гидроксила и алкокси; каждый Rx независимо представляет собой алкилен или простую связь;
Rv представляет собой водород, алкил, алкенил или алкинил;
Rw независимо представляет собой водород, алкил, алкенил, алкинил или галогеналкил;
Ry и Rz выбраны следующим образом:
(i) Ry и Rz, каждый независимо, представляет собой водород, алкил, алкенил, алкинил, циклоалкил или галогеналкил;
(ii) Ry и Rz, вместе с атомом азота, с которым они связаны, образуют гетероциклил или гетероарил, который необязательно замещен 1-2 группами, каждая из которых независимо выбрана из галогена, алкила, галогеналкила, гидроксила и алкокси;
p имеет значение 0-5; и
каждый q, независимо, имеет значение 0, 1 или 2; и при условии, что, когда R1 и R2 вместе образуют =О, тогда ни R7c ни R7d не может представлять собой -ORW. В одном варианте воплощения, R7d представляет собой водород. В одном варианте воплощения, R7d представляет собой водород, и R7b и R7c, каждый независимо, выбраны из водорода, галогена, алкила, галогеналкила и -RXORW. В другом варианте воплощения, R7b представляет собой галоген, R7b выбран из водорода, галогена, алкила, галогеналкила и -RXORW, и R7c представляет собой водород. В другом варианте воплощения, R7c и R7d представляют собой водород.
В другом варианте воплощения, в настоящей заявке представлены соединения формулы (XIV)
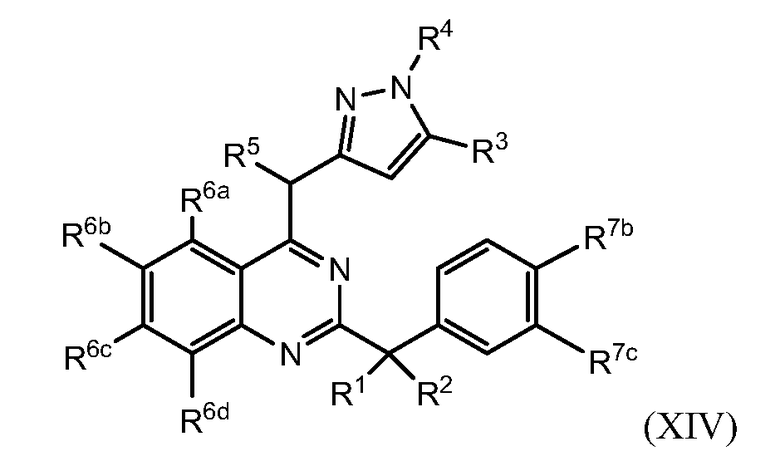
или их фармацевтически приемлемые соли, сольваты или гидраты, где:
R1 и R2 выбраны из (i), (ii), (iii), (iv) и (v) и имеют следующие значения:
(i) R1 и R2 вместе образуют =О, =S, =NR9 или =CR10R11;
(ii) R1 и R2 оба представляют собой -OR8, или R1 и R2, вместе с атомом углерода, с которым они связаны, образуют диоксациклоалкил;
(iii) R1 представляет собой водород или галоген; и R2 представляет собой галоген; и
(iv) R1 представляет собой алкил, алкенил, алкинил или циклоалкил, где алкил, алкенил, алкинил или циклоалкил необязательно замещен одним или несколькими, в одном варианте воплощения одним-четырьмя, в одном варианте воплощения одним-тремя, в одном варианте воплощения одним, двумя или тремя заместителями, выбранными из галогена, циано, алкила, -RXORW, -RxS(O)qRv, -RxNRyRz и -C(O)ORW; и R2 представляет собой водород, галоген или -OR8; и
(v) R1 представляет собой галоген, дейтеро, -OR12, -NR13R14 или -S(O)qR15; и R2 представляет собой водород, дейтеро, алкил, алкенил, алкинил или циклоалкил, где алкил, алкенил, алкинил или циклоалкил, необязательно замещен одним или несколькими, в одном варианте воплощения одним-четырьмя, в одном варианте воплощения одним-тремя, в одном варианте воплощения одним, двумя или тремя заместителями, выбранными из галогена, циано, алкила, -RXORW, -RxS(O)qRv и -RxNRyRz;
R3 представляет собой водород, галоген, алкил, галогеналкил, циклоалкил, гидрокси или алкокси;
R4 и R5, каждый независимо, представляет собой водород или алкил;
R6a, R6b, R6d представляют собой водород;
R6c представляет собой водород, галоген, алкил, гидрокси, гидроксиалкил, алкоксиалкил, алкилсульфонилалкил, алкокси, гидроксиалкокси или алкоксиалкокси,
R7b представляет собой галоген, и R7c представляет собой водород, галоген, гидрокси или алкокси;
R8 представляет собой алкил, алкенил или алкинил;
R9 представляет собой водород, алкил, галогеналкил, гидрокси, алкокси или амино;
R10 представляет собой водород или алкил;
R11 представляет собой водород, алкил, галогеналкил или -C(O)OR8;
R12 выбран из водорода, алкила, алкенила, алкинила, циклоалкила, циклоалкилалкила, гетероциклила, гетероциклилалкила, арила, аралкила, гетероарила, гетероаралкила, -C(O)RV, -C(O)ORW и -C(O)NRyRz, где алкил, алкенил, алкинил, циклоалкил, циклоалкилалкил, гетероциклил, гетероциклилалкил, арил, аралкил, гетероарил и гетероаралкил, каждый, необязательно замещен одним или несколькими, в одном варианте воплощения одним-четырьмя, в одном варианте воплощения одним-тремя, в одном варианте воплощения одним, двумя или тремя заместителями, независимо выбранными из галогена, оксо, алкила, гидрокси, алкокси, амино и алкилтио;
R13 и R14 выбраны следующим образом:
(i) R13 представляет собой водород или алкил; и R14 выбран из водорода, алкила, алкенила, алкинила, циклоалкила, циклоалкилалкила, гетероциклила, гетероциклилалкила, арила, аралкила, гетероарила, гетероаралкила, алкокси, -C(O)RV, -C(O)ORW, -C(O)NRyRz и -S(O)qRv, где алкил, алкенил, алкинил, циклоалкил, циклоалкилалкил, гетероциклил, гетероциклилалкил, арил, аралкил, гетероарил или гетероаралкил, каждый, необязательно замещен одним или несколькими, в одном варианте воплощения одним-четырьмя, в одном варианте воплощения одним-тремя, в одном варианте воплощения одним, двумя или тремя заместителями, независимо выбранными из галогена, оксо, алкила, гидрокси, алкокси, амино и алкилтио; или
(ii) R13 и R14, вместе с атомом азота, с которым они связаны, образуют гетероциклил или гетероарил, где гетероциклил или гетероарил необязательно замещен одним или несколькими, в одном варианте воплощения одним-четырьмя, в одном варианте воплощения одним-тремя, в одном варианте воплощения одним, двумя или тремя заместителями, независимо выбранными из галогена, алкила, гидрокси, алкокси, амино и алкилтио, и где гетероциклил также необязательно замещен группой оксо;
R15 представляет собой алкил, алкенил, алкинил, циклоалкил, циклоалкилалкил, гетероциклил, гетероциклилалкил, арил, аралкил, гетероарил, гетероаралкил, -C(O)NRyRz или -NRyRz, где алкил, алкенил, алкинил, циклоалкил, циклоалкилалкил, гетероциклил, гетероциклилалкил, арил, аралкил, гетероарил или гетероаралкил, каждый, необязательно замещен одним или несколькими, в одном варианте воплощения одним-четырьмя, в одном варианте воплощения одним-тремя, в одном варианте воплощения одним, двумя или тремя заместителями, независимо выбранными из галогена, оксо, алкила, гидрокси, алкокси, амино и алкилтио; каждый Rx независимо представляет собой алкилен или простую связь;
Rv представляет собой водород, алкил, алкенил или алкинил;
Rw независимо представляет собой водород, алкил, алкенил, алкинил или галогеналкил;
Ry и Rz выбраны следующим образом:
(i) Ry и Rz, каждый независимо, представляет собой водород, алкил, алкенил, алкинил, циклоалкил или галогеналкил;
(ii) Ry и Rz, вместе с атомом азота, с которым они связаны, образуют гетероциклил или гетероарил, который необязательно замещен 1-2 группами, каждая из которых независимо выбрана из галогена, алкила, галогеналкила, гидроксила и алкокси; при условии, что, когда R3 представляет собой водород, тогда R6c является отличным от галогена.
В другом варианте воплощения, в настоящей заявке представлены соединения формулы (XIV) или их фармацевтически приемлемые соли, сольваты или гидраты, где
(i) R1 и R2 вместе образуют =О, =S, =NR9 или =CR10R11;
(ii) R1 и R2, вместе с атомом углерода, с которым они связаны, образуют диоксациклоалкил;
(iii) R1 представляет собой водород или галоген; и R2 представляет собой галоген; и
(iv) R1 представляет собой галоген, дейтеро, гидроксил, и R2 представляет собой водород или дейтеро;
R3 представляет собой водород, галоген, алкил, галогеналкил, циклоалкил, гидрокси или алкокси;
R4 и R5, каждый независимо, представляет собой водород или алкил;
R6a, R6b, R6d представляют собой водород;
R6c представляет собой водород, галоген, алкил, гидрокси, гидроксиалкил, алкоксиалкил, алкилсульфонилалкил, алкокси, гидроксиалкокси или алкоксиалкокси,
R7b представляет собой галоген и R7c представляет собой водород; при условии, что, когда R3 представляет собой водород, тогда R6c является отличным от галогена.
В другом варианте воплощения, R3 представляет собой галоген, алкил, циклоалкил, галогеналкил, гидрокси или алкокси. В следующем варианте воплощения, R3 представляет собой алкил, галогеналкил, циклоалкил, гидрокси или алкокси. В следующем варианте воплощения, R3 представляет собой алкил, циклоалкил или алкокси. В другом варианте воплощения, R6c представляет собой водород, фтор, хлор, гидрокси, алкил, гидроксиалкил, алкоксиалкил, алкилсульфонилалкил, алкокси, гидроксиалкокси или алкоксиалкокси. В другом варианте воплощения, R6c представляет собой водород, фтор, хлор, гидрокси, метил, гидроксиметил, гидроксиэтил, метоксиметил, этоксиметил, метилсульфонилметил, этилсульфонилметил, метокси, этокси, пропилокси, гидроксипропилокси, гидроксиэтокси, гидроксиметокси, метоксиметокси или метоксиэтокси. В следующем варианте воплощения, R6c представляет собой водород, алкил, гидрокси, гидроксиалкил, алкоксиалкил, алкилсульфонилалкил, гидроксиалкокси или алкоксиалкокси.
В одном варианте воплощения, в настоящем изобретении представлено соединение, выбранное из следующих:
(4-хлорхиназолин-2-ил)(3-фторфенил)метанон;
(4-(1Н-пиразол-3-иламино)хиназолин-2-ил)(3-фторфенил)метанон;
(4-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанон;
(4-(1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанон;
(4-(1Н-пиразол-3-иламино)хиназолин-2-ил)(2-метоксифенил)метанон;
(4-(1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанол;
2-(фтор(4-фторфенил)метил)-N-(1Н-пиразол-3-ил)хиназолин-4-амин;
2-(дифтор(4-фторфенил)метил)-N-(5-метил-1Н-пиразол-3-ил)хиназолин-4-амин;
2-(дифтор(4-фторфенил)метил)-N-(1Н-пиразол-3-ил)хиназолин-4-амин;
N-(5-циклопропил-1Н-пиразол-3-ил)-2-(дифтор(4-фторфенил)метил)хиназолин-4-амин;
3-(2-(4-фторбензоил)хиназолин-4-иламино)-1Н-пиразол-5-карбонитрил;
(4-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанол;
2-((4-фторфенил)(метокси)метил)-N-(5-метил-1Н-пиразол-3-ил)хиназолин-4-амин;
2-(амино(4-фторфенил)метил)-N-(5-метил-1Н-пиразол-3-ил)хиназолин-4-амин;
3-(2-((4-фторфенил)(гидрокси)метил)хиназолин-4-иламино)-1Н-пиразол-5-карбонитрил;
(5-фтор-4-(5-метил-1H-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанол;
(4-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)-7-(трифторметил)хиназолин-2-ил)метанон;
(4-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)-7-(трифторметил)хиназолин-2-ил);
(7-фтор-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанон;
2-(дифтор(4-фторфенил)метил)-7-фтор-N-(5-метил-1Н-пиразол-3-ил)хиназолин-4-амин;
2-(дифтор(4-фторфенил)метил)-7-фтор-N-(1Н-пиразол-3-ил)хиназолин-4-амин;
(4-(1Н-пиразол-3-иламино)-7-иодхиназолин-2-ил)(4-фторфенил)метанон;
(4-(1Н-пиразол-3-иламино)-7-иодхиназолин-2-ил)(4-фторфенил)метанол;
(4-фторфенил)(7-метил-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанон;
(4-фторфенил)(7-метил-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанол;
2-(дифтор(4-фторфенил)метил)-7-метил-N-(5-метил-1Н-пиразол-3-ил)хиназолин-4-амин;
2-(дифтор(4-фторфенил)метил)-7-метил-N-(1Н-пиразол-3-ил)хиназолин-4-амин;
(4-(1Н-пиразол-3-иламино)-7-метоксихиназолин-2-ил)(4-фторфенил)метанон;
(4-(1Н-пиразол-3-иламино)-7-метоксихиназолин-2-ил)(4-фторфенил)метанол;
(4-фторфенил)(7-метокси-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанон;
(4-фторфенил)(7-метокси-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанол;
2-(дифтор(4-фторфенил)метил)-7-метокси-N-(5-метил-1Н-пиразол-3-ил)хиназолин-4-амин;
2-(дифтор(4-фторфенил)метил)-7-метокси-N-(1Н-пиразол-3-ил)хиназолин-4-амин;
2-(дифтор(4-фторфенил)метил)-8-фтор-N-(5-метил-1Н-пиразол-3-ил)хиназолин-4-амин;
(4-(1Н-пиразол-3-иламино)-8-метоксихиназолин-2-ил)(4-фторфенил)метанон;
2-((4-фторфенил)(гидрокси)метил)-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-7-ол;
(4-фторфенил)(7-гидрокси-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанон;
(4-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)-7-(2-морфолиноэтокси)хиназолин-2-ил)метанол;
2-(2-((4-фторфенил)(гидрокси)метил)-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-7-илокси)этанол;
3-(2-((4-фторфенил)(гидрокси)метил)-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-7-илокси)пропан-1-ол;
(4-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)-7-(пиперидино-4-илокси)хиназолин-2-ил)метанол;
(4-фторфенил)(7-(2-метоксиэтокси)-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанол;
трет-бутил 2-(2-((4-фторфенил)(гидрокси)метил)-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-7-илокси)ацетат;
2-(2-((4-фторфенил)(гидрокси)метил)-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-7-илокси)уксусная кислота;
метиловый эфир {(4-фтор-фенил)-[4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил]метил}карбаминовой кислоты; и
бис-(4-фтор-фенил)-[4-(5-метил-1Н-пиразол-3-иламино)-хиназолин-2-ил]метанол.
В одном варианте воплощения, в настоящем изобретении представлено соединение, выбранное из следующих:
(R,S)-метил-(4-фторфенил)(4-(5-метил-4H-пиразол-3-иламино)хиназолин-2-ил)метилкарбамат;
(R,S)-(4-фторфенил)(8-метил-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанол;
(R,S)-(7-фтор-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанол;
(4-(1Н-пиразол-3-иламино)хиназолин-2-ил)бис(4-фторфенил)метанол;
(2-(дифтор(4-фторфенил)метил)-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-7-ил)метанол;
2-(дифтор(4-фторфенил)метил)-N-(5-метил-1Н-пиразол-3-ил)-7-(метилсульфонилметил)хиназолин-4-амин;
2-(Дифтор(4-фторфенил)метил)-7-(этоксиметил)-N-(5-метил-1Н-пиразол-3-ил)хиназолин-4-амин;
(R,S)(7-хлор-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанол;
(6-фтор-4-(5-метил-1H-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанон(R,S)-(6-фтор-4-(5-метил-1H-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанол;
(R,S)-(4-(1Н-пиразол-3-иламино)-6-фторхиназолин-2-ил)(4-фторфенил)метанол;
(7-бром-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанон;
(7-бром-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанол;
(R,S)-(4-(1Н-пиразол-3-иламино)-7-бромхиназолин-2-ил)(4-фторфенил)метанол;
2-(2-(4-фторфенил)-1,3-диоксолан-2-ил)-N-(5-метил-1Н-пиразол-3-ил)хиназолин-4-амин;
(8-фтор-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанон;
(R,S)-(8-фтор-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанол;
(2-метоксифенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанон;
(R,S)-(2-метоксифенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанол;
(3-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанол;
N-((4-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метил)формамид;
(R,S)-(3,4-дифторфенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанол;
(3-хлор-4-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанол;
3-(4-фторфенил)-3-(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)пропаннитрил;
2-((циклопропиламино)(4-фторфенил)метил)-N-(5-метил-1Н-пиразол-3-ил)хиназолин-4-амин;
2-(1-(4-фторфенил)-2-(метилсульфонил)этил)-N-(5-метил-1Н-пиразол-3-ил)хиназолин-4-амин;
2-(3-амино-1-(4-фторфенил)пропил)-N-(5-метил-1Н-пиразол-3-ил)хиназолин-4-амин;
(R,S)(4-фторфенил)(4-(5-метил-1H-пиразол-3-иламино)хиназолин-2-ил)метанол-1-d;
(4-фторфенил)(4-(5-метокси-1Н-пиразол-3-иламино)хиназолин-2-ил)метанон;
(R,S)-(4-(5-этил-1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанол;
(4-Фторфенил)(4-(5-метокси-1Н-пиразол-3-иламино)хиназолин-2-ил)метанол;
(4-фтор-3-метоксифенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанон;
(4-фтор-3-гидроксифенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанон; и
(R,S)-(2-фтор-5-(гидрокси(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метил)фенолацетат.
Также в настоящей заявке представлены изотопно обогащенные аналоги соединений, представленных в настоящем изобретении. Изотопное обогащение (например, дейтерирование) фармацевтических веществ для улучшения фармакокинетических свойств ("PK"), фармакодинамических свойств ("PD") и профилей токсичности было продемонстрировано ранее с некоторыми классами лекарственных средств. См., например, Lijinsky et al., Food Cosmet. Toxicol, 20: 393 (1982); Lijinsky et al., J. Nat. Cancer Inst., 69: 1127 (1982); Mangold et al., Mutation Res. 308: 33 (1994); Gordon et al., Drug Metab. Dispos., 15: 589 (1987); Zello et al., Metabolism, 43: 487 (1994); Gately et al., J. Nucl Med., 27: 388 (1986); Wade D, Chem. Biol Interact. 117: 191 (1999).
Изотопное обогащение лекарственного средства можно использовать, например, для (1) снижения уровня или устранения нежелательных метаболитов, (2) увеличения периода полужизни исходного лекарственного средства, (3) уменьшения количества доз, необходимых для достижения желаемого эффекта, (4) уменьшения дозируемого количества, необходимого для достижения желаемого эффекта, (5) увеличения образования активных метаболитов, если они образуются, и/или (6) уменьшения продукции вредных метаболитов в специфических тканях и/или создания более эффективного лекарственного средства и/или более безопасного лекарственного средства для комбинированной терапии, независимо от того, является ли комбинированная терапия желательной или нет.
Замещение атома одним из его изотопов часто приводит к изменению скорости реакции при химическом взаимодействии. Это явление известно как Кинетический Изотопный Эффект ("KIE"). Например, если C-H связь разрывается на определяющей скорость стадии в химической реакции (т.е. стадия с наивысшей энергией перехода), замещение дейтерием такого водорода вызовет уменьшение скорости реакции и замедление процесса. Это явление известно как Кинетический Изотопный Эффект Дейтерия ("DKIE"). (См., например, Foster et al., Adv. Drug Res., vol. 14, pp. 1-36 (1985); Kushner et al., Can. J. Physiol. Pharmacol, vol. 77, pp. 79-88 (1999)).
Тритий ("T") представляет собой радиоактивный изотоп водорода, используемый в научных экспериментах, термоядерных реакторах, генераторах нейтронов и радиофармацевтических средствах. Тритий представляет собой атом водорода, который имеет 2 нейтрона в ядре и имеет атомную массу близкую к 3. В природной среде он встречается в очень низких концентрациях, наиболее часто присутствует как T2O. Тритий распадается медленно (период полужизни = 12,3 лет) и испускает бета-частицу с низкой энергией, которая не может проникать через внешний слой человеческой кожи. Основной опасностью является внутреннее воздействие, связанное с этим изотопом, хотя его нужно было бы принять внутрь в больших количествах, чтобы вызвать существенный риск для здоровья. По сравнению с дейтерием, требуется меньшее количество трития для достижения опасного эффекта. Замещение водорода тритием ("T") приводит к более сильной связи, чем при замещении дейтерием, и дает значительно большие изотопные эффекты. Подобным образом, изотопное замещение других элементов, включая, но не ограничиваясь этим, 13C или 14C для углерода, 33S, 34S или 36S для серы, 15N для азота и 17O или 18O для кислорода, даст аналогичные кинетические изотопные эффекты.
C. Формулирование фармацевтический композиций
В настоящей заявке представлены фармацевтические композиции, включающие соединение, представленное в настоящем изобретении, например, соединение формулы I, в качестве активного ингредиента или его фармацевтически приемлемую соль, сольват или гидрат; в сочетании с фармацевтически приемлемым наполнителем, носителем, разбавителем или эксципиентом или их смесью.
Соединение, представленное в настоящем изобретении, можно вводить отдельно или в сочетании с одним или несколькими другими соединениями, представленными в настоящем изобретении. Фармацевтические композиции, которые включают соединение, представленное в настоящем изобретении, например, соединение формулы I, могут быть сформулированы в различные лекарственные формы для перорального, парентерального и местного введения. Фармацевтические композиции также могут быть сформулированы в виде лекарственных форм с модифицированным высвобождением, включая лекарственные формы замедленного, продленного, пролонгированного, длительного, пульсирующего, контролируемого, ускоренного и быстрого, направленного, программируемого высвобождения и удерживаемые в желудке. Эти лекарственные формы можно получить в соответствии с традиционными способами и процедурами, которые известны специалистам в данной области (см., Remington: The Science and Practice of Pharmacy, supra; Modified-Release Drug Deliver Technology, Rathbone et al., Eds., Drugs and the Pharmaceutical Science, Marcel Dekker, Inc.: New York, NY, 2003; Vol. 126).
В одном варианте воплощения, обеспечиваются фармацевтические композиции в лекарственной форме для перорального введения, которые включают соединение, представленное в настоящем изобретении, например, соединение формулы I или его фармацевтически приемлемую соль, сольват или гидрат; и один или несколько фармацевтически приемлемых эксципиентов или носителей.
В другом варианте воплощения, обеспечиваются фармацевтические композиции в лекарственной форме для парентерального введения, которые включают соединение, представленное в настоящем изобретении, например, соединение формулы I или его фармацевтически приемлемую соль, сольват или гидрат; и один или несколько фармацевтически приемлемых эксципиентов или носителей.
В следующем варианте воплощения, обеспечиваются фармацевтические композиции в лекарственной форме для местного введения, которые включают соединение, представленное в настоящем изобретении, например, соединение формулы I, или его фармацевтически приемлемую соль, сольват или гидрат; и один или несколько фармацевтически приемлемых эксципиентов или носителей.
Фармацевтические композиции, представленные в настоящем изобретении, могут обеспечиваться в лекарственной форме, представляющей собой стандартную дозу, или в многодозовой лекарственной форме. Лекарственная форма, представляющая собой стандартную дозу, как это используется в настоящей заявке, относится к физически дискретной единице, подходящей для введения субъекту, такому как человек или животное, и индивидуально упакованной, как это известно из предшествующего уровня техники. Каждая стандартная доза содержит предварительно определенное количество активного ингредиента(ингредиентов), достаточное для получения желаемого терапевтического эффекта, в ассоциации с необходимыми фармацевтическими носителями или эксципиентами. Примеры лекарственной формы, включающей стандартную дозу, включают ампулу, шприц и индивидуально упакованную таблетку и капсулу. Лекарственную форму, представляющую собой стандартную дозу, можно вводить как разделенную на части или доли. Многодозовая лекарственная форма представляет собой несколько идентичных стандартных доз, упакованных в одном контейнере, для введения в виде отдельных единиц лекарственной формы. Примеры многодозовой лекарственной формы включают флакон, бутыль с таблетками или капсулами или бутыль, содержащую пинты или галлоны лекарственного средства.
Фармацевтические композиции, представленные в настоящем изобретении, можно вводить одноразово или в несколько приемов с определенными интервалами времени. Должно быть понятно, что конкретная доза и продолжительность лечения могут варьировать в зависимости от возраста, массы тела и состояния пациента, принимающего лечение, и могут быть определены эмпирическим путем с использованием известных протоколов испытаний или путем экстраполирования из данных in vivo или in vitro испытаний или диагностических данных. Кроме того, должно быть понятно, что для любого конкретного индивидуума конкретную схему введения лекарственного средства следует со временем регулировать в соответствии с потребностями индивидуума и профессиональным суждением лица, осуществляющим введение или контролирующим введение препаратов.
В одном варианте воплощения, терапевтически эффективная доза составляет от около 0,1 мг до около 2000 мг в день соединения, представленного в настоящем изобретении. Поэтому фармацевтические композиции должны обеспечивать дозу от около 0,1 мг до около 2000 мг соединения. В некоторых вариантах воплощения фармацевтические стандартные лекарственные формы получают таким образом, чтобы они обеспечивали от около 1 мг до около 2000 мг, от около 10 мг до около 1000 мг, от около 20 мг до около 500 мг или от около 25 мг до около 250 мг действующего активного ингредиента или сочетания действующих ингредиентов на стандартную дозируемую единицу. В некоторых вариантах воплощения фармацевтические стандартные лекарственные формы получают таким образом, чтобы они обеспечивали около 10 мг, 20 мг, 25 мг, 50 мг, 100 мг, 250 мг, 500 мг, 1000 мг или 2000 мг действующего активного ингредиента.
Пероральное введение
Фармацевтические композиции, обспечиваемые в настоящем изобретении, могут быть представлены в твердой, полутвердой или жидкой лекарственной форме для перорального введения. Как это используется в настоящей заявке, пероральное введение также включает буккальное, лингвальное и сублингвальное введение. Подходящие пероральные лекарственные формы включают, но не ограничиваются этим, таблетки, быстротающие таблетки, жевательные таблетки, капсулы, пилюли, драже, лепешки, пастилки, саше, гранулы, содержащие лекарственное средство жевательные резинки, сыпучие порошки, шипучие или нешипучие порошки или гранулы, растворы, эмульсии, суспензии, облатки, присыпки, эликсиры и сиропы. В добавление к активному ингредиенту(ингредиентам), фармацевтические композиции могут содержать один или несколько фармацевтически приемлемых носителей или эксципиентов, включая, но не ограничиваясь этим, связующие, наполнители, разбавители, разрыхлители, смачивающие вещества, смазывающие вещества, агенты скольжения, красители, ингибиторы миграции красителя, подсластители и отдушки.
Связующие или агенты гранулирования придают когезионные свойства таблетке, чтобы таблетка не разрушалась в результате прессования. Подходящие связующие или агенты гранулирования включают, но не ограничиваются этим, крахмалы, такие как кукурузный крахмал, картофельный крахмал и предварительно желатинизированный крахмал (например, КРАХМАЛ 1500); желатин; сахара, такие как сахароза, глюкоза, декстроза, меласса и лактоза; природные и синтетические камеди, такие как аравийская камедь, альгиновая кислота, альгинаты, экстракт карагена, камедь panwar, камедь гхатти, клейкая выжимка из шелуха isabgol, карбоксиметилцеллюлоза, метилцеллюлоза, поливинилпирролидон (PVP), Veegum, арабогалактан из лиственницы, порошкообразный трагакант и гуаровая камедь; целлюлозы, такие как этилцеллюлоза, ацетат целлюлозы, кальциевая соль карбоксиметилцеллюлозы, натриевая соль карбоксиметилцеллюлозы, метилцеллюлоза, гидроксиэтилцеллюлоза (HEC), гидроксипропилцеллюлоза (HPC), гидроксипропилметилцеллюлоза (HPMC); микрокристаллические целлюлозы, такие как AVICEL-PH-101, AVICEL-PH-103, AVICEL RC-581, AVICEL-PH-105 (FMC Corp., Marcus Hook, PA); и их смеси. Подходящие наполнители включают, но не ограничиваются этим, тальк, карбонат кальция, микрокристаллическую целлюлозу, порошкообразную целлюлозу, декстраты, каолин, маннит, кремневую кислоту, сорбит, крахмал, предварительно желатинизированный крахмал и их смеси. В фармацевтических композициях, обспечиваемых в настоящем изобретении, связующее или наполнитель может присутствовать в количестве от около 50 до около 99% масс.
Подходящие разбавители включают, но не ограничиваются этим, дикальцийфосфат, сульфат кальция, лактозу, сорбит, сахарозу, инозит, целлюлозу, каолин, маннит, хлорид натрия, сухой крахмал и порошкообразный сахар. Некоторые разбавители, такие как маннит, лактоза, сорбит, сахароза и инозит, когда они присутствуют в достаточном количестве, могут придавать свойства некоторым прессованным таблеткам, которые делают возможным разложение во рту при жевании. Такие прессованные таблетки можно использовать в качестве жевательных таблеток.
Подходящие разрыхлители включают, но не ограничиваются этим, агар; бентонит; целлюлозы, такие как метилцеллюлоза и карбоксиметилцеллюлоза; продукты из древесины; природную губку; катионообменные смолы; альгиновую кислоту; камеди, такие как гуаровая камедь и Veegum HV; цитрусовую пульпу; сшитые целлюлозы, такие как кроскармелоза; сшитые полимеры, такие как кросповидон; сшитые крахмалы; карбонат кальция; микрокристаллическую целлюлозу, такую как натрия крахмалгликолят; полакрилин калий; крахмалы, такие как кукурузный крахмал, картофельный крахмал, тапиоковый крахмал и предварительно желатинизированный крахмал; глины; водоросли; и их смеси. Количество разрыхлителя в фармацевтических композициях, обспечиваемых в настоящем изобретении, варьирует в зависимости от типа препарата, и его может легко определить специалист со средней квалификацией. Фармацевтические композиции, обспечиваемые в настоящем изобретении, могут содержать от около 0,5 до около 15% или от около от 1 до около 5% масс. разрыхлителя.
Подходящие смазывающие вещества включают, но не ограничиваются этим, стеарат кальция; стеарат магния; минеральное масло; светлое минеральное масло; глицерин; сорбит; маннит; гликоли, такие как глицеринбегенат и полиэтиленгликоль (PEG); стеариновую кислоту; лаурилсульфат натрия; тальк; гидрогенизированное растительное масло, включая арахисовое масло, масло семян хлопчатника, подсолнечное масло, кунжутное масло, оливковое масло, кукурузное масло и соевое масло; стеарат цинка; этилолеат; этиллауреат; агар; крахмал; ликоподий; диоксид кремния или силикагели, такие как AEROSIL® 200 (W.R. Grace Co., Baltimore, MD) и CAB-O-SIL® (Cabot Co. of Boston, MA); и их смеси. Фармацевтические композиции, обеспечиваемые в настоящем изобретении, могут содержать около 0,1 до около 5% масс. смазывающего вещества.
Подходящие агенты скольжения включают коллоидный диоксид кремния, CAB-O-SIL® (Cabot Co. of Boston, MA) и не содержащий асбеста тальк. Красители включают любые из утвержденных для применения сертифицированных водорастворимых FD&C красителей и не растворимых в воде FD&C красителей, суспендированных в гидрате алюминия, и цветных лаков, и их смесей. Цветной лак представляет собой сочетание путем адсорбции водорастворимого красителя с гидроокисью тяжелого металла, дающее нерастворимую форму красителя. Отдушки включают природные отдушки, экстрагированные из растений, таких как фрукты, и синтетические смеси соединений, которые дают приятный вкусовые ощущения, такие как перечная мята и метилсалицилат. Подсластители включают сахарозу, лактозу, маннит, сиропы, глицерин и искусственные подсластители, такие как сахарин и аспартам. Подходящие эмульгаторы включают желатин, аравийскую камедь, трагакант, бентонит и поверхностно-активные вещества, такие как полиоксиэтиленсорбитанмоноолеат (TWEEN® 20), полиоксиэтиленсорбитанмоноолеат 80 (TWEEN® 80) и триэтаноламинолеат. Суспендирующие и диспергирующие вещества включают натриевую соль карбоксиметилцеллюлозы, пектин, трагакант, Veegum, аравийскую камедь, натриевую соль карбометилцеллюлозы, гидроксипропилметилцеллюлозу и поливинилпирролидон. Консерванты включают глицерин, метил- и пропилпарабен, бензойную кислоту, бензоат натрия и спирт. Смачивающие вещества включают пропиленгликоль моностеарат, сорбитанмоноолеат, диэтиленгликоль монолаурат и лауриловый эфир полиоксиэтилена. Растворители включают глицерин, сорбит, этиловый спирт и сироп. Примеры неводных жидкостей, используемых в эмульсиях, включают минеральное масло и масло семян хлопчатника. Органические кислоты включают лимонную и винную кислоту. Источники диоксида углерода включают бикарбонат натрия и карбонат натрия.
Должно быть понятно, что многие носители и эксципиенты могут выполнять несколько функций, даже в одной и той же композиции.
Фармацевтические композиции, обспечиваемые в настоящем изобретении, могут быть представлены в виде прессованных таблеток, растираемых в порошок таблеток, жевательных лепешек, быстрорастворимых таблеток, мультипрессованных таблеток или таблеток с энетеросолюбильным покрытием, сахарным покрытием или пленочным покрытием. Таблетки с энетеросолюбильным покрытием представляют собой прессованные таблетки, покрытые веществами, которые являются стойкими к действию желудочной кислоты, но растворяются или разлагаются в кишечнике, таким образом, защищая активные ингредиенты от кислотной среды желудка. Энетеросолюбильные покрытия включают, но не ограничиваются этим, жирные кислоты, жиры, фенилсалицилат, воски, щеллак, аммиачный щеллак и ацетатфталаты целлюлозы. Таблетки с сахарным покрытием представляют собой прессованные таблетки, окруженные сахарным покрытием, которое может выгодным образом маскировать неприятные вкусы или запахи и защищать таблетки от окисления. Таблетки с пленочным покрытием представляют собой прессованные таблетки, которые покрыты тонким слоем или пленкой из водорастворимого материала. Пленочные покрытия включают, но не ограничиваются этим, гидроксиэтилцеллюлозу, натриевую соль карбоксиметилцеллюлозы, полиэтиленгликоль 4000 и ацетатфталат целлюлозы. Пленочное покрытие придает такие же общие свойства, как и сахарное покрытие. Мультипрессованные таблетки представляют собой прессованные таблетки, полученные с использованием более чем одного цикла прессования, включая многослойные таблетки и таблетки с пресс-покрытием или сухим покрытием.
Лекарственные формы в виде таблеток можно получить из активного ингредиента в порошкообразной, кристаллической или гранулированной форме, отдельно или в сочетании с одним или несколькими носителями или эксципиентами, описанными в настоящей заявке, включая связующие, разрыхлители, полимеры, обеспечивающие контролируемое высвобождение, смазывающие вещества, разбавители и/или красители. Отдушки и подсластители являются особенно полезными для получения жевательных таблеток и лепешек.
Фармацевтические композиции, обспечиваемые в настоящем изобретении, могут быть представлены в виде мягких или твердых капсул, которые могут быть выполнены из желатина, метилцеллюлозы, крахмала или альгината кальция. Твердая желатиновая капсула, также известная как капсула сухого заполнения (DFC), состоит из двух секций, где одна секция окружает другую, таким образом, полностью заключая в себе активный ингредиент. Мягкая эластичная капсула (SEC) представляет собой мягкую сферическую оболочку, такую как желатиновая оболочка, которая пластифицирована добавлением глицерина, сорбита или подобного полиола. Мягкие желатиновые оболочки могут содержать консервант для предотвращения роста микроорганизмов. Подходящие консерванты представляют собой такие, которые описаны в настоящей заявке, включая метил- и пропилпарабены и сорбиновую кислоту. Жидкие, полутвердые и твердые лекарственные формы, представленные в настоящем изобретении, могут быть инкапсулированы в капсуле. Подходящие жидкие и полутвердые лекарственные формы включают растворы и суспензии в пропиленкарбонате, растительных маслах или триглицеридах. Капсулы, содержащие такие растворы, можно получить, как описано в Патентах США №№ 4328245; 4409239; и 4410545. Капсулы также могут иметь покрытие, нанесенное способом, известным специалистам в данной области, для модификации или задержки растворения активного ингредиента.
Фармацевтические композиции, обспечиваемые в настоящем изобретении, могут быть представлены в жидких и полутвердых лекарственных формах, включая эмульсии, растворы, суспензии, эликсиры и сиропы. Эмульсия представляет собой двухфазную систему, в которой одна жидкость диспергирована в форме небольших капелек в другой жидкости, которая может представлять собой эмульсию масло-в-воде или вода-в-масле. Эмульсии могут включать фармацевтически приемлемую неводную жидкость или растворитель, эмульгатор и консервант. Суспензии могут включать фармацевтически приемлемый суспендирующий агент и консервант. Водные спиртовые растворы могут включать фармацевтически приемлемый ацеталь, такой как ди(низший алкил)ацеталь низшего алкилальдегида, например, ацетальдегид диэтилацеталь; и смешиваемый с водой растворитель, содержащий одну или несколько гидроксильных групп, такой как пропиленгликоль и этанол. Эликсиры представляют собой прозрачные, подслащенные и водоспиртовые растворы. Сиропы представляют собой концентрированные водные растворы сахара, например, сахарозы, и также могут содержать консервант. Что касается жидкой лекарственной формы, например, раствор в полиэтиленгликоле можно разбавить достаточным количеством фармацевтически приемлемого жидкого носителя, например, водой, для более удобного отмеривания дозы для введения.
Другие полезные жидкие и полутвердые лекарственные формы включают, но не ограничиваются этим, лекарственные формы, содержащие активный ингредиент(ингредиенты), представленный в настоящем изобретении, и диалкилированный моно- или полиалкиленгликоль, включая, 1,2-диметоксиметан, диглим, триглим, тетраглим, полиэтиленгликоль-350-диметиловый эфир, полиэтиленгликоль-550-диметиловый эфир, полиэтиленгликоль-750-диметиловый эфир, где 350, 550 и 750 относятся к приблизительной средней молекулярной массе полиэтиленгликоля. Эти композиции могут дополнительно включать один или несколько антиоксидантов, таких как бутилированный гидрокситолуол (BHT), бутилированный гидроксианизол (BHA), пропилгаллат, витамин E, гидрохинон, гидроксикумарины, этаноламин, лецитин, цефалин, аскорбиновая кислота, яблочная кислота, сорбит, фосфорная кислота, бисульфит, метабисульфит натрия, тиодипропионовая кислота и ее эфиры и дитиокарбаматы.
Фармацевтические композиции, обспечиваемые в настоящем изобретении для перорального введения, также могут быть представлены в формах липосом, мицелл, микросфер или наносистем. Лекарственные формы в виде мицелл можно получить, как описано в Патенте США № 6350458.
Фармацевтические композиции, обспечиваемые в настоящем изобретении, могут быть представлены в виде нешипучих или шипучих гранул и порошков для реструктурирования в жидкую лекарственную форму. Фармацевтически приемлемые носители и эксципиенты, используемые в нешипучих гранулах или порошках, могут включать разбавители, подсластители и смачивающие вещества. Фармацевтически приемлемые носители и эксципиенты, используемые в шипучих гранулах или порошках, могут включать органические кислоты и источник диоксида углерода.
Красители и отдушки можно использовать во всех описанных выше лекарственных формах.
Фармацевтические композиции, обспечиваемые в настоящем изобретении, могут быть сформулированы в виде лекарственных форм немедленного или модифицированного высвобождения, включая формы замедленного, длительного, пульсирующего, контролируемого, направленного и программируемого высвобождения.
Фармацевтические композиции, обспечиваемые в настоящем изобретении, могут быть сформулированы совместно с другими активными ингредиентами, которые не сообщают желаемое терапевтическое действие, или с веществами, которые дополняют желаемое действие.
Парентеральное введение
Фармацевтические композиции, обспечиваемые в настоящем изобретении, можно вводить парентерально путем инъекци, инфузии или имплантации, для местного или системного введения. Парентеральное введение, как этот термин используется в настоящей заявке, включает внутривенное, внутриартериальное, интраперитонеальное, интратекальное, интравентрикулярное, интрауретральное, интрастернальное, интракраниальное, внутримышечное, интрасиновиальное, интравезикальное и подкожное введение.
Фармацевтические композиции, обспечиваемые в настоящем изобретении, могут быть сформулированы в любые лекарственные формы, которые являются подходящими для парентерального введения, включая растворы, суспензии, эмульсии, мицеллы, липосомы, микросферы, наносистемы и твердые формы, подходящие для растворения или суспендирования в жидкости перед инъекцией. Такие лекарственные формы можно получить в соответствии с традиционными способами, которые известны специалистам в области фармацевтики (см., Remington: The Science and Practice of Pharmacy, supra).
Фармацевтические композиции, предназначенные для парентерального введениея, могут включать один или несколько фармацевтически приемлемых носителей и эксципиентов, включая, но не ограничиваясь этим, водные носители, смешиваемые с водой носители, неводные носители, противомикробные средства или консерванты против роста микроорганизмов, стабилизаторы, вещества, усиливающие солюбилизацию, изотонические агенты, буферные вещества, антиоксиданты, местные анестетики, суспендирующие и диспергирующие вещества, смачивающие вещества или эмульгаторы, комплексообразующие вещества, секвестранты или хелатообразующие вещества, криозащитные вещества, лиозащитные вещества, загустители, вещества, регулирующие рН, и инертные газы.
Подходящие водные носители включают, но не ограничиваются этим, воду, солевой раствор, физиологический солевой раствор или фосфатно-буферный солевой раствор (PBS), раствор хлорида натрия для инъекций, раствор Рингера для инъекций, изотонический раствор декстрозы для инъекций, стерильную воду для инъекций, раствор декстрозы и содержащий лактат раствор Рингера для инъекций. Неводные носители включают, но не ограничиваются этим, нелетучие масла растительного происхождения, касторовое масло, кукурузное масло, масло семян хлопчатника, оливковое масло, арахисовое масло, масло мяты перечной, сафлоровое масло, кунжутное масло, соевое масло, гидрогенизированное растительное масла, гидрогенизированное соевое масло и имеющие среднюю длину цепи триглицериды масла кокосового ореха и масла из семян пальмы. Смешиваемые с водой носители включают, но не ограничиваются этим, этанол, 1,3-бутандиол, жидкий полиэтиленгликоль (например, полиэтиленгликоль 300 и полиэтиленгликоль 400), пропиленгликоль, глицерин, N-метил-2-пирролидон, N,N-диметилацетамид и диметилсульфоксид.
Подходящие противомикробные средства или консерванты включают, но не ограничиваются этим, фенолы, крезолы, соединения ртути, бензиловый спирт, хлорбутанол, метил- и пропил п-гидроксибензоаты, тимеросал, бензалконий хлорид (например, бензетонийхлорид), метил- и пропилпарабены и сорбиновую кислоту. Подходящие изотонические агенты включают, но не ограничиваются этим, хлорид натрия, глицерин и декстрозу. Подходящие буферные вещества включают, но не ограничиваются этим, фосфат и цитрат. Подходящими антиоксидантами являются такие, которые описаны в настоящей заявке, включая бисульфит и метабисульфит натрия. Подходящие местные анестетики включают, но не ограничиваются этим, прокаин гидрохлорид. Подходящими суспендирующими и диспергирующими веществами являются такие, которые описаны в настоящей заявке, включая натриевую соль карбоксиметилцеллюлозы, гидроксипропилметилцеллюлозу и поливинилпирролидон. Подходящие эмульгаторы включают вещества, описанные в настоящей заявке, включая полиоксиэтиленсорбитанмонолаурат, полиоксиэтиленсорбитанмоноолеат 80 и триэтаноламинолеат. Подходящие секвестранты или хелатообразующие вещества включают, но не ограничиваются этим EDTA. Подходящие вещества, регулирующие рН, включают, но не ограничиваются этим, гидроксид натрия, хлористоводородную кислоту, лимонную кислоту и молочную кислоту. Подходящие комплексообразующие вещества включают, но не ограничиваются этим, циклодекстрины, включая α-циклодекстрин, β-циклодекстрин, гидроксипропил-β-циклодекстрин, сульфобутиловый эфир-β-циклодекстрин и сульфобутиловый эфир 7-β-циклодекстрин (CAPTISOL®, CyDex, Lenexa, KS).
Фармацевтические композиции, обспечиваемые в настоящем изобретении, могут быть сформулированы для введения с использованием разовой дозы или нескольких доз. Композиции для разовой дозы упакованы в ампулу, флакон или шприц. Многодозовые парентеральные композиции должны содержать противомикробное средство в бактериостатической или фунгистатической концентрации. Все парентеральные композиции должны быть стерильными, как это известно и практикуется в данной области.
В одном варианте воплощения, фармацевтические композиции обеспечиваются в виде готовых для использования стерильных растворов. В другом варианте воплощения, фармацевтические композиции обеспечиваются в виде стерильных сухих растворимых продуктов, включая лиофилизированные порошки и гиподермические таблетки, для реструктурирования носителем перед использованием. В следующем варианте воплощения, фармацевтические композиции обеспечиваются в виде готовых для использования стерильных суспензий. В следующем варианте воплощения, фармацевтические композиции обеспечиваются в виде стерильных сухих растворимых продуктов для реструктурирования носителем перед использованием. Еще в одном варианте воплощения, фармацевтические композиции обеспечиваются в виде готовых для использования стерильных эмульсий.
Фармацевтические композиции, обспечиваемые в настоящем изобретении, могут быть сформулированы в виде лекарственных форм немедленного или модифицированного высвобождения, включая формы замедленного, длительного, пульсирующего, контролируемого, направленного и программируемого высвобождения.
Фармацевтические композиции могут быть сформулированы в виде суспензии, твердого вещества, полутвердого вещества или тиксотропной жидкости, для введения в виде имплантируемого депо препарата. В одном варианте воплощения, фармацевтические композиции, представленные в настоящей заявке, диспергированы в твердой внутренней матрице, которая окружена внешней полимерной оболочкой, которая нерастворима в жидкостях организма, но обеспечивает возможность диффузии через нее активного ингредиента, содержащегося в фармацевтических композициях.
Подходящие внутренние матрицы включают полиметилметакрилат, полибутилметакрилат, пластифицированный или непластифицированный поливинилхлорид, пластифицированный найлон, пластифицированный полиэтилентерефталат, природный каучук, полиизопрен, полиизобутилен, полибутадиен, полиэтилен, этилен-винилацетатные сополимеры, силиконовые каучуки, полидиметилсилоксаны, силикон-карбонатные сополимеры, гидрофильные полимеры, такие как гидрогели сложных эфиров акриловой и метакриловой кислоты, коллаген, сшитый поливиниловый спирт и сшитый частично гидролизованный поливинилацетат.
Подходящие внешние полимерные оболочки включают полиэтилен, полипропилен, этилен/пропиленовые сополимеры, этилен/этилакрилатные сополимеры, этилен/винилацетатные сополимеры, силиконовые каучуки, полидиметилсилоксаны, неопреновый каучук, хлорированный полиэтилен, поливинилхлорид, сополимеры винилхлорида с винилацетатом, винилиденхлоридом, этиленом и пропиленом, иономерный полиэтилентерефталат, бутилкаучук, эпихлоргидриновые каучуки, сополимер этилена/винилового спирта, терполимер этилена/винилацетата/винилового спирта и сополимер этилена/винилоксиэтанола.
Местное введение
Фармацевтические композиции, обспечиваемые в настоящем изобретении, можно вводить местно на кожу, отверстия или слизистую оболочку. Местное введение, как этот термин используется в настоящей заявке, включает (интра)дермальное, в конъюнктиву, интракорнеальное, внутриглазное, офтальмическое, внутриушное, чрескожное, назальное, вагинальное, уретральное, респираторное и ректальное введение.
Фармацевтические композиции, обспечиваемые в настоящем изобретении, могут быть сформулированы в любые лекарственные формы, которые являются подходящими для местного введения для обеспечения локального или системного эффекта, включая эмульсии, растворы, суспензии, кремы, гели, гидрогели, мази, тонкодисперсные порошки, повязки, эликсиры, лосьоны, суспензии, настойки, пасты, пены, пленки, аэрозоли, препараты для орошения, спреи, суппозитории, бандажи, кожные пластыри. Фармацевтические композиции, сформулированные для местного введения, обспечиваемые в настоящем изобретении, также могут включать липосомы, мицеллы, микросферы, наносистемы и их смеси.
Фармацевтически приемлемые носители и эксципиенты, подходящие для использования в композициях для местного введения, представленных в настоящем изобретении, включают, но не ограничиваются этим, водные носители, смешиваемые с водой носители, неводные носители, противомикробные средства или консерванты против роста микроорганизмов, стабилизаторы, вещества, усиливающие солюбилизацию, изотонические агенты, буферные вещества, антиоксиданты, местные анестетики, суспендирующие и диспергирующие средства, смачивающие вещества или эмульгаторы, комплексообразующие вещества, секвестранты или хелатообразующие вещества, усилители пенетрации, криозащитные вещества, лиозащитные вещества, загустители и инертные газы.
Фармацевтические композиции также можно вводить местным путем с использованием электропорации, ионофореза, фонофореза, сонофореза или микроигл или безигловой инъекции, такой как POWDERJECT™ (Chiron Corp., Emeryville, CA) и BIOJECT™ (Bioject Medical Technologies Inc., Tualatin, OR).
Фармацевтические композиции, обспечиваемые в настоящем изобретении, могут быть представлены в форме мазей, кремов и гелей. Подходящие носители для мазей включают масляные или углеводородные носители, включая лярд, бензоинированный лярд, оливковое масло, масло семян хлопчатника и другие масла, вазелин; эмульгируемые или абсорбирующие носители, такие как гидрофильный петролатум, гидроксистеаринсульфат и безводный ланолин; водоудаляемые носители, такие как гидрофильная мазь; водорастворимые носители для мазей, включая полиэтиленгликоли с различной молекулярной массой; эмульгированные носители, либо в виде эмульсии вода-в-масле (W/O), либо в виде эмульсии масло-в-воде (O/W), включая цетиловый спирт, глицерилмоностеарат, ланолин и стеариновую кислоту (см., Remington: The Science and Practice of Pharmacy, supra). Эти носители представляют собой смягчающие вещества, но, как правило, требуют добавления антиоксидантов и консервантов.
Подходящая основа для крема может представлять собой масло-в-воде или вода-в-масле. Носители для кремов могут быть водо-смываемыми и содержат масляную фазу, эмульгатор и водную фазу. Масляную фазу также называют "внутренней" фазой, которая обычно состоит из петролатума и жирного спирта, такого как цетиловый или стеариловый спирт. Водная фаза обычно, хотя необязательно, превышает масляную фазу по объему и обычно содержит увлажняющее вещество. Эмульгатор в композиции крема может представлять собой неионное, анионное, катионное или амфотерное поверхностно-активное вещество.
Гели представляют собой полутвердые системы типа суспензии. Однофазные гели содержат органические макромолекулы, распределенные, по существу, однородно в жидком носителе. Подходящие гелеобразующие вещества включают сшитые полимеры акриловой кислоты, такие как карбомеры, карбоксиполиалкилены, CARBOPOL®; гидрофильные полимеры, такие как полиэтиленоксиды, полиоксиэтилен-полиоксипропиленовые сополимеры и поливиниловый спирт; целюлозные полимеры, такие как гидроксипропилцеллюлоза, гидроксиэтилцеллюлоза, гидроксипропилметилцеллюлоза, фталат гидроксипропилметилцеллюлозы и метилцеллюлозы; камеди, такие как трагакант и ксантановая камедь; альгинат натрия; и желатин. Для получения однородного геля можно добавить диспергирующие вещества, такие как спирт или глицерин, или гелеобразующее вещество можно диспергировать путем растирания в порошок, механического смешивания и/или перемешивания.
Фармацевтические композиции, обспечиваемые в настоящем изобретении, можно вводить ректально, уретрально, вагинально или перивагинально в форме суппозиториев, пессариев, палочек, припарок или компресса, паст, порошков, повязок, кремов, пластырей, контрацептивов, мазей, растворов, эмульсий, суспензий, тампонов, гелей, пен, спреев или клизьм. Эти лекарственные формы можно получить с использованием традиционных способов, как описано в Remington: The Science and Practice of Pharmacy, supra.
Ректальные, уретральные и вагинальные суппозитории представляют собой твердые препараты для введения в отверстия тела, которые являются твердыми при обычных температурах, но плавятся или размягчаются при температуре тела с высвобождением активного ингредиента(ингредиентов) внутри этих отверстий. Фармацевтически приемлемые носители, используемые в ректальных и вагинальных суппозиториях, включают основы или носители, такие как отвердители, которые обеспечивают точку плавления близкую к температуре тела, при формулировании их с фармацевтическими композициями, обспечиваемыми в настоящем изобретении; и антиоксиданты, описанные в настоящей заявке, включая бисульфит и метабисульфит натрия. Подходящие носители включают, но не ограничиваются этим, масло какао (какао-масло), глицерин-желатин, carbowax (полиоксиэтиленгликоль), спермацет, парафин, белый и желтый воск и подходящие смеси моно-, ди- и триглицеридов жирных кислот, гидрогели, такие как поливиниловый спирт, гидроксиэтилметакрилат, полиакриловая кислота; глицеринированный желатин. Можно использовать комбинации различных носителей. Ректальные и вагинальные суппозитории можно получить способом прессования или формования. Типичная масса ректального и вагинального суппозитория составляет от около 2 до около 3 г.
Фармацевтические композиции, обспечиваемые в настоящем изобретении, можно вводить офтальмическим путем в форме растворов, суспензий, мазей, эмульсий, гелеобразующих растворов, порошков для растворения, гелей, глазных вставок и имплантатов.
Фармацевтические композиции, обспечиваемые в настоящем изобретении, можно вводить интраназально или путем ингаляции в дыхательные пути. Фармацевтические композиции могут обеспечиваться в форме аэрозоля или раствора для доставки с использованием контейнера, в котором создано давление, насоса, спрея, распылителя, такого как распылитель с использованием электрогидродинамики для получения тонкого распыления или небулайзер, отдельно или в сочетании с подходящим пропеллентом, таким как 1,1,1,2-тетрафторэтан или 1,1,1,2,3,3,3-гептафторпропан. Фармацевтические композиции также могут быть представлены в виде сухого порошка для инсуффляции, отдельно или в сочетании с инертным носителем, таким как лактоза или фосфолипиды; и назальных капель. Для интраназального применения, порошок может включать биоадгезивное вещество, включая хитозан или циклодекстрин.
Растворы или суспензии для использования в контейнере под давлением, насосе, спрее, распылителе или небулайзере могут быть сформулированы таким образом, чтобы они содержали этанол, водный раствор этанола или подходящее альтернативное вещество для диспергирования, солюбилизации или продления высвобождения активного ингредиента, представленного в настоящем изобретении, пропеллент в качестве растворителя; и/или поверхностно-активное вещество, такое как сорбитантриолеат, олеиновая кислота или олигомолочная кислота.
Фармацевтические композиции, обспечиваемые в настоящем изобретении, можно измельчить до размера частиц, подходящего для доставки путем ингаляции, такого как около 50 мкм или меньше, или около 10 мкм или меньше. Частицы таких размеров можно получить с использованием способа измельчения, известного специалистам в данной области, такого как спиральное струйное измельчение, струйное измельчение в псевдоожиженном слое, сперхкритическая жидкостная обработка с образованием наночастиц, гомогенизация высокого давления или распылительная сушка.
Капсулы, блистеры и картриджи для использования в ингаляторе или инсуффляторе могут быть сформулированы так, чтобы они содержали порошкообразную смесь, включающую фармацевтические композиции, обспечиваемые в настоящем изобретении; подходящую основу для порошка, такую как лактоза или крахмал; и модифицирующую добавку, такую как L-лейцин, маннит или стеарат магния. Лактоза может быть безводной или в форме моногидрата. Другие подходящие эксципиенты или носители включают декстран, глюкозу, мальтозу, сорбит, ксилит, фруктозу, сахарозу и трегалозу. Фармацевтические композиции, обспечиваемые в настоящем изобретении, для введения путем ингаляции/интраназального введения могут дополнительно включать подходящую отдушку, такую как ментол и левоментол, или подсластители, такие как сахарин или сахарин натрий.
Фармацевтические композиции, обспечиваемые в настоящем изобретении для местного введения, могут быть сформулированы для немедленного высвобождения или модифицированного высвобождения, включая замедленное, длительное, пульсирующее, контролируемое, направленное и программируемое высвобождение.
Модифицированное высвобождение
Фармацевтические композиции, обспечиваемые в настоящем изобретении, могут бытьсформулированы в виде лекарственной формы модифицированного высвобождения. Как он используется в настоящей заявке, термин "модифицированное высвобождение" относится к лекарственной форме, в которой скорость или место высвобождения активного ингредиента(ингредиентов) отличаются от лекарственной формы немедленного высвобождения при введении таким же путем. Лекарственные формы модифицированного высвобождения включают формы замедленного, продленного, пролонгированного, длительного, пульсирующего, контролируемого, ускоренного и быстрого, направленного, программируемого высвобождения и удерживаемые в желудке лекарственные формы. Фармацевтические композиции для лекарственных форм модифицированного высвобождения можно получить с использованием различных устройств и способов модифицированного высвобождения, которые известны специалистам в данной области, включая, но не ограничиваясь этим, матричные устройства контролируемого высвобождения, устройства осмотически контролируемого высвобождения, устройства контролируемого высвобождения, включающие частицы различной формы и размера, ионообменные смолы, энетеросолюбильные покрытия, многослойные покрытия, микросферы, липосомы и их сочетания. Скорость высвобождения активного ингредиента(ингредиентов) также можно модифицировать путем изменения размера частиц и полиморфизма активного ингредиента(ингредиентов).
Примеры модифицированного высвобождения включают, но не ограничиваются этим, описанные в Патентах США №№: 3845770; 3916899; 3536809; 3598123; 4008719; 5674533; 5059595; 5591767; 5120548; 5073543; 5639476; 5354556; 5639480; 5733566; 5739108; 5891474; 5922356; 5972891; 5980945; 5993855; 6045830; 6087324; 6113943; 6197350; 6248363; 6264970; 6267981; 6376461; 6419961; 6589548; 6613358; и 6699500.
1. Матричные устройства контролируемого высвобождения
Фармацевтические композиции, обспечиваемые в настоящем изобретении для лекарственной формы модифицированного высвобождения, можно получить с использованием матричного устройства контролируемого высвобождения, известного специалистам в данной области (см., Takada et al. "Encyclipedia of Controlled Drug Delivery," Vol. 2, Mathiowitz Ed., Wiley, 1999).
В одном варианте воплощения, фармацевтические композиции, обспечиваемые в настоящем изобретении для лекарственной формы модифицированного высвобождения, получают с использованием разрушаемого матричного устройства, которое представляет собой набухаемые, разрушаемые или растворимые в воде полимеры, включая синтетические полимеры и природные полимеры и производные, такие как полисахариды и белки.
Вещества, полезные для образования разрушаемой матрицы, включают, но не ограничиваются этим, хитин, хитозан, декстран и пуллулан; смолистый агар, аравийскую камедь, камедь карайи, камедь плодов робинии, камедь трагаканта, каррагены, камедь гхатти, гуаровую камедь, ксантановую камедь и склероглюкан; крахмалы, такие как декстрин и мальтодекстрин; гидрофильные коллоиды, такие как пектин; фосфатиды, такие как лецитин; альгинаты; пропиленгликоль альгинат; желатин; коллаген; и производные целюлозы, такие как этилцеллюлоза (EC), метилэтилцеллюлоза (MEC), карбоксиметилцеллюлоза (CMC), CMEC, гидроксиэтилцеллюлоза (HEC), гидроксипропилцеллюлоза (HPC), ацетат целлюлозы (CA), пропионат целлюлозы (CP), бутират целлюлозы (CB), ацетатбутират целлюлозы (CAB), CAP, CAT, гидроксипропилметилцеллюлоза (HPMC), HPMCP, HPMCAS, ацетат гидроксипропилметилцеллюлоза тримеллитат (HPMCAT) и этилгидроксиэтилцеллюлоза (EHEC); поливинилпирролидон; поливиниловый спирт; поливинилацетат; сложные эфиры жирных кислот глицерина; полиакриламид; полиакриловая кислота; сополимеры этакриловой кислоты или метакриловой кислоты (EUDRAGIT®, Rohm America, Inc., Piscataway, NJ); поли(2-гидроксиэтил-метакрилат); полилактиды; сополимеры L-глутаминовой кислоты и этил-L-глутамата; разлагаемые сополимеры молочной кислоты-гликолевой кислоты; поли-D-(-)-3-гидроксимасляная кислота; и другие производные акриловой кислоты, такие как гомополимеры и сополимеры бутилметакрилата, метилметакрилата, этилметакрилата, этилакрилата, (2-диметиламиноэтил)метакрилата и (триметиламиноэтил)метакрилатхлорида.
В следующих вариантах воплощения, фармацевтические композиции формулируют с неразрушаемым матричным устройством. Активный ингредиент(ингредиенты) растворяют или диспергируют в инертной матрице, и он высвобождается преимущественно путем диффузии через инертную матрицу после введения. Вещества, подходящие для использования в качестве неразрушаемого матричного устройства, включают, но не ограничиваются этим, нерастворимые пластики, такие как полиэтилен, полипропилен, полиизопрен, полиизобутилен, полибутадиен, полиметилметакрилат, полибутилметакрилат, хлорированный полиэтилен, поливинилхлорид, сополимеры метилакрилата-метилметакрилата, сополимеры этилена-винилацетата, этилен/пропиленовые сополимеры, сополимеры этилена/этилакрилата, сополимеры винилхлорида с винилацетатом, винилиденхлоридом, этиленом и пропиленом, иономерный полиэтилентерефталат, бутилкаучук, эпихлоргидриновые каучуки, сополимер этилена/винилового спирта, терполимер этилена/винилацетата/винилового спирта и сополимер этилена/винилоксиэтанола, поливинилхлорид, пластифицированный найлон, пластифицированный полиэтилентерефталат, природный каучук, силиконовые каучуки, полидиметилсилоксаны, силиконкарбонатные сополимеры и; гидрофильные полимеры, такие как этилцеллюлоза, ацетат целлюлозы, кросповидон и сшитый частично гидролизованный поливинилацетат; и жирные соединения, такие как воск карнаубы, микрокристаллический воск и триглицериды.
В матричной системе контролируемого высвобождения желаемую кинетику высвобождения можно контролировать, например, посредством используемого типа полимера, вязкости полимера, размера частиц полимера и/или активного ингредиента(ингредиентов), отношения активного ингредиента(ингредиентов) к полимеру и другим эксципиентам или носителям в композициях.
Фармацевтические композиции, обспечиваемые в настоящем изобретении для лекарственной формы модифицированного высвобождения, можно получить способами, которые известны специалистам в данной области, включая непосредственное прессование, сухое или мокрое гранулирование с последующим прессованием, гранулирование из расплава с последующим прессованием.
2. Устройства осмотически контролируемого высвобождения
Фармацевтические композиции, обспечиваемые в настоящем изобретении для лекарственной формы модифицированного высвобождения, можно получить с использованием устройства осмотически контролируемого высвобождения, включающее однокамерную систему, двухкамерную систему, метод асимметрической мембраны (AMT) и систему экструдирования сердцевины (ECS). Как правило, такие устройства имеют, по меньшей мере, два компонента: (a) сердцевина, которая содержит активный ингредиент(ингредиенты); и (b) частично проницаемую оболочку с, по меньшей мере, одним отверстием для доставки, которая инкапсулирует сердцевину. Частично проницаемая оболочка контролирует приток воды к сердцевине из водного окружения, вызывая, таким образом, высвобождение лекарственного средства путем экструзии через отверстие(отверстия) для доставки.
Помимо активного ингредиента(ингредиентов), сердцевина устройства для осмотически контролируемого высвобождения необязательно включает осмотический агент, который создает движущую силу для транспорта воды из данного окружения в сердцевину устройства. Один класс осмотических агентов представляют водо-набухаемые гидрофильные полимеры, которые также называют "осмополимерами" и "гидрогелями", включая, но не ограничиваясь этим, гидрофильные виниловые и акриловые полимеры, полисахариды, такие как альгинат кальция, полиэтиленоксид (PEO), полиэтиленгликоль (PEG), полипропиленгликоль (PPG), поли(2-гидроксиэтилметакрилат), поли(акриловая)кислота, поли(метакриловая) кислота, поливинилпирролидон (PVP), сшитый PVP, поливиниловый спирт (PVA), PVA/PVP сополимеры, PVA/PVP сополимеры с гидрофобными мономерами, такими как метилметакрилат и винилацетат, гидрофильные полиуретаны, содержащие большое количество PEO блоков, натрий кроскармелозу, карраген, гидроксиэтилцеллюлозу (HEC), гидроксипропилцеллюлозу (HPC), гидроксипропилметилцеллюлозу (HPMC), карбоксиметилцеллюлозу (CMC) и карбоксиэтилцеллюлозу (CEC), альгинат натрия, поликарбофил, желатин, ксантановую камедь и натрийкрахмалгликолят.
Другой класс осмотических агентов представляют осмогены, которые способны поглощать воду, влияя на градиент осмотического давления через барьер окружающего покрытия. Подходящие осмогены включают, но не ограничиваются этим, неорганические соли, такие как сульфат магния, хлорид магния, хлорид кальция, хлорид натрия, хлорид лития, сульфат калия, фосфаты калия, карбонат натрия, сульфит натрия, сульфат лития, хлорид калия и сульфат натрия; сахара, такие как декстроза, фруктоза, глюкоза, инозит, лактоза, мальтоза, маннит, раффиноза, сорбит, сахароза, трегалоза и ксилит; органические кислоты, такие как аскорбиновая кислота, бензойная кислота, фумаровая кислота, лимонная кислота, малеиновая кислота, себациновая кислота, сорбиновая кислота, адипиновая кислота, эдетиновая кислота, глутаминовая кислота, п-толуолсульфоновая кислота, янтарная кислота и винная кислота; мочевину; и их смеси.
Осмотические агенты с разными скоростями растворения можно использовать для влияния на то, насколько быстро активный ингредиент(ингредиенты) изначально доставляется из лекарственной формы. Например, аморфные сахара, такие как MANNOGEM EZ (SPI Pharma, Lewes, DE) можно использовать для обеспечения ускоренной доставки в течение первых двух часов, чтобы сразу обеспечить желаемый терапевтический эффект, и постепенного и непрерывного высвобождения остального количества для поддержания желаемого уровня терапевтического или профилактического эффекта в течение длительного периода времени. В этом случае, активный ингредиент(ингредиенты) высвобождаются с такой скоростью, чтобы возместить метаболизированное или выведенное из организма количество активного ингредиента.
Сердцевина также включает большое разнообразие других эксципиентов и носителей, описанных в настоящей заявке, для усиления действия лекарственной формы или улучшения стабильности или обрабатываемости.
Вещества, полезные для получения частично проницаемой оболочки, включают различные сорта акрилов, винилов, эфиров, полиамидов, полиэфиров и целюлозных производных, которые являются водопроницаемыми и нерастворимыми в воде при физиологически релевантных значениях pH, или их можно сделать нерастворимыми в воде путем химического изменения, такого как сшивка. Примеры подходящих полимеров, полезных для получения покрытия, включают пластифицированный, непластифицированный и усиленный ацетат целлюлозы (CA), диацетат целлюлозы, триацетат целлюлозы, CA пропионат, нитрат целлюлозы, ацетатбутират целлюлозы (CAB), CA этилкарбамат, CAP, CA метилкарбамат, CA сукцинат, целлюлоза ацетат тримеллитат (CAT), CA диметиламиноацетат, CA этилкарбонат, CA хлорацетат, CA этилоксалат, CA метилсульфонат, CA бутилсульфонат, CA p-толуолсульфонат, агар ацетат, триацетат амилозы, бетаглюканацетат, бетаглюкантриацетат, ацетальдегид диметилацетат, триацетат камедь плодов робинии, гидроксилированный этилен-винилацетат, EC, PEG, PPG, PEG/PPG сополимеры, PVP, HEC, HPC, CMC, CMEC, HPMC, HPMCP, HPMCAS, HPMCAT, поли(акриловые) кислоты и сложные эфиры и поли-(метакриловые) кислоты и сложные эфиры и их сополимеры, крахмал, декстран, декстрин, хитозан, коллаген, желатин, полиалкены, полиэфиры, полисульфоны, полиэфирсульфоны, полистиролы, поливинилгалогениды, сложные и простые поливиниловые эфиры, природные воски и синтетические воски.
Частично проницаемая оболочка также может представлять собой гидрофобную микропористую мембрану, где поры, по существу, заполнены газом и не смачиваются водной средой, но являются проницаемыми для водяного пара, как раскрыто в Патенте США № 5798119. Такая гидрофобная, но проницаемая для водяного пара мембрана типично состоит из гидрофобных полимеров, таких как полиалкены, полиэтилен, полипропилен, политетрафторэтилен, производные полиакриловой кислоты, полиэфиры, полисульфоны, полиэфирсульфоны, полистиролы, поливинилгалогениды, поливинилиденфторид, простые и сложные поливиниловые эфиры, природные воски и синтетические воски.
Имеющееся на частично проницаемой оболочке отверстие(отверстия) для доставки может быть образовано после образования покрытия путем механического или лазерного просверливания. Отверстие(отверстия) для доставки также может быть образовано in situ путем эрозии слоя водорастворимого вещества или разрыва более тонкой части оболочки над внедренным в нее ядром. Кроме того, отверстие для доставки может быть образовано в процессе нанесения покрытия, как в случае асимметрических мембранных покрытий типа, раскрытого в Патентах США №№ 5612059 и 5698220.
Общее количество активного ингредиента(ингредиентов), которое высвобождается и скорость высвобождения, по существу, можно модулировать с учетом толщины и пористости частично проницаемой оболочки, композиции сердцевины и количества, размера и положения отверстий для доставки.
Фармацевтические композиции для лекарственной формы осмотически контролируемого высвобождения могут дополнительно включать дополнительные традиционные эксципиенты или носители, описанные в настоящей заявке, для улучшения действия или обрабатываемости композиции.
Лекарственные формы с осмотически контролируемым высвобождением можно получить в соответствии с традиционными способами и процедурами, которые известны специалистам в данной области (см., Remington: The Science and Practice of Pharmacy, supra; Santus and Baker, J. Controlled Release 1995, 35, 1-21; Verma et al., Drug Development and Industrial Pharmacy 2000, 26, 695-708; Verma et al., J. Controlled Release 2002, 79, 7-27).
В некоторых вариантах воплощения фармацевтические композиции, представленные в настоящей заявке, сформулированы в виде лекарственной формы с AMT контролируемым высвобождением, которая включает асимметрическую осмотическую мембрану, которая покрывает сердцевину, включающую активный ингредиент(ингредиенты), и другие фармацевтически приемлемые эксципиенты или носители. См., Патент США № 5612059 и WO 2002/17918. Лекарственные формы с AMT контролируемым высвобождением можно получить в соответствии с традиционными способами и процедурами, которые известны специалистам в данной области, включая непосредственное прессование, сухое гранулирование, мокрое гранулирование и способ нанесения покрытия окунанием.
В некоторых вариантах воплощения фармацевтические композиции, представленные в настоящей заявке, сформулированы в виде лекарственной формы с ESC контролируемым высвобождением, которая покрывает сердцевину, включающую активный ингредиент(ингредиенты), гидроксилэтилцеллюлозу и другие фармацевтически приемлемые эксципиенты или носители.
3. Устройства контролируемого высвобождения, включающие частицы различной формы и размера
Фармацевтические композиции, обспечиваемые в настоящем изобретении для лекарственной формы модифицированного высвобождения, можно получить в виде устройства контролируемого высвобождения с частицами различной формы и размера, которое включает множество частицы, гранул или шариков с размером от около 10 мкм до около 3 мм, от около 50 мкм до около 2,5 мм или от около 100 мкм до около 1 мм в диаметре. Такие композиции в виде множества различных частиц можно получить способами, которые известны специалистам в данной области, включая мокрое и сухое гранулирование, экструзию/сферонизацию, роликовое уплотнение, отверждение расплава и путем нанесения покрытия методом спрея на зерна сердцевины. См., например, Multiparticulate Oral Drug Delivery; Marcel Dekker: 1994; и Pharmaceutical Pelletization Technology; Marcel Dekker: 1989.
Другие эксципиенты или носители, описанные в настоящей заявке, можно смешивать с фармацевтическими композициями для облегчения обработки и образования множества частиц. Полученные частицы могут сами образовывать устройство, включающее множество различных частиц, или на них можно нанести покрытие с использованием различных пленкообразующих веществ, таких как энтеросолюбильные полимеры, водонабухаемые и водорастворимые полимеры. Композиции, состоящие из множества различных частиц, могут быть далее переработаны с получением капсул или таблеток.
4. Направленная доставка
Фармацевтические композиции, обспечиваемые в настоящем изобретении, также могут быть сформулированы для нацеливания на конкретную ткань, рецептор или другую область организма субъекта, подлежащего лечению, включая липосомные системы доставки, системы доставки на основе высвобождения эритроцитов и на основе антител. Примеры включают, но не ограничиваются этим, Патенты США №№ 6316652; 6274552; 6271359; 6253872; 6139865; 6131570; 6120751; 6071495; 6060082; 6048736; 6039975; 6004534; 5985307; 5972366; 5900252; 5840674; 5759542; и 5709874.
D. Оценка активности соединений
Стандартные физиологические, фармакологические и биохимические процедуры являются доступными для испытания соединений, чтобы определить те из них, которые обладают биологической активностью, которая модулирует активность JAK киназ, включая дикого типа и мутантные JAK киназы. Такие анализы включают, например, биохимические анализы, такие как анализы связывания, см., Fabian et al., Nature Biotechnology 2005, 23,329-336, анализы включения радиоактивности, а также различные клеточные анализы.
Примеры методов клеточного анализа включают измерение STAT5A фосфорилирования, например, методом твердофазного иммуноферментного анализа (ELISA), или измерение пролиферации в лейкозных клеточных линиях, таких как TF-1 или HEL-2, например, посредством включения BrdU, методом флуоресцентного мечения или при помощи анализа репортера, активированного транскрипционным фактором STAT5. Клетки, полезные для таких анализов, включают клетки с JAK дикого типа, такие как TF-1, или с мутированной JAK, такие как клеточная линия HEL-2, которые экспрессируют конститутивно активную JAK2, имеющую V617F мутацию. Подходящие клетки включают клетки, полученные путем культивирования клеток из образцов, взятых у пациента, а также клетки, полученные с использованием рутинных методов молекулярной биологии, например, ретровирусной трансдукции, трансфекции, мутагенеза и т.п.
E. Способы применения соединений и композиций
Также в настоящей заявке представлены способы использования раскрываемых соединений и композиций или их фармацевтически приемлемых солей, сольватов или гидратов для лечения, профилактики или облегчения заболевания или расстройства, которое опосредовано или на которое иным образом влияет активность JAK киназы, включая JAK2 киназу, или одного или нескольких симптомов заболеваний или расстройств, которые опосредованы или на которые иным образом влияет активность JAK киназы, включая JAK2 киназу. JAK киназа может представлять собой форму дикого типа и/или мутантную форму JAK2 киназы. В соответствии с описанным выше, такие заболевания или расстройства включают, без ограничения: миелопролиферативные расстройства, такие как истинная полицитемия (PCV), эссенциальная тромбоцитемия и идиопатический миелофиброз (IMF); лейкоз, такой как миелоидный лейкоз, включая хронический миелоидный лейкоз (CML), иматиниб-резистентные формы CML, острый миелоидный лейкоз (AML) и подтип AML, острый мегакариобластный лейкоз (AMKL); лимфопролиферативные заболевания, такие как миелома; рак, включая рак головы и шеи, рак предстательной железы, рак молочной железы, рак яичника, меланому, рак легкого, опухоль головного мозга, рак поджелудочной железы и карциному почки; и воспалительные заболевания или расстройства, связанные с иммунной дисфункцией, иммунодефицитом, иммуномодуляцией, аутоиммунный заболевания, отторжение тканевого трансплантата, болезнь трансплантат-против-хозяина, заживление ран, заболевание почек, рассеянный склероз, тиреоидит, диабет типа 1, саркоидоз, псориаз, аллергический ринит, воспалительное заболевание кишечника, включая болезнь Крона и язвенный колит (UC), системную красную волчанку (SLE), артрит, остеоартрит, ревматоидный артрит, остеопороз, астму и хроническое обструктивное заболевание легких (COPD) и синдром сухих глаз (или сухой кератоконъюнктивит (KCS)).
В некоторых вариантах воплощения в настоящей заявке представлены способы использования раскрываемых соединений и композиций или их фармацевтически приемлемых солей, сольватов или гидратов для лечения, профилактики или облегчения заболевания или расстройства, выбранного из миелопролиферативных расстройств, таких как истинная полицитемия (PCV), эссенциальная тромбоцитемия и идиопатический миелофиброз (IMF) и гиперэозинофильный синдром (HES); лейкоз, такой как миелоидный лейкоз, включая хронический миелоидный лейкоз (CML), иматиниб-резистентные формы CML, острый миелоидный лейкоз (AML), острый лимфобластный лейкоз (ALL) и подтип AML, острый мегакариобластный лейкоз (AMKL); лимфопролиферативные заболевания, такие как миелома; рак, включая рак головы и шеи, рак предстательной железы, рак молочной железы, рак яичника, меланома, рак легкого, рак головного мозга, рак поджелудочной железы, рак желудка, рак щитовидной железы, карцинома почки, саркома Капоши, болезнь Кастлмана, меланома; и воспалительные заболевания или расстройства, связанные с иммунной дисфункцией, иммунодефицитом или иммуномодуляцией, такие как отторжение тканевого трансплантата, болезнь трансплантат-против-хозяина, заживление ран, заболевание почек; аутоиммунные заболевания, такие как рассеянный склероз, тиреоидит, диабет типа 1, саркоидоз, псориаз, аллергический ринит, атопический дерматит, злокачественная миастения, воспалительное заболевание кишечника, включая болезнь Крона и язвенный колит (UC), системная красная волчанка (SLE), артрит, остеоартрит, ревматоидный артрит, остеопороз, астма и хроническое обструктивное заболевание легких (COPD), воспалительные заболевания глаз, включая коонъюнктивит, увеит, ирит, склерит, воспалительные заболевания верхних дыхательных путей, включая верхние дыхательные пути, такие как ринит и синусит, и воспалительные заболевания нижних дыхательных путей, включая бронхит; воспалительная миопатия, такая как миокардит, другие воспалительные заболевания, такие как ишемические реперфузионные поражения, связанные с воспалительным ишемическим событием, таким как удар или остановка сердца, и другие воспалительные состояния, такие как синдром системного воспалительного ответа (SIRS) и сепсис.
В некоторых вариантах воплощения JAK-опосредованые заболевания и расстройства включают рестеноз, фиброз и склеродерму. В некоторых вариантах воплощения JAK-опосредованые заболевания включают вирусные заболевания, такие как вирус Эпштейна-Барра (EBV), гепатит (гепатит B или гепатит C), вирус иммунодефицита человека (ВИЧ), T-лимфотропный вирус типа 1 человека (HTLV-I), вирус ветряной оспы и папиллома-вирус человека (HPV).
F. Комбинированная терапия
Кроме того, специалистам в данной области должно быть понятно, что соединения, изомеры и фармацевтически приемлемые соли, сольваты или гидраты, представленные в настоящем изобретении, включая фармацевтические композиции и композиции, содержащие эти соединения, можно использовать в различных способах комбинированной терапии для лечения состояний и заболеваний, описанных выше. Таким образом, настоящим изобретением также предусматривается использование соединений, изомеров и фармацевтически приемлемых солей, сольватов или гидратов, представленных в настоящем изобретении, в сочетании с другими активными фармацевтическими средствами для лечения заболеваний/состояний, описанных в настоящей заявке.
В одном варианте воплощения, такие дополнительные фармацевтические средства включают, без ограничения противораковые средства, включая химиотерапевтические средства и антипролиферативные средства; противовоспалительные средства и иммуномодуляторные средства или иммуносупрессивные средства.
В некоторых вариантах воплощения противораковые средства включают анти-метаболиты (например, 5-фторурацил, цитарабин, метотрексат, флударабин и другие), средства против образования микротрубочек (например, алкалоиды барвинка, такие как винкристин, винбластин; таксаны, такие как паклитаксел и доцетаксел), алкилирующие средства (например, циклофосфамид, мелфалан, кармустин, средства на основе нитрозомочевины, такие как бисхлорэтилнитрозомочевина и гидроксимочевина), средства на основе платины (например, цисплатин, карбоплатин, оксалиплатин, сатраплатин и CI-973), антрациклины (например, доксрубицин и даунорубицин), противоопухолевые антибиотики (например, митомицин, идарубицин, адриамицин и дауномицин), ингибиторы топоизомеразы (например, этопозид и камптотецины), средства против ангиогенеза (например, Sutent®, сорафениб и Бевацизумаб) или любые другие цитотоксические средства, (например, эстрамустин фосфат, преднимустин), гормоны или агонисты, антагонисты, частичные агонисты или частичные антагонисты гормонов, ингибиторы киназы (такие как иматиниб) и лучевую терапию.
В некоторых вариантах воплощения противовоспалительные средства включают ингибиторы металлопротеиназы матрикса, ингибиторы провоспалительных цитокинов (например, анти-TNF молекулы, растворимые рецепторы TNF и IL1), нестероидные противовоспалительные лекарственные средства (NSAIDs), такие как ингибиторы простагландинсинтазы (например, холинмагний салицилат и салицилсалициловая кислота), ингибиторы COX-1 или COX-2 или агонисты глюкокортикоидного рецептора, такие как кортикостероиды, метилпреднизон, преднизон или кортизон.
Соединение или композицию, представленные в настоящем изобретении, или их фармацевтически приемлемые соли, сольваты или гидраты, можно вводить одновременно с введением, до или после введения одного или нескольких из указанных выше средств.
Также обеспечиваются фармацевтические композиции, содержащие соединение, представленное в настоящем изобретении, или его фармацевтически приемлемые соли, сольваты или гидраты и одно или несколько из указанных выше средств.
Также обеспечивается комбинированная терапия, которая обеспечивает лечение или препятствует возникновению симптомов или соответствующих осложнений рака и подобных заболеваний и расстройств, включающая введение субъекту, нуждающемуся в этом, одного из соединений или композиций, раскрытых в настоящей заявке, или их фармацевтически приемлемых солей, сольватов или гидратов, с одним или несколькими противораковыми средствами.
G. Получение соединений
Исходные вещества в примерах синтеза, представленных в настоящей заявке, либо являются доступными из коммерческих источников, либо их можно получить с использованием известных из литературы процедур (например, March Advanced Organic Chemistry: Reactions, Mechanisms and Structure, (1992) 4th Ed.; Wiley Interscience, New York). Все коммерчески доступные соединения использовали без дополнительной очистки, если не указано иное. Спектры 300 МГц протонного (1H) ядерного магнитного резонанса (ЯМР) записывали на Bruker Avance 300 ЯМР спектрометре. Значимые пики представляли в виде табличных данных, и они типично включали: количество протонов и мультиплетность (с - синглет; д - дублет; т - триплет; кв. - квартет; м - мультиплет; ушир.с - уширенный синглет). Химические сдвиги указаны в виде миллионных долей (δ) относительно тетраметилсилана. Масс-спектры низкого разрешения (MS) получали в виде масс-спектров ионизации электроспреем (ESI), которые записывали на устройстве Shimadzu HPLC/MS с использованием обращено-фазовых условий (ацетонитрил/вода, 0,05% уксусной кислоты). Препаративную обращено-фазовую ВЭЖХ обычно осуществляли с использованием ВЭЖХ системы Varian, снабженной колонкой с обращенной фазой Phenomenex фенилгексил, Phenomenex Luna C18 или Varian Pursuit дифенил; типичные условия элюирования включали использование градиента, включающего увеличивающуюся композицию органического сорастворителя (0,05% HOAc/CH3CN или 0,05% HOAc/MeOH) до водного со-растворителя (0,05% водного раствора HOAc). Хроматографию на силикагеле осуществляли либо вручную, обычно следуя опубликованной процедуре для флэш-хроматографии (Still et al. (1978) J. Org. Chem. 43:2923), либо на автоматизированной системе (например, Biotage SP instrument) с использованием колонок с насадкой из силикагеля.
Должно быть понятно, что в следующем далее описании, сочетания заместителей и/или переменных в представленных формулах допустимы только в том случае, когда это приводит к подходящим соединениям в стандартных условиях.
Также, специалистам в данной области должно быть понятно, что в способе, описанном ниже, может быть необходима защита функциональных групп промежуточных соединений подходящими защитными группами. Такие функциональные группы включают гидрокси, амино, меркапто и карбоновую кислоту. Подходящие защитные группы для гидрокси включают триалкилсилил или диарилалкилсилил (например, трет-бутилдиметилсилил, трет-бутилдифенилсилил или триметилсилил), тетрагидропиранил, бензил и подобные. Подходящие защитные группы для амино, амидино и гуанидино включают трет-бутоксикарбонил, бензилоксикарбонил и подобные. Подходящие защитные группы для меркапто включают -C(O)-R (где R представляет собой алкил, арил или аралкил), п-метоксибензил, тритил и подобные. Подходящие защитные группы для карбоновой кислоты включают алкиловый, ариловый или аралкиловый эфиры.
Защитные группы могут быть добавлены или удалены в соответствии со стандартными способами, которые хорошо известны специалистам в данной области, как указано в настоящей заявке. Использование защитных групп подробно описано в Green, T. W. and P.G.M. Wutz, Protective Groups in Organic Synthesis (1991), 2nd Ed., Wiley-Interscience.
Специалист в данной области со средней квалификацией сможет легко определить, какие варианты для каждого заместителя являются подходящими для условий реакции каждой Схемы. Кроме того, заместители выбраны из компонентов, как указано в описании выше, и могут быть присоединены к исходным веществам, промежуточным соединениям и/или конечным продуктам, в соответствии со схемами, известными специалистам со средней квалификацией.
Также должно быть понятно, что соединения, представленные в настоящем изобретении, могут существовать в виде одного или нескольких изомеров, которые представляют собой E/Z изомеры, энантиомеры и/или диастереомеры.
Соединения формулы (I) могут быть получены, как показано на представленных ниже схемах, если не указано иное, и различные заместители определены в настоящей заявке в других ее разделах.
В настоящей заявке использованы стандартные аббревиатуры и акронимы, определенные в J. Org. Chem. 2007 72(1): 23A-24A. Другие аббревиатуры и акронимы, используемые в настоящей заявке, имеют следующее значение:
Соединения, представленные в настоящей заявке, синтезировали в соответствии со следующими схемами и описаниями. Схема 1 иллюстрирует общий путь к ключевым синтетическим промежуточным соединениям и соединениям, представленным в настоящем изобретении, через замещенные антранилимиды 5. Исходя из замещенной бензойной кислоты, нитрование в стандартных условиях, например, обработка азотной кислотой в кислотных условиях при нагревании, если это необходимо, дает соответствующую нитробензойную кислоту 2, которую отделяют от каких-либо нежелательных региоизомеров при помощи хроматографии или кристаллизации. Восстановление нитрогруппы в стандартных условиях, например, с использованием газообразного водорода и катализатора на основе благородного металла в растворителе, таком как вода, низший спирт, EtOAc или DMF; металлический Sn или Fe в кислотных условиях; или SnCl2 в растворителе, таком как EtOH или DMF, дает соответствующую антраниловую кислоту 4. Преобразование карбоксильной группы 4 в карбоксамидную группу 5 осуществляют любым из многочисленных стандартных способов, включая обработку аммиаком или хлоридом аммония в присутствии связывающих реагентов, таких как HATU, EDCI, (гексафторфосфат бензотриазол-1-илокси)трис(диметиламино)фосфония, дициклогексилкарбодиимид и подобные, или, альтернативно, через хлорангидрид кислоты при обработке кислотой с тионилхлоридом или фосфорилхлоридом или т.п., с последующим добавлением подходящей формы аммиака, такой как аммиак в MeOH или гидроксид аммония. Антраниламид 5 затем конденсируют с подходяще активированным производным карбоновой кислоты 6 с последующей дегидратирующей циклизацией, промотируемой, например, нагреванием или TMSCl, в присутствии третичного аминового основания, такого как TEA, DIEA или пиридин, с получением 4-гидроксихиназолинов 8. Гидроксильную группу 4-гидроксихиназолина 8 затем преобразовывают в удаляемую группу. Примеры такого преобразования включают обработку галогенирующим агентом, таким как фосфорилгалогенид, с получением хиназолина 9 (Z = галоген), или сульфонилхлоридом с получением хиназолина 9 (Z = сульфонилокси производное), или галогенирующим агентом, затем органическим меркаптаном с последующим окислением серы с получением соединения 7 (Z = сульфинильное или сульфонильное производное). Хиназолин 9 затем подвергают взаимодействию с подходящим пиразоламином (пиразол-NH2) в подходящем растворителе, таком как DMF или диметилацетамид, необязательно в присутствии источника ионов иода, например, иодида калия или иодида тетрабутиламмония, необязательно при нагревании, с получением, после выделения, соединения 10.
Схема 1
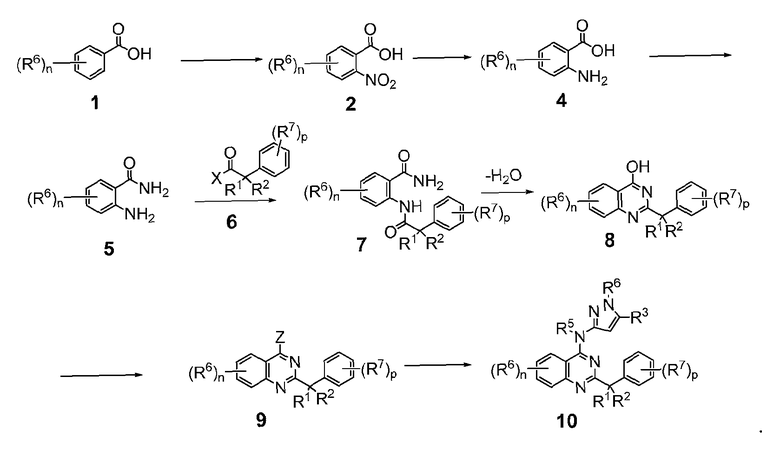
Должно быть понятно, что на подходящих стадиях способа синтеза, таких, которые показаны на Схеме 1, одна или несколько R6 групп формул 1-10 может служить в качестве предшественника модифицированной R6 группы в конечном соединении, обеспечиваемом в настоящем изобретении. Например, когда в соединении 1 R6=CO2Me, метоксикарбонильная группа может быть преобразована на подходящей стадии синтеза, например, в карбоксильную группу путем гидролиза, в амид путем гидролиза с последующей активацией карбоксигруппы и обработкой амином, в гидроксиметильную группу путем восстановления, в третичный карбинол путем обработки двумя эквивалентами реагента Гриньяра, в аминометильную группу путем восстановления до гидроксиметильной группы с последующим окислением до альдегида и последующим восстановительным аминированием подходящим амином в присутствии селективного восстановителя, такого как триацетоксиборогидрид натрия. Подобным образом, когда R6=OCH2Ph, тогда R6 может быть преобразован в OH путем гидролитического расщепления бензильной группы, с последующим алкилированием полученной фенольной гидроксильной группы при помощи необязательно замещенного алкилгалогенида или необязательно замещенного алкилсульфоната с получением соответствующего ароматического эфира.
Подобным образом, некоторые R группы в промежуточных соединениях 8, 9 или 10 могут быть включены, как показано на Схеме 1, и затем преобразованы в альтернативные R группы.
На Схеме 2, антраниламид 5, полученный в соответствии со Схемой 1, обрабатывают активированным производным щавелевой кислоты, таким как диалкилоксалат, либо в чистом виде, либо в подходящем растворителе, таком как EtOH или HOAc; или антраниламид 5 обрабатывают хлоридом моноалкилового эфира щавелевой кислоты в подходящем растворителе, таком как DCM, в присутствии основания, такого как TEA, и, необязательно, в присутствии катализатора, такого как DMAP; или антраниламид 5 обрабатывают моноалкиловым эфиром цианооксоацетата при нагревании в подходящем растворителе, таком как ацетонитрил или DMF, в присутствии основания, такого как TEA. Аналогично способам, описанным на Схеме 1, последующая обработка полученных продуктов в дегидратирующих условиях, например, при нагревании с или без TMSCl, в присутствии подходящего основания, такого как DIEA, в подходящем растворителе, таком как DCE, дает бициклический продукт 11. Кроме того, незначительно модифицированная процедура выхода на промежуточные соединения 11 описана в патентной заявке WO2004/20441, включенной посредством ссылки во всей полноте. Условия для преобразования соединения 11 в соединение 12, преобразования соединения 12 в соединение 13 или соединение 14 и для преобразования соединения 13 в соединение 15 аналогичны тем, которые описаны на Схеме 1 для аналогичных преобразований. Алкоксикарбонильную группу в 2-положении хиназолинового кольца соединения 12 или соединения 14 можно обработать металлоареном, например, ариллитиевым соединением или арильным реагентом Гриньяра в подходящем растворителе, таком диэтиловый эфир, ТГФ или другой эфирный растворитель, с получением кетона 13 или 15, соответственно. Хотя это не показано на Схеме 2, промежуточное соединение 11 также можно аналогичным образом обработать металлоареном с получением соответствующего арилкетона, который затем может быть преобразован в соединение 15 путем преобразования 4-гидроксигруппы в удаляемую группу, например, хлор, с последующим замещением группы хлора подходяще замещенным 3-аминопиразолом с использованием реагентов и условий, аналогичных тем, которые описаны для аналогичных преобразований на Схеме 1.
Схема 2
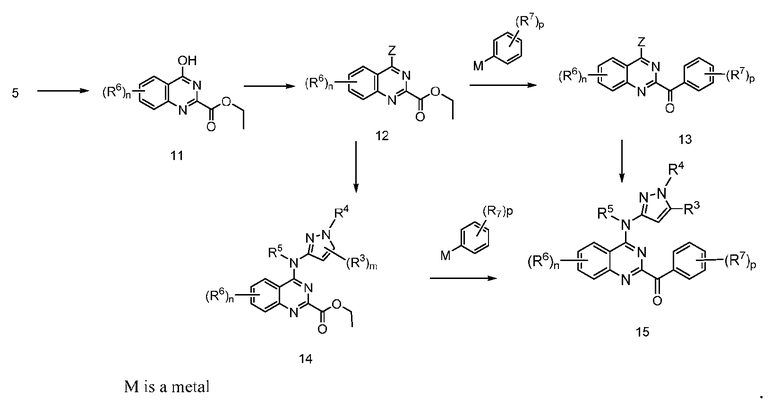
М представляет собой металл
На Схеме 3 проиллюстрированы способы для преобразования кетонов 15, полученных в соответствии со Схемой 2, в следующие соединения, обеспечиваемые в настоящем изобретении. Обработка соединения 15 подходящим восстановителем, например, гидридным реагентом, таким как борогидрид натрия в спиртовом растворителе или борогидрид лития в эфирном растворителе или соответствущий борогидридный или алюминийгидридный реагент в подходящей системе растворителя, восстанавливает кетон до соответствующего карбинола 16. Обработка кетона 15 алкил- или ариллитиевым или магнийгалогенидным реагентом дает третичный карбинол 17. Альтернативно, обработка кетона 15 O-замещенным или O-незамещенным гидроксиламином в подходящем растворителе, таком как спирт или смесь спирт/вода, необязательно в присутствии кислотного или щелочного катализатора, дает оксим 18. Оксим можно подвергнуть дальнейшей обработке в восстановительных условиях, например, боран-аминовым комплексом в присутствии сильной кислоты при нагревании в течение пролонгированного времени реакции или в условиях гидрогенолиза (H2, катализатор на основе благородного металла, необязательно в присутствии органической или минеральной кислоты) в течение длительных периодов времени с получением амина 19. Альтернативно, использование более мягких условий, таких как более низкая температура, меньшее время реакции или более мягкая кислота, если она присутствует, дает алкоксиламин или гидроксиламин 20.
Схема 3
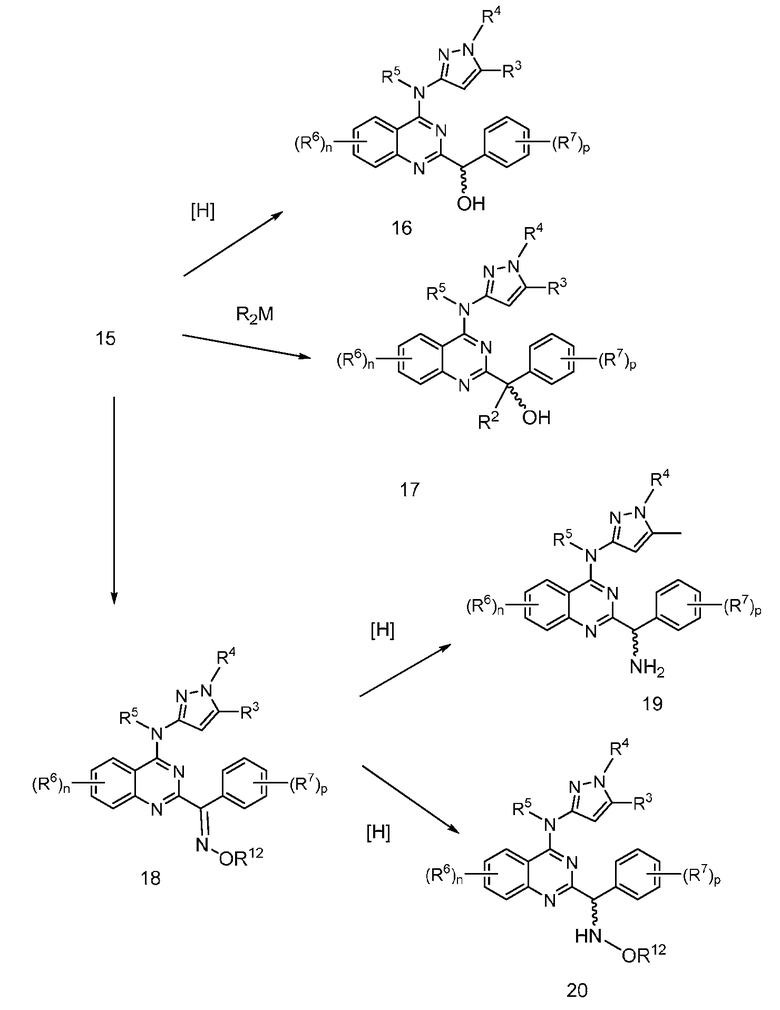
Альтернативно, амины 19 можно получить в соответствии с последовательностью синтеза, проиллюстрированной на Схеме 4. На Схеме 4, гидроксильную группу в карбиноле 16 преобразовывают в удаляемую группу Z путем обработки, например, галогенидом фосфора с получением соединения 21 (Z = галоген), или путем обработки сульфонилгалогенидом в подходящем растворителе, таком как DCM, и в присутствии галогенводородного акцептора, такого как третичный амин, например DIEA или пиридин, с получением соединения 21 (Z = сульфонилокси производное). Для последней реакции, в случае случайного сульфонилирования на одном или нескольких других участках молекулы, посторонние сульфонильные группы удаляют на поздей стадии путем обработки нуклеофилом, таким как гидроксид, аммиак или гидразин.
Как показано на Схеме 4, промежуточное соединение 21 подвергают дальнейшему преобразованию в азид 22 путем замещения удаляемой группы Z азидным ионом, например, путем обработки соединения 21 азидом щелочного металла в подходящем растворителе, таком как диполярный апротонный растворитель, например, DMF или DMSO, при температуре в пределах от около 0°C до около 100°C. Восстановление азида при помощи системы восстановителя, например, трифенилфосфина с последующей обработкой в условиях гидролиза или гидрогенолиза (H2, катализатор на основе благородного металла) в подходящем растворителе, таком как спирт или DMF, дает амин 23.
Схема 4 также иллюстрирует, что амины 23 могут быть далее модифицированы с получением продуктов 24 по настоящему изобретению, где один из атомов водорода аминогруппы замещен группой R14. Обработка амина 23 ацилирующим агентом, таким как хлорангидрид кислоты или ангидрид кислоты, обычно в присутствии основания и, необязательно, в присутствии катализатора ацилирования, такого как DMAP или пиридин, в подходящем растворителе, таком как EtOAc, DCM, DMF или ТГФ, дает продукты 24 (R14 = ацил). Альтернативно, амин 23 обрабатывают алкилхлороформиатом, например, этил- или изопропилхлороформиатом, в присутствии основания и, необязательно, в присутствии катализатора ацилирования, такого как DMAP или пиридин, в подходящем растворителе, таком как EtOAc, DCM, DMF или ТГФ, с получением соответствующего карбамата 24 (R14=-C(O)OR12). Альтернативно, амин 23 обрабатывают сульфонилгалогенидом, например, метан- или бензолсульфонилхлоридом, в присутствии основания и, необязательно, в присутствии катализатора ацилирования, такого как DMAP или пиридин, в подходящем растворителе, таком как EtOAc, DCM, DMF или ТГФ, с получением соответствующего сульфонамида 23 (R14=-SO2R12). Альтернативно, амин 23 в подходящем растворителе, таком как MeOH, EtOH или DME, обрабатывают альдегидом в дегидратирующих условиях, например, в присутствии молекулярных сит или триметилортоформиата, необязательно в присутствии кислотного катализатора, такого как уксусная кислота или хлористоводородная кислота, с получением промежуточного иминового соединения, и смесь обрабатывают, либо одновременно, либо последовательно, селективным восстановителем, таким как цианоборогидрид натрия или триацетоксиборогидрид натрия или (особенно в случае предварительной обработки альдегидом) борогидрид натрия, с получением нового амина 23 (R14 = алкил или арил, каждый из которых необязательно замещен).
Схема 4
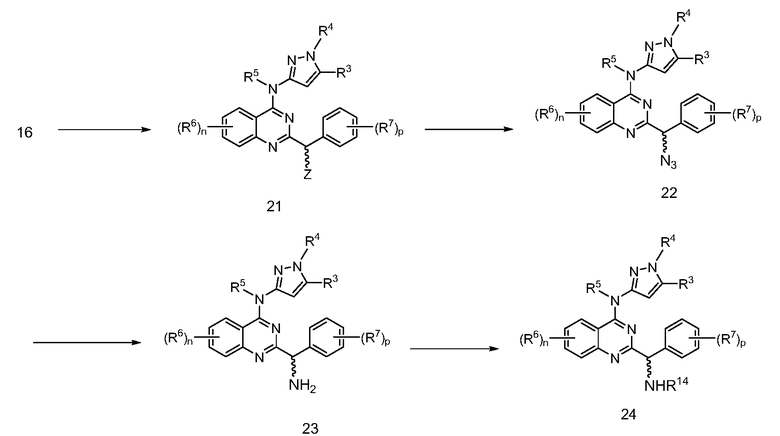
Репрезентативный способ проиллюстрирован на Схеме 5 для преобразования кетона 15 в дополнительные соединения, обеспечиваемые в настоящем изобретении. Кетон 15 в подходящем растворителе, таком как ТГФ, DME, диглим или DMSO, обрабатывают анионом, образованным в результате обработки триалкилфосфоноацетата сильным основанием, таким как гидрид натрия, амид лития, диметилсульфоксидный (DMSO) анион или т.п., при подходящей температуре в пределах от около 0°C до около 100°C с получением, после окончательной обработки и выделения, α,β-ненасыщенного сложного эфира 25. Обработка соединения 25 в подходящих восстановительных условиях, например, H2 в присутствии катализатора на основе благородного металла в подходящем растворителе, таком как спирт или DMF, дает сложный эфир 26. Восстановление сложноэфирной группы соединения 19 можно осуществить путем обработки гидридной восстановительной системой, такой как LiAlH4/ТГФ, LiBH4/ТГФ или Ca(BH4)2/EtOH/ТГФ, с получением первичного спирта 27. Гидроксильную группу спирта 27 преобразовывают в удаляемую группу Z с использованием способа, хорошо известного из уровня техники, например, путем обработки соединения 20 фосфорилгалогенидом с получением соединения 28 (Z = галогенид), или путем обработки соединения 27 сульфонилгалогенидом с получением соединения 28 (Z = сульфонилокси производное). Промежуточное соединение 28 затем может быть обработано нуклеофилом с получением соединения 29. Например, обработка соединения 28 меркаптидным нуклеофилом дает соединение 29 (Y=S); обработка соединения 28 алкоксидным нуклеофилом дает соединение 29 (Y=O); обработка соединения 28 аминовым нуклеофилом дает соединение 29 (Y=NH или NRX).
Схема 5
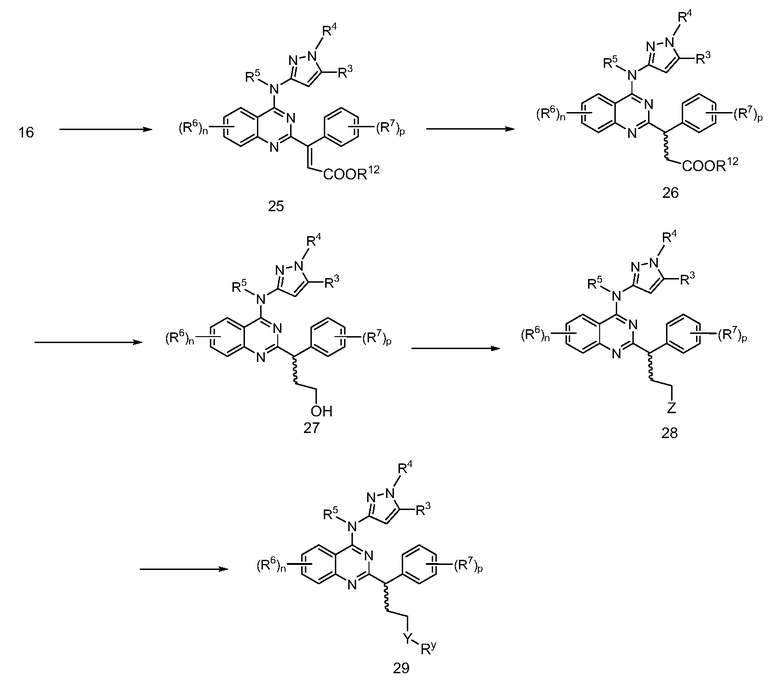
Комплементарный подход к преобразованию спирта 27 в соединения, обеспечиваемые в настоящем изобретении, проиллюстрирован на Схеме 6. Спирт 27 сначала обрабатывают подходящей системой окисления, такой как пиридиний хлорхромат/DCM или реагент Сверна (DMSO/оксалилхлорид/TEA/DCM) или DMSO/комплекс пиридин-триоксид серы/TEA или периодинан Dess-Martin (1,1,1-трисацетокси)-1,1-дигидро-1,2-бензиодоксол-3-(1H)-он/DCM), с получением альдегида 30. Обработка альдегида 30 первичным или вторичным амином в присутствии селективного восстановителя, такого как триацетоксиборогидрид натрия или цианоборогидрид натрия, в подходящем растворителе, таком как спирт, необязательно в присутствии каталитического количества кислоты, такой как уксусная кислота, дает амин 31.
Схема 6
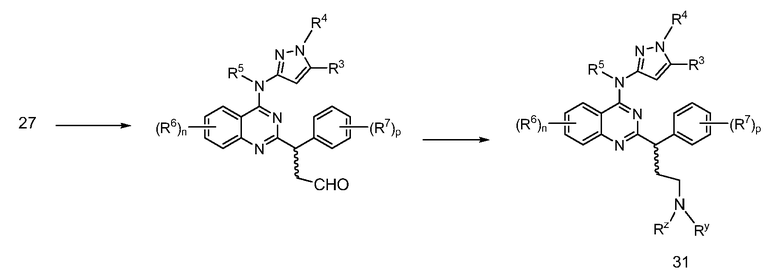
Изобретение было описано иллюстративным образом, и должно быть понятно, что используемая терминология предназначена для раскрытия сути изобретения, а не для ограничения. Таким образом, специалистам в данной области должно быть понятно, что условия, такие как выбор растворителя, температура реакции, объемы, время реакции, могут варьировать, при условии, что они обеспечивают получение желаемых соединений. Кроме того, специалистам в данной области также должно быть понятно, что многие из реагентов, представленных в следующих далее примерах, могут быть замещены другими подходящими реагентами. См., например, Smith & March, Advanced Organic Chemistry, 5th ed. (2001). Такие изменения и модификации, включая, без ограничения, те, которые относятся к используемым химическим структурам, заместителям, производным, промежуточным соединениям, синтезу, композициям и/или способам, обеспечиваем в настоящем изобретении, можно осуществить без отступления от сути и объема настоящего изобретения. Патенты США и публикации, на которые ссылаются в настоящем описании, включены посредством ссылки.
ПРИМЕРЫ
Варианты воплощения, описанные выше, следует рассматривать только в качестве примеров, и специалистам в данной области должны быть известны, или они смогут определить с использованием не более чем рутинного экспериментирования, различные эквиваленты конкретных соединений, веществ и процедур. Все такие эквиваленты предусматриваются как включенные в объем заявленного изобретения и охватываются прилагаемой формулой изобретения.
Пример 1
Получение (3-фторфенил)(4-(5-метил-1H-пиразол-3-иламино)хиназолин-2-ил)метанон
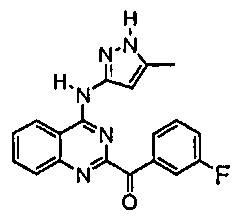
Стадия А: К суспензии этил 4-хлорхиназолин-2-карбоксилата (237 мг, 1 ммоль) в ТГФ (5 мл) добавляли по каплям 1М раствор 3-фторфенилмагнийбромида в ТГФ (2 мл, 2 ммоль) при -20°C. Реакционную смесь перемешивали при -20°C в течение 4 часов. Смесь гасили добавлением 0,5 н. раствора HCl (5 мл) и двухфазную смесь экстрагировали при помощи EtOAc (2×10 мл). Объединенные органические слои промывали насыщенным солевым раствором и сушили над MgSO4. (4-хлорхиназолин-2-ил)(3-фторфенил)метанон получали в виде желтого твердого вещества (190 мг, 66%). LC-MS (ESI) m/z 287 (M+H)+.
Стадия B: (3-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанон получали, следуя аналогичной процедуре, как описанно для синтеза (4-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанона в Примере 3, с использованием (4-хлорхиназолин-2-ил)(3-фторфенил)метанона в качестве исходного вещества. Очистку осуществляли при помощи ВЭЖХ без окончательной обработки (26% выход). 1H ЯМР (300 МГц, DMSO-d6) δ 2,19 (с, 3H), 6,54 (с, 1H), 7,60 (м, 2H), 7,70 (м, 1H), 7,83-7,92 (м, 4H), 8,75 (м, 1H), 10,73 (с, 1H), 12,24 (с, 1H); LC-MS (ESI) m/z 348 (M+H)+.
Пример 2
Получение (4-(1Н-пиразол-3-иламино)хиназолин-2-ил)(3-фторфенил)метанона
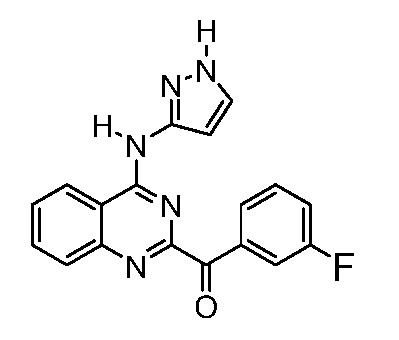
К раствору (4-хлорхиназолин-2-ил)(3-фторфенил)метанона из Примера 1 (57 мг, 0,20 ммоль) в DMF (3 мл) добавляли DIEA (0,069 мл, 0,4 ммоль) и 1H-пиразол-3-амин (83 мг, 1 ммоль). Смесь перемешивали при 50°C в течение 2 часов. Реакцию гасили добавлением воды и осадок фильтровали. Неочищенное твердое вещество очищали при помощи препаративной ТСХ с использованием DCM/MeOH в качестве подвижной фазы с получением (4-(1Н-пиразол-3-иламино)хиназолин-2-ил)(3-фторфенил)метанона (18 мг, 27%). 1H ЯМР (300 МГц, DMSO-d6) δ 6,80 (с, 1H), 7,67-7,61 (м, 4H), 7,92-7,84 (м, 4H), 8,78 (м, 1H), 10,82 (с, 1H), 12,54 (с, 1H); LC-MS (ESI) m/z 334 (M+H)+.
Пример 3
Получение (4-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанона
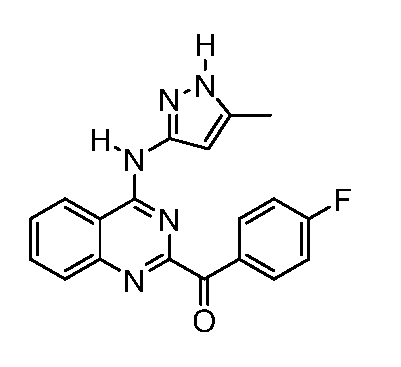
Стадия A: К раствору этил 4-хлорхиназолин-2-карбоксилата (0,6 г, 2,53 ммоль) в ТГФ (6 мл) при -40°C, добавляли по каплям 1 M раствор 4-фторфенилмагнийбромида в ТГФ (3 мл, 3,0 ммоль, 1,2 экв.). Смесь перемешивали при -40°C в течение 4 часов. Реакцию гасили добавлением 0,5 н. раствора HCl (5 мл) и смесь экстрагировали при помощи EtOAc (2×10 мл). Объединенные органические слои промывали насыщенным солевым раствором и сушили над MgSO4. Неочищенный продукт очищали на колонке с силикагелем с использованием смеси EtOAc-гексан в качестве элюента. (4-хлорхиназолин-2-ил)(4-фторфенил)метанон получали в виде светло-желтого твердого вещества (440 мг, 60%). 1H ЯМР (300 МГц, DMSO-d6) δ 7,45-7,40 (м, 2H), 8,07-8,03 (м, 1H), 8,17-8,13 (м, 2H), 8,23 (м, 2H), 8,42 (д, 1H); LC-MS (ESI) m/z 287 (M+H)+.
Стадия B: К раствору (4-хлорхиназолин-2-ил)(4-фторфенил)метанона (84 мг, 0,30 ммоль) в DMF (3 мл) добавляли DIEA (0,103 мл, 0,6 ммоль) и 5-метил-1Н-пиразол-3-амин (88 мг, 0,9 ммоль) при комнатной температуре. Реакционную смесь нагревали при 40°C в течение ночи. Реакцию гасили добавлением воды и желтый осадок собирали фильтрованием и промывали водой. Неочищенный продукт очищали хроматографией на силикагеле, элюируя смесью DCM/MeOH с получением (4-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанона (30 мг, 29%). 1H ЯМР (300 МГц, DMSO-d6) δ 2,19 (с, 3H), 6,54 (с, 1H), 7,40 (м, 2H), 7,68 (т, 1H), 7,9-7,7 (м, 2H), 8,08 (м, 2H), 8,74 (д, 1H), 10,66 (с, 1H), 12,20 (с, 1H); LC-MS (ESI) m/z 348 (M+H)+.
Пример 4
Получение (4-(1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанона
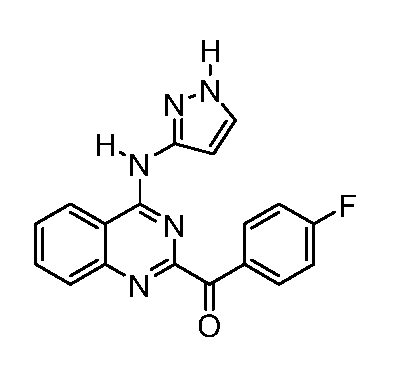
(4-(1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанон получали, следуя процедуре, описанной в Примере 3 для синтеза (4-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанона, замещая 5-метил-1Н-пиразол-3-амин в Примере 3 1H-пиразол-3-амином (30% выход). 1H ЯМР (300 МГц, DMSO-d6) δ 6,78 (с, 1H), 7,39 (т, 2H), 7,70 (м, 2H), 7,90 (м, 2H), 8,10 (м, 2H), 8,77 (д, 1H), 10,84 (с, 1H), 12,56 (с, H); LC-MS (ESI) m/z 334,3 (M+H)+.
Пример 5
Получение (4-(1Н-пиразол-3-иламино)хиназолин-2-ил)(2-метоксифенил)метанона
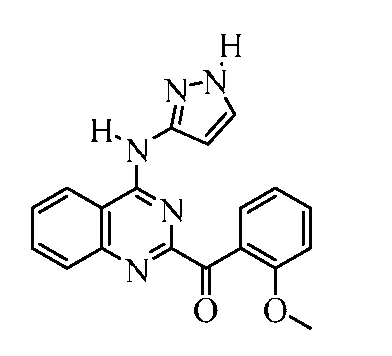
Стадия A: К раствору этил 4-хлорхиназолин-2-карбоксилата (0,250 г, 1,05 ммоль) в DMF (2,5 мл) при комнатной температуре в атмосфере аргона добавляли иодид калия (0,192 г, 1,16 ммоль), DIEA (0,238 мл, 1,37 ммоль) и 1Н-пиразол-3-амин (0,113 г, 1,37 ммоль). Смесь перемешивали при комнатной температуре в течение 5 часов и разбавляли при помощи H2O (5 мл). Осадок собирали фильтрованием, промывали при помощи H2O и сушили в условиях высокого вакуума в течение нескольких часов с получением этил 4-(1Н-пиразол-3-иламино)хиназолин-2-карбоксилата в виде ржаво-коричневого твердого вещества (0,190 г, 64%). 1H ЯМР (300 МГц, DMSO-d6) δ 12,52 (с, 1H), 10,58 (с, 1H), 8,72 (д, 1H), 7,90 (д, 2H), 7,78 (с, 1H), 7,68 (м, 1H), 7,18 (с, 1H), 4,48 (кв., 2H), 1,48 (т, 3H); LC-MS (ESI) m/z 284 (M+H)+.
Стадия B: К суспензии этил 4-(1Н-пиразол-3-иламино)хиназолин-2-карбоксилата (0,110 г, 0,39 ммоль) в ТГФ (5 мл) в атмосфере аргона при -40°C добавляли (2-метоксифенил)магнийбромид (0,5 M раствор в ТГФ; 2,32 мл, 1,16 ммоль). Смесь перемешивали при -40°C в течение 3 часов и гасили 0,5 н. раствором HCl (10 мл). Органический слой отделяли. Водный слой промывали при помощи 10% MeOH/CH2Cl2 (50 мл ×2). Объединенные органические слои промывали насыщенным солевым раствором (25 мл), сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали при помощи препаративной ВЭЖХ (колонка Phenomenex фенилгексил с обращенной фазой, элюировали с использованием градиента растворителя B=0,05% HOAC/CH3CN и растворителя А=0,05% HOAc/H2O) с получением (4-(1Н-пиразол-3-иламино)хиназолин-2-ил)(2-метоксифенил)метанона в виде желтого твердого вещества (0,023 г, 17%). 1H ЯМР (300 МГц, DMSO-d6) δ 3,50 (с, 3H), 6,58 (с, 1H), 7,15 (м, 2H), 7,70-7,50 (м, 4H), 7,88 (м, 2H), 8,75 (д, 1H), 10,68 (с, 1H), 12,42 (с, 1H); LC-MS (ESI) m/z 346 (M+H)+.
Пример 6
Получение (R,S)-(4-(1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанола
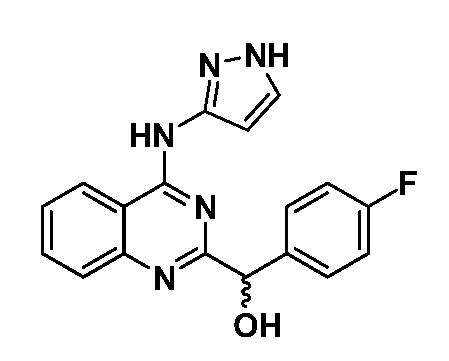
К раствору (4-(1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанона из Примера 4 (375 мг, 1,12 ммоль) в смеси 1:1 MeOH/ТГФ (10 мл) при 0°C добавляли NaBH4 (64 мг, 1,69 ммоль). Реакционную смесь перемешивали при 0°C в течение 3 часов. Реакционную смесь гасили добавлением воды и твердое вещество собирали фильтрованием. Неочищенный продукт очищали при помощи обращенно-фазовой ВЭЖХ с получением (R,S)-(4-(1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанола в виде белого твердого вещества (130 мг, 34%). 1H ЯМР (300 МГц, DMSO-d6) δ 5,67 (м, 1H), 5,79 (м, 1H), 6,85 (с, 1H), 7,11 (т, 2H), 7,55 (м, 3H), 7,70 (с, 1H), 7,80 (м, 2H), 8,61 (д, 1H), 10,50 (с, 1H), 12,46 (с, 1H); LC-MS (ESI) m/z 336 (M+H)+.
Пример 7
Получение (R,S)-2-(фтор(4-фторфенил)метил)-N-(1Н-пиразол-3-ил)хиназолин-4-амина
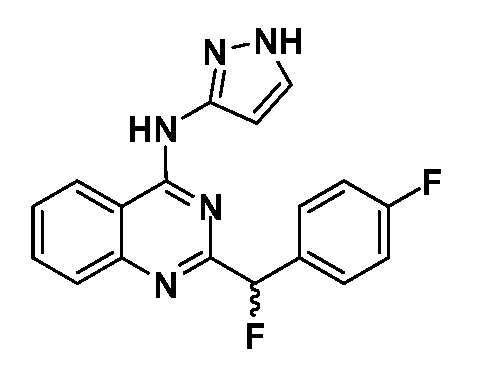
К раствору (R,S)-(4-(1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанола из Примера 6 (88 мг, 0,238 ммоль) в смеси DCM/ТГФ (18 мл, 2:1) добавляли трифторид бис(2-метоксиэтил)-амино)серы (0,066 мл, 0,22 ммоль) при комнатной температуре. Реакционную смесь перемешивали при 50°C в течение ночи. Реакционную смесь гасили добавлением ацетона (0,1 мл), растворитель выпаривали и остаток очищали при помощи ВЭЖХ. (R,S)-2-(Фтор(4-фторфенил)метил)-N-(1Н-пиразол-3-ил)хиназолин-4-амин получали в виде белого порошка (12 мг, 15%). 1H ЯМР (300 МГц, DMSO-d6) δ 6,46 (с, 1H), 6,61 (с, 1H), 6,86 (с, 1H), 7,22 (т, 2H), 7,64-7,56 (м, 2H), 7,70 (с, 1H), 7,82 (м, 2H), 8,65 (д, 1H), 10,63 (с, 1H), 12,50 (с, 1H); LC-MS (ESI) m/z 338 (M+H)+.
Пример 8
Получение 2-(дифтор(4-фторфенил)метил)-N-(5-метил-1Н-пиразол-3-ил)хиназолин-4-амина
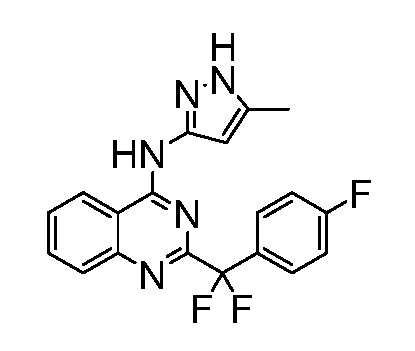
Стадия A: 2,2-Дифтор-2-(4-фторфенил)уксусную кислоту получали в соответствии с Middleton et al., J. Org. Chem., 1980, 45(14): 2883-2887) путем взаимодействия этил 2-(4-фторфенил)-2-оксоацетата с трифторидом (диэтиламино)серы с последующим омылением сложного эфира.
Стадия B: К раствору 2,2-дифтор-2-(4-фторфенил)уксусной кислоты (1,33 г, 7,0 ммоль) в DCM (50 мл) добавляли оксалилхлорид (0,640 мл, 7,5 ммоль) и каталитическое количество DMF. После перемешивания в течение 3 часов смесь концентрировали при пониженном давлении с получением 2,2-дифтор-2-(4-фторфенил)ацетилхлорида. К раствору 2-аминобензамида (0,857 г, 6,3 ммоль) и TEA (1,04 мл, 0,0075 моль) в DCE (100 мл) при комнатной температуре добавляли 2,2-дифтор-2-(4-фторфенил)ацетилхлорид, указанный выше, в DCE (100 мл) и реакционную смесь перемешивали в течение ночи. После добавления EtOAc (200 мл) смесь промывали 1 н. раствором HCl, насыщенным раствором NaHCO3 и насыщенным солевым раствором. Органический раствор концентрировали с получением не совсем белого твердого вещества (0,989 мг, 51%). 1H ЯМР (300 МГц, DMSO-d6) δ 7,15 (т, 1H), 7,27 (м, 2H), 7,54 (м, 1H), 7,74 (м, 2H), 7,92 (м, 2H), 8,44 (д, 2H), 13,37 (с, 1H).
Стадия C: К раствору 2-(2,2-дифтор-2-(4-фторфенил)ацетамидо)бензамида (0,95 г, 3,08 ммоль) в DCE (25 мл) добавляли TEA (17,2 мл, 0,123 моль) и хлортриметилсилан (5,84 мл, 0,0462 моль) при комнатной температуре. Реакционную смесь перемешивали при 85°C в течение ночи. После охлаждения до комнатной температуры твердое вещество фильтровали и фильтрат концентрировали досуха. Остаток очищали на колонке с силикагелем с использованием DCM/MeOH в качестве элюента. 2-(Дифтор(4-фторфенил)метил)хиназолин-4-ол получали в качестве не совсем белого твердого вещества (0,668 г, 75%). 1H ЯМР (300 МГц, DMSO-d6) δ 7,39 (т, 2H), 7,62 (м, 1H), 7,78-7,71 (м, 3H), 7,84 (м, 1H), 8,16 (м, 1H), 13,11 (с, 1H).
Стадия D: 4-Хлор-2-(дифтор(4-фторфенил)метил)хиназолин получали в соответствии с процедурой, описанной в Примере 26 для получения 4-хлор-2-(дифтор(4-фторфенил)метил)-7-метилхиназолина, замещая 2-(дифтор(4-фторфенил)метил)-7-метилхиназолин-4-ол в Примере 26 2-(дифтор(4-фторфенил)метил)хиназолин-4-олом (95% выход). LC-MS (ESI) m/z 308 (M+H)+.
Стадия E: К раствору 4-хлор-2-(дифтор(4-фторфенил)метил)хиназолина (0,150 г, 0,487 ммоль) в DMF (2 мл) при комнатной температуре добавляли иодид калия (0,081 г, 0,487 ммоль), DIEA (0,093 мл, 0,535 ммоль) и 5-метил-1Н-пиразол-3-амин (0,048 г, 0,487 ммоль). После перемешивания реакционной смеси при комнатной температуре в течение ночи реакцию гасили добавлением воды (15 мл). Осадок собирали фильтрованием и промывали при помощи H2O. Неочищенное твердое вещество растирали в порошок с MeOH. 2-(Дифтор(4-фторфенил)метил)-N-(5-метил-1Н-пиразол-3-ил)хиназолин-4-амин получали в виде не совсем белого твердого вещества (0,125 г, 69%). 1H ЯМР (300 МГц, DMSO-d6) δ 2,24 (с, 3H), 6,31 (с, 1H), 7,34 (м, 2H), 7,68 (м, 3H), 7,87 (м, 2H), 8,68 (м, 1H), 10,69 (с, 1H), 12,20 (с, 1H); LC-MS (ESI) m/z 370 (M+H)+.
Пример 9
Получение 2-(дифтор(4-фторфенил)метил)-N-(1Н-пиразол-3-ил)хиназолин-4-амина
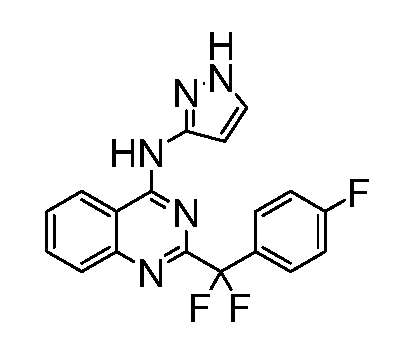
2-(Дифтор(4-фторфенил)метил)-N-(1Н-пиразол-3-ил)хиназолин-4-амин получали с использованием процедуры, аналогичной описанной в Примере 8, используя 1Н-пиразол-3-амин вместо 5-метил-1Н-пиразол-3-амина, используемого в Примере 8 Стадия E (61% выход). 1H ЯМР (300 МГц, DMSO-d6) δ 6,77 (с, 1H), 7,32 (т, 2H), 7,77-7,63 (м, 4H), 7,88 (м, 2H), 8,71 (д, 1H), 10,82 (с, 1H), 12,55 (с, 1H); LC-MS (ESI) m/z 356 (M+H)+.
Пример 10
Получение N-(5-циклопропил-1Н-пиразол-3-ил)-2-(дифтор(4-фторфенил)метил)хиназолин-4-амина
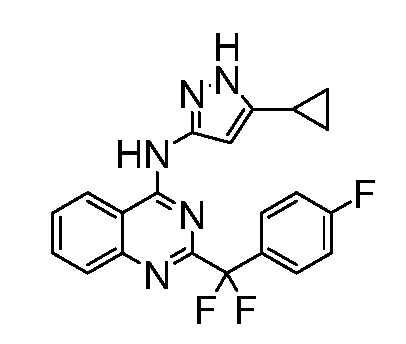
N-(5-циклопропил-1Н-пиразол-3-ил)-2-(дифтор(4-фторфенил)метил)хиназолин-4-амин получали с использованием процедуры, аналогичной описанной в Примере 8, используя 5-циклопропил-1Н-пиразол-3-амин вместо 5-метил-1Н-пиразол-3-амина, используемого в Примере 8 Стадия E (68% выход). 1H ЯМР (300 МГц, DMSO-d6) δ 0,637 (м, 2H), 0,96 (м, 2H), 1,91 (м, 1H), 6,20 (с, 1H), 7,70 (м, 2H), 7,80 (м, 3H), 7,90 (м, 4H), 8,70 (д, 1H), 10,68 (с, 1H), 12,20 (с, 1H); LC-MS (ESI) m/z 396 (M+H)+.
Пример 11
Получение 3-(2-(4-фторбензоил)хиназолин-4-иламино)-1Н-пиразол-5-карбонитрила
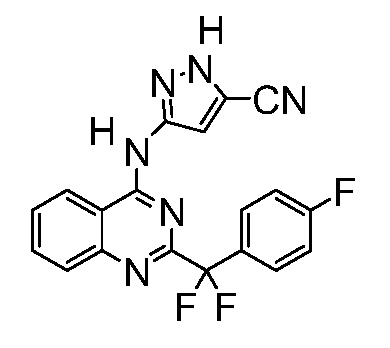
Стадия A: К раствору 5-нитро-3-пиразолкарбоновой кислоты (6,28 мг, 40 ммоль) в DMF (30 мл) добавляли карбонилдиимидазол (12,96 мг, 80 ммоль). Смесь оставляли для перемешивания в течение 30 минут и добавляли раствор аммиака в MeOH (2M, 60 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Смесь концентрировали при пониженном давлении с получением неочищенного продукта, который затем промывали эфиром, с получением 3-нитро-1Н-пиразол-5-карбоксамида (3,0 г, 48%), который использовали непосредственно на следующей стадии без дополнительной очистки. LC-MS (ESI) m/z 155 (M-H)-.
Стадия B: 3-нитро-1Н-пиразол-5-карбоксамид (3,0 г, 19,2 ммоль) в пиридине (30 мл) обрабатывали оксихлоридом фосфора (5,9 г) и полученный раствор перемешивали в течение 3 часов при комнатной температуре. Реакционную смесь разбавляли льдом, затем экстрагировали при помощи DCM (100 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением неочищенного 3-нитро-1Н-пиразол-5-карбонитрил, который использовали непосредственно на следующей стадии без дополнительной очистки. LC-MS (ESI) m/z 137 (M-H)-.
Стадия C: К раствору 3-нитро-1Н-пиразол-5-карбонитрила (1000 мг, 7,24 ммоль) в AcOH (10 мл) и H2O (2 мл) добавляли цинковый порошок (2,35 мг, 36,24 ммоль) при 0°C. Полученный раствор перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь фильтровали, рН доводили до 8 при помощи гидроксида аммония и затем смесь экстрагировали при помощи EtOAc (30 мл). Органический слой сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением неочищенного продукта 3-амино-1Н-пиразол-5-карбонитрила (200 мг, 28%), который использовали непосредственно на следующей стадии без дополнительной очистки. LC-MS (ESI) m/z 107 (M+H)+.
Стадия D: Смесь (4-хлорхиназолин-2-ил)(4-фторфенил)метанона из Примера 3 (580 мг, 2,02 ммоль) и 3-амино-1Н-пиразол-5-карбонитрила (218 мг,2,02 ммоль) в DMF (5 мл) перемешивали при комнатной температуре в течение ночи. Затем к этой смеси добавляли MeOH (10 мл) и осадок фильтровали, промывали при помощи MeOH и сушили с получением 3-(2-(4-фторбензоил)хиназолин-4-иламино)-1Н-пиразол-5-карбонитрила (170 мг, 23,4%). 1H ЯМР (300 МГц, DMSO-d6) δ 6,89 (с, 1H), 7,40 (д, 2H), 7,83 (с, 1H), 7,98 (м, 2H), 8,11 (м, 2H), 8,56 (с, 1H), 11,18 (с, 1H), 13,84 (с, 1H); LC-MS (ESI) m/z 359 (M+H)+.
Пример 12
Получение гидрохлорида (R,S)-(4-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанола
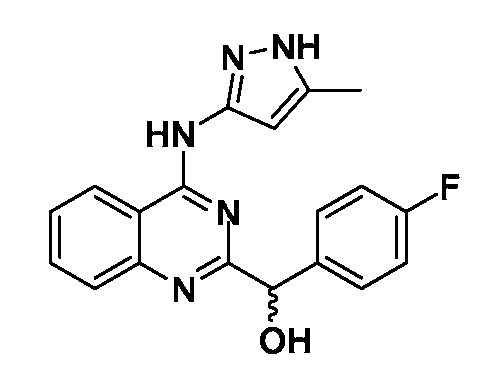
К раствору (4-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанона из Примера 3 (60 мг, 0,172 ммоль) в смеси 1:1 MeOH/ТГФ (10 мл) при 0°C добавляли NaBH4 (64 мг, 1,69 ммоль). Реакционную смесь перемешивали при 0°C в течение 1,5 часов. Реакционную смесь гасили добавлением нескольких капель ацетона и концентрировали досуха. Неочищенное твердое вещество очищали при помощи ВЭЖХ с получением (4-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанола (18 мг, 30%); 1H ЯМР (300 МГц, DMSO-d6) δ 2,25 (с, 3H), 5,67 (с, 1H), 5,83 (ушир.с, 1H), 6,40 (ушир.с, 1H), 7,13 (м, 2H), 7,55-7,53 (м, 3H), 7,79 (с, 2H), 8,57 (ушир.с, 1H), 10,43 (с, 1H), 12,12 (ушир.с, 1H); LC-MS (ESI) m/z 350 (M+H)+.
К суспензии (4-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанона (2,3 г) в 30% MeOH/DCM (60 мл) при 0°C добавляли по каплям раствор 4M HCl/l,4-диоксан (10 мл). После полного растворения всех твердых веществ, смесь концентрировали при пониженном давлении и к остатку добавляли 30% раствор CH3CN/H2O (80 мл) и смесь обрабатывали ультразвуком до полного растворения всех твердых веществ. Смесь замораживали и лиофилизировали в течение ночи с получением гидрохлорида (4-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанола (100%). 1H ЯМР (300 МГц, DMSO-d6) δ 2,25 (с, 3H), 6,02 (с, 1H), 6,20 (с, 1H), 7,27 (т, 2H), 7,60 (кв.т, 2H), 7,80 (т, 1H), 8,08 (т, 1H), 8,23 (д, 1H), 8,83 (д, 1H), 12,16 (с, 1H), 14,51 (ушир., 1H); LC-MS (ESI) m/z 350 (M+H)+.
Пример 13
Получение (R,S)-2-((4-фторфенил)(метокси)метил)-N-(5-метил-1Н- пиразол-3-ил)хиназолин-4-амина
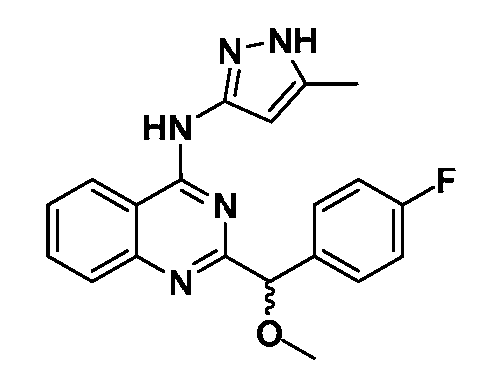
Стадия A: К раствору (R,S)-2-бром-2-(4-фторфенил)уксусной кислоты (2,02 г, 8,66 ммоль) в DCM (15 мл) и DMF (0,15 мл) добавляли оксалилхлорид (0,8 мл, 9,1 ммоль), затем смесь оставляли для перемешивания в течение 30 минут при комнатной температуре. Реакционную смесь затем охлаждали до 0°C и медленно добавляли 2-аминобензамид (1,12 г, 8,23 ммоль) в пиридин (2 мл). Смесь доводили до комнатной температуры в течение ~ 1 часа и затем упаривали. Растирание в порошок со смесью 2н HCl/метанол/вода давало неочищенный (R,S)-2-(2-бром-2-(4-фторфенил)ацетамидо)бензамид, который использовали без дополнительной очистки (2,13 г, 73%). LC-MS (ESI) m/z 351 (M+H)+.
Стадия B: К (R,S)-2-(2-бром-2-(4-фторфенил)ацетамидо)бензамиду (0,52 г, 1,48 ммоль) в MeOH (4 мл) добавляли метоксид натрия в MeOH (25%, 0,64 мл, 2,96 ммоль) и полученный раствор перемешивали в течение ночи при 65°C. Реакционную смесь распределяли между EtOAc и 2 н. раствором HCl, EtOAc слой сушили при помощи сульфата натрия и затем упаривали. Неочищенный продукт затем растирали в порошок с эфиром с получением (R,S)-2-((4-фторфенил)(метокси)метил)хиназолин-4-ола, который использовали без дополнительной очистки (260 мг, 62%). LC-MS (ESI) m/z 285 (M+H)+.
Стадия C: К раствору (R,S)-2-((4-фторфенил)(метокси)метил)хиназолин-4-ола (200 мг, 0,7 ммоль) в DCM (2 мл) добавляли DMAP (8 мг, 0,07 ммоль) и TEA (0,39 мл, 2,8 ммоль), с последующим добавлением 2,4,6-триизопропилбензол-1-сульфонилхлорида (211 мг, 0,91 ммоль) и реакционную смесь перемешивали в течение 30 минут при комнатной температуре. Неочищенную смесь очищали при помощи хроматографии на силикагеле, элюируя при помощи 0-50% EtOAc и гексана, с получением (R,S)-2-((4-фторфенил)(метокси)метил)хиназолин-4-ил 2,4,6-триизопропилбензолсульфоната (320 мг, 83%). LC-MS (ESI) m/z 573 (M+Na)+.
Стадия D: К (R,S)-2-((4-фторфенил)(метокси)метил)хиназолин-4-ил-2,4,6-триизопропилбензолсульфонату (77 мг, 0,14 ммоль) в DMF (2 мл) добавляли 5-метил-1Н-пиразол-3-амин (20 мг, 0,2 ммоль), TEA (0,02 мл, 0,14 ммоль) и иодид калия (33 мг, 0,2 ммоль) и реакционную смесь перемешивали при 50°C в течение 1 часа, с последующим нагреванием при 70°C в течение 2 часов. Затем добавляли дополнительное количество 5-метил-1Н-пиразол-3-амина (45 мг) и смесь нагревали при 50°C в течение ночи. Смесь упаривали и очищали при помощи хроматографии на силикагеле, элюируя при помощи 0-12% MeOH в DCM. Очищенные фракции упаривали и затем подвергали дополнительной очистке при помощи препаративной ВЭЖХ (колонка Phenomenex C-18 с обращенной фазой, элюировали с использованием градиента растворителя B=0,05% HOAc/CH3CN и растворителя А=0,05% HOAc/H2O), с последующим использованием препаративной тонкослойной хроматографии (10% MeOH в DCM) с получением (R,S)-2-((4-фторфенил)(метокси)метил)-N-(5-метил-1Н-пиразол-3-ил)хиназолин-4-амина (5 мг, 10%). 1H ЯМР (300 МГц, DMSO-d6) δ 2,28 (с, 3H), 3,32 (с, 3H), 5,37 (с, 1H), 6,56 (с, 1H), 7,15 (д, 2H), 7,57 (м, 3H), 7,81 (м, 2H), 8,59 (д, 1H), 10,48 (с, 1H), 12,09 (с, 1H); LC-MS (ESI) m/z 364 (M+H)+.
Пример 14
Получение (R,S)-2-(амино(4-фторфенил)метил)-N-(5-метил-1Н-пиразол-3-ил)хиназолин-4-амина
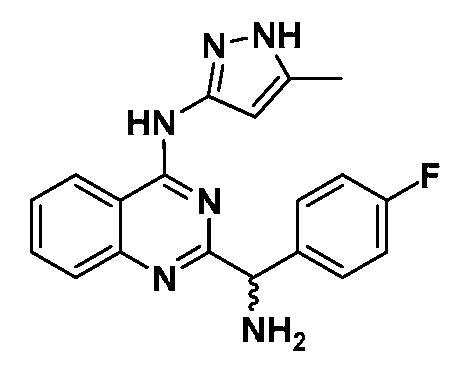
Стадия A: К раствору 2-амино-2-(4-фторфенил)уксусной кислоты (5 г, 29,5 ммоль) в ТГФ (50 мл) при 50°C добавляли трифосген (8,77 г, 29,5 ммоль), затем продолжали нагревание в течение 3 часов. Реакционную смесь затем фильтровали и упаривали до объема около 10 мл, с последующим добавлением 150 мл гексана. Смесь слегка нагревали и затем охлаждали до -20°C в течение 1 часа. Неочищенную суспензию фильтровали с получением 4-(4-фторфенил)оксазолидин-2,5-диона (5,03 г, 87%), который использовали без дополнительной очистки.
Стадия B: К раствору 4-(4-фторфенил)оксазолидин-2,5-диона (2,5 г, 12,8 ммоль) в ТГФ (30 мл), охлажденному до -25°C, добавляли бензилхлороформиат (2,3 мл, 16,6 ммоль) с последующим медленным добавлением (~10 минут) N-метилморфолина (2,11 мл, 19,2 ммоль) в виде раствора в ТГФ (5 мл). Раствор перемешивали при этой температуре в течение 1 часа и затем давали нагреться до комнатной температуры в течение ночи. Полученный раствор фильтровали через Целит и фильтрат концентрировали. Полученное неочищенное вещество перекристаллизовывали из смеси 2:2:1 трет-бутилметиловый эфир:гексан:ТГФ с получением бензил 4-(4-фторфенил)-2,5-диоксооксазолидин-3-карбоксилата (2,7 г, 64%), который использовали без дополнительной очистки.
Стадия C: К раствору 2-аминобензамида (591 мг, 4,34 ммоль) в ТГФ (10 мл) добавляли бензил 4-(4-фторфенил)-2,5-диоксооксазолидин-3-карбоксилат (1,43 г, 4,34 ммоль) и реакционную смесь нагревали при 50°C в течение 2 часов. Добавляли дополнительное количество 5 мл ТГФ и продолжали нагревание еще в течение 0,5 часов. Затем добавляли метоксид натрия в MeOH (25%, 1,87 мл, 8,68 ммоль) и реакционную смесь нагревали до 65°C в течение 2 часов. Затем добавляли HOAc (0,4 мл), раствор упаривали и неочищенную смесь очищали хроматографией на силикагеле, элюируя смесью 0-10% MeOH в DCM, с получением (R,S)-(4-фторфенил)(4-гидроксихиназолин-2-ил)метилкарбамата (1,1 г, 63%). LC-MS (ESI) m/z 404 (M+Na)+.
Стадия D: К раствору (R,S)-(4-фторфенил)(4-гидроксихиназолин-2-ил)метилкарбамата (451 мг, 1,11 ммоль) в DCM (5 мл) добавляли DMAP (7 мг, 0,05 ммоль), TEA (0,61 мл, 4,4 ммоль) и 2,4,6-триизопропилбензол-1-сульфонилхлорид (440 мг, 1,45 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 0,5 часов и затем упаривали. Остаток очищали хроматографией на силикагеле, элюируя смесью 0-50% EtOAc в гексане, с получением (R,S)-2-((бензилоксикарбониламино)(4-фторфенил)метил)хиназолин-4-ил 2,4,6-триизопропилбензолсульфоната (580 мг, 75%). LC-MS (ESI) m/z 692 (M+Na)+.
Стадия E: К (R,S)-2-((бензилоксикарбониламино)(4-фторфенил)метил)хиназолин-4-ил-2,4,6-триизопропилбензолсульфонату (216 мг, 0,32 ммоль), в DMA (2 мл) добавляли 5-метил-1Н-пиразол-3-амин (198 мг, 2,04 ммоль) и иодид калия (140 мг, 0,83 ммоль) и смесь перемешивали при 55°C в течение 4 часов. Неочищенную смесь распределяли между EtOAc и насыщенным раствором гидрокарбоната натрия. EtOAc слой сушили при помощи сульфата натрия и затем упаривали до получения масла. Половину этого неочищенного масла растворяли в MeOH (5 мл) и добавляли 10% гидроксид палладия на углероде (50 мг). Полученный раствор перемешивали в атмосфере водорода в течение 6 часов, затем фильтровали. Очистка при помощи препаративной ВЭЖХ (колонки с обращенной фазой Varian дифенил и затем Phenomenex C-18, элюировали с использованием градиента растворителя B=0,05% HOAC/CH3CN и растворителя А=0,05% HOAc/H2O) давала (R,S)-2-(амино(4-фторфенил)метил)-N-(5-метил-1Н-пиразол-3-ил)хиназолин-4-амин в виде его ацетатной соли (5 мг, 9%). 1H ЯМР (300 МГц, DMSO-d6) δ 1,89 (с, 3H), 2,25 (с, 3H), 5,07 (с, 1H), 6,35 (с, 1H), 7,12 (т, 2H), 7,51 (м, 3H), 7,77 (м, 2H), 8,56 (д, 1H), 10,55 (с, 1H); LC-MS (ESI) m/z 349 (M+H)+.
Пример 15
Получение (R,S)-3-(2-((4-фторфенил)(гидрокси)метил)хиназолин-4-иламино)-1Н-пиразол-5-карбонитрила
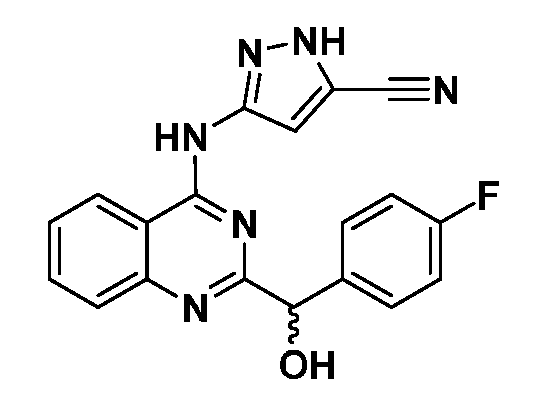
К раствору 3-(2-(4-фторбензоил)хиназолин-4-иламино)-1Н-пиразол-5-карбонитрила из Примера 11 (107 мг, 0,3 ммоль) в MeOH (4 мл) и ТГФ (4 мл) добавляли борогидрид натрия (22,7 мг, 0,6 ммоль) при 0°C и смесь перемешивали в течение ночи при комнатной температуре. Смесь выливали в H2O (20 мл), в результате чего происходило образование осадка. После фильтрования получали твердое вещество, которое очищали при помощи препаративной ВЭЖХ, с получением (R,S)-3-(2-((4-фторфенил)(гидрокси)метил)хиназолин- 4-иламино)-1Н-пиразол-5-карбонитрила (30 мг, 29%). 1H ЯМР (300 МГц, DMSO-d6) δ 5,81 (с, 1H), 6,34 (ушир.с, 1H), 6,88 (с, 1H), 7,17 (т, 2H), 7,58 (м, 3H), 7,81 (с, 2H), 8,37 (м, 1H); LC-MS (ESI) m/z 361 (M+H)+.
Пример 16
Получение (R,S)-(5-фтор-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанола
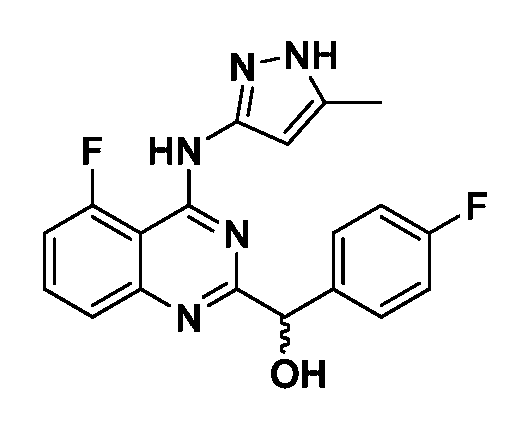
Стадия A: 5-(4-фторфенил)-1,3-диоксолан-2,4-дион получали в соответствии с JACS, 2002 2870-2871. К 2-амино-6-фторбензамиду (550 мг, 3,5 ммоль) в ТГФ (15 мл) добавляли 5-(4-фторфенил)-1,3-диоксолан-2,4-дион (1049 мг, 5,35 ммоль) и смесь нагревали при 50°C в течение ночи. Растворитель выпаривали с получением неочищенного 2-(2,2-дифтор-2-(4-фторфенил)ацетамидо)-4-метоксибензамида и неочищенную смесь растворяли в этаноле (12 мл), добавляли водный раствор карбоната калия и реакционную смесь нагревали при 80°C в течение ночи. Неочищенную смесь экстрагировали при помощи EtOAc и воды и EtOAc слой концентрировали в вакууме с получением (R,S)-5-фтор-2-((4-фторфенил)(гидроксил)метил)хиназолин-4-ола (650 мг, %). LC-MS (ESI) m/z 290 (M+Na)-.
Стадия B: К (R,S)-5-фтор-2-((4-фторфенил)(гидрокси)метил)хиназолин-4-олу (650 мг, 2,25 ммоль) добавляли периодинан Dess-Martin (1140 мг, 2,7 ммоль) в ацетонитриле (15 мл) и смесь перемешивали при комнатной температуре в течение 30 минут. К неочищенной смеси добавляли водный раствор бикарбоната натрия и смесь перемешивали в течение 0,5 часов. Полученный коричневый осадок собирали и промывали диэтиловый эфиром (650 мг, количественный); LC-MS (ESI) m/z 287 (M+Na)+.
Стадия C: К оксихлориду фосфора (7 мл) добавляли (5-фтор-4-гидроксихиназолин-2-ил)(4-фторфенил)метанон (650 мг, 2,26 ммоль) с последующим добавлением DMA (1 капля). Раствор нагревали при 85°C в течение 3 часов и затем смесь концентрировали. Остаток охлаждали в -20°C охлаждающей бане и разбавляли холодным EtOAc. Раствор промывали насыщенным водный раствором бикарбоната натрия и насыщенным солевым раствором. Удаление растворителя давало коричневое твердое вещество. Очистка хроматографиией (градиент элюирования 0-40% EtOAc в гексане) давала желтое твердое вещество (300 мг, 44%); LC-MS (ESI) m/z 305 (M+Na)+.
Стадия D: К (4-хлор-5-фторхиназолин-2-ил)(4-фторфенил)метанону (300 мг, 0,98 ммоль) в диметилформамиде (8,0 мл) добавляли DIEA (0,17 мл, 0,98 ммоль), 5-метил-1Н-пиразол-3-амин (240 мг, 2,5 ммоль) и иодид калия (162 мг, 0,98 ммоль) и смесь перемешивали при комнатной температуре в течение 1 часа. К реакционной смеси добавляли воду и осадок собирали фильтрованием. Осадок промывали диэтиловый эфиром с получением желтого твердого вещества (280 мг, 78%); LC-MS (ESI) m/z 366 (M+Na)+.
Стадия E: К (5-фтор-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанону (280 мг, 0,76 ммоль) в 1:1 смеси MeOH и ТГФ (8 мл) при 0°C добавляли NaBH4 (43 мг, 1,14 ммоль). После перемешивания в течение 1 часа при 0°C добавляли 10 капель воды. Растворители удаляли в условиях вакуума и остаток растворяли в EtOAc (15 мл), промывали насыщенным солевым раствором и сушили над Na2SO4. Неочищенный продукт очищали при помощи обращенно-фазовой препаративной ВЭЖХ (градиент элюирования 40-90% ацетонитрила в воде с 0,05% уксусной кислоты) с получением (R,S)-(5-фтор-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанола в виде белого твердого вещества. 1H ЯМР (300 МГц, DMSO-d6) δ м.д. 2,2 (с, 3 H) 5,7 (с, 1H) 5,9 (с,lH) 6,55 (с, 1 H) 7,1-7,2 (м, 2H) 7,35-7,9 (м, 4H) 8,9 (с, 1H) 12,25 (с, 1 H) LC-MS (ESI) m/z 368 (M+H)+.
Пример 17
Получение (4-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)-7-(трифторметил)хиназолин-2-ил)метанона
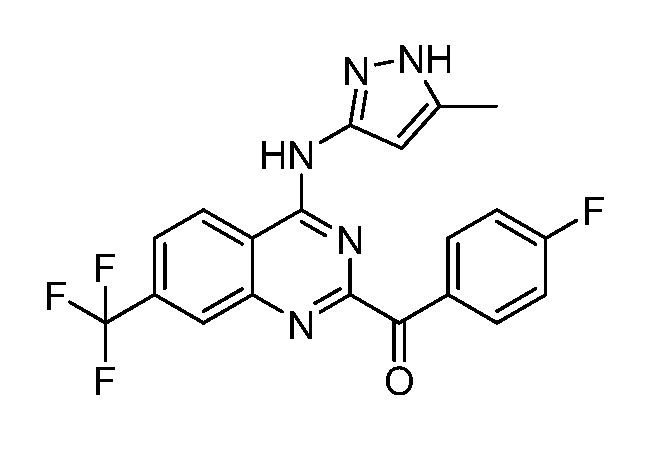
Стадия A: К 2-нитро-4-(трифторметил)бензамиду (1000 мг, 4,27 ммоль) в MeOH (15 мл) добавляли гидроксид палладия 20% масс. (230 мг) и смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь фильтровали через Целит с промывкой при помощи MeOH. Неочищенную смесь концентрировали в вакууме с получением 2-амино-4-(трифторметил)бензамида (840 мг, 96%). LC-MS (ESI) m/z 205 (M+Na)+.
Стадия B: К 2-амино-4-(трифторметил)бензамиду (840 мг, 4,16 ммоль) в ТГФ (15 мл) добавляли 5-(4-фторфенил)-1,3-диоксолан-2,4-дион из Примера 16 (1225 мг, 6,24 ммоль) и смесь нагревали при 50°C в течение 4 часов. Растворитель выпаривали и неочищенный 2-(2-(4-фторфенил)-2-гидроксиацетамидо)-4-(трифторметил)бензамид растворяли в MeOH (10 мл), добавляли раствор 0,5 M метоксида натрия в MeOH (2,5 мл, 1,25 ммоль) и реакционную смесь нагревали при 50°C в течение 1 часа. Растворитель выпаривали и затем добавляли 1 н. раствор хлористоводородной кислоты. Смесь экстрагировали при помощи EtOAc и органическую фазу сушили над сульфатом натрия и концентрировали в вакууме с получением неочищенного 2-((4-фторфенил)(гидрокси)метил)-7-(трифторметил)хиназолин-4-ола, который использовали в следующей реакции без очистки. LC-MS (ESI) m/z 339 (M+Na)+.
Стадия D: К 2-((4-фторфенил)(гидрокси)метил)-7-(трифторметил)хиназолин-4-олу (2000 мг, 5,89 ммоль) добавляли периодинан Dess-Martin (3000 мг, 7,07 ммоль) в ацетонитриле (25 мл) и смесь перемешивали при комнатной температуре в течение 2 часов. К неочищенной смеси добавляли водный раствор бикарбоната натрия и смесь перемешивали в течение 0,5 часов. Образовавшийся коричневый осадок собирали, промывали диэтиловый эфиром и сушили в условиях высокого вакуума с получением (4-фторфенил)(4-гидрокси-7-(трифторметил)хиназолин-2-ил)метанона (2,57 г, количественный выход); LC-MS (ESI) m/z 336 (M+Na)+.
Стадия E: К оксихлориду фосфора (6 мл) добавляли (4-фторфенил)(4-гидрокси-7-(трифторметил)хиназолин-2-ил)метанон (1280 мг, 3,80 ммоль) с последующим добавлением DMA (1 капля). Раствор нагревали при 85°C в течение ночи и затем смесь концентрировали. Неочищенный (4-хлор-7-(трифторметил)хиназолин-2-ил)(4-фторфенил)метанон использовали на следующей стадии без очистки. LC-MS (ESI) m/z 305 (M+Na)+.
Стадия F: К (4-хлор-7-(трифторметил)хиназолин-2-ил)(4-фторфенил)метанону (1 г, 2,82 ммоль) в DMF (10 мл) добавляли DIEA (0,49 мл, 2,82 ммоль), 5-метил-1Н-пиразол-3-амин (823 мг, 8,47 ммоль) и иодид калия (468 мг, 2,82 ммоль) и смесь перемешивали при комнатной температуре в течение 2 часов. К реакционной смеси добавляли воду с последующей экстракцией при помощи EtOAc. Органические фазы сушили над сульфатом натрия. Растворитель концентрировали и остаток сушили в условиях высокого вакуума в течение ночи. Неочищенное твердое вещество (240 мг) очищали при помощи обращенно-фазовой препаративной ВЭЖХ (градиентнное элюирование 40-90% ацетонитрила в воде с 0,05% уксусной кислоты) с получением (4-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)-7-(трифторметил)хиназолин-2-ил)метанона в виде желтого твердого вещества (40 мг, 16%). 1H ЯМР (300 МГц, DMSO-d6) δ м.д. 2,2 (с, 3H) 6,55 (с, 1H) 7,35-7,5(м, 3H) 7,9-8,0 (м, 1H) 8,05-8,3 (м, 4H) 11,1 (с, 1H) 12,25 (с, 1H); LC-MS (ESI) m/z 416 (M+H)+.
Пример 18
Получение (R,S)-(4-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)-7-(трифторметил)хиназолин-2-ил)а
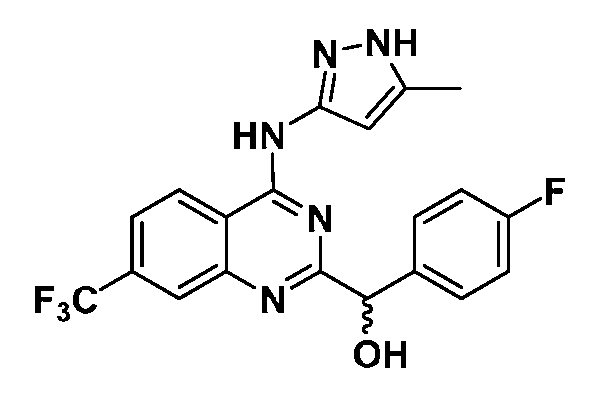
К (4-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)-7-(трифторметил)хиназолин-2-ил)метанону (500 мг, 1,2 ммоль) в 1:1 смеси MeOH/ТГФ (10 мл) при 0°C добавляли NaBH4 (68 мг, 1,79 ммоль). После перемешивания в течение 10 минут при 0°C добавляли 10 капель воды. Растворители удаляли в условиях вакуума и остаток растворяли в смеси 1:1 воды и EtOAc (20 мл), промывали насыщенным солевым раствором и сушили над сульфатом натрия. Неочищенный продукт (360 мг) очищали при помощи обращенно-фазовой препаративной ВЭЖХ (градиент элюирования 40-90% ацетонитрила в воде с 0,05% уксусной кислоты) с получением (4-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)-7-(трифторметил)-хиназолин-2-ил)метанола в виде белого твердого вещества (140 мг, 40%). 1H ЯМР (300 МГц, DMSO-d6) δ м.д. 2,2 (с, 3H) 5,9(с, 1H) 6,55 (с, 1H) 7,35-7,5(м, 3H) 7,9-8,0(м, 1H) 8,05-8,3 (м, 4H) 11,1 (ушир.с, 1H) 12,25 (ушир.с, 1H); LC-MS (ESI) m/z 418 (M+H)+.
Пример 19
Получение (7-фтор-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанона
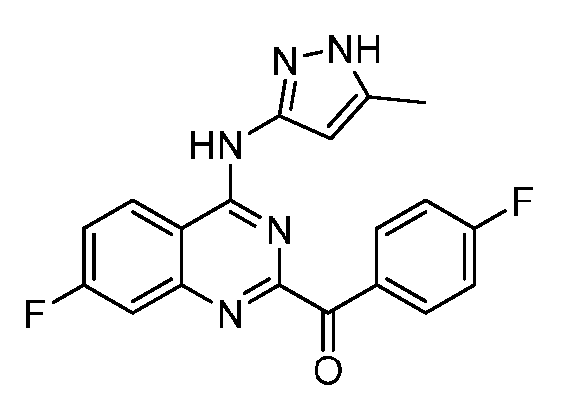
Стадия A: К раствору 2-бром-2-(4-фторфенил)уксусной кислоты (425 мг, 1,82 ммоль) в DCM (5 мл) и DMF (0,05 мл) добавляли оксалилхлорид (0,17 мл, 1,91 ммоль) и раствор перемешивали в течение 0,75 часов. Раствор затем охлаждали до 0°C и добавляли раствор 2-амино-4-фторбензамида (267 мг, 1,73 ммоль) в пиридине (1 мл). Раствор перемешивали при комнатной температуре в течение 1 часа и затем упаривали. Неочищенный остаток распределяли между EtOAc и 2 н. раствором HCl. EtOAc слой упаривали с получением 2-(2-бром-2-(4-фторфенил)ацетамидо)-4-фторбензамида в виде неочищенного масла, которое использовали без дополнительной очистки. (420 мг, 62%). LC-MS (ESI) m/z 369 (M-H)-.
Стадия B: К 2-(2-бром-2-(4-фторфенил)ацетамидо)-4-фторбензамиду (420 мг, 1,1 ммоль) в диглиме (5 мл), добавляли 1 мл 10% водного раствора карбоната калия и раствор нагревали при 95°C в течение 6 часов, затем при 60°C в течение ночи. Неочищенный остаток распределяли между EtOAc и 2 н. раствором HCl. EtOAc слой упаривали и неочищенную смесь очищали при помощи хроматографии на силикагеле (0-10% MeOH в DCM) с получением бензил 7-фтор-2-((4-фторфенил)(гидрокси)метил)хиназолин-4-ола (98 мг, 31%). LC-MS (ESI) m/z 289 (M+H)+.
Стадия C: К раствору 7-фтор-2-((4-фторфенил)(гидрокси)метил)хиназолин-4-ола (98 мг, 0,33 ммоль) в ацетонитриле (4 мл) добавляли периодинан Dess-Martin (168 мг, 0,4 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 0,75 часов. Затем добавляли насыщенный раствор гидрокарбоната натрия и раствор перемешивали в течение 1 часа. Этот раствор затем фильтровали и полученное твердое вещество сушили. К этому неочищенному твердому веществу добавляли оксихлорид фосфора (2 мл) и DMA (0,02 мл) и полученный раствор нагревали при 85°C в течение 0,75 часов. Растворитель выпаривали и затем добавляли DCM и раствор фильтровали через пробку силикагеля с промывкой при помощи DCM. Растворитель выпаривали с получением (4-хлор-7-фторхиназолин-2-ил)(4-фторфенил)метанона (27 мг, 27%), который использовали без дополнительной очистки. LC-MS (ESI) m/z 305 (M+H)+.
Стадия D: раствор 5-метил-1Н-пиразол-3-амина (13 мг, 0,13 ммоль), иодида калия (15 мг, 0,088 ммоль) и DIEA (0,016 мл, 0,088 ммоль) в DMF (2 мл) добавляли к (4-хлор-7-фторхиназолин-2-ил)(4-фторфенил)метанону (0,027 мг, 0,088 ммоль). Полученный раствор перемешивали при комнатной температуре в течение ночи и затем очищали при помощи препаративной ВЭЖХ (колонка Varian дифенил с обращенной фазой, элюировали с использованием градиента растворителя B=0,05% HOAC/CH3CN и растворителя А=0,05% HOAc/H2O) с получением (7-фтор-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанона (10 мг, 31%). 1H ЯМР (300 МГц, DMSO-d6) δ 2,18 (с, 3H), 6,48 (с, 1H), 7,36 (т, 2H), 7,60 (м, 2H), 8,09 (м, 2H), 8,29 (т, 1H), 10,78 (с, 1H), 12,23 (с, 1H); LC-MS (ESI) m/z 366 (M+H)+.
Пример 20
Получение 2-(дифтор(4-фторфенил)метил)-7-фтор-N-(5-метил-1Н- пиразол-3-ил)хиназолин-4-амина
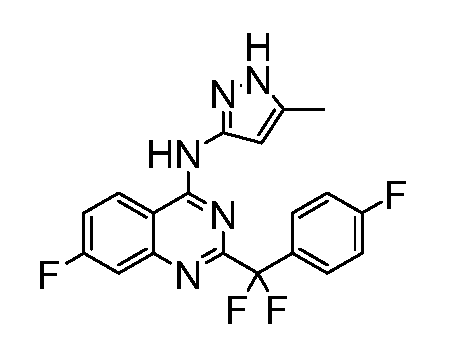
Стадия A: 2,2-дифтор-2-(4-фторфенил)ацетилхлорид получали, как описано в Примере 8 Стадия B. К раствору 2-амино-4-фторбензамида (0,330 г, 2,14 ммоль) и TEA (0,395 мл, 2,83 ммоль) в DCE (15 мл) добавляли раствор 2,2-дифтор-2-(4-фторфенил)ацетилхлорида (0,460 мг, 2,2 ммоль) в DCE (4 мл) при комнатной температуре и реакционную смесь перемешивали в течение ночи. После добавления EtOAc (20 мл) смесь промывали водой, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором. Органический раствор концентрировали с получением не совсем белого твердого вещества (0,650 г, 84%). LC-MS (ESI) m/z 327 (M+H)+.
Стадия B: К раствору 2-(2,2-дифтор-2-(4-фторфенил)ацетамидо)-4-фторбензамида (0,650 г, 1,9 ммоль) в DCE (14 мл) добавляли TEA (10,6 мл, 76 ммоль) и хлортриметилсилан (3,78 мл, 29,9 ммоль) при комнатной температуре. Реакционную смесь перемешивали при 85°C в течение ночи. После охлаждения до комнатной температуры твердое вещество фильтровали и фильтрат концентрировали досуха. Остаток растворяли в смеси EtOAc/ТГФ (1:1) и промывали водой и насыщенным солевым раствором. Органическую фазу сушили над MgSO4. Неочищенный продукт очищали на колонке с силикагелем с использованием смеси DCM/MeOH в качестве элюента с получением 2-(дифтор(4-фторфенил)-7-фторхиназолин-4-ола (0,668 г, 75%). 1H ЯМР (300 МГц, DMSO-d6) δ 7,40 (т, 2H), 7,48 (дт, 1H), 7,56 (дд, 1H), 7,77 (дд, 2H), 8,21 (дд, 1H), 13,25 (с, 1H).
Стадия C: раствор 2-(дифтор(4-фторфенил)-7-фторхиназолин-4-ола (0,350 г, 1,13 ммоль) в POCl3 (5 мл) нагревали при 105°C в течение 4 часов. Реакционную смесь концентрировали досуха при пониженном давлении и остаток растворяли в безводном толуоле. Толуол концентрировали при пониженном давлении. Остаток растворяли в небольшом объеме DCM и пропускали через короткую пробку силикагеля, элюируя при помощи DCM. 4-Хлор-2-(дифтор(4-фторфенил)метил)-7-фторхиназолин получали в виде бледно-желтого твердого вещества (325 мг, 88,5%). LC-MS (ESI) m/z 327 (M+H)+
Стадия D: К раствору 4-хлор-2-(дифтор(4-фторфенил)метил)-7-фторхиназолина (0,160 г, 0,492 ммоль) в DMF (2 мл) при комнатной температуре добавляли иодид калия (0,082 г, 0,492 ммоль), DIEA (0,094 мл, 0,541 ммоль) и 5-метил-1Н-пиразол-3-амин (0,048 г, 0,492 ммоль). После перемешивания реакционной смеси при 50°C в течение ночи смесь охлаждали до комнатной температуры и добавляли H2O (15 мл). Осадок собирали фильтрованием и промывали при помощи H2O. Неочищенный продукт очищали при помощи ВЭЖХ (колонка с обращенной фазой Phenomenex фенилгексил, элюировали с использованием градиента растворителя B=0,05% HOAc/CH3CN и растворителя А=0,05% HOAc/H2O) с получением 2-(дифтор(4-фторфенил)метил)-7-фтор-N-(5-метил-1Н-пиразол-3-ил)хиназолин-4-амина в виде белого порошка (36 мг, 19%). 1H ЯМР (300 МГц, DMSO-d6) δ 2,26 (с, 3H), 6,27 (с, 1H), 7,35 (т, 2H), 7,56 (м, 1H), 7,72-763 (м, 3H), 8,78 (м, 1H), 10,82 (с, 1H), 12,23 (с, 1H); LC-MS (ESI) m/z 388 (M+H)+.
Пример 21
Получение 2-(дифтор(4-фторфенил)метил)-7-фтор-N-(1Н-пиразол-3-ил)хиназолин-4-амина
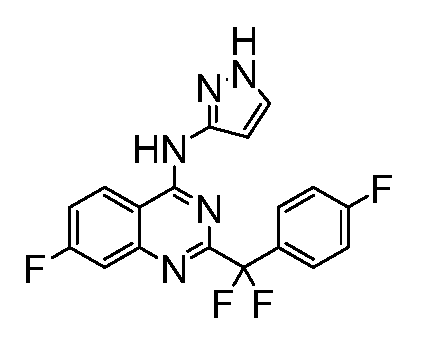
2-(Дифтор(4-фторфенил)метил)-7-фтор-N-(1Н-пиразол-3-ил)хиназолин-4-амин получали в соответствии с процедурой, описанной в Примере 20 для получения 2-(дифтор(4-фторфенил)метил)-7-фтор-N-(5-метил-1Н-пиразол-3-ил)хиназолин-4-амина, замещая 5-метил-1Н-пиразол-3-амин в Примере 20 1Н-пиразол-3-амином (11% выход). 1H ЯМР (300 МГц, DMSO-d6) δ 6,74 (с, 1H), 7,32 (т, 2H), 7,72-7,55 (м, 5H), 8,81 (м, 1H), 10,95 (с, 1H), 12,58 (с, 1H); LC-MS (ESI) m/z 374 (M+H)+.
Пример 22
Получение (4-(1Н-пиразол-3-иламино)-7-иодхиназолин-2-ил)(4-фторфенил)метанона
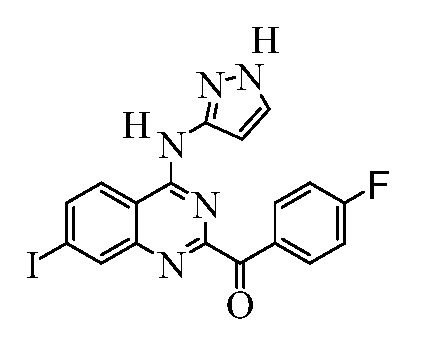
Стадия A: К раствору 2-амино-4-иодбензойной кислоты (2,5 г, 9,50 ммоль) в DMF (10 мл) при комнатной температуре в атмосфере аргона добавляли EDCI (2,18 г, 11,40 ммоль), 1-гидроксибензотриазол (1,54 г, 11,40 ммоль), DIEA (1,98 мл, 11,40 ммоль) и аммиак (7,0 н. раствор в MeOH; 1,90 мл, 13,30 ммоль). Темный раствор перемешивали при комнатной температуре в течение ночи и разбавляли при помощи H2O до образования осадка. Осадок выделяли фильтрованием, промывали при помощи H2O и сушили в условиях высокого вакуума в течение нескольких часов с получением 2-амино-4-иодбензамида в виде ржаво-коричневого твердого вещества (1,3 г, 52%). LC-MS (ESI) m/z 263 (M+H)+.
Стадия B: К раствору 2-амино-4-иодбензамида (1,0 г, 3,61 ммоль) в ледяной уксусной кислоте (10 мл) при комнатной температуре добавляли диэтилоксалат (5 мл). Смесь нагревали при 120°C в течение 24 часов. Смесь охлаждали до комнатной температуры и разбавляли при помощи H2O до образования осадка. Осадок удаляли фильтрованием, промывали при помощи H2O и сушили в условиях высокого вакуума в течение нескольких часов с получением этил 7-иод-4-оксо-3,4-дигидрохиназолин-2-карбоксилата (1,0 г, 76%) в виде ржаво-коричневого твердого вещества. 1H ЯМР (300 МГц, DMSO-d6) δ 1,36 (т, 3H), 4,48 (кв., 2H), 7,90 (д, 1H), 7,95 (д, 1H), 8,20 (д, 1H), 8,28 (с, 1H), 12,78 (с, 1H); LC-MS (ESI) m/z 330 (M+H)+.
Стадия C: Суспензию этил 7-иод-4-оксо-3,4-дигидрохиназолин-2-карбоксилата (1,0 г, 2,90 ммоль) в оксихлориде фосфора (10 мл) нагревали при 110°C в атмосфере аргона в течение 12 часов. Реакционную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью 30% EtOAc/гексан, с получением этил 4-хлор-7-иодхиназолин-2-карбоксилата в виде белого твердого вещества (0,510 г, 48%). 1H ЯМР (300 МГц, DMSO-d6) δ 1,32 (т, 3H), 4,48 (кв., 2H), 7,88 (д, 1H), 7,92 (д, 1H), 8,25 (с, 1H); LC-MS (ESI) m/z 363 (M+H)+.
Стадия D: К раствору этил 4-хлор-7-иодхиназолин-2-карбоксилата (0,138 г, 0,38 ммоль) в DMF (2 мл) при комнатной температуре в атмосфере аргона добавляли иодид калия (0,069 г, 0,42 ммоль), DIEA (0,079 мл, 0,45 ммоль) и 1Н-пиразол-3-амин (0,038 г, 0,46 ммоль). Смесь перемешивали при комнатной температуре в течение ночи, затем разбавляли при помощи H2O (15 мл). Осадок собирали фильтрованием, промывали при помощи H2O и сушили в условиях высокого вакуума в течение нескольких часов с получением этил 4-(1Н-пиразол-3-иламино)-7-иодхиназолин-2-карбоксилата в виде желтого твердого вещества (0,130 г, 84%). 1H ЯМР (300 МГц, DMSO-d6) δ 1,34 (т, 3H), 4,38 (кв., 2H), 7,14 (м, 1H), 7,72 (м, 1H), 7,94 (д, 1H), 8,28 (д, 1H), 8,48 (д, 1H), 10,92 (с, 1H), 12,58 (с, 1H); LC-MS (ESI) m/z 410 (M+H)+.
Стадия E: К суспензии этил 4-(lH-пиразол-3-иламино)-7-иодхиназолин-2-карбоксилата (0,130 г, 0,31 ммоль) в ТГФ (5 мл) при -40°C добавляли (4-фторфенил)магнийбромид (1,0 M раствор в ТГФ, 0,797 мл, 0,79 ммоль). Смесь перемешивали при -40°C в течение 5 часов, гасили 1,0 н. раствором HCl (2,0 мл) и концентрировали при пониженном давлении. Остаток очищали при помощи препаративной ВЭЖХ (колонка Phenomenex фенилгексил с обращенной фазой, элюировали с использованием градиента растворителя B=0,05% HOAc/CH3CN и растворителя А=0,05% HOAc/H2O) с получением (4-(1Н-пиразол-3-иламино)-7-иодхиназолин-2-ил)(4-фторфенил)метанона в виде желтого твердого вещества (0,050 г, 34%). 1H ЯМР (300 МГц, DMSO-d6) δ 6,72 (с, 1H), 7,35 (т, 2H), 7,64 (с, 1H), 8,00 (д, 1H), 8,08 (дд, 2H), 8,28 (с, 1H), 8,55 (д, 1H), 10,88 (с, 1H), 12,55 (с, 1H); LC-MS (ESI) m/z 460 (M+H)+.
Пример 23
Получение (R,S)-(4-(1Н-пиразол-3-иламино)-7-иодхиназолин-2-ил)(4-фторфенил)метанола
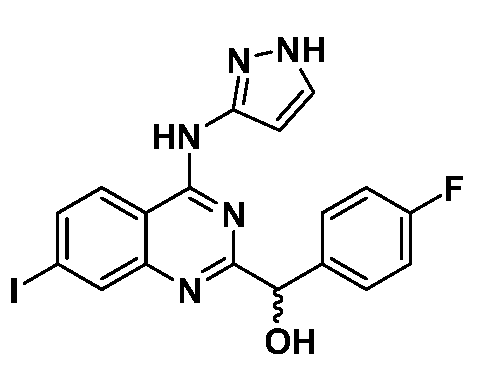
К суспензии (4-(1Н-пиразол-3-иламино)-7-иодхиназолин-2-ил)(4-фторфенил)метанона из Примера 22 (0,032 г, 0,07 ммоль) в смеси 1:1 ТГФ/MeOH (2 мл) при 0°C в атмосфере аргона добавляли NaBH4 (0,004 г, 0,10 ммоль). Смесь перемешивали при 0°C в течение 3 часов, гасили добавлением двух капель ацетона и концентрировали при пониженном давлении. Остаток очищали при помощи препаративной ВЭЖХ (колонка Phenomenex фенилгексил с обращенной фазой, элюировали с использованием градиента растворителя B=0,05% HOAc/CH3CN и растворителя А=0,05% HOAc/H2O) с получением (R,S)-(4-(1H-пиразол-3-иламино)-7-иодхиназолин-2-ил)(4-фторфенил)метанола в виде белого твердого вещества (0,020 г, 63%). 1H ЯМР (300 МГц, DMSO-d6) δ 5,64 (с, 1H), 5,88 (ушир.с, 1H), 6,80 (ушир.с, 1H), 7,15 (т, 2H), 7,52 (м, 2H), 7,65 (с, 1H), 7,80 (д, 1H), 8,12 (ушир.с, 1H), 8,35 (ушир.с, 1H), 10,62 (ушир.с, 1H), 12,50 (ушир.с, 1H); LC-MS (ESI) m/z 462 (M+H)+.
Пример 24
Получение (4-фторфенил)(7-метил-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанона
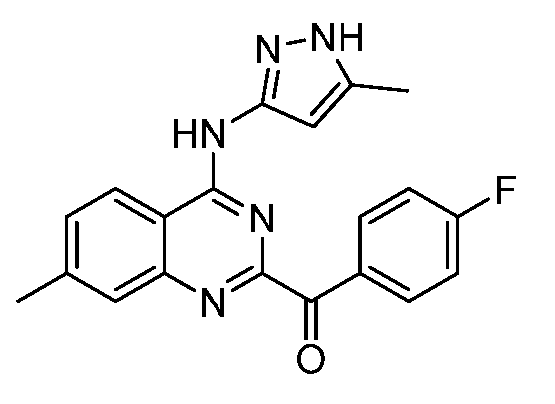
Стадия A: К раствору 2-амино-4-метилбензамида (3 г, 20,0 ммоль) в ТГФ (40 мл) добавляли 5-(4-фторфенил)-1,3-диоксолан-2,4-дион из Примера 16 (4,7 г, 24 ммоль) и раствор перемешивали в течение 2 часов при 50°C. Затем добавляли метоксид натрия в MeOH (25%, 5,2 мл, 24 ммоль) и раствор перемешивали при 50°C в течение ночи. Реакционный раствор концентрировали, добавляли 2 н. раствор HCl и смесь фильтровали. Собранное твердое вещество сушили с получением 2-((4-фторфенил)(гидрокси)метил)-7-метилхиназолин-4-ола (5,14 г, 91%), который использовали без дополнительной очистки. LC-MS (ESI) m/z 285 (M+H)+.
Стадия B: К раствору 2-((4-фторфенил)(гидрокси)метил)-7-метилхиназолин-4-ола (3 г, 10,56 ммоль) в ацетонитриле (45 мл) добавляли периодинан Dess-Martin (5,37 г, 12,67 ммоль) и смесь перемешивали при комнатной температуре в течение 5 часов. Затем добавляли насыщенный раствор гидрокарбоната натрия и смесь перемешивали при комнатной температуре в течение ночи. Полученную суспензию затем фильтровали и полученное твердое вещество сушили с получением неочищенного (4-фторфенил)(4-гидрокси-7-метилхиназолин-2-ил)метанона (2,65 г, 89%). LC-MS (ESI) m/z 283 (M+H)+.
Стадия C: К раствору (4-фторфенил)(4-гидрокси-7-метилхиназолин-2-ил)метанона (650 мг, 2,3 ммоль) в DCM (4 мл) добавляли TEA (1,23 мл, 9,2 ммоль), DMAP (15 мг, 0,05 ммоль) и 2,4,6-триизопропилбензол-1-сульфонилхлорид (905 мг, 3,0 ммоль) и смесь перемешивали при комнатной температуре в течение 0,5 часов. Неочищенную смесь концентрировали и остаток очищали хроматографией на силикагеле, элюируя смесью 0-50% EtOAc в гексане, с получением 2-(4-фторбензоил)-7-метилхиназолин-4-ил 2,4,6-триизопропилбензолсульфоната (790 мг, 63%), который использовали без дополнительной очистки. LC-MS (ESI) m/z 571 (M+Na)+.
Стадия D: раствор 5-метил-1Н-пиразол-3-амина (225 мг, 2,31 ммоль), иодида калия (188 мг, 0,088 ммоль) и 2-(4-фторбензоил)-7-метилхиназолин-4-ил 2,4,6-триизопропилбензолсульфоната (380 мг, 0,69 ммоль) в DMA (2 мл) перемешивали при 50°C в течение 6 часов, затем добавляли воду и раствор фильтровали. Твердое вещество затем сушили и растирали в порошок с ацетонитрилом с получением (4-фторфенил)(7-метил-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанона (36 мг, 14%). 1H ЯМР (300 МГц, DMSO-d6) δ 2,18 (с, 3H), 2,50 (с, 3H), 6,53 (с, 1H), 7,38 (т, 2H), 7,51 (д, 2H), 7,66 (с,lH), 8,08 (м, 2H), 8,62 (д, 1H), 10,57 (с, 1H), 12,18 (с, 1H); LC-MS (ESI) m/z 362 (M+H)+.
Пример 25
Получение (R,S)-(4-фторфенил)(7-метил-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанола
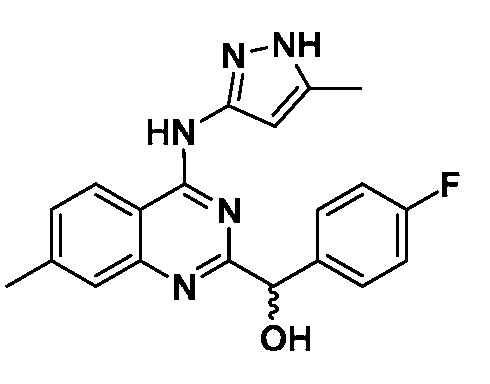
К суспензии (4-фторфенил)(7-метил-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанона (40 мг, 0,11 ммоль) в MeOH (2 мл), охлажденному до 0°C, добавляли борогидрид натрия (30 мг, 0,8 ммоль). Раствору давали медленно нагреться до комнатной температуры и перемешивали в течение 2 часов. Затем добавляли 1 н. раствор HCl, раствор перемешивали в течение 10 минут и затем фильтровали. Неочищенное твердое вещество очищали при помощи препаративной ВЭЖХ (колонка Varian дифенил с обращенной фазой, элюировали с использованием градиента растворителя B=0,05% HOAC/CH3CN и растворителя А=0,05% HOAc/H2O) с получением (R,S)-(4-фторфенил)(7-метил-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанола (17 мг, 42%). 1H ЯМР (300 МГц, DMSO-d6) δ 2,24 (с, 3H), 2,47 (с, 3H), 5,64, (с, 1H), 5,82 (ушир.с, 1H), 6,36 (с, 1H), 7,14 (т, 2H), 7,34 (д, 1H), 7,54 (м, 3H), 8,45 (д, 1H), 10,39 (с, 1H), 12,18 (с, 1H); LC-MS (ESI) m/z 364 (M+H)+.
Пример 26
Получение 2-(дифтор(4-фторфенил)метил)-7-метил-N-(5-метил-1Н-пиразол-3-ил)хиназолин-4-амина
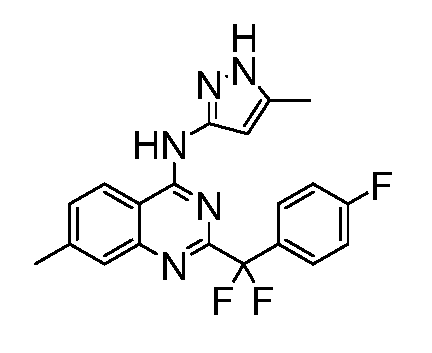
Стадия A: 2,2-дифтор-2-(4-фторфенил)ацетилхлорид получали, как описано в Примере 8 Стадия B. К раствору 2-амино-4-метилбензамида (4,0 г, 0,026 моль) и TEA (4,35 мл, 0,0312 моль) в DCE (60 мл) добавляли раствор 2,2-дифтор-2-(4-фторфенил)ацетилхлорида (4,90 г, 0,025 моль) в DCE (10 мл) при комнатной температуре и реакционную смесь перемешивали в течение ночи. После добавления EtOAc (200 мл) смесь промывали водой, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором. Органический раствор концентрировали с получением не совсем белого твердого вещества (5,85 г, 69%). LC-MS (ESI) m/z 305 (M+H)+.
Стадия B: К раствору 2-(2,2-дифтор-2-(4-фторфенил)ацетамидо)-4-метилбензамида (5,85 г, 0,0181 моль) в DCE (120 мл) добавляли TEA (91,5 мл, 0,724 моль) и хлортриметилсилан (34,4 мл, 0,272 моль) при комнатной температуре. Реакционную смесь перемешивали при 85°C в течение ночи. После охлаждения до комнатной температуры твердое вещество фильтровали и фильтрат концентрировали досуха. Осуществляли поглощение осадка в смесь EtOAc/ТГФ (1:1) и промывали водой и насыщенным солевым раствором. Чистый продукт получали после кристаллизации из горячего EtOAc (2,02 г, 37%); LC-MS (ESI) m/z 305 (M+H)+.
Стадия C: Раствор 2-(дифтор(4-фторфенил)метил)-7-метилхиназолин-4-ола (0,304 г, 1 ммоль) в POCl3 (5 мл) нагревали при 105°C в течение ночи. Реакционную смесь концентрировали досуха при пониженном давлении и остаток растворяли в безводном толуоле. Толуол концентрировали при пониженном давлении. Остаток растворяли в небольшом объеме DCM и пропускали через короткую подушку силикагеля, с использованием DCM в качестве растворителя. 4-Хлор-2-(дифтор(4-фторфенил)метил)-7-фторхиназолин получали в виде бледно-желтого твердого вещества (308 мг, 95%). 1H ЯМР (300 МГц, DMSO-d6) δ 2,61 (с, 3H), 7,33 (т, 2H), 7,73 (м, 2H), 7,82 (дд, 1H), 8,01 (с, 1H), 8,23 (д, 1H).
Стадия D: 2-(Дифтор(4-фторфенил)метил)-7-метил-N-(5-метил-1Н-пиразол-3-ил)хиназолин-4-амин получали в соответствии с процедурой, описанной в Примере 20 для получения 2-(дифтор(4-фторфенил)метил)-7-фтор-N-(5-метил-1Н-пиразол-3-ил)хиназолин-4-амина, замещая 4-хлор-2-(дифтор(4-фторфенил)метил)-7-фторхиназолин в Примере 20 4-хлор-2-(дифтор(4-фторфенил)метил)-7-метилхиназолином (13% выход). 1H ЯМР (300 МГц, DMSO-d6) δ 2,26 (с, 3H), 2,50 (с, 3H), 6,30 (с, 1H), 7,34 (т, 2H), 7,47 (м, 1H), 7,71-7,66 (м, 3H), 8,56 (д, 1H), 10,59 (с, 1H), 12,20 (ушир.с, 1H); LC-MS (ESI) m/z 384 (M+H)+.
Пример 27
Получение 2-(дифтор(4-фторфенил)метил)-7-метил-N-(1Н-пиразол-3-ил)хиназолин-4-амина
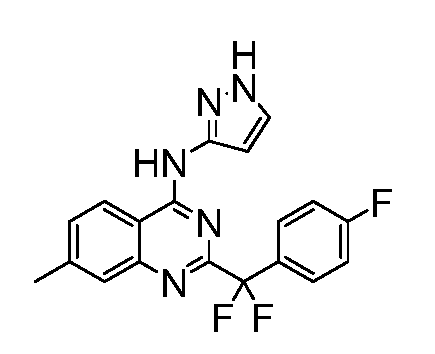
2-(Дифтор(4-фторфенил)метил)-7-метил-N-(1Н-пиразол-3-ил)хиназолин-4-амин получали в соответствии с процедурой, описанной в Примере 20 для получения 4-хлор-2-(дифтор(4-фторфенил)метил)-7-фторхиназолина, замещая 4-хлор-2-(дифтор(4-фторфенил)метил)-7-фторхиназолин в Примере 20 4-хлор-2-(дифтор(4-фторфенил)метил)-7-метилхиназолином и замещая 5-метил-1Н-пиразол-3-амин в Примере 20 1Н-пиразол-3-амином (6% выход). 1H ЯМР (300 МГц, DMSO-d6) δ 2,50 (с, 3H), 6,75 (с, 1H), 7,32 (т, 2H), 7,48 (м, 1H), 7,71-7,66 (м, 4H), 8,69 (д, 1H), 10,72 (с, 1H), 12,51 (с, 1); LC-MS (ESI) m/z 370 (M+H)+.
Пример 28
Получение (4-(1Н-пиразол-3-иламино)-7-метоксихиназолин-2-ил)(4-фторфенил)метанона
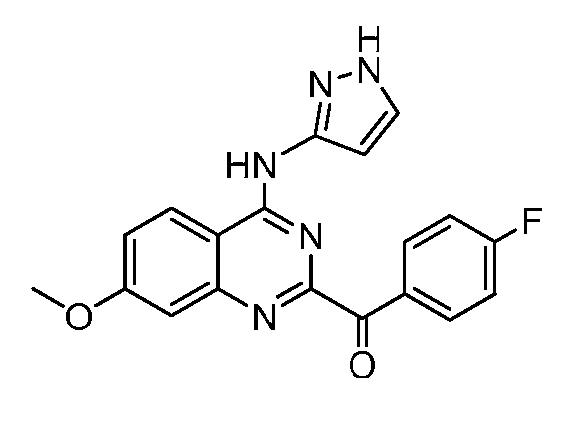
Стадия A: К 2-амино-4-метоксибензойной кислоте (10,00 г, 59,82 ммоль) в DMF (150 мл) при комнатной температуре добавляли DIEA (16,2 мл, 71,79 ммоль), 2 н. раствор аммиака в MeOH (41,8 мл, 83,75 ммоль), 1-EDCI (13,76 г, 71,79 ммоль) и 1-гидроксибензотриазол (9,70 г, 71,79 ммоль). Раствор перемешивали при комнатной температуре в атмосфере аргона. Через 20 часов раствор концентрировали, разбавляли водой и экстрагировали семь раз при помощи EtOAc. EtOAc концентрировали для уменьшения объема и раствор промывали насыщенным солевым раствором. EtOAc фракцию концентрировали и разбавляли диэтиловый эфиром. Полученный осадок собирали и сушили в вакууме с получением ржаво-коричневого твердого вещества (9,8 грамм, 91%). 1H ЯМР (300 МГц, DMSO-d6) δ м.д. 3,69 (с, 3H) 6,06 (дд, J=8,76, 2,54 Гц, 1H) 6,19 (д, J=2,64 Гц, 1H) 6,72 (ушир.с, 2H) 7,48 (д, J=8,67 Гц, 1H); LC-MS (ESI) m/z 167 (M+H)+.
Стадия B: К 2-амино-4-метоксибензамиду (6,0 г, 36,11 ммоль) в DCM (200 мл) добавляли DIEA (8,2 мл, 46,94 ммоль). Раствор охлаждали до 0°C с последующим добавлением по каплям этилхлорглиоксилата(4,44 мл, 39,72 ммоль) в DCM (50 мл). Затем добавляли DMAP (20 мг) с последующим удалением охлаждающей бани. После перемешивания в течение 20 часов при комнатной температуре в атмосфере Ar, смесь концентрировали и добавление воды приводило к образованию осадка, который фильтровали и промывали водой. Сушка в вакууме давала твердое вещество (6,9 г, 72%). 1H ЯМР (300 МГц, DMSO-d6) δ м.д. 1,31 (т, 3H) 3,82 (с, 3H) 4,30 (кв., 7=7,10 Гц, 2H) 6,80 (дд, J=8,85, 2,64 Гц, 1H) 7,64 (ушир.с, 1H) 7,89 (д, J=8,85 Гц, 1H) 8,20 (д, J=2,64 Гц, 2H) 13,53 (с, 1H); LC-MS (ESI) m/z 250, 289, 330.
Стадия C: К этил 2-(2-карбамоил-5-метоксифениламино)-2-оксоацетату (6,9 г, 25,92 ммоль) в DCE (300 мл) при комнатной температуре добавляли TEA (144 мл, 1,04 моль) с последующим добавлением триметилсилилхлорида (49 мл, 388,7 ммоль). Гетерогенную смесь нагревали до температуры кипения с обратным холодильником в атмосфере Ar. Через 20 часов раствор охлаждали и выливали в лед/воду. Органический слой отделяли и концентрировали, затем добавляли к водной фракции. Смесь подкисляли до pH 4 и осадок собирали и сушили в вакууме с получением ржаво-коричневого белого твердого вещества (5,4 г, 85%). 1H ЯМР (300 МГц, DMSO-d6) δ м.д. 1,35 (т, 7=7,16 Гц, 3H) 3,92 (с, 3H) 4,38 (кв., 7=7,16 Гц, 2H) 7,21 (дд, 7=8,76, 2,54 Гц, 1H) 7,30 (д, 7=2,64 Гц, 1H) 8,07 (д, 7=8,67 Гц, 1H) 12,48 (ушир.с, 1H); LC-MS (ESI) m/z 249 (M+H)+.
Стадия D: К оксихлориду фосфора (5 мл) добавляли этил 7-метокси-4-оксо-3,4-дигидрохиназолин-2-карбоксилат (1,0 г, 4,03 ммоль) с последующим добавлением диметилформамида (4 капли). Раствор нагревали до 85°C в течение 2 часов и затем концентрировали. Остаток охлаждали в -20°C охлаждающей бане и разбавляли холодным EtOAc. Охлажденный раствор промывали холодной водой, насыщенным водным раствором бикарбоната натрия и насыщенным солевым раствором. Удаление растворителя давало белое твердое вещество (1,2 г, 100%.). LC-MS (ESI) m/z 267 (M+H)+.
Стадия E: К этил 4-хлор-8-метоксихиназолин-2-карбоксилату (500 мг, 1,88 ммоль) в DMF (20 мл) добавляли DIEA (0,720 мл, 4,14 ммоль), 3-аминопиразол (309 мг, 3,76 ммоль) и иодид калия (312 мг, 1,88 ммоль) при комнатной температуре. После перемешивания в течение 18 часов и в течение 6 часов при 40°C раствор концентрировали. Добавление воды приводило к образованию осадка, который собирали и промывали водой. Сушка в вакууме давала твердое вещество (475 мг, 81%). 1H ЯМР (300 МГц, DMSO-d6) δ м.д. 1,36 (т, 3H) 3,94 (с, 3H) 4,39 (кв., 2H) 7,16 (с, 1H) 7,28 (м, 1H) 7,34 (м, 1H) 7,74 (ушир.с, 1H) 8,65 (м, 1H) 10,65 (с, 1H) 12,50 (с, 1H); LC-MS (ESI) m/z 314 (M+H)+.
Стадия F: К этил 4-(1Н-пиразол-3-иламино)-7-метоксихиназолин-2-карбоксилату (30 мг, 0,10 ммоль) в безводном DMA (2,5 мл), охлажденном в -20°C охлаждающей бане, добавляли по каплям в 4-фторфенилмагнийбромид в ТГФ (0,306 мл, 0,306 ммоль). Через 2 часа добавляли дополнительное количество 1 н. раствора 4-фторфенилмагнийбромида (0,050 мл). Через 2 часа реакционную смесь гасили путем добавления насыщенного раствора хлорида аммония. Раствор концентрировали и добавляли H2O. Осадок промывали водой и очищали при помощи препаративной тонкослойной хроматографии на силикагеле, элюируя при помощи 10% MeOH/DCM, с получением твердого вещества (21 мг, 60%). 1H ЯМР (300 МГц, DMSO-d6) δ м.д. 3,93 (с, 3H) 6,73 (ушир.с, 1H) 7,29 (м, 2H) 7,38 (м, 2H) 7,63 (ушир.с, 1H) 8,08 (м, 2H) 8,64 (м, 1H) 10,63 (ушир.с, 1H) 12,47 (ушир.с, 1H); LC-MS (ESI) m/z 364 (M+H)+.
Пример 29
Получение (R,S)-(4-(1Н-пиразол-3-иламино)-7-метоксихиназолин-2-ил)(4-фторфенил)метанола
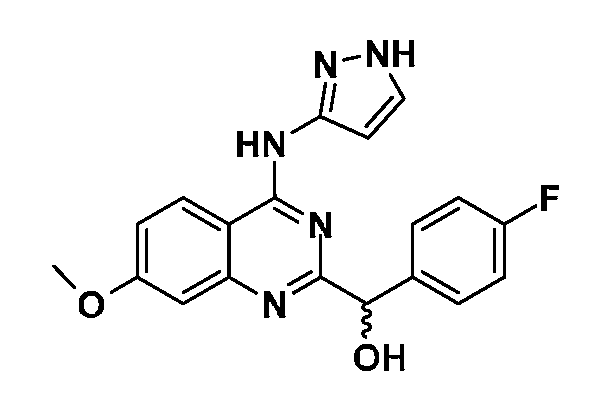
К (4-(1Н-пиразол-3-иламино)-7-метоксихиназолин-2-ил)(4-фторфенил)метанону (50 мг, 0,14 ммоль) в смеси 2:1 MeOH/DMF (4,5 мл) при комнатной температуре добавляли борогидрид натрия (8 мг, 0,21 ммоль) одной порцией. После перемешивания в течение 40 минут добавляли раствор LiOH (60 мг) в H2O (1 мл) и перемешивание продолжали в течение 45 минут. Раствор концентрировали и разбавляли водой, что приводило к образованию белого осадка (32 мг). Осадок собирали и очищали препаративной тонкослойной хроматографией на диоксиде кремния, элюируя при помощи 10% MeOH/DCM, с получением (R,S)-(4-(1Н-пиразол-3-иламино)-7-метоксихиназолин-2-ил)(4-фторфенил)метанола в виде белого твердого вещества (10 мг, 20%). 1H ЯМР (300 МГц, DMSO-d6) δ м.д. 3,91 (с, 3H) 5,64 (м, 1H) 5,77 (ушир.с, 1H) 6,80 (ушир.с, 1H) 7,12 (м, 4H) 7,19 (ушир.с, 1H) 7,54 (м, 2H) 7,66 (ушир.с, 1H) 8,49 (м, 1H) 10,38 (ушир.с, 1H) 12,44 (ушир.с, 1H); LC-MS (ESI) m/z 366 (M+H)+.
Пример 30
Получение (4-фторфенил)(7-метокси-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанона
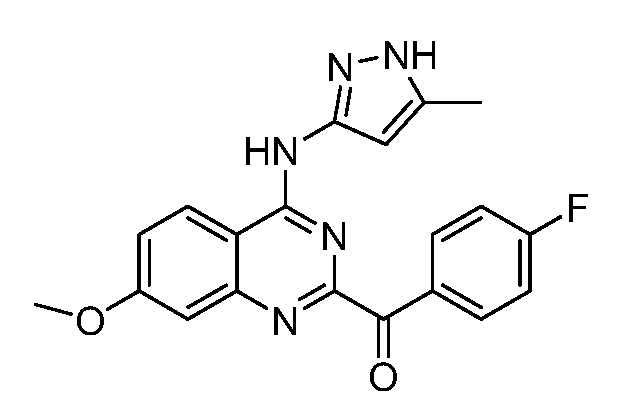
Стадия A: К этил 4-хлор-7-метоксихиназолин-2-карбоксилату (600 мг, 2,26 ммоль) в DMF (8 мл) добавляли DIEA (0,864 мл, 4,96 ммоль), 5-метил-1Н-пиразол-3-амин (657 мг, 6,77 ммоль) и иодид калия (374 мг, 2,26 ммоль) при комнатной температуре. После перемешивания в течение 18 часов при 40°C раствор концентрировали, и добавление воды приводило к образованию осадка, который собирали и промывали водой. Сушка в вакууме давала твердое вещество (570 мг, 77%). 1H ЯМР (300 МГц, DMSO-d6) δ м.д. 1,37 (т, 3H) 2,27 (с, 3) 3,93 (с, 3H) 4,36 (кв., 2H) 6,93 (с, 1H) 7,25 (м, 1H) 7,32 (ушир.с, 1H) 8,62 (м, 1H) 10,53 (с, 1H) 12,18 (с, 1H); LC-MS (ESI) m/z 328 (M+H)+.
Стадия B: К этил 7-метокси-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-карбоксилату (379 мг, 1,16 ммоль) в безводном DMA (16 мл), охлажденном в -30°C бане, добавляли по каплям 1 н. раствор 4-фторфенилмагнийбромида в ТГФ (4,05 мл, 4,05 ммоль). Через 4 часа реакционную смесь гасили добавлением насыщенного раствора хлорида аммония. Раствор концентрировали и добавляли H2O. Осадок промывали водой и диэтиловым эфиром с получением желтого твердого вещества (415 мг, 95%). 1H ЯМР (300 МГц, DMSO-d6) δ м.д. 2,18 (ушир.с, 3H) 3,92 (ушир.с, 3H) 6,48 (ушир.с, 1H) 7,27 (ушир.с, 2H) 7,39 (ушир.с, 2H) 8,07 (ушир.с, 2H) 8,62 (ушир.с, 1H) 10,50 (ушир.с, 1H) 12,15 (ушир.с, 1H); LC-MS (ESI) m/z 378 (M+H)+.
Пример 31
Получение (R,S)-(4-фторфенил)(7-метокси-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанола
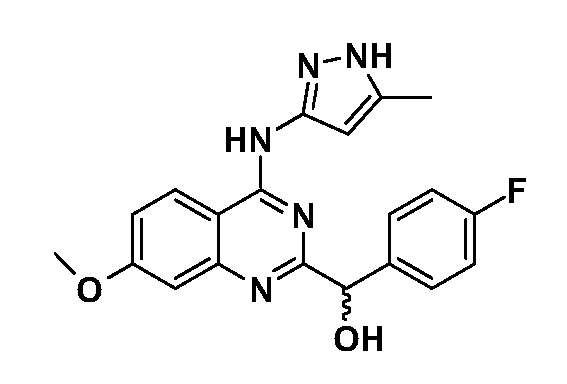
К (4-фторфенил)(7-метокси-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанону (150 мг, 0,40 ммоль) в растворе 2:1 MeOH/DMF (8 мл), охлажденном до 0°C, добавляли борогидрид натрия (23 мг, 0,60 ммоль) одной порцией. После перемешивания в течение двух часов при комнатной температуре раствор охлаждали до 0°C и гасили путем добавления 1 н. раствора HCl. Раствор концентрировали и разбавляли водой, что приводило к образованию белого осадка (130 мг). Осадок собирали и очищали на диоксиде кремния, элюируя при помощи 3-15% MeOH/DCM, с получением белого твердого вещества (20 мг, 13%). 1H ЯМР (300 МГц, DMSO-d6) δ м.д. 2,25 (с, 3H) 3,91 (с, 3H) 5,62 (м, 1H) 5,74 (м, 1H) 6,43 (с, 1H) 6,96-7,19 (м, 4H) 7,51-7,55 (м, 2H) 8,49 (м, 1H) 10,22 (с, 1H) 12,08 (ушир.с, 1H); LC-MS (ESI) m/z 380 (M+H)+.
Пример 32
Получение 2-(дифтор(4-фторфенил)метил)-7-метокси-N-(5-метил-1Н-пиразол-3-ил)хиназолин-4-амина
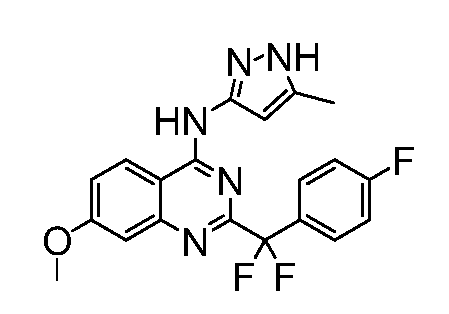
Стадия A: 2,2-дифтор-2-(4-фторфенил)ацетилхлорид получали, как описано в Примере 8 Стадия B. К раствору 2-амино-4-метоксибензамида (0,415 г, 2,5 ммоль) и TEA (0,418 мл, 3 ммоль) в DCE (15 мл) добавляли раствор 2,2-дифтор-2-(4-фторфенил)ацетилхлорида (0,579 мг, 2,78 ммоль) в DCE (5 мл) при комнатной температуре и реакционную смесь перемешивали в течение ночи. После добавления EtOAc (200 мл) смесь промывали 1 н. раствором HCl, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором. Органический раствор концентрировали с получением не совсем белого твердого вещества (371 г, 44%). LC-MS (ESI) m/z 339 (M+H)+.
Стадия B: 2-(Дифтор(4-фторфенил)метил)-7-метоксихиназолин-4-ол получали в соответствии с процедурой, описанной в Примере 20 для получения 2-(дифтор(4-фторфенил)-7-фторхиназолин-4-ола, замещая 2-(2,2-дифтор-2-(4-фторфенил)ацетамидо)-4-фторбензамид в Примере 20 2-(2,2-дифтор-2-(4-фторфенил)ацетамидо)-4-метоксибензамидом. Неочищенный продукт (~100% выход) использовали непосредственно на следующей стадии. 1H ЯМР (300 МГц, DMSO-d6) δ 3,89 (с, 3H), 7,16 (м, 2H), 7,39 (т, 2H), 7,75 (м, 2H), 8,04 (д, 1H), 12,96 (с, 1H); LC-MS (ESI) m/z 321 (M+H)+.
Стадия C: 4-Хлор-2-(дифтор(4-фторфенил)метил)-7-метоксихиназолин получали в соответствии с процедурой, описанной в Примере 26 для синтеза 4-хлор-2-(дифтор(4-фторфенил)метил)-7-метилхиназолина, замещая 2-(дифтор(4-фторфенил)метил)-7-метилхиназолин-4-ол в Примере 26 2-(дифтор(4-фторфенил)метил)-7-метоксихиназолин-4-олом. 4-Хлор-2-(дифтор(4-фторфенил)метил)-7-метоксихиназолин выделяли в виде светло-желтого твердого вещества (0,290 г, 89%). LC-MS (ESI) m/z 339 (M+H)+.
Стадия D: 2-(Дифтор(4-фторфенил)метил)-7-метокси-N-(5-метил-1Н-пиразол-3-ил)-хиназолин-4-амин получали в соответствии с процедурой, описанной в Примере 20 для получения 2-(дифтор(4-фторфенил)метил)-7-фтор-N-(5-метил-1Н-пиразол-3-ил)хиназолин-4-амина, замещая 4-хлор-2-(дифтор(4-фторфенил)метил)-7-фторхиназолин в Примере 20 4-хлор-2-(дифтор(4-фторфенил)метил)-7-метоксихиназолином (36% выход). 1H ЯМР (300 МГц, DMSO-d6) δ 2,27 (с, 3H), 3,93 (с, 3H), 6,28 (с, 1H), 7,37-7,20 (м, 4H), 7,71-7,66 (м, 1H), 8,58 (д, 2H), 10,53 (с, 1H); LC-MS (ESI) m/z 400 (M+H)+.
Пример 33
Получение 2-(дифтор(4-фторфенил)метил)-7-метокси-N-(1Н-пиразол-3-ил)хиназолин-4-амина
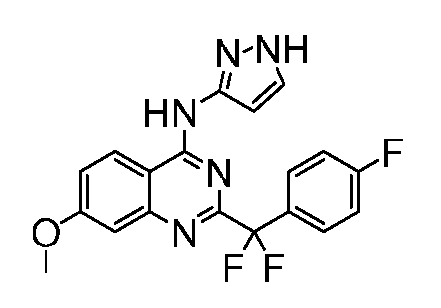
2-(дифтор(4-фторфенил)метил)-7-метокси-N-(1Н-пиразол-3-ил)хиназолин-4-амин получали в соответствии с процедурой, описанной в Примере 20 для получения 2-(дифтор(4-фторфенил)метил)-7-фтор-N-(5-метил-1Н-пиразол-3-ил)хиназолин-4-амина, замещая 4-хлор-2-(дифтор(4-фторфенил)метил)-7-фторхиназолин в Примере 20 4-хлор-2-(дифтор(4-фторфенил)метил)-7-метоксихиназолином и замещая 5-метил-1Н-пиразол-3-амин в Примере 20 1Н-пиразол-3-амином (24% выход). 1H ЯМР (300 МГц, DMSO-d6) δ 4,17 (с, 3H), 6,75 (с, 1H), 7,43-7,22 (м, 4H), 7,71-7,67 (м, 3H), 8,60 (д, 1H), 10,70 (с, 1H), 12,50 (с, 1H); LC-MS (ESI) m/z 386 (M+H)+.
Пример 34
Получение 2-(дифтор(4-фторфенил)метил)-8-фтор-N-(5-метил-1Н- пиразол-3-ил)хиназолин-4-амина
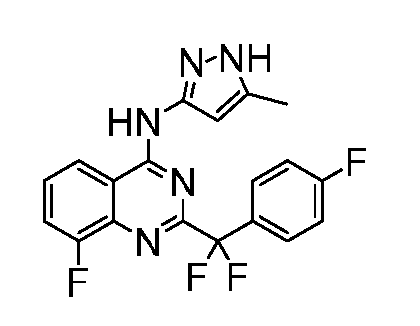
Стадия A: 2-(2,2-Дифтор-2-(4-фторфенил)ацетамидо)-3-фторбензамид получали в соответствии с процедурой, описанной в Примере 32 для получения 2-(2,2-дифтор-2-(4-фторфенил)ацетамидо)-4-метоксибензамида, замещая 2-амино-4-метоксибензамид в Примере 32 2-амино-3-фторбензамидом. Продукт очищали на колонке с силикагелем с использованием DCM/MeOH в качестве элюента (20%); LC-MS (ESI) m/z 327 (M+H)+.
Стадия B: раствор 2-(2,2-дифтор-2-(4-фторфенил)ацетамидо)-3-фторбензамида (0,235 г, 0,72 ммоль) в уксусной кислоте (2 мл) нагревали при 120°C в течение 3 часов. Реакционной смеси давали нагреться до комнатной температуры и затем добавляли воду. Твердое вещество собирали фильтрованием и промывали при помощи H2O. 2-(Дифтор(4-фторфенил)метил-8-фторхиназолин-4-ол получали в виде не совсем белого твердого вещества (0,135 г, 61%). 1H ЯМР (300 МГц, DMSO-d6) δ 7,38 (м, 2H), 7,61 (м, 1H), 7,80-7,74 (м, 3H), 7,97 (м, 1H), 13,43 (с, 1H); LC-MS (ESI) m/z 309 (M+H)+.
Стадия C: 4-Хлор-2-(дифтор(4-фторфенил)метил)-8-фторхиназолин получали в соответствии с процедурой, описанной в Примере 26 для получения 4-хлор-2-(дифтор(4-фторфенил)метил)-7-метилхиназолина, замещая 2-(дифтор(4-фторфенил)метил)-7-метилхиназолин-4-ол в Примере 26 2-(дифтор(4-фторфенил)метил-8-фторхиназолин-4-олом (94% выход). LC-MS (ESI) m/z 327 (M+H)+.
Стадия D: 2-(Дифтор(4-фторфенил)метил)-8-фтор-N-(5-метил-1Н-пиразол-3-ил)хиназолин-4-амин получали в соответствии с процедурой, описанной в Примере 20 для получения 2-(дифтор(4-фторфенил)метил)-7-фтор-N-(5-метил-1Н-пиразол-3-ил)хиназолин-4-амина, замещая 4-хлор-2-(дифтор(4-фторфенил)метил)-7-фторхиназолин в Примере 20 4-хлор-2-(дифтор(4-фторфенил)метил)-8-фторхиназолином. Чистое соединение получали после растирания в порошок с MeOH (34%). 1H ЯМР (300 МГц, DMSO-d6) δ 2,24 (с, 3H), 6,27 (с, 1H), 7,34 (т, 2H), 7,64 (м, 1H), 7,78-7,69 (м, 3H), 8,51 (д, 1H), 10,85 (с, 1H), 12,25 (с, 1H); LC-MS (ESI) m/z 388 (M+H)+.
Пример 35
Получение (4-(1Н-пиразол-3-иламино)-8-метоксихиназолин-2-ил)(4-фторфенил)метанона
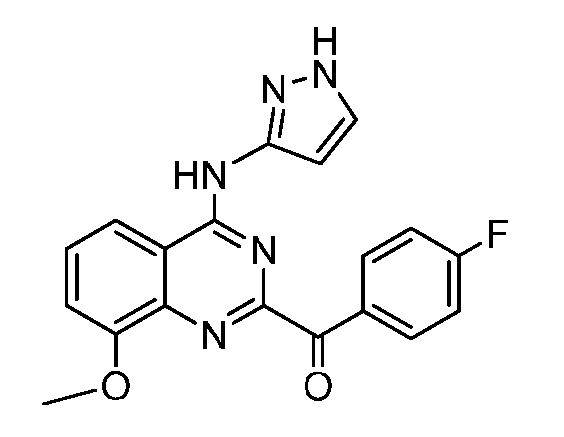
Стадия A: К 2-амино-3-метоксибензойной кислоте (8,11 г, 48,52 ммоль) в DMF (150 мл) при комнатной температуре добавляли DIEA (13,2 мл, 58,22 ммоль), 2 н. раствор аммиака в MeOH (33,96 мл, 67,92 ммоль), EDCI (11,16 г, 58,22 ммоль) и 1-гидроксибензотриазол (7,87 г, 58,22 ммоль). Раствор перемешивали при комнатной температуре в атмосфере аргона. Через 20 часов раствор разбавляли водой и экстрагировали десять раз при помощи EtOAc. EtOAc концентрировали для уменьшения объема и промывали насыщенным солевым раствором. EtOAc фракцию концентрировали и разбавляли диэтиловый эфиром. Полученное ржаво-коричневого твердое вещество собирали и сушили в вакууме с получением 2-амино-3-метоксибензамида (6,08 г, 76%). 1H ЯМР (300 МГц, DMSO-d6) δ 3,79 (с, 3H), 6,26 (ушир.с, 2H), 6,48 (м, 1H), 6,88 (д, J=7,9 Гц, 1H), 7,12 (ушир.с, 1H), 7,19 (дд, J=8,2, 1,0 Гц, 1H), 7,70 (ушир.с, 1H); LC-MS (ESI) m/z 167 (M+H)+.
Стадия B: К 2-амино-3-метоксибензамиду (1 г, 6,02 ммоль) в DCM (20 мл) добавляли DIEA (1,37 мл, 7,82 ммоль). Раствор охлаждали до 0°C с последующим добавлением этилхлорглиоксалата (0,808 мл, 7,22 ммоль) в DCM (5 мл) по каплям. По завершении этого добавления добавляли диметиламинопиридин (10 мг) с последующим удалением охлаждающей бани. После перемешивания в течение 20 часов при комнатной температуре в атмосфере Ar смесь промывали водой и очищали при помощи хроматографии на диоксиде кремния, элюируя при помощи EtOAc/DCM (20 до 60%) и MeOH/DCM (2 до 15%)с получением белого твердого вещества (770 мг, количеств.). 1H ЯМР (300 МГц, DMSO-d6) δ м.д. 1,30 (т, J=1,16 Гц, 3H) 3,79 (с, 3H) 4,29 (кв., J=7,03 Гц, 2H) 7,16 (дд, J=17,2, 1,2 Гц, 1H) 7,21 (дд, J=17,8, 1,2 Гц, 1H) 7,27-7,39 (м, 1H) 7,44 (ушир.с, 1H) 7,67 (ушир.с, 1H) 10,14 (ушир.с, 1H); LC-MS (ESI) m/z 250 (M-16)-.
Стадия C: К этил 2-(2-карбамоил-6-метоксифениламино)-2-оксоацетату (3,4 г, 12,77 ммоль) в DCE (50 мл) при комнатной температуре добавляли TEA (71 мл, 511 ммоль) с последующим быстрым добавлением триметилсилилхлорида (21 мл, 191 ммоль) в течение двадцати секунд. Гетерогенный раствор нагревали до температуры кипения с обратным холодильником в атмосфере Ar. Через 18 часов раствор охлаждали и выливали в лед/воду. Полученную смесь подкисляли до pH 3-4 и осажденный продукт собирали фильтрованием. Кислотный слой экстрагировали четыре раза при помощи EtOAc. Водный слой подщелачивали до pH 7 при помощи насыщенного раствора бикарбоната натрия и экстрагировали при помощи EtOAc. Органические экстракты объединяли, промывали насыщенным солевым раствором и концентрировали до объема 50 мл. Добавляли диэтиловый эфир (10 мл) и полученный осадок собирали. Объединение обоих осадков давало ржаво-коричневое твердое вещество (3,85 г, количеств.). 1H ЯМР (300 МГц, DMSO-d6) δ м.д. 1,36 (т, J=7,06 Гц, 3H) 3,94 (с, 3H) 4,39 (кв., J=6,97 Гц, 2H) 7,44 (д, J=8,10 Гц, 1H) 7,58 (т, J=8,01 Гц, 1H) 7,72 (д, J=7,91 Гц, 1H) 12,56 (ушир.с, 1H); LC-MS (ESI) m/z 249 (M+H)+.
Стадия D: К оксихлориду фосфора (2 мл) добавляли этил 8-метокси-4-оксо-3,4-дигидрохиназолин-2-карбоксилат (100 мг, 0,403 ммоль) с последующим добавлением диметилформамида (2 капли). Раствор нагревали при 80°C в течение 1,5 часов и затем концентрировали. Остаток охлаждали в -20°C охлаждающей бане и разбавляли холодным EtOAc. Охлажденный раствор промывали холодной водой, насыщенным водным раствором бикарбоната натрия и насыщенным солевым раствором. Удаление растворителя давало белое твердое вещество (98 мг, 91%). 1H ЯМР (300 МГц, DMSO-d6) δ м.д. 1,38 (т, J=7,06 Гц, 3H) 4,06 (с, 3H) 4,45 (кв., J=7,03 Гц, 2H) 7,51-7,76 (м, 1H) 7,76-8,12 (м, 2H).
Стадия E: К этил 4-хлор-8-метоксихиназолин-2-карбоксилату (550 мг, 2,07 ммоль) в диметилформамиде (6 мл) добавляли DIEA (0,468 мл, 2,69 ммоль), 3-аминопиразол (221 мг, 2,69 ммоль) и иодид калия (343 мг, 2,07 ммоль) при комнатной температуре. После перемешивания в течение 18 часов добавляли дополнительное количество 3-аминопиразола (100 мг) и перемешивание продолжали в течение 5 часов. Раствор выливали в воду и фильтровали и твердое вещество промывали диэтиловый эфиром с получением желтого твердого вещества (510 мг, 79% выход). 1H ЯМР (300 МГц, DMSO-d6) δ м.д. 137 (т, 3H) 3,97 (с, 3H) 4,38 (кв., J=I,16 Гц, 2H) 7,18 (ушир.с, 1H) 7,38 (д, J=7,72 Гц, 1H) 7,60 (м, 1H) 7,75 (ушир.с, 1H) 8,25 (д, J=8,29 Гц, 1H) 10,65 (с, 1H) 12,53 (ушир.с, 1H); LC-MS (ESI) m/z 314 (M+H)+.
Стадия F: К этил 4-(1Н-пиразол-3-иламино)-8-метоксихиназолин-2-карбоксилату (200 мг, 0,64 ммоль) в безводном ТГФ (8 мл), охлажденном до -40°C, добавляли по каплям в течение 2 минут в 4-фторфенилмагнийбромид в ТГФ (2,17 мл, 2,17 ммоль). Через 1,5 часа реакционную смесь гасили путем добавления насыщенного водного раствора хлорида аммония. Раствор концентрировали и добавляли H2O. Осадок промывали водой и диэтиловым эфиром с получением(4-(1Н-пиразол-3-иламино)-8-метоксихиназолин-2-ил)(4-фторфенил)метанона в виде желтого твердого вещества (74 мг, 87% чистота по данным LC/MS). 1H ЯМР (300 МГц, DMSO-d6) δ м.д. 3,94 (с, 3H) 6,75 (с, 1H) 7,35-7,41 (м, 3H) 7,58-7,65 (м, 2H) 8,07-8,12 (м, 2H) 8,2 (д, J=8,48 Гц, 1H) 10,64 (ушир.с, 1H) 12,51 (ушир.с, 1H); LC-MS (ESI) m/z 364 (M+H)+.
Пример 36
Получение (R,S)-2-((4-фторфенил)(гидрокси)метил)-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-7-ола
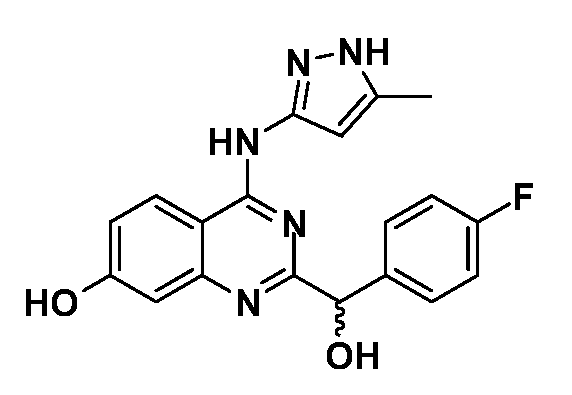
Стадия A: К 2-амино-4-метоксибензамиду (7,0 г, 42 ммоль) в 1,2-дихлорэтане (100 мл) добавляли трибромид бора (25 г, 100 ммоль) при комнатной температуре. После нагревания при 40°C в течение 20 часов добавляли 1 н. раствор трибромида бора в ТГФ (40 мл) и реакционную смесь нагревали до 50°C в течение 20 часов. Смесь охлаждали и гасили путем добавления водного раствора бикарбоната натрия. Полученный осадок собирали фильтрованием с получением 2-амино-4-гидроксибензамида в виде белого твердого вещества (2,0 г). Маточные растворы концентрировали, разбавляли при помощи MeOH, фильтровали и концентрировали. Остаток снова разбавляли при помощи MeOH, фильтровали и концентрировали и полученный остаток очищали хроматографией на силикагеле, элюируя при помощи 5-15% MeOH/DCM, с получением 2-амино-4-гидроксибензамида в виде белого твердого вещества (4,7 грамм). Твердые вещества объединяли с получением общего выхода 6,7 г (количественный). 1H ЯМР (300 МГц, DMSO-d6) δ м.д. 5,91 (дд, J=8,67, 2,26 Гц, 1H) 6,03 (д, J=2,45 Гц, 1H) 6,62 (ушир.с, 2H) 7,38 (д, J=8,67 Гц, 1H) 9,45 (с, 1H), LC-MS (ESI) m/z 153 (M+H)+.
Стадия B: К раствору 95% NaH (1,82 г, 72,30 ммоль) в DMF (100 мл) при 10°C добавляли 2-амино-4-гидроксибензамид (10,0 г, 66,72 ммоль) по порциям, поддерживая внутреннюю температуру примерно при 15°C. Охлаждающую баню удаляли и раствору давали нагреться до 40°C в течение 25 минут. Смесь охлаждали до 10°C и добавляли по каплям раствор бензилбромида (7,8 мл, 66,72 ммоль) в DMF (20 мл) и смеси давали нагреться до комнатной температуры. После перемешивания в течение 20 часов при комнатной температуре смесь охлаждали на ледяной бане, и гасили путем добавления водного раствора хлорида аммония. Раствор концентрировали и разбавляли водой. Осадок собирали фильтрованием и фильтрат экстрагировали при помощи EtOAc. Осадок, указанный выше, и этилацетатные экстракты объединяли и очищали при помощи хроматографии на силикагеле, элюируя при помощи 20-80% EtOAc/DCM, с получением 2-амино-4-(бензилокси)бензамида в виде твердого вещества (6,8 г, 43%). 1H ЯМР (300 МГц, DMSO-d6) δ м.д. 5,04 (с, 2H) 6,14 (дд, J=8,76, 2,54 Гц, 1H) 6,27 (д, J=2,64 Гц, 1H) 6,71 (ушир.с, 2H) 7,26-7,58 (м, 6H); LC-MS (ESI) m/z 243 (M+H)+.
Стадия C: К раствору 2-амино-4-(бензилокси)бензамида (4,0 г, 16,5 ммоль) в ТГФ (60 мл) добавляли раствор 5-(4-фторфенил)-1,3-диоксолан-2,4-диона (3,65 г, 18,6 ммоль) из Примера 16 в ТГФ (20 мл) по порциям при комнатной температуре. После нагревания до 63°C в течение 18 часов раствор охлаждали и концентрировали. После добавления H2O раствор экстрагировали два раза при помощи DCM. Объединенные органические фазы промывали насыщенным солевым раствором и сушили над сульфатом натрия. Хроматография на силикагеле с элюированием при помощи 10 до 80% EtOAc/DCM давала 4-(бензилокси)-2-(2-(4-фторфенил)-2-гидроксиацетамидо)бензамид в виде пенистого твердого вещества (4,1 г, 64%). 1H ЯМР (300 МГц, DMSO-d6) δ м.д. 5,06 (м, 1H), 5,12 (с, 2H) 6,68-6,74 (м, 1H) 7,14-7,20 (м, 2H) 7,32-7,51 (м, 6H) 7,77 (д, J=8,85 Гц, 1H) 8,05 (ушир.с, 1H) 8,30 (д, J=2,64 Гц, 1H) 12,73 (с, 1H), LC-MS (ESI) m/z 392 (M-2).
Стадия D: К 4-(бензилокси)-2-(2-(4-фторфенил)-2-гидроксиацетамидо)бензамиду (4,1 г, 10,4 ммоль) в абсолютном EtOH (50 мл) добавляли 20% водный раствор карбоната калия (5 мл). После нагревания и перемешивания при 80°C в течение 20 часов раствор охлаждали и концентрировали с получением твердого вещества. Твердое вещество промывали водой и сушили в условиях вакуума с получением 7-(бензилокси)-2-((4-фторфенил)(гидрокси)метил)хиназолин-4(3H)-она в виде белого твердого вещества (3,51 г, 90%). 1H ЯМР (300 МГц, DMSO-d6) δ м.д. 5,25 (с, 2H) 5,55 (с, 1H) 7,10-7,21 (м, 4H) 7,36-7,48 (м, 5H) 7,56-7,61 (м, 2H) 7,97 (д, J=8,67 Гц, 1H), LC-MS (ESI) m/z 377 (M+H)+.
Стадия E: К 4-(бензилокси)-2-((4-фторфенил)(гидрокси)метил)хиназолин-4(3H)-ону (3,5 г, 9,3 ммоль) в DMSO (15 мл) и CHCl3 (30 мл) при 0°C добавляли по порциям периодинан Dess-Martin (5,52 г, 13,02 ммоль). После перемешивания в течение 6 часов добавляли 1:1 смесь 10% водного раствора тиосульфата натрия пентагидрата и насыщенного водного раствора бикарбоната натрия. После встряхивания с DCM образовывался осадок, который собирали фильтрованием. Фильтрат экстрагировали три раза при помощи DCM и объединенные органические фракции промывали насыщенным солевым раствором и сушили над сульфатом магния. Концентрирование и объединение с первоначально образованным осадком давали 7-(бензилокси)-2-(4-фторбензоил)хиназолин-4(3H)-он в виде белого твердого вещества (3,0 г, 86%). LC-MS (ESI) m/z 375 (M+H)+.
Стадия F: К оксихлориду фосфора (10 мл), охлажденному до 5°C, добавляли по порциям 7-(бензилокси)-2-(4-фторбензоил)хиназолин-4(3H)-он (500 мг, 1,34 ммоль) с последующим добавлением DMF (4 капель). Смесь нагревали до 56°C в течение 10 минут и выдерживали при этой температуре в течение 2 минут, затем нагревающую баню удаляли. Смесь концентрировали и остаток разбавляли при помощи EtOAc, затем раствор промывали холодной водой, насыщенным раствором бикарбоната натрия (водн.), насыщенным солевым раствором и сушили над сульфатом натрия. Раствор концентрировали с получением (7-(бензилокси)-4-хлорхиназолин-2-ил)(4-фторфенил)метанона в виде не совсем белого твердого вещества (380 мг, 72%). 1H ЯМР (300 МГц, DMSO-d6) δ м.д. 5,40 (с, 2H) 7,37-7,45 (м, 5H) 7,52-7,55 (м, 2H) 7,66-7,71 (м, 2H) 8,09-8,14 (м, 2H) 8,31 (д, J=9,04 Гц, 1H), LC-MS (ESI) m/z 393 (M+H)+.
Стадия G: К (7-(бензилокси)-4-хлорхиназолин-2-ил)(4-фторфенил)метанону (380 мг, 0,97 ммоль) в DMF (10 мл) при комнатной температуре добавляли DIEA (500 мкл, 2,90 ммоль), 5-метил-1Н-пиразол-3-амин (280 мг, 2,90 ммоль) и KI (161 мг, 0,97 ммоль). После перемешивания при 40°C в течение 18 часов раствор охлаждали и разбавляли водой. После выстаивания при 0°C в течение 1 часа осадок собирали фильтрованием и сушили при пониженном давлении с получением (7-(бензилокси)-4-(5-метил-1Н- пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанона в виде желтого твердого вещества (365 мг, 83%). 1H ЯМР (300 МГц, DMSO-d6) δ м.д. 2,18 (с, 3H) 5,30 (с, 2H) 6,48 (с, 1H) 7,33-7,44 (м, 8H) 7,50-7,52 (м, 2H) 8,07 (м, 2H) 8,65 (м, 1H) 10,53 (с, 1H) 12,18 (с, 1H), LC-MS (ESI) m/z 454 (M+H)+.
Стадия H: К 10% Pd/C (200 мг) добавляли раствор (7-(бензилокси)-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанона (390 мг, 0,99 ммоль) в DMF (30 мл). После перемешивания в атмосфере H2 при 1 атм в течение 18 часов смесь фильтровали и фильтрат концентрировали. Остаток пропускали через короткую колонку с силикагелем, элюируя сначала при помощи 10-30% EtOAc/DCM, затем 1-5% AcOH/9-5% MeOH/90% DCM. Фракции, содержащие продукт, объединяли и промывали раствором бикарбоната натрия с последующим упариванием с получением (R,S)-2-((4-фторфенил)(гидрокси)метил)-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-7-ола в виде белого твердого вещества (130 мг, 36%). 1H ЯМР (300 МГц, DMSO-d6) δ м.д. 2,24 (с, 3H) 5,59 (ушир.с, 1H) 5,73 (м, 1H) 6,38 (м, 1H) 6,99 (ушир.с, 2H) 7,14 (м, 2H) 7,53 (м, 2H) 8,39 (ушир.с, 1H) 10,11 (ушир.с, 1H) 12,06 (ушир.с, 1H), LC-MS (ESI) m/z 366 (M+H)+.
Пример 37
Получение (4-фторфенил)(7-гидрокси-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанона
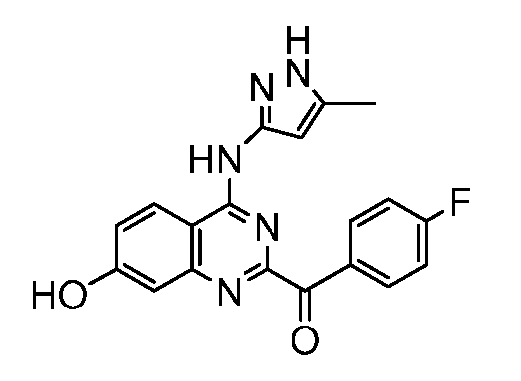
К 2-((4-фторфенил)(гидрокси)метил)-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-7-олу (50 мг, 0,14 ммоль) в 1:3 DMSO/DCM добавляли периодинан Dess-Martin (81 мг, 0,19 ммоль) одной порцией. После перемешивания в течение 1 часа при комнатной температуре раствор охлаждали до 0°C и гасили путем добавления 1:1 смеси 10% раствора тиосульфата натрия пентагидрата и насыщенного раствора бикарбоната натрия. Полученный темный осадок собирали и очищали при помощи хроматографии на диоксиде кремния, элюируя смесью 2-10% MeOH/DCM. Растирание в порошок окрашенного твердого вещества с MeOH давало (4-фторфенил)(7-гидрокси-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанон в виде белого твердого вещества (7 мг, 14%). 1H ЯМР (300 МГц, DMSO-d6) δ м.д. 2,27 (с, 3H), 6,47 (ушир.с, 1H), 7,06 (м, 2H), 7,13 (м, 1H), 7,38 (м, 2H), 8,06 (м, 2H), 8,57 (м, 2H), 10,41 (ушир.с, 1H), 10,60 (ушир.с, 1H), 12,15 (ушир.с, 1H), LC-MS (ESI) m/z 364 (M+H)+.
Пример 38
Получение (R,S)-(4-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)-7-(2-морфолиноэтокси)хиназолин-2-ил)метанола
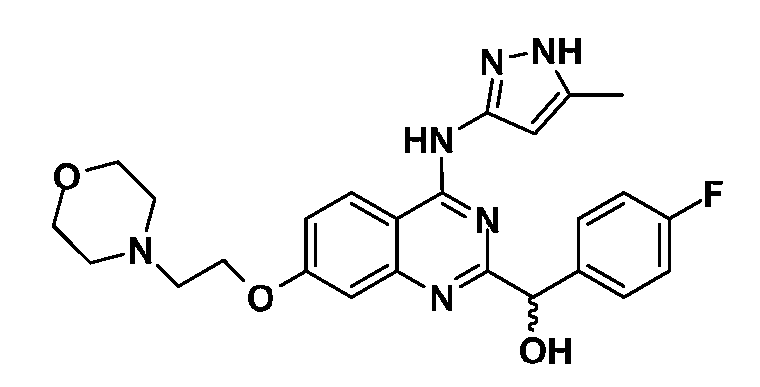
К (R,S)-2-((4-фторфенил)(гидрокси)метил)-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-7-олу (50 мг, 0,14 ммоль) в DMF (2 мл) добавляли 4-(2-хлорэтил)морфолин (51 мг, 0,27 ммоль) и карбонат цезия (134 мг, 0,41 ммоль) при комнатной температуре. После нагревания при 40°C в течение 18 часов раствор разбавляли при помощи EtOAc и промывали водой и насыщенным солевым раствором и сушили над сульфатом натрия. Хроматография на силикагеле с элюированием смесью 2-10% MeOH/DCM давала (R,S)-(4-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)-7-(2-морфолиноэтокси)хиназолин-2-ил)метанол в виде твердого вещества (26 мг, 40%). 1H ЯМР (300 МГц, DMSO-d6) δ м.д. 2,25 (ушир.с, 3H) 2,75 (m 3H) 3,59 (м, 6H) 4,26 (м, 3H) 5,63 (м, 1H) 5,75 (м, 1H) 6,43 (ушир.с, 1H) 7,10-7,20 (м, 4H) 7,53 (м, 2H) 8,47 (м, 1H) 10,23 (ушир.с, 1H) 12,09 (ушир.с, 1H), LC-MS (ESI) m/z 479 (M+H)+.
Пример 39
Получение (R,S)-2-(2-((4-фторфенил)(гидрокси)метил)-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-7-илокси)этанола
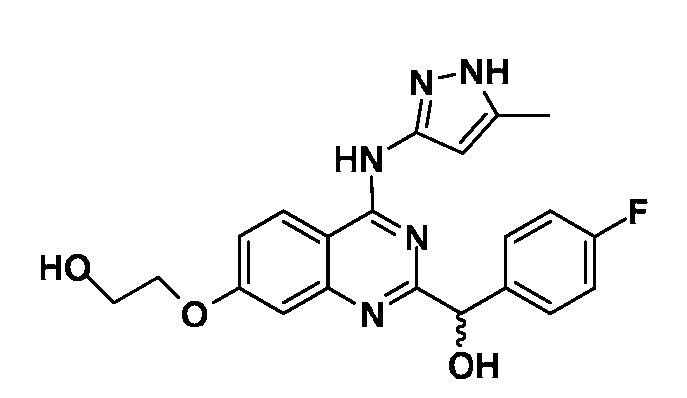
(R,S)-(7-(2-(трет-бутилдиметилсилилокси)этокси)-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанол получали, следуя процедуре, описанной в Примере 38 для синтеза (R,S)-(4-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)-7-(2-морфолиноэтокси)хиназолин-2-ил)метанола, замещая 4-(2-хлорэтил)морфолин в Примере 38 (2-бромэтокси)(трет- бутил)диметилсиланом, с получением 150 мг неочищенного содержащего примеси твердого вещества. К этому неочищенному твердому веществу (150 мг) в ТГФ (1 мл) добавляли тетрабутиламмонийфторид (1,0 мл) по каплям при комнатной температуре. Через 18 часов раствор концентрировали, разбавляли при помощи EtOAc и промывали водой. Хроматография остатка на силикагеле с элюированием смесью 2-8% 1%NH4OH.9%MeOH)/DCM давала (R,S)-2-(2-((4-фторфенил)(гидрокси)метил)-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-7-илокси)этанол в виде твердого вещества (31 мг, 18%). 1H ЯМР (300 МГц, DMSO-d6) δ м.д. 2,31 (с, 3H) 3,84 (кв., J=5,09 Гц, 2H) 4,22 (т, 7=4,71 Гц, 2H) 5,01 (т, J=5,46 Гц, 1H) 5,69 (м, 1H) 5,83 (ушир.с, 1H) 6,47 (ушир.с, 1H) 7,17-7,25 (м, 4H) 7,59 (м, 2H) 8,54 (д, J=9,04 Гц, 1H) 10,30 (ушир.с, 1H) 12,15 (ушир.с, 1H), LC-MS (ESI) m/z 410 (M+H)+.
Пример 40
Получение (R,S)-3-(2-((4-фторфенил)(гидрокси)метил)-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-7-илокси)пропан-1-ола
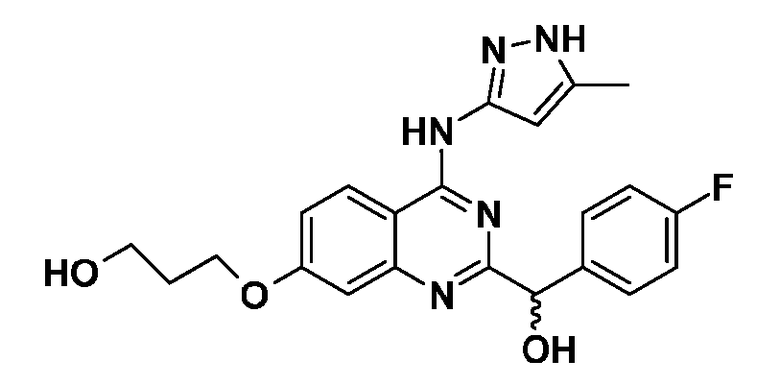
(R,S)-3-(2-((4-фторфенил)(гидрокси)метил)-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-7-илокси)пропан-1-ол получали, следуя процедуре, описанной в Примере 38 для синтеза (R,S)-(4-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)-7-(2-морфолиноэтокси)хиназолин-2-ил)метанола, замещая 4-(2-хлорэтил)морфолин в Примере 38 3-хлорпропан-1-олом (35 мг, 30%). 1H ЯМР (300 МГц, DMSO-d6) δ м.д. 1,96 (м, 2H) 2,24 (с, 3H) 3,60 (м, 2H) 4,26 (т, J=6,41 Гц, 2H) 5,97 (с, 1H) 6,15 (с, 1H) 7,24-7,41 (м, 4H) 7,60 (м, 2H) 7,70 (ушир.с, 1H) 8,73 (д, J=9,42 Гц, 4H) 11,87 (ушир.с, 1H) 12,56 (ушир.с, 1H) 14,21 (ушир.с, 1H); LC-MS (ESI) m/z 424 (M+H)+.
Пример 41
Получение (4-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)-7-(пиперидино-4-илокси)хиназолин-2-ил)метанола
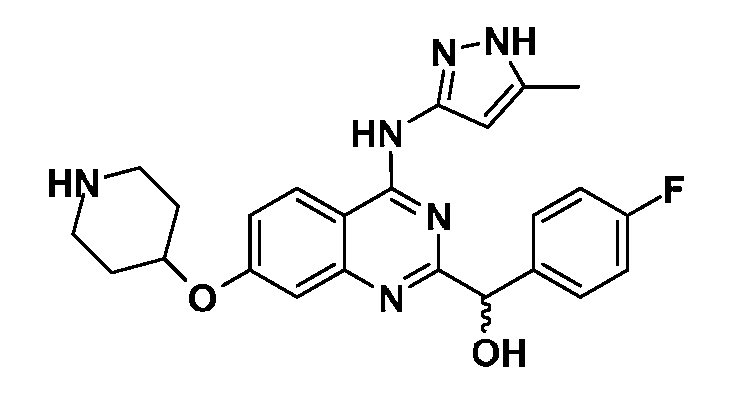
Промежуточное соединение (R,S)-трет-бутил 4-(2-((4-фторфенил)(гидрокси)метил)-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-7-илокси)пиперидино-1-карбоксилат получали (105 мг), следуя процедуре, описанной в Примере 38 для синтеза (R,S)-(4-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)-7-(2-морфолиноэтокси)хиназолин-2-ил)метанола, замещая 4-(2-хлорэтил)морфолин в Примере 38 трет-бутил 4-(метилсульфонилокси)пиперидино-1-карбоксилатом. К неочищенному трет-бутил 4-(2-((4-фторфенил)(гидрокси)метил)-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-7-илокси)пиперидино-1-карбоксилату (100 мг, 18,2 ммоль) в колбе при 0°C добавляли 4 н. раствор HCl/диоксан (5 мл). После перемешивания в течение 20 часов растворитель удаляли и остаток взбалтывали с DCM и водным раствором бикарбоната натрия. Растворители удаляли и оставшийся остаток экстрагировали при помощи MeOH/DCM. Экстракты концентрировали и остаток очищали при помощи обращенно-фазовой ВЭЖХ с получением (4-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)-7-(пиперидин-4-илокси)хиназолин-2-ил)метанола в виде белого твердого вещества (32 мг, 21%). 1H ЯМР (300 МГц, DMSO-d6) δ м.д. 1,54 (м, 2H) 1,89 (с, 13H) 1,89 (с, 3H) 1,98 (м, 2H) 2,68 (м, 2H) 2,99 (м, 2H) 4,66 (м, 4H) 5,62 (с, 1H) 5,76 (с, 1H) 6,36 (ушир., 1H) 7,08-7,18 (м, 4H) 7,52 (м, 2H) 8,45 (м, 1H), 10,29 (ушир.с, 1H), LC-MS (ESI) m/z 449 (M+H)+.
Пример 42
Получение (R,S)-(4-фторфенил)(7-(2-метоксиэтокси)-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанола
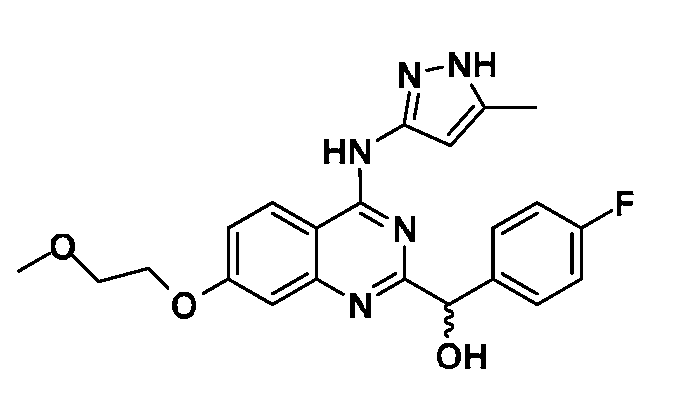
(R,S)-(4-фторфенил)(7-(2-метоксиэтокси)-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанол получали (35 мг, 25%), следуя процедуре, описанной в Примере 38 для синтеза (4-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)-7-(2-морфолиноэтокси)хиназолин-2-ил)метанола, замещая 4-(2-хлорэтил)морфолин в Примере 38 1-бром-2-метоксиэтаном. 1H ЯМР (300 МГц, DMSO-d6) δ м.д. 2,24 (с, 3H), 3,71 (м, 2H), 4,26 (м, 2H), 5,63 (ушир.с, 1H), 5,77 (ушир.с, 1H), 6,40 (ушир.с, 1H), 7,14 (м, 4H), 7,53 (м, 2H), 8,47 (м, 1H), 10,25 (ушир.с, 1H), 12,09 (ушир.с, 1H), LC-MS (ESI) m/z 424 (M+H)+.
Пример 43
Получение (R,S)-трет-бутил 2-(2-((4-фторфенил)(гидрокси)метил)-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-7-илокси)ацетата
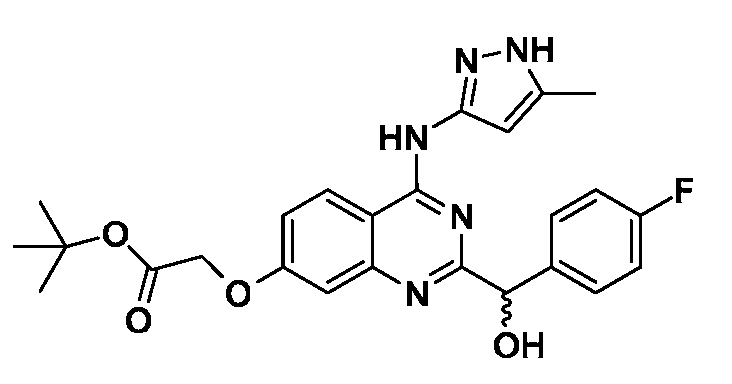
(R,S)-Трет-бутил 2-(2-((4-фторфенил)(гидрокси)метил)-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-7-илокси)ацетат получали (70 мг, 30%), следуя процедуре, описанной в Примере 38 для синтеза (4-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)-7-(2-морфолиноэтокси)хиназолин-2-ил)метанола, замещая 4-(2-хлорэтил)морфолин в Примере 38 трет-бутил 2-бромацетатом. 1H ЯМР (300 МГц, DMSO-d6) δ м.д. 1,43 (с, 9H), 2,25 (с, 3H), 4,86 (с, 2H), 5,62 (м, 1H), 5,77 (м, 1H), 7,09-7,16 (м, 4H), 7,52 (м, 2H), 8,50 (м, 1H), 10,26 (ушир.с, 1H), 12,09 (ушир.с, 1H), LC-MS (ESI) m/z 424 (M+H)+.
Пример 44
Получение (R,S)-2-(2-((4-фторфенил)(гидрокси)метил)-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-7-илокси)уксусной кислоты
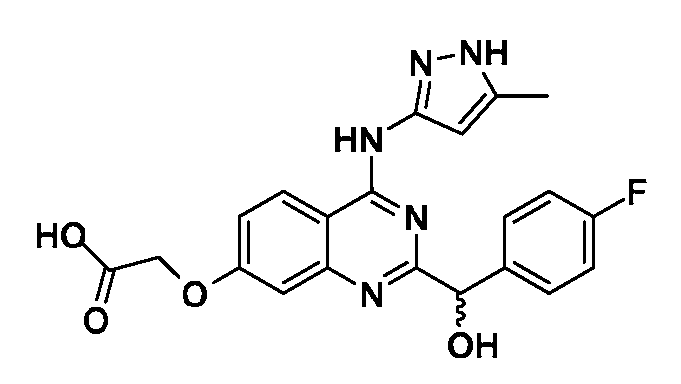
К (R,S)-трет-бутил 2-(2-((4-фторфенил)(гидрокси)метил)-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-7-илокси)ацетату (26 мг, 0,05 ммоль) из Примера 43 в DCM (1 мл) при 0°C добавляли TFA (1 мл). После перемешивания в течение 18 часов при 0°C растворитель удаляли и остаток растирали в порошок с DCM с получением (R,S)-2-(2-((4-фторфенил)(гидрокси)метил)-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-7-илокси)уксусной кислоты в виде твердого вещества (20 мг, 87%). 1H ЯМР (300 МГц, DMSO-d6) δ м.д. 2,27 (с, 3H), 5,93 (с, 1H), 6,15 (с, 1H), 7,22-7,43 (м, 4H), 7,59 (м, 3H), 8,71 (м, 1H); LC-MS (ESI) m/z 424 (M+H)+.
Пример 45
Получение (R,S)-метил (4-фторфенил)(4-(5-метил-4H-пиразол-3-иламино)хиназолин-2-ил)метилкарбамата
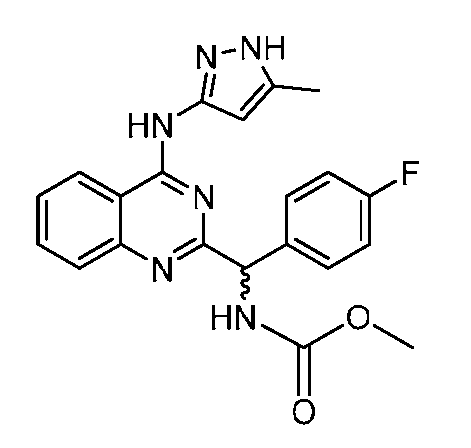
Стадия A: К (4-фторфенил)(4-(5-метил-4H-пиразол-3-иламино)хиназолин-2-ил)метанону из Примера 3 (0,700 г, 2,15 ммоль) в EtOH (10 мл) добавляли гидрохлорид метоксиламина (0,336 г, 4,02 ммоль) и смесь нагревали до 60°C в течение 30 минут. Добавляли воду и желтый осадок собирали фильтрованием и промывали при помощи MeOH с получением (4-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанон O-метилоксима (0,88 г). LC-MS (ESI) m/z 377 (M+H)+.
Стадия B: К раствору (4-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанон O-метилоксима (0,88 г, 2,33 ммоль) в уксусной кислоте (25 мл) добавляли цинковую пыль (3,0 г, 46 ммоль) и смесь перемешивали при комнатной температуре в течение ночи затем фильтровали через Целит. Фильтрат концентрировали и остаток очищали при помощи обращенно-фазовой препаративной ВЭЖХ, элюируя смесью 30-50% CH3CN/H2O, содержащей 0,05% HOAc, с получением (R,S)-2-(амино(4-фторфенил)метил)-N-(5-метил-1Н-пиразол-3-ил)хиназолин-4-амина в виде светло-желтого твердого вещества (105 мг, 40%). LC-MS (ESI) m/z 349 (M+H)+.
Стадия C: К раствору (R,S)-2-(амино(4-фторфенил)метил)-N-(5-метил-1Н-пиразол-3-ил)хиназолин-4-амина (0,105 г, 0,3 ммоль) в безводном ТГФ (3 мл) добавляли по каплям метилхлороформиат (0,02 мл, 0,3 ммоль). Добавляли DIEA (0,06 мл, 0,36 ммоль) и смесь перемешивали при 0°C в течение 10 минут. Смеси давали нагреться до комнатной температуры и перемешивали в течение 5 минут. Смесь распределяли между водой и EtOAc и органический слой сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали при помощи обращенно-фазовой препаративной ВЭЖХ, элюируя смесью CH3CN/H2O, содержащей 0,05% HOAc, с получением (R,S)-метил (4-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метилкарбамата в виде белого порошка (35 мг, 29% выход). 1H ЯМР (300 МГц, DMSO-d6) δ 2,2 (с, 3H) 3,57(с, 3H) 5,8 (с, 1H) 6,4 (с, 1H) 7,1-7,2 (м, 2H) 7,4-7,5 (м, 3H) 7,7-7,9 (м, 3H) 8,5 (с, 1H) 10,42 (с, 1H) 12,25 (с, 1H); LC-MS (ESI) m/z 407 (M+H)+.
Пример 46
Получение (R,S)-(4-фторфенил)(8-метил-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанола
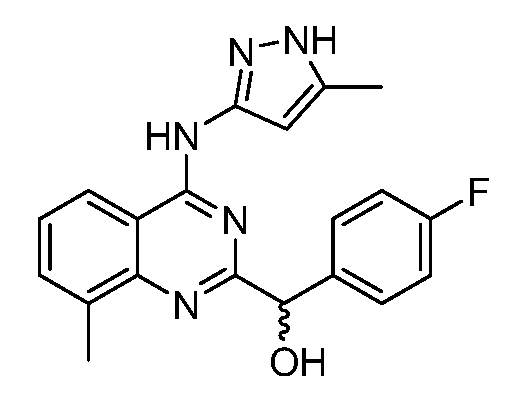
Стадия A: К раствору 2-амино-3-метилбензойной кислоты (4,0 г, 26,5 ммоль) в дегазированном DMF (40 мл) добавляли HOBt (4,28 г, 31,7 ммоль), DIEA (5,52 мл, 31,7 ммоль) и 2 н. раствор NH3/MeOH (19 мл, 37,1 ммоль). Раствор перемешивали при комнатной температуре в течение 16 часов, затем смесь концентрировали при пониженном давлении и остаток очищали хроматографией на силикагеле, элюируя смесью 10-60% EtOAc/гексан, с получением 2-амино-3-метилбензамида в виде твердого вещества (2,52 г, 63%). 1H ЯМР (300 МГц, DMSO-d6) δ 2,07 (с, 3H), 6,40-6,47 (м, 3H), 7,06 (м, 2H), 7,42 (д, 1H), 7,72 (ушир.д, 1H).
Стадия B: К раствору 2-амино-3-метилбензамида (1,50 г, 10,0 ммоль) и DIEA (2,61 мл, 15 ммоль) в ТГФ (50 мл) при 0°C добавляли этилхлороксоацетат (1,23 мл, 11,0 ммоль). Раствору давали нагреться до комнатной температуры и перемешивали в течение 16 часов, затем концентрировали при пониженном давлении. Остаток очищали хроматографией на силикагеле, элюируя смесью 20-100% EtOAc/гексан, с получением этил 2-(2-карбамоил-6-метилфениламино)-2-оксоацетата в виде твердого вещества (490 мг, 20%). 1H ЯМР (300 МГц, DMSO-d6) δ 1,31 (т, J=7,1 Гц, 3H), 2,17 (с, 3H), 4,30 (кв., J=7,1 Гц, 2H), 7,28 (т, 1H), 7,33-7,57 (м, 3H), 7,82 (с, 1H), 10,67 (с, 1H).
Стадия C: К раствору этил 2-(2-карбамоил-6-метилфениламино)-2-оксоацетата (490 мг, 1,96 ммоль) и TEA (10,4 мл, 75 ммоль) в DCE (20 мл) добавляли TMS-Cl (3,6 мл, 29 ммоль) и раствор перемешивали при 80°C в течение 16 часов. Смесь концентрировали при пониженном давлении и остаток распределяли между DCM (100 мл) и насыщенным водным раствором NaHCO3. Отделенную водную фазу экстрагировали при помощи DCM (2×100 мл) и объединенные органические слои сушили над MgSO4, фильтровали и концентрировали. Остаток очищали хроматографией на силикагеле, элюируя смесью 10-40% EtOAc/гексан, с получением этил 4-гидрокси-8-метилхиназолин-2-карбоксилата в виде твердого вещества (330 мг, 72%). 1H ЯМР (300 МГц, DMSO-d6) δ 1,36 (т, J=7,2 Гц, 3H), 2,58 (с, 3H), 4,39 (кв., J=7,2 Гц, 2H), 7,46-7,64 (м, 1H), 7,64 (м, 1H), 8,02 (м, 1H), 12,62 (ушир.с, 1H).
Стадия D: Смесь этил 4-гидрокси-8-метилхиназолин-2-карбоксилата (330 мг, 1,39 ммоль), POCl3 (20 мл) и DMF (3 капли) перемешивали при 80°C в течение 48 часов. Смесь концентрировали при пониженном давлении и твердый остаток распределяли между DCM (100 мл) и холодной H2O (100 мл). Отделенную водную фазу экстрагировали при помощи DCM (2×100 мл) и объединенные органические слои сушили над MgSO4, фильтровали и концентрировали. Остаток очищали хроматографией на силикагеле, элюируя смесью EtOAc/гексан, с получением этил 4-хлор-8-метилхиназолин-2-карбоксилата в виде твердого вещества (320 мг, 90%). 1H ЯМР (300 МГц, DMSO-d6) δ 1,39 (т, J=7,1 Гц, 3H), 2,76 (с, 3H), 4,45 (кв., J=7,1 Гц, 2H), 7,90 (м, 1H), 8,1 (м, 1H), 8,2 (м, 1H).
Стадия E: К раствору этил 4-хлор-8-метилхиназолин-2-карбоксилата (320 мг, 0,43 ммоль) в ТГФ (10 мл) при -40°C добавляли раствор 1М 4-фторфенилмагнийбромида (1,55 мл, 1,55 ммоль) и смесь перемешивали при -40°C в течение 1 часа. Реакцию гасили насыщенным водным раствором NH4Cl и смесь концентрировали при пониженном давлении и остаток распределяли между DCM (100 мл) и H2O (100 мл). Отделенную водную фазу экстрагировали при помощи DCM (2×100 мл) и объединенные органические слои сушили над MgSO4, фильтровали и концентрировали с получением твердого вещества (370 мг). Раствор полученного твердого вещества (370 мг, 1,22 ммоль) и KI (224 мг, 1,35 ммоль) в DMF (10 мл) перемешивали при комнатной температуре в течение 30 минут и затем добавляли 5-метил-1Н-пиразол-3-амин (257 мг, 2,63 ммоль) и DIEA (275 мкл, 1,60 ммоль) и смесь перемешивали при 50°C в течение 24 часов. Добавляли воду и осажденное твердое вещество собирали фильтрованием и промывали несколько раз водой. Неочищенный продукт очищали при помощи ВЭЖХ, элюируя смесью 10-80% ACN/H2O, содержащей 0,05% HOAc, с получением (4-фторфенил)(8-метил-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанона в виде твердого вещества (64 мг, 14%). 1H ЯМР (300 МГц, DMSO-d6) δ 2,20 (с, 3H), 2,59 (с, 3H), 6,56 (ушир.с, 1H), 7,40 (т, J=8,7 Гц, 2H), 7,56 (т, J=7,6 Гц, 1H), 7,77 (д, J=6,8 Гц, 1H), 8,16 (т, J=6,3 Гц, 2H), 8,56 (д, J=7,7 Гц, 1H), 10,59 (ушир.с, 1H), 12,20 (ушир.с, 1H).
Стадия F: Смесь (4-фторфенил)(8-метил-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанона (64 мг, 0,177 ммоль) и NaBH4 (15 мг, 0,355 ммоль) в MeOH (3 мл) перемешивали при комнатной температуре в течение 24 часов. Добавляли 1М HCl по каплям до получения гомогенной смеси и смесь перемешивали в течение 5 минут. Затем добавляли насыщенный водный раствор NaHCO3 до тех пор, пока не происходило образование твердого осадка. Твердое вещество собирали фильтрованием с промывкой при помощи H2O с получением (R,S)-(4-фторфенил)(8-метил-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанола в виде твердого вещества (42 мг, 66%). 1H ЯМР (300 МГц, DMSO-d6) δ 2,25 (с, 3H), 2,65 (с, 3H), 5,68 (с, 1H), 5,78 (с, 1H), 6,41 (с, 1H), 7,15 (м, 2H), 7,41 (м, 1H), 7,56 (м, 2H), 7,68 (м, 1H), 8,42 (м, 1H), 10,31 (ушир.с, 1H), 12,11 (ушир.с, 1H); LC-MS (ESI) m/z 364 (M+H)+.
Пример 47
Получение (R,S)-(7-фтор-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанола
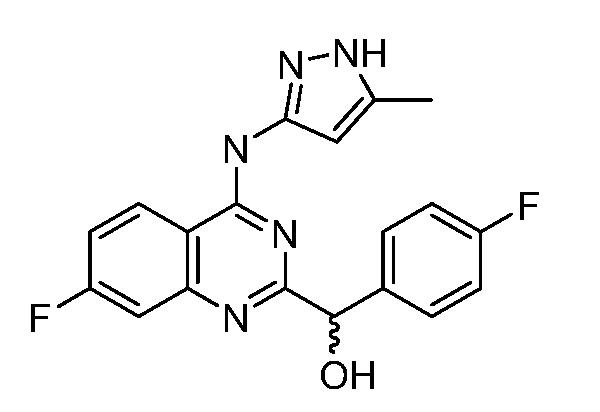
Стадия A: К раствору 2-амино-4-фторбензойной кислоты (4,0 г, 25,8 ммоль) в дегазированном DMF (50 мл) добавляли HOBt (4,19 г, 31 ммоль), DIEA (5,4 мл, 31 ммоль), EDCI (5,94 г, 31 ммоль) и 2 н. раствор NH3/MeOH (18 мл, 36,1 ммоль). Раствор перемешивали при комнатной температуре в течение 48 часов и затем концентрировали при пониженном давлении. Остаток очищали при помощи хроматографии на силикагеле с получением 2-амино-4-фторбензамида в виде твердого вещества (2,89 г, 66%). 1H ЯМР (300 МГц, DMSO-d6) δ 6,28 (м, 1H), 6,44 (м, 1H), 6,90 (ушир.с, 2H), 7,08 (м, 1H), 7,58 (м, 1H), 7,71 (ушир.д, 1H).
Стадия B: Раствор 2-амино-4-фторбензамида (750 мг, 4,40 ммоль), диэтилоксалата (20 мл) и AcOH (8 мл) перемешивали при 140°C в течение 16 часов. Твердое вещество собирали фильтрованием и сушили при пониженном давлении с получением этил 7-фтор-4-гидроксихиназолин-2-карбоксилата в виде твердого вещества (235 мг, 22%). 1H ЯМР (300 МГц, DMSO-d6) δ 1,36 (т, J=7,08 Гц, 2H), 4,39 (кв., J=7,08 Гц, 2H), 7,52 (м, 1H), 7,67 (м, 1H), 8,25 (м, 1H), 11,97 (ушир.с, 1H), 12,77 (ушир.с, 1H).
Стадия C: Смесь этил 7-фтор-4-гидроксихиназолин-2-карбоксилата (235 мг, 1,38 ммоль), POCl3 (15 мл) и DMF (3 капли) перемешивали при 80°C в течение 24 часов. Смесь концентрировали при пониженном давлении и остаток распределяли между холодной H2O (100 мл) и DCM (50 мл). Отделенную водную фазу экстрагировали при помощи DCM (2×50 мл) и объединенные органические слои сушили над MgSO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали хроматографией на силикагеле, элюируя смесью 20-80% EtOAc/гексан, с получением, этил 4-хлор-7-фторхиназолин-2-карбоксилата в виде твердого вещества (128 мг, 37%). 1H ЯМР (300 МГц, DMSO-d6) δ 1,38 (т, 7=7,10 Гц, 3H), 4,45 (кв., J=7,10 Гц, 2H), 7,92 (м, 1H), 8,15 (м, 1H), 8,37-8,65 (м, 1H).
Стадия D: К раствору этил 4-хлор-7-фторхиназолин-2-карбоксилата (122 мг, 0,479 ммоль) в 5 мл ТГФ при -40°C добавляли раствор 1М 4-фторфенилмагнийбромид/ТГФ (0,58 мл, 0,58 ммоль) и смесь перемешивали при -40°C в течение 2 часов. Добавляли насыщенный водный раствор NH4Cl и смесь концентрировали при пониженном давлении. Остаток распределяли между H2O (100 мл) и DCM (50 мл). Отделенную водную фазу экстрагировали при помощи DCM (2×50 мл) и объединенные органические слои сушили над MgSO4, фильтровали и концентрировали при пониженном давлении с получением твердого вещества (130 мг), содержащего (4-хлор-7-фторхиназолин-2-ил)(4-фторфенил)метанон. Раствор этого твердого вещества (130 мг, 0,43 ммоль) и KI (78 мг, 0,47 ммоль) в DMF (6 мл) перемешивали при комнатной температуре в течение 30 минут и затем добавляли 5-метил-1Н-пиразол-3-амин (88 мг, 0,88 ммоль) и DIEA (96 мкл, 0,56 ммоль). Смесь перемешивали при комнатной температуре в течение 24 часов и затем добавляли воду. Осажденное твердое вещество собирали фильтрованием с промывкой водой. Твердое вещество очищали при помощи препаративной обращенно-фазовой ВЭЖХ, элюируя смесью 10-80% ACN/H2O, содержащей 0,05% HOAC, с получением (7-фтор-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанона в виде твердого вещества (96 мг, 45%). 1H ЯМР (300 МГц, DMSO-d6) δ м.д. 2,18 (с, 3H), 6,48 (с, 1H), 7,39 (м, 2H), 7,63 (м, 2H), 8,13 (м, 2H), 8,84 (м, 1H), 10,80 (с, 1H), 12,08 (ушир.с, 1H).
Стадия E: раствор (7-фтор-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанона (96 мг, 0,26 ммоль) и NaBH4 (21 мг, 0,53 ммоль) в MeOH (3 мл) перемешивали при комнатной температуре в течение 24 часов и затем добавляли 1М HCl по каплям до получения гомогенной смеси. Смесь перемешивали в течение 5 минут и затем добавляли насыщенный водный раствор NaHCO3 вплоть до образования осадка. Твердое вещество собирали фильтрованием и промывали при помощи H2O с получением (R,S)-(7-фтор-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанола в виде твердого вещества (62 мг, 64%). 1H ЯМР (300 МГц, DMSO-d6) (представленная одной или несколькими примесями) δ 2,25 (с), 5,66 (1H), 5,84 (1H), 6,44 (ушир.с, 1H), 7,14 (м), 7,37-7,50 (м), 7,47-7,68 (м), 8,56-8,91 (м, 1H), 10,50 (ушир.с, 1H), 12,15 (ушир.с, 1H); LC-MS (ESI) m/z 368 (M+H)+.
Пример 48
Получение (4-(1Н-пиразол-3-иламино)хиназолин-2-ил)бис(4-фторфенил)метанола
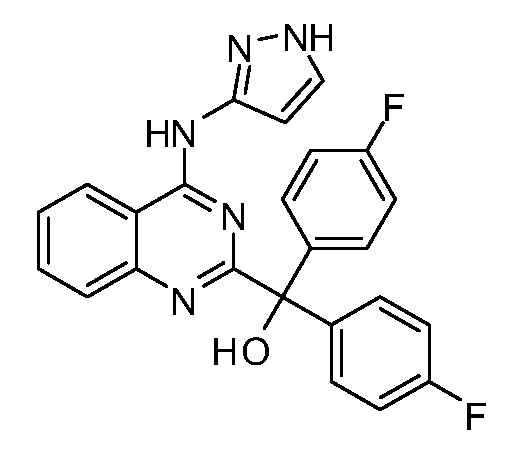
Стадия A: Перемешиваемую смесь этил 4-хлорхиназолин-2-карбоксилата (709 мг, 3 ммоль), 3-аминопиразола (274 мг, 3,3 ммоль), иодида калия (498 мг, 3 ммоль) и DIEA (574 мкл, 3,3 ммоль) в N,N-диметилформамиде (5 мл) нагревали при 50°C в течение 2 часов и затем перемешивали при комнатной температуре в течение ночи. К смеси добавляли воду и осажденное твердое вещество фильтровали, промывали водой и сушили в условиях высокого вакуума при 50°C в течение 3 часов с получением этил 4-(1Н-пиразол-3-иламино)хиназолин-2-карбоксилата (595 мг, 70%). LC-MS (ESI) m/z 284 (M+H)+.
Стадия B: К перемешиваемому раствору этил 4-(1Н-пиразол-3-иламино)хиназолин-2-карбоксилата (595 мг, 2,1 ммоль) в ТГФ (15 мл) при -40°C добавляли по каплям 2 M раствор 4-фторфенилмагнийбромида в ТГФ (4,2 мл, 8,4 ммоль). Смесь перемешивали при -40°C в течение 2 часов и затем хранили при -30°C в течение 18 часов. Реакцию гасили добавлением 0,5 н. раствор HCl при 0°C и смесь экстрагировали при помощи EtOAc (2×20 мл). Твердый осадок их объединенных органических слоев удаляли фильтрованием и полученный фильтрат промывали насыщенным солевым раствором. Органическую фазу сушили над MgSO4, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали хроматографией на силикагеле, элюируя смесью 0-5% MeOH/DCM, с получением (4-(1Н-пиразол-3-иламино)хиназолин-2-ил)бис(4-фторфенил)метанола в виде твердого вещества (75 мг, 8%). 1H ЯМР (300 МГц, DMSO-d6) δ 12,50 (с, 1H), 10,66 (с, 1H), 8,67 (д, J=8,1, 1H), 7,81-7,87 (м, 2H), 7,60-7,62 (м, 2H), 7,42-7,47 (м, 4H), 7,07-7,13 (м, 4H), 6,66 (с, 1H), 6,26 (с, 1H); LC-MS (ESI) m/z 430 (M+H)+.
Пример 49
Получение (2-(дифтор(4-фторфенил)метил)-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-7-ил)метанола
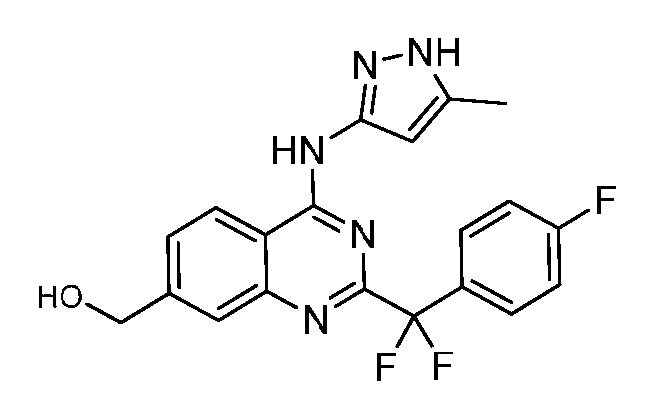
Стадия A: Смесь 4-(метоксикарбонил)-3-нитробензойной кислоты (200 мг) и концентрированного NH4OH (30 мл) в герметично закрытой пробирке нагревали при 105°C в течение ночи. После охлаждения до комнатной температуры смесь концентрировали при пониженном давлении и затем добавляли 2 н. раствор HCl (5 мл). Смесь экстрагировали при помощи EtOAc (3×50 мл) и объединенные органические экстракты промывали насыщенным солевым раствором (50 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. К остатку в MeOH (20 мл) добавляли по каплям тионилхлорид (0,2 мл) и смесь нагревали при температуре кипения с обратным холодильником в течение 6 часов. Смесь концентрировали при пониженном давлении и остаток распределяли между насыщенным водным раствором NaHCO3 (50 мл) и EtOAc (50 мл), отделенную водную фазу экстрагировали при помощи EtOAc (2×50 мл). Объединенные органические слои промывали насыщенным солевым раствором (50 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. К остатку в EtOH (30 мл) добавляли 10% Pd/C (10 мг) и смесь перемешивали при комнатной температуре в атмосфере H2 (1 атм) в течение 4 часов. Смесь фильтровали через Целит с промывкой при помощи MeOH. Фильтрат концентрировали при пониженном давлении и остаток очищали хроматографией на силикагеле, элюируя смесью 5% MeOH/DCM, с получением метил 3-амино-4-карбамоилбензоата в виде белого твердого вещества (142 мг, 82,5%). 1H ЯМР (300 МГц, DMSO-d6) δ 3,82 (с, 3H), 6,75 (с, 2H), 7,01 (д, 1H), 7,28 (с, 1H), 7,34 (с, 1H), 7,62 (д, 1H) 7,89 (с, 1H); LC-MS (ESI) m/z 211 (M+H)+.
Стадия B: К раствору метил 4-карбамоил-3-(2,2-дифтор-2-(4-фторфенил)ацетамидо)бензоата (1,3 г, 3,5 ммоль) в DCE (20 мл) добавляли триэтиламин (20 мл, 142 ммоль) и триметилсилилхлорид (6,7 мл, 53,2 ммоль) и смесь нагревали при 85°C в течение ночи. Смеси давали охладиться до комнатной температуры и затем концентрировали при пониженном давлении. Остаток распределяли между EtOAc (150 мл) и H2O (100 мл) и отделенную водную фазу экстрагировали при помощи EtOAc (3×150 мл). Объединенные органические слои промывали насыщенным солевым раствором (100 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток обрабатывали при помощи MeOH с использованием ультразвуковой обработки и твердое вещество собирали фильтрованием с получением метил 2-(дифтор(4-фторфенил)метил)-4-гидроксихиназолин-7-карбоксилата (1,1 г, 89%). 1H ЯМР (300 МГц, DMSO-d6) δ 3,91 (с, 3H), 7,39 (т, 2H), 7,78 (т, 2H), 8,06 (д, 1H), 8,16 (с, 1H), 8,27 (д, 1H), 13,34 (с, 1H); LC-MS (ESI) m/z 349 (M+H)+.
Стадия C: Смесь метил 2-(дифтор(4-фторфенил)метил)-4-гидроксихиназолин-7-карбоксилата (1,1 г, 3,2 ммоль) и оксихлорида фосфора (15 мл) нагревали при температуре кипения с обратным холодильником в течение ночи. Смесь концентрировали при пониженном давлении и затем добавляли толуол (20 мл) и упаривали при пониженном давлении (2×). Остаток в DCM фильтровали через слой силикагеля, элюируя при помощи DCM. Фильтрат концентрировали при пониженном давлении с получением метил 4-хлор-2-(дифтор(4-фторфенил)метил)хиназолин-7-карбоксилата (1 г, 86%). 1H ЯМР (300 МГц, DMSO-d6) δ 3,98 (с, 3H), 7,37 (т, 2H), 7,75 (т, 2H), 8,39 (д, 1H), 8,49 (с, 1H), 8,64 (д, 1H).
Стадия D: Смесь метил 4-хлор-2-(дифтор(4-фторфенил)метил)хиназолин-7-карбоксилата (1 г, 2,7 ммоль), 5-метил-1Н-пиразол-3-амина (0,32 г, 3,27 ммоль), DIEA (0,62 мл, 3,5 ммоль) и KI (0,5 г, 3 ммоль) в DMF (20 мл) перемешивали при комнатной температуре в течение 20 часов. Смесь разбавляли при помощи H2O и перемешивали в течение 1 часа и затем осажденное твердое вещество собирали фильтрованием, промывали при помощи H2O и сушили с получением метил 2-(дифтор(4-фторфенил)метил)-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-7-карбоксилата (1,17 г, 100%). 1H ЯМР (300 МГц, DMSO-d6) δ 2,24 (с, 3H), 3,97 (с, 3H), 6,31 (с, 1H), 7,15-7,50 (м, 2H), 7,62-7,90 (м, 2H), 7,99-8,13 (м, 1H), 8,20-8,55 (м, 1H), 8,69-9,04 (м, 1H), 10,96 (с, 1H), 12,28 (с, 1H); LC-MS (ESI) m/z 428 (M+H)+.
Стадия E: К суспензии LAH (0,26 г, 6,84 ммоль) в ТГФ (50 мл) при 0°C медленно добавляли суспензию метил 2-(дифтор(4-фторфенил)метил)-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-7-карбоксилата (1,17 г, 2,74 ммоль) в ТГФ (30 мл). Смесь перемешивали при 0°C в течение 0,5 часов и затем при комнатной температуре в течение 4 часов. Смесь охлаждали до 0°C и добавляли по каплям воду (0,26 мл) и смесь перемешивали в течение 30 минут. Затем добавляли 15% раствор NaOH (0,39 мл) и смесь перемешивали в течение 1 часа. Затем добавляли воду (1,3 мл) и смесь перемешивали при комнатной температуре в течение ночи. Смесь фильтровали через Целит с промывкой при помощи 20% MeOH/DCM (500 мл) и фильтрат концентрировали при пониженном давлении. Остаток распределяли между водой (200 мл) и EtOAc (150 мл) и отделенную водную фазу экстрагировали при помощи EtOAc (2×150 мл). Объединенные органические слои промывали насыщенным солевым раствором (200 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали при помощи препаративной обращенно-фазовой ВЭЖХ с получением (2-(дифтор(4-фторфенил)метил)-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-7-ил)метанола в виде белого твердого вещества (901 мг, 82%). 1H ЯМР (300 МГц, DMSO-d6) δ 2,24 (с, 3H), 4,70 (с, 2H), 5,49 (с, 1H), 6,31 (с, 1H), 7,35 (т, 2H), 7,56 (д, 2H), 7,70 (т, 2H), 7,77 (с, 1H), 8,63 (д, 1H), 10,64 (с, 1H), 12,18 (с, 1H); LC-MS (ESI) m/z 400 (M+H)+.
Пример 50
Получение 2-(дифтор(4-фторфенил)метил)-N-(5-метил-1Н-пиразол-3-ил)-7-(метилсульфонилметил)хиназолин-4-амина
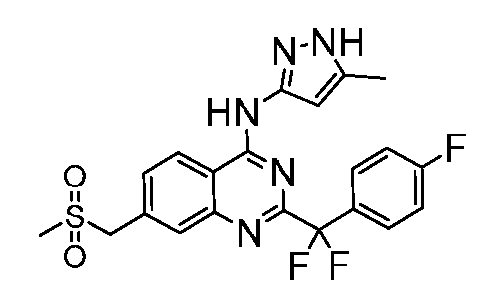
К суспензии (2-(дифтор(4-фторфенил)метил)-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-7-ил)метанола из Примера 49 (150 мг, 0,38 ммоль) в DCM (20 мл) добавляли PBr3 (203 мг, 0,75 ммоль) при нагревании смеси при 60°C. Полученную смесь перемешивали при 60°C в течение 30 минут, затем охлаждали до комнатной температуры и концентрировали при пониженном давлении. Затем добавляли тиометоксид натрия (133 мг, 1,90 ммоль) и DMF (10 мл) и смесь перемешивали при комнатной температуре в течение 2 дней. Добавляли насыщенный водный раствор NaHCO3 (50 мл) и смесь экстрагировали при помощи EtOAc (3×50 мл). Объединенные органические слои промывали насыщенным солевым раствором (3×80 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. К остатку в DCM (50 мл) добавляли 4-хлорпербензойную кислоту (655 мг, 3,80 ммоль) и смесь перемешивали при комнатной температуре в течение 4 часов. Добавляли насыщенный водный раствор NaHCO3 (50 мл) и смесь экстрагировали при помощи DCM (3×50 мл). Объединенные органические слои промывали насыщенным солевым раствором (60 мл), сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали при помощи препаративной обращенно-фазовой ВЭЖХ с получением 2-(дифтор(4-фторфенил)метил)-N-(5-метил-1Н-пиразол-3-ил)-7-(метилсульфонилметил)хиназолин-4-амина в виде белого твердого вещества (11,3 мг, 6,5%). 1H ЯМР (300 МГц, DMSO-d6) δ 2,24 (с, 3H), 2,96 (с, 3H), 4,73 (с, 2H), 6,32 (с, 1H), 7,35 (т, 2H), 7,63-7,73 (м, 3H), 7,92 (с, 1H), 8,70 (д, 1H), 10,79 (с, 1H), 12,23 (с, 1H); LC-MS (ESI) m/z 462 (M+H)+.
Пример 51
Получение 2-(дифтор(4-фторфенил)метил)-7-(этоксиметил)-N-(5-метил-1Н-пиразол-3-ил)хиназолин-4-амина
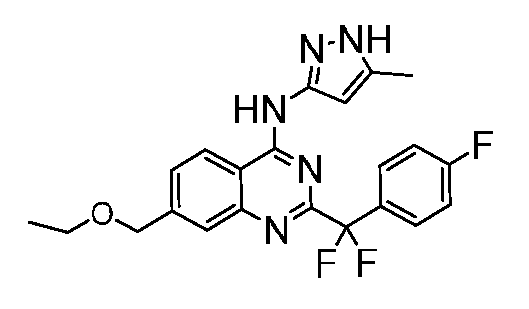
К суспензии (2-(дифтор(4-фторфенил)метил)-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-7-ил)метанола из Примера 49 (150 мг, 0,38 ммоль) в DCM (20 мл) добавляли PBr3 (203 мг, 0,75 ммоль) при нагревании смеси при 60°C. Смесь перемешивали при 60°C в течение 30 минут, давали охладиться до комнатной температуры и концентрировали при пониженном давлении. К остатку добавляли EtOH (20 мл) и 21% раствор NaOEt/EtOH (3 мл) и смесь нагревали при температуре кипения с обратным холодильником в течение 15 часов. Смеси давали охладиться до комнатной температуры и затем смесь концентрировали при пониженном давлении. Добавляли воду (50 мл) и затем добавляли медленно 1 н. раствор HCl для доведения pH до <4 и затем добавляли насыщенный водный раствор NaHCO3 (50 мл). Смесь экстрагировали при помощи EtOAc (3×80 мл) и объединенные органические слои промывали насыщенным солевым раствором (80 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали при помощи препаративной обращенно-фазовой ВЭЖХ с получением 2-(дифтор(4-фторфенил)метил)-7-(этоксиметил)-N-(5-метил-1Н-пиразол-3-ил)хиназолин-4-амина в виде белого твердого вещества (11,3 мг, 6,5%). 1H ЯМР (300 МГц, DMSO-d6) δ 1,21 (т, 3H), 2,23 (с, 3H), 3,56 (кв.т, 2H), 4,66 (с, 2H), 6,31 (с, 1H), 7,38 (т, 2H), 7,56 (д, 1H), 7,70 (кв.т, 2H), 7,76 (с, 1H), 8,65 (д, 1H), 10,69 (с, 1H), 12,20 (с, 1H); LC-MS (ESI) m/z 428 (M+H)+.
Пример 52
Получение (R,S)(7-хлор-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанола
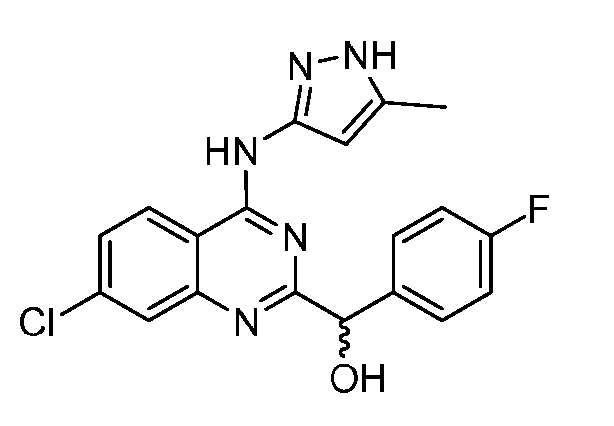
Стадия A: К раствору 2-амино-4-хлорбензойной кислоты (4,4 г, 25,8 ммоль) в дегазированном DMF (75 мл) добавляли последовательно HOBt (4,19 г, 31 ммоль), DIEA (5,4 мл, 31 ммоль), EDCI (5,37 г, 28 ммоль) и 2 н. раствор NH3/MeOH (18 мл, 36 ммоль) и раствор перемешивали при комнатной температуре в течение 4 дней. Смесь концентрировали при пониженном давлении и остаток распределяли между H2O (200 мл) и DCM (200 мл). Отделенную водную фазу экстрагировали при помощи DCM (2×200 мл) и объединенные органические слои сушили над MgSO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали хроматографией на силикагеле, элюируя смесью 10-50% EtOAc/гексан, с получением 2-амино-4-хлорбензамида в виде твердого вещества (2,91 г, 66%). 1H ЯМР (300 МГц, DMSO-d6) δ 6,50 (дд, J=8,48, 2,26 Гц, 1H), 6,75 (д, J=2,26 Гц, 1H), 6,84 (ушир.с, 2H), 7,18 (ушир.с, 1H), 7,48-7,63 (м, 1H), 7,80 (ушир.с, 1H).
Стадия B: К раствору 2-амино-4-хлорбензамида (393 мг, 2,30 ммоль) и DIEA (0,60 мл, 3,45 ммоль) в ТГФ (15 мл) при 0°C добавляли этил хлороксоацетат (0,28 мл, 2,53 ммоль). Раствору давали нагреться до комнатной температуры и перемешивали в течение 2 часов. Смесь концентрировали при пониженном давлении и остаток очищали хроматографией на силикагеле, элюируя смесью 10-50% EtOAc/гексан, с получением этил 2-(2-карбамоил-5-хлорфениламино)-2-оксоацетата в виде твердого вещества (545 мг, 88%). 1H ЯМР (300 МГц, DMSO-d6) δ 1,32 (т, J=6,97 Гц, 3H), 4,31 (кв., J=6,97 Гц, 2H), 7,34 (д, J=8,48 Гц, 1H), 7,86-8,03 (м, 2H), 8,44 (ушир.с, 1H), 8,62 (с, 1H), 13,24 (с, 1H).
Стадия C: К раствору этил 2-(2-карбамоил-5-хлорфениламино)-2-оксоацетата (545 мг, 2,02 ммоль) и TEA (11 мл, 80 ммоль) в DCE (20 мл) добавляли триметилсилилхлорид (3,8 мл, 30 ммоль) и раствор перемешивали при 80°C в течение 16 часов. Смесь концентрировали при пониженном давлении и остаток распределяли между насыщенным водным раствором NaHCO3 и DCM (100 мл). Отделенную водную фазу экстрагировали при помощи DCM (2×100 мл) и объединенные органические слои сушили над MgSO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали хроматографией на силикагеле, элюируя смесью 10-60% EtOAc/гексан, с получением этил 7-хлор-4-гидроксихиназолин-2-карбоксилата в виде твердого вещества (321 мг, 63%). 1H ЯМР (300 МГц, DMSO-d6) δ 1,36 (т, J=7,06 Гц, 3H), 4,39 (кв., J=7,10 Гц, 2H), 7,60-7,74 (м, 1H), 7,85-7,98 (м, 1H), 8,11-8,23 (м, 1H), 12,82 (ушир.с, 1H).
Стадия D: (R,S)-(7-Хлор-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанол получали в виде твердого вещества с использованием процедур, аналогичных тем, которые описаны в Примере 46 Стадии D-F, используя этил 7-хлор-4-гидроксихиназолин-2-карбоксилат вместо этил 4-гидрокси-8-метилхиназолин-2-карбоксилата, используемого в Примере 46 Стадия D. 1H ЯМР (300 МГц, DMSO-d6) δ 2,25 (с, 3H), 5,69 (д, J=1,00 Гц, 1H), 5,82 (д, J=1,00 Гц, 1H), 6,43 (с, 1H), 7,07-7,23 (м, 2H), 7,46-7,62 (м, 3H), 7,81 (с, 1H), 8,57-8,69 (м, 1H), 10,53 (ушир.с, 1H), 12,18 (ушир.с, 1H), LC-MS (ESI) m/z 384 (M+H)+.
Пример 53
Получение (6-фтор-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанона
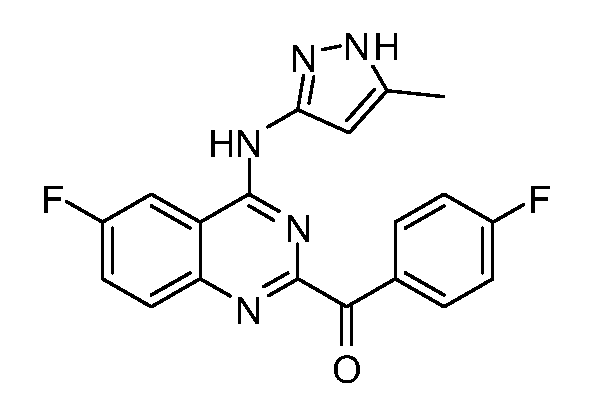
Стадия A: Перемешиваемую смесь метил 2-амино-5-фторбензоата (1 г, 5,92 ммоль) и этилкарбоноцианидата (1,17 г, 11,8 ммоль) в HOAc (8 мл) обрабатывали 12 н водным раствором хлористоводородной кислоты (0,8 мл). Полученную смесь нагревали при 70°C в течение 3 часов. После охлаждения до комнатной температуры растворитель удаляли при пониженном давлении. Остаток суспендировали в воде и обрабатывали насыщенным водным раствором NaHCO3 до достижения pH=7. Твердое вещество фильтровали, промывали водой/диэтиловым эфиром и сушили при пониженном давлении с получением этил 6-фтор-4-оксо-3,4-дигидрохиназолин-2-карбоксилата (1,2 г, 86%). 1H ЯМР (300 МГц, DMSO-d6) δ 12,79 (с, 1H), 7,76-7,95 (м, 3H), 4,39 (кв., J=7,0 Гц, 2H), 1,36 (т, J=7,0 Гц, 3H); LC-MS (ESI) m/z 237 (M+H)+.
Стадия B: Перемешиваемый раствор этил 6-фтор-4-оксо-3,4-дигидрохиназолин-2-карбоксилата (1,2 г, 5,08 ммоль) в оксихлориде фосфора (15 мл) нагревали при 105°C в течение 6 часов. После охлаждения до комнатной температуры реакционную смесь концентрировали досуха при пониженном давлении и остаток растворяли в безводном толуоле. Толуол концентрировали при пониженном давлении. Остаток растворяли в небольшом объеме DCM и пропускали через короткую пробку силикагеля, элюируя при помощи DCM. Этил 4-хлор-6-фторхиназолин-2-карбоксилат получали в виде твердого вещества (1,3 г, 100%). LC-MS (ESI) m/z 255 (M+H)+.
Стадия C: К перемешиваемому раствору этил 4-хлор-6-фторхиназолин-2-карбоксилата (1,06 г, 4,5 ммоль) в ТГФ (15 мл) при -40°C добавляли по каплям 1 M раствор 4-фторфенилмагнийбромид/ТГФ (5,85 мл, 5,85 ммоль). Смесь перемешивали при -40°C в течение 2 часов. Реакцию гасили добавлением 0,5 н. раствора HCl при 0°C и затем смесь экстрагировали при помощи EtOAc (2×20 мл). Твердое вещество, которое осаждалось из объединенных органических слоев, удаляли фильтрованием и фильтрат промывали насыщенным солевым раствором. Органическую фазу сушили над MgSO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали хроматографией на силикагеле с получением (4-хлор-6-фторхиназолин-2-ил)(4-фторфенил)метанона в виде твердого вещества (950 мг, 69%). 1H ЯМР (300 МГц, DMSO-d6) δ 8,32-8,36 (м, 1H), 8,13-8,21 (м, 4H), 7,43 (т, J=8,2 Гц, 2H); LC-MS (ESI) m/z 305 (M+H)+.
Стадия D: (6-Фтор-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанон получали из (4-хлор-6-фторхиназолин-2-ил)(4-фторфенил)метанона (306 мг, 1 ммоль) и 5-метил-1Н-пиразол-3-амина (194 мг, 2 ммоль) с использованием процедуры, аналогичной описанной в Примере 48 Стадия A. Неочищенный продукт растирали в порошок с MeOH с получением (6-фтор-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанона (310 мг, 85%). 1H ЯМР (300 МГц, DMSO-d6) δ 12,24 (с, 1H), 10,71 (с, 1H), 8,64 (д, J=9,6 Гц, 1H), 7,95-8,09 (м, 2H), 7,93-7,95 (м, 1H), 7,80-7,86 (м, 1H), 7,39 (т, J=8,6 Гц, 2H), 6,53 (с, 1H), 1,91 (с, 3H); LC-MS (ESI) m/z 366 (M+H)+.
Пример 54
Получение (R,S)-(6-фтор-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанола
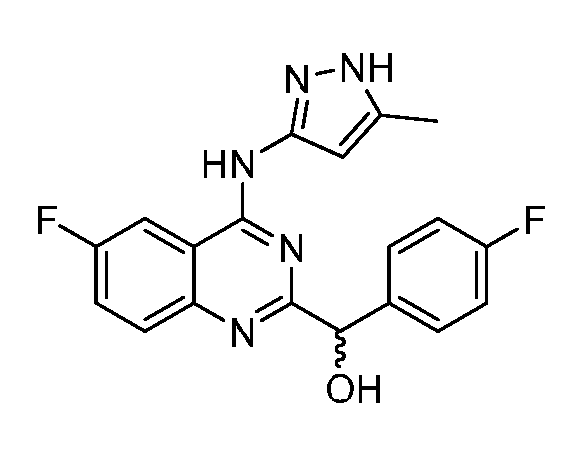
К перемешиваемой суспензии (6-фтор-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанона из Примера 53 Стадия D (200 мг, 0,55 ммоль) в смеси 4:1 MeOH/ТГФ (10 мл) добавляли борогидрид натрия (33 мг, 0,88 ммоль) и смесь перемешивали при комнатной температуре в течение 2 часов. Добавляли воду (8 мл) и осажденное твердое вещество собирали фильтрованием, промывали при помощи MeOH и очищали два раза при помощи препаративной обращенно-фазовой ВЭЖХ с получением (R,S)-(6-фтор-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанола в виде твердого вещества (72,5 мг, 36%). 1H ЯМР (300 МГц, DMSO-d6) δ 12,15 (с, 1H), 10,41 (с, 1H), 8,49 (д, J=9,8 Гц, 1H), 7,84-7,88 (м, 1H), 7,70-7,76 (м, 1H), 7,52-7,56 (м, 2H), 7,14 (т, J=8,5 Гц, 2H), 6,45 (с, 1H), 5,84 (ушир.с, 1H), 5,68 (ушир.с, 1H), 2,25 (с, 3H); LC-MS (ESI) m/z 368 (M+H)+.
Пример 55
Получение (R,S)-(4-(1Н-пиразол-3-иламино)-6-фторхиназолин-2-ил)(4-фторфенил)метанола
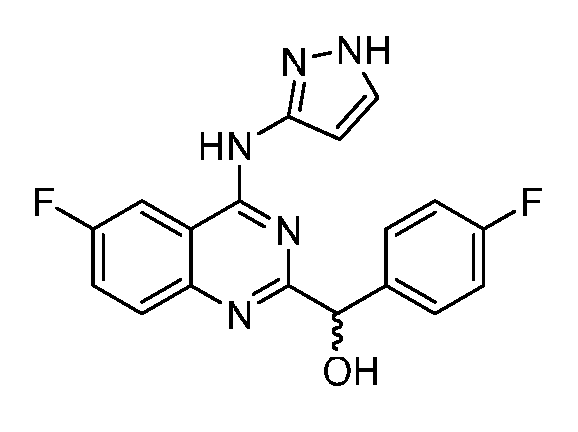
Стадия A: (4-(1Н-Пиразол-3-иламино)-6-фторхиназолин-2-ил)(4-фторфенил)метанон получали из (4-хлор-6-фторхиназолин-2-ил)(4-фторфенил)метанона из Примера 53 Стадия C (304 мг, 1 ммоль) и 1Н-пиразол-3-амина (166 мг, 2 ммоль) с использованием процедуры, аналогичной описанной в Примере 48 Стадия A. Неочищенный продукт растирали в порошок с MeOH с получением (4-(1Н-пиразол-3-иламино)-6-фторхиназолин-2-ил)(4-фторфенил)метанона в виде твердого вещества (275 мг, 78%). 1H ЯМР (300 МГц, DMSO-d6) δ 12,56 (с, 1H), 10,84 (с, 1H), 8,67 (д, J=9,9 Гц, 1H), 8,08-8,13 (м, 2H), 7,95-7,99 (м, 1H), 7,82-7,87 (м, 1H), 7,67 (с, 1H), 7,36-7,41 (м, 2H), 6,78 (с, 1H); LC-MS (ESI) m/z 352 (M+H)+.
Стадия B: (4-(1Н-Пиразол-3-иламино)-6-фторхиназолин-2-ил)(4-фторфенил)метанол получали из (4-(1Н-пиразол-3-иламино)-6-фторхиназолин-2-ил)(4-фторфенил)метанона (200 мг, 0,57 ммоль) с использованием процедуры, аналогичной описанной в Примере 54, с получением (R,S)-(4-(1Н-пиразол-3-иламино)-6-фторхиназолин-2-ил)(4-фторфенил)метанола в виде твердого вещества (59 мг, 29%). 1H ЯМР (300 МГц, DMSO-d6) δ 12,50 (с, 1H), 10,53 (с, 1H), 8,53 (д, J=9,8 Гц, 1H), 7,85-7,89 (м, 1H), 7,71-7,77 (м, 2H), 7,52-7,57 (м, 2H), 7,12 (т, J=8,5 Гц, 2H), 6,88 (с, 1H), 5,84 (ушир.с, 1H), 5,69 (ушир.с, 1H); LC-MS (ESI) m/z 354 (M+H)+.
Пример 56
Получение (7-бром-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанона
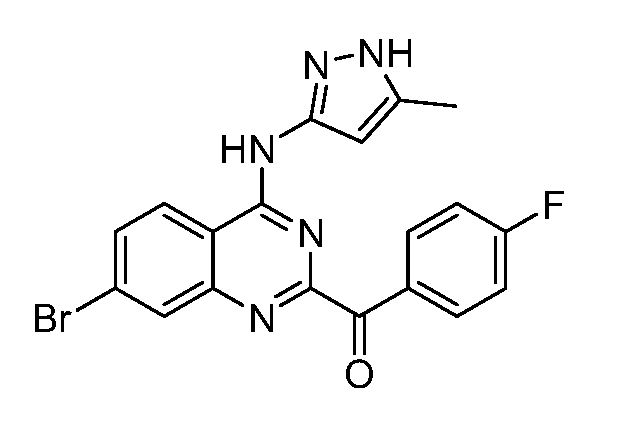
Стадия A: Метил 2-амино-4-бромбензоат получали из соответствующей кислоты путем этерификации по методу Фишера. Смесь метил 2-амино-4-бромбензоата (1,1 г, 4,8 ммоль) и этилкарбоноцианидата (0,95 г, 9,66 ммоль) в HOAc (4 мл) обрабатывали 2 н. раствором HCl (0,4 мл) и полученную смесь перемешивали при 70°C в течение 3 часов. После охлаждения до комнатной температуры добавляли воду с последующим добавлением водного раствора гидрокарбоната натрия до достижения pH~5. Осажденное твердое вещество фильтровали и промывали тщательно водой и диэтиловым эфиром с получением этил 7-бром-4-оксо-3,4-дигидрохиназолин-2-карбоксилата (825 мг, 58%). 1H ЯМР (300 МГц, DMSO-d6) δ 12,82 (с, 1H), 8,06-8,09 (м, 2H), 7,81 (д, J=8,4 Гц, 1H), 4,38 (кв., J=7,0 Гц, 2H), 1,35 (т, J=7,0 Гц, 3H); LC-MS (ESI) m/z 296 (M+H)+.
Стадия B: Этил 7-бром-4-хлорхиназолин-2-карбоксилат получали с 69% выходом с использованием процедуры, аналогичной описанной в Примере 53 Стадия B, используя этил 7-бром-4-оксо-3,4-дигидрохиназолин-2-карбоксилат (825 мг, 2,78 ммоль) вместо этил 6-фтор-4-оксо-3,4-дигидрохиназолин-2-карбоксилата, используемого в Примере 53. LC-MS (ESI) m/z 315 и 317 (M+H)+.
Стадия C: (7-Бром-4-хлорхиназолин-2-ил)(4-фторфенил)метанон получали с 43% выходом с использованием процедуры, аналогичной описанной в Примере 53 Стадия C, используя этил 7-бром-4-оксо-3,4-дигидрохиназолин-2-карбоксилат (605 мг, 1,92 ммоль) вместо этил 4-хлор-6-фторхиназолин-2-карбоксилата, используемого в Примере 53. LC-MS (ESI) m/z 387 (M+Na)+.
Стадия D: (7-Бром-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанон получали с использованием процедуры, аналогичной описанной в Примере 48 Стадия A, используя (7-бром-4-хлорхиназолин-2-ил)(4-фторфенил)метанон (300 мг, 0,82 ммоль) вместо этил 4-хлорхиназолин-2-карбоксилата, используемого в Примере 48. Часть неочищенного продукта (90 мг) очищали при помощи препаративной обращенно-фазовой ВЭЖХ с получением (7-бром-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанона в виде твердого вещества (12 мг, 11%). 1H ЯМР (300 МГц, DMSO-d6) δ 12,23 (с, 1H), 10,85 (с, 1H), 8,69 (д, J=8,0 Гц, 1H), 8,07-8,11 (м, 3H), 7,84 (д, J=9,0 Гц, 1H), 7,36-7,42 (м, 2H), 6,49 (с, 1H), 2,18 (с, 3H); LC-MS (ESI) m/z 426 (M+H)+.
Пример 57
Получение (7-бром-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанола
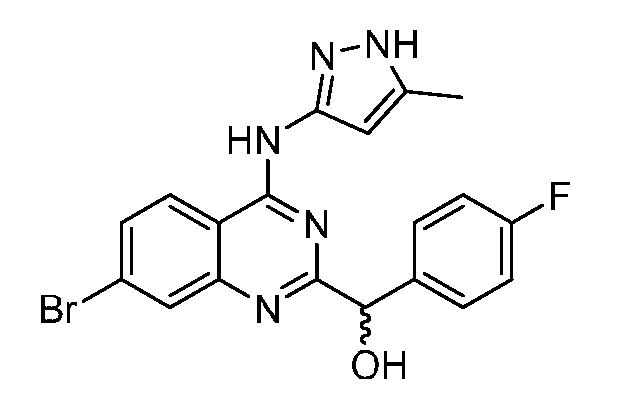
(7-Бром-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанол получали с использованием процедуры, аналогичной описанной в Примере 54 используя (7-бром-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанон из Примера 56 (240 мг, 0,56 ммоль) вместо (6-фтор-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанона, используемого в Примере 54 (31 мг, 13%). 1H ЯМР (300 МГц, DMSO-d6) δ 12,17 (с, 1H), 10,55 (с, 1H), 8,54 (ушир.с, 1H), 7,97 (ушир.с, 1H), 7,67 (д, J=8,4 Гц, 1H), 7,52-7,56 (м, 2H), 7,12-7,17 (м, 2H), 6,44 (с, 1H), 5,84 (с, 1H), 5,66 (с, 1H), 2,25 (с, 3H); LC-MS (ESI) m/z 428 (M+H)+.
Пример 58
Получение (R,S)-(4-(1Н-пиразол-3-иламино)-7-бромхиназолин-2-ил)(4-фторфенил)метанола
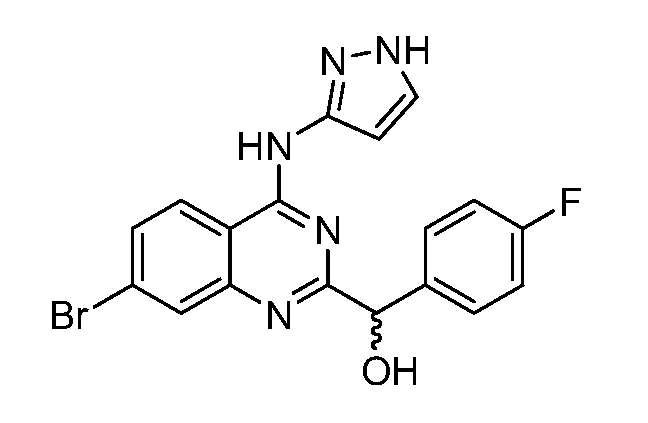
Стадия A: (4-(1Н-Пиразол-3-иламино)-7-бромхиназолин-2-ил)(4-фторфенил)метанон (270 мг, 80%) получали с использованием процедуры, аналогичной описанной в Примере 48 Стадия A, используя (7-бром-4-хлорхиназолин-2-ил)(4-фторфенил)метанон из Примера 56 (300 мг, 0,82 ммоль) вместо этил 4-хлорхиназолин-2-карбоксилата, используемого в Примере 48. 1H ЯМР (300 МГц, DMSO-d6) δ 12,57 (с, 1H), 10,97 (с, 1H), 8,72 (д, J=8,3 Гц, 1H), 8,10 (т, J=8,3 Гц, 3H), 7,86 (д, J=8,5 Гц, 1H), 7,67 (ушир.с, 1H), 7,36 (т, J=8,0 Гц, 2H), 6,75 (ушир.с, 1H); LC-MS (ESI) m/z 412 (M+H)+.
Стадия B: (R,S)-(4-(1Н-Пиразол-3-иламино)-7-бромхиназолин-2-ил)(4-фторфенил)метанол (37 мг, 18%) получали с использованием процедуры, аналогичной описанной в Примере 54, используя (4-(1Н-пиразол-3-иламино)-7-бромхиназолин-2-ил)(4-фторфенил)метанон (200 мг, 0,49 ммоль) вместо (6-фтор-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанона, используемого в Примере 54. 1H ЯМР (300 МГц, DMSO-d6) δ 12,47 (с, 1H), 10,70 (с, 1H), 8,57 (д, J=8,9 Гц, 1H), 7,99 (с, 1H), 7,69-7,72 (м, 2H), 7,54 (т, J=6,4 Гц, 2H), 7,12 (т, J=8,2 Гц, 2H), 6,82 (с, 1H), 5,86 (ушир.с, 1H), 5,68 (ушир.с, 1H); LC-MS (ESI) m/z 414 и 416 (M+H)+.
Пример 59
Получение 2-(2-(4-фторфенил)-1,3-диоксолан-2-ил)-N-(5-метил-1Н-пиразол-3-ил)хиназолин-4-амина
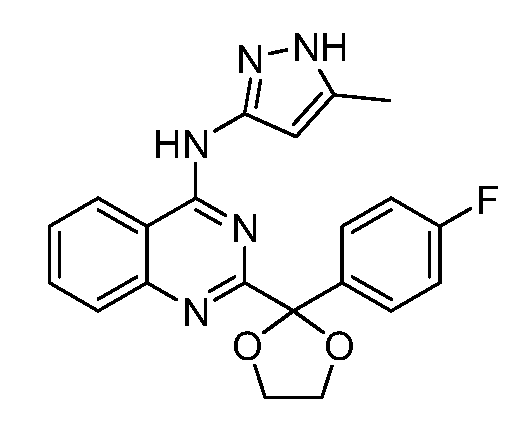
К (4-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанону из Примера 3 (0,20 г, 0,58 ммоль) в толуоле (20 мл) добавляли этиленгликоль (0,2 мл) и моногидрат п-толуолсульфоновой кислоты (каталитическое количество). Смесь нагревали при температуре кипения с обратным холодильником в течение ночи, собирая при этом воду при помощи ловушки Дина-Старка. Добавляли дополнительное количество этиленгликоля (1,5 мл) и моногидрата п-толуолсульфоновой кислоты (50 мг) и смесь нагревали при температуре кипения с обратным холодильником в течение ночи, собирая при этом воду. После охлаждения смесь упаривали досуха и затем растворяли в DMSO (8 мл). 5-мл аликвоту этого раствора очищали при помощи препаративной обращенно-фазовой ВЭЖХ (колонка Varian дифенил с обращенной фазой, элюирование с использованием градиента растворителя B=0,05% HOAC/ACN и растворителя А=0,05% HOAc/H2O) с последующей дополнительной очисткой при помощи препаративной обращенно-фазовой ВЭЖХ (колонка Phenomenex C-18 с обращенной фазой, элюирование с использованием градиента растворителя B=0,05% HOAC/ACN и растворителя А=0,05% HOAc/H2O) с получением 2-(2-(4-фторфенил)-1,3-диоксолан-2-ил)-N-(5-метил-1Н-пиразол-3-ил)хиназолин-4-амина (2 мг, 1%). 1H ЯМР (300 МГц, DMSO-d6) δ 2,20 (с, 3H) 4,05-4,22 (м, 4H) 6,2 (с,lH) 7,19 (т, 2H) 7,5-7,65 (м, 3H) 7,81 (м, 2H) 8,59 (д, 1H) 10,38 (с, 1H) 12,08 (с, 1H); LC-MS (ESI) m/z 392 (M+H)+.
Пример 60
Получение (8-фтор-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанона
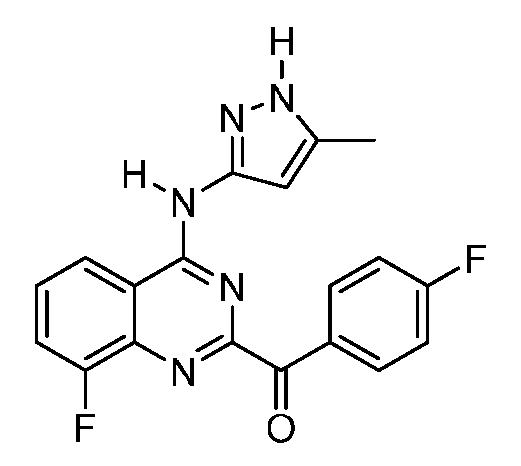
Стадия A: Этил 2-(2-карбамоил-6-фторфениламино)-2-оксоацетат получали со ссылкой на процедуры, аналогичные тем, которые описаны в Примере 46 Стадии А и B, где можно использовать 2-амино-3-фторбензойную кислоту вместо 2-амино-3-метилбензойной кислоты, используемой в Примере 46.
Стадия B: Смесь этил 2-(2-карбамоил-6-фторфениламино)-2-оксоацетата (600 мг, 2,4 ммоль) и трет-бутоксида калия (300 мг, 6,7 ммоль) в EtOH (10 мл) перемешивали при комнатной температуре в течение 5 часов. Добавляли насыщенный водный раствор NaCl (50 мл) и HOAc (0,5 мл) и смесь экстрагировали при помощи EtOAc (3×80 мл). Объединенные органические слои промывали насыщенным солевым раствором (80 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением этил 8-фтор-4-гидроксихиназолин-2-карбоксилата (530 мг, 93%). 1H ЯМР (300 МГц, DMSO-d6) δ 1,39 (т, J=7,2 Гц, 3H), 4,40 (кв., J=7,2 Гц, 2H), 7,57-7,70 (м, 1H), 7,78 (т, J=9,2 Гц, 1H), 7,99 (д, 7=8,1 Гц, 1H), 12,86 (с, 1H); LC-MS (ESI) m/z 237 (M+H)+.
Стадия C: Смесь этил 8-фтор-4-гидроксихиназолин-2-карбоксилата (530 мг, 2,2 ммоль) и POCl3 (3 мл) нагревали при температуре кипения с обратным холодильником в течение 5 часов. Смесь концентрировали при пониженном давлении и затем добавляли толуол два раза и концентрировали при пониженном давлении. Остаток очищали хроматографией на силикагеле, элюируя смесью 0-25% EtOAc/гексан, с получением этил 4-хлор-8-фторхиназолин-2-карбоксилата (365 мг, 65%). 1H ЯМР (300 МГц, DMSO-d6) δ 1,39 (т, J=7,1 Гц, 3H), 4,46 (кв., J=7,1 Гц, 2H), 7,97-8,06 (м, 1H), 8,07-8,16 (м, 1H), 8,18-8,24 (м, 1H).
Стадия D: К раствору этил 4-хлор-8-фторхиназолин-2-карбоксилата (360 мг, 1,42 ммоль) в ТГФ (20 мл) при -40°C добавляли раствор 1М 4-фторфенилмагнийбромид/ТГФ (1,7 мл, 1,7 ммоль) и смесь перемешивали при -40°C в течение 3 часов. По прошествии этого времени, добавляли дополнительную порцию раствора 1М 4-фторфенилмагнийбромид/ТГФ (0,3 мл, 0,3 ммоль) и смесь перемешивали при -40°C в течение 2 часов. К смеси добавляли 0,5 н. раствор HCl для достижения рН~2 и затем добавляли насыщенный водный раствор NaCl (50 мл) и смесь экстрагировали при помощи EtOAc (3×50 мл). Объединенные органические слои промывали насыщенным солевым раствором (80 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением (4-хлор-8-фторхиназолин-2-ил)(4-фторфенил)метанона (0,4 г, 93%). 1H ЯМР (300 МГц, DMSO-d6) δ 7,04-7,25 (м, 1H), 7,31-7,58 (м, 2H), 7,70-8,36 (м, 4H); LC-MS (ESI) m/z 305 (M+H)+, 327 (M+Na)+.
Стадия E: Смесь (4-хлор-8-фторхиназолин-2-ил)(4-фторфенил)метанона (0,4 г, 1,32 ммоль), 5-метил-1Н-пиразол-3-амина (0,15 г, 1,58 ммоль), DIEA (0,69 мл, 4,0 ммоль) и KI (0,24 г, 1,45 ммоль) в DMF (8 мл) перемешивали при комнатной температуре в течение 20 часов. Смесь разбавляли при помощи H2O (35 мл) и осажденное твердое вещество собирали фильтрованием, промывали при помощи H2O и растирали в порошок с MeOH при 0°C. Часть полученного твердого вещества очищали при помощи препаративной обращенно-фазовой ВЭЖХ с получением (8-фтор-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанона 1H ЯМР (300 МГц, DMSO-d6) δ 2,19 (с, 3H), 6,52 (с, 1H), 7,41 (т, 2H), 7,64-7,80 (м, 2H), 8,11 (т, 2H), 8,57 (д, 1H), 10,84 (с, 1H), 12,27 (ушир.с, 1H); LC-MS (ESI) m/z 366 (M+H)+.
Пример 61
Получение (R,S)-(8-фтор-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанола
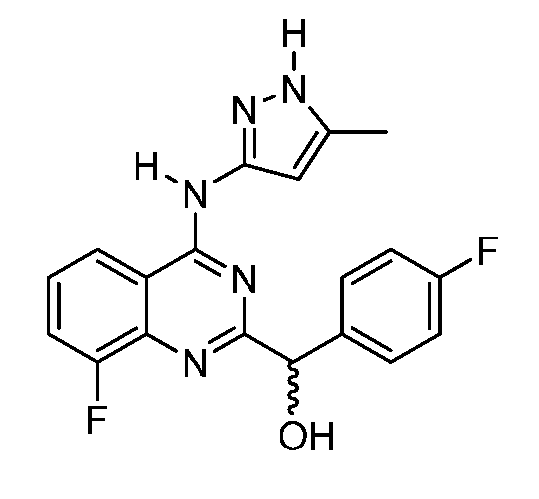
К суспензии (8-фтор-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанона (233 мг, 0,64 ммоль) в смеси 2:1 MeOH/ТГФ (15 мл) при 0°C добавляли борогидрид натрия (36 мг, 0,96 ммоль) и смесь перемешивали при 0°C в течение 1 часа и при комнатной температуре в течение ночи. Смесь охлаждали до 0°C и добавляли 1 н. раствор HCl для достижения рН<2, затем добавляли насыщенный водный раствор NaHCO3 и насыщенный солевой раствор. Осажденное твердое вещество собирали фильтрованием, промывали при помощи H2O и очищали при помощи препаративной обращенно-фазовой ВЭЖХ с получением (R,S)-(8-фтор-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанола (141 мг, 60%). 1H ЯМР (300 МГц, DMSO-d6) δ 2,24 (с, 3H), 5,69 (с, 1H), 5,86 (ушир.с, 1H), 6,41 (ушир.с, 1H), 7,15 (т, 2H), 7,47-7,65 (м, 4H), 8,34 (ушир.с, 1H), 10,53 (ушир.с, 1H), 12,18 (ушир.с, 1H); LC-MS (ESI) m/z 368 (M+H)+.
Пример 62
Получение (2-метоксифенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанона
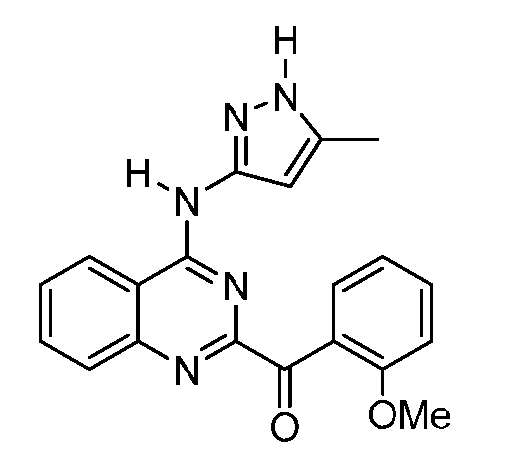
К раствору этил 4-хлорхиназолин-2-карбоксилата (500 мг, 2,11 ммоль) в ТГФ (20 мл) при -40°C добавляли по каплям 1М раствор 2-метоксифенилмагнийбромида (2,6 мл, 2,5 ммоль) и смесь перемешивали при -40°C в течение 4 часов. К смеси добавляли 1 н. раствор HCl для доведения до pH<2 и насыщенный водный раствор NaCl (50 мл) и смесь экстрагировали при помощи EtOAc (3×80 мл). Объединенные органические слои промывали насыщенным солевым раствором (50 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. К остатку (673 мг) добавляли раствор 5-метил-1Н-пиразол-3-амина (247 мг, 2,84 ммоль) в DMF (8 мл), иодид калия (387 мг, 2,33 ммоль) и DIEA (822 мг, 6,36 ммоль) и смесь перемешивали при комнатной температуре в течение ночи и затем при 50°C в течение 5 часов и затем при 80°C в течение 2 часов. Смеси давали охладиться до комнатной температуры и затем добавляли H2O (30 мл). Смесь перемешивали и охлаждали до 0°C и осажденное твердое вещество собирали фильтрованием и промывали при помощи MeOH. Часть твердого вещества очищали при помощи препаративной обращенно-фазовой ВЭЖХ с получением (2-метоксифенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанона. 1H ЯМР (300 МГц, DMSO-d6) δ 2,11 (с, 3H), 3,48 (с, 3H), 6,15 (с, 1H), 7,11-7,18 (м, 2H), 7,56-7,64 (м, 3H), 7,56 (ушир.с, 2H), 8,70 (д, 1H), 10,58 (с, 1H), 12,11 (с, 1H); LC-MS (ESI) m/z 360 (M+H)+.
Пример 63
Получение (R,S)-(2-метоксифенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанола
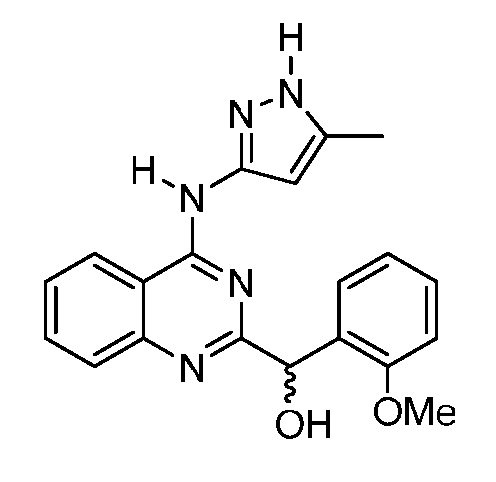
К суспензии (2-метоксифенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанона (420 мг, 1,17 ммоль) в смеси 2:1 MeOH/ТГФ (15 мл) при 0°C добавляли NaBH4 (53 мг, 1,40 ммоль) и смесь перемешивали при 0°C в течение 1 часа и при комнатной температуре в течение ночи. Смесь охлаждали до 0°C и добавляли 1 н. раствор HCl для доведения до pH<2. Затем добавляли насыщенный водный раствор NaHCO3 (50 мл) и смесь экстрагировали при помощи EtOAc (3×80 мл). Объединенные органические слои промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали при помощи препаративной обращенно-фазовой ВЭЖХ с получением (R,S)-(2-метоксифенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанола (55,3 мг, 15%). 1H ЯМР (300 МГц, DMSO-d6) δ 2,19 (с, 3H), 3,76 (с, 3H), 5,65 (с, 1H), 6,00 (с, 1H), 6,24 (с, 1H), 6,92-7,01 (м, 3H), 7,25 (т, 1H), 7,45-7,52 (м, 2H), 7,80 (с, 2H), 8,57 (д, 1H), 10,36 (с, 1H), 12,05 (с, 1H); LC-MS (ESI) m/z 362 (M+H)+.
Пример 64
Получение (3-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанола
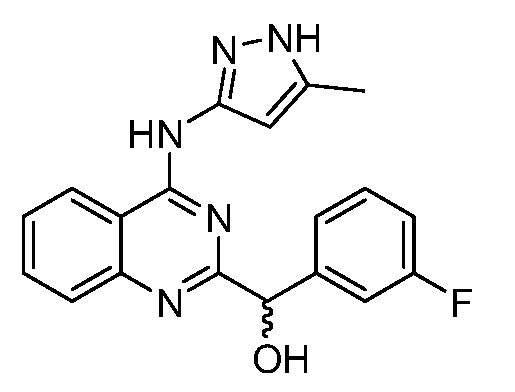
К (3-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанона из Примера 1 (0,15 г, 0,43 ммоль) в смеси 1:1 ТГФ/MeOH (4 мл) при 0°C добавляли борогидрид натрия (0,02 г, 0,58 ммоль) и смесь перемешивали при 0°C в течение 30 минут. К смеси добавляли 6 н. раствор HCl и DMSO (3 мл) и смесь очищали при помощи препаративной ВЭЖХ (колонка Varian дифенил с обращенной фазой, элюировали с использованием градиента растворителя B=0,05% HOAC/MeOH и растворителя А=0,05% HOAc/H2O) с получением (3-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанола (50 мг, 32%). 1H ЯМР (300 МГц, DMSO-d6) δ 2,26 (с, 3H) 5,7 (с, 1H) 5,93 (с,lH) 6,44 (с, 1H) 7,07 (м, 1H) 7,34-7,39 (м, 3H) 7,54 (м, 1H) 7,81 (с, 2H) 8,59 (д, 1H) 10,42 (с, 1H) 12,15 (с, 1H); LC-MS (ESI) m/z 350 (M+H)+.
Пример 65
Получение N-((4-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метил)формамида
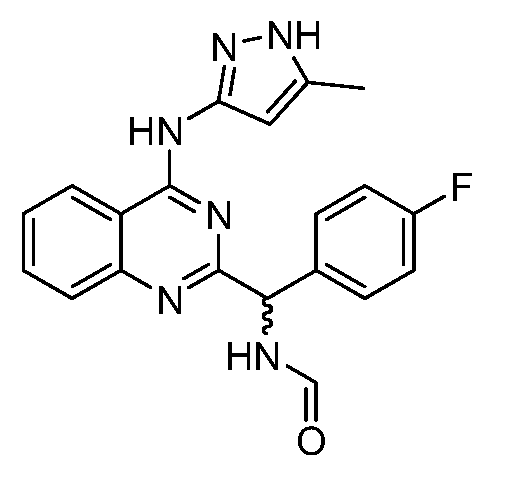
К 2-(амино(4-фторфенил)метил)-N-(5-метил-1Н-пиразол-3-ил)хиназолин-4-амину из Примера 45 Стадия B (0,1 г, 0,28 ммоль) в этилформиате (4 мл) добавляли TEA (0,2 мл) и EtOH (0,5 мл) и смесь нагревали до 120°C в течение 30 минут в микроволновом реакторе Biotage и затем концентрировали при пониженном давлении. Остаток разбавляли при помощи DMSO (5 мл) и очищали при помощи обращенно-фазовой препаративной ВЭЖХ (колонка Varian дифенил с обращенной фазой, элюировали с использованием градиента растворителя B=0,05% HOAC/MeOH и растворителя А=0,05% HOAc/H2O) с получением N-((4-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метил)формамида (8 мг, 8%). 1H ЯМР (300 МГц, DMSO-d6) δ 2,26 (с, 3H) 6,07 (д, 1H) 6,44 (с, 1H) 7,16 (т, 2H) 7,4-7,6 (м, 3H) 7,8 (м, 2H) 8,21 (с, 1H) 8,62 (д, 1H) 9,06 (д, 1H) 10,48 (ушир.с, 1H) 12,17 (ушир.с, 1H); LC-MS (ESI) m/z 377 (M+H)+.
Пример 66
Получение (R,S)-(3,4-дифторфенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанола
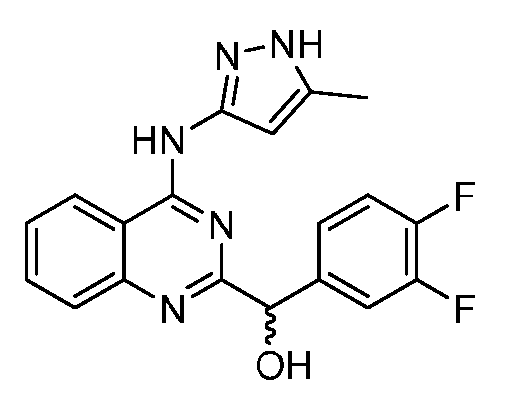
Стадия A: Смесь этил 4-хлорхиназолин-2-карбоксилата (1 г, 4,2 ммоль) в ТГФ (50 мл) фильтровали и к фильтрату при -30°C в атмосфере Ar добавляли раствор 0,5M (3,4-дифторфенил)магнийбромид/ТГФ (10,4 мл, 5,2 ммоль). Смесь перемешивали при -30°C в течение 1,5 часов, и затем добавляли дополнительное количество раствора 0,5M (3,4-дифторфенил)магнийбромид/ТГФ (3 мл). Еще через 1,5 часа добавляли дополнительное количество раствора 0,5M (3,4-дифторфенил)магнийбромид/ТГФ (3 мл). К смеси добавляли насыщенный водный раствор хлорида аммония и смеси давали нагреться до комнатной температуры. Смесь экстрагировали при помощи EtOAc (2×) и объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением (4-хлорхиназолин-2-ил)(3,4-дифторфенил)метанона (330 мг, 26%), который использовали непосредственно на следующей стадии.
Стадия B: К смеси 5-метил-1Н-пиразол-3-амина (0,18 г, 1,8 ммоль), иодида калия (0,18 г, 1,1 ммоль) и TEA (0,16 мл, 1,1 ммоль) в DMF (5 мл) добавляли раствор (4-хлорхиназолин-2-ил)(3,4-дифторфенил)метанона (0,33 г, 1,1 ммоль) в DMF (5 мл) и смесь перемешивали при комнатной температуре в течение ночи. Добавляли воду и осажденное твердое вещество собирали фильтрованием. Желтое твердое вещество (553 мг), содержащее (3,4-дифторфенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанон, использовали непосредственно на следующей стадии. LC-MS (ESI) m/z 366 (M+H)+.
Стадия C: К неочищенному (3,4-дифторфенил)(4-(5-метил-1H-пиразол-3-иламино)хиназолин-2-ил)метанону (553 мг, 1,5 ммоль) в смеси 1:1 MeOH/ТГФ (40 мл) при 0°C добавляли борогидрид натрия (0,09 г, 2,4 ммоль) и раствор перемешивали в течение 30 минут, после чего добавляли 6 н. раствор HCl. Смесь концентрировали досуха и примерно 2/3 остатка очищали хроматографией на силикагеле, элюируя смесью 0-15% MeOH/DCM, с получением (3,4-дифторфенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанола (34 мг). 1H ЯМР (300 МГц, DMSO-d6) δ 2,26 (с, 3H) 6,03 (с, 1H) 6,22 (с, 1H) 7,45-7,55 (м, 2H) 7,66 (т, 1H) 7,78 (т, 1H) 8,07 (т, 1H) 8,19 (т, 1H) 8,82 (д, 1H) 12,09 (ушир.с, 1H) 12,67 (ушир.с, 1H); LC-MS (ESI) m/z 368 (M+H)+.
Пример 67
Получение (3-хлор-4-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанола
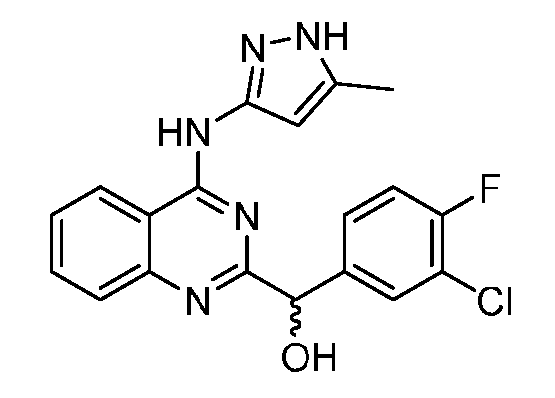
Стадия A: Смесь этил 4-хлорхиназолин-2-карбоксилата (1 г, 4,2 ммоль) в ТГФ (50 мл) фильтровали и к фильтрату при -30°C в атмосфере Ar добавляли 0,5 M раствор (3-хлор-4-фторфенил)магнийбромид/ТГФ (10,4 мл, 5,2 ммоль). После перемешивания при -30°C в течение 0,75 часов добавляли дополнительное количество 0,5 M раствора (3-хлор-4-фторфенил)магнийбромид/ТГФ (4,2 мл). Через 1 час реакцию гасили добавлением насыщенного водного раствора хлорида аммония и давали нагреться до комнатной температуры. Смесь экстрагировали при помощи EtOAc и экстракты сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением (3-хлор-4-фторфенил)(4-хлорхиназолин-2-ил)метанона (495 мг, 37%), который использовали непосредственно на следующей стадии. LC-MS (ESI) m/z 321 (M+H)+.
Стадия B: К смеси 5-метил-1Н-пиразол-3-амина (0,26 г, 2,68 ммоль), иодида калия (0,26 г, 1,57 ммоль) и TEA (0,45 мл, 3,23 ммоль) в DMF (30 мл) добавляли (3-хлор-4-фторфенил)(4-хлорхиназолин-2-ил)метанон (0,495 г, 1,54 ммоль) и смесь перемешивали при комнатной температуре в течение ночи. Добавляли воду и осажденное твердое вещество собирали фильтрованием с получением неочищенного (3-хлор-4-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанона (458 мг), который использовали непосредственно на следующей стадии. LC-MS (ESI) m/z 382 (M+H)+.
Стадия C: К неочищенному (3-хлор-4-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанону (458 мг, 1,2 ммоль) в смеси 1:1 MeOH/ТГФ (60 мл) при 0°C добавляли борогидрид натрия (0,048 г, 1,3 ммоль) и раствор перемешивали в течение 30 минут. Реакцию гасили добавлением 6 н. раствора HCl и смесь концентрировали досуха. Остаток очищали хроматографией на силикагеле, элюируя смесью 0-15% MeOH/DCM. Полученное твердое вещество растирали в порошок с метанолом с получением (3-хлор-4-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанола (42 мг). 1H ЯМР (300 МГц, DMSO-d6) δ 2,27 (с, 3H) 5,70 (с, 1H) 6,01 (с, 1H) 6,41(с, 1H) 7,36 (т, 1H) 7,47-7,52 (м, 2H) 7,79-7,82 (м, 3H) 8,59 (д, 1H) 10,42 (ушир.с, 1H) 12,16 (ушир.с, 1H); LC-MS (ESI) m/z 384 (M+H)+.
Пример 68
Получение 3-(4-фторфенил)-3-(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)пропаннитрила
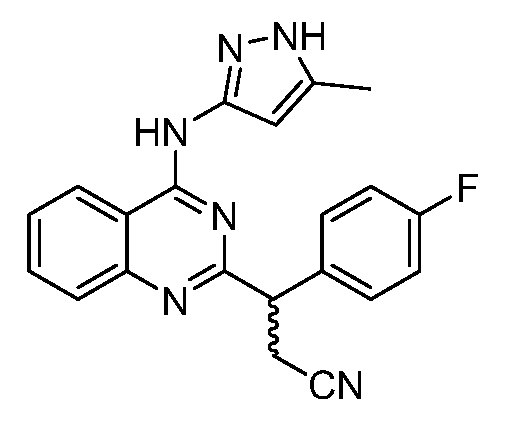
К суспензии 60% гидрид натрия/минеральное масло (173 мг, 4,32 ммоль) в ТГФ (10 мл) при 0°C в атмосфере Ar добавляли диэтилцианометилфосфонат (0,68 мл, 4,32 ммоль) и смесь перемешивали в течение 10 минут. Затем добавляли суспензию (4-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанона из Примера 3 (500 мг, 1,44 ммоль) в ТГФ (20 мл) и смесь перемешивали при комнатной температуре в течение 30 минут. Затем добавляли AcOH (0,5 мл) и Целит и смесь концентрировали при пониженном давлении. Смесь элюировали на колонке с силикагелем и затем элюировали при помощи EtOAc/гексан. К выделенному веществу добавляли EtOH (100 мл) и 10% Pd-C (180 мг) и смесь нагревали при 70°C в течение ночи в атмосфере водорода. Смесь концентрировали и подвергали хроматографии на силикагеле, элюируя смесью EtOAc/гексан, с получением 360 мг неочищенного вещества. Половину этого вещества подвергали дополнительной очистке при помощи препаративной ВЭЖХ (колонка Varian дифенил с обращенной фазой, элюировали с использованием градиента растворителя B=0,05% HOAc/MeOH и растворителя А=0,05% HOAc/H2O) с получением 3-(4-фторфенил)-3-(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)пропаннитрила (65 мг, 24%). 1H ЯМР (300 МГц, DMSO-d6) δ 2,25 (с, 3H) 3,24-3,48 (м, 2H) 4,57 (м, 1H) 6,36 (с, 1H) 7,17 (м, 2H) 7,46-7,55 (м, 3H) 7,77-7,81 (м, 2H) 8,60 (д, 1H) 10,41 (с, 1H) 12,13 (с, 1H); LC-MS (ESI) m/z 373 (M+H)+.
Пример 69
Получение 2-((циклопропиламино)(4-фторфенил)метил)-N-(5-метил-1Н-пиразол-3-ил)хиназолин-4-амина
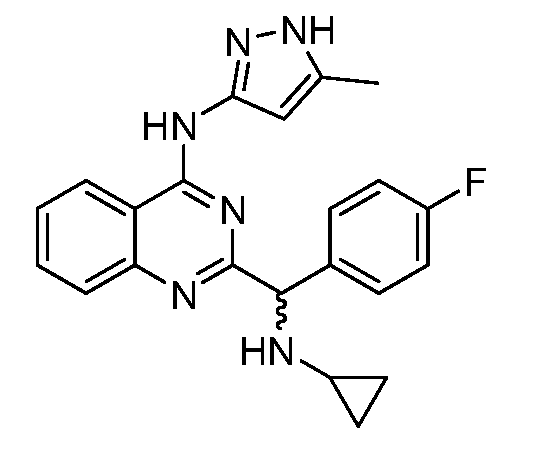
К (4-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанону из Примера 3 (100 мг, 0,28 ммоль) в 2-пропаноле (3 мл) добавляли циклопропиламин (0,1 мл) и 3 Ǻ (8-12 меш) молекулярные сита и смесь нагревали при 140°C в микроволновом реакторе Biotage. Добавляли дополнительное количество циклопропиламина (0,15 мл) и смесь в герметично закрытом флаконе нагревали обычным способом при 90°C в течение 4 дней. Затем добавляли по каплям суспензию борогидрида натрия (140 мг) в 2-пропаноле и смесь перемешивали при комнатной температуре в течение 1 часа. Затем добавляли MeOH (0,1 мл), борогидрид натрия (100 мг) и снова MeOH (2 мл) и смесь фильтровали и фильтрат концентрировали. К остатку добавляли ТГФ (5 мл), MeOH (2 мл) и борогидрид натрия (100 мг) и смесь перемешивали в течение 1 часа при комнатной температуре. Затем добавляли AcOH (0,4 мл) и смесь концентрировали. Добавляли DMSO (3 мл) и смесь очищали при помощи препаративной ВЭЖХ (колонка Varian дифенил с обращенной фазой, элюировали с использованием градиента растворителя B=0,05% HOAc/MeOH и растворителя А=0,05% HOAc/H2O) с последующей дополнительной очисткой при помощи препаративной ВЭЖХ (колонка Phenomonex C-18 с обращенной фазой, элюировали с использованием градиента растворителя B=0,05% HOAc/MeOH и растворителя А=0,05% HOAc/H2O) с получением 2-((циклопропиламино)(4-фторфенил)метил)-N-(5-метил-1Н-пиразол-3-ил)хиназолин-4-амина (5,5 мг, 9%). 1H ЯМР (300 МГц, DMSO-d6) δ 0,34 (м, 4H) 1,24 (м, 1H) 2,03 (м, 1H) 2,26 (с, 3H) 4,91 (с, 1H) 6,41 (м, 1H) 7,11 (т, 2H) 7,4-7,6 (м, 3H) 7,76 (м, 2H) 8,56 (д, 1H) 10,38 (ушир.с, 1H) 12,15 (ушир.с, 1H); LC-MS (ESI) m/z 389 (M+H)+.
Пример 70
Получение 2-(1-(4-фторфенил)-2-(метилсульфонил)этил)-N-(5-метил-1Н-пиразол-3-ил)хиназолин-4-амина
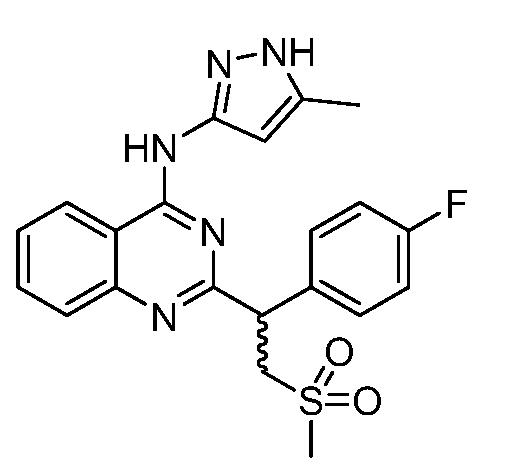
Стадия A: К 3-хлорбензопероксокислоте (77%, 11,21 г, 50 ммоль) в DCM (150 мл) добавляли диэтил метилтиометилфосфонат (4,4 мл, 25 ммоль) и смесь оставляли для перемешивания при комнатной температуре в течение ночи. Затем добавляли дополнительное количество 3-хлорбензопероксокислоты (5,6 г) и перемешивание продолжали в течение 4 часов при комнатной температуре. Раствор промывали насыщенным водным раствором карбоната калия и концентрировали. Остаток растворяли в DCM и промывали снова насыщенным раствором карбоната калия. Органический слой концентрировали с получением диэтил метилсульфонилметилфосфоната (4,51 г, 39%). 1H ЯМР (300 МГц, DMSO-d6) δ 1,25 (т, 6H) 3,13 (с, 3H) 4,09 (м, 4H) 4,20 (д, 2H); LC-MS (ESI) m/z 231 (M+H)+.
Стадия B: К диэтилметилсульфонилметил-фосфонату (746 мг, 3,24 ммоль) в ТГФ (20 мл) при 0°C добавляли трет-бутоксид калия (1,0 M в ТГФ, 3,25 мл, 3,25 ммоль) и смесь перемешивали в течение 5 минут. Затем добавляли (4-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанон из Примера 3 (375 мг, 1,08 ммоль) и смесь перемешивали при комнатной температуре в течение 4 часов. Смесь обрабатывали 1 н. раствором HCl и полученную смесь экстрагировали при помощи EtOAc. Органический слой промывали насыщенным солевым раствором и затем концентрировали с получением остатка (700 мг), который подвергали хроматографии на силикагеле, элюируя при помощи 1-10% MeOH/DCM, с получением неочищенного вещества. К части этого вещества (166 мг) в EtOAc (5 мл) добавляли 10% Pd-C (166 мг) и смесь перемешивали в атмосфере водорода при комнатной температуре в течение ночи. Смесь фильтровали и к фильтрату добавляли палладий на углероде (10%). Смесь перемешивали в атмосфере водорода в течение нескольких часов и затем фильтровали. Фильтрат концентрировали с получением твердого вещества, которое растирали в порошок с простым эфиром, с получением 2-(1-(4-фторфенил)-2-(метилсульфонил)этил)-N-(5-метил-1Н-пиразол-3-ил)хиназолин-4-амина (22 мг). 1H ЯМР (300 МГц, DMSO-d6) δ 2,28 (с, 3H) 2,87 (с, 3H) 3,85 (м, 1H) 4,34 (м, 1H) 4,68 (ушир.с, 1H) 6,45 (с, 1H) 7,15 (т, 2H) 7,49 (м, 3H) 7,76 (м, 2H) 8,56 (д, 1H) 10,36 (с, 1H) 12,17 (с, 1H); LC-MS (ESI) m/z 426 (M+H)+.
Пример 71
Получение 2-(3-амино-1-(4-фторфенил)пропил)-N-(5-метил-1Н-пиразол-3-ил)хиназолин-4-амина
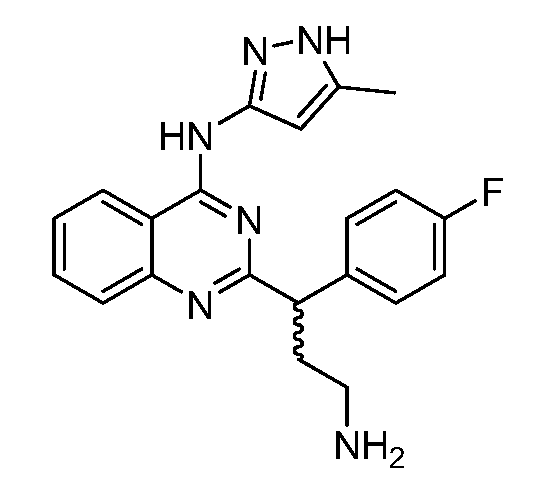
К 3-(4-фторфенил)-3-(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)пропаннитрилу из Примера 68 (80 мг, 0,21 ммоль) в ТГФ (3 мл) при 0°C добавляли литийалюминийгидрид (20 мг) и смесь перемешивали в течение 5 минут затем давали нагреться до комнатной температуры и перемешивали в течение 2-3 часов. Затем добавляли дополнительное количество литийалюминийгидрида (40 мг) и перемешивание продолжали в течение 45 минут. К раствору медленно добавляли 1 н. раствор NaOH (0,5 мл) с последующим добавлением MeOH (2 мл). Смесь перемешивали в течение 5 минут и затем добавляли AcOH (0,5 мл) с последующим добавлением DMSO (2 мл). Полученный раствор очищали при помощи препаративной ВЭЖХ (колонка Varian Дифенил с обращенной фазой, элюировали с использованием градиента растворителя B=0,05% HOAc/ACN и растворителя А=0,05% HOAc/H2O) с получением 2-(3-амино-1-(4-фторфенил)пропил)-N-(5-метил-1Н-пиразол-3-ил)хиназолин-4-амина в виде ацетатной соли (21 мг, 25%). 1H ЯМР (300 МГц, DMSO-d6) δ 1,81 (с, 3H) 2,2-2,6 (м, 7H) 4,26 (т, 1H) 6,46 (с, 1H) 7,12 (т, 2H) 7,46-7,51 (м, 3H) 7,73-7,78 (м, 2H) 8,57 (д, 1H); LC-MS (ESI) m/z 377 (M+H)+.
Пример 72
Получение (R,S)(4-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанола-1-d
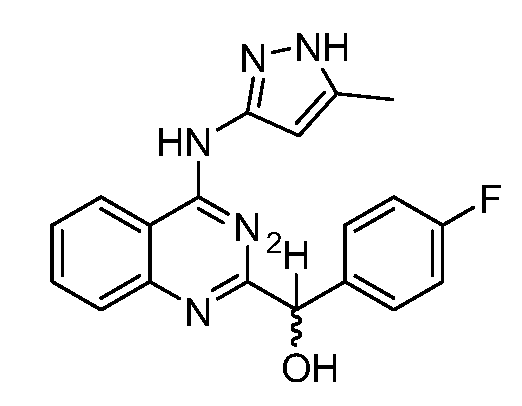
К (4-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанону из Примера 3 (200 мг, 0,57 ммоль) в ТГФ (5 мл) и MeOH-d4 (2 мл) при 0°C добавляли бородейтерид натрия (50 мг, 98%). Раствор перемешивали при 0°C в течение 1 часа и затем давали нагреться до комнатной температуры в течение 30 минут. Затем добавляли 2 н. раствор HCl (0,4 мл) и раствор концентрировали. Остаток очищали при помощи препаративной ВЭЖХ (колонка Varian дифенил с обращенной фазой, элюировали с использованием градиента растворителя B=0,05% HOAc/ACN и растворителя А=0,05% HOAc/H2O) с получением (R,S)(4-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанола-1-d (60 мг, 30%). 1H ЯМР (300 МГц, DMSO-d6) δ 2,25 (с, 3H) 5,80 (с, 1H) 6,41 (с, 1H) 7,14 (т, 2H) 7,53-7,55 (м, 3H) 7,79 (м, 2H) 8,58 (д, 1H) 10,41 (ушир.с, 1H) 12,15 (ушир.с, 1H); LC-MS (ESI) m/z 351 (M+H)+.
Пример 73
Получение (4-фторфенил)(4-(5-метокси-1Н-пиразол-3-иламино)хиназолин-2-ил)метанона
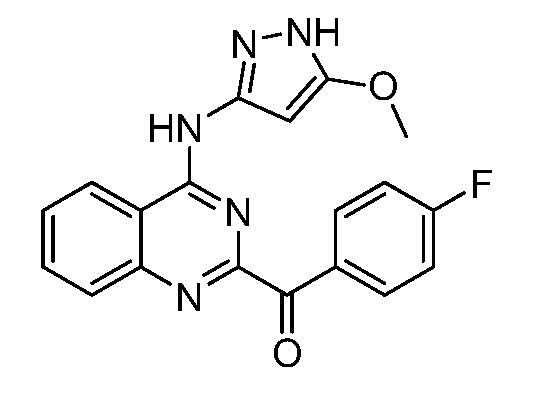
Стадия A: К 3,5-динитро-1Н-пиразолу (517 мг, 3,27 ммоль) в DMF (15 мл) добавляли карбонат калия (903 мг, 6,54 ммоль) и (2-(хлорметокси)этил)триметилсилан (0,64 мл, 3,59 ммоль) и смесь перемешивали при комнатной температуре в течение 2 часов. Смесь концентрировали и остаток очищали хроматографией на силикагеле, элюируя смесью 0-20% этилацетат/гексан, с получением масла (840 мг). К полученному маслу в MeOH (20 мл) добавляли 25% раствор NaOMe/MeOH (1,25 мл, 5,83 ммоль) и смесь нагревали до 60°C в течение 3-4 часов. Затем добавляли AcOH (0,3 мл) и смесь очищали при помощи хроматографии на силикагеле (этилацетат/гексан 0-30%) с получением единственного соединения (3-метокси-5-нитро-1-((2-триметилсилил)этокси)метил)-1H-пиразола или 5-метокси-3-нитро-1-((2-(триметилсилил)этокси)метил)-1Н-пиразол)(610 мг, 68%). 1H ЯМР (300 МГц, CDCl3) δ 0,00 (с, 9H) 0,94 (т, 2H) 3,67 (т, 2H) 4,02 (с, 3H) 5,41 (с, 2H), 6,23 (с, 1H); LC-MS (ESI) m/z 296 (M+H)+.
Стадия B: К продукту Примера 73 Стадии А (150 мг, 0,55 ммоль) в EtOH (10 мл) добавляли палладий на углероде (10%, 30 мг) и смесь перемешивали в атмосфере водорода в течение 1-2 часов. Смесь фильтровали и фильтрат концентрировали до получения масла. К этому маслу в DMF (4 мл) добавляли DIEA (0,96 мл), иодид калия (65 мг) и (4-хлорхиназолин-2-ил)(4-фторфенил)метанон из Примера 3 Стадия А (112 мг, 0,39 ммоль). Смесь нагревали при 65°C в течение ночи и затем разбавляли при помощи EtOAc и промывали водой (3×), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток растворяли в ТГФ (5 мл) и часть (3 мл) концентрировали, обрабатывали при помощи TFA (3 мл) при комнатной температуре в течение 45 минут. Смесь концентрировали и затем добавляли MeOH и упаривали. Остаток растворяли в DMSO и снова объединяли со второй частью образца, которая была обработана аналогичным образом. Смесь очищали при помощи препаративной ВЭЖХ (колонка Varian дифенил с обращенной фазой, элюировали с использованием градиента растворителя B=0,05% HOAC/ACN и растворителя А=0,05% HOAc/H2O) с получением (4-фторфенил)(4-(5-метокси-1Н-пиразол-3-иламино)хиназолин-2-ил)метанона (12 мг). 1H ЯМР (300 МГц, DMSO-d6) δ 3,73 (с, 3H) 5,78 (ушир.с, 1H) 7,40 (т, 2H) 7,78 (м, 1H) 7,94 (м, 2H) 8,12 (м, 2H) 8,59 (ушир.с, 1H) 10,88 (ушир.с, 1H) 11,77 (ушир.с, 1H); LC-MS (ESI) m/z 364 (M+H)+.
Пример 74
Получение (R,S)-(4-(5-этил-1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанола
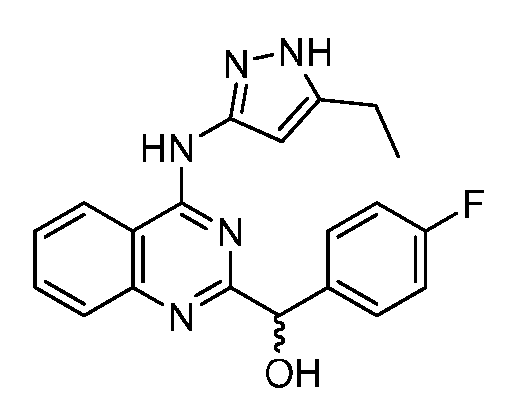
Стадия A: К смеси 5-этил-1Н-пиразол-3-амина (101 мг, 0,91 ммоль), иодида калия (117 мг, 0,7 ммоль) и DIEA (0,15 мл, 0,84 ммоль) в DMF (4 мл) добавляли (4-хлорхиназолин-2-ил)(4-фторфенил)метанон из Примера 3 Стадия А (200 мг, 0,7 ммоль) и смесь перемешивали при комнатной температуре в течение ночи. Добавляли воду и осажденное твердое вещество собирали фильтрованием. Желтое твердое вещество (226 мг), содержащее (4-(5-этил-1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанон, использовали непосредственно на следующей стадии. LC-MS (ESI) m/z 362 (M+H)+.
Стадия B: К неочищенному (4-(5-этил-1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанону (104 мг, 0,28 ммоль) в смеси 1:1 MeOH/ТГФ (4 мл) при комнатной температуре добавляли борогидрид натрия (22 мг, 0,57 ммоль) и раствор перемешивали в течение 30 минут, затем добавляли 4 н. раствор HCl (0,1 мл). Смесь концентрировали досуха и остаток очищали при помощи препаративной ВЭЖХ (колонка Varian дифенил с обращенной фазой, элюировали с использованием градиента растворителя B=0,05% HOAc/ACN и растворителя А=0,05% HOAc/H2O) с получением (4-(5-этил-1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанола (7 мг). 1H ЯМР (300 МГц, DMSO-d6) δ 1,24 (т, 3H) 2,62 (кв., 2H) 5,67 (м, 1H) 5,83 (с, 1H) 6,45 (с, 1H) 7,13 (т, 2H) 7,53-7,57 (м, 3H) 7,81 (с, 2H) 8,59 (д, 1H) 10,42 (ушир.с, 1H) 12,15 (ушир.с, 1H); LC-MS (ESI) m/z 364 (M+H)+.
Пример 75
Получение (4-Фторфенил)(4-(5-метокси-1Н-пиразол-3-иламино)хиназолин-2-ил)метанола
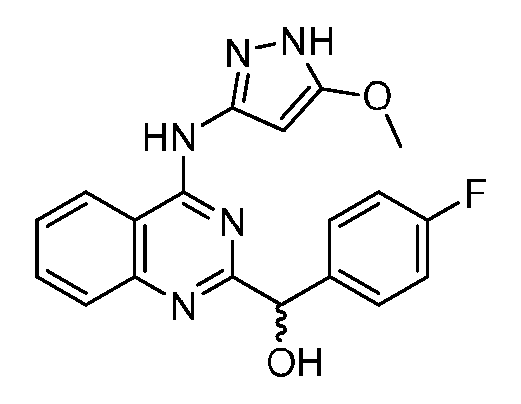
(4-Фторфенил)(4-(5-метокси-1Н-пиразол-3-иламино)хиназолин-2-ил)метанол получали с использованием процедуры, аналогичной описанной в Примере 6, используя (4-фторфенил)(4-(5-метокси-1Н-пиразол-3-иламино)хиназолин-2-ил)метанон из Примера 75 вместо (4-(1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанона, используемого в Примере 6.
Пример 76
Получение (4-фтор-3-метоксифенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанона
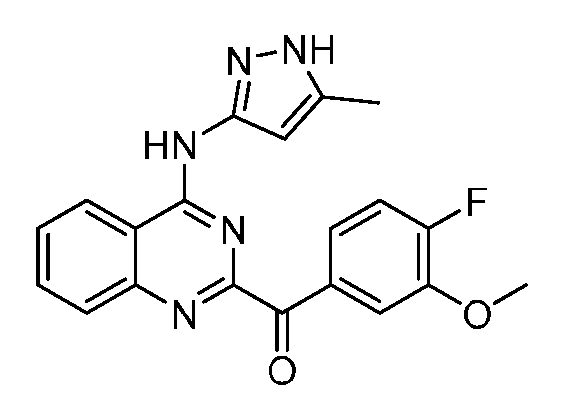
Стадия A: К перемешиваемой смеси этил 4-хлорхиназолин-2-карбоксилата (500 мг, 2,11 ммоль) в ТГФ (17 мл) при -40°C добавляли по каплям 1 M раствор 4-фтор-3-метоксифенилмагнийбромид/2-метилтетрагидрофуран (2,53 мл, 2,53 ммоль). Реакционную смесь перемешивали при температуре в пределах от - 30 до - 40°C в течение 3 часов. Добавляли дополнительное количество 1 M раствора 4-фтор-3-метоксифенилмагнийбромид/2-метилтетрагидрофуран (1,05 мл, 1,05 ммоль) и перемешивание продолжали при температуре в пределах от - 30 до - 40°C еще в течение 1,5 часа. К смеси добавляли насыщенный водный раствор хлорида аммония (25 мл) и смеси давали нагреться до комнатной температуры. Смесь экстрагировали при помощи EtOAc (2×) и объединенные органические слои сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении. Остаток растирали в порошок с диэтиловым эфиром и полученное твердое вещество собирали фильтрованием и сушили с получением (4-хлорхиназолин-2-ил)(4-фтор-3-метоксифенил)метанона в виде бесцветного твердого вещества (417 мг, 62%). 1H ЯМР (300 МГц, DMSO-d6) δ м.д. 3,92 (с, 3H), 7,42 (дд, J=11,1, 8,7 Гц, 1H), 7,64 (м, 1H), 7,85 (д, J=8,4 Гц, 1H), 8,04 (м, 1H), 8,23-8,25 (м, 2H), 8,43 (д, J=8,4 Гц, 1H); LC-MS (ESI) m/z 317 (M+H)+.
Стадия B: Смесь (4-хлорхиназолин-2-ил)(4-фтор-3-метоксифенил)метанона (337 мг, 1,06 ммоль), 5-метил-1Н-пиразол-3-амина (206 мг, 2,12 ммоль), иодида калия (528 мг, 3,18 ммоль) и DIEA (0,37 мл, 2,13 ммоль) в DMF (10 мл) перемешивали при комнатной температуре в течение 20 часов. К смеси добавляли воду (80 мл) и полученное твердое вещество собирали фильтрованием и промывали водой и затем диэтиловым эфиром. Твердое вещество сушили с получением (4-фтор-3-метоксифенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанона в виде желтого твердого вещества (300 мг, 75%). 1H ЯМР (300 МГц, DMSO-d6) δ м.д. 2,19 (с, 3H), 3,90 (с, 3H), 6,55 (с, 1H), 7,38 (дд, J=11,1, 8,4 Гц, 1H), 7,59 (м, 1H), 7,68 (дд, 7=7,8, 7,8 Гц, 1H), 7,77-7,94 (м, 3H), 8,75 (д, J=7,8 Гц, 1H), 10,69 (ушир.с, 1H), 12,23 (ушир.с, 1H); LC-MS (ESI) m/z 378 (M+H)+.
Пример 77
Получение (4-фтор-3-гидроксифенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанона
К перемешиваемой суспензии (4-фтор-3-метоксифенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанона из Примера 76 (100 мг, 0,265 ммоль) в DCM (4 мл) при 0°C добавляли по каплям 1,0 M раствор трибромид бора/DCM (2,12 мл, 2,12 ммоль). Смесь перемешивали при 0°C в течение 30 минут. Добавляли дополнительное количество 1,0 M раствора трибромид бора/DCM (1,50 мл, 1,50 ммоль) и смеси давали нагреться до комнатной температуры и перемешивали еще в течение 2,5 часов. Добавляли воду (10 мл) и смесь экстрагировали при помощи 25% раствора 2-пропанола/DCM (2×). Объединенные органические слои сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении. Остаток растирали в порошок с диэтиловым эфиром и полученное твердое вещество собирали фильтрованием и сушили. Твердое вещество подвергали дополнительной очистке при помощи обращенно-фазовой ВЭЖХ, элюируя смесью воды и ацетонитрила, с получением (4-фтор-3-гидроксифенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанона в виде желтого твердого вещества (20 мг, 21%). 1H ЯМР (300 МГц, DMSO-d6) δ м.д. 2,19 (с, 3H), 6,51 (с, 1H), 7,28 (дд, J=10,8, 8,4 Гц, 1H), 7,39 (м, 1H), 7,58 (дд, J=8,7, 1,8 Гц, 1H), 7,67 (ддд, J=8,1, 8,1, 1,5 Гц, 1H), 7,83-7,92 (м, 2H), 8,73 (д, J=8,4 Гц, 1H), 10,65 (ушир.с, 1H), 12,22 (ушир.с, 1H); LC-MS (ESI) m/z 364 (M+H)+.
Пример 78
Получение (R,S)-(2-фтор-5-(гидрокси(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метил)фенолацетата
К перемешиваемому раствору (4-фтор-3-гидроксифенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанона из Примера 77 (20 мг, 0,055 ммоль) в смеси ТГФ (0,3 мл) и MeOH (0,3 мл) при 0°C добавляли борогидрид натрия (3 мг, 0,079 ммоль) и смесь перемешивали при 0°C в течение 15 минут. К смеси добавляли концентрированный раствор хлористоводородной кислоты до достижения pH 1. Эту смесь объединяли со смесью, полученный аналогичным образом, исходя из (4-фтор-3-гидроксифенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанона (60 мг, 0,165 ммоль). Полученную смесь очищали при помощи обращенно-фазовой ВЭЖХ, элюируя смесью вода в ацетонитриле, с получением (R,S)-(2-фтор-5-(гидрокси(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метил)фенолацетата в виде твердого вещества (26 мг, 28%). 1H ЯМР (300 МГц, DMSO-d6) δ м.д. 1,89 (с, 3H), 2,25 (с, 3H), 5,55 (с, 1H), 5,75 (ушир.с, 1H), 6,43 (ушир.с, 1H), 6,92 (м, 1H), 7,02-7,11 (м, 2H), 7,52 (м, 1H), 7,70-7,79 (ушир.м, 2H), 8,58 (м, 1H), 10,44 (ушир.с, 1H), 12,10 (ушир.с, 1H); LC-MS (ESI) m/z 366 (M+H)+.
Пример 79
Анализ конкурентного связывания для определения констант связывания (K d ) соединений против JAK киназ
Анализы конкурентного связывания, которые использовали, были разработаны, утверждены и осуществлены, как описано в Fabian et al., Nature Biotechnology 2005, 23,329-336. Киназы были получены в виде слияний с T7 фагом (См., Fabian et al. или WO04/015142), или, альтернативно, киназы экспрессировали в HEK-293 клетках и затем метили при помощи ДНК для ПЦР-детекции (См., WO08/005310). Для анализов связывания, покрытые стрептавидином магнитные шарики обрабатывали биотинилированными аффинными лигандами в течение 30 минут при комнатной температуре для образования аффинных смол. Обработанные лигандами шарики блокировали избыточным количеством биотина и промывали блокирующим буфером (SeaBlock (Pierce), 1% BSA, 0,05% Tween 20, 1 мМ DTT) для удаления несвязанного лиганда и для уменьшения неспецифического связывания. Реакции связывания объединяли путем объединения киназы, связанных с лигандом аффинных шариков и испытываемых соединений в 1× буфере для связывания (20% SeaBlock, 0,17× PBS, 0,05% Tween 20, 6 мМ DTT). Испытываемые соединения подготавливали в виде 100× исходных растворов в DMSO и быстро разбавляли в водной среде. DMSO добавляли для контрольных анализов без испытываемого соединения. Взаимодействия для первичного скрининга осуществляли в полипропиленовых 384-луночных планшетах в конечном объеме 34 мкл, а определения Kd осуществляли в полистирольных 96-луночных планшетах в конечном объеме 135 мкл. Аналитические планшеты инкубировали при комнатной температуре при встряхивании в течение 1 часа, этого времени было достаточно для того, чтобы реакции связывания достигли равновесия, и аффинные шарики тщательно промывали промывочным буфером (1× PBS, 0,05% Tween 20) для удаления несвязанного белка. Шарики затем ресуспендировали в элюирующем буфере (1× PBS, 0,05% Tween 20, 2 мкМ небиотинилированного аффинного лиганда) и инкубировали при комнатной температуре при встряхивании в течение 30 минут. Концентрацию киназы в элюатах измеряли при помощи количественного ПЦР анализа. Каждую киназу испытывали индивидуально против каждого соединения. Kd определяли с использованием одиннадцати серийных трехкратных разведений. Показатель селективности, который представляет собой количественное измерение селективности соединения против ряда ферментов, можно рассчитать для соединения путем деления количества ферментов, в отношении которых соединение удовлетворяет установленным критериям (например, константа связывания 100 нМ или меньше), на общее количество испытываемых ферментов. Показатель селективности в отношении киназы, S10, например, расчитывают для каждого соединения путем деления количества киназ, в отношении которых соединение при определенной концентрации (например, 10 мкМ) демонстрирует ингибирование 90% или больше по сравнению с отрицательным контролем без ингибиторов (только DMSO), на 321, т.е. количество отдельных испытываемых киназ, за исключением мутантных вариантов.
В одном варианте воплощения соединения, обеспечиваемые в настоящем изобретении, как было обнаружено, имеют Kd меньше чем около 20 мкМ против JAK2. В другом варианте воплощения соединения, обеспечиваемые в настоящем изобретении, как было обнаружено, имеют Kd меньше чем около 10 мкМ против JAK2. В другом варианте воплощения соединения, обеспечиваемые в настоящем изобретении, как было обнаружено, имеют Kd меньше чем около 1 мкМ против JAK2.
В другом варианте воплощения соединения, обеспечиваемые в настоящем изобретении, как было обнаружено, имеют Kd меньше чем около 20 мкМ против JAK3. В другом варианте воплощения соединения, обеспечиваемые в настоящем изобретении, как было обнаружено, имеют Kd меньше чем около 10 мкМ против JAK3. В другом варианте воплощения соединения, обеспечиваемые в настоящем изобретении, как было обнаружено, имеют Kd меньше чем около 1 мкМ против JAK3.
Пример 80
Анализ репортера на основе клеток csTF-1
csTF-1 клетки выделяли из человеческой эритролейкозной клеточной линии, рост которой является зависимым от GM-CSF и которая имеет интактный GM-CSFR/JAK2/STAT5 путь. Клеточная линия содержит стабильно интегрированный репортерный ген бета-лактамазы под контролем респонсивного элемента регуляторного фактора 1 (irf 1), распознаваемого активированным транскрипционным фактором STAT5. csTF-1 клетки (Invitrogen K1219) промывали средой для анализа (97% OPTIMEM/0,5% диализованного FBS/0,1 мМ NEAA/1 мМ Na pyr/ P/S) и высевали в эту же среду при плотности 5×105 клеток/мл в T150 колбе. После 16 часов инкубации клетки высевали при плотности 2×105 клеток/лунка в объеме 50 мкл 96-луночные аналитические планшеты Costar с прозрачным дном. Серийные разведения соединений добавляли в планшеты при конечной концентраци DMSO 0,5% и GM-CSF 2 нг/мл и планшеты затем инкубировали при 30°C и 5% CO2 в течение 4 часов. Планшеты доводили до комнатной температуры перед добавлением Смеси Субстрата, в соответствии с протоколом изготовителя (Invitrogen, Catalog # K1085). Аналитические планшеты, содержащие субстратную смесь, инкубировали в темноте при комнатной температуре в течение 2 часов. Синюю и зеленую флуоресценцию измеряли при длине волны возбуждения при 409 нм и эмиссии при 460 нм (для синей) и при длине волны возбуждения при 409 нм и эмиссии при 530 нм (для зеленой) с использованием устройства Spectra Max Gemini EM. Соединения, обеспечиваемые в настоящем изобретении, как было обнаружено, имеют ИК50 меньше чем около 5 мкМ. В другом варианте воплощения соединения, обеспечиваемые в настоящем изобретении, как было обнаружено, обладают активностью при ИК50 меньше чем около 500 нМ.
Соединения, обеспечиваемые в настоящем изобретении, как было обнаружено, имеют следующую активность, показанную Таблице 1:
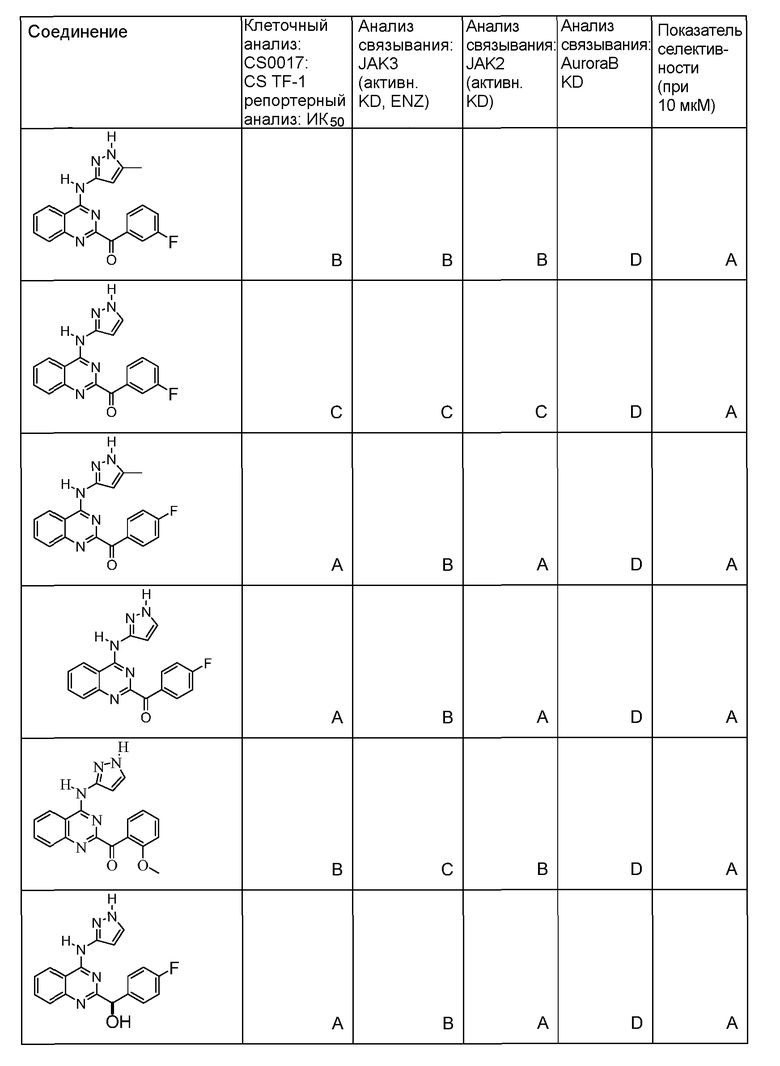
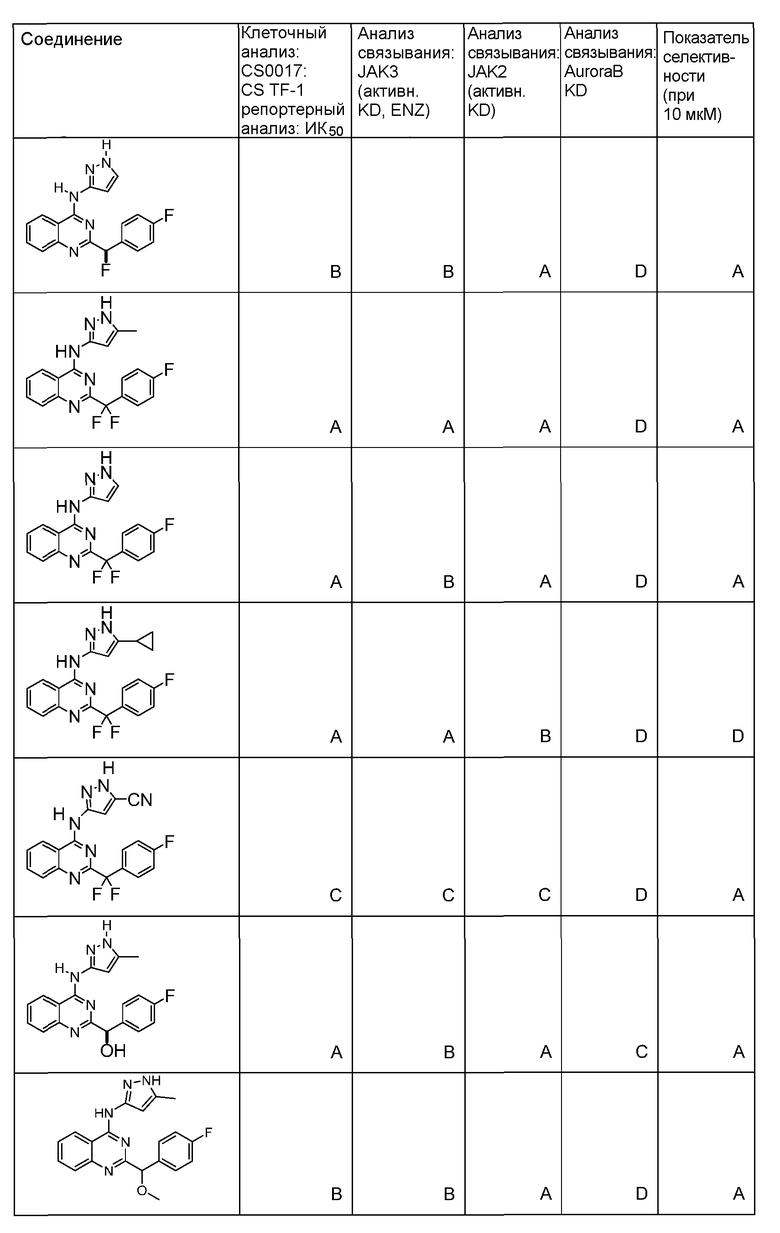
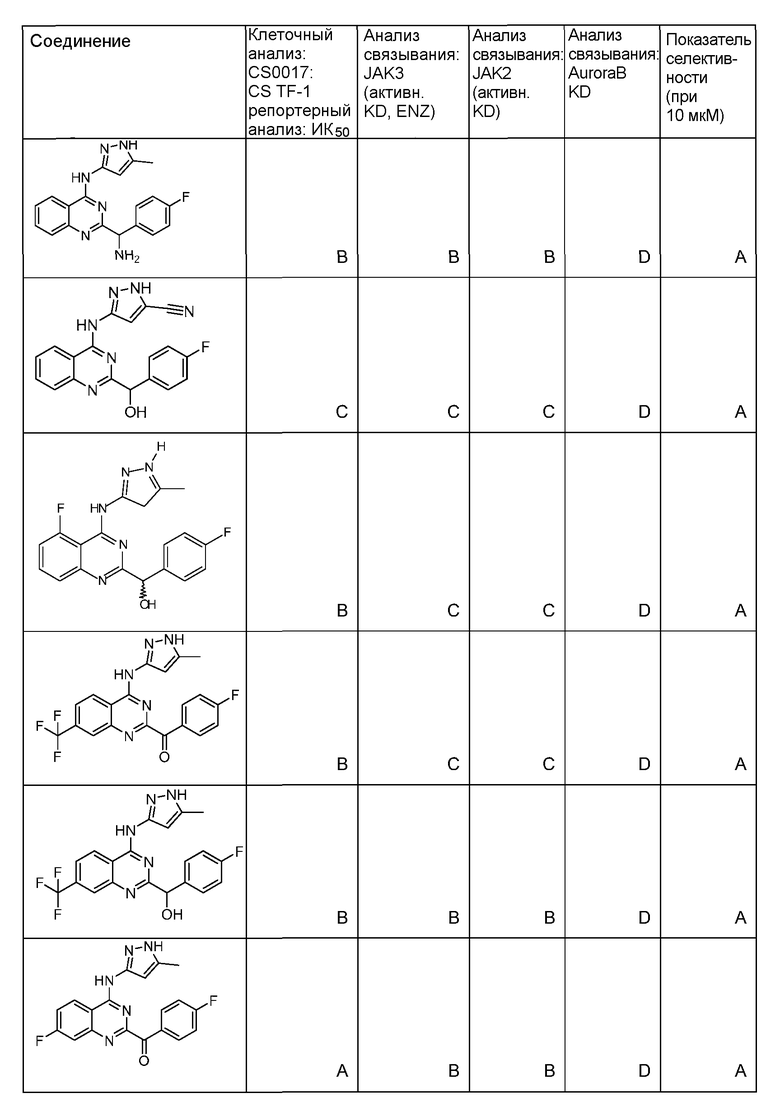
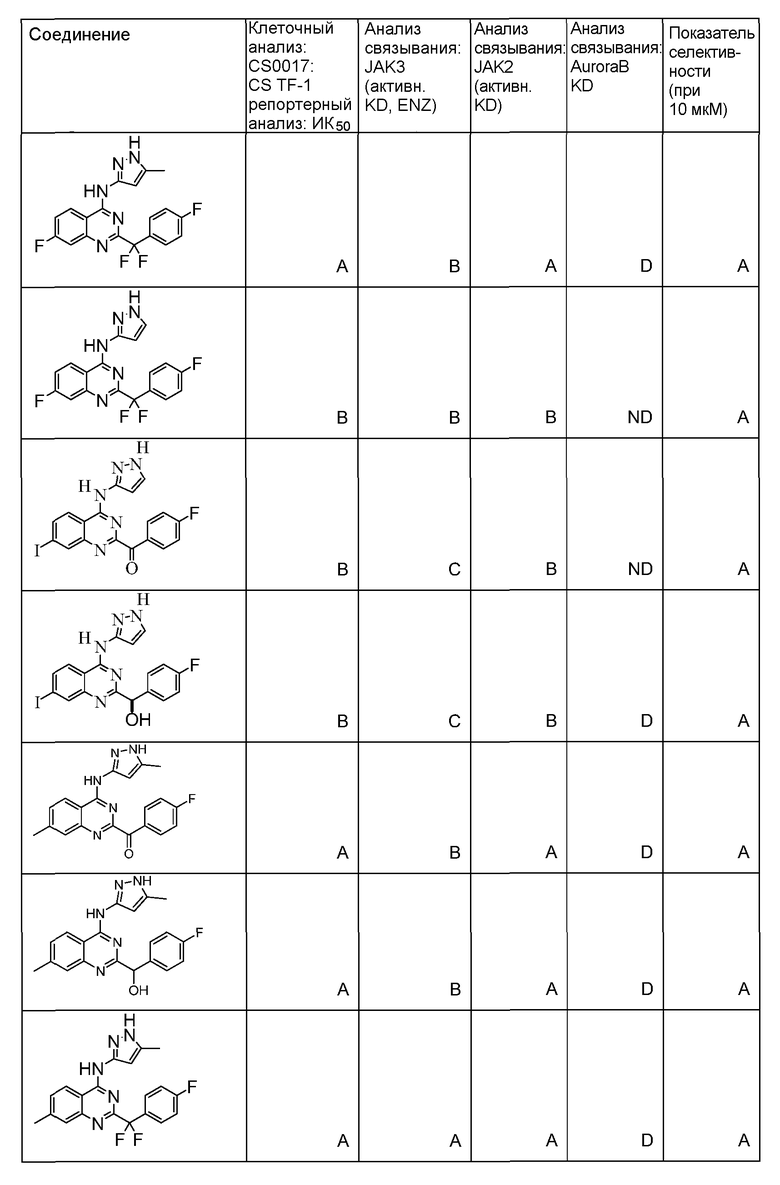
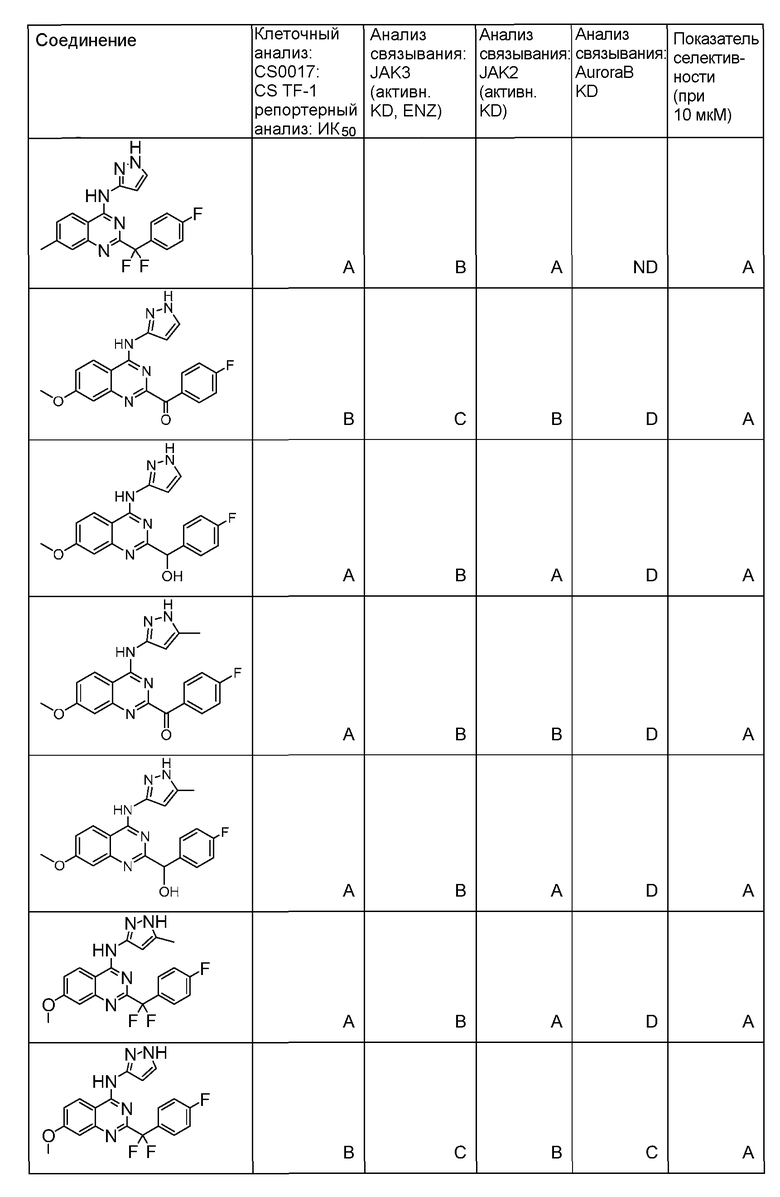
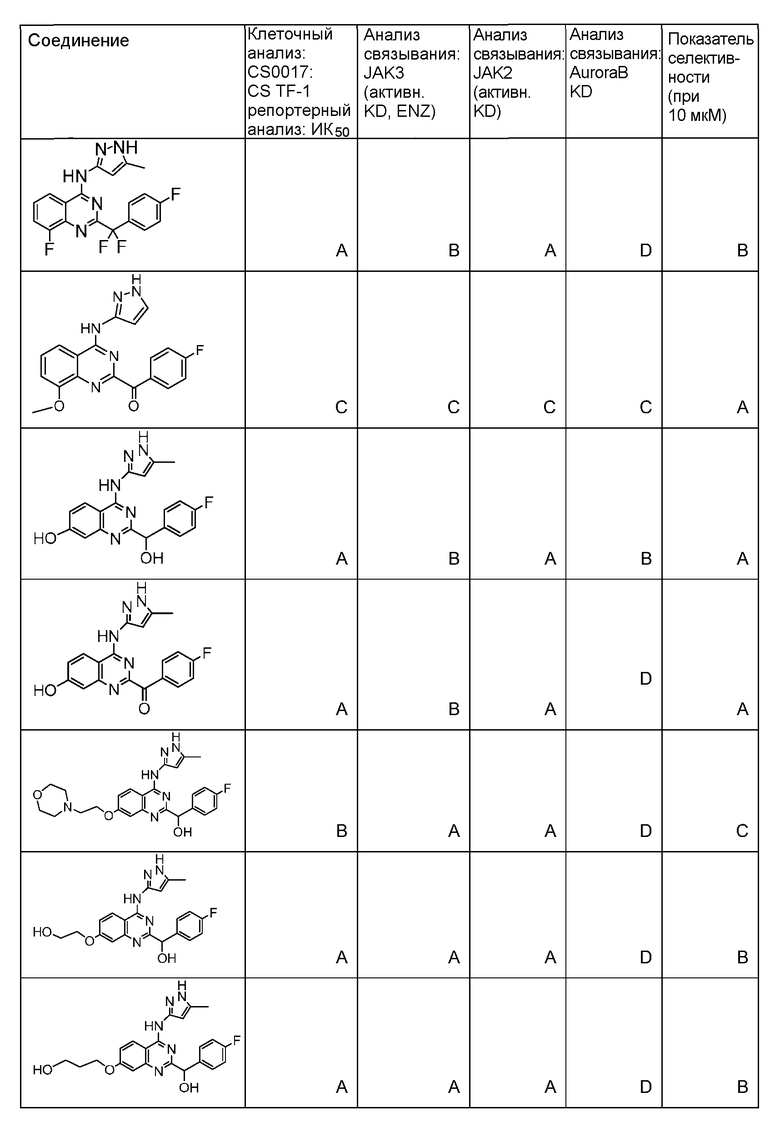
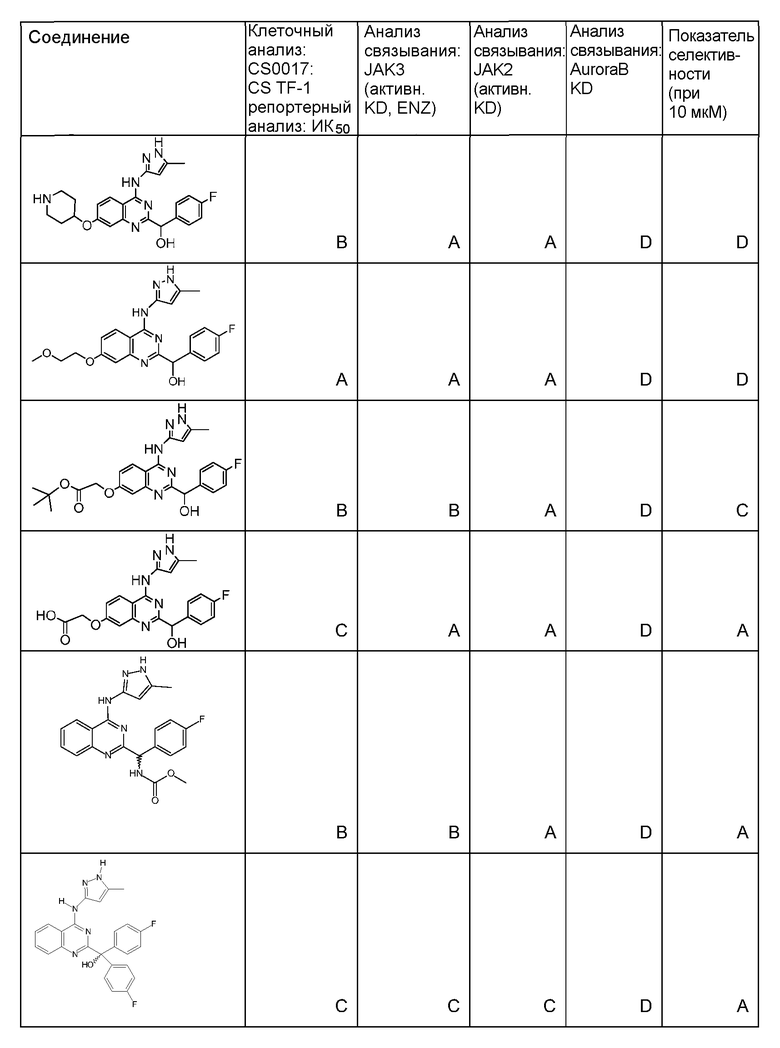
Следующие иллюстративные соединения, обеспечиваемые в настоящем изобретении, как было обнаружено, имеют следующую активность, показанную Таблице 2:
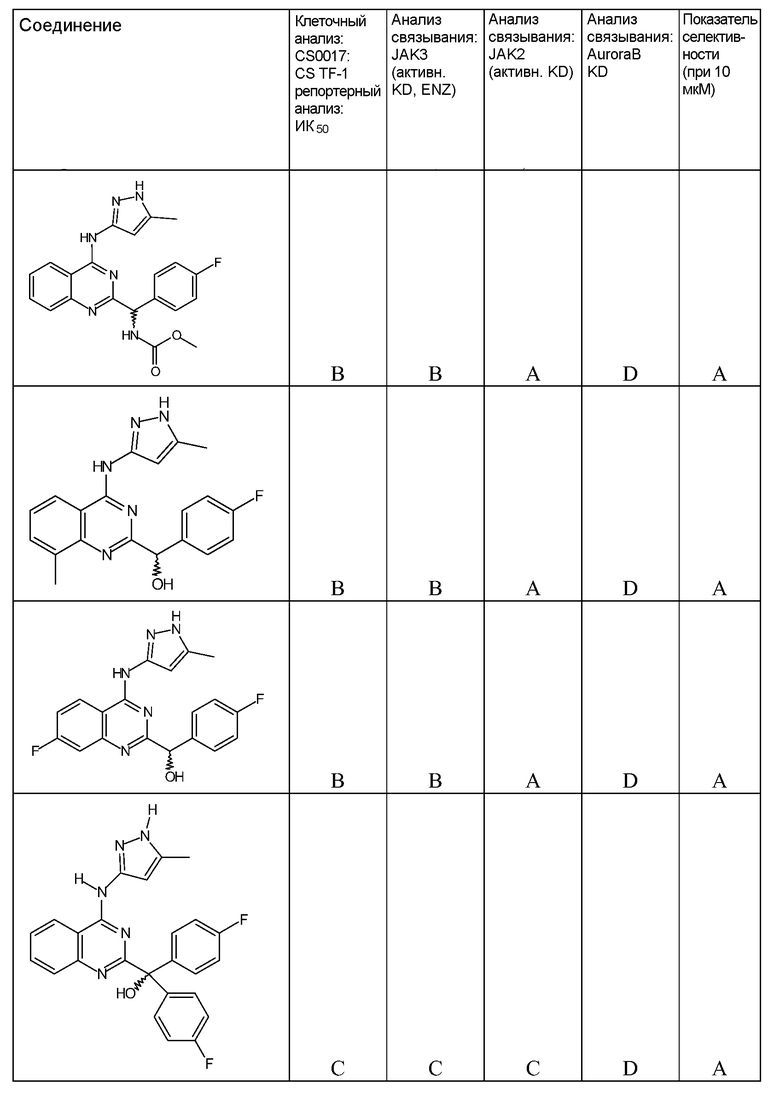
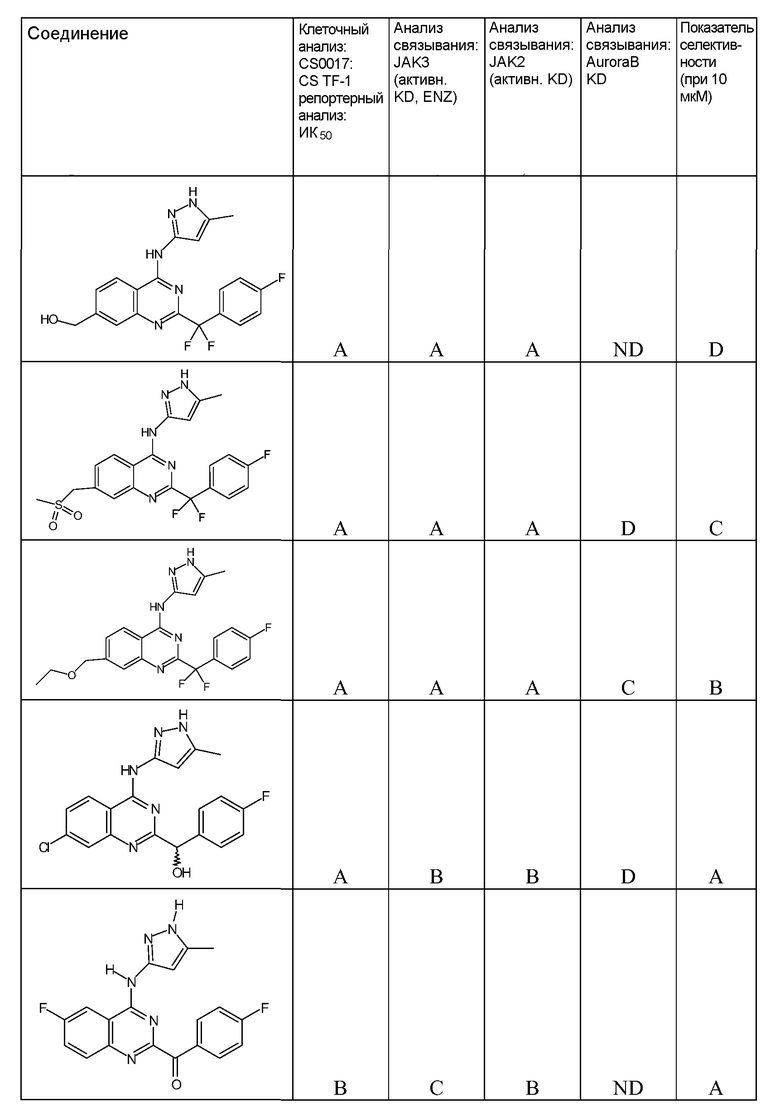
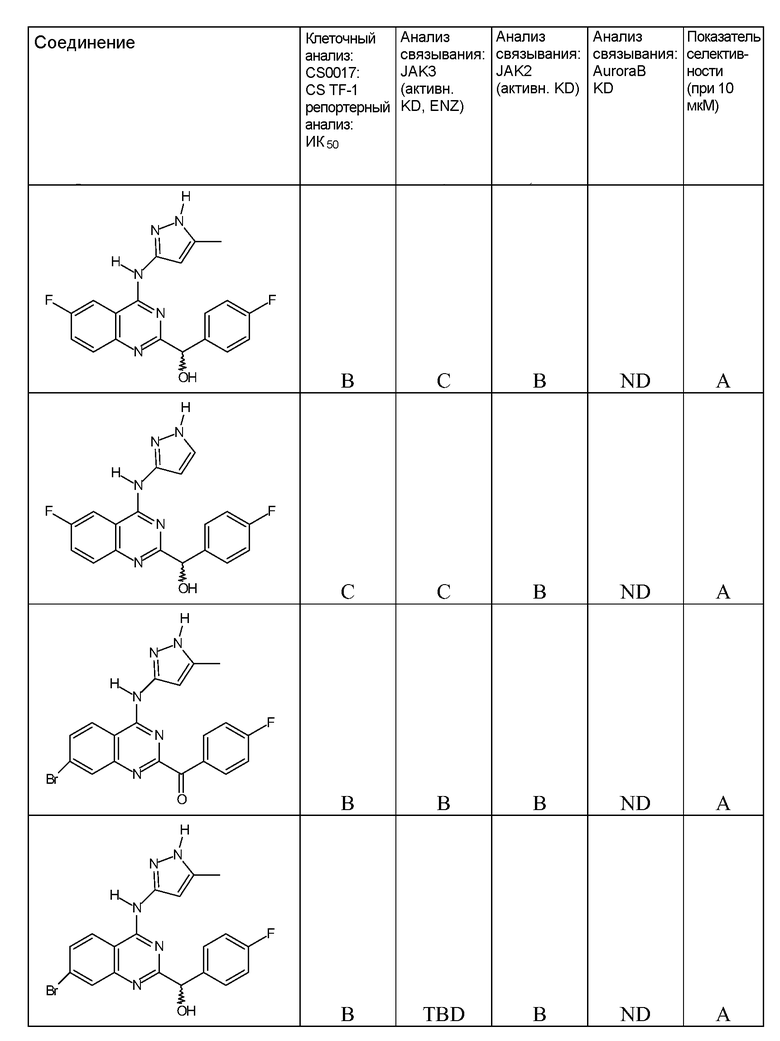
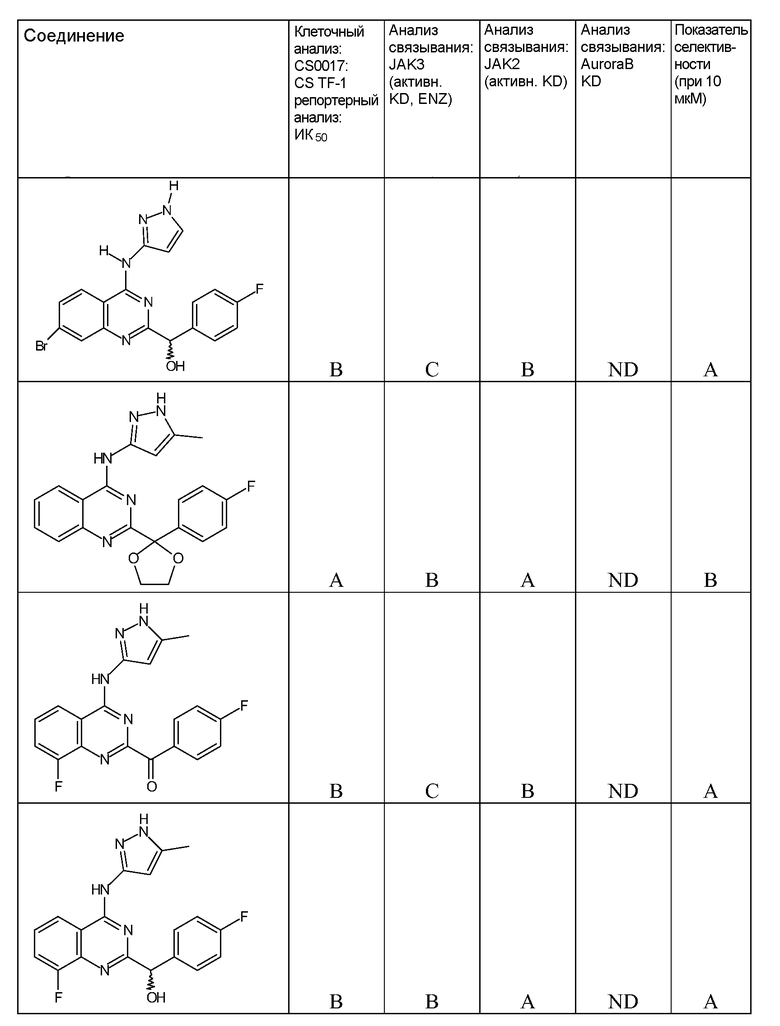
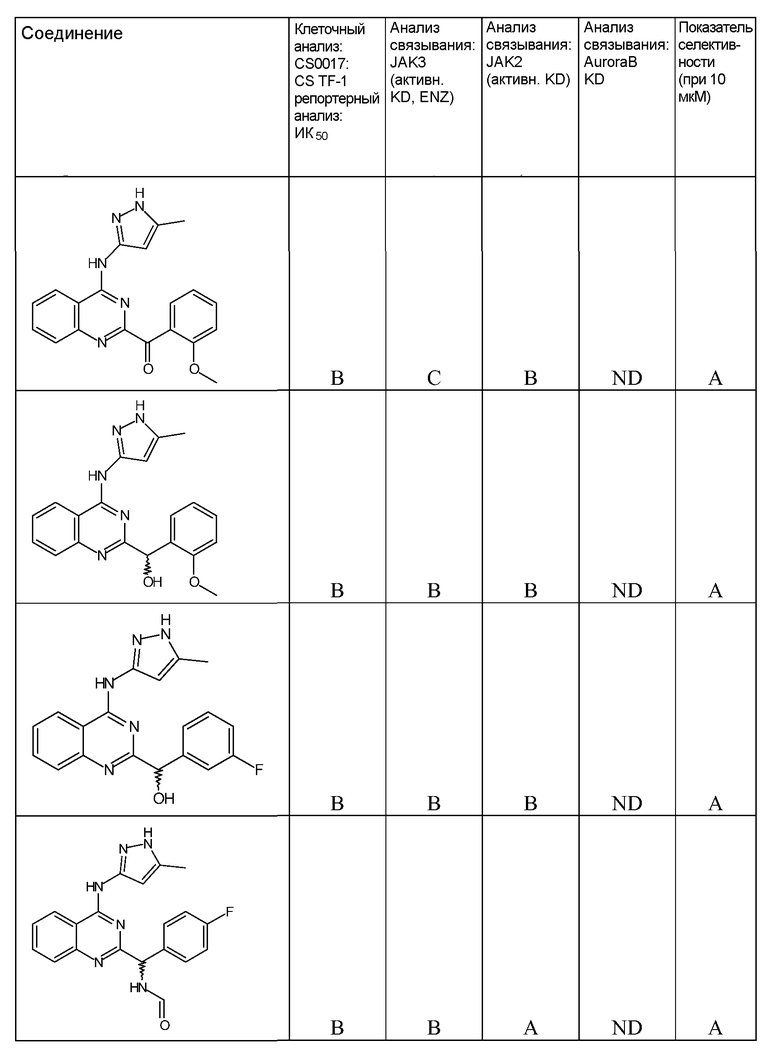
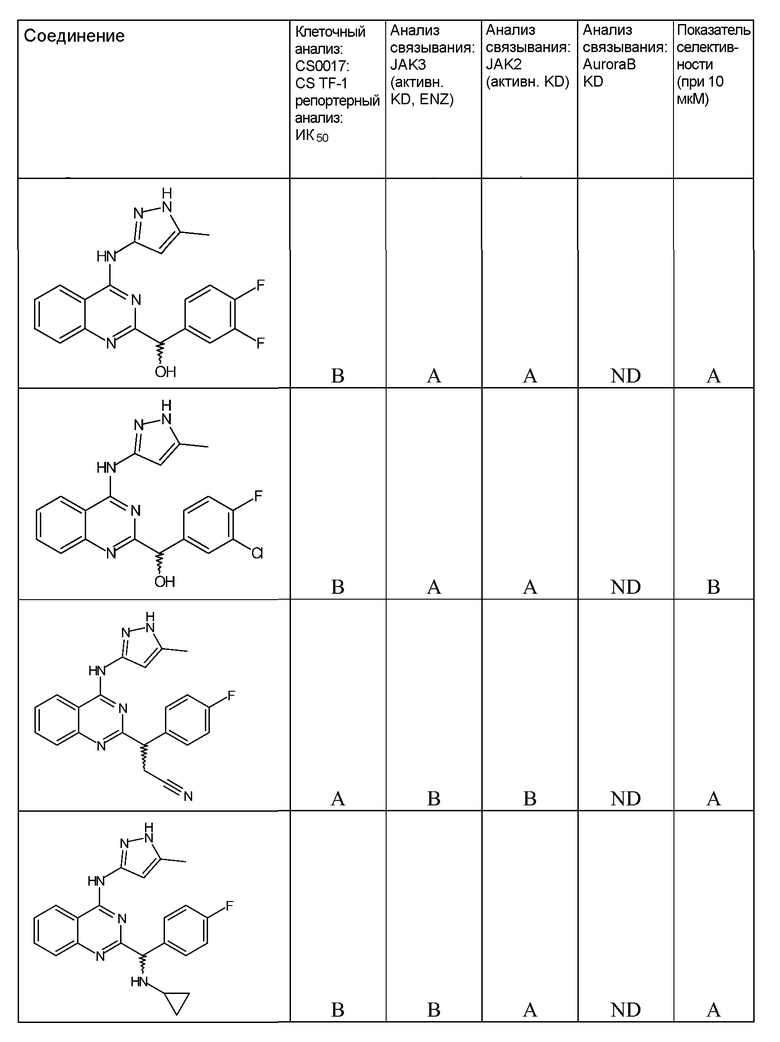
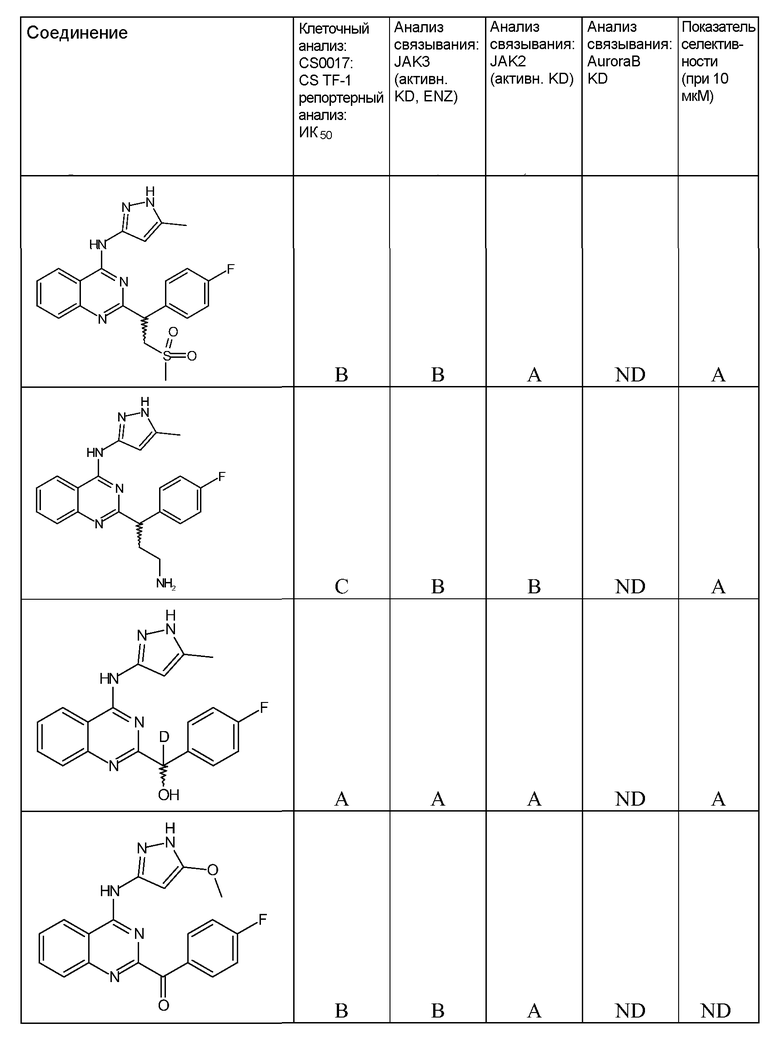
В Таблицах 1 и 2,
анализ репортера на основе CSTF-1, ИК50 (нМ): А≤100, 100<B≤500, С<500;
JAK2 Kd (нМ): А≤1, 1<B≤10, C>10; JAK3 Kd (нМ): A≤10, 10<B≤100, C>100;
AuroraB Kd (нМ) A≤20, 20<B≤50, 50<C≤200 D>200
Показатель селективности: А≤0,3, 0,3<B≤0,4, 0,4<C≤0,5, D>0,5; и ND = нет данных.
В некоторых вариантах воплощения соединения, обеспечиваемые в настоящем изобретении, связываются с JAK2 киназой с более высокой специфичностью по сравнению с немутантными и не относящимися к JAK семейству киназами. Для некоторых соединений, обеспечиваемых в настоящем изобретении, константы связывания в отношении менее 10 немутантных и не относящихся к JAK семейству киназ находятся в диапазоне 100-кратных значений от константы связывания в отношении JAK2 киназы для соединений, обеспечиваемых в настоящем изобретении. Для некоторых соединений, обеспечиваемых в настоящем изобретении, константы связывания в отношении менее 8 немутантных и не относящихся к JAK семейству киназ находятся в диапазоне 100-кратных значений от константы связывания в отношении JAK2 киназы для соединений, обеспечиваемых в настоящем изобретении. Для некоторых соединений, обеспечиваемых в настоящем изобретении, константы связывания в отношении менее 6 немутантных и не относящихся к JAK семейству киназ находятся в диапазоне 100-кратных значений от константы связывания в отношении JAK2 киназы для соединений, обеспечиваемых в настоящем изобретении.
Пример 81
Доза-ответ эффекты соединений формулы I в модели тип II коллаген-индуцированного артрита (CIA) у крыс
В День 0 эксперимента, самкам крыс Lewis (Charles River) вводили путем подкожной инъекции на трех разных участках спины по 300 мкл бычьего коллагена типа II, эмульгированного в неполном адъюванте Фрейнда, всего кажому животному вводили 1,2 мг коллагена. Животные получали бустер-дозу в День 6. Измерения при помощи циркуля (до заболевания) правого и левого голеностопного суставов осуществляли в День 8. Сразу после начала развития артрита (День 9) крыс рандомизированно распределяли на группы обработки, по 8 животных на каждую артритную группу (при этом 4 животных изначально были оставлены для нормальной контрольной группы). Начиная в День 9, соединение формулы I разбавляли в Pharmatek#6 и обработку начинали с перорального введения соединения формулы I при 5 мг/кг, 20 мг/кг или 60 мг/кг два раза в день (BID) с 12-часовыми интервалами или при 60 мг/кг QD. Контрольные артритные группы включали контрольную группу, которой вводили носитель, контрольную группу, которой вводили воду, контрольную группу, не получающую никакой обработки, и положительную контрольную группу, которой перорально вводили дексаметазон при 0,03 мг/кг QD. Обработку продолжали в течение 6 дней. Измерения при помощи циркуля голеностопных суставов осуществляли каждый день, начиная с Дня 9 по День 16. Результаты анализа представлены на Фиг. 1.
Иллюстративное соединение формулы I обеспечивало статистически значимое улучшение толщины голеностопного сустава при >5 мг/кг BID, начиная даже с Дня 2 обработки.
Максимальную эффективность соединения в крысиной модели CIA наблюдали при 60 мг/кг (QD или BID). Наблюдали взаимосвязь между клинической оценкой опухания лапы и гистологией.
Эффект обработки на массу тела крыс измеряли с Дня 9 по День 16 как процент изменения массы тела от базовой линии. Результаты представлены на Фиг. 2.
Пример 82
In vivo испытание эффективности у мышей с использованием мышиной модели TELJAK
Это испытание осуществляли для определения эффекта выбранного соединения формулы I на развитие опухоли и выживание. Мышей CB17 SCID (Harlan Laboratories) заражали путем прививки им 5e5 TEL-JAK клеток через хвостовую вену в День 0. Клеткам давали адаптироваться в организме животного и в День 3 начинали введение следующим образом: носитель (Pharmatek#6) вводили при 50 мкл BID первой группе, соединение фирмы Ambit, предназначенное только для внутреннего использования, подготавливали для введения в Pharmatek #6 и вводили при 50мг/кг BID второй группе, и TGEN101348 подготавливали для введения в Pharmatek #6 и вводили третьей группе при 100 мг/кг BID. Каждой группе обработки (по 16 животных в каждой группе) осуществляли введение два раза в день в течение двух недель. Необработанная группа из 10 животных также служила в качестве контроля. Фиг. 3 показывает результаты анализа выживания Каплана-Мейера.
Второе испытание осуществляли с использованием этого же протокола с теми же контролями и следующими группами обработки: соединение фирмы Ambit, предназначенное только для внутреннего использования, подготавливали для введения в Pharmatek #6 и вводили при 60 мг/кг BID, и выбранное соединение формулы I подготавливали для введения в Pharmatek #6 и вводили при 60 мг/кг BID. Фиг. 4 показывает результаты анализа выживания Каплана-Мейера.
Данные показывают, что соединение фирмы Ambit, предназначенное только для внутреннего использования, обеспечивает больше чем 70% увеличение времени выживания (ILS), тогда как TGEN101348 обеспечивает 30% ILS. Было показано, что выбранное соединение формулы I действует лучше, чем внутреннее соединени Ambit, и поэтому ожидают, что оно будет действовать лучше, чем TGEN101348 предклинически.
Пример 83
In vivo испытание эффективности у мышей с использованием мышиной модели HELV617F жидкой опухоли
Это испытание осуществляли для определения эффекта выбранного соединения формулы I на прогрессирование опухоли и выживание. CB 17 SCID мышей (Charles River Labs) предварительно обрабатывали циклофосфамидом интраперитонеально QD при 150 мг/кг в течение двух последовательных дней, после чего им вводили клетки путем инъекции. В День 0, 75 мышей заражали путем прививки им внутривенно 5e6 HEL 92.1.7 клеток, суспендированных в стерильном солевом растворе. Животных взвешивали в День 8 и распределяли по группам так, чтобы каждая группа включала животных с одинаковой средней массой тела и одинаковыми стандартными отклонениями от среднего. Животным осуществляли введение в День 8 в течение 21-дневного периода введения. Группы обработки были следующими (по 10 животных в каждой группе): первой группе вводили носитель (Pharmatek#6), перорально, BID; второй группе вводили фирменное соединение Ambit для внутреннего использования, подготовленное для введения в Pharmatek#6, при 50 мг/кг, перорально, BID; и третьей группе вводили TGEN101348, подготовленное для введения в Pharmatek#6, при 120 мг/кг, перорально, BID. Необработанная группа из 10 животных также служила в качестве контроля. Фиг. 5 показывает результаты анализа Каплана-Мейера.
Еще одно испытание осуществляли с использованием этого же протокола с теми же контролями и следующими группами обработки: фирменное соединение Ambit для внутреннего использования, подготовленное для введения в Pharmatek#6, вводили при 60 мг/кг BID, и выбранное соединение формулы I, подготовленное для введения в Pharmatek#6, вводили при 60 мг/кг BID. Фиг. 6 показывает результаты анализа Каплана-Мейера. Фиг. 5 и 6 показывают, что фирменное соединение Ambit для внутреннего использования и выбранное соединение формулы I обеспечивают приблизительно 30% увеличение периода выживания (ILS), тогда как TGEN101348 не обеспечивает никакого выгодного эффекта, связанного с периодом выживания (приблизительно 5% ILS).
Пример 84
Другие клеточные анализы
Соединения формулы I также испытывали в других клеточных анализах, например, pSTAT5 электрохемилюминесцентных иммуноанализах (Meso Scale Discovery) в клеточных линиях csTF-1 и HEL, и было обнаружено, что соединения являются активными в этих анализах. Эффекты соединений формулы I на клеточную пролиферацию BaF3 также испытывали с использованием анализа CellTiter-Blue® (Promega).
Ингибирование DTH ответа на овальбумин у CD-1 мышей
Испытание осуществляли для оценки эффекта соединения формулы I мышиной модели овальбумин-индуцированной аллергической реакции замедленного типа (аллергическая реакция типа IV, DTH) с использованием двух схем введения. Аллергическая реакция замедленного типа характеризуется антиген-специфической продукцией цитокинов T-клетками, приводящей к увеличению сосудистой проницаемости, инфильтрации моноядерными и полиморфоядерными клетками, отеку и индурации. Первичное воздействие антигена (фаза сенсибилизации) вызывает развитие антиген-специфических T-клеток памяти, которые активируются при вторичном воздействии (провокация).
Двухфазная природа аллергических реакций типа IV обеспечивает модель для дифференцирования иммунофармакологической активности соединений с иммуномодуляторными свойствами. Ожидают, что соединения, которые являются иммуносупрессорами (подавляют первичный иммунный ответ), будут эффективными при введении в фазе сенсибилизации. Соединения, которые являются противовоспалительными, как ожидают, будут эффективными при введении в фазе повторного воздействия антигена путем предотвращения или даун-регуляции распознавания антигена и/или активации T-клеток памяти или путем прерывания вторичных сигнальных каскадов, индуцированных продуцируемыми T-клетками цитокинами и ростовыми факторами (таким образом, уменьшая/предотвращая сосудистую проницаемость и подвижность/рекрутмент воспалительных клеток). Ожидают, что соединения, которые являются иммуностимулирующими, будут усиливать DTH ответ при введении в фазе сенсибилизации (повышенная антигенная презентация или экспансия T-клеток памяти) или в фазе повторного воздействия антигена (повышенная T-клеточная активация и продукция цитокинов/хемокинов, и/или подвижность/рекрутмент/активация воспалительных клеток).
Поскольку модификации должны быть очевидны для специалистов в данной области, заявленное изобретение должно ограничиваться только объемом прилагаемой формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВОЕ ПИПЕРИДИНОВОЕ СОЕДИНЕНИЕ ИЛИ ЕГО СОЛЬ | 2013 |
|
RU2581834C1 |
| ПИРИМИДИНАМИДНЫЕ СОЕДИНЕНИЯ КАК ИНГИБИТОРЫ PGDS | 2006 |
|
RU2420519C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛА, ИСПОЛЬЗУЕМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗЫ | 2001 |
|
RU2340611C2 |
| ТРИАЗОЛОПИРИДИНОВЫЕ СОЕДИНЕНИЯ-ИНГИБИТОРЫ JAK И СПОСОБЫ | 2009 |
|
RU2561104C2 |
| ИНГИБИТОРЫ КИНАЗ, ПРИМЕНИМЫЕ ДЛЯ ЛЕЧЕНИЯ МИЕЛОПРОЛИФЕРАТИВНЫХ ЗАБОЛЕВАНИЙ И ДРУГИХ ПРОЛИФЕРАТИВНЫХ ЗАБОЛЕВАНИЙ | 2007 |
|
RU2482112C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛА, ПОЛЕЗНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗЫ | 2001 |
|
RU2355688C2 |
| ИНГИБИТОРЫ СЕРИН/ТРЕОНИНОВЫХ КИНАЗ | 2013 |
|
RU2650501C2 |
| СПИРО-АМИНОСОЕДИНЕНИЯ, ПРИГОДНЫЕ ДЛЯ ЛЕЧЕНИЯ НАРУШЕНИЙ СНА И ЛЕКАРСТВЕННОГО ПРИВЫКАНИЯ | 2010 |
|
RU2562609C2 |
| КОНДЕНСИРОВАННЫЕ С ГЕТЕРОАРИЛКЕТОНАМИ АЗАДЕКАЛИНЫ - МОДУЛЯТОРЫ ГЛЮКОКОРТИКОИДНЫХ РЕЦЕПТОРОВ | 2013 |
|
RU2639867C2 |
| РЕГУЛЯТОРЫ ПРОИЗВОДНЫХ СТЕРОИДОВ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2803499C1 |
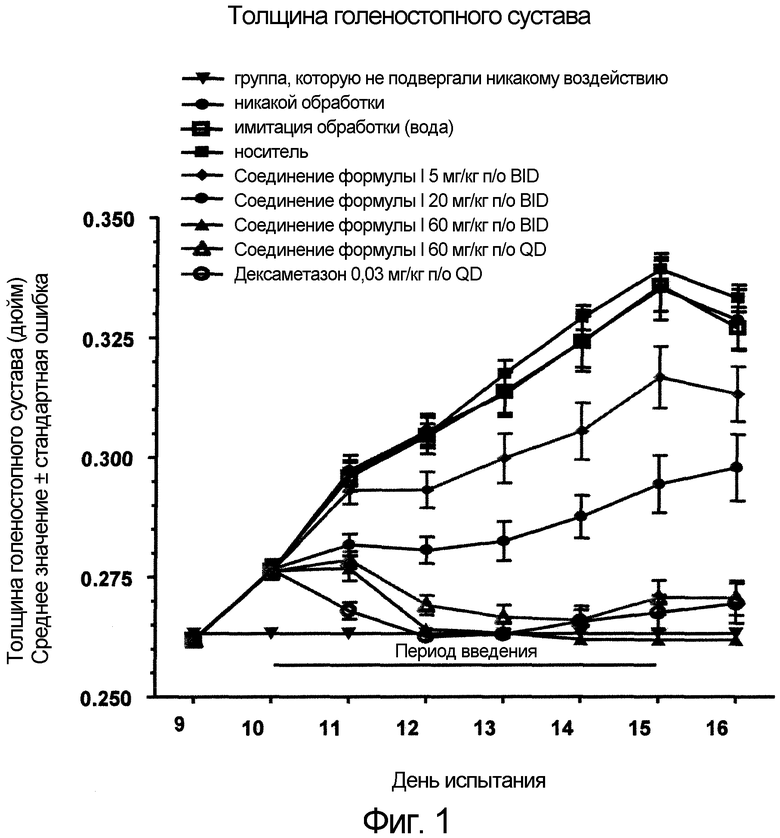
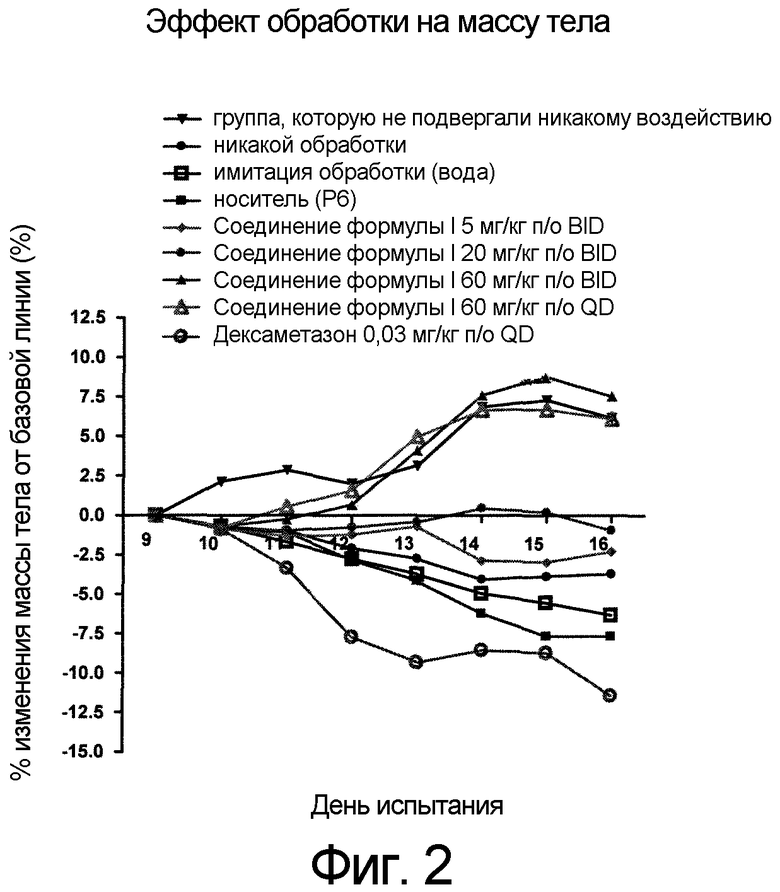
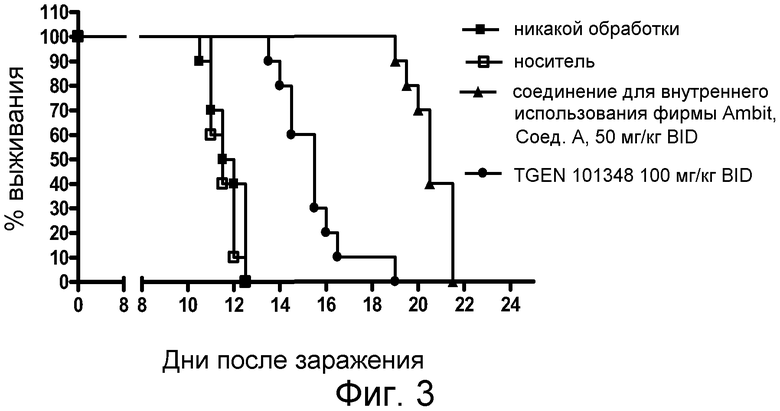
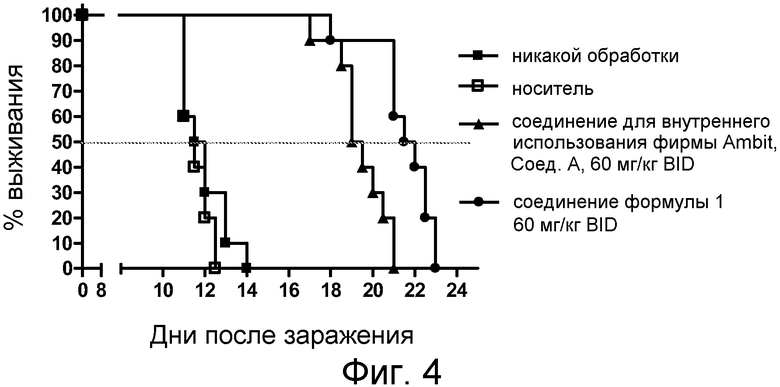
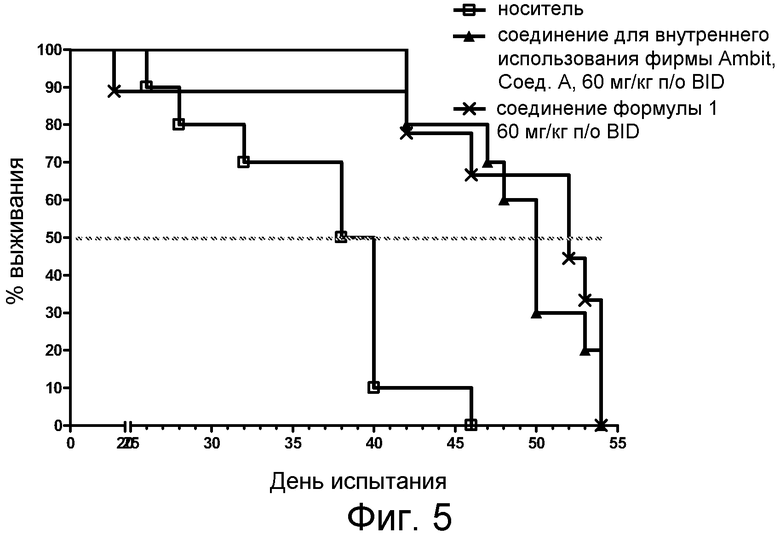
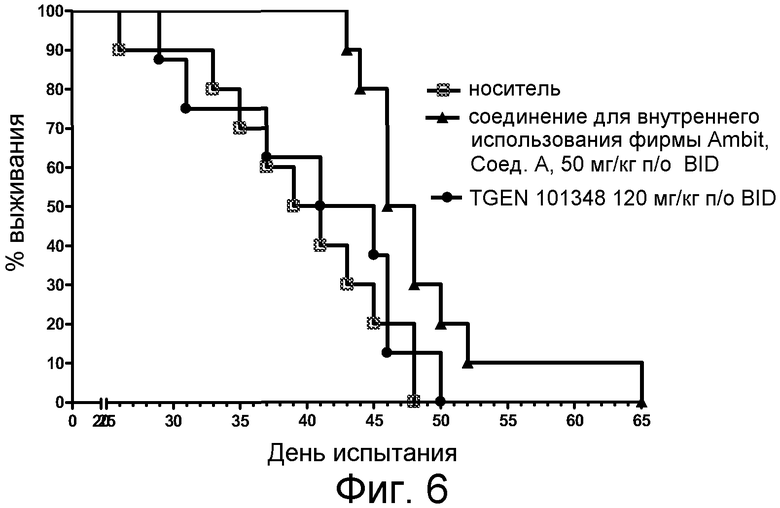
Изобретение относится к соединениям формулы (I), где R1 и R2 имеют следующие значения: (i) R1 и R2 вместе образуют =O; (ii) R1 и R2, вместе с атомом углерода, с которым они связаны, образуют диоксациклоалкил; (iii) R1 представляет собой водород или галоген; и R2 представляет собой галоген; (iv) R1 представляет собой C1-6алкил, где алкил необязательно замещен циано, -RxS(O)qRv или -RxNRyRz; и R2 представляет собой водород; (v) R1 представляет собой -OR12 или -NR13R14; и R2 представляет собой водород, дейтеро или фенил, который необязательно замещен галогеном; R3 представляет собой водород, галоген, C1-6алкил, циано, галоген C1-6алкил, C3-10циклоалкил или C1-6алкокси; R4 и R5 представляют собой водород; R6 независимо выбран из галогена, C1-6алкила, галогенC1-6алкила, -RxOR18 и -RxS(O)qRv; R7 независимо представляет собой галоген или -RxORw; R12 выбран из водорода и C1-6алкила; R13 представляет собой водород; R14 выбран из водорода, C3-10циклоалкила, -C(O)Rv и -C(O)ORw; R18 представляет собой водород, C1-6алкил или пиперидинил; где R18 необязательно замещен 1-3 группами Q1, каждый Q1 независимо выбран из гидроксила, C1-6алкокси, C1-6алкоксикарбонила, карбоксила и морфолинила; Rx независимо представляет собой C1-6алкилен или простую связь; Rv и Rw представляют собой водород или C1-6алкил; Ry и Rz представляют собой водород; n имеет значение 0-4; p имеет значение 0-5; и каждый q, независимо, имеет значение 0, 1 или 2. Также изобретение относится к соединениям формулы (II), где заместители имеют значения, указанные в формуле изобретения, к фармацевтической композиции, обладающей ингибирующей активностью в отношении JAK киназ, содержащей соединения формулы (I) или (II), способам лечения JAK-модулируемых заболеваний, и применению соединений формулы (I) или (II). Технический результат - соединения формулы (I) или (II) в качестве ингибиторов JAK киназ. 6 н. и 26 з.п. ф-лы, 6 ил., 2 табл., 84 пр.


1. Соединение, имеющее формулу (I):
или его фармацевтически приемлемые соли, где
R1 и R2 выбраны из (i), (ii), (iii), (iv) и (v) и имеют следующие значения:
(i) R1 и R2 вместе образуют =O;
(ii) R1 и R2, вместе с атомом углерода, с которым они связаны, образуют диоксациклоалкил;
(iii) R1 представляет собой водород или галоген; и R2 представляет собой галоген;
(iv) R1 представляет собой C1-6алкил, где алкил необязательно замещен циано, -RxS(O)qRv или -RxNRyRz; и R2 представляет собой водород; и
(v) R1 представляет собой -OR12 или -NR13R14; и R2 представляет собой водород, дейтеро или фенил, который необязательно замещен галогеном;
R3 представляет собой водород, галоген, C1-6алкил, циано, галоген C1-6алкил, C3-10циклоалкил или C1-6алкокси;
R4 и R5 представляют собой водород;
каждый R6 независимо выбран из галогена, C1-6алкила, галогенC1-6алкила, -RxOR18 и -RxS(O)qRv;
каждый R7 независимо представляет собой галоген или -RxORw;
R12 выбран из водорода и C1-6алкила;
R13 представляет собой водород;
R14 выбран из водорода, C3-10циклоалкила, -C(O)Rv и -C(O)ORw;
R18 представляет собой водород, C1-6алкил или пиперидинил; где R18 необязательно замещен 1-3 группами Q1, каждый Q1 независимо выбран из гидроксила, C1-6алкокси, C1-6алкоксикарбонила, карбоксила и морфолинила;
Rx независимо представляет собой C1-6алкилен или простую связь;
Rv представляет собой водород или C1-6алкил;
Rw независимо представляет собой водород или C1-6алкил;
Ry и Rz представляют собой водород;
n имеет значение 0-4;
p имеет значение 0-5; и
каждый q, независимо, имеет значение 0, 1 или 2.
2. Соединение по п.1, имеющее формулу (II)
или его фармацевтически приемлемая соль, где
R1 и R2 выбраны из (i), (iii) и (v) и имеют следующие значения:
(i) R1 и R2 вместе образуют =O;
(iii) R1 представляет собой водород, и R2 представляет собой галоген; и
(v) R1 представляет собой -OR12, и R2 представляет собой водород;
R3 представляет собой водород или C1-6алкил,
R4 и R5 представляют собой водород;
R6a, R6b, R6c и R6d, каждый независимо, выбраны из галогена, C1-6алкила, галогенC1-6алкила и -RxOR18;
R7 представляет собой галоген;
R12 представляет собой водород;
R18 представляет собой водород, C1-6алкил или пиперидинил; где R18 необязательно замещен 1-3 группами Q1, где каждый Q1 независимо выбран из гидроксила, C1-6алкокси, C1-6алкоксикарбонила, карбоксила или морфолинила;
каждый Rx независимо представляет собой C1-6алкилен или простую связь; и
каждый q, независимо, имеет значение 0, 1 или 2.
3. Соединение по п.1 или 2, где R1 и R2 вместе образуют =O
4. Соединение по п.1, где R1 представляет собой водород или фтор и R2 представляет собой фтор.
5. Соединение по п.1, где R1 представляет собой гидроксил, метокси, амино или метоксикарбониламино и R2 представляет собой водород, фенил или фторфенил.
6. Соединение по п.1, где R3 представляет собой водород, C1-6алкил или C1-6алкокси.
7. Соединение по п.1, где каждый R6 независимо выбран из галогена, C1-6алкила, гидроксила и C1-6алкокси.
8. Соединение по п.1, где R7 представляет собой галоген.
9. Соединение по п.1, где n имеет значение 0 или 1.
10. Соединение по п.1, где p имеет значение 1 или 2.
11. Соединение по п.1, имеющее формулу (III) или (IIIа)
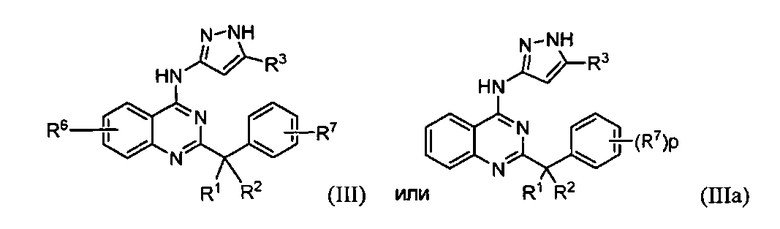
или его фармацевтически приемлемая соль, где
R1 и R2 выбраны из (i) и (ii) и имеют следующие значения:
(i) R1 и R2 вместе образуют =O; и
(ii) R1 представляет собой -OR12 или -NR13R14 и R2 представляет собой водород или фенил;
R12 выбран из водорода и C1-6алкила;
R13 представляет собой водород; и R14 выбран из водорода, C3-10циклоалкила, -C(O)Rv и -C(O)ORw;
R3 представляет собой водород или C1-6алкил;
R6 выбран из галогена, C1-6алкила, галогенC1-6алкила и -RxOR18; где R18 представляет собой водород, C1-6алкил и пиперидинил; где R18 необязательно замещен группой Q1, где Q1 выбран из гидроксила, Q1алкокси, C1-6алкоксикарбонила, карбоксила и морфолинила;
R7 представляет собой галоген, гидроксил или C1-6алкокси;
Rv представляет собой водород или C1-6алкил;
Rw представляет собой водород или C1-6алкил;
Ry и Rz представляют собой водород; и
q имеет значение 0, 1 или 2.
12. Соединение по п.1, имеющее формулу (IV) или (IVa)
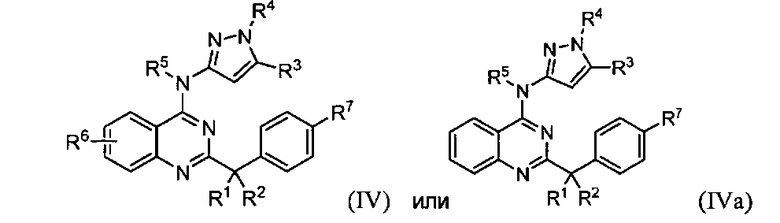
или его фармацевтически приемлемая соль.
13. Соединение по п.1, имеющее формулу (VI)
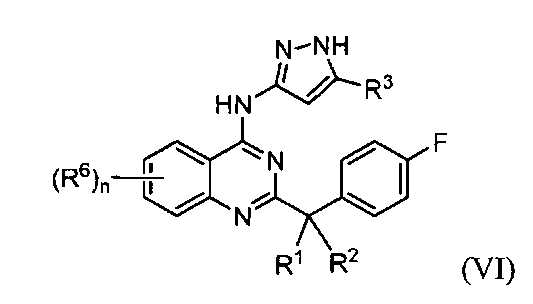
или его фармацевтически приемлемая соль.
14. Соединение по п.1, имеющее формулу (VII)
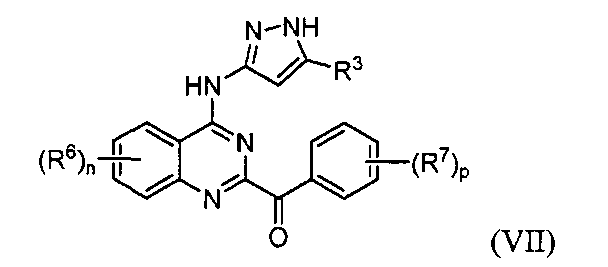
или его фармацевтически приемлемая соль.
15. Соединение по п.1, имеющее формулу (VIII)
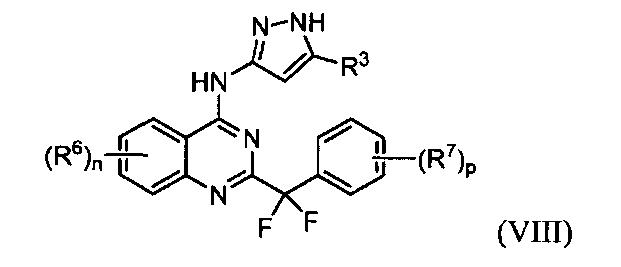
или его фармацевтически приемлемая соль.
16. Соединение по п.1, имеющее формулу (IX)
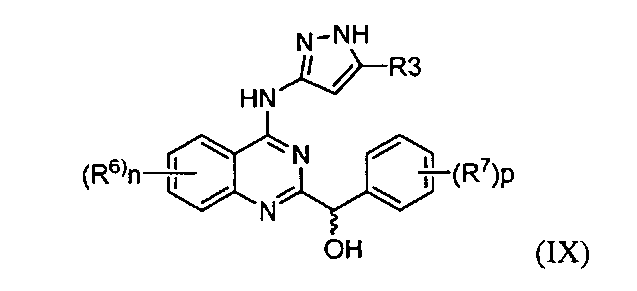
или его фармацевтически приемлемая соль.
17. Соединение по п.1, имеющее формулу (X)
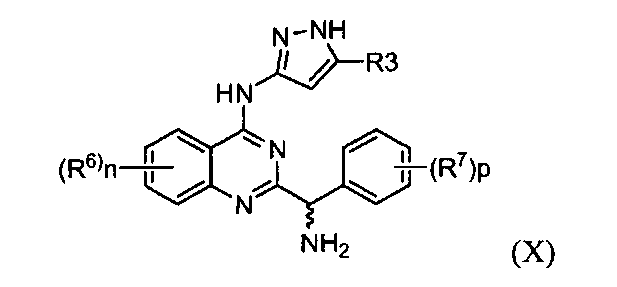
или его фармацевтически приемлемая соль.
18. Соединение по п.1, выбранное из следующих:
(3-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанон;
(4-(1Н-пиразол-3-иламино)хиназолин-2-ил)(3-фторфенил)метанон;
(4-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанон;
(4-(1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанон;
(4-(1Н-пиразол-3-иламино)хиназолин-2-ил)(2-метоксифенил)метанон;
(4-(1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанол;
2-(фтор(4-фторфенил)метил)-N-(1Н-пиразол-3-ил)хиназолин-4-амин;
2-(дифтор(4-фторфенил)метил)-N-(5-метил-1Н-пиразол-3- ил)хиназолин-4-амин;
2-(дифтор(4-фторфенил)метил)-N-(1Н-пиразол-3-ил)хиназолин-4-амин;
N-(5-циклопропил-1Н-пиразол-3-ил)-2-(дифтор(4-фторфенил)метил)хиназолин-4-амин;
3-(2-(4-фторбензоил)хиназолин-4-иламино)-1Н-пиразол-5-карбонитрил;
(4-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанол;
2-((4-фторфенил)(метокси)метил)-N-(5-метил-1Н-пиразол-3-ил)хиназолин-4-амин;
2-(амино(4-фторфенил)метил)-N-(5-метил-1Н-пиразол-3-ил)хиназолин-4-амин;
3-(2-((4-фторфенил)(гидрокси)метил)хиназолин-4-иламино)-1Н-пиразол-5-карбонитрил;
(5-фтор-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанол;
(4-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)-7-(трифторметил)хиназолин-2-ил)метанон;
(4-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)-7-(трифторметил)хиназолин-2-ил);
(7-фтор-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанон;
2-(дифтор(4-фторфенил)метил)-7-фтор-N-(5-метил-1Н-пиразол-3-ил)хиназолин-4-амин;
2-(дифтор(4-фторфенил)метил)-7-фтор-N-(1Н-пиразол-3-ил)хиназолин-4-амин;
(4-(1Н-пиразол-3-иламино)-7-иодхиназолин-2-ил)(4-фторфенил)метанон;
(4-(1Н-пиразол-3-иламино)-7-иодхиназолин-2-ил)(4-фторфенил)метанол;
(4-фторфенил)(7-метил-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанон;
(4-фторфенил)(7-метил-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанол;
2-(дифтор(4-фторфенил)метил)-7-метил-N-(5-метил-1Н-пиразол-3-ил)хиназолин-4-амин;
2-(дифтор(4-фторфенил)метил)-7-метил-N-(1Н-пиразол-3-ил)хиназолин-4-амин;
(4-(1Н-пиразол-3-иламино)-7-метоксихиназолин-2-ил)(4-фторфенил)метанон;
(4-(1Н-пиразол-3-иламино)-7-метоксихиназолин-2-ил)(4-фторфенил)метанол;
(4-фторфенил)(7-метокси-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанон;
(4-фторфенил)(7-метокси-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанол;
2-(дифтор(4-фторфенил)метил)-7-метокси-N-(5-метил-1Н-пиразол-3-ил)хиназолин-4-амин;
2-(дифтор(4-фторфенил)метил)-7-метокси-1H-(1Н-пиразол-3-ил)хиназолин-4-амин;
2-(дифтор(4-фторфенил)метил)-8-фтор-N-(5-метил-1Н-пиразол-3-ил)хиназолин-4-амин;
(4-(1Н-пиразол-3-иламино)-8-метоксихиназолин-2-ил)(4-фторфенил)метанон;
2-((4-фторфенил)(гидрокси)метил)-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-7-ол;
(4-фторфенил)(7-гидрокси-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанон;
(4-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)-7-(2-морфолиноэтокси)хиназолин-2-ил)метанол;
2-(2-((4-фторфенил)(гидрокси)метил)-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-7-илокси)этанол;
3-(2-((4-фторфенил)(гидрокси)метил)-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-7-илокси)пропан-1-ол;
(4-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)-7-(пиперидино-4-илокси)хиназолин-2-ил)метанол;
(4-фторфенил)(7-(2-метоксиэтокси)-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанол;
трет-бутил 2-(2-((4-фторфенил)(гидрокси)метил)-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-7-илокси)ацетат;
2-(2-((4-фторфенил)(гидрокси)метил)-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-7-илокси)уксусная кислота;
метиловый эфир {(4-фторфенил)-[4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил]метил}карбаминовой кислоты;
бис-(4-фторфенил)-[4-(5-метил-1Н-пиразол-3-иламино)-хиназолин-2-ил]метанол;
метил (4-фторфенил)(4-(5-метил-4Н-пиразол-3-иламино)хиназолин-2-ил)метилкарбамат;
(4-фторфенил)(8-метил-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанол;
(7-фтор-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанол;
(4-(1Н-пиразол-3-иламино)хиназолин-2-ил)бис(4-фторфенил)метанол;
(2-(дифтор(4-фторфенил)метил)-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-7-ил)метанол;
2-(дифтор(4-фторфенил)метил)-N-(5-метил-1Н-пиразол-3-ил)-7-(метилсульфонилметил)хиназолин-4-амин;
2-(дифтор(4-фторфенил)метил)-7-(этоксиметил)-N-(5-метил-1Н-пиразол-3-ил)хиназолин-4-амин;
(7-хлор-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанол;
(6-фтор-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанон;
(6-фтор-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанол;
(4-(1Н-пиразол-3-иламино)-6-фторхиназолин-2-ил)(4-фторфенил)метанол;
(7-бром-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанон;
(7-бром-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанол;
(4-(1Н-пиразол-3-иламино)-7-бромхиназолин-2-ил)(4-фторфенил)метанол;
2-(2-(4-фторфенил)-1,3-диоксолан-2-ил)-N-(5-метил-1Н-пиразол-3-ил)хиназолин-4-амин;
(8-фтор-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанон;
(8-фтор-4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанол;
(2-метоксифенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанон;
(2-метоксифенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанол;
N-((4-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метил)фтормамид;
(3,4-дифторфенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанол;
(3-хлор-4-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанол;
3-(4-фторфенил)-3-(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)пропаннитрил;
2-((циклопропиламино)(4-фторфенил)метил)-N-(5-метил-1Н-пиразол-3-ил)хиназолин-4-амин;
2-(1-(4-фторфенил)-2-(метилсульфонил)этил)-N-(5-метил-1Н-пиразол-3-ил)хиназолин-4-амин;
2-(3-амино-1-(4-фторфенил)пропил)-N-(5-метил-1Н-пиразол-3-ил)хиназолин-4-амин;
(4-фторфенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанол-1-d;
(4-фторфенил)(4-(5-метокси-1Н-пиразол-3-иламино)хиназолин-2-ил)метанон;
(4-(5-этил-1Н-пиразол-3-иламино)хиназолин-2-ил)(4-фторфенил)метанол;
(4-фторфенил)(4-(5-метокси-1Н-пиразол-3-иламино)хиназолин-2-ил)метанол;
(4-фтор-3-метоксифенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанон;
(4-фтор-3-гидроксифенил)(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метанон; и
(2-фтор-5-(гидрокси(4-(5-метил-1Н-пиразол-3-иламино)хиназолин-2-ил)метил)фенолацетат.
19. Соединение по п.1 для применения в лечении JAK-модулируемого заболевания.
20. Фармацевтическая композиция, обладающая ингибирующей активностью в отношении JAK киназ, содержащая терапевтически эффективное количество соединения по любому из пп.1-18 и фармацевтически приемлемый носитель, разбавитель или эксципиент.
21. Способ лечения JAK-модулируемого заболевания, включающий введение терапевтически эффективного количества соединения по любому из пп.1-18.
22. Способ лечения JAK2-модулируемого заболевания, включающий введение терапевтически эффективного количества соединения по любому из пп.1-18.
23. Способ по п.22, где JAK2 представляет собой JAK2 дикого типа или мутантный JAK2.
24. Способ по п.21, где заболевание представляет собой рак, миелопролиферативное расстройство, воспаление или аутоиммунное заболевание.
25. Способ по п.21, где заболевание выбирают из истинной полицитемии, эссенциальной тромбоцитемии, идиопатического миелофиброза, хронического миеломоноцитарного лейкоза, хронического эозинофильного лейкоза, миеломы и системного мастоцитоза.
26. Способ по п.24, где рак представляет собой лейкоз.
27. Способ по п.26, где лейкоз представляет собой хронический миелоидный лейкоз, иматиниб-резистентный хронический миелоидный лейкоз, острый миелоидный лейкоз, острый лимфобластный лейкоз или острый мегакариобластный лейкоз.
28. Способ по п.24, где рак выбирают из рака головы и шеи, рака предстательной железы, рака молочной железы, рака яичника, меланомы, рака легкого, рака головного мозга, рака поджелудочной железы, рака желудка, рака щитовидной железы, карциномы почки, саркомы Капоши, болезни Кастлмана и меланомы.
29. Способ по п.21, где заболевание выбирают из гиперэозинофильного синдрома, болезни трансплантат-против-хозяина, заживления ран, заболевания почек, рассеянного склероза, тиреоидита, диабета типа 1, саркоидоза, псориаза, аллергического ринита, атопического дерматита, злокачественной миастении, воспалительного заболевания кишечника, болезни Крона, язвенного колита, системной красной волчанки, артрита, остеоартрита, ревматоидного артрита, остеопороза, астмы, хронического обструктивного заболевания легких, коонъюнктивита, увеита, ирита, склерита, синдрома сухих глаз, сухого кератоконъюнктивита, ринита, синусита, бронхита, воспалительной миопатии, миокардита, ишемических реперфузионных поражений, синдрома системного воспалительного ответа и сепсиса.
30. Способ по п.21, где заболевание выбирают из рестеноза, фиброза и склеродермы.
31. Способ по п.21, где заболевание выбирают из вируса Эпштейна-Барра, гепатита B, гепатита C, вируса иммунодефицита человека, Т-лимфотропного вируса типа 1 человека, вируса ветряной оспы и папиллома-вируса человека.
32. Применение соединения по любому из пп.1-18 для получения лекарственного средства для лечения JAK-модулируемого заболевания.
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| Приспособление для точного наложения листов бумаги при снятии оттисков | 1922 |
|
SU6A1 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛА, ПОЛЕЗНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗЫ | 2001 |
|
RU2332415C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛА, ИСПОЛЬЗУЕМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗЫ | 2001 |
|
RU2340611C2 |

