Область изобретения
Настоящее изобретение относится к новым C-арилглюкозидным производным формулы (I) или их фармацевтическим солям, или стереоизомерам, способам их получения, содержащим их фармацевтическим композициям и их терапевтическим применениям, в частности их фармацевтическому применению в качестве ингибитора натрий-зависимого переносчика глюкозы SGLT.
Предшествующий уровень техники
На начальных стадиях лечения диабета контроль над диетой и лечебная физкультура способствуют предпочтительному гликемическому контролю. Когда эти способы утрачивают контроль над гликемической регуляцией, тогда для лечения срочно необходимы инсулин или гипогликемические лекарства для перорального введения. Существуют различные гипогликемические лекарства для клинического лечения, включающие бигуаниды, соединения сульфонилмочевины, агенты, улучшающие резистентность к инсулину и ингибиторы α-глюкозидазы и т.д. Вследствие различных отрицательных воздействий вышеупомянутых лекарств, соответственно, указанные лекарства не удовлетворяют требованиям длительной терапии. Например бигуаниды могут увеличивать риск лактацидоза; соединения сульфонилмочевины могут приводить к симптому гипогликемии; агенты, улучшающие резистентность к инсулину, могут быть склонны вызывать отек и сердечную недостаточность; а ингибиторы α-глюкозидазы могут вызывать боль в животе, раздувание, диарею и т.д. Следовательно, разработка новых более безопасных и намного более эффективных гипогликемических агентов, как очень ожидают, будет удовлетворять требованиям лечения диабета.
Исследование указывает на то, что перенос клеточной глюкозы осуществляется как освобожденными ("пассивными") переносчиками глюкозы (GLUT), так и связанными с натрием ("активными") переносчиками глюкозы (SGLT). Члены SGLT, действующие как переносчики глюкозы, главным образом, распределены в кишечнике и в проксимальных почечных канальцах, свидетельствуя о том, что SGLT ответственны за большую часть обратного захвата глюкозы в кишечнике и в почках. SGLT рассматривают в качестве потенциальных и идеальных антидиабетических мишеней.
Более подробно, SGLT-1 преимущественно экспрессируется в клетках слизистой оболочки тонкой кишки и некоторое количество экспрессируется в миокарде и почках. SGLT-1 модулирует абсорбцию глюкозы в кишечнике в кооперации с GLUT. Второй Nа+-зависимый переносчик глюкозы SGLT-2 ответственен за обратный захват глюкозы в почках, о чем свидетельствует высокий уровень его экспрессии в почках. Глюкоза в моче активно захватывается эпителиальными клетками почечных канальцев из клубочкового фильтрата и повторно используется в клетках посредством переносчиков SGLT2. На такой стадии SGLT-2 отвечает за 90% реабсорбции, в то время как SGLT1 переносит оставшиеся 10%. Заключение о том, что SGLT2 представляет собой основной переносчик глюкозы, дополнительно подтверждено в исследованиях на животных. Почечный гликемический повторный захват у крыс значительно подавляется, когда экспрессию мРНК SGLT-2 в клетках коры почек ингибируют с использованием специфического для SGLT-2 антисмыслового олигонуклеотида. Показано, что новые ингибиторы SGLT (SGLT-1/SGLT-2), которые могут реализовать контроль над абсорбцией глюкозы в кишечнике, а также ингибирование обратного захвата глюкозы в почках посредством регуляции функции транспорта глюкозы, могут представлять собой идеальные потенциальные противодиабетические лекарства с улучшением уровня экскреции глюкозы с мочой и систематическим действием по уменьшению уровня сахара в крови.
Кроме того, применение ингибиторов SGLT также встречали при лечении осложнений диабета, включая ретинопатию, нейропатию, нефропатию и связанные заболевания, такие как метаболизм глюкозы (нарушение глюкозного гомеостаза), гиперинсулинемия, гипергликемия и ожирение и т.д. В то же время, ингибиторы SGLT позволяли избежать или ослабить ответную неблагоприятную реакцию и улучшали соблюдение пациентом схемы лечения в сочетании с существующими терапевтическими лекарствами, включающими сульфонамид, тиазолидиндион, метформин и инсулин, и т.д, не влияя на эффективность и уменьшение количества лекарств.
В заключение, оказалось, что ингибиторы SGLT, особенно ингибиторы SGLT-2, представляют собой перспективные кандидаты для использования в качестве противодиабетических препаратов и нового противодиабетического средства. Хотя в патентах CN 1989132 A, CN 1671682 A, CN 1829729 A и WO 2010023594 A1 раскрыты серии C-арилглюкозида и производные для применения в качестве ингибиторов SGLT-2, новые соединения, обладающие улучшенной эффективностью, фармакокинетикой и безопасностью, все же срочно требуются для лечения диабета и связанных с ним метаболических расстройств. В настоящем изобретении раскрыты соединения формулы (I) и обнаружено, что такие соединения обладают превосходным ингибирующим SGLT-2 и гипогликемическим действием.
Описание изобретения
Настоящее изобретение направлено на то, чтобы предложить соединение формулы (I) и его таутомер, энантиомер, диастереоизомер, рацемат и его фармацевтически приемлемые соли, и его метаболиты, предшественники или пролекарство.
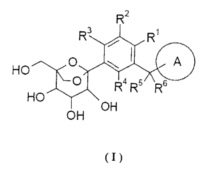
где:
кольцо A выбрано из группы, состоящей из арила и гетероарила, где каждый из указанного арила или гетероарила независимо возможно дополнительно замещен одной или более чем одной группой, состоящей из галогена, алкила, алкенила, алкинила, циклоалкила, гетероциклила, арила, гетероарила, -OR7, -S(O)mR7, -C(O)R7, -C(O)OR7, -NR8R9 и -C(O)NR8R9, где каждый из указанного алкила, циклоалкила, гетероциклила, арила или гетероарила независимо возможно замещен одной или более чем одной группой, выбранной из группы, состоящей из дейтерия, галогена, алкенила, алкинила, нитро, циано, алкокси, циклоалкила, -OR7, -S(O)mR7, -C(O)R7, -C(O)OR7, -NR8R9 и -C(O)NR8R9;
каждый из R1, R2, R3 или R4 независимо выбран из группы, состоящей из водорода, галогена, циано, гидроксила, амино, алкила, алкокси, циклоалкила, арила и гетероарила, где указанный алкил, алкокси, циклоалкил, арил или гетероарил независимо возможно дополнительно замещен одной или более чем одной группой, выбранной из группы, состоящей из дейтерия, галогена, гидроксила, аминогруппы, алкила, алкокси, карбоксила и эфира карбоновой кислоты;
или R2 и R3 вместе с фенилом конденсируются в кольцо, возможно выбранное из группы, состоящей из циклоалкила, гетероциклила, арила и гетероарила, где каждый из указанного циклоалкила, гетероциклила, арила или гетероарила независимо возможно замещен одной или более чем одной группой, выбранной из группы, состоящей из галогена, гидроксила, амино, алкила, алкокси, алкенила, алкинила, циклоалкила, гетероциклила, арила, гетероарила, карбоксила и эфира карбоновой кислоты;
при условии, что
когда кольцо A представляет собой фенил, R2, R3, R4 представляют собой водород, R1 выбран из группы, состоящей из водорода, С1-4 алкила, F, Cl, циано и -OR10, то кольцо A не может быть замещено одной группой, выбранной из C1-4алкила, F, Cl, циано, гидроксила, -OR11, C1-2 алкила, замещенного F, -S(O)2R11, С3-6 циклоалкила и С5-6 насыщенного гетероциклила, замещенного 1-2 N, O или S;
каждый из R5 или R6 независимо выбран из группы, состоящей из водорода и дейтерия;
R7 выбран из группы, состоящей из водорода, дейтерия, алкила, циклоалкила, гетероциклила, арила и гетероарила, где каждый из указанного алкила, циклоалкила, гетероциклила, арила или гетероарила независимо возможно дополнительно замещен одной или более чем одной группой, выбранной из группы, состоящей из дейтерия, алкила, галогена, гидроксила, аминогруппы, алкокси, циклоалкила, гетероциклила, арила, гетероарила, карбоновой кислоты и эфира карбоновой кислоты;
каждый из R8 или R9 независимо выбран из водорода, алкила, циклоалкила, гетероциклила, арила и гетероарила, где каждый из указанного алкила, циклоалкила, гетероциклила, арила или гетероарила независимо возможно дополнительно замещен одной или более чем одной группой, выбранной из группы, состоящей из алкила, галогена, гидроксила, аминогруппы, алкокси, циклоалкила, гетероциклила, арила, гетероарила, карбоновой кислоты и эфира карбоновой кислоты;
или R8 и R9 взяты вместе с присоединенным атомом азота с образованием гетероциклила, где указанный гетероциклил содержит один или более чем один гетероатом N, O или S(O)m, и указанный гетероциклил возможно дополнительно замещен одной или более чем одной группой, выбранной из группы, состоящей из алкила, галогена, гидроксила, алкокси, циклоалкила, гетероциклила, арила, гетероарила, карбоновой кислоты и эфира карбоновой кислоты;
R10 представляет собой С1-4алкил;
R11 выбран из C1-4алкила, 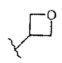 и
и  , и
, и
m равен 0, 1 или 2.
Предпочтительно, соединение формулы (I), его фармацевтически приемлемые соли или стереоизомеры, включающие соединение следующей формулы (II) или его фармацевтически приемлемые соли:
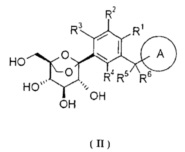
где кольцо A и R1-R6 определены как в формуле (I).
Предпочтительно, настоящее изобретение относится к соединению формулы (I), его фармацевтически приемлемым солям или стереоизомерам, где: кольцо A представляет собой арил, где указанный арил может быть возможно дополнительно замещен одной или более чем одной группой, выбранной из группы, состоящей из галогена, алкила, алкенила, алкинила, циклоалкила, гетероциклила, арила, гетероарила, -OR7, -S(O)mR7, -C(O)R7, -C(O)OR7, -NR8R9 и -C(O)NR8R9, где каждый из указанного алкила, циклоалкила, гетероциклила, арила или гетероарила независимо возможно дополнительно замещен одной или более чем одной группой, выбранной из группы, состоящей из дейтерия, галогена, алкенила, алкинила, нитро, циано, алкокси, циклоалкила, -OR7, -S(O)mR7, -C(O)R7, -C(O)OR7, -NR8R9 и -C(O)NR8R9.
Предпочтительно, настоящее изобретение относится к соединению формулы (I), его фармацевтически приемлемым солям или стереоизомерам,
где:
кольцо A представляет собой арил, где указанный арил возможно дополнительно замещен одной или более чем одной группой, выбранной из группы, состоящей из галогена, алкила, алкенила, алкинила, циклоалкила, гетероциклила, арила, гетероарила, -OR7, -S(O)mR7, -C(O)R7, -C(O)OR7, -NR8R9 и -C(O)NR8R9, где каждый из указанного алкила, циклоалкила, гетероциклила, арила или гетероарила независимо возможно дополнительно замещен одной или более чем одной группой, выбранной из группы, состоящей из дейтерия, галогена, алкенила, алкинила, нитро, циано, алкокси, циклоалкила, -OR7, -S(O)mR7, -C(O)R7, -C(O)OR7, -NR8R9 и -C(O)NR8R9;
каждый из R2, R3 и R4 независимо представляет собой водород; и
R1 представляет собой галоген.
Предпочтительно, настоящее изобретение относится к соединению формулы (I), его фармацевтически приемлемым солям или стереоизомерам,
где:
кольцо A представляет собой фенил, где указанный фенил возможно дополнительно замещен 1-5 группами, выбранными из группы, состоящей из галогена и -OR7,
R7 представляет собой алкил, где указанный алкил возможно дополнительно замещен 1-3 группами, выбранными из группы, состоящей из дейтерия, галогена, алкокси и циклоалкокси.
Предпочтительно, настоящее изобретение относится к соединению формулы (II), его фармацевтически приемлемым солям или стереоизомерам, где:
кольцо A представляет собой гетероарил, где указанный гетероарил может быть дополнительно возможно замещен одной или более чем одной группой, выбранной из группы, состоящей из галогена, алкила, алкенила, алкинила, циклоалкила, гетероциклила, арила, гетероарила, -OR7, -S(O)mR7, -C(O)R7, -C(O)OR7, -NR8R9 и -C(O)NR8R9, где каждый из указанного алкила, циклоалкила, гетероциклила, арила или гетероарила независимо возможно дополнительно замещен одной или более чем одной группой, выбранной из группы, состоящей из дейтерия, галогена, алкенила, алкинила, нитро, циано, алкокси, циклоалкила, -OR7, -S(O)mR7, -C(O)R7, -C(O)OR7, -NR8R9 и -C(O)NR8R9.
Предпочтительно, настоящее изобретение относится к соединению формулы (II), его фармацевтически приемлемым солям или стереоизомерам, где:
кольцо A представляет собой гетероарил, где указанный гетероарил возможно дополнительно замещен одной или более чем одной группой, выбранной из группы, состоящей из галогена, алкила, алкенила, алкинила, циклоалкила, гетероциклила, арила, гетероарила, -OR7, -S(O)mR7, -C(O)R7, -C(O)OR7, -NR8R9 и -C(O)NR8R9, где каждый из указанных алкила, циклоалкила, гетероциклила, арила или гетероарила независимо возможно дополнительно замещен одной или более чем одной группой, выбранной из группы, состоящей из дейтерия, галогена, алкенила, алкинила, нитро, циано, алкокси, циклоалкила, -OR7, -S(O)mR7, -C(O)R7, -C(O)OR7, -NR8R9 и -C(O)NR8R9;
каждый из R2, R3 и R4 независимо представляет собой водород; и
R1 представляет собой галоген.
Предпочтительно, настоящее изобретение относится к соединению формулы (I), его фармацевтически приемлемым солям или стереоизомерам, где кольцо A представляет собой 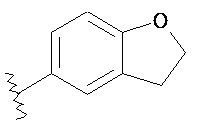 или тиенил.
или тиенил.
Предпочтительно, настоящее изобретение относится к соединению формулы (I), его фармацевтически приемлемым солям или стереохимическим изомерам, где:
кольцо A возможно замещено одной или более чем одной группой, выбранной из группы, состоящей из арила, галогена и -OR7, где указанный арил возможно дополнительно замещен одним или более чем одним атомом галогена; при условии, что когда кольцо A замещено на -OR7, где R7 представляет собой C1-4 алкил, то кольцо A должно быть одновременно замещено одним или более чем одним атомом галогена.
Предпочтительно, настоящее изобретение относится к соединению формулы (I), его фармацевтически приемлемым солям или стереоизомерам, где R5 или R6 представляет собой дейтерий.
Предпочтительно, настоящее изобретение относится к соединению формулы (I), его фармацевтически приемлемым солям или стереоизомерам, где R7 представляет собой алкил, указанный алкил возможно дополнительно замещен одним или более чем одним атомом дейтерия.
Соединение формулы (I) может содержать асимметрические атомы углерода, поэтому оно может существовать в форме оптически чистого диастереоизомера, диастереоизомерной смеси, диастереоизомерного рацемата, смеси диастереоизомерного рацемата или в виде мезо-соединения. Настоящее изобретение включает все эти формы. Диастереоизомерная смесь, диастереоизомерный рацемат или смесь диастереоизомерного рацемата могут быть выделены при помощи обычных способов, таких как колоночная хроматография, тонкослойная хроматография и высокоэффективная жидкостная хроматография.
Предпочтительные соединения по настоящему изобретению включают, но не ограничиваются следующим:

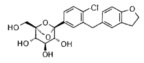

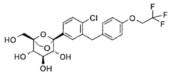
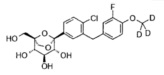
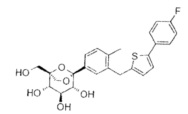
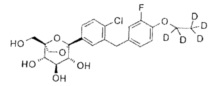
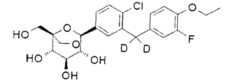
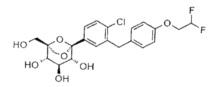
или их фармацевтически приемлемые соли, или любые их стереохимические изомеры.
Данное изобретение относится к способу получения соединения формулы (I), включающему следующие стадии:
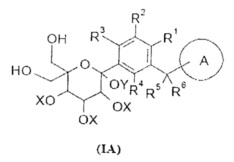
превращения соединения формулы (IA) в соединение формулы (IB);
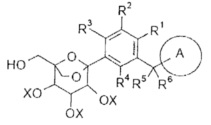
удаления защиты с соединения формулы (IB) в соединение формулы (I);
где:
R1-R6 и кольцо A являются такими, как определено в формуле (I);
X и Y представляют собой защитные группы гидроксила, предпочтительно алкил или бензил.
Настоящее изобретение относится к применению соединений формулы (I), их физиологических солей и стереоизомеров в изготовлении ингибитора натрий-зависимого переносчика глюкозы.
Кроме того, настоящее изобретение относится к применению соединений формулы (I), их физиологических солей и их стереоизомеров в изготовлении лекарственного средства для лечения или замедления развития или возникновения следующего заболевания, где указанное заболевание выбрано из группы, состоящей из сахарного диабета, ретинопатии, нейропатии, нефропатии, инсулинорезистентности, гипергликемии, гиперинсулинемии, повышенных уровней жирных кислот или глицерина, гиперлипидемии, ожирения, гипертриглицеридемии, синдрома X, диабетических осложнений, атеросклероза и гипертензии.
Настоящее изобретение также относится к способу лечения заболеваний, выбранных из группы, состоящей из сахарного диабета, ретинопатии, нейропатии, нефропатии, инсулинорезистентности, гипергликемии, гиперинсулинемии, повышенных уровней жирных кислот или глицерина, гиперлипидемии, ожирения, гипертриглицеридемии, синдрома X, диабетических осложнений, атеросклероза и гипертензии, при котором субъекту, нуждающемуся в таком лечении, вводят терапевтически эффективное количество соединения формулы (I) или его фармацевтических солей или его стереоизомеров.
Настоящее изобретение также относится к соединению формулы (I) или его фармацевтическим солям или его стереоизомерам для применения в качестве лекарственного средства для лечения или замедления развития или возникновения следующих заболеваний, где указанные заболевания выбраны из группы, состоящей из сахарного диабета, ретинопатии, нейропатии, нефропатии, инсулинорезистентности, гипергликемии, гиперинсулинемии, повышенных уровней жирных кислот или глицерина, гиперлипидемии, ожирения, гипертриглицеридемии, синдрома X, диабетических осложнений, атеросклероза и гипертензии.
Кроме того, настоящее изобретение относится к фармацевтической композиции, содержащей терапевтически эффективное количество соединения формулы (I), его фармацевтически приемлемых солей или его стереоизомеров, и фармацевтически приемлемые носители. Также настоящее изобретение относится к применению указанной фармацевтической композиции в изготовлении лекарственного средства для лечения или замедления развития или возникновения следующих заболеваний, где указанные заболевания выбраны из группы, состоящей из сахарного диабета, ретинопатии, нейропатии, нефропатии, инсулинорезистентности, гипергликемии, гиперинсулинемии, повышенных уровней жирных кислот или глицерина, гиперлипидемии, ожирения, гипертриглицеридемии, синдрома X, диабетических осложнений, атеросклероза и гипертензии.
Настоящее изобретение также относится к способу лечения заболеваний, выбранных из группы, состоящей из сахарного диабета, ретинопатии, нейропатии, нефропатии, инсулинорезистентности, гипергликемии, гиперинсулинемии, повышенных уровней жирных кислот или глицерина, гиперлипидемии, ожирения, гипертриглицеридемии, синдрома X, диабетических осложнений, атеросклероза и гипертензии, при котором субъекту, нуждающемуся в таком лечении, вводят терапевтически эффективное количество указанной фармацевтической композиции.
Настоящее изобретение также относится к применению указанной фармацевтической композиции в качестве лекарственного средства для лечения или замедления развития или возникновения следующих заболеваний, где указанные заболевания выбраны из группы, выбранной из сахарного диабета, ретинопатии, нейропатии, нефропатии, инсулинорезистентности, гипергликемии, гиперинсулинемии, повышенных уровней жирных кислот или глицерина, гиперлипидемии, ожирения, гипертриглицеридемии, синдрома X, диабетических осложнений, атеросклероза и гипертензии.
Подробное описание изобретения
Если не указано иное, то следующие термины, используемые в описании и формуле изобретения, имеют значения, описанные ниже.
"Алкил" относится к насыщенной алифатической углеводородной группе, включающей С1-C20 прямоцепочечные и имеющие разветвленную цепь группы. Предпочтительно, алкильная группа представляет собой алкил, имеющий от 1 до 12 атомов углерода. Типичные примеры включают, но не ограничиваются метилом, этилом, н-пропилом, изопропилом, н-бутилом, изобутилом, трет-бутилом, втор-бутилом, н-пентилом, 1,1-диметил пропилом, 1,2-диметилпропилом, 2,2-диметилпропилом, 1-этилпропилом, 2-метилбутилом, 3-метилбутилом, н-гексилом, 1-этил-2-метилпропилом, 1,1,2-триметилпропилом, 1,1-диметилбутилом, 1,2-диметилбутилом, 2,2-диметилбутилом, 1,3-диметилбутилом, 2-этилбутилом, 2-метилпентилом, 3-метилпентилом, 4-метилпентилом, 2,3-диметилбутилом, н-гептилом, 2-метилгексилом, 3-метилгексилом, 4-метилгексилом, 5-метилгексилом, 2,3-диметилпентилом, 2,4-диметилпентилом, 2,2-диметилпентилом, 3,3-диметилпентилом, 2-этилпентилом, 3-этилпентилом, н-октилом, 2,3-диметилгексилом, 2,4-диметилгексилом, 2,5-диметилгексилом, 2,2-диметилгексилом, 3,3-диметилгексилом, 4,4-диметилгексилом, 2-этилгексилом, 3-этилгексилом, 4-этилгексилом, 2-метил-2-этилпентилом, 2-метил-3-этилпентилом, н-нонилом, 2-метил-2-этилгексилом, 2-метил-3-этилгексилом, 2,2-диэтилпентилом, н-децилом, 3,3-диэтилгексилом, 2,2-диэтилгексилом, и изомеры его разветвленной цепи. Более предпочтительно, алкильная группа представляет собой низший алкил, имеющий от 1 до 6 атомов углерода. Типичные примеры включают, но не ограничиваются метилом, этилом, н-пропилом, изопропилом, н-бутилом, изобутилом, трет-бутилом, втор-бутилом, н-пентилом, 1,1-диметилпропилом, 1,2-диметилпропилом, 2,2-диметилпропилом, 1-этилпропилом, 2-метилбутилом, 3-метилбутилом, н-гексилом, 1-этил-2-метилпропилом, 1,1,2-триметилпропилом, 1,1-диметилбутилом, 1,2-диметилбутилом, 2,2-диметилбутилом, 1,3-диметилбутилом, 2-этилбутилом, 2-метилпентилом, 3-метилпентилом, 4-метилпентилом, 2,3-диметилбутилом и т.д. Алкильная группа может быть замещенной или незамещенной. В случае замещения группа(ы) заместителя предпочтительно представляет собой одну или более чем одну группу, независимо выбранную из группы, состоящей из алкила, алкенила, алкинила, алкилокси, алкилсульфо, алкиламино, галогена, тиола, гидроксила, нитро, циано, циклоалкила, гетероциклического алкила, арила, гетероарила, циклоалкилокси, гетероциклического алкилокси, циклоалкилтио, гетероциклического алкилтио, оксо, -OR7, -S(O)mR7, -C(O)R7, -C(O)OR7, -NR8R9 и -C(O)NR8R9.
"Циклоалкил" относится к насыщенной и/или частично ненасыщенной моноциклической или полициклической углеводородной группе и имеет от 3 до 20 атомов углерода. Предпочтительно, циклоалкильная группа представляет собой циклоалкил, имеющий от 3 до 12 атомов углерода. Более предпочтительно, циклоалкильная группа представляет собой циклоалкил, имеющий от 3 до 10 атомов углерода. Типичные примеры моноциклического циклоалкила включают, но не ограничиваются циклопропилом, циклобутилом, циклопентилом, циклопентенилом, циклогексилом, циклогексенилом, циклогексадиенилом, циклогептилом, циклогептатриенилом, циклооктилом и т.д. Полициклический циклоалкил включает циклоалкил, имеющий спирокольцо, конденсированное кольцо и связанное мостиковой связью кольцо.
"Спироциклоалкил" относится к 5-20-членной полициклической углеводородной группе, причем кольца связаны через один общий атом углерода (также названный спироатомом), где одно или более чем одно кольцо может содержать одну или более чем одну двойную связь, но ни одно из колец не имеет полностью конъюгированной системы π-электронов. Предпочтительно, спироциклоалкил является от 6 до 14-членным, более предпочтительно является 7-10 членным. В соответствии с количеством общих спироатомов между спирокольцами спироциклоалкил разделяется на моноциклическое спирокольцо, бициклическое спирокольцо или мультициклическое спирокольцо, предпочтительно относится к моноциклическому спирокольцу или бициклическому спирокольцу. Более предпочтительно, спироциклоалкил представляет собой 4-членное/4-членное, 4-членное/5-членное, 4-членное/6-членное, 5-членное/5-членное или 5-членное/6-членное моноциклическое спирокольцо. Типичные примеры спироциклоалкила включают, но не ограничиваются следующими группами:
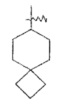 ,
, 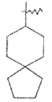 ,
, 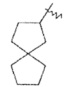 ,
, 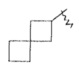 и
и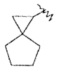 .
.
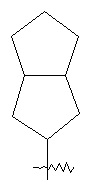
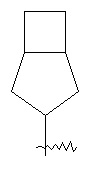
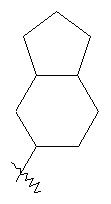
"Конденсированный циклоалкил" относится к 5-20 членной полициклической углеводородной группе, где каждое кольцо в системе имеет общую соседнюю пару атомов углерода с другим кольцом, где одно или более чем одно кольцо может содержать одну или более чем одну двойную связь, но ни одно из колец не обладает полностью конъюгированной системой π-электронов. Предпочтительно, конденсированная циклоалкильная группа является 6-14-членной, более предпочтительно, 7-10 членной. В соответствии с количеством кольцевых членов конденсированный циклоалкил разделяется на конденсированное бициклическое кольцо, трициклическое кольцо, тетрациклическое кольцо или мультициклическое кольцо, предпочтительно конденсированное бициклическое кольцо или трициклическое кольцо. Более предпочтительно, конденсированный циклоалкил представляет собой 5-членное/5-членное или 5-членное/6-членное конденсированное бициклическое кольцо. Типичные примеры конденсированного циклоалкила включают, но не ограничиваются следующими группами:
 ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
, 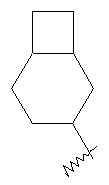 и
и 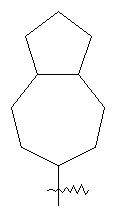
"Связанный мостиковой связью циклоалкил" относится к 5-20-членной полициклической углеводородной группе, где каждые два кольца в системе являются общими с двумя разъединенными атомами углерода. Указанные кольца могут иметь одну или более чем одну двойную связь, но не обладают полностью конъюгированной системой π-электронов.
Предпочтительно, связанный мостиковой связью циклоалкил является 6-14-членным, более предпочтительно 7-10-членным. В соответствии с количеством кольцевых членов, связанный мостиковой связью циклоалкил разделяется на связанное мостиковой связью бициклическое кольцо, трициклическое кольцо, тетрациклическое кольцо или мультициклическое кольцо, предпочтительно связанный мостиковой связью с бициклическим кольцом, трициклическим кольцом или тетрациклическим кольцом циклоалкил, более предпочтительно, связанный мостиковой связью с бициклическим кольцом или трициклическим кольцом циклоалкил. Типичные примеры связанного мостиковой связью циклоалкила включают, но не ограничиваются следующими группами:
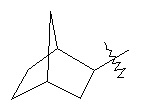 ,
,  ,
, 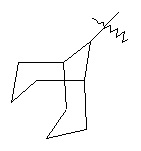 ,
, 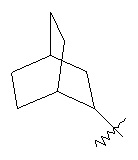 ,
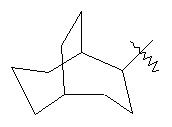
, 
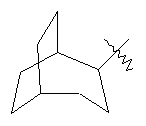 ,
, 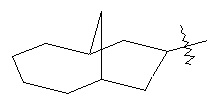 и
и  .
.
Указанный циклоалкил может быть конденсирован с арилом, гетероарилом или гетероциклическим алкилом, где кольцо, связанное с родительской структурой, представляет собой циклоалкил. Типичные примеры связанного мостиковой связью циклоалкила включают, но не ограничиваются инданилуксусным, тетрагидронафталином, бензоцидогептилом и т.п. Указанный циклоалкил может быть замещенным или незамещенным. В случае замещенного циклоалкила группа(ы) заместителя предпочтительно представляет собой одну или более чем одну группу, независимо выбранную из группы, состоящей из алкила, алкенила, алкинила, алкилокси, алкилсульфо, алкиламино, галогена, тиола, гидроксила, нитро, циано, циклоалкила, гетероциклического алкила, арила, гетероарила, циклоалкилокси, гетероциклического алкилокси, циклоалкилтио, гетероциклического алкилтио, оксо, -OR7, -S(O)mR7, -C(O)R7, -C(O)OR7, -NR8R9 и -C(O)NR8R9.
"Алкенил" относится к определенному выше алкилу, который имеет по меньшей мере два атома углерода и по меньшей мере одну углерод-углеродную двойную связь. Например, он относится к винилу, 1-пропенилу, 2-пропенилу, 1-, 2- или 3-бутенилу и т.д. Алкенильная группа может быть замещенной или незамещенной. Когда алкенильная группа является замещенной, то группа(ы) заместителя предпочтительно представляет собой одну или более чем одну группу, независимо выбранную из группы, состоящей из алкила, алкенила, алкинила, алкилокси, алкилсульфо, алкиламино, галогена, тиола, гидроксила, нитро, циано, циклоалкила, гетероциклического алкила, арила, гетероарила, циклоалкилокси, гетероциклического алкилокси, циклоалкилтио, гетероциклического алкилтио, оксо, -OR7, -S(O)mR7, -C(O)R7, -C(O)OR7, -NR8R9 и -C(O)NR8R9.
"Алкинил" относится к определенному выше алкилу, имеющему по меньшей мере два атома углерода и по меньшей мере одну углерод-углеродную тройную связь. Например, он относится к этинилу, 1-пропинилу, 2-пропинилу, 1-, 2- или 3-бутинилу и т.п. Алкинильная группа может быть замещенной или незамещенной. Когда алкинильная группа является замещенной, тогда группа(ы) заместителя предпочтительно представляет собой одну или более чем одну, независимо выбранную из группы, состоящей из алкила, алкенила, алкинила, алкилокси, алкилсульфо, алкиламино, галогена, тиола, гидроксила, нитро, циано, циклоалкила, гетероциклического алкила, арила, гетероарила, циклоалкилокси, гетероциклического алкилокси, циклоалкилтио, гетероциклического алкилтио, оксо, -OR7, -S(O)mR7, -C(O)R7, -C(O)OR7, -NR8R9 и -C(O)NR8R9.
"Гетероциклический алкил" относится к 3-20 членной насыщенной и/или частично ненасыщенной моноциклической или полициклической углеводородной группе, имеющей один или более чем один гетероатом, выбранный из группы, состоящей из N, O и S(O)m (где m равен 0, 1 или 2), в качестве кольцевых атомов, но за исключением -O-O-, -O-S- или -S-S- в кольце, причем оставшиеся кольцевые атомы представляют собой C. Предпочтительно, гетероциклический алкил является 3-12-членным, имеющим от 1 до 4 указанных гетероатомов; более предпочтительно, является 3-10-членным. Типичные примеры моноциклического гетероциклического алкила включают, но не ограничиваются пирролидилом, пиперидилом, пиперазинилом, морфолинилом, сульфо-морфолинилом, гомопиперазинилом и т.п. Полициклический гетероциклический алкил включает гетероциклический алкил, имеющий спирокольцо, конденсированное кольцо и связанное мостиковой связью кольцо. "Спирогетероциклоалкил" относится к 5-20-членной полициклической гетероциклической алкильной группе с кольцами, связанными посредством одного общего атома углерода (названного спироатомом), где указанные кольца имеют один или более чем один гетероатом, выбранный из группы, состоящей из N, О и S(O)p (где р равен 0, 1 или 2) в качестве кольцевых атомов, причем оставшиеся кольцевые атомы представляют собой С, где одно или более чем одно кольцо может содержать одну или более чем одну двойную связь, но ни одно из колец не обладает полностью конъюгированной системой π-электронов. Предпочтительно спирогетероциклический алкил является 6-14-членным, более предпочтительно 7-10-членным. В соответствии с количеством общих атомов спирогетероциклический алкил разделяется на моноциклический спирогетероциклоалкил, бициклический спирогетероциклический алкил или мультициклический спирогетероциклоалкил, предпочтительно моноциклический спирогетероциклический алкил или бициклический спирогетероциклоалкил. Более предпочтительно, спирогетероциклический алкил представляет собой 4-членный/4-членный, 4-членный/5-членный, 4-членный/6-членный, 5-членный/5-членный или 5-членный/6-членный моноциклический спирогетероциклоалкил. Типичные примеры спирогетероциклического алкила включают, но не ограничиваются следующими группами:
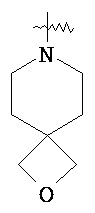 ,
, 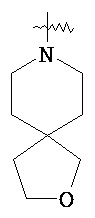 ,
, 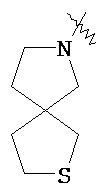 ,
, 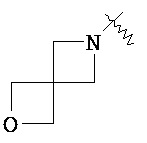 и
и 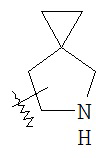 .
.
"Конденсированный гетероциклический алкил" относится к 5-20 членной полициклической гетероциклической алкильной группе, где каждое кольцо в системе имеет общую соседнюю пару атомов углерода с другим кольцом, где одно или более чем одно кольцо может содержать одну или более чем одну двойную связь, но ни одно из колец не имеет полностью конъюгированной системы π-электронов, и где указанные кольца имеют один или более чем один гетероатом, выбранный из группы, состоящей из N, O и S(O)p (где p равен 0, 1 или 2), в качестве кольцевых атомов, причем оставшиеся кольцевые атомы представляют собой C.
Предпочтительно, конденсированный гетероциклический алкил является 6-14 членным, более предпочтительно 7-10 членным. В соответствии с количеством кольцевых членов конденсированный гетероциклический алкил разделяется на конденсированное бициклическое кольцо, трициклическое кольцо, тетрациклическое кольцо или мультициклическое кольцо, предпочтительно конденсированное бициклическое кольцо или трициклическое кольцо. Более предпочтительно, конденсированный гетероциклический алкил представляет собой 5-членное/5-членное или 5-членное/6-членное конденсированное бициклическое кольцо. Типичные примеры конденсированного гетероциклического алкила включают, но не ограничиваются следующими группами:
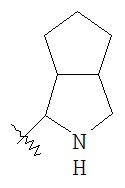 ,
,  ,
,  ,
,  ,
,  ,
,  ,
, 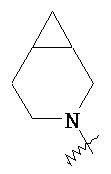 ,
, 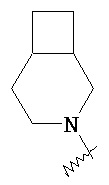 и
и 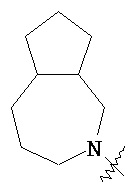 ,
, 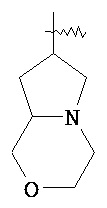 ,
,  и
и 
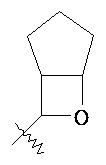
"Связанный мостиковой связью гетероциклический алкил" относится к 5-14-членной полициклической гетероциклической алкильной группе, где каждые два кольца в системе имеют общими два разомкнутых атома углерода, где указанные кольца могут иметь одну или более чем одну двойную связь, но не обладать полностью конъюгированной системой π-электронов, и указанные кольца имеют один или более чем один гетероатом, выбранный из группы, состоящей из N, О и S(O)m (где m равен 0, 1 или 2) в качестве кольцевых атомов, причем оставшиеся кольцевые атомы представляют собой C. Предпочтительно связанный мостиковой связью гетероциклический алкил является 6-14 членным, более предпочтительно 7-10 членным. В соответствии с количеством кольцевых членов связанный мостиковой связью гетероциклический алкил разделяется на связанное мостиковой связью бициклическое кольцо, трициклическое кольцо, тетрациклическое кольцо или мультициклическое кольцо, предпочтительно связанный мостиковой связью с бициклическим кольцом, трициклическим кольцом или тетрациклическим кольцом гетероциклоалкил, более предпочтительно связанный мостиковой связью с бициклическим кольцом или трициклическим кольцом гетероциклоалкил. Типичные примеры связанного мостиковой связью гетероциклического алкила включают, но не ограничиваются следующими группами:
 ,
,  ,
,  ,
,  ,
,  ,
,  ,
, 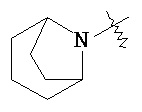 и
и 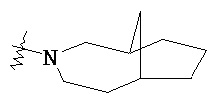 .
.
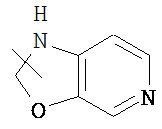
Указанный гетероциклический алкил может быть конденсирован с арилом, гетероциклическим алкилом или циклоалкилом, где кольцо, связанное с родительской структурой, представляет собой гетероциклический алкил. Типичные примеры гетероциклического алкила включают, но не ограничиваются следующими группами:
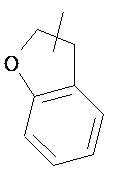 ,
,  ,
, 
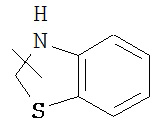 и т.п. Гетероциклический алкил может быть замещенным или незамещенным. Когда гетероциклический алкил является замещенным, тогда группа(ы) заместителя предпочтительно представляет собой одну или более чем одну группу, независимо выбранную из группы, состоящей из алкила, алкенила, алкинила, алкилокси, алкилсульфо, алкиламино, галогена, тиола, гидроксила, нитро, циано, циклоалкила, гетероциклического алкила, арила, гетероарила, циклоалкилокси, гетероциклического алкилокси, циклоалкилтио, гетероциклического алкилтио, оксо, -OR7, -S(O)mR7, -C(O)R7, -C(O)OR7, -NR8R9 и -C(O)NR8R9.
и т.п. Гетероциклический алкил может быть замещенным или незамещенным. Когда гетероциклический алкил является замещенным, тогда группа(ы) заместителя предпочтительно представляет собой одну или более чем одну группу, независимо выбранную из группы, состоящей из алкила, алкенила, алкинила, алкилокси, алкилсульфо, алкиламино, галогена, тиола, гидроксила, нитро, циано, циклоалкила, гетероциклического алкила, арила, гетероарила, циклоалкилокси, гетероциклического алкилокси, циклоалкилтио, гетероциклического алкилтио, оксо, -OR7, -S(O)mR7, -C(O)R7, -C(O)OR7, -NR8R9 и -C(O)NR8R9.
"Арил" относится к 6-14-членной полностью состоящей из углеродов моноциклической кольцевой или мультициклической конденсированной кольцевой ("конденсированная" кольцевая система означает, что каждое кольцо в системе имеет общую соседнюю пару атомов углерода с другим кольцом в системе) группе, и обладает полностью конъюгированной системой π-электронов. Предпочтительно арил является 6-10-членным, таким как фенил и нафтил. Указанный арил может быть конденсирован с гетероарилом, гетероциклическим алкилом или циклоалкилом, где кольцо, связанное с родительской структурой, представляет собой арил. Типичные примеры арила включают, но не ограничиваются следующими группами:
 ,
,  ,
,  ,
,  ,
, 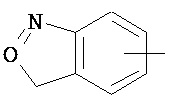 ,
, 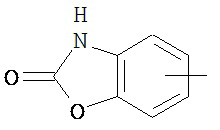 ,
, 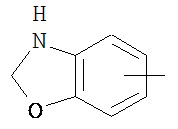 ,
, 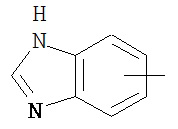 ,
, 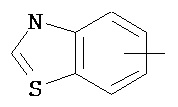 ,
, 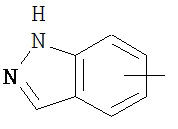 ,
, 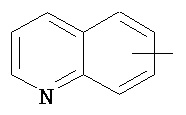 ,
, 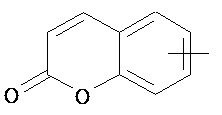 и
и  .
.
Арильная группа может быть замещенной или незамещенной. Когда арильная группа является замещенной, тогда группа(ы) заместителя предпочтительно представляет собой одну или более чем одну группу, независимо выбранную из группы, состоящей из алкила, алкенила, алкинила, алкилокси, алкилсульфо, алкиламино, галогена, тиола, гидроксила, нитро, циклоалкила, гетероциклического алкила, арила, гетероарила, циклоалкилокси, гетероциклического алкилокси, циклоалкилтио, гетероциклического алкилтио, оксо, -OR7, -S(O)mR7, -C(O)R7, -C(O)OR7, -NR8R9 и -C(O)NR8R9.
"Гетероарил" относится к гетероарилу, имеющему от 1 до 4 гетероатомов, выбранных из группы, состоящей из N, O и S, в качестве кольцевых атомов, и имеет от 5 до 14 кольцевых атомов, предпочтительно 5-10-членное кольцо, более предпочтительно 5- или 6-членное кольцо. Примеры гетероарильных групп включают фурил, тиенил, пиридил, пирролил, N-алкилпирролил, пиримидинил, пиразинил, имидазолил и т.п. Указанный гетероарил может быть конденсирован с кольцом арила, гетероциклической группой или циклоалкилом, где кольцо, связанное с родительской структурой, представляет собой гетероарил. Типичные примеры включают, но не ограничиваются следующими группами:
 ,
,  ,
,  ,
,  ,
,  ,
, 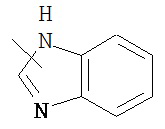 ,
, 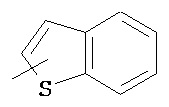 ,
, 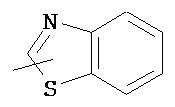 ,
, 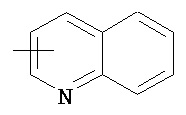 , и
, и  .
.
Гетероарильная группа может быть замещенной или незамещенной. Когда гетероарильная группа является замещенной, тогда группа(ы) заместителя представляет собой предпочтительно одну или более чем одну группу, независимо выбранную из группы, состоящей из алкила, алкенила, алкинила, алкилокси, алкилсульфо, алкиламино, галогена, тиола, гидроксила, нитро, циано, циклоалкила, гетероциклического алкила, арила, гетероарила, циклоалкилокси, гетероциклического алкилокси, циклоалкилтио, гетероциклического алкилтио, оксо, -OR7, -S(O)mR7, -C(O)R7, -C(O)OR7, -NR8R9 и -C(O)NR8R9.
"Алкоксил" относится к -О-(алкильной) группе, где алкил определен выше. Типичные примеры включают, но не ограничиваются метокси, этокси, пропокси, бутокси и т.п. Алкоксил может быть замещенным или незамещенным. Когда алкоксил является замещенным, тогда заместитель предпочтительно представляет собой одну или более чем одну группу, независимо выбранную из группы, состоящей из алкила, алкенила, алкинила, алкилокси, алкилсульфо, алкиламино, галогена, тиола, гидроксила, нитро, циано, циклоалкила, гетероциклического алкила, арила, гетероарила, циклоалкилокси, гетероциклического алкилокси, циклоалкилтио, гетероциклического алкилтио, оксо, -OR7, -S(O)mR7, -C(O)R7, -C(O)OR7, -NR8R9 или -C(O)NR8R9.
"Циклоалкокси" относится к -O-(циклоалкильной) группе, где циклоалкил определен выше. Типичные примеры включают, но не ограничиваются циклопропилокси, циклобутилокси, циклопентилокси, циклогексилокси и т.п. Циклоалкокси может быть замещенным или незамещенным. Когда циклоалкокси является замещенным, тогда заместитель предпочтительно представляет собой одну или более чем одну группу, независимо выбранную из группы, состоящей из алкила, алкенила, алкинила, алкилокси, алкилсульфо, алкиламино, галогена, тиола, гидроксила, нитро, циано, циклоалкила, гетероциклического алкила, арила, гетероарила, циклоалкилокси, гетероциклического алкилокси, циклоалкилтио, гетероциклического алкилтио, оксо, -OR7, -S(O)mR7, -C(O)R7, -C(O)OR7, -NR8R9 или -C(O)NR8R9.
"Гидрокси" относится к группе -OH.
"Галоген" относится к фтору, хлору, брому или йоду.
"Амино" относится к группе -NH2.
"Циано" относится к группе -CN.
"Нитро" относится к группе -NO2.
"Бензил" относится к -СН2-фенильной группе.
"Оксо" относится к группе=O.
"Карбоновая кислота" относится к группе -С(O)ОН.
"Эфир карбоновой кислоты" относится к группе -С(O)O(алкил) или (циклоалкильной) группе.
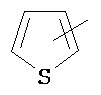
"Тиенил" относится к группе
"Возможный" или "возможно" означает, что далее описанное событие или явление может произойти или не произойти, и что описание включает примеры, когда событие или явление может произойти или не произойти. Например, "гетероциклическая группа, возможно, дополнительно замещенная алкильной группой" означает, что алкил может присутствовать или отсутствовать, и описание включает ситуации, когда гетероциклическая группа замещена алкильной группой, и ситуации, когда гетероциклическая группа не замещена алкильной группой.
"Замещенный" относится к одному или более чем одному атому водорода в группе, предпочтительно максимально к 5, более предпочтительно 1-3, независимо замещенному соответствующим количеством заместителей. Безусловно, что заместители располагаются только в возможном химическом положении. Специалисты в данной области техники способны определить (при помощи экспериментов или теоретически) возможные или невозможные заместители, не прилагая избыточных усилий. Например, группы являются нестабильными при связывании аминогруппы или гидроксильной группы, имеющей свободный водород, с атомами углерода, имеющими ненасыщенную связь (такую как олефиновая).
"Фармацевтическая композиция" относится к смеси одного или более чем одного описанного в данном документе соединения, или его физиологически/фармацевтически приемлемых солей или пролекарств с другими химическими компонентами, такими как физиологически/фармацевтически приемлемые носители и эксципиенты. Задача фармацевтической композиции заключается в том, чтобы облегчить введение соединения в организм, что делает поглощение активного ингредиента более эффективным.
Состояния, заболевания и расстройства, собирательно названные "Синдромом X" (также известным как метаболический синдром), подробно описаны в Johannsson, J.Clin. Endocrinol. Metab., 1997;82, 727-734, включенном в данный документ путем ссылки.
m и R7-R9 являются такими, как определено в формуле (I).
Конкретные способы воплощения
Для реализации задачи изобретения в нем применяется следующее техническое решение.
Способ получения соединений формулы (I) или их фармацевтически приемлемых солей или их стереохимических изомеров по настоящему изобретению включает следующие стадии:
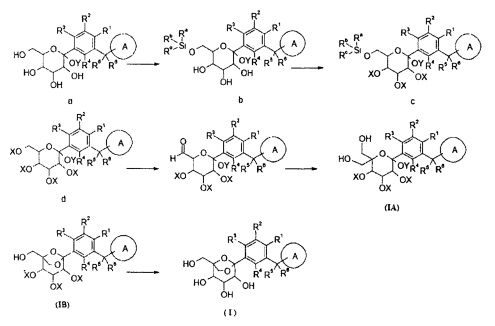
В основных условиях соединение а приводили во взаимодействие с хлорсилановым реагентом с получением при комнатной температуре защищенного силилом соединения b, которое обрабатывают NaH и бензилбромидом с получением соединения c. В растворе метанола силильную защитную группу соединения c удаляют при помощи хлористого ацила с получением гидроксильного соединения d, которое окисляют с получением альдегида e. Затем соединение е приводили во взаимодействие с гидроксидом натрия в растворе формальдегида с получением двойного гидроксильного соединения f (также названного соединением (IA)). После обработки трифторуксусной кислотой (TFA) соединение (IA) превращали в соединение (IB), и бензильные защитные группы соединения (IB) восстанавливали путем каталитического гидрирования с палладием на угле с получением соединения формулы (I), где:
R1-R6 являются такими, как определено в формуле (I) выше;
X и Y представляют собой защитные группы гидроксила, предпочтительно алкильную или бензильную группу.
Примеры воплощения изобретения
Настоящее изобретение дополнительно описано при помощи следующих примеров, которые не предназначены для того, чтобы ограничивать объем изобретения.
Примеры
Структуры всех соединений идентифицировали при помощи ядерного магнитного резонанса (1Н ЯМР) и/или масс-спектрометрии (МС). Химические сдвиги в 1Н ЯМР регистрировали в виде млн-1 (10-6). 1Н ЯМР осуществляли на спектрометре Bruker AVANCE-400. Подходящие растворители включали дейтерированный метанол (CD3OD), дейтерированный хлороформ (CDCl3) и дейтерированный диметилсульфоксид (DMSO-d6) с тетраметилсиланом (TMS) в качестве внутреннего стандарта.
МС определяли на масс-спектрометре FINNIGAN LCQ Ad (ESI) (Thermo, Model: Finnigan LCQ advantage MAX).
ВЭЖХ (высокоэффективная жидкостная хроматография) проводили на спектрометре системы жидкостной хроматографии высокого давления Agilent 1200DAD (хроматографическая колонка Sunfire C18 150*4,6 мм) и системы жидкостной хроматографии высокого давления Waters 2695-2996 (хроматографическая колонка Gimini C18 150*4,6 мм).
Используемый для тонкослойной хроматографии силикагель представлял собой пластину силикагеля Yantai Huanghai HSGF254 или Qingdao GF254. Размер используемых пластин для ТСХ (тонкослойной хроматографии) составлял от 0,15 мм до 0,2 мм, и размер пластин, используемых при очистке продукта, составлял от 0,4 мм до 0,5 мм.
Используемая колоночная хроматография, как правило, представляла собой Yantai Huanghai с силикагелем от 200 до 300 меш в качестве носителя.
Исходные вещества по настоящему изобретению являлись известными или были приобретены в ABCR GmbH & Co. KG, Acros Organics, Aldrich Chemical Company, Acceia ChemBio Inc, Darui Finechemical Co., Ltd и т.д, или они могли быть получены при помощи обычных для предшествующего уровня техники способов синтеза.
Термин "атмосфера аргона" или "атмосфера азота" относился к такой атмосфере, для создания которой реакционную колбу оборудовали баллоном, заполненным приблизительно 1 л аргона или азота.
Термин "атмосфера водорода" относился к такой атмосфере, для создания которой реакционную колбу оборудовали баллоном, заполненным приблизительно 1 л водорода.
Реакции гидрогенизации под давлением осуществляли с использованием спектрометра гидрогенизации Parr 3916ЕКХ и генератора водорода QL-500, или спектрометра гидрогенизации HC2-SS.
В реакциях гидрогенизации реакционную систему, как правило, вакуумировали и заполняли водородом; в то же время вышеприведенную операцию повторяли трижды.
Микроволновую реакцию осуществляли в микроволновом реакторе типа СЕМ Discover-S-908860.
Если не указано иное, то реакцию проводили в атмосфере азота или аргона.
Если не указано иное, то раствор, используемый в примерах, относился к водному раствору.
Если не указано иное, то температура реакции представляла собой комнатную температуру.
Комнатная температура представляла собой наиболее нормальную температуру реакции, которая составляла 20°C-30°C.
Развитие реакции в примерах контролировали при помощи тонкослойной хроматографии (ТСХ). Хроматографическая система растворителей включает систему дихлорметана и метанола, систему гексана и этилацетата, систему петролейного эфира и этилацетата, и ацетон. Объемное отношение растворителей корректировали в соответствии с полярностью соединений.
Элюирующая система колоночной хроматографии и система растворителей тонкослойной хроматографии включает: A: систему дихлорметана и метанола, В: систему н-гексана и этилацетата, C: систему дихлорметана и ацетона. Объемное отношение растворителей корректировали в соответствии с полярностью соединений, и иногда его также корректировали путем добавления основного агента, такого как триэтиламин, или кислотного агента, такого как уксусная кислота.
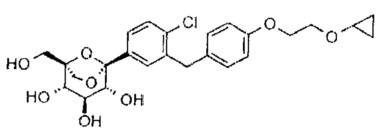
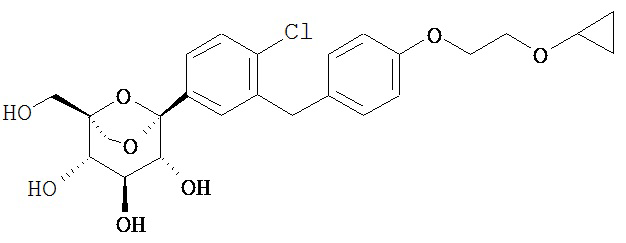
Пример 1
(1S,2S,3S,4R,5R)-5-[4-хлор-3-[[4-[2-(циклопропокси)этокси]фенил]метил]фенил]-1-(гидроксиметил)-6,8-диоксабицикло[3.2.1]октан-2,3,4-триол
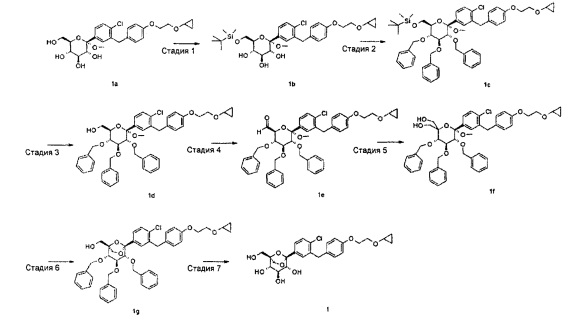
Стадия 1
(2S,3R,4S,5S,6R)-6-[(трет-бутил(диметил)силил)оксиметил]-2-[4-хлор-3-[[4-[2-(циклопропокси)этокси]фенил]метил]фенил]-2-метокси-тетрагидропиран-3,4,5-триол
(2S,3R,4S,5S,6R)-2-[4-хлор-3-[[4-[2-(циклопропокси)этокси]фенил]метил]фенил]-6-(гидроксиметил)-2-метокси-тетрагидропиран-3,4,5-триол 1a (Полученный согласно способу в WO 2010022313) (3,0 г, 6,06 ммоль) растворяли в 20 мл пиридина с последующим добавлением один за другим 4-диметиламино пиридина (148 мг, 1,21 ммоль) и TBSCl (1,1 г, 7,27 ммоль). Реакционную смесь перемешивали в течение 16 часов, затем концентрировали при пониженном давлении. Остаток растворяли в 100 мл этилацетата и 100 мл воды, и он распределялся. Водную фазу экстрагировали этилацетатом (100 мл), и органический слой промывали водой (50 мл), объединяли, сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении. Получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой A с получением указанного в заголовке соединения (2S,3R,4S,5S,6R)-6-[(трет-бутил(диметил)силил)оксиметил]-2-[4-хлор-3-[[4-[2-(циклопропокси)этокси]фенил]метил]фенил]-2-метокси-тетрагидропиран-3,4,5-триола 1b (3,0 г, желтое твердое вещество), выход: 81,1%.
1Н ЯМР (400 МГц, CD3OD): δ 7.50 (dd, 1Н), 7.38 (m, 2Н), 7.08 (d, 2Н), 6.82 (d, 2Н), 4.04 (m, 5Н), 3.88 (m, 1Н), 3.82 (m, 2Н), 3.75 (m, 1Н), 3.59 (m, 1Н), 3.40(m, 2Н), 3.07 (m, 1Н), 3.06 (s, 3H), 0.90 (s, 9Н), 0.53 (m, 4Н), 0.10 (s, 3H), 0.07 (s, 3H).
Стадия 2
[[(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[4-[2-(циклопропокси)этокси]фенил]метил]фенил]-6-метокси-тетрагидропиран-2-ил]метокси]-трет-бутил-диметил-силан
(2S,3R,4S,5S,6R)-6-[(трет-бутил(диметил)силил)оксиметил]-2-[4-хлор-3-[[4-[2-(циклопропокси)этокси]фенил]метил]фенил]-2-метокси-тетрагидропиран-3,4,5-триол 1b (3,0 г, 4,92 ммоль) растворяли в 50 мл N.N-диметилформамида и охлаждали до 0°C. Добавляли 60% NaH (984 мг, 24,6 ммоль). Затем реакционную смесь нагревали до комнатной температуры и перемешивали в течение 15 минут перед тем, как добавляли бензилбромид (2,95 мл, 24,6 ммоль). Смесь перемешивали в течение 16 часов. Реакционную смесь концентрировали при пониженном давлении после добавления 5 мл метанола. Остаток растворяли в 100 мл этилацетата, и он распределялся. Органические экстракты промывали водой (50 мл × 2) и объединяли, сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения [[(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[4-[2-(циклопропокси)этокси]фенил]метил]фенил]-6-метокси-тетрагидропиран-2-ил]метокси]-трет-бутил-диметил-силана 1c (4,26 г, желтое жироподобное вещество), который использовали непосредственно на следующей стадии без очистки.
Стадия 3
[(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[4-[2-(циклопропокси)этокси]фенил]метил]фенил]-6-метокси-тетрагидропиран-2-ил]метанол
Неочищенный [[(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[4-[2-(циклопропокси)этокси]фенил]метил]фенил]-6-метокси-тетрагидропиран-2-ил]метокси]-трет-бутил-диметил-силан 1c (4,26 г, 4,92 ммоль) растворяли в 30 мл метанола с последующим добавлением ацетилхлорида (52 мкл, 0,74 ммоль). Реакционную смесь перемешивали в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении и очищали при помощи хроматографии на силикагеле с элюирующей системой В с получением указанного в заголовке соединения [(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[4-[2-(циклопропокси)этокси]фенил]метил]фенил]-6-метокси-тетрагидропиран-2-ил]метанола 1d (2,3 г, желтое жироподобное вещество), выход: 62,2%.
1H ЯМР (400 МГц, CDCl3): δ 7.35 (m, 13Н), 7.20 (m, 3H), 7.03 (m, 4Н), 6.80 (d, 2Н), 4.92 (m, 3H), 4.70 (m, 1Н), 4.50 (m, 1Н), 4.17 (m, 1Н), 4.05 (m, 3H), 3.85 (m, 6Н), 3.70 (m, 2Н), 3.40 (m, 1Н), 3.30 (m, 1Н), 3.07 (s, 3H), 0.63 (m, 2Н), 0.48 (m, 2Н).
Стадия 4
(2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[4-[2-(циклопропокси)этокси]фенил]метил]фенил]-6-метокси-тетрагидропиран-2-карбальдегид
Оксалилхлорид (0,19 мл, 2,2 ммоль) растворяли в 10 мл метиленхлорида, охлаждали до -78°C, а затем по каплям добавляли 5 мл раствора диметилсульфоксида в метиленхлориде и 10 мл [(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[4-[2-(циклопропокси)этокси]фенил]метил]фенил]-6-метокси-тетрагидропиран-2-ил]метанола 1d (1,3 г, 1,7 ммоль) в метиленхлориде. Смесь перемешивали в течение 30 минут при -78°C. После этого смесь нагревали до комнатной температуры и перемешивали в течение 1-2 часов, после чего добавляли триэтиламин (1,18 мл, 8,5 ммоль). Реакционная смесь распределялась после добавления 10 мл 1 М соляной кислоты. Органическую фазу промывали насыщенным раствором хлорида натрия (10 мл × 2), объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения (2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[4-[2-(циклопропокси)этокси]фенил]метил]фенил]-6-метокси-тетрагидропиран-2-карбальдегида 1e (1,3 г, бесцветное масло), который использовали непосредственно на следующей стадии без очистки. МС m/z (ESI (ионизация путем распыления электронов)): 780,3 [M+18]
Стадия 5
[(3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[4-[2-(циклопропокси)этокси]фенил]метил]фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-ил]метанол
Неочищенный (2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[4-[2-(циклопропокси)этокси]фенил]метил]фенил]-6-метокси-тетрагидропиран-2-карбальдегид 1е (1,3 г, 1,7 ммоль) растворяли в 15 мл 1,4-диоксана с последующим добавлением в реакционную смесь 37% раствора формальдегида (2,6 мл, 34 ммоль) и раствора гидроксида натрия (204 мг, 5,1 ммоль) в 5,1 мл воды. Реакционную смесь перемешивали в течение 4 часов при 70°C, затем охлаждали до 50°C и перемешивали в течение 16 часов. Реакционную смесь экстрагировали этилацетатом (20 мл × 3) после того, как добавляли 20 мл насыщенного раствора хлорида натрия. Органический экстракт промывали насыщенным раствором бикарбоната натрия (20 мл), насыщенным раствором хлорида натрия (20 мл), объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении. Затем 20 мл смешанного раствора (THF (тетрагидрофуран) и MeOH, v:v=1:1) добавляли к остатку перед тем, как добавляли боргидрид натрия (130 мг, 3,4 ммоль). Реакционную смесь перемешивали в течение 30 минут и концентрировали при пониженном давлении. Остаток растворяли в этилацетате (50 мл), и он распределялся. Органический экстракт промывали насыщенным раствором хлорида натрия (10 мл × 2), объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении, затем получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой В с получением указанного в заголовке соединения [(3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[4-[2-(циклопропокси)этокси]фенил]метил]фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-ил]метанола 1f (320 мг, бесцветное масло), выход: 23,7%.
Стадия 6
[(1S,2S,3S,4R,5S)-2,3,4-трибензилокси-5-[4-хлор-3-[[4-[2-(циклопропокси)этокси]фенил]метил]фенил]-6,8-диоксабицикло[3.2.1]октан-1-ил]метанол
[(3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[4-[2-(циклопропокси)этокси]фенил]метил]фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-ил]метанол 1f (320 мг, 0,4 ммоль) растворяли в 10 мл метиленхлорида и охлаждали до -10°C с последующим добавлением трифторуксусной кислоты (62 мл, 0,8 ммоль). Смесь перемешивали в течение 1 часа. После этого реакционная смесь распределялась после добавления 10 мл насыщенного раствора бикарбоната натрия. Водную фазу экстрагировали дихлорметаном (10 мл), и органический экстракт промывали насыщенным раствором хлорида натрия (10 мл), объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении, и получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой В с получением указанного в заголовке соединения [(1S,2S,3S,4R,5R)-2,3,4-трибензилокси-5-[4-хлор-3-[[4-[2-(циклопропокси)этокси]фенил]метил]фенил]-6,8-диоксабицикло[3.2.1]октан-1-ил]метанола 1g (230 мг, бесцветное масло), выход: 75,3%. МС m/z (ESI): 780,3 [М+18]
1Н ЯМР (400 МГц, CDCl3): δ 7.33 (m, 12Н), 7.15 (m, 4Н), 7.05 (m, 2Н), 6.86 (d, 2Н), 6.76 (d, 2Н), 4.77 (m, 4Н), 4.27 (m, 2Н), 4.00 (m, 6Н), 3.83 (m, 3H), 3.70 (m, 4Н), 3.38 (m, 1Н), 0.63 (m, 2Н), 0.48 (m, 2Н).
Стадия 7
(1S,2S,3S,4R,5R)-5-[4-хлор-3-[[4-[2-(циклопропокси)этокси]фенил]метил]фенил]-1-(гидроксиметил)-6,8-диоксабицикло[3.2.1]октан-2,3,4-триол
[(1S,2S,3S,4R,5R)-2,3,4-трибензилокси-5-[4-хлор-3-[[4-[2-(циклопропокси)этокси]фенил]метил]фенил]-6,8-диоксабицикло[3.2.1]октан-1-ил]метанол 1g (220,3 мг, 0,29 ммоль) растворяли в 10 мл смешанного раствора (THF и MeOH, v:v=1:1) с последующим добавлением 1,2-дихлорбензола (0,34 мл, 3 ммоль) и палладия на угле (90 мг, 10%). Смесь трижды продували H2 и перемешивали в течение 3 часов, фильтровали. Фильтрат концентрировали при пониженном давлении, и получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой A с получением указанного в заголовке соединения (1S,2S,3S,4R,5R)-5-[4-хлор-3-[[4-[2-(циклопропокси)этокси]фенил]метил]фенил]-1-(гидроксиметил)-6,8-диоксабицикло[3.2.1]октан-2,3,4-триола 1 (140 мг, белое твердое вещество), выход: 100%.
МС m/z (ESI): 510,2 [М+18]
1Н ЯМР (400 МГц, CD3OD): δ 7.45 (d, 1Н), 7.36 (m, 2Н), 7.10 (d, 2Н), 6.82 (m, 2Н), 4.14 (d, 1Н), 4.05 (m, 4Н), 3.83 (m, 3H), 3.78 (m, 1Н), 3.66 (m, 2Н), 3.57 (m, 2Н), 3.40 (m, 1Н), 0.56 (m, 2Н), 0.48 (m, 2Н).
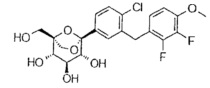
Пример 2
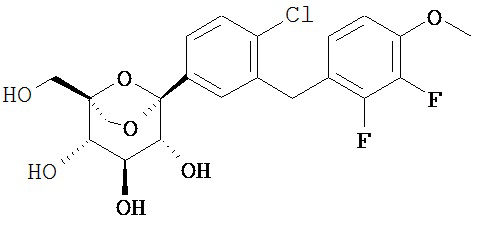
(1R,2R,3S,4S,5S)-5-[4-хлор-3-[(2,3-дифтор-4-метокси-фенил)метил]фенил]-1-(гидроксиметил)-6,8-диоксабицикло[3.2.1]октан-2,3,4-триол
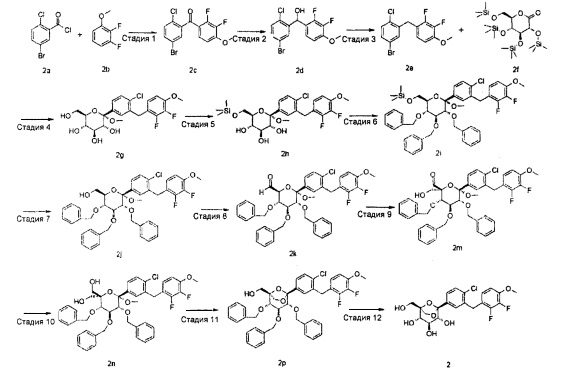
Стадия 1
(5-бром-2-хлор-фенил)-(2,3-дифтор-4-метокси-фенил)он
5-бром-2-хлор-бензоилхлорид 2a (7,22 г, 28,45 ммоль) и 1,2-дифтор-3-метокси-бензол 2b (4,1 г, 28,45 ммоль, полученный согласно способу в CN 2003468 A) растворяли в 50 мл метиленхлорида и охлаждали до 0°C с последующим добавлением порции трихлорида алюминия (3,4 г, 25,6 ммоль). Смесь перемешивали в течение 16 часов. Затем смесь распределялась после добавления 20 мл 1 M соляной кислоты, и ее экстрагировали дихлорметаном (50 мл × 2). Органический экстракт промывали насыщенным раствором карбоната натрия (50 мл) и насыщенным раствором хлорида натрия (50 мл), объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (5-бром-2-хлор-фенил)-(2,3-дифтор-4-метокси-фенил)она 2c (5,1 г, желтое твердое вещество), выход: 49,5%.
МС m/z (ESI): 362,9 [М+18]
Стадия 2
(5-бром-2-хлор-фенил)-(2,3-дифтор-4-метокси-фенил)метанол
(5-бром-2-хлор-фенил)-(2,3-дифтор-4-метокси-фенил)он 2c (5,1 г, 14,1 ммоль) растворяли в 40 мл смешанного раствора (THF и МеОН, v:v=1:1) и охлаждали до 0°C с последующим добавлением порции боргидрида натрия (1,07 г, 28,2 ммоль). Реакционную смесь перемешивали в течение 30 минут. После этого реакционную смесь концентрировали при пониженном давлении после того, как добавляли 10 мл ацетона. Получающийся в результате остаток растворяли в этилацетате (100 мл), и он распределялся. Органический экстракт промывали водой (20 мл × 2), объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (5-бром-2-хлор-фенил)-(2,3-дифтор-4-метокси-фенил)метанола 2d (5,1 г, желтое жироподобное вещество), выход: 99,6%.
Стадия 3
1-[(5-бром-2-хлор-фенил)метил]-2,3-дифтор-4-метокси-бензол
(5-бром-2-хлор-фенил)-(2,3-дифтор-4-метокси-фенил)метанол 2d (5,1 г, 14,1 ммоль) растворяли в 40 мл метиленхлорида с последующим добавлением триэтилсилана (6,75 мл, 42,3 ммоль) и добавлением по каплям диэтилового эфира трифторида бора (3,57 мл, 28,2 ммоль). Реакционную смесь перемешивали в течение 16 часов. После этого реакционная смесь распределялась после добавления 20 мл насыщенного раствора карбоната натрия. Водную фазу экстрагировали дихлорметаном (20 мл × 3), и органический экстракт объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении, и получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой B с получением указанного в заголовке соединения 1-[(5-бром-2-хлор-фенил)метил]-2,3-дифтор-4-метокси-бензола 2e (3,55 г, белое твердое вещество), выход: 72,4%.
1Н ЯМР (400 МГц, CDCl3): δ 7.36-7.33 (m, 1Н), 7.30-7.27 (m, 2Н), 6.81-6.79 (m, 1Н), 6.73-6.69 (m, 1Н), 4.00 (s, 2Н), 3.92 (s, 3H).
Стадия 4
(2S,3R,4S,5S,6R)-2-[4-хлор-3-[(2,3-дифтор-4-метокси-фенил)метил]фенил]-6-(гидроксиметил)-2-метокси-тетрагидропиран-3,4,5-триол
1-[(5-Бром-2-хлор-фенил)метил]-2,3-дифтор-4-метокси-бензол 2е (3,55 г, 10,2 ммоль) растворяли в 30 мл смешанного раствора (THF и толуол, v:v=1:2) и охлаждали до -78°C, а затем по каплям добавляли раствор nBuLi в н-гексане (4,9 мл, 12,26 ммоль). После перемешивания в течение 1 часа при -78°C добавляли раствор (3R,4S,5R,6R)-3,4,5-трис(триметилсилилокси)-6-(триметилсилилоксиметил)тетрагидропиран-2-она 2f (5,24 г, 11,22 ммоль, полученный согласно способу в WO 2010048358) в толуоле (30 мл). Реакционную смесь перемешивали в течение 3 часов при -78°C. После этого реакционную смесь концентрировали при пониженном давлении после добавления 30 мл насыщенного раствора карбоната натрия. Остаток растворяли в 30 мл насыщенного раствора хлорида натрия и экстрагировали этилацетатом (50 мл × 3), и органический экстракт объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении, и получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой A с получением указанного в заголовке соединения (2S,3R,4S,5S,6R)-2-[4-хлор-3-[(2,3-дифтор-4-метокси-фенил)метил]фенил]-6-(гидроксиметил)-2-метокси-тетрагидропиран-3,4,5-триола 2g (1,31 г, белое твердое вещество), выход: 27,9%.
МС m/z (ESI): 429,1 [М-31]
Стадия 5
(2S,3R,4S,5S,6R)-2-[4-хлор-3-[(2,3-дифтор-4-метокси-фенил)метил]фенил]-2-метокси-6-(триметилсилилоксиметил)тетрагидропиран-3,4,5-триол
(2S,3R,4S,5S,6R)-2-[4-хлор-3-[(2,3-дифтор-4-метокси-фенил)метил]фенил]-6-(гидроксиметил)-2-метокси-тетрагидропиран-3,4,5-триол 2g (1,31 г, 2,84 ммоль) растворяли в 15 мл пиридина с последующим добавлением 4-диметиламинопиридина (70 мг, 0,57 ммоль) и триметил-хлор-силана (514 мг, 3,4 ммоль). Реакционную смесь перемешивали в течение 16 часов и затем концентрировали при пониженном давлении. Получающийся в результате остаток растворяли в 150 мл этилацетата и промывали по очереди пиридином (50 мл × 2), водой (30 мл) и насыщенным раствором хлорида натрия (30 мл). Органический экстракт объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (2S,3R,4S,5S,6R)-2-[4-хлор-3-[(2,3-дифтор-4-метокси-фенил)метил]фенил]-2-метокси-6-(триметилсилилоксиметил)тетрагидропиран-3,4,5-триола 2h (1,63 г, бледно-желтое твердое вещество), выход: 100%.
Стадия 6
[[(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[(2,3-дифтор-4-метокси-фенил)метил]фенил]-6-метокси-тетрагидропиран-2-ил]метокси]триметилсилан
(2S,3R,4S,5S,6R)-2-[4-хлор-3-[(2,3-дифтор-4-метокси-фенил)метил]фенил]-2-метокси-6-(триметилсилилоксиметил)тетрагидропиран-3,4,5-триол 2h (1,63 г, 2,84 ммоль) растворяли в 30 мл DMF (диметилформамид) и охлаждали до 0°C с последующим добавлением 60% NaH (570 мг, 14,2 ммоль). Затем реакционную смесь нагревали до комнатной температуры и перемешивали в течение 45 минут. После этого добавляли бензилбромид (1,7 мл, 14,2 ммоль) перед тем, как реакционную смесь перемешивали в течение ночи. Реакционную смесь концентрировали при пониженном давлении после добавления 5 мл метанола. После добавления к остатку 150 мл этилацетата и 50 мл воды получающийся в результате остаток распределялся, и органический экстракт промывали водой (50 мл), объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения [[(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[(2,3-дифтор-4-метокси-фенил)метил]фенил]-6-метокси-тетрагидропиран-2-ил]метокси]триметилсилана 2i (2,4 г, желтое жироподобное вещество) с выходом:100%.
Стадия 7
[(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[(2,3-дифтор-4-метокси-фенил)метил]фенил]-6-метокси-тетрагидропиран-2-ил]метанол
[[(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[(2,3-дифтор-4-метокси-фенил)метил]фенил]-6-метокси-тетрагидропиран-2-ил]метокси]триметилсилан 2i (2,4 г, 2,84 ммоль) растворяли в 20 мл метанола и перемешивали в течение 1 часа после добавления ацетилхлорида (30 мкл, 0,43 ммоль). Реакционную смесь концентрировали при пониженном давлении, и получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой В с получением указанного в заголовке соединения [(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[(2,3-дифтор-4-метокси-фенил)метил]фенил]-6-метокси-тетрагидропиран-2-ил]метанола 2j (1,25 г, желтое твердое вещество) с выходом: 60,4%.
Стадия 8
(2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[(2,3-дифтор-4-метокси-фенил)метил]фенил]-6-метокси-тетрагидропиран-2-карбальдегид
Оксалилхлорид растворяли в 5 мл метиленхлорида и охлаждали до -78°C, а затем по каплям добавляли 3 мл раствора диметилсульфоксида ((0,26 мл, 3,59 ммоль) в метиленхлориде. Реакционную смесь перемешивали в течение 15 минут перед тем, как по каплям добавляли 5 мл раствора [(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[(2,3-дифтор-4-метокси-фенил)метил]фенил]-6-метокси-тетрагидропиран-2-ил]метанола 2j (1,25 г, 1,71 ммоль) в метиленхлориде. Смесь перемешивали в течение 40 минут. По каплям добавляли триэтиламин (1,19 мл, 8,55 ммоль) перед тем, как реакционную смесь нагревали до комнатной температуры и перемешивали в течение 1,5 часов. После этого реакционную смесь промывали 5 мл 1 M соляной кислоты, и органический экстракт собирали, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[(2,3-дифтор-4-метокси-фенил)метил]фенил]-6-метокси-тетрагидропиран-2-карбальдегида 2k (1,24 г, желтое твердое вещество), который использовали на следующей стадии без очистки.
Стадия 9
(2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[(2,3-дифтор-4-метокси-фенил)метил]фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-карбальдегид
(2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[(2,3-дифтор-4-метокси-фенил)метил]фенил]-6-метокси-тетрагидропиран-2-карбальдегид 2k (1,24 г, 1,71 ммоль) растворяли в 15 мл 1,4-диоксана с последующим добавлением 37%-40% водного раствора формальдегида (2,8 мл, 34,2 ммоль) и 1,5 мл раствора гидроксида натрия (205 мг, 5,13 ммоль). Реакционную смесь перемешивали в течение ночи при 70°C. После этого реакционную смесь концентрировали при пониженном давлении, и она распределялась после добавления 30 мл этилацетата и 15 мл насыщенного раствора хлорида натрия. Водную фазу экстрагировали этилацетатом (15 мл × 2), и органический экстракт объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[(2,3-дифтор-4-метокси-фенил)метил]фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-карбальдегида 2m (1,29 г, желтое жироподобное вещество), который использовали непосредственно на следующей стадии без очистки.
Стадия 10
[(3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[(2,3-дифтор-4-метокси-фенил)метил]фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-ил]метанол
(2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[(2,3-дифтор-4-метокси-фенил)метил]фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-карбальдегид 2m (1,29 г, 1,71 ммоль) растворяли в 15 мл смешанного раствора (THF и MeOH, v:v=1:2) с последующим добавлением боргидрида натрия (129 мг, 3,42 ммоль). Реакционную смесь перемешивали в течение 20 минут. После этого реакционную смесь концентрировали при пониженном давлении, и она распределялась после добавления 30 мл этилацетата и 15 мл насыщенного раствора хлорида натрия. Водную фазу экстрагировали этилацетатом (15 мл × 2), и органический экстракт объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении, и получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой В с получением указанного в заголовке соединения [(3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[(2,3-дифтор-4-метокси-фенил)метил]фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-ил]метанола 2n (520 мг, белое твердое вещество) с выходом: 40%.
Стадия 11
[(1R,2R,3S,4S,5S)-2,3,4-трибензилокси-5-[4-хлор-3-[(2,3-дифтор-4-метокси-фенил)метил]фенил]-6,8-диоксабицикло[3.2.1]октан-1-ил]метанол
[(3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[(2,3-дифтор-4-метокси-фенил)метил]фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-ил]метанол 2n (500 мг, 0,66 ммоль) растворяли в 10 мл дихлорметана, а затем по каплям добавляли трифторуксусную кислоту (0,2 мл, 2,62 ммоль). Реакционную смесь перемешивали в течение 1,5 часов. После этого реакционную смесь промывали 10 мл насыщенного раствор бикарбоната натрия, и органический экстракт объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении, и получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой В с получением указанного в заголовке соединения [(1R,2R,3S,4S,5S)-2,3,4-трибензилокси-5-[4-хлор-3-[(2,3-дифтор-4-метокси-фенил)метил]фенил]-6,8-диоксабицикло[3.2.1]октан-1-ил]метанола 2p (360 мг, белое твердое вещество) с выходом: 75,3%.
МС m/z(ESI): 746,2 [М+18]
Стадия 12
(1R,2R,3S,4S,5S)-5-[4-хлор-3-[(2,3-дифтор-4-метокси-фенил)метил]фенил]-1-(гидроксиметил)-6,8-диоксабицикло[3.2.1]октан-2,3,4-триол
[(1R,2R,3S,4S,5S)-2,3,4-трибензилокси-5-[4-хлор-3-[(2,3-дифтор-4-метокси-фенил)метил]фенил]-6,8-диоксабицикло[3.2.1]октан-1-ил]метанол 2p (350 мг, 0,48 ммоль) растворяли в 10 мл смешанного раствора (THF и МеОН, v:v=1:1) с последующим добавлением по очереди о-дихлорбензола (0,55 мл, 4,8 ммоль) и палладия на угле (300 мг, 10%). Смесь трижды продували Н2 и перемешивали в течение 3 часов, фильтровали через силикагель, и фильтрат концентрировали при пониженном давлении, и получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой А с получением указанного в заголовке соединения (1R,2R,3S,4S,5S)-5-[4-хлор-3-[(2,3-дифтор-4-метокси-фенил)метил]фенил]-1-(гидроксиметил)-6,8-диоксабицикло[3.2.1]октан-2,3,4-триола 2 (210 мг, белое твердое вещество), выход: 95,5%.
МС m/z (ESI): 459,1 [М+1]
1Н ЯМР (400 МГц, CD3OD): δ 7.42 (m, 3H), 6.81 (m, 2Н), 4.16 (d, 1Н), 4.10 (s, 2Н), 3.87 (s, 3H), 3.82 (m, 2Н), 3.68 (m, 2Н), 3.55 (m, 1Н), 3.61 (m, 1Н).
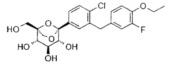
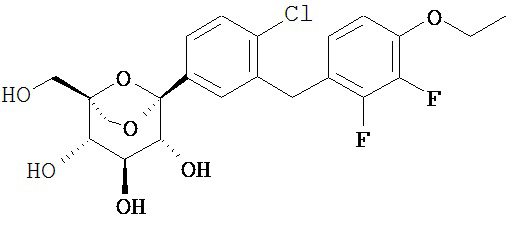
Пример 3
(1S,2S,3S,4R,5S)-5-[4-хлор-3-[(4-этокси-2,3-дифтор-фенил)метил]фенил]-1-(гидроксиметил)-6,8-диоксабицикло[3.2.1]октан-2,3,4-триол
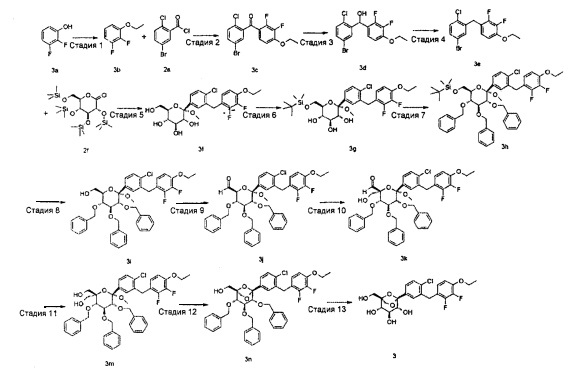
Стадия 1
1-Этокси-2,3-дифтор-бензол
2,3-дифторфенол 3a (4 г, 30,7 ммоль) растворяли в 60 мл ацетона с последующим добавлением карбоната калия (6,36 г, 46,1 ммоль) и этилйодида (3,19 мл, 39,9 ммоль). Реакционную смесь перемешивали в течение 5 часов при 70°C. После этого реакционную смесь фильтровали. Фильтрат концентрировали при пониженном давлении. Получающийся в результате остаток растворяли в 100 мл этилацетата и промывали водой (100 мл) и насыщенным раствором хлорида натрия (100 мл), и органический экстракт объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения 1-этокси-2,3-дифтор-бензола 3b (4,82 г, желтое жироподобное вещество), выход: 99,4%.
1Н ЯМР (400 МГц, CDCl3): δ 6.95-6.89 (m, 1Н), 6.78-6.71 (m, 1Н), 4.12 (q, 2Н), 1.45 (t, 3H)
Стадия 2
(5-Бром-2-хлор-фенил)-(4-этокси-2,3-дифтор-фенил)метанон
5-Бром-2-хлор-бензоилхлорид 2а (7,74 г, 30,5 ммоль) растворяли в 200 мл дихлорметана с последующим добавлением 1-этокси-2,3-дифтор-бензола 3b (4,82 г, 30,5 ммоль) и трихлорида алюминия (4,07 г, 30,5 ммоль). Реакционную смесь перемешивали в течение 16 часов. После этого смесь распределялась после добавления 100 мл 2 М соляной кислоты. Органический экстракт промывали насыщенным раствором хлорида натрия (100 мл) и объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (5-бром-2-хлор-фенил)-(4-этокси-2,3-дифтор-фенил)метанона 3с (8,0 г, желтое жироподобное вещество), выход: 70,2%.
МС m/z (ESI): 376,9 [М+1]
Стадия 3
(5-Бром-2-хлор-фенил)-(4-этокси-2,3-дифтор-фенил)метанол
(5-Бром-2-хлор-фенил)-(4-этокси-2,3-дифтор-фенил)метанон 3c (8,0 г, 21,3 ммоль) растворяли в 240 мл смешанного раствора (THF и МеОН, v:v=1:1) с последующим добавлением боргидрида калия (1,73 г, 32,0 ммоль) в ледяной бане. Реакционную смесь перемешивали в течение 16 часов при комнатной температуре. После этого добавляли 50 мл 1 М соляной кислоты. Реакционную смесь концентрировали при пониженном давлении, и экстрагировали дихлорметаном (100 мл × 2). Органический экстракт объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (5-бром-2-хлор-фенил)-(4-этокси-2,3-дифтор-фенил)метанола 3d (8,0 г, желтое жироподобное вещество), который использовали непосредственно на следующей стадии без очистки.
Стадия 4
1-[(5-Бром-2-хлор-фенил)метил]-4-этокси-2,3-дифтор-бензол
(5-Бром-2-хлор-фенил)-(4-этокси-2,3-дифтор-фенил)метанол 3d (8,0 г, 21.2 ммоль) растворяли в 150 мл смешанного раствора (ацетонитрил и дихлорметан, v:v=2:1) с последующим добавлением триэтилсилана (10,1 мл, 63,6 ммоль) и эфирата трифторида бора (5,3 мл, 42,4 ммоль). Реакционную смесь перемешивали в течение 3 часов перед тем, как добавляли 100 мл 2 М гидроксида калия. Органический экстракт объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении, и получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой С с получением указанного в заголовке соединения 1-[(5-бром-2-хлор-фенил)метил]-4-этокси-2,3-дифтор-бензола 3е (5,5 г, белое твердое вещество), выход: 72,4%.
МС m/z (ESI): 360,5 [М+1]
1Н ЯМР (400 МГц, CDCl3): δ 7.36-7.27 (m, 3H), 6.81-6.76 (dd, 1Н), 6.72-6.68 (dd, 1Н), 4.17-4.13 (q, 2Н), 4.10 (s, 2Н), 1.50-1.47 (t, 3H)
Стадия 5
(2S,3R,4S,5S,6R)-2-[4-хлор-3-[(4-этокси-2,3-дифтор-фенил)метил]фенил]-6-(гидроксиметил)-2-метокси-тетрагидропиран-3,4,5-триол
1-[(5-бром-2-хлор-фенил)метил]-4-этокси-2,3-дифтор-бензол 3е (5,5 г, 15.3 ммоль) растворяли в 20 мл THF и охлаждали до -78°C, а затем по каплям добавляли раствор н-BuLi в н-гексане (7,3 мл, 18,3 ммоль). Реакционную смесь перемешивали в течение 1 часа при -78°C. Добавляли 30 мл раствора (3R,4S,5R,6R)-3,4,5-трис-(триметилсилилокси)-6-(триметилсилилоксиметил)тетрагидропиран-2-она 2f (7,5 г, 16,1 ммоль) в THF при -78°C перед тем, как реакционную смесь перемешивали в течение 2 часов при -78°C. Добавляли раствор (51 мл) 0,6 М метансульфоновой кислоты в метаноле перед тем, как реакционную смесь нагревали и перемешивали в течение 16 часов при комнатной температуре. После этого реакционную смесь концентрировали при пониженном давлении, растворяли в воде (50 мл) и экстрагировали этилацетатом (100 мл × 4). Органический экстракт объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении, и получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой A с получением указанного в заголовке соединения (2S,3R,4S,5S,6R)-2-[4-хлор-3-[(4-этокси-2,3-дифтор-фенил)метил]фенил]-6-(гидроксиметил)-2-метокси-тетрагидропиран-3,4,5-триола 3f (3,05 г, белое твердое вещество), выход: 45,5%.
Стадия 6
(2S,3R,4S,5S,6R)-6-[(трет-бутил(диметил)силил)оксиметил]-2-[4-хлор-3-[(4-этокси-2,3-дифтор-фенил)метил]фенил]-2-метокси-тетрагидропиран-3,4,5-триол
(2S,3R,4S,5S,6R)-2-[4-хлор-3-[(4-этокси-2,3-дифтор-фенил)метил]фенил]-6-(гидроксиметил)-2-метокси-тетрагидропиран-3,4,5-триол 3f (3,0 г, 6,8 ммоль) растворяли в 30 мл пиридина с последующим добавлением по очереди 4-диметиламинопиридина (166 мг, 1,36 ммоль) и TBSCI (трет-бутилдиметилсилилхлорид) (1,23 г, 8,2 ммоль). Реакционную смесь перемешивали в течение 16 часов. После этого реакционную смесь концентрировали при пониженном давлении перед тем, как добавляли 150 мл этилацетата. Реакционную смесь по очереди промывали насыщенным раствором сульфата меди (100 мл) и насыщенным раствором хлорида натрия (100 мл). Органический экстракт объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (2S,3R,4S,5S,6R)-6-[(трет-бутил(диметил)силил)оксиметил]-2-[4-хлор-3-[(4-этокси-2,3-дифтор-фенил)метил]фенил]-2-метокси-тетрагидропиран-3,4,5-триола 3g (3,75 г, белое твердое вещество), выход: 99,7%.
Стадия 7
[[(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[(4-этокси-2,3-дифтор-фенил)метил]фенил]-6-метокси-тетрагидропиран-2-ил]трет-бутил-диметил-метокси]силан
(2S,3R,4S,5S,6R)-6-[(трет-бутил(диметил)силил)оксиметил]-2-[4-хлор-3-[(4-этокси-2,3-дифтор-фенил)метил]фенил]-2-метокси-тетрагидропиран-3,4,5-триол 3g (3,75 г, 6,8 ммоль) растворяли в 30 мл DMF и охлаждали до 0°C с последующим добавлением 60% NaH (1,36 г, 34 ммоль). Реакционную смесь нагревали и перемешивали в течение 45 минут при комнатной температуре перед тем, как по очереди добавляли тетрабутиламмоний йодид (125 мг, 0,34 ммоль) и бензилбромид (4,01 мл, 34 ммоль). После перемешивания в течение 16 часов при комнатной температуре реакционную смесь концентрировали при пониженном давлении после добавления 10 мл метанола. Получающийся в результате остаток растворяли в 100 мл этилацетата, и он распределялся. Органический экстракт промывали водой (50 мл × 2) и объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения [[(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[(4-этокси-2,3-дифтор-фенил)метил]фенил]-6-метокси-тетрагидропиран-2-ил]трет-бутил-диметил-метокси]силана 3h (4,2 г, бесцветное жироподобное вещество), выход: 75,0%.
Стадия 8
[(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[(4-этокси-2,3-дифтор-фенил)метил]фенил]-6-метокси-тетрагидропиран-2-ил]метанол
[[(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[(4-этокси-2,3-дифтор-фенил)метил]фенил]-6-метокси-тетрагидропиран-2-ил]трет-бутил-диметил-метокси]силан 3h (4,7 г, 5,46 ммоль) растворяли в 50 мл метанола с последующим добавлением ацетилхлорида (80 мг, 0,82 ммоль). Реакционную смесь перемешивали в течение 1 часа при комнатной температуре. После этого реакционную смесь концентрировали при пониженном давлении, и получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой В с получением указанного в заголовке соединения [(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[(4-этокси-2,3-дифтор-фенил)метил]фенил]-6-метокси-тетрагидропиран-2-ил]метанола 3i (2,5 г, желтое жироподобное вещество), выход: 61,4%.
Стадия 9
(2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[(4-этокси-2,3-дифтор-фенил)метил]фенил]-6-метокси-тетрагидропиран-2-карбальдегид
Оксалилхлорид (0,37 мл, 4,37 ммоль) растворяли в 20 мл дихлорметана и охлаждали до -78°C, а затем по очереди по каплям добавляли раствор (10 мл) диметилсульфоксида (0,5 мл, 7,05 ммоль) в дихлорметане и раствор (20 мл) [(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[(4-этокси-2,3-дифтор-фенил)метил]фенил]-6-метокси-тетрагидропиран-2-ил]метанола 3i (2,5 г, 3,36 ммоль) в дихлорметане. Реакционную смесь перемешивали в течение 30 минут при -78°C. Добавляли триэтиламин (2,33 мл, 16,8 ммоль) перед тем, как реакционную смесь нагревали и перемешивали в течение 16 часов при комнатной температуре. После этого реакционная смесь распределялась после добавления 15 мл 1 М соляной кислоты. Органический экстракт промывали насыщенным раствором хлорида натрия (20 мл × 2) и объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[(4-этокси-2,3-дифтор-фенил)метил]фенил]-6-метокси-тетрагидропиран-2-карбальдегида 3j (2,4 г, желтое жироподобное вещество), выход: 96,0%.
Стадия 10
(2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[(4-этокси-2,3-дифтор-фенил)метил]фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-карбальдегид
(2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[(4-этокси-2,3-дифтор-фенил)метил]фенил]-6-метокси-тетрагидропиран-2-карбальдегид 3j (2,4 г, 3,23 ммоль) растворяли в 20 мл 1,4-диоксана с последующим добавлением 5 мл 37% раствора формальдегида и 13 мл 1 М гидроксида натрия. Реакционную смесь перемешивали в течение 21 часа при 70°C. После этого реакционную смесь экстрагировали этилацетатом (50 мл × 3) после добавления 20 мл насыщенного раствора хлорида натрия. Органический экстракт объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[(4-этокси-2,3-дифтор-фенил)метил]фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-карбальдегида 3k (1,6 г, желтое жироподобное вещество), выход: 64,0%.
Стадия 11
[(3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[(4-этокси-2,3-дифтор-фенил)метил]фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-ил]метанол
(2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[(4-этокси-2,3-дифтор-фенил)метил]фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-карбальдегид 3k (1,6 г, 2,1 ммоль) растворяли в 35 мл смешанного раствора (THF и MeOH, v:v=2:5) с последующим добавлением боргидрида натрия (78 мг, 4,2 ммоль). Реакционную смесь перемешивали в течение 2 часов. После этого реакционную смесь концентрировали при пониженном давлении, и получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой В с получением указанного в заголовке соединения [(3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[(4-этокси-2,3-дифтор-фенил)метил]фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-ил]метанола 3m (1,0 г, желтое жироподобное вещество), выход: 62,5%.
Стадия 12
[(1S,2S,3S,4R,5S)-2,3,4-трибензилокси-5-[4-хлор-3-[(4-этокси-2,3-дифтор-фенил)-метил]фенил]-6,8-диоксабицикло[3.2.1]октан-1-ил]метанол[(3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[(4-этокси-2,3-дифтор-фенил)метил]фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-ил]метанол 3m (1,0 г, 1,29 ммоль) растворяли в 10 мл дихлорметана с последующим добавлением трифторуксусной кислоты (0,19 мл, 2,58 ммоль). Реакционную смесь перемешивали в течение 4 часов. После этого реакционную смесь концентрировали при пониженном давлении, и получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой B с получением указанного в заголовке соединения [(1S,2S,3S,4R,5S)-2,3,4-трибензилокси-5-[4-хлор-3-[(4-этокси-2,3-дифтор-фенил)метил]фенил]-6,8-диоксабицикло[3.2.1]октан-1-ил]метанола 3n (500 мг, желтое жироподобное вещество), выход: 52,1%.
МС m/z (ESI): 760,3 [М+18]
1Н ЯМР (400 МГц, CD3OD): δ 7.37-7.30 (m, 13Н), 7.23-7.18 (m, 3H), 6.91-6.89 (m, 2Н), 6.68 (dd, 1Н), 6.55 (dd, 1Н), 4.93-4.90 (m, 2Н), 4.79-4.56 (m, 4Н), 4.30-4.28 (m, 2Н), 4.18-4.05 (m, 4Н), 3.83-3.49 (m, 5Н), 1.33 (t, 3H).
Стадия 13
(1S,2S,3S,4R,5S)-5-[4-хлор-3-[(4-этокси-2,3-дифтор-фенил)метил]фенил]-1-(гидроксиметил)-6,8-диоксабицикло[3.2.1]октан-2,3,4-триол
[(1S,2S,3S,4R,5S)-2,3,4-трибензилокси-5-[4-хлор-3-[(4-этокси-2,3-дифтор-фенил)метил]фенил]-6,8-диоксабицикло[3.2.1]октан-1-ил]метанол 3n (550 мг, 0,74 ммоль) растворяли в 20 мл смешанного раствора (THF и МеОН, v:v=1:1) с последующим добавлением о-дихлорбензола (0,84 мл, 7,4 ммоль) и палладия на угле (300 мг, 10%). Смесь трижды продували Н2 и перемешивали в течение 3 часов. После этого реакционную смесь фильтровали после добавления небольшого количества этилацетата. Фильтрат концентрировали при пониженном давлении, и получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой A с получением указанного в заголовке соединения (1S,2S,3S,4R,5S)-5-[4-хлор-3-[(4-этокси-2,3-дифтор-фенил)метил]фенил]-1-(гидроксиметил)-6,8-диоксабицикло[3.2.1]октан-2,3,4-триола 3 (160 мг, белое твердое вещество), выход: 45,7%.
МС m/z (ESI): 472,2 [М+1]
1Н ЯМР (400 МГц, CDCl3): δ 7.43-7.37 (m, 3H), 6.80-6.76 (m, 2Н), 4.16-4.07 (m, 5Н), 3.85-3.70 (m, 2Н), 3.70-3.51 (m, 4Н), 1.34 (t, 3H).
Пример 4
(1S,2S,3S,4R,5S)-5-[4-хлор-3-[(4-этокси-2,3-дифтор-фенил)метил]фенил]-1-(гидроксиметил)-6,8-диоксабицикло[3.2.1]октан-2,3,4-триол
Стадия 1
1-Этокси-2-фтор-бензол
2-Фторфенол 4а (6,7 г, 60 ммоль) растворяли в 66 мл ацетона с последующим добавлением этилйодида (6,3 мл, 78 ммоль) и карбоната калия (12,4 г, 90 ммоль). Реакционную смесь кипятили с обратным холодильником в масляной бане в течение 5 часов. После этого реакционную смесь концентрировали при пониженном давлении, и она распределялась после добавления 100 мл этилацетата и 60 мл воды. Водную фазу экстрагировали этилацетатом (30 мл × 2), и органический экстракт объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения 1-этокси-2-фтор-бензола 4b (6,9 г, красное жироподобное вещество), выход: 82,1%.
МС m/z (ESI): 280,2 [2М+1]
Стадия 2
(5-Бром-2-хлор-фенил)-(4-этокси-3-фтор-фенил)метанон
5-Бром-2-хлор-бензоилхлорид 2а (12,4 г, 48,8 ммоль) и 1-этокси-2-фтор-бензол 4b (6,84 г, 48,8 ммоль) растворяли в 100 мл дихлорметана и охлаждали до 0°C с последующим добавлением порции трихлорида алюминия (5,86 г, 44 ммоль). Реакционную смесь перемешивали в течение 16 часов. После этого реакционная смесь распределялась после добавления по каплям 20 мл 2 М соляной кислоты в ледяной бане. Водную фазу экстрагировали 30 мл дихлорметана, и органический экстракт объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (5-бром-2-хлор-фенил)-(4-этокси-3-фтор-фенил)метанона 4c (12,7 г, желтое твердое вещество), выход: 72,6%.
МС m/z (ESI): 358,9 [М+1]
Стадия 3
(5-Бром-2-хлор-фенил)-(4-этокси-3-фтор-фенил)метанол
(5-Бром-2-хлор-фенил)-(4-этокси-3-фтор-фенил)метанон 4c (12,7 г, 35,5 ммоль) растворяли в 100 мл смешанного раствора (THF и MeOH, v:v=1:1) с последующим добавлением порции боргидрида натрия (2,68 г, 70 ммоль) в ледяной бане. Реакционную смесь нагревали и перемешивали в течение 30 минут при комнатной температуре. После этого реакционную смесь концентрировали при пониженном давлении после добавления 15 мл ацетона. Остаток растворяли в 150 мл этилацетата и промывали насыщенным раствором хлорида натрия (50 мл × 2). Органический экстракт объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (5-бром-2-хлор-фенил)-(4-этокси-3-фтор-фенил)метанола 4d (12,7 г, оранжевое жироподобное вещество), который использовали непосредственно на следующей стадии без очистки.
Стадия 4
4-[(5-Бром-2-хлор-фенил)метил]-1-этокси-2-фтор-бензол
(5-Бром-2-хлор-фенил)-(4-этокси-3-фтор-фенил)метанол 4d (12,7 г, 35,3 ммоль) растворяли в 100 мл дихлорметана с последующим добавлением триэтилмоносилана (16,9 мл, 106 ммоль) и добавлением по каплям эфирата трифторида бора (8,95 мл, 70,6 ммоль). Реакционную смесь перемешивали в течение 3 часов. После этого реакционная смесь распределялась после добавления 50 мл насыщенного раствор бикарбоната натрия. Водную фазу экстрагировали этилацетатом (100 мл × 2), и органический экстракт объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении, и получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой В с получением указанного в заголовке соединения 4-[(5-бром-2-хлор-фенил)метил]-1-этокси-2-фтор-бензола 4e (10 г, бледно-желтое жироподобное вещество), выход: 82,4%.
1Н ЯМР (400 МГц, CDCl3): δ 7.33-7.27 (m, 3H), 6.95-6.90 (m, 3H), 4.14 (q, 2Н), 4.01 (s, 2Н), 1.49 (t, 3H).
Стадия 5
(2S,3R,4S,5S,6R)-2-[4-хлор-3-[(4-этокси-3-фтор-фенил)метил]фенил]-6-(гидроксиметил)-2-метокси-тетрагидропиран-3,4,5-триол
4-[(5-Бром-2-хлор-фенил)метил]-1-этокси-2-фтор-бензол 4е (7,36 г, 21,4 ммоль) растворяли в 30 мл THF и охлаждали до -78°C, а затем по каплям добавляли раствор nBuLi в н-гексане (10,27 мл, 25,7 ммоль). После перемешивания в течение 1 часа при -78°C добавляли раствор (20 мл) (3R,4S,5R,6R)-3,4,5-трис(риметилсилилокси)-6-(триметилсилилоксиметил)тетрагидропиран-2-она 2f (11 г, 23,6 ммоль) в THF перед тем, как реакционную смесь перемешивали в течение 2 часов при -78°C. Затем реакционную смесь нагревали и перемешивали в течение 16 часов при комнатной температуре после добавления 2,8 мл метилсульфоновой кислоты и 71 мл метанола. После этого реакционную смесь концентрировали при пониженном давлении после добавления 100 мл насыщенного раствора карбоната натрия. Остаток растворяли в 50 мл насыщенного хлорида натрия, экстрагировали этилацетатом (100 мл × 3). Органический экстракт объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении, и получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой A с получением указанного в заголовке соединения (2S,3R,4S,5S,6R)-2-[4-хлор-3-[(4-этокси-3-фтор-фенил)метил]фенил]-6-(гидроксиметил)-2-метокси-тетрагидропиран-3,4,5-триола 4f (5,7 г, белое твердое вещество), выход: 58,3%.
1Н ЯМР (400 МГц, CD3OD): δ 7.56 (s, 1Н), 7.48 (dd, 1Н), 7.37 (dd, 1Н), 6.95-6.87 (m, 3H), 4.08-4.07 (m, 4Н), 3.91 (m, 1Н), 3.93-3.73 (m, 2Н), 3.56-3.53 (m, 1Н), 3.45-3.43 (m, 1Н), 3.30 (s, 2Н), 3.08 (s, 3H), 1.35 (t, 3H).
Стадия 6
(2S,3R,4S,5S,6R)-6-[(трет-бутил(диметил)силил)оксиметил]-2-[4-хлор-3-[(4-этокси-3-фтор-фенил)метил]фенил]-2-метокси-тетрагидропиран-3,4,5-триол
(2S,3R,4S,5S,6R)-2-[4-хлор-3-[(4-этокси-3-фтор-фенил)метил]фенил]-6-(гидроксиметил)-2-метокси-тетрагидропиран-3,4,5-триол 4f (5,7 г, 12,5 ммоль) растворяли в 50 мл пиридина с последующим добавлением по очереди TBSCI (2,26 г, 15 ммоль) и 4-диметиламинопиридина (305 мг, 2,5 ммоль). Реакционную смесь перемешивали в течение 16 часов. После этого реакционную смесь концентрировали при пониженном давлении. Остаток растворяли в 200 мл этилацетата и промывали насыщенным раствором сульфата меди (50 мл × 3). Органический экстракт объединяли, сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении с получением указанного в заголовке соединения (2S,3R,4S,5S,6R)-6-[(трет)-бутил(диметил)силил)оксиметил]-2-[4-хлор-3-[(4-этокси-3-фтор-фенил)метил]фенил]-2-метокси-тетрагидропиран-3,4,5-триола 4g (7,14 г, бесцветное жироподобное вещество), который использовали непосредственно на следующей стадии без очистки.
Стадия 7
[[(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[(4-этокси-3-фтор-фенил)метил]фенил]-6-метокси-тетрагидропиран-2-ил]метокси]трет-бутил-диметилсилан
(2S,3R,4S,5S,6R)-6-[(трет-бутил(диметил)силил)оксиметил]-2-[4-хлор-3-[(4-этокси-3-фтор-фенил)метил]фенил]-2-метокси-тетрагидропиран-3,4,5-триол 4g (7,14 г, 12,5 ммоль) растворяли в 100 мл N,N-диметилформамида с последующим добавлением 60% NaH (2,5 г, 62,5 ммоль) в ледяной бане. Реакционную смесь нагревали и перемешивали в течение 40 минут при комнатной температуре. Смесь перемешивали в течение 16 часов после добавления бензилбромида (7,5 мл, 62,5 ммоль). После этого реакционную смесь концентрировали при пониженном давлении после добавления 20 мл метанола. Остаток растворяли в 200 мл этилацетата и 50 мл воды, и он распределялся. Водную фазу экстрагировали этилацетатом (50 мл). Органические экстракты промывали водой (50 мл), затем насыщенным хлоридом натрия (50 мл), объединяли, сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении с получением указанного в заголовке соединения [[(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[(4-этокси-3-фтор-фенил)метил]фенил]-6-метокси-тетрагидропиран-2-ил]метокси]трет-бутил-диметил-силана 4h (10,5 г, желтое жироподобное вещество), выход: 99,8%.
Стадия 8
[(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[(4-этокси-3-фторфенил)метил]фенил]-6-метокси-тетрагидропиран-2-ил]метанол
[[(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[(4-этокси-3-фтор-фенил)метил]фенил]-6-метокси-тетрагидропиран-2-ил]метокси]трет-бутил-диметил-силан 4h (10,52 г, 12,5 ммоль) растворяли в 50 мл метанола, а затем по каплям добавляли ацетилхлорид (0,13 мл, 1,9 ммоль). Реакционную смесь перемешивали в течение 1 часа. После этого реакционную смесь концентрировали при пониженном давлении, и получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой В с получением указанного в заголовке соединения [(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[(4-этокси-3-фтор-фенил)метил]фенил]-6-метокси-тетрагидропиран-2-ил]метанола 4i (7,6 г, желтое жироподобное вещество), выход: 83,6%.
Стадия 9
(2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[(4-этокси-3-фтор-фенил)метил]фенил]-6-метокси-тетрагидропиран-2-карбальдегид
Оксалилхлорид (1,17 мл, 13,6 ммоль) растворяли в 20 мл дихлорметана и охлаждали до -78°C, а затем по каплям добавляли раствор (20 мл) диметилсульфоксида (1,56 мл, 21,9 ммоль) в дихлорметане и раствор (50 мл) [(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[(4-этокси-3-фтор-фенил)метил]фенил]-6-метокси-тетрагидропиран-2-ил]метанола 4i (7,6 г, 10,45 ммоль) в дихлорметане. Реакционную смесь перемешивали в течение 30 минут при -78°C. Добавляли триэтиламин (7,25 мл, 52,3 ммоль) перед тем, как реакционную смесь нагревали и перемешивали в течение 2 часов при комнатной температуре. После этого реакционная смесь распределялась после добавления 50 мл 1 М соляной кислоты. Органический экстракт промывали насыщенным раствором хлорида натрия (50 мл × 2). Водную фазу экстрагировали дихлорметаном (50 мл). Органический экстракт объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[(4-этокси-3-фтор-фенил)метил]фенил]-6-метокси-тетрагидропиран-2-карбальдегида 4j (7,58 г, бесцветное жироподобное вещество), который использовали непосредственно на следующей стадии без очистки.
Стадия 10
(2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[(4-этокси-3-фтор-фенил)метил]фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-карбальдегид
(2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[(4-этокси-3-фтор-фенил)метил]фенил]-6-метокси-тетрагидропиран-2-карбальдегид 4j (7,6 г, 10,45 ммоль) растворяли в 80 мл 1,4-диоксана с последующим добавлением по очереди 15,8 мл 37% раствора формальдегида и гидроксида натрия (31,35 мл, 31,35 ммоль). Реакционную смесь перемешивали в течение 16 часов при 70°C. После этого реакционную смесь экстрагировали этилацетатом (50 мл × 4) после добавления 50 мл насыщенного раствора хлорида натрия. Органический экстракт промывали насыщенным раствором бикарбоната натрия (50 мл) и насыщенным раствором хлорида натрия (50 мл), и объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[(4-этокси-3-фтор-фенил)метил]фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-карбальдегида 4k (7,9 г, бесцветное жироподобное вещество), который использовали непосредственно на следующей стадии без очистки.
Стадия 11
[(3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[(4-этокси-3-фтор-фенил)метил]-фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-ил]метанол
(2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[(4-этокси-3-фтор-фенил)метил]фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-карбальдегид 4k (7,9 г, 10,45 ммоль) растворяли в 50 мл смешанного раствора (THF и MeOH, v:v=2:3) с последующим добавлением боргидрида натрия (794 мг, 20,9 ммоль). Реакционную смесь перемешивали в течение 30 минут. После этого реакционную смесь концентрировали при пониженном давлении после добавления небольшого количества ацетона, и получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой А с получением указанного в заголовке соединения [(3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[(4-этокси-3-фторфенил)метил]фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-ил]метанола 4m (1,11 г, бесцветное жироподобное вещество), выход: 14,1%.
Стадия 12
[(1S,2S,3S,4R,5S)-2,3,4-трибензилокси-5-[4-хлор-3-[(4-этокси-3-фтор-фенил)метил]фенил]-6,8-диоксабицикло[3.2.1]октан-1-ил]метанол
[(3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[(4-этокси-3-фтор-фенил)метил]фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-ил]метанол 4m (1,11 г, 1,46 ммоль) растворяли в 20 мл дихлорметана и охлаждали до -10°C с последующим добавлением трифторуксусной кислоты (0,23 мл, 3 ммоль). Реакционную смесь нагревали и перемешивали в течение 2 часов, и она распределялась после добавления 20 мл насыщенного раствора бикарбоната натрия. Водную фазу экстрагировали дихлорметаном (20 мл × 2). Органический экстракт объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения [(1S,2S,3S,4R,5S)-2,3,4-трибензилокси-5-[4-хлор-3-[(4-этокси-3-фтор-фенил)метил]фенил]-6,8-диоксабицикло[3.2.1]октан-1-ил]метанола 4n (830 мг, бесцветное жироподобное вещество), выход: 78,3%.
МС m/z (ESI): 742,3 [М+18]
Стадия 13
(1S,2S,3S,4R,5S)-5-[4-хлор-3-[(4-этокси-3-фтор-фенил)метил]фенил]-1-(гидроксиметил)-6,8-диоксабицикло[3.2.1]октан-2,3,4-триол
[(1S,2S,3S,4R,5S)-2,3,4-трибензилокси-5-[4-хлор-3-[(4-этокси-3-фтор-фенил)метил]фенил]-6,8-диоксабицикло[3.2.1]октан-1-ил]метанол 4n (830 мг, 1,14 ммоль) растворяли в 20 мл смешанного раствора (THF и MeOH, v:v=1:1) с последующим добавлением o-дихлорбензола (1,3 мл, 11,4 ммоль) и палладия на угле (500 мг, 10%). Смесь трижды продували H2 и перемешивали в течение 3 часов, и фильтровали. После этого реакционную смесь элюировали небольшим количеством этилацетата. Фильтрат концентрировали при пониженном давлении, и получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой А с получением указанного в заголовке соединения (1S,2S,3S,4R,5S)-5-[4-хлор-3-[(4-этокси-2,3-дифтор-фенил)метил]фенил]-1-(гидроксиметил)-6,8-диоксабицикло[3.2.1]октан-2,3,4-триола 4 (420 мг, белое твердое вещество), выход: 81,0%.
МС m/z (ESI): 472,2 [М+18]
1Н ЯМР (400 МГц, CD3OD): δ 7.47 (s, 1Н), 7.42-7.35 (m, 2Н), 6.95-6.87 (m, 3H), 4.16-4.14 (m, 1Н), 4.06-4.02 (m, 4Н), 3.85-3.70 (m, 2Н), 3.67-3.54 (m, 4Н), 1.37 (t, 3H).
Пример 5
(1S,2S,3S,4R,5S)-5-[4-хлор-3-(2,3-дигидробензофуран-5-илметил)фенил]-1-(гидроксиметил)-6,8-диоксабицикло[3.2.1]октан-2,3,4-триол
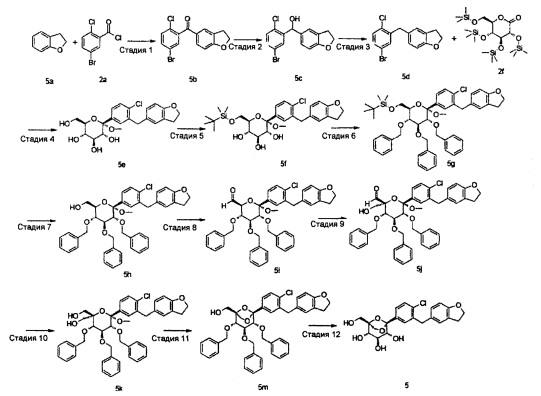
Стадия 1
(5-Бром-2-хлор-фенил)-(2,3-дигидробензофуран-5-ил)метанон
5-Бром-2-хлор-бензоилхлорид 2a (10,8 г, 42,5 ммоль) растворяли в 100 мл дихлорметана с последующим добавлением 2,3-дигидробензофуран 5a (5,11 г, 42,5 ммоль) и добавлением порции трихлорида алюминия (6,8 г, 51,0 ммоль). Реакционную смесь перемешивали в течение 2 часов. После этого реакционную смесь концентрировали при пониженном давлении, и получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой D с получением указанного в заголовке соединения (5-бром-2-хлор-фенил)-(2,3-дигидробензофуран-5-ил)метанона 5b (10,47 г, белое твердое вещество), выход: 72,9%.
МС m/z (ESI): 339,0 [М+1]
1Н ЯМР (400 МГц, CDCl3): δ 7.73 (d, 1Н), 7.58 (dd, 1Н), 7.53 (dd, 1Н), 7.47 (d, 1Н), 7.32 (d, 1Н), 6.81 (d, 1Н), 4.68 (t, 2Н), 3.26 (t, 2Н).
Стадия 2
(5-Бром-2-хлор-фенил)-(2,3-дигидробензофуран-5-ил)метанол
(5-Бром-2-хлор-фенил)-(2,3-дигидробензофуран-5-ил)метанон 5b (10,47 г, 31,0 ммоль) растворяли в 100 мл смешанного раствора (THF и МеОН, v:v=1:1) и охлаждали до 0°C с последующим добавлением порции боргидрида натрия (2,35 г, 62,0 ммоль). Реакционную смесь перемешивали в течение 30 минут при 0°C. После этого реакционную смесь концентрировали при пониженном давлении после добавления 20 мл ацетона. Получающийся в результате остаток растворяли в 250 мл этилацетата, и он распределялся. Органический экстракт промывали водой (100 мл × 2), объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (5-бром-2-хлор-фенил)-(2,3-дигидробензофуран-5-ил)метанола 5с (10,5 г, бледно-желтое жироподобное вещество), выход: 99,7%.
1Н ЯМР (400 МГц, CDCl3): δ 7.88 (d, 1Н), 7.34 (dd, 1Н), 7.18 (d, 1Н), 7.16 (s, 1Н), 7.11 (dd, 1Н), 6.73 (d, 1Н), 6.05 (s, 1Н), 4.56 (t, 2Н), 3.18 (t, 2Н), 2.27 (s, 1Н)
Стадия 3
5-[(5-Бром-2-хлор-фенил)метил]-2,3-дигидробензофуран
(5-Бром-2-хлор-фенил)-(2,3-дигидробензофуран-5-ил)метанол 5с (10,5 г, 30,9 ммоль) растворяли в 100 мл дихлорметана с последующим добавлением триэтилсилана (14,8 мл, 92,7 ммоль) и добавлением по каплям эфирата трифторида бора (7,8 мл, 61,8 ммоль). Реакционную смесь перемешивали в течение 16 часов. После этого реакционную смесь концентрировали при пониженном давлении, и получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой D с получением указанного в заголовке соединения 5-[(5-бром-2-хлор-фенил)метил]-2,3-дигидробензо-фурана 5d (10,0 г, бледно-желтое жироподобное вещество), выход: 100%.
1Н ЯМР (400 МГц, CDCl3): δ 7.29-7.21 (m, 3H), 7.00 (s, 1Н), 6.93 (d, 1Н), 6.73 (d, 1Н), 4.56 (t, 2Н), 3.98 (s, 2Н), 3.18 (t, 2Н)
Стадия 4
(2S,3R,4S,5S,6R)-2-[4-хлор-3-(2,3-дигидробензофуран-5-илметил)фенил]-6-(гидроксиметил)-2-метокси-тетрагидропиран-3,4,5-триол
5-[(5-Бром-2-хлор-фенил)метил]-2,3-дигидробензофуран 5d (10,0 г, 30,9 ммоль) растворяли в 90 мл смешанного раствора (THF и толуол, v:v=1:2) и охлаждали до -78°C, а затем по каплям добавляли раствор nBuLi в н-гексане (14,83 мл, 37,1 ммоль). После перемешивания в течение 1 часа при -78°C по каплям добавляли раствор (3R,4S,5R,6R)-3,4,5-трис(триметилсилилокси)-6-(триметилсилилоксиметил)тетрагидропиран-2-она 2f (15,87 г, 33,9 ммоль) в толуоле (90 мл) перед тем, как реакционную смесь перемешивали в течение 3 часов при -78°C. Добавляли раствор 0,6 М метансульфоновой кислоты в метаноле (103 мл) перед тем, как реакционную смесь перемешивали в течение 16 часов. После этого реакционную смесь концентрировали при пониженном давлении после добавления 100 мл насыщенного раствора карбоната натрия. Остаток экстрагировали этилацетатом (100 мл × 3) после добавления 50 мл насыщенного раствора хлорида натрия, и органический экстракт объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении, и получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой E с получением указанного в заголовке соединения (2S,3R,4S,5S,6R)-2-[4-хлор-3-(2,3-дигидробензофуран-5-илметил)фенил]-6-(гидроксиметил)-2-метокси-тетрагидропиран-3,4,5-триола 5е (6,3 г, белое твердое вещество), выход: 46,7%.
1Н ЯМР (400 МГц, CD3OD): δ 7.53 (d, 1Н), 7.45 (dd, 1Н), 7.35 (d, 1Н), 7.03 (s, 1Н), 6.91 (d, 1Н), 6.60 (d, 1Н), 4.48 (t, 2Н), 4.13-3.91 (m, 3H), 3.84-3.73 (m, 2Н), 3.61-3.56 (m, 1Н), 3.44-3.39 (m, 1Н), 3.11 (dd, 3H), 3.07 (s, 3H)
Стадия 5
(2S,3R,4S,5S,6R)-6-[(трет-бутил(диметил)силил)оксиметил]-2-[4-хлор-3-(2,3-дигидробензофуран-5-илметил)фенил]-2-метокси-тетрагидропиран-3,4,5-триол
(2S,3R,4S,5S,6R)-2-[4-хлор-3-(2,3-дигидробензофуран-5-илметил)фенил]-6-(гидроксиметил)-2-метокси-тетрагидропиран-3,4,5-триол 5е (6,3 г, 14,44 ммоль) растворяли в 60 мл пиридина с последующим добавлением по очереди 4-диметиламинопиридина (353 мг, 2,89 ммоль) и трет-бутил-диметил-хлор-силана (2,61 г, 17,32 ммоль). Реакционную смесь перемешивали в течение 16 часов и концентрировали при пониженном давлении. Получающийся в результате остаток растворяли в 200 мл этилацетата, и он распределялся. Органический экстракт промывали водой (50 мл) и насыщенным раствором хлорида натрия (50 мл), объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (2S,3R,4S,5S,6R)-6-[(трет-бутил(диметил)силил)оксиметил]-2-[4-хлор-3-(2,3-дигидробензофуран-5-илметил)фенил]-2-метокси-тетрагидропиран-3,4,5-триола 5f (7,96 г, бледно-желтое твердое вещество), который использовали непосредственно на следующей стадии без очистки.
Стадия 6
[[(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-(2,3-дигидробензофуран-5-илметил)фенил]-6-метокси-тетрагидропиран-2-ил]трет-бутил-диметил-метокси]силан
(2S,3R,4S,5S,6R)-6-[(трет-бутил(диметил)силил)оксиметил]-2-[4-хлор-3-(2,3-дигидробензофуран-5-илметил)фенил]-2-метокси-тетрагидропиран-3,4,5-триол 5f (7,96 г, 14,4 ммоль) растворяли в 80 мл DMF и охлаждали до 0°C с последующим добавлением 60% NaH (2,89 г, 72,21 ммоль). Затем реакционную смесь нагревали до комнатной температуры и перемешивали в течение 15 минут. После этого добавляли бензилбромид (8,58 мл, 72,21 ммоль) перед тем, как смесь перемешивали в течение 16 часов. Реакционную смесь концентрировали при пониженном давлении после добавления 10 мл метанола. С последующим добавлением 200 мл этилацетата реакционную смесь промывали водой (50 мл × 2), и органический экстракт объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения [[(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-(2,3-дигидро-бензофуран-5-илметил)фенил]-6-метокси-тетрагидропиран-2-ил]трет-бутил-диметил-метокси]силана 5g (11,86 г, черное жироподобное вещество), который использовали непосредственно на следующей стадии без очистки.
Стадия 7
[(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-(2,3-дигидробензофуран-5-илметил)фенил]-6-метокси-тетрагидропиран-2-ил]метанол
[[(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-(2,3-дигидробензофуран-5-илметил)фенил]-6-метокси-тетрагидропиран-2-ил]трет-бутил-диметил-метокси]силан 5g (11,86 г, 14,4 ммоль) растворяли в 100 мл метанола с последующим добавлением ацетилхлорида (152 мкл, 2,17 ммоль). Реакционную смесь перемешивали в течение 1 часа и концентрировали при пониженном давлении, и получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой D с получением указанного в заголовке соединения [(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-(2,3-дигидробензофуран-5-илметил)фенил]-6-метокси-тетрагидропиран-2-ил]метанола 5h (9,0 г, желтая жидкость), выход: 88,1%.
Стадия 8
(2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-(2,3-дигидробензофуран-5-илметил)фенил]-6-метокси-тетрагидропиран-2-карбальдегид
Оксалилхлорид (0,76 мл, 8,91 ммоль) растворяли в 10 мл метиленхлорида и охлаждали до -78°C, а затем по каплям добавляли 10 мл смешанного раствора (метиленхлорид и диметилсульфоксид, v:v=10:0,85). Реакционную смесь перемешивали в течение 15 минут. Затем по каплям добавляли 25 мл раствора [(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-(2,3-дигидробензофуран-5-илметил)фенил]-6-метокси-тетрагидропиран-2-ил]метанола 5h (4,2 г, 5,94 ммоль) в метиленхлориде перед тем, как смесь перемешивали в течение 40 минут. Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 2 часов после того, как по каплям добавляли триэтиламин (4,29 мл, 29,69 ммоль). После этого реакционная смесь распределялась после добавления 35 мл 1 М соляной кислоты, органический экстракт промывали насыщенным раствором хлорида натрия (35 мл × 2), объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-(2,3-дигидробензофуран-5-илметил)фенил]-6-метокси-тетрагидропиран-2-карбальдегида 5i (4,19 г, бледно-желтое жироподобное вещество), который использовали непосредственно на следующей стадии без очистки.
Стадия 9
(2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-(2,3-дигидробензофуран-5-илметил)фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-карбальдегид
(2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-(2,3-дигидробензофуран-5-илметил)фенил]-6-метокси-тетрагидропиран-2-карбальдегид 5i (4,19 г, 5,9 ммоль) растворяли в 45 мл 1,4-диоксана с последующим добавлением 9,6 мл 37% раствора формальдегида и добавлением по каплям 17,82 мл 1 М раствора гидроксида натрия. Реакционную смесь перемешивали в течение 16 часов при 70°C. После этого реакционную смесь концентрировали при пониженном давлении, и она распределялась после добавления 100 мл этилацетата и 50 мл насыщенного раствора хлорида натрия. Водную фазу экстрагировали этилацетатом (100 мл × 2), и органический экстракт объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-(2,3-дигидробензофуран-5-илметил)фенил]-2-(гидрокcиметил)-6-метокси-тетрагидропиран-2-карбальдегида 5j (4,36 г, бледно-желтое жироподобное вещество), который использовали непосредственно на следующей стадии без очистки.
Стадия 10
[(3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-(2,3-дигидробензофуран-5-ил-метил)фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-ил]метанол
(2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-(2,3-дигидробензофуран-5-ил-метил)фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-карбальдегид 5j (4,36 г, 5,93 ммоль) растворяли в 50 мл смешанного раствора (THF и МеОН, v:v=1:1) с последующим добавлением боргидрида натрия (0,45 г, 11,9 ммоль). Реакционную смесь перемешивали в течение 30 минут. После этого реакционную смесь концентрировали при пониженном давлении, и получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой D с получением указанного в заголовке соединения [(3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-(2,3-дигидробензофуран-5-илметил)фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-ил]метанола 5k (1,26 г, белое твердое вещество), выход: 28,8%.
1Н ЯМР (400 МГц, CD3OD): δ 7.53 (dd, 1Н), 7.44 (d, 1Н), 7.35 (d, 1Н), 7.31-7.26 (m, 5Н), 7.25-7.18 (m, 8Н), 7.04-7.02 (m, 2Н), 6.90 (s, 1Н), 6.80 (d, 1Н), 6.54 (d, 1H), 4.90-4.79 (m, 4H), 4.73 (d, 1H), 4.54 (d, 1H), 4.46-4.41 (m, 2H), 4.20 (t, 1H), 4.10-4.00 (m, 5H), 3.88 (dd, 2H), 3.74 (d, 1H), 3.15 (s, 3H), 3.06-3.00 (m, 2H).
Стадия 11
[(1S,2S,3S,4R,5S)-2,3,4-трибензилокси-5-[4-хлор-3-(2,3-дигидробензофуран-5-илметил)фенил]-6,8-диоксабицикло[3.2.1]октан-1-ил]метанол
[(3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-(2,3-дигидробензофуран-5-илметил)фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-ил]метанол 5k (1,2 г, 1,63 ммоль) растворяли в 25 мл дихлорметана, а затем по каплям добавляли трифторуксусную кислоту (0,5 мл, 6,52 ммоль). Реакционную смесь перемешивали в течение 1,5 часов. После этого реакционную смесь концентрировали при пониженном давлении, и получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой D с получением указанного в заголовке соединения [(1S,2S,3S,4R,5S)-2,3,4-трибензилокси-5-[4-хлор-3-(2,3-дигидробензофуран-5-илметил)фенил]-6,8-диоксабицикло[3.2.1]октан-1-ил]метанола 5m (580 мг, белое твердое вещество), выход: 50,4%.
МС m/z (ESI): 722,3 [М+18]
1Н ЯМР (400 МГц, CD3OD): δ 7.48-7.39 (m, 3H), 7.35-7.23 (m, 10H), 7.21-7.11 (m 3H), 6.97 (s, 1Н), 6.90 (d, 1Н), 6.84 (d, 2Н), 6.57 (d, 1Н), 4.83 (d, 4Н), 4.44 (t, 2Н), 4.23 (m, 2Н), 4.07-4.00 (m, 3H), 3.95 (dd, 1Н), 3.87 (d, 1Н), 3.79 (d, 1Н), 3.73-3.69 (m, 2Н), 3.56 (d, 1Н), 3.01 (t, 2Н).
Стадия 12
(1S,2S,3S,4R,5S)-5-[4-хлор-3-(2,3-дигидробензофуран-5-илметил)фенил]-1-(гидроксиметил)-6,8-диоксабицикло[3.2.1]октан-2,3,4-триол
[(1S,2S,3S,4R,5S)-2,3,4-трибензилокси-5-[4-хлор-3-(2,3-дигидробензофуран-5-илметил)фенил]-6,8-диоксабицикло[3.2.1]октан-1-ил]метанол 5m (100 мг, 0,14 ммоль) растворяли в 5 мл смешанного раствора (THF и МеОН, v:v=1:1) с последующим добавлением о-дихлорбензола (208 мг, 1,42 ммоль) и палладия на угле (10 мг, 10%). Смесь трижды продували Н2 и перемешивали в течение 1,5 часов, фильтровали, и фильтрат концентрировали при пониженном давлении. Получающийся в результате остаток очищали при помощи тонкослойной хроматографии с хроматографической системой растворителей A с получением указанного в заголовке соединения (1S,2S,3S,4R,5S)-5-[4-хлор-3-(2,3-дигидробензофуран-5-илметил)фенил]-1-(гидроксиметил)-6,8-диоксабицикло[3.2.1]октан-2,3,4-триола 5 (58 мг, белое твердое вещество), выход: 93,5%.
МС m/z (ESI): 435,1 [М+1]
1Н ЯМР (400 МГц, CD3OD): δ 7.47 (d, 1Н), 7.39-7.35 (m, 2Н), 7.04 (s, 1Н), 6.93 (d, 1Н), 6.62 (d, 1Н), 4.50 (t, 2Н), 4.17 (d, 1Н), 4.03 (s, 2Н), 3.88-3.79 (m, 2Н), 3.71-3.65 (m, 2Н), 3.62-3.56 (m, 2Н), 3.15 (t, 2Н).
Пример 6
(1S,2S,3S,4R,5S)-5-[4-хлор-3-[[4-(трифторметокси)фенил]метил]фенил]-1-(гидроксиметил)-6,8-диоксабицикло[3.2.1]октан-2,3,4-триол
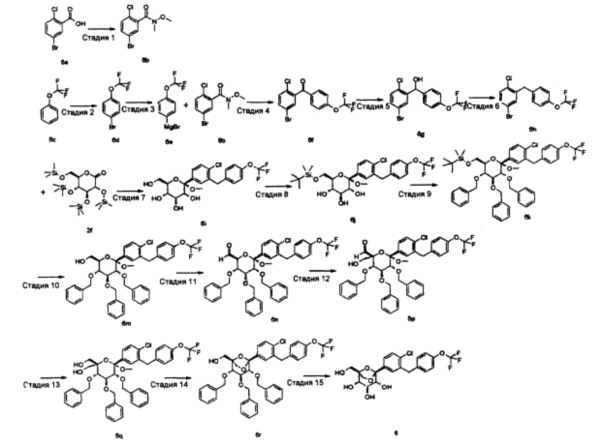
N-метил-N-метоксиламин гидрохлорид (3,41 г, 35 ммоль) растворяли в 140 мл метиленхлорида. Добавляли триэтиламин (14,6 мл, 105 ммоль), и реакционную смесь перемешивали в течение 10 минут. Затем реакционную смесь перемешивали в течение 16 часов после добавления по очереди 5-бром-2-хлор-бензойной кислоты 6а (8,24 г, 35 ммоль) и бис(2-оксо-3-оксазолидинил)фосфинового хлорида (10,69 г, 42 ммоль). После этого реакционную смесь экстрагировали этилацетатом (80 мл × 3) после добавления 120 мл воды. Органический экстракт промывали насыщенным раствором хлорида натрия (60 мл) и объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении, и получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой B с получением указанного в заголовке соединения 5-бром-2-хлор-N-метокси-N-метил-бензамида 6b (7,50 г, бледно-желтое твердое вещество), выход: 76,9%.
МС m/z (ESI): 280,0 [М+1]
1Н ЯМР (400 МГц, CDCl3): δ 7.46-7.44 (m, 2Н), 7.29-7.26 (m, 1Н), 3.49 (s, 3H), 3.37 (s, 3H)
Стадия 2
1-Бром-4-(трифторметокси)бензол
Трифторметоксибензол 6c (11,35 г, 70 ммоль) растворяли в 3,57 мл жидкого брома, а затем добавляли железо (0,24 г). Реакционную смесь перемешивали в течение 16 часов при 100°C. Добавляли 450 мл дихлорметана, и органический экстракт промывали 6 M соляной кислоты (140 мл), 10% раствором гидросульфита натрия (140 мл) и насыщенным раствором хлорида натрия (140 мл), объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения 1-бром-4-(трифторметокси)бензола 6d (14,1 г, желтое твердое вещество), выход: 83,8%.
1Н ЯМР (400 МГц, CDCl3): δ 7.54-7.50 (m, 2Н), 7.11-7.09 (m, 2H)
Стадия 3
1-Бром-магний-4-(трифторметокси)бензол
Магний (0,12 г, 5 ммоль) и йод (I2, каталитическое количество) добавляли в колбу реактора, а затем по каплям добавляли раствор (5 мл) 1-бром-4-(трифторметокси)бензола 6d (1,2 г, 5 ммоль) в THF. Реакционную смесь кипятили с обратным холодильником в течение 1 часа с получением указанного в заголовке соединения 1-бром-магний-4-(трифторметокси)бензола 6e (1,32 г), который использовали непосредственно на следующей стадии без очистки.
Стадия 4
(5-Бром-2-хлор-фенил)-[4-(трифторметокси)фенил]метанон
5-Бром-2-хлор-N-метокси-N-метил-бензамид 6b (5,16 г, 18,5 ммоль) растворяли в 50 мл THF, а затем по каплям добавляли 1-бром-магний-4-(трифторметокси)бензол 6e (13,21 г, 49,8 ммоль). Реакционную смесь перемешивали в течение 2 часов. После этого реакционная смесь распределялась после добавления 200 мл насыщенного раствора хлорида натрия, 200 мл воды и 150 мл этилацетата. Водную фазу экстрагировали этилацетатом (150 мл), и органический экстракт промывали насыщенным раствором хлорида натрия (200 мл), объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении. Получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой В с получением указанного в заголовке соединения (5-бром-2-хлор-фенил)-[4-(трифтор-метокси)фенил]метанона 6f (4,08 г, бледно-желтое твердое вещество), выход: 58,1%.
МС m/z(ESI): 380,9 [М+1]
1Н ЯМР (400 МГц, CDCl3): δ 7.88-7.84 (m, 2Н), 7.58 (dd, 1Н), 7.51 (d, 1Н), 7.35 (d, 1Н), 7.32-7.30 (m,2Н)
Стадия 5
(5-Бром-2-хлор-фенил)-[4-(трифторметокси)фенил]метанол
(5-Бром-2-хлор-фенил)-[4-(трифторметокси)фенил]метанон 6f (4,35 г, 11,5 ммоль) растворяли в 40 мл смешанного раствора (THF и MeOH, v:v=1:1) и охлаждали до 0°C с последующим добавлением порции боргидрида натрия (0,87 г, 23,0 ммоль). Реакционную смесь перемешивали в течение 30 минут при 0°С. После этого реакционную смесь концентрировали при пониженном давлении после добавления 10 мл ацетона. Реакционная смесь распределялась после добавления 100 мл этилацетата и 50 мл воды, и органический экстракт промывали насыщенным раствором хлорида натрия (50 мл × 2), объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (5-бром-2-хлор-фенил)-[4-(трифторметокси)фенил]метанола 6g (4,4 г, бледно-желтое жироподобное вещество), который использовали непосредственно на следующей стадии без очистки.
1Н ЯМР (400 МГц, CDCl3): δ 7.80 (d, 1Н), 7.43-7.40 (m, 2Н), 7.36 (dd, 1Н), 7.20 (t, 3H), 6.16 (s, 1Н).
Стадия 6
4-Бром-1-хлор-2-[[4-(трифторметокси)фенил]метил]бензол
(5-Бром-2-хлор-фенил)-[4-(трифторметокси)фенил]метанол 6g (4,37 г, 11,5 ммоль) растворяли в 35 мл дихлорметана с последующим добавлением триэтилсилана (5,49 мл, 34,4 ммоль) и добавлением по каплям эфирата трифторида бора (2,9 мл, 22,9 ммоль). Реакционную смесь перемешивали в течение 16 часов. После этого реакционную смесь концентрировали при пониженном давлении, и она распределялась после добавления 20 мл насыщенного раствора бикарбоната натрия. Водную фазу экстрагировали дихлорметаном (25 мл × 3), и органический экстракт объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении. Получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой C с получением указанного в заголовке соединения 4-бром-1-хлор-2-[[4-(трифторметокси)фенил]метил]бензола 6h (3,0 г, бесцветное жироподобное вещество), выход: 71,6%.
1Н ЯМР (400 МГц, CDCl3): δ 7.33-7.29 (m, 2Н), 7.26-7.24 (m, 1Н), 7.21-7.18 (m, 2Н), 7.15 (d, 2Н), 4.06 (s, 2Н).
Стадия 7
(2S,3R,4S,5S,6R)-2-[4-хлор-3-[[4-(трифторметокси)фенил]метил]фенил]-6-(гидроксиметил)-2-метокси-тетрагидропиран-3,4,5-триол
4-Бром-1-хлор-2-[[4-(трифторметокси)фенил]метил]бензол 6h (2,33 г, 6,38 ммоль) растворяли в 40 мл смешанного раствора (THF и н-гексан, v:v=1:3) и охлаждали до -78°C, а затем по каплям добавляли раствор nBuLi в н-гексане (3,83 мл, 9,57 ммоль). После перемешивания в течение 1,5 часов при -78°C по каплям добавляли раствор (30 мл) (3R,4S,5R,6R)-3,4,5-трис(триметилсилилокси)-6-(триметилсилилоксиметил)тетрагидропиран-2-она 2f (4,47 г, 9,57 ммоль) в смешанном растворе (THF и н-гексан, v:v=1:3) перед тем, как реакционную смесь перемешивали в течение 2 часов при -78°C. Добавляли раствор (32 мл) 0,6 М метансульфоновой кислоты в метаноле перед тем, как реакционную смесь нагревали и перемешивали в течение 16 часов при комнатной температуре. После этого реакционная смесь распределялась после добавления 150 мл насыщенного раствора карбоната натрия. Водную фазу экстрагировали этилацетатом (100 мл × 3), и органические экстракты объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении. Получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой A с получением указанного в заголовке соединения (2S,3R,4S,5S,6R)-2-[4-хлор-3-[[4-(трифторметокси)фенил]метил]фенил]-6-(гидроксиметил)-2-метокси-тетрагидропиран-3,4,5-триола 6i (1,38 г, белое твердое вещество), выход: 45,2%.
1Н ЯМР (400 МГц, CD3OD): δ 7.61 (d, 1Н), 7.51 (dd, 1Н), 7.40 (d, 1Н), 7.31-7.29 (m, 2Н), 7.17 (d, 2Н), 4.23-4.12 (m, 2Н), 3.94 (d, 1Н), 3.86-3.75 (m, 2Н), 3.95-3.59 (m, 2Н), 3.47-3.42 (m, 1Н), 3.10 (s, 3H)
Стадия 8
(2S,3R,4S,5S,6R)-6-[(трет-бутил(диметил)силил)оксиметил]-2-[4-хлор-3-[[4-(трифторметокси)фенил]метил]фенил]-2-метокси-тетрагидропиран-3,4,5-триол
(2S,3R,4S,5S,6R)-2-[4-хлор-3-[[4-(трифторметокси)фенил]метил]фенил]-6-(гидроксиметил)-2-метокси-тетрагидропиран-3,4,5-триол 6i (1,33 г, 2,78 ммоль) растворяли в 12 мл пиридина с последующим добавлением по очереди 4-диметиламинопиридина (67,93 мг, 0,55 ммоль) и TBSCI (0,50 г, 3,34 ммоль). Реакционную смесь перемешивали в течение 16 часов. После этого реакционную смесь концентрировали при пониженном давлении, и она распределялась после добавления 75 мл этилацетата и 75 мл воды. Водную фазу экстрагировали этилацетатом (50 мл × 2), и органические экстракты объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (2S,3R,4S,5S,6R)-6-[(трет-бутил(диметил)силил)оксиметил]-2-[4-хлор-3-[[4-(трифторметокси)фенил]метил]фенил]-2-метокси-тетрагидропиран-3,4,5-триола 6j (1,60 г, белое твердое вещество), выход: 97,6%.
1Н ЯМР (400 МГц, CD3OD): δ 7.55 (d, 1Н), 7.46 (d, 1Н), 7.41 (d, 1Н), 7.26 (d, 2Н), 7.17 (d, 2Н), 4.17 (s, 2Н), 4.04 (d, 1Н), 3.92-3.88 (m, 1Н), 3.77 (t, 1Н), 3.61-3.59 (m, 1Н), 3.09 (s, 3H)
Стадия 9
[[(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[4-(трифторметокси)фенил]метил]фенил]-6-метокси-тетрагидропиран-2-ил]метокси]трет-бутил-диметил-силан
(2S,3R,4S,5S,6R)-6-[(трет-бутил(диметил)силил)оксиметил]-2-[4-хлор-3-[[4-(трифторметокси)фенил]метил]фенил]-2-метокси-тетрагидропиран-3,4,5-триол 6j (1,60 г, 2,69 ммоль) растворяли в 15 мл DMF и охлаждали до 0°C с последующим добавлением 60% NaH (0,54 г, 13,5 ммоль). Затем реакционную смесь нагревали и перемешивали в течение 15 минут при комнатной температуре, а затем добавляли бензилбромид (1,6 мл, 13,5 ммоль). После перемешивания в течение 16 часов реакционную смесь концентрировали при пониженном давлении после добавления 5 мл метанола. Затем получающийся в результате остаток растворяли в этилацетате (100 мл) и промывали водой (50 мл × 2). Органический экстракт объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения [[(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[4-(трифторметокси)фенил]метил]фенил]-6-метокси-тетрагидропиран-2-ил]метокси]трет-бутил-диметил-силана 6k (2,32 г, желтое жироподобное вещество), который использовали непосредственно на следующей стадии без очистки.
Стадия 10
[(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[4-(трифторметокси)фенил]метил]фенил]-6-метокси-тетрагидропиран-2-ил]метанол
[[(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[4-(трифторметокси)фенил]метил]фенил]-6-метокси-тетрагидропиран-2-ил]метокси]трет-бутил-диметил-силан 6k (2,32 г, 2,69 ммоль) растворяли в 12 мл метанола с последующим добавлением ацетилхлорида (16 мкл, 0,40 ммоль). Реакционную смесь перемешивали в течение 1,5 часов. После этого реакционную смесь концентрировали при пониженном давлении, и получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой с получением указанного в заголовке соединения [(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[4-(трифторметокси)фенил]метил]фенил]-6-метокси-тетрагидропиран-2-ил]метанола 6m (1,22 г, бледно-желтое жироподобное вещество), выход: 60,7%.
1Н ЯМР (400 МГц, CD3OD): δ 7.54-7.52 (m, 2Н), 7.41 (d, 1Н), 7.32-7.22 (m, 13Н), 7.18 (d, 2Н), 7.08-7.04 (m, 4Н), 4.91-4.85 (m, 3H), 4.75 (d, 1Н), 4.52 (d, 1Н), 4.17-4.09 (m, 3H), 4.00 (d, 1Н), 3.94-3.83 (m, 3H), 3.75-3.69 (m, 2Н), 3.09 (m, 3H).
Стадия 11
(2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[4-(трифторметокси)фенил]метил]фенил]-6-метокси-тетрагидропиран-2-карбальдегид
Оксалилхлорид (0,21 мл, 2,45 ммоль) растворяли в 5 мл дихлорметана и охлаждали до -78°C. Смесь перемешивали в течение 15 минут после добавления 5 мл раствора диметилсульфоксида (0,24 мл, 3,26 ммоль) в дихлорметане. Затем добавляли 10 мл раствора [(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[4-(трифторметокси)фенил]метил]фенил]-6-метокси-тетрагидропиран-2-ил]метанола 6m (1,22 г, 1,63 ммоль) в дихлорметане перед тем, как смесь перемешивали в течение 40 минут. Затем реакционную смесь нагревали и перемешивали в течение 1,5 часов при комнатной температуре после добавления триэтиламина (1,18 мл, 8,15 ммоль), и она распределялась после добавления 10 мл 1 М соляной кислоты. Органический экстракт промывали насыщенным раствором хлорида натрия (10 мл × 2) и объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[4-(трифторметокси)фенил]метил]фенил]-6-метокси-тетрагидропиран-2-карбальдегида 6n (1,21 г, желтое жироподобное вещество), который использовали непосредственно на следующей стадии без очистки.
Стадия 12
(2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[4-(трифторметокси)фенил]метил]фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-карбальдегид
(2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[4-(трифторметокси)фенил]метил]фенил]-6-метокси-тетрагидропиран-2-карбальдегид 6n (1,21 г, 1,63 ммоль) растворяли в 12 мл 1,4-диоксана с последующим добавлением 2,65 мл 37% раствора формальдегида и добавлением по каплям 4,89 мл 1 М гидроксида натрия. Реакционную смесь перемешивали в течение 16 часов при 70°C. После этого реакционную смесь концентрировали при пониженном давлении, и она распределялась после добавления 30 мл насыщенного раствора хлорида натрия. Водную фазу экстрагировали этилацетатом (30 мл × 3), и органический экстракт объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[4-(трифторметокси)фенил]метил]фенил]-2-(гидроксиметил)-6-метокси-тетрагидро-пиран-2-карбальдегида 6р (1,27 г, бледно-желтое жироподобное вещество), который использовали непосредственно на следующей стадии без очистки.
Стадия 13
[(3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[4-(трифторметокси)фенил]метил]фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-ил]метанол
(2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[4-(трифторметокси)фенил]метил]фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-карбальдегид 6р (1,27 г, 1,63 ммоль) растворяли в 10 мл смешанного раствора (THF и MeOH, v:v=1:1) и с последующим добавлением порции боргидрида натрия (0,12 г, 3,26 ммоль). Реакционную смесь перемешивали в течение 1,5 часов. После этого реакционную смесь концентрировали при пониженном давлении, и она распределялась после добавления 30 мл этилацетата. Органический экстракт промывали насыщенным раствором хлорида натрия (15 мл × 2), объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении, и получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой E с получением указанного в заголовке соединения [(3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[4-(трифторметокси)фенил]метил]фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-ил]метанола 6q (400 мг, белое твердое вещество), выход: 31,5%.
1Н ЯМР (400 МГц, CD3OD): δ 7.58 (dd, 1Н), 7.54 (d, 1Н), 7.39 (d, 1Н), 7.31-7.24 (m, 13Н), 7.16 (d, 2Н), 7.11-7.06 (m, 4Н), 4.92 (d, 1Н), 4.76 (d, 1Н), 4.66-4.59 (m, 1Н), 4.27-4.22 (m, 2Н), 4.17-4.10 (m, 2Н), 4.07-4.04 (m, 3H), 3.99-3.95 (m, 2Н), 3.77 (d, 1Н), 3.65-3.60 (m, 2Н), 3.35 (s, 2Н), 3.18 (s, 3H)
Стадия 14
[(1S,2S,3S,4R,5S)-2,3,4-трибензилокси-5-[4-хлор-3-[[4-(трифторметокси)фенил]метил]фенил]-6,8-диоксабицикло[3.2.1]октан-1-ил]метанол
[(3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[4-(трифторметокси)фенил]метил]фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-ил]метанол 6q (400 мг, 0,51 ммоль) растворяли в 10 мл дихлорметана с последующим добавлением трифторуксусной кислоты (0,15 мл, 2,05 ммоль). Реакционную смесь перемешивали в течение 1,5 часов. После этого реакционную смесь концентрировали при пониженном давлении, и получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой B с получением указанного в заголовке соединения [(1S,2S,3S,4R,5S)-2,3,4-трибензилокси-5-[4-хлор-3-[[4-(трифторметокси)фенил]метил]фенил]-6,8-диоксабицикло[3.2.1]октан-1-ил]метанола 6r (290 мг, белое твердое вещество), выход: 76,1%.
1Н ЯМР (400 МГц, DMSO-d6): δ 7.59 (dd, 1Н), 7.50-7.44 (m, 2Н), 7.34-7.15 (m, 17Н), 6.83 (d, 2Н), 5.22 (t, 1Н), 4.81-4.74 (m, 4Н), 4.32 (d, 1Н), 4.11-4.08 (m, 3H), 4.07-4.04 (m, 1Н), 3.93 (d, 1Н), 3.87 (t, 1Н), 3.79-3.71 (m, 3H), 3.59 (dd, 1Н), 3.52 (d, 1Н)
Стадия 15
(1S,2S,3S,4R,5S)-5-[4-хлор-3-[[4-(трифторметокси)фенил]метил]фенил]-1-(гидроксиметил)-6,8-диоксабицикло[3.2.1]октан-2,3,4-триол
[(1S,2S,3S,4R,5S)-2,3,4-трибензилокси-5-[4-хлор-3-[[4-(трифторметокси)фенил]метил]фенил]-6,8-диоксабицикло[3.2.1]октан-1-ил]метанол 6r (150 мг, 0,20 ммоль) растворяли в 10 мл смешанного раствора (THF и МеОН, v:v=1:1) с последующим добавлением о-дихлорбензола (0,23 мл, 2 ммоль) и палладия на угле (60 мг, 10%). Смесь трижды продували H2 и перемешивали в течение 1 часа. После этого реакционную смесь фильтровали. Фильтрат концентрировали при пониженном давлении, и получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой E с получением указанного в заголовке соединения (1S,2S,3S,4R,5S)-5-[4-хлор-3-[[4-(трифторметокси)фенил]метил]фенил]-1-(гидроксиметил)-6,8-диоксабицикло-[3.2.1]октан-2,3,4-триола 6 (82 мг, белое твердое вещество), выход: 86,0%.
МС m/z (ESI): 477,1 [М+1]
1Н ЯМР (400 МГц, CD3OD): δ 7.54 (d, 1Н), 7.46-7.39 (m, 2Н), 7.30 (d, 2Н), 7.17 (d, 2Н), 4.17 (d, 3H), 3.86 (d, 1Н), 3.80 (d, 1Н), 3.72-3.68 (m, 2Н), 3.62 (dd, 1Н), 3.58 (d, 1Н)
Пример 7
(1S,2S,3S,4R,5S)-5-[4-хлор-3-[[4-(2,2,2-трифторэтокси)фенил]метил]фенил]-1-(гидроксиметил)-6,8-диоксабицикло[3.2.1]октан-2,3,4-триол

Стадия 1
2,2,2-Трифторэтил 4-метилбензолсульфонат
2,2,2-Трифторэтанол 7a (7,2 мл, 100 ммоль) растворяли в 300 мл дихлорметана с последующим добавлением триэтиламина (28 мл, 200 ммоль). Реакционную смесь охлаждали до 0°C. Реакционную смесь нагревали и перемешивали в течение 16 часов при комнатной температуре после добавления по каплям раствора (100 мл) пара-толуолсульфонилхлорида (TsCl) (30 г, 150 ммоль) в дихлорметане. После этого реакционную смесь экстрагировали дихлорметаном (100 мл × 3) после добавления 100 мл воды. Органический экстракт промывали насыщенным раствором хлорида натрия (50 мл) и объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении, и получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой D с получением указанного в заголовке соединения 2,2,2-трифторэтил 4-метилбензолсульфоната 7b (25 г, бесцветная жидкость), выход: 98,4%.
1Н ЯМР (400 МГц, CDCl3): δ 7.88-7.76 (m, 2Н), 7.44-7.33 (m, 2Н), 4.35 (d, 2Н), 2.47 (s, 3H).
Стадия 2
4-Бром-1-хлор-2-[[4-(2,2,2-трифторэтокси)фенил]метил]бензол
4-[(5-Бром-2-хлор-фенил)метил]фенол 7c (14,5 г, 48,7 ммоль, полученный согласно способу в WO 2009026537) растворяли в 300 мл DMF с последующим добавлением карбоната цезия (31,7 г, 97,5 ммоль). Реакционную смесь перемешивали в течение 10 минут. Реакционную смесь перемешивали в течение 8 часов при 80°С после добавления 2,2,2-трифторэтил-4-метилбензолсульфоната 7b (12,4 г, 48,7 ммоль). После этого реакционную смесь фильтровали и промывали небольшим количеством этилацетата перед тем, как фильтрат концентрировали при пониженном давлении. Получающийся в результате остаток распределялся после добавления 100 мл воды и 50 мл насыщенного раствора хлорида натрия. Водную фазу экстрагировали этилацетатом (100 мл × 3), и органический экстракт промывали насыщенным раствором хлорида натрия (100 мл), объединяли и концентрировали. Получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой В с получением указанного в заголовке соединения 4-бром-1-хлор-2-[[4-(2,2,2-трифторэтокси)фенил]метил]бензола 7d (7,26 г, белое твердое вещество), выход: 39,2%.
1Н ЯМР (400 МГц, CDCl3): δ 7.32 (s, 1Н), 7.30-7.24 (m, 2Н), 7.21-7.14 (m, 2Н), 6.98-6.87 (m, 2Н), 4.38 (d, 2Н), 4.06 (s, 2Н)
Стадия 3
(2S,3R,4S,5S,6R)-2-[4-хлор-3-[[4-(2,2,2-трифторэтокси)фенил]метил]фенил]-6-(гидроксиметил)-2-метокси-тетрагидропиран-3,4,5-триол
4-Бром-1-хлор-2-[[4-(2,2,2-трифторэтокси)фенил]метил]бензол 7d (6,15 г, 16,2 ммоль) растворяли в 150 мл смешанного раствора (THF и н-гексан, v:v=2:3) и охлаждали до -78°C, а затем по каплям добавляли раствор nBuLi в н-гексане (10 мл, 24,3 ммоль). Реакционную смесь перемешивали в течение 1 часа при -78°C перед добавлением по каплям раствора (35 мл) (3R,4S,5R,6R)-3,4,5-трис(триметилсилилокси)-6-(триметилсилил-оксиметил)тетрагидропиран-2-она 2f (8,32 г, 17,8 ммоль) в н-гексане. Затем реакционную смесь перемешивали в течение 2 часов при -78°C перед добавлением 50 мл метанола и 3,2 мл метилсульфоновой кислоты. Реакционную смесь нагревали и перемешивали в течение 16 часов при комнатной температуре. После этого реакционную смесь концентрировали при пониженном давлении, и она распределялась после добавления 30 мл насыщенного раствора бикарбоната натрия и 10 мл воды. Водную фазу экстрагировали этилацетатом (100 мл × 3), и органический экстракт промывали насыщенным раствором хлорида натрия (20 мл) и объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении, и получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой D с получением указанного в заголовке соединения (2S,3R,4S,5S,6R)-2-[4-хлор-3-[[4-(2,2,2-трифторэтокси)фенил]метил]фенил]-6-(гидроксиметил)-2-метокси-тетрагидропиран-3,4,5-триола 7e (3,93 г, белое твердое вещество), выход: 49,2%.
1Н ЯМР (400 МГц, CD3OD): δ 7.57 (d, 1Н), 7.49 (dd, 1Н), 7.38 (d, 1Н), 7.18 (d, 2Н), 6.99-6.88 (m, 2Н), 4.49 (q, 2Н), 4.19-4.01 (m, 3H), 3.99-3.91 (m, 1Н), 3.89-3.71 (m, 2Н), 3.66-3.55 (m, 1Н), 3.49-3.39 (m, 1Н), 3.14-3.04(s, 3H).
Стадия 4
(2S,3R,4S,5S,6R)-6-[(трет-бутил(диметил)силил)оксиметил]-2-[4-хлор-3-[[4-(2,2,2-трифторэтокси)фенил]метил]фенил]-2-метокси-тетрагидропиран-3,4,5-триол
(2S,3R,4S,5S,6R)-2-[4-хлор-3-[[4-(2,2,2-трифторэтокси)фенил]метил]фенил]-6-(гидроксиметил)-2-метокси-тетрагидропиран-3,4,5-триол 7е (3,8 г, 7,71 ммоль) растворяли в 100 мл DMF с последующим добавлением по очереди DMAP (4-диметиламинопиридин) (188 мг, 1,54 ммоль), TBSCl (1,39 г, 9,25 ммоль) и пиридина (50 мл), реакционную смесь перемешивали в течение 36 часов. После этого реакционную смесь концентрировали при пониженном давлении, и она распределялась после добавления 150 мл воды. Водную фазу экстрагировали этилацетатом (100 мл × 3), и органический экстракт промывали насыщенным раствором хлорида натрия (30 мл) и объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении и получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой D с получением указанного в заголовке соединения (2S,3R,4S,5S,6R)-6-[(трет-бутил(диметил)силил)окси-метил]-2-[4-хлор-3-[[4-(2,2,2-трифторэтокси)фенил]метил]фенил]-2-метокси-тетрагидропиран-3,4,5-триола 7f (2,94 г, желтое жироподобное вещество), выход: 62,8%.
1Н ЯМР (400 МГц, DMSO-d6): δ 7.94 (s, 2Н), 7.45 (d, 1Н), 7.40 (d, 1Н), 7.35-7.28 (m, 1Н), 7.17-7.05 (m, 2Н), 7.01-6.90 (m, 2Н), 5.00 (d, 1Н), 4.84-4.74 (m, 2Н), 4.68 (q, 2Н), 4.11-3.87 (m, 3H), 3.73 (dd, 1Н), 3.60-3.46 (m, 1Н),3.42 (ddd, 1Н), 3.14 (td, 1Н), 2.92 (s, 3H), 0.05-0.02 (m, 3H).
Стадия 5
[[(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[4-(2,2,2-трифторэтокси)фенил]метил]фенил]-6-метокси-тетрагидропиран-2-ил]метокси]трет-бутил-диметил-силан
(2S,3R,4S,5S,6R)-6-[(трет-бутил(диметил)силил)оксиметил]-2-[4-хлор-3-[[4-(2,2,2-трифторэтокси)фенил]метил]фенил]-2-метокси-тетрагидропиран-3,4,5-триол 7f (2,90 г, 4,78 ммоль) растворяли в 70 мл DMF и охлаждали до 0°C с последующим добавлением 60% NaH (955 мг, 23,9 ммоль). Реакционную смесь нагревали и перемешивали в течение 1 часа при комнатной температуре перед тем, как добавляли бензилбромид (3,0 мл, 23,9 ммоль). После перемешивания в течение 3 часов добавляли 5 мл метанола и 10 мл воды. Реакционная смесь распределялась после добавления 100 мл воды и 30 мл насыщенного раствора хлорида натрия. Водную фазу экстрагировали этилацетатом (50 мл × 3), и органический экстракт промывали насыщенным раствором хлорида натрия (30 мл) и объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения [[(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[4-(2,2,2-трифторэтокси)фенил]метил]фенил]-6-метокси-тетрагидропиран-2-ил]метокси]трет-бутил-диметил-силана 7g (4,60 г, бледно-желтая жидкость), который использовали непосредственно на следующей стадии без очистки.
Стадия 6
[(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[4-(2,2,2-трифторэтокси)фенил]метил]фенил]-6-метокси-тетрагидропиран-2-ил]метанол
[[(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[4-(2,2,2-трифторэтокси)фенил]метил]фенил]-6-метокси-тетрагидропиран-2-ил]метокси]трет-бутил-диметил-силан 7g (4,19 г, 4,78 ммоль) растворяли в 30 мл метанола с последующим добавлением ацетилхлорида (51 мкл, 0,72 ммоль). Реакционную смесь перемешивали в течение 1 часа. После этого реакционную смесь концентрировали при пониженном давлении, и получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой D с получением указанного в заголовке соединения [(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[4-(2,2,2-трифторэтокси)фенил]метил]фенил]-6-метокси-тетрагидропиран-2-ил]метанола 7h (1,1 г, белое жироподобное вещество), выход: 30,1%.
1Н ЯМР (400 МГц, DMSO-d6): δ 7.55-7.43 (m, 2Н), 7.39-7.28 (m, 6Н), 7.28-7.16 (m, 9Н), 7.11-7.03 (m, 1Н), 7.01 (dd, 2Н), 6.90 (d, 2Н), 4.91-4.73 (m, 4Н), 4.73-4.58 (m, 3H), 4.42 (d, 1Н), 4.17-3.87 (m, 4Н), 3.83-3.62 (m, 4Н), 3.53 (dd, 1Н), 3.24 (d, 1Н), 3.08-2.86 (m, 3H)
Стадия 7
(2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[4-(2,2,2-трифторэтокси)фенил]метил]фенил]-6-метокси-тетрагидропиран-2-карбальдегид
Оксалилхлорид (0,16 мл, 1,87 ммоль) растворяли в 5 мл дихлорметана и охлаждали до -78°C, а затем по каплям добавляли диметилсульфоксид (0,2 мл, 2,88 ммоль) в дихлорметане (3 мл). Реакционную смесь перемешивали в течение 15 минут перед тем, как добавляли 10 мл раствора [(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[4-(2,2,2-трифторэтокси)фенил]метил]фенил]-6-метокси-тетрагидропиран-2-ил]метанола 7h (1,10 г, 1,44 ммоль) в дихлорметане, и перемешивали в течение 40 минут. Затем реакционную смесь нагревали и перемешивали в течение 3 часов при комнатной температуре после добавления по каплям триэтиламина (1,0 мл, 7,21 ммоль). После этого реакционная смесь распределялась после добавления 5 мл 1 М соляной кислоты, водную фазу экстрагировали этилацетатом (10 мл × 2), и органические экстракты объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[4-(2,2,2-трифторэтокси)фенил]метил]фенил]-6-метокси-тетрагидропиран-2-карбальдегида 7i (1,06 г, желтое жироподобное вещество), который использовали непосредственно на следующей стадии без очистки.
Стадия 8
(2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[4-(2,2,2-трифторэтокси)фенил]метил]фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-карбальдегид
(2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[4-(2,2,2-трифторэтокси)фенил]метил]фенил]-6-метокси-тетрагидропиран-2-карбальдегид 7i (1,06 г, 1,39 ммоль) растворяли в 20 мл 1,4-диоксана с последующим добавлением 2,3 мл 37% раствора формальдегида и 4 мл 2,9 М гидроксида натрия. Реакционную смесь перемешивали в течение 25 часов при 70°C. После этого реакционную смесь концентрировали при пониженном давлении перед тем, как добавляли 20 мл воды и 10 мл насыщенного раствора хлорида натрия. Реакционную смесь экстрагировали этилацетатом (30 мл × 3). Органический экстракт объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[4-(2,2,2-трифторэтокси)фенил]-метил]фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-карбальдегида 7j (1,1 г, желтое жироподобное вещество), который использовали непосредственно на следующей стадии без очистки.
Стадия 9
[(3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[4-(2,2,2-трифторэтокси)фенил]метил]фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-ил]метанол
(2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[4-(2,2,2-трифторэтокси)фенил]метил]фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-карбальдегид 7j (1,1 г, 1,39 ммоль) растворяли в 30 мл смешанного раствора (THF и МеОН, v:v=1:2) с последующим добавлением порции боргидрида натрия (106 мг, 2,78 ммоль). Реакционную смесь перемешивали в течение 30 минут перед тем, как добавляли 20 мл воды, затем 30 мл воды. Реакционную смесь экстрагировали этилацетатом (50 мл × 3). Органический экстракт промывали насыщенным раствором хлорида натрия (30 мл) и объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении, и получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой D с получением указанного в заголовке соединения [(3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[4-(2,2,2-трифторэтокси)фенил]метил]фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-ил]метанола 7k (100 мг, желтое жироподобное вещество), выход: 9,1%.
Стадия 10
[(1S,2S,3S,4R,5S)-2,3,4-трибензилокси-5-[4-хлор-3-[[4-(2,2,2-трифторэтокси)фенил]метил]фенил]-6,8-диоксабицикло[3.2.1]октан-1-ил]метанол
[(3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[4-(2,2,2-трифторэтокси)фенил]метил]фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-ил]метанол 7k (100 мг, 0,13 ммоль) растворяли в 10 мл дихлорметана с последующим добавлением 0,1 мл трифторуксусной кислоты. Реакционную смесь перемешивали в течение 1 часа. После этого реакционную смесь концентрировали при пониженном давлении, и получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой D с получением указанного в заголовке соединения [(1S,2S,3S,4R,5S)-2,3,4-трибензилокси-5-[4-хлор-3-[[4-(2,2,2-трифторэтокси)фенил]метил]фенил]-6,8-диоксабицикло[3.2.1]октан-1-ил]метанола 7m (22 мг, белое твердое вещество), выход: 22,9%.
МС m/z(ESI): 778,3 [М+18]
Стадия 11
(1S,2S,3S,4R,5S)-5-[4-хлор-3-[[4-(2,2,2-трифторэтокси)фенил]метил]фенил]-1-(гидроксиметил)-6,8-диоксабицикло[3.2.1]октан-2,3,4-триол
[(1S,2S,3S,4R,5S)-2,3,4-трибензилокси-5-[4-хлор-3-[[4-(2,2,2-трифторэтокси)фенил]метил]фенил]-6,8-диоксабицикло[3.2.1]октан-1-ил]метанол 7m (20 мг, 0,03 ммоль) растворяли в 10 мл смешанного раствора (THF и МеОН, v:v=1:1) с последующим добавлением по очереди о-дихлорбензола (31 мкл, 0,27 ммоль) и палладия на угле (20 мг, 10%). Смесь трижды продували H2 и перемешивали в течение 2 часов. После этого реакционную смесь фильтровали. Фильтрат концентрировали при пониженном давлении, и получающийся в результате остаток очищали при помощи тонкослойной хроматографит с системой хроматографических растворителей A с получением указанного в заголовке соединения (1S,2S,3S,4R,5S)-5-[4-хлор-3-[[4-(2,2,2-трифторэтокси)фенил]метил]фенил]-1-(гидроксиметил)-6,8-диоксабицикло[3.2.1]октан-2,3,4-триола 7 (9 мг, белое твердое вещество), выход: 69,2%.
МС m/z(ESI): 491,1 [М+1]
1Н ЯМР (400 МГц, CD3OD): δ 7.48 (m, 1Н), 7.40-7.38 (m,2Н), 7.18-7.16 (d, 2Н), 6.93-6.91 (d, 2Н), 4.51-4.45 (q, 2Н), 4.6-4.15 (d, 1Н), 4.08 (s, 2Н), 3.81-3.78 (d, 1Н), 3.78-3.71 (d, 1Н), 3.69-3.67 (m, 2Н), 3.62-3.57 (m, 2Н)
Пример 8
(1S,2S,3S,4R,5S)-5-[4-хлор-3-[[3-фтор-4-(тридейтериометокси)фенил]метил]фенил]-1-(гидроксиметил)-6,8-диоксабицикло[3.2.1]октан-2,3,4-триол
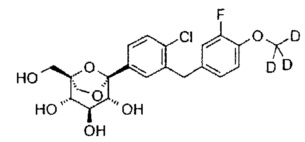
Стадия 1
4-[(5-Бром-2-хлор-фенил)метил]-2-фтор-фенол
4-[(5-Бром-2-хлор-фенил)метил]-1-этокси-2-фтор-бензол 4e (31,7 г, 92,3 ммоль) растворяли в 300 мл дихлорметана с последующим добавлением трибромида бора (11,4 мл, 120 ммоль) в ледяной бане, затем реакционную смесь нагревали и перемешивали в течение 4 часов. После этого реакционную смесь экстрагировали этилацетатом (100 мл × 4) после медленного добавления по каплям 300 мл насыщенного раствора карбоната натрия. Органический экстракт промывали насыщенным раствором хлорида натрия (30 мл) и объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении. Получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой D с получением указанного в заголовке соединения 4-[(5-бром-2-хлор-фенил)метил]-2-фтор-фенола 8а (28,1 г, белое твердое вещество), выход: 96,7%.
1Н ЯМР (400 МГц, CDCl3): δ 7.36-7.18 (m, 3 Н), 7.01-6.80 (m, 3H), 5.08 (br, 1Н), 3.99 (s, 2H)
Стадия 2
4-[(5-Бром-2-хлор-фенил)метил]-2-фтор-1-(тридейтериометокси)бензол
4-[(5-Бром-2-хлор-фенил)метил]-2-фтор-фенол 8а (10,0 г, 31,75 ммоль) растворяли в 150 мл THF с последующим добавлением трифенилфосфина (16,6 г, 63,5 ммоль) и диизопропилового эфира азодикарбоновой кислоты (12,6 мл, 63,5 ммоль). Реакционную смесь перемешивали в течение 30 минут. Реакционную смесь перемешивали в течение 18 часов после добавления 3 мл CD3OD. После этого реакционную смесь концентрировали при пониженном давлении, и получающийся в результате остаток перекристаллизовывали с петролейным эфиром, фильтровали, собирали в виде продукта. Продукт перерастворяли в 50 мл метанола перед добавлением 2 мл перекиси водорода. И после перемешивания в течение 1 минуты реакционная смесь распределялась после добавления 5 г тиосульфата натрия, 40 мл воды и 50 мл этилацетата. Водную фазу экстрагировали этилацетатом (30 мл × 3), и органический экстракт промывали насыщенным раствором хлорида натрия (30 мл), объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении, и остаток очищали при помощи хроматографии на силикагеле с элюирующей системой D с получением указанного в заголовке соединения 4-[(5-бром-2-хлор-фенил)метил]-2-фтор-1-(тридейтериометокси)бензола 8b (8,2 г, белое твердое вещество), выход: 77,4%.
1Н ЯМР (400 МГц, CDCl3): δ 7.37-7.32 (m, 1H), 7.32-7.28 (m, 2H), 6.99-6.89 (m, 3H), 4.02 (s, 2H)
Стадия 3
(2S,3R,4S,5S,6R)-2-[4-хлор-3-[[3-фтор-4-(тридейтериометокси)фенил]метил]фенил]-6-(гидроксиметил)-2-метокси-тетрагидропиран-3,4,5-триол
4-[(5-Бром-2-хлор-фенил)метил]-2-фтор-1-(тридейтериометокси)бензол 8b (8,2 г, 24,6 ммоль) растворяли в 150 мл смешанного раствора (THF и н-гексан, v:v=2:3) и охлаждали до -78°C, а затем по каплям добавляли раствор nBuLi (14,8 мл, 36,9 ммоль) в н-гексане. После перемешивания в течение 2 часов при -78°C по каплям добавляли раствор (40 мл) (3R,4S,5R,6R)-3,4,5-трис(триметилсилилокси)-6-(триметилсилилоксиметил)тетра-гидропиран-2-она 2f (12,6 г, 27,1 ммоль) в н-гексане. Затем, реакционную смесь перемешивали в течение 3 часов при -78°C. Добавляли 40 мл метанола и 4,79 мл метансульфоновой кислоты перед тем, как реакционную смесь нагревали и перемешивали в течение 16 часов при комнатной температуре. После этого реакционную смесь концентрировали при пониженном давлении, и она распределялась после добавления 40 мл насыщенного раствор бикарбоната натрия, 100 мл этилацетата и 100 мл воды. Водную фазу экстрагировали этилацетатом (50 мл × 4), и органические экстракты объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении, и получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой D с получением указанного в заголовке соединения (2S,3R,4S,5S,6R)-2-[4-хлор-3-[[3-фтор-4-(тридейтериометокси)фенил]метил]фенил]-6-(гидроксиметил)-2-метокситетрагидропиран-3,4,5-триола 8с (8,2 г, белое твердое вещество), выход: 83,6%.
1Н ЯМР (400 МГц, CDCl3): δ 7.47-7.29 (m, 3H), 6.93-6.78 (m, 3H), 4.08-3.98 (m, 2H), 3.98-3.78 (m, 5H), 3.73 (d, 1H), 3.47 (s, 6H)
Стадия 4
(2S,3R,4S,5S,6R)-6-[(трет-бутил(диметил)силил)оксиметил]-2-[4-хлор-3-[[3-фтор-4-(тридейтериометокси)фенил]метил]фенил]-2-метокси-тетрагидропиран-3,4,5-триол
(2S,3R,4S,5S,6R)-2-[4-хлор-3-[[3-фтор-4-(тридейтериометокси)фенил]метил]фенил]-6-(гидроксиметил)-2-метокси-тетрагидропиран-3,4,5-триол 8с (9,2 г, 20,6 ммоль) растворяли в 80 мл пиридина с последующим добавлением 4-диметиламинопиридина (502 мг, 4,11 ммоль) и трет-бутил-диметил-хлор-силана (3,72 г, 24,7 ммоль). Реакционную смесь перемешивали в течение 24 часов и концентрировали при пониженном давлении, и она распределялась после добавления 80 мл этилацетата и 80 мл воды. Водную фазу экстрагировали этилацетатом (30 мл × 3), и органический экстракт объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении, и получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой D с получением указанного в заголовке соединения (2S,3R,4S,5S,6R)-6-[(трет-бутил(диметил)силил)оксиметил]-2-[4-хлор-3-[[3-фтор-4-(тридейтериометокси)фенил]метил]фенил]-2-метокси-тетрагидропиран-3,4,5-триола 8d (2,4 г, желтое жироподобное вещество), выход: 20,9%.
Стадия 5
[[(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[3-фтор-4-(тридейтериометокси)фенил]метил]фенил]-6-метокси-тетрагидропиран-2-ил]метокси]трет-бутил-диметил-силан
(2S,3R,4S,5S,6R)-6-[(трет-бутил(диметил)силил)оксиметил]-2-[4-хлор-3-[[3-фтор-4-(тридейтериометокси)фенил]метил]фенил]-2-метокси-тетрагидропиран-3,4,5-триол 8d (2,4 г, 4,29 ммоль) растворяли в 70 мл DMF и охлаждали до 0°C с последующим добавлением 60% NaH (857 мг, 21,4 ммоль). Затем реакционную смесь нагревали до комнатной температуры и перемешивали в течение 1 часа. После этого добавляли бензилбромид (2,56 мл, 21,4 ммоль) перед тем, как смесь перемешивали в течение 3 часов. Добавляли 5 мл метанола и 100 мл воды перед тем, как реакционную смесь экстрагировали этилацетатом (50 мл × 3). Органический экстракт промывали насыщенным раствором хлорида натрия (30 мл) и объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения [[(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[3-фтор-4-(тридейтериометокси)фенил]метил]фенил]-6-метокси-тетрагидропиран-2-ил]метокси]трет-бутил-диметил-силана 8е (3,56 г, желтое твердое вещество), который использовали непосредственно на следующей стадии без очистки.
Стадия 6
[(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[3-фтор-4-(тридейтериометокси)фенил]метил]фенил]-6-метокси-тетрагидропиран-2-ил]метанол
[[(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[3-фтор-4-(тридейтериометокси)фенил]метил]фенил]-6-метокси-тетрагидропиран-2-ил]метокси]трет-бутил-диметил-силан 8е (3,56, 4,29 ммоль) растворяли в 30 мл метанола и перемешивали в течение 4 часов после добавления ацетилхлорида (95,5 мкл, 1,34 ммоль). После этого реакционную смесь концентрировали при пониженном давлении. Реакционную смесь экстрагировали этилацетатом (30 мл × 3) после добавления 30 мл воды. Органический экстракт объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении, и получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой D с получением указанного в заголовке соединения [(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[3-фтор-4-(тридейтериометокси)фенил]метил]фенил]-6-метокси-тетрагидропиран-2-ил]метанола 8f (2,2 г, желтое твердое вещество), выход: 73,3%.
1Н ЯМР (400 МГц, CDCl3): δ 7.42-7.27 (m, 13Н), 7.24-7.14 (m, 3H), 6.98 (d, 2H), 6.89-6.72 (m, 3H), 4.96-4.87 (m, 3H), 4.71-4.67 (m, 1H), 4.51 (d, 1H), 4.18 (t, 1H), 4.07 (d, 1H), 3.95-3.84 (m, 3H), 3.83-3.77 (m, 1H), 3.76-3.64 (m, 2H), 3.30 (d, 1H), 3.07 (s, 3H).
Стадия 7
(2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[3-фтор-4-(тридейтериометокси)фенил]метил]фенил]-6-метокси-тетрагидропиран-2-карбальдегид
Оксалилхлорид (0,34 мл, 3,93 ммоль) растворяли в 5 мл дихлорметана и охлаждали до -78°C. Добавляли раствор (10 мл) диметилсульфоксида (0,43 мл, 6,04 ммоль) в дихлорметане перед тем, как смесь перемешивали в течение 15 минут. Затем добавляли 15 мл раствора [(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[3-фтор-4-(тридейтериометокси)фенил]метил]фенил]-6-метокси-тетрагидропиран-2-ил]метанола 8f (2,2 г, 3,02 ммоль) в метиленхлориде перед тем, как смесь перемешивали в течение 45 минут. После этого добавляли триэтиламин (2,1 мл, 15,1 ммоль) перед тем, как смесь нагревали до комнатной температуры и перемешивали в течение 2,5 часов. Добавляли 5 мл 1 М раствора соляной кислоты перед тем, как смесь экстрагировали дихлорметаном (15 мл × 2). Органические экстракты объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[3-фтор-4-(тридейтериометокси)фенил]метил]фенил]-6-метокси-тетрагидропиран-2-карбальдегида 8g (2,1 г, желтая жидкость), который использовали непосредственно на следующей стадии без очистки.
Стадия 8
(2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[3-фтор-4-(тридейтериометокси)фенил]метил]фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-карбальдегид
(2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[3-фтор-4-(тридейтериометокси)-фенил]метил]фенил]-6-метокси-тетрагидропиран-2-карбальдегид 8g (2,1 г, 2,94 ммоль) растворяли в 50 мл 1,4-диоксана с последующим добавлением раствора формальдегида (5,4 мл, 72,2 ммоль) и 3,94 мл 3 М гидроксида натрия. Реакционную смесь перемешивали в течение 6 часов при 50°C. После этого реакционную смесь экстрагировали этилацетатом (20 мл × 3) после добавления 20 мл воды. Органические экстракты объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[3-фтор-4-(тридейтериометокси)фенил]метил]фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-карбальдегида 8h (2,5 г, желтая жидкость), который использовали непосредственно на следующей стадии без очистки.
Стадия 9
[(3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[3-фтор-4-(тридейтериометокси)фенил]метил]фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-ил]метанол
(2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[3-фтор-4-(тридейтериометокси)фенил]метил]фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-карбальдегид 8h (2,5 г, 3,36 ммоль) растворяли в 30 мл смешанного раствора (THF и МеОН, v:v=1:2) с последующим добавлением порции боргидрида натрия (269 мг, 6,72 ммоль). Реакционную смесь перемешивали в течение 2 часов, затем гасили 40 мл воды. Реакционную смесь экстрагировали этилацетатом (40 мл × 3). Органический экстракт промывали насыщенным раствором хлорида натрия (30 мл) и объединяли, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения [(3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[3-фтор-4-(тридейтериометокси)фенил]метил]фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-ил]метанола 8i (350 мг, желтая жидкость), выход: 15,9%.
1Н ЯМР (400 МГц, CDCl3): δ 7.41-7.19 (m, 16H), 7.04 (dd, 2H), 6.86-6.76 (m, 3H), 5.01-4.87 (m, 3H), 4.71-4.59 (m, 2H), 4.44-4.31 (m, 2H), 4.06-3.92 (m, 3H), 3.87-3.76 (m, 3H), 3.68 (d, 1H), 3.25 (d, 1H), 3.07 (s, 3H).
Стадия 10
[(1S,2S,3S,4R,5S)-2,3,4-трибензилокси-5-[4-хлор-3-[[3-фтор-4-(тридейтериометокси)фенил]метил]фенил]-6,8-диоксабицикло[3.2.1]октан-1-ил]метанол
[(3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[3-фтор-4-(тридейтериометокси)фенил]метил]фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-ил]метанол 8i (350 мг, 0,47 ммоль) растворяли в 20 мл дихлорметана, а затем добавляли 0,5 мл трифторуксусной кислоты. Реакционную смесь перемешивали в течение 2 часов. После этого реакционную смесь концентрировали при пониженном давлении, и получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой D с получением указанного в заголовке соединения [(1S,2S,3S,4R,5S)-2,3,4-трибензилокси-5-[4-хлор-3-[[3-фтор-4-(тридейтериометокси)фенил]метил]фенил]-6,8-диоксабицикло[3.2.1]октан-1-ил]метанола 8j (200 мг, бесцветная жидкость), выход: 59,7%.
МС m/z (ESI): 731,3 [М+18]
Стадия 11
(1S,2S,3S,4R,5S)-5-[4-хлор-3-[[3-фтор-4-(тридейтериометокси)фенил]метил]фенил]-1-(гидроксиметил)-6,8-диоксабицикло[3.2.1]октан-2,3,4-триол
[(1S,2S,3S,4R,5S)-2,3,4-трибензилокси-5-[4-хлор-3-[[3-фтор-4-(тридейтериометокси)фенил]метил]фенил]-6,8-диоксабицикло[3.2.1]октан-1-ил]метанол 8j (190 мг, 0,27 ммоль) растворяли в 30 мл смешанного раствора (THF и МеОН, v:v=1:1) с последующим добавлением по очереди о-дихлорбензола (391 мг, 2,66 ммоль) и палладия на угле (20 мг, 10%). Смесь трижды продували H2 и перемешивали в течение 3 часов. После этого реакционную смесь фильтровали. Фильтрат концентрировали при пониженном давлении, и получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой F с получением указанного в заголовке соединения (1S,2S,3S,4R,5S)-5-[4-хлор-3-[[3-фтор-4-(тридейтериометокси)фенил]метил]фенил]-1-(гидроксиметил)-6,8-диоксабицикло[3.2.1]октан-2,3,4-триола 8 (74 мг, белое твердое вещество), выход: 62,7%.
МС m/z (ESI): 461,1 [М+18]
1Н ЯМР (400 МГц, CD3OD): δ 7.59-7.45 (m, 1H), 7.45-7.33 (m, 2H), 7.06-6.84 (m, 3H), 4.16 (d, 1H), 4.05 (d, 2H), 3.93-3.76 (m, 2H), 3.75-3.52 (m, 4H)
Пример 9
(1S,2S,3S,4R,5S)-5-[3-[[5-(4-фторфенил)-2-тиенил]метил]-4-метил-фенил]-1-(гидроксиметил)-6,8-диоксабицикло[3.2.1]октан-2,3,4-триол
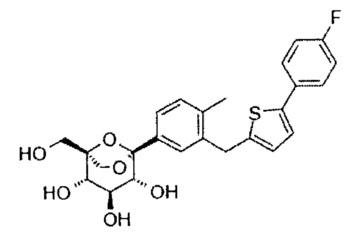
Стадия 1
2-(4-Фторфенил)тиофен
2-Йодтиофен (1,05 г, 5 ммоль) растворяли в 6 мл смешанного раствора (диметиловый эфир и вода, v:v=2:1) с последующим добавлением 1-фтор-4-метил-бензола 9а (700 мг, 5 ммоль), карбоната калия (1,38 г, 10 ммоль) и тетракис(трифенилфосфин)палладия (173 мг, 0,15 ммоль). Смесь подвергали обработке в микроволновом реакторе в течение 30 минут при 100°C. После этого реакционную смесь экстрагировали этилацетатом (20 мл × 3) после добавления 10 мл воды. Органический экстракт промывали насыщенным раствором хлорида натрия (20 мл) и объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении, и получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой C с получением указанного в заголовке соединения 2-(4-фторфенил)тиофена 9b (767 мг, белое твердое вещество), выход: 86,1%.
Стадия 2
(5-Бром-2-хлор-фенил)-[5-(4-фторфенил)-2-тиенил]метанон
AlCl3 (1,5 г, 11 ммоль) растворяли в 10 мл метиленхлорида и охлаждали до -10°С с последующим добавлением 5-бром-2-хлор-бензоилхлорида 2а (2,54 г, 10 ммоль) и 2-(4-фторфенил)тиофена 9b (1,78 г, 10 ммоль). Реакционную смесь перемешивали в течение 30 минут и затем нагревали до комнатной температуры и перемешивали в течение еще 16 часов. После этого реакционную смесь охлаждали до -10°C. Реакционную смесь экстрагировали этилацетатом (50 мл × 2) после добавления небольшого количества воды и 20 мл 1 М соляной кислоты. Органические экстракты объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении, и получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой В с получением указанного в заголовке соединения (5-бром-2-хлор-фенил)-[5-(4-фторфенил)-2-тиенил]метанона 9с (1,5 г, желтое твердое вещество), выход: 37,9%.
Стадия 3
(2S,3R,4S,5S,6R)-2-[3-[[5-(4-фторфенил)-2-тиенил]метил]-4-метил-фенил]-6-(гидроксиметил)-2-метокси-тетрагидропиран-3,4,5-триол
(5-Бром-2-хлор-фенил)-[5-(4-фторфенил)-2-тиенил]метанон 9с (3,7 г, 10,4 ммоль) растворяли в 40 мл THF и охлаждали до -78°C, а затем по каплям добавляли раствор nBuLi в н-гексане (5 мл, 12,5 ммоль). Реакционную смесь перемешивали в течение 1 часа при -78°C. Добавляли 30 мл раствора (3R,4S,5R,6R)-3,4,5-трис(триметилсилилокси)-6-(триметилсилилоксиметил)тетрагидропиран-2-она 2f (4,85 г, 10,4 ммоль) в THF перед тем, как реакционную смесь перемешивали в течение 2 часов при -78°С. Добавляли раствор (60 мл) 0,6 М метансульфоновой кислоты в метаноле перед тем, как реакционную смесь нагревали и перемешивали в течение 16 часов при комнатной температуре. После этого реакционную смесь концентрировали при пониженном давлении и экстрагировали этилацетатом (40 мл × 3) после добавления 30 мл насыщенного раствора карбоната натрия. Органические экстракты объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении, и получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой A с получением указанного в заголовке соединения (2S,3R,4S,5S,6R)-2-[3-[[5-(4-фторфенил)-2-тиенил]метил]-4-метил-фенил]-6-(гидроксиметил)-2-метокси-тетрагидропиран-3,4,5-триола 9d (1,8 г, оранжевое твердое вещество), выход: 36,7%.
Стадия 4
(2S,3R,4S,5S,6R)-6-[(трет-бутил(диметил)силил)оксиметил]-2-[3-[[5-(4-фторфенил)-2-тиенил]метил]-4-метил-фенил]-2-метокси-тетрагидропиран-3,4,5-триол
(2S,3R,4S,5S,6R)-2-[3-[[5-(4-фторфенил)-2-тиенил]метил]-4-метил-фенил]-6-(гидроксиметил)-2-метокси-тетрагидропиран-3,4,5-триол 9d (1,8 г, 3,8 ммоль) растворяли в 20 мл пиридина с последующим добавлением по очереди 4-диметиламинопиридина (93 мг, 0,76 ммоль) и TBSCl (686 мг, 4,55 ммоль). Реакционную смесь перемешивали в течение 16 часов. После этого реакционную смесь концентрировали при пониженном давлении, растворяли в 30 мл этилацетата, и она распределялась после добавления 30 мл воды. Водную фазу экстрагировали этилацетатом (30 мл × 3), и органические экстракты объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении, и получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой A с получением указанного в заголовке соединения (2S,3R,4S,5S,6R)-6-[(трет-бутил(диметил)силил)оксиметил]-2-[3-[[5-(4-фторфенил)-2-тиенил]метил]-4-метил-фенил]-2-метокси-тетрагидропиран-3,4,5-триола 9е (2,0 г, оранжевое твердое вещество), выход: 89,7%.
Стадия 5
[[(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[3-[[5-(4-фторфенил)-2-тиенил]метил]-4-метил-фенил]-6-метокси-тетрагидропиран-2-ил]метокси]трет-бутил-диметил-силан
(2S,3R,4S,5S,6R)-6-[(трет-бутил(диметил)силил)оксиметил]-2-[3-[[5-(4-фторфенил)-2-тиенил]метил]-4-метил-фенил]-2-метокси-тетрагидропиран-3,4,5-триол 9е (2,0 г, 3,4 ммоль) растворяли в 20 мл DMF и охлаждали до 0°C с последующим добавлением 60% NaH (680 мг, 17 ммоль). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 30 минут. После этого добавляли бензилбромид (2,0 мл, 17 ммоль) перед тем, как смесь перемешивали в течение 16 часов. Реакционную смесь концентрировали при пониженном давлении после добавления 10 мл метанола, и она распределялась после добавления 30 мл этилацетата и 30 мл воды. Водную фазу экстрагировали этилацетатом (20 мл × 3), и органические экстракты объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения [[(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[3-[[5-(4-фторфенил)-2-тиенил]метил]-4-метил-фенил]-6-метокси-тетрагидропиран-2-ил]метокси]трет-бутил-диметил-силана 9f (2,9 г, желтое жироподобное вещество), который использовали непосредственно на следующей стадии без очистки.
Стадия 6
[(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[3-[[5-(4-фторфенил)-2-тиенил]метил]-4-метил-фенил]-6-метокси-тетрагидропиран-2-ил]метанол
[[(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[3-[[5-(4-фторфенил)-2-тиенил]метил]-4-метил-фенил]-6-метокси-тетрагидропиран-2-ил]метокси]трет-бутил-диметил-силан 9f (2,9 г, 3,37 ммоль) растворяли в 20 мл метанола с последующим добавлением ацетилхлорида (38 мкл, 0,5 ммоль). Реакционную смесь перемешивали в течение 2 часов, и она распределялась после добавления 30 мл этилацетата и 30 мл воды. Водную фазу экстрагировали этилацетатом (30 мл × 2), и органические экстракты объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении, и затем получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой В с получением указанного в заголовке соединения [(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[3-[[5-(4-фторфенил)-2-тиенил]метил]-4-метил-фенил]-6-метокси-тетрагидропиран-2-ил]метанола 9g (1,9 г, желтое твердое вещество), выход: 76,0%.
Стадия 7
(2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[3-[[5-(4-фторфенил)-2-тиенил]метил]-4-метил-фенил]-6-метокси-тетрагидропиран-2-карбальдегид
Оксалилхлорид (0,27 мл, 3,12 ммоль) растворяли в 8 мл дихлорметана и охлаждали до -78°C. Реакционную смесь перемешивали в течение 15 минут перед тем, как добавляли 4 мл раствора диметилсульфоксида (0,36 мл, 5,04 ммоль) в дихлорметане. Затем по каплям добавляли 8 мл раствора [(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[3-[[5-(4-фторфенил)-2-тиенил]метил]-4-метил-фенил]-6-метокси-тетрагидропиран-2-ил]метанола 9g (1,8 г, 2,4 ммоль) в дихлорметане. Реакционную смесь перемешивали в течение 30 минут. Затем добавляли триэтиламин (1,66 мл, 12 ммоль) перед тем, как реакционную смесь нагревали до комнатной температуры и перемешивали в течение 1 часа. После этого реакционная смесь распределялась после добавления 12 мл 1 М соляной кислоты, органический экстракт объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[3-[[5-(4-фторфенил)-2-тиенил]метил]-4-метил-фенил]-6-метокси-тетрагидропиран-2-карбальдегида 9h (1,8 г, желтое жироподобное вещество), который использовали непосредственно на следующей стадии без очистки.
Стадия 8
(2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[3-[[5-(4-фторфенил)-2-тиенил]метил]-4-метил-фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-карбальдегид
(2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[3-[[5-(4-фторфенил)-2-тиенил]метил]-4-метил-фенил]-6-метокси-тетрагидропиран-2-карбальдегид 9h (1,53 г, 2,1 ммоль) растворяли в 15 мл 1,4-диоксана с последующим добавлением 3,4 мл 37% раствора формальдегида и 6,3 мл 1 М гидроксида натрия. Реакционную смесь перемешивали в течение 16 часов при 70°C, и она распределялась после добавления 50 мл насыщенного раствора хлорида натрия. Водную фазу экстрагировали этилацетатом (50 мл × 3), и органические экстракты объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[3-[[5-(4-фторфенил)-2-тиенил]метил]-4-метил-фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-карбальдегида 9i (2,0 г, бледно-желтое жироподобное вещество), который использовали непосредственно на следующей стадии без очистки.
Стадия 9
[(3S,4S,5R,6S)-3,4,5-трибензилокси-6-[3-[[5-(4-фторфенил)-2-тиенил]метил]-4-метил-фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-ил]метанол
(2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[3-[[5-(4-фторфенил)-2-тиенил]метил]-4-метил-фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-карбальдегид 9i (2,0 г, 2,1 ммоль) растворяли в 30 мл смешанного раствора (THF и МеОН, v:v=1:20) с последующим добавлением порции боргидрида натрия (238 мг, 6,3 ммоль). Реакционную смесь перемешивали в течение 1 часа. После этого реакционную смесь концентрировали при пониженном давлении, и получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой B и E с получением указанного в заголовке соединения [(3S,4S,5R,6S)-3,4,5-трибензилокси-6-[3-[[5-(4-фторфенил)-2-тиенил]метил]-4-метил-фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-ил]метанола 9j (420 мг, бледно-желтое твердое вещество), выход: 25,8%.
1Н ЯМР (400 МГц, CD3OD): δ 7.55 (d, 2Н), 7.48-7.45 (m, 3H), 7.32-7.28 (m, 5Н), 7.25-7.21 (m, 9Н), 7.16 (d, 2Н), 7.07-7.03 (m, 3H), 6.63 (d, 1Н), 4.91 (d, 1Н), 4.83 (d, 1Н), 4.75 (d, 1Н), 4.54 (d, 1Н), 4.26-4.17 (m, 2Н), 4.10-4.04 (m, 5Н), 3.94 (d, 1Н), 3.78 (d, 1Н), 3.36 (d, 1Н), 3.26 (s, 3H), 2.34 (s, 3H)
Стадия 10
(1S,2S,3S,4R,5S)-5-[3-[[5-(4-фторфенил)-2-тиенил]метил]-4-метил-фенил]-1-(гидроксиметил)-6,8-диоксабицикло[3.2.1]октан-2,3,4-триол
[(3S,4S,5R,6S)-3,4,5-трибензилокси-6-[3-[[5-(4-фторфенил)-2-тиенил]метил]-4-метил-фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-ил]метанол 9j (130 мг, 0,17 ммоль) растворяли в 10 мл метанола с последующим добавлением раствора (4 мл) 2 M соляной кислоты в этилацетате и палладия на угле (260 мг, 20%). Смесь трижды продували H2 и перемешивали в течение 16 часов. После этого реакционную смесь фильтровали. Фильтрат концентрировали при пониженном давлении, и получающийся в результате остаток очищали при помощи ВЭЖХ с получением указанного в заголовке соединения (1S,2S,3S,4R,5S)-5-[3-[[5-(4-фторфенил)-2-тиенил]метил]-4-метил-фенил]-1-(гидроксиметил)-6,8-диоксабицикло[3.2.1]октан-2,3,4-триола 9 (11 мг, белое твердое вещество), выход: 13,8%.
МС m/z (ESI): 473,2 [М+1]
1Н ЯМР (400 МГц, CD3OD): δ 7.57-7.56 (m, 2Н), 7.48 (d, 1Н), 7.39-7.37 (m, 1Н), 7.19 (d, 1Н), 7.15 (d, 1Н), 7.10-7.06 (m, 2Н), 6.67 (d, 1Н), 4.18 (d, 3H), 3.88-3.81 (m, 2Н), 3.73-3.68 (m, 2Н), 3.65-3.62 (m, 2Н), 2.33 (s, 3H)
Пример 10
(1S,2S,3S,4R,5S)-5-[4-хлор-3-[[3-фтор-4-(1,1,2,2,2-пентадейтериоэтокси)фенил]метил]фенил]-1-(гидроксиметил)-6,8-диоксабицикло[3.2.1]октан-2,3,4-триол
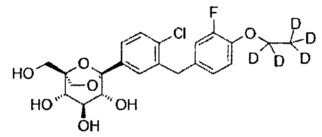
Стадия 1
4-[(5-Бром-2-хлор-фенил)метил]-2-фтор-1-(1,1,2,2,2-пентадейтериоэтокси)бензол
4-[(5-Бром-2-хлор-фенил)метил]-2-фтор-фенол 8a (6,0 г, 19,05 ммоль) растворяли в 100 мл THF с последующим добавлением трифенилфосфина (9,98 г, 38,1 ммоль) и диизопропилового эфира азодикарбоновой кислоты (7,7 мл, 38,1 ммоль). Реакционную смесь перемешивали в течение 30 минут. Добавляли 2,5 мл этанола-d6 перед тем, как реакционную смесь перемешивали в течение 18 часов. После этого реакционную смесь концентрировали при пониженном давлении, и получающийся в результате остаток перекристаллизовывали с петролейным эфиром, фильтровали и собирали в виде осадка на фильтре. Осадок на фильтре растворяли в 50 мл метанола перед тем, как добавляли 2 мл перекиси водорода. После перемешивания в течение 1 минуты смесь распределялась после добавления 5 г тиосульфата натрия, 40 мл воды и 50 мл этилацетата. Водную фазу экстрагировали этилацетатом (30 мл × 3), и органический экстракт промывали насыщенным раствором хлорида натрия (30 мл), объединяли, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении, и получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой D с получением указанного в заголовке соединения 4-[(5-бром-2-хлор-фенил)метил]-2-фтор-1-(1,1,2,2,2-пентадейтериоэтокси)бензола 10а (5,78 г, бесцветная жидкость), выход: 86%.
1Н ЯМР (400 МГц, CDCl3): δ 7.38-7.24 (m, 3H), 6.99-6.83 (m, 3H), 4.02 (s, 2Н)
Стадия 2
(2S,3R,4S,5S,6R)-2-[4-хлор-3-[[3-фтор-4-(1,1,2,2,2-пентадейтериоэтокси)фенил]метил]фенил]-6-(гидроксиметил)-2-метокси-тетрагидропиран-3,4,5-триол
4-[(5-Бром-2-хлор-фенил)метил]-2-фтор-1-(1,1,2,2,2-пентадейтериоэтокси)бензол 10a (5,78 г, 16,6 ммоль) растворяли в 125 мл смешанного раствора (THF и н-гексан, v:v=2:3) и охлаждали до -78°C, а затем по каплям добавляли раствор nBuLi (8,53 г, 18,26 ммоль) в н-гексане. После перемешивания в течение 2 часов при -78°C по каплям добавляли раствор (40 мл) (3R,4S,5R,6R)-3,4,5-трис(триметилсилилокси)-6-(триметилсилилоксиметил)тетрагидропиран-2-она 2f (12,6 г, 27,1 ммоль) в н-гексане. Реакционную смесь перемешивали в течение 3 часов при -78°C. Добавляли 40 мл метанола и 4,79 мл метансульфоновой кислоты перед тем, как реакционную смесь нагревали и перемешивали в течение 16 часов при комнатной температуре. После этого реакционную смесь концентрировали при пониженном давлении, и она распределялась после добавления 40 мл насыщенного раствор бикарбоната натрия, 100 мл этилацетата и 100 мл воды. Водную фазу экстрагировали этилацетатом (50 мл × 4), и органические экстракты объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении, и получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой D с получением указанного в заголовке соединения (2S,3R,4S,5S,6R)-2-[4-хлор-3-[[3-фтор-4-(1,1,2,2,2-пентадейтериоэтокси)фенил]метил]фенил]-6-(гидроксиметил)-2-метокси-тетрагидропиран-3,4,5-триола 10b (1,6 г, желтое твердое вещество), выход: 20,9%.
Стадия 3
(2S,3R,4S,5S,6R)-6-[(трет-бутил(диметил)силил)оксиметил]-2-[4-хлор-3-[[3-фтор-4-(1,1,2,2,2-пентадейтериоэтокси)фенил]метил]фенил]-2-метокси-тетрагидропиран-3,4,5-триол
(2S,3R,4S,5S,6R)-2-[4-хлор-3-[[3-фтор-4-(1,1,2,2,2-пентадейтериоэтокси)фенил]метил]фенил]-6-(гидроксиметил)-2-метокси-тетрагидропиран-3,4,5-триол 10b (1,6 г, 3,47 ммоль) растворяли в 40 мл пиридина с последующим добавлением по очереди 4-диметиламинопиридина (64 мг, 0,52 ммоль) и TBSCl (0,57 г, 3,8 ммоль). Реакционную смесь перемешивали в течение 24 часов и концентрировали при пониженном давлении, и она распределялась после добавления 50 мл этилацетата и 50 мл воды. Водную фазу экстрагировали этилацетатом (30 мл × 3), и органический экстракт объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении, и получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой D с получением указанного в заголовке соединения (2S,3R,4S,5S,6R)-6-[(трет-бутил(диметил)силил)оксиметил]-2-[4-хлор-3-[[3-фтор-4-(1,1,2,2,2-пентадейтериоэтокси)фенил]метил]фенил]-2-метокси-тетрагидропиран-3,4,5-триола 10с (1,7 г, желтая жидкость), выход: 85%.
Стадия 4
[[(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[3-фтор-4-(1,1,2,2,2-пентадейтериоэтокси)фенил]метил]фенил]-6-метокси-тетрагидропиран-2-ил]метокси]трет-бутил-диметил-силан
(2S,3R,4S,5S,6R)-6-[(трет-бутил(диметил)силил)оксиметил]-2-[4-хлор-3-[[3-фтор-4-(1,1,2,2,2-пентадейтериоэтокси)фенил]метил]фенил]-2-метокси-тетрагидропиран-3,4,5-триол 10с (1,7 г, 2,90 ммоль) растворяли в 50 мл DMF и охлаждали до 0°C с последующим добавлением 60% NaH (620 мг, 14,0 ммоль). Затем реакционную смесь нагревали до комнатной температуры и перемешивали в течение 1 часа. После этого добавляли бензилбромид (1,90 мл, 14 ммоль) перед тем, как смесь перемешивали в течение 3 часов. Реакционную смесь экстрагировали этилацетатом (50 мл × 3) после добавления 5 мл метанола и 100 мл воды. Органический экстракт промывали насыщенным раствором хлорида натрия (30 мл) и объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения [[(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[3-фтор-4-(1,1,2,2,2-пентадейтериоэтокси)фенил]метил]фенил]-6-метокси-тетрагидропиран-2-ил]метокси]трет-бутил-диметил-силана 10d (2,45 г, желтая жидкость), который использовали непосредственно на следующей стадии без очистки.
Стадия 5
[(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[3-фтор-4-(1,1,2,2,2-пентадейтериоэтокси)фенил]метил]фенил]-6-метокси-тетрагидропиран-2-ил]метанол
[[(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[3-фтор-4-(1,1,2,2,2-пентадейтериоэтокси)фенил]метил]фенил]-6-метокси-тетрагидропиран-2-ил]метокси]трет-бутил-диметил-силан 10d (2,45 г, 2,90 ммоль) растворяли в 30 мл метанола с последующим добавлением ацетилхлорида (50 мкл, 0,4 ммоль). Реакционную смесь перемешивали в течение 4 часов. После этого реакционную смесь концентрировали при пониженном давлении перед тем, как добавляли 30 мл воды. Затем получающийся в результате остаток экстрагировали этилацетатом (30 мл × 3), и органические экстракты объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении, и затем получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой D с получением указанного в заголовке соединения [(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[3-фтор-4-(1,1,2,2,2-пентадейтериоэтокси)фенил]метил]фенил]-6-метокси-тетрагидропиран-2-ил]метанола 10e (1,2 г, желтая жидкость), выход: 56,8%.
Стадия 6
(2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[3-фтор-4-(1,1,2,2,2-пентадейтериоэтокси)фенил]метил]фенил]-6-метокси-тетрагидропиран-2-карбальдегид
Оксалилхлорид (0,36 мл, 3,93 ммоль) растворяли в 5 мл дихлорметана и охлаждали до -78°C. Добавляли 10 мл раствора диметилсульфоксида (0,35 мл, 4,92 ммоль) в дихлорметане и перемешивали в течение 15 минут, затем добавляли 15 мл раствора [(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[3-фтор-4-(1,1,2,2,2-пентадейтериоэтокси)фенил]метил]фенил]-6-метокси-тетрагидропиран-2-ил]метанола 10е (1,2 г, 1,64 ммоль) в дихлорметане и перемешивали в течение 45 минут. Затем добавляли триэтиламин (1,2 мл, 8,2 ммоль) перед тем, как реакционную смесь нагревали и перемешивали в течение 2,5 часов при комнатной температуре. После этого реакционную смесь экстрагировали дихлорметаном (15 мл × 2) после добавления 5 мл 1 М соляной кислоты. Органические экстракты объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[3-фтор-4-(1,1,2,2,2-пентадейтериоэтокси)фенил]метил]фенил]-6-метокси-тетрагидропиран-2-карбальдегида 10f (1,2 г, желтая жидкость), который использовали непосредственно на следующей стадии без очистки.
Стадия 7
[(3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[3-фтор-4-(1,1,2,2,2-пентадейтериоэтокси)фенил]метил]фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-ил]метанол
(2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[3-фтор-4-(1,1,2,2,2-пентадейтериоэтокси)фенил]метил]фенил]-6-метокси-тетрагидропиран-2-карбальдегид 10f (1,2 г, 1,64 ммоль) растворяли в 40 мл 1,4-диоксана с последующим добавлением раствора формальдегида (2,2 мл, 7,54 ммоль), гидроксида калия (0,27 г, 4,92 ммоль) и бензилового спирта (177 мг, 1,64 ммоль). Реакционную смесь перемешивали в течение 6 часов при 50°C. После этого реакционную смесь экстрагировали этилацетатом (20 мл × 3) после добавления 20 мл воды. Органические экстракты объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения [(3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[3-фтор-4-(1,1,2,2,2-пентадейтериоэтокси)фенил]метил]фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-ил]метанола 10g (0,64 г, желтая жидкость), который использовали непосредственно на следующей стадии без очистки.
Стадия 8
[(1S,2S,3S,4R,5S)-2,3,4-трибензилокси-5-[4-хлор-3-[[3-фтор-4-(1,1,2,2,2-пентадейтериоэтокси)фенил]метил]фенил]-6,8-диоксабицикло[3.2.1]октан-1-ил]метанол
[(3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[3-фтор-4-(1,1,2,2,2-пентадейтериоэтокси)фенил]метил]фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-ил]метанол 10g (640 мг, 0,83 ммоль) растворяли в 20 мл метиленхлорида, а затем по каплям добавляли трифторуксусную кислоту (0,5 мл). Смесь перемешивали в течение 2 часов. После этого реакционную смесь концентрировали при пониженном давлении, и получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой D с получением указанного в заголовке соединения [(1S,2S,3S,4R,5S)-2,3,4-трибензилокси-5-[4-хлор-3-[[3-фтор-4-(1,1,2,2,2-пентадейтериоэтокси)фенил]метил]фенил]-6,8-диоксабицикло[3.2.1]октан-1-ил]метанола 10h (300 мг, бледно-желтое твердое вещество), выход: 41,0%.
МС m/z(ESI): 747,3 [М+18]
Стадия 9
(1S,2S,3S,4R,5S)-5-[4-хлор-3-[[3-фтор-4-(1,1,2,2,2-пентадейтериоэтокси)фенил]метил]фенил]-1-(гидроксиметил)-6,8-диоксабицикло[3.2.1]октан-2,3,4-триол
[(1S,2S,3S,4R,5S)-2,3,4-трибензилокси-5-[4-хлор-3-[[3-фтор-4-(1,1,2,2,2-пентадейтериоэтокси)фенил]метил]фенил]-6,8-диоксабицикло[3.2.1]октан-1-ил]метанол 10h (300 мг, 0,41 ммоль) растворяли в 30 мл смешанного раствора (THF и МеОН, v:v=1:1) с последующим добавлением по очереди 1,2-дихлорбензола (600 мг, 4,10 ммоль) и палладия на угле (30 мг, 10%). Смесь трижды продували H2 и перемешивали в течение 3 часов. После этого реакционную смесь фильтровали. Фильтрат концентрировали при пониженном давлении, и получающийся в результате остаток очищали при помощи обращенно-фазовой колоночной хроматографии с элюирующей системой F с получением указанного в заголовке соединения (1S,2S,3S,4R,5S)-5-[4-фтор-3-[[3-фтор-4-(1,1,2,2,2-пентадейтериоэтокси)фенил]метил]фенил]-1-(гидроксиметил)-6,8-диоксабицикло[3.2.1]октан-2,3,4-триола 10 (134 мг, белое твердое вещество), выход: 70,0%.
МС m/z(ESI): 477,1 [М+18]
1Н ЯМР (400 МГц, CD3OD): δ 7.49 (d, 1Н), 7.46-7.30 (m, 2Н), 7.05-6.81 (m, 3H), 4.17 (d, 1Н), 4.06 (s, 2Н), 3.86(d, 1Н), 3.79 (s, 1Н), 3.75-3.51 (m, 4Н).
Пример 11
(1S,2S,3S,4R,5S)-5-[4-хлор-3-[дидейтерио-(4-этокси-3-фтор-фенил)метил]фенил]-1-(гидроксиметил)-6,8-диоксабицикло[3.2.1]октан-2,3,4-триол
Стадия 1
(5-Бром-2-хлор-фенил)-дейтерио-(4-этокси-3-фтор-фенил)метанол
(5-Бром-2-хлор-фенил)-(4-этокси-3-фторфенил)метанон 4c (15,0 г, 41,96 ммоль) растворяли в 120 мл THF с последующим добавлением AlCl3 (12,3 г, 92 ммоль) и добавлением порции бордейтерида натрия (7,00 г, 167,2 ммоль) и 150 мл трифторуксусной кислоты в ледяной бане. Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 2 часов. После этого реакцию гасили 15 мл ацетона, и реакционную смесь концентрировали при пониженном давлении. Остаток растворяли в 150 мл этилацетата и промывали насыщенным раствором хлорида натрия (50 мл × 2). Органические экстракты объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (5-бром-2-хлор-фенил)-дейтерио-(4-этокси-3-фтор-фенил)метанола 11а (15,6 г, оранжевое жироподобное вещество), который использовали непосредственно на следующей стадии без очистки.
Стадия 2
4-[(5-бром-2-хлор-фенил)-дидейтерио-метил]-1-этокси-2-фтор-бензол
(5-бром-2-хлор-фенил)-дейтерио-(4-этокси-3-фтор-фенил)метанол 11а (6,4 г, 18,3 ммоль) растворяли в 40 мл трифторуксусной кислоты с последующим добавлением бордейтерида натрия (1,6 г, 38 ммоль). Реакционную смесь перемешивали в течение 3 часов. После этого смесь гасили 50 мл насыщенного раствора бикарбоната натрия, и она распределялась. Водную фазу экстрагировали этилацетатом (100 мл × 2), и органический экстракт объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении, и получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой В с получением указанного в заголовке соединения 4-[(5-бром-2-хлор-фенил)-дидейтерио-метил]-1-этокси-2-фтор-бензола 11b (4,3 г, бесцветное жироподобное вещество), выход: 67,8%.
1Н ЯМР (400 МГц, CDCl3): δ 7.43-7.23 (m, 3H), 7.03-6.85 (m, 3H), 4.21-4.08 (m, 2Н), 1.54-1.44 (m, 3H)
Стадия 3
(2S,3R,4S,5S,6R)-2-[4-хлор-3-[дидейтерио-(4-этокси-3-фтор-фенил)метил]фенил]-6-(гидроксиметил)-2-метокси-тетрагидропиран-3,4,5-триол
4-[(5-бром-2-хлор-фенил)-дидейтерио-метил]-1-этокси-2-фтор-бензол 11b (5,00 г, 16,4 ммоль) растворяли в смешанном растворе 50 мл THF и 75 мл н-гексана и охлаждали до -78°C, а затем по каплям добавляли nBuLi (9,00 мл, 21,6 ммоль). После перемешивания в течение 2 часов при -78°C добавляли раствор (30 мл) (3R,4S,5R,6R)-3,4,5-трис(триметилсилилокси)-6-(триметилсилилоксиметил)тетрагидропиран-2-она 2f (7,4 г, 15,8 ммоль) в н-гексане, и реакционную смесь перемешивали в течение 2 часов при -78°С. Добавляли 3,5 мл метансульфоновой кислоты и 40 мл метанола перед тем, как реакционную смесь нагревали и перемешивали в течение 16 часов при комнатной температуре. После этого смесь гасили 100 мл насыщенного раствора карбоната натрия и концентрировали при пониженном давлении. Остаток растворяли после добавления 50 мл насыщенного раствора хлорида натрия и экстрагировали этилацетатом (100 мл × 3). Органические экстракты объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении, и получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой A с получением указанного в заголовке соединения (2S,3R,4S,5S,6R)-2-[4-хлор-3-[дидейтерио-(4-этокси-3-фтор-фенил)метил]фенил]-6-(гидроксиметил)-2-метокси-тетрагидропиран-3,4,5-триола 11c (2,4 г, бесцветная жидкость), выход: 36,4%.
1Н ЯМР (400 МГц, CDCl3): δ 7.57-7.34 (m, 3H), 7.04-6.81 (m, 3H), 4.20-3.80 (m, 8Н), 3.52 (s, 3H), 2.54 (br. s., 4Н), 1.41-1.24 (m, 3H)
Стадия 4
(2S,3R,4S,5S,6R)-6-[(трет-бутил(диметил)силил)оксиметил]-2-[4-хлор-3-[дидейтерио-(4-этокси-3-фтор-фенил)метил]фенил]-2-метокси-тетрагидропиран-3,4,5-триол
(2S,3R,4S,5S,6R)-2-[4-хлор-3-[дидейтерио-(4-этокси-3-фтор-фенил)метил]фенил]-6-(гидроксиметил)-2-метокси-тетрагидропиран-3,4,5-триол 11c (2,4 г, 5,24 ммоль) растворяли в 40 мл дихлорметана с последующим добавлением по очереди DMAP (97 мг, 0,79 ммоль), имидазола (1,07 мг, 15,7 ммоль) и TBSCl (0,87 г, 5,76 ммоль). Реакционную смесь перемешивали в течение 16 часов. После этого реакционную смесь концентрировали при пониженном давлении. Реакционную смесь промывали насыщенным раствором сульфата меди (50 мл × 3) после добавления 200 мл этилацетата. Органические экстракты объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (2S,3R,4S,5S,6R)-6-[(трет-бутил(диметил)силил)оксиметил]-2-[4-хлор-3-[дидейтерио-(4-этокси-3-фтор-фенил)метил]фенил]-2-метокси-тетрагидропиран-3,4,5-триола 11d (2,87 г, бледно-желтое твердое вещество), который использовали непосредственно на следующей стадии без очистки.
Стадия 5
[[(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[дидейтерио-(4-этокси-3-фтор-фенил)метил]фенил]-6-метокси-тетрагидропиран-2-ил]метокси]трет-бутил-диметил-силан
(2S,3R,4S,5S,6R)-6-[(трет-бутил(диметил)силил)оксиметил]-2-[4-хлор-3-[дидейтерио-(4-этокси-3-фтор-фенил)метил]фенил]-2-метокси-тетрагидропиран-3,4,5-триол 11d (2,87 г, 5,02 ммоль) растворяли в 60 мл DMF с последующим добавлением 60% NaH (1,00 г, 25,1 ммоль) в ледяной бане. Затем реакционную смесь нагревали до комнатной температуры и перемешивали в течение 40 минут перед тем, как добавляли бензилбромид (3,00 мл, 25,1 ммоль), и перемешивали в течение 3 часов. Реакционную смесь гасили 20 мл метанола, и реакционную смесь концентрировали при пониженном давлении. Получающийся в результате остаток растворяли в 200 мл этилацетата и 50 мл воды, и он распределялся. Водную фазу экстрагировали 50 мл этилацетата. Органические экстракты по очереди промывали 50 мл воды и 50 мл насыщенного раствора хлорида натрия, и объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения [[(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[дидейтерио-(4-этокси-3-фтор-фенил)метил]фенил]-6-метокси-тетрагидропиран-2-ил]метокси]трет-бутил-диметил-силана 11е (3,00 г, желтое жироподобное вещество), выход: 99,8%.
Стадия 6
[(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[дидейтерио-(4-этокси-3-фтор-фенил)метил]фенил]-6-метокси-тетрагидропиран-2-ил]метанол
[[(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[дидейтерио-(4-этокси-3-фтор-фенил)метил]фенил]-6-метокси-тетрагидропиран-2-ил]метокси]трет-бутил-диметил-силан 11e (4,22 г, 5,02 ммоль) растворяли в 30 мл метанола и перемешивали в течение 1 часа после добавления ацетилхлорида (0,05 мл, 0,75 ммоль). После этого реакционную смесь концентрировали при пониженном давлении, и получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой В с получением указанного в заголовке соединения [(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[дидейтерио-(4-этокси-3-фтор-фенил)метил]фенил]-6-метокси-тетрагидропиран-2-ил]метанола 11f (1,45 г, желтое жироподобное вещество), выход: 55,0%.
Стадия 7
(2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[дидейтерио-(4-этокси-3-фтор-фенил)метил]фенил]-6-метокси-тетрагидропиран-2-карбальдегид
Оксалилхлорид (0,3 мл, 3,57 ммоль) растворяли в 20 мл дихлорметана и охлаждали до -78°C. Добавляли 10 мл раствора диметилсульфоксида (0,39 мл, 5,48 ммоль) в дихлорметане перед тем, как по очереди добавляли 15 мл раствора [(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[дидейтерио-(4-этокси-3-фтор-фенил)метил]фенил]-6-метокси-тетрагидропиран-2-ил]метанола 11f (2 г, 2,74 ммоль) в метиленхлориде и перемешивали в течение 30 минут при -78°C. После этого смесь нагревали до комнатной температуры и перемешивали в течение 2 часов после добавления триэтиламина (1,9 мл, 13,7 ммоль). Смесь гасили 5 мл 1 M соляной кислоты, и она распределялась. Водную фазу экстрагировали дихлорметаном (20 мл), и органический экстракт промывали насыщенным раствором хлорида натрия (20 мл × 2), объединяли, сушили над безводным сульфатом магния, и фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[дидейтерио-(4-этокси-3-фторфенил)метил]фенил]-6-метокси-тетрагидропиран-2-карбальдегида 11g (2,00 г, желтое жироподобное вещество), который использовали непосредственно на следующей стадии без очистки.
Стадия 8
(2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[дидейтерио-(4-этокси-3-фторфенил)метил]фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-карбальдегид
(2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[дидейтерио-(4-этокси-3-фторфенил)метил]фенил]-6-метокси-тетрагидропиран-2-карбальдегид 11g (2,00 г, 2,74 ммоль) растворяли в 30 мл 1,4-диоксана с последующим добавлением по очереди 4,1 мл 37% раствора формальдегида и раствора гидроксида натрия (330 мг, 2,74 ммоль). Реакционную смесь перемешивали в течение 6 часов при 70°C. После этого реакционную смесь экстрагировали этилацетатом (20 мл × 4) после добавления 20 мл насыщенного раствора хлорида натрия. Органический экстракт промывали насыщенным бикарбонатом натрия (20 мл) и насыщенным раствором хлорида натрия (20 мл) и объединяли, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[дидейтерио-(4-этокси-3-фтор-фенил)метил]фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-карбальдегида 11h (2,1 г, желтое жироподобное вещество), который использовали непосредственно на следующей стадии без очистки.
Стадия 9
[(3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[дидейтерио-(4-этокси-3-фтор-фенил)метил]фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-ил]метанол
(2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[дидейтерио-(4-этокси-3-фтор-фенил)метил]фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-карбальдегид 11h (2,07 г, 2,74 ммоль) растворяли в 30 мл смешанного раствора (THF и МеОН, v:v=2:3) с последующим добавлением боргидрида натрия (200 мг, 5,48 ммоль). Реакционную смесь перемешивали в течение 2 часов. После этого реакционную смесь гасили небольшим количеством ацетона, и реакционную смесь концентрировали при пониженном давлении. Получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой A с получением указанного в заголовке соединения [(3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[дидейтерио-(4-этокси-3-фтор-фенил)метил]фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-ил]метанола 11i (0,20 г, бесцветное жироподобное вещество), выход: 10%.
1Н ЯМР (400 МГц, CDCl3): δ 7.39-7.19 (m, 16Н), 7.04 (dd, 2Н), 6.89-6.74 (m, 3H), 5.03-4.86 (m, 3H), 4.72-4.59 (m, 2Н), 4.45-4.30 (m, 2Н), 4.05 (q, 2Н), 3.98 (dd, 2Н), 3.90-3.80 (m, 2Н), 3.75-3.62 (m, 1Н), 3.25 (d, 1Н), 3.06 (s, 3H), 1.42 (t, 3H)
Стадия 10
[(1S,2S,3S,4R,5S)-2,3,4-трибензилокси-5-[4-хлор-3-[дидейтерио-(4-этокси-3-фтор-фенил)метил]фенил]-6,8-диоксабицикло[3.2.1]октан-1-ил]метанол
[(3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[дидейтерио-(4-этокси-3-фтор-фенил)метил]фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-ил]метанол 11i (0,50 г, 6,60 ммоль) растворяли в 2 мл дихлорметана и охлаждали до -10°C перед тем, как добавляли 1 мл трифторуксусной кислоты. Реакционную смесь нагревали и перемешивали в течение 2 часов при комнатной температуре. После этого реакционную смесь гасили 5 мл насыщенного бикарбоната натрия, и она распределялась. Реакционную смесь концентрировали при пониженном давлении, и получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой B с получением указанного в заголовке соединения [(1S,2S,3S,4R,5S)-2,3,4-трибензилокси-5-[4-хлор-3-[дидейтерио-(4-этокси-3-фтор-фенил)метил]фенил]-6,8-диоксабицикло[3.2.1]октан-1-ил]метанола 11j (300 мг, белое твердое вещество), выход: 62,6%.
МС m/z(ESI): 744,0 [М+18]
Стадия 11
(1S,2S,3S,4R,5S)-5-[4-хлор-3-[дидейтерио-(4-этокси-3-фтор-фенил)метил]фенил]-1-(гидроксиметил)-6,8-диоксабицикло[3.2.1]октан-2,3,4-триол
[(1S,2S,3S,4R,5S)-2,3,4-трибензилокси-5-[4-хлор-3-[дидейтерио-(4-этокси-3-фтор-фенил)метил]фенил]-6,8-диоксабицикло[3.2.1]октан-1-ил]метанол 11j (300 мг, 0,41 ммоль) растворяли в 10 мл смешанного раствора (THF и МеОН, v:v=1:1) с последующим добавлением о-дихлорбензола (600 мг, 0,41 ммоль) и палладия на угле (30 мг, 10%). Смесь трижды продували H2 и перемешивали в течение 3 часов. После этого реакционную смесь фильтровали и элюировали небольшим количеством этилацетата. Фильтрат концентрировали при пониженном давлении. Получающийся в результате остаток очищали при помощи хроматографии на силикагеле с элюирующей системой A с получением указанного в заголовке соединения (1S,2S,3S,4R,5S)-5-[4-хлор-3-[дидейтерио-(4-этокси-3-фтор-фенил)метил]фенил]-1-(гидроксиметил)-6,8-диоксабицикло[3.2.1]октан-2,3,4-триола 11 (143 мг, белое твердое вещество), выход: 76,0%.
MC m/z (ESI): 474,1 [M+18]
1H ЯМР (400 МГц, CD3OD): δ 7.49 (d, 1Н), 7.45-7.36 (m, 2Н), 7.00-6.88 (m, 3H), 4.17 (d, 1Н), 4.08 (q, 2Н), 3.89-3.77 (m, 2Н), 3.73-3.54 (m, 4Н), 1.40 (t, 3H)
Пример 12
(1S,2S,3S,4R,5S)-5-[4-хлор-3-[[4-(2,2-дифторэтокси)фенил]метил]фенил]-1-(гидроксиметил)-6,8-диоксабицикло[3.2.1]октан-2,3,4-триол
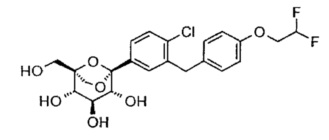

Стадия 1
2,2-Дифторэтоксибензол
В атмосфере N2 в реакционную колбу объемом 500 мл добавляли 60% NaH (10,2 г, 254,27 ммоль) и 40 мл DMF и охлаждали до 0°C. 2,2-Дифторэтанол 12а (23 г, 280,3 ммоль) растворяли в 40 мл DMF и затем по каплям добавляли в смесь в течение 4 часов при 0°C. Реакционную смесь затем нагревали до комнатной температуры. Через 30 минут по очереди добавляли раствор бромбензола (39,92 г, 254,25 ммоль) в DMF (40 мл) и CuBr (0,35 г, 2,43 ммоль) перед тем, как реакционную смесь нагревали и перемешивали в течение 16 часов при 160°C. После этого реакционную смесь охлаждали до комнатной температуры и фильтровали. Осадок на фильтре промывали н-гексаном. 5% соляную кислоту (160 мл) добавляли в фильтрат перед тем, как получающийся в результате остаток экстрагировали н-гексаном (160 мл × 3). Органический экстракт промывали насыщенным раствором хлорида натрия (50 мл) и объединяли, сушили над безводным сульфатом магния, и фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения 2,2-дифторэтоксибензола 12c (32,97 г, желтая жидкость), выход: 82,0%.
Стадия 2
2-Хлор-5-бром-бензоилхлорид
Под защитой Ar 5-бром-2-хлор-бензойную кислоту (35 г, 148,6 ммоль) растворяли в толуоле (230 мл) с последующим добавлением DMF (0,5 мл) при комнатной температуре, затем реакционную смесь охлаждали до 0°C. Реакционную смесь нагревали до 100°C после добавления по каплям тионилхлорида (44 г, 372 ммоль). Через 5 часов реакционную смесь концентрировали при пониженном давлении с получением указанного в заголовке соединения 5-бром-2-хлор-бензоилхлорида 12n (35,5 г, бледно-желтое жироподобное вещество), выход: 94,0%.
Стадия 3
(5-Бром-2-хлор-фенил)-[4-(2,2-дифторэтокси)фенил]метанон
Под защитой Ar 5-бром-2-хлор-бензоилхлорид 12n (37,7 г, 148,6 ммоль) растворяли в 350 мл дихлорметана, и добавляли 2,2-дифторэтоксибензол 12с (25 г, 158,6 ммоль) и перемешивали для растворения, и охлаждали до 0°C с последующим добавлением порциями AlCl3 (19,1 г,142,4 ммоль). Реакционную смесь перемешивали в течение 2 часов при 0°C, затем выливали в 300 мл ледяной воды и перемешивали в течение 30 минут, и она распределялась. Водный слой экстрагировали дихлорметаном (100 мл). Органический слой объединяли, и он распределялся после добавления метанола (50 мл), дихлорметана (100 мл) и воды (200 мл). Органический слой промывали насыщенным раствором NaCl (200 мл) и объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (5-бром-2-хлор-фенил)-[4-(2,2-дифторэтокси)фенил]метанона 12о (56 г, желтое жироподобное вещество), выход: 99,0%.
Стадия 4
4-Бром-1-хлор-2-[[4-(2,2-дифторэтокси)фенил]метил]бензол
Под защитой Ar (5-бром-2-хлор-фенил)-[4-(2,2-дифторэтокси)фенил]метанон 12o (55,7 г, 148,6 ммоль) растворяли в 400 мл ацетонитрила, добавляли триэтил силан (46,54 г, 401,22 ммоль) и охлаждали до 0°C, а затем медленно добавляли по каплям эфират трифторида бора (57 г, 401,22 ммоль), затем реакционную смесь нагревали до 50°C и перемешивали в течение 16 часов. После этого реакционную смесь охлаждали до комнатной температуры перед добавлением по каплям МТВЕ (200 мл) и 300 мл насыщенного раствора бикарбоната натрия, и она распределялась. Органические экстракты промывали 200 мл насыщенного раствора NaCl и объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении, и получающийся в результате остаток очищали путем колоночной хроматографии с получением указанного в заголовке соединения 4-бром-1-хлор-2-[[4-(2,2-дифторэтокси)фенил]метил]бензола 12p (20 г, бесцветное жироподобное вещество), выход: 20,0%.
МС m/z(ESI): 362,0 [М+1]
Стадия 5
(2S,3R,4S,5S,6R)-2-[4-хлор-3-[[4-(2,2-дифторэтокси)фенил]метил]фенил]-6-(гидроксиметил)-2-метокси-тетрагидропиран-3,4,5-триол
Под защитой Ar 4-бром-1-хлор-2-[[4-(2,2-дифторэтокси)фенил]метил]бензол 12p (26,5 г, 73,3 ммоль), МТВЕ (266 мл) и н-гексан (133 мл) добавляли в реакционную колбу объемом 1 л, перемешивали до однородности и охлаждали до -78°C, а затем по каплям добавляли 2,4 М nBuLi (52 мл, 124,6 ммоль) в течение 30 минут. После перемешивания в течение 50 минут при -78°C смешанный раствор (3R,4S,5R,6R)-3,4,5-трис(триметилсилилокси)-6-(триметилсилилоксиметил)тетрагидропиран-2-она 2f (55 г, 117,3 ммоль) в МТВЕ и н-гексане (60 мл:30 мл) добавляли в течение 20 минут при -78°C перед тем, как реакционную смесь перемешивали в течение 4 часов при -78°С. После этого добавляли 130 мл метанола перед тем, как перемешивали в течение 20 минут. Затем реакционную смесь нагревали до комнатной температуры и перемешивали в течение 16 часов после добавления метансульфоновой кислоты (25 г, 256,55 ммоль). 500 мл насыщенного бикарбоната натрия добавляли к реакционной смеси, перемешивали в течение 1 часа, и она распределялась. Водную фазу экстрагировали МТВЕ (100 мл × 2), и органический экстракт объединяли, концентрировали и очищали при помощи колоночной хроматографии (элюант:дихлорметан:метанол=100:1-10:1) с получением указанного в заголовке соединения (2S,3R,4S,5S,6R)-2-[4-хлор-3-[[4-(2,2-дифторэтокси)фенил]метил]фенил]-6-(гидроксиметил)-2-метокси-тетрагидропиран-3,4,5-триола 12е (5 г, бледно-желтое твердое вещество), выход: 10%.
МС m/z (ESI): 492,46 [М+18]
Стадия 6
(2S,3R,4S,5S,6R)-6-[(трет-бутил(диметил)силил)оксиметил]-2-[4-хлор-3-[[4-(2,2-дифторэтокси)фенил]метил]фенил]-2-метокси-тетрагидропиран-3,4,5-триол
(2S,3R,4S,5S,6R)-2-[4-хлор-3-[[4-(2,2-дифторэтокси)фенил]метил]фенил]-6-(гидроксиметил)-2-метокси-тетрагидропиран-3,4,5-триол 12e (4 г, 8,43 ммоль) растворяли в 40 мл дихлорметана с последующим добавлением по очереди DMAP (103 мг, 0,84 ммоль), TBSCl (1,4 г, 9,27 ммоль) и имидазола (1,72 г, 25,3 ммоль). Реакционную смесь перемешивали в течение 16 часов. Добавляли 40 мл насыщенного бикарбоната натрия перед тем, как реакционную смесь перемешивали, и она распределялась. После этого органический экстракт промывали 0,1 н. соляной кислотой (20 мл) и насыщенным раствором хлорида натрия (40 мл), сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (2S,3R,4S,5S,6R)-6-[(трет-бутил(диметил)силил)оксиметил]-2-[4-хлор-3-[[4-(2,2-дифторэтокси)фенил]метил]фенил]-2-метокси-тетрагидропиран-3,4,5-триола 12f (4,96 г, бледно-желтое твердое вещество), выход: 100%.
Стадия 7
[[2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[4-(2,2-дифторэтокси)фенил]метил]фенил]-6-метокси-тетрагидропиран-2-ил]метокси]трет-бутил-диметил-силан
60% NaH (1,9 г, 47,21 ммоль) и 15 мл THF добавляли в реакционную колбу объемом 100 мл и охлаждали до 0°C, а затем по каплям добавляли (2S,3R,4S,5S,6R)-6-[(трет-бутил(диметил)силил)оксиметил]-2-[4-хлор-3-[[4-(2,2-дифторэтокси)фенил]метил]фенил]-2-метокси-тетрагидропиран-3,4,5-триол 12f (4,96 г, 8,43 ммоль) в THF (18 мл) в течение 10 минут при 0°C. Реакционную смесь перемешивали в течение 30 минут. Затем по каплям добавляли раствор бензилбромида (7,21 г, 42,15 ммоль) в N,N-диметилформамиде (10 мл) перед тем, как реакционную смесь нагревали и перемешивали в течение 16 часов при комнатной температуре. После этого реакционная смесь распределялась после добавления 200 мл этилацетата, насыщенного бикарбоната натрия (70 мл) и воды (50 мл). Органический экстракт промывали 0,01 н. соляной кислотой (60 мл) и насыщенным раствором хлорида натрия, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения [[(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[4-(2,2-дифторэтокси)фенил]метил]фенил]-6-метокси-тетрагидропиран-2-ил]метокси]трет-бутил-диметил-силана 12g (7,25 г, бледно-желтая жидкость), который использовали непосредственно на следующей стадии без очистки.
Стадия 8
[(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[4-(2,2-дифторэтокси)фенил]метил]фенил]-6-метокси-тетрагидропиран-2-ил]метанол
[[2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[4-(2,2-дифторэтокси)фенил]метил]фенил]-6-метокси-тетрагидропиран-2-ил]метокси]трет-бутил-диметил-силан 12g (4,10 г, 4,78 ммоль) растворяли в 30 мл метанола с последующим добавлением ацетилхлорида (51 мкл, 0,72 ммоль). Реакционную смесь перемешивали в течение 1 часа. После этого реакционную смесь концентрировали при пониженном давлении, и получающийся в результате остаток очищали путем колоночной хроматографии с получением указанного в заголовке соединения [(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[4-(2,2-дифтор-этокси)фенил]метил]фенил]-6-метокси-тетрагидропиран-2-ил]метанола 12h (1,23 г, белое жироподобное вещество), выход: 34,6%.
Стадия 9
(2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[4-(2,2-дифторэтокси)фенил]метил]фенил]-6-метокси-тетрагидропиран-2-карбальдегид
Оксалилхлорид (0,18 мл, 2,16 ммоль) растворяли в 5 мл метиленхлорида и охлаждали до -78°C, а затем по каплям добавляли раствор (3 мл) диметилсульфоксида (0,23 мл, 3,31 ммоль) в метиленхлориде, реакционную смесь перемешивали в течение 15 минут. По каплям добавляли 10 мл раствора [(2R,3R,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[4-(2,2-дифторэтокси)фенил]метил]фенил]-6-метокси-тетрагидропиран-2-ил]метанола 12h (1,23 г, 1,66 ммоль) в метиленхлориде перед тем, как смесь перемешивали в течение 40 минут. Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 3 часов после добавления по каплям триэтиламина (1,2 мл, 8,31 ммоль). После этого реакционная смесь распределялась после добавления 5 мл 1 М соляной кислоты. Водную фазу экстрагировали этилацетатом (20 мл × 2), и органический экстракт объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[4-(2,2-дифтор-этокси)фенил]метил]фенил]-6-метокси-тетрагидропиран-2-карбальдегида 12i (1,10 г, желтое жироподобное вещество), который использовали непосредственно на следующей стадии без очистки.
Стадия 10
(2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[4-(2,2-дифторэтокси)фенил]метил]фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-карбальдегид
(2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[4-(2,2-дифторэтокси)фенил]метил]фенил]-6-метокси-тетрагидропиран-2-карбальдегид 12i (1,10 г, 1,48 ммоль) растворяли в 20 мл 1,4-диоксана с последующим добавлением 2,5 мл 37% раствора формальдегида и добавлением по каплям 4 мл 2,9 M раствора гидроксида натрия. Реакционную смесь перемешивали в течение 25 часов при 70°C. После этого реакционную смесь концентрировали при пониженном давлении перед тем, как добавляли 20 мл воды и 10 мл насыщенного раствора хлорида натрия. Водную фазу экстрагировали этилацетатом (30 мл × 3), и органический экстракт объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[4-(2,2-дифторэтокси)фенил]метил]фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-карбальдегида 12j (1,09 г, желтое жироподобное вещество), который использовали непосредственно на следующей стадии без очистки.
Стадия 11
[(3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[4-(2,2-дифторэтокси)фенил]метил]фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-ил]метанол
(2S,3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[4-(2,2-дифторэтокси)фенил]метил]фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-карбальдегид 12j (1,09 г, 1,42 ммоль) растворяли в 30 мл смешанного раствора (THF и MeOH, v:v=1:2) с последующим добавлением порции боргидрида натрия (108 мг, 2,83 ммоль). Реакционную смесь перемешивали в течение 30 минут перед тем, как добавляли 20 мл воды и 30 мл воды. Водную фазу экстрагировали этилацетатом (30 мл × 3), и органический экстракт промывали насыщенным раствором хлорида натрия (30 мл) и объединяли, сушили над безводным сульфатом магния, фильтровали. Фильтрат концентрировали при пониженном давлении, и получающийся в результате остаток очищали путем колоночной хроматографии с получением указанного в заголовке соединения [(3S,4S,5R,6S)-3,4,5-трибензил-окси-6-[4-хлор-3-[[4-(2,2-дифторэтокси)фенил]метил]фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-ил]метанола 12k (293 мг, желтое жироподобное вещество), выход: 27,0%.
Стадия 12
[(1S,2S,3S,4R,5S)-2,3,4-трибензилокси-5-[4-хлор-3-[[4-(2,2-дифторэтокси)фенил]метил]фенил]-6,8-диоксабицикло[3.2.1]октан-1-ил]метанол
[(3S,4S,5R,6S)-3,4,5-трибензилокси-6-[4-хлор-3-[[4-(2,2-дифторэтокси)фенил]метил]фенил]-2-(гидроксиметил)-6-метокси-тетрагидропиран-2-ил]метанол 12k (293 мг, 0,38 ммоль) растворяли в 10 мл дихлорметана, а затем по каплям добавляли трифторуксусную кислоту (0,1 мл). Реакционную смесь перемешивали в течение 1 часа. После этого реакционную смесь концентрировали при пониженном давлении, и получающийся в результате остаток очищали путем колоночной хроматографии с получением указанного в заголовке соединения [(1S,2S,3S,4R,5S)-2,3,4-трибензилокси-5-[4-хлор-3-[[4-(2,2-дифторэтокси)фенил]метил]фенил]-6,8-диоксабицикло[3.2.1]октан-1-ил]метанола 12m (76 мг, белое твердое вещество), выход: 27,6%.
Стадия 13
(1S,2S,3S,4R,5S)-5-[4-хлор-3-[[4-(2,2-дифторэтокси)фенил]метил]фенил]-1-(гидроксиметил)-6,8-диоксабицикло[3.2.1]октан-2,3,4-триол
[(1S,2S,3S,4R,5S)-2,3,4-трибензилокси-5-[4-хлор-3-[[4-(2,2-дифторэтокси)фенил]метил]фенил]-6,8-диоксабицикло[3.2.1]октан-1-ил]метанол 12m (76 мг, 0,10 ммоль) растворяли в 10 мл смешанного раствора (THF и MeOH, v:v=1:1) с последующим добавлением o-дихлорбензола (103 мкл, 0,9 ммоль) и палладия на угле (180 мг, 10%). Смесь трижды продували H2 и перемешивали в течение 2 часов, затем фильтровали. Фильтрат концентрировали при пониженном давлении, и получающийся в результате остаток очищали путем колоночной хроматографии с получением указанного в заголовке соединения (1S,2S,3S,4R,5S)-5-[4-хлор-3-[[4-(2,2-дифторэтокси)фенил]метил]фенил]-1-(гидроксиметил)-6,8-диоксабицикло[3.2.1]октан-2,3,4-триола 12 (17 мг, бледно-желтое твердое вещество), выход: 34,0%.
МС m/z (ESI): 490,24 [М+18]
1Н ЯМР (400 МГц, CD3OD): δ 7.48-7.49 (m, 1Н), 7.37-7.39 (m, 2Н), 7.15-7.17 (d, 2Н), 6.88-6.91 (d, 2Н), 6.02-6.29 (ddt, 1Н), 4.23-4.24 (d, 1Н), 4.20-4.21 (d, 1Н), 4.16-4.17 (d, 1Н), 4.08 (s, 2Н), 3.78-3.88 (m, 2Н), 3.67-3.72 (m, 2Н), 3.55-3.57 (m, 2Н)
ПРИМЕР ТЕСТИРОВАНИЯ
Измерение активности SGLT1 и SGLT2
Следующие способы могут быть использованы для определения ингибирующей активности соединений по настоящему изобретению в отношении SGLT1 и SGLT2. Далее кратко описаны экспериментальные способы:
Штамм с немедленным переносом SGLT1 или SGLT2 (плотность клеток: 1-1,5×104) вносили в каждую лунку 96-луночного планшета (подготовлен согласно существующей литературе "Diabetes, 57, 1723-1729, 2008", где кДНК SGLT1 и SGLT2 была приобретена в Origene) и инкубировали в увлажненной среде, содержащей 5% СO2 при 37°C в течение 48 часов. Затем каждую лунку 96-луночного планшета дважды промывали 200 мкл не содержащего натрий буфера, и добавляли 90 мкл содержащего натрий буферного раствора, содержащего тестируемые соединения в различных концентрациях, причем тестирование каждого из соединений в соответствующих концентрациях повторяли в трех лунках. Соединения инкубировали при 37°C в течение 15 минут, и затем каждую лунку 96-луночного планшета инкубировали с [14C]метил α-D-глюкопиранозидом (10 мкл, в общем 0,1 мКи) в течение еще 2 часов при 37°С. После этого супернатант удаляли; клеточный осадок дважды промывали предварительно охлажденным не содержащим натрий буфером и лизировали в 200 мМ NaOH (100 мкл). Добавляли 100 мкл сцинтилляционной жидкости и смешивали, и 14C количественно определяли с использованием жидкостной сцинтилляции.
Значения IC50 для соединений могут быть рассчитаны в соответствии со скоростью агрегации в различных концентрациях.
Заключение: Соединения по настоящему изобретению обладают высокой избирательностью и значимым ингибированием SGLT2.
Предварительная оценка гипогликемического действия
1. Задача
Обнаружение действия тестируемых соединений на уровень глюкозы в крови мышей с глюкозной нагрузкой. Определение и анализ содержания сахара в крови, отобранной из хвостовой вены мышей, в различные моменты времени в течение 2 часов после введения. Предварительная оценка гипогликемической активности in vivo.
2. Тестируемые соединения
Соединения Примера 1, Примера 2 и Примера 4
3. Экспериментальные животные
24 здоровых мыши ICR (массой 20-24 г), 12 самок, 12 самцов, приобретенных в Shanghai Super - B&K laboratory animal Corp.Ltd., номер лицензии для разведения животных: SCXK (Shanghai) 2008-0016.
4. Приготовление лекарства
Некоторое количество соединений взвешивали и растворяли в воде (чистая вода OWN), и готовили водный раствор 0,1 мг/мл (5% DMSO (диметилсульфоксид) для растворимости).
5. Способы тестирования
5.1 Установки дозы
Вводимая доза составляла 1 мг/кг, группы, которым вводили холостой образец и воду (содержащие 5% DMSO).
5.2 Способ введения
Ig, 20% раствор глюкозы (4 г/кг, 0,8 мл на каждую мышь) давали после 15 минут введения.
5.3 Определение уровня глюкозы в крови
Дозу вводили и измеряли уровень глюкозы в крови (- 15 минут). Давали 20% раствор глюкозы (4 г/кг, 0,8 мл на каждую мышь) после 15 минут введения, затем измеряли уровень глюкозы в крови для каждой мыши на 0, 15, 30, 45, 60, 120 минуте с использованием Roche ACCU-CHEK, и рассчитывали скорость уменьшения площади под кривой (AUC) под действием лекарства.
6. Результаты теста:
Заключение: при рассмотрении соединений по настоящему изобретению через 15 минут после введения уровень глюкозы в крови значительно уменьшался.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ МАННОЗЫ ДЛЯ ЛЕЧЕНИЯ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ | 2013 |
|
RU2667060C2 |
| КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ИНГИБИТОРЫ КОТРАНСПОРТЕРОВ НАТРИЯ-ГЛЮКОЗЫ 1 И 2, И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2012 |
|
RU2669921C2 |
| ПРОИЗВОДНЫЕ МАННОЗЫ ДЛЯ ЛЕЧЕНИЯ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ | 2014 |
|
RU2678327C2 |
| БЕТА-ЛАКТАМЫ, СПОСОБ ПОЛУЧЕНИЯ УКАЗАННЫХ СОЕДИНЕНИЙ И СЫВОРОТОЧНЫЕ ГИПОХОЛЕСТЕРИНЕМИЧЕСКИЕ СРЕДСТВА, СОДЕРЖАЩИЕ ТАКИЕ СОЕДИНЕНИЯ | 2002 |
|
RU2301799C2 |
| КОНЪЮГАТ ЛИГАНДА С ЦИТОТОКСИЧЕСКИМ ЛЕКАРСТВЕННЫМ СРЕДСТВОМ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2016 |
|
RU2708461C2 |
| ПРОИЗВОДНЫЕ БЕНЗИЛФЕНИЛЦИКЛОГЕКСАНА И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2008 |
|
RU2505521C2 |
| ПОЛИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2012 |
|
RU2621039C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО ДИФЕНИЛМЕТАНА | 2017 |
|
RU2797392C2 |
| С-ГЛИКОЗИДНЫЕ ПРОИЗВОДНЫЕ, СОДЕРЖАЩИЕ КОНДЕНСИРОВАННОЕ ФЕНИЛЬНОЕ КОЛЬЦО, ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБ ПОЛУЧЕНИЯ ТАКОВЫХ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ТАКОВЫЕ | 2017 |
|
RU2739024C2 |
| СПОСОБ ПОЛУЧЕНИЯ БЕНЗИЛБЕНЗОЛЬНЫХ ИНГИБИТОРОВ SGLT2 | 2013 |
|
RU2625795C2 |
Настоящее изобретение относится к соединениям формулы (I), их фармацевтически приемлемым солям или стереоизомерам, способу их получения и применению в медицине:
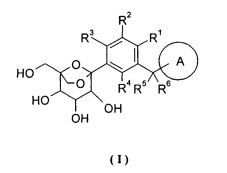
где А представляет собой фенил,  , или тиенил, где фенил замещен 1-5 атомами галогена и -OR7; R7 представляет собой C1-6 алкил, где C1-6 алкил возможно замещен одной или более группами, выбранными из дейтерия, галогена, C1-6 алкокси и C1-6 циклоалкокси; или указанный фенил замещен только -OR7, где R7 представляет собой C1-6 алкил, и указанный C1-6 алкил дополнительно замещен 1-3 группами, выбранными из галогена и С3-10 циклоалкокси; или, если А представляет собой
, или тиенил, где фенил замещен 1-5 атомами галогена и -OR7; R7 представляет собой C1-6 алкил, где C1-6 алкил возможно замещен одной или более группами, выбранными из дейтерия, галогена, C1-6 алкокси и C1-6 циклоалкокси; или указанный фенил замещен только -OR7, где R7 представляет собой C1-6 алкил, и указанный C1-6 алкил дополнительно замещен 1-3 группами, выбранными из галогена и С3-10 циклоалкокси; или, если А представляет собой  или тиенил, тиенил возможно замещен одной или более группами, выбранными из галогена, C1-6 алкила и фенила, необязательно замещенного дейтерием или галогеном; R1, R2, R3 или R4 выбраны из водорода, C1-6 алкила или галогена; R5 и R6 выбраны из водорода и дейтерия. Предложенный способ получения соединения формулы (I) включает превращение соединения формулы (IA) в соединение формулы (IB)
или тиенил, тиенил возможно замещен одной или более группами, выбранными из галогена, C1-6 алкила и фенила, необязательно замещенного дейтерием или галогеном; R1, R2, R3 или R4 выбраны из водорода, C1-6 алкила или галогена; R5 и R6 выбраны из водорода и дейтерия. Предложенный способ получения соединения формулы (I) включает превращение соединения формулы (IA) в соединение формулы (IB)
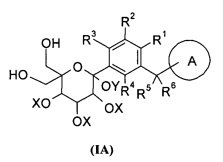
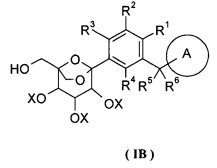
удаление защиты с соединения формулы (IB) с получением соединения формулы (I); где R1-R6 и А определены выше, X и Y представляют собой защитные группы гидроксила. Предложены новые ингибиторы натрий-зависимого переносчика глюкозы (SGLT)-1 и эффективный способ их получения. 7 н. и 9 з.п. ф-лы, 13 пр., 2 табл.
1. Соединение формулы (I), его фармацевтически приемлемые соли или стереоизомеры:
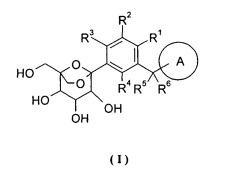
где
кольцо А представляет собой фенил,  , или тиенил, где указанный фенил дополнительно одновременно замещен 1-5 атомами галогена и -OR7; R7 представляет собой C1-6 алкил, где C1-6 алкил возможно дополнительно замещен одной или более группами, выбранными из группы, состоящей из дейтерия, галогена, C1-6 алкокси и C1-6 циклоалкокси; или в случае, если указанный фенил замещен только -OR7, где R7 представляет собой C1-6 алкил, указанный C1-6 алкил дополнительно замещен 1-3 группами, выбранными из группы, состоящей из галогена и С3-10 циклоалкокси;
, или тиенил, где указанный фенил дополнительно одновременно замещен 1-5 атомами галогена и -OR7; R7 представляет собой C1-6 алкил, где C1-6 алкил возможно дополнительно замещен одной или более группами, выбранными из группы, состоящей из дейтерия, галогена, C1-6 алкокси и C1-6 циклоалкокси; или в случае, если указанный фенил замещен только -OR7, где R7 представляет собой C1-6 алкил, указанный C1-6 алкил дополнительно замещен 1-3 группами, выбранными из группы, состоящей из галогена и С3-10 циклоалкокси;
если кольцо А представляет собой  или тиенил, указанный тиенил возможно дополнительно замещен одной или более группами, выбранными из группы, состоящей из галогена, C1-6 алкила и фенила, где указанный фенил возможно дополнительно замещен дейтерием или галогеном;
или тиенил, указанный тиенил возможно дополнительно замещен одной или более группами, выбранными из группы, состоящей из галогена, C1-6 алкила и фенила, где указанный фенил возможно дополнительно замещен дейтерием или галогеном;
каждый из R1, R2, R3 или R4 независимо выбран из водорода, C1-6 алкила или галогена;
каждый из R5 и R6 независимо выбран из группы, состоящей из водорода и дейтерия.
2. Соединение формулы (I), его фармацевтически приемлемые соли или стереоизомеры по п. 1, включающее соединение формулы (II), его фармацевтически приемлемые соли или стереоизомеры:
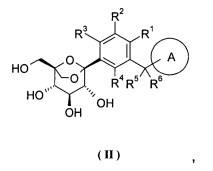
R1-R6 и кольцо А определены в п. 1.
3. Соединение формулы (I), его фармацевтически приемлемые соли или стереоизомеры по п. 1, где:
каждый из R2, R3 и R4 независимо представляет собой водород; и
R1 представляет собой галоген или C1-6 алкил.
4. Соединение формулы (I), его фармацевтически приемлемые соли или стереоизомеры по п. 1, где
кольцо А представляет собой фенил, где указанный фенил дополнительно одновременно замещен 1-5 галогенами и -OR7;
R7 представляет собой C1-6 алкил, где указанный C1-6 алкил возможно дополнительно замещен 1-3 группами, выбранными из группы, состоящей из дейтерия, галогена, C1-6 алкокси и С3-10 циклоалкокси.
5. Соединение формулы (I), его фармацевтически приемлемые соли или стереоизомеры по п. 1, где
кольцо А представляет собой  или тиенил, где указанный тиенил дополнительно возможно замещен одной или более чем одной группой, выбранной из группы, состоящей из галогена, C1-6 алкила и фенила, где указанный фенил возможно дополнительно замещен дейтерием или галогеном.
или тиенил, где указанный тиенил дополнительно возможно замещен одной или более чем одной группой, выбранной из группы, состоящей из галогена, C1-6 алкила и фенила, где указанный фенил возможно дополнительно замещен дейтерием или галогеном.
6. Соединение формулы (I), его фармацевтически приемлемые соли или стереоизомеры по п. 4, где R7 представляет собой С1-4алкил.
7. Соединение формулы (I), его фармацевтически приемлемые соли или стереоизомеры по п. 1, где R5 или R6 представляет собой атом дейтерия.
8. Соединение формулы (I), его фармацевтически приемлемые соли или стереоизомеры по п. 4, где R7 представляет собой C1-6 алкил, где указанный C1-6 алкил возможно дополнительно замещен 1-3 атомами дейтерия.
9. Соединение формулы (I), его фармацевтически приемлемые соли или стереоизомеры по любому из пп. 1-8, где соединение выбрано из группы, состоящей из:
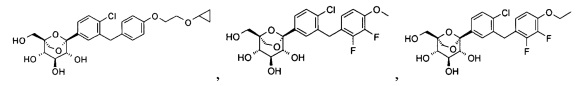
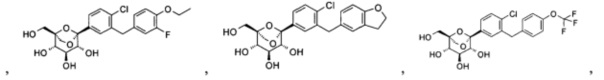
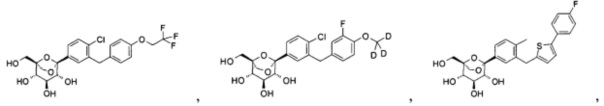
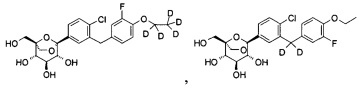 и
и 
10. Способ получения соединения формулы (I) по п. 1, где указанный способ включает превращение соединения формулы (IA)
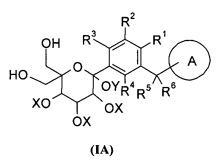
в соединение формулы (IB)
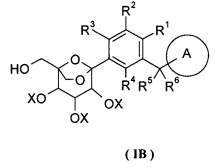
дополнительное удаление защиты с соединения формулы (IB) с превращением в соединение формулы (I);
где
R1-R6 и кольцо А определены в п. 1;
X и Y представляют собой защитные группы гидроксила.
11. Способ получения соединения формулы (I) по п. 10, где указанные X и Y представляют собой алкил и бензил.
12. Фармацевтическая композиция, обладающая активностью ингибитора натрий-зависимого переносчика глюкозы, содержащая терапевтически эффективное количество соединения формулы (I) или его фармацевтически приемлемых солей или стереоизомеров по любому из пп. 1-9 и фармацевтически приемлемые носители.
13. Применение соединения формулы (I) или его фармацевтически приемлемых солей или стереоизомеров по любому из пп. 1-9 или фармацевтической композиции по п. 12 в получении ингибитора натрий-зависимого переносчика глюкозы.
14. Применение соединения формулы (I) или его фармацевтически приемлемых солей или стереоизомеров по любому из пп. 1-9 или фармацевтической композиции по п. 12 при получении лекарственного средства для лечения или замедления развития или возникновения следующих заболеваний, где указанные заболевания выбраны из группы, состоящей из сахарного диабета, ретинопатии, нейропатии, нефропатии, инсулинорезистентности, гипергликемии, гиперинсулинемии, повышенных уровней жирных кислот или глицерина, гиперлипидемии, ожирения, гипертриглицеридемии, синдрома X, диабетических осложнений, атеросклероза и гипертензии.
15. Способ лечения заболевания, выбранного из группы, состоящей из сахарного диабета, ретинопатии, нейропатии, нефропатии, инсулинорезистентности, гипергликемии, гиперинсулинемии, повышенных уровней жирных кислот или глицерина, гиперлипидемии, ожирения, гипертриглицеридемии, синдрома X, диабетических осложнений, атеросклероза и гипертензии, при котором субъекту, нуждающемуся в таком лечении, вводят терапевтически эффективное количество соединения формулы (I) или его фармацевтических солей или стереоизомеров по любому из пп. 1-9 или фармацевтической композиции по п. 12.
16. Применение соединения формулы (I) или его фармацевтически приемлемых солей или стереоизомеров по любому из пп. 1-9 или фармацевтической композиции по п. 12 в качестве лекарственного средства для лечения или замедления развития или возникновения следующих заболеваний, где указанные заболевания выбраны из группы, состоящей из сахарного диабета, ретинопатии, нейропатии, нефропатии, инсулинорезистентности, гипергликемии, гиперинсулинемии, повышенных уровней жирных кислот или глицерина, гиперлипидемии, ожирения, гипертриглицеридемии, синдрома X, диабетических осложнений, атеросклероза и гипертензии.
| EA 201290267 A1, 28.12.2012 | |||
| WO 2010023594 A1, 04.03.2010 | |||
| Vincent Mascitti, Organic letters, 2010, 12, 13, 2940-2943. |

