ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к новым полициклическим производным, способам их получения, содержащим их фармацевтическим композициям, и их применению в качестве терапевтического средства, в частности, в качестве агониста GPR40 (G-protein receptor) и к получению лекарственного средства для лечения диабета, метаболического синдрома и т.д.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Диабет II типа, известный также как инсулиннезависимый сахарный диабет или диабет взрослых, в основном показывает, что секреция инсулина у пациентов слишком мала или организм не может эффективно использовать инсулин (а именно резистентность к инсулину). В настоящее время во всем мире существует около 185 миллионов диабетиков, причем, доля больных диабетом II типа составляет примерно от 90 до 95% всех больных сахарным диабетом, и она растет на 6% в год. В 2010 году заболеваемость диабетом II типа среди взрослых китайцев старше 20 лет достигла порядка 9,7%.
В настоящее время методы лечения диабета II типа включают в себя: использование стимуляторов секреции инсулина, таких как, сульфонилмочевина, стимулирующая β клетки поджелудочной железы для выработки большего количества инсулина; противодиабетические средства, такие как метформин, снижающий образование глюкозы в печени; активаторы рецептора PPAR-γ (peroxisome proliferator-activated receptors - рецепторы, активируемые пролифераторами пероксисом), такие как тиазолидиндионы, повышающие чувствительность к инсулину и его биодоступность; и ингибиторы α-глюкозидазы, которые препятствуют получению глюкозы. Однако современное лечение существующими методами имеет определенные недостатки. Например, использование сульфонилмочевины и инъекции инсулина могут быть связаны с гипогликемическими эпизодами и увеличением веса. Более того, часто пациенты со временем перестают отвечать на сульфонилмочевину и возникает привыкание. Применение метформина и ингибиторов α-глюкозидазы часто приводит к желудочно-кишечным проблемам, а применение агонистов PPAR-γ, как правило, приводит к увеличению веса и отекам.
Чтобы обеспечить появление на лекарственном рынке новых, более эффективных противодиабетических препаратов, исследования ведут в нескольких областях. Например, авторы настоящего изобретения изучают способы снижения чрезмерной продукции глюкозы в печени, способы улучшения внутриклеточных путей передачи сигнала инсулин-индуцированного усвоения глюкозы, пути улучшения глюкозостимулированной секреции инсулина (GSIS - glucose-stimulated insulin secretion) в β клетках поджелудочной железы, а также изучают ожирение и жировой обмен, накопление аномалий, и тому подобное.
Свободные жирные кислоты (FFA - free fatty acid) играют ключевую роль в нескольких аспектах обмена веществ, например, они являются “стимулом”, который повышает количество инсулина, вырабатываемого β-клетками поджелудочной железы в ответ на глюкозу в голодном состоянии, и они являются отправными точками для липогенеза. Первоначально GPR40 был найден в виде рецепторов в геноме человека. GPR40 активно экспрессируется в β-клетках поджелудочной железы и клеточных линиях, секретирующих инсулин. GPR40, также известный как рецептор свободных жирных кислот 1 (FFAR1 - free fatty acid receptor 1), является членом суперсемейства рецепторов, сопряженных с G-белками (GPCR, G-protein coupled receptors). GPCRs представляют собой мембранные белки, имеющие семь трансмембранных доменов, способных реагировать с различными молекулами, тем самым активируя внутриклеточные пути передачи сигнала, и имеют решающее значение для выполнения разнообразных физиологических функций.
Активация GPR40 связана с регулированием Gq семейства внутриклеточных сигнальных белков и сопровождается индуцированием увеличения уровня ионов кальция. GPR40 был первым рецептором жирных кислот, который был идентифицирован на поверхности клетки, способным связывать наиболее распространенные жирные кислоты в плазме крови, такие как пальмитат, олеат, стеарат, линолеат и линоленат и т.д. GPR40 может считаться рецептором, «распознавания питательных веществ», играющим несколько тканеспецифичных ролей, что может влиять на общую утилизацию глюкозы и/или жировой обмен. Например, длинноцепочечные FFAs усиливают GSIS в β-клетках поджелудочной железы посредством активации GPR40.
Регуляторы GPR40 выполняют роль инкретина для продвижения GSIS, кроме того, их можно комбинировать с различными противодиабетическими препаратами. Исходя из вышеизложенного, агонисты GPR40 могут быть использованы для лечения диабета и связанных с ним состояний, в частности, диабета II типа, ожирения, нарушения толерантности к глюкозе, инсулинорезистентности, метаболического синдрома X, гиперлипидемии, гиперхолестеринемии, атеросклероза, нейродегенеративных заболеваний (например болезни Альцгеймера), и других состояний, таких как инсульт. Рассматривая GPR40 как потенциальную терапевтическую мишень, поиск соединения для обнаружения и модификации GPR40 имеет очень важную исследовательскую ценность и перспективы применения.
К настоящему времени целая серия агонистов GPR40 был описан в ряде патентных заявок, таких как WO 2005087710, WO 2005051890, WO 2004106276 и т.д.
Однако, хотя ряд агонистов GPR40 для лечения заболеваний, таких как диабет и метаболический синдром X и т.д. в настоящее время уже описаны, сохраняется потребность в разработке новых более эффективных соединений. Настоящее изобретение предлагает соединения Формулы (I) и демонстрирует, что соединения такой структуры обладают большей эффективностью.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение направлено на получение соединения формулы (I) и/или его таутомера, мезомера, рацемата, энантиомера, диастереомера и их смеси, и его фармацевтически приемлемой соли, а также их метаболитов, метаболических предшественников или пролекарств.
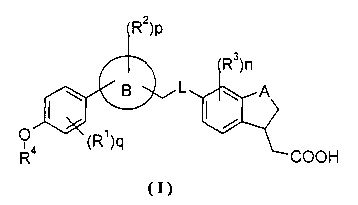
где
А выбран из группы, состоящей из -О-, -СН2- и -СН2СН2-;
L выбран из группы, состоящей из -О- и -NH-;
кольцо В выбрано из группы, состоящей из арила и гетероарила;
R1, R2 и R3, каждый независимо, выбран из группы, состоящей из галогена, гидроксила, циано, нитро, алкила, алкокси, циклоалкила, гетероциклила, арила, гетероарила, -C(O)OR5, -OC(O)R5, -C(O)R5, -NHC(O)R5, -NR6R7 и -S(O)mR5, где алкил, алкокси, циклоалкил, гетероциклил, арил или гетероарил, каждый, возможно, замещен одной или несколькими группами, выбранными из группы, состоящей из галогена, гидроксила, циано, нитро, алкил, алкокси, циклоалкила, гетероциклила, арила, гетероарила, -C(O)OR5, -OC(O)R5, -C(O)R5, -NHC(O)R5, -NR6R7 и -S(O)mR5;
R4 выбран из группы, состоящей из циклоалкила, гетероциклила, арила и гетероарила, где циклоалкил, гетероциклил, арил или гетероарил, каждый, возможно, замещен одной или несколькими группами, выбранными из группы, состоящей из галогена, гидроксила, циано, нитро, алкила, алкокси, циклоалкила, гетероциклила, арила, гетероарила, -C(O)OR5, -OC(O)R5, -C(O)R5, -NHC(O)R5, -NR6R7 and -S(O)mR5;
R5 выбирают из группы, состоящей из водорода, алкила, циклоалкила, гетероциклила, арила и гетероарила, где алкил, циклоалкил, гетероциклил, арил или гетероарил, каждый, возможно, замещен одной или несколькими группами, выбранными из группы, состоящей из алкила, галогена, гидроксила, циано, алкокси, циклоалкила, гетероциклила, арила, гетероарила, карбоксила и алкоксикарбонила;
R6 и R7 каждый, независимо, выбраны из группы, состоящей из водорода, алкила, циклоалкила, гетероциклила, арила и гетероарила, где алкил, циклоалкил, гетероциклил, арил или гетероарил, каждый, возможно, замещен одной или несколькими группами, выбранными из группы, состоящей из алкила, галогена, гидроксила, алкокси, циклоалкила, гетероциклила, арила, гетероарила, карбоксила и алкоксикарбонила;
m представляет собой 0,1 или 2;
n представляет собой 0, 1, 2 или 3;
p представляет собой 0, 1, 2, 3 или 4; и
q представляет собой 0, 1, 2, 3 или 4;
при условии, что: когда p и n представляют собой 0, q представляет собой 2, R1 представляет собой метил, А представляет собой -О-, и R4 представляет собой гетероциклил, гетероциклил не содержит атома S.
В одном варианте осуществления изобретения, соединение формулы (I) или его таутомер, мезомер, рацемат, энантиомер, диастереомер и их смеси, и его фармацевтически приемлемая соль, где кольцо B представляет собой арил, предпочтительно, фенил.
В другом варианте осуществления настоящего изобретения соединение формулы (I) или его таутомер, мезомер, рацемат, энантиомер, диастереомер, и их смеси, и его фармацевтически приемлемую соль, выбиранное из соединения формулы (II) или его таутомера, рацемата, энантиомера, диастереомера, их смеси, и его фармацевтически приемлемой соли:

где A, L, R1-R4, n, p и q являются такими, как определено в формуле (I).
В другом варианте осуществления изобретения соединение формулы (I) или формулы (II) или его таутомер, мезомер, рацемат, энантиомер, диастереомер и их смесь, и его фармацевтически приемлемая соль, где L представляет собой -O-.
В другом варианте осуществления изобретения, соединения формулы (I) или формулы (II) или его таутомер, рацемат, мезомер, энантиомер, диастереомер и их смесь, и его фармацевтически приемлемая соль, где R1 выбран из группы, состоящей из алкила, галогена и гидроксиалкила.
В другом варианте осуществления изобретения, соединение формулы (I) или формулы (II) или его таутомер, мезомер, рацемат, энантиомер, диастереомер и их смеси, и его фармацевтически приемлемая соль, где R2 выбран из группы, состоящей из водорода, алкила и галогена.
В другом варианте осуществления изобретения соединение формулы (I) или формулы (II) или его таутомер, мезомер, рацемат, энантиомер, диастереомер, и их смесь, и его фармацевтически приемлемая соль, где R3
представляет собой водород.
В другом варианте осуществления изобретения, соединение формулы (I) или формулы (II) или его таутомер, мезомер, рацемат, энантиомер, диастереомер и их смесь, и его фармацевтически приемлемая соль, где R4 выбран из группы, состоящей из циклоалкила, гетероциклила и гетероарила, где циклоалкил, гетероциклил или гетероарил каждый, возможно, замещен одной или несколькими группами, выбранными из группы, состоящей из галогена, гидроксила, циано, нитро, алкила, алкокси, циклоалкила, гетероциклила, арила, гетероарила, -C(O)OR5, -OC(O)R5, -C(O)R5, -NHC(O)R5, -NR6R7 и -S(O)mR5; и R5, R6, R7 и m являются такими, как определено в формуле (I).
В другом варианте осуществления изобретения, соединение формулы (I) или формулы (II) или его таутомер, рацемат, мезомер, энантиомер, диастереомер и их смесь, и его фармацевтически приемлемая соль, где R4 выбран из группы, состоящей из следующих циклоалкила, гетероциклила и гетероарила:
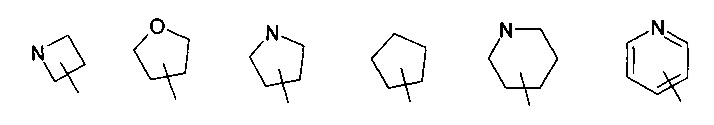
где циклоалкил, гетероциклил или гетероарил, каждый, возможно, замещен одной или несколькими группами, выбранными из группы, состоящей из галогена, гидроксила, алкила, алкокси, -C(O)OR5, -OC(O)R5, -C(O)R5, -NHC(O)R5, -NR6R7 и -S(O)mR5; и R5, R6, R7 и m являются такими, как определено в формуле (I).
Более того, в другом варианте осуществления настоящего изобретения соединение формулы (II) или его таутомер, мезомер, рацемат, энантиомер, диастереомер и их смесь, и его фармацевтически приемлемая соль, выбраны из соединения формулы (III) или его таутомера, мезомера, рацемата, энантиомера, диастереомера и их смеси, и его фармацевтически приемлемой соли:

где кольцо E выбрано из группы, состоящей из циклоалкила, гетероциклила и гетероарила;
R1 выбрано из группы, состоящей из галогена, гидроксила, циано, нитро, алкила, алкокси, циклоалкила, гетероциклила, арила, гетероарила, -C(O)OR5, -OC(O)R5, -C(O)R5, -NHC(O)R5, -NR6R7 and -S(O)mR5, где алкил, циклоалкил, алкокси, гетероциклил, арил или гетероарил, каждый, возможно замещен одной или несколькими группами, выбранными из группы, состоящей из галогена, гидроксила, циано, нитро, алкила, алкокси, циклоалкила, гетероциклила, арила, гетероарила, -C(O)OR5, -OC(O)R5, -C(O)R5, -NHC(O)R5, -NR6R7 и -S(O)mR5;
R5 выбран из группы, состоящей из водорода, алкила, циклоалкила, гетероциклила, арила и гетероарила, где алкил, циклоалкил, гетероциклил, арил или гетероарил, каждый, возможно замещены одной или несколькими группами, выбранными из группы, состоящей из алкила, галогена, гидроксила, циано, алкокси, циклоалкила, гетероциклила, арила, гетероарила, карбоксила и алкоксикарбонила;
R6 и R7 каждый независимо выбран из группы, состоящей из водорода, алкила, циклоалкила, гетероциклила, арила и гетероарила, где алкил, циклоалкил, гетероциклил, арил или гетероарил, каждый, возможно замещены одной или несколькими группами, выбранными из группы, состоящей из алкила, галогена, гидроксила, алкокси, циклоалкила, гетероциклила, арила, гетероарила, карбоксила и алкоксикарбонила;
R8 выбран из группы, состоящей из галогена, гидроксила, циано, нитро, алкила, алкокси, циклоалкила, гетероциклила, арила, гетероарила, -C(O)OR5, -OC(O)R5, -C(O)R5, -NHC(O)R5, -NR6R7 и -S(O)mR5;
m представляет собой 0, 1 или 2;
q представляет собой 0, 1, 2, 3 или 4; и
s представляет собой 0, 1, 2 или 3;
при условии, что: когда q представляет собой 2, R1 представляет собой метил, E представляет собой гетероциклил, гетероциклил не содержит атома S.
В другом варианте осуществления изобретения, соединение формулы (III) или его таутомер, рацемат, мезомер, энантиомер, диастереомер и их смесь, и его фармацевтически приемлемая соль, где кольцо E выбрано из группы, состоящей из следующих циклоалкила, гетероциклила и гетероарила:

где циклоалкил, гетероциклил или гетероарил, каждый, возможно замещены одной или несколькими группами, выбранными из группы, включающей галоген, гидроксил, алкил, алкокси, -C(O)OR5, -OC(O)R5, -C(O)R5, -NHC(O)R5, -NR6R7 и -S(O)mR5; и R5, R6, R7 и m являются такими, как определено в формуле (III).
Кроме того, в другом варианте осуществления настоящего изобретения соединение формулы (III), или его таутомер, мезомер, рацемат, энантиомер, диастереомер и их смесь, и их фармацевтически приемлемая соль, выбраны из соединения формулы (IV) или его таутомера, мезомера, рацемата, энантиомера, диастереомера, и их смеси, и его фармацевтически приемлемой соли:
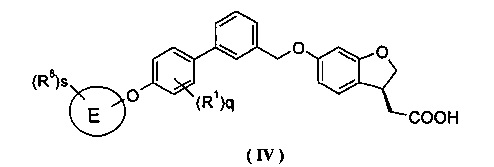
где E, R1, R8, s и q являются такими, как определено в формуле (III).
Соединение формулы (I) по настоящему изобретению, предпочтительно включает, но не ограничено ими:



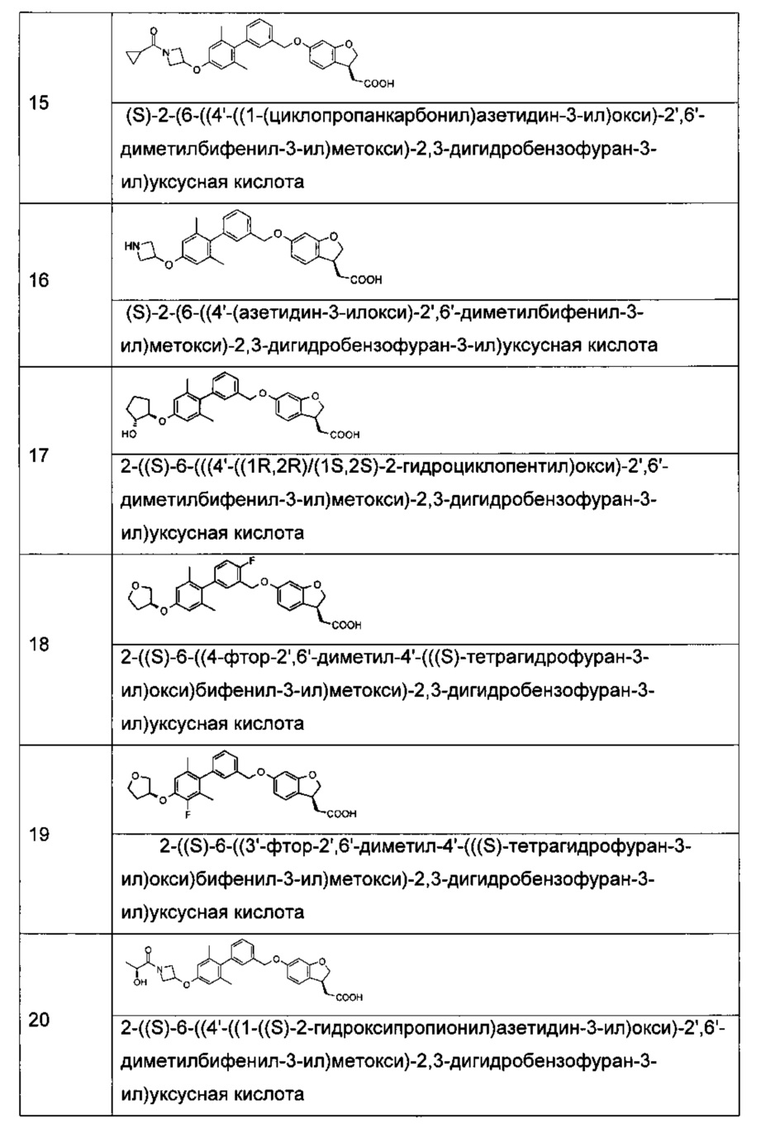



или его таутомер, мезомер, рацемат, энантиомер, диастереоизомер, и их смесь, и его фармацевтически приемлемая соль.
Настоящее изобретение относится к соединению формулы (IA) в качестве промежуточных продуктов синтеза соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереоизомера, и их смеси, и его фармацевтически приемлемой соли:
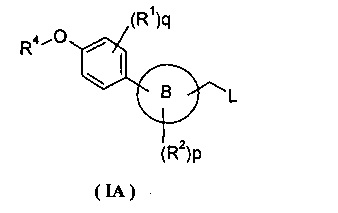
где кольцо B, L, R1, R2, R4, p и q являются такими, как определено в формуле (I).
Настоящее изобретение относится к способу получения соединения формулы (I) или его таутомера, рацемата, мезомера, энантиомера, диастереомера и их смеси, и его фармацевтически приемлемой соли, включающему стадии:

конденсации соединения формулы (IA) с гидроксил-замещенным соединением с бензольным кольцом в растворителе; дополнительно, возможно, проведение гидролиза сложного эфира в щелочных условиях с получением соединения формулы (I); щелочные условия обеспечиваются гидроксидом щелочного металла, предпочтительно, гидроксидом натрия или гидроксидом калия;
где кольцо B, A, L, R1-R4, n, p и q являются такими, как определено в формуле (I).
Настоящее изобретение относится к соединению формулы (IB):

где кольцо B, A, L, R1-R3, n, p и q являются такими, как определено в формуле (I).
Настоящее изобретение относится к способу получения соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера и их смеси, и его фармацевтически приемлемой соли, включающему стадии:

взаимодействия соединения формулы (IB) и R4-замещенного алкил сульфоната в растворителе, дополнительно, возможно, проведение гидролиза сложного эфира в щелочных условиях с получением соединения формулы (I); щелочные условия обеспечиваются гидроксидом щелочного металла, предпочтительно гидроксидом натрия или гидроксидом калия;
где кольцо B, A, L, R1-R4, n, p и q являются такими, как определено в формуле (I), и R9 представляет собой алкил, предпочтительно, метил.
Кроме того, в другом аспекте, настоящее изобретение относится к фармацевтической композиции, содержащей терапевтически эффективное количество соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера и их смеси, и его фармацевтически приемлемой соли или его пролекарству и фармацевтически приемлемому носителю или наполнителю.
В еще одном аспекте, настоящее изобретение также относится к применению соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереоизомера и их смеси, и его фармацевтически приемлемой соли или фармацевтической композиции, содержащей то же самое, для получения лекарственного средства, служащего в качестве регулятора рецептора GPR40.
В еще одном аспекте настоящее изобретение относится к соединению формулы (I) или его таутомеру, мезомеру, рацемату, энантиомеру, диастереомеру и их смеси, и его фармацевтически приемлемой соли, или фармацевтической композиции, содержащей то же самое, для применения в качестве регулятора рецептора GPR40.
фармацевтической композиции, содержащей то же самое, для применения в качестве регулятора рецептора GPR40.
В еще одном аспекте, настоящее изобретение относится к способу регулирования рецептора GPR40, включающему этап введения пациенту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомер, и их смеси, и его фармацевтически приемлемой соли, или фармацевтической композиции, содержащей то же самое.
Настоящее изобретение относится к применению соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера и их смеси, и его фармацевтически приемлемой соли или фармацевтической композиции, содержащей то же самое, при получении лекарственного средства, являющегося агонистом GPR40.
В еще одном аспекте настоящее изобретение относится к соединению формулы (I) или его таутомеру, мезомеру, рацемату, энантиомеру, диастереомеру и их смеси, и его фармацевтически приемлемой соли, или фармацевтической композиции, содержащей то же самое, для использования в качестве агониста GPR40.
В еще одном аспекте настоящее изобретение относится к способу возбуждения рецептора GPR40, включающему этап введения пациенту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы (I) или его таутомера, рацемата, мезомера, энантиомера, диастереомера, и их смеси, и его фармацевтически приемлемой соли, или фармацевтической композиции, содержащей то же самое.
Настоящее изобретение также относится к применению соединения формулы (I) или его таутомера, рацемата, мезомера, энантиомера, диастереомера и их смеси, и его фармацевтически приемлемой соли, или фармацевтической композиции, содержащей то же самое, в получении лекарственного средства для лечения диабета и метаболического синдрома, предпочтительно, диабет является диабетом II типа.
В еще одном аспекте настоящее изобретение относится к способу профилактики и лечения диабета и метаболического синдрома, включающему стадию введения пациенту, нуждающемуся в этом, терапевтически фармацевтически приемлемой соли, или фармацевтической композиции, содержащей то же самое, предпочтительно, диабет является диабетом II типа.
В еще одном аспекте настоящее изобретение относится к применению соединения формулы (I) или его таутомера, рацемата, мезомера, энантиомера, диастереомера и их смеси, и его фармацевтически приемлемой соли, или фармацевтической композиции, содержащей то же самое, в качестве лекарственного средства для профилактики и лечения диабета и метаболического синдрома, предпочтительно, диабет является диабетом II типа.
В еще одном аспекте настоящее изобретение относится к способу регулирования инсулина, включающему этап введения пациенту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы (I) или его таутомера, рацемата, мезомера, энантиомера, диастереомера, и их смеси, и его фармацевтически приемлемой соли, содержащей то же самое.
В еще одном аспекте настоящее изобретение относится к использованию соединения формулы (I) или его таутомера, рацемата, мезомера, энантиомера, диастереоизомера и их смеси, и его фармацевтически приемлемой соли, или фармацевтической композиции, содержащей то же самое, в качестве лекарственного средства для регулирования уровня инсулина. Настоящее изобретение относится к применению соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера и их смеси, и его фармацевтически приемлемой соли или фармацевтической композиции, содержащей то же самое, при приготовлении лекарственного средства для регулирования инсулина.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Если не указано иное, следующие определения, используемые в описании и формуле изобретения, имеют значения, приведенные ниже.
“Алкил” означает насыщенную алифатическую углеводородную группу, включающую группы С1-С20 с неразветвленной и разветвленной цепями. Предпочтительно алкильная группа представляет собой алкил, имеющий от 1 до 10 атомов углерода, более предпочтительно, имеющий от 1 до 6 атомов углерода. Типичные примеры включают, но не ограничиваются ими, метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, втор-бутил, н-пентил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, н-гексил, 1-этил-2-метилпропил, 1,1,2-триметилпропил, 1,1-диметилбутил, 1,2-диметилбутил, 2,2-диметилбутил, 1,3-диметилбутил, 2-этилбутил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 2,3-диметилбутил, n-гептил, 2-метилгексил, 3-метилгексил, 4-метилгексил, 5-метилгексил, 2,3-диметилпентил, 2,4-диметилпентил, 2,2-диметилпентил, 3,3-диметилпентил, 2-этилпентил, 3-этилпентил, n-октил, 2,3-диметилгексил, 2,4-диметилгексил, 2,5-диметилгексил, 2,2-диметилгексил, 3,3-диметилгексил, 4,4-диметилгексил, 2-этилгексил, 3-этилгексил, 4-этилгексил, 2-метил-2-этилпентил, 2-метил-3-этилпентил, н-нонил, 2-метил-2-этилгексил, 2-метил-3-этилгексил, 2,2-диэтилпентил, н-децил, 3,3-диэтилгексил, 2,2-диэтилгексил и изомеры их разветвленных цепей. Более предпочтительно, алкильная группа представляет собой низший алкил, имеющий от 1 до 6 атомов углерода. Типичные примеры включают, но не ограничиваются ими, метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, втор-бутил, н-пентил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, н-гексил, 1-этил-2-метил пропил, 1,1,2-триметилпропил, 1,1-диметилбутил, 1,2-диметилбутил, 2,2-диметилбутил, 1,3-диметилбутил, 2-этилбутил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 2,3-диметилбутил и т.п. Алкильная группа может быть замещенной или незамещенной. В случае замещения, замещающая группа(ы) может быть замещена в любом доступном месте соединения, предпочтительно, замещающая группа(ы) представляет собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилсульфо, алкиламино, галогена, тиола, гидроксила, нитро, циано, циклоалкила, гетероциклоалкила, арила, гетероарила, циклоалкокси, гетероциклического алкокси, циклоалкилтио, гетероциклического алкилтио, карбонила, -C(O)OR5, -OC(O)R5, -C(O)R5, -NHC(O)R5, -NR6R7 and -S(O)mR5.
“Циклоалкил” означает насыщенную или частично ненасыщенную моноциклическую или полициклическую углеводородную группу, содержащую от 3 до 20 атомов углерода, предпочтительно от 3 до 12 атомов углерода, более предпочтительно от 3 до 10 атомов углерода, и наиболее предпочтительно от 3 до 6 атомов углерода. Типичные примеры моноциклического циклоалкила включают, но не ограничиваются ими, циклопропил, циклобутил, циклопентил, циклопентенил, циклогексил, циклогексенил, циклогексадиенил, циклогептил, циклооктил, циклогептатриенил и т.д. Полициклический циклоалкил включает циклоалкил, имеющий спиро-кольцо, конденсированное кольцо и мостиковое кольцо.
“Спиро-циклоалкил” означает 5-20-членную полициклическую углеводородную группу с кольцами, соединенными через один общий атом углерода (называемый спиро-атом), в которой одно или более колец может содержать одну или несколько двойных связей, но ни одно из колец не имеет полностью сопряженной пиэлектронной системы. Предпочтительно, спиро-циклоалкил является 6-14-членным, более предпочтительно 7-10-членным. По количеству общих спиро-атомов, спиро-циклоалкилы делятся на моно-спиро-циклоалкилы, ди-спиро-циклоалкилы или поли-спиро-циклоалкилы, предпочтительно, являются моно-спиро-циклоалкилами или ди-спиро-циклоалкилами, более предпочтительно 4-членным/4-членным, 4-членным/5-членным, 4-членным/6-членным, 5-членным/5-членным или 5-членным/6-членным моно-спиро-циклоалкилом. Типичные примеры спиро-циклоалкила включают, но не ограничиваются ими, следующие группы:
 .
.
“Конденсированный циклоалкил” обозначает 5-20-членную полициклическую углеводородную группу, где каждое кольцо (цикл) в системе имеет общую пару атомов углерода с другим кольцом (циклом), где одно или более колец может содержать одну или несколько двойных связей, но ни одно из колец не имеет полностью сопряженной пи-электронной системы. Предпочтительно конденсированная циклоалкильная группа является 6-14-членной, более предпочтительно 7-10-членной. В зависимости от числа колец, конденсированные циклоалкилы делятся на бициклические, трициклические, тетрациклические или полициклические конденсированные циклоалкилы, предпочтительно, конденсированный циклоалкил является бициклическим или три циклически м конденсированным циклоалкилом, более предпочтительно, 5-членным/5-членным или 5-членным/6-членным бициклическим конденсированным циклоалкилом Типичные примеры конденсированных циклоалкилов включают, но не ограничиваются ими, следующие группы:
 .
.
“Мостиковый циклоалкил” обозначает 5-20 членную пол и циклическую углеводородную группу, в которой каждые два кольца в системе имеют два общих несвязанных атома углерода. Указанные кольца могут иметь одну или несколько двойных связей, но не имеют полностью сопряженной пи-электронной системы. Предпочтительно, мостиковый циклоалкил является 6-14-членным, более предпочтительно 7-10-членным. В соответствии с числом колец, мостиковые циклоалкилы делятся на бициклические, трициклические, тетрациклические или полициклические мостиковые циклоалкилы, предпочтительно, мостиковый циклоалкил является бициклическим, трициклическим или тетрациклическим мостиковым циклоалкилом, более предпочтительно, бициклическим или трициклическим мостиковым циклоалкилом. Типичные примеры мостиковых циклоалкилов включают, но не ограничиваются ими, следующие группы:
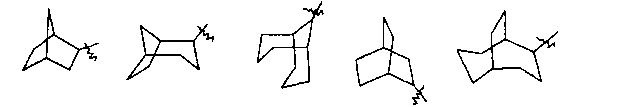
 .
.
Указанный циклоалкил может быть конденсирован с арилом, гетероарилом или кольцом гетероциклоалкила, где кольцо, связаное с материнской структурой, представляет собой циклоалкил. Типичные примеры включают, но не ограничиваются ими инданил уксусной кислоты, тетрагидронафталин, бензоциклогептил и так далее. Указанный циклоалкил, возможно может быть замещенным или незамещенным. В случае замещения замещающей группой (группами) являются, предпочтительно, одна или несколько групп, независимо выбранных из группы, включающей алкил, алкенил, алкинил, алкокси, алкилсульфо, алкиламино, галоген, тиол, гидроксил, нитро, циано, циклоалкил, гетероциклоалкил, арил, гетероарил, циклоалкокси, гетероциклический алкокси, циклоалкилтио, гетероциклический алкилтио, карбонил, -C(O)OR5, -OC(O)R5, -C(O)R5, -NHC(O)R5, -NR6R7 and -S(O)mR5.
“Гетероциклил” означает 3-20-членную насыщенную или частично ненасыщенную моноциклическую или полициклическую углеводородную группу, содержащую один или более гетероатомов, выбранных из группы, состоящей из N, О и S(O)m (где m представляет собой целое число_от 0 до 2) в качестве кольцевых атомов, за исключением -O-O-, -O-S- или -S-S- в кольце, остальные кольцевые атомы представляют собой С. Предпочтительно, гетероциклил является 3-12-членным, содержащим от 1 до 4 гетероатомов; более предпочтительно 3-10-членным; наиболее предпочтительно 4-6-членным. Типичные примеры моноциклического гетероциклила включают, но не ограничиваются ими, пирролидил, пиперидил, пиперазинил, морфолинил, сульфо-морфолинил, гомопиперазинил, фурил, азетидинил и так далее. Полициклический гетероциклил включает гетероциклил, имеющий спиро-кольцо, конденсированное кольцо и мостиковое кольцо.
“Спиро-гетероциклил” означает 5-20-членную полициклическую гетероциклическую группу с кольцами, соединенными через один общий атом углерода (называемый спиро-атом), отличающуюся тем, что кольца имеют один или несколько гетероатомов, выбранных из группы, состоящей из N, О и S(O)m (где m представляет собой целое число от 0 до 2) в качестве кольцевых атомов, а остальные кольцевые атомы представляют собой C, где одно или более колец может содержать одну или несколько двойных связей, но ни одно из колец не имеет полностью сопряженной пи-электронной системы. Предпочтительно, спиро-гетероциклил является 6-14-членным, более предпочтительно 7-10-членным. В соответствии с числом общих спиро-атомов, спиро-гетероциклилы делится на моно-спиро-гетероциклилы, ди-спиро-гетероциклилы или поли-спиро-гетероциклилы, предпочтительно, представляют собой моно-спиро-гетероциклилы или ди-спиро-гетероциклилы, более предпочтительно, 4-членный/4-членный, 4-членный/5-членный, 4-членный/6-членный, 5-членный/5-членный или 5-членный/6-членный моно-спиро-гетероцикло-алкил. Типичные примеры спиро-гетероциклила включают, но не ограничиваются ими, следующие группы:
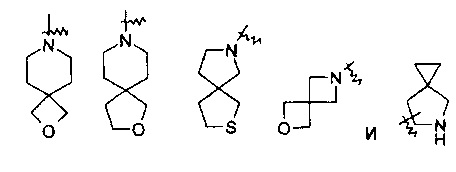 .
.
“Конденсированный гетероциклил” означает 5-20-членную полициклическую гетероциклическую группу, где каждое кольцо в системе имеет общую пару атомов углерода с другим кольцом, где одно или более колец могут содержать одну или несколько двойных связей, но ни одно из колец не имеет полностью сопряженной пи-электронной системы, и где указанные кольца имеют один или несколько гетероатомов, выбранных из группы, состоящей из N, O и S(O)m (где m представляет собой целое число от 0 до 2) в качестве кольцевых атомов, остальные кольцевые атомы представляют собой С. Предпочтительно, конденсированный гетероциклил является 6-14-членным, более предпочтительно 7-10-членным. В соответствии с числом колец-членов, конденсированные гетероциклилы делятся на бициклические, трициклические, тетрациклические или полициклические конденсированные гетероциклилы, предпочтительно, конденсированный гетероциклил представляет собой бициклический или трициклический конденсированный гетероциклил, более предпочтительно, 5-членный/5-членный или 5-членный/6-членный бициклический конденсированный гетероциклил. Типичные примеры конденсированного гетероциклила включают, но не ограничиваются ими, следующие группы:
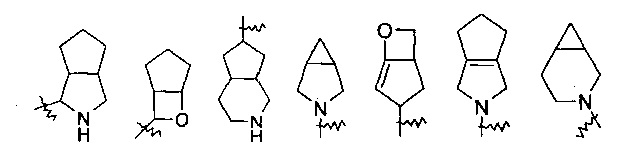
 .
.
“Мостиковый Гетероциклил» означает 5-14-членную полициклическую гетероциклильную группу, в которой каждые два кольца в системе имеют два общих несвязанных атома, указанные кольца могут иметь одну или несколько двойных связей, но не имеют полностью сопряженной пи-электронной системы, и указанные кольца имеют один или несколько гетероатомов, выбранных из группы, состоящей из N, О и S(O)m (где т представляет собой целое число от 0 до 2) в качестве кольцевых атомов, оставшиеся кольцевые атомы представляют собой С. Предпочтительно, мостиковый гетероциклил является 6-4-членным, более предпочтительно, 7-10-членным. В соответствии с числом колец-членов, мостиковые гетероциклилы делятся на бициклические, трициклические, тетрациклические или полициклические мостиковые гетероциклилы, предпочтительно, являются бициклическими, трициклическими или тетрациклическими мостиковыми гетероциклилами, более предпочтительно, бициклическими или трициклическими мостиковыми гетероциклилами. Типичные примеры мостиковых гетероциклилов включают, но не ограничиваются ими, следующие группы:
 .
.
Указанное кольцо гетероциклила может быть конденсировано с арилом, гетероарилом или циклоалкилом, где кольцо, связанное с материнской структурой представляет собой гетероциклил. Типичные примеры включают, но не ограничиваются ими, следующие группы:
 .
.
Гетероциклил, возможно может быть замещенным или незамещенным. В случае замещения замещающая группа(ы) представляет собой, предпочтительно, одну или несколько групп, независимо выбранных из группы, включающей алкил, алкенил, алкинил, алкокси, alkylsulfo, алкиламино, галоген, тиол, гидроксил, нитро, циано, циклоалкил, гетероциклоалкил, арил, гетероарил, циклоалкокси, гетероциклический алкокси, циклоалкилтио, гетероциклический алкилтио, карбонил, -C(O)OR5, -OC(O)R5, -C(O)R5, -NHC(O)R5, -NR6R7 и -S(O)mR5.
“Циклоалкилиден” означает насыщенный или частично ненасыщенный моноциклический или полициклический углеводород, из которого два водорода равномерно устранены, имеющий от 3 до 20 атомов углерода, предпочтительно, от 3 до 12 атомов углерода, более предпочтительно, моноциклический циклоалкилиден, имеющий от 3 до 10 атомов углерода, наиболее предпочтительно, от 3 до 6 атомов углерода. Типичные примеры моноциклического циклоалкилидена включают, но не ограничиваются ими, циклопропилиден, циклобутилиден, циклопентилиден, циклогексилиден, циклогептилиден, циклооктилиден, 1,2-циклопропилиден, 1,3-циклобутилиден, 1,4-циклогексилиден и т.д. полициклические циклоалкилидены включают суб-спиро, суб-конденсированные и суб-мостиковые циклоалкилидены.
“Арил” означает 6-14-членное углеродное моноциклическое кольцо или полициклическое конденсированное кольцо (“конденсированная” циклическая система означает, что каждое кольцо (цикл) в указанной системе имеет общую пару соседних атомов углерода с другим кольцом (циклом) в указанной циклической системе) группы, и имеет полностью сопряженную пи-электронную систему. Предпочтительно, арил является 6-10-членным, таким как фенил и нафтил, наиболее предпочтительно, является фенилом. Указанный арил может быть конденсирован с гетероарилом, гетероциклилом или циклоалкилом, где кольцо связанное с материнской структурой представляет собой арил. Типичные примеры включают, но не ограничиваются ими, следующие группы:
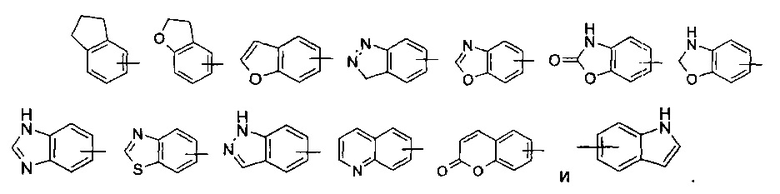
Арильная группа может быть замещенной или незамещенной. В случае замещения замещающая группа(ы) представляет собой, предпочтительно, одну или несколько групп, независимо выбранных из группы, включающей алкил, алкенил, алкинил, алкокси, alkylsulfo, алкиламино, галоген, тиол, гидроксил, нитро, циано, циклоалкил, гетеро циклоалкил, арил, гетероарил, циклоалкокси, гетероциклический алкокси, циклоалкилтио, гетероциклический алкилтио, -C(O)OR5, -OC(O)R5, -C(O)R5, -NHC(O)R5, -NR6R7 и -S(O)mR5.
“Гетероарил” означает гетероарил, содержащий от 1 до 4 гетероатомов, выбранных из группы, состоящей из N, О и S в качестве кольцевых атомов и содержащий от 5 до 14 циклических атомов, предпочтительно 5-10-членное кольцо и, более предпочтительно, 5-6-членное кольцо. Примеры включают фурил, тиенил, пиридил, пирролил, N-алкил пирролил, пиримидинил, пиразинил, имидазолил, тетразолил и тому подобное. Указанный гетероарил может быть конденсирован с кольцом арила, гетероциклила или циклоалкила, где кольцо, связанное с материнской структурой, представляет собой гетероарильное кольцо. Типичные примеры включают, но не ограничиваются ими, следующие группы:

Гетероарильная группа может быть, возможно замещенной или незамещенной. В случае замещения, замещающая группа (ы), представляет собой, предпочтительно, одну или несколько групп, независимо выбранных из группы, включающей алкил, алкенил, алкинил, алкокси, алкилсульфо, алкиламино, галоген, тиол, гидроксил, нитро, циано, циклоалкил, гетероциклоалкил, арил, гетероарил, циклоалкокси, гетероциклического алкокси, циклоалкилтиогруппы, гетероциклического алкилтио, -C(O)OR5, -OC(O)R5, -C(O)R5, -NHC(O)R5, -NR6R7 и -S(O)mR5.
“Алкокси” обозначает как -О-(алкил) так и -O-(незамещенный циклоалкил) группу, где алкил такой, как определено выше. Типичные примеры включают, но не ограничиваются ими, метокси, этокси, пропокси, бутокси, циклопропилокси, циклобутилокси, циклопентилокси, циклогексилокси, и тому подобное. Алкокси может быть, возможно замещенным или незамещенным. В случае замещения, замещающая группа представляет собой, предпочтительно, одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, alkylsulfo, алкиламино, галогена, тиола, гидроксила, нитро, циано, циклоалкила, гетероциклоалкила, арила, гетероарила, циклоалкокси, гетероциклического алкокси, циклоалкилтио, гетероциклического алкилтио, -C(O)OR5, -OC(O)R5, -C(O)R5, -NHC(O)R5, -NR6R7 и -S(O)mR5.
“Галогеналкил” означает алкил, замещенный одним или более галогенами, где алкил такой, как определено выше.
“Гидрокси” обозначает группу -ОН.
“Гидроксиалкил” обозначает алкильню группу, замещенную гидроксильной группой, где алкил такой, как определено выше. “Галоген” означает фтор, хлор, бром или йод.
“Амино” означает группу -NH2.
“Циано” означает группу -CN.
“Нитро” означает группу-NO2.
“Бензил” означает группу -CH2-(penyl).
“Карбонил” означает =O.
“Карбоксил” означает группу -C(O)OH.
“Алкоксикарбонил” означает -C(O)O(алкил) или (циклоалкил) группу, где алкил и циклоалкил такие, как определено выше.
“Возможный” или “возможно” означает, что описанное событие или обстоятельство, может, но не обязательно, иметь место, и описание включает в себя примеры, когда событие или обстоятельство могут возникнуть или могут не возникнуть. Например, фраза “гетероциклическая группа, возможно, замещенная алкилом” означает, что алкильная группа может присутствовать, но не обязательно присутствует, и описание включает случай, когда гетероциклическая группа может быть замещена алкильной и случай, когда гетероциклическая группа может быть не замещена алкилом.
“Замещенный” означает один или более атомов водорода в группе, предпочтительно, до 5, более предпочтительно от 1 до 3 атомов водорода, независимо замещенных соответствующим количеством заместителей. Само собой разумеется, что заместители существуют только в единственно возможном для них химическом положении. Специалист в данной области техники без особых экспериментальных и теоретических изысканий может определить, является или не является ли возможным замещение. Например, сочетание амино или гидроксильной групп, имеющих свободные атомы водорода и углерода, с ненасыщенными связями (например, олефиновыми) может быть неустойчивым.
“Фармацевтическая композиция” означает смесь одного или более из описанных здесь соединений или их физиологически/фармацевтически приемлемых солей или пролекарств, а также других химических компонентов, таких как физиологически/фармацевтически приемлемые носители и наполнители. Целью фармацевтической композиции является облегчение введения соединения в организм, что способствует поглощению активного ингредиента и, таким образом, отображает биологическую активность.
m и R5-R7 являются такими, как определено в формуле (I).
Способ синтеза соединения по настоящему изобретению
Для достижения конечной цели изобретения, предполагают следующее техническое решение:
Процесс получения соединений формулы (I) или их таутомеров, рацематов, энантиомеров, диастереомеров и их смесей, и их фармацевтически приемлемых солей по изобретению, включающий следующие схемы:

Взаимодействие R4-замещенного гидроксильного соединения с алкил-сульфонилгалогенидом (предпочтительно метансульфонилхлоридом) в щелочной среде с получением R4-замещенного алкил сульфоната; нагревание R4-замещенного алкил-сульфоната с гидроксил-замещенным фенильным соединением в растворителе в присутствии карбоната цезия с получением соединения формулы (IA); конденсация соединения формулы (IA) с гидроксил-замещенным бензо-циклическим соединением в растворителе в присутствии
трифенилфосфина и диизопропил азодикарбоксилата, с получением соединения формулы (I).
где кольцо B, L, A, R1-R4, n, p и q являются такими, как определено в формуле (I), и R9 представляет собой алкил, предпочтительно метил.
Щелочные условия включают органические основания, и неогранические основания, где указанные органические основания включают, но не ограничиваются ими, триэтиламин, N,N-диизопропилэтиламин, N,N-диметилформамид, н-бутиллитий, трет-бутоксид калия, бромид тетрабутил аммония, предпочтительно триэтиламин; и где указанные неогранические основания включают, но не ограничиваются ими, гидрид натрия, гидроксид натрия, гидроксид калия, карбонат натрия, бикарбонат натрия, карбонат калия, бикарбонат калия и карбонат цезия, предпочтительно карбонат натрия, карбонат калия, гидроксид натрия или гидроксид калия.
Растворитель включает, но не ограничивается ими: уксусную кислоту, метанол, этанол, ацетонитрил, тетрагидрофуран, дихлорметан, диметилсульфоксид, 1,4-диоксан, воду или N,N-диметилформамид.

Конденсация алкилсилокси-замещенного фенильного соединения с гидроксил-замещенным соединением с бензольным кольцом в растворителе в присутствии трифенилфосфина и диизопропилазодикарбоксилата с последующим снятием защиты с получением соединения формулы (IB); нагревание соединения формулы (IB) с R4-замещенным алкил-сульфонатом (предпочтительно R4-замещенный метилсульфонат) в растворителе в присутствии карбоната цезия с получением соединения формулы (I);
где кольцо B, L, A, R1-R4, n, p и q являются такими, как определено выше, и R9 представляет собой алкил, предпочтительно метил.
Щелочные условия включают органические щелочи, и неорганические щелочи, где указанные органические щелочи включают, но не ограничиваются ими, триэтиламин, N,N-диизопропилэтиламин, N,N-диметилформамид, n-бутиллитий, трет-бутоксид калия, бромид тетрабутиламмония, предпочтительно, триэтиламин, где указанные неорганическки щелочи включают, но не ограничиваются ими, гидрид натрия, гидроксид натрия, гидроксид калия, карбонат натрия, бикарбонат натрия, карбонат калия, бикарбонат калия и карбонат цезия, предпочтительно карбонат натрия, карбонат калия, гидроксид натрия или гидроксид калия.
Растворитель включает, но не ограничивается ими: уксусную кислоту, метанол, этанол, ацетонитрил, тетрагидрофуран, дихлорметан, диметилсульфоксид, 1,4-диоксан, воду или N,N-диметилформамид.

Взаимодействие кольца Е-замещенного гидроксильного соединения с алкил сульфонилгалогенидом (предпочтительно, метансульфонил хлоридом) в щелочных условиях в растворителе с получением кольца Е-замещенного алкильного сульфоната; нагревание кольца Е-замещенного алкил сульфоната, в растворителе, с гидроксил-замещенным бифенил спиртовым соединением в присутствии карбоната цезия, с получением соединения формулы (III-A); конденсация соединения формулы (III-А) с гидроксил-замещенным дигидробензофурановым соединением в растворителе в присутствии трифенилфосфина и диизопропилазодикарбоксилата с получением соединения формулы (III);
где кольцо E, R1, R8, s и q являются такими, как определено в формуле (III), a R9 представляет собой алкил, предпочтительно метил.
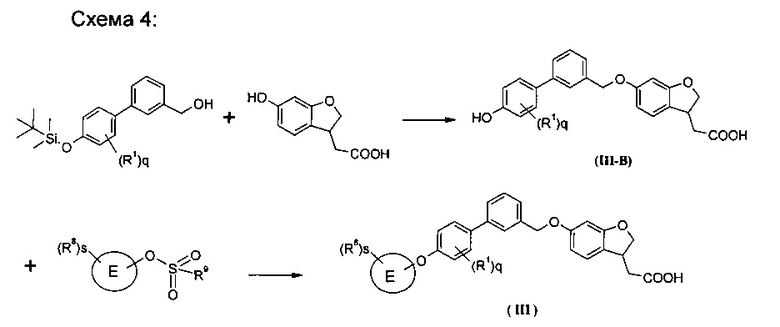
Конденсация алкилсилилокси-замещенного бифенил спиртового соединения с гидроксил-замещенным дигидробензофурановым соединением в растворителе в присутствии трифенилфосфина и диизопропилазодикарбоксилата с последующим снятием защиты с получением соединения формулы (III-B); нагревание соединения формулы (III-B) с кольцом Е-замещенного алкил сульфоната (предпочтительно кольцом Е-замещенного метансульфоната) в растворителе в присутствии карбоната цезия, с получением соединения формулы (III);
где R8 представляет собой гидроксил, гидроксил, возможно дополнительно защищен защитной группой, и затем защитную группу удаляют, с получением соединения формулы (III);
где кольцо Е, R1, R8, s и q имеют значения, определенные в формуле (III), a R9 представляет собой алкил, предпочтительно метил.
Щелочные условия включают органические щелочи и неорганические щелочи, где указанные органические щелочи включают, но не ограничиваются ими, триэтиламин, N,N-диизопропилэтиламин, N,N-диметилформамид, n-бутиллитий, трет-бутоксид калия, бромид тетрабутиламмония, предпочтительно триэтиламин, где указанные неорганические щелочи включают, но не ограничиваются ими, гидрид натрия, гидроксид натрия, гидроксид калия, карбонат натрия, бикарбонат натрия, карбонат калия, бикарбонат калия и карбонат цезия, предпочтительно карбонат натрия, карбонат калия, гидроксид натрия или гидроксид калия.
Растворитель включает, но не ограничивается ими: уксусную кислоту, метанол, этанол, ацетонитрил, тетрагидрофуран, дихлорметан, диметилсульфоксид, 1,4-диоксан, воду или N,N-диметилформамид.

Нагревание соединения формулы (III-B) и кольца Е-замещенного алкил сульфоната (предпочтительно, кольца Е-замещенного метансульфоната) в растворителе в присутствии карбоната цезия, с получением соединения формулы (III-C); взаимодействие соединения формулы (III-C) с замещенным R8 (предпочтительно, сульфонилхлоридом или ангидридом), с получением соединения формулы (III);
где кольцо E, R1, R8, s и q являются такими, как определено в формуле (III).
Щелочные условия включают в себя органические щелочи и неорганические щелочи, где указанные органические щелочи включают, но не ограничиваются ими, триэтиламин, N,N-диизопропилэтиламин, N,N-диметилформамид, н-бутиллитий, трет-бутоксид калия, бромид тетрабутиламмония, предпочтительно, триэтиламин, где указанные неорганические щелочи включают, но не ограничиваются ими, гидрид натрия, гидроксид натрия, гидроксид калия, карбонат натрия, бикарбонат натрия, карбонат калия, бикарбонат калия и карбонат цезия, предпочтительно, карбонат натрия, карбонат калия, гидроксид натрия или гидроксид калия.
Растворитель включает, но не ограничивается ими: уксусную кислоту, метанол, этанол, ацетонитрил, тетрагидрофуран, дихлорметан, диметилсульфоксид, 1,4-диоксан, воду или N,N-диметилформамид.
Предпочтительные варианты
Следующие примеры служат для иллюстрации настоящего изобретения, но эти примеры не следует рассматривать как ограничивающие объем изобретения.
ПРИМЕРЫ
Структура соединения была идентифицирована с помощью ЯМР (ядерного магнитного резонанса) или МС (масс-спектрометрии). ЯМР определяли с помощью оборудования Bruker Avance-400. Использовали растворители дейтерированный-диметилсульфоксид (DMCO-d6), дейтерированный хлороформ - (CDCl3) и дейтерированный метанол (CD3OD) с тетраметилсиланом (ТМС) в качестве внутреннего стандарта. ЯМР химические сдвиги (δ) были приведены в 10-6 (м.д.).
МС определяли с использованием масс-спектрометра FINNIGAN LCQAd (ИЭР - ионизация электрораспылением) (изготовитель: Thermo, type: Finnigan LCQ advantage MAX).
ВЭЖХ (высокоэффективная жидкостная хроматография) проводили с использованием спектрометра жидкостной хроматографии высокого давления Agilent 1200DAD (Sunfire С18 150×4,6 мм хроматографическая колонка) и спектрометра жидкостной хроматографии высокого давления Waters 2695-2996 (Gimini С18 150×4,6 мм хроматографическая колонка).
Среднюю степень ингибирования киназы и IC50 определяли с помощью Novostar ELIASA (BMG Co., Германия).
Для тонкослойного силикагеля использовали Yantai Huanghai HSGF254 или Qingdao GF254 пластинки с силикагелем. Размер пластин, используемых в ТСХ (тонкослойной хроматографии), был от 0,15 мм до 0,2 мм, а размер пластин, использованных для очистки продукта, был от 0,4 мм до 0,5 мм. После колоночной хроматографии обычно использовали Yantai Huanghai силикагель от 200 до 300 меш в качестве носителя.
Известный исходный материал по изобретению может быть получен с помощью обычного метода синтеза, известного из предшествующего уровня техники, или приобретен у ABCR GmbH & Co. KG, Acros Organics, Aldrich Chemical Company, Accela ChemBio Inc or Dari chemical Company, etc.
Если не указано иное, следующие реакции были помещены в атмосферу азота или аргона.
Определение “атмосфера азота” или “атмосфера аргона” обозначает, что реакционная колба оснащена 1 л шаром с азотом или аргоном.
Определение “атмосфера водорода” обозначает, что, реакционная колба оснащена 1 л шаром с водородом.
Реакции гидрирования под давлением проводились с использованием спектрометра гидрирования Parr 3916ЕКХ и водородного генератора QL-500.
В реакциях гидрирования, в реакционной системе создавал вакуум, и наполняли систему водородом, и повторяли описанную выше операцию три раза.
Микроволновые реакции проводили с использованием микроволнового реактора СЕМ Discover-S 908860.
Если не указано иное, раствор, использованный в следующей реакции, представляет собой водный раствор. Если не указано иное, температура реакции в следующей реакции является комнатной температурой, а диапазон температуры составляет 20°C-30°C.
За ходом реакции следили с помощью тонкослойной хроматографии (ТСХ), система проявления растворителя включала: А: дихлорметан и метанол, В: н-гексан и этилацетат, С: петролейный эфир и этилацетат, D: ацетон. Объем растворителя был скорректирован в соответствии с полярностью соединений.
Элюционные системы очистки соединений путем колоночной хроматографии и тонкослойной хроматографии включали: дихлорметан и метанол, В: н-гексан и этилацетат-систему, С: н-гексан и ацетон, D: н-гексан, Е: этилацетат. Объем растворителя регулировали в зависимости от полярности соединений, и иногда добавляли немного щелочного реагента, такого как триэтиламин а также кислотного реагента.
Пример 1
2-((S)-6-((2',6'-диметил-4'-(((S)-тетрагидрофуран-3-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусная кислота


Стадия 1
(R)-тетрагидрофуран-3-ил метансульфонат
(R)-тетрагидрофуран-3-ол 1а (300 мг, 3.40 ммоль) и триэтиламин (687 мг, 6.80 ммоль) растворяли в 20 мл дихлорметана в бане со льдом с последующим добавлением метансульфонилхлорида (468 мг, 4.10 ммоль). Реакционный раствор нагревали до комнатной температуры и перемешивли в течение 2 часов. В реакционный раствор добавляли 10 мл дихлорметана, промывали водой (30 мл × 3), сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (R)-тетрагидрофуран-3-ил метансульфоната 1b (520 мг) в виде желтого масла, которое непосредственно использовали на следующей стадии без дополнительной очистки.
Стадия 2
(S)-(2',6'-диметил-4'-((тетрагидрофуран-3-ил)окси)бифенил-3-ил)метанол
Неочищенный (R)-тетрагидрофуран-3-ил метансульфонат 1b (520 мг, 3.13 ммоль), 3' (гидроксиметил)-2,6-диметилбифенил-4-ол(714 мг, 3.13 ммоль, получали способом, описанным в патентной заявке CN 101616913) и карбонат цезия (3.10 г, 9.39 ммоль) растворяли в 30 мл N,N-диметилформамида. Реакционную смесь нагревали до 80°C и перемешивали в течение 7 часов. Полученный раствор концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле с системой элюирования В, получая указанное в заголовке соединение (S)-(2',6'-диметил-4'-((тетрагидрофуран-3-ил)окси)бифенил-3-ил)метанол 1 с (500 мг, выход 54.0%) в виде бесцветного масла.
МС m/z (ИЭР): 299,3 [М+1]
Стадия 3
Метил 2-((S)-6-((2',6'-диметил-4'-(((S)-тетрагидрофуран-3-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетат
(S)-(2',6'-диметил-4'-((тетрагидрофуран-3-ил)окси)бифенил-3-ил)метанол 1 с: (143 мг, 0,48 ммоль), метил-(S)-2-(6-гидроксил-2,3-дигидробензофуран-3-ил)ацетат (100 мг, 0.48 ммоль, получали способом, описанным в патентной заявке CN 101616913. Рацемат выделяли с помощью хиральной ВЭЖХ с использованием метода ВЭЖХ, условия разделения: хиральная колонка Chiralpak IA, подвижная фаза: н-гексан: тетрагидрофуран = 80: 20, скорость потока: 1.0 мл/мин. Соответствующий компонент собирали и ротационно выпаривали, чтобы удалить растворитель, и трифенилфосфин (189 мг, 0,72 ммоль) растворяли в 20 мл дихлорметана. Реакционный раствор охлаждали до 0°C с последующим добавлением диизопропилазодикарбоксилата (146 мг, 0,72 ммоль), затем нагревали до комнатной температуры и перемешивали в течение 2 часов. Полученный раствор концентрировали при пониженном давлении, остаток очищали колоночной хроматографией на силикагеле с системой элюирования В, с получением указанного в заголовке соединения метил 2-((S)-6-((2',6-диметил-4'-(((S)-тетрагидрофуран-3-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетата 1d (220 мг, выход 94,0%) в виде белого твердого вещества.
МС m/z (ИЭР): 489,4 [М+1]
Стадия 4
2-((S)-6-((2',6'-диметил-4'-(((S)-тетрагидрофуран-3-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусная кислота
Метил 2-((S)-6-((2',6'-диметил-4'-(((S)-тетрагидрофуран-3-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетат 1d (220 мг, 0.45 ммоль) растворяли в 15 мл метанола, с последующим добавлением 3 M водного раствора гидроксида натрия (1,5 мл, 4,50 ммоль). Реакционную смесь подвергали взаимодействию в течение 3 часов. Смесь концентрировали при пониженном давлении, добавляли 10 мл воды и добавляли по каплям 1 M раствор соляной кислоты для доведения рН до 2. Полученный раствор экстрагировали этилацетатом (20 мл × 2). Объединенные органические экстракты промывали насыщенным раствором хлорида натрия (30 мл), сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения 2-((S)-6-((2',6'-диметил-4'-(((S)-тетрагидрофуран-3-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусной кислоты 1 (210 мг, выход 100%) в виде белого твердого вещества.
МС m/z (ИЭР): 473,2 [М-1]
1Н ЯМР (400 МГц, CDCl3) δ 7,45 (дт, 2Н), 7,21 (с, 1Н), 7,11 (дд, 2Н), 6,66 (с, 2Н), 6,58-6,46 (м, 2Н), 5,10 (с, 2Н), 5,04-4,94 (м, 1Н), 4.80 (т, 1Н), 4.33 (дд 1Н), 4,11-4,01 (м, 3Н), 4,01-3,92 (м, 1Н), 3,85 (ддд, 1Н), 2.85 (дд, 1Н), 2.66 (дд 1Н), 2,30-2,19 (м, 2Н), 2,03 (с, 6Н).
Пример 2
2-((S)-6-((2',6'-диметил-4'-(((R)-тетрагидрофуран-3-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусная кислота

Стадия 1
(S)-тетрагидрофуран-3-ил метансульфонат
(S)-тетрагидрофуран-3-ол 2а (300 мг, 3,40 ммоль) и триэтиламин (687 мг, 6,80 ммоль) растворяли в 20 мл дихлорметана в бане со льдом с последующим добавлением метансульфонилхлорида (908 мг, 7,93 ммоль). Реакционный раствор нагревали до комнатной температуры и перемешивали в течение 2 часов. В полученный раствор добавляли 10 мл дихлорметана, промывали водой (30 мл × 2), сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке неочищенного соединения (S)-тетрагидрофуран-3-ил метансульфоната 2b в виде желтого масла (522 мг, выход 92,5%).
Стадия 2
(R)-(2',6'-диметил-4'-((тетрагидрофуран-3-ил)окси)бифенил-3-ил)метанол
Неочищенный (S)-тетрагидрофуран-3-ил метансульфонат 2b (522 мг, 3,14 ммоль), 3'-(гидроксиметил)-2,6-диметилбифенил-4-ола (714 мг, 3,13 ммоль, получали способом, описанным в патентной заявке CN 101616913) и карбонат цезия (3,10 г, 9,39 ммоль) растворяли в 30 мл N,N-диметилформамида. Реакционную смесь нагревали до 80°C и перемешивали в течение 12 часов. Полученный раствор концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле с системой элюирования В, с получением указанного в заголовке соединения (R)-(2',6'-диметил-4'-((тетрагидрофуран-3-ил)окси)бифенил-3-ил)метанола 2с (540 мг, выход 58,0%) в виде бесцветного масла.
МС m/z (ИЭР): 299,2 [М+1]
Стадия 3
Метил 2-((S)-6-((2',6'-диметил-4'-(((R)-тетрагидрофуран-3-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетат
(R)-(2',6'-диметил-4'-((тетрагидрофуран-3-ил)окси)бифенил-3-ил)метанол 2с (143 мг, 0,48 ммоль), метил-(S)-2-(6-гидроксил-2,3-дигидробензофуран-3-ил) ацетат (100 мг, 0,48 ммоль) и трифенилфосфин (189 мг, 0,72 ммоль) растворяли в 20 мл дихлорметана. Реакционный раствор охлаждали до 0°C с последующим добавлением диизопропилазодикарбоксилата (146 мг, 0,72 ммоль). Реакционный раствор нагревали до комнатной температуры и перемешивали в течение 12 часов. Полученный раствор концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле с системой элюирования В, с получением указанного в заголовке соединения метил 2-((S)-6-((2',6'-диметил-4'-(((R)-тетрагидрофуран-3-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетата 2d (201 мг, выход 85,9%) в виде бесцветного масла.
МС m/z (ИЭР): 489,4 [М+1]
Стадия 4
2-((S)-6-((2',6'-диметил-4'-(((R)-тетрагидрофуран-3-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусная кислота.
Метил 2-((S)-6-((2',6’-диметил-4'-(((R)-тетрагидрофуран-3-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетат 2d (201 мг, 0,41 ммоль) растворяли в 15 мл метанола. В реакционный раствор добавляли 2 M водный раствор гидроксида натрия (2 мл, 4,10 ммоль) и перемешивали в течение 3 часов. Полученный раствор концентрировали при пониженном давлении, добавляли 10 мл воды и добавляли по каплям 1 M раствор соляной кислоты для доведения рН до 2-3. Раствор экстрагировали этилацетатом (20 мл × 2). Объединенные органические экстракты промывали насыщенным раствором хлорида натрия (30 мл), сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения 2-((S)-6-((2',6'-диметил-4'-(((R)-тетрагидрофуран-3-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусной кислоты 2 (150 мг, выход 77,1%) в виде белого твердого вещества.
МС m/z (ИЭР): 473,4 [М-1]
1Н ЯМР (400 МГц, CDCl3) δ 7,45 (дт, 2Н), 7,21 (с, 1Н), 7,11 (дд, 2Н), 6,66 (с, 2Н), 6,58-6,48 (м, 2Н), 5,10 (с, 2Н), 5.04-4.94 (м, 1Н), 4,80 (т, 1Н), 4,33 (дд, 1Н), 4,11-4,01 (м, 3Н), 4,00-3,91 (м, 1Н), 3,86 (дд, 1Н), 2,85 (дд, 1Н), 2,66 (дд, 1Н), 2,33-2,16 (м, 2Н), 2,03 (с, 6Н).
Пример 3
(S)-2-(6-((4'-((1-(трет-бутоксикарбонил)пиперидин-4-ил)окси)-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусная кислота
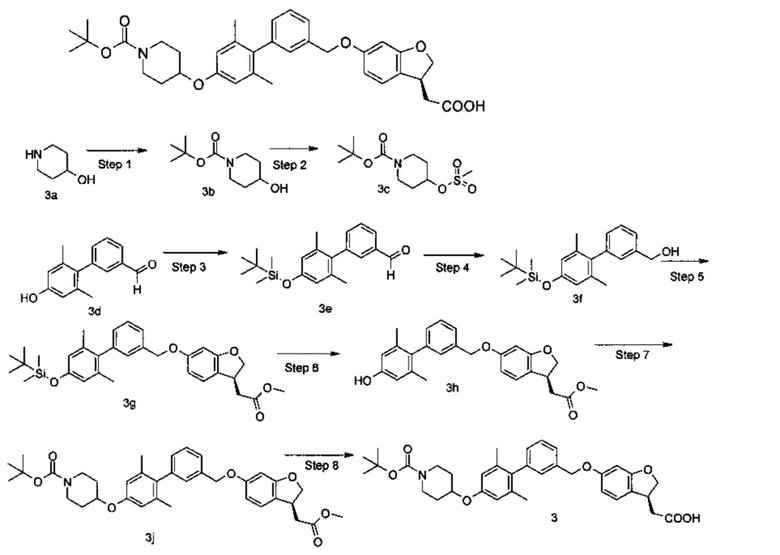
Стадия 1
Трет-бутил-4-гидроксипиперидин-1-карбоксилат
4-гидроксил-пиперидин 3а (1,01 г, 10 ммоль) и триэтиламина (2,02 г, 20 ммоль) растворяли в 20 мл дихлорметана в бане со льдом с последующим добавлением ди-трет-бутилдикарбоната (2,18 г, 10 ммоль). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 3 часов. Раствор промывали водой (20 мл × 2), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения трет-бутил-4-гидроксипиперидин-1-карбоновой кислоты 3b (2 г) в виде бесцветного масла, которое непосредственно использовали на следующей стадии без дополнительной очистки.
Стадия 2
Трет-бутил 4-((метилсульфонил)окси)пиперидин-1-карбоксилат
Неочищенный трет-бутил 4-гидроксипиперидин-1-карбоксилат 3b (2,01 г, 10 ммоль) растворяли в 30 мл дихлорметана в бане со льдом с последующим добавлением триэтиламина (2.02 г, 20 ммоль) и метансульфонил хлорида (1,26 г, 11 ммоль). Реакционный раствор нагревали до комнатной температуры и перемешивали в течение 3 часов. Полученный раствор промывали водой (20 мл × 2), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного целевого соединения трет-бутил 4-((метилсульфонил)окси)пиперидин-1-карбоксилата 3c (2.20 г) в виде светло-желтого твердого вещества, которое непосредственно использовали на следующей стадии без дополнительной очистки.
Стадия 3
4'-((трет-бутилдиметилсилил)окси)-2',6'-диметилбифенил-3-карбальдегид 4'-гидрокси-2',6'-диметилбифенил-3-карбальдегид 3d (7.20 г, 31,80 ммоль, полученный способом, описанным в патентной заявке CN 101616913), трет-бутилдиметилсилил (5,27 г, 34,90 ммоль) и имидазол (2,60 г, 38,10 ммоль) растворяли в 60 мл N,N-диметилформамида. Реакционный раствор нагревали до 60°C и перемешивали в течение 12 часов. Полученный раствор концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле с системой элюирования В, с получением указанного в заголовке соединения 4'-((трет-бутилдиметилсилил)окси)-2',6'-диметилбифенил-3-карбальдегида 3е (10 г, выход 92.0%) в виде коричневого масла.
МС m/z (ИЭР): 341,2 [М+1]
Стадия 4
(4'-((трет-бутилдиметилсилил)окси)-2',6'-диметилбифенил-3-ил)метанол
4'-((трет-бутилдиметилсилил)окси)-2',6'-диметилбифенил-3-карбальдегид 3е (2.60 г, 38.10 ммоль) растворяли в 80 мл метанола с последующим добавлением боргидрида натрия (1.66 г, 43.95 ммол). Реакционную смесь перемешивали в течение 3 часов. В полученную смесь добавляли 50 мл ацетона и концентрировали при пониженном давлении. К остатку добавляли 200 мл этилацетата, промывали водой (60 мл × 2), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения (4'-((трет-бутилдиметилсилил)окси)-2',6'-диметилбифенил-3-ил)метанола 3f (9.80 г, выход 98.0%) в виде бесцветного масла.
МС m/z (ИЭР): 343,2 [М+1]
Стадия 5
(S)-метил 2-(6-((4'-((трет-бутилдиметилсилил)окси)-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетат
(4'-((трет-бутилдиметилсилил)окси)-2',6'-диметилбифенил-3-ил)метанол 3f (1,19 г, 3,48 ммоль), метил (S)-2-(6-гидроксил-2,3-дигидробензофуран-3-ил)ацетат (660 мг, 3,17 ммоль) и трифенилфосфин (1.34 г, 4,75 ммоль) растворяли в 10 мл дихлорметана. Реакционный раствор охлаждали до 0°C с последующим добавлением диизопропилазодикарбоксилата (960 мг, 4,75 ммоль), затем нагревали до комнатной температуры и перемешивали в течение 4 часов. Полученный раствор концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле с системой элюирования В, с получением указанного в заголовке соединения (S)-метил 2-(6-((4'-((трет-бутилдиметилсилил)окси)-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетата 3g (1,28 г, выход 76,0%) в виде белого твердого вещества.
МС m/z (ИЭР): 533,3 [М+1]
Стадия 6
(S)-метил 2-(6-((4'-гидрокси-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетат
(S)-Метил 2-(6-((4'-((трет-бутилдиметилсилил)окси)-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетат 3g (1,28 г, 2,40 ммоль) растворяли в 50 мл тетрагидрофурана, с последующим добавлением 10 мл 6 M хлористоводородной кислоты. Реакционный раствор перемешивали при комнатной температуре в течение 12 часов. Полученный раствор концентрировали при пониженном давлении и к остатку добавляли 100 мл этилацетата. Органическую фазу промывали водой (20 × 2 мл) и насыщенным раствором хлорида натрия (20 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. К остатку добавляли 60 мл метанола и 1 мл концентрированной серной кислоты. Полученный раствор нагревали до 70°C и перемешивали в течение 1,5 часов. Раствор концентрировали при пониженном давлении, добавляли 100 мл этилацетата, промывали водой (20 мл × 2), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и остаток очищали с помощью системы элюирования В, с получением указанного в заголовке соединения (S)-метил 2-(6-((4'-гидрокси-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетата 3h (670 мг, выход 67,0%) в виде бесцветного масла.
МС m/z (ИЭР): 419,2 [М+1]
Стадия 7
Метил-(S)-2-(6-((4'-((1-(трет-бутоксикарбонил)пиперидин-4-ил)окси)-2',6'-диметилбифенил-3-ил)метокси)-2,3дигидробензофуран-3-ил)ацетат
4-[(метилсульфонил)окси]пиперидин-1-карбоновой кислоты трет-бутиловый эфир (134 мг, 0,48 ммоль), (S)-метил 2-(6-((4'-гидрокси-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетат 3h (200 мг, 0,48 ммоль) и карбонат цезия (470 мг, 1,43 ммоль) растворяли в 10 мл N,N-диметилформамида. Реакционную смесь нагревали до 80°C и перемешивали в течение 12 часов. В полученный раствор добавляли 30 мл этилацетата. Органическую фазу промывали водой (20 × 2 мл) и насыщенным раствором хлорида натрия (20 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и остаток очищали с помощью системы элюирования В, с получением указанного в заголовке соединения метил (S)-2-(6-((4'-((1-(трет-бутоксикарбонил)пиперидин-4-ил)окст)-2',6'-димеилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетата 3j (101 мг, выход 35,0%) в виде бесцветной слизи.
МС m/z (ИЭР): 502,3 [М-99]
Стадия 8
(S)2-(6-((4'-((1-(трет-бутоксикарбонил)пиперидин-4-ил)окси)-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусная кислота
Метил-(S)-2-(6-((4'-((1-(трет-бутоксикарбонил)пиперидин-4-ил)окси)-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетат 3j (26 мг, 0,04 ммоль) растворяли в 5 мл метанола с последующим добавлением 1 M водного раствора гидроксида натрия (0,5 мл, 0,43 ммоль). Реакцию проводили в течение 1,5 часов. Раствор концентрировали при пониженном давлении, добавляли 3 мл воды, добавляли по каплям 1 M лимонную кислоту для доведения рН до 4-5 и экстрагировали этилацетатом (20 мл × 2).
Объединенные органические экстракты промывали насыщенным раствором хлорида натрия (30 мл), сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (S)-2-(6-((4'-((1-(трет-бутоксиарбонил)пиперидин-4-илу)окси)-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусной кислоты 3 (12 мг, выход 47,4%) в виде белого твердого вещества.
МС m/z (ИЭР): 586,3 [М-1]
1Н ЯМР (400 МГц, CDCl3) δ 7,51-7,38 (м, 2Н), 7,21 (с, 1Н), 7,11 (дд, 2Н), 6,70 (с, 2Н), 6,51-6,54 (м, 2Н), 5,10 (с, 2Н), 4,80 (т, 1Н), 4,52 (с, 1Н), 4,33 (т, 1Н), 3,76-3,85 (м, 3Н), 3,41 (с, 2Н), 2,85 (д, 1Н), 2,66 (дд, 1Н), 2,03 (с, 6Н), 1,90 (д, 4Н), 1,52 (с, 9Н).
Пример 4
(S)-2-(6-((2',6'-диметил-4'-(пиперидин-4-илокси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусная кислота

Стадия 1
(S)-Метил 2-(6-((2',6'-диметил-4'-(пиперидин-4-илокси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетат
Метил (S)-2-(6-((4'-((1-(трет-бутоксикарбонил)пиперидин-4-ил)окси)-2',6'-диметил-бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетат 3j (75 мг, 0.13 ммоль) растворяли в 15 мл хлористоводородной кислоты в диоксане. Реакцию проводили в течение 1 часа. Полученный раствор концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения (S)-метил 2-(6-((2',6'-диметил-4'-(пиперидин-4-илокси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетата 4а (75 мг), в виде белого твердого вещества, которое непосредственно использовали на следующей стадии без дополнительной очистки.
Стадия 2
(S)-2-(6-((2',6'-диметил-4'-(пиперидин-4-илокси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусная кислота
Неочищенный (S)-метил 2-(6-((2',6'-диметил-4'-(пиперидин-4-илокси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетат 4а (31 мг, 0.06 ммоль) растворяли в 5 мл метанола с последующим добавлением 1 M водного раствора гидроксида натрия (0.6 мл, 0.62 ммоль). Реакционный раствор подвергали взаимодействию в течение 2 часов. Полученный раствор концентрировали при пониженном давлении, добавляли 3 мл воды, добавляли по каплям 1 M раствор лимонной кислоты для доведения рН до 5 и экстрагировали этилацетатом (20 мл × 2). Объединенные органические экстракты промывали насыщенным раствором хлорида натрия (30 мл), сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (S)-2-(6-((2',6'-диметил-4'-(пиперидин-4-илокси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусной кислоты 4 (31 мг, выход 100%) в виде белого твердого вещества.
МС m/z (ИЭР): 486.3 [М-1]
1Н-ЯМР (400 МГц, DMCO-d6) δ 7,51-7,38 (м, 2Н), 7,21 (с, 1Н), 7,11 (дд, 2Н), 6,70 (с, 2Н), 6,51-6,54 (м, 2Н), 5,10 (с, 2Н), 4,80 (т, 1Н), 4,52 (с, 1Н), 4,33 (т, 1Н), 3,81-3,86 (м, 1Н), 3,15 (с, 2Н), 2,81 (с, 2Н), 2,85 (д, 1Н), 2,66 (дд, 1Н), 2,03 (с, 6Н), 1,90 (д, 4Н).
Пример 5
(S)-2-(6-((2',6'-диметил-4'-((1-(метилсульфонил)пиперидин-4-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусная кислота


Стадия 1
(S)-Метил 2-(6-((2',6'-диметил-4'-((1-(метилсульфонил)пиперидин-4-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетат
(S)-Метил 2-(6-((2',6'-диметил-4'-(пиперидин-4-илокси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетат 4а (26 мг, 0.05 ммоль) растворяли в 5 мл дихлорметана с последующим добавлением метансульфонилхлорида (6 мг, 0,05 ммоль) и триэтиламина (15 мг, 0,15 ммоль). Реакцию проводили течение 1 часа. В полученный раствор добавляли 10 мл воды, концентрировали при пониженном давлении с получением неочищенного целевого соединения (выметил 2-(6-((2',6'-диметил-4'-((1-(метилсульфонил)пиперидин-4-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетата 5а (40 мг) в виде белого твердого вещества, которое непосредственно использовали на следующей стадии без дополнительной очистки.
МС m/z (ИЭР): 578,3 [М-1]
Стадия 2
(S)-2-(6-((2',6'-диметил-4'-((1-(метилсульфонил)пиперидин-4-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусная кислота
Неочищенный (S)-метил 2-(6-((2',6'-диметил-4'-((1-(метилсульфонил)пиперидин-4-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетат 5а (28 мг, 0,05 ммоль) растворяли в 5 мл этанола с последующим добавлением 1 M водного раствора гидроксида натрия (0,5 мл, 0,50 ммоль). Реакцию проводили в течение 1 часа. В полученный раствор добавляли по каплям 1 M раствор соляной кислоты для доведения рН до 5 и экстрагировали этил ацетатом (20 мл × 2). Объединенные органические экстракты промывали насыщенным раствором хлорида натрия (30 мл), сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении и остаток очищали с методом препаративной сепарации, с получением указанного в заголовке соединения (S)-2-(6-((2',6'-диметил-4'-((1-(метилсульфонил)пиперидин-4-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусной кислоты 5 (12 мг, выход 44.0%) в виде белого твердого вещества.
МС m/z (ИЭР): 564.3 [М-1]
1Н ЯМР (400 МГц, CDCl3) δ 7,38-7,45 (м, 2Н), 7,18 (с, 1Н), 7,05-7,10 (м, 2Н), 6,67 (с, 2Н), 6,47-6,51 (м, 2Н), 5.07 (с, 2Н), 4,77 (т, 1Н), 4,57 (с, 1Н), 4,30 (т, 1Н), 3,82 (т, 1Н), 3,36-3,41 (м, 4Н), 2,79-2,84 (м, 4Н), 2,59-2,66 (м, 1Н), 2,00-2,04 (м, 10Н).
Пример 6
(S)-2-(6-((4'-((1-ацетилпиперидин-4-ил)окси)-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусная кислота

Стадия 1
(S)-метил 2-(6-((4'-((1-ацетилпиперидин-4-ил)окси)-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетат
(S)-Метил 2-(6-((2',6'-диметил-4'-(пиперидин-4-илокси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетат 4а (20 мг, 0,04 ммоль) растворяли в 2 мл дихлорметана с последующим добавлением уксусного ангидрида (5 мг, 0,05 ммоль) и триэтиламина (12 мг, 0,12 ммоль). Реакционный раствор перемешивали в течение 2 часов. Полученный раствор концентрировали при пониженном давлении и остаток очищали с помощью тонкослойной хроматографии с системой элюирования А, с получением указанного в заголовке соединения (S)-метил 2-(6-((4'-((1-ацетилпиперидин-4-ил)окси)-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетата 6а (10 мг, выход 47,0%) в виде бесцветного масла.
МС m/z (ИЭР): 544,3 [М+1]
Стадия 2
(S)-2-(6-((4'-((1-ацетилпиперидин-4-ил)окси)-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусная кислота
(S)-Метил 2-(6-((4'-((1-ацетилпиперидин-4-ил)окси)-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетат 6а (10 мг, 0,02 ммоль) растворяли в 2 мл смеси растворителей тетрагидрофурана и метанола (об./об. = 1:1) с последующим добавлением 1 M водного раствора гидроксида лития (0,2 мл, 0,20 ммоль). Реакционный раствор перемешивали в течение 2 часов. Полученный раствор концентрировали при пониженном давлении, добавляли по каплям 1 M соляной кислоты для доведения рН до 5 и экстрагировали этилацетатом (20 мл × 2). Объединенные органические экстракты промывали насыщенным раствором хлорида натрия (30 мл), сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (S)-2-(6-((4'-((1-ацетилпиперидин-4-ил)окси)-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусной кислоты 6 (8 мг, выход 84,2%) в виде белого твердого вещества.
МС m/z (ИЭР): 530,4 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 7,37-7.45 (м, 2Н), 7,18 (с, 1Н), 7,05-7,10 (м, 2Н), 6,67 (с, 2Н), 6,47-6,51 (м, 2Н), 5,07 (с, 2Н), 4,75-4,79 (м, 1Н), 4,57 (с, 1Н), 4,28-4,31 (м, 1Н), 3,73-3,83 (м, 4Н), 2,78-2,83 (м, 1Н), 2,58-2,64 (м, 1Н), 2,22-2,26 (м, 1Н), 2,15 (м, 3Н), 1,94-2.06 (м, 10Н).
Пример 7
2-((S)-6-((4'-(((S)-1-(трет-бутоксикарбонил)пирролидин-3-ил)окси)-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусная кислота

Стадия 1
(R)-трет-бутил-3-гидроксипирролидин-1 -карбоновая кислота
(R)-пирролидин-3-ол 7а (348 мг, 4 ммоль) и триэтиламина (808 мг, 8 ммоль) растворяли в 20 мл дихлорметана с последующим добавлением ди-трет-бутилдикарбоната (959 мг, 4,40 ммоль) на бане со льдом. Реакционный раствор нагревали до комнатной температуры и перемешивали в течение 3 часов. В полученный раствор добавляли 50 мл дихлорметана, промывали насыщенным раствором хлорида натрия (5 мл × 3), сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле с системой элюирования В, с получением указанного в заголовке соединения (R)-трет-бутил-3-гидроксипирролидин-1-карбоксилат 7b (400 мг, выход 53,4%) в виде бесцветного масла.
Стадия 2
(R)-трет-бутил 3-((метилсульфонил)окси)пирролидин-1-карбоновая кислота
(R)-трет-бутил 3-гидроксипирролидин-1-карбоксилат 7b (375 мг, 2 ммоль) растворяли в 10 мл дихлорметана в бане со льдом с последующим добавлением триэтиламина (404 мг, 4 ммоль) и метансульфонилхлорида (274 мг, 2,40 ммоль). Реакционный раствор нагревали до комнатной температуры и перемешивали в течение 3 часов. В полученный раствор добавляли 5 мл воды и экстрагировали дихлорметаном (20 мл × 3). Объединенные органические экстракты промывали насыщенным раствором хлорида натрия (30 мл), сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения (R)-трет-бутил 3-((метилсульфонил)окси)пирролидин-1-карбоксилат 7 с (530 мг) в виде желтого масла, которое непосредственно использовали на следующей стадии без дополнительной очистки.
Стадия 3
Метил 2-((S)-6-((4'-((S)-1-(трет-бутоксикарбонил)пирролидин-3-ил)окси)-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетат
(R)-трет-бутил 3-((метилсульфонил)окси)пирролидин-1-карбоксилат 7с (190 мг, 0,72 ммоль), метил (S)-2-(6-((4-гидроксил-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетат 3Н (200 мг, 0,48 ммоль) и карбонат цезия (310 мг, 0,96 ммоль) растворяли в 10 мл N,N-диметилформамида. Реакционный раствор нагревали до 80°C и перемешивали в течение 12 часов. В полученный раствор добавляли 5 мл воды и 50 мл этилацетата. Органическую фазу промывали насыщенным раствором хлорида натрия (20 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и остаток очищали системой элюирования В с получением указанного в заголовке соединения метил 2-(((S)-6-((4'-((S)-1-(трет-бутоксикарбонил)пирролидин-3-ил)окси)-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетата 7d (130 мг, выход 46,4%) в виде бесцветного масла.
МС m/z (ИЭР): 488,2 [М-99]
Стадия 4
2-((S)-6-((4'-(((S)-1-(трет-бутоксикарбонил)пирролидин-3-ил)окси)-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусная кислота
Метил 2-(((S)-6-((4'-((S)-1-(трет-бутоксикарбонил)пирролидин-3-ил)окси)-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетат 7d (30 мг, 0,05 ммоль) растворяли в 4 мл смеси растворителей тетрагидрофурана и метанола (об./об. = 1:1), с последующим добавлением 1 M водного раствора гидроксида лития (0,1 мл, 0,10 ммоль). Реакционный раствор перемешивали в течение 2 часов. Полученный раствор концентрировали при пониженном давлении, добавляли по каплям 1 M соляной кислоты для доведения рН до 5 и экстрагировали этилацетатом (20 мл × 2). Объединенные органические экстракты промывали насыщенным раствором хлорида натрия (30 мл), сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении и остаток очищали с помощью тонкослойной хроматографии с системой элюирования А, с получением указанного в заголовке соединения 2-((S)-6-((4'-(((S)-1-(трет-бутоксикарбонил)пирролидин-3-ил)окси)-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусной кислоты 7 (10 мг, выход 34,4%) в виде белого твердого вещества.
МС m/z (ИЭР): 572,3 [М-1]
1Н ЯМР (400 МГц, CDCl3) δ 7,37-7,45 (м, 2Н), 7,18 (с, 1Н), 7,05-7,10 (м, 2Н), 6,63 (с, 2Н), 6,47-6,51 (м, 2Н), 5,07 (с, 2Н), 4,91 (с, 1Н), 4,75-4,79 (м, 1Н), 4,28-4,32 (м, 1Н), 3,80-3,86 (м, 1Н), 3,52-3,67 (м, 3Н), 2,78-2,84 (м, 1Н), 2,59-2,65 (м, 1Н), 2,59-2,66 (м, 1Н), 2,22-2,24 (м, 1Н), 2,00-2,03 (м, 7Н), 1,49 (с, 9Н).
Пример 8
2-((S)-6-((2',6'-диметил-4'-(((S)-1-(метилсульфонил)пирролидин-3-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусная кислота

Стадия 1
Метил 2-(S)-6-((2',6'-диметил-4'-((S)-пирролидин-3-илокси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетат
Метил 2-((S)-6-((4'-((S)-1-(трет-бутоксикарбонил)пирролидин-3-ил)окси)-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетат 7d (100 мг, 0,17 ммоль) растворяли в 3 мл дихлорметана с последующим добавлением 2 мл трифторуксусной кислоты в бане со льдом. Реакционный раствор перемешивали в течение 1 часа. Полученный раствор концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения метил 2-((S)-6-((2',6'-диметил-4'-((S)-пирролидин-3-илокси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетата 8а (90 мг) в виде бесцветного масла, которое непосредственно использовали на следующей стадии без дополнительной очистки.
Стадия 2
Метил 2-((S)-6-((2',6'-диметил-4'-(((S)-1-(метилсульфонил)пирролидин-3-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетат
Неочищенный метил 2-((S)-6-((2',6'-диметил-4'-((S)-пирролидин-3-илокси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетат 8а (45 мг, 0.09 ммоль) растворяли в 3 мл дихлорметана с последующим добавлением метансульфонилхлорида (13 мг, 0,11 ммоль) и триэтиламина (18 мг, 0,18 ммоль). Реакционный раствор перемешивали в течение 2 часов. В полученный раствор добавляли 30 мл этилацетата. Органическую фазу промывали насыщенным раствором хлорида натрия (30 мл), сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении и остаток очищали с помощью тонкослойной хроматографии с системой элюирования А с получением указанного в заголовке соединения метил 2-((S)-6-((2',6'-диметил-4'-(((S)-1-(метилсульфонил)пирролидин-3-ил)окси) бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетата 8b (40 мг, выход 77,0%) в виде бесцветного масла.
МС m/z (ИЭР): 583,4 [М+18]
Стадия 3
2-((S)-6-((2',6'-диметил-4'-(((S)-1-(метилсульфонил)пирролидин-3-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусная кислота
Метил 2-((S)-6-((2',6'-диметил-4'-(((S)-1-(метилсульфонил)пирролидин-3-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетат 8b (30 мг, 0.05 ммоль) растворяли в 2 мл смеси растворителей тетрагидрофурана и метанола (об./об. = 1:1) с последующим добавлением 1 M водного раствора гидроксида лития (0,1 мл, 0,10 ммоль). Реакционный раствор перемешивали в течение 2 часов. Полученный раствор концентрировали при пониженном давлении, добавляли по каплям 1 M раствор соляной кислоты для доведения рН до 5 и экстрагировали этилацетатом (20 мл × 2). Объединенные органические экстракты промывали насыщенным раствором хлорида натрия (30 мл), сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения 2-((S)-6-((2',6'-диметил-4'-(((S)-1-(метилсульфонил)пирролидин-3-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусной кислоты 8 (28 мг, выход 94,2%) в виде белого твердого вещества.
МС m/z (ИЭР): 550,2 [М-1]
1Н ЯМР (400 МГц, CDCl3) δ 7,38-7,46 (м, 2Н), 7,17 (с, 1Н), 7,05-7,09 (м, 2Н), 6,59 (с, 2Н), 6,47-6,52 (м, 2Н), 5,07 (с, 2Н), 4,96 (с, 1Н), 4,75-4,79 (м, 1Н), 4,28-4,32 (м, 1Н), 3,80-3,86 (м, 1Н), 3,61-3,67 (м, 3Н), 3,45-3,52 (м, 1Н), 2,88 (с, 3Н), 2,79-2,84 (м, 1Н), 2,59-2,66 (м, 1Н), 2,33-2,38 (м, 1Н), 2,17-2,25 (м, 1Н), 1,99 (с, 6Н).
Пример 9
2-((S)-6-((4'-(((S)-1-ацетилпирролидон-3-ил)окси)-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)-уксусная кислота

Стадия 1
Метил 2-((S)-6-((4'-(((S)-1-ацетилпирролидин-3-ил)окси)-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетат
Неочищенный метил 2-((S)-6-((2',6-диметил-4'-((S)-пирролидин-3-илокси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетат 8а (45 мг, 0,09 ммоль) растворяли в 3 мл дихлорметана с последующим добавлением уксусного ангидрида (13 мг, 0,11 ммоль) и триэтиламина (18 мг, 0,18 ммоль). Реакционный аствор перемешивали в течение 2 часов. В полученный раствор добавляли 20 мл дихлорметана. Органическую фазу промывали насыщенным раствором хлорида натрия (30 мл), сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке неочищенного соединения метил 2-((S)-6-((4'-(((S)-1-ацетилпирролидин-3-ил)окси)-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетат 9а (45 мг, выход 92,5%) в виде бесцветного масла.
МС m/z (ИЭР): 530,4 [М+1]
Стадия 2
2-((S)-6-((4'-(((S)-1-ацетилпирролидин-3-ил)окси)-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусная кислота
Неочищенный метил 2-((S)-6-((4'-(((S)-1-ацетил пирролидин-3-ил)окси)-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетат 9а (40 мг, 0,08 ммоль) растворяли в 2 мл смеси растворителей тетрагидрофурана и метанола (об./об. = 1:1) с последующим добавлением 1 M водного раствора гидроксида лития (0,4 мл, 0,40 ммоль). Реакционный раствор перемешивали в течение 3 часов. Полученный раствор концентрировали при пониженном давлении. К остатку добавляли 10 мл воды, добавляли по каплям 1 M раствор соляной кислоты для доведения рН до 3 и экстрагировали этилацетатом (20 мл × 2). Объединенные органические экстракты промывали насыщенным раствором хлорида натрия (30 мл), сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении и остаток очищали с помощью тонкослойной хроматографии с системой элюирования А, с получением указанного в заголовке соединения 2-((S)-6-((4'-(((S)-1-ацетилпирролидин-3-ил)окси)-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусной кислоты 9 (20 мг, выход 51,3%) в виде белого твердого вещества.
МС m/z (ИЭР): 514,3 [М-1]
1Н ЯМР (400 МГц, CDCl3) δ 7,41 (дд, 2Н), 7,15 (д, 1Н), 7,05 (д, 2Н), 6,61 (д, 2Н), 6,52-6,42 (м, 2Н), 5,06 (с, 2Н), 4,97 (д, 1Н), 4,76 (дд, 1Н), 4,28 (дд, 1Н), 3,93-3,56 (м, 5Н), 2,79 (дд, 1Н), 2,60 (дд, 1Н), 2,40-2,17 (м, 2Н), 2,16-2,07 (м, 3Н), 2,02-1,92 (м, 6H).
Пример 10
(S)-2-(6-((4'-(циклопентилокси)-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусная кислота

Стадия 1
Циклопентанол
Циклопентанон 10а (1 г, 0,01 моль) растворяли в 10 мл метанола с последующим добавлением боргидрида натрия (542 мг, 0,01 ммоль) в бане со льдом. Реакционный раствор нагревали до комнатной температуры и перемешивали в течение 2 часов. В полученный раствор добавляли 10 мл воды и экстрагировали этилацетатом (20 мл × 2). Объединенные органические экстракты промывали насыщенным раствором хлорида натрия (30 мл), сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного целевого соединения 10b циклопентанола (1 г) в виде бесцветного масла, которое непосредственно использовали на следующей стадии без дополнительной очистки.
Стадия 2
Циклопентил метансульфонат
Неочищенный циклопентанол 10b (50 мг, 0.58 ммоль), метансульфонилхлорид (80 мг, 0,70 ммоль) и триэтиламин (117 мг, 1,16 ммоль) растворяли в 10 мл дихлорметана. Реакционный раствор перемешивали в течение 2 часов. В полученный раствор добавляли 10 мл воды и экстрагировали этилацетатом (20 мл × 2). Объединенные органические экстракты промывали насыщенным раствором хлорида натрия (30 мл), сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного целевого соединения циклопентил метансульфоната 10 с (90 мг) в виде желтого масла, которое непосредственно использовали на следующей стадии без дополнительной очистки.
Стадия 3
(S)-Метил 2-(6-((4'-(циклопентилокси)-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетат
Циклопентил метансульфонат 10 с (50 мг, 0,28 ммоль), (S)-метил 2-(6-((4'-гидрокси-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетат 3h (60 мг, 0,14 ммоль) и карбонат цезия (100 мг, 0,30 ммоль) растворяли в 10 мл N,N-диметилформамида. Реакционную смесь нагревали до 80°C и перемешивали в течение 12 часов. В полученную смесь добавляли 10 мл воды и экстрагировали этилацетатом (20 мл × 2). Органическую фазу промывали водой (20 × 2 мл) и насыщенным раствором хлорида натрия (20 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и остаток очищали с помощью системы элюирования В, с получением указанного в заголовке соединения (S)-метил 2-(6-((4'-(циклопентилокси)-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетат 10d (40 мг, выход 30.0%) в виде бесцветной слизи.
МС m/z (ИЭР): 487,3 [М+1]
Стадия 4
(S)-2-(6-((4'-(циклопентилокси)-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусная кислота
(S)-Метил 2-(6-((4'-(циклопентилокси)-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетат 10d (40 мг, 0,08 ммоль) растворяли в 7 мл смеси растворителей тетрагидрофурана и метанола (об./об. = 2:5) с последующим добавлением 1 M водного раствора гидроксида лития (0.8 мл, 0.80 ммоль). Реакционный раствор перемешивали в течение 2 часов. Полученный раствор концентрировали при пониженном давлении. К остатку добавляли 10 мл воды, добавляли по каплям 1 M раствор соляной кислоты для доведения рН до 4-5 и экстрагировали этилацетатом (20 мл × 2).
Объединенные органические экстракты промывали насыщенным раствором хлорида натрия (30 мл), сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (S)-2-(6-((4'-(циклопентилокси)-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусной кислоты 10 (30 мг, выход 80,0%) в виде белого твердого вещества.
МС m/z (ИЭР): 471,2 [М-1]
1Н ЯМР (400 МГц, CDCl3) δ 7,38-7,42 (м, 2Н), 7,18 (с, 1Н), 7,05-7,11 (м, 2Н), 6,63 (с, 2Н), 6,47-6,51 (м, 2Н), 5,07 (с, 2Н), 4,74-4,79 (м, 2Н), 4,27-4,31 (м, 1Н), 3,75-4,90 (м, 1Н), 2,79-2,81 (м, 1Н), 2,63-2,66 (м, 2Н), 1,99 (с, 6Н), 1,89-1,92 (м, 6Н), 1,62-1,82 (м, 2Н).
Пример 11
2-((S)-6-((2',6'-диметил-4'-(((R)-1-(метилсульфонил)пирролидин-3-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусная кислота

Стадия 1
(S)-3-гидроксил-пирролидин гидрохлорид
(S)-трет-бутил-3-гидроксипирролидин-1-карбоксилат 11а (1,87 г, 10 ммоль) растворяли в 20 мл хлористоводородной кислоты в диоксане. Реакционный раствор перемешивали в течение 0,5 часов. Полученный раствор концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения (S)-3-гидроксил-пирролидина гидрохлорид 11b (0,98 г) в виде белого твердого вещества, которое непосредственно использовали на следующей стадии без дополнительной очистки.
Стадия 2
(S)-1-(метилсульфонил)пирролидин-3-ил метансульфонат
Неочищенный (S)-3-гидроксил-пирролидина гидрохлорид 11b (124 мг, 1 ммоль) растворяли в 15 мл дихлорметана с последующим добавлением триэтиламина (303 мг, 3 ммоль). Реакционный раствор охлаждали до 0°C с последующим добавлением метансульфонилхлорида (264 мг, 2,30 ммоль), затем нагревали до комнатной температуры и перемешивали еще в течение 2,5 часов. В полученный раствор добавляли 10 мл дихлорметана, добавляли по каплям 1 M раствор лимонной кислоты для доведения рН до 4-5, органическую фазу сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения (S)-1-(метилсульфонил)пирролидин-3-ил метансульфоната 11с (153 мг) в виде желтого твердого вещества, которое непосредственно использовали на следующей стадии без дополнительной очистки.
Стадия 3
Метил 2-((S)-6-((2',6-диметил-4'-(((R)-1-(метилсульфонил)пирролидин-3-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетат
(S)-1-(метилсульфонил)пирролидин-3-ил метансульфонат 11с (41 мг, 0,17 ммоль), (S)-метил 2-(6-((4'-гидрокси-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетат 3h (70 мг, 0,17 ммоль) и карбонат цезия (165 мг, 0,50 ммоль) растворяли в 10 мл N,N-диметилформамида. Реакционный раствор нагревали до 80°C и перемешивали в течение 12 часов. Полученный раствор концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле с системой элюирования В, с получением указанного в заголовке соединения метил 2-((S)-6-((2',6'-диметил-4'-(((R)-1-(метилсульфонил)пирролидин-3-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетат 11d (71 мг, выход 75,1%) в виде светло-желтой слизи.
МС m/z (ИЭР): 583,3 [М+18]
Стадия 4
2-((S)-6-((2',6'-диметил-4'-(((R)-1-(метилсульфонил)пирролидин-3-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусная кислота
Метил 2-((S)-6-((2',6'-диметил-4'-(((R)-1-(метилсульфонил)пирролидин-3-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетат 11d (71 мг, 0,13 ммоль) растворяли в 10 мл метанола, с последующим добавлением 2 водного раствора гидроксида натрия M (0,5 мл, 1 ммоль). Реакционный раствор перемешивали в течение 2 часов. В полученный раствор добавляли по каплям 1 M раствор лимонной кислоты для доведения рН до 4-5. Органическую фазу сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения 2-((S)-6-((2',6'-диметил-4'-(((R)-1-(метилсульфонил)пирролидин-3-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробнзофуран-3-ил)уксусной кислоты 11 (70 мг, выход 100%) в виде желтого твердого вещества.
МС m/z (ИЭР): 550,3 [М-1]
1Н ЯМР (400 МГц, CDCl3) δ 7,53-7,37 (м, 2Н), 7,20 (с, 1Н), 7,11 (т, 2Н), 6,62 (с, 2Н), 6,58-6,47 (м, 2Н), 5,10 (с, 2Н), 4,99 (с, 1Н), 4,81 (т, 1Н), 4,33 (дд, 1Н), 3,85 (дт, 1Н), 3,75-3,61 (м, 3Н), 3,58-3,45 (м, 1Н), 2,92 (с, 3Н), 2,85 (дд, 1Н), 2,66 (дд, 1Н), 2,39 (дд, 1Н), 2,32-2,15 (м, 1Н), 2,02 (д, 6Н).
Пример 12
2-((S)-6-((2,2',6'-триметил-4'-(((S)-тетрагидрофуран-3-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусная кислота

Стадия 1
(4-(бензилокси)-2,6-диметилфенил)бороновая кислота
5-(бензилокси)-2-бром-1,3-диметилбензол 12а (2,91 г, 10 ммоль, полученный способом, описанным в патентной заявке WO 2005019151) растворяли в 35 мл тетрагидрофурана в бане с сухим льдом с последующим добавлением н-бутиллития (4,8 мл, 12 ммоль). Реакционный раствор перемешивали 1,5 часа, добавляли трипропил борат (5,64 г, 30 ммоль), затем нагревали до 30°C и перемешивали в течение 12 часов. В полученный раствор добавляли по каплям 10 мл раствора 2 M соляной кислоты, затем охлаждали до комнатной температуры и перемешивали еще в течение 2 часов. Полученный раствор концентрировали при пониженном давлении и фильтровали. Осадок на фильтре последовательно промывали водой (10 мл) и н-гексаном (10 мл) с получением указанного в заголовке соединения (4-(бензилокси)-2,6-диметилфенил) 12b бороновой кислоты (1,54 г, выход 60,2%) в виде белого твердого вещества.
МС m/z (ИЭР): 257,2 [М+1]
Стадия 2
(4'-(бензилокси)-2,2',6'-триметилбифенил-3-ил)метанол 2-метил-3-бром-фенилметанол (201 мг, 1 ммоль, полученный способом, описанным в патентной заявке WO 2010143733), (4-(бензилокси)-2,6-диметилфенил) 12b бороновую кислоту (300 мг, 1.20 ммоль), 1 мл 2 M водный раствор карбоната натрия, 2-дициклогексилфосфино-2',6'-диметокси-1,1'-бифенил (33 мг, 0,08 ммоль) и трис(дибензилиденацетон)дипалладий (18 мг, 0,02 ммоль) растворяли в 1 мл N,N-диметилформамида. Реакционную смесь подвергали реакции при 120°C в условиях микроволн в течение 1 часа. В полученную смесь добавляли 10 мл воды и экстрагировали этилацетатом (20 мл × 2). Объединенные органические экстракты промывали насыщенным раствором хлорида натрия (30 мл), сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле с системой элюирования В, с получением указанного в заголовке соединения (4'-(бензилокси)-2,2',6'-триметилбифенил-3-ил)метанола 12 с (190 мг, выход 57,2%) в виде красной слизи.
МС m/z (ИЭР): 333,3 [М+1]
Стадия 3
3'-(гидроксиметил)-2,2',6-триметилбифенил-4-ол
(4'-(бензилокси)-2,2',6'-триметилбифенил-3-ил)метанол 12с (190 мг, 0,57 ммоль) растворяли в 5 мл метанола, с последующим добавлением Pd/C (20 мг, 10%) в атмосфере водорода. Реакционный раствор перемешивали в течение 12 часов. Полученный раствор фильтровали и фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 3'-(гидроксиметил)-2,2',6-триметилбифенил-4-ол 12d (128 мг) в виде желтого твердого вещества, которое непосредственно использовали на следующей стадии без дополнительной очистки.
МС m/z (ИЭР): 241,2 [М-1]
Стадия 4
(S)-(2,2',6'-триметил-4'-((тетрагидрофуран-3-ил)окси)бифенил-3-ил)метанол
Неочищенный (R)-тетрагидрофуран-3-ил метансульфонат 1b (69 мг, 0,41 ммоль), неочищенный 3'-(гидроксиметил)-2,2',6-триметилбифенил-4-ол 12d (100 мг, 3,13 ммоль) и карбонат цезия (403 мг, 1,24 ммоль) растворяли в 315 мл N,N-диметилформамида. Реакционную смесь нагревали до 80°C и перемешивали в течение 12 часов. Полученный раствор концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле с системой элюирования В, с получением указанного в заголовке соединения (S)-(2,2',6'-триметил-4'-((тетрагидрофуран-3-ил)окси)бифенил-3-ил) метанола 12е (110 мг, выход 86,0%) в виде не совсем белого твердого вещества.
МС m/z (ИЭР): 313,2 [М+1]
Стадия 5
Метил 2-((S)-6-((2,2',6'-триметил-4'-(((S)-тетрагидрофуран-3-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетат
(S)-(2,2',6'-триметил-4'-((тетрагидрофуран-3-ил)окси)бифенил-3-ил)метанол 12е (75 мг, 0,24 ммоль), метил (S)-2-(6-гидроксил-2,3-дигидробензофуран-3-ил)ацетат (50 мг, 0,24 ммоль) и трифенилфосфин (94 мг, 0,36 ммоль) растворяли в 20 мл дихлорметана. Реакционный раствор охлаждали до 0°C с последующим добавлением диизопропилазодикарбоксилата (73 мг, 0,36 ммоль), затем нагревали до комнатной температуры и перемешивали в течение 2 часов. Полученный раствор концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле с системой элюирования В, с получением указанного в заголовке соединения метил 2-((S)-6-((2,2',6'-триметил-4'-(((S)-тетрагидрофуран-3-ил)окси) бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетата 12f (100 мг, выход 83,0%) в виде белого твердого вещества.
МС m/z (ИЭР): 503,4 [М+1]
Стадия 6
2-((S)-6-((2,2','-триметил-4'-(((S)-тетрагидрофуран-3-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусная кислота
Метил 2-((S)-6-((2,2',6'-триметил-4'-(((S)-тетрагидрофуран-3-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетат 12f (100 мг, 0.20 ммоль) растворяли в 4 мл смеси растворителей тетрагидрофурана и метанола (об./об. = 1:1), с последующим добавлением 2 водного раствора гидроксида лития M (1 мл, 2 ммоль). Реакционный раствор перемешивали в течение 2 часов. Полученный раствор концентрировали при пониженном давлении. К остатку добавляли 10 мл воды, добавляли по каплям 1 M раствор соляной кислоты для доведения рН до 1-2 и экстрагировали этилацетатом (20 мл × 2). Объединенные органические экстракты промывали насыщенным раствором хлорида натрия (30 мл), сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали, используя метод препаративного разделения с получением указанного в заголовке соединения 2-((S)-6-((2,2',6'-триметил-4'-(((S)-тетрагидрофуран-3-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусной кислоты 12 (80 мг, выход 83,0%) в виде белого твердого вещества.
МС m/z (ИЭР): 487,2 [М-1]
1Н ЯМР (400 МГц, CDCl3) δ 7,41-7,43 (д, 1Н), 7,24-7,26 (д, 1Н), 7,08-7,10 (д, 1Н), 7,02-7,04 (д, 1Н), 6,64 (с, 2Н), 6,51-6,56 (м, 2Н), 5,05 (с, 2Н), 4,96-4.,97 (м, 1Н), 4,77-4,81 (м, 1Н), 4,30-4,31 (м, 1Н), 4,03-4,07 (м, 3Н), 3,92-3,97 (м, 1Н), 3,83-3,85 (м, 1Н), 2,81-2,87 (м, 1Н), 2,61-2,68 (м, 1Н), 2,20-2,25 (м, 2Н), 1,97 (с, 3Н), 1,93 (с, 6Н), 1,27-1,30 (м, 1Н).
Пример 13
2-((S)-6-((4'-(((3R,4R)/(3S,4S)-4-гидрокситетрагидрофурана-3-ил)окси)-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусная кислота
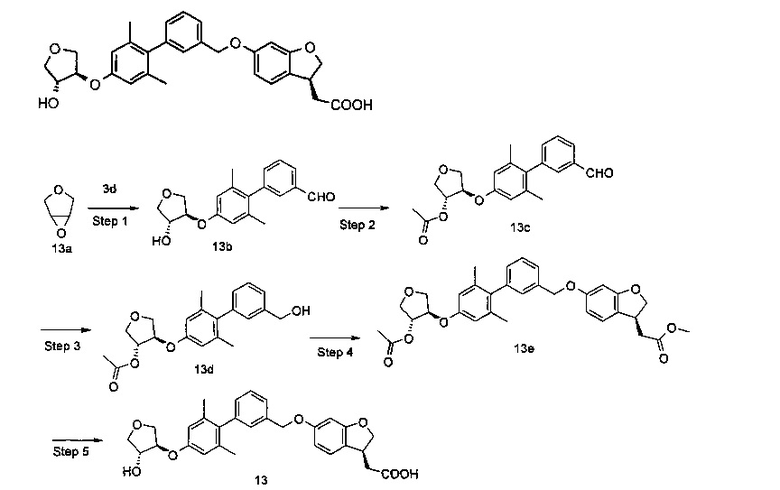
Стадия 1
4'-((3R,4R)/(3S,4S)-4-гидрокситетрагидрофуран-3-ил)окси)-2',6'-диметилбифенил-3-карбальдегид
4'-гидрокси-2',6'-диметилбифенил-3-карбальдегид 3d (226 мг, 1 ммоль), 3,4-эпокситетрагидрофуран 13а (600 мг, 6,97 ммоль) и карбонат калия (180 мг, 1,30 ммоль) растворяли в 1 мл этанола. Реакционную смесь подвергали реакции при 100°C в течение 90 минут в условиях микроволн. В полученную смесь добавляли 10 мл этилацетата, фильтровали и промывали этилацетатом (10 мл × 3). Фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле с системой элюирования В, с получением указанного в заголовке соединения 4'-(((3R,4R)/(3S,4S)-4-гидрокситетрагидрофуран-3-ил)окси)-2',6'-диметилбифенил-3-карбальдегида 13b (312 мг, выход 100%) в виде желтой слизи.
МС m/z (ИЭР): 313,1 [М+1]
Стадия 2
(3R,4R)/(3S,4S)-4-((3-формил-2,6-диметилбифенил-4-ил)окси)-тетрагидрофуран-3-ил ацетат
4'-(((3R,4R)/(3S,4S)-4-гидрокситетрагидрофуран-3-ил)окси)-2',6'-диметилбифенил-3-карбальдегид 13b (312 мг, 1 ммоль) растворяли в 5 мл дихлорметана в бане со льдом с последующим добавлением триэтиламина (0,3 мл, 2 ммоль) и ацетилхлорид (0,1 мл, 1,50 ммоль) последовательно. Реакционный раствор нагревают до комнатной температуры и перемешивают в течение 30 минут. В полученный раствор добавляли 10 мл воды, экстрагировали дихлорметаном (10 мл × 3). Объединенную органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле с системой элюирования В, с получением указанного в заголовке соединения (3R,4R)/(3S,4S)-4-((3-формил-2,6-диметилбифенил-4-ил)окси)-тетрагидрофуран-3-илацетата 13с (280 мг, выход 79.1%) в виде бесцветной слизи.
МС m/z (ИЭР): 372,3 [М+18]
Стадия 3
(3R,4R)/(3S,4S)-4-((3-(гидроксиметил)-2,6-диметилбифенил-4-ил)окси)-тетрагидрофуран-3-ил ацетат
(3R,4R)-4-((3-формил-2,6-диметилбифенил-4-ил)окси)тетрагидрофуран-3-ил ацетат 13 с (280 мг, 0,79 ммоль) растворяли в 5 мл метанола. Реакционную смесь охлаждали до 0°C с последующим добавлением боргидрида натрия (45 мг, 1,20 ммоль), затем нагревали до комнатной температуры и перемешивали в течение 30 минут. В полученную смесь добавляли 5 мл ацетона и концентрировали при пониженном давлении. К остатку добавляли 10 мл воды, разделяли и водную фазу экстрагировали этилацетатом (20 мл × 3). Объединенную органическую фазу промывали насыщенным раствором хлорида натрия (10 мл × 3), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле с системой элюирования В, получая указанное в заголовке соединение (3R,4R)/(3S,4S)-4-((3-(гидроксиметил)-2,6-диметилбифенил-4-ил)окси)тетрагидрофуран-3-ил ацетат 13d (200 мг, выход 70,9%) в виде бесцветной слизи.
МС m/z (ИЭР): 357,2 [М+1]
Стадия 4
Метил 2-((S)-6-((4'((3R,4R)/(3S,4S)-4-ацетокситетрагидрофуран-3-ил)окси)-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил) ацетат
(3R,4R)/(3S,4S)-4-((3-(гидроксиметил)-2,6-диметилбифенил-4-ил)окси)тетрагидрофуран-3-ил ацетат 13d (200 мг, 0,56 ммоль), метил (S)-2-(6-гидроксил-2,3-дигидробензофуран-3-ил)ацетат (117 мг, 0,56 ммоль) и трифенилфосфин (221 мг, 0,84 ммоль) растворяли в 5 мл дихлорметана. Реакционный раствор охлаждали до 0°C с последующим добавлением диизопропилазодикарбоксилата (170 мг, 0,84 ммоль). Реакционный раствор нагревали до комнатной температуры и перемешивали в течение 3 часов. Полученный раствор концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле с системой элюирования В, с получением указанного в заголовке соединения метил 2-((S)-6-((4'-(((3R,4R)/(3S,4S)-4-ацетокситетрагидрофуран-3-ил)окси)-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил) ацетата 13е (200 мг, выход 65,1%) в виде бесцветной слизи.
МС m/z (ИЭР): 564,3 [М+18]
Стадия 5
2-((S)-6-((4'-(((3R,4R)/(3S,4S)-4-гидрокситетрагидрофуран-3-ил)окси)-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусная кислота
Метил 2-((S)-6-((4'-(((3R,4R)/(3S,4S)-4-ацетокситетрагидрофуран-3-ил)окси)-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил) ацетат 13е (200 мг, 0,37 ммоль) растворяли в 4 мл смеси растворителей из дихлорметана и метанола (об.:об. = 1:3) с последующим добавлением 3 водного раствора гидроксида лития M (0,5 мл, 1,50 ммоль). Реакционный раствор перемешивали в течение 12 часов. Полученный раствор концентрировали при пониженном давлении. К остатку добавляли 10 мл воды, добавляли по каплям 1 M раствор соляной кислоты для доведения рН до 2-3 и экстрагировали этилацетатом (20 мл × 2). Объединенные органические экстракты промывали насыщенным раствором хлорида натрия (30 мл), сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле с системой элюирования А, с получением указанного в заголовке соединения 2-((S)-6-((4'-(((3R,4R)/(3S,4S)-4-гидрокситетрагидрофурана-3-ил)окси)-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусной кислоты 13 (110 мг, выход 61,4%) в виде белого твердого продукта.
МС m/z (ИЭР): 508,3 [М+18]
1Н ЯМР (400 МГц, CDCl3) δ 7,40 (м, 2Н), 7,16 (с, 1Н), 7,06 (м, 2Н), 6,66 (с, 2Н), 6,48 (м, 2Н), 5,06 (с, 2Н), 4,76 (м, 2Н), 4,47 (д, 1Н), 4,29 (м, 2Н), 4,07 (дд, 1Н), 3,95 (дд, 1Н), 3,86 (д, 1Н), 3,80 (м, 1Н), 2,80 (дд, 1Н), 2,61 (дд, 1Н), 1,99 (с, 6Н).
Пример 14
(S)-2-(6-((2',6'-диметил-4'-((1-пропионилазетидин-3-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусная кислота

Стадия 1
(S)-трет-бутил 3-((3'-(((3-(2-метокси-2-оксоэтил)-2,3-дигидробензофуран-6-ил)окси)метил)-2,6-диметилбифенил-4-ил)окси)азетидин-1-карбоксилат
Трет-бутил 3-((метилсульфонил))окси)азетидин-1-карбоксилат 14а (100 мг, 0,40 ммоль, полученный способом, описанным в WO 2011097958), (S)-метил 2-(6-((4'-гидрокси-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетат 3h (80 мг, 0,19 ммоль) и карбонат цезия (130 мг, 0,40 ммоль) растворяли в 10 мл из N,N-диметилформамида. Реакционный раствор нагревали до 80°C и перемешивали в течение 12 часов. В полученный раствор добавляли 10 мл воды, экстрагировали этилацетатом (30 мл × 2). Объединенные органические экстракты промывали насыщенным раствором хлорида натрия (30 мл), сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле с системой элюирования В, с получением указанного в заголовке соединения (S)-трет-бутил 3-((3'-(((3-(2-метокси-2-оксоэтил)-2,3-дигидробензофуран-6-ил)окси)метил)-2,6-диметилбифенил-4-ил)окси)азетидин-1-карбоксилата 14b (60 мг, выход 50,0%) в виде бесцветного масла.
Стадия 2
(S)-Метил 2-(6-((4'-(азетидин-3-илокси)-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил) ацетат 2,2,2-трифторацетат
(S)-трет-бутил 3-((3'-(((3-(2-метокси-2-оксоэтил)-2,3-дигидробензофуран-6-ил)окси)метил)-2,6-диметилбифенил-4-ил)окси)азетидин-1-карбоксилат 14b (100 мг, 0,17 ммоль) растворяли в 5 мл дихлорметана с последующим добавлением 1 мл трифторуксусной кислоты. Реакционный раствор перемешивали в течение 2 часов. Полученный раствор концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения (S)-метил 2-(6-((4'-(азетидин-3-илокси)-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил) ацетат 2,2,2-трифторацетата 14с (80 мг) в виде коричневого масла, которое непосредственно использовали на следующей стадии без дополнительной очистки.
МС m/z (ИЭР): 474.2 [М+1]
Стадия 3
(S)-метил 2-(6-((2',6'-диметил-4'-((1-пропионилазетидин-3-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетат
Неочищенный (S)-метил 2-(6-((4'-(азетидин-3-илокси)-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетат 2,2,2-трифторацетат 14с (40 мг, 0,09 ммоль) растворяли в 3 мл дихлорметана с последующим добавлением последовательно триэтиламина (0,1 мл, 0.26 ммоль) и хлорида пропионовой кислоты (11 мл, 0,13 ммоль). Реакционный раствор перемешивали в течение 12 часов. Полученный раствор добавляли в 10 мл дихлорметана, промывали насыщенным раствором хлорида натрия (5 мл), сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения (S)-метил 2-(6-((2',6'-диметил-4'-((1-пропионилазетидин-3-ил)окси)-бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетат 14d (45 мг) в виде желтого масла, которое непосредственно использовали на следующей стадии без дополнительной очистки.
МС m/z (ИЭР): 474,2 [М+1]
Стадия 4
(S)-2-(6-((2',6'-диметил-4'-((1-пропионилазетидин-3-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусная кислота
Неочищенный (S)-метил 2-(6-((2',6'-диметил-4'-((1-пропионилазетидин-3-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетат 14d (45 мг, 0,09 ммоль) растворяли в 3 мл метанола с последующим добавлением 1 M водного раствора гидроксида лития (0,5 мл, 0,50 ммоль). Реакционный раствор перемешивали в течение 3 часов. В полученный раствор добавляли по каплям 1 M раствор соляной кислоты для доведения рН до 3, добавляли 3 мл воды, концентрировали при пониженном давлении и экстрагировали этилацетатом (20 мл × 2). Объединенные органические экстракты промывали насыщенным раствором хлорида натрия (30 мл), сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (S)-2-(6-((2',6'-диметил-4'-((1-пропионилазетидин-3-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусной кислоты 14 (20 мг, выход 43,0%) в виде желтого твердого вещества.
МС m/z (ИЭР): 514,3 [М-1]
1Н ЯМР (400 МГц, CDCl3) δ 7,43-7,34 (м, 2Н), 7,16 (с, 1Н), 7,08-7,05 (м, 2Н), 6,50-6,46 (м, 4Н), 5,05 (с, 2Н), 5,06-5,03 (м, 1Н), 4,82-4,70 (м, 1Н), 4,65-4,38 (м, 2Н), 4,28-4,16 (м, 3Н); 3,87-3,77 (м, 1Н); 2,79-2,78 (м, 1Н), 2,65-2,62 (м, 1Н), 2,19-2,17 (д, 2Н), 1,99 (с, 6Н), 1,16 (т, 3Н).
Пример 15
(S)-2-(6-((4'-((1-(циклопропанкарбонил)азетидин-3-ил)окси)-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусная кислота

Стадия 1
(S)-Метил 2-(6-((4'-((1-(циклопропанкарбонил)азетидин-3-ил)окси)-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетат
Неочищенный (S)-метил 2-(6-((4'-(азетидин-3-илокси)-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетат 2,2,2-трифторацетат 14с (40 мг, 0.09 ммоль) растворяли в 3 мл дихлорметана с последующим добавлением триэтиламина (0.1 мл, 0.26 ммоль) и хлорида циклопропанкарбонилхлорида 15а (14 мг, 0.13 ммоль). Реакционный раствор перемешивали в течение 12 часов. В полученный раствор добавляли 10 мл дихлорметана, промывали насыщенным раствором хлорида натрия (5 мл), сушили над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения (S)-метил 2-(6-((4'-((1-(циклопропанкарбонил)азетидин-3-ил)окси)-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетата 15b (46 мг) в виде красного масла, которое непосредственно использовали на следующей стадии без дополнительной очистки.
МС m/z (ИЭР): 542,2 [М+1]
Стадия 2
(S)-2-(6-((4'-((1-(циклопропанкарбонил)азетидин-3-ил)окси)-2',6'-диметил-бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусная кислота
Неочищенный (S)-метил 2-(6-((4'-((1-(циклопропанкарбонил)азетидин-3-ил)окси)-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетат 15b (46 мг, 0,09 ммоль) растворяли в 3 мл метанола с последующим добавлением 1 M водного раствора гидроксида лития (0,5 мл, 0,50 ммоль). Реакционный раствор перемешивали в течение 3 часов. Полученный раствор концентрировали при пониженном давлении, добавляли по каплям 1 M раствор соляной кислоты для доведения рН до 3, добавляли 3 мл воды и экстрагировали этилацетатом (20 мл × 2). Объединенные органические экстракты промывали насыщенным раствором хлорида натрия (30 мл), сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (S)-2-(6-((4'-((1-(циклопропанкарбонил)азетидин-3-ил)окси)-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусной кислоты 15 (20 мг, выход 44,0%) в виде желтого твердого вещества.
МС m/z (ИЭР): 526.3 [М-1]
1Н ЯМР (400 МГц, CDCl3) δ 7,43-7,34 (м, 2Н), 7,16 (с, 1Н), 7,08-7,05 (м, 2Н), 6,50-6,46 (м, 4Н), 5,05 (с, 2Н), 5,06-5,03 (м, 1Н), 4,82-4,70 (м, 1Н), 4,65-4,38 (м, 2Н), 4,28-4,16 (м, 3Н), 3,87-3,77 (м, 1Н), 2,79-2,78 (м, 1Н), 2,65-2,62 (м, 1Н), 1,99 (с, 6Н), 1,16 (м, 1Н), 0,89 (м, 2Н), 0,64 (м, 2Н).
Пример 16
(S)-2-(6-((4'-(азетидин-3-илокси)-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусная кислота

Стадия 1
2-цианоацетил хлорид
2-цианоуксусную кислоту, 16а (25 мг, 0,30 ммоль) растворяли в 10 мл дихлорметана с последующим добавлением оксалилхлорида (47 мг, 0,45 ммоль), капли N,N-диметилформамида. Реакционный раствор перемешивали в течение 30 минут. Полученный раствор концентрировали при пониженном давлении с получением неочищенного 2-цианоацетил хлорида 16b (30 мг) в виде красного масла, которое непосредственно использовали на следующей стадии без дополнительной очистки.
Стадия 2
(S)-Метил 2-(6-((4'-((1-(2-цианоацетил)азетидин-3-ил)окси)-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетат
(S)-Метил 2-(6-((4'-(азетидин-3-илокси)-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетат 2,2,2-трифторацетат 14с (150 мг, 0,32 ммоль) растворяли в 10 мл дихлорметана с последующим добавлением триэтиламина (65 мг, 0,64 ммоль) и неочищенного 2-цианоацетил хлорида 16b (49 мг, 0,48 ммоль). Реакционный раствор перемешивали в течение 16 часов. В полученный раствор добавляли 10 мл воды и экстрагировали этилацетатом (20 мл × 2). Объединенные органические экстракты промывали насыщенным раствором хлорида натрия (5 мл), сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении и остаток очищали с помощью тонкослойной хроматографии с системой элюирования А с получением неочищенного указанного в заголовке соединения (S)-метил 2-(6-((4'-((1-(2-цианоацетил)азетидин-3-ил)окси)-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетата 16с (80 мг, выход 46,2%) в виде желтого твердого вещества.
МС m/z (ИЭР): 558,3 [М+18]
Стадия 3
(S)-2-(6-((4'-(азетидин-3-илокси)-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусная кислота
(S)-Метил 2-(6-((4'-((1-(2-цианоацетил)азетидин-3-ил)окси)-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетат 16с (80 мг, 0.15 ммоль) растворяли в 2 мл смеси растворителей тетрагидрофурана и метанола (об./об. = 1:1) с последующим добавлением 1М водного раствора гидроксида лития (1 мл, 1 ммоль). Реакционный раствор перемешивали в течение 2 часов. Полученный раствор концентрировали при пониженном давлении. К остатку добавляли 10 мл воды, добавляли по каплям 1 M раствор соляной кислоты для доведения рН до 3 и экстрагировали этилацетатом (20 мл × 2). Объединенные органические экстракты промывали насыщенным раствором хлорида натрия (30 мл), сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле с системой элюирования В, получая указанное в заголовке соединение (S)-2-(6-((4'-(азетидин-3-илокси)-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусной кислоты 16 (65 мг, выход 95,6%) в виде светло-желтого твердого вещества.
МС m/z (ИЭР): 458,2 [М-1]
1Н ЯМР (400 МГц, DMSO) δ 7,47-7,43 (м, 1Н), 7,41-7,37 (м, 1Н), 7,15-7,09 (м, 2Н), 7,05 (д, 1Н), 6,63 (с, 2Н), 6,50-6,43 (м, 2Н), 5,10 (с, 2Н), 5,07-5,03 (м, 1Н), 4,73-4,65 (м, 1Н), 4,50-4,41 (м, 2Н), 4,22-4,15 (м, 1Н), 4,01-3,96 (м, 2Н), 3,74-3,62 (м, 1Н), 2,75-2,65 (м, 1Н), 2,50-2,43 (м, 1Н), 1,93 (с, 6Н).
Пример 17
2-((S)-6-((4'-(((1R,2R)/(1S,2S)-2-гидроксициклопентил)окси)-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусная кислота
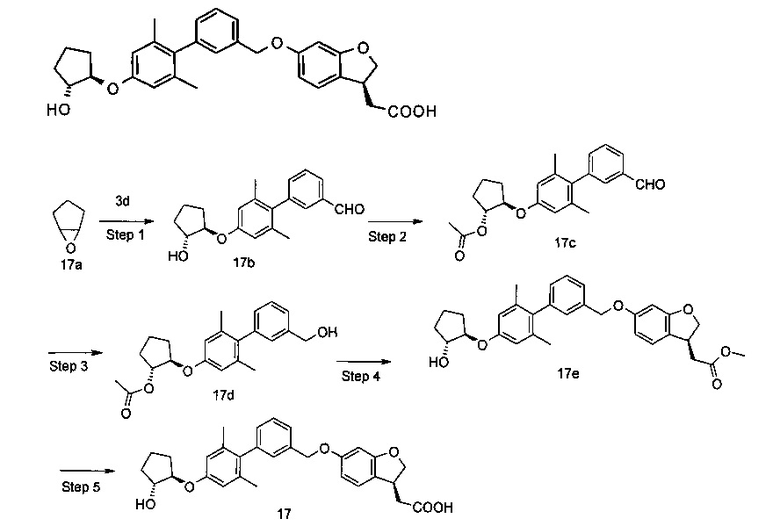
Стадия 1
4'-(((1R,2R)/(1S,2S)-2-гидроксициклопентил)окси)-2',6'-диметилбифенил-3-карбальдегид
4'-Гидрокси-2',6'-диметилбифенил-3-карбальдегид 3d (113 мг, 0.50 ммоль), 1,2-эпоксициклопентан 17а (300 мг, 3.57 ммоль) и карбонат калия (90 мг, 0.65 ммоль) растворяли в 0,5 мл этанола. Реакционную смесь подвергали реакции при 100 C в течение 75 минут в условиях микроволн. В полученную смесь добавляли 15 мл этилацетата и фильтровали. Фильтрат промывали водой (10 мл × 3), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 4'-(((1R,2R)/(1S,2S)-2-гидроксициклопентил)окси)-2',6'-диметилбифенил-3-карбальдегид 17b (180 мг) в виде желтой слизи, которую непосредственно использовали на следующей стадии без дополнительной очистки.
МС m/z (ИЭР): 311,3 [М+1]
Стадия 2
(1R,2R)/(1S,2S)-2-((3-формил-2,6-диметилбифенил-4-ил)окси)циклопентил ацетат
Неочищенный 4'-(((1R,2R)/(1S,2S)-2-гидроксициклопентил)окси)-2',6'-диметилбифенил-3-карбальдегид 17b (155 мг, 0,50 ммоль) растворяли в 15 мл дихлорметана. В реакционный раствор последовательно добавляли триэтиламин (0,1 мл, 1 ммоль) и ацетилхлорид (61 мг, 0,60 ммоль). Реакционный раствор перемешивали в течение 2 часов. В полученный раствор добавляли 10 мл дихлорметана, промывали водой (10 мл × 3), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и остаток очищали с помощью системы элюирования В, с получением указанного в заголовке соединения (1R,2R)/(1S,2S)-2-((3'-формил-2,6-диметилбифенил-4-ил)окси)циклопентил ацетата 17 с (71 мг, выход 40,3%) в виде белого твердого вещества.
Стадия 3
(1R,2R)/(1S,2S)-2-((3'-(гидроксиметил)-2,6-диметилбифенил-4-ил)окси)циклопентил ацетат
(1R,2R)/(1S,2S)-2-((3'-формил-2,6-диметилбифенил-4-ил)окси)циклопентил ацетат 17с (71 мг, 0,20 ммоль) растворяли в 10 мл метанола с последующим добавлением боргидрида натрия (15 мг, 0,40 ммоль). Реакционный раствор перемешивали в течение 2 часов. В полученный раствор добавляли 6 мл ацетона и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле с системой элюирования В, с получением указанного в заголовке соединения (1R,2R)/(1S,2S)-2-((3'-(гидроксиметил)-2,6-диметилбифенил-4-ил)окси)циклопентил ацетата 17d (61 мг, выход 85,2%) в виде бесцветной слизи.
МС m/z (ИЭР): 355,3 [М+1]
Стадия 4
Метил 2-((S)-6-((4'-(((1R,2R)/(1S,2S)-2-гидроксициклопентил)окси)-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетат
(1R,2R)/(1S,2S)-2-((3'-(гидроксиметил)-2,6-диметилбифенил-4-ил)окси)циклопентил ацетат 17d (61 мг, 0,17 ммоль), метил (S)-2-(6-гидроксил-2,3-дигидробензофуран-3-ил)ацетат (36 мг, 0,17 ммоль) и трифенилфосфин (68 мг, 0.26 ммоль) растворяли в 10 мл дихлорметана. Реакционный раствор охлаждали до 0°C с последующим добавлением диизопропилазодикарбоксилата (53 мг, 0,26 ммоль). Реакционный раствор нагревали до комнатной температуры и перемешивали в течение 1.5 часов. Полученный раствор концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле с системой элюирования В, с получением указанное в заголовке соединения метил 2-((S)-6-((4'-((1R,2R)/(1S,2S)-2-гидроксициклопентил)окси)-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусная кислота 17е (64 мг, выход 69,0%) в виде бесцветной слизи.
МС m/z (ИЭР): 520,4 [М+18]
Стадия 5
2-((S)-6-((4'-(((1R,2R)/(1S,2S)-2-гидроксициклопентил)окси)-2',6'-иметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусная кислота
Метил 2-((S)-6-((4'-(((1R,2R)/(1S,2S)-2-гидроксициклопентил)окси)-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетат 17е (54 мг, 0,10 ммоль) растворяли в 10 мл метанола, с последующим добавлением 2 M водного раствора гидроксида лития (0,5 мл, 1 ммоль). Реакционный раствор перемешивали в течение 2 часов. Полученный раствор концентрировали при пониженном давлении. К остатку добавляли 3 мл воды, добавляли по каплям 1 M раствор лимонной кислоты для доведения рН до 2-3 и экстрагировали этилацетатом (20 мл × 2). Объединенные органические экстракты промывали насыщенным раствором хлорида натрия (30 мл), сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения 2-((S)-6-(((4'-((1R,2R)/(1S,2S)-2-гидроксициклопентил)окси)-2',6'-диметилбифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусной кислоты 17 (21 мг, выход 43,0%) в виде светло- желтого твердого вещества.
МС m/z (ИЭР): 506,4 [М+18]
1Н ЯМР (400 МГц, CDCl3) δ 7,41-7,48 (м, 2Н), 7,.22 (с, 1Н), 7,04-7,14 (м, 2Н), 6,66 (с, 2Н), 6,51-6,55 (м, 2Н), 5,10 (с, 2Н), 4,78-4,83 (с, 1Н), 3,96-3,99 (м, 1Н), 3,72-3,86 (м, 1Н), 2,83-2,88 (м, 1Н), 2,63-2,69 (м, 1Н), 2,24-2,76 (м, 2Н), 2,03 (с, 6Н), 1,96-2,06 (м, 2Н), 1,61-1,86 (м, 2Н), 1,46-1,56 (м, 2Н).
Пример 18
2-((S)-6-((4-фтор-2',6'-диметил-4'-(((S)-тетрагидрофуран-3-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусная кислота

Стадия 1
4'-(бензилокси)-4-фтор-2',6'-диметилбифенил-3-карбальдегид
(4-(бензилокси)-2,6-диметилфенил) 12b бороновую кислоту (256 мг, 1 ммоль), 2-фтор-5-бром-бензальдегид (203 мг, 1 ммоль), 2 M водный раствор карбоната натрия (1 мл, 2 ммоль), 2-дициклогексилфосфино-2',6'-диметоксибифенил (37 мг, 0,08 ммоль) и трис(дибензилиденацетон)дипалладий (18 мг, 0,02 ммоль) растворяли в 3 мл смеси растворителей из диметилового эфира этиленгликоля и толуола (об./об. = 1:2). Реакционную смесь нагревали при 100°C в течение 2 часов, а затем при 120°C в течение 0,5 часов в условиях микроволн. В полученную смесь добавляли 10 мл воды и экстрагировали этилацетатом (10 мл × 3). Объединенные органические экстракты промывали насыщенным раствором хлорида натрия (30 мл), сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле с системой элюирования В с получением указанного в заголовке соединения 4'-(бензилокси)-4-фтор-2',6'-диметилбифенил-3-карбальдегида 18а (390 мг, выход 58,4%) в виде бесцветного масла.
МС m/z (ИЭР): 335,2 [М+1]
Стадия 2
(4'-(бензилокси)-4-фтор-2',6'-диметилбифенил-3-ил)метанол
4'-(бензилокси)-4-фтор-2',6'-диметилбифенил-3-карбальдегид 18а (390 мг, 1,17 ммоль) растворяли в 5 мл метанола с последующим добавлением боргидрида натрия (66 мг, 1,75 ммоль). Реакционный раствор перемешивали в течение 30 минут. В полученный раствор добавляли 10 мл ацетона и концентрировали при пониженном давлении. К остатку добавляли 10 мл воды и экстрагировали этилацетатом (10 мл × 3). Объединенные органические экстракты промывали насыщенным раствором хлорида натрия (30 мл), сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения (4'-(бензилокси)-4-фтор-2',6'-диметилбифенил-3-ил)метанол 18b (370 мг) в виде бесцветной слизи, который был непосредственно использовали на следующей стадии без дополнительной очистки.
Стадия 3
4'-фтор-3'-(гидроксиметил)-2,6-диметилбифенил-4-ол
Неочищенный(4'-(бензилокси)-4-фтор-2',6'-диметилбифенил-3-ил)метанол 18b (370 мг, 1.10 ммоль) растворяли в 5 мл метанола, с последующим добавлением Pd/C (40 мг, 10%) в атмосфере водорода. Реакционную смесь перемешивали в течение 12 часов. Полученную смесь фильтровали. Фильтрат концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле с системой элюирования В, с получением указанного в заголовке соединения 4-фтор-3'-(гидроксиметил)-2,6-диметилбифенил-4-ола 18с (225 мг, выход 83,3%) в виде белого твердого вещества.
МС m/z (ИЭР): 245,2 [М-1]
Стадия 4
(S)-(4-фтор-2',6'-диметил-4'-((тетрагидрофуран-3-ил)окси)бифенил-3-ил)метанол
Неочищенный (R)-тетрагидрофуран-3-ил метансульфонат 1b (83 мг, 0.50 ммоль), 4'-фтор-3'-(гидроксиметил)-2,6-диметилбифенил-4-ол 18с (110 мг, 0,45 ммоль) и карбонат цезия (293 мг, 0,90 ммоль) растворяли в 2 мл N,N-диметилформамида. Реакционную смесь нагревали до 90°C и перемешивали в течение 12 часов. Полученную смесь концентрировали при пониженном давлении, добавляли 10 мл воды и экстрагировали этилацетатом (10 мл × 3). Объединенные органические экстракты промывали насыщенным раствором хлорида натрия (30 мл), сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении с получением сырого указанного в заголовке соединения (S)-(4-фтор-2',6'-диметил-4'-((тетрагидрофуран-3-ил)окси)бифенил-3-ил)метанола 18d (150 мг) в виде бесцветной слизи, который непосредственно использовали на следующей стадии без дополнительной очистки.
МС m/z (ИЭР): 317,1 [М+1]
Стадия 5
Метил 2-((S)-6-((4-фтор-2',6'-диметил-4,-(((S)-тетрагидрофуран-3-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил) ацетат
Неочищенный (S)-(4-фтор-2',6'-диметил-4'-((тетрагидрофуран-3-ил)окси)бифенил-3-ил)метанол 18d (150 мг, 0.45 ммоль), метил(S)-2-(6-гидроксил-2,3-дигидробензофуран-3-ил)ацетат (94 мг, 0.45 ммоль) и трифенилфосфин (177 мг, 0,68 ммоль) растворяли в 5 мл дихлорметана. Реакционный раствор охлаждали до 0°C с последующим добавлением диизопропилазодикарбоксилата (136 мг, 0,68 ммоль). Реакционный раствор нагревали до комнатной температуры и перемешивали в течение 12 часов. Полученный раствор концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле с системой элюирования В, с получением указанного в заголовке соединения метил 2-((S)-6-((4-фтор-2',6'-диметил-4'-(((S)-тетрагидрофуран-3-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетата 18е (150 мг, выход 65.8%) в виде бесцветной слизи.
МС m/z (ИЭР): 507,2 [М+1]
Стадия 6
2-((S)-6-((4-фтор-2',6'-диметил-4'-(((S)-тетрагидрофуран-3-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусная кислота
Метил 2-((S)-6-((4-фтор-2',6'-диметил-4'-(((S)-тетрагидрофуран-3-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетат 18е (150 мг, 0,30 ммоль) растворяли в 4,5 мл смеси растворителя из тетрагидрофурана, метанола и воды (об./об. = 1:3:0,5) с последующим добавлением 1 M водного раствора гидроксида лития (1 мл, 1 ммоль). Реакционный раствор перемешивали в течение 12 часов. Полученный раствор концентрировали при пониженном давлении. К остатку добавляли 10 мл воды, добавляли по каплям 2 M раствор соляной кислоты для доведения рН до 2-3 и экстрагировали этилацетатом (20 мл × 2). Объединенные органические экстракты промывали насыщенным раствором хлорида натрия (30 мл), сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении и остаток очищали с помощью тонкослойной хроматографии с системой элюирования А, с получением указанного в заголовке соединения 2-((S)-6-((4-фтор-2',6'-диметил-4'-(((S)-тетрагидрофуран-3-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусной кислоты 18 (100 мг, выход 66,7%) в виде белого твердого вещества.
МС m/z (ИЭР): 491,3 [М-1]
1Н ЯМР (400 МГц, CDCl3) δ 7,23 (дд, 1Н), 7,14-7,04 (м, 3Н), 6,60 (с, 2Н), 6,50-6,45 (м, 2Н), 5,13 (с, 2Н), 4,94 (м, 1Н), 4,76 (т, 1Н), 4,28 (дд, 1Н), 4,02-3,89 (м, 4Н), 3,80 (м, 1Н), 2,80 (дд, 1Н), 2,61 (дд, 1Н), 2,20 (м, 2Н), 1,96 (с, 6Н).
Пример 19
2-((S)-6-((3'-фтор-2',6'-диметил-4'-(((S)-тетрагидрофуран-3-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусная кислота

Стадия 1
3-фтор-3'-(гидроксиметил)-2,6-диметилбифенил-4-ол
3'-фтор-4'-гидрокси-2',6'-диметилбифенил-3-карбальдегид 19а (700 мг, 2.87 ммоль, полученный способом, описанным в патентной заявке WO 2008001931) растворяли в 10 мл метанола с последующим добавлением боргидрида натрия (130 мг, 3,44 ммоль). Реакционный раствор перемешивали в течение 2 часов. Полученный раствор концентрировали при пониженном давлении. К остатку добавляли 10 мл воды и экстрагировали этилацетатом (50 мл × 2). Объединенные органические экстракты промывали насыщенным раствором хлорида натрия (30 мл), сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 3-фтор-3'-(гидроксиметил)-2,6-диметилбифенил-4-ола 19b (700 мг) в виде белого твердого вещества, которое непосредственно использовали на следующей стадии без дополнительной очистки МС m/z (ИЭР): 247,1 [М+1]
Стадия 2
(S)-(3'-фтор-2',6'-диметил-4'-((тетрагидрофуран-3-ил)окси)бифенил-3-ил)метанол
Неочищенный (R)-тетрагидрофуран-3-ил метансульфонат 1b (202 мг, 1.22 ммоль), неочищенный 3-фтор-3'-(гидроксиметил)-2,6-диметилбифенил-4-ол 19b (200 мг, 0,81 ммоль) и карбонат цезия (527 мг, 1,62 ммоль) растворяли в 5 мл N,N-диметилформамида. Реакционную смесь нагревали до 80°C и перемешивали в течение 12 часов. В полученную смесь добавляли 20 мл воды и экстрагировали этилацетатом (30 мл × 2). Объединенную органическую фазу промывали водой (30 мл), насыщенным раствором хлорида натрия (30 мл), сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле с системой элюирования В, с получением указанного в заголовке соединения (S)-(3'-фтор-2',6'-диметил-4'-((тетрагидрофуран-3-ил)окси)бифенил-3-ил)метанола 19с (130 мг, выход 50,8%) в виде бесцветной слизи.
МС m/z (ИЭР): 334,2 [М+18]
Стадия 3
Метил 2-((S)-6-((3'-фтор-2',6'-диметил-4'-(((S)-тетрагидрофуран-3-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетат
(S)-(3'-фтор-2',6'-диметил-4'-((тетрагидрофуран-3-ил)окси)бифенил-3-ил)метанол 19с (130 мг, 0.41 ммоль), метил-(S)-2-(6-гидроксил-2,3-дигидробензофуран-3-ил)ацетат (85 мг, 0.41 ммоль) и трифенилфосфин (161 мг, 0.62 ммоль) растворяли в 10 мл дихлорметана с последующим добавлением диизопропилазодикарбоксилата (125 мг, 0.62 ммоль). Реакционный раствор перемешивали в течение 2 часов. Полученный раствор концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле с системой элюирования В, с получением указанного в заголовке соединения метил 2-((S)-6-((3'-фтор-2',6'-диметил-4'-(((S)-тетрагидрофуран-3-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетата 19d (150 мг, выход 72.5%) в виде бесцветного масла.
МС m/z (ИЭР): 507,2 [М+1]
Стадия 4
2-((S)-6-((3'-фтор-2',6'-диметил-4'-(((S)-тетрагидрофуран-3-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусная кислота
Метил 2-((S)-6-((3'-фтор-2',6'-диметил-4'-(((S)-тетрагидрофуран-3-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетат 19d (150 мг, 0.30 ммоль) растворяли в 4 мл смеси растворителей из метанола и тетрагидрофурана (об./об. = 1:1) с последующим добавлением 1 M водного раствора гидроксида лития (1,5 мл, 1,50 ммоль). Реакционный раствор перемешивали в течение 2 часов. Полученный раствор концентрировали при пониженном давлении, добавляли 10 мл воды, добавляли по каплям 1 M соляной кислоты для доведения рН до 2-3 и экстрагировали этилацетатом (20 мл × 2). Объединенные органические экстракты промывали насыщенным раствором хлорида натрия (30 мл), сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении и остаток очищали с помощью тонкослойной хроматографии с системой элюирования А, с получением указанного в заголовке соединения 2-((S)-6-((3'-фтор-2',6'-диметил-4'-(((S)-тетрагидрофуран-3-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусной кислоты 19 (85 мг, выход 57.8%) в виде белого твердого вещества.
МС m/z (ИЭР): 491,2 [М-1]
1Н ЯМР (400 МГц, CDCl3) δ 7,52-7,41 (м, 2Н), 7,19 (с, 1Н), 7,10 (д, 2Н), 6,70 (д, 1Н), 6,58-6,52 (м, 1Н), 6,51 (с, 1Н), 5,10 (с, 2Н), 5,07-4,96 (м, 2Н), 4.83-4,78 (м, 1Н), 4,38-4,29 (м, 1Н), 4,14-4,05 (м, 3Н), 4,02-3.95 (м, 1Н), 2,90-2.79 (м, 1Н), 2,70-2,60 (м, 1Н), 2,31-2,20 (м, 2Н), 2,00 (с, 3Н), 1,96 (д, 3Н).
Соединения из примеров 20-24 были получены с использованием соответствующих реагентов в соответствии со способами, описанными в примере 3 и 5.
№ примера, структура и характеристика данных перечислены ниже:



Соединения из примеров 25-34 были получены с использованием соответствующих реагентов в соответствии со способами, описанными в примерах 1 и 18.
№ примера, структура и характеристика данных перечислены ниже:



Пример 35
2-((S)-6-((2'-(гидроксиметил)-6'-метил-4'-(((R)-тетрагидрофуран-3-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил) уксусная кислота

Стадия 1
(R)-3-(4-бром-3,5-диметилфенокси)тетрагидрофуран
(S)-тетрагидрофуран-3-ол 2а (1 г, 11,35 ммоль) растворяли в 30 мл дихлорметана с последующим добавлением 4-бром-3,5-диметилфенола (2,28 г, 11,35 ммоль). Реакционный раствор охлаждали до 0°C с последующим последовательным добавлением трифенилфосфина (3,57 г, 13,62 ммоль) и диизопропилазодикарбоксилата (2,75 г, 13,62 ммоль). Реакционный раствор нагревали до комнатной температуры и перемешивали в течение 12 часов. Полученный раствор концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле с системой элюирования В, с получением указанного в заголовке соединения (R)-3-(4-бром-3,5-диметилфенокси)тетрагидрофурана 35а (2,27 г, выход 73,9%) в виде бесцветного масла.
Стадия 2
(R)-3-(4-бром-3-(бромметил)-5-метилфенокси)тетрагидрофуран
(R)-3-(4-бром-3,5-диметилфенокси)тетрагидрофуран 35а (2.27 г, 8.37 ммоль), N-бромсукцинимид (1,79 г, 10,04 ммоль) и азобисизобутиронитрил (64 мг, 0,42 ммоль) растворяли в 50 мл четыреххлористого углерода. Реакционный раствор нагревали до 60°C и перемешивали в течение 12 часов. Полученный раствор концентрировали при пониженном давлении, добавляли 50 мл трет-бутил метилового эфира, перемешивали в течение 10 минут, фильтровали и промывали трет-бутилметиловым эфиром (20 мл × 2). Объединенный фильтрат концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле с системой элюирования В, с получением указанного в заголовке соединения (R)-3-(4-бром-3-(бромметил)-5-метилфенокси)тетрагидрофуран 35b (707 мг, выход 24,2%) в виде светло-желтого твердого вещества.
Стадия 3
(R)-2-бром-3-метил-5-((тетрагидрофуран-3-ил)окси)бензил ацетат
(R)-3-(4-бром-3-(бромметил)-5-метилфенокси)тетрагидрофуран 35b (700 мг, 2 ммоль) растворяли в 15 мл N,N-диметилформамида, с последующим добавлением ацетата калия (197 мг, 2 ммоль). Реакционный раствор нагревали до 50°C и перемешивали в течение 1 часа. Полученный раствор охлаждали до комнатной температуры, добавляли 100 мл воды и экстрагировали этилацетатом (20 мл × 2). Объединенные органические экстракты промывали насыщенным раствором хлорида натрия (30 мл), сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения (R)-2-бром-3-метил-5-((тетрагидрофуран-3-ил)окси)-бензил ацетата 35 с (658 мг) в виде желтого масла, которое непосредственно использовали на следующей стадии без дополнительной очистки.
МС m/z (ИЭР): 346,3 [М+18]
Стадия 4
(R)-(3'-гидроксиметил)-6-метил-4-((тетрагидрофуран-3-ил)окси)бифенил-2-ил)метил ацетат
Неочищенный (R)-2-бром-3-метил-5-((тетрагидрофуран-3-ил)окси)-бензил ацетат 35с (658 мг, 2 ммоль) растворяли в 50 мл диоксана с последующим добавлением 3-гидроксиметил фенилбороновой кислоты (608 мг, 4 ммоль) и 6 мл водного раствора карбоната калия (636 мг, 4 ммоль), а затем [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия (II) (74 мг, 0,10 ммоль). Реакционный раствор нагревали до 70°C и перемешивали в течение 4 часов. Полученный раствор охлаждали, выливали в 100 мл 1 M водного раствора хлористоводородной кислоты и экстрагировали этилацетатом (30 мл × 3). Объединенные органические экстракты промывали насыщенным раствором хлорида натрия (30 мл), сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле с системой элюирования В, с получением указанного в заголовке соединения (R)-(3'-(гидроксиметил)-6-метил-4-((тетрагидрофуран-3-ил)окси)бифенил-2-ил)метил ацетата 35d (476 мг, выход 66,8%) в виде желтого масла.
МС m/z (ИЭР): 374,4 [М+18]
Стадия 5
Метил 2-((S)-6-((2'-(ацетоксиметил)-6'-метил-4'-(((R)-тетрагидрофуран-3-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил) ацетат
(R)-(3'-(гидроксиметил)-6-метил-4-((тетрагидрофуран-3-ил)окси)бифенил-2-ил)метил ацетат 35d (127 мг, 0,36 ммоль) растворяли в 10 мл дихлорметана, с последующим добавлением метилового эфира (S)-2-(6-гидроксил-2,3-дигидробензофуран-3-ил) ацетата (89 мг, 0,43 ммоль) и трифенилфосфина (112 мг, 0,43 ммоль). Реакционный раствор охлаждали до 0°C с последующим добавлением диизопропилазодикарбоксилата (87 мг, 0,43 ммоль). Реакционный раствор нагревали до комнатной температуры и перемешивали в течение 12 часов. Полученный раствор концентрировали при пониженном давлении с получением указанного в заголовке неочищенного соединения метил 2-((S)-6-((2'-(ацетоксиметил)-6'-метил-4'-(((R)-тетрагидрофуран-3-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетата 35е (189 мг) в виде желтого масла, которое непосредственно использовали на следующей стадии без дополнительной очистки.
Стадия 6
2-((S)-6-((2'-(гидроксиметил)-6'-метил-4'-(((R)-тетрагидрофуран-3-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусная кислота
Неочищенный метил 2-((S)-6-((2'-(ацетоксиметил)-6'-метил-4'-(((R)-тетрагидрофуран-3-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетат 35е (189 мг, 0.36 ммоль) растворяли в 10 мл смеси растворителей из метанола и тетрагидрофурана (об./об. = 1:4), с последующим добавлением 1 M водного раствора гидроксида лития (4 мл, 4 ммоль). Реакционный раствор перемешивали в течение 3 часов. В полученный раствор добавляли 50 мл воды и 5 мл 2 водного раствора гидроксида натрия М. Водную фазу промывали этилацетатом (30 мл), затем добавляли по каплям 1 M раствор соляной кислоты для доведения рН до 2-3 и экстрагировали этилацетатом (20 мл × 2). Объединенные органические экстракты промывали насыщенным раствором хлорида натрия (30 мл), сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении и остаток очищали с помощью тонкослойной хроматографии с системой элюирования А, с получением указанного в заголовке соединения 2-((S)-6-((2'-(гидроксиметил)-6'-метил-4'-(((R)-тетрагидрофуран-3-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусной кислоты 35 (15 мг, выход 8,6%) в виде белого твердого вещества.
МС m/z (ИЭР): 489,2 [М-1]
1Н-ЯМР (400 МГц, DMSO-d6) δ 7,35-7,48 (м, 2Н), 7,03-7,19 (м, 3Н), 6,93 (шир, 1Н), 6,73 (шир., 1Н), 6,.42-6,52 (м, 2Н), 5,08 (с, 2Н), 5,03 (шир., 1Н), 4,68 (т, 1Н), 4,14-4,24 (м, 1Н), 4,07 (с, 2Н), 3,91 (дд, 1Н), 3,73-3,88 (м, 3Н), 3,61-3,73 (м, 1Н), 2,64-2,77 (м, 1Н), 2,46 (д, 1Н), 2,16-2,30 (м, 1Н), 1,95-2,06 (м, 1Н), 1,91 (с, 3Н).
Пример 36
2-((S)-6-((4-гидрокси-2',6'-диметил-4'-(((R)-тетрагидрофуран-3-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусная кислота

Стадия 1
(R)-(4-(бензилокси)-2',6'-диметил-4'-((тетрагидрофуран-3-ил)окси)бифенил-3-ил)метанол
(R)-3-(4-бром-3,5-диметилфенокси)тетрагидрофуран 35а (731 мг, 2,69 ммоль) растворяли в 20 мл диоксана, с последующим добавлением (2-(бензилокси)-5-(4,4,5,5-тетраметил-диоксаборолан-2-ил)фенил)метанола (1,28 г, 3,76 ммоль, полученного способом, описанным в заявке на патент US 2011275797) и 4 мл водного раствора бикарбоната натрия (855 мг, 8,07 ммоль) и [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия (II) (98 мг, 0,14 ммоль). Реакционный раствор нагревали до 70°C и перемешивали в течение 4 часов. Полученный раствор охлаждали и выливали в 100 мл воды, добавляли по каплям 1 M раствор соляной кислоты для доведения рН до 2-3 и экстрагировали этилацетатом (30 мл × 3). Объединенные органические экстракты промывали насыщенным раствором хлорида натрия (30 мл), сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле с системой элюирования В, с получением указанного в заголовке соединения (R)-(4-(бензилокси)-2',6'-диметил-4'-((тетрагидрофуран-3-ил)окси)бифенил-3-ил)метанола 36а (637 мг, выход 63,7%) в виде желтого масла.
Стадия 2
Метил 2-((S)-6-((4-(бензилокси)-2',6'-диметил-4'-(((R)-тетрагидрофуран-3-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетат
(R)-(4-(бензилокси)-2',6'-диметил-4'-((тетрагидрофуран-3-ил)окси)бифенил-3-ил)метанол 36а (637 мг, 1.69 ммоль) растворяли в 20 мл дихлорметана с последующим добавлением метилового эфира (S)-2-(6-гидроксил-2,3-дигидробензофуран-3-ил)ацетата (423 мг, 2.03 ммоль) и трифенилфосфина (532 мг, 2,03 ммоль). Реакционный раствор охлаждали до 0°C с последующим добавлением диизопропилазодикарбоксилата (411 мг, 2,03 ммоль), затем нагревали до комнатной температуры и перемешивали в течение 12 часов. Полученный раствор концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения метил 2-((S)-6-((4-(бензилокси)-2',6'-диметил-4'-(((R)-тетрагидрофуран-3-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетата 36b (1 г) в виде коричневого масла, которое непосредственно использовали на следующей стадии без дополнительной очистки.
Стадия 3
2-((S)-6-((4-(бензилокси)-2',6'-диметил-4'-(((R)-тетрагидрофуран-3-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусная кислота
Неочищенный метил 2-((S)-6-((4-(бензилокси)-2',6'-диметил-4'-(((R)-тетрагидрофуран-3-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)ацетат 36b (1 г, 1.69 ммоль) растворяли в 50 мл смеси растворителей из метанола и тетрагидрофурана (об./об. = 1:4) с последующим добавлением 1 M водного раствора гидроксида лития (20 мл, 20 ммоль). Реакционный раствор перемешивали в течение 2 часов. Полученный раствор концентрировали при пониженном давлении. К остатку добавляли 150 мл воды и 30 мл этилацетата, добавляли по каплям 1 M раствор соляной кислоты для доведения рН до 2-3 и экстрагировали этилацетатом (20 мл × 2). Объединенные органические экстракты промывали насыщенным раствором хлорида натрия (30 мл), сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 2-((S)-6-((4-(бензилокси)-2',6'-диметил-4'-(((R)-тетрагидрофуран-3-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил) уксусной кислоты 36 с (152 мг) в виде коричневого масла, которое непосредственно использовали на следующей стадии без дополнительной очистки.
МС m/z (ИЭР): 579,4 [М-1]
Стадия 4
2-((S)-6-((4-гидрокси-2',6'-диметил-4'-(((R)-тетрагидрофуран-3-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусная кислота
2-((S)-6-((4-(Бензилокси)-2',6'-диметил-4'-(((R)-тетрагидрофуран-3-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусную кислоту 36 с (152 мг, 0.26 ммоль) растворяли в 5 мл этилацетата, с последующим добавлением Pd/C (30 мг, 10%). Реакционный раствор перемешивали в атмосфере водорода в течение 12 часов. Полученный раствор фильтровали. Фильтрат концентрировали при пониженном давлении и остаток очищали с помощью тонкослойной хроматографии с системой элюирования А с получением неочищенного указанного в заголовке соединения 2-((S)-6-((4-гидрокси-2',6'-диметил-4'-(((R)-тетрагидрофуран-3-ил)окси)бифенил-3-ил)метокси)-2,3-дигидробензофуран-3-ил)уксусной кислоты 36 (18 мг, выход 14,1%) в виде белого твердого вещества.
МС m/z (ИЭР): 491,4 [М+1]
1Н-ЯМР (400 МГц, DMSO-d6) δ 12.34 (шир., 1Н), 9,68 (с, 1Н), 7,08 (д, 1Н), 6.96 (с, 1Н), 6.82-6.93 (м, 2Н), 6.63 (с, 2Н), 6.37-6.49 (м, 2Н), 5,00 (с, 3Н), 4,67 (т, 1Н), 4,12-4,22 (м, 1Н), 3.89 (дд, 1Н), 3,71-3,85 (м, 3Н), 3,66 (т, 1Н), 2,68 (дд, 1Н), 2,45 (д, 1Н), 2,19 (дд, 1Н), 1,96 (дд, 1Н), 1,89 (с, 6Н).
Примеры испытаний
Биологический анализ
Пример испытания 1:
Агонистическая активность соединений по настоящему изобретению в клетках CHO-K1/GPR40.
Для определения агонистической активности соединений по настоящему изобретению в GPR40 был использован следующий метод. Процесс эксперимента описан следующим образом:
CHO-K1/GPR40 клетки (клеточная линия СНО-K1 экспрессирующая вектор GPR40, который был сконструирован с использованием метода ретровирусной инфекции, клетки CHO-K1/GPR40, где СНО-K1 клетки были приобретены в Клеточном банке Китайской Академии Наук, каталожный номер GNHa 7; кДНК GPR40 была приобретена в Гуанчжоу FulenGen, Ltd, каталожный номер EX-U0270-M02) засевали на 96-луночный планшет с плотностью инокуляции 25000/на лунку. Луночный планшет инкубировали при 37°C и 5% СО2 в течение 24 часов. Культуральную жидкость удаляли в ходе эксперимента, клетки единожды промывали буферным раствором (1×HBSS (Hank's buffered salt solution - сбалансированный солевой раствор Хенкса) + 20 мМ HEPES (N-2-hydroxyethyl-piperazine-N-2-ethanesulfonic acid - N-2-гидроксиэтилпиперазин-N-2-этансульфоновая кислота) рН 7,4), быстро добавляли в каждую лунку 100 мкл ионного красителя Fluo-4 кальция, инкубировали в течение 30 минут в темноте при 37°C и затем еще в течение 30 минут при комнатной температуре. В ходе детекции, определяли сначала базовое значение для каждой лунки, а затем в лунки добавляли разные концентрации препарата (50 мкл/лунку), определяли уровень флуоресценции. Длина волны возбуждения флуоресценции была 494 нм и длина волны излучения была 516 нм. Увеличение интенсивности флуоресценции было пропорционально уровню ионов кальция в клетках. Уровень клеточного ответа в каждой лунке (максимальное значение флуоресценции - минимальное значение флуоресценции)/Минимальное значение флуоресценции, величины ЕС50 соединений были определены.


Заключение: соединения по настоящему изобретению обладали значительной агонистической активностью в отношении GPR40. Фармакокинетический анализ
1. Цель
Соединения по настоящему изобретению вводили внутрижелудочно для определения концентрации лекарственного средства в плазме крови в различные моменты времени. Фармакокинетическое поведение соединений по настоящему изобретению изучали и оценивали на крысах.
2. Протокол
2.1 Образцы
Соединения из примера 1, примера 2, примера 13 и примера 30
2.2 Экспериментальные животные
16 взрослых здоровых крыс SD, 4 в каждой группе, пополам самцы и самки, разделенные на 4 группы, были приобретены у SINO-BRITSH SIPPR/BK LAB. ANIMAL LTD., СО, License number: SCXK (Шанхай) 2008-0016.
2.3 Приготовление испытываемых соединений
Нужное количество испытываемых соединений были взвешены и добавлены к 0.5% натрий-карбоксиметилцеллюлозе, для приготовления 0.5 мг/мл суспензии с использованием микроволновой печи.
2.4 Введение и сбор образцов
Крысам, после голодания в течение ночи, вводили внутрижелудочно в дозе 5.0 мг/кг, в объеме 10 мл/кг. Образцы крови брали перед введением и после введения в различные моменты времени в течение 24.0 часов, в качестве антикоагулянта использовали гепарин. Отделенные образцы плазмы хранили при -20°C. Крыс кормили через 2 часа после введения.
3. Аналитические методы
Образцы крови (0,1 мл) брали перед введением и через 1,0, 2,0, 3,0, 4,0, 6,0, 8,0, 11,0, 24,0 и 48,0 ч после введения, образцы хранились в пробирках с гепарином, которые центрифугировали в течение 5 минут при 3500 об./мин для отделения плазмы крови. Образцы плазмы хранили при -20°C. Крыс кормили через 2 часа после введения.
Содержание различных тестируемых соединений в образцах плазмы крови крыс после внутрижелудочного введения определяли методом жидкостной хромотографии с тандемной масс-спектрометрией. Линейность метода составляет 1.00-2000 нг/мл. Образец крови анализировали после осаждения белка путем добавления метанола.
4. Фармакокинетические параметры
Фармакокинетические параметры соединений по настоящему изобретению были показаны следующим образом:


Заключение: соединения по настоящему изобретению имели высокую концентрацию и влияние в плазме крови, длительный период полураспада, и хорошие фармакокинетические профили после внутрижелудочного введения крысам.
Фармакодинамический анализ
1. Цель
В качестве подопытных животных были использованы мыши линии ICR (Institute of cancer research - институт раковых исследований), находящиеся на диете с высоким содержанием жиров. Был изучено действие перорально введенной разовой дозы соединений по настоящему изобретению на ПТГ тест (ПТГ - проба на толерантность к глюкозе) у мышей.
2. Испытываемые соединения
Соединения из примера 1, примера 2, примера 13, примера 16, примера 18, примера 19 и примера 26
3. Экспериментальные животные
Здоровые мыши ICR, самцы, были приобретены у SINO-BRITSH SIPPR/BK LAB. ANIMAL LTD., СО, Номер лицензии: SCXK (Шанхай) 2008-0016
4. Подготовка испытываемых соединений
Тестируемые соединения добавляли к водному 0,5%-ному раствору CMC-Na (Sodium carboxymethyl cellulose - карбоксиметилцеллюлоза), для получения суспензии соответствующей концентрации с использованием микроволновой печи.
5. Метод тестирования
5.1 Группирование
После ночного голодания в течение 16 часов, самцов мышей, находящихся на диете с высоким содержанием жиров, взвешивали и определяли базовый уровень глюкозы в крови. Мыши были произвольно разделены на контрольную группу и группу каждого тестируемого соединения по настоящему изобретению в соответствии с уровнем глюкозы в крови.
5.2 Установление дозировки
Дозировка для введения была 20 мг/кг или 40 мг/кг, контрольной группе вводили водный 0,5% раствор CMC-Na.
5.3 Введение
20% раствор глюкозы вводили через 15 минут после внутрижелудочного введения (каждой мыши вводили 0.4 мл)
5.4 Определение уровня глюкозы в крови
Соединения вводили в соответствии с вышеуказанной дозировкой для определения уровня глюкозы в крови (-15 минут).
Через 15 минут после введения, 20% раствор глюкозы вводили в дозе 2 г/кг и определяли уровень глюкозы в крови через 0, 15, 30, 45, 60 и 120 минут у каждой мыши с помощью глюкозометра Roche Accu-Chek.
5.5 Статистические Данные
С использованием программы Excel: среднее значение было рассчитано с использованием AVG; Значение стандартного отклонения рассчитывали с использованием STDEV, значение P рассчитывали с использованием ТТЕСТ.
Формула расчета AUC (площаль под кривой):
AUC=(t15min+t0min)×0,25/2+(t30min+t15min)×0,25/2+(t45min+t30min)×0,25/2+(t60min+t45min)×0,25/2+(t120min+t60min)×1/2, где t0min, t15min, t30min, t45min, t60min, t120min представляли собой уровень глюкозы в крови, определенные в различные моменты времени.
6. Результат эксперимента
Степень снижения AUC, %

Вывод: при пероральной дозировке 20 мг/кг или 40 мг/кг соединения по настоящему изобретению, значительно снижается возрастание уровня глюкозы в крови, вызванное введением глюкозы мышам, получающим корм с высоким содержанием жиров.
Настоящее изобретение относится к полициклическим производным, представленным соединением общей формулы (I), где А представляет собой -О-; L представляет собой -О-; кольцо В представляет собой фенил; R1, R2 и R3 каждый независимо выбран из группы, состоящей из галогена, гидроксила, алкила, где алкил возможно замещен гидроксилом; R4 выбран из группы, состоящей из С3-12циклоалкила, гетероциклила и гетероарила, таких как  , где циклоалкил, гетероциклил или гетероарил каждый возможно замещен одной или несколькими группами, выбранными из группы, состоящей из гидроксила, циано, нитро, алкила, алкокси, циклоалкила, гетероциклила, арила, гетероарила, -C(O)OR5, -OC(O)R5, -C(O)R5 и -S(O)mR5; R5 выбран из группы, состоящей из водорода, алкила, С3-12 циклоалкила, где алкил, циклоалкил каждый возможно замещен гидроксилом; m представляет собой 2; n представляет собой 0; р представляет собой 0 или 1 и q представляет собой 2, 3 или 4, или его рацематам, энантиомерам, диастереомерам и их смеси, и его фармацевтически приемлемым солям, а также к способам его получения, содержащим его фармацевтическим композициям и его применению в качестве терапевтических средств, в частности в качестве агониста GPR40, и к способам лечения и профилактики заболеваний, подобных диабету, метаболическому синдрому и т.д. 12 н. и 4 з.п. ф-лы, 5 табл., 36 пр.
, где циклоалкил, гетероциклил или гетероарил каждый возможно замещен одной или несколькими группами, выбранными из группы, состоящей из гидроксила, циано, нитро, алкила, алкокси, циклоалкила, гетероциклила, арила, гетероарила, -C(O)OR5, -OC(O)R5, -C(O)R5 и -S(O)mR5; R5 выбран из группы, состоящей из водорода, алкила, С3-12 циклоалкила, где алкил, циклоалкил каждый возможно замещен гидроксилом; m представляет собой 2; n представляет собой 0; р представляет собой 0 или 1 и q представляет собой 2, 3 или 4, или его рацематам, энантиомерам, диастереомерам и их смеси, и его фармацевтически приемлемым солям, а также к способам его получения, содержащим его фармацевтическим композициям и его применению в качестве терапевтических средств, в частности в качестве агониста GPR40, и к способам лечения и профилактики заболеваний, подобных диабету, метаболическому синдрому и т.д. 12 н. и 4 з.п. ф-лы, 5 табл., 36 пр.

1. Соединение формулы (I) или его рацемат, энантиомер, диастереомер и их смесь, и его фармацевтически приемлемая соль:

где
А представляет собой -О-;
L представляет собой -О-;
кольцо В представляет собой фенил;
R1, R2 и R3 каждый независимо выбран из группы, состоящей из галогена, гидроксила, алкила, где алкил возможно замещен гидроксилом;
R4 выбран из группы, состоящей из С3-12циклоалкила, гетероциклила и гетероарила, таких как  , где циклоалкил, гетероциклил или гетероарил каждый возможно замещен одной или несколькими группами, выбранными из группы, состоящей из гидроксила, циано, нитро, алкила, алкокси, циклоалкила, гетероциклила, арила, гетероарила, -C(O)OR5, -OC(O)R5, -C(O)R5 и -S(O)mR5;
, где циклоалкил, гетероциклил или гетероарил каждый возможно замещен одной или несколькими группами, выбранными из группы, состоящей из гидроксила, циано, нитро, алкила, алкокси, циклоалкила, гетероциклила, арила, гетероарила, -C(O)OR5, -OC(O)R5, -C(O)R5 и -S(O)mR5;
R5 выбран из группы, состоящей из водорода, алкила, С3-12 циклоалкила, где алкил, циклоалкил каждый возможно замещен гидроксилом;
m представляет собой 2;
n представляет собой 0;
р представляет собой 0 или 1 и
q представляет собой 2, 3 или 4.
2. Соединение формулы (I) или его рацемат, энантиомер, диастереомер и их смесь, и его фармацевтически приемлемая соль, по п. 1, выбранное из соединения формулы (II) или его рацемата, энантиомера, диастереомера и их смеси, и его фармацевтически приемлемой соли:

где А, L, R1-R4, n, р и q являются такими, как определено в п. 1.
3. Соединение формулы (I) или его рацемат, энантиомер, диастереомер и их смесь, и его фармацевтически приемлемая соль, по п. 2, выбранное из соединения формулы (III) или рацемата, энантиомера, диастереомера и их смеси, и его фармацевтически приемлемой соли:

где
кольцо Е выбрано из группы, состоящей из С3-12циклоалкила, гетероциклила и гетероарила, таких как  ;
;
R1 выбран из группы, состоящей из галогена, гидроксила, алкила, где алкил возможно замещен гидроксилом;
R5 выбран из группы, состоящей из водорода, алкила, С3-12циклоалкила, где алкил, циклоалкил каждый возможно замещен гидроксилом;
R8 выбран из группы, состоящей из гидроксила, циано, нитро, алкила, алкокси, циклоалкила, гетероциклила, арила, гетероарила, -C(O)OR5, -OC(O)R5, -C(O)R5 и -S(O)mR5;
m представляет собой 2;
q представляет собой 2, 3 или 4 и
s представляет собой 0, 1, 2 или 3.
4. Соединение формулы (I) или его рацемат, энантиомер, диастереомер и их смесь, и его фармацевтически приемлемая соль, по п. 3, выбранное из соединения формулы (IV) или рацемата, энантиомера, диастереомера и их смеси, и его фармацевтически приемлемой соли:
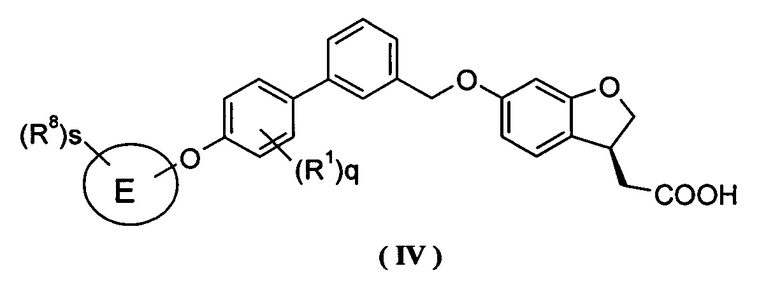
где кольцо Е, R1, R8, s и q являются такими, как определено в п. 3.
5. Соединение формулы (I) или его рацемат, энантиомер, диастереомер и их смесь, и его фармацевтически приемлемая соль, по п. 1, где соединение выбрано из группы, состоящей из:


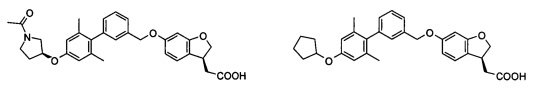

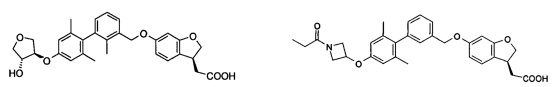
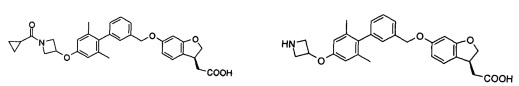






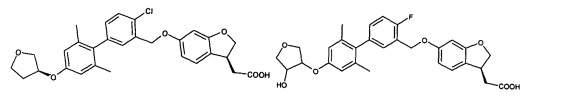

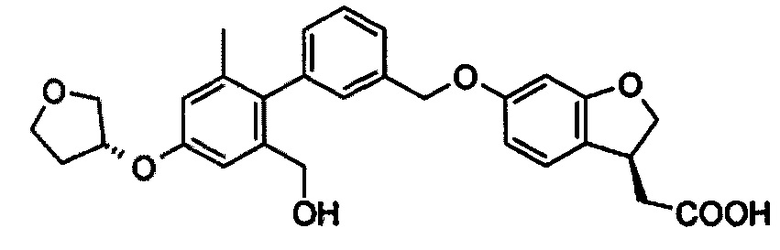 или
или 
6. Соединение формулы (IA):

где R4 выбран из группы, состоящей из С3-12циклоалкила, гетероциклила и гетероарила, таких как 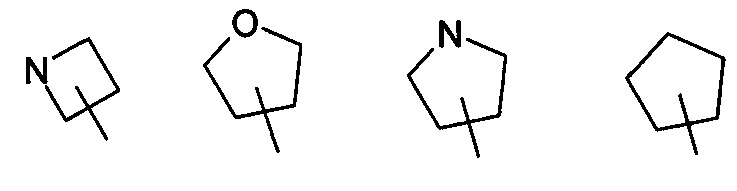 , где циклоалкил, гетероциклил или гетероарил каждый возможно замещен одной или несколькими группами, выбранными из группы, состоящей из гидроксила, циано, нитро, алкила, алкокси, циклоалкила, гетероциклила, арила, гетероарила, -C(O)OR5, -OC(O)R5, -C(O)R5 и -S(O)mR5;
, где циклоалкил, гетероциклил или гетероарил каждый возможно замещен одной или несколькими группами, выбранными из группы, состоящей из гидроксила, циано, нитро, алкила, алкокси, циклоалкила, гетероциклила, арила, гетероарила, -C(O)OR5, -OC(O)R5, -C(O)R5 и -S(O)mR5;
кольцо В, L, R1, R2, R5, m, р и q являются такими, как определено в п. 1.
7. Способ получения соединения формулы (I) или его рацемата, энантиомера, диастереомера и их смеси, и его фармацевтически приемлемой соли, по п. 1, включающий стадии:
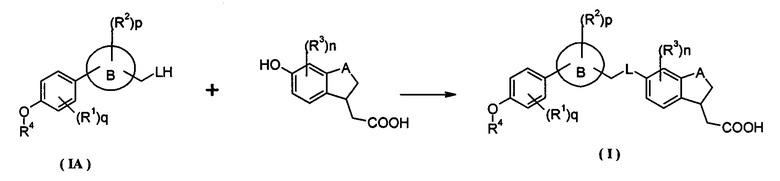
конденсации соединения формулы (IA) с гидроксил-замещенным соединением с бензольным кольцом в растворителе с получением соединения формулы (I);
где R4 является таким, как определено в п. 6;
кольцо В, A, L, R1-R3, n, р и q являются такими, как определено в п. 1.
8. Соединение формулы (IB):

где кольцо В, A, L, R1-R3, n, р и q являются такими, как определено в п. 1.
9. Способ получения соединения формулы (I) или его рацемата, энантиомера, диастереомера и их смеси, и его фармацевтически приемлемой соли, по п. 1, включающий стадии:
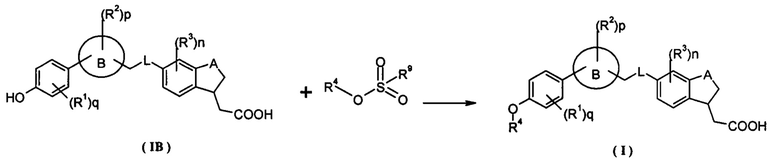
взаимодействия соединения формулы (IB) и R4-замещенного алкилсульфоната в растворителе с получением соединения формулы (I);
где R4 является таким, как определено в п. 6;
кольцо В, A, L, R1-R3, n, р и q являются такими, как определено в п. 1, и R9 представляет собой алкил.
10. Фармацевтическая композиция, обладающая активностью возбуждения рецептора GPR40, содержащая терапевтически эффективное количество соединения формулы (I) или его рацемата, энантиомера, диастереомера и их смеси, и его фармацевтически приемлемой соли, по любому из пп. 1-5, и фармацевтически приемлемый носитель.
11. Применение соединения формулы (I) или его рацемата, энантиомера, диастереоизомера и их смеси, и его фармацевтически приемлемой соли, по любому из пп. 1-5, или фармацевтической композиции по п. 10, в качестве регулятора функции рецептора GPR40.
12. Применение соединения формулы (I) или его рацемата, энантиомера, диастереоизомера и их смеси, и его фармацевтически приемлемой соли, по любому из пп. 1-5, или фармацевтической композиции по п. 10, для предотвращения и лечения диабета и метаболического синдрома, предпочтительно диабет является диабетом II типа.
13. Способ регулирования рецептора GPR40, включающий стадию введения пациенту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы (I) или его рацемата, энантиомера, диастереомера и их смеси, или его фармацевтически приемлемой соли, по любому из пп. 1-5, или фармацевтической композиции по п. 10.
14. Способ профилактики и лечения диабета и метаболического синдрома, включающий стадию введения пациенту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы (I) или его рацемата, энантиомера, диастереомера и их смеси, и его фармацевтически приемлемой соли по любому из пп. 1-5, или фармацевтической композиции по п. 10.
15. Применение соединения формулы (I) или его рацемата, энантиомера, диастереомера и их смеси, и его фармацевтически приемлемой соли, по любому из пп. 1-5, или фармацевтической композиции по п. 10, в качестве агониста GPR40.
16. Способ возбуждения рецептора GPR40, включающий стадию введения пациенту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы (I) или его рацемата, энантиомера, диастереомера и их смеси, и его фармацевтически приемлемой соли, по любому из пп. 1-5, или фармацевтической композиции по п. 10.
| US 20060258722 A1 16.11.2006 | |||
| US 20110313003 A1 23.12.2011 | |||
| US 20070244155 A1 18.10.2007 | |||
| ДИГИДРОБЕНЗОФУРАНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1995 |
|
RU2138498C1 |
| US 20060258722 A1 16.11.2006 | |||
| US 20110313003 A1 23.12.2011 | |||
| US 20070244155 A1 18.10.2007 | |||
| ДИГИДРОБЕНЗОФУРАНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1995 |
|
RU2138498C1 |

