Настоящее изобретение относится к замещенным N-{3-[4-(1-метил-1Н-индол-3-ил)пиримидин-2-иламино]-4-метоксифенил}амидам, их фармацевтически приемлемым солям, сольватам и гидратам, которые могут быть полезны при лечении или профилактике заболевания или медицинского состояния, обусловленных определенными мутантными формами рецептора эпидермального фактора роста (EGFR), например мутанты L858R, Exon19 и Т790М. Такие соединения и их соли могут быть пригодны в лечении или профилактике ряда различных видов рака. Изобретение также относится к фармацевтическим композициям, содержащим указанные соединения и их соли, сольваты и гидраты, полиморфные формы этих соединений и их солей, к способам их получения и к способам лечения заболеваний, обусловленных различными формами EGFR, с помощью указанных соединений и их солей.
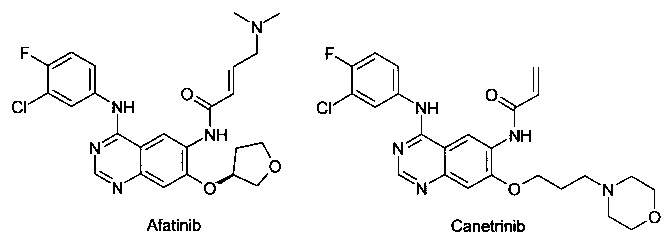
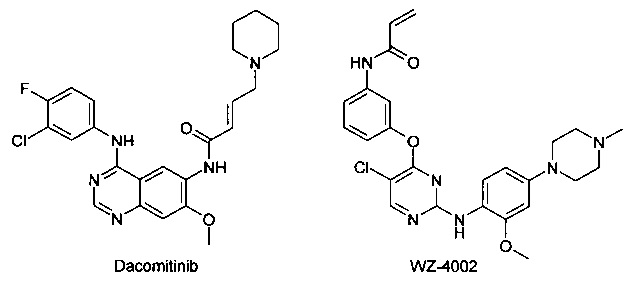
Известен ряд необратимых ингибиторов второго поколения, содержащих аминохиназолиновый фрагмент Гефитиниба. Эти ингибиторы содержат акриламидный заместитель, который ковалентно связывается с Cys-797, который расположен в канале растворителя киназы. К этому типу ингибиторов относятся Афатиниб, Канертиниб и Докомитиниб, WZ-4002. Ковалентный механизм связывания, как полагают, способствует преодолению АТФ сродства двойного мутанта, в результате чего эти соединения в клеточных моделях демонстрируют высокую активность. Тем не менее, мутант Cys-797 присутствует во всех соответствующих формах EGFR. Эти соединения второго поколения обладают не только повышенной активностью по отношению к устойчивым мутантам, но и по отношению к EGFR дикого типа. Ингибирование дикого типа EGFR не вносит свой вклад в их клиническую эффективность, но, как полагают, снижает побочные эффекты (кожная сыпь и диарея). Поскольку эти побочные эффекты, как правило, ограничивают дозу у пациентов, то активность этих ингибиторов против дикого типа EGFR ограничит их активность у пациентов против мутации Т790М.
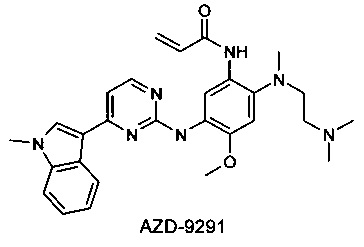
Компания АстраЗенека недавно запатентовала 2-(2,4,5-замещенные анилино)пиримидины в качестве модуляторов EGFR, полезных для лечения рака, включая модулятор AZD-9291 [Butterworrth, S.; Раймонд, М.; Финлей В. и др. 2-(2,4,5-Замещенные анилино)пиримидины как модуляторы EGFR, полезные для лечения рака [WO 2013014448]. Модулятор AZD-9291 наиболее продвинут в клинических исследованиях, и в настоящее время он проходит несколько Фаз III клинических испытаний [AstraZeneca, NCT02151981; AstraZeneca, NCT02296125].
До сих пор остается потребность в соединениях, которые могут демонстрировать благоприятный профиль активности против дикого EGFR по сравнению с активностью против мутантных форм EGFR (например, мутанта L858R EGFR) и/или устойчивой мутантной формы EGFR (например, Т790М EGFR мутанта). В этой связи существует потребность в соединениях, которые показывают более высокое ингибирование определенных активирующих или мутантных форм EGFR, в то же время показывая относительно низкое ингибирование дикого EGFR. Такие соединения, как ожидается, могут больше подходить в качестве терапевтических агентов, особенно для лечения рака, за счет уменьшения токсикологии, связанной с ингибированием дикого EGFR. Такие токсикологии, как известно, проявляются в человеке как сыпь и/или диареи кожи.
Заявители неожиданно обнаружили, что новые замещенные N-{3-[4-(1-метил-1Н-индол-3-ил)пиримидин-2-иламино]-4-метоксифенил}-амиды имеют более высокую эффективность против нескольких мутантных форм EGFR по сравнению с активностью против дикого типа EGFR, а также более высокую растворимость в водных растворах.
Новые соединения могут быть особенно полезными в лечении болезненных состояний, в которых вовлечены EGFR, и/или активации мутаций EGFR, и/или ингибирования мутаций EGFR, например в лечении рака.
Ниже приведены определения терминов, которые использованы в описании этого изобретения.
«Алкил» означает алифатическую углеводородную линейную или разветвленную группу с 1-12 атомами углерода в цепи. Разветвленная означает, что алкильная цепь имеет один или несколько «низших алкильных» (С1-С6)алкильных заместителей. Предпочтительными алкильными группами являются (С1-С6)алкил, еще более предпочтительными (С1-С3)алкил, метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, циклопропилметил, циклобутилметил, циклопентилметил, н-пентил, 2-пентил, 3-пентил, нео-пентил, н-гексил, циклогексил. Алкил может иметь заместители.
«Замещенный алкил» - замещенный алкил может иметь один или несколько одинаковых или различных заместителей, включая галоген, алкенилокси, циклоалкил, арил, гетероарил, гетероциклил, ароил, гетероароил, циано, гидрокси, алкокси, карбокси, алкинилокси, аралкокси, арилокси, арилоксикарбонил, алкилтио, гетероарилтио, аралкилтио, арилсульфонил, алкилсульфонил, гетероаралкилокси или RkaRk+1aN-, где Rka и Rk+1a независимо друг от друга представляют собой «заместители аминогруппы», значение которых определено в данном разделе, например атом водорода, алкил, арил, аралкил, гетероаралкил, гетероциклил или гетероарил, или Rka и Rk+1a вместе с атомом N, с которым они связаны, образуют через Rka и Rk+1a 4-7-членный гетероциклил или гетероцикленил. Предпочтительными «алкильными заместителями» являются арил, гетероарил, гетероциклил, гидрокси, С1-С5 алкокси, С1-С5 алкоксикарбонил, аралкокси, арилокси, алкилтио, гетероарилтио, аралкилтио, алкилсульфонил, арилсульфонил, алкоксикарбонил, аралкоксикарбонил, гетероаралкилоксикарбонил или RkaRk+1aN-, RkaRk+1aNC(=O)-, аннелированный арилгетероцикленил, аннелированный арилгетероциклил.
«Алкокси» означает (С1-С3)алкил-O-группу, в которой алкил определен в данном разделе. Предпочтительным алкилокси группами являются метокси, этокси, н-пропокси, изопропокси и н-бутокси.
«Аминогруппа» означает R1R2N-группу, замещенную или незамещенную необязательно одинаковыми заместителями R1 и R2. Аминогруппа может иметь заместители.
«Активный компонент» (лекарственное вещество, лекарственная субстанция, drug-substance) означает физиологически активное вещество синтетического или иного (биотехнологического, растительного, животного, микробного и прочего) происхождения, обладающее фармакологической активностью и являющееся активным началом фармацевтической композиции, используемой для производства и изготовления лекарственного препарата (средства).
«Галоген» означает фтор, хлор, бром и йод. Предпочтительными являются фтор, хлор и бром.
«Заместитель» означает химический радикал, который присоединяется к молекулярному остову (скэффолду, фрагменту), например «заместитель алкильный», «заместитель аминогруппы», «заместитель карбамоильный», «заместитель циклической системы», значения которых определены в данном разделе.
«Лекарственное средство (препарат)» - вещество (или смесь веществ в виде фармацевтической композиции) в виде таблеток, капсул, инъекций, мазей и других готовых форм, предназначенное для восстановления, исправления или изменения физиологических функций у человека и животных, а также для лечения и профилактики болезней, диагностики, анестезии, контрацепции, косметологии и прочего.
«Фармацевтическая композиция» обозначает композицию, включающую в себя активный компонент и, по крайней мере, один из компонентов, выбранных из группы, состоящей из фармацевтически приемлемых и фармакологически совместимых наполнителей, растворителей, разбавителей, носителей, вспомогательных, распределяющих и воспринимающих средств, средств доставки, таких как консерванты, стабилизаторы, наполнители, измельчители, увлажнители, эмульгаторы, суспендирующие агенты, загустители, подсластители, отдушки, ароматизаторы, антибактериальные агенты, фунгициды, лубриканты, регуляторы пролонгированной доставки, выбор и соотношение которых зависит от природы и способа назначения и дозировки. Примерами суспендирующих агентов являются этоксилированный изостеариловый спирт, полиоксиэтилен, сорбитол и сорбитовый эфир, микрокристаллическая целлюлоза, метагидроксид алюминия, бентонит, агар-агар и трагакант, а также смеси этих веществ. Защита от действия микроорганизмов может быть обеспечена с помощью разнообразных антибактериальных и противогрибковых агентов, например таких, как парабены, хлорбутанол, сорбиновая кислота и подобные им соединения. Композиция может включать также изотонические агенты, например сахара, хлористый натрий и им подобные. Пролонгированное действие композиции может быть обеспечено с помощью агентов, замедляющих абсорбцию активного начала, например моностеарат алюминия и желатин. Примерами подходящих носителей, растворителей, разбавителей и средств доставки являются вода, этанол, полиспирты, а также их смеси, растительные масла (такие, как оливковое масло) и инъекционные органические сложные эфиры (такие как этилолеат). Примерами наполнителей являются лактоза, молочный сахар, цитрат натрия, карбонат кальция, фосфат кальция и им подобные. Примерами измельчителей и распределяющих средств являются крахмал, альгиновая кислота и ее соли, силикаты. Примерами лубрикантов являются стеарат магния, лаурилсульфат натрия, тальк, а также полиэтиленгликоль с высоким молекулярным весом. Фармацевтическая композиция для перорального, сублингвального, трансдермального, внутримышечного, внутривенного, подкожного, местного или ректального введения активного начала, одного или в комбинации с другим активным началом, может быть введена животным и людям в стандартной форме введения в виде смеси с традиционными фармацевтическими носителями. Пригодные стандартные формы введения включают пероральные формы, такие как таблетки, желатиновые капсулы, пилюли, порошки, гранулы, жевательные резинки и пероральные растворы или суспензии, сублингвальные и трансбуккальные формы введения, аэрозоли, имплантаты, местные, трансдермальные, такие как мази и кремы, подкожные, внутримышечные, внутривенные, интраназальные или внутриглазные формы введения и ректальные формы введения.
«Фармацевтически приемлемая соль» означает относительно нетоксичные органические и неорганические соли кислот и оснований, заявленных в настоящем изобретении. Эти соли могут быть получены in situ в процессе синтеза, выделения или очистки соединений или приготовлены специально. В частности, соли оснований могут быть получены специально исходя из очищенного свободного основания заявленного соединения и подходящей органической или неорганической кислоты. Примерами полученных таким образом солей являются гидрохлориды, гидробромиды, сульфаты, бисульфаты, фосфаты, нитраты, ацетаты, оксалаты, валериаты, олеаты, пальмитаты, стеараты, лаураты, бораты, бензоаты, лактаты, тозилаты, цитраты, малеаты, фумараты, сукцинаты, тартраты, мезилаты, малонаты, салицилаты, пропионаты, этансульфонаты, бензолсульфонаты, сульфаматы и им подобные (Подробное описание свойств таких солей дано в Berge S.M., et al., "Pharmaceutical Salts" J. Pharm. Sci. 1977, 66: 1-19). Соли заявленных кислот также могут быть специально получены реакцией очищенной кислоты с подходящим основанием, при этом могут быть синтезированы соли металлов и аминов. К металлическим относятся соли натрия, калия, кальция, бария, цинка, магния, лития и алюминия, наиболее желательными из которых являются соли натрия и калия. Подходящими неорганическими основаниями, из которых могут быть получены соли металлов, являются гидроксид, карбонат, бикарбонат и гидрид натрия, гидроксид и бикарбонат калия, поташ, гидроксид лития, гидроксид кальция, гидроксид магния, гидроксид цинка. В качестве органических оснований, из которых могут быть получены соли заявленных кислот, выбраны амины и аминокислоты, обладающие достаточной основностью, чтобы образовать устойчивую соль, и пригодные для использования в медицинских целях (в частности, они должны обладать низкой токсичностью). К таким аминам относятся аммиак, метиламин, диметиламин, триметиламин, этиламин, диэтиламин, триэтиламин, бензиламин, дибензиламин, дициклогексиламин, пиперазин, этилпиперидин, трис(гидроксиметил)аминометан и подобные им. Кроме того, для солеобразования могут быть использованы гидроокиси тетраалкиламмония, например такие, как холин, тетраметиламмоний, тетраэтиламмоний и им подобные. В качестве аминокислот могут быть использованы основные аминокислоты - лизин, орнитин и аргинин.
Цель настоящего изобретения заключается в создании новых модуляторов EGFR для лечения рака.
Поставленная цель достигается новыми замещенными N-{3-[4-(1-метил-1Н-индол-3-ил)пиримидин-2-иламино]-4-метоксифенил}-амидами общей формулы 1, их полиморфными формами, солями, сольватами и гидратами.
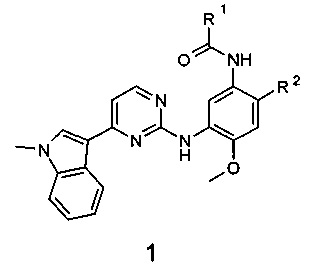
где:
R1 представляет собой радикал, выбранный из CH2=C(R3)-, R3CH=CH- и R4C≡C-;
R2 представляет собой (2-диметиламиноэтил)-метиламино, 3-диметиламинопирролидин-1-ил, 3-диметиламинопиперидин-1-ил и 3-диметиламиноазепан-1-ил, 4-метилпиперазин-1-ил;
R3 представляет собой метил, трифторметил, фтор, хлор, метоксикарбонил и диметилкарбамоил;
R4 представляет собой водород или метил;
или R1 представляет собой винил, a R2 представляет собой 3-диметиламинопирролидин-1-ил;
R4 представляет собой С1-С4алкил, С3-С6циклоалкил.
Более предпочтительным являются ингибиторы EGFR общей формулы 1, выбранные из:
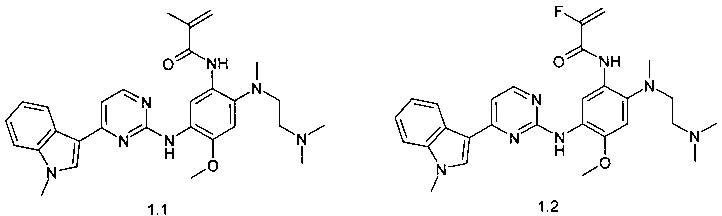
N-{2-[(2-диметиламиноэтил)-метиламино]-5-[4-(1-метил-1Н-индол-3-ил)-пиримидин-2-иламино]-4-метоксифенил}-2-метил-акриламид (1.1);
N-{2-[(2-диметиламиноэтил)-метиламино]-5-[4-(1-метил-1Н-индол-3-ил)-пиримидин-2-иламино]-4-метоксифенил}-2-фтор-акриламид (1.2);
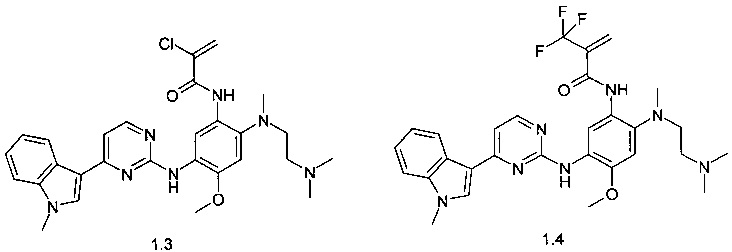
N-{2-[(2-диметиламиноэтил)-метиламино]-5-[4-(1-метил-1Н-индол-3-ил)-пиримидин-2-иламино]-4-метоксифенил}-2-хлор-акриламид (1.3);
N-{2-[(2-диметиламиноэтил)-метиламино]-5-[4-(1-метил-1Н-индол-3-ил)-пиримидин-2-иламино]-4-метоксифенил}-2-трифторметил-акриламид (1.4);
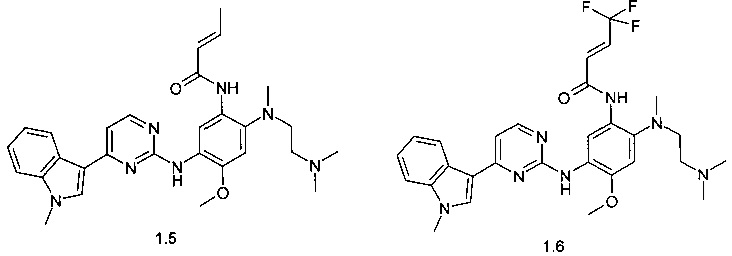
N-{2-[(2-диметиламиноэтил)-метиламино]-5-[4-(1-метил-1Н-индол-3-ил)-пиримидин-2-иламино]-4-метоксифенил}-2-бутенамид (1.5);
N-{2-[(2-диметиламиноэтил)-метиламино]-5-[4-(1-метил-1Н-индол-3-ил)-пиримидин-2-иламино]-4-метоксифенил}-4,4,4-трифторбут-2-енамид (1.6);
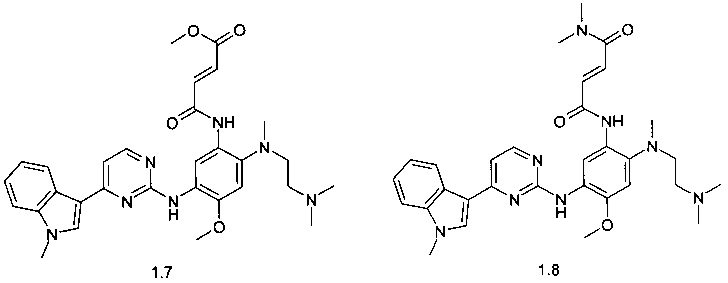
(E)-3-{2-[(2-диметиламиноэтил)-метиламино]-5-[4-(1-метил-1Н-индол-3-ил)-пиримидин-2-иламино]-4-метоксифенилкарбамоил}-акриловой кислоты метиловый эфир (1.7);
(E)-3-{2-[(2-диметиламиноэтил)-метиламино]-5-[4-(1-метил-1Н-индол-3-ил)-пиримидин-2-иламино]-4-метоксифенилкарбамоил}-N,N-диметилакриламид (1.8);
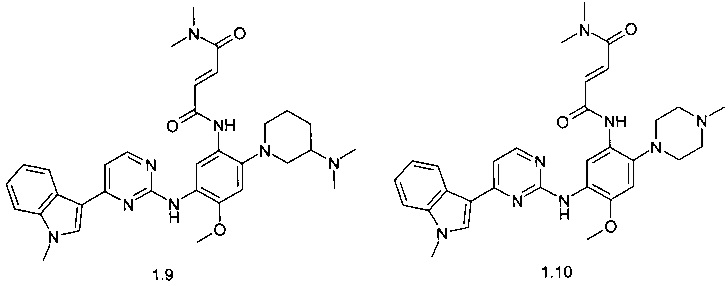
(E)-N,N-диметил-3-{2-(3-диметиламинопиперидин-1-ил)-5-[4-(1-метил-1Н-индол-3-ил)-пиримидин-2-иламино]-4-метоксифенилкарбамоил}-акриламид (1.9);
(E)-N,N-диметил-3-{5-[4-(1-метил-1Н-индол-3-ил)-пиримидин-2-иламино]-2-(4-метил-пиперазин-1-ил)-4-метоксифенилкарбамоил}-акриламид (1.10);
N-{2-(3-диметиламинопиперидин-1-ил)-5-[4-(1-метил-1Н-индол-3-ил)-пиримидин-2-иламино]-4-метоксифенил}-акриламид (1.11);
N-{2-[(2-диметиламиноэтил)-метиламино]-5-[4-(1-метил-1Н-индол-3-ил)-пиримидин-2-иламино]-4-метоксифенил}-пропинамид (1.12);
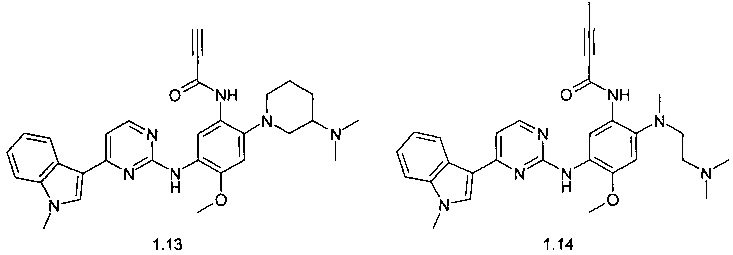
N-{2-(3-диметиламинопиперидин-1-ил)-5-[4-(1-метил-1Н-индол-3-ил)-пиримидин-2-иламино]-4-метоксифенил}-пропинамид (1.13);
N-{2-[(2-диметиламиноэтил)-метиламино]-5-5-[4-(1-метил-1Н-индол-3-ил)-пиримидин-2-иламино]-4-метоксифенил}-2-бутинамид (1.14)
или их фармацевтически приемлемых солей.

Настоящее изобретение также включает фармацевтически приемлемые соли соединений общей формулы 1. Примеры фармацевтически приемлемых солей включают, но не ограничиваются ими, соли минеральных или органических кислот соединений формулы 1, таких как карбоновые кислоты, например, дихлоруксусная кислота, а также соли хлористого водорода, фосфорной кислоты или сульфокислоты. Фармацевтически приемлемые соли по настоящему изобретению включают обычные нетоксичные соли исходного соединения, образованные, например, из нетоксичных неорганических или органических кислот. Фармацевтически приемлемые соли по настоящему изобретению могут быть синтезированы из исходного соединения, которое содержит основную или кислотную группу, обычными химическими способами. Как правило, такие соли могут быть получены взаимодействием свободных кислотных и основных форм соединений общей формулы 1 со стехиометрическим количеством соответствующего основания или кислоты в воде, или в органическом растворителе, или в смеси двух растворителей. Как правило, предпочтительны неводные среды, такие как эфир, этилацетат, этанол, изопропанол, ацетон или ацетонитрил (ACN). Списки подходящих солей можно найти в справочнике фармацевтических солей [Р.Н. Stahl, C.G. Wermuth (Eds.). Handbook of Pharmaceutical Salts, Properties, Selection, and Use. VHCA, Verlag Helvetica Chimica Acta, Zurich, Switzerland, and Wiley-VCH, Weinheim, Germany. 2002].
Соединения общей формулы 1 и их фармацевтически приемлемые соли могут существовать в сольватированных и несольватированных формах. Например, в виде гидрата. Настоящее изобретение охватывает все такие сольватированные и несольватированные формы.
Безусловным преимуществом новых киназных ингибиторов общей формулы 1 является, в первую очередь, их высокая растворимость в водных средах, которая в ряде случаев на порядки превышает растворимость известного AZD-9291 (Таблица 1). Так, например, растворимость ингибитора 1.12⋅2MeSO3H в воде при рН2 более чем в 10 раз выше растворимости AZD-9291, а при рН7 - более чем в 10900 раз.

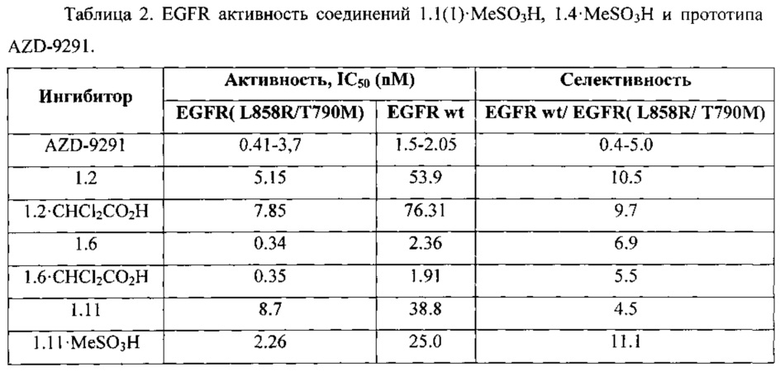
Соединения общей формулы 1 и их фармацевтически приемлемые соли в сопоставимых условиях против двойного мутанта EGFR(L858R/T790M) имеют, как правило, ингибирующую активность, сравнимую с соответствующей активностью известного ингибитора AZD-9291. Так, например, (Таблица 2) новые ингибиторы общей формулы 1 имеют значение ингибирующей активности IC50 менее 10 nM (IC50 от 0,34 nM до 8,7 nM), а AZD-9291 имеет соответствующую IC50=0,41÷3,7 nM. В то же время, в этих условиях новые ингибиторы уступают AZD-9291 по активности против дикого EGFR wt, что является их безусловным преимуществом, поскольку в этом случае их селективность, как правило, выше селективности AZD-9291 (Таблица 2).
Данное изобретение относится к соединениям общей формулы 1, которые ингибируют одну или несколько активирующих или пассивирующих EGFR мутаций, например L858R мутант, Exon 19 удаленный EGFR мутант и резистентный Т790М мутант. Предпочтительно такие соединения могут быть использованы для лечения рака, устойчивого к существующей терапии. Согласно настоящему изобретению соединения общей формулы 1 показывают более высокое ингибирование или пассивирование мутантных форм EGFR, чем дикого EGFR.
Такие соединения, как ожидается, будут больше подходить в качестве терапевтических агентов, особенно для лечения рака, за счет уменьшения токсикологии, связанного с ингибированием дикого EGFR. Такие токсикологии, как известно, проявляются в человеке как сыпь и/или диареи кожи.
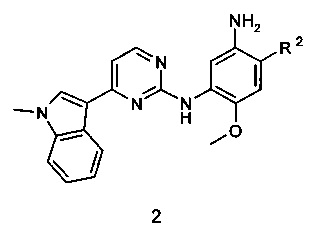
Соединения общей формулы 1 могут быть получены реакцией соединения формулы 2, в котором R2 имеет вышеуказанное значение, или его соли с активированным производным органической кислоты (например, хлорангидридом или соответствующим сложным эфиром или ангидридом органической кислоты) в растворителе, таком как CH2Cl2, тетрагидрофуране, N,N-диметилформамиде или N,N-диметилацетамиде.
Предметом данного изобретения является также фармацевтическая композиция, которая включает соединение общей формулы 1 или его фармацевтически приемлемую соль, как определено выше, в сочетании с фармацевтически приемлемым разбавителем или носителем.
Композиции по изобретению могут быть в форме, подходящей для перорального применения (например, в виде таблеток, пластинок, твердых или мягких капсул, водных или масляных суспензий, эмульсий, диспергируемых порошков или гранул, сиропов или эликсиров), для местного применения (например, в виде кремов, мазей, гелей или водных или масляных растворов или суспензий), для введения путем ингаляции (например, в виде тонкоизмельченного порошка или жидкого аэрозоля), для введения путем инсуффляции (например, в виде тонкоизмельченного порошка) или для парентерального введения (например, в виде стерильного водного или масляного раствора для внутривенного, подкожного, внутримышечного введения дозы или в виде суппозиториев для ректального введения дозы).
Композиции по изобретению могут быть получены обычными способами с использованием обычных фармацевтических наполнителей, хорошо известных в данной области. Таким образом, композиции, предназначенные для перорального применения, могут содержать, например, один или несколько окрашивающих, подслащивающих, вкусовых агентов и/или консервантов.
Соединение общей формулы 1 обычно вводят пациенту в единичной дозе в диапазоне 5-5000 мг/м2 площади тела, то есть примерно 0,1-100 мг/кг, и это обычно обеспечивает терапевтически эффективную дозу. Форма единичной дозы, такой как таблетка или капсула, обычно содержит, например, 1-250 мг активного ингредиента. Суточная доза обязательно будет варьироваться в зависимости от пациента, конкретного пути введения и тяжести заболевания, которое лечат. Соответственно, доктор, который лечит конкретного пациента, может определить оптимальную дозу.
Соединения общей формулы 1 и их фармацевтически приемлемые соли, ингибирующие активности мутантов L858R EGFR, Т790М EGFR и Exon19 EGFR, как ожидается, будут полезны при лечении заболеваний или медицинских состояний, обусловленных полностью или частично активностью EGFR мутанта, например рака. Типы рака, которые могут быть восприимчивы к лечению с использованием соединения общей формулы 1 или его фармацевтически приемлемой соли, включают, но не ограничиваются ими, рак яичников, рак шейки матки, колоректальный рак, рак молочной железы, рак поджелудочной железы, глиому, глиобластомы, меланомы, рак простаты, лейкоз, лимфому не-Ходжкина лимфомы, рак желудка, рак легких, гепатоцеллюлярный рак, желудочно-кишечных стромальных опухолей (GIST), рак щитовидной железы, рак желчного протока, рак эндометрия, рак почки, анапластическую крупноклеточную клеточную лимфому, острый миелоидный лейкоз (ОМЛ), множественные миеломы, меланомы и мезотелиомы.
Метод лечения указанных здесь типов рака заключается во введении соединения формулы 1 млекопитающему, более предпочтительно человеку.
Аналогично, использование соединения общей формулы 1 для лечения указанных здесь типов рака предполагает введение соединение общей формулы 1 млекопитающему, более предпочтительно человеку.
Предметом данного изобретения является также применения соединения общей формулы 1 или его фармацевтически приемлемой соли, гидрата или сольвата в качестве лекарственного средства.
Предметом данного изобретения является также применение соединения общей формулы 1, как определено выше, или его фармацевтически приемлемой соли, гидрата или сольвата для лечения заболевания, обусловленного мутантом L858R EGFR, и/или мутантом Т790М EGFR, и/или мутантом Exon19, например, для лечения рака.
Предметом данного изобретения является также использование соединения общей формулы 1, как определено выше, или его фармацевтически приемлемой соли, гидрата или сольвата, для изготовления лекарственного средства для лечения заболевания, обусловленного мутантом L858R EGFR, и/или мутантом Т790М EGFR, и/или мутантом Exon19, например, для лечения рака.
Предметом данного изобретения является также использование соединения общей формулы 1, как определено выше, или его фармацевтически приемлемой соли, гидрата или сольвата для лечения рака.
Предметом данного изобретения является также способ получения противоракового эффекта в теплокровном животном, таком как человек, нуждающемся в таком лечении, который включает введение ему эффективного количества соединения общей формулы 1 или его фармацевтически приемлемой соли, гидрата или сольвата.
В любом из аспектов или вариантов, упомянутых здесь, указанный рак может быть выбран из рака яичников, рака шейки матки, колоректального рака, рака молочной железы, рака поджелудочной железы, глиомы, глиобластомы, меланомы, рака простаты, лейкемии, лимфомы не-Ходжкина лимфомы, рака желудка, рака легких, гепатоцеллюлярного рака, желудочно-кишечных стромальных опухолей (GIST), рака щитовидной железы, рака желчного протока, рака эндометрия, рака почки, анапластической крупноклеточной лимфомы, острого миелоидного лейкоза (ОМЛ), множественной миеломы, меланомы и мезотелиомы.
Описанное выше лечение рака может быть как единственная терапия или может включать в дополнение соединение общей формулы 1 по настоящему изобретению к обычной хирургии, или лучевой терапии, или химиотерапии, или иммунотерапии. Химиотерапия может использоваться одновременно, последовательно или раздельно с лечением соединением общей формулы 1 по изобретению и может включать один или более противоопухолевых агентов, в том числе: антипролиферативные/противоопухолевые препараты и их комбинации, используемые в медицинской онкологии, такие как алкилирующие агенты, антиметаболиты, противоопухолевые антибиотики, антимитотические агенты и ингибиторы топоизомеразы; цитостатические агенты, такие как антиэстрогены, антиандрогены, антагонисты или агонисты LHRH, ингибиторы ароматазы и ингибиторы 5а-редуктазы; ингибиторы функции фактора роста и др. препараты.
Таким образом, предметом настоящего изобретения является фармацевтическая композиция, включающая соединение общей формулы 1, как определено выше, или его фармацевтически приемлемую соль, гидрат или сольват и дополнительное противоопухолевое вещество для совместного лечения рака в виде таблеток, капсул или инъекций. Здесь, где термин "совместное лечение" используют по отношению к комбинированной терапии, следует понимать, что это может относиться к одновременному, раздельному или последовательному введения препаратов.
Таким образом, в одном из вариантов осуществления изобретения предлагается использование соединения общей формулы 1 или его фармацевтически приемлемой соли, гидрата или сольвата и дополнительно противоопухолевого вещества для совместного лечения рака.
Предметом настоящего изобретения является способ получения противоракового эффекта в теплокровном животном, таком как человек, который нуждается в таком лечении, который включает введение указанному млекопитающему соединения общей формулы 1 или его фармацевтически приемлемой соли, гидрата или сольвата и одновременное, раздельное или последовательное введение дополнительного противоопухолевого вещества указанному млекопитающему, где количества соединения общей формулы 1 или его фармацевтически приемлемой соли и дополнительного противоопухолевого вещества совместно обеспечивают производство противоракового эффекта.
Ниже изобретение иллюстрируется с помощью конкретных примеров. Следующие примеры представлены с целью иллюстрации и не предназначены для ограничения изобретения каким-либо образом. Специалисты в данной области техники легко поймут различие некритических параметров, которые могут быть изменены или модифицированы, чтобы получить те же результаты.
Пример 1. N-{2-[(2-диметиламиноэтил)-метиламино]-5-[4-(1-метил-1Н-индол-3-ил)-пиримидин-2-иламино]-4-метоксифенил}-2-метил-акриламид (1.1) получают согласно схеме 1.
Схема 1.
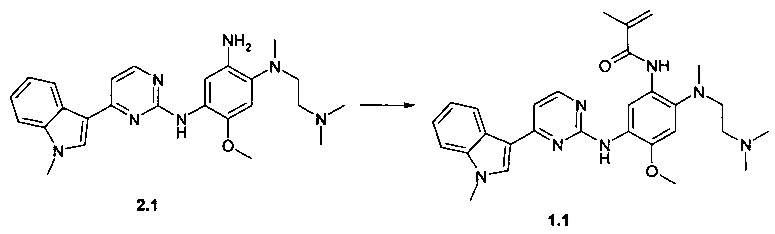
К перемешиваемому раствору N1-[2-(диметиламино)этил]-N1-метил-N4-[4-(1-метил-1Н-индол-3-ил)пиримидин-2-ил]-5-метоксибензол-1,2,4-триамина (2.1, 1.0 г, 2.2 ммоль), DCC (0,51 г, 2,4 ммоль) и 2-метилакриловой кислоты (193 мг, 2,2 ммоль) в CH2Cl2 (30 мл) добавляли DMAP (27 мг, 0,22 ммоль) при комнатной температуре. Полученную смесь перемешивали в течение ночи. Осадок отфильтровывали, маточный раствор концентрировали и очищали с помощью колоночной хроматографии с использованием ацетона в качестве элюента. Соответствующие фракции объединяли и концентрировали в вакууме. Получали 400 мг ингибитора 1.1. 1H-NMR (DMSO-d6, 400 MHz): 2.03 (s, 3Н), 2.12 (s, 6H), 2.20 (t, J=5.5 Hz, 2H), 2.68 (s, 3H), 2.96 (t, J=5.5 Hz, 2H), 3.86 (s, 3H), 3.92 (s, 3H), 5.54 (s, 1H), 5.85 (s, 1H), 7.08 (s, 1H), 7.15 (t, J=7.1 Hz, 1H), 7.20-7.29 (m, 2H), 7.52 (d, J=8.1 Hz, 1H), 7.89 (s, 1H), 8.23 (d, J=8.1 Hz, 1H), 8.34 (d, J=5.5 Hz, 1H), 8.71 (s, 1H), 9.19 (s, 1H), 9.96 (s, 1H).
Пример 2. N-{2-[(2-диметиламиноэтил)-метиламино]-5-[4-(1-метил-1H-индол-3-ил)-пиримидин-2-иламино]-4-метоксифенил}-2-фтор-акриламид (1.2) и его дихлорацетат 1.2⋅CHCl2CO2H получают согласно схеме 2.
Схема 2.
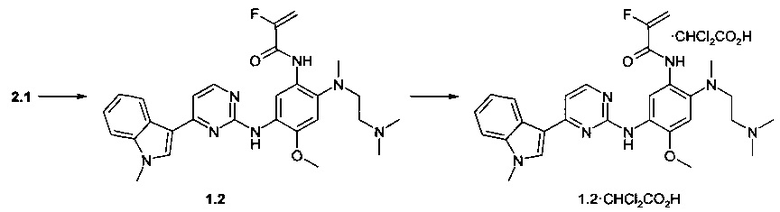
К перемешиваемому раствору N1-[2-(диметиламино)этил]-N1-метил-N4-[4-(1-метил-1Н-индол-3-ил)пиримидин-2-ил]-5-метоксибензол-1,2,4-триамина (2.1, 0,5 г, 1,2 ммоль), DCC (0,255 г, 1,2 ммоль) и 2-фторакриловой кислоты (101 мг, 1,1 ммоль) в CH2Cl2 (20 мл) добавляли DMAP (14 мг, 0,11 ммоль) при комнатной температуре. Полученную смесь перемешивали в течение ночи. Осадок отфильтровывали, маточный раствор концентрировали и очищали с помощью колоночной хроматографии с использованием ацетона в качестве элюента. Соответствующие фракции объединяли и концентрировали в вакууме. Получали 117 мг ингибитора 1.2. 1H-NMR (DMSO-d6, 400 MHz): 2.14 (s, 3Н), 2.17 (t, J=5.7 Hz, 2H), 2.69 (s, 3H), 2.97 (t, J=5.7 Hz, 2H), 3.88 (s, 3H), 3.90 (s, 3H), 5.42 (dd, J1=15.7 Hz, J2=3.3 Hz, 1H), 5.67 (dd, J1=48.2 Hz, J2=3.3 Hz, 1H), 7.09-7.19 (m, 2H), 7.21-7.28 (m, 2H), 7.52 (d, J=8.3 Hz, 1H), 7.93 (s, 1H), 8.25 (d, J=8.3 Hz, 1H), 8.34 (d, J=5.3 Hz, 1H), 8.59 (s, 1H), 9.08 (s, 1H), 10.68 (s, 1H).
К перемешиваемому раствору ингибитора 1.2 (40 мг, 0.077 ммоль) в ацетоне (1 мл) при 25°С добавляли дихлоруксусную кислоту (9.9 мг, 0.077 ммоль) в виде раствора в ацетоне (1 мл). Полученную смесь перемешивают в течение 15 ч. Осадок отфильтровывали и сушили на воздухе в течение ночи. Получали 17 мг ингибитора 1.2⋅CHCl2CO2H в виде белых кристаллов. 1H-NMR (DMSO-d6, 400 MHz): 2.66 (s, 3Н), 2.70 (s, 6H), 2.96-3.11 (m, 2H), 3.24-3.33 (m, 2H), 3.90 (s, 3Н), 3.92 (s, 3Н), 5.48 (dd, J1=15.5 Hz, J2=3.3 Hz, 1H), 5.75 (dd, J1=48.4 Hz, J2=3.3 Hz, 1H), 5.98 (s, 1H), 7.06 (s, 1H), 7.17 (t, J=7.5 Hz, 1H), 7.22-7.30 (m, 2H), 7.53 (d, J=8.3 Hz, 1H), 7.95 (s, 1H), 8.31 (d, J=8.3 Hz, 1H), 8.34 (d, J=5.3 Hz, 1H), 8.52 (s, 1H), 8.87 (s, 1H), 9.87 (s, 1H).
Пример 3. N-{2-[(2-диметиламиноэтил)-метиламино]-5-[4-(1-метил-1Н-индол-3-ил)-пиримидин-2-иламино]-4-метоксифенил}-2-бутенамид (1.5) и его мезилат (1.5⋅CH3SO3H) получают согласно схеме 3.
Схема 3.
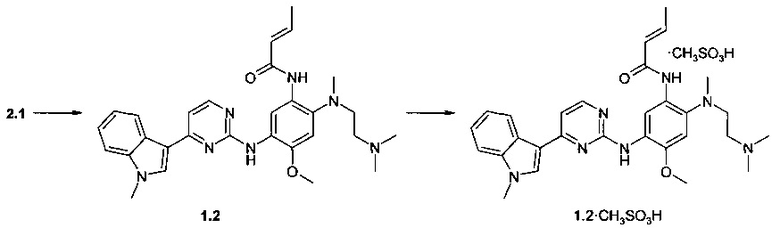
К перемешиваемому раствору N1-[2-(диметиламино)этил]-N1-метил-N4-[4-(1-метил-1Н-индол-3-ил)пиримидин-2-ил]-5-метоксибензол-1,2,4-триамина (2.1, 70 мг, 1,57 ммоль) и TEA (238 мг, 2,36 ммоль) в CH2Cl2 (5 мл) добавляли акрилоилхлорид (142 мг, 1,57 ммоль) в CH2Cl2 (1 мл) при -20°С. Полученную смесь нагревали до комнатной температуры, перемешивали в течение 3 ч, затем промывали водой (5 мл). Органическую фазу концентрировали и очищали с помощью колоночной хроматографии с использованием смеси EtOAc и МеОН (1:1). Соответствующие фракции объединяли и концентрировали в вакууме. Получали 200 мг ингибитора 1.5. 1H-NMR (DMSO-d6, 400 MHz): 1.90 (d, J=6.7 Hz, 3H), 2.22 (s, 6H), 2.31 (t, J=5.5 Hz, 2H), 2.71 (s, 3H), 2.88 (t, J=5.5 Hz, 2H), 3.85 (s, 3H), 3.92 (s, 3Н), 6.10 (d, J=15.3 Hz, 1H), 6.80 (dt, J1=6.7 Hz, J2=15.3 Hz, 1H), 7.03 (s, 1H), 7.15 (t, J=7.1 Hz, 1H), 7.20-7.27 (m, 2H), 7.52 (d, J=8.1 Hz, 1H), 7.88 (s, 1H), 8.23 (d, J=8.1 Hz, 1H), 8.32 (d, J=5.5 Hz, 1H), 8.68 (s, 1H), 9.14 (s, 1H), 9.97 (s, 1H).
К перемешиваемому раствору ингибитора 1.5 (8, 55 мг, 0.11 ммоль) в ацетоне (3 мл) при 25°С добавляли метансульфоновую кислоту (10 мг, 0.11 ммоль) в виде раствора в ацетоне (1 мл). Полученную смесь перемешивают в течение 15 ч. Осадок отфильтровывали и сушили на воздухе в течение ночи. Получали 36 мг ингибитора 1.5⋅CH3SO3H в виде светло-желтых кристаллов. 1H-NMR (DMSO-d6, 400 MHz): 1.90 (d, J=6.7 Hz, 3H), 2.33 (s, 3H), 2.63 (s, 3H), 2.81 (s, 6H), 3.22-3.30 (m, 4H), 3.89 (s, 3H), 3.90 (s, 3H), 6.36 (d, J=15.3 Hz, 1H), 6.86 (dt, J1=6.7 Hz, J2=15.3 Hz, 1H), 7.00 (s, 1H), 7.16 (t, J=7.1 Hz, 1H), 7.22-7.29 (m, 2H), 7.53 (d, J=8.1 Hz, 1H), 8.14 (br.s, 1H), 7.22-7.29 (m, 2H), 8.54 (s, 1H), 8.64 (s, 1H), 9.26 (s, 1H), 9.37 (s, 1H).
Пример 4. N-{2-[(2-диметиламиноэтил)-метиламино]-5-[4-(1-метил-1Н-индол-3-ил)-пиримидин-2-иламино]-4-метоксифенил}-4,4,4-трифторбут-2-енамид (1.6) и его дихлорацетат 1.6⋅CHCl2CO2H получают согласно схеме 4.
Схема 4.
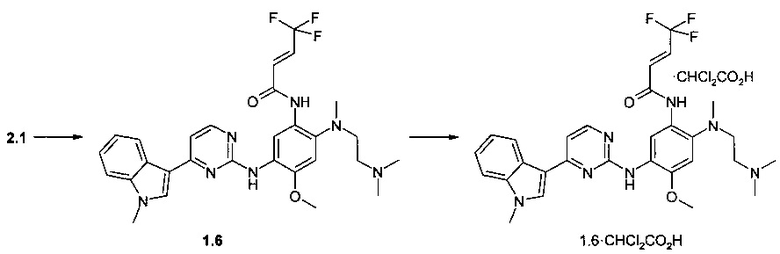
К перемешиваемому раствору N1-[2-(диметиламино)этил]-N1-метил-N4-[4-(1-метил-1Н-индол-3-ил)пиримидин-2-ил]-5-метоксибензол-1,2,4-триамина (2.1, 1,0 г, 2,2 ммоль), DCC (0,51 г, 2,4 ммоль) и (2E)-4,4,4-трифторбут-2-еновой кислоты (314 мг, 2,2 ммоль) в CH2Cl2 (30 мл) добавляли при комнатной температуре DMAP (27 мг, 0,22 ммоль). Полученную смесь перемешивали в течение ночи. Осадок отфильтровывали, маточный раствор концентрировали и очищали с помощью колоночной хроматографии с использованием ацетона в качестве элюента. Соответствующие фракции объединяли и концентрировали в вакууме. Получали 233 мг ингибитора 1.6. 1H-NMR (DMSO-d6, 400 MHz): 2.19 (s, 3Н), 2.36 (t, J=5.7 Hz, 2H), 2.74 (s, 3H), 2.88 (t, J=5.7 Hz, 2H), 3.87 (s, 3H), 3.90 (s, 3H), 6.85-6.96 (m, 1H), 6.97-7.08 (m, 2H), 7.13 (t, J=7.6 Hz, 1H), 7.19-7.27 (m, 2H), 7.52 (d, J=8.3 Hz, 1H), 7.93 (s, 1H), 8.26 (d, J=8.3 Hz, 1H), 8.31 (d, J=5.3 Hz, 1H), 8.55 (s, 1H), 9.07 (s, 1H), 10.45 (s, 1H).
К перемешиваемому раствору ингибитора 1.6 (40 мг, 0,07 ммоль) в ацетоне (1 мл) при 25°С добавляли дихлоруксусную кислоту (9 мг, 0,07 ммоль) в виде раствора в ацетоне (1 мл). Полученную смесь перемешивали в течение 15 ч. Осадок отфильтровали и сушили на воздухе в течение ночи. Получали 23 мг ингибитора 1.6⋅CHCl2CO2H в виде кристаллов бежевого цвета. 1H-NMR (DMSO-d6, 400 MHz): 2.63 (s, 3Н), 2.75 (s, 6H), 3.11-3.28 (m, 4H), 3.89 (s, 3Н), 3.90 (s, 3Н), 6.02 (s, 1H), 6.86-6.98 (m, 1H), 7.05 (s, 1H), 7.13 (t, J=7.5 Hz, 1H), 7.19-7.27 (m, 2H), 7.43 (d, J=14.3 Hz, 1H), 7.52 (d, J=8.3 Hz, 1H), 7.96 (s, 1H), 8.30 (d, J=8.3 Hz, 1H), 8.32 (d, J=5.3 Hz, 1H), 8.50 (s, 1H), 9.00 (s, 1H), 9.99 (s, 1H), 10.48 (br.s, 1H).
Пример 5. (E)-3-{2-[(2-диметиламиноэтил)-метиламино]-5-[4-(1-метил-1Н-индол-3-ил)-пиримидин-2-иламино]-4-метоксифенилкарбамоил}-акриловой кислоты метиловый эфир (1.7) и его мезилат 1.7⋅CH3SO3H получают согласно схеме 5.
Схема 5.
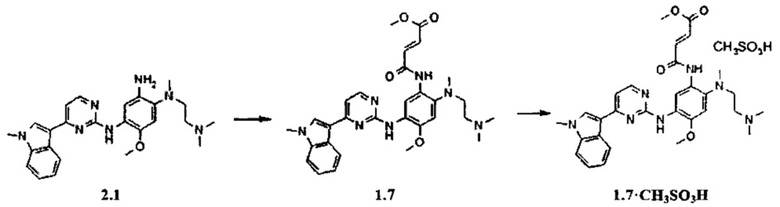
К перемешиваемому раствору N1-[2-(диметиламино)этил]-N1-метил-N4-[4-(1-метил-1Н-индол-3-ил)пиримидин-2-ил]-5-метоксибензол-1,2,4-триамина (2.1, 300 мг, 0,67 ммоль) и TEA (81 мг, 0,80 ммоль) в CH2Cl2 (5 мл) добавляли метил (2Z)-4-хлор-4-оксобут-2-еновой кислоты (110 мг, 0,73 ммоль) в CH2Cl2 (1 мл) при 0°С. Полученную смесь нагревали до комнатной температуры, перемешивали в течение 3 ч, затем промывали водой (5 мл). Органическую фазу концентрировали и очищали с помощью колоночной хроматографии с использованием смеси EtOAc: МеОН 1:1. Соответствующие фракции объединяли и концентрировали в вакууме и получали 144 мг (E)-3-{2-[(2-диметиламиноэтил)-метил-амино]-5-[4-(1-метил-1Н-индол-3-ил)-пиримидин-2-иламино]-4-метоксифенилкарбамоил}-акриловой кислоты метилового эфира (1.7), 1H-NMR (DMSO-d6, 400 MHz): 2.24 (s, 6Н), 2.36 (t, J=5.5 Hz, 2H), 2.74 (s, 3H), 2.88 (t, J=5.5 Hz, 2H), 3.77 (s, 3H), 3.87 (s, 3H), 3.91 (s, 3H), 6.73 (d, J=15.5 Hz, 1H), 7.06 (s, 1H), 7.13 (t, J=7.1 Hz, 1H), 7.17-7.29 (m, 3H), 7.53 (d, J=8.1 Hz, 1H), 7.92 (s, 1H), 8.25 (d, J=8.1 Hz, 1H), 8.32 (d, J=5.5 Hz, 1H), 8.63 (s, 1H), 9.17 (s, 1H), 10.59 (s, 1H). К перемешиваемому раствору ингибитора 1.7 (144 мг, 0,26 ммоль) в ацетоне (6 мл) при 25°С добавляли метансульфоновую кислоту (20 мг, 0,21 ммоль) в виде раствора в ацетоне (1 мл). Полученную смесь перемешивали в течение 15 ч. Осадок отфильтровывали и сушили на воздухе в течение ночи, получали 75 мг мезилата (E)-3-{2-[(2-диметиламиноэтил)-метиламино]-5-[4-(1-метил-1Н-индол-3-ил)-пиримидин-2-иламино]-4-метоксифенилкарбамоил}-акриловой кислоты метилового эфира (1.7⋅CH3SO3H) в виде желтых кристаллов, 1H-NMR (DMSO-d6, 400 MHz): 2.32 (s, 3Н), 2.63 (s, 3Н), 2.80 (br.s, 6H), 3.29 (br.s, 4H), 3.78 (s, 3Н), 3.89 (s, 3Н), 3.90 (s, 3Н), 6.78 (d, J=15.3 Hz, 1H), 7.03 (s, 1H), 7.14 (t, J=7.5 Hz, 1H), 7.20-7.27 (m, 2H), 7.50-7.60 (m, 2H), 7.95 (s, 1H), 8.28-8.35 (m, 2H), 8.52 (s, 1H), 8.89 (s, 1H), 9.32 (br.s, 2H), 9.88 (br.s, 1H).
Пример 6. (E)-3-{2-[(2-диметиламиноэтил)-метиламино]-5-[4-(1-метил-1Н-индол-3-ил)-пиримидин-2-иламино]-4-метоксифенилкарбамоил}-N,N-диметилакриламид (1.8) и его бис-дихлорацетат 1.8⋅2CHCl2CO2H получают согласно схеме 6.
Схема 6.
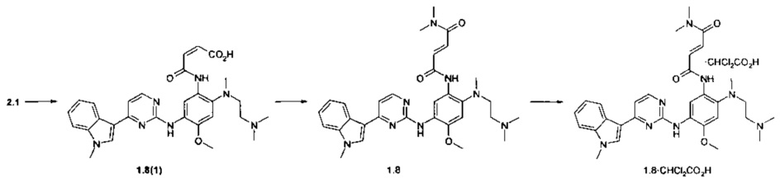
К перемешиваемому раствору N1-[2-(диметиламино)этил]-N1-метил-N4-[4-(1-метил-1Н-индол-3-ил)пиримидин-2-ил]-5-метоксибензол-1,2,4-триамина (2.1, 500 мг, 1.12 ммоль) в диоксане (4 мл) добавляли фуран-2,5-дион (110 мг, 1.12 ммоль) в диоксане (1 мл) при комнатной температуре. Полученную смесь перемешивали в течение 15 ч. Осадок отфильтровывали и сушили на воздухе в течение ночи. Получали 570 мг (93%) (Z)-3-{2-[(2-диметиламиноэтил)-метиламино]-5-[4-(1-метил-1Н-индол-3-ил)-пиримиин-2-иламино]-4-метоксифенилкарбамоил}-акриловой кислоты (1.8(1)). 1H-NMR (DMSO-d6, 400 MHz): 2.54 (s, 3Н), 2.72 (s, 6H), 3.25-3.30 (m, 4H), 3.85 (s, 3Н), 3.90 (s, 3Н), 5.80 (d, J=13.1 Hz, 1H), 6.26 (d, J=13.1 Hz, 1H), 6.92 (s, 1H), 7.12 (t, J=7.5 Hz, 1H), 7.19 (d, J=5.4 Hz, 1H), 7.22 (t, J=7.5 Hz, 1H), 7.51 (d, J=8.1 Hz, 1H), 7.90 (s, 1H), 8.24 (d, J=8.1 Hz, 1H), 8.29 (d, J=5.4 Hz, 1H), 8.66 (s, 1H), 9.30 (s, 1H), 12.76-13.17 (m, 3Н). Этилхлорформиат (120 мг, 1,1 ммоль) в безводном ТГФ (10 мл) добавляли к суспензии полученной кислоты 1.8(1) (600 мг, 1,1 ммоль) и триэтиламин (223 мг, 2,2 ммоль) в безводном ТГФ (50 мл) при -15°С в потоке азота. Через 30 мин добавляли 20%-ный раствор диметиламина в ТГФ (2,5 мл) и полученную смесь перемешивали в течение ночи при комнатной температуре. Осадок отфильтровывали, а маточник упаривали в вакууме. Получали масло, которое подвергали хроматографии на колонке с силикагелем (ацетоном-МеОН, 20:1). Получали 73 мг продукта 1.8, который перекристаллизовывали из диэтилового эфира. 1H-NMR (DMSO-d6, 400 MHz): 2.29 (s, 6Н), 2.41 (br.s, 2Н), 2.73 (s, 3Н), 2.89 (br.s, 2Н), 2.95 (s, 3Н), 3.12 (s, 3Н), 3.87 (s, 3Н), 3.93 (s, 3Н), 7.01-7.11 (m, 2Н), 7.15 (t, J=7.5 Hz, 1H), 7.20-7.29 (m, 2H), 7.39 (d, J=14.7 Hz, 1H), 7.52 (d, J=8.3 Hz, 1H), 7.90 (s, 1H), 8.24 (d, J=8.3 Hz, 1H), 8.33 (d, J=5.3 Hz, 1H), 8.68 (s, 1H), 9.22 (s, 1H), 10.51 (br.s, 1H).
К перемешиваемому раствору ингибитора 1.8 (26 мг, 0.046 ммоль) в ацетоне (1 мл) при 25°С добавляли дихлоруксусную кислоту (5.9 мг, 0.046 ммоль) в виде раствора в ацетоне (1 мл). Полученную смесь перемешивали в течение 15 ч. Осадок отфильтровали и сушили на воздухе в течение ночи. Получали 24 мг ингибитора 1.8⋅2CHCl2CO2H в виде светло-желтых кристаллов. 1H-NMR (DMSO-d6, 400 MHz): 2.61 (s, 3Н), 2.78 (s, 6H), 2.95 (s, 3Н), 3.11 (s, 3Н), 3.22-3.32 (m, 4H), 3.90 (s, 3Н), 3.91 (s, 3Н), 6.12 (s, 1H), 7.01 (s, 1H), 7.15 (t, J=7.5 Hz, 1H), 7.20-7.27 (m, 2H), 7.40 (s, 1H), 7.52 (d, J=8.3 Hz, 1H), 7.94 (s, 1H), 8.30 (d, J=8.3 Hz, 1H), 8.32 (d, J=5.3 Hz, 1H), 8.57 (s, 1H), 8.96 (s, 1H), 9.81 (s, 1H), 10.10 (br.s, 1H).
Пример 7. N-(2-[3-(Диметиламино)пиперидин-1-ил]-5-{{4-(1-метил-1Н-индол-3-ил)пиримидин-2-ил]амино}-4-метоксифенил)акриламид (1.11) и его мезилат 1.11⋅CH3SO3H получают в соответствии со схемой 7.
Схема 7.
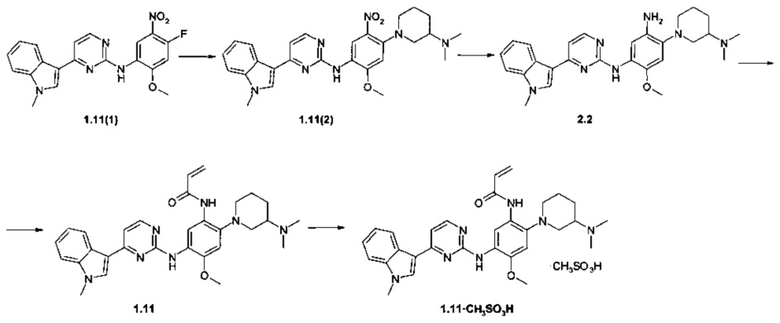
К перемешиваемому раствору N-(2-метокси-5-нитрофенил-4-фтор)-4-(1-метил-1Н-индол-3-ил)-пиримидин-2-амина (1.11 (1): 2,0 г, 5,1 ммоль) и N,N-диметилпиперидин-3-амина (1,2 г, 6,1 ммоль) в ДМФ (60 мл) добавляли при комнатной температуре TEA (2,1 г, 20,3 ммоль). Полученную смесь перемешивали при 85°С в течение 6 ч, затем охлаждали до комнатной температуры, добавляли воду (250 мл) и полученную смесь перемешивали в течение 3 ч. Твердое вещество собирали фильтрацией, промывали водой (50 мл) и сушили на воздухе в течение ночи. Получали [4-(3-диметиламинопиперидин-1-ил)-2-метокси-5-нитрофенил]-[4-(1-метил-1Н-индол-3-ил)-пиримидин-2-ил]-амин (1.11(2): 5,6 г, 82%), 1Н-NMR (DMSO-d6, 400 MHz): 1.22-1.35 (m, 1Н), 1.57-1.70 (m, 1H), 1.76-1.85 (m, 1H), 1.91-2.01 (m, 1H), 2.23 (s, 6H), 2.41-2.48 (m, 1H), 2.63-2.71 (m, 1H), 2.74-2.83 (m, 1H), 3.17-3.24 (m, 1H), 3.33-3.38 (m, 1H), 3.88 (s, 3H), 3.99 (s, 3H), 6.85 (s, 1H), 7.12 (t, J=7.5 Hz, 1H), 7.23 (d, J=5.4 Hz, 1H), 7.25 (t, J=7.5 Hz, 1H), 7.52 (d, J=8.1 Hz, 1H), 8.12 (s, 1H), 8.31-8.39 (m, 3H), 8.79 (s, 1H). К смеси соединения 1.11(2) (2,1 г, 4,2 ммоль), железа (1,4 г, 25,2 ммоль), NH4Cl (0,16 г, 2,9 ммоль) и этанола (40 мл) добавляли в один прием воду (5,5 мл). Полученную смесь нагревали при 80°С в течение 3 ч, затем фильтровали через слой целита. Фильтрат концентрировали и очищали с помощью колоночной хроматографии с использованием смеси хлороформа и метанола в соотношении 10:1. Соответствующие фракции объединяли и концентрировали в вакууме. Получали 4-[3-(диметиламино)пиперидин-1-ил]-6-метокси-N-[4-(1-метил-1Н-индол-3-ил)пиримидин-2-ил]бензол-1,3-диамина (2.2: 1,7 г, 86%), 1H-NMR (DMSO-d6, 400 MHz): 1.00-1.16 (m, 1H), 1.29-1.51 (m, 1H), 1.52-1.74 (m, 1H), 1.74-2.01 (m, 1Н), 2.38 (s, 6H), 2.52-2.63 (m, 1H), 2.82-3.00 (m, 1H), 3.12-3.28 (m, 1H), 3.32-3.45 (m, 1H), 3.75 (s, 3H), 3.87 (s, 3H), 6.72 (br.s, 1H), 7.09-7.31 (m, 3H), 7.45-7.62 (m, 2H), 7.76 (br.s, 1H), 8.22-8.34 (m, 2H), 8.43 (br.s, 1H). К перемешиваемой суспензии соединения 2.2 (700 мг, 1,48 ммоль) и K2CO3 (143 мг, 1,04 ммоль) в ацетоне (15 мл) добавляли метил-3-хлорпропаноил хлорид (226 мг, 1,78 ммоль) в ацетоне (1 мл) при -50°С. Полученную смесь нагревали до -20°С и перемешивали в течение 30 мин. Затем добавляли МеОН (2 мл) и раствор NaOH (53 мг, 1,33 ммоль в 7 мл воды). Полученную смесь перемешивают в течение 3 ч при комнатной температуре. Твердое вещество собирали фильтрацией и сушили на воздухе в течение ночи. Получали 150 мг N-(2-[3-(диметиламино)пиперидин-1-ил]-5-{[4-(1-метил-1Н-индол-3-ил)пиримидин-2-ил]амино}-4-метоксифенил)акриламида (1.11), 1H-NMR (DMSO-d6, 400 MHz): 1.21-1.41 (m, 1H), 1.67-1.86 (m, 2H), 1.87-1.97 (m, 1H), 2.21 (s, 6H), 2.41-2.48 (m, 1H), 2.58-2.70 (m, 1H), 2.90-2.99 (m, 1H), 3.08-3.15 (m, 1H), 3.33-3.38 (m, 1H), 3.87 (s, 3H), 3.91 (s, 3H), 5.75 (d, J=10.5 Hz, 1H), 6.24 (d, J=17.0 Hz, 1H), 6.65 (dd, J1=10.5 Hz, J2=17.0 Hz, 1H), 6.88 (s, 1H), 7.14-7.28 (m, 3H), 7.52 (d, J=8.1 Hz, 1H), 7.86 (s, 1H), 8.26 (d, J=8.1 Hz, 1H), 8.31 (d, J=5.5 Hz, 1H), 8.58 (s, 1H), 8.84 (s, 1H), 9.09 (s, 1H). К перемешиваемому раствору ингибитора 1.11 (100 мг, 0,19 ммоль) в ацетоне (4 мл) при 25°С добавляли метансульфоновую кислоту (18 мг, 0,19 ммоль) в виде раствора в ацетоне (1 мл). Полученную смесь перемешивали в течение 15 ч. Полученное твердое вещество собирали фильтрованием и сушили на воздухе в течение ночи. Получали 80 мг N-(2-[3-(диметиламино)пиперидин-1-ил]-5-{[4-(1-метил-1Н-индол-3-ил)пиримидин-2-ил]амино}-4-метоксифенил)акриламида мезилат (1.11⋅CH3SO3H) в виде светло-желтых кристаллов, 1Н-NMR (DMSO-d6, 400 MHz): 1.69-1.85 (m, 2Н), 1.85-1.96 (m, 1Н), 1.98-2.06 (m, 1H), 2.34 (s, 6H), 2.74-2.93 (m, 8H), 3.02-3.12 (m, 1H), 3.23-3.29 (m, 1H), 3.49-3.59 (m, 1H), 3.89 (s, 3H), 3.91 (s, 3H), 5.77 (d, J=10.5 Hz, 1H), 6.28 (d, J=17.0 Hz, 1H), 6.85 (dd, J1=10.5 Hz, J2=17.0 Hz, 1H), 6.93 (s, 1H), 7.13-7.32 (m, 3H), 7.52 (d, J=8.1 Hz, 1H), 7.91 (s, 1H), 8.28 (d, J=8.1 Hz, 1H), 8.32 (d, J=5.5 Hz, 1H), 8.57 (s, 1H), 8.87 (s, 1H), 9.22 (s, 1H), 9.67 (br.s, 1H).
Пример 8. N-{2-[(2-диметиламиноэтил)-метиламино]-5-[4-(1-метил-1Н-индол-3-ил)-пиримиин-2-иламино]-4-метоксифенил}-пропинамид (1.12) и его димезилат (1.12⋅2CH3SO3H) получают в соответствии со схемой 8.

К перемешиваемому раствору N1-[2-(диметиламино)этил]-N1-метил-N4-[4-(1-метил-1Н-индол-3-ил)пиримидин-2-ил]-5-метоксибензол-1,2,4-триамина (2.1: 1,0 г, 2,2 ммоль), DCC (0,51 г, 2,4 ммоль) и пропиоловой кислоты (157 мг, 2,2 ммоль) в CH2Cl2 (30 мл) добавляли DMAP (27 мг, 0,22 ммоль) при комнатной температуре. Полученную смесь перемешивали в течение ночи. Твердое вещество собирали фильтрацией, маточный раствор концентрировали и очищали с помощью колоночной хроматографии с использованием смеси ацетон в качестве элюента. Соответствующие фракции объединяли и концентрировали в вакууме. Получали 63 мг N-{2-[(2-диметиламиноэтил)-метиламино]-5-[4-(1-метил-1Н-индол-3-ил)-пиримиин-2-иламино]-4-метоксифенил}-пропинамид (1.12), 1Н-NMR (DMSO-d6, 400 MHz): 2.28 (s, 8Н), 2.73 (s, 3Н), 2.85-2.93 (m, 2Н), 3.85 (s, 3Н), 3.90 (s, 3Н), 4.31 (s, 1Н), 7.07 (s, 1H), 7.16 (t, J=7.3 Hz, 1H), 7.22 (d, J=5.5 Hz, 1H), 7.25 (t, J=7.3 Hz, 1H), 7.52 (d, J=8.1 Hz, 1H), 7.91 (s, 1H), 8.26 (d, J=8.1 Hz, 1H), 8.31 (d, J=5.5 Hz, 1H), 8.52 (s, 1H), 8.93 (s, 1H), 11.41 (s, 1H). К раствору ингибитора 1.12 (63 мг, 0,13 ммоль) в ацетоне (3 мл) при 25°С добавляли метансульфоновую кислоту (24 мг, 0,26 ммоль) в виде раствора в ацетоне (1 мл). Полученную смесь перемешивали в течение 15 ч. Осадок отфильтровывали и сушили на воздухе в течение ночи. Получали 31 мг N-{2-[(2-диметиламиноэтил)-метиламино]-5-[4-(1-метил-1Н-индол-3-ил)-пиримиин-2-иламино]-4-метоксифенил}-пропинамид димезилат (1.12⋅2CH3SO3H) в виде светло-желтых кристаллов, 1H-NMR (DMSO-d6, 400 MHz): 2.34 (s, 6H), 2.69 (s, 3Н), 2.86 (s, 6H), 3.28-3.39 (m, 2H), 3.86 (s, 3Н), 3.93 (s, 3Н), 4.47 (s, 1H), 7.05 (s, 1H), 7.21 (t, J=7.3 Hz, 1H), 7.32 (t, J=7.3 Hz, 1H), 7.39 (d, J=5.5 Hz, 1H), 7.60 (d, J=8.1 Hz, 1H), 8.16 (br.s, 1H), 8.24 (br.s, 2H), 8.72 (s, 1H), 9.13 (br.s, 1H), 9.97 (s, 1H).
Пример 9. N-{2-[(2-диметиламино-этил)-метиламино]-5-5-[4-(1-метил-1H-индол-3-ил)-пиримидин-2-иламино]-4-метоксифенил}-2-бутинамид (1.14) и его мезилат 1.14⋅CH3SO3H получают в соответствии со схемой 9.
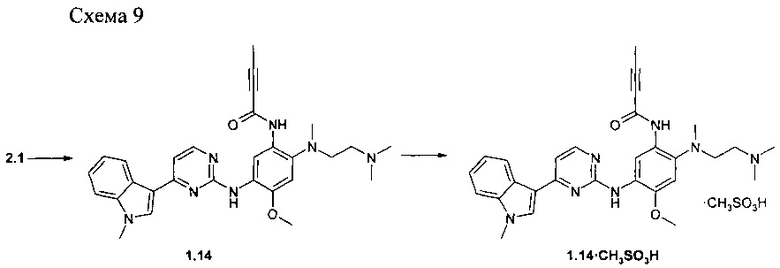
К перемешиваемому раствору N1-[2-(диметиламино)этил]-N1-метил-N4-[4-(1-метил-1Н-индол-3-ил)пиримидин-2-ил]-5-метоксибензол-1,2,4-триамина (2.1: 1,0 г, 2,2 ммоль), DCC (0,51 г, 2,4 ммоль) и бут-2-иновой кислоты (189 мг, 2,2 ммоль) в CH2Cl2 (30 мл) добавляли при комнатной температуре DMAP (27 мг, 0,22 ммоль). Полученную смесь перемешивали в течение ночи. Осадок отфильтровывали, маточный раствор концентрировали и очищали с помощью колоночной хроматографии с использованием ацетона в качестве элюента. Соответствующие фракции объединяли и концентрировали в вакууме. Получали 440 мг ингибитора 1.14. 1H-NMR (DMSO-d6, 400 MHz): 2.05 (s, 3Н), 2.28 (s, 8H), 2.72 (s, 3Н), 2.88 (t, J=5.3 Hz, 2H), 3.84 (s, 3H), 3.90 (s, 3H), 7.05 (s, 1H), 7.15 (t, J=7.3 Hz, 1H), 7.21 (d, J=5.5 Hz, 1H), 7.25 (t, J=7.3 Hz, 1H), 7.52 (d, J=8.1 Hz, 1H), 7.92 (s, 1H), 8.26 (d, J=8.1 Hz, 1H),8.31 (d, J=5.5 Hz, 1H), 8.52 (s, 1H), 8.92 (s, 1H), 11.08 (s, 1H).
К перемешиваемому раствору ингибитора 1.14 (300 мг, 0,13 ммоль) в ацетоне (5 мл) при 25°С добавляли метансульфоновую кислоту (48 мг, 0.11 ммоль) в виде раствора в ацетоне (1 мл). Полученную смесь перемешивали в течение 15 ч. Осадок отфильтровали и сушили на воздухе в течение ночи. Получали 245 мг ингибитора 1.14⋅CH3SO3H в виде светло-желтых кристаллов. 1H-NMR (DMSO-d6, 400 MHz): 2.07 (s, 3Н), 2.33 (s, 3Н), 2.63 (s, 3Н), 2.83 (s, 6H), 3.21-3.32 (m, 4H), 3.89 (s, 6H), 6.97 (s, 1H), 7.20 (t, J=7.3 Hz, 1H), 7.22 (d, J=5.5 Hz, 1H), 7.26 (t, J=7.3 Hz, 1H), 7.53 (d, J=8.1 Hz, 1H), 7.95 (s, 1H), 8.28-8.38 (m, 2H), 8.43 (s, 1H), 8.57 (s, 1H), 9.08 (br.s, 1H), 9.78 (br.s, 2H).
Пример 10. Определение термодинамической растворимости соединения формулы 1 и прототипа AZD-9291. Смешивали 5 мг исследуемого соединения с 1 мл универсального буфера (pION) с рН 2,0, 4,0 или 7,0 в течение 15 мин при 25°С. Дополнительные количества веществ добавляли до тех пор, пока раствор не становился мутным. Виалы с раствором инкубировали при перемешивании в течение 24 ч при 25°С для достижения равновесия между раствором и осадком при насыщении. После уравновешивания 200 мкл раствора (в 2-х повторах) фильтровали через 96-луночный фильтровальный планшет (Millipore) для отделения осадка. Концентрацию соединений в фильтрате определяли спектрофотометрически с помощью стандартной калибровочной кривой. Проводили измерение спектра оптического поглощения вещества и построение калибровочной кривой при выбранной длине волны (обычно, соответствующей максимуму поглощения вещества λmax). Концентрацию вещества в фильтрате (т.е. растворимость) рассчитывали по нижеприведенной формуле:
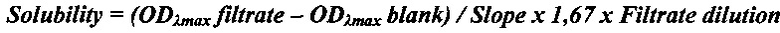 ,
,
где:
ODλmax filtrate - оптическая плотность фильтрата,
ODλmax blank - оптическая плотность холостого раствора без вещества,
Slope - наклон калибровочной кривой,
1,67 - фактор разведения фильтрата ацетонитрилом,
Filtrate dilution - фактор разведения фильтрата буфером.
Полученные результаты представлены в Таблице 1.
Пример 11. Анализ активности. Соединения формулы 1 и их соли тестировали по влиянию на активность киназ EGFR(L858R/T790M) и EGFR wt (все ферменты предоставлены Invitrogen Corp., каталожные номера PV3872, PV4128 and PV4803, соответственно) с помощью скрининговой платформы Z'-LYTE (производство Life Technologies). Концентрация ДМСО в реакционной смеси составляла 1%. 5 мкл 2-кратной смеси Субстрат/Киназа (Tyr4/EGFRwt или EGFR-L858R или EGFR-T790M, конечные концентрации - 0.5 мкМ для субстрата Tyr 4 и 1000, 250, 1000 нг/мл для EGFRwt или EGFR-L858R или EGFR-T790M соответственно) были добавлены в 384-луночные планшеты (черные, малого объема, производство Corning, кат. #3676). 1.5 мкл 100-кратных стоков исследуемых веществ в 100% ДМСО были разбавлены в 10 раз в 13.5 мкл киназного буфера (50 мМ HEPES pH 6.5, 0.01% BRIJ-35, 10 мМ MgCl2, 1 мМ EGTA, 0.02% NaN3) и затем 1 мкл разбавленных веществ добавляли к смеси Субстрат/Киназа. Вещества преинкубировали с киназами в течение 10 мин при комнатной температуре. После этого для начала реакции добавляли 4 мкл 2.5-кратного раствора АТФ (конечная концентрация АТФ в реакционной смеси была 180 или 100 или 40 мкМ для EGFRwt, EGFR-L858R и EGFR-T790M соответственно). После 30 сек инкубации на шейкере реакцию инкубировали в течение 60 мин при комнатной температуре. После этого добавляли 5 мкл Реагента В (разбавленного 1:500) и инкубировали еще 60 мин при комнатной температуре. Измеряли флуоресценцию при возбуждении длиной волны 400 нм и эмиссии при 445 и 520 нм. Степень фосфорилирования пептидного субстрата рассчитывали по формуле ниже (если отношение эмиссии низкое - пептид фосфорилирован, т.е. нет ингибирования активности киназы, если отношение высокое - пептид не фосфорилирован, т.е. киназа ингибирована)
% фосфорилирования рассчитывается по формуле, указанной на странице 6 в стандартном протоколе скрининга Z'-LYTE (Life Technologies).
Концентрационная кривая зависимости активности киназы от концентрации тестируемых веществ построена с использованием сигмоидной модели. Результаты сравнительного анализа активности некоторых соединений формулы (1) и прототипа AZD-9291 представлены в Таблице 2.
Пример 12. Получение лекарственного средства в виде таблеток. Спрессовывали смесь 1600 мг крахмала, 1600 мг измельченной лактозы, 400 мг талька и 1000 мг ингибитора 1.1(1). Полученный брусок измельчали в гранулы и просеивали через сито, собирая гранулы размером 14-16 меш. Полученные гранулы таблетировали в таблетки, пригодных форм весом 500 мг каждая.
Пример 13. Получение лекарственного средства в форме капсул. Ингибитор 1.1(1) тщательно смешивали с порошком лактозы в соотношении 2:1. Полученную порошкообразную смесь упаковывали по 600 мг в желатиновые капсулы подходящего размера.
Пример 14. Получение лекарственного средства в виде композиций для внутримышечных, внутрибрюшинных или подкожных инъекций. Смешали 500 мг ингибитора 1.1(1) 300 мг хлорбутанола, 2 мл пропиленгликоля и 100 мл инъекционной воды. Раствор фильтровали и помещали в 1 мл в ампулы и укупоривали.
Специалисты в данной области техники легко поймут различие некритических параметров, которые могут быть изменены или модифицированы, чтобы получить те же результаты. Различные модификации изобретения, в дополнение к описанным здесь, станут очевидными специалистам в данной области техники из приведенного выше описания.
Изобретение относится к новым замещенным N-{3-[4-(1-метил-1Н-индол-3-ил)пиримидин-2-иламино]-4-метоксифенил}-амидам общей формулы 1 и их фармацевтически приемлемым солям, обладающим свойствами модуляторов EGFR и пригодным для лечения рака, например мелкоклеточного рака легких. В общей формуле 1
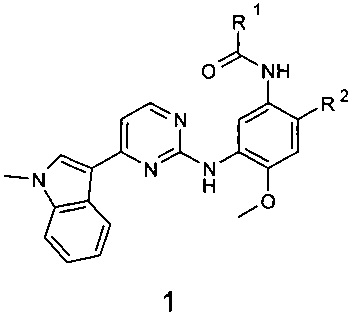
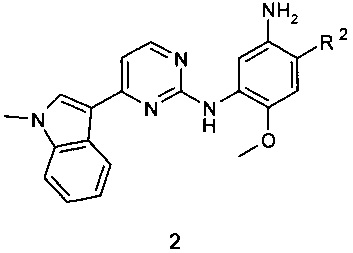
R1 представляет собой из CH2=C(R3)-, R3CH=CH-; R2 представляет собой (2-диметиламиноэтил)-метиламино, 3-диметиламинопиперидин-1-ил; R3 представляет собой фтор, трифторметил, диметилкарбамоил; или R2 представляет собой 3-диметиламинопиперидин-1-ил; R3 представляет собой Н; или мезилат соединения 1, где R1 представляет собой С(СН3)≡С-, R2 представляет собой (2-диметиламиноэтил)-метиламино. Предпочтительными соединениями являются N-{2-[(2-диметиламиноэтил)-метиламино]-5-[4-(1-метил-1Н-индол-3-ил)-пиримидин-2-иламино]-4-метоксифенил}-2-фтор-акриламид (1.2); N-{2-[(2-диметиламиноэтил)-метиламино]-5-[4-(1-метил-1Н-индол-3-ил)-пиримидин-2-иламино]-4-метоксифенил}-4,4,4-трифторбут-2-енамид (1.6); (E)-3-{2-[(2-диметиламиноэтил)-метиламино]-5-[4-(1-метил-1Н-индол-3-ил)-пиримидин-2-иламино]-4-метоксифенилкарбамоил}-N,N-диметилакриламид (1.8); N-{2-(3-диметиламинопиперидин-1-ил)-5-[4-(1-метил-1Н-индол-3-ил)-пиримидин-2-иламино]-4-метоксифенил}-акриламид (1.11); мезилат N-{2-[(2-диметиламиноэтил)-метиламино]-5-[4-(1-метил-1Н-индол-3-ил)-пиримидин-2-иламино]-4-метоксифенил}-2-бутинамида (1.14⋅CH3SO3H) или мезилат N-{2-(3-диметиламинопиперидин-1-ил)-5-[4-(1-метил-1Н-индол-3-ил)-пиримидин-2-иламино]-4-метоксифенил}-акриламида (1.11⋅CH3SO3H). Изобретение также относится к способу получения соединений формулы 1. Способ заключается во взаимодействии соединения формулы 2 или его соли с активированным производным соответствующей органической кислоты, таким как хлорангидрид, сложный эфир или ангидрид органической кислоты. 11 н. и 4 з.п. ф-лы, 2 табл., 14 пр.
1. Замещенные N-{3-[4-(1-метил-1Н-индол-3-ил)пиримидин-2-иламино]-4-метоксифенил}-амиды общей формулы 1 и их фармацевтически приемлемые соли
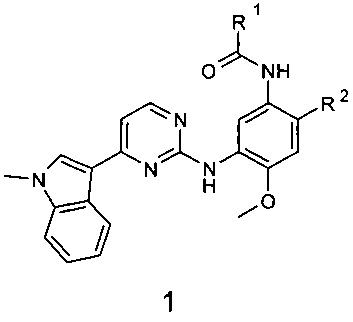
где: R1 представляет собой из CH2=C(R3)-, R3CH=CH-;
R2 представляет собой (2-диметиламиноэтил)-метиламино, 3-диметиламинопиперидин-1-ил;
R3 представляет собой фтор, трифторметил, диметилкарбамоил; или
R2 представляет собой 3-диметиламинопиперидин-1-ил; R3 представляет собой Н;
или мезилат соединения 1, где R1 представляет собой С(СН3)≡С-, R2 представляет собой (2-диметиламиноэтил)-метиламино.
2. Соединения по п. 1, выбранные из: N-{2-[(2-диметиламиноэтил)-метиламино]-5-[4-(1-метил-1Н-индол-3-ил)-пиримидин-2-иламино]-4-метоксифенил}-2-фтор-акриламида (1.2);
N-{2-[(2-диметиламиноэтил)-метиламино]-5-[4-(1-метил-1Н-индол-3-ил)-пиримидин-2-иламино]-4-метоксифенил}-4,4,4-трифторбут-2-енамида (1.6);
(E)-3-{2-[(2-диметиламиноэтил)-метиламино]-5-[4-(1-метил-1Н-индол-3-ил)-пиримидин-2-иламино]-4-метоксифенилкарбамоил}-N,N-диметилакриламида (1.8);
N-{2-(3-диметиламинопиперидин-1-ил)-5-[4-(1-метил-1Н-индол-3-ил)-пиримидин-2-иламино]-4-метоксифенил}-акриламида (1.11);
или их фармацевтически приемлемых солей.
3. Соединения по п. 1, выбранные из мезилата N-{2-[(2-диметиламиноэтил)-метиламино]-5-[4-(1-метил-1Н-индол-3-ил)-пиримидин-2-иламино]-4-метоксифенил}-2-бутинамида (1.14⋅CH3SO3H) или мезилата N-{2-(3-диметиламинопиперидин-1-ил)-5-[4-(1-метил-1Н-индол-3-ил)-пиримидин-2-иламино]-4-метоксифенил}-акриламида (1.11⋅CH3SO3H).
4. Активный компонент, обладающий противораковой активностью, для приготовления фармацевтической композиции и лекарственных форм, представляющий собой соединение по любому из пп. 1, 2 или 3.
5. Лекарственное средство, представляющее собой соединение по любому из пп. 1, 2 или 3 для применения для лечения рака.
6. Фармацевтическая композиция, обладающая противораковой активностью, в форме таблеток, капсул или инъекций, помещенных в фармацевтически приемлемую упаковку, включающая активный компонент по п. 4 в терапевтически эффективном количестве в сочетании с фармацевтически приемлемым разбавителем и/или носителем.
7. Способ профилактики и/или лечения рака, включающий введение в эффективном количестве соединения по пп. 1, или 2, или 3.
8. Способ профилактики и/или лечения рака, включающий введение в эффективном количестве активного компонента по п. 4 или лекарственного средства по п. 5.
9. Способ профилактики и/или лечения рака, включающий введение в эффективном количестве фармацевтической композиции по п. 6.
10. Способ по любому из пп. 7, 8 или 9, предусматривающий профилактику и/или лечение мелкоклеточного рака легкого.
11. Способ получения противоракового эффекта у млекопитающего животного и человека, нуждающегося в таком лечении, который включает введение указанному млекопитающему животному и человеку эффективного количества соединения по пп. 1, 2 или 3.
12. Способ изготовления лекарственного средства смешением соединения по пп. 1, 2 или его фармацевтически приемлемой соли или соединения по п. 3 с фармацевтически приемлемым разбавителем и/или носителем.
13. Способ изготовления лекарственного средства по п. 12, пригодного для лечения мелкоклеточного рака легкого.
14. Применение соединения по любому из пп. 1, 2 или 3 для использования с дополнительным противоопухолевым веществом путем одновременного, раздельного или последовательного введения для лечения рака.
15. Способ получения соединения общей формулы 1 или его фармацевтически приемлемой соли взаимодействием соединения формулы 2 или его соли с активированным производным соответствующей органической кислоты, таким как хлорангидрид, сложный эфир или ангидрид органической кислоты

где R2 представляет собой (2-диметиламиноэтил)-метиламино, 3-диметиламинопиперидин-1-ил с выделением полученного соединения формулы 1 в свободном виде или в виде фармацевтически приемлемой соли.
| CN 105001208 A, 28.10.2015 | |||
| CN 104761544 A, 08.07.2015 | |||
| EA 201391491 А1, 29.08.2014. |

