Настоящее изобретение относится к комбинированной терапии для лечения пациента, страдающего пролиферативным заболеванием, в частности солидной опухолью, например, колоректальным раком, меланомой и раком щитовидной железы, включающей введение пациенту {3-[5-(4-хлор-фенил)-1Н-пирроло [2,3-b] пиридин-3-карбонил]-2,4-дифтор-фенил}-амида пропан-1-сульфоновой кислоты и интерферона.
Нормально функционирующая b-Raf киназа, которая вовлечена в передачу сигналов от клеточной мембраны к ядру, активируется только при необходимости передачи таких сигналов. Однако мутантная b-Raf, имеющая мутацию V600E, активирована постоянно, вследствие чего опосредует развитие опухолей. Такая мутантная b-Raf связана с различными опухолями, например колоректальным раком, меланомой и раком щитовидной железы.
{3-[5-(4-хлор-фенил)-1Н-пирроло [2,3-b] пиридин-3-карбонил]-2,4-дифтор-фенил}-амид пропан-1-сульфоновой кислоты (далее также обозначается «Соединение I») представляет собой ингибитор b-raf киназы, специфично воздействующий на мутантную b-Raf, имеющую мутацию V600E. Это соединение описано в WO 2007/002325. Соответственно, этот ингибитор применяется для ингибирования опухолей, в частности солидных опухолей, например колоректального рака, меланомы и рака щитовидной железы, с наличием b-Raf с мутацией V600E.
Интерфероны (IFNs) представляют собой белки естественного происхождения, обладающие антивирусной, антипролиферативной и иммунорегуляторной активностью. Семейство IFNα представляет преобладающий класс интерферонов, продуцируемых активированными лейкоцитами периферической крови и клетками лимфобластоидной и миелобластоидной линий. Интерфероны подавляют экспрессию bFGF. Эти препараты могут вводиться пациентами самостоятельно путем подкожной инъекции и при этом обладают хорошей фармакокинетикой. В целях данного описания термин «интерферон» будет также использоваться для обозначения модифицированных интерферонов и/или интерферонов, полученных рекомбинантым способом, таких как пегинтерферон альфа-2а.
Пегинтерферон альфа-2а (продаваемый компанией Genentech, South San Francisco, USA под названием Pegasys®) представляет собой ковалентный конъюгат рекомбинантного альфа-2а интерферона (имеющего приблизительный молекулярный вес 20,000 дальтон) с одной цепью разветвленного бис-монометокси полиэтиленгликоля (ПЭГ) (имеющего приблизительный молекулярный вес 40,000 дальтон). Компонент ПЭГ связан в одном участке с компонентом альфа-интерферона посредством стабильной амидной связи с лизином. Пегинтерферон альфа-2a имеет приблизительный молекулярный вес 60,000 дальтон. Преимущество пегинтерферона по сравнению с другими интерферонами, не содержащими компонента ПЭГ, заключается в том, что пегинтерферон альфа-2a обладает большим периодом полужизни и требует менее частого дозирования.
Настоящее изобретение касается способа лечения пациентов, страдающих пролиферативными заболеваниями, включающего введение пациенту: (А) первого компонента, содержащего в качестве активного агента Соединение I или его фармацевтически приемлемую соль; и (Б) второй компонент, содержащий в качестве активного агента интерферон; при этом количества указанных активных агентов являются таковыми, что их комбинация является терапевтически эффективной в лечении указанного пролиферативного заболевания.
Настоящее изобретение также касается набора, содержащего: (А) первый компонент, содержащий в качестве активного агента Соединение I или его фармацевтически приемлемую соль; и (Б) второй компонент, содержащий в качестве активного агента интерферон.
Настоящее изобретение также касается композиции, содержащей: (А) первый компонент, содержащий в качестве активного агента Соединение I или его фармацевтически приемлемую соль; и (Б) второй компонент, содержащий в качестве активного агента интерферон.
Кроме того, настоящее изобретение относится к применению Соединения I или его фармацевтически приемлемой соли и интерферона для лечения пролиферативного заболевания.
Другим аспектом настоящего изобретения является применение Соединения I или его фармацевтически приемлемой соли и интерферона для изготовления лекарственного средства для лечения пролиферативного заболевания.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
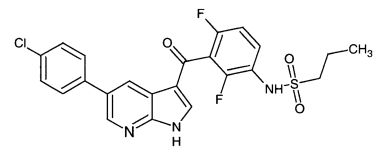
На Фигуре 1 показана переносимость, представленная в виде изменения веса тела в процентах, при монотерапии Соединением I 75 мг/кг дважды в сутки, монотерапии пегинтерфероном альфа-2a 900 мкг 1 раз в неделю и комбинированной терапии Соединением I 75 мг/кг дважды в сутки/пегинтерфероном альфа-2a 900 мкг 1 раз в неделю.
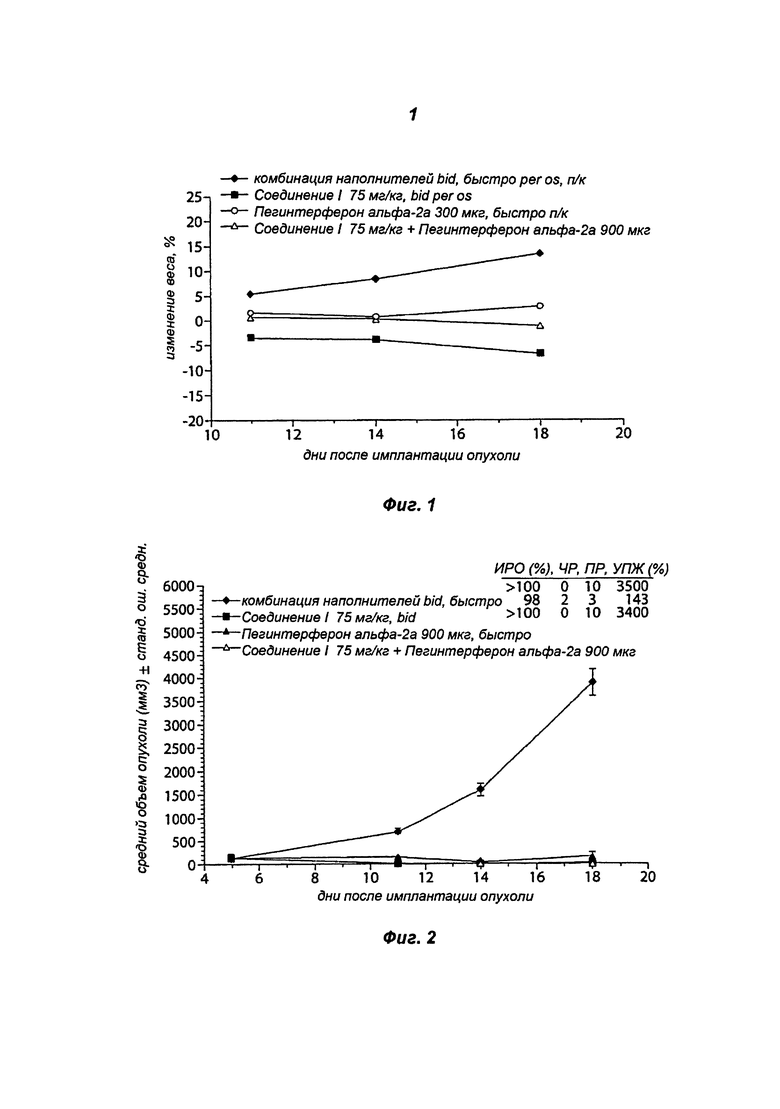
На Фигуре 2 показана противоопухолевая активность, представленная в виде объема опухоли, при монотерапии Соединением I 75 мг/мг дважды в сутки, монотерапии пегинтерфероном альфа-2а 900 мкг 1 раз в неделю и комбинированной терапии Соединением I 75 мг/кг дважды в сутки/пегинтерфероном альфа-2а 900 мкг 1 раз в неделю.
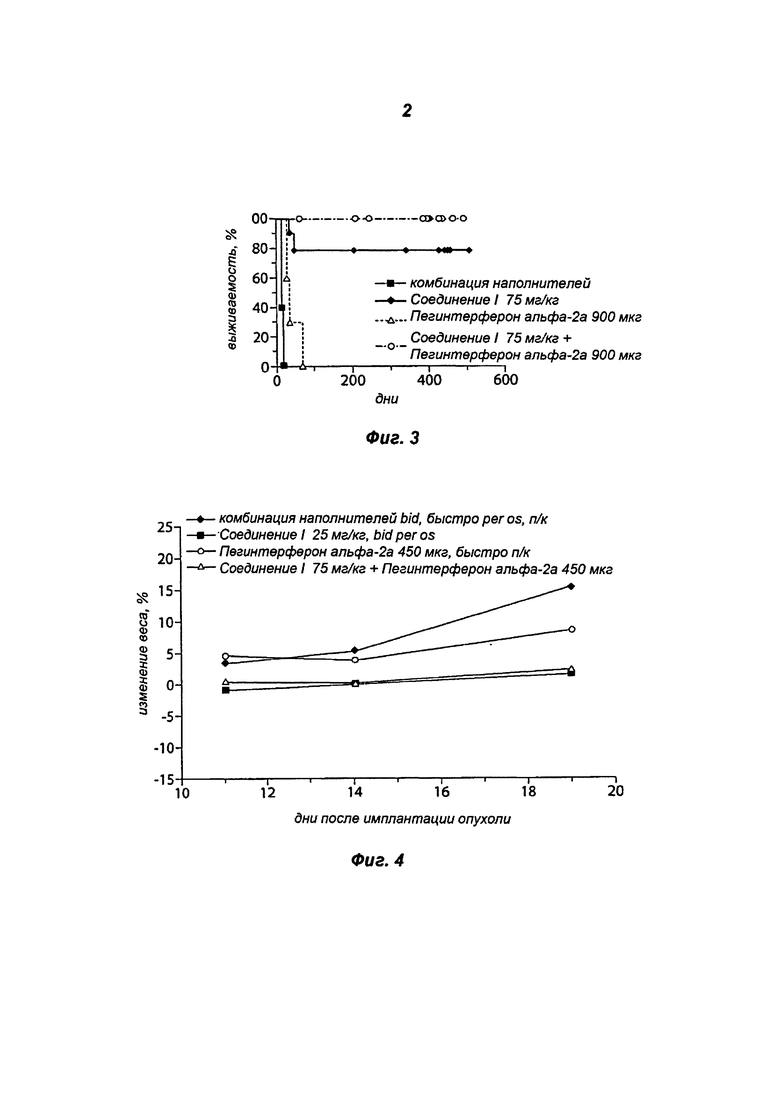
На Фигуре 3 показан эффект на выживаемость, представленный в виде процентного отношения выживших мышей с течением времени, при монотерапии Соединением I 75 мг/кг дважды в сутки, монотерапии пегинтерфероном альфа-2а 900 мкг 1 раз в неделю и комбинированной терапии Соединением I 75 мг/кг дважды в сутки/пегинтерфероном альфа-2а 900 мкг 1 раз в неделю.
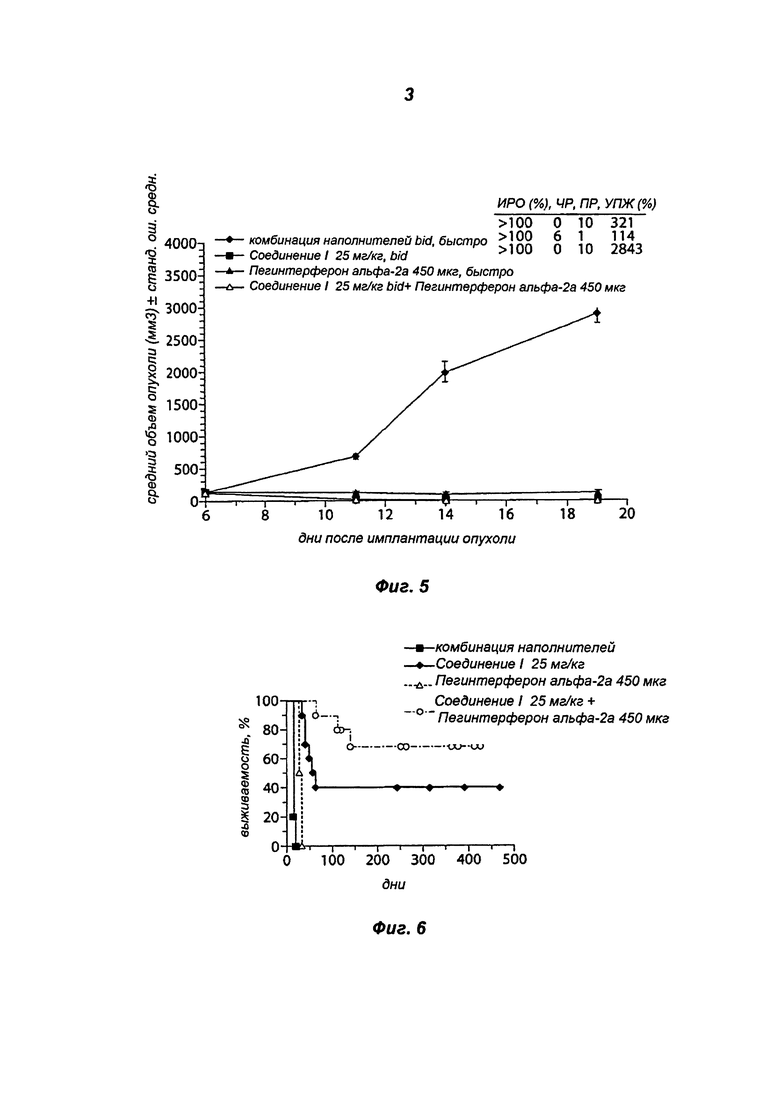
На Фигуре 4 показана переносимость, представленная в виде изменения веса тела в процентах при монотерапии Соединением I 25 мг/кг дважды в сутки, монотерапии пегинтерфероном альфа-2а 450 мкг 1 раз в неделю и комбинированной терапии Соединением I 25 мг/кг дважды в сутки/пегинтерфероном альфа-2а 450 мкг 1 раз в неделю.
На Фигуре 5 показана противоопухолевая активность, представленная в виде объема опухоли, при монотерапии Соединением I 25 мг/кг дважды в сутки, монотерапии пегинтерфероном альфа-2а 450 мкг 1 раз в неделю и комбинированной терапии Соединением I 25 мг/кг дважды в сутки/пегинтерфероном альфа-2а 450 мкг 1 раз в неделю.
На Фигуре 6 показан эффект на выживаемость, представленный в виде процентного отношения выживших мышей с течением времени, при монотерапии Соединением I 25 мг/кг дважды в сутки, монотерапии пегинтерфероном альфа-2а 450 мкг 1 раз в неделю и комбинированной терапии Соединением I 25 мг/кг дважды в сутки/пегинтерфероном альфа-2а 450 мкг 1 раз в неделю.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Как указано выше, "Соединение I" здесь обозначает {3-[5-(4-хлор-фенил)-1Н-пирроло [2,3-b] пиридин-3-карбонил-2,4-дифтор-фенил]-амид пропан-1-сульфоновой кислоты. Это соединение имеет следующую структуру.
Соединение I представляет собой ингибитор b-Raf киназы, специфично воздействующий на b-Raf с мутацией V600E.
Мутация b-Raf «V600E» здесь обозначает мутацию белка b-Raf, при которой остаток валина в положении 600 b-Raf заменен глутаминовой кислотой.
В данном документе термин «фармацевтически приемлемый носитель» указывает, что указанный носитель не имеет свойств, которые бы заставили в должной степени добросовестного практикующего врача избегать их введения пациенту, с учетом заболевания или состояний, требующих лечения, и соответствующего пути введения.
В данном документе термин «фармацевтически приемлемая соль» соединения обозначает любую стандартную соль или основно-аддитивную соль, сохраняющую биологическую эффективность и свойства соединения, которая образована при участии подходящей нетоксичной органической или неорганической кислоты или органического или неорганического основания.
В данном документе термин «терапевтически эффективный» означает количество препарата, или комбинации, или композиции, которое является эффективным для получения желаемого терапевтического эффекта при введении пациенту, например, для задержки роста или уменьшения размеров злокачественной опухоли или увеличения продолжительности жизни пациента.
Термины «заболевание с пролиферацией клеток» и «пролиферативное заболевание» обозначают заболевания, связанные в той или иной степени с аномальной пролиферацией клеток. В одном воплощении пролиферативное заболевание представляет собой рак.
Термин «рак» и «раковый» обозначают или описывают физиологическое состояние млекопитающих, которое, как правило, характеризуется неконтролируемым ростом/пролиферацией клеток. Примеры раковых заболеваний включают колоректальный рак, меланому и рак щитовидной железы, но не ограничиваются ими.
Термин «колоректальная опухоль» или «колоректальный рак» обозначает любую опухоль или рак толстого кишечника, включая толстую кишку (толстый кишечник от слепой кишки до прямой кишки) и прямую кишку, включая, например, аденокарциномы и менее распространенные формы, такие как лимфомы и плоскоклеточные карциномы.
«Ингибирование роста или пролиферации клеток» означает снижение роста или пролиферации клеток по меньшей мере на 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 95% или 100%, и включает индукцию клеточной гибели.
Словосочетание «по существу сниженный» или «по существу отличающийся» в данном документе обозначает достаточно высокую степень различия между двумя численными значениями (как правило, одно из них относится к молекуле, а другое относится к референсной молекуле/молекуле сравнения), благодаря чему любой специалист в данной области сможет считать различия между двумя значениями статистически значимыми в контексте биологических характеристик, оцениваемых при помощи указанных значений.
Термин «опухоль» обозначает все виды неопластического роста и пролиферации клеток, как злокачественные, так и доброкачественные, и все предраковые и раковые клетки и ткани. Термин «рак», «раковый», «заболевание с пролиферацией клеток», «пролиферативное заболевание» и «опухоль» в данном документе не являются взаимоисключающими.
Считается, что «регрессия» опухоли после лечения происходит, когда уменьшается объем указанной опухоли. Если опухоль продолжает существовать (объем опухоли > 0 мм3), но ее объем снижается по сравнению с ее объемом до начала лечения, считается что происходит ее «частичная регрессия» (ЧР). Если после лечения опухоль не обнаруживается при пальпации, считается что происходит ее «полная регрессия» (ПР).
В одном воплощении настоящее изобретение относится к фармацевтическому продукту, содержащему (А) первый компонент, содержащий в качестве активного агента Соединение I или его фармацевтически приемлемую соль; и (Б) второй компонент, содержащий в качестве активного агента интерферон; в качестве комбинированного препарата для одновременного или последовательного применения в лечении пролиферативного заболевания, в частности рака, более конкретно колоректального рака, меланомы и рака щитовидной железы с наличием b-Raf с мутацией V600.
Следует понимать, что лечение пролиферативного заболевания включает удерживание или снижение размера опухоли, включая регрессию опухоли (частичную либо полную), ингибирование роста опухоли и/или увеличение продолжительности жизни пациента, страдающего указанным заболеванием.
Настоящее изобретение также относится к набору или композиции, содержащей: (А) первый компонент, содержащий в качестве активного агента Соединение I или его фармацевтически приемлемую соль; и (Б) второй компонент, содержащий в качестве активного агента интерферон. Набор или композиция могут применяться, например, в лечении пролиферативного заболевания.
Кроме того, в настоящем изобретении предложено применение Соединения I или его фармацевтически приемлемой соли и интерферона для лечения пролиферативного заболевания.
В изобретении также предложено применение Соединения I или его фармацевтически приемлемой соли и интерферона для изготовления лекарственного средства для лечения пролиферативного заболевания.
В воплощении настоящего изобретения пациентом является человек.
В воплощении изобретения пролиферативное заболевание представляет собой солидную опухоль.
В другом воплощении изобретения пролиферативное заболевание представляет собой опухоль, имеющую b-Raf с мутацией V600E.
В следующем воплощении изобретения опухоль представляет собой солидную опухоль, выбранную из группы, состоящей из: колоректального рака, меланомы и рака щитовидной железы, а рак подразумевает опухоль, содержащую b-Raf с мутацией V600E.
В еще одном воплощении изобретения пролиферативное заболевание представляет собой солидную опухоль, содержащую b-Raf с мутацией V600E.
В еще одном воплощении изобретения опухоль представляет собой солидную опухоль, имеющую b-Raf с мутацией V600E, и указанная опухоль выбрана из группы, состоящей из: колоректального рака, меланомы и рака щитовидной железы.
В еще одном воплощении изобретения опухоль представляет собой меланому.
В еще одном воплощении изобретения опухоль представляет собой меланому, имеющую b-Raf с мутацией V600E.
В еще одном воплощении изобретения интерферон выбран из группы, состоящей из: пегинтерферона альфа-2a, интерферона альфа-2a, пегинтерферона альфа-2b и интерферона альфа-2b.
В еще одном воплощении изобретения интерферон представляет собой пегинтерферон альфа-2a.
В еще одном воплощении изобретения интерферон представляет собой пегинтерферон альфа-2b.
В еще одном воплощении изобретения настоящее изобретение относится к способу лечения пациента, страдающего меланомой с наличием b-Raf с мутацией V600E, включающему введение пациенту: (А) первого компонента, содержащего в качестве активного агента Соединение I или его фармацевтически приемлемую соль; и (Б) второго компонента, содержащего в качестве активного агента интерферон, предпочтительно пегинтерферон альфа-2a; количество указанных активных агентов является таковым, что их комбинация является терапевтически эффективной для лечения указанной меланомы.
Количество каждого компонента, вводимого в соответствии с данным способом, может, но не обязательно должно быть терапевтически эффективным само по себе. Иными словами, в данном изобретении, в частности, предусматриваются комбинации, в которых количество Соединения I или его фармацевтически приемлемой соли и/или количество интерферона в комбинации могут быть меньшими, чем количество, являющееся терапевтически эффективным для каждого активного агента, когда указанный агент вводят в виде монотерапии.
Соединение I или его фармацевтически приемлемую соль можно, например, вводить орально. Пегинтерферон альфа-2a можно, например, вводить подкожно.
Первый компонент и второй компонент по настоящему изобретению можно вводить в любом количестве и с любой продолжительностью, так чтобы их суммарное количество было терапевтически эффективным для лечения пролиферативного заболевания.
В воплощениях настоящего изобретения Соединение I вводят ежедневно в дозировке приблизительно от 200 мг/сут до приблизительно 3000 мг/сут, приблизительно от 800 мг/сут до приблизительно 2500 мг/сут, приблизительно от 1400 мг/сут до приблизительно 2100 мг/сут, приблизительно 960 мг/сут, приблизительно 1440 мг/сут, или приблизительно 1920 мг/сут.
В воплощении настоящего изобретения вышеупомянутые количества Соединения I можно вводить в виде однократной дозы в сутки или разделять, например, на равные дозы (хотя это не обязательно) и вводить дважды в сутки (bid). Например, Соединение I можно вводить ежедневно в дозировке приблизительно от 100 мг до приблизительно 1500 мг bid, приблизительно от 400 мг до приблизительно 1250 мг bid, приблизительно от 700 мг до приблизительно 1050 мг bid, приблизительно 480 мг bid, приблизительно 720 мг bid, или приблизительно 960 мг bid.
В воплощении настоящего изобретения введение Соединения I или его фармацевтически приемлемой соли осуществляют до наступления прогрессирования заболевания или до развития нежелательной токсичности.
В воплощении настоящего изобретения пегинтерферон альфа-2a вводят в дозировке от приблизительно 1 мкг/неделю до приблизительно 1000 мкг/неделю, от приблизительно 50 мкг/неделю до приблизительно 800 мкг/неделю, или от приблизительно 90 мкг/неделю до приблизительно 630 мкг/неделю. В другом воплощении дозировка составляет приблизительно 180 мкг/неделю.
В воплощении настоящего изобретения введение пегинтерферон альфа-2a осуществляют до наступления обострения заболевания, прогрессирования заболевания или до развития нежелательной токсичности. В другом воплощении пегинтерферон альфа-2a вводят в дозировках, описанных выше, в течение периода времени до 12 месяцев, до 24 месяцев, до 36 месяцев или до 60 месяцев.
В настоящем изобретении также предложен фармацевтический продукт, согласно описанию выше, для лечения пролиферативного заболевания, характеризующийся тем, что указанный компонент (А) вводят в количестве от приблизительно 200 мг/сут до приблизительно 3000 мг/сут, от приблизительно 800 мг/сут до приблизительно 2500 мг/сут, от приблизительно 1400 мг/сут до приблизительно 2100 мг/сут, приблизительно 960 мг/сут, приблизительно 1440 мг/сут или приблизительно 1920 мг/сут; а указанный компонент (Б) вводят в количестве от приблизительно 1 мкг/неделю до приблизительно 1000 мкг/неделю, от приблизительно 50 мкг/неделю до приблизительно 800 мкг/неделю, от приблизительно 90 мкг/неделю до приблизительно 630 мкг/неделю или приблизительно 180 мкг/неделю. В этом воплощении изобретения пролиферативное заболевание представляет собой солидную опухоль, в частности, заболевание, выбранное из группы, состоящей из: колоректального рака, меланомы и рака щитовидной железы. Более конкретно, пролиферативное заболевание подразумевает опухоль, содержащую b-Raf с мутацией V600E. В другом воплощении данного изобретения пролиферативное заболевание представляет собой меланому, содержащую b-Raf с мутацией V600E.
В настоящем изобретении также предложено применение любого фармацевтического продукта, согласно описанию выше, для производства лекарственных средств для лечения пролиферативного заболевания, характеризующегося тем, что указанный компонент (А) вводят в количестве от приблизительно 200 мг/сут до приблизительно 3000 мг/сут, от приблизительно 800 мг/сут до приблизительно 2500 мг/сут, от приблизительно 1400 мг/сут до приблизительно 2100 мг/сут, приблизительно 960 мг/сут, приблизительно 1440 мг/сут, или приблизительно 1920 мг/сут; а указанный компонент (Б) вводят в количестве от приблизительно 1 мкг/неделю до приблизительно 1000 мкг/неделю, от приблизительно 50 мкг/неделю до приблизительно 800 мкг/неделю, от приблизительно 90 мкг/неделю до приблизительно 630 мкг/неделю или приблизительно 180 мкг/неделю. В этом воплощении изобретения пролиферативное заболевание представляет собой солидную опухоль, в частности заболевание, выбранное из группы, состоящей из: колоректального рака, меланомы и рака щитовидной железы. Более конкретно, пролиферативное. заболевание подразумевает опухоль, содержащую b-Raf с мутацией V600E. В другом воплощении данного изобретения пролиферативное заболевание представляет собой меланому, содержащую b-Raf с мутацией V600E.
В настоящем изобретении также предложен способ лечения пациента, страдающего пролиферативным заболеванием, включающий введение пациенту: (А) первого компонента, включающего в качестве активного агента Соединение I в количестве от приблизительно 200 мг/сут до приблизительно 3000 мг/сут, от приблизительно 800 мг/сут до приблизительно 2500 мг/сут, от приблизительно 1400 мг/сут до приблизительно 2100 мг/сут, приблизительно 960 мг/сут, приблизительно 1440 мг/сут, или приблизительно 1920 мг/сут; и (Б) второго компонента, содержащего в качестве активного агента пегинтерферон альфа-2a в количестве от приблизительно 1 мкг/неделю до приблизительно 1000 мкг/неделю, от приблизительно 50 мкг/неделю до приблизительно 800 мкг/неделю, от приблизительно 90 мкг/неделю до приблизительно 630 мкг/неделю, или приблизительно 180 мкг/неделю. В воплощении данного изобретения пролиферативное заболевание представляет собой солидную опухоль, в частности, заболевание, выбранное из группы, состоящей из: колоректального рака, меланомы и рака щитовидной железы. В другом воплощении данного изобретения пролиферативное заболевание подразумевает опухоль, содержащую b-Raf с мутацией V600E. В конкретном воплощении данного изобретения пролиферативное заболевание представляет собой меланому, содержащую b-Raf с мутацией V600E.
В другом аспекте данного изобретения компоненты, описанные выше, вводят в сочетании с лучевой терапией и/или в сочетании с другим активным агентом.
Соединение I в обычном состоянии существует в кристаллической форме. Однако, аморфная форма соединения обладает более высокой растворимостью в воде по сравнению с кристаллической формой и, следовательно, улучшенной биодоступностью по сравнению с кристаллической формой. Таким образом, аморфная форма соединения является предпочтительной. Соответственно, в предпочтительных воплощениях данного изобретения Соединение I находится в по существу аморфной форме и, более предпочтительно, в аморфной форме. В данном документе термин «по существу аморфный» материал включает в себя материал, не более чем приблизительно 10% которого находятся в кристаллическом состоянии; а «аморфный» материал включает в себя материал, не более чем приблизительно 2% которого находятся в кристаллическом состоянии.
В воплощении настоящего изобретения Соединение I находится в виде прочного молекулярного комплекса, образованного с ацетат-сукцинатом гидроксипропилметилцеллюлозы (HPMC-AS). В данном документе «прочный молекулярный комплекс» означает композицию, в которой Соединение I случайным образом распределено («диспергировано на молекулярном уровне») в составе матрикса, образованного HPMC-AS. В некоторых воплощениях Соединение I в полимере имеет наивысшую степень дисперсности. В некоторых воплощениях Соединение I диспергировано на молекулярном уровне в составе матрикса HPMC-AS, так что оно иммобилизовано в своей аморфной форме. Под «иммобилизованным» понимают, что молекулы Соединения I взаимодействуют с молекулами HPMC-AS таким образом, что они удерживаются вышеупомянутым матриксом, и образование центров кристаллизации в них предупреждается за счет недостаточной подвижности. В некоторых воплощениях полимер может препятствовать образованию внутримолекулярных водородных связей или слабым дисперсионным взаимодействиям между двумя или более молекулами Соединения I.
В некоторых воплощениях соотношение между весовым количеством Соединения I в составе прочного молекулярного комплекса и весовым количеством HPMC-AS составляет приблизительно от 1:9 до приблизительно 5:5. В воплощении указанное соотношение составляет приблизительно от 2:8 до приблизительно 4:6. В другом воплощении указанное соотношение составляет приблизительно 3:7.
В некоторых воплощениях способа и набора по настоящему изобретению первый компонент содержит вышеупомянутый прочный молекулярный комплекс Соединения I и HPMC-AS, смешанный с коллоидным диоксидом кремния. В некоторых воплощениях смесь по меньшей мере на 0,5% по весу состоит из диоксида кремния. В воплощении данного изобретения смесь приблизительно на 97% представляет собой комплекс и приблизительно на 3% диоксид кремния.
В другом воплощении первый компонент включает композицию, содержащую вышеупомянутый прочный молекулярный комплекс, либо смешанный, либо не смешанный с диоксидом кремния, согласно описанию выше, и фармацевтически приемлемый носитель. В некоторых воплощениях вышеупомянутый комплекс или смесь, содержащая его, ресуспендированы в носителе. Примером носителя является гидроксипропилцеллюлоза (HPC). В воплощении наполнитель содержит приблизительно 2% по весу HPC.
Каждый компонент может также содержать дополнительные агенты, такие как консервирующие агенты, растворители, стабилизирующие агенты, смачивающие агенты, эмульгирующие агенты, подсластители, красители, ароматизаторы, соли для модификации осмотического давления, буферы, покровные вещества и антиоксиданты.
В некоторых воплощениях первый компонент может содержать прочный молекулярный комплекс Соединения I и HPMC-AS, смешанный с коллоидным диоксидом кремния, гидроксипропилцеллюлозой, кросповидоном (дезинтегрирующим агентом), стеаратом магния (любрикантом, который может применяться в производстве таблеток и при капсулировании), и/или кроскармеллозой натрия (дезинтегрирующим агентом).
В воплощении первый компонент представляет собой твердую желатиновую капсулу, содержащую прочный молекулярный комплекс Соединения I и HPMC-AS, смешанный с коллоидным диоксидом кремния, гидроксипропилцеллюлозой, стеаратом магния и кроскармеллозой натрия.
В воплощении первый компонент представляет собой таблетку, содержащую Соединение I или его фармацевтически приемлемую соль. В воплощении таблетка содержит прочный молекулярный комплекс Соединения I или его фармацевтически приемлемой соли и HPMC-AS. Комплекс может, например, быть смешан с коллоидным диоксидом кремния, гидроксипропилцеллюлозой, стеаратом магния и кроскармеллозой натрия. Таблетка может, например, быть покрыта пленочной оболочкой. Пленочная оболочка может, например, содержать поливиниловый спирт, диоксид титана, полиэтиленгликоль 3350, тальк и краситель оксид железа красный.
В некоторых воплощениях второй компонент может содержать пегинтерферон альфа-2a в виде инъекционного раствора.
Пегасис® имеется в продаже в виде инъекционного раствора в ампулах и предварительно заполненных шприцах. Каждая ампула 180 мкг/1,0 мл содержит приблизительно 1,2 мл раствора, обеспечивающего доставку 1,0 мл лекарственного продукта. Подкожное (п/к) введение 1,0 мл обеспечивает доставку 180 мкг лекарственного продукта (выраженное в количестве интерферона альфа-2a), 8,0 мг хлорида натрия, 0,05 мг полисорбата 80, 10,0 мг бензилового спирта, 2,62 мг натрия ацетата тригидрата и 0,0462 мг уксусной кислоты. Раствор от бесцветного до светло-желтого, имеет pH 6,0±0,5. Каждый предварительно заполненный шприц 180 мкг/0,5 мл содержит 0,6 мл раствора, обеспечивающего доставку 0,5 мл лекарственного продукта. Подкожное (п/к) введение 0,5 мл обеспечивает доставку 180 мкг лекарственного продукта (выраженное в количестве интерферона альфа-2а), 4,0 мг хлорида натрия, 0,025 мг полисорбата 80, 5,0 мг бензилового спирта, 1,3085 мг натрия ацетата тригидрата и 0,0231 мг уксусной кислоты. Раствор от бесцветного до светло-желтого, имеет pH 6,0±0,5.
Заявители провели исследования на мышах с ксенографтами меланомы человека.
Заявители обнаружили, что комбинация Соединения I по 75 мг/кг bid и пегинтерферона альфа-2a по 900 мкг 1х/нед обеспечивала существенное увеличение продолжительности жизни (УПЖ) мышей по сравнению с монотерапией пегинтерфероном альфа-2a 900 мкг 1х/нед, показатели УПЖ были статистически сопоставимы с эффектом монотерапии Соединением I по 75 мг/кг bid.
Для выявления эффекта комбинированной терапии заявители провели исследования, в которых Соединение I вводили по 25 мг/кг bid, а пегинтерферон альфа-2a вводили по 450 мкг 1х/нед. 25 мг/кг Соединения I bid позволяло достичь 321% увеличения продолжительности жизни (УПЖ) у мышей, а пегинтерферон альфа-2a по 450 мкг 1х/нед позволял достичь 114% УПЖ. Напротив, введение мышам Соединения I по 25 мг/кг bid и пегинтерферона альфа-2a по 450 мкг 1 раз в неделю в составе комбинированной терапии позволяло достичь 2843% УПЖ. Таким образом, показатель УПЖ в результате комбинированной терапии существенно превосходил соответствующие результаты монотерапии (p<0,05).
Важно отметить, что в группах, получавших комбинированную терапию, не отмечалось повышения токсичности, и между двумя агентами не наблюдалось антагонизма.
Данные исследования показали, что лечение пациентов комбинацией Соединения I и пегинтерферона альфа-2a превосходит лечение каким-либо из агентов в отдельности, и что комбинация двух агентов позволяет снизить дозу каждого агента, необходимую для получения сопоставимых или превосходящих результатов.
ОПИСАНИЕ ПРИМЕРОВ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Следующие ниже примеры дают возможность понять изобретение в более полной мере. Однако они не должны рассматриваться как ограничивающие рамки изобретения.
В документе использованы следующие сокращения:
Пример 1
В данном примере описано получение суспензии, содержащей Соединение I.
Вначале был получен прочный молекулярный комплекс, содержащий Соединение I и ацетат-сукцинат гидроксипропилметилцеллюлозы (HPMC-AS).
Соединение I и HPMC-AS в соотношении приблизительно 3:7, соответственно, растворяли в диметилацетамиде (DMA). Полученный раствор затем добавляли при перемешивании к очень холодному раствору разбавленной соляной кислоты, что приводило к ко-преципитации Соединения I и HPMC-AS в виде прочного молекулярного комплекса, в котором Соединение I входило в состав частиц, размер которых лежал в нанометровом диапазоне. Соотношение DMA к кислоте находилось в диапазоне от 1:5 до 1:10.
Затем ко-преципитат отмывали водой для удаления DMA, фильтровали, высушивали так, чтобы содержание влаги не превышало <2% и пропускали через сито # 30 перед анализом. Полученный прочный молекулярный комплекс на 30% по весу состоял из Соединения I и на 70% по весу из НРМС.
Затем комплекс смешивали с коллоидным диоксидом кремния (предоставляется под названием Aerosil® 200 компанией Evonik Industries AG, Essen, Germany) так, чтобы на 100 г смеси приходилось 97 г комплекса и 3 г коллоидного диоксида кремния.
Затем подготавливали водный наполнитель, содержащий 2% гидроксипропилцеллюлозы (предоставляется под названием Klucel® LF компанией Aqualon, Wilmington, Delaware, USA) и 1N HCL q.s. для получения pH4. 23,2 мл наполнителя уравновешивали до комнатной температуры и медленно переносили к 773,2 мг вышеупомянутой смеси. Полученный препарат затем медленно перемешивали до получения гомогенной суспензии. Суспензию хранили при 2-8°С, защищая от света.
Суспензия содержала 9,375 мг/мл Соединения I.
Пример 2
В данном примере описан инъекционный раствор пегинтерферона альфа-2a.
Раствор хранили при 2-8°C.
Пример 3
Мышам имплантировали ксенографты клеток меланомы человека LOX-IMVI. Мыши, использованная клеточная линия и процедура имплантации описаны ниже.
Для тестирования эффективности [препаратов] использовали самок бестимусных мышей Crl:NU-Foxn1nu (Charles River, Wilmington, MA, USA). Возраст мышей составлял 10-12 недель, вес 23-25 граммов. Состояние здоровья мышей оценивали ежедневно путем визуального осмотра и анализа образцов крови, которые брали у индикаторных животных, находящихся на общих полках стеллажей. Всем животным была предоставлена возможность на протяжении одной недели акклиматизироваться и восстановиться после стресса, связанного с перевозкой. Автоклавированную воду и облученный корм (5058-ms Pico Lab mouse chow, Purina Mills, Richmond, IN, USA) давали ad libitum, животных содержали при чередовании 12 часовых фаз света и темноты. Клетки, подстилки и емкости для воды перед применением автоклавировали и меняли ежедневно. Все эксперименты с животными проводили в соответствии с Руководством по содержанию и использованию лабораторных животных (Guide for the Care and Use of Laboratory Animals), местными правилами и протоколами, одобренными комитетом Roche по содержанию и использованию животных в нашем центре, аккредитованном AAALAC.
Клетки LOX-IMVI (также известные под названием LOX, National Cancer Institute - Bethesda, MD) культивировали в среде RPMI-1640 с добавлением 10% эмбриональной телячьей сыворотки (FBS) и 1% 200 нМ L-глутамина, наращивали, собирали и подготавливали таким образом, чтобы каждая мышь получала 2×106 клеток в 0,2 мл фосфатно-солевого буфера (PBS), не содержащего кальция и магния. Клетки имплантировали подкожно в правый бок каждой мыши.
Мышей с имплантированными человеческими ксенографтами рандомизировали в 8 групп по 10 мышей в каждой, в соответствии с объемом опухоли, так что во всех группах исходные средние размеры опухолей были одинаковыми. Приблизительный исходный средний объем опухоли в данном исследовании составлял 130 мм3.
Пример 4
Соединение I находилось в виде суспензии, как описано в Примере 1. Пегинтерферон альфа-2a находился в виде инъецируемого раствора, как описано в Примере 2.
Лечение начинали на 5 день после имплантации клеток и завершали на 18 день после имплантации клеток. Использовали четыре группы мышей, описанных в Примере 3. Каждая группа получала различную терапию:
(1) мыши получали носитель Соединения I bid per os и носитель пегинтерферона альфа-2a, 1х/нед п/к;
(2) мыши получали Соединение I по 75 мг/кг bid bid per os;
(3) мыши получали пегинтерферон альфа-2a по 900 мкг 1х/нед п/к; и
(4) мыши получали Соединение I по 75 мг/кг bid bid per os и пегинтерферон альфа-2a по 900 мкг 1х/нед п/к.
Суспензию Соединения I и его соответствующий носитель дозировали при помощи стерильного 1 см3 шприца и зонд-иглы 18 калибра (0,2 мл/животное) дважды в день. Раствор пегинтерферона альфа-2a и его соответствующий носитель дозировали при помощи стерильного 1 см3 шприца и иглы 26 калибра иглы (0,2 мл/животное) один раз в неделю на 5 и 12 день после имплантации клеток, т.е. суммарно две инъекции.
Опухоли измеряли один или два раза в неделю. За каждым животным в отдельности наблюдали в течение всего эксперимента.
Уменьшение веса представлено графически как изменение среднего по группе веса тела, выраженное в процентах, рассчитанное по формуле: ((W-W0)/W0)×100, где 'W' обозначает средний вес тела леченной группы в конкретный день, a 'W0' представляет средний вес тела той же леченной группы на начало эксперимента. Максимальное уменьшение веса также было рассчитано с применением указанной выше формулы и указывало максимальное уменьшение веса, выраженное в процентах, наблюдавшееся в конкретной группе в любое время на протяжении всего эксперимента.
Данные по эффективности представлены графически как средний объем опухоли ± стандартная ошибка среднего (SEM). Кроме того, объем опухолей в леченных группах выражали в процентном отношении к объему опухолей контрольных групп (%T/C), с применением формулы: 100×((T-T0)/(C-C0)), где T обозначает средний объем опухоли в леченной группе в определенный день эксперимента, T0 обозначает средний объем опухоли в той же леченной группе в первый день лечения; C обозначает средний объем опухоли в контрольной группе в определенный день эксперимента, а C0 обозначает средний объем опухоли в той же леченной группе в первый день лечения.
Объем опухоли (в кубических миллиметрах) рассчитывали с применением формулы: (D×(d2))/2, где "D" обозначает большой диаметр опухоли, a "d" обозначает малый диаметр.
Кроме того, рассчитывали регрессию опухоли и/или изменение объема опухоли в процентах при помощи формулы: ((T-T0)/T0)×100, где T обозначает средний объем опухоли в леченной группе в конкретный день, а 'T0' обозначает средний объем опухоли в той же леченной группе при начале лечения.
Статистическую обработку проводили при помощи критерия суммы рангов и однофакторного дисперсионного анализа и апостериорного t-теста Бонферрони (SigmaStat, версия 2.0, Jandel Scientific, San Francisco, CA, USA). Различия между группами считались достоверными при уровне значимости (p)≤0,05.
Для оценки выживаемости рассчитывали увеличение продолжительности жизни (УПЖ) в процентах: 100×[(медиана количества прожитых дней в леченной группе - медиана количества прожитых дней в контрольной группе)/медиана количества прожитых дней в контрольной группе]. Медиану количества прожитых дней определяли при помощи анализа кривых дожития Каплана-Мейера. Выживаемость в леченных группах сравнивали с группой, получавшей носитель, сравнение выживаемости между группами выполняли при помощи лог-рангового критерия (Graph Pad Prism, La Jolla, CA, USA). Различия между группами считали достоверными при уровне значимости (p)≤0,05.
Токсичность
Во всех описанных экспериментах ни при одной из дозировок в группах не было отмечено признаков токсичности, которую оценивали по изменению веса тела и общего осмотра отдельных животных. Эти результаты представлены в Таблице 1 и на Фиг.1.
День 18
Ингибирование роста опухоли (ИРО)
Группа, получавшая Соединение I по 75 мг/кг bid в виде монотерапии, продемонстрировала более чем 100% ИРО, в 10 случаях из 10 наблюдалась полная регрессия (ПР). Группа, получавшая пегинтерферон альфа-2a по 900 мкг 1 раз в неделю в виде монотерапии, продемонстрировала 98% ИРО, в 2 случаях из 10 наблюдалась частичная регрессия (ЧР) и в 3 из 10 - полная регрессия. Группа, получавшая Соединение I по 75 мг/кг bid и пегинтерферон альфа-2a по 900 мкг 1 раз в неделю в составе комбинированной терапии, продемонстрировала более чем 20 100% ИРО, в 10 случаях из 10 наблюдалась полная регрессия (ПР). См. Таблицы 2 и 3 и Фиг.2.
Начало исследования
День: 5
Окончание исследования
День: 18
День 18
Окончание исследования
День: 18
Окончание исследования
День: 18
%
Оценка выживаемости
Группа, получавшая Соединение I по 75 мг/кг bid в виде монотерапии, продемонстрировала 3500% увеличение продолжительности жизни (УПЖ). Группа, получавшая пегинтерферон альфа-2a по 900 мкг 1 раз в неделю в виде монотерапии, продемонстрировала 143% УПЖ. Группа, получавшая Соединение I по 75 мг/кг bid и пегинтерферон альфа-2a по 900 мкг 1 раз в неделю в составе комбинированной терапии, продемонстрировала 3400% УПЖ. См. Таблицу 4 и Фиг.3.
Дни
Дни
Статистический анализ
В группе, получавшей 75 мг/кг Соединения I bid/900 мкг пегинтерферона альфа-2a 1х/нед в составе комбинированной терапии, % ИРО было достоверно выше, чем в группе, получавшей 900 мкг пегинтерферона альфа-2a 1х/нед в виде монотерапии, но сопоставимо с группой, получавшей 75 мг/кг Соединения I bid в виде монотерапии. В группе, получавшей 75 мг/кг Соединения I bid /900 мкг пегинтерферона альфа-2a 1х/нед в составе комбинированной терапии, % УПЖ был достоверно выше, чем в группе, получавшей 900 мкг пегинтерферона альфа-2a 1х/нед в виде монотерапии, но сопоставимо с группой, получавшей 75 мг/кг Соединения I bid в виде монотерапии. См. Таблицу 5.
**критерий Бреслоу-Гехана-Вилкоксона
Пример 5
Соединение I находилось в форме суспензии, как описано в Примере 1. Наполнитель Соединения I представлял собой 2,0 грамма Klucel LF в Пегинтерфероне альфа-2a и находился в форме инъекционного раствора, как описано в Примере 2.
Терапию начинали на 6 день после имплантации клеток и завершали на 19 день после имплантации клеток. Использовали четыре группы мышей, полученных, как описано в Примере 3. Были сформированы следующие экспериментальные группы:
(1) мыши, получавшие наполнитель Соединения I bid per os и наполнитель пегинтерферона альфа-2a 1х/нед п/к;
(2) мыши, получавшие по 25 мг/кг Соединения I bid per os;
(3) мыши, получавшие по 450 мкг пегинтерферона альфа-2a 1х/нед п/к; и
(4) мыши, получавшие по 25 мг/кг Соединения I bid и по 450 мкг пегинтерферона альфа-2a 1х/нед.
Суспензию Соединения I и его соответствующий наполнитель дозировали стерильным 1 см3 шприцом и зонд-иглой 18 калибра (0,2 мл/животное) дважды в день. Раствор пегинтерферона альфа-2a и его соответствующий наполнитель дозировали при помощи стерильного 1 см3 шприца и иглы 26 калибра (0,2 мл/животное) один раз в неделю на 6 и 13 дни после имплантации клеток, т.е. суммарно две инъекции.
Опухоли измеряли один или два раза в неделю. За каждым животным в отдельности наблюдали в течение всего эксперимента.
Уменьшение веса представлено графически как изменение среднего по группе веса тела, выраженное в процентах, рассчитанное по формуле: ((W-W0)/W0)×100, где 'W' обозначает средний вес тела леченной группы в конкретный день, а 'W0' представляет средний вес тела той же леченной группы на начало эксперимента. Максимальное уменьшение веса также было рассчитано с применением указанной выше формулы и указывало максимальное уменьшение веса, выраженное в процентах, наблюдавшееся в конкретной группе в любое время на протяжении всего эксперимента.
Данные по эффективности представлены графически как средний объем опухоли ± стандартная ошибка среднего (SEM). Кроме того, объем опухолей в леченных группах выражали в процентном отношении к объему опухолей контрольных групп (%T/C), с применением формулы: 100×((T-T0)/(C-C0)), где Т обозначает средний объем опухоли в леченной группе в определенный день эксперимента, Т0 обозначает средний объем опухоли в той же леченной группе в первый день лечения; С обозначает средний объем опухоли в контрольной группе в определенный день эксперимента, а С0 обозначает средний объем опухоли в той же леченной группе в первый день лечения.
Объем опухоли (в кубических миллиметрах) рассчитывали с применением формулы: (D×(d2))/2, где "D" обозначает большой диаметр опухоли, a "d" обозначает малый диаметр.
Кроме того, рассчитывали регрессию опухоли и/или изменение объема опухоли в процентах при помощи формулы: ((T-T0)/T0)×100, где 'Т' обозначает средний объем опухоли в леченной группе в конкретный день, а 'Т0' обозначает средний объем опухоли в той же леченной группе при начале лечения.
Статистическую обработку проводили при помощи критерия суммы рангов и однофакторного дисперсионного анализа и апостериорного t-теста Бонферрони (SigmaStat, версия 2.0, Jandel Scientific, San Francisco, CA, USA). Различия между группами считались достоверными при уровне значимости (p)≤0,05.
Для оценки выживаемости рассчитывали увеличение продолжительности жизни (УПЖ) в процентах: 100×[(медиана количества прожитых дней в леченной группе - медиана количества прожитых дней в контрольной группе)/медиана количества прожитых дней в контрольной группе]. Медиану количества прожитых дней определяли при помощи анализа кривых дожития Каплана-Мейера. Выживаемость в леченных группах сравнивали с группой, получавшей наполнитель, сравнение выживаемости между группами выполняли при помощи лог-рангового критерия (Graph Pad Prism, La Jolla, CA, USA). Различия между группами считали достоверными при уровне значимости (р)≤0,05.
Токсичность
Во всех описанных экспериментах ни при одной из дозировок в группах не было отмечено признаков токсичности, которую оценивали по изменению веса тела и общего осмотра отдельных животных. Эти результаты представлены в Таблице 6 и на Фиг.4.
День 18
Ингибирование роста опухоли (ИРО)
Группа, получавшая по 25 мг/кг Соединения I bid в виде монотерапии, продемонстрировала более чем 100% ИРО, с полной регрессией в 10 случаях из 10. Группа, получавшая по 450 мкг пегинтерферона альфа-2a 1х/нед в виде монотерапии, продемонстрировала более чем 100% ИРО, с частичной регрессией в 6 случаях из 10 и полной регрессией в 1 случае из 10. Группа, получавшая по 25 мг/кг Соединения I bid и по 450 мкг пегинтерферона альфа-2a 1х/нед в виде комбинированной терапии, продемонстрировала более чем 100% ИРО, с полной регрессией (ПР) в 10 случаях из 10. См. Таблицы 7 и 8 и Фиг.5.
Начало исследования
День: 6
Окончание исследования
День: 19
День: 19
Окончание исследования
День: 19
Окончание исследования
День: 19
Оценка выживаемости
Группа, получавшая 25 мг/кг Соединения I bid в виде монотерапии, продемонстрировала 321% увеличение продолжительности жизни (УПЖ). Группа, получавшая 450 мкг пегинтерферона альфа-2a 1х/нед в виде монотерапии, продемонстрировала 114% УПЖ. Группа, получавшая 25 мг/кг Соединения I bid и 450 мкг пегинтерферона альфа-2a 1х/нед в виде комбинированной терапии, продемонстрировала 2843% УПЖ. См. Таблицу 9 и Фиг.6.
Дни
Дни
В группе, получавшей 25 мг/кг Соединения I bid/450 мкг пегинтерферона альфа-2a 1х/нед в виде комбинированной терапии, % ИРО был достоверно выше, чем в группе, получавшей 450 мкг пегинтерферона альфа-2a 1х/нед в виде монотерапии, но сопоставимо с группой, получавшей 25 мг/кг Соединения I bid в виде монотерапии. В группе, получавшей 25 мг/кг Соединения I bid/450 мкг пегинтерферона альфа-2a 1х/нед в виде комбинированной терапии, % УПЖ был достоверно выше, чем в обеих группах, получавших монотерапию. См. Таблицу 10.
**критерий Бреслоу-Гехана-Вилкоксона
| название | год | авторы | номер документа |
|---|---|---|---|
| КОМБИНИРОВАННАЯ ПРОТИВОРАКОВАЯ ТЕРАПИЯ | 2011 |
|
RU2607596C2 |
| КОМБИНИРОВАННАЯ ТЕРАПИЯ | 2018 |
|
RU2815400C2 |
| ТЕРАПЕВТИЧЕСКИЕ КОМБИНАЦИИ, СОДЕРЖАЩИЕ ИНГИБИТОР RAF И ИНГИБИТОР ERK | 2017 |
|
RU2774612C2 |
| Фармацевтические комбинации | 2017 |
|
RU2759669C2 |
| Фармацевтическая композиция для лечения рака, содержащая конъюгат интерферона-альфа | 2012 |
|
RU2622077C2 |
| КОМБИНИРОВАННАЯ ТЕРАПИЯ ДЛЯ ЛЕЧЕНИЯ ИНФЕКЦИЙ ВГВ | 2015 |
|
RU2702109C1 |
| Способ адъювантного лечения рака | 2013 |
|
RU2640180C2 |
| СОЕДИНЕНИЕ ДЛЯ ЛЕЧЕНИЯ РАКА | 2014 |
|
RU2708247C2 |
| ЛЕКАРСТВЕННЫЕ ФОРМЫ | 2005 |
|
RU2393863C2 |
| СПОСОБ ВВЕДЕНИЯ ПРОТИВООПУХОЛЕВОГО АГЕНТА | 2013 |
|
RU2638795C2 |
Группа изобретений относится к комбинированной терапии солидной опухоли или рака. Предложены: фармацевтический продукт в виде комбинированного препарата указанного назначения, содержащий в качестве активного агента {3-[5-(4-хлор-фенил)-1H-пирроло[2,3-b]пиридин-3-карбонил-2,4-дифтор-фенил]-амид}пропан-1-сульфоновой кислоты или его фармацевтически приемлемую соль и пег-интерферон альфа-2а; набор того же назначения, включающий вышеперечисленные активные агенты; применение {3-[5-(4-хлор-фенил)-1H-пирроло[2,3-b]пиридин-3-карбонил-2,4-дифтор-фенил]-амид}пропан-1-сульфоновой кислоты или его соли и пег-интерферона альфа-2а в лечении солидной опухоли или рака; применение {3-[5-(4-хлор-фенил)-1H-пирроло[2,3-b]пиридин-3-карбонил-2,4-дифтор-фенил]-амид}пропан-1-сульфоновой кислоты или его соли и пег-интерферона альфа-2а для производства лекарственного средства для лечения солидной опухоли или рака. Технический результат: увеличение продолжительности жизни больных. 4 н. и 14 з.п. ф-лы, 6 ил., 10 табл.
1. Фармацевтический продукт, содержащий (А) первый компонент, который содержит в качестве активного агента {3-[5-(4-хлор-фенил)-1Н-пирроло[2,3-b]пиридин-3-карбонил-2,4-дифтор-фенил]-амид}пропан-1-сульфоновой кислоты или его фармацевтически приемлемую соль; и (Б) второй компонент, который содержит в качестве активного агента пег-интерферон альфа-2а; в виде комбинированного препарата для одновременного или последовательного применения в лечении солидной опухоли или рака.
2. Фармацевтический продукт по п. 1 для одновременного или последовательного применения в лечении меланомы, содержащей b-Raf с мутацией V600E.
3. Фармацевтический продукт по п. 1, в котором {3-[5-(4-хлор-фенил)-1Н-пирроло[2,3-b]пиридин-3-карбонил-2,4-дифтор-фенил]-амид}пропан-1-сульфоновой кислоты или его фармацевтически приемлемая соль, по существу, находится в аморфной форме.
4. Фармацевтический продукт по п. 1, в котором {3-[5-(4-хлор-фенил)-1Н-пирроло[2,3-b]пиридин-3-карбонил-2,4-дифтор-фенил]-амид}пропан-1-сульфоновой кислоты или его фармацевтически приемлемая соль находится в аморфной форме.
5. Фармацевтический продукт по п. 1, в котором {3-[5-(4-хлор-фенил)-1Н-пирроло[2,3-b]пиридин-3-карбонил-2,4-дифтор-фенил]-амид}пропан-1-сульфоновой кислоты или его фармацевтически приемлемая соль находятся в составе прочного молекулярного комплекса, образованного с участием ацетат-сукцината гидроксипропилметилцеллюлозы, благодаря чему они иммобилизованы в своей аморфной форме.
6. Фармацевтический продукт по п. 5, в котором количество {3-[5-(4-хлор-фенил)-1Н-пирроло[2,3-b]пиридин-3-карбонил-2,4-дифтор-фенил]-амида}пропан-1-сульфоновой кислоты или его фармацевтически приемлемой соли и ацетат-сукцината гидроксипропилметилцеллюлозы в указанном комплексе находятся в соотношении от приблизительно 1:9 до приблизительно 5:5 соответственно.
7. Фармацевтический продукт по п. 5, в котором количество {3-[5-(4-хлор-фенил)-1Н-пирроло[2,3-b]пиридин-3-карбонил-2,4-дифтор-фенил]-амида}пропан-1-сульфоновой кислоты или его фармацевтически приемлемой соли и ацетат-сукцината гидроксипропилметилцеллюлозы в указанном комплексе находятся в соотношении приблизительно 3:7 соответственно.
8. Фармацевтический продукт по п. 1, в котором указанный первый компонент (А) содержит смесь, в которой приблизительно 97% смеси по весу представляет собой прочный молекулярный комплекс по п. 5 и приблизительно 3% смеси по весу представляет собой диоксид кремния.
9. Фармацевтический продукт по п. 1, в котором указанный первый компонент (А) содержит суспензию прочного молекулярного комплекса по п. 5 в фармацевтически приемлемом носителе.
10. Продукт по п. 1, в котором первый указанный компонент (А) содержит таблетку, содержащую прочный молекулярный комплекс {3-[5-(4-хлор-фенил)-1Н-пирроло[2,3-b]пиридин-3-карбонил-2,4-дифтор-фенил]-амида}пропан-1-сульфоновой кислоты или его фармацевтически приемлемую соль и HPMC-AS.
11. Продукт по п. 10, в котором указанный второй компонент (Б) содержит раствор, содержащий пег-интерферон альфа-2а.
12. Набор для лечения солидной опухоли или рака, содержащий: (А) первый компонент, содержащий в качестве активного агента {3-[5-(4-хлор-фенил)-1Н-пирроло[2,3-b]пиридин-3-карбонил-2,4-дифтор-фенил]-амид}пропан-1-сульфоновой кислоты или его фармацевтически приемлемую соль; и (Б) второй компонент, который содержит в качестве активного агента пег-интерферон альфа-2а.
13. Применение {3-[5-(4-хлор-фенил)-1Н-пирроло[2,3-b]пиридин-3-карбонил-2,4-дифтор-фенил]-амида}пропан-1-сульфоновой кислоты или его фармацевтически приемлемой соли и пег-интерферона альфа-2а в лечении солидной опухоли или рака.
14. Применение по п. 13, в котором {3-[5-(4-хлор-фенил)-1Н-пирроло[2,3-b]пиридин-3-карбонил-2,4-дифтор-фенил]-амид}пропан-1-сульфоновой кислоты или его фармацевтически приемлемую соль вводят в количестве приблизительно от 200 мг/сут до приблизительно 3000 мг/сут.
15. Применение по п. 13, в котором {3-[5-(4-хлор-фенил)-1Н-пирроло[2,3-b]пиридин-3-карбонил-2,4-дифтор-фенил]-амид}пропан-1-сульфоновой кислоты или его фармацевтически приемлемую соль вводят в количестве приблизительно от 1700 мг/сут до приблизительно 2100 мг/сут.
16. Применение по п. 13, в котором указанный пег-интерферон альфа-2а вводят в количестве приблизительно от 1 мкг/неделю до приблизительно 1000 мкг/неделю.
17. Применение по п. 16, в котором указанный пег-интерферон альфа-2а вводят в количестве приблизительно от 90 мкг/неделю до приблизительно 630 мкг/неделю.
18. Применение {3-[5-(4-хлор-фенил)-1Н-пирроло[2,3-b]пиридин-3-карбонил-2,4-дифтор-фенил]-амида}пропан-1-сульфоновой кислоты или его фармацевтически приемлемой соли и пег-интерферона альфа-2а для изготовления лекарственного средства для лечения солидной опухоли или рака.
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| УСТРОЙСТВО ДЛЯ ИЗМЕРЕНИЯ СКРУЧИВАНИЯ ВРАЩАЮЩИХСЯ ВАЛОВ | 1928 |
|
SU8766A1 |
| Автоматическая масленка для крейцкопфов | 1928 |
|
SU9668A1 |
| СПОСОБ ЛЕЧЕНИЯ ПОЧЕЧНО-КЛЕТОЧНОГО РАКА | 2001 |
|
RU2179859C1 |
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| В.В.МАЙСКИЙ Элементарная фармакология М., Фарма Диалог 2009 с.391-392 статья Препараты интерферонов. | |||

