Область изобретения
Данное изобретение относится к комбинированной терапии для лечения пациентов, страдающих от пролиферативного нарушения, в частности солидной опухоли, например от колоректального рака, меланомы и рака щитовидной железы, включающей введение пациенту {3-[5-(4-хлор-фенил)-1Н-пирроло[2,3-b]пиридин-3-карбонил-2,4-дифторфенил}амида пропан-1-сульфокислоты и ингибитора EGFR.
Уровень изобретения
Нормально функционирующая b-Raf представляет собой киназу, которая участвует в передаче сигналов от клеточной мембраны к ядру, и является активной только тогда, когда это необходимо для передачи таких сигналов. Тем не менее, мутантная b-Raf, имеющая мутацию V600E, постоянно активна и, таким образом, играет роль в развитии опухоли. Такая мутантная b-Raf была найдена при различных опухолях, например, колоректальном раке, меланоме и раке щитовидной железы.
Пропан-1-сульфокислоты {3-[5-(4-хлор-фенил)-1Н-пирроло[2,3-b]пиридин-3-карбонил-2,4-дифторфенил]амид} (далее также называемый «Соединение I») представляет собой ингибитор киназы b-Raf, который специфически нацелен на мутантную b-Raf с мутацией V600E. Это соединение описано в WO 2007/002325. Соответственно, такой ингибитор используется для ингибирования опухолей, особенно солидных опухолей, например, колоректального рака, меланомы и рака щитовидной железы, которые содержат b-Raf с мутацией V600, предпочтительно мутацией V600E.
Белковые тирозинкиназы (protein tyrosine kinases, PTK) катализируют фосфорилирование остатков тирозина в различных белках, участвующих в регуляции роста и дифференцировки клеток (Wilks et al., Progress in Growth Factor Research 97 (1990) 2; Chan, А.С., and Shaw, A.S., Curr. Opin. Immunol. 8 (1996) 394-401). Такие PTK можно разделить на рецепторные тирозинкиназы (например, EGFR/HER-1, c-erbB-2/HER-2, c-met, PDGFr, FGFr) и нерецепторные тирозинкиназы (например, src, Ick).
Известно, что рецепторные тирозинкиназы из HER-семейства, такие как HER-2 и EGFR (HER-1), часто аберрантно экспрессированы в распространенных человеческих раках, таких как рак молочной железы, рака желудочно-кишечного тракта (рак толстой кишки, прямой кишки и желудка), рака щитовидной железы, лейкемия, рак яичников, бронхов и поджелудочной железы и меланома. Высокие уровни этих рецепторов коррелируют с плохим прогнозом и ответом на лечение (Wright, С., et al., Br.J.Cancer 65 (1992) 118-121).
Ингибиторы РТК и, в частности, EGFR были разработаны. Тем не менее, также известно, что опухоли, содержащие b-Raf с мутацией V600E, устойчивы к лечению ингибиторами EGFR. См. Prewett et al., Clin. Cancer Res. (2002), 8:994-1003 и Ouchi et al., Cancer Chemother. Pharmacol. (2006), 57:693-702. Тем не менее, заявители неожиданно обнаружили, что комбинация соединения I с ингибитором EGFR не только способна снизить такую устойчивость, но также приводит к улучшенным противоопухолевым эффектам, которые значительно превосходят результаты, полученные для каждого соединения в отдельности, без значительного увеличения токсичности.
В дополнение к ингибиторам EGFR, ингибиторы топоизомеразы также являются антипролиферативными агентами. Тем не менее, также известно, что опухоли с мутацией V600E устойчивы к лечению ингибиторами топоизомеразы. См. Prewett et al., Clin. Cancer Res. (2002), 8:994-1003 и Abal et al., Oncogene (2004), 23:1737-44. Тем не менее, заявители неожиданно обнаружили, что комбинация соединения I с ингибитором EGFR и ингибитором топоизомеразы не только способна снизить такую устойчивость, но также приводит к улучшенным противоопухолевым эффектам, которые значительно превосходят результаты, полученные для каждого соединения в отдельности или для упомянутой выше комбинации соединения I с ингибитором EGFR, без значительного увеличения токсичности.
Сущность изобретения
Данное изобретение относится к фармацевтическому продукту;
включающему: (А) первый компонент, который содержит в качестве активного агента {3-[5-(4-хлор-фенил)-1Н-пирроло[2,3-b]пиридин-3-карбонил-2,4-дифторфенил}амид пропан-1-сульфокислоты или его фармацевтически приемлемую соль; и (В) второй компонент, который включает в качестве активного агента ингибитор EGFR; в виде комбинированного препарата для одновременного или последовательного применения в лечении пролиферативного нарушения, в частности, рака, более конкретно колоректального рака, меланомы и рака щитовидной железы, содержащего b-Raf с мутацией V600 и, в частности, с мутацией V600E.
Данное изобретение также относится к способу лечения пациента, страдающего от пролиферативного нарушения, который включает введение комбинации, упомянутой выше.
Данное изобретение также относится к набору, включающему: (А) первый компонент, который содержит в качестве активного агента соединение 1 или его фармацевтически приемлемую соль; и (В) второй компонент, который содержит в качестве активного агента ингибитор EGFR.
Данное изобретение также относится к набору, включающему: (А) первый компонент, который содержит в качестве активного агента соединение 1 или его фармацевтически приемлемую соль; и (В) второй компонент, который содержит в качестве активного агента ингибитор EGFR; и возможно (С) третий компонент, который содержит ингибитор топоизомеразы или его фармацевтически приемлемую соль; в виде комбинированного препарата для одновременного или последовательного применения в лечении пролиферативного нарушения, в частности, рака, более конкретно колоректального рака, меланомы и рака щитовидной железы, содержащего b-Raf с мутацией V600 и, в частности, с мутацией V600E.
Кроме того, данное изобретение относится к применению соединения 1 или его фармацевтически приемлемой соли и ингибитора EGFR для лечения пролиферативного нарушения.
Еще одним аспектом данного изобретения является применение соединения 1 или его фармацевтически приемлемой соли и ингибитора EGFR для изготовления лекарственного средства для лечения пролиферативного нарушения.
Краткое описание графических материалов
На фиг.1 приведена переносимость, показанная как % изменения массы тела при монотерапии соединением I в дозе 75 мг/кг 2 раза/день, при монотерапии эрлотиниба гидрохлоридом в дозе 67 мг/кг 1 раз/день, при монотерапии эрлотиниба гидрохлоридом в дозе 100 мг/кг 1 раз/день, и при комбинированной терапии соединением I в дозе 75 мг/кг 2 раза/день и эрлотиниба гидрохлоридом в дозе 67 мг/кг 1 раз/день.
На фиг.2 приведена противоопухолевая активность, показанная как изменение среднего объема опухоли с течением времени при монотерапии соединением I в дозе 75 мг/кг 2 раза/день, при монотерапии эрлотиниба гидрохлоридом в дозе 67 мг/кг 1 раз/день, при монотерапии эрлотиниба гидрохлоридом в дозе 100 мг/кг 1 раз/день, и при комбинированной терапии соединением I в дозе 75 мг/кг 2 раза/день и эрлотиниба гидрохлоридом в дозе 67 мг/кг 1 раз/день.
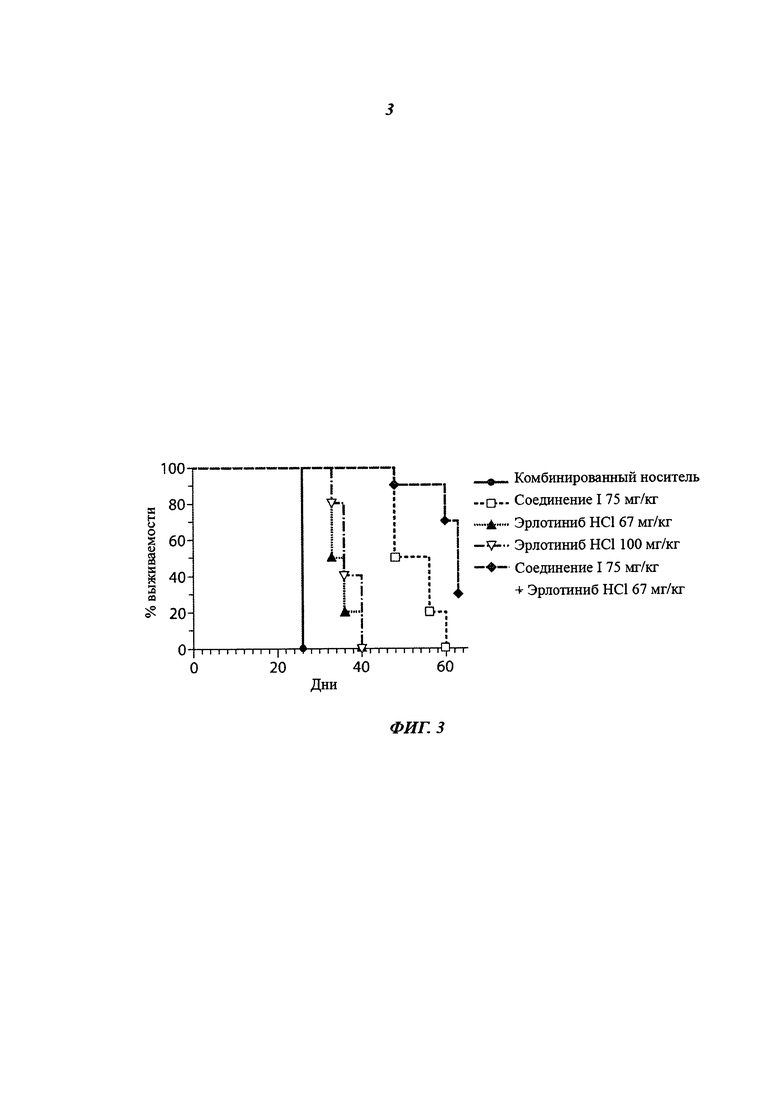
На фиг.3 приведено влияние на выживаемость, показанное как процент выживших мышей с течением времени при монотерапии соединением I в дозе 75 мг/кг 2 раза/день, при монотерапии эрлотиниба гидрохлоридом в дозе 67 мг/кг 1 раз/день, при монотерапии эрлотиниба гидрохлоридом в дозе 100 мг/кг 1 раз/день, и при комбинированной терапии соединением I в дозе 75 мг/кг 2 раза/день и эрлотиниба гидрохлоридом в дозе 67 мг/кг 1 раз/день.
На фиг.4 приведена переносимость, показанная как % изменения массы тела при монотерапии соединением I в дозе 75 мг/кг 2 раза/день, при монотерапии соединением I в дозе 25 мг/кг 2 раза/день, при монотерапии цетуксимабом в дозе 40 мг/кг 2 раза/нед, при комбинированной терапии соединением I в дозе 25 мг/кг 2 раза/день и цетуксимабом в дозе 40 мг/кг 2 раза/нед, и при комбинированной терапии соединением I в дозе 75 мг/кг 2 раза/день и цетуксимабом в дозе 40 мг/кг 2 раза/нед.
На фиг.5 приведена противоопухолевая активность, показанная как изменение среднего объема опухоли с течением времени при монотерапии соединением I в дозе 75 мг/кг 2 раза/день, при монотерапии соединением I в дозе 25 мг/кг 2 раза/день, при монотерапии цетуксимабом в дозе 40 мг/кг 2 раза/нед, при комбинированной терапии соединением I в дозе 25 мг/кг 2 раза/день и цетуксимабом в дозе 40 мг/кг 2 раза/нед, и при комбинированной терапии соединением I в дозе 75 мг/кг 2 раза/день и цетуксимабом в дозе 40 мг/кг 2 раза/нед.
На фиг.6 приведено влияние на выживаемость, показанное как процент выживших мышей с течением времени при монотерапии соединением I в дозе 75 мг/кг 2 раза/день, при монотерапии соединением I в дозе 25 мг/кг 2 раза/день, при монотерапии цетуксимабом в дозе 40 мг/кг 2 раза/нед, при комбинированной терапии соединением I в дозе 25 мг/кг 2 раза/день и цетуксимабом в дозе 40 мг/кг 2 раза/нед, и при комбинированной терапии соединением I в дозе 75 мг/кг 2 раза/день и цетуксимабом в дозе 40 мг/кг 2 раза/нед.
На фиг.7 приведена переносимость, показанная как % изменения массы тела при монотерапии соединением I в дозе 25 мг/кг 2 раза/день, при монотерапии цетуксимабом в дозе 40 мг/кг 2 раза/нед, при монотерапии иринотеканом HCl в дозе 40 мг/кг q4dx5, при комбинированной терапии соединением I в дозе 25 мг/кг 2 раза/день и иринотеканом HCl в дозе 40 мг/кг q4dx5, при комбинированной терапии цетуксимабом в дозе 40 мг/кг 2 раза/неделя и иринотеканом HCl в дозе 40 мг/кг q4dx5, при комбинированной терапии соединением 1 в дозе 25 мг/кг 2 раза/день и цетуксимабом в дозе 40 мг/кг 2 раза/нед, и при комбинированной терапии соединением 1 в дозе 25 мг/кг 2 раза/день, цетуксимабом в дозе 40 мг/кг 2 раза/нед и иринотеканом HCl в дозе 40 мг/кг q4dx5.
На фиг.8 приведена противоопухолевая активность, показанная как изменение среднего объема опухоли с течением времени при монотерапии соединением 1 в дозе 25 мг/кг 2 раза/день, при монотерапии цетуксимабом в дозе 40 мг/кг 2 раза/нед, при монотерапии иринотеканом HCl в дозе 40 мг/кг q4dx5, при комбинированной терапии соединением 1 в дозе 25 мг/кг 2 раза/день и иринотеканом HCl в дозе 40 мг/кг q4dx5, при комбинированной терапии цетуксимабом в дозе 40 мг/кг 2 раза/нед и иринотеканом HCl в дозе 40 мг/кг q4dx5, при комбинированной терапии соединением 1 в дозе 25 мг/кг 2 раза/день и цетуксимабом в дозе 40 мг/кг 2 раза/нед, и при комбинированной терапии соединением 1 в дозе 25 мг/кг 2 раза/день, цетуксимабом в дозе 40 мг/кг 2 раза/неделя и иринотеканом HCl в дозе 40 мг/кг q4dx5.
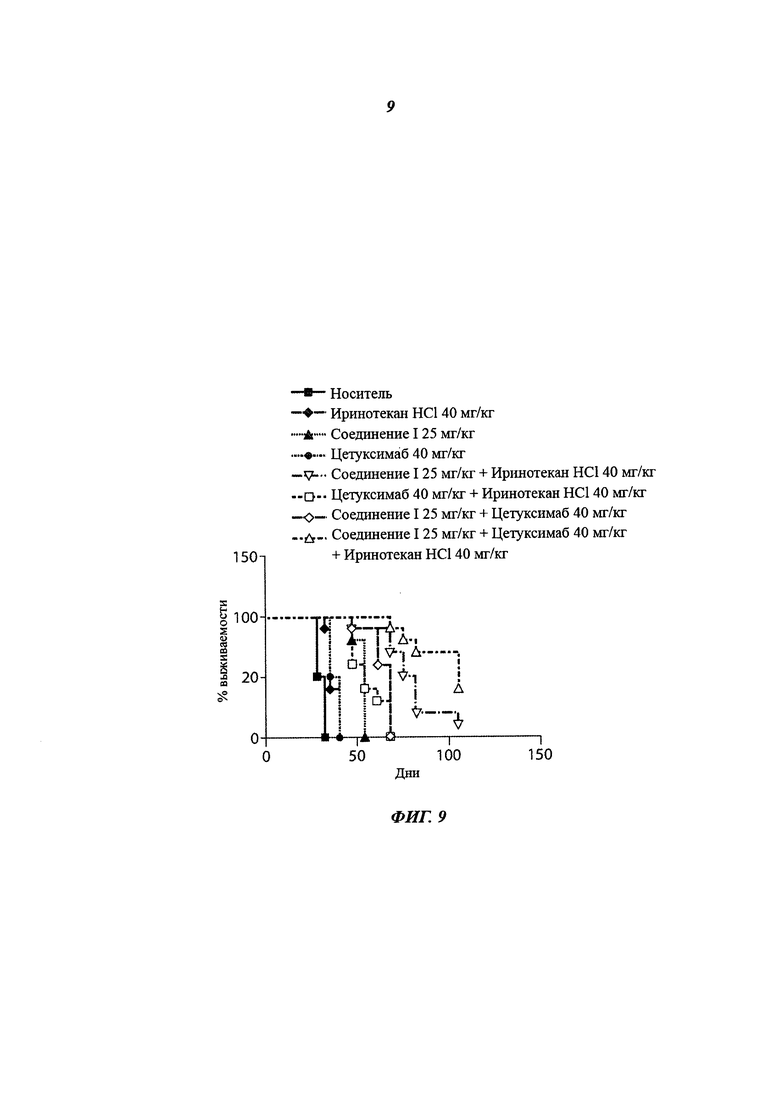
На фиг.9 приведено влияние на выживаемость, показанное как процент выживших мышей с течением времени при монотерапии соединением 1 в дозе 25 мг/кг 2 раза/день, при монотерапии цетуксимабом в дозе 40 мг/кг 2 раза/неделя, при монотерапии иринотеканом HCl в дозе 40 мг/кг q4dx5, при комбинированной терапии соединением 1 в дозе 25 мг/кг 2 раза/день и иринотеканом HCl в дозе 40 мг/кг q4dx5, при комбинированной терапии цетуксимабом в дозе 40 мг/кг 2 раза/нед и иринотеканом HCl в дозе 40 мг/кг q4dx5, при комбинированной терапии соединением 1 в дозе 25 мг/кг 2 раза/день и цетуксимабом в дозе 40 мг/кг 2 раза/нед, и при комбинированной терапии соединением 1 в дозе 25 мг/кг 2 раза/день, цетуксимабом в дозе 40 мг/кг 2 раза/нед и иринотеканом HCl в дозе 40 мг/кг q4dx5.
Подробное описание изобретения
Как определено выше, «Соединение I» в данном документе относится к {3-[5-(4-хлорфенил)-1Н-пирроло[2,3-b]пиридин-3-карбонил-2,4-дифторфенил]амиду}пропан-1-сульфокислоты. Это соединение имеет следующую структуру.

Соединение I представляет собой ингибитор киназы b-Raf, который специфически нацелен на b-Raf с мутацией V600E.
Понятие мутации «V600E» b-Raf, используемое в данном документе, относится к мутации в белке b-Raf, где остаток валина в позиции остатка 600 в b-Raf заменен глутаминовой кислотой.
Используемая в данном документе аббревиатура «HER», когда речь идет о рецепторных тирозинкиназах из HER-семейства, таких как HER-2 и EGFR (HER-1), относится к человеческому эпидермальному рецептору, а аббревиатура «EGFR» относится к рецептору эпидермального фактора роста.
Термин «комбинированный препарат», используемый в данном документе, означает одновременную или последовательную комбинацию. Активные агенты, используемые в данных комбинациях, например, соединение I или ингибитор топоизомеразы, такой как, например, иринотекан, или ингибитор EGFR, такой как эрлотиниб или его фармацевтически приемлемая соль или цетуксимаб, могут быть использованы в любой применимой лекарственной форме, такой как, например, таблетки, капсулы, растворы, суспензии и т.п., в зависимости от свойств выбранного активного ингредиента. Соединение I или его фармацевтически приемлемая соль могут быть введены, например, перорально. Эрлотиниб или его фармацевтически приемлемая соль могут быть введены, например, перорально. Цетуксимаб может быть введен, например, внутрибрюшинно или внутривенно. Иринотекан HCl или его фармацевтически приемлемая соль могут быть введены, например, внутрибрюшинно или внутривенно. Некоторые комбинации, описанные в данном документе, могут продемонстрировать терапевтический эффект, который является более чем аддитивным (синергетическим).
Используемый в данном документе термин «фармацевтически приемлемый носитель» означает, что указанный носитель не обладает свойствами, которые могут быть причиной того, что достаточно предусмотрительный врач будет избегать их введения пациенту с учетом заболевания или состояний, которые подвергают лечению, и соответствующих путей введения.
Используемый в данном документе термин «фармацевтически приемлемая соль» соединения относится к любой обычной соли или соли присоединения основания, которая сохраняет биологическую эффективность и свойства соединения и которая формируется из подходящей нетоксичной органической или неорганической кислоты, либо органического или неорганического основания. Используемый в данном документе термин «терапевтически эффективное» означает количество лекарственного средства или комбинации или композиции, которое является эффективным для получения желаемого терапевтического эффекта при введении пациенту, например, чтобы остановить рост или привести к сокращению раковой опухоли или увеличить продолжительность жизни пациента.
Термины «нарушение клеточной пролиферации» и «пролиферативное нарушение» относятся к нарушениям, связанным с некоторой степенью аномальной пролиферации клеток. В одном воплощении пролиферативное нарушение представляет собой рак.
Термины «рак» и «раковый» относятся к или описывают физиологическое состояние у млекопитающих, которое обычно характеризуется неконтролируемым ростом/пролиферацией клеток. Примеры рака включают, но не ограничиваясь ими, колоректальный рак, меланому и рак щитовидной железы.
Термины «колоректальная опухоль» или «колоректальный рак» относятся к любой опухоли или раку толстого кишечника, который включает толстую кишку (толстый кишечник от слепой кишки до прямой кишки) и прямую кишку, в том числе, например, к аденокарциномам и менее распространенным формам, таким как лимфомы и плоскоклеточные карциномы.
«Ингибирование клеточного роста или пролиферации» означает уменьшение роста или пролиферации клеток по меньшей мере на 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 95% или 100% и включает гибель клетки.
Фраза «существенно уменьшенный» или «существенно отличающийся», используемая в данном документе, относится к достаточно высокой степени различия между двумя числовыми значениями (как правило, одно связано с молекулой, а другое связано с референсной/сравниваемой молекулой), так что специалист в данной области будет рассматривать разницу между двумя значениями как статистически значимую в контексте биологической характеристики, измеряемой указанными значениями.
Термин «опухоль» относится к росту и пролиферации всех опухолевых клеток, независимо от того, злокачественные они или доброкачественные, и ко всем предраковым и раковым клеткам и тканям. Термины «рак», «раковый», «нарушение клеточной пролиферации», «пролиферативное нарушение» и «опухоль» не являются взаимоисключающими при упоминании в данном документе.
О «регрессии» опухоли говорят после лечения, когда объем указанной опухоли уменьшается. Если опухоль сохраняется (объем опухоли > 0 мм3), но ее объем уменьшается по сравнению с началом лечения, то говорят о «частичной регрессии» (partial регрессия, PR). Если после лечения опухоль пальпаторно не выявляется, то говорят о «полной регрессии» (complete регрессия,CR).
Термин «малая молекула», используемый в данном документе, представляет собой химическое соединение с молекулярной массой менее 1000 г/моль, предпочтительно менее 700 г/моль. Малые молекулы в соответствии с данным изобретением могут быть получены в результате химических реакций, известных специалистам в области органической химии и/или медицинской химии. Примерами малых молекул могут быть, но не ограничиваясь ими, соединение I или соединение, известное как эрлотиниб, предпочтительно эрлотиниба гидрохлорид.
Термин «большая молекула», используемый в данном документе, относится к химическим соединениям с молекулярной массой более 1000 г/моль. Предпочтительно «большие молекулы» представляют собой соединения, которые могут быть получены с помощью биотехнологических процессов производства, таких как ферментация. Более предпочтительно термин «большая молекула» относится к полипептидам, таким как, например, антитела, более конкретно моноклональные антитела. Одним из примеров такой большой молекулы в соответствии с данным изобретением является цетуксимаб.
Данное изобретение относится к фармацевтическому продукту, включающему: (А) первый компонент, который содержит в качестве активного агента соединение I или его фармацевтически приемлемую соль; и (В) второй компонент, который содержит в качестве активного агента ингибитор EGFR; при этом количества указанных активных агентов таковы, что их комбинация является терапевтически эффективной в лечении указанного пролиферативного нарушения. Данное изобретение также относится к упомянутому выше продукту в виде комбинированного препарата для одновременного или последовательного применения в лечении пролиферативного нарушения, в частности рака, более конкретно солидной опухоли, конкретно колоректального рака, меланомы и/или рака щитовидной железы, содержащего b-Raf с мутацией V600E.
Данное изобретение также относится к способу лечения пациента, страдающего от пролиферативного нарушения, который включает введение пациенту комбинации или фармацевтических препаратов, упомянутых выше.
Следует понимать, что «лечение пролиферативного нарушения» включает сохранение или уменьшение размера опухоли, индукцию регрессии опухоли (частичной или полной), ингибирование роста опухоли и/или увеличение продолжительности жизни пациента, страдающего от указанного нарушения.
Данное изобретение также относится к набору или композиции, содержащей: (А) первый компонент, который содержит в качестве активного агента соединение I или его фармацевтически приемлемую соль; и (В) второй компонент, который включает в качестве активного агента ингибитор EGFR. Набор или композиция могут быть использованы, например, для лечения пролиферативного нарушения.
В одном воплощении изобретения пролиферативным нарушением является солидная опухоль, в частности, колоректальный рак, меланома и/или рак щитовидной железы.
В другом воплощении изобретения пролиферативным нарушением является опухоль, содержащая b-Raf с мутацией V600, предпочтительно с мутацией V600E.
В еще одном воплощении изобретения пролиферативное нарушение выбрано из группы, состоящей из колоректального рака, меланомы и рака щитовидной железы, и рак включает опухоль, содержащую b-Raf с мутацией V600, предпочтительно с мутацией V600E.
В еще одном воплощении изобретения пролиферативным нарушением является солидная опухоль, содержащая b-Raf с мутацией V600, предпочтительно с мутацией V600E.
В еще одном воплощении изобретения пролиферативным нарушением является колоректальный рак.
В еще одном воплощении изобретения пролиферативным нарушением является колоректальный рак, включающий опухоль, содержащую b-Raf с мутацией V600, предпочтительно с мутацией V600E.
В еще одном воплощении изобретения ингибитор EGFR является малой молекулой ингибитором EGFR. В одном таком воплощении ингибитор EGFR представляет собой эрлотиниб или его фармацевтически приемлемую соль, например, эрлотиниба гидрохлорид (эрлотиниб HCl). Эрлотиниб HCl также известен под торговым названием Tarceva® и продается, например, в США компанией Genentech, Саут-Сан-Франциско, США.
В еще одном воплощении изобретения ингибитор EGFR является большой молекулой ингибитором EGFR, например, антителом, которое нацелено на EGFR. В одном таком воплощении ингибитор EGFR может быть моноклональным антителом, которое нацелено на EGFR, например, цетуксимабом. Цетуксимаб также известен под торговым названием Erbitux® и продается, например, в США компанией ImClone Systems, Inc., Нью-Йорк, США.
В некоторых воплощениях данное изобретение относится к фармацевтическому продукту для лечения колоректального рака, включающего опухоль, содержащую b-Raf с мутацией V600E, где указанный продукт включает: (А) первый компонент, который содержит в качестве активного агента соединение 1 или его фармацевтически приемлемую соль; и (В) второй компонент, который содержит в качестве активного агента эрлотиниб или его фармацевтически приемлемую соль; при этом количества указанных активных веществ таковы, что их комбинация является терапевтически эффективной в лечении указанного рака.
В другом воплощении данное изобретение относится к фармацевтическому продукту для лечения колоректального рака, включающего опухоль, содержащую b-Raf с мутацией V600E, где указанный продукт включает: (А) первый компонент, который содержит в качестве активного агента соединение 1 или его фармацевтически приемлемую соль; и (В) второй компонент, который содержит в качестве активного агента цетуксимаб; при этом количества указанных активных веществ таковы, что их комбинация является терапевтически эффективной в лечении указанного рака.
Количество каждого компонента, вводимого в данных комбинациях или композициях, может, но не должно быть терапевтически эффективно само по себе. Т.е. это изобретение, в частности, предусматривает комбинации, в которых количество соединения 1 или его фармацевтически приемлемой соли и/или количество ингибитора EGFR в комбинации могут быть меньше, чем сумма количеств, которые терапевтически эффективны для каждого активного агента, когда указанные агенты вводятся в виде монотерапии.
Первый компонент (А) и второй компонент (В) данного изобретения вводят в любом количестве и в течение любого периода времени, чтобы их комбинированное количество было терапевтически эффективным в лечении пролиферативного нарушения.
В некоторых воплощениях данного изобретения соединение 1 или его фармацевтически приемлемую соль вводят в дозировке от примерно 200 мг/сут до примерно 3000 мг/сут, от примерно 1000 мг/день до примерно 2500 мг/день, или от примерно 1700 мг/день до примерно 2100 мг/сут. В еще одном воплощении дозировка составляет примерно 1920 мг/сут.
В другом воплощении изобретения указанные выше количества соединения I или его фармацевтически приемлемой соли могут быть введены в виде разовой дозы ежедневно или разделены, например, на равные дозы (хотя это не обязательно) и введены дважды в день. Например, соединение I или его фармацевтически приемлемая соль могут быть введены в дозировке от примерно 100 мг до примерно 1500 мг 2 раза/день, от примерно 500 мг до примерно 1250 мг 2 раза/день, от примерно 850 мг до примерно 1050 мг 2 раза/день, или примерно 960 мг 2 раза/день.
В одном воплощении данного изобретения введение соединения I или его фармацевтически приемлемой соли осуществляют до прогрессирования заболевания или развития неприемлемой токсичности.
В некоторых воплощениях данного изобретения эрлотиниб или его фармацевтически приемлемую соль вводят в дозировке от примерно 20 мг/день до примерно 500 мг/день, от примерно 100 мг/день до примерно 400 мг/день, или от примерно 100 мг/день до примерно 200 мг/день.
В другом воплощении данного изобретения введение эрлотиниба или его фармацевтически приемлемой соли осуществляют до прогрессирования заболевания или развития неприемлемой токсичности.
В еще одном воплощении данного изобретения цетуксимаб вводят в дозировке от примерно от 50 мг/м2/нед до примерно 700 мг/м2/нед, от примерно 100 мг/м2/нед до примерно 600 мг/м2/нед, или от примерно 200 мг/м2/нед до примерно 500 мг/м2/нед.
В еще одном воплощении цетуксимаб вводят раз в неделю, при первом введении в количестве от примерно 400 мг/м2 до примерно 500 мг/м2, и при каждом последующем введении в количестве от примерно 200 мг/м2 до примерно 300 мг/м2.
В еще одном воплощении цетуксимаб вводят раз в неделю, при первом введении в количестве примерно 450 мг/м2, и при каждом последующем введении в количестве примерно 250 мг/м2.
В еще одном воплощении данного изобретения введение цетуксимаба осуществляют до прогрессирования заболевания или развития неприемлемой токсичности.
Таким образом, в другом воплощении данное изобретение предусматривает фармацевтической продукт, содержащий (А) первый компонент, который содержит в качестве активного агента соединение I или его фармацевтически приемлемую соль; и (В) второй компонент, который содержит в качестве активного агента эрлотиниб или его фармацевтически приемлемую соль, в виде комбинированного препарата для одновременного или последовательного применения в лечении пролиферативного нарушения, где
(A) вводят в количестве от примерно 200 мг/сут до примерно 3000 мг/сут, от примерно 1000 мг/сут до примерно 2500 мг/сут, от примерно 1700 мг/сут до примерно 2100 мг/сут, или примерно 1920 мг/сут, и
(B) вводят в количестве от примерно 20 до примерно 500 мг/день, от примерно 100 до примерно 400 мг/день, или от примерно 100 до примерно 200 мг/день.
В этом воплощении соединение I или его фармацевтически приемлемая соль могут быть введены два раза в день. Пролиферативное нарушение, которое лечат, представляет собой солидную опухоль, в частности, колоректальный рак, меланому и рак щитовидной железы, включающий опухоль, содержащую b-Raf с мутацией V600E. Более конкретно Пролиферативное нарушение представляет собой колоректальный рак с участием опухоли, содержащей b-Raf с мутацией V600E.
Также в этом воплощении продукт данного изобретения может содержать соединение I или его фармацевтически приемлемую соль, которую вводят перорально в дозировке от примерно 850 мг 2 раза/день до примерно 1050 мг 2 раза/день, или примерно 960 мг 2 раза/день, а эрлотиниб или его фармацевтически приемлемую соль вводят перорально в дозировке от примерно 100 мг/день до примерно 400 мг/день, или от примерно 100 мг/день до примерно 200 мг/день. Оба агента могут вводить, например, до прогрессирования заболевания или развития неприемлемой токсичности.
Данное изобретение также предусматривает фармацевтической продукт, содержащий (А) первый компонент, который содержит в качестве активного агента соединение I или его фармацевтически приемлемую соль; и (В) второй компонент, который содержит в качестве активного агента цетуксимаб; в виде комбинированного препарата для одновременного или последовательного применения в лечении пролиферативного нарушения, где
(A) вводят в количестве от примерно 200 мг/сут до примерно 3000 мг/сут, от примерно 1000 мг/сут до примерно 2500 мг/сут, от примерно 1700 мг/сут до примерно 2100 мг/сут, или примерно 1920 мг/сут, и
(B) вводят в количестве от 50 мг/м2/нед до примерно 700 мг/м2/нед, от примерно 100 мг/м2/нед до примерно 600 мг/м2/нед, или от примерно 200 мг/м2/нед до примерно 500 мг/м2/нед.
В этом воплощении Пролиферативное нарушение, которое лечат, представляет собой солидную опухоль, в частности, колоректальный рак, меланому и рак щитовидной железы, содержащий b-Raf с мутацией V600E. Более конкретно пролиферативное нарушение представляет собой колоректальный рак, включающий опухоль, содержащую b-Raf с мутацией V600E.
Также в этом воплощении продукт данного изобретения может содержать соединение 1 или его фармацевтически приемлемую соль, которую вводят перорально в дозировке от примерно 850 мг 2 раза/день до примерно 1050 мг 2 раза/день, или примерно 960 мг 2 раза/день; и цетуксимаб, который вводят внутривенно в дозировке от примерно 200 мг/м2/нед до примерно 500 мг/м2/нед. В одном воплощении цетуксимаб вводят вначале в дозировке 400 мг/м2 в виде 120-минутной внутривенной инфузии, а затем в дозировке 250 мг/м2 внутривенно в виде 60-минутной инфузии один раз в неделю. Оба агента могут вводиться, например, до прогрессирования заболевания или развития неприемлемой токсичности.
Данное изобретение также предусматривает набор или композицию, включающую: (А) первый компонент, который содержит в качестве активного агента соединение I или его фармацевтически приемлемую соль; и (В) второй компонент, который включает в качестве активного агента эрлотиниб или его фармацевтически приемлемую соль или пролекарство.
Данное изобретение также предусматривает набор или композицию, включающую: (А) первый компонент, который содержит в качестве активного агента соединение I или его фармацевтически приемлемую соль; и (В) второй компонент, который включает в качестве активного агента цетуксимаб.
В другом аспекте данного изобретения описанный выше фармацевтический продукт, включающий описанные выше компоненты (А) и (В), вводят в сочетании с лучевой терапией и/или в сочетании с другим активным агентом.
Таким образом, в некоторых воплощениях данное изобретение предусматривает фармацевтический продукт, включающий компоненты (А) и (В), описанные выше, и третий компонент (С), который содержит в качестве активного агента ингибитор типоизомеразы; в виде комбинированного препарата для одновременного или последовательного применения в лечении пролиферативного нарушения, такого как солидные опухоли, которые содержат b-Raf с мутацией V600, предпочтительно мутацией V600E. Как указывалось выше, количество каждого компонента, вводимого в соответствии с данным комбинированным продуктом, может, но не должно быть терапевтически эффективно само по себе, и это изобретение, в частности, предусматривает комбинации, в которых количества всех активных агентов в комбинации могут быть меньше, чем сумма количеств, которые терапевтически эффективны для каждого активного агента, когда указанные агенты вводятся в виде монотерапии.
В одном воплощении данного изобретения ингибитор топоизомеразы представляет собой ингибитор топоизомеразы 1 типа. В одном воплощении изобретения ингибитор топоизомеразы представляет собой иринотекан или его фармацевтически приемлемую соль, например, гидрохлорид иринотекана (иринотекан HCl). Иринотекан HCl коммерчески доступен как Camptosar® от компании Pfizer Inc, Нью-Йорк, США. Иринотекан или его фармацевтически приемлемая соль могут быть введены, например, внутрибрюшинно или внутривенно.
В одном воплощении данного изобретения иринотекан или его фармацевтически приемлемую соль вводят в дозировке от примерно 1 до примерно 400 мг/м2/нед или от примерно 1 до примерно 250 мг/м2/нед. В другом воплощении иринотекан или его фармацевтически приемлемую соль вводят в дозировке от примерно 50 до примерно 200 мг/м2/нед. В еще одном воплощении иринотекан или его фармацевтически приемлемую соль вводят в дозировке примерно 125 мг/м2/нед.
В еще одном воплощении дозировка иринотекана или его фармацевтически приемлемой соли составляет шестинедельный цикл от примерно 75 до примерно 175 мг/м2/нед, например, примерно 125 мг/м2/нед, в течение первых четырех недель, например, в 1, 8, 15 и 22 дни. В другом воплощении дозировка иринотекана составляет шестинедельный цикл от примерно 130 до примерно 230 мг/м2/нед, например, примерно 180 мг/м2/нед, раз в две недели, начиная с первой недели, например, в дни 1, 15 и 29. В еще одном воплощении дозировка составляет один раз в три недели от примерно 300 до примерно 400 мг/м2, например, примерно 350 мг/м2. В еще одном воплощении дозировка составляет раз в две недели от примерно 130 до примерно 230 мг/м2, например, примерно 180 мг/м2. Дозировка может осуществляться путем инфузии, например, в течение примерно 90 минут. Лечение может продолжаться до прогрессирования заболевания или до развития неприемлемой токсичности.
Уровни доз каждого из компонентов (А) и (В) и (С) могут быть изменены врачом до значения выше или ниже, чем было оговорено в данном документе, в зависимости от потребностей пациента и реакции пациента на лечение. Дозировки могут быть введены в соответствии с любым дозирующим графиком, определенным врачом в соответствии с потребностями пациента. Например, дозировки каждого из компонентов могут вводиться в одной или в отдельных дозах в течение нескольких дней или по графику с переменными днями.
Данное изобретение также предусматривает фармацевтический продукт, содержащий (А) первый компонент, который содержит в качестве активного агента соединение 1 или его фармацевтически приемлемую соль; (В) второй компонент, который содержит в качестве активного агента цетуксимаб; и (С) третий компонент, который содержит в качестве активного агента иринотекан или его фармацевтически приемлемую соль; в виде комбинированного препарата для одновременного или последовательного применения в лечении указанного пролиферативного нарушения, где
(A) вводят в количестве от примерно 200 мг/день до примерно 3000 мг/день, от примерно 1000 мг/день до примерно 2500 мг/день, от примерно 1700 мг/день до примерно 2100 мг/день, или примерно 1920 мг/день;
(B) вводят в количестве от примерно 50 мг/м2/нед до примерно 700 мг/м2/нед, от примерно 100 мг/м2/нед до примерно 600 мг/м2/нед, или от примерно 200 мг/м2/нед до примерно 500 мг/м2/нед; и
(C) вводят в количестве от примерно 1 до примерно 250 мг/м2/нед, от примерно 50 до примерно 200 мг/м2/нед, или примерно 125 мг/м2/нед.
В этом воплощении пролиферативное нарушение, которое подвергают лечению таким образом, представляет собой солидную опухоль, в частности, колоректальный рак, меланому и рак щитовидной железы, содержащий b-Raf с мутацией V600E. Более конкретно пролиферативное нарушение представляет собой колоректальный рак, который включает опухоль, содержащую b-Raf с мутацией V600E.
Также в этом воплощении продукт данного изобретения содержит соединение I или его фармацевтически приемлемую соль, которую вводят перорально в дозировке от примерно 850 мг до примерно 1050 мг 2 раза/день, или примерно 960 мг 2 раза/день; цетуксимаб, который вводят внутривенно в дозировке от примерно 200 мг/м2/нед до примерно 500 мг/м2/нед; и иринотекан, который вводят внутривенно в дозировке от примерно 50 до примерно 200 мг/м2/нед, или примерно 125 мг/м2/нед. Все агенты могут вводить, например, до прогрессирования заболевания или развития неприемлемой токсичности.
Данное изобретение также предусматривает набор или композицию, включающую: (А) первый компонент, который содержит в качестве активного агента соединение I или его фармацевтически приемлемую соль; (В) второй компонент, который содержит в качестве активного агента цетуксимаб; и (С) третий компонент, который содержит в качестве активного агента иринотекан или его фармацевтически приемлемую соль.
Соединение I существует в природном состоянии в кристаллической форме. Тем не менее, аморфная форма соединения имеет большую растворимость в воде по сравнению с кристаллической формой и, следовательно, имеет повышенную скорость растворения и, следовательно, повышенную биодоступность по сравнению с кристаллической формой. Таким образом, аморфная форма соединения является предпочтительной. Соответственно, в предпочтительных воплощениях способа и набора данного изобретения соединение I находится по существу в аморфной форме и более предпочтительно в аморфной форме. Используемый в данном документе термин «по существу аморфный материал» охватывает материал, который не более чем на 10% является кристаллическим; а термин «аморфный материал» охватывает материал, который является не более чем на 2% кристаллическим.
В одном воплощении данного изобретения соединение I содержится в твердом молекулярном комплексе, образованном гидроксипропилметилцеллюлозы ацетата сукцинатом (HPMC-AS). Используемый в данном документе термин «твердый молекулярный комплекс» означает композицию, в которой соединение I случайным образом распределено («молекулярно рассеяно») в матрице, образованной HPMC-AS. Предпочтительно такая композиция соединения I и HPMC-AS формирует однофазовую систему, которая может быть охарактеризована порошковой картиной рентгеновской дифракции, которая по существу свободна или свободна от кристаллических сигналов, связанных с кристаллической формой соединения I. В некоторых воплощениях соединение I присутствует в полимере в конечном более мелком состоянии. В некоторых воплощениях соединение I молекулярно диспергировано в матрице HPMC-AS так, что оно будет иммобилизовано в его аморфной форме. Под «иммобилизованным» понимается, что молекулы соединения I взаимодействуют с молекулами HPMC-AS таким образом, что они удерживаются в вышеупомянутой матрице, и образование кристаллов предотвращается в связи с отсутствием подвижности. В некоторых воплощениях полимер может предотвращать внутримолекулярные водородные связи или слабые дисперсионные силы между двумя или более молекулами соединения I.
В некоторых воплощениях соотношение количества по весу соединения I в твердом молекулярном комплексе и количества по весу HPMC-AS в нем составляет от примерно 1:9 до примерно 5:5. В одном воплощении указанное соотношение составляет от примерно 2:8 до примерно 4:6. В другом воплощении указанное соотношение составляет примерно 3:7.
В некоторых воплощениях способа и набора данного изобретения первый компонент включает вышеупомянутый твердый молекулярный комплекс соединения I и HPMC-AS, смешанный с коллоидным диоксидом кремния. В некоторых воплощениях смесь составляет по меньшей мере 0,5% от массы диоксида кремния. В одном воплощении изобретения смесь состоит примерно из 97% комплекса и примерно 3% диоксида кремния.
В другом воплощении первый компонент включает композицию, содержащую вышеупомянутый твердый молекулярный комплекс, смешанный или не смешанный с диоксидом кремния, как описано выше, и фармацевтически приемлемый носитель. В некоторых воплощениях вышеупомянутый комплекс или смесь, содержащая то же самое, суспендированы в носителе. Примером носителя является гидроксипропилцеллюлоза (НРС). В одном воплощении носитель составляет примерно 2% от массы НРС.
Каждый компонент может также содержать дополнительные агенты, такие как консерванты, солюбилизирующие агенты, стабилизирующие агенты, увлажняющие агенты, эмульгаторы, подсластители, красители, ароматизаторы, соли для изменения осмотического давления, буферы, покрывающие агенты и антиоксиданты.
В некоторых воплощениях первый компонент может содержать твердый молекулярный комплекс соединения I и HPMC-AS, смешанный с коллоидным диоксидом кремния, гидроксипропилцеллюлозой, кросповидоном (дезинтегрирующий агент), стеаратом магния (смазка, которая может быть использована в таблетках и капсулах) и/или кроскармеллозой натрия (дезинтегрирующий агент).
В одном воплощении первым компонентом является твердая желатиновая капсула, содержащая твердый молекулярный комплекс соединения I и HPMC-AS, смешанный с коллоидным диоксидом кремния, гидроксипропилцеллюлозой, стеаратом магния и кроскармеллозой натрия.
В одном воплощении первым компонентом является таблетка, содержащая соединение I или его фармацевтически приемлемую соль. В одном воплощении таблетка содержит твердый молекулярный комплекс соединения I или его фармацевтически приемлемой соли и HPMC-AS. Комплекс, например, может быть смешан с коллоидным диоксидом кремния, гидроксипропилцеллюлозой, стеаратом магния и кроскармеллозой натрия. Таблетка, например, может быть покрыта пленочным покрытием. Пленочное покрытие может содержать, например, поливиниловый спирт, диоксид титана, полиэтиленгликоль 3350, тальк и красный оксид железа.
В некоторых воплощениях второй компонент может содержать цетуксимаб в растворе. В одном воплощении раствор содержит примерно 2 мг/мл цетуксимаба.
В некоторых воплощениях второй компонент может включать таблетку, содержащую эрлотиниб или его фармацевтически приемлемую соль, например, гидрохлорид эрлотиниба.
В некоторых воплощениях третий компонент может включать раствор, содержащий иринотекан или его фармацевтически приемлемую соль, например, гидрохлорид иринотекана. В одном воплощении раствор представляет собой примерно 5% раствор декстрозы. В одном воплощении каждый мл раствора содержит примерно 20 мг гидрохлорида иринотекана, примерно 45 мг сорбита и примерно 0,9 мг молочной кислоты. В одном воплощении раствор имеет, рН примерно от 3,0 до примерно 3,8, например, примерно 3,5.
Кроме того, данное изобретение предусматривает применение соединения 1 или его фармацевтически приемлемой соли и ингибитора EGFR, возможно также в комбинации с ингибитором топоизомеразы, в лечении пролиферативного нарушения, в частности, солидной опухоли, более конкретно колоректального рака, меланомы и/или рака щитовидной железы, каждый из которых содержит b-Raf с мутацией V600E.
Данное изобретение предусматривает применение соединения I или его фармацевтически приемлемой соли и ингибитора EGFR, возможно также в комбинации с ингибитором топоизомеразы, для изготовления лекарственного препарата для лечения пролиферативного нарушения, в частности, солидной опухоли, более конкретно колоректального рака, меланомы и/или рака щитовидной железы, каждый из которых содержит b-Raf с мутацией V600E.
Данное изобретение также предусматривает способ лечения пациента, страдающего от пролиферативного нарушения, в частности, солидной опухоли, более конкретно колоректального рака, меланомы и/или рака щитовидной железы, каждый из которых содержит b-Raf с мутацией V600E, который включает введение указанному пациенту комбинации (А) и (В) и (С) в дозировках и соответствии с дозирующими графиками, описанными выше в данном документе.
Заявители проводили исследования с использованием мышей, содержащих ксенотрансплантат человеческого колоректального рака. Заявители обнаружили, что комбинация соединения 1 в дозе 75 мг/кг 2 раза/день и эрлотиниба гидрохлорида в дозе 67 мг/кг 1 раз/день давала результаты по ингибированию роста опухоли (tumor growth inhibition, TGI) и увеличению продолжительности жизни (increased life span, ILS), которые были значительно лучше, чем результаты при монотерапии эрлотиниба гидрохлоридом в дозе 100 мг/кг 1 раз/день. Кроме того, 9 из 10 мышей, получавших комбинированную терапию, имели частичную регрессию, в то время как в группах с монотерапией регрессия (частичная или полная) не наблюдалась. Эти исследования показывают, что лечение пациентов комбинацией соединения I и эрлотиниба гидрохлорида превосходит лечение каждым агентом в отдельности. Кроме того, исследования показывают, что комбинация двух агентов дает по меньшей мере уменьшение дозировки эрлотиниба гидрохлорида с получением таких же или улучшенных результатов.
Заявители обнаружили, что комбинация соединения I в дозе 25 мг/кг 2 раза/день и цетуксимаба в дозе 40 мг/кг 2 раза/нед давала результаты по ингибированию роста опухоли (TGI) и увеличению продолжительности жизни (ILS), которые были значительно лучше, чем результаты при соответствующей монотерапии с р<0,05, а также лучше, чем результаты, полученные при монотерапии соединением I в дозе 75 мг/кг 2 раза/день. Заявители также обнаружили, что комбинация соединения I в дозе 75 мг/кг 2 раза/день и цетуксимаба в дозе 40 мг/кг 2 раза/нед давала результаты по TGI и ILS, которые были значительно лучше, чем результаты при соответствующей монотерапии с р<0,05, а также лучше, чем результаты, полученные при монотерапии соединением I в дозе 25 мг/кг 2 раза/день. Кроме того, 7 из 9 мышей, получавших комбинированную терапию соединением I в дозе 25 мг/кг 2 раза/день и цетуксимабом в дозе 40 мг/кг 2 раза/нед, имели частичную регрессию, а 10 из 10 мышей, получавших комбинированную терапию соединением I в дозе 75 мг/кг 2 раза/день и цетуксимабом в дозе 40 мг/кг 2 раза/нед, демонстрировали регрессию, 7 из которых были частичными и 3 из которых были полными. Напротив, в группах с монотерапией регрессия (частичная или полная) не наблюдалась.
В дополнение к вышесказанному, заявители обнаружили, что комбинация соединения I в дозе 25 мг/кг 2 раза/день, цетуксимаба в дозе 40 мг/кг 2 раза/нед и иринотекана гидрохлорида в дозе 40 мг/кг q4dx5 давала результаты по ингибированию роста опухоли (TGI) и увеличению продолжительности жизни (ILS), которые были значительно лучше, чем результаты при соответствующей монотерапии с р<0,05, а также лучше, чем результаты, полученные при комбинированной терапии соединением I в дозе 25 мг/кг 2 раза/день и иринотекана гидрохлоридом в дозе 40 мг/кг q4dx5 и при комбинированной терапии соединением 1 в дозе 25 мг/кг 2 раза/день и цетуксимабом в дозе 40 мг/кг 2 раза/нед. В исследовании комбинированная терапия соединением 1 в дозе 25 мг/кг 2 раза/день и иринотекана гидрохлоридом в дозе 40 мг/кг q4dx5 приводила к 4 из 10 частичным регрессиям и не приводила к полной регрессии, а комбинированная терапия соединением I в дозе 25 мг/кг 2 раза/день и цетуксимабом в дозе 40 мг/кг 2 раза/нед приводила к 5 из 10 частичным регрессиям и не приводила к полной регрессии. Комбинированная терапия цетуксимабом в дозе 40 мг/кг 2 раза/нед и иринотекана гидрохлоридом в дозе 40 мг/кг q4dx5 и соответствующие монотерапии соединением I, цетуксимабом и иринотекана гидрохлоридом не приводили к регрессии. Напротив, терапия соединением I в дозе 25 мг/кг 2 раза/день, цетуксимабом в дозе 40 мг/кг 2 раза/нед и иринотекана гидрохлоридаом в дозе 40 мг/кг q4dx5 приводила к 10 из 10 регрессий, 9 частичным и одной полной.
Эти исследования показывают, что лечение пациентов комбинацией соединения I и цетуксимаба превосходит лечение каждым агентом в отдельности. Также они показывают, что комбинация двух агентов позволяет по меньшей мере уменьшить дозировку соединения I с получением таких же или улучшенных результатов. Кроме того, исследования показывают, что лечение пациентов комбинацией соединения I, цетуксимаба и иринотекана гидрохлорида дает еще более хорошие результаты.
Примеры
Изобретение будет более понятным со ссылкой на следующие примеры. Тем не менее, они не должны рассматриваться как ограничивающие объем изобретения.
Следующие сокращения используются в данном документе:
В примерах, приведенных ниже, потерю веса графически представляли как процентное изменение средней массы тела группы, используя формулу: ((W-W0)/W0)×100, где «W» представляет собой средний вес тела в группе, получавшей лечение, в определенный день, a «W0» представляет собой среднюю массу тела в той же группе, получавшей лечение, в начале лечения. Также была представлена максимальная потеря веса по приведенной выше формуле и указанный максимальный процент потери веса тела, который наблюдался в любое время в течение всего эксперимента для конкретной группы.
Данные эффективности графически представляли как средний объем опухоли ± стандартная ошибка среднего (SEM). Кроме того, объемы опухолей в группах, получавших лечение, были представлены в процентах от объемов опухолей в контрольных группах (% Т/С) по формуле: 100×((Т-Т0)/(С-С0)), где Т представляет собой средний объем опухоли в группе, получавшей лечение, в определенный день в ходе эксперимента, Т0 представляет собой средний объем опухоли в той же группе, получавшей лечение, в первый день лечения; С представляет собой средний объем опухоли в контрольной группе в определенный день в ходе эксперимента, а С0 представляет собой средний объем опухоли в той же группе, получавшей лечение, в первый день лечения.
Объем опухоли (в кубических миллиметрах) рассчитывали по формуле эллипсоида: (D×(d2))/2, где «D» представляет собой большой диаметр опухоли, а «d» представляет собой малый диаметр.
Кроме того, регрессию опухоли и/или процентное изменение объема опухоли рассчитывали по формуле: ((Т-Т0)/T0)×100, где «Т» представляет собой средний объем опухоли в группе, получавшей лечение, в определенный день, а «Т0» представляет собой средний объем опухоли в той же группе, получавшей лечение, в начале лечения.
Статистический анализ проводили с помощью критерия суммы рангов и однофакторного дисперсионного анализа (One Way Anova) с последующим t-тестом Бонферрони (SigmaStat, версия 2.0, Jandel Scientific, Сан-Франциско, Калифорния, США). Различия между группами считались существенными, когда значение вероятности (р) составляло ≤ 0,05.
Для оценки выживаемости процент увеличения продолжительности жизни (ILS) рассчитывали как: 100×[(средняя выживаемость в днях в группе, получавшей лечение - средняя выживаемость в днях в контрольной группе)/средняя выживаемость в днях в контрольной группе]. Среднюю выживаемость определяли с использованием анализа выживаемости Каплана-Мейера. Выживаемость в группах, получавших лечение, статистически сравнивали с группой, получавшей носители, а также сравнивали выживаемость между группами с помощью логарифмического рангового критерия (Graph Pad Prism, Ла-Хойя, Калифорния, США). Различия между группами считались значимыми, когда значение вероятности (р) составляло ≤0,05.
Пример 1
Этот пример описывает формирование суспензии, содержащей соединение I.
Вначале формировали твердый молекулярный комплекс, состоящий из соединения I и гидроксипропилметилцеллюлозы ацетата сукцината (HPMC-AS).
Соединение I и HPMC-AS в соотношении примерно 3:7, соответственно, растворяли в диметилацетамиде (DMA). Затем полученный раствор добавляли при перемешивании к очень холодной разбавленной соляной кислоте, вызывая копреципитацию соединения I и HPMC-AS в виде твердого молекулярного комплекса, в котором соединение I находилось в диапазоне размеров наночастиц. Отношение DMA к кислоте находилось в диапазоне от 1:5 до 1:10.
Затем копреципитат промывали водой, чтобы удалить DMA, фильтровали, сушили до содержания влаги <2% и пропускали через сетку №30 перед оценкой. Полученный твердый молекулярный комплекс состоял на 30% по весу из соединения I и на 70% по весу из НРМС.
Затем комплекс смешивали с коллоидным диоксидом кремния (доступен как Aerosil® 200 от Evonik Industries AG, Эссен, Германия), так что на 100 г смеси приходилось 97 г комплекса и 3 г коллоидного диоксида кремния.
Затем готовили водный носитель, содержащий 2% гидроксипропилцеллюлозы (доступна как Klucel® LF от Aqualon, Уилмингтон, Делавэр, США), и доводили рН до 4 с помощью 1N HCl.
23,2 мл носителя доводили до комнатной температуры, постепенно переносили в 773,2 мг вышеупомянутой смеси. Полученный препарат медленно смешивали до получения однородной суспензии. Таким образом получали суспензию, содержащую 9,375 мг/мл соединения I. Суспензию хранили при температуре 2-8°С в защищенном от света месте.
Пример 2
Мышам имплантировали ксенотрансплантаты человеческих клеток НТ-29. Используемые мыши, клеточная линия и имплантация описаны ниже.
Самок бестимусных мышей Crl:NU-Foxn1nu использовали для анализа эффективности (Charles River, Уилмингтон, Массачусетс, США). Мыши были в возрасте 10-12 недель и весили 23-25 граммов. Здоровье мышей оценивали ежедневно путем наблюдения и анализа образцов крови, взятых от сторожевых (контрольных) животных с общей полки. В течение одной недели всем животным позволяли акклиматизироваться и оправиться от стресса, связанного с доставкой. Автоклавированную воду и облученные пищевые продукты (5058-ms Pico Lab mouse chow, Purina Mills, Ричмонд, Индиана, США) предоставляли без ограничения, и содержали животных в условиях чередования 12-часовых периодов света и темноты. Клетки, подстилку и бутылки с водой автоклавировали перед применением и меняли еженедельно. Все эксперименты на животных проводили в соответствии с Руководством по уходу и использованию лабораторных животных, локальным нормативным актам и протоколам, утвержденным комитетом по уходу и использованию животных Roche Animal Care and Use Committee в нашей лаборатории, аккредитованной AAALAC.
Клетки НТ-29 (American Type Culture Collection, Роквилль, Мэриленд, США) выращивали в среде МсСоу-5 с добавлением 10% эмбриональной телячьей сыворотки (FBS) и 1% 200 нМ L-глутамина, выращивали, собирали и подготавливали таким образом, чтобы каждая мышь получила 3×106 клеток в 0,2 мл фосфатно-солевого буфера (PBS) без кальция и магния. Клетки имплантировали подкожно в правый бок каждой мыши.
Пример 3
Этот пример описывает приготовление суспензии эрлотиниба гидрохлорида. Один грамм Tween 80 добавляли приблизительно к 950 мл воды. При перемешивании на высокой скорости в раствор добавляли 3 грамма натрия карбоксиметилцеллюлозы. Перемешивание продолжали до растворения натрия карбоксиметилцеллюлозы. Затем добавляли воду q.s. до 1 литра. Затем 12,5 граммов эрлотиниба гидрохлорида (получен от Genentech как Tarceva®) суспендировали в растворе и пропускали через растворитель. Затем раствор деаэрировали азотом.
Состав окончательной суспензии является следующим:
Таким образом получали раствор с содержанием эрлотиниба гидрохлорида 12,5 мг/мл. Раствор хранили при температуре 2-8°С.
Пример 4
Суспензию, содержащую соединение I, получали, как описано в примере 1.
Раствор эрлотиниба гидрохлорида 12,5 мг/мл получали, как описано в примере 3. Также готовили раствор 8,30 мг/мл аналогично тому, как описано в примере 3, за исключением того, что вместо 12,5 г использовали 8,30 г эрлотиниба гидрохлорида.
Мышей, содержащих ксенотрансплантаты НТ-29, полученные в соответствии с описанием в примере 2, рандомизировали в группы по 10 мышей в каждой в соответствии с объемом опухоли таким образом, чтобы все группы имели похожие исходные значения объемов опухолей. Приблизительное исходное среднее значение объема опухоли для данного исследования составляло 136 мм3.
Лечение начинали на 12-й день после имплантации клеток и заканчивали на 29 день после имплантации клеток. Каждую группу подвергали различному лечению следующим образом:
(1) мыши, получавшие носитель соединения I 2 раза/день перорально и носитель эрлотиниба гидрохлорида 1 раз/день перорально;
(2) мыши, получавшие соединение I в дозе 75 мг/кг 2 раза/день перорально;
(3) мыши, получавшие эрлотиниба гидрохлорид в дозе 67 мг/кг 1 раз/день перорально;
(4) мыши, получавшие эрлотиниба гидрохлорид в дозе 100 мг/кг 1 раз/день перорально;
(5) мыши, получавшие соединение I в дозе 75 мг/кг 2 раза/день перорально и эрлотиниба гидрохлорид в дозе 67 мг/кг 1 раз/день перорально.
Суспензию соединения I и соответствующий носитель вводили с помощью стерильного шприца объемом 1 см3 и зонда-иглы размера 18 (0,2 мл/животное) дважды в день. Раствор эрлотиниба гидрохлорида и соответствующий носитель вводили с помощью стерильного шприца объемом 1 см3 и зонда-иглы размера 18 (0,2 мл/животное) один раз в день с 12-го дня по 29-1 день после имплантации. Раствор 12,5 мг/мл использовали для групп с дозой эрлотиниба гидрохлорида 100 мг/кг, а раствор 8,30 мг/мл использовали для групп с дозой эрлотиниба гидрохлорида 67 мг/кг 1 раз/день. Все дозировки были основаны на среднем весе мыши 25 граммов.
Измерения опухолей проводили один или два раза в неделю. Все животные были индивидуально проведены через весь эксперимент.
Токсичность
В целом, ни в одной группе данного исследования ни при каких дозах не было отмечено никаких серьезных признаков токсичности, которые оценивали путем измерения изменений массы тела и общего наблюдения за отдельными животными. Эрлотиниба гидрохлорид в дозе 100 мг/кг один раз в день исторически не очень хорошо переносится в комбинации (Higgins et al., Anticancer Drugs, 15:503-12 (2004)), поэтому в группах с комбинированной терапией для улучшения переносимости использовали 67 мг/кг один раз в день. Эрлотиниба гидрохлорид в дозе 100 мг/кг один раз в день был включен в качестве группы с монотерапией для сравнения. Тем не менее, соединение I очень хорошо переносилось и вводилось в дозе 75 мг/кг 2 раза/день даже в комбинации с эрлотиниба гидрохлоридом. У мышей, получавших эрлотиниба гидрохлорид, была распространена связанная с ингибитором EGFR сыпь на коже с самоограничивающимся характером даже при продолжающемся лечении. См. Таблицу 1 и фиг.1.
Ингибирование роста опухоли (TGI)
В группе, получавшей монотерапию соединением I в дозе 75 мг/кг 2 раза/день, было показано TGI 91%, в то время как в группе, получавшей монотерапию эрлотиниба гидрохлоридом в дозе 100 мг/кг один раз в день, было показано TGI 51%, а в группе, получавшей монотерапию эрлотиниба гидрохлоридом в дозе 67 мг/кг один раз в день, было показано TGI 38%. Отсутствие регрессии опухоли наблюдалось во всех перечисленных выше группах. В группе, получавшей комбинированную терапию соединением I в дозе 75 мг/кг 2 раза/день и эрлотиниба гидрохлоридом в дозе 67 мг/кг один раз в день, наблюдалось TGI более 100% с 9 частичными регрессиями (PR) из 10. См. Таблицы 2 и 3 и фиг.2.
Оценка выживаемости
В группе, получавшей монотерапию соединением I в дозе 75 мг/кг, было показано увеличение продолжительности жизни (ILS) 100%. В группе, получавшей монотерапию эрлотиниба гидрохлоридом в дозе 100 мг/кг 1 раз/день, было показано ILS 38%. В группе, получавшей монотерапию эрлотиниба гидрохлоридом в дозе 67 мг/кг 1 раз/день, было показано ILS 35%. В группе, получавшей комбинированную терапию соединением I в дозе 75 мг/кг 2 раза/день и эрлотиниба гидрохлоридом в дозе 67 мг/кг 1 раз/день, было показано ILS 142%. См. таблицу 4 и фиг.3.
Статистический анализ
% TGI при комбинированной терапии соединением I в дозе 75 мг/кг 2 раза/день и эрлотиниба гидрохлоридом в дозе 67 мг/кг 1 раз/день был статистически выше, чем во всех группах, получавших монотерапию (р<0,05). % ILS при комбинированной терапии соединением I в дозе 75 мг/кг 2 раза/день и эрлотиниба гидрохлоридом в дозе 67 мг/кг 1 раз/день был статистически выше, чем во всех группах, получавших монотерапию (р 0,05 для всех сравнений). См. Таблицу 5.
Пример 5
Две суспензии, содержащие соединение I, получали, как описано в примере 1, за исключением того, что при приготовлении 20 мл суспензии 9,375 мг/мл использовали 19,4 мл носителя и 644 мг смеси, а при приготовлении 20 мл суспензии 3,125 мг/мл использовали 19,8 мл носителя и 214,8 мг смеси.
Цетуксимаб был приобретен у компании ImClone Systems, Inc. (поставляется как Erbitux®) в виде раствора 2 мг/мл.
Мышей, содержащих ксенотрансплантаты НТ-29, полученные в соответствии с описанием в примере 2, рандомизировали в группы по 10 мышей в каждой в соответствии с объемом опухоли таким образом, чтобы все группы имели похожие исходные значения объемов опухолей. Приблизительное исходное среднее значение объема опухоли для данного исследования составляло 135 мм3.
Лечение начинали на 12-й день после имплантации клеток и заканчивали на 34 день после имплантации клеток. Каждую группу подвергали различному лечению следующим образом:
(1) мыши, получавшие носитель соединения I 2 раза/день перорально и носитель цетуксимаба 2 раза/нед внутрибрюшинно;
(2) мыши, получавшие цетуксимаб в дозе 40 мг/кг 2 раза/нед внутрибрюшинно;
(3) мыши, получавшие соединение I в дозе 25 мг/кг 2 раза/день перорально;
(4) мыши, получавшие соединение I в дозе 75 мг/кг 2 раза/день перорально;
(5) мыши, получавшие соединение I в дозе 25 мг/кг 2 раза/день перорально и цетуксимаб в дозе 40 мг/кг 2 раза/нед внутрибрюшинно;
(6) мыши, получавшие соединение I в дозе 75 мг/кг 2 раза/день перорально и цетуксимаб в дозе 40 мг/кг 2 раза/нед внутрибрюшинно.
Суспензию соединения I и соответствующий носитель вводили с помощью стерильного шприца объемом 1 см3 и зонда-иглы размера 18 (0,2 мл/животное) дважды в день. Суспензию 9,375 мг/кг использовали для групп с дозировкой соединения I 75 мг/кг 2 раза/день, а суспензию 3,125 мг/кг использовали для групп с дозировкой соединения I 25 мг/кг 2 раза/день. Цетуксимаб и соответствующий носитель вводили внутрибрюшинно с помощью стерильного шприца объемом 1 см3 и зонда-иглы размера 26 (0,2 мл/животное) два раза в неделю по графику понедельник/четверг или вторник/пятница. Все дозировки были основаны на среднем весе мыши 25 граммов.
Измерения опухолей проводили один или два раза в неделю. Все животные были индивидуально проведены через весь эксперимент.
Токсичность
В целом, ни в одной группе данного исследования ни при каких дозах не было отмечено никаких серьезных признаков токсичности, которые оценивали путем измерения изменений массы тела и общего наблюдения за отдельными животными. См. Таблицу 6 и фиг.4. У мышей, получавших цетуксимаб, была распространена связанная с ингибитором EGFR сыпь на коже с самоограничивающимся характером даже при продолжающемся лечении. У одной мыши, по-видимому, развилась бактериальная инфекция как осложнение сыпи, что привело к прогрессирующей потере веса >20% и тем самым к необходимости умерщвления. Эту мышь удалили из общего анализа ингибирования роста опухоли и выживаемости.
Ингибирование роста опухоли (TGI)
В группе, получавшей монотерапию соединением I в дозе 25 мг/кг 2 раза/день, было показано TGI 74%, а в группе, получавшей монотерапию соединением I в дозе 75 мг/кг 2 раза/день, было показано TGI 93%. В группе, получавшей цетуксимаб в дозе 40 мг/кг 2 раза/нед, было получено TGI 51%. Отсутствие регрессии опухоли наблюдалось во всех перечисленных выше группах. Тем не менее, в обеих группах с комбинированной терапией наблюдалось TGI более 100%. В группе, получавшей соединение I в дозе 25 мг/кг 2 раза/день и цетуксимаб в дозе 40 мг/кг 2 раза/нед, наблюдалось 7 из 10 частичных регрессий (PR), но не наблюдалось полной регрессии (CR). В группе, получавшей соединение I в дозе 75 мг/кг 2 раза/день и цетуксимаб в дозе 40 мг/кг 2 раза/нед, наблюдалось 7 из 10 PR и 3 из 10 CR.
См. Таблицы 7 и 8 и фиг.5.
Оценка выживаемости
В группе, получавшей монотерапию соединением I в дозе 25 мг/кг 2 раза/день, было показано ILS 44%, а в группе, получавшей монотерапию соединением I в дозе 75 мг/кг 2 раза/день, было показано ILS 75%. В группе, получавшей цетуксимаб в дозе 40 мг/кг 2 раза/нед, было получено ILS 16%. В группе, получавшей соединение I в дозе 25 мг/кг 2 раза/день и цетуксимаб в дозе 40 мг/кг 2 раза/нед, было показано ILS 97%. В группе, получавшей соединение I в дозе 75 мг/кг 2 раза/день и цетуксимаб в дозе 40 мг/кг 2 раза/нед, было показано ILS 122%. См. таблицу 9 и фиг.6.
Статистический анализ
% TGI при комбинированной терапии соединением I в дозе 25 мг/кг 2 раза/день и цетуксимабом в дозе 40 мг/кг 2 раза/нед и при комбинированной терапии соединением I в дозе 75 мг/кг 2 раза/день и цетуксимабом в дозе 40 мг/кг 2 раза/нед был статистически выше, чем во всех группах, получавших монотерапию (р<0,05). % ILS, полученный при сочетании монотерапий, был статистически выше (р<0,05 для всех сравнений), чем во всех группах, получавших монотерапию (р<0,05 для всех сравнений). См. Таблицу 10.
Пример 6
Суспензию, содержащую соединение I, получали, как описано в примере 1, за исключением того, что при приготовлении 40 мл суспензии 3,125 мг/кг использовали 39,6 мл носителя и 429,6 мг смеси.
Цетуксимаб был приобретен у компании ImClone Systems, Inc. (поставляется как Erbitux®) в виде раствора 2 мг/мл. Иринотекана гидрохлорид был приобретен у компании Pfizer Inc. (поставляется как Camptosar®) в виде стокового стерильного раствора 20 мг/мл, который разводили при необходимости стерильным физиологическим раствором до 2 мг/мл.
Мышей, содержащих ксенотрансплантаты НТ-29, полученные в соответствии с описанием в примере 2, рандомизировали в группы по 10 мышей в каждой в соответствии с объемом опухоли таким образом, чтобы все группы имели похожие исходные значения объемов опухолей. Приблизительное исходное среднее значение объема опухоли для данного исследования составляло 135 мм3.
Лечение начинали на 11-й день после имплантации клеток и заканчивали на 32 день после имплантации клеток. Каждую группу подвергали различному лечению следующим образом:
(1) мыши, получавшие носитель соединения I 2 раза/день перорально, носитель цетуксимаба 2 раза/нед внутрибрюшинно и носитель Иринотекана HCl q4dx5 внутрибрюшинно;
(2) мыши, получавшие иринотекан HCl в дозе 40 мг/кг q4dx5 внутрибрюшинно;
(3) мыши, получавшие соединение I в дозе 25 мг/кг 2 раза/день перорально;
(4) мыши, получавшие цетуксимаб в дозе 40 мг/кг 2 раза/нед внутрибрюшинно;
(5) мыши, получавшие соединение I в дозе 25 мг/кг 2 раза/день перорально и иринотекан HCl в дозе 40 мг/кг q4dx5 внутрибрюшинно;
(6) мыши, получавшие цетуксимаб в дозе 40 мг/кг 2 раза/нед внутрибрюшинно и иринотекан HCl в дозе 40 мг/кг q4dx5 внутрибрюшинно;
(7) мыши, получавшие соединение I в дозе 25 мг/кг 2 раза/день перорально и цетуксимаб в дозе 40 мг/кг 2 раза/нед внутрибрюшинно;
(8) мыши, получавшие соединение I в дозе 25 мг/кг 2 раза/день перорально, цетуксимаб в дозе 40 мг/кг 2 раза/нед внутрибрюшинно и иринотекан HCl в дозе 40 мг/кг q4dx5 внутрибрюшинно.
Суспензию соединения 1 и соответствующий носитель вводили с помощью стерильного шприца объемом 1 см3 и зонда-иглы размера 18 (0,2 мл/животное) дважды в день. Цетуксимаб и соответствующий носитель вводили внутрибрюшинно с помощью стерильного шприца объемом 1 см3 и зонда-иглы размера 26 (0,2 мл/животное) два раза в неделю по графику понедельник/четверг или вторник/пятница. Иринотекан HCl и соответствующий носитель вводили внутрибрюшинно с помощью стерильного шприца объемом 1 см3 и зонда-иглы размера 26 (0,2 мл/животное) по графику q4dx5. Все дозировки были основаны на среднем весе мыши 25 граммов.
Измерения опухолей проводили один или два раза в неделю. Все животные были индивидуально проведены через весь эксперимент.
Токсичность
В целом, ни в одной группе данного исследования ни при каких дозах не было отмечено никаких серьезных признаков токсичности, которые оценивали путем измерения изменений массы тела и общего наблюдения за отдельными животными. См. Таблицу 11 и фиг.7. У мышей, получавших цетуксимаб, была распространена связанная с ингибитором EGFR сыпь на коже с самоограничивающимся характером даже при продолжающемся лечении.
Ингибирование роста опухоли (TGI)
В группе, получавшей монотерапию соединением I в дозе 25 мг/кг 2 раза/день, было показано TGI 76%. В группе, получавшей монотерапию цетуксимабом в дозе 40 мг/кг 2 раза/нед, было показано TGI 58%. В группе, получавшей монотерапию иринотеканом HCl в дозе 40 мг/кг q4dx5, было показано TGI 59%. В группе, получавшей соединение I в дозе 25 мг/кг 2 раза/день и иринотекан HCl в дозе 40 мг/кг q4dx5, было показано TGI 98%. В группе, получавшей цетуксимаб в дозе 40 мг/кг 2 раза/нед и иринотекан HCl в дозе 40 мг/кг q4dx5, было показано TGI 92%. В группе, получавшей соединение I в дозе 25 мг/кг 2 раза/день и цетуксимаб в дозе 40 мг/кг 2 раза/нед, было показано TGI>100%. В группе, получавшей соединение I в дозе 25 мг/кг 2 раза/день, цетуксимаб в дозе 40 мг/кг 2 раза/нед и иринотекан HCl в дозе 40 мг/кг q4dx5, было показано TGI>100%. Отсутствие регрессии опухоли наблюдалось в группах с любой монотерапией. В группе, получавшей соединение I в дозе 25 мг/кг 2 раза/день и цетуксимаб в дозе 40 мг/кг 2 раза/нед, наблюдалось 5 из 10 частичных регрессий (PR), но не наблюдалось полной регрессии (CR). В группе, получавшей соединение I в дозе 25 мг/кг 2 раза/день, цетуксимаб в дозе 40 мг/кг 2 раза/нед и иринотекан HCl в дозе 40 мг/кг q4dx5, наблюдалось 9 из 10 PR и 1 из 10 CR. См. Таблицы 12 и 13 и фиг.8.
Оценка выживаемости
В группе, получавшей монотерапию соединением I в дозе 25 мг/кг 2 раза/день, было показано ILS 80%. В группе, получавшей монотерапию цетуксимабом в дозе 40 мг/кг 2 раза/нед, было показано ILS 27%. В группе, получавшей монотерапию иринотеканом HCl в дозе 40 мг/кг q4dx5, было показано ILS 17%. В группе, получавшей соединение I в дозе 25 мг/кг 2 раза/день и иринотекан HCl в дозе 40 мг/кг q4dx5, было показано ILS 163%. В группе, получавшей цетуксимаб в дозе 40 мг/кг 2 раза/нед и иринотекан HCl в дозе 40 мг/кг q4dx5, было показано ILS 80%. В группе, получавшей соединение I в дозе 25 мг/кг 2 раза/день и цетуксимаб в дозе 40 мг/кг 2 раза/нед, было показано ILS 127%. В группе, получавшей соединение I в дозе 25 мг/кг 2 раза/день, цетуксимаб в дозе 40 мг/кг 2 раза/нед и иринотекан HCl в дозе 40 мг/кг q4dx5, было показано ILS 259%. См. таблицу 14 и фиг.9.
Статистический анализ
% TGI при комбинированной терапии соединением I/цетуксимабом, соединением I/иринотеканом HCl и соединением I/цетуксимабом/иринотеканом НС1 был статистически выше, чем во всех группах, получавших монотерапию (р<0,05). % TGI при комбинированной терапии соединением I/цетуксимабом/иринотеканом HCl был также статистически выше, чем при комбинированной терапии соединением I/иринотеканом HCl и цетуксимабом/иринотеканом HCl (р<0,05).
% ILS при комбинированной терапии соединением I/цетуксимабом, соединением I/иринотеканом HCl и соединением I/цетуксимабом/иринотеканом HCl был статистически выше, чем во всех группах, получавших монотерапию (р<0,05 для всех сравнений). % ILS при комбинированной терапии соединением I/цетуксимабом/иринотеканом HCl был также статистически выше, чем при комбинированной терапии соединением I/иринотеканом HCl и соединением I/цетуксимабом.
См.Таблицу 15.
| название | год | авторы | номер документа |
|---|---|---|---|
| КОМБИНИРОВАННАЯ ТЕРАПИЯ, ВКЛЮЧАЮЩАЯ ВЕМУРАФЕНИБ И ИНТЕРФЕРОН, ДЛЯ ПРИМЕНЕНИЯ В ЛЕЧЕНИИ РАКА | 2011 |
|
RU2592983C2 |
| Фармацевтические комбинации | 2017 |
|
RU2759669C2 |
| СПОСОБ ВВЕДЕНИЯ ПРОТИВООПУХОЛЕВОГО АГЕНТА | 2013 |
|
RU2638795C2 |
| КОМБИНИРОВАННАЯ ТЕРАПИЯ | 2018 |
|
RU2815400C2 |
| КОМБИНАЦИИ (СОСТАВЫ), ВКЛЮЧАЮЩИЕ DMXAA ДЛЯ ЛЕЧЕНИЯ РАКА | 2006 |
|
RU2404765C2 |
| СОКРИСТАЛЛЫ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2014 |
|
RU2675270C2 |
| СО-КРИСТАЛЛЫ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2014 |
|
RU2823603C2 |
| ТЕРАПЕВТИЧЕСКИЕ КОМБИНАЦИИ, СОДЕРЖАЩИЕ ИНГИБИТОР RAF И ИНГИБИТОР ERK | 2017 |
|
RU2774612C2 |
| ИСПОЛЬЗОВАНИЕ ПРОИЗВОДНЫХ 2-КАРБОКСАМИД-ЦИКЛОАМИНО МОЧЕВИНЫ В ЛЕЧЕНИИ EGFR-ЗАВИСИМЫХ ЗАБОЛЕВАНИЙ ИЛИ ЗАБОЛЕВАНИЙ С ПРИОБРЕТЕННОЙ РЕЗИСТЕНТНОСТЬЮ К АГЕНТАМ, НАЦЕЛЕННЫМ НА ЧЛЕНЫ EGFR-СЕМЕЙСТВА | 2011 |
|
RU2589695C2 |
| СПОСОБЫ ЛЕЧЕНИЕ РАКА ПОДЖЕЛУДОЧНОЙ ЖЕЛЕЗЫ | 2011 |
|
RU2576609C2 |







Данное изобретение относится к комбинированной терапии {3-[5-(4-хлорфенил)-1Н-пирроло[2,3-b]пиридин-3-карбонил]-2,4-дифторфенил}амидом пропан-1-сульфокислоты (соединение 1) или его фармацевтически приемлемой солью и ингибитором EGFR, выбранный из эрлотиниба и цетуксимаба для лечения рака, содержащего b-Raf с мутацией V600, более конкретно колоректального рака, меланомы и рака щитовидной железы. Комбинация соединения 1 с ингибитором EGFR приводит к улучшенным противоопухолевым эффектам, которые значительно превосходят результаты, полученные для каждого соединения в отдельности без увеличения токсичности. 6 н. и 14 з.п. ф-лы,6 пр., 15 табл., 9 ил.
1. Фармацевтический продукт, включающий (А) первый компонент, который содержит в качестве активного агента {3-[5-(4-хлорфенил)-1Н-пирроло[2,3-b]пиридин-3-карбонил]-2,4-дифторфенил}амид пропан-1-сульфокислоты или его фармацевтически приемлемую соль; и (В) второй компонент, который содержит в качестве активного агента ингибитор EGFR, выбранный из эрлотиниба и цетуксимаба; в виде комбинированного препарата для одновременного или последовательного применения в лечении рака, содержащего b-Raf с мутацией V600, более конкретно колоректального рака, меланомы и рака щитовидной железы, содержащего b-Raf с мутацией V600.
2. Фармацевтический продукт по п. 1 для лечения колоректального рака, меланомы и рака щитовидной железы, содержащего b-Raf с мутацией V600E.
3. Фармацевтический продукт по п. 1, где указанный ингибитор EGFR представляет собой эрлотиниб или его фармацевтически приемлемую соль.
4. Фармацевтический продукт по п. 1, где указанный ингибитор EGFR представляет собой цетуксимаб.
5. Фармацевтический продукт по п. 1, также включающий третий компонент (С), который содержит в качестве активного агента ингибитор топоизомеразы, в частности ингибитор топоизомеразы I типа.
6. Фармацевтический продукт по п. 5, где указанный ингибитор топоизомеразы представляет собой иринотекан или его фармацевтически приемлемую соль.
7. Фармацевтический продукт по любому из пп. 1-6, где {3-[5-(4-хлорфенил)-1Н-пирроло[2,3-b]пиридин-3-карбонил]-2,4-дифторфенил}-амид пропан-1-сульфокислоты или его фармацевтически приемлемая соль находится в аморфной или по существу аморфной форме.
8. Набор для лечения рака, содержащего b-Raf с мутацией V600, более конкретно колоректального рака, меланомы и рака щитовидной железы, содержащего b-Raf с мутацией V600, состоящий из: (А) первого компонента, который содержит в качестве активного агента {3-[5-(4-хлор-фенил)1Н-пирроло[2,3-b]пиридин-3-карбонил]-2,4-дифторфенил}-амид пропан-1-сульфокислоты или его фармацевтически приемлемую соль; и (В) второго компонента, который содержит в качестве активного агента ингибитор EGFR, выбранный из эрлотиниба и цетуксимаба.
9. Набор по п. 8, также включающий третий компонент, который содержит в качестве активного агента ингибитор топоизомеразы, более конкретно иринотекан.
10. Набор по п. 8 или 9 для применения в лечении пролиферативного нарушения, в частности рака, более конкретно колоректального рака, меланомы и рака щитовидной железы, содержащего b-Raf с мутацией V600E.
11. Фармацевтический продукт по п. 3, где указанный {3-[5-(4-хлор-фенил)1Н-пирроло[2,3-b]пиридин-3-карбонил]-2,4-дифторфенил}-амид пропан-1-сульфокислоты или его фармацевтически приемлемую соль вводят 2 раза/день в количестве от примерно 850 мг до примерно 1050 мг; а указанный эрлотиниб или его фармацевтически приемлемую соль вводят ежедневно в количестве от примерно 100 мг до примерно 200 мг.
12. Фармацевтический продукт по п. 4, где указанный {3-[5-(4-хлор-фенил)1Н-пирроло[2,3-b]пиридин-3-карбонил]-2,4-дифторфенил}-амид пропан-1-сульфокислоты или его фармацевтически приемлемую соль вводят 2 раза/день в количестве от примерно 850 мг до примерно 1050 мг; а указанный цетуксимаб вводят раз в неделю, при первом введении в количестве от примерно 400 мг/м2 до примерно 500 мг/м2, и при каждом последующем введении в количестве от примерно 200 мг/м2 до примерно 300 мг/м2.
13. Фармацевтический продукт по п. 11 или 12, также включающий иринотекан или его фармацевтически приемлемую соль.
14. Фармацевтический продукт по п. 13, где указанный иринотекан или его фармацевтически приемлемую соль вводят раз в неделю в количестве от примерно 50 мг/м2 до примерно 200 мг/м2.
15. Фармацевтический продукт по п. 13, где иринотекан или его фармацевтически приемлемую соль вводят по шестинедельному циклу в количестве от примерно 75 до примерно 175 мг/м2/нед еженедельно в течение первых четырех недель.
16. Фармацевтический продукт по п. 13, где иринотекан или его фармацевтически приемлемую соль вводят раз в три недели в количестве от примерно 300 до примерно 400 мг/м2; или раз в две недели в количестве от примерно 130 до примерно 230 мг/м2.
17. Фармацевтический продукт, включающий (А) первый компонент, который содержит в качестве активного агента {3-[5-(4-хлорфенил)-1Н-пирроло[2,3-b]пиридин-3-карбонил]-2,4-дифторфенил}амид пропан-1-сульфокислоты или его фармацевтически приемлемую соль; и (В) второй компонент, который содержит в качестве активного агента эрлотиниб или его фармацевтически приемлемую соль;
в виде комбинированного препарата для одновременного или последовательного применения в лечении рака, содержащего b-Raf с мутацией V600, более конкретно колоректального рака, меланомы и рака щитовидной железы, содержащего b-Raf с мутацией V600E, где
(A) вводят в количестве от примерно 200 мг/день до примерно 3000 мг/день, от примерно 1000 мг/день до примерно 2500 мг/день, от примерно 1700 мг/день до примерно 2100 мг/день, или примерно 1920 мг/день; а
(B) вводят в количестве от примерно 20 мг/день до примерно 500 мг/день, от примерно 100 мг/день до примерно 400 мг/день, или от примерно 100 мг/день до примерно 200 мг/день.
18. Фармацевтический продукт, включающий (А) первый компонент, который содержит в качестве активного агента {3-[5-(4-хлорфенил)-1Н-пирроло[2,3-b]пиридин-3-карбонил]-2,4-дифторфенил}амид пропан-1-сульфокислоты или его фармацевтически приемлемую соль; и (В) второй компонент, который содержит в качестве активного агента цетуксимаб; в виде комбинированного препарата для одновременного или последовательного применения в лечении рака, содержащего b-Raf с мутацией V600, где
(A) вводят в количестве от примерно 200 мг/день до примерно 3000 мг/день, от примерно 1000 мг/день до примерно 2500 мг/день, от примерно 1700 мг/день до примерно 2100 мг/день, или примерно 1920 мг/день; а
(B) вводят в количестве от примерно 50 мг/м2/нед до примерно 700 мг/м2/нед, от примерно 100 мг/м2/нед до примерно 600 мг/м2/нед, или от примерно 200 мг/м2/нед до примерно 500 мг/м2/нед.
19. Фармацевтический продукт, включающий (А) первый компонент, который содержит в качестве активного агента {3-[5-(4-хлорфенил)-1Н-пирроло[2,3-b]пиридин-3-карбонил]-2,4-дифторфенил}амид пропан-1-сульфокислоты или его фармацевтически приемлемую соль; (В) второй компонент, который содержит в качестве активного агента цетуксимаб; и (С) третий компонент, который содержит в качестве активного агента иринотекан или его фармацевтически приемлемую соль; в виде комбинированного препарата для одновременного или последовательного применения в лечении рака, содержащего b-Raf с мутацией V600, где
(А) вводят в количестве от примерно 200 мг/день до примерно 3000 мг/день, от примерно 1000 мг/день до примерно 2500 мг/день, от примерно 1700 мг/день до примерно 2100 мг/день, или примерно 1920 мг/день;
(B) вводят в количестве от примерно 50 мг/м2/нед до примерно 700 мг/м2/нед, от примерно 100 мг/м2/нед до примерно 600 мг/м2/нед, или от примерно 200 мг/м2/нед до примерно 500 мг/м2/нед; и
(C) вводят в количестве от примерно 1 до примерно 250 мг/м2/нед, от примерно 50 до примерно 200 мг/м2/нед, или примерно 125 мг/м2/нед.
20. Применение {3-[5-(4-хлорфенил)-1Н-пирроло[2,3-b]пиридин-3-карбонил]-2,4-дифторфенил}-амида пропан-1-сульфокислоты или его фармацевтически приемлемой соли и ингибитора EGFR, выбранного из эрлотиниба и цетуксимаба, для изготовления лекарственных средств для лечения рака, содержащего b-Raf с мутацией V600, более конкретно колоректального рака, меланомы и рака щитовидной железы, каждый из которых содержит b-Raf с мутацией V600, в частности с мутацией V600E.
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Колосоуборка | 1923 |
|
SU2009A1 |
| Иротекан | |||
| Справочник лекарств: лекарственные препараты и лекарственные средства | |||
| Видоизменение прибора с двумя приемами для рассматривания проекционные увеличенных и удаленных от зрителя стереограмм | 1919 |
|
SU28A1 |
| М.Д | |||
| Машковский | |||
| Лекарственные средства | |||
| Пособие для врачей | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Дивов, 2001, стр | |||
| Походная разборная печь для варки пищи и печения хлеба | 1920 |
|
SU11A1 |

