ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
[0001] Настоящая заявка испрашивает приоритет предварительной заявки на патент США 62/345,389, поданной 3 июня 2016 года, которая полностью включена в настоящий документ посредством ссылки.
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
[0002] Настоящее изобретение относится к фармацевтической комбинации, независимо содержащей терапевтически эффективные количества: (a) ингибитора BRAF или его фармацевтически приемлемой соли, (b) по меньшей мере одного ингибитора митоген-активируемой протеинкиназы (MEK) или его фармацевтически приемлемой соли и (c) ингибитора рецептора эпидермального фактора роста (EGFR) или его фармацевтически приемлемой соли; и, необязательно, по меньшей мере один фармацевтически приемлемый носитель; способам получения фармацевтических комбинаций и применениям фармацевтических комбинаций в лечении пролиферативных заболеваний, таких как рак.
[0003] Настоящее изобретение также относится к фармацевтической комбинации, независимо содержащей терапевтически эффективные количества: (a) ингибитора BRAF или его фармацевтически приемлемой соли, (b) по меньшей мере одного ингибитора митоген-активируемой протеинкиназы (MEK) или его фармацевтически приемлемой соли и (c) антитела против EGFR, и, необязательно, по меньшей мере один фармацевтически приемлемый носитель; применениям такой комбинации в лечении пролиферативных заболеваний, таких как рак; и способам лечения субъекта, страдающего пролиферативным заболеванием, таким как рак, включающим введение терапевтически эффективного количества такой комбинации.
УРОВЕНЬ ТЕХНИКИ
[0004] Путь митоген-активируемой протеинкиназы (MAPK) опосредует активность ряда эффекторных молекул, которые согласованно осуществляют регуляцию клеточной пролиферации, выживания, дифференцировки и миграции. Стимуляция клеток, например, факторами роста, цитокинами или гормонами приводит к тому, что Ras, ассоциированная с плазматической мембраной, связывается с ГТФ и, таким образом, активируется, вызывая рекрутинг RAF. Это взаимодействие индуцирует киназную активность RAF, что приводит к прямому фосфорилированию MAPK/ERK (MEK), которая, в свою очередь, фосфорилирует киназу, связанную с внеклеточными сигналами (ERK). Затем активированная ERK фосфорилирует широкий спектр эффекторных молекул, например, киназы, фосфатазы, факторы транскрипции и цитоскелетные белки. Таким образом, сигнальный путь RAS-RAF-MEK-ERK передает сигналы от рецепторов клеточной поверхности к ядру и важен, например, для пролиферации и выживания клеток. Регуляция этого сигнального каскада также дополняется множеством изоформ RAS (включая KRAF, NRAS и HRAS), RAF (ARAF, BRAF, CRAF/RAF-1), MEK (MEK-1 и MEK-2) и ERK (ERK-1 и ERK-2). Исследования показали, что данный путь регулирует несколько ключевых клеточных активностей, включая пролиферацию, дифференцировку, выживание и ангиогенез. Было показано, что неправильная активация белков по этому пути встречается при многих формах рака, таких как меланома, немелкоклеточный рак легкого, рак толстой и прямой кишки и рак щитовидной железы. Поскольку 10-20% раковых опухолей человека содержат онкогенные мутации Ras, и во многих раковых опухолях человека активированы рецепторы факторов роста, этот путь является идеальной мишенью для вмешательства.
[0005] Важная роль и положение RAF во многих сигнальных путях были продемонстрированы в исследованиях с использованием дерегулированных и доминантных ингибирующих мутантов RAF в клетках млекопитающих, а также в исследованиях с использованием биохимических и генетических методик моделирования организмов. Ранее внимание к RAF как противоопухолевому средству было сосредоточено на его функции в качестве нижестоящего эффектора RAS. Однако последние данные свидетельствуют о том, что RAF может играть заметную роль в формировании некоторых опухолей без необходимости в онкогенном аллеле Ras. В частности, активирующие аллели BRAF и NRAS были обнаружены приблизительно в 70% меланом, 40% папиллярных карцином щитовидной железы, 30% карцином яичников низкой степени злокачественности и в 10% случаев рака толстой и прямой кишки. Мутации в K-Ras встречаются примерно в 90% случаев рака поджелудочной железы. Большинство мутаций BRAF присутствуют в киназном домене, при этом одна замена (V600E) составляет не менее 80%. Мутантные белки BRAF активируют путь RAF-MEK-ERK либо посредством повышенной киназной активности по отношению к MEK, либо в результате активации CRAF. Данные показывают, что ингибиторы Raf-киназы могут в значительной степени ингибировать сигнализацию по пути MAPK, что приводит к резкому уменьшению опухолей BRAF (V600E). Рак толстой и прямой кишки является третьим по распространенности раком у мужчин и женщин в США, с более чем 134000 новыми случаями и почти 50000 случаев смерти от этого заболевания, спрогнозированными в 2016 году. В США мутации BRAF встречаются у 8-15% больных раком толстой и прямой кишки и представляют плохой прогноз для этих пациентов. Опубликованные ретроспективные результаты по выживаемости без прогрессирования заболевания (PFS) и общей выживаемости (OS) после лечения препаратами первой линии колеблются от 1,8 до 2,5 месяцев и от 4 до 6 месяцев, соответственно, и опубликованные данные по проценту ответа в различных исследованиях терапии на основе EGFR в этой совокупности пациентов колеблются от 6 до 8 процентов. Несмотря на значительный прогресс в лечении метастатического рака толстой и прямой кишки, в течение последних 2 десятилетий прогноз у пациентов с метастатическим раком толстой и прямой кишки (mCRC) остается неутешительным. Системная химиотерапия продолжает оставаться основным методом лечения пациентов с mCRC (James J. Lee, MD, PhD, and Weijing Sun, MD, Clinical Advances in Hematology & Oncology, January 2016, Vol 14, Issue 1). Управление по контролю качества пищевых продуктов и лекарственных препаратов США (FDA) одобрило несколько цитотоксических средств и средств направленного действия для лечения mCRC, включающих иринотекан, оксалиплатин и капецитабин (S-1 был одобрен в Японии и ряде других стран, но не в США). Комбинация фторпиримидина (5-фторурацила [5-ФУ] или перорального капецитабина) с оксалиплатином или иринотеканом широко применяется в качестве стандартной цитотоксической химиотерапии при mCRC, в качестве терапии первого или второго ряда. Эти схемы включают фолиевую кислоту/5-ФУ/оксалиплатин (FOLFOX), капецитабин/оксалиплатин (XELOX), фолиновую кислоту/5-ФУ/иринотекан (FOLFIRI) и капецитабин/иринотекан (XELIRI). Недавно, в сентябре 2015 года, FDA одобрило комбинацию трифлуридина и типирацила (Lonsurf, Taiho Oncology) для примнения при рефрактерном mCRC (James J. Lee, MD, PhD, and Weijing Sun, MD, Clinical Advances in Hematology & Oncology, January 2016, Vol 14, Issue 1).
[0006] Рецепторы EGFR - трансмембранные рецепторы, присутствующие на клеточных мембранах. Они имеют внеклеточный связывающий компонент, трансмембранный компонент и внутриклеточный тирозинкиназный компонент. EGFR играют важную роль в регуляции нормального роста клеток, апоптоза и других клеточных функций. Нарушение активности EGFR может привести к постоянной или патологической активации рецепторов, вызывающей нерегулируемое деление клеток. В уровне техники известны ингибиторы рецептора эпидермального фактора роста. На EGFR воздействуют две группы лекарственных средств: моноклональные антитела и ингибиторы тирозинкиназы (TKI). Примеры моноклональных антител включают панитумумаб и цетуксимаб, и их механизм действия заключается во внеклеточном связывании с последующим ингибированием сигнальных путей EGFR. Примеры ингибиторов тирозинкиназы включают эрлотиниб, гефитиниб и лапатиниб, и их механизм действия заключается во внутриклеточном связывании и последующем ингибировании сигнальных путей EGFR.
[0007] Несмотря на многочисленные варианты лечения пациентов с онкологическими заболеваниями, сохраняется потребность в эффективных и безопасных терапевтических средствах и в новых способах комбинированной терапии, которые можно применять для эффективного долговременного лечения рака.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
[0008] В настоящем документе предложена фармацевтическая комбинация, независимо содержащая терапевтически эффективные количества: (a) ингибитора BRAF или его фармацевтически приемлемой соли, (b) по меньшей мере одного ингибитора митоген-активируемой протеинкиназы (МЕК) или его фармацевтически приемлемой соли, и (c) ингибитора рецептора эпидермального фактора роста (EGFR) или его фармацевтически приемлемой соли; и, необязательно, по меньшей мере, один фармацевтически приемлемый носитель.
[0009] Также в настоящем документе предложена фармацевтическая комбинация, независимо содержащая терапевтически эффективные количества: (a) ингибитора BRAF или его фармацевтически приемлемой соли, (b) по меньшей мере одного ингибитора MEK или его фармацевтически приемлемой соли и (c) антитела против EGFR; и, необязательно, по меньшей мере один фармацевтически приемлемый носитель.
[0010] Также в настоящем документе предложена фармацевтическая комбинация, независимо содержащая терапевтически эффективные количества: (a) ингибитора BRAF, которым является метил-N-[(2S)-1-({4-[3-(5-хлор-2-фтор-3-метансульфонамидофенил)-1-(пропан-2-ил)-1H-пиразол-4-ил]пиримидин-2-ил}амино)пропан-2-ил]карбамат или его фармацевтически приемлемая соль, (b) по меньшей мере одного ингибитора MEK или его фармацевтически приемлемой соли и (c) антитела против EGFR, и, необязательно, по меньшей мере один фармацевтически приемлемый носитель. В одном варианте осуществления ингибитор BRAF, ингибитор MEK и антитело против EGFR, включены в отдельные стандартные лекарственные формы для одновременного, раздельного или последовательного введения.
[0011] Также в настоящем документе предложена фармацевтическая комбинация, независимо содержащая терапевтически эффективные количества: (a) ингибитора BRAF, которым является метил-N-[(2S)-1-({4-[3-(5-хлор-2-фтор-3-метансульфонамидофенил)-1-(пропан-2-ил)-1H-пиразол-4-ил]пиримидин-2-ил}амино)пропан-2-ил]карбамат или его фармацевтически приемлемая соль, (b) по меньшей мере одного ингибитора MEK, где по меньшей мере одним из указанных ингибиторов MEK является (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты или его фармацевтически приемлемая соль и (c) антитела против EGFR, и, необязательно, по меньшей мере один фармацевтически приемлемый носитель. В одном варианте осуществления ингибитор BRAF, ингибитор MEK и антитело против EGFR сформулированы в виде единичных стандартных лекарственных форм для одновременного, раздельного или последовательного введения.
[0012] Также в настоящем документе предложена фармацевтическая комбинация, независимо содержащая терапевтически эффективные количества: (a) ингибитора BRAF, которым является метил-N-[(2S)-1-({4-[3-(5-хлор-2-фтор-3-метансульфонамидофенил)-1-(пропан-2-ил)-1H-пиразол-4-ил]пиримидин-2-ил}амино)пропан-2-ил]карбамат или его фармацевтически приемлемая соль, (b) ингибитора MEK, которым является (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты или его фармацевтически приемлемая соль и (c) антитела против EGFR, которым является цетуксимаб, и, необязательно, по меньшей мере один фармацевтически приемлемый носитель. В одном варианте осуществления ингибитор BRAF, ингибитор MEK и антитело против EGFR сформулированы в виде единичных стандартных лекарственных форм для одновременного, раздельного или последовательного введения.
[0013] Также предложены способы лечения пролиферативного заболевания, включающие введение нуждающемуся в этом пациенту терапевтически эффективного количества фармацевтической комбинации, описанной в настоящем документе.
[0014] Также предложена фармацевтическая комбинация, независимо содержащая терапевтически эффективные количества: (a) ингибитора BRAF, которым является метил-N-[(2S)-1-({4-[3-(5-хлор-2-фтор-3-метансульфонамидофенил)-1-(пропан-2-ил)-1H-пиразол-4-ил]пиримидин-2-ил}амино)пропан-2-ил]карбамат или его фармацевтически приемлемая соль, (b) по меньшей мере одного ингибитора MEK и (c) ингибирующего антитела против EGFR, и, необязательно, по меньшей мере один фармацевтически приемлемый носитель, для применения в лечении пролиферативного заболевания. В одном варианте осуществления ингибитором MEK является (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты или его фармацевтически приемлемая соль. В одном варианте осуществления ингибиующим антителом против EGFR является цетуксимаб.
[0015] Также предложен способ уменьшения размера опухоли, включающий контакт опухоли с фармацевтической комбинацией согласно изобретению.
[0016] Также предложены способы лечения рака у нуждающегося в этом пациента, включающие: (a) определение, является ли рак у пациента BRAF-ассоциированным раком; и (b) если рак определен как BRAF-ассоциированный рак, введение пациенту терапевтически эффективного количества фармацевтической комбинации согласно изобретению.
[0017] Также предложен способ лечения рака у нуждающегося в этом пациента, включающий: (a) определение, имеет ли рак мутантную BRAF-киназу, и (b) если определено, что рак имеет мутантную BRAF-киназу, введение указанному субъекту терапевтически эффективного количества фармацевтической комбинации согласно изобретению.
[0018] Также предложен способ лечения рака у нуждающегося в этом пациента, включающий: (a) обнаружение мутантной BRAF-киназы при раке и (b) введение указанному субъекту терапевтически эффективного количества фармацевтической комбинации согласно изобретению.
[0019] В одном варианте осуществления в настоящем документе предложены способы лечения пациента с раком толстой и прямой кишки, которому назначено лечение цетуксимабом, включающие введение указанному пациенту: (а) терапевтически эффективного количества ингибитора BRAF, которым является метил-N-[(2S)-1-({4-[3-(5-хлор-2-фтор-3-метансульфонамидофенил)-1-(пропан-2-ил)-1H-пиразол-4-ил]пиримидин-2-ил}амино)пропан-2-ил]карбамат (СОЕДИНЕНИЕ A) или его фармацевтически приемлемая соль, и (b) терапевтически эффективного количества ингибитора MEK (СОЕДИНЕНИЯ B) или его фармацевтически приемлемой соли.
[0020] В одном варианте осуществления в настоящем документе предложены способы лечения пациента, имеющего рак толстой и прямой кишки, где указанный способ включает: a) ежедневное введение первой дозы: (a) терапевтически эффективного количества ингибитора BRAF, которым является метил-N-[(2S)-1-({4-[3-(5-хлор-2-фтор-3-метансульфонамидофенил)-1-(пропан-2-ил)-1H-пиразол-4-ил]пиримидин-2-ил}амино)пропан-2-ил]карбамат (СОЕДИНЕНИЕ A) или его фармацевтически приемлемая соль, и (b) терапевтически эффективного количества ингибитора MEK (СОЕДИНЕНИЯ B) или его фармацевтически приемлемой соли; b) через по меньшей мере 30 минут после этапа a), введение первого терапевтически эффективного количества дозы цетуксимаба; c) ежедневное введение второй дозы указанного СОЕДИНЕНИЯ B, где указанную вторую дозу вводят через 10-14 часов после введения указанной первой дозы СОЕДИНЕНИЯ B; и d) еженедельное введение второго терапевтически эффективного количества дозы цетуксимаба, где указанное введение указанного второго количества дозы цетуксимаба производят через одну неделю после введения указанного первого количества дозы цетуксимаба.
[0021] Также предложены способы (например, способы in vitro) подбора лечения для пациента, у которого идентифицировано или диагностировано наличие BRAF-ассоциированного рака.
[0022] Также в настоящем документе предложены способы подбора лечения для пациента, где способы включают этап проведения анализа образца, полученного от пациента, с целью определения, имеет ли пациент дисрегуляцию гена BRAF, BRAF-киназы или экспрессии или активности, или уровня любого из них, и идентификации или диагностики пациента, у которого определена наличие дисрегуляции гена BRAF, BRAF-киназы или экспрессии или активности, или уровня любого из них, как имеющего BRAF-ассоциированный рак.
[0023] Также предложены способы отбора пациента для лечения, где способы включают отбор, идентификацию или диагностику пациента, имеющего BRAF-ассоциированный рак, и отбор пациента для лечения, включающего введение терапевтически эффективного количества фармацевтической комбинации согласно изобретению.
[0024] Также предложены способы определения вероятности того, что субъект, имеющий рак, будет иметь положительный ответ на лечение фармацевтической комбинацией согласно изобретению, включающие: определение, имеет ли раковая клетка в образце, полученном от субъекта, одну или более мутаций BRAF; и определение того, что субъект, имеющий раковую клетку, которая имеет одну или более мутаций BRAF, имеет повышенную вероятность положительного ответа на лечение фармацевтической комбинацией согласно изобретению.
[0025] Также предложены способы прогнозирования эффективности лечения фармацевтической комбинацией согласно изобретению у субъекта, имеющего рак, включающие: определение, имеет ли раковая клетка в образце, полученном от субъекта, одну или более мутаций BRaf; и определение, что лечение фармацевтической комбинацией согласно изобретению с большей вероятностью будет эффективным у субъекта, имеющего раковую клетку в образце, полученном от субъекта, который имеет одну или более мутаций BRAF.
[0026] Также предложена коммерческая упаковка, содержащая в качестве терапевтических средств комбинацию, содержащую: (a) ингибитор BRAF, которым является метил-N-[(2S)-1-({4-[3-(5-хлор-2-фтор-3-метансульфонамидофенил)-1-(пропан-2-ил)-1H-пиразол-4-ил]пиримидин-2-ил}амино)пропан-2-ил]карбамат или его фармацевтически приемлемая соль, (b) по меньшей мере один ингибитор MEK и (c) антитело против EGFR, и, необязательно, по меньшей мере один фармацевтически приемлемый носитель, вместе с инструкциями по их одновременному, раздельному или последовательному введению для применения в лечении пролиферативного заболевания. В одном варианте осуществления ингибитором MEK является (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты или его фармацевтически приемлемая соль. В одном варианте осуществления ингибирующим антителом против EGFR является цетуксимаб.
[0027] Также предложена коммерческая упаковка, включающая: (a) ингибитор BRAF, которым является метил-N-[(2S)-1-({4-[3-(5-хлор-2-фтор-3-метансульфонамидофенил)-1-(пропан-2-ил)-1H-пиразол-4-ил]пиримидин-2-ил}амино)пропан-2-ил]карбамат или его фармацевтически приемлемая соль, (b) по меньшей мере один ингибитор MEK, вместе с инструкциями по их одновременному, раздельному или последовательному введению вместе с (СОЕДИНЕНИЕМ C) при лечении пролиферативного заболевания. В одном варианте осуществления ингибитором MEK является (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты или его фармацевтически приемлемая соль. В одном варианте осуществления ингибирующим антителом против EGFR является цетуксимаб. В некоторых вариантах осуществления в настоящем документе предложены способы снижения токсичности (например, кожной токсичности, желудочно-кишечной токсичности, миелосупрессии, кардиотоксичности, нейроцеребральной токсичности или фототоксичности) комбинации биниметиниба и цетуксимаба у субъекта, включающие введение энкорафениба в комбинации с биниметинибом и цетуксимабом. В некоторых таких вариантах осуществления субъект выбран как имеющий мутацию BRAF V600 (например, V600E).
[0028] В некоторых вариантах осуществления в настоящем документе предложены способы снижения токсичности (например, кожной токсичности, желудочно-кишечной токсичности, миелосупрессии, кардиотоксичности, нейроцеребральной токсичности или фототоксичности) комбинации энкорафениба и цетуксимаба у субъекта, включающие введение биниметиниба в комбинации с энкорафенибом и цетуксимабом. В некоторых таких вариантах осуществления субъект выбран как имеющий мутацию BRAF V600 (например, V600E).
[0029] В некоторых вариантах осуществления в настоящем документе предложены способы снижения нежелательных явлений (например, снижения остроты зрения, диареи, ладонно-подошвенного синдрома, раздражения кожи (например, сыпи, зудящих поражений, трещин, шелушения), остановки сердца, отека, усталости, астении, гипертензии, пневмонита, тромбоза (например, тромбоза легких), острой почечной недостаточности, пареза лица, головной боли или вторичных новообразований), связанных с первым терапевтическим средством, выбранным из группы, состоящей из энкорафениба, цетуксимаба или биниметиниба, у субъекта, включающие введение двух других терапевтических средств, выбранных не из группы первого терапевтического средства, в комбинации с первым терапевтическим средством. В некоторых таких вариантах осуществления первым терапевтическим средством является энкорафениб, а двумя другими терапевтическими средствами являются цетуксимаб и биниметиниб. В некоторых таких вариантах осуществления первым терапевтическим средством является цетуксимаб, а двумя другими терапевтическими средствами являются энкорафениб и биниметиниб. В некоторых таких вариантах осуществления первым терапевтическим средством является биниметиниб, а двумя другими терапевтическими средствами являются цетуксимаб и энкорафениб. В некоторых таких вариантах осуществления субъект выбран как имеющий мутацию BRAF V600 (например, V600E).
[0030] В некоторых вариантах осуществления в настоящем документе предложены способы снижения одного или более нежелательных явлений (например, кожной токсичности, ладонно-подошвенного синдрома (ЛПС), сыпи, акнеформной сыпи, дерматита, ретинопатии или отслоения сетчатки), связанных с противоопухолевой терапевтической схемой у субъекта, идентифицированного как имеющего BRAF V600E рак толстой и прямой кишки, включающие: включение биниметиниба в схему лечения рака, которая включает энкорафениб и антитело против EGFR. В некоторых таких вариантах осуществления антитело против EGFR выбрано из группы, состоящей из цетуксимаба, гефитиниба, эрлотиниба, лапатиниба, дакомитиниба, нератиниба, вандетаниба и панитумумаба. В некоторых таких вариантах осуществления антителом против EGFR является цетуксимаб. В некоторых таких вариантах осуществления субъект выбран как имеющий мутацию BRAF V600 (например, V600E).
[0031] В некоторых вариантах осуществления в настоящем документе предложены способы снижения одного или более нежелательных явлений (например, кожной токсичности, ладонно-подошвенного синдрома (ЛПС), сыпи, акнеформной сыпи, дерматита, ретинопатии или отслоения сетчатки), связанных с противоопухолевой терапевтической схемой у субъекта, идентифицированного как имеющего BRAF V600E рак толстой и прямой кишки, включающие: включение энкорафениба в схему лечения рака, которая включает биниметиниб и антитело против EGFR. В некоторых таких вариантах осуществления антитело против EGFR выбрано из группы, состоящей из цетуксимаба, гефитиниба, эрлотиниба, лапатиниба, дакомитиниба, нератиниба, вандетаниба и панитумумаба. В некоторых таких вариантах осуществления антителом против EGFR является цетуксимаб. В некоторых таких вариантах осуществления субъект выбран как имеющий мутацию BRAF V600 (например, V600E).
[0032] В некоторых вариантах осуществления в настоящем документе предложены способы повышения безопасной дозы энкорафениба у субъекта, включающие введение энкорафениба в комбинации с биниметинибом и цетуксимабом. В некоторых таких вариантах осуществления субъект выбран как имеющий мутацию BRAF V600 (например, V600E).
[0033] В некоторых вариантах осуществления в настоящем документе предложены способы повышения безопасной дозы биниметиниба у субъекта, включающие введение биниметиниба в комбинации с энкорафенибом и цетуксимабом. В некоторых таких вариантах осуществления субъект выбран как имеющий мутацию BRAF V600 (например, V600E).
[0034] В некоторых вариантах осуществления в настоящем документе предложены способы повышения безопасной дозы цетуксимаба у субъекта, включающие введение цетуксимаба в комбинации с биниметинибом и энкорафенибом. В некоторых таких вариантах осуществления субъект выбран как имеющий мутацию BRAF V600 (например, V600E).
[0035] В некоторых вариантах осуществления в настоящем документе предложены способы повышения фармакологической эффективности комбинации биниметиниба и цетуксимаба у субъекта, включающие введение энкорафениба в сочетании с биниметинибом и цетуксимабом. В некоторых таких вариантах осуществления повышение фармакологической эффективности определяют с помощью магнитно-резонансной томографии (МРТ) (например, МРТ всего тела), компьютерной томографии, рентгеновского исследования, остеосцинтиграфии с Tc99m, позитронно-эмиссионной томографии с фтордезоксиглюкозой (ФДГ-ПЭТ) или позитронно-эмиссионной томографии с фторидом натрия (NaF-ПЭТ). В некоторых таких вариантах осуществления субъект выбран как имеющий мутацию BRAF V600 (например, V600E).
[0036] В некоторых вариантах осуществления в настоящем документе предложены способы повышения фармакологической эффективности комбинации энкорафениба и цетуксимаба у субъекта, включающие введение биниметиниба в сочетании с энкорафенибом и цетуксимабом. В некоторых таких вариантах осуществления повышение фармакологической эффективности определяют с помощью магнитно-резонансной томографией (МРТ) (например, МРТ всего тела), компьютерной томографии, рентгеновского исследования, остеосцинтиграфии с Tc99m, позитронно-эмиссионной томографии с фтордезоксиглюкозой (ФДГ-ПЭТ) или позитронно-эмиссионной томографии с фторидом натрия (NaF-ПЭТ). В некоторых таких вариантах осуществления субъект выбран как имеющий мутацию BRAF V600 (например, V600E).
[0037] В некоторых вариантах осуществления в настоящем документе предложены способы повышения фармакологической эффективности комбинации энкорафениба и биниметиниба у субъекта, включающие введение цетуксимаба в сочетании с энкорафенибом и биниметинибом. В некоторых таких вариантах осуществления повышение фармакологической эффективности определяют с помощью магнитно-резонансной томографии (МРТ) (например, МРТ всего тела), компьютерной томографии, рентгеновского исследования, остеосцинтиграфии с Tc99m, позитронно-эмиссионной томографии с фтордезоксиглюкозой (ФДГ-ПЭТ) или позитронно-эмиссионной томографии с фторидом натрия (NaF-ПЭТ). В некоторых таких вариантах осуществления субъект выбран как имеющий мутацию BRAF V600 (например, V600E).
[0038] В некоторых вариантах осуществления в настоящем документе предложены способы повышения максимальной переносимой дозы (МПД) энкорафениба у субъекта, проходящего лечение рака энкорафенибом, включающие введение субъекту энкорафениба в комбинации с ингибитором MEK. В некоторых таких вариантах осуществления максимальная переносимая доза составляет больше чем приблизительно 50 мг (например, больше чем приблизительно 100 мг, больше чем приблизительно 200 мг, больше чем приблизительно 300 мг, больше чем приблизительно 400 мг, больше чем приблизительно 500 мг, больше чем приблизительно 600 мг или больше чем приблизительно 700 мг, или больше чем приблизительно 800 мг). В некоторых таких вариантах осуществления максимальная переносимая доза составляет приблизительно 50 мг (например, приблизительно 100 мг, 200 мг, 300 мг, 400 мг, 500 мг, 600 мг, 700 мг или 800 мг). В некоторых таких вариантах осуществления рак связан с BRAF мутантом V600E. В некоторых таких вариантах осуществления рак является раком толстой и прямой кишки (например, метастатическим раком толстой и прямой кишки). В некоторых таких вариантах осуществления ингибитор MEK выбран из группы, состоящей из биниметиниба, траметиниба, кобиметиниба, селуметиниба и PD-325901 (например, биниметиниба). В некоторых таких вариантах осуществления субъект выбран как имеющий мутацию BRAF V600 (например, V600E).
[0039] В некоторых вариантах осуществления в настоящем документе предложены способы повышения максимальной переносимой дозы (МПД) энкорафениба у субъекта, проходящего лечение рака энкорафенибом, включающие введение субъекту энкорафениба в комбинации с ингибитором EGFR. В некоторых таких вариантах осуществления максимальная переносимая доза составляет больше чем приблизительно 50 мг (например, больше чем приблизительно 100 мг, больше чем приблизительно 200 мг, больше чем приблизительно 300 мг, больше чем приблизительно 400 мг, больше чем приблизительно 500 мг, больше чем приблизительно 600 мг, больше чем приблизительно 700 мг или больше чем приблизительно 800 мг). В некоторых таких вариантах осуществления максимальная переносимая доза составляет приблизительно 50 мг (например, приблизительно 100 мг, 200 мг, 300 мг, 400 мг, 500 мг, 600 мг, 700 мг или 800 мг). В некоторых таких вариантах осуществления рак связан с BRAF мутантом V600E. В некоторых таких вариантах осуществления рак является раком толстой и прямой кишки (например, метастатическим раком толстой и прямой кишки). В некоторых таких вариантах осуществления ингибитор EGFR выбран из группы, состоящей из цетуксимаба, гефитиниба, эрлотиниба, лапатиниба, дакомитиниба, нератиниба, вандетаниба и панитумумаба (например, цетуксимаба). В некоторых таких вариантах осуществления субъект выбран как имеющий мутацию BRAF V600 (например, V600E).
[0040] В некоторых вариантах осуществления в настоящем документе предложены способы увеличения максимальной переносимой дозы (МПД) энкорафениба у субъекта, проходящего лечение рака энкорафенибом, включающие введение энкорафениба в комбинации с ингибитором MEK и ингибитором EGFR субъекту. В некоторых таких вариантах осуществления максимальная переносимая доза составляет больше чем приблизительно 50 мг (например, больше чем приблизительно 100 мг, больше чем приблизительно 200 мг, больше чем приблизительно 300 мг, больше чем приблизительно 400 мг, больше чем приблизительно 500 мг, больше чем приблизительно 600 мг, больше чем приблизительно 700 мг или больше чем приблизительно 800 мг). В некоторых таких вариантах осуществления максимальная переносимая доза составляет приблизительно 50 мг (например, приблизительно 100 мг, 200 мг, 300 мг, 400 мг, 500 мг, 600 мг, 700 мг или 800 мг). В некоторых таких вариантах осуществления рак связан с BRAF мутантом V600E. В некоторых таких вариантах осуществления рак является раком толстой и прямой кишки (например, метастатическим раком толстой и прямой кишки). В некоторых таких вариантах осуществления ингибитор MEK выбран из группы, состоящей из биниметиниба, траметиниба, кобиметиниба, селуметиниба и PD-325901 (например, биниметиниба). В некоторых таких вариантах осуществления ингибитор EGFR выбран из группы, состоящей из цетуксимаба, гефитиниба, эрлотиниба, лапатиниба, дакомитиниба, нератиниба, вандетаниба и панитумумаба (например, цетуксимаба). В некоторых таких вариантах осуществления субъект выбран как имеющий мутацию BRAF V600 (например, V600E).
[0041] В некоторых вариантах осуществления в настоящем документе предложены способы повышения безопасной дозы комбинации биниметиниба и цетуксимаба у субъекта, включающие введение энкорафениба в сочетании с биниметинибом и цетуксимабом. В некоторых таких вариантах осуществления повышение безопасной дозы биниметиниба и/или цетуксимаба независимо составляет по меньшей мере приблизительно по 2% (например, по меньшей мере приблизительно 4%, по меньшей мере приблизительно 5%, по меньшей мере приблизительно 6%, по меньшей мере приблизительно 8%, по меньшей мере приблизительно 10%, по меньшей мере приблизительно 15%, по меньшей мере приблизительно 20%, по меньшей мере приблизительно 25%, по меньшей мере приблизительно 30%, по меньшей мере приблизительно 35%, по меньшей мере приблизительно 40%, по меньшей мере приблизительно 45%, по меньшей мере приблизительно 50%, по меньшей мере приблизительно 60%, по меньшей мере приблизительно 70%, по меньшей мере приблизительно 80%, по меньшей мере приблизительно 90%, по меньшей мере приблизительно 100%). В некоторых таких вариантах осуществления субъект выбран как имеющий мутацию BRAF V600 (например, V600E).
[0042] В некоторых вариантах осуществления в настоящем документе предложены способы повышения безопасной дозы комбинации энкорафениба и цетуксимаба у субъекта, включающие введение биниметиниба в сочетании с энкорафенибом и цетуксимабом. В некоторых таких вариантах осуществления повышение безопасной дозы энкорафениба и/или цетуксимаба независимо составляет по меньшей мере приблизительно по 2% (например, по меньшей мере приблизительно 4%, по меньшей мере приблизительно 5%, по меньшей мере приблизительно 6%, по меньшей мере приблизительно 8%, по меньшей мере приблизительно 10%, по меньшей мере приблизительно 15%, по меньшей мере приблизительно 20%, по меньшей мере приблизительно 25%, по меньшей мере приблизительно 30%, по меньшей мере приблизительно 35%, по меньшей мере приблизительно 40%, по меньшей мере приблизительно 45%, по меньшей мере приблизительно 50%, по меньшей мере приблизительно 60%, по меньшей мере приблизительно 70%, по меньшей мере приблизительно 80%, по меньшей мере приблизительно 90%, по меньшей мере приблизительно 100%). В некоторых таких вариантах осуществления субъект выбран как имеющий мутацию BRAF V600 (например, V600E).
[0043] В некоторых вариантах осуществления в настоящем документе предложены способы повышения безопасной дозы комбинации энкорафениба и биниметиниба у субъекта, включающие введение цетуксимаба в сочетании с энкорафенибом и биниметинибом. В некоторых таких вариантах осуществления повышение безопасной дозы энкорафениба и/или биниметиниба независимо составляет по меньшей мере приблизительно по 2% (например, по меньшей мере приблизительно 4%, по меньшей мере приблизительно 5%, по меньшей мере приблизительно 6%, по меньшей мере приблизительно 8%, по меньшей мере приблизительно 10%, по меньшей мере приблизительно 15%, по меньшей мере приблизительно 20%, по меньшей мере приблизительно 25%, по меньшей мере приблизительно 30%, по меньшей мере приблизительно 35%, по меньшей мере приблизительно 40%, по меньшей мере приблизительно 45%, по меньшей мере приблизительно 50%, по меньшей мере приблизительно 60%, по меньшей мере приблизительно 70%, по меньшей мере приблизительно 80%, по меньшей мере приблизительно 90%, по меньшей мере приблизительно 100%). В некоторых таких вариантах осуществления субъект выбран как имеющий мутацию BRAF V600 (например, V600E).
[0044] В некоторых вариантах осуществления в настоящем документе предложены способы ингибирования передачи сигнала MAPK у субъекта, которому была введена комбинация энкорафениба и цетуксимаба, включающие введение субъекту биниметиниба. В некоторых таких вариантах осуществления субъект выбран как имеющий мутацию BRAF V600 (например, V600E).
[0045] В некоторых вариантах осуществления в настоящем документе предложены способы супрессии активации EGFR у субъекта, которому была введена комбинация энкорафениба и биниметиниба, включающие введение антитела против EGFR (например, цетуксимаба, гефитиниба, эрлотиниба, лапатиниба, дакомитиниба, нератиниба, вандетаниба или панитумумаба). В некоторых таких вариантах осуществления субъект выбран как имеющий мутацию BRAF V600 (например, V600E).
[0046] В некоторых вариантах осуществления в настоящем документе предложены способы ингибирования B-Raf-киназы (например, мутанта V600E) у субъекта, включающие введение терапевтически эффективного количества биниметиниба или его фармацевтически приемлемой соли, цетуксимаба или его фармацевтически приемлемой соли и энкорафениба или его фармацевтически приемлемой соли. В некоторых таких вариантах осуществления белок B-Raf кодируется мутантным BRAF. В некоторых таких вариантах осуществления BRAF включает мутацию в киназном домене. В некоторых таких вариантах осуществления BRAF включает мутацию V600 (например, V600E, V600K или V600G).
[0047] В некоторых вариантах осуществления в настоящем документе предложены способы снижения активации пути Raf-MEK-ERK у субъекта, включающие введение терапевтически эффективного количества биниметиниба или его фармацевтически приемлемой соли, цетуксимаба или его фармацевтически приемлемой соли и энкорафениба или его фармацевтически приемлемой соли субъекту. В некоторых таких вариантах осуществления субъект выбран как имеющий мутацию BRAF V600 (например, V600E).
[0048] В некоторых вариантах осуществления в настоящем документе предложены способы снижения активации пути Raf-MEK-ERK у субъекта, подвергаемого схеме с цетуксимабом и энкорафенибом, включающие введение субъекту биниметиниба. В некоторых таких вариантах осуществления активация связана с повышенной киназной активностью в отношении MEK. В некоторых таких вариантах осуществления активация связана с активацией C-Raf. В некоторых таких вариантах осуществления субъект имеет B-Raf-ассоциированный рак. В некоторых таких вариантах осуществления субъект имеет рак толстой и прямой кишки (например, метастатический рак толстой и прямой кишки). В некоторых таких вариантах осуществления введение облегчает один или более симптомов рака толстой и прямой кишки (например, метастатического рака толстой и прямой кишки), вызывает положительный ответ (например, частичный или полный) и/или уменьшает размер опухоли и/или количество раковых клеток у субъекта.
[0049] В некоторых вариантах осуществления в настоящем документе предложены способы повышения эффективности ингибирования B-Raf энкорафенибом у субъекта, включающие введение биниметиниба и цетуксимаба в комбинации с энкорафенибом. В некоторых таких вариантах осуществления субъект выбран как имеющий мутацию BRAF V600 (например, V600E).
[0050] В некоторых вариантах осуществления в настоящем документе предложены способы повышения эффективности ингибирования MAPK энкорафенибом и цетуксимабом у субъекта, включающие введение биниметиниба в комбинации с энкорафенибом. В некоторых таких вариантах осуществления субъект выбран как имеющий мутацию BRAF V600 (например, V600E).
[0051] В некоторых вариантах осуществления в настоящем документе предложены способы лечения рака толстой и прямой кишки (например, метастатического рака толстой и прямой кишки), который прогрессировал после первичного лечения рака у субъекта, включающие введение субъекту комбинации энкорафениба, цетуксимаба и биниметиниба. В некоторых таких вариантах осуществления первичное лечение рака производило частичный положительный ответ. В некоторых таких вариантах осуществления первичное лечение рака не производило ответ. В некоторых таких вариантах осуществления первичное лечение рака включало химиотерапию. В некоторых таких вариантах осуществления первичное лечение рака включало введение одного или более терапевтических средств. В некоторых таких вариантах осуществления одно или более терапевтических средств вводили в комбинации. В некоторых таких вариантах осуществления одно или более терапевтических средств выбраны из группы, состоящей из цетуксимаба, бевацизумаба, афлиберцепта, панитумумаба, иринотекана, лейковорина и фторурацила. В некоторых таких вариантах осуществления первичное лечение рака включает введение иринотекана одновременно с фолиновой кислотой, с последующим введением фторурацила путем внутривенного струйного введения, с последующим введением фторурацила путем внутривенной инфузии. В некоторых таких вариантах осуществления иринотекан вводят в дозе приблизительно 180 мг/м2 приблизительно в течение 90 минут. В некоторых таких вариантах осуществления фолиновую кислоту вводят в дозе приблизительно 400 мг/м2 (или 2×250 мг/м2) приблизительно в течение 120 минут. В некоторых таких вариантах осуществления фторурацил вводят путем внутривенного струйного введения в дозе от приблизительно 400 мг/м2 до 500 мг/м2. В некоторых таких вариантах осуществления фторурацил вводят путем внутривенной инфузии в дозе от приблизительно 2400 мг/м2 до 3000 мг/м2. В некоторых таких вариантах осуществления рак толстой и прямой кишки (например, метастатический рак толстой и прямой кишки) связан с мутацией BRAF V600E.
[0052] В некоторых вариантах осуществления в настоящем документе предложены способы лечения рака толстой и прямой кишки (например, метастатического рака толстой и прямой кишки), который прогрессировал после первичного лечения рака у субъекта, включающие независимое введение субъекту терапевтически эффективных доз энкорафениба, цетуксимаба и биниметиниба. В некоторых таких вариантах осуществления первичное лечение рака производило частичный положительный ответ. В некоторых таких вариантах осуществления первичное лечение рака не производило ответа. В некоторых таких вариантах осуществления первичное лечение рака включало химиотерапию. В некоторых таких вариантах осуществления первичное лечение рака включало введение одного или более терапевтических средств. В некоторых таких вариантах осуществления одно или более терапевтических средств вводили в комбинации. В некоторых таких вариантах осуществления одно или более терапевтических средств выбраны из группы, состоящей из цетуксимаба, бевацизумаба, афлиберцепта, панитумумаба, иринотекана, лейковорина и фторурацила. В некоторых таких вариантах осуществления первичное лечение рака включает введение иринотекана одновременно с фолиновой кислотой, с последующим введением фторурацила путем внутривенного струйного введения, с последующим введением фторурацила путем внутривенной инфузии. В некоторых таких вариантах осуществления иринотекан вводят в дозе приблизительно 180 мг/м2 приблизительно в течение 90 минут. В некоторых таких вариантах осуществления фолиновую кислоту вводят в дозе приблизительно 400 мг/м2 (или 2×250 мг/м2) приблизительно в течение 120 минут. В некоторых таких вариантах осуществления фторурацил вводят путем внутривенного струйного введения в дозе от приблизительно 400 мг/м2 до 500 мг/м2. В некоторых таких вариантах осуществления фторурацил вводят путем внутривенной инфузии в дозе от приблизительно 2400 мг/м2 до 3000 мг/м2. В некоторых таких вариантах осуществления рак толстой и прямой кишки (например, метастатический рак толстой и прямой кишки) связан с мутацией BRAF V600E.
[0053] В некоторых вариантах осуществления в настоящем документе предложены способы лечения рака у субъекта, включающие введение комбинации энкорафениба, биниметиниба и цетуксимаба; где энкорафениб и биниметиниб вводят по меньшей мере за 30 минут до введения цетуксимаба. В некоторых таких вариантах осуществления энкорафениб и биниметиниб вводят утром. В некоторых таких вариантах осуществления биниметиниб вводят вечером. В некоторых таких вариантах осуществления субъект не употреблял пищу (например, твердую или жидкую) в течение одного или более (например, двух или более, трех или более, четырех или более, пяти или более или 6 или более) часов до введения биниметиниба. В некоторых таких вариантах осуществления энкорафениб вводят один раз в день. В некоторых таких вариантах осуществления энкорафениб вводят в одно и то же время каждый день, когда его вводят. В некоторых таких вариантах осуществления доза вводимого энкорафениба составляет от приблизительно 100 мг до приблизительно 400 мг (например, приблизительно 120 мг, приблизительно 150 мг, приблизительно 175 мг, приблизительно 200 мг, приблизительно 225 мг, приблизительно 250 мг, приблизительно 275 мг, приблизительно 300 мг, приблизительно 325 мг, приблизительно 350 мг, приблизительно 375 мг или приблизительно 400 мг). В некоторых таких вариантах осуществления энкорафениб вводят перорально. В некоторых таких вариантах осуществления биниметиниб вводят два раза в день. В некоторых таких вариантах осуществления биниметиниб вводят два раза в одно и то же время каждый день, когда его вводят. В некоторых таких вариантах осуществления доза вводимого биниметиниба составляет от приблизительно 10 мг до приблизительно 85 мг (например, приблизительно 15 мг, приблизительно 20 мг, приблизительно 25 мг, приблизительно 30 мг, приблизительно 35 мг, приблизительно 40 мг, приблизительно 45 мг, приблизительно 50 мг, приблизительно 55 мг, приблизительно 60 мг, приблизительно 65 мг, приблизительно 70 мг, приблизительно 75 мг, приблизительно 80 мг или приблизительно 85 мг). В некоторых таких вариантах осуществления биниметиниб вводят перорально. В некоторых таких вариантах осуществления цетуксимаб вводят один раз в неделю. В некоторых таких вариантах осуществления доза вводимого цетуксимаба составляет от приблизительно 150 мг до приблизительно 500 мг (например, приблизительно 175 мг, приблизительно 200 мг, приблизительно 225 мг, приблизительно 250 мг, приблизительно 275 мг, приблизительно 300 мг, приблизительно 325 мг, приблизительно 350 мг, приблизительно 375 мг, приблизительно 400 мг, приблизительно 425 мг, приблизительно 450 мг или приблизительно 475 мг), если это первая доза, или от приблизительно 100 мг до приблизительно 350 мг (например, приблизительно 125 мг, приблизительно 150 мг, приблизительно 175 мг, приблизительно 200 мг, приблизительно 225 мг, приблизительно 250 мг, приблизительно 275 мг, приблизительно 300 мг или приблизительно 325 мг), если это не первая доза. В некоторых таких вариантах осуществления цетуксимаб вводят путем внутривенной инфузии в течение от приблизительно 30 минут до приблизительно 180 минут (например, 40 минут, 50 минут, 60 минут, 70 минут, 80 минут, 90 минут, 100 минут, 110 минут, 120 минут, 130 минут, 140 минут, 150 минут, 160 минут или 170 минут). В некоторых таких вариантах осуществления субъекту предписывают избегать употребления грейпфрутов, гранатов, карамболы или померанцев. В некоторых таких вариантах осуществления субъект выбран как имеющий мутацию BRAF V600 (например, V600E).
КРАТКОЕ ОПИСАНИЕ ФИГУР
[0054] Фигура 1A - кривая роста опухоли в ксенотрансплантатной модели HT29 через 21 день введения растворителя (контроль IgG), цетуксимаба, LGX818 и LGX818+цетуксимаб, представленная в виде объема опухоли (мм3) в зависимости от количества дней после имплантации, где закрашенный ромб обозначает растворитель (контроль IgG), закрашенный треугольник обозначает цетуксимаб, закрашенный перевернутый треугольник обозначает LGX818, и закрашенный квадрат обозначает LGX818+цетуксимаб.
[0055] Фигура 1B - каскадная диаграмма, на которой показаны ответы у отдельных животных (регрессия опухоли) на LGX818+цетуксимаб. Каскадная диаграмма отражает наилучший ответ у животных в любой момент времени.
[0056] Фигура 1C - каскадная диаграмма, на которой показаны ответы у отдельных животных (регрессия опухоли) на LGX818+цетуксимаб+биниметиниб. Каскадная диаграмма отражает наилучший ответ у животных в любой момент времени.
[0057] Фигура 2 - кривая изменения массы тела в ксенотрансплантатной модели HT29 через 21 день введения растворителя (контроль IgG), цетуксимаба, LGX818 и LGX818+цетуксимаб, представленного как объем опухоли (мм3) в зависимости от количества дней после имплантации, где закрашенный ромб обозначает растворитель (контроль IgG), закрашенный треугольник обозначает цетуксимаб, закрашенный перевернутый треугольник обозначает LGX818, и закрашенный квадрат обозначает LGX818+цетуксимаб.
[0058] Фигура 3 - кривая роста опухоли в ксенотрансплантатной модели HT29 через 21 день введения растворителя (контроль IgG), LGX818, MEK162 и LGX818+MEK162, представленного как объем опухоли (мм3) в зависимости от количества дней после имплантации, где закрашенный ромб обозначает растворитель, незакрашенный треугольник обозначает LGX818, закрашенный перевернутый треугольник обозначает MEK162, и незакрашенный ромб обозначает LGX818+MEK162.
[0059] Фигура 4 - кривая изменения массы тела в ксенотрансплантатной модели HT29 через 21 день введения растворителя (контроль IgG), LGX818, MEK162 и LGX818+MEK162, представленного как объем опухоли (мм3) в зависимости от количества дней после имплантации, где закрашенный ромб обозначает растворитель (контроль IgG), закрашенный треугольник обозначает LGX818, закрашенный перевернутый треугольник обозначает MEK162, и закрашенный квадрат обозначает LGX818+MEK162.
[0060] Фигура 5 - кривая роста опухоли в ксенотрансплантатной модели HT29 через 21 день введения растворителя (контроль IgG), цетуксимаба, MEK162 и MEK162+цетуксимаба, представленного как объем опухоли (мм3) в зависимости от количества дней после имплантации, где закрашенный ромб обозначает растворитель, (контроль IgG), закрашенный треугольник обозначает цетуксимаб, закрашенный перевернутый треугольник обозначает MEK162, и закрашенный квадрат обозначает MEK162+цетуксимаб.
[0061] Фигура 6 - кривая изменения массы тела в ксенотрансплантатной модели HT29 через 21 день введения растворителя (контроль IgG), цетуксимаба, MEK162 и MEK162+цетуксимаб, представленного как объем опухоли (мм3) в зависимости от количества дней после имплантации, где закрашенный ромб обозначает растворитель, (контроль IgG), закрашенный треугольник обозначает цетуксимаб, закрашенный перевернутый треугольник обозначает MEK162, и закрашенный квадрат обозначает MEK162+цетуксимаб.
[0062] Фигура 7 - кривая возобновления роста опухоли в ксенотрансплантатной модели HT29 после завершения введения растворителя (контроль IgG), цетуксимаба, LGX818, MEK162, LGX818+MEK162, LGX818+цетуксимаб, MEK162+цетуксимаб и LGX818 +, MEK162+цетуксимаб, представленного как объем опухоли (мм3) в зависимости от количества дней после имплантации, где незакрашенный квадрат обозначает растворитель, заштрихованный треугольник обозначает цетуксимаб, закрашенный перевернутый треугольник обозначает LGX818, закрашенный круг обозначает MEK162, закрашенный ромб обозначает LGX818+MEK162, закрашенный квадрат обозначает LGX818+цетуксимаб, незакрашенный круг обозначает MEK162+цетуксимаб, и незакрашенный треугольник обозначает LGX818+MEK162+цетуксимаб.
[0063] На Фигуре 8 показана порошковая дифракционная рентгенограмма кристаллизованного (2-гидроксиэтокси)амида 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты, полученного согласно Примеру 3.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
[0064] В настоящем изобретении предложены фармацевтические комбинации, независимо содержащие терапевтически эффективные количества: (a) ингибитора BRAF или его фармацевтически приемлемой соли, (b) по меньшей мере одного ингибитора митоген-активируемой протеинкиназы (MEK) или его фармацевтически приемлемой соли и (c) ингибитора рецептора эпидермального фактора роста (EGFR) или его фармацевтически приемлемой соли; и, необязательно, по меньшей мере один фармацевтически приемлемый носитель, для применения в лечении пролиферативного заболевания.
[0065] В одном варианте осуществления в настоящем документе предложена фармацевтическая комбинация, независимо содержащая терапевтически эффективные количества: (a) ингибитора BRAF, которым является метил-N-[(2S)-1-({4-[3-(5-хлор-2-фтор-3-метансульфонамидофенил)-1-(пропан-2-ил)-1H-пиразол-4-ил]пиримидин-2-ил}амино)пропан-2-ил]карбамат (СОЕДИНЕНИЕ A) или его фармацевтически приемлемая соль, (b) по меньшей мере одного ингибитора MEK (СОЕДИНЕНИЯ B) и (c) антитела против EGFR (СОЕДИНЕНИЯ C), и, необязательно, по меньшей мере один фармацевтически приемлемый носитель, для применения в лечении пролиферативного заболевания.
[0066] В одном варианте осуществления в настоящем документе предложена фармацевтическая комбинация, независимо содержащая терапевтически эффективные количества: (a) ингибитора BRAF, которым является метил-N-[(2S)-1-({4-[3-(5-хлор-2-фтор-3-метансульфонамидофенил)-1-(пропан-2-ил)-1H-пиразол-4-ил]пиримидин-2-ил}амино)пропан-2-ил]карбамат (СОЕДИНЕНИЕ A) или его фармацевтически приемлемая соль, (b) ингибитора MEK (СОЕДИНЕНИЯ B), которым является (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты или его фармацевтически приемлемая соль, и (c) антитела против EGFR (СОЕДИНЕНИЯ C), и, необязательно, по меньшей мере один фармацевтически приемлемый носитель.
[0067] В одном варианте осуществления в настоящем документе предложена фармацевтическая комбинация, независимо содержащая терапевтически эффективные количества: (a) ингибитора BRAF, которым является метил-N-[(2S)-1-({4-[3-(5-хлор-2-фтор-3-метансульфонамидофенил)-1-(пропан-2-ил)-1H-пиразол-4-ил]пиримидин-2-ил}амино)пропан-2-ил]карбамат (СОЕДИНЕНИЕ A) или его фармацевтически приемлемая соль, (b) ингибитора MEK (СОЕДИНЕНИЯ B), которым является (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты или его фармацевтически приемлемая соль, и (c) антитела против EGFR (СОЕДИНЕНИЯ C), которым является цетуксимаб, и, необязательно, по меньшей мере один фармацевтически приемлемый носитель.
[0068] Фармацевтические комбинации настоящего изобретения включают ингибитор BRAF, которым является метил-N-[(2S)-1-({4-[3-(5-хлор-2-фтор-3-метансульфонамидофенил)-1-(пропан-2-ил)-1H-пиразол-4-ил]пиримидин-2-ил}амино)пропан-2-ил]карбамат (именуемый в настоящем документе как "СОЕДИНЕНИЕ A"). СОЕДИНЕНИЕ A является соединением Формулы I:
 .
.
[0069] СОЕДИНЕНИЕ A также известно как метил-[(2S)-1-{[4-(3-{5-хлор-2-фтор-3-[(метилсульфонил)амино]фенил}-1-изопропил-1H-пиразол-4-ил)-2-пиримидинил]амино}-2-пропанил]карбамат, метил-N-{(2S)-1-[(4-{3-[5-хлор-2-фтор-3-(метансульфонамидо)фенил]-1-(пропан-2-ил)-1H-пиразол-4-ил}пиримидин-2-ил)амино]пропан-2-ил}карбамат, LGX818, NVP-LGX818 и энкорафениб. СОЕДИНЕНИЕ A описано в WO 2011/025927 и патенте США 8,501,758. Синтез СОЕДИНЕНИЯ A описан в Примерах 5 и 6 заявки WO 2011/025927, которая настоящим полностью включена посредством ссылки. В одном варианте осуществления СОЕДИНЕНИЕ A находится в аморфной форме.
[0070] При указании СОЕДИНЕНИЯ A, термин "соль" или "соли" следует понимать, как соль СОЕДИНЕНИЯ A, которая может присутствовать отдельно или в смеси со свободным соединением Формулы (I) и предпочтительно является фармацевтически приемлемой солью. Такие соли образуются, например, в виде соли присоединения кислоты в реакции формы свободного основания СОЕДИНЕНИЯ A с фармацевтически приемлемой неорганической или органической кислотой. Соли СОЕДИНЕНИЯ A предпочтительно являются фармацевтически приемлемыми солями.
[0071] СОЕДИНЕНИЕ A обладает асимметричным центром и может быть получено в виде отдельных (R)- или (S)-стереоизомеров, или в виде их смеси, как описано в WO 2011/025927. Если не указаано иное, описание или обозначение СОЕДИНЕНИЯ A в описании и формуле изобретения должно включать как отдельные энантиомеры, так и их рацемические смеси. Таким образом, настоящее изобретение также включает все такие изомеры, в том числе диастереомерные смеси и разделенные энантиомеры СОЕДИНЕНИЯ A. Энантиомеры могут быть разделены путем превращения энантиомерной смеси в диастереомерную смесь при взаимодействии с подходящим оптически активным соединением (например, спиртом), разделении диастереомеров и превращении (например, гидролизе) отдельных диастереомеров с получением соответствующих чистых энантиомеров. Способы определения стереохимии и разделения стереоизомеров хорошо известны в уровне техники (см. обсуждение в Главе 4 "Advanced Organic Chemistry", 4-е издание, J. March. John Wiley and Sons, New York, 1992). В одном варианте осуществления СОЕДИНЕНИЕ A является (S)-стереоизомером, то есть метил-[(2S)-1-{[4-(3-{5-хлор-2-фтор-3-[(метилсульфонил)амино]фенил}-1-изопропил-1H-пиразол-4-ил)-2-пиримидинил]амино}-2-пропанил]-карбаматом.
[0072] Фармацевтические комбинации настоящего изобретения включают по меньшей мере один ингибитор MEK (именуемый в настоящем документе как "СОЕДИНЕНИЕ B"). В одном варианте осуществления ингибитор MEK выбран из (2-гидроксиэтокси)амида 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты, траметиниба, кометиниба (GDC-0973) и селуметиниба (AZD6244). В одном варианте осуществления СОЕДИНЕНИЕ B является (2-гидроксиэтокси)амидом 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты, который является соединением формулы (II):
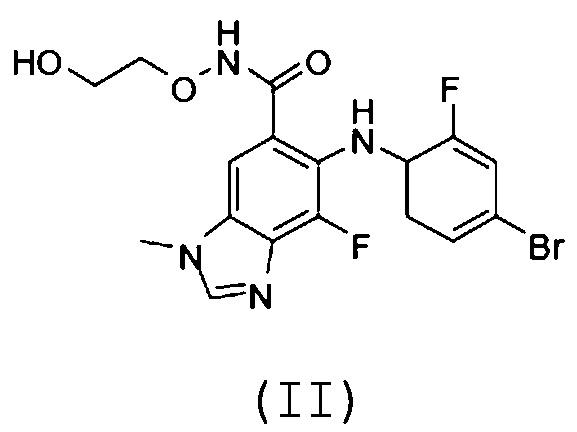 .
.
[0073] Ингибитор MEK (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты описан в публикации PCT WO 03/077914, в которой описаны способы его получения, например, в Примере 18 (соединение 29lll). (2-Гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты также известен как 5-[(4-бром-2-фторфенил)амино]-4-фтор-N-(2-гидроксиэтокси)-1-метил-1H-бензимидазол-6-карбоксамид, ARRY-162, MEK162 и биниметиниб. В одном варианте осуществления (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты представляет собой кристаллизованный (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты. Кристаллизованный (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты и способы получения кристаллизованного (2-гидроксиэтокси)амида 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты описаны в публикации PCT WO 2014/063024, которая включена в настоящий документ посредством ссылки.
[0074] В одном варианте осуществления ингибитор MEK представляет собой кристаллизованный (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты. В одном варианте осуществления кристаллизованный (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты может быть получен согласно Примеру 3. Порошковая дифракционная рентгенограмма кристаллизованного (2-гидроксиэтокси)амида 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты имеет характеристические дифракционные пики, выраженные в градусах 2тета (2θ). Анализ методом порошковой рентгеновской дифракции (XRPD) выполняли при использовании рентгеновского порошкового дифрактометра Shimadzu XRD-6000 с использованием Cu Kα излучения. Прибор оборудован длинной тонкофокусной рентгеновской трубкой. Напряжение трубки и силу тока устанавливали на 40 кВ и 40 мА, соответственно. Щели расходимости и рассеивания устанавливали на 1°, и приемную щель устанавливали на 0,15 мм. Дифрагированное излучение детектировали с помощью NaI сцинтилляционного детектора. Использовали непрерывное сканирование θ-2θ при 1°/мин (величина шага 0,02°) от 2 до 42° 2θ (время счета=1200 секунд; длина волны=1,540562; число точек=2001; разрешение файла данных=1600). Оптика дифрактометра: фиксированная щель; без конфигурации X2. Трубка: фиксированная щель; без конфигурации X2. Кремниевый стандарт анализировали для проверки выравнивания инструмента. Данные регистрировали и обрабатывали при использовании XRD-6100/7000 v.5.0. Образцы подготавливали для анализа, помещая их в держатель образцов из нержавеющей стали.
[0075] В некоторых вариантах осуществления кристаллизованный (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты имеет характеристический пик, выраженный в градусах 2θ, при 20,38. В некоторых вариантах осуществления кристаллизованный (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты имеет характеристический пик, выраженный в градусах 2θ, при 28,39. В некоторых вариантах осуществления кристаллизованный (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты имеет характеристический пик, выраженный в градусах 2θ, при 11,18. В некоторых вариантах осуществления кристаллизованный (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты имеет характеристический пик, выраженный в градусах 2θ, при 29,18. В некоторых вариантах осуществления кристаллизованный (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты имеет характеристический пик, выраженный в градусах 2θ, при 22,43. В некоторых вариантах осуществления кристаллизованный (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты имеет характеристический пик, выраженный в градусах 2θ, при 22,75. В некоторых вариантах осуществления кристаллизованный (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты имеет характеристический пик, выраженный в градусах 2θ, при 25,23. В некоторых вариантах осуществления кристаллизованный (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты имеет характеристический пик, выраженный в градусах 2θ, при 16,05. В некоторых вариантах осуществления кристаллизованный (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты имеет характеристический пик, выраженный в градусах 2θ, при 11,82. В некоторых вариантах осуществления кристаллизованный (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты имеет характеристический пик, выраженный в градусах 2θ, при 23,74. В некоторых вариантах осуществления кристаллизованный (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты имеет характеристический пик, выраженный в градусах 2θ, при 16,33. В некоторых вариантах осуществления кристаллизованный (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты имеет характеристический пик, выраженный в градусах 2θ, при 19,00.
[0076] В некоторых вариантах осуществления кристаллизованный (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты имеет порошковую дифракционную рентгенограмму, включающую 2-12 следующих характеристических пиков, выраженных в градусах 2θ, при 20,38, 28,39, 11,18, 29,18, 22,43, 22,75, 25,23, 16,05, 11,82, 23,74, 16,33 и 19,00. Например, кристаллизованный (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты может иметь порошковую дифракционную рентгенограмму, включающую характеристические пики, выраженные в градусах 2θ, при 20,38 или 28,39; или порошковую дифракционную рентгенограмму, включающую характеристические пики, выраженные в градусах 2θ, при 20,38, 28,39 и 11,18; или порошковую дифракционную рентгенограмму, включающую характеристические пики, выраженные в градусах 2θ, при 20,38, 28,39, 11,18 и 29,18; или порошковую дифракционную рентгенограмму, включающую характеристические пики, выраженные в градусах 2θ, при 20,38, 28,39, 11,18, 29,18, 22,43 и 22,75; или порошковую дифракционную рентгенограмму, включающую характеристические пики, выраженные в градусах 2θ, при 20,38, 28,39, 11,18, 29,18, 22,43, 22,75, 25,23, 16,05 и 11,82; или порошковую дифракционную рентгенограмму, включающую характеристические пики, выраженные в градусах 2θ, при 20,38, 28,39, 11,18, 29,18, 22,43, 22,75, 25,23, 16,05, 11,82, 23,74, 16,33 и 19,00.
[0077] В некоторых вариантах осуществления кристаллизованный (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты имеет порошковую дифракционную рентгенограмму, включающую характеристические пики, выраженные в градусах 2θ, при 20,38, 28,39, 11,18, 29,18, 22,43, 22,75, 25,23, 16,05, 11,82, 23,74, 16,33, 19,00, 30,01, 25,98, 37,35, 6,79, 9,47, 34,21, 30,43 и 31,40.
[0078] В некоторых вариантах осуществления кристаллизованный (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты имеет характеристический пик, выраженный в градусах 2θ, при 20,4±0,2. В некоторых вариантах осуществления кристаллизованный (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты имеет характеристический пик, выраженный в градусах 2θ, при 28,4±0,2. В некоторых вариантах осуществления кристаллизованный (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты имеет характеристический пик, выраженный в градусах 2θ, при 11,2±0,2. В некоторых вариантах осуществления кристаллизованный (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты имеет характеристический пик, выраженный в градусах 2θ, при 29,2±0,2. В некоторых вариантах осуществления кристаллизованный (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты имеет характеристический пик, выраженный в градусах 2θ, при 22,4±0,2. В некоторых вариантах осуществления кристаллизованный (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты имеет характеристический пик, выраженный в градусах 2θ, при 22,7±0,2. В некоторых вариантах осуществления кристаллизованный (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты имеет характеристический пик, выраженный в градусах 2θ, при 25,2±0,2. В некоторых вариантах осуществления кристаллизованный (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты имеет характеристический пик, выраженный в градусах 2θ, при 16,0±0,2. В некоторых вариантах осуществления кристаллизованный (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты имеет характеристический пик, выраженный в градусах 2θ, при 11,8±0,2. В некоторых вариантах осуществления кристаллизованный (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты имеет характеристический пик, выраженный в градусах 2θ, при 23,7±0,2. В некоторых вариантах осуществления кристаллизованный (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты имеет характеристический пик, выраженный в градусах 2θ, при 16,3±0,2. В некоторых вариантах осуществления кристаллизованный (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты имеет характеристический пик, выраженный в градусах 2θ, при 19,0±0,2.
[0079] В некоторых вариантах осуществления кристаллизованный (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты имеет порошковую дифракционную рентгенограмму, включающую 2-12 следующих характеристических пиков, выраженных в градусах 2θ, при 20,4±0,2, 28,4±0,2, 11,2±0,2, 29,2±0,2, 22,4±0,2, 22,7±0,2, 25,2±0,2, 16,0±0,2, 11,8±0,2, 23,7±0,2, 16,3±0,2, и 19,0±0,2. Например, кристаллизованный (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты может иметь порошковую дифракционную рентгенограмму, включающую характеристические пики, выраженные в градусах 2θ, при 20,4±0,2 и 28,4±0,2; или порошковую дифракционную рентгенограмму, включающую характеристические пики, выраженные в градусах 2θ, при 20,4±0,2, 28,4±0,2 и 11,2±0,2; или порошковую дифракционную рентгенограмму, включающую характеристические пики, выраженные в градусах 2θ, при 20,4±0,2, 28,4±0,2, 11,2±0,2 и 29,2±0,2; или порошковую дифракционную рентгенограмму, включающую характеристические пики, выраженные в градусах 2θ, при 20,4±0,2, 28,4±0,2, 11,2±0,2, 29,2±0,2, 22,4±0,2 и 22,7±0,2; или порошковую дифракционную рентгенограмму, включающую характеристические пики, выраженные в градусах 2θ, при 20,4±0,2, 28,4±0,2, 11,2±0,2, 29,2±0,2, 22,4±0,2, 22,7±0,2, 25,2±0,2, 16,0±0,2 и 11,8±0,2; или порошковую дифракционную рентгенограмму, включающую характеристические пики, выраженные в градусах 2θ, при 20,4±0,2, 28,4±0,2, 11,2±0,2, 29,2±0,2, 22,4±0,2, 22,7±0,2, 25,2±0,2, 16,0±0,2, 11,8±0,2, 23,7±0,2, 16,3±0,2 и 19,0±0,2.
[0080] В некоторых вариантах осуществления кристаллизованный (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты имеет порошковую дифракционную рентгенограмму, включающую характеристические пики, выраженные в градусах 2θ, при 20,4±0,2, 28,4±0,2, 11,2±0,2, 29,2±0,2, 22,4±0,2, 22,7±0,2, 25,2±0,2, 16,0±0,2, 11,8±0,2, 23,7±0,2, 16,3±0,2, 19,0±0,2, 30,0±0,2, 26,0±0,2, 37,3±0,2, 6,8±0,2, 9,5±0,2, 34,2±0,2, 30,4±0,2 и 31,4±0,2.
[0081] В некоторых вариантах осуществления кристаллизованный (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты имеет порошковую дифракционную рентгенограмму по меньшей мере с 20 характеристическими пиками (в градусах 2θ), перечисленными в Таблице A.
Таблица A. Пики XRPD кристаллизованного (2-гидроксиэтокси)амида 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты
[0082] В отношении СОЕДИНЕНИЯ B, термин "соль" или "соли", если не указано иное, включает соли кислотных и основных групп, которые могут присутствовать в соединениях настоящего изобретения. СОЕДИНЕНИЕ B способно образовывать соли с различными неорганическими и органическими кислотами. Кислоты, которые могут использоваться для получения фармацевтически приемлемых солей присоединения кислот таких основных соединений настоящего изобретения, являются кислотами, которые образуют нетоксичные соли присоединения кислот, то есть соли, содержащие фармацевтически приемлемые анионы, такие как ацетат, бензоат, бромид, хлорид, цитрат, фумарат, гидробромид, гидрохлорид, иодид, лактат, малеат, манделат, нитрат, оксалат, салицилат, сукцинат и тартрат. Примером соли такого типа является гидрохлорид или сульфат СОЕДИНЕНИЯ B согласно настоящему изобретению.
[0083] Дополнительные фармацевтически приемлемые соли СОЕДИНЕНИЯ B, подходящие в рамках настоящего изобретения, включают соли, раскрытые в заявке PCT WO 03/077914, которая включена в настоящий документ посредством ссылки.
[0084] Фармацевтические комбинации настоящего изобретения включают ингибитор рецептора эпидермального фактора роста (EGFR) (именуемый в настоящем документе как СОЕДИНЕНИЕ C). В одном варианте осуществления ингибитором EGFR является ингибитор тирозинкиназы (например, эрлотиниб, гефитиниб или лапатиниб). В одном варианте осуществления ингибитором EGFR является моноклональное антитело, также именуемое в настоящем документе как антитело против EGFR (например, панитумумаб или цетуксимаб). Термин "антитело против EGFR" определен в настоящем документе для обозначения антитела или его антигенсвязывающего фрагмента, который способен связываться с EGFR (т.е. антагонист EGFR). Антитело против EGFR конкурирует за связывание лиганда с рецептором, блокируя лигандсвязывающую область. Антитело против EGFR может вызвать интернализацию рецептора и последующую деградацию без фосфорилирования рецептора. В одном варианте осуществления антитело против EGFR распознает тот же эпитоп, что и C225 (также известный как цетуксимаб), описанный в WO 96/40210. В другом варианте осуществления антитело против EGFR имеет те же 6 CDR-областей (определяющих комплементарность областей), что и C225 (цетуксимаб), описанный в WO 96/40210. В одном варианте осуществления СОЕДИНЕНИЕ C является антителом против EGFR цетуксимабом (также известным как C225 и Эрбитукс®). Цетуксимаб представляет собой моноклональное антитело, и его последовательности и способы его получения и применения для лечения пролиферативных заболеваний раскрыты в патенте США 6,217,866 и в WO 96/40210, которые включены в настоящий документ посредством ссылки. Так как цетуксимаб является моноклональным антителом, его механизм действия отличается от стандартных неселективных химиотерапевтических терапий тем, что он специфично взаимодействует и связывается с EGFR. Такое связывание вызывает ингибирование активации рецептора и последующего пути передачи сигнала, что приводит к уменьшению инвазии нормальных тканей опухолевыми клетками и распространения опухолей в новые области. Как предполагают, он также ингибирует способность опухолевых клеток восстанавливаться после химиотерапии и лучевой терапии и ингибирует образование новых кровеносных сосудов в опухолях, что, по-видимому, приводит к полной супрессии роста опухоли.
[0085] В дальнейшем, тройные комбинации СОЕДИНЕНИЯ A, СОЕДИНЕНИЯ B и СОЕДИНЕНИЯ C будут именоваться КОМБИНАЦИЕЙ ИЗОБРЕТЕНИЯ, где каждое СОЕДИНЕНИЕ A, СОЕДИНЕНИЕ B и СОЕДИНЕНИЕ C может быть указано как партнер по комбинации, компонент комбинации или терапевтическое средство.
[0086] Настоящее изобретение относится к КОМБИНАЦИИ ИЗОБРЕТЕНИЯ, применимой для раздельного, одновременного или последовательного введения компонентов комбинации нуждающемуся в этом пациенту для лечения пролиферативного заболевания.
[0087] В настоящем изобретении также предложена коммерческая упаковка, содержащая в качестве терапевтических средств КОМБИНАЦИЮ ИЗОБРЕТЕНИЯ, вместе с инструкциями по раздельному, одновременному или последовательному введению для применения при лечении пролиферативного заболевания.
[0088] В настоящем изобретении также предложена коммерческая упаковка, содержащая в качестве терапевтических средств СОЕДИНЕНИЕ A (метил-N-[(2S)-1-({4-[3-(5-хлор-2-фтор-3-метансульфонамидофенил)-1-(пропан-2-ил)-1H-пиразол-4-ил]пиримидин-2-ил}амино)пропан-2-ил]карбамат или его фармацевтически приемлемую соль) и СОЕДИНЕНИЕ B, вместе с инструкциями по раздельному, одновременному или последовательному введению СОЕДИНЕНИЯ A и СОЕДИНЕНИЯ B вместе с СОЕДИНЕНИЕМ C. В одном варианте осуществления ингибитором MEK является (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты или его фармацевтически приемлемая соль. В одном варианте осуществления ингибирующим антителом против EGFR является цетуксимаб.
[0089] Общие термины, используемые в настоящем документе, определены согласно следующим значениям, если прямо не указано иное:
[0090] Термины "содержащий" и "включающий" используются в настоящем документе в своем открытом и неограничивающем смысле, если не указано иное.
[0091] При использовании формы множественного числа в отношении соединений, солей и т.п., это также означает одно соединение, соль и т.п.
[0092] Термин "комбинация" или "фармацевтическая комбинация" определен в настоящем документе для обозначения либо фиксированной комбинации в одной единичной лекарственной форме, либо нефиксированной комбинации или набора компонентов для комбинированного введения назначенной схемы лечения с тройной терапией, где ингибитор BRAF, т.е. СОЕДИНЕНИЕ A или его фармацевтически приемлемая соль, ингибитор MEK, т.е. СОЕДИНЕНИЕ B или его фармацевтически приемлемая соль, и ингибитор EGFR, т.е. СОЕДИНЕНИЕ C, например антитело против EGFR, могут вводить одновременно или независимо в одно и то же время, или раздельно в пределах некоторых временных интервалов. В одном варианте осуществления партнеры по комбинации (т.е. СОЕДИНЕНИЕ A, СОЕДИНЕНИЕ B и СОЕДИНЕНИЕ C) вводят одновременно, независимо в одно и то же время или раздельно, необязательно в пределах некоторых временных интервалов, что позволяет партнерам по комбинации проявлять совместное, например, синергическое, действие. Термин "фиксированная комбинация" означает, что партнеры по комбинации, СОЕДИНЕНИЕ A и СОЕДИНЕНИЕ B, вводят пациенту одновременно в форме одной единицы или единичной лекарственной формы, тогда как СОЕДИНЕНИЕ C вводят в виде отдельной единицы или единичной лекарственной формы. Термин "нефиксированная комбинация" означает, что СОЕДИНЕНИЕ A или его фармацевтически приемлемую соль, СОЕДИНЕНИЕ B или его фармацевтически приемлемую соль и СОЕДИНЕНИЕ C вводят пациенту в виде отдельных единичных лекарственных форм, одновременно, раздельно или последовательно, необязательно в пределах определенных временных интервалов, при этом такое введение обеспечивает терапевтически эффективные уровни трех соединений в теле пациента.
[0093] Термин "фармацевтическая композиция" определен в настоящем документе для обозначения смеси или раствора, содержащего по меньшей мере одно терапевтическое средство (т.е. СОЕДИНЕНИЕ A, СОЕДИНЕНИЕ B или СОЕДИНЕНИЕ C), вводимое субъекту, например, млекопитающему или человеку, для лечения конкретного заболевания или состояния, поражающего млекопитающее.
[0094] Термин "фармацевтически приемлемый" определен в настоящем документе для обозначения таких соединений, материалов, композиций и/или лекарственных форм, которые, в рамках обоснованной клинической оценки, подходят для контакта с тканями субъекта, например, млекопитающего или человека, без чрезмерной токсичности, раздражающей аллергической реакции и других проблемных осложнений, соизмеримых с приемлемым отношением пользы/риска.
[0095] Термины "совместное введение" или "комбинированное введение" при использовании в настоящем документе определены как охватывающие введение выбранных терапевтических средств одному пациенту и должны включать схемы лечения, в которых средства не вводят обязательно одним и тем же путем введения или в одно и то же время.
[0096] Термин "лечение" при использовании в настоящем документе включает лечение, ослабляющее, уменьшающее или облегчающее по меньшей мере один симптом у субъекта, или вызывающее задержку прогрессирования заболевания. Например, лечение может быть уменьшением одного или более симптомов нарушения или полным устранением нарушения, такого как рак. В рамках настоящего изобретения термин "лечить" также означает остановку, задержку начала (то есть периода до клинической манифестации заболевания) и/или уменьшение риска развития или ухудшения заболевания. "Лечение" также может означать увеличение выживаемости по сравнению с выживаемостью, ожидаемой в отсутствие лечения, например, увеличение общей выживаемости (OS) по сравнению с субъектом, не получающим лечение, как описано в настоящем документе, и/или увеличение выживаемости без прогрессирования заболевания (PFS) по сравнению с субъектом, не получающим лечение, как описано здесь. Термин "лечение" может также означать улучшение состояния субъекта, имеющего рак, например, одно или более из уменьшения размера одной или более опухолей у субъекта, уменьшение или отсутствие существенного изменения скорости роста одной или более опухолей у субъекта, уменьшение метастаза у субъекта и увеличение периода ремиссии у субъекта (например, по сравнению с одним или более показателями у субъекта, имеющего подобную форму рака, не получающего лечение или получающего другое лечение, или по сравнению с одним или более показателями у одного и того же субъекта до лечения). Дополнительные показатели для оценки ответа на лечение у субъекта, имеющего рак, раскрыты в настоящем документе ниже.
[0097] Термин "совместно терапевтически активный" или "совместный терапевтический эффект" при использовании в настоящем документе означает, что терапевтические средства КОМБИНАЦИИ ИЗОБРЕТЕНИЯ можно давать пациенту одновременно или отдельно (например, в хронологическом ступенчатом порядке, например, в определенной последовательности), в таких временных интервалах, что они проявляют взаимодействие (например, совместный терапевтический эффект, например, синергический эффект). Так это или нет, в частности, можно определить путем отслеживания уровней в крови и подтверждения того, что компоненты комбинации присутствуют в крови человека, проходящего лечение, по меньшей мере, в течение некоторых временных интервалов.
[0098] Термин "фармацевтически эффективное количество" или "клинически эффективное количество" или "терапевтически эффективное количество" комбинации терапевтических средств КОМБИНАЦИИ ИЗОБРЕТЕНИЯ является количеством, достаточным для обеспечения заметного улучшения по сравнению с исходными, клинически наблюдаемыми признаками и симптомами нарушения, подвергаемого лечению комбинацией.
[0099] Термины "антитело" и "антитела" используются попеременно в самом широком смысле и включают моноклональные антитела, выделенные, модифицированные, химически синтезированные или рекомбинантные антитела (например, полноразмерные или интактные моноклональные антитела), поликлональные антитела, мультивалентные антитела или мультиспецифичные антитела (например, биспецифичные антитела), а также фрагменты антител, при условии, что они демонстрируют требуемую биологическую активность. В одном варианте осуществления антитело является рекомбинантным антителом.
[00100] При использовании в настоящем документе выражение "антитело к EGFR" должно интерпретироваться аналогично "антителу против EGFR" и означает антитело, способное связываться с EGFR.
[00101] Под "EGFR-связывающим фрагментом" или "антигенсвязывающим фрагментом" антитела подразумевается любой пептид, полипептид или белок, сохраняющий способность связываться с EGFR в качестве мишени (также обычно называемой антигеном) антитела. В варианте осуществления такие "антигенсвязывающие фрагменты" выбраны из группы, состоящей из Fv, scFv (sc означает одноцепочечный), Fab, F(ab')2, Fab', scFv-Fc фрагментов или диател, или любого фрагмента, период полувыведения которого может быть увеличен путем химической модификации, такой как присоединение полиалкиленгликоля, такого как полиэтиленгликоль ("ПЭГилирование") (ПЭГилированные фрагменты называются FV-ПЭГ, SCFV-ПЭГ, Fab-ПЭГ, F(ab')2-ПЭГ или Fab'-ПЭГ) ("ПЭГ" означает полиэтиленгликоль), или путем включения в липосому, причем указанные фрагменты обладают по меньшей мере одной из характеристик CDR-областей антитела согласно изобретению. В одном варианте осуществления "антигенсвязывающие фрагменты" будут состоять или будут включать неполную последовательность вариабельной области тяжелой или легкой цепи антитела, из которого они получены, при этом указанная неполная последовательность является достаточной для сохранения такой же специфичности связывания, как у антитела, из которого она получена, и достаточной аффинности по отношению к мишени, например, равной по меньшей мере 1/100, в качестве другого примера равной по меньшей мере 1/10, аффинности антитела, из которого она получена.
[00102] Термин "ингибитор BRAF" при использовании в настоящем документе относится к соединению, которое направленно воздействует, уменьшает или ингибирует киназную активность белка BRAF, в том числе мутантных форм белка BRAF, таких как мутации BRAF V600, такие как V600E, V600D и V600K. В одном варианте осуществления мутантный белок BRAF включает мутацию V600E.
[00103] Термин "ингибитор MEK" при использовании в настоящем документе относится к соединению, которое направленно воздействует, уменьшает или ингибирует киназную активность MAP-киназы (MEK), в том числе мутантных форм MEK. В одном варианте осуществления ингибитор MEK выбран из (2-гидроксиэтокси)амида 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты (биниметиниба; MEK162; ARRY-162), траметиниба, кометиниба (GDC-0973) и селуметиниба (AZD6244). В одном варианте осуществления ингибитором MEK является (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты. В одном варианте осуществления ингибитор MEK является кристаллизованным (2-гидроксиэтокси)амидом 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты.
[00104] Термин "ингибитор EGFR" при использовании в настоящем документе относится к соединению, которое направленно воздействует, уменьшает или ингибирует активность EGFR, в том числе мутантных форм EGFR, включая (EGFRvIII). В одном варианте осуществления ингибитором EGFR является антитело против EGFR.
[00105] Термин "антитело против EGFR" при использовании в настоящем документе относится к антителу или его антигенсвязывающему фрагмент, который способен связываться с EGFR (т.е. к антагонисту EGFR), который направленно воздействует, уменьшает или ингибирует киназную активность активности EGFR, в том числе мутантных форм EGFR. В одном варианте осуществления антителом против EGFR является цетуксимаб, гефитиниб, эрлотиниб, лапатиниб, дакомитиниб, нератиниб, вандетаниб или панитумумаб. В одном варианте осуществления антителом против EGFR является цетуксимаб.
[00106] При использовании в настоящем документе термины "субъект", "индивид" или "пациент" используются попеременно для обозначения любого животного, включая млекопитающих, таких как люди, приматы, мыши, крысы, другие грызуны, кролики, собаки, кошки, свиньи, рогатый скот, овцы и лошади. В некоторых вариантах осуществления субъект - человек. В некоторых вариантах осуществления субъект испытал и/или продемонстрировал по меньшей мере один симптом заболевания или нарушения, подлежащего лечению. В некоторых вариантах осуществления у субъекта было определено или диагностировано наличие BRAF-ассоциированного заболевания или нарушения. В некоторых вариантах осуществления у субъекта было определено или диагностировано наличие рака с дисрегуляцией гена BRAF, белка BRAF или их экспрессии или активности, или уровня (например, BRAF-ассоциированного рака) (например, определенного при использовании одобренного регулирующим органом, например одобренного FDA, анализа или набора). В некоторых вариантах осуществления субъект имеет опухоль, которая является положительной на дисрегуляцию гена BRAF, белка BRAF или их экспрессии или активности, или уровня (например, определенного при использовании одобренного регулирующим органом анализа или набора). Субъект может быть субъектом с опухолью(ями), которая является положительной на дисрегуляцию гена BRAF, белка BRAF или их экспрессии или активности, или уровня (например, идентифицированной как положительная при использовании одобренного регулирующим органом, например одобренного FDA, анализа или набора). Субъект может быть субъектом, в опухоли которого присутствует дисрегуляция гена BRAF, белка BRAF или их экспрессии или активности, или уровня (например, где опухоль определена такой при использовании одобренного регулирующим органом, например одобренного FDA, набора или анализа). В некоторых вариантах осуществления у субъекта подозревают наличие BRAF-ассоциированного рака. В некоторых вариантах осуществления субъект имеет запись в медицинской карте, указывающую, что субъект имеет опухоль, в которой присутствует дисрегуляция гена BRAF, белка BRAF или их экспрессии или активности, или уровня (и, необязательно, запись в медицинской карте указывает, что субъект подлежит лечению любой из предложенных в настоящем документе комбинаций). В некоторых вариантах осуществления у субъекта было определено или диагностировано наличие MEK-ассоциированного заболевания или нарушения. В некоторых вариантах осуществления у субъекта было определено или диагностировано наличие рака с дисрегуляцией гена MEK, белка MEK или их экспрессии или активности, или уровня (например, MEK-ассоциированного рака) (например, определенного при использовании одобренного регулирующим органом, например одобренного FDA, анализа или набора). В некоторых вариантах осуществления субъект имеет опухоль, которая является положительной на дисрегуляцию гена MEK, белка MEK или их экспрессии или активности, или уровня (например, определенного при использовании одобренного регулирующим органом анализа или набора). Субъект может быть субъектом с опухолью(ями), которая является положительной на дисрегуляцию гена MEK, белка MEK или их экспрессии или активности, или уровня (например, идентифицированной как положительная при использовании одобренного регулирующим органом, например одобренного FDA, анализа или набора). Субъект может быть субъектом, в опухоли которого присутствует дисрегуляция гена MEK, белка MEK или их экспрессии или активности, или уровня (например, где опухоль определена такой при использовании одобренного регулирующим органом, например одобренного FDA, набора или анализа). В некоторых вариантах осуществления у субъекта подозревают наличие MEK-ассоциированного рака. В некоторых вариантах осуществления субъект имеет запись в медицинской карте, указывающую, что субъект имеет опухоль, в которой присутствует дисрегуляция гена MEK, белка MEK или их экспрессии или активности, или уровня (и, необязательно, запись в медицинской карте указывает, что субъект подлежит лечению любой из предложенных в настоящем документе комбинаций). В некоторых вариантах осуществления у субъекта было определено или диагностировано наличие EGFR-ассоциированного заболевания или нарушения. В некоторых вариантах осуществления у субъекта было определено или диагностировано наличие рака с дисрегуляцией гена EGFR, белка EGFR или их экспрессии или активности, или уровня (например, EGFR-ассоциированного рака) (например, определенного при использовании одобренного регулирующим органом, например одобренного FDA, анализа или набора). В некоторых вариантах осуществления субъект имеет опухоль, которая является положительной на дисрегуляцию гена EGFR, белка EGFR или их экспрессии или активности, или уровня (например, определенного при использовании одобренного регулирующим органом анализа или набора). Субъект может быть субъектом с опухолью(ями), которая является положительной на дисрегуляцию гена EGFR, белка EGFR или их экспрессии или активности, или уровня (например, идентифицированной как положительная при использовании одобренного регулирующим органом, например одобренного FDA, анализа или набора). Субъект может быть субъектом, в опухоли которого присутствует дисрегуляция гена EGFR, белка EGFR или их экспрессии или активности, или уровня (например, где опухоль определена такой при использовании одобренного регулирующим органом, например одобренного FDA, набора или анализа). В некоторых вариантах осуществления у субъекта подозревают наличие EGFR-ассоциированного рака. В некоторых вариантах осуществления субъект имеет запись в медицинской карте, указывающую, что субъект имеет опухоль, в которой присутствует дисрегуляция гена EGFR, белка EGFR или их экспрессии или активности, или уровня (и, необязательно, запись в медицинской карте указывает, что субъект подлежит лечению любой из предложенных в настоящем документе комбинаций). В некоторых вариантах осуществления субъект является педиатрическим пациентом. В одном варианте осуществления субъект имеет рак толстой и прямой кишки. В одном варианте осуществления субъект имеет метастатический рак толстой и прямой кишки. В одном варианте осуществления субъект имеет метастатический рак толстой и прямой кишки, имеющий мутацию BRAF V600E. В одном варианте осуществления субъект имеет метастатический рак толстой и прямой кишки IV стадии, имеющий мутацию BRAF V600E.
[00107] Термин "регулирующий орган" является государственным органом, осуществляющим регламентирование медицинского применения фармацевтических средств в стране. Например, неограничивающим примером регулирующего органа является Управление по контролю качества пищевых продуктов и лекарственных препаратов США (FDA).
[00108] Термин "педиатрический пациент" при использовании в настоящем документе относится к пациенту возрастом до 16 лет на момент проведения диагностики или лечения. Термин "педиатрический" может дополнительно подразделить на различные подгруппы, включающие: новорожденные (от рождения до первого месяца жизни); грудных детей (возрастом от 1 месяца до двух лет); детей младшего возраста (возрастом от двух лет до 12 лет); и подростков (возрастом от 12 лет до 21 года (до, но не включая, двадцать второй день рождения)). Berhman RE, Kliegman R, Arvin AM, Nelson WE. Nelson Textbook of Pediatrics, 15th Ed. Philadelphia: W.B. Saunders Company, 1996; Rudolph AM, et al. Rudolph's Pediatrics, 21st Ed. New York: McGraw-Hill, 2002; и Avery MD, First LR. Pediatric Medicine, 2nd Ed. Baltimore: Williams & Wilkins; 1994.
[00109] Термин "BRAF-ассоциированное заболевание или нарушение" при использовании в настоящем документе относится к заболеваниям или нарушениям, ассоциированным с дисрегуляцией или имеющим дисрегуляцию гена BRAF, BRAF-киназы (также именуемой в настоящем документе белок BRAF-киназы или BRAF-киназа) или экспрессии или активности, или уровня (например, одного или более) указанных (например, любого из типов дисрегуляции гена BRAF, BRAF-киназы, домена BRAF-киназы или их экспрессии или активности, или уровня, описанных в настоящем документе). Неограничивающие примеры BRAF-ассоциированного заболевания или нарушения включают, например, BRAF-ассоциированный рак.
[00110] Термин "BRAF-ассоциированный рак" при использовании в настоящем документе относится к раковым опухолям, ассоциированным с дисрегуляцией или имеющим дисрегуляцию гена BRAF, BRAF-киназы (также именуемой в настоящем документе белок BRAF-киназы или BRAF), или их экспрессии или активности, или уровня. В одном варианте осуществления BRAF-ассоциированный рак является раком, имеющим мутацию BRAF V600. В одном варианте осуществления BRAF-ассоциированный рак является раком, имеющим мутацию BRAF V600E. В одном варианте осуществления BRAF-ассоциированный рак является раком толстой и прямой кишки. В одном варианте осуществления рак толстой и прямой кишки является метастатическим раком толстой и прямой кишки. В одном варианте осуществления BRAF-ассоциированный рак является метастатическим раком толстой и прямой кишки, имеющим мутацию BRAF V600E. В одном варианте осуществления BRAF-ассоциированный рак является метастатическим раком толстой и прямой кишки, имеющим мутацию BRAF V600.
[00111] Фраза "дисрегуляция гена BRAF, BRAF-киназы или их экспрессии или активности, или уровня" относится к генетической мутации (например, транслокации гена BRAF, которая приводит к экспрессии слитого белка, делеции в гене BRAF, которая приводит к экспрессии белка BRAF, который включает делецию по меньшей мере одной аминокислоты по сравнению с белком BRAF дикого типа, мутацию в гене BRAF, которая приводит к экспрессии белка BRAF с одной или более точечными мутациями или альтернативно сплайсированному варианту мРНК BRAF, которая приводит к белку BRAF, который приводит к делеции по меньшей мере одной аминокислоты в белке BRAF по сравнению с белком BRAF дикого типа), амплификацию гена BRAF, которая приводит к оверэкспрессии белка BRAF или аутокринной активности в результате оверэкспрессии гена BRAF клетки, что приводит к патогенному увеличению активности киназного домена белка BRAF (например, конститутивно активного киназного домена белка BRAF) в клетке. Например, дисрегуляция гена BRAF, белка BRAF или его экспрессии, или активности, или уровня может быть мутацией в гене BRAF, который кодирует белок BRAF, который является конститутивно активным или обладает повышенной активностью по сравнению с белком, кодируемым геном BRAF, который не включает мутацию. В качестве другого примера, дисрегуляция гена BRAF, белка BRAF или его экспрессии или активности, или уровня может быть результатом транслокации гена или хромосомы, что приводит к экспрессии слитого белка, который содержит первую часть BRAF, которая включает функциональный киназный домен, и вторую часть белка-партнера (т.е. не являющегося BRAF). В некоторых примерах дисрегуляция гена BRAF, белка BRAF или экспрессии или активности может быть результатом трансляции гена одного гена BRAF с другим геном BRAF. Примеры мутаций BRAF включают мутации V600E, V600K и V600G. В одном варианте осуществления мутация представляет собой мутацию BRAF V600E.
[00112] Термин "MEK-ассоциированное заболевание или нарушение" при использовании в настоящем документе относится к заболеваниям или нарушениям, ассоциированным с путем Ras/Raf/MEK, например заболеваниям или нарушениям, ассоциированным с дисрегуляцией или имеющим дисрегуляцию гена MEK, MEK-киназы (также именуемой в настоящем документе белок MEK-киназы или MEK-киназа), или экспрессии или активности, или уровню любого (например, одного или более) из перечисленных (например, любого из типов дисрегуляции гена MEK, MEK-киназы, домена MEK-киназы или экспрессии или активности, или уровня любого из них, как описано в настоящем документе). Неограничивающие примеры MEK-ассоциированного заболевания или нарушения включают, например, рак.
[00113] Термин "MEK-ассоциированный рак" при использовании в настоящем документе относится к раковым опухолям, ассоциированным с дисрегуляцией или имеющим дисрегуляцию гена MEK, MEK-киназы (также именуемой в настоящем документе белок MEK-киназы или MEK), или их экспрессии или активности, или уровню. Неограничивающие примеры MEK-ассоциированного рака описаны в настоящем документе. В одном варианте осуществления MEK-ассоциированный рак является раком толстой и прямой кишки. В одном варианте осуществления рак толстой и прямой кишки является метастатическим раком толстой и прямой кишки.
[00114] Фраза "дисрегуляция гена MEK, MEK-киназы или их экспрессии или активности, или уровня" относится к генетической мутации (например, транслокации гена MEK, которая приводит к экспрессии слитого белка, делеции в гене MEK, которая приводит к экспрессии белка MEK, который включает делецию по меньшей мере одной аминокислоты по сравнению с белком MEK дикого типа, мутацию в гене MEK, которая приводит к экспрессии белка MEK с одной или более точечными мутациями или альтернативно сплайсированному варианту мРНК MEK, которая приводит к белку MEK, который приводит к делеции по меньшей мере одной аминокислоты в белке MEK по сравнению с белком MEK дикого типа), амплификацию гена MEK, которая приводит к оверэкспрессии белка MEK или аутокринной активности в результате оверэкспрессии гена MEK клетки, которая приводит к патогенному увеличению активности киназного домена белка MEK (например, конститутивно активному киназному домену белка MEK) в клетке. Например, дисрегуляция гена MEK, белка MEK или их экспрессии или активности, или уровня может быть мутацией в гене MEK, который кодирует белок MEK, который является конститутивно активным или обладает повышенной активностью по сравнению с белком, кодируемым геном MEK, который не включает мутацию. В качестве другого примера, дисрегуляция гена MEK, белка MEK или их экспрессии или активности, или уровня может быть результатом транслокации гена или хромосомы, что приводит к экспрессии слитого белка, который содержит первую часть MEK, которая включает функциональный киназный домен, и вторую часть белка-партнера (т.е. не являющегося MEK). В некоторых примерах дисрегуляция гена MEK, белка MEK или экспрессии или активности может быть результатом трансляции одного гена MEK с другим геном MEK.
[00115] Термин "EGFR-ассоциированное заболевание или нарушение" при использовании в настоящем документе относится к заболеваниям или нарушениям, ассоциированным с дисрегуляцией или имеющим дисрегуляцию гена EGFR, EGFR-киназы (также именуемой в настоящем документе белок EGFR-киназы или EGFR-киназа), или экспрессии или активности, или уровня любого (например, одного или более) из перечисленного (например, любого из типов дисрегуляции гена EGFR, EGFR-киназы, домена EGFR-киназы или их экспрессии или активности, или уровня, как описано в настоящем документе). Неограничивающие примеры EGFR-ассоциированного заболевания или нарушения включают, например, рак, например, рак толстой и прямой кишки, например метастатический рак толстой и прямой кишки.
[00116] Термин "EGFR-ассоциированный рак" при использовании в настоящем документе относится к раковым образованиям, ассоциированным с дисрегуляцией или имеющим дисрегуляцию гена EGFR, EGFR-киназы (также именуемой в настоящем документе белок EGFR-киназы или EGFR), или их экспрессии или активности, или уровня. В одном варианте осуществления EGFR-ассоциированный рак является раком толстой и прямой кишки. В одном варианте осуществления рак толстой и прямой кишки является метастатическим раком толстой и прямой кишки.
[00117] Фраза "дисрегуляция гена EGFR, EGFR-киназы или их экспрессии или активности, или уровня" относится к генетической мутации (например, транслокации гена EGFR, которая приводит к экспрессии слитого белка, делеции в гене EGFR, которая приводит к экспрессии белка EGFR, который включает делецию по меньшей мере одной аминокислоты по сравнению с белком EGFR дикого типа, мутации в гене EGFR, которая приводит к экспрессии белка EGFR с одной или более точечными мутациями или альтернативному сплайсированному варианту мРНК EGFR, которая приводит к белку EGFR, который приводит к делеции по меньшей мере одной аминокислоты в белке EGFR по сравнению с белком EGFR дикого типа), амплификации гена EGFR, которая приводит к оверэкспрессии белка EGFR или аутокринной активности в результате оверэкспрессии гена EGFR клетки, которая приводит к патогенному увеличению активности киназного домена белка EGFR (например, конститутивно активного киназного белка EGFR) в клетке. Например, дисрегуляция гена EGFR, белка EGFR или их экспрессии или активности, или уровня может быть мутацией в гене EGFR, который кодирует белок EGFR, который является конститутивно активным или обладает увеличенной активностью по сравнению с белком, кодируемым геном EGFR, который не включает мутацию. В качестве другого примера, дисрегуляция гена EGFR, белка EGFR или их экспрессии или активности, или уровня может быть результатом транслокации гена или хромосомы, что приводит к экспрессии слитого белка, который содержит первую часть EGFR, которая включает функциональный киназный домен, и вторую часть белка-партнера (т.е. не являющегося EGFR). В некоторых примерах, дисрегуляция гена EGFR, белка EGFR или экспрессии или активности может быть результатом трансляции одного гена EGFR с другим геном EGFR.
[00118] При одновременном, последовательном или раздельном введении ингибитор BRAF (СОЕДИНЕНИЕ A), ингибитор MEK (СОЕДИНЕНИЕ B) и антитело против EGFR (СОЕДИНЕНИЕ C) могут взаимодействовать синергически, вызывая сильное ингибирование пролиферации клеток. Такая неожиданная синергическая реакция может позволить снизить дозу, требуемую для каждого соединения, что приведет к уменьшению побочных эффектов и улучшению долговременной клинической эффективности соединений при лечении.
[00119] Термин "синергический эффект" при использовании в настоящем документе относится к действию двух или трех терапевтических средств, таких как, например, ингибитор BRAF (СОЕДИНЕНИЕ A) и/или ингибитор MEK (СОЕДИНЕНИЕ B), и/или антитело против EGFR (СОЕДИНЕНИЕ C), производящих эффект, например, замедляющий симптоматическую прогрессию пролиферативного заболевания, в частности рака или его симптомов, который превышает простой аддитивный эффект каждого лекарственного средства (т.е. терапевтического средства), вводимого самостоятельно. Синергический эффект может быть вычислен, например, при использовании подходящих методов, такие как уравнение сигмоидальной функции Emax (Holford, N. H. G. and Scheiner, L. B., Clin. Pharmacokinet. 6: 429-453 (1981)), уравнение аддитивности Леве (Loewe, S. and Muischnek, H., Arch. Exp. Pathol Pharmacol. 114: 313-326 (1926)) и уравнение медианного эффекта (Chou, T. C. and Talalay, P., Adv. Enzyme Regul. 22: 27-55 (1984)). Каждое уравнение, указанное выше, может быть применено к экспериментальным данным для получения соответствующего графика, удобного при оценке эффектов комбинации лекарственных средств. Соответствующие графики, связанные с уравнениями, указанными выше, являются кривой зависимости эффекта от концентрации, кривой изоболограммы и кривой индекса комбинации, соответственно.
[00120] Также предложен способ лечения пролиферативного заболевания у нуждающегося в этом субъекта, включающий введение субъекту КОМБИНАЦИИ ИЗОБРЕТЕНИЯ, где СОЕДИНЕНИЕ A, СОЕДИНЕНИЕ B и СОЕДИНЕНИЕ C вводят в терапевтически эффективных дозах, которые, при объединении в комбинацию, обеспечивают благоприятное воздействие. Введение компонентов комбинации может быть одновременным или последовательным. В одном варианте осуществления СОЕДИНЕНИЕ A, СОЕДИНЕНИЕ B и СОЕДИНЕНИЕ C сформулированные каждое отдельно в единичные стандартные лекарственные формы для одновременного, раздельного или последовательного введения.
[00121] В одном варианте осуществления пролиферативным заболеванием, которое лечат КОМБИНАЦИЕЙ ИЗОБРЕТЕНИЯ, является рак. Примеры раковых опухолей, которые можно лечить КОМБИНАЦИЕЙ ИЗОБРЕТЕНИЯ, включают рак толстой и прямой кишки (CRC) (включая метастатический рак толстой и прямой кишки), меланому (включая метастатическую меланому), рак легкого (включая немелкоклеточный рак легкого (НМРЛ)), рак молочной железы, рак почки, такой как, например, почечно-клеточную карциному (RCC), рак печени, рак эндометрия, острый миелогенный лейкоз (ОМЛ), миелодиспластические синдромы (MDS), рак щитовидной железы, в частности папиллярный рак щитовидной железы, рак поджелудочной железы, нейрофиброматоз или гепатоцеллюлярную карциному. Рак толстой и прямой кишки является третьим по распространенности раком у мужчин и женщин в США. Рак толстой и прямой кишки является третьим по распространенности раком у мужчин и женщин в США, с более чем 134000 новыми случаями и почти 50000 случаев смерти от этого заболевания, спрогнозированными в 2016 году. В США мутации BRAF встречаются у 8-15% больных раком толстой и прямой кишки и представляют плохой прогноз для этих пациентов. Опубликованные ретроспективные результаты по выживаемости без прогрессирования заболевания (PFS) и общей выживаемости (OS) после лечения препаратами первой линии колеблются от 1,8 до 2,5 месяцев и от 4 до 6 месяцев, соответственно, и опубликованные данные по проценту ответа в различных исследованиях терапии на основе EGFR в этой совокупности пациентов колеблются от 6 до 8 процентов. В одном варианте осуществления рак является BRAF-ассоциированным раком. В одном варианте осуществления рак является раком, имеющим мутацию BRAF V600. В одном варианте осуществления рак является раком, имеющим мутацию BRAF V600E. В одном варианте осуществления рак является колоректальным раком. В одном варианте осуществления колоректальный рак является метастатическим колоректальным раком. В одном варианте осуществления рак является колоректальным раком, имеющим мутацию BRAF V600E. В одном варианте осуществления рак является метастатическим колоректальным раком, имеющим мутацию BRAF V600E. В одном варианте осуществления рак является MEK-ассоциированным раком. В одном варианте осуществления MEK-ассоциированный рак является колоректальным раком. В одном варианте осуществления рак является EGFR-ассоциированным раком. В одном варианте осуществления EGFR-ассоциированный рак является колоректальным раком.
[00122] В соответствии с настоящим изобретением, терапевтически эффективное количество каждого из партнеров по комбинации в КОМБИНАЦИИ ИЗОБРЕТЕНИЯ могут вводить одновременно, раздельно или последовательно и в любом порядке, и компоненты могут вводить раздельно или в фиксированной комбинации. В одном варианте осуществления способ лечения пролиферативного заболевания может включать: (i) введение СОЕДИНЕНИЯ A в свободной форме или форме фармацевтически приемлемой соли, (ii) введение СОЕДИНЕНИЯ B в свободной форме или форме фармацевтически приемлемой соли, и (iii) введение СОЕДИНЕНИЯ C, одновременно или последовательно, в любом порядке, в совместно терапевтически эффективных количествах, (например, в синергически эффективных количествах), например, в ежедневных или периодически вводимых дозах, соответствующих количествам, описанным в настоящем документе. Отдельные партнеры по комбинации в КОМБИНАЦИИ ИЗОБРЕТЕНИЯ могут вводить раздельно, в разное время в течение терапии, или параллельно, в разделенных или единых комбинированных формах. В одном варианте осуществления СОЕДИНЕНИЕ A могут вводить ежедневно, один раз в день или два раза в день, СОЕДИНЕНИЕ B могут вводить ежедневно, один раз в день или два раза в день, и СОЕДИНЕНИЕ C могут вводить еженедельно. Таким образом, следует понимать, что настоящее изобретение охватывает все такие схемы одновременного или чередующегося лечения, и термин "введение" следует интерпретировать соответствующим образом.
[00123] Эффективная доза каждого из партнеров по комбинации, применяемых в КОМБИНАЦИИ ИЗОБРЕТЕНИЯ, может изменяться в зависимости от конкретного используемого соединения или фармацевтической композиции, способа введения, подвергаемого лечению состояния и тяжести подвергаемого лечению состояния. Таким образом, схему применения КОМБИНАЦИИ ИЗОБРЕТЕНИЯ выбирают в соответствии с различными факторами, включающими путь введения и функцию почек и печени пациента.
[00124] Оптимальные соотношения, индивидуальные и комбинированные дозы и концентрации компонентов комбинации (СОЕДИНЕНИЕ A, СОЕДИНЕНИЕ B и СОЕДИНЕНИЯ C) КОМБИНАЦИИ ИЗОБРЕТЕНИЯ, которые обеспечивают эффективность без токсичности, основаны на кинетике доступности терапевтических средств для сайтов-мишеней и могут быть определены при использовании способов, известных специалистам в данной области.
[00125] Эффективная доза каждого из партнеров по комбинации может потребовать более частого введения одного из соединений по сравнению с другими соединениями в комбинации. Таким образом, для обеспечения правильного дозирования, в одном варианте осуществления упакованные фармацевтические продукты могут содержать одну или более лекарственных форм, которые содержат комбинацию соединений, и одну или более лекарственных форм, которые содержат одно соединение из комбинации соединений, но не содержат другое соединение(я) комбинации. В еще одном варианте осуществления упакованные фармацевтические продукты могут содержать одну или более лекарственных форм одного, двух и/или всех трех компонентов комбинации.
[00126] В случае, когда партнеры по комбинации, применяемые в КОМБИНАЦИИ ИЗОБРЕТЕНИЯ, применяют в форме, доступной на рынке, в виде отдельных лекарственных средств, их доза и способ введения могут соответствовать информации, представленной на вкладыше в упаковку соответствующего коммерческого лекарственного средства, если в настоящем документе не указано иное.
[00127] Ингибитор BRAF (СОЕДИНЕНИЕ A) могут вводить подходящему субъекту ежедневно в однократных или разделенных дозах, в эффективной дозе в диапазоне от приблизительно 0,05 до приблизительно 50 мг на кг массы тела в день (например, приблизительно 0,10 мг/кг/день, 0,30 мг/кг/день, 0,50 мг/кг/день, 0,80 мг/кг/день, 1 мг/кг/день, 5 мг/кг/день, 10 мг/кг/день, 15 мг/кг/день, 20 мг/кг/день, 25 мг/кг/день, 30 мг/кг/день, 35 мг/кг/день, 40 мг/кг/день, 50 мг/кг/день, 0,1-25 мг/кг/день или 0,5-10 мг/кг/день). Например, дозы могут составлять от приблизительно 35 мг до приблизительно 700 мг (например, приблизительно 50 мг, приблизительно 75 мг, приблизительно 100 мг, приблизительно 120 мг, приблизительно 150 мг, приблизительно 175 мг, приблизительно 200 мг, приблизительно 225 мг, приблизительно 250 мг, приблизительно 275 мг, приблизительно 300 мг, приблизительно 325 мг, приблизительно 350 мг, приблизительно 375 мг, приблизительно 400 мг, приблизительно 450 мг, приблизительно 500 мг, приблизительно 550 мг, приблизительно 600 мг или приблизительно 650 мг). В однократных или разделенных дозах. Для человека весом 70 кг такое количество будет составлять предпочтительный диапазон дозы приблизительно 35-700 мг в день, например, 300 мг в день. В одном варианте осуществления СОЕДИНЕНИЕ A вводят перорально. В одном варианте осуществления СОЕДИНЕНИЕ A находится в форме капсул. В одном варианте осуществления капсулированная лекарственная форма СОЕДИНЕНИЯ A включает 50 мг СОЕДИНЕНИЯ A. В одном варианте осуществления капсулированная лекарственная форма СОЕДИНЕНИЯ A включает 75 мг СОЕДИНЕНИЯ A. В одном варианте осуществления капсулированная лекарственная форма СОЕДИНЕНИЯ A включает 100 мг СОЕДИНЕНИЯ A. В одном варианте осуществления капсулированная лекарственная форма СОЕДИНЕНИЯ A включает аморфное СОЕДИНЕНИЕ A. В одном варианте осуществления 300 мг СОЕДИНЕНИЯ A вводят перорально один раз в день. В одном варианте осуществления 225 мг СОЕДИНЕНИЯ A вводят перорально один раз в день. В одном варианте осуществления 150 мг СОЕДИНЕНИЯ A вводят перорально один раз в день. В одном варианте осуществления 300 мг СОЕДИНЕНИЯ A первоначально вводят перорально один раз в день для наблюдения нежелательных явлений, после чего 225 мг СОЕДИНЕНИЯ A вводят один раз в день. В одном варианте осуществления доза пациентов, дозу которых уменьшили до 225 мг СОЕДИНЕНИЯ A один раз в день, может быть снова повышена до 300 мг СОЕДИНЕНИЯ A один раз в день, если нежелательные явления, которые привели к снижению дозы, уменьшаются до исходного уровня и остаются стабильными в течение, например, до 6 дней или до 10 дней, или до 14 дней, или до 15 дней, или до 4 недель, или до 6 недель, в зависимости от тяжести нежелательной реакции, если отсутствуют какие-либо иные сопутствующие токсические явления, связанные с СОЕДИНЕНИЕМ A, которые будут препятствовать возобновлению применения более высокой дозы лекарственного средства. В одном варианте осуществления 300 мг СОЕДИНЕНИЯ A первоначально вводят перорально один раз в день для наблюдения нежелательных явлений, после чего 150 мг СОЕДИНЕНИЯ A вводят один раз в день. В одном варианте осуществления доза пациентов, дозу которых уменьшили до 150 мг СОЕДИНЕНИЯ A один раз в день, может быть снова повышена до 300 мг СОЕДИНЕНИЯ A один раз в день, если нежелательные явления, которые привели к снижению дозы, уменьшаются до исходного уровня и остаются стабильными в течение, например, до 6 дней или до 10 дней, или до 14 дней, или до 15 дней, или до 4 недель, или до 6 недель, в зависимости от тяжести нежелательной реакции, если отсутствуют какие-либо иные сопутствующие токсические явления, связанные с СОЕДИНЕНИЕМ A, которые будут препятствовать возобновлению применения более высокой дозы лекарственного средства.
[00128] Ингибитор MEK (СОЕДИНЕНИЕ B) могут вводить подходящему субъекту ежедневно в однократных или разделенных дозах, в эффективной дозе в диапазоне от приблизительно 0,001 до приблизительно 100 мг на кг массы тела в день (например, приблизительно 0,01 мг/кг/день, приблизительно 0,10 мг/кг/день, 0,30 мг/кг/день, 0,50 мг/кг/день, 0,80 мг/кг/день, 1 мг/кг/день, 5 мг/кг/день, 10 мг/кг/день, 15 мг/кг/день, 20 мг/кг/день, 25 мг/кг/день, 30 мг/кг/день, 35 мг/кг/день, 40 мг/кг/день, 50 мг/кг/день, 60 мг/кг/день, 70 мг/кг/день, 80 мг/кг/день, 90 мг/кг/день, 100 мг/кг/день, 0,1-25 мг/кг/день, 0,5-10 мг/кг/день или 1-35 мг/кг/день). Например, дозы могут составлять от приблизительно 10 мг до приблизительно 85 мг (например, приблизительно 15 мг, приблизительно 20 мг, приблизительно 25 мг, приблизительно 30 мг, приблизительно 35 мг, приблизительно 40 мг, приблизительно 45 мг, приблизительно 50 мг, приблизительно 55 мг, приблизительно 60 мг, приблизительно 65 мг, приблизительно 70 мг, приблизительно 75 мг, приблизительно 80 мг или приблизительно 85 мг). Для человека весом 70 кг такое количество будет составлять предпочтительный диапазон дозы приблизительно 0,05-7 г/день, предпочтительно от приблизительно 0,05 до приблизительно 2,5 г/день, в частности 0,9 г/день. В одном варианте осуществления СОЕДИНЕНИЕ B вводят перорально. В одном варианте осуществления СОЕДИНЕНИЕ B включено в таблетированную лекарственную форму. В одном варианте осуществления таблетированная лекарственная форма СОЕДИНЕНИЯ B включает 15 мг СОЕДИНЕНИЯ B. В одном варианте осуществления СОЕДИНЕНИЕ B вводят перорально два раза в день. В одном варианте осуществления СОЕДИНЕНИЕ B вводят перорально два раза в день, где вторую дозу СОЕДИНЕНИЯ B вводят через приблизительно 12 часов после первой дозы СОЕДИНЕНИЯ B. В одном варианте осуществления 45 мг СОЕДИНЕНИЯ B вводят перорально два раза в день. В одном варианте осуществления СОЕДИНЕНИЕМ B является (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты или его фармацевтически приемлемая соль. В одном варианте осуществления СОЕДИНЕНИЕМ B является кристаллизованный (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты. В одном варианте осуществления таблетированная лекарственная форма СОЕДИНЕНИЯ B включает 15 мг кристаллизованного (2-гидроксиэтокси)амида 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты. В одном варианте осуществления 45 мг кристаллизованного (2-гидроксиэтокси)амида 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты вводят два раза в день для наблюдения нежелательных явлений, после чего 30 мг кристаллизованного (2-гидроксиэтокси)амида 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты вводят два раза в день. В одном варианте осуществления доза пациентов, дозу которых уменьшили до 30 мг два раза в день, может быть снова повышена до 45 мг два раза в день, если нежелательные явления, которые привели к снижению дозы, уменьшаются до исходного уровня и остаются стабильными в течение, например, до 14 дней или до трех недель, или до 4 недель, если отсутствуют какие-либо иные сопутствующие токсические явления, связанные с (2-гидроксиэтокси)амидом 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты, которые будут препятствовать возобновлению применения более высокой дозы лекарственного средства.
[00129] В одном варианте осуществления 45 мг кристаллизованного (2-гидроксиэтокси)амида 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты вводят два раза в день для наблюдения нежелательных явлений, после чего 15 мг кристаллизованного (2-гидроксиэтокси)амида 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты вводят два раза в день. В одном варианте осуществления доза пациентов, дозу которых уменьшили до 15 мг два раза в день, может быть снова повышена до 45 мг два раза в день, если нежелательные явления, которые привели к снижению дозы, уменьшаются до исходного уровня и остаются стабильными в течение, например, до 14 дней или до трех недель, или до 4 недель, если отсутствуют какие-либо иные сопутствующие токсические явления, связанные с (2-гидроксиэтокси)амидом 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты, которые будут препятствовать возобновлению применения более высокой дозы лекарственного средства.
[00130] Антитело против EGFR (СОЕДИНЕНИЕ C) могут вводить ежедневно или еженедельно подходящему субъекту в однократных или разделенных дозах, в эффективной дозе в диапазоне от приблизительно 0,001 до приблизительно 100 мг на кг массы тела в день (например, приблизительно 0,01 мг/кг/день, приблизительно 0,10 мг/кг/день, 0,30 мг/кг/день, 0,50 мг/кг/день, 0,80 мг/кг/день, 1 мг/кг/день, 5 мг/кг/день, 10 мг/кг/день, 15 мг/кг/день, 20 мг/кг/день, 25 мг/кг/день, 30 мг/кг/день, 35 мг/кг/день, 40 мг/кг/день, 50 мг/кг/день, 60 мг/кг/день, 70 мг/кг/день, 80 мг/кг/день, 90 мг/кг/день, 100 мг/кг/день, 0,1-25 мг/кг/день, 0,5-10 мг/кг/день или 1-35 мг/кг/день). Например, дозы могут составлять от приблизительно 50 мг до приблизительно 1000 мг, например, от приблизительно 150 мг до приблизительно 500 мг (например, приблизительно 175 мг, приблизительно 200 мг, приблизительно 225 мг, приблизительно 250 мг, приблизительно 275 мг, приблизительно 300 мг, приблизительно 325 мг, приблизительно 350 мг, приблизительно 375 мг, приблизительно 400 мг, приблизительно 425 мг, приблизительно 450 мг или приблизительно 475 мг) или от приблизительно 100 мг до приблизительно 350 мг (например, приблизительно 125 мг, приблизительно 150 мг, приблизительно 175 мг, приблизительно 200 мг, приблизительно 225 мг, приблизительно 250 мг, приблизительно 275 мг, приблизительно 300 мг или приблизительно 325 мг). Для человека весом 70 кг такое количество будет составлять предпочтительный диапазон дозы приблизительно 0,07-2,45 г/день, предпочтительно от приблизительно 0,05 до приблизительно 1,0 г/день. В одном варианте осуществления антитело против EGFR вводят в виде внутривенной инфузии. В одном варианте осуществления антителом против EGFR является цетуксимаб (Эрбитукс®), где цетуксимаб вводят еженедельно в виде внутривенной инфузии. В одном варианте осуществления антителом против EGFR является цетуксимаб (Эрбитукс®), где цетуксимаб вводят в виде внутривенной инфузии в количестве 400 мг/м2 в течение 120 минут в качестве начальной дозы, с последующими еженедельными внутривенными инфузиями в количестве 250 мг/м2 в течение 30 минут, согласно инструкции по медицинскому применению лекарственного препарата. В одном варианте осуществления антителом против EGFR является цетуксимаб (Эрбитукс®), где цетуксимаб вводят в виде внутривенной инфузии в количестве 400 мг/м2 в течение 120 минут в качестве начальной дозы, с последующими еженедельными внутривенными инфузиями в количестве 250 мг/м2 в течение 60 минут согласно инструкции по медицинскому применению лекарственного препарата. Однако также возможно снижение дозы. Таким образом, согласно одному варианту осуществления цетуксимаб первоначально вводят в виде внутривенной инфузии в дозе от 200 до 400 мг/м2 в течение 120 минут, с последующими еженедельными внутривенными инфузиями в дозе от 125 до 250 мг/м2. Например, в одном варианте осуществления дозу цетуксимаба, вводимого после начальной внутривенной инфузии в количестве 400 мг/м, снижают до 200 мг/м2 (60-минутная инфузия) один раз в неделю. В другом варианте осуществления дозу цетуксимаба, вводимую после начальной внутривенной инфузии в количестве 400 мг/м2, снижают до 150 мг/м2 (60-минутная инфузия) один раз в неделю.
[00131] В одном варианте осуществления схема применения КОМБИНАЦИИ ИЗОБРЕТЕНИЯ включает пероральное введение терапевтически эффективного количества СОЕДИНЕНИЯ A, пероральное введение терапевтически эффективного количества СОЕДИНЕНИЯ B и внутривенное введение терапевтически эффективного количества СОЕДИНЕНИЯ C. В одном варианте осуществления схема применения КОМБИНАЦИИ ИЗОБРЕТЕНИЯ включает пероральное введение СОЕДИНЕНИЯ (300 мг, вводимые перорально два раза в день), пероральное введение СОЕДИНЕНИЯ B (45 мг, вводимые перорально один раз в день) и внутривенное введение СОЕДИНЕНИЯ C. В одном варианте осуществления схема применения КОМБИНАЦИИ ИЗОБРЕТЕНИЯ включает пероральное введение СОЕДИНЕНИЯ (300 мг, вводимые перорально два раза в день), пероральное введение СОЕДИНЕНИЯ B (45 мг, вводимые перорально один раз в день) и еженедельное внутривенное введение СОЕДИНЕНИЯ C. В одном варианте осуществления СОЕДИНЕНИЕ A находится в аморфной форме. В одном варианте осуществления СОЕДИНЕНИЕМ B является (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты. В одном варианте осуществления СОЕДИНЕНИЕМ B является кристаллизованным (2-гидроксиэтокси)амидом 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты. В одном варианте осуществления СОЕДИНЕНИЕ B вводят перорально два раза в день, где вторую дозу СОЕДИНЕНИЯ B вводят через приблизительно 12 часов после первой дозы СОЕДИНЕНИЯ B. В одном варианте осуществления СОЕДИНЕНИЕМ C является цетуксимаб. В одном варианте осуществления цетуксимаб вводят в виде внутривенной инфузии в количестве 400 мг/м2 в течение 120 минут в качестве начальной дозы, с последующими еженедельными внутривенными инфузиями в количестве 250 мг/м2 в течение 30 минут. В одном варианте осуществления цетуксимаб вводят первоначально в виде внутривенной инфузии в дозе от 200 до 400 мг/м2 в течение 120 минут в качестве начальной дозы, с последующими еженедельными внутривенными инфузиями в дозе от 125 до 250 мг/м2 в течение 30 минут. В одном варианте осуществления цетуксимаб вводят в виде внутривенной инфузии в количестве 400 мг/м2 в течение 120 минут в качестве начальной дозы, с последующими еженедельными внутривенными инфузиями в количестве 250 мг/м2 в течение 60 минут. В одном варианте осуществления цетуксимаб вводят первоначально в виде внутривенной инфузии в дозе от 200 до 400 мг/м2 в течение 120 минут в качестве начальной дозы, с последующими еженедельными внутривенными инфузиями в дозе от 125 до 250 мг/м2 в течение 60 минут. В одном варианте осуществления схема применения КОМБИНАЦИИ ИЗОБРЕТЕНИЯ включает пероральное введение СОЕДИНЕНИЯ (300 мг в капсулированной лекарственной форме вводят перорально один раз в день), пероральное введение СОЕДИНЕНИЯ B (45 мг вводят перорально два раза в день, где введение включает пероральное введение трех таблеток таблетированной лекарственной формы СОЕДИНЕНИЯ B, где каждая таблетка включает 15 мг СОЕДИНЕНИЯ B) и внутривенное введение СОЕДИНЕНИЯ C. В одном варианте осуществления СОЕДИНЕНИЕ B вводят перорально два раза в день, где вторую дозу СОЕДИНЕНИЯ B вводят через приблизительно 12 часов после первой дозы СОЕДИНЕНИЯ B. В одном варианте осуществления, в дни, когда вводят СОЕДИНЕНИЕ C, СОЕДИНЕНИЕ A и СОЕДИНЕНИЕ B вводят по меньшей мере за 30 минут до введения СОЕДИНЕНИЯ C.
[00132] КОМБИНАЦИЮ ИЗОБРЕТЕНИЯ могут вводить в качестве КОМБИНИРОВАННОЙ ТЕРАПИИ. Термин "комбинированная терапия" при использовании в настоящем документе относится к схеме применения терапевтически активных средств, СОЕДИНЕНИЯ A, СОЕДИНЕНИЯ B и СОЕДИНЕНИЯ C, содержащихся в одной или нескольких композициях, где терапевтически активные средства вводят вместе или отдельно (каждое или в их любых комбинациях), как предписано лицом, осуществляющим медицинское обслуживание, или в соответствии с нормативами регулирующего органа, как определено в настоящем документе. Таким образом, в одном варианте осуществления в настоящем документе предложен способ лечения пролиферативного заболевания, включающий введение нуждающемуся в этом пациенту комбинированной терапии, независимо включающей терапевтически эффективные количества: (a) ингибитора BRAF, которым является метил-N-[(2S)-1-({4-[3-(5-хлор-2-фтор-3-метансульфонамидофенил)-1-(пропан-2-ил)-1H-пиразол-4-ил]пиримидин-2-ил}амино)пропан-2-ил]карбамат (СОЕДИНЕНИЕ A) или его фармацевтически приемлемая соль, (b) по меньшей мере одного ингибитора MEK (СОЕДИНЕНИЯ B) или его фармацевтически приемлемой соли и (c) антитела против EGFR (СОЕДИНЕНИЯ C).
[00133] В одном варианте осуществления любой из схем применения КОМБИНИРОВАННОЙ ТЕРАПИИ, описанной в настоящем документе, терапевтически эффективное количество СОЕДИНЕНИЯ A принимают вместе с первой терапевтически эффективной дозой СОЕДИНЕНИЯ B. При использовании в настоящем документе фраза "принимают вместе с" означает, что не больше 5 минут или не больше 10 минут, или не больше 15 минут, или не больше 20 минут, или не больше 25 минут или не больше 30 минут прошло между введением СОЕДИНЕНИЯ A и СОЕДИНЕНИЯ B.
[00134] В одном варианте осуществления любой из схем применения КОМБИНИРОВАННОЙ ТЕРАПИИ, описанной в настоящем документе, вторую терапевтически эффективную дозу СОЕДИНЕНИЯ B вводят через приблизительно 12 часов после введения первой дозы СОЕДИНЕНИЯ B. При использовании в настоящем документе, фраза "через приблизительно 12 часов после введения первой дозы СОЕДИНЕНИЯ B" означает, что вторую дозу СОЕДИНЕНИЯ B вводят через 10-14 часов после введения первой дозы СОЕДИНЕНИЯ B.
[00135] В одном варианте осуществления любой из схем применения КОМБИНИРОВАННОЙ ТЕРАПИИ, описанной в настоящем документе, в дни, когда вводят СОЕДИНЕНИЕ C, СОЕДИНЕНИЕ C вводят через по меньшей мере 30 минут после последнего введения терапевтически эффективного количества СОЕДИНЕНИЯ A и первой терапевтически эффективной дозы СОЕДИНЕНИЯ B. При использовании в настоящем документе, фраза "через по меньшей мере 30 минут после" означает, что СОЕДИНЕНИЕ C вводят через по меньшей мере 30 минут или по меньшей мере 35 минут, или по меньшей мере 40 минут, или по меньшей мере 45 минут, или по меньшей мере 50 минут, или по меньшей мере 55 минут, или по меньшей мере 60 минут, или по меньшей мере 65 минут, или по меньшей мере 70 минут, или по меньшей мере 75 минут, или по меньшей мере 80 минут, или по меньшей мере 85 минут, или по меньшей мере 90 минут после последнего введения СОЕДИНЕНИЯ A и первой дозы СОЕДИНЕНИЯ B.
[00136] В одном варианте осуществления любой из схем применения КОМБИНИРОВАННОЙ ТЕРАПИИ, описанной в настоящем документе, в дни, когда вводят СОЕДИНЕНИЕ C, терапевтически эффективное количество СОЕДИНЕНИЯ C вводят по меньшей мере за 30 минут до введения терапевтически эффективного количества СОЕДИНЕНИЯ A и первой терапевтически эффективной дозы СОЕДИНЕНИЯ B, в зависимости от того, что произойдет раньше. При использовании в настоящем документе фраза "по меньшей мере за 30 минут до" означает, что СОЕДИНЕНИЕ C вводят за по меньшей мере 30 минут или по меньшей мере 35 минут, или по меньшей мере 40 минут, или по меньшей мере 45 минут, или по меньшей мере 50 минут, или по меньшей мере 55 минут, или по меньшей мере 60 минут до введения СОЕДИНЕНИЯ A и первой дозы СОЕДИНЕНИЯ B, в зависимости от того, что произойдет раньше.
[00137] В одном варианте осуществления схема применения КОМБИНИРОВАННОЙ ТЕРАПИИ включает:
аморфный метил-N-[(2S)-1-({4-[3-(5-хлор-2-фтор-3-метансульфонамидофенил)-1-(пропан-2-ил)-1H-пиразол-4-ил]пиримидин-2-ил}амино)пропан-2-ил]карбамат
кристаллический (2-гидроксиэтокси)-амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты
где первую дозу СОЕДИНЕНИЯ B могут вводить:
(i) вместе с СОЕДИНЕНИЕМ A;
(ii) до введения СОЕДИНЕНИЯ A; или
(iii) после введения СОЕДИНЕНИЯ A
и где вторую дозу СОЕДИНЕНИЯ B вводят через 12 часов ±2 часа после первой дозы СОЕДИНЕНИЯ B
цетуксимаб
[00138] В одном варианте осуществления схема применения включает:
аморфный метил-N-[(2S)-1-({4-[3-(5-хлор-2-фтор-3-метансульфонамидо-фенил)-1-(пропан-2-ил)-1H-пиразол-4-ил]пиримидин-2-ил}амино)пропан-2-ил]карбамат
кристаллический (2-гидроксиэтокси)-амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты
где первую дозу СОЕДИНЕНИЯ B могут вводить:
(i) вместе с СОЕДИНЕНИЕМ A;
(ii) до введения СОЕДИНЕНИЯ A; или
(iii) после введения СОЕДИНЕНИЯ A;
и где вторую дозу СОЕДИНЕНИЯ B вводят через 12 часов ±2 часа после первой дозы СОЕДИНЕНИЯ B
цетуксимаб
где СОЕДИНЕНИЕ C вводят через по меньшей мере 30 минут после введения дозы СОЕДИНЕНИЯ A или СОЕДИНЕНИЯ B, которую вводили последней
[00139] В одном варианте осуществления схема применения включает:
аморфный метил-N-[(2S)-1-({4-[3-(5-хлор-2-фтор-3-метансульфонамидофенил)-1-(пропан-2-ил)-1H-пиразол-4-ил]пиримидин-2-ил}амино)пропан-2-ил]карбамат
кристаллический (2-гидроксиэтокси)-амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты
где первую дозу могут вводить:
(i) вместе с СОЕДИНЕНИЕМ A;
(ii) до введения СОЕДИНЕНИЯ A; или
(iii) после введения СОЕДИНЕНИЯ A;
и где вторую дозу СОЕДИНЕНИЯ B вводят через 12 часов ±2 часа после первой дозы СОЕДИНЕНИЯ B
цетуксимаб
где СОЕДИНЕНИЕ C вводят через по меньшей мере 60 минут после введения дозы СОЕДИНЕНИЯ A или СОЕДИНЕНИЯ B, которую вводили последней
[00140] В одном варианте осуществления схема применения включает:
аморфный метил-N-[(2S)-1-({4-[3-(5-хлор-2-фтор-3-метансульфонамидофенил)-1-(пропан-2-ил)-1H-пиразол-4-ил]пиримидин-2-ил}амино)пропан-2-ил]карбамат
кристаллический (2-гидроксиэтокси)-амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты
где первую дозу Соединения B могут вводить:
(i) вместе с СОЕДИНЕНИЕМ A;
(ii) до введения СОЕДИНЕНИЯ A; или
(iii) после введения СОЕДИНЕНИЯ A
и где вторую дозу СОЕДИНЕНИЯ B вводят через 12 часов ±2 часа после первой дозы СОЕДИНЕНИЯ B
цетуксимаб
где СОЕДИНЕНИЕ C вводят по меньшей мере за 30 минут до введения СОЕДИНЕНИЯ A и первой дозы СОЕДИНЕНИЯ B
[00141] В одном варианте осуществления любая КОМБИНИРОВАННАЯ ТЕРАПИЯ, описанная в настоящем документе, необязательно дополнительно включает введение одного или более лекарственных средств для предварительной медикаментозной подготовки перед введением СОЕДИНЕНИЯ C. В одном варианте осуществления одно или более лекарственных средств для предварительной медикаментозной подготовки вводят не раньше чем через 1 час после введения СОЕДИНЕНИЯ A и биниметиниба, СОЕДИНЕНИЯ B. В одном варианте осуществления одно или более лекарственных средств для предварительной медикаментозной подготовки вводят за 30-60 минут до введения СОЕДИНЕНИЯ C. В одном варианте осуществления одно или более лекарственных средств для предварительной медикаментозной подготовки вводят за 30 минут до введения СОЕДИНЕНИЯ C. В одном варианте осуществления одно или более лекарственных средств для предварительной медикаментозной подготовки выбраны из одного или более H1-антагонистов (например, антигистаминных средств, таких как дифенгидрамин) и системных кортикостероидов (например, низкой дозы системного кортикостероида, например, дексаметазона или преднизолона).
[00142] В одном варианте осуществления любая КОМБИНИРОВАННАЯ ТЕРАПИЯ, описанная в настоящем документе, дополнительно включает указание пациенту избегать употребления грейпфрута, гранатов, карамболы, померанцев или продуктов, содержащих любой из этих соков.
[00143] Оптимальная доза каждого партнера по комбинации для лечения пролиферативного заболевания может быть определена эмпирически для каждого пациента при использовании известных методов и будет зависеть от множества факторов, включающих, без ограничения перечисленными, степень прогрессирования заболевания; возраст, массу тела, общее состояние здоровья, пол и питание пациента; время и путь введения; а также другие лекарственные препараты, применяемые пациентом. Оптимальные дозы могут быть установлены при использовании стандартных анализов и процедур, которые известны в уровне техники.
[00144] Количество каждого партнера по комбинации, который может быть объединен с материалами-носителями с получением единой лекарственной формы, будет изменяться в зависимости от проходящего лечение пациента и конкретного способа введения. В некоторых вариантах осуществления единичные лекарственные формы, содержащие комбинацию средств, как описано в настоящем документе, будут содержать количества каждого средства в комбинации, которые, как правило, применяются, когда средства вводят по отдельности.
[00145] Частота введения может изменяться в зависимости от используемого соединения и конкретного состояния, подвергаемого лечению. Как правило, предпочтительно использование минимальной дозы, которая достаточна для обеспечения эффективной терапии. Пациентов обычно могут наблюдать на предмет терапевтической эффективности при использовании анализов, подходящих для подвергаемого лечению состояния, которые будут знакомы средним специалистам в данной области.
[00146] Также в настоящем документе предложен способ лечения субъекта, имеющего пролиферативное заболевание, включающий введение указанному субъекту КОМБИНАЦИИ ИЗОБРЕТЕНИЯ в количестве, которое является совместно терапевтически эффективным против пролиферативного заболевания. В одном варианте осуществления пролиферативным заболеванием, подлежащим лечению с применением КОМБИНАЦИИ ИЗОБРЕТЕНИЯ, является рак. В одном варианте осуществления рак выбран из рака толстой и прямой кишки (CRC) (включая метастатический рак толстой и прямой кишки), меланомы (включая метастатическую и неоперабельную меланому), рака легкого (включая немелкоклеточный рак легкого (НМРЛ)), рака молочной железы, рака почки, такого как, например, почечно-клеточная карцинома (RCC), рака печени, рака эндометрия, острого миелогенного лейкоза (ОМЛ), миелодиспластических синдромов (MDS), рака щитовидной железы, в частности папиллярного рака щитовидной железы, рака поджелудочной железы, нейрофиброматоза и гепатоцеллюлярной карциномы. В одном варианте осуществления рак является раком толстой и прямой кишки (CRC). В одном варианте осуществления рак толстой и прямой кишки является метастатическим раком толстой и прямой кишки. В одном варианте осуществления рак является BRAF-ассоциированным раком. В одном варианте осуществления рак является раком, имеющим мутацию BRAF V600. В одном варианте осуществления рак является раком, имеющим мутацию BRAF V600E. В одном варианте осуществления рак является раком толстой и прямой кишки. В одном варианте осуществления рак является раком толстой и прямой кишки, имеющим мутацию BRAF V600E. В одном варианте осуществления рак является метастатическим раком толстой и прямой кишки, имеющим мутацию BRAF V600E. В одном варианте осуществления рак является MEK-ассоциированным раком. В одном варианте осуществления рак является EGFR-ассоциированным раком.
[00147] В одном варианте осуществления в настоящем документе предложен способ лечения рака у нуждающегося в этом пациента, включающий: (a) определение, является ли рак у пациента BRAF-ассоциированным раком; и (b) если рак определен как BRAF-ассоциированный рак, введение пациенту терапевтически эффективного количества КОМБИНАЦИИ ИЗОБРЕТЕНИЯ. В некоторых вариантах осуществления наличие BRAF-ассоциированного рака у пациента определено с помощью одобренного регулирующим органом, например, одобренного FDA, теста или анализа для идентификации дисрегуляции гена BRAF, BRAF-киназы или их экспрессии или активности, или уровня у пациента или в образце биопсии пациента, или путем выполнения любого из неограничивающих примеров анализов, описанных в настоящем документе. В некоторых вариантах осуществления тест или анализ предложен в виде набора. В некоторых вариантах осуществления рак является BRAF-мутантным раком толстой и прямой кишки. В одном варианте осуществления рак является метастатическим раком толстой и прямой кишки, имеющим мутацию BRAF. В одном варианте осуществления рак является метастатическим раком толстой и прямой кишки, имеющим мутацию BRAF V600E.
[00148] В одном варианте осуществления в настоящем документе предложен способ лечения рака у нуждающегося в этом субъекта, включающий: (a) определение, имеет ли рак мутантную BRAF-киназу и (b) если рак определен как имеющий мутантную BRAF-киназу, введение указанному субъекту терапевтически эффективного количества КОМБИНАЦИИ ИЗОБРЕТЕНИЯ.
[00149] В одном варианте осуществления в настоящем документе предложен способ (например, способ in vitro) подбора лечения для пациента, у которого идентифицировано или диагностировано наличие BRAF-ассоциированного рака. Некоторые варианты осуществления могут дополнительно включать введение подобранного лечения пациенту, у которого идентифицировано или диагностировано наличие BRAF-ассоциированного рака. Например, подобранное лечение может включать введение терапевтически эффективного количества КОМБИНАЦИИ ИЗОБРЕТЕНИЯ. Некоторые варианты осуществления могут дополнительно включать этап проведения анализа образца, полученного у пациента, для определения наличия у пациента дисрегуляции гена BRAF, BRAF-киназы или их экспрессии или активности, или уровня, и идентификацию и диагностику пациента, у которого определена дисрегуляция гена BRAF, BRAF-киназы или их экспрессии или активности, или уровня, как имеющего BRAF-ассоциированный рак. В некоторых вариантах осуществления у пациента было идентифицировано или диагностировано наличие BRAF-ассоциированного рака при использовании одобренного регулирующим агентством, например, одобренного FDA, набора для идентификации нарушения регуляции гена BRAF, BRAF-киназы или их экспрессии или активности, или уровня, у пациента или в образце биопсии пациента. В некоторых вариантах осуществления BRAF-ассоциированный рак является раком, описанным здесь или известным в данной области. В некоторых вариантах осуществления BRAF-ассоциированный рак является раком толстой и прямой кишки. В некоторых вариантах осуществления BRAF-ассоциированный рак является раком толстой и прямой кишки, имеющим мутацию BRAF V600E. В некоторых вариантах осуществления BRAF-ассоциированный рак является метастатическим раком толстой и прямой кишки. В некоторых вариантах осуществления BRAF-ассоциированный рак является метастатическим раком толстой и прямой кишки, имеющим мутацию BRAF V600E. В некоторых вариантах осуществления анализ является анализом in vitro, например, анализом, в котором используется секвенирование нового поколения, иммуногистохимия или анализ break-apart FISH. В некоторых вариантах осуществления анализом является набор, одобренный регулирующим органом, например, одобренный FDA.
[00150] В одном варианте осуществления в настоящем документе предложен способ отбора пациента для лечения, где способы включают отбор, идентификацию или диагностику пациента с наличием BRAF-ассоциированного рака, и отбор пациента для лечения, включающего введение терапевтически эффективного количества КОМБИНАЦИИ ИЗОБРЕТЕНИЯ. В некоторых вариантах осуществления идентификация или диагностика пациента, имеющего BRAF-ассоциированный рак, может включать этап проведения анализа образца, полученного у пациента (например, образца биопсии), с целью определения наличия у пациента дисрегуляции гена BRAF, BRAF-киназы или их экспрессии или активности, или уровня, и идентификацию или диагностику пациента, у которого определено наличие дисрегуляции гена BRAF, BRAF-киназы или их экспрессии или активности, или уровня, как имеющего BRAF-ассоциированный рак. В некоторых вариантах осуществления BRAF-ассоциированный рак является раком толстой и прямой кишки, имеющим мутацию BRAF V600E. В некоторых вариантах осуществления BRAF-ассоциированный рак является метастатическим раком толстой и прямой кишки. В некоторых вариантах осуществления BRAF-ассоциированный рак является метастатическим раком толстой и прямой кишки, имеющим мутацию BRAF V600E. В некоторых вариантах осуществления анализ является анализом in vitro, например, анализом, в котором используется секвенирование нового поколения, иммуногистохимия или анализ break-apart FISH. В некоторых вариантах осуществления анализом является набор, одобренный регулирующим органом, например, одобренный FDA.
[00151] Также предложены способы определения вероятности, что субъект, имеющий рак, будет иметь положительный ответ на лечение с применением КОМБИНАЦИИ ИЗОБРЕТЕНИЯ, которые включают: определение, имеет ли раковая клетка в образце, полученном у субъекта, одну или более мутаций BRAF; и определение, что субъект, имеющий раковую клетку, которая имеет одну или более мутаций BRAF, имеет повышенную вероятность наличия положительного ответа на лечение с применением КОМБИНАЦИИ ИЗОБРЕТЕНИЯ.
[00152] Также предложены способы прогноза эффективности лечения с применением фармацевтической комбинации согласно изобретению у субъекта, имеющего рак, которые включают: определение, имеет ли раковая клетка в образце, полученном у субъекта, одну или более мутаций BRAF; и определение, что лечение с применением КОМБИНАЦИИ ИЗОБРЕТЕНИЯ с более высокой вероятностью будет эффективным у субъекта, имеющего раковую клетку в образце, полученном у субъекта, которая имеет одну или более мутаций BRAF. В одном варианте осуществления мутация BRAF является мутацией V600E.
[00153] В одном варианте осуществления в настоящем документе предложен способ лечения BRAF-ассоциированного рака (например, рака, имеющего мутацию BRAF-киназы) у нуждающегося в этом субъекта, включающий: (a) определение, имеет ли рак мутантную BRAF-киназу и, (b) если рак определен как имеющий мутантную BRAF-киназу, введение указанному субъекту терапевтически эффективного количества КОМБИНАЦИИ ИЗОБРЕТЕНИЯ. В одном варианте осуществления КОМБИНАЦИЯ ИЗОБРЕТЕНИЯ включает аморфный метил-N-[(2S)-1-({4-[3-(5-хлор-2-фтор-3-метансульфон-амидофенил)-1-(пропан-2-ил)-1H-пиразол-4-ил]пиримидин-2-ил}амино)пропан-2-ил]карбамат или его фармацевтически приемлемую соль (СОЕДИНЕНИЕ A), (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты или его фармацевтически приемлемую соль (СОЕДИНЕНИЕ B), и цетуксимаб (СОЕДИНЕНИЕМ C). В одном варианте осуществления СОЕДИНЕНИЕМ B является кристаллизованный (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты. В одном варианте осуществления рак является раком, имеющим мутацию BRAF V600. В одном варианте осуществления рак является раком, имеющим мутацию BRAF V600E. В одном варианте осуществления рак является раком толстой и прямой кишки. В одном варианте осуществления рак является метастатическим раком толстой и прямой кишки. В одном варианте осуществления рак является раком толстой и прямой кишки, имеющим мутацию BRAF V600E. В одном варианте осуществления рак является метастатическим раком толстой и прямой кишки, имеющим мутацию BRAF V600E. В одном варианте осуществления субъект имеет рак толстой и прямой кишки IV стадии. В одном варианте осуществления субъект имеет рак толстой и прямой кишки IV стадии, имеющий мутацию BRAF V600E. В некоторых вариантах осуществления субъект ранее проходил лечение с применением другой противоопухолевой терапии, например, системной противоопухолевой терапии первой или второй линии (например, лечение с применением одного или более цитотоксических средств), резекции опухоли или лучевой терапии. В одном варианте осуществления субъект имеет метастатический рак толстой и прямой кишки, и ранее проходил лечение по одной или двум схемам лечения с применением системной противоопухолевой терапии первой или второй линии (например, лечение с применением одного или более цитотоксических средств). В одном варианте осуществления субъект имеет метастатический рак толстой и прямой кишки, который прогрессировал после одной или двух схем лечения с применением системной противоопухолевой терапии первой или второй линии (например, лечения с применением одного или более цитотоксических средств). В одном варианте осуществления цитотоксические средства выбраны из иринотекана, оксалиплатина, капецитабина, фолиновой кислоты и 5-фторурацила. В одном варианте осуществления субъект имеет рак, который также экспрессирует KRAS дикого типа (KRASwt).
[00154] В одном варианте осуществления в настоящем документе предложен способ лечения BRAF-ассоциированного рака (например, рака, имеющего мутацию BRAF-киназы) у нуждающегося в этом субъекта, включающий: (a) обнаружение мутантной BRAF-киназы при раке и (b) введение указанному субъекту терапевтически эффективного количества КОМБИНАЦИИ ИЗОБРЕТЕНИЯ. В одном варианте осуществления КОМБИНАЦИЯ ИЗОБРЕТЕНИЯ включает аморфный метил-N-[(2S)-1-({4-[3-(5-хлор-2-фтор-3-метансульфонамидофенил)-1-(пропан-2-ил)-1H-пиразол-4-ил]пиримидин-2-ил}амино)пропан-2-ил]карбамат или его фармацевтически приемлемую соль (СОЕДИНЕНИЕ A), (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты или его фармацевтически приемлемую соль (СОЕДИНЕНИЕ B) и цетуксимаб (СОЕДИНЕНИЕ C). В одном варианте осуществления СОЕДИНЕНИЕМ B является кристаллизованный (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты. В одном варианте осуществления рак является раком, имеющим мутацию BRAF V600. В одном варианте осуществления рак является раком, имеющим мутацию BRAF V600E. В одном варианте осуществления рак является раком толстой и прямой кишки. В одном варианте осуществления рак является метастатическим раком толстой и прямой кишки. В одном варианте осуществления рак является раком толстой и прямой кишки, имеющим мутацию BRAF V600E. В одном варианте осуществления рак является метастатическим раком толстой и прямой кишки, имеющим мутацию BRAF V600E. В некоторых вариантах осуществления субъект ранее проходил лечение с применением другой противоопухолевой терапии, например, системной противоопухолевой терапии первой или второй линии (например, лечение с применением одного или более цитотоксических средств), резекции опухоли или лучевой терапии. В одном варианте осуществления субъект имеет метастатический рак толстой и прямой кишки, и ранее проходил лечение по одной или двум схемам лечения с применением системной противоопухолевой терапии первой или второй линии (например, лечение с применением одного или более цитотоксических средств). В одном варианте осуществления субъект имеет метастатический рак толстой и прямой кишки, который прогрессировал после одного или двух режимов лечения с применением системной противоопухолевой терапии первой или второй линии (например, лечения с применением одного или более цитотоксических средств). В одном варианте осуществления цитотоксические средства выбраны из иринотекана, оксалиплатина, капецитабина, фолиновой кислоты и 5-фторурацила. В одном варианте осуществления субъект имеет рак, который также экспрессирует KRAS дикого типа (KRASwt).
[00155] В одном варианте осуществления в настоящем документе предложен способ лечения субъекта, имеющего рак толстой и прямой кишки, включающий введение указанному субъекту КОМБИНАЦИИ ИЗОБРЕТЕНИЯ в количестве, которое является совместно терапевтически эффективным против указанного рака толстой и прямой кишки. В одном варианте осуществления субъект имеет рак толстой и прямой кишки, имеющий мутацию BRAF V600E. В одном варианте осуществления у субъекта определено наличие рака толстой и прямой кишки, имеющего мутацию BRAF V600E, как определено при использовании анализа или набора, одобренного регулирующим органом, например, одобренного FDA. В одном варианте осуществления рак толстой и прямой кишки является метастатическим раком толстой и прямой кишки. В одном варианте осуществления субъект имеет рак толстой и прямой кишки IV стадии. В одном варианте осуществления субъект имеет рак толстой и прямой кишки IV стадии, имеющий мутацию BRAF V600E. В некоторых вариантах осуществления субъект ранее проходил лечение с применением другой противоопухолевой терапии, например, системной противоопухолевой терапии первой или второй линии (например, лечение с применением одного или более цитотоксических средств), резекции опухоли или лучевой терапии. В одном варианте осуществления субъект имеет метастатический рак толстой и прямой кишки, и ранее проходил лечение по одной или двум схемам лечения с применением системной противоопухолевой терапии первой или второй линии (например, лечение с применением одного или более цитотоксических средств). В одном варианте осуществления субъект имеет метастатический рак толстой и прямой кишки, который прогрессировал после одной или двух схем лечения с применением системной противоопухолевой терапии первой или второй линии (например, лечения с применением одного или более цитотоксических средств). В одном варианте осуществления цитотоксические средства выбраны из иринотекана, оксалиплатина, капецитабина, фолиновой кислоты и 5-фторурацила. В одном варианте осуществления субъект имеет рак, который также экспрессирует KRAS дикого типа (KRASwt).
[00156] Также предложены способы лечения пациента, которые включают проведение анализа образца, полученного у пациента, с целью определения, имеет ли пациент BRAF-ассоциированный рак (например, рак, имеющий мутацию BRAF-киназы), и введение терапевтически эффективного количества КОМБИНАЦИИ ИЗОБРЕТЕНИЯ пациенту, у которого определено наличие BRAF-ассоциированного рака (например, рака, имеющего мутацию BRAF-киназы). В некоторых вариантах осуществления субъект ранее проходил лечение с применением другой противоопухолевой терапии, например, системной противоопухолевой терапии первой или второй линии (например, лечение с применением одного или более цитотоксических средств), резекции опухоли или лучевой терапии. В одном варианте осуществления субъект имеет метастатический рак толстой и прямой кишки, и ранее проходил лечение по одной или двум схемам лечения с применением системной противоопухолевой терапии первой или второй линии (например, лечение с применением одного или более цитотоксических средств). В одном варианте осуществления субъект имеет метастатический рак толстой и прямой кишки, который прогрессировал после одной или двух схем лечения с применением системной противоопухолевой терапии первой или второй линии (например, лечения с применением одного или более цитотоксических средств). В одном варианте осуществления цитотоксические средства выбраны из иринотекана, оксалиплатина, капецитабина, фолиновой кислоты и 5-фторурацила. В некоторых вариантах осуществления пациент является пациентом, подозреваемым на наличие BRAF-ассоциированного рака (например, рака, имеющего мутацию BRAF-киназы), пациентом, демонстрирующим один или более симптомов BRAF-ассоциированного рака (например, рака, имеющего мутацию BRAF-киназы), или пациентом с повышенным риском заболевания BRAF-ассоциированным раком (например, рака, имеющего мутацию BRAF-киназы). В одном варианте осуществления рак является раком, имеющим мутацию BRAF V600. В одном варианте осуществления рак является раком, имеющим мутацию BRAF V600E. В некоторых вариантах осуществления рак является раком толстой и прямой кишки. В одном варианте осуществления рак толстой и прямой кишки является метастатическим раком толстой и прямой кишки. В одном варианте осуществления рак является раком толстой и прямой кишки, имеющим мутацию BRAF V600. В одном варианте осуществления рак является раком толстой и прямой кишки, имеющим мутацию BRAF V600E. В одном варианте осуществления субъект имеет рак толстой и прямой кишки IV стадии. В одном варианте осуществления способ дополнительно включает проведение анализа с целью определения, имеет ли пациент рак, который также экспрессирует KRAS дикого типа (KRASwt).
[00157] В одном варианте осуществления в настоящем документе предложен способ лечения субъекта, имеющего BRAF-ассоциированный рак (например, рак, имеющий мутацию BRAF-киназы), где указанный способ включает введение указанному субъекту терапевтически эффективного количества КОМБИНАЦИИ ИЗОБРЕТЕНИЯ, где субъект проходил лечение с применением системной противоопухолевой терапии первой или второй линии до лечения указанной КОМБИНАЦИЕЙ ИЗОБРЕТЕНИЯ. В одном варианте осуществления субъект получал системную противоопухолевую терапию по одной или двум схемам лечения до лечения КОМБИНАЦИЕЙ ИЗОБРЕТЕНИЯ. В одном варианте осуществления субъект имеет метастатический рак толстой и прямой кишки, и ранее проходил лечение по одной или двум схемам лечения с применением системной противоопухолевой терапии первой или второй линии (например, лечение с применением одного или более цитотоксических средств). В одном варианте осуществления субъект имеет метастатический рак толстой и прямой кишки, который прогрессировал после одной или двух схем лечения с применением системной противоопухолевой терапии первой или второй линии (например, лечения с применением одного или более цитотоксических средств). В одном варианте осуществления системные терапии первой или второй линии включают лечение с применением одного или более цитотоксических средств. В одном варианте осуществления одно или более цитотоксических средств выбраны из иринотекана, оксалиплатина, капецитабина, фолиновой кислоты и 5-фторурацила. В одном варианте осуществления системная противоопухолевая терапия не включает ингибитор BRAF, ингибитор MEK или ингибитор EGFR. В одном варианте осуществления рак является раком, имеющим мутацию BRAF V600. В одном варианте осуществления рак является раком, имеющим мутацию BRAF V600E. В одном варианте осуществления рак является раком толстой и прямой кишки. В одном варианте осуществления рак является метастатическим раком толстой и прямой кишки. В одном варианте осуществления рак является раком толстой и прямой кишки, имеющим мутацию BRAF V600E. В одном варианте осуществления рак является метастатическим раком толстой и прямой кишки, имеющим мутацию BRAF V600E. В одном варианте осуществления субъект имеет рак, который также экспрессирует KRAS дикого типа (KRASwt).
[00158] В одном варианте осуществления в настоящем документе предложен способ лечения субъекта, имеющего рак толстой и прямой кишки, где указанный рак толстой и прямой кишки имеет мутацию BRAF V600E, где способ включает: (i) введение по меньшей мере одного цитотоксического средства указанному субъекту в течение некоторого периода времени; и (ii) прекращение введения указанного по меньшей мере одного цитотоксического средства и начало лечения с применением КОМБИНАЦИИ ИЗОБРЕТЕНИЯ, где указанное лечение с применением указанной КОМБИНАЦИИ ИЗОБРЕТЕНИЯ включает любую из схем лечения, описанных в настоящем документе. В одном варианте осуществления субъект имеет рак толстой и прямой кишки IV стадии, имеющий мутацию BRAF V600E. В одном варианте осуществления цитотоксические средства выбраны из иринотекана, оксалиплатина, капецитабина, фолиновой кислоты и 5-фторурацила. В одном варианте осуществления субъект имеет рак, который также экспрессирует KRAS дикого типа (KRASwt).
[00159] В одном варианте осуществления в настоящем документе предложен способ лечения субъекта, имеющего рак толстой и прямой кишки, имеющий мутацию BRAF V600E, где способ включает введение указанному субъекту КОМБИНАЦИИ ИЗОБРЕТЕНИЯ в количестве, которое является совместно терапевтически эффективным против указанного рака толстой кишки, где субъект не проходил предварительное лечение ингибитором BRAF до лечения КОМБИНАЦИЕЙ ИЗОБРЕТЕНИЯ. В одном варианте осуществления рак также экспрессирует KRAS дикого типа (KRASwt).
[00160] В одном варианте осуществления в настоящем документе предложен способ лечения субъекта, имеющего рак толстой и прямой кишки, имеющий мутацию BRAF V600E, включающий введение указанному субъекту КОМБИНАЦИИ ИЗОБРЕТЕНИЯ в количестве, которое является совместно терапевтически эффективным против указанного рака толстой кишки, где субъект не проходил предварительное лечение ингибитором MEK до лечения КОМБИНАЦИЕЙ ИЗОБРЕТЕНИЯ. В одном варианте осуществления рак также экспрессирует KRAS дикого типа (KRASwt).
[00161] В одном варианте осуществления в настоящем документе предложен способ лечения субъекта, имеющего рак толстой и прямой кишки, имеющий мутацию BRAF V600E, где способ включает введение указанному субъекту КОМБИНАЦИИ ИЗОБРЕТЕНИЯ в количестве, которое является совместно терапевтически эффективным против указанного рака толстой кишки, где субъект не проходил предварительное лечение ингибитором EGFR до лечения КОМБИНАЦИЕЙ ИЗОБРЕТЕНИЯ. В одном варианте осуществления субъект не проходил лечение ингибитором EGFR, выбранным из цетуксимаба и панитумумаба, до лечения КОМБИНАЦИЕЙ ИЗОБРЕТЕНИЯ. В одном варианте осуществления рак также экспрессирует KRAS дикого типа (KRASwt).
[00162] В одном варианте осуществления в настоящем документе предложен способ лечения субъекта, имеющего рак толстой и прямой кишки, имеющий мутацию BRAF V600E, где способ включает введение указанному субъекту КОМБИНАЦИИ ИЗОБРЕТЕНИЯ в количестве, которое является совместно терапевтически эффективным против указанного рака толстой кишки, где субъект не проходил лечение ингибитором BRAF, ингибитором MEK и/или ингибитором EGFR до лечения КОМБИНАЦИЕЙ ИЗОБРЕТЕНИЯ. В одном варианте осуществления рак также экспрессирует KRAS дикого типа (KRASwt).
[00163] В одном варианте осуществления в настоящем документе предложен способ уменьшения размера злокачественной опухоли, включающий контакт опухоли с КОМБИНАЦИЕЙ ИЗОБРЕТЕНИЯ. В одном варианте осуществления контакт осуществляют in vitro. В одном варианте осуществления контакт осуществляют in vivo. В одном варианте осуществления опухоль выбрана из рака толстой и прямой кишки (CRC) (включая метастатический рак толстой и прямой кишки), меланомы (включая метастатическую меланому), рака легкого (включая немелкоклеточный рак легкого (НМРЛ)), рака молочной железы, рака почки, такого как, например, почечно-клеточная карцинома (RCC), рака печени, рака эндометрия, острого миелогенного лейкоза (ОМЛ), миелодиспластических синдромов (MDS), рака щитовидной железы, в частности папиллярного рака щитовидной железы, рака поджелудочной железы, нейрофиброматоза и гепатоцеллюлярной карциномы. В одном варианте осуществления рак является раком толстой и прямой кишки (CRC). В одном варианте осуществления рак толстой и прямой кишки является метастатическим раком толстой и прямой кишки. В одном варианте осуществления рак является BRAF-ассоциированным раком. В одном варианте осуществления рак является раком, имеющим мутацию BRAF V600. В одном варианте осуществления рак является раком, имеющим мутацию BRAF V600E. В одном варианте осуществления рак является раком толстой и прямой кишки, имеющим мутацию BRAF V600E. В одном варианте осуществления рак является метастатическим раком толстой и прямой кишки, имеющим мутацию BRAF V600E. В одном варианте осуществления рак также экспрессирует KRAS дикого типа (KRASwt).
[00164] Улучшение при раке или подобных онкологических заболеваниях может быть охарактеризовано как полный или частичный ответ. "Полный ответ" или "CR" относится к отсутствию клинически обнаружимого заболевания с нормализацией любых, ранее патологических рентгенологических исследований, костного мозга и спинномозговой жидкости (СМЖ) или измерений аномальных моноклональных белков. "Частичный ответ" относится по меньшей мере приблизительно к 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80% или 90% уменьшению всей поддающейся измерению опухолевой нагрузке (т.е. количества злокачественных клеток, присутствующих у субъекта, или измеренной опухолевой массы, или количества аномального моноклонального белка) в отсутствие новых очагов.
[00165] Лечение можно оценивать по ингибированию прогрессирования заболевания, ингибированию роста опухоли, уменьшению первичной опухоли, ослаблению симптомов, связанных с опухолью, ингибированию секретируемых опухолью факторов (включая уровни экспрессии белков контрольных точек, как определено в настоящем документе), задержке появления первичных или вторичных опухолей, замедленному развитию первичных или вторичных опухолей, уменьшению возникновения первичных или вторичных опухолей, замедлению или уменьшению тяжести вторичных эффектов заболевания, остановке роста опухоли и регрессии опухолей, увеличению времени до прогрессирования (TTP), увеличению выживаемости без прогрессирования (PFS), увеличению общей выживаемости (OS), помимо прочего. При использовании в настоящем документе OS означает время от начала лечения до смерти вследствие любой причины. Термин "TTP" в настоящем описании означает время от начала лечения до прогрессирования опухоли; TTP не включает смерть. При использовании в настоящем документе термин PFS означает время от начала лечения до прогрессирования опухоли или смерти. В крайнем случае, полное ингибирование именуется в настоящем документе предотвращением или химиопрофилактикой.
[00166] В некоторых вариантах осуществления способов, описанных в настоящем документе, лечение можно оценивать по одному или нескольким клиническим конечным показателям, выбранным из положительного ответа опухоли, полного ответа, частичного ответа или стабильного заболевания, увеличения выживаемости без прогрессирования опухоли, ингибирования прогрессирования заболевания, ингибирования роста опухоли, уменьшения первичной опухоли, ослабления связанных с опухолью симптомов, ингибирования секретируемых опухолью факторов, замедленного появления первичных или вторичных опухолей, замедленного развития первичных или вторичных опухолей, уменьшения возникновения первичных или вторичных опухолей, замедления или уменьшения тяжести вторичных эффектов заболевания, остановки роста опухоли и регрессии опухолей, увеличения времени до прогрессирования (TTP), увеличения выживаемости без прогрессирования (PFS), увеличения общей выживаемости (OS) или увеличения продолжительности ответа (DOR)
[00167] Таким образом, в настоящем документе предложены способы достижения одного или более клинических результатов, связанных с лечением гемобластоза (или солидной опухоли), описанным в настоящем документе. В одном варианте осуществления пациент, описанный в настоящем документе, может демонстрировать положительный ответ опухоли, такой как ингибирование роста опухоли или уменьшение размера опухоли после лечения комбинацией, описанной в настоящем документе. В некоторых вариантах осуществления пациент, описанный в настоящем документе, может достигать таких Критериев оценки ответа при солидных опухолях (например, RECIST 1.1), как полный ответ, частичный ответ или стабилизация заболевания после введения эффективного количества КОМБИНАЦИИ ИЗОБРЕТЕНИЯ. В некоторых вариантах осуществления пациент, описанный в настоящем документе, может демонстрировать увеличение выживаемости без прогрессирования опухоли. В некоторых вариантах осуществления пациент, описанный в настоящем документе, может демонстрировать ингибирование прогрессирования заболевания, ингибирование роста опухоли, уменьшение первичной опухоли, облегчение связанных с опухолью симптомов, ингибирование секретируемых опухолью факторов (включающих секретируемые опухолевые гормоны, такие как гормоны, которые способствуют развитию карциноидного синдрома), задержку появления первичных или вторичных опухолей, замедленное развитие первичных или вторичных опухолей, уменьшение возникновения первичных или вторичных опухолей, замедление или уменьшение тяжести вторичных эффектов заболевания, остановку роста опухоли и регрессию опухолей, увеличение времени до прогрессирования (TTP), увеличение выживаемости без прогрессирования (PFS) и/или увеличение общей выживаемости (OS), помимо прочего.
[00168] В другом варианте осуществления предложены способы увеличения общей выживаемости, процента объективных ответов, времени до прогрессирования, выживаемости без прогрессирования и/или времени до констатации неэффективности лечения у пациента, имеющего рак, описанный в настоящем документе, включающие введение эффективного количества КОМБИНАЦИИ ИЗОБРЕТЕНИЯ, как описано в настоящем документе. В одном варианте осуществления предложен способ увеличения общей выживаемости пациента, имеющего рак, описанный в настоящем документе, включающий введение эффективного количества КОМБИНАЦИИ ИЗОБРЕТЕНИЯ, как описано в настоящем документе. В одном варианте осуществления предложен способ увеличения процента объективных ответов у пациента, имеющего рак, описанный в настоящем документе, включающий введение эффективного количества КОМБИНАЦИИ ИЗОБРЕТЕНИЯ, как описано в настоящем документе. В одном варианте осуществления предложен способ увеличения времени до прогрессирования у пациента, имеющего рак, описанный в настоящем документе, включающий введение эффективного количества КОМБИНАЦИИ ИЗОБРЕТЕНИЯ, как описано в настоящем документе. В одном варианте осуществления предложен способ увеличения выживаемости без прогрессирования у пациента, имеющего рак, описанный в настоящем документе, включающий введение эффективного количества КОМБИНАЦИИ ИЗОБРЕТЕНИЯ, как описано в настоящем документе. В одном варианте осуществления предложен способ увеличения времени до констатации неэффективности лечения у пациента, имеющего рак, описанный в настоящем документе, включающий введение эффективного количества КОМБИНАЦИИ ИЗОБРЕТЕНИЯ, как описано в настоящем документе. В одном варианте осуществления рак является раком, имеющим мутацию BRAF V600. В одном варианте осуществления рак является раком, имеющим мутацию BRAF V600E. В некоторых вариантах осуществления рак является раком толстой и прямой кишки. В одном варианте осуществления рак толстой и прямой кишки является метастатическим раком толстой и прямой кишки. В одном варианте осуществления рак является раком толстой и прямой кишки, имеющим мутацию BRAF V600. В одном варианте осуществления рак является раком толстой и прямой кишки, имеющим мутацию BRAF V600E.
[00169] В некоторых вариантах осуществления любого из способов или применений, описанных в настоящем документе, анализ, используемый для определения наличия у пациента BRAF-ассоциированного рака с использованием образца, полученного у пациента, может включать, например, секвенирование нового поколения, иммуногистохимию, флуоресцентную микроскопию, анализ break-apart FISH, Саузерн-блоттинг, Вестерн-блоттинг, FACS анализ, Нозерн-блоттинг и амплификацию на основе ПЦР (например, ОТ-ПЦР и количественную ОТ-ПЦР в реальном времени). Как известно в уровне техники, анализы, как правило, выполняют, например, по меньшей мере с одним меченым зондом на основе нуклеиновой кислоты или по меньшей мере одним меченым антителом или его антигенсвязывающим фрагментом. В анализах могут использовать другие методы обнаружения, известные в уровне техники, для обнаружения дисрегуляции гена BRAF, BRAF-киназы или экспрессии или активности, или уровней любого из них (см., например, источники, цитируемые в настоящем документе). В некоторых вариантах осуществления образец является биологическим образцом или образцом биопсии (например, залитый парафином образец биопсии) пациента. В некоторых вариантах осуществления пациент является пациентом, подозреваемым на наличие BRAF-ассоциированного рака, пациентом, имеющим один или более симптомов BRAF-ассоциированного рака, и/или пациентом с повышенным риском заболевания BRAF-ассоциированным раком.
[00170] В одном варианте осуществления способы лечения рака согласно изобретению также включают хирургию или лучевую терапию. Неограничивающие примеры хирургии включают, например, открытое хирургическое вмешательство или минимально инвазивное хирургическое вмешательство. Хирургия может включать, например, полное удаление опухоль, уменьшение объема опухоли или удаление опухоль, которая причиняет боль или создает давление у субъекта. Способы проведения открытого хирургического вмешательства и минимально инвазивного хирургического вмешательства у субъекта, имеющего рак, известны в уровне техники. Неограничивающие примеры лучевой терапии включают наружную лучевую терапию (например, наружную лучевую терапию с использованием киловольтного пучка рентгеновского излучения или мегавольтного пучка рентгеновского излучения) или внутреннюю лучевую терапию. Внутренняя лучевая терапия (также называемая брахитерапией) может включать, например, внутреннюю лучевую терапию низкой дозы облучения или внутреннюю лучевую терапию высокой дозы облучения. Внутренняя лучевая терапия низкой дозы облучения включает, например, введение небольших радиоактивных капсул (также называемых зернами) в раковую ткань у субъекта или вблизи от нее. Внутренняя лучевая терапия высокой дозы облучения включает, например, введение тонкой трубки (например, катетера) или имплантата в раковую ткань субъекта или вблизи от нее и доставку высокой дозы излучения в тонкую трубку или имплантат с использованием установки, генерирующей радиоактивное излучение. Способы проведения лучевой терапии у субъекта, имеющего рак, известны в уровне техники.
[00171] Настоящее изобретение также относится к КОМБИНАЦИИ ИЗОБРЕТЕНИЯ для применения в лечении пролиферативного заболевания. В одном варианте осуществления пролиферативным заболеванием является рак. В одном варианте осуществления рак выбран из рака толстой и прямой кишки (CRC) (включая метастатический рак толстой и прямой кишки), меланомы (включая метастатическую и неоперабельную меланому), рака легкого (включая немелкоклеточный рак легкого (НМРЛ)), рака молочной железы, рака почки, такого как, например, почечно-клеточная карцинома (RCC), рака печени, рака эндометрия, острого миелогенного лейкоза (ОМЛ), миелодиспластических синдромов (MDS), рака щитовидной железы, в частности папиллярного рака щитовидной железы, рака поджелудочной железы, нейрофиброматоза и гепатоцеллюлярной карциномы. В одном варианте осуществления рак является раком толстой и прямой кишки (CRC). В одном варианте осуществления рак толстой и прямой кишки является метастатическим раком толстой и прямой кишки. В одном варианте осуществления рак является BRAF-ассоциированным раком. В одном варианте осуществления рак является раком, имеющим мутацию BRAF V600. В одном варианте осуществления рак является раком, имеющим мутацию BRAF V600E. В одном варианте осуществления рак является раком толстой и прямой кишки. В одном варианте осуществления рак является раком толстой и прямой кишки, имеющим мутацию BRAF V600E. В одном варианте осуществления рак является MEK-ассоциированным раком. В одном варианте осуществления рак является EGFR-ассоциированным раком.
[00172] Также предложена КОМБИНАЦИЯ ИЗОБРЕТЕНИЯ, подходящая для лечения пролиферативного заболевания у нуждающегося в этом субъекта. В одном варианте осуществления пролиферативным заболеванием является рак. В одном варианте осуществления рак выбран из рака толстой и прямой кишки (CRC) (включая метастатический рак толстой и прямой кишки), меланомы (включая метастатическую и неоперабельную меланому), рака легкого (включая немелкоклеточный рак легкого (НМРЛ)), рака молочной железы, рака почки, такого как, например, почечно-клеточная карцинома (RCC), рака печени, рака эндометрия, острого миелогенного лейкоза (ОМЛ), миелодиспластических синдромов (MDS), рака щитовидной железы, в частности папиллярного рака щитовидной железы, рака поджелудочной железы, нейрофиброматоза и гепатоцеллюлярной карциномы. В одном варианте осуществления рак является раком толстой и прямой кишки (CRC). В одном варианте осуществления рак толстой и прямой кишки является метастатическим раком толстой и прямой кишки. В одном варианте осуществления рак является BRAF-ассоциированным раком. В одном варианте осуществления рак является раком, имеющим мутацию BRAF V600. В одном варианте осуществления рак является раком, имеющим мутацию BRAF V600E. В одном варианте осуществления рак является раком толстой и прямой кишки. В одном варианте осуществления рак является раком толстой и прямой кишки, имеющим мутацию BRAF V600E. В одном варианте осуществления рак является MEK-ассоциированным раком. В одном варианте осуществления MEK-связанный рак является раком толстой и прямой кишки. В одном варианте осуществления рак является EGFR-ассоциированным раком. В одном варианте осуществления EGFR-ассоциированный рак является раком толстой и прямой кишки.
[00173] Комбинированная терапия, включающая КОМБИНАЦИЮ ИЗОБРЕТЕНИЯ, может приводить к неожиданному улучшению лечения пролиферативных заболеваний по сравнению с монотерапией. В одном варианте осуществления, при одновременном, последовательном или раздельном введении, ингибитор BRAF (СОЕДИНЕНИЕ A), ингибитор MEK, (СОЕДИНЕНИЕ B) и (c) антитело против EGFR (СОЕДИНЕНИЕ C) взаимодействуют синергически, вызывая ингибирование пролиферации клеток.
[00174] Природа пролиферативных заболеваний является многофакторной. При некоторых обстоятельствах лекарственные средства с разными механизмами действия можно комбинировать. Однако просто рассмотрение любой комбинации терапевтических средств, имеющих разный механизм действия, не обязательно приводит к комбинациям с полезными эффектами.
[00175] Введение КОМБИНАЦИИ ИЗОБРЕТЕНИЯ может приводить не только к благоприятному воздействию, например синергическому терапевтическому эффекту, например, в отношении облегчения, задержки прогрессирования (например, увеличения выживаемости без прогрессирования (PFS)), увеличения общей выживаемости (OS) или ингибирования симптомов, но также и к более неожиданным благоприятным эффектам, например, уменьшению побочных эффектов, улучшению качества жизни или уменьшению тяжести заболевания, по сравнению с монотерапией, в которой применяют только одно из фармацевтически терапевтических средств, применяемых в КОМБИНАЦИИ ИЗОБРЕТЕНИЯ.
[00176] Дополнительная выгода состоит в том, что могут использоваться более низкие дозы терапевтических средств КОМБИНАЦИИ ИЗОБРЕТЕНИЯ, например, что дозы часто могут быть не только меньше, но также их можно применять реже или можно использовать для уменьшения частоты побочных эффектов, наблюдаемых отдельно с одним из партнеров по комбинации. Это соответствует пожеланиям и потребностям пациентов, которые проходят лечение.
[00177] Стандартные экспериментальные модели могут показать, что КОМБИНАЦИЯ ИЗОБРЕТЕНИЯ приводит к благоприятным эффектам, описанным выше. Специалист в данной области обладает всеми возможностями для выбора подходящей экспериментальной модели для подтверждения таких полезных эффектов. Фармакологическая активность КОМБИНАЦИИ ИЗОБРЕТЕНИЯ может быть, например, продемонстрирована в клиническом исследовании или в экспериментальной процедуре, например, как описано ниже.
[00178] Подходящими клиническими исследованиями являются, например, открытые исследования с повышением дозы у больных пролиферативным заболеванием. Такие исследования могут продемонстрировать, в частности, синергизм терапевтических средств КОМБИНАЦИИ ИЗОБРЕТЕНИЯ. Благоприятные эффекты в отношении пролиферативных заболеваний могут быть определены непосредственно по результатам этих исследований. Такие исследования, в частности, могут подходить для сравнения эффектов монотерапии с применением любого из СОЕДИНЕНИЯ A, СОЕДИНЕНИЯ B или СОЕДИНЕНИЯ C с эффектами КОМБИНАЦИИ ИЗОБРЕТЕНИЯ, или для сравнения эффектов двойной терапии с применением двух любых из СОЕДИНЕНИЯ A, СОЕДИНЕНИЯ B или СОЕДИНЕНИЯ C с эффектами КОМБИНАЦИИ ИЗОБРЕТЕНИЯ.
[00179] В одном варианте осуществления дозу ингибитора BRAF (СОЕДИНЕНИЕ A) повышают, пока не будет достигнута максимальная переносимая доза, при этом ингибитор MEK (СОЕДИНЕНИЕ B) и антитело против EGFR (СОЕДИНЕНИЕ C) вводят в фиксированной дозе. В альтернативе СОЕДИНЕНИЕ A и СОЕДИНЕНИЕ B могут вводить в фиксированной дозе, а дозу СОЕДИНЕНИЯ C могут повышать, пока не будет достигнута максимальная переносимая доза. В альтернативе СОЕДИНЕНИЕ A и СОЕДИНЕНИЕ C могут вводить в фиксированной дозе, а дозу СОЕДИНЕНИЯ B могут повышать, пока не будет достигнута максимальная переносимая доза.
[00180] Каждый пациент может получать дозы ингибитора BRAF (СОЕДИНЕНИЕ A) и/или ингибитора MEK (СОЕДИНЕНИЕ B), и/или ингибитора EGFR (СОЕДИНЕНИЯ C) либо ежедневно, либо периодически. Эффективность лечения могут определять в таких исследованиях, например, через 6, 12, 18 или 24 недели, при помощи шкалы оценки симптомов, например, каждые 6 недель.
[00181] Определение синергического взаимодействия между одним или более компонентами КОМБИНАЦИИ ИЗОБРЕТЕНИЯ, оптимального диапазона действия и абсолютных диапазонов дозы каждого компонента для действия может быть окончательно измерено при введении компонентов в различных диапазонах весовых отношений и различных дозах нуждающимся в лечении пациентам.
[00182] В одном варианте осуществления настоящего изобретения предложена синергическая комбинация для медицинского применения, предназначенная для лечения пролиферативного заболевания, такого как рак, включающая: (a) ингибитор BRAF (СОЕДИНЕНИЕ A) или его фармацевтически приемлемую соль, (b) ингибитор MEK (СОЕДИНЕНИЕ B) или его фармацевтически приемлемую соль и (c) ингибитор EGFR (СОЕДИНЕНИЕ C) в диапазоне комбинаций вес:вес:вес, например, в в/в/в диапазоне компонентов комбинации, который соответствует диапазонам, наблюдаемым в модели опухоли, используемой для обнаружения синергического взаимодействия.
[00183] В КОМБИНАЦИИ ИЗОБРЕТЕНИЯ партнеры по комбинации (СОЕДИНЕНИЕ A), (СОЕДИНЕНИЕ B) и (СОЕДИНЕНИЕ C) могут вводить в одном составе или единичной лекарственной форме, или могут вводить параллельно, но раздельно, в виде единичных лекарственных форм, или могут вводить последовательно в виде единичных стандартных лекарственных форм, в любом порядке и с определенными или неопределенными временными интервалами, любым подходящим путем. Единичная лекарственная форма также может быть фиксированной комбинацией, например, СОЕДИНЕНИЯ A и СОЕДИНЕНИЯ B.
[00184] СОЕДИНЕНИЕ A, СОЕДИНЕНИЕ B и СОЕДИНЕНИЕ C могут вводить в виде фармацевтических композиций любым стандартным путем, в частности энтерально, например, перорально, например, в форме таблеток или капсул, парентерально, например, в форме растворов или суспензий для инъекций, наружно, например, в форме лосьонов, гелей, мазей или кремов, или в форме для назального применения или в форме суппозитория. Фармацевтические композиции, включающие СОЕДИНЕНИЕ A, СОЕДИНЕНИЕ B или СОЕДИНЕНИЕ C в свободной форме или в форме фармацевтически приемлемой соли, в сочетании по меньшей мере с одним фармацевтически приемлемым носителем или разбавителем, могут быть получены стандартным образом, с помощью способов смешивания, грануляции или нанесения оболочки. Например, композиции для приема внутрь могут быть таблетками или желатиновыми капсулами, включающими действующее вещество вместе с: a) разбавителями, например, лактозой, декстрозой, сахарозой, маннитом, сорбитом, целлюлозой и/или глицином; b) смазывающими веществами, например, диоксидом кремния, тальком, стеариновой кислотой, ее магниевой или кальциевой солью и/или полиэтиленгликолем; также для таблеток, c) связующими веществами, например, силикатом алюминия магния, пастой крахмала, желатином, трагакантом, метилцеллюлозой, натрий-карбоксиметилцеллюлозой и/или поливинилпирролидоном; при необходимости, d) разрыхлителями, например, крахмалами, агаром, альгиновой кислотой или ее натриевой солью, или шипучими смесями; и/или e) абсорбентами, красителями, ароматизаторами и подсластителями. Композиции для инъекций могут быть водными изотоническими растворами или суспензиями, и суппозитории могут быть изготовлены из жировых эмульсий или суспензий. Композиции могут быть стерильными и/или содержать вспомогательные вещества, такие как консерванты, стабилизаторы, смачивающие или эмульгирующие вещества, вещества, способствующие образованию раствора, соли для регуляции осмотического давления и/или буферы. Кроме того, они также могут содержать другие терапевтически ценные вещества. Например, соединения изобретения могут быть включены в концентрат микроэмульсии (MEPC).
[00185] Единичные лекарственные формы СОЕДИНЕНИЯ A, СОЕДИНЕНИЯ B и СОЕДИНЕНИЯ C необязательно могут также включать дополнительные стандартные носители или вспомогательные вещества, используемые в фармацевтических препаратах. Примеры таких носителей включают, без ограничения перечисленными, разрыхлители, связующие вещества, смазывающие вещества, скользящие вещества, стабилизаторы и наполнители, разбавители, красители, ароматизаторы и консерванты. Средний специалист в данной области сможет выбрать один или более вышеуказанных носителей в зависимости от конкретных требуемых свойств лекарственной формы с помощью стандартных экспериментов и без каких-либо излишних трудностей. Количество каждого использованного носителя может изменяться в стандартных диапазонах. В следующих источниках, которые настоящим включены посредством ссылки, раскрыты способы и вспомогательные вещества, используемые при изготовлении лекарственных форм для перорального применения. См. The Handbook of Pharmaceutical Excipients, 4th edition, Rowe et al., Eds., American Pharmaceuticals Association (2003); и Remington: the Science and Practice of Pharmacy, 20th edition, Gennaro, Ed., Lippincott Williams & Wilkins (2003).
[00186] Такие необязательные дополнительные стандартные носители могут быть включены в лекарственную форму для перорального применения либо путем включения одного или более стандартных носителей в исходную смесь до или во время гранулирования, либо путем объединения одного или более стандартных носителей с гранулами, содержащими комбинацию средств или отдельные средства комбинации средств в лекарственной форме для перорального применения. В последнем варианте осуществления комбинированную смесь могут дополнительно смешивать, например, с помощью V-образного смесителя, а затем прессовать или формовать в таблетку, например, монолитную таблетку, заключать в капсулу или заполнять саше.
[00187] Примеры фармацевтически приемлемых разрыхлителей включают, без ограничения перечисленными, крахмалы; глины; целлюлозы; альгинаты; камеди; поперечно сшитые полимеры, например, поперечно сшитый поливинилпирролидон или кросповидон, например, POLYPLASDONE XL производства International Specialty Products (Wayne, NJ); поперечно сшитую натрий-карбоксиметилцеллюлозу или натрий-кроскармеллозу, например, AC-DI-SOL производства FMC; и поперечно сшитую кальций карбоксиметилцеллюлозу; полисахариды сои; и гуаровую камедь. Разрыхлитель может присутствовать в количестве от приблизительно 0% до приблизительно 10% от веса композиции. В одном варианте осуществления разрыхлитель присутствует в количестве от приблизительно 0,1% до приблизительно 5% от веса композиции.
[00188] Примеры фармацевтически приемлемых связующих веществ включают, без ограничения перечисленными, крахмалы; целлюлозы и их производные, например, микрокристаллическую целлюлозу, например, AVICEL PH производства FMC (Philadelphia, PA), гидроксипропилцеллюлозу, гидроксиэтилцеллюлозу и гидроксипропилметилцеллюлозу METHOCEL производства Dow Chemical Corp. (Midland, MI); сахарозу; декстрозу; кукурузный сироп; полисахариды; и желатин. Связующее вещество может присутствовать в количестве от приблизительно 0% до приблизительно 50%, например 2-20%, от веса композиции.
[00189] Примеры фармацевтически приемлемых смазывающих веществ и фармацевтически приемлемых скользящих веществ включают, без ограничения перечисленными, коллоидный диоксид кремния, трисиликат магния, крахмалы, тальк, фосфат кальция, стеарат магния, стеарат алюминия, стеарат кальция, карбонат магния, оксид магния, полиэтиленгликоль, порошок целлюлозы и микрокристаллическую целлюлозу. Смазывающее вещество может присутствовать в количестве от приблизительно 0% до приблизительно 10% от веса композиции. В одном варианте осуществления смазывающее вещество может присутствовать в количестве от приблизительно 0,1% до приблизительно 1,5% от веса композиции. Скользящее вещество может присутствовать в количестве от приблизительно 0,1% до приблизительно 10% по весу.
[00190] Примеры фармацевтически приемлемых наполнителей и фармацевтически приемлемых разбавителей включают, без ограничения перечисленными, сахарную пудру, кусковой сахар, декстраты, декстрин, декстрозу, лактозу, маннит, микрокристаллическую целлюлозу, порошок целлюлозы, сорбит, сахарозу и тальк. Наполнитель и/или разбавитель, например, может присутствовать в количестве от приблизительно 0% до приблизительно 80% от веса композиции.
[00191] В одном варианте осуществления ингибитор BRAF (СОЕДИНЕНИЕ A) сформулирован для перорального применения.
[00192] В одном варианте осуществления ингибитор BRAF (СОЕДИНЕНИЕ A) сформулирован в виде таблетки или капсулы. Способы изготовления лекарственных форм для перорального применения СОЕДИНЕНИЯ A описаны в публикации PCT WO 2013/078264, которая включена в настоящий документ посредством ссылки.
[00193] В одном варианте осуществления СОЕДИНЕНИЕ A включено в лекарственную форму в виде твердой фармацевтической композиции для перорального применения, которая включает: (i) внутреннюю фазу, которая является твердой дисперсией, включающей аморфный (S)-метил-(1-((4-(3-(5-хлор-2-фтор-3-(метилсульфон-амидо)фенил)-1-изопропил-1H-пиразол-4-ил)пиримидин-2-ил)амино)пропан-2-ил)карбамат (СОЕДИНЕНИЕ A), гидрофильньное связующее вещество, поверхностно-активное вещество и (ii) внешнюю фазу, которая включает подкислитель, наполнитель и смазывающее вещество.
[00194] В одном варианте осуществления твердых лекарственных форм для перорального применения СОЕДИНЕНИЯ A, может присутствовать количество СОЕДИНЕНИЯ A в диапазонах 1-1500 мг, 2,5-800 мг или 5-400 мг. В одном варианте осуществления твердая лекарственная форма для перорального применения включает 10 мг, 20 мг, 25 мг, 50 мг, 75 мг, 100 мг, 200 мг, 300 мг, 400 мг или 500 мг СОЕДИНЕНИЯ A. В одном варианте осуществления твердая лекарственная форма для перорального применения включает 50 мг аморфного СОЕДИНЕНИЯ A. В одном варианте осуществления твердая лекарственная форма для перорального применения включает 75 мг аморфного СОЕДИНЕНИЯ A. В одном варианте осуществления твердая лекарственная форма для перорального применения аморфного СОЕДИНЕНИЯ A изготовлена в виде капсулы.
[00195] В одном варианте осуществления СОЕДИНЕНИЕ A включено в твердую фармацевтическую композицию для перорального применения, которая включает: (i) внутреннюю фазу, которая является твердой дисперсией, включающей аморфное СОЕДИНЕНИЕ A; коповидон; и полоксамер 188 или сорбит; и (ii) внешнюю фазу, которая включает янтарную кислоту, микрокристаллическую целлюлозу, кросповидон, коллоидный диоксид кремния и стеарат магния. В одном варианте осуществления внутренняя фаза включает от 5% до 40% по весу аморфного СОЕДИНЕНИЯ A, от 50% до 80% по весу коповидона и от 5% до 20% по весу полоксамера 188 или сорбита. В одном варианте осуществления внешняя фаза включает от 2% до 60% по весу янтарной кислоты, от 30% до 70% по весу микрокристаллической целлюлозы, от 5% до 20% по весу кросповидона, от 0,5% до 5% по весу коллоидного диоксида кремния и от 0,5% до 5% по весу стеарата магния. В одном варианте осуществления твердая фармацевтическая композиция для перорального применения включает смесь внутренней и внешней фаз в соотношении от 80:20 до 40:60. В одном варианте осуществления твердая фармацевтическая композиция для перорального применения включает смесь внутренней и внешней фаз в соотношении от 75:25 до 50:50. В одном варианте осуществления твердая фармацевтическая композиция для перорального применения включает 0 мг, 25 мг, 50 мг, 75 мг или 100 мг аморфного СОЕДИНЕНИЯ A. В одном варианте осуществления твердая фармацевтическая композиция для перорального применения включает 10 мг, 25 мг, 50 мг, 75 мг или 100 мг аморфного СОЕДИНЕНИЯ A. В одном варианте осуществления твердая лекарственная форма для перорального применения включает 50 мг аморфного СОЕДИНЕНИЯ A. В одном варианте осуществления твердая лекарственная форма для перорального применения включает 75 мг аморфного СОЕДИНЕНИЯ A. В одном варианте осуществления твердая фармацевтическая композиция для перорального применения включает 15% по весу аморфного СОЕДИНЕНИЯ A. Фармацевтические композиции СОЕДИНЕНИЯ A описаны в публикации PCT WO 2013/078264.
[00196] В одном варианте осуществления твердая фармацевтическая композиция аморфного СОЕДИНЕНИЯ A для перорального применения выбрана из:
A)
и
B)
[00197] В одном варианте осуществления твердая фармацевтическая композиция аморфного СОЕДИНЕНИЯ A для перорального применения выбрана из:
и
[00198] В одном варианте осуществления твердая лекарственная форма для перорального применения СОЕДИНЕНИЯ A изготовлена способом, включающим: (i) смешивание смеси, включающей аморфное СОЕДИНЕНИЕ A; коповидон; и полоксамер 188 или сорбит, с получением первой смеси; (ii) экструзию первой смеси с получением экструдата; (iii) измельчение экструдата с получением молотого экструдата; (iv) смешивание молотого экструдата по меньшей мере с одним из янтарный кислоты, микрокристаллической целлюлозы, кросповидона, коллоидного диоксида кремния и стеарата магния с получением второй смеси; (v) необязательно, повтор этапа (iv), при необходимости, с получением третьей смеси, включающей янтарную кислоту, микрокристаллическую целлюлозу, кросповидон, коллоидный диоксид кремния, стеарат магния и молотый экструдат; и, (vi) необязательно, таблетирование или инкапсулировние третьей смеси.
[00199] В одном варианте осуществления ингибитор MEK (СОЕДИНЕНИЕ B) сформулирован для перорального применения. В одном варианте осуществления ингибитор MEK (СОЕДИНЕНИЕ B) сформулирован в виде таблетки или капсулы. В одном варианте осуществления ингибитор MEK (СОЕДИНЕНИЕ B) сформулирован в виде таблетки. В одном варианте осуществления таблетка является таблеткой, покрытой оболочкой. В одном варианте осуществления ингибитор MEK (СОЕДИНЕНИЕ B) является (2-гидроксиэтокси)амидом 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты. В одном варианте осуществления ингибитор MEK (СОЕДИНЕНИЕ B) является кристаллизованным (2-гидроксиэтокси)амидом 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты. Способы получения лекарственных форм (2-гидроксиэтокси)амида 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты для перорального применения описаны в публикации PCT WO 2014/063024.
[00200] В одном варианте осуществления фармацевтическая композиция СОЕДИНЕНИЯ B включает кристаллизованное СОЕДИНЕНИЕ B, по меньшей мере один сахар и по меньшей мере одно вспомогательное вещество, производное целлюлозы. В одном варианте осуществления фармацевтической композиции, предложенной в настоящем документе, фармацевтическая композиция включает приблизительно 5-35% кристаллизованного СОЕДИНЕНИЯ B от веса композиции. В другом варианте осуществления фармацевтическая композиция включает приблизительно 5-11% кристаллизованного СОЕДИНЕНИЯ B от веса композиции. В другом предпочтительном варианте осуществления фармацевтическая композиция включает приблизительно 6,25% кристаллизованного СОЕДИНЕНИЯ B от веса композиции. В другом предпочтительном варианте осуществления фармацевтическая композиция включает приблизительно 10% кристаллизованного СОЕДИНЕНИЯ B от веса композиции. В другом варианте осуществления фармацевтическая композиция включает приблизительно 15 мг или 45 мг кристаллизованного СОЕДИНЕНИЯ B. В другом варианте осуществления фармацевтическая композиция включает приблизительно 15 мг кристаллизованного СОЕДИНЕНИЯ B. Подходящий сахар для применения в фармацевтических композициях включает, без ограничения, лактозу (например, высушенную распылением лактозу, моногидрат лактозы), мальтозу, фруктозу, галактозу, сахарную пудру, кусковой сахар, декстраты, декстрин, декстрозу, маннит, Nu-Tab, Di-Pac, Emdex и сахарозу. В предпочтительном варианте осуществления сахар, применяемый в фармацевтической композиции, является лактозой, в частности моногидратом лактозы. В одном варианте осуществления фармацевтической композиции, предложенной в настоящем документе, фармацевтическая композиция включает приблизительно 30-70% по меньшей мере одного сахара от веса композиции. В другом варианте осуществления фармацевтическая композиция включает приблизительно 50-60% лактозы от веса композиции. В другом варианте осуществления фармацевтическая композиция включает приблизительно 50-60% моногидрата лактозы от веса композиции. В предпочтительном варианте осуществления фармацевтическая композиция включает приблизительно 55-56% моногидрата лактозы от веса композиции. Подходящие вспомогательные вещества, производные целлюлозы, включают, без ограничения перечисленными, микрокристаллическую целлюлозу, микродисперсную целлюлозу, порошок целлюлозы, метилцеллюлозу, гидроксиэтилцеллюлозу, гидроксиметилцеллюлозу, гидроксипропилцеллюлозу и гидроксипропилметилцеллюлозу. В предпочтительном варианте осуществления вспомогательным веществом на основе целлюлозы является микрокристаллическая целлюлоза. В одном варианте осуществления фармацевтической композиции, предложенной в настоящем документе, фармацевтическая композиция включает приблизительно 20-40% вспомогательного вещества, производного целлюлозы, от веса композиции. В другом варианте осуществления фармацевтическая композиция включает приблизительно 20-40% микрокристаллической целлюлозы, от веса композиции. В другом варианте осуществления фармацевтическая композиция включает приблизительно 30-40% микрокристаллической целлюлозы, от веса композиции. В одном варианте осуществления фармацевтическая композиция включает приблизительно 30-36% микрокристаллической целлюлозы, от веса композиции. В одном варианте осуществления фармацевтическая композиция СОЕДИНЕНИЯ B включает (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты. В одном варианте осуществления фармацевтическая композиция СОЕДИНЕНИЯ B включает кристаллизованный (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты. В одном варианте осуществления фармацевтическая композиция СОЕДИНЕНИЯ B включает 15 мг кристаллизованного (2-гидроксиэтокси)амида 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты.
[00201] Фармацевтическая композиция СОЕДИНЕНИЯ B может включать дополнительные вспомогательные вещества или носители, включающие, без ограничения, разрыхлители, смазывающее вещества, скользящие вещества, связующие веществами, стабилизаторы и наполнители, разбавители, красители, ароматизаторы и консерванты. Средний специалист в данной области сможет выбрать один или более вышеуказанных носителей в зависимости от конкретных требуемых свойств лекарственной формы с помощью стандартных экспериментов и без каких-либо излишних трудностей. Количество каждого используемого носителя может изменяться в диапазонах, обычных в уровне техники. В следующих источниках, которые настоящим включены посредством ссылки, раскрыты способы и вспомогательные вещества, используемые для изготовления лекарственных форм для перорального применения. См. The Handbook of Pharmaceutical Excipients, 4th edition, Rowe et al., Eds., American Pharmaceuticals Association (2003); и Remington: the Science and Practice of Pharmacy, 20th edition, Gennaro, Ed., Lippincott Williams & Wilkins (2003). Эти необязательные дополнительные стандартные носители могут быть включены в состав лекарственной формы для перорального применения либо путем включения одного или более обычных носителей в исходную смесь, либо добавлены во время фаз смешивания.
[00202] Примеры фармацевтически приемлемых разрыхлителей для композиций СОЕДИНЕНИЯ B включают, без ограничения перечисленными, крахмалы; глины; целлюлозы; альгинаты; камеди; поперечно сшитые полимеры, например поперечно сшитый поливинилпирролидон или кросповидон, например, POLYPLASDONE XL производства International Specialty Products (Wayne, NJ); поперечно сшитую натрий-карбоксиметилцеллюлозу или натрий-кроскармеллозу (например, AC-DI-SOL производства FMC); и поперечно сшитую кальций-карбоксиметилцеллюлозу; полисахариды сои; и гуаровую камедь. Разрыхлитель может присутствовать в количестве от приблизительно 0% до приблизительно 10% от веса композиции. В одном варианте осуществления разрыхлитель присутствует в количестве от приблизительно 0,1-5% или приблизительно 1-3%, или приблизительно 1,5-2,5% от веса композиции.
[00203] В одном варианте осуществления фармацевтическая композиция СОЕДИНЕНИЯ B включает разрыхлитель натрий-кроскармеллозу. В другом варианте осуществления фармацевтическая композиция настоящего изобретения включает приблизительно 2% натрий-кроскармеллозы от веса композиции.
[00204] Примеры фармацевтически приемлемых смазывающих веществ и фармацевтически приемлемых скользящих веществ для композиций СОЕДИНЕНИЯ B включают, без ограничения перечисленными, коллоидный диоксид кремния/коллоидный безводный кремнезем (например, Aerosil 200®), трисиликат магния, крахмалы, тальк, фосфат кальция, стеарат магния, стеарат алюминия, стеарат кальция, карбонат магния, оксид магния, полиэтиленгликоль, порошок целлюлозы и микрокристаллическую целлюлозу. Смазывающее вещество может присутствовать в количестве от приблизительно 0% до приблизительно 10% от веса композиции. В одном варианте осуществления смазывающее вещество может присутствовать в количестве от приблизительно 0,1-1,5%, приблизительно 0,1-1% или приблизительно 0,5-0,9% от веса композиции. Скользящее вещество может присутствовать в количестве от приблизительно 0,1-10%, приблизительно 0,1-5% или приблизительно 0,1-1% от веса композиции.
[00205] В одном варианте осуществления фармацевтическая композиция СОЕДИНЕНИЯ B включает скользящее вещество коллоидный диоксид кремния/коллоидный безводный кремнезем. В другом варианте осуществления фармацевтическая композиция СОЕДИНЕНИЯ B включает приблизительно 0,25% (от веса композиции) коллоидного диоксида кремния/коллоидного кремнезема.
[00206] В другом варианте осуществления фармацевтическая композиция СОЕДИНЕНИЯ B включает смазывающее вещество, стеарат магния. В другом варианте осуществления фармацевтическая композиция СОЕДИНЕНИЯ B включает приблизительно 0,75% стеарата магния от веса композиции.
[00207] В другом варианте осуществления фармацевтическая композиция СОЕДИНЕНИЯ B включает коллоидный диоксид кремния/коллоидный безводный кремнезем и стеарат магния. В другом варианте осуществления фармацевтическая композиция СОЕДИНЕНИЯ B включает приблизительно 0,25% коллоидного диоксида кремния/коллоидного безводного кремнезема от веса композиции и приблизительно 0,75% стеарата магния от веса композиции.
[00208] Примеры фармацевтически приемлемых связующих веществ для композиций СОЕДИНЕНИЯ B включают, без ограничения перечисленными, крахмалы; целлюлозы и их производные, например, микрокристаллическую целлюлозу, например, AVICEL PH производства FMC (Philadelphia, PA), гидроксипропилцеллюлозу, гидроксиэтилцеллюлозу и гидроксипропилметилцеллюлозу METHOCEL производства Dow Chemical Corp. (Midland, MI); сахарозу; декстрозу; кукурузный сироп; полисахариды; и желатин. Связующее вещество может присутствовать в количестве от приблизительно 0-50% или приблизительно 2-20% от веса композиции.
[00209] Примеры фармацевтически приемлемых разбавителей для композиций СОЕДИНЕНИЯ B включают, без ограничения перечисленными, сахарную пудру, кусковой сахар, декстраты, декстрин, декстрозу, лактозу, маннит, микрокристаллическую целлюлозу, порошок целлюлозы, сорбит, сахарозу и тальк. Разбавитель, например, может присутствовать в количестве от приблизительно 0-80% или приблизительно 0-50%, или приблизительно 1-40%, или приблизительно 1-10% от веса композиции.
[00210] В одном варианте осуществления фармацевтическая композиция СОЕДИНЕНИЯ B дополнительно включает одно или более из натрий-кроскармеллозы, стеарата магния и диоксида кремния.
[00211] Если водные суспензии или настойки предпочтительны для приема внутрь, активное соединение в них может быть объединено с различным подсластителями или ароматизаторами, окрашивающими веществами или красителями и, при необходимости, с эмульгаторами или суспендирующими веществами, вместе с разбавителями, такими как вода, этанол, пропиленгликоль, глицерин или их комбинации.
[00212] В одном варианте осуществления фармацевтическая композиция СОЕДИНЕНИЯ B включает приблизительно 5-11% кристаллизованного СОЕДИНЕНИЯ B от веса композиции, приблизительно 55-56% моногидрата лактозы от веса композиции и приблизительно 30-36% микрокристаллической целлюлозы от веса композиции. В одном варианте осуществления фармацевтическая композиция СОЕДИНЕНИЯ B включает (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты.
[00213] В одном варианте осуществления фармацевтическая композиция СОЕДИНЕНИЯ B включает приблизительно 5-11% кристаллизованного СОЕДИНЕНИЯ B, приблизительно 55-56% моногидрата лактозы, приблизительно 30-36% микрокристаллической целлюлозы, от веса композиции, приблизительно 1,5-2,5% натрий-кроскармеллозы, приблизительно 0,5-0,9% стеарата магния и приблизительно 0,1-1% процент коллоидного диоксида кремния/коллоидного безводного кремнезема, от веса композиции. В одном варианте осуществления фармацевтическая композиция СОЕДИНЕНИЯ B включает (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты.
[00214] В одном варианте осуществления фармацевтическая композиция СОЕДИНЕНИЯ B включает приблизительно 5-11% кристаллизованного СОЕДИНЕНИЯ B от веса композиции, приблизительно 55-56% моногидрата лактозы от веса композиции, приблизительно 30-36% микрокристаллической целлюлозы от веса композиции, приблизительно 2% натрий-кроскармеллозы от веса композиции, приблизительно 0,75 процента стеарата магния от веса композиции и приблизительно 0,25 процента коллоидного диоксида кремния/коллоидного безводного кремнезема от веса композиции. В одном варианте осуществления фармацевтическая композиция СОЕДИНЕНИЯ B включает (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты.
[00215] В одном варианте осуществления фармацевтическая композиция СОЕДИНЕНИЯ B включает 15 мг кристаллизованного СОЕДИНЕНИЯ B. В одном варианте осуществления фармацевтическая композиция СОЕДИНЕНИЯ B включает кристаллическое СОЕДИНЕНИЕ B, моногидрат лактозы, микрокристаллическую целлюлозу, коллоидный диоксид кремния, натрий-кроскармеллозу и стеарат магния. В одном варианте осуществления фармацевтическая композиция СОЕДИНЕНИЯ B включает (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты.
[00216] В одном варианте осуществления фармацевтическая композиция СОЕДИНЕНИЯ B включает:
(% по весу)
(в мг/ единица)
(% по весу)
(в мг/ единица)
[00217] В одном варианте осуществления вышеуказанной лекарственной формы, кристаллизованное СОЕДИНЕНИЕ B является кристаллизованным (2-гидроксиэтокси)амидом 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты.
[00218] В вариантах осуществления, где ингибитор EGFR (СОЕДИНЕНИЯ C) является ингибитором тирозинкиназы, соединение может быть включено в лекарственную форму для перорального применения.
[00219] В вариантах осуществления, где ингибитор EGFR (СОЕДИНЕНИЯ C) является моноклональным антителом (т.е. антителом против EGFR), антитело включено в лекарственную форму для внутривенного введения. В одном варианте осуществления антитело против EGFR включено в лекарственную форму в виде раствора без консервантов, который содержит 8,48 мг/мл натрия хлорида, 1,88 мг/мл двухосновного натрия фосфата гептагидрата, 0,41 мг/мл одноосновного натрия фосфата моногидрат и воду для инъекций.
[00220] Также предложена коммерческая упаковка, включающая в качестве терапевтических средств КОМБИНАЦИЮ ИЗОБРЕТЕНИЯ, вместе с инструкциями по их одновременному, раздельному или последовательному введению для применения в задержке прогрессии или лечении пролиферативного заболевания у нуждающегося в этом субъекта.
[00221] Также предложена коммерческая упаковка, включающая: (a) ингибитор BRAF, которым является метил-N-[(2S)-1-({4-[3-(5-хлор-2-фтор-3-метансульфонамидофенил)-1-(пропан-2-ил)-1H-пиразол-4-ил]пиримидин-2-ил}амино)пропан-2-ил]карбамат или его фармацевтически приемлемая соль, (b) по меньшей мере один ингибитор MEK, вместе с инструкциями по их одновременному, раздельному или последовательному введению вместе с (СОЕДИНЕНИЕМ C) при лечении пролиферативного заболевания. В одном варианте осуществления ингибитором MEK является (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты или его фармацевтически приемлемая соль. В одном варианте осуществления ингибирующим антителом против EGFR является цетуксимаб.
[00222] Следующие Примеры иллюстрируют изобретение, описанное выше; однако они никоим образом не предназначены для ограничения объема изобретения. Полезные эффекты фармацевтической комбинации настоящего изобретения также могут быть определены с помощью других экспериментальных моделей, известных в качестве таковых специалисту в соответствующей области.
ПРИМЕР 1
Противоопухолевое действие тройной комбинации LGX818 (энкорафениба; ингибитора BRAF), MEK162 (биниметиниба; ингибитора MEK) и цетуксимаба (ингибитора EGFR) в модели HT-29 CRC
[00223] LGX818 (СОЕДИНЕНИЕ A), MEK162 (СОЕДИНЕНИЕ B) и цетуксимаб (СОЕДИНЕНИЯ C) тестировали в комбинациях в ксенотрансплантатной BRAF мутантной модели опухоли HT-29 рака толстой и прямой кишки (CRC). LGX818, MEK162 и цетуксимаб, в качестве отдельных средств, демонстрировали T/C 42%, 28% и 95%, соответственно. Комбинация LGX818 или MEK162 с цетуксимабом привела к T/C 22%. Тройная комбинация LGX818, MEK162 и цетуксимаба привела к регрессии опухоли с T/T0 -14%. Эти результаты продемонстрировали пользу комбинации LGX818 с MEK162 и цетуксимабом в BRAF V600E мутантном CRC.
1. СОКРАЩЕНИЯ
[00224] 2. МЕТОДЫ
[00225] 2.1 Материалы
Таблица 1 Характеристики животных
(Mus musculus)
[00226] 2.1.1 Животные и условия содержания
[00227] Для всех экспериментов, голых мышей содержали в виварии в условиях светового цикла 12 часов света/темноты и обеспечивали доступ к пище и воде без ограничения.
[00228] 2.1.2 Заявление о надлежащем обращении с животными
[00229] Животным позволяли акклиматизироваться в виварии Novartis NIBRI в течение по меньшей мере 3 дней до начала экспериментов. С животными обращались в соответствии с инструкциями и рекомендациями IACUC Novartis.
[00230] 2.1.3 Клетки и условия культивирования клеток
[00231] Клетки HT-29 приобретали в ATCC, и основные стоки были получены в CLE. Рабочий сток получали от CLE и культивировали в среде МакКоя 5А, содержащей 10% термоинактивированной эмбриональной бычьей сыворотки без антибиотиков до момента имплантации. Протестированные клетки не содержали микоплазмы и вирусной контаминации в панели тестов ПЦР IMPACT VIII (IDEXX RADIL, IDEXX Laboratories INC. Westbrook, ME). Клетки HT-29 были получены после пассажа 10 для исследования TRP-377-HT-29-XEF.
[00232] 2.1.4 Композиция тестируемого соединения
[00233] LGX818 растворяли в 0,5% КМЦ/0,5% Tween 80. Оно оставалось стабильным в течение по меньшей мере одной недели при комнатной температуре; и его вводили в дозе 20 мг/кг, п/о, qd×21.
[00234] MEK162 растворяли в 1% КМЦ/0,5% Tween 80. Оно оставалось стабильным в течение по меньшей мере одной недели при комнатной температуре; и его вводили в дозе 3,5 мг/кг, п/о, bid×21.
[00235] Цетуксимаб (Эрбитукс) - продукт ImClone LLC, подразделения Eli Lilly. Он поставляется в концентрации 2 мг/мл. Его вводили в дозе 20 мг/кг, в/б, 2qw×10.
[00236] 2.2 МЕТОДЫ
[00237] 2.2.1 Ксенотрансплантатная модель рака толстой кишки HT-29 на самках голых мышей
[00238] Клетки HT-29 собирали в фазе экспоненциального роста. Пять миллионов клеток в 200 мкл PBS подкожно имплантировали в верхнюю область на правом боку 112 самкам голых мышей. Опухоли достигли размера приблизительно 200 мм3 в день 22 после имплантации клеток. В День 22 мышей-опухоленосителей рандомизировали в контрольные группы, и лечение начинали в День 22 и заканчивали в День 43. Опухоли наблюдали на предмет роста после завершения лечения.
[00239] 2.2.2 Наблюдение животных
[00240] Состояние и поведение животных, включая груминг и способность передвигаться, наблюдали два раза в день. Контролировали общее состояние здоровья мышей и ежедневно регистрировали смертность. Агонизирующих животных умерщвляли.
[00241] 2.2.3 Критерии исключения
[00242] Животных исключали из конечного анализа на основе следующих критериев: (1) умерщвление на основе критериев тяжести заболевания, включая потерю массы тела 20% или больше по сравнению с контролем, (2) снижение массы тела на 15% или больше в течение 3 последовательных дней по сравнению с контролем по растворителю или (3) объем опухоли, больше или равный 10% массы тела, или изъязвление опухоли.
[00243] Доклинические фармакологические исследования проводили в относительно крупном масштабе, при этом нередко регистрировали единичные случаи смерти. Это могло быть связано с индивидуальными изменениями в когорте животных или с неизвестными причинами. Редкие и единичные события не указывают обязательно на какой-либо повышенный риск недостаточной безопасности рассматриваемого соединения(й). Все исследования проводили в соответствии с установленными руководствами по определению приемлемой токсичности для максимальной переносимой дозы (MTD).
[00244] 2.2.4 Схема исследования эффективности
[00245] Схема TRP-377-HT29-XEF, включая схему введения доз во всех группах лечения, представлена в Таблице 2. Животных взвешивали в день (дни) введения дозы и корректировали дозу в соответствии с массой тела, вводимый объем составлял 10 мл/кг. Размеры опухоли и массу тела регистрировали при рандомизации, а затем два раза в неделю в течение всего исследования. Следующие данные получали после каждого дня сбора данных: число случаев смерти, индивидуальные и средние по группам показатели массы тела, и индивидуальные и среднего по группам объемы опухоли.
Таблица 2. Доза и схема введения в исследовании TRP-377-HT29-XEF
(1% КМЦ/0,5% Tween 80)
IgG контроль
20 мг/кг, в/б, 2qw
MEK162
3,5 мг/кг, п/о, bid
цетуксимаб
20 мг/кг, в/б, 2qw
цетуксимаб
20 мг/кг, в/б, 2qw
MEK162
цетуксимаб
3,5 мг/кг, п/о, bid
20 мг/кг, в/б, 2qw
В исследовании TRP-377-HT29-XEF лечение начинали в день 22 после имплантации пяти миллионов опухолевых клеток HT29, когда средний объем опухоли составил 220 мм3. Лечение продолжали в течение 21 дня.
[00246] 2.2.5 Анализ данных
[00247] 2.2.5.1 Масса тела
[00248] % изменение в теле вычисляли как (BWтекущий-BWисходный)/(BWисходный)×100. Данные представлены как процентное изменение массы тела со дня начала лечения.
[00249] 2.2.5.2 Объем опухоли
[00250] Процентные значения лечения/контроля (T/C) вычисляли при использовании следующих формул:
[00251] % T/C=100×ΔT/ΔC, если ΔT>0
[00252] % Регрессии=100×ΔT/ΔT0, если ΔT<0
[00253] где:
[00254] T=средний объем опухоли в группе лечения лекарственным средством в последний день исследования или в последний день лечения;
[00255] ΔT=средний объем опухоли в группе лечения лекарственным средством в последний день исследования или в последний день лечения - средний объем опухоли в группе лечения лекарственным средством в день введения первой дозы;
[00256] T0=средний объем опухоли в группе лечения лекарственным средством в день распределения в когорты;
[00257] C=средний объем опухоли в контрольной группе в последний день исследования или в последний день лечения; и
[00258] ΔC=средний объем опухоли в контрольной группе в последний день исследования или в последний день лечения - средний объем опухоли в контрольной группе в день введения первой дозы.
[00259] 2.2.5.3 Статистический анализ
[00260] Все данные были выражены как среднее значение±среднеквадратическое отклонение (SEM). Для статистического анализа использовали изменение объема опухоли и процентные изменения массы тела. Сравнения между группами проводили с использованием ANOVA Краскела-Уоллиса с последующим апостериорным тестом Данна. Для всех статистических оценок уровень значимости был установлен на уровне p<0,05. Приведена значимость в сравнении с контрольной группой, получавшей растворитель, если не указано иное.
[00261] Стандартные протоколы, используемые в фармакологических исследованиях, не позволяют продемонстрировать статистически значимое превосходство комбинации над соответствующими монотерапиями. Статистическая мощность часто ограничена мощными ответами одного средства и/или вариабельностью модели. Тем не менее, представлены p-значения для комбинаций в сравнении с монотерапией.
[00262] 3. РЕЗУЛЬТАТЫ
[00263] 3.1 Переносимость
[00264] Исходная средняя масса тела (BW) и максимальные изменения массы тела в день 43 приведены в Таблице 3. Среднее изменение массы тела показано на Фигурах 2, 4 и 6. Все лечение переносили с максимальной потерей массы тела -7,7%. Никаких других признаков нежелательных явлений в этом исследовании не наблюдали.
Таблица 3. Средняя исходная масса тела и максимальные изменения массы тела во время лечения (TRP-377-HT-29-XEF)
Контроль IgG
MEK162
цетуксимаб
цетуксимаб
MEK162
цетуксимаб
[00265] 3.2 Эффективность in vivo
[00266] Противоопухолевое действие и процентное изменение массы тела в день 43 показано на Фигурах 1-6.
[00267] LGX818 в дозе 20 мг/кг, MEK162 в дозе 3,5 мг/кг и цетуксимаб в дозе 20 мг/кг оказали статистически незначимое противоопухолевое действие с T/C 42%, 28% и 95%, соответственно. LGX818 в комбинации с MEK162 привело к T/C 22% (p>0,05 в сравнении с группой, получавшей растворитель); LGX818 в комбинации с цетуксимабом привело к T/C 6% (p<0,05 в сравнении с группами, получавшими растворитель или цетуксимаб); MEK162 в комбинации с цетуксимабом привело к T/C 5% (p<0,05 в сравнении с группами, получавшими растворитель или цетуксимаб). Тройная комбинация LGX818+MEK162+цетуксимаб привела к регрессии опухоли с T/T0 -14%. Лечение тройной комбинацией является статистически значимым (p<0,05) при сравнении с монотерапией растворителем, LGX818 или цетуксимабом.
[00268] Последнюю дозу давали в день 43 и наблюдали опухоли в течение еще 3 недель. После завершения лечения опухоли возобновили рост во всех группах лечения (Фигура 7), что указывает на то, что долговременное лечение требуется для получения продолжительной противоопухолевой эффективности.
[00269] ИСТОЧНИКИ
[00270] Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, O'Dwyer PJ, Lee RJ, Grippo JF, Nolop K, Chapman PB. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010 Aug 26; 363(9), 809-19.
[00271] Kefford R, Arkenau H, Brown JP, et al. Phase I/II study of GSK118436, a selective inhibitor of oncogenic mutant BRAF kinase, in patients with metastatic melanoma and other solid tumours. J Clin Oncol. 2010; 28(15s); abstr 8503.
[00272] Prahallad A. Sun C. Huang S, Di Nicolantonio F, Salazar R, Zecchin D, Beijersbergen RL, Bardelli A, Bernards R. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012 Jan 26; 483 (7387):100-3.
[00273] Corcoran RB, Ebi H, Turke AB, Coffee EM, Nishino M, Cogdill AP, Brown RD, Della Pelle P, Dias-Santagata D, Hung KE, Flaherty KT, Piris A, Wargo JA, Settleman J, Mino-Kenudson M, Engelman JA. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2012 Mar; 2(3):227-35.
ПРИМЕР 2
Клиническое исследование энкорафениба (ингибитора BRAF), биниметиниба (ингибитора MEK) и цетуксимаба (ингибитора EGFR) у больных с BRAF-мутантным метастатическим раком толстой и прямой кишки (CRC).
[00274] Краткое описание: Данное исследование представляет собой рандомизированное открытое исследование 3-й фазы, разработанное для демонстрации эффективности и безопасности энкорафениба (СОЕДИНЕНИЯ A), биниметиниба (СОЕДИНЕНИЯ B) и цетуксимаба (СОЕДИНЕНИЯ C) у больных BRAF-мутантным CRC (BRAFV600E), которые ранее получали системную противоопухолевую терапию первой линии. Пациенты, включенные в исследование, имели возраст по меньшей мере 18 лет, IV стадию CRC с подтвержденной мутацией BRAF, проходили лечение по одной или двум схемам с применением системной противоопухолевой терапии в условиях локально распространенной или метастатической опухоли и ранее не проходили терапию с применением ингибитора MEK, BRAF или EGFR.
[00275] Пациентов рандомизировали 1:1:1 с распределением в группу тройной терапии (биниметиниб, энкорафениб и цетуксимаб), двойной терапии (энкорафениб и цетуксимаб) или контрольную группу (терапия на основе иринотекана с цетуксимабом).
[00276] Основным конечным показателем исследования являлась общая выживаемость (OS) в группе тройной терапии по сравнению с контрольной группой. Вторичные конечные показатели были направлены на эффективность двойной терапии по сравнению с контрольной группой и тройной терапии по сравнению с двойной терапией. Другие ключевые вторичные конечные показатели включали выживаемость без прогрессирования (PFS), процент пациентов с объективным ответом (ORR), длительность ответа, безопасность и переносимость. Также оценивали качество жизни, связанное с состоянием здоровья.
СПИСОК СОКРАЩЕНИЙ И ОПРЕДЕЛЕНИЕ ТЕРМИНОВ
[00277] Вводная фаза оценки безопасности
[00278] Основная цель Вводной фазы оценки безопасности состоит в оценки безопасности/переносимости комбинации энкорафениба+биниметиниба+цетуксимаба. Основные результаты:
процент пациентов с дозолимитирующими токсическими явлениями (DLT)
процент пациентов с нежелательными явлениями (AE) и их тяжесть, оцениваемая согласно Общим терминологическим критериям оценки нежелательных явлений (CTCAE), версии 4.03, Национального института онкологии (NCI), и изменения показателей клинических лабораторных исследований, основных физиологических показателей, изменения на электрокардиограммах, эхокардиограммах/мультисинхронизизированных радионуклидных ангиограммах и при офтальмологическом обследовании
процент пациентов с приостановкой терапии, коррекцией дозы и досрочным прекращением терапии вследствие Нежелательных явлений (AE)
[00279] Вводную фазу оценки безопасности будут проводить в ограниченном количестве центров. Дозолимитирующие токсические явления и переносимость комбинации биниметиниба, энкорафениба и цетуксимаба будут оценивать Спонсор и Исследователь при (приблизительно) еженедельных контактах. Комитет по мониторингу данных (DMC) будет оценивать данные по безопасности в установленных интервалах и в дополнительные моменты в течение поведения Вводной фазы оценки безопасности, при необходимости. Первые 9 подлежащих оценке пациентов будут зарегистрированы в исследовании на постоянной основе в одну когорту для оценки комбинации 300 мг энкорафениба один раз в день (QD)+45 мг биниметиниба два раза в день (BID)+400 мг/м2 цетуксимаба, с последующим в/в введением 250 мг/м2 еженедельно. Дополнительные пациенты будут зарегистрированы на основе оценки данных по безопасности, выполненной DMC во время Вводной фазы оценки безопасности. Дозы для группы тройной терапии в рандомизированной части Фазы 3 исследования будут определены после того, как в общей сложности 25-30 пациентов пройдут лечение в предложенных дозах, и их данных будут оценены DMC.
[00280] Рандомизированное исследование Фазы 3
[00281] Основная цель Рандомизированного исследования Фазы 3 будет состоять в сравнении активности энкорафениба+биниметиниба+цетуксимаба (Группа тройной терапии) с иринотеканом/цетуксимабом или 5-фторурацилом (5-ФУ)/фолиновой кислотой (FA)/иринотеканом (FOLFIRI)/цетуксимабом (Контрольная группа) при измерении общей выживаемости (OS). Основным конечным показателем является Общая выживаемость (OS), определяемая как время от рандомизации до наступления смерти вследствие любой причины, в Группе тройной терапии по сравнению с Контрольной группой. Ключевым вторичным конечным показателем является Общая выживаемость (OS) в группе энкорафениба+биниметиниба (Группе двойной терапии) по сравнению с Контрольной группой. Другие вторичные конечные показатели являются следующими:
Подтвержденная процентная доля пациентов с объективным ответом (ORR) по RECIST (Критериям оценки ответа при солидных опухолях), v1.1, в Группе тройной терапии по сравнению с Контрольной группой, определенная как число пациентов, достигших наилучшего общего ответа, из пациентов с полным ответом (CR) или частичным ответом (PR), деленное на общее количество пациентов
Подтвержденная процентная доля пациентов с объективным ответом (ORR) по RECIST, v1.1, Группе двойной терапии по сравнению с Контрольной группой, определенная как число пациентов, достигших наилучшего общего ответа, из пациентов с полным ответом (CR) или частичным ответом (PR), деленное на общее количество пациентов
Определенная исследователями выживаемость без прогрессирования (PFS), определенная как время от рандомизации до наиболее раннего задокументированного прогрессирования заболевания или наступления смерти вследствие любой причины, в Группе тройной терапии по сравнению с Контрольной группой
Определенная исследователями выживаемость без прогрессирования (PFS) в Группе двойной терапии по сравнению с Контрольной группой
Общая выживаемость (OS) в Группе тройной терапии по сравнению с Группой двойной терапии
Подтвержденная ORR по RECIST, v1.1, В Группе тройной терапии по сравнению с Группой двойной терапии
PFS в Группе тройной терапии по сравнению с Группой двойной терапии
Длительность ответа (DOR) в Группе тройной терапии по сравнению с Контрольной группой, в Группе двойной терапии по сравнению с Контрольной группой и в Группе тройной терапии по сравнению с Группой двойной терапии
Время до ответа, определенное как время от рандомизации до первого рентгенологического подтверждения ответа, в Группе тройной терапии по сравнению с Контрольной группой, в Группе двойной терапии по сравнению с Контрольной группой и в Группе тройной терапии по сравнению с Группой двойной терапии
Уровень и тяжесть AE, классифицированных по CTCAE NCI, v.4.03, и изменения показателей клинических лабораторных исследований, основных физиологических показателей, ЭКГ, ECHO/MUGA и при офтальмологическом обследовании
Изменение относительно исходных показателей по разработанным Европейской организацией по исследованию и лечению онкологических заболеваний (EORTC) опросникам оценки качества жизни для онкобольных (QLQ-C30), функциональной оценки противоопухолевой терапии при раке толстой кишки (FACT-C), EuroQol-5D-5L (EQ-5D-5L) и общего впечатления пациента об изменении собственного состояния (PGIC) в Группе тройной терапии по сравнению с Контрольной группой, в Группе двойной терапии по сравнению с Контрольной группой и в Группе тройной терапии по сравнению с Группой двойной терапии
Полученные с использованием моделей параметры ФК энкорафениба, цетуксимаба, биниметиниба и активного метаболита биниметиниба
[00282] Основанная на модели ФК оценка лекарственных взаимодействий между энкорафенибом, цетуксимабом, биниметинибом и активным метаболитом биниметиниба (AR00426032)
[00283] Лечение будут назначать в 28-дневных курсах до прогрессирования заболевания, неприемлемых токсических явлений, отзыва согласия, начала следующей противоопухолевой терапии или смерти.
[00284] Тестирование BRAF
[00285] Пациенты смогут участвовать в исследовании при идентификации мутации BRAF V600E в опухоли, выполненной в центральной лаборатории, в качестве части Молекулярного скрининга для исследования, или по результату локального лабораторного исследования, полученному в любое время до скрининга.
[00286] Предварительный молекулярный скрининг
[00287] Перед оценкой соответствия критериям для включения/рандомизации в исследовании, пациенты могут проходить предварительный молекулярный скрининг опухоли в центральной лаборатории с использованием анализа на мутацию BRAF в любое время до Скрининга, при условии, что они соответствуют всем критериям включения/исключения для Предварительного молекулярного скрининга.
[00288] Совокупность
[00289] a) Группа пациентов
[00290] Исследование будут проводить у больных с метастатическим раком толстой и прямой кишки BRAFV600E (mCRC), у которых заболевание прогрессировало после предыдущего применения 1 или 2 схем лечения метастатической формы.
[00291] b) Критерии включения
[00292] Для участия в исследовании пациент должен соответствовать всем следующим критериям включения:
1. Представить подписанный документ об информированном согласии с проставленной датой при Скрининге
2. Возраст≥18 лет на момент подписания информированного согласия
3. Гистологически или цитологически подтвержденный CRC, который является метастатическим
4. Наличие BRAFV600E в опухолевой ткани, предварительно определенное при анализе в локальном центре, в любое время до скрининга, или в центральной лаборатории
Примечания:
a. Принимаются только результаты локальных лабораторных исследований, полученные с помощью ПЦР и NGS.
b. Повторное лабораторное исследование в центральной лаборатории не допускается в целях устранения расхождений с локальным результатом, как только центральная лаборатория предоставляет определенный результат (положительный или отрицательный).
c. Если результат из центральной лаборатории неопределенный, или образец считают непригодным к исследованию, может быть представлен второй образец.
d. Если в какое-либо время будет присутствовать расхождение в результатах между локальным анализом и центральной лабораторией (потенциальный ложноположительный локальный результат) или отсутствие подтверждения BRAFV600E у 18 пациентов, все последующие пациенты должны будут иметь BRAFV600E, определенную в центральной лабораторией, до включения в исследование.
e. Результаты локальных лабораторий более чем с 1 расходящимся результатом, приводящим к включению пациента, не будут приняты при включении следующих пациентов.
5. Возможность предоставить достаточное количество репрезентативного образца опухоли (первичной или метастатической, архивной или только что полученной) для подтверждения при исследовании в центральной лаборатории мутационного статуса BRAF и KRAS (минимум 6 микропрепаратов; оптимально до 15 микропрепаратов)
Примечание: Образцы опухоли должны быть представлены в центральную лабораторию для анализа BRAF как можно быстрее после подписания информированного согласия при Предварительном молекулярном скрининге. Статус BRAF должен быть подтвержден не позднее, чем через 30 дней после введения первой дозы исследуемого лекарственного препарата.
6. Разрешение применения цетуксимаба в соответствии с локально одобренными показаниями с учетом RAS статуса опухоли
7. Прогрессирование заболевания после предыдущего применения 1 или 2 схем лечения метастатической формы
Примечания:
a. Рецидив заболевания во время лечения или в течение 6 месяцев после адъювантной терапии будут считать метастатическим процессом.
b. Пациенты, которые ранее проходили лечение с 2 схемами (т.е. пациенты, которые были включены в исследование в условиях применения терапии 3-й линии), должны были получить, или им должны предложить, предварительный оксалиплатин, если он не был противопоказан из-за первопричинных заболеваний.
c. Поддерживающая терапия, применяемая при лечении метастатической формы, не будет считаться отдельной схемой.
d. В части Фазы 3 исследования число пациентов, получавших 2 предыдущих схемы лечения, будет ограничено 215 (35% от общего количества рандомизированных пациентов). Пациентам с 2 предыдущими схемами, которые были зарегистрированы в Скрининге на момент достижения данного предельного числа, разрешат продолжить участие в исследовании, если они будут соответствовать другим критериям включения.
8. Подтверждение поддающегося измерению или поддающегося оценке, но неподдающегося измерению заболевания по RECIST, v1.1
9. ECOG PS 0 или 1
10. Достаточная функция костного мозга, характеризуемая следующими показателями при скрининге:
a. Абсолютное число нейтрофилов (ANC) ≥1,5×109/л;
b. Тромбоциты≥100×109/л;
c. Гемоглобин≥9,0 г/дл
Примечание: Переливания будут допускаться для достижения таких значений. Переливания будут разрешены, если пациент не получал больше 2 единиц эритроцитов за 4 предыдущие недели, чтобы достигнуть этого критерия.
11. Достаточная функция почек, характеризуемая креатинином сыворотки≤1,5× верхней границы диапазона нормальных значений (ULN) или вычисленная по формуле Кокрофта-Голта, или непосредственно измеренный клиренс креатинина≥50 мл/мин при скрининге.
12. Достаточная функция печени, характеризуемая следующими показателями при скрининге:
a. Общий билирубин сыворотки≤1,5×ULN и <2 мг/дл
Примечание: Пациентам, которые имеют уровень общего билирубина >1,5×ULN, разрешат участвовать, если их уровень несвязанного билирубина составит≤1,5×ULN
b. Аланинаминотрансфераза (АЛТ) и/или аспартатаминотрансфераза (АСТ) ≤2,5×ULN или≤5×ULN при наличии метастазов в печени
13. Достаточная функция сердца, характеризуемая следующими показателями при скрининге:
a. Фракция выброса левого желудочка (ФВЛЖ) ≥50%, определенная при исследовании MUGA или ЭхоКГ;
b. Среднее значение интервала QT при тройной регистрации с коррекцией по частоте сердечных сокращений, при использовании формулы Фридерисиа (QTcF), ≤480 мс
14. Возможность приема лекарственных препаратов внутрь
15. Желание и способность соблюдать график запланированных посещений, лечения, лабораторных исследований и других процедур в исследовании
16. Пациентки либо в постменопаузе в течение по меньшей мере 1 года, либо стерильны в результате хирургического вмешательства в течение по меньшей мере 6 недель, либо должны согласиться предпринять надлежащие меры предосторожности, чтобы избежать беременности, начиная со скрининга до последующего наблюдения включительно, в случае способности к деторождению
У всех женщин результат теста на беременность должен быть отрицательным при скрининге. Примечание: Пациентам должны сообщить о разрешенных способах контрацепции и получить подтверждение их понимания.
17. Мужчины должны согласиться предпринять надлежащие меры предосторожности, чтобы избежать рождения ребенка после скрининга до последующего наблюдения включительно.
Примечание: Пациентам должны сообщить о разрешенных способах контрацепции и получить подтверждение их понимания.
[00293] c) Критерии исключения
[00294] Пациенты, соответствующие любому из следующих критериев, не будут включены в исследование:
1. Предшествующее лечение любым ингибитором RAF, ингибитором MEK, цетуксимабом, панитумумабом или другими ингибиторами EGFR
2. Предшествующая гиперчувствительность или токсичность при применении иринотекана, которая предполагает непереносимость иринотекана в дозе 180 мг/м2 раз в 2 недели
3. Клинические симптомы метастазов в головном мозге
Примечания: Допускаются пациенты, которые ранее проходили лечение или не проходили лечение по поводу этого состояния, не имеющие симптомов в отсутствие терапии кортикостероидами и противосудорожными средствами. Метастазы в головном мозге должны быть стабильными в течение≥4 недель, с подтверждением на снимке (например, магнитно-резонансной томографии [МРТ] или компьютерной томографии [КТ]) текущего отсутствия прогрессирующих метастазов в головном мозге при скрининге.
4. Лаптоменингеальный канцероматоз
5. Наличие в анамнезе или текущее подтверждение RVO или текущие факторы риска RVO (например, неконтролируемая глаукома или повышение внутриглазного давления, синдромы повышенной вязкости или повышенной свертываемости в анамнезе)
6. Применение каких-либо лекарственных препаратов/добавок растительного происхождения или любых лекарственных средств или продуктов, которые являются сильными ингибиторами или индукторами цитохрома P450 (CYP) 3A4/5 за≤1 неделю до начала лечения в исследовании
7. Сведения о перенесенном остром или хроническом панкреатите
8. Хроническое воспалительное заболевание кишечника или болезнь Крона в анамнезе, требующие медицинского вмешательства (иммуномодулирующей или иммунодепрессивной терапии или хирургии) за≤12 месяцев до рандомизации
9. Нарушение сердечно-сосудистой функции или клинически значимые сердечно-сосудистые заболевания, включая любое из следующего:
a. Перенесенный острый инфаркт миокарда, острые коронарные синдромы (включая нестабильную стенокардию, аортокоронарное шунтирование [АКШ], коронарную ангиопластику или стентирование) ≤6 месяцев до начала лечения в исследовании;
b. Клинические симптомы застойной сердечной недостаточности (т.е. 2 степени или выше), случай в анамнезе или текущее проявление клинически значимой аритмии сердца и/или нарушения проводимости за≤6 месяцев до начала лечения в исследовании, за исключением мерцательной аритмии и пароксизмальной наджелудочковой тахикардии
10. Неконтролируемая артериальная гипертензия, определяемая как стойкое систолическое артериальное давление≥150 мм рт.ст. или диастолическое артериальное давление≥100 мм рт.ст. несмотря на текущую терапию
11. Нарушение функции печени, определяемое как класс B или C по классификации Чайлда-Пью
12. Нарушение функции или заболевание желудочно-кишечного тракта (ЖКТ), которые может значимо изменять абсорбцию энкорафениба или биниметиниба (например, язвенные болезни, неконтролируемая рвота, синдром мальабсорбции, резекция тонкой кишки с нарушением кишечной абсорбции)
13. Сопутствующее или предыдущее другое злокачественное новообразование в течение 5 лет с момента включения в исследование, за исключением вылеченного базальноклеточного или плоскоклеточного рака кожи, поверхностного рака мочевого пузыря, интраэпителиальной неоплазии предстательной железы, карциномы in situ шейки матки или другого неинвазивного или медленно растущего злокачественного новообразования без одобрения Спонсора
14. Перенесенные тромбоэмболические или цереброваскулярные нарушения за≤6 месяцев до начала лечения в исследовании, включая транзиторные ишемические атаки, инсульты, тромбоз глубоких вен или легочную эмболию
15. Сопутствующее нервно-мышечное нарушение, которое связано с возможным повышением КК (например, воспалительные миопатии, мышечная дистрофия, боковой амиотрофический склероз, спинальная мышечная артрофия)
16. Лечение любым из следующего:
a. Циклическая химиотерапия в течение некоторого периода времени, который имел меньшую длительность, чем цикл, используемый для такого лечения (например, 6 недель для нитрозомочевины, митомицина-C), до начала лечения в исследовании
b. Терапия биопрепаратами (например, антителами) за исключением бевацизумаба или афлиберцепта, непрерывное или периодическое применение низкомолекулярных терапевтических средств или любых других исследуемых средств в течение периода времени, который≤5 периодов полувыведения (t1/2) или≤4 недель (в зависимости от того, какой период короче) до начала лечения в исследовании
c. Терапия бевацизумабом или афлиберцептом за≤3 недели до начала лечения в исследовании
d. Лучевая терапия, которая включала >30% костного мозга
17. Остаточные CTCAE≥2 степени токсичности после применения любой предыдущей противоопухолевой терапии, за исключением алопеции 2 степени или невропатии 2 степени
18. Известный положительный статус ВИЧ-инфекции
19. Активный гепатит B или гепатит С
20. Синдром Жильбера или наличие любого из следующих генотипов: UGT1A1*6/*6, UGT1A1*28/*28 или UGT1A1*6/*28.
21. Известное противопоказание к применению цетуксимаба или иринотекан в запланированных дозах.
22. Параллельное лечение препаратом не для наружного применения, который, как известно, является сильным ингибитором CYP3A4. Впрочем, допускаются к участию пациенты, которые прекратили такое лечение или перешли на другое лечение по меньшей мере за 7 дней до начала лечения в исследовании.
23. Параллельное применение зверобоя (Hypericum perforatum).
24. Другое тяжелое, острое или хроническое заболевание или расстройство психики, или отклонение от нормы результатов лабораторных исследований, которые могут увеличить риск, связанный с участием в исследовании или введением исследуемого лекарственного препарата, или могут препятствовать интерпретации результатов исследования, и, по мнению Исследователя, могут не позволить пациенту участвовать в исследовании.
25. Беременность, подтвержденная положительным результатом лабораторного исследования на хорионический гонадотропин человека (ХГЧ), или кормление ребенка (грудью).
26. Лица, находящиеся под опекой или временным опекунством, должны быть исключены, за исключением случаев, когда предложить участие требуется в местной юрисдикции, в соответствии с местными законами или нормативными актами.
27. Предыдущее включение в это клиническое исследование.
[00295] Схемы лечения
[00296] Исследуемыми продуктами в данном исследовании являлись энкорафениб (СОЕДИНЕНИЕ A) и биниметиниб (СОЕДИНЕНИЕ B), которые вводят перорально (п/о) в комбинации с цетуксимабом (СОЕДИНЕНИЯ C) (т.е. энкорафениб+биниметиниб+цетуксимаб [Вводная фаза оценки безопасности и Группа тройной терапии] и энкорафениб+цетуксимаб [Группа двойной терапии]). Комбинированной терапией сравнения в зависимости от выбора Исследователя будет либо иринотекан/цетуксимаб, либо FOLFIRI/цетуксимаб (Контрольная группа).
[00297] Пациенты будут получать следующие препараты в течение 28-дневного цикла. Начальные дозы и Схема лечения показаны в Таблице 5.
Таблица 5
[00298] Для отдельного пациента доза исследуемого лекарственного средства (средств) может быть снижена или приостановлено применение, при необходимости, в зависимости от определяемых протоколом модификаций лечения.
[00299] Введение энкорафениба или энкорафениба+биниметиниба
[00300] Энкорафениб будут вводить по схеме QD, а биниметиниб будут вводить по схеме BID, оба препарата перорально, в постоянной фиксированной дозе, а не в расчете на массу тела или площадь поверхности тела (BSA) (Таблица 5). Биниметиниб и энкорафениб следует принимать независимо от приема пищи. Пациентам дают инструкции глотать целые капсулы/таблетки, а не жевать или разламывать их.
Введение QD: Пациентам указывают ежедневно принимать капсулы энкорафениба с большим стаканом воды (~250 мл) утром, приблизительно в одно и то же время ежедневно. Дозы энкорафениба, пропущенные из-за AE или по любой другой причине, можно принимать не позднее, чем за 12 часов до приема следующей дозы.
Введение BID: Пациентам указывают принимать таблетки биниметиниба с интервалом 12±2 часа с большим стаканом воды (~250 мл) утром и вечером, приблизительно в одно и то же время ежедневно. Дозы биниметиниба, пропущенные из-за AE или по любой другой причине, не следует принимать позже в этот же день или в конце периода применения.
[00301] Во Вводной фазе оценки безопасности и в группе тройной терапии оба пероральных исследуемых препарата (энкорафениб+биниметиниб) следует принимать вместе утром, и только средство, вводимое BID (биниметиниб), следует принимать вечером, независимо от приема пищи.
[00302] В дни, когда забор крови запланирован в исследовательском центре, пациенты будут принимать утреннюю дозу энкорафениба и биниметиниба (в зависимости от требований) в центре, после забора, под наблюдением Исследователя или уполномоченного лица. Вечером, в день посещения, пациенты будут принимать биниметиниб (в зависимости от требований) дома (Вводная фаза оценки безопасности и Группа тройной терапии). Во все остальные дни пациенты будут принимать энкорафениб и биниметиниб (в зависимости от требований) дома. Забор образцов до введения дозы для исследования ФК энкорафениба и биниметиниба следует производить только до приема энкорафениба и биниметиниба (в зависимости от требований).
[00303] Если у пациента возникает рвота в любое время после приема дозы, дозу исследуемого лекарственного средства не следует вводить повторно.
[00304] Пациенты должны избегать употребления грейпфрута, гранатов, карамболы, померанцев или продуктов, содержащих их сок, в течение всего исследования и предпочтительно за 7 дней до введения первой дозы исследуемых препаратов, из-за потенциального взаимодействия CYP3A4 с исследуемыми препаратами. Допускается употребление апельсинового сока.
[00305] Энкорафениб и биниметиниб (в соответствующих случаях) будут вводить по меньшей мере за 30 минут до цетуксимаба.
[00306] Введение цетуксимаба
[00307] Цетуксимаб будут вводить в/в, раз в неделю, в Дни 1, 8, 15 и 22 (±3 дня) каждого 28-дневного цикла в исследовательском центре (Таблица 5) согласно внутренним стандартам центра. Начальная доза цетуксимаба (День 1 Цикла 1) составляет 400 мг/м2, вводимых путем 120-минутной в/в инфузии, с последующим введением доз 250 мг/м2, вводимых путем 60-минутной в/в инфузии. Скорость инфузии должна соответствовать местной инструкции по применению, но не должна превышать 10 мг/мин. Тщательный мониторинг необходимо вести во время инфузии и в течение по меньшей мере 1 часа после завершения инфузии. Если на участке инфузии наблюдается реакция во время введения цетуксимаба, инфузию следует немедленно остановить, при этом пациентов нужно тщательно наблюдать и лечить в соответствии с внутренними стандартами центра. Лекарственные средства для медикаментозной подготовки к стандартным инфузиям цетуксимаба могут использоваться в соответствии с инструкциями по применению, а также с национальными и/или внутренними стандартами. Лекарственные средства для медикаментозной подготовки следует вводить не раньше, чем через 1 час после введения энкорафениба и биниметиниба (в соответствующих случаях) и за 30 минут до инфузии цетуксимаба.
[00308] Введение иринотекана
[00309] Иринотекан будут вводить в/в раз в две недели, в Дни 1 и 15 (±3 дня) каждого 28-дневного цикла в исследовательском центре согласно внутренним стандартам. Начальная доза иринотекана (День 1 Цикла 1) составляет 180 мг/м2, вводимые путем 90-минутной в/в инфузии или согласно внутренним стандартам центра.
[00310] Введение FOLFIRI
[00311] Начальные дозы 5-ФУ, FA и иринотекана представлены в Таблице 5. Пациенты, которым потребовалось снижение дозы 5-ФУ и FA в предшествующих схемах (например, в качестве части схем FOLFOX или FOLFOXIRI), должны начинать лечение с применения доз, перечисленных в Таблице 5, которые наиболее приближены, без превышения, к ранее переносимой дозе. При этих обстоятельствах начальную дозу иринотекана не следует снижать.
[00312] Фолиновую кислоту будут вводить в/в раз в две недели, в Дни 1 и 15 (±3 дня) каждого 28-дневного цикла в исследовательском центре (Таблица 5) согласно внутренним стандартам. Начальная доза FA составляет 400 мг/м2, вводимых путем 120-минутной в/в инфузии или согласно внутренним стандартам центра. В альтернативе, FA могут вводить (через разные инфузионные системы) одновременно с иринотеканом. Дозы FA, пропущенные из-за AE или по любой другой причине, не следует вводить дополнительно.
[00313] 5-Фторурацил будут вводить в/в раз в две недели, в Дни 1 и 15 (±3 дня) каждого 28-дневного цикла в исследовательском центре (Таблица 5), сразу после завершения инфузии FA, согласно внутренним стандартам. Начальная доза 5-ФУ составляет 400 мг/м2 струйно (не больше 15 минут), вводимых в/в в Дни 1 и 15, с последующим переходом на непрерывную в/в инфузию 1200 мг/м2/день ×2 дня (в общей сложности 2400 мг/м2 за 46-48 часов), или согласно внутренним стандартам цетра. Дозы 5-ФУ, пропущенные из-за AE или по любой другой причине, не следует вводить дополнительно.
[00314] КОРРЕКЦИЯ ДОЗЫ
[00315] Пациентов будут непрерывно наблюдать на AE. Тяжесть AE будут оценивать при использовании CTCAE NCI, v.4.03. Если у пациента развивается токсическое действие, дозу могут корректировать, как описано в Таблице 6. Все коррекции дозы должны быть основаны на предшествующем ухудшении токсических явлений.
[00316] В группах Вводной оценки безопасности и Тройной терапии, если пациент постоянно прерывает прием биниметиниба из-за AE или клинически значимого показателя лабораторного исследования, он может продолжать прием энкорафениба в комбинации с цетуксимабом. Из-за недостаточной эффективности биниметиниба, энкорафениба или цетуксимаба при отдельном применении в качестве монотерапии у пациентов с BRAF-мутантным mCRC, пациенты, которые не могут переносить эти средства в комбинации по меньшей мере с еще одним средством, должны полностью прекратить лечение в исследовании, совершить заключительно посещение для лечения и продолжить последующее наблюдение для оценки выживаемости (и прогрессирования заболевания, в соответствующем случае).
[00317] Цетуксимаб можно снизить до 2 уровня дозы до минимального значения 150 мг/м2 вследствие развития AE или отклоняющихся от нормы результатов лабораторных исследований (Таблица 6). Если снижение дозы потребуется из-за AE, последующее повышение дозы цетуксимаба до предыдущего уровня для того пациента не будет разрешено в течение всего лечения в исследовании. Если после разрешения AE, лечение возобновляют в той же дозе, и при этом снова наблюдается такое же токсическое действие с такой же тяжестью, любое возобновление лечения следует начинать при следующем более низком уровне дозы, независимо от продолжительности, за некоторыми исключениями в случае кожной токсичности. Кроме того, пациент должен прекратить лечение в исследовании, если после возобновления лечения с более низкой дозой цетуксимаба повторно развивается такое же токсическое явление с такой же или более высокой тяжестью.
[00318] Если пациент пропускает >28 последовательных доз энкорафениба или биниметиниба, >4 последовательных доз цетуксимаба или 2 последовательных дозы иринотекана, 5-ФУ или FA в результате AE или клинически значимого отклонения от нормы результата лабораторного исследования, то применение соответствующего вещества следует прекратить. Как отмечено ранее, из-за отсутствия эффективности у пациентов с BRAF-мутантным mCRC, пациентам не разрешат продолжить отдельное применение биниметиниба, энкорафениба или цетуксимаба, полностью прекратят их лечение в исследовании, и после совершения заключительного посещения для лечения, продолжат их наблюдение для оценки выживаемости (и прогрессирования заболевания, в соответствующем случае).
Таблица 6: Уровни доз для коррекции дозы
Сокращения: BID=два раза в день; 5-ФУ=5-фторурацил; QD=один раз в день.
aНачальная доза 400 мг/м2 (120-минутная инфузия), затем 250 мг/м2 (60-минутная инфузия).
[00319] Коррекция дозы энкорафениба и/или биниметиниба
[00320] Дозы энкорафениба и/или биниметиниба следует корректировать при AE в течение исследования (Таблица 12). Обычно дозы не нужно снижать или прекращать лечение при развитии AE 1 степени, если AE не является определенным глазным AE, указанным в Таблице 12, однако лечение для контроля симптомов следует обеспечить при необходимости, в соответствующем случае.
[00321] У отдельного пациента дозу энкорафениба и/или биниметиниба можно снижать до уровней, указанных в Таблице 6. Минимальный рекомендуемый уровень дозы энкорафениба составляет 150 мг QD, а минимальный рекомендуемый уровень дозы биниметиниба составляет 15 мг BID. Если АЕ, которое привело к снижению дозы, уменьшается и остается стабильным на уровне, наблюдаемом у пациента до начала лечения, в течение минимум 14 дней, дозу можно снова увеличить до следующего уровня дозы по усмотрению Исследователя, при условии, что отсутствуют другие сопутствующие токсические явления, которые препятствуют повторному повышению дозы лекарственного препарата. Ограничения в отношении количества раз, когда доза у пациента может быть снижена или повторно повышена, отсутствуют (с шагом, указанным в Таблице 6); тем не менее:
[00322] - повторное повышение дозы энкорафениба не допускается после снижения дозы из-за увеличения QTcF≥501 мс;
[00323] - повторное повышение дозы биниметиниба не допускается после снижения дозы из-за ФВЛЖ дисфункции или увеличения QTcF≥501 мс;
[00324] - повторное повышение дозы биниметиниба или энкорафениба не допускается после снижения дозы из-за токсического воздействия на сетчатку≥2 степени.
[00325] См. Таблицу 12 по поводу рекомендуемых коррекций доз энкорафениба и/или биниметиниба, при необходимости, связанных с возникновением AE, вызванных лечением энкорафенибом и/или биниметинибом.
[00326] Глазные нарушения, степень которых нельзя точно определить согласно CTCAE NCI, v.4.03, следует классифицировать согласно Таблице 7. Серозное отcлоение сетчатки следует классифицировать согласно Таблице 8. Увеит следует классифицировать согласно CTCAE NCI, v.4.03, как описано в Таблице 9. Ладонно-подошвенный синдром следует классифицировать согласно CTCAE NCI, v.4.03, как описано в Таблице 10. Диарею следует классифицировать согласно модифицированным CTCAE NCI, v.4.03, как описано в Таблице 11.
Таблица 7: Модифицированные критерии CTCAE NCI, версия 4.03, определение степени глазных нарушений
Таблица 8: CTCAE NCI, версия 4.03, определение степени серозного отслоения сетчатки
Примечание: В случае регматогенного отслоения сетчатки, определение степени согласно CTCAE NCI v.4.03 Отслоение сетчатки.
Таблица 9: CTCAE NCI, версия 4.03, оценка увеита
Таблица 10: CTCAE NCI, версия 4.03, определение степени ладонно-подошвенного синдрома (ЛПС)a
Сокращения: ADL=действия ежедневной активности.
aЛПС или синдром ладонно-подошвенной эритродизестезии, нарушение, характеризуемое покраснением, заметным дискомфортом, опуханием и покалыванием в ладонях или подошвах ног;
bДополнительные конкретные примеры 1 степени и 3 степени добавлены для облегчения надлежащей оценки [из инструкции по применению в упаковке сорафениба (West Haven, CT: Bayer Pharmaceuticals Corporation; 2007).
Таблица 11: Модифицированные критерии CTCAE NCI, версии 4.03, определение степени диареи
• Умеренный или тяжелый спазм
• Тошнота/рвота ≥2 степени
• Пониженный функциональный статус
• Лихорадка
• Сепсис
• Нейтропения
• Выраженное кровотечение
• Обезвоживание
• Некупируемая диарея после 48 часов лечения лоперамидом (включая применение в высокой дозе) и начало лечения второй линии
Таблица 12: Рекомендуемые коррекции дозы при связанных с энкорафенибом и/или связанных с биниметинибом нежелательных явлениях
и биниметиниба (Группа тройной терапии)
Примечание: Результаты и снимки оптической когерентной томографии (ОКТ) должны быть доступны по запросу.
Любое ухудшение остроты зрения при скрининге требуется задокументировать и рассматривать как исходный уровень.
• Если пациент остается бессимптомным (1 степень), величину дозы энкорафениба и биниметиниба сохраняют и продолжают запланированные проверки зрения, установленные в протоколе
• Если у пациента появляются симптомы (нечеткое зрение, светобоязнь и т.д.), или оценка остроты зрения показывает 2 степень, нужно следовать рекомендациям по дозам при 2 степени, представленным ниже
• При улучшении до исходного уровня или Степени ≤1, возобновляют лечение с текущей величиной дозы энкорафениба и биниметиниба и продолжают запланированные проверки зрения, установленные в протоколе
• При отсутствии улучшения до исходного уровня или Степени ≤1, возобновляют лечение при 1 пониженной величине дозыb энкорафениба и биниметиниба и продолжают запланированные проверки зрения, установленные в протоколе
• При улучшении до исходного уровня или Степени ≤2, возобновляют лечение при 1 пониженной величине дозыb энкорафениба и биниметиниба и продолжают запланированные проверки зрения, установленные в протоколе
• При отсутствии улучшения до исходного уровня или Степени ≤2, лечение не возобновляют и повторяют офтальмологическое обследование в течение 10 дней.
ο При улучшении до исходного уровня или Степени ≤2, возобновляют лечение при 1 пониженной величине дозыb энкорафениба и биниметиниба и продолжают запланированные проверки зрения, установленные в протоколе
ο При сохранении 3 степени, энкорафениб и биниметиниб отменяют на неопределенный период
Примечание: Результаты и снимки офтальмологических обследований должны быть доступны по запросу. Это включает сканы/снимки флуоресцентной ангиографии, если пациента обследуют с применением данной методики.
• При улучшении до Степени ≤1 за ≤21 день, лечение возобновляют при 1 пониженной величине дозыb энкорафениба и биниметиниба
• При отсутствии улучшения до Степени ≤1 за ≤21 день, энкорафениб и биниметиниб отменяют на неопределенный период и ведут тщательное наблюдение с офтальмологическим контролем до стабилизации или улучшенияc
АСТ или АЛТ от >3 до 5,0×ULN или 3× повышение относительно исходного значенияd И билирубин в кровиg
≤2,0×ULN
• При улучшении за ≤14 дней, сохранение величины дозы энкорафениба и биниметиниба
• При отсутствии улучшения за ≤14 дней, приостановка применения энкорафениба (в дополнение к предыдущей приостановке биниметиниба) до улучшения до Степени ≤1 (или Степени ≤2 в случае метастаза печени), затем лечение возобновляют при текущей величине дозы энкорафениба и 1 пониженной величине дозыb биниметиниба
При дополнительном случае:
• Приостановка применения энкорафениба и биниметиниба до улучшения до Степени ≤1 (или Степени ≤2 в случае метастаза печени), затем лечение возобновляют при 1 пониженной величине дозыb энкорафениба и биниметиниба
Лечение энкорафенибом и биниметинибом может быть затем возобновлено по решению Исследователя, при этом применение одного энкорафениба возобновляют на одну неделю раньше, чем возобновляют лечение биниметинибом
• При улучшении за ≤7 дней, возобновляют лечение при 1 пониженной величине дозыb энкорафениба и биниметиниба
• При отсутствии улучшения за ≤7 дней, энкорафениб и биниметиниб отменяют на неопределенный период
Лечение энкорафенибом и биниметинибом может быть затем возобновлено по решению Исследователя, при этом применение одного энкорафениба возобновляют на одну неделю раньше, чем возобновляют лечение биниметинибом
• При улучшении за ≤14 дней, лечение возобновляют при текущей величине дозы энкорафениба и биниметиниба
• При отсутствии улучшения за ≤14 дней, лечение возобновляют при 1 пониженной величине дозыb энкорафениба и биниметиниба
Лечение энкорафенибом и биниметинибом может быть затем возобновлено по решению Исследователя, при этом применение одного энкорафениба возобновляют на одну неделю раньше, чем возобновляют лечение биниметинибом
При дополнительном случае:
• Приостановка применения энкорафениба и биниметиниба до улучшения до Степени ≤1 (или Степени ≤2 в случае метастаза печени), затем лечение возобновляют при 1 пониженной величине дозыb энкорафениба и биниметиниба
АСТ или АЛТ >20,0×ULN
• При восстановлении значения ФВЛЖ (определяемого как ФВЛЖ ≥50% или ≥LLN и абсолютное уменьшение ≤10% по сравнению с исходным уровнем) за ≤21 день, лечение возобновляют при 1 пониженной величине дозыb биниметиниба после одобрения наблюдателем, назначенным Спонсором. Мониторинг ФВЛЖ 2 недели после возобновления приема биниметиниба, каждые 4 недели в течение 12 недель, и затем согласно протоколу
• Если значение ФВЛЖ не восстанавливается за ≤21 день, биниметиниб отменяют на неопределенный период. Ведут тщательный мониторинг ФВЛЖ до улучшения или до 16 недель
Примечание: У пациентов можно запросить копии ЭхоКГ и/или сканов MUGA для доступа Спонсора, для пациентов с абсолютным уменьшением ФВЛЖ >10% по сравнению с исходным уровнем и ФВЛЖ <50% или LLN
• Если общая КК ≥3×ULN, измеряют изоферменты КК и миоглобин в крови или моче
>5,0-10,0×ULN без нарушения функции почек (т.е. креатинин сыворотки <1,5×ULN или 1,5× исходного значения)
При появлении симптомов (боль в мышцах/спазмы/мышечная слабость), продолжение применения энкорафениба и приостановка применения биниметиниба до улучшения до Степени CTCAE ≤1, и тщательное наблюдение, затем:
• При улучшении за ≤21 день, сохранение дозы энкорафениба и продолжение лечения при 1 пониженной величине дозыb биниметиниба
• При отсутствии улучшения за ≤21 день, сохранение дозы энкорафениба и отмена биниметиниба на неопределенный период
без нарушения функции почек (т.е. креатинин сыворотки <1,5×ULN или 1,5× исходного значения)
• При улучшении за ≤21 день, сохранение дозы энкорафениба и продолжение лечения при 1 пониженной величине дозыb биниметиниба
• При отсутствии улучшения за ≤21 день, сохранение дозы энкорафениба и отмена биниметиниба на неопределенный период
При появлении симптомов (боль в мышцах/спазмы/мышечная слабость), сохранение дозы энкорафениба и отмена биниметиниба на неопределенный период
с нарушением функции почек (т.е. креатинин сыворотки ≥1,5×ULN или 1,5× исходного значения)
• При улучшении за ≤21 день, рассматривают возможность возобновления лечения при 1 пониженной величине дозыb энкорафениба и биниметиниба
• При отсутствии улучшения за ≤21 день, энкорафениб и биниметиниб отменяют на неопределенный период
2-й случай:
Энкорафениб и биниметиниб отменяют на неопределенный период
• Временная приостановка применения энкорафениба и биниметиниба до QTcF <500 мс. Затем лечение возобновляют при 1 пониженной величине дозыb энкорафениба и биниметиниба
2-й случай:
• Временная приостановка лечения энкорафенибом и биниметинибом до QTcF <500 мс. Затем лечение возобновляют при 1 пониженной величине дозыb энкорафениба и биниметиниба
3-й случай:
• Энкорафениб и биниметиниб отменяют на неопределенный период
Применение Схемы первичного лечения сыпи, если ее еще не применили, при этом необходимо вести тщательное наблюдение сыпи
• Сохранение величины дозы энкорафениба и биниметиниба
• Применение Схемы первичного лечения сыпи, если ее еще не применили, при этом необходимо вести тщательное наблюдение сыпи
• Повторная оценка в течение ≤14 дней. Если сыпь усилилась или не уменьшилась, Приостановка применения энкорафениба и биниметиниба до улучшения до Степени ≤1. Затем лечение возобновляют при текущей величине дозы энкорафениба и биниметиниба. В случае акнеформного дерматита, лечение энкорафенибом могут продолжать, если исследователь сочтет, что сыпь не связана с энкорафенибом. Если лечение энкорафенибом продолжали, и при этом в течение 8 дней не наблюдалось улучшение, приостановка применения энкорафениба
2-й случай:
• Повторная оценка в течение ≤14 дней. Если сыпь усилилась или не уменьшилась, приостановка применения энкорафениба и биниметиниба до улучшения до Степени ≤1. Затем лечение возобновляют при текущей величине дозы энкорафениба и 1 пониженной величине дозыb биниметиниба. В случае сыпи по типу акнеформного дерматита, лечение энкорафенибом могут продолжать, если исследователь сочтет, что сыпь не связана с энкорафенибом. Если лечение энкорафенибом продолжали, и при этом в течение 8 дней не наблюдалось никакого улучшения, приостановка применения энкорафениба
• Приостановка применения энкорафениба и биниметиниба до улучшения до Степени ≤1. Повторную оценку проводят еженедельно. Затем лечение возобновляют при текущей величине дозы энкорафениба и биниметиниба.
• Рассматривают возможность направления к дерматологу и лечение сыпи проводят согласно рекомендации дерматолога.
2-й случай:
• Приостановка применения энкорафениба и биниметиниба до улучшения до Степени ≤1. Затем лечение возобновляют при 1 пониженной величине дозыb энкорафениба и биниметиниба. Лечение энкорафенибом возобновляют при той же величине дозы, если исследователь сочтет, что сыпь не связана с энкорафенибом
• Рассматривают возможность направления к дерматологу и лечение сыпи проводят согласно рекомендации дерматолога
• Сохранение дозы энкорафениба, при этом необходимо вести тщательное наблюдение ЛПС. Быстро вводят поддерживающие меры, такие как наружную терапию, для облегчения симптомов. Дают инструкцию по изменению образа жизни.
• При отсутствии улучшения ≤14 дней, приостановка применения энкорафениба до улучшения до Степени ≤1. Лечение энкорафенибом возобновляют при текущей величине дозы. Продолжают применение поддерживающих мер, таких как наружной терапии, для облегчения симптомов. Дают инструкцию по изменению образа жизни.
Дополнительный случай:
• Лечение энкорафенибом могут продолжать или приостанавливать по решению Исследователя. Продолжают применение поддерживающих мер, таких как наружной терапии, для облегчения симптомов. Дают инструкцию по изменению образа жизни.
• Если применение энкорафениба приостановлено по решению Исследователя, приостановку сохраняют до улучшения до Степени ≤1. Лечение энкорафенибом возобновляют при той же величине дозы или при 1 пониженной величине дозыb по выбору Исследователя.
• Приостановка применения энкорафениба до улучшения до Степени ≤1. Быстро вводят поддерживающие меры, такие как наружную терапию, для облегчения симптомов. Дают инструкцию по изменению образа жизни. Повторную оценку пациента проводят еженедельно. Затем лечение возобновляют при одной пониженной величине дозыb энкорафениба
• Рассматривают возможность направления к дерматологу и проводят лечение ЛПС согласно рекомендации дерматолога
>3-го случая:
• Приостановка применения энкорафениба до улучшения до Степени ≤1, решение о возобновлении лечения энкорафенибом при одной пониженной величине дозыb или отмену энкорафениба на неопределенный период должно быть основано на решении Исследователя.
Применение биниметиниба приосттанавливают до улучшения до Степени ≤1. Затем лечение возобновляют при 1 пониженной величине дозыb биниметиниба
Примечание: Приостановка применения энкорафениба и биниметиниба до рвоты ≥3 степени или тошноты 3 степени, только если рвоту или тошноту не удается контролировать оптимальными противорвотными средствами (согласно местной практике)
При улучшении до Степени 0 или 1, лечение возобновляют при 1 пониженной величине дозы биниметиниба.
При отсутствии улучшения в течение 3 недель, биниметиниб отменяют на неопределенный период.
b Снижение дозы ниже 75 мг QD для энкорафениба, и ниже 15 мг BID для биниметиниба не допускается.
c Офтальмологическое обследование по причине ретинальных нарушений, заднего увеита, RVO: дальнейшая оценка с применением специализированного метода визуализации сетчатки (например, оптической когерентной томографии, флуоресцентной ангиографии). Любой диагноз ретинальных нарушений должен быть подтвержден наличием или отсутствием симптомов, оценкой остроты зрения и результатов ОКТ.
d Для пациентов, включенных в исследование с метастазами печени и исходным повышением ФПП.
e Нарушение, характеризуемое покраснением, заметным дискомфортом, опуханием и покалыванием в ладонях или подошвах ног.
f Пациент с AE 4 степени может возобновить лечение при более низкой дозе, если AE улучшается до Степени ≤1 в течение 28 дней после отмены лекарственного средства и, если по мнению Исследователя и назначенного Спонсором наблюдателя, событие не представляет угрозы для жизни и пациент может продолжать лечение и наблюдение рецидива AE. Любые пациенты, требующие приостановки лечения на >28 дней, должны прекратить применение исследуемого лекарственного средства.
g Относится к общему билирубину.
[00327] Коррекция дозы цетуксимаба
[00328] Рекомендованные коррекции дозы цетуксимаба при возникновении AE, связанных с лечением цетуксимабом, приведены в Таблице 1.
Таблица 13: Рекомендованные коррекции дозы при связанных с цетуксимабом нежелательных явлениях
Все последующие инфузии также следует проводить с более низкой скоростью.
• При улучшении за ≤7 дней (или ≤14 дней в случае акнеформной сыпи), то сохранение величины дозы
• При отсутствии улучшения за ≤7 дней несмотря на соответствующую терапию при кожной токсичности (или ≤14 дней в случае акнеформной сыпи), то отмена цетуксимаба на неопределенный период
• При улучшении за ≤7 дней (или ≤14 дней в случае акнеформноц сыпи), то уменьшение на 1 уровень дозы
• При отсутствии улучшения за ≤7 дней несмотря на соответствующую терапию при кожной токсичности (или ≤14 дней в случае акнеформной сыпи), то отмена цетуксимаба на неопределенный период
• Отмена цетуксимаба на неопределенный период после 3-го рецидива (при 4-м случае)
Пример 3
Получение кристаллизованного (2-гидроксиэтокси)амида 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты
[00329] Получение кристаллизованного (2-гидроксиэтокси)-амида 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимида-зол-5-карбоновой кислоты описано в публикации PCT WO 2014/063024 следующим образом.
[00330] В сухом сосуде при комнатной температуре (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты добавляли к предварительно смешанному раствору растворителей метанола/ТГФ/воды (35/35/30 w/w/w). Суспензию нагревали до внутренней температуры 53-55°C, и полученный раствор фильтровали в горячем состоянии путем глубинной и мембранной фильтрации (через бумажный фильтр и ПТФЭ мембрану) при внутренней температуре 53-56°C. Прозрачный раствор перемешивали и охлаждали до 47-48°C, и добавляли суспензию затравочных кристаллов (т.е. затравочных кристаллов кристаллизованного (2-гидроксиэтокси)амида 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты в воде, 10% м/м) (0,2-0,5% кристаллизованного (2-гидроксиэтокси)амида 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты от массы ожидаемого выхода). Приблизительно через 20 минут воду медленно добавляли в течение 25 часов (33,3% в течение 15 часов и 66,6% в течение 10 часов, с перемешиванием в течение по меньшей мере 10 минут после добавления воды), с получением конечного отношения метанола/ТГФ/воды (20/20/60 в/в). После добавления воды суспензию охлаждали до внутренней температуры 3-5°C в течение 10 часов и перемешивали в течение 0,5 часа. Белую суспензию фильтровали через вакуумный нутч-фильтр из пористого стекла (75 мл, диаметр=6 см, поры 3) и один раз промывали охлажденным во льду метанолом/ТГФ/водой (15/15/70 в/в при 2-4°C) и два раза охлажденной во льду (2-4°C) водой. Сушку проводили в вакуумном сушильном шкафу при 20°C в течение 10 часов, затем при 40°C в течение 10 часов, а затем при 60°C в течение по меньшей мере 12 часов при давлении <10 мбар, получив кристаллизованный (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты, который можно определить по структуре XRPD на Фигуре 8.
Пример 4
Фармацевтическая композиция кристаллизованного (2-гидроксиэтокси)амида 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты
[00331] Кристаллизованный (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты получали, как указано в Таблице 14:
Таблица 14
(% по весу)
(в мг/ единица)
(%по весу)
(в мг/ единица)
* Вес лекарственной субстанции взят в расчете на сухое вещество (100%) на основе анализируемого значения. Разницу в весе доводили количеством лактозы моногидрата.
** Opadry II объединяли со стерилизованной водой, получив 12% в/в суспензию пленочной оболочки Opadry II (85F), которую затем наносили распылением на ядро таблетки.
*** Удаляли в процессе обработки
[00332] При смешивании компонентов ядра таблетки фармацевтическую композицию превращали в таблетированную форму путем прямого прессования. Сформированная таблетка может быть затем покрыта оболочкой для таблеток, представленной выше.
| название | год | авторы | номер документа |
|---|---|---|---|
| КОМПЛЕКСНАЯ ТЕРАПИЯ | 2013 |
|
RU2677245C2 |
| СОЕДИНЕНИЯ 4-ОКСО-3,4-ДИГИДРОХИНАЗОЛИНОНА ДЛЯ ЛЕЧЕНИЯ BRAF-АССОЦИИРОВАННЫХ ЗАБОЛЕВАНИЙ И НАРУШЕНИЙ | 2021 |
|
RU2814662C1 |
| 3,4-ДИГИДРО-2,7-НАФТИРИДИН-1,6(2H,7H)-ДИОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ MEK | 2022 |
|
RU2826000C1 |
| ИНГИБИТОРЫ MDM2 И ИХ КОМБИНАЦИИ | 2016 |
|
RU2740091C2 |
| СИНЕРГИЧЕСКИЕ КОМПОЗИЦИИ ИНГИБИТОРОВ PI3K И МЕК | 2012 |
|
RU2607944C2 |
| ТЕРАПЕВТИЧЕСКИЕ КОМБИНАЦИИ, СОДЕРЖАЩИЕ ИНГИБИТОР RAF И ИНГИБИТОР ERK | 2017 |
|
RU2774612C2 |
| ПРОИЗВОДНЫЕ ХИНАЗОЛИН-4-ОНА, ПОЛЕЗНЫЕ ДЛЯ ЛЕЧЕНИЯ BRAF-АССОЦИИРОВАННЫХ ЗАБОЛЕВАНИЙ И НАРУШЕНИЙ | 2020 |
|
RU2797606C1 |
| СПОСОБЫ И КОМБИНИРОВАННОЕ ТЕРАПЕВТИЧЕСКОЕ СРЕДСТВО ДЛЯ ЛЕЧЕНИЯ РАКА ЖЕЛЧНЫХ ПРОТОКОВ | 2019 |
|
RU2793543C2 |
| СОЕДИНЕНИЯ ПИРАЗОЛО[3,4-b]ПИРИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ ТАМ И МЕТ | 2019 |
|
RU2773611C1 |
| КОМБИНИРОВАННАЯ ТЕРАПИЯ | 2018 |
|
RU2815400C2 |
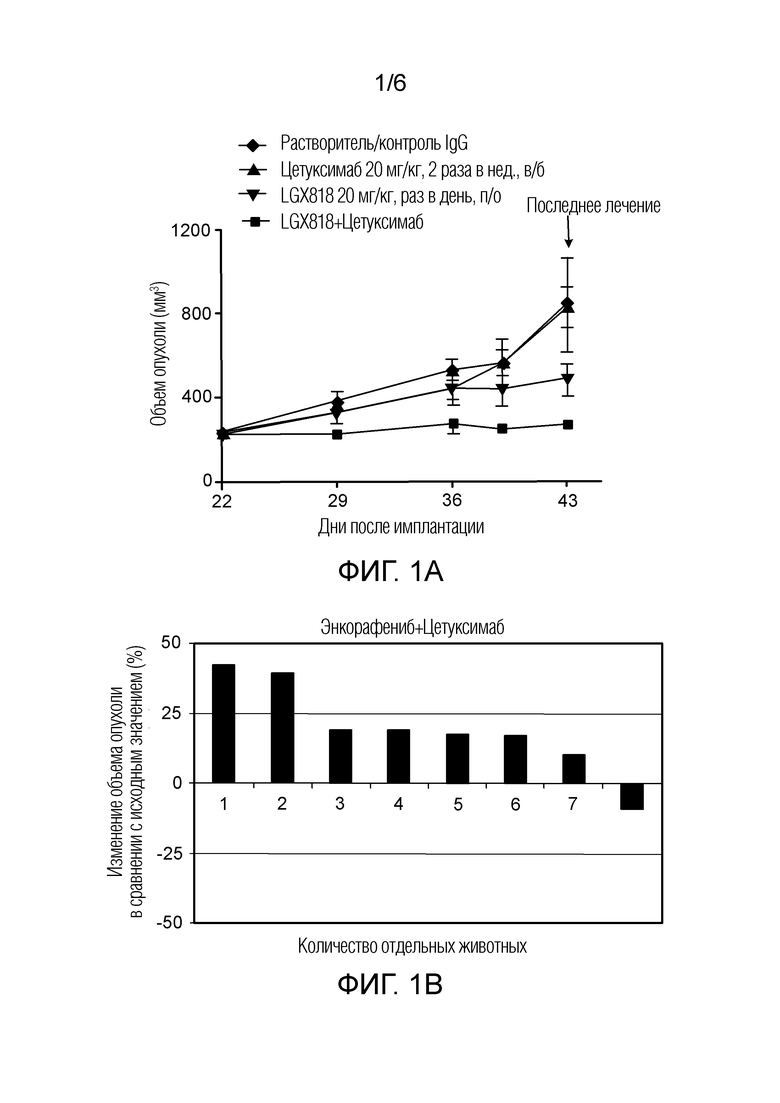
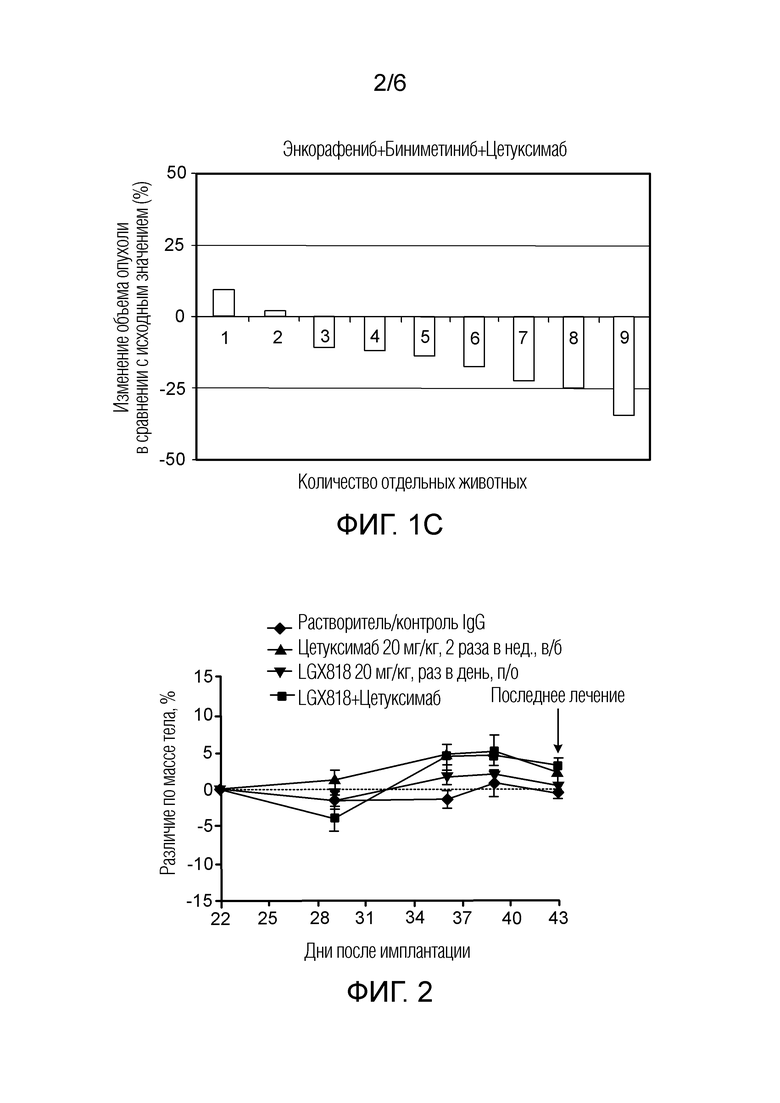
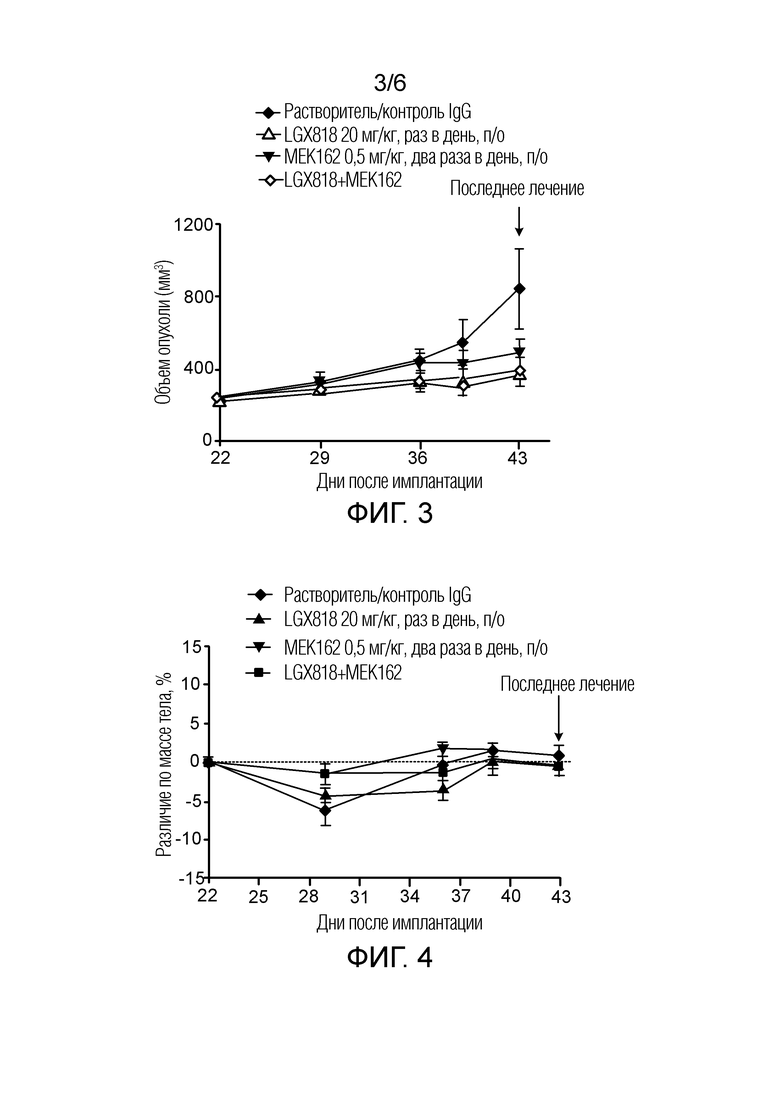
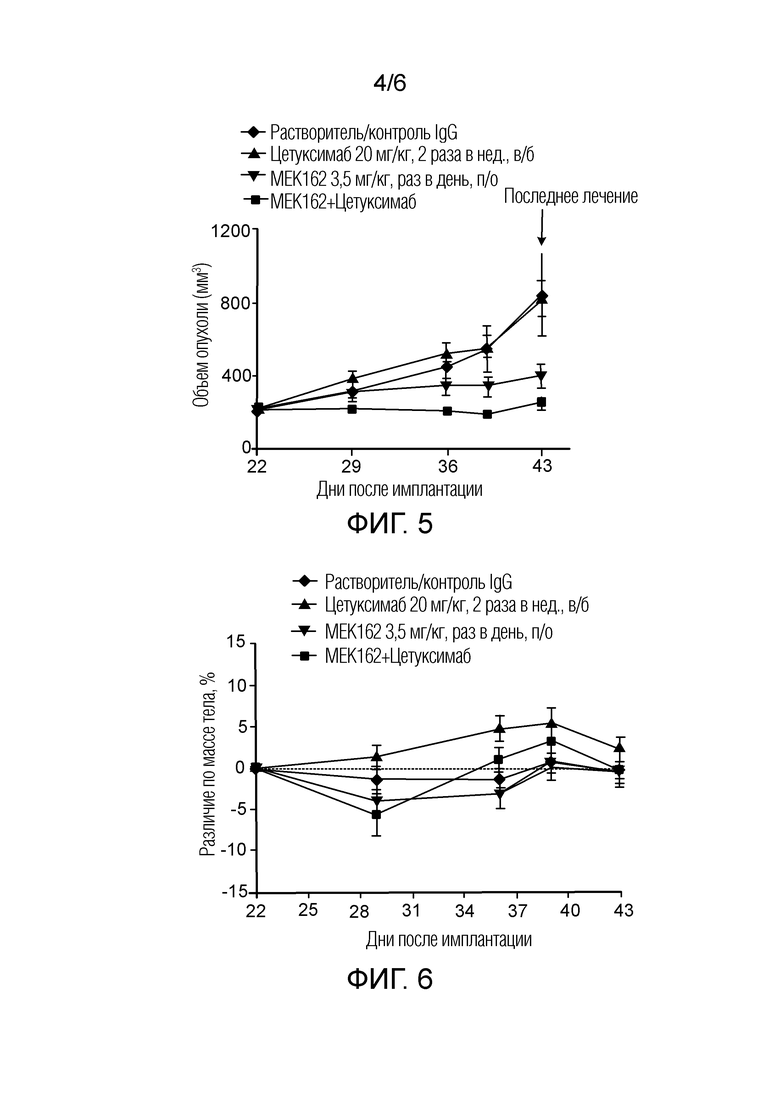
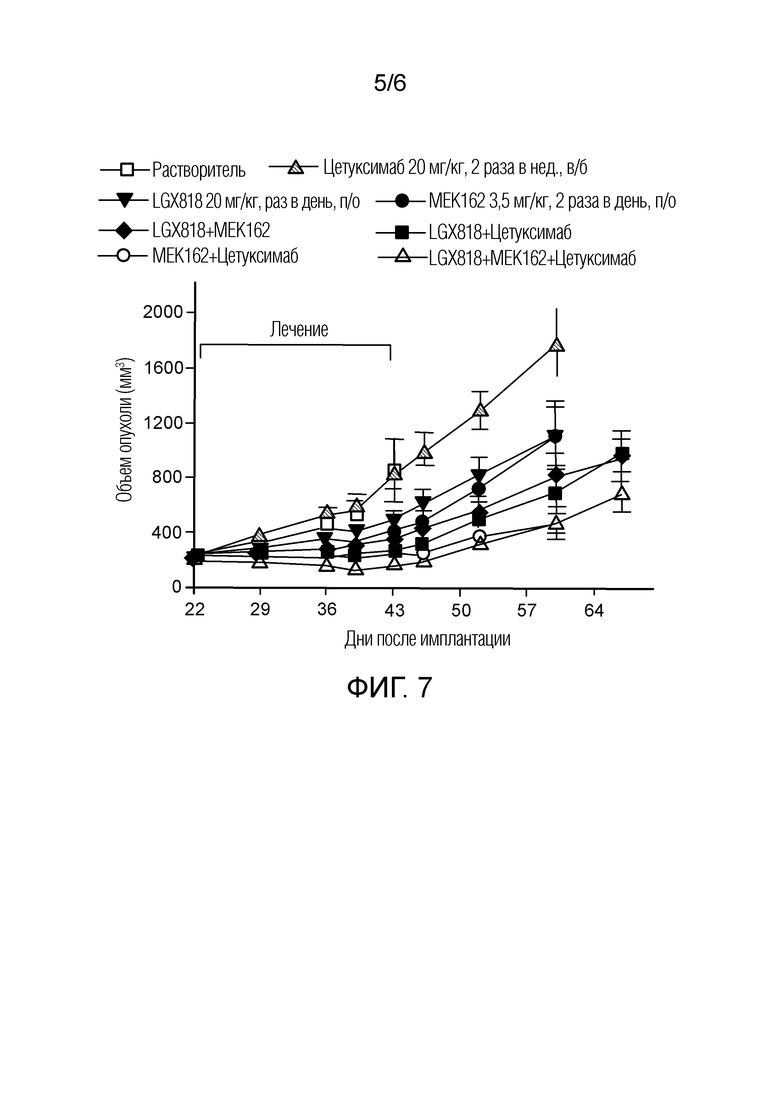
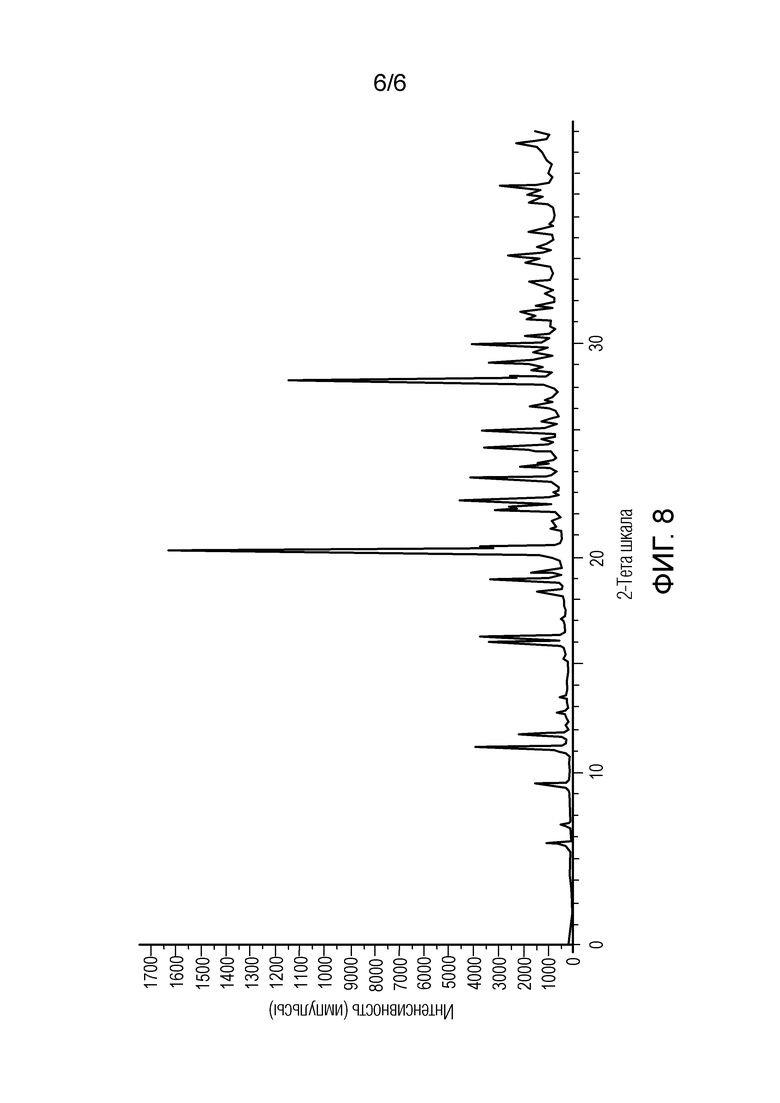
Настоящее изобретение относится к области медицины, а именно к онкологии, и предназначено для лечения BRAF-ассоциированного рака, при этом включает введение нуждающемуся в этом пациенту комбинированной терапии, независимо включающей терапевтически эффективные количества: (a) ингибитора BRAF, которым является метил-N-[(2S)-1-({4-[3-(5-хлор-2-фтор-3-метансульфонамидофенил)-1-(пропан-2-ил)-1H-пиразол-4-ил]пиримидин-2-ил}амино)пропан-2-ил]карбамат (СОЕДИНЕНИЕ A), (b) ингибитора MEK (СОЕДИНЕНИЯ B), который является (2-гидроксиэтокси)амидом 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты, и (c) антитела против EGFR (СОЕДИНЕНИЯ C), которое является цетуксимабом. Техническим результатом настоящего изобретения является эффективное и долговременное лечение рака при использовании способа комбинированной терапии. 29 з.п. ф-лы, 14 табл., 8 ил., 4 пр.
1. Способ лечения BRAF-ассоциированного рака, включающий введение нуждающемуся в этом пациенту комбинированной терапии, независимо включающей терапевтически эффективные количества:
(a) ингибитора BRAF, которым является метил-N-[(2S)-1-({4-[3-(5-хлор-2-фтор-3-метансульфонамидофенил)-1-(пропан-2-ил)-1H-пиразол-4-ил]пиримидин-2-ил}амино)пропан-2-ил]карбамат (СОЕДИНЕНИЕ A),
(b) ингибитора MEK (СОЕДИНЕНИЯ B), который является (2-гидроксиэтокси)амидом 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты, и
(c) антитела против EGFR (СОЕДИНЕНИЯ C), которое является цетуксимабом.
2. Способ по п.1, где ингибитором BRAF является аморфный метил-N-[(2S)-1-({4-[3-(5-хлор-2-фтор-3-метансульфонамидофенил)-1-(пропан-2-ил)-1H-пиразол-4-ил]пиримидин-2-ил}амино)пропан-2-ил]карбамат.
3. Способ по любому из пп.1, 2, где указанный ингибитор MEK является кристаллизованным (2-гидроксиэтокси)амидом 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты.
4. Способ по п.3, где указанный кристаллизованный (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты характеризуется наличием дифракционных пиков XRPD (в градусах 2θ) при 20,38 и 28,39.
5. Способ по п.3, где указанный кристаллизованный (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты характеризуется наличием дифракционных пиков XRPD (в градусах 2θ) при 20,38, 28,39 и 11,18.
6. Способ по п.3, где указанный кристаллизованный (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты характеризуется наличием дифракционных пиков XRPD (в градусах 2θ) при 20,38, 28,39, 11,18 и 29,18.
7. Способ по п.3, где указанный кристаллизованный (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты характеризуется наличием дифракционных пиков XRPD (в градусах 2θ) при 20,38, 28,39, 11,18, 29,18, 22,43 и 22,75.
8. Способ по п.3, где указанный кристаллизованный (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты характеризуется наличием дифракционных пиков XRPD (в градусах 2θ) при 20,38, 28,39, 11,18, 29,18, 22,43, 22,75, 25,23, 16,05 и 11,82.
9. Способ по п.3, где указанный кристаллизованный (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты характеризуется наличием дифракционных пиков XRPD (в градусах 2θ) при 20,38, 28,39, 11,18, 29,18, 22,43, 22,75, 25,23, 16,05, 11,82, 23,74, 16,33 и 19,00.
10. Способ по п.3, где указанный кристаллизованный (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты имеет XRPD рентгенограмму по существу, как показано на Фигуре 8.
11. Способ по любому из пп.1-10, где рак выбран из рака толстой и прямой кишки, меланомы, рака легкого, рака молочной железы, рака почки, рака печени, рака эндометрия, острого миелогенного лейкоза, миелодиспластических синдромов, рака щитовидной железы, в частности папиллярного рака щитовидной железы, рака поджелудочной железы, нейрофиброматоза и гепатоцеллюлярной карциномы.
12. Способ по п.11, где рак является раком толстой и прямой кишки.
13. Способ по п.12, где рак толстой и прямой кишки является метастатическим раком толстой и прямой кишки.
14. Способ по любому из пп.1-13, где рак является раком, имеющим мутацию BRAF V600.
15. Способ по п.14, где рак является раком, имеющим мутацию BRAF V600E.
16. Способ по любому из пп.1-15, где рак дополнительно экспрессирует KRAS дикого типа (KRASwt).
17. Способ по любому из пп.1-16, где терапевтически эффективное количество СОЕДИНЕНИЯ A вводят перорально один раз в день.
18. Способ по п.17, где терапевтически эффективное количество СОЕДИНЕНИЯ A составляет 300 мг, которые вводят перорально один раз в день.
19. Способ по любому из пп.1-18, где терапевтически эффективное количество СОЕДИНЕНИЯ B вводят перорально два раза в день в качестве первой и второй терапевтически эффективных доз, где первую терапевтически эффективную дозу СОЕДИНЕНИЯ B принимают вместе с терапевтически эффективным количеством СОЕДИНЕНИЯ A.
20. Способ по п.19, где каждая указанная первая и вторая терапевтически эффективная доза СОЕДИНЕНИЯ B включает 45 мг СОЕДИНЕНИЯ B.
21. Способ по п.19 или 20, где указанную первую терапевтически эффективную дозу СОЕДИНЕНИЯ B вводят в течение 30 минут после введения терапевтически эффективного количества СОЕДИНЕНИЯ A.
22. Способ по любому из пп.1-21, где терапевтически эффективное количество СОЕДИНЕНИЯ C вводят в виде внутривенной инфузии в количестве 400 мг/м2 за 120 минут в качестве начальной дозы, с последующими еженедельными внутривенными инфузиями в количестве 250 мг/м2 за 60 минут.
23. Способ по любому из пп.19-22, где терапевтически эффективное количество СОЕДИНЕНИЯ C вводят по меньшей мере через 30-90 минут после введения терапевтически эффективного количества СОЕДИНЕНИЯ A и первой терапевтически эффективной дозы СОЕДИНЕНИЯ B.
24. Способ по любому из пп.1-23, где способ необязательно дополнительно включает введение одного или более лекарственных средств для предварительной медикаментозной подготовки указанному субъекту до лечения СОЕДИНЕНИЕМ C.
25. Способ по п.24, где указанное лекарственное средство для предварительной медикаментозной подготовки вводят за 30-60 минут до введения СОЕДИНЕНИЯ C.
26. Способ по п.25, где указанное одно или более лекарственное средство для предварительной медикаментозной подготовки выбрано из одного или более из H1-антагониста и системного кортикостероида.
27. Способ по любому из пп.1-26, где указанный субъект проходил лечение по меньшей мере одним средством для системной противоопухолевой терапии в течение некоторого периода времени до лечения указанной комбинированной терапией.
28. Способ по п.27, где системная противоопухолевая терапия включает лечение с применением одного или более цитотоксических средств.
29. Способ по п.28, где указанное одно или более цитотоксическое средство выбрано из иринотекана, оксалиплатина, капецитабина, фолиновой кислоты и 5-фторурацила.
30. Способ по любому из пп.1-29, где способ дополнительно включает оценку лечения указанной комбинированной терапией путем определения одного или более из ингибирования прогрессирования заболевания, ингибирования роста опухоли, уменьшения первичной опухоли, облегчения связанных с опухолью симптомов, ингибирования секретируемых опухолью факторов, задержки появления первичных или вторичных опухолей, замедления развития первичных или вторичных опухолей, уменьшения возникновения первичных или вторичных опухолей, замедления возникновения или уменьшения тяжести вторичных эффектов заболевания, остановки роста опухоли и регрессии опухолей, увеличения времени до прогрессирования (TTP), увеличения выживаемости без прогрессирования (PFS), увеличения общей выживаемости (OS) или увеличения длительности ответа (DOR).
| WO 2014066606 A2, 01.05.2014 | |||
| WO 2015087279 A1, 18.06.2015 | |||
| WO 2011025927 A1, 03.03.2011 | |||
| N3-АЛКИЛИРОВАННЫЕ БЕНЗИМИДАЗОЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ MEK | 2003 |
|
RU2307831C9 |
| US 20150164897 A1, 18.06.2015 | |||
| СНЕГООЧИСТИТЕЛЬ ДЛЯ ЖЕЛЕЗНЫХ ДОРОГ | 1923 |
|
SU818A1 |
| Найдено в Интернет: URL: | |||

