Область техники, к которой относится изобретение
Настоящее изобретение относится к применению ингибитора RAF, в частности ингибитора c–RAF (C–RAF или CRAF), для лечения рака, который представляет собой солидную опухоль на поздней стадии, которая содержит изменения митоген–активируемой протеинкиназы (MAPK), как например KRAS–мутантные опухоли, и в частности KRAS–мутантный NSCLC (немелкоклеточный рак легкого), меланома, рак поджелудочной железы, колоректальный рак и рак яичника. В частности, оно относится к терапевтическим комбинациям с применением по меньшей мере одного ингибитора RAF для лечения форм рака.
Настоящее изобретение также относится к применению ингибитора ERK (ERKi) для лечения рака, особенно рака, характеризующегося мутацией KRAS, и в частности мутацией KRAS с приобретением функции, в том числе рака легкого (особенно NSCLC), меланомы, рака поджелудочной железы и рака яичника.
Настоящее изобретение дополнительно относится к фармацевтической комбинации, которая содержит (а) по меньшей мере один ингибитор ERK (ERKi) и (b) ингибитор Raf, который предпочтительно представляет собой ингибитор c–RAF (CRAF), который также может ингибировать b–Raf, где два соединения получают и/или применяют для одновременного, раздельного или последовательного введения для лечения пролиферативного заболевания, и к фармацевтической композиции, содержащей такую комбинацию; способу лечения субъекта, у которого имеется пролиферативное заболевание, предусматривающему введение указанной комбинации нуждающемуся в этом субъекту; применению такой комбинации для лечения пролиферативного заболевания и коммерческой упаковке, содержащей такую комбинацию. В настоящем изобретении указанное пролиферативное заболевание часто представляет собой солидную опухоль, которая содержит изменения митоген–активируемой протеинкиназы (MAPK), как например KRAS–мутантные опухоли, и в частности KRAS–мутантный NSCLC (немелкоклеточный рак легкого), меланома, рак поджелудочной железы, колоректальный рак и рак яичника. Как правило, как ингибитор CRAF, так и ингибитор ERK представляют собой низкомолекулярные соединения, и в частности настоящее изобретение относится к комбинациям соединения A и соединения B для применения, как описано в данном документе.
В частности, предусмотрена фармацевтическая комбинация, содержащая соединение A или его фармацевтически приемлемую соль и соединение B или его фармацевтически приемлемую соль. Эта фармацевтическая комбинация может быть особенно применима при лечении KRAS– или BRAF–мутантного NSCLC, в том числе прогрессирующего или метастатического KRAS– или BRAF–мутантного NSCLC.
УРОВЕНЬ ТЕХНИКИ
Сигнальный путь RAS/RAF/MEK/ERK или MAPK является ключевым сигнальным каскадом, который управляет пролиферацией, дифференцировкой и выживаемостью клеток. Нарушение регуляции этого сигнального пути лежит в основе многих случаев онкогенеза. Нарушенная передача сигнала или недостаточная активация пути MAPK была продемонстрирована при многих типах опухолей, в том числе меланоме, раке легкого и поджелудочной железы, и может возникать посредством нескольких различных механизмов, в том числе активирующих мутаций в RAS и RAF. RAS представляет собой суперсемейство GTPаз и включает в себя KRAS (гомолог вирусного онкогена саркомы крысы Кирстена v–Ki–ras2), который представляет собой регулируемый сигнальный белок, который может быть включен (активирован) посредством различных одноточечных мутаций, которые известны как мутации с приобретением функции. Сигнальный путь MAPK часто мутирует при раке человека, при этом мутации KRAS и BRAF являются одними из наиболее частых (примерно 30%). Мутации RAS, в частности мутации с приобретением функции, были обнаружены в 9–30% всех форм рака, причем KRAS–мутации характеризуются наибольшей распространенностью (86%), за которыми следуют NRAS (11%) и в редких случаях HRAS (3%) (Cox AD, Fesik SW, Kimmelman AC, et al. (2014), Nat Rev Drug Discov. Nov; 13(11):828–51.). Хотя селективные ингибиторы BRAF (BRAFi) и, в меньшей степени, ингибиторы MEK (MEKi) продемонстрировали хорошую активность в BRAF–мутантных опухолях, в настоящее время не существует каких–либо эффективных видов терапии для KRAS–мутантных опухолей (Cantwell–Dorris ER, O'Leary JJ, Sheils OM (2011) Mol Cancer Ther. Mar;10(3):385–94.).
Новые данные о роли CRAF в опосредовании передачи сигналов KRAS и в развитии KRAS–мутантного немелкоклеточного рака легкого (NSCLC) делают его подходящей мишенью для терапевтического вмешательства (Blasco RB, Francoz S, Santamaría D, et al (2011) c–Raf, but not B–Raf, is essential for development of K–Ras oncogene–driven non–small cell lung carcinoma. Cancer Cell. 2011 May 17;19(5):652–63.). Было показано, что CRAF способствует реактивации сигнального пути, опосредованной обратной связью, после лечения с помощью MEKi при KRAS–мутантных формах рака (Lito P, Saborowski A, Yue J, et al. (2014) Disruption of CRAF–Mediated MEK Activation Is Required for Effective MEK Inhibition in KRAS Mutant Tumors. Cancer Cell 25, 697–710., Lamba et al 2014). Кроме того, CRAF играет важную роль в опосредовании парадоксальной активации после лечения с помощью BRAFi (Poulikakos PI, Zhang C, Bollag G, et al. (2010), Nature. Mar 18; 464(7287):427–30., Hatzivassiliou et al 2010, Heidorn et al 2010). Таким образом, селективные ингибиторы всех форм RAF, которые эффективно ингибируют активность CRAF и BRAF, могут быть эффективными в блокировании развития BRAF–мутантных опухолей и опосредованного RAS–мутациями онкогенеза, а также могут снижать активацию по типу обратной связи. Описанное в данном документе соединение А является эффективным ингибитором как CRAF, так и BRAF.
Рак легкого представляет собой распространенный тип рака, который поражает мужчин и женщин по всему миру. NSCLC является наиболее распространенным типом (ориентировочно 85%) рака легкого, причем у примерно 70% из этих пациентов на момент постановки диагноза присутствует поздняя стадия заболевания (стадия IIIB или стадия IV). Приблизительно 30% опухолей NSCLC содержат активирующие мутации KRAS, и эти мутации связаны с устойчивостью к ингибиторам тирозинкиназы EGFR (TKI) (Pao W, Wang TY, Riely GJ, et al. (2005) PLoS Med; 2(1): e17). Активирующие мутации KRAS также обнаружены при меланоме (British J. Cancer 112, 217–26 (2015)), раке поджелудочной железы (Gastroenterology vol. 144(6), 1220–29 (2013)) и раке яичника (British J. Cancer 99 (12), 2020–28 (2008)). Мутации BRAF наблюдались в не более чем 3% случаев NSCLC и также были описаны как механизм устойчивости при положительном по мутации EGFR NSCLC.
Непосредственное ингибирование KRAS оказалось труднодостижимым. На сегодняшний день нет утвержденных способов направленной терапии для пациентов с KRAS–мутантными формами рака, как например NSCLC. Таким образом, в медицине существует высокая нереализованная потребность для пациентов, страдающих KRAS–мутантным NSCLC, и для пациентов, страдающих BRAF–мутантным NSCLC. Существует необходимость в направленной терапии, которая была бы безопасной и/или хорошо переносимой. Терапия, которая приводит к продолжительным и устойчивым ответным реакциям в клинических условиях, также была бы полезной.
КРАТКОЕ ОПИСАНИЕ
Настоящее изобретение также предусматривает фармацевтическую комбинацию, которая содержит (a) ингибитор CRAF, который представляет собой соединение А,
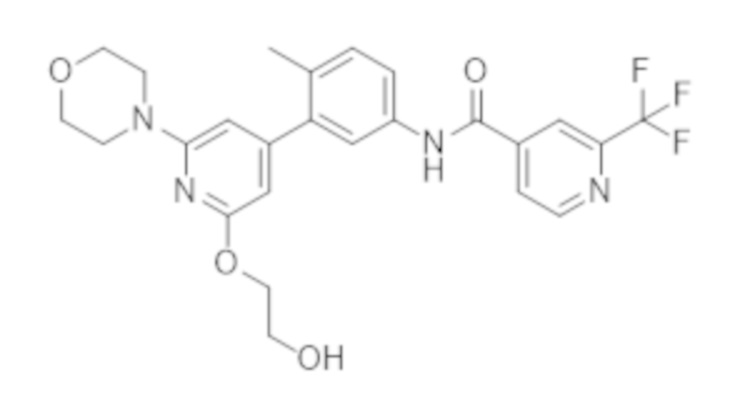
или его фармацевтически приемлемую соль, и
(b) ингибитор ERK, который представляет собой соединение B,
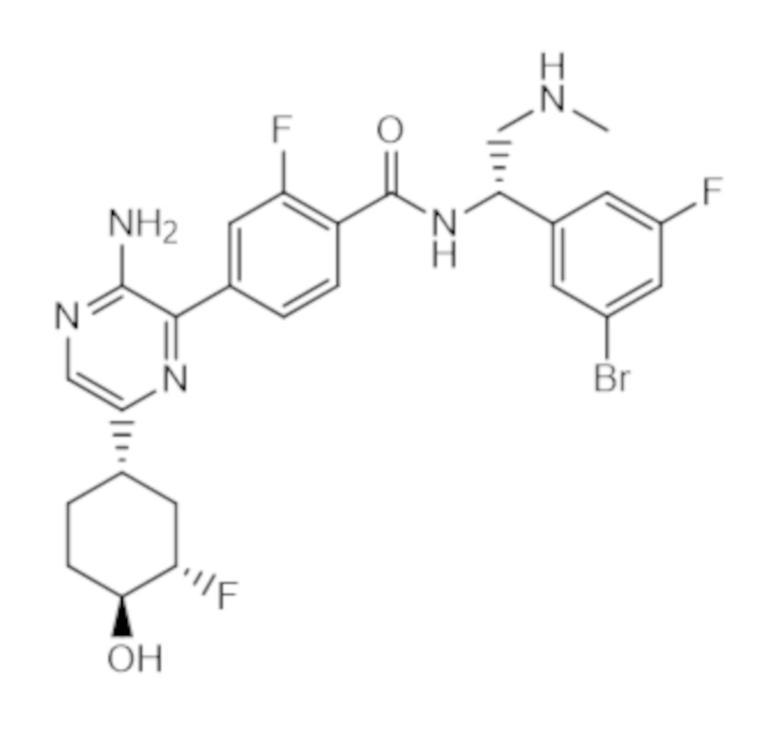
или его фармацевтически приемлемую соль. Эта комбинация названа в данном документе как "комбинация по настоящему изобретению".
Настоящее изобретение дополнительно предусматривает фармацевтическую комбинацию, содержащую ингибитор киназы c–Raf, который представляет собой соединение A или его фармацевтически приемлемую соль, и ингибитор ERK, который представляет собой соединение B или его фармацевтически приемлемую соль, как описано в данном документе, для одновременного, раздельного или последовательного (в любом порядке) введения для применения в лечении пролиферативного заболевания. В частности, настоящее изобретение относится к комбинации по настоящему изобретению для применения в лечении пролиферативного заболевания, характеризующегося активирующими мутациями в сигнальном пути MAPK и, в частности, одной или несколькими мутациями в KRAS или BRAF. В частности, настоящее изобретение относится к лечению KRAS–мутантного NSCLC (немелкоклеточного рака легкого), BRAF–мутантного NSCLC, KRAS–мутантного рака поджелудочной железы, KRAS–мутантного колоректального рака (CRC) и KRAS–мутантного рака яичника.
Соединение А представляет собой аденозинтрифосфат–конкурентный (ATP–конкурентный) ингибитор протеинкиназ BRAF (также называемой в данном документе b–RAF или b–Raf) и CRAF (также называемой в данном документе c–RAF или c–Raf). По всему тексту настоящего описания соединение A также упоминается как ингибитор c–RAF (или CRAF) или ингибитор киназы C–RAF/c–Raf.
Соединение A представляет собой N–(3–(2–(2–гидроксиэтокси)–6–морфолинoпиридин–4–ил)–4–метилфенил)–2–(трифторметил)изоникотинамид и представляет собой соединение следующей структуры:
 соединение A.
соединение A.
В клеточных анализах соединение A продемонстрировало антипролиферативную активность в клеточных линиях, которые содержат множество мутаций, активирующих передачу сигнала посредством MAPK. In vivo обработка соединением А приводила к регрессии опухоли в нескольких KRAS–мутантных моделях, в том числе полученных из NSCLC Calu–6 (KRAS Q61K) и NCI–H358 (KRAS G12C). В совокупности, подавление сигнального пути MAPK in vitro и in vivo и антипролиферативная активность, наблюдаемые для соединения A в хорошо переносимых дозах, дают основания предполагать, что соединение A может характеризоваться противоопухолевой активностью у пациентов с опухолями, содержащими активирующие нарушения в сигнальном пути MAPK. Кроме того, соединение A является ATP–конкурентным ингибитором типа 2 как B–Raf, так и C–Raf, который удерживает карман киназы в неактивной конформации, уменьшая тем самым парадоксальную активацию, наблюдаемую при использовании многих ингибиторов B–Raf, и блокируя управляемые мутантным RAS передачу сигналов и пролиферацию клеток. Соединение А продемонстрировало эффективность в многочисленных человеческих линиях клеток MAPK–опосредованного рака и в отношении ксенотрансплантатных опухолей, представляющих собой модельные опухоли, несущие нарушения в онкогенах KRAS, NRAS и BRAF человека.
Соединение B является ингибитором регулируемых внеклеточными сигналами киназ 1 и 2 (ERK 1/2). Соединение B известно под названием 4–(3–амино–6–((1S,3S,4S)–3–фтор–4–гидроксициклогексил)пиразин–2–ил)–N–((S)–1–(3–бром–5–фторфенил)–2–(метиламино)этил)–2–фторбензамид и представляет собой соединение следующей структуры:
соединение B.
Было показано, что соединение B является активным в качестве средства для монотерапии в моделях различных солидных опухолей и было особенно эффективным при использовании в сочетании со вторым противораковым терапевтическим средством. Например, в моделях аденокарциномы протоков поджелудочной железы (PDAC), особенно трудно поддающейся лечению формы рака (5–летняя выживаемость для PDAC составляет 7% в соответствии с Cancers (Basel), vol. 8(4), 45 (April 2016)), соединение B в сочетании с несколькими различными противоопухолевыми средствами демонстрировало значительное уменьшение размеров опухоли и было более эффективным, чем ожидалось исходя из активности отдельных средств, являющихся компонентами определенных комбинаций. В частности, комбинации соединения B с соединением A продемонстрировали повышенную интенсивность и продолжительность ответной реакции опухоли по сравнению со средством для монотерапии на ксенотрансплантатной модели NSCLC человека Calu–6.
Таким образом, ожидается, что вертикальное ингибирование MAPK (митоген–активируемой протеинкиназы) при объединении ингибитора всех форм RAF, такого как соединение A, с ингибитором киназы ERK1/2, такого как соединение B, будет оптимизировать подавление передачи сигналов посредством MAPK в KRAS– и BRAF–мутантном NSCLC. Эта комбинация может также способствовать предотвращению возникновения устойчивости к комбинации ингибиторов BRAF и MEK (митоген–активируемой протеинкиназы) в BRAFV600E–мутантном NSCLC.
Соответственно, настоящее изобретение предусматривает композиции и способы, в которых применяется соединение B в комбинации с ингибитором RAF, и в частности с соединением A, для лечения солидных опухолей, в частности опухолей, которые экспрессируют мутации KRAS, в том числе NSCLC, и особенно KRAS–мутантный NSCLC, а также BRAF–мутантный NSCL, в том числе BRAFV600E–мутантный NSCLC.
В данном документе раскрыты фармацевтические комбинации, которые содержат (a) ингибитор c–RAF, такой как соединение A или его фармацевтически приемлемая соль, и (b) ингибитор ERK, такой как соединение B или его фармацевтически приемлемая соль, для одновременного, раздельного или последовательного введения для лечения пролиферативного заболевания, в частности солидной опухоли, которая содержит изменения митоген–активируемой протеинкиназы (MAPK), как например KRAS–мутантные опухоли и некоторые BRAF–мутантные опухоли. Таковые включают KRAS–мутантный NSCLC (немелкоклеточный рак легкого), BRAF–мутантный NSCLC (немелкоклеточный рак легкого), KRAS–мутантный и BRAF–мутантный NSCLC (немелкоклеточный рак легкого), KRAS–мутантный рак поджелудочной железы, KRAS–мутантный колоректальный рак (CRC) и KRAS–мутантный рак яичника. Настоящее изобретение также предусматривает фармацевтическую комбинацию по настоящему изобретению для применения в лечении BRAF V600–мутантной меланомы, в том числе рецидивирующей или рефракторной BRAF V600–мутантной меланомы. Также раскрыты фармацевтическая композиция, содержащая такую комбинацию; способ лечения субъекта, у которого имеется пролиферативное заболевание, предусматривающий введение такой комбинации нуждающемуся в этом субъекту; применение такой комбинации для лечения пролиферативного заболевания; и коммерческая упаковка, содержащая такую комбинацию. Другие признаки, цели и преимущества настоящего изобретения будут очевидны из описания и графических материалов, а также из формулы изобретения.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
На фигуре 1A изображена эффективность соединения A и соединения B, применяемых по отдельности и совместно, в ксенотрансплантатных моделях опухоли Calu–6 NSCLC у мышей. Как указано, соединения вводили перорально либо один раз в сутки (qd), либо один раз в двое суток (q2d).
На фигуре 1B изображена продолжительность ответной реакции на обработки, показанные на фигуре 1А, демонстрируя тем самым, что комбинированные обработки превосходили любую обработку одним средством.
На фигуре 1C изображена переносимость обработок, приведенных на фигуре 1A, о чем свидетельствует изменение веса тела с течением времени.
ПОДРОБНОЕ ОПИСАНИЕ
Фармацевтическая комбинация по настоящему изобретению представляет собой фармацевтическую комбинацию соединения A или его фармацевтически приемлемой соли и соединения B или его фармацевтически приемлемой соли. В предпочтительном варианте осуществления предусмотрена фармацевтическая комбинация по настоящему изобретению для применения в лечении пролиферативного заболевания, выбранного из KRAS–мутантного NSCLC (немелкоклеточного рака легкого), BRAF–мутантного NSCLC (в том числе BRAFV600E–мутантного NSCLC), KRAS–мутантного рака поджелудочной железы, KRAS–мутантного колоректального рака (CRC) и KRAS–мутантного рака яичника. Пациенты, которым фармацевтическая комбинации с большей вероятностью принесет пользу, включают пациентов с прогрессирующим или метастатическим заболеванием, например пациенты, страдающие NSCLC, с диагнозом прогрессирующий или метастатический KRAS– или BRAF–мутантный NSCLC, у которых он мог прогрессировать после получения стандартного лечения.
Ингибитор киназы CRAF
Ингибиторы киназы CRAF по настоящему изобретению включают соединение A. Соединение А раскрыто в качестве примера 1156 в WO2014/151616. В WO2014/151616 также описывается его получение и фармацевтические композиции, содержащие соединение A.
В предпочтительном варианте осуществления способов, видов лечения, комбинации и композиций, описанных в данном документе, ингибитор CRAF представляет собой соединение А или его фармацевтически приемлемую соль.
Соединение А характеризуется следующей структурой:
Соединение А (соединение А) также известно под названием N–(3–(2–(2–гидроксиэтокси)–6–морфолинопиридин–4–ил)–4–метилфенил)–2–(трифторметил)изоникотинамид.
В клеточных анализах соединение A продемонстрировало антипролиферативную активность в клеточных линиях, которые содержат множество мутаций, активирующих передачу сигнала посредством MAPK. In vivo обработка соединением А приводила к регрессии опухоли в нескольких KRAS–мутантных моделях, включая полученные из NSCLC Calu–6 (KRAS Q61K) и NCI–H358 (KRAS G12C). В совокупности, подавление сигнального пути MAPK in vitro и in vivo и антипролиферативная активность, наблюдаемые для соединения A в хорошо переносимых дозах, дают основания предполагать, что соединение A может характеризоваться противоопухолевой активностью у пациентов с опухолями, содержащими активирующие нарушения в сигнальном пути MAPK. Кроме того, соединение A является ATP–конкурентным ингибитором типа 2 как B–Raf, так и C–Raf, который удерживает карман киназы в неактивной конформации, уменьшая тем самым парадоксальную активацию, наблюдаемую при использовании многих ингибиторов B–Raf, и блокируя управляемые мутантным RAS передачу сигналов и пролиферацию клеток. Соединение А продемонстрировало эффективность в многочисленных человеческих линиях клеток MAPK–опосредованного рака и в отношении ксенотрансплантатных опухолей, представляющих собой модельные опухоли, несущие нарушения в онкогенах KRAS, NRAS и BRAF человека.
Основываясь на механизме действия соединения A, доклинических данных и опубликованной литературе в отношении важности CRAF в регуляции сигнального пути MAPK, соединение A в сочетании с по меньшей мере одним другим ингибитором сигнального пути MAPK, таким как соединение B, может быть применимо в лечении пациентов с солидными опухолями на поздней стадии, у которых имеются изменения пути MAPK. Соединение А может применяться для лечения (например, одного или нескольких из уменьшения, ингибирования или задержки прогрессирования) пролиферативного заболевания, которое представляет собой солидную опухоль на поздней стадии, которая содержит изменения митоген–активируемой протеинкиназы (MAPK), как например KRAS–мутантные опухоли, и в частности опухоли, экспрессирующие по меньшей мере одну мутацию с приобретением функции RAS или RAF, в том числе рак легкого, NSCLC (немелкоклеточный рак легкого), рак яичника, рак поджелудочной железы, колоректальный рак или меланома, у субъекта, нуждающегося в таком лечении.
Ингибиторы ERK
Соединение ERKi, применяемое согласно настоящему изобретению в данном документе, как правило, представляет собой соединение B либо в свободной форме, либо в виде фармацевтически приемлемой соли.
Соединение B является ингибитором регулируемых внеклеточными сигналами киназ 1 и 2 (ERK 1/2). Это соединение раскрыто, а его получение и фармацевтические композиции, содержащие это соединение, описаны в опубликованной заявке согласно PCT на патент WO 2015/066188. Соединение B характеризуется следующей структурой:
соединение B.
В некоторых вариантах осуществления применяют хлористоводородную соль соединения B.
Варианты терапевтического применения
В одном варианте осуществления настоящее изобретение относится к способу лечения (например, ингибированию, уменьшению, снижению тяжести или предупреждению) нарушения, например гиперпролиферативного состояния или нарушения (например, рака) у субъекта. Способ включает введение субъекту ингибитора ERK в комбинации с ингибитором c–RAF; в некоторых вариантах осуществления ингибитор c–RAF представляет собой соединение A, а ингибитор ERK представляет собой соединение B. В данном документе описаны подходящие дозировки и схемы введения для применения этих соединений в таких способах.
В некоторых вариантах осуществления пролиферативное нарушение представляет собой KRAS–мутантную опухоль, такую как опухоль, экспрессирующую по меньшей мере одну KRAS–мутацию с приобретением функции, как описано в данном документе, и в частности KRAS–мутантные формы рака, как например NSCLC (немелкоклеточный рак легкого). Включены опухоли, содержащие мутации BRAF, в том числе V600E и другие, например NSCLC, характеризующийся по меньшей мере одной мутацией V600E или другой мутацией BRAF, независимо от того, является ли она типичной или атипичной. Ингибитор CRAF для применения в способах, видах лечения и комбинациях, раскрытых в данном документе, является эффективным ингибитором по меньшей мере CRAF и необязательно также BRAF. В некоторых вариантах осуществления ингибитор CRAF или его фармацевтически приемлемую соль вводят перорально. В некоторых вариантах осуществления ингибитор CRAF представляет собой соединение A или его фармацевтически приемлемую соль.
В случае если указанное количество дозы в данном документе описывают с использованием ‘приблизительно', фактическая доза может варьировать не более чем на 10%, например 5% от указанного количества, причем такое использование выражения ‘приблизительно' означает, что точное количество в данной лекарственной форме может незначительно отличаться от предполагаемого количества по разным причинам, что не оказывает существенного влияния на эффект, оказываемый вводимым соединением in vivo. Специалист поймет, что если в данном документе указана доза или дозирование терапевтического соединения, то это количество относится к количеству терапевтического соединения в его свободной форме.
Единичную дозу ингибитора CRAF можно вводить один раз в сутки, или два раза в сутки, или три раза в сутки, или четыре раза в сутки, при этом фактическая доза и время введения определяются по таким критериям, как возраст, вес и пол пациента; степень и тяжесть рака, подлежащего лечению; и решение лечащего врача.
В одном варианте осуществления соединение А получают для перорального введения и вводят перорально в дозе, составляющей 100 мг, 150 мг, 200 мг, 250 мг, 300 мг или 400 мг, доставляемой не более четырех раз в сутки, причем прогнозируется, что доза 100 мг один или два раза в сутки обеспечивает концентрацию в плазме крови у людей, которая может быть эффективной у людей, исходя из аллометрического масштабирования соответствующих уровней содержания в плазме у животных, и может вводиться доза, составляющая 200 мг не более четырех раз в сутки, для достижения большей эффективности, обеспечивая в то же время удовлетворительный терапевтический индекс. В некоторых вариантах осуществления соединение А вводят один раз в сутки в дозе, составляющей 100 мг, 200 мг, 250 мг, 300 мг или 400 мг. Аллометрическое масштабирование доклинических моделей указывает на то, что суточная доза соединения А, составляющая 300 мг или выше, которая может вводиться один раз в сутки или в виде двух, трех или четырех отдельных доз в течение суток в качестве монотерапии, должна обеспечивать терапевтический эффект по многим показаниям, включая солидные опухоли, которые содержат или экспрессируют мутации KRAS.
Ожидается, что в комбинациях по настоящему изобретению терапевтические дозы соединения А у этих субъектов будут ниже, поэтому при комбинированном лечении для таких субъектов обычно используют суточные дозы соединения А, составляющее 100 мг, 200 мг, 250 мг или 300 мг. Соответствующим образом, в комбинациях и способах по настоящему изобретению дозу, составляющую 100 мг, или 200 мг, или 250 мг, или 300 мг соединения А, вводят один раз в сутки.
В одном варианте осуществления соединение B получают для введения посредством пероральной доставки и может применяться в виде его гидрохлоридной соли. В некоторых вариантах осуществления соединение или его HCl соль просто инкапсулированы в фармацевтически приемлемый контейнер, такой как твердая или мягкая желатиновая капсула для перорального введения. Желатиновые капсулы можно получать с различными размерами доз для обеспечения адаптируемого введения; например, можно получать желатиновые капсулы, содержащие приблизительно 5 мг, приблизительно 20 мг, приблизительно 50 мг или приблизительно 100 мг соединения B или его HCl соли.
В комбинациях по настоящему изобретению и в вариантах терапевтического применения, описанных в данном документе, соединение А или его фармацевтически приемлемую соль можно вводить в суточной дозе, составляющей приблизительно 100 мг, или приблизительно 150 мг, или приблизительно 200 мг, или приблизительно 250 мг, в комбинации с соединением B или его фармацевтически приемлемой солью, которое можно вводить в суточной дозе, составляющей приблизительно 50 мг, или приблизительно 75 мг, или приблизительно 100 мг, или приблизительно 150 мг, или приблизительно 200 мг. Например, соединение А или его фармацевтически приемлемую соль можно вводить в общей дозе, составляющей приблизительно 100 мг соединения А один раз в сутки, и соединение В или его фармацевтически приемлемую соль в общей дозе, составляющей приблизительно 100 мг один раз в сутки, причем дозы предпочтительно назначают один раз в сутки. Пациенты, нуждающиеся в этом, могут также получать общую дозу, составляющую приблизительно 200 мг соединения A или его фармацевтически приемлемой соли один раз в сутки, и общую дозу, составляющую приблизительно 100 мг соединения B или его фармацевтически приемлемой соли один раз в сутки, причем дозы предпочтительно вводят один раз в сутки.
Соединение A и соединение B можно применять совместно в соответствии со способами, раскрытыми в данном документе. Два соединения можно вводить совместно или по отдельности в любом порядке, в зависимости от предполагаемой величины дозы и частоты введения, поскольку предполагается, что способы лечения по настоящему изобретению можно осуществлять в течение 2 дней, 3 дней, 4 дней, 5 дней, 6 дней, 1 недели, 2 недель, 3 недель, 4 недель или более 4 недель согласно тому, что лечащий врач будет считать целесообразным, и дополнительно руководствуясь описанными в данном документе способами для определения подходящих дозы и частоты введения. В способах и вариантах применения, раскрытых в данном документе, соединение A и/или соединение B можно вводить один раз в сутки в течение по меньшей мере пяти последовательных дней.
В другом аспекте настоящее изобретение предусматривает способ уменьшения активности (например, роста, выживаемости или жизнеспособности или всего из перечисленного) гиперпролиферативной (например, раковой) клетки. В другом аспекте настоящее изобретение предусматривает способы и композиции с применением соединения B для лечения солидных опухолей, вводят или получают для введения по отдельности, одновременно или последовательно с ингибитором CRAF. Оно также предусматривает ингибитор CRAF для применения в лечении солидной опухоли, экспрессирующей мутацию с приобретением функции в сигнальном пути MAPK, такой как KRAS–мутантный NSCLC (немелкоклеточный рак легкого), BRAF–мутантный NSCLC (немелкоклеточный рак легкого), KRAS–мутантный и BRAF–мутантный NSCLC (немелкоклеточный рак легкого), KRAS–мутантный рак поджелудочной железы, KRAS–мутантный колоректальный рак (CRC) и KRAS–мутантный рак яичника и BRAF V600–мутантная меланома, где ингибитор CRAF вводят или получают для введения по отдельности, одновременно или последовательно с ингибитором ERK, таким как соединение B. Как правило, соединение B вводят перорально и вводят по отдельности, одновременно или последовательно с ингибитором CRAF, который также часто вводят перорально. Подходящие способы, пути введения, дозы и частота введения соединения А и соединения В для применения в этих способах и композициях описаны в данном документе.
В другом аспекте настоящее изобретение предусматривает ингибитор ERK 1/2 для применения в лечении KRAS–мутантного NSCLC (немелкоклеточного рака легкого) и для применения в лечении BRAF–мутантного NSCLC, где ингибитор ERK 1/2 вводят или получают для введения по отдельности, одновременно или последовательно с ингибитором CRAF. Оно также предусматривает ингибитор CRAF для применения в лечении KRAS–мутантного NSCLC (немелкоклеточного рака легкого) и для применения в лечении BRAF–мутантного NSCLC, где ингибитор CRAF вводят или получают для введения по отдельности, одновременно или последовательно с ингибитором ERK 1/2. Как правило, ингибитор ERK 1/2 вводят перорально, и его можно вводить по отдельности или последовательно с ингибитором CRAF, который также вводят перорально. Подходящие способы, пути, дозы и частота введения ингибитора CRAF и ингибитора ERK 1/2 описаны в данном документе. В некоторых вариантах осуществления ингибитор CRAF представляет собой соединение A; в некоторых вариантах осуществления ингибитор ERK 1/2 представляет собой соединение B.
Раскрытые в данном документе комбинации можно вводить совместно в одной композиции или вводить по отдельности в двух или более различных композициях, например в композициях или лекарственных формах, описанных в данном документе. Фармацевтические комбинации, описанные в данном документе, в частности фармацевтическая комбинация по настоящему изобретению, могут представлять собой продукт на основе свободной комбинации, т. е. комбинацию из двух или более активных ингредиентов, например соединения А и соединения B, которые вводят одновременно, по отдельности или последовательно в виде двух или более отдельных лекарственных форм. Введение терапевтических средств можно осуществлять в любом порядке. Первое средство и дополнительные средства (например, второе, третье средства) можно вводить посредством одного и того же пути введения или посредством разных путей введения.
В другом аспекте настоящее изобретение относится к композиции, содержащей соединение A и соединение B, необязательно также содержащее по меньшей мере одно, и необязательно более чем одно, фармацевтически приемлемое вспомогательное вещество или носитель. Такую композицию применяют для лечения солидной опухоли, как правило солидной опухоли, экспрессирующей мутацию KRAS или мутацию RAF, часто для лечения NSCLC, и в частности для лечения пациента, у которого имеется NSCLC, который характеризуется по меньшей мере одной мутацией KRAS, особенно мутацией с приобретением функции, такой как те, что описаны в данном документе.
Немелкоклеточный рак легкого (NSCLC)
Рак легкого представляет собой распространенный тип рака, который поражает мужчин и женщин по всему миру. Немелкоклеточная карцинома (NSCLC) является наиболее распространенным типом (ориентировочно 85%) рака легкого, причем у примерно 70% таких пациентов на момент постановки диагноза имеется поздняя стадия заболевания (стадия IIIB или стадия IV). В то время как рак легкого обычно связан с курением, некурящие также подвержены раку легкого, в частности NSCLC, причем постановка диагноза у некурящих часто затягивается из–за высокой ассоциации с курением, тем не менее 10–15% пациентов с раком легкого никогда не курили. В приблизительно 30% случаев NSCLC содержит активирующие мутации KRAS, и эти мутации связаны с устойчивостью к TKI EGFR (Pao W, Wang TY, Riely GJ et al (2005) PLoS Med; 2 (1): e17).
Способы иммунотерапии, которые разрабатываются в настоящее время, начали приносить значительную пользу пациентам с раком легкого, в том числе тем, для кого обычные методы лечения являются неэффективными. Недавно пембролизумаб (Keytruda®) и ниволумаб (Opdivo®), два ингибитора взаимодействия PD–1/PD–L1, были одобрены для применения при NSCLC. Однако результаты показывают, что многие пациенты, получавшие ингибиторы PD–1 в виде отдельного средства, не получают достаточного положительного эффекта от лечения.
Прямое ингибирование KRAS оказалось труднодостижимым, и KRAS–мутантный NSCLC остается неуловимой мишенью для терапии рака. На сегодняшний день нет утвержденных способов направленной терапии для пациентов с KRAS– или BRAF–мутантным NSCLC.
Мутации BRAF наблюдались в не более чем 3% случаев NSCLC и также были описаны как механизм устойчивости при положительном по мутации EGFR NSCLC (Paik PK, Arcila ME, Fara M, et al (2011). J Clin Oncol. May 20; 29(15):2046–51).
Рак яичника
Рак яичника является наиболее смертельной гинекологической формой рака и представляет собой гетерогенное заболевание, состоящее из совокупности различных гистологических и молекулярных подтипов с варьирующим прогнозом. Эпителиальный подтип составляет 90% случаев рака яичника.
Наиболее распространенным гистологическим подтипом эпителиального рака яичника является серозная карцинома, охватывающая от 60 до 70% случаев эпителиального рака яичника. Двухуровневая система классификации разделяет серозную карциному на серозную карциному низкой степени злокачественности (LGS) и серозную карциному высокой степени злокачественности (HGS), которые характеризуются разными молекулярными характеристиками, иммуногистохимическим профилем, эпидемиологическими особенностями и клиническим поведением. LGS–карцинома составляет до 10% случаев серозного эпителиального рака яичника, и карциномы яичника с KRAS– (до 40%) или BRAF–мутациями (2–6%) представляют собой преимущественно LGS–карциномы. LGS–карцинома является устойчивой к химическим воздействиям не только к средствам первой линии, но также и при лечении рецидивирующего заболевания.
Рак поджелудочной железы
Используемый в данном документе термин "рак поджелудочной железы" включает аденокарциному протоков поджелудочной железы (PDAC). PDAC является наиболее распространенным типом рака поджелудочной железы.
KRAS–мутантный рак и BRAF–мутантный NSCLC
Настоящее изобретение предусматривает фармацевтическую комбинацию, содержащую (а) ингибитор CRAF, который представляет собой соединение А или его фармацевтически приемлемую соль, и (b) ингибитор ERK, который представляет собой соединение В или его фармацевтически приемлемую соль, для применения в лечении KRAS–мутантного NSCLC.
В другом аспекте настоящее изобретение предусматривает фармацевтическую комбинацию, содержащую (а) ингибитор CRAF, который представляет собой соединение А или его фармацевтически приемлемую соль, и (b) ингибитор ERK, который представляет собой соединение В или его фармацевтически приемлемую соль, для применения в лечении KRAS–мутантного колоректального рака (CRC).
В другом аспекте настоящее изобретение предусматривает фармацевтическую комбинацию, содержащую (а) ингибитор CRAF, который представляет собой соединение А или его фармацевтически приемлемую соль, и (b) ингибитор ERK, который представляет собой соединение В или его фармацевтически приемлемую соль, для применения в лечении KRAS–мутантного рака яичника.
В другом аспекте настоящее изобретение предусматривает фармацевтическую комбинацию, содержащую (а) ингибитор CRAF, который представляет собой соединение А или его фармацевтически приемлемую соль, и (b) ингибитор ERK, который представляет собой соединение В или его фармацевтически приемлемую соль, для применения в лечении KRAS–мутантного рака поджелудочной железы.
В другом аспекте настоящее изобретение предусматривает фармацевтическую комбинацию, содержащую (а) ингибитор CRAF, который представляет собой соединение А или его фармацевтически приемлемую соль, и (b) ингибитор ERK, который представляет собой соединение В или его фармацевтически приемлемую соль, для применения в лечении BRAF–мутантного NSCLC.
Термин "BRAF–мутантная" опухоль или рак включает любую опухоль которая характеризуется мутировавшим белком BRAF. Примеры мутаций B–Raf включают без ограничения V600E и V600K. Большинство мутаций B–Raf сконцентрированы в двух областях: богатой глицином P–петле N–доли, а также сегменте активации и фланкирующих областях. Мутация V600E была выявлена при многих формах рака и обусловлена заменой тимина аденином в нуклеотиде 1799. Это приводит к тому, что валин (V) замещается глутаматом (E) в кодоне 600 (теперь называемом V600E).
Термин "KRAS–мутантная" опухоль или рак включает любую опухоль, которая характеризуется мутировавшим белком KRAS, в частности KRAS–мутацией с приобретением функции; в особенности, любой KRAS–мутант G12X, G13X, Q61X или A146X, где X представляет собой любую аминокислоту, отличную от той, которая естественно встречается в этом положении. Например, мутация G12V означает, что глицин замещен валином в кодоне 12. Примеры мутаций KRAS в опухолях включают Q61K, G12V, G12C и A146T. Таким образом, KRAS–мутантные NSCLC, CRC, рак яичника и рак поджелудочной железы включают без ограничения Q61K–, G12V–, G12C– и A146T–мутантный NSCLC, Q61K–, G12V–, G12C– и A146T–мутантный CRC, рак яичника или рак поджелудочной железы. Например, формы рака, подлежащие лечению с помощью комбинированной терапии, раскрытой в данном документе, включают рак легкого KRASQ61K, рак яичника KRASG12D, рак поджелудочной железы KRASG12D и рак поджелудочной железы KRASG12R.
Термин "BRAF–мутантная" опухоль или рак включает любую опухоль, которая характеризуется мутировавшим белком BRAF. Примеры мутаций B–Raf включают без ограничения V600E и V600K. Большинство мутаций B–Raf сконцентрированы в двух областях: богатой глицином P–петле N–доли, а также сегменте активации и фланкирующих областях. Мутация V600E была выявлена при многих формах рака и обусловлена заменой тимина аденином в нуклеотиде 1799. Это приводит к тому, что валин (V) замещается глутаматом (E) в кодоне 600 (теперь называемом V600E).
Формы рака, которые лечат с помощью фармацевтических комбинаций, описанных в данном документе, могут находиться на ранней, промежуточной или поздней стадиях.
Пути применения способов комбинированной терапии
В одном аспекте предусмотрен способ лечения (например, одно или несколько из уменьшения, ингибирования или замедления прогрессирования) пролиферативного заболевания, которое представляет собой солидную опухоль на поздней стадии, которая содержит одно или несколько изменений митоген–активируемой протеинкиназы (MAPK), как например KRAS–мутантная опухоль, и в частности KRAS–мутантный NSCLC (немелкоклеточный рак легкого), у субъекта. Способ предусматривает введение субъекту комбинации, раскрытой в данном документе (например, комбинации, содержащей терапевтически эффективное количество ингибитора ERK 1/2 и терапевтически эффективное количество соединения А или его фармацевтически приемлемой соли).
Описанные в данном документе комбинации можно вводить субъекту системно (например, перорально, парентерально, подкожно, внутривенно, ректально, внутримышечно, внутрибрюшинно, интраназально, трансдермально или путем ингаляции или внутриполостной установки), местно или путем нанесения на слизистые оболочки, как например носа, горла и бронхов. В некоторых вариантах осуществления ингибитор ERK 1/2 для применения в таких комбинациях и способах вводят перорально. В некоторых вариантах осуществления ингибитор CRAF для применения в комбинациях и способах по настоящему изобретению вводят перорально. При применении ингибитора ERK 1/2 и ингибитора CRAF в комбинации оба можно вводить перорально и можно вводить совместно (одновременно) или по отдельности в любом порядке в соответствии со схемами дозирования, определяемыми лечащим врачом, причем подходящие дозы и схемы дозирования раскрыты в данном документе.
Дополнительные варианты комбинированной терапии
В некоторых вариантах осуществления способы и композиции, описанные в данном документе, применяют в комбинации с одним или несколькими другими средствами для терапии рака, такими как молекулы антител, химиотерапия, другие средства противораковой терапии (например, средства направленной терапии, генная терапия, вирусная терапия, РНК–терапия c трансплантацией костного мозга, нанотерапия или онколитические лекарственные средства), цитотоксические средства, средства иммунотерапии (например, цитокины, иммуностимуляторы или средства иммунотерапии на основе клеток), хирургические процедуры (например, лампэктомия или мастэктомия) или лучевые процедуры или комбинация любого из вышеперечисленного. Дополнительная терапия может быть в форме адъювантной или неоадъювантной терапии. В некоторых вариантах осуществления дополнительная терапия представляет собой ферментный ингибитор (например, низкомолекулярный ферментный ингибитор) или метастатический ингибитор. Иллюстративные цитотоксические средства, которые можно вводить в сочетании с комбинацией по настоящему изобретению, включают антимикротубулиновые средства, ингибиторы топоизомераз, антиметаболиты, митотические ингибиторы, алкилирующие средства, антрациклины, алкалоиды барвинка, интеркалирующие средства, средства, способные нарушать путь передачи сигнала, средства, которые стимулируют апоптоз, ингибиторы протеосом и радиацию (например, местное облучение или общее облучение тела (например, гамма–облучение). В других вариантах осуществления дополнительная терапия представляет собой хирургическое вмешательство, или лучевую терапию, или их комбинацию. В других вариантах осуществления дополнительная терапия представляет собой терапию, направленную на одно или несколько из сигнального пути PI3K/AKT/mTOR, ингибитора HSP90 или ингибитора тубулина.
В качестве альтернативы или в сочетании с вышеупомянутыми комбинациями способы и композиции, описанные в данном документе, можно вводить в сочетании с одним или несколькими из: иммуномодулятора (например, активатора костимулирующей молекулы или ингибитора ингибирующей молекулы, например молекулы иммунной контрольной точки); вакцины, например терапевтической противораковой вакцины; или других форм клеточной иммунотерапии.
Любую комбинацию и последовательность других терапевтических средств, процедур или приемов (например, как описано в данном документе) можно использовать в сочетании со способами лечения по настоящему изобретению. Композиции и комбинации по настоящему изобретению можно вводить перед другими способами лечения, одновременно с другими способами лечения, между циклами таких других способов лечения или во время ремиссии нарушения.
В данном документе раскрыты способы, комбинации и композиции, предусматривающие ингибитор ERK и/или ингибитор C–Raf для применения при лечении форм рака, особенно солидных опухолей, экспрессирующих по меньшей мере одну мутацию с приобретением функции в сигнальном пути MAPK.
Выбранные термины определены ниже и по всему тексту данной заявке.
Используемые в данном документе формы единственного числа относятся к одному или более чем одному (например, по меньшей мере к одному) грамматическому объекту формы.
Термин "или" используется в данном документе для обозначения и используется взаимозаменяемо с термином "и/или", если контекст явно не указывает на иное.
Термины "приблизительно" и "примерно" будут обычно означать приемлемую степень погрешности для измеряемого количества с учетом природы или точности измерений. Иллюстративные степени погрешности находятся в пределах 20 процентов (%), как правило в пределах 10%, и в большинстве случаев в пределах 5% от заданного значения или диапазона значений. В частности, когда доза упоминается как ‘приблизительное' определенное значение, то предполагается, что она включает диапазон около указанного значения, составляющий плюс или минус 10%. Как принято в данной области техники, дозы относятся к количеству терапевтического средства в его свободной форме. Например, в случае если упоминается доза, составляющая 100 мг соединения B, и соединение B применяется в качестве его гидрохлоридной соли, количество применяемого терапевтического средства эквивалентно 100 мг свободной формы соединения B.
Термины "комбинация" или "в комбинации с" не предназначены для обозначения того, что терапия или терапевтические средства должны быть физически смешаны или введены в одно и то же время и/или составлены для доставки вместе, хотя эти способы доставки находятся в пределах объема, описанного в данном документе. Терапевтическое средство в этих комбинациях можно вводить одновременно с одним или несколькими дополнительными методами терапии или терапевтическими средствами, до или после них. Терапевтические средства или терапевтический протокол можно применять в любом порядке. Как правило, каждое средство будет вводиться в дозе и/или по временной схеме, определенным для этого средства. Кроме того, следует также принять во внимание, что дополнительное терапевтическое средство, применяемое в этой комбинации, можно вводить совместно в одной композиции или вводить по отдельности в разных композициях. В общем, ожидается, что дополнительные терапевтические средства, применяемые в комбинации, будут применяться при уровнях, которые не превышают уровни, при которых они применяются по отдельности. В некоторых вариантах осуществления уровни, используемые в комбинации, будут ниже уровней, применяемых в отношении лекарственных препаратов для монотерапии.
Используемый в данном документе термин "синергетический" относится к действию двух терапевтических средств, таких как, например, ингибитор c–RAF, представляющий собой соединение A, и ингибитор ERK 1/2, представляющий собой соединение B, проявляющих эффект, например, в отношении замедления симптоматического прогрессирования пролиферативного заболевания, в частности рака, или его симптомов, который сильнее, чем простая сумма эффектов каждого лекарственного средства, вводимого отдельно. Синергетический эффект может быть рассчитан, например, с применением подходящих способов, таких как способы, описанные в (Lehar et al 2009).
В вариантах осуществления дополнительное терапевтическое средство (например, ингибитор CRAF) вводят в терапевтической или ниже терапевтической дозе относительно уровня дозы отдельно взятого средства. В определенных вариантах осуществления концентрация второго терапевтического средства, которая требуется для достижения ингибирования, например ингибирования роста или уменьшения размеров опухоли, является более низкой в случае, когда второе терапевтическое средство, например ингибитор ERK 1/2, применяют или вводят в комбинации с первым терапевтическим средством, в отличие от случая, когда второе терапевтическое средство вводят отдельно. В некоторых вариантах осуществления концентрация или доза первого терапевтического средства, которая необходима для достижения ингибирования, например ингибирования роста, является более низкой в случае, когда первое терапевтическое средство вводят в комбинации со вторым терапевтическим средством, в отличие от в случая, когда первое терапевтическое средство вводят отдельно. В некоторых вариантах осуществления в комбинированной терапии концентрация или доза второго терапевтического средства, которая необходима для достижения ингибирования, например ингибирования роста, является более низкой, чем терапевтическая доза второго терапевтического средства в качестве монотерапии, например ниже на 10–20%, 20–30%, 30–40%, 40–50%, 50–60%, 60–70%, 70–80% или 80–90%. В некоторых вариантах осуществления в комбинированной терапии концентрация или доза первого терапевтического средства, которая необходима для достижения ингибирования, например ингибирования роста, является более низкой, чем терапевтическая доза первого терапевтического средства в качестве монотерапии, например ниже на 10–20%, 20–30%, 30–40%, 40–50%, 50–60%, 60–70%, 70–80% или 80–90%.
Термины "ингибирование", "ингибитор" или "антагонист" включают снижение определенного параметра, например активности данной молекулы или сигнального пути. Например, данный термин включает ингибирование активности целевой киназы (CRAF или ERK 1/2) на 5%, 10%, 20%, 30%, 40% или более. Таким образом, ингибирование может, но не обязательно должно составлять 100%.
Термин "рак" относится к заболеванию, которое характеризуется нежелательным и неконтролируемым ростом аберрантных клеток. Раковые клетки могут распространяться локально или через кровоток и лимфатическую систему в другие части тела. Используемые в данном документе термины "рак" или "опухоль" включают предраковые, а также злокачественные формы рака и опухолей.
Используемые в данном документе термины "лечить", "лечение" и "осуществление лечения" относятся к уменьшению или ослаблению прогрессирования, тяжести и/или длительности нарушения, например пролиферативного нарушения, или уменьшению проявления одного или нескольких симптомов (предпочтительно одного или нескольких явных симптомов) нарушения в результате применения одного или нескольких средств для терапии. В конкретных вариантах осуществления термины "лечить", "лечение" и "осуществление лечения" относятся к уменьшению по меньшей мере одного измеряемого физического параметра пролиферативного нарушения, такого как рост опухоли, необязательно явного для пациента. В других вариантах осуществления термины "лечить", "лечение" и "осуществление лечения" относятся к ингибированию прогрессирования пролиферативного нарушения либо с физической точки зрения, например, посредством стабилизации явного симптома, физиологической точки зрения, например посредством стабилизации физического параметра, либо с обоих точек зрения. В других вариантах осуществления термины "лечить", "лечение" и "осуществление лечения" относятся к снижению или стабилизации размера опухоли или количества раковых клеток.
Фармацевтические композиции и наборы
Режимы дозирования корректируют для обеспечения оптимальной требуемой ответной реакции (например, терапевтической ответной реакции). Например, можно вводить один болюс, можно вводить несколько разделенных доз в течение некоторого времени, или дозу можно пропорционально уменьшать или увеличивать в соответствии с потребностями терапевтической ситуации.
Единичная дозированная форма, применяемая в данном документе, относится к физически дискретным единицам, подходящим в качестве единичных доз для субъектов, подлежащих лечению; при этом каждая единица содержит заранее заданное количество активного соединения, рассчитанное для получения требуемого терапевтического эффекта, в сочетании с необходимым фармацевтическим носителем. Параметры единичных дозированных форм по настоящему изобретению продиктованы и непосредственно зависят от (a) уникальных характеристик активного соединения и конкретного терапевтического эффекта, который должен быть достигнут, и (b) ограничений, свойственных в данной области техники в отношении составления такого активного соединения для лечения чувствительности у индивидуумов.
Фармацевтические композиции по настоящему изобретению могут включать "терапевтически эффективное количество" или "профилактически эффективное количество" соединения по настоящему изобретению. "Терапевтически эффективное количество" относится к количеству, эффективному в дозах и в течение периодов времени, необходимых для достижения требуемого терапевтического результата. Терапевтически эффективное количество может варьировать в зависимости от таких факторов, как стадия заболевания, возраст, пол и вес индивидуума. Терапевтически эффективное количество также является таким, в котором любые токсические или вредные эффекты ингибитора CRAF и/или ингибитора ERK 1/2 перевешиваются терапевтически полезными эффектами. "Терапевтически эффективная доза" предпочтительно модулирует измеряемый параметр требуемым образом, например скорость роста опухоли, на по меньшей мере приблизительно 20%, более предпочтительно на по меньшей мере приблизительно 40%, еще более предпочтительно на по меньшей мере приблизительно 60% и даже еще более предпочтительно на по меньшей мере приблизительно 80% по сравнению с субъектами, не подвергавшимися лечению. Способность соединения требуемым образом модулировать измеряемый параметр, например в случае рака, может быть оценена на модельной системе животного, прогнозирующей эффективность в опухолях человека, для содействия установлению подходящих уровней и схем дозирования. В качестве альтернативы это свойство композиции может быть оценено путем изучения способности соединения модулировать нежелательный параметр с использованием анализов in vitro, известных специалисту–практику.
"Профилактически эффективное количество" относится к количеству, эффективному в дозах и в течение периодов времени, необходимых для достижения требуемого профилактического результата. Как правило, поскольку профилактическую дозу применяют у субъектов до или на более ранней стадии заболевания, то профилактически эффективное количество будет меньше, чем терапевтически эффективное количество.
Также в объем настоящего изобретения входит набор, содержащий одно или несколько соединений, описанных в данном документе. Набор также может включать один или несколько других элементов: инструкцию по применению; другие реагенты для применения с соединением(–ями); устройства или другие материалы для подготовки соединения к введению, такие как контейнер для смешивания; фармацевтически приемлемые носители и устройства или другие материалы для введения субъекту, как например шприц.
Комбинации по настоящему изобретению обладают терапевтическими, или защитными, или и теми и другими функциями и могут применяться in vivo или ex vivo. Например, эти молекулы можно вводить в клетки в культуре in vitro или ex vivo или человеку для лечения, предупреждения и/или диагностики ряда нарушений, таких как формы рака, как описано в данном документе.
Соответственно, в одном аспекте настоящее изобретение предусматривает способ повышения эффективности противоракового соединения путем применения его в комбинации с другим противораковым соединением, в частности способ с применением соединения A совместно с соединением B для обеспечения повышенной эффективности, которую невозможно безопасно достичь путем введения аналогичных доз любого из соединений в виде отдельного средства. Эти комбинации особенно применимы для лечения форм рака, экспрессирующих одну или несколько мутаций с приобретением функции в сигнальном пути MAPK, в частности мутации в генах RAS и/или Raf.
ПРИМЕРЫ
Приведенные ниже примеры изложены в целях облегчения понимания настоящего изобретения, но не предназначены и не должны толковаться как ограничивающие его объем каким–либо образом.
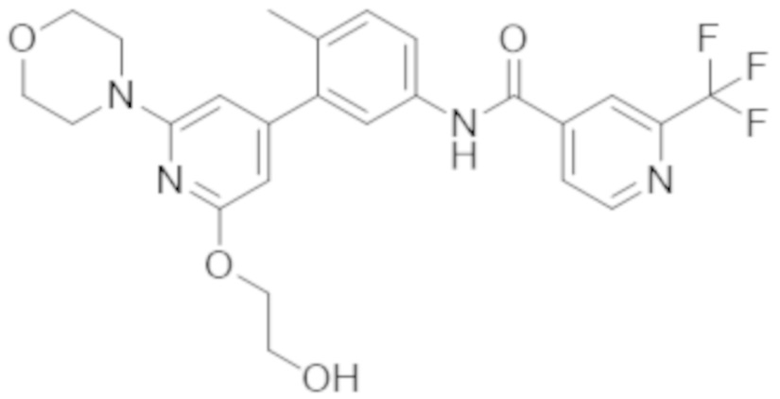
Пример 1. N–(3–(2–(2–гидроксиэтокси)–6–морфолинопиридин–4–ил)–4–метилфенил)–2–(трифторметил)изоникотинамид
Соединение А (соединение А) представляет собой морфолинзамещенное биарильное соединение следующей структуры:
.
Соединение A представляет собой пример 1156 в опубликованный заявке согласно PCT WO2014/151616. Получение соединения А, фармацевтически приемлемых солей соединения А и фармацевтических композиций, содержащих соединение А, также описано в РСТ–заявке, например, см. страницы 739–741.
Пример 1A
Определение in vitro активности Raf
Все ферменты RAF и субстрат, представляющий собой каталитически неактивный белок MEK1, были получены своими силами с применением общепринятых способов. Субклонировали кДНК CRAF в виде полноразмерного белка с активирующими мутациями Y340E и Y341E в бакуловирусный экспрессионный вектор для экспрессии в клетках насекомых Sf9. Субклонировали кДНК h14–3–3 типа дзета в бакуловирусный экспрессионный вектор для экспрессии в клетках насекомых Sf9. Клетки Sf9, совместно экспрессирующие оба белка, лизировали, и подвергали хроматографии с иммобилизованным никелем, и элюировали имидазолом. Применяли вторую колонку (StrepII–связывающую колонку), и элюировали дестиобиотином. Белковые метки удаляли с применением фермента Prescission, и белок дополнительно очищали с применением проточной стадии для удаления меток.
C–Raf FL относится к полноразмерному белку C–Raf.
В качестве субстрата RAF применяли полноразмерный MEK1 с инактивирующей мутацией сайта связывания ATP K97R. кДНК MEK1 вместе с N–концевой (his)6–меткой субклонировали в вектор для экспрессии в E. Coli. Субстрат MEK1 очищали от лизата E. Coli с помощью никель–аффинной хроматографии с последующим анионным обменом. Конечный препарат MEK1 биотинилировали (Pierce EZ–Link Sulfo–NHS–LC–Biotin) и концентрировали.
Материалы для анализа
Аналитический буфер: 50 мМ Трис, pH 7,5, 15 мМ MgCl2, 0,01% бычий сывороточный альбумин (BSA), 1 мМ дитиотреитол (DTT).
Останавливающий буфер: 60 мМ этилендиаминтетрауксусная кислота (EDTA), 0,01% Tween® 20.
b–Raf(V600E), активный;
биотинилированный Mek с подавленной киназной активностью;
набор для выявления Alpha Screen (доступный от PerkinElmer™, № 6760617R);
антитело к фосфо–MEK1/2 (доступно от Cell Signalling Technology, Inc. № 9121);
384–луночные малообъемные аналитические планшеты (планшеты White Greiner®).
Условия анализа
b–Raf(V600E), примерно 4 пМ;
c–Raf, примерно 4 нМ;
биотинилированный Mek с подавленной киназной активностью, примерно 10 нМ;
ATP, 10 мкМ для BRAF(V600E) и 1 мкМ для CRAF;
время предварительной инкубации с соединениями составляло 60 минут при комнатной температуре;
время реакции составляло 1 или 3 часа при комнатной температуре.
Протокол анализа
Raf и биотинилированный Mek с подавленной киназной активностью объединяли при 2X конечных концентрациях в аналитическом буфере (50 мМ Трис, рН 7,5, 15 мМ MgCl2, 0,01% BSA и 1 мМ DTT) и вносили по 5 мл на лунку в аналитические планшеты (белые 384–луночные аналитические планшеты Greiner, № 781207), содержащие 0,25 мл 40Х тестируемого соединения, ингибирующего киназу Raf, разбавленного в 100% DMSO. Планшет инкубировали в течение 60 минут при комнатной температуре.
Реакцию киназной активности Raf инициировали добавлением 5 мл на лунку 2X ATP, разбавленного в аналитическом буфере. Через 3 часа (b–Raf(V600E)) или 1 час (c–Raf). Реакции останавливали и фосфорилированный продукт измеряли с использованием антитела к p–MEK кролика (Cell Signaling, № 9121) и набора для выявления Alpha Screen IgG (ProteinA) (PerkinElmer № 6760617R) путем добавления 10 мл на лунку смеси нагруженных антителами гранул (разбавление 1:2000) и гранул для определения (разбавление обоих типов гранул в пропорции 1:2000) в останавливающем буфере для гранул (25 мМ EDTA, 50 мМ Трис, рН 7,5, 0,01% Tween20). Добавления осуществляли в темных условиях для защиты гранул для определения от воздействия света. Крышку помещали на верхнюю часть планшета, и инкубировали в течение 1 часа при комнатной температуре, затем люминесценцию считывали на приборе PerkinElmer Envision. Концентрацию каждого соединения для 50% ингибирования (IC 50) рассчитывали с помощью нелинейной регрессии с использованием программного обеспечения для анализа данных XL Fit.
При применении вышеописанных анализов соединение А демонстрировало ингибирующую эффективность, как указано ниже.
Соединение А представляет собой ингибитор типа II, ингибирующий как b–Raf, так и c–Raf.
Пример 1B
Соединение А характеризуется активностью в отношении многочисленных линий раковых клеток человека, которые экспрессируют мутации в сигнальном пути МАРК, как показано в нижеследующей таблице. Следует обратить внимание, что активность является особенно сильной в отношении клеточных линий, которые содержат по меньшей мере одну мутацию в BRAF или RAS.
Таблица 1. Эффект соединения А в отношении пролиферации в панели линий раковых клеток человека
Пример 1C
Для исследования активности соединения A в BRAF V600–мутантных клетках меланомы, устойчивых к ингибиторам BRAF и/или MEK, оценивали антипролиферативную активность соединения A в механистических моделях, полученных из клеточной линии A375 меланомы BRAF V600, экспрессирующей мутации MEK1/2, NRAS или вариант сплайсинга BRAF. Эти мутации, как было продемонстрировано как в доклинических исследованиях, так и в клинических образцах, вызывают устойчивость к ингибиторам BRAF и/или MEK. Эффекты соединения A в отношении ингибирования роста в исходной клеточной линии A375 и ее производных, экспрессирующих различные мутантные аллели, по сравнению с эффективностью ингибитора BRAF вемурафениба и ингибитора MEK селуметиниба приведены ниже. Мутации обеспечивали устойчивость как к ингибиторам BRAF, так и к ингибиторам MEK, что приводило к более чем 50–кратному увеличению значений IC50. В то же время, резистентные модели с устойчивостью, тем не менее, были чувствительны к соединению А, характеризуясь только 2–3–кратным увеличением IC50. Эти данные свидетельствуют в пользу применения соединения А у пациентов с меланомой BRAF V600, которые стали невосприимчивыми к ингибиторам BRAF и/или MEK.
Антипролиферативный эффект соединения A в отношении механистических моделей A375, устойчивых к ингибиторам BRAF и MEK
Пример 1D
Соединение А составляли с гидроксипропилметилцеллюлозой/гипромеллозой, коллоидным диоксидом кремния, микрокристаллической целлюлозой, поливинилпирролидоном/повидоном и стеаратом магния и формировали в таблетки, содержащие приблизительно 50 мг соединения А. Определенное количество таблеток, достаточное для достижения требуемой дозы, вводили субъектам натощак один раз в сутки. Субъектов обрабатывали при дозах, составляющих 100 мг один раз в сутки или 200 мг один раз в сутки. Серийные образцы крови для оценки PK собирали не более чем через 48 часов после введения первой дозы соединения А (цикл 1, день 1) и не более чем через 24 часа после введения многократных доз (цикл 1, день 15). Максимальные концентрации в плазме крови (Cmax), составляющие 447 нг/мл и 889 нг/мл, достигали в течение 4 часов после введения однократной дозы, составляющей 100 мг, и однократной дозы, составляющей 200 мг, соответственно. Среднее содержание в плазме крови на протяжении интервала между введением доз, составляющем 24 часа (AUCtau), на 1–й день дозирования составило 5679 нг⋅ч./мл и 10019 нг⋅ч./мл после введения доз соединения А, составляющих 100 мг и 200 мг, соответственно. По расчетам, период полувыведения у пациентов составлял около 23–24 часов. Дозировка, составляющая 100 мг один раз в сутки, приводила к незначительному накоплению соединения А в плазме крови с коэффициентом накопления, составляющим 1,8. На основании этих данных была составлена схема дозирования с частотой один раз в сутки. Поскольку данное исследование продолжается, то все представленные данные считаются предварительными.
Пример 2. Противоопухолевая активность соединения А в моделях KRAS–мутантного NSCLC
Модель H358
Опухоленесущих самок SCID–мышей NCI–H358 бежевого цвета, n=8 на группу, рандомизировали на 3 группы через 14 дней после инокуляции опухолевых клеток со средним объемом опухоли, находящимся в диапазоне 259,44–262,47 мм3.
Животным во время курса обработки вводили пероральную дозу либо среды–носителя, либо соединения А в количестве 30 мг/кг или 200 мг/кг один раз в сутки в течение 14 последовательных дней при объеме дозирования, составляющем 10 мл/кг веса тела животного. Объемы опухолей измеряли с помощью цифрового штангенциркуля 3 раза в неделю, а вес тела каждого животного регистрировали на протяжении курса обработки.
Модель Calu6
Самок голых мышей, несущих опухоль Calu6, n=6 на группу, рандомизировали на группы обработки на 17–й день после имплантации опухоли, когда средний объем опухоли составлял 180 мм3. Обработки соединением А начинали на 17–й день и продолжали в течение 16 дней. Объем дозирования составлял 10 мл/кг. Объемы опухолей измеряли во время рандомизации и затем два раза в неделю в течение всего периода проведения исследования.
Модель H727
Самок голых мышей, несущих опухоль NCI–H358, n=8 на группу, рандомизировали на 2 группы со средним объемом опухоли в пределах 275,74 мм3. Животным во время курса обработки вводили пероральную дозу либо среды–носителя, либо соединения А в количестве 100 мг/кг один раз в сутки в течение 14 последовательных дней при объеме дозирования, составляющем 10 мл/кг веса тела животного. Объемы опухолей измеряли с помощью цифрового штангенциркуля 3 раза в неделю, а вес тела каждого животного регистрировали на протяжении курса обработки. Как показано на фигурах 1A, 1B и 1C, соединение А в качестве отдельно взятого средства продемонстрировало активность в моделях NSCLC KRASmt.
В клеточных анализах соединение A продемонстрировало антипролиферативную активность в клеточных линиях, которые содержат множество мутаций, активирующих передачу сигнала посредством MAPK. Например, соединение А ингибировало пролиферацию клеточной линии немелкоклеточного рака легкого Calu–6 (KRAS Q61K), колоректальной клеточной линии HCT116 (KRAS G13D со значениями IC50 в диапазоне от 0,2 до 1,2 мкМ. In vivo обработка соединением A приводила к опухолевой регрессии в нескольких человеческих KRAS–мутантных моделях, в том числе ксенотрансплантатах на основе полученных из NSCLC Calu–6 (KRAS Q61K) и NCI–H358 (KRAS G12C). Во всех случаях противоопухолевые эффекты являлись дозозависимыми и хорошо переносимыми, о чем свидетельствовало отсутствие значительной потери веса тела. Модель Calu–6 проявляла чувствительность к соединению А при имплантации как у голых мышей, так и у голых крыс с регрессией, наблюдаемой при дозах 100, 200 и 300 мг/кг один раз в сутки (QD) у мышей и 75 и 150 мг/кг QD у крыс. Остановку роста опухоли в этой модели наблюдали при 30 мг/кг QD и 35 мг/кг QD у мышей и крыс соответственно. Регрессию также наблюдали во второй модели NSCLC человека NCI–H358 при дозе, составляющей 200 мг/кг QD у мышей, и в ксенотрансплантате рака яичника человека hey–A8 при дозе, составляющей всего лишь 30 мг/кг QD у мышей. Кроме того, данные исследования эффективности фракционирования дозы в ксенотрансплантатах Calu–6 продемонстрировали, что при разных уровнях дозирования соединение А, дозированное QD и фракционированное дважды в сутки (BID), демонстрировало аналогичные уровни противоопухолевой активности. Эти результаты свидетельствуют в пользу апробирования режимов дозирования QD или BID в клинической практике. В совокупности, подавление сигнального пути MAPK и антипролиферативная активность in vitro и in vivo, наблюдаемые для соединения А в хорошо переносимых дозах, дают основание предполагать, что соединение А может характеризоваться противоопухолевой активностью у пациентов с опухолями, содержащими активирующие нарушения в сигнальном пути MAPK, и поэтому, в частности, может быть применимо в качестве отдельного средства или в комбинации со вторым средством, таким как ингибитор, влияющий на другой участок сигнального пути MAPK, для лечения пациентов с NSCLC, имеющих мутации KRAS. Было показано, что соединение A в качестве отдельного средства характеризуется активностью против различных других видов рака, которые экспрессируют мутации с приобретением функции в сигнальном пути MAPK, например в RAS или RAF, в том числе рака яичника, рака поджелудочной железы и меланомы, и в модельных системах было показано, что оно является более эффективным в отношении этих состояний в случае применения в комбинации с ингибитором ERK, таким как соединение B.
Пример 3
Соединение B представляет собой ингибитор ERK 1/2. Соединение раскрыто и его получение описано в опубликованной заявке согласно РСТ на патент WO2015/066188.
Соединение B
В некоторых вариантах осуществления это соединение применяют в виде его гидрохлоридной соли.
В 3–дневном анализе пролиферации соединение B продемонстрировало устойчивое ингибирование роста клеток со значениями IC50, составляющими менее 1 мкМ, в подгруппе клеточных линий, содержащих мутации KRAS, в том числе клеточной линии рака легкого Calu6 (KRASQ61K), клеточной линии рака яичника HeyA8 (KRASG12D, клеточных линиях рака поджелудочной железы AsPC–1 (KRASG12D) и PSN1 (KRASG12R).
Пример 4. Эффект комбинации соединения A и соединения B в отношении KRAS–мутантных клеточных линий
Эффект комбинации соединения A и соединения B в отношении пролиферации оценивали in vitro на панели из четырнадцати (14) KRAS–мутантных клеточных линий, полученных из NSCLC (4), колоректального рака (CRC) (4) и аденокарциномы протоков поджелудочной железы (PDAC) (6).
Посредством набора для люминесцентного анализа жизнеспособности клеток CellTiter–Glo® (CTG) (Promega, Мэдисон, Висконсин, США) измеряли количество ATP, присутствующего в лунке после лизиса клеток. ATP, высвобождаемый при лизисе, измеряли в ходе ферментативной реакции, которая включала в себя люциферазу и ее субстрат люциферин. Количество излучаемого света пропорционально количеству ATP, которое в свою очередь пропорционально количеству живых клеток в лунке. Этот анализ применяли для определения доли жизнеспособных клеток после обработки лекарственным средством.
Реагенты, используемые для анализа CTG, в том числе 14 клеточных линий, среды и плотности клеток, описаны в таблицах ниже.
Клетки высевали в дублированных группах в соответствии с информацией, приведенной в таблице ниже, при 50 мкл/лунка в белых 384–луночных планшетах для культивирования тканей (№ 3707, Corning, Нью–Йорк, США). На следующий день соединения разбавляли в DMSO в планшете для соединений (№ 788876, Greiner, Монро, Северная Каролина, США) с получением 7–точечной серии разбавлений 1:3 (в 1000 раз превышающую необходимые конечные концентрации). Соединения вносили в планшеты с клетками для достижения разбавления 1:1000 с применением акустического дозатора (ATS100, EDC Biosystems, Калифорния, США). Например, одна лунка планшета для соединений содержала 10 мМ соединения A, и посредством переноса 50 нл соединения в 50 мкл клеток достигали конечной концентрации, составляющей 10 мМ, для данного соединения в данной лунке. В разделе 2.3 изложена схема и значения концентрации сетки комбинаций, которая была создана в аналитических планшетах авторов настоящего изобретения с применением ATS100. Затем аналитические планшеты возвращали в увлажненный инкубатор с CO2 при 37°C.
После 5 дней инкубации соединений в каждую лунку добавляли по 25 мкл/лунка реагента для определения жизнеспособности CTG (общий объем 75 мкл), и через 15 минут инкубации при комнатной температуре планшеты считывали на считывающем устройстве для микропланшетов с применением 0,1–секундного протокола считывания люминесценции с низким пределом чувствительности (Envision, Perkin Elmer, Хопкингтон, Массачусетс, США).
Данные нормализовали относительно среднего значения для необработанных лунок (обработанных только DMSO) для данной рассматриваемой клеточной линии. Затем значения вычитали из 1 и умножали на 100 для получения % ингибирования. Затем нормализованные данные подгоняли к кривым с применением регрессионной логистической модели подбора кривой по четырем параметрам (4PL) с помощью коммерческого программного обеспечения (HELIOS). Значения IC50 (полумаксимальной ингибирующей концентрации) получали для кривых для отдельного средства, где соединение ингибировало рост клеток на 50%.
Таблица. Реагенты и материалы для анализа
Реагент
Таблица. Среды и значения густоты посева для клеточных линий
Во всех случаях комбинации оценивали в отформатированной в шахматном порядке матрице с применением концентраций соединения A в диапазоне от 0,014 до 10 мкМ и концентраций соединения B в диапазоне от 0,011 до 8,0 мкМ. Являлась ли комбинация синергетической в отношении конкретной клеточной линии или нет, определяли с помощью двух параметров синергии; показатель синергии и показатель аддитивности (CI) при уровне ингибирования, составляющем 50 или 75 процентов (Lehar et al. 2009).
Комбинация соединения А и соединения В характеризовалась умеренной или сильной синергией в 7/14 протестированных клеточных линиях. Краткое описание этих значений для каждой клеточной линии показано в таблице ниже.
Таблица. Краткое описание показателей синергии и значений CI50 для комбинации соединения A и соединения B
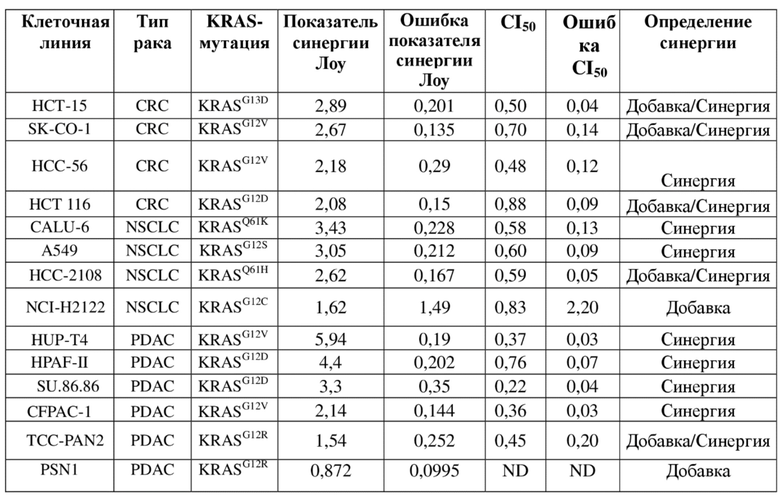
Общее руководство по интерпретации показателей/значений приведено в таблице ниже.
Таблица. Интерпретация активности комбинации

В группе протестированных клеточных линий синергия преимущественно наблюдалась в моделях, полученных из линий NSCLC и PDAC, причем модели Calu–6 (KRASQ61K) и HUPT4 (KRASG12V) являлись наиболее восприимчивыми для двух линий соответственно.
Пример 5. Соединение А в комбинации с соединением В в ксенотрансплантатной модели заболевания
Опухоли Calu–6 NSCLC приживляли бестимусным самкам мышей с помощью подкожной инъекции 10 миллионов клеток в 50% Matrigel™ в боковую область каждой мыши. Когда опухоли достигли приблизительно 250 мм3, мышей рандомизировали в соответствии с объемом опухоли на группы обработки (n=7). Тестируемые средства вводили перорально один раз в сутки (qd) или один раз в двое суток (q2d) при указанных уровнях доз. На A–B) приведены объемы опухолей в группах обработки относительно количества дней после рандомизации. На C) приведено среднее процентное изменение веса тела от исходного. Данные для этих обработок показаны на фигурах 1А, 1В и 1С.
Комбинированная обработка соединением A и соединением B приводила к увеличению интенсивности и продолжительности ответной реакции опухоли по сравнению с любым отдельным средством в ксенотрансплантате Calu–6 NSCLC человека. Соединение А, вводимое в дозе 30 мг/кг qd, достигало 26% Т/С, в то время как соединение В, вводимое в дозе 75 мг/кг q2d или 50 мг/кг qd, достигало 4% Т/С и 22% регрессии соответственно через 17 дней после дозирования. Объединение соединения А, вводимого в дозе 30 мг/кг в сутки, с соединением В, вводимым в дозе 50 мг/кг в сутки или 75 мг/кг в сутки, достигало 66% и 51% регрессии соответственно через 17 дней после дозирования. В дополнение к повышенной интенсивности ответной реакции комбинация соединения A и соединения B также приводила к увеличению продолжительности ответной реакции. В то время как опухоли у мышей, которым вводили соединение А и соединение В в виде отдельных средств, прогрессировали при лечении, совместная комбинация соединения А и соединения В (независимо от дозы соединения В) поддерживала регрессию опухоли через 42 дня после дозирования. В совокупности, эти данные предполагают, что комбинированное лечение соединением A и соединением B может достигать более сильных и более продолжительных ответных реакций у пациентов с активированным сигнальным путем MAPK из–за мутаций с приобретением функции в сигнальном пути MAPK. Кроме того, такие результаты свидетельствуют в пользу потенциального исследования применения прерывистого дозирования соединения B в комбинации с соединением A. Тестируемые комбинации были хорошо переносимы, о чем свидетельствовало отсутствие потери веса тела.
Пример 6. Фаза I исследования по подбору дозы соединения A у взрослых пациентов с солидными опухолями на поздней стадии, содержащими изменения сигнального пути MAPK, отдельно или в сочетании с соединением B
Соединение А в качестве отдельного средства
Рекомендуемая начальная доза и режим приема соединения А как отдельного средства в настоящем исследовании составляет 100 мг QD перорально исходя из доклинических данных о безопасности, переносимости, данных PK/PD, полученных в доклинических исследованиях, а также прогноза исследуемого эффективного диапазона дозы для человека. Можно применять начальные дозы, составляющие 100 мг, 200 мг, 250 мг, 300 мг или 400 мг; предварительные данные свидетельствуют о том, что начальная доза, составляющая 250 мг один раз в сутки, может быть эффективной в отношении солидных опухолей. Для максимальной гибкости дозирования соединение А можно получать в виде 50 мг и/или 100 мг таблеток для перорального введения. Предложенный состав для клинического применения соединения A включает соединение A в сочетании с одним или несколькими вспомогательными веществами, выбранными из гидроксипропилметилцеллюлозы (гипромеллозы), коллоидного диоксида кремния, микрокристаллической целлюлозы, поливинилпирролидона (повидона) и стеарата магния, и может быть получен в форме таблеток для перорального введения, соответственно содержащих 50 мг или 100 мг соединения А.
Предварительные уровни доз для осуществления повышения дозы можно найти в таблице ниже.
Таблица. Уровни предварительных доз для соединения А
**Уровень дозы –1 представляет терапевтические дозы для пациентов, нуждающихся в снижении дозы относительно начального уровня дозы.
В части исследования с расширением дозы пациенты в группе с соединением А, применяемым в качестве отдельного средства, получали соединение А в соответствии с рекомендуемой дозой и режимом, выбранными на основании данных повышения дозы. Ожидается, что такая доза будет безопасной и переносимой у взрослых пациентов по всем показаниям, включенным в исследование.
Клинический режим для данного испытания, впервые проводимого на человеке, представляет собой непрерывную схему дозирования соединения A один раз в сутки. В доклинических исследованиях было продемонстрировано, что схема QD является эффективной и переносимой. В ксенотрансплантатах Calu6 аналогичные уровни эффективности достигались либо с использованием QD, либо с использованием фракционированных режимов BID, свидетельствуя о том, что эффективность связана с общим воздействием. Прогнозирование PK и прогнозирование периода полувыведения (~9 ч.) у человека также дает основания предполагать, что эффективное воздействие может достигаться при использовании дозирования QD.
Кроме того, эффективную дозировку можно определить с помощью отслеживания биомаркеров, указывающих на ингибирование сигнального пути MAP–киназы. В частности, DUSP6 (фосфатаза 6 с двойной специфичностью) является известным биомаркером для данного сигнального пути, и было показано, что уровни DUSP6 in vivo снижаются у субъекта, получающего дозу соединения A, что ассоциируют с эффективными уровнями соединения A в плазме крови. DUSP6 может применяться в качестве фармакодинамического биомаркера у субъектов, получающих соединение А либо в виде отдельного средства, либо в комбинации с другим терапевтическим средством.
Соединение A в комбинации с соединением B
Соединение B может применяться в виде его гидрохлоридной соли. Для целей тестирования его можно вводить перорально либо в составе с одним или несколькими вспомогательными веществами, либо в виде одного соединения, содержащегося в фармацевтически приемлемой капсуле, такой как мягкая или твердая желатиновая капсула, или смешанного с подходящей средой для введения через желудочный зонд.
Повышение дозы соединения А в комбинации с соединением В начинали с режима дозирования, определенного для соединения А в качестве отдельного средства: начальная доза соединения А была ниже, чем его доза при применении в качестве отдельного средства. Таким образом, выбор этой дозы должен минимизировать воздействие потенциально токсичных уровней лекарственного средства, сокращая при этом количество пациентов, которые могли бы получать дозы, слишком низкие для обеспечения надлежащей эффективности.
Режим дозирования соединения А был таким же, как и выбранный для соединения А в виде отдельного средства. В случае, если обе схемы для соединения А в виде отдельного средства будут изучаться в ходе части исследования с расширением дозы отдельного средства, один предпочтительный режим будет выбран для комбинации исходя из всех доступных данных, включая данные о безопасности и воздействии. Решение об изменении режима дозирования для соединения А в группе комбинированной терапии на более поздней стадии может быть принято на основе получаемых данных.
Соединение B вводили при уровне дозировки, прогнозируемом для обеспечения воздействия, достаточного для обеспечения эффективного подавления опухоли, исходя из активности в доклинических моделях. Исходя их доклинического тестирования, эффективность, по–видимому, определяется временем сохранения концентрации выше эффективного уровня в плазме крови, таким образом, в одном варианте осуществления дозу можно определять путем отслеживания уровней соединения В в плазме крови. Согласно прогнозированию требуемый минимальный остаточный уровень в плазме крови для обеспечения эффективности составляет приблизительно 600 нМ, или приблизительно 350 нг/мл; соответственно, дозировку и режим (частоту) введения можно определять, руководствуясь целью поддержания концентрации соединения В в плазме выше данного минимального остаточного уровня в плазме крови в течение по меньшей мере суток, или по меньшей мере недели, или в течение цикла лечения, такого как 1, 2, 3 или 4 недели, как будет сочтено целесообразным лечащим врачом.
Клиническим режимом для данного испытания являлась непрерывная схема дозирования один раз в сутки для соединения A и соединения B.
Это дополнительно подтверждалось предварительными результатами, полученными в ходе клинических испытаний. Было показано, что субъект с немелкоклеточным раком легкого (NSCLC), получавший 1200 мг QD соединения А, проявлял частичную ответную реакцию, составляющую –35%, в соответствии с критериями оценки ответной реакции при солидных опухолях (RECIST).
В части с расширением дозы пациентов в группе комбинированной терапии лечили в соответствии с рекомендуемыми дозой и режимом дозирования для комбинации лекарственных средств исходя из данных о повышении дозы. Для обеспечения подходящей гибкости дозирования соединение B можно получать в форме желатиновых капсул, содержащих различные количества соединения B (необязательно в виде его гидрохлорида), как например дозы, составляющие приблизительно 5 мг, 20 мг, 50 мг или 100 мг на желатиновую капсулу.
Пациентов в группе комбинированной терапии можно лечить соединением А в суточной дозе, составляющей приблизительно 100 мг, или приблизительно 150 мг, или приблизительно 200 мг, или приблизительно 250 мг, и соединением В в суточной дозе, составляющей приблизительно 50 мг, или приблизительно 75 мг, или приблизительно 100 мг. Например, пациентам можно вводить общую дозу соединения A, составляющую приблизительно 100 мг в сутки, и общую дозу соединения B, составляющую приблизительно 100 мг в сутки, причем дозы предпочтительно вводить один раз в сутки. Пациенты могут также получать общую дозу, составляющую приблизительно 200 мг соединения A в сутки, и общую дозу, составляющую приблизительно 100 мг соединения B в сутки, причем дозы предпочтительно вводят один раз в сутки.
Пациенты, получающие комбинированную терапию, включают пациентов с NSCLC, например взрослых пациентов с прогрессирующим или метастатическим KRAS–мутантным или BRAF–мутантным (например, BRAF V600E–мутантным) NSCLC. Такие пациенты могут характеризоваться прогрессированием после проведения стандартного лечения, или для них не существует эффективной стандартной терапии.
Эффективность испытания может быть оценена путем измерения общей частоты ответной реакции (ORR), частоты контроля заболевания (DCR), продолжительности ответной реакции (DOR), выживаемости без прогрессирования (PFS) согласно версии RECIST 1.1 и общей выживаемости (OS).
ЭКВИВАЛЕНТЫ
Хотя были обсуждены конкретные варианты осуществления настоящего изобретения, приведенное выше описание является иллюстративным, а не ограничивающим. Многие вариации настоящего изобретения будут очевидны для специалистов в данной области техники после ознакомления с настоящим описанием и нижеприведенной формулой изобретения. Полный объем настоящего изобретения должен определяться посредством ссылки на пункты формулы изобретения наряду с полным объемом их эквивалентов, а также описанием наряду с такими вариациями.
| название | год | авторы | номер документа |
|---|---|---|---|
| КОМБИНИРОВАННАЯ ТЕРАПИЯ | 2018 |
|
RU2815400C2 |
| КОМПОЗИЦИИ НА ОСНОВЕ ИНГИБИТОРА SHP2 И СПОСОБЫ ЛЕЧЕНИЯ РАКА | 2018 |
|
RU2805355C2 |
| Фармацевтические комбинации | 2017 |
|
RU2759669C2 |
| НОВЫЕ МАЛЫЕ МОЛЕКУЛЫ ДЛЯ ЦЕЛЕНАПРАВЛЕННОГО РАСЩЕПЛЕНИЯ НЕ ПОДДАЮЩЕГОСЯ ЦЕЛЕНАПРАВЛЕННОМУ ВОЗДЕЙСТВИЮ KRAS В ТЕРАПИИ РАКА | 2021 |
|
RU2839932C1 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМБИНАЦИИ ИНГИБИТОРА CDK4/6 И ИНГИБИТОРА В-Raf | 2013 |
|
RU2685250C2 |
| КОМБИНАЦИИ ИНГИБИТОРА PI3K И ИНГИБИТОРА МЕК | 2010 |
|
RU2563193C2 |
| ОПРЕДЕЛЕНИЕ ЧУВСТВИТЕЛЬНОСТИ КЛЕТОК К ОБРАБОТКЕ ИНГИБИТОРОМ B-Raf ПУТЕМ ДЕТЕКЦИИ МУТАЦИИ K-ras И УРОВНЕЙ ЭКСПРЕССИИ RTK | 2010 |
|
RU2553379C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ МЕТИЛХИНАЗОЛИНОНА | 2020 |
|
RU2802968C1 |
| ИНГИБИТОРЫ MDM2 И ИХ КОМБИНАЦИИ | 2016 |
|
RU2740091C2 |
| КОМБИНАЦИЯ (А) ИНГИБИТОРА ФОСФОИНОЗИТ-3-КИНАЗЫ И (Б) МОДУЛЯТОРА ПУТИ Ras/Raf/Mek | 2009 |
|
RU2508110C2 |
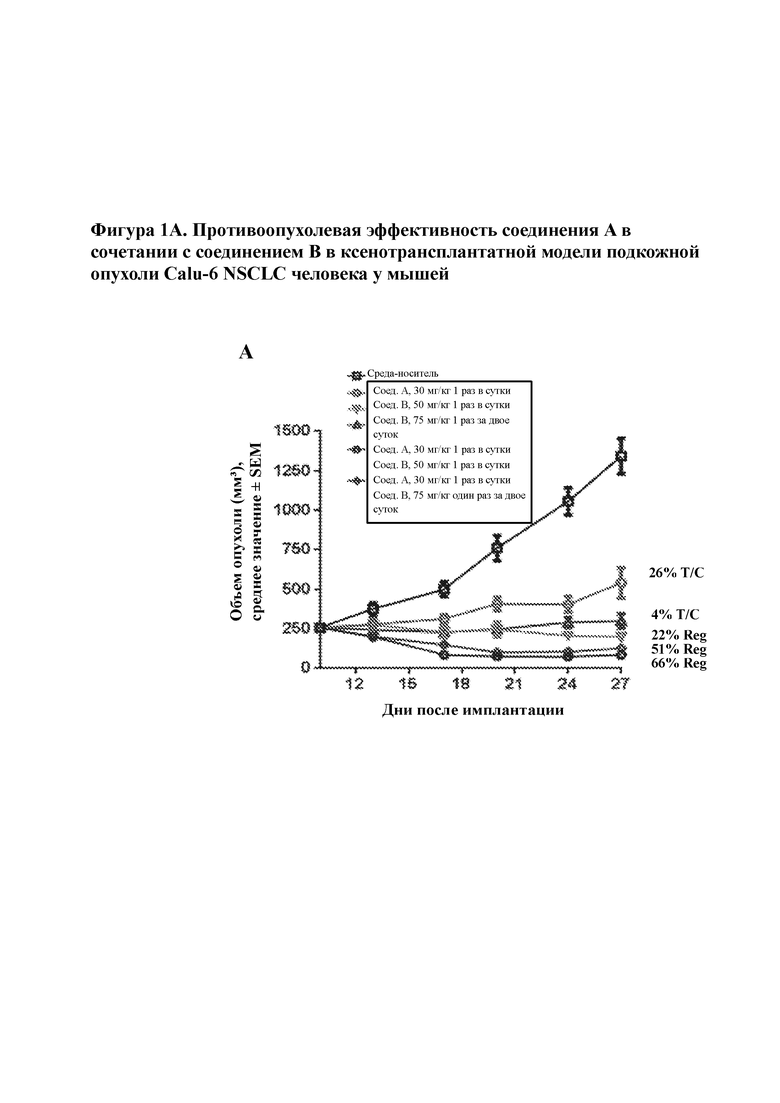
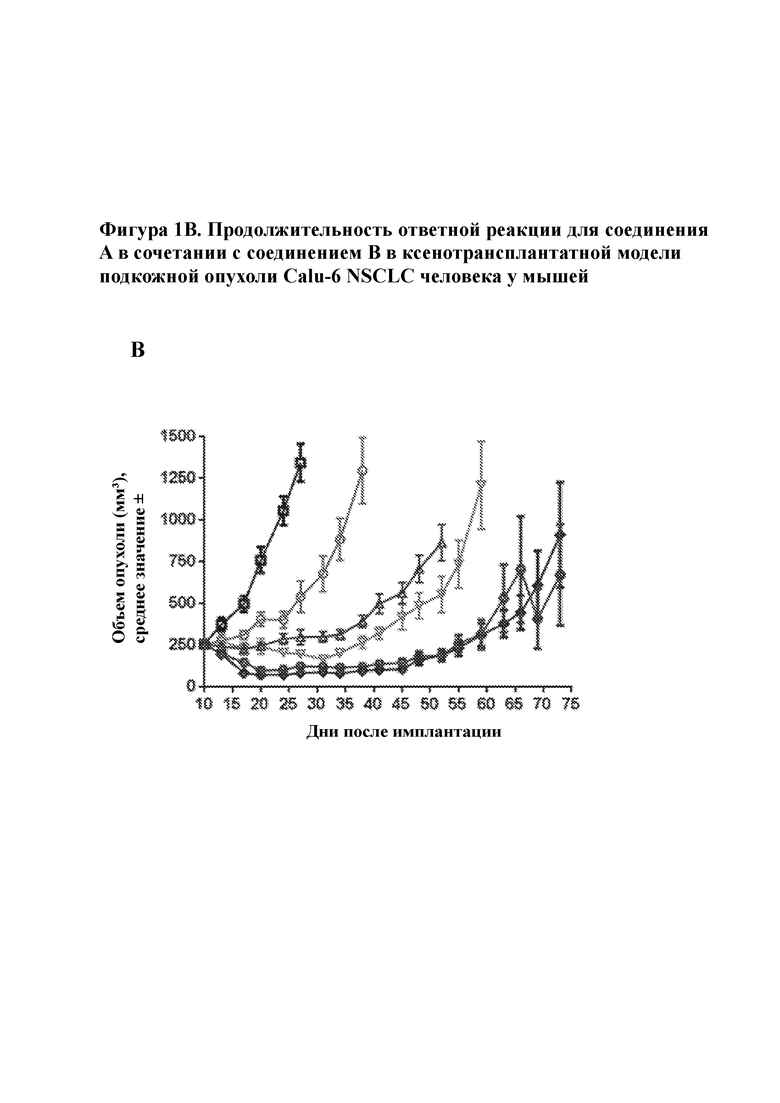
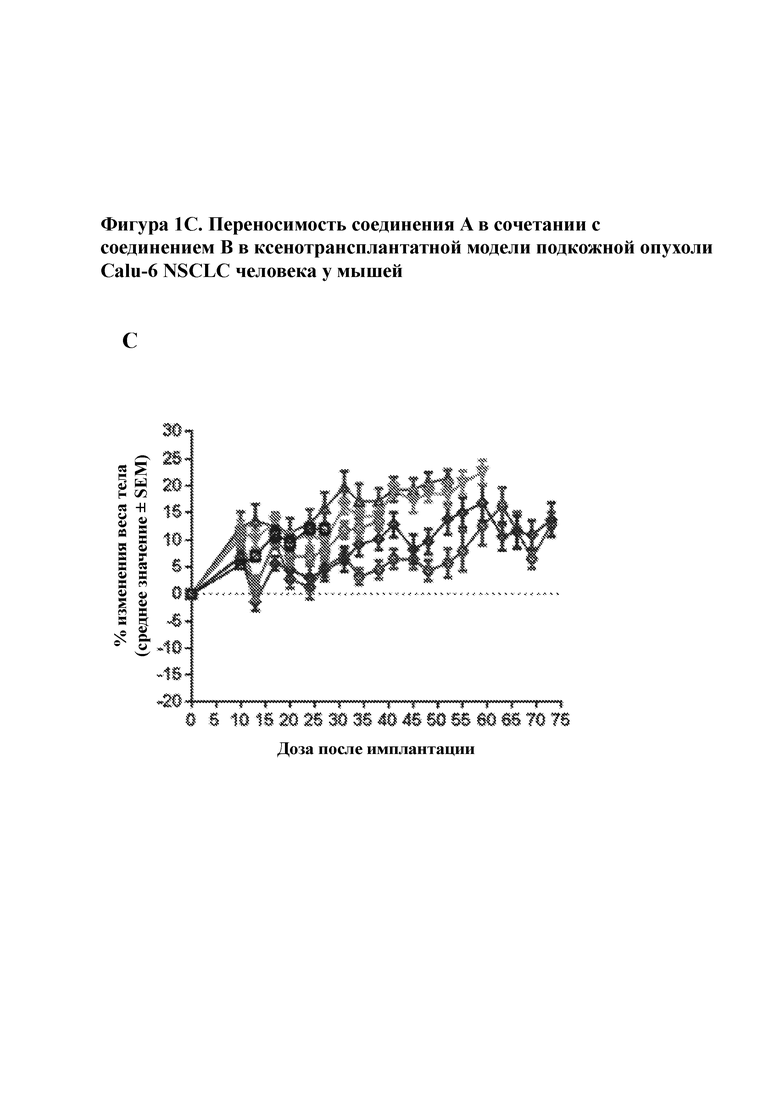
Группа изобретений относится к области медицины и фармацевтики, в частности к лечению KRAS–мутантного NSCLC (немелкоклеточного рака легкого), BRAF-мутантного NSCLC, KRAS-мутантного рака поджелудочной железы, KRAS-мутантного колоректального рака (CRC) и KRAS-мутантного рака яичника. Для этого разработана фармацевтическая комбинация, содержащая ингибитор CRAF который представляет собой соединение A N-(3-(2-(2–гидроксиэтокси)-6-морфолинoпиридин-4-ил)-4–метилфенил)-2-(трифторметил)изоникотинамид или его фармацевтически приемлемую соль и ингибитор ERK, который представляет собой соединение B 4-(3-амино-6-((1S,3S,4S)-3-фтор-4-гидроксициклогексил)пиразин-2-ил)-N-((S)-1-(3-бром-5-фторфенил)-2-(метиламино)этил)-2-фторбензамид или его фармацевтически приемлемую соль. Также раскрыты фармацевтическая композиция, применение фармацевтической комбинации и способ лечения. Группа изобретений обеспечивает создание комбинации соединения A и соединения B, объединение которых будет оптимизировать подавление передачи сигналов посредством MAPK в KRAS- и BRAF-мутантном NSCLC. Эта комбинация может также способствовать предотвращению возникновения устойчивости к комбинации ингибиторов BRAF и MEK (митоген-активируемой протеинкиназы) в BRAFV600E-мутантном NSCLC. 4 н. и 8 з.п. ф-лы, 3 ил., 6 табл., 6 пр.
1. Фармацевтическая комбинация для применения в лечении KRAS-мутантного NSCLC (немелкоклеточного рака легкого), BRAF-мутантного NSCLC, KRAS-мутантного рака поджелудочной железы, KRAS-мутантного колоректального рака (CRC) и KRAS-мутантного рака яичника, где фармацевтическая комбинация содержит
(i) ингибитор CRAF, который представляет собой соединение A,
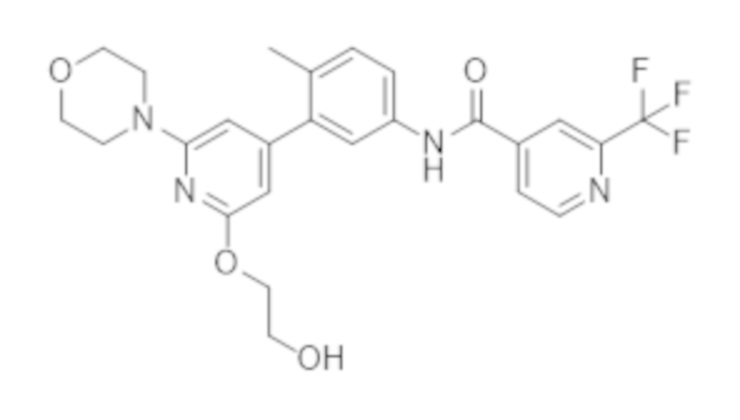 ,
,
или его фармацевтически приемлемую соль;
и
(ii) ингибитор ERK, который представляет собой соединение B,
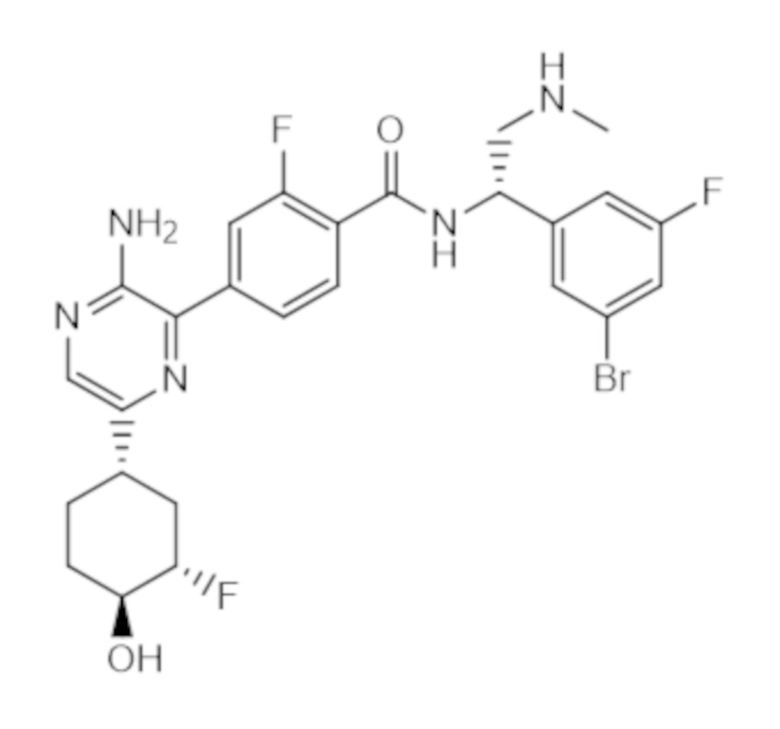 ,
,
или его фармацевтически приемлемую соль.
2. Фармацевтическая комбинация по п. 1, где ингибитор CRAF или его фармацевтически приемлемая соль и ингибитор ERK или его фармацевтически приемлемая соль вводятся по отдельности, одновременно или последовательно.
3. Фармацевтическая композиция для применения в лечении KRAS-мутантного NSCLC (немелкоклеточного рака легкого), BRAF-мутантного NSCLC, KRAS-мутантного рака поджелудочной железы, KRAS-мутантного колоректального рака (CRC) и KRAS-мутантного рака яичника, содержащая фармацевтическую комбинацию по п. 1 или п. 2 и по меньшей мере один фармацевтически приемлемый носитель.
4. Фармацевтическая комбинация по п. 1 или 2 или фармацевтическая композиция по п. 3, где опухоль или рак экспрессирует по меньшей мере одну мутацию, выбранную из группы, состоящей из RAF-мутаций V600E, V600D и G464E и RAS-мутаций A146T, Q61L, Q61K, G12D, G12C, G13D, G12V и G12R.
5. Фармацевтическая комбинация по любому из пп. 1-4 или фармацевтическая композиция по п. 3 или 4, где ингибитор CRAF или его фармацевтически приемлемая соль вводятся в суточной дозе, составляющей приблизительно 100 мг, или приблизительно 150 мг, или приблизительно 200 мг, или приблизительно 250 мг.
6. Фармацевтическая комбинация по любому из пп. 1-5 или фармацевтическая композиция по любому из пп. 3-5, где ингибитор ERK или его фармацевтически приемлемая соль вводятся в дозе, составляющей приблизительно 50 мг в сутки, или приблизительно 75 мг в сутки, или приблизительно 100 мг в сутки.
7. Фармацевтическая комбинация по любому из пп. 1–4 или фармацевтическая композиция по п. 3 или 4, где (i) соединение A вводится в суточной дозе, составляющей 100 мг (например, один раз в сутки), и соединение B вводится в дозе, составляющей 100 мг (например, один раз в сутки).
8. Фармацевтическая комбинация по любому из пп. 1-4 или фармацевтическая композиция по п. 3 или 4, где соединение A вводится в суточной дозе, составляющей 200 мг (например, один раз в сутки), и соединение B вводится в дозе, составляющей 100 мг (например, один раз в сутки).
9. Применение фармацевтической комбинации, включающей ингибитор CRAF, который представляет собой соединение A,
,
или его фармацевтически приемлемую соль, и
ингибитор ERK, который представляет собой соединение B
или его фармацевтически приемлемую соль, для лечения пролиферативного заболевания, которое выбрано из группы, состоящей из KRAS-мутантного NSCLC (немелкоклеточного рака легкого), BRAF-мутантного NSCLC, KRAS-мутантного рака поджелудочной железы, KRAS–мутантного колоректального рака (CRC) и KRAS-мутантного рака яичника.
10. Применение по п. 9, где пролиферативное заболевание представляет собой рак, который экспрессирует по меньшей мере одну мутацию, выбранную из группы, состоящей из RAF–мутаций V600E, V600D и G464E и RAS–мутаций A146T, Q61L, Q61K, G12D, G12C, G13D, G12V и G12R.
11. Способ лечения пролиферативного заболевания, которое выбрано из группы, состоящей из KRAS-мутантного NSCLC (немелкоклеточного рака легкого), BRAF-мутантного NSCLC, KRAS-мутантного рака поджелудочной железы, KRAS-мутантного колоректального рака (CRC) и KRAS-мутантного рака яичника, включающей введение фармацевтической комбинации, содержащей эффективное количество ингибитора ERK, который представляет собой соединение B,
,
или его фармацевтически приемлемой соли и эффективное количество ингибитора CRAF, который представляет собой соединение A,
или его фармацевтически приемлемой соли.
12. Способ по п. 11, где пролиферативное заболевание представляет собой рак, который экспрессирует по меньшей мере одну мутацию, выбранную из группы, состоящей из RAF-мутаций V600E, V600D и G464E и RAS-мутаций A146T, Q61L, Q61K, G12D, G12C, G13D, G12V и G12R.
| WO 2014151616 A1, 25.09.2014 | |||
| WO 2015066188 A1, 07.05.2015 | |||
| WO 2016038581 A1, 17.03.2016 | |||
| EA 201591727 A1, 31.05.2016 | |||
| WO 2015126490 A1, 07.05.2015 | |||
| WHITTAKER S | |||
| R | |||
| et al., Combined pan-RAF and MEK inhibition overcomes multiple resistance mechanisms to selective RAF inhibitors, Author Manuscript Published Online, 2015, p.1-30. |

