Настоящее изобретение относится к новым производным 1-(3-аминофенил)-6,8-диметил-5-(4-иод-2-фтор-фениламино)-3-циклопропил-1H,6H-пиридо[4,3-d]пиримидин-2,4,7-триона и их фармацевтически приемлемым солям, представляющим интерес в качестве средства для лечения заболеваний, вызванных нежелательной пролиферацией клеток, в частности, в качестве противоопухолевого средства. Более конкретно настоящее изобретение относится к MEK1, MEK2 и MEK1/2 ингибиторам представляющим интерес в качестве противоопухолевых препаратов, в том числе для лечения злокачественных меланом.
Известны многочисленные ингибиторы к MEK1, MEK2 и MEK1/2, структура которых включает фрагмент 1H,6H-пиридо[4,3-d]пиримидин-2,4,7-триона [WO 2005/121142, WO 2012/088033, США 7378423], среди которых наиболее продвинутым препаратом является Mekinist (Trametinib dimethyl sulfoxide, GSK1120212) [H. Abe, S. Kikuchi, K. Hayakawa et al. ACS Med. Chem. Lett. 2011, 2, 320-324]. GSK1120212 является эффективным ингибитором MEK1/2, который показывает более высокую эффективность против u-MEK1/2 до активации C-Raf (u-MEK1: IC50=0,7 нМ) по сравнению с предварительно активированным (рр-MEK1: IC50=14.9 нМ) [http://clincancerres.aacrjournals.org/content/17/5/989.full].

Однако Траметиниб и Мекинист практически нерастворимы в водных средах в интервале от pH 2 до pH 8. В частности, растворимость Мекиниста в водной среде в диапазоне от pH 2 до pH 8 при температуре 37°С составляет всего 0,0002-0,0003 мг/мл [http://www.accessdata.fda.gov/drugsatfda_docs/nda/2013/204114Origls000ClinPharmR.pdf; http://www.gsk.ca/english/docs-pdf/product-monographs/mekinist.pdf].
Поэтому до сих пор остается потребность в соединениях, которые могут демонстрировать благоприятные профили эффективности по отношению к MEK1, MEK2 и MEK1/2 и иметь хорошую растворимость в водных средах. Такие соединения будут, как ожидается, больше подходить в качестве терапевтических агентов для лечения рака.
Заявители неожиданно обнаружили, что новые производные 1-(3-аминофенил)-6,8-диметил-5-(4-иод-2-фтор-фениламино)-3-циклопропил-1H,6H-пиридо[4,3-d]пиримидин-2,4,7-триона более эффективны по отношению к MEK1, MEK2 и MEK1/2 и имеют более высокую растворимость в водных средах.
Таким образом, новые соединения могут быть особенно полезны при лечении болезненных состояний, в которых участвуют MEK1, MEK2 и MEK1/2, например при лечении рака, в том числе при лечении злокачественных меланом.
Ниже приведены определения терминов, которые использованы в описании этого изобретения:«Алкил» означает алифатическую углеводородную линейную или разветвленную группу с 1-12 атомами углерода в цепи. Разветвленная означает, что алкильная цепь имеет один или несколько «низших алкильных» (C1-C6)алкильных заместителей. Предпочтительными алкильными группами являются (C1-C6)алкил, еще более предпочтительными являются (C1-C3)алкил, метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, циклопропилметил, циклобутилметил, циклопентилметил, н-пентил, 2-пентил, 3-пентил, нео-пентил, н-гексил, циклогексил. Алкил может иметь заместители.
«Алкокси» означает (C1-C3)алкил-O- группу, в которой алкил определен в данном разделе. Предпочтительным алкилокси группами являются метокси, этокси, н-пропокси, изопропокси и н-бутокси.
«Аминогруппа» означает R1R2N- группу, замещенную или незамещенную необязательно одинаковыми заместителями R1 и R2. Аминогруппа может иметь заместители.
«Активный компонент» (лекарственное вещество, лекарственная субстанция, drug-substance) означает физиологически активное вещество синтетического или иного (биотехнологического, растительного, животного, микробного и прочего) происхождения, обладающее фармакологической активностью и являющееся активным началом фармацевтической композиции, используемой для производства и изготовления лекарственного препарата (средства).
«Галоген» означает фтор, хлор, бром и йод. Предпочтительными являются фтор, хлор и бром.
«Гетероцикл» означает ароматическую или неароматическую насыщенную или частично насыщенную моноциклическую или полициклическую систему, включающую от 3 до 10 атомов углерода, преимущественно от 3 до 6 атомов углерода, в которой один или несколько атомов углерода заменены на гетероатом, такой как азот, кислород, сера, фосфор. Приставка «аза», «окса» или «тиа» перед гетероциклилом означает наличие в циклической системе атома азота, атома кислорода или атома серы соответственно. Гетероциклил может иметь один или несколько заместителей, которые могут быть одинаковыми или разными. Атомы азота и серы, находящиеся в гетероциклиле, могут быть окислены до N-оксида, S-оксида или S-диоксида. Представителями гетероциклилов являются пиперидинил, пирролидинил, пиперазинил, морфолинил, тиоморфолинил, тиазолидинил, 1,4-диоксан-2-ил, тетрагидрофурил, тетрагидротиенил и др.
«Замещенный алкил» - замещенный алкил может иметь один или несколько одинаковых или различных заместителей, включая галоген, алкенилокси, циклоалкил, арил, гетероарил, гетероциклил, ароил, гетероароил, циано, гидрокси, алкокси, карбокси, алкинилокси, аралкокси, арилокси, арилоксикарбонил, алкилтио, гетероарилтио, аралкилтио, арилсульфонил, алкилсульфонил, гетероаралкилокси или Rk aRk+1 aN-, где Rk a и Rk+1 a независимо друг от друга представляют собой «заместители аминогруппы», значение которых определено в данном разделе, например, атом водорода, алкил, арил, аралкил, гетероаралкил, гетероциклил или гетероарил, или Rk a и Rk+1 a вместе с атомом N, с которым они связаны, образуют через Rk a и Rk+1 a 4-7-ленный гетероциклил или гетероцикленил. Предпочтительными «алкильными заместителями» являются арил, гетероарил, гетероциклил, гидрокси, C1-C5 алкокси, C1-C5 алкоксикарбонил, аралкокси, арилокси, алкилтио, гетероарилтио, аралкилтио, алкилсульфонил, арилсульфонил, алкоксикарбонил, аралкоксикарбонил, гетероаралкилоксикарбонил или Rk aRk+1 aN-, Rk aRk+1 aNC(=O)-, аннелированный арилгетероцикленил, аннелированный арилгетероциклил.
«Заместители циклической системы» могут быть представителями арильных групп, предпочтительно фенил или нафтил, замещенный фенил или замещенный нафтил. Арил может быть аннелирован с неароматической циклической системой или гетероциклом. Предпочтительными заместителями циклической системы являются водород, галогены (хлор, фтор, бром), необязательно замещенный C1-C5алкил, необязательно замещенный циклоС1-C5алкил, C1-C5алкен, гидроксигруппа, C1-C5алкилоксигруппа (метокси, этокси, пропокси, диэфир этиленгликоль, диэфир метандиола), цианогруппа, C1-C5алкилоксикарбонил (метил, этил), алкилтиогруппа (метилтио), карбоксигруппа, аминокарбонил.
«Заместитель» означает химический радикал, который присоединяется к молекулярному остову (скэффолду, фрагменту), например «заместитель алкильный», «заместитель аминогруппы», «заместитель карбамоильный», «заместитель циклической системы», значения которых определено в данном разделе.
«Лекарственное средство (препарат)» - вещество (или смесь веществ в виде фармацевтической композиции) в виде таблеток, капсул, инъекций, мазей и других готовых форм, предназначенное для восстановления, исправления или изменения физиологических функций у человека и животных, а также для лечения и профилактики болезней, диагностики, анестезии, контрацепции, косметологии и прочего.
«Терапия» - процесс, целью которого является профилактика, облегчение, снятие или устранение симптомов и проявлений того или иного заболевания, патологического состояния или иного нарушения жизнедеятельности, нормализация нарушенных процессов жизнедеятельности и выздоровление, восстановление здоровья.
«Фармацевтическая композиция» обозначает композицию, включающую в себя соединение формулы 1 и, по крайней мере, один из компонентов, выбранных из группы, состоящей из фармацевтически приемлемых и фармакологически совместимых наполнителей, растворителей, разбавителей, носителей, вспомогательных, распределяющих и воспринимающих средств, средств доставки, таких как консерванты, стабилизаторы, наполнители, измельчители, увлажнители, эмульгаторы, суспендирующие агенты, загустители, подсластители, отдушки, ароматизаторы, антибактериальные агенты, фунгициды, лубриканты, регуляторы пролонгированной доставки, выбор и соотношение которых зависит от природы и способа назначения и дозировки. Примерами суспендирующих агентов являются этоксилированный изостеариловый спирт, полиоксиэтилен, сорбитол и сорбитовый эфир, микрокристаллическая целлюлоза, метагидроксид алюминия, бентонит, агар-агар и трагакант, а также смеси этих веществ. Защита от действия микроорганизмов может быть обеспечена с помощью разнообразных антибактериальных и противогрибковых агентов, например, таких как, парабены, хлорбутанол, сорбиновая кислота и подобные им соединения. Композиция может включать также изотонические агенты, например сахара, хлористый натрий и им подобные. Пролонгированное действие композиции может быть обеспечено с помощью агентов, замедляющих абсорбцию активного начала, например моностеарат алюминия и желатин. Примерами подходящих носителей, растворителей, разбавителей и средств доставки являются вода, этанол, полиспирты, а также их смеси, растительные масла (такие как оливковое масло) и инъекционные органические сложные эфиры (такие как этилолеат). Примерами наполнителей являются лактоза, молочный сахар, цитрат натрия, карбонат кальция, фосфат кальция и им подобные. Примерами измельчителей и распределяющих средств являются крахмал, альгиновая кислота и ее соли, силикаты. Примерами лубрикантов являются стеарат магния, лаурилсульфат натрия, тальк, а также полиэтиленгликоль с высоким молекулярным весом. Фармацевтическая композиция для перорального, сублингвального, трансдермального, внутримышечного, внутривенного, подкожного, местного или ректального введения активного начала, одного или в комбинации с другим активным началом, может быть введена животным и людям в стандартной форме введения в виде смеси с традиционными фармацевтическими носителями. Пригодные стандартные формы введения включают пероральные формы, такие как таблетки, желатиновые капсулы, пилюли, порошки, гранулы, жевательные резинки и пероральные растворы или суспензии, сублингвальные и трансбуккальные формы введения, аэрозоли, имплантаты, местные, трансдермальные, такие как мази и кремы, подкожные, внутримышечные, внутривенные, интраназальные или внутриглазные формы введения и ректальные формы введения.
«Фармацевтически приемлемая соль» означает относительно нетоксичные органические и неорганические соли кислот и оснований, заявленных в настоящем изобретении. Эти соли могут быть получены in situ в процессе синтеза, выделения или очистки соединений или приготовлены специально. В частности, соли оснований могут быть получены специально, исходя из очищенного свободного основания заявленного соединения и подходящей органической или неорганической кислоты. Примерами полученных таким образом солей являются гидрохлориды, гидробромиды, сульфаты, бисульфаты, фосфаты, нитраты, ацетаты, оксалаты, валериаты, олеаты, пальмитаты, стеараты, лаураты, бораты, бензоаты, лактаты, тозилаты, цитраты, малеаты, фумараты, сукцинаты, тартраты, мезилаты, малонаты, салицилаты, пропионаты, этансульфонаты, бензолсульфонаты, сульфаматы и им подобные (Подробное описание свойств таких солей дано в Berge S.M., et al., "Pharmaceutical Salts" J. Pharm. Sci. 1977, 66: 1-19). Соли заявленных кислот также могут быть специально получены реакцией очищенной кислоты с подходящим основанием, при этом могут быть синтезированы соли металлов и аминов. К металлическим относятся соли натрия, калия, кальция, бария, цинка, магния, лития и алюминия, наиболее желательными из которых являются соли натрия и калия. Подходящими неорганическими основаниями, из которых могут быть получены соли металлов, являются гидроксид, карбонат, бикарбонат и гидрид натрия, гидроксид и бикарбонат калия, поташ, гидроксид лития, гидроксид кальция, гидроксид магния, гидроксид цинка. В качестве органических оснований, из которых могут быть получены соли заявленных кислот, выбраны амины и аминокислоты, обладающие достаточной основностью, чтобы образовать устойчивую соль, и пригодные для использования в медицинских целях (в частности, они должны обладать низкой токсичностью). К таким аминам относятся аммиак, метиламин, диметиламин, триметиламин, этиламин, диэтиламин, триэтиламин, бензиламин, дибензиламин, дициклогексиламин, пиперазин, этилпиперидин, трис(гидроксиметил)аминометан и подобные им. Кроме того, для солеобразования могут быть использованы гидроокиси тетраалкиламмония, например, такие как холин, тетраметиламмоний, тетраэтиламмоний и им подобные. В качестве аминокислот могут быть использованы основные аминокислоты - лизин, орнитин и аргинин.
Предметом настоящего изобретения являются производные 1-(3-аминофенил)-6,8-диметил-5-(4-иод-2-фтор-фениламино)-3-циклопропил-1H,6H-пиридо[4,3-d]пиримидин-2,4,7-триона общей формулы 1, их энантиомеры, фармацевтически приемлемые соли и/или сольваты:

где R1 представляет собой необязательно замещенный у атома азота радикал, выбранный из группы, которая включает в себя аминометил, 1-аминоэтил, (R)-1-аминоэтил, (S)-1-аминоэтил, 2-аминоэтил, 3-аминопропил, пирролидин-2-ил, (R)-пирролидин-2-ил и (S)-пирролидин-2-ил;
R2 представляет собой атом водорода, атом галогена или гидроксил или
R1 представляет собой метил, a R2 представляет собой атом галогена или гидроксил.
Предпочтительными являются соединения, в которых R1 замещен по азоту C1-C3 алкилом.
Предпочтительными являются также фармацевтически приемлемые соли, выбранные из гидрохлоридов, мезилатов, дихлорацетатов или фосфатов.
Предметом настоящего изобретения являются также ингибиторы MEK1, MEK2 и MEK1/2, представляющие собой соединения общей формулы 1.
Более предпочтительным ингибитором MEK1, MEK2 и MEK1/2 является соединение, выбранное из:
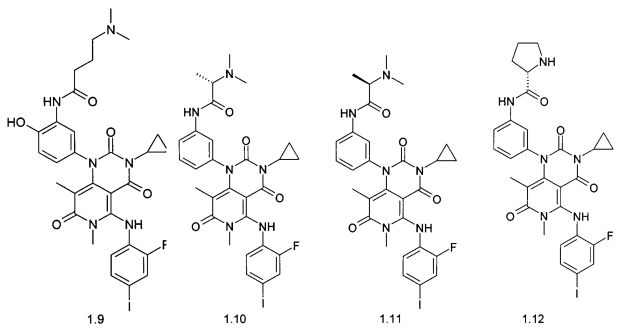
N-{3-[6,8-диметил-5-(4-иод-2-фтор-фениламино)-3-циклопропил-2,4,7-триоксо-3,4,6,7-тетрагидро-2H-пиридо[4,3-d]пиримидин-1-ил]-фенил}-2-диметиламиноацетамида (1.1),
N-{5-[6,8-диметил-5-(4-иод-2-фтор-фениламино)-3-циклопропил-2,4,7-триоксо-3,4,6,7-тетрагидро-2H-пиридо[4,3-d]пиримидин-1-ил]-2-фторфенил}-2-диметиламиноацетамида (1.2),
N-{2-гидрокси-5-[6,8-диметил-5-(4-иод-2-фтор-фениламино)-3-циклопропил-2,4,7-триоксо-3,4,6,7-тетрагидро-2H-пиридо[4,3-d]пиримидин-1-ил]-фенил}-2-диметиламиноацетамида (1.3),
N-{3-[6,8-диметил-5-(4-иод-2-фтор-фениламино)-3-циклопропил-2,4,7-триоксо-3,4,6,7-тетрагидро-2H-пиридо[4,3-d]пиримидин-1-ил]-фенил}-2-диметиламинопропионамида (1.4),
N-{5-[6,8-диметил-5-(4-иод-2-фтор-фениламино)-3-циклопропил-2,4,7-триоксо-3,4,6,7-тетрагидро-2H-пиридо[4,3-d]пиримидин-1-ил]-2-фторфенил}-2-диметиламинопропионамида (1.5),
N-{2-гидрокси-5-[6,8-диметил-5-(4-иод-2-фтор-фениламино)-3-циклопропил-2,4,7-триоксо-3,4,6,7-тетрагидро-2H-пиридо[4,3-d]пиримидин-1-ил]-фенил}-2-диметиламинопропионамида (1.6),
N-{3-[6,8-диметил-5-(4-иод-2-фтор-фениламино)-3-циклопропил-2,4,7-триоксо-3,4,6,7-тетрагидро-2H-пиридо[4,3-d]пиримидин-1-ил]-фенил}-2-диметиламинобутирамида (1.7),
N-{5-[6,8-диметил-5-(4-иод-2-фтор-фениламино)-3-циклопропил-2,4,7-триоксо-3,4,6,7-тетрагидро-2H-пиридо[4,3-d]пиримидин-1-ил]-2-фторфенил}-2-диметиламинобутирамида (1.8),
N-{2-гидрокси-5-[6,8-диметил-5-(4-иод-2-фтор-фениламино)-3-циклопропил-2,4,7-триоксо-3,4,6,7-тетрагидро-2H-пиридо[4,3-d]пиримидин-1-ил]-фенил}-2-диметиламинобутирамида (1-9),
(R)-N-{3-[6,8-диметил-5-(4-иод-2-фтор-фениламино)-3-циклопропил-2,4,7-триоксо-3,4,6,7-тетрагидро-2H-пиридо[4,3-d]пиримидин-1-ил]-фенил}-2-диметиламинопропионамида (1.10),
(S)-N-{3-[6,8-диметил-5-(4-иод-2-фтор-фениламино)-3-циклопропил-2,4,7-триоксо-3,4,6,7-тетрагидро-2H-пиридо[4,3-d]пиримидин-1-ил]-фенил}-2-диметиламинопропионамида (1.11),
{3-[6,8-диметил-5-(4-иод-2-фтор-фениламино)-3-циклопропил-2,4,7-триоксо-3,4,6,7-тетрагидро-2H-пиридо[4,3-d]пиримидин-1-ил]-фенил}-амид (R)-пирролидин-2-карбоновой кислоты (1.12),
{3-[6,8-диметил-5-(4-иод-2-фтор-фениламино)-3-циклопропил-2,4,7-триоксо-3,4,6,7-тетрагидро-2H-пиридо[4,3-d]пиримидин-1-ил]-фенил}-амид (S)-пирролидин-2-карбоновой кислоты (1.13),
{3-[6,8-диметил-5-(4-иод-2-фтор-фениламино)-3-циклопропил-2,4,7-триоксо-3,4,6,7-тетрагидро-2Н-пиридо[4,3-d]пиримидин-1-ил]-фенил}-амид (R)-1-метилпирролидин-2-карбоновой кислоты (1.14),
{3-[6,8-диметил-5-(4-иод-2-фтор-фениламино)-3-циклопропил-2,4,7-триоксо-3,4,6,7-тетрагидро-2Н-пиридо[4,3-d]пиримидин-1-ил]-фенил}-амид (S)-1-метилпирролидин-2-карбоновой кислоты (1.15),
N-{5-[6,8-диметил-5-(4-иод-2-фтор-фениламино)-3-циклопропил-2,4,7-триоксо-3,4,6,7-тетрагидро-2Н-пиридо[4,3-d]пиримидин-1-ил]-2-фторфенил}-ацетамида (1.16) или их фармацевтически приемлемых солей и/или сольватов:



Соединения общей формулы (1) и их фармацевтически приемлемые соли могут существовать в сольватированных и несольватированных формах. Например, сольватированная форма может быть гидратом или диметилсульфатом. Настоящее изобретение охватывает все сольватированные и несольватированные формы.
Безусловным преимуществом новых соединений общей формулы (1) является их высокая растворимость в водных средах. Так, например, расворимость ингибитора 1.1·HCl в водной среде при pH 7 более чем в 3000 раз выше, чем растворимость Траметиниба и Мекиниста, а растворимость других новых ингибиторов общей формулы (1) в водной среде при pH 7 еще выше (Таблица 1). В кислой среде (pH 2) новые ингибиторы растворяются еще лучше. Например, растворимость ингибиторов 1.1·HCl и 1.7·HCl имеет значение 5.0 мг/мл и 7.55 мг/мл, которые более чем в 16000 раз и в 25000 раз соответственно выше растворимости Траметиниба и Мекиниста. Ингибирующая активность новых соединений общей формулы (1) сопоставима с ингибирующей активностью Траметиниба и Мекиниста по отношению к MEK1. Так, например, ингибиторы 1.1·HCl и 1.4·HCl имеют IC50=9.02 nM и IC50=5.96 nM соответственно. Соответствующая активность известных ингибиторов, измеренная в идентичных условиях, имеет значение IC50=8.14 nM в случае Траметиниба и IC50=6.17 nM в случае Мекиниста.

Предметом настоящего изобретения является также ингибитор MEK1, MEK2 и MEK1/2, представляющий собой соединение общей формулы 1, его энантиомеры, фармацевтически приемлемые соли и/или сольваты. Предпочтительно такой ингибитор используется для лечения у пациента рака, в том числе для лечения злокачественных меланом.
Предметом настоящего изобретения является также способ получения соединений общей формулы (1), который заключается во взаимодействии соединения формулы 2 или его соли с органической кислотой или ее активированным производным (например, хлорангидридом или соответствующим активированным сложным эфиром или ангидридом органической кислоты) в растворителе, например, таком как CH2Cl2, тетрагидрофуран, N,N-диметилформамид, N,N-диметилацетамид или N-метилпирролидон:

где R2 имеет вышеуказанное значение.
Предметом настоящего изобретения является противораковый ингредиент для приготовления фармацевтических композиций и лекарственных форм, представляющий собой соединение общей формулы 1 или его фармацевтически приемлемую соль и/или сольват.
Предметом настоящего изобретения является фармацевтическая композиция, которая содержит соединение общей формулы 1 или его фармацевтически приемлемую соль и/или сольват и фармацевтически приемлемые наполнители.
Композиции по изобретению могут быть в форме, подходящей для перорального применения (например, в виде таблеток, пастилок, твердых или мягких капсул, водных или масляных суспензий, эмульсий, диспергируемых порошков или гранул, сиропов или эликсиров), для местного применения (например, в виде кремов, мазей, гелей или водных или масляных растворов или суспензий), для введения путем ингаляции (например, в виде тонкоизмельченного порошка или жидкого аэрозоля), для введения путем инсуффляции (например, в виде тонкоизмельченного порошка) или для парентерального введения (например, в виде стерильного водного или масляного раствора для внутривенного, подкожного, внутримышечного или внутримышечного введения дозы или в виде суппозиториев для ректального введения).
Композиции по изобретению могут быть получены обычными способами с использованием обычных фармацевтических наполнителей, хорошо известных в данной области. Таким образом, композиция, предназначенная для перорального применения, может содержать, например, одну или несколько окрашивающих, подслащивающих, вкусовых и/или консервант добавок.
Соединения общей формулы 1 обычно вводят пациенту в единичной дозе в диапазоне 5-5000 мг/м2 площади тела, то есть примерно 0,1-100 мг/кг, и это обычно обеспечивает терапевтически эффективную дозу. Форма единичной дозы, такой как таблетка или капсула, обычно содержит, например, 1-250 мг для активного ингредиента. Суточная доза обязательно будет варьировать в зависимости от пациента, конкретного пути введения и тяжести заболевания, которое лечат. Соответственно доктор, который лечит конкретного пациента, может определить оптимальную дозу.
В данном описании термин "терапия" также включает "профилактику.
Соединения общей формулы 1 и их фармацевтически приемлемые соли и сольваты, обладающие ингибирующей активностью по отношению к MEK1, MEK2 и MEK1/2, могут применяться при лечении заболеваний или медицинских состояний, опосредованных с MEK1, MEK2 и MEK1/2 активностью, например при лечении рака. Типы рака, которые могут быть восприимчивы к лечению с помощью соединений общей формулы 1, или их фармацевтически приемлемых солей и/или сольватов, включают в себя, но не ограничиваются, злокачественные меланомы.
Предметом настоящего изобретения является также лекарственное средство, включающее соединение формулы 1, как определено выше, или его фармацевтически приемлемую соль и/или сольват.
Предметом настоящего изобретения является также способ лечения заболевания, опосредованного через MEK1, MEK2 и MEK1/2, включающий использование соединения общей формулы 1, как определено выше, или его фармацевтически приемлемой соли и/или сольвата. Один из вариантов настоящего изобретения заключается в лечении заболевания, опосредованного через MEK1, MEK2 и MEK1/2, представляющего собой рак.
Предметом настоящего изобретения является также лекарственное средство для лечения заболевания, опосредованного MEK1, MEK2 и MEK1/2, включающее соединение общей формулы 1, как определено выше, или его фармацевтически приемлемую соль и/или сольват. Один из вариантов настоящего изобретения заключается в том, что указанное лекарственное средство предназначено для лечения заболевания, опосредованного через MEK1, MEK2 и MEK1/2, представляющего собой рак.
В соответствии с другим аспектом настоящего изобретения, предлагается использование соединения общей формулы 1, как определено выше, или его фармацевтически приемлемой соли и/или сольвата для изготовления лекарственного средства для лечения рака.
В соответствии с другим аспектом настоящего изобретения предложен также способ получения противоракового эффект в пациенте, нуждающемся в таком лечении, который включает введение указанному пациенту эффективного количества соединения общей формулы 1 или его фармацевтически приемлемой соли и/или сольвата.
В соответствии с другим аспектом настоящего изобретения предложен также способ лечения человека, страдающего от заболевания, при котором ингибирование MEK1/2 является полезным, содержащим этапы по введению человеку, нуждающемуся в этом, терапевтически эффективного количества соединения общей формулы 1 или его фармацевтически приемлемой соли и/ил сольвата. В частности, заболеванием, в котором полезно ингибирование MEK1, MEK2 и MEK1/2, является рак.
В любом из аспектов или вариантов, упомянутых здесь, где рак указан в общем смысле, указанный рак может быть выбран из опухоли головного мозга (нейроглиома, имеющая компонент злокачественной астроглиомы и олигодендроглиом и т.п.), рак пищевода, рак желудка, рак печени, рак поджелудочной железы, колоректальный рак (рак толстой кишки, рак прямой кишки и т.д.), рак легкого (немелкоклеточный рак легкого, мелкоклеточный рак легкого, первичных и метастатических раковых плоскоклеточный и т.д.), рак почки, рак молочной железы, рак яичников, рак предстательной железы, рак кожи, нейробластомы, саркомы, остеохондрома, остеому, остеосаркомы, семиномы, внегонадные опухоли, опухоли яичка, рак матки (рак шейки матки, рак эндометрия и т.п.), рак головы и опухоли шеи (рак гайморовой полости, рак гортани, рак глотки, рак языка, рак внутриротовой и т.п.), множественная миелома, злокачественная лимфома (ретикулосаркомы, лимфосаркома, болезнь Ходжкина и т.п.), истинная полицитемия, лейкоз (острый миелоидный лейкоз, хронический миелоидный лейкоз, острый лимфолейкоз, хронический лимфоцитарный лейкоз и т.д.), зоб, рак таза, опухоль мочеточника, опухоль мочевого пузыря, рак желчного пузыря, рак желчных протоков, злокачественная меланома, детская опухоль (саркома семьи Юинга, опухоли Вильмса, рабдомиосаркомы, сосудистая саркома, эмбриональный рак яичка, нейробластомы, ретинобластомы, гепатобластомы, нефробластома и т.д.) и тому подобное.
Кроме того, возможно лечение хронической боли, в частности невропатической боли, катаплексической боли, боли, связанной с хроническим алкоголизмом, дефицитом витамина, уремии и гипотиреоз. Также возможно лечение нейтрофил-опосредованного заболевания или симптомов, в частности ишемии реперфузионного повреждения, хронически обструктивного заболевания легких, синдрома острого респираторного заболевания, муковисцидоза, легочного фиброза, сепсиса, эндотоксемии, эмфиземы легких и легочного асбестоза. Также возможно лечение отторжения трансплантата, артрита, особенно ревматоидного артрита и остеоартрита. Кроме того, возможно лечение астмы, вирусных заболеваний, в частности вируса герпеса (ВПГ-1), цитомегаловируса человека (HCMV), вируса иммунодефицита человека (ВИЧ), заболевания, вызванного денатурацией или травмой хряща, в частности остеоартроза, ревматоидного артрита, остеохондроза и болезни, требующей хондрогенез. Помимо вышеизложенного, возможно лечение рестеноза, псориаза, атеросклероза, сердечной недостаточности, апоплексии и т.п.
Описанное выше лечение раковых заболеваний может применяться в качестве единственной терапии или может включать в дополнение к соединениям по настоящему изобретению, обычную хирургию, или лучевую терапию, или химиотерапию, или иммунотерапию. Такая химиотерапия может вводиться одновременно, последовательно или раздельно и может включать дополнительно один или более противоопухолевых агентов из следующих категорий: антипролиферативные / противоопухолевые препараты и их комбинации, используемые в медицинской онкологии; цитостатические агенты; агенты антивторжения; ингибиторы функции фактора роста; антиангиогенные агенты; сосудистые средства; повреждающие антагонисты рецепторов эндотелина; антисмысловые терапии; подходы генной терапии; и (J) подходы иммунотерапии.
Таким образом, согласно настоящему изобретению предложена фармацевтическая композиция, включающая соединения общей формулы 1 или их фармацевтически приемлемые соли и/или сольваты, как определено выше, и дополнительное противоопухолевое вещество, как определено выше, для совместного лечения рака.
Согласно настоящему изобретению предложен фармацевтический продукт, содержащий соединения формулы 1, или его фармацевтически приемлемую соль и/или сольват, и дополнительно противоопухолевое вещество, как определено выше, для комбинированного лечения рака.
Термин "совместное лечение", используемый по отношению к комбинированной терапии, следует понимать как одновременное, раздельное или последовательное введение.
Согласно изобретению предлагается использование соединения общей формулы 1 или его фармацевтически приемлемой соли и/или сольвата, как определено здесь, и дополнительно противоопухолевого вещества для совместного лечения рака.
Далее изобретение будет описано более подробно с помощью конкретных примеров. Следующие примеры представлены с целью иллюстрации и не предназначены для ограничения изобретения каким-либо образом. Специалисты в данной области техники легко поймут различные некритические параметры, которые могут быть изменены или модифицированы, чтобы дать существу те же результаты.
Пример 1. Общий способ получения игибиторов 1.1-1.9, 1.16 и их солей
Смесь 2,62 ммоль гидрохлорида диметиламино-уксусной кислоты, 3-диметиламино-пропионовой кислоты, 4-диметиламиномасляной кислоты, или уксусной кислоты, 1,74 ммоль соответствующего 1-(3-аминофенил)-6,8-диметил-5-(4-иод-2-фтор-фениламино)-3-циклопропил-1H,6H-пиридо[4,3-d]пиримидин-2,4,7-триона (2), 0,787 г (2,44 ммоль) N,N,N`,N`-тетраметил-O-(бензотриазол-1-ил)урония тетрафторбората и 0,56 г (4,36 ммоль) диизопропилэтиламина в 15 мл диметилформамида перемешивают 48 ч при комнатной температуре. По завершении реакции (ТСХ контроль) летучие вещества выпаривают в вакууме, остаток промывали дважды по 10 мл водой и сушат при пониженном давлении. Полученный продукт подвергали флэш-хроматографии, элюируя этилацетатом. Получают соответствующий ингибитор 1.1-1.9. Соли этих ингибиторов получают добавлением избытка HCl в этилацетате, фосфорной кислоты, метансульфокислоты или дихлоруксусной кислоты в ацетоне к раствору основания 1.1-1.9 в дихлорметане.
N-{3-[6,8-диметил-5-(4-иод-2-фтор-фениламино)-3-циклопропил-2,4,7-триоксо-3,4,6,7-тетрагидро-2H-пиридо[4,3-d]пиримидин-1-ил]-фенил}-2-диметиламино-ацетамида гидрохлорид (1.1·HCl): LC-MS (ESI) (m/z): 659 (М+Н)+; 1H-NMR (DMSO-d6, 400 MHz): 0.66 (m, 2H), 0.95 (m, 2H), 1.25 (s, 3H), 2.62 (m, 1H), 2.87 (s, 6H), 3.08 (s, 3H), 4.15 (s, 2H), 6.92 (t, J=8.7 Hz, 1H), 7.12 (d, J=8.3 Hz, 1H), 7.44 (t, J=8.2 Hz, 1H), 7.55 (d, J=8.2 Hz, 1H), 7.61-7.67 (m, 2H), 7.78 (d, J=10.5 Hz, 1H), 9.86 (br, 1H), 10.88 (s, 1H), 11.07 (s, 1H). MS(+) 658.5 (M+1). N-{5-[6,8-диметил-5-(4-иод-2-фтор-фениламино)-3-циклопропил-2,4,7-триоксо-3,4,6,7-тетрагидро-2H-пиридо[4,3-d]пиримидин-1-ил]-2-фторфенил}-2-диметиламиноацетамида гидрохлорид (1.2 HCl): LC-MS (ESI) (m/z): 677 (M+H)+. N-{2-гидрокси-5-[6,8-диметил-5-(4-иод-2-фтор-фениламино)-3-циклопропил-2,4,7-триоксо-3,4,6,7-тетрагидро-2H-пиридо[4,3-d]пиримидин-1-ил]-фенил}-2-диметиламиноацетамида гидрохлорид (1.3·HCl): LC-MS (ESI) (m/z): 675 (M+H)+. N-{3-[6,8-диметил-5-(4-иод-2-фтор-фениламино)-3-циклопропил-2,4,7-триоксо-3,4,6,7-тетрагидро-2H-пиридо[4,3-d]пиримидин-1-ил]-фенил}-2-диметиламинопропионамида гидрохлорид (1.4·HCl), фосфат (1.4·H3PO4), мезилат (1.4·CH3SO3H) и дихлорацетат (1.4·CHCl2CO2H) имеют в LC-MS (ESI) (m/z): 673 (М+Н)+. Гидрохлорид 1.4 HCl: 1H-NMR (DMSO-d6, 400 MHz): 0.65 (m, 2H), 0.95 (m, 2H), 1.25 (s, 3H), 2.62 (m, 1H), 2.77 (m, 6H), 2.9 (t, 2H), 3.02-3.1 (m, 5H), 6.92 (t, J=9 Hz, 1H), 7.06 (d, J=8 Hz, 1H), 7.39 (t, J=8 Hz, 1H), 7.55 (d, J=8 Hz, 1H), 7.62 (d, J=9 Hz, 1H), 7.66 (m, 1H), 7.78 (d, J=10 Hz, 1H), 10.1 (br, 1H), 10.55 (s, 1H), 11.07 (s, 1H). N-{5-[6,8-диметил-5-(4-иод-2-фтор-фениламино)-3-циклопропил-2,4,7-триоксо-3,4,6,7-тетрагидро-2H-пиридо[4,3-d]пиримидин-1-ил]-2-фторфенил}-2-диметиламинопропионамид гидрохлорид (1.5·HCl): LC-MS (ESI) (m/z): 691 (M+H)+. N-{3-[6,8-диметил-5-(4-иод-2-фтор-фениламино)-3-циклопропил-2,4,7-триоксо-3,4,6,7-тетрагидро-2H-пиридо[4,3-d]пиримидин-1-ил]-фенил}-2-диметиламинобутирамид гидрохлорид (1.7·HCl): LC-MS (ESI) (m/z): 687 (M+H)+. N-{5-[6,8-диметил-5-(4-иод-2-фтор-фениламино)-3-циклопропил-2,4,7-триоксо-3,4,6,7-тетрагидро-2H-пиридо[4,3-d]пиримидин-1-ил]-2-фторфенил}-2-диметиламинобутирамид гидрохлорид (1.8·HCl): LC-MS (ESI) (m/z): 705 (M+H)+; N-{5-[6,8-диметил-5-(4-иод-2-фтор-фениламино)-3-циклопропил-2,4,7-триоксо-3,4,6,7-тетрагидро-2H-пиридо[4,3-d]пиримидин-1-ил]-2-фторфенил}-2-ацетамид гидрохлорид (1.16·HCl): LC-MS (ESI) (m/z): 634 (M+H)+.
Пример 2. Общий способ получения игибиторов 1.10-1.15 и их солей
Смесь 0,70 ммоль N,N-диметил-L-аланина, N,N-диметил-D-аланина, N-Boc-L-пролина, N-Boc-D-пролина, N-метил-L-пролина или N-метил-D-пролина, 48 мг (0,35 ммоль) 7-аза-1-гидроксибензотриазол, 100 мг (0,52 ммоль) гидрохлорида 1-этил-(3-диметиламинопропил)карбодиимида и 0,34 ммоль соответствующего 1-(3-аминофенил)-6,8-диметил-5-(4-иод-2-фтор-фенламино)-3-циклопропил-1H,6H-пиридо[4,3-d]пиримидин-2,4,7-триона (2), в 5 мл ДМФА перемешивают при 0°С в течение 12 ч, затем при комнатной температуре в течение ночи. Смесь выливают в воду и продукт экстрагируют дихлорметаном. Органический слой промывают насыщенным раствором соли, сушат над Na2SO4 и концентрируют в вакууме. Остаток очищают флэш-хроматографией, элюент - этилацетат. Получают соответствующий ингибитор 1.10-1.15. Соли этих ингибиторов получают добавлением избытка HCl в этилацетате, фосфорной кислоты, метансульфокислоты или дихлоруксусной кислоты в ацетоне к раствору основания 1.10-1.15 в дихлорметане.
(S)-N-{3-[6,8-Диметил-5-(4-иод-2-фтор-фениламино)-3-циклопропил-2,4,7-триоксо-3,4,6,7-тетрагидро-2H-пиридо[4,3-d]пиримидин-1-ил]-фенил}-2-диметиламинопропионамид гидрохлорид (1.10·HCl): LC-MS (ESI) (m/z): 673 (М+Н)+; 1H-NMR (DMSO-d6, 400 MHz): 0.67 (m, 2H), 0.95 (m, 2H), 1.26 (s, 3H), 1.57 (d, 3H), 2.62 (m, 1H), 2.84 (s, 6H), 3.08 (s, 3H), 4.18 (m, 1H), 6.92 (t, J=9 Hz, 1H), 7.14 (d, J=8 Hz, 1H), 7.44 (t, J=8 Hz, 1H), 7.55 (d, J=8 Hz, 1H), 7.65 (br, 1H), 7.70 (d, J=8 Hz, 1H), 7.78 (d, J=10 Hz, 1H), 10.35 (br, 1H), 11.07 (br, 2H); (R)-N-{3-[6,8-диметил-5-(4-иод-2-фтор-фениламино)-3-циклопропил-2,4,7-триоксо-3,4,6,7-тетрагидро-2H-пиридо[4,3-d]пиримидин-1-ил]-фенил}-2-диметиламинопропионамид гидрохлорид (1.11·HCl): LC-MS (ESI) (m/z): 673 (M+H)+; 1H-NMR (DMSO-d6, 400 MHz): 0.67 (m, 2H), 0.95 (m, 2H), 1.26 (s, 3H), 1.57 (d, 3H), 2.62 (m, 1H), 2.84 (s, 6H), 3.08 (s, 3H), 4.18 (m, 1H), 6.92 (t, J=9 Hz, 1H), 7.14 (d, J=8 Hz, 1H), 7.44 (t, J=8 Hz, 1H), 7.55 (d, J=8 Hz, 1H), 7.65 (br, 1H), 7.70 (d, J=8 Hz, 1H), 7.78 (d, J=10 Hz, 1H), 10.35 (br, 1H), 11.07 (br, 2H); {3-[6,8-диметил-5-(4-иод-2-фтор-фениламино)-3-циклопропил-2,4,7-триоксо-3,4,6,7-тетрагидро-2H-пиридо[4,3-d]пиримидин-1-ил]-фенил}-амид (S)-пирролидин-2-карбоновой кислоты гидрохлорид (1.6·HCl): LC-MS (ESI) (m/z): 671 (M+H)+; 1H-NMR (DMSO-d6, 400 MHz): 0.66 (m, 2H), 0.95 (m, 2H), 1.26 (s, 3H), 1.9-2.05 (m, 3H), 2.35-2.45 (m, 2H), 2.61 (m, 1H), 3.07 (s, 3H), 3.2-3.3 (m, 1H), 3.5-3.7 (m, 1H), 4.4 (m, 1H), 6.92 (t, J=9 Hz, 1H), 7.12 (d, J=8 Hz, 1H), 7.43 (t, J=8 Hz, 1H), 7.55 (d, J=8 Hz, 1H), 7.6-7.75 (br, 2H), 7.78 (d, J=10 Hz, 1H), 8.67 (br, 1H), 9.91 (br, 1H), 11.02 (s, 1H), 11.07 (s, 1H); {3-[6,8-диметил-5-(4-иод-2-фтор-фениламино)-3-циклопропил-2,4,7-триоксо-3,4,6,7-тетрагидро-2H-пиридо[4,3-d]пиримидин-1-ил]-фенил}-амид (R)-пирролидин-2-карбоновой кислоты гидрохлорид (1.13·HCl): LC-MS (ESI) (m/z): 671 (M+H)+; 1H-NMR (DMSO-d6, 400 MHz): 0.66 (m, 2H), 0.95 (m, 2H), 1.26 (s, 3H), 1.9-2.05 (m, 3H), 2.35-2.45 (m, 1H), 2.61 (m, 1H), 3.07 (s, 3H), 3.15-3.3 (m, 2H), 4.36 (m, 1H), 6.92 (t, J=9 Hz, 1H), 7.14 (d, J=8 Hz, 1H), 7.44 (t, J=8 Hz, 1H), 7.56 (d, J=8 Hz, 1H), 7.6-7.7 (br, 2H), 7.78 (d, J=10 Hz, 1H), 8.65 (br, 1H), 9.5 (br, 1H), 10.83 (s, 1H), 11.07 (s, 1H); {3-[6,8-диметил-5-(4-иод-2-фтор-фениламино)-3-циклопропил-2,4,7-триоксо-3,4,6,7-тетрагидро-2H-пиридо[4,3-d]пиримидин-1-ил]-фенил}-амид (S)-1-метилпирролидин-2-карбоновой кислоты гидрохлорид (1.14·HCl): LC-MS (ESI) (m/z): 685 (М+Н)+; 1H-NMR (DMSO-d6, 400 MHz): 0.66 (m, 2H), 0.95 (m, 2H), 1.26 (s, 3H), 1.9-2.15 (m, 3H), 2.58-2.72 (m, 2H), 2.89 (s, 3H), 3.08 (s, 3H), 3.24 (m, 1H), 3.62-3.75 (m, 1H), 4.35 (m, 1H), 6.92 (t, J=9 Hz, 1H), 7.12 (d, J=8 Hz, 1H), 7.43 (t, J=8 Hz, 1H), 7.55 (d, J=8 Hz, 1H), 7.64-7.77 (br, 2H), 7.78 (d, J=10 Hz, 1H), 9.86 (br, 1H), 11.06 (s, 1H), 11.32 (s, 1H); {3-[6,8-диметил-5-(4-иод-2-фтор-фениламино)-3-циклопропил-2,4,7-триоксо-3,4,6,7-тетрагидро-2H-пиридо[4,3-d]пиримидин-1-ил]-фенил}-амид LC-MS (ESI) (m/z): 685 (M+H)+; (R)-1-метилпирролидин-2-карбоновой кислоты гидрохлорид (1.15·HCl): 1H-NMR (DMSO-d6, 400 MHz): 0.66 (m, 2H), 0.95 (m, 2H), 1.26 (s, 3H), 1.9-2.15 (m, 3H), 2.58-2.72 (m, 2H), 2.89 (s, 3H), 3.08 (s, 3H), 3.24 (m, 1H), 3.62-3.75 (m, 1H), 4.35 (m, 1H), 6.92 (t, J=9 Hz, 1H), 7.12 (d, J=8 Hz, 1H), 7.43 (t, J=8 Hz, 1H), 7.55 (d, J=8 Hz, 1H), 7.64-7.77 (br, 2H), 7.78 (d, J=10 Hz, 1H), 9.86 (br, 1H), 11.06 (s, 1H), 11.32(s, 1H).
Пример 3. Определение термодинамической растворимости соединения формулы 1 и прототипа Trametinib
Смешивали 5 мг исследуемого соединения с 1 мл универсального буфера (pION) с pH 2,0, 4,0 или 7,0 в течение 15 мин при 25°С. Дополнительные количества веществ добавляли до тех пор, пока раствор не становился мутным. Виалы с раствором инкубировали при перемешивании в течение 24 ч при 25°С для достижения равновесия между раствором и осадком при насыщении. После уравновешивания 200 мкл раствора (в 2-х повторах) фильтровали через 96-луночный фильтровальный планшет (Millipore) для отделения осадка. Концентрацию соединений в фильтрате определяли спектрофотометрически с помощью стандартной калибровочной кривой. Проводили измерение спектра оптического поглощения вещества и построение калибровочной кривой при выбранной длине волны (обычно соответствующей максимуму поглощения вещества λmax). Концентрацию вещества в фильтрате (т.е. растворимость) рассчитывали по нижеприведенной формуле:
 ,
,
где ODλmax filtrate - оптическая плотность фильтрата,
ODλmax blank - оптическая плотность холостого раствора без вещества,
Slope - наклон калибровочной кривой,
1,67 - фактор разведения фильтрата ацетонитрилом,
Filtrate dilution - фактор разведения фильтрата буфером.
Полученные результаты представлены в таблице 1.
Пример 4. Эссей
Вещества тестировали по влиянию на активность киназы MEK1 с помощью скрининговой платформы Z`-LYTE (Life Technologies). Концентрация ДМСО в реакционной смеси составляла 1%. 100 нл 100-кратных стоков исследуемых веществ в 100% ДМСО были разбавлены в 2.4 мкл киназного буфера (50 мМ HEPES pH 7.5, 0.01% BRIJ-35, 10 мМ MgCl2, 1 мМ EGTA) и добавлены к 5 мкл 2-кратной смеси Субстрат/Киназа (MEK1/инактивированная MAPK1(ERK2)/ Ser/Thr03, финальные концентрации - 0.08-0.31 нг MEK1, 105 нг инактивированной MAPK1 (ERK2), и 2 мкМ Ser/Thr03) в 384-луночном планшете (черные, малого объема, производство Corning, кат.#3676). Вещества проинкубировали с киназами в течение 10 мин при комнатной температуре. После этого для начала реакции добавляли 2.5 мкл 4-кратного раствора АТФ (конечная концентрация АТФ в реакционной смеси 100 мкМ). После 30 сек инкубации на шейкере реакцию инкубировали в течение 60 мин при комнатной температуре. После этого добавляли 5 мкл Реагента В (разбавленного 1:1024) и инкубировали еще 60 мин при комнатной температуре. Измеряли флуоресценцию при возбуждении длиной волны 400 нм и эмиссии при 445 и 520 нм. Степень фосфорилирования пептидного субстрата рассчитывали по формуле ниже (если отношение эмиссии низкое - пептид фосфорилирован, т.е. нет ингибирования активности киназы, если отношение высокое - пептид не фосфорилирован, т.е. киназа ингибирована). % фосфорилирования рассчитывается как:

% ингибирования рассчитывается как:

где С100% = средний сигнал эмиссии кумарина, контроль 100% фосфорилирования,
С0% = средний сигнал эмиссии кумарина, контроль 0% фосфорилирования,
F100% = средний сигнал эмиссии флуоресцеина, контроль 100% фосфорилирования,
F0% = средний сигнал эмиссии флуоресцеина, контроль 0% фосфорилирования.
Пример 5. Приготовление лекарственного средства в виде таблеток
Смешивают 1600 мг крахмала, 1600 мг размолотой лактозы, 400 мг талька и 1000 мг ингибитора 1.2 и прессуют в брусок. Полученный брусок измельчают в гранулы и просеивают через сита, собирая гранулы размером 14-16 меш. Полученные гранулы таблетируют в таблетки пригодных форм весом 500 мг каждая.
Пример 6. Приготовление лекарственного средства в виде капсул
Ингибитор 1.2 тщательно смешивают с лактозой в соотношении 2:1. Полученную порошкообразную смесь упаковывают по 600 мг в желатиновые капсулы подходящего размера.
Пример 7. Приготовление лекарственного средства в виде композиций для внутримышечных, внутрибрюшинных или подкожных инъекций
Смешивают 500 мг ингибитора 1.2 и 300 мг хлорбутанол, 2 мл пропиленгликоля и 100 мл воды для инъекций. Раствор фильтруют и помещали в 1 мл ампулы, которые укупоривают.
Специалисты в данной области техники легко поймут различные некритические параметры, которые могут быть изменены или модифицированы, чтобы получить те же результаты. Различные модификации изобретения в дополнение к описанным здесь станут очевидными специалистам в данной области техники из приведенного выше описания.
Настоящее изобретение относится к новым производным 1-(3-аминофенил)-6,8-диметил-5-(4-иод-2-фтор-фениламино)-3-циклопропил-1H,6H-пиридо[4,3-d]пиримидин-2,4,7-триона, которые обладают активностью в отношении киназы МЕК1 и могут быть использованы для лечения и профилактики рака, в частности злокачественной меланомы. Указанные производные выбраны из следующей группы соединений: N-{3-[6,8-диметил-5-(4-иод-2-фтор-фениламино)-3-циклопропил-2,4,7-триоксо-3,4,6,7-тетрагидро-2Н-пиридо[4,3-d]пиримидин-1-ил]-фенил}-2-диметиламиноацетамида гидрохлорида (1.1), N-{3-[6,8-диметил-5-(4-иод-2-фтор-фениламино)-3-циклопропил-2,4,7-триоксо-3,4,6,7-тетрагидро-2Н-пиридо[4,3-d]пиримидин-1-ил]-фенил}-2-диметиламинопропионамида гидрохлорида (1.4), N-{3-[6,8-диметил-5-(4-иод-2-фтор-фениламино)-3-циклопропил-2,4,7-триоксо-3,4,6,7-тетрагидро-2Н-пиридо[4,3-d]пиримидин-1-ил]-фенил}-2-диметиламинобутирамида гидрохлорида (1.7), (R)-N-{3-[6,8-диметил-5-(4-иод-2-фтор-фениламино)-3-циклопропил-2,4,7-триоксо-3,4,6,7-тетрагидро-2Н-пиридо[4,3-d]пиримидин-1-ил]-фенил}-2-диметиламинопропионамида гидрохлорида (1.10), {3-[6,8-диметил-5-(4-иод-2-фтор-фениламино)-3-циклопропил-2,4,7-триоксо-3,4,6,7-тетрагидро-2Н-пиридо[4,3-d]пиримидин-1-ил]-фенил}-амида (R)-1-метилпирролидин-2-карбоновой кислоты гидрохлорида (1.14). Изобретение также относится к способу получения указанных соединений. Способ заключается в том, что соединение формулы 2 или его соль:
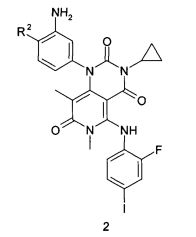
где R2 представляет собой атом водорода, подвергают взаимодействию с соответствующей органической кислотой или ее активированным производным, с последующей обработкой полученного продукта раствором хлористого водорода. 8 н. и 3 з.п. ф-лы, 1 табл., 7 пр.
1. Соединения, выбранные из:
N-{3-[6,8-диметил-5-(4-иод-2-фтор-фениламино)-3-циклопропил-2,4,7-триоксо-3,4,6,7-тетрагидро-2Н-пиридо[4,3-d]пиримидин-1-ил]-фенил}-2-диметиламиноацетамида гидрохлорида (1.1),
N-{3-[6,8-диметил-5-(4-иод-2-фтор-фениламино)-3-циклопропил-2,4,7-триоксо-3,4,6,7-тетрагидро-2Н-пиридо[4,3-d]пиримидин-1-ил]-фенил}-2-диметиламинопропионамида гидрохлорида (1.4),
N-{3-[6,8-диметил-5-(4-иод-2-фтор-фениламино)-3-циклопропил-2,4,7-триоксо-3,4,6,7-тетрагидро-2Н-пиридо[4,3-d]пиримидин-1-ил]-фенил}-2-диметиламинобутирамида гидрохлорида (1.7),
(R)-N-{3-[6,8-диметил-5-(4-иод-2-фтор-фениламино)-3-циклопропил-2,4,7-триоксо-3,4,6,7-тетрагидро-2Н-пиридо[4,3-d]пиримидин-1-ил]-фенил}-2-диметиламинопропионамида гидрохлорида (1.10),
{3-[6,8-диметил-5-(4-иод-2-фтор-фениламино)-3-циклопропил-2,4,7-триоксо-3,4,6,7-тетрагидро-2Н-пиридо[4,3-d]пиримидин-1-ил]-фенил}-амида (R)-1-метилпирролидин-2-карбоновой кислоты гидрохлорида (1.14).
2. Противораковый ингредиент для приготовления фармацевтических композиций и лекарственных форм, представляющий собой соединение по п. 1.
3. Фармацевтическая композиция, обладающая активностью в отношении киназы МЕК1 в виде таблеток, капсул или инъекций, помещенных в фармацевтически приемлемую упаковку, содержащая противораковый ингредиент по п. 2 в терапевтически эффективном количестве.
4 Применение соединений по п. 1 для получения лекарственного средства для профилактики или лечения рака.
5. Способ профилактики и/или лечения рака, опосредованного активностью МЕК1, включающий введение пациенту эффективного количества соединения по п. 1.
6. Способ профилактики и/или лечения рака, включающий введение пациенту эффективного количества фармацевтической композиции по п. 3.
7. Способ по п. 6, где рак представляет собой злокачественную меланому.
8. Способ изготовления лекарственного средства для лечения рака, опосредованного активностью МЕК1, путем смешения соединения по п. 1 с фармацевтически приемлемым наполнителем.
9. Способ по п. 8, согласно которому получают лекарственное средство для лечения рака, в частности для лечения злокачественной меланомы.
10. Способ получения соединения по п. 1, включающий взаимодействие соединения формулы 2 или его соли с соответствующей органической кислотой или ее активированным производным с последующим взаимодействием полученного продукта с раствором хлористого водорода:
где R2 представляет собой атом водорода.
11. Способ по п. 10, отличающийся тем, что взаимодействие с раствором хлористого водорода проводят в органическом растворителе, например в диоксане или тетрагидрофуране.
| WO 2005121142 A1,22.12.2005 & RU 2364596 C2 | |||
| WO 2005121142 A1, 22.12.2005 & RU2364596 (C2) | |||
| US 7378423 B2, 27.05.2008 | |||
| US 7378423 B2, 27.05.2008 | |||
| MAY Y.K et al.,Trametinib,a first -in-class oral MEK inhibitor mass balance study with limited entrollimentto two male subjects with advanced cancers, HENOBIOTICA, 2014, 44(4), 352-366 | |||
| Видоизменение устройства для движения судна реакцией выходящей из трубчатых насадок кожуха смеси пара с топочными газами парового котла, воздухом и водой | 1927 |
|
SU19951A1 |

