Область, к которой относится изобретение
[0001] Настоящая заявка относится к соединениям, которые селективно ингибируют гликозидазы, и способам их применения.
Уровень техники
[0002] Широкий диапазон клеточных белков, как ядерных так и цитоплазматических, посттрансляционно модифицируется добавлением моносахарида 2-ацетамидо-2-дезокси-β-D-глюкопиранозида (β-Ν-ацетилглюкозамина), который присоединен посредством O-гликозидной связи.1 Эта модификация в целом именуется O-связанным N-ацетилглюкозамином или O-GlcNAc. Ферментом, ответственным за посттрансляционное связывание β-Ν-ацетилглюкозамина (GlcNAc) со специфическими остатками серина и треонина многочисленных нуклеоцитоплазматических белков, является O-GlcNAc-трансфераза (OGT).2-5 Второй фермент, известный как гликопротеин 2-ацетамидо-2-дезокси-β-D-глюкопиранозидаза (O-GlcNAcase)6,7, устраняет эту посттрансляционную модификацию для освобождения белков, делая O-GlcNAc-модификацию динамическим циклом, происходящим несколько раз в течение продолжительности жизни белка.8
[0003] O-GlcNAc-модифицированные белки регулируют широкий диапазон жизненно важных клеточных функций, включая, например, транскрипцию,9-12 протеосомальную деградацию,13 и клеточную передачу сигналов.14 O-GlcNAc также обнаруживается на многих структурных белках.15-17 Например, он был обнаружен на ряде белков цитоскелета, включая нейрофибриллярные белки,18,19 синапсины,6,20 синапсин-специфический белок сборки клатрина AP-37 и анкирина G.14 Было обнаружено, что модификация O-GlcNAc часто встречается в головном мозге.21,22 Она также была обнаружена на белках, отчетливо вовлеченных в этиологию нескольких заболеваний, включая болезнь Альцгеймера (AD) и рак.
[0004] Например, хорошо установлено, что AD и ряд связанных таутопатий, включая синдром Дауна, болезнь Пика, болезнь Ньюманна-Пика типа C и боковой амиотрофический склероз (ALS) частично характеризуются развитием нейрофибриллярных сплетений (NFT). Эти NFT представляют собой агрегаты парных спиральных волокон (PHF) и составлены из аномальной формы белка цитоскелета «тау». В норме, тау стабилизирует ключевую клеточную сеть микротрубочек, которая существенна для распределения белков и питательных веществ внутри нейронов. Однако у пациентов с AD тау становится гиперфосфорилированным, нарушая его нормальные функции, образуя PHF, и, в конечном счете, агрегируясь для образования NFT. Шесть изоформ тау обнаруживаются в мозге человека. У пациентов с AD все шесть изоформ тау обнаруживаются в NFT, и все являются заметно гиперфосфорилированными.23,24 Тау в здоровой мозговой ткани несет лишь 2 или 3 фосфатные группы, тогда как тау, обнаруживаемые в мозге пациентов с AD, несут в среднем 8 фосфатных групп.25,26 Отчетливая параллель между уровнями NFT в мозге пациентов с AD и тяжестью деменции, веско подтверждает ключевую роль дисфункции тау при AD.27,28 Точные причины этого гиперфосфорилирования тау остаются неясными. Соответственно, значительные усилия были посвящены: a) выяснению молекулярной физиологической основы гиперфосфорилирования тау;29 и b) идентификации стратегий, которые могут ограничить гиперфосфорилирование тау в надежде, что это может остановить или даже вызвать обратное развитие прогрессирования болезни Альцгеймера.30-33 К настоящему времени, несколько линий доказательств свидетельствуют о том, что стимулирующая регуляция ряда киназ может быть вовлечена в гиперфосфорилирование тау,21,34,35, хотя совсем недавно была выдвинута альтернативная основа для этого гиперфосфорилирования.21
[0005] В частности, оказалось, что уровни фосфатов в тау регулируются уровнями O-GlcNAc на тау. Присутствие O-GlcNAc на тау стимулировало исследования, которые определяют корреляцию уровней O-GlcNAc с уровнями форсфорилирования тау. Представляется, что интерес к этой области происходит из наблюдения, что, как было обнаружено, модификация O-GlcNAc происходит на многих белках в аминокислотных остатках, которые, как известно, также являются фосфорилированными.36-38 Согласованно с этим наблюдением, было обнаружено, что увеличение уровней фосфорилирования приводит к снижению уровней O-GlcNAc и наоборот, повышенные уровни O-GlcNAc коррелируются со сниженными уровнями фосфорилирования.39 Эта взаимная связь между O-GlcNAc и фосфорилированием была названа «гипотезой Йин-Янг»40, и получила веское биохимическое подтверждение обнаружением того, что фермент OGT4 образует функциональный комплекс с фосфатазами, которые действуют для удаления фосфатных групп из белков.41 Подобно фосфорилированию, O-GlcNAc представляет собой динамическую модификацию, которая может быть удалена и вновь вставлена несколько раз в течение срока жизни белка. Предположительно, ген, кодирующий O-GlcNAcase, был картирован в хромосомный локус, который связан с AD.7,42 Гиперфосфорилированный тау в мозге людей, страдающих AD, имеет значительно более низкие уровни O-GlcNAc, чем обнаруживаемые в мозге здоровых людей.21 Было показано, что уровни O-GlcNAc растворимого белка тау из мозга людей, пораженных AD, значительно ниже, чем уровни O-GlcNAc растворимого белка тау из здорового мозга.21 Кроме того, предполагали, что PHF из пораженного мозга полностью лишен какой-либо модификации O-GlcNAc.21 Молекулярная основа этого гипогликозилирования тау неизвестна, хотя оно может происходить из повышенной активности киназ и/или дисфункции одного из ферментов, участвующих в процессинге O-GlcNAc. Подтверждая эту последнюю точку зрения, и в нейронных клетках PC-12, и срезах ткани мозга мышей, не селективный ингибитор N-ацетилглюкозаминидазы использовали для повышения уровней O-GlcNAc, после чего наблюдали снижение фосфорилирования.21 Сущность этих объединенных результатов заключается в том, что путем поддержания нормальных уровней O-GlcNAc у пациентов с AD, например, ингибированием действия O-GlcNAcase, может быть блокировано гиперфосфорилирование тау и все эффекты, связанные с гиперфосфорилированием тау, включая образование NFT, и эффекты ниже по ходу транскрипции. Однако ввиду того, что должное функционирование β-гексозаминидаз имеет решающее значение, любое потенциальное терапевтическое вмешательство для лечения AD, которое блокирует O-GlcNAcase, должно было бы избегать сопутствующего ингибирования обеих гексозаминидаз A и B.
[0006] Нейроны не накапливают глюкозу, и поэтому мозг зависит от глюкозы, доставляемой кровью, для поддержания его существенных метаболических функций. В частности, было показано, что со старением внутри мозга уменьшается захват и метаболизм глюкозы.43 Внутри мозга пациентов с AD происходит выраженное уменьшение степени утилизации глюкозы, и это считают возможной причиной нейродегенерации.44 Основой этой сниженной доставки глюкозы в мозг при AD45-47 считают любой сниженный транспорт глюкозы,48,49 нарушенная инсулиновая сигнализация,50,51 и сниженный кровоток.52
[0007] В свете этого нарушенного метаболизма глюкозы, следует отметить, что из всей глюкозы, поступающей в клетки, 2-5% шунтируется в биосинтетический путь гексозамина, посредством этого, регулируя клеточные концентрации конечного продукта этого пути, уридиндифосфат-N-ацетилглюкозамина (UDP-GlcNAc).53 UDP-GlcNAc представляет собой субстрат нуклеоцитоплазматического фермента O-GlcNAc трансферазы (OGT)2-5, который действует для посттрансляционного добавления GlcNAc к специфическим сериновым и треониновым остаткам многочисленных нуклеоцитоплазматических белков. OGT распознает многие из его субстратов54,55 и партнеров связывания41,56 посредством его доменов, содержащих тетратрикопептидные повторы (TPR).57,58 Как описано выше, O-GlcNAcase6,7 устраняет эту посттрансляционную модификацию для освобождения белков, делая модификацию O-GlcNAc динамическим циклом, происходящим несколько раз в течение времени жизни белка.8 O-GlcNAc была обнаружена в нескольких белках на известных участках фосфорилирования10,37,38,59, включая тау и нервные волокна60. Кроме того, OGT проявляет необычное кинетическое поведение, делая ее исключительно чувствительной к внутриклеточным концентрациям субстрата UDP-GlcNAc, и поэтому к доставке глюкозы.41
[0008] Согласованно с известными свойствами биосинтетического пути гексозамина, ферментными свойствами OGT и взаимной связью между O-GlcNAc и фосфорилированием, было показано, что уменьшенная доступность глюкозы в мозге ведет к гиперфосфорилированию44 тау. Поэтому, постепенное нарушение транспорта и метаболизма глюкозы, независимо от его причины, ведет к уменьшению O-GlcNAc и гиперфосфорилированию тау (и других белков). Соответственно, ингибирование O-GlcNAcase должно компенсировать связанное с возрастом нарушение метаболизма глюкозы внутри мозга здоровых индивидов, а также пациентов, страдающих AD, или родственными нейродегенеративными заболеваниями.
[0009] Эти результаты свидетельствуют о том, что нарушение функции в механизмах, регулирующих уровни O-GlcNAc тау, может быть жизненно важным в образовании NFT и связанной с ним нейродегенерации. Надежное подтверждение блокировки гиперфосфорилирования тау в качестве терапевтически полезного вмешательства61 исходит из недавних исследований, показывающих, что когда трансгенных мышей, несущих человеческий тау, лечат ингибиторами киназы, у них не развиваются типичные двигательные дефекты33, и в другом случае,32 проявляются сниженные уровни нерастворимого тау. Эти исследования ясно свидетельствуют о связи между снижением уровней фосфорилирования тау и облегчением подобных AD поведенческих симптомов на мышиной модели этого заболевания. Действительно, фармакологическая модуляция гиперфосфорилирования тау широко признана в качестве обоснованной терапевтической стратегии для лечения AD и других нейродегенеративных расстройств.62
[0010] Низкомолекулярные ингибиторы O-GlcNAcase для ограничения гиперфосфорилирования тау рассматривали как средства для лечения AD и родственных таупатий.63 В частности, с воздействием ингибитора O-GlcNAcase тиамета-G связывали снижение фосфорилирования тау в культивируемых клетках PC-12 в патологически релевантных участках.63 Кроме того, с пероральным введением тиамета-G здоровым крысам Sprague-Dawley связывали снижение фосфорилирования в Thr231, Ser396 и Ser422 и в коре, и в гиппокампе крыс.63
[0011] Имеется также множество доказательств, указывающих на то, что повышенный уровень модификации белка O-GlcNAc обеспечивает защиту против патогенных воздействий стресса в сердечной ткани, включая вызванную стрессом ишемию, кровотечение, гиперволемический шок и кальциевый парадокс. Например, было продемонстрировано, что активация пути биосинтеза гексозамина (HBP) введением глюкозамина оказывает защитный эффект на экспериментальных моделях ишемии/реперфузии,64-70 травматического кровотечения,71-73 гиперволемического шока74 и кальциевого парадокса64,75 у животных. Кроме того, веское доказательство указывает на то, что эти кардиопротективные эффекты опосредуются повышенными уровнями модификации белка O-GlcNAc modification.64,65,67,70,72,75-78 Имеется также доказательство того, что модификация O-GlcNAc играет роль при разнообразных нейродегенеративных заболеваниях, включая болезнь Паркинсона и болезнь Хантингтона.79
[0012] У людей имеется три гена, кодирующих ферменты, которые отщепляют концевые остатки β-Ν-ацетилглюкозамина от гликоконъюгатов. Первый из них кодирует O-GlcNAcase. O-GlcNAcase является членом семейства 84 гликозидных гидролаз, которое включает ферменты из организмов, таких же разнообразных как прокариотические патогены для людей (классификацию семейства гликозидных гидролаз см. на сайте интернета Coutinho, P.M. & Henrissat, B. (1999) Carbohydrate-Active Enzymes server at URL: http://afmb.cnrs-mrs. fr/CAZY/.27,28 O-GlcNAcase действует для гидролитического отсоединения O-GlcNAc от сериновых и треониновых остатков посттрансляционно модифицированных белков.1,6,7,80,81 Согласовано с присутствием O-GlcNAc на многих внутриклеточных белках, представляется, что фермент O-GlcNAcase играет роль в этиологии нескольких заболеваний, включая сахарный диабет II типа,14,82 AD,16,21,83 и рак.22,84 Хотя O-GlcNAcase была, вероятно, выделена ранее,18,19 прошло примерно 20 лет перед тем как поняли её биохимическую роль в действии для отщепления O-GlcNAc от сериновых и треониновых остатков белков.6 Позднее, была клонирована,7 частично охарактеризована20 O-GlcNAcase и было высказано предположение о ее дополнительной активности в качестве гистон-ацетилтрансферазы.20 Однако было мало информации о каталитическом механизме этого фермента.
[0013] Два других гена, HEXA и HEXB, кодируют ферменты, катализирующие гидролитическое отщепление концевых остатков β-Ν-ацетилглюкозамина от гликоконъюгатов. Генные продукты HEXA и HEXB преимущественно обеспечивают выход двух димерных изоферментов, соответственно, гексозаминидазы A и гексозаминидазы B. Гексозаминидаза A (αβ), гетеродимерный фермент, составлен из α- и β-субъединицы. Гексозаминидаза B (ββ), гомодимерный фермент, составлен из двух β-субъединиц. Две субъединицы, α- и β-, имеют высокий уровень идентичности последовательностей. Оба эти фермента классифицируются как члены семейства 20 гликозидных гидролаз, и обычно локализуются внутри лизосом. Должное функционирование этих лизосомальных β-гексозаминидаз играет ключевую роль для развития организма человека, обстоятельство, которое недооценивается и приводит к трагическим генетическим заболеваниям, болезням Тея-Сакса и Сандхоффа, которые возникают соответственно в результате дисфункции гексозаминидазы A и гексозаминидазы B.85 Эти ферментные недостаточности вызывают накопление гликолипидов и гликоконъюгатов в лизосомах, приводя к неврологическому нарушению и деформации. Вредные воздействия накопления ганглиозидов на уровне организма еще продолжают выявляться.86
[0014] В результате биологической важности этих β-N-ацетилглюкозаминидаз, низкомолекулярные ингибиторы гликозидаз87-90 привлекли большое внимание,91 и как инструменты для выяснения роли этих ферментов в биологических процессах, и в разработке потенциальных видов терапевтического применения. Регуляция функции гликозидазы с использованием мелких молекул обеспечивает несколько преимуществ перед исследованиями генетического нокаута, включая способность быстрого варьирования доз или полную отмену лечения.
[0015] Однако, основной проблемой при разработке ингибиторов для блокировки функции гликозидаз млекопитающих, включая O-GlcNAcase, является большое число функционально связанных ферментов, присутствующих в тканях высших эукариотов. Соответственно, использование неселективных ингибиторов при исследовании физиологической роли одного конкретного фермента на клеточном уровне и на уровне организма осложнено, потому что в результате сопутствующего ингибирования таких функционально связанных ферментов возникают сложные фенотипы. В случае β-N-ацетилглюкозаминидаз, многие соединения, которые действуют для блокировки функции O-GlcNAcase, являются неспецифичными и потенциально действуют для ингибирования лизосомальных β-гексозаминидаз.
[0016] Некоторыми из лучше охарактеризованных ингибиторов β-N-ацетилглюкозаминидаз, которые использовались в исследованиях посттрансляционной модификации O-GlcNAc внутри и клеток, и тканей, являются стрептозотоцин (STZ), 2'-метил-α-D-глюкопирано-[2,l-d]-Δ2'-тиазолин (NAG-тиазолин) и O-(2-ацетамидо-2-дезокси-D-глюкопираносилиден)амино N-фенилкарбамат (PUGNAc).14,92-95
[0017] STZ в течение длительного времени использовали в качестве диабетогенного соединения, потому что он оказывает особенно повреждающий эффект на β-островковые клетки.96 STZ оказывает свои цитотоксические эффекты посредством и алкилирования клеточной ДНК96,97, а также генерации видов радикалов, включая оксид азота.98 Происходящий в результате разрыв нити ДНК стимулирует активацию поли(АДФ-рибоза)-полимеразы (PARP)99 с чистым эффектом истощения уровней клеточного NAD+, и, в конечном счете, ведущим к гибели клеток.100,101 Другие исследователи вместо этого предположили, что токсичность STZ является следствием необратимого ингибирования O-GlcNAcase, которая высоко экспрессирована внутри β-островковых клеток.92,102 Однако эта гипотеза была подвергнута сомнению двумя независимыми исследовательскими группами.103,104 Ввиду того, что уровни клеточного O-GlcNAc на белках увеличиваются в ответ на многие формы клеточного стресса105, представляется возможным, что STZ приводит к повышенным уровням модификации O-GlcNAc на белках путем индукции клеточного стресса, а не посредством какого-либо специфического и прямого действия на O-GlcNAcase. Действительно, Hanover и сотрудники показали, что STZ функционирует в качестве слабого и, в некоторой степени, селективного ингибитора O-GlcNAcase106, и, хотя другие исследователи предположили, что STZ действует для необратимого ингибирования O-GlcNAcase,107 этот тип действия не был ясно продемонстрирован. Позднее, было показано, что STZ не вызывает необратимое ингибирование O-GlcNAcase.108
[0018] Было обнаружено, что NAG-тиазолин является сильнодействующим ингибитором семейства 20 гексозаминидаз,90,109, и, позднее, семейства 84 O-GlcNAcases.108 Несмотря на силу его действия, негативным аспектом использования NAG-тиазолина в сложном биологическом контексте является то, что он лишен селективности и поэтому нарушает множественные клеточные процессы.
[0019] PUGNAc является другим соединением, недостатком которого является та же проблема отсутствия селективности, и тем не менее ее использовали в качестве ингибитора и человеческой O-GlcNAcase6,110, и семейства 20 человеческих β-гексоаминидаз.111 Было обнаружено, что эта молекула, разработанная Vasella и сотрудниками, является сильнодействующим конкурентным ингибитором β-N-ацетилглюкозаминидаз из Canavalia ensiformis, Mucor rouxii, и β-гексозаминидазы из коровьей почки.88 Было продемонстрировано, что введение PUGNAc на крысиной модели травматического кровотечения уменьшает циркулирующие уровни провоспалительных цитокинов TNF-α и IL-6.112 Было также показано, что введение PUGNAc на модели активации лимфоцитов, основанной на клетках, уменьшает продукцию цитокина 1L-2.113 Последующие исследования показали, что PUGNAc можно использовать на экспериментальной модели у животных для уменьшения размера инфаркта миокарда после окклюзий левой коронарной артерии.114 Особое значение имеет то, что повышение уровней O-GlcNAc введением PUGNAc, ингибитора O-GlcNAcase, на модели травматического кровотечения у крыс, улучшает сердечную функцию.112,115 Кроме того, повышение уровней O-GlcNAc обработкой PUGNAc на клеточной модели ишемии/реперфузионного повреждения с использованием желудочковых миоцитов новорожденных крыс улучшало жизнеспособность клеток и уменьшало некроз и апоптоз, по сравнению с необработанными клетками.116
[0020] Позднее, было высказано предположение, что селективный ингибитор O-GlcNAcase NButGT проявляет защитную активность на моделях ишемии/реперфузии и клеточных стрессов, включая окислительный стресс, на клеточной основе.117 В этом исследовании предлагают применять ингибиторы O-GlcNAcase для повышения уровней белка O-GlcNAc и, посредством этого, предотвращения патогенных эффектов стресса на сердечную ткань.
[0021] В заявках на Международные патенты PCT/CA2006/000300, поданной 1 марта 2006 г, опубликованной под № W0 2006/092049 8 сентября 2006 г; PCT/CA2007/001554, поданной 31 августа 2007 г, опубликованной под № WO 2008/025170 6 марта 2008 г; PCT/CA2009/001087, поданной 31 июля 2009 г, опубликованной под № WO 2010/012106 4 февраля 2010 г; PCT/CA2009/001088, поданной 31 июля 2009 г, опубликованной под № WO 2010/012107 4 февраля 2010 г и PCT/CA2009/001302, поданной 16 сентября 2009 г, опубликованной под № WO 2010/037207 8 апреля 2010 г, описаны селективные ингибиторы O-GlcNAcase.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
[0022] Изображение частично относится к соединениям для селективного ингибирования гликозидаз, пролекарствам этих соединений, способам применения этих соединений и пролекарств, фармацевтическим композициям, включающим эти соединения или пролекарства этих соединений и к способам лечения заболеваний и расстройств, связанных с дефицитом или сверхэкспрессией O-GlcNAcase, и/или накоплением или дефицитом O-GlcNAc.
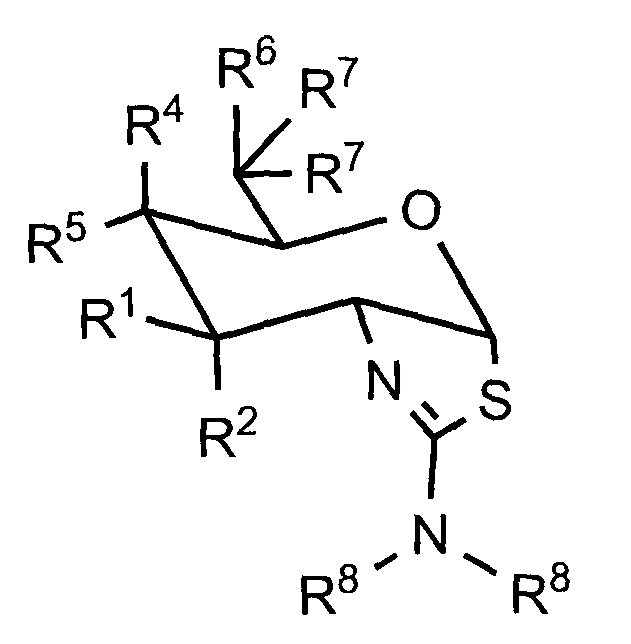
[0023] В одном аспекте, изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли:
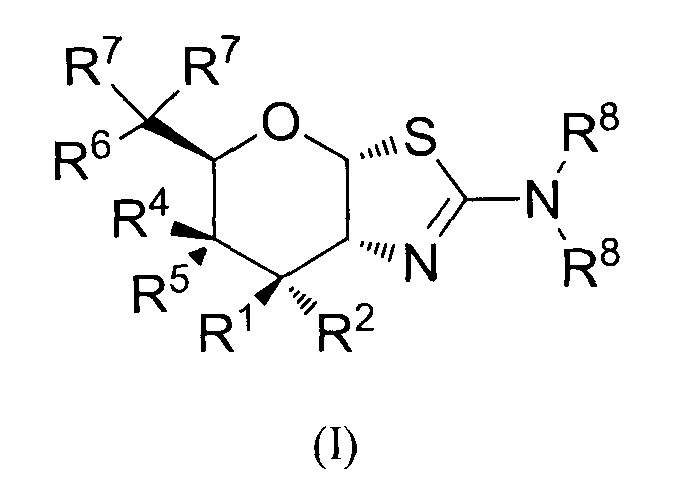
где R1 и R2 могут обозначать H, или R1 может обозначать H и R2 может обозначать F, или R1 может обозначать F и R2 может обозначать H, или R1 может обозначать OR3 и R2 может обозначать H; каждая R3 может обозначать независимо H или C1-6 ацил; R4 может обозначать H и R5 может обозначать OR3, или R4 может обозначать OR3 и R5 может обозначать H; R6 может обозначать H, F или OR3; каждая R7 может обозначать независимо H или F; каждая R8 может быть независимо выбрана из группы, состоящей из: H, C1-6 алкила, C3-6 алкенила, C3-6 алкинила и C1-6 алкокси, где C1-6 алкил, C3-6 алкенил, C3-6 алкинил или C1-6 алкокси могут быть необязательно замещены числом заместителей от одного до максимального одним или более из фтор-, OH или метила, или две группы R8 могут быть соединены вместе с атомом азота, с которым они соединены, для образования кольца, причем указанное кольцо необязательно независимо замещено числом заместителей от одного до максимального одним или более из фтор-, OH, или метила; при условии, что когда R6 обозначает OR3, то каждая R7 обозначает H; и при условии, что или R1, или R6 не обозначает OR3.
[0024] В альтернативных вариантах осуществления, изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли:
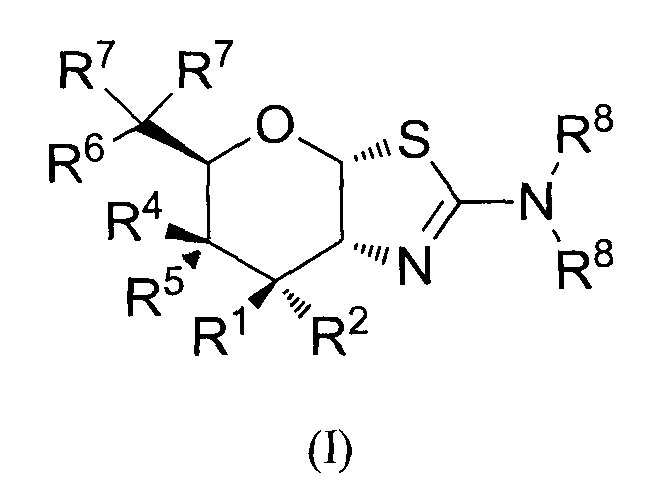
где R1 и R2 могут обозначать H и R6 может обозначать H, F или OR3; или R1 может обозначать H и R2 может обозначать F и R6 может обозначать H, F или OR3; или R1 может обозначать F и R2 может обозначать H и R6 может обозначать H, F или OR3; или R1 может обозначать OR3 и R2 может обозначать H и R6 может обозначать H или F; каждая R3 может обозначать независимо H или C1-6 ацил; R4 может обозначать H и R5 может обозначать OR3; или R4 может обозначать OR3 и R5 может обозначать H; каждая R7 может обозначать независимо H или F; каждая R8 может быть независимо выбрана из группы, состоящей из: H, C1-6 алкила, C3-6 алкенила, C3-6 алкинила и C1-6 алкокси, где C1-6 алкил, C3-6 алкенил, C3-6 алкинил или C1-6 алкокси могут быть необязательно замещены числом заместителей от одного до максимального одним или более из фтор-, OH или метила, или две группы R8 могут быть соединены вместе с атомом азота, с которым они соединены, для образования кольца, причем указанное кольцо необязательно независимо замещено числом заместителей от одного до максимального одним или более из фтор-, OH или метила; при условии, что когда R6 обозначает OR3, то каждая R7 обозначает H.
[0025] В альтернативных вариантах осуществления, изобретение относится к соединению формулы (la) или его фармацевтически приемлемой соли:
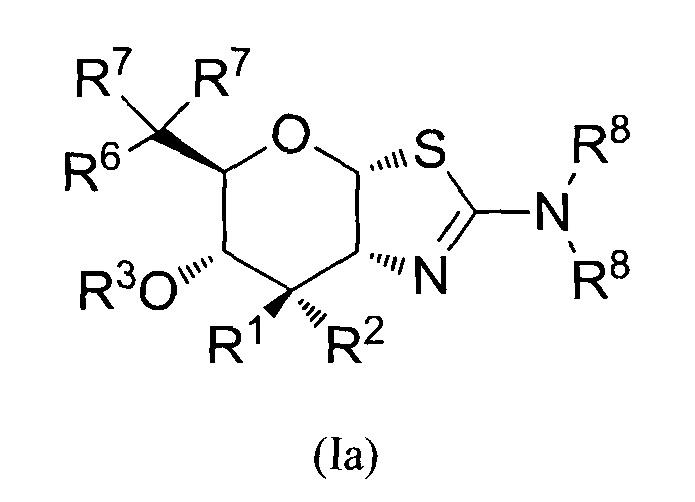
где R1 и R2 могут обозначать H и R6 может обозначать F; или R1 может обозначать H и R2 может обозначать F и R6 может обозначать H, F или OR3; или R1 может обозначать F и R2 может обозначать H и R6 может обозначать H, F или OR3; или R1 может обозначать OR3 и R2 может обозначать H и R6 может обозначать F; каждая R3 может обозначать независимо H или C1-6 ацил; каждая R7 может обозначать независимо H или F; каждая R8 может быть независимо выбрана из группы, состоящей из: H, C1-6 алкила, C3-6 алкенила, C3-6 алкинила и C1-6 алкокси, где C1-6 алкил, C3-6 алкенил, C3-6 алкинил и C1-6 алкокси могут быть необязательно замещены числом заместителей от одного до максимального одним или более из фтор-, OH, или метила, или две группы R8 могут быть соединены вместе с атомом азота, с которым они соединены, для образования кольца, причем указанное кольцо необязательно независимо замещено числом заместителей от одного до максимального одним или более из фтор-, OH или метила; при условии, что когда R6 обозначает OR3, то каждая R7 обозначает H.
[0026] В альтернативных вариантах осуществления, изобретение относится к соединению формулы (lb) или его фармацевтически приемлемой соли:
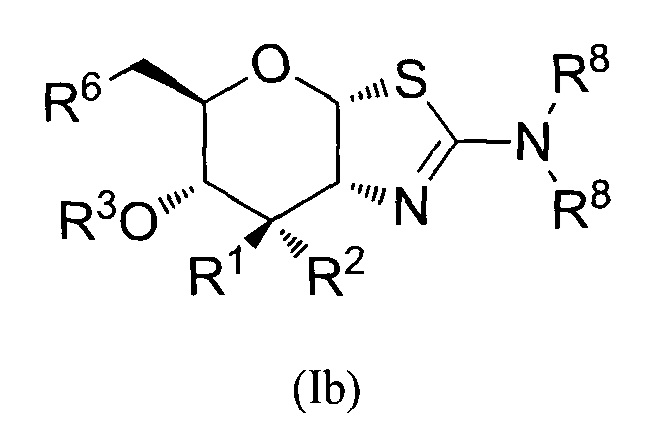
где R1 и R2 могут обозначать H и R6 может обозначать H или OR3; или R1 может обозначать OR3 и R2 может обозначать H и R6 может обозначать H; каждая R3 может обозначать независимо H или C1-6 ацил; каждая R8 может быть независимо выбрана из группы, состоящей из: H, C1-6 алкила, C3-6 алкенила, C3-6 алкинила и C1-6 алкокси, где C1-6 алкил, C3-6 алкенил, C3-6 алкинил или C1-6 алкокси могут быть необязательно замещены числом заместителей от одного до максимального одним или более из фтор-, OH или метила, или две группы R8 могут быть соединены вместе с атомом азота, с которым они связаны, для образования кольца, причем указанное кольцо необязательно независимо замещено числом заместителей от одного до максимального одним или более из фтор-, OH, или метила.
[0027] В альтернативных вариантах осуществления, изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли:
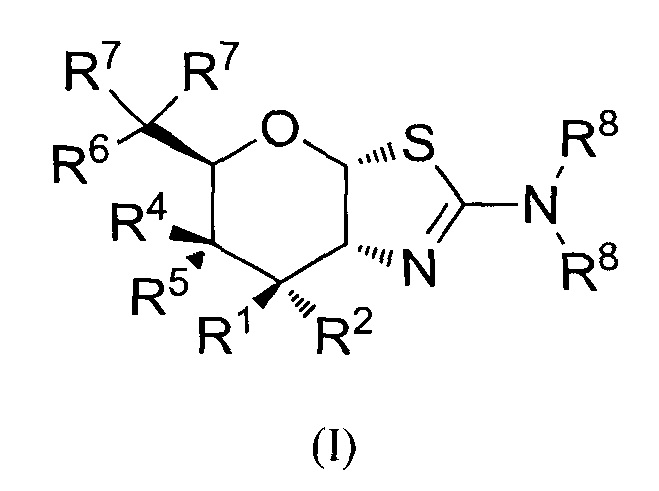
где R1 может обозначать H, F или OR3; R2 может обозначать H или F; каждая R3 может обозначать независимо H или C1-6 ацил; R4 может обозначать H; R5 может обозначать OR3; R6 может обозначать H, F или OR3; каждая R7 может обозначать независимо H или F; каждая R8 может быть независимо выбрана из группы, состоящей из: H, C1-6 алкила, C1-6 алкенила, C3-6 алкинила и C1-6 алкокси, где C1-6 алкил, C3-6 алкенил, C3-6 алкинил или C1-6 алкокси могут быть необязательно замещены числом заместителей от одного до максимального одним или более из фтор-, OH или метила, или две группы R8 могут быть соединены вместе с атомом азота, с которым они связаны, для образования кольца, причем указанное кольцо необязательно независимо замещено числом заместителей от одного до максимального одним или более из фтор-, OH или метила; при условии, что когда R1 обозначает OR3, R2 обозначает H; и при условии, что когда R6 обозначает OR3, каждая R7 обозначает H; и при условии, что или R1, или R6 не обозначает OR3; и при условии, что или R1, или R2 не обозначает F.
[0028] В альтернативных вариантах осуществления, соединение может представлять собой пролекарство; соединение может селективно ингибировать O-гликопротеин 2-ацетамидо-2-дезокси-β-D-глюкопиранозидазу (O-GlcNAcase); соединение может селективно связывать O-GlcNAcase (например, O-GlcNAcase млекопитающих); соединение может селективно ингибировать расщепление 2-ацетамидо-2-дезокси-β-D-глюкопиранозида (O-GlcNAc); соединение может по существу не ингибировать β-гексозаминидазу млекопитающих.
[0029] В альтернативных вариантах осуществления, соединение в соответствии с формулой (la) или формулой (lb) может иметь увеличенную проникающую способность.
[0030] В альтернативных аспектах, изобретение относится к фармацевтической композиции, включающей соединение по изобретению, в комбинации с фармацевтически приемлемым носителем.
[0031] В альтернативных аспектах, изобретение относится к способам селективного ингибирования O-GlcNAcase или ингибирования O-GlcNAcase у нуждающегося в них индивида, или повышения уровня O-GlcNAc, или лечения нейродегенеративного заболевания, таупатии, рака или стресса у нуждающегося в них индивида, введением индивиду эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли:
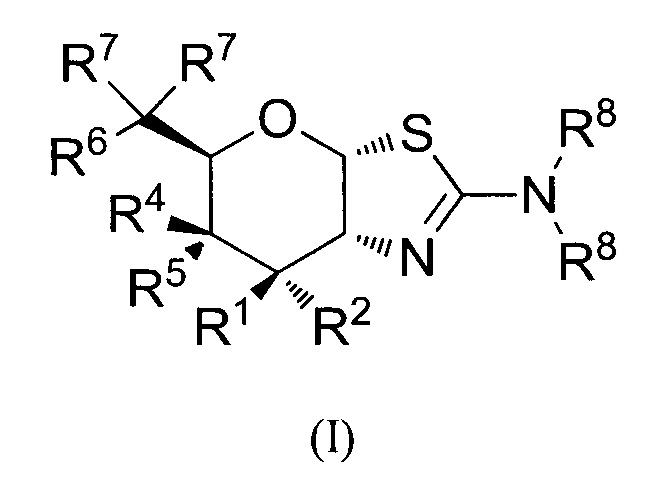
где R1 и R2 могут обозначать H, или R1 может обозначать H и R2 может обозначать F, или R1 может обозначать F и R2 может обозначать H, или R1 может обозначать OR3 и R2 может обозначать H; каждая R3 может обозначать независимо H или C1-6 ацил; R4 может обозначать H, и R5 может обозначать OR3, или R4 может обозначать OR3 и R5 может обозначать H; R6 может обозначать H, F или OR3; каждая R7 может обозначать независимо H или F; каждая R8 может быть независимо выбрана из группы, состоящей из: H, C1-6 алкила, C3-6 алкенила, C3-6 алкинила и C1-6 алкокси, где C1-6 алкил, C3-6 алкенил, C3-6 алкинил или C1-6 алкокси могут быть необязательно замещены числом заместителей от одного до максимального одним или более из фтор-, OH или метила, или две группы R8 могут быть соединены вместе с атомом азота, с которым они связаны, для образования кольца, причем указанное кольцо необязательно независимо замещено числом заместителей от одного до максимального одним или более из фтор-, OH или метила; при условии, что когда R6 обозначает OR3, каждая R7 обозначает H; и при условии, что или R1, или R6 не обозначает OR3. Состояние может представлять собой болезнь Альцгеймера, боковой амиотрофический склероз (ALS), боковой амиотрофический склероз с когнитивным нарушением (ALSci), деменцию с аргирофильными зернами, болезнь Блюита, кортикобазальную дегенерацию (CBD), деменцию боксеров, диффузные нейрофибриллярные сплетения с обызвествлением, синдром Дауна, семейную британскую деменцию, семейную датскую деменцию, лобно-височную деменцию с паркинсонизмом, связанную с хромосомой 17 (FTDP-17), болезнь Герстманна-Страусслера-Шейнкера, гваделупский паркинсонизм, болезнь Халлевордена-Спатца (нейродегенерацию с накоплением железа в мозге I типа), множественную системную атрофию, миотоническую дистрофию, болезнь Ниманна-Пика (типа C), паллидо-понто-нигральную дегенерацию, комплекс паркинсонизма-деменции Гуама, болезнь Пика (PiD), постэнцефалитический паркинсонизм (PEP), прионные болезни (включая болезнь Крейтцфелдта-Якоба (CJD), вариантную болезнь Крейтцфелдта-Якоба (vCJD), летальную семейную бессонницу и куру), прогрессирующий суперкортикальный глиоз, прогрессирующий супрануклеарный паралич (PSP), синдром Ричардсона, подострый склерозирующий панэнцефалит, деменцию только со сплетениями, болезнь Хантингтона, болезнь Паркинсона, шизофрению, легкое когнитивное нарушение (MCI), нейропатию (включая периферическую нейропатию, вегетативную нейропатию, неврит и диабетическую нейропатию) или глаукому. Стресс может представлять собой сердечное расстройство, например, ишемию; кровотечение; гиповолемический шок; инфаркт миокарда; инвазивную кардиологическую процедуру; аортокоронарное шунтирование; фибринолитическую терапию; ангиопластику; или размещение стента.
[0032] В альтернативных аспектах, изобретение относится к способу лечения состояния, опосредованного O-GlcNAcase, за исключением нейродегенеративного заболевания, таупатии, рака или стресса, у нуждающегося в нем индивида введением индивиду эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли:
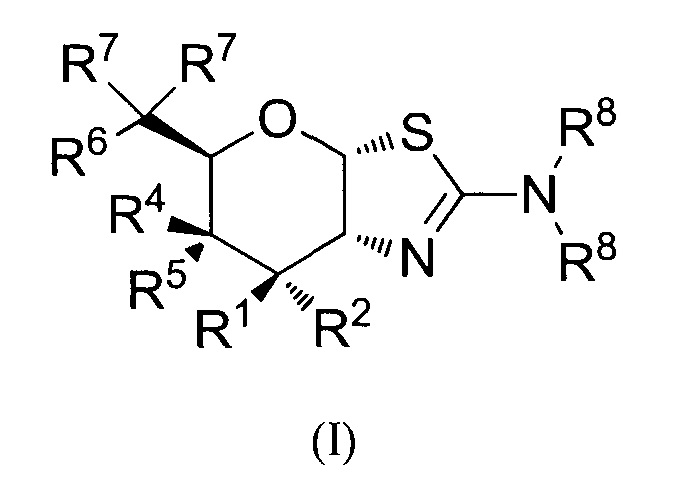
где R1 и R2 могут обозначать H, или R1 может обозначать H и R2 может обозначать F, или R1 может обозначать F и R2 может обозначать H, или R1 может обозначать OR3 и R2 может обозначать H; каждая R3 может обозначать независимо H или C1-6 ацил; R4 может обозначать H и R5 может обозначать OR3, или R4 может обозначать OR3 и R5 может обозначать H; R6 может обозначать H, F или OR3; каждая R7 может обозначать независимо H или F; каждая R8 может быть независимо выбрана из группы, состоящей из: H, C1-6 алкила, C3-6 алкенила, C3-6 алкинила и C1-6 алкокси, где C1-6 алкил, C3-6 алкенил, C3-6 алкинил или C1-6 алкокси могут быть необязательно замещены числом заместителей от одного до максимального одним или более из фтор-, OH или метила, или две группы R8 могут быть соединены вместе с атомом азота, с которым они связаны, для образования кольца, причем указанное кольцо необязательно независимо замещено числом заместителей от одного до максимального одним или более из фтор-, OH или метила; при условии, что когда R6 обозначает OR3, каждая R7 обозначает H; и при условии, что или R1, или R6 не обозначает OR3. В некоторых вариантах осуществления, состояние может представлять собой воспалительные или аллергические заболевания, такие как астма, аллергический ринит, связанные с гиперчувствительностью легочные заболевания, связанный с гиперчувствительностью пневмонит, эозинофильные пневмонии, гиперчувствительность отсроченного типа, атеросклероз, интерстициальное легочное заболевание (ILD) (например, идиопатический легочный фиброз, или ILD, связанное с ревматоидным артритом, системная красная волчанка, анкилозирующий спондилит, системный склероз, сидром Шегрена, полимиозит или дерматомиозит); системная анафилаксия или реакции гиперчувствительности, лекарственные аллергии, аллергии к укусам насекомых; аутоиммуные заболевания, такие как ревматоидный артрит, псориатический артрит, рассеянный склероз, синдром Жиллена-Барре, системная красная волчанка, генерализованная миастения, гломерулонефрит, аутоиммунный тироидит, отторжение трансплантата, включая отторжение аллотрансплантата или болезнь трансплантат против хозяина; воспалительные кишечные заболевания, такие как болезнь Крона и язвенный колит; спондилоартропатии; склеродермию; псориаз (включая псориаз, опосредованный T-клетками) и воспалительные дерматозы, такие как дерматит, экзема, атопический дерматит, аллергический контактный дерматит, сыпь; васкулит (например, некротизирующий, кожный и связанный гиперчувствительностью васкулит); эозинофильный миозит и эозинофильный фасциит; отторжение трансплантата, в частности, без ограничения трансплантатов сỏлидных органов, таких как трансплантаты сердца, легких, печени, почек и поджелудочной железы (например, аллотрансплантатов почек и легких); эпилепсия; боль; фибромиалгия; инсульт, например, нейропротекция после инсульта.
[0033] В альтернативных вариантах осуществления, R1 и R2 могут обозначать H, или R1 может обозначать H и R2 может обозначать F, или R1 может обозначать F и R2 может обозначать H, или R1 может обозначать OR3 и R2 может обозначать H; каждая R3 может обозначать независимо H или C1-6 ацил; R4 может обозначать H и R5 может обозначать OR3, или R4 может обозначать OR3 и R5 может обозначать H; R6 может обозначать H, F или OR3; каждая R7 может обозначать независимо H или F; каждая R8 может быть независимо выбрана из группы, состоящей из: H, C1-6 алкила, C3-6 алкенила, C3-6 алкинила и C1-6 алкокси, где C1-6 алкил, C3-6 алкенил, C3-6 алкинил или C1-6 алкокси может быть необязательно замещен числом заместителей от одного до максимального одним или более из фтор-, OH или метила, или две группы R могут быть соединены вместе с атомом азота, с которым они связаны, для образования кольца, причем указанное кольцо необязательно независимо замещено числом заместителей от одного до максимального одним или более из фтор-, OH или метила; при условии, что когда R6 обозначает OR3, то ждая R7 обозначает H; и при условии, что или R1, или R6 не обозначает OR3. Введение может увеличить уровень O-GlcNAc у индивида. Индивид может представлять собой человека.
[0034] В альтернативных аспектах, изобретение относится к применению соединения или эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли:

где R1 и R2 могут обозначать H, или R1 может обозначать H и R2 может обозначать F, or R1 может обозначать F и R2 может обозначать H, или R1 может обозначать OR3 и R2 может обозначать H; каждая R3 может обозначать независимо H или C1-6 ацил; R4 может обозначать H и R5 может обозначать OR3, или R4 может обозначать OR3 и R5 может обозначать H; R6 может обозначать H, F или OR3; каждая R7 может обозначать независимо H или F; каждая R8 может быть независимо выбрана из группы, состоящей из: H, C1-6 алкила, C3-6 алкенила, C3-6 алкинила и C1-6 алкокси, где C1-6 алкил, C3-6 алкенил, C3-6 алкинил или C1-6 алкокси может быть необязательно замещен числом заместителей от одного до максимального одним или более из фтор-, OH или метила, или где группы R8 могут быть соединены вместе с атомом азота, с которым они связаны, для образования кольца, причем указанное кольцо необязательно независимо замещено числом заместителей от одного до максимального одним или более из фтор-, OH или метила; при условии, что когда R6 обозначает OR3, то каждая R7 обозначает H; и при условии, что или R1, или R6 не обозначает OR3, при получении лекарственного препарата. Лекарственный препарат может быть предназначен для селективного ингибирования O-GlcNAcase, для повышения уровня O-GlcNAc, для лечения состояния, модулируемого O-GlcNAcase, для лечения нейродегенеративного заболевания, таупатии, рака или стресса.
[0035] В альтернативных аспектах, изобретение относится к способу скрининга для выявления селективного ингибитора O-GlcNAcase a) обеспечением контакта первого образца с тестируемым соединением; b) обеспечением контакта второго образца с соединением формулы (I)
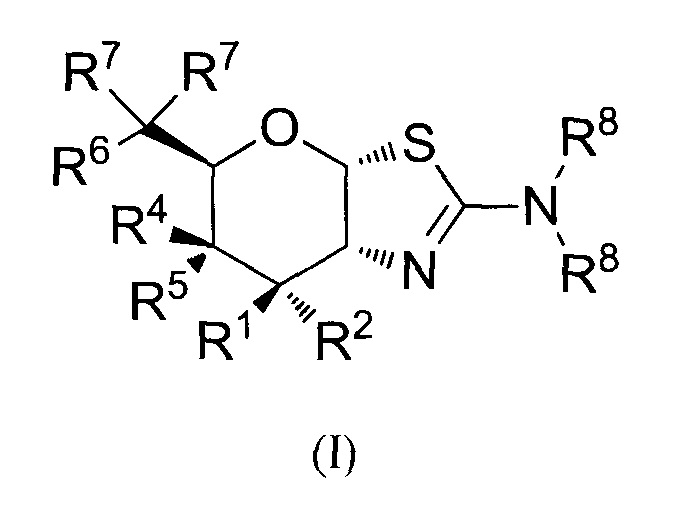
где R1 и R2 могут обозначать H, или R1 может обозначать H и R2 может обозначать F, или R1 может обозначать F и R2 может обозначать H, или R1 может обозначать OR3 и R2 может обозначать H; каждая R3 может обозначать независимо H или C1-6 ацил; R4 может обозначать H и R5 может обозначать OR3, или R4 может обозначать OR3 и R5 может обозначать H; R6 может обозначать H, F или OR3; каждая R7 может обозначать независимо H или F; каждая R8 может быть независимо выбрана из группы, состоящей из: H, C1-6 алкила, C3-6 алкенила, C3-6 алкинила и C1-6 алкокси, где C1-6 алкил, C3-6 алкенил, C3-6 алкинил или C1-6 алкокси может быть необязательно замещен числом заместителей от одного до максимального одним или более из фтор-, OH или метила, или две группы R8 могут быть соединены вместе с атомом азота, с которым они связаны, для образования кольца, причем указанное кольцо необязательно независимо замещено числом заместителей от одного до максимального одним или более из фтор-, OH или метила; при условии, что когда R6 обозначает OR3, то каждая R7 обозначает H; и при условии, что или R1, или R6 не обозначает OR3; c) определением уровня ингибирования O-GlcNAcase в первом и втором образцах, где тестируемое соединение представляет собой селективный ингибитор O-GlcNAcase, если тестируемое соединение проявляет такое же или большее ингибирование O-GlcNAcase, по сравнению с соединением формулы (I).
[0036] В данном кратком изложении сущности изобретения необязательно описаны все признаки изобретения.
ПОДРОБНОЕ ОПИСАНИЕ
[0037] Изобретение частично относится к новым соединениям, которые способны ингибировать O-гликопротеин 2-ацетамидо-2-дезокси-β-D-глюкопиранозидазу (O-GlcNAcase). В некоторых вариантах осуществления, O-GlcNAcase представляет собой O-GlcNAcase млекопитающих, такую как крысиная, мышиная или человеческая O-GlcNAcase.
[0038] В некоторых вариантах осуществления, одно или более соединений в соответствии с изобретением проявляют повышенную проникающую способность. Проникающую способность можно оценить, используя разнообразные стандартные экспериментальные методики, включая без ограничения перфузию in situ, тканевую диффузию ex vivo, клеточные монослои in vitro (например, клетки Caco-2, клетки MDCK, клетки LLC-PK1), и искусственные клеточные мембраны (например, анализ PAMPA (параллельный анализ проникновения через искусственную мембрану); обзор подходящих методик для измерения эффективной проникающей способности (Peff) или видимой проникающей способности (Papp) написан, например, Volpe в журнале The AAPS Journal, 2010, 12(4), 670-678. В некоторых вариантах осуществления, одно или более соединений в соответствии с изобретением проявляют повышенную проникающую способность при тестировании в одном или более из этих анализов для определения Peff или Papp. В некоторых вариантах осуществления, соединение, которое проявляет повышенную проникающую способность, проявляет большее пероральное всасывание. В некоторых вариантах осуществления, соединение, которое проявляет повышенную проникающую способность, проявляет большее проникновение в мозг при введении in vivo. В некоторых вариантах осуществления, соединение, которое проявляет повышенную проникающую способность, достигает более высоких концентраций в мозге при введении in vivo. В некоторых вариантах осуществления, соединение, которое проявляет повышенную проникающую способность, проявляет более высокое отношение концентрации в мозге/плазме при введении in vivo. В некоторых вариантах осуществления, «повышенная проникающая способность» означает увеличение измеренной Peff или Papp на любую величину от 10% до 100%, или любую целую величину от 10% до 100%, например, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 100% или более 100%), или увеличение в 1, 2 или 3 раза или более, по сравнению с подходящим эталонным соединением, описанным, например, в опубликованных заявках на международные патенты WO 2006/092049 или WO 2008/025170. Подходящее эталонное соединение может представлять собой, например, (3aR,5R,6S,7R,7aR)-5-(гидроксиметил)-2-пропил-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диол, или (3aR,5R,6S,7R,7aR)-2-(этиламино)-5-(гидроксиметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диол, или (3aR,5R,6S,7R,7aR)-2-(диметиламино)-5-(гидроксиметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диол. В некоторых вариантах осуществления, «повышенная проникающая способность» означает измеряемую величину Papp (т.е., величину больше нуля) в анализе, описанном ниже для определения Papp, в клетках LLC-PK1. В некоторых вариантах осуществления, «повышенная проникающая способность» означает величину Papp, больше чем 2×10-6 см/с, в анализе, описанном ниже, для определения Papp в клетках LLC-PK1. В некоторых вариантах осуществления, «повышенная проникающая способность» означает величину Papp, больше чем 1×10-6 см/с, в анализе, описанном ниже, для определения Papp в клетках LLC-PK1. В альтернативных вариантах осуществления, «повышенная проникающая способность» означает величину Papp в диапазоне от 2×10-6 см/с до 30×10-6 см/с в анализе, описанном ниже, для определения Papp в клетках LLC-PK1.
[0039] В некоторых вариантах осуществления, соединение в соответствии с изобретением проявляет превосходящую селективность при ингибировании O-GlcNAcase. В некоторых вариантах осуществления, одно или более соединений в соответствии с изобретением являются более селективными к O-GlcNAcase, чем β-гексозаминидаза. В некоторых вариантах осуществления, одно или более соединений селективно ингибируют активность O-GlcNAcase млекопитающих, чем β-гексозаминидаза млекопитающих. В некоторых вариантах осуществления, селективный ингибитор O-GlcNAcase по существу не ингибируют β-гексозаминидазу. В некоторых вариантах осуществления, β-гексозаминидаза представляет собой β-гексозаминидазу млекопитающих, такую как крысиная, мышиная или человеческая β-гексозаминидаза. Соединение, которое «селективно» ингибирует O-GlcNAcase, представляет собой соединение, которое ингибирует активность или биологическую функцию O-GlcNAcase, но по существу не ингибирует активность или биологическую функцию β-гексозаминидазы. Например, в некоторых вариантах осуществления, селективный ингибитор O-GlcNAcase селективно ингибирует отщепление 2-ацетамидо-2-дезокси-β-D-глюкопиранозида (O-GlcNAc) от полипептидов. В некоторых вариантах осуществления, селективный ингибитор O-GlcNAcase селективно связывается с O-GlcNAcase. В некоторых вариантах осуществления, селективный ингибитор O-GlcNAcase ингибирует гиперфосфорилирование белка тау и/или ингибирует образование NFT. Под терминами «ингибирует», «ингибирование» или «ингибирующее» означает уменьшение на любую величину от 10% до 90%, или любую целую величину от 30% до 60%, или более 100%, или уменьшение в 1, 2, 5, 10 или более раз. Следует понимать, что ингибирование не требует полного ингибирования. В некоторых вариантах осуществления, селективный ингибитор O-GlcNAcase повышает или увеличивает уровни O-GlcNAc, например, уровни модифицированного O-GlcNAc полипептида, в клетках, тканях ии органах (например, в мозге, мышцах или сердце (сердечной ткани) и у животных. Под «повышением» или «увеличением» подразумевается увеличение на любую величину от 10% до 90%, или на любую целую величину от 30% до 60%, или более 100%), или увеличение в 1, 2, 5, 10, 15, 25, 50, 100 или более раз. В некоторых вариантах осуществления, селективный ингибитор O-GlcNAcase проявляет относительную селективность, как описано в настоящей заявке, в диапазоне от 10 до 100000, или в диапазоне от 100 до 100000, или в диапазоне от 1000 до 100000, или по меньшей мере 10, 20, 50, 100, 200, 500, 1000, 1500, 2000, 2500, 3000, 3500, 4000, 4500, 5000, 6000, 7000, 10000, 25000, 50000, 75000, или любую величину в пределах или около описанного диапазона.
[0040] Одно или более соединений по настоящему изобретению повышают уровни O-GlcNAc на модифицированных O-GlcNAc полипептидах или белках in vivo, в частности, посредством взаимодействия с ферментом O-GlcNAcase, и эффективны при лечении состояний, которые требуют или реагируют на ингибирование активности O-GlcNAcase.
[0041] В некоторых вариантах осуществления, одно или более соединений по настоящему изобретению могут применяться в качестве средств, которые вызывают уменьшение фосфорилирования тау и образования NFT. Поэтому, в некоторых вариантах осуществления, одно или более соединений могут применяться для лечения болезни Альцгеймера и родственных таупатий. Таким образом, в некоторых вариантах осуществления, одно или более соединений способны лечить болезнь Альцгеймера и родственные таупатии снижением фосфорилирования тау и уменьшением образования NFT в результате увеличения уровней O-GlcNAc тау. В некоторых вариантах осуществления, одно или более соединений вызывают увеличение уровней модификации O-GlcNAc на модифицированных O-GlcNAc полипептидах или белках, и поэтому они могут применяться для лечения расстройств, реагирующих на такие увеличения модификации O-GlcNAc; эти расстройства включают без ограничения нейродегенеративные, воспалительные, сердечнососудистые и иммунорегуляторные заболевания. В некоторых вариантах осуществления, соединение может также применяться в результате других видов биологической активности гликозидазных ферментов. В альтернативных вариантах осуществления, одно или более соединений по изобретению являются ценными инструментами при исследовании физиологической роли O-GlcNAc на уровне клеток и организма.
[0042] В альтернативных вариантах осуществления, изобретение относится к способам увеличения или повышения уровней модификации белка O-GlcNAc у животных, таких как животные в области ветеринарии и люди. В альтернативных вариантах осуществления, изобретение относится к способам селективного ингибирования фермента O-GlcNAcase у животных, таких как, животные в области ветеринарии и люди. В альтернативных вариантах осуществления, изобретение относится к способам ингибирования фосфорилирования полипептидов тау, или ингибирования образования NFT, у животных, таких как животные в области ветеринарии и люди.
[0043] В определенных вариантах осуществления, изобретение относится к соединениям, описываемым в целом формулой (I), и к их солям, пролекарствам и энантиомерным формам:
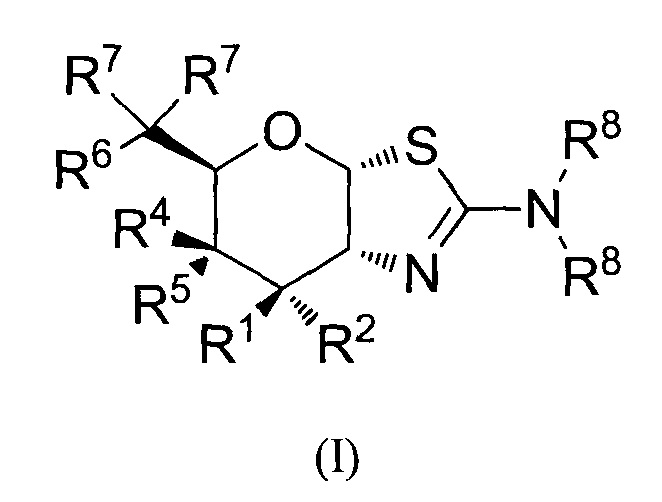
[0044] Как указано в формуле (I): R1 и R2 могут обозначать H, или R1 может обозначать H и R2 может обозначать F, или R1 может обозначать F и R2 может обозначать H, или R1 может обозначать OR3 и R2 может обозначать H; каждая R3 может обозначать независимо H или C1-6 ацил; R4 может обозначать H и R5 может обозначать OR3, или R4 может обозначать OR3 и R5 может обозначать H; R6 может обозначать H, F or OR3; каждая R7 может обозначать независимо H or F; каждая R8 может быть независимо выбрана из группы, состоящей из: H, C1-6 алкила, C3-6 алкенила, C3-6 алкинила и C1-6 алкокси, где C1-6 алкил, C3-6 алкенил, C3-6 алкинил и C1-6 алкокси могут быть необязательно замещены числом заместителей от одного до максимального одним или более из фтор-, OH или метила, или две группы R8 могут быть соединены вместе с атомом азота, с которым они связаны, для образования кольца, причем указанное кольцо необязательно независимо замещено числом заместителей от одного до максимального одним или более из фтор-, OH или метила; при условии, что когда R6 обозначает OR3, каждая R7 обозначает H; и при условии, что или R1, или R6 не обозначает OR3.
[0045] В некоторых вариантах осуществления, R1, как указано в формуле (I), может обозначать H, F, OH или OC(O)R9, где R9 может обозначать H, C1-6 алкил или C3-6 циклоалкил. В некоторых вариантах осуществления, R1 может обозначать H, F или OH.
[0046] В некоторых вариантах осуществления, R2, как указано в формуле (I), может обозначать H или F.
[0047] В некоторых вариантах осуществления, R3, как указано в формуле (I), может обозначать H или C(O)R9, где R9 может обозначать H, C1-6 алкил или C3-6 циклоалкил. В некоторых вариантах осуществления, R3 может обозначать H.
[0048] В некоторых вариантах осуществления, R4, как указано в формуле (I), может обозначать H, OH или OC(O)R9, где R9 может обозначать H, C1-6 алкил или C3-6 циклоалкил. В некоторых вариантах осуществления, R4 может обозначать H или OH.
[0049] В некоторых вариантах осуществления, R5, как указано в формуле (I), может обозначать H, OH или OC(O)R9, где R9 может обозначать H, C1-6 алкил или C3-6 циклоалкил. В некоторых вариантах осуществления, R5 может обозначать H или OH.
[0050] В некоторых вариантах осуществления, R6, как указано в формуле (I), может обозначать H, F, OH или OC(O)R9, где R9 может обозначать H, C1-6 алкил или C3-6 циклоалкил. В некоторых вариантах осуществления, R6 может обозначать H, F или OH.
[0051] В некоторых вариантах осуществления, каждая R7, как указано в формуле (I), может обозначать независимо H или F.
[0052] В некоторых вариантах осуществления, каждая R8, как указано в формуле (I), может обозначать независимо H, C1-6 алкил, C3-6 алкенил, C3-6 алкинил или Ci-6 алкокси, где C1-6 алкил, C3-6 алкенил, C3-6 алкинил или Ci-6 алкокси могут быть необязательно замещены числом заместителей от одного до максимального одним или более из фтор-, OH или метила. В некоторых вариантах осуществления, каждая R8 может обозначать независимо H, CH3, CH2CH3, (CH2)2CH3, CH2CH=CH2, or CH2C≡CH.
[0053] В некоторых вариантах осуществления, две группы R8, как указано в формуле (I), могут быть соединены вместе с атомом азота, с которым они связаны, для образования кольца, причем указанное кольцо необязательно независимо замещено числом заместителей от одного до максимального одним или более из фтор-, OH или метила.
[0054] В некоторых вариантах осуществления, NR82, как указано в формуле (I), может быть необязательно замещена
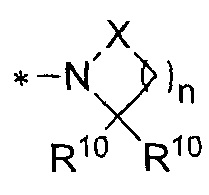 , где X может обозначать CR102, NR10, O, C=O, O(C=O), (C=O)O, NR10(C=O), или (C=O)NR10; где каждая R10 может обозначать независимо H или C1-4 алкил; и n может обозначать целое число от 0 до 3. В некоторых вариантах осуществления, NR82 может быть необязательно замещена 1-азиридинилом, 1-азетидинилом, 1-пирролидинилом, 1-пиперидинилом, морфолин-4-илом, 1-пиперазинилом, азетидин-2-он-l-илом, пирролдин-2-он-l-илом или пиперид-2-он-l-илом. В некоторых вариантах осуществления, NR82 может обозначать
, где X может обозначать CR102, NR10, O, C=O, O(C=O), (C=O)O, NR10(C=O), или (C=O)NR10; где каждая R10 может обозначать независимо H или C1-4 алкил; и n может обозначать целое число от 0 до 3. В некоторых вариантах осуществления, NR82 может быть необязательно замещена 1-азиридинилом, 1-азетидинилом, 1-пирролидинилом, 1-пиперидинилом, морфолин-4-илом, 1-пиперазинилом, азетидин-2-он-l-илом, пирролдин-2-он-l-илом или пиперид-2-он-l-илом. В некоторых вариантах осуществления, NR82 может обозначать  или
или 
[0055] В определенных вариантах осуществления изобретения, соединения в соответствии с формулой (I) включают соединения, описанные в таблице 1.
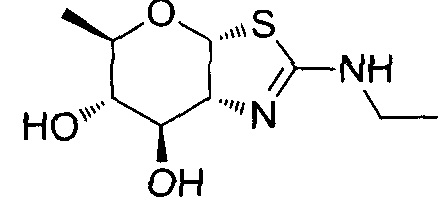
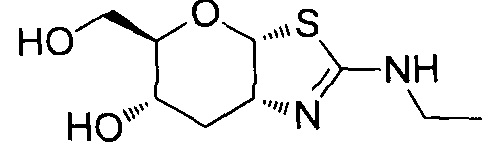
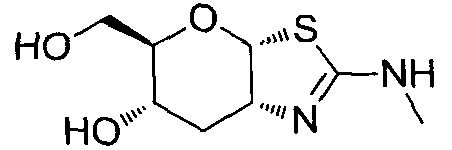
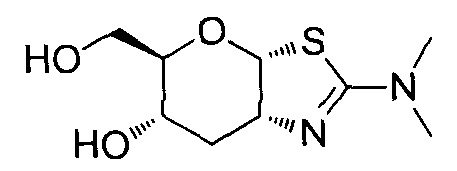
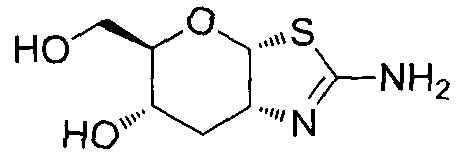
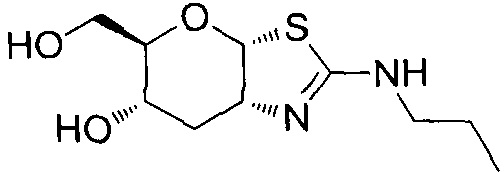
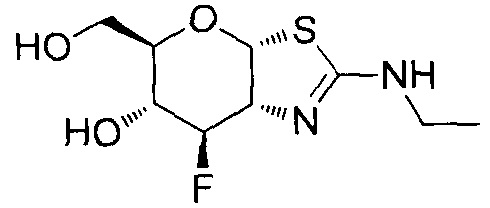
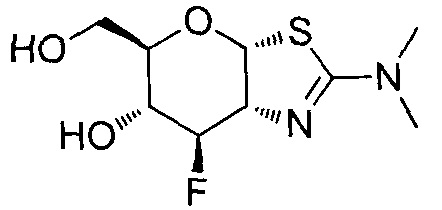
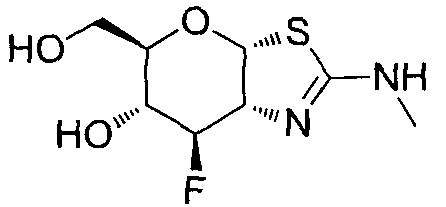
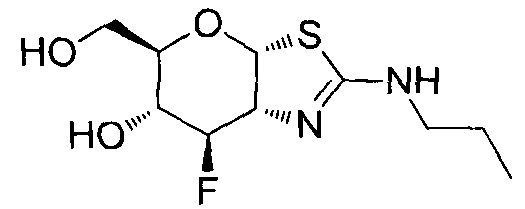
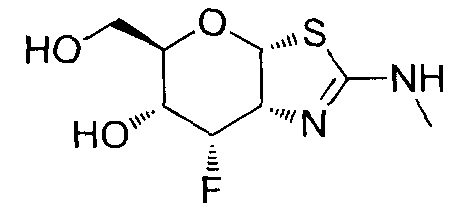
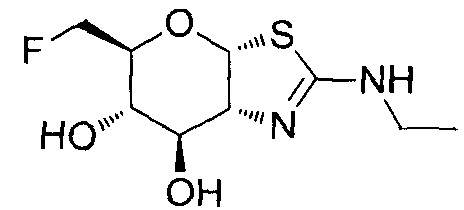
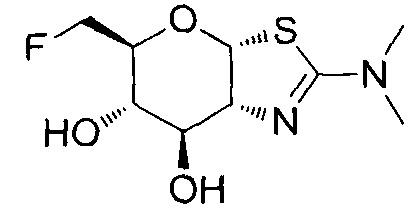
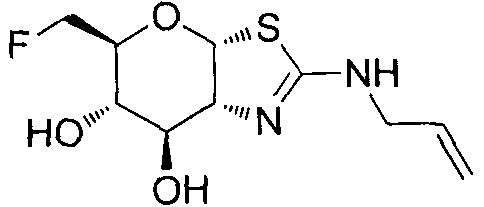
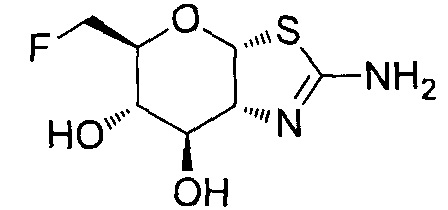
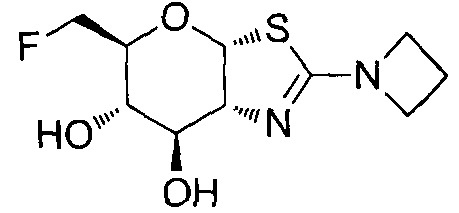
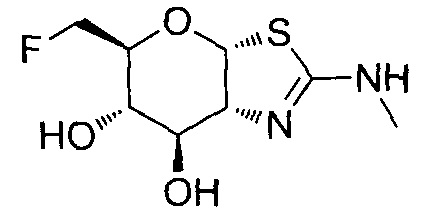
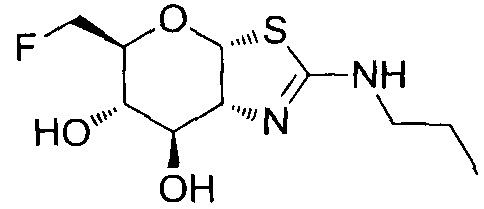
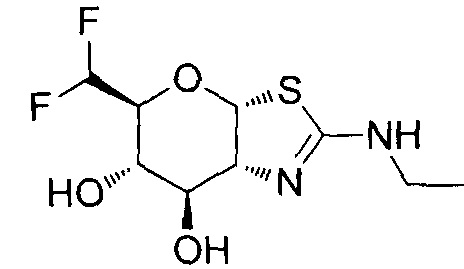
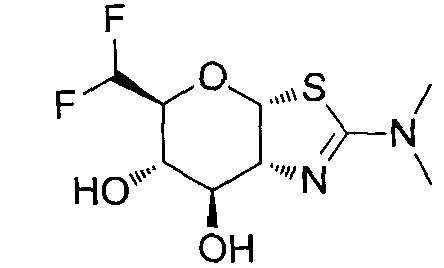
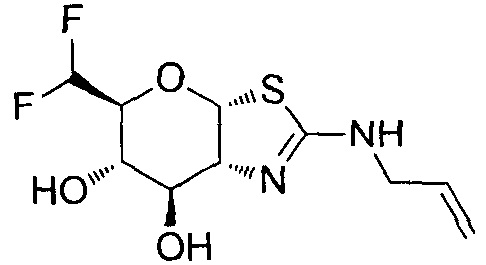
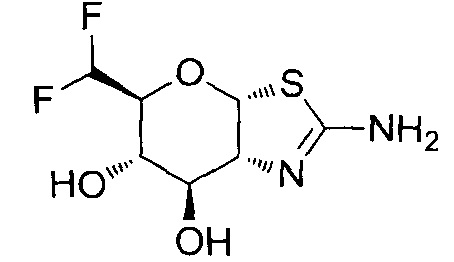
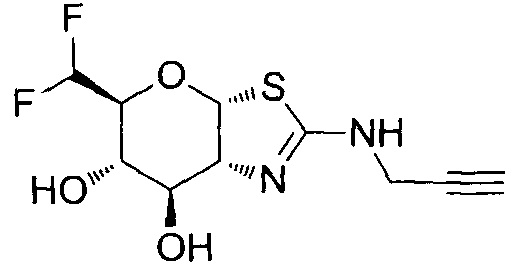
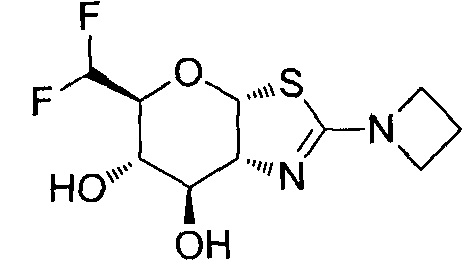
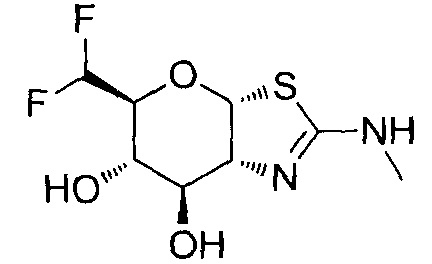
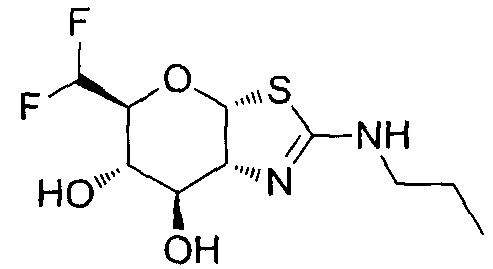
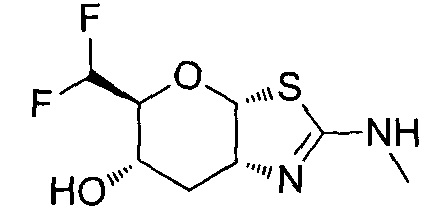
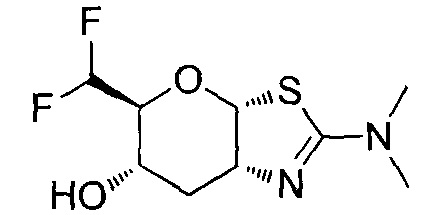
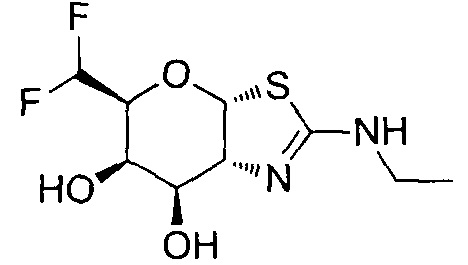
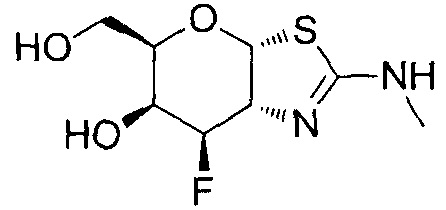
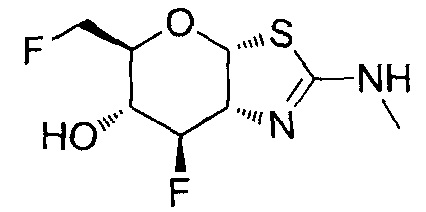
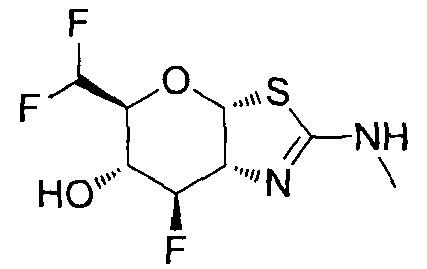
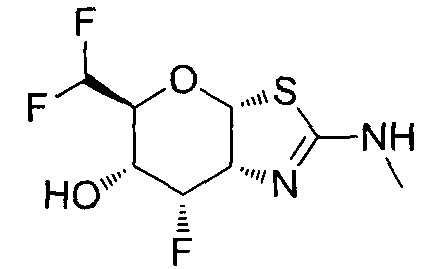
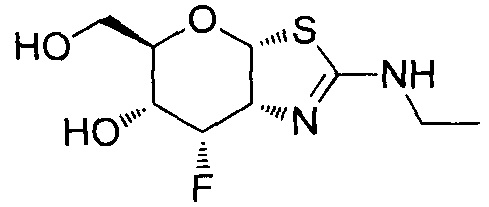
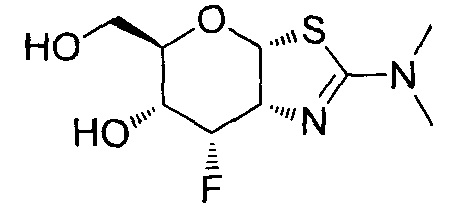
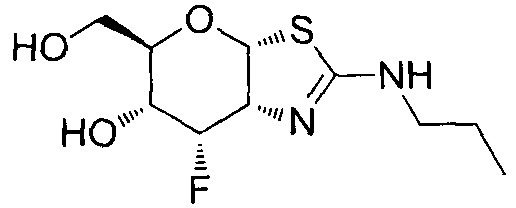
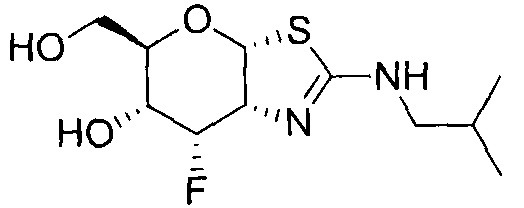
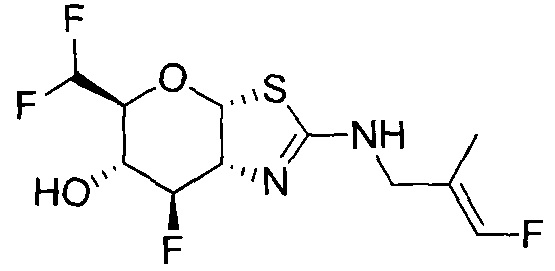
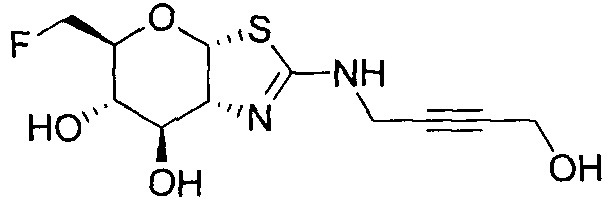
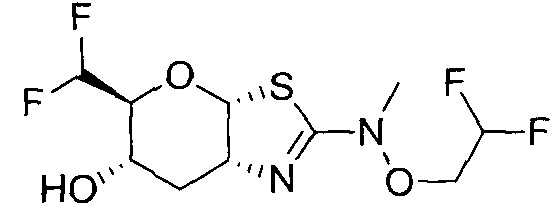
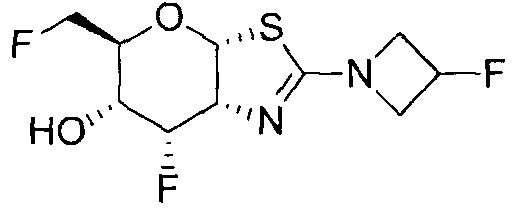
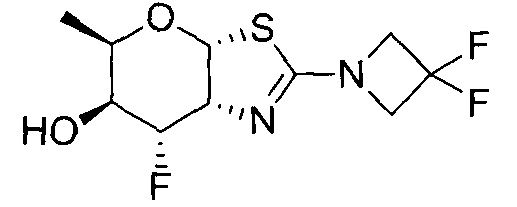
[0056] Специалистам в данной области понятно, что указанная выше формула (I) может быть также альтернативно представлена следующим образом:
[0057] Пока контекст ясно не требует иного, используемые в настоящем описании формы единственного числа с неопределенным и определенным артиклями включают соответствующие формы множественного числа. Например, «соединение» относится к одному или более таких соединений, тогда как «фермент» включает конкретный фермент, а также других членов семейства и их эквиваленты, как известно специалистам в данной области.
[0058] По всему тексту настоящей заявки предусматривается, что термин «соединение» или «соединения» относится к соединениям, включая производные с защищенными ацильными группами и фармацевтически приемлемые соли соединений, предшественники и производные. Изобретение также включает пролекарства соединений, фармацевтические композиции, содержащие соединения и фармацевтически приемлемый носитель, и фармацевтические композиции, содержащие пролекарства и фармацевтически приемлемый носитель.
[0059] Соединения по настоящему изобретению могут содержать один или более асимметричных центров, и, таким образом, встречаются в виде рацематов и рацемических смесей, одиночных энантиомеров, диастреомерных смесей и отдельных диастереомеров. Могут присутствовать дополнительные асимметричные центры, в зависимости от природы различных заместителей на молекуле. Каждый такой асимметричный центр независимо продуцирует два оптических изомера и подразумевается, что все возможные оптические изомеры и диастереомеры в смесях и в виде чистых или частично очищенных соединений включены в объем настоящего изобретения. Любые формулы, структуры или названия соединений, описанных в настоящей заявке, которые не уточняют конкретную стереохимию, предназначены для включения любого и всех существующих изомеров, как описано выше, и их смесей в любой пропорции. Когда стереохимия определена, то изобретение предназначено для включения указанного конкретного изомера в чистой форме или как часть смеси с другими изомерами в любой пропорции.
[0060] Определенные группы могут быть необязательно замещены, как описано в настоящей заявке. Подходящие заместители включают: H, алкил(C1-6), алкенил(C2-6) или алкинил(C2-6), каждый из которых может необязательно содержать один или более гетероатомов, выбранных из O, S, P, N, F, Cl, Br, I или B, и каждый из которых может быть дополнительно замещен, например, =O; или необязательно замещенные формы ацила, алкила, алкенила и алкинила и их формы, которые содержат гетероатомы в алкильной, алкенильной или алкинильных составляющих. Другие подходящие заместители включают =O, =NR, галоид, CN, CF3, CHF2, NO2, OR, SR, NR2, N3, COOR и CONR2, где R обозначает H или алкил, циклоалкил, алкенил или алкинил. Когда замещенный атом представляет собой C, то заместители могут включать, в дополнение к перечисленным выше заместителям, галоид, OOCR, NROCR, где R обозначает H или указанный выше заместитель.
[0061] Например, в указанной выше формуле (I), каждая C1-6 ацильная, C1-6 алкильная, C3-6 алкенильная, C3-6 алкинильная или C1-6 алкокси группа может быть независимо необязательно замещена одним или более заместителями. В некоторых вариантах осуществления каждая C1-6 ацильная, C1-6 алкильная, C3-6 алкенильная, C3-6 алкинильная или C1-6 алкокси группа в формуле (I) может быть замещена одним или более неорганическими заместителями; фосфорилом; галоидом; =O; =NR9; OR; C1-6 алкилом или C2-6 алкенилом, необязательно содержащими один или более P, N, O, S, N, F, Cl, Br, I или B, и необязательно замещена галоидом; CN; необязательно замещенным карбонилом; NR92; C=NR9; необязательно замещенным карбоциклическим или гетероциклическим кольцом, где R9 может обозначать H, C1-6 алкил или C3-6 циклоалкил. В альтернативных вариантах осуществления, каждая C1-6 ацильная, C1-6 алкильная, C3-6 алкенильная, C3-6 алкинильная или C1-6 алкокси группа может быть независимо необязательно замещена числом заместителей от одного до максимального одним или более из фтор-, OH, или метила. В альтернативных вариантах осуществления, каждая необязательно замещенная C1-6 ацильная, C1-6 алкильная, C3-6 алкенильная, C3-6 алкинильная или C1-6 алкокси группа формулы (I) может независимо включать один или более гетероатомов, выбранных из P, O, S, N, F, Cl, Br, I или B.
[0062] «Алкил» относится к группе с прямой или разветвленной цепью, состоящей исключительно из атомов углерода или водорода, не содержащей насыщение, и включающей, от одного до десяти атомов углерода, например, 1, 2, 3, 4, 5, 6, 7, 8, 9 или 10 атомов углерода, и которая присоединена к остальной части молекулы одинарной связью. Пока в описании нет конкретных иных указаний, алкильная группа может быть необязательно замещена одним или более заместителями, как описано в настоящей заявке. Пока в настоящем описании нет конкретных иных указаний, подразумевается, что замещение может происходить на любом атоме углерода алкильной группы.
[0063] «Алкенил» относится к группе с прямой или разветвленной цепью, состоящей исключительно из атомов углерода и водорода, содержащей по меньшей мере одну двойную связь, и включающей, например, от двух до десяти атомов углерода, например, 2, 3, 4, 5, 6, 7, 8, 9 или 10 атомов углерода, и которая присоединена к остальной части молекулы одинарной связью или двойной связью. Пока в описании нет конкретных иных указаний, алкенильная группа может быть необязательно замещена одним или более заместителями, как описано в настоящей заявке. Пока в настоящем описании нет конкретных иных указаний, подразумевается, что замещение может происходить на любом атоме углерода алкенильной группы.
[0064] «Алкинил» относится к группе с прямой или разветвленной цепью, состоящей исключительно из атомов углерода и водорода, содержащей по меньшей мере одну тройную связь, и включающей от двух до десяти атомов углерода. Пока в описании нет конкретных иных указаний, алкинильная группа может быть необязательно замещена одним или более заместителями, как описано в настоящей заявке.
[0065] «Арил» относится к фенильной группе, ароматическому кольцу, включающему 6 атомов углерода. Пока в настоящем описании нет иных конкретных указаний, термин «арил» предназначен для включения арильных групп, необязательно замещенных одним или более заместителями, как описано в настоящей заявке.
[0066] «Гетероарил» относится к одной ароматической кольцевой группе, содержащей один или более гетероатомов в кольце, например N, O, S, включая, например, 5-6 членов, например, 5 или 6 членов. Примеры гетероарильных групп включают фуран, тиофен, пиррол, оксазол, тиазол, имидазол, пиразол, изоксазол, изотиазол, 1,2,3-оксадиазол, 1,2,3-триазол, 1,2,4-триазол, 1,3,4-тиадиазол, тетразол, пиридин, пиридазин, пиримидин, пиразин, 1,3,5-триазин, имидазол. Пока в настоящем описании нет иных конкретных указаний, термин «гетероарил» предназначен для включения гетероарильных групп, необязательно замещенных одним или более заместителей, как описано в настоящей заявке.
[0067] "Ацил" относится к группе формулы -C(O)Ra, где Ra обозначает C1-10 алкильную или C3-15 циклоалкильную группу, как описано в настоящей заявке. Алкильная или циклоалкильная группа(ы) могут быть необязательно замещены, как описано в настоящей заявке.
[0068] «Алкокси» относится к группе формулы -ORb, где Rb обозначает C1-10 алкильную группу, как описано в настоящей заявке. Алкильная группа(ы) может быть необязательно замещена, как описано в настоящей заявке.
[0069] «Циклоалкил» относится к устойчивой моновалентной моноциклической, бициклической или трициклической углеводородной группе, состоящей из атомов углерода и водорода, имеющей, например, от 3 до 15 атомов углерода, и которая является насыщенной и присоединена к остальной части молекулы одинарной связью. Пока в настоящем описании нет иного конкретного указания, термин «циклоалкил» предназначен для включения циклоалкильных групп, которые необязательно замещены, как описано в настоящей заявке.
[0070] «Галоид» относится к бром-, хлор-, фтор-, йод- и т.д. В некоторых вариантах осуществления, подходящие галогены включают фтор или хлор.
[0071] В некоторых вариантах осуществления, две группы R8, как указано в формуле (I), могут быть соединены вместе с атомом азота, с которым они связаны, для образования кольца. В этих вариантах осуществления, «кольцо» относится к устойчивой, содержащей азот моноциклической группе, имеющей от 3 до 6 членов, которая может быть насыщенной или мононенасыщенной. В альтернативных вариантах осуществления, кольцо может включать атомы C, H и N. В других вариантах осуществления, кольцо может включать гетероатомы, например O и S. Примеры кольца в этих вариантах осуществления включают 1-азиридинил, 1-азетидинил, 1-пирролидинил, 2,5-дигидро-1H-пиррол-l-ил, 1-пиперидинил, 1,2,3,6-тетрагидропиридин-l-ил, морфолин-4-ил, тиоморфолин-4-ил, 1-пиперизинил, азетидин-2-он-l-ил, пирролидин-2-он-l-ил, пиперид-2-он-l-ил, 1,2-оксазетидин-2-ил, изоксазолидин-2-ил и 1,2-оксазинан-2-ил. Кольцо в этих вариантах осуществления может быть необязательно замещено, как описано в настоящей заявке.
[0072] «Необязательное» или «необязательно» означает, что описанное в последующем событие обстоятельств может произойти или не произойти, и что описание включает случаи, когда указанное событие происходит один или более раз, и случаи, в которых оно не происходит. Например, «необязательно замещенный алкил» значит, что алкильная группа может быть или не быть замещенной, и что описание включает и замещенные алкильные группы, и алкильные группы, не имеющие замещений, и что указанные алкильные группы могут быть замещены один или более раз. Примеры необязательно замещенных алкильных групп включают без ограничения метил, этил, пропил и т.д., и включая циклоалкилы, такие как циклопропил, циклобутил, циклопентил, циклогексил, циклогептил и т.д.; примеры необязательно замещенных алкенильных групп включают аллил, кротил, 2-пентенил, 3-гексенил, 2-циклопентенил, 2-циклогексенил, 2-циклопентенилметил, 2-циклогексенилметил и т.д. В некоторых вариантах осуществления, необязательно замещенные алкильные и алкенильные группы включают C1-6 алкилы или алкенилы.
Терапевтические показания
[0073] Изобретение относится к способам лечения состояний, которые модулируются, прямо или косвенно, ферментом O-GlcNAcase, или модифицированными O-GlcNAc уровнями белка, например, состояния, при котором благоприятный эффект оказывает ингибирование фермента O-GlcNAcase или повышение уровней модифицированного O-GlcNAc белка. Такие состояния включают без ограничения глаукому, шизофрению, таупатии, такие как болезнь Альцгеймера, нейродегенеративные заболевания, сердечнососудистые заболевания, заболевания, связанные с воспалением, заболевания, связанные с подавлением иммунитета и различными формами рака. Одно или более соединений по изобретению могут также применяться при лечении заболеваний или расстройств, связанных с дефицитом или сверхэкспрессией O-GlcNAcase или накоплением или истощением запасов O-GlcNAc, или любого заболевания или расстройства, реагирующего на терапию ингибированием гликозидазы. Такие заболевания и расстройства включают без ограничения глаукому, шизофрению, нейродегенеративные расстройства, такие как болезнь Альцгеймера (AD) или рак. Такие заболевания и расстройства могут также включать заболевания и расстройства, связанные с накоплением или дефицитом фермента OGT. Включен также способ защиты или лечения клеток-мишеней, экспрессирующих белки, которые модифицированы остатками O-GlcNAc, нарушение регуляции которых приводит к заболеванию или патологии. Используемый в настоящем описании термин «лечение» включает лечение, предотвращение и облегчение течения.
[0074] В альтернативных вариантах осуществления, изобретение относится к способам увеличения или повышения уровней модификации белка O-GlcNAc у животных в области ветеринарии и у людей. Этот подъем уровней O-GlcNAc может применяться для предотвращения или лечения болезни Альцгеймера; предотвращения или лечения других нейродегенеративных заболеваний (например, болезни Паркинсона, болезни Хантингтона); обеспечения нейропротективных эффектов; предотвращения повреждения сердечной ткани и лечения заболеваний, связанных с воспалением или иммуносупрессией.
[0075] В альтернативных вариантах осуществления, изобретение относится к способам селективного ингибирования фермента O-GlcNAcase у животных в области ветеринарии и у людей.
[0076] В альтернативных вариантах осуществления, изобретение относится к способам ингибирования фосфорилирования полипептидов тау или ингибирования образования NFT у животных в области ветеринарии и у людей. Соответственно, соединение по изобретению может применяться для исследования и лечения болезни Альцгеймера и других таупатий.
[0077] В целом, способы по изобретению осуществляются введением соединения в соответствии с изобретением нуждающемуся в нем индивиду, или обеспечением контакта клетки или образца с соединением в соответствии с изобретением, например, фармацевтической композиции, содержащей терапевтически эффективное количество соединения в соответствии с формулой (I). Конкретнее, они могут применяться при лечении расстройства, при котором вовлекается регуляция модификация белка O-GlcNAc, или любого состояния, как описано в настоящей заявке. Представляющие интерес патологические состояния включают болезнь Альцгеймера (AD) и родственные нейродегенеративные таупатии, при которых в патогенез заболевания вовлечено патологическое гиперфосфорилирование связанного с микротрубочками белка тау. В некоторых вариантах осуществления, соединение может применяться для блокировки гиперфосфорилирования тау поддержанием повышенных уровней O-GlcNAc на тау, посредством этого, обеспечивая терапевтический благоприятный эффект.
[0078] Эффективность соединения при лечении патологии, связанной с накоплением токсических видов тау (например, болезни Альцгеймера и других таупатий) может быть подтверждена тестированием способности соединения блокировать образование токсических видов тау на принятых клеточных118-120 и/или экспериментальных моделях заболеваний у животных.32,33
[0079] Таупатии, которые можно лечить соединением по изобретению, включают: болезнь Альцгеймера, боковой амиотрофический склероз (ALS), боковой амиотрофический склероз с когнитивным нарушением (ALSci), деменцию с аргирофильными зернами, болезнь Блюита, кортикобазальную дегенерацию (CBD), деменцию боксеров, диффузные нейрофибриллярные сплетения с обызвествлением, синдром Дауна, семейную британскую деменцию, семейную датскую деменцию, лобно-височную деменцию с паркинсонизмом, сцепленным с хромосомой 17 (FTDP-17), болезнь Герстманна-Штрауслера-Шейнкера, гваделупский паркинсонизм, болезнь Галлевордена-Спаца (нейродегенерацию с накоплением железа в мозге 1 типа), множественную системную атрофию, миотоническую дистрофию, болезнь Найманна-Пика (типа C), паллидо-понто-нигральную дегенерацию, комплекс паркинсонизма-деменции Гуама, болезнь Пика (PiD), постэнцефалический паркинсонизм (PEP), прионные болезни (включая болезнь Крейтцфелдта-Якоба (CJD), вариантную болезнь Крейтцфелдта-Якоба (vCJD), летальную семейную бессонницу и куру), прогрессирующий суперкортикальный глиоз, прогрессирующий супрануклеарный паралич (PSP), синдром Ричардсона, подострый склерозирующий панэнцефалит, деменцию только со сплетениями, и глаукому.
[0080] Одно или более соединений по настоящему изобретению могут также применяться при лечении состояний, связанных с повреждением ткани или стрессом, стимуляции клеток или содействия дифференциации клеток. Соответственно, в некоторых вариантах осуществления, соединение по настоящему изобретению может применяться для обеспечения терапевтического благоприятного эффекта при разнообразных состояниях или медицинских процедурах, вовлекающих напряжение сердечной ткани, включая без ограничения: ишемию; кровотечение; гиповолемический шок; инфаркт миокарда; инвазивную кардиологическую процедуру; аортокоронарное шунтирование; фибринолитическую терапию; ангиопластику и размещение стента.
[0081] Эффективность соединения при лечении патологии, связанной с клеточным стрессом (включая ишемию, кровотечение, гиповолемический шок, инфаркт миокарда и другие сердечнососудистые расстройства), может быть подтверждена тестированием способности соединения предотвратить клеточное повреждение в принятых анализах клеточного стресса,105,116,117 и для предотвращения повреждения и стимуляции функционального восстановления на экспериментальных моделях у животных ишемии-реперфузии70,114 и травмы-кровотечения.72,112,115
[0082] Соединения, которые селективно ингибируют активность O-GlcNAcase, могут применяться для лечения заболеваний, которые связаны с воспалением, включая без ограничения воспалительные или аллергические заболевания, такие как астма, аллергический ринит, связанные с гиперчувствительностью легочные заболевания, связанный с гиперчувствительностью пневмонит, эозинофильные пневмонии, гиперчувствительность замедленного типа, атеросклероз, интерстициальное легочное заболевание (ILD) (например, идиопатический легочный фиброз или ILD, связанное с ревматоидным артритом, системную красную волчанку, анкилозирующий спондилит, системный склероз, сидром Шегрена, полимиозит или дерматомиозит); системную анафилаксию или реакции гиперчувствительности, лекарственные аллергии, аллергии к укусам насекомых; аутоиммунные заболевания, такие как ревматоидный артрит, псориатический артрит, рассеянный склероз, синдром Жиллена-Барре, системную красную волчанку, генерализованную миастению, гломерулонефрит, аутоиммунный тироидит, отторжение трансплантата, включая отторжение аллотрансплантата или болезнь трансплантат против хозяина; воспалительные кишечные заболевания, такие как болезнь Крона и язвенный колит; спондилоартропатии; склеродермию; псориаз (включая псориаз, опосредованный T-клетками) и воспалительные дерматозы, такие как дерматит, экзема, атопический дерматит, аллергический контактный дерматит, сыпь; васкулит (например, некротизирующий, кожный и связанный гиперчувствительностью васкулит); эозинофильный миозит, эозинофильный фасциит и различные формы рака.
[0083] Кроме того, соединения, которые воздействуют на уровни модификации белка O-GlcNAc, могут применяться для лечения заболеваний, связанных с иммуносупрессией, таких как индивиды, получающие химиотерапию, лучевую терапию, усиленного заживления ран и лечения ожогов, лечения по поводу аутоиммунных заболеваний, или другой лекарственной терапии (например, терапии кортикостероидами) или в комбинации с обычными лекарственными препаратами, применяемыми при лечении аутоиммунных заболеваний и отторжения трансплантата, которое вызывает иммуносупрессию; или иммуносупрессии, вследствие врожденного дефицита функции рецепторов или других причин.
[0084] Одно или более из соединений по изобретению могут применяться для лечения нейродегенеративных заболеваний, включая болезнь Паркинсона и болезнь Хантингтона. Другие состояния, которые можно лечить, представляют собой те, которые запускаются, подвергаются воздействию или любым другим образом коррелируют с уровнями посттрансляционной модификации O-GlcNAc на белке. Ожидают, что одно или более соединений по настоящему изобретению могут применяться для лечения таких состояний и, в частности без огранения, следующих, для которых была установлена связь уровней O-GlcNAc на белках: отторжение трансплантата, в частности без ограничения трансплантатов солидных органов, таких как трансплантаты сердца, легких, печени, почки и поджелудочной железы (например, аллотрансплантаты почки и легких); рак, в частности, без ограничения рак молочной железы, легких, предстательной железы, ободочной кишки, прямой кишки, мочевого пузыря, почек, яичников; а также неходжкинской лимфомы и меланомы; эпилепсии, боли, фибромиалгии или инсульта, например, для нейропротекции после инсульта.
Фармакологические и ветеринарные композиции, дозировки и введение
[0085] Фармацевтические композиции, включающие соединения в соответствии с изобретением, или для применения в соответствии с изобретением, предусматриваются как входящие в объем изобретения. В некоторых вариантах осуществления, изобретение относится к фармацевтическим композициям, включающим эффективное количество соединения формулы (I).
[0086] Соединения формулы (I) и их фармацевтически приемлемые соли, энантиомеры, сольваты и производные могут применяться, потому что они обладают фармакологической активностью у животных, включая людей. В некоторых вариантах осуществления, одно или более из соединений в соответствии с изобретением устойчивы в плазме при введении индивиду.
[0087] В некоторых вариантах осуществления, соединение в соответствии с изобретением или для применения в соответствии с изобретением может вводиться в комбинации с любыми другими активными средствами или фармацевтическими композициями, где такая комбинированная терапия может применяться для модуляции активности O-GlcNAcase, например, для лечения нейродегенеративных, воспалительных, сердечнососудистых или иммунорегуляторных заболеваний или любого состояния, описанного в настоящей заявке. В некоторых вариантах осуществления, соединение в соответствии с изобретением или для применения в соответствии с изобретением может использоваться в комбинации с одним или более средств, применяемых при профилактике или лечении болезни Альцгеймера. Примеры таких средств включают без ограничения,
• ингибиторы ацетилхолинэстеразы (AChEI), такие как Арицепт® (донепезил), экселон® (ривастигмин), разадин® (разадин ER®, реминил®, нивалин®, галантамин), когнекс® (такрин), димебон, гуперзин A, фенсерин, дебио-9902 SR (ZT-1 SR), занапезил (TAK 0147), ганстигмин, NP7557 и т.д.;
• антагонисты рецепторов NMDA (N-Метил-D-аспартата), такие как наменда® (аксура®, акатинол®, эбикса®, мемантин), димебон, SGS-742, нерамексан, дебио-9902 SR (ZT-1 SR) и т.д.;
• ингибиторы и/или модуляторы гамма-секретазы, такие как флуризан™ (таренфлурбил, MPC-7869, R-флурбипрофен), LY450139, MK 0752, E2101, BMS-289948, BMS-299897, BMS-433796, LY-411575, GSI-136, и т.д;
• ингибиторы бета-секретазы, такие как ATG-Z1, CTS-21166 и т.д.;
• активаторы альфа-секретазы, такие как NGX267, и т.д;
• ингибиторы агрегации и/или фибриллизации амилоида-β, такие как алцхемед (3APS, трамипрозат, 3-амино-l-пропансульфоновую кислоту), AL-108, AL-208, AZD-103, PBT2, цереакт, ONO-2506PO, PPI-558 и т.д.;
• ингибиторы агрегации тау, такие как метиленовый синий, и т. д.;
• стабилизаторы микротрубочек, такие как AL-108, AL-208, паклитаксел, и т.д.;
• ингибиторы RAGE (рецепторов конечных продуктов позднего гликирования), такие как TTP488, и т.д.;
• антагонисты рецепторов 5-HTla, такие как ксалипроден, лекозотан, и т.д.;
• антагонисты рецепторов 5-HT4, такие как PRX-03410, и т.д.;
• Ингибиторы киназы, такие как SRN-003-556, амфуриндамид, LiCl, AZD1080, NP031112, SAR-502250, и т.д.;
• гуманизированные моноклональные антитела к Αβ, такие как бапинейзумаб (AAB-001),LY2062430, RN1219, ACU-5A5, и т.д.;
• амилоидные вакцины, такие как AN-1792, ACC-001, и т.д.;
• нейропротективные средства, такие как церебролезин, AL-108, AL-208, гуперзин A, и т.д.;
• антагонисты кальциевых каналов типа L, такие как MEM-1003, и т.д.;
• антагонисты никотиновых рецепторов, такие как AZD3480, GTS-21, и т.д.;
• агонисты никотиновых рецепторов, такие как MEM 3454, нефирацетам, и т.д.;
• агонисты активированных пролифераторами пероксисом гамма-рецепторов (PPAR), такие как авандиа® (росглитазон), и т.д.;
• ингибиторы фосфодиэстеразы IV (PDE4), такие как MK-0952, и т.д.;
• гормональная заместительная терапия, такая как эстроген (премарин), и т.д.;
• ингибиторы моноаминоксидазы (MAO), такие как NS2330, расагилин (азилект®), TVP-1012, и т.д.;
• модуляторы рецепторов AMPA (A-Амино-3-гидрокси-5-метил-4-изоксазолепропионовой кислоты), такие как ампалекс (CX 516), и т.д.;
• нервные факторы роста или стимуляторы NGF, такие как CERE-110 (AAV-NGF), T-588, T-817MA, и т.д.;
• средства, которые предотвращают высвобождение лютеинизирующего гормона (LH) гипофизом, такие как лейопролид (VP-4896), и т.д.;
• модуляторы рецепторов GABA (гамма-аминомасляной кислоты, ГАМК), такие как AC-3933, NGD 97-1, CP-457920, и т.д.;
• инверсионные агонисты бензодиазепиновых рецепторов, такие как SB-737552 (S-8510), AC-3933, и т.д.;
• средства, высвобождающие норадреналин, такие как T-588, T-817MA, и т.д.
[0088] Следует понимать, что комбинация соединений в соответствии с изобретением или для применения в соответствии с изобретением со средствами против болезни Альцгеймера, не должна ограничиваться примерами, описанными в настоящей заявке, но включает комбинацию с любым средством, применяемым для лечения болезни Альцгеймера. Комбинация соединений в соответствии с изобретением или для применения в соответствии с изобретением и других средств против болезни Альцгеймера может вводиться отдельно или в сочетании. Одно средство можно вводить перед, одновременно или после введения другого средства (средств).
[0089] В альтернативных вариантах осуществления, соединение может доставляться в виде «пролекарства» или защищенных форм, которые высвобождают соединение после введения индивиду. Например, соединение может нести защитную группу, которая отщепляется гидролизом в биологических жидкостях, например, в кровотоке, таким образом, высвобождая активное соединение, или окисляется или восстанавливается в биологических жидкостях для высвобождения соединения. Соответственно, «пролекарство» предназначено для указания соединения, которое может быть превращено в физиологических условиях или сольволизом в биологически активное соединение по изобретению. Таким образом, термин «пролекарство» относится к метаболическому предшественнику соединения по изобретению, которое является фармацевтически приемлемым. Пролекарство может быть неактивным при введении нуждающемуся в нем индивиду, но превращается in vivo в активное соединение по изобретению. Пролекарства обычно быстро трансформируются in vivo для выхода материнского соединения по изобретению, например, гидролизом в крови. Пролекарственное соединение часто обеспечивает преимущества растворимости, тканевой совместимости или отсроченного высвобождения у индивида.
[0090] Термин «пролекарство» также предназначен для включения любых ковалентно связанных носителей, которые высвобождают активное соединение по изобретению in vivo, когда такое пролекарство вводится индивиду. Пролекарства соединения по изобретению могут быть получены модификацией функциональных групп, присутствующих в соединении по изобретению, таким образом, что модификации расщепляются или обычной манипуляцией, или in vivo, в материнское соединение по изобретению. Пролекарства включают соединения по изобретению, где гидрокси, амино или меркапто группа связана с любой группой, которая, когда пролекарство соединения по изобретению вводится млекопитающему индивиду, расщепляется для образования соответственно свободной гидрокси, свободной амино или свободной меркапто группы. Примеры пролекарств включают без ограничения ацетатные, формиатные и бензоатные производные спирта и ацетамидных, формамидных и бензамидных производных аминофункциональных групп в одном или более соединений по изобретению и тому подобных.
[0091] Обсуждение пролекарств можно найти в руководствах «Smith and Williams' Introduction to the Principles of Drug Design,» H.J. Smith, Wright, Second Edition, London (1988); Bundgard, H., Design of Peodrugs (1985), pp. 7-9, 21-24 (Elsevier, Amsterdam); The Practice of Medicinal Chemistry, Camille G. Wermuth et al., Ch 31, (Academic Press, 1996); A Textbook of Drug Design and Development, P. Krogsgaard-Larson and H. Bundgaard, eds. Ch 5, pgs 113 191 (Harwood Academic Publishers, 1991); Higuchi, T., et al, «Pro-drugs as Novel Delivery Systems,» A.C.S. Symposium Series, Vol. 14; или в руководстве Bioreversible Carriers in Drug Design, ed. Edward B. Roche, American Pharmaceutical Association and Pergamon Press, 1987, которые все полностью включены в настоящее описание путем ссылки.
[0092] Подходящие пролекарственные формы одного или более соединений по изобретению включают варианты осуществления, при которых одна или более R3, как указано в формуле (I), обозначает (CO)R, где R обозначает необязательно замещенный алкил, алкенил, алкинил, арил или гетероарил. В этих случаях сложноэфирные группы могут быть гидролизированы in vivo (например, в биологических жидкостях), высвобождая активные соединения, в которых каждая R3 обозначает H. Предпочтительные пролекарственные варианты осуществления изобретения включают соединения формулы (I), где одна или более R3 обозначает C(O)CH3.
[0093] Соединения в соответствии с изобретением или для применения в соответствии с изобретением могут быть предоставлены отдельно или в комбинации с другими соединениями в присутствии липосомы, адъюванта или любого фармацевтически приемлемого носителя, разбавителя или эксципиента в форме, подходящей для введения индивиду, такому как млекопитающее, например, люди, крупный рогатый скот, овцы и т.д. При желании, лечение соединением в соответствии с изобретением может комбинироваться с более традиционными и существующими средствами лечения по терапевтическим показаниям, описанным в настоящей заявке. Соединения в соответствии с изобретением можно вводить хронически или периодически. «Хроническое» введение относится к введению соединения (соединений) непрерывным образом, в отличие от «острого» режима, с тем, чтобы поддерживать исходный терапевтический эффект в течение продолжительного периода времени. «Периодическое» введение представляет собой лечение, которое не проводится последовательно без перерыва, но скорее является циклическим по природе. Используемые в настоящем описании термины «введение», «вводимое» следует понимать как обозначающие доставку соединения по изобретению нуждающемуся в лечении индивиду.
[0094] «Фармацевтически приемлемый носитель, разбавитель или эксципиент» включает без ограничения любой адъювант, носитель, эксципиент, глянцеватель, подслащивающий агент, разбавитель, консервант, краситель, усилитель аромата, поверхностно-активное вещество, смачивающий агент, диспергирующий агент, суспендирующий агент, стабилизатор, изотонический агент, растворитель или эмульгатор, которые были утверждены для применения, например, Администрацией Пищевых Продуктов и Лекарственных Средств США или другим правительственным агентством как приемлемые для применения у людей и домашних животных.
[0095] Соединение по настоящему изобретению может вводиться в форме фармацевтически приемлемой соли. В таких случаях, фармацевтические композиции в соответствии с настоящим изобретением могут содержать соль такого соединения, предпочтительно, физиологически приемлемую соль, которые известны в данной области. В некоторых вариантах осуществления, термин «фармацевтически приемлемая соль», используемый в настоящем описании, означает активный ингредиент, содержащий соединения формулы I, применяемые в форме их соли, в частности, когда солевая форма придает активному ингредиенту улучшенные фармакокинетические свойства, по сравнению со свободной формой активного ингредиента или другой ранее описанной солевой формой.
[0096] «Фармацевтически приемлемая соль» включает и кислотно-аддитивные и основно-аддитивные соли. «Фармацевтически приемлемая кислотно-аддитивная соль» относится к тем солям, которые сохраняют биологическую эффективность и свойства свободных оснований, которые не являются биологически или иным образом нежелательными, и которые образованы с неорганическими кислотами, такими как хлористоводородная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и тому подобные, и органическими кислотами, такими как уксусная кислота, трифторуксусная кислота, пропионовая кислота, гликолевая кислота, пировиноградная кислота, щавелевая кислота, малеиновая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, винная кислота, лимонная кислота, бензойная кислота, коричная кислота, миндальная кислота, метансульфоновая кислота, этансульфоновая кислота, п-толуолсульфоновая кислота, салициловая кислота и тому подобные.
[0097] «Фармацевтически приемлемая основно-аддитивная соль» относится к тем солям, которые сохраняют биологическую эффективность и свойства свободных кислот, которые не являются биологически или иным образом нежелательными. Эти соли получают в результате добавления неорганического основания или органического основания к свободной кислоте. Соли, происходящие из неорганических оснований, включают без ограничения, соли натрия, калия, лития, аммония, кальция, магния, железа, цинка, меди, марганца, алюминия и тому подобные. Предпочтительными неорганическими солями являются соли аммония, натрия, калия, кальция и магния. Соли, происходящие из органических оснований, включают без ограничения соли первичных, вторичных и третичных аминов, замещенных аминов, включая естественно встречающиеся замещенные амины, циклические амины и основные ионообменные смолы, такие как изопропиламин, триметиламин, диэтиламин, триэтиламин, трипропиламин, этаноламин, 2-диметиламиноэтанол, 2-диэтиламиноэтанол, дициклогексиламин, лизин, аргинин, гистидин, кофеин, прокаин, гидрабамин, холин, бетаин, этилендиамин, глюкозамин, метилглюкамин, теобромин, пурины, пиперазин, пиперидин, N-этилпиперидин, аминовые смолы и тому подобные. Особенно предпочтительными органическими основаниями являются изопропиламин, диэтиламин, этаноламин, триметиламин, дициклогексиламин, холин и кофеин.
[0098] Таким образом, термин «фармацевтически приемлемая соль» охватывает все приемлемые соли, включая без ограничения ацетат, лактобионат, бензолсульфонат, лаурат, бензоат, малат, бикарбонат, малеат, бисульфат, манделат, битартрат, мезилат, борат, метилбромид, бромид, метилнитрит, эдетат кальция, метилсульфат, кампсилат, мукат, карбонат, напсилат, хлорид, нитрат, клавуланат, N-метилглюкамин, цитрат, соль аммония, дигидрохлорид, олеат, эдетат, оксалат, эдисилат, памоат (эмбонат), эстолат, пальмитат, эзилат, пантотенат, фумарат, фосфат/дифосфат, глюкептат, полигалактуронат, глюконат, салицилат, глутамат, стеарат, гликоллиларсанилат, сульфат, гексилрезорцинат, субацетат, гидрадамин, сукцинат, гидробромид, таннат, гидрохлорид, тартрат, гидроксинафтоат, теоклат, йодид, тозилат, изотионат, триэтйодид, лактат, паноат, валерат и тому подобные.
[0099] Фармацевтически приемлемые соли соединения по настоящему изобретению могут применяться в виде дозировки для модификации характеристик растворимости или гидролиза, или могут применяться в препаративных формах длительного высвобождения или пролекарства. Фармацевтически приемлемые соли соединения по настоящему изобретению могут также включать соли, образованные из катионов, таких как натрий, калий, алюминий, кальций, литий, магний, цинк, и из оснований, таких как аммиак, этилендиамин, N-метилглутамин, лизин, аргинин, орнитин, холин, N,N'-дибензилэтилендиамин, хлорпрокаин, диэтаноламин, прокаин, N-бензилфенэтиламин, диэтиламин, пиперазин, трис(гидроксиметил)аминометан и тетраметиламмоний гидроксид.
[00100] Фармацевтические препаративные формы обычно включают один или более носителей, приемлемых для способа введения препарата, осуществляется ли он инъекцией, ингаляцией, топическим введением, лаважем, или другими способами, подходящими для выбранного лечения. Подходящими носителями являются те, которые известны в данной области для таких способов введения.
[00101] Подходящие фармацевтические композиции могут составляться средствами, известными в данной области, и в соответствии с путем их введения и дозой, определяемыми опытным специалистом в данной области. Для парентерального введения, соединение может быть растворено в стерильной воде или солевом растворе или в фармацевтически приемлемой основе, используемой для введения нерастворимых в воде соединений, таких как основы, используемые для витамина K. Для энтерального введения соединение может вводиться в таблетке, капсуле или в растворенном виде в жидкой форме. Таблетка или капсула может быть покрыта энтеросолюбильным покрытием или представлена в виде препаративной формы для продолжительного высвобождения. Известно много подходящих препаративных форм, включая полимерные или белковые микрокапсулы, инкапсулирующие подлежащее высвобождению соединение, мази, гели, гидрогели или растворы, которые могут использоваться топически или местно для введения соединения. Трансдермальная система или имплантат длительного высвобождения могут использоваться для обеспечения высвобождения в течение продолжительного периода времени. Множество технологий, известных опытным специалистам в данной области, описаны в руководстве Remington: the Science & Practice of Pharmacy by Alfonso Gennaro, 20th ed., Williams & Wilkins, (2000). Препаративные формы для парентерального введения могут, например, содержать эксципиенты, полиалкиленгликоли, такие как полиэтиленгликоль, масла растительного происхождения или гидрогенизированные нафталины. Биосовместимый, биоразлагаемый лактидный полимер, сополимер лактида/гликолида или сополимеры полиоксиэтилена-полиоксипропилена могут использоваться для регулирования высвобождения соединения. Другие потенциально полезные системы парентеральной доставки для модуляторных соединений включают частицы сополимера этилена-винилацетата, осмотические насосы, имплантируемые инфузионные устройства и липосомы. Препаративные формы для ингаляции могут содержать эксципиенты, например, лактозу, или могут представлять собой водные растворы, содержащие, например, простой полиоксиэтилен-9-лауриловый эфир, гликохолат и дезоксихолат, или могут представлять собой масляные растворы для введения в форме носовых капель или в виде геля.
[00102] Соединение или фармацевтическая композиция в соответствии с настоящим изобретением может вводиться пероральным или не пероральным путем, например, внутримышечной, внутрибрюшинной, внутривенной, интрацистернальной инъекцией или инфузией, трансдермальным путем или через слизистые оболочки. В некоторых вариантах осуществления, соединение или фармацевтическая композиция в соответствии с настоящим изобретением или для применения в настоящем изобретении может вводиться посредством медицинского устройства или приспособления, такого как имплантат, трансплантат, протез, стент, и т.д. Могут быть сконструированы имплантаты, которые предназначены для содержания и высвобождения таких соединений или композиций. Примером мог бы быть имплантат, выполненный из полимерного материала, приспособленный для высвобождения соединения в течение периода времени. Соединение может вводиться отдельно или в виде смеси с фармацевтически приемлемым носителем, например, в виде твердых препаративных форм, таких как таблетки, капсулы, гранулы, порошки и т.д.; жидких препаративных форм, таких как сиропы, инъекционные препараты и т.д.; растворы для инъекций, капли, суппозитории, пессарии. В некоторых вариантах осуществления, соединения или фармацевтические композиции в соответствии с настоящим изобретением или для применения в настоящем изобретении могут вводиться ингаляционным распылением, интраназальным, вагинальным, ректальным, сублингвальным или топическим путями и могут включаться в состав отдельно или вместе, в подходящие препаративные формы в виде стандартной лекарственной формы, содержащей обычные нетоксичные фармацевтически приемлемые носители, адъюванты и основы, целесообразные для каждого пути введения.
[00103] Соединение по изобретению может применяться для лечения животных, включая мышей, крыс, лошадей, крупный рогатый скот, овец, собак, кошек и обезьян. Однако соединение по изобретению может также применяться в других организмах, таких как птицы (например, куры). Одно или более соединений по изобретению могут также быть эффективными для применения у людей. Термин «индивид» или альтернативно используемый в настоящем описании термин «пациент» предназначен для ссылки на животное, предпочтительно, млекопитающее, наиболее предпочтительно, человека, который был объектом лечения, наблюдения или эксперимента. Однако одно или более соединений, способов и фармацевтических композиций по настоящему изобретению могут применяться при лечении животных. Соответственно, используемый в настоящем описании термин «индивид» может обозначать человека, не человекообразного примата, крысу, мышь, корову, лошадь, свинью, овцу, козу, собаку, кошку и т.д. У индивида может подозреваться наличие или риск наличия состояния, требующего модуляцию активности O-GlcNAcase.
[00104] «Эффективное количество» соединения в соответствии с изобретением включает терапевтически эффективное количество или профилактически эффективное количество. «Терапевтически эффективное количество» относится к количеству, эффективному в дозировках и в течение периодов времени, необходимых для достижения желательного терапевтического результата, такого как ингибирование O-GlcNAcase, подъем уровней O-GlcNAc, ингибирование фосфорилирования тау, или любого состояния, описанного в настоящей заявке. Терапевтически эффективное количество соединения может варьироваться в соответствии с такими факторами как патологическое состояние, возраст, пол и масса тела индивида, и способность соединения вызвать желательную реакцию у индивида. Схемы дозировки могут подбираться для обеспечения оптимальной терапевтической реакции. Терапевтически эффективное количество также представляет собой количество, при котором любые токсические или повреждающие эффекты соединение перевешиваются терапевтически благоприятными эффектами. «Профилактически эффективное количество» относится к количеству, эффективному в дозировках и в течение периодов времени, необходимых для достижения желательного профилактического результата, такого как ингибирование O-GlcNAcase, подъем уровней O-GlcNAc, ингибирование фосфорилирования тау, или любое состояние, описанное в настоящей заявке. Обычно, профилактическая доза применяется у индивидов перед или на ранней стадии заболевания, так что профилактически эффективное количество может быть меньше, чем терапевтически эффективное количество. Подходящий диапазон для терапевтически или профилактически эффективных количеств соединения может составлять любое целое число от 0,1 нМ до 0,1 М, от 0,1 нМ до 0,05 М, от 0,05 нМ до 15 мкМ или от 0,01 нМ до 10 мкМ.
[00105] В альтернативных вариантах осуществления, при лечении или профилактике состояний, которые требуют модуляции активности O-GlcNAcase, целесообразный уровень дозировки в целом составляет примерно от 0,01 до 500 мг на 1 кг массы тела индивида в день, и может вводиться в одной или множественных дозах. В некоторых вариантах осуществления, уровень дозировки составляет от примерно 0,1 до примерно 250 мг/кг в день. Следует понимать, что определенный уровень дозы и частота введения для любого конкретного пациента может варьироваться и зависит от разнообразных факторов, включая активность определенного используемого соединения, метаболическую устойчивость и длительность действия этого соединения, возраст, массу тела, общее состояние здоровья, пол, рацион питания, путь и время введения, скорость выведения, лекарственная комбинация, тяжесть конкретного состояния и пациент, получающий лечение.
[00106] Следует отметить, что величины дозировки могут варьироваться в зависимости от тяжести состояния, которое предстоит облегчить. Для любого конкретного индивида определенные схемы дозировки могут подбираться с течением времени в соответствии с индивидуальной потребностью и профессиональным суждением лица, вводящего или контролирующего введение композиций. Указанные в настоящем описании диапазоны дозировки являются лишь иллюстративными и не ограничивают диапазоны дозировки, которые может выбрать медицинский работник. Количество активного соединения (соединений) в композиции может варьироваться в соответствии с такими факторами как патологическое состояние, возраст, пол и масса тела индивида. Схемы дозировки могут подбираться для обеспечения оптимальной терапевтической реакции. Например, может вводиться один болюс, несколько дробных доз могут вводиться в течение времени, или доза может пропорционально уменьшаться или увеличиваться по показаниям острой необходимости терапевтической ситуации. Может иметь преимущество составление парентеральных композиций в стандартную лекарственную форму для легкости введения и однородности дозировки. В целом, соединения по изобретению должны применяться, не вызывая существенной токсичности, и, как описано в настоящей заявке, одно или более соединений проявляют подходящий профиль безопасности для терапевтического применения. Токсичность соединения по изобретению можно определить, используя стандартные методики, например, тестированием на клеточных культурах или экспериментальных животных и определением терапевтического индекса, т.е., соотношения между LD50 (дозой, летальной для 50% популяции) и LD100 (дозой, летальной для 100% популяции). Однако в некоторых ситуациях, таких как тяжелые патологические состояния, может быть необходимо введение существенных избытков композиций.
[00107] В соединениях общей формулы (I) атомы могут проявлять естественное изотопное относительное содержание или один или более из атомов может быть искусственно обогащен определенным изотопом, имеющим тот же атомный номер, но атомную массу или атомное число, отличные от атомной массы или атомного числа, преимущественно обнаруживаемых в природе. Настоящее изобретение предназначено для включения всех подходящих изотопных вариантов соединений генерической формулы (I). Например, различные изотопные формы водорода (H) включают протий (1H), дейтерий (2H) и тритий (3H). Протий является преобладающим изотопом водорода, обнаруживаемым в природе. Обогащение дейтерием может обеспечить определенные терапевтические преимущества, такие как увеличение периода полувыведения in vivo или уменьшение требуемых дозировок, или может обеспечить получение соединения, полезного в качестве стандарта для характеристики биологических образцов. Изотопно обогащенные соединения в пределах общей формулы (I) могут быть получены без ненужного экспериментирования обычными методиками, хорошо известными специалистам в данной области, или способами, аналогичными способам, описанным в схемах и примерах в настоящей заявке, с использованием соответствующих изотопно обогащенных реагентов и/или промежуточных соединений.
Другие способы применения и анализы
[00108] Соединение формулы (I) можно применять в скрининговых анализах для выявления соединений, которые модулируют активность гликозидазных ферментов, предпочтительно, фермента O-GlcNAcase. Способность тестируемого соединения ингибировать O-GlcNAcase-зависимое отщепление O-GlcNAc от модельного субстрата можно измерить, используя анализы, как описано в настоящей заявке, или известно среднему специалисту в данной области. Например, можно применять флуоресценцию или анализ на основе УФ, известные в данной области. «Тестируемое соединение» представляет собой любое естественно встречающееся или искусственно полученное химическое соединение. Тестируемые соединения могут включать без ограничения пептиды, полипептиды, синтезированные органические молекулы, естественно встречающиеся органические молекулы и молекулы нуклеиновых кислот. Тестируемое соединение может «конкурировать» с известным соединением, таким как соединение формулы (I), например, препятствуя ингибированию O-GlcNAcase-зависимого отщепления O-GlcNAc или препятствуя любой биологической реакции, вызванной соединением формулы (I).
[00109] В целом, тестируемое соединение может проявлять любую величину модуляции от 10% до 200% или более 500%, по сравнению с соединением формулы (I) или другим эталонным соединением. Например, тестируемое соединение может проявлять по меньшей мере любую величину модуляции в виде целого числа от 10% до 200%, или величину модуляции по меньшей мере в виде любого положительного или отрицательного целого числа от 30% до 150%, или по меньшей мере величину модуляции по меньшей мере в виде любого положительного или отрицательного целого числа от 60% до 100%, или величину модуляции по меньшей мере в виде любого положительного или отрицательного целого числа более 100%. Соединение, которое является отрицательным модулятором, в целом уменьшает модуляцию относительно известного соединения, тогда как соединение, которое является положительным модулятором, увеличивает модуляцию относительно известного соединения.
[00110] В целом, тестируемые соединения идентифицируются из больших библиотек и натуральных продуктов, или синтетических (или полусинтетических) экстрактов, или химических библиотек в соответствии со способами, известными в данной области. Специалистам в области выявления и разработки лекарственных средств понятно, что точный источник тестируемых экстрактов или соединений не имеет решающего значения для способа (способов) по изобретению. Соответственно, по существу может проводиться скрининг любого числа химических экстрактов или соединений с использованием иллюстративных способов, описанных в настоящей заявке. Примеры таких экстрактов или соединений включают без ограничения экстракты на растительной, грибковой, прокариотической или животной основе, ферментационные бульоны и синтетические соединения, а также модификацию существующих соединений. Доступны также многочисленные способы получения случайным или направленным синтезом (например, полусинтезом или полным синтезом) любого числа химических соединений, включая без ограничения соединения на сахаридной, липидной, пептидной и нуклеиновокислотной основе. Библиотеки синтетических соединений имеются в продаже. Альтернативно, библиотеки натуральных соединений в форме бактериальных, грибковых, растительных и животных экстрактов имеются в продаже из ряда источников, включая компании Biotics (Sussex, UK), Xenova (Slough, UK), Harbor Branch Oceanographic Institute (Ft. Pierce, FL, USA) и PharmaMar, MA, USA. Кроме того, при желании натуральные и синтетически продуцируемые библиотеки получают в соответствии со способами, известными в данной области, например, стандартными способами экстракции и фракционирования. Кроме того, при желании, любая библиотека или соединение легко модифицируется с использованием стандартных химических, физических или биохимических способов.
[00111] Когда обнаруживается, что неочищенный экстракт модулирует ингибирование O-GlcNAcase-зависимого отщепления O-GlcNAc, или любая биологическая реакция, вызванная соединением формулы (I), для выделения химических ингредиентов, ответственных за наблюдаемой эффект, необходимо дополнительное фракционирование положительного основного экстракта. Таким образом, целью процесса экстракции, фракционирования и очистки является тщательная характеристика и идентификация химического соединения внутри неочищенного экстракта, обладающего ингибирующей активностью в отношении O-GlcNAcase. Те же анализы, описанные в настоящей заявке для выявления видов активности в смесях соединений, могут применяться для очистки активного компонента и для тестирования его производных. Способы фракционирования и очистки таких гетерогенных экстрактов известны в данной области. При желании, соединения, которые, как показано, являются полезными средствами для лечения, химически модифицируются в соответствии со способами, известными в данной области. Соединения, идентифицированные как имеющие терапевтическую, профилактическую, диагностическую или другую ценность, могут в последующем анализироваться с использованием подходящей экспериментальной модели на животных, как описано в настоящей заявке или на основании общеизвестных данных в этой области.
[00112] В некоторых вариантах осуществления, одно или более из соединений могут применяться при разработке экспериментальных моделей на животных для исследования заболеваний или расстройств, связанных с дефицитами O-GlcNAcase, сверхэкспрессией O-GlcNAcase, накоплением O-GlcNAc, истощением запасов O-GlcNAc и для изучения лечения заболеваний и расстройств, связанных с дефицитом или сверхэкспрессией O-GlcNAcase, или накоплением или истощением запасов O-GlcNAc. Такие заболевания и расстройства включают нейродегенеративные заболевания, включая болезнь Альцгеймера и рак.
[00113] В настоящей заявке описаны различные альтернативные варианты осуществления и примеры изобретения. Эти варианты осуществления и примеры являются иллюстративными и не должны рассматриваться как ограничивающие объем изобретения.
ПРИМЕРЫ
[00114] Следующие примеры предназначены для иллюстрации вариантов осуществления изобретения и не предназначены для рассмотрения как ограничивающие объем изобретения.
Аббревиатуры
ABCN = 1,1'-азобис(циклогексанкарбонитрил)
AcCl = ацетилхлорид
AcOH = уксусная кислота
BC13 = трихлорид бора
BnBr = бензилбромид
Boc2O = ди-трет-бутилдикарбонат
BzCl = бензоилхлорид
DAST = трифторид диэтиламиносеры
DCM = дихлорметан
DIPEA = диизопропилэтиламин
DMAP = 4-диметиламинопиридин
DMF = N,N-диметилформамид
DMP = перйодинат Десс-Мартина
Et3N = триэтиламин
Et2O = простой диэтилэфир
TBDMSC1 = трет-бутилдиметилсилил хлорид
TBAF = тетра-н-бутиламмоний фторид
TFA = 2,2,2-трифторуксусная кислота
THF = тетрагидрофуран
тио-CDI = 1,1'-тиокарбонилдиимидазол
Пример 1
(3aR,5R,6S,7R,7aR)-2-(этиламино)-5-метил-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диол
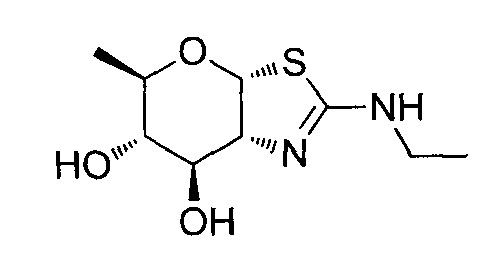
[00115] К суспензии (3aR,5R,6S,7R,7aR)-2-(этиламино)-5-(гидроксиметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диола (35,0 г, 141 ммоль) в DMF (300 мл), охлажденной при 15°C, добавляли DIPEA (6,0 мл), Boc2O (61,5 г, 282 ммоль) и MeOH (6,0 мл). Смесь перемешивали при комнатной температуре в течение 16 ч, и затем добавляли MeOH (50 мл). Реакционную смесь концентрировали при пониженном давлении при ~35°C. Остаток очищали колоночной флэш-хроматографией на силикагеле (EtOAc/гексаны 1:1, затем MeOH/DCM, 1:5), с последующей перекристаллизацией из EtOAc/гексанов, для получения трет-бутил-((3aR,5R,6S,7R,7aR)-6,7-дигидрокси-5-(гидроксиметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-2-ил)(этил)карбамата в виде белого твердого вещества (31,5 г, выход 64%). 1H ЯМР (400 МГц, CDCl3) δ 6,12 (д, J=6,8 Гц, 1H), 4,23-4,22 (м, 1H), 4,17-4,14 (м, 1H), 3,91-3,86 (м, 2H), 3,81-3,77 (м, 3H), 3,59-3,55 (м, 1H), 3,17-3,16 (м, 1H, OH), 1,53 (с, 9H), 1,16 (т, J=7,0 Гц, 3H).
[00116] К раствору указанного выше соединения (5,0 г, 14,4 ммоль) в DMF (25 мл) добавляли имидазол (1,57 г, 23,1 ммоль) и TBDMSC1 (2,82 г, 18,7 ммоль). Реакционную смесь, перемешивавшуюся при комнатной температуре в течение 30 ч, разбавляли EtOAc (100 мл). Органические вещества промывали насыщенным NH4C1, солевым раствором, сушили над безводным Na2SO4 и концентрировали. Остаток очищали колоночной флэш-хроматографией на силикагеле (EtOAc/гексаны, 1:1), получая трет-бутил- ((3aR,5R,6S,7R,7aR)-5-(((трет-бутил-диметилсилил)окси)метил)-6,7-дигидрокси-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-2-ил)(этил)карбамат в виде белого твердого вещества (5,08 г, 76%). 1H ЯМР (400 МГц, CDCl3) δ 6,12 (д, J=6,7 Гц, 1H), 4,25 (т, J=6,2 Гц, 1H), 4,16 (т, J=6,4 Гц, 1H), 4,10-4,04 (м, 2H), 3,91-3,85 (м, 3H), 3,65-3,62 (м, 1H), 1,55 (с, 9H), 1,26 (т, J=7,0 Гц, 3H), 0,89 (с, 9H), 0,08 (с, 6H).
[00117] К раствору указанного выше соединения (1,15 г, 2,5 ммоль), тетрабутиламмония йодида (0,092 г, 0,25 ммоль) в DMF (25 мл) при 0°C добавляли NaH (60%, 0,3 г, 7,5 ммоль) с последующим добавлением метоксибензил хлорида (1,02 мл, 7,5 ммоль). Затем реакционную смесь перемешивали при комнатной температуре в течение 6 ч, разбавляли Et2O (50 мл) и гасили водой (5 мл). Эфирный слой дополнительно промывали насыщенным NH4C1, солевым раствором, сушили над безводным Na2SO4 и концентрировали. Смесь двух продуктов, трет-бутил-((3aR,5R,6S,7R,7aR)-6-((трет-бутил-диметилсилил)окси)-7-((4-метоксибензил)окси)-5-((4-метоксифенокси)метил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-2-ил)(этил)карбамата и трет-бутил-((3aR,5R,6S,7R,7aR)-5-(((трет-бутил-диметилсилил)окси)метил)-6,7-бис((4-метоксибензил)окси)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-2-ил)(этил)карбамата отделяли (1,6 г, 66%) в отношении 1:2 соответственно, как указано 1Ή ЯМР.
[00118] К раствору указанной выше смеси (1,58 г, 2,26 ммоль) в THF (15 мл) добавляли 1M раствор TBAF (4,0 мл, 4 ммоль) при 0°C и затем смесь перемешивали при комнатной температуре в течение 2 ч. Разбавляли EtOAc (50 мл), органический слой промывали насыщенным NH4Cl, сушили над безводным Na2SO4 и концентрировали. Неочищенный остаток очищали колоночной флэш-хроматографией на силикагеле (EtOAc/гексаны, 1:2), получая трет-бутил-этил-((3aR,5R,6S,7R,7aR)-5-(гидроксиметил)-6,7-бис((4-метоксибензил)окси)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-2-ил)карбамат (0,75 г, 56%) в виде смолянистого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 7,30 (д, J=8,6, 2H), 7,15 (д, J=8,6, 2H), 6,88-6,80 (м, 4H), 6,01 (д, J=6,9 Гц, 1H), 4,64 (д, J=11,8 Гц, 1H), 4,59 (д, J=11,8 Гц, 1H), 4,48-4,45 (м, 1H), 4,37-4,34 (ддд, J=6,8, 3,6, 1,28 Гц, 1H), 4,27 (д, J=11,2, 1H), 4,24-4,23 (дд, J=1,8, 1,76 Гц, 1H), 3,87-3,80 (м, 2H), 3,78 (с, 3H), 3,77 (с, 3H), 3,70-3,65 (м, 1H), 3,59-3,56 (дт, J=9,0, 1,44 Гц, 1H), 3,55-3,49 (м, 1H), 3,41-3,37 (ддд, J=8,5, 5,3, 2,8 Гц, 1H), 1,78 (т, J=6,4 Гц, 1H), 1,50 (с, 9H), 1,18 (т, J=6,9 Гц, 3H).
[00119] К раствору трифенилфосфина (0,179 г, 0,68 ммоль) в сухом толуоле (5 мл) добавляли йод (0,145 г, 0,57 ммоль), пиридин (0,1 мл, 1,2 ммоль) и раствор указанного выше соединения (0,210 г, 0,357 ммоль), предварительно растворенного в толуоле (3 мл). Реакционную смесь перемешивали при 65°C в течение 3,5 ч. Нерастворимый желтоватый материал отфильтровывали, и фильтрат разбавляли EtOAc (50 мл), промывали 1N HCl (20 мл), насыщенным NaHCO3 (20 мл) и солевым раствором. Слой EtOAc отделяли, сушили над Na2SO4 и концентрировали в неочищенный продукт колоночной флэш-хроматографией на силикагеле (EtOAc/гексаны, 3:7), получая трет-бутил-этил-((3aR,5S,6S,7R,7aR)-5-(йодметил)-6,7-бис((4-метоксибензил)окси)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-2-ил)карбамат в виде белого твердого вещества (0,23 г, 92,4%). 1H ЯМР (400 МГц, CDCl3) δ 7,33 (д, J=8,4, 2H), 7,18 (д, J=8,4, 2H), 6,89-6,82 (м, 4H), 6,07 (д, J=7 Гц, 1H), 4,67 (д, J=11,6 Гц, 1H), 4,61 (д, J=11,6 Гц, 1H), 4,54-4,51 (м, 1H), 4,36-4,31 (м, 2H), 4,19 (т, J=2,64 Гц, 1H), 3,87 (д, J=6,9 Гц, 1H), 3,83 (д, J=6,9 Гц, 1H), 3,77 (с, 6H), 3,50-3,48 (дд, J=6,8, 1,3 Гц, 1H), 3,37-3,34 (м, 1H), 3,23-3,20 (м, 2H), 1,51 (с, 9H), 1,10 (т, J=6,9 Гц, 3H).
[00120] К раствору указанного выше соединения (0,120 г, 0,172 ммоль) в этаноле (4 мл) и Et3N (0,1 мл) добавляли Pd/C (10% масс., 0,060 г). Смесь гидрогенизировали при 50 фунтах/дюйм2 (2585,745 мм рт.ст.) и при комнатной температуре в течение 24 ч. Катализатор удаляли фильтрацией, и растворители выпаривали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле, элюировали 3:7 EtOAc/гексаны для получения трет-бутил-((3aR,5R,6R,7R,7aR)-6,7-бис((4-метоксибензил)окси)-5-метил-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-2-ил)(этил)карбамата (0,077 г, 78,4%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 7,31 (д, J=6,8, 2H), 7,17 (д, J=6,8, 2H), 6,87-6,81 (м, 4H), 6,07 (д, J=5,6 Гц, 1H), 4,67 (д, J=9,4 Гц, 1H), 4,58 (д, J=9,4 Гц, 1H), 4,50 (д, J=8,9 Гц, 1H), 4,34 (м, 1H), 4,27 (д, J=8,9 Гц, 1H), 4,23-4,17 (м, 1H), 3,87-3,84 (м, 2H), 3,78 (с, 3H), 3,77 (с, 3H), 3,44-3,39 (м, 1H), 3,30-3,28 (м, 1H), 1,50 (с, 9H), 1,18 (д, J=4,9 Гц, 3H), 1,10 (т, J=5,5 Гц, 3H).
[00121] Указанное выше соединение (0,075 г, 0,131 ммоль) растворяли в 30% растворе TFA/DCM (10 мл) и перемешивали при комнатной температуре в течение 2,5 ч. Реакционную смесь концентрировали и совместно выпаривали с Et2O (20 мл). К остатку добавляли 2M раствор NH3/MeOH (3 мл) и концентрировали при пониженном давлении. Полученный продукт очищали колоночной флэш-хроматографией на силикагеле, элюировали 5% MeOH в DCM и 94:4:2 DCM-MeOH-NH4OH (28% водном) для получения (3aR,5R,6S,7R,7aR)-2-(этиламино)-5-метил-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диола в виде белого твердого вещества (0,021 г, 68,7%). 1H ЯМР (400 МГц, CD3OD) δ 6,60 (д, J=6,5 Гц, 1H), 4,08 (т, J=7,04 Гц, 1H), 3,77 (т, J=7,2 Гц, 1H), 3,75-3,72 (м, 1H), 3,44-3,38 (ддд, J=14,6, 7,28, 2,08 Гц, 2H), 3,25-3,20 (дд, J=7,7, 1,12 Гц, 1H), 1,33 (д, J=6,2 Гц, 3H), 1,28 (т, J=7,2 Гц, 3H); 13C ЯМР (100 МГц, CD3OD) δ 160,40, 90,08, 75,69, 75,66, 74,21, 66,27, 55,65, 18,91, 14,51; MS, m/z=233 (M+1).
Пример 2
(3aR,5R,6S,7aR)-2-(этиламино)-5-(гидроксиметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d ]тиазол-6-ол
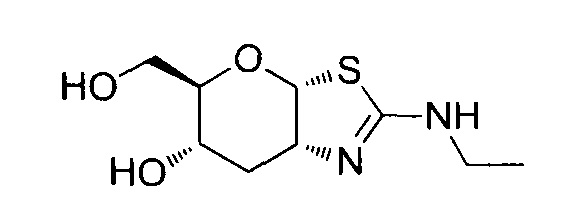
[00122] К раствору трет-бутил-((3aR,5R,6S,7R,7aR)-6,7-дигидрокси-5-(гидроксиметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-2-ил)(этил)карбамата (1,64 г, 4,73 ммоль), DIPEA (1,34 г, 10,4 ммоль) и DMAP (0,010 г, 0,082 ммоль) в DCM (50 мл) при 0°C медленно добавляли BzCl (1,33 г, 9,50 ммоль). После добавления смесь перемешивали при комнатной температуре в течение ночи. Добавляли насыщенный водный раствор NH4C1 (50 мл), и органический слой собирали. Водный слой дополнительно экстрагировали DCM (2×40 мл). Объединенный экстракт сушили над безводным Na2SO4. После фильтрации растворитель выпаривали при пониженном давлении, и остаток отделяли колоночной флэш-хроматографией на силикагеле (EtOAc/гексаны, от 1:4 до 1:2), получая ((3aR,5R,6S,7R,7aR)-6-(бензоилокси)-2-((трет-бутоксикарбонил)(этил)амино)-7-гидрокси-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-5-ил)метил бензоат в виде белого твердого вещества (0,67 г, 26%). 1H ЯМР (400 МГц, CDCl3) δ 8,08 (д, J=8,1 Гц, 4H), 7,57-7,35 (м, 6H), 6,19 (д, J=7,1 Гц, 1H), 5,21 (дд, J=2,8, 9,2 Гц, 1H), 4,56-4,51 (м, 2H), 4,47-4,42 (м, 2H), 4,14-4,10 (м, 1H), 3,99-3,92 (м, 2H), 1,55 (с, 9H), 1,19 (т, J=7,2 Гц, 3H).
[00123] Смесь указанного выше соединения (0,346 г, 0,623 ммоль) и тио-CDI (90% тех., 0,20 г, 1,0 ммоль) в безводном толуоле (10 мл) перемешивали при 95°C в течение 4 ч. После охлаждения растворитель удаляли при пониженном давлении, и остаток очищали колоночной флэш-хроматографией на силикагеле (EtOAc/гексаны, 2:3), получая (3aR,5R,6S,7R,7aR)-7-((1H-Имидазол-1-карбонотиоил)окси)-5-((бензоилокси)метил)-2-((трет-бутоксикарбонил)(этил)амино)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6-ил бензоат в виде желтого твердого вещества (0,343 г, 83%). 1H ЯМР (400 МГц, CDCl3) δ 8,76 (с, 1H), 8,08-7,94 (м, 4H), 7,70 (с, 1H), 7,57-7,37 (м, 6H), 7,18 (с, 1H), 6,36 (дд, J=1,9, 3,7 Гц, 1H), 6,17 (д, J=7,1 Гц, 1H), 5,54 (тд, J=1,2, 9,2 Гц, 1H), 4,70-4,67 (м, 1H), 4,60 (дд, J=3,2, 12,1 Гц, 1H), 4,42 (дд, J=5,1, 12,2 Гц, 1H), 4,11-4,08 (м, 1H), 4,05-3,97 (м, 2H), 1,56 (с, 9H), 1,22 (т, J=7,2 Гц, 3H).
[00124] Смесь указанного выше соединения (0,343 г, 0,515 ммоль), Bu3SnH (0,291 г, 1,00 ммоль) и ABCN (8,0 мг, 0,033 ммоль) в безводном толуоле (10 мл) перемешивали при 90°C в течение 3 ч. После охлаждения растворитель удаляли при пониженном давлении, и остаток очищали колоночной флэш-хроматографией на силикагеле (EtOAc/гексаны, 1:4), получая ((3aR,5R,6S,7aR)-6-(бензоилокси)-2-((трет-бутоксикарбонил)(этил)амино)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-5-ил)метил бензоат в виде белого твердого вещества (0,141 г, 51%). 1H ЯМР (400 МГц, CDCl3) 8,03-7,94 (м, 4H), 7,57-7,52 (м, 2H), 7,44-7,35 (м, 4H), 6,07 (д, J=7,2 Гц, 1H), 5,44-5,40 (м, 1H), 4,52-4,41 (м, 3H), 4,06-3,96 (м, 3H), 2,70-2,64 (м, 1H), 2,47-2,40 (м, 1H), 1,56 (с, 9H), 1,18 (т, J=7,2 Гц, 3H).
[00125] Смесь указанного выше соединения (0,140 г, 0,260 ммоль) и K2CO3 (0,030 г, 0,22 ммоль) в безводном MeOH (6 мл) перемешивали при комнатной температуре в течение 3 ч. Растворитель удаляли при пониженном давлении, и остаток очищали колоночной флэш-хроматографией на силикагеле (EtOAc), получая белое твердое вещество. Твердое вещество растворяли в MeOH (3 мл), через который пробулькивали HC1 (г) в течение 30 сек. Затем раствор перемешивали при комнатной температуре в течение 3 ч. Растворитель удаляли, и остаток нейтрализовали 1,0 M NH3 в MeOH и в последующем очищали колоночной флэш-хроматографией на силикагеле (1,0 M NH3 в MeOH/DCM, 1:8), получая (3aR,5R,6S,7aR)-2-(этиламино)-5-(гидроксиметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6-ол в виде не совсем белого твердого вещества (0,044 г, 73%). 1H ЯМР (500 МГц, CD3OD) δ 6,20 (д, J=7,1 Гц, 1H), 4,28-4,25 (м, 1H), 3,75 (дд, J=2,6, 14,4 Гц, 1H), 3,73-3,69 (м, 1H), 3,61 (дд, J=6,4, 14,4 Гц, 1H), 3,54-3,51 (м, 1H), 3,30-3,22 (м, 2H), 2,16-2,11 (м, 1H), 2,06-2,00 (м, 1H), 1,18 (т, J=7,2 Гц, 3H); 13C ЯМР (125 МГц, CD3OD) δ 163,24, 91,35, 77,13, 69,15, 65,79, 63,36, 39,60, 34,75, 14,87; MS, m/z=233 (M+1).
Пример 3
(3aR,5R,6S,7aR)-5-(гидроксиметил)-2-(метиламино)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6-ол
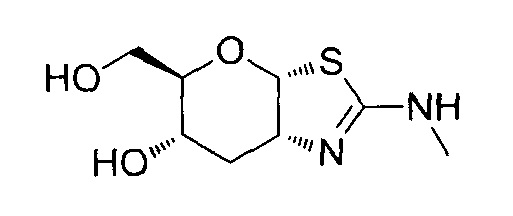
[00126] К суспензии (3aR,5R,6S,7R,7aR)-5-(гидроксиметил)-2-(метиламино)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диола (1,50 г, 6,40 ммоль) и NaHCO3 (1,01 г, 12,0 ммоль) в THF/воде (40 мл, 1:3) добавляли бензил хлорформиат (1,70 г, 10,0 ммоль). Смесь перемешивали при комнатной температуре в течение 4 дней. Органический растворитель удаляли при пониженном давлении для получения осадка. Осажденное твердое вещество собирали фильтрацией, промывали Et2O и сушили в вакууме, получая бензил ((3aR,5R,6S,7R,7aR)-6,7-дигидрокси-5-(гидроксиметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-2-ил)(метил)карбамат в виде белого твердого вещества (1,72 г, 73%). 1H ЯМР (500 МГц, CD3OD) 7,44-7,32 (м, 5H), 6,13 (д, J=6,9 Гц, 1H), 5,27-5,20 (м, 2H), 4,14 (т, J=6,1 Гц, lH), 4,02 (т, J=4,9 Гц, 1H), 3,74 (дд, J=2,4, 12,0 Гц, 1H), 3,63 (дд, J=6,1, 12,0 Гц, 1H), 3,53 (дд, J=4,5, 9,2 Гц, 1H), 3,47-3,43 (м, 1H), 3,39 (с, 3H).
[00127] Смесь указанного выше соединения (1,50 г, 4,08 ммоль), бензальдегиддиметилацеталя (3 мл) и моногидрата п-толуолсульфоновой кислоты (0,058 г, 0,30 ммоль) в DMF (10 мл) перемешивали при 50°C в легком вакууме в течение ночи. После охлаждения раствор нейтрализовали твердым NaHCO3, и затем концентрировали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (EtOAc/DCM/гексаны, 2:4:1), получая бензил-((3aR,4aR,8aS,9R,9aR)-9-гидрокси-7-фенил-3a,4a,5,8a,9,9a-гексагидро-[l,3]диоксин[4',5':5,6]пирано[3,2-d]тиазол-2-ил)(метил)карбамат в виде белого твердого вещества (0,91 г, 53%). 1H ЯМР (400 МГц, CDCl3) δ 7,49-7,47 (м, 2H), 7,40-7,35 (м, 8H), 6,28 (д, J=7,5 Гц, 1H), 5,78 (с, 1H), 5,29-5,21 (м, 2H), 4,34 (дд, J=5,1, 10,4 Гц, 1H), 4,15-4,06 (м, 2H), 3,89-3,84 (м, 1H), 3,76 (т, J=10,4 Гц, 1H), 3,58 (т, J=9,4, 1H), 3,38 (с, 3H), 2,62 (д, J=2,4 Гц, 1H).
[00128] Смесь указанного выше соединения (0,700 г, 1,54 ммоль) и тио-CDI (90% тех., 0,80 г, 4,04 ммоль) в безводном DMF (20 мл) перемешивали при 90°C в течение ночи. После охлаждения растворитель удаляли при пониженном давлении, и остаток очищали колоночной флэш-хроматографией на силикагеле (EtOAc/DCM, 2:1), получая O-((3aR,4aR,8aS,9R,9aR)-2-(((бензилокси)карбонил)(метил)амино)-7-фенил-3a,4a,5,8a,9,9a-гексагидро-[l,3]диоксин[4',5':5,6]пирано[3,2-d]тиазол-9-ил)-1H-имидазол-l-карботиоат в виде желтого твердого вещества (0,71 г, 81%). 1H ЯМР (400 МГц, CDCl3) δ 8,36 (с, 1H), 7,66 (с, 1H), 7,42-7,37 (м, 7H), 7,35-7,32 (м, 3H), 7,03 (с, 1H), 6,28 (д, J=7,2 Гц, 1H), 6,20 (дд, J=7,1, 8,6 Гц, 1H), 5,56 (с, 1H), 5,26 (с, 2H), 4,43 (т, J=7,1 Гц, 1H), 4,39 (дд, J=5,1, 10,4 Гц, 1H), 4,19-4,13 (м, 1H), 3,93 (т, J=9,0 Гц, 1H), 3,79 (т, J=10,4 Гц, 1H), 3,32 (с, 3H).
[00129] Смесь указанного выше соединения (0,560 г, 1,00 ммоль), Bu3SnH (0,87 г, 3,0 ммоль) и ABCN (24 мг, 0,10 ммоль) в безводном THF (20 мл) перемешивали при кипячении в сосуде с обратным холодильником в течение 3 ч. После охлаждения растворитель удаляли при пониженном давлении, и остаток очищали колоночной флэш-хроматографией на силикагеле (EtOAc/гексаны/DCM, 1:3:1), получая бензил-метил-((3aR,4aR,8aS,9aR)-7-фенил-3a,4a,5,8a,9,9a-гексагидро-[l,3]диоксино[4',5':5,6]пирано[3,2-d]тиазол-2-ил)карбамата в виде белого твердого вещества (0,32 г, 73%). 1H ЯМР (400 МГц, CDCl3) δ 7,49-7,47 (м, 2H), 7,39-7,33 (м, 8H), 6,21 (д, J=6,5 Гц, 1H), 5,56 (с, 1H), 5,29-5,20 (м, 2H), 4,52-4,25 (м, 2H), 4,08-4,02 (м, 1H), 3,72 (т, J=10,3 Гц, 1H), 3,61-3,54 (м, 1H), 3,33 (с, 3H), 2,41-2,35 (м, 1H), 1,76-1,68 (м, 1H).
[00130] К раствору указанного выше соединения (0,20 г, 0,45 ммоль) в безводном DCM (10 мл), охлажденному при -78°C в атмосфере N2, добавляли трихлорид бора в DCM (1,0 M, 2,0 мл, 2,0 ммоль). Смесь перемешивали в течение 3 ч, в то время как температуру реакционной смеси постепенно доводили до ~0°C. Затем смесь снова охлаждали при -78°C и осторожно добавляли MeOH (5 мл). После перемешивания при комнатной температуре в течение 30 мин смесь концентрировали при пониженном давлении. Остаток нейтрализовали 1,0 M NH3 в MeOH, и в последующем очищали колоночной флэш-хроматографией на силикагеле (1,0 M NH3 в MeOH:DCM, 1:7), получая (3aR,5R,6S,7aR)-5-(гидроксиметил)-2-(метиламино)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6-ол в виде не совсем белого твердого вещества (0,070 г, 72%). 1H ЯМР (500 МГц, CD3OD) δ 6,20 (д, J=6,4 Гц, 1H), 4,27-4,23 (м, 1H), 3,74 (дд, J=2,5, 12,0 Гц, 1H), 3,71-3,67 (м, 1H), 3,63 (дд, J=6,2, 12,0 Гц, 1H), 3,54-3,49 (м, 1H), 2,83 (с, 3H), 2,16-2,10 (м, 1H), 2,04-1,97 (м, 1H); 13C ЯМР (100 МГц, CD3OD) δ 163,99, 91,81, 77,11, 69,31, 65,82, 63,32, 34,93, 30,49; MS, m/z=219 (M+1).
Пример 4
(3aR,5R,6S,7aR)-5-(гидроксиметил)-2-(диметиламино)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6-ол
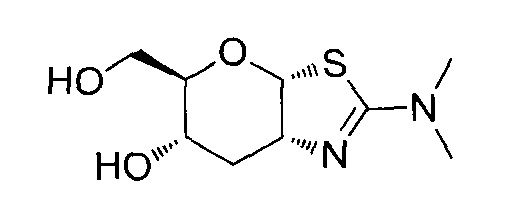
[00131] Суспензию (3aR,5R,6S,7R,7aR)-2-(диметиламино)-5-(гидроксиметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диола (0,800 г, 3,23 ммоль) и безводного ZnCl2 (1,00 г, 7,35 ммоль) в бензальдегиде (5 мл) перемешивали при комнатной температуре в течение ночи. Добавляли DCM (50 мл) и насыщенный водный NaHCO3 (50 мл), и смесь перемешивали в течение 20 мин. Твердое вещество отфильтровывали, и органический слой собирали из фильтрата. Водный слой экстрагировали DCM (2×50 мл), и объединенный экстракт сушили над безводным Na2SO4. После фильтрации растворитель выпаривали при пониженном давлении, и остаток очищали колоночной флэш-хроматографией на силикагеле (1,0 M NH3 в MeOH/DCM, 3:100), получая (3aR,4aR,8aS,9R,9aR)-2-(диметиламино)-7-фенил-3a,4a,5,8a,9,9a-гексагидро-[l,3]диоксин[4',5':5,6]пирано[3,2-d]тиазол-9-ол в виде бледно-желтого твердого вещества (0,76 г, 70%). 1H ЯМР (400 МГц, CDCl3) δ 7,51-7,48 (м, 2H), 7,40-7,34 (м, 3H), 6,39 (д, J=6,6 Гц, 1H), 5,58 (с, 1H), 4,31 (дд, J=5,1, 10,4 Гц, 1H), 4,10-4,03 (м, 2H), 3,86 (дд, J=8,2, 9,5 Гц, 1H), 3,76 (т, J=10,4 Гц, 1H), 3,59 (т, J=9,5 Гц, 1H), 3,00 (с, 6H).
[00132] Смесь указанного выше соединения (0,67 г, 2,0 ммоль) и тио-CDI (90% тех., 1,6 г, 8,0 ммоль) в безводном DMF (20 мл) перемешивали при 95°C в течение ночи. После охлаждения растворитель удаляли при пониженном давлении, и остаток очищали колоночной флэш-хроматографией на силикагеле (MeOH/DCM, от 1:100 до 5:100), получая смесь, содержащую O-((3aR,4aR,8aS,9R,9aR)-2-(диметиламино)-7-фенил-3a,4a,5,8a,9,9a-гексагидро-[l,3]диоксин[4',5':5,6]пирано[3,2-d]тиазол-9-ил)-lH-имидазол-l-карботиоат. Эту смесь растворяли в безводном THF (30 мл) и добавляли Bu3SnH (1,0 г, 3,4 ммоль) и ABCN (0,060 мг, 0,24 ммоль), и смесь перемешивали при кипячении в сосуде с обратным холодильником в течение ночи. Затем растворитель удаляли при пониженном давлении, и остаток очищали колоночной флэш-хроматографией на силикагеле (1,0 M NH3 в MeOH:DCM, 3:100), получая бледно-желтое твердое вещество, содержащее (3aR,4aR,8aS,9aR)-N,N-диметил-7-фенил-3a,4a,5,8a,9,9a-гексагидро-[l,3]диоксин[4',5':5,6]пирано[3,2-d]тиазол-2-амин (0,17 г, с примесями). Твердое вещество обрабатывали 2% HCl в MeOH (10 мл) при комнатной температуре в течение 2 ч, и затем растворитель удаляли при пониженном давлении. Остаток нейтрализовали 1,0 M NH3 в MeOH, и в последующем очищали колоночной флэш-хроматографией на силикагеле (1,0 M NH3 в MeOH:DCM, 1:10), получая чистый (3aR,5R,6S,7aR)-5-(гидроксиметил)-2-(диметиламино)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6-ол в виде не совсем белого твердого вещества (0,050 г, общий выход 11%). 1H ЯМР (400 МГц, CD3OD) δ 6,23 (д, J=6,4 Гц, 1H), 4,28-4,24 (м, 1H), 3,74 (дд, J=2,5, 12,0 Гц, 1H), 3,70-3,65 (м, 1H), 3,59 (дд, J=6,2, 12,0 Гц, 1H), 3,53-3,48 (м, 1H), 2,99 (с, 6H), 2,14-2,08 (м, 1H), 2,02-1,95 (м, 1H); 13C ЯМР (100 МГц, CD3OD) δ 165,60, 92,43, 77,19, 69,67, 65,79, 63,27, 40,15, 34,93; MS, m/z=255 (M+23).
[00133] Соединения в следующих примерах были синтезированы в соответствии с процедурами, аналогичными схемам и описанным выше примерам.
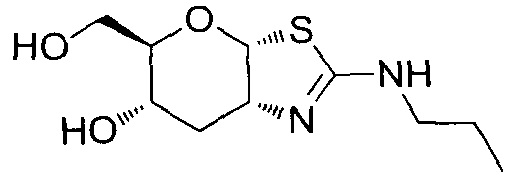
Пример 7
(3aR,5R,6R,7R,7aR)-2-(этиламино)-7-фтор-5-(гидроксиметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6-ол
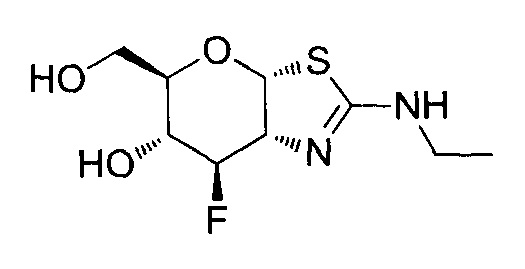
[00134] К раствору ((3aR,5R,6S,7R,7aR)-6-(бензоилокси)-2-((трет-бутоксикарбонил)(этил)амино)-7-гидрокси-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-5-ил)метил бензоата (0,49 г, 0,88 ммоль) в безводном DCM (3 мл) при -40°C в атмосфере N2, добавляли DAST (1,0 г, 6,2 ммоль). После добавления смесь перемешивали при комнатной температуре в течение ночи. После того как реакционную смесь снова охлаждали при -40°C, ее разбавляли DCM (20 мл), и затем гасили добавлением по каплям насыщенного водного NaHCO3. Органический слой собирали, а водный экстрагировали DCM (2×15 мл). Объединенный экстракт сушили над безводным Na2SO4. После фильтрации растворитель выпаривали при пониженном давлении, и остаток очищали колоночной флэш-хроматографией на силикагеле (EtOAc/гексаны, от 1:5 до 2:7), получая ((3aR,5R,6R,7R,7aR)-6-(бензоилокси)-2-((трет-бутоксикарбонил)(этил)амино)-7-фтор-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-5-ил)метил бензоат в виде белого твердого вещества (0,14 г, с примесями, ≈28%).
[00135] Превращение указанного выше соединения в указанное в заголовке соединение осуществляли удалением бензоата и Boc защитных групп с использованием соответственно K2CO3 в MeOH и насыщенной HC1 в MeOH, посредством процедур, описанных для примера 2. После очистки колоночной флэш-хроматографией на силикагеле (1,0 M NH3 в MeOH/DCM, 1:7), получали (3aR,5R,6R,7R,7aR)-2-(этиламино)-7-фтор-5-(гидроксиметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6-ол в виде не совсем белого твердого вещества (0,051 г, в целом, 23%). 1H ЯМР (400 МГц, CD3OD) δ 6,33 (д, J=6,6 Гц, 1H), 4,77 (тд, J=5,0, 48,2 Гц, 1H), 4,34-4,27 (м, 1H), 3,81 (дд, J=2,0, 12,0 Гц, 1H), 3,77-3,72 (м, 1H), 3,68 (дд, J=5,7, 12,0 Гц, 1H), 3,63-3,59 (м, 1H), 3,31-3,23 (м, 2H), 1,18 (т, J=7,2 Гц, 3H); 13C ЯМР (100 МГц, CD3OD) δ 163,40, 96,25 (д, J=177,7 Гц), 90,55 (д, J=3,9 Гц), 75,26 (д, J=4,6 Гц), 73,58 (д, J=24,5 Гц), 68,91 (д, J=22,8 Гц), 62,85, 39,54, 14,82; MS, m/z=251 (M+1).
Пример 8
(3aR,5R,6R,7R,7aR)-2-(диметиламино)-7-фтор-5-(гидроксиметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6-ол
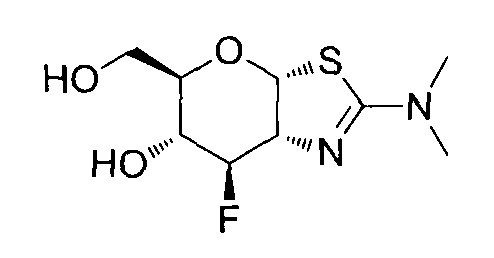
[00136] К раствору (3aR,5R,6S,7R,7aR)-2-(диметиламино)-5-(гидроксиметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диола (5,20 г, 21,0 ммоль) и имидазола (12,0 г, 176 ммоль) в DMF (60 мл) добавляли TBDMSCl (трет-бутилдиметилсилил хлорид) (15,0 г, 99,5 ммоль). Смесь перемешивали при комнатной температуре в течение 24 ч, и затем разбавляли солевым раствором (200 мл). Смесь экстрагировали Et2O (3×100 мл). Объединенный экстракт промывали солевым раствором (100 мл) и водой (100 мл), и затем сушили над безводным Na2SO4. После фильтрации растворитель выпаривали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (EtOAc/гексаны, от 1:4 до 1:1), получая (3aR,5R,6R,7R,7aR)-7-((трет-бутил-диметилсилил)окси)-5-(((трет-бутил-диметилсилил])окси)метил)-2-(диметиламино)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6-ол в виде белого твердого вещества (5,95 г, 60%). 1H ЯМР (500 МГц, CDCl3) δ 6,15 (д, J=5,8 Гц, 1H), 4,34-4,33 (м, 1H), 4,20 (т, J=5,0 Гц, 1H), 3,80-3,72 (м, 2H), 3,48 (шир.с, 2H), 3,01 (с, 6H), 0,90 (с, 9H), 0,89 (с, 9H), 0,13 (с, 3H), 0,12 (с, 3H), 0,07 (с, 6H).
[00137] К раствору указанного выше соединения (5,95 г, 12,5 ммоль) и DMAP (0,12 г, 0,98 ммоль) в пиридине (50 мл) при 0°C медленно добавляли BzCl (3,00 г, 21,3 ммоль). После добавления смесь перемешивали при комнатной температуре в течение 24 ч. Добавляли насыщенный водный раствор NaHCO3 (100 мл), и смесь экстрагировали EtOAc (3×100 мл). Объединенный экстракт сушили над безводным Na2SO4. После фильтрации растворитель выпаривали при пониженном давлении, и остаток очищали колоночной флэш-хроматографией на силикагеле (EtOAc/гексаны, от 1:9 до 1:3), получая (3aR,5R,6R,7R,7aR)-7-((трет-бутил-диметилсилил)окси)-5-(((трет-бутил-диметилсилил)окси)метил)-2-(диметиламино)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6-ил бензоат в виде прозрачного масла (6,85 г, 94%). 1H ЯМР (400 МГц, CDCl3) δ 8,05-8,02 (м, 2H), 7,56-7,52 (м, 1H), 7,43-7,39 (м, 2H), 6,26 (д, J=6,4 Гц, 1H), 5,06-5,03 (м, 1H), 4,40 (дд, J=2,2, 3,8 Гц, 1H), 3,81-3,78 (м, 1H), 3,71-3,70 (м, 2H), 3,03 (с, 6H), 0,88 (с, 9H), 0,85 (с, 9H), 0,17 (с, 3H), 0,13 (с, 3H), 0,02 (с, 3H), 0,00 (3H).
[00138] Через раствор указанного выше соединения (9,30 г, 16,8 ммоль) в сухом MeOH (100 мл) пробулькивали HC1 в течение 2 мин, и затем смесь перемешивали при комнатной температуре в течение 2 ч. После концентрации при пониженном давлении смесь обрабатывали насыщенным водным раствором NaHCO3 (150 мл), и затем экстрагировали EtOAc (6×80 мл). Объединенный экстракт сушили над безводным Na2SO4. После фильтрации растворитель выпаривали при пониженном давлении, получая (3aR,5R,6S,7R,7aR)-2-(диметиламино)-7-гидрокси-5-(гидроксиметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6-ил бензоат в виде белого кристаллического твердого вещества (5,40 г, 96%). 1H ЯМР (400 МГц, CDCl3) δ 8,03-8,01 (м, 2H), 7,58-7,53 (м, 1H), 7,44-7,39 (м, 2H), 6,37 (д, J=4,5 Гц, 1H), 5,12-5,09 (м, 1H), 4,41-4,37 (м, 2H), 3,94-3,85 (м, 1H), 3,78-3,74 (м, 1H), 3,67 (дд, J=4,9, 12,5 Гц, 1H), 3,00 (с, 6H).
[00139] К раствору указанного выше соединения (5,35 г, 15,2 ммоль) и DMAP (0,050 г, 0,41 ммоль) в пиридине (50 мл) при 0°C медленно добавляли BzCl (2,22 г, 15,8 ммоль). После добавления смесь перемешивали при комнатной температуре в течение 4 ч. Добавляли насыщенный водный раствор NaHCO3 (100 мл), и смесь экстрагировали EtOAc (2×50 мл). Объединенный экстракт сушили над безводным Na2SO4. После фильтрации растворитель выпаривали при пониженном давлении, и остаток очищали колоночной флэш-хроматографией на силикагеле (EtOAc/гексаны, от 1:1 до 10:1), получая ((3aR,5R,6S,7R,7aR)-6-(бензоилокси)-2-(диметиламино)-7-гидрокси-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-5-ил)метил бензоат в виде белого твердого вещества (4,45 г, 64%). 1H ЯМР (400 МГц, CDCl3) δ 8,04-8,00 (м, 4H), 7,58-7,51 (м, 2H), 7,44-7,37 (м, 4H), 6,38 (д, J=6,6 Гц, 1H), 5,23-5,20 (м, 1H), 4,55 (дд, J=3,2, 12,0 Гц, 1H), 4,48-4,40 (м, 3H), 4,27-4,22 (м, 1H), 3,00 (с, 6H).
[00140] К раствору указанного выше соединения (0,410 г, 0,898 ммоль) в безводном DCM (6 мл) при -78°C в атмосфере N2 добавляли DAST (0,87 г, 5,4 ммоль). После добавления смесь перемешивали при комнатной температуре в течение 16 ч. Затем реакционную смесь охлаждали при -78°C, разбавляли DCM (20 мл) и гасили добавлением насыщенного водного NaHCO3 (20 мл). Органический слой собирали, а водный экстрагировали DCM (2×30 мл). Объединенный экстракт сушили над безводным Na2SO4. После фильтрации растворитель выпаривали при пониженном давлении, и остаток очищали колоночной флэш-хроматографией на силикагеле (EtOAc/гексаны, от 1:3 до 1:2), получая ((3aR,5R,6R,7R,7aR)-6-(бензоилокси)-2-(диметиламино)-7-фтор-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-5-ил)метил бензоат) в виде белого твердого вещества (0,380 г, 92%). 1H ЯМР (400 МГц, CDCl3) δ 8,03-8,00 (м, 4H), 7,58-7,55 (м, 1H), 7,54-7,51 (м, 1H), 7,44-7,41 (м, 2H), 7,39-7,36 (м, 2H), 6,35 (д, J=5,4 Гц, 1H), 5,47 (дд, J=7,3, 16,3 Гц, 1H), 5,23 (ддд, J=1,1, 2,8, 35,2 Гц, 1H), 4,70-4,66 (м, 1H), 4,52 (дд, J=2,8, 9,6 Гц), 4,40 (дд, J=4,7, 9,6 Гц, 1H), 4,14-4,10 (м, 1H), 3,05 (с, 6H).
[00141] Смесь указанного выше соединения (0,375 г, 0,818 ммоль) и K2CO3 (0,113 г, 0,818 ммоль) в безводном MeOH (8 мл) перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь нейтрализовали сухим льдом, и затем растворитель удаляли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (1,0 M NH3 в MeOH/DCM, 1:12), получая (3aR,5R,6R,7R,7aR)-2-(диметиламино)-7-фтор-5-(гидроксиметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6-ол в виде белого твердого вещества (0,190 г, 93%). 1H ЯМР (400 МГц, CD3OD) δ 6,35 (д, J=6,7 Гц, 1H), 4,74 (дт, J=48,1, 5,0 Гц, 1H), 4,31 (дт, J=13,8, 6,0 Гц, 1H), 3,79 (дд, J=2,0, 12,0 Гц, 1H), 3,76-3,69 (м, 1H), 3,66 (дд, J=5,8, 12,0 Гц), 3,61-3,57 (м, 1H), 3,01 (с, 6H); 13C ЯМР (100 МГц, CD3OD) δ 166,25, 96,36 (д, J=176,8 Гц), 91,60 (д, J=3,9 Гц), 75,45 (д, J=4,6 Гц), 73,99 (д, J=24,7 Гц), 68,98 (д, J=22,9 Гц), 62,91, 40,33; MS, (ES, m/z) [M+H]+ 251,1.
[00142] В следующих примерах соединения синтезировали в соответствии с процедурами, аналогичными указанным выше схемам и примерам.
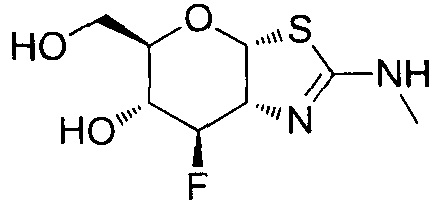
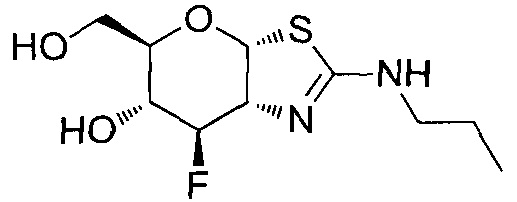
Пример 11
(3aR,5R,6R,7S,7aR)-7-фтор-5-(гидроксиметил)-2-(метиламино)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6-ол
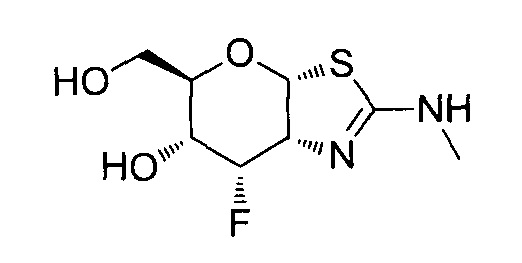
[00143] Через раствор трет-бутил-((3aR,5R,6R,7S,7aR)-5-(((трет-бутилдиметилсилил)окси)метил)-7-фтор-6-гидрокси-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-2-ил)(метил)карбамата (0,200 г, 0,444 ммоль) в сухом MeOH (6 мл) пробулькивали газообразную HC1 в течение 30 сек. Смесь перемешивали при комнатной температуре в течение 5 ч. После выпаривания растворителя при пониженном давлении остаток нейтрализовали 1,0 M NH3 в MeOH и очищали колоночной флэш-хроматографией на силикагеле (1,0 M NH3 в MeOH/DCM, 1:8) с последующей перекристаллизацией из MeOH/Et2O, получая (3aR,5R,6R,7S,7aR)-7-фтор-5-(гидроксиметил)-2-(метиламино)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6-ол в виде белого твердого вещества (0,093 г, 89%). 1H ЯМР (400 МГц, CD3OD) δ 6,62 (д, J=6,5 Гц, 1H), 4,89 (тд, J=3,5 Гц, 51,0 Гц, 1H), 4,54 (ддд, J=3,5, 6,5, 20,6, 1H), 3,98-3,94 (м, 1H), 3,90-3,84 (м, 1H), 3,83-3,81 (м, 1H), 3,75 (дд, J=5,4, 12,2 Гц, 1H), 2,99 (с, 3H); 13C ЯМР (100 МГц, CD3OD) δ 90,63 (д, J=181,3 Гц), 88,53 (д, J=2,3 Гц), 73,60 (д, J=4,3 Гц), 66,02 (д, J=17,3 Гц), 64,80-64,50 (м), 62,44, 31,38; MS, (ES, m/z) [M+H]+ 237,1.
Пример 12
(3aR,5S,6S,7R,7aR)-2-(этиламино)-5-(фторметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диол
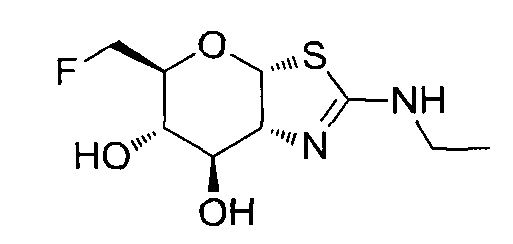
[00144] Метансульфонил хлорид (0,038 мл, 0,495 ммоль) порциями добавляли к перемешанному раствору трет-бутил-этил((3aR,5R,6S,7R,7aR)-5-(гидроксиметил)-6,7-бис((4-метоксибензил)окси)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-2-ил)карбамата (0,190 г, 0,323 ммоль) в сухом пиридине (5 мл) при -20°C. Через 3 ч, реакционную смесь разбавляли DCM (20 мл) и экстракт DCM промывали насыщенным NaHCO3, солевым раствором, сушили над Na2SO4 и концентрировали. Остаточный пиридин удаляли совместным выпариванием с гексанами. Неочищенный продукт очищали колоночной хроматографией на силикагеле (3:7 EtOAc/гексаны) для получения ((3aR,5R,6S,7R,7aR)-2-((трет-бутоксикарбонил)(этил)амино)-6,7-бис((4-метоксибензил)окси)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-5-ил)метил метансульфоната (0,215 г, 99,6%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 7,32 (д, J=8,5 Гц, 2H), 7,17 (д, J=8,5 Гц, 2H), 6,91-6,83 (м, 4H), 6,01 (д, J=6,88 Гц, 1H), 4,66 (д, J=11,6 Гц, 1H), 4,60 (д, J=11,6 Гц, 1H), 4,52 (д, J=11,1 Гц, 1H), 4,42-4,36 (м, 2H), 4,27-4,23 (м, 3H), 3,96-3,84 (м, 2H), 3,8 (с, 3H), 3,79 (с, 3H), 3,55 (м, 2H), 2,98 (с, 3H), 1,52 (с, 9H), 1,20 (т, J=6,9 Гц, 3H).
[00145] К раствору указанного выше соединения (0,520 г, 0,78 ммоль) в CH3CN (6 мл) добавляли раствор тетраэтиламмония фторида (0,640 г, 4,28 ммоль) в CH3CN (4 мл), и смесь нагревали до дефлегмации в течение 2,5 ч. После выпаривания растворителя остаток очищали колоночной хроматографией на силикагеле (1:1 EtOAc/гексаны) для получения трет-бутил-этил((3aR,5S,6S,7R,7aR)-5-(фторметил)-6,7-бис((4-метоксибензил)окси)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-2-ил)карбамата (0,335 г, 72,7%) в виде пенистого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 7,35 (д, J=8,5 Гц, 2H), 7,20 (д, J=8,5 Гц, 2H), 6,93-6,86 (м, 4H), 6,08 (д, J=6,88 Гц, 1H), 4,72 (д, J=11,7 Гц, 1H), 4,65 (д, J=11,7 Гц, 1H), 4,56 (д, J=11,6 Гц, 1H), 4,52-4,51 (м, 1H), 4,41-4,38 (м, 2H), 4,33 (д, J=11,6 Гц, 1H), 4,26 (т, J=2,88 Гц, 1H), 3,93-3,87 (м, 2H), 3,83 (с, 3H), 3,82 (с, 3H), 3,69-3,66 (м, 1H), 3,61-3,50 (м, 1H), 1,55 (с, 9H), 1,15 (т, J=6,9 Гц, 3H).
[00146] Указанное выше соединение (0,115 г, 0,195 ммоль), растворенное в 30% TFA/DCM, при 0°C перемешивали в течение 3 ч при комнатной температуре. TFA/DCM затем полностью выпаривали, и полученный остаток суспендировали в 2M NH3/MeOH (3 мл). Метанольный раствор аммиака снова концентрировали, и неочищенный остаток очищали колоночной хроматографией на силикагеле, используя 10% MeOH в DCM, для получения (3aR,5S,6S,7R,7aR)-2-(этиламино)-5-(фторметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диол в виде светло-желтоватого твердого вещества (0,035 г, 72,8%). 1H ЯМР (400 МГц, CD3OD) 6,49 (д, J=6,48 Гц, 1H), 4,66-4,52 (дт, J=47,6, 10,5, 4,3 Гц, 2H), 4,12 (т, J=6,6 Гц, 1H), 3,90 (т, J=6,9 Гц, 1H), 3,80-3,73 (м, 2H), 3,55 (дд, J=6,3, 3,2 Гц, 1H), 3,4-3,3 (м, 2H), 1,23 (т, J=7,2 Гц, 3H). 13C ЯМР (100 МГц, MeOD) δ 163,78, 89,63, 83,90 (д, JC6,F 171,7 Гц, C-6), 76,83 (д, JC5,F 17,8 Гц, C-5), 75,43, 69,51 (д, JC4,F 7,0 Гц, C-4), 67,71, 42,13, 14,74. ES/MS: 251,0 [M+1].
Пример 13
(3aR,5S,6S,7R,7aR)-2-(диметиламино)-5-(фторметил)-5,6,7,7a- тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диол
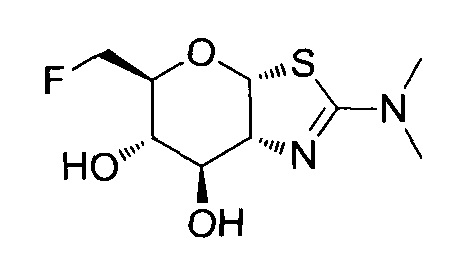
[00147] К перемешанному раствору (3aR,5R,6S,7R,7aR)-5-(((трет-бутил-диметилсилил)окси)метил)-2-(диметиламино)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диола (1,19 г, 3,3 ммоль) в DMF (18 мл) при 0°C небольшими порциями добавляли NaH (60%, 0,5 г, 13 ммоль). Через 20 мин добавляли BnBr (1,17 мл, 9,9 ммоль), и затем реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли DCM (100 мл), и экстракт DCM промывали насыщенным NaHCO3, солевым раствором, сушили над безводным Na2SO4 и концентрировали. Неочищенный продукт очищали колоночной флэш-хроматографией на силикагеле (EtOAc/гексаны, 1:1), получая (3aR,5R,6S,7R,7aR)-6,7-бис(бензилокси)-5-(((трет-бутил-диметилсилил)окси)метил)-N,N-диметил-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-2-амин метанол в виде смолянистого твердого вещества (0,805 г, 45%). 1H ЯМР (400 МГц, CDCl3) δ 7,31-7,16 (м, 10H), 6,19 (д, J=6,7 Гц, 1H), 4,72 (д, J=12 Гц, 1H), 4,60 (д, J=12 Гц, 1H), 4,57 (д, J=11,4 Гц, 1H), 4,37-4,32 (м, 2H), 4,06-4,00 (м, 1H), 3,69-3,62 (м, 2H), 3,60-3,52 (м, 2H), 2,89 (с, 6H), 0,80 (с, 9H), 0,04 (с, 6H).
[00148] К раствору указанного выше соединения (0,80 г, 1,48 ммоль) в THF (6 мл) добавляли 1M раствор TBAF (2,25 мл, 2,25 ммоль) при 0°C, и затем смесь перемешивали при комнатной температуре в течение ночи. Разбавляли EtOAc (50 мл), органический слой промывали насыщенным NH4Cl, сушили над безводным Na2SO4 и концентрировали. Неочищенный остаток очищали колоночной флэш-хроматографией на силикагеле (100% EtOAc), получая ((3aR,5R,6S,7R,7aR)-6,7-бис(бензилокси)-2-(диметиламино)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-5-ил)метанол (0,57 г, 90%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 7,38-7,28 (м, 10H), 6,29 (д, J=6, Гц, 1H), 4,79 (д, J=12,1 Гц, 1H), 4,69 (д, J=12,0 Гц, lH), 4,65 (д, J=11,7 Гц, 1H), 4,55-4,53 (м, 1H), 4,43 (д, J=11,5 Гц, 1H), 4,24 (м, 1H), 3,77 (д, J=12,1 Гц, 1H), 3,69-3,59 (м, 3H), 3,00 (с, 6H).
[00149] Метансульфонил хлорид (0,180 мл, 2,32 ммоль) порциями добавляли к перемешанному раствору указанного выше соединения (0,673 г, 1,57 ммоль) в сухом пиридине (8 мл) при -20°C. Через 3 ч реакционную смесь разбавляли DCM (20 мл) и экстракт DCM промывали насыщенным NaHCO3 (30 мл), солевым раствором, сушили над Na2SO4 и концентрировали. Остаточный пиридин удаляли совместным выпариванием с гексанами. Неочищенный продукт очищали колоночной хроматографией на силикагеле, элюировали 100% EtOAc для получения ((3aR,5R,6S,7R,7aR)-6,7-бис(бензилокси)-2-(диметиламино)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-5-ил)метил метансульфоната (0,610 г, 76,4%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) 7,32-7,18 (м, 10H), 6,15 (д, J=6,48 Гц, 1H), 4,68 (д, J=12 Гц, 1H), 4,57 (д, J=12 Гц, 1H), 4,53 (д, J=11,5 Гц, 1H), 4,48-4,45 (дд, J=3,8, 2,0 Гц, 1H), 4,29 (д, J=11,4 Гц, 1H), 4,24-4,16 (м, 3H), 3,73-3,69 (м, 1H), 3,53-3,50 (дт, J=8,9, 1,7 Гц, 1H), 2,91 (с, 6H), 2,90 (с, 3H).
[00150] К раствору указанного выше соединения (0,7 г, 1,38 ммоль) в CH3CN (15 мл) добавляли раствор TBAF (1,13 г, 7,59 ммоль) в CH3CN (5 мл), и смесь нагревали до температуры дефлегмации в течение 2,5 ч. После выпаривания растворителя остаток очищали колоночной хроматографией на силикагеле (7:3 EtOAc/гексаны) для получения (3aR,5S,6S,7R,7aR)-6,7-бис(бензилокси)-5-(фторметил)-N,N-диметил-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-2-амина (0,567 г, 95%) в виде пенистого твердого вещества. 1H ЯМР (400 МГц, CDCl3) 7,33-7,18 (м, 10H), 6,20 (д, J=6,5 Гц, 1H), 4,72 (д, J=12 Гц, 1H), 4,60 (д, J=12,6 Гц, 1H), 4,57 (д, J=11,9 Гц, 1H), 4,46-4,41 (м, 2H), 4,34-4,30 (м, 2H), 4,14 (т, J=3 Гц, 1H), 3,74-3,64 (м, 1H), 3,59-3,55 (м, 1H), 2,91 (с, 6H).
[00151] К раствору указанного выше соединения (0,56 г, 1,3 ммоль) в сухом DCM (8 мл) при -78°C по каплям добавляли раствор BC13 (1,0 M в DCM, 6,5 мл, 6,5 ммоль). Смесь перемешивали при -78°C до 0°C в течение 3 ч. Затем реакцию гасили добавлением раствора 1:1 DCM-MeOH (5 мл) при -78°C Смесь медленно нагревали до комнатной температуры. Растворители выпаривали при пониженном давлении. К остатку добавляли 1,3 M раствор NH3/MeOH (10 мл) и выпаривали. Это повторяли один или более раз. Остаток очищали колоночной хроматографией на силикагеле, элюировали 90:10 DCM-1,3M раствором NH3-MeOH для получения (3aR,5S,6S,7R,7aR)-2-(диметиламино)-5-(фторметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диола в виде белого твердого вещества (0,25 г, 76,7%). 1H ЯМР (400 МГц, CD3OD) 6,31 (д, J=6,44 Гц, 1H), 4,63-4,50 (дд, J=44,4, 3,4 Гц, 2H), 4,09 (т, J=6,0 Гц, 1H), 3,92 (т, J=6,9 Гц, 1H), 3,79-3,69 (м, 1H), 3,55-3,51 (дд, J=5,4, 4,04 Гц, 1H), 3,01 (с, 6H). 13C ЯМР (100 МГц, MeOD) δ 166,34, 92,58, 84,50 (д, JC6,F 171,2 Гц, C-6), 77,09, 76,56, 76,55, 75,72 (д, JC5,F 17,7 Гц, C-5), 70,73 (д, JC4,F 7,12 Гц, C-4), 40,97. ES/MS: 251,6 [M+1].
Пример 14
(3aR,5S,6S,7R,7aR)-2-(аллиламино)-5-(фторметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диол
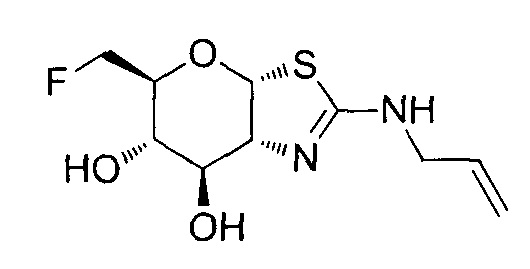
[00152] Раствор (3aR,5R,6S,7R,7aR)-2-(аллил(трет-бутоксикарбонил)амино)-5-(гидроксиметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диил дибензоата (1,7 г, 3,0 ммоль) в DCM (40 мл) обрабатывали DAST (2,5 г, 15,4 ммоль) в течение 30 мин при -78°C. После перемешивания в течение ночи при комнатной температуре реакцию гасили насыщенным водным NaHCO3 (30 мл). Органический слой отделяли, и водный слой экстрагировали DCM (3×20 мл). Объединенный органический слой промывали солевым раствором (20 мл), сушили над безводным Na2SO4 и концентрировали в вакууме. Остаток очищали на колонке с силикагелем с 5%-10% EtOAc в простом петролейном эфире для получения (3aR,5S,6S,7R,7aR)-2-(аллил(трет-бутоксикарбонил)амино)-5-(фторметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диил дибензоата в виде белого твердого вещества (580 мг, 32%). (ES, m/z): [M+H]+ 571,0; 1H ЯМР (300 МГц, CDCl3) δ 8,09 (д, J=7,5 Гц, 4H), 7,60 (т, J=7,8 Гц, 2H), 7,45 (т, J=7,8 Гц, 4H), 6,27 (д, J=6,3 Гц, 1H), 6,08 (с, 1H), 5,86-5,92 (м, 1H), 5,40 (д, J=9,6 Гц, lH), 5,12 (д, J=9,3 Гц, 2H), 4,81-4,89 (дд, J=10,0, 2,8 Гц, 1H), 4,63-4,69 (м, 2H), 4,50-4,54 (м, 2H), 3,82-3,98 (м, 1H), 1,63 (с, 9H).
[00153] Раствор указанного выше соединения (540 мг, 0,94 ммоль) в MeOH (10 мл) обрабатывали K2CO3 (26 мг, 0,18 ммоль) в течение 2 ч при комнатной температуре, затем нейтрализовали AcOH. Летучие вещества удаляли для получения остатка, который очищали колоночной хроматографией на силикагеле, элюировали 5% MeOH в DCM для получения трет-бутил-аллил-((3aR,5S,6S,7R,7aR)-5-(фторметил)-6,7-дигидрокси-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-2-ил)карбамата в виде белого твердого вещества (310 мг, 86%). (ES, m/z): [M+H]+ 363,0; 1H ЯМР (300 МГц, CDCl3) δ 6,10 (д, J=6,9Гц, 1H), 5,85-5,96 (м, 1H), 5,11-5,18 (м, 2H), 4,61 (д, J=2,4 Гц, 1H), 4,43-4,58 (м, 3H), 4,13-4,16 (м, 1H), 4,04-4,07 (м, 1H), 3,53-3,63 (м, 2H), 1,53 (с, 9H).
[00154] Через раствор указанного выше соединения (100 мг, 0,27 ммоль) в MeOH (6 мл) пробулькивали сухой газообразный HCl при комнатной температуре до насыщения. Еще через 4 ч летучие вещества удаляли для получения остатка, который повторно растворяли в MeOH (5 мл) и нейтрализовали концентрированным NH4OH, концентрировали, и очищали на колонке с силикагелем, элюировали 5%-10% MeOH в DCM для получения (3aR,5S,6S,7R,7aR)-2-(аллиламино)-5-(фторметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диола в виде белого твердого вещества (42,7 мг, 58%). (ES, m/z): [M+H]+ 263,0; 1H ЯМР (300 МГц, D2O) δ 6,29 (д, J=6,3 Гц, 1H), 5,85-5,96 (м, 1H), 5,19-5,26 (м, 1H), 5,09-5,14 (м, 1H), 4,63 (д, J=3,3 Гц, 1H), 4,47 (д, J=3,6 Гц, 1H), 4,07-4,11 (м, 1H), 3,95-3,99 (м, 1H), 3,86-3,89 (м, 2H), 3,68-3,79 (м, 1H), 3,51-3,56 (м, 1H).
Пример 15
(3aR,5S,6S,7R,7aR)-2-амино-5-(фторметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диол
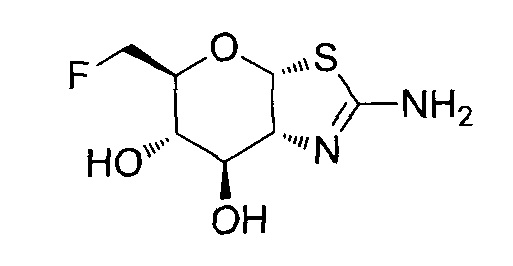
[00155] К раствору (3aR,5S,6S,7R,7aR)-2-(аллил(трет-бутоксикарбонил)амино)-5-(фторметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диил дибензоата (500 мг, 0,88 ммоль) в 1,4-диоксане (30 мл) добавляли Pd(PPh3)4 (200 мг, 0,17 ммоль), HCO2H (84 мг, 1,76 ммоль) и Et3N (177 мг, 1,76 ммоль) при комнатной температуре в атмосфере N2. Через 20 мин при 60°C в реакционную смесь добавляли дополнительное количество HCO2H (404 мг, 8,8 ммоль). Реакционную смесь перемешивали в течение дополнительных 2 ч при 60°C, и затем реакцию гасили H2O (40 мл), нейтрализовали NaHCO3, экстрагировали DCM (3×50 мл). Объединенный органический слой концентрировали и очищали на колонке с силикагелем, элюировали 10% EtOAc в простом петролейном эфире для получения (3aR,5S,6S,7R,7aR)-2-(трет-бутоксикарбониламино)-5-(фторметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диил дибензоата в виде белого сиропа (330 мг, 71%). (ES, m/z): [M+H]+ 531,0; 1H ЯМР (300 МГц, CDCl3) δ 8,30 (д, J=7,2 Гц, 2H), 8,06 (д, J=7,2 Гц, 2H), 7,46-7,66 (м, 5H), 6,46 (д, J=7,5 Гц, 1H), 5,85 (с, 1H), 5,53 (д, J=6,6 Гц, 1H), 4,71-4,79 (м, 2H), 4,62 (т, J=2,1 Гц, 1H), 3,95-4,03 (м, 1H), 1,57 (с, 9H).
[00156] Раствор указанного выше соединения (330 мг, 0,62 ммоль) в MeOH (10 мл) обрабатывали K2CO3 (24 мг, 0,17 ммоль) в течение 2 ч при комнатной температуре, затем нейтрализовали AcOH. Летучие вещества удаляли для получения остатка, который очищали колоночной хроматографией на силикагеле, элюировали 2%-5% MeOH в DCM для получения трет-бутил-(3aR,5S,6S,7R,7aR)-5-(фторметил)-6,7-дигидрокси-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-2-илкарбамата в виде белого твердого вещества (170 мг, 85%). (ES, m/z): [M+H]+ 323,0; 1H ЯМР (300 МГц, CDCl3) δ 6,22 (д, J=6,6 Гц, 1H), 4,67-4,79 (м, 1H), 4,57-4,66 (м, 2H), 3,84-3,96 (м, 2H), 3,70 (т, J=8,4 Гц, 1H), 1,52 (с, 9H).
[00157] Через раствор указанного выше соединения (110 мг, 0,34 ммоль) в MeOH (5 мл) пробулькивали сухой газообразный HCl до насыщения при комнатной температуре. Еще через 3 ч, летучие вещества удаляли для получения остатка, который нейтрализовали концентрированным NH4OH и очищали на колонке с силикагелем, элюировали 10%-20% MeOH в DCM для получения (3aR,5S,6S,7R,7aR)-2-амино-5-(фторметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диола в виде белого твердого вещества (15 мг, 20%). (ES, m/z): [M+H]+ 222,9; 1H ЯМР (300 МГц, D2O) δ 6,31 (д, J=6,3 Гц, 1H), 4,63 (д, J=3,3 Гц, 1H), 4,47 (д, J=3,6 Гц, 1H), 4,07 (т, J=5,7 Гц, 1H), 3,93 (т, J=4,8 Гц, 1H), 3,66-3,80 (м, 2H), 3,51-3,58 (м, 1H).
Пример 16
(3aR,5S,6S,7R,7aR)-2-(азетидин-1-ил)-5-(фторметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диол
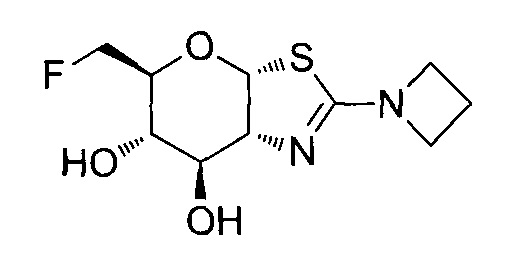
[00158] Раствор (3aR,5R,6S,7R,7aR)-2-(азетидин-1-ил)-5-(гидроксиметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диил дибензоата (100 мг, 0,2 ммоль) в DCM (10 мл) обрабатывали DAST (137 мг, 0,8 ммоль) в течение 30 мин при -78°C. После перемешивания в течение ночи при комнатной температуре, реакцию гасили насыщенным водным NaHCO3 (10 мл), экстрагировали DCM (3×10 мл), промывали солевым раствором (10 мл), сушили над безводным Na2SO4 и концентрировали в вакууме для получения неочищенного продукта в виде белого сиропа (80 мг), который растворяли в MeOH (5 мл) и обрабатывали K2CO3 (10 мг, 0,07 ммоль) в течение 3 ч при комнатной температуре. Реакционную смесь нейтрализовали AcOH и концентрировали. Остаток очищали на колонке с силикагелем, элюировали 5%-l0% MeOH в DCM для получения (3aR,5S,6S,7R,7aR)-2-(азетидин-l-ил)-5-(фторметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диола в виде белого твердого вещества (10 мг, 17%, две стадии). (ES, m/z): [M+H]+ 263,0; 1H ЯМР (300 МГц, D2O) δ 6,30 (д, J=6,3 Гц, 1H), 4,49-4,69 (дд, J=57, 3,3 Гц, 1H), 4,11-4,15 (м, 1H), 3,93-4,01 (м, 5H), 3,70-3,82 (м, 1H), 3,59-3,69 (м, 1H), 2,24-2,34 (м, 2H).
[00159] Соединения в следующих примерах синтезировали в соответствии с процедурами, аналогичными указанным выше схемам и примерам.
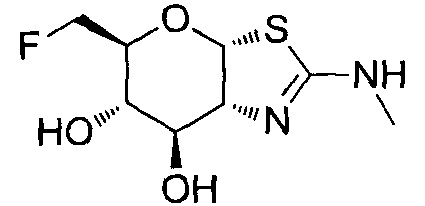
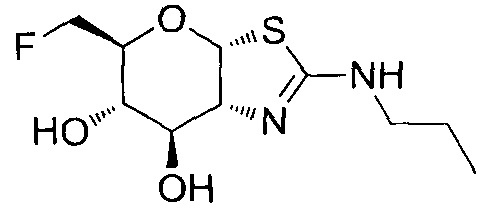
Пример 19
(3aR,5S,6S,7R,7aR)-5-(дифторметил)-2-(этиламино)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диол
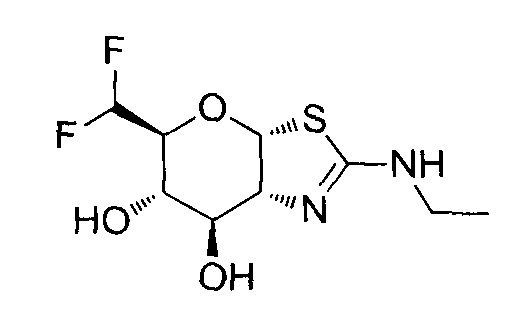
[00160] К раствору трет-бутил-((3aR,5R,6S,7R,7aR)-6,7-дигидрокси-5-(гидроксиметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-2-ил)(этил)карбамата (5,01 г, 14,4 ммоль) в DMF (25 мл) добавляли имидазол (1,57 г, 23,1 ммоль) и TBDMSCl (2,82 г, 21,7 ммоль). Реакционную смесь, перемешанную при комнатной температуре в течение 30 ч, разбавляли EtOAc (100 мл). Органические вещества промывали насыщенным NH4C1, солевым раствором, сушили над безводным Na2SO4 и концентрировали. Остаток очищали колоночной флэш-хроматографией на силикагеле (EtOAc/гексаны, 1:1), получая трет-бутил-((3aR,5R,6S,7R,7aR)-5-(((трет-бутилдиметилсилил)окси)метил)-6,7-дигидрокси-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-2-ил)(этил)карбамат в виде белого твердого вещества (5,08 г, 76%). 1H ЯМР (400 МГц, CDCl3) δ 6,12 (д, J=6,7 Гц, 1H), 4,25 (т, J=6,2 Гц, 1H), 4,16 (т, J=6,4 Гц, 1H), 4,10-4,04 (м, 2H), 3,91-3,85 (м, 3H), 3,65-3,62 (м, 1H), 1,55 (с, 9H), 1,26 (т, J=7 Гц, 3H), 0,89 (с, 9H), 0,08 (с, 6H).
[00161] К раствору указанного выше соединения (1,0 г, 2,2 ммоль) в пиридине (20 мл) при 0°C добавляли DMAP (0,024 г, 0,20 ммоль) с последующим медленным добавлением BzCl (2,0 мл, 17,6 ммоль). Смесь нагревали до комнатной температуры и перемешивали в течение ночи. Реакционную смесь разбавляли EtOAc (50 мл), промывали насыщенным раствором NaHCO3 и солевым раствором. Органический экстракт сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Остаточный пиридин совместно выпаривали с гексанами, и неочищенный остаток отделяли колоночной флэш-хроматографией на силикагеле (EtOAc/гексаны, 1:5) для получения (3aR,5R,6S,7R,7aR)-2-((трет-бутоксикарбонил)(этил)амино)-5-(((трет-бутил-диметилсилил)окси)метил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диил дибензоата (1,05 г, 71,3%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,08 (м, 2H), 7,99 (м, 2H), 7,61-7,52 (м, 2H), 7,47-7,38 (м, 4H), 6,12 (д, J=5,6 Гц, 1H), 5,89 (дд, J=1,5, 1,4 Гц, 1H), 5,39 (м, 1H), 4,46 (ддд, J=5,5, 2,9, 0,96 Гц, 1H), 3,96 (м, 2H), 3,80 (м, 1H), 3,75-3,70 (м, 2H), 1,54 (с, 9H), 1,18 (т, J=5,5 Гц, 3H), 0,83 (с, 9H), 0,01 (с, 3H), 0,03 (с, 3H).
[00162] К раствору указанного выше соединения (3,2 г, 4,8 ммоль) в сухом MeOH (30 мл) добавляли AcCl (0,07 мл, 1,0 ммоль) при 0°C. Реакционную смесь перемешивали при этой температуре в течение 30 мин и затем при комнатной температуре в течение ночи. Реакционную смесь разбавляли DCM (30 мл) и нейтрализовали 10% водным раствором NaHCO3. Слой DCM дополнительно промывали солевым раствором, сушили над безводным Na2SO4 и концентрировали. Неочищенный осадок очищали колоночной хроматографией на силикагеле (EtOAc/гексаны, 3:7) для получения (3aR,5R,6S,7R,7aR)-2-((трет-бутоксикарбонил)(этил)амино)-5-(гидроксиметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диил дибензоата (2,4 г, 90%) в виде пенистого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,06 (м, 2H), 8,01 (м, 2H), 7,58-7,53 (м, 2H), 7,47-7,39 (м, 4H), 6,14 (д, J=7,0 Гц, 1H), 5,93 (дд, J=1,9, 1,8 Гц, 1H), 5,34 (м, 1H), 4,49 (ддд, J=6,8, 3,6, 0,96 Гц, 1H), 4,03-3,92 (м, 2H), 3,80-3,65 (м, 3H), 1,53 (с, 9H), 1,19 (т, J=6,9 Гц, 3H).
[00163] К раствору указанного выше соединения (1,0 г, 1,8 ммоль) в сухом DCM (25 мл) при 0°C добавляли сухой пиридин (0,30 мл, 3,7 ммоль), с последующим добавлением DMP (1,14 г, 2,69 ммоль). Реакционную смесь перемешивали при 0°C в течение 10 мин и при комнатной температуре в течение следующих 1,5 ч. Реакционную смесь разбавляли 1 M Na2S203/насыщенным NaHCO3 (30 мл, 1:1) и перемешивали в течение 10 мин. Слой DCM отделяли, сушили над безводным Na2SO4 и концентрировали для выхода неочищенного твердого вещества (3aR,5S,6S,7R,7aR)-2-((трет-бутоксикарбонил)(этил)амино)-5-формил-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диил дибензоата (1,0 г неочищенного соединения). Продукт переносили для следующей реакции без какой-либо дополнительной очистки. 1H ЯМР (400 МГц, CDCl3) δ 9,71 (с, 1H), 8,04 (м, 4H), 7,58 (м, 2H), 7,47-7,39 (м, 4H), 6,14 (д, J=6,24 Гц, 1H), 6,01 (дд, J=1,8, 1,3 Гц, 1H), 5,53-5,51 (м, 1H), 4,38 (ддд, J=6,2, 3,2, 1,2 Гц, 1H), 4,27 (д, J=7,3 Гц, 1H), 4,0-3,9 (м, 2H), 1,53 (с, 9H), 1,15 (т, J=6,8 Гц, 3H).
[00164] Указанное выше соединение (1,0 г неочищенного соединения) собирали в DCM (30 мл) и охлаждали до -78°C. По каплям добавляли DAST (1 мл, 7,7 ммоль) с перемешиванием при -78°C. После добавления охлаждающую баню удаляли, и реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли насыщенным раствором NaHCO3 (15 мл). Слой DCM отделяли, сушили над безводным Na2SO4 и концентрировали. Неочищенный остаток очищали колоночной хроматографией на силикагеле (EtOAc/гексаны, 1:4) для получения (3aR,5S,6S,7R,7aR)-2-((трет-бутоксикарбонил)(этил)амино)-5-(дифторметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диил дибензоата (0,413 г, 40%) в виде пенистого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,08 (м, 2H), 8,0 (м, 2H), 7,60-7,53 (м, 2H), 7,46-7,39 (м, 4H), 6,13 (д, J=7,0 Гц, 1H), 5,94 (м, 1H), 5,93 (тд, J=54,08, 2,9 Гц, 1H), 5,59-5,56 (м, 1H), 4,55-4,52 (ддд, J=7,0, 3,4, 1,3 Гц, 1H), 3,98-3,86 (м, 3H), 1,55 (с, 9H), 1,17 (т, J=6,9 Гц, 3H).
[00165] К перемешанному раствору указанного выше соединения (0,41 г, 0,71 ммоль) в сухом MeOH (20 мл) добавляли K2CO3 (0,050 г, 0,36 ммоль) при 0°C. Реакционную смесь согревали при комнатной температуре и перемешивали 1,5 ч. К реакционной смеси добавляли AcOH (0,5 мл), и содержимое концентрировали. Неочищенный остаток очищали колоночной хроматографией на силикагеле (EtOAc/гексаны, 1:1) для получения трет-бутил-((3aR,5S,6S,7R,7aR)-5-(дифторметил)-6,7-дигидрокси-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-2-ил)(этил)карбамата (0,24 г, 91,6%) в виде пенистого твердого вещества. 1H ЯМР (400 МГц, MeOD) δ 6,03 (д, J=6,6 Гц, 1H), 5,93 (тд, J=54,5, 2,6 Гц, lH), 4,19 (м, 1H), 4,11 (т, J=4,3 Гц, 1H), 3,96-3,87 (м, 2H), 3,82 (дд, J=4,7, 4,4 Гц, 1H), 3,56-3,47 (м, 1H), 1,53 (с, 9H), 1,16 (т, J=6,9 Гц, 3H).
[00166] Указанное выше соединение (0,24 г, 0,65 ммоль) собирали в 30% TFA/DCM (10 мл) при 0°C и перемешивали при этой температуре в течение 1 ч и медленно нагревали до комнатной температуры в течение следующего 1 ч. Реакционную смесь выпаривали до сухости. Остаток нейтрализовали 2M раствором NH3/MeOH (5 мл) и очищали колоночной хроматографией на силикагеле (DCM/MeOH, 95:5) для получения (3aR,5S,6S,7R,7aR)-5-(дифторметил)-2-(этиламино)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диола в виде белого твердого вещества (0,166 г, 95,2%). 1H ЯМР (400 МГц, MeOD) δ 6,41 (д, J=6,4 Гц, 1H), 6,00 (тд, J=54,4, 1,9 Гц, 1H), 4,22 (т, J=5,9 Гц, 1H), 4,01 (т, J=4,6 Гц, 1H), 3,76-3,70 (м, 2H), 3,37-3,33 (м, 2H), 1,21 (т, J=7,2 Гц, 3H), 13C ЯМР (100 МГц, MeOD) δ 167,35, 116,25 (т, JC6,F 242,0 Гц, C-6), 89,10, 75,42 (т, J=21,0 Гц), 74,74, 71,24, 69,96 (т, J=4,0 Гц, C-4), 41,61, 15,09. ES/MS: 269,1 [M+1].
[00167] Образец указанного выше соединения (13,0 г, 48,5 ммоль с чистотой 97% по данным ВЭЖХ с УФ выявлением при 220 нм) растворяли в кипящем EtOAc (70 мл), затем по каплям добавляли гексан (примерно 20 мл) до появления некоторого количества твердых веществ. Полученному раствору давали возможность охладиться до комнатной температуры. Твердые вещества собирали для получения (3aR,5S,6S,7R,7aR)-5-(дифторметил)-2-(этиламино)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диола (10,6 г, с чистотой 98,3% по данным ВЭЖХ с УФ выявлением при 220 нм). Этот материал проявлял спектральные характеристики, идентичные характеристикам, описанным выше для этого соединения.
Пример 20
(3aR,5S,6S,7R,7aR)-5-(дифторметил)-2-(диметиламино)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диол
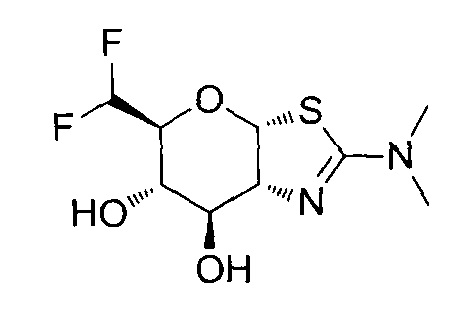
[00168] К раствору (3aR,5R,6S,7R,7aR)-2-(диметиламино)-5-(гидроксиметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диола (0,944 г, 3,8 ммоль) в DMF (5 мл) добавляли имидазол (0,8 г, 12 ммоль) и TBDMSC1 (0,75 г, 4,97 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 18 ч и концентрировали, используя высокий вакуум. Неочищенный остаток очищали колоночной флэш-хроматографией на силикагеле (DCM/MeOH, 95:5), получая (3aR,5R,6S,7R,7aR)-5-(((трет-бутил-диметилсилил)окси)метил)-2-(диметиламино)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диол в виде белого твердого вещества (0,76 г, 55,2%). 1H ЯМР (400 МГц, MeOD) δ 6,31 (д, J=6,4 Гц, 1H), 4,06 (т, J=6,1 Гц, 1H), 3,92-3,88 (м, 2H), 3,81-3,76 (дд, J=11,4, 5,7 Гц, 1H), 3,62-3,58 (м, 1H), 3,52-3,48 (дд, J=9,3, 5,5 Гц, 1H), 3,02 (с, 6H), 0,93 (с, 9H), 0,10 (с, 6H).
[00169] К раствору указанного выше соединения (0,84 г, 2,31 ммоль) в пиридине (10 мл) при 0°C добавляли DMAP (0,028 г, 0,23 ммоль) с последующим медленным добавлением BzCl (1,6 мл, 13,8 ммоль). Смесь нагревали до комнатной температуры и перемешивали в течение ночи. Реакционную смесь разбавляли EtOAc (50 мл), промывали насыщенным раствором NaHCO3 и солевым раствором. Органический экстракт сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Остаточный пиридин совместно выпаривали с гексанами, и неочищенный остаток отделяли колоночной флэш-хроматографией на силикагеле (EtOAc/гексаны, 2:3) для получения (3aR,5R,6S,7R,7aR)-5-(((трет-бутил-диметилсилил)окси)метил)-2-(диметиламино)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диил дибензоата (0,9 г, 68,4%) в виде пенистого белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,04-8,01 (м, 4H), 7,53-7,47 (м, 2H), 7,40-7,32 (м, 4H), 6,36 (д, J=5,2 Гц, 1H), 5,81 (т, J=2,4 Гц, 1H), 5,35-5,33 (дд, J=7,1, 0,9 Гц, 1H), 4,59 (т, J=4,2 Гц, 1H), 3,92-3,89 (м, 1H), 3,80-3,73 (м, 2H), 3,05 (с, 6H), 0,83 (с, 9H), 0,01 (с, 6H).
[00170] К раствору указанного выше соединения (0,88 г, 1,54 ммоль) в сухом MeOH (6 мл) добавляли AcCl (0,054 мл, 0,77 ммоль) при 0°C. Реакционную смесь перемешивали при этой температуре в течение 30 мин и затем при комнатной температуре в течение ночи. Реакционную смесь разбавляли DCM (30 мл) и нейтрализовали 10% водным раствором NaHCO3. Слой DCM дополнительно промывали солевым раствором, сушили над безводным Na2SO4 и концентрировали. Неочищенный осадок очищали колоночной хроматографией на силикагеле (EtOAc/гексаны, 1:1) для получения (3aR,5R,6S,7R,7aR)-2-(диметиламино)-5-(гидроксиметил)-5,6,7,7a-тетрагидро-3aH-пирано-[3,2-d]тиазол-6,7-диил дибензоата (0,57 г, 81%) в виде пенистого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,03-8,01 (м, 4H), 7,55-7,49 (м, 2H), 7,41-7,37 (м, 4H), 6,36 (д, J=6,5 Гц, 1H), 5,84-5,83 (дд, J=4,3, 3,0 Гц, 1H), 5,32-5,28 (м, 1H), 4,57 (т, J=5,2 Гц, 1H), 3,92-3,88 (м, 1H), 3,78-3,75 (дд, J=12,4, 2,5 Гц, 1H), 3,70-3,65 (дд, J=12,4, 5,4 Гц, 1H), 3,03 (с, 6H).
[00171] К раствору указанного выше соединения (0,57 г, 1,25 ммоль) в сухом DCM (10 мл) при 0°C добавляли DMP (0,8 г, 1,88 ммоль). Реакционную смесь перемешивали при 0°C в течение 10 мин и при комнатной температуре в течение следующих 1,5 ч, когда исходный материал был полностью израсходован. Реакционную смесь разбавляли 1:1 1M Na2S2O3:насыщенным NaHCO3 (30 мл) и перемешивали в течение 10 мин. Слой DCM отделяли, сушили над безводным Na2SO4 и концентрировали для выхода неочищенного твердого вещества, содержащего (3aR,5S,6S,7R,7aR)-2-(диметиламино)-5-формил-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диил дибензоат (0,56 г неочищенного соединения). Неочищенный продукт переносили для следующей реакции без какой-либо дополнительной очистки.
[00172] Указанное выше соединение (0,56 г неочищенного соединения) собирали в DCM (10 мл) и охлаждали до -78°C. По каплям добавляли DAST (0,72 мл, 5,5 ммоль) с перемешиванием при -78°C. После добавления охлаждающую баню удаляли, и реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли насыщенным раствором NaHCO3 (15 мл). Слой DCM отделяли, сушили над безводным Na2SO4 и концентрировали. Неочищенный остаток очищали колоночной хроматографией на силикагеле (EtOAc гексаны, 1:4) для получения (3aR,5S,6S,7R,7aR)-5-(дифторметил)-2-(диметиламино)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диил дибензоата (0,330 г, 55,2% в 2 стадии) в виде пенистого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,06-8,01 (м, 4H), 7,57-7,53 (м, 2H), 7,44-7,39 (м, 4H), 6,38 (д, J=5,3 Гц, 1H), 5,99-5,77 (м, 1H), 5,85 (м, 1H), 5,54 (д, J=6,8 Гц, 1H), 4,66-4,64 (м, 1H), 4,06-4,01 (м, 1H), 3,06 (с, 6H).
[00173] К перемешанному раствору указанного выше соединения (0,33 г, 0,69 ммоль) в сухом MeOH (8 мл) добавляли K2CO3 (0,073 г, 0,53 ммоль) при 0°C. Реакционную смесь согревали при комнатной температуре и перемешивали 1,5 ч. К реакционной смеси добавляли AcOH (0,5 мл), и содержимое концентрировали. Полученную таким образом соль AcOH обрабатывали 2M раствором NH3/MeOH 8 мл) и снова концентрировали. Остаток очищали колоночной хроматографией на силикагеле (DCM/MeOH, 9:1) для получения (3aR,5S,6S,7R,7aR)-5-(дифторметил)-2-(диметиламино)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диола в виде пенистого твердого вещества (0,1 г, 54%). 1H ЯМР (400 МГц, MeOD) δ 6,31 (д, J=6,4 Гц, 1H), 5,98 (тд, J=54,1, 2,0 Гц, 1H), 4,20 (т, J=5,6 Гц, 1H), 4,04 (т, J=4,7 Гц, 1H), 3,78-3,66 (м, 2H), 3,03 (с, 6H), 13C ЯМР (100 МГц, MeOD) δ 166,20, 116,56 (т, JC6,F 241,2 Гц, C-6), 91,41, 77,21, 75,57, 75,04 (т, J=21,0 Гц), 70,68 (т, J=3,3 Гц, C-4), 41,17. ES/MS: 269,1 [M+1].
Пример 21
(3aR,5S,6S,7R,7aR)-2-(аллиламино)-5-(дифторметил)-5,6,7,7a-тетрагидро-3aH-пирано(3,2-d]тиазол-6,7-диол
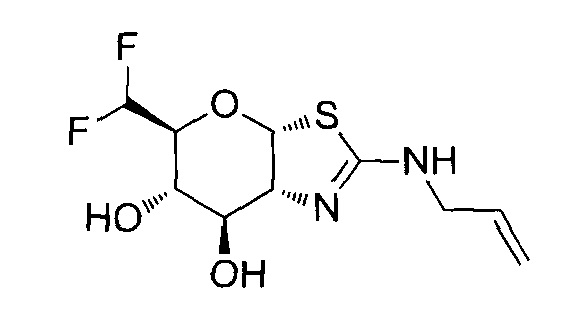
[00174] Смесь проп-2-ен-1-амина (9,2 г, 0,16 моль) и (2S,3R,4R,5S,6R)-6-(ацетоксиметил)-3-изотиоцианаттетрагидро-2H-пиран-2,4,5-триил триацетата (60 г, 0,154 моль) в DCM (300 мл) перемешивали в течение 1 ч при комнатной температуре с последующим добавлением TFA (88 г, 0,77 моль). Полученный раствор перемешивали в течение ночи при комнатной температуре и гасили водным NaHCO3 до pH 8 и экстрагировали DCM (3×300 мл). Объединенный органический слой сушили над безводным Na2SO4 и конденсировали для получения остатка, который очищали колоночной хроматографией на силикагеле с 1%~2% MeOH в DCM для получения (3aR,5R,6S,7R,7aR)-5-(ацетоксиметил)-2-(аллиламино)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диил диацетата в виде желтой жидкости (58 г, 84%). (ES, m/z): [M+H]+ 387,0; 1H ЯМР (300 МГц, CDCl3) δ 6,33 (д, J=6,6 Гц, 1H), 5,84-5,96 (м, 1H), 5,32-5,44 (м, 4H), 4,98-5,04 (м, 1H), 4,37 (т, J=5,7 Гц, 1H), 4,28-4,22 (м, 2H), 3,92-3,95 (м, 3H), 2,14 (с, 3H), 2,12 (с, 3H), 2,11 (с, 3H).
[00175] Раствор указанного выше соединения (50 г, 0,13 моль) в MeOH (300 мл) обрабатывали K2CO3 (3,6 г, 0,26 mol) в течение ночи при комнатной температуре и с последующим добавлением Boc2O (56 г, 0,26 моль) и Et3N (19,6 г, 0,19 моль). После дополнительных 2 ч полученный раствор концентрировали для получения остатка, который очищали колоночной хроматографией на силикагеле, элюировали 5% MeOH в DCM для получения трет-бутил-аллил((3aR,5R,6S,7R,7aR)-6,7-дигидрокси-5-(гидроксиметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-2-ил)карбамата в виде масла (42 г, 90% в две стадии). (ES, m/z): [M+H]+ 361,0; 1H ЯМР (300 МГц, CDCl3) δ 6,14 (д, J=6,9 Гц, 1H), 5,84-5,96 (м, 1H), 5,22-5,44 (м, 4H), 4,37-4,38 (м, 1H), 4,22-4,28 (м, 2H), 3,92-3,95 (м, 3H), 1,54 (с, 9H).
[00176] Смесь TBDMSC1 (26 г, 0,17 ммоль), DMAP (1,4 г, 0,01 моль), Et3N (23,5 г, 0,23 ммоль), и указанное выше соединение (42 г, 0,12 моль) в DCM (300 мл) перемешивали в течение ночи при 40°C. Реакционную смесь гасили насыщенным водным NaHCO3 (20 мл) и конденсировали для получения остатка, который очищали колоночной хроматографией на силикагеле, элюировали 1%~2% MeOH в DCM для получения трет-бутил-аллил-((3aR,5R,6S,7R,7aR)-5-((трет-бутил-диметилсилилокси)метил)-6,7-дигидрокси-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-2-ил)карбамата в виде светло-желтого сиропа (48 г, 87%). (ES, m/z): [M+H]+ 474,9; 1H ЯМР (300 МГц, CDCl3) δ 6,16 (д, J=6,6 Гц, 1H), 5,84-5,93 (м, 1H), 5,22-5,32 (м, 4H), 4,65-4,67 (м, 1H), 4,26-4,31 (м, 1H), 4,20-4,22 (м, 1H), 3,84-3,88 (м, 3H), 1,54 (с, 9H), 0,93 (с, 9H), 0,09 (с, 6H).
[00177] Смесь BzCl (11,1 г, 79 ммоль), DMAP (322 мг, 2,63 ммоль), пиридина (20,8 г, 263 ммоль) и указанного выше соединения (12,5 г, 26,3 ммоль) в DCM (150 мл) при 0°C перемешивали в течение ночи при комнатной температуре. Реакционную смесь гасили насыщенным водным NaHCO3 (200 мл), экстрагировали DCM (3×100 мл), промывали солевым раствором (3×50 мл), сушили над безводным MgSO4 и конденсировали для получения остатка, который очищали колоночной хроматографией на силикагеле 10% EtOAc в простом петролейном эфире для получения (3aR,5R,6S,7R,7aR)-2-(аллил(трет-бутоксикарбонил)амино)-5-((трет-бутил--диметилсилилокси)метил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диил дибензоата в виде белого твердого вещества (16,5 г, 78%). (ES, m/z): [M+H]+ 683,1; 1H ЯМР (300 МГц, CDCl3) δ 8,05-8,10 (м, 4H), 7,56-7,61 (м, 2H), 7,42-7,47 (м, 4H), 6,27 (д, J=7,2 Гц, 1H), 5,99-6,02 (м, 2H), 5,86-5,92 (м, 1H), 5,43-5,46 (м, 1H), 5,10-5,18 (м, 2H), 4,81-4,89 (м, 1H), 4,56-4,62 (м, 2H), 3,76-3,87 (м, 3H), 1,56 (с, 9H), 0,88 (с, 9H), 0,04 (с, 6H).
[00178] Раствор указанного выше соединения (7,0 г, 10,3 ммоль) в MeOH (50 мл) обрабатывали AcCl (0,5 мл, 0,07 ммоль) в течение 3 ч при комнатной температуре. Реакционную смесь гасили насыщенным водным NaHCO3 (10 мл) и водой (200 мл), экстрагировали DCM (3×50 мл), сушили над безводным MgSO4 и концентрировали в вакууме для получения остатка, который очищали колоночной хроматографией на силикагеле 10% EtOAc в простом петролейном эфире для получения (3aR,5R,6S,7R,7aR)-2-(аллил(трет-бутоксикарбонил)амино)-5-(гидроксиметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диил дибензоата в виде белого сиропа (1,7 г, 29%). (ES, m/z): [M+H]+ 569,0; 1H ЯМР (300 МГц, CDCl3) δ 8,06-8,09 (м, 4H), 7,60-7,63 (м, 2H), 7,43-7,58 (м, 4H), 6,26 (д, J=6,9 Гц, 1H), 6,06-6,27 (м, 2H), 5,37-5,41 (д, J=8,4 Гц, 1H), 5,11-5,28 (м, 2H), 4,82-4,84 (м, 1H), 4,56-4,62 (м, 2H), 3,71-3,86 (м, 3H), 1,56 (с, 9H).
[00179] Раствор указанного выше соединения (1,5 г, 2,6 ммоль) в DCM (30 мл) обрабатывали DMP (1,7 г, 4,0 ммоль) в течение 2 ч при комнатной температуре. Полученный раствор гасили насыщенным водным NaHCO3 (10 мл) и насыщенным водным Na2S2O3 (10 мл), экстрагировали DCM (3×50 мл), сушили над Na2SO4, и концентрировали в вакууме для получения остатка, который очищали на короткой колонке с силикагелем (элюент 30% EtOAc в петролейном эфире) для получения неочищенного альдегида (1,3 г), который растворяли в DCM (20 мл) и обрабатывали DAST (1,5 г, 9,3 ммоль) при -78°C. После перемешивания в течение ночи при комнатной температуре, полученный раствор гасили насыщенным водным NaHCO3 (50 мл), экстрагировали DCM (3×50 мл), сушили над Na2SO4 и концентрировали в вакууме для получения остатка, который очищали колоночной хроматографией на силикагеле (элюент 10% EtOAc в петролейном эфире) для получения (3aR,5S,6S,7R,7aR)-2-(аллил(трет-бутоксикарбонил)амино)-5-(дифторметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диил дибензоата в виде белого твердого вещества (650 мг, 43%). (ES, m/z): [M+H]+ 589,0; 1H ЯМР (300 МГц, CDCl3) δ 8,03-8,13 (м, 4H), 7,56-7,61 (м, 2H), 7,48-7,51 (м, 4H), 6,11-6,14 (д, J=8,7 Гц, 1H), 5,95-6,09 (тд, J=54,3 Гц, 2,8 Гц, 1H), 5,19-5,25 (м, 2H), 4,44-4,45 (м, 2H), 4,31-4,36 (м, 2H), 4,10-4,18 (м, 2H), 1,53 (с, 9H).
[00180] Раствор указанного выше соединения (200 мг, 0,34 ммоль) в MeOH (15 мл) обрабатывали K2CO3 (10 мг, 0,07 ммоль) в течение 3 ч при комнатной температуре. Реакционную смесь нейтрализовали уксусной кислотой и конденсировали для получения остатка, который очищали колоночной хроматографией на силикагеле, элюировали 1%~2% MeOH в DCM для получения трет-бутил-аллил-((3aR,5S,6S,7R,7aR)-5-(дифторметил)-6,7-дигидрокси-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-2-ил)карбамата в виде желтого масла (150 мг, 76%). (ES, m/z): [M+H]+ 381,0; 1H ЯМР (300 МГц, CDCl3) δ 6,13 (д, J=8,7 Гц, 1H), 5,95-6,09 (м, 1H), 5,19-5,25 (м, 2H), 4,44-4,45 (м, 2H), 4,31-4,36 (м, 2H), 4,10-4,18 (м, 2H), 1,53 (с, 9H).
[00181] Раствор указанного выше соединения (150 мг, 0,39 ммоль) в DCM (18 мл) обрабатывали TFA (1,8 мл) в течение ночи при комнатной температуре. Реакционную смесь конденсировали для получения остатка, который нейтрализовали NH4OH (0,5 мл, 25%~28%, масс./об.) для очистки препаративной ВЭЖХ в следующих условиях [(препаративная ВЭЖХ Agilent 1200; Колонка: Sun Fire Prep C 18, 19*50мм 5 мкм; подвижная фаза: Вода с 0,03% NH4OH и CH3CN (10% CH3CN до 45% через 10 мин; Детектор: УФ 220 нм) для получения (3aR,5S,6S,7R,7aR)-2-(аллиламино)-5-(дифторметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диола в виде белого твердого вещества (16,6 мг, 15%). [M+H]+ 281,0; 1H ЯМР (300 МГц, D2O) δ 6,20 (д, J=6,3 Гц, 1H), 6,14-6,16 (м, 1H), 5,74-5,78 (т, J=2,7 Гц, 1H), 5,04-5,10 (м, 2H), 4,20-4,24 (т, J=6,0 Гц, 1H), 4,04-4,07 (т, J=4,2 Гц, 1H), 3,66-3,86 (м, 4H).
Пример 22
3aR,5S,6S,7R,7aR)-2-амино-5-(дифторметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диол
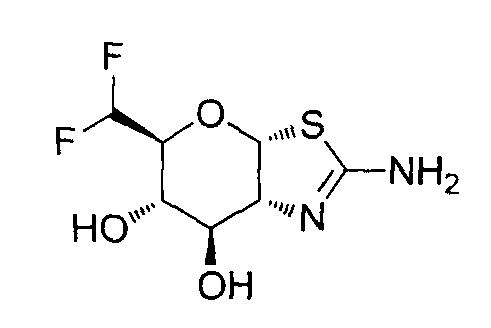
[00182] К раствору (3aR,5S,6S,7R,7aR)-2-(аллил(трет-бутоксикарбонил)амино)-5-(дифторметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диил дибензоата (900 мг, 1,53 ммоль) в 1,4-диоксане (30 мл) добавляли Pd(PPh3)4 (347 мг, 0,30 ммоль), HCO2H (384 мг, 8,35 ммоль) и Et3N (846 мг, 8,38 ммоль) в атмосфере N2 при 10°C. Через 20 мин при 60°C добавляли дополнительный HCO2H (1,4 г, 30,3 ммоль). После перемешивания в течение ночи при 60°C реакционную смесь гасили насыщенным водным NaHCO3, экстрагировали DCM (3×30 мл), сушили безводным MgSO4, и концентрировали при пониженном давлении для получения остатка, который очищали колоночной хроматографией на силикагеле, элюировали 10% EtOAc в гексане для получения (3aR,5S,6S,7R,7aR)-2-(трет-бутоксикарбониламино)-5-(дифторметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диил дибензоата в виде белого сиропа (600 мг, 72%). (ES, m/z): [M+H]+ 549,0; 1H ЯМР (300 МГц, CDCl3) δ 8,07-8,14 (м, 4H), 7,60-7,65 (м, 2H), 7,51-7,59 (м, 4H), 6,34 (д, J=8,7 Гц, 1H), 5,82-6,20 (тд, J=54,0 Гц, 2,7 Гц, 1H), 5,83-5,85 (м, 1H), 5,61-5,64 (м, 1H), 4,59-4,63 (м, 1H), 4,05-4,18 (м, 1H), 1,53 (с, 9H).
[00183] Раствор указанного выше соединения (600 мг, 1,1 ммоль) в MeOH (50 мл) обрабатывали K2CO3 (45 мг, 0,33 ммоль) в течение 3 ч при комнатной температуре. Реакционную смесь нейтрализовали добавлением AcOH и конденсировали для получения остатка, который очищали колоночной хроматографией на силикагеле, элюировали 1% ~ 5% MeOH в DCM для получения трет-бутил-(3aR,5S,6S,7R,7aR)-5-(дифторметил)-6,7-дигидрокси-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-2-илкарбамата в виде твердого вещества (300 мг, 81%). (ES, m/z): [M+H]+ 341,0; 1H ЯМР (300 МГц, CDCl3) δ 6,17 (д, J=6,6 Гц, 1H), 5,85-6,21 (тд, J=51,6 Гц, 2,7 Гц, 1H), 3,84-4,16 (м, 4H), 1,56 (с, 9H).
[00184] Раствор указанного выше соединения (135 мг, 0,4 ммоль) в DCM (10 мл) обрабатывали TFA (l мл) в течение ночи при комнатной температуре. Удаление растворителя давало остаток, который растворяли в MeOH (5 мл) и нейтрализовали концентрированным NH4OH (0,5 мл, 25% ~ 28%, масс./об.). Концентрация и очистка препаративной ВЭЖХ в следующих условиях: [Колонка (Agilent 1200):, X-Bridge C 18; подвижная фаза, 50 ммоль/л NH4HCO3 в воде с 0,05% NH4OH и CH3CN (CH3CN от 5% до 20% через 10 мин); Детектор, 220 нм УФ] давали (3aR,5S,6S,7R,7aR)-2-амино-5-(дифторметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диол в виде белого твердого вещества (26,7 мг, 27%). (ES, m/z): [M+H]+ 240,9; 1H ЯМР (300 МГц, CDOD) δ 6,28 (д, J=6,3 Гц, 1H), 5,50-6,14 (тд, J=54,3 Гц, 2,1 Гц, 1H), 4,16-4,20 (м, 1H), 4,02-4,05 (т, J=4,5 Гц, 1H), 3,74-3,80 (м, 1H), 3,63-3,71 (м, 1H).
Пример 23
(3aR,SS,6S,7R,7aR)-5-(дифторметил)-2-(проп-2-иниламино)-5,6,7,7а-тетрагидро-3аН-пирано[3,2-d]тиазол-6,7-диол
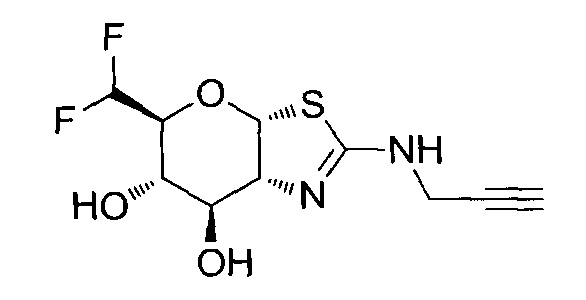
[00186] Раствор (3aR,5S,6S,7R,7aR)-2-(трет-бутоксикарбониламино)-5-(дифторметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диил дибензоата (800 мг, 1,46 ммоль) и 3-бромпроп-1-ина (538 мг, 4,56 ммоль) в DMF (15 мл) обрабатывали K2CO3 (125 мг, 0,91 ммоль) в течение ночи при комнатной температуре. Реакционную смесь гасили насыщенным водным NH4C1 (30 мл), экстрагировали DCM (3×10 мл), промывали солевым раствором (3×10 мл), сушили над безводным MgSO4, и конденсировали для получения остатка, который очищали колоночной хроматографией на силикагеле 10% EtOAc в простом петролейном эфире для получения смеси, содержащей (3aR,5S,6S,7R,7aR)-2-(трет-бутоксикарбонил(проп-2-инил)амино)-5-(дифторметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диил дибензоат в виде светло-желтого твердого вещества (210 мг), которое использовали на следующей стадии без дополнительной очистки. (ES, m/z): [M+H]+ 587,0.
[00187] Раствор указанной выше смеси (210 мг, 0,36 ммоль) в MeOH (10 мл) обрабатывали K2CO3 (14,8 мг, 0,11 ммоль) в течение 2 ч при 25°C и с последующей нейтрализацией AcOH. Удаление летучих веществ давало остаток, который очищали колоночной хроматографией на силикагеле 20% EtOAc в простом петролейном эфире для получения смеси, содержащей трет-бутил-(3aR,5S,6S,7R,7aR)-5-(дифторметил)-6,7-дигидрокси-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-2-ил(проп-2-инил)карбамат (125 мг). (ES, m/z): [M+H]+ 379,0.
[00188] Раствор указанной выше смеси (50 мг, 0,1 ммоль) в THF (10 мл) обрабатывали MeMgCl (0,3 мл, 1 ммоль, 3 M в THF) в течение ночи при 10°C. Реакцию гасили насыщенным водным NH4C1 (1 мл), и конденсировали для получения остатка, который растворяли в 10% MeOH в DCM и фильтровали через короткую колонку силикагеля. Концентрация и очистка препаративной ВЭЖХ в следующих условиях: [(Препаративная ВЭЖХ Agilent 1200): Колонка, Sun Fire Prep C 18*50 мм 5 мкм; подвижная фаза, H2O с CH3CN (от 40% CH3CN до 60% через 5 мин); Детектор, УФ, 220 нм] давала (3aR,5S,6S,7R,7aR)-5-(дифторметил)-2-(проп-2-иниламино)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диол (22 мг, быстрее передвигающийся изомер ВЭЖХ). (ES, m/z): [M+H]+ 279,0; 1H ЯМР (300 МГц, DMSO) δ 7,37 (шир., 1H), 6,21 (д, J=6,3 Гц, 1H), 5,76-6,28 (тд, J=53,7 Гц, 2,1 Гц, 1H), 5,27(д, J=4,5 Гц, 1H), 4,96-4,98(д, J=6,3 Гц, 1Η), 4,05-4,09 (м, 1H), 3,85-3,99 (м, 3H), 3,51-3,60 (м, 2H), 3,13 (с, 1H).
Пример 24
(3aR,5S,6S,7R,7aR)-2-(азетидин-1-ил)-5-(дифторметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диол
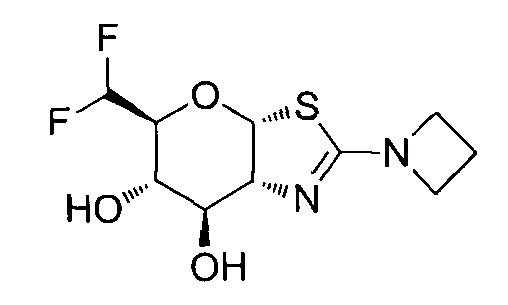
[00190] Смесь Et3N (18,7 г, 185 ммоль), азетидина гидрохлорида (12 г, 129 ммоль) и (2S,3R,4R,5S,6R)-6-(ацетоксиметил)-3-изотиоцианаттетрагидро-2H-пиран-2,4,5-триил триацетата (48 г, 123 ммоль) в DCM (500 мл) перемешивали в течение 2 ч при комнатной температуре и с последующим добавлением TFA (56 г, 493 ммоль). Полученный раствор перемешивали в течение ночи при комнатной температуре, нейтрализовали NaHCO3, экстрагировали DCM (3×100 мл), сушили над безводным Na2SO4, и конденсировали для получения остатка, который очищали колоночной хроматографией на силикагеле, элюировали 30% EtOAc в петролейном эфире для получения (3aR,5R,6S,7R,7aR)-5-(ацетоксиметил)-2-(азетидин-l-ил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диил диацетата в виде светло-желтого сиропа (36 г, 75%). (ES, m/z): [M+H]+: 386,9; 1H ЯМР (300 МГц, CDCl3) δ 6,29 (д, J=6,6 Гц, 1H), 5,44-5,47 (м, 1H), 4,95-4,99 (м, 1H), 4,36 (т, J=5,4 Гц, 1H), 4,05-4,18 (м, 6H), 3,86-3,92 (м, 1H), 2,34-2,44 (м, 2H), 2,07-2,14 (м, 9H).
[00191] Раствор указанного выше соединения (36 г, 93 ммоль) в MeOH (200 мл) обрабатывали K2CO3 (5,14 г, 37 ммоль) в течение 4 ч при комнатной температуре. Полученный раствор фильтровали через короткую колонку силикагеля для получения (3aR,5R,6S,7R,7aR)-2-(азетидин-l-ил)-5-(гидроксиметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диол в виде светло-желтого сиропа (21 г, 87%). (ES, m/z) [M+H]+: 261,0; 1H ЯМР (300 МГц, DMSO) δ 6,27 (д, J=6,3 Гц, 1H), 3,94 (т, J=6,3 Гц, 1H), 3,85 (т, J=7,5 Гц, 4H), 3,70 (т, J=4,8 Гц, 1H), 3,55-3,59 (м, 1H), 3,33-3,42 (м, 3H), 2,21-2,51 (м, 2H).
[00192] К раствору указанного выше соединения (34 г, 131 ммоль), Et3N (20,2 г, 0,2 моль) и DMAP (0,5 г, 4 ммоль) в DCM (200 мл) добавляли TBDMSC1 (21,6 г, 143 ммоль). После перемешивания в течение ночи при 15°C реакционную смесь гасили добавлением насыщенного водного NaHCO3 (200 мл), экстрагировали DCM (3×100 мл), сушили над безводным Na2SO4, и концентрировали в вакууме для получения остатка, который очищали на колонке с силикагелем 2% MeOH в DCM для получения (3aR,5R,6S,7R,7aR)-2-(азетидин-l-ил)-5-((трет-бутил-диметилсилилокси)метил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диола в виде твердого вещества (32 г, 65%). (ES, m/z): [M+H]+: 375,0; 1H ЯМР (300 МГц, CDCl3) δ 6,38 (д, J=6,3 Гц, 1H), 4,24 (т, J=6,3 Гц, 1H), 4,04-4,09 (м, 5H), 3,85 (д, J=3,6 Гц, 2H), 3,70-3,78 (м, 1H), 3,66-3,69 (м, 1H), 2,33-2,43 (м, 2H), 0,91 (с, 9H), 0,09 (с, 6H).
[00193] Раствор указанного выше соединения (3,0 г, 8,0 ммоль) в DMF (40 мл) обрабатывали NaH (1,5 г, 37,50 ммоль) при комнатной температуре в течение 30 мин и с последующим добавлением BzCl (3,36 г, 24 ммоль). Полученный раствор перемешивали в течение ночи при 15°C, гасили водой/льдом (100 мл), экстрагировали DCM (3×50 мл), промывали солевым раствором (3×20 мл), сушили над безводным Na2SO4, и конденсировали для получения остатка, который очищали колоночной хроматографией на силикагеле 10% EtOAc в простом петролейном эфире для получения (3aR,5R,6S,7R,7aR)-2-(азетидин-l-ил)-5-((трет-бутил-диметилсилилокси)метил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диил дибензоата в виде светло-желтого масла (1,38 г, 30%). (ES, m/z): [M+H]+: 583,0; 1H ЯМР (300 МГц, CDCl3) δ 8,05-8,09 (м, 4H), 7,54-7,59 (м, 2H), 7,28-7,39 (м, 4H), 6,43 (д, J=6,3 Гц, 1H), 5,84-5,96 (м, 1H), 5,37 (дд, J=2,1, 1,5 Гц, 1H), 4,59-4,61 (м, 1H), 4,04-4,09 (м, 5H), 3,78-3,93 (м, 2H), 2,38-2,43 (м, 2H), 0,91 (с, 9H), 0,09 (с, 6H).
[00194] Раствор указанного выше соединения (1,3 г, 2,2 ммоль) в MeOH (10 мл) обрабатывали AcCl (1 мл, 0,45 ммоль) в течение ночи при 15°C. Реакцию гасили насыщенным водным NaHCO3 (20 мл), экстрагировали DCM (3×40 мл), сушили над безводным Na2SO4, и концентрировали в вакууме для получения остатка, который очищали колоночной хроматографией на силикагеле, элюировали 20% EtOAc в простом петролейном эфире для получения (3aR,5R,6S,7R,7aR)-2-(азетидин-l-ил)-5-(гидроксиметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диил дибензоат в виде твердого вещества (0,42 г, 40%). (ES, m/z): [M+H]+: 469,0; 1H ЯМР (300 МГц, CDCl3) δ 8,04-8,11 (м, 4H), 7,55-7,62 (м, 2H), 7,42-7,48 (м, 4H), 6,48 (д, J=6,6 Гц, 1H), 5,92 (т, J=3,6 Гц, 1H), 5,35 (дд, J=8,0, 3,6 Гц, 1H), 4,63 (т, J=5,4 Гц, 1H), 4,11-4,18 (м, 4H), 3,92-3,96 (м, 1H), 3,80-3,84 (м, 1H), 3,69-3,68 (м, 1H), 2,38-2,48 (м, 2H).
[00195] Раствор указанного выше соединения (400 мг, 0,85 ммоль) в DCM (20 мл) обрабатывали DMP (600 мг, 1,41 ммоль) при 0°C в течение 10 мин и дополнительно 2 ч при 25°C. Полученный раствор гасили насыщенным водным NaHCO3 (10 мл) и насыщенным водным Na2S203 (10 мл), экстрагировали DCM (3×20 мл), сушили над безводным Na2SO4 и концентрировали в вакууме для получения остатка, который растворяли в DCM (20 мл) и обрабатывали DAST (5 мл, 4,48 ммоль) при -78°C. Полученный раствор перемешивали в течение ночи при 15°C и гасили насыщенным водным NaHCO3 (10 мл). Органический слой отделяли, и полученный водный слой экстрагировали DCM (2×20 мл). Объединенные органические слои сушили над безводным Na2SO4 и концентрировали в вакууме. Остаток очищали на колонке с силикагелем с 20% EtOAc в простом петролейном эфире для получения (3aR,5S,6S,7R,7aR)-2-(азетидин-l-ил)-5-(дифторметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диил дибензоата в виде светло-желтого масла (240 мг, 46%). (ES, m/z): [M+H]+: 488,9; 1H ЯМР (300 МГц, CDCl3) δ 8,03-8,13 (м, 4H), 7,56-7,63 (м, 2H), 7,42-7,49 (м, 4H), 6,48 (д, J=6,6 Гц, 1H), 5,94-5,96 (м, 2H), 5,58 (д, J=8,7 Гц, 1H), 4,73 (с, 1H), 4,07-4,17 (м, 4H), 2,43-2,51 (м, 2H).
[00196] Раствор указанного выше соединения (110 мг, 0,23 ммоль) в MeOH (10 мл) обрабатывали K2CO3 (20 мг, 0,14 ммоль) в течение 2 ч при 25°C и затем нейтрализовали AcOH. Удаление летучих веществ давало остаток, который очищали колоночной хроматографией на силикагеле 10% MeOH в DCM для получения (3aR,5S,6S,7R,7aR)-2-(азетидин-l-ил)-5-(дифторметил)-5,6,7,7a- тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диола в виде белого твердого вещества (24 мг, 22%). (ES, m/z): [M+H]+: 280,9; 1H ЯМР (300 МГц, D2O) δ 6,29 (д, J=6,3 Гц, 1H), 5,82-6,00 (тд, J=54,0 Гц, 1,8 Гц, 1H), 4,24 (т, J=5,7 Гц, 1H), 3,97-4,04 (м, 5H), 3,82-3,87 (м, 1H), 3,69-3,80 (м, 1H), 2,29 (м, 2H).
[00197] Соединения следующих примеров были синтезированы в соответствии с процедурами, аналогичными указанным выше схемам и примерам.

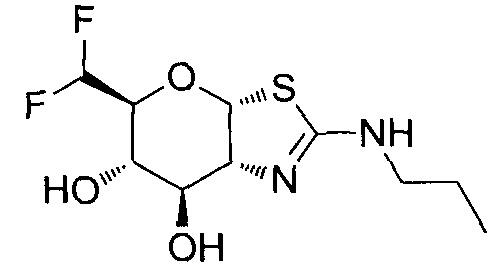
Пример 27
(3aR,5S,6S,7aR)-5-(дифторметил)-2-(метиламино)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6-ол
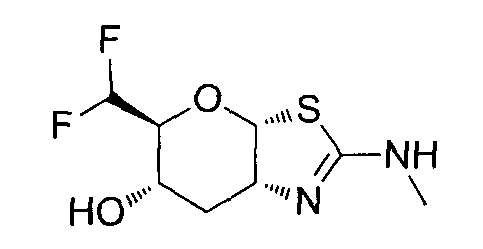
[00198] Смесь ((3aR,5R,6S,7R,7aR)-6-(бензоилокси)-2-((трет-бутоксикарбонил)(метил)амино)-7-гидрокси-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-5-ил)метил бензоата (5,00 г, 9,24 ммоль) и тио-CDI (90% тех., 3,40 г, 19,1 ммоль) в безводном DMF (30 мл) перемешивали при 95°C в течение 4 ч. После охлаждения растворитель удаляли при пониженном давлении, и остаток очищали колоночной флэш-хроматографией на силикагеле (EtOAc/гексаны, от 1:10 до 2:3), получая (3aR,5R,6S,7R,7aR)-7-((1H-имидазол-l-карбонотиоил)окси)-5-((бензоилокси)метил)-2-((трет-бутоксикарбонил)(метил)амино)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6-ил бензоат в виде бледно-желтого твердого вещества (5,60 г, 93%). 1H ЯМР (400 МГц, CDCl3) δ 8,76 (с, 1H), 8,03-8,01 (м, 2H), 7,97-7,95 (м, 2H), 7,64-7,60 (м, 1H), 7,54-7,50 (м, 1H), 7,45 (т, J=7,7 Гц, 2H), 7,34 (т, J=7,7 Гц, 2H), 7,02 (с, 1H), 6,38-6,37 (м, 1H), 6,15 (д, J=7,1 Гц, 1H), 5,56 (тд, J=1,2, 9,2 Гц, 1H), 4,70-4,67 (м, 1H), 4,58 (дд, J=3,2, 12,1 Гц, 1H), 4,42 (дд, J=5,1, 12,1 Гц, 1H), 4,08-4,03 (м, 1H), 3,43 (с, 3H), 1,56 (с, 9H).
[00199] Смесь указанного выше соединения (5,60 г, 8,59 ммоль), Bu3SnH (5,84 г, 17,0 ммоль) и ABCN (0,15 г, 0,60 ммоль) в смешанном безводном толуоле/THF (50/50 мл) перемешивали при 90°C в течение 16 ч. После охлаждения растворитель удаляли при пониженном давлении, и остаток очищали колоночной флэш-хроматографией на силикагеле (EtOAc/гексаны, от 1:10 до 1:2), получая ((3aR,5R,6S,7aR)-6-(бензоилокси)-2-((трет-бутоксикарбонил)(метил)амино)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-5-ил)метил бензоат в виде белого твердого вещества (3,20 г, 70%). 1H ЯМР (400 МГц, CDCl3) δ 8,03-7,98 (м, 4H), 7,58-7,49 (м, 2H), 7,44-7,40 (м, 4H), 6,08 (д, J=7,3 Гц, 1H), 5,44-5,40 (м, 1H), 4,49-4,40 (м, 3H), 4,07-4,03 (м, 1H), 3,35 (с, 3H), 2,64-2,59 (м, 1H), 2,44-2,37 (м, 1H), 1,56 (с, 9H).
[00200] Соблюдая процедуру, описанную для примера 32, указанное выше соединение (3,20 г, 6,10 ммоль) был подвергнут снятию защиты бензоила с использованием K2CO3. После очистки колоночной флэш-хроматографией на силикагеле (MeOH/DCM, от 1:50 до 1:20) получали трет-бутил-((3aR,5R,6S,7aR)-6-гидрокси-5-(гидроксиметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-2-ил)(метил)карбамат в виде белого твердого вещества (1,82 г, 94%). 1H ЯМР (400 МГц, CDCl3) δ 5,91 (д, J=6,9 Гц, 1H), 4,36-4,32 (м, 1H), 3,89-3,85 (м, 1H), 3,81-3,75 (м, 1H), 3,65-3,59 (м, 1H), 3,38-3,34 (м, 1H), 3,33 (с, 3H), 2,48-2,43 (м, 1H), 2,32 (д, J=10,7 Гц, 1H), 2,17-2,11 (м, 1H), 1,84 (т, J=6,3 Гц, 1H), 1,54 (с, 9H).
[00201] Соблюдая процедуру, описанную для примера 32, указанное выше соединение (1,82 г, 5,74 ммоль) было защищено моно-TBDMS. После очистки колоночной флэш-хроматографией на силикагеле (EtOAc/гексаны, от 1:10 до 1:2) получали трет-бутил-((3aR,5R,6S,7aR)-5-(((трет-бутил-диметилсилил)окси)метил)-6-гидрокси-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-2-ил)(метил)карбамат в виде бесцветного липкого масла (2,30 г, 93%). 1H ЯМР (400 МГц, CDCl3) δ 5,92 (д, J=6,8 Гц, 1H), 4,31-4,28 (м, 1H), 3,92-3,90 (м, 1H), 3,73 (д, J=4,6 Гц, 2H), 3,35-3,31 (м, 1H), 3,33 (с, 3H), 2,41 (д, J=9,4 Гц, 1H), 2,41-2,36 (м, 1H), 2,18-2,12 (м, 1H), 1,54 (с, 9H), 0,89 (с, 9H), 0,06 (с, 6H).
[00202] Соблюдая процедуру, описанную для примера 32, указанное выше соединение (2,78 г, 6,45 ммоль) было подвергнуто защите бензила с использованием BriBr. После очистки колоночной флэш-хроматографией на силикагеле (EtOAc/гексаны, от 1:10 до 1:4) получали трет-бутил-((3aR,5R,6S,7aR)-6-(бензилокси)-5-(((трет-бутил-диметилсилил)окси)метил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-2-ил)(метил)карбамат в виде бесцветного липкого масла (2,7 г, 80%). 1H ЯМР (400 МГц, CDCl3) δ 7,36-7,27 (м, 5H), 6,02 (д, J=7,1 Гц, 1H), 4,67 (д, J=11,6 Гц, 1H), 4,40 (д, J=11,6 Гц, 1H), 4,34-4,30 (м, 1H), 3,83-3,78 (м, 1H), 3,77-3,69 (м, 2H), 3,53-3,50 (м, 1H), 3,29 (с, 3H), 2,44-2,39 (м, 1H), 2,14-2,08 (м, 1H), 1,52 (с, 9H), 0,88 (с, 9H), 0,04 (с, 6H).
[00203] Соблюдая процедуру, описанную для примера 32, указанное выше соединение (2,70 г, 5,30 ммоль) было подвергнуто снятию защиты силила с использованием TBAF. После очистки колоночной флэш-хроматографией на силикагеле (EtOAc/гексаны, от 1:5 до 1:1) получали трет-бутил-((3aR,5R,6S,7aR)-6-(бензилокси)-5-(гидроксиметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-2-ил)(метил)карбамат в виде бесцветной липкой пены (2,0 г, 93%). 1H ЯМР (400 МГц, CDCl3) δ 7,37-7,27 (м, 5H), 6,01 (д, J=7,2 Гц, 1H), 4,69 (д, J=11,6 Гц, 1H), 4,40 (д, J=11,6 Гц, 1H), 4,36-4,34 (м, 1H), 3,77-3,72 (м, 2H), 3,62-3,54 (м, 2H), 3,30 (с, 3H), 2,53-2,48 (м, 1H), 2,09-2,02 (м, 1H), 1,71 (т, J=6,3 Гц, 1H), 1,53 (с, 9H).
[00204] Соблюдая процедуру, описанную для примера 32, указанное выше соединение (0,663 г, 1,62 ммоль) окисляли в альдегид, используя DMP. После очистки колоночной флэш-хроматографией на силикагеле (EtOAc/гексаны, от 1:10 до 2:3) получали трет-бутил-((3aR,5S,6S,7aR)-6-(бензилокси)-5-формил-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-2-ил)(метил)карбамат в виде белой пены (0,57 г, 86%). 1H ЯМР (400 МГц, CDCl3) δ 9,63 (с, 1H), 7,36-7,27 (м, 5H), 6,04 (д, J=4,3 Гц, 1H), 4,69 (д, J=9,2 Гц, 1H), 4,50 (д, J=9,2 Гц, 1H), 4,43-4,39 (м, 1H), 4,07 (д, J=6,4 Гц), 4,02-3,99 (м, 1H), 3,29 (с, 3H), 2,64-2,59 (м, 1H), 2,10-2,03 (м, 1H), 1,53 (с, 9H).
[00205] Соблюдая процедуру, описанную для примера 32, указанное выше соединение (0,550 г, 1,35 ммоль) обрабатывали DAST. После очистки колоночной флэш-хроматографией на силикагеле (EtOAc/гексаны, от 1:10 до 1:3) получали трет-бутил-((3aR,5S,6S,7aR)-6-(бензилокси)-5-(дифторметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-2-ил)(метил)карбамат в виде бледно-желтого липкого масла (0,48 г, 83%). 1H ЯМР (400 МГц, CDCl3) δ 7,34-7,27 (м, 5H), 6,04 (д, J=7,4 Гц, 1H), 5,79 (дт, J=2,2, 54,7 Гц, 1H), 4,67 (д, J=11,4 Гц, 1H), 4,43 (д, J=11,4 Гц, 1H), 4,43-3,40 (м, 1H), 4,01-3,97 (м, 1H), 3,82-3,73 (м, 1H), 3,27 (с, 3H), 2,59-2,54 (м, 1H), 2,10-2,05 (м, 1H), 1,53 (с, 9H).
[00206] Соблюдая процедуру, описанную для примера 32, указанное выше соединение (0,480 г, 1,12 ммоль подвергали снятию защиты, используя BC13. После очистки колоночной флэш-хроматографией на силикагеле (1,0 M NH3 в MeOH/DCM, 1:12) получали (3aR,5S,6S,7aR)-5-(дифторметил)-2-(метиламино)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6-ол в виде бледно-желтого липкого твердого вещества (0,24 г, 87%). 1H ЯМР (400 МГц, CD3OD) δ 6,17 (д, J=6,4 Гц, 1H), 5,91 (дт, J=2,6, 54,4 Гц, 1H), 4,38-4,34 (м, 1H), 4,03-3,98 (м, 1H), 3,64-3,61 (м, 1H), 2,84 (с, 3H), 2,16-2,13 (м, 2H);, 13C ЯМР (100 МГц, CD3OD) δ 163,35, 116,00 (т, J=241,0 Гц), 91,12, 74,97 (т, J=42,7 Гц), 70,00, 64,54 (т, J=3,6 Гц), 34,19, 30,79; MS, m/z=239,0 (M+1).
Пример 28
(3aR,5S,6S,7aR)-5-(дифторметил)-2-(диметиламино)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6-ол
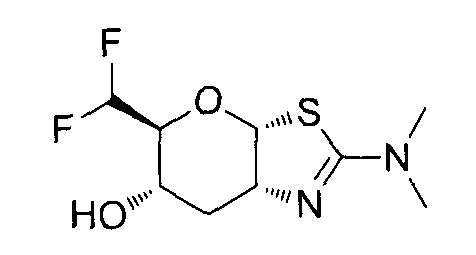
[00207] К раствору (3aR,5R,6R,7R,7aR)-7-((трет-бутил-диметилсилил)окси)-5-(((трет-бутил-диметилсилил)окси)метил)-2-(диметиламино)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6-ола (4,26 г, 8,95 ммоль) в безводном DMF (30 мл) добавляли BnBr (1,68 г, 9,84 ммоль) и тетрабутиламмоний йодид (0,330 г, 0,895 ммоль). При 0°C порциями добавляли NaH (60%, 0,430 г, 10,744 ммоль) и затем реакционную смесь перемешивали при этой температуре в течение 2 ч. Смесь разбавляли водой (200 мл), экстрагировали Et2O (2×100 мл). Объединенный экстракт промывали солевым раствором (100 мл) и сушили над безводным Na2SO4. The растворитель выпаривали при пониженном давлении для получения смеси, содержащей ≈80% (3aR,5R,6R,7R,7aR)-6-(бензилокси)-7-((трет-бутил-диметилсилил)окси)-5-(((трет-бутил-диметилсилил)окси)метил)-N,N-диметил-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-2-амина (5,06 г, 100%), который был неотделим на колонке силикагеля. Эту смесь непосредственно использовали в следующей реакции.
[00208] К раствору указанного выше соединения (9,89 г, 17,5 ммоль) в MeOH (100 мл) при 0°C добавляли AcCl (6,21 мл, 87,4 ммоль). Смесь перемешивали при комнатной температуре в течение 16 ч. Растворитель выпаривали при пониженном давлении. Остаток очищали на колонке силикагеля, элюировали 2%-5% 2M раствора NH3MeOH в DCM для получения (3aR,5R,6S,7R,7aR)-6-(бензилокси)-2-(диметиламино)-5-(гидроксиметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-7-ола (4,58 г, 78%). MS m/z 339.1 (M+1, 100%).
[00209] К раствору указанного выше соединения (4,58 г, 13,6 ммоль) и имидазола (2,76 г, 40,7 ммоль) в безводном DMF (30 мл) при 0°C добавляли TBDMSC1 (2,45 г, 16,3 ммоль). Смесь перемешивали при комнатной температуре в течение 21 ч. Реакционную смесь разбавляли водой (200 мл), экстрагировали Et2O (2×100 мл). Объединенный экстракт промывали солевым раствором (100 мл) и сушили над безводным Na2SO4. Растворитель выпаривали при пониженном давлении. Остаток очищали на колонке силикагеля, элюировали l%-3% 2M раствора NH3MeOH в DCM для получения (3aR,5R,6S,7R,7aR)-6-(бензилокси)-5-(((трет-бутил-диметилсилил)окси)метил)-2-(диметиламино)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-7-ола (5,80 г, 95%). MS m/z 453,2 (M+1, 100%); 1H ЯМР (400 МГц, CDCl3) δ 7,27-7,37 (м, 5H), 6,37 (д, J=6,6 Гц, 1H), 4,88 (д, J=11,4 Гц, 1H), 4,63 (д, J=11,4 Гц, 1H), 4,22 (т, J=6,4 Гц, 1H), 4,09-4,13 (м, 1H), 3,80-3,82 (м, 2H), 3,74-3,78 (м, 1H), 3,64 (дд, J=8,7 Гц, 6,4 Гц, 1H), 2,98 (с, 6H), 0,897 (с, 9H), 0,90 (с, 9H), 0,052 (с, 3H), 0,056 (с, 3H).
[00210] Смесь указанного выше соединения (5,80 г, 12,8 ммоль) и тио-CDI (90% тех., 4,57 г, 23,1 ммоль) в безводном DMF (30 мл) перемешивали при 90°C в течение 2,5 ч. После охлаждения смесь разбавляли водой (200 мл), экстрагировали Et2O (2×100 мл). Объединенные экстракты промывали солевым раствором (100 мл) и сушили над безводным Na2SO4. Растворитель выпаривали при пониженном давлении. Остаток очищали на колонке силикагеля, элюировали 30%-100% EtOAc в гексанах для получения O-((3aR,5R,6S,7R,7aR)-6-(бензилокси)-5-(((трет-бутил-диметилсилил)окси)метил)-2-(диметиламино)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-7-ил)1H-имидазол-l-карботиоата (6,51 г, 90%). MS m/z 563,2 (M+1, 100%); 1H ЯМР (400 МГц, CDCl3) δ 8,30 (с, 1H), 7,59 (с, 1H), 7,23-7,34 (м, 5H), 7,03 (с, 1H), 6,36 (шир.с, 1H), 6,31 (д, J=6,7 Гц, 1H), 4,91 (д, J=11,6 Гц, 1H), 4,62 (м, 1H), 4,58 (д, J=11,6 Гц, 1H), 3,85 (д, J=8,7 Гц, 1H), 3,64-3,74 (м, 3H), 3,03 (с, 6H), 0,82 (с, 9H), 0,02 (с, 3H), 0,00 (с, 3H).
[00211] Смесь указанного выше соединения (6,51 г, 11,6 ммоль), гидрида трибутилолова (7,47 г, 25,7 ммоль) и ABCN (0,313 г, 1,28 ммоль) в безводном THF (100 мл) нагревали в сосуде с обратным холодильником в течение 17 ч. После охлаждения растворитель выпаривали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле, элюировали 50%-80% EtOAc в гексанах для получения (3aR,5R,6S,7aR)-6-(бензилокси)-5-(((трет-бутил-диметилсилил)окси)метил)-N,N-диметил-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-2-амина в виде белого твердого вещества (4,07 г, 81%). MS m/z 437,2 (M+1, 100%); 1H ЯМР (400 МГц, CDCl3) δ 7,24-7,37 (м, 5H), 6,25 (д, J=6,6 Гц, 1H), 4,71 (д, J=11,6 Гц, 1H), 4,42 (д, J=11,6 Гц, 1H), 4,34-4,39 (м, 1H), 3,65-3,80 (м, 4H), 2,99 (с, 6H), 2,25 (шир.с, 1H), 2,10-2,16 (м, 1H), 0,89 (с, 9H), 0,05 (с, 6H).
[00212] К раствору указанного выше соединения (4,07 г, 9,33 ммоль) в MeOH (50 мл) при 0°C добавляли AcCl (1,33 мл, 18,7 ммоль). Смесь перемешивали при комнатной температуре в течение 16 ч. Растворитель выпаривали при пониженном давлении. Остаток очищали на колонке силикагеля, элюировали 3%-5% 2M раствором NH3 MeOH в DCM для получения ((3aR,5R,6S,7aR)-6-(бензилокси)-2-(диметиламино)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-5-ил)метанола (2,78 г, 91%). MS m/z 323,1 (M+1, 100%); 1H ЯМР (400 МГц, CDCl3) δ 7,25-7,36 (м, 5H), 6,22 (д, J=6,6 Гц, 1H), 4,70 (д, J=11,6 Гц, 1H), 4,42 (д, J=11,6 Гц, 1H), 4,32-4,37 (м, 1H), 3,71-3,79 (м, 2H), 3,60-3,67 (м, 2H), 2,97 (с, 6H), 2,19-2,25 (м, 1H), 2,09-2,15 (м, 1H), 1,86 (шир.с, 1H).
[00213] К раствору DMSO (диметилсульфоксида) (0,875 г, 11,2 ммоль) в безводном DCM (15 мл) при -78°C в атмосфере N2 по каплям добавляли оксалилхлорид (1,316 г, 10,36 ммоль). Смесь перемешивали при ~ -30°C в течение 30 мин и снова охлаждали до -78°C. По каплям добавляли раствор указанного выше соединения (1,39 г, 4,32 ммоль) в безводном DCM (15 мл). После перемешивания при ~ -30°C в течение 2 ч реакционную смесь снова охлаждали до -78°C и добавляли Et3N (1,74 г, 17,3 ммоль). Смесь перемешивали при- -30°C в течение еще 30 мин и затем гасили водой (50 мл). Органический слой собирали, а водный экстрагировали DCM (2×20 мл). Объединенные экстракты сушили над безводным Na2SO4. Растворитель выпаривали при пониженном давлении для получения неочищенного (3aR,5S,6S,7aR)-6-(бензилокси)-2-(диметиламино)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-5-карбальдегида (1,24 г) в виде желтой пены. MS m/z 353,1 (M+23, 100%).
[00214] К раствору указанного выше неочищенного соединения (200 мг) в безводном DCM (5 мл) при 0°C добавляли трифторид бис(2-метоксиэтил)аминосеры (0,553 г, 2,50 ммоль). Смесь перемешивали при комнатной температуре в течение 27 ч. Реакцию гасили насыщенным водным NaHCO3(10 мл) и затем экстрагировали EtOAc (2×10 мл). Объединенный экстракт сушили над безводным Na2SO4. Растворители выпаривали при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (EtOAc/гексаны, от 2:1 до 5:1) для получения (3aR,5S,6S,7aR)-6-(бензилокси)-5-(дифторметил)-N,N-диметил-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-2-амина в виде бледно-желтой пены (0,050 г, 23%, 2 стадии). MS m/z 343,1 (M+1, 100%).
[00215] К раствору указанного выше соединения (0,049 г, 0,14 ммоль) в DCM (2 мл) при -78°C добавляли раствор BC13 в DCM (1,0 M, 0,19 мл, 0,19 ммоль). Смесь медленно нагревали до комнатной температуры и перемешивали в течение 18 ч. Реакционную смесь снова охлаждали до -78°C и по каплям добавляли смесь 1:1 MeOH-DCM (2 мл) для гашения реакции.
Растворители выпаривали, и остаток еще три раза обрабатывали MeOH. Неочищенный продукт очищали колоночной хроматографией на силикагеле, элюировали l%-2% 2M раствора NH3MeOH в DCM для получения продукта (3aR,5S,6S,7aR)-5-(дифторметил)-2-(диметиламино)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6-ола (0,0193 г, 53%) в виде белого твердого вещества. MS m/z 253,1 (M+1, 100%); 1H ЯМР (400 МГц, MeOD) δ 6,22 (д, J=6,5 Гц, 1H), 5,93 (тд, J=54,4 Гц, 2,6 Гц, 1H), 4,36-4,40 (м, 1H), 3,99-4,03 (м, 1H), 3,59-3,68 (м, 1H), 3,03 (с, 6H), 2,12-2,16 (м, 2H); 13C ЯМР (100 МГц, MeOD) δ 165,06, 115,86 (т, J=192,8 Гц), 91,48, 74,91 (т, J=17,1 Гц), 70,02, 64,40 (т, J=2,8 Гц), 40,31, 34,09.
Пример 29
(3aR,5S,6R,7R,7aR)-5-(дифторметил)-2-(этиламино)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диол
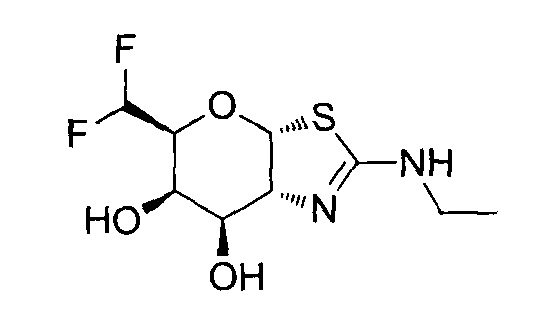
[00216] Раствор трет-бутил-((3aR,5S,6S,7R,7aR)-5-(дифторметил)-6,7-дигидрокси-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-2-ил)(этил)карбамата (500 мг, 1,3 ммоль) и имидазола (277 мг, 4 ммоль) в DMF (20 мл) обрабатывали TBDMSCl (611 мг, 4 ммоль) в течение 3 ч при 40°C. Затем полученный раствор охлаждали до комнатной температуры, гасили насыщенным водным раствором NaHCO3 (50 мл), экстрагировали DCM (3×20 мл), промывали солевым раствором (3×10 мл), сушили над безводным MgSO4 и концентрировали в вакууме для получения остатка, который очищали колоночной хроматографией на силикагеле, элюировали 5%-10% EtOAc в простом петролейном эфире для получения трет-бутил-(3aR,5S,6R,7R,7aR)-7-(трет-бутил-диметилсилилокси)-5-(дифторметил)-6-гидрокси-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-2-ил(этил)карбамата в виде светло-желтого сиропа (420 мг, 64%). (ES, m/z): [M+1]+ 483,0; 1H ЯМР (300 МГц, CDCl3) δ 5,87 (д, J=5,4 Гц, 1H), 5,98 (тд, J=55,2 Гц, 3,6 Гц, 1H), 4,10-3,91 (м, 5H), 3,77-3,70 (м, 1H), 1,56 (с, 9H), 1,21 (т, J=6,9 Гц, 3H), 0,94 (с, 9H), 0,19 (с, 3H), 0,15 (с, 3H).
[00217] Раствор указанного выше соединения (440 мг, 0,9 ммоль) в DCM (20 мл) обрабатывали DMP (587 мг, 1,4 ммоль) в течение 1 ч при комнатной температуре. Реакционную смесь гасили смесью насыщенного водного NaHCO3 (5 мл) и гипосульфита натрия (5 мл). Органический слой собирали, а водный слой экстрагировали DCM (3×20 мл). Объединенный органический слой промывали солевым раствором (10 мл) и сушили над безводным MgSO4. После фильтрации фильтраты концентрировали для получения остатка, который очищали колоночной хроматографией на силикагеле, элюировали 5% EtOAc в простом петролейном эфире для получения трет-бутил-(3aR,5S,7R,7aR)-7-(трет-бутил-диметилсилилокси)-5-(дифторметил)-6-оксо-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-2-ил(этил)карбамата в виде белого сиропа (370 мг, 84%). (ES, m/z): [M+1]+ 481,0; 1H ЯМР (300 МГц, CDCl3) δ 6,20 (д, J=6,9 Гц, 1H), 6,08 (тд, J=52,8 Гц, 1,5 Гц, 1H), 4,69 (д, J=3,6 Гц, 1H), 4,63 (дд, J=6,9 Гц, 3,9 Гц, 1H), 4,16-4,09 (м, 1H), 3,98-3,89 (м, 2H), 1,57 (с, 9H), 1,14 (т, J=6,9 Гц, 3H), 0,93 (с, 9H), 0,20 (с, 3H), 0,13 (с, 3H).
[00218] Раствор указанного выше соединения (400 мг, 0,8 ммоль) в MeOH (5 мл) обрабатывали NaBH4 (63 мг, 1,7 ммоль) в течение 2 ч при комнатной температуре. Реакционную смесь гасили водой (20 мл) и экстрагировали DCM (3×10 мл). Объединенный органический слой промывали солевым раствором (10 мл), сушили над безводным MgSO4, и концентрировали в вакууме для получения остатка, который очищали колоночной хроматографией на силикагеле, элюировали 10% EtOAc в простом петролейном эфире для получения трет-бутил-(3aR,5S,6S,7R,7aR)-7-(трет-бутил-диметилсилилокси)-5-(дифторметил)-6-гидрокси-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-2-ил(этил)карбамата в виде белого сиропа (103 мг, 25%). (ES, m/z): [M+1]+ 483,0; 1H ЯМР (300 МГц, CDCl3) δ 6,04 (д, J=6,3 Гц, 1H), 6,06 (тд, J=70,2 Гц, 6,6 Гц, 1H), 4,37-4,31 (м, 1H), 4,25-4,20 (м, 1H), 4,12-4,06 (м, 1H), 4,01-3,92 (м, 2H), 3,87 (м, 1H), 3,85-3,76 (м, 1H), 1,56 (с, 9H), 1,21 (т, J=6,9 Гц, 3H), 0,96 (с, 9H), 0,22 (с, 3H), 0,18 (с, 3H).
[00219] Через раствор указанного выше соединения (20 мг, 0,04 ммоль) в MeOH (10 мл) пробулькивали сухой газообразной HCl до его насыщения при 0°C. Полученный раствор перемешивали в течение 3 ч при комнатной температуре. Удаление летучих веществ давало остаток, который растворяли в MeOH (2 мл) и нейтрализовали концентрированным NH4OH (1 мл). После концентрации при пониженном давлении, неочищенный продукт очищали на короткой колонке с силикагелем, элюировали 10% MeOH в DCM для получения (3aR,5S,6R,7R,7aR)-5-(дифторметил)-2-(этиламино)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диола в виде белого твердого вещества (10 мг, 90%). (ES, m/z): [M+1]+ 269,0; 1H ЯМР (100 МГц, CD3OD) δ 6,31 (д, J=6,0 Гц, 1H), 5,97 (тд, J=56,1, 6,3Гц, 1H), 4,12 (т, J=6,3 Гц, 1H), 4,03 (т, J=3,0 Гц, 1Η), 3,95-3,90 (м, 1H), 3,88-3,85 (м, 1H), 3,32-3,21 (м, 2H), 1,17 (т, J=7,5 Гц, 3H).
Пример 30
(3aR,5R,6S,7R,7aR)-7-фтор-5-(гидроксиметил)-2-(метиламино)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6-ол
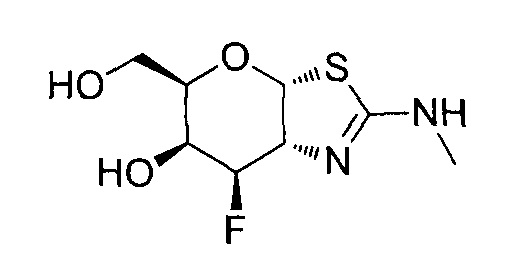
[00220] К раствору трет-бутил-((3aR,5R,7R,7aR)-5-(((трет-бутил-диметилсилил)окси)метил)-7-фтор-6-оксо-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-2-ил)(метил)карбамата (2,57 г, 5,72 ммоль) в сухом MeOH (50 мл) при 0°C добавляли NaBH4 (0,295 г, 7,80 ммоль). После перемешивания смеси при 0°C в течение 20 мин добавляли осколок сухого льда, и растворитель выпаривали. Остаток растворяли в DCM (50 мл) и промывали насыщенным водным NaHCO3 (50 мл). Органический слой собирали, а водный экстрагировали DCM (2×30 мл). Объединенный экстракт сушили над безводным Na2SO4. После фильтрации растворитель выпаривали при пониженном давлении, и остаток очищали колоночной флэш-хроматографией на силикагеле (EtOAc/гексаны, от 1:10 до 1:3), получая трет-бутил-((3aR,5R,6S,7R,7aR)-5-(((трет-бутил-диметилсилил)окси)метил)-7-фтор-6-гидрокси-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-2-ил)(метил)карбамат (0,95 г, 37%), в виде липкого масла. 1H ЯМР (400 МГц, CDCl3) δ 6,11 (д, J=6,7 Гц, 1H), 4,84 (ддд, J=3,2, 6,7, 48,2 Гц, 1H), 4,45 (тд, J=6,7, 16,6 Гц, 1H), 4,32-4,29 (м, 1H), 4,00-3,93 (м, 2H), 3,90-3,86 (м, 1H), 3,36 (с, 3H), 3,19 (шир.с, 1 H, (OH)), 1,53 (с, 9H), 0,90 (с, 9H), 0,093 (с, 3H), 0,087 (с, 3H).
[00221] Через раствор указанного выше соединения (0,110 г, 0,244 ммоль) в сухом MeOH (4 мл) пробулькивали сухой газообразной HCl в течение 30 сек. Смесь перемешивали при комнатной температуре в течение 2 ч. После выпаривания растворителя при пониженном давлении остаток нейтрализовали 1,0 M NH3 в MeOH и очищали колоночной флэш-хроматографией на силикагеле (1,0 M NH3 в MeOH/DCM, 1:9), получая (3aR,5R,6S,7R,7aR)-7-фтор-5-(гидроксиметил)-2-(метиламино)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6-ол в виде белого твердого вещества (0,043 г, 89%). 1H ЯМР (400 МГц, CD3OD) δ 6,38 (дд, J=1,0, 6,7 Гц, 1H), 4,65 (ддд, J=3,3, 7,8, 48,2 Гц, 1H), 4,30-4,22 (м, 1H), 4,15-4,11 (м, 1H), 4,01-3,98 (м, 1H), 3,78-3,72 (м, 2H), 2,83 (с, 3H); 13C ЯМР (100 МГц, CD3OD) δ 164,87, 94,77 (д, J=182,5 Гц), 91,73 (д, J=7,8 Гц), 75,66 (д, J=5,8 Гц), 69,89 (д, J=21,0 Гц), 67,01 (д, J=27,6 Гц), 61,81 (д, J=3,1 Гц), 30,30; MS, (ES, m/z) [M+H]+ 237,1.
Пример 31
(3aR,5S,6R,7R,7aR)-7-фтор-5-(фторметил)-2-(метиламино)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6-ол
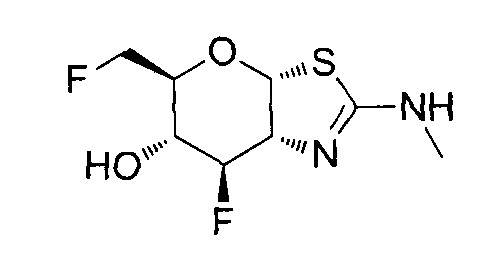
[00222] К суспензии (3aR,5R,6S,7R,7aR)-5-(гидроксиметил)-2-(метиламино)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диола (80,00 г, 341,6 ммоль) в DMF (500 мл) последовательно добавляли DIPEA (8 мл), MeOH (8 мл) и Boc2O (100,0 г, 458,7 ммоль). Суспензию перемешивали при комнатной температуре в течение 6 ч, и она становилась прозрачным раствором. После уменьшения объема раствора при пониженном давлении при комнатной температуре примерно на 100 мл для удаления MeOH и трет-бутанола, раствор охлаждали на ледяной бане и последовательно добавляли имидазол (92,9 г, 1,36 моль) и TBDMSCl (155 г, 1,03 моль). После перемешивания при комнатной температуре в течение ночи смесь разбавляли солевым раствором (1,5 л). Экстракцию выполняли Et2O три раза (500 мл и 2×300 мл). Объединенный эфирный экстракт промывали солевым раствором (1 л) и сушили над безводным Na2SO4. После фильтрации растворитель выпаривали при пониженном давлении, и остаток двух партий объединяли и очищали колоночной флэш-хроматографией на силикагеле (EtOAc/гексаны, 2:11) для получения трет-бутил-((3aR,5R,6R,7R,7aR)-7-((трет-бутил-диметилсилил)окси)-5-(((трет-бутил-диметилсилил)окси)метил)-6-гидрокси-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-2-ил)(метил)карбамата в виде белого кристаллического твердого вещества (136 г, 71%). 1H ЯМР (500 МГц, CDCl3) δ 6,15 (д, J=6,2 Гц, 1H), 4,29 (шир.с, 1H), 4,06-4,05 (м, 1H), 3,81-3,75 (м, 2H), 3,71-3,68 (м, 1H), 3,47-3,43 (м, 1H), 3,36 (с, 3H), 1,54 (с, 9H), 0,915 (с, 9H), 0,893 (с, 9H), 0,161 (с, 3H), 0,149 (с, 3H), 0,068 (с, 6H).
[00223] При 0°C к раствору указанного выше соединения (94,30 г, 167,5 ммоль) и DMAP (0,50 г, 4,1 ммоль) в пиридине (350 мл) добавляли BzCl (28,3 г, 201 ммоль). Смесь перемешивали при комнатной температуре в течение ночи, добавляли еще одну порцию BzCl (6,90 г, 49,1 ммоль), и смесь перемешивали при комнатной температуре в течение еще 3 ч. Добавляли MeOH (20 мл), и смесь перемешивали в течение еще 30 мин. Смесь разбавляли солевым раствором (1 л) и три раза экстрагировали смесью EtOAc/гексанами (1:4; 600 мл, 200 мл и 100 мл). Объединенный экстракт сушили над безводным Na2SO4. После фильтрации растворитель выпаривали при пониженном давлении, и объединенные остатки всех трех партий обрабатывали 1,5 M HC1 в MeOH (1,0 л) при комнатной температуре в течение 16 ч. Затем растворитель удаляли при комнатной температуре при пониженном давлении для получения белого твердого вещества. К белому твердому веществу в DCM (1,6 ч) последовательно добавляли DIPEA (100 мл) и Boc2O (109 г, 500 ммоль). Затем смесь перемешивали при комнатной температуре в течение 4 дней. Реакционную смесь промывали насыщенным водным NH4C1 (1 л) и солевым раствором (1 л) и сушили над безводным Na2SO4. (Объединенный водный промывной раствор ощелачивали до pH = ~9 насыщенным водным раствором Na2CO3, и экстрагировали DCM (5×150 мл). Объединенные экстракты DCM сушили над безводным Na2SO4. После фильтрации раствор обрабатывали Boc2O (30 г) в течение 5 ч. После этого промывали насыщенным водным NH4C1 (1 л) и солевым раствором (1 л) и сушили над безводным Na2SO4). Все экстракты DCM объединяли. После фильтрации растворитель выпаривали при пониженном давлении, и остаток очищали колоночной флэш-хроматографией на силикагеле (EtOAc/гексаны, от 1:4 до 3:2) для получения (3aR,5R,6S,7R,7aR)-2-((трет-бутоксикарбонил)(метил)амино)-7-гидрокси-5-(гидроксиметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6-ил бензоата в виде белого твердого вещества (56,5 г, 77%). 1H ЯМР (400 МГц, CDCl3) δ 8,00-7,98 (м, 2H), 7,59-7,54 (м, 1H), 7,44-7,40 (м, 2H), 6,20 (д, J=7,0 Гц, 1H), 5,15-5,12 (м, 1H), 4,55-4,50 (м, 1H), 4,41-4,39 (м, 1H), 3,80-3,76 (м, 1H), 3,70-3,66 (м, 1H), 3,49 (шир.с, 1H), 3,34 (с, 3H), 1,56 (с, 9H).
[00224] К раствору указанного выше соединения (500 мг, 1,1 ммоль) в DCM (15 мл) добавляли DAST (1,1 г, 6,8 ммоль) при -78°C в атмосфере азота. После перемешивания в течение ночи при комнатной температуре, реакционную смесь гасили насыщенным водным раствором NaHCO3 (30 мл), экстрагировали DCM (3×15 мл), сушили над безводным Na2SO4 и конденсировали для получения остатка, который очищали колоночной хроматографией на силикагеле, элюировали 10%-30% EtOAc в простом петролейном эфире для получения (3aR,5S,6R,7R,7aR)-2-(трет-бутоксикарбонил(метил)амино)-7-фтор-5-(фторметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6-ил бензоата (370 мг, 73%) в виде светло-желтого масла. (ES, m/z): [M+H]+ 443,0; 1H ЯМР (300 МГц, CDCl3) δ 8,02-8,01 (м, 2H), 7,64-7,62 (м, 1H), 7,59-7,44 (м, 2H), 6,19 (д, J=7,2 Гц, 1H), 5,44-5,26 (м, 2H), 4,62-4,60 (м, 2H), 4,58-4,46 (м, 1H), 3,90-3,80 (м, 1H), 3,39 (с, 3H), 1,58 (с, 9H).
[00225] Раствор указанного выше соединения (170 мг, 0,4 ммоль) в THF (10 мл) обрабатывали MeMgCl (3 ммоль, 1 мл, 3 M в THF) в течение 1 ч при комнатной температуре. Реакционную смесь гасили насыщенным водным раствором NH4C1 (30 мл), экстрагировали EtOAc (3×20 мл), промывали солевым раствором (10 мл), сушили над безводным Na2SO4, и конденсировали для получения остатка, который очищали колоночной хроматографией на силикагеле, элюировали 2%-5% MeOH в DCM для получения (3aR,5S,6R,7R,7aR)-7-фтор-5-(фторметил)-2-(метиламино)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6-ола в виде белого твердого вещества (35 мг, 38%). (ES, m/z): [M+H]+ 239,0; 1H ЯМР (300 МГц, CDCl3) δ 6,27 (д, J=6,3 Гц, 1H), 5,14 (тд, J=45,6 Гц, 2,1 Гц, 1H), 4,70-4,44 (м, 3H), 3,90-3,79 (м, 1H), 3,73-3,61 (м, 1H), 2,95 (с, 3H).
Пример 32
(3aR,5S,6R,7R,7aR)-5-(дифторметил)-7-фтор-2-(метиламино)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6-ол
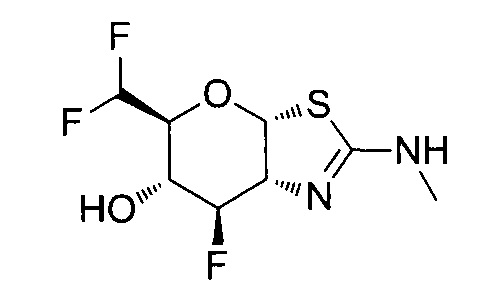
[00226] К суспензии (3aR,5R,6S,7R,7aR)-2-(метиламино)-5-(гидроксиметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диола (8,50 г, 37,0 ммоль) в DMF (60 мл) добавляли DIPEA (2,0 мл), Boc2O (23,0 г, 105 ммоль) и MeOH (2,0 мл). Смесь перемешивали при комнатной температуре в течение 3 ч и затем добавляли MeOH (50 мл). Реакционную смесь концентрировали при пониженном давлении при ~35°C. Остаток очищали колоночной флэш-хроматографией на силикагеле (MeOH/DCM, 1:8), с последующей перекристаллизацией из EtOAc/гексанов для получения трет-бутил-((3aR,5R,6S,7R,7aR)-6,7-дигидрокси-5-(гидроксиметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-2-ил)(метил)карбамата в виде белого твердого вещества (11,8 г, 96%). 1H ЯМР (400 МГц, CDCl3) δ 6,14 (д, J=6,9 Гц, 1H), 4,20 (д, J=6,4 Гц, 1H), 4,11 (д, J=5,6 Гц, 1H), 3,85-3,70 (м, 2H), 3,63-3,55 (м, 1H), 3,31 (с, 3H), 1,53 (с, 9H).
[00227] К раствору указанного выше соединения (11,7 г, 35,1 ммоль), DIPEA (10,3 г, 80,0 ммоль) и DMAP (0,040 г, 0,33 ммоль) в DCM (180 мл) при 0°C медленно добавляли BzCl (10,1 г, 72,0 ммоль). После добавления смесь перемешивали при комнатной температуре в течение 5 ч. Добавляли насыщенный водный раствор NH4Cl (100 мл), и органический слой собирали. Водный слой дополнительно экстрагировали DCM (3×50 мл). Объединенный экстракт сушили над безводным Na2SO4. После фильтрации растворитель выпаривали при пониженном давлении, и остаток отделяли колоночной флэш-хроматографией на силикагеле (EtOAc/гексаны, от 1:4 до 1:1), получая ((3aR,5R,6S,7R,7aR)-6-(бензоилокси)-2-((трет-бутоксикарбонил)(метил)амино)-7-гидрокси-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-5-ил)метил бензоат в виде белого твердого вещества (4,20 г, 22%). 1H ЯМР (400 МГц, CDCl3) δ 8,01-7,99 (м, 4H), 7,60-7,55 (м, 1H), 7,54-7,50 (м, 1H), 7,45-7,41 (м, 2H), 7,37-7,35 (м, 2H), 6,21 (д, J=7,1 Гц, 1H), 5,23-5,20 (м, 1H), 4,55-4,51 (м, 2H), 4,48-4,42 (м, 2H), 4,15-4,07 (м, 2H), 3,36 (с, 3H), 1,56 (с, 9H).
[00228] К раствору указанного выше соединения (7,91 г, 14,6 ммоль) в безводном DCM (100 мл) при -78°C в атмосфере N2, добавляли DAST (11,8 г, 73,0 ммоль). После добавления смесь перемешивали при комнатной температуре в течение 72 ч. Затем реакционную смесь охлаждали при -78°C, разбавляли DCM (100 мл), и затем гасили насыщенным водным NaHCO3 (150 мл). Органический слой собирали, а водный экстрагировали DCM (2×100 мл). Объединенный экстракт сушили над безводным Na2SO4. После фильтрации растворитель выпаривали при пониженном давлении, и остаток очищали колоночной флэш-хроматографией на силикагеле (EtOAc/гексаны, от 1:10 до 1:4), получая ((3aR,5R,6R,7R,7aR)-6-(бензоилокси)-2-((трет-бутоксикарбонил)(метил)амино)-7-фтор-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-5-ил)метил бензоата в виде белого твердого вещества (6,10 г, 77%). 1H ЯМР (400 МГц, CDCl3) δ 8,01-7,98 (м, 4H), 7,60-7,56 (м, 1H), 7,56-7,52 (м, 1H), 7,45-7,41 (м, 2H), 7,38-7,35 (м, 2H), 6,19 (д, J=7,2 Гц, 1H), 5,52-5,46 (м, 1H), 5,40-5,28 (м, 1H), 4,61-4,56 (м, 1H), 4,52 (дд, J=3,6, 12,0 Гц, 1H), 4,43 (дд, J=5,7, 12,0 Гц, 1H), 4,03-3,99 (м, 1H), 3,36 (с, 3H), 1,56 (с, 9H).
[00229] Смесь указанного выше соединения (6,10 г, 11,2 ммоль) и K2C03 (1,00 г, 7,25 ммоль) в безводном MeOH (50 мл) перемешивали при комнатной температуре в течение 3 ч. Добавляли сухой лед, и растворитель удаляли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (EtOAc/гексаны, от 1:1 до 10:1), получая трет-бутил-((3aR,5R,6R,7R,7aR)-7-фтор-6-гидрокси-5-(гидроксиметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-2-ил)(метил)карбамат в виде белого твердого вещества (3,25 г, 86%). 1H ЯМР (400 МГц, CDCl3) δ 6,06 (д, J=6,8 Гц, 1H), 5,15 (ддд, J=2,4, 4,4, 45,7 Гц, 1H), 4,46-4,41 (м, 1H), 3,96-3,89 (м, 1H), 3,83 (дд, J=3,2, 11,8 Гц, 1H), 3,73 (дд, J=5,4, 11,8 Гц, 1H), 3,46-3,42 (м, 1H), 3,32 (с, 3H), 1,54 (с, 9H).
[00230] При 0°C к раствору указанного выше соединения (0,880 г, 2,61 ммоль) и имидазола (0,354 г, 5,20 ммоль) в безводном DMF (15 мл) добавляли TBDMSC1 (0,452 г, 3,00 ммоль). Смесь перемешивали при комнатной температуре в течение 72 ч и разбавляли Et2O (100 мл) и солевым раствором (100 мл). Органический слой собирали, а водный экстрагировали Et2O (50 мл). Объединенный экстракт промывали водой (50 мл) и сушили над безводным Na2SO4. После фильтрации растворитель выпаривали при пониженном давлении, и остаток очищали колоночной флэш-хроматографией на силикагеле (EtOAc/гексаны, от 1:10 до 1:3), получая трет-бутил-((3aR,5R,6R,7R,7aR)-5-(((трет-бутил-диметилсилил)окси)метил)-7-фтор-6-гидрокси-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-2-ил)(метил)карбамат в виде белой пены (1,10 г, 93%). 1H ЯМР (400 МГц, CDCl3) δ 6,06 (д, J=6,8 Гц, 1H), 5,19-5,02 (м, 1H), 4,43-4,38 (м, 1H), 3,98-3,93 (м, 1H), 3,85 (дд, J=5,0, 10,6 Гц, 1H), 3,73 (дд, J=5,2, 10,6 Гц, 1H), 3,45-3,43 (м, 1H), 3,34 (с, 3H), 1,54 (с, 9H), 0,89 (с, 9H), 0,08 (с, 6H).
[00231] При 0°C к раствору указанного выше соединения (1,06 г, 2,35 ммоль) и Bu4NI (0,087 г, 0,24 ммоль) в безводном DMF (15 мл) добавляли NaH (60% в минеральном масле, 0,118 г, 2,94 ммоль). После добавления NaH, к реакционной смеси добавляли BnBr (0,703 г, 4,11 ммоль). Смесь перемешивали при комнатной температуре в течение 16 ч и разбавляли Et2O (60 мл) и насыщенным NH4Cl (50 мл). Органический слой собирали, а водный экстрагировали Et2O (2×30 мл). Объединенный экстракт промывали солевым раствором (40 мл) и сушили над безводным Na2SO4. После фильтрации растворитель выпаривали при пониженном давлении, и остаток очищали колоночной флэш-хроматографией на силикагеле (EtOAc/гексаны, от 1:10 до 1:4), получая трет-бутил-((3aR,5R,6R,7R,7aR)-6-(бензилокси)-5-(((трет-бутил-диметилсилил)окси)метил)-7-фтор-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-2-ил)(метил)карбамат в виде липкого масла (1,22 г, 96%). 1H ЯМР (400 МГц, CDCl3) δ 7,37-7,27 (м, 5H), 6,10 (д, J=7,0 Гц, 1H), 5,30-5,16 (м, 1H), 4,80 (д, J=11,0 Гц, 1H), 4,55 (д, J=11,0 Гц, 1H), 4,48-4,42 (м, 1H), 3,88-3,80 (м, 1H), 3,78-3,69 (м, 2H), 3,46-3,44 (м, 1H), 3,31 (с, 3H), 1,53 (с, 9H), 0,89 (с, 9H), 0,04 (с, 6H).
[00232] При 0°C, к раствору указанного выше соединения (1,22 г, 2,25 ммоль) в THF (15 мл) добавляли TBAF (1,0 M в THF, 5,0 мл, 5,0 ммоль). После добавления реакционную смесь перемешивали при комнатной температуре в течение 2 ч и разбавляли EtOAc (20 мл) и солевым раствором (50 мл). Органический слой собирали, а водный экстрагировали EtOAc (2×50 мл). Объединенный экстракт сушили над безводным Na2SO4. После фильтрации растворитель выпаривали при пониженном давлении, и остаток очищали колоночной флэш-хроматографией на силикагеле (EtOAc/гексаны, от 1:5 до 1:2), получая трет-бутил-((3aR,5R,6R,7R,7aR)-6-(бензилокси)-7-фтор-5-(гидроксиметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-2-ил)(метил)карбамат в виде белого твердого вещества (0,96 г, 100%). 1H ЯМР (400 МГц, CDCl3) δ 7,37-7,29 (м, 5H), 6,09 (д, J=6,7 Гц, 1H), 5,32 (ддд, J=1,8, 3,6, 45,4 Гц, 1H), 4,80 (д, J=11,6 Гц, lH), 4,55 (д, J=11,6 Гц, 1H), 4,53-4,48 (м, 1H), 3,81-3,72 (м, 2H), 3,61-3,55 (м, 1H), 3,49-3,45 (м, 1H), 3,31 (с, 3H), 1,53 (с, 9H).
[00233] К раствору указанного выше соединения (1,50 г, 3,52 ммоль) в DCM (40 мл) добавляли DMP (2,20 г, 5,20 ммоль). После перемешивания при комнатной температуре в течение 1 ч реакционную смесь разбавляли Et2O (20 мл) и затем концентрировали до сухого состояния. Добавляли насыщенный водный NaHCO3 (30 мл) с Na2S2O3 (2 г), и смесь экстрагировали EtOAc (2×50 мл). Объединенный экстракт сушили над безводным Na2SO4. После фильтрации растворитель выпаривали при пониженном давлении, и остаток очищали колоночной флэш-хроматографией на силикагеле (EtOAc/гексаны, от 1:5 до 1:2), получая трет-бутил-((3aR,5S,6R,7R,7aR)-6-(бензилокси)-7-фтор-5-формил-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-2-ил)(метил)карбамат в виде белого твердого вещества (1,02 г, 68%). 1H ЯМР (400 МГц, CDCl3) δ 9,60 (с, 1H), 7,35-7,29 (м, 5H), 6,12 (д, J=7,0 Гц, 1H), 5,39-5,27 (м, 1H), 4,78 (д, J=11,4 Гц, 1H), 4,66 (д, J=11,4 Гц, 1H), 4,57-4,51 (м, 1H), 4,00-3,95 (м, 2H), 3,31 (с, 3H), 1,53 (с, 9H).
[00234] К раствору указанного выше соединения (0,156 г, 0,367 ммоль) в безводном DCM (6 мл) при -78°C в атмосфере N2 добавляли DAST (0,354 г, 2,20 ммоль). После добавления смесь перемешивали при комнатной температуре в течение 16 ч. Затем реакционную смесь охлаждали при -78°C, разбавляли DCM (10 мл), и затем гасили насыщенным водным NaHCO3 (10 мл). Органический слой собирали, а водный экстрагировали DCM (2×15 мл). Объединенный экстракт сушили над безводным Na2SO4. После фильтрации растворитель выпаривали при пониженном давлении, и остаток очищали колоночной флэш-хроматографией на силикагеле (EtOAc/гексаны, от 1:10 до 1:4), получая трет-бутил-((3aR,5S,6R,7R,7aR)-6-(бензилокси)-5-(дифторметил)-7-фтор-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-2-ил)(метил)карбамат в виде бледно-желтого масла (0,125 г, 76%). 1H ЯМР (400 МГц, CDCl3) δ 7,38-7,29 (м, 5H), 6,11 (д, J=7,2 Гц, 1H), 5,79 (дт, J=2,4, 54,4 Гц, 1H), 5,37-5,25 (м, 1H), 4,78 (д, J=11,3 Гц, 1H), 4,59 (д, J=11,3 Гц, 1H), 4,58-4,53 (м, 1H), 4,00-3,92 (м, 1H), 3,72-3,63 (м, 1H), 3,29 (с, 3H), 1,53 (с, 9H).
[00235] К раствору указанного выше соединения (0,240 г, 0,537 ммоль) и пентаметилбензола (0,26 г, 1,7 ммоль) в безводном DCM (10 мл) при -78°C в атмосфере N2 добавляли BC13 (1,0 M в DCM, 1,6 мл, 1,6 ммоль). Смесь перемешивали при комнатной температуре в течение ~3 ч, пока температура охлаждающей ловушки не достигала 0°C. Реакционную смесь охлаждали при -78°C, гасили смешанным MeOH/DCM и затем концентрировали до сухости. Остаток очищали колоночной флэш-хроматографией на силикагеле (1,0 M NH3 в MeOH/DCM, 1:15), получая (3aR,5S,6R,7R,7aR)-5-(дифторметил)-7-фтор-2-(метиламино)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6-ол в виде не совсем белого твердого вещества (0,104 г, 76%). 1H ЯМР (400 МГц, CD3OD) δ 6,30 (д, J=6,6 Гц, 1H), 5,96 (дт, J=2,4, 54,1 Гц, 1H), 4,89 (тд, J=3,9, 46,5 Гц, 1H), 4,48-4,42 (м, 1H), 4,02-3,94 (м, 1H), 3,72-3,63 (м, 1H), 2,85 (с, 3H); 13C ЯМР (100 МГц, CD3OD) δ 164,08 (д, J=2,3 Гц), 115,63 (дт, J=7,9, 241,6 Гц), 94,73 (д, J=177,6 Гц), 89,90 (д, J=2,0 Гц), 74,04 (д, J=26,1 Гц), 72,92 (дт, J=3,9, 21,3 Гц), 67,87 (тд, J=3,8, 25,0 Гц), 30,74; MS, m/z=257,0 (M+1).
Пример 33
(3aR,5S,6R,7S,7aR)-5-(дифторметил)-7-фтор-2-(метиламино)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6-ол
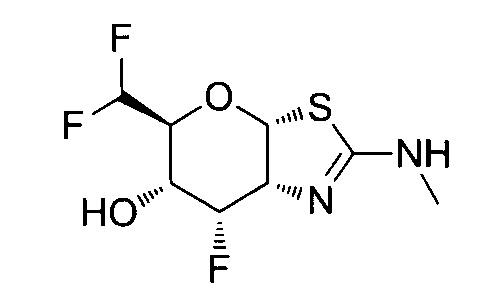
[00236] При 0°C к раствору трет-бутил-((3aR,5R,6R,7R,7aR)-7-фтор-6-гидрокси-5-(гидроксиметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-2-ил)(метил)карбамата (18,7 г, 55,5 ммоль) и имидазола (11,4 г, 167 ммоль) в безводном DMF (150 мл) добавляли TBDMSC1 (9,19 г, 61,0 ммоль). Смесь перемешивали при комнатной температуре в течение 16 ч, разбавляли Et2O (300 мл) и промывали солевым раствором (2×300 мл). Органический слой собирали, а водный экстрагировали Et2O (2×150 мл). Объединенный экстракт промывали солевым раствором (100 мл) и сушили над безводным Na2SO4. После фильтрации растворитель выпаривали при пониженном давлении, и остаток очищали колоночной флэш-хроматографией на силикагеле (EtOAc/гексаны, от 1:10 до 1:3), получая трет-бутил-((3aR,5R,6R,7R,7aR)-5-(((трет-бутил-диметилсилил)окси)метил)-7-фтор-6-гидрокси-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-2-ил)(метил)карбамат в виде белой пены (24,1 г, 96%). 1H ЯМР (400 МГц, CDCl3) δ 6,06 (д, J=6,8 Гц, 1H), 5,19-5,02 (м, 1H), 4,43-4,38 (м, 1H), 3,98-3,93 (м, 1H), 3,85 (дд, J=5,0, 10,6 Гц, 1H), 3,73 (дд, J=5,2, 10,6 Гц, 1H), 3,45-3,43 (м, 1H), 3,34 (с, 3H), 1,54 (с, 9H), 0,89 (с, 9H), 0,08 (с, 6H).
[00237] К раствору указанного выше соединения (9,28 г, 20,6 ммоль) в DCM (150 мл) добавляли DMP (13,1 г, 30,9 ммоль). После перемешивания при комнатной температуре в течение 1 ч реакционную смесь разбавляли Et2O (400 мл). Полученную суспензию фильтровали через слой целита, и фильтрат концентрировали до сухости при комнатной температуре. Остаток загружали на слоистую пробку NaHCO3/силикагеля, и продукт элюировали (EtOAc/гексаны, 1:4), получая трет-бутил-((3aR,5R,7R,7aR)-5-(((трет-бутил-диметилсилил)окси)метил)-7-фтор-6-оксо-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-2-ил)(метил)карбамат в виде белого кристаллического твердого вещества (8,96 г, 97%). 1H ЯМР (400 МГц, CDCl3) δ 6,29 (д, J=7,0 Гц, 1H), 5,09 (дд, J=4,7, 48,4 Гц, 1H), 4,75-4,69 (м, 1H), 4,12-4,05 (м, 2H), 3,96-3,93 (м, 1H), 3,28 (с, 3H), 1,54 (с, 9H), 0,86 (с, 9H), 0,056 (с, 3H), 0,050 (с, 3H).
[00238] К раствору указанного выше соединения (8,96 г, 20,0 ммоль) в сухом MeOH (250 мл) добавляли NaH (60% в минеральном масле, 0,158 г, 3,95 ммоль), и смесь перемешивали при комнатной температуре в течение 15 мин. Затем реакционную смесь охлаждали при 0°C и добавляли NaBH4 (1,32 г, 34,9 ммоль). После перемешивания смеси при 0°C в течение 20 мин добавляли осколок сухого льда, и растворитель выпаривали. Указанный выше остаток растворяли в DCM (100 мл) и промывали насыщенным водным NH4C1 (100 мл). Органический слой собирали, а водный экстрагировали DCM (2×50 мл). Объединенный экстракт сушили над безводным Na2SO4. После фильтрации растворитель выпаривали при пониженном давлении, и остаток очищали колоночной флэш-хроматографией на силикагеле (EtOAc/гексаны, от 1:10 до 2:5), получая трет-бутил-((3aR,5R,6R,7S,7aR)-5-(((трет-бутил-диметилсилил)окси)метил)-7-фтор-6-гидрокси-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-2-ил)(метил)карбамат в виде белой пены (6,84 г, 76% в 2 стадии). 1H ЯМР (400 МГц, CDCl3) δ 6,06 (д, J=6,7 Гц, 1H), 5,01 (тд, J=4,3, 46,8 Гц, 1H), 4,49-4,44 (м, 1H), 4,17-4,13 (м, 1H), 3,80-3,79 (м, 2H), 3,66-3,63 (м, 1H), 3,38 (с, 3H), 2,72 (шир.с, 1H, (OH)), 1,54 (с, 9H), 0,89 (с, 9H), 0,062 (с, 3H), 0,057 (с, 3H).
[00239] При 0°C к раствору указанного выше соединения (1,30 г, 2,89 ммоль) и Bu4NI (0,107 г, 0,290 ммоль) в безводном DMF (12 мл) добавляли NaH (60% в минеральном масле, 0,145 г, 3,63 ммоль). После добавления NaH добавляли BnBr (0,989 г, 5,78 ммоль). После перемешивания при 0°C в течение 30 мин и затем при комнатной температуре в течение ночи смесь разбавляли Et2O (100 мл). Смесь промывали насыщенным водным NH4C1 (2×50 мл). Водный слой экстрагировали Et2O (2×40 мл). Объединенный экстракт промывали солевым раствором (50 мл) и сушили над безводным Na2SO4. После фильтрации растворитель выпаривали при пониженном давлении, и остаток очищали колоночной флэш-хроматографией на силикагеле (EtOAc/гексаны, от 1:20 до 1:4), получая трет-бутил-((3aR,5R,6R,7S,7aR)-6-(бензилокси)-5-(((трет-бутил-диметилсилил)окси)метил)-7-фтор-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-2-ил)(метил)карбамат в виде липкого масла (1,44 г, 92%). 1H ЯМР (400 МГц, CDCl3) δ 7,36-7,27 (м, 5H), 6,21 (д, J=7,2 Гц, 1H), 5,30-5,16 (м, 1H), 4,80 (д, J=11,4 Гц, 1H), 4,56 (д, J=11,4 Гц, 1H), 4,50-4,42 (м, 1H), 3,95-3,78 (м, 4H), 3,44 (с, 3H), 1,54 (с, 9H), 0,89 (с, 9H), 0,049 (с, 6H).
[00240] При 0°C к раствору указанного выше соединения (1,44 г, 2,66 ммоль) в THF (25 мл) добавляли TBAF (1,0 M в THF, 3,5 мл, 3,5 ммоль). После добавления реакционную смесь перемешивали при комнатной температуре в течение 2 ч и разбавляли солевым раствором (50 мл). Смесь экстрагировали EtOAc (2×40 мл). Объединенный экстракт сушили над безводным Na2SO4. После фильтрации растворитель выпаривали при пониженном давлении, и остаток очищали колоночной флэш-хроматографией на силикагеле (EtOAc гексаны, от 1:2 до 1:1), получая трет-бутил-((3aR,5R,6R,7S,7aR)-6-(бензилокси)-7-фтор-5-(гидроксиметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-2-ил)(метил)карбамат в виде белого твердого вещества (1,08 г, 95%). 1H ЯМР (400 МГц, CDCl3) δ 7,37-7,27 (м, 5H), 6,18 (д, J=7,4 Гц, 1H), 5,17-5,04 (м, 1H), 4,84 (д, J=11,6 Гц, 1H), 4,55 (д, J=11,6 Гц, 1H), 4,50-4,43 (м, 1H), 3,95-3,91 (м, 1H), 3,88-3,82 (м, 1H), 3,79-3,75 (м, 1H), 3,71-3,67 (м, 1H), 3,37 (с, 3H), 1,53 (с, 9H).
[00241] К раствору указанного выше соединения (2,57 г, 6,03 ммоль) в DCM (60 мл) при 0°C добавляли DMP (3,82 г, 9,00 ммоль). После перемешивания при комнатной температуре в течение 1 ч реакционную смесь разбавляли Et2O (100 мл). Полученную суспензию фильтровали через слой целита, и фильтрат концентрировали до сухости при комнатной температуре. Остаток экстрагировали EtOAc (3×50 мл), и твердое вещество отфильтровывали. Экстракт промывали смешанным насыщенным водным NaHCO3 (30 мл) и Na2S2O3 (5 мл). Экстракт собирали и сушили над безводным MgSO4. После фильтрации растворитель выпаривали при пониженном давлении для получения неочищенного альдегида трет-бутил-((3aR,5S,6R,7S,7aR)-6-(бензилокси)-7-фтор-5-формил-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-2-ил)(метил)карбамата. Этот материал использовали на следующей стадии без дополнительной очистки. 1H ЯМР (400 МГц, CDCl3) δ 9,65 (с, 1H), 7,39-7,29 (м, 5H), 6,04 (д, J=7,0 Гц, 1H), 5,08 (тд, J=4,2, 46,7 Гц, 1H), 4,84 (д, J=11,4 Гц, 1H), 4,64 (д, J=11,4 Гц, 1H), 4,55-4,49 (м, 1H), 4,31 (д, J=7,5 Гц, 1H), 4,19-4,15 (м, 1H), 3,30 (с, 3H), 1,52 (с, 9H).
[00242] К раствору указанного выше соединения (0,19 г, чистота ≈80%, ≈0,36 ммоль) в безводном DCM (6 мл) при -78°C в атмосфере N2 добавляли DAST (0,37 г, 2,3 ммоль). После добавления смесь перемешивали при комнатной температуре в течение 24 ч. Затем реакционную смесь охлаждали при -78°C и гасили насыщенным водным NaHCO3 (10 мл). Органический слой собирали, а водный экстрагировали DCM (2×10 мл). Объединенный экстракт сушили над безводным Na2SO4. После фильтрации растворитель выпаривали при пониженном давлении, и остаток очищали колоночной флэш-хроматографией на силикагеле (EtOAc/гексаны, от 1:20 до 1:4), получая трет-бутил-((3aR,5S,6R,7S,7aR)-6-(бензилокси)-5-(дифторметил)-7-фтор-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-2-ил)(метил)карбамат в виде белой пены (0,097 г, 60%). 1H ЯМР (500 МГц, CDCl3) δ 7,37-7,28 (м, 5H), 6,12 (д, J=6,9 Гц, 1H), 5,85 (дт, J=1,6, 54,5 Гц, 1H), 5,11 (тд, J=4,0, 47,3 Гц, 1H), 4,83 (д, J=11,2 Гц, 1H), 4,57 (д, J=11,2 Гц, 1H), 4,55-4,50 (м, 1H), 4,12-4,04 (м, 2H), 3,28 (с, 3H), 1,52 (с, 9H).
[00243] К раствору указанного выше соединения (0,097 г, 0,22 ммоль) и пентаметилбензола (0,050 г, 0,34 ммоль) в безводном DCM (3 мл) при -78°C в атмосфере N2 добавляли BCl3 (1,0 M в DCM, 1,0 мл, 1,0 ммоль). Смесь перемешивали при комнатной температуре в течение ~5 ч, пока температура охлаждающей ловушки ни достигала комнатной температуры. Реакционную смесь охлаждали при -78°C, гасили смешанным раствором MeOH/DCM и затем концентрировали до сухого состояния. Остаток очищали колоночной флэш-хроматографией на силикагеле (1,0 M NH3 в MeOH/CH2Cl2, 1:12), получая (3aR,5S,6R,7S,7aR)-5-(дифторметил)-7-фтор-2-(метиламино)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6-ол в виде белого твердого вещества (0,041 г, 72%). 1H ЯМР (400 МГц, CD3OD) δ 6,37 (д, J=6,6 Гц, 1H), 6,04 (дт, J=1,5, 54,0 Гц, 1H), 4,86 (тд, J=3,3, 50,9 Гц, 1H), 4,39 (ддд, J=3,8, 6,6, 20,9 Гц, 1H), 4,05-3,95 (м, 2H), 2,86 (с, 3H); 13C ЯМР (100 МГц, CD3OD) δ 165,00, 115,89 (т, J=242,6 Гц), 90,65 (д, J=185,7 Гц), 90,25 (д, J=3,5 Гц), 72,22-71,71 (м), 71,71 (д, J=16,1 Гц), 66,22-65,97 (м), 30,48; MS, m/z=257,1 (M+1).
[00244] Соединения из следующих примеров могут быть синтезированы в соответствии с процедурами, аналогичными схемам и примерам, указанным выше.
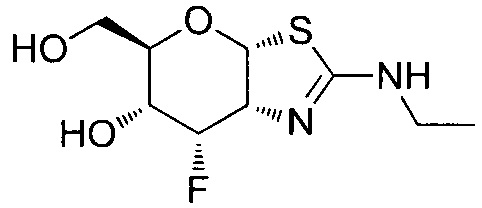
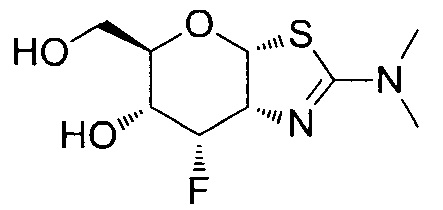
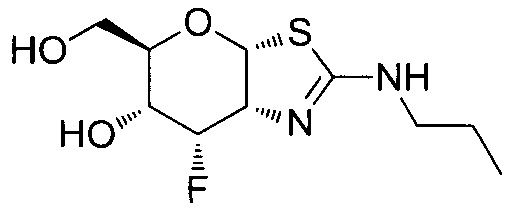
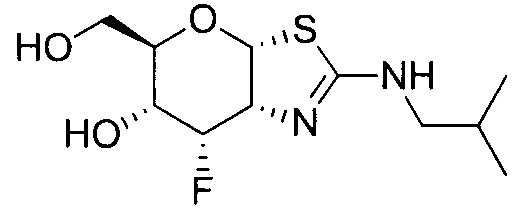

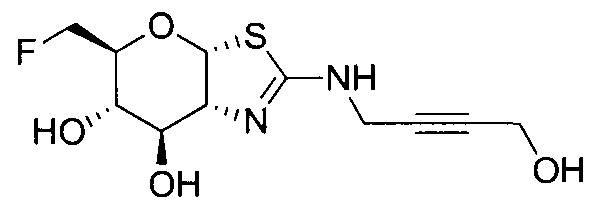
Биологическая активность
Анализ для определения величин K1 активности ингибирования O-GlcNAcase
[00245] Экспериментальная процедура для кинетических анализов: Ферментативные реакции проводили в реакционной смеси, содержащей 50 мМ NaH2PO4, 100 мМ NaCl и 0,1% BSA (pH 7,0), используя 2 мМ 4-Метилумбеллиферил-N-ацетил-β-D-глюкозаминид дигидрат (Sigma M2133), растворенный в ddH2O (дважды дистиллированной воде) в качестве субстрата. Количество очищенного фермента O-GlcNAcase, использованное в реакции, составляло 0,7 нМ. Тестируемое соединение в варьируемых концентрациях добавляли к ферменту перед инициацией реакции. Реакцию выполняли при комнатной температуре в 96-луночном планшете и инициировали добавлением субстрата. Продукцию флуоресцентного продукта измеряли через каждые 60 сек в течение 45 мин планшетным ридером Tecan Infinite M200 с возбуждением при 355 нМ и эмиссией, выявляемой при 460 нМ, с 4-Метилумбеллифероном (Sigma M1381), используемым для построения стандартной кривой. Наклон кривой выхода продукта определяли для каждой концентрации тестируемого соединения и наносили на график, используя алгоритмы подгонки к стандартной кривой для сигмоидальных кривых зависимости реакции от дозы. Определяли согласие величин для четырехпараметрической логистической кривой данных.
[00246] Величины K1 определяли, используя уравнение Ченга-Прусоффа; величина Km O-GlcNAcase для субстрата составила 0,2 мМ.
[00247] Соединения примеров с 1 по 33 тестировали в описанном выше анализе и проявили величины K1 для ингибирования O-GlcNAcase в диапазоне от 0,1 нМ до 10 мкМ.
Анализ для определения величин Κ1 для ингибирования активности β-гексозаминидазы
[00248] Экспериментальная процедура для кинетических анализов: Ферментативные реакции проводили в реакционной смеси, содержащей 50 мМ NaH2P04, 100 мМ NaCl и 0,1% BSA (pH 7,0), используя 2 мМ 4-Метилумбеллиферил-N-ацетил-β-D-глюкозаминид дигидрат (Sigma M2133), растворенный в ddH2O в качестве субстрата. Количество очищенного человеческого фермента β-гексозаминидазы, использованное в реакции, составило 24 нМ. Реакцию выполняли при комнатной температуре в 96-луночном планшете и инициировали добавлением субстрата. Выход флуоресцентного продукта измеряли через каждые 60 сек в течение 45 мин планшетным ридером Tecan Infinite M200 с возбуждением при 355 нМ и эмиссией, выявляемой при 460 нМ, с 4-Метилумбеллифероном (Sigma M1381), используемым для построения стандартной кривой. Наклон кривой выхода продукта определяли для каждой концентрации тестируемого соединения и наносили на график, используя алгоритмы подгонки к стандартной кривой для сигмоидальных кривых зависимости реакции от дозы. Определяли согласие величин для четырехпараметрической логистической кривой данных.
[00249] Величины K1 определяли, используя уравнение Ченга-Прусоффа.
[00250] При тестировании в этом анализе, многие из соединений, описанных в настоящей заявке, проявляли величины K1 для ингибирования β-гексозаминидазы в диапазоне от 10 нМ до более чем 100 мкМ.
[00251] Относительную селективность для ингибирования O-GlcNAcase относительно β-гексозаминидазы определяется в настоящем описании как:
K1 (β-гексозаминидазы)/ K1 (O-GlcNAcase)
[00252] В целом, соединения, описанные в настоящей заявке, проявляли относительную селективность в диапазоне примерно от 10 до 100000. Таким образом, многие соединения по изобретению проявляют высокую селективность ингибирования O-GlcNAcase относительно β-гексозаминидазы.
Анализ для определения клеточной активности для соединений, которые ингибируют активность O-GlcNAcase
[00253] Ингибирование O-GlcNAcase, которое удаляет O-GlcNAc из клеточных белков, приводит к увеличению уровня O-GlcNАцилированного белка в клетках. Увеличение содержания O-GlcNАцилированного белка можно измерить антителом, таким как RL-2, которое связывается с O-GlcNАцилированным белком. Количественное взаимодействие O-GlcNАцилированного белка:антитела RL2 можно измерить процедурами иммуноферментного анализа (ELISA).
[00254] Могут использоваться разнообразные линии клеток тканевых культур, экспрессирующие эндогенные уровни O-GlcNAcase; примеры включают крысиные клетки PC-12 и человеческие клетки U-87 или клетки SK-N-SH. В этом анализе крысиные клетки PC-12 высевали в 96-луночные планшеты при плотности приблизительно 10000 клеток/лунку. Соединения, подлежащие тестированию, растворяли в DMSO, или 2, или 10 мМ основного раствора и затем разбавляли DMSO и водой в двухстадийном способе, используя автоматизированную энзим-иммунизационную рабочую станцию Tecan. Клетки обрабатывали разбавленными соединениями в течение 24 ч (5,4 мкл, в 200 мкл объема 1 лунки) для достижения конечной концентрации ингибитора, желательной для измерения зависимой от концентрации соединения реакции; обычно, стадии 3-кратного разведения, начиная при 10 мкМ, использовали для определения кривой зависимости реакции от концентрации. Для получения клеточного лизата удаляли среды клеток, обработанных соединением, клетки промывали однократно солевым раствором, забуференным фосфатом (PBS), и затем лизировали в течение 5 минут при комнатной температуре в 50 мкл реагента Phosphosafe (Novagen Inc, Madison, WI) с ингибиторами протеазы и PMSF. Клеточный лизат собирали и переносили в новый планшет, который затем или покрывали для непосредственного анализа планшетов, или замораживали при -80°C до использования в процедуре ELISA. При желании, общую концентрацию белка образцов определяли, используя 20 мкл образца, с использованием способа BCA (анализа состава образца).
[00255] Относящуюся к ELISA часть анализа выполняли в черном 96-луночном планшете Maxisorp, который покрывали в течение ночи при 4°C с 100 мкл клеточного лизата /лунку (разведение 1:10 лизата PBS, содержащим ингибиторы протеазы, ингибиторы фосфатазы и PMSF (фенилметансульфонил фторид)). На следующий день лунки промывали 3 раза 300 мкл/лунку промывного буфера (солевого раствора, забуференного Трис-буфером, с 0,1% Твин 20). Лунки блокировали 100 мкл/лунку блокирующего буфера (солевого раствора, забуференного Трис-буфером/0,05% Твин 20 и 2,5% бычьего сывороточного альбумина на лунку). Затем каждую лунку промывали два раза 300 мкл/лунку промывного буфера. Антитело RL-2 (Abeam, Cambridge, MA) к O-GlcNAc, разбавленное 1:1000 в блокирующем буфере, добавляли в концентрации 100 мкл/лунку. Планшет герметично укупоривали и инкубировали при 37°C в течение 2 ч при легком встряхивании. Затем лунки промывали 3 раза 300 мкл/лунку промывного буфера. Для выявления количества связанного RL-2 добавляли конъюгированное с пероксидазой хрена (HRP) козье антимышиное вторичное антитело (разведенное 1:3000 в блокирующем буфере) в концентрации 100 мкл/лунку. Планшет инкубировали в течение 60 мин при 37°C при легком встряхивании. Затем каждую лунку промывали 3 раза 300 мкл/лунку промывного буфера. Добавляли реагент выявления, 100 мкл/лунку реагента Amplex Ultra RED (полученного добавлением 30 мкл 10 мМ основного раствора Amplex Ultra Red к 10 мл PBS с 18 мкл 3% пероксида водорода, H2O2). Реакцию выявления проводили в условиях инкубации в течение 15 минут при комнатной температуре и затем считывали с возбуждением при 530 нм и эмиссией при 590 нм.
[00256] Количество O-GlcNАцетилированного белка, по данным выявления анализом ELISA, наносили на график для каждой концентрации тестируемого соединения, используя алгоритмы подгонки к стандартной кривой для сигмоидальных кривых зависимости реакции от дозы. Определяли величины данных для четырехпараметрической логистической кривой, причем точка перегиба кривой представляла собой величину активности тестируемого соединения.
Анализ для определения видимой проникающей способности (Papp)
[00257] Двунаправленный транспорт оценивали на клетках LLC-PK1 для определения видимой проникающей способности (Papp). Клетки LLC-PK1 могут образовывать плотный монослой, и поэтому могут использоваться для оценки векторного транспорта соединений из базолатерального к апикальному отделению (B → A) и от апикального к базолатеральному отделению (A → B).
[00258] Для определения Papp клетки LLC-PK1 культивировали в 96-луночных транслуночных культуральных планшетах (Millipore). Растворы, содержащие тестируемые соединения (1 мкМ), получали в сбалансированном солевом растворе Хэнкса с 10 мМ HEPES. Раствор субстрата (150 мкл) добавляли или в апикальное (A), или в базолатеральное (B) отделение культурального планшета, и буфер (150 мкл) добавляли в отделение, находящееся напротив отделения, содержащего соединение. Во время t=3 ч, 50 мкл образцов удаляли из обеих сторон монослоев, в которые вводили тестируемое соединение, и помещали в 96-луночные планшеты, к образцам добавляли сцинтиллирующее вещество (200 мкл) или внутренний стандарт (100 мкл лабетлола 1 мкМ), и концентрацию определяли подсчетом сцинтилляции жидкости в сцинтилляционном счетчике MicroBeta Wallac Trilux scintillation (Perkin Elmer Life Sciences, Boston, MA) или LCMS/MS (ЖХМС/МС) (масс-спектрометр с тройным квадруполем) Applied Biosystems SCIEX API 5000). [3H]Верапамил (1 мкМ) использовали в качестве положительного контроля. Эксперимент выполняли в трех повторениях.
[00259] Видимую проникающую способность, Papp, рассчитывали по следующей формуле для образцов, взятых во время t=3 ч:
Где: Объем приемной камеры составил 0,15 мл; Площадь мембраны составила 0,11 см2; Исходная концентрация представляет сумму концентрации, измеренной в донорском отделении плюс концентрация, измеренная в приемных отделениях во время t=3 ч; Δ Концентрации представляет концентрацию в приемном отделении через 3 ч; и Δ Времени представляет время инкубации (3×60×60=10800 с). Papp выражали в виде 10-6 см/с. Papp (клеток LLC-PK1) представляют среднюю величину Papp для транспорта из A в B и Papp для транспорта из B в A во время t=3 ч:
Репрезентативные данные описанных выше анализов связывания, на основе клеток и проникающей способности показаны в следующей таблице. Определенные соединения по изобретению проявили превосходящую активность и проникающую способность в одном или более из этх анализов, по сравнению с соединениями, описанными в опубликованных заявках на Международные патенты WO 2006/092049 и WO 2008/025170. Для сравнения, в первых двух строках таблицы показаны данные для соединений (3aR,5R,6S,7R,7aR)-2-(этиламино)-5-(гидроксиметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диол и (3aR,5R,6S,7R,7aR)-2-(диметиламино)-5-(гидроксиметил)-5,6,7,7a-тетрагидро-3aH-пирано[3,2-d]тиазол-6,7-диол, описанным в опубликованной заявке на Международный патент WO 2008/025170.
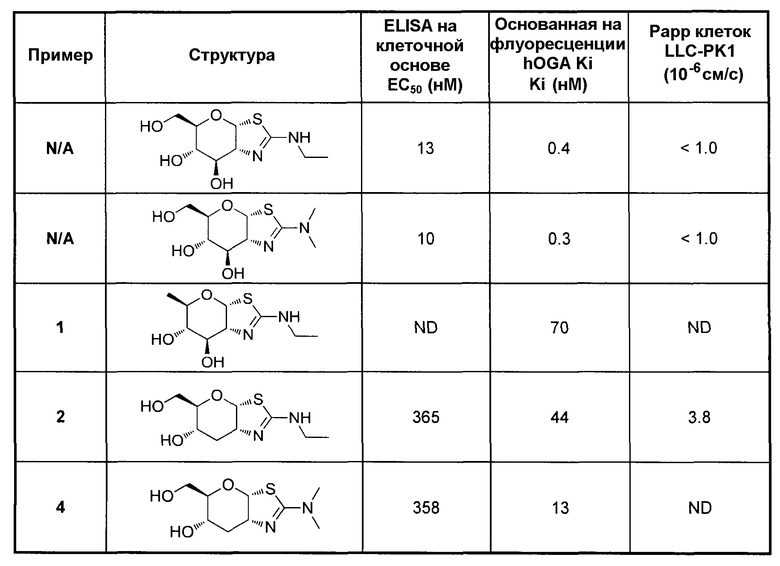
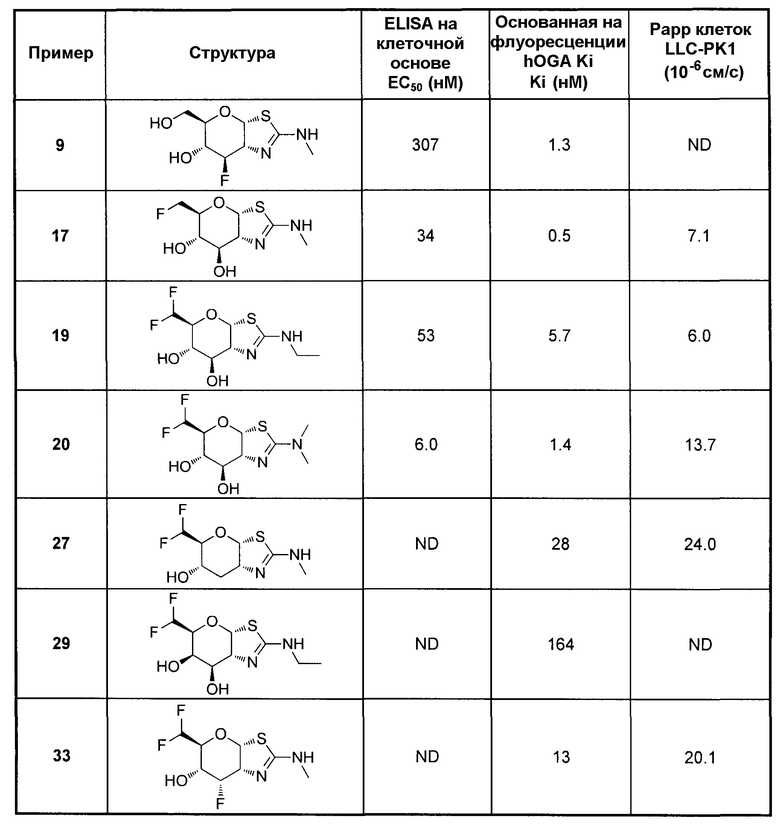
[00260] Настоящее изобретение было описано в отношении одного или более вариантов осуществления. Однако для специалистов в данной области очевидно, что может быть внесен ряд изменений и модификаций без отхода от объема изобретения, определенного в формуле изобретения.
ССЫЛКИ
1. C. R. Torres, G. W. Hart, J Biol Chem 1984, 259, 3308.
2. R. S. Haltiwanger, G. D. Holt, G. W. Hart, J Biol Chem 1990, 265, 2563.
3. L. K. Kreppel, M. A. Blomberg, G. W. Hart, J Biol Chem 1997, 272, 9308.
4. W. A. Lubas, D. W. Frank, M. Krause, J. A. Hanover, J Biol Chem 1997, 272, 9316.
5. W. A. Lubas, J. A. Hanover, J Biol Chem 2000, 275, 10983.
6. D. L. Dong, G. W. Hart, J Biol Chem 1994, 269, 19321.
7. Y. Gao, L. Wells, F. I. Comer, G. J. Parker, G. W. Hart, J Biol Chem 2001, 276, 9838.
8. E. P. Roquemore, M. R. Chevrier, R. J. Cotter, G. W. Hart, Biochemistry 1996, 35, 3578.
9. S. P. Jackson, R. Tjian, Cell 1988, 55, 125.
10. W. G. Kelly, M. E. Dahmus, G. W. Hart, J Biol Chem 1993, 268, 10416.
11. M. D. Roos, K. Su, J. R. Baker, J. E. Kudlow, Mol Cell Biol 1997, 17, 6472.
12. N. Lamarre-Vincent, L. C. Hsieh-Wilson, J Am Chem Soc 2003, 125, 6612.
13. F. Zhang, K. Su, X. Yang, D. B. Bowe, A. J. Paterson, J. E. Kudlow, Cell 2003, 115, 715.
14. K. Vosseller, L. Wells, M. D. Lane, G. W. Hart, Proc Natl Acad Sci US A 2002, 99, 5313.
15. W. A. Lubas, M. Smith, C. M. Starr, J. A. Hanover, Biochemistry 1995, 34, 1686.
16. L. S. Griffith, B. Schmitz, Biochem Biophys Res Commun 1995, 213, 424.
17. R. N. Cole, G. W. Hart, J Neurochem 1999, 73, 418.
18. I. Braidman, M. Carroll, N. Dance, D. Robinson, Biochem J 1974, 143, 295.
19. R. Ueno, C. S. Yuan, Biochim Biophys Acta 1991, 1074, 79.
20. C. Toleman, A. J. Paterson, T. R. Whisenhunt, J. E. Kudlow, J Biol Chem 2004.
21. F. Liu, K. Iqbal, I. Grundke-Iqbal, G. W. Hart, C. X. Gong, Proc Natl Acad Sci USA 2004, 101, 10804.
22. T. Y. Chou, G. W. Hart, Adv Exp Med Biol 2001, 491, 413.
23. M. Goedert, M. G. Spillantini, N. J. Cairns, R. A. Crowther, Neuron 1992, 8, 159.
24. M. Goedert, M. G. Spillantini, R. Jakes, D. Rutherford, R. A. Crowther, Neuron 1989, 3, 519.
25. E. Kopke, Y. C. Tung, S. Shaikh, A. C. Alonso, K. Iqbal, I. Grundke-Iqbal, J Biol Chem 1993, 268, 24374.
26. H. Ksiezak-Reding, W. K. Liu, S. H. Yen, Brain Res 1992, 597, 209.
27. B. Henrissat, A. Bairoch, Biochem J 1996, 316 ( Pt 2), 695.
28. B. Henrissat, A. Bairoch, Biochem J 1993, 293 (Pt 3), 781.
29. C. X. Gong, F. Liu, I. Grundke-Iqbal, K. Iqbal, J Neural Transm 2005, 112, 813.
30. K. Iqbal, C. Alonso Adel, E. El-Akkad, C. X. Gong, N. Haque, S. Khatoon, I. Tsujio, I. Grundke-Iqbal, J Neural Transm Suppl 2002, 309.
31. K. Iqbal, C. Alonso Adel, E. El-Akkad, C. X. Gong, N. Haque, S. Khatoon, J. J. Pei, H. Tanimukai, I. Tsujio, et al., J Mol Neurosci 2003, 20, 425.
32. W. Noble, E. Planel, C. Zehr, V. Olm, J. Meyerson, F. Suleman, K. Gaynor, L. Wang, J. LaFrancois, et al., Proc Natl Acad Sci US A 2005, 102, 6990.
33. S. Le Corre, H. W. Klafki, N. Plesnila, G. Hubinger, A. Obermeier, H. Sahagun, B. Monse, P. Seneci, J. Lewis, et al, Proc Natl Acad Sci USA 2006, 103, 9673.
34. S. J. Liu, J. Y. Zhang, H. L. Li, Z. Y. Fang, Q. Wang, H. M. Deng, C. X. Gong, I. Grundke-Iqbal, K. Iqbal, et al, J Biol Chem 2004, 279, 50078.
35. G. Li, H. Yin, J. Kuret, J Biol Chem 2004, 279, 15938.
36. T. Y. Chou, G. W. Hart, C. V. Dang, J Biol Chem 1995, 270, 18961.
37. X. Cheng, G. W. Hart, J Biol Chem 2001, 276, 10570.
38. X. Cheng, R. N. Cole, J. Zaia, G. W. Hart, Biochemistry 2000, 39, 11609.
39. L. S. Griffith, B. Schmitz, Eur J Biochem 1999, 262, 824.
40. K. Kamemura, G. W. Hart, Prog Nucleic Acid Res Mol Biol 2003, 73, 107.
41. L. Wells, L. K. Kreppel, F. I. Comer, B. E. Wadzinski, G. W. Hart, J Biol Chem 2004, 279, 38466.
42. L. Bertram, D. Blacker, K. Mullin, D. Keeney, J. Jones, S. Basu, S. Yhu, M. G. Mclnnis, R. C. Go, et al., Science 2000, 290, 2302.
43. S. Hoyer, D. Blum-Degen, H. G. Bernstein, S. Engelsberger, J. Humrich, S. Laufer, D. Muschner, A. Thalheimer, A. Turk, et al., Journal of Neural Transmission 1998, 105, 423.
44. C. X. Gong, F. Liu, I. Grundke-Iqbal, K. Iqbal, Journal of Alzheimers Disease 2006, 9, 1.
45. W. J. Jagust, J. P. Seab, R. H. Huesman, P. E. Valk, C. A. Mathis, B. R. Reed, P. G. Coxson, T. F. Budinger, Journal of Cerebral Blood Flow and Metabolism 1991, 11, 323.
46. S. Hoyer, Experimental Gerontology 2000, 35, 1363.
47. S. Hoyer, in Frontiers in Clinical Neuroscience: Neurodegeneration and Neuroprotection, Vol. 541, 2004, pp. 135.
48. R. N. Kalaria, S. I. Harik, Journal of Neurochemistry 1989, 53, 1083.
49. I. A. Simpson, K. R. Chundu, T. Davieshill, W. G. Honer, P. Davies, Annals of Neurology 1994, 35, 546.
50. S. M. de la Monte, J. R. Wands, Journal of Alzheimers Disease 2005, 7, 45.
51. X. W. Zhu, G. Perry, M. A. Smith, Journal of Alzheimers Disease 2005, 7, 81.
52. J. C. de la Torre, Neurological Research 2004, 26, 517.
53. S. Marshall, W. T. Garvey, R. R. Traxinger, Faseb J 1991, 5, 3031.
54. S. P. Iyer, Y. Akimoto, G. W. Hart, J Biol Chem 2003, 278, 5399.
55. K. Brickley, M. J. Smith, M. Beck, F. A. Stephenson, J Biol Chem 2005, 280, 14723.
56. S. Knapp, C. H. Yang, T. Haimowitz, Tetrahedron Letters 2002, 43, 7101.
57. S. P. Iyer, G. W. Hart, J Biol Chem 2003, 278, 24608.
58. M. Jinek, J. Rehwinkel, B. D. Lazarus, E. Izaurralde, J. A. Hanover, E. Conti, Nat Struct Mol Biol 2004, 77, 1001.
59. K. Kamemura, B. K. Hayes, F. I. Comer, G. W. Hart, J Biol Chem 2002, 277, 19229.
60. Y. Deng, B. Li, F. Liu, K. Iqbal, I. Grundke-Iqbal, R. Brandt, C.-X. Gong, FASEB J, 2007, fj.07.
61. L. F. Lau, J. B. Schachter, P. A. Seymour, M. A. Sanner, Curr Top Med Chem 2002, 2, 395.
62. M. P. Mazanetz, P. M. Fischer, Nature Reviews Drug Discovery 2007, 6, 464.
63. S. A. Yuzwa, M. S. Macauley, J. E. Heinonen, X. Shan, R. J. Dennis, Y. He, G. E. Whitworth, K. A. Stubbs, E. J. McEachern, et al., Nat Chem Biol 2008, 4, 483.
64. P. Bounelis, J. Liu, Y. Pang, J. C. Chatham, R. B. Marchase, Shock 2004, 21 170 Suppl. 2, 58.
65. N. Fulop, V. Champattanachal, R. B. Marchase, J. C. Chatham, Circulation Research 2005, 97, E28.
66. J. Liu, R. B. Marchase, J. C. Chatham, Faseb Journal 2006, 20, A317.
67. R. Marchase, P. Bounelis, J. Chatham, I. Chaudry, Y. Pang, PCT Int. Appl. WO 2006016904 2006.
68. N. Fulop, P. P. Wang, R. B. Marchase, J. C. Chatham, Journal of Molecular and Cellular Cardiology 2004, 37, 286.
69. N. Fulop, P. P. Wang, R. B. Marchase, J. C. Chatham, Faseb Journal 2005, 19, A689.
70. J. Liu, R. B. Marchase, J. C. Chatham, Journal of Molecular and Cellular Cardiology 2007, 42, 177.
71. L. G. Not, C. A. Brocks, N. Fulop, R. B. Marchase, J. C. Chatham, Faseb Journal 2006, 20, A 1471.
72. S. L. Yang, L. Y. Zou, P. Bounelis, I. Chaudry, J. C. Chatham, R. B. Marchase, Shock 2006, 25, 600.
73. L. Y. Zou, S. L. Yang, P. Bounelis, I. H. Chaudry, J. C. Chatham, R. B. Marchase, Faseb Journal 2005, 19, A 1224.
74. R. B. Marchase, J. Liu, L. Y. Zou, V. Champattanachai, Y. Pang, N. Fulop, P. P. Wang, S. L. Yang, P. Bounelis, et al., Circulation 2004, 110, 1099.
75. J. Liu, Y. Pang, T. Chang, P. Bounelis, J. C. Chatham, R. B. Marchase, Journal of Molecular and Cellular Cardiology 2006, 40, 303.
76. J. Liu, J. C. Chatham, R. B. Marchase, Faseb Journal 2005, 19, A691.
77. T. Nagy, V. Champattanachai, R. B. Marchase, J. C. Chatham, American Journal of Physiology-Cell Physiology 2006, 290, C57.
78. N. Fulop, R. B. Marchase, J. C. Chatham, Cardiovascular Research 2007, 73, 288.
79. T. Lefebvre, C. Guinez, V. Dehennaut, O. Beseme-Dekeyser, W. Morelle, J. C. Michalski, Expert Review of Proteomics 2005, 2, 265.
80. L. Wells, K. Vosseller, G. W. Hart, Science 2001, 291, 2376.
81. J. A. Hanover, FASEB J 2001, 15, 1865.
82. D. A. McClain, W. A. Lubas, R. C. Cooksey, M. Hazel, G. J. Parker, D. C. Love, J. A. Hanover, Proc Natl Acad Sci USA 2002, 99, 10695.
83. P. J. Yao, P. D. Coleman, J Neurosci 1998, 75, 2399.
84. W. H. Yang, J. E. Kim, H. W. Nam, J. W. Ju, H. S. Kim, Y. S. Kim, J. W. Cho, Nature Cell Biology 2006, 8, 1074.
85. B. Triggs-Raine, D. J. Mahuran, R. A. Gravel, Adv Genet 2001, 44, 199.
86. D. Zhou, J. Mattner, C. Cantu Iii, N. Schrantz, N. Yin, Y. Gao, Y. Sagiv, K. Hudspeth, Y. Wu, et al., Science 2004.
87. G. Legler, E. Lullau, E. Kappes, F. Kastenholz, Biochim Biophys Acta 1991, 1080, 89.
88. M. Horsch, L. Hoesch, A. Vasella, D. M. Rast, Eur J Biochem 1991, 197, 815.
89. J. Liu, A. R. Shikhman, M. K. Lotz, C. H. Wong, Chem Biol 2001, 8, 701.
90. S. Knapp, D. J. Vocadlo, Z. N. Gao, B. Kirk, J. P. Lou, S. G. Withers, J. Am. Chem. Soc. 1996, 118, 6804.
91. V. H. Lillelund, H. H. Jensen, X. Liang, M. Bols, Chem Rev 2002, 102, 515.
92. R. J. Konrad, I. Mikolaenko, J. F. Tolar, K. Liu, J. E. Kudlow, Biochem J 2001, 356, 31.
93. K. Liu, A. J. Paterson, F. Zhang, J. McAndrew, K. Fukuchi, J. M. Wyss, L. Peng, Y. Hu, J. E. Kudlow, J Neurochem 2004, 89, 1044.
94. G. Parker, R. Taylor, D. Jones, D. McClain, J Biol Chem 2004, 279, 20636.
95. E. B. Arias, J. Kim, G. D. Cartee, Diabetes 2004, 53, 921.
96. A. Junod, A. E. Lambert, L. Orci, R. Pictet, A. E. Gonet, A. E. Renold, Proc Soc Exp Biol Med 1967, 126, 201.
97. R. A. Bennett, A. E. Pegg, Cancer Res 1981, 41, 2786.
98. K. D. Kroncke, K. Fehsel, A. Sommer, M. L. Rodriguez, V. Kolb-Bachofen, Biol Chem Hoppe Seyler 1995, 376, 179.
99. H. Yamamoto, Y. Uchigata, H. Okamoto, Nature 1981, 294, 284.
100. K. Yamada, K. Nonaka, T. Hanafusa, A. Miyazaki, H. Toyoshima, S. Tarui, Diabetes 1982, 31, 749.
101. V. Burkart, Z. Q. Wang, J. Radons, B. Heller, Z. Herceg, L. Stingl, E. F. Wagner, H. Kolb, Nat Med 1999, 5, 314.
102. M. D. Roos, W. Xie, K. Su, J. A. Clark, X. Yang, E. Chin, A. J. Paterson, J. E. Kudlow, Proc Assoc Am Physicians 1998, 110, 422.
103. Y. Gao, G. J. Parker, G. W. Hart, Arch Biochem Biophys 2000, 383, 296.
104. R. Okuyama, M. Yachi, Biochem Biophys Res Commun 2001, 287, 366.
105. Ν. E. Zachara, Ν. O'Donnell, W. D. Cheung, J. J. Mercer, J. D. Marth, G. W. Hart, J Biol Chem 2004, 279, 30133.
106. J. A. Hanover, Z. Lai, G. Lee, W. A. Lubas, S. M. Sato, Arch Biochem Biophys 1999, 362, 38.
107. K. Liu, A. j. Paterson, R. J. Konrad, A. F. Parlow, S. Jimi, M. Roh, E. Chin, Jr., J. E. Kudlow, Mol Cell Endocrinol 2002, 194, 135.
108. M. S. Macauley, G. E. Whitworth, A. W. Debowski, D. Chin, D. J. Vocadlo, J Biol Chem 2005, 280, 25313.
109. B. L. Mark, D. J. Vocadlo, S. Knapp, B. L. Triggs-Raine, S. G. Withers, M. Ν. James, J Biol Chem 2001, 276, 10330.
110. R. S. Haltiwanger, K. Grove, G. A. Philipsberg, J Biol Chem 1998, 273, 361 1.
111. D. J. Miller, X. Gong, B. D. Shur, Development 1993, 118, 1279.
112. L. Y. Zou, S. L. Yang, S. H. Hu, I. H. Chaudry, R. B. Marchase, J. C. Chatham, Shock 2007, 27, 402.
113. J. B. Huang, A. J. Clark, H. R. Petty, Cellular Immunology 2007, 245, 1.
114. U. J. G. Conference, in US/Japan Glyco 2004 Conference, Honolulu, Hawaii, 2004.
115. L. Y. Zou, S. L. Yang, S. H. Hu, I. H. Chaudry, R. B. Marchase, J. C. Chatham, Faseb Journal 2006, 20, A 1471.
116. V. Champattanachai, R. B. Marchase, J. C. Chatham, American Journal of Physiology-Cell Physiology 2007, 292, C 178.
117. V. Champattanachai, R. B. Marchase, J. C. Chatham, American Journal of Physiology-Cell Physiology 2008, 294, C I 509.
118. I. Khlistunova, M. Pickhardt, J. Biernat, Y. P. Wang, E. M. Mandelkow, E. Mandelkow, Current Alzheimer Research 2007, 4, 544.
119. P. Friedhoff, A. Schneider, E. M. Mandelkow, E. Mandelkow, Biochemistry 1998, 37, 10223.
120. M. Pickhardt, Z. Gazova, M. von Bergen, I. Khlistunova, Y. P. Wang, A. Hascher, E. M. Mandelkow, J. Biernat, E. Mandelkow, Journal of Biological Chemistry 2005, 280, 3628.
| название | год | авторы | номер документа |
|---|---|---|---|
| СЕЛЕКТИВНЫЕ ИНГИБИТОРЫ ГЛИКОЗИДАЗЫ И ИХ ПРИМЕНЕНИЕ | 2011 |
|
RU2592285C2 |
| ПРОНИЦАЕМЫЕ ИНГИБИТОРЫ ГЛИКОЗИДАЗЫ И ИХ ПРИМЕНЕНИЯ | 2013 |
|
RU2707292C2 |
| СЕЛЕКТИВНЫЕ ИНГИБИТОРЫ ГЛИКОЗИДАЗЫ И ИХ ПРИМЕНЕНИЕ | 2012 |
|
RU2625308C2 |
| ИНГИБИТОРЫ ГЛИКОЗИДАЗ И ИХ ПРИМЕНЕНИЯ | 2013 |
|
RU2672873C2 |
| РЕАКЦИИ МАКРОЦИКЛИЗАЦИИ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И ДРУГИЕ ФРАГМЕНТЫ, ПРИГОДНЫЕ В ПОЛУЧЕНИИ АНАЛОГОВ ХАЛИХОНДРИНА B | 2014 |
|
RU2710545C2 |
| ИНГИБИТОРЫ RMT5 | 2019 |
|
RU2814198C2 |
| АМИДЫ КОНДЕНСИРОВАННОГО ПИПЕРИДИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ ИОННЫХ КАНАЛОВ | 2014 |
|
RU2741810C2 |
| РЕАКЦИЯ ПРИНСА И ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ, ПРИМЕНИМЫЕ ПРИ СИНТЕЗЕ МАКРОЛИДОВ ГАЛИХОНДРИНОВОГО РЯДА И ИХ АНАЛОГОВ | 2017 |
|
RU2777913C2 |
| КОНДЕНСИРОВАННОЕ ПРОИЗВОДНОЕ АМИНОДИГИДРОТИАЗИНА | 2009 |
|
RU2476431C2 |
| ЗАМЕЩЕННЫЕ ПУРИНОВЫЕ И 7-ДЕАЗАПУРИНОВЫЕ СОЕДИНЕНИЯ | 2011 |
|
RU2606514C2 |
Изобретение относится к соединениям, представленным формулами (1), (2) или (3), или их фармацевтически приемлемой соли. Изобретение также относится к фармацевтической композиции, включающей в качестве активного ингредиента соединение, представленное формулами (1), (2) или (3), обладающей ингибирующей активностью в отношении O-GlcNAcase, или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель. Соединения или их фармацевтически приемлемая соль предназначены для лечения Альцгеймера, болезни Паркинсона или прогрессирующего супрануклеарного паралича (PSP) у пациента человека, нуждающегося в этом. Технический результат – соединения, являющиеся селективными ингибиторами гликозидазы. 12 н. и 33 з.п. ф-лы, 6 табл., 33 пр.
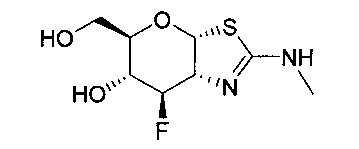 (1),
(1),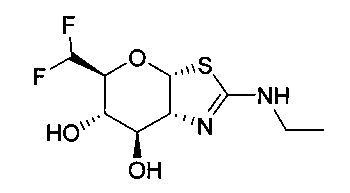 (2),
(2),
 (3).
(3).
1. Соединение, представляющее собой
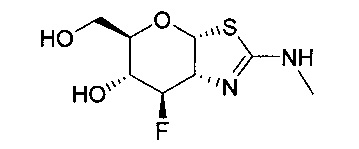
или его фармацевтически приемлемая соль.
2. Соединение, представляющее собой
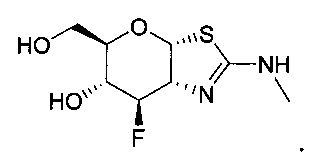
3. Соединение, представляющее собой
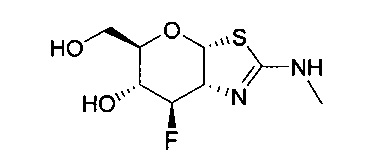
в форме фармацевтически приемлемой соли.
4. Соединение, представляющее собой
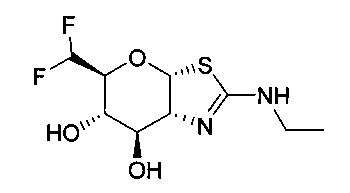
или его фармацевтически приемлемая соль.
5. Соединение, представляющее собой
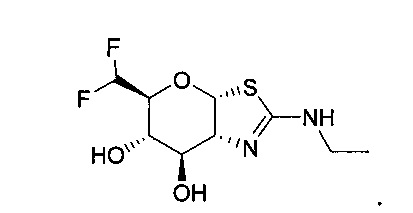
6. Соединение, представляющее собой
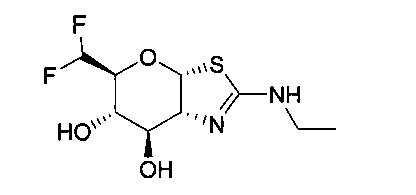
в форме фармацевтически приемлемой соли.
7. Соединение, представляющее собой
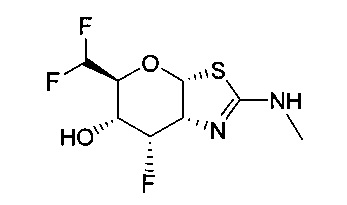
или его фармацевтически приемлемая соль.
8. Соединение, представляющее собой
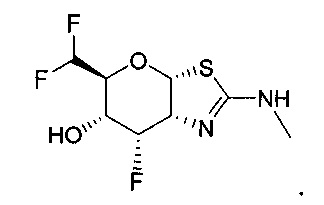
9. Соединение, представляющее собой
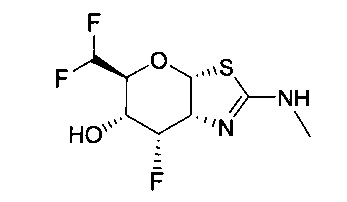
в форме фармацевтически приемлемой соли.
10. Фармацевтическая композиция, включающая в качестве активного ингредиента соединение, обладающее ингибирующей активностью в отношении O-GlcNAcase, представляющее собой
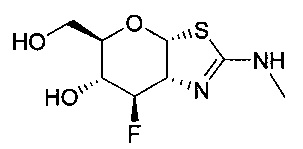
или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
11. Фармацевтическая композиция по п.10, где соединение представляет собой
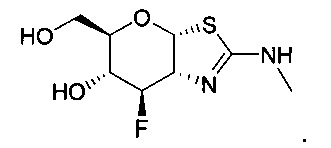
12. Фармацевтическая композиция по п.10, где соединение представляет собой
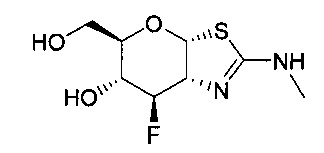
в форме фармацевтически приемлемой соли.
13. Фармацевтическая композиция, включающая в качестве активного ингредиента соединение, обладающее ингибирующей активностью в отношении O-GlcNAcase, представляющее собой
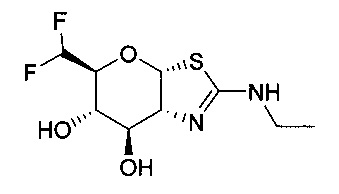
или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
14. Фармацевтическая композиция по п.13, где соединение представляет собой
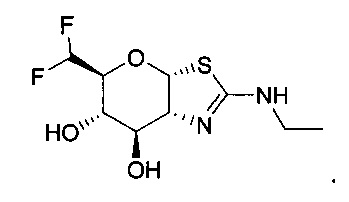
15. Фармацевтическая композиция по п.13, где соединение представляет собой
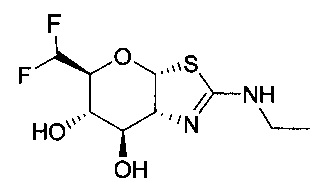
в форме фармацевтически приемлемой соли.
16. Фармацевтическая композиция, включающая в качестве активного ингредиента соединение, обладающее ингибирующей активностью в отношении O-GlcNAcase, представляющее собой
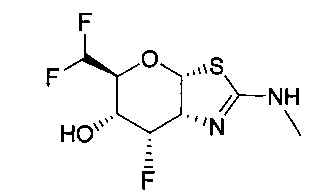
или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
17. Фармацевтическая композиция по п.16, где соединение представляет собой
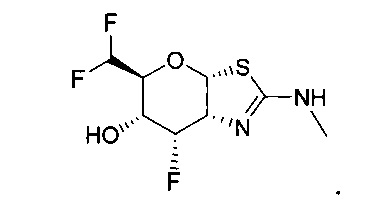
18. Фармацевтическая композиция по п.16, где соединение представляет собой
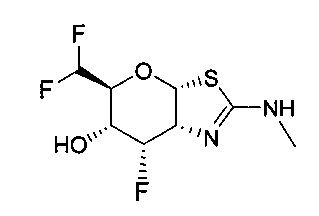
в форме фармацевтически приемлемой соли.
19. Соединение по п.1 или его фармацевтически приемлемая соль для лечения болезни Альцгеймера у пациента человека, нуждающегося в этом.
20. Соединение по п.2 для лечения болезни Альцгеймера у пациента человека, нуждающегося в этом.
21. Соединение по п.3 в форме фармацевтически приемлемой соли для лечения болезни Альцгеймера у пациента человека, нуждающегося в этом.
22. Соединение по п.4 или его фармацевтически приемлемая соль для лечения болезни Альцгеймера у пациента человека, нуждающегося в этом.
23. Соединение по п.5 для лечения болезни Альцгеймера у пациента человека, нуждающегося в этом.
24. Соединение по п.6 в форме фармацевтически приемлемой соли для лечения болезни Альцгеймера у пациента человека, нуждающегося в этом.
25. Соединение по п.7 или его фармацевтически приемлемая соль для лечения болезни Альцгеймера у пациента человека, нуждающегося в этом.
26. Соединение по п.8 для лечения болезни Альцгеймера у пациента человека, нуждающегося в этом.
27. Соединение по п.6 в форме фармацевтически приемлемой соли для лечения болезни Альцгеймера у пациента человека, нуждающегося в этом.
28. Соединение по п.1 или его фармацевтически приемлемая соль для лечения болезни Паркинсона у пациента человека, нуждающегося в этом.
29. Соединение по п.2 для лечения болезни Паркинсона у пациента человека, нуждающегося в этом.
30. Соединение по п.3 в форме фармацевтически приемлемой соли для лечения болезни Паркинсона у пациента человека, нуждающегося в этом.
31. Соединение по п.4 или его фармацевтически приемлемая соль для лечения болезни Паркинсона у пациента человека, нуждающегося в этом.
32. Соединение по п.5 для лечения болезни Паркинсона у пациента человека, нуждающегося в этом.
33. Соединение по п.6 в форме фармацевтически приемлемой соли для лечения болезни Паркинсона у пациента человека, нуждающегося в этом.
34. Соединение по п.7 или его фармацевтически приемлемая соль для лечения болезни Паркинсона у пациента человека, нуждающегося в этом.
35. Соединение по п.8 для лечения болезни Паркинсона у пациента человека, нуждающегося в этом.
36. Соединение по п.9 в форме фармацевтически приемлемой соли для лечения болезни Паркинсона у пациента человека, нуждающегося в этом.
37. Соединение по п.1 или его фармацевтически приемлемая соль для лечения прогрессирующего супрануклеарного паралича (PSP) у пациента человека, нуждающегося в этом.
38. Соединение по п.2 для лечения прогрессирующего супрануклеарного паралича (PSP) у пациента человека, нуждающегося в этом.
39. Соединение по п.3 в форме фармацевтически приемлемой соли для лечения супрануклеарного паралича (PSP) у пациента человека, нуждающегося в этом.
40. Соединение по п.4 или его фармацевтически приемлемая соль для лечения прогрессирующего супрануклеарного паралича (PSP) у пациента человека, нуждающегося в этом.
41. Соединение по п.5 для лечения прогрессирующего супрануклеарного паралича (PSP) у пациента человека, нуждающегося в этом.
42. Соединение по п.6 в форме фармацевтически приемлемой соли для лечения супрануклеарного паралича (PSP) у пациента человека, нуждающегося в этом.
43. Соединение по п.7 или его фармацевтически приемлемая соль для лечения прогрессирующего супрануклеарного паралича (PSP) у пациента человека, нуждающегося в этом.
44. Соединение по п.8 для лечения прогрессирующего супрануклеарного паралича (PSP) у пациента человека, нуждающегося в этом.
45. Соединение по п.9 в форме фармацевтически приемлемой соли для лечения супрануклеарного паралича (PSP) у пациента человека, нуждающегося в этом.
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| Tracey Gloster et al., Mechanism, Structure, and Inhibition of O-GlcNAc Processing Enzymes, Current Signal Transduction Therapy, 2010, vol.5, no.1, p.74-91;Macauley M | |||
| S | |||
| et al., Increasing O-GlcNAc levels: An overview of small-molecule inhibitors of O-GlcNAcase, Biochimica et Biophysica Acta, 2010, vol.1800, p.107-121;Scott A Yuzwa et al., A potent mechanism-inspired O-GlcNAcase inhibitor that blocks phosphorylation of tau in vivo, Nature Chemical Biology, 2008, vol.4, no.8, p.483-490;Macauley M | |||
| S | |||
| et al., Enzymatic characterization and inhibition of the nuclear variant of human O-GlcNAcase, Carbohydrate Research, 2009, vol.344, p.1079-1084;Whitworth G | |||
| E | |||
| et al., Analysis of PUGNAc and NAG-thiazoline as Transition State Analogues for Human O-GlcNAcase: Mechanistic and Structural Insights into Inhibitor Selectivity and Transition State Poise, Journal of the American Chemical Society, 2007, vol.129, p.635-644;Macauley M | |||
| S | |||
| et al., O-GlcNAcase Uses Substrate-assisted Catalysis, Journal of Biological Chemistry, 2005, vol.280, no.27, p.25313-25322;Spencer Knapp et al., Tautomeric Modification of GlcNAc-Thiazoline, Organic Letters, 2007, vol.9, no.12, p.2321-2324. | |||

