Перекрестная ссылка на родственную заявку
Настоящая заявка испрашивает приоритет и преимущества предварительной заявки США № 61/419661, поданной 3 декабря 2010 года, содержание которой включено в настоящую заявку посредством ссылки во всей своей полноте.
Предпосылки изобретения
В эукариотных клетках ДНК упакована с гистонами с образованием хроматина. Приблизительно 150 пар оснований ДНК обвиваются два раза вокруг октамера гистонов (двух из каждых следующих гистонов 2A, 2B, 3, и 4) с образованием нуклеосомы, базовой единицы хроматина. Изменения в упорядоченной структуре хроматина могут привести к изменениям в транскрипции соответствующих генов. Этот процесс является высоко контролируемым, поскольку изменения картин генной экспрессии может существенно влиять на фундаментальные клеточные процессы, такие как дифференциация, пролиферация и апоптоз. Контроль изменений структуры хроматина (и, следовательно, транскрипции) опосредован ковалентными модификациями гистонов, особенно концевых групп на их N-конце. Эти модификации часто называют эпигенетическими, поскольку они могут привести к наследственным изменениям генной экспрессии, но не влияют на последовательность ДНК как таковую. Ковалентные модификации (например, метилирование, ацетилирование, фосфорилирование и убихитинирование) боковых цепей аминокислот являются ферментативно опосредованными.
Селективное присоединение метильных групп к специфическим аминокислотным сайтам на гистонах контролируется действием уникального семейства ферментов, известного как гистонметилтрансферазы (HMT). На уровень экспрессии конкретного гена влияет присутствие или отсутствие метильной группы на соответствующем сайте гистона. Специфический эффект метильной группы на определенном сайте гистона сохраняется вплоть до удаления метильной группы под действием гистондеметилазы, или до тех пор, пока модифицированный гистон не заменяется через обновление нуклеосомы. Подобным образом, другие классы ферментов могут декорировать ДНК и гистоны другими типами химических групп, а еще некоторые ферменты могут удалять эти группы с обеспечением временного контроля генной экспрессии.
Согласованно функционирующая совокупность биохимических систем вслед за транскрипционной регуляцией должна строго контролироваться для оптимального осуществления клеточного роста и дифференциации. Болезненные состояния возникают, когда эти контроли нарушаются путем аберрантной экспрессии и/или активности ферментов, ответственных за модификацию ДНК и гистона. Что касается рака человека, например, постоянно увеличивается совокупность доказательств, говорящих о том, что нарушенная регуляция активности эпигенетических ферментов способствует неконтролируемой клеточной пролиферации, связанной с раком, а также другими имеющими отношение к раку фенотипами, такими как повышенная клеточная миграция и инвазия. Помимо рака, появляется все больше доказательств роли эпигенетических ферментов в ряде других заболеваний человека, включая метаболические заболевания (такие как диабет), воспалительные заболевания (такие как болезнь Крона), нейродегенеративные заболевания (такие как болезнь Альцгеймера) и сердечно-сосудистые заболевания. Поэтому селективная модуляция аберрантного действия эпигенетических ферментов является многообещающей для лечения ряда заболеваний.
В настоящее время существует потребность в новых средствах, которые модулируют аберрантное действие эпигенетических ферментов. Настоящее изобретение обеспечивает соединения, которые отвечают этим требованиям.
Краткое описание изобретения
Настоящее изобретение обеспечивает соединения, полезные для модуляции аберрантного действия эпигенетических ферментов. Настоящее изобретение также обеспечивает фармацевтически приемлемые соли, сложные эфиры и/или N-оксиды, этих соединений.
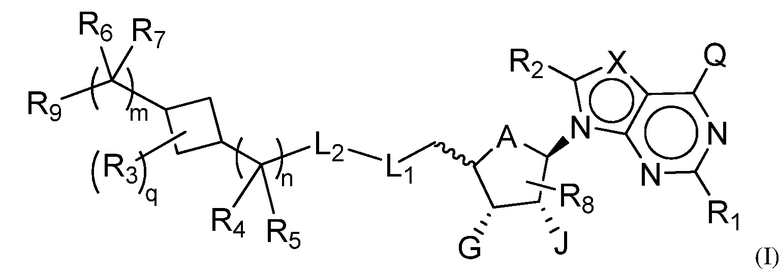
В одном аспекте, настоящее изобретение представляет замещенное пуриновое или 7-деазапуриновое соединение формулы (I) ниже или его фармацевтически приемлемую соль или сложный эфир.
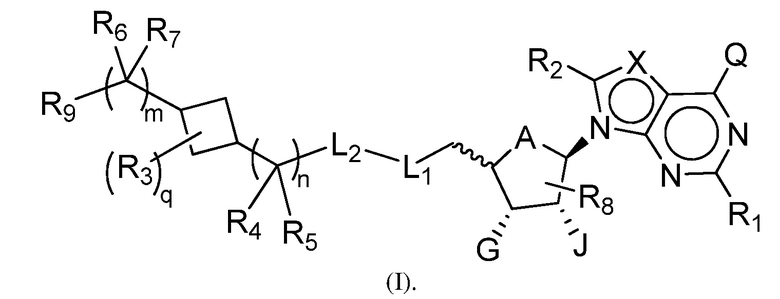
В этой формуле
A представляет собой O или CH2;
каждый из G и J независимо представляет собой H, галоген, C(O)OH, C(O)O-C1-C6 алкил или ORa, Ra представляет собой H, C1-C6 алкил или C(O)-C1-C6 алкил, где C(O)O-C1-C6 алкил, C1-C6 алкил или C(O)-C1-C6 алкил необязательно замещен одним или несколькими заместителями, выбранными из группы, включающей галоген, циано, гидроксил, карбоксил, C1-C6 алкоксил, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино и C3-C8 циклоалкил;
Q представляет собой H, NH2, NHRb, NRbRc, Rb или ORb, где каждый из Rb и Rc независимо представляет собой C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, C6-C10 арил, 4-7-членный гетероциклоалкил, 5-10-членный гетероарил или -M1-T1, где M1 представляет собой связь или C1-C6 алкильный линкер, необязательно замещенный галогеном, циано, гидроксилом или C1-C6 алкоксилом, и T1 представляет собой C3-C8 циклоалкил, C6-C10 арил, 4-6-членный гетероциклоалкил или 5-10-членный гетероарил, или Rb и Rc, вместе с N атомом, с которым они связаны, образуют 4-7-членный гетероциклоалкил, содержащий 0 или 1 дополнительный гетероатом помимо N атома, который необязательно замещен C1-C6 алкилом, C2-C6 алкенилом, C2-C6 алкинилом, галогеном, гидроксилом, карбоксилом, C(O)OH, C(O)O-C1-C6 алкилом, OC(O)-C1-C6 алкилом, циано, C1-C6 алкоксилом, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино, C3-C8 циклоалкилом, C6-C10 арилом, 4-6-членным гетероциклоалкилом или 5-6-членным гетероарилом, и каждый из Rb, Rc и T1 необязательно замещен одним или несколькими заместителями, выбранными из группы, включающей C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, галоген, гидроксил, карбоксил, циано, C1-C6 алкоксил, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино, C3-C8 циклоалкил, C6-C10 арил, 4-6-членный гетероциклоалкил и 5-6-членный гетероарил;
X представляет собой N или CRx, где Rx представляет собой H, галоген, гидроксил, карбоксил, циано или RS1, RS1 представляет собой амино, C1-C6 алкоксил, C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, C6-C10 арил, 4-6-членный гетероциклоалкил или 5-6-членный гетероарил, и RS1 необязательно замещен одним или несколькими заместителями, выбранными из группы, включающей галоген, гидроксил, карбоксил, циано, C1-C6 алкоксил, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино, C3-C8 циклоалкил, C6-C10 арил, 4-6-членный гетероциклоалкил и 5-6-членный гетероарил;
L1 представляет собой N(Y), S, SO или SO2;
L2 представляет собой CO или отсутствует, когда L1 представляет собой N(Y), или L2 отсутствует, когда L1 представляет собой S, SO или SO2, где Y представляет собой H, Rd, SO2Rd или CORd, когда L2 отсутствует, или Y представляет собой H или Rd, когда L2 представляет собой CO, где Rd представляет собой C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, C6-C10 арил, 4-6-членный гетероциклоалкил или 5-6-членный гетероарил, и Rd необязательно замещен одним или несколькими заместителями, выбранными из группы, включающей C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, галоген, гидроксил, карбоксил, циано, C1-C6 алкоксил, C1-C6 алкилсульфонил, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино, C3-C8 циклоалкил, C6-C10 арил, 4-6-членный гетероциклоалкил и 5-6-членный гетероарил, и при этом C3-C8 циклоалкил, C6-C10 арил, 4-6-членный гетероциклоалкил или 5-6-членный гетероарил, необязательно, дополнительно замещен C1-C6 алкилом, C2-C6 алкенилом, C2-C6 алкинилом, галогеном, гидроксилом, карбоксилом, C(O)OH, C(O)O-C1-C6 алкилом, OC(O)-C1-C6 алкилом, циано, C1-C6 алкоксилом, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино, C3-C8 циклоалкилом, C6-C10 арилом, 4-6-членным гетероциклоалкилом или 5-6-членным гетероарилом;
каждый из R1, R2, R3, R4, R5, R6 и R7 независимо представляет собой H, галоген, гидроксил, карбоксил, циано, RS2, где RS2 представляет собой амино, C1-C6 алкоксил, C1-C6 алкил, C2-C6 алкенил или C2-C6 алкинил, и каждый RS2 необязательно замещен одним или несколькими заместителями, выбранными из группы, включающей галоген, гидроксил, карбоксил, циано, C1-C6 алкоксил, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино, C3-C8 циклоалкил, C6-C10 арил, 4-6-членный гетероциклоалкил и 5-6-членный гетероарил;
R8 представляет собой H, галоген или RS3, где RS3 представляет собой C1-C6 алкил, C2-C6 алкенил или C2-C6 алкинил, и RS3 необязательно замещен одним или несколькими заместителями, выбранными из группы, включающей галоген, гидроксил, карбоксил, циано, амино, C1-C6 алкоксил, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино и C3-C8 циклоалкил;
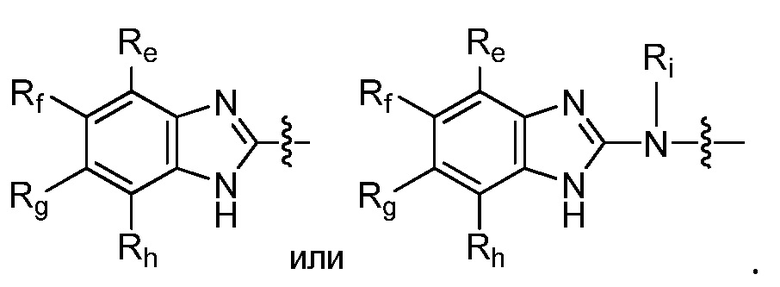
R9 представляет собой
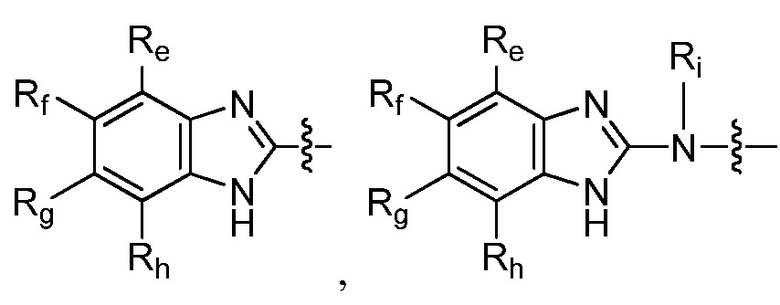 или
или  ,
,
где каждый из Re, Rf, Rg и Rh, независимо представляет собой группу -M2-T2, в которой M2 представляет собой связь, SO2, SO, S, CO, CO2, O, O-C1-C4 алкильный линкер, C1-C4 алкильный линкер, NH или N(Rt), где Rt представляет собой C1-C6 алкил, и T2 представляет собой H, галоген или RS4, где RS4 представляет собой C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, C6-C10 арил, 4-8-членный гетероциклоалкил или 5-10-членный гетероарил, и каждый из O-C1-C4 алкильного линкера, C1-C4 алкильного линкера, Rt и RS4 необязательно замещен одним или несколькими заместителями, выбранными из группы, включающей галоген, гидроксил, карбоксил, циано, C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C1-C6 алкоксил, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино, C3-C8 циклоалкил, C6-C10 арил, 4-6-членный гетероциклоалкил и 5-6-членный гетероарил, Ri представляет собой H или C1-C6 алкил, необязательно замещенный одним или несколькими заместителями, выбранными из группы, включающей галоген, гидроксил, карбоксил, циано, C1-C6 алкоксил, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино, C3-C8 циклоалкил, C6-C10 арил, 4-6-членный гетероциклоалкил и 5-6-членный гетероарил, D представляет собой O, NRj или CRjRk, каждый из Rj и Rk независимо представляет собой H или C1-C6 алкил, или Rj и Rk, взятые вместе с атомом углерода, с которым они связаны, образуют C3-C10 циклоалкильное кольцо, и E представляет собой -M3-T3, где M3 представляет собой связь или C1-C6 алкильный линкер, необязательно замещенный галогеном или циано, T3 представляет собой C3-C10 циклоалкил, C6-C10 арил, 5-10-членный гетероарил или 4-10-членный гетероциклоалкил, и T3 необязательно замещен одним или несколькими заместителями, выбранными из группы, включающей галоген, гидроксил, тиол, карбоксил, циано, нитро, C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C1-C6 алкоксил, C1-C6 галогеналкил, C1-C6 галогеналкоксил, C1-C6 алкилтио, C1-C6 алкилсульфонил, C1-C6 галогеналкилсульфонил, C1-C6 алкилкарбонил, C1-C6 алкоксикарбонил, оксо, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино, C3-C8 циклоалкил, C4-C12 алкилциклоалкил, C6-C10 арил, C6-C10 арилоксил, C7-C14 алкиларил, C6-C10 аминоарилоксил, C6-C10 арилтио, 4-6-членный гетероциклоалкил, необязательно замещенный галогеном, C1-C4 алкилом, C1-C4 галогеналкилом, 5-6-членный гетероарил, необязательно замещенный галогеном, C1-C4 алкилом, и C1-C6 алкил, который замещен гидрокси, галогеном, C1-C6 алкоксикарбонилом, C3-C8 циклоалкилом, C6-C10 арилом, 4-6-членным гетероциклоалкилом или 5-6-членным гетероарилом, который необязательно дополнительно замещен галогеном, гидроксилом или C1-C6 алкоксилом;
q имеет значение 0, 1, 2, 3 или 4;
m имеет значение 0, 1 или 2; и
n имеет значение 0, 1 или 2.
Одна подгруппа соединений формулы (I) включает соединения формулы (II):
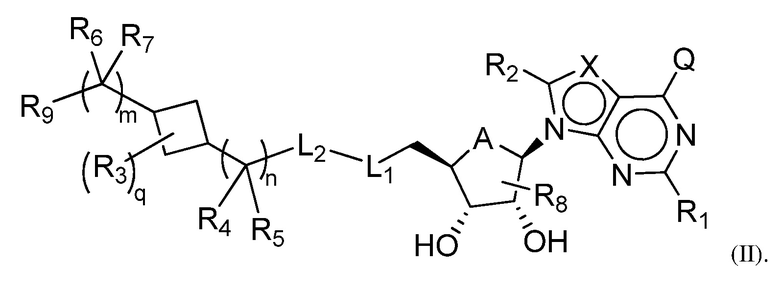
Другая подгруппа соединений формулы (I) включает соединения формулы (IIIa), (IIIb) или (IIIc):
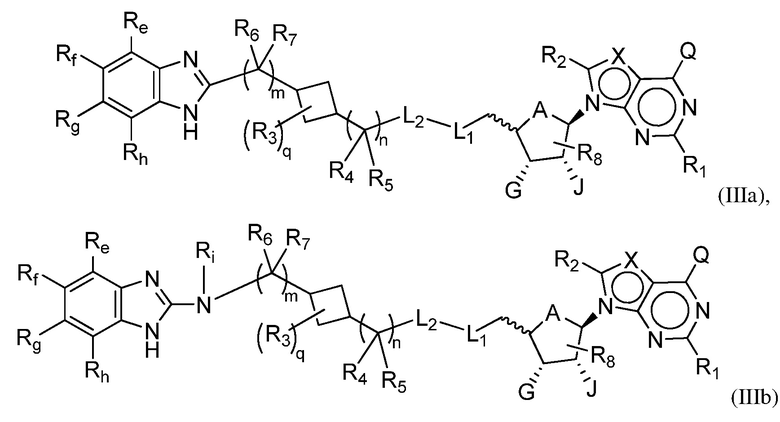 или
или
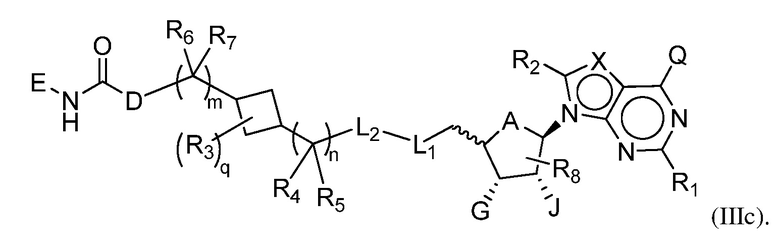
Соединения формул (I), (II), (IIIa), (IIIb), (IIIc) и (IV) могут включать один или несколько из следующих отличительных признаков.
Сумма m и n составляет по меньшей мере 1.
m имеет значение 1 или 2, и n имеет значение 0.
m имеет значение 2, и n имеет значение 0.
A представляет собой CH2.
A представляет собой O.
L1 представляет собой N(Y).
L1 представляет собой SO или SO2.
Y представляет собой Rd.
Rd представляет собой C1-C6 алкил.
L2 отсутствует.
По меньшей мере один из Re, Rf, Rg и Rh представляет собой галоген (такой как F, Cl и Br), C1-C6 алкоксил, необязательно замещенный одним или несколькими атомами галогена (такой как OCH3, OCH2CH3, O-iPr и OCF3), C1-C6 алкилсульфонил, необязательно замещенный одним или несколькими атомами галогена (такой как SO2CF3), или C1-C6 алкил, необязательно замещенный одним или несколькими атомами галогена (такой как CH3, изо-Pr, трет-Bu и CF3).
Ri представляет собой H или C1-C6 алкил.
R9 представляет собой 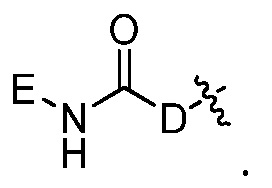
D представляет собой O.
D представляет собой NRj, например, NH.
D представляет собой CRjRk, например, CH2, CHCH3 или C(CH3)2.
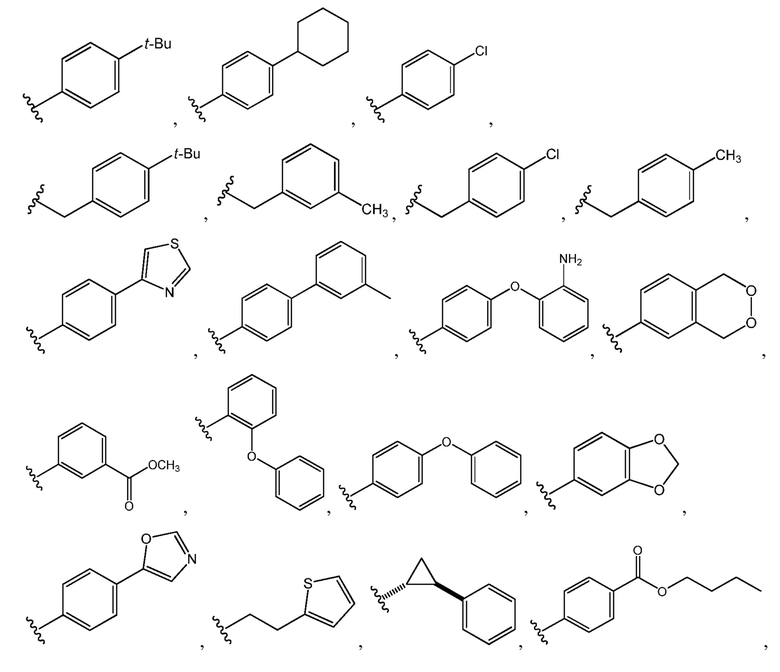
E представляет собой группу -M3-T3, в которой M3 представляет собой связь или C1-C3 алкильный линкер, T3 представляет собой фенил, нафтил, тиенил, циклопропил или циклогексил, и T3 необязательно замещен одним или несколькими заместителями, выбранными из группы, включающей галоген, гидроксил, тиол, карбоксил, циано, нитро, C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C1-C6 алкоксил, C1-C6 галогеналкил, C1-C6 галогеналкоксил, C1-C6 алкилтио, C1-C6 алкилсульфонил, C1-C6 алкилкарбонил, C1-C6 алкоксикарбонил, оксо, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино, C3-C8 циклоалкил, C4-C12 алкилциклоалкил, C6-C10 арил, C6-C10 арилоксил, C7-C14 алкиларил, C6-C10 аминоарилоксил, C6-C10 арилтио, 4-6-членный гетероциклоалкил, необязательно замещенный C1-C4 алкилом, 5-6-членный гетероарил, необязательно замещенный C1-C4 алкилом, и C1-C6 алкил, который замещен гидрокси, C1-C6 алкоксикарбонилом, C3-C8 циклоалкилом, C6-C10 арилом, 4-6-членным гетероциклоалкилом или 5-6-членным гетероарилом.
T3 представляет собой фенил, необязательно замещенный одним или несколькими заместителями, выбранными из группы, включающей галоген, гидроксил, карбоксил, циано, нитро, C1-C6 алкил, C1-C6 алкоксил, C1-C6 галогеналкил, C1-C6 галогеналкоксил, C1-C6 алкилсульфонил, C6-C10 арил и C6-C10 арилоксил и C7-C14 алкиларил.
X представляет собой N.
X представляет собой CRx, например, CH.
Q представляет собой NH2 или NHRb, где Rb представляет собой -M1-T1, M1 представляет собой связь или C1-C6 алкильный линкер, и T1 представляет собой C3-C8 циклоалкил.
Q представляет собой H.
R9 представляет собой
По меньшей мере один из Re, Rf, Rg и Rh выбран из группы, включающей F, Cl, CF3, OCF3, SO2CF3, C1-C4 алкил и C1-C4 алкоксил.
R1, R2, R3, R4, R5, R6, R7 и R8, каждый, представляют собой H.
Настоящее изобретение также относится к соединению формулы (IV) или его N-оксиду или фармацевтически приемлемой соли такого соединения:
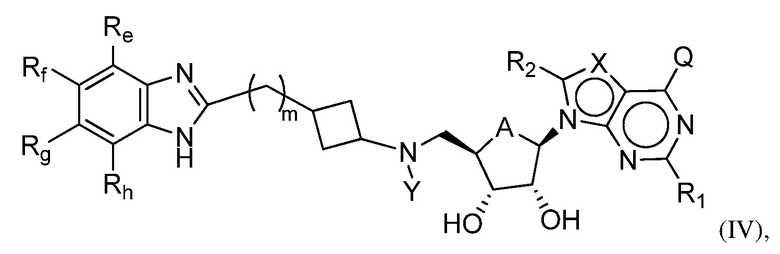
где, A представляет собой O или CH2;
Q представляет собой H, NH2, NHRb, NRbRc, OH, Rb или ORb, где каждый из Rb и Rc независимо представляет собой C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, C6-C10 арил, 4-7-членный гетероциклоалкил, 5-10-членный гетероарил или группу -M1-T1, в которой M1 представляет собой связь или C1-C6 алкильный линкер, необязательно замещенный галогеном, циано, гидроксилом или C1-C6 алкоксилом, и T1 представляет собой C3-C8 циклоалкил, C6-C10 арил, 4-6-членный гетероциклоалкил или 5-10-членный гетероарил, или Rb и Rc, вместе с N атомом, с которым они связаны, образуют 4-7-членный гетероциклоалкил, содержащий 0 или 1 дополнительный гетероатом помимо N атома, который необязательно замещен C1-C6 алкилом, C2-C6 алкенилом, C2-C6 алкинилом, галогеном, гидроксилом, карбоксилом, C(O)OH, C(O)O-C1-C6 алкилом, OC(O)-C1-C6 алкилом, циано, C1-C6 алкоксилом, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино, C3-C8 циклоалкилом, C6-C10 арилом, 4-6-членным гетероциклоалкилом или 5-6-членным гетероарилом, и каждый из Rb, Rc и T1 необязательно замещен одним или несколькими заместителями, выбранными из C1-C6 алкила, C2-C6 алкенила, C2-C6 алкинила, галогена, гидроксила, карбоксила, циано, C1-C6 алкоксила, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино, C3-C8 циклоалкила, C6-C10 арила, 4-6-членного гетероциклоалкила и 5-6-членного гетероарила;
X представляет собой N или группу CRx, в которой Rx представляет собой H, галоген, гидроксил, карбоксил, циано или RS1, где RS1 представляет собой амино, C1-C6 алкоксил, C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, C6-C10 арил, 4-6-членный гетероциклоалкил или 5-6-членный гетероарил, и RS1 необязательно замещен одним или несколькими заместителями, выбранными из галогена, гидроксила, карбоксила, циано, C1-C6 алкоксила, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино, C3-C8 циклоалкила, C6-C10 арила, 4-6-членного гетероциклоалкила и 5-6-членного гетероарила;
Y представляет собой H, Rd, SO2Rd или CORd, где Rd представляет собой C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, C6-C10 арил, 4-6-членный гетероциклоалкил или 5-6-членный гетероарил, и Rd необязательно замещен одним или несколькими заместителями, выбранными из C1-C6 алкила, C2-C6 алкенила, C2-C6 алкинила, галогена, гидроксила, карбоксила, циано, C1-C6 алкоксила, C1-C6 алкилсульфонила, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино, C3-C8 циклоалкила, C6-C10 арила, 4-6-членного гетероциклоалкила и 5-6-членного гетероарила, и при этом C3-C8 циклоалкил, C6-C10 арил, 4-6-членный гетероциклоалкил или 5-6-членный гетероарил, необязательно, дополнительно замещен C1-C6 алкилом, C2-C6 алкенилом, C2-C6 алкинилом, галогеном, гидроксилом, карбоксилом, C(O)OH, C(O)O-C1-C6 алкилом, OC(O)-C1-C6 алкилом, циано, C1-C6 алкоксилом, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино, C3-C8 циклоалкилом, C6-C10 арилом, 4-6-членным гетероциклоалкилом или 5-6-членным гетероарилом;
каждый из R1 и R2 независимо представляет собой H, галоген, гидроксил, карбоксил, циано, RS2, где RS2 представляет собой амино, C1-C6 алкоксил, C1-C6 алкил, C2-C6 алкенил или C2-C6 алкинил, и каждый RS2 необязательно замещен одним или несколькими заместителями, выбранными из галогена, гидроксила, карбоксила, циано, C1-C6 алкоксила, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино, C3-C8 циклоалкила, C6-C10 арила, 4-6-членного гетероциклоалкила и 5-6-членного гетероарила;
каждый из Re, Rf, Rg и Rh независимо представляет собой группу -M2-T2, в которой M2 представляет собой связь, SO2, SO, S, CO, CO2, O, O-C1-C4 алкильный линкер, C1-C4 алкильный линкер, NH или N(Rt), где Rt представляет собой C1-C6 алкил, и T2 представляет собой H, галоген или RS4, где RS4 представляет собой C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, C6-C10 арил, 4-8-членный гетероциклоалкил или 5-10-членный гетероарил, и каждый из O-C1-C4 алкильного линкера, C1-C4 алкильного линкера, Rt и RS4 необязательно замещен одним или несколькими заместителями, выбранными из галогена, гидроксила, карбоксила, циано, C1-C6 алкила, C2-C6 алкенила, C2-C6 алкинила, C1-C6 алкоксила, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино, C3-C8 циклоалкила, C6-C10 арила, 4-6-членного гетероциклоалкила и 5-6-членного гетероарила, и
m имеет значение 0, 1 или 2.
Например, A представляет собой O. В некоторых соединениях формулы (IV) A представляет собой O, и m имеет значение 2.
В некоторых соединениях формулы (IV) X представляет собой N.
Например, в некоторых соединениях Q представляет собой группу NH2 или NHRb, в которой Rb представляет собой -M1-T1, где M1 представляет собой связь или C1-C6 алкильный линкер, и T1 представляет собой C3-C8 циклоалкил.
Например, в некоторых соединениях формулы (IV) R1 и R2, каждый, представляют собой H.
В некоторых соединениях формулы (IV) Y представляет собой Rd. Например, Rd представляет собой C1-C6 алкил, необязательно замещенный C3-C8 циклоалкилом или галогеном. Например, Rd представляет собой C3-C8 циклоалкил, необязательно замещенный C1-C6 алкилом или галогеном.
Настоящее изобретение также относится к соединению Формулы (IV), где по меньшей мере один из Re, Rf, Rg и Rh представляет собой галоген, C1-C6 алкоксил, необязательно замещенный одним или несколькими атомами галогена; C1-C6 алкилсульфонил, необязательно замещенный одним или несколькими атомами галогена; C1-C6 алкил, необязательно замещенный одним или несколькими заместителями, выбранными из CN, галогена, C3-C8 циклоалкила, гидрокси и C1-C6 алкоксила; C3-C8 циклоалкил, необязательно замещенный одним или несколькими заместителями, такими как C1-C6 алкил или CN; или 4-8-членный гетероциклоалкил, необязательно замещенный одним или несколькими заместителями, выбранными из CN, галогена, гидрокси, C1-C6 алкила и C1-C6 алкоксил. Например, соединение формулы (IV) содержит по меньшей мере один из Re, Rf, Rg и Rh, выбранный из F; Cl; Br; CF3; OCF3; SO2CF3; оксетанила, необязательно замещенного одним или несколькими заместителями, выбранными из CN, галогена, гидрокси, C1-C6 алкила и C1-C6 алкоксила; C3-C8 циклоалкила, необязательно замещенного одним или несколькими заместителями, выбранными из C1-C4 алкила; и C1-C4 алкила, необязательно замещенного одним или несколькими заместителями, выбранными из галогена, C3-C8 циклоалкила, гидрокси и C1-C6 алкоксила.
Например, настоящее изобретение относится к соединениям формулы (IV), где по меньшей мере один из Rf и Rg представляет собой алкил, необязательно замещенный гидроксилом. Например, настоящее изобретение относится к соединениям, где по меньшей мере один из Rf и Rg представляет собой трет-бутил, замещенный гидроксилом.
Настоящее изобретение относится к соединению, выбранному из Соединений 1-140. Настоящее изобретение также относится к соли соединения, выбранного из Соединений 1-140. Настоящее изобретение также относится к N-оксиду соединения, выбранного из Соединений 1-140. Настоящее изобретение также относится к соли N-оксида соединения, выбранного из Соединений 1-140. Например, настоящее изобретение относится к соединению, выбранному из Соединений 1-7, 9-109 и 111-140.
Настоящее изобретение также относится к фармацевтической композиции, включающей терапевтически эффективное количество соединения формулы (IV) и фармацевтически приемлемый носитель.
Настоящее изобретение также относится к фармацевтической композиции, включающей терапевтически эффективное количество соли соединения формулы (IV) и фармацевтически приемлемый носитель.
Настоящее изобретение также относится к фармацевтической композиции, включающей терапевтически эффективное количество гидрата соединения формулы (IV) и фармацевтически приемлемый носитель.
Настоящее изобретение также относится к фармацевтической композиции, включающей терапевтически эффективное количество соединения, выбранного из Соединений 1-140, и фармацевтически приемлемый носитель. Настоящее изобретение также относится к фармацевтической композиции, включающей терапевтически эффективное количество соли соединения, выбранного из Соединений 1-140, и фармацевтически приемлемый носитель. Настоящее изобретение также относится к фармацевтической композиции, включающей терапевтически эффективное количество N-оксида соединения, выбранного из Соединений 1-140, и фармацевтически приемлемый носитель. Настоящее изобретение также относится к фармацевтической композиции, включающей терапевтически эффективное количество N-оксида соли соединения, выбранного из Соединений 1-140, и фармацевтически приемлемый носитель. Настоящее изобретение также относится к фармацевтической композиции, включающей терапевтически эффективное количество гидрата соединения, выбранного из Соединений 1-140, и фармацевтически приемлемый носитель.
Настоящее изобретение обеспечивает фармацевтические композиции, включающие одно или несколько соединений формулы (I), (II), (IIIa), (IIIb), (IIIc) или (IV) и один или несколько фармацевтически приемлемых носителей.
Настоящее изобретение обеспечивает способы лечения или профилактики рака. Настоящее изобретение обеспечивает способы лечения рака. Настоящее изобретение также обеспечивает способы профилактики рака. Способ включает введение субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы (I), (II), (IIIa), (IIIb) или (IIIc). Рак может представлять собой гематологический рак. Предпочтительно, рак представляет собой лейкоз. Более предпочтительно, рак представляет собой острый миелогенный лейкоз, острый лимфоцитарный лейкоз или лейкоз смешанного происхождения.
Настоящее изобретение обеспечивает способы лечения или профилактики заболевания или расстройства, опосредованного транслокацией гена на хромосоме 11q23. Настоящее изобретение обеспечивает способы лечения заболевания или расстройства, опосредованного транслокацией гена на хромосоме 11q23. Настоящее изобретение также обеспечивает способы профилактики заболевания или расстройства, опосредованного транслокацией гена на хромосоме 11q23. Способ включает введение субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы (I), (II), (IIIa), (IIIb), (IIIc) или (IV).
Настоящее изобретение обеспечивает способы лечения или профилактики заболевания или расстройства, в котором играет роль DOT1-опосредованное метилирование белка, или заболевания или расстройства, опосредованного DOT1-опосредованным метилированием белка. Настоящее изобретение обеспечивает способы лечения заболевания или расстройства, в котором играет роль DOT1-опосредованное метилирование белка, или заболевания или расстройства, опосредованного DOT1-опосредованным метилированием белка. Настоящее изобретение также обеспечивает способы профилактики заболевания или расстройства, в котором играет роль DOT1-опосредованное метилирование белка, или заболевания или расстройства, опосредованного DOT1-опосредованным метилированием белка. Способ включает введение субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы (I), (II), (IIIa), (IIIb), (IIIc) или (IV).
Настоящее изобретение обеспечивает способы ингибирования активности DOT1L в клетке. Способ включает контактирование клетки с эффективным количеством одного или нескольких соединений формулы (I), (II), (IIIa), (IIIb), (IIIc) или (IV).
Еще один аспект настоящего изобретения относится к способу снижения уровня метилирования Лизинового остатка 79 Гистона H3 (H3-K79) в клетке. Способ включает контактирование клетки с соединением по настоящему изобретению. Такой способ можно использовать для уменьшения интенсивности симптомов любого состояния, которое вызвано или потенциировано активностью DOT1 через метилирование H3-K79.
Настоящее изобретение относится к применению соединений, раскрытых в настоящем изобретении, для получения лекарственного средства для лечения или профилактики рака. Применение включает соединение формулы (I), (II), (IIIa), (IIIb), (IIIc) или (IV) для введения субъекту, нуждающемуся в этом, в терапевтически эффективном количестве. Рак может представлять собой гематологический рак. Предпочтительно, рак представляет собой лейкоз. Более предпочтительно, рак представляет собой острый миелогенный лейкоз, острый лимфоцитарный лейкоз или лейкоз смешанного происхождения.
Настоящее изобретение обеспечивает применение соединений, раскрытых в настоящем изобретении, для получения лекарственного средства для лечения или профилактики заболевания или расстройства, опосредованного транслокацией гена на хромосоме 11q23. Применение включает соединение формулы (I), (II), (IIIa), (IIIb), (IIIc) или (IV) для введения субъекту, нуждающемуся в этом, в терапевтически эффективном количестве.
Настоящее изобретение обеспечивает применение соединений, раскрытых в настоящем изобретении, для получения лекарственного средства для лечения или профилактики заболевания или расстройства, в котором играет роль DOT1-опосредованное метилирование белка, или заболевания или расстройства, опосредованного DOT1-опосредованным метилированием белка. Применение включает соединение формулы (I), (II), (IIIa), (IIIb), (IIIc) или (IV) для введения субъекту, нуждающемуся в этом, в терапевтически эффективном количестве.
Настоящее изобретение обеспечивает применение соединений, раскрытых в настоящем изобретении, для ингибирования активности DOT1L в клетке. Применение включает контактирование клетки с эффективным количеством одного или нескольких соединений формулы (I), (II), (IIIa), (IIIb), (IIIc) или (IV).
Еще один аспект настоящего изобретения относится к применению соединений, раскрытых в настоящем изобретении, для снижения уровня метилирования Лизинового остатка 79 Гистона H3 (H3-K79) в клетке. Применение включает контактирование клетки с соединением по настоящему изобретению. Такое применение может уменьшить интенсивности симптомов любого состояния, которое вызвано или потенциировано активностью DOT1 через метилирование H3-K79.
В формулах, представленных в настоящей заявке, переменные можно выбрать из соответствующих групп химических составляющих, которые определены ниже в подробном описании изобретения.
Кроме того, настоящее изобретение обеспечивает способы синтеза указанных выше соединений. После синтеза терапевтически эффективное количество одного или нескольких соединений можно сформулировать с фармацевтически приемлемым носителем для введения млекопитающему, в частности, человеку для использования в модуляции эпигенетического фермента. В некоторых вариантах воплощения, соединения по настоящему изобретению являются полезными для лечения, профилактики или снижения риска рака или для получения лекарственного средства для лечения, профилактики или снижения риска рака. Соответственно, соединения или композиции можно вводить, например, пероральным, парентеральным, ушным, глазным, назальным или местным путем для обеспечения эффективного количества соединения млекопитающему.
Если не определено иное, все технические и научные термины, используемые в настоящей заявке, имеют значение, традиционно известное рядовым специалистам в области, к которой относится данное изобретение. В описании изобретения, формы единственного числа также включают множественное число, если из контекста явно не следует иное. Хотя способы и вещества, подобные или эквивалентные тем, которые описаны в настоящей заявке, можно использовать для практического осуществления или испытания настоящего изобретения, подходящие способы и вещества описаны ниже. Все публикации, патентные заявки, патенты и другие ссылочные документы, указанные в настоящей заявке, включены посредством ссылки. Ссылочные документы, на которые ссылаются в настоящей заявке, не являются признанными в качестве прототипов заявленного изобретения. В случае конфликта, преимущество имеет настоящее описание, включая определения. Кроме того, вещества, способы и примеры являются только иллюстративными и не предназначены для ограничения.
Другие характерные признаки и преимущества настоящего изобретения будут очевидны из следующего далее подробного описания и формулы изобретения.
Описание рисунков
Фиг. 1A и 1B соответственно представляют таблицу и график, демонстрирующие уровень активности и селективность анти-пролиферативной активности Соединения 2 с использованием панели MLL-реаранжированных и не-MLL-реаранжированных клеточных линий лейкоза человека. Клеточные линии, используемые в исследовании, указаны на Фиг. 1A
Фиг. 2 представляет график, показывающий рост опухоли в течение 21 дня введения средства.
Фиг. 3A представляет график, показывающий установившееся, согласно оценке, состояние концентраций в плазме Соединения 2 в Группах 4 и 5, как было определено на основании усредненных образцов крови, взятых в дни 7, 14 и 21.
Фиг. 3B представляет график, показывающий концентрации в плазме соединения 2 против времени после интраперитонеальной инъекции.
Подробное описание изобретения
Настоящее изобретение обеспечивает семейство соединений, которые можно использовать для селективной модуляции аберрантного действия эпигенетического фермента. Кроме того, соединения можно использовать для лечения или профилактики болезненного состояния у млекопитающего, вызванного или опосредованного аберрантным действием эпигенетического фермента. Настоящее изобретение включает фармацевтически приемлемые соли, сложные эфиры, таутомеры и N-оксиды этих соединений.
Настоящее изобретение обеспечивает новые замещенные пуриновые и 7-деазапуриновые соединения, способы синтеза для получения соединений, фармацевтические композиции, содержащие такие соединения, и различные применения соединений.
1. Замещенные Пуриновые Соединения и Замещенные 7-Деазапуриновые Соединения
Настоящее изобретение обеспечивает соединения формулы (I):
или их фармацевтически приемлемые соли или сложные эфиры, где:
A представляет собой O или CH2;
каждый из G и J независимо представляет собой H, галоген, C(O)OH, C(O)O-C1-C6 алкил или ORa, где Ra представляет собой H, C1-C6 алкил или C(O)-C1-C6 алкил, при этом C(O)O-C1-C6 алкил, C1-C6 алкил или C(O)-C1-C6 алкил необязательно замещен одним или несколькими заместителями, выбранными из группы, включающей галоген, циано, гидроксил, карбоксил, C1-C6 алкоксил, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино и C3-C8 циклоалкил;
Q представляет собой H, NH2, NHRb, NRbRc, Rb или ORb, где каждый из Rb и Rc независимо представляет собой C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, C6-C10 арил, 4-7-членный гетероциклоалкил, 5-10-членный гетероарил или группу -M1-T1, в которой M1 представляет собой связь или C1-C6 алкильный линкер, необязательно замещенный галогеном, циано, гидроксилом или C1-C6 алкоксилом, и T1 представляет собой C3-C8 циклоалкил, C6-C10 арил, 4-6-членный гетероциклоалкил или 5-10-членный гетероарил, или Rb и Rc вместе с N атомом, с которым они связаны, образуют 4-7-членный гетероциклоалкил, содержащий 0 или 1 дополнительный гетероатом помимо N атома, который необязательно замещен C1-C6 алкилом, C2-C6 алкенилом, C2-C6 алкинилом, галогеном, гидроксилом, карбоксилом, C(O)OH, C(O)O-C1-C6 алкилом, OC(O)-C1-C6 алкилом, циано, C1-C6 алкоксилом, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино, C3-C8 циклоалкилом, C6-C10 арилом, 4-6-членным гетероциклоалкилом или 5-6-членным гетероарилом, и каждый из Rb, Rc и T1 необязательно замещен одним или несколькими заместителями, выбранными из группы, включающей C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, галоген, гидроксил, карбоксил, циано, C1-C6 алкоксил, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино, C3-C8 циклоалкил, C6-C10 арил, 4-6-членный гетероциклоалкил и 5-6-членный гетероарил;
X представляет собой N или группу CRx, в которой Rx представляет собой H, галоген, гидроксил, карбоксил, циано или RS1, где RS1 представляет собой амино, C1-C6 алкоксил, C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, C6-C10 арил, 4-6-членный гетероциклоалкил или 5-6-членный гетероарил, и RS1 необязательно замещен одним или несколькими заместителями, выбранными из группы, включающей галоген, гидроксил, карбоксил, циано, C1-C6 алкоксил, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино, C3-C8 циклоалкил, C6-C10 арил, 4-6-членный гетероциклоалкил и 5-6-членный гетероарил;
L1 представляет собой N(Y), S, SO или SO2;
L2 представляет собой CO или отсутствует, когда L1 представляет собой N(Y), или L2 отсутствует, когда L1 представляет собой S, SO или SO2, где Y представляет собой H, Rd, SO2Rd или CORd, когда L2 отсутствует, или Y представляет собой H или Rd, когда L2 представляет собой CO, где Rd представляет собой C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, C6-C10 арил, 4-6-членный гетероциклоалкил или 5-6-членный гетероарил, и Rd необязательно замещен одним или несколькими заместителями, выбранными из группы, включающей C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, галоген, гидроксил, карбоксил, циано, C1-C6 алкоксил, C1-C6 алкилсульфонил, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино, C3-C8 циклоалкил, C6-C10 арил, 4-6-членный гетероциклоалкил и 5-6-членный гетероарил, и при этом C3-C8 циклоалкил, C6-C10 арил, 4-6-членный гетероциклоалкил или 5-6-членный гетероарил, необязательно, дополнительно замещен C1-C6 алкилом, C2-C6 алкенилом, C2-C6 алкинилом, галогеном, гидроксилом, карбоксилом, C(O)OH, C(O)O-C1-C6 алкилом, OC(O)-C1-C6 алкилом, циано, C1-C6 алкоксилом, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино, C3-C8 циклоалкилом, C6-C10 арилом, 4-6-членным гетероциклоалкилом или 5-6-членным гетероарилом;
каждый из R1, R2, R3, R4, R5, R6 и R7 независимо представляет собой H, галоген, гидроксил, карбоксил, циано, RS2, где RS2 представляет собой амино, C1-C6 алкоксил, C1-C6 алкил, C2-C6 алкенил или C2-C6 алкинил, и каждый RS2 необязательно замещен одним или несколькими заместителями, выбранными из группы, включающей галоген, гидроксил, карбоксил, циано, C1-C6 алкоксил, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино, C3-C8 циклоалкил, C6-C10 арил, 4-6-членный гетероциклоалкил и 5-6-членный гетероарил;
R8 представляет собой H, галоген или RS3, где RS3 представляет собой C1-C6 алкил, C2-C6 алкенил или C2-C6 алкинил, и RS3 необязательно замещен одним или несколькими заместителями, выбранными из группы, включающей галоген, гидроксил, карбоксил, циано, амино, C1-C6 алкоксил, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино и C3-C8 циклоалкил;
R9 представляет собой
 или
или  ,
,
где каждый из Re, Rf, Rg и Rh независимо представляет собой группу -M2-T2, в которой M2 представляет собой связь, SO2, SO, S, CO, CO2, O, O-C1-C4 алкильный линкер, C1-C4 алкильный линкер, NH или N(Rt), где Rt представляет собой C1-C6 алкил, и T2 представляет собой H, галоген или RS4, где RS4 представляет собой C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, C6-C10 арил, 4-8-членный гетероциклоалкил или 5-10-членный гетероарил, и каждый из O-C1-C4 алкильного линкера, C1-C4 алкильного линкера, Rt и RS4 необязательно замещен одним или несколькими заместителями, выбранными из группы, включающей галоген, гидроксил, карбоксил, циано, C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C1-C6 алкоксил, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино, C3-C8 циклоалкил, C6-C10 арил, 4-6-членный гетероциклоалкил и 5-6-членный гетероарил, Ri представляет собой H или C1-C6 алкил, необязательно замещенный одним или несколькими заместителями, выбранными из группы, включающей галоген, гидроксил, карбоксил, циано, C1-C6 алкоксил, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино, C3-C8 циклоалкил, C6-C10 арил, 4-6-членный гетероциклоалкил и 5-6-членный гетероарил, D представляет собой O, NRj или CRjRk, где каждый из Rj и Rk независимо представляет собой H или C1-C6 алкил, или Rj и Rk, взятые вместе с атомом углерода, с которым они связаны, образуют C3-C10 циклоалкильное кольцо, и E представляет собой -M3-T3, где M3 представляет собой связь или C1-C6 алкильный линкер, необязательно замещенный галогеном или циано, T3 представляет собой C3-C10 циклоалкил, C6-C10 арил, 5-10-членный гетероарил или 4-10-членный гетероциклоалкил, и T3 необязательно замещен одним или несколькими заместителями, выбранными из группы, включающей галоген, гидроксил, тиол, карбоксил, циано, нитро, C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C1-C6 алкоксил, C1-C6 галогеналкил, C1-C6 галогеналкоксил, C1-C6 алкилтио, C1-C6 алкилсульфонил, C1-C6 галогеналкилсульфонил, C1-C6 алкилкарбонил, C1-C6 алкоксикарбонил, оксо, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино, C3-C8 циклоалкил, C4-C12 алкилциклоалкил, C6-C10 арил, C6-C10 арилоксил, C7-C14 алкиларил, C6-C10 аминоарилоксил, C6-C10 арилтио, 4-6-членный гетероциклоалкил, необязательно замещенный галогеном, C1-C4 алкилом, C1-C4 галогеналкилом, 5-6-членный гетероарил, необязательно замещенный галогеном, C1-C4 алкилом, и C1-C6 алкил, который замещен гидрокси, галогеном, C1-C6 алкоксикарбонилом, C3-C8 циклоалкилом, C6-C10 арилом, 4-6-членным гетероциклоалкилом или 5-6-членным гетероарилом, который необязательно дополнительно замещен галогеном, гидроксилом или C1-C6 алкоксилом;
q имеет значение 0, 1, 2, 3 или 4;
m имеет значение 0, 1 или 2; и
n имеет значение 0, 1 или 2.
Например, сумма m и n составляет по меньшей мере 1.
Например, m имеет значение 1 или 2, и n имеет значение 0.
Например, m имеет значение 2, и n имеет значение 0.
Например, A представляет собой CH2.
Например, A представляет собой O.
Например, L1 представляет собой N(Y).
Например, L1 представляет собой SO или SO2.
Например, Y представляет собой Rd.
Например, Rd представляет собой C1-C6 алкил.
Например, L2 отсутствует.
Например, каждый из G и J независимо представляет собой ORa.
Например, Ra представляет собой H.
Например, R9 представляет собой  . Например, R9 представляет собой
. Например, R9 представляет собой  .
.
Например, по меньшей мере один из Re, Rf, Rg и Rh представляет собой галоген (такой как F, Cl и Br), C1-C6 алкоксил, необязательно замещенный одним или несколькими атомами галогена (такой как OCH3, OCH2CH3, O-изо-Pr и OCF3), C1-C6 алкилсульфонил, необязательно замещенный одним или несколькими атомами галогена (такой как SO2CF3), или C1-C6 алкил, необязательно замещенный одним или несколькими атомами галогена (такой как CH3, изопропил, н-бутил и CF3).
Например, Ri представляет собой H или C1-C6 алкил (например, метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, трет-бутил, н-пентил, втор-пентил и н-гексил).
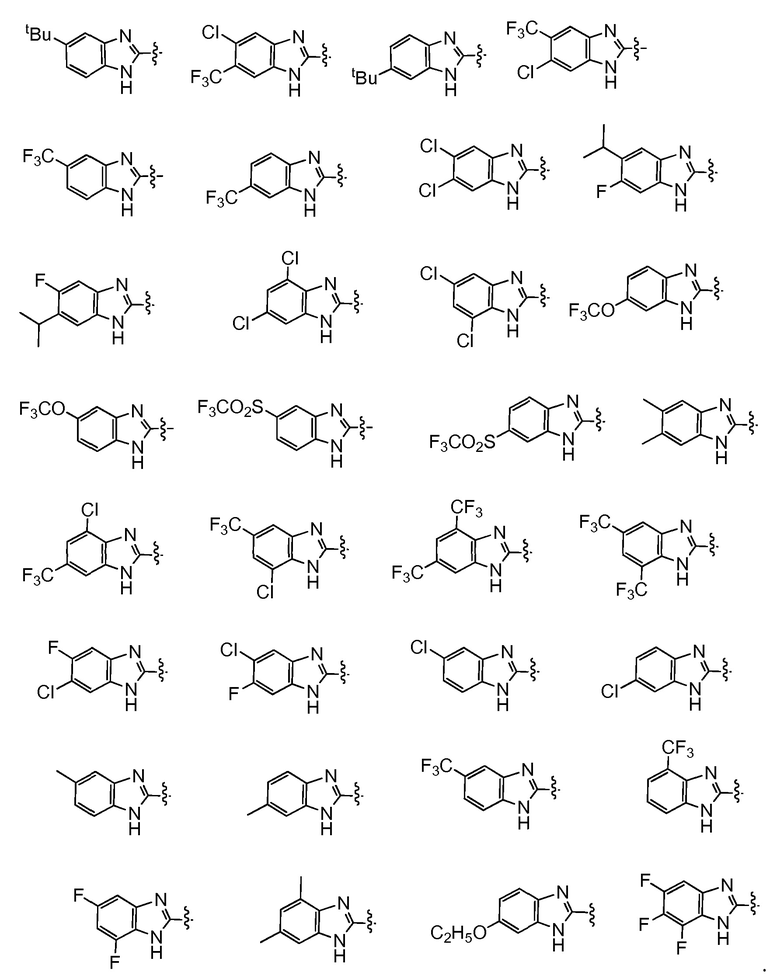
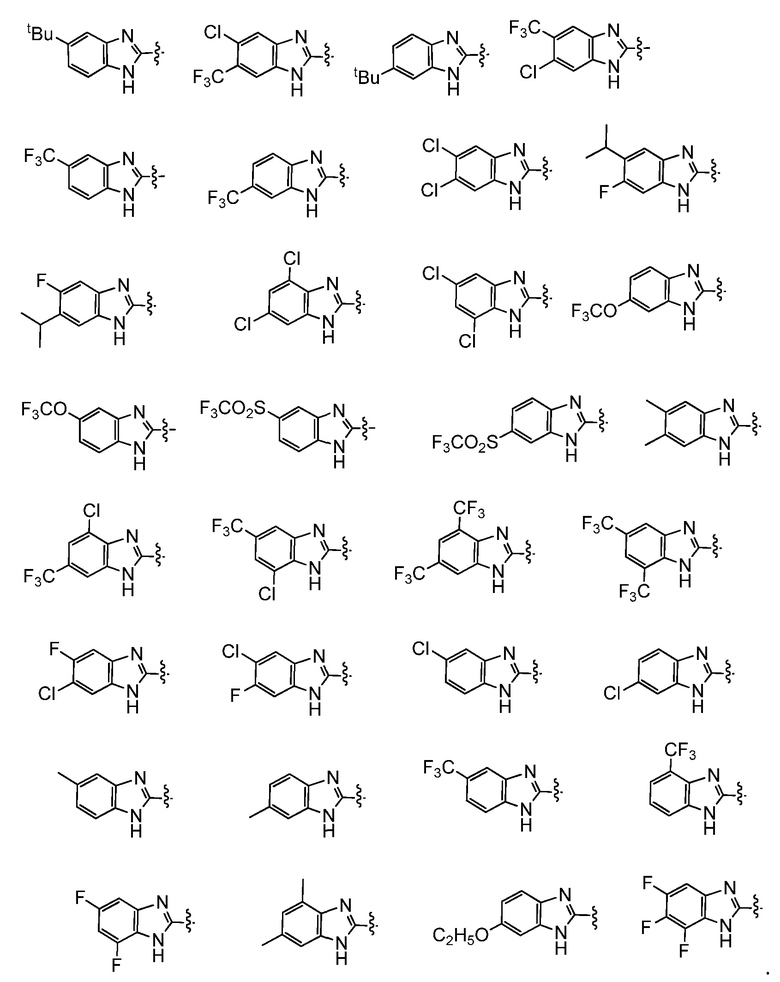
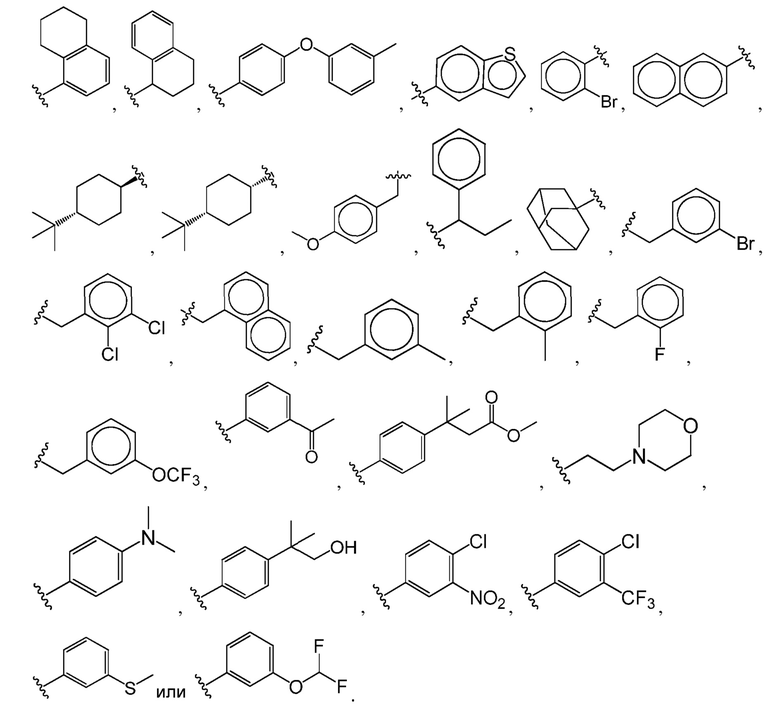
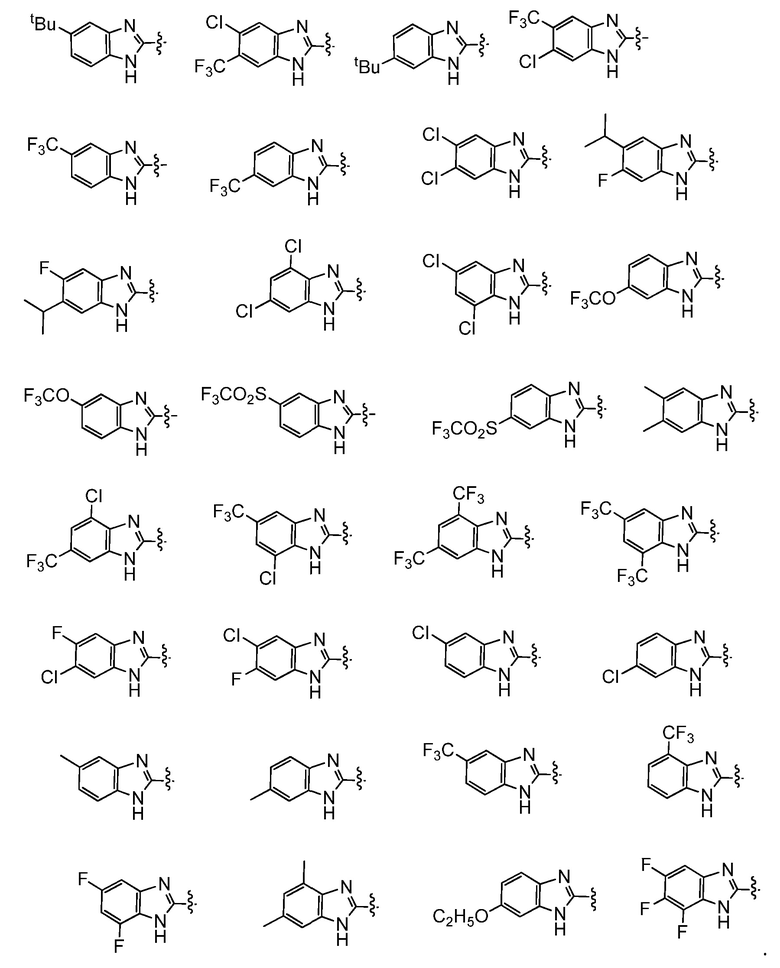
Например,  представляет собой незамещенный бензимидазолил или одну из следующих групп:
представляет собой незамещенный бензимидазолил или одну из следующих групп:
Например, R9 представляет собой 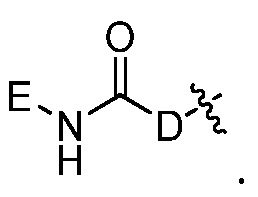
Например, D представляет собой O.
Например, D представляет собой NRj.
Например, Rj представляет собой H.
Например, D представляет собой CRjRk.
Например, каждый из Rj и Rk представляет собой H.
Например, E представляет собой группу -M3-T3, в которой M3 представляет собой связь или C1-C3 алкильный линкер, T3 представляет собой фенил, нафтил, тиенил, циклопропил или циклогексил, и T3 необязательно замещен одним или несколькими заместителями, выбранными из группы, включающей галоген, гидроксил, тиол, карбоксил, циано, нитро, C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C1-C6 алкоксил, C1-C6 галогеналкил, C1-C6 галогеналкоксил, C1-C6 алкилтио, C1-C6 алкилсульфонил, C1-C6 алкилкарбонил, C1-C6 алкоксикарбонил, оксо, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино, C3-C8 циклоалкил, C4-C12 алкилциклоалкил, C6-C10 арил, C6-C10 арилоксил, C7-C14 алкиларил, C6-C10 аминоарилоксил, C6-C10 арилтио, 4-6-членный гетероциклоалкил, необязательно замещенный C1-C4 алкилом, 5-6-членный гетероарил, необязательно замещенный C1-C4 алкилом, и C1-C6 алкил, который замещен гидрокси, C1-C6 алкоксикарбонилом, C3-C8 циклоалкилом, C6-C10 арилом, 4-6-членным гетероциклоалкилом или 5-6-членным гетероарилом.
Например, T3 представляет собой фенил, необязательно замещенный одним или несколькими заместителями, выбранными из группы, включающей галоген, гидроксил, карбоксил, циано, нитро, C1-C6 алкил (например, метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, трет-бутил, н-пентил, втор-пентил и н-гексил), C1-C6 алкоксил, C1-C6 галогеналкил, C1-C6 галогеналкоксил, C1-C6 алкилсульфонил, C6-C10 арил (например, фенил или нафтил) и C6-C10 арилоксил и C7-C14 алкиларил.
Например, E представляет собой
Например, X представляет собой N.
Например, X представляет собой CRx.
Например, X представляет собой CH.
Например, Q представляет собой NH2 или группу NHRb, в которой Rb представляет собой -M1-T1, где M1 представляет собой связь или C1-C6 алкильный линкер, и T1 представляет собой C3-C8 циклоалкил.
Например, Q представляет собой H.
Например, R1, R2, R3, R4, R5, R6, R7 и R8, каждый, представляют собой H.
Например, когда R8 представляет собой галоген и связан с тем же самым атомом углерода, с которым связан J, тогда J не является гидроксилом.
Например, когда R8 представляет собой галоген и связан с тем же самым атомом углерода, с которым связан G, тогда G не является гидроксилом.
Например, T2 не является галогеном, когда M2 представляет собой SO2, SO, S, CO или O.
Например, T2 представляет собой 4-8-членный гетероциклоалкил, который связан с M2 через гетероатом.
Например, T2 представляет собой 4-8-членный гетероциклоалкил, который связан с M2 через N атом.
Например, T2 представляет собой 4-8-членный гетероциклоалкил, который связан с M2 через C атом.
Настоящее изобретение обеспечивает соединения формулы (II):
или их фармацевтически приемлемые соли или сложные эфиры, где:
A представляет собой O или CH2;
Q представляет собой H, NH2, NHRb, NRbRc, Rb или ORb, где каждый из Rb и Rc независимо представляет собой C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, C6-C10 арил, 4-7-членный гетероциклоалкил, 5-10-членный гетероарил или группу-M1-T1, в которой M1 представляет собой связь или C1-C6 алкильный линкер, необязательно замещенный галогеном, циано, гидроксилом или C1-C6 алкоксилом, и T1 представляет собой C3-C8 циклоалкил, C6-C10 арил, 4-6-членный гетероциклоалкил или 5-10-членный гетероарил, или Rb и Rc, вместе с N атомом, с которым они связаны, образуют 4-7-членный гетероциклоалкил, содержащий 0 или 1 дополнительный гетероатом помимо N атома, который необязательно замещен C1-C6 алкилом, C2-C6 алкенилом, C2-C6 алкинилом, галогеном, гидроксилом, карбоксилом, C(O)OH, C(O)O-C1-C6 алкилом, OC(O)-C1-C6 алкилом, циано, C1-C6 алкоксилом, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино, C3-C8 циклоалкилом, C6-C10 арилом, 4-6-членным гетероциклоалкилом или 5-6-членным гетероарилом, и каждый из Rb, Rc и T1 необязательно замещен одним или несколькими заместителями, выбранными из группы, включающей C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, галоген, гидроксил, карбоксил, циано, C1-C6 алкоксил, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино, C3-C8 циклоалкил, C6-C10 арил, 4-6-членный гетероциклоалкил и 5-6-членный гетероарил;
X представляет собой N или группу CRx, в которой Rx представляет собой H, галоген, гидроксил, карбоксил, циано или RS1, где RS1 представляет собой амино, C1-C6 алкоксил, C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, C6-C10 арил, 4-6-членный гетероциклоалкил или 5-6-членный гетероарил, и RS1 необязательно замещен одним или несколькими заместителями, выбранными из группы, включающей галоген, гидроксил, карбоксил, циано, C1-C6 алкоксил, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино, C3-C8 циклоалкил, C6-C10 арил, 4-6-членный гетероциклоалкил и 5-6-членный гетероарил;
L1 представляет собой N(Y), S, SO или SO2;
L2 представляет собой CO или отсутствует, когда L1 представляет собой N(Y), или L2 отсутствует, когда L1 представляет собой S, SO или SO2, где Y представляет собой H, Rd, SO2Rd или CORd, когда L2 отсутствует, или Y представляет собой H или Rd, когда L2 представляет собой CO, где Rd представляет собой C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, C6-C10 арил, 4-6-членный гетероциклоалкил или 5-6-членный гетероарил, и Rd необязательно замещен одним или несколькими заместителями, выбранными из группы, включающей C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, галоген, гидроксил, карбоксил, циано, C1-C6 алкоксил, C1-C6 алкилсульфонил, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино, C3-C8 циклоалкил, C6-C10 арил, 4-6-членный гетероциклоалкил и 5-6-членный гетероарил, и при этом C3-C8 циклоалкил, C6-C10 арил, 4-6-членный гетероциклоалкил или 5-6-членный гетероарил, необязательно, дополнительно замещен C1-C6 алкилом, C2-C6 алкенилом, C2-C6 алкинилом, галогеном, гидроксилом, карбоксилом, C(O)OH, C(O)O-C1-C6 алкилом, OC(O)-C1-C6 алкилом, циано, C1-C6 алкоксилом, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино, C3-C8 циклоалкилом, C6-C10 арилом, 4-6-членным гетероциклоалкилом или 5-6-членным гетероарилом;
каждый из R1, R2, R3, R4, R5, R6 и R7 независимо представляет собой H, галоген, гидроксил, карбоксил, циано, RS2, где RS2 представляет собой амино, C1-C6 алкоксил, C1-C6 алкил, C2-C6 алкенил или C2-C6 алкинил, и каждый RS2 необязательно замещен одним или несколькими заместителями, выбранными из группы, включающей галоген, гидроксил, карбоксил, циано, C1-C6 алкоксил, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино, C3-C8 циклоалкил, C6-C10 арил, 4-6-членный гетероциклоалкил и 5-6-членный гетероарил;
R8 представляет собой H, галоген или RS3, где RS3 представляет собой C1-C6 алкил, C2-C6 алкенил или C2-C6 алкинил, и RS3 необязательно замещен одним или несколькими заместителями, выбранными из группы, включающей галоген, гидроксил, карбоксил, циано, амино, C1-C6 алкоксил, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино и C3-C8 циклоалкил;
R9 представляет собой
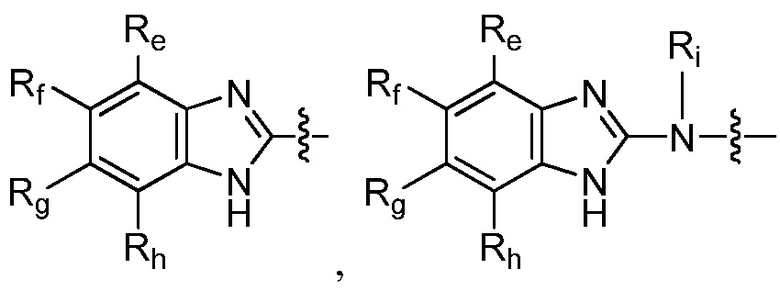 или
или 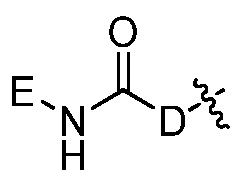 ,
,
где каждый из Re, Rf, Rg и Rh независимо представляет собой группу -M2-T2, в которой M2 представляет собой связь, SO2, SO, S, CO, CO2, O, O-C1-C4 алкильный линкер, C1-C4 алкильный линкер, NH или N(Rt), где Rt представляет собой C1-C6 алкил, и T2 представляет собой H, галоген или RS4, где RS4 представляет собой C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, C6-C10 арил, 4-8-членный гетероциклоалкил или 5-10-членный гетероарил, и каждый из O-C1-C4 алкильного линкера, C1-C4 алкильного линкера, Rt и RS4 необязательно замещен одним или несколькими заместителями, выбранными из группы, включающей галоген, гидроксил, карбоксил, циано, C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C1-C6 алкоксил, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино, C3-C8 циклоалкил, C6-C10 арил, 4-6-членный гетероциклоалкил и 5-6-членный гетероарил, Ri представляет собой H или C1-C6 алкил, необязательно замещенный одним или несколькими заместителями, выбранными из группы, включающей галоген, гидроксил, карбоксил, циано, C1-C6 алкоксил, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино, C3-C8 циклоалкил, C6-C10 арил, 4-6-членный гетероциклоалкил и 5-6-членный гетероарил, D представляет собой O, NRj или CRjRk, каждый из Rj и Rk независимо представляет собой H или C1-C6 алкил, или Rj и Rk, взятые вместе с атомом углерода, с которым они связаны, образуют C3-C10 циклоалкильное кольцо, и E представляет собой -M3-T3, где M3 представляет собой связь или C1-C6 алкильный линкер, необязательно замещенный галогеном или циано, T3 представляет собой C3-C10 циклоалкил, C6-C10 арил, 5-10-членный гетероарил или 4-10-членный гетероциклоалкил, и T3 необязательно замещен одним или несколькими заместителями, выбранными из группы, включающей галоген, гидроксил, тиол, карбоксил, циано, нитро, C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C1-C6 алкоксил, C1-C6 галогеналкил, C1-C6 галогеналкоксил, C1-C6 алкилтио, C1-C6 алкилсульфонил, C1-C6 галогеналкилсульфонил, C1-C6 алкилкарбонил, C1-C6 алкоксикарбонил, оксо, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино, C3-C8 циклоалкил, C4-C12 алкилциклоалкил, C6-C10 арил, C6-C10 арилоксил, C7-C14 алкиларил, C6-C10 аминоарилоксил, C6-C10 арилтио, 4-6-членный гетероциклоалкил, необязательно замещенный галогеном, C1-C4 алкилом, C1-C4 галогеналкилом, 5-6-членный гетероарил, необязательно замещенный галогеном, C1-C4 алкилом, и C1-C6 алкил, который замещен гидрокси, галогеном, C1-C6 алкоксикарбонилом, C3-C8 циклоалкилом, C6-C10 арилом, 4-6-членным гетероциклоалкилом или 5-6-членным гетероарилом, который необязательно дополнительно замещен галогеном, гидроксилом или C1-C6 алкоксилом;
q имеет значение 0, 1, 2, 3 или 4;
m имеет значение 0, 1 или 2; и
n имеет значение 0, 1 или 2.
Например, сумма m и n составляет по меньшей мере 1.
Например, m имеет значение 1 или 2, и n имеет значение 0.
Например, m имеет значение 2, и n имеет значение 0
Например, A представляет собой CH2.
Например, A представляет собой O.
Например, L1 представляет собой N(Y).
Например, L1 представляет собой SO или SO2.
Например, Y представляет собой Rd.
Например, Rd представляет собой C1-C6 алкил.
Например, L2 отсутствует.
Например, R9 представляет собой 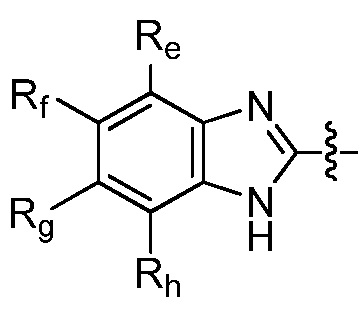 . Например, R9 представляет собой
. Например, R9 представляет собой 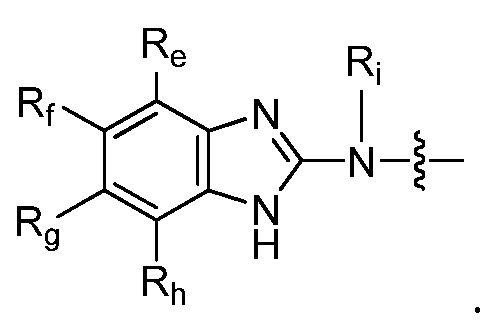
Например, по меньшей мере один из Re, Rf, Rg и Rh представляет собой галоген (такой как F, Cl и Br), C1-C6 алкоксил, необязательно замещенный одним или несколькими атомами галогена (такой как OCH3, OCH2CH3, O-изо-Pr и OCF3), C1-C6 алкилсульфонил, необязательно замещенный одним или несколькими атомами галогена (такой как SO2CF3), или C1-C6 алкил, необязательно замещенный одним или несколькими атомами галогена (такой как CH3, изопропил, н-бутил и CF3).
Например, Ri представляет собой H или C1-C6 алкил (например, метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, трет-бутил, н-пентил, втор-пентил или н-гексил).
Например, 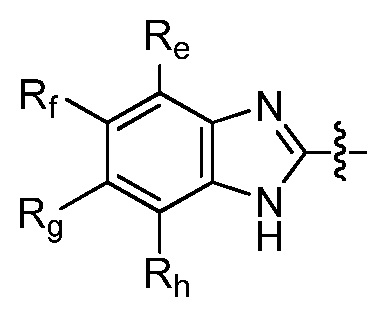 представляет собой незамещенный бензимидазолил или одну из следующих групп:
представляет собой незамещенный бензимидазолил или одну из следующих групп:
Например, R9 представляет собой 
Например, D представляет собой O.
Например, D представляет собой NRj.
Например, Rj представляет собой H.
Например, D представляет собой CRjRk.
Например, каждый из Rj и Rk представляет собой H.
Например, E представляет собой группу -M3-T3, в которой M3 представляет собой связь или C1-C3 алкильный линкер, T3 представляет собой фенил, нафтил, тиенил, циклопропил или циклогексил, и T3 необязательно замещен одним или несколькими заместителями, выбранными из группы, включающей галоген, гидроксил, тиол, карбоксил, циано, нитро, C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C1-C6 алкоксил, C1-C6 галогеналкил, C1-C6 галогеналкоксил, C1-C6 алкилтио, C1-C6 алкилсульфонил, C1-C6 алкилкарбонил, C1-C6 алкоксикарбонил, оксо, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино, C3-C8 циклоалкил, C4-C12 алкилциклоалкил, C6-C10 арил, C6-C10 арилоксил, C7-C14 алкиларил, C6-C10 аминоарилоксил, C6-C10 арилтио, 4-6-членный гетероциклоалкил, необязательно замещенный C1-C4 алкилом, 5-6-членный гетероарил, необязательно замещенный C1-C4 алкилом, и C1-C6 алкил, который замещен гидрокси, C1-C6 алкоксикарбонилом, C3-C8 циклоалкилом, C6-C10 арилом, 4-6-членным гетероциклоалкилом или 5-6-членным гетероарилом.
Например, T3 представляет собой фенил, необязательно замещенный одним или несколькими заместителями, выбранными из группы, включающей галоген, гидроксил, карбоксил, циано, нитро, C1-C6 алкил (например, метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, трет-бутил, н-пентил, втор-пентил и н-гексил), C1-C6 алкоксил, C1-C6 галогеналкил, C1-C6 галогеналкоксил, C1-C6 алкилсульфонил, C6-C10 арил (например, фенил или нафтил) и C6-C10 арилоксил и C7-C14 алкиларил.
Например, E представляет собой
Например, X представляет собой N.
Например, X представляет собой CRx.
Например, X представляет собой CH.
Например, Q представляет собой NH2 или группу NHRb, в которой Rb представляет собой -M1-T1, где M1 представляет собой связь или C1-C6 алкильный линкер, и T1 представляет собой C3-C8 циклоалкил.
Например, Q представляет собой H.
Например, R1, R2, R3, R4, R5, R6, R7 и R8, каждый, представляют собой H.
Например, когда R8 представляет собой галоген и связан с тем же самым атомом углерода, с которым связан J, тогда J не является гидроксилом.
Например, когда R8 представляет собой галоген и связан с тем же самым атомом углерода, с которым связан G, тогда G не является гидроксилом.
Например, T2 не является галогеном, когда M2 представляет собой SO2, SO, S, CO или O.
Например, T2 представляет собой 4-8-членный гетероциклоалкил, который связан с M2 через гетероатом.
Например, T2 представляет собой 4-8-членный гетероциклоалкил, который связан с M2 через N атом.
Например, T2 представляет собой 4-8-членный гетероциклоалкил, который связан с M2 через C атом.
Настоящее изобретение обеспечивает соединения формулы (IIIa) или (IIIb):
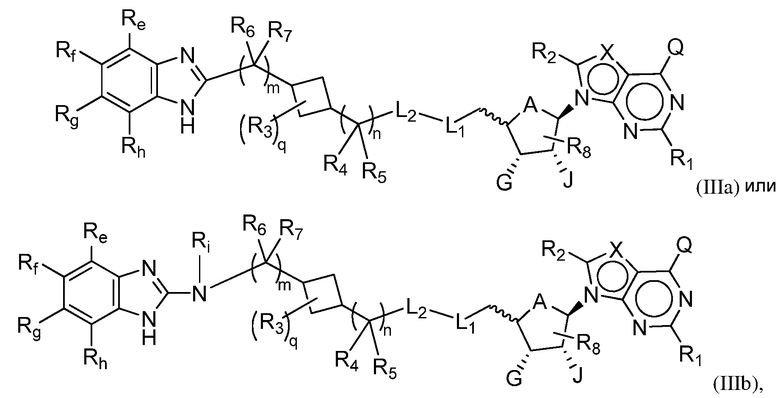
или их фармацевтически приемлемые соли или сложные эфиры, где:
A представляет собой O или CH2;
каждый из G и J независимо представляет собой H, галоген, C(O)OH, C(O)O-C1-C6 алкил или ORa, где Ra представляет собой H, C1-C6 алкил или C(O)-C1-C6 алкил, при этом C(O)O-C1-C6 алкил, C1-C6 алкил или C(O)-C1-C6 алкил необязательно замещен одним или несколькими заместителями, выбранными из группы, включающей галоген, циано, гидроксил, карбоксил, C1-C6 алкоксил, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино и C3-C8 циклоалкил;
Q представляет собой H, NH2, NHRb, NRbRc, Rb или ORb, где каждый из Rb и Rc независимо представляет собой C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, C6-C10 арил, 4-7-членный гетероциклоалкил, 5-10-членный гетероарил или группу -M1-T1, в которой M1 представляет собой связь или C1-C6 алкильный линкер, необязательно замещенный галогеном, циано, гидроксилом или C1-C6 алкоксилом, и T1 представляет собой C3-C8 циклоалкил, C6-C10 арил, 4-6-членный гетероциклоалкил или 5-10-членный гетероарил, или Rb и Rc, вместе с N атомом, с которым они связаны, образуют 4-7-членный гетероциклоалкил, содержащий 0 или 1 дополнительный гетероатом помимо N атома, который необязательно замещен C1-C6 алкилом, C2-C6 алкенилом, C2-C6 алкинилом, галогеном, гидроксилом, карбоксилом, C(O)OH, C(O)O-C1-C6 алкилом, OC(O)-C1-C6 алкилом, циано, C1-C6 алкоксилом, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино, C3-C8 циклоалкилом, C6-C10 арилом, 4-6-членным гетероциклоалкилом или 5-6-членным гетероарилом, и каждый из Rb, Rc и T1 необязательно замещен одним или несколькими заместителями, выбранными из группы, включающей C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, галоген, гидроксил, карбоксил, циано, C1-C6 алкоксил, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино, C3-C8 циклоалкил, C6-C10 арил, 4-6-членный гетероциклоалкил и 5-6-членный гетероарил;
X представляет собой N или группу CRx, в которой Rx представляет собой H, галоген, гидроксил, карбоксил, циано или RS1, где RS1 представляет собой амино, C1-C6 алкоксил, C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, C6-C10 арил, 4-6-членный гетероциклоалкил или 5-6-членный гетероарил, и RS1 необязательно замещен одним или несколькими заместителями, выбранными из группы, включающей галоген, гидроксил, карбоксил, циано, C1-C6 алкоксил, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино, C3-C8 циклоалкил, C6-C10 арил, 4-6-членный гетероциклоалкил и 5-6-членный гетероарил;
L1 представляет собой N(Y), S, SO или SO2;
L2 представляет собой CO или отсутствует, когда L1 представляет собой N(Y), или L2 отсутствует, когда L1 представляет собой S, SO или SO2, где Y представляет собой H, Rd, SO2Rd или CORd, когда L2 отсутствует, или Y представляет собой H или Rd, когда L2 представляет собой CO, где Rd представляет собой C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, C6-C10 арил, 4-6-членный гетероциклоалкил или 5-6-членный гетероарил, и Rd необязательно замещен одним или несколькими заместителями, выбранными из группы, включающей C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, галоген, гидроксил, карбоксил, циано, C1-C6 алкоксил, C1-C6 алкилсульфонил, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино, C3-C8 циклоалкил, C6-C10 арил, 4-6-членный гетероциклоалкил и 5-6-членный гетероарил, и при этом C3-C8 циклоалкил, C6-C10 арил, 4-6-членный гетероциклоалкил или 5-6-членный гетероарил, необязательно, дополнительно замещен C1-C6 алкилом, C2-C6 алкенилом, C2-C6 алкинилом, галогеном, гидроксилом, карбоксилом, C(O)OH, C(O)O-C1-C6 алкилом, OC(O)-C1-C6 алкилом, циано, C1-C6 алкоксилом, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино, C3-C8 циклоалкилом, C6-C10 арилом, 4-6-членным гетероциклоалкилом или 5-6-членным гетероарилом;
каждый из R1, R2, R3, R4, R5, R6 и R7 независимо представляет собой H, галоген, гидроксил, карбоксил, циано, RS2, где RS2 представляет собой амино, C1-C6 алкоксил, C1-C6 алкил, C2-C6 алкенил или C2-C6 алкинил, и каждый RS2 необязательно замещен одним или несколькими заместителями, выбранными из группы, включающей галоген, гидроксил, карбоксил, циано, C1-C6 алкоксил, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино, C3-C8 циклоалкил, C6-C10 арил, 4-6-членный гетероциклоалкил и 5-6-членный гетероарил;
R8 представляет собой H, галоген или RS3, где RS3 представляет собой C1-C6 алкил, C2-C6 алкенил или C2-C6 алкинил, и RS3 необязательно замещен одним или несколькими заместителями, выбранными из группы, включающей галоген, гидроксил, карбоксил, циано, амино, C1-C6 алкоксил, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино и C3-C8 циклоалкил;
каждый из Re, Rf, Rg и Rh независимо представляет собой группу -M2-T2, в которой M2 представляет собой связь, SO2, SO, S, CO, CO2, O, O-C1-C4 алкильный линкер, C1-C4 алкильный линкер, NH или N(Rt), где Rt представляет собой C1-C6 алкил, и T2 представляет собой H, галоген или RS4, где RS4 представляет собой C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, C6-C10 арил, 4-8-членный гетероциклоалкил или 5-10-членный гетероарил, и каждый из O-C1-C4 алкильного линкера, C1-C4 алкильного линкера, Rt и RS4 необязательно замещен одним или несколькими заместителями, выбранными из группы, включающей галоген, гидроксил, карбоксил, циано, C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C1-C6 алкоксил, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино, C3-C8 циклоалкил, C6-C10 арил, 4-6-членный гетероциклоалкил и 5-6-членный гетероарил,
Ri представляет собой H или C1-C6 алкил, необязательно замещенный одним или несколькими заместителями, выбранными из группы, включающей галоген, гидроксил, карбоксил, циано, C1-C6 алкоксил, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино, C3-C8 циклоалкил, C6-C10 арил, 4-6-членный гетероциклоалкил и 5-6-членный гетероарил;
q имеет значение 0, 1, 2, 3 или 4;
m имеет значение 0, 1 или 2; и
n имеет значение 0, 1 или 2.
Например, сумма m и n составляет по меньшей мере 1.
Например, m имеет значение 1 или 2, и n имеет значение 0.
Например, m имеет значение 2, и n имеет значение 0
Например, A представляет собой CH2.
Например, A представляет собой O.
Например, L1 представляет собой N(Y).
Например, L1 представляет собой SO или SO2.
Например, Y представляет собой Rd.
Например, Rd представляет собой C1-C6 алкил.
Например, L2 отсутствует.
Например, каждый из G и J независимо представляет собой ORa.
Например, Ra представляет собой H.
Например, по меньшей мере один из Re, Rf, Rg и Rh представляет собой галоген (такой как F, Cl и Br), C1-C6 алкоксил, необязательно замещенный одним или несколькими атомами галогена (такой как OCH3, OCH2CH3, O-изо-Pr и OCF3), C1-C6 алкилсульфонил, необязательно замещенный одним или несколькими атомами галогена (такой как SO2CF3), или C1-C6 алкил, необязательно замещенный одним или несколькими атомами галогена (такой как CH3, изопропил, н-бутил и CF3).
Например, Ri представляет собой H или C1-C6 алкил (например, метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, трет-бутил, н-пентил, втор-пентил и н-гексил).
Например,  представляет собой незамещенный бензимидазолил или одну из следующих групп:
представляет собой незамещенный бензимидазолил или одну из следующих групп:
Например, X представляет собой N.
Например, X представляет собой CRx.
Например, X представляет собой CH.
Например, Q представляет собой NH2 или группу NHRb, в которой Rb представляет собой -M1-T1, где M1 представляет собой связь или C1-C6 алкильный линкер, и T1 представляет собой C3-C8 циклоалкил.
Например, Q представляет собой H.
Например, R1, R2, R3, R4, R5, R6, R7 и R8, каждый, представляют собой H.
Например, когда R8 представляет собой галоген и связан с тем же самым атомом углерода, с которым связан J, тогда J не является гидроксилом.
Например, когда R8 представляет собой галоген и связан с тем же самым атомом углерода, с которым связан G, тогда G не является гидроксилом.
Например, T2 не является галогеном, когда M2 представляет собой SO2, SO, S, CO или O.
Например, T2 представляет собой 4-8-членный гетероциклоалкил, который связан с M2 через гетероатом.
Например, T2 представляет собой 4-8-членный гетероциклоалкил, который связан с M2 через N атом.
Например, T2 представляет собой 4-8-членный гетероциклоалкил, который связан с M2 через C атом.
Настоящее изобретение обеспечивает соединения формулы (IIIc):
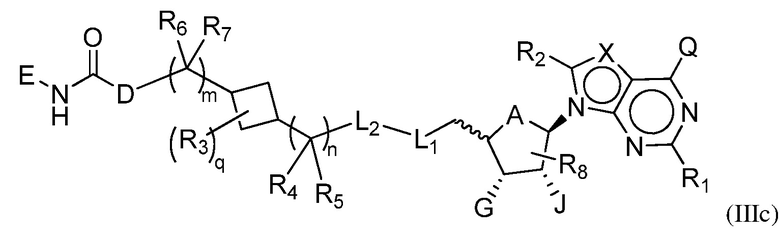
или их фармацевтически приемлемые соли или сложные эфиры, где:
A представляет собой O или CH2;
каждый из G и J независимо представляет собой H, галоген, C(O)OH, C(O)O-C1-C6 алкил или ORa, где Ra представляет собой H, C1-C6 алкил или C(O)-C1-C6 алкил, при этом C(O)O-C1-C6 алкил, C1-C6 алкил или C(O)-C1-C6 алкил необязательно замещен одним или несколькими заместителями, выбранными из группы, включающей галоген, циано, гидроксил, карбоксил, C1-C6 алкоксил, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино и C3-C8 циклоалкил;
Q представляет собой H, NH2, NHRb, NRbRc, Rb или ORb, где каждый из Rb и Rc независимо представляет собой C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, C6-C10 арил, 4-7-членный гетероциклоалкил, 5-10-членный гетероарил или группу -M1-T1, в которой M1 представляет собой связь или C1-C6 алкильный линкер, необязательно замещенный галогеном, циано, гидроксилом или C1-C6 алкоксилом, и T1 представляет собой C3-C8 циклоалкил, C6-C10 арил, 4-6-членный гетероциклоалкил или 5-10-членный гетероарил, или Rb и Rc, вместе с N атомом, с которым они связаны, образуют 4-7-членный гетероциклоалкил, содержащий 0 или 1 дополнительный гетероатом помимо N атома, который необязательно замещен C1-C6 алкилом, C2-C6 алкенилом, C2-C6 алкинилом, галогеном, гидроксилом, карбоксилом, C(O)OH, C(O)O-C1-C6 алкилом, OC(O)-C1-C6 алкилом, циано, C1-C6 алкоксилом, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино, C3-C8 циклоалкилом, C6-C10 арилом, 4-6-членным гетероциклоалкилом или 5-6-членным гетероарилом, и каждый из Rb, Rc и T1 необязательно замещен одним или несколькими заместителями, выбранными из группы, включающей C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, галоген, гидроксил, карбоксил, циано, C1-C6 алкоксил, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино, C3-C8 циклоалкил, C6-C10 арил, 4-6-членный гетероциклоалкил и 5-6-членный гетероарил;
X представляет собой N или группу CRx, в которой Rx представляет собой H, галоген, гидроксил, карбоксил, циано или RS1, где RS1 представляет собой амино, C1-C6 алкоксил, C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, C6-C10 арил, 4-6-членный гетероциклоалкил или 5-6-членный гетероарил, и RS1 необязательно замещен одним или несколькими заместителями, выбранными из группы, включающей галоген, гидроксил, карбоксил, циано, C1-C6 алкоксил, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино, C3-C8 циклоалкил, C6-C10 арил, 4-6-членный гетероциклоалкил и 5-6-членный гетероарил;
L1 представляет собой N(Y), S, SO или SO2;
L2 представляет собой CO или отсутствует, когда L1 представляет собой N(Y), или L2 отсутствует, когда L1 представляет собой S, SO или SO2, где Y представляет собой H, Rd, SO2Rd или CORd, когда L2 отсутствует, или Y представляет собой H или Rd, когда L2 представляет собой CO, где Rd представляет собой C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, C6-C10 арил, 4-6-членный гетероциклоалкил или 5-6-членный гетероарил, и Rd необязательно замещен одним или несколькими заместителями, выбранными из группы, включающей C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, галоген, гидроксил, карбоксил, циано, C1-C6 алкоксил, C1-C6 алкилсульфонил, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино, C3-C8 циклоалкил, C6-C10 арил, 4-6-членный гетероциклоалкил и 5-6-членный гетероарил, и при этом C3-C8 циклоалкил, C6-C10 арил, 4-6-членный гетероциклоалкил или 5-6-членный гетероарил, необязательно, дополнительно замещен C1-C6 алкилом, C2-C6 алкенилом, C2-C6 алкинилом, галогеном, гидроксилом, карбоксилом, C(O)OH, C(O)O-C1-C6 алкилом, OC(O)-C1-C6 алкилом, циано, C1-C6 алкоксилом, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино, C3-C8 циклоалкилом, C6-C10 арилом, 4-6-членным гетероциклоалкилом или 5-6-членным гетероарилом;
каждый из R1, R2, R3, R4, R5, R6 и R7 независимо представляет собой H, галоген, гидроксил, карбоксил, циано, RS2, где RS2 представляет собой амино, C1-C6 алкоксил, C1-C6 алкил, C2-C6 алкенил или C2-C6 алкинил, и каждый RS2 необязательно замещен одним или несколькими заместителями, выбранными из группы, включающей галоген, гидроксил, карбоксил, циано, C1-C6 алкоксил, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино, C3-C8 циклоалкил, C6-C10 арил, 4-6-членный гетероциклоалкил и 5-6-членный гетероарил;
R8 представляет собой H, галоген или RS3, где RS3 представляет собой C1-C6 алкил, C2-C6 алкенил или C2-C6 алкинил, и RS3 необязательно замещен одним или несколькими заместителями, выбранными из группы, включающей галоген, гидроксил, карбоксил, циано, амино, C1-C6 алкоксил, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино и C3-C8 циклоалкил;
D представляет собой O, NRj или CRjRk, каждый из Rj и Rk независимо представляет собой H или C1-C6 алкил, или Rj и Rk, взятые вместе с атомом углерода, с которым они связаны, образуют C3-C10 циклоалкильное кольцо;
E представляет собой -M3-T3, где M3 представляет собой связь или C1-C6 алкильный линкер, необязательно замещенный галогеном или циано, T3 представляет собой C3-C10 циклоалкил, C6-C10 арил, 5-10-членный гетероарил или 4-10-членный гетероциклоалкил, и T3 необязательно замещен одним или несколькими заместителями, выбранными из группы, включающей галоген, гидроксил, тиол, карбоксил, циано, нитро, C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C1-C6 алкоксил, C1-C6 галогеналкил, C1-C6 галогеналкоксил, C1-C6 алкилтио, C1-C6 алкилсульфонил, C1-C6 галогеналкилсульфонил, C1-C6 алкилкарбонил, C1-C6 алкоксикарбонил, оксо, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино, C3-C8 циклоалкил, C4-C12 алкилциклоалкил, C6-C10 арил, C6-C10 арилоксил, C7-C14 алкиларил, C6-C10 аминоарилоксил, C6-C10 арилтио, 4-6-членный гетероциклоалкил, необязательно замещенный галогеном, C1-C4 алкилом, C1-C4 галогеналкилом, 5-6-членный гетероарил, необязательно замещенный галогеном, C1-C4 алкилом, и C1-C6 алкил, который замещен гидрокси, галогеном, C1-C6 алкоксикарбонилом, C3-C8 циклоалкилом, C6-C10 арилом, 4-6-членным гетероциклоалкилом или 5-6-членным гетероарилом, который, необязательно, дополнительно замещен галогеном, гидроксилом или C1-C6 алкоксилом;
q имеет значение 0, 1, 2, 3 или 4;
m имеет значение 0, 1 или 2; и
n имеет значение 0, 1 или 2.
Например, сумма m и n составляет по меньшей мере 1.
Например, m имеет значение 1 или 2, и n имеет значение 0.
Например, m имеет значение 2 и n имеет значение 0.
Например, A представляет собой CH2.
Например, A представляет собой O.
Например, L1 представляет собой N(Y).
Например, L1 представляет собой SO или SO2.
Например, Y представляет собой Rd.
Например, Rd представляет собой C1-C6 алкил.
Например, L2 отсутствует.
Например, каждый из G и J независимо представляет собой ORa.
Например, Ra представляет собой H.
Например, D представляет собой O.
Например, D представляет собой NRj.
Например, Rj представляет собой H.
Например, D представляет собой CRjRk.
Например, каждый из Rj и Rk представляет собой H.
Например, E представляет собой группу -M3-T3, в которой M3 представляет собой связь или C1-C3 алкильный линкер, T3 представляет собой фенил, нафтил, тиенил, циклопропил или циклогексил, и T3 необязательно замещен одним или несколькими заместителями, выбранными из группы, включающей галоген, гидроксил, тиол, карбоксил, циано, нитро, C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C1-C6 алкоксил, C1-C6 галогеналкил, C1-C6 галогеналкоксил, C1-C6 алкилтио, C1-C6 алкилсульфонил, C1-C6 алкилкарбонил, C1-C6 алкоксикарбонил, оксо, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино, C3-C8 циклоалкил, C4-C12 алкилциклоалкил, C6-C10 арил, C6-C10 арилоксил, C7-C14 алкиларил, C6-C10 аминоарилоксил, C6-C10 арилтио, 4-6-членный гетероциклоалкил, необязательно замещенный C1-C4 алкилом, 5-6-членный гетероарил, необязательно замещенный C1-C4 алкилом, и C1-C6 алкил, который замещен гидрокси, C1-C6 алкоксикарбонилом, C3-C8 циклоалкилом, C6-C10 арилом, 4-6-членным гетероциклоалкилом или 5-6-членным гетероарилом.
Например, T3 представляет собой фенил, необязательно замещенный одним или несколькими заместителями, выбранными из группы, включающей галоген, гидроксил, карбоксил, циано, нитро, C1-C6 алкил (например, метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, трет-бутил, н-пентил, втор-пентил и н-гексил), C1-C6 алкоксил, C1-C6 галогеналкил, C1-C6 галогеналкоксил, C1-C6 алкилсульфонил, C6-C10 арил (например, фенил или нафтил) и C6-C10 арилоксил и C7-C14 алкиларил.
Например, E представляет собой
Например, X представляет собой N.
Например, X представляет собой CRx.
Например, X представляет собой CH.
Например, Q представляет собой NH2 или группу NHRb, в которой Rb представляет собой -M1-T1, где M1 представляет собой связь или C1-C6 алкильный линкер, и T1 представляет собой C3-C8 циклоалкил.
Например, Q представляет собой H.
Например, R1, R2, R3, R4, R5, R6, R7 и R8, каждый, представляют собой H.
Например, когда R8 представляет собой галоген и связан с тем же самым атомом углерода, с которым связан J, тогда J не является гидроксилом.
Например, когда R8 представляет собой галоген и связан с тем же самым атомом углерода, с которым связан G, тогда G не является гидроксилом.
Например, T2 не является галогеном, когда M2 представляет собой SO2, SO, S, CO или O.
Например, T2 представляет собой 4-8-членный гетероциклоалкил, который связан с M2 через гетероатом.
Например, T2 представляет собой 4-8-членный гетероциклоалкил, который связан с M2 через N атом.
Например, T2 представляет собой 4-8-членный гетероциклоалкил, который связан с M2 через C атом.
Настоящее изобретение также относится к соединению формулы (IV) или его N-оксиду или фармацевтически приемлемой соли такого соединения:
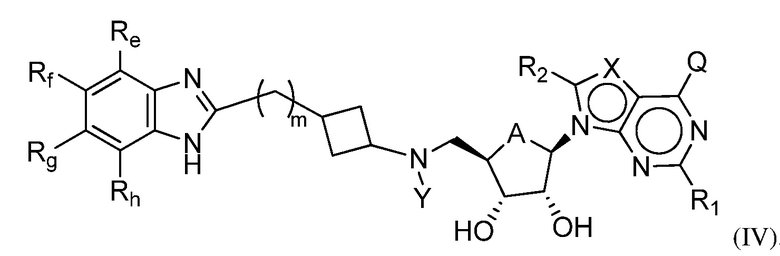
где A представляет собой O или CH2;
Q представляет собой H, NH2, NHRb, NRbRc, OH, Rb или ORb, где каждый из Rb и Rc независимо представляет собой C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, C6-C10 арил, 4-7-членный гетероциклоалкил, 5-10-членный гетероарил или группу -M1-T1, в которой M1 представляет собой связь или C1-C6 алкильный линкер, необязательно замещенный галогеном, циано, гидроксилом или C1-C6 алкоксилом, и T1 представляет собой C3-C8 циклоалкил, C6-C10 арил, 4-6-членный гетероциклоалкил или 5-10-членный гетероарил, или Rb и Rc, вместе с N атомом, с которым они связаны, образуют 4-7-членный гетероциклоалкил, содержащий 0 или 1 дополнительный гетероатом помимо N атома, который необязательно замещен C1-C6 алкилом, C2-C6 алкенилом, C2-C6 алкинилом, галогеном, гидроксилом, карбоксилом, C(O)OH, C(O)O-C1-C6 алкилом, OC(O)-C1-C6 алкилом, циано, C1-C6 алкоксилом, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино, C3-C8 циклоалкилом, C6-C10 арилом, 4-6-членным гетероциклоалкилом или 5-6-членным гетероарилом, и каждый из Rb, Rc и T1 необязательно замещен одним или несколькими заместителями, выбранными из C1-C6 алкила, C2-C6 алкенила, C2-C6 алкинила, галогена, гидроксила, карбоксила, циано, C1-C6 алкоксила, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино, C3-C8 циклоалкила, C6-C10 арила, 4-6-членный гетероциклоалкила и 5-6-членного гетероарила;
X представляет собой N или группу CRx, в которой Rx представляет собой H, галоген, гидроксил, карбоксил, циано или RS1, где RS1 представляет собой амино, C1-C6 алкоксил, C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, C6-C10 арил, 4-6-членный гетероциклоалкил или 5-6-членный гетероарил, и RS1 необязательно замещен одним или несколькими заместителями, выбранными из галогена, гидроксила, карбоксила, циано, C1-C6 алкоксила, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино, C3-C8 циклоалкила, C6-C10 арила, 4-6-членного гетероциклоалкила и 5-6-членного гетероарила;
Y представляет собой H, Rd, SO2Rd или CORd, где Rd представляет собой C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, C6-C10 арил, 4-6-членный гетероциклоалкил или 5-6-членный гетероарил, и Rd необязательно замещен одним или несколькими заместителями, выбранными из C1-C6 алкила, C2-C6 алкенила, C2-C6 алкинила, галогена, гидроксила, карбоксила, циано, C1-C6 алкоксила, C1-C6 алкилсульфонила, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино, C3-C8 циклоалкила, C6-C10 арила, 4-6-членного гетероциклоалкила и 5-6-членного гетероарила, и при этом C3-C8 циклоалкил, C6-C10 арил, 4-6-членный гетероциклоалкил или 5-6-членный гетероарил, необязательно, дополнительно замещен C1-C6 алкилом, C2-C6 алкенилом, C2-C6 алкинилом, галогеном, гидроксилом, карбоксилом, C(O)OH, C(O)O-C1-C6 алкилом, OC(O)-C1-C6 алкилом, циано, C1-C6 алкоксилом, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино, C3-C8 циклоалкилом, C6-C10 арилом, 4-6-членным гетероциклоалкилом или 5-6-членным гетероарилом;
каждый из R1 и R2 независимо представляет собой H, галоген, гидроксил, карбоксил, циано, RS2, где RS2 представляет собой амино, C1-C6 алкоксил, C1-C6 алкил, C2-C6 алкенил или C2-C6 алкинил, и каждый RS2 необязательно замещен одним или несколькими заместителями, выбранными из галогена, гидроксила, карбоксила, циано, C1-C6 алкоксила, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино, C3-C8 циклоалкила, C6-C10 арила, 4-6-членного гетероциклоалкила и 5-6-членного гетероарила;
каждый из Re, Rf, Rg и Rh независимо представляет собой группу -M2-T2, в которой M2 представляет собой связь, SO2, SO, S, CO, CO2, O, O-C1-C4 алкильный линкер, C1-C4 алкильный линкер, NH или N(Rt), где Rt представляет собой C1-C6 алкил, и T2 представляет собой H, галоген или RS4, RS4 представляет собой C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, C6-C10 арил, 4-8-членный гетероциклоалкил или 5-10-членный гетероарил, и каждый из O-C1-C4 алкильного линкера, C1-C4 алкильного линкера, Rt и RS4 необязательно замещен одним или несколькими заместителями, выбранными из галогена, гидроксила, карбоксила, циано, C1-C6 алкила, C2-C6 алкенила, C2-C6 алкинила, C1-C6 алкоксила, амино, моно-C1-C6 алкиламино, ди-C1-C6 алкиламино, C3-C8 циклоалкила, C6-C10 арила, 4-6-членного гетероциклоалкила и 5-6-членного гетероарила, и
m имеет значение 0, 1 или 2.
Например, A представляет собой O. В некоторых соединениях формулы (IV), A представляет собой O, и m имеет значение 2.
В некоторых соединениях формулы (IV), X представляет собой N.
Например, в некоторых соединениях, Q представляет собой NH2 или группу NHRb, в которой Rb представляет собой -M1-T1, M1 представляет собой связь или C1-C6 алкильный линкер, и T1 представляет собой C3-C8 циклоалкил.
Например, в некоторых соединениях формулы (IV), R1 и R2, каждый, представляют собой H.
В некоторых соединениях формулы (IV), Y представляет собой Rd. Например, Rd представляет собой C1-C6 алкил, необязательно замещенный C3-C8 циклоалкилом или галогеном. Например, Rd представляет собой C3-C8 циклоалкил, необязательно замещенный C1-C6 алкилом или галогеном.
Настоящее изобретение также относится к соединению Формулы (IV), где по меньшей мере один из Re, Rf, Rg и Rh представляет собой галоген, C1-C6 алкоксил, необязательно замещенный одним или несколькими атомами галогена; C1-C6 алкилсульфонил, необязательно замещенный одним или несколькими атомами галогена; C1-C6 алкил, необязательно замещенный одним или несколькими заместителями, выбранными из CN, галогена, C3-C8 циклоалкила, гидрокси и C1-C6 алкоксила; C3-C8 циклоалкил, необязательно замещенный одним или несколькими заместителями, выбранными из C1-C6 алкила или CN; или 4-8-членный гетероциклоалкил, необязательно замещенный одним или несколькими заместителями, выбранными из CN, галогена, гидрокси, C1-C6 алкила и C1-C6 алкоксила. Например, соединение формулы (IV) содержит по меньшей мере один из Re, Rf, Rg и Rh, выбран из группы, включающей F; Cl; Br; CF3; OCF3; SO2CF3; оксетанил, необязательно замещенный одним или несколькими заместителями, выбранными из CN, галогена, гидрокси, C1-C6 алкила и C1-C6 алкоксила; C3-C8 циклоалкил, необязательно замещенный одним или несколькими заместителями, выбранными из C1-C4 алкила; и C1-C4 алкил, необязательно замещенный одним или несколькими заместителями, выбранными из галогена, C3-C8 циклоалкила, гидрокси и C1-C6 алкоксила.
Например, настоящее изобретение относится к соединениям формулы (IV), где по меньшей мере один из Rf и Rg представляет собой алкил, необязательно замещенный гидроксилом. Например, настоящее изобретение относится к соединениям, где по меньшей мере один из Rf и Rg представляет собой трет-бутил, замещенный гидроксилом.
Настоящее изобретение относится к соединению, выбранному из Соединений 1-140. Настоящее изобретение также относится к соли соединения, выбранного из Соединений 1-140. Настоящее изобретение также относится к N-оксиду соединения, выбранного из Соединений 1-140. Настоящее изобретение также относится к соли N-оксида соединения, выбранного из Соединений 1-140. Например, настоящее изобретение относится к соединению, выбранному из Соединений 1-7, 9-109 и 111-140.
Настоящее изобретение также относится к фармацевтической композиции, включающей терапевтически эффективное количество соединения формулы (IV) и фармацевтически приемлемый носитель.
Настоящее изобретение также относится к фармацевтической композиции, включающей терапевтически эффективное количество соли соединения формулы (IV) и фармацевтически приемлемый носитель.
Настоящее изобретение также относится к фармацевтической композиции, включающей терапевтически эффективное количество гидрата соединения формулы (IV) и фармацевтически приемлемый носитель.
Настоящее изобретение также относится к фармацевтической композиции, включающей терапевтически эффективное количество соединения, выбранного из Соединений 1-140, и фармацевтически приемлемый носитель. Настоящее изобретение также относится к фармацевтической композиции, включающей терапевтически эффективное количество соли соединения, выбранного из Соединений 1-140, и фармацевтически приемлемый носитель. Настоящее изобретение также относится к фармацевтической композиции, включающей терапевтически эффективное количество N-оксида соединения, выбранного из Соединений 1-140, и фармацевтически приемлемый носитель. Настоящее изобретение также относится к фармацевтической композиции, включающей терапевтически эффективное количество N-оксида соли соединения, выбранного из Соединений 1-140, и фармацевтически приемлемый носитель. Настоящее изобретение также относится к фармацевтической композиции, включающей терапевтически эффективное количество гидрата соединения, выбранного из Соединений 1-140, и фармацевтически приемлемый носитель.
Настоящее изобретение обеспечивает фармацевтическую композицию, включающую одно или несколько соединений формулы (I), (II), (IIIa), (IIIb) или (IIIc) и один или несколько фармацевтически приемлемых носителей.
Настоящее изобретение также относится к фармацевтической композиции, включающей терапевтически эффективное количество соли соединения формулы (I), (II), (IIIa), (IIIb) или (IIIc) и фармацевтически приемлемый носитель.
Настоящее изобретение также относится к фармацевтической композиции, включающей терапевтически эффективное количество гидрата соединения формулы (I), (II), (IIIa), (IIIb) или (IIIc) и фармацевтически приемлемый носитель.
Настоящее изобретение обеспечивает способы лечения или профилактики рака. Настоящее изобретение обеспечивает способы лечения рака. Настоящее изобретение также обеспечивает способы профилактики рака. Способ включает введение субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы (I), (II), (IIIa), (IIIb) или (IIIc). Рак может представлять собой гематологический рак. Предпочтительно, рак представляет собой лейкоз. Более предпочтительно, рак представляет собой острый миелогенный лейкоз, острый лимфоцитарный лейкоз или лейкоз смешанного происхождения.
Настоящее изобретение обеспечивает способы лечения или профилактики заболевания или расстройства, опосредованного транслокацией гена на хромосоме 11q23. Настоящее изобретение обеспечивает способы лечения заболевания или расстройства, опосредованного транслокацией гена на хромосоме 11q23. Настоящее изобретение также обеспечивает способы профилактики заболевания или расстройства, опосредованного транслокацией гена на хромосоме 11q23. Способ включает введение субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы (I), (II), (IIIa), (IIIb), (IIIc) или (IV).
Настоящее изобретение обеспечивает способы лечения или профилактики заболевания или расстройства, в котором играет роль DOT1-опосредованное метилирование белка, или заболевания или расстройства, опосредованного DOT1-опосредованным метилированием белка. Настоящее изобретение обеспечивает способы лечения заболевания или расстройства, в котором играет роль DOT1-опосредованное метилирование белка, или заболевания или расстройства, опосредованного DOT1-опосредованным метилированием белка. Настоящее изобретение также обеспечивает способы профилактики заболевания или расстройства, в котором играет роль DOT1-опосредованное метилирование белка, или заболевания или расстройства, опосредованного DOT1-опосредованным метилированием белка. Способ включает введение субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы (I), (II), (IIIa), (IIIb), (IIIc) или (IV).
Настоящее изобретение обеспечивает способы ингибирования активности DOT1L в клетке. Способ включает контактирование клетки с эффективным количеством одного или нескольких соединений формулы (I), (II), (IIIa), (IIIb), (IIIc) или (IV).
Еще один аспект настоящего изобретения относится к способу снижения уровня метилирования Лизинового остатка 79 Гистона H3 (H3-K79) в клетке. Способ включает контактирование клетки с соединением по настоящему изобретению. Такой способ можно использовать для уменьшения интенсивности симптомов любого состояния, которое вызвано или потенциировано активностью DOT1 через метилирование H3-K79.
Настоящее изобретение относится к применению соединений, раскрытых в настоящем изобретении, для получения лекарственного средства для лечения или профилактики рака. Применение включает соединение формулы (I), (II), (IIIa), (IIIb), (IIIc) или (IV) для введения субъекту, нуждающемуся в этом, в терапевтически эффективном количестве. Рак может представлять собой гематологический рак. Предпочтительно, рак представляет собой лейкоз. Более предпочтительно, рак представляет собой острый миелогенный лейкоз, острый лимфоцитарный лейкоз или лейкоз смешанного происхождения.
Настоящее изобретение обеспечивает применение соединений, раскрытых в настоящем изобретении, для получения лекарственного средства для лечения или профилактики заболевания или расстройства, опосредованного транслокацией гена на хромосоме 11q23. Применение включает соединение формулы (I), (II), (IIIa), (IIIb), (IIIc) или (IV) для введения субъекту, нуждающемуся в этом, в терапевтически эффективном количестве.
Настоящее изобретение обеспечивает применение соединений, раскрытых в настоящем изобретении, для получения лекарственного средства для лечения или профилактики заболевания или расстройства, в котором играет роль DOT1-опосредованное метилирование белка, или заболевания или расстройства, опосредованного DOT1-опосредованным метилированием белка. Применение включает соединение формулы (I), (II), (IIIa), (IIIb), (IIIc) или (IV) для введения субъекту, нуждающемуся в этом, в терапевтически эффективном количестве.
Настоящее изобретение обеспечивает применение соединений, раскрытых в настоящем изобретении, для ингибирования активности DOT1L в клетке. Применение включает контактирование клетки с эффективным количеством одного или нескольких соединений формулы (I), (II), (IIIa), (IIIb), (IIIc) или (IV).
Еще один аспект настоящего изобретения относится к применению соединений, раскрытых в настоящем изобретении, для снижения уровня метилирования Лизинового остатка 79 Гистона H3 (H3-K79) в клетке. Применение включает контактирование клетки с соединением по настоящему изобретению. Такое применение уменьшает интенсивность симптомов любого состояния, которое вызвано или потенциировано активностью DOT1 через метилирование H3-K79.
В формулах, представленных в настоящей заявке, переменные можно выбрать из соответствующих групп химических составляющих, которые определены ниже в подробном описании изобретения.
Кроме того, настоящее изобретение обеспечивает способы синтеза указанных выше соединений. После синтеза терапевтически эффективное количество одного или нескольких соединений можно сформулировать с фармацевтически приемлемым носителем для введения млекопитающему, в частности, человеку для использования в модуляции эпигенетического фермента. В некоторых вариантах воплощения, соединения по настоящему изобретению являются полезными для лечения, профилактики или снижения риска рака или для получения лекарственного средства для лечения, профилактики или снижения риска рака. Соответственно, соединения или композиции можно вводить, например, пероральным, парентеральным, ушным, глазным, назальным или местным путем для обеспечения эффективного количества соединения млекопитающему.
Репрезентативные соединения по настоящему изобретению включают соединения, указанные в Таблице 1 (см. графическую часть).
Как используется в настоящей заявке, “алкил”, “C1, C2, C3, C4, C5 или C6 алкил” или “C1-C 6 алкил” включает C1, C2, C3, C4, C5 или C6 насыщенные алифатические углеводородные группы с прямой цепью (линейные) и C3, C4, C5 или C6 разветвленные насыщенные алифатические углеводородные группы. Например, C1-C6 алкил включает C1, C2, C3, C4, C5 и C6 алкильные группы. Примеры алкила включают, группы, содержащие от одного до шести атомов углерода, такие как, но не ограничиваясь этим, метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, трет-бутил, н-пентил, втор-пентил или н-гексил.
В некоторых вариантах воплощения, алкил с прямой или разветвленной цепью содержит шесть или меньше атомов углерода (например, C1-C6 для прямой цепи, C3-C6 для разветвленной цепи), и в другом варианте воплощения алкил с прямой или разветвленной цепью содержит четыре или меньше атомов углерода.
Как используется в настоящей заявке, термин “циклоалкил” относится к насыщенной или ненасыщенной неароматической углеводородной моно- или мульти-кольцевой системе, содержащей от 3 до 30 атомов углерода (например, C3-C10). Примеры циклоалкила включают, но не ограничиваются этим, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил, циклопентенил, циклогексенил, циклогептенил и адамантил. Термин "гетероциклоалкил" относится к насыщенной или ненасыщенной неароматической 5-8-членной моноциклической, 8-12 членной бициклической или 11-14-членной трициклической кольцевой системе, содержащей один или несколько гетероатомов (таких как O, N, S или Se). Примеры гетероциклоалкильных групп включают, но не ограничиваются этим, пиперазинил, пирролидинил, диоксанил, морфолинил и тетрагидрофуранил.
Термин “необязательно замещенный алкил” относится к незамещенному алкилу или алкилу, содержащему указанные заместители, замещающие один или несколько атомов водорода на одном или нескольких атомах углерода основной углеводородной цепи. Такие заместители могут включать, например, алкил, алкенил, алкинил, галоген, гидроксил, алкилкарбонилокси, арилкарбонилокси, алкоксикарбонилокси, арилоксикарбонилокси, карбоксилат, алкилкарбонил, арилкарбонил, алкоксикарбонил, аминокарбонил, алкиламинокарбонил, диалкиламинокарбонил, алкилтиокарбонил, алкоксил, фосфат, фосфонато, фосфинато, амино (включая алкиламино, диалкиламино, ариламино, диариламино и алкилариламино), ациламино (включая алкилкарбониламино, арилкарбониламино, карбамоил и уреидо), амидино, имино, сульфгидрил, алкилтио, арилтио, тиокарбоксилат, сульфаты, алкилсульфинил, сульфонато, сульфамоил, сульфонамидо, нитро, трифторметил, циано, азидо, гетероциклил, алкиларил или ароматическую или гетероароматическую группу.
“Арилалкил” или “аралкил” группа представляет собой алкил, замещенный арилом (например, фенилметил (бензил)). “Алкиларильная” группа представляет собой арил, замещенный алкилом (например, метилфенил).
Как используется в настоящей заявке, “алкильный линкер” включает C1, C2, C3, C4, C5 или C6 насыщенные двухвалентные алифатические углеводородные группы с прямой цепью (линейные) и C3, C4, C5 или C6 разветвленные насыщенные алифатические углеводородные группы. Например, C1-C6 алкильный линкер включает C1, C2, C3, C4, C5 и C6 алкильные линкерные группы. Примеры алкильного линкера включают, группы, содержащие от одного до шести атомов углерода, такие как, но не ограничиваясь этим, метил (-CH2-), этил (-CH2CH2-), н-пропил (-CH2CH2CH2-), изопропил (-CHCH3CH2-), н-бутил (-CH2CH2CH2CH2-), втор-бутил (-CHCH3CH2CH2-), изобутил (-C(CH3)2CH2-), н-пентил (-CH2CH2CH2CH2CH2-), втор-пентил (-CHCH3CH2CH2CH2-) или н-гексил (-CH2CH2CH2CH2CH2CH2-).
“Алкенил” включает ненасыщенные алифатические группы, аналогичные по длине и возможному замещению алкильными группами, описанным выше, но которые содержат по меньшей мере одну двойную связь. Например, термин “алкенил” включает линейные алкенильные группы (например, этенил, пропенил, бутенил, пентенил, гексенил, гептенил, октенил, ноненил, деценил) и разветвленные алкенильные группы. В некоторых вариантах воплощения, линейная алкенильная группа содержит шесть или меньше атомов углерода в своей основной цепи (например, C2-C6 для линейной, C3-C6 для разветвленной цепи). Термин “C2-C6” включает алкенильные группы, содержащие от двух до шести атомов углерода. Термин “C3-C6” включает алкенильные группы, содержащие от трех до шести атомов углерода.
Термин “необязательно замещенный алкенил” относится к незамещенному алкенилу или алкенилу, содержащему указанные заместители, заменяющие один или несколько атомов водорода на одном или нескольких атомах углерода основной углеводородной цепи. Такие заместители могут включать, например, алкил, алкенил, алкинил, галоген, гидроксил, алкилкарбонилокси, арилкарбонилокси, алкоксикарбонилокси, арилоксикарбонилокси, карбоксилат, алкилкарбонил, арилкарбонил, алкоксикарбонил, аминокарбонил, алкиламинокарбонил, диалкиламинокарбонил, алкилтиокарбонил, алкоксил, фосфат, фосфонато, фосфинато, амино (включая алкиламино, диалкиламино, ариламино, диариламино и алкилариламино), ациламино (включая алкилкарбониламино, арилкарбониламино, карбамоил и уреидо), амидино, имино, сульфгидрил, алкилтио, арилтио, тиокарбоксилат, сульфаты, алкилсульфинил, сульфонато, сульфамоил, сульфонамидо, нитро, трифторметил, циано, гетероциклил, алкиларил или ароматическую или гетероароматическую группу.
“Алкинил” включает ненасыщенные алифатические группы, аналогичные по длине и возможному замещению алкильным группам, описанным выше, но которые содержат по меньшей мере одну тройную связь. Например, “алкинил” включает линейные алкинильные группы (например, этинил, пропинил, бутинил, пентинил, гексинил, гептинил, октинил, нонинил, децинил) и разветвленные алкинильные группы. В некоторых вариантах воплощения, алкинильная группа с прямой или разветвленной цепью содержит шесть или меньше атомов углерода в своей основной цепи (например, C2-C6 для линейной, C3-C6 для разветвленной цепи). Термин “C2-C6” включает алкинильные группы, содержащие от двух до шести атомов углерода. Термин “C3-C6” включает алкинильные группы, содержащие от трех до шести атомов углерода.
Термин “необязательно замещенный алкинил” относится к незамещенному алкинилу или алкинилу, содержащему указанные заместители, заменяющие один или несколько атомов водорода на одном или нескольких атомах углерода основной углеводородной цепи. Такие заместители могут включать, например, алкил, алкенил, алкинил, галоген, гидроксил, алкилкарбонилокси, арилкарбонилокси, алкоксикарбонилокси, арилоксикарбонилокси, карбоксилат, алкилкарбонил, арилкарбонил, алкоксикарбонил, аминокарбонил, алкиламинокарбонил, диалкиламинокарбонил, алкилтиокарбонил, алкоксил, фосфат, фосфонато, фосфинато, амино (включая алкиламино, диалкиламино, ариламино, диариламино и алкилариламино), ациламино (включая алкилкарбониламино, арилкарбониламино, карбамоил и уреидо), амидино, имино, сульфгидрил, алкилтио, арилтио, тиокарбоксилат, сульфаты, алкилсульфинил, сульфонато, сульфамоил, сульфонамидо, нитро, трифторметил, циано, азидо, гетероциклил, алкиларил или ароматическую или гетероароматическую группу.
Другие необязательно замещенные группы (такие как необязательно замещенный циклоалкил, гетероциклоалкил, арил или гетероарил) включают как замещенные группы, так и группы, содержащие один или несколько из указанных заместителей.
“Арил” включает группы с ароматичностью, включая “сопряженные” или полициклические системы с по меньшей мере одним ароматическим кольцом, и не содержат никаких гетероатомов в кольцевой структуре. Примеры включают фенил, бензил, 1,2,3,4-тетрагидронафталенил и т.п.
“Гетероарильные” группы представляют собой арильные группы, определенные выше, за исключением того, что они содержат от одного до четырех гетероатомов в кольцевой структуре, и также могут быть указаны как “арильные гетероциклы” или “гетероароматические группы”. Как используется в настоящей заявке, термин “гетероарил” включает стабильное 5- или 6-членное моноциклическое или 7-, 8-, 9-, 10-, 11- или 12-членное бициклическое ароматическое гетероциклическое кольцо, которое состоит из атомов углерода и одного или нескольких гетероатомов, например, 1 или 1-2 или 1-3 или 1-4 или 1-5 или 1-6 гетероатомов или, например, 1, 2, 3, 4, 5 или 6 гетероатомов, независимо выбранных из группы, включающей азот, кислород и серу. Атом азота может быть замещенным или незамещенным (т.е. N или NR, где R представляет собой H или другие заместители, которые определены). Гетероатомы азота и серы необязательно могут быть окислены (т.е. N→O и S(O)p, где p=1 или 2). Необходимо отметить, что общее количество S и O атомов в ароматическом гетероцикле составляет не более 1.
Примеры гетероарильных групп включают пиррол, фуран, тиофен, тиазол, изотиазол, имидазол, триазол, тетразол, пиразол, оксазол, изоксазол, пиридин, пиразин, пиридазин, пиримидин и т.п.
Кроме того, термины “арил” и “гетероарил” включают полициклические арильные и гетероарильные группы, например, трициклические, бициклические, например, нафталин, бензоксазол, бензодиоксазол, бензотиазол, бензоимидазол, бензотиофен, метилендиоксифенил, хинолин, изохинолин, нафтиридин, индол, бензофуран, пурин, бензофуран, деазапурин, индолизин.
В случае полициклических ароматических колец, только одно из колец должно быть ароматическим (например, 2,3-дигидроиндол), хотя все кольца могут быть ароматическими (например, хинолин). Второе кольцо также может быть конденсированным или связанным мостиковой связью.
Арильное или гетероарильное ароматическое кольцо может быть замещено в одном или нескольких положениях в кольце заместителями, которые описаны выше, такими как, например, алкил, алкенил, алкинил, галоген, гидроксил, алкокси, алкилкарбонилокси, арилкарбонилокси, алкоксикарбонилокси, арилоксикарбонилокси, карбоксилат, алкилкарбонил, алкиламинокарбонил, аралкиламинокарбонил, алкениламинокарбонил, алкилкарбонил, арилкарбонил, аралкилкарбонил, алкенилкарбонил, алкоксикарбонил, аминокарбонил, алкилтиокарбонил, фосфат, фосфонато, фосфинато, амино (включая алкиламино, диалкиламино, ариламино, диариламино и алкилариламино), ациламино (включая алкилкарбониламино, арилкарбониламино, карбамоил и уреидо), амидино, имино, сульфгидрил, алкилтио, арилтио, тиокарбоксилат, сульфаты, алкилсульфинил, сульфонато, сульфамоил, сульфонамидо, нитро, трифторметил, циано, азидо, гетероциклил, алкиларил или ароматическая или гетероароматическая группа. Арильные группы также могут быть конденсированными или связанными мостиковой связью с алициклическими или гетероциклическими кольцами, которые не являются ароматическими, с образованием, таким образом, полициклической системы (например, тетралин, метилендиоксифенил).
Как используется в настоящей заявке, “карбоцикл” или “карбоциклическое кольцо” включает любое стабильное моноциклическое, бициклическое или трициклическое кольцо, содержащее указанное количество атомов углерода, любое из которых может быть насыщенным, ненасыщенным или ароматическим. Например, C3-C14 карбоцикл включает моноциклическое, бициклическое или трициклическое кольцо, содержащее 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13 или 14 атомов углерода. Примеры карбоциклов включают, но не ограничиваются этим, циклопропил, циклобутил, циклобутенил, циклопентил, циклопентенил, циклогексил, циклогептенил, циклогептил, циклогептенил, адамантил, циклооктил, циклооктенил, циклооктадиенил, флуоренил, фенил, нафтил, инданил, адамантил и тетрагидронафтил. Связанные мостиковой связью кольца также включены в определение “карбоцикл”, включая, например, [3.3.0]бициклооктан, [4.3.0]бициклононан, [4.4.0]бициклодекан и [2.2.2]бициклооктан. Связанное мостиковой связью кольцо образуется, когда один или несколько атомов углерода связываются с двумя не-смежными атомами углерода. В одном варианте воплощения, кольца с мостиком включают один или два атома углерода. Следует отметить, что мостик всегда преобразует моноциклическое кольцо в трициклическое кольцо. Когда кольцо связанно мостиковой связью, заместители, указанные для кольца, также могут присутствовать на мостике. Также включены конденсированные (например, нафтил, тетрагидронафтил) и спиро кольца.
Как используется в настоящей заявке, “гетероцикл” включает любую кольцевую структуру (насыщенную или частично ненасыщенную), которая содержит по меньшей мере один кольцевой гетероатом (например, N, O или S). Примеры гетероциклов включают, но не ограничиваются этим, морфолин, пирролидин, тетрагидротиофен, пиперидин, пиперазин и тетрагидрофуран.
Примеры гетероциклических групп включают, но не ограничиваются этим, акридинил, азоцинил, бензимидазолил, бензофуранил, бензотиофуранил, бензотиофенил, бензоксазолил, бензоксазолинил, бензтиазолил, бензтриазолил, бензтетразолил, бензизоксазолил, бензизотиазолил, бензимидазолинил, карбазолил, 4aH-карбазолил, карболинил, хроманил, хроменил, циннолинил, декагидрохинолинил, 2H,6H-1,5,2-дитиазинил, дигидрофуро[2,3-b]тетрагидрофуран, фуранил, фуразанил, имидазолидинил, имидазолинил, имидазолил, 1H-индазолил, индоленил, индолинил, индолизинил, индолил, 3H-индолил, изатионил, изобензофуранил, изохроманил, изоиндазолил, изоиндолинил, изоиндолил, изохинолинил, изотиазолил, изоксазолил, метилендиоксифенил, морфолинил, нафтиридинил, октагидроизохинолинил, оксадиазолил, 1,2,3-оксадиазолил, 1,2,4-оксадиазолил, 1,2,5-оксадиазолил, 1,3,4-оксадиазолил, 1,2,4-оксадиазол(4H)-он, оксазолидинил, оксазолил, оксиндолил, пиримидинил, фенантридинил, фенантролинил, феназинил, фенотиазинил, феноксатинил, феноксазинил, фталазинил, пиперазинил, пиперидинил, пиперидонил, 4-пиперидонил, пиперонил, птеридинил, пуринил, пиранил, пиразинил, пиразолидинил, пиразолинил, пиразолил, пиридазинил, пиридооксазол, пиридоимидазол, пиридотиазол, пиридинил, пиридил, пиримидинил, пирролидинил, пирролинил, 2H-пирролил, пирролил, хиназолинил, хинолинил, 4H-хинолизинил, хиноксалинил, хинуклидинил, тетрагидрофуранил, тетрагидроизохинолинил, тетрагидрохинолинил, тетразолил, 6H-1,2,5-тиадиазинил, 1,2,3-тиадиазолил, 1,2,4-тиадиазолил, 1,2,5-тиадиазолил, 1,3,4-тиадиазолил, тиантренил, тиазолил, тиенил, тиенотиазолил, тиенооксазолил, тиеноимидазолил, тиофенил, триазинил, 1,2,3-триазолил, 1,2,4-триазолил, 1,2,5-триазолил, 1,3,4-триазолил и ксантенил.
Термин “замещенный”, как используется в настоящей заявке, означает, что любые один или несколько атомов водорода на указанном атоме заменены заместителями, выбранными из указанной группы, при условии, что указанная нормальная валентность атома не превышена, и что такое замещение приводит к стабильному соединению. Когда заместитель представляет собой оксо или кето (т.е. =O), тогда заменены 2 атома водорода на таком атоме. Кето заместители не присутствуют в ароматической группе. Двойные связи в кольце, как используется в настоящей заявке, представляют собой двойные связи, которые образованы между двумя смежными кольцевыми атомами (например, C=C, C=N или N=N). “Стабильное соединение” и “стабильная структура” означают соединение, которое является достаточно устойчивым, чтобы пережить выделение из реакционной смеси до полезной степени чистоты и формулирование в эффективное терапевтическое средство.
Когда связь с заместителем показана как пересекающая связь, соединяющую два атома в кольце, тогда такой заместитель может быть связан с любым атомом в кольце. Когда заместитель указан без указания атома, через который такой заместитель связан с остальной частью соединения указанной формулы, тогда такой заместитель может быть связан через любой атом в указанной формуле. Комбинации заместителей и/или переменных возможны, но только если такие комбинации приводят к стабильным соединениям.
Когда какая-либо переменная (например, R1) встречается более одного раза в любой составляющей или формуле для соединения, ее определение в каждом случае ее присутствия является независимым от ее определения в каждом другом случае. Так, например, если группа показана как замещенная 0-2 группами R1, тогда группа, необязательно, может иметь в качестве заместителей до двух групп R1, и R1 в каждом случае выбран независимо от определения R1. Также, комбинации заместителей и/или переменных возможны, но только если такие комбинации приводят к стабильным соединениям.
Термин “гидрокси” или “гидроксил” включает группы с -OH или -O-.
Как используется в настоящей заявке, “гало” или “галоген” относится к фтору, хлору, брому и иоду. Термин “пергалогенированный”, как правило, относится к группе, где все атомы водорода заменены атомами галогена. Термин “галогеналкил” или “галогеналкоксил” относится к алкилу или алкоксилу, замещенному одним или несколькими атомами галогена.
Термин “карбонил” включает соединения и группы, которые содержат атом углерода, связанный двойной связью с атомом кислорода. Примеры групп, содержащих карбонил, включают, но не ограничиваются этим, альдегиды, кетоны, карбоновые кислоты, амиды, сложные эфиры, ангидриды и т.п.
Термин “карбоксил” относится к -COOH или его C1-C6 алкиловому эфиру.
“Ацил” включает группы, которые содержат ацильный радикал (R-C(O)-) или карбонильную группу. “Замещенный ацил” включает ацильные группы, где один или несколько атомов водорода заменены, например, алкильными группами, алкинильными группами, галогеном, гидроксилом, алкилкарбонилокси, арилкарбонилокси, алкоксикарбонилокси, арилоксикарбонилокси, карбоксилатом, алкилкарбонилом, арилкарбонилом, алкоксикарбонилом, аминокарбонилом, алкиламинокарбонилом, диалкиламинокарбонилом, алкилтиокарбонилом, алкоксилом, фосфатом, фосфонато, фосфинато, амино (включая алкиламино, диалкиламино, ариламино, диариламино и алкилариламино), ациламино (включая алкилкарбониламино, арилкарбониламино, карбамоил и уреидо), амидино, имино, сульфгидрилом, алкилтио, арилтио, тиокарбоксилатом, сульфатами, алкилсульфинилом, сульфонато, сульфамоилом, сульфонамидо, нитро, трифторметилом, циано, азидо, гетероциклилом, алкиларилом или ароматической или гетероароматической группой.
“Ароил” включает группы с арильной или гетероароматической группой, связанной с карбонильной группой. Примеры ароильных групп включают фенилкарбокси, нафтилкарбокси и т.п.
“Алкоксиалкил”, “алкиламиноалкил” и “тиоалкоксиалкил” включают алкильные группы, описанные выше, где атомы кислорода, азота или серы замещают один или несколько атомов углерода основной углеводородной цепи.
Термин “алкокси” или “алкоксил” включает замещенные и незамещенные алкильные, алкенильные и алкинильные группы, ковалентно связанные с атомом кислорода. Примеры алкокси групп или алкоксильных радикалов включают, но не ограничиваются этим, метокси, этокси, изопропилокси, пропокси, бутокси и пентокси группы. Примеры замещенных алкокси групп включают галогенированные алкокси группы. Алкокси группы могут быть замещены группами, такими как алкенил, алкинил, галоген, гидроксил, алкилкарбонилокси, арилкарбонилокси, алкоксикарбонилокси, арилоксикарбонилокси, карбоксилат, алкилкарбонил, арилкарбонил, алкоксикарбонил, аминокарбонил, алкиламинокарбонил, диалкиламинокарбонил, алкилтиокарбонил, алкоксил, фосфат, фосфонато, фосфинато, амино (включая алкиламино, диалкиламино, ариламино, диариламино и алкилариламино), ациламино (включая алкилкарбониламино, арилкарбониламино, карбамоил и уреидо), амидино, имино, сульфгидрил, алкилтио, арилтио, тиокарбоксилат, сульфаты, алкилсульфинил, сульфонато, сульфамоил, сульфонамидо, нитро, трифторметил, циано, азидо, гетероциклил, алкиларил или ароматические или гетероароматические группы. Примеры галоген-замещенных алкокси групп включают, но не ограничиваются этим, фторметокси, дифторметокси, трифторметокси, хлорметокси, дихлорметокси и трихлорметокси.
Термин “простой эфир” или “алкокси” включает соединения или группы, которые содержат атом кислорода, связанный с двумя атомами углерода или гетероатомами. Например, термин включает “алкоксиалкил”, который относится к алкильной, алкенильной или алкинильной группе, ковалентно связанной с атомом кислорода, который ковалентно связан с алкильной группой.
Термин “сложный эфир” включает соединения или группы, которые содержат атом углерода или гетероатом, связанный с атомом кислорода, который связан с углеродом карбонильной группы. Термин “сложный эфир” включает алкоксикарбокси группы, такие как метоксикарбонил, этоксикарбонил, пропоксикарбонил, бутоксикарбонил, пентоксикарбонил и т.п.
Термин “тиоалкил” включает соединения или группы, которые содержат алкильную группу, связанную с атомом серы. Тиоалкильные группы могут быть замещены группами, такими как алкил, алкенил, алкинил, галоген, гидроксил, алкилкарбонилокси, арилкарбонилокси, алкоксикарбонилокси, арилоксикарбонилокси, карбоксилат, карбоновая кислота, алкилкарбонил, арилкарбонил, алкоксикарбонил, аминокарбонил, алкиламинокарбонил, диалкиламинокарбонил, алкилтиокарбонил, алкоксил, амино (включая алкиламино, диалкиламино, ариламино, диариламино и алкилариламино), ациламино (включая алкилкарбониламино, арилкарбониламино, карбамоил и уреидо), амидино, имино, сульфгидрил, алкилтио, арилтио, тиокарбоксилат, сульфаты, алкилсульфинил, сульфонато, сульфамоил, сульфонамидо, нитро, трифторметил, циано, азидо, гетероциклил, алкиларил или ароматические или гетероароматические группы.
Термин “тиокарбонил” или “тиокарбокси” включает соединения и группы, которые содержат атом углерода, связанный двойной связью с атомом серы.
Термин “тиоэфир” включает группы, которые содержат атом серы, связанный с двумя атомами углерода или гетероатомами. Примеры тиоэфиров включают, но не ограничиваются этим алктиоалкилы, алктиоалкенилы и алктиоалкинилы. Термин “алктиоалкилы” включают группы, содержащие алкильную, алкенильную или алкинильную группу, связанную с атомом серы, который связан с алкильной группой. Аналогичным образом, термин “алктиоалкенилы” относится к группам, где алкильная, алкенильная или алкинильная группа связана с атомом серы, который ковалентно связан с алкенильной группой; и алктиоалкинилы” относится к группам, где алкильная, алкенильная или алкинильная группа связана с атомом серы, который ковалентно связан с алкинильной группой.
Как используется в настоящей заявке, “амин” или “амино” относится к незамещенной или замещенной группе -NH2. “Алкиламино” включает группы соединений, где азот -NH2 связан с по меньшей мере одной алкильной группой. Примеры алкиламино групп включают бензиламино, метиламино, этиламино, фенетиламино и т.п. “Диалкиламино” включает группы, где азот -NH2 связан с по меньшей мере двумя дополнительными алкильными группами. Примеры диалкиламино групп включают, но не ограничиваются этим, диметиламино и диэтиламино. “Ариламино” и “диариламино” включают группы, где азот связан с по меньшей мере одной или двумя арильными группами, соответственно. “Аминоарил” и “аминоарилокси” относятся к арилу и арилокси, замещенному амино группой. “Алкилариламино”, “алкиламиноарил” или “ариламиноалкил” относится к амино группе, которая связана с по меньшей мере одной алкильной группой и по меньшей мере одной арильной группой. “Алкаминоалкил” относится к алкильной, алкенильной или алкинильной группе, связанной с атомом азота, который также связан с алкильной группой. “Ациламино” включает группы, где азот связан с ацильной группой. Примеры ациламино включают, но не ограничиваются этим, алкилкарбониламино, арилкарбониламино, карбамоильную и уреидо группы.
Термин “амид” или “аминокарбокси” включает соединения или группы, которые содержат атом азота, который связан с углеродом карбонильной или тиокарбонильной группы. Термин включает “алкаминокарбокси” группы, которые включают группы алкил, алкенил или алкинил, связанные с амино группой, которая связана с углеродом карбонильной или тиокарбонильной группы. Термин также включает “ариламинокарбокси” группы, которые включают арильные или гетероарильные группы, связанные с амино группой, которая связана с углеродом карбонильной или тиокарбонильной группы. Термины “алкиламинокарбокси”, “алкениламинокарбокси”, “алкиниламинокарбокси” и “ариламинокарбокси” включают группы, где группы алкил, алкенил, алкинил и арил, соответственно, связаны с атомом азота, который, в свою очередь, связан с углеродом карбонильной группы. Амиды могут быть замещены заместителями, такими как линейный алкил, разветвленный алкил, циклоалкил, арил, гетероарил или гетероцикл. Заместители в амидной группе могут быть дополнительно замещены.
Соединения по настоящему изобретению, которые содержат атомы азота, можно преобразовать в N-оксиды путем обработки окислителем (например, 3-хлорпероксибензойной кислотой (mCPBA) и/или пероксидами водорода) с получением других соединений по настоящему изобретению. Таким образом, все показанные и заявленные азот-содержащие соединения рассматриваются, когда это позволяет валентность и структура, включающие как представленное соединение, так и его N-оксидное производное (которое может быть указано как N→O или N+-O-). Кроме того, в других случаях, атомы азота в соединениях по настоящему изобретению можно преобразовать в N-гидрокси или N-алкокси соединения. Например, N-гидрокси соединения можно получить путем окисления исходного амина при помощи окислителя, такого как m-CPBA. Все показанные и заявленные азот-содержащие соединения рассматриваются, когда это позволяет валентность и структура, как представленное соединение, так и его N-гидрокси (т.е. N-OH) и N-алкокси (т.е. N-OR, где R представляет собой замещенный или незамещенный C1-C6 алкил, C1-C6 алкенил, C1-C6 алкинил, 3-14-членный карбоцикл или 3-14-членный гетероцикл) производные.
В настоящем описании, структурная формула соединения в некоторых случаях для удобства представляет определенный изомер, но настоящее изобретение включает все изомеры, такие как геометрические изомеры, оптические изомеры на основании асимметрического углерода, стереоизомеры, таутомеры и т.п. Кроме того, может присутствовать кристаллический полиморфизм для соединений, представленных этой формулой. Следует отметить, что любая кристаллическая форма, смесь кристаллических форм или их ангидрид или гидрат включены в объем настоящего изобретения. Кроме того, так называемый метаболит, который образуется путем разложения соединения по настоящему изобретению in vivo, включен в объем настоящего изобретения.
“Изомерия” означает соединения, которые имеют одинаковые молекулярные формулы, но отличаются друг от друга последовательностью связывания их атомов или расположением их атомов в пространстве. Изомеры, которые отличаются по расположению их атомов в пространстве, называют “стереоизомерами”. Стереоизомеры, которые не являются зеркальным отображением друг друга, называют “диастереоизомерами”, и стереоизомеры, которые не являются совмещаемыми зеркальными отображениями друг друга, называют “энантиомерами” или иногда оптическими изомерами. Смесь, содержащую равные количества отдельных энантиомерных форм противоположной хиральности, называют “рацемической смесью”.
Атом углерода, связанный с четырьмя неодинаковыми заместителями, называют “хиральным центром”.
“Хиральный изомер” означает соединение с по меньшей мере одним хиральным центром. Соединения с более чем одним хиральным центром могут существовать или в виде индивидуального диастереомера или в виде смеси диастереомеров, которая называется “диастереомерной смесью”. Когда присутствует один хиральный центр, стереоизомер может быть охарактеризован абсолютной конфигурацией (R или S) этого хирального центра. Абсолютная конфигурация относится к расположению в пространстве заместителей, связанных с хиральным центром. Порядок очередности рассматриваемых заместителей, связанных с хиральным центром, определяют в соответствии с Правилом Последовательности Cahn, Ingold and Prelog. (Cahn et al., Angew. Chem. Inter. Edit. 1966, 5, 385; errata 511; Cahn et al., Angew. Chem. 1966, 78, 413; Cahn and Ingold, J. Chem. Soc. 1951 (London), 612; Cahn et al., Experientia 1956, 12, 81; Cahn, J. Chem. Educ. 1964, 41, 116).
“Геометрический изомер” означает диастереомеры, которые существуют, благодаря затрудненному вращению вокруг двойных связей или циклоалкильного линкера (например, 1,3-циклобутил). Эти конфигурация различают по их названиям при помощи префиксов цис и транс или Z и E, которые показывают, что группы расположены на одной и той же или на противоположной стороне двойной связи в молекуле в соответствии с правилом Cahn-Ingold-Prelog.
Должно быть понятно, что соединения по настоящему изобретению могут быть описаны как хиральные изомеры или геометрические изомеры. Также должно быть понятно, что, когда соединения имеют хиральные изомерные или геометрические изомерные формы, предполагается, что все изомерные формы включены в объем настоящего изобретения, и название соединений не исключает никакие изомерные формы.
Например, соединения формулы (I) включают следующие хиральные изомеры и геометрические изомеры.
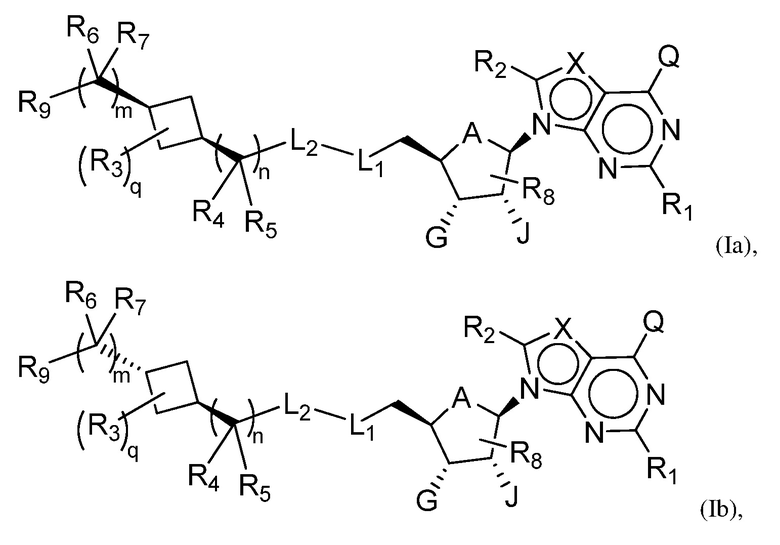
Кроме того, структуры и другие соединения, обсуждаемые в настоящем изобретении, включают все их атропические изомеры. “Атропические изомеры” представляют собой тип стереоизомера, в котором атомы двух изомеров расположены по-разному в пространстве. Атропические изомеры существуют, благодаря ограниченному вращению, вызванному затруднением вращения большой группы вокруг центральной связи. Такие атропические изомеры типично существуют в виде смеси, однако, как результат современных достижений в хроматографии, появилась возможность разделения смесей двух атропических изомеров в некоторых случаях.
“Таутомер” представляет собой один из двух или более структурных изомеров, которые существуют в равновесии, и легко преобразуется из одной изомерной формы в другую. Это преобразование приводит к формальной миграции атома водорода, сопровождаемой переключением смежных сопряженных двойных связей. Таутомеры существуют в виде смеси таутомерного ряда в растворе. В растворах, где возможна таутомеризация, будет достигаться химическое равновесие таутомеров. Точное соотношение таутомеров зависит от различных факторов, включая температуру, растворитель и pH. Когда таутомеры являются взаимопревращаемыми путем таутомеризации, это называют таутомерией.
Из различных типов таутомерии, которые возможны, обычно наблюдают два типа. В кето-енольной таутомерии происходит одновременный сдвиг электронов и атома водорода. Кольцевая цепочечная таутомерия возникает в результате взаимодействия альдегидной группы (-CHO) в сахарной цепи молекулы с одной из гидрокси групп (-OH) в той же молекуле с приданием ей циклической (кольцевой) формы, как показано на примере глюкозы.
Обычными таутомерными парами являются следующие: кетон-енол, амид-нитрил, лактам-лактим, амид-имидиновая кислота таутомерия в гетероциклических кольцах (например, в нуклеооснованиях, таких как гуанин, тимин и цитозин), амин-енамин и енамин-енамин. Бензимидазолы также демонстрируют таутомерию, когда бензимидазол содержит один или несколько заместителей в 4, 5, 6 или 7 положениях, возникает возможность различных изомеров. Например, 2,5-диметил-1H-бензо[d]имидазол может существовать в равновесии с его изомером 2,6-диметил-1H-бензо[d]имидазолом через таутомеризацию.
Другой пример таутомерии показан ниже.
Должно быть понятно, что соединения по настоящему изобретению могут быть описаны как различные таутомеры. Также должно быть понятно, что, когда соединения имеют таутомерные формы, предполвагается, что все таутомерные формы включены в объем настоящего изобретения, и название соединений не исключает никакую таутомерную форму.
Термин “кристаллические полиморфы”, “полиморфы” или “кристаллические формы” означает кристаллические структуры, в которых соединение (или его соль или сольват) может кристаллизоваться в различных кристаллических упаковках, которые все имеют одинаковую композицию элементов. Различные кристаллические формы обычно имеют разные картины дифракции рентгеновких лучей, инфракрасные спектры, температуры плавления, плотность, твердость, форму кристаллов, оптические и электрические свойства, стабильность и растворимость. Растворитель для перекристаллизации, скорость кристаллизации, температура хранения и другие факторы могут привести к тому, что одна кристаллическая форма будет доминировать. Кристаллические полиморфы соединений можно получить путем кристаллизации в различных условиях.
Соединения по настоящему изобретению могут быть кристаллическими, полукристаллическими, некристаллическими, аморфными, мезоморфными и т.п.
Соединения формулы (I), (II), (IIIa), (IIIb), (IIIc) или (IV) включают соединения как таковые, а также их N-оксиды, соли, их сольваты и их пролекарства, если это является подходящим. Соль, например, может быть образована между анионом и положительно заряженной группой (например, амино) в замещенном пуриновом или 7-деазапуриновом соединении. Подходящие анионы включают хлорид, бромид, иодид, сульфат, бисульфат, сульфамат, нитрат, фосфат, цитрат, метансульфонат, трифторацетат, глутамат, глюкуронат, глутарат, малат, малеат, сукцинат, фумарат, тартрат, тозилат, салицилат, лактат, нафталинсульфонат и ацетат. Подобным образом, соль также может быть образована между катионом и отрицательно заряженной группой (например, карбоксилат) в замещенном пуриновом или 7-деазапуриновом соединении. Подходящие катионы включают ион натрия, ион калия, ион магния, ион кальция и катион аммония, такой как тетраметиламмониевый ион. Замещенные пуриновые или 7-деазапуриновые соединения также включают соли, содержащие четвертичные атомы азота. Примеры пролекарств включают сложные эфиры и другие фармацевтически приемлемые производные, которые при введении субъекту могут обеспечивать активные замещенные пуриновые или 7-деазапуриновые соединения.
Кроме того, соединения по настоящему изобретению, например, соли соединений, могут существовать как в гидратированной, так и в негидратированной (безводной) форме или в виде сольватов с другими молекулами растворителя. Неограничивающие примеры гидратов включают полугидраты, моногидраты, дигидраты, тригидраты и т.п. Неограничивающие примеры сольватов включают этанольные сольваты, ацетоновые сольваты и т.п.
“Сольват” означает формы присоединения растворителя, которые содержат либо стехиометрические, либо не-стехиометрические количества растворителя. Некоторые соединения имеют тенденцию захватывать фиксированное молярное количество молекул растворителя в кристаллическом твердом состоянии с образованием, таким образом, сольвата. Если растворителем является вода, образуемый сольват представляет собой гидрат; и если растворителем является спирт, образуемый сольват представляет собой алкоголят. Гидраты образуются путем объединения одной или нескольких молекул воды с одной молекулой вещества, в котором вода сохраняет свое молекулярное состояние как H2O. Полугидрат образуется путем объединения одной молекулы воды с более чем одной молекулой вещества, в котором вода сохраняет свое молекулярное состояние как H2O.
Как используется в настоящей заявке, термин “аналог” относится к химическому соединению, которое является структурно схожим с другим, но имеет небольшое отличие в композиции (например, замещение одного атома атомом другого элемента, или присутствие конкретной функциональной группы, или замещение одной функциональной группы другой функциональной группой). Таким образом, аналог представляет собой соединение, которое является схожим или сопоставимым по функции и внешнему виду, но не по структуре или происхождению, со ссылочным соединением.
Как определено в настоящей заявке, термин “производное” относится к соединениям, которые имеют общую структуру ядра и замещены различными группами, как описано в настоящей заявке. Например, все соединения, представленные Формулой (I), представляет собой замещенные пуриновые соединения или замещенные 7-деазапуриновые соединения и имеют Формулу (I) в качестве общего ядра.
Термин “биоизостера” относится к соединению, образованному в результате обмена атома или группы атомов с другими, в широком смысле подобными, атомом или группой атомов. Целью биоизостерного замещения является создание нового соединения с биологическими свойствами, подобными свойствам исходного соединения. Биоизостерное замещение может происходить на физико-химической или топологической основе. Примеры карбоновокислотных биоизостер включают, но не ограничиваются этим, ацилсульфонимиды, тетразолы, сульфонаты и фосфонаты. См., например, Patani and LaVoie, Chem. Rev. 96, 3147-3176, 1996.
Настоящее изобретение включает все изотопы атомов, присутствующих в соединениях по настоящему изобретению. Изотопы включают атомы, имеющие одинаковый атомный номер, но разные массовые числа. В качестве общего примера и без ограничения, изотопы водорода включают тритий и дейтерий, а изотопы углерода включают C-13 и C-14.
2. Синтез замещенных пуриновых соединений и замещенных 7-деазапуриновых соединений
Настоящее изобретение обеспечивает способы синтеза соединений формул (I), (II), (IIIa), (IIIb), (IIIc) и (IV). Настоящее изобретение также обеспечивает подробные способы синтеза различных раскрываемых соединений по настоящему изобретению в соответствии со следующими схемами, как показано в примерах.
В различных разделах настоящего описания, где композиции описаны как содержащие, включающие или состоящие из конкретных компонентов, предполагается, что композиции также состоят по существу из, или состоят из, указанных компонентов. Аналогичным образом, когда способы или процессы описаны как содержащие, включающие или состоящие из конкретных технологических операций, эти способы также состоят по существу из, или состоят из, указанных технологических операций. Кроме того, должно быть понятно, что порядок стадий или порядок осуществления определенных действий не имеет значения, при условии, что настоящее изобретение является осуществимым. Более того, две или более стадий или действий можно осуществлять одновременно.
Способы синтеза по настоящему изобретению могут работать с множеством различных функциональных групп, поэтому можно использовать различные замещенные исходные вещества. Способы, как правило, обеспечивают желаемое конечное соединение в конце или близко к концу способа в целом, хотя в некоторых случаях может быть желательным дальнейшее преобразование соединения в его фармацевтически приемлемую соль, сложный эфир или пролекарство.
Соединения по настоящему изобретению можно получить различными путями с использованием коммерчески доступных исходных веществ, соединений, известных из литературы, или из легко получаемых промежуточных соединений, с использованием стандартных способов синтеза и процедур, которые либо известны специалистам в данной области, либо которые будут очевидны для специалистов в данной области в свете раскрытия настоящей заявки. Стандартные способы синтеза и процедуры для получения органических молекул и преобразований функциональных групп и манипуляций с ними можно найти в соответствующей научной литературе или в стандартных справочниках, относящихся к данной области техники. Не ограничиваясь каким-либо одним или несколькими источниками, классические справочники, такие как Smith, M. B., March, J., March’s Advanced Organic Chemistry: Reactions, Mechanisms and Structure, 5th edition, John Wiley & Sons: New York, 2001; Greene, T.W., Wuts, P.G. M., Protective Groups in Organic Synthesis, 3rd edition, John Wiley & Sons: New York, 1999; R. Larock, Comprehensive Organic Traansformations, VCH Publishers (1989); L. Fieser and M. Fieser, Fieser and Fieser’s Reagents for Organic Synthesis, John Wiley and Sons (1994); и L. Paquette, ed., Encyclopedia of Reagents for Organic Synthesis Синтез, John Wiley and Sons (1995), включенные в настоящую заявку посредством ссылки, являются полезной и общепризнанной справочной литературой в области органического синтеза, известной специалистам. Следующие описания способов синтеза предназначены для иллюстрации, но не для ограничения, общих процедур получения соединений по настоящему изобретению.
Соединения по настоящему изобретению удобным образом могут быть получены различными способами, известными специалистам в данной области. Соединения по настоящему изобретению, имеющие Формулы (I), (II), (IIIa), (IIIb), (IIIc) и (IV), можно получить в соответствии с процедурами, проиллюстрированными на Схемах A-W ниже, из коммерчески доступных исходных веществ или из исходных веществ, которые можно получить с использованием известных из литературы процедур. R группы (такие как R, R’ и Ra) на Схемах A-P могут соответствовать переменным (т.е. R1, R2, Rb и Rc), определенным в Формуле (I), (II), (IIIa), (IIIb), (IIIc) или (IV), если не указано иное. “PG” на Схемах относится к защитной группе.
Рядовые специалисты в данной области могут заметить, что в процессе последовательного осуществления реакций и схем синтеза, описанных в настоящей заявке, порядок некоторых стадий может быть изменен, например, стадий введения и удаления защитных групп.
Рядовым специалистам в данной области должно быть понятно, что некоторые группы могут нуждаться в защите от реакционных условий через применение защитных групп. Защитные группы также можно использовать для дифференцирования схожих функциональных групп в молекулах. Перечень защитных групп и способы введения и удаления этих групп можно найти в Greene, T.W., Wuts, P.G. M., Protective Groups in Organic Synthesis, 3rd edition, John Wiley & Sons: New York, 1999.
Предпочтительные защитные группы включают, но не ограничиваются этим:
Для гидроксильной группы: TBS, бензил, THP, Ac
Для карбоновой кислоты: бензиловый эфир, метиловый эфир, этиловый эфир, аллиловый эфир
Для аминов: Cbz, BOC, DMB
Для диолов: Ac (×2) TBS (×2) или, когда взяты вместе, ацетониды
Для тиолов: Ac
Для бензимидазолов: SEM, бензил, PMB, DMB
Для альдегидов: ди-алкилацетали, такие как диметоксиацеталь или диэтилацетил.
В схемах реакций, описанных в настоящей заявке, можно получить различные стереоизомеры. Когда не указан какой-либо конкретный стереоизомер, имеются в виду все возможные стереоизомеры, которые могут быть получены в результате реакции. Рядовым специалистам в данной области должно быть понятно, что реакции можно оптимизировать с получением, предпочтительно, одного изомера, или могут быть разработаны новые схемы для получения единственного изомера. Если получают смеси, можно использовать такие процедуры, как препаративная тонкослойная хроматография, препаративная ВЭЖХ, препаративная хиральная ВЭЖХ или препаративная SFC, для разделения изомеров.
Следующие аббревиатуры используют повсеместно в тексте описания изобретения, и их определение представлено ниже:
Настоящее изобретение обеспечивает способы получения соединений по настоящему изобретению. Следующие схемы представляют иллюстративные химические приемы, которые можно использовать для синтеза соединений по настоящему изобретению.
Схема A: Синтез 5’-Амино Пурин Рибозы (A-V)
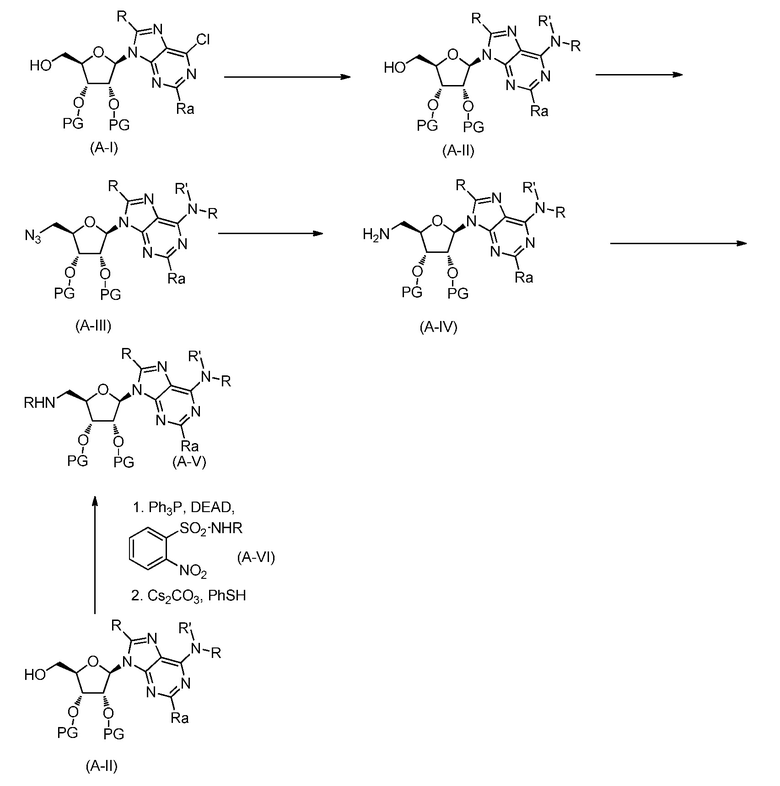
5’-Амино пурин-рибозы - промежуточные соединения (A-V) можно синтезировать, как показано на Схеме A выше. Подходящее защищенное 6-Cl аденозиновое производное (A-I) преобразуют в 6-амино-производное (A-II) путем обработки подходящим амином (включая аммиак) в присутствии основания, такого как Et3N, K2CO3 или основание Хунига, в растворителе, таком как MeCN или DMF, ТГФ, iPrOH или их смесь. Если необходимо, реакционную смесь можно нагреть до 100°C (когда необходимая температура выше чем температура кипения одного или нескольких компонентов в смеси, реакцию можно осуществить в герметично закрытой пробирке). R группы в схеме могут представлять алкильные защитные группы (например, 2, 4 диметоксибензил). 6-амино продукт (A-II) можно преобразовать в 5’-азидо промежуточное соединение (A-III) путем преобразования 5’-гидроксильной группы в удаляемую группу, такую как MsO (т.е. CH3S(O)2O) путем обработки метансульфонилхлоридом (MsCl) в присутствии основания, такого как Et3N, пиридин или K2CO3, в инертном растворителе, таком как CH2Cl2, ТГФ, MeCN, DMF или их смесь. 5’-удаляемую группу затем замещают азидным анионом из NaN3 в инертном растворителе, таком как DMF. Альтернативно, (A-II) можно непосредственно преобразовать в (A-III) путем обработки при помощи DPPA, Ph3P и DIAD в растворителе, таком как ТГФ. Азидо группу в (A-III) можно восстановить до первичного амина (A-IV) путем восстановления при помощи H2 в присутствии металлического катализатора (например, Pd/C, PtO2) или при помощи реакции Staudinger с фосфином, таким как Ph3P или PMe3. Первичный амин (A-IV) можно преобразовать во вторичный амин (A-V) путем обработки подходящим кетоном или альдегидом в присутствии подходящего восстановителя, такого как NaBH(OAc)3 или NaCNBH3. Могут быть добавлены дополнительные реагенты, такие как Ti(OiPr)4.
Альтернативно, 5’-гидрокси промежуточное соединение (A-II) можно обработать сульфонамидом (A-VI), DEAD и Ph3P в инертном растворителе, таком как ТГФ. Полученный сульфонамидный продукт затем можно обработать бензолтиолом в присутствии основания, такого как K2CO3, Cs2CO3, с получением вторичного амина (A-V).
Эти показанные выше последовательности реакций также могут быть применимы к производным ликсозы, исходя из (A-VII), с получением диастереомера с противоположной конфигурацией в 5’ положении.
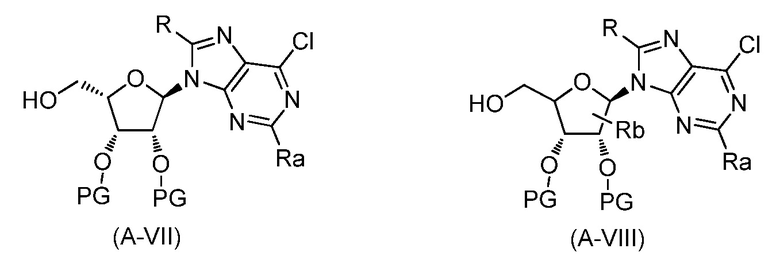
Подобный ряд последовательностей реакций можно использовать для 2’-дезокси или 3’-дезокси или замещенной рибозы или ликсозы (A-VIII), представленной выше, с получением промежуточных соединений 5’-амино пурин-рибозы/ликсозы.
Альтернативный способ для введения 6-NH2 группы, как показано ниже, включает обработку (A-IX) производных при помощи NaN3 с получением 6-азидо промежуточного соединения, с последующим восстановлением до NH2 группы (A-X) с использованием триалкилфосфина, такого как PMe3 или PPh3.
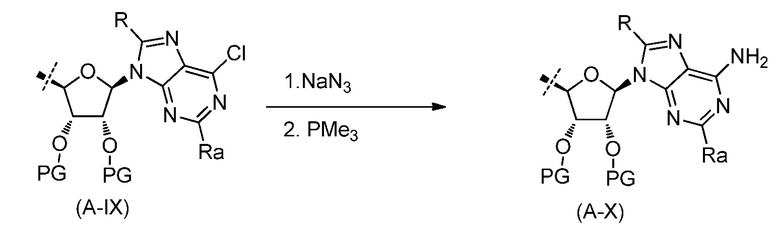
Схема B: Синтез 5’ Амино 7-Деазапурин Рибозы
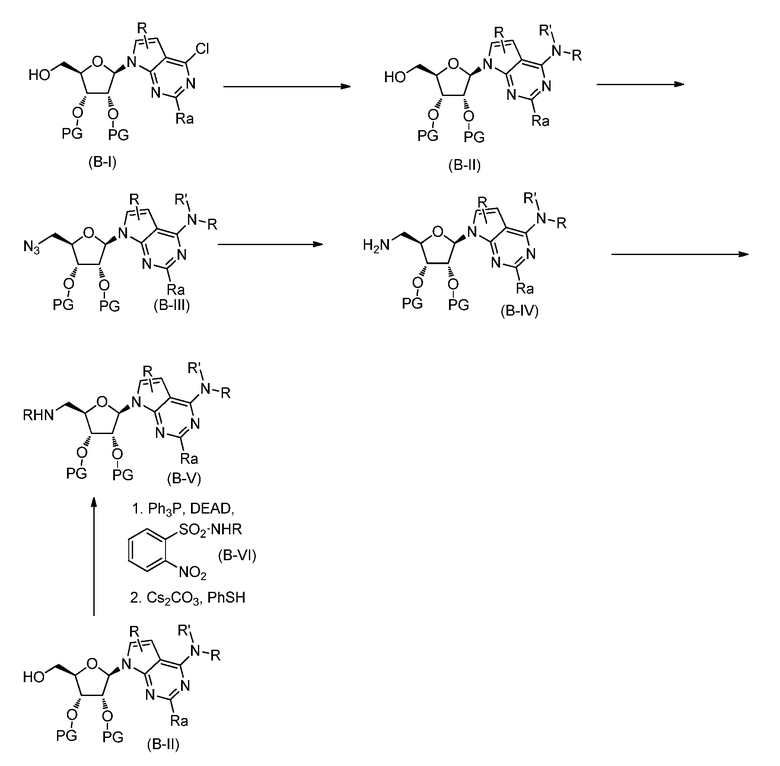
Промежуточные соединения 5’-амино-7-деазапурин-рибозы (B-V) можно синтезировать, как показано на Схеме B выше. Подходящее защищенное промежуточное соединение 7-деазапурин-рибозы, содержащее 6-хлор заместитель (B-I), можно преобразовать в соответствующее 6-амино-производное (B-II) через обработку подходящим амином (включая аммиак) в присутствии основания, такого как Et3N, K2CO3 или основание Хунига, в растворителе, таком как MeCN или DMF, ТГФ, iPrOH или их смесь. Если необходимо, реакционную смесь можно нагреть до 100°C (когда необходимая температура выше, чем температура кипения одного или нескольких компонентов в смеси, реакцию можно осуществить в герметично закрытой пробирке). R группы в схеме могут представлять алкильные защитные группы (например, 2, 4 диметоксибензил). 6-амино продукт (B-II) можно преобразовать в 5’-азидо промежуточное соединение (B-III) путем преобразования 5’-гидроксильной группы в удаляемую группу, такую как MsO, путем обработки при помощи MsCl в присутствии основания, такого как Et3N, пиридин или K2CO3, в инертном растворителе, таком как CH2Cl2, ТГФ, MeCN, DMF или их смесь. 5’-удаляемую группу затем замещают азидным анионом из азидного источника, такого как NaN3, в инертном растворителе, таком как DMF. Альтернативно, (B-II) можно непосредственно преобразовать в (B-III) путем обработки при помощи DPPA, Ph3P и DIAD в растворителе, таком как ТГФ. Азидо группу в (B-III) можно восстановить до первичного амина (B-IV) путем восстановления при помощи H2 в присутствии металлического катализатора (например, Pd/C, PtO2) или при помощи реакции Staudinger с фосфином, таким как Ph3P или PMe3. Первичный амин (B-IV) можно преобразовать во вторичный амин (B-V) путем обработки подходящим кетоном или альдегидом в присутствии подходящего восстановителя, такого как NaBH(OAc)3 или NaCNBH3. Могут быть добавлены дополнительные реагенты, такие как Ti(OiPr)4. Альтернативно 5’-гидрокси промежуточное соединение (B-II) можно обработать сульфонамидом (B-VI), DEAD и Ph3P в инертном растворителе, таком как ТГФ. Полученный сульфонамидный продукт затем можно обработать бензолтиолом в присутствии основания, такого как K2CO3, Cs2CO3, с получением вторичного амина (B-V). Эти последовательности реакций также могут быть применимы к производным ликсозы, исходя из (B-VII) с получением диастереоизомера с противоположной конфигурацией в 5’-положении
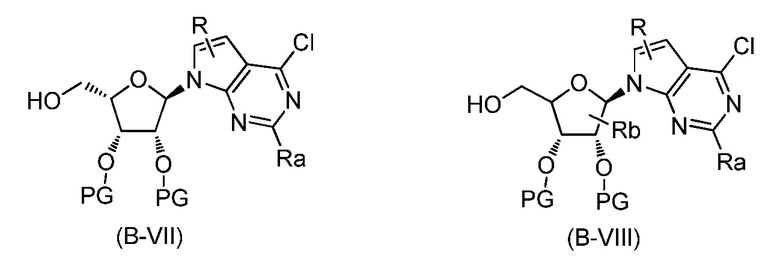
Подобный ряд последовательности реакций можно использовать для 2’-дезокси или 3’-дезокси или замещенной рибозы или ликсозы (B-VIII), показанной выше, с получением промежуточных соединений 5’-амино 7-деазапурин-рибозы/ликсозы.
Альтернативный способ для введения 6-NH2 группы, как показано ниже, включает обработку (B-IX) производных при помощи NaN3 с получением 6-азидо промежуточного соединения, с последующим восстановлением до NH2 группы (B-X) с использованием триалкилфосфина, такого как PMe3 или PPh3.
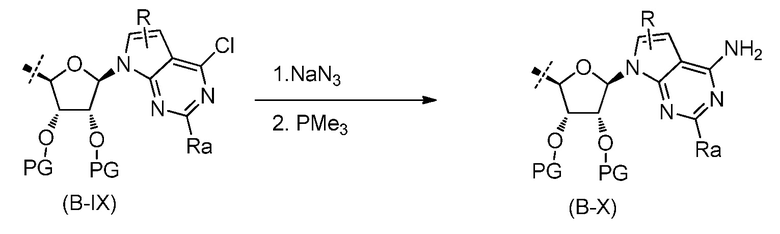
Схема C: Синтез пурин-карбоциклических промежуточных соединений
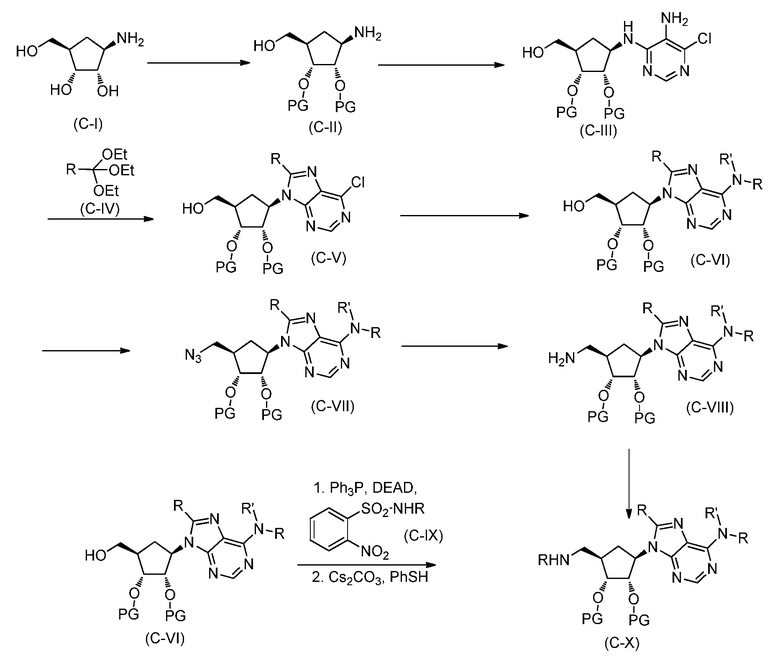
5’-аминопуриновые карбоциклические промежуточные соединения (C-X) можно получить, как показано на Схеме C. Циклопентан (C-I), необязательно, может быть защищен способами, известными рядовым специалистам в данной области, с получением соединения (C-II). (C-II) обрабатывают подходящим 4,6-дихлорпиримидин-5-амином в присутствии основания, такого как Et3N, в протонном растворителе, таком как н-бутанол. Реакционную смесь нагревают или подвергают условиям микроволнового облучения с получением промежуточного соединения (C-III). Пуриновое промежуточное соединение (C-V) получают путем обработки (C-III) ортоэфиром (C-IV) в присутствии кислоты, такой как AcOH. Реакционную смесь обычно нагревают. 6-амино заместитель можно ввести путем обработки подходящим амином (включая аммиак) в присутствии основания, такого как Et3N, K2CO3 или основание Хунига, в растворителе, таком как MeCN или DMF, ТГФ, iPrOH или их смесь. Если необходимо, реакционную смесь можно нагреть до 100°C (когда необходимая температура выше, чем температура кипения одного или нескольких компонентов в смеси, реакцию можно осуществить в герметично закрытой пробирке). R группы в схеме могут представлять алкильные защитные группы (например, 2, 4 диметоксибензил). 6-амино продукт (C-VI) можно преобразовать в 5’-азидо промежуточное соединение (C-VII) путем преобразования 5’-гидроксильной группы в удаляемую группу, такую как MsO, путем обработки при помощи MsCl в присутствии основания, такого как Et3N, пиридин или K2CO3, в инертном растворителе, таком как CH2Cl2, ТГФ, MeCN, DMF или их смесь. 5’-удаляемую группу затем замещают азидным анионом из NaN3 в инертном растворителе, таком как DMF. Альтернативно, (C-VI) можно непосредственно преобразовать в (C-VII) путем обработки при помощи DPPA, Ph3P и DIAD в растворителе, таком как ТГФ. Азидо группу в (C-VII) можно восстановить до первичного амина (C-VIII) путем восстановления при помощи H2 в присутствии металлического катализатора (например, Pd/C, PtO2) или при помощи реакции Staudinger с фосфином, таким как Ph3P или PMe3. Первичный амин (C-VIII) можно преобразовать во вторичный амин (C-X) путем обработки подходящим кетоном или альдегидом в присутствии подходящего восстановителя, такого как NaBH(OAc)3 или NaCNBH3. Могут быть добавлены дополнительные реагенты, такие как Ti(OiPr)4. Альтернативно, 5’-гидрокси промежуточное соединение (C-VI) можно обработать сульфонамидом (C-IX), DEAD и Ph3P в инертном растворителе, таком как ТГФ. Полученный сульфонамидный продукт затем можно обработать бензолтиолом в присутствии основания, такого как K2CO3, Cs2CO3, с получением вторичного амина (C-X).
Схема D: Синтез 7-деазапурин-карбоциклических промежуточных соединений
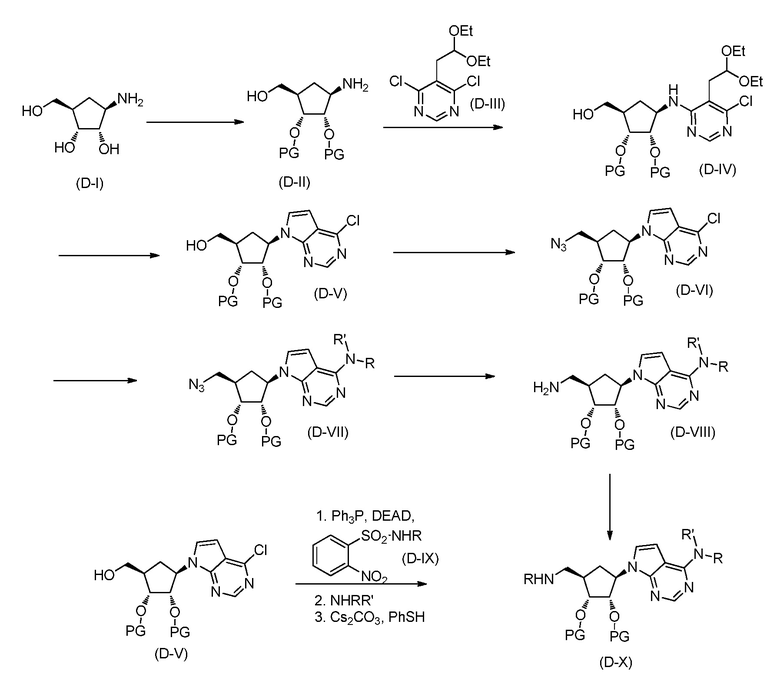
5’-амино 7-деазапуриновые карбоциклические промежуточные соединения (D-X) можно получить, как показано на Схеме D. Циклопентан (D-I), необязательно, может быть защищен способами, известными рядовым специалистам в данной области, с получением соединения (D-II). (D-II) обрабатывают подходящим 4,6-дихлорпиримидином (D-III) в присутствии основания, такого как Et3N, в протонном растворителе, таком как EtOH, н-бутанол. Реакционную смесь нагревают с получением промежуточного соединения (D-IV). Промежуточное соединение (D-V) получают путем обработки (D-IV) кислотой, такой как HCl или AcOH.
5’-гидроксильное соединение (D-V) можно преобразовать в 5’-азидо промежуточное соединение (D-VI) сначала путем преобразования 5’-гидроксильной группы в удаляемую группу, такую как MsO, путем обработки при помощи MsCl в присутствии основания, такого как Et3N, пиридин или K2CO3, в инертном растворителе, таком как CH2Cl2, ТГФ, MeCN, DMF или их смесь, и затем замещения удаляемой группы азидным анионом из NaN3 в инертном растворителе, таком как DMF. Альтернативно, (D-V) можно непосредственно преобразовать в (D-VI) путем обработки при помощи DPPA, Ph3P и DIAD в растворителе, таком как ТГФ.
6-Амино заместитель можно ввести путем обработки соединения (D-VI) подходящим амином (включая аммиак) в присутствии основания, такого как Et3N, K2CO3 или основание Хунига, в растворителе, таком как MeCN или DMF, ТГФ, iPrOH или их смесь. Если необходимо, реакционную смесь можно нагреть до 100°C (когда необходимая температура выше, чем температура кипения одного или нескольких компонентов в смеси, реакцию можно осуществить в герметично закрытой пробирке). R группы в схеме могут представлять алкильные защитные группы (например, 2, 4 диметоксибензил). Азидо группу в (D-VII) можно восстановить до первичного амина (D-VIII) путем восстановления при помощи H2 в присутствии металлического катализатора (например, Pd/C, PtO2) или при помощи реакции Staudinger с фосфином, таким как Ph3P или PMe3. Первичный амин (D-VIII) можно преобразовать во вторичный амин (D-X) путем обработки подходящим кетоном или альдегидом в присутствии подходящего восстановителя, такого как NaBH(OAc)3 или NaCNBH3. Могут быть добавлены дополнительные реагенты, такие как Ti(OiPr)4. Альтернативно, 5’-гидрокси промежуточное соединение (D-V) можно обработать сульфонамидом (D-IX), DEAD и Ph3P в инертном растворителе, таком как ТГФ. 6-Амино группу затем можно ввести с использованием условий, аналогичных тем, которые использовали для преобразования (D-VI) в (D-VII). Полученный сульфонамидный продукт затем можно обработать бензолтиолом в присутствии основания, такого как K2CO3, Cs2CO3, с получением вторичного амина (D-X).
Специалистам, имеющим среднюю квалификацию, должно быть понятно, что использование подходяще замещенного дихлорпиримидина (D-III) обеспечивает возможность замещения по 7-деазапуриновой группе.
Схема E: Инверсия стереохимии в 5’-положении
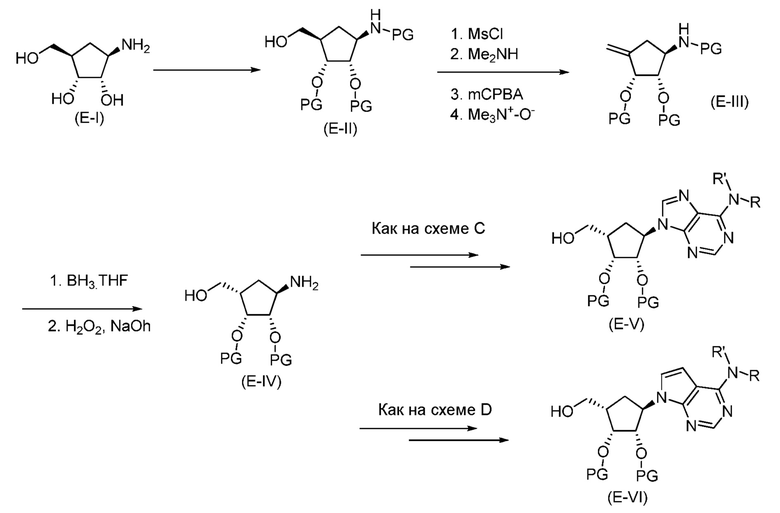
Для получения соответствующих промежуточных соединений противоположной стереохимии в 5’-положении можно использовать последовательность реакций, как показано на Схеме E выше. Циклопентан (E-I), необязательно, может быть защищен, после чего 5’-гидроксил преобразуют в удаляемую группу через обработку при помощи MsCl в присутствии Et3N в растворителе, таком CH2Cl2. 5’-удаляемую группу замещают группой Me2NH, путем обработки при помощи Me2NH в виде 2,0M раствора в ТГФ в герметично закрытой пробирке. Реакционную смесь нагревают до 40-80°C. Полученный третичный амин окисляют при помощи окислителя, такого как mCPBA, в растворителе, таком CH2Cl2, с получением соответствующего N-оксида. N-оксид затем нагревают при 50-120°C в инертном растворителе, таком N,N-диметилацетамид, с получением алкена (E-III). Алкен подвергают гиброборированию/окислительной обработке с получением инвертированного 5’ стереоизомера (E-IV). Подходящие реагенты для гиброборирования включают BH3-ТГФ, и подходящие условия окислительной обработки включают H2O2/NaOH. Промежуточное соединение (E-IV) затем можно подвергнуть последовательностям реакций, которые показаны на Схемах C и D, для получения промежуточных соединений (E-V) и (E-VI).
Схема F: 2’ и 3’ Дезоксициклопентаны и рибозы
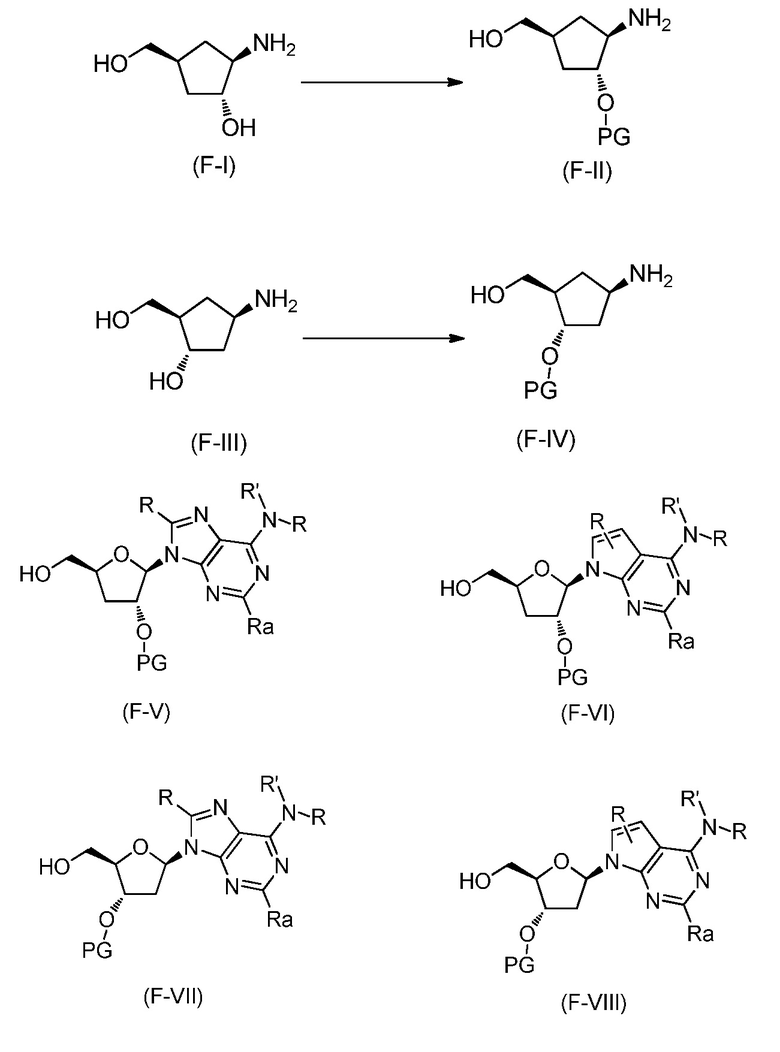
Использование промежуточных соединений (F-I) и (F-III) в процедурах, показанных на Схемах C и D, позволяет осуществить синтез пуриновых и 7-деазапуриновых дезоксикарбоциклических производных. Промежуточные соединения на основе рибозы (F-V), (F-VI), (F-VIII) и (F-IX) можно использовать в реакционных процедурах, подобных тем, которые показаны на Схемах A и B выше.
Схема G: Синтез циклобутанов
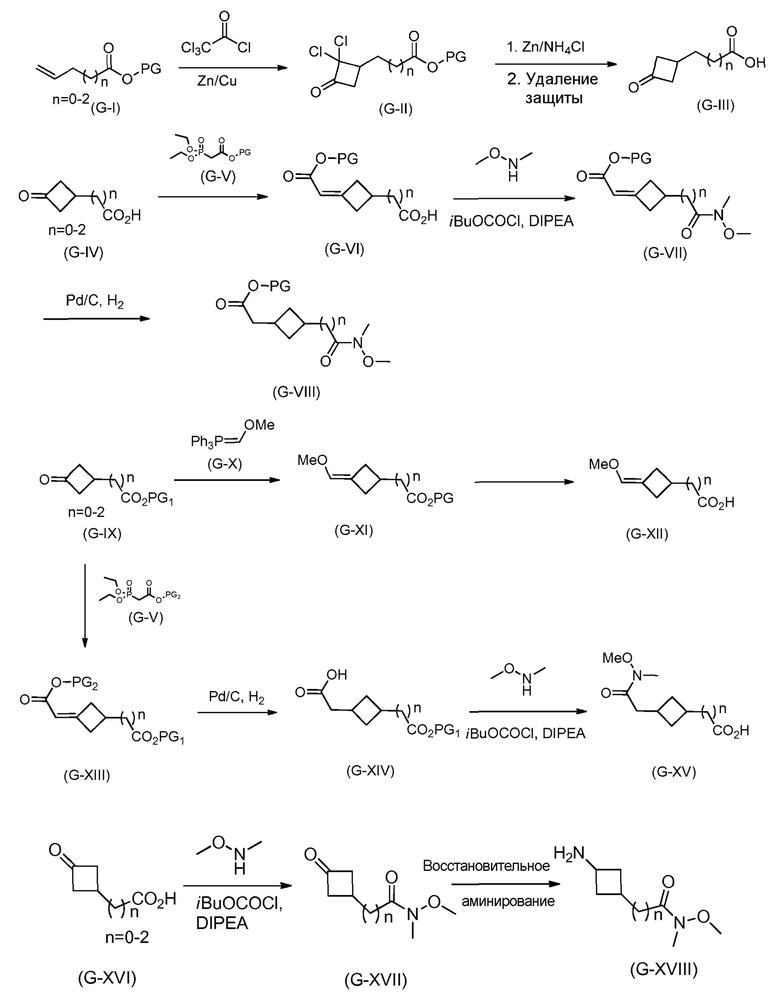
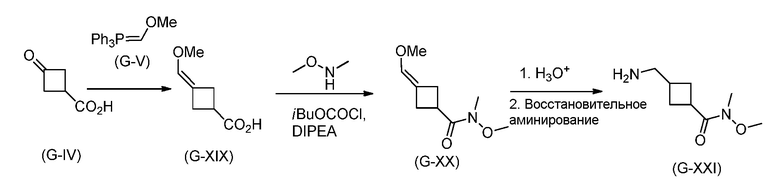
Циклобутаны формул (G-VIII), (G-XV) и (G-XXI) можно синтезировать, как показано на Схеме G. Алкениловые сложные эфиры (G-I) можно подвергнуть реакции [2+2] циклоприсоединения с трихлорацетилхлоридом в присутствии пары Zn/Cu в инертном растворителе, таком как Et2O, DME, ТГФ или их смесь. Альтернативно, реакцию [2+2] циклоприсоединения можно осуществить с использованием Zn пыли в условиях обработки ультразвуком. Дихлориды (G-II) восстанавливают через обработку Zn порошком в присутствии протонного донора, такого как NH4Cl, в растворителе, таком как MeOH. Циклобутаноны (G-IV) (которые включают (G-III)) можно далее переработать путем обработки фосфонатом (G-V) с получением α, β ненасыщенных сложных эфиров (G-VI). Кислоту (VI) преобразуют в амид Вайнреба (G-VII) в стандартных условиях (например, изобутилхлорформиат, основание Хунига, N,O-диметилгидроксиламин). Двойную связь затем можно восстановить через гидрирование с использованием H2 в присутствии металлического катализатора, такого как Pd/C, PtO2 или Pd(OH)2, с получением циклобутановых промежуточных соединений (G-VIII).
Циклобутаноны (G-IX) можно обработать реагентом Виттига (G-X) с получением циклобутаненолового простого эфира (G-XI), который после удаления защиты дает соответствующую кислоту (G-XII).
Циклобутаноны (G-IX) также можно обработать стабилизированным фосфонатом (G-V) в присутствии основания, такого как KOtBu, LDA, NaHMDS, KHMDS или LiHMDS, или обработать при помощи Et3N в присутствии LiCl в инертном растворителе с получением α, β ненасыщенного сложного эфира (G-XIII), который можно восстановить до (G-XIV) путем обработки при помощи H2 в присутствии металлического катализатора, такого как Pd/C, Pd(OH)2 или PtO2, в инертном растворителе. Кислотную функциональную группу в (G-XIV) можно преобразовать в соответствующий амид Вайнреба путем обработки N,O-диметилгидроксиламином в присутствии подходящего связывающего агента, такого как изобутилхлорформиат, и основания, такого как основание Хунига, с получением соединения (G-XV).
Циклобутаноны (G-XVI) также можно обработать N,O-диметилгидроксиламином в присутствии подходящего связывающего агента, такого как изобутилхлорформиат, и основания, такого как основание Хунига, с получением соответствующего амида Вайнреба (G-XVII), который после восстановительного аминирования эквивалентом аммиака с последующим удалением защиты, при необходимости, дает амин (G-XVIII). Подходящие эквиваленты аммиака включают бензгидриламин, NH3, NH4Cl, BnNH2, PMB-NH2, 2,4 DMB-NH2, которые можно обработать кетоном (G-XVII) и подходящим восстановителем, таким как NaCN(BH3) или Na(OAc)3BH, в присутствии кислоты, если необходимо, такой как HCl или AcOH. Защитные группы в продуктах восстановительного аминирования можно удалить способами, известными рядовым специалистам в данной области. Альтернативно, кетон (G-XVII) можно обработать гидроксиламином с образованием соответствующего оксима, который затем можно восстановить при помощи H2 в присутствии металлического катализатора, такого как Pd/C, PtO2 или Pd(OH)2, с получением промежуточного соединения (G-XVIII).
Циклобутан (G-IV) можно преобразовать в амин (G-XXI) через несколько последовательно осуществляемых стадий, включающих обработку (G-IV) фосфораном (G-V) с получением енольного простого эфира (G-XIX). Обработка соединения (G-XIX) N,O-диметилгидроксиламином в присутствии подходящего связывающего агента, такого как изобутилхлорформиат, и основания, такого как основание Хунига, дает соответствующий амид Вайнреба (G-XX), который после водного гидролиза енольного простого эфира (например, TsOH/H2O, HCl/H2O) и восстановительного аминирования эквивалентом аммиака с последующим удалением защиты, при необходимости, дает амин (G-XXI). Подходящие эквиваленты аммиака включают бензгидриламин, NH3, NH4Cl, BnNH2, PMB-NH2, 2,4 DMB-NH2. Подходящие восстановители для восстановительного аминирования включают NaCN(BH3) или Na(OAc)3BH, используемый в присутствии кислоты, если необходимо, такой как HCl или AcOH. Защитные группы в продуктах восстановительного аминирования можно удалить способами, известными рядовым специалистам в данной области.
Схема H: Преобразование циклобутанов
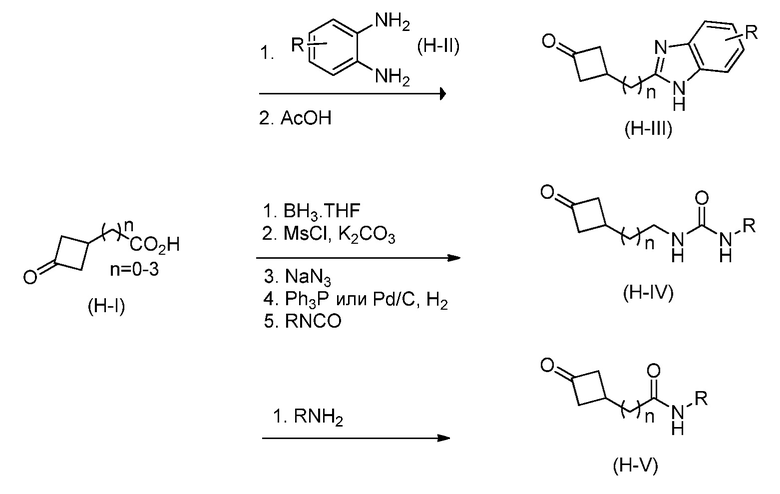
Циклобутаноны (H-I) можно преобразовать в бензимидазолы (H-III), мочевины (H-IV) и амиды (H-V) через последовательности реакций, показанные на Схеме H. Бензимидазолы (H-III) могут быть образованы путем обработки кислот (H-I) подходящим бензолдиамином (H-II) в присутствии подходящего связывающего агента (например, HATU, PPAA, COMU, EDC, EDCI), в присутствии основания (например, Et3N, основание Хунига, K2CO3). Могут быть добавлены дополнительные реагенты, такие как HOAT, HOBt или HOSu, при необходимости. Полученные амино-амиды затем подвергают циклизации до бензимидазолов в присутствии кислоты, например, AcOH, которая также может служить в качестве растворителя. Реакцию обычно осуществляют при температурах в пределах от комнатной температуры до 80°C. Мочевины (H-IV) можно получить путем преобразования кислоты (H-I) следующим образом. Кислоту восстанавливают до соответствующего первичного спирта с использованием реагента, такого как LiAlH4 или BH3.ТГФ. При необходимости, функциональную группу кетона сначала можно защитить (например, в виде кеталя) до восстановления и затем удалить защиту. Первичный спирт затем преобразовывают в удаляемую группу, такую как мезилат. Полученную удаляемую группу замещают азидом из источника, такого как NaN3. Продукт, представляющий собой азидосоединение, восстанавливают до соответствующего аминосоединения с использованием H2 в присутствии металлического катализатора, такого как Pd/C, или через реакцию Staudinger с фосфином, таким как PMe3 или PPh3. Мочевину (H-IV) затем получают путем обработки первичного амина подходящим изоцианатом R-C=N=O в присутствии основания, такого как Et3N или K2CO3, в инертном растворителе, таком как CH2Cl2. Амиды (H-V) получают путем обработки кислоты (H-I) подходящим амином R-NH2 в присутствие подходящего связывающего агента (например, HATU, PPAA, COMU, EDC, EDCI), в присутствии основания (например, Et3N, основание Хунига, K2CO3). Могут быть добавлены дополнительные реагенты, такие как HOAT, HOBt или HOSu, при необходимости.
Схема I: Преобразование циклобутан амидов Вайнреба
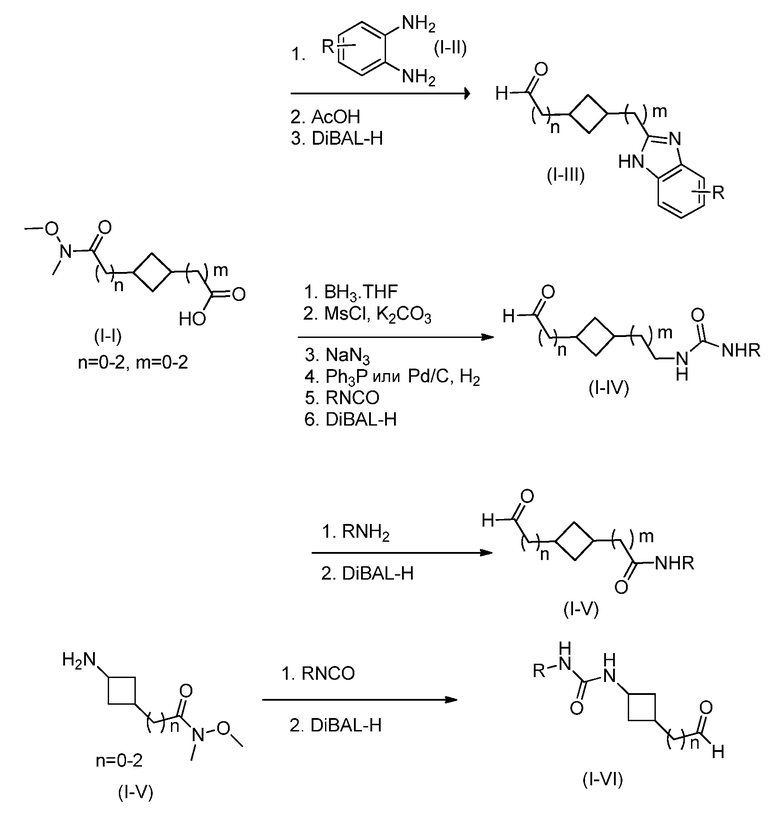
Как показано на Схеме I выше, амиды Вайнреба (I-I) и (I-V) можно преобразовать в бензимидазолальдегиды (I-III), мочевино-альдегидные соединения (I-IV) и (I-VI) и амидальдегиды (I-V) с использованием процедур, аналогичных описанным для Схемы H, с последующим восстановлением амида Вайнреба. Восстановление амида Вайнреба можно осуществить с использованием DiBAL-H в инертном растворителе, таком как CH2Cl2, ТГФ, при температуре от 10 до -78°C. Аминоамид Вайнреба (I-V) также можно преобразовать в мочевину (I-VI) через обработку подходящим изоцианатом, с последующим восстановлением с использованием DiBAL-H.
Схема J: Преобразование циклобутана
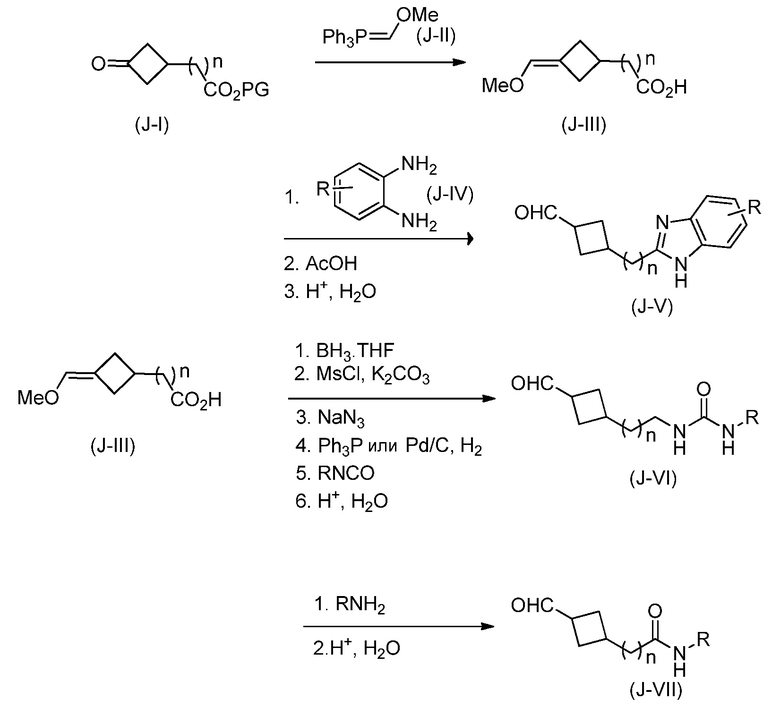
Бензимидазольные (J-V), мочевинные (J-VI) и амидные (J-VII) промежуточные соединения можно синтезировать, как показано на Схеме J. Циклобутаноны (J-I) можно подвергнуть реакции Виттига с фосфораном (J-II) с получением енольного простого эфира (J-III). Кислотное енолэфирное соединение затем подвергают реакционным условиям, подобным тем, которые описаны для схемы H, с получением соответствующих енолэфирных бензимидазолов, мочевин и амидов, которые при обработке водным раствором кислоты, такой как TsOH/H2O, HCl, образуют соответствующие альдегидные промежуточные соединения (J-V), (J-VI) и (J-VII), соответственно.
Схема K: Взаимодействие циклобутанов с аминами
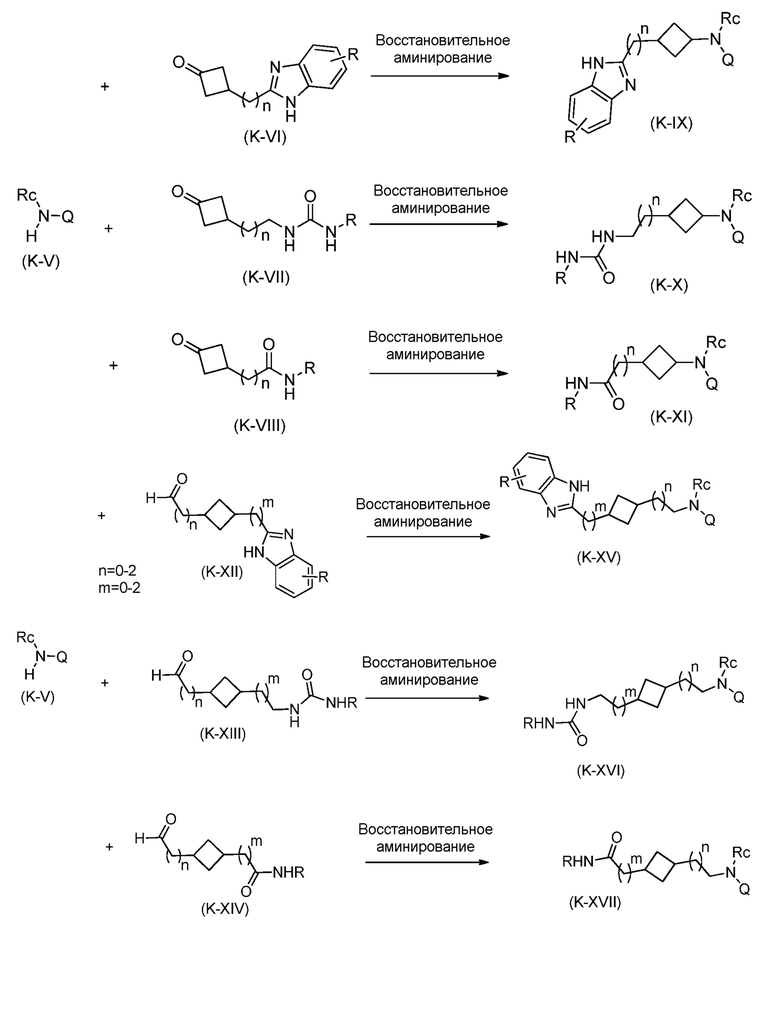
Формула (K-V) 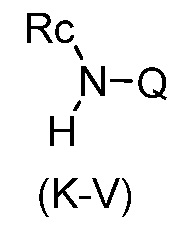 на Схеме K выше представляет промежуточные соединения (K-I - K-IV), показанные ниже, и их соответствующие 2'- или 3'-дезокси промежуточные соединения, синтез которых описан на Схемах A-F.
на Схеме K выше представляет промежуточные соединения (K-I - K-IV), показанные ниже, и их соответствующие 2'- или 3'-дезокси промежуточные соединения, синтез которых описан на Схемах A-F.
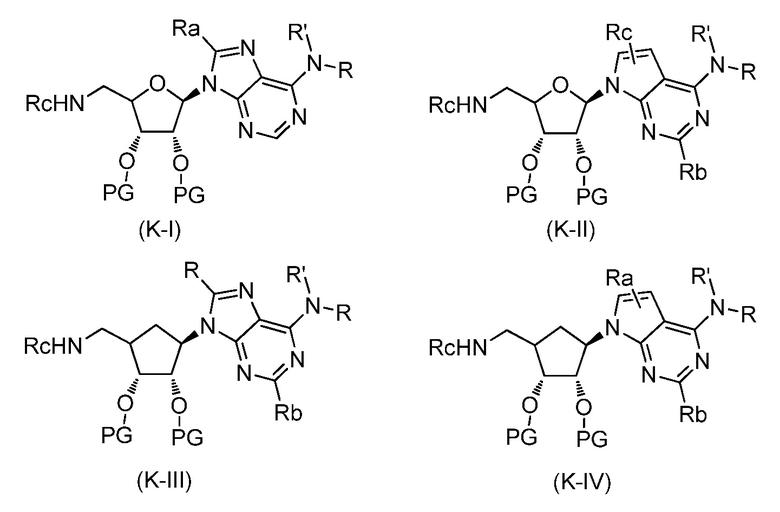
Как показано на Схеме K, кетоны (K-VI), (K-VII) и (K-VIII) и альдегиды (K-XII), (K-XIII) и (K-XIV) преобразовывают в соответствующие бензимидазолы (K-IX) и (K-XV), мочевины (K-X) и (K-XVI) и амиды (K-XI) и (K-XVII) через восстановительное аминирование при помощи (K-V). Восстановительное аминирование можно осуществить с использованием подходящего восстановителя, такого как NaCN(BH3) или Na(OAc)3BH, в присутствии кислоты, если необходимо, такой как HCl или AcOH, или кислоты Льюиса/агента дегидратации, такого как Ti(OiPr)4 или MgSO4.
Схема L: Альтернативное взаимодействие
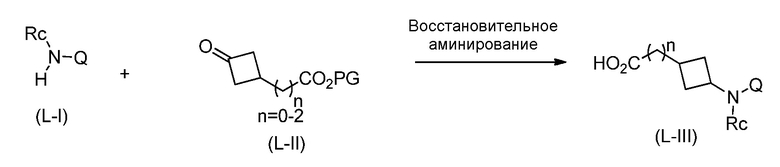
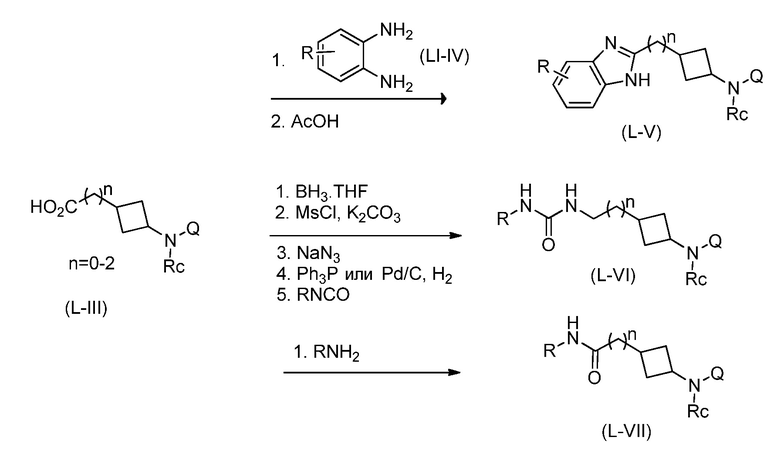
В альтернативной последовательности реакций, целевые бензимидазолы (L-V), мочевины (L-VI) и амиды (L-VII) можно получить путем осуществления последовательности реакций, показанной на Схеме L. Амины (L-I), где Q и RC имеют значения, определенные для Схемы K, обрабатывают циклобутанами (L-II) в условиях восстановительного аминирования с использованием восстановителя, такого как NaCNBH3 или Na(OAc)3BH, в присутствии кислоты, такой как HCl или AcOH, или кислоты Льюиса, такой как Ti(OiPr)4, с получением кислоты (L-III). Эту кислоту можно преобразовать в соответствующие целевые бензимидазолы (L-V), мочевины (L-VI) и амиды (L-VII) с использованием реакционных условий, аналогичных тем, которые показаны на Схеме H.
Схема M: Альтернативное взаимодействие
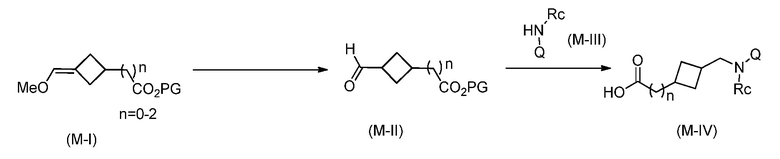
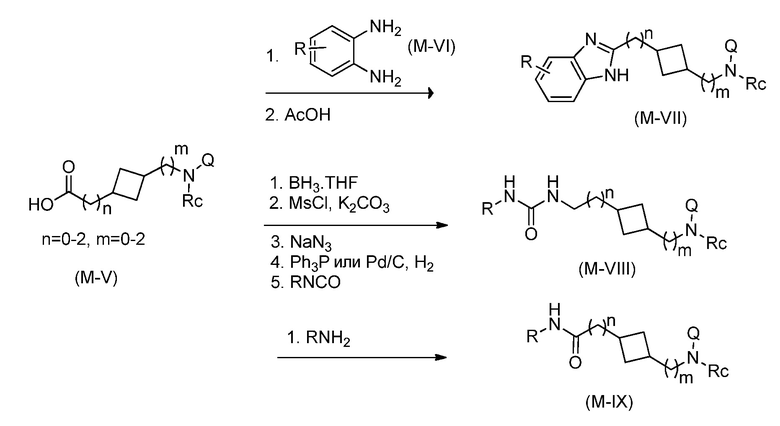
Енольные эфиры (M-1) могут быть гидролизованы в кислотных условиях (например, TsOH/H2O, HCl/H2O) с получением альдегидов (M-II). Восстановительное аминирование соединения (M-II) осуществляют с использованием аминов (M-III), где Q и RC имеют значения, определенные для схемы K, с использованием восстановителя, такого как NaCNBH3 или Na(OAc)3BH, в присутствии кислоты, такой как HCl или AcOH, или кислоты Льюиса/агенты дегидратации, такого как Ti(OiPr)4 или MgSO4. Затем осуществляют удаление сложноэфирной защитной группы с получением кислоты (M-IV). Кислоты (M-V) (которые включают кислоты (M-IV)) можно преобразовать в соответствующие целевые бензимидазолы (M-VII), мочевины (M-VIII) и амиды (M-IX) с использованием реакционных условий, подобных тем, которые описаны для схемы H.
Схема N: Синтез амидов
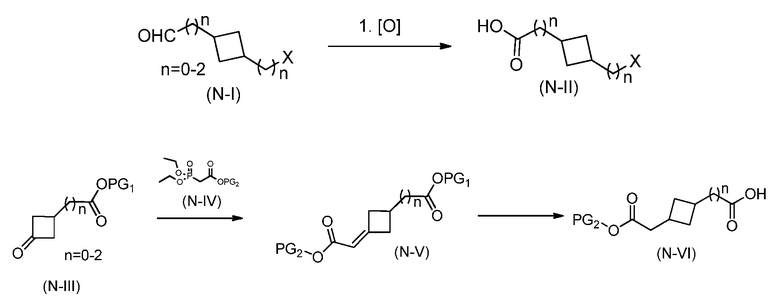
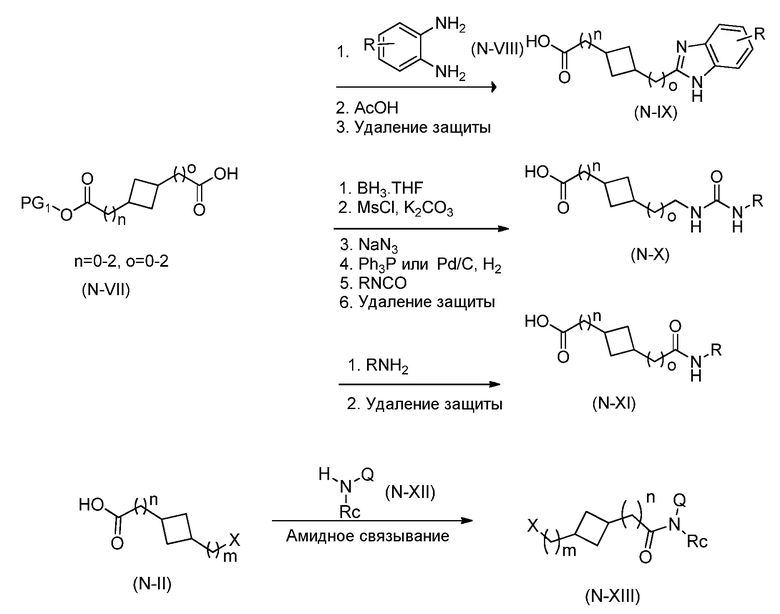
Бензимидазолы, мочевины и амиды формулы (N-XIII) можно синтезировать, как показано на Схеме N. Альдегид (N-I), где X представляет собой бензимидазольную, мочевинную или амидную функциональную группу, можно преобразовать в соответствующую кислоту через окисление с использованием реагента, такого как NaClO2. Циклобутаноны формулы (N-III) можно обработать фосфонатами формулы (N-IV) и подходящим основанием, таким как LiHMDS, NaHMDS, KHMDS, KOtBu, LDA или Et3N/LiCl, в инертном растворителе с получением циклобутанов (N-V), которые в результате восстановления через обработку при помощи H2 в присутствии подходящего металлического катализатора, такого как Pd/C, PtO2 или Pd(OH)2, дают кислоту (N-VI).
Кислоты формулы (N-VII), которые включают кислоты формулы (N-VI), можно преобразовать в соответствующие бензимидазолы (N-IX), мочевины (N-X) и амиды (N-XI) через серию реакций, подобных тем, которые показаны на схеме H. Бензимидазолы, мочевины и амиды формулы (N-II) затем можно преобразовать в бензимидазолы, мочевины и амиды формулы (N-XIII) через реакцию амидного связывания с амином (N-XIII), где Rc и Q имеют значения, определенные для Схемы K, с использованием подходящего связывающего агента (например, HATU, PPAA, COMU, EDC, EDCI), в присутствии основания (например, Et3N, основание Хунига, K2CO3). Могут быть добавлены дополнительные реагенты, такие как HOAT, HOBt или HOSu, при необходимости.
Схема O: 5’-Сера-содержащие аналоги
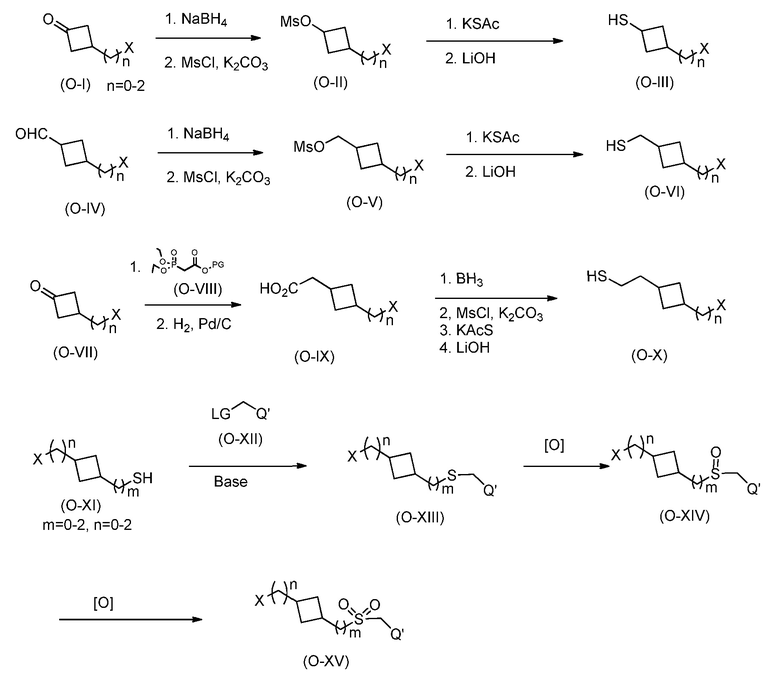
Тиоэфиры (O-XIII), сульфоксиды (O-XIV) и сульфоны (O-XV) можно синтезировать, как показано на Схеме O. Циклобутаноны (O-I), где X представляет собой бензимидазольную, мочевинную или амидную функциональную группу, можно восстановить с использованием восстановителя, такого как NaBH4, с получением соответствующего спирта, который, в свою очередь, можно преобразовывать в удаляемую группу, такую MsO, путем обработки при помощи MsCl, с использованием основания, такого как K2CO3, в инертном растворителе. Циклобутан (O-II) затем преобразовывают в соответствующий тиол (O-III) сначала путем обработки нуклеофильным соединением на основе серы, таким как KSAc, с последующим гидролизом сложного тиоэфира в щелочных условиях, например, LiOH/H2O/MeOH.
Альдегиды (O-IV) можно преобразовать в соответствующие тиолы через последовательность реакций, подобную той, которая описана для преобразования (O-I) в (O-III).
Циклобутаноны (O-VII) можно преобразовать в кислоты (O-IX) путем обработки фосфонатом формулы (O-VIII) в присутствии подходящего основания (Cs2CO3, K2CO3, LiHMDS, NaHMDS, KHMDS, KOtBu, LDA), с последующим восстановлением с использованием H2, в присутствии подходящего металлического катализатора, такого как Pd/C, PtO2 или Pd(OH)2. Кислоту селективно восстанавливают до первичного спирта с использованием реагента, такого как BH3:ТГФ, или путем взаимодействия с карбонилдиимидазолом, Et3N и затем NaBH4. Первичный спирт преобразуют в соответствующий тиол с использованием реакции, подобных тем, которые использовали для синтеза (O-III) и (O-VI) из их вторичных спиртовых промежуточных соединений.
Тиолы (O-XI) затем можно обработать промежуточными соединениями (O-XII), где Q’ представляет собой промежуточные соединения (O-XVI), (O-XVII), (O-XVIII) и (O-XIX), показанные ниже (и также соответствующие 2'- или 3'-дезокси промежуточным соединениям), и LG представляет собой удаляемую группу, такую как Cl, TfO или MsO. Реакцию можно осуществить в присутствии подходящего основания, такого как K2CO3, Cs2CO3, Et3N или основание Хунига, с получением тиоэфиров (O-XIII), которые можно преобразовать в соответствующие сульфоксиды (O-XIV) или сульфоны (O-XV) через обработку подходящими окислителями, такими как H2O2 или mCPBA.
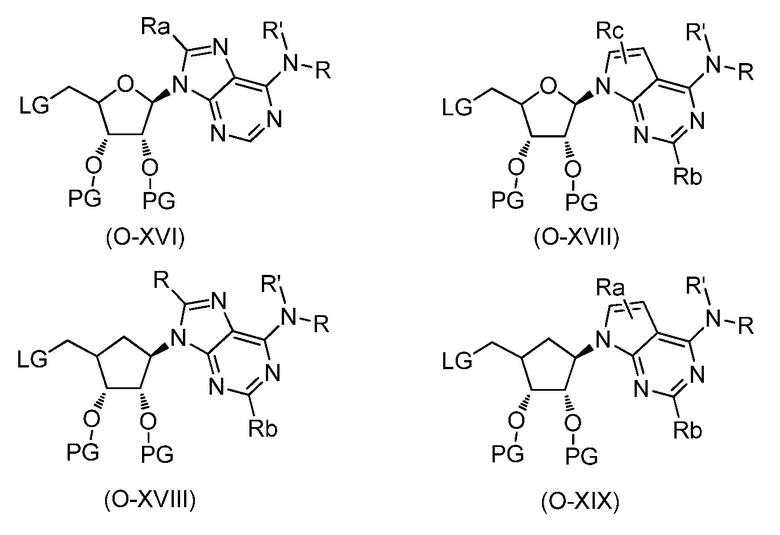
Схема P: Аминовые, амидные и сульфонамидные кэппирующие группы
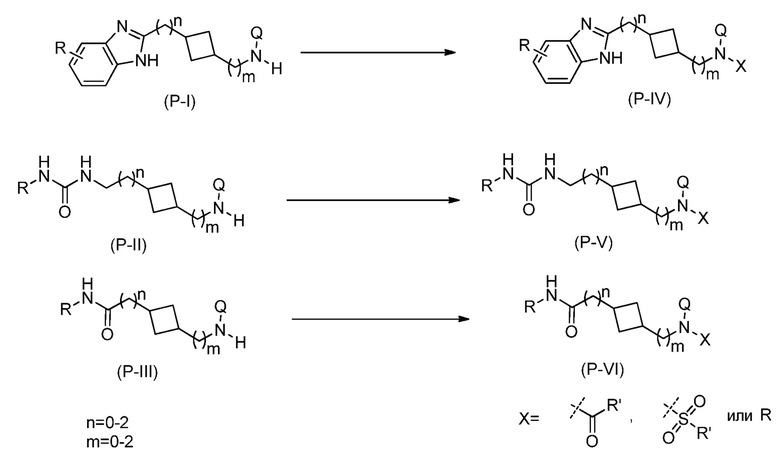
Аминовые, амидные и сульфонамидные целевые молекулы можно синтезировать из аминов (P-I), (P-II) и (P-III) с использованием стандартных реакционных условий. Амидные целевые молекулы можно получить путем обработки аминов подходящей карбоновой кислотой в присутствии подходящего связывающего агента (например, HATU, PPAA, COMU, EDC, EDCI), в присутствии основания (например, Et3N, основание Хунига, K2CO3). Могут быть добавлены дополнительные реагенты, такие как HOAt, HOBt или HOSu, при необходимости. Сульфонамидные целевые молекулы можно получить путем обработки аминов подходящим сульфонилхлоридом в присутствии основания, такого как K2CO3 или Et3N. Аминовые целевые молекулы можно получить путем восстановительного аминирования с использованием подходящего альдегида или кетона в присутствии подходящего восстановителя, такого как NaBH(OAc)3 или NaCNBH3. Могут быть добавлены дополнительные реагенты, такие как кислоты AcOH или HCl, или кислота Льюиса/агенты дегидратации Ti(OiPr)4 или MgSO4.
Схема Q: Получение 4-(1-метокси-2-метилпропан-2-ил)бензол-1,2-диамина
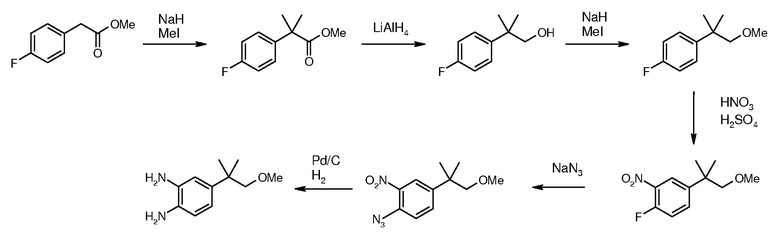
Схема R: Получение 4-циклобутилбензол-1,2-диамина
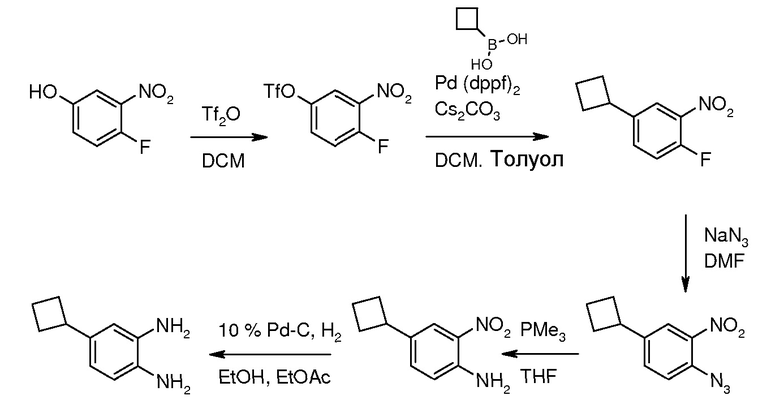
Схема S: Получение 1-(3,4-диаминофенил)циклобутанкарбонитрила
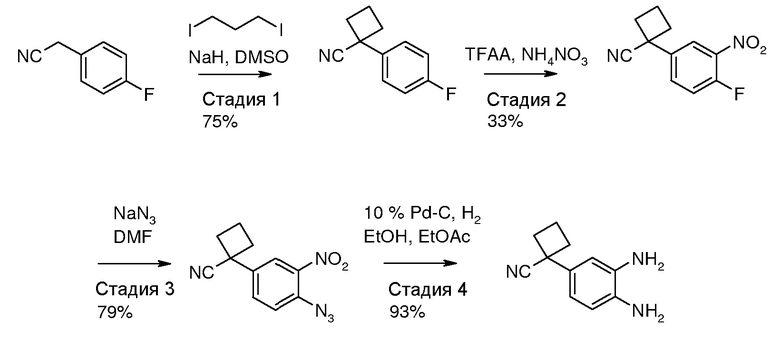
Схема T: Получение 4-циклопропилбензол-1,2-диамина
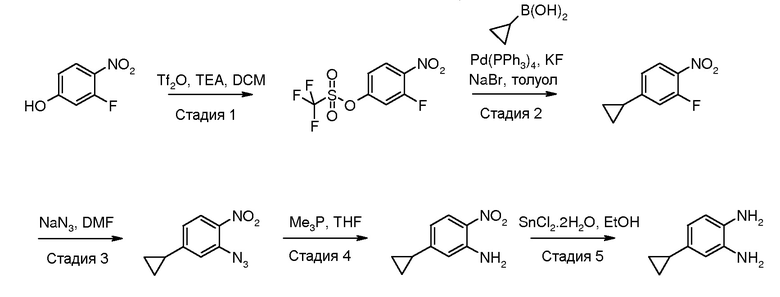
Схема U: Получение 1-(3,4-диаминофенил)циклопропанкарбонитрила

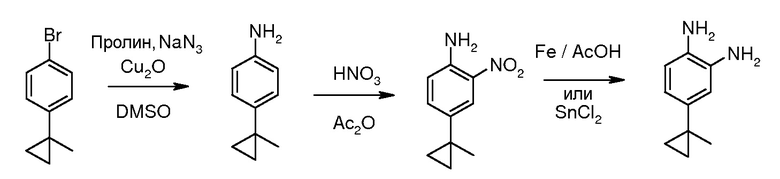
Схема V: Получение 4-(2,2,2-трифторэтил)бензол-1,2-диамина
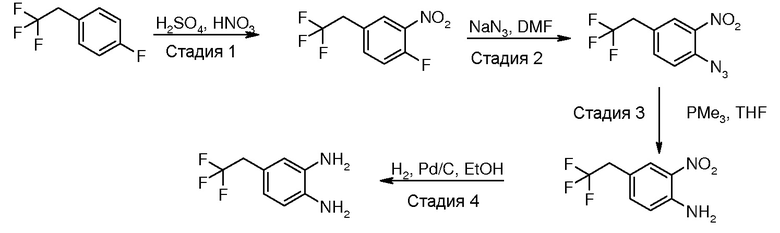
Схема W: Получение 2-(3,4-диаминофенил)-2-метилпропаннитрила
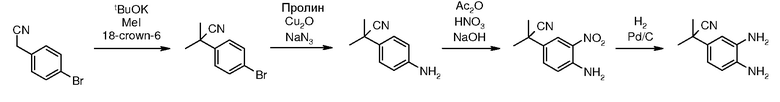
В различных разделах настоящего описания, где композиции описаны как содержащие, включающие или состоящие из конкретных компонентов, или где способы описаны как содержащие, включающие или состоящие из конкретных технологических операций, предполагается, что композиции по настоящему изобретению также состоят по существу из, или состоят из, указанных компонентов, и что способы по настоящему изобретению также состоят по существу из, или состоят из, указанных технологических операций. Кроме того, должно быть понятно, что порядок стадий или порядок осуществления определенных действий не имеет значения, при условии, что настоящее изобретение является осуществимым. Более того, две или более стадий или действий можно осуществлять одновременно.
Соединения, полученные, выбранные и/или оптимизированные способами, описанными выше, после их получения могут быть охарактеризованы с использованием различных анализов, которые известны специалистам в данной области как определяющие, обладают или нет соединения биологической активностью. Например, молекулы могут быть охарактеризованы с использованием традиционных анализов, включая, но не ограничиваясь этим, анализы, которые описаны ниже, для определения обладают они или нет предполагаемой активностью, активностью связывания и/или специфичностью связывания.
Кроме того, можно использовать высокопродуктивный скрининг для ускорения анализа с использованием таких методов. Как результат, возможно быстрое скринирование молекул, описанных в настоящей заявке, на активность с использованием методов, известных из уровня техники. Общие методики для осуществления высокопродуктивного скрининга описаны, например, в Devlin (1998) High Throughput Screening, Marcel Dekker; и в Патенте США № 5763263. В высокопродуктивных анализах можно использовать один или несколько различных методов оценки, включая, но не ограничиваясь этим, методы, описанные в настоящей заявке.
Для дополнительной оценки лекарственных свойств соединения, измерения активности ингибирования ферментов цитохром P450 и фермента, метаболизирующего фазу II, также можно осуществить либо с использованием рекомбинантных ферментных систем человека, либо более сложных систем, таких как микросомы печени человека. Кроме того, соединения также могут быть оценены как субстраты для активности этих метаболических ферментов. Эти активности являются полезными для определения потенциала соединения вызывать межлекарственные взаимодействия или образовывать метаболиты, которые сохраняют или не обладают полезной антимикробной активностью.
Для оценки потенциала соединения быть перорально биодоступным, необходимо также осуществить анализы растворимости и Caco-2. Последний представляет собой клеточную линию из человеческого эпителия, которая делает возможным измерение поглощения лекарственного средства и его прохождения через Caco-2 клеточный монослой, часто растущий в лунках 24-луночного микротитровального планшета, снабженного 1-микронной мембраной. Концентрации свободного лекарственного средства можно измерить на бозолатеральной стороне монослоя, определяя количество лекарственного средства, которое может проходить через интестинальный монослой. Необходимы подходящие контроли для обеспечения целостности монослоя и плотности щелевых контактов. С использованием этой же самой системы можно определить P-гликопротеин-опосредованный отток. P-гликопротеин представляет собой насос, который относится к апикальной мембране клетки, образуя поляризованные монослои. Этот насос упраздняет активное или пассивное поглощение через Caco-2 клеточную мембрану, приводя к тому, что меньшее количество лекарственного средства проходит через слой кишечного эпителия. Эти результаты часто получают вместе с измерениями растворимости, и известно, что оба эти фактора способствуют пероральной биодоступности у млекопитающих. Путем измерения пероральной биодоступности у животных и, в конечном счете, у человека с использованием традиционных фармакокинетических экспериментов можно определить абсолютную пероральную биодоступность.
Результаты экспериментов также можно использовать для построения моделей, которые помогают предсказать физико-химические параметры, которые полезны для получения лекарственных свойств. Когда такая модель принята, экспериментальную методологию можно уменьшить, больше полагаясь при этом на предсказуемость модели.
3. Способы лечения
Лейкоз смешанного происхождения (MLL) представляет собой генетически особую форму острого лейкоза, которая составляет более 70% детских лейкозов и приблизительно 10% острых миелогенных лейкозов взрослых (AML) (Hess, J. L. (2004), Trends Mol Med 10, 500-507; Krivtsov, A. V. and Armstrong, S. A. (2007), Nat Rev Cancer 7, 823-833). MLL представляет особенно агрессивную форму лейкоза, и пациенты с этим заболеванием, как правило, имеют плохие прогнозы; эти пациенты часто страдают от раннего рецидива после лечения существующими химиотерапевтическими средствами. Таким образом, существует настоятельная потребность в новых способах терапевтического воздействия на пациентов, страдающих MLL.
Универсальным признаком MLL заболевания являеься хромосомная транслокация, затрагивающая MLL ген на хромосоме 11q23 (Hess, 2004; Krivtsov and Armstrong, 2007). Обычно, MLL ген кодирует SET-домен гистонметилтрансферазу, которая катализирует метилирование лизина 4 гистона H3 (H3K4) в специфических локусах (Milne et al. (2002) Mol Cell 10, 1107-1117; Nakamura et al. (2002), Mol Cell 10, 1119-1128). Локализация гена сообщается посредством специфических взаимодействий с распознающими элементами в MLL, внешними для SET-домена (Ayton et al. (2004) Mol Cell Biol 24, 10470-10478; Slany et al., (1998) Mol Cell Biol 18, 122-129; Zeleznik-Le et al. (1994) Proc Natl Acad Sci U S A 91, 10610-10614). В связанных с заболеванием транслокациях, каталитический SET-домен утрачивается, и оставшийся MLL белок гибридизуется с рядом партнеров, в том чисте членами AF и ENL семейства белков, такими как AF4, AF9, AF10 и ENL (Hess, 2004; Krivtsov and Armstrong, 2007; Slany (2009) Haematologica 94, 984-993). Эти гибридизующиеся партнеры способны к взаимодействию непосредственно или опосредованно, с другой гистонметилтрансферазой, DOT1L (Bitoun et al. (2007) Hum Mol Genet 16, 92-106; Mohan et al. (2010) Genes Dev. 24, 574-589; Mueller et al. (2007) Blood 110, 4445-4454; Mueller et al. (2009) PLoS Biol 7, e1000249; Okada et al. (2005) Cell 121, 167-178; Park et al. (2010) Protein J 29, 213-223; Yokoyama et al. (2010) Cancer Cell 17, 198-212; Zhang et al. (2006) J Biol Chem 281, 18059-18068). Как результат, продукты транслокации сохраняют ген-специфическое распознавание элементов в остальной части MLL белка, но также приобретают способность рекрутинга DOT1L, к этим участкам (Monroe et al. (2010) Exp Hematol. 2010 Sep18. [Epub ahead of print] Pubmed PMID: 20854876; Mueller et al., 2007; Mueller et al., 2009; Okada et al., 2005). DOT1L катализирует метилирование H3K79, модификацию хроматина, связанную с активно транскрибируемыми генами (Feng et al. (2002) Curr Biol 12, 1052-1058; Steger et al. (2008) Mol Cell Biol 28, 2825-2839). Метилирование эктопического H3K79, которое является результатом осуществляемого MLL гибридным белком рекрутинга DOT1L, приводит к повышенной экспрессии лейкемогенных генов, включая HOKCA9 и MEIS1 (Guenther et al. (2008) Genes & Development 22, 3403-3408; Krivtsov et al. (2008) Nat Rev Cancer 7, 823-833; Milne et al. (2005) Cancer Res 65, 11367-11374; Monroe et al., 2010; Mueller et al., 2009; Okada et al., 2005; Thiel et al.(2010) Cancer Cell 17, 148-159). Следовательно, хотя DOT1L не является генетически измененным в заболевании per se, смещение направленности его ферментативной активности является прямым последствием хромосомной транслокации, имеющей место у MLL пациентов; таким образом, было сделано предположение, что DOT1L является каталитическим драйвером возникновения и развития лейкоза в этом заболевании (Krivtsov et al., 2008; Monroe et al., 2010; Okada et al., 2005; Yokoyama et al. (2010) Cancer Cell 17, 198-212). Дальнейшее подтверждение патогенной роли DOT1L в MLL получают в модельных системах, которые демонстрируют необходимость DOT1L в распространении трансформирующей активности MLL гибридных белков (Mueller et al., 2007; Okada et al., 2005).
Данные показывают, что ферментативная активность DOT1L является критической для патогенеза в MLL, и ингибирование DOT1L может обеспечить фармакологическую основу для терапевтического вмешательства в это заболевание. Лечение соединениями приводит к селективному зависимому от концентрации лизису лейкозных клеток с MLL-транслокацией, без какого-либо эффекта на клетки, не трансформированные MLL. Анализ генной экспрессии обработанных ингибитором клеток показывает даун-регуляцию генов, аберрантно чрезмерно экспрессируемых в MLL-реаранжированных лейкозах, и картины, сходные с изменениями генной экспрессии, которые вызваны генетическим нокаутом Dot1L гена в мышиной модели MLL-AF9 лейкоза.
Настоящее изобретение обеспечивает способы лечения клеточно-пролиферативного расстройства у субъекта, нуждающегося в этом, путем введения субъекту, нуждающемуся в таком лечении, терапевтически эффективного количества соединения по настоящему изобретению или его фармацевтически приемлемой соли, пролекарства, метаболита, полиморфа или сольвата. Клеточно-пролиферативное расстройство может представлять собой рак или предраковое состояние. Настоящее изобретение, кроме того, обеспечивает применение соединения по настоящему изобретению или его фармацевтически приемлемой соли, пролекарства, метаболита, полиморфа или сольвата для получения лекарственного средства, полезного для лечения клеточно-пролиферативного расстройства.
Настоящее изобретение обеспечивает способы лечения гематологического рака или гематологических опухолей у субъекта, нуждающегося в этом, путем введения субъекту, нуждающемуся в таком лечении, терапевтически эффективного количества соединения по настоящему изобретению или его фармацевтически приемлемой соли, пролекарства, метаболита, полиморфа или сольвата. Настоящее изобретение, кроме того, обеспечивает применение соединения по настоящему изобретению или его фармацевтически приемлемой соли, пролекарства, метаболита, полиморфа или сольвата для получения лекарственного средства, полезного для лечения гематологического рака или гематологических опухолей.
Настоящее изобретение обеспечивает способы лечения лейкоза у субъекта, нуждающегося в этом, путем введения субъекту, нуждающемуся в таком лечении, терапевтически эффективного количества соединения по настоящему изобретению или его фармацевтически приемлемой соли, пролекарства, метаболита, полиморфа или сольвата. Лейкоз может представлять собой острый или хронический лейкоз. Предпочтительно, лейкоз представляет собой острый миелогенный лейкоз, острый лимфоцитарный лейкоз или лейкоз смешанного происхождения. Настоящее изобретение, кроме того, обеспечивает применение соединения по настоящему изобретению или его фармацевтически приемлемой соли, пролекарства, метаболита, полиморфа или сольвата, для получения лекарственного средства, полезного для лечения лейкоза.
Настоящее изобретение обеспечивает способы лечения заболевания или расстройства, опосредованного транслокацией гена на хромосоме 11q23, у субъекта, нуждающегося в этом, путем введения субъекту, нуждающемуся в таком лечении, терапевтически эффективного количества соединения по настоящему изобретению или его фармацевтически приемлемой соли, пролекарства, метаболита, полиморфа или сольвата. Ген может представлять собой MLL ген. Настоящее изобретение, кроме того, обеспечивает применение соединения по настоящему изобретению или его фармацевтически приемлемой соли, пролекарства, метаболита, полиморфа или сольвата для получения лекарственного средства, полезного для лечения заболевания или расстройства, опосредованного транслокацией гена на хромосоме 11q23.
Настоящее изобретение обеспечивает способы лечения заболевания или расстройства, опосредованного DOT1 (например, DOT1L)-опосредованным метилированием белка у субъекта, нуждающегося в этом, путем введения субъекту, нуждающемуся в таком лечении, терапевтически эффективного количества соединения по настоящему изобретению или его фармацевтически приемлемой соли, пролекарства, метаболита, полиморфа или сольвата. Настоящее изобретение, кроме того, обеспечивает применение соединения по настоящему изобретению или его фармацевтически приемлемой соли, пролекарства, метаболита, полиморфа или сольвата, для получения лекарственного средства, полезного для лечения заболевания или расстройства, опосредованного DOT1L-опосредованным метилированием белка.
Настоящее изобретение обеспечивает способы лечения расстройства, на развитие которого влияет модуляция состояния метилирования гистонов или других белков, при этом указанное состояние метилирования опосредовано, по меньшей мере, частично, активностью DOT1L. Модуляция состояния метилирования гистонов, в свою очередь, может влиять на уровень экспрессии представляющих интерес генов, активируемых метилированием, и/или представляющих интерес генов, подавляемых метилированием. Способ включает введение субъекту, нуждающемуся в таком лечении, терапевтически эффективного количества соединения по настоящему изобретению или его фармацевтически приемлемой соли, пролекарства, метаболита, полиморфа, сольвата или стереоизомера.
Расстройство, в котором играет роль DOT1L-опосредованное метилирование белка, может представлять собой рак или предраковое состояние или неврологическое заболевание. Настоящее изобретение, кроме того, обеспечивает применение соединения по настоящему изобретению или его фармацевтически приемлемой соли, пролекарства, метаболита, полиморфа или сольвата для получения лекарственного средства, полезного для лечения рака или неврологического заболевания.
Настоящее изобретение также обеспечивает способы защиты против расстройства, в котором играет роль DOT1L-опосредованное метилирование белка, у субъекта, нуждающегося в этом, путем введения терапевтически эффективного количества соединения по настоящему изобретению или его фармацевтически приемлемой соли, пролекарства, метаболита, полиморфа или сольвата субъекту, нуждающемуся в таком лечении. Расстройство может представлять собой рак или неврологическое заболевание. Настоящее изобретение также обеспечивает применение соединения по настоящему изобретению или его фармацевтически приемлемой соли, пролекарства, метаболита, полиморфа, сольвата или стереоизомера для получения лекарственного средства, полезного для профилактики клеточно-пролиферативного расстройства.
Соединения по настоящему изобретению можно использовать для модуляции метилирования белка (например, гистона), например, для модуляции ферментативной активности гистонметилтрансферазы или гистондеметилазы. Сообщалось, что метилирование гистона вовлечено в аберрантную экспрессию некоторых генов в раках и в сайленсинг нейрональных генов в не-нейрональных клетках. Соединения, описанные в настоящей заявке, можно использовать для лечения этих заболеваний, т.е. для снижения уровня метилирования или восстановления метилирования примерно до его уровня в аналогичных нормальных клетках.
Как правило, соединения, которые являются модуляторами метилирования, можно использовать для модуляци клеточной пролиферации, в основном. Например, в некоторых случаях чрезмерную пролиферацию можно уменьшить при помощи средств, которые уменьшают метилирование, тогда как недостаточную пролиферацию можно стимулировать при помощи средств, которые увеличивают метилирование. Соответственно, заболевания, которые можно лечить соединениями по настоящему изобретению, включают гиперпролиферативные заболевания, такие как доброкачественный клеточный рост и злокачественный клеточный рост.
Как используется в настоящей заявке, “субъект, нуждающийся в этом”, представляет собой субъекта, имеющего клеточно-пролиферативное расстройство, или субъекта, имеющего повышенный риск развития клеточно-пролиферативного расстройства относительно популяции в целом. У субъекта может быть рак или предраковое состояние. Предпочтительно, у субъекта, нуждающегося в этом, диагностирован рак. Более предпочтительно, гематологический рак или лейкоз. “Субъект” включает млекопитающих. Млекопитающее может представлять собой, например, любое млекопитающее, например, человека, обезьяну, птицу, мышь, крысу, дикую птицу, собаку, кошку, корову, лошадь, козу, верблюда, овцу или свинью. Предпочтительно, млекопитающим является человек.
Как используется в настоящей заявке, термин “клеточно-пролиферативное расстройство” относится к состояниям, при которых нерегулируемый или аномальный рост, или и то и другое, клеток может привести к развитию нежелательного состояния или заболевания, которое может быть раковым или нет. Примеры клеточно-пролиферативных расстройств по настоящему изобретению охватывают различные состояния, где клеточное деление является нерегулируем. Примеры клеточно-пролиферативных расстройств включают, но не ограничиваются этим, неоплазмы, доброкачественные опухоли, злокачественные опухоли, предраковые состояния, in situ опухоли, инкапсулированные опухоли, метастатические опухоли, жидкие опухоли, солидные опухоли, иммунологические опухоли, гематологические опухоли, раковые заболевания, карциномы, лейкозы, лимфомы, саркомы и быстро делящиеся клетки. Термин “быстро делящаяся клетка”, как это используется в настоящей заявке, означает любую клетку, которая делится со скоростью, которая превышает или больше, чем та, которая является ожидаемой или наблюдаемой среди соседних или сопредельных клеток в той же самой ткани. Клеточно-пролиферативное расстройство включает пред-рак или предраковое состояние. Клеточно-пролиферативное расстройство включает рак. Предпочтительно, способы обеспечиваемые в настоящей заявке, используют для лечения или облегчения симптома рака. Термин “рак” включает солидные опухоли, а также, гематологические опухоли и/или злокачественные заболевания. “Предраковая клетка” или “пред-злокачественная клетка” представляет собой клетку, демонстрирующую клеточно-пролиферативное расстройство, которое представляет собой пред-раковое или пред-злокачественное состояние. “Раковая клетка” или “злокачественная клетка” представляет собой клетку, демонстрирующую клеточно-пролиферативное расстройство, которое представляет собой рак. Для идентификации раковых клеток или пред-злокачественных клеток можно использовать любое воспроизводимое средство измерения. Раковые клетки или пред-злокачественные клетки можно идентифицировать при помощи гистологического типирования или классификации образца ткани (например, образца биопсии). Раковые клетки или пред-злокачественные клетки можно идентифицировать путем использования подходящих молекулярных маркеров.
Примеры не-злокачественных состояний или расстройств включают, но не ограничиваются этим, ревматоидный артрит; воспаление; аутоиммунное заболевание; лимфопролиферативные состояния; акромегалию; ревматоидный спондилит; остеоартрит; подагру, другие артритные состояния; сепсис; септический шок; эндотоксический шок; грам-отрицательный сепсис; синдром токсического шока; астму; респираторный дистресс-синдром взрослых; хроническое обструктивное легочное заболевание; хроническое воспаление легких; воспалительное заболевание кишечника; болезнь Крона; псориаз; экзему; язвенный колит; панкреатический фиброз; фиброз печени; острое и хроническое почечное заболевание; синдром раздраженной толстой кишки; парез; рестеноз; церебральную малярию; инсульт и ишемическое поражение; неврологическую травму; болезнь Альцгеймера; болезнь Гентингтона; болезнь Паркинсона; острую и хроническую боль; аллергический ринит; аллергический конъюнктивит; хроническую сердечную недостаточность; острый коронарный синдром; кахексию; малярию; лепру; лейшманиаз; болезнь Лайма; синдром Рейтера; острый синовит; мышечную дегенерацию, бурсит; тендонит; теносиновит; грыжу, синдром прободения или выпадения межпозвонкового диска; остеопетрозит; тромбоз; рестеноз; силикоз; патологическое разрастание мягких тканей легких; заболевания, связанные с резорбцией кости, такие как остеопороз; реакция “трансплантат-против-хозяина”; рассеянный склероз; волчанку; фибромиалгию; СПИД и другие вирусные заболевания, такие как опоясывающий лишай, простой герпес I или II, вирус гриппа и цитомегаловирус; и сахарный диабет.
Примеры раковых заболеваний включают, но не ограничиваются этим, адренокортикальную карциному, связанные со СПИДом раковые заболевания, связанную со СПИДом лимфому, анальный рак, аноректальный рак, рак анального канала, рак аппендикса, детскую мозжечковую астроцитому, детскую церебральную астроцитому, базально-клеточную карциному, рак кожи (отличный от меланомы), билиарный рак, рак экстрагепатических желчных протоков, рак интрагепатических желчных протоков, рак мочевого пузыря, рак костей и суставов, остеосаркому и злокачественную фиброзную гистиоцитому, рак головного мозга, опухоль головного мозга, глиому ствола головного мозга, мозжечковую астроцитому, церебральную астроцитому/злокачественную глиому, эпендиому, медуллобластому, расположенные над мозжечковым наметом примитивные нейроэктодермальные опухоли, глиому зрительного пути и гипоталамуса, рак молочной железы, бронхиальные аденомы/карциноиды, карциноидную опухоль, желудочно-кишечный рак, рак нервной системы, лимфому нервной систем, рак центральной нервной систем, лимфому центральной нервной систем, цервикальный рак, детские раковые заболевания, хронический лимфоцитарный лейкоз, хронический миелогенный лейкоз, хронический миелопролиферативные расстройства, рак толстой кишки, колоректальный рак, кожную T-клеточную лимфому, лимфоидное новообразование, грибковидный микоз, синдром Seziary, эндометриальный рак, рак пищевода, экстракраниальную зародышево-клеточную опухоль, экстрагонадальную зародышево-клеточную опухоль, рак экстрагепатических желчных протоков, рак глаза, внутриглазную меланому, ретинобластому, рак желчного пузыря, гастральный рак (рак желудка), желудочно-кишечную карциноидную опухоль, желудочно-кишечную стромальную опухоль (GIST), зародышево-клеточную опухоль, зародышево-клеточную опухоль яичника, гестационную трофобластную опухолевую глиому, рак головы и шеи, гепатоклеточный рак (рак печени), лимфому Ходжкина, гипофарингеальный рак, внутриглазную меланому, глазной рак, островково-клеточные опухоли (эндокринные опухоли поджелудочной железы), саркому Капоши, рак почки, ренальный рак, рак почки, ларингеальный рак, острый лимфобластный лейкоз, острый лимфоцитарный лейкоз, острый миелогенный лейкоз, хронический лимфоцитарный лейкоз, хронический миелогенный лейкоз, волосисто-клеточный лейкоз, рак губы и полости рта, рак печени, рак легкого, немелкоклеточный рак легкого, мелкоклеточный рак легкого, связанную со СПИДом лимфому, не-ходжкинскую лимфому, первичную лимфому центральной нервной системы, макроглобулинемию Валденстрема, медуллобластому, меланому, внутриглазную (глазную) меланому, меркель-клеточную карциному, злокачественную мезотелиому, мезотелиому, метастатический сквамозный рак шеи, рак полости рта, рак языка, множественный эндокринный неопластический синдром, грибковидный микоз, миелодиспластические синдромы, миелодиспластические/миелопролиферативные заболевания, хронический миелогенный лейкоз, острый миелогенный лейкоз, множественную миелому, хронические миелопролиферативные расстройства, назофарингеальный рак, нейробластому, оральный рак, рак полости рта, орофарингеальный рак, рак яичника, рак эпителия яичника, опухоль яичника с низким злокачественным потенциалом, панкреатический рак, островково-клеточный панкреатический рак, рак параназального синуса и носовой полости, рак паращитовидной железы, рак полового члена, фарингеальный рак, феохромоцитому, пинеобластому и расположенные над мозжечковым наметом примитивные нейроэктодермальные опухоли, опухоль гипофиза, плазменно-клеточное новообразование/множественную миелому, плевропульмонарную бластому, рак предстательной железы, рак прямой кишки, рак почечной лоханки и уретры, переходно-клеточный рак, ретинобластому, рабдомиосаркому, рак слюнных желез, саркому семейства опухолей Юинга, саркому Капоши, саркому мягких тканей, рак матки, сарком матки, рак кожи (не-меланому), рак кожи (меланому), клеточную карциному кожи Маркеля, рак тонкого кишечника, саркому мягких тканей, сквамозно-клеточную карциному, рак желудка (гастральный рак), расположенные над мозжечковым наметом примитивные нейроэктодермальные опухоли, тестикулярный рак, рак горла, тиому, тиому и тимусную карциному, рак щитовидной железы, переходно-клеточный рак почечной лоханки и уретры и других органов мочевыделения, гестационную трофобластную опухоль, уретральный рак, эндометриальный рак матки, саркому матки, рак тела матки, вагинальный рак, рак вульвы и опухоль Вильмса.
“Клеточно-пролиферативное расстройство гематологической системы” представляет собой клеточно-пролиферативное расстройство, включающее клетки гематологической системы. Клеточно-пролиферативное расстройство гематологической системы может включать лимфому, лейкоз, миелогенные неоплазмы, мастоцитарные неоплазмы, миелодисплазию, доброкачественную моноклональную гаммопатию, лимфоматоидный грануломатоз, лимфоматоидный папулез, истинную полицитемию, хронический миелоцитарный лейкоз, идиопатическую миелогенную метаплазию и эссенциальную тромбоцитемию. Клеточно-пролиферативное расстройство гематологической системы может включать гиперплазию, дисплазию и метаплазию клеток гематологической системы. Предпочтительно, композиции по настоящему изобретению можно использовать для лечения рака, выбранного из группы, включающей гематологический рак по настоящему изобретению или гематологическое клеточно-пролиферативное расстройство по настоящему изобретению. Гематологический рак по настоящему изобретению может включать множественную миелому, лимфому (включая лимфому Ходжкина, неходжкинскую лимфому, детские лимфомы и лимфомы лимфоцитарного и кожного происхождения), лейкоз (включая детский лейкоз, волосисто-клеточный лейкоз, острый лимфоцитарный лейкоз, острый миелоцитарный лейкоз, хронический лимфоцитарный лейкоз, хронический миелоцитарный лейкоз, хронический миелогенный лейкоз и мастоцитарный лейкоз), миелогенные неоплазмы и мастоцитарные неоплазмы.
“Клеточно-пролиферативное расстройство легких” представляет собой клеточно-пролиферативное расстройство, включающее клетки легких. Клеточно-пролиферативные расстройства легких могут включать все формы клеточно-пролиферативных расстройств, поражающих клетки легких. Клеточно-пролиферативные расстройства легких могут включать рак легкого, предраковое или предканцерогенное состояние легких, доброкачественный рост или поражения в легких и злокачественный рост или поражения в легких и метастатические поражения в тканях и органах в организме, отличных от легких. Предпочтительно, композиции по настоящему изобретению можно использовать для лечения рака легкого или клеточно-пролиферативных расстройств легких. Рак легкого может включать все формы рака легких. Рак легкого может включать злокачественные легочные неоплазмы, карциному in situ, типичные карциноидные опухоли и атипичные карциноидные опухоли. Рак легкого может включать мелкоклеточный рак легкого (“SCLC”), немелкоклеточный рак легкого (“NSCLC”), сквамозно-клеточную карциному, аденокарциному, мелкоклеточную карциному, крупно-клеточную карциному, аденосквамозно-клеточную карциному и мезотелиому. Рак легкого может включать “рубцовую карциному”, бронхиоальвеолярную карциному, гиганто-клеточную карциному, веретенообразно-клеточную карциному и крупно-клеточную нейроэндокринную карциному. Рак легкого может включать легочные неоплазмы, имеющие гистологическую и ультраструктурную гетерогенность (например, смешанные клеточные типы).
Клеточно-пролиферативные расстройства легких могут включать все формы клеточно-пролиферативных расстройств, поражающих клетки легких. Клеточно-пролиферативные расстройства легких могут включать рак легкого, пред-злокачественные состояния легких. Клеточно-пролиферативные расстройства легких могут включать гиперплазию, метаплазию и дисплазию легких. Клеточно-пролиферативные расстройства легких могут включать асбест-индуцированную гиперплазию, сквамозную метаплазию и доброкачественную реактивную мезотелиальную метаплазию. Клеточно-пролиферативные расстройства легких могут включать замещение цилиндрического эпителия слоистым сквамозным эпителием и дисплазию слизистой оболочки. Субъекты, подверженные воздействию вдыхаемых вредных окружающих веществ, таких как сигаретный дым и асбест, могут иметь повышенный риск развития клеточно-пролиферативного расстройства легких. Предшествующие легочные заболевания, которые могут способствовать развитию у субъекта клеточно-пролиферативных расстройств легких, могут включать хроническое интерстициальное легочное заболевание, некротизирующее пульмонарное заболевание, склеродерму, ревматоидное заболевание, саркоидоз, интерстициальный пневмонит, туберкулез, повторные пневмонии, идиопатический пульмонарный фиброз, гранулему, асбестоз, фиброзирующий альвеолит и болезнь Ходжкина.
“Клеточно-пролиферативное расстройство толстой кишки” представляет собой клеточно-пролиферативное расстройство, включающее клетки толстой кишки. Предпочтительно, клеточно-пролиферативное расстройство толстой кишки представляет собой рак толстой кишки. Предпочтительно, композиции по настоящему изобретению можно использовать для лечения рака толстой кишки или клеточно-пролиферативных расстройств толстой кишки. Рак толстой кишки может включать все формы рака толстой кишки. Рак толстой кишки может включать спорадический и наследственный рак толстой кишки. Рак толстой кишки может включать злокачественные неоплазмы толстой кишки, карциному in situ, типичные карциноидные опухоли и атипичные карциноидные опухоли. Рак толстой кишки может включать аденокарциному, сквамозно-клеточную карциному и аденосквамозно-клеточную карциному. Рак толстой кишки может быть связан с наследственным синдромом, выбранным из группы, включающей наследственный не-полипозный колоректальный рак, семейный аденомитозный полипоз, синдром Гарднера, синдром Пейтца-Егерса, синдром Туркота и ювенильный полипоз. Рак толстой кишки может быть вызван наследственным синдромом, выбранным из группы, включающей наследственный не-полипозный колоректальный рак, семейный аденомитозный полипоз, синдром Гарднера, синдром Пейтца-Егерса, синдром Туркота и ювенильный полипоз.
Клеточно-пролиферативные расстройства толстой кишки могут включать все формы клеточно-пролиферативных расстройств, поражающих клетки толстой кишки. Клеточно-пролиферативные расстройства толстой кишки могут включать рак толстой кишки, предраковые состояния толстой кишки, аденомитозный полипоз толстой кишки и метахронозные поражения толстой кишки. Клеточно-пролиферативное расстройство толстой кишки может включать аденому. Клеточно-пролиферативные расстройства толстой кишки могут характеризоваться гиперплазией, метаплазией и дисплазией толстой кишки. Предшествующие заболевания толстой кишки, которые могут способствовать развитию у субъекта клеточно-пролиферативных расстройств толстой кишки, могут включать предраковое состояние толстой кишки. Распространенные в настоящее время заболевания, которые могут способствовать развитию у субъекта клеточно-пролиферативных расстройств толстой кишки, могут включать болезнь Крона и язвенный колит. Клеточно-пролиферативное расстройство толстой кишки может быть связано с мутацией в гене, выбранном из группы, включающей p53, ras, FAP и DCC. У субъекта может быть повышенный риск развития клеточно-пролиферативного расстройства толстой кишки из-за наличия мутации в гене, выбранном из группы, включающей p53, ras, FAP и DCC.
“Клеточно-пролиферативное расстройство поджелудочной железы” представляет собой клеточно-пролиферативное расстройство, включающее клетки поджелудочной железы. Клеточно-пролиферативные расстройства поджелудочной железы могут включать все формы клеточно-пролиферативных расстройств, поражающих панкреатические клетки. Клеточно-пролиферативные расстройства поджелудочной железы могут включать рак поджелудочной железы, предраковое или предканцерогенное состояние поджелудочной железы, гиперплазию поджелудочной железы и дисплазию поджелудочной железы, доброкачественный рост или поражения поджелудочной железы и злокачественный рост или поражения поджелудочной железы и метастатические поражения в тканях и органах в организме, отличных от поджелудочной железы. Панкреатический рак включает все формы рака поджелудочной железы. Панкреатический рак может включать дуктальную аденокарциному, аденосквамозную карциному, плеоморфную гиганто-клеточную карциному, слизистую аденокарциному, остеокласто-подобную гиганто-клеточную карциному, слизистую цистаденокарциному, ацинозную карциному, неклассифицированную крупно-клеточную карциному, мелкоклеточную карциному, панкреатобластому, папиллярную неоплазму, слизистую цистаденому, папиллярную кистозную неоплазму и серозную цистаденому. Панкреатический рак также может включать панкреатические неоплазмы, имеющие гистологическую и ультраструктурную гетерогенность (например, смешанные клеточные типы).
“Клеточно-пролиферативное расстройство предстательной железы” представляет собой клеточно-пролиферативное расстройство, включающее клетки предстательной железы. Клеточно-пролиферативные расстройства предстательной железы могут включать все формы клеточно-пролиферативных расстройств, поражающих клетки предстательной железы. Клеточно-пролиферативные расстройства предстательной железы могут включать рак предстательной железы, предраковое или предканцерогенное состояние предстательной железы, доброкачественный рост или поражения предстательной железы и злокачественный рост или поражения предстательной железы и метастатические поражения в тканях и органах в организме, отличных от предстательной железы. Клеточно-пролиферативные расстройства предстательной железы могут включать гиперплазию, метаплазию и дисплазию предстательной железы.
“Клеточно-пролиферативное расстройство кожи” представляет собой клеточно-пролиферативное расстройство, включающее клетки кожи. Клеточно-пролиферативные расстройства кожи могут включать все формы клеточно-пролиферативных расстройств, поражающих клетки кожи. Клеточно-пролиферативные расстройства кожи могут включать предраковое или предканцерогенное состояние кожи, доброкачественный рост или поражения кожи, меланому, злокачественную меланому и другие расстройства, относящиеся к злокачественному росту или поражению кожи, и метастатические поражения в тканях и органах в организме, отличных от кожи. Клеточно-пролиферативные расстройства кожи могут включать гиперплазию, метаплазию и дисплазию кожи.
“Клеточно-пролиферативное расстройство яичника” представляет собой клеточно-пролиферативное расстройство, включающее клетки яичника. Клеточно-пролиферативные расстройства яичника могут включать все формы клеточно-пролиферативных расстройств, поражающих клетки яичника. Клеточно-пролиферативные расстройства яичника могут включать предраковое или предканцерогенное состояние яичника, доброкачественный рост или поражения яичника, рак яичника, злокачественный рост или поражения яичника и метастатические поражения в тканях и органах в организме, отличных от яичника. Клеточно-пролиферативные расстройства яичника могут включать гиперплазию, метаплазию и дисплазию клеток яичника.
“Клеточно-пролиферативное расстройство молочной железы” представляет собой клеточно-пролиферативное расстройство, включающее клетки молочной железы. Клеточно-пролиферативные расстройства молочной железы могут включать все формы клеточно-пролиферативных расстройств, поражающих клетки молочной железы. Клеточно-пролиферативные расстройства молочной железы могут включать рак молочной железы, предраковое или предканцерогенное состояние молочной железы, доброкачественный рост или поражения молочной железы и злокачественный рост или поражения молочной железы и метастатические поражения в тканях и органах в организме, отличных от молочной железы. Клеточно-пролиферативные расстройства молочной железы могут включать гиперплазию, метаплазию и дисплазию молочной железы.
Клеточно-пролиферативное расстройство молочной железы может представлять собой предраковое состояние молочной железы. Композиции по настоящему изобретению можно использовать для лечения предракового состояния молочной железы. Предраковое состояние молочной железы может включать атипичную гиперплазию молочной железы, дуктальную карциному in situ (DCIS), интрадуктальную карциному, лобулярную карциному in situ (LCIS), лобулярную неоплазию и стадию 0 или уровень 0 роста или поражения молочной железы (например, стадию 0 или уровень 0 рака молочной железы или карциномы in situ). Предраковое состояние молочной железы можно классифицировать по стадиям в соответствии с классификационной схемой TNM, принятой the American Joint Committee on Cancer (AJCC), где первичная опухоль (T) соответствует стадии T0 или Tis; и где региональные лимфатические узлы (N) соответствуют стадии N0; и где отдаленный метастаз (M) соответствует стадии M0.
Клеточно-пролиферативное расстройство молочной железы может представлять собой рак молочной железы. Предпочтительно, композиции по настоящему изобретению можно использовать для лечения рака молочной железы. Рак молочной железы включает все формы рака молочной железы. Рак молочной железы может включать первичный эпителиальный рак молочной железы. Рак молочной железы может включать раковые заболевания, в которых молочная железа вовлечена за счет других опухолей, таких как лимфома, саркома или меланома. Рак молочной железы может включать карциному молочной железы, дуктальную карциному молочной железы, лобулярную карциному молочной железы, недифференцированную карциному молочной железы, листовидную цистосаркому молочной железы, ангиосаркому молочной железы и первичную лимфому молочной железы. Рак молочной железы может включать Стадию I, II, IIIA, IIIB, IIIC и IV рака молочной железы. Дуктальная карцинома молочной железы может включать инвазивную карциному, инвазивную карцином in situ с преобладающим интрадуктальным компонентом, воспалительный рак молочной железы и дуктальную карциному молочной железы с гистологическим типом, выбранным из группы, включающей комедон, слизистый (коллоидный), медуллярный, медуллярный с лимфоцитарным инфильтратом, папиллярный, скиррозный и тубулярный. Лобулярная карцинома молочной железы может включать инвазивную лобулярную карциному с преобладающим in situ компонентом, инвазивную лобулярную карциному и инфильтрирующую лобулярную карциному. Рак молочной железы может включать болезнь Пэджета, болезнь Пэджета с интрадуктальной карциномой и болезнь Пэджета с инвазивной дуктальной карциномой. Рак молочной железы может включать неоплазмы молочной железы, имеющие гистологическую и ультраструктурную гетерогенность (например, смешанные клеточные типы).
Предпочтительно, соединение по настоящему изобретению или его фармацевтически приемлемую соль, пролекарство, метаболит, полиморф или сольват, можно использовать для лечения рака молочной железы. Рак молочной железы, который можно лечить, может включать семейный рак молочной железы. Рак молочной железы, который можно лечить, может включать спорадический рак молочной железы. Рак молочной железы, который можно лечить, может возникать у субъекта мужского пола. Рак молочной железы, который можно лечить, может возникать у субъекта женского пола. Рак молочной железы, который можно лечить, может возникать у предклимактерического субъекта женского пола или у постклимактерического субъекта женского пола. Рак молочной железы, который можно лечить, может возникать у субъекта в возрасте 30 лет или старше, или у субъекта моложе 30 лет. Рак молочной железы, который можно лечить, может возникать у субъекта в возрасте 50 лет или старше, или у субъекта моложе 50 лет. Рак молочной железы, который можно лечить, может возникать у субъекта в возрасте 70 лет или старше, или у субъекта моложе 70 лет.
Рак молочной железы, который можно лечить, можно типировать для идентификации семейной или спонтанной мутации в BRCA1, BRCA2 или p53. Рак молочной железы, который можно лечить, можно типировать, такие как включающий HER2/neu генную амплификацию, как сверхэкспрессирующий HER2/neu или как включающий низкий, средний или высокий уровень экспрессии HER2/neu. Рак молочной железы, который можно лечить, можно типировать для определения маркера, выбранного из группы, включающей эстрогеновый рецептор (ER), прогестероновый рецептор (PR), рецептор-2 человеческого эпидермального ростового фактора, Ki-67, CA15-3, CA 27-29 и c-Met. Рак молочной железы, который можно лечить, можно типировать как ER-неизвестный, ER-обогащенный или ER-обедненный. Рак молочной железы, который можно лечить, можно типировать как ER-отрицательный или ER-положительный. ER-типирование рака молочной железы можно осуществить любыми воспроизводимыми средствами. ER-типирование рака молочной железы можно осуществить, как описано в Onkologie 27: 175-179 (2004). Рак молочной железы, который можно лечить, можно типировать как PR-неизвестный, PR- обогащенный или PR- обедненный. Рак молочной железы, который можно лечить, можно типировать как PR-отрицательный или PR-положительный. Рак молочной железы, который можно лечить, можно типировать как рецептор-положительный или рецептор-отрицательный. Рак молочной железы, который можно лечить, можно типировать как связанный с повышенными уровнями в крови CA 15-3 или CA 27-29 или обоих.
Рак молочной железы, который можно лечить, может включать локализованную опухоль молочной железы. Рак молочной железы, который можно лечить, может включать опухоль молочной железы, которая связана с отрицательной биопсией сторожевого лимфоузла (SLN). Рак молочной железы, который можно лечить, может включать опухоль молочной железы, которая связана с положительной биопсией сторожевого лимфоузла (SLN). Рак молочной железы, который можно лечить, может включать опухоль молочной железы, которая связана с одним или несколькими положительными подмышечными лимфатическиими узлами, где стадии подмышечных лимфатических узлов определены любым подходящим способом. Рак молочной железы, который можно лечить, может включать опухоль молочной железы, которая была типирована как имеющая нодальный отрицательный статус (например, узел-отрицательная) или нодальный положительный статус (например, узел-положительная). Рак молочной железы, который можно лечить, может включать опухоль молочной железы, которая метастазировала в другие участки в организме. Рак молочной железы, который можно лечить, можно классифицировать как метастазировавший на участке, выбранном из группы, включающей кость, легкие, печень или головной мозг. Рак молочной железы, который можно лечить, можно классифицировать в соответствии с характеристикой, выбранной из группы, включающей метастатический, локализованный, региональный, локально-региональный, локально прогрессирующий, отдаленный, многоцентровой, билатеральный, ипсилатеральный, контралатеральный, вновь диагностированный, рецидивирующий в настоящее время и неоперабельный.
Соединение по настоящему изобретению или его фармацевтически приемлемую соль, пролекарство, метаболит, полиморф или сольват можно использовать для лечения или профилактики клеточно-пролиферативного расстройства молочной железы или для лечения или профилактики рака молочной железы у субъекта, имеющего повышенный риск развития рака молочной железы относительно популяции в целом. Субъект с повышенным риском развития рака молочной железы относительно популяции в целом представляет собой субъекта женского пола с семейной историей или персональной историей рака молочной железы. Субъект с повышенным риском развития рака молочной железы относительно популяции в целом представляет собой субъекта женского пола, имеющего мутацию в зародышевой линии или спонтанную мутацию в BRCA1 или BRCA2, или обе. Субъект с повышенным риском развития рака молочной железы относительно популяции в целом представляет собой субъекта женского пола с семейной историей рака молочной железы и с мутацией в зародышевой линии или спонтанной мутацией в BRCA1 или BRCA2, или обеими. Субъект с повышенным риском развития рака молочной железы относительно популяции в целом представляет собой женщину старше 30 лет, старше 40 лет, старше 50 лет, старше 60 лет, старше 70 лет, старше 80 лет или старше 90 лет. Субъект с повышенным риском развития рака молочной железы относительно популяции в целом представляет собой субъекта с атипичной гиперплазией молочной железы, дуктальной карциномой in situ (DCIS), интрадуктальной карциномой, лобулярной карциномой in situ (LCIS), лобулярной неоплазией или стадией 0 роста или поражения молочной железы (например, стадией 0 или уровнем 0 рака молочной железы или карциномы in situ).
Рак молочной железы, который можно лечить, может гистологически классифицироваться в соответствии с системой Скарфа-Блюма-Ричардсона, где опухоли молочной железы оценивают по баллам на основании митоза 1, 2 или 3; на основании нуклеарного прейоморфизма 1, 2 или 3; на основании образования трубочек 1, 2 или 3; и общей оценки по системе Скарфа-Блюма-Ричардсона от 3 до 9. Рак молочной железы, который можно лечить, можно определить как имеющий степень развития опухоли в соответствии с International Consensus Panel on the Treatment of Breast Cancer, выбранную из группы, включающей степень 1, степень 1-2, степень 2, степень 2-3 или степень 3.
Рак, который можно лечить, можно классифицировать по стадиям в соответствии с системой классификации American Joint Committee on Cancer (AJCC) TNM, где опухоль (T) определяют как имеющую стадию TX, T1, T1mic, T1a, T1b, T1c, T2, T3, T4, T4a, T4b, T4c или T4d; и где региональные лимфатические узлы (N) определяют как имеющие стадию NX, N0, N1, N2, N2a, N2b, N3, N3a, N3b или N3c; и где отдаленный метастаз (M) определяют как имеющую стадию MX, M0 или M1. Рак, который можно лечить, можно классифицировать по стадиям, в соответствии с классификацией American Joint Committee on Cancer (AJCC), как Стадия I, Стадия IIA, Стадия IIB, Стадия IIIA, Стадия IIIB, Стадия IIIC или Стадия IV. Степень рака, который можно лечить, можно определить в соответствии с классификацией AJCC как степень GX (например, степень нельзя определить), степень 1, степень 2, степень 3 или степень 4. Рак, который можно лечить, можно классифицировать по стадиям, в соответствии с патологической классификацией(pN) AJCC, как pNX, pN0, PN0 (I-), PN0 (I+), PN0 (mol-), PN0 (mol+), PN1, PN1(mi), PN1a, PN1b, PN1c, pN2, pN2a, pN2b, pN3, pN3a, pN3b или pN3c.
Рак, который можно лечить, может включать опухоль, которая была определена как меньше чем или равная примерно 2 сантиметрам в диаметре. Рак, который можно лечить, может включать опухоль, которая была определена как от около 2 до около 5 сантиметров в диаметре. Рак, который можно лечить, может включать опухоль, которая была определена как больше чем или равная примерно 3 сантиметрам в диаметре. Рак, который можно лечить, может включать опухоль, которая была определена как больше чем 5 сантиметров в диаметре. Рак, который можно лечить, можно классифицировать также в соответствии с микроскопическим изображением как дифференцируемый, умеренно дифференцируемый, плохо дифференцируемый или недифференцируемый. Рак, который можно лечить, можно классифицировать на основании микроскопического изображения в соответствии с оценкой митоза (например, количество клеточного деления) или нуклеарного плейоморфизма (например, изменение в клетках). Рак, который можно лечить, можно классифицировать в соответствии с микроскопическим изображением как связанный с областями некроза (например, области умирания или дегенерации клеток). Рак, который можно лечить, можно классифицировать как имеющий аномальный кариотип, имеющий аномальное количество хромосом или имеющий одну или несколько хромосом, которые имеют аномальный внешний вид. Рак, который можно лечить, можно классифицировать как анэуплоидный, триплоидный, тетраплоидный или как имеющий измененную плоидность. Рак, который можно лечить, можно классифицировать как имеющий хромосомную транслокацию или делецию или дупликацию целой хромосомы, или область делеции, дупликации или амплификации части хромосомы.
Рак, который можно лечить, можно оценить методом ДНК цитометрии, проточной цитометрии или визуализационной цитометрии. Рак, который можно лечить, можно типировать как имеющий 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80% или 90% клеток в стадии синтеза клеточного деления (например, в S фазе клеточного деления). Рак, который можно лечить, можно типировать как имеющий низкую долю S-фазы или высокую долю S-фазы.
Как используется в настоящей заявке, “нормальная клетка” представляет собой клетку, которая не может классифицироваться как часть “клеточно-пролиферативного расстройства”. У нормальной клетки отсутствует нерегулируемый или аномальный рост, или и то и другое, который может привести к развитию нежелательного состояния или заболевания. Предпочтительно, нормальная клетка обладает нормально функционирующими механизмами контроля контрольных точек клеточного цикла.
Как используется в настоящей заявке, “контактирование клетки” относится к состоянию, где соединение или другая композиция вещества находится в непосредственном контакте с клеткой или находится достаточно близко, чтобы индуцировать желаемый биологический эффект в клетке.
Как используется в настоящей заявке, “соединение-кандидат ” относится к соединению по настоящему изобретению или к фармацевтически приемлемой соли, пролекарству, метаболиту, полиморфу или сольвату такого соединения, которое было или будет испытано в одном или нескольких in vitro или in vivo биологических анализах, для определения, может или нет такое соединение вызывать желаемый биологический или медицинский ответ в клетке, ткани, системе, у животного или человека, получение которого добивается исследователь или клиницист. Соединение-кандидат представляет собой соединение по настоящему изобретению или его фармацевтически приемлемую соль, пролекарство, метаболит, полиморф или сольват. Биологический или медицинский ответ может представлять собой лечение рака. Биологический или медицинский ответ может представлять собой лечение или профилактику клеточно-пролиферативного расстройства. In vitro или in vivo биологические анализы могут включать, но не ограничиваются этим, анализы ферментативной активности, анализы электрофоретической подвижности, анализы репортерных генов, in vitro анализы жизнеспособности клеток и анализы, описанные в настоящей заявке.
Как используется в настоящей заявке, “монотерапия” относится к введению одного активного или терапевтического соединения субъекту, нуждающемуся в этом. Предпочтительно, монотерапия включает введение терапевтически эффективного количества одного активного соединения. Например, монотерапия рака с использованием одного единственного соединения по настоящему изобретению или его фармацевтически приемлемой соли, пролекарства, метаболита, аналога или производного для введения субъекту, нуждающемуся в лечении рака. В одном аспекте, единственное активное соединение представляет собой соединение по настоящему изобретению или его фармацевтически приемлемую соль, пролекарство, метаболит, полиморф или сольват.
Как используется в настоящей заявке, “лечение” или “лечить” означает лечение и уход за пациентом с целью борьбы с заболеванием, состоянием или расстройством, и включает введение соединения по настоящему изобретению или его фармацевтически приемлемой соли, пролекарства, метаболита, полиморфа или сольвата для облегчения симптомов или осложнений заболевания, состояния или расстройства или для устранения заболевания, состояния или расстройства.
Соединение по настоящему изобретению или его фармацевтически приемлемую соль, пролекарство, метаболит, полиморф или сольват также можно использовать для профилактики заболевания, состояния или расстройства. Как используется в настоящей заявке, “профилактика” или “осуществлять профилактику” означает уменьшение или устранение развития симптомов или осложнений заболевания, состояния или расстройства.
Как используется в настоящей заявке, термин "облегчение" означает способ, посредством которого уменьшается тяжесть признака или симптома расстройства. Важно отметить, что признак или симптом можно облегчить без его устранения. В предпочтительном варианте воплощения, введение фармацевтических композиций по настоящему изобретению приводит к устранению признака или симптома, однако, устранение не является необходимым. Ожидают, что эффективные дозы должны снижать тяжесть признака или симптома. Например, признак или симптом расстройства, такого как рак, который может возникать на нескольких участках, облегчается, если тяжесть рака снижается по меньшей мере на одном из нескольких участков.
Как используется в настоящей заявке, термин "тяжесть" означает потенциал рака к трансформации из пред-злокачественного или доброкачественного состояния в злокачественное состояние. Альтернативно или в дополнение к этому, тяжесть означает стадию рака, например, в соответствии с TNM системой (принятой International Union Against Cancer (UICC) и American Joint Committee on Cancer (AJCC)) или другими принятыми в данной области способами. Стадия рака относится к степени или тяжести рака, на основании факторов, таких как локализация первичной опухоли, размер опухоли, количество опухолей и вовлечение лимфатических узлов (распространение рака в лимфатические узлы). Альтернативно или в дополнение к этому, тяжесть означает стадию опухоли, определенную признанными в данной области способами (см., National Cancer Institute, www.рак.gov). Определение стадии опухоли представляет собой систему, используемую для классификации раковых клеток в соответствии с тем, насколько аномально они выглядят под микроскопом и насколько быстрым может быть рост и распространение опухоли. При определении стадии опухоли учитывают многие факторы, включая структуру и картину роста клеток. Специфические факторы, используемые для определения стадии опухоли, варьируются в зависимости от типа рака. Тяжесть также характеризует гистологическую оценку, также называемую дифференциацией, которая отражает сколько опухолевых клеток напоминает нормальные клетки этого же самого типа ткани (см. National Cancer Institute, www.cancer.gov). Кроме того, тяжесть характеризует ядерный уровень, который относится к размеру и форме ядра в опухолевых клетках и к проценту опухолевых клеток, которые делятся (см. National Cancer Institute, www.cancer.gov).
В другом аспекте настоящего изобретения, тяжесть характеризует степень, до которой имела место секреция ростовых факторов опухоли, разрушение внеклеточного матрикса, опухоль стала васкуляризованной, утратила адгезию с сопредельными тканями или метастазировала. Более того, тяжесть характеризует количество участков, в которые метастазировала первичная опухоль. В конечном счете, тяжесть включает трудность лечения опухолей различных типов и локализаций. Например, неоперабельные опухоли, такие раковые опухоли, которые имеют больший доступ к многим системам организма (гематологические и иммунологические опухоли), и такие, которые являются наиболее резистентными к традиционным лечения, считаются наиболее тяжелыми. В этих ситуациях, продление ожидаемой продолжительности жизни субъекта и/или уменьшение боли, уменьшение количества раковых клеток или ограничение клеток до одной системы и улучшение таких оценок, как стадия рака/стадия опухоли/гистологическая оценка/нуклеарная стадия, рассматривают как облегчение признака или симптома рака.
Как используется в настоящей заявке термин "симптом" определяется как признак заболевания, болезни, поражения или того, что что-то неправильно в организме. Симптомы ощутимы или заметны для субъекта, испытывающего такой симптом, но могут быть незаметными для других. Другие определяются как отличные от специалистов в области медицины.
Как используется в настоящей заявке, термин "признак" также определяется как признак того, что что-то неправильно в организме. Но признаки определяются как такие вещи, которые может заметить врач, медицинская сестра или другие специалисты в области медицины.
Рак представляет собой группу заболеваний, которые могут вызывать почти любой признак или симптом. Признаки и симптомы зависят от места расположения рака, размера рака и от того, насколько он затрагивает ближайшие органы или структуры. Если рак распространяется (метастазирует), тогда симптомы могут возникать в различных частях организма.
По мере роста рака, он начинает давить на ближайшие органы, кровеносные сосуды и нервы. Это давление создает некоторые признаки и симптомы рака. Если рак находится в критической области, такой как некоторые участки головного мозга, даже самая маленькая опухоль может вызвать ранние симптомы.
Но иногда раковые заболевания начинаются в местах, где они не могут вызвать какие-либо симптомы до тех пор, пока рак не выростет до достаточного большого размера. Рак поджелудочной железы, например, обычно не вырастает до достаточного большого размера, чтобы его можно было ощатить снаружи. Некоторые панкреатические раковые заболевания не вызывают симптомов до тех пор, пока они не начинают расти вокруг ближайших нервов (это вызывает боль в спине). Другие растут вокруг желчных протоков, что блокирует поток желчи и приводит к пожелтению кожи, известному как желтуха. К тому времени, когда панкреатический рак вызывает эти признаки или симптомы, он обычно достигает поздней стадии.
Рак также может вызывать симптомы, такие как лихорадка, усталость или потеря веса. Это может происходить потому, что раковые клетки потребляют большую часть энергии организма или высвобождают вещества, которые изменяют обмен веществ в организме. Или рак может вызвать реакцию иммунной системы таким путем, который вызывает эти симптомы.
Иногда раковые клетки высвобождают вещества в кровоток, что вызывает симптомы, которые обычно не считают результатом раковых заболеваний. Например, некоторые раковые заболевания поджелудочной железы могут выделять вещества, которые вызывают развитие сгустков крови в венах ног. Некоторые раки легких образуют гормоно-подобные вещества, которые влияют на уровни кальция в крови, поражая нервы и мышцы и вызывая слабость и головокружение.
Рак имеет некоторые основные признаки или симптомы, которые возникают, когда присутствует множество подтипов раковых клеток. Большинство людей, страдающих раком, теряют вес в определенный период их заболевания. Необъяснимая (непреднамеренная) потеря веса 10 фунтов (4536 г) или более может быть первым признаком рака, в частности, раковых заболеваний поджелудочной железы, желудка, пищевода или легких.
Лихорадка является обычным явлением при раке, но более часто наблюдается при запущенном заболевании. Почти у всех пациентов с раком в какое-то время будет лихорадка, особенно если рак или его лечение затрагивает иммунную систему и затрудняет борьбу организма с инфекциями. Менее часто лихорадка может быть ранним признаком рака, например, при лейкозе или лимфоме.
Усталость может быть важным симптомом прогрессирования рака. Она может возникать рано, хотя при раковых заболеваниях, например, в случае лейкоза, или если рак вызывает постоянную потерю крови, как в некоторых случаях рака толстой кишки или желудка.
Боль может быть ранним симптомом при некоторых раковых заболеваниях, таких как рак кости или тестикулярный рак. Но наиболее часто боль представляет собой симптом запущенного заболевания.
Наряду с раковыми заболеваниями кожи (см. следующий раздел), некоторые внутренние раковые заболевания могут вызывать кожные признаки, которые можно видеть. Эти изменения включают такие, когда кожа выглядит темнее (гиперпигментация), желтой (желтуха) или красной (эритема); зуд; или чрезмерный рост волос.
Альтернативно или в дополнение к этому, подтипы рака имеют специфические признаки или симптомы. Изменения работы кишечника или функции мочевого пузыря могут быть признаками рака. Длительный запор, диарея или изменение размера стула могут быть признаками рака толстой кишки. Боль при мочеиспускании, кровь в моче или изменение функции мочевого пузыря (такое как более частое или менее частое мочеиспускание) могут быть связаны с раком мочевого пузыря или предстательной железы.
Изменения состояния кожи или появление нового состояния кожи могут быть признаками рака. Кожные раковые заболевания могут кровоточить и выглядеть как болячки, которые не залечиваются. Язвы во рту в течение длительного периода могут быть признаками рака полости рта, особенно у пациентов, которые курят, жуют табак или часто принимают алкоголь. Язвы в области полового члена или вагины могут быть либо признаками инфекции, либо ранней стадии рака.
Необычное кровотечение или выделение могут быть признаками рака. Необычное кровотечение может быть как на ранней, так и на поздней стадии рака. Кровь в выделяемой мокроте (слизи) может быть признаком рака легкого. Кровь в стуле (или темный или черный стул) может быть признаком рака толстой кишки или прямой кишки. Рак шейки матки или эндометрия (внутренней оболочки матки) могут вызывать вагинальное кровотечение. Кровь в моче может быть признаком рака мочевого пузыря или почки. Кровянистые выделения из соска могут быть признаками рака молочной железы.
Утолщение или припухлость в области молочной железы или в других частях организма могут указывать на наличие рака. Многие раковые опухоли могут ощущаться через кожу, большей частью в области молочной железы, мужских половых желез, лимфатических узлов (шейные железы) и мягких тканей организма. Припухлость или утолщение могут быть ранними или поздними признаками рака. Любая припухлость или утолщение могут быть признаками рака, особенно если образование является новым или увеличилось в размере.
Нарушение пищеварения или проблемы с глотанием могут быть признаками рака. Хотя эти симптомы часто имеют другие причины, нарушение пищеварения или проблемы с глотанием могут быть признаками рака пищевода, желудка или глотки (горла).
Свежие изменения бородавок или родинок могут быть признаками рака. Любая бородавка, родинка или пятно на коже, у которых изменился цвет, размер или форма, или которые потеряли свои четкие границы, указывают на потенциальное развитие рака. Например, поражение кожи может представлять собой меланому.
Постоянный кашель или хрипота могут быть признаками рака. Кашель, который не прекращается, может быть признаком рака легкого. Хрипота может представлять собой признак рака гортани или щитовидной железы.
Хотя признаки и симптомы, перечисленные выше, являются наиболее распространенными, которые наблюдаются при раке, могут быть и другие, которые являются менее распространенными и не указаны в настоящей заявке. Однако все признанные в данной области признаки и симптомы рака рассматриваются и охватываются настоящим изобретением.
Лечения рака может приводить к уменьшению размера опухоли. Уменьшение размера опухоли также может быть указано как “регрессия опухоли”. Предпочтительно, после лечения, размер опухоли уменьшается на 5% или больше по сравнению с ее размером до лечения; более предпочтительно, размер опухоли уменьшается на 10% или больше; более предпочтительно, уменьшается на 20% или больше; более предпочтительно, уменьшается на 30% или больше; более предпочтительно, уменьшается на 40% или больше; еще более предпочтительно, уменьшается на 50% или больше; и наиболее предпочтительно, уменьшается на больше чем 75% или больше. Размер опухоли можно измерить любыми воспроизводимыми средствами измерения. Размер опухоли можно измерить как диаметр опухоли.
Лечения рака может приводить к уменьшению объема опухоли. Предпочтительно, после лечения, объем опухоли уменьшается на 5% или больше по сравнению с ее размером до лечения; более предпочтительно, объем опухоли уменьшается на 10% или больше; более предпочтительно, уменьшается на 20% или больше; более предпочтительно, уменьшается на 30% или больше; более предпочтительно, уменьшается на 40% или больше; еще более предпочтительно, уменьшается на 50% или больше; и наиболее предпочтительно, уменьшается на больше чем 75% или больше. Объем опухоли можно измерить любыми воспроизводимыми средствами измерения.
Лечение рака приводит к уменьшению количества опухолей. Предпочтительно, после лечения, количество опухолей уменьшается на 5% или больше по сравнению с количеством до лечения; более предпочтительно, количество опухолей уменьшается на 10% или больше; более предпочтительно, уменьшается на 20% или больше; более предпочтительно, уменьшается на 30% или больше; более предпочтительно, уменьшается на 40% или больше; еще более предпочтительно, уменьшается на 50% или больше; и наиболее предпочтительно, уменьшается на больше чем 75%. Количество опухолей можно измерить любыми воспроизводимыми средствами измерения. Количество опухолей можно измерить путем подсчета опухолей, видимых невооруженным глазом или при определенном увеличении. Предпочтительно, определенное увеличение означает 2×, 3×, 4×, 5×, 10× или 50×.
Лечения рака может привести к уменьшению количества метастатических поражений в других тканях или органах, отдаленных от участка первичной опухоли. Предпочтительно, после лечения, количество метастатических поражений уменьшается на 5% или больше по сравнению с количеством до лечения; более предпочтительно, количество метастатических поражений уменьшается на 10% или больше; более предпочтительно, уменьшается на 20% или больше; более предпочтительно, уменьшается на 30% или больше; более предпочтительно, уменьшается на 40% или больше; еще более предпочтительно, уменьшается на 50% или больше; и наиболее предпочтительно, уменьшается на больше чем 75%. Количество метастатических поражений можно измерить любыми воспроизводимыми средствами измерения. Количество метастатических поражений можно измерить путем подсчета метастатических поражений, видимых невооруженным глазом или при определенном увеличении. Предпочтительно, определенное увеличение означает 2×, 3×, 4×, 5×, 10× или 50×.
Лечения рака может привести к увеличению среднего времени выживания популяции субъектов, которые принимают лечение, по сравнению с популяцией, принимающей только носитель. Предпочтительно, среднее время выживания увеличивается больше чем на 30 дней; более предпочтительно, больше чем на 60 дней; более предпочтительно, больше чем на 90 дней; и наиболее предпочтительно, больше чем на 120 дней. Увеличение среднего времени выживания популяции можно измерить любыми воспроизводимыми средствами. Увеличение среднего времени выживания популяции можно измерить, например, путем подсчета для популяции средней продолжительности выживания после начала лечения активным соединением. Увеличение среднего времени выживания популяции также можно измерить, например, путем подсчета для популяции средней продолжительности выживания после завершения первого цикла лечения активным соединением.
Лечение рака может привести к увеличению среднего времени выживания популяции субъектов, которые принимают лечение, по сравнению с популяцией субъектов, на принимающих лечение. Предпочтительно, среднее время выживания увеличивается больше чем на 30 дней; более предпочтительно, больше чем на 60 дней; более предпочтительно, больше чем на 90 дней; и наиболее предпочтительно, больше чем на 120 дней. Увеличение среднего времени выживания популяции можно измерить любыми воспроизводимыми средствами. Увеличение среднего времени выживания популяции можно измерить, например, путем подсчета для популяции средней продолжительности выживания после начала лечения активным соединением. Увеличение среднего времени выживания популяции также можно измерить, например, путем подсчета для популяции средней продолжительности выживания после завершения первого цикла лечения активным соединением.
Лечение рака может привести к увеличению среднего времени выживания популяции субъектов, которые принимают лечение по сравнению с популяцией, принимающей монотерапию с лекарственным средством, которое не является соединением по настоящему изобретению или его фармацевтически приемлемой солью, пролекарством, метаболитом, аналогом или производным. Предпочтительно, среднее время выживания увеличивается больше чем на 30 дней; более предпочтительно, больше чем на 60 дней; более предпочтительно, больше чем на 90 дней; и наиболее предпочтительно, больше чем на 120 дней. Увеличение среднего времени выживания популяции можно измерить любыми воспроизводимыми средствами. Увеличение среднего времени выживания популяции можно измерить, например, путем подсчета для популяции средней продолжительности выживания после начала лечения активным соединением. Увеличение среднего времени выживания популяции также можно измерить, например, путем подсчета для популяции средней продолжительности выживания после завершения первого цикла лечения активным соединением.
Лечение рака может привести к уменьшению процента смертности в популяции субъектов, которые принимают лечение, по сравнению с популяцией, принимающей только носитель. Лечение рака может привести к уменьшению процента смертности в популяции субъектов, которые принимают лечение, по сравнению с популяцией, не принимающей лечение. Лечение рака может привести к уменьшению процента смертности в популяции субъектов, которые принимают лечение, по сравнению с популяцией, принимающей монотерапию с лекарственным средством, которое не является соединением по настоящему изобретению или его фармацевтически приемлемой солью, пролекарством, метаболитом, аналогом или производным. Предпочтительно, процент смертности уменьшается больше чем на 2%; более предпочтительно, больше чем на 5%; более предпочтительно, больше чем на 10%; и наиболее предпочтительно, больше чем на 25%. Уменьшение процента смертности в популяции субъектов, которые принимают лечение, можно измерить любыми воспроизводимыми средствами. Уменьшение процента смертности в популяции можно измерить, например, путем подсчета для популяции среднего количества связанных с заболеванием случаев смерти на единицу времени после начала лечения активным соединением. Уменьшение процента смертности в популяции можно измерить, например, путем подсчета для популяции среднего количества связанных с заболеванием случаев смерти на единицу времени после завершения первого цикла лечения активным соединением.
Лечение рака может привести к уменьшению скорости роста опухоли. Предпочтительно, после лечения, скорость роста опухоли уменьшается по меньшей мере на 5% по сравнению с показателем до лечения; более предпочтительно, скорость роста опухоли уменьшается по меньшей мере на 10%; более предпочтительно, уменьшается по меньшей мере на 20%; более предпочтительно, уменьшается по меньшей мере на 30%; более предпочтительно, уменьшается по меньшей мере на 40%; более предпочтительно, уменьшается по меньшей мере на 50%; еще более предпочтительно, уменьшается по меньшей мере на 50%; и наиболее предпочтительно, уменьшается по меньшей мере на 75%. Скорость роста опухоли можно измерить любыми воспроизводимыми средствами измерения. Скорость роста опухоли можно измерить в соответствии с изменением диаметра опухоли в расчете на единицу времени.
Лечение рака может привести к уменьшению возобновления роста опухоли. Предпочтительно, после лечения, возобновление роста опухоли составляет меньше чем 5%; более предпочтительно, возобновление роста опухоли составляет меньше чем 10%; более предпочтительно, меньше чем 20%; более предпочтительно, меньше чем 30%; более предпочтительно, меньше чем 40%; более предпочтительно, меньше чем 50%; еще более предпочтительно, меньше чем 50%; и наиболее предпочтительно, меньше чем 75%. Возобновление роста опухоли можно измерить любыми воспроизводимыми средствами измерения. Возобновление роста опухоли измеряют, например, путем измеряния увеличения диаметра опухоли после предшествующего сжатия опухоли, которое имело место после лечения. Уменьшение возобновления роста опухоли является показателем неспособности опухоли к повторному возникновению после прекращения лечения.
Лечение или профилактика клеточно-пролиферативного расстройства может приводить к уменьшению скорости клеточной пролиферации. Предпочтительно, после лечения, скорость клеточной пролиферации уменьшается по меньшей мере на 5%; более предпочтительно, по меньшей мере на 10%; более предпочтительно, по меньшей мере на 20%; более предпочтительно, по меньшей мере на 30%; более предпочтительно, по меньшей мере на 40%; более предпочтительно, по меньшей мере на 50%; еще более предпочтительно, по меньшей мере на 50%; и наиболее предпочтительно, по меньшей мере на 75%. Скорость клеточной пролиферации можно измерить любыми воспроизводимыми средствами измерения. Скорость клеточной пролиферации измеряют, например, путем измеряния количества делящихся клеток в образце ткани на единицу времени.
Лечение или профилактика клеточно-пролиферативного расстройства может приводить к уменьшению пропорции пролиферирующих клеток. Предпочтительно, после лечения, пропорция пролиферирующих клеток уменьшается по меньшей мере на 5%; более предпочтительно, по меньшей мере на 10%; более предпочтительно, по меньшей мере на 20%; более предпочтительно, по меньшей мере на 30%; более предпочтительно, по меньшей мере на 40%; более предпочтительно, по меньшей мере на 50%; еще более предпочтительно, по меньшей мере на 50%; и наиболее предпочтительно, по меньшей мере на 75%. Пропорцию пролиферирующих клеток можно измерить любыми воспроизводимыми средствами измерения. Предпочтительно, пропорцию пролиферирующих клеток измеряют, например, путем подсчета количества делящихся клеток относительно количества неделящихся клеток образца ткани. Пропорция пролиферирующих клеток может быть эквивалентной митотическому индексу.
Лечение или профилактика клеточно-пролиферативного расстройства может привести к уменьшению размера площади или зоны клеточной пролиферации. Предпочтительно, после лечения, размер площади или зоны клеточной пролиферации уменьшается по меньшей мере на 5% по сравнению с ее размером до лечения; более предпочтительно, уменьшается по меньшей мере на 10%; более предпочтительно, уменьшается по меньшей мере на 20%; более предпочтительно, уменьшается по меньшей мере на 30%; более предпочтительно, уменьшается по меньшей мере на 40%; более предпочтительно, уменьшается по меньшей мере на 50%; еще более предпочтительно, уменьшается по меньшей мере на 50%; и наиболее предпочтительно, уменьшается по меньшей мере на 75%. Размер площади или зоны клеточной пролиферации можно измерить любыми воспроизводимыми средствами измерения. Размер площади или зоны клеточной пролиферации можно измерить как диаметр или ширину площади или зоны клеточной пролиферации.
Лечение или профилактика клеточно-пролиферативного расстройства может привести к уменьшению количества или пропорции клеток, имеющих аномальный внешний вид или морфологию. Предпочтительно, после лечения, количество клеток, имеющих аномальную морфологию, уменьшается по меньшей мере на 5% по сравнению с количеством таких клеток до лечения; более предпочтительно, уменьшается по меньшей мере на 10%; более предпочтительно, уменьшается по меньшей мере на 20%; более предпочтительно, уменьшается по меньшей мере на 30%; более предпочтительно, уменьшается по меньшей мере на 40%; более предпочтительно, уменьшается по меньшей мере на 50%; еще более предпочтительно, уменьшается по меньшей мере на 50%; и наиболее предпочтительно, уменьшается по меньшей мере на 75%. Аномальный клеточный внешний вид или морфологию можно измерить любыми воспроизводимыми средствами измерения. Аномальную клеточную морфологию можно измерить при помощи микроскопии, например, с использованием инвертированного микроскопа для тканевых культур. Аномальная клеточная морфология может принимать форму нуклеарного плейоморфизма.
Как используется в настоящей заявке, термин “селективно” означает тенденцию возникновения с большей частотой в одной популяции по сравнению с другой популяцией. Сравниваемые популяции могут представлять собой клеточные популяции. Предпочтительно, соединение по настоящему изобретению или его фармацевтически приемлемая соль, пролекарство, метаболит, полиморф или сольват действует селективно на раковую или предраковую клетку, но не действует на нормальную клетку. Предпочтительно, соединение по настоящему изобретению или его фармацевтически приемлемая соль, пролекарство, метаболит, полиморф или сольват действует селективно для модуляции одной молекулярной мишени (например, являющейся мишенью протеинметилтрансферазы), но не осуществляет никакой существенной модуляции другой молекулярной мишени (например, не являющейся мишенью протеинметилтрансферазы). Настоящее изобретение также обеспечивает способ селективного ингибирования активности фермента, такого как протеинметилтрансфераза. Предпочтительно, событие происходит селективно в популяции А относительно популяции B, если оно происходит более чем в два раза чаще в популяции А по сравнению с популяцией B. Событие происходит селективно, если оно происходит более чем в пять раз чаще в популяции A. Событие происходит селективно, если оно происходит более чем в десять раз чаще в популяции A; более предпочтительно, более чем в пятьдесят раз; еще более предпочтительно, более чем в 100 раз; и наиболее предпочтительно, более чем в 1000 раз чаще в популяции А по сравнению с популяцией B. Например, можно сказать, что клеточная гибель происходит селективно в раковых клетках, если это происходит более чем в два раза чаще в раковых клетках по сравнению с нормальными клетками.
Соединение по настоящему изобретению или его фармацевтически приемлемая соль, пролекарство, метаболит, полиморф или сольват могут модулировать активность молекулярной мишени (например, являющейся мишенью протеинметилтрансферазы). Модуляция относится к стимулированию или ингибированию активности молекулярной мишени. Предпочтительно, соединение по настоящему изобретению или фармацевтически приемлемая соль, пролекарство, метаболит, полиморф или сольват такого соединения модулирует активность молекулярной мишени, если оно стимулирует или ингибирует активность молекулярной мишени по меньшей мере 2-кратно по сравнению с активностью молекулярной мишени в тех же условиях, но только без присутствия указанного соединения. Более предпочтительно, соединение по настоящему изобретению или его фармацевтически приемлемая соль, пролекарство, метаболит, полиморф или сольват модулирует активность молекулярной мишени, если оно стимулирует или ингибирует активность молекулярной мишени по меньшей мере 5-кратно, по меньшей мере 10-кратно, по меньшей мере 20-кратно, по меньшей мере 50-кратно, по меньшей мере 100-кратно по сравнению с активностью молекулярной мишени в тех же условиях, но только без присутствия указанного соединения. Активность молекулярной мишени можно измерить любыми воспроизводимыми средствами. Активность молекулярной мишени можно измерить in vitro или in vivo. Например, активность молекулярной мишени можно измерить in vitro при помощи анализа ферментативной активности или анализа связывания ДНК, или активность молекулярной мишени можно измерить in vivo путем анализа экспрессии репортерного гена.
Соединение по настоящему изобретению или его фармацевтически приемлемая соль, пролекарство, метаболит, полиморф или сольват не модулирует каким-либо существенным образом активность молекулярной мишени, если добавление соединения не стимулирует и не ингибирует активность молекулярной мишени больше чем на 10% по сравнению с активностью молекулярной мишени в тех же условиях, но только без присутствия указанного соединения.
Как используется в настоящей заявке, термин “изофермент-селективный” означает преимущественное ингибирование или стимуляцию первой изоформы фермента по сравнению с второй изоформой фермента (например, преимущественное ингибирование или стимуляцию протеинметилтрансферазного изофермента альфа по сравнению с протеинметилтрансферазным изоферментом бета). Предпочтительно, соединение по настоящему изобретению или его фармацевтически приемлемая соль, пролекарство, метаболит, полиморф или сольват демонстрирует минимально четырех-кратную разницу, предпочтительно десяти-кратную разницу, более предпочтительно пятнадцати-кратную разницу в дозе, необходимой для достижения биологического эффекта. Предпочтительно, соединение по настоящему изобретению или его фармацевтически приемлемая соль, пролекарство, метаболит, полиморф или сольват демонстрирует эту разницу в диапазоне ингибирования, и разница проиллюстрирована при ИК50, т.е. 50% ингибирования, для представляющей интерес молекулярной мишени.
Введение соединение по настоящему изобретению или его фармацевтически приемлемой соли, пролекарства, метаболита, полиморфа или сольвата в клетку или субъекту, нуждающемуся в этом, может привести к модуляции (т.е. стимуляции или ингибированию) активности представляющей интерес протеинметилтрансферазы.
Настоящее изобретение обеспечивает способы оценки биологической активности соединения по настоящему изобретению или его фармацевтически приемлемой соли, пролекарства, метаболита, полиморфа или сольвата или способы идентификации испытываемого соединения в качестве модулятора (например, ингибитора) DOT1L. DOT1L полипептиды и нуклеиновые кислоты можно использовать для тестирования соединений, которые связываются с и/или модулируют (например, увеличивают или уменьшают) одну или несколько биологических активностей DOT1L, включая, но не ограничиваясь этим, H3K79 HMTазную активность, SAM-связывающую активность, гистон и/или нуклеосома-связывающую активность, AF10-связывающую активность, активность связывания с AF10-MLL или другим MLL гибридным белком, и/или любую другую биологическую активность, представляющую интерес. DOT1L полипептид может представлять собой функциональный фрагмент полноразмерного DOT1L полипептида или его функционального эквивалента, и может включать любой представляющий интерес DOT1 домен, включая, но не ограничиваясь этим, каталитический домен, SAM-связывающий домен и/или положительно заряженный домен, взаимодействующий с AF10 домен и/или ядерный экспортируемый сигнал.
Способы оценки DOT1L связывания с гистонами, нуклеосомами, нуклеиновыми кислотами или полипептидами можно осуществить с использованием стандартных способов, которые должны быть известны специалистам в данной области (иллюстративные способы см. в разделе Примеры). Такие способы включают анализы с использованием дрожжей и двухгибридных клеток млекопитающих и методы ко-иммунопреципитации.
Например, соединение, которое модулирует активность DOT1L H3K79 HMTазы, можно контролировать путем: контактирования DOT1L полипептида с гистоновым или пептидным субстратом, включающим H3, в присутствии испытываемого соединения; определения уровня H3K79 метилирования гистонового или пептидного субстрата в условиях, достаточных для обеспечения H3K79 метилирования, где увеличение или уменьшение H3K79 метилирования в присутствии испытываемого соединения по сравнению с уровнем H3K79 метилирования гистона в отсутствии испытываемого соединения указывает на то, что испытываемое соединение модулирует активность DOT1L H3K79 HMTазы.
Способы испытания по настоящему изобретению можно осуществить в клеточной или бесклеточной системе. В качестве еще одной альтернативы, анализ можно осуществить с использованием интактного животного (включая трансгенных животных, отличных от человека). Кроме того, что касается клеточных систем, DOT1L полипептид (или любой другой полипептид, используемый в анализе) можно добавить непосредственно к клетке, или он может продуцироваться из нуклеиновой кислоты в клетке. Нуклеиновая кислота может быть эндогенной для клетки или может быть чужеродной (например, генетически модифицированная клетка).
В некоторых анализах используют иммунологические реагенты, например, антитела и антигены. В некоторых анализах для измерения ферментативной активности можно использовать флуоресценцию. Как используется в настоящей заявке, “флуоресценция” относится к способу, посредством которого молекула испускает фотон как результат абсорбирования входящего фотона более высокой энергии этой же самой молекулой. Конкретные способы оценки биологической активности раскрытых соединений описаны в примерах.
Введение соединения по настоящему изобретению или его фармацевтически приемлемой соли, пролекарства, метаболита, полиморфа или сольвата в клетку или субъекту, нуждающемуся в этом, приводит к модуляции (т.е. стимуляции или ингибированию) активности внутриклеточной мишени (например, субстрата). Некоторые внутриклеточные мишени можно модулировать соединениями по настоящему изобретению, включая, но не ограничиваясь этим, протеинметилтрансферазу.
Активирование относится к приведению композиции вещества (например, белка или нуклеиновой кислоты) в состояние, подходящее для осуществления желаемой биологической функции. Композиция вещества, которая может быть активирована, также имеет неактивированное состояние. Активированная композиция вещества может иметь ингибиторную или стимуляторную биологическую функцию, или обе такие функции.
Увеличение относится к повышению желаемой биологической активности композиции вещества (например, белка или нуклеиновой кислоты). Увеличение может происходить через повышение концентрации композици ивещества.
Как используется в настоящей заявке, “путь, включающий контрольную точку клеточного цикла” относится к биохимическому пути, который вовлечен в модуляцию контрольной точки клеточного цикла. Путь, включающий контрольную точку клеточного цикла, может иметь стимулирующие или ингибирующие эффекты, или и те и другие, в отношении одной или нескольких функций, включающих контрольную точку клеточного цикла. Путь, включающий контрольную точку клеточного цикла, состоит из, по меньшей мере, двух композиций вещества, предпочтительно белков, которые обе способствуют модуляции контрольной точки клеточного цикла. Путь, включающий контрольную точку клеточного цикла, может активироваться через активацию одного или нескольких членов пути, включающего контрольную точку клеточного цикла. Предпочтительно, путь, включающий контрольную точку клеточного цикла, представляет собой биохимический сигнальный путь.
Как используется в настоящей заявке, “регулятор контрольной точки клеточного цикла” относится к композиции вещества, которая может иметь функцию, по меньшей мере, частично, модуляции контрольной точки клеточного цикла. Регулятор контрольной точки клеточного цикла может иметь стимулирующие или ингибирующие эффекты, или и те и другие, в отношении одной или нескольких функций, включающих контрольную точку клеточного цикла. Регулятор контрольной точки клеточного цикла может представлять собой белок или небелковое вещество.
Лечение рака или клеточно-пролиферативного расстройства может привести к клеточной гибели, и, предпочтительно, клеточная гибель приводит к уменьшению по меньшей мере на 10% количества клеток в популяции. Более предпочтительно, клеточная гибель означает уменьшение по меньшей мере на 20%; более предпочтительно, уменьшение по меньшей мере на 30%; более предпочтительно, уменьшение по меньшей мере на 40%; более предпочтительно, уменьшение по меньшей мере на 50%; наиболее предпочтительно, уменьшение по меньшей мере на 75%. Количество клеток в популяции можно измерить любыми воспроизводимыми средствами. Количество клеток в популяции можно измерить методом активируемого флуоресценцией клеточного сортинга (FACS), иммунофлуоресцентной микроскопии и световой микроскопии. Способы измерения клеточной гибели описаны в Li et al., Proc Natl Acad Sci U S A. 100(5): 2674-8, 2003. В одном аспекте, клеточная гибель происходит посредством апоптоза.
Предпочтительно, эффективное количество соединения по настоящему изобретению или его фармацевтически приемлемой соли, пролекарства, метаболита, полиморфа или сольвата не является существенно цитотоксичным для нормальных клеток. Терапевтически эффективное количество соединения не является существенно цитотоксичным для нормальных клеток, если введение соединения в терапевтически эффективном количестве не индуцирует клеточную гибель в более чем 10% нормальных клеток. Терапевтически эффективное количество соединения не влияет существенным образом на жизнеспособность нормальных клеток, если введение соединения в терапевтически эффективном количестве не индуцирует клеточную гибель в более чем 10% нормальных клеток. В одном аспекте, клеточная гибель происходит посредством апоптоза.
Контактирование клетки с соединением по настоящему изобретению или его фармацевтически приемлемой солью, пролекарством, метаболитом, полиморфом или сольватом может индуцировать или активировать клеточную гибель селективно в раковых клетках. Введение субъекту, нуждающемуся в этом, соединения по настоящему изобретению или его фармацевтически приемлемой соли, пролекарства, метаболита, полиморфа или сольвата может индуцировать или активировать клеточную гибель селективно в раковых клетках. Контактирование клетки с соединением по настоящему изобретению его фармацевтически приемлемой солью, пролекарством, метаболитом, полиморфом или сольватом может индуцировать клеточную гибель селективно в одной или нескольких клетках, пораженных клеточно-пролиферативным расстройством. Предпочтительно, введение субъекту, нуждающемуся в этом, соединения по настоящему изобретению или его фармацевтически приемлемой соли, пролекарства, метаболита, полиморфа или сольвата индуцирует клеточную гибель селективно в одной или нескольких клетках, пораженных клеточно-пролиферативным расстройство.
Настоящее изобретение относится к способу лечения или профилактики рака путем введения соединения по настоящему изобретению или его фармацевтически приемлемой соли, пролекарства, метаболита, полиморфа или сольвата субъекту, нуждающемуся в этом, где введение соединения по настоящему изобретению или его фармацевтически приемлемой соли, пролекарства, метаболита, полиморфа или сольвата приводит к одному или нескольким следующим эффектам: аккумуляция клеток в G1 и/или S фазе клеточного цикла, цитотоксичность через клеточную гибель в раковых клетках без существенного количества клеточной гибели в нормальных клетках, притивоопухолевая активность у животных с терапевтическим индексом по меньшей мере 2 и активация контрольной точки клеточного цикла. Как используется в настоящей заявке, “терапевтический индекс” представляет собой максимально переносимую дозу, деленную на эффективную дозу.
Специалисты в данной области могут обратиться к общей справочной литературе, где можно найти подробные описания известных способов, обсуждаемых в настоящей заявке, или эквивалентных способов. Эти справочники включают Ausubel et al., Current Protocols in Molecular Biology, John Wiley and Sons, Inc. (2005); Sambrook et al., Molecular Cloning, Laboratory Manual (3rd edition), Cold Spring Harbor Press, Cold Spring Harbor, New York (2000); Coligan et al., Current Protocols in Immunology, John Wiley & Sons, N.Y.; Enna et al., Current Protocols in Pharmacology, John Wiley & Sons, N.Y.; Fingl et al., The Pharmaceutical Basis of Therapeutics (1975), Remington's Pharmaceutical Sciences, Mack Publishing Co., Easton, PA, 18th edition (1990). Конечно, на эти тексты можно ссылаться также при осуществлении или использовании аспекта настоящего изобретения.
Соединения по настоящему изобретению также можно использовать для лечения или профилактики неврологических заболеваний или расстройств. Неврологические заболевания или расстройства, которые можно лечить соединениями по настоящему изобретению, включают эпилепсию, шизофрению, биполярное расстройство или другие физиологические и/или психиатрические расстройства, невропатии, скелетно-мышечную атрофию и нейродегенеративные заболевания, например, нейродегенеративное заболевание. Примеры нейродегенеративных заболеваний включают: болезнь Альцгеймера, амиотрофический боковой склероз (ALS) и болезнь Паркинсона. Другой класс нейродегенеративных заболеваний включает заболевания, вызванные, по меньшей мере, частично, агрегацией поли-глутамина. Заболевания этого класса включают: болезнь Гентингтона, спинально-бульбарную мышечную атрофию (SBMA или болезнь Кеннеди), дентатопаллидорубральную атрофию (DRPLA), спино-мозжечковую атаксию 1 (SCA1), спино-мозжечковую атаксию 2 (SCA2), болезнь Machado-Joseph (MJD; SCA3), спино-мозжечковую атаксию 6 (SCA6), спино-мозжечковую атаксию 7 (SCA7) и спино-мозжечковую атаксию 12 (SCA12).
Любое другое заболевание, в котором играет роль эпигенетическое метилирование, которое опосредовано DOT1, можно лечить или предотвратить с использованием соединений и способов, описанных в настоящей заявке.
4. Фармацевтические композиции
Настоящее изобретение также обеспечивает фармацевтические композиции, включающие соединение формул (I), (II), (IIIa), (IIIb), (IIIc) и (IV) в сочетании с, по меньшей мере, одним фармацевтически приемлемым эксципиентом или носителем.
“Фармацевтическая композиция” представляет собой композицию, содержащую соединения по настоящему изобретению в форме, подходящей для введения субъекту. В одном варианте воплощения, фармацевтическая композиция представлена без упаковки или в виде стандартных доз. Стандартная доза может быть представлена в любой из различных форм, включая, например, капсулу, содержащий внутривенную дозу резервуар, таблетку, одну впрыскиваемую дозу в аэрозольном ингаляторе или ампулу. Количество активного ингредиента (например, композиции раскрытого соединения или его соли, гидрата, сольвата или изомера) в стандартной дозе композиции представляет собой эффективное количество, и оно варьируется в соответствии с конкретным лечением. Специалистам в данной области должно быть понятно, что иногда необходимы рутинные изменения дозы в зависимости от возраста и состояния пациента. Доза также зависит от пути введения. Рассматриваются различные пути, включая пероральный, пульмонарный, ректальный, парентеральный, чрескожный, подкожный, внутривенный, внутримышечный, интраперитонеальный, путем ингаляции, буккальный, сублингвальный, интраплевральный, интратекальный, интраназальный и т.п. Лекарственные формы для местного или чрескожного введения соединения по настоящему изобретению включают порошки, спреи, мази, пасты, кремы, лосьоны, гели, растворы, пластыри и препараты для ингаляций. В одном варианте воплощения, активное соединение смешивают в стерильных условиях с фармацевтически приемлемым носителем и с любыми консервантами, буферами или пропеллентами, которые необходимы.
Как используется в настоящей заявке, фраза “фармацевтически приемлемый” относится к таким соединениям, веществам, композициям, носителям и/или лекарственным формам, которые, согласно взвешенной медицинской оценке, являются подходящими для использования в контакте с тканями человека и животных, без чрезмерной токсичности, раздражения, аллергических реакций или других проблем или осложнений, и соответствуют разумному отношению польза/риск.
“Фармацевтически приемлемый эксципиент” означает эксципиент, который является полезным для получения фармацевтической композиции, которая в целом является безопасной, нетоксичной и не является ни биологически ни каким-либо иным образом нежелательной, и включает эксципиент, который является приемлемым для применения в ветеринарии, а также фармацевтически приемлемый для человека. “Фармацевтически приемлемый эксципиент”, как это используется в описании изобретения и в формуле изобретения, включает как один, так и более одного таких эксципиентов.
Фармацевтическую композицию по настоящему изобретению формулируют таким образом, чтобы она была совместима с предполагаемым путем ее введения. Примеры путей введения включают парентеральный, например, внутривенный, внутрикожный, подкожный, пероральный (например, путем ингаляции), чрескожный (местный) и введение через слизистую оболочку. Растворы или суспензии, используемые для парентерального, внутрикожного или подкожного применения, могут включать следующие компоненты: стерильный разбавитель, такой как вода для инъекций, физиологический раствор, нелетучие масла, полиэтиленгликоли, глицерин, пропиленгликоль или другие синтетические растворители; антибактериальные средства, такие как бензиловый спирт или метилпарабены; антиокислители, такие как аскорбиновая кислота или бисульфит натрия; хелатообразующие вещества, такие как этилендиаминтетрауксусная кислота; буферы, такие как ацетаты, цитраты или фосфаты, и средства для регулирования тоничности, такие как хлорид натрия или декстроза. pH можно регулировать при помощи кислот или оснований, таких как хлористоводородная кислота или гидроксид натрия. Парентеральный препарат может быть заключен в ампулы, одноразовые шприцы или многодозовые флаконы из стекла или пластика.
Соединение или фармацевтическую композицию по настоящему изобретению можно вводить субъекту различными хорошо известными способами, которые в настоящее время используют для химиотерапевтического лечения. Например, для лечения рака, соединение по настоящему изобретению можно вводить путем инъекции непосредственно в опухоли, вводить путем инъекции в кровоток или полости организма или принимать перорально или наносить при помощи пластырей для введения через кожу. Выбранная доза должна быть достаточной для обеспечения эффективного лечения, но не такой высокой, чтобы вызывать неприемлемые побочные эффекты. Следует внимательно наблюдать за состоянием заболевания (например, рак, предраковое состояние и т.п.) и здоровьем пациента, предпочтительно в течение всего периода лечения и в течение разумного периода после лечения.
Термин “терапевтически эффективное количество”, как используется в настоящей заявке, относится к количеству фармацевтического средства для лечения, уменьшения интенсивности симптомов или профилактики идентифицированного заболевания или состояния или для проявления заметного терапевтического или ингибиторного эффекта. Эффект можно определить при помощи любого анализа, известного из уровня техники. Точное эффективное количество для субъекта зависит от массы тела субъекта, его размера и состояния здоровья; природы и степени тяжести состояния; и терапевтического средства, выбранного для введения. Терапевтически эффективные количества для конкретной ситуации можно определить путем рутинного экспериментирования, и это вполне может осуществить клиницист на основании своих знаний и оценок. В предпочтительном аспекте, заболевание или состояние, подлежащее лечению, представляет собой рак. В другом аспекте, заболевание или состояние, подлежащее лечению, представляет собой клеточно-пролиферативное расстройство.
Для любого соединения, терапевтически эффективное количество может быть определено сначала либо в анализах клеточной культуры, например, опухолевых клеток, либо в животных моделях, обычно крыс, мышей, кроликов, собак или свиней. Животную модель также можно использовать для определения подходящего диапазона концентраций и пути введения. Такую информацию затем можно использовать для определения полезных доз и путей для введения человеку. Терапевтическую/профилактическую эффективность и токсичность можно определить при помощи стандартных фармацевтических процедур с использованием клеточных культур или экспериментальных животных, например, ED50 (доза, являющаяся терапевтически эффективной в 50% популяции) и LD50 (доза, являющаяся летальной для 50% популяции). Отношение доз, вызывающих токсический и терапевтический эффекты, представляет собой терапевтический индекс и может быть выражено как отношение LD50/ED50. Фармацевтические композиции, которые демонстрируют высокие терапевтические индексы, являются предпочтительными. Доза может варьировать в этих пределах, в зависимости от используемой лекарственной формы, чувствительности пациента и пути введения.
Дозу и введение регулируют для обеспечения достаточных уровней активного вещества(веществ) или для поддержания желаемого эффекта. Факторы, которые могут быть приняты во внимание, включают тяжесть заболевания, общее состояние здоровья субъекта, возраст, массу тела и пол субъекта, режим питания, время и частоту введения, лекарственное взаимодействие(взаимодействия), аллергические реакции и переносимость/ответ на терапию. Долго действующие фармацевтические композиции можно вводить через каждые 3-4 дня, через неделю или раз в две недели, в зависимости от периода полужизни и скорости выведения из организма конкретной композиции.
Фармацевтические композиции, содержащие активные соединения по настоящему изобретению, можно получить способом, который, как правило, является известным, например, способами обычного смешивания, растворения, гранулирования, получения драже, растирания в порошок, эмульгирования, инкапсулирования, заключения в оболочку или лиофилизации. Фармацевтические композиции можно формулировать обычным способом с использованием одного или нескольких фармацевтически приемлемых носителей, включая эксципиенты и/или вспомогательные вещества, которые способствуют переработке активных соединений в препараты, которые можно использовать фармацевтически. Конечно, подходящая композиция зависит от выбранного пути введения.
Фармацевтические композиции, подходящие для введения путем инъекции, включают стерильные водные растворы (когда композиция является водорастворимой) или дисперсии и стерильный порошки для приготовления непосредственно перед применением стерильных растворов или дисперсии для инъекций. Для внутривенного введения, подходящие носители включают физиологический раствор, бактериостатическую воду, Cremophor EL™ (BASF, Parsippany, N.J.) или фосфатно-буферный физиологический раствор(PBS). Во всех случаях, композиция должна быть стерильной и должна быть жидкой до такой степени, чтобы ее можно было вводить через шприц. Она должна быть стабильной в условиях ее изготовления и хранения и должна быть защищена против загрязняющего действия микроорганизмов, таких как бактерии и грибы. Носитель может представлять собой растворитель или диспергирующую среду, содержащую, например, воду, этанол, полиол (например, глицерин, пропиленгликоль и жидкий полиэтиленгликоль и т.п.), и подходящие смеси таких веществ. Необходимую текучесть можно поддерживать, например, путем использования покрытия, такого как лецитин, путем поддержания нужного размера частиц в случае дисперсии и путем использования поверхностно-активных веществ. Предотвращение действия микроорганизмов достигается при помощи различных антибактериальных и противогрибковых средств, например, парабенов, хлорбутанола, фенола, аскорбиновой кислоты, тимеросала и т.п. Во многих случаях предпочтительно включать в композицию изотонические вещества, например, сахара, полиспирты, такие как маннит и сорбит, и хлорид натрия. Пролонгированную абсорбцию композиций для инъекций можно получить путем включения в композицию вещества, которое замедляет абсорбцию, например, алюминиймоностеарат и желатин.
Старильные растворы для инъекций можно получить путем включения активного соединения в необходимом количестве в подходящий растворитель, с использованием одного или комбинации ингредиентов, перечисленных выше, при необходимости, с последующей стерилизацией фильтрованием. Как правило, дисперсии получают путем включения активного соединения в стерильный носитель, который содержит щелочную диспергирующую среду и другие необходимые ингредиенты из перечисленных выше. В случае стерильных порошков для получения стерильных растворов для инъекций, способы их получения включают вакуумную сушку и сушку замораживанием, которая дает порошок активного ингредиента плюс любого дополнительного желаемого ингредиента из его ранее стерильно отфильтрованного раствора.
Пероральные композиции, как правило, включают инертный разбавитель или пищевой фармацевтически приемлемый носитель. Они могут быть заключены в желатиновые капсулы или спрессованы в таблетки. Для целей перорального терапевтического введения, активное соединение может быть включено с эксципиентами и использоваться в форме таблеток, пастилок или капсул. Пероральные композиции также можно получить с использованием жидкого носителя для применения в виде жидкости для полоскания рта, где соединение в жидком носителе применяют перорально и после полоскания сплевывают или проглатывают. Фармацевтически совместимые связующие вещества и/или адъюванты могут быть включены как часть композиции. Таблетки, пилюли, капсулы, пастилки и т.п. могут содержать любой из следующих ингредиентов или имеющие подобную природу соединения: связующее, такое как микрокристаллическая целлюлоза, камедь трагаканта или желатин; эксципиент, такой как крахмал или лактоза, разрыхлитель, такой как альгиновая кислота, Primogel или кукурузный крахмал; смазывающее вещество, такое как стеарат магния или Sterotes; агент скольжения, такой как коллоидный диоксид кремния; подсластитель, такой как сахароза или сахарин; или отдушка, такая как перечная мята, метилсалицилат или апельсиновая отдушка.
Для введения путем ингаляции соединения доставляют в форме аэрозольного спрея из находщегося под давлением контейнера или дозатора, который содержит подходящий пропеллент, например, газ, такой как диоксид углерода, или небулайзера.
Системное введение также можно осуществить через слизистую оболочку или чрескожным путем. Для введения через слизистую оболочку или чрескожного введения в композиции используют агенты пенетрации, подходящие для барьера, через который происходит проникновение композиции. Такие агенты пенетрации, как правило, известны из уровня техники и включают, например, для введения через слизистую оболочку, детергенты, соли желчных кислот и производные фусидовой кислоты. Введение через слизистую оболочку можно осуществить с использованием назальных спреей или суппозиториев. Для чрескожного введения активные соединения формулируют в виде мазей, бальзамов, гелей или кремов, как в основном известно из уровня техники.
Активные соединения могут быть сформулированы с фармацевтически приемлемыми носителями, которые защищают соединение от быстрого выведения из организма, например, композиция контролируемого высвобождения, включая имплантаты и микроинкапсулированные системы доставки. Можно использовать биоразлагаемые, биосовместимые полимеры, такие как этиленвинилацетат, полиангидриды, полигликолевая кислота, коллаген, сложные полиортоэфиры и полимолочная кислота. Способы получения таких композиций должны быть известны специалистам в данной области. Такие вещества также можно получить коммерческим путем от Alza Corporation и Nova Pharmaceuticals, Inc. Липосомные суспензии (включая липосомы, нацеленные на инфицированные клетки, с моноклональными антителами к вирусным антигенам) также можно использовать в качестве фармацевтически приемлемых носителей. Их можно получить в соответствии со способами, известными специалистам в данной области, например, как описано в патенте США № 4522811.
Особенно выгодно формулировать пероральные или парентеральные композиции в виде стандартных лекарственных форм для простоты введения и равномерного дозирования. Стандартная лекарственная форма, как это используется в настоящей заявке, относится к физически дискретным единицам, подходящим в качестве унитарных доз для субъекта, подлежащего лечению; при этом каждая единица содержит предварительно определенное количество активного соединения, рассчитанное для обеспечения желаемого терапевтического эффекта, в ассоциации с необходимым фармацевтическим носителем. Требования к стандартным лекарственным формам по настоящему изобретению продиктованы и непосредственно зависят от уникальных характеристик активного соединения и конкретного терапевтического эффекта, который нужно получить.
Для терапевтического применения дозы фармацевтических композиций, используемых в соответствии с настоящим изобретением, варьируются в зависимости от средства, возраста, массы тела и клинического состояния пациента, который является реципиентом, а также опыта и мнения клинициста или специалиста, осуществляющего терапию, среди других факторов, влияющих на выбор дозы. Как правило, доза должна быть достаточной для обеспечения замедления, и предпочтительно регрессии, роста опухолей, а также предпочтительно вызывать полную регрессию рака. Дозы могут находиться в пределах от около 0,01 мг/кг в день до около 5000 мг/кг в день. В предпочтительных аспектах, дозы могут находиться в пределах от около 1 мг/кг в день до около 1000 мг/кг в день. В одном аспекте, доза будет находиться в пределах около 0,1 мг/день до около 50 г/день; от около 0,1 мг/день до около 25 г/день; от около 0,1 мг/день до около 10 г/день; от около 0,1 мг до около 3 г/день; или от около 0,1 мг до около 1 г/день, в виде одной дозы, раздельных или непрерывных доз (такую дозу можно отрегулировать в расчете на массу тела пациента в кг, площадь поверхности тела в м2 и возраст в годах). Эффективное количество фармацевтического средства является таким, которое обеспечивает объективно идентифицируемое улучшение, которое отмечает клиницист или другой квалифицированный наблюдающий специалист. Например, регрессию опухоли у пациента можно измерить по диаметру опухоли. Уменьшение опухоли в диаметре указывает на регрессию. На регрессию также указывает неспособность опухолей снова возникать после прекращения лечения. Как используется в настоящей заявке, термин “дозо-эффективным образом” относится к количеству активного соединения для получения желаемого биологического эффекта у субъекта или в клетке.
Фармацевтические композиции могут быть заключены в контейнер, упаковку или дозирующее устройство вместе с инструкциями по введению.
Соединения по настоящему изобретению способны к дальнейшему образованию солей. Все из этих форм также предусматриваются как охватываемые объемом заявленного изобретения.
Как используется в настоящей заявке, “фармацевтически приемлемые соли” относятся к производным соединений по настоящему изобретению, где исходное соединение является модифицированным с образованием его кислотных или основных солей. Примеры фармацевтически приемлемых солей включают, но не ограничиваются этим, соли минеральных или органических кислот основных остатков, таких как амины, щелочные или органические соли кислотных остатков, таких как карбоновые кислоты и т.п. Фармацевтически приемлемые соли включают традиционные нетоксичные соли или четвертичные аммониевые соли исходного соединения, образованные, например, из нетоксичных неорганических или органических кислот. Например, такие традиционные нетоксичные соли включают, но не ограничиваются этим, соли, образованные из неорганических и органических кислот, выбранных из 2-ацетоксибензойной, 2-гидроксиэтансульфоновой, уксусной, аскорбиновой, бензолсульфоновой, бензойной, биугольной, угольной, лимонной, эдетиновой, этандисульфоновой, 1,2-этансульфоновой, фумаровой, глюкогептоновой, глюконовой, глутаминовой, гликолевой, гликоллиарсаниловой, гексилрезорциновой, гидрабамовой, бромистоводородной, хлористоводородной, иодистоводородной, гидроксималеиновой, гидроксинафтойной, изетионовой, молочной, лактобионовой, лаурилсульфоновой, малеиновой, яблочной, миндальной, метансульфоновой, напсиловой, азотной, щавелевой, памовой, пантотеновой, фенилуксусной, фосфорной, полигалактуроновой, пропионовой, салициловой, стеариновой, субуксусной, янтарной, сульфаминовой, сульфаниловой, серной, дубильной, винной, толуолсульфоновой, и широко распространенных аминокислот, например, глицина, аланина, фенилаланина, аргинина и т.п.
Другие примеры фармацевтически приемлемых солей включают соли гексановой кислоты, циклопентанпропионовой кислоты, пирувиновой кислоты, малоновой кислоты, 3-(4-гидроксибензоил)бензойной кислоты, коричной кислоты, 4-хлорбензолсульфоновой кислоты, 2-нафталинсульфоновой кислоты, 4-толуолсульфоновой кислоты, камфорсульфоновой кислоты, 4-метилбицикло-[2,2,2]-окт-2-ен-1-карбоновой кислоты, 3-фенилпропионовой кислоты, триметилуксусной кислоты, третичной бутилуксусной кислоты, муконовой кислоты и т.п. Настоящее изобретение также охватывает соли, образованные, когда кислотный протон, присутствующий в исходном соединении, либо замещается металлическим ионом, например, ионом щелочного металла, ионом щелочно-земельного металла или ионом алюминия; или координируется с органическим основанием, таким как этаноламин, диэтаноламин, триэтаноламин, трометамин, N-метилглюкамин и т.п.
Должно быть понятно, что все ссылки на фармацевтически приемлемые соли включают формы добавления растворителя (сольваты) или кристаллические формы (полиморфы), определенные в настоящей заявке, этой же самой соли.
Соединения по настоящему изобретению также можно получить в форме сложных эфиров, например, фармацевтически приемлемых сложных эфиров. Например, карбоновокислотную функциональную группу в соединении можно преобразовать в ее соответствующий сложный эфир, например, метиловый, этиловый или другой эфир. Также, спиртовую группу в соединении можно преобразовать в ее соответствующий сложный эфир, например, ацетатный, пропионатный или другой эфир.
Соединения по настоящему изобретению также можно получить в форме пролекарств, например, фармацевтически приемлемых пролекарств. Термины “про-лекарственный” и “пролекарство” используют взаимозаменяемо в настоящей заявке, и они относятся к любому соединению, которое высвобождает активное исходное лекарственное средство in vivo. Поскольку известно, что пролекарства усиливают различные желательные качества фармацевтических средств (например, растворимость, биодоступность, технологические свойства и т.п.), доставку соединений по настоящему изобретению можно осуществлять в форме пролекарств. Таким образом, предполагается, что настоящее изобретение охватывает пролекарства заявленных соединений, способы их доставки и композиции, содержащие такие пролекарства. “Пролекарства” включают любые ковалентно-связанные носители, которые высвобождают активное исходное лекарственное средство по настоящему изобретению in vivo, когда такое пролекарство вводят субъекту. Пролекарства в настоящем изобретении получают путем модификации функциональной группы, присутствующей в соединении, таким образом, чтобы такие модификации расщеплялись in vivo до исходного соединения. Пролекарства включают соединения по настоящему изобретению, где гидрокси, амино, сульфгидрильная, карбокси или карбонильная группа связана с любой группой, которая может отщепляться in vivo с образованием свободной гидроксильной, свободной амино, свободной сульфгидрильной, свободной карбокси или свободной карбонильной группы, соответственно.
Примеры пролекарств включают, но не ограничиваются этим, сложные эфиры (например, ацетаты, диалкиламиноацетаты, формиаты, фосфаты, сульфаты и бензоатные производные) и карбаматы (например, N,N-диметиламинокарбонил) функциональной гидроксигруппы, сложные эфиры (например, этиловые сложные эфиры, морфолиноэтанольные сложные эфиры) карбоксильной функциональной группы, N-ацил-производные (например, N-ацетил) N-оснований Манниха, оснований Шиффа и енаминов функциональной аминогруппы, оксимы, ацетали, кетали и енольные сложные эфиры функциональных групп кетона и альдегида в соединениях по настоящему изобретению и т.п., см. Bundegaard, H., Design of Prodrugs, p1-92, Elesevier, New York-Oxford (1985).
Соединения или их фармацевтически приемлемые соли, сложные эфиры или пролекарства вводят перорально, назально, чрескожно, пульмонарно, путем ингаляции, буккально, сублингвально, интраперитонеально, подкожно, внутримышечно, внутривенно, ректально, интраплеврально, интратекально и парентерально. В одном варианте воплощения, соединение вводят перорально. Специалистам в данной области должны быть понятны преимущества некоторых путей введения.
Схему приема лекарственных средств, включающих соединения по настоящему изобретению, выбирают с учетом различных факторов, включая тип, вид, возраст, массу тела, пол и медицинское состояние пациента; тяжесть состояния, подлежащего лечению; путь введения; почечную и печеночную функцию пациента; и конкретное используемое соединение или его соль. Имеющий среднюю квалификацию лечащий врач или ветеринар легко сможет определить и прописать эффективное количество лекарственного средства, необходимое для профилактики, противодействия или остановки прогрессирования состояния.
Методы формулирования и введения раскрытых соединений по настоящему изобретению можно найти в Remington: the Science and Practice of Pharmacy, 19th edition, Mack Publishing Co., Easton, PA (1995). В одном варианте воплощения, соединения, описанные в настоящей заявке, и их фармацевтически приемлемые соли используют в фармацевтических препаратах в сочетании с фармацевтически приемлемым носителем или разбавителем. Подходящие фармацевтически приемлемые носители включают инертные твердые наполнители или разбавители и стерильные водные или органические растворы. Соединения могут присутствовать в таких фармацевтических композициях в количествах, достаточных для обеспечения желаемого дозируемого количества в пределах, описанных в настоящей заявке.
Все проценты и отношения, используемые в настоящей заявке, если не указано иное, указаны в расчете на массу. Другие характеристики и преимущества настоящего изобретения очевидны из различных примеров. Представленные примеры иллюстрируют различные компоненты и приемы, полезные при осуществлении настоящего изобретения. Примеры не ограничивают заявленное изобретение. На основании представленного раскрытия, специалист в данной области может определить и использовать другие компоненты и приемы, полезные для осуществления настоящего изобретения.
В схемах синтеза, описанных в настоящей заявке, соединения могут быть представлены в одной конкретной конфигурации, для простоты. Такие конкретные конфигурации не следует рассматривать как ограничивающие настоящее изобретение тем или иным изомером, таутомером, региоизомером или стереоизомером, также это не исключает смеси изомеров, таутомеров, региоизомеров или стереоизомеров.
Соединения, описанные в настоящей заявке, анализируют на модуляцию активности, например, метилирования гистона, модуляцию клеточного роста и/или ИК50, как описано в примерах ниже. Значения ИК50 представлены как = <0,1 мкМ; B = >0,1 мкМ и <1 мкМ; C = >1 мкМ и <10 мкМ; и D = >10 мкМ и <50 мкМ.
ИК50
(мкМ)
Все публикации и патентные документы, на которые ссылаются в настоящей заявке, включены в настоящую заявку посредством ссылки, как если бы каждая такая публикация или документ были специально и отдельно указаны как включенные в настоящую заявку посредством ссылки. Ссылка на публикации и патентные документы не предполагает допущения, что какой-либо из этих документов является известным уровнем техники, имеющим отношение к предмету настоящей заявки, это также не служит каким-либо признанием того, что касается их содержания или даты. Настоящее изобретение описано выше путем письменного описания, специалистам в данной области должно быть понятно, что настоящее изобретение можно осуществить в различных вариантах воплощения, и что представленное выше описание и следующие далее примеры предназначены для иллюстрации, а не для ограничения формулы изобретения, которая представлена ниже.
5. Примеры
Спектры ядерного магнитного резонанса (ЯМР) получали на спектрометре Bruker Avance 400, работающем при напряженности поля 400,130 МГц, или Bruker DRX 500 МГц ЯМР, или HЯМР спектры получали на спектрометре 500 МГц Bruker AVANCE III. Обычно используемые в реакциях растворители имели чистоту, либо соответствующую требованиям для использования в высоко-эффективной жидкостной хроматографии (ВЭЖХ), либо соответствующую требованиям Американского Химического Общества (ACS), и были безводными, как их получали от изготовителя, если не указано иное. ЖХ/MS анализ осуществляли на Waters Micromass ZMD с разделительным модулем Waters 2795 Separations Module и с фотодиодным матричным детектором Waters Micromass 996, и на Waters Micromass ZQ с разделительным модулем Waters 2695 Separations Module и с фотодиодным матричным детектором Waters 996, или на индивидуальном квадрупольном масс-спектрометре Waters Micromass Platform LCZ с модулем подачи растворителя Waters 600, вспомогательными насосами Waters 515, УФ-детектором Waters 2487 и автоматическим пробоотборником и сборником фракций Gilson 215. Или ЖХ/MS анализ осуществляли с использованием SQ масс- спектрометра, объединенного с ВЭЖХ-устройством AGILENT 1200 Series HPLC. Данные ЖХ/MS, если они имеются, представлены в примерах ниже, а также в Таблице 1. Данные MS представлены с использованием условных обозначений для m/z в формате [M + H]+.
Соединения по настоящему изобретению можно получить с использованием известных химических преобразований, адаптированных для данной конкретной ситуации.
Препаративный Пример 1: Исходные вещества или промежуточные соединения
Стадия 1: (1R,2S,3R,5R)-3-((5-амино-6-хлорпиримидин-4-ил)амино)-5-(гидроксиметил)циклопентан-1,2-диол
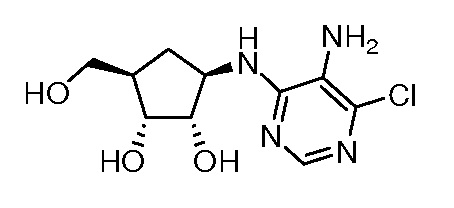
Смесь гидрохлорида (1R,2S,3R,5R)-3-амино-5-(гидроксиметил)циклопентан-1,2-диола (16,9 г, 45,1 ммоль) и 4,6-дихлорпиримидин-5-амина (5,7 г, 35 ммоль) в этаноле (45 мл) равномерно распределяли по трем пробиркам и подвергали условиям микроволнового облучения (CEM устройство, 300 Вт макс., 150°C макс., 250 ф/дюйм2 макс.(17,6 кг/см2), 3 мин. линейное изменение, 30 мин. удерживание) с получением коричневых растворов; ВЭЖХ/ЖХ-MS анализ показал преобразование в желаемый продукт. Три реакционные смеси объединяли и концентрировали в вакууме с получением неочищенного указанного в заголовке соединения в виде темно-коричневого масла, которое концентрировали из толуола (2×30 мл) и использовали далее без очистки: MS (ESI+) для C10H15ClN4O3 m/z 275,0 (M+H)+; MS (ESI-) для C10H15ClN4O3 m/z 273,0 (M-H)-.
Стадия 2: ((3aR,4R,6R,6aS)-6-(6-хлор-9H-пурин-9-ил)-2-этокситетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метанол
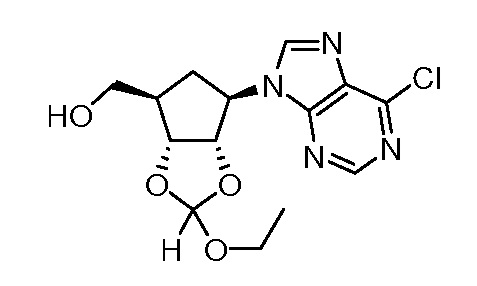
Полученный на предыдущей стадии неочищенный (1R,2S,3R,5R)-3-((5-амино-6-хлорпиримидин-4-ил)амино)-5-(гидроксиметил)циклопентан-1,2-диол обрабатывали этилортоформиатом (120 мл, 720 ммоль) и 10-камфорсульфоновой кислотой (8,11 г, 34,9 ммоль). Гетерогенную коричневую смесь интенсивно перемешивали с получением почти гомогенного коричневого раствора через 10 минут. Через 5 часов ЖХ-MS анализ показал желаемый продукт в виде основного продукта, и реакцию гасили насыщенным водным раствором NaHCO3 (120 мл). Смесь разбавляли водой (75 мл), экстрагировали при помощи CH2Cl2 (3×200 мл) и объединенные органические слои сушили (Na2SO4) и концентрировали в вакууме с получением неочищенного указанного в заголовке соединения в виде темно-коричневой жидкого вещества, которое использовали далее без дополнительных манипуляций: MS (ESI+) для C14H17ClN4O4 m/z 341,0 (M+H)+.
Стадия 3: ((3aR,4R,6R,6aS)-6-(6-хлор-9H-пурин-9-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метанол
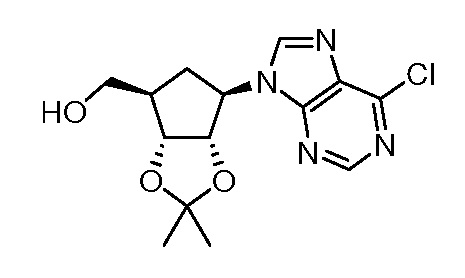
Полученный на предыдущей стадии неочищенный ((3aR,4R,6R,6aS)-6-(6-хлор-9H-пурин-9-ил)-2-этокситетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метанол брали для поглощения в 2,2-диметоксипропан (214 мл, 1740 ммоль) и обрабатывали моногидратом п-толуолсульфоновой кислоты (13,2 г, 69,5 ммоль) с получением коричневого масла, частично суспендированного в мутном растворе, который перемешивали при комнатной температуре в течение 1 часа 20 минут; ВЭЖХ/ЖХ-MS анализ показал полное преобразование в желаемый продукт. Реакцию гасили путем осторожного добавления бикарбоната натрия (8,76 г, 104 ммоль) и минимального количества воды. Летучие вещества удаляли в вакууме и оставшийся водный слой разбавляли водой (100 мл) и экстрагировали при помощи CH2Cl2 (3×400 мл). Объединенные органические слои сушили (Na2SO4) и концентрировали в вакууме с получением коричневого масла. Очистка колоночной хроматографией (7×16 см диоксид кремния; 0-5% MeOH/CH2Cl2) давала указанное в заголовке соединение (8,30 г, 74% от 3 стадий) в виде желтого пенистого вещества: MS (ESI+) для C14H17ClN4O3 m/z 325,1 (M+H)+; MS (ESI-) для C14H17ClN4O3 m/z 369,0 (M+HCO2)-; ВЭЖХ чистота >95% площади.
Стадия 4: 9-((3aS,4R,6R,6aR)-6-(азидометил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-6-хлор-9H-пурин
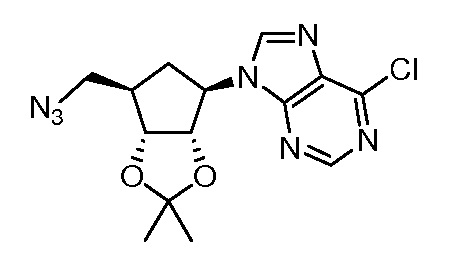
Смесь((3aR,4R,6R,6aS)-6-(6-хлор-9H-пурин-9-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метанола (7,9 г, 24 ммоль) и трифенилфосфина на полимерном носителе (3 ммоль/г нагрузка; 11 г, 34 ммоль) в ТГФ (100 мл) охлаждали до 0°C (баня лед/насыщенный солевой раствор) и обрабатывали по каплям диизопропилазодикарбоксилатом (6,7 мл, 34 ммоль). Желто-коричневую суспензию перемешивали в течение 15 минут и обрабатывали по каплям раствором азида дифенилфосфоновой кислоты (7,3 мл, 34 ммоль) в ТГФ (24 мл). Коричневую реакционную смесь перемешивали в течение 18,5 часов пока ледяная баня не растаяла; ВЭЖХ анализ показал преобразование в желаемый продукт. По прошествии 21,5 часов реакционную смесь фильтровали, твердые вещества промывали при помощи CH2Cl2 и фильтрат концентрировали в вакууме. Красно-коричневый остаток брали для поглощения в CH2Cl2 (300 мл) и промывали насыщенным водным раствором NaHCO3 (1×100 мл), водой (1×100 мл) и насыщенным солевым раствором (1×150 мл). Отделенный органический слой сушили (Na2SO4) и концентрировали в вакууме с получением красно-оранжевого масла. Очистка колоночной хроматографией (7×16 см диоксид кремния; 0-10% ацетона/CH2Cl2) давала указанное в заголовке соединение (4,82 г, 57%) в виде желтого маслянистого/пенообразного вещества MS (ESI+) для C14H16ClN7O2 m/z 350,1 (M+H)+; MS (ESI-) для C14H16ClN7O2 m/z 394,1 (M+HCO2)-; ВЭЖХ чистота >95% площади.
Стадия 5: 9-((3aS,4R,6R,6aR)-6-(азидометил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-N-(2,4-диметоксибензил)-9H-пурин-6-амин
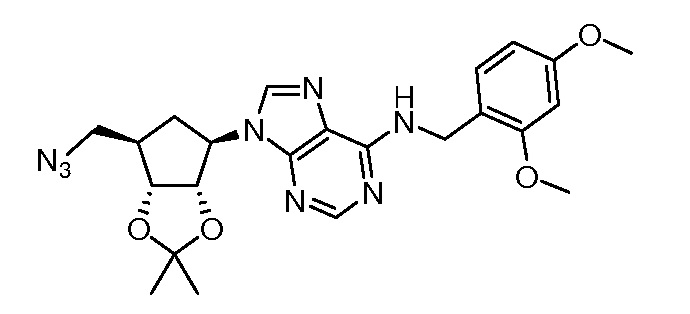
Раствор 9-((3aS,4R,6R,6aR)-6-(азидометил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-6-хлор-9H-пурина (1,29 г, 3,69 ммоль) и (2,4-диметоксифенил)метанамина (0,71 мл, 4,7 ммоль) в 1-бутаноле (10 мл) обрабатывали N,N-диизопропилэтиламином (0,93 мл, 5,3 ммоль) и нагревали при 80°C в течение 16,5 часов; ВЭЖХ/ЖХ-MS анализ показал преобразование в желаемый продукт. Реакционной смеси давали охладиться до комнатной температуры и летучие вещества удаляли под потоком воздуха с получением коричневато-оранжевого пастообразного вещества. Очистка колоночной хроматографией (2×8 см диоксид кремния; 0-10% ацетона/CH2Cl2) давала указанное в заголовке соединение (1,72 г, 97%) в виде желтовато-оранжевого пенистого/маслянистого вещества: MS (ESI+) для C23H28N8O4 m/z 481,2 (M+H)+; ВЭЖХ чистота >95% площади.
Стадия 6: 9-((3aS,4R,6R,6aR)-6-(аминометил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-N-(2,4-диметоксибензил)-9H-пурин-6-амин
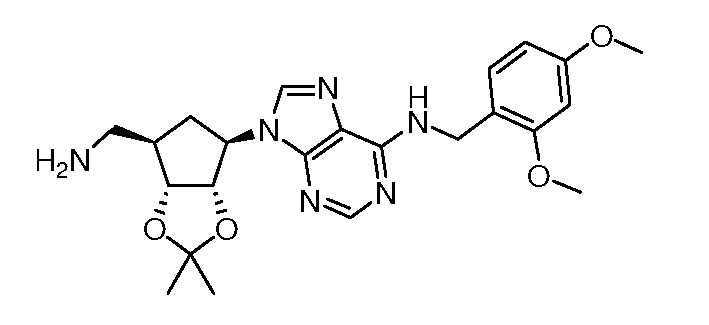
Раствор 9-((3aS,4R,6R,6aR)-6-(азидометил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-N-(2,4-диметоксибензил)-9H-пурин-6-амина (1,72 г, 3,58 ммоль) в ТГФ (16 мл) охлаждали до 0°C (баня лед/насыщенный солевой раствор) и обрабатывали по каплям 1,0 M раствором триметилфосфина в ТГФ (6,30 мл, 6,30 ммоль). Охлаждающую баню удаляли через 30 минут и реакционную смесь перемешивали в течение 1,5 часов; ВЭЖХ/ЖХ-MS анализ показал, что исходный азид полностью израсходован. К оранжевому раствору добавляли воду (2,84 мл, 157 ммоль) (заметное газовыделение) и реакционную смесь перемешивали в течение 2,75 часов при комнатной температуре; ВЭЖХ анализ показал полное преобразование в желаемый амин. Реакционную смесь концентрировали в вакууме с получением оранжевого масла. Остаток брали для поглощения в CH2Cl2 (150 мл) и промывали водой (2×50 мл) и насыщенным солевым раствором (1×75 мл). Отделенный органический слой сушили (Na2SO4) и концентрировали в вакууме с получением указанного в заголовке соединения (1,6 г, 98%) в виде бледно-желтого пенообразного вещества MS (ESI+) для C23H30N6O4 m/z 455,2 (M+H)+; ВЭЖХ чистота >95% площади.
Стадия 1: этил 3-(3-((((3aR,4R,6R,6aS)-6-(6-((2,4-диметоксибензил)амино)-9H-пурин-9-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)амино)циклобутил)пропаноат
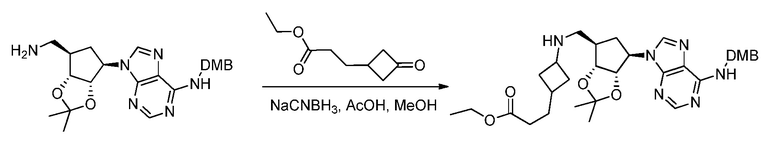
Триацетоксиборогидрид натрия (0,839 г, 3,96 ммоль) добавляли к раствору 9-((3aS,4R,6R,6aR)-6-(аминометил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-N-(2,4-диметоксибензил)-9H-пурин-6-амина (1,5 г, 3,3 ммоль), этил 3-(3-оксоциклобутил)пропаноата (0,562 г, 3,30 ммоль) и уксусной кислоты (0,188 мл, 3,30 ммоль) в 1,2-дихлорэтане (26,0 мл, 3,30E2 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение ночи. На следующее утро по данным ВЭЖХ исходное вещество было израсходовано, поэтому добавляли NaHCO3 и водный слой экстрагировали 3× при помощи DCM. Объединенные органические слои сушили при помощи MgSO4 и очищали жидкостной хроматографией (DCM/7н NH3 в MeOH 95:5) с получением этил 3-3-((((3aR,4R,6R,6aS)-6-(6-((2,4-диметоксибензил)амино)-9H-пурин-9-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)амино)циклобутил)пропаноата (1,5 г; 75%) в виде желтого смолистого/пенистого вещества. MS (ESI+) для C32H44N6O6 m/z 609,3 [M+H]+.
Стадия 2: этил 3-(3-((((3aR,4R,6R,6aS)-6-(6-((2,4-диметоксибензил)амино)-9H-пурин-9-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)(изопропил)амино)-циклобутил)пропаноат
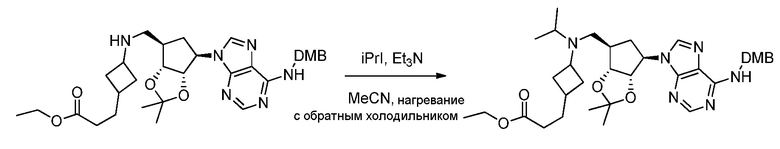
Этил 3-(3-((((3aR,4R,6R,6aS)-6-(6-((2,4-диметоксибензил)-амино)-9H-пурин-9-ил)-2,2-диметилтетрагидро-3aH-циклопента-[d][1,3]диоксол-4-ил)метил)амино)циклобутил)пропаноат (0,06 г, 0,1 ммоль) брали для поглощения в ацетонитрил (2,6 мл, 50 ммоль) и добавляли изопропилиодид (0,098 мл, 0,98 ммоль) и триэтиламин (0,21 мл, 1,5 ммоль). Реакционную смесь нагревали до 80°C в течение 12 часов, к этому времени оказалось, что реакция остановилась. Добавляли еще 15 эквивалентов триэтиламина и еще 15 эквивалентов изопропилиодида и реакция продолжалась еще в течение 8 часов. Оказалось, что реакция снова остановилась, поэтому снова добавляли по 15 эквивалентов каждого из изопропилиодида и триэтиламина. Когда исходное вещество было израсходовано, реакционную смесь концентрировали и добавляли насыщенный раствор Na2CO3 (20 мл) и DCM (20 млs). Остаток распределяли между органическим слоем и водным слоем. Водный слой экстрагировали 3 раза при помощи DCM, затем объединенные органические слои сушили и очищали жидкостной хроматографией (DCM/7н NH3 в MeOH 97:3). Продукт все еще содержал примеси TEA-H+I-, поэтому к 30 мл раствора продукта в DCM добавляли 20 мл насыщенного раствора NaHCO3 и 10 мл 1н раствора NaOH. Смесь перемешивали в течение 15 минут, затем водный слой экстрагировали при помощи DCM 3 раза. Объединенные органические слои сушили при помощи MgSO4 и растворитель удаляли с получением чистого этил 3-(3-((((3aR,4R,6R,6aS)-6-(6-((2,4-диметоксибензил)амино)-9H-пурин-9-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)(изопропил)амино)циклобутил)пропаноата (0,045 г; 70%) в виде коричневого пенистого/твердого вещества, без присутствия какой-либо аминовой соли, согласно данным ЯМР. MS (ESI+) для C35H50N6O6 m/z 651,3 [M+H]+.
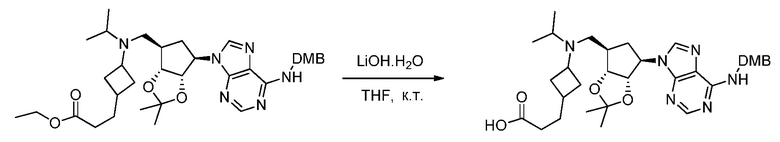
Гидроксид лития моногидрат (0,838 г, 20,0 ммоль) добавляли к раствору этил 3-(3-((((3aR,4R,6R,6aS)-6-(6-((2,4-диметоксибензил)амино)-9H-пурин-9-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)(изопропил)амино)циклобутил)пропаноата (1,3 г, 2,0 ммоль) в тетрагидрофуране (30 мл, 300 ммоль) и метаноле (6,5 мл, 160 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре и к следующему утру исходное вещество было израсходовано и преобразовано в кислоту. Реакционную смесь подкисляли при помощи 1н раствора HCl до pH=6. Летучие вещества удаляли в вакууме и оставшуюся воду удаляли путем азеотропной перегонки с этанолом с последующей лиофилизацией в течение 24 часов. Полученное коричневое твердое вещество использовали без дополнительной очистки.
Этил 3-(3-((((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)(метил)амино)циклобутил)пропаноат
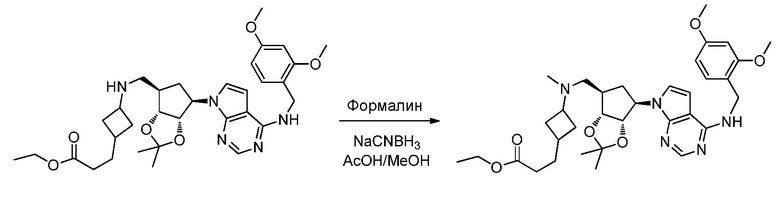
Амин этил 3-[3-({[(3aR,4R,6R,6aS)-6-{4-[(2,4-диметоксибензил)амино]-7H-пирроло[2,3-d]пиримидин-7-ил}-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил]метил}амино)циклобутил]пропаноат (1,8 г, 3,0 ммоль) брали для поглощения в метанол (20 мл, 600 ммоль) и добавляли цианоборогидрид натрия (0,19 г, 3,0 ммоль). pH доводили до приблизительно 6 с использованием 10% раствора AcOH в MeOH, затем добавляли формалин (0,29 мл, 3,9 ммоль) одной порцией. Реакцию осуществляли в течение 3 часов, к этому времени MS анализ показал, что исходное вещество полностью израсходовано. К реакционной смеси добавляли NaHCO3 (насыщенный), затем экстрагировали 3 раза при помощи DCM. Объединенные органические слои сушили при помощи MgSO4 и концентрировали с получением желтого смолистого вещества. Этот остаток очищали жидкостной хроматографией (DCM/7н NH3 в MeOH 93:7) с получением этил 3-(3-((((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)(метил)амино)циклобутил)пропаноата (1,6 г; 87%) в виде бесцветного пенистого вещества. MS (ESI+) для C34H47N5O6 m/z 622,3 [M+H]+.
1H ЯМР(400 МГц, d3-хлороформ) δH 8,282 (с, 1H), 7,203-7,168 (м, 1H), 6,877-6,865 (м, 1H), 6,399-6,334 (м, 2H), 6,242-6,236 (м, 1H), 5,330 (с, 1H), 4,890-4,835 (м, 2H), 4,664-4,650 (д, J=5,6 Гц, 2H), 4,391-4,354 (м, 1H), 4,067-4,000 (м, 2H), 3,757 (с, 3H), 3,710 (с, 3H), 2,864-2,784 (м, 0,5H (метин транс изомера)), 2,553-2,474 (м, 0,5H (метин цис изомера), 2,432-2,370 (м, 1H), 2,322-2,278 (м, 2H), 2,212-2,089 (м, 4H), 2,022 & 2,018 (с, 3H (перекрывающие синглеты из-за N-метила цис и транс изомеров), 1,964-1,908 (м, 3H), 1,778-1,584 (м, 4H), 1,486 (с, 3H), 1,363-1,296 (м, 1H), 1,219 (с, 3H), 1,182-1,146 (м, 3H).
((3aR,4R,6R,6aS)-6-(6-хлор-9H-пурин-9-ил)-2-этокситетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метанол
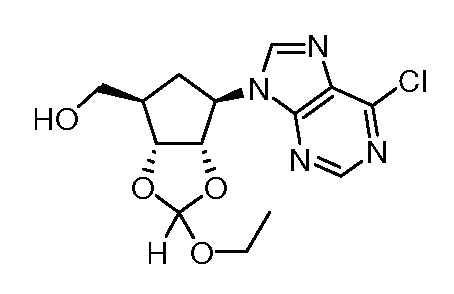
Полученный на предыдущей стадии неочищенный (1R,2S,3R,5R)-3-((5-амино-6-хлорпиримидин-4-ил)амино)-5-(гидроксиметил)циклопентан-1,2-диол обрабатывали этилортоформиатом (120 мл, 720 ммоль) и 10-камфорсульфоновой кислотой (8,11 г, 34,9 ммоль). Гетерогенную коричневую смесь интенсивно перемешивали с получением через 10 минут почти гомогенного коричневого раствора. По прошествии 5 часов ЖХ-MS анализ показал желаемый продукт в виде основного продукта, и реакцию гасили насыщенным водным раствором NaHCO3 (120 мл). Смесь разбавляли водой (75 мл), экстрагировали при помощи CH2Cl2 (3×200 мл) и объединенные органические слои сушили (Na2SO4) и концентрировали в вакууме с получением неочищенного указанного в заголовке соединения в виде темно-коричневого жидкого вещества, которое использовали далее без дополнительных манипуляций: MS (ESI+) для C14H17ClN4O4 m/z 341,0 (M+H)+.
Стадия 3: ((3aR,4R,6R,6aS)-6-(6-хлор-9H-пурин-9-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метанол
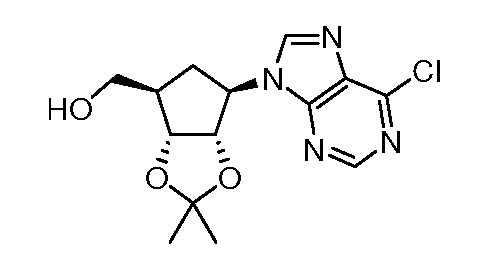
Полученный на предыдущей стадии неочищенный ((3aR,4R,6R,6aS)-6-(6-хлор-9H-пурин-9-ил)-2-этокситетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метанол брали для поглощения в 2,2-диметоксипропан (214 мл, 1740 ммоль) и обрабатывали моногидратом п-толуолсульфоновой кислоты (13,2 г, 69,5 ммоль) с получением коричневого масла, частично суспендированного в мутном растворе, который перемешивали при комнатной температуре в течение 1 часа 20 минут; ВЭЖХ/ЖХ-MS анализ показал полное преобразование в желаемый продукт. Реакцию гасили путем осторожного добавления бикарбоната натрия (8,76 г, 104 ммоль) и минимального количества воды. Летучие вещества удаляли в вакууме и оставшийся водный слой разбавляли водой (100 мл) и экстрагировали при помощи CH2Cl2 (3×400 мл). Объединенные органические слои сушили (Na2SO4) и концентрировали в вакууме с получением коричневого масла. Очистка колоночной хроматографией (7×16 см диоксид кремния; 0-5% MeOH/CH2Cl2) давала указанное в заголовке соединение (8,30 г, 74% от 3 стадий) в виде желтого пенообразного вещества MS (ESI+) для C14H17ClN4O3 m/z 325,1 (M+H)+; MS (ESI-) для C14H17ClN4O3 m/z 369,0 (M+HCO2)-.
Стадия 4: 9-((3aS,4R,6R,6aR)-6-(азидометил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-6-хлор-9H-пурин
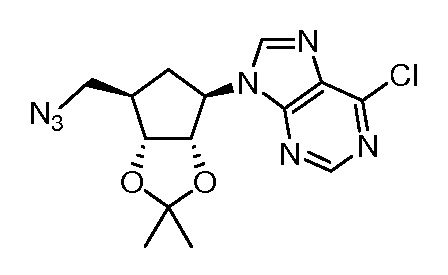
Смесь((3aR,4R,6R,6aS)-6-(6-хлор-9H-пурин-9-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метанола (7,9 г, 24 ммоль) и трифенилфосфина на полимерном носителе (3 ммоль/г нагрузка; 11 г, 34 ммоль) в ТГФ (100 мл) охлаждали до 0°C (баня лед/насыщенный солевой раствор) и обрабатывали по каплям диизопропилазодикарбоксилатом (6,7 мл, 34 ммоль). Желто-коричневую суспензию перемешивали в течение 15 минут и обрабатывали по каплям раствором азида дифенилфосфоновой кислоты (7,3 мл, 34 ммоль) в ТГФ (24 мл). Коричневую реакционную смесь перемешивали в течение 18,5 часов, пока ледяная баня не растаяла; ВЭЖХ анализ показал преобразование в желаемый продукт. По прошествии 21,5 часов реакционную смесь фильтровали, твердые вещества промывали при помощи CH2Cl2 и фильтрат концентрировали в вакууме. Красно-коричневый остаток брали для поглощения в CH2Cl2 (300 мл) и промывали насыщенным водным раствором NaHCO3 (1×100 мл), водой (1×100 мл) и насыщенным солевым раствором (1×150 мл). Отделенный органический слой сушили (Na2SO4) и концентрировали в вакууме с получением красно-оранжевого масла. Очистка колоночной хроматографией (7×16 см диоксид кремния; 0-10% ацетона/CH2Cl2) давала указанное в заголовке соединение (4,82 г, 57%) в виде желтого маслянистого/пенообразного вещества MS (ESI+) для C14H16ClN7O2 m/z 350,1 (M+H)+; MS (ESI-) для C14H16ClN7O2 m/z 394,1 (M+HCO2)-.
Стадия 5: 9-((3aS,4R,6R,6aR)-6-(азидометил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-N-(2,4-диметоксибензил)-9H-пурин-6-амин
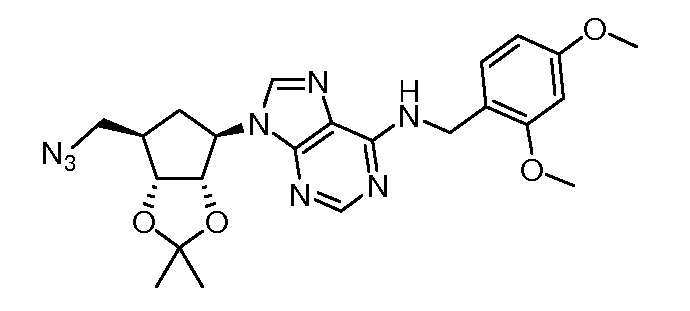
Раствор 9-((3aS,4R,6R,6aR)-6-(азидометил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-6-хлор-9H-пурина (1,29 г, 3,69 ммоль) и (2,4-диметоксифенил)метанамина (0,71 мл, 4,7 ммоль) в 1-бутаноле (10 мл) обрабатывали N,N-диизопропилэтиламином (0,93 мл, 5,3 ммоль) и нагревали при 80°C в течение 16,5 часов; ВЭЖХ/ЖХ-MS анализ показал преобразование в желаемый продукт. Реакционной смеси давали охладиться до комнатной температуры и летучие вещества удаляли под потоком воздуха с получением коричневато-оранжевого пастообразного вещества. Очистка колоночной хроматографией (2×8 см диоксид кремния; 0-10% ацетона/CH2Cl2) давала указанное в заголовке соединение (1,72 г, 97%) в виде желтовато-оранжевого пенистого/маслянистого вещества: MS (ESI+) для C23H28N8O4 m/z 481,2 (M+H)+.
Стадия 6: 9-((3aS,4R,6R,6aR)-6-(аминометил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-N-(2,4-диметоксибензил)-9H-пурин-6-амин
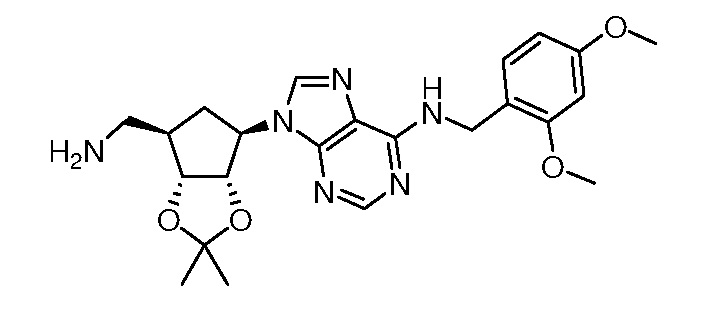
Раствор 9-((3aS,4R,6R,6aR)-6-(азидометил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-N-(2,4-диметоксибензил)-9H-пурин-6-амина (1,72 г, 3,58 ммоль) в ТГФ (16 мл) охлаждали до 0°C (баня лед/насыщенный солевой раствор) и обрабатывали по каплям 1,0 M раствором триметилфосфина в ТГФ (6,30 мл, 6,30 ммоль). Охлаждающую баню удаляли через 30 минут и реакционную смесь перемешивали в течение 1,5 часов; ВЭЖХ/ЖХ-MS анализ показал, что исходный азид полностью израсходован. К оранжевому раствору добавляли воду (2,84 мл, 157 ммоль) (заметное газовыделение) и реакционную смесь перемешивали в течение 2,75 часов при комнатной температуре; ВЭЖХ анализ показал полное преобразование в желаемый амин. Реакционную смесь концентрировали в вакууме с получением оранжевого масла. Остаток брали для поглощения в CH2Cl2 (150 мл) и промывали водой (2×50 мл) и насыщенным солевым раствором (1×75 мл). Отделенный органический слой сушили (Na2SO4) и концентрировали в вакууме с получением указанного в заголовке соединения (1,6 г, 98%) в виде бледно-желтого пенообразного вещества MS (ESI+) для C23H30N6O4 m/z 455,2 (M+H)+.
Стадия 1: этил 3-(3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)амино)циклобутил)пропаноат
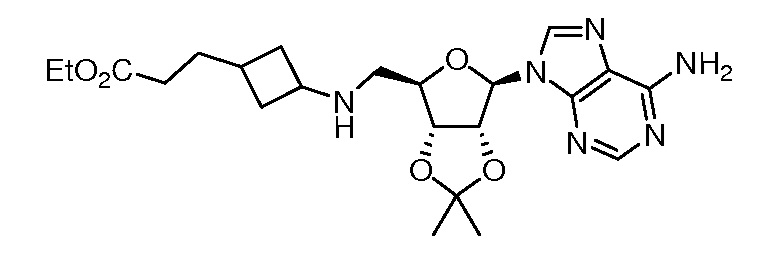
Смесь 9-((3aR,4R,6R,6aR)-6-(аминометил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)-9H-пурин-6-амина (0,50 г, 1,6 ммоль) и этил 3-(3-оксоциклобутил)пропаноата (0,27 г, 1,6 ммоль) в метаноле (10 мл) обрабатывали уксусной кислотой (0,09 мл, 2 ммоль) при комнатной температуре и в колбе создавали вакуум и продували азотом (×3). Реакционную смесь обрабатывали при комнатной температуре цианоборогидридом натрия (0,26 г, 4,1 ммоль), что приводило к мгновенному газовыделению и получению почти бесцветного прозрачного раствора в течение нескольких минут. Реакционную смесь перемешивали в течение 1 часа при комнатной температуре; ВЭЖХ/ЖХ-MS анализ показал ~2:1 смесь продукта и исходного амина. По прошествии 1,5 часа добавляли дополнительное количество этил 3-(3-оксоциклобутил)пропаноата (66 мг, 0,39 ммоль) в MeOH (1,0 мл) и реакционную смесь перемешивали при комнатной температуре в течение 30 минут; ВЭЖХ/ЖХ-MS анализ показал ~70% преобразование и некоторое диалкилирование. По прошествии 2 часов 15 минут добавляли воду (4,0 мл) и смесь концентрировали в вакууме. Остаточный водный слой разбавляли насыщенным водным раствором бикарбоната натрия (10 мл, до pH 9) и экстрагировали при помощи CH2Cl2 (3×15 мл). Объединенные органические слои сушили (Na2SO4) и концентрировали в вакууме с получением продукта восстановительного аминирования в виде белого пенистого вещества, которое использовали далее без дополнительной очистки: MS (ESI+) для C22H32N6O5 m/z 461,1 (M+H)+, 483,1 (M+Na)+.
Стадия 2: этил 3-(3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(метил)амино)циклобутил)пропаноат
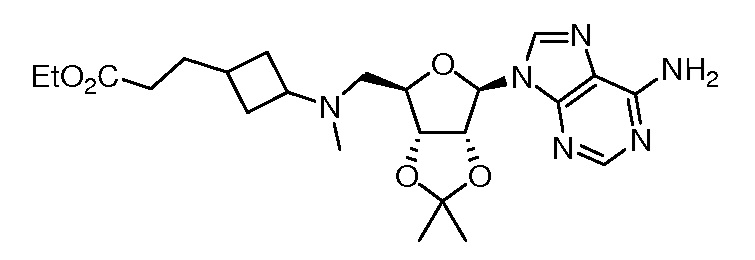
Полученный на предыдущей стадии неочищенный вторичный амин брали для поглощения в метанол (10 мл) и обрабатывали цианоборогидридом натрия (0,30 г, 4,8 ммоль). Добавляли раствор 10% об/об уксусной кислоты в метаноле для доведения pH до ~6, с последующим добавлением по каплям 37% водного раствора формальдегида (0,65 мл, 6,3 ммоль), что приводило к газовыделению. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа; ВЭЖХ/ЖХ-MS анализ показал полное преобразование в желаемый продукт. По прошествии 1,5 часов добавляли воду (5,0 мл) и реакционную смесь концентрировали в вакууме. Остаток разбавляли насыщенным водным раствором NaHCO3 (10 мл, до pH ~9) и экстрагировали при помощи CH2Cl2 (3×15 мл). Объединенные органические слои разбавляли небольшим количеством EtOH с получением прозрачного раствора, сушили (Na2SO4) и концентрировали в вакууме с получением почти бесцветного масла. Очистка колоночной хроматографией (4×17 см диоксид кремния; 0-5% 7н метанольный раствор NH3/CH2Cl2) давала указанное в заголовке соединение (0,50 г, 60%) в виде белого пенистого вещества/бесцветного масла: MS (ESI+) для C23H34N6O5 m/z 475,1 (M+H)+, 497,1 (M+Na)+.
Стадия 1: этил 3-(3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)амино)циклобутил)пропаноат
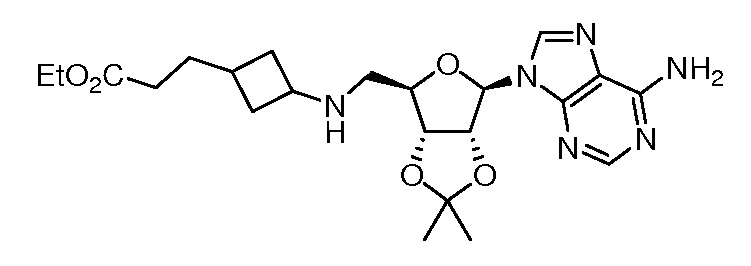
Смесь 9-((3aR,4R,6R,6aR)-6-(аминометил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)-9H-пурин-6-амина (2,04 г, 6,66 ммоль) и этил 3-(3-оксоциклобутил)пропаноата (1,2 г, 7,0 ммоль) в метаноле (41 мл) обрабатывали уксусной кислотой (0,37 мл, 6,5 ммоль) при комнатной температуре и в колбе создавали вакуум и продували азотом (×3). Реакционную смесь обрабатывали при комнатной температуре цианоборогидридом натрия (1,0 г, 16 ммоль), что приводило к мгновенному газовыделению и получению почти бесцветного прозрачного раствора в течение нескольких минут. Реакционную смесь перемешивали в течение 1 часа при комнатной температуре; ВЭЖХ/ЖХ-MS анализ показал, что осталось исходное вещество. По прошествии 1 часа 20 минут добавляли дополнительное количество этил 3-(3-оксоциклобутил)пропаноата (0,50 г, 2,93 ммоль) в MeOH (3 мл). Реакционную смесь перемешивали в течение 30 минут и обрабатывали водой (12 мл). Смесь концентрировали в вакууме и остаточный водный слой разбавляли насыщенным водным раствором бикарбоната натрия (40 мл, до pH 9) и экстрагировали при помощи CH2Cl2 (3×60 мл). Объединенные органические слои сушили (Na2SO4) и концентрировали в вакууме с получением неочищенного указанного в заголовке соединения в виде белого пенистого вещества/очень бледно-желтого масла, которое использовали далее без дополнительной очистки: MS (ESI+) для C22H32N6O5 m/z 461,2 (M+H)+ и 483,1 (M+Na)+.
Стадия 2: этил 3-(3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(изопропил)амино)циклобутил)пропаноат
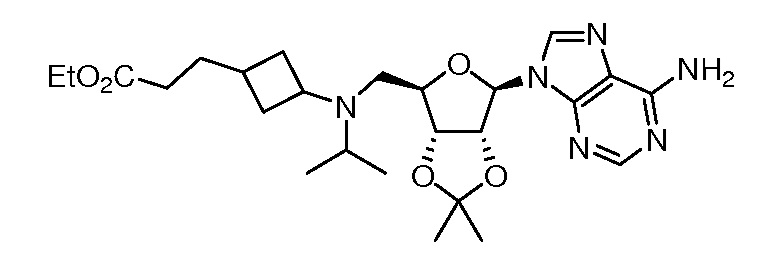
Раствор полученного на предыдущей стадии неочищенного этил 3-(3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)амино)циклобутил)пропаноата в ацетонитриле (30 мл) обрабатывали карбонатом калия (6,3 г, 46 ммоль) и изопропилиодидом (3,9 мл, 39 ммоль). Реакционную смесь нагревали при 90°C в герметично закрытой пробирке в течение 6,5 часов; ВЭЖХ анализ показал 4:1 смесь продукта с исходным веществом. Реакционную смесь перемешивали в течение ночи (17,5 часа) при комнатной температуре, обрабатывали дополнительным количеством изопропилиодида (2,0 мл, 20 ммоль) и нагревали при 90°C в течение 3 часов; ВЭЖХ/ЖХ-MS анализ показал почти полное преобразование. Реакционную смесь охлаждали до комнатной температуры и твердые вещества удаляли при помощи вакуумной фильтрации, с промывкой при помощи CH3CN и фильтрат концентрировали в вакууме с получением оранжеватого масла с осадком. Очистка колоночной хроматографией (5×14,5 см диоксид кремния; 0-10% 7н метанольный раствор NH3/CH2Cl2) давала указанное в заголовке соединение (0,49 г, 15%) в виде белого пенистого вещества/бесцветного масла. Смешанные фракции, содержащие продукт, снова очищали колоночной хроматографией (4×10,5 см диоксид кремния; 0-5% 7н метанольный раствор NH3/CH2Cl2) с получением указанного в заголовке соединения (1,66 г, 40%) в виде белого пенистого вещества/бесцветного масла, загрязненного побочным продуктом повторного восстановительного аминирования со Стадии 1: MS (ESI+) для C25H38N6O5 m/z 503,2 (M+H)+.
этил 3-(3-((((1R,2R,3S,4R)-4-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,3-дигидроксициклопентил)метил)амино)циклобутил)пропаноат
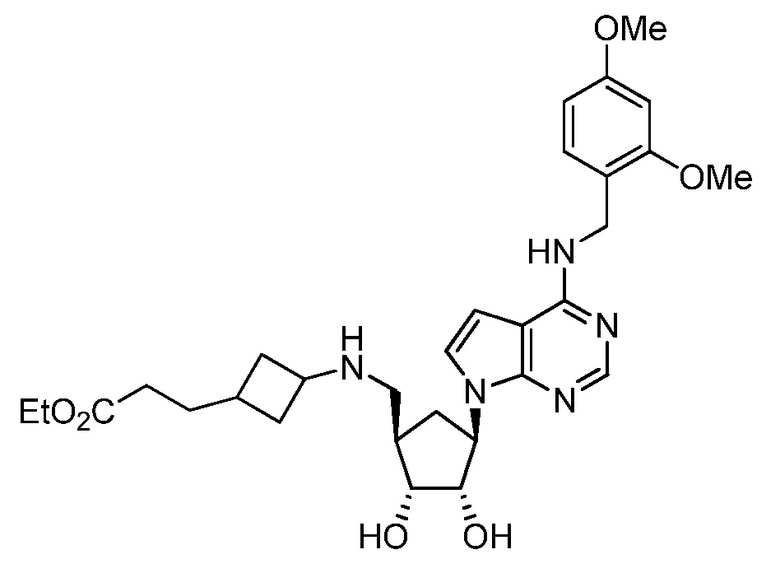
Триацетоксиборогидрид натрия (2,43 г, 11,5 ммоль) добавляли к раствору (1S,2R,3R,5R)-3-(аминометил)-5-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)циклопентан-1,2-диола (2,6 г, 5,7 ммоль) и этил 3-(3-оксоциклобутил)пропаноата (0,976 г, 5,73 ммоль) и уксусной кислоты (0,326 мл, 5,73 ммоль) в 1,2-дихлорэтане (20 мл) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Добавляли NaHCO3 и водный слой экстрагировали 3× при помощи DCM. Объединенные органические слои сушили при помощи MgSO4, фильтровали, концентрировали и очищали флэш-хроматографией (DCM/7н NH3 в MeOH 90:10) с получением желаемого соединения (1,8 г) в виде вязкого желтого смолистого вещества.
N-(2,4-диметоксибензил)-7-((3aS,4R,6R,6aR)-6-((изопропиламино)метил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-7H-пирроло[2,3-d]пиримидин-4-амин
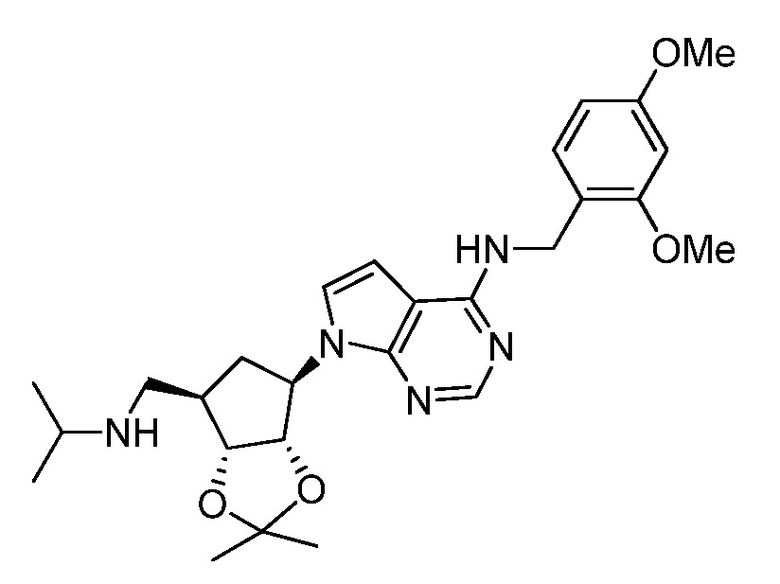
Раствор 7-((3aS,4R,6R,6aR)-6-(аминометил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-N-(2,4-диметоксибензил)-7H-пирроло[2,3-d]пиримидин-4-амина (7,50 г, 16,5 ммоль) в 1,2-дихлорэтане (140 мл, 1800 ммоль) обрабатывали по каплям ацетоном (1,34 мл, 18,2 ммоль;) и уксусной кислотой (0,94 мл, 16 ммоль), затем триацетоксиборогидридом натрия (4,20 г, 19,8 ммоль) и смесь перемешивали при комнатной температуре в течение 4 часов. ВЭЖХ анализ показал, что реакция завершилась. Реакционную смесь разбавляли при помощи 200 мл CH2Cl2 и промывали при помощи 150 мл насыщенного раствора NaHCO3. Водную фазу промывали при помощи 100 мл CH2Cl2 и объединенную органическую фазу сушили над Na2SO4, фильтровали и концентрировали с получением масла, которое образовывало жесткую пену, когда его помещали в условия высокого вакуума. Это неочищенное вещество (9,3 г) использовали непосредственно на следующей стадии.
этил 3-(3-((((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)(изопропил)амино)циклобутил)пропаноат
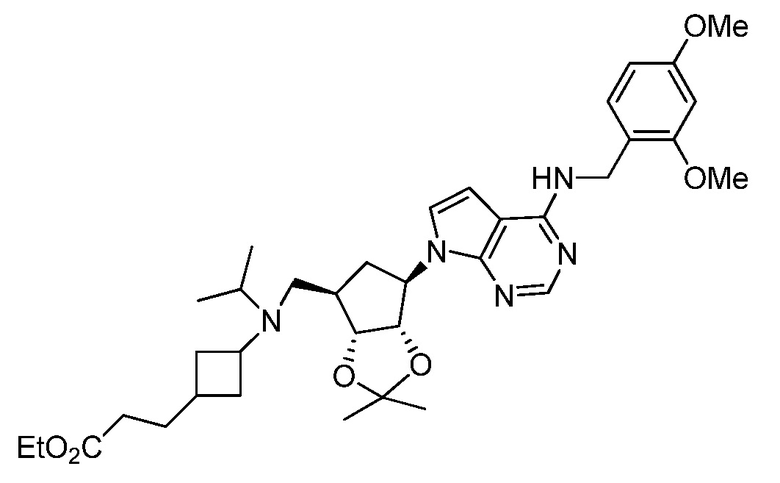
Раствор N-(2,4-диметоксибензил)-7-((3aS,4R,6R,6aR)-6-((изопропиламино)метил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-7H-пирроло[2,3-d]пиримидин-4-амина (9,50 г, 15,3 ммоль) в 1,2-дихлорэтане (75 мл, 950 ммоль) обрабатывали по каплям этил 3-(3-оксоциклобутил)пропаноатом (3,92 г, 23,0 ммоль) и уксусной кислотой (1,0 мл, 18 ммоль), затем триацетоксиборогидридом натрия (4,58 г, 21,6 ммоль) и смесь перемешивали при комнатной температуре в течение 6 дней. Реакционную смесь разбавляли при помощи 150 мл CH2Cl2 и промывали при помощи 100 мл насыщенного раствора NaHCO3. Водную фазу промывали при помощи 100 мл CH2Cl2 и объединенную органическую фазу сушили над Na2SO4, фильтровали и концентрировали с получением светло-коричневого вязкого стеклообразного вещества.
Неочищенное вещество очищали флэш-хроматографией (SiO2, элюировали при помощи 2-3% 7н NH3 в CH3OH/CH2Cl2) с получением слегка стеклообразного вещества/жесткой пены (7,10 г).
этил 3-(3-((((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)амино)циклобутил)пропаноат
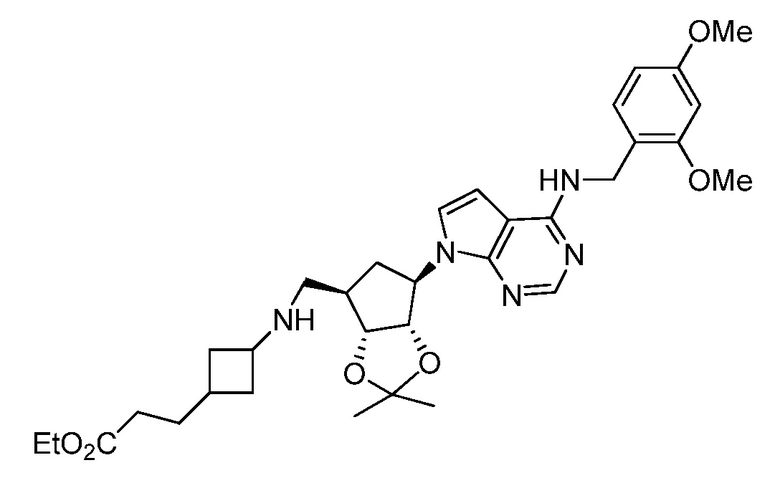
Раствор 7-((3aS,4R,6R,6aR)-6-(аминометил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-N-(2,4-диметоксибензил)-7H-пирроло[2,3-d]пиримидин-4-амина (8,00 г, 15,2 ммоль) в 1,2-дихлорэтане (119,5 мл, 1517 ммоль) обрабатывали по каплям этил 3-(3-оксоциклобутил)пропаноатом (2,58 г, 15,2 ммоль) и уксусной кислотой (0,86 мл, 15 ммоль), затем триацетоксиборогидридом натрия (3,86 г, 18,2 ммоль) и смесь перемешивали при комнатной температуре в течение 19 часов. Реакционную смесь разбавляли при помощи 150 мл CH2Cl2 и промывали при помощи 150 мл насыщенного раствора NaHCO3. Водную фазу промывали при помощи 70 мл CH2Cl2 и объединенную органическую фазу сушили над Na2SO4, фильтровали и концентрировали с получением желто-коричневого стеклообразного вещества, которое образовывало липкую пену, когда его помещали в условия высокого вакуума. Это неочищенное вещество очищали флэш-хроматографией (SiO2, элюировали при помощи 3-4% 7н NH3 в CH3OH/CH2Cl2) с получением светло-желтого вязкого масла, которое образовывало липкую пену в условиях высокого вакуума (5,03 г). MS 608,3 (M+H).
Пример 1: Синтез 1-((3-((((2R,3S,4R,5R)-5-(6-амино-9H-пурин-9-ил)-3,4-дигидрокситетрагидрофуран-2-ил)метил)(метил)амино)циклобутил)метил)-3-(4-(трет-бутил)фенил)мочевины (Соединение 110)
Стадия 1: Синтез метил 3-оксоциклобутанкарбоксилата
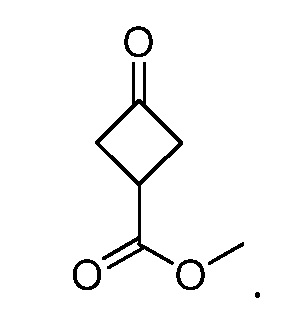
К раствору DCC (5,96 г, 28,95 ммоль) в DCM (20 мл) добавляли по каплям смесь 3-оксоциклобутанкарбоновой кислоты (3,0 г, 26,31 ммоль), MeOH (1,68 г, 52,62 ммоль) и DMAP (2,57 г, 21,05 ммоль) в DCM (30 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Смесь фильтровали. Фильтрат промывали 0,5 M раствором HCl (50 мл). Органический слой сушили над Na2SO4 и концентрировали. Остаток очищали при помощи SGC (PE:EA=5:1) с получением указанного в заголовке соединения (4,0 г).
1H ЯМР (500 МГц, CDCl3): δ 3,77 (с, 3H), 3,42-3,26 (м, 5H) м.д.
Стадия 2: Синтез метил 3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(метил)амино)циклобутанкарбоксилата
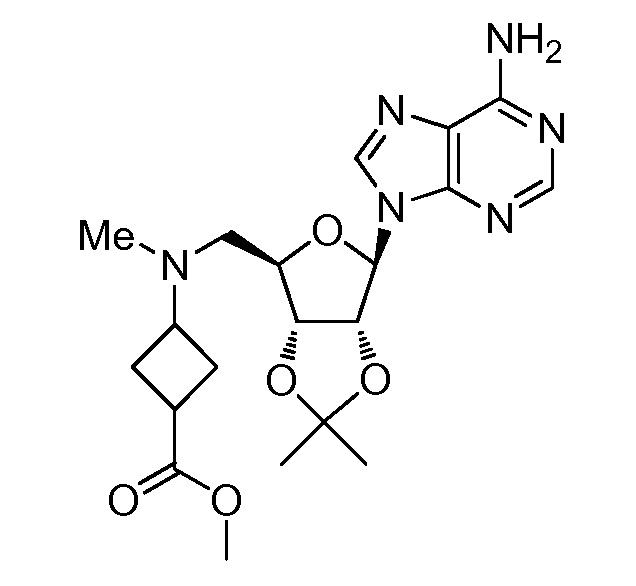
Раствор метил 3-оксоциклобутанкарбоксилата (1,28 г, неочищенный), 9-((3aR,4R,6R,6aR)-2,2-диметил-6-((метиламино)метил)тетрагидрофуро[3,4-d][1,3]диоксол-4-ил)-9H-пурин-6-амина (2,0 г, 6,25 ммоль) (Townsend et al Org Lett 2009, 11, 2976-2679) и Ti(iPrO)4 (1,78 г, 6,25 ммоль) в MeOH (50 мл) перемешивали при 45oC в течение 2 часов, затем добавляли NaCNBH3 (0,79 г, 12,50 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакцию гасили насыщенным водным раствором NaHCO3 (40 мл), фильтровали, экстрагировали при помощи DCM (40 мл ×3), сушили над Na2SO4 и концентрировали. Остаток очищали при помощи SGC (DCM:MeOH=12:1) с получением указанного в заголовке соединения (1,7 г, выход 63%).
1H ЯМР (500 МГц, MeOD): δH 8,28-8,27 (м, 1H), 8,21 (с, 1H), 6,20-6,18 (м, 1H), 5,52 (дд, J=1,5, 6,0 Гц, 1H), 5,00 (дд, J=3,0, 6,0 Гц, 1H), 5,33 (шир.с, 1H), 3,65-3,63 (м, 3H), 2,77-2,55 (м, 4H), 2,19-2,11 (м, 5H), 2,00-1,82 (м, 2H), 1,59 (с, 3H), 1,38 (с, 3H) м.д.; ESI-MS (m/z): 433,2 [M+1]+.
Стадия 3: Синтез (3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(метил)амино)циклобутил)метанола
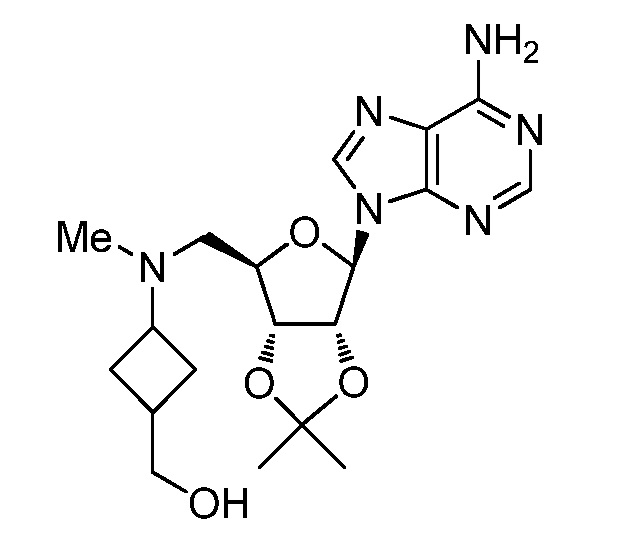
К раствору метил 3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(метил)амино)циклобутанкарбоксилата (1,0 г, 2,31 ммоль) в ТГФ (40 мл) добавляли LiAlH4 (0,53 г, 13,89 ммоль) при 0oC и смесь перемешивали в течение ночи. К смеси медленно добавляли воду (1,0 г) и 15% раствор NaOH (3,0 г) и после перемешивания в течение 15 минут смесь фильтровали. Фильтрат концентрировали с получением неочищенного указанного в заголовке соединения, которое использовали непосредственно на следующей стадии.
Стадия 4: Синтез (3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(метил)амино)циклобутил)метилметансульфоната
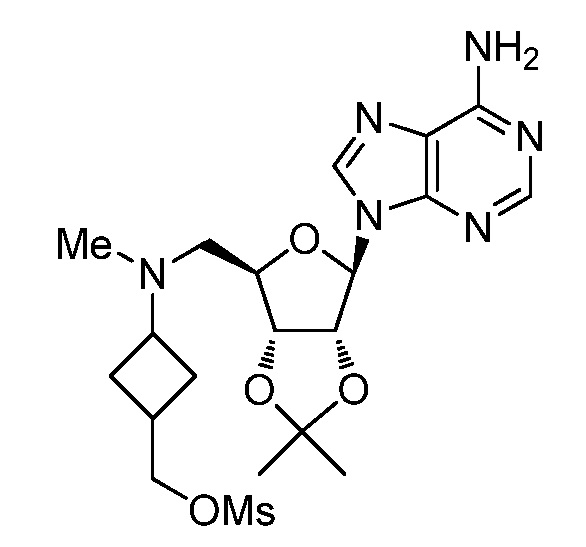
К раствору (3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2- диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(метил)амино)циклобутил)метанола, взятого непосредственно с предыдущей стадии, в DCM (25 мл) добавляли Et3N (467 мг, 4,62 ммоль) и MsCl (264 мг, 2,31 ммоль) в виде раствора в DCM (5 мл). Смесь перемешивали в течение 2 часов. Добавляли воду (20 мл) и DCM (30 мл ×2). Органический слой сушили над Na2SO4 и концентрировали, очищали методом препаративной ТСХ (DCM:MeOH=10:1) с получением указанного в заголовке соединения (390 мг, выход 35% для двух стадий).
1H ЯМР (500 МГц, MeOD): δH 8,27 (с, 1H), 8,21 (с, 1H), 6,203-6,200 (м, 1H), 5,529 (дд, J=2,0, 7,0 Гц, 1H), 5,010 (дд, J=3,0, 6,0 Гц, 1H), 4,354 (дд, J=3,5, 8,0 Гц, 1H), 4,197-4,182 (м, 2H), 3,585 (шир.с, 1H), 3,073-2,948 (м, 5H), 2,595-2,513 (м, 2H), 2,394 (шир.с, 1H), 2,207 (шир.с, 1H), 2,107 (с, 3H), 2,030-1,989 (м, 1H), 1,840-1,811 (м, 2H), 1,586 (с, 3H), 1,390 (шир.с, 1H), 1,329-1,280 (м, 6H), 0,905-0,878 (м, 1H) м.д.; ESI-MS (m/z): 483,3 [M+1]+.
Стадия 5: Синтез 9-((3aR,4R,6R,6aR)-6-(((3-(азидометил)циклобутил)(метил)амино)метил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)-9H-пурин-6-амина

К раствору (3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(метил)амино)циклобутил)метилметансульфоната (150 мг, 0,31 ммоль) в DMF (3 мл) добавляли NaN3 (81 мг, 1,24 ммоль). Смесь нагревали при 70oC в течение 3 часов. Добавляли воду (30 мл) и смесь экстрагировали этилацетатом (20 мл ×3). Объединенные органические слои сушили над Na2SO4 и концентрировали. Остаток очищали методом препаративной ТСХ (DCM:MeOH=30:1) с получением указанного в заголовке соединения (90 мг, выход 67%). ESI-MS (m/z): 430,2[M+1]+.
Стадия 6: Синтез 9-((3aR,4R,6R,6aR)-6-(((3-(аминометил)циклобутил)(метил)амино)метил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)-9H-пурин-6-амина
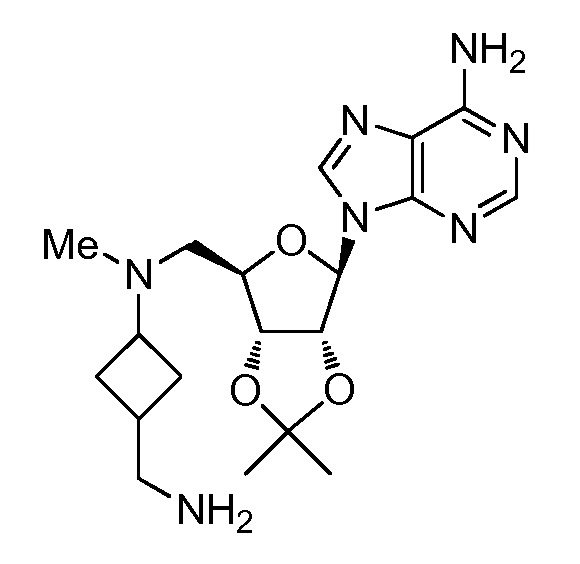
Pd/C (10 мг) добавляли к раствору 9-((3aR,4R,6R,6aR)-6-(((3-(азидометил)циклобутил)(метил)амино)метил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)-9H-пурин-6-амина (90 мг, 0,21 ммоль) в MeOH (6 мл). Смесь перемешивали при комнатной температуре в течение ночи в атмосфере H2. Смесь фильтровали и фильтрат концентрировали с получением указанного в заголовке соединения, которое использовали непосредственно на следующей стадии.
Стадия 7: Синтез 1-((3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(метил)амино)циклобутил)метил)-3-(4-(трет-бутил)фенил)мочевины
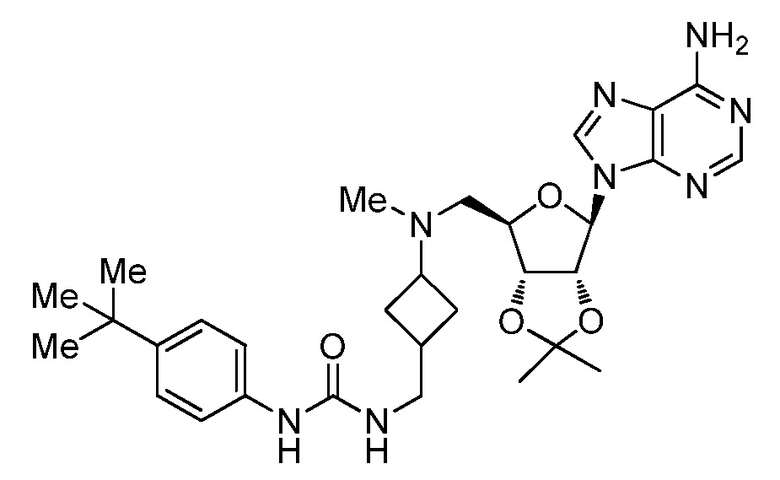
К раствору 9-((3aR,4R,6R,6aR)-6-(((3-(аминометил)циклобутил)(метил)амино)метил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)-9H-пурин-6-амина в DCM (4 мл) добавляли 1-трет-бутил-4-изоцианатобензол (37 мг). Смесь перемешивали при комнатной температуре в течение 1 часа. Смесь концентрировали и очищали методом препаративной ТСХ (DCM:MeOH=10:1) с получением указанного в заголовке соединения (55 мг, выход 45% для двух стадий). ESI-MS (m/z): 578,3[M+1]+.
Стадия 8: Синтез соединения 110
Раствор 1-((3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(метил)амино)циклобутил)метил)-3-(4-(трет-бутил)фенил)мочевины (55 мг) в HCl/MeOH (2,5 моль/л) (2 мл) перемешивали при комнатной температуре в течение 2 часов и затем концентрировали досуха. Добавляли K2CO3 (52 мг) в воде (0,5 мл) и MeOH (5 мл). Полученную смесь перемешивали еще в течение 10 минут при комнатной температуре, фильтровали и фильтрат концентрировали. Остаток очищали методом препаративной ВЭЖХ с получением соединения 110 (10 мг, выход: 25%) в виде белого твердого вещества.
1H ЯМР (500 МГц, MeOD): δH 8,26 (с, 1H), 8,18 (с, 1H), 7,26-7,19 (м, 4H), 5,96 (д, J=4,5 Гц, 1H), 4,674-4,655 (м, 1H), 4,24-4,16 (м, 2H), 3,15 (д, J=5,0 Гц, 2H), 2,83-2,73 (м, 3H), 2,20-1,59 (м, 8H), 1,26 (с, 9H) м.д.; ESI-MS (m/z): 539,3 [M+1]+.
Пример 2: Синтез (2R,3R,4S,5R)-2-(6-амино-9H-пурин-9-ил)-5-((((1r,3S)-3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(изопропил)амино)метил)тетрагидрофуран-3,4-диола (Соединение 2)
Стадия 1: Синтез цис и транс метил 3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)амино)циклобутанкарбоксилата
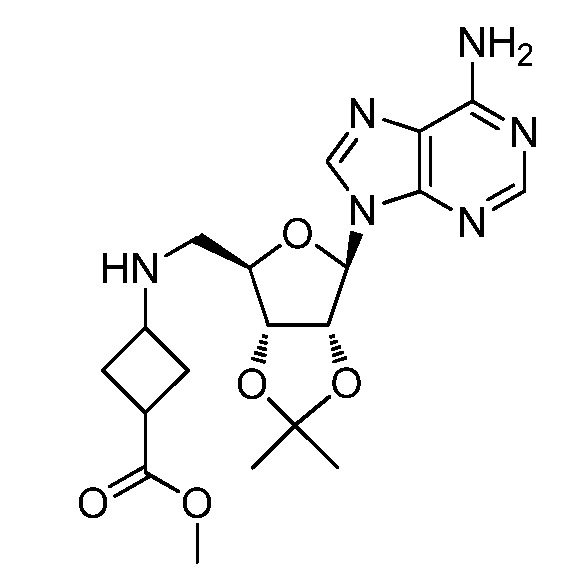
Раствор метил 3-оксоциклобутанкарбоксилата (4,60 г, 35,94 ммоль), 9-((3aR,4R,6R,6aR)-6-(аминометил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)-9H-пурин-6-амина (11,0 г, 35,94 ммоль) и Ti(iPrO)4 (4,0 г, 14,08 ммоль) в MeOH (80 мл) перемешивали при 45oC в течение 2 часов, затем добавляли NaCNBH3 (4,5 г, 71,87 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакцию гасили насыщенным водным раствором NaHCO3 (40 мл) и фильтровали, экстрагировали при помощи DCM (80 мл ×3), сушили над Na2SO4 и концентрировали. Остаток очищали методом препаративной ВЭЖХ с получением указанного в заголовке соединения (6,2 г, выход 41%).
1H ЯМР (500 МГц, CDCl3): δH 8,38-8,34 (м, 1H), 7,90 (с, 1H), 5,98 (д, J=3,0 Гц, 1H), 5,75 (щир.с, 2H), 5,48-5,46 (м, 1H), 5,03-5,01 (м, 1H), 4,35-4,33 (м, 1H), 3,69-3,66 (м, 3H), 3,50-3,17 (м, 1H), 3,05-2,73 (м, 3H), 2,48-2,44 (м, 2H), 1,95-1,91 (м, 2H), 1,62 (с, 3H), 1,39 (с, 3H) м.д.; ESI-MS (m/z): 419,2 [M+1]+.
Цис/транс смесь метил 3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)амино)циклобутанкарбоксилата (6,2 г) разделяли методом хиральной ВЭЖХ (CHIRALCEL AD-H 20×250 мм, 5 мкм (Daicel), Температура колонки: 35°C, Подвижные фазы: CO2/Метанол (0,1% DEA)=70/30, Скорость потока: 50 г/мин) с получением чистого цис продукта (3,5 г) и чистого транс продукта (1,7 г).
Стадия 2: Синтез (1S,3s)-метил 3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(изопропил)амино)циклобутанкарбоксилата
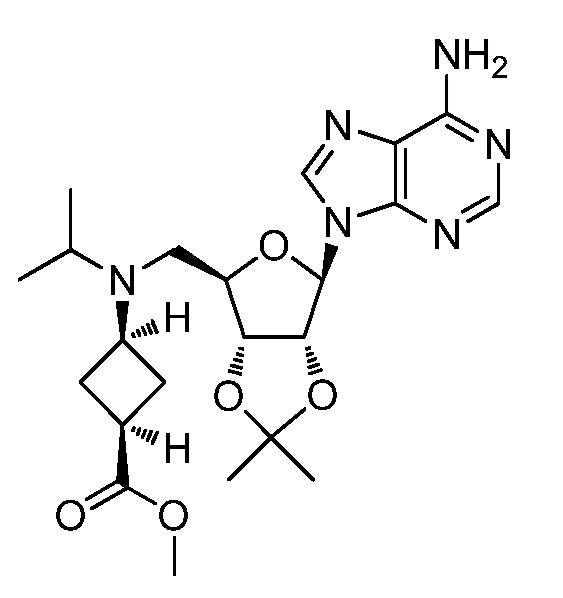
К раствору цис метил 3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)амино)циклобутанкарбоксилата (2,0 г, 4,78 ммоль) в CH3CN (15 мл) добавляли 2-иодпропан (4,0 г, 23,92 ммоль) и K2CO3 (1,0 г, 7,18 ммоль). Реакционную смесь нагревали до 95oC в течение ночи в герметично закрытой пробирке. Смесь фильтровали, фильтрат концентрировали и очищали при помощи SGC (DCM:MeOH=12:1) с получением указанного в заголовке соединения (1,9 г, выход 86%).
1H ЯМР (500 МГц, CDCl3): δH 8,37 (с, 1H), 7,89 (с, 1H), 6,03 (д, J=1,5 Гц, 1H), 5,53-5,48 (м, 3H), 5,00 (шир.с, 1H), 4,25 (шир.с, 1H), 3,66 (с, 3H), 3,19-3,18 (м, 1H), 2,96 (шир.с, 1H), 2,80-2,78 (м, 1H), 2,67-2,58 (м, 2H), 2,20-2,12 (м, 4H), 1,62 (с, 3H), 1,39 (с, 3H), 1,00 (д, J=6,0 Гц, 3H), 0,84 (д, J=6,0 Гц, 3H) м.д.; ESI-MS (m/z): 461,4 [M+1]+.
Стадия 3: Синтез (1S,3s)-3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(изопропил)амино)циклобутанкарбальдегид
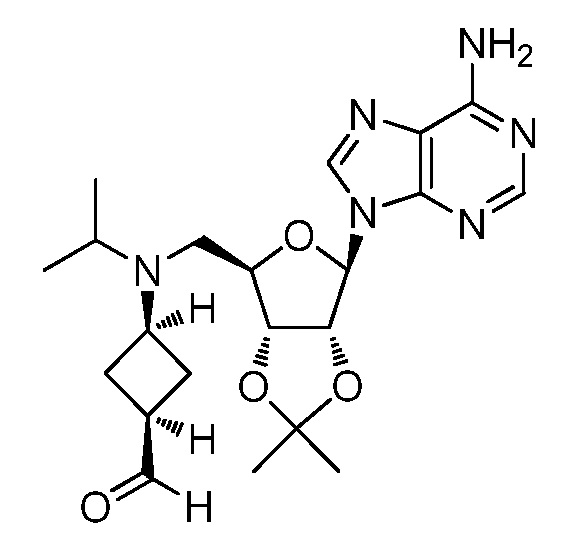
К раствору (1S,3s)-метил 3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(изопропил)амино)циклобутанкарбоксилата (1,2 г, 2,60 ммоль) в DCM (50 мл) добавляли DIBAL-H по каплям при -78oC, пока все исходное вещество не было израсходовано, как было определено при помощи ТСХ. Добавляли MeOH (2 мл) и смесь перемешивали при комнатной температуре в течение 30 минут, после чего добавляли воду (50 мл) и смесь экстрагировали при помощи DCM (50 мл ×2). Органический слой сушили над Na2SO4 и концентрировали с получением неочищенного указанного в заголовке соединения (1,0 7, которое использовали непосредственно на следующей стадии).
1H ЯМР (500 МГц, CDCl3): δH 9,56 (д, J=2,5 Гц, 1H), 8,36 (с, 1H), 7,88 (с, 1H), 6,03 (д, J=2,5 Гц, 1H), 5,66 (шир.с, 2H), 5,50 (дд, J=2,0, 6,5 Гц, 1H), 5,01 (дд, J=3,5, 6,5 Гц, 1H), 3,331-3,337 (м, 1H), 2,96-2,97 (м, 1H), 2,77-2,59 (м, 3H), 2,14-2,05 (м, 4H), 1,60 (с, 3H), 1,39 (с, 3H), 1,01 (д, J=6,5 Гц, 3H), 0,85 (д, J=6,0 Гц, 3H) м.д.
Стадия 4: Синтез (E)-этил 3-((1S,3s)-3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(изопропил)амино)циклобутил)акрилата
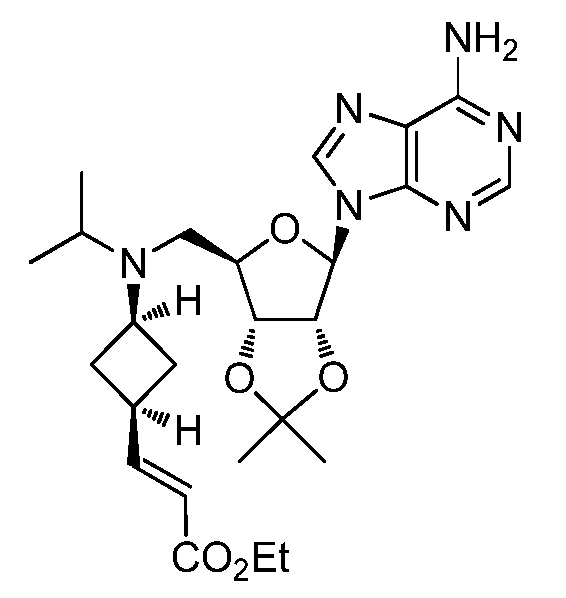
К раствору (1S,3s)-3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(изопропил)амино)циклобутанкарбальдегида (930 мг, 2,16 ммоль) в CH3CN:DCM=5:1 (50 мл) добавляли этил 2-(диэтоксифосфорил)ацетат (484 мг, 2,16 ммоль), DBU (328 мг, 2,16 ммоль) и LiCl (91 мг, 2,16 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа и затем концентрировали. Добавляли воду (20 мл) и смесь экстрагировали при помощи DCM (25 мл ×3). Объединенные органические слои сушили над Na2SO4, концентрировали и остаток очищали при помощи SGC (DCM:MeOH=30:1) с получением указанного в заголовке соединения (900 мг, выход 83%).
1H ЯМР (500 МГц, CDCl3): δH 8,36 (с, 1H), 7,89 (с, 1H), 6,94-6,90 (м, 1H), 6,03 (с, 1H), 5,72-5,89 (м, 1H), 5,57 (с, 2H), 5,52 (д, J=4,5 Гц, 1H), 5,00 (дд, J=3,5, 6,0 Гц, 1H), 4,25 (д, J=3,0 Гц, 1H), 4,21-4,17 (м, 2H), 3,14 (шир.с, 1H), 2,961-2,936 (м, 1H), 2,74-2,52 (м, 3H), 2,22-2,14 (м, 2H), 1,79-1,76 (м, 2H), 1,60 (с, 3H), 1,40 (с, 3H), 1,30-1,27 (м, 3H), 1,00 (д, J=7,0 Гц, 3H), 0,82 (д, J=6,5 Гц, 3H) м.д.; ESI-MS (m/z): 501,4 [M+1]+.
Стадия 5: Синтез этил 3-((1S,3r)-3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(изопропил)амино)циклобутил)пропаноата
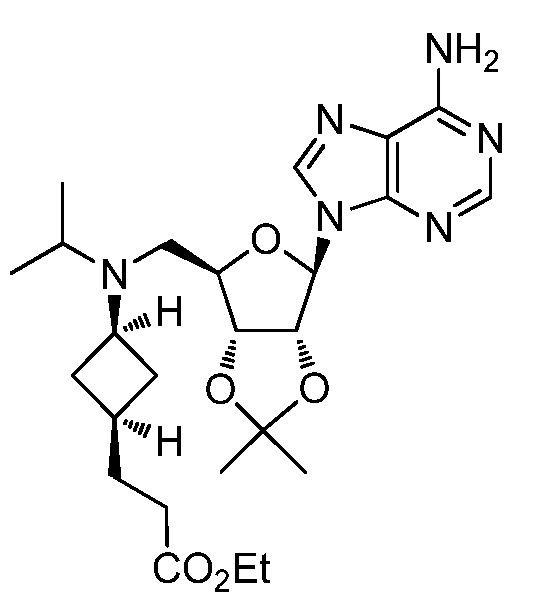
К раствору (E)-этил 3-((1S,3s)-3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(изопропил)амино)циклобутил)акрилата (900 мг, 1,8 ммоль) в MeOH (50 мл) добавляли Pd/C (20 мг). Смесь перемешивали при комнатной температуре в течение ночи в атмосфере водорода. Смесь фильтровали и фильтрат концентрировали с получением указанного в заголовке соединения (700 мг, выход 78%).
1H ЯМР (500 МГц, CDCl3): δH 8,36 (с, 1H), 7,89 (с, 1H), 6,03 (д, J=2,5 Гц, 1H), 5,69 (с, 2H), 5,51 (дд, J=2,5, 8,0 Гц, 1H), 4,99 (дд, J=4,0, 7,5 Гц, 1H), 4,26 (шир.с, 1H), 4,13-4,08 (м, 2H), 2,99-2,92 (м, 2H), 2,706-2,655 (м, 1H), 2,539-2,486 (м, 1H), 2,18-2,02 (м, 4H), 1,76 (шир.с, 1H), 1,65-1,60 (м, 5H), 1,43-1,37 (м, 5H), 1,26-1,23 (м, 2H), 0,97 (д, J=9,0 Гц, 3H), 0,79 (д, J=8,5 Гц, 3H) м.д.; ESI-MS (m/z): 503,4 [M+1]+.
Стадия 6: Синтез 3-((1S,3r)-3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(изопропил)амино)циклобутил)пропановой кислоты
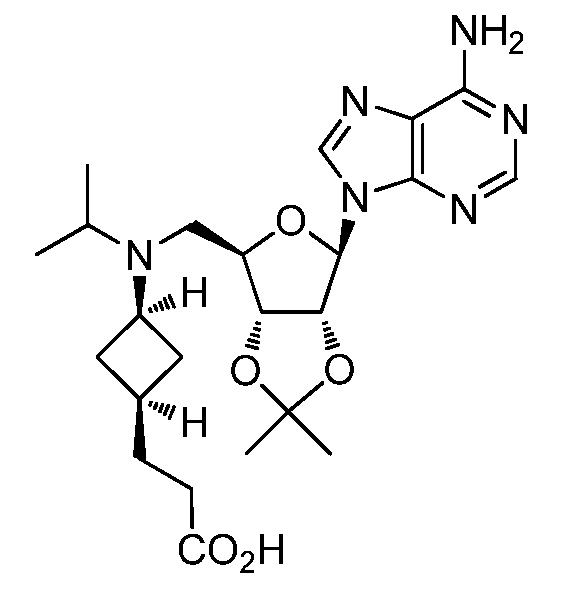
К раствору этил 3-((1S,3r)-3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(изопропил)амино)циклобутил)пропаноата (650 мг, 1,29 ммоль) в ТГФ:MeOH=5:1 (30 мл) добавляли LiOH·H2O (543 мг, 1,29 ммоль). Смесь перемешивали при комнатной температуре в течение ночи, концентрировали и затем ее переносили для поглощения в MeOH (10 мл). Добавляли по каплям 1M HCl раствор при 0°C до pH=7. Смесь концентрировали и очищали методом препаративной ВЭЖХ с получением указанного в заголовке соединения (170 мг).
Стадия 7: Синтез N-(2-амино-4-(трет-бутил)фенил)-3-((1S,3r)-3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(изопропил)амино)циклобутил)пропанамида
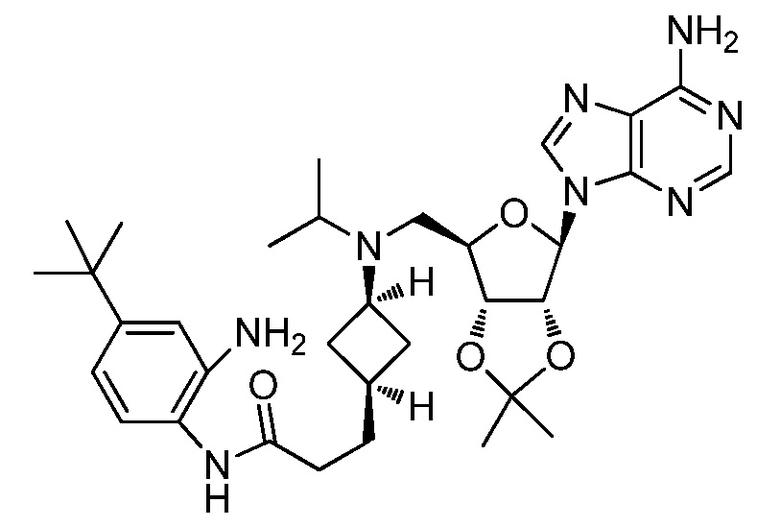
К раствору 3-((1S,3r)-3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(изопропил)амино)циклобутил)пропановой кислоты (170 мг, 0,36 ммоль) в DCM (15 мл) добавляли 4-трет-бутилбензол-1,2-диамин (117 мг, 0,72 ммоль), EDCI (137 мг, 0,72 ммоль), HOBT (97 мг, 0,72 ммоль) и TEA (217 мг, 2,15 ммоль). Смесь перемешивали при комнатной температуре в течение ночи и концентрировали. Добавляли насыщенный раствор NaHCO3 (20 мл) и смесь экстрагировали при помощи DCM (20 мл ×3). Органические слои сушили над Na2SO4 и концентрировали. Неочищенное вещество очищали методом препаративной ТСХ (DCM:MeOH=12:1) с получением указанного в заголовке соединения (110 мг неочищенного вещества).
Стадия 8: Синтез 9-((3aR,4R,6R,6aR)-6-((((1r,3S)-3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(изопропил)амино)метил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)-9H-пурин-6-амин
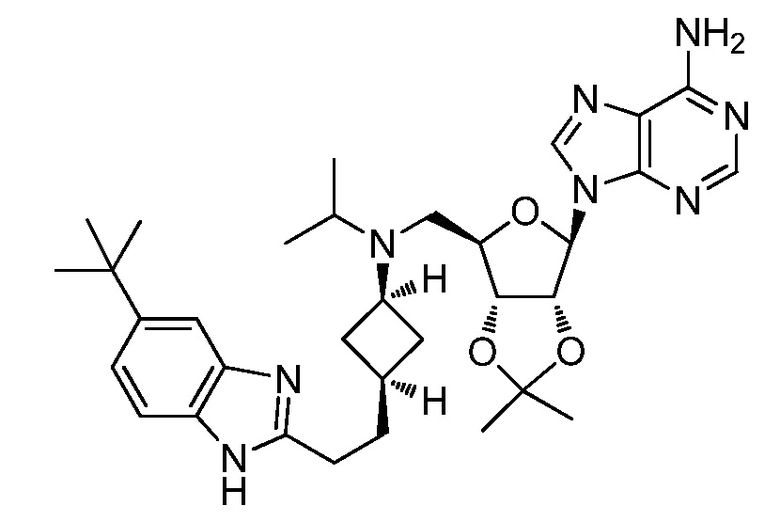
Раствор N-(2-амино-4-(трет-бутил)фенил)-3-((1S,3r)-3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(изопропил)амино)циклобутил)пропанамида (110 мг) в AcOH (10 мл) нагревали до 65°C в течение ночи. Смесь концентрировали, добавляли насыщенный раствор NaHCO3 (20 мл) и смесь экстрагировали при помощи DCM (20 мл ×3). Объединенные органические слои сушили над Na2SO4 и концентрировали с получением указанного в заголовке соединения (105 мг неочищенного вещества).
1H ЯМР (500 МГц, CDCl3): δH 8,36 (с, 1H), 7,89 (с, 1H), 7,48-7,24 (м, 3H), 6,01 (д, J=1,5 Гц, 1H), 5,60-5,53 (м, 3H), 4,98 (дд, J=3,0, 6,5 Гц, 1H), 4,22 (шир.с, 1H), 2,97 (шир.с, 1H), 2,874-2,847 (м, 1H), 2,56-2,50 (м, 3H), 1,87-1,78 (м, 2H), 1,70-1,54 (м, 7H), 1,35-1,17 (м, 14H), 0,90 (д, J=6,5 Гц, 3H), 0,80 (д, J=6,5 Гц, 3H) м.д.; ESI-MS (m/z): 603,5 [M+1]+.
Стадия 9: Синтез соединения 2
Раствор 9-((3aR,4R,6R,6aR)-6-((((1r,3S)-3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(изопропил)амино)метил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)-9H-пурин-6-амина (105 мг) в HCl/MeOH (2,5 моль/л) (10 мл) перемешивали при комнатной температуре в течение 2 часов, затем концентрировали досуха. Добавляли K2CO3 (96 мг) в воде (0,5 мл) и MeOH (5 мл) и полученную смесь перемешивали еще в течение 10 минут при комнатной температуре и затем фильтровали. Фильтрат концентрировали и остаток очищали методом препаративной ВЭЖХ (xbrige 30мм ×150мм, Подвижные фазы: A: вода (10 мМ NH4HCO3) B: CAN, Градиент: 35-45% B в течение 10 минут, 45-45% B в течение 6 минут, остановка в момент времени 20 минут, Скорость потока: 50 мл/мин) с получением соединения 2 (50 мг, выход: 51%) в виде белого твердого вещества.
1H ЯМР (500 МГц, MeOD): δH 8,29 (с, 1H), 8,20 (с, 1H), 7,47-7,39 (м, 3H), 5,96 (д, J=4,0 Гц, 1H), 4,70-4,75 (м, 1H), 4,26-4,27 (м, 1H), 4,05-4,06 (м, 1H), 3,140-3,155 (м, 1H), 3,00-2,76 (м, 5H), 2,18-2,16 (м, 2H), 1,87-1,85 (м, 2H), 1,57-1,55 (м, 2H), 1,36 (с, 9H), 1,01 (д, J=6,5 Гц, 3H), 0,94 (д, J=6,5 Гц, 3H) м.д.; ESI-MS (m/z): 563,4 [M+1]+.
Пример 3: Синтез (2R,3R,4S,5R)-2-(6-амино-9H-пурин-9-ил)-5-((((1s,3R)-3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(изопропил)амино)метил)тетрагидрофуран-3,4-диола (Соединение 3)
Стадия 1: Синтез (1R,3r)-метил 3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(изопропил)амино)-циклобутанкарбоксилата
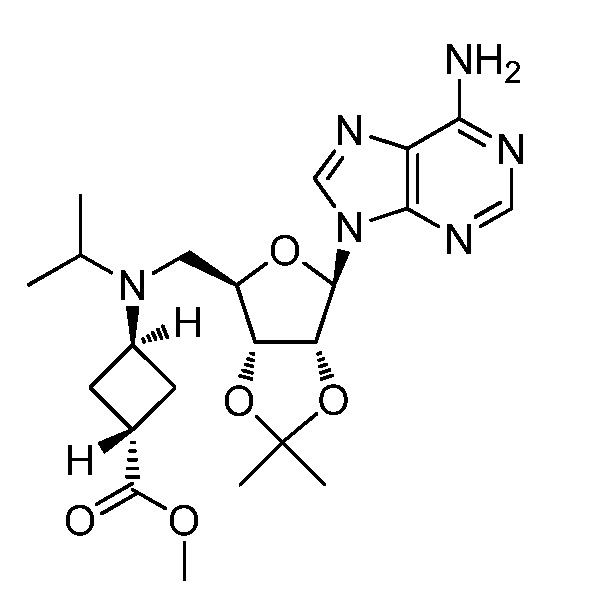
К раствору (1R,3r)-метил 3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)амино)циклобутанкарбоксилата (1,7 г, 4,07 ммоль) в CH3CN (15 мл) добавляли 2-иодпропан (3,5 г, 20,3 ммоль) и K2CO3 (0,84 г, 6,10 ммоль). Реакционную смесь нагревали до 95oC в течение ночи в герметично закрытой пробирке. Смесь фильтровали и фильтрат концентрировали и очищали при помощи SGC (DCM:MeOH=12:1) с получением указанного в заголовке соединения (1,35 г, выход 72%).
1H ЯМР (500 МГц, CDCl3): δH 8,36 (с, 1H), 7,88 (с, 1H), 6,03 (д, J=2,0 Гц, 1H), 5,55 (м, 2H), 5,49 (дд, J=1,5, 6,0 Гц, 1H), 5,01 (дд, J=3,5, 6,0 Гц, 1H), 4,254-4,247 (м, 1H), 3,68 (с, 3H), 3,60-3,50 (м, 1H), 2,930-2,917 (м, 1H), 2,79-2,74 (м, 2H), 2,59-2,57 (м, 1H), 2,25-2,12 (м, 4H), 1,60 (с, 3H), 1,39 (с, 3H), 1,00 (д, J=6,5 Гц, 3H), 0,83 (д, J=7,0 Гц, 3H) м.д.; ESI-MS (m/z): 461,3 [M+1]+.
Стадия 2: Синтез (1R,3r)-3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(изопропил)амино)циклобутанкарбальдегида
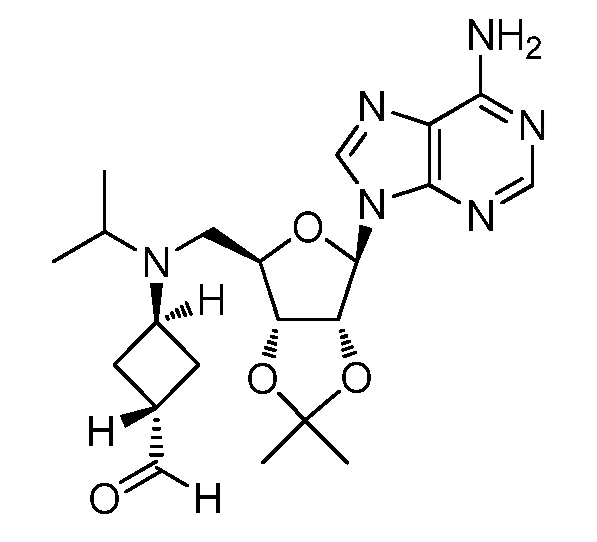
К раствору (1R,3r)-метил 3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(изопропил)амино)циклобутанкарбоксилата (1,35 г, 2,93 ммоль) в DCM (50 мл) при -78oC добавляли DiBAL-H по каплям, до тех пор, пока исходное вещество не было полностью израсходовано, как было определено при помощи ТСХ. Добавляли MeOH (2 мл) и смесь перемешивали при комнатной температуре (RT) в течение 30 минут. Добавляли воду (50 мл) и смесь экстрагировали при помощи DCM (50 мл ×2). Объединенные органические слои сушили над Na2SO4 и концентрировали с получением неочищенного указанного в заголовке соединения (1,1 г), которое использовали непосредственно на следующей стадии
1H ЯМР (500 МГц, CDCl3): δH 9,80 (с, 1H), 8,35 (с, 1H), 7,88 (с, 1H), 6,04 (с, 1H), 5,56 (с, 2H), 5,50 (д, J=6,5 Гц, 1H), 5,028-5,026 (м, 1H), 4,26 (шир.с, 1H), 3,33-3,30 (м, 1H), 2,956-2,930 (м, 1H), 2,80-2,55 (м, 3H), 2,27-2,07 (м, 4H), 1,60 (с, 3H), 1,39 (с, 3H), 1,00 (д, J=7,0 Гц, 3H), 0,82 (д, J=6,5 Гц, 3H) м.д.
Стадия 3: Синтез (E)-этил 3-((1R,3r)-3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(изопропил)амино)циклобутил)акрилата
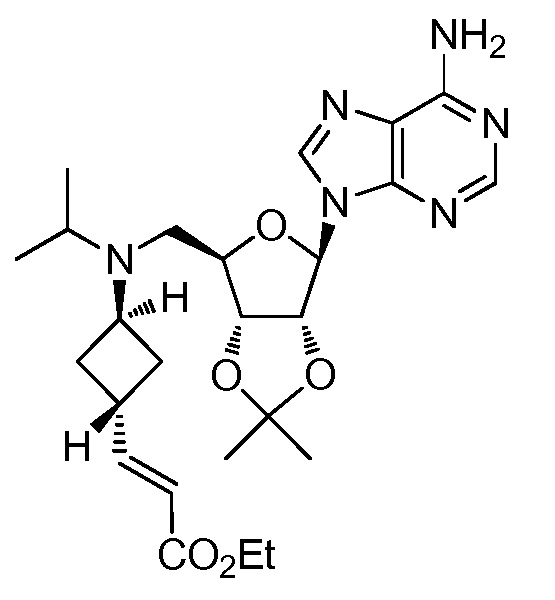
К раствору (1R,3r)-3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(изопропил)амино)циклобутанкарбальдегида (1,1 г, 2,56 ммоль) в CH3CN:DCM=5:1 (50 мл) добавляли этил 2-(диэтоксифосфорил)ацетат (573 мг, 2,56 ммоль), DBU (389 мг, 2,56 ммоль) и LiCl (107 мг, 2,56 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа и концентрировали, затем добавляли воду (20 мл) и смесь экстрагировали при помощи DCM (25 мл ×3). Объединенные органические слои сушили над Na2SO4 и концентрировали. Остаток очищали при помощи SGC (DCM:MeOH=30:1) с получением указанного в заголовке соединения (1,0 г, выход 78%).
1H ЯМР (500 МГц, CDCl3): δH 8,35 (с, 1H), 7,89 (с, 1H), 7,16-7,11 (м, 1H), 6,03 (д, J=2,0 Гц, 1H), 5,79-5,76 (м, 1H), 5,56 (с, 2H), 5,51 (дд, J=1,5, 6,0 Гц, 1H), 5,02 (дд, J=3,0, 6,0 Гц, 1H), 4,25 (д, J=8,0 Гц, 1H), 4,22-4,17 (м, 2H), 3,44 (шир.с, 1H), 2,93 (шир.с, 1H), 2,78-2,56 (м, 3H), 2,27-2,16 (м, 2H), 1,93-1,91 (м, 2H), 1,60 (с, 3H), 1,40 (с, 3H), 1,31-1,27 (м, 3H), 0,98 (д, J=6,5 Гц, 3H), 0,82 (д, J=6,5 Гц, 3H) м.д.; ESI-MS (m/z): 501,4 [M+1]+.
Стадия 4: Синтез этил 3-((1R,3s)-3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(изопропил)амино)циклобутил)пропаноата
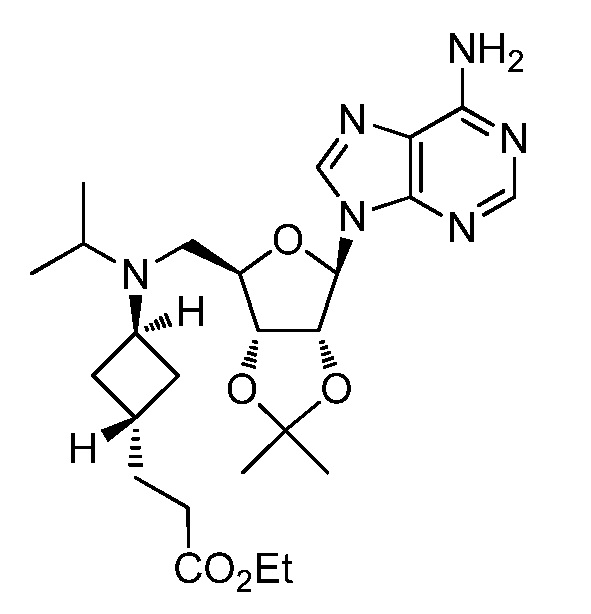
К смеси (E)-этил 3-((1R,3r)-3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(изопропил)амино)циклобутил)акрилата (1,0 г, 2,0 ммоль) и 10% Pd/C (30 мг) в MeOH (50 мл) добавляли Pd/C (30 мг). Смесь перемешивали при комнатной температуре в течение ночи в атмосфере водорода. Полученную смесь фильтровали и фильтрат концентрировали с получением указанного в заголовке соединения (1,0 г, выход 100%).
1H ЯМР (500 МГц, CDCl3): δH 8,36 (с, 1H), 7,89 (с, 1H), 6,03 (д, J=2,5 Гц, 1H), 5,58 (с, 2H), 5,51 (дд, J=2,0, 6,5 Гц, 1H), 5,00 (дд, J=3,5, 6,0 Гц, 1H), 4,276-4,269 (м, 1H), 4,13-4,09 (м, 2H), 3,38-3,37 (м, 1H), 2,94-2,54 (м, 3H), 2,22-1,97 (м, 5H), 1,79-1,62 (м, 4H), 1,60 (с, 3H), 1,40 (с, 3H), 1,28-1,23 (м, 2H), 0,97 (д, J=7,0 Гц, 3H), 0,79 (д, J=7,0 Гц, 3H) м.д.; ESI-MS (m/z): 503,4 [M+1]+.
Стадия 5: Синтез 3-((1R,3s)-3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(изопропил)амино)циклобутил)пропановой кислоты
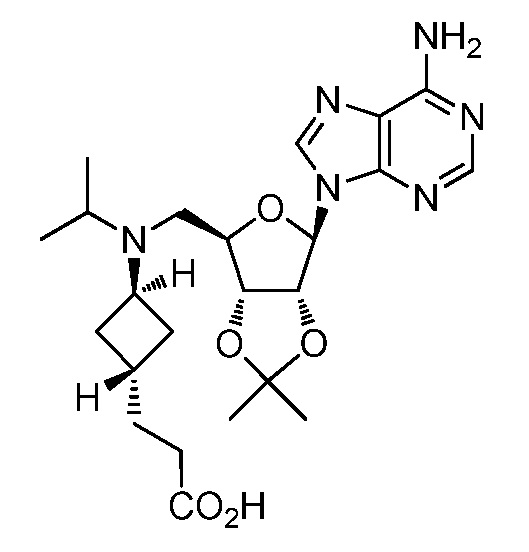
К раствору этил 3-((1R,3s)-3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(изопропил)амино)циклобутил)пропаноата (360 мг, 0,72 ммоль) в ТГФ:MeOH=5:1 (30 мл) добавляли LiOH·H2O (301 мг, 7,20 ммоль). Смесь перемешивали при комнатной температуре в течение ночи, концентрировали, затем растворяли в MeOH (10 мл). Добавляли раствор 1 M HCl по каплям при 0°C до pH=7. Смесь концентрировали с получением неочищенного указанного в заголовке соединения, которое использовали непосредственно на следующей стадии.
Стадия 6: Синтез N-(2-амино-4-(трет-бутил)фенил)-3-((1R,3s)-3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(изопропил)амино)циклобутил)пропанамида
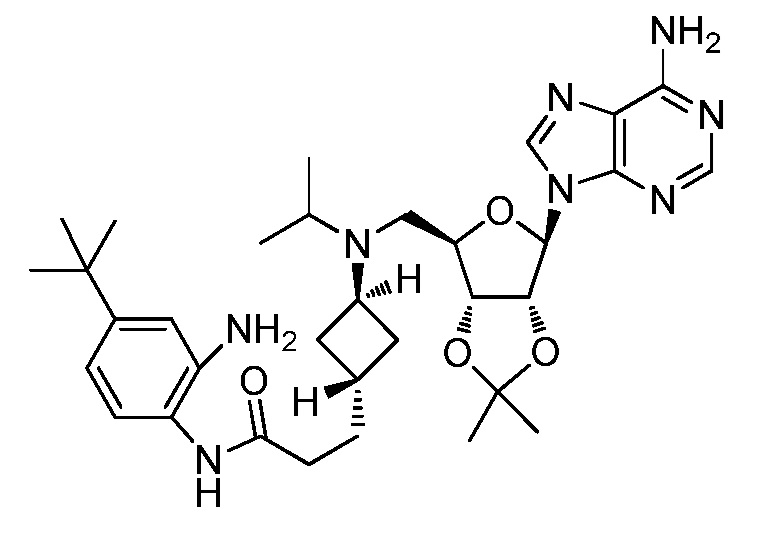
К раствору 3-((1R,3s)-3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(изопропил)амино)циклобутил)пропановой кислоты в DMF (5 мл) добавляли 4-(трет-бутил)бензол-1,2-диамин (235 мг, 1,43 ммоль), EDCI (274 мг, 1,43 ммоль), HOBT (193 мг, 1,43 ммоль) и TEA (435 мг, 4,30 ммоль). Смесь нагревали до 45oC в течение ночи и концентрировали. Добавляли насыщенный раствор NaHCO3 раствор (20 мл) и смесь экстрагировали при помощи DCM (20 мл ×3). Органические слои сушили над Na2SO4 и концентрировали. Неочищенное вещество очищали методом препаративной ТСХ (DCM:MeOH=12:1) с получением указанного в заголовке соединения (110 мг), которое использовали на следующей стадии без дополнительной очистки.
Стадия 7: Синтез 9-((3aR,4R,6R,6aR)-6-((((1s,3R)-3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(изопропил)амино)метил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)-9H-пурин-6-амина
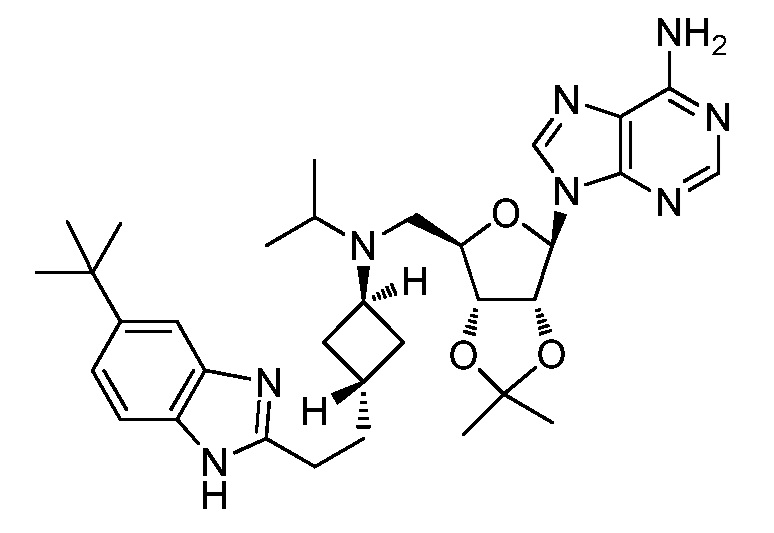
Раствор N-(2-амино-4-(трет-бутил)фенил)-3-((1R,3s)-3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(изопропил)амино)циклобутил)пропанамида (110 мг) в AcOH (15 мл) нагревали при 65°C в течение ночи. Смесь концентрировали, добавляли насыщенный раствор NaHCO3 (20 мл) и смесь экстрагировали при помощи DCM (20 мл ×3). Объединенные органические слои сушили над Na2SO4 и концентрировали с получением указанного в заголовке соединения (100 мг неочищенного вещества).
1H ЯМР (500 МГц, CDCl3): δH 8,36 (с, 1H), 7,94 (с, 1H), 7,48-7,27 (м, 3H), 6,07 (д, J=1,5 Гц, 1H), 5,64-5,58 (м, 3H), 5,02 (дд, J=3,0, 6,0 Гц, 1H), 4,30 (шир.с, 1H), 3,38-3,37 (м, 1H), 2,97-2,95 (м, 1H), 2,76-2,55 (м, 3H), 1,97-1,74 (м, 5H), 1,67-1,57 (м, 5H), 1,45-1,40 (м, 12H), 0,99 (д, J=6,5 Гц, 3H), 0,83 (д, J=6,5 Гц, 3H) м.д.; ESI-MS (m/z): 603,5 [M+1]+.
Стадия 8: Синтез соединения 3
Раствор 9-((3aR,4R,6R,6aR)-6-((((1s,3R)-3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(изопропил)амино)метил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)-9H-пурин-6-амина (190 мг) в HCl/MeOH (2,5 моль/л) (15 мл) перемешивали при комнатной температуре в течение 2 часов и концентрировали досуха. Добавляли K2CO3 (161 мг) в воде (0,5 мл) и MeOH (5 мл). Полученную смесь перемешивали еще в течение 10 минут при комнатной температуре, затем фильтровали. Фильтрат концентрировали и остаток очищали методом препаративной ВЭЖХ (xbrige 30 мм ×150мм, Подвижные фазы: A: вода(10 мМ NH4HCO3) B: CAN, Градиент: 35-45% B в течение 10 минут, 45-45% B в течение 6 минут, остановка в момент времени 20 минут, Скорость потока: 50 мл/мин) с получением соединения 3 (65 мг, выход: 70%) в виде белого твердого вещества.
1H ЯМР (500 МГц, MeOD): δH 8,29 (с, 1H), 8,19 (с, 1H), 7,47-7,28 (м, 3H), 5,95 (д, J=4,5 Гц, 1H), 4,744-4,724 (м, 1H), 4,27-4,26 (м, 1H), 4,07-4,06 (м, 1H), 3,56 (шир.с, 1H), 3,01-2,78 (м, 5H), 2,17 (шир.с, 2H), 2,00-1,93 (м, 2H), 1,80-1,79 (м, 2H), 1,36 (с, 9H), 1,02 (д, J=5,5 Гц, 3H), 0,95 (д, J=6,0 Гц, 3H) м.д.; ESI-MS (m/z): 563,5 [M+1]+.
Пример 4: Синтез (2R,3R,4S,5R)-2-(6-амино-9H-пурин-9-ил)-5-((((1s,3R)-3-(2-(5-хлор-6-(трифторметил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(изопропил)амино)метил)тетрагидрофуран-3,4-диола (Соединение 4)
Стадия 1: Синтез N-(2-амино-4-хлор-5-(трифторметил)фенил)-3-((1R,3s)-3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(изопропил)амино)циклобутил)пропанамида
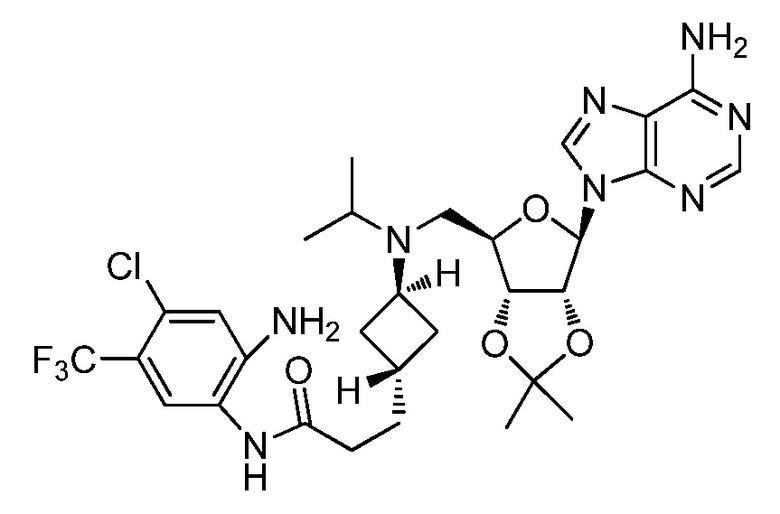
К раствору 3-((1R,3s)-3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(изопропил)амино)циклобутил)пропановой кислоты (250 мг, 0,53 ммоль) в DCM (30 мл) добавляли 4-хлор-5-(трифторметил)бензол-1,2-диамин (221 мг, 1,05 ммоль), EDCI (201 мг, 1,05 ммоль), HOBT (142 мг, 1,05 ммоль) и TEA (320 мг, 3,15 ммоль). Смесь перемешивали при комнатной температуре в течение ночи, затем добавляли насыщенный раствор NaHCO3 (20 мл) и смесь экстрагировали при помощи DCM (20 мл ×3). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали. Неочищенное вещество очищали методом препаративной ТСХ (DCM:MeOH=12:1) с получением указанного в заголовке соединения (250 мг неочищенного вещества).
Стадия 2: Синтез 9-((3aR,4R,6R,6aR)-6-((((1s,3R)-3-(2-(5-хлор-6-(трифторметил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(изопропил)амино)метил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)-9H-пурин-6-амина
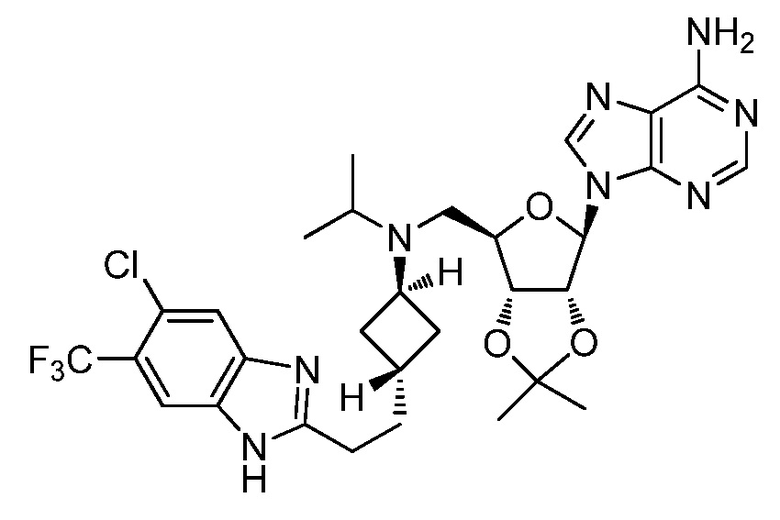
Раствор N-(2-амино-4-хлор-5-(трифторметил)фенил)-3-((1R,3s)-3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(изопропил)амино)циклобутил)пропанамида (250 мг) в AcOH (15 мл) нагревали до 65°C в течение ночи. Смесь концентрировали, добавляли насыщенный раствор NaHCO3 (20 мл) и смесь экстрагировали при помощи DCM (20 мл ×3). Объединенные органические слои сушили над Na2SO4 и концентрировали с получением указанного в заголовке соединения (200 мг неочищенного вещества).
Стадия 3: Синтез соединения 4
Раствор 9-((3aR,4R,6R,6aR)-6-((((1s,3R)-3-(2-(5-хлор-6-(трифторметил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)-(изопропил)амино)метил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)-9H-пурин-6-амина (200 мг) в HCl/MeOH (2,5 моль/л) (15 мл) перемешивали при комнатной температуре в течение 2 часов, затем концентрировали досуха. Добавляли K2CO3 (166 мг) в воде (0,5 мл) и MeOH (5 мл) и полученную смесь перемешивали еще в течение 10 минут при комнатной температуре. Смесь фильтровали и фильтрат концентрировали. Остаток очищали методом препаративной ВЭЖХ с получением соединения 4 (80 мг, выход: 43%) в виде белого твердого вещества.
1H ЯМР (500 МГц, MeOD): δH 8,29 (с, 1H), 8,19 (с, 1H), 7,88 (с, 1H), 7,68 (с, 1H), 5,96 (д, J=4,0 Гц, 1H), 4,748-4,730 (м, 1H), 4,284-4,263 (м, 1H), 4,09 (шир.с, 1H), 3,65-3,50 (м, 1H), 3,03-2,85 (м, 5H), 2,191-2,176 (м, 2H), 2,03-2,00 (м, 2H), 1,80 (шир.с, 2H), 1,02 (д, J=6,0 Гц, 3H), 0,96 (д, J=6,6 Гц, 3H) м.д.; ESI-MS (m/z): 609,2 [M+1]+.
Пример 5: Синтез (2R,3R,4S,5R)-2-(6-амино-9H-пурин-9-ил)-5-((((1r,3S)-3-(2-(5-хлор-6-(трифторметил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(изопропил)амино)метил)тетрагидрофуран-3,4-диола (Соединение 5)
Стадия 1: Синтез N-(2-амино-4-хлор-5-(трифторметил)фенил)-3-((1S,3r)-3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(изопропил)амино)циклобутил)пропанамида
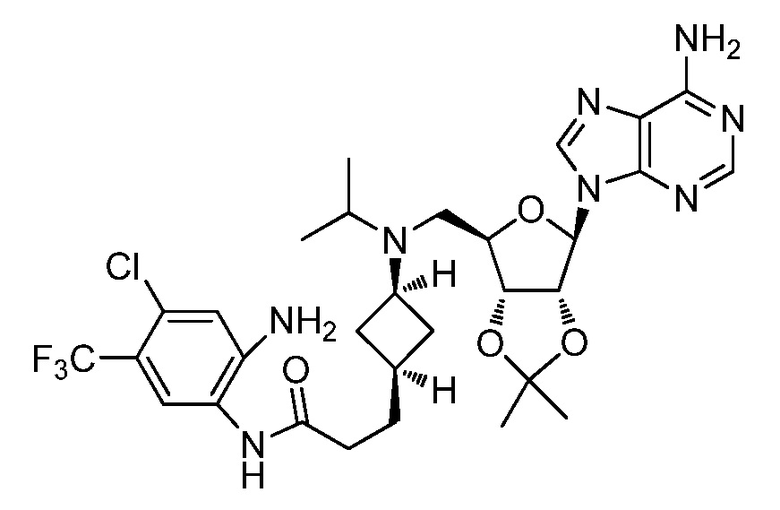
К раствору 3-((1S,3r)-3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(изопропил)амино)циклобутил)пропановой кислоты в DCM:DMF=15:1 (30 мл) добавляли 4-хлор-5-(трифторметил)бензол-1,2-диамин (334 мг, 1,60 ммоль), EDCI (304 мг, 1,60 ммоль), HOBT (215 мг, 1,60 ммоль) и TEA (483 мг, 4,80 ммоль). Смесь перемешивали в течение ночи при комнатной температуре. Смесь концентрировали, добавляли насыщенный раствор NaHCO3 (20 мл) и полученную смесь экстрагировали при помощи DCM (20 мл ×3). Объединенные органические слои сушили над Na2SO4 и концентрировали. Неочищенный остаток очищали методом препаративной ТСХ (DCM:MeOH=12:1) с получением указанного в заголовке соединения (220 мг).
Стадия 2: Синтез 9-((3aR,4R,6R,6aR)-6-((((1r,3S)-3-(2-(5-хлор-6-(трифторметил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(изопропил)амино)метил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)-9H-пурин-6-амин
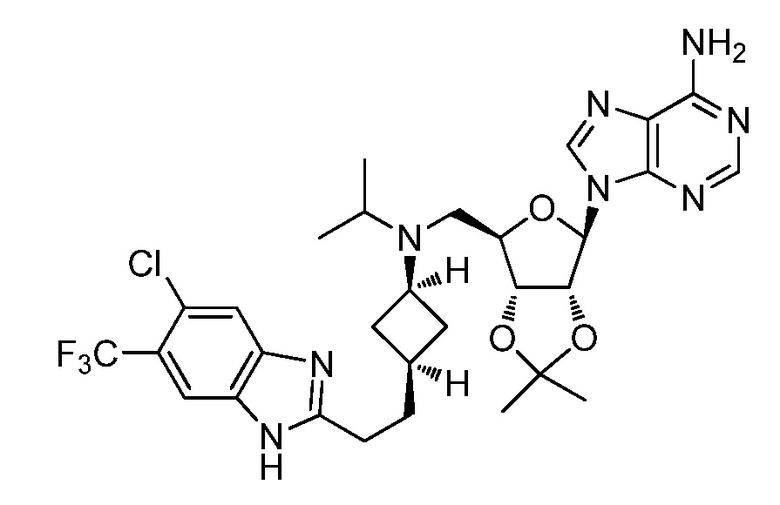
Раствор N-(2-амино-4-хлор-5-(трифторметил)фенил)-3-((1S,3r)-3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(изопропил)амино)циклобутил)пропанамида (220 мг) в AcOH (15 мл) нагревали при 65°C в течение ночи. Смесь концентрировали, добавляли насыщенный раствор NaHCO3 (20 мл) и смесь экстрагировали при помощи DCM (20 мл ×3). Объединенные органические слои сушили над Na2SO4 и концентрировали с получением указанного в заголовке соединения (190 мг).
Стадия 3: Синтез соединения 5
Раствор 9-((3aR,4R,6R,6aR)-6-((((1r,3S)-3-(2-(5-хлор-6-(трифторметил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(изопропил)амино)метил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)-9H-пурин-6-амина (190 мг) в HCl/MeOH (2,5 моль/л) (15 мл) перемешивали при комнатной температуре в течение 2 часов, затем концентрировали досуха. Добавляли K2CO3 (161 мг) в воде (0,5 мл) и MeOH (5 мл) и полученную смесь перемешивали еще в течение 10 минут при комнатной температуре. Смесь фильтровали и фильтрат концентрировали. Остаток очищали методом препаративной ВЭЖХ с получением соединения 5 (90 мг, выход: 51%) в виде белого твердого вещества.
1H ЯМР (500 МГц, MeOD): δH 8,29 (с, 1H), 8,19 (с, 1H), 7,88 (с, 1H), 7,67 (с, 1H), 5,95 (д, J=5,0 Гц, 1H), 4,736-4,716 (м, 1H), 4,268-4,246 (м, 1H), 4,070-4,051 (м, 1H), 3,15 (шир.с, 1H), 3,00-2,71 (м, 5H), 2,17 (шир.с, 2H), 1,93-1,88 (м, 2H), 1,58-1,56 (м, 2H), 1,01 (д, J=5,5 Гц, 3H), 0,95 (д, J=6,0 Гц, 3H) м.д.; ESI-MS (m/z): 609,2 [M+1]+.
Пример 6: Синтез (2R,3R,4S,5R)-2-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-(((3-((5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)метил)циклобутил)(метил)амино)метил)тетрагидрофуран-3,4-диола (Соединение 6)
Стадия 1: Синтез 7-((3aR,4R,6R,6aR)-6-(азидометил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)-N-(2,4-диметоксибензил)-7H-пирроло[2,3-d]пиримидин-4-амина
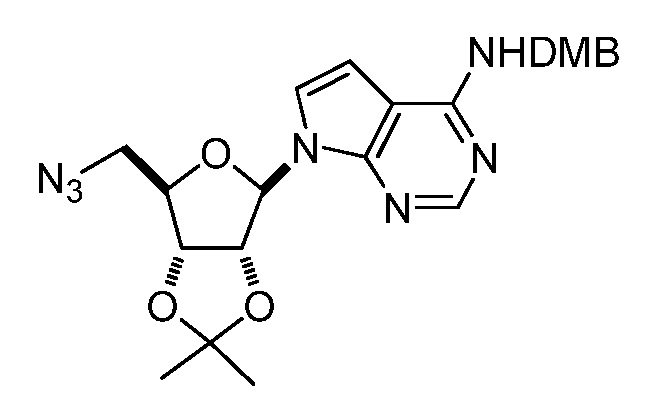
Раствор ((3aR,4R,6R,6aR)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метанола (2,83 г, 6,20 ммоль) и трифенилфосфина (2,28 г, 8,68 ммоль) в безводном тетрагидрофуране (32 мл) охлаждали при 0°C на бане лед/вода. Добавляли по каплям диизопропилазодикарбоксилат (1,71 мл, 8,68 ммоль), с последующим добавлением раствора азида дифенилфосфоновой кислоты (1,87 мл, 8,68 ммоль) в тетрагидрофуране (5,3 мл, 66 ммоль). После добавления DPPA раствора происходило образование белого молочного осадка. Примерно через 30 минут реакционной смеси давали нагреться до комнатной температуры и перемешаться в течение ночи. Через 24 часа ВЭЖХ анализ показал, что все исходное вещество было израсходовано. Реакционную смесь концентрировали до около 1/2 исходного объема и очищали флэш-хроматографией (175 г силикагеля, 10-55% EA/гептан) с получением указанного в заголовке соединения (2,49 г, 83%) в виде слегка желтой жесткой пены: MS (ESI+) для C23H27N7O5 m/z 482,2 (M+H)+; (ESI-) для C23H27N7O5 m/z 480,1 (M+H)-, m/z 526,1 (M+CO2H)-; ВЭЖХ чистота 97%.
Стадия 2: Синтез 7-((3aR,4R,6R,6aR)-6-(аминометил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)-N-(2,4-диметоксибензил)-7H-пирроло[2,3-d]пиримидин-4-амин
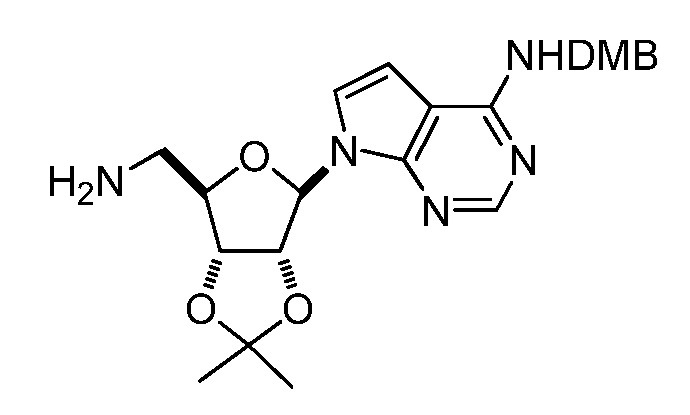
Раствор ((3aR,4R,6R,6aR)-6-(азидометил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)-N-(2,4-диметоксибензил)-7H-пирроло[2,3-d]пиримидин-4-амина (2,49 г, 5,17 ммоль) в тетрагидрофуране (50 мл, 600 ммоль) обрабатывали по каплям раствором 1,0 M триметилфосфина в тетрагидрофуране (7,24 мл, 7,24 ммоль) и смесь перемешивали в течение 20 часов. Реакционную смесь обрабатывали водой (1,80 мл, 99,9 ммоль) и перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь концентрировали, неочищенный продукт брали для поглощения в 90 мл CH2Cl2 и промывали четырьмя 30 мл порциями H2O и 15 мл насыщенного солевого раствора. Раствор сушили над Na2SO4, фильтровали и концентрировали. Неочищенное вещество очищали флэш-хроматографией (120 г силикагеля, 3-10% 7н NH3 в CH3OH/CH2Cl2) с получением указанного в заголовке соединения (1,76 г, 75%) в виде пенообразного вещества MS (ESI+) для C23H29N5O5 m/z 456,2 (M+H)+; (ESI-) для C26H35N5O5 m/z 454,1 (M-H)-; ВЭЖХ чистота 92% (время удерживания, 2,65 мин).
Стадия 3: Синтез метил 2-(3-((((3aR,4R,6R,6aR)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)амино)циклобутил)ацетата
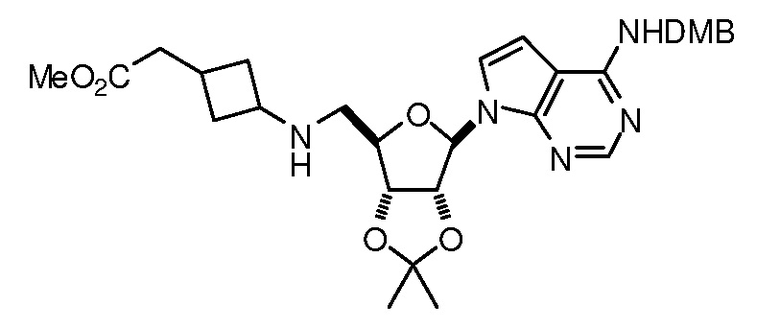
Раствор 7-((3aR,4R,6R,6aR)-6-(аминометил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)-N-(2,4-диметоксибензил)-7H-пирроло[2,3-d]пиримидин-4-амина (400 мг, 0,88 ммоль) и метил 2-(3-оксоциклобутил)ацетата (100 мг, 0,70 ммоль) [получен с использованием процедур, описанных в Публикаци патентной заявки США 2009/0118287] в 1,2-дихлорэтане (12 мл) обрабатывали по каплям уксусной кислотой (50 мкл, 0,88 ммоль). Раствор обрабатывали триацетоксиборогидридом натрия (260 мг, 1,2 ммоль) одной порцией и оставляли для перемешивания при комнатной температуре до завершения реакции по данным ВЭЖХ. Через 4 часа ВЭЖХ анализ показал, что реакция завершена примерно на 80%. Добавляли дополнительные 20 мг кетона и перемешивание продолжали в течение 2,5 часов. Реакционную смесь разбавляли при помощи 30 мл CH2Cl2 и промывали при помощи 15 мл насыщенного раствора NaHCO3. Водную фазу промывали при помощи 15 мл CH2Cl2 и объединенную органическую фазу сушили над Na2SO4. Органическую фазу фильтровали и концентрировали с получением светло-желтого стеклообразного вещества, которое очищали флэш-хроматографией (70 г силикагеля; 2% 7н NH3 в CH3OH/CH2Cl2) с получением указанного в заголовке соединения (270 мг, 66%) в виде бесцветного стеклообразного вещества: MS (ESI+) для C30H39N5O7 m/z 582,2 (M+H)+; ВЭЖХ чистота >95% (время удерживания, 2,88 мин).
Стадия 4: Синтез метил 2-(3-((((3aR,4R,6R,6aR)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(метил)амино)циклобутил)ацетата
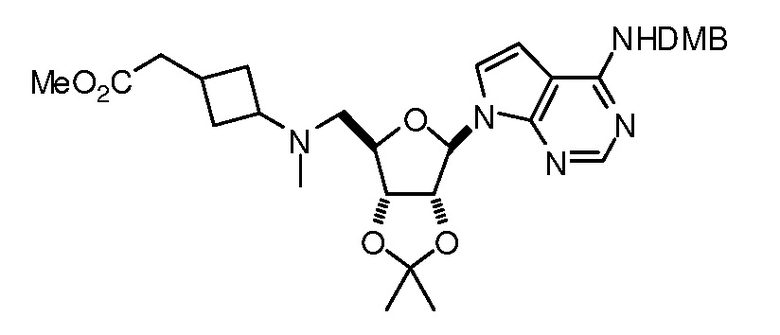
Раствор метил 2-(3-((((3aR,4R,6R,6aR)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)амино)циклобутил)ацетата (267 мг, 0,459 ммоль) в метаноле (12 мл) обрабатывали цианоборогидридом натрия (380 мг, 6,1 ммоль). pH раствора доводили до ~6 путем добавления по каплям 10% (об/об) раствора ледяной уксусной кислоты в метаноле. Смесь обрабатывали по каплям 37% раствором формальдегида (0,57 мл, 7,6 ммоль) и смесь перемешивали при комнатной температуре в течение 1 часа, к этому времени ВЭЖХ анализ показал, что исходное вещество было израсходовано. Реакционную смесь концентрировали для удаления метанола. Водный раствор, который оставался, разбавляли при помощи 25 мл NaHCO3 и водную фазу экстрагировали тремя 20 мл порциями CH2Cl2. Органическую фазу промывали при помощи 20 мл насыщенного раствора NaHCO3, сушили над Na2SO4, фильтровали и концентрировали с получением указанного в заголовке соединения (272 мг, 100%) в виде бесцветной жесткой пены, которая оказалась достаточно чистой для использования на следующей стадии: MS (ESI+) для C31H41N5O7 m/z 596,5 (M+H)+; ВЭЖХ чистота >95% (время удерживания, 2,89 мин).
Стадия 5: Синтез 2-(3-((((3aR,4R,6R,6aR)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(метил)амино)циклобутил)уксусной кислоты
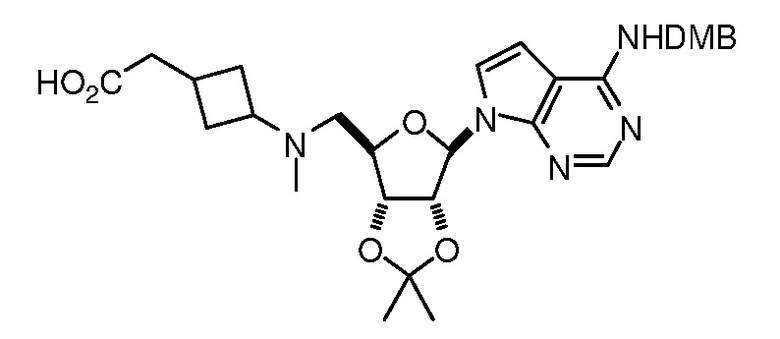
Раствор метил 2-(3-((((3aR,4R,6R,6aR)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(метил)амино)циклобутил)ацетата (270 мг, 0,453 ммоль) в метаноле (8,6 мл) обрабатывали по каплям раствором гидроксида натрия (36 мг, 0,91 ммоль) в воде (0,9 мл, 50 ммоль) и смесь нагревали при 50°C. Через 17 часов ВЭЖХ анализ показал, что реакция заверщена. Реакционную смесь охлаждали до комнатной температуры и обрабатывали при помощи 0,91 мл 1,0н раствора HCl для доведения pH до ~7. Раствор концентрировали для удаления метанола и полученную водную суспензию лиофилизировали с получением белого твердого вещества. Это вещество использовали как таковое на следующей стадии, предполагая количественный выход: MS (ESI+) для C30H39N5O7 m/z 582,4 (M+H)+; MS (ESI-) для C30H39N5O7 m/z 580,4 (M-H)-; ВЭЖХ чистота >95% (время удерживания, 2,72 мин).
Стадия 6: Синтез N-(2-амино-4-(трет-бутил)фенил)-2-(3-((((3aR,4R,6R,6aR)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(метил)амино)циклобутил)ацетамида
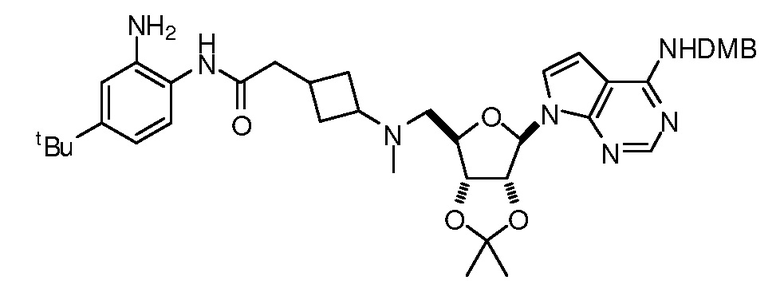
Раствор 2-(3-((((3aR,4R,6R,6aR)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(метил)амино)циклобутил)уксусной кислоты и 4-трет-бутилбензол-1,2-диамина (89,4 мг, 0,545 ммоль) в N,N-диметилформамиде (4,5 мл) обрабатывали N,N-диизопропилэтиламином (0,261 мл, 1,50 ммоль) по каплям, затем гексафторфосфатом N,N,N',N'-тетраметил-O-(7-азабензотриазол-1-ил)урония (259 мг, 0,681 ммоль). Раствор оставляли для перемешивания при комнатной температуре в течение 18 часов, в этот период ЖХ-MS анализ показал, что исходное вещество было израсходовано. Реакционную смесь концентрировали в условиях высокого вакуума. Остаток брали для поглощения в 30 мл этилацетата и 20 мл смеси 1/1 H2O/насыщенный раствор NaHCO3. Смесь экстрагировали и водную фазу промывали при помощи 35 мл этилацетата. Объединенную органическую фазу промывали двумя 20 мл порциями H2O и 20 мл насыщенного солевого раствора. Органическую фазу сушили над Na2SO4, фильтровали и концентрировали с получением желтовато-коричневого стеклообразного вещества/жесткой пены. Неочищенное вещество очищали флэш-хроматографией (35 г силикагеля; 4% 7н NH3 в CH3OH/CH2Cl2) с получением указанного в заголовке соединения (272 мг, 82%) в виде светло-желто-коричневого стеклообразного вещества/жесткой пены, при этом вещество представляло собой смесь региоизомерных амидов: MS (ESI+) для C41H53N7O6 m/z 728,8 (M+H)+; MS (ESI-) для C41H53N7O6 m/z 726,9 (M-H)-; ВЭЖХ чистота >95%, (время удерживания, 3,14, 3,17 мин) наблюдали два пика из-за присутствия амидных региоизомеров.
Стадия 7: Синтез 7-((3aR,4R,6R,6aR)-6-(((3-((5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)метил)циклобутил)(метил)амино)метил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)-N-(2,4-диметоксибензил)-7H-пирроло[2,3-d]пиримидин-4-амина
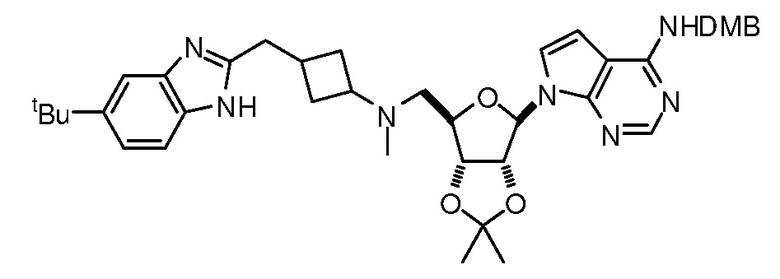
N-(2-амино-4-(трет-бутил)фенил)-2-(3-((((3aR,4R,6R,6aR)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(метил)амино)циклобутил)ацетамид (272 мг, 0,374 ммоль) брали для поглощения в уксусную кислоту (7,2 мл) и раствор нагревали при 65°C. Через 1,5 часа ВЭЖХ анализ показал, что реакция заверщена. Реакционную смесь охлаждали до комнатной температуры и растворитель удаляли в условиях высокого вакуума. Остаток брали для поглощения в 35 мл CH2Cl2 и органическую фазу промывали при помощи 25 мл насыщенного раствора NaHCO3 и 20 мл 2% раствора Na2CO3. Органическую фазу сушили над Na2SO4, фильтровали и концентрировали с получением светло-желто-коричневого стеклообразного вещества/жесткой пены. Неочищенное вещество очищали флэш-хроматографией (30 г силикагеля; 4% 7н NH3 в CH3OH/CH2Cl2) с получением указанного в заголовке соединения (224 мг, 84%) в виде светло-желто-коричневого стеклообразного вещества, которое представляло собой смесь цис и транс диастереомеров вокруг циклобутильного кольца: MS (ESI+) для C40H51N7O5 m/z 710,6 (M+H)+; MS (ESI-) для C41H51N7O5 m/z 708,7 (M-H)-; ВЭЖХ чистота >95% (время удерживания, 3,29, 3,33 мин), наблюдали два пика из-за присутствия диастереомеров вокруг циклобутильного кольца.
Стадия 8: Синтез соединения 6
7-((3aR,4R,6R,6aR)-6-(((3-((5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)метил)циклобутил)(метил)амино)метил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)-N-(2,4-диметоксибензил)-7H-пирроло[2,3-d]пиримидин-4-амин (170 мг, 0,24 ммоль) растворяли в смеси трифторуксусной кислоты (5,0 мл) и воды (0,5 мл), которая была предварительно охлаждена при 0°C на ледяной бане. Раствор перемешивали при 0°C в течение 30 минут и нагревали до комнатной температуры. По прошествии 5 часов при комнатной температуре ставшую очень ярко-розовой реакционную смесь концентрировали. Остаток брали для поглощения в 10 мл MeOH и концентрировали. Эту процедуру повторяли два раза и остаток помещали в условия высокого вакуума на 1 час. Это вещество брали для поглощения в 7 мл MeOH и обрабатывали при помощи 130 мг K2CO3 и пяти капель воды. Смесь оставляли для перемешивания в течение 1 часа, в течение этого времени раствор становился щелочным. Смесь фильтровали через тонкую фритту, твердые вещества промывали при помощи 10 мл MeOH и фильтрат концентрировали с получением почти бесцветного твердого вещества. Неочищенное вещество очищали флэш-хроматографией (30 г силикагеля; 12% 7н NH3 в CH3OH/CH2Cl2) с получением соединения 6 (81 мг, 65%) в виде бесцветного стеклообразного вещества/жесткой пены: MS (ESI+) для C28H37N7O3 m/z 520,4 (M+H)+; MS (ESI-) для C28H37N7O3 m/z 518,5 (M-H)-; ВЭЖХ чистота >95% (время удерживания, 2,51 мин);
1H ЯМР (400 МГц, d4-MeOH) δH м.д. 8,08 (с, 1H), 7,48 (шир.с, 1H), 7,39 (д, J=8,50 Гц, 1H), 7,29 (дд, J=8,40, 4,87 Гц, 1H), 6,63 (м, 1H), 6,12 (д, J=4,15 Гц, 1H), 4,40 (м, 1H), 4,09 (м, 2H), 3,15 (м, 0,5H), 3,02 (д, J=8,09 Гц, 1H), 2,92 (д, J=7,26 Гц, 1H), 2,84 (м, 0,5H), 2,65 (м, 2H), 2,43 (м, 1H), 2,29 (м, 1H), 2,20 (д, J=5,80 Гц, 3H), 2,13 (м, 1H), 1,99 (шир.с, 1H), 1,67 (м, 1H), 1,37 (д, J=3,94 Гц, 9H), 1,30 (дд, J=13,99, 4,66 Гц, 1H).
Пример 7: Синтез (1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-(((3-(2-(6-хлор-5-(трифторметил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(метил)амино)метил)циклопентан-1,2-диола (Соединение 7)
Стадия 1: Синтез 4,6-дихлор-5-(2,2-диэтоксиэтил)пиримидина
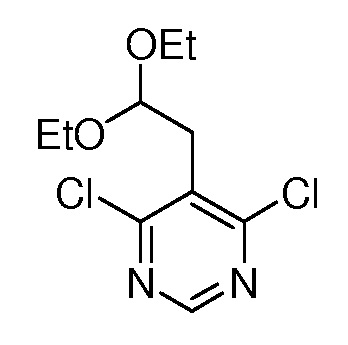
Указанное в заголовке соединение получали по методу Монтгомери, см.: Montgomery, J. A.; Hewson, K. J. Med. Chem. 10, 665 (1967).
Стадия 2: Синтез (1R,2S,3R,5R)-3-((6-хлор-5-(2,2-диэтоксиэтил)пиримидин-4-ил)амино)-5-(гидроксиметил)циклопентан-1,2-диола
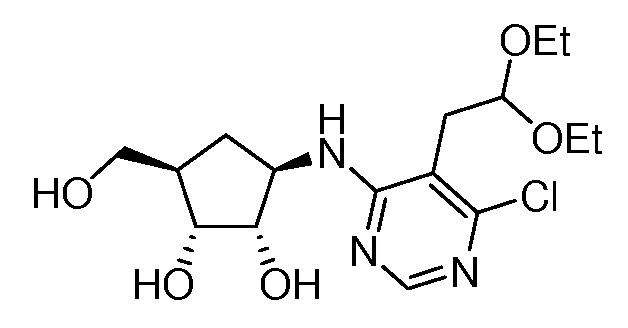
Смесь 4,6-дихлор-5-(2,2-диэтоксиэтил)пиримидина (5,35 г, 20,2 ммоль) и (1R,2S,3R,4R)-2,3-дигидрокси-4-(гидроксиметил)циклопентанаминийхлорида (9,29 г, 24,3 ммоль) брали для поглощения в этанол (236 мл), обрабатывали при помощи Et3N (11,2 мл, 80,8 ммоль) и нагревали при температуре кипения с обратным холодильником в течение 23 часов; ВЭЖХ/ЖХ-MS анализ показал, что исходные вещества израсходованы и присутствует продукт. Реакционную смесь концентрировали с получением желто-коричневой суспензии, которую использовали далее неочищенной: MS (ESI+) для C16H26ClN3O5 m/z 376,2 (M+H)+; MS (ESI-) для C16H26ClN3O5 m/z 374,2 (M-H)-; ВЭЖХ чистота >95% (время удерживания, 2,436 мин). Variation on route from J. Med. Chem. 10, 665 (1967).
Стадия 3: Синтез (1R,2S,3R,5R)-3-(4-хлор-7H-пирроло[2,3-d]пиримидин-7-ил)-5-(гидроксиметил)циклопентан-1,2-диола
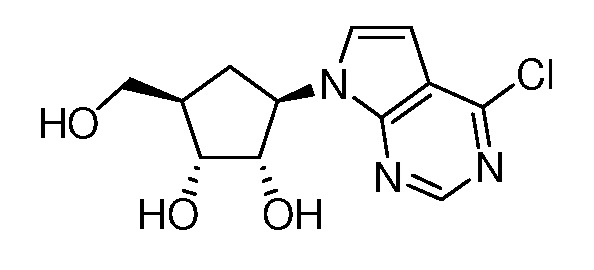
Суспензию неочищенного (1R,2S,3R,5R)-3-((6-хлор-5-(2,2-диэтоксиэтил)пиримидин-4-ил)амино)-5-(гидроксиметил)циклопентан-1,2-диола в 1,4-диоксане (160 мл) обрабатывали 1 M водным раствором HCl (30 мл, 30 ммоль) и перемешивали при комнатной температуре в течение 69,5 часов; ВЭЖХ анализ показал чистое преобразование в один продукт, ЖХ-MS анализ показал массу для желаемого продукта. Реакционную смесь найтрализовали концентрированным водным раствором NH4OH (до pH 7) и летучие вещества удаляли в вакууме с получением коричневой суспензии, которую использовали далее без дополнительной очистки: MS (ESI+) для C12H14ClN3O3 m/z 284,1 (M+H)+; MS (ESI-) для C12H14ClN3O3 m/z 282,2 (M-H)-, 328,2 (M+HCO2)-; ВЭЖХ чистота >95% (время удерживания, 1,947 мин). Variation on route from J. Med. Chem. 10, 665 (1967).
Стадия 4: Синтез ((3aR,4R,6R,6aS)-6-(4-хлор-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метанола
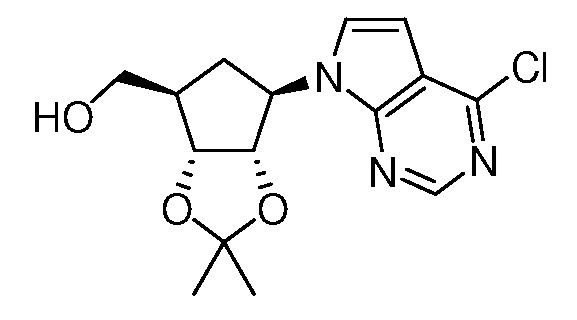
Смесь неочищенного (1R,2S,3R,5R)-3-(4-хлор-7H-пирроло[2,3-d]пиримидин-7-ил)-5-(гидроксиметил)циклопентан-1,2-диола (10 г, ~20 ммоль, чистота 54% согласно данным ЯМР) и 2,2-диметоксипропана (100 мл, 800 ммоль) обрабатывали моногидратом п-толуолсульфоновой кислоты (7,28 г, 38,3 ммоль) и желтовато-коричневую реакционную смесь интенсивно перемешивали в течение 1,25 часов, к этому времени только что образовавшиеся твердые вещества представляли собой тонкий желто-коричневый осадок. ВЭЖХ анализ показал, что исходное вещество почти полностью израсходовано. Реакционную смесь разбавляли водой (30 мл) и найтрализовали при помощи твердого NaHCO3 (4,80 г, 57,1 ммоль). Летучие вещества осторожно удаляли в вакууме и полученный коричневый водный раствор экстрагировали при помощи EtOAc (3×100 мл). Объединенные органические слои сушили (Na2SO4) и концентрировали в вакууме с получением желто-коричневого пастообразного вещества. Очистка колоночной хроматографией (4×22 см диоксид кремния; 0-66% EtOAc/гексан) давала указанное в заголовке соединение (4,38 г, 70%, одна стадия) в виде бесцветного пенистого вещества/стеклообразного вещества: MS (ESI+) для C15H18ClN3O3 m/z 324,2 (M+H)+; MS (ESI-) для C15H18ClN3O3 m/z 368,2 (M+HCO2)-; ВЭЖХ чистота >95% (время удерживания, 3,034 мин).
Стадия 5: Синтез 7-((3aS,4R,6R,6aR)-6-(азидометил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-4-хлор-7H-пирроло[2,3-d]пиримидина
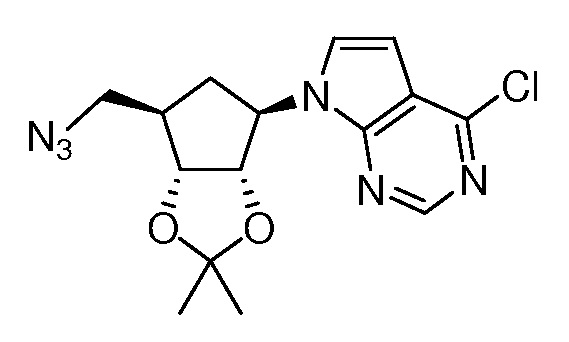
((3aR,4R,6R,6aS)-6-(4-хлор-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метанол (2,68 г, 8,28 ммоль) растворяли в ТГФ (32 мл), обрабатывали при помощи PPh3 (3,05 г, 11,6 ммоль) и реакционный сосуд охлаждали на бане лед-насыщенный солевой раствор. Добавляли по каплям через шприц диизопропилазодикарбоксилат [DIAD] (2,3 мл, 12 ммоль) и смесь перемешивали в течение 10 минут. Раствор азида дифенилфосфоновой кислоты [DPPA] (2,50 мл, 11,6 ммоль) в ТГФ (7,8 мл) добавляли по каплям через шприц с получением не совсем белой смеси, которую перемешивали в течение 21 часа, давая ледяной бане нагреться до комнатной температуры; ВЭЖХ/ЖХ-MS анализ показал, что исходное вещество полностью израсходовано и произошло образование продукта. По прошествии 22,5 часов реакционную смесь концентрировали в вакууме и очищали колоночной хроматографией (4×22 см диоксид кремния; 0-25% EtOAc/гексан) с получением указанного в заголовке соединения (2,27 г, 78%) в виде прозрачного бесцветного масла: MS (ESI+) для C15H17ClN6O2 m/z 349,2 (M+H)+; MS (ESI-) для C15H17ClN6O2 m/z 393,2 (M+HCO2)-; ВЭЖХ чистота >95% (время удерживания, 4,169 мин).
Стадия 6: Синтез 7-((3aS,4R,6R,6aR)-6-(азидометил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-N-(2,4-диметоксибензил)-7H-пирроло[2,3-d]пиримидин-4-амина
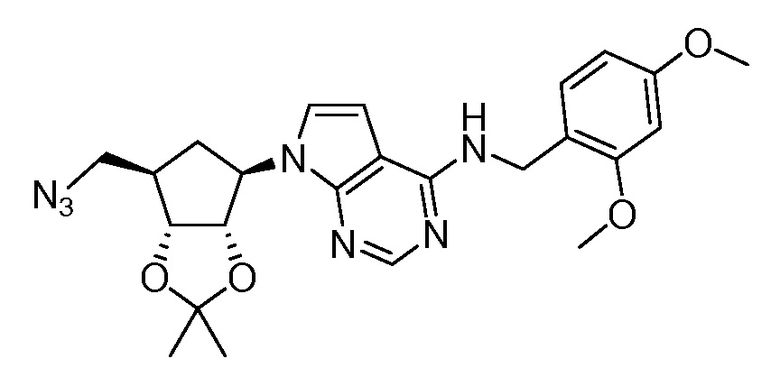
Раствор 7-((3aS,4R,6R,6aR)-6-(азидометил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-4-хлор-7H-пирроло[2,3-d]пиримидина и 2,4-диметоксибензиламина (1,2 мл, 7,8 ммоль) в 1-бутаноле (18,6 мл) обрабатывали N,N-диизопропилэтиламином (1,4 мл, 7,8 ммоль) и нагревали при 80°C в течение 22 часов; ВЭЖХ/ЖХ-MS анализ показал ~90% преобразование в желаемый продукт. Летучие вещества удаляли и желтовато-коричневое пастообразное вещество брали для поглощения в CH2Cl2 (90 мл) и промывали водой (2×30 мл) и насыщенным солевым раствором (1×45 мл). Отделенный органический слой сушили (Na2SO4) и концентрировали в вакууме с получением оранжевого масла. Очистка колоночной хроматографией (2×22 см диоксид кремния; 0-50% EtOAc/гексан) давала указанное в заголовке соединение (2,23 г, 72%) в виде бледно-желтого стеклообразного вещества/пенообразного вещества MS (ESI+) для C24H29N7O4 m/z 480,5 (M+H)+; MS (ESI-) для C24H29N7O4 m/z 524,3 (M+HCO2)-; ВЭЖХ чистота >95% (время удерживания, 3,551 мин).
Стадия 7: Синтез 7-((3aS,4R,6R,6aR)-6-(аминометил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-N-(2,4-диметоксибензил)-7H-пирроло[2,3-d]пиримидин-4-амина
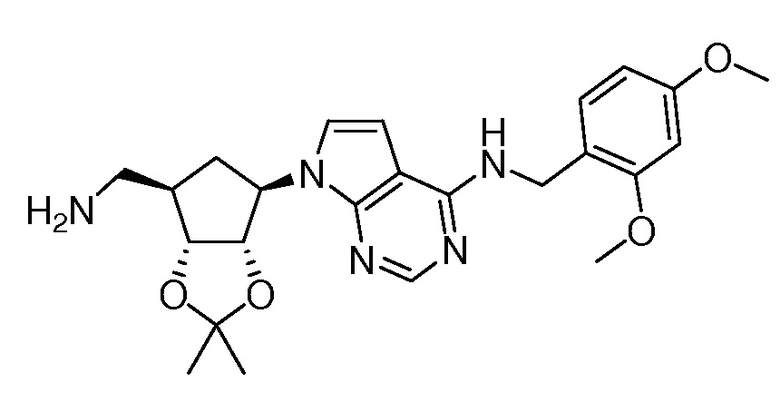
Раствор 7-((3aS,4R,6R,6aR)-6-(азидометил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-N-(2,4-диметоксибензил)-7H-пирроло[2,3-d]пиримидин-4-амина (2,23 г, 4,65 ммоль) в ТГФ (33 мл, 410 ммоль) охлаждали до 0°C и обрабатывали по каплям 1,0 M раствором триметилфосфина в ТГФ (9,3 мл, 9,3 ммоль). Охлаждающую баню удаляли и реакционной смеси давали нагреться до комнатной температуры при перемешивании в течение 1 часа; по данным ВЭЖХ не было никакого оставшегося исходного вещества. По прошествии 1,5 часов добавляли воду (4,3 мл, 240 ммоль) и реакционную смесь перемешивали в течение 1 часа 15 минут; ТСХ анализ показал один продукт. Реакционную смесь концентрировали в вакууме с получением светло-оранжевого пастообразного вещества. Остаток разбавляли при помощи CH2Cl2 (120 мл) и промывали водой (2×40 мл) и насыщенным солевым раствором (1×40 мл). Органический слой сушили (Na2SO4) и концентрировали в вакууме с получением оранжевого масла. Очистка колоночной хроматографией (2×22 см диоксид кремния; 0-5% 7н NH3 в CH3OH/CH2Cl2) давала указанное в заголовке соединение (1,97 г, 53% от 3 стадий) в виде бесцветного пенообразного вещества MS (ESI+) для C24H31N5O4 m/z 454,3 (M+H)+; ВЭЖХ чистота >95% (время удерживания, 2,541 мин).
Стадия 8: Синтез этил 3-(2,2-дихлор-3-оксоциклобутил)пропаноата
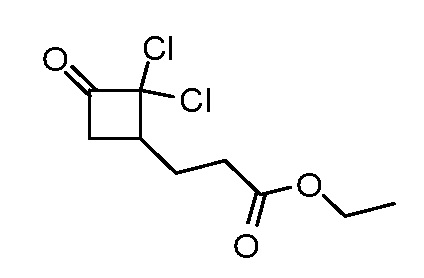
Смесь этилового эфира 4-пентеновой кислоты (7,07 г, 55,2 ммоль) и пары цинк-медь (10,2 г, 140 ммоль) в диэтиловом эфире (170 мл) и 1,2-диметоксиэтане (25 мл) обрабатывали по каплям трихлорацетилхлоридом (25 г, 140 ммоль). Смесь перемешивали при комнатной температуре в течение 3 дней. Красноватую гетерогенную реакционную смесь фильтровали через слой целита и этот слой промывали при помощи 300 мл Et2O. Фильтрат концентрировали до около половины исходного объема и органическую фазу промывали двумя 150 мл порциями H2O и одной 150 мл порцией насыщенного раствора NaHCO3. Органическую фазу сушили над MgSO4, фильтровали и концентрировали с получением коричневой жидкости. Это вещество очищали вакуумной дистилляцией (90-100°C @ 0,044 тор) с получением указанного в заголовке соединения (10,49 г, 80%) в виде светло-желтой жидкости: ГХ чистота 95,8% (время удерживания, 4,92 мин).
Стадия 9: Синтез этил 3-(3-оксоциклобутил)пропаноата
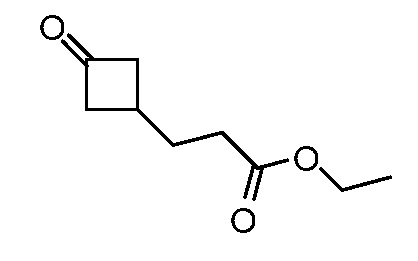
Раствор этил 3-(2,2-дихлор-3-оксоциклобутил)пропаноата (10,49 г, 43,87 ммоль) и хлорида аммония (12 г, 220 ммоль) в метаноле (310 мл, 7600 ммоль) обрабатывали небольшими порциями цинкового порошка (14 г, 220 ммоль). Реакционную смесь нагревали при температуре кипения с обратным холодильником в течение 3 часов, после этого ГХ анализ показал, что реакция завершена. Реакционную смесь охлаждали до комнатной температуры и фильтровали через Целит, промывая этот слой при помощи Et2O. Фильтрат концентрировали в вакууме с получением бледно-желтого раствора. Раствор разбавляли при помощи 200 мл Et2O и промывали при помощи 100 мл воды. Отделенный водный слой обратно экстрагировали при помощи 100 мл Et2O и объединенную органическую фазу промывали при помощи 100 мл смеси 1:1 вода/насыщенный солевой раствор, 50 мл воды и 150 мл насыщенного водного раствора NaHCO3. Органический слой сушили над MgSO4, фильтровали и концентрировали в вакууме с получением указанного в заголовке соединения (4,49 г, 60%) в виде бледно-желтого масла, которое было достаточно чистым для использования на следующей стадии: ГХ чистота >95% (время удерживания, 4,24 мин).
Стадия 10: Синтез 3-(3-оксоциклобутил)пропановой кислоты
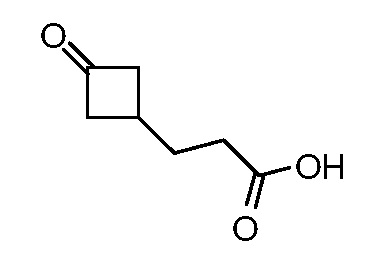
Раствор этил 3-(3-оксоциклобутил)пропаноата (200 мг, 1,18 ммоль) в метаноле (4 мл) обрабатывали водой (0,75 мл) и 2н раствором гидроксида натрия (0,75 мл, 1,41 ммоль) и раствор нагревали при 55°C до тех пор, пока исходное вещество не было израсходовано по данным ТСХ (25% EA/гептан). Через 1 час было обнаружено, что исходное вещество израсходовано. Реакционную смесь охлаждали до комнатной температуры и концентрировали для удаления MeOH. Водную фазу разбавляли при помощи 2 мл H2O и подкисляли до pH~2 1н раствором HCl. Раствор насыщали NaCl и экстрагировали тремя 10 мл порциями этилацетата. Органическую фазу сушили над Na2SO4, фильтровали и концентрировали с получением указанного в заголовке соединения (157 мг, 94 %) в виде светло-оранжевого вязкого масла, которое использовали как таковое на следующей стадии: ГХ чистота 63,2% (время удерживания, 4,27 мин).
Стадия 11: Синтез N-(2-амино-4-хлор-5-(трифторметил)фенил)-3-(3-оксоциклобутил)пропанамида
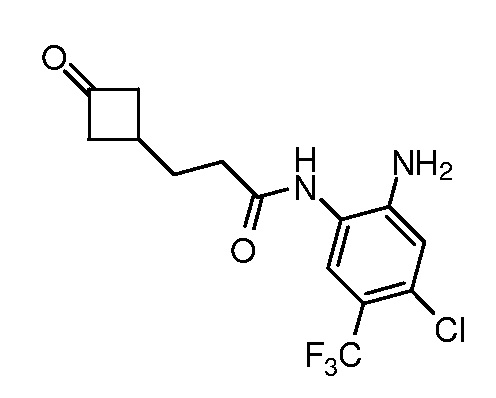
Раствор 3-(3-оксоциклобутил)пропановой кислоты (157 мг, 0,696 ммоль) и 4-хлор-5-(трифторметил)бензол-1,2-диамина (146 мг, 0,696 ммоль) в N,N-диметилформамиде (2,5 мл) охлаждали при 0°C. Раствор обрабатывали по каплям N,N-диизопропилэтиламином (0,364 мл, 2,09 ммоль), с последующим добавлением гексафторфосфата N,N,N',N'-тетраметил-O-(7-азабензотриазол-1-ил)урония (291 мг, 0,765 ммоль) одной порцией. Раствор оставляли для перемешивания и медленного нагревания до комнатной температуры. Через 40 часов реакционную смесь концентрировали частично в условиях высокого вакуума. Оставшуюся коричневую жидкость брали для поглощения в 25 мл EA и 15 мл 1/1 смеси насыщенный NaHCO3/H2O и экстрагировали. Водную фазу промывали двумя 15 мл порциями этилацетата и объединенную органическую фазу промывали 30 мл порциями H2O и насыщенного солевого раствора. Органическую фазу сушили над MgSO4, фильтровали и концентрировали с получением желтовато-коричневого вязкого маслянистого/стеклообразного вещества. Неочищенное вещество очищали флэш-хроматографией (40 г силикагеля, 50-80% EA/гептан) с получением указанного в заголовке соединения (72 мг, 31%) в виде слегка желтовато-коричневого стеклообразного вещества/жесткой пены: MS (ESI+) для C14H14ClF3N2O2 m/z 335,2 (M+H)+; MS (ESI-) для C14H14ClF3N2O2 m/z 333,3 (M-H)-; ВЭЖХ чистота 78,2% (время удерживания, 3,56 мин).
Стадия 12: Синтез 3-(2-(6-хлор-5-(трифторметил)-1H-бензо[d]имидазол-2-ил)этил)циклобутанона
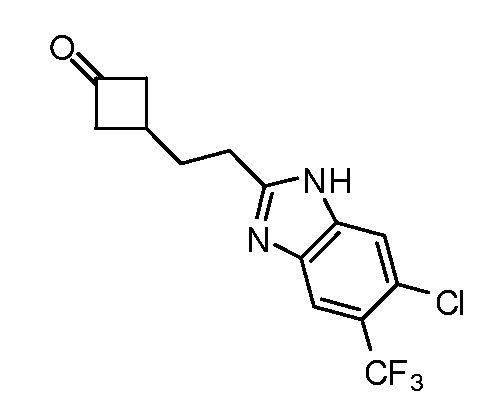
N-(2-амино-4-хлор-5-(трифторметил)фенил)-3-(3-оксоциклобутил)пропанамид (72 мг, ммоль) брали для поглощения в уксусную кислоту (3,2 мл) и раствор нагревали при 65°C в течение 26 часов, затем ВЭЖХ анализ показал, что исходное вещество было израсходовано и образовался новый продукт. Реакционную смесь охлаждали и растворитель удаляли в условиях высокого вакуума. Светло-коричневый остаток брали для поглощения в 20 мл этилацетата и органическую фазу промывали 10 мл порциями насыщенного раствора NaHCO3 и H2O. Органическую фазу сушили над Na2SO4, фильтровали и концентрировали с получением светло-коричневого стеклообразного вещества. Неочищенное вещество очищали методом препаративной ТСХ (20см × 20см × 1,0мм пластина для препаративной ТСХ, 3% MeOH/EA) с получением указанного в заголовке соединения (45 мг, 66%) в виде желто-коричневого стеклообразного вещества: MS (ESI+) для C14H12ClF3N2O m/z 317,2 (M+H)+; MS (ESI-) для C14H12ClF3N2O m/z 315,2 (M-H)-; ВЭЖХ чистота 84,1% (время удерживания, 2,98 мин).
Стадия 13: Синтез 7-((3aS,4R,6R,6aR)-6-(((3-(2-(6-хлор-5-(трифторметил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)амино)метил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-N-(2,4-диметоксибензил)-7H-пирроло[2,3-d]пиримидин-4-амина
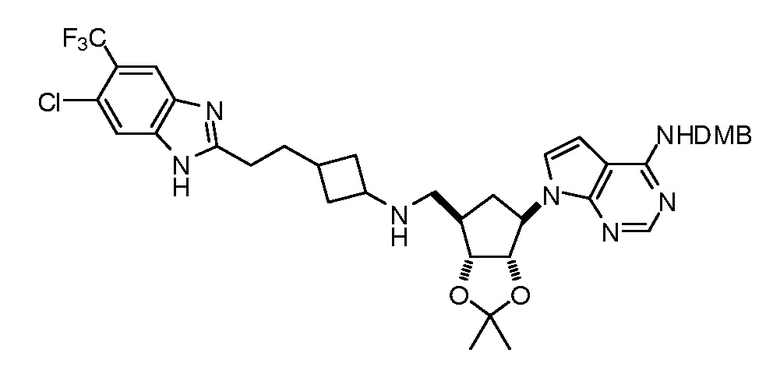
Раствор 7-((3aS,4R,6R,6aR)-6-(аминометил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-N-(2,4-диметоксибензил)-7H-пирроло[2,3-d]пиримидин-4-амина (80 мг, 0,18 ммоль) и 3-(2-(6-хлор-5-(трифторметил)-1H-бензо[d]имидазол-2-ил)этил)циклобутанона (45 мг, 0,14 ммоль) в 1,2-дихлорэтане (2,4 мл) обрабатывали по каплям уксусной кислотой (10 мкл, 0,18 ммоль). Раствор обрабатывали триацетоксиборогидридом натрия (53 мг, 0,25 ммоль) одной порцией и оставляли для перемешивания при комнатной температуре до тех пор, пока реакция не завершалась, по данным ВЭЖХ. Через 4 часа реакционную смесь разбавляли при помощи 10 мл CH2Cl2 и промывали при помощи 10 мл насыщенного раствора NaHCO3. Водную фазу промывали при помощи 10 мл CH2Cl2 и объединенную органическую фазу сушили над Na2SO4. Раствор фильтровали и концентрировали с получением светло-желтовато-коричневого стеклообразного вещества/жесткой пены. Неочищенное вещество очищали флэш-хроматографией (25 г силикагеля; 5% 7н NH3 в CH3OH/CHCl3) с получением указанного в заголовке соединения (76 мг, 71%) в виде бесцветного стеклообразного вещества/жесткой пены: MS (ESI+) для C38H43ClF3N7O4 m/z 754,3 (M+H)+; MS (ESI-) для C38H43ClF3N7O4 m/z 752,3 (M-H)-; ВЭЖХ чистота 90,5% (время удерживания, 3,24 мин).
Стадия 14: Синтез 7-((3aS,4R,6R,6aR)-6-(((3-(2-(6-хлор-5-(трифторметил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(метил)амино)метил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-N-(2,4-диметоксибензил)-7H-пирроло[2,3-d]пиримидин-4-амина
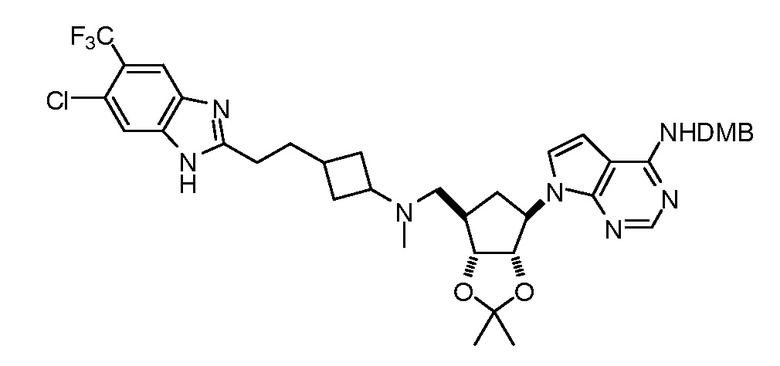
Раствор 7-((3aS,4R,6R,6aR)-6-(((3-(2-(6-хлор-5-(трифторметил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)амино)метил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-N-(2,4-диметоксибензил)-7H-пирроло[2,3-d]пиримидин-4-амина (76 мг, 0,10 ммоль) в метаноле (2,5 мл) обрабатывали цианоборогидридом натрия (84 мг, 1,3 ммоль). pH раствора доводили до ~6 путем добавления по каплям 10% (об/об) раствора ледяной уксусной кислоты в метаноле. Смесь обрабатывали по каплям 37% водным раствором формальдегида (0,12 мл, 1,7 ммоль) и смесь перемешивали при комнатной температуре до тех пор, пока реакция не завершалась, по данным ЖХ-MS. Через 2 часа реакция была завершена, и реакционную смесь концентрировали для удаления метанола. Оставшийся водный раствор разбавляли при помощи 7 мл NaHCO3 и водную фазу экстрагировали тремя 10 мл порциями CH2Cl2. Органическую фазу сушили над Na2SO4, фильтровали и концентрировали с получением бесцветной жесткой пены/стеклообразного вещества. Неочищенное вещество очищали флэш-хроматографией (20 г силикагеля; 4% 7н NH3 в CH3OH/CH2Cl2) с получением указанного в заголовке соединения (62 мг, 80%) в виде бесцветного стеклообразного вещества: MS (ESI+) для C39H45ClF3N7O4 m/z 768,0 (M+H)+; MS (ESI-) для C39H45ClF3N7O4 m/z 766,3 (M-H)-; ВЭЖХ чистота 92,1% (время удерживания, 3,29 мин).
Стадия 15: Синтез соединения 7
7-((3aS,4R,6R,6aR)-6-(((3-(2-(6-хлор-5-(трифторметил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(метил)амино)метил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-N-(2,4-диметоксибензил)-7H-пирроло[2,3-d]пиримидин-4-амин (60 мг, 0,078 ммоль) растворяли в смеси трифторуксусной кислоты (3,6 мл) и воды (0,4 мл), которая была предварительно охлаждена при 0°C на ледяной бане. Раствор перемешивали при 0°C в течение 30 минут и затем нагревали до комнатной температуры. После выдерживания в течение 3 часов при комнатной температуре ВЭЖХ анализ показал, что реакция заверщена. Образовавшуюся ярко-розовую реакционную смесь концентрировали. Остаток брали для поглощения в 10 мл MeOH и концентрировали. Эту процедуру повторяли два раза и остаток помещали в условия высокого вакуума на 1 час. Это вещество брали для поглощения в 7 мл MeOH и обрабатывали при помощи 120 мг K2CO3 и десяти капель воды. Смесь оставляли для перемешивания в течение 1 часа. Смесь фильтровали через тонкую фритту, твердые вещества промывали при помощи 10 мл MeOH и фильтрат концентрировали с получением почти бесцветного твердого вещества. Неочищенное вещество очищали флэш-хроматографией (30 г силикагеля; 10-15% 7н NH3 в CH3OH/CH2Cl2) с получением соединения 7 (31 мг, 69%) в виде бесцветного стеклообразного вещества/жесткой пены: MS (ESI+) для C27H31ClF3N7O2 m/z 578,3 (M+H)+; MS (ESI-) для C27H31ClF3N7O2 m/z 576,4 (M-H)-; ВЭЖХ чистота >95% (время удерживания, 2,57 мин);
1H ЯМР (400 МГц, d4-MeOD) δH 8,06 (с, 1H), 7,89 (д, J=2,07 Гц, 1H), 7,69 (д, J=2,28 Гц, 1H), 7,21 (дд, J=3,42, 1,76 Гц, 1H), 6,59 (д, J=3,52 Гц, 1H), 4,33 (т, J=6,84 Гц, 1H), 3,89 (кв., J=5,25 Гц, 1H), 3,03 (м, 0,5H), 2,89 (м, 2H), 2,70 (м, 0,5H), 2,49 (м, 1H), 2,40 (м, 2H), 2,27 (шир.с, 2H), 2,16 (д, J=7,26 Гц, 4H), 2,06 (м, 2H), 1,91 (м, 2H), 1,62 (м, 1H), 1,52 (м, 1H).
Пример 8: Синтез соединений 8-140
Соединения 8-140 были синтезированы способами, аналогичными тем, которые описаны для примеров 1-7, или в соответствии со схемами реакций, представленными на общих схемах. Подробные описания, как некоторые из них были получены, представлены ниже. MS и ЯМР данные для соединений 2-140 представлены в Таблице 1 или в Примерах, описанных в настоящей заявке.
Соединение 8: 1-(3-((((2R,3S,4R,5R)-5-(6-амино-9H-пурин-9-ил)-3,4-дигидрокситетрагидрофуран-2-ил)метил)(метил)амино)циклобутил)-3-(4-(трет-бутил)фенил)мочевина
Бензил (3-оксоциклобутил)карбамат
К раствору 3-оксоциклобутанкарбоновой кислоты (1,0 г, 8,77 ммоль) и DIEA (1,92 г, 14,92 ммоль) в толуоле (8 мл) добавляли DPPA (2,89 г, 10,52 ммоль) при комнатной температуре. Смесь нагревали до 60°C в атмосфере аргона в течение 3 часов, затем добавляли бензиловый спирт (1,14 г, 10,52 ммоль). Смесь перемешивали при 60°C в течение ночи. Реакционную смесь концентрировали, остаток очищали при помощи SGC (PE:EA=8:1) с получением желаемого соединения (240 мг, выход 50%).
1H ЯМР (500 МГц, CDCl3): δH 7,38-7,33 (м, 5H), 5,12 (д, J=7,5 Гц, 2H), 4,34-4,33 (шир.с, 1H), 3,44-3,39 (м, 2H), 3,10-3,07 (шир.с, 2H) м.д.; ESI-MS (m/z): 220,2 [M+1]+.
Бензил (3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(метил)амино)циклобутил)карбамат
К раствору 9-((3aR,4R,6R,6aR)-2,2-диметил-6-((метиламино)метил)тетрагидрофуро[3,4-d][1,3]диоксол-4-ил)-9H-пурин-6-амина (190 мг, 0,59 ммоль) и бензил (3-оксоциклобутил)карбамата (240 мг, 1,37 ммоль) в MeOH (5 мл) добавляли Ti[OCH(CH3)2]4 (216 мг, 0,59 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа. Затем добавляли NaCNBH3 (95 мг, 1,52 ммоль), реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь фильтровали и упаривали, остаток очищали методом препаративной ТСХ (DCM:MeOH=20:1) с получением желаемого соединения (90 мг, выход 29%).
1H ЯМР (500 МГц, MeOD): δH 8,28 (с, 1H), 8,21 (с, 1H), 7,33-7,28 (м, 5H), 6,19 (д, J=2,0 Гц, 1H), 5,52-5,51 (м, 1H), 5,03 (с, 1H), 5,00-4,98 (м, 1H), 4,34 (т, J=3,5 Гц, 1H), 3,70 (м, 1H), 2,58-2,47 (м, 4H), 2,38-2,26 (м, 2H), 2,09 (с, 3H), 1,69-1,67 (м, 1H), 1,58 (с, 3H), 1,37 (с, 3H) м.д.; ESI-MS (m/z): 524,3 [M+1]+.
N1-(((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)-N1-метилциклобутан-1,3-диамин
В раствор бензил (3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(метил)амино)циклобутил)карбамата (190 мг, 0,17 ммоль) и Pd(OH)2 (14 мг, 0,1 ммоль) в MeOH (5 мл) загружали H2. Реакционную смесь перемешивали при 35°C в течение 5 часов. Реакционную смесь фильтровали через Целит и концентрировали досуха. Остаток очищали методом препаративной ТСХ (DCM:MeOH=10:1) с получением желаемого соединения (28 мг, выход 42%).
1H ЯМР (500 МГц, MeOD): δH 8,29 (с, 1H), 8,21 (с, 1H), 6,19 (д, J=2,0 Гц, 1H), 5,51 (дд, J=6,5 и 2,0 Гц, 1H), 5,00 (дд, J=6,0 и 3,5 Гц, 1H), 4,34 (д, J=8,5 Гц, 1H), 3,14-3,11 (м, 1H), 2,60-2,57 (м, 1H), 2,52-2,48 (м, 2H), 2,34-2,31 (м, 2H), 2,10 (с, 3H), 1,66 (кв., J=10,0 Гц, 1H), 1,58 (с, 3H), 1,48 (кв., J=10,0 Гц, 1H), 1,37 (с, 3H) м.д.; ESI-MS (m/z): 390,2 [M+1]+.
1-(3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(метил)амино)циклобутил)-3-(4-(трет-бутил)фенил)мочевина
Раствор N1-(((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)-N1-метилциклобутан-1,3-диамина (28 мг, 0,072 ммоль) и TEA (22 мг, 0,22 ммоль) в ТГФ (3 мл) добавляли по каплям к 1-трет-бутил-4-изоцианатобензолу (18 мг, 0,11 ммоль) в DCM (0,5 мл). Реакционную смесь перемешивали в течение 1 часа при комнатной температуре. Реакционную смесь концентрировали и очищали методом препаративной ТСХ (два раза, DCM:MeOH:NH4OH=300:30:8, об/об) с получением желаемого соединения (28 мг, выход: 88%) в виде бледно-белого твердого вещества.
1H ЯМР (500 МГц, MeOD): δH 8,29 (с, 1H), 8,24 (с, 1H), 7,28-7,22 (м, 4H), 6,25 (д, J=2,5 Гц, 1H), 5,52-5,50 (м, 1H), 5,07-5,05 (м, 1H), 4,46-4,44 (м, 1H), 3,88-3,85 (м, 1H), 2,97 (шир.с, 1H), 2,80-2,78 (м, 2H), 2,48-2,42 (м, 2H), 2,30 (с, 3H), 1,84-1,82 (м, 1H), 1,60 (с, 3H), 1,58-1,56 (м, 1H), 1,39 (с, 3H), 1,28 (с, 9H) м.д.; ESI-MS (m/z): 565,3 [M+1]+.
1-(3-((((2R,3S,4R,5R)-5-(6-амино-9H-пурин-9-ил)-3,4-дигидрокситетрагидрофуран-2-ил)метил)(метил)амино)циклобутил)-3-(4-(трет-бутил)фенил)мочевина
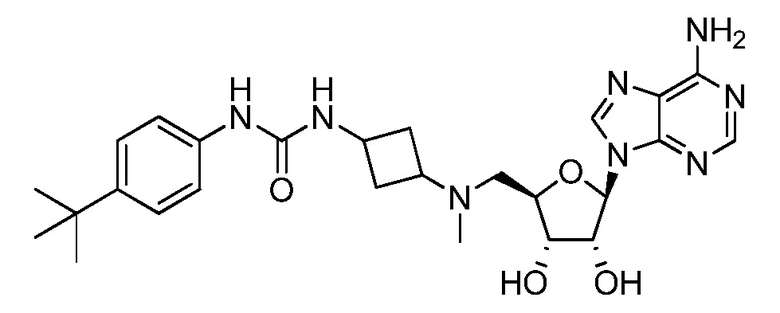
Раствор 1-(3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(метил)амино)циклобутил)-3-(4-(трет-бутил)фенил)мочевины (125 мг, 0,23 ммоль) в TFA (0,90 мл) и 0,10 мл воды перемешивали в течение 1 часа при комнатной температуре. Реакционную смесь концентрировали досуха, растворяли в MeOH (5 мл) и добавляли по каплям K2CO3 (60 мг) в 0,5 мл воды. Реакционную смесь перемешивали при комнатной температуре в течение 0,5 часа и концентрировали с получением остатка, который очищали методом препаративной ТСХ (DCM:MeOH:NH4OH=300:30:8, об/об) с получением желаемого соединения (75 мг, выход: 65%) в виде бледно-белого твердого вещества.
1H ЯМР (500 МГц, MeOD): δH 8,27 (с, 1H), 8,21 (с, 1H), 7,28-7,20 (м, 4H), 6,00-5,99 (м, 1H), 4,77-4,75 (м, 1H), 4,28-4,23 (м, 2H), 3,92-3,88 (м, 1H), 2,92 (шир.с, 1H), 2,83-2,81 (м, 2H), 2,59-2,56 (м, 2H), 2,32 (с, 3H), 1,74-1,64 (м, 2H), 1,27 (с, 9H) м.д.; ESI-MS (m/z): 525,3 [M+1]+.
Соединения 9 и 12
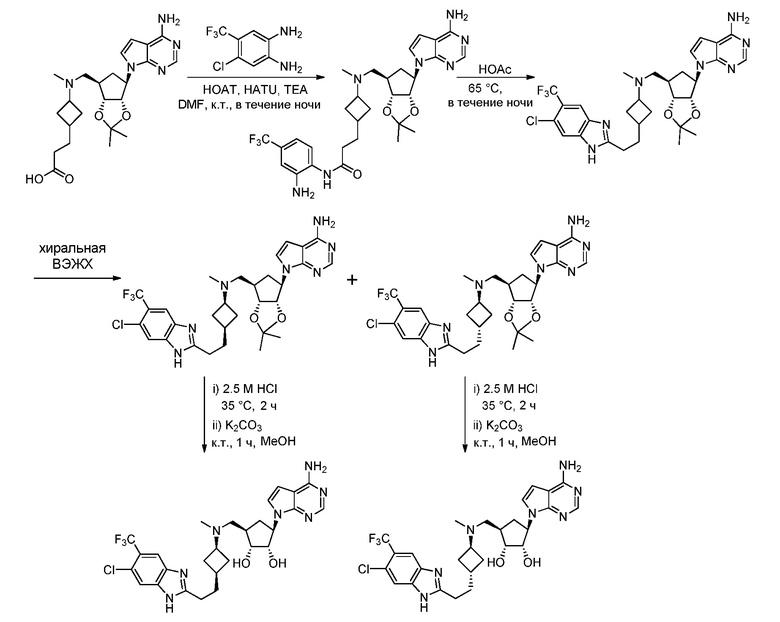
Соединение 9: (1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-((((1r,3S)-3-(2-(6-хлор-5-(трифторметил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(метил)амино)метил)циклопентан-1,2-диол:
1H ЯМР (500 МГц, MeOD): δH 8,07 (с, 1H), 7,90 (с, 1H), 7,69 (с, 1H), 7,22 (д, J=3,5 Гц, 1H), 6,61 (д, J=3,5 Гц, 1H), 4,34 (т, J=6,5 Гц, 1H), 3,89 (т, J=5,0 Гц, 1H), 2,88 (т, J=7,0 Гц, 2H), 2,74-2,68 (м, 1H), 2,55-2,49 (м, 1H), 2,46-2,35 (м, 2H), 2,32-2,22 (м, 3H), 2,17 (с, 3H), 2,00-1,90 (м, 3H), 1,68-1,60 (м, 1H), 1,58-1,48 (м, 2H) м.д.; LC-MS (m/z): 578,3 [M+1]+.
Соединение 12: (1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-((((1s,3R)-3-(2-(6-хлор-5-(трифторметил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(метил)амино)метил)циклопентан-1,2-диол:
1H ЯМР (500 МГц, MeOD): δH 8,07 (с, 1H), 7,91 (с, 1H), 7,70 (с, 1H), 7,22 (д, J=3,5 Гц, 1H), 6,61 (д, J=4,0 Гц, 1H), 4,34 (дд, J=7,0 и 6,0 Гц, 1H), 3,90 (т, J=5,0 Гц, 1H), 3,05-3,00 (м, 1H), 2,92 (т, J=7,5 Гц, 2H), 2,55-2,49 (м, 1H), 2,47-2,35 (м, 2H), 2,32-2,22 (м, 1H), 2,20-2,02 (м, 8H), 1,93-1,86 (м, 2H), 1,70-1,60 (м, 1H) м.д.; LC-MS (m/z): 578,3 [M+1]+.
Соединения 10 и 11
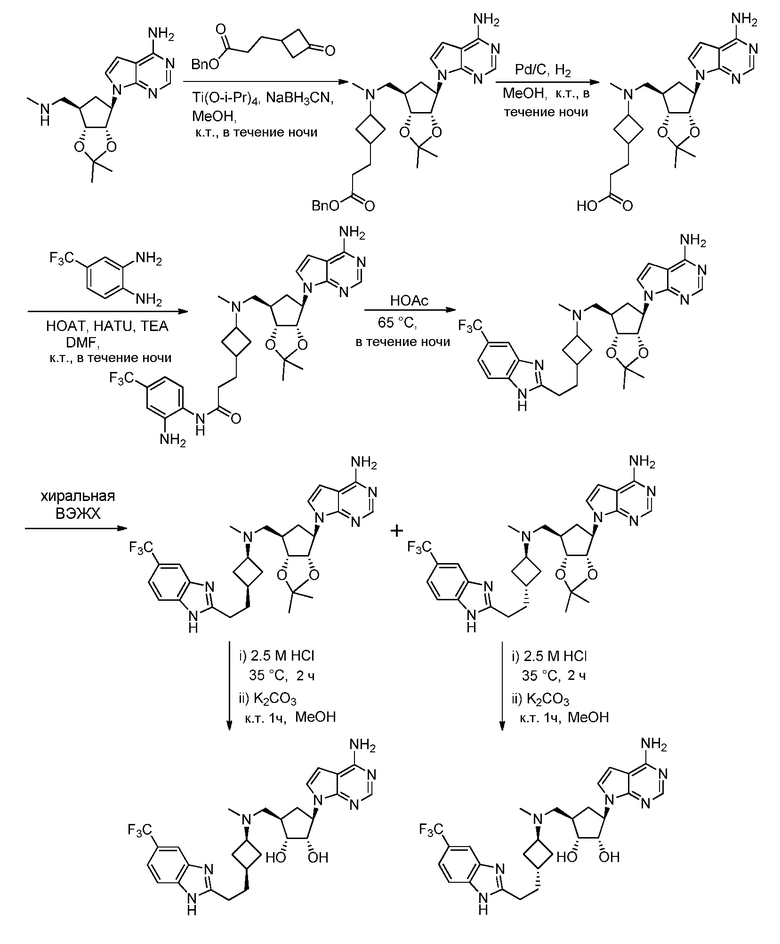
Соединение 10: (1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-((метил((1r,3S)-3-(2-(5-(трифторметил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)амино)метил)циклопентан-1,2-диол:
1H ЯМР (500 МГц, MeOD): δH 8,06 (с, 1H), 7,79 (с, 1H), 7,62 (д, J=9,0 Гц, 1H), 7,47 (д, J=8,5 Гц, 1H), 7,20 (д, J=3,5 Гц, 1H), 6,59 (д, J=3,0 Гц, 1H), 4,33-4,30 (м, 1H), 3,88-3,86 (м, 1H), 3,32-3,31 (м, 1H), 2,89-2,86 (м, 2H), 2,67-2,66 (м, 1H), 2,48-2,26 (м, 6H), 2,14 (с, 3H), 1,95-1,93 (м, 3H), 1,62-1,48 (м, 3H) м.д.; LC-MS (m/z): 544,3 [M+1]+.
Соединение 11: (1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-((метил((1s,3R)-3-(2-(5-(трифторметил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)амино)метил)циклопентан-1,2-диол:
1H ЯМР (500 МГц, MeOD): δH 8,06 (с, 1H), 7,79 (с, 1H), 7,62 (д, J=8,5 Гц, 1H), 7,47 (д, J=8,5 Гц, 1H), 7,20 (д, J=3,0 Гц, 1H), 6,59 (д, J=3,5 Гц, 1H), 4,33-4,31 (м, 1H), 3,90-3,87 (м, 1H), 3,01-3,0 (м, 1H), 2,92-2,89 (м, 2H), 2,48-2,03 (м, 13H), 1,93-1,89 (м, 2H), 1,63-1,61 (м, 1H) м.д.; LC-MS (m/z): 544,3 [M+1]+.
Соединение 13: (1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-((метил(3-(2-(5-(трифторметил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)амино)метил)циклопентан-1,2-диол
N-(2-амино-4-(трифторметил)фенил)-3-(3-((((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)(метил)амино)циклобутил)пропанамид
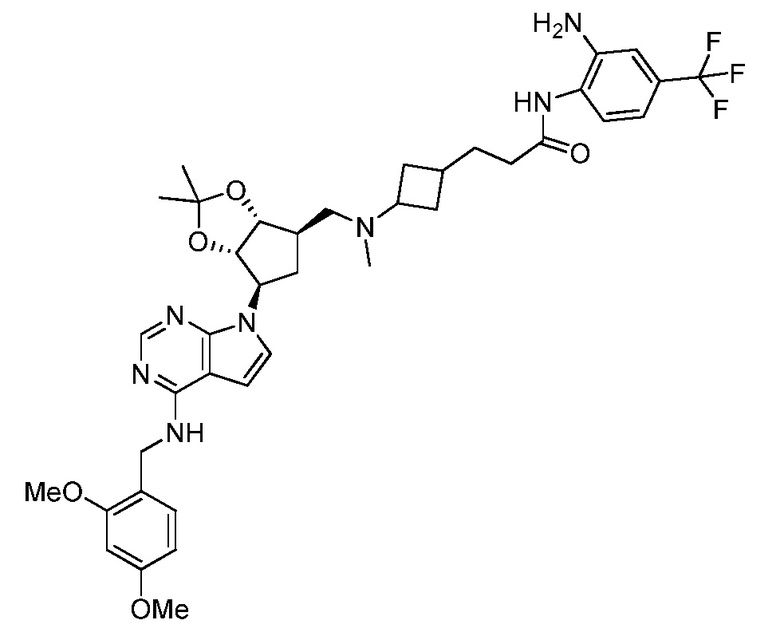
Гексафторфосфат N,N,N',N'-тетраметил-0-(7-азабензотриазол-1-ил)урония (0,44 г, 1,2 ммоль) добавляли к раствору 3-(3-((((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)(метил)амино)циклобутил)пропановой кислоты (460 мг, 0,77 ммоль), N,N-диизопропилэтиламина (0,44 мл, 2,6 ммоль) в N,N-диметилформамиде (5 мл). Реакционную смесь перемешивали в течение ночи при комнатной температуре, частично концентрировали, затем добавляли раствор NaHCO3 (насыщенный). Водный слой экстрагировали (3×) при помощи EtOAc и объединенные органические слои сушили при помощи MgSO4, фильтровали, концентрировали и очищали флэш-хроматографией (DCM/7н NH3 в MeOH 95:5) с получением желаемого соединения (0,34 г) в виде твердого вещества.
N-(2,4-диметоксибензил)-7-((3aS,4R,6R,6aR)-2,2-диметил-6-((метил(3-(2-(5-(трифторметил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)амино)метил)тетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-7H-пирроло[2,3-d]пиримидин-4-амин
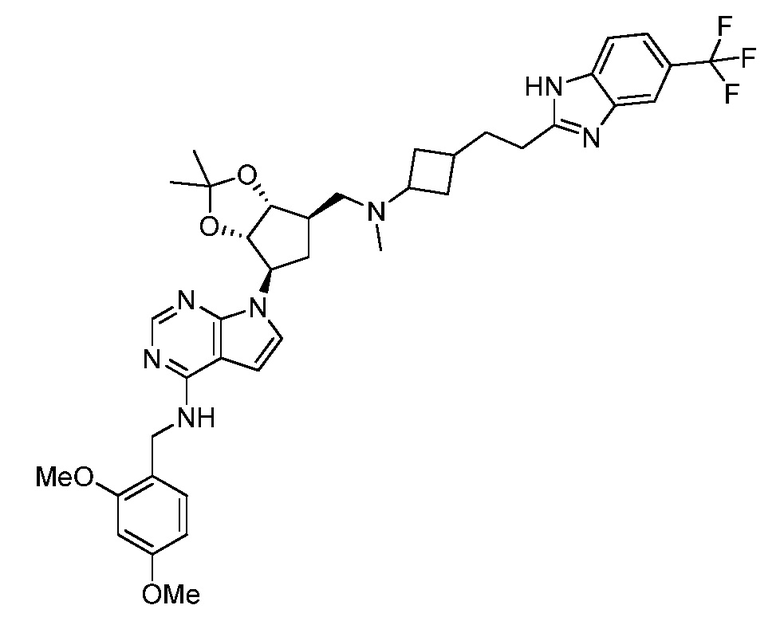
N-(2-амино-4-(трифторметил)фенил)-3-(3-((((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)(метил)амино)циклобутил)пропанамид (0,38 г, 0,50 ммоль) и уксусную кислоту (5 мл) перемешивали в течение ночи при 65°C. Летучие вещества удаляли в вакууме и оставшуюся воду удаляли путем азеотропной перегонки с этанолом, затем в условиях высокого вакуума в течение 1 часа. Полученный остаток распределяли между раствором NaHCO3 (насыщенный) и DCM. Водный слой экстрагировали (3×) и объединенные органические слои сушили при помощи MgSO4, фильтровали, концентрировали и затем очищали флэш-хроматографией (DCM/7н NH3 в MeOH 93:7) с получением не совсем белого пенистого вещества.
(1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-((метил(3-(2-(5-(трифторметил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)амино)метил)циклопентан-1,2-диол
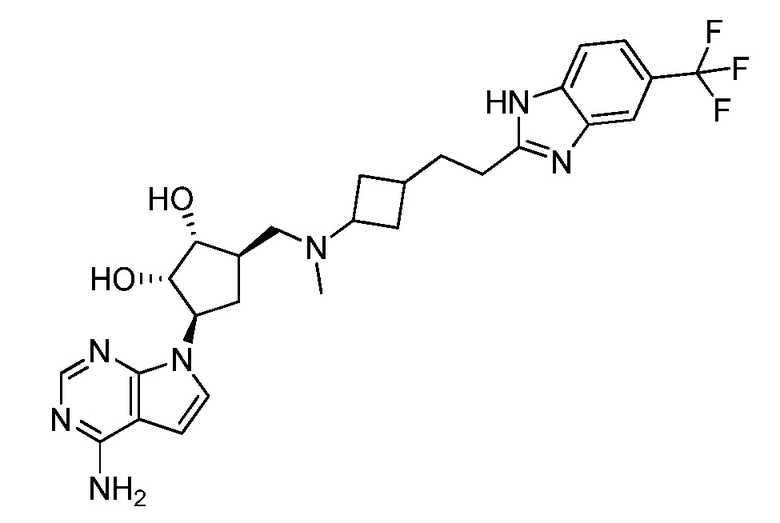
Трифторуксусную кислоту (5 мл) добавляли к смеси воды (0,5 мл) и N-(2,4-диметоксибензил)-7-((3aS,4R,6R,6aR)-2,2-диметил-6-((метил(3-(2-(5-(трифторметил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)амино)метил)тетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-7H-пирроло[2,3-d]пиримидин-4-амина (0,31 г, 0,42 ммоль) при комнатной температуре. Реакцию осуществляли в течение ночи, затем ее гасили триэтилсиланом (0,13 мл, 0,84 ммоль). Летучие вещества удаляли в вакууме и полученный остаток распределяли между насыщенным раствором NaHCO3 и DCM/MeOH (10:1). Водный слой экстрагировали (3×) снова при помощи DCM/MeOH (10:1) и объединенные органические слои сушили над MgSO4, фильтровали и концентрировали. Остаток очищали флэш-хроматографией (DCM/7н NH3 в MeOH 87:13) с получением желаемого соединения (0,12 г) в виде не совсем белого пенистого/смолистого вещества. MS (ESI+) для C27H32F3N7O2 m/z 544,5 [M+H]+; MS (ESI-) для C27H32F3N7O2 m/z 542,3 [M-H]-; ВЭЖХ чистота >85% (время удерживания, 2,418 минут.).
1H ЯМР (400 МГц, d4-MeOH) δH 8,078 (с, 1H), 7,812 (с, 1H), 7,654-7,634 (м, 1H), 7,507-7,487 (м, 1H), 7,229-7,214 (м, 1H), 6,617-6,608 (д, J=3,6 Гц, 1H), 4,361-4,322 (м, 1H), 3,927-3,887 (м, 1H), 3,062-3,024 (м, 0,5H (метин транс изомера)), 2,944-2,873 (м, 2H), 2,758-2,554 (м, 0,5H (метин цис изомера)), 2,554-2,507 (м, 1H), 2,447-2,351 (м, 2H), 2,291-2,263 (м, 2H), 2,194-2,054 (м, 6H), 1,960-1,887 (м, 3H), 1,686-1,480 (м, 2H).
Время удерживания: 2,418 ВЭЖХ Условия: колонка Agilent Zorbax Exlipse XDB-C18, 4,6×50 мм (1,8 мкм наполнителя), Растворитель A- Вода (0,1% TFA), Растворитель B- Ацетонитрил (0,07% TFA), 6 мин. градиент от 5 до 95% B; 1 мин. удерживание; затем повторный цикл.
Соединение 14: (1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-(((3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(метил)амино)метил)циклопентан-1,2-диол
N-(2-амино-4-(трет-бутил)фенил)-3-(3-((((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)(метил)амино)циклобутил)пропанамид
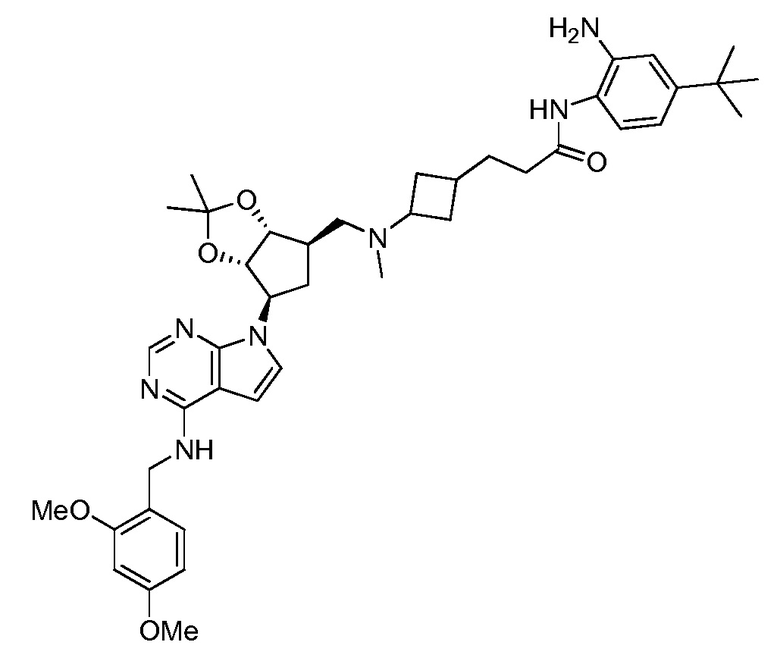
Гексафторфосфат N,N,N',N'-тетраметил-0-(7-азабензотриазол-1-ил}урония (0,44 г, 1,2 ммоль) добавляли к раствору 3-(3-((((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)(метил)амино)циклобутил)пропановой кислоты (460 мг, 0,77 ммоль) и N,N-Диизопропилэтиламина (0,44 мл, 2,6 ммоль) и 4-трет-бутилбензол-1,2-диамина (0,15 г, 0,93 ммоль) в N,N-диметилформамиде (5 мл, 60 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре, частично концентрировали примерно до 2 мл и затем добавляли NaHCO3 (насыщенный). Смесь экстрагировали при помощи EtOAc (3×) и объединенные органические слои сушили при помощи MgSO4 и концентрировали. Очищали флэш-хроматографией (DCM/7н NH3 в MeOH 95:5) с получением твердого вещества (0,24 г).
7-((3aS,4R,6R,6aR)-6-(((3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(метил)амино)метил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-N-(2,4-диметоксибензил)-7H-пирроло[2,3-d]пиримидин-4-амин
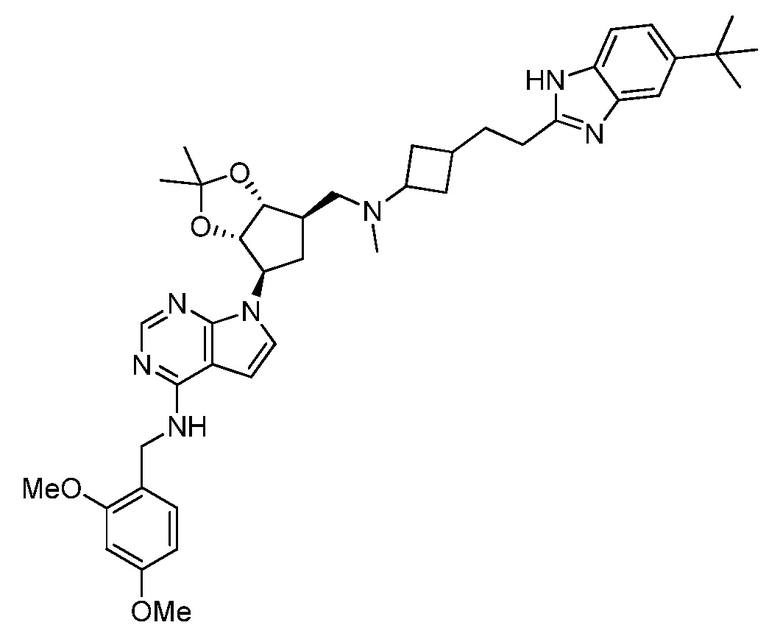
Раствор N-(2-амино-4-(трет-бутил)фенил)-3-(3-((((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)(метил)амино)циклобутил)пропанамида (0,24 г, 0,32 ммоль) в уксусной кислоте (5 мл, 90 ммоль) перемешивали в течение ночи при 60°C. Летучие вещества удаляли в вакууме и оставшийся остаток распределяли между Na2CO3 (2н) и DCM. Водный слой экстрагировали (3×) при помощи DCM и объединенные органические слои сушили при помощи MgSO4, фильтровали и концентрировали. Остаток очищали флэш-хроматографией (DCM/7н NH3 в MeOH 94:6) с получением желаемого соединения (0,20 г) в виде твердого вещества.
(1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-(((3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(метил)амино)метил)циклопентан-1,2-диол
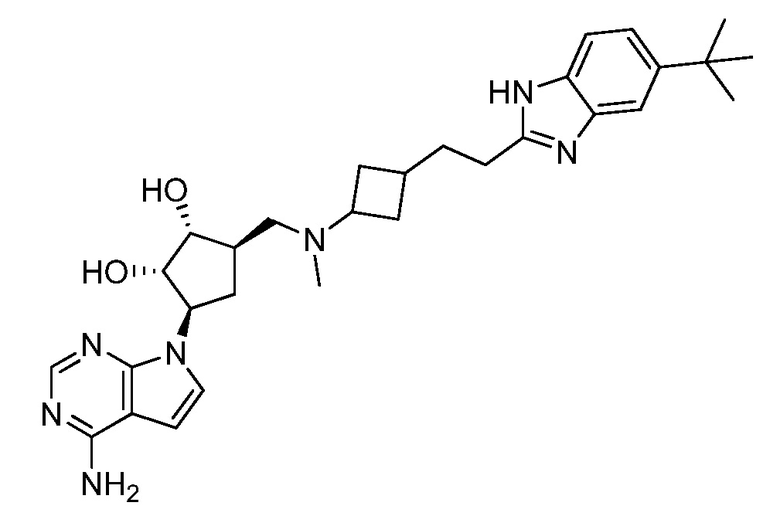
Трифторуксусную кислоту (5 мл) добавляли к смеси воды (0,5 мл) и 7-((3aS,4R,6R,6aR)-6-(((3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(метил)амино)метил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-N-(2,4-диметоксибензил)-7H-пирроло[2,3-d]пиримидин-4-амина (0,20 г, 0,28 ммоль) при комнатной температуре. Реакции давали осуществиться в течение ночи, затем гасили триэтилсиланом (0,088 мл, 0,55 ммоль). Летучие вещества удаляли в вакууме и полученный остаток распределяли между насыщенным раствором NaHCO3 и DCM/MeOH (10:1). Водный слой экстрагировали (3×) снова при помощи DCM/MeOH (10:1) и объединенные органические слои сушили над MgSO4, фильтровали, концентрировали и очищали флэш-хроматографией (DCM/7н NH3 в MeOH 87:13) с получением желаемого продукта в виде не совсем белого пенистого вещества (0,060 г). MS (ESI+) для C30H41N7O2 m/z 532,3 [M+H]+; MS (ESI-) для C30H41N7O2 m/z 530,4 [M-H]-; ВЭЖХ чистота >94% (время удерживания, 2,723 минут).
1H ЯМР (400 МГц, d4-MeOH) δH 8,079 (с, 1H), 7,500 (с, 1H), 7,418-7,398 (м, 1H), 7,310-7,307 (м, 1H), 7,230-7,216 (м, 1H), 6,619-6,610 (м, 1H), 4,355-4,316 (м, 1H), 3,926-3,887 (м, 1H), 3,088-3,017 (м, 0,5H (метин транс изомера)), 2,879-2,809 (м, 2H), 2,745-2,685 (м, 0,5H (метин цис изомера), 2,532-2,512 (м, 1H), 2,446-2,373 (м, 2H), 2,294-2,276 (м, 2H), 2,202-2,012 (м, 5H), 1,685-1,603 (м, 1H), 1,545-1,504 (м, 1H), 1,383 (с, 1H).
Время удерживания: 2,723 минут. ВЭЖХ Условия: колонка Agilent Zorbax Exlipse XDB-C18, 4,6×50 мм (1,8 мкм наполнителя), Растворитель A- Вода (0,1% TFA), Растворитель B- Ацетонитрил (0,07% TFA) 6 мин. градиент от 5 до 95% B; 1 мин. удерживание; затем повторный цикл.
Соединение 15: (1R,2S,3R,5R)-3-{4-амино-7H-пирроло[2,3-d]пиримидин-7-ил}-5-{[пропан-2-ил({4-[5-(трифторметил)-1H-1,3-бензодиазол-2-ил]бутил})амино]метил}циклопентан-1,2-диол
Стадия 1: 7-[(3aS,4R,6R,6aR)-2,2-диметил-6-{[пропан-2-ил({4-[5-(трифторметил)-1-{[2-(триметилсилил)этокси]метил}-1H-1,3-бензодиазол-2-ил]бутил})амино]метил}-гексагидроциклопента[d][1,3]диоксол-4-ил]-7H-пирроло[2,3-d]пиримидин-4-амин
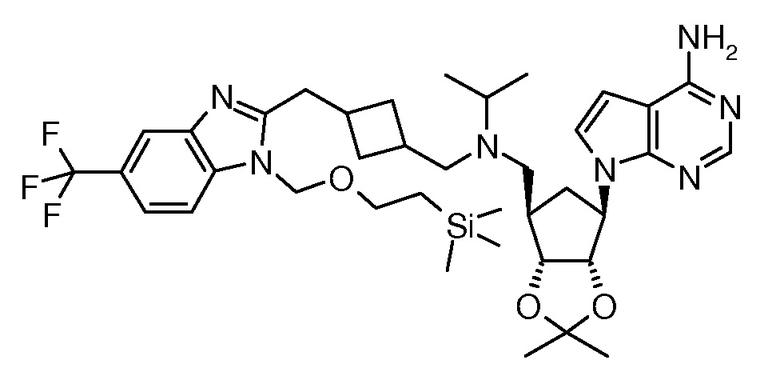
Раствор 3-{[5-(трифторметил)-1-{[2-(триметилсилил)этокси]метил}-1H-1,3-бензодиазол-2-ил]метил}циклобутан-1-карбальдегида (243 мг, 0,59 ммоль), 7-[(3aS,4R,6R,6aR)-2,2-диметил-6-[(пропан-2-иламино)метил]-гексагидроциклопента[d][1,3]диоксол-4-ил]-7H-пирроло[2,3-d]пиримидин-4-амина (170 мг, 0,49 ммоль) и MgSO4 (710 мг, 5,90 ммоль) в DCE (10 мл) перемешивали в течение 15 минут. Затем к реакционной смеси добавляли STAB (175 мг, 0,83 ммоль) и перемешивали в течение 1 часа при комнатной температуре. Реакцию отслеживали при помощи ЖХ-MS, никакого амина не наблюдали по прошествии 1 часа. К реакционной смеси добавляли насыщенный раствор NaHCO3 (20 мл) и перемешивали в течение 5 минут. Затем к реакционной смеси добавляли насыщенный солевой раствор (10 мл). Продукт экстрагировали при помощи DCM (2×30 мл), сушили над Na2SO4, фильтровали и упаривали. Очистка колоночной хроматографией на силикагеле с элюированием при помощи 7н NH3 в MeOH:DCM (1:99-4:96) давала желаемый продукт (170 мг, 47 %) в виде масла; MS (ESI+) для C38H54F3N7O3Si m/z 742,40 [M+H]+; ВЭЖХ чистота 100% (время удерживания, 1,78 мин);
1H ЯМР (500 МГц, ХЛОРОФОРМ-d) δH м.д. -0,25-0,11 (9H, м), 0,66-1,04 (8H, м), 1,17-1,48 (5H, м), 1,49-1,61 (3H, м), 1,77-2,04 (2H, м), 2,19-2,37 (5H, м), 2,37-2,51 (2H, м), 2,52-2,80 (2H, м), 2,81-2,91 (1H, м), 2,91-3,22 (2H, м), 3,34-3,79 (2H, м), 4,29-4,52 (1H, м), 4,78-5,12 (2H, м), 5,28-5,70 (4H, м), 6,34 (1H, д, J=3,63 Гц), 6,80-7,16 (1H, м), 7,29-7,71 (2H, м), 7,71-8,11 (1H, м), 8,11-8,46 (1H, м).
Стадия 2. (1R,2S,3R,5R)-3-{4-амино-7H-пирроло[2,3-d]пиримидин-7-ил}-5-{[пропан-2-ил({4-[5-(трифторметил)-1H-1,3-бензодиазол-2-ил]бутил})амино]метил}циклопентан-1,2-диол
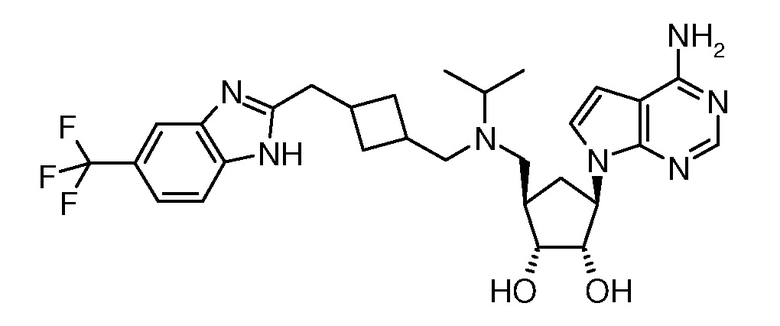
12н раствор HCl (3 мл) медленно добавляли к раствору 7-[(3aS,4R,6R,6aR)-2,2-диметил-6-({пропан-2-ил[(3-{[5-(трифторметил)-1-{[2-(триметилсилил)этокси]метил}-1H-1,3-бензодиазол-2-ил]метил}циклобутил)метил]амино}метил)-гексагидроциклопента[d][1,3]диоксол-4-ил]-7H-пирроло[2,3-d]пиримидин-4-амина (170 мг, 0,23 ммоль) в MeOH (3 мл) и перемешивали при 40o C в течение 2,5 часов. Реакцию отслеживали при помощи ЖХ-MS, через 2,5 часа никакого исходного вещества не наблюдали. Реакционную смесь концентрировали в вакууме, затем подщелачивали при помощи 7н раствора NH3 в MeOH. Затем смесь упаривали досуха. Очистка колоночной хроматографией на силикагеле с элюированием при помощи 7н NH3 в MeOH:DCM (1:9) давала желаемый продукт (100 мг, 76 %) в виде белого твердого вещества; MS (ESI+) для C29H36F3N7O3 m/z 572,40 [M+H]+; ВЭЖХ чистота 99% (время удерживания, 2,17 мин);
1H ЯМР (500 МГц, ХЛОРОФОРМ-d) δH м.д. 0,78-1,19 (6H, м), 1,35-1,69 (2H, м), 1,80-2,04 (1H, м), 2,11-2,87 (10H, м), 2,88-3,18 (3H, м), 3,79-4,06 (1H, м), 4,15-4,47 (1H, м), 4,82-5,12 (1H, м), 6,43-6,77 (1H, м), 7,19 (1H, д, J=3,47 Гц), 7,47 (1H, д, J=8,35 Гц), 7,63 (1H, д, J=8,51 Гц), 7,79 (1H, с), 7,95-8,24 (1H, м).
Соединение 18: (1R,2S,3R,5R)-3-{4-амино-7H-пирроло[2,3-d]пиримидин-7-ил}-5-({метил[(3-{[5-(трифторметил)-1H-1,3-бензодиазол-2-ил]метил}циклобутил)метил]амино}метил)циклопентан-1,2-диол
Стадия 1: 3-[2-(бензилокси)-2-оксоэтилиден]циклобутан-1-карбоновая кислота
Смесь циклобутанон-3-карбоновой кислоты (5 г, 43,82 ммоль), бензил-2-(диметоксифосфорил)ацетата (13,58 г, 52,59 ммоль), LiOH (4,20 г, 23,95 ммоль) и 3Å активированных молекулярных сит (25 г, в порошкообразной форме) в ТГФ (250 мл) нагревали до температуры кипения с обратным холодильником в атмосфере азота в течение 4 часов. Реакционной смеси давали охладиться до комнатной температуры и добавляли EtOAc (100 мл) с последующим добавлением HCl (1н, 100 мл). Эту смесь фильтровали через целит. Фазы разделяли, и водный слой экстрагировали при помощи EtOAc (4×50 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали с получением бесцветного масла. Флэш-хроматография на «сухих» колонках с SiO2 c элюированием смесью гептан:EtOAc от 7:3 до 1:1 давала желаемый продукт в виде бесцветного масла (5,2 г, 39 %); MS (ESI+) для C14H14O4 m/z 269,05 [M+Na]+; MS (ESI-) для C14H14O4 m/z 245,15 [M-H]-; ВЭЖХ чистота 81% (время удерживания, 1,85 мин);
1H ЯМР (250 МГц, ХЛОРОФОРМ-d) δH м.д. 2,95-3,62 (5H, м), 4,96-5,31 (2H, м), 5,75 (1H, т, J=2,21 Гц), 7,27-7,45 (5H, м).
Стадия 2. бензил 2-{3-[метокси(метил)карбамоил]-циклобутилиден}ацетат
К охлажденному льдом раствору 3-[2-(бензилокси)-2-оксоэтилиден]циклобутан-1-карбоновой кислоты (2,0 г, 8,12 ммоль), N-метилморфолина (2,70 мл, 24,36 ммоль) в DCM (50 мл) добавляли изобутилхлорформиат (1,70 мл, 12,99 ммоль) по каплям в течение 5 минут. Еще через 5 минут добавляли гидрохлорид метокси(метил)амина (1,58 г, 16,24 ммоль) и смесь перемешивали в течение ночи, давая ей при этом нагреться до комнатной температуры. Реакционную смесь затем разбавляли при помощи DCM (30 мл), промывали 0,1н раствором HCl (50 мл), затем насыщенным раствором NaHCO3 (50 мл), сушили над Na2SO4, фильтровали и упаривали. Очистка колоночной хроматографией на силикагеле с элюированием при помощи EtOAc:гептан от 1:9 до 3:7 давала желаемый продукт (1,54 г, 65 %); MS (ESI+) для C16H19NO4 m/z 290,10 [M+H]+; ВЭЖХ чистота 100% (время удерживания, 1,86 мин);
1H ЯМР (500 МГц, ХЛОРОФОРМ-d) δH м.д. 2,96 (1H, ддд, J=16,98, 8,79, 1,81 Гц), 3,17-3,30 (4H, м), 3,30-3,48 (2H, м), 3,51-3,63 (1H, м), 3,64-3,75 (3H, м), 5,15 (2H, с), 5,73 (1H, квин., J=2,25 Гц), 7,29-7,42 (5H, м).
Стадия 3. 2-{3-[метокси(метил)карбамоил]циклобутил}уксусная кислота
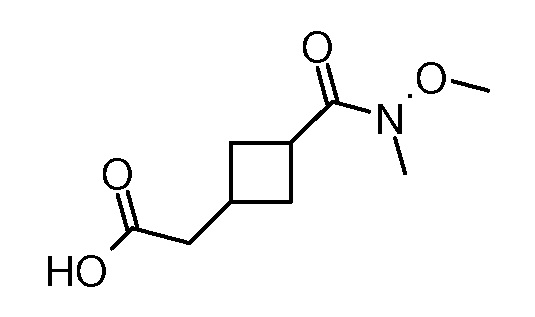
Палладий на угле (10%, 0,1 г) добавляли к раствору бензил 2-{3-[метокси(метил)карбамоил]циклобутилиден}ацетата (1,54 г, 5,32 ммоль) в EtOH (20 мл) и перемешивали в атмосфере водорода при комнатной температуре в течение 6 часов. Реакционную смесь фильтровали через целит и упаривали досуха с получением бесцветного масла (1,04 г, 89%); MS (ESI+) для C9H15NO4 m/z 202,00 [M+H]+; MS (ESI)- для C9H15NO4 m/z 200,05 [M-H]-; ВЭЖХ чистота 92% (время удерживания, 1,10 мин);
1H ЯМР (500 МГц, MeOD) δH м.д. 1,84-2,08 (2H, м), 2,28-2,53 (4H, м), 2,56-2,72 (1H, м), 3,06-3,22 (3H, м), 3,38-3,56 (1H, м), 3,68 (3H, д, J=8,04 Гц).
Стадия 4i. 3-({[2-амино-5-(трифторметил)фенил]-карбамоил}метил)-N-метокси-N-метилциклобутан-1-карбоксамид
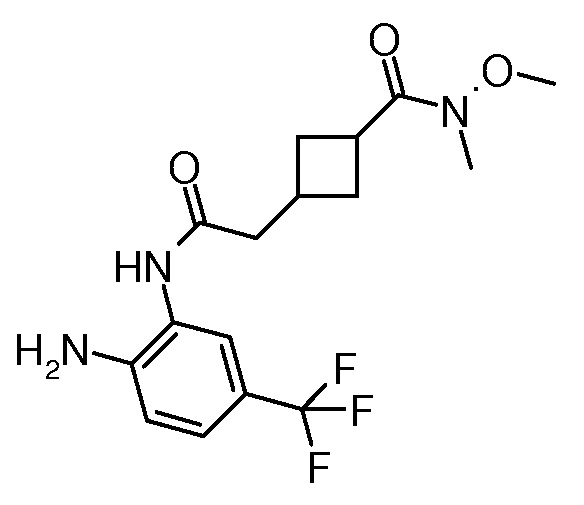
TEA (1,49 мл, 10,70 ммоль) добавляли к суспензии 2-{3-[метокси(метил)карбамоил]циклобутил}уксусной кислоты, EDC·HCl 1,18 г, 6,20 ммоль), HOBt.xH2O (0,77 г, 5,69 ммоль) в DCM (20 мл) при 0oC и перемешивали в течение 5 минут перед добавлением 4-(трифторметил)бензол-1,2-диамина (1,04 г, 10,34 ммоль). Смесь перемешивали еще в течение 20 минут при 0oC, затем давали нагреться до комнатной температуры. Реакцию отслеживали при помощи ЖХ-MS, через 2 часа реакционную смесь промывали 1н раствором HCl (50 мл), затем насыщенным раствором NaHCO3 (50 мл). Смесь сушили над Na2SO4, фильтровали и упаривали досуха. Очистка колоночной хроматографией на силикагеле с элюированием при помощи EtOAc давала желаемый продукт (0,83 г, 44%) в виде бежевого твердого вещества; MS (ESI+) для C16H20N3O3 m/z 360,00 [M+H]+; ВЭЖХ чистота 90% (время удерживания, 1,64 мин);
1H ЯМР (500 МГц, MeOD) δH м.д. 2,02-2,16 (2H, м), 2,31-2,68 (4H, м), 2,69-2,88 (1H, м), 3,18 (3H, д, J=4,41 Гц), 3,36-3,59 (1H, м), 3,65-3,76 (3H, м), 6,81-6,97 (1H, м), 7,02-7,27 (1H, м), 7,28-7,48 (1H, м).
Стадия 4ii. N-метокси-N-метил-3-{[5-(трифторметил)-1H-1,3-бензодиазол-2-ил]метил}-циклобутан-1-карбоксамид
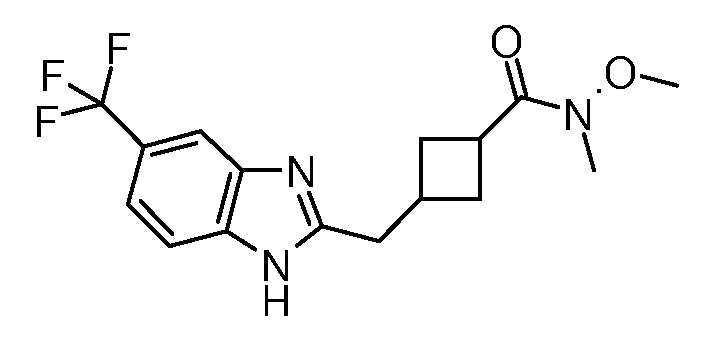
Раствор 3-({[2-амино-5-(трифторметил)фенил]карбамоил}метил)-N-метокси-N-метилциклобутан-1-карбоксамида (0,82 г, 2,29 ммоль) в AcOH (10 мл) нагревали до температуры кипения с обратным холодильником (~125oC) при перемешивании в течение 2,5 часов. Реакцию отслеживали при помощи ЖХ-MS. Реакционной смеси давали охладиться до комнатной температуры и затем упаривали в вакууме. Остаток растворяли в DCM (30 мл) и промывали насыщенным раствором NaHCO3 (50 мл), сушили над Na2SO4, фильтровали и упаривали. Неочищенное вещество очищали колоночной хроматографией на силикагеле с элюированием при помощи MeOH:DCM (2:98-5:95) с получением желтовато-коричневого масла (0,75 г, 94%); MS (ESI+) для C16H18F3N3O2 m/z 342,10 [M+H]+; ВЭЖХ чистота 97% (время удерживания, 1,40 мин);
1H ЯМР (500 МГц, MeOD) δH м.д. 1,96-2,17 (2H, м), 2,27-2,55 (2H, м), 2,66-2,92 (1H, м), 2,97-3,15 (2H, м), 3,15-3,22 (3H, м), 3,32-3,62 (1H, м), 3,63-3,73 (3H, м), 7,37-7,53 (1H, м), 7,53-8,00 (2H, м).
Стадия 5. N-метокси-N-метил-3-{[5-(трифторметил)-1-{[2-(триметилсилил)этокси]метил}-1H-1,3-бензодиазол-2-ил]метил}-циклобутан-1-карбоксамид
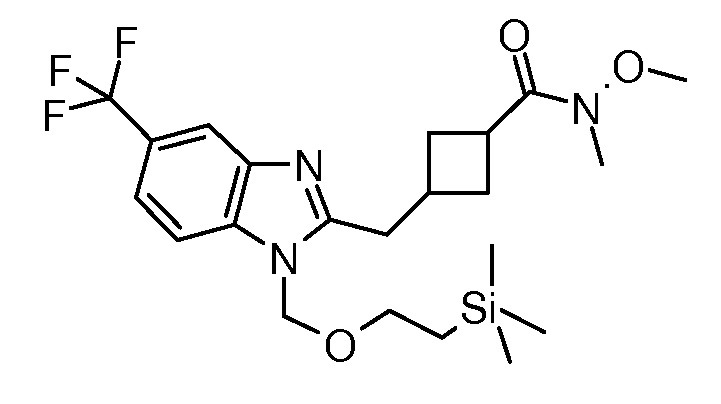
K2CO3 (381 мг, 2,76 ммоль), затем SEM-Cl (430 мкл, 2,43 ммоль) добавляли к раствору N-метокси-N-метил-3-{[5-(трифторметил)-1H-1,3-бензодиазол-2-ил]метил}циклобутан-1-карбоксамида (754 мг, 2,21 ммоль) в DMF (10 мл) при комнатной температуре и перемешивали в течение ночи. Реакцию отслеживали при помощи ЖХ-MS. Реакционную смесь разбавляли водой (3 мл), насыщенным солевым раствором (30 мл) и затем экстрагировали при помощи EtOAc (2×50 мл). Смесь сушили над Na2SO4, фильтровали и упаривали с получением прозрачного оранжевого масла. Очистка колоночной хроматографией на силикагеле с элюированием смесью EtOAc:гептан (1:1-1) давала желаемые продукты в виде бежевого масла, в виде одного региоизомера (217 мг, 21%); MS (ESI+) для C22H32F3N3O3Si m/z 472,55 [M+H]+; ВЭЖХ чистота 99% (время удерживания, 2,37 мин);
1H ЯМР (250 МГц, ХЛОРОФОРМ-d) δH м.д. -0,31-0,22 (9H, м), 0,83-1,00 (2H, м), 2,03-2,25 (2H, м), 2,30-2,74 (2H, м), 2,85-3,14 (3H, м), 3,18 (3H, с), 3,32-3,60 (3H, м), 3,61-3,72 (3H, м), 5,52 (2H, с), 7,51 (1H, дд, J=8,38, 1,22 Гц), 7,70 (1H, с), 7,79 (1H, д, J=8,53 Гц) и в виде смеси региоизомеров (280 мг, 27%); C22H32F3N3O3Si m/z 472,55 [M+H]+; ВЭЖХ чистота 72% & 21% (время удерживания, 2,39 & 2,36 мин); 1H ЯМР (250 МГц, ХЛОРОФОРМ-d) δ м.д. -0,08 - -0,01 (9H, м), 0,81-0,99 (2H, м), 1,94-2,24 (2H, м), 2,38-2,74 (2H, м), 2,90-3,14 (3H, м), 3,14-3,22 (3H, м), 3,33-3,63 (3H, м), 3,63-3,70 (3H, м), 5,40-5,64 (2H, м), 7,40-7,74 (2H, м), 7,75-8,14 (1H, м).
Стадия 6. 3-{[5-(трифторметил)-1-{[2-(триметилсилил)этокси]-метил}-1H-1,3-бензодиазол-2-ил]метил}циклобутан-1-карбальдегид
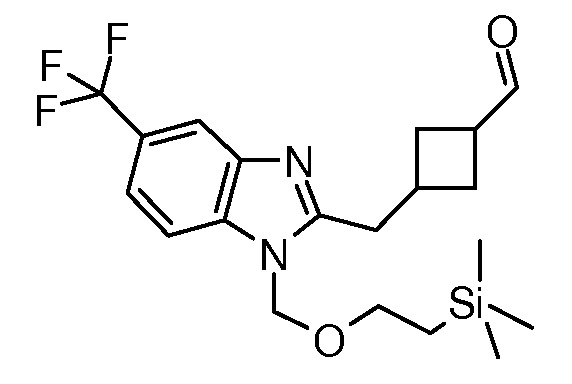
DIBAL (0,69 мл, 0,69 ммоль, 1M в ТГФ) добавляли по каплям к раствору N-метокси-N-метил-3-{[5-(трифторметил)-1-{[2-(триметилсилил)этокси]метил}-1H-1,3-бензодиазол-2-ил]метил}циклобутан-1-карбоксамида в ТГФ при -10oC при перемешивании. Реакцию продолжали в течение 3 часов при -10oC. Реакционную смесь выливали в насыщенный водный раствор сегнетовой соли (20 мл), разбавляли при помощи Et2O (50 мл) и перемешивали в течение 30 минут. Смесь затем разделяли и органический слой промывали сегнетовой солью (30 мл), насыщенным раствором NaHCO3, (30 мл) и насыщенным солевым раствором (30 мл). Смесь сушили над Na2SO4, фильтровали и упаривали с получением бесцветного смолистого вещества (190 мг, 87%); MS (ESI+) для C20H27F3N2O2Si m/z 413,6 [M+H]+; ВЭЖХ чистота 87% (время удерживания, 2,25 мин);
1H ЯМР (500 МГц, ХЛОРОФОРМ-d) δH м.д. -0,10-0,09 (9H, м), 0,88-1,00 (4H, м), 1,63-1,97 (2H, м), 2,07-2,23 (2H, м), 2,24-2,37 (0H, м), 2,37-2,42 (1H, м), 2,43-2,66 (2H, м), 2,93-3,34 (4H, м), 5,49-5,61 (2H, м), 7,16-7,31 (2H, м), 7,32-7,32 (2H, м), 7,49-7,61 (1H, м), 7,73 (1H, с), 7,83 (1H, д, J=8,35 Гц).
Стадия 7. 7-[(3aS,4R,6R,6aR)-2,2-диметил-6-({[(3-{[5-(трифторметил)-1-{[2-(триметилсилил)этокси]метил}-1H-1,3-бензодиазол-2-ил]метил}циклобутил)метил]амино}метил)-гексагидроциклопента[d][1,3]диоксол-4-ил]-7H-пирроло[2,3-d]пиримидин-4-амин
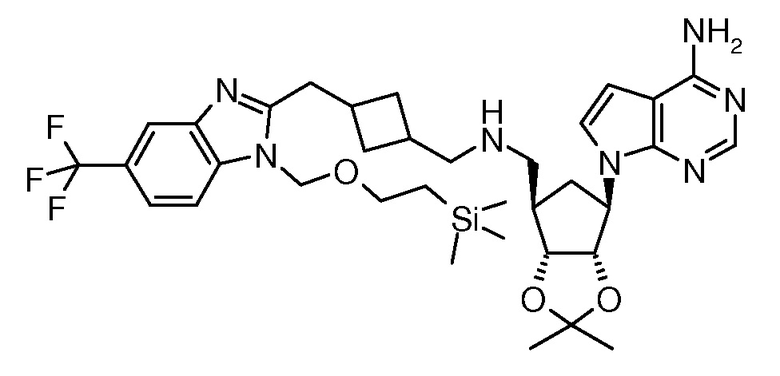
Раствор 3-{[5-(трифторметил)-1-{[2-(триметилсилил)этокси]-метил}-1H-1,3-бензодиазол-2-ил]метил}циклобутан-1-карбальдегида (190 мг, 0,46 ммоль), (1R,2S,3R,5R)-3-{4-амино-7H-пирроло[2,3-d]пиримидин-7-ил}-5-(аминометил)циклопентан-1,2-диола (140 мг, 0,46 ммоль) и MgSO4 (554 мг, 4,61 ммоль) в DCE (10 мл) перемешивали в течение 15 минут. Затем к реакционной смеси добавляли STAB (137 мг, 0,645 ммоль) и перемешивали. Реакцию отслеживали при помощи ЖХ-MS, после этого никакого амина не наблюдали. К реакционной смеси добавляли насыщенный раствор NaHCO3 (20 мл) и перемешивали в течение 5 минут. К реакционной смеси добавляли насыщенный солевой раствор (10 мл) и продукт экстрагировали при помощи DCM (2×30 мл), сушили над Na2SO4, фильтровали и упаривали. Очистка колоночной хроматографией на силикагеле с элюированием смесью 7н NH3 в MeOH:DCM (1:99-5:95) давала желаемый продукт в виде розового пенистого твердого вещества. Смешанные фракции объединяли и очищали на пластинке для препаративной ТСХ, элюировали смесью 7н NH3 в MeOH:DCM (2×4:96) с получением в целом 170 мг, 53%. MS (ESI+) для C35H48F3N7O3Si m/z 700 [M+H]+; ВЭЖХ чистота 100% (время удерживания, 1,79 мин);
1H ЯМР (250 МГц, ХЛОРОФОРМ-d) δH м.д. -0,26-0,16 (9H, м), 0,72-0,97 (2H, м), 1,30 (3H, с), 1,38-1,60 (5H, м), 1,91-2,59 (8H, м), 2,60-3,27 (6H, м), 3,35-3,70 (2H, м), 4,52 (1H, т, J=5,94 Гц), 4,78-5,20 (4H, м), 5,39-5,66 (2H, м), 6,36 (1H, д, J=3,65 Гц), 7,03 (1H, д, J=3,65 Гц), 7,42-7,59 (1H, м), 7,69 (1H, с), 7,79 (1H, д, J=8,53 Гц), 8,32 (1H, с).
Стадия 8. 7-[(3aS,4R,6R,6aR)-2,2-диметил-6-({метил[(3-{[5-(трифторметил)-1-{[2-(триметилсилил)этокси]метил}-1H-1,3-бензодиазол-2-ил]метил}циклобутил)метил]амино}метил)-гексагидроциклопента[d][1,3]диоксол-4-ил]-7H-пирроло[2,3-d]пиримидин-4-амин
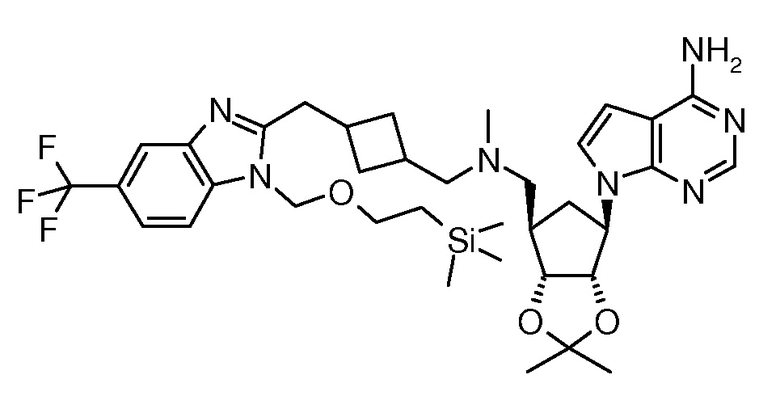
Формальдегид (36 мкл, 0,49 ммоль, 37%(водн.)) добавляли к раствору 7-[(3aS,4R,6R,6aR)-2,2-диметил-6-({[(3-{[5-(трифторметил)-1-{[2-(триметилсилил)этокси]метил}-1H-1,3-бензодиазол-2-ил]метил}циклобутил)метил]амино}метил)-гексагидроциклопента[d][1,3]диоксол-4-ил]-7H-пирроло[2,3-d]пиримидин-4-амина в MeOH (5 мл) и ТГФ (5 мл) и реакционную смесь перемешивали при комнатной температуре в течение 30 минут. Добавляли по порциям NaCNBH3 (18 мг, 0,29 ммоль) и реакционную смесь перемешивали еще в течение 2 часов при комнатной температуре, после чего ЖХ-MS анализ показал, что реакция завершена. Реакционную смесь концентрировали в вакууме и остаток распределяли между водой (20 мл) и DCM (20 мл) и слои разделяли. Водный слой экстрагировали при помощи DCM (2×20 мл), объединенные органические слои затем сушили над Na2SO4 и концентрировали. Очистка методом препаративной ТСХ c элюированием смесью 7н NH3 в MeOH:DCM (5:95) давала желаемый продукт (114 мг, 66%) в виде бесцветного масла; MS (ESI+) для C36H50F3N7O3Si m/z 714,45 [M+H]+; ВЭЖХ чистота 100% (время удерживания, 1,77 мин);
1H ЯМР (500 МГц, ХЛОРОФОРМ-d) δH м.д. -0,14-0,05 (9H, м), 0,85-0,99 (2H, м), 1,18-1,35 (3H, м), 1,37-1,53 (2H, м), 1,52-1,64 (3H, м), 1,89-2,15 (3H, м), 2,18-2,28 (2H, м), 2,29-2,70 (6H, м), 2,72-2,96 (1H, м), 2,97-3,20 (2H, м), 3,48 (3H, с), 3,50-3,58 (2H, м), 4,37-4,58 (1H, м), 4,85-5,05 (2H, м), 5,11-5,38 (2H, м), 5,41-5,57 (2H, м), 6,35 (1H, д, J=3,63 Гц), 6,84-7,19 (1H, м), 7,50 (1H, д, J=8,35 Гц), 7,68 (1H, с), 7,78 (1H, д, J=8,35 Гц), 8,13-8,52 (1H, м).
Стадия 9. (1R,2S,3R,5R)-3-{4-амино-7H-пирроло[2,3-d]пиримидин-7-ил}-5-({метил[(3-{[5-(трифторметил)-1H-1,3-бензодиазол-2-ил]метил}циклобутил)метил]амино}метил)циклопентан-1,2-диол
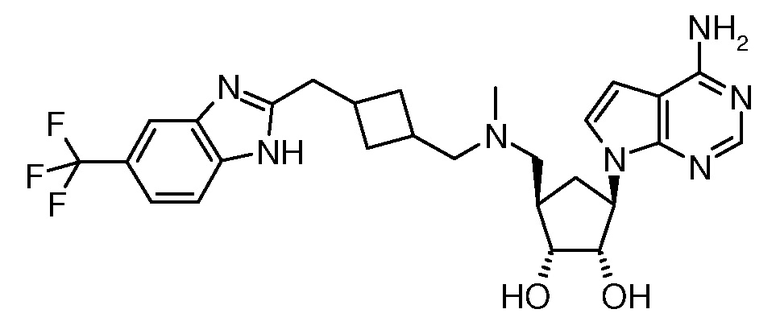
Раствор HCl в MeOH (1:1, 3 мл) добавляли к 7-[(3aS,4R,6R,6aR)-2,2-диметил-6-({метил[(3-{[5-(трифторметил)-1-{[2-(триметилсилил)этокси]метил}-1H-1,3-бензодиазол-2-ил]метил}циклобутил)метил]амино}метил)-гексагидроциклопента[d][1,3]диоксол-4-ил]-7H-пирроло[2,3-d]пиримидин-4-амину при 0oC и перемешивали. Сразу после этого смесь перемешивали при 40oC в течение 4 часов (реакцию отслеживали при помощи ЖХ-MS). Реакцию продолжали еще в течение 1 часа. ЖХ-MS анализ все еще показывал 30% SM с удаленной защитной группой ацеталя. К реакционной смеси добавляли еще 1 мл HCl (36% водный раствор) и реакцию продолжали при 40°C еще в течение 1 часа. Реакционную смесь затем упаривали в вакууме. Остаток растворяли в DCM (100 мл) + MeOH (1 мл) и промывали насыщенным раствором NaHCO3 (2×50 мл), сушили над Na2SO4, фильтровали и упаривали. Очистку осуществляли методом препаративной ТСХ c элюированием смесью 7н NH3 в MeOH:DCM (1:9); MS (ESI+) для C27H32F3N7O2 m/z 544 [M+H]+; ВЭЖХ чистота 96% (время удерживания, 2,03 мин);
1H ЯМР (500 МГц, MeOD) δH м.д. 1,47-1,58 (1H, м), 1,58-1,69 (1H, м), 1,87-2,12 (1H, м), 2,12-2,54 (10H, м), 2,53-2,91 (3H, м), 2,93-3,15 (2H, м), 3,88-4,08 (1H, м), 4,32 (1H, дд, J=7,57, 6,15 Гц), 4,88-5,01 (1H, м), 6,59 (1H, д, J=3,63 Гц), 7,20 (1H, д, J=3,63 Гц), 7,42-7,53 (1H, м), 7,63 (1H, д, J=8,20 Гц), 7,79 (1H, с), 7,95-8,22 (1H, м).
Соединение 19: 7-((3aS,4R,6R,6aR)-6-(((3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(изопропил)-амино)метил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]-диоксол-4-ил)-N-(2,4-диметоксибензил)-7H-пирроло[2,3-d]пиримидин-4-амин
Раствор 3-(3-((((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)(изопропил)амино)циклобутил)пропановой кислоты (490 мг, 0,79 ммоль) и 4-трет-бутилбензол-1,2-диамина (155 мг, 0,946 ммоль) в N,N-диметилформамиде (8,1 мл) обрабатывали N,N-диизопропилэтиламином (0,453 мл, 2,60 ммоль) по каплям с последующим добавлением гексафторфосфата N,N,N',N'-тетраметил-0-(7-азабензотриазол-1-ил)урония (449 мг, 1,18 ммоль) одной порцией. Реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали в условиях высокого вакуума и остаток распределяли между 50 мл EtOAc и 50 мл смеси 1/1 H2O/насыщенный раствор NaHCO3. Водную фазу экстрагировали при помощи 30 мл EtOAc и объединенную органическую фазу промывали 30 мл порциями H2O и насыщенного солевого раствора. Органическую фазу сушили над Na2SO4, фильтровали и концентрировали с получением стеклообразного вещества/жесткой пены. Это неочищенное вещество очищали флэш-хроматографией (SiO2 c элюированием смесью 4% 7н NH3 в CH3OH/CH2Cl2) с получением желаемого промежуточного соединения в виде смеси амидных региоизомеров (400 мг). Раствор промежуточного соединения (0,40 г) в уксусной кислоте (15 мл) нагревали при 65°C в течение 2,5 часов и реакционную смесь охлаждали и помещали в условия высокого вакуума для удаления уксусной кислоты. Остаток брали для поглощения в 60 мл CH2Cl2 и промывали 40 мл порциями насыщенного раствора NaHCO3 и 2% раствора Na2CO3. Органическую фазу сушили над Na2SO4, фильтровали и концентрировали с получением стеклообразного вещества/жесткой пены. Это вещество помещали в условия высокого вакуума и использовали непосредственно на следующей стадии (380 мг).
(1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-(((3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(изопропил)амино)метил)циклопентан-1,2-диол
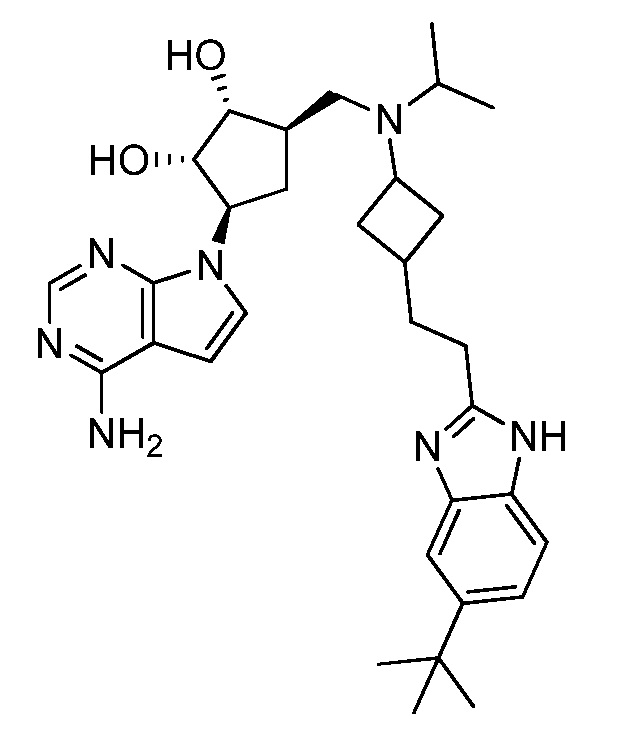
7-((3aS,4R,6R,6aR)-6-(((3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(изопропил)амино)метил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-N-(2,4-диметоксибензил)-7H-пирроло[2,3-d]пиримидин-4-амин (390 мг, 0,52 ммоль) растворяли в смеси трифторуксусной кислоты (7,2 мл) и воды (0,8 мл), которая была предварительно охлаждена при 0°C на ледяной бане. Раствор перемешивали при 0°C в течение 30 минут, затем нагревали до комнатной температуры. После выдерживания в течение 2,5 часов при комнатной температуре остаток брали для поглощения в 15 мл MeOH и концентрировали. Эту процедуру повторяли два раза и остаток помещали в условия высокого вакуума. Это вещество брали для поглощения в 15 мл MeOH (происходило образование суспензии) и обрабатывали при помощи 500 мг K2CO3 и 8 капель воды. Смесь оставляли для перемешивания в течение 1 часа, в течение этого времени раствор становился щелочным. Смесь фильтровали через тонкую фритту, твердые вещества промывали при помощи 10 мл MeOH и фильтрат концентрировали с получением не совсем белого твердого вещества. Это вещество оставляли в условиях высокого вакуума в течение ночи. Это неочищенное вещество очищали флэш-хроматографией (SiO2 c элюированием смесью 8-10% 7н NH3 в CH3OH/CH2Cl2) с получением стеклообразного вещества/жесткой пены (0,22 г).
1H ЯМР (400 МГц, MeOD) δH м.д. 8,06 (с, 1H), 7,48 (шир.с, 1H), 7,39 (м, 1H), 7,27 (м, 1H), 7,20 (д, J=3,52 Гц, 1H), 6,60 (м, 1H), 4,32 (т, J=6,43 Гц, 1H), 3,93 (т, J=5,29 Гц, 1H), 3,54 (м, 0,2H), 3,11 (т, J=9,33 Гц, 1H), 3,02 (м, 1H), 2,82 (м, 2H), 2,66 (дд, J=1 3,68, 8,09 Гц, 1H), 2,46 (м, 1H), 2,36 (м, 1H), 2,23 (м, 3H), 2,05 (м, 1H), 1,91 (м, 3H), 1,59 (м, 3H), 1,36 (с, 9H), 1,02 (м, 6H).
Соединение 20: (1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-(((3-(2-(6-хлор-5-(трифторметил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(изопропил)амино)метил)циклопентан-1,2-диол
7-((3aS,4R,6R,6aR)-6-(((3-(2-(6-хлор-5-(трифторметил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(изопропил)амино)метил)- 2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-N-(2,4-диметоксибензил)-7H-пирроло[2,3-d]пиримидин-4-амин
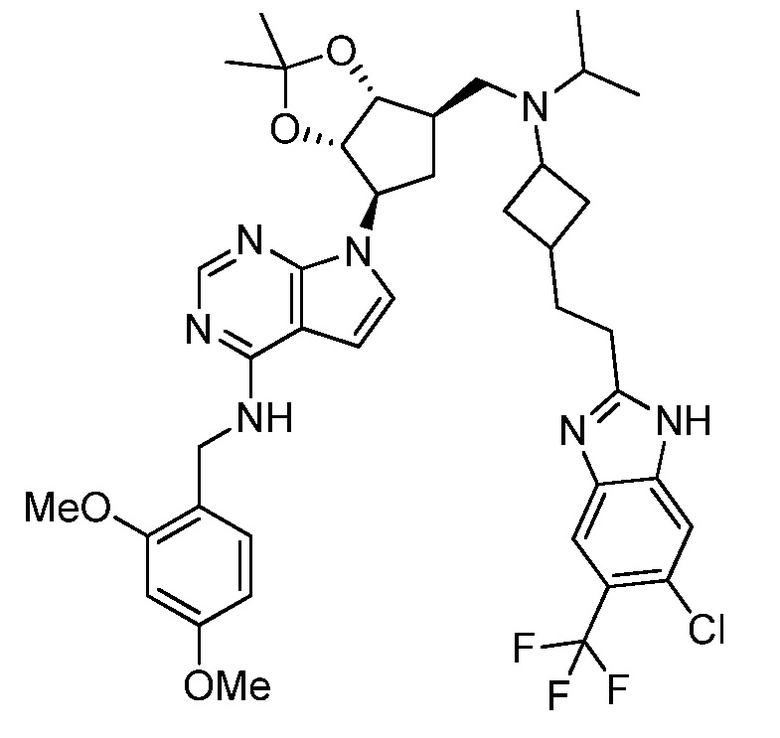
Раствор 3-(3-((((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)(изопропил)амино)циклобутил)пропановой кислоты (78,5 мг, 0,126 ммоль) и 4-хлор-5-(трифторметил)бензол-1,2-диамина (31,9 мг, 0,151 ммоль) в N,N-диметилформамиде (1,3 мл) обрабатывали N,N-диизопропилэтиламином (72,5 мкл, 0,416 ммоль по каплям с последующим добавлением гексафторфосфата N,N,N’,N’-тетраметил-0-(7-азабензотриазол-1-ил)урония (72,0 мг, 0,189 ммоль) одной порцией. Реакционную смесь перемешивали при комнатной температуре в течение 7,5 часов. Реакционную смесь помещали в холодильник и выдерживали в течение ночи. Реакционную смесь концентрировали в условиях высокого вакуума и остаток распределяли между 20 мл EtOAc и 20 мл смеси 1/1 H2O/насыщенный раствор NaHCO3. Водную фазу экстрагировали при помощи 10 мл EtOAc и объединенную органическую фазу промывали 10 мл порциями H2O и насыщенного солевого раствора. Органическую фазу сушили над Na2SO4, фильтровали и концентрировали. Это неочищенное вещество очищали флэш-хроматографией (SiO2, c элюированием смесью 3% 7н NH3 в CH3OH/CH2Cl2) с получением желаемого промежуточного соединения в виде стеклообразного вещества/жесткой пены (региоизомерные амиды и цис/транс диастереомеры, 87 мг). Промежуточное соединение (0,087 г) брали для поглощения в уксусную кислоту (4,5 мл), нагревали при 65°C в течение 6 часов и охлаждали до комнатной температуры и перемешивали при комнатной температуре в течение 48 часов. Реакционную смесь нагревали при 65°C в течение 8 часов, затем при комнатной температуре в течение ночи, затем при 65°C еще в течение 6,5 часов. Смесь охлаждали и помещали в условия высокого вакуума для удаления уксусной кислоты. Остаток брали для поглощения в 15 мл CH2Cl2 и промывали 10 мл порциями насыщенного раствора NaHCO3 и 2% раствора Na2CO3. Органическую фазу сушили над Na2SO4, фильтровали и концентрировали с получением стеклообразного вещества/жесткой пены. Это вещество помещали в условия высокого вакуума и выдерживали в течение ночи. Это неочищенное вещество очищали методом препаративной ТСХ на 20 см × 20 см × 1,0 мм пластине для препаративной ТСХ, элюируя два раза смесью 4% 7н NH3 в CH3OH/CH2Cl2. Фракцию с продуктом выделяли с получением продукта в виде белого твердого вещества (48 мг).
(1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-(((3-(2-(6-хлор-5-(трифторметил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(изопропил)амино)метил)циклопентан-1,2-диол
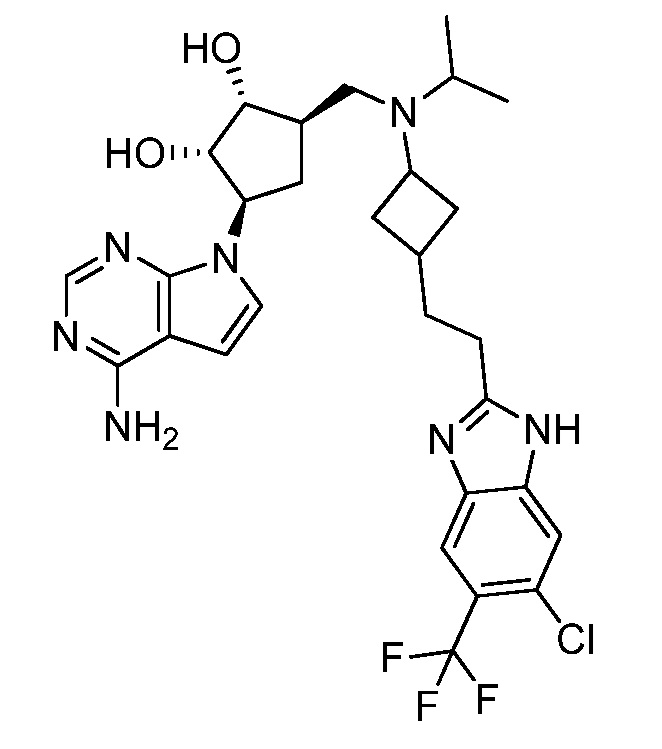
7-((3aS,4R,6R,6aR)-6-(((3-(2-(6-хлор-5-(трифторметил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(изопропил)амино)метил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-N-(2,4-диметоксибензил)-7H-пирроло[2,3-d]пиримидин-4-амин (525 мг, 0,683 ммоль) растворяли в смеси трифторуксусной кислоты (9 мл) и воды (1 мл), которая была предварительно охлаждена при 0°C на ледяной бане. Раствор перемешивали при 0°C в течение 30 минут, затем нагревали до комнатной температуры. После выдерживания в течение 4 часов при комнатной температуре смесь концентрировали. Остаток брали для поглощения в 20 мл MeOH и концентрировали. Эту процедуру повторяли два раза и остаток помещали в условия высокого вакуума. Это вещество брали для поглощения в 15 мл MeOH (происходило образование суспензии) и обрабатывали при помощи 500 мг K2CO3 и 15 капель воды. Смесь оставляли для перемешивания в течение 1 часа, в течение этого времени раствор становился щелочным. Смесь фильтровали через тонкую фритту, твердые вещества промывали при помощи 10 мл MeOH и фильтрат концентрировали с получением не совсем белого твердого вещества. Это вещество оставляли в условиях высокого вакуума в течение ночи. Это неочищенное вещество очищали флэш-хроматографией (SiO2, c элюированием смесью 12% 7н NH3 в CH3OH/CH2Cl2) с получением бесцветного стеклообразного вещества/жесткой пены.
1H ЯМР (400 МГц, MeOD) δH м.д. 8,06 (с, 1H), 7,89 (д, J=2,07 Гц, 1H), 7,69 (д, J=2,28 Гц, 1H), 7,21 (дд, J=3,42, 1,76 Гц, 1H), 6,59 (д, J=3,52 Гц, 1H), 4,33 (т, J=6,84 Гц, 1H), 3,89 (кв., J=5,25 Гц, 1H), 3,03 (м, 0,5H), 2,89 (м, 2H), 2,70 (м, 0,5H), 2,49 (м, 1H), 2,40 (м, 2H), 2,27 (шир.с, 2H), 2,16 (д, J=7,26 Гц, 4H), 2,06 (м, 2H), 1,91 (м, 2H), 1,62 (м, 1H), 1,52 (м, 1H).
Соединение 21: (1R,2S,3R,5R)-3-{4-амино-7H-пирроло[2,3-d]пиримидин-7-ил}-5-({[(3-{[6-хлор-5-(трифторметил)-1H-1,3-бензодиазол-2-ил]метил}циклобутил)метил](пропан-2-ил)амино}метил)циклопентан-1,2-диол
Стадия 1: 7-[(3aS,4R,6R,6aR)-6-({[(3-{[6-хлор-5-(трифторметил)-1-{[2-(триметилсилил)этокси]метил}-1H-1,3-бензодиазол-2-ил]метил}циклобутил)метил](пропан-2-ил)амино}метил)-2,2-диметил-гексагидроциклопента[d][1,3]диоксол-4-ил]-7H-пирроло[2,3-d]пиримидин-4-амин
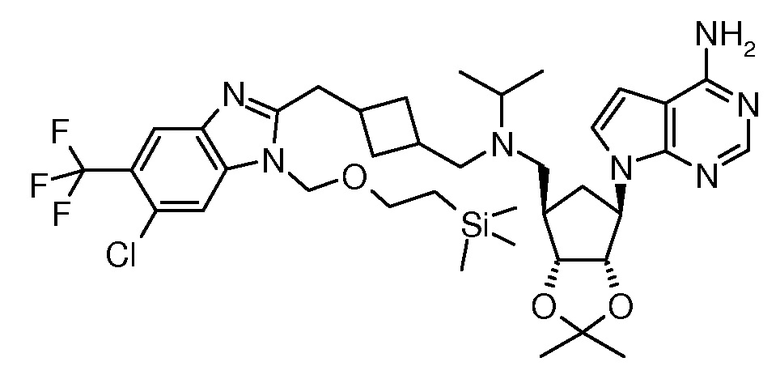
Раствор 3-{[6-хлор-5-(трифторметил)-1-{[2-(триметилсилил)этокси]метил}-1H-1,3-бензодиазол-2-ил]метил}циклобутан-1-карбальдегида (223 мг, 0,50 ммоль), 7-[(3aS,4R,6R,6aR)-2,2-диметил-6-[(пропан-2-иламино)метил]- гексагидроциклопента[d][1,3]диоксол-4-ил]-7H-пирроло[2,3-d]пиримидин-4-амина (172 мг, 0,50 ммоль) и MgSO4 (600 мг, 5,00 ммоль) в DCE (10 мл) перемешивали в течение 15 минут. К реакционной смеси затем добавляли STAB (148 мг, 0,70 ммоль) и перемешивали в течение 1 часа при комнатной температуре. Реакцию отслеживали при помощи ЖХ-MS, никакого амина не наблюдали по прошествии 2,5 часов. К реакционной смеси добавляли насыщенный раствор NaHCO3 (20 мл) и перемешивали в течение 5 минут. К реакционной смеси затем добавляли насыщенный солевой раствор (20 мл). Продукт экстрагировали при помощи DCM (2×30 мл), сушили над Na2SO4, фильтровали и упаривали. Очистка методом препаративной ВЭЖХ давала желаемый продукт (174 мг, 45%) в виде белого твердого вещества; MS (ESI+) для C38H53ClF3N7O3Si m/z 776,30 [M+H]+; ВЭЖХ чистота 100% (время удерживания, 1,68 мин);
1H ЯМР (500 МГц, MeOD) δH м.д. -0,25-0,11 (9H, м), 0,90 (2H, тд, J=7,88, 3,47 Гц), 1,30 (3H, с), 1,37 (6H, т, J=7,49 Гц), 1,55 (3H, с), 1,63-1,84 (1H, м), 1,99-2,37 (2H, м), 2,37-3,03 (6H, м), 3,04-3,28 (3H, м), 3,34-3,50 (1H, м), 3,61 (2H, тд, J=7,92, 2,29 Гц), 3,79 (1H, шир.с), 4,68 (1H, т, J=6,70 Гц), 4,95-5,27 (2H, м), 5,55-5,81 (1H, м), 6,88 (1H, д, J=3,63 Гц), 7,06-7,62 (2H, м), 7,61-7,94 (1H, м), 7,94-8,16 (1H, м), 8,22 (1H, с).
Стадия 2. (1R,2S,3R,5R)-3-{4-амино-7H-пирроло[2,3-d]пиримидин-7-ил}-5-({[(3-{[6-хлор-5-(трифторметил)-1H-1,3-бензодиазол-2-ил]метил}циклобутил)метил](пропан-2-ил)амино}метил)циклопентан-1,2-диол
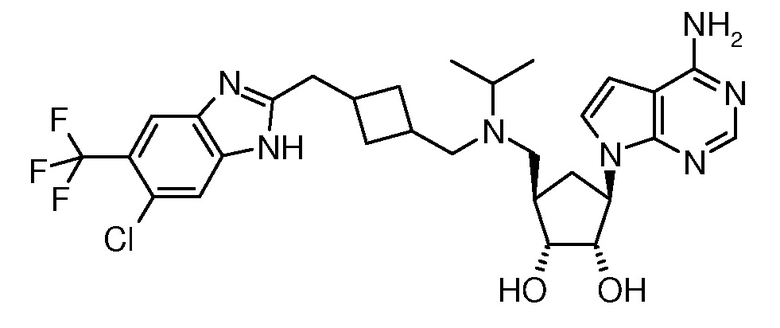
12н раствор HCl (1,5 мл) добавляли медленно к раствору 7-[(3aS,4R,6R,6aR)-6-({[(3-{[6-хлор-5-(трифторметил)-1-{[2-(триметилсилил)этокси]метил}-1H-1,3-бензодиазол-2-ил]метил}циклобутил)метил](пропан-2-ил)амино}метил)-2,2-диметил-гексагидроциклопента[d][1,3]диоксол-4-ил]-7H-пирроло[2,3-d]пиримидин-4-амина (174 мг, 0,22 ммоль) в MeOH (1,5 мл) и перемешивали при 40oC в течение 6 часов. Реакцию отслеживали при помощи ЖХ-MS, через 2,5 часа никакого исходного вещества не наблюдали. Реакционную смесь концентрировали в вакууме, затем подщелачивали при помощи 7н раствора NH3 в MeOH (5 мл). Смесь затем упаривали досуха. Очистка колоночной хроматографией на силикагеле с элюированием смесью 7н NH3 в MeOH:DCM (1:9) давала желаемый продукт (36 мг, 27%) в виде белого твердого вещества; MS (ESI+) для C29H35ClF3N7O2 m/z 606,30 [M+H]+; ВЭЖХ чистота 100% (время удерживания, 2,59 мин);
1H ЯМР (500 МГц, MeOD) δH м.д. 0,93-1,09 (6H, м), 1,41-1,64 (2H, м), 1,76-2,06 (1H, м), 2,15-2,65 (9H, м), 2,65-2,88 (1H, м), 2,91-3,13 (3H, м), 3,80-4,09 (1H, м), 4,19-4,46 (1H, м), 4,88-4,94 (1H, м), 6,46-6,77 (1H, м), 7,19 (1H, д, J=3,63 Гц), 7,69 (1H, с), 7,89 (1H, с), 8,06 (1H, с).
Соединение 22: (1R,2S,3R,5R)-3-{4-амино-7H-пирроло[2,3-d]пиримидин-7-ил}-5-{[({3-[(5-трет-бутил-1H-1,3-бензодиазол-2-ил)метил]циклобутил}метил)(пропан-2-ил)амино]метил}циклопентан-1,2-диол
Стадия 1: 3-{[(2-амино-4-трет-бутилфенил)карбамоил]метил}-N-метокси-N-метилциклобутан-1-карбоксамид
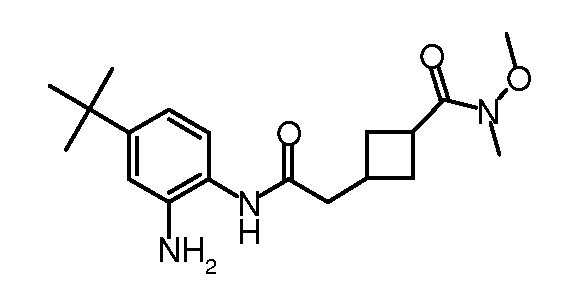
N,N-Диизопропилэтиламин (5,19 мл, 29,82 ммоль) добавляли к суспензии 2-{3-[метокси(метил)карбамоил]циклобутил}уксусной кислоты (3 г, 14,91 ммоль), 4-tBu фенилендиамина (2,69 г, 16,4 ммоль) и гексафторфосфата (1-циано-2-этокси-2-оксоэтилиденаминоокси)диметиламино-морфолино-карбения (7,02 г, 16,4 ммоль) в дихлорметане (60 мл) при 0oC и перемешивали в течение 20 минут, затем смеси давали нагреться до комнатной температуры. Реакционную смесь оставляли при комнатной температуре в течение 4 часов. Реакционную смесь концентрировали и остаток снова растворяли в EtOAc (60 мл). Раствор промывали водой (3×60 мл), затем насыщенным солевым раствором (60 мл), сушили (MgSO4) и концентрировали при пониженном давлении. Это неочищенное вещество очищали флэш-хроматографией на «сухих» колонках c элюированием при помощи 100% EtOAc с получением указанного в заголовке соединения (3,74 г, 46%) в виде коричневого масла: MS (ESI+) для C19H29N3O3 m/z 348,5 [M+H]+; LC чистота 26% и 44% (УФ), 18% и 68% (ELS), (время удерживания, 1,65 и 1,71 мин).
Стадия 2: 3-[(5-трет-бутил-1H-1,3-бензодиазол-2-ил)метил]-N-метокси-N-метилциклобутан-1-карбоксамид
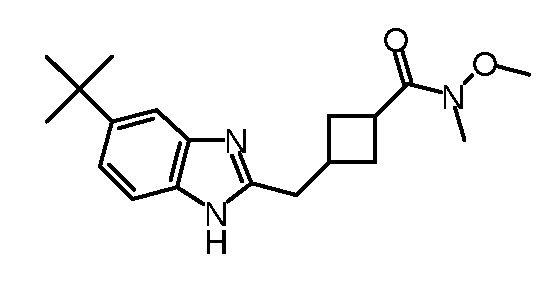
Перемешиваемый раствор 3-{[(2-амино-4-трет-бутилфенил)карбамоил]метил}-N-метокси-N-метилциклобутан-1-карбоксамида (70%, 2,62 г, 5,28 ммоль) в уксусной кислоте (25 мл) нагревали до температуры кипения с обратным холодильником в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении и остаток распределяли между насыщенным раствором NaHCO3 (водн.) (25 мл) и EtOAc (25 мл) и слои разделяли. Водный слой экстрагировали при помощи EtOAc (2×25 мл), объединенные органические слои промывали насыщенным солевым раствором (50 мл), сушили (MgSO4) и концентрировали. Это неочищенное вещество очищали колоночной флэш-хроматографией на силикагеле c элюированием при помощи 1-10% 2M NH3 в MeOH в DCM с получением указанного в заголовке соединения (2,02 г, 93%) в виде желтого смолистого вещества: MS (ESI+) для C19H27N3O2 m/z 330,5 [M+H]+; LC чистота 80% (УФ), 100% (ELS), (время удерживания, 1,51 мин);
1H ЯМР (500 МГц, ХЛОРОФОРМ-d) δH м.д. 7,56 (шир.с, 1H), 7,48 (д, J=8,5 Гц, 1H), 7,30 (дд, J=8,5, 1,7 Гц, 1H), 3,80-3,90 (м, 1H), 3,65 (с, 3H), 3,47-3,57 (м, 1H), 3,20 (с, 3H), 2,94-3,13 (м, 2H), 2,88 (дт, J=16,1, 8,1 Гц, 1H), 2,32-2,60 (м, 2H), 2,01-2,17 (м, 2H), 1,38 (с, 9H).
Стадия 3: 3-[(5-трет-бутил-1-{[2-(триметилсилил)этокси]метил}-1H-1,3-бензодиазол-2-ил)метил]-N-метокси-N-метилциклобутан-1-карбоксамид
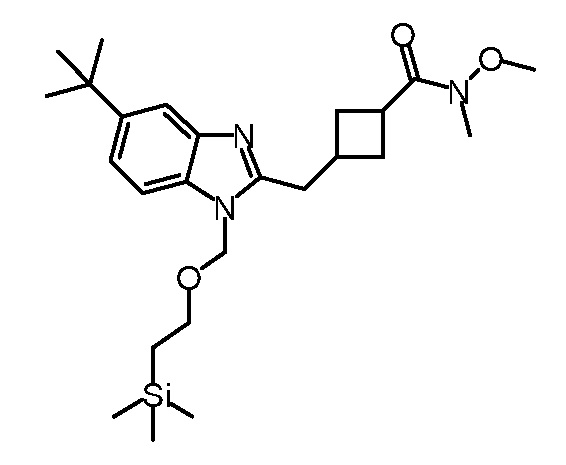
К раствору 3-[(5-трет-бутил-1H-1,3-бензодиазол-2-ил)метил]-N-метокси-N-метилциклобутан-1-карбоксамида (80%, 2,02 г, 4,91 ммоль) в N,N-диметилформамиде (40 мл) в атмосфере N2 добавляли карбонат калия (1,36 г, 9,81 ммоль) и реакционную смесь перемешивали в течение 1 часа. Медленно добавляли 2-(триметилсилил)этоксиметилхлорид (1,31 мл, 7,36 ммоль) и перемешивание поддерживали в течение 20 часов. Реакционную смесь фильтровали и концентрировали при пониженном давлении. Остаток брали для поглощения в EtOAc (50 мл) и промывали водой (3×50 мл), затем насыщенным солевым раствором (50 мл), затем сушили (MgSO4) и концентрировали. Неочищенное вещество очищали колоночной флэш-хроматографией на силикагеле c элюированием при помощи 50-100% EtOAc в гептане с получением указанного в заголовке соединения (1,21 г, 54%) в виде бесцветного смолистого вещества: MS (ESI+) для C25H41N3O3Si m/z 461,0 [M+H]+; LC чистота 97% (УФ), 100% (ELS), (время удерживания, 1,98 мин);
1H ЯМР (500 МГц, ХЛОРОФОРМ-d) δ м.д. 7,58-7,89 (м, 1H), 7,33 (с, 2H), 5,39-5,53 (м, 2H), 3,65 (с, 3H), 3,49-3,58 (м, 2H), 3,41 (шир.с, 1H), 3,10-3,23 (м, 3H), 3,03 (с, 3H), 2,32-2,63 (м, 2H), 2,00-2,18 (м, 2H), 1,32-1,45 (м, 9H), 0,91 (тд, J=8,1, 4,9 Гц, 2H), -0,13-0,07 (м, 9H).
Стадия 4: 3-[(5-трет-бутил-1-{[2-(триметилсилил)этокси]метил}-1H-1,3-бензодиазол-2-ил)метил]циклобутан-1-карбальдегид
К раствору 3-[(5-трет-бутил-1-{[2-(триметилсилил)этокси]метил}-1H-1,3-бензодиазол-2-ил)метил]-N-метокси-N-метилциклобутан-1-карбоксамида (0,61 г, 1,32 ммоль) в тетрагидрофуране (15 мл) добавляли по каплям раствор 1M диизобутилалюминийгидрида в толуоле (3,29 мл) в атмосфере N2 при -10°C. Реакционную смесь перемешивали при этой температуре в течение 2,5 часов, затем реакцию гасили путем добавления метанола (2 мл) и перемешивали в течение 5 минут. Раствор выливали в насыщенный водный раствор сегнетовой соли (20 мл), разбавляли при помощи Et2O (30 мл) и перемешивали в течение 30 минут. Смесь затем разделяли и органический слой промывали сегнетовой солью (30 мл), насыщенным раствором NaHCO3 (30 мл) и насыщенным солевым раствором (30 мл). Смесь сушили (MgSO4) и концентрировали с получением указанного в заголовке соединения (0,69 г, 130%) в виде бесцветного смолистого вещества, которое использовали неочищенным в следующей реакции.
Стадия 5: 7-[(3aS,4R,6R,6aR)-6-{[({3-[(5-трет-бутил-1-{[2-(триметилсилил)этокси]метил}-1H-1,3-бензодиазол-2-ил)метил]циклобутил}метил)(пропан-2-ил)амино]метил}-2,2-диметил-гексагидроциклопента[d][1,3]диоксол-4-ил]-7H-пирроло[2,3-d]пиримидин-4-амин
3-[(5-Трет-бутил-1-{[2-(триметилсилил)этокси]метил}-1H-1,3-бензодиазол-2-ил)метил]циклобутан-1-карбальдегид (282,3 мг, 0,7 ммоль), 7-[(3aS,4R,6R,6aR)-2,2-диметил-6-[(пропан-2-иламино)метил]-гексагидроциклопента[d][1,3]диоксол-4-ил]-7H-пирроло[2,3-d]пиримидин-4-амин (243,41 мг, 0,7 ммоль) и сульфат магния (127,22 мг, 1,06 ммоль) перемешивали в 1,2-дихлорэтане (10 мл) в течение 15 минут. Добавляли триацетоксиборогидрид натрия (179,21 мг, 0,85 ммоль) и реакционную смесь перемешивали в течение ночи. Реакцию гасили путем добавления насыщенного раствора Na2CO3 (10 мл) и раствор экстрагировали при помощи DCM (3×10 мл). Объединенные органические слои сушили (MgSO4) и концентрировали. Это неочищенное вещество очищали масс-направленной препаративной ВЭЖХ (кислотный метод). После объединения фракций добавляли небольшое количество 7M NH3 в MeOH для подщелачивания раствора. После концентрирования остаток распределяли между водой (5 мл) и DCM (5 мл) и слои разделяли. Водный слой экстрагировали при помощи DCM (2×3 мл) и объединенные органические слои сушили (MgSO4) и концентрировали с получением указанного в заголовке соединения (58,7 мг, 11%) в виде бесцветного смолистого вещества: MS (ESI+) для C41H63N7O3Si m/z 730,2 [M+H]+; ЖХ чистота 95% (УФ), 100% (ELS), (время удерживания, 1,65 мин);
1H ЯМР (500 МГц, ХЛОРОФОРМ-d) δH 8,30 (с, 1H), 7,75 (с, 1H), 7,32 (с, 2H), 7,04 (д, J=3,6 Гц, 1H), 6,37 (д, J=3,6 Гц, 1H), 5,40-5,50 (м, 2H), 5,24 (шир.с, 2H), 4,89-5,04 (м, 2H), 4,45 (д, J=5,2 Гц, 1H), 3,50 (с, 3H), 2,11-3,09 (м, 13H), 2,01 (с, 6H), 1,34-1,49 (м, 6H), 1,29 (с, 3H), 0,85-1,02 (м, 8H), -0,11-0,05 (м, 9H).
Стадия 6: (1R,2S,3R,5R)-3-{4-амино-7H-пирроло[2,3-d]пиримидин-7-ил}-5-{[({3-[(5-трет-бутил-1H-1,3-бензодиазол-2-ил)метил]циклобутил}метил)(пропан-2-ил)амино]метил}циклопентан-1,2-диол
7-[(3aS,4R,6R,6aR)-6-{[({3-[(5-трет-бутил-1-{[2-(триметилсилил)этокси]метил}-1H-1,3-бензодиазол-2-ил)метил]циклобутил}метил)(пропан-2-ил)амино]метил}-2,2-диметил-гексагидроциклопента[d][1,3]диоксол-4-ил]-7H-пирроло[2,3-d]пиримидин-4-амин (58,7 мг, 0,08 ммоль) растворяли в концентрированном растворе HCl (5 мл) и метаноле (5 мл) и нагревали до 40°C в течение 2 часоа. Реакционную смесь концентрировали при пониженном давлении и остаток распределяли между насыщенным раствором NaHCO3 (водн.) (10 мл) и EtOAc (10 мл). Слои разделяли и водный слой экстрагировали при помощи EtOAc (2×10 мл), объединенные органические слои затем сушили (MgSO4) и концентрировали. Это неочищенное вещество очищали методом препаративной ТСХ c элюированием при помощи 10% 2M NH3 в MeOH в DCM с получением указанного в заголовке соединения (24,7 мг, 55%) в виде бесцветного смолистого вещества: MS (ESI+) для C32H45N7O2 m/z 560,4 [M+H]+; ЖХ чистота 100% (УФ), (время удерживания, 5,10 мин);
1H ЯМР (500 МГц, Ацетон) δH 8,12 (с, 1H), 7,30-7,60 (м, 2H), 7,13-7,28 (м, 2H), 6,54 (д, J=3,5 Гц, 1H), 6,33 (шир.с, 1H), 4,85-5,07 (м, 1H), 4,36 (т, J=6,2 Гц, 1H), 4,08 (т, J=5,2 Гц, 1H), 3,01 (д, J=7,7 Гц, 1H), 2,93 (д, J=7,4 Гц, 2H), 2,78 (шир.с, 2H), 2,59-2,73 (м, 2H), 2,15-2,56 (м, 7H), 1,68 (дт, J=12,4, 9,7 Гц, 1H), 1,44-1,62 (м, 2H), 1,35 (с, 9H), 0,90-1,07 (м, 6H).
Соединение 23: (1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-(((3-(2-(5,6-дихлор-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(метил)амино)метил)циклопентан-1,2-диол
7-((3aS,4R,6R,6aR)-6-(((3-(2-(5,6-дихлор-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(метил)амино)метил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-N-(2,4-диметоксибензил)-7H-пирроло[2,3-d]пиримидин-4-амин
Раствор 3-(3-((((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)(метил)амино)циклобутил)пропановой кислоты (450 мг, 0,76 ммоль) и 4,5-дихлор-1,2-фенилендиамина (161 мг, 0,910 ммоль) в N,N-диметилформамиде (7,8 мл) обрабатывали по каплям N,N-диизопропилэтиламином (0,44 мл, 2,5 ммоль) с последующим добавлением гексафторфосфата N,N,N’,N’-тетраметил-0-(7-азабензотриазол-1-ил}урония (432 мг, 1,14 ммоль) одной порцией. Реакционную смесь перемешивали при комнатной температуре 5,5 часов. Реакционную смесь концентрировали в условиях высокого вакуума и остаток распределяли между 50 мл EtOAC и 50 мл смеси 1/1 H2O/насыщенный раствор NaHCO3. Водную фазу экстрагировали при помощи 30 мл EtOAc и объединенную органическую фазу промывали 30 мл порциями H2O и насыщенного солевого раствора. Органическую фазу сушили над Na2SO4, фильтровали и концентрировали с получением пенистого вещества. Это неочищенное вещество очищали флэш-хроматографией (SiO2, элюировали смесью 5% 7н NH3 в CH3OH/CH2Cl2) с получением промежуточного амидного соединения (в виде смеси амидных региоизомеров, 520 мг).
Промежуточный амид (0,52 г) в уксусной кислоте (16 мл) нагревали при 65°C в течение 5,5 часов, реакционную смесь охлаждали и помещали в условия высокого вакуума для удаления уксусной кислоты. Остаток брали для поглощения в 70 мл CH2Cl2 и промывали 50 мл порциями насыщенного раствора NaHCO3 и 2% раствора Na2CO3. Органическую фазу сушили над Na2SO4, фильтровали и концентрировали с получением пенистого вещества. Это вещество помещали в условия высокого вакуума и выдерживали в течение ночи и остаток очищали два раза флэш-хроматографией (SiO2 c элюированием смесью 4% 7NH NH3 в CH3OH/CH2Cl2, вторую колонку элюировали смесью 2-6% насыщенный водный раствор EtOH/NH3/CH2Cl2) с получением желаемого соединения (377 мг).
1H ЯМР (400 МГц, MeOD) δH м.д. 8,10 (с, 1H), 7,63 (д, J=1,66 Гц, 2H), 7,21 (м, 1H), 7,13 (д, J=8,29 Гц, 1H), 6,62 (д, J=3,32 Гц, 1H), 6,54 (д, J=2,07 Гц, 1H), 6,43 (дд, J=8,40, 2,38 Гц, 1H), 4,96 (м, 2H), 4,65 (с, 2H), 4,51 (м, 1H), 3,84 (д, J=1,04 Гц, 3H), 3,76 (с, 3H), 2,99 (м, 0,5H), 2,83 (м, 2H), 2,66 (м, 0,5H), 2,38 (м, 4H), 2,22 (м, 1H), 2,15 (д, J=7,67 Гц, 3H), 2,08 (м, 2H), 2,00 (м, 2H), 1,90 (м, 1H), 1,84 (м, 1H), 1,53 (с, 3H), 1,46 (м, 1H), 1,29 (с, 3H).
(1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-(((3-(2-(5,6-дихлор-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(метил)амино)метил)циклопентан-1,2-диол
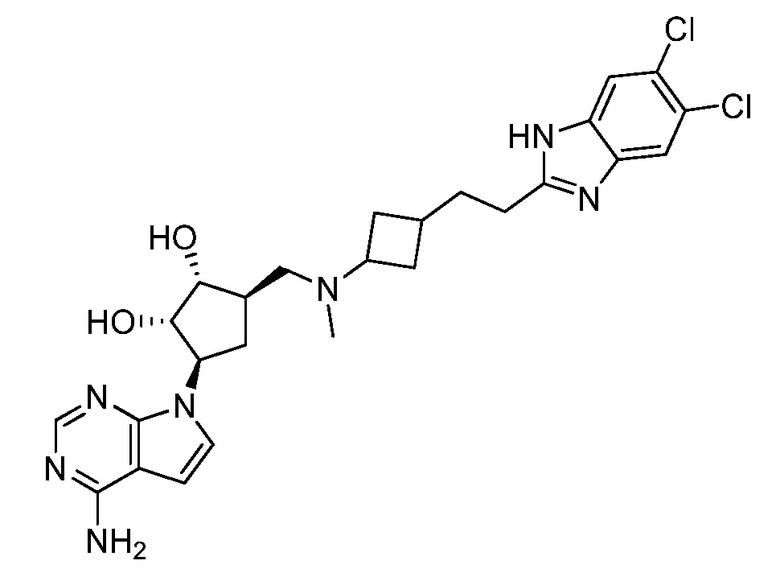
7-((3aS,4R,6R,6aR)-6-(((3-(2-(5,6-Дихлор-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(метил)амино)метил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-N-(2,4-диметоксибензил)-7H-пирроло[2,3-d]пиримидин-4-амин (377 мг, 0,513 ммоль) растворяли в смеси трифторуксусной кислоты (7,1 мл) и воды (0,8 мл), которая была предварительно охлаждена при 0°C на ледяной бане. Раствор перемешивали при 0°C в течение 30 минут, затем нагревали до комнатной температуры и перемешивание продолжали в течение 3 часов. Полученную суспензию концентрировали и остаток брали для поглощения в 15 мл MeOH и концентрировали. Эту процедуру повторяли два раза и остаток помещали в условия высокого вакуума. Это вещество брали для поглощения в 10 мл MeOH (происходило образование суспензии) и обрабатывали при помощи 500 мг K2CO3 и 0,2 мл воды. Смесь оставляли для перемешивания в течение 1,5 часов, в течение этого времени pH раствора достигал уровня ~9. Смесь фильтровали через тонкую фритту, твердые вещества промывали при помощи 20 мл MeOH и фильтрат концентрировали с получением не совсем белого твердого вещества. Это вещество оставляли в условиях высокого вакуума в течение ночи и очищали флэш-хроматографией (SiO2, c элюированием смесью 10-12% 7н NH3 в CH3OH/CH2Cl2) с получением желаемого продукта (227 мг).
1H ЯМР (400 МГц, MeOD) δH м.д. 8,06 (с, 1H), 7,63 (м, 2H), 7,21 (дд, J=3,63, 2,38 Гц, 1H), 6,59 (д, J=3,52 Гц, 1H), 4,32 (м, 1H), 3,88 (м, 1H), 3,01 (м, 0,5H), 2,84 (м, 2H), 2,70 (м, 0,5H), 2,51 (м, 1H), 2,40 (м, 2H), 2,26 (м, 2H), 2,17 (д, J=7,05 Гц, 3H), 2,11 (м, 2H), 2,02 (м, 1H), 1,90 (м, 3H), 1,62 (м, 1H), 1,49 (м, 1H).
Соединение 24: (1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-((метил(3-(2-(5-(трифторметокси)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)амино)метил)циклопентан-1,2-диол
N-(2-амино-4-(трифторметокси)фенил)-3-(3-((((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)(метил)амино)циклобутил)пропанамид
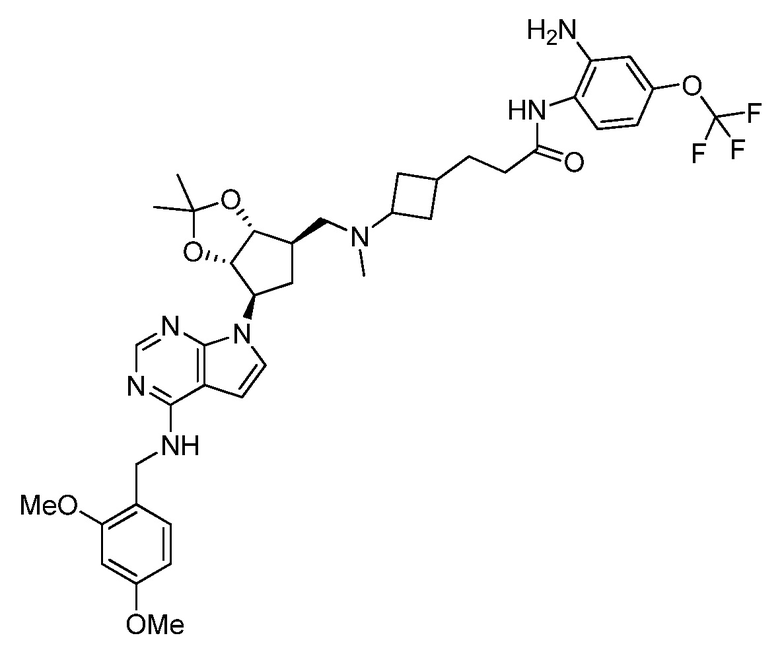
Гексафторфосфат N,N,N',N'-тетраметил-0-(7-азабензотриазол-1-ил)урония (1,2 г, 3,2 ммоль) добавляли к раствору 3-(3-((((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)(метил)амино)циклобутил)пропановой кислоты (1,25 г, 2,10 ммоль) и N,N-диизопропилэтиламина (1,2 мл, 6,9 ммоль) и 4-(трифторметокси)бензол-1,2-диамина (0,48 г, 2,5 ммоль) в N,N-диметилформамиде (10 мл). Смесь перемешивали в течение ночи при комнатной температуре, частично концентрировали примерно до 2 мл и затем добавляли NaHCO3 (насыщенный раствор). Смесь экстрагировали при помощи EtOAc (3×) и объединенные органические слои сушили при помощи MgSO4, фильтровали и концентрировали. Остаток очищали флэш-хроматографией (DCM/7н NH3 в MeOH 95:5) с получением желаемого соединения (2 г) в виде масла.
N-(2,4-диметоксибензил)-7-((3aS,4R,6R,6aR)-2,2-диметил-6-((метил(3-(2-(5-(трифторметокси)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)амино)метил)тетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-7H-пирроло[2,3-d]пиримидин-4-амин
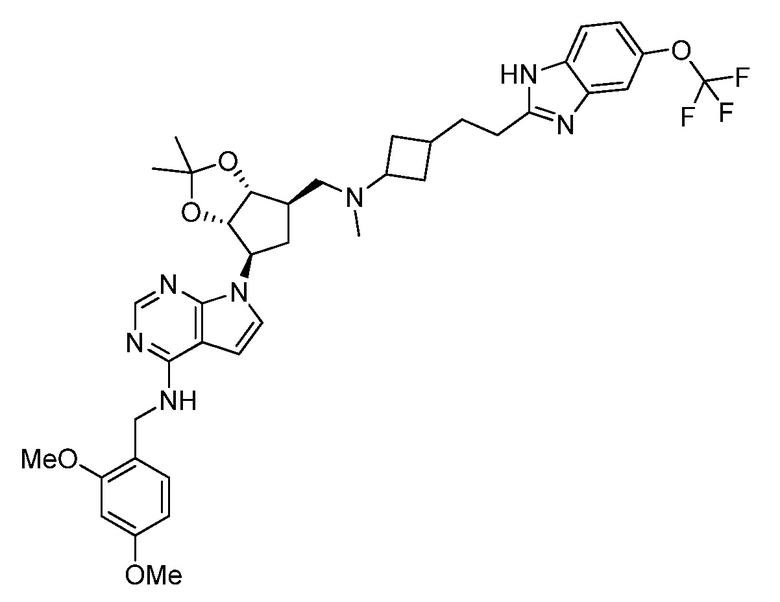
Раствор N-(2-амино-4-(трифторметокси)фенил)-3-(3-((((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)(метил)амино)циклобутил)пропанамида (2 г, 2 ммоль) в уксусной кислоте (4 мл) перемешивали в течение ночи при 60°C. Летучие вещества удаляли в вакууме и оставшийся остаток очищали флэш-хроматографией (DCM/7н NH3 в MeOH 92:8) с получением желаемого соединения (1 г) в виде твердого вещества.
(1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-((метил(3-(2-(5-(трифторметокси)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)амино)метил)циклопентан-1,2-диол
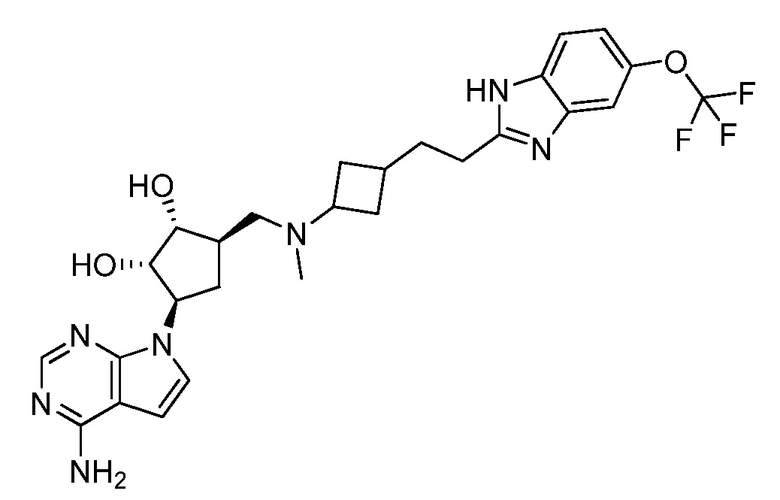
Трифторуксусную кислоту (20 мл) добавляли к смеси воды (2 мл) и N-(2,4-диметоксибензил)-7-((3aS,4R,6R,6aR)-2,2-диметил-6-((метил(3-(2-(5-(трифторметокси)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)амино)метил)тетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-7H-пирроло[2,3-d]пиримидин-4-амина (1 г, 1 ммоль) при комнатной температуре.
Реакционную смесь перемешивали в течение 1,5 часа, затем гасили триэтилсиланом (0,43 мл, 2,7 ммоль). Летучие вещества удаляли в вакууме и полученный остаток очищали два раза флэш-хроматографией (DCM/7н NH3 в MeOH 87:13) с получением желаемого продукта (0,28 г) в виде пенистого вещества. MS (ESI+) для C27H32F3N7O3 m/z 560,2 [M+H]+; MS (ESI-) для C27H32F3N7O3 m/z 558,2 [M-H]-; ВЭЖХ чистота >93% (время удерживания, 2,504 минут.)
1H ЯМР (400 МГц, d4-MeOH) δH 8,081 (с, 1H), 7,551-7,530 (м, 1H), 7,414 (с, 1H), 7,230-7,216 (м, 1H), 7,156-7,134 m, 1H), 6,621-6,613 (д, J=3,2 Гц, 1H), 4,362-4,326 (м, 1H), 3,952-3,915 (м, 1H), 3,237-3,182 (м, 0,5H (метин транс изомера), 2,927-2,857 (м, 2,5H (содержит метин цис изомера)), 2,697-2,646 (м, 1H), 2,590-2,515 (м, 1H), 2,479-2,408 (м, 1H), 2,343-2,305 (м, 5H), 2,210-2,174 (м, 2H), 2,086-1,949 (м, 4H), 1,721-1,545 (м, 2H).
Время удерживания: 2,52 мин. 1ВЭЖХ Условия: колонка Agilent Zorbax Exlipse XDB-C18, 4,6×50 мм (1,8 мкм наполнителя), Растворитель A- Вода (0,1% TFA), Растворитель B- Ацетонитрил (0,07% TFA) 6 мин. градиент от 5 до 95% B; 1 мин. удерживание; затем повторный цикл.
Соединение 25: (1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-(((3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(этил)амино)метил)циклопентан-1,2-диол
N-(2-амино-4-(трет-бутил)фенил)-3-(3-((((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)(этил)амино)циклобутил)пропанамид
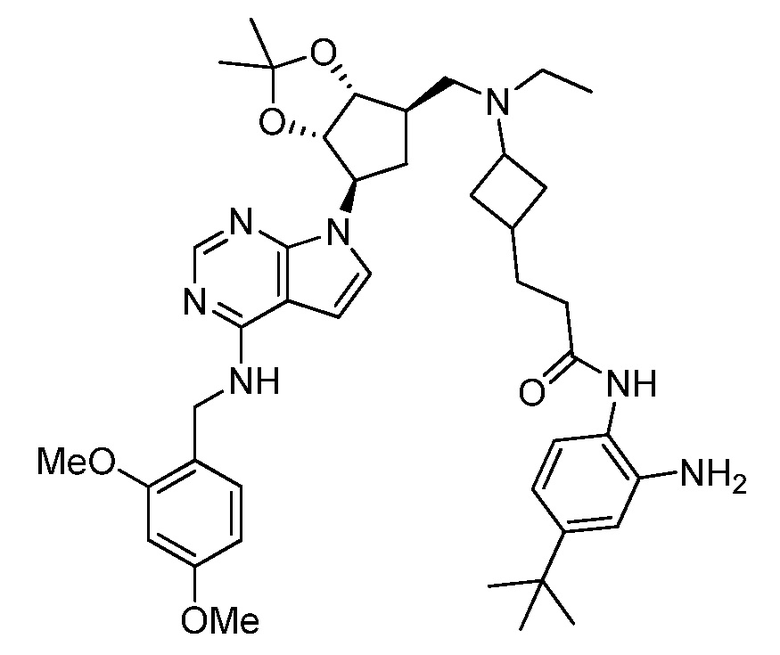
Гексафторфосфат N,N,N',N'-тетраметил-0-(7-азабензотриазол-1-ил)урония (0,84 г, 2,2 ммоль) добавляли к раствору 3-(3-((((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)(этил)амино)циклобутил)пропановой кислоты (0,89 г, 1,5 ммоль) и N,N-Диизопропилэтиламина (0,84 мл, 4,8 ммоль) и 4-трет-бутилбензол-1,2-диамин (0,29 г, 1,8 ммоль) в N,N-диметилформамиде (9 мл). Реакционную смесь перемешивали в течение ночи при комнатной температуре, частично концентрировали примерно до 2 мл и затем добавляли NaHCO3 (насыщенный раствор). Смесь экстрагировали при помощи EtOAc (3×) и объединенные органические слои сушили при помощи MgSO4, фильтровали и концентрировали. Остаток очищали флэш-хроматографией (DCM I 7н NH3 в MeOH 94:6) с получением желаемого соединения (0,88 г) в виде бесцветного твердого вещества. Время удерживания C: 3,363 минут.
7-((3aS,4R,6R,6aR)-6-(((3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(этил)амино)метил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-N-(2,4-диметоксибензил)-7H-пирроло[2,3-d]пиримидин-4-амин
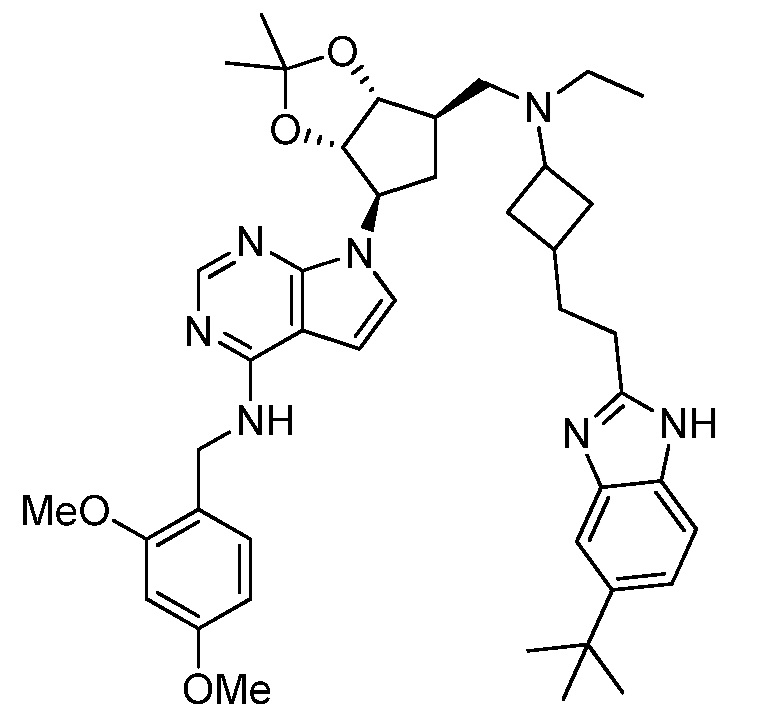
Раствор N-(2-амино-4-(трет-бутил)фенил)-3-(3-((((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)(этил)амино)циклобутил)пропанамида (0,88 г, 1,2 ммоль) в уксусной кислоте (3 мл) перемешивали в течение ночи при 60°C, летучие вещества удаляли в вакууме и оставшийся остаток очищали флэш-хроматографией (DCM/7н NH3 в MeOH 93:7) с получением желаемого соединения (0,85 г) в виде пенистого вещества.
(1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-(((3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(этил)амино)метил)циклопентан-1,2-диол
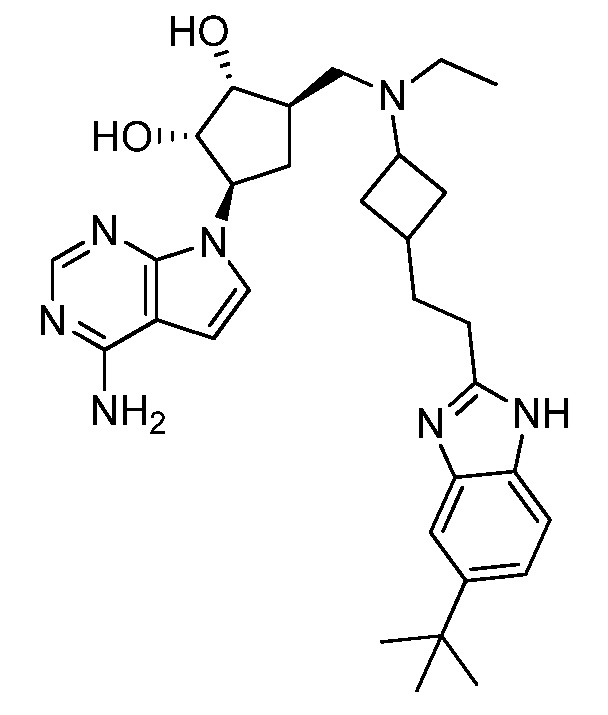
Трифторуксусную кислоту (20 мл) добавляли к смеси воды (2 мл) и 7-((3aS,4R,6R,6aR)-6-(((3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(этил)амино)метил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-N-(2,4-диметоксибензил)-7H-пирроло[2,3-d]пиримидин-4-амина (0,85 г, 1,2 ммоль) при комнатной температуре. Реакцию осуществляли в течение одного часа, в это время добавляли триэтилсилан (0,37 мл, 2,3 ммоль). Летучие вещества удаляли в вакууме и полученный остаток брали для поглощения в MeOH (3 млs), добавляли 1 мл K2CO3 (насыщенный раствор) и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Смесь распределяли между H2O и DCM/MeOH (9: 1). Водный слой экстрагировали (3×) и объединенные органические слои сушили при помощи MgSO4, фильтровали и концентрировали. Полученный остаток очищали флэш-хроматографией (DCM/7н NH3 в MeOH 90:10) с получением желаемого продукта (0,150 г) в виде не совсем белого пенистого вещества. MS (ESI+) для C31H43N7O2 m/z 546,3 [M+H]+; MS (ESI-) для C31H43N7O2 m/z 544,3 [M-H]-; ВЭЖХ чистота >91% (время удерживания, 2,734 минут).
1H ЯМР (400 МГц, d4-MeOH) δH 8,081 (с, 1H), 7,493 (с, 1H), 7,414-7,393 (м, 1H), 7,291-7,266 (м, 1H), 7,215-7,202 (м, 1H), 6,619-6,609 (д, J=4,0 Гц, 1H), 4,345-4,312 (м, 1H), 3,923-3,885 (м, 1H), 2,994-2,915 (м, 0,5 H (метин транс изомера)), 2,860-2,793 (м, 2H), 2,701-2,578 (м, 3H), 2,501-2,380 (м, 2H), 2,259-2,234 (м, 2H), 2,109- 2,008 (м, 3H), 1,920-1,880 (м, 3H), 1,658-1,499 (м, 2H), 1,364 (с, 9H), 1,036-0,991 (м, 3H).
Время удерживания: 2,734 минут. ВЭЖХ Условия: колонка Agilent Zorbax Exlipse XDB-C18, 4,6×50 мм (1,8 мкм наполнителя), Растворитель A- Вода (0,1% TFA), Растворитель B- Ацетонитрил (0,07% TFA). 6 мин. градиент от 5 до 95% B; 1 мин. удерживание; затем повторный цикл.
Соединение 26: (1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-(((3-(2-(5-бром-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(метил)амино)метил)циклопентан-1,2-диол
N-(2-амино-4-бромфенил)-3-(3-((((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)(метил)амино)циклобутил)пропанамид
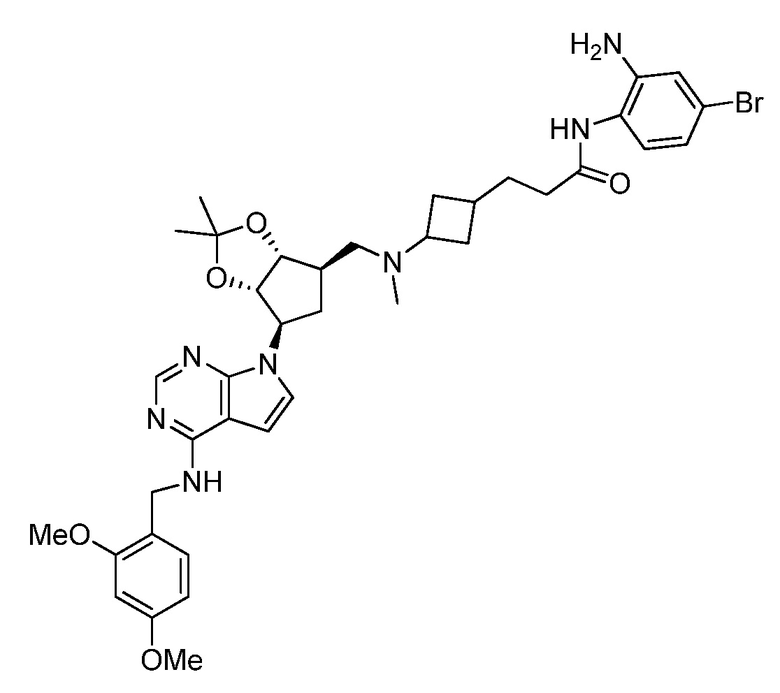
Гексафторфосфат N,N,N',N'-тетраметил-0-(7-азабензотриазол-1-ил)урония (1,20 г, 3,16 ммоль) добавляли к раствору 3-(3-((((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)(метил)амино)циклобутил)пропановой кислоты (1,25 г, 2,10 ммоль) и N,N-Диизопропилэтиламина (1,21 мл, 6,95 ммоль) и 4-бромбензол-1,2-диамина (0,472 г, 2,53 ммоль) в N,N-диметилформамиде (13,0 мл). Реакционную смесь перемешивали в течение ночи при комнатной температуре, частично концентрировали примерно до 2 мл и затем добавляли NaHCO3 (насыщенный раствор). Смесь экстрагировали при помощи EtOAc (3×) и объединенные органические слои сушили при помощи MgSO4, фильтровали и концентрировали. Остаток очищали флэш-хроматографией (DCM/7н NH3 в MeOH 95:5) с получением желаемого соединения (1,2 г) в виде твердого вещества.
7-((3aS,4R,6R,6aR)-6-(((3-(2-(5-бром-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(метил)амино)метил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-N-(2,4-диметоксибензил)-7H-пирроло[2,3-d]пиримидин-4-амин
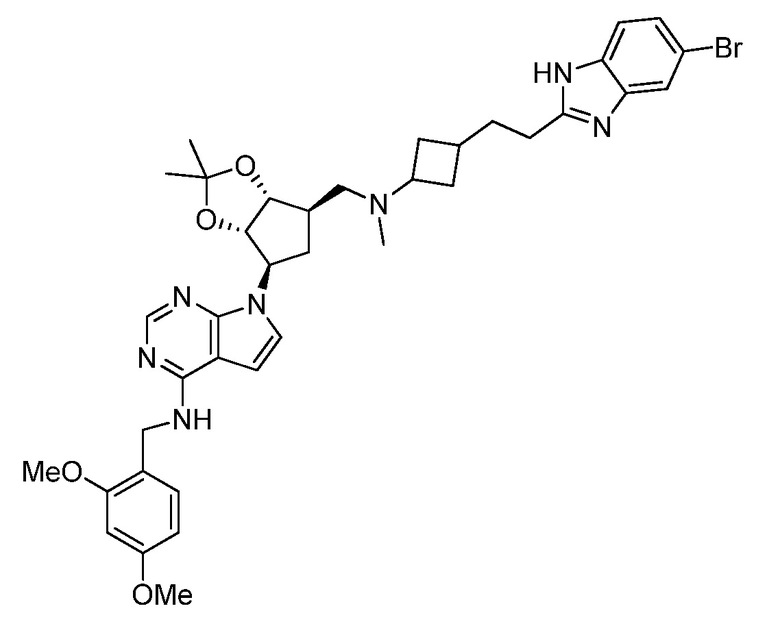
Раствор N-(2-амино-4-бромфенил)-3-(3-((((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)(метил)амино)циклобутил)пропанамида (1,2 г, 1,6 ммоль) в уксусной кислоте (4 мл, 70 ммоль) перемешивали в течение ночи при 60°C, летучие вещества удаляли в вакууме и оставшийся остаток очищали непосредственно флэш-хроматографией (DCM/7н NH3 в MeOH 91 :9) с получением (0,9 г) в виде пенистого вещества.
(1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-(((3-(2-(5-бром-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(метил)амино)метил)циклопентан-1,2-диол
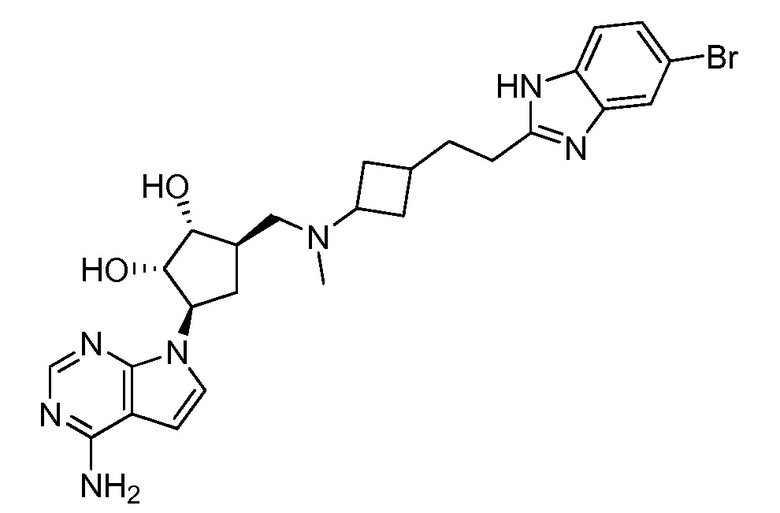
Трифторуксусную кислоту (20 мл) добавляли к смеси воды (2 мл) и 7-((3aS,4R,6R,6aR)-6-(((3-(2-(5-бром-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(метил)амино)метил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-N-(2,4-диметоксибензил)-7H-пирроло[2,3-d]пиримидин-4-амина (0,9 г, 1 ммоль) при комнатной температуре. Реакционную смесь перемешивали в течение одного часа, добавляли триэтилсилан (0,39 мл, 2,4 ммоль). Летучие вещества удаляли в вакууме и полученный остаток очищали два раза флэш-хроматографией (DCM/7н NH3 в MeOH 87:13). Остаток брали для поглощения в MeOH/H2O (5:0,5 мл) и добавляли K2CO3 (100 мг). Смесь перемешивали в течение 1 часа, затем концентрировали и очищали флэш-хроматографией (DCM/7н NH3 в MeOH 87:13) с получением желаемого продукта (0,15 г) в виде не совсем белого пенистого/смолистого вещества. циклобутил)(метил)амино)-метил)циклопентан-1,2-диол (0,15 г; 20%) в виде не совсем белого пенистого/смолистого вещества. MS (ESI+) для C26H32BrN7O2 m/z 554,1 [M+H]+; MS (ESI-) для C26H32BrN7O2 m/z 552,1 [M-H]-; ВЭЖХ чистота >90% (время удерживания, 2,298 минут).
1H ЯМР (400 МГц, d4-MeOH) δH 8,080 (с, 1H), 7,652 (с, 1H), 7,425-7,403 (м, 1H), 7,342-7,7,312 (м, 1H), 7,232-7,217 (м, 1H), 6,620-6,611 (д, J=3,6 Гц, 1H), 4,358-4,318 (м, 1H), 3,928-3,888 (м, 1H), 3,099-3,039 (м, 0,5H (метин транс изомера)), 2,898-2,829 (м, 2H), 2,777-2,726 (м, 0,5H (метин цис изомера)), 2,580-2,529 (м, 1H), 2,453-2,397 (м, 2H), 2,307 -2,141 (7H), 2,068-2,017 (м, 1H), 1,955-1,891 (м, 3H), 1,695-1,506 (м, 2H).
Соединение 27: (2R,3R,4S,5R)-2-(6-амино-9H-пурин-9-ил)-5-((изопропил(3-(2-(5-(1-метилциклобутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)амино)метил)тетрагидрофуран-3,4-диол
9-((3aR,4R,6R,6aR)-6-((изопропил(3-(2-(5-(1-метилциклобутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)-амино)метил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)-9H-пурин-6-амин
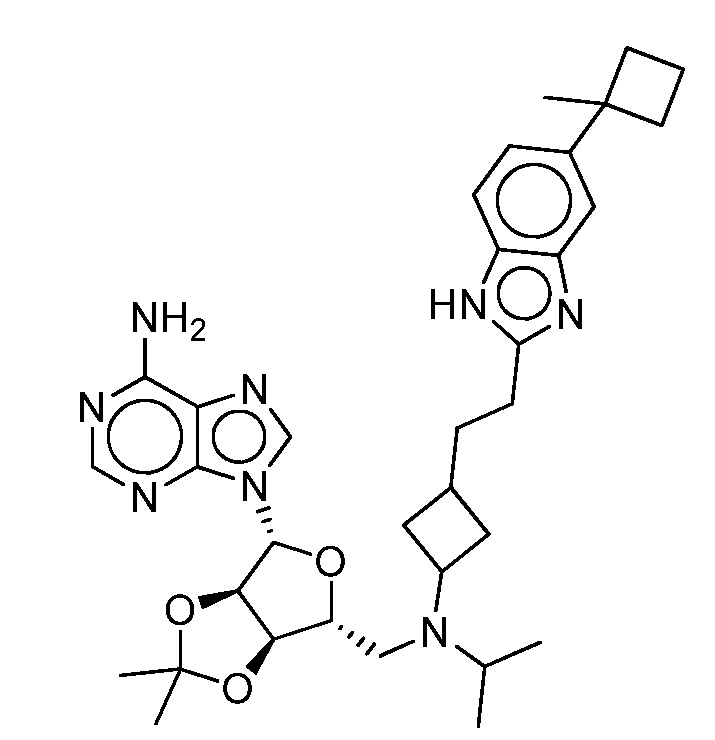
Раствор 5'-{[3-(2-карбоксиэтил)циклобутил](изопропил)амино}-5'-дезокси-2',3'-0-изопропилиденаденозина (0,463 г, 0,976 ммоль) и 4-(1-метилциклобутил)бензол-1,2-диамина (0,184 г, 1,04 ммоль) в N,N-диметилформамиде (10 мл,) охлаждали при 0°C.
Раствор обрабатывали N,N-диизопропилэтиламином (0,462 мл, 2,65 ммоль) по каплям с последующим добавлением гексафторфосфата N,N,N’,N’-тетраметил-0-(7-азабензотриазол-1-ил)урония (0,367 г, 0,965 ммоль) одной порцией. Смесь перемешивали при 0°C в течение 1 часа, затем медленно нагревали до комнатной температуры. После выдерживания в течение 5 часов при комнатной температуре реакционную смесь хранили в холодильнике в течение ночи, реакционную смесь помещали в условия высокого вакуума. Полученное стеклообразное вещество брали для поглощения в 30 мл H2O и экстрагировали 30 мл порцией смеси 10% MeOH/EtOAc. Водную фазу снова экстрагировали 30 мл порцией EtOAc. Объединенные органические фазы промывали 25 мл порциями насыщенного раствора NaHCO3 и насыщенного солевого раствора и сушили над Na2SO4. Смесь фильтровали и концентрировали с получением стеклообразного/пенистого вещества (700 мг). Это неочищенное вещество очищали флэш-хроматографией (SiO2, 4-5% 7н NH3 в MeOH/CH2C12) с получением промежуточного амидного соединения (~80% чистота, 390 мг).
Промежуточное амидное соединение (110 мг, 0,174 ммоль) брали для поглощения в 4 мл уксусной кислоты и раствор нагревали при 65°C. Реакционную смесь охлаждали и летучие вещества удаляли в условиях высокого вакуума с получением стеклообразного вещества. Неочищенный продукт брали для поглощения в 25 мл CH2Cl2 и промывали при помощи 20 мл насыщенного раствора NaHCO3 и 2% раствора Na2CO3, сушили над Na2SO4, фильтровали и концентрировали с получением стеклообразного вещества/жесткой пены. Это неочищенное вещество очищали методом препаративной ТСХ (SiO2, c элюированием смесью 7% 7н NH3 в CH3OH/CH2Cl2) с получением желаемого продукта в виде жесткой пены (54 мг). Описанную выше процедуру повторяли (за исключением того, что очистку осуществляли методом флэш-хроматографии, SiO2, элюировали смесью 4-5% 7н NH3 в CH3OH/CH2Cl2) на следующей партии промежуточного амидного соединения (389 мг) с получением дополнительных 368 мг желаемого соединения, которое объединяли с указанным выше бензимидазолом.
(2R,3R,4S,5R)-2-(6-амино-9H-пурин-9-ил)-5-((изопропил(3-(2-(5-(1-метилциклобутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)амино)метил)тетрагидрофуран-3,4-диол
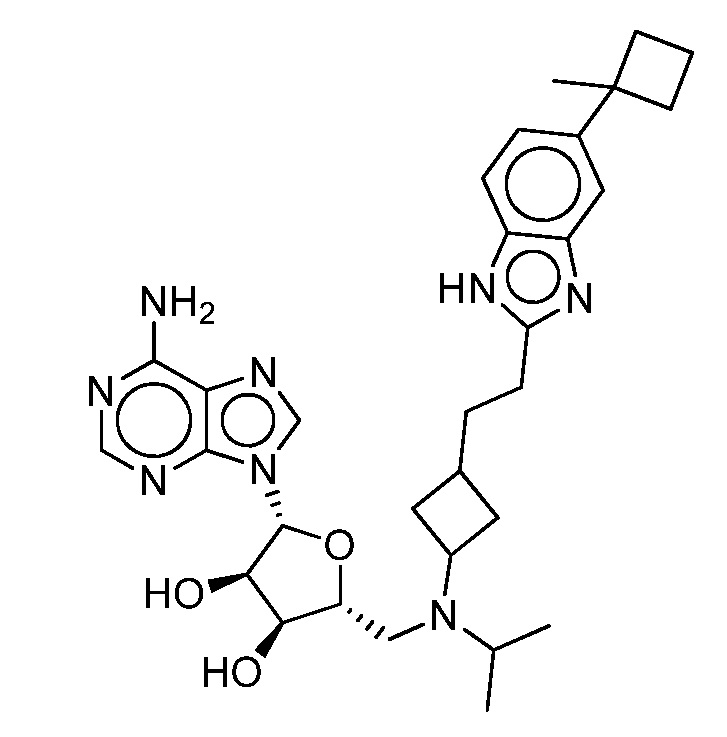
9-((3aR,4R,6R,6aR)-6-((изопропил(3-(2-(5-(1-метилциклобутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)амино)метил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)-9H-пурин-6-амин (422 мг, 0,686 ммоль) растворяли в смеси трифторуксусной кислоты (6,3 мл, 82 ммоль) и воды (0,7 мл, 40 ммоль), которая была предварительно охлаждена при 0°C на ледяной бане. Раствор перемешивали при 0°C в течение 30 минут, затем ледяную баню удаляли и смесь нагревали до комнатной температуры. Смесь перемешивали при комнатной температуре в течение 2,5 часов, затем остаток брали для поглощения в 12 мл MeOH, концентрировали досуха. Это повторяли два раза, полученное стеклообразное вещество помещали в условия высокого вакуума. Неочищенный остаток разбавляли при помощи 11 мл MeOH, обрабатывали при помощи 600 мг K2CO3 и 0,5 мл H2O и оставляли для перемешивания при комнатной температуре до тех пор, пока раствор не становился щелочным, как было определено при помощи pH бумаги. Смесь фильтровали и твердые вещества промывали при помощи 20 мл MeOH. Раствор концентрировали с получением остатка, который помещали в условия высокого вакуума. Это неочищенное вещество очищали флэш-хроматографией (SiO2, c элюированием смесью 10-12% 7н NH3 в CH3OH/CH2Cl2) с получением желаемого соединения в виде жесткой пены/стеклообразного вещества (256 мг).
1H ЯМР (400 МГц, MeOD) δH м.д. 8,30 (м, 1H), 8,20 (д, J=1,04 Гц, 1H), 7,38 (д, J=8,09 Гц, 1H), 7,22 (шир.с, 1H), 6,98 (дд, J=8,50, 1,66 Гц, 1H), 5,96 (м, 1H), 4,74 (т, J=4,87 Гц, 1H), 4,27 (д, J=3,11 Гц, 1H), 4,08 (м, 1H), 3,56 (м, 1H), 3,13 (м, 1H), 3,00 (м, 1H), 2,90 (дд, J=14,51, 4,35 Гц, 1H), 2,75 (м, 3H), 2,41 (м, 2H), 2,12 (м, 5H), 2,00 (м, 1H), 1,84 (м, 3H), 1,58 (м, 1H), 1,46 (с, 3H), 1,02 (м, 3H), 0,95 (д, J=6,63 Гц, 3H).
Соединение 28: (2R,3R,4S,5R)-2-(6-амино-9H-пурин-9-ил)-5-((изопропил((1r,3S)-3-(2-(5-(1-метилциклобутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)амино)метил)тетрагидрофуран-3,4-диол
Диастереомеры разделяли при помощи SFC. Вещество брали для поглощения в MeOH/H2O и лиофилизировали с получением белого порошка (132 мг).
1H ЯМР (400 МГц, MeOD) δ м.д. 8,30 (с, 1H), 8,20 (с, 1H), 7,38 (д, J=8,09 Гц, 1H), 7,22 (с, 1H), 6,99 (дд, J=8,40, 1,55 Гц, 1H), 5,96 (д, J=4,56 Гц, 1H), 4,73 (м, 1H), 4,26 (т, J=5,29 Гц, 1H), 4,07 (м, 1H), 3,13 (м, 1H), 3,00 (м, 1H), 2,90 (дд, J=14,41, 4,46 Гц, 1H), 2,76 (т, J=7,15 Гц, 2H), 2,70 (м, 1H), 2,42 (м, 2H), 2,18 (м, 2H), 2,11 (м, 3H), 1,85 (м, 4H), 1,57 (кв., J=8,85 Гц, 2H), 1,47 (с, 3H), 1,02 (д, J=6,84 Гц, 3H), 0,95 (д, J=6,63 Гц, 3H).
Соединение 29: (1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-((метил(3-(2-(5-(1-метилциклобутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)амино)метил)циклопентан-1,2-диол
(1-метилциклобутил)бензол
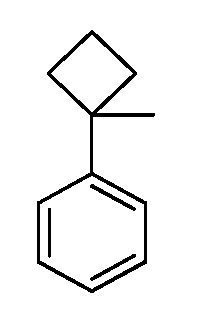
Перемешиваемую смесь бензола (5,0 мл, 56 ммоль) и серной кислоты (1,17 мл, 21,9 ммоль) охлаждали до 0°C и обрабатывали по каплям раствором метиленциклобутана (1,00 мл, 10,8 ммоль) в бензоле (3,0 мл, 34 ммоль) в течение 1 часа. После завершения добавления реакционную смесь перемешивали еще в течение 1 часа, нагревая ее при этом до комнатной температуры. Смесь экстрагировали при помощи 15 мл гексана. Органическую фазу промывали при помощи 10 мл H2O и 10 мл насыщенного раствора NaHCO3, сушили над Na2SO4, фильтровали и концентрировали с получением бесцветной жидкости. Жидкость очищали вакуумной дистилляцией (5-10 тор) с получением желаемого соединения в виде бесцветной жидкости. Первую фракцию собирали при 75-85°C в виде бесцветного жидкого продукта (330 мг).
1-(1-метилциклобутил)-4-нитробензол
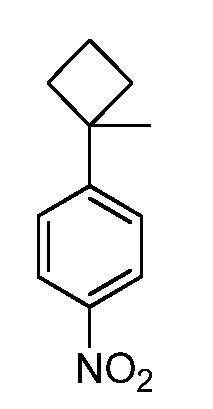
70% Азотную кислоту (7:3, азотная кислота:вода, 0,375 мл, 5,92 ммоль) добавляли по каплям в течение 60 минут к раствору (1-метилциклобутил)бензола (346 мг. 2,37 ммоль) в уксусном ангидриде (1,4 мл, 15 ммоль), охлажденному при 0°C. Температуру раствора поддерживали ниже 5°C в процессе добавления. После завершения добавления реакционную смесь перемешивали в течение 60 минут при олаждении. Реакционную смесь выливали в 40 мл ледяной воды и льду давали растаять. Водную фазу экстрагировали тремя ×20 мл порциями Et2O и объединенную органическую фазу промывали при помощи 25 мл H2O, затем двумя 20 мл порциями насыщенного раствора NaHCO3. Органическую фазу сушили над Na2SO4, фильтровали и концентрировали с получением светлого продукта в виде масла (418 мг), которое использовали как таковое на следующей стадии.
1H ЯМР (400 МГц, CDCI3) δ м.д. 8,16 (д, J=8,71 Гц, 2H), 7,29 (д, J=8,71 Гц, 2H), 2,41 (м, 2H), 2,15 (м, 3H), 1,88 (м, 1H), 1,48 (с, 3H)
4-(1-метилциклобутил)анилин
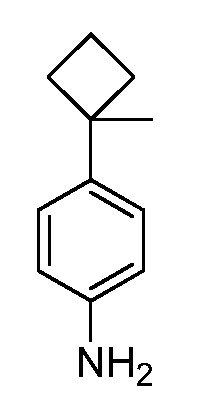
Раствор 1-(1-метилциклобутил)-4-нитробензола (708 мг, 3,70 ммоль) в этаноле (24 мл, 410 ммоль) осторожно обрабатывали при помощи 5% Pd на углероде (87 мг, 0,041 ммоль). Из реакционной колбы откачивали газ и колбу заполняли газообразным водородом три раза и реакционную смесь перемешивали в атмосфере водорода в течение 19 часов. Реакционную смесь фильтровали через слой solka floc® и этот слой промывали при помощи 25 мл EtOH. Растворитель удаляли с получением масла, которое быстро помещали в условия высокого вакуума, с получением желаемого соединения (609 мг), которое использовали непосредственно на следующей стадии.
1H ЯМР (400 МГЦ, CDCl3) δ м.д. 6,99 (м, 2H), 6,66 (м, 2H), 3,39 (шир.с, 2H), 2,35 (м, 2H), 2,05 (м, 3H), 1,82 (м, 1H), 1,42 (с, 3H).
2,2,2-трифтор-N-(4-(1-метилциклобутил)-2-нитрофенил)ацетамид
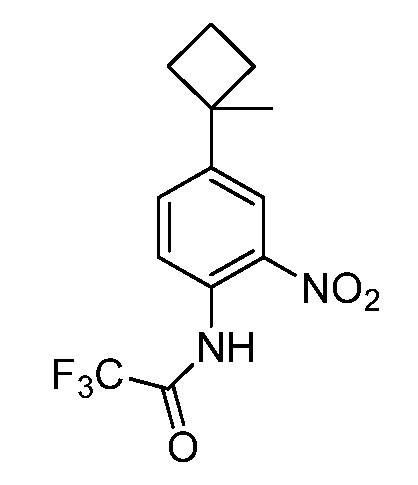
4-(1-Метилциклобутил)анилин (500 мг, 2,79 ммоль) и нитрат аммония (220 мг, 2,8 ммоль) обрабатывали трифторуксусным ангидридом (1,97 мл, 14,0 ммоль), затем хлороформом (10 мл, 120 ммоль). Реакционную смесь оставляли для перемешивания при комнатной температуре в течение периода до 5 часов, за это время все твердые вещества растворяли. Реакционную смесь выливали в 50 мл H2O и экстрагировали тремя 25 мл порциями CH2Cl2. Объединенную органическую фазу промывали при помощи 10 мл насыщенного раствора NaHCO3, сушили над Na2SO4, фильтровали и концентрировали с получением масла. Это неочищенное вещество очищали флэш-хроматографией (SiO2, c элюированием смесью 2,5-3,5% этилового эфира/гексан) с получением желаемого соединения (800 мг).
4-(1-метилциклобутил)-2-нитроанилин
Раствор 2,2,2-трифтор-N-[4-(1-метилциклобутил)-2-нитрофенил]ацетамида (580 мг, 1,9 ммоль) в метаноле (18 мл, 440 ммоль) обрабатывали раствором карбоната калия (788 мг, 5,70 ммоль) в воде (4,5 мл, 250 ммоль) и смесь нагревали при 45°C в течение 50 минут. Реакционную смесь охлаждали до комнатной температуры и метанол удаляли в вакууме. Оставшуюся водную фазу разбавляли при помощи 10 мл H2O и экстрагировали тремя 20 мл порциями EtOAc. Объединенную органическую фазу сушили над Na2SO4, фильтровали и концентрировали с получением масла. Это вещество помещали в условия высокого вакуума, где при отверждении оно давало желаемое соединение (400 мг). Это вещество использовали непосредственно на следующей стадии.
1H ЯМР (400 МГц, CDCl3) δ м.д. 7,90 (д, J=2,07 Гц, 1H), 7,23 (дд, J=8,50, 2,28 Гц, 1H), 6,77 (д, J=8,71 Гц, 1H), 5,96 (шир.с, 2), 2,33 (м, 2H), 2,13 (м, 1H), 2,04 (м, 2H), 1,84 (м, 1H), 1,43 (с, 3H).
4-(1-метилциклобутил)бензол-1,2-диамин
Раствор 4-(1-метилциклобутил)-2-нитроанилина (138 мг, 0,668 ммоль) в этаноле (8,5 мл, 140 ммоль) осторожно обрабатывали 10% палладием на углероде (14,2 мг, 0,0134 ммоль) в виде суспензии в этаноле. Из реакционной колбы откачивали газ и колбу заполняли газообразным водородом три раза и реакционную смесь перемешивали в атмосфере водорода в течение 4 часов. Реакционную смесь фильтровали через слой solka floc® и этот слой промывали при помощи 20 мл MeOH. Фильтрат концентрировали с получением масла, которое помещали в условия высокого вакуума, с получением желаемого соединения в виде твердого вещества (119 мг), которое использовали непосредственно на следующей стадии.
1H ЯМР (400 МГЦ, CDCl3) δH м.д. 6,66 (м, 1H), 6,53 (м, 2H), 3,34 (шир.с, 4H), 2,33 (м, 2H), 2,08 (м, 1H), 1,99 (м, 2H), 1,80 (м, 1H), 1,42 (с, 3H).
N-(2,4-диметоксибензил)-7-((3aS,4R,6R,6aR)-2,2-диметил-6-((метил(3-(2-(5-(1-метилциклобутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)амино)метил)тетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-7H-пирроло[2,3-d]пиримидин-4-амин
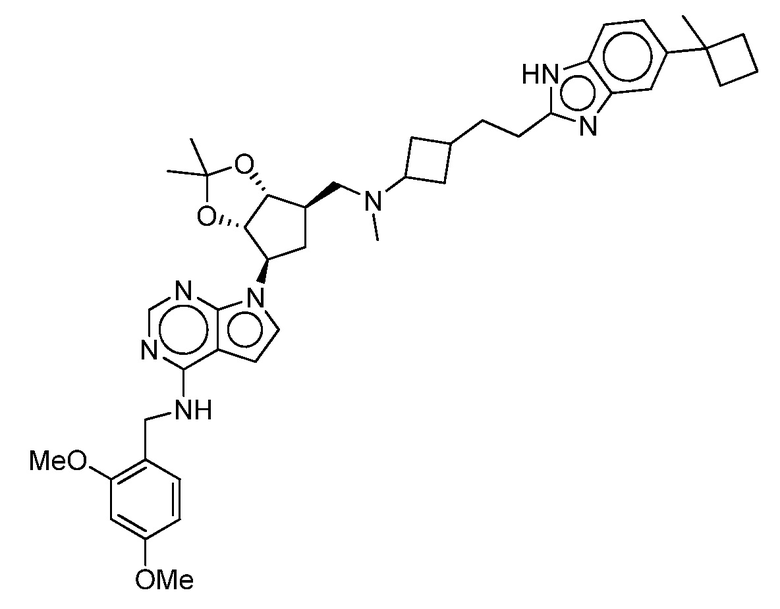
Раствор 3-(3-((((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)(метил)амино)циклобутил)пропановой кислоты (387 мг, 0,652 ммоль) и [8]4-(1-метилциклобутил)бензол-1,2-диамина (120 мг, 0,68 ммоль) в N,N-диметилформамиде (6,7 мл, 87 ммоль) обрабатывали по каплям N,N-диизопропилэтиламином (0,38 мл, 2,2 ммоль) с последующим добавлением гексафторфосфата N,N,N’,N’-тетраметил-0-(7-азабензотриазол-1-ил)урония (372 мг, 0,978 ммоль) одной порцией. Реакционную смесь перемешивали при комнатной температуре в течение 2,5 часов, реакционную смесь затем концентрировали в условиях высокого вакуума. Остаток распределяли между 40 мл EtOAc (добавляли некоторое количество CH2Cl2 для облегчения солюбилизации продукта) и 40 мл смеси 1/1 H2O/насыщенный раствор NaHCO3. Водную фазу экстрагировали при помощи 30 мл 1/1 смеси EA/CH2Cl2 и объединенную органическую фазу сушили над Na2SO4, фильтровали и концентрировали. Это неочищенное вещество очищали флэш-хроматографией (SiO2, c элюированием смесью 5-6% 7н NH3 в CH3OH/CH2Cl2) с получением желаемого промежуточного соединения в виде смеси амидных региоизомеров.
Промежуточное соединение брали для поглощения в уксусную кислоту (5,4 мл, 95 ммоль) и раствор нагревали при 65°C в течение 3 часов. Уксусную кислоту удаляли в условиях высокого вакуума с использованием теплой водяной бани. Неочищенный продукт брали для поглощения в 30 мл CH2Cl2 и органическую фазу промывали 10 мл порциями насыщенного раствора NaHCO3 и 2% раствора K2CO3, сушили над Na2SO4, фильтровали и концентрировали с получением стеклообразного вещества, которое образовывало пенистое вещество в условиях высокого вакуума. Это неочищенное вещество очищали флэш-хроматографией (SiO2, элюировали смесью 5% 7н NH3 в CH3OH/CH2Cl2, с получением желаемого продукта (140 мг).
(1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-((метил(3-(2-(5-(1-метилциклобутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)амино)метил)циклопентан-1,2-диол
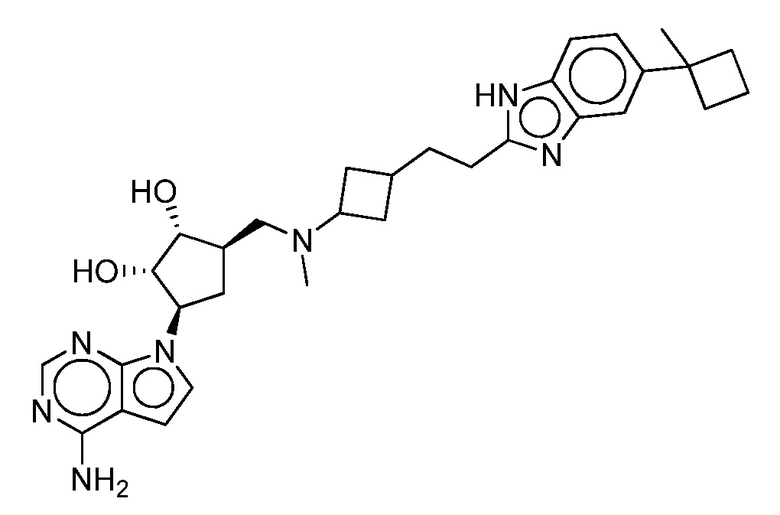
N-(2,4-диметоксибензил)-7-((3aS,4R,6R,6aR)-2,2-диметил-6-((метил(3-(2-(5-(1-метилциклобутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)амино)метил)тетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-7H-пирроло[2,3-d]пиримидин-4-амин (128 мг, 0,174 ммоль) растворяли в смеси трифторуксусной кислоты (3,60 мл, 46,7 ммоль) и воды (0,4 мл, 20 ммоль), которая была предварительно охлаждена при 0°C на ледяной бане. Раствор перемешивали при 0°C в течение 30 минут, затем ледяную баню удаляли и смесь нагревали до комнатной температуры, и эту температуру поддерживали еще в течение 2,5 часов. Реакционную смесь концентрировали в вакууме. Остаток брали для поглощения в 3 мл MeOH и концентрировали, этот способ повторяли два раза. Полученный белый остаток помещали в условия высокого вакуума. Неочищенный остаток объединяли с другой партией неочищенного вещества (полученной идентичным способом, ~1/3 количества использовали в этой реакции), разбавляли при помощи 5 мл MeOH, обрабатывали при помощи 140 мг K2CO3 и 10 капель H2O и оставляли для перемешивания при комнатной температуре до тех пор, пока раствор не становился щелочным, как было определено при помощи pH бумаги. Смесь фильтровали и твердые вещества промывали при помощи 15 мл MeOH. Раствор концентрировали с получением масла, которое помещали в условия высокого вакуума. Неочищенное вещество очищали флэш-хроматографией (SiO2, c элюированием смесью 10-15% 7н NH3 в CH3OH/CH2Cl2, с получением желаемого продукта в виде стеклообразного вещества/жесткой пены (68 мг).
1H ЯМР (400 МГц, MeOD) δH м.д. 8,06 (с, 1H), 7,39 (д, J=7,88 Гц, 1H), 7,23 (с, 1H), 7,21 (дд, J=3,63, 1,76 Гц, 1H), 6,99 (м, 1H), 6,60 (д, J=3,52 Гц, 1H), 4,93 (м, 1H), 4,32 (м, 1H), 3,89 (м, 1H), 3,03 (м, 1H), 2,83 (м, 2H), 2,70 (кв., J=8,15 Гц, 1H), 2,52 (м, 1H), 2,40 (м, 4H), 2,27 (м, 2H), 2,18 (д, J=6,22 Гц, 3H), 2,11 (м, 4H), 2,03 (м, 1H), 1,86 (м, 4H), 1,62 (м, 1H), 1,51 (м, 1H), 1,47 (с, 3H).
Соединение 30: (2R,3R,4S,5R)-2-(6-амино-9H-пурин-9-ил)-5-((метил((1r,3S)-3-(2-(5-(1-метилциклобутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)амино)метил)тетрагидрофуран-3,4-диол
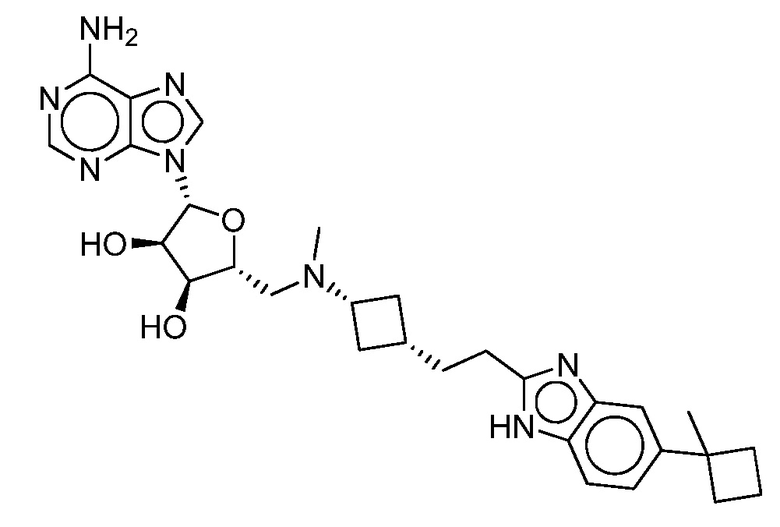
Диастереомеры разделяли при помощи SFC. Это вещество брали для поглощения в MeOH/H2O и лиофилизировали с получением белого порошка (67 мг).
1H ЯМР (400 МГц, MeOD) δH м.д. 8,27 (с, 1H), 8,20 (с, 1H), 7,38 (д, J=8,29 Гц, 1H), 7,22 (шир.с, 1H), 6,99 (дд, J=8,40, 1,55 Гц, 1H), 5,97 (д, J=4,15 Гц, 1H), 4,69 (дд, J=5,18, 4,15 Гц, 1H), 4,22 (т, J=5,60 Гц, 1H), 4,16 (м, 1H), 2,77 (м, 2H), 2,72 (д, J=8,09 Гц, 1H), 2,67 (м, 2H), 2,42 (м, 2H), 2,21 (м, 2H), 2,15 (с, 3H), 2,10 (м, 3H), 1,85 (м, 4H), 1,47 (с, 3H), 1,46 (м, 2H).
Соединение 31: (2R,3R,4S,5R)-2-(6-амино-9H-пурин-9-ил)-5-((изопропил((1s,3R)-3-(2-(5-(1-метилциклобутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)амино)метил)тетрагидрофуран-3,4-диол
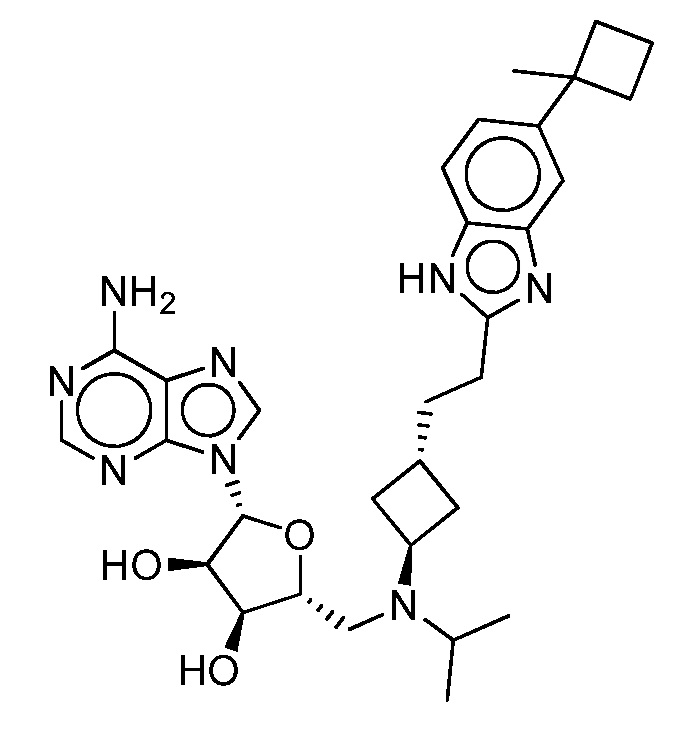
Диастереомеры разделяли при помощи SFC. Это вещество, как было определено, представляло собой транс диастереомер, согласно данным ЯМР. Это вещество брали для поглощения в MeOH/H2O и лиофилизировали с получением белого порошка (63 мг).
1H ЯМР (400 МГц, MeOD) δH м.д. 8,31 (с, 1H), 8,20 (с, 1H), 7,38 (д, J=8,29 Гц, 1H), 7,22 (с, 1H), 6,99 (дд, J=8,29, 1,66 Гц, 1H), 5,97 (д, J=4,56 Гц, 1H), 4,74 (м, 1H), 4,27 (т, J=5,39 Гц, 1H), 4,09 (м, 1H), 3,53 (м, 1H), 3,01 (м, 1H), 2,93 (дд, J=14,72, 4,35 Гц, 1H), 2,80 (т, J=7,46 Гц, 2H), 2,73 (дд, J=1 4,51, 7,46 Гц, 1H), 2,42 (м, 2H), 2,13 (м, 5H), 2,01 (м, 3H), 1,82 (м, 3H), 1,47 (с, 3H), 1,02 (д, J=6,63 Гц, 3H), 0,95 (д, J=6,63 Гц, 3H).
Соединение 32: (1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-((((1r,3S)-3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(метил)амино)метил)циклопентан-1,2-диол
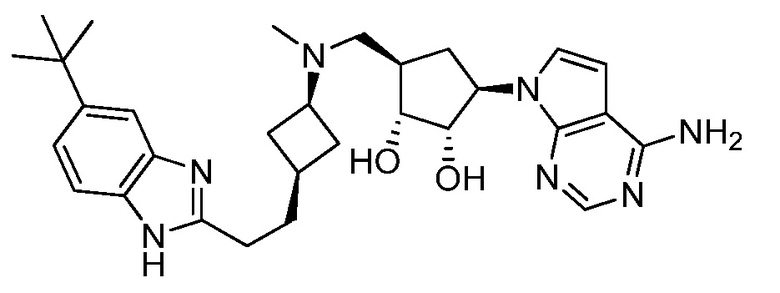
Диастереоизомеры разделяли при помощи SFC. (условия указаны ниже) с получением 120 мг. Препаративный метод: IC (2×15 см), 35% изопропанола(0,2% DEA))/CO2, 100 бар, 60 мл/мин 220 нм., объем вводимой пробы: 0,75 мл, 4 мг/мл метанола Пик 1: 5,27 минут
1H ЯМР (400 МГц, d4-MeOH) δH 8,080 (с, 1H), 7,495 (с, 1H), 7,417-7,395 (м, 1H), 7,303-7,281 (м, 1H), 7,220-7,212 (м, 1H), 6,619-6,610 (м, 1H), 4,349-4,315 (м, 1H), 2,837-2,802 (м, 2H), 2,718-2,641 (м, 1H), 2,508-2,365 (м, 3H), 2,284-2,258 (м, 3H), 2,156 (с, 3H), 1,954-1,906 (м, 1H), 1,549-1,460 (м, 2H), 1,375 (с, 9H).
Соединение 33: (2R,3R,4S,5R)-2-(6-амино-9H-пурин-9-ил)-5-((метил(3-(2-(5-(1-метилциклобутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)амино)метил)тетрагидрофуран-3,4-диол
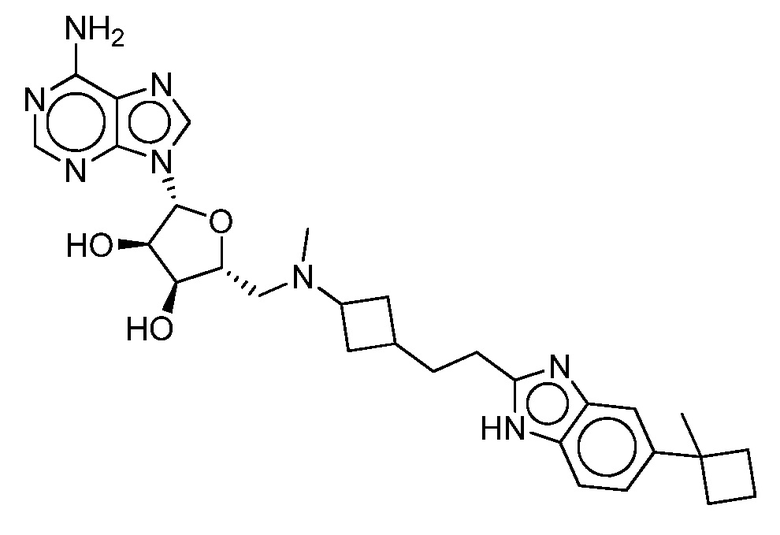
9-((3aR,4R,6R,6aR)-2,2-диметил-6-((метил(3-(2-(5-(1-метилциклобутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)амино)метил)тетрагидрофуро[3,4-d][1,3]диоксол-4-ил)-9H-пурин-6-амин
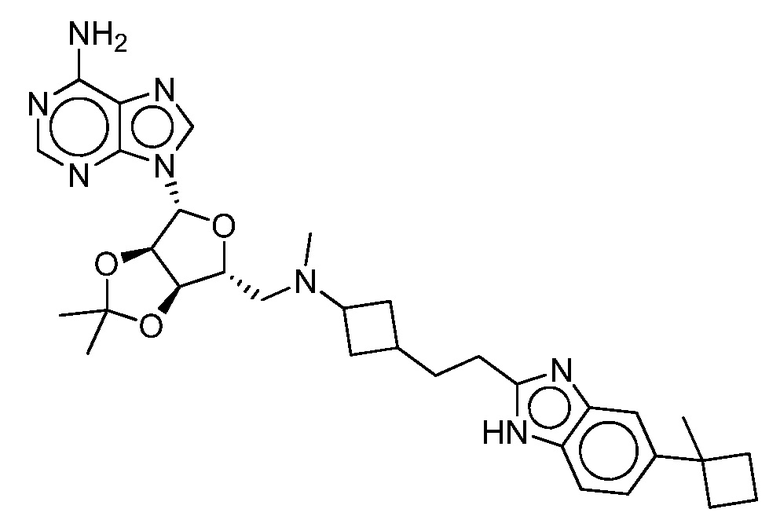
Раствор 3-(3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(метил)амино)циклобутил)пропановой кислоты (0,461 г, 1,03 ммоль) и 4-(1-метилциклобутил)бензол-1,2-диамина (0,150 г, 0,851 ммоль) в N,N-диметилформамиде (11 мл, 140 ммоль) охлаждали при 0°C. Раствор обрабатывали по каплям N,N-диизопропилэтиламином (0,489 мл, 2,81 ммоль) с последующим добавлением гексафторфосфата N,N,N’,N’-тетраметил-0-(7-азабензотриазол-1-ил)урония (0,388 г, 1,02 ммоль) одной порцией. Смесь перемешивали при 0°C в течение 30 минут, затем медленно нагревали до комнатной температуры, перемешивание продолжали при комнатной температуре в течение 6 часов. Реакционную смесь разбавляли при помощи 30 мл H2O и экстрагировали 25 мл порциями 10% MeOH/EtOAc. Водную фазу снова экстрагировали двумя 20 мл порциями EtOAc. Объединенные органические фазы промывали 25 мл порциями насыщенного раствора NaHCO3 и насыщенного солевого раствора и сушили над Na2SO4. Раствор фильтровали и концентрировали с получением стеклообразного вещества. Это неочищенное вещество очищали флэш-хроматографией (SiO2, c элюированием смесью 5% 7н NH3 в MeOH/CH2C12.
Промежуточное амидное соединение брали для поглощения в уксусную кислоту (7,0 мл, 120 ммоль) и раствор нагревали при 65°C в течение 2,5 часов, реакционную смесь охлаждали и уксусную кислоту удаляли в условиях высокого вакуума с получением стеклообразного вещества. Неочищенный продукт брали для поглощения в 25 мл CH2Cl2 и промывали при помощи 20 мл насыщенного раствора NaHCO3, 2% раствора Na2CO3, сушили над Na2SO4, фильтровали и концентрировали в вакууме с получением стеклообразного вещества/жесткой пены. Это неочищенное вещество очищали флэш-хроматографией (SiO2, c элюированием смесью 5-7% 7н NH3 в CH3OH/CH2Cl2) с получением желаемого соединения (214 мг).
(2R,3R,4S,5R)-2-(6-амино-9H-пурин-9-ил)-5-((метил(3-(2-(5-(1-метилциклобутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)амино)метил)тетрагидрофуран-3,4-диол
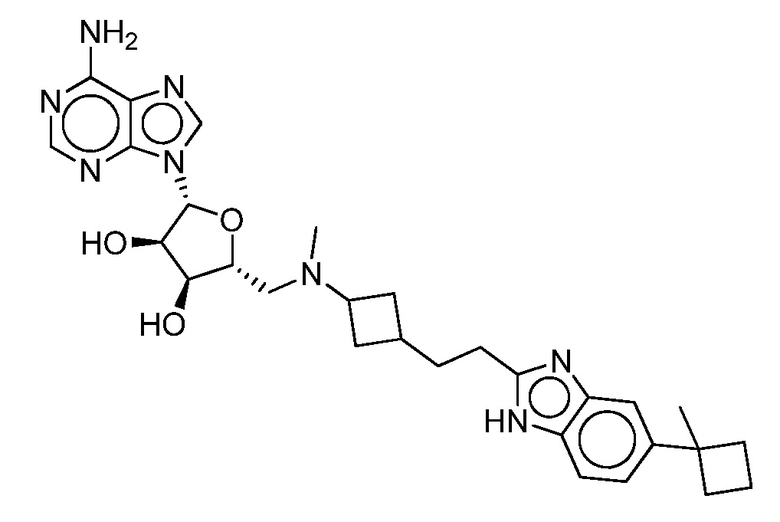
9-((3aR,4R,6R,6aR)-2,2-диметил-6-((метил(3-(2-(5-(1-метилциклобутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)амино)метил)тетрагидрофуро[3,4-d][1,3]диоксол-4-ил)-9H-пурин-6-амин (188 мг, 0,320 ммоль) растворяли в смеси трифторуксусной кислоты (4,00 мл, 51,9 ммоль) и воды (0,4 мл, 20 ммоль), которая была предварительно охлаждена при 0°C на ледяной бане. Раствор перемешивали при 0°C. Реакционную смесь перемешивали в течение 30 минут при 0°C, затем ледяную баню удаляли и смесь нагревали до комнатной температуры, при этом перемешивание продолжали еще в течение 2 часов. Реакционную смесь концентрировали в вакууме. Остаток брали для поглощения в 10 мл MeOH и концентрировали и способ повторяли два раза. Полученное стеклообразное вещество помещали в условия высокого вакуума и выдерживали в течение 1 часа. Неочищенный остаток разбавляли при помощи 7 мл MeOH, обрабатывали при помощи 150 мг K2CO3 и 10 капель H2O и оставляли для перемешивания при комнатной температуре до тех пор, пока раствор не становился щелочным, как это определяли при помощи pH бумаги. Смесь фильтровали и твердые вещества промывали при помощи 10 мл MeOH. Раствор концентрировали с получением остатка, который помещали в условия высокого вакуума. Это неочищенное вещество очищали флэш-хроматографией (SiO2, c элюированием смесью 10-15% 7н NH3 в CH3OH/CH2Cl2) с получением желаемого соединения в виде стеклообразного вещества/жесткой пены (66%).
1H ЯМР (400 МГц, MeOD) δH м.д. 8,28 (м, 1H), 8,20 (м, 1H), 7,38 (д, J=8,29 Гц, 1H), 7,23 (с, 1H), 7,00 (дд, J=8,40, 1,55 Гц, 1H), 5,98 (т, J=3,21 Гц, 1H), 4,70 (м, 1H), 4,24 (кв., J=5,18 Гц, 1H), 4,17 (м, 1H), 3,10 (м, 0,4H), 2,80 (м, 3H), 2,71 (д, J=5,60 Гц, 2H), 2,43 (м, 2H), 2,23 (дд, J=11,71, 6,12 Гц, 1H), 2,19 (м, 3H), 2,12 (м, 4H), 1,99 (м, 1H), 1,85 (м, 4H), 1,48 (с, 3H), 1,48 (м, 1H).
Соединение 34: (2R,3R,4S,5R)-2-(6-амино-9H-пурин-9-ил)-5-((метил((1s,3R)-3-(2-(5-(1-метилциклобутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)амино)метил)тетрагидрофуран-3,4-диол
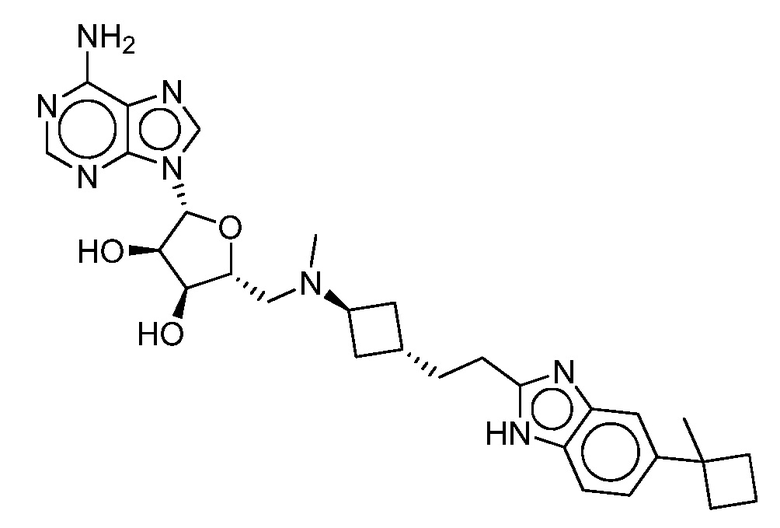
Диастереомеры разделяли при помощи SFC. Это вещество брали для поглощения в MeOH/H2O и лиофилизировали с получением белого порошка (30 мг).
1H ЯМР (400 МГц, MeOD) δH м.д. 8,28 (с, 1H), 8,19 (с, J=4,15 Гц, 1H), 4,69 (м, 1H), 4,23 (т, J=5,49 Гц, 1H), 4,17 (м, 1H), 3,07 (м, 1H), 2,81 (т, J=7,57 Гц, 2H), 2,68 (м, 2H), 2,42 (м, 2H), 2,17 (с, 3H), 2,09 (м, 6H), 1,97 (м, 2H), 1,84 (м, 3H), 1,47 (с, 3H).
35: (1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-((((1s,3R)-3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(метил)амино)метил)циклопентан-1,2-диол
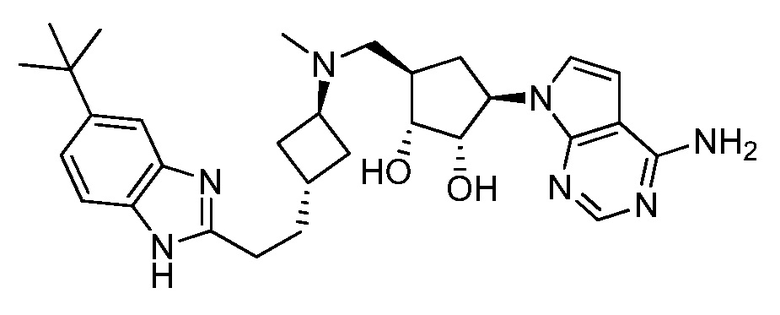
Диастереоизомеры разделяли при помощи SFC. Осуществляли следующее SFC разделение (120 мг).
Препаративный метод: IC (2×15 см), 35% изопропанола(0,2% DEA))/CO2, 100 бар, 60 мл/мин 220 нм, объем вводимой пробы: 0,75 мл, 4 мг/мл метанола. Пик 2: 6,24 минут.
1H ЯМР (400 МГц, d4-MeOH) δH 8,078 (с, 1H), 7,501 (с, 1H), 7,422-7,401 (м, 1H), 7,310-7,285 (м, 1H), 7,228-7,219 (м, 1H), 6,618-6,609 (м, 1H), 4,355-4,320 (м, 1H), 3,053-2,977 (м, 1H), 2,874-2,836 (м, 2H), 2,535-2,268 (м, 4H), 2,177-2,003 (м, 8H), 1,909-1,869 (м, 2H), 1,677-1,595 (м, 1H), 1,381 (с, 9H).
Соединение 36: (1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-((метил((1r,3S)-3-(2-(5-(1-метилциклобутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)амино)метил)циклопентан-1,2-диол
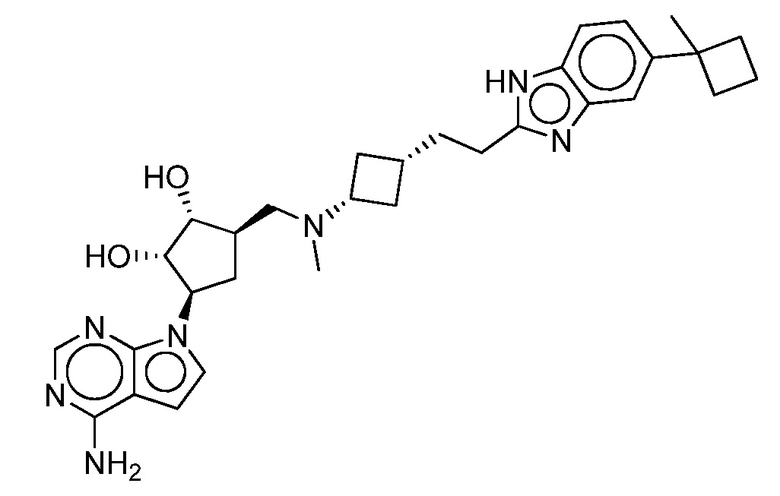
Диастереоизомеры разделяли при помощи SFC. Это вещество брали для поглощения в MeOH/H2O и лиофилизировали с получением белого твердого вещества (15 мг).
1H ЯМР (400 МГц, MeOD) δH м.д. 8,06 (с, 1H), 7,38 (д, J=8,09 Гц, 1H), 7,23 (шир.с, 1H), 7,20 (д, J=3,32 Гц, 1H), 6,99 (м, 1H), 6,60 (д, J=3,52 Гц, 1H), 4,95 (м, 1H), 4,31 (т, J=6,74 Гц, 1H), 3,88 (м, 1H), 2,81 (м, 2H), 2,66 (м, 1H), 2,40 (м, 5H), 2,25 (м, 3H), 2,14 (шир.с, 3H), 2,11 (м, 3H), 1,91 (м, 2H), 1,83 (м, 1H), 1,61 (м, 1H), 1,51 (м, 1H), 1,47 (с, 3H).
Соединение 37: (1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-((метил((1r,3S)-3-(2-(5-(трифторметокси)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)амино)метил)циклопентан-1,2-диол
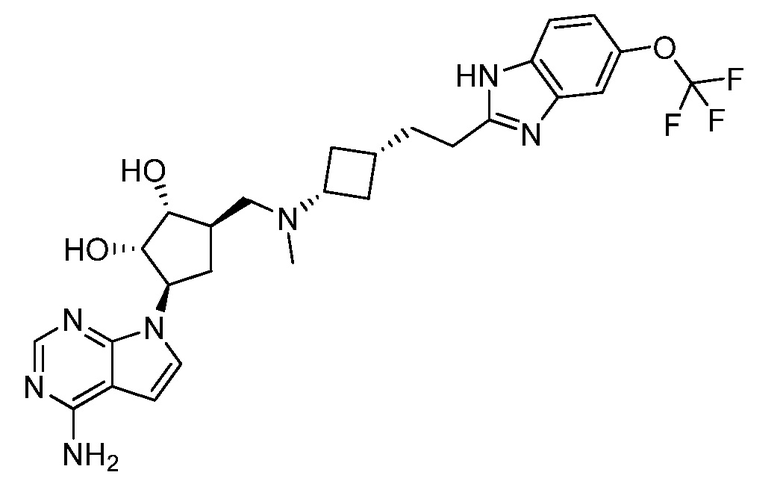
Диастереоизомеры разделяли при помощи SFC. Лиофилизация давала желаемый продукт в виде бесцветного твердого вещества (0,060 г).
1H ЯМР (400 МГц, d4-MeOH) δH 8,079 (с, 1H), 7,546-7,524 (м, 1H), 7,409 (с, 1H), 7,226-7,217 (м, 1H), 7,150-7,124 (м, 1H), 6,619-6,610 (м, 1H), 4,355-4,320 (м, 1H), 3,912-3,885 (м, 1H), 2,881-2,845 (м, 2H), 2,727-2,674 (м, 1H), 2,538-2,262 (м, 6H), 2,172 (с, 3H), 1,955-1,916 (м, 3H), 1,670-1,492 (м, 3H).
Соединение 38: (1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-((((1r,3S)-3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(этил)амино)метил)циклопентан-1,2-диол
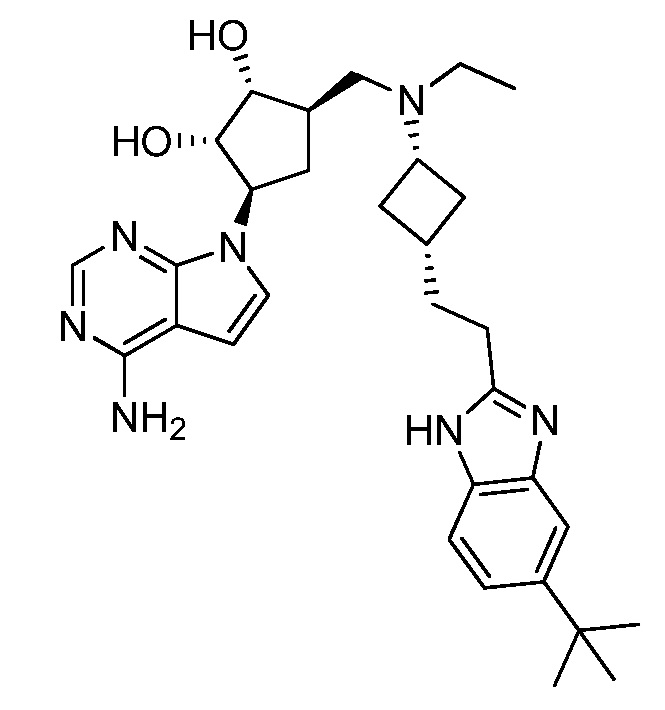
Диастереоизомеры разделяли при помощи SFC. После лиофилизации желаемый продукт выделяли в виде бесцветного твердого вещества (32 мг).
Препаративный метод: Lux-3 (2×15 см), 30% этанола (0,2% DEA)/CO2, 100 бар, 65 мл/мин 220 нм, объем вводимой пробы: 0,4 мл, 6,2 мг/мл метанола.
1H ЯМР (400 МГц, d4-MeOH) δH 8,082 (с, 1H), 7,493 (с, 1H), 7,415-7,393 (м, 1H), 7,295-7,270 (м, 1H), 7,215-7,206 (м, 1H), 6,621-6,612 (м, 1H), 4,343-4,309 (м, 1H), 3,924-3,897 (м, 1H), 3,044-2,962 (м, 1H), 2,834-2,798 (м, 2H), 2,728-2,695 (м, 1H), 2,660-2,607 (м, 2H), 2,543-2,380 (м, 2H), 2,281-2,257 (м, 3H), 1,932-1,906 (м, 3H), 1,660-1,523 (м, 3H), 1,368 (с, 9H), 1,050-1,014 (т, J=7,2 Гц, 3H).
Соединение 39: (1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-((метил((1s,3R)-3-(2-(5-(1-метилциклобутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)амино)метил)циклопентан-1,2-диол
Диастереоизомеры разделяли при помощи SFC. Это вещество брали для поглощения в MeOH/H2O и лиофилизировали с получением белого порошка (19 мг).
1H ЯМР (400 МГц, MeOD) δH м.д. 8,06 (с, 1H), 7,39 (д, J=8,50 Гц, 1H), 7,21 (м, 2H), 6,99 (дд, J=8,29, 1,45 Гц, 1H), 6,59 (д, J=3,52 Гц, 1H), 4,94 (м, 1H), 4,32 (дд, J=7,77, 5,91 Гц, 1H), 3,89 (м, 1H), 3,00 (м, 1H), 2,83 (т, J=7,57 Гц, 2H), 2,49 (м, 1H), 2,38 (м, 4H), 2,23 (м, 1H), 2,16 (с, 3H), 2,11 (м, 5H), 2,01 (м, 2H), 1,84 (м, 3H), 1,61 (м, 1H), 1,46 (с, 3H).
Соединение 40: (1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-((((1r,3S)-3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(циклопропилметил)амино)метил)циклопентан-1,2-диол
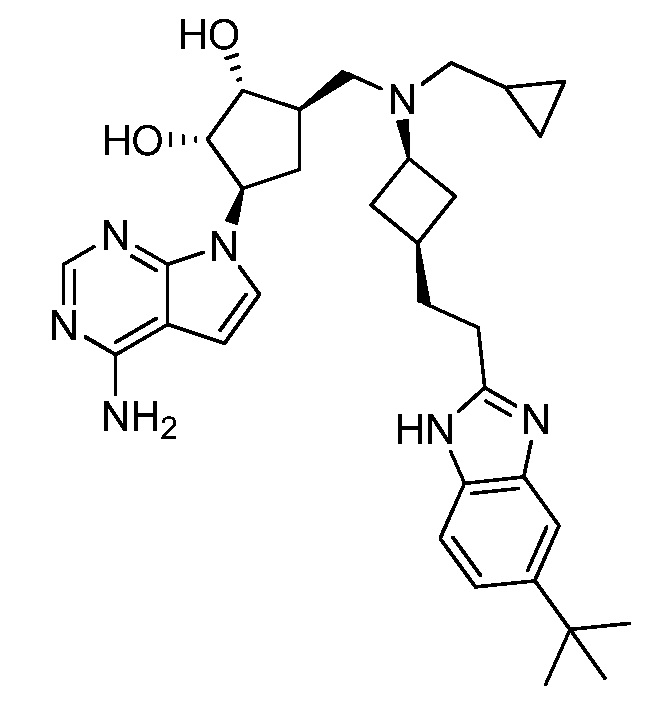
Диастереоизомеры разделяли при помощи SFC. Это вещество брали для поглощения в MeOH/H2O и лиофилизировали с получением белого порошка (45 мг).
1H ЯМР (400 МГц, MeOD) δH м.д. 8,06 (с, 1H), 7,48 (шир.с, 1H), 7,39 (д, J=8,29 Гц, 1H), 7,27 (м, 1H), 7,20 (д, J=3,73 Гц, 1H), 6,60 (д, J=3,52 Гц, 1H), 4,32 (дд, J=7,36, 6,12 Гц, 1H), 3,90 (м, 1H), 3,06 (м, 1H), 2,81 (т, J=6,84 Гц, 2H), 2,74 (м, 1H), 2,55 (дд, J=12,75, 7,77 Гц, 1H), 2,41 (м, 1H), 2,37 (д, J=6,84 Гц, 2H), 2,29 (м, 3H), 1,91 (м, 3H), 1,60 (м, 1H), 1,50 (м, 2H), 1,36 (с, 9H), 0,87 (м, 1H), 0,48 (д, J=8,09 Гц, 2H), 0,10 (м, 2H).
Соединение 41: (1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-((((1r,3S)-3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(изопропил)амино)метил)циклопентан-1,2-диол
Диастереоизомеры разделяли при помощи SFC с последующей лиофилизацией из H2O/MeOH/CH3CN с получением белого порошка (100 мг).
1H ЯМР (400 МГц, MeOD) δH м.д. 8,06 (с, 1H), 7,48 (шир.с, 1H), 7,39 (м, 1H), 7,27 (м, 1H), 7,20 (д, J=3,52 Гц, 1H), 6,60 (м, 1H), 4,32 (т, J=6,43 Гц, 1H), 3,93 (т, J=5,29 Гц, 1H), 3,54 (м, 0,2H), 3,11 (т, J=9,33 Гц, 1H), 3,02 (м, 1H), 2,82 (м, 2H), 2,66 (дд, J=13,68, 8,09 Гц, 1H), 2,46 (м, 1H), 2,36 (м, 1H), 2,23 (м, 3H), 2,05 (м, 1H), 1,91 (м, 3H), 1,59 (м, 3H), 1,36 (с, 9H), 1,02 (м, 6H).
Соединение 42: (1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-((((1r,3S)-3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(циклобутилметил)-амино)метил)циклопентан-1,2-диол
Стадия 1: этил 3-((1S,3r)-3-((циклобутилметил)-(((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)амино)циклобутил)пропаноат
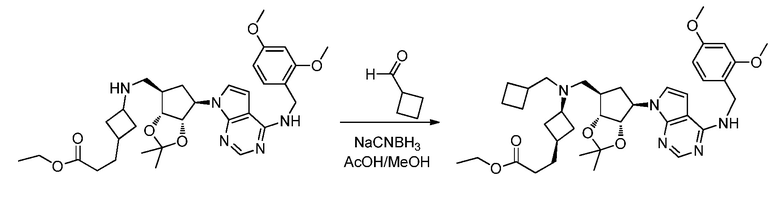
Амин этил 3-[3-({[(3aR,4R,6R,6aS)-6-{4-[(2,4-диметоксибензил)амино]-7H-пирроло[2,3-d]пиримидин-7-ил}-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил]метил}амино)циклобутил]пропаноат (1,8 г, 3,0 ммоль) брали для поглощения в метанол (20 мл, 600 ммоль) и добавляли цианоборогидрид натрия (0,37 г, 5,9 ммоль). pH доводили до приблизительно 6 с использованием 10% раствора AcOH в метаноле, затем добавляли одной порцией циклобутанкарбоксальдегид (0,32 г, 3,8 ммоль). Реакцию осуществляли в течение 5 часов, к этому времени ВЭЖХ анализ показал, что реакция остановилась. Добавляли еще 1,3 эквивалента циклобутанкарбоксальдегида, и реакция продолжалась в течение ночи. К реакционной смеси добавляли NaHCO3 (насыщенный раствор), затем экстрагировали 3 раза при помощи DCM. Объединенные органические слои сушили при помощи MgSO4 и концентрировали с получением желтого смолистого вещества. Цис и транс- изомеры можно было разделить на диоксиде кремния. Осуществляли очистку жидкостной хроматографией (DCM/7н NH3 в MeOH 96:4) с получением 2 отдельных партий продукта, каждая из которых была обогащена одним соответствующим изомером до около 90%. Верхний изомер: 0,38 г (11:1 смесь, цис). Нижний изомер: 0,31 г (6:1 смесь, транс). MS (ESI+) для C35H49N5O6 m/z 676,7 [M+H]+; ВЭЖХ чистота > 69% (время удерживания, 3,791).
Стадия 2: N-(2-амино-5-(трет-бутил)фенил)-3-((1S,3r)-3-((циклобутилметил)(((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)амино)циклобутил)пропанамид
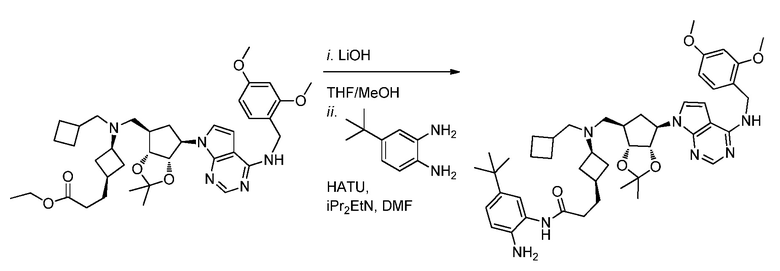
Верхний изомер (цис): Гидроксид лития моногидрат (0,236 г, 5,62 ммоль) добавляли к раствору этил 3-((1S,3r)-3-((циклобутилметил)(((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)амино)циклобутил)пропаноата (6 мл, 70 ммоль) и метанола (1,5 мл, 37 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре, и к следующему утру исходное вещество было израсходовано и преобразовано в кислоту. Реакционную смесь подкисляли при помощи 1н раствора HCl до pH=6. Летучие вещества удаляли в вакууме и оставшуюся воду удаляли путем азеотропной перегонки с этанолом с последующей лиофилизацией в течение 72 часов. Полученное не совсем белое твердое вещество использовали без дополнительной очистки. Время удерживания: 3,330 минут MS (ESI+) для C36H49N5O6 m/z 648,4 [M+H]+; MS (ESI-) для C36H49N5O6 m/z 646,4 [M-H]-; ВЭЖХ чистота >97% (время удерживания, 3,329).
Гексафторфосфат N,N,N',N'-тетраметил-O-(7-азабензотриазол-1-ил)урония (0,334 г, 0,880 ммоль) добавляли к раствору 3-{цис-3-[(циклобутилметил){[(3aR,4R,6R,6aS)-6-{4-[(2,4-диметоксибензил)-амино]-7H-пирроло[2,3-d]пиримидин-7-ил}-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил]метил}амино]циклобутил}-пропановой кислоты (0,38 г, 0,59 ммоль) и N,N-диизопропилэтиламина (0,337 мл, 1,94 ммоль) и 4-трет-бутилбензол-1,2-диамина (0,116 г, 0,704 ммоль) в N,N-диметилформамиде (3,63 мл, 46,9 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре, и к следующему утру исходное вещество было израсходовано. Реакционную смесь частично концентрировали примерно до 2 мл и затем добавляли NaHCO3 (насыщенный раствор). Смесь экстрагировали при помощи EtOAc 3 раза и объединенные органические слои сушили при помощи MgSO4 и концентрировали. Полученный остаток очищали жидкостной хроматографией (DCM/7н NH3 в MeOH 95:5) с получением N-(2-амино-5-(трет-бутил)фенил)-3-((1S,3r)-3-((циклобутилметил)(((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)амино)циклобутил)пропанамида (0,30 г; 64%) в виде фиолетово-коричневого аморфного твердого вещества. ВЭЖХ чистота >19% (время удерживания, 3,574 минут).
Стадия 3: 7-((3aS,4R,6R,6aR)-6-((((1r,3S)-3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)-(циклобутилметил)амино)метил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-N-(2,4-диметоксибензил)-7H-пирроло[2,3-d]пиримидин-4-амин
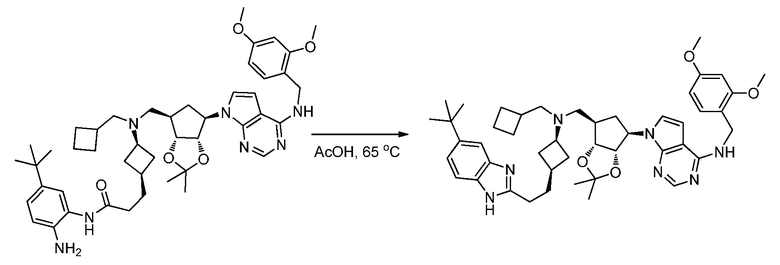
Раствор N-(2-амино-4-трет-бутилфенил)-3-{цис-3-[(циклобутилметил){[(3aR,4R,6R,6aS)-6-{4-[(2,4-диметоксибензил)амино]-7H-пирроло[2,3-d]пиримидин-7-ил}-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил]метил}амино]циклобутил}пропанамида (0,3 г, 0,4 ммоль) в уксусной кислоте (1,0 мл, 20 ммоль) перемешивали в течение ночи при 65oC, и к следующему утру исходное вещество было израсходовано. Летучие вещества удаляли в вакууме и полученный остаток очищали жидкостной хроматографией (DCM/7н NH3 в MeOH 93:7) с получением 7-((3aS,4R,6R,6aR)-6-((((1r,3S)-3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)-(циклобутилметил)амино)метил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-N-(2,4-диметоксибензил)-7H-пирроло[2,3-d]пиримидин-4-амина в виде не совсем белого твердого вещества. MS (ESI+) для C46H61N7O4 m/z 777,7 [M+H]+; ВЭЖХ чистота >64% (время удерживания, 3,690 минут).
Стадия 4: (1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-((((1r,3S)-3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(циклобутилметил)- амино)метил)циклопентан-1,2-диол
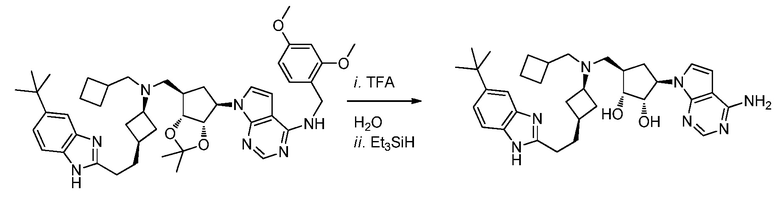
Трифторуксусную кислоту (5 мл, 60 ммоль) добавляли к смеси воды (0,5 мл, 20 ммоль) и 7-((3aS,4R,6R,6aR)-6-((((1r,3S)-3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)-(циклобутилметил)амино)метил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-N-(2,4-диметоксибензил)-7H-пирроло[2,3-d]пиримидин-4-амина (0,2 г, 0,2 ммоль) при комнатной температуре. Реакцию осуществляли в течение ночи, затем ярко-розовую суспензию гасили триэтилсиланом (0,082 мл, 0,52 ммоль). Летучие вещества удаляли в вакууме и полученный остаток брали для поглощения в метанол (15 млs). Добавляли 500 мг K2CO3 и 8 капель H2O и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Смесь фильтровали и фильтровальную лепешку промывали 10 мл метанола. Фильтрат концентрировали и полученный остаток очищали жидкостной хроматографией (DCM/7н NH3 в MeOH 90:10) с получением (1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-((((1r,3S)-3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(циклобутилметил)амино)метил)циклопентан-1,2-диола (0,037 г; 20%) в виде бесцветного твердого вещества. MS (ESI+) для C34H47N7O2 m/z 586,3 [M+H]+; ВЭЖХ чистота >89% (время удерживания 2,970 минут).
1H ЯМР (400 МГц, d4-MeOH) δH 8,083 (с, 1H), 7,498 (с, 1H), 7,417-7,396 (м, 1H), 7,302-7,277 (м, 1H), 7,206-7,197 (м, 1H), 6,621-6,612 (м, 1H), 4,347-4,314 (м, 1H), 3,912-3,885 (м, 1H), 2,973-2,922 (м, 1H), 2,836-2,800 (м, 2H), 2,662-2,366 (м, 6H), 2,282-2,241 (м, 3H), 2,061-2,034 (м, 2H), 1,912-1,494 (м, 10H), 1,374 (с, 9H).
Соединение 43: (1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-(((3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(циклобутил)амино)метил)циклопентан-1,2-диол
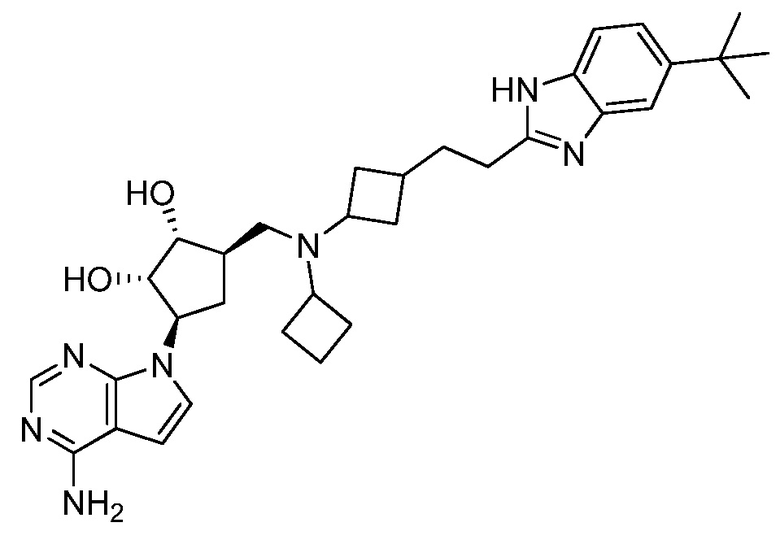
этил 3-(3-(циклобутил(((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)амино)циклобутил)пропаноат
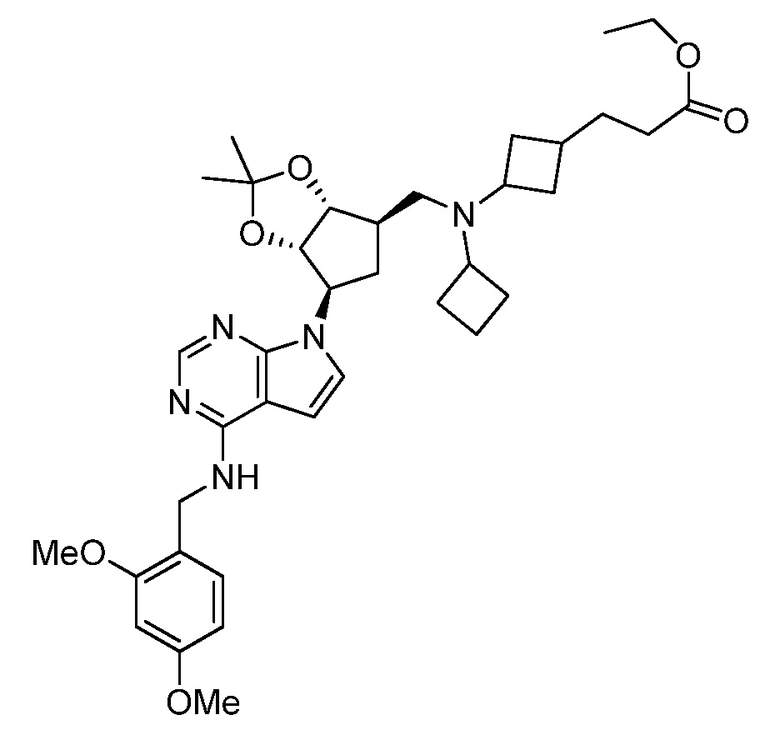
Этил 3-(3-((((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)амино)циклобутил)пропаноат (0,84 г, 1,4 ммоль) брали для поглощения в метанол (10 мл) и добавляли цианоборогидрид натрия (0,087 г, 1,4 ммоль). pH доводили до приблизительно 6 с использованием 10% раствора AcOH в MeOH, затем добавляли одной порцией циклобутанон (0,15 мл, 2,1 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 дней. К реакционной смеси добавляли NaHCO3 (насыщенный раствор), затем экстрагировали (3×) при помощи DCM. Объединенные органические слои сушили при помощи MgSO4, фильтровали и концентрировали. Полученное вещество использовали без дополнительной очистки.
3-(3-(циклобутил(((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)амино)циклобутил)пропановая кислота
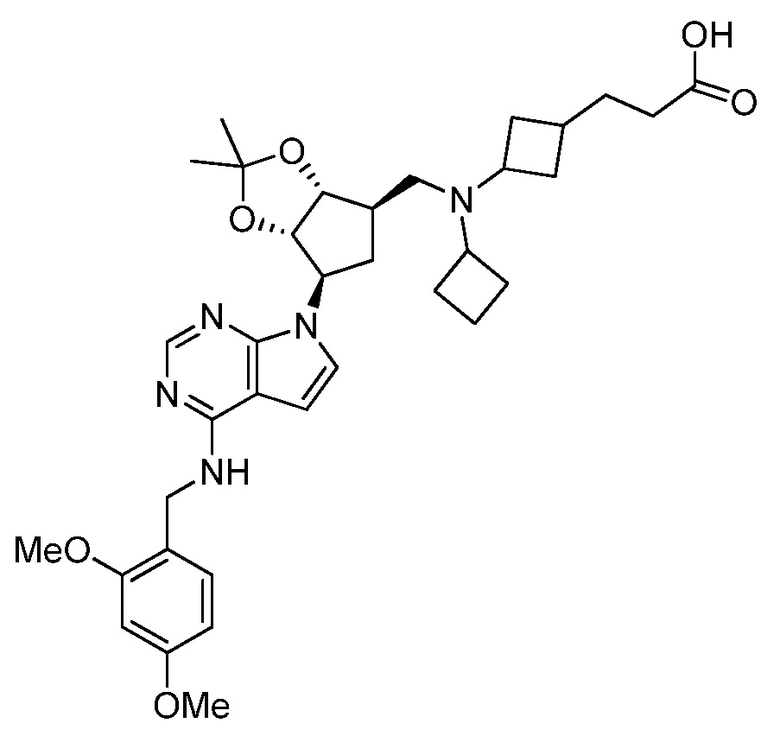
Гидроксид лития моногидрат (0,58 г, 14 ммоль) добавляли к раствору этил 3-(3-(циклобутил(((3aR,4R,6R,6aS)-6-(4-((2,4-
диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)амино)циклобутил)пропаноата (0,91 г, 1,4 ммоль) в тетрагидрофуране (12 мл, 150 ммоль) и метаноле (3 мл, 60 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре, затем подкисляли 1н раствором HCl до pH=6. Летучие вещества удаляли в вакууме и оставшуюся воду удаляли путем азеотропной перегонки с этанолом с последующей лиофилизацией в течение 18 часов. Полученное не совсем белое твердое вещество использовали без дополнительной очистки.
N-(2-амино-4-(трет-бутил)фенил)-3-(3-(циклобутил(((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)амино)циклобутил)пропанамид
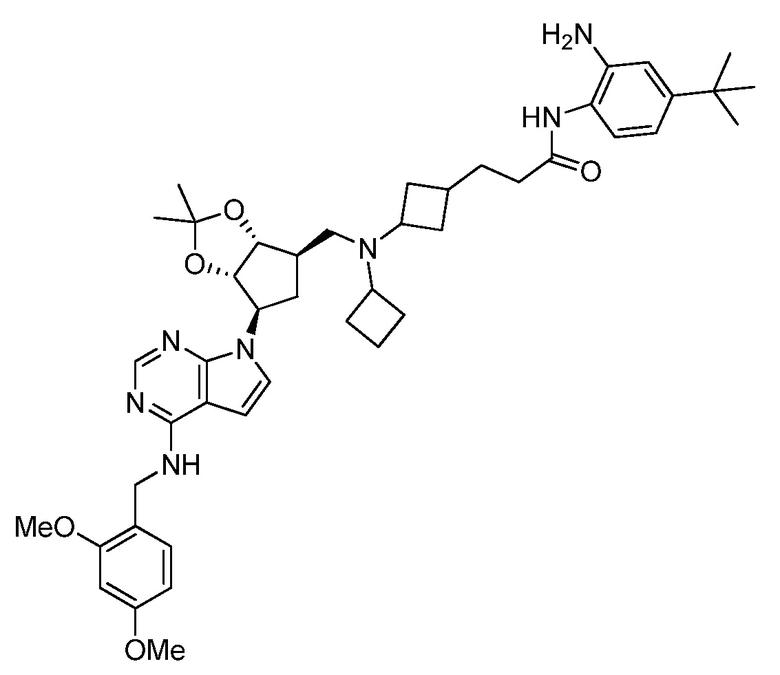
Гексафторфосфат N,N,N',N'-тетраметил-0-(7-азабензотриазол-1-ил)урония (0,783 г, 2,06 ммоль) добавляли к раствору 3-(3-(циклобутил(((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)амино)циклобутил)пропановой кислоты (0,87 г, 1,4 ммоль) и N,N-диизопропилэтиламина (0,789 мл, 4,53 ммоль) и [8]4-трет-бутилбензол-1,2-диамина (0,270 г, 1,65 ммоль) в N,N-диметилформамиде (8,50 мл). Реакционную смесь перемешивали в течение ночи при комнатной температуре, затем смесь частично концентрировали примерно до 2 мл и затем добавляли NaHCO3 (насыщенный раствор). Смесь экстрагировали при помощи EtOAc (3×) и объединенные органические слои сушили при помощи MgSO4, фильтровали и концентрировали. Остаток очищали флэш-хроматографией (DCM/7н NH3 в MeOH 95:5) с получением желаемого соединения (0,76 г) в виде твердого вещества.
7-((3aS,4R,6R,6aR)-6-(((3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(циклобутил)амино)метил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-N-(2,4-диметоксибензил)-7H-пирроло[2,3-d]пиримидин-4-амин
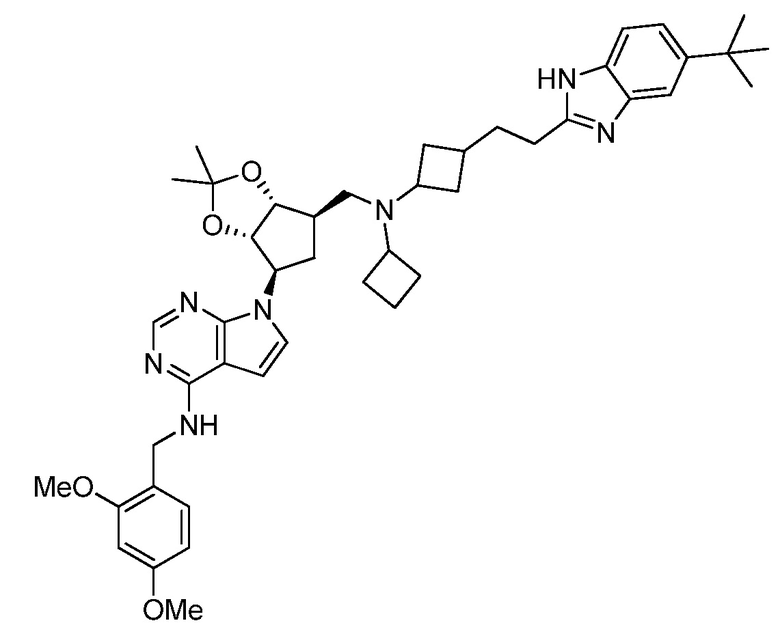
Раствор N-(2-амино-4-(трет-бутил)фенил)-3-(3-(циклобутил-(((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)амино)циклобутил)-пропанамида (0,76 г, 0,97 ммоль) в уксусной кислоте (2 мл) перемешивали в течение ночи при 60°C. Летучие вещества удаляли в вакууме и остаток очищали непосредственно флэш-хроматографией (DCM/7н NH3 в MeOH 91:9) с получением желаемого соединения (0,61 г) в виде пенистого вещества.
(1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-(((3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(циклобутил)амино)метил)циклопентан-1,2-диол
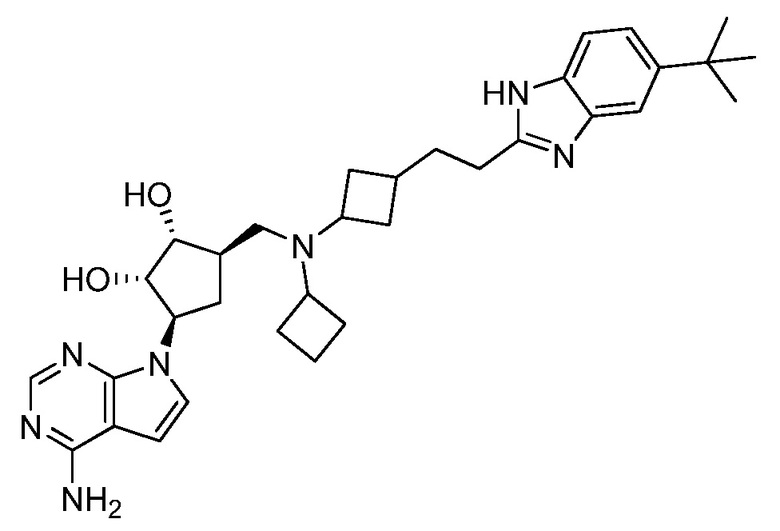
Трифторуксусную кислоту (10 мл, 200 ммоль) добавляли к смеси воды (1 мл, 80 ммоль) и 7-((3aS,4R,6R,6aR)-6-(((3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(циклобутил)амино)метил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-N-(2,4-диметоксибензил)-7H-пирроло[2,3-d]пиримидин-4-амина (0,61 г, 0,80 ммоль) при комнатной температуре. Реакционную смесь перемешивали в течение ночи при комнатной температуре и гасили путем добавления триэтилсилана (0,26 мл, 1,6 ммоль). Летучие вещества удаляли в вакууме и полученный остаток брали для поглощения в MeOH (15 мл), добавляли 500 мг K2CO3 и 8 капель воды и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Смесь фильтровали и фильтровальную лепешку промывали при помощи MeOH (10 мл). Фильтрат концентрировали и полученный остаток очищали флэш-хроматографией (DCM/7н NH3 в MeOH 90:10) с получением желаемого продукта (0,13 г) в виде бесцветного пенистого вещества. MS (ESI+) для C33H45N7O2 m/z 572,2 [M+H]+; MS (ESI-) для C33H45N7O2 m/z 570,2 [M-H]-; ВЭЖХ чистота >90% (время удерживания, 2,850 минут).
1H ЯМР (400 МГц, d4-MeOH) δH 8,083 (с, 1H), 7,492 (с, 1H), 7,412-7,392 (м, 1H), 7,309-7,286 (м, 1H), 7,220-7,205 (м, 1H), 6,620-6,610 (д, J=4,0 Гц, 1H), 4,321-4,283 (м, 1H), 3,888-3,848 (м, 1H), 3,505-3,417 (м, 0,5H (метин транс изомера)), 3,231-3,147 (м, 0,5H) (метин цис изомера)), 3,051-2,953 (м, 1H), 2,871-2,732 (м, 3H), 2,583-2,501 (м, 1H), 2,441-2,368 (м, 1H), 2,244-2,205 (м, 3H), 2,170-1,833 (м, 9H), 1,695-1,560 (м, 4H), 1,384 (с, 9H).
Соединение 44: (1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-(((3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(циклопропилметил)амино)метил)циклопентан-1,2-диол
Этил 3-(3-((циклопропилметил)(((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)амино)циклобутил)пропаноат
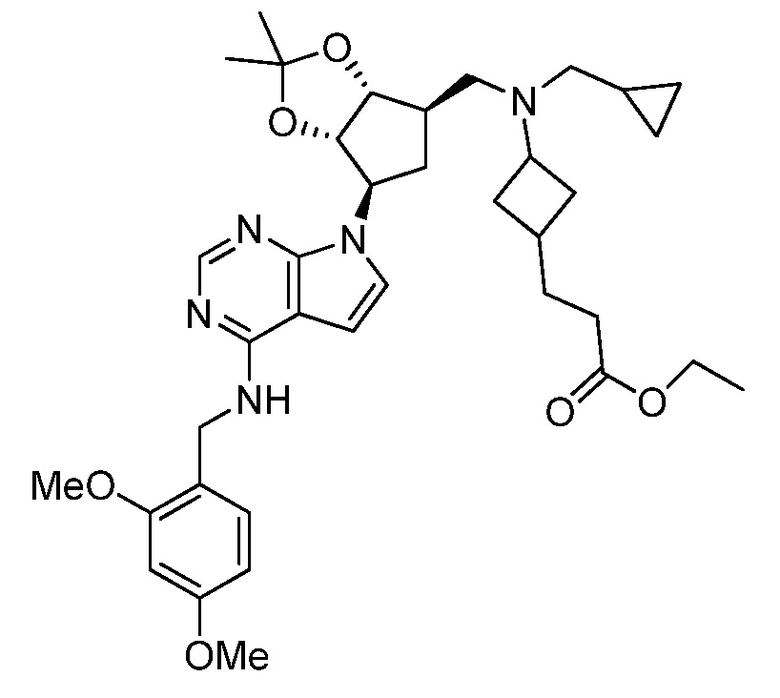
Амин этил 3-(3-((((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)амино)циклобутил)пропаноат (0,90 г, 1,5 ммоль) брали для поглощения в метанол (10 мл) и добавляли цианоборогидрид натрия (0,093 г, 1,5 ммоль). pH доводили до приблизительно 6 с использованием 10% раствора AcOH в MeOH. Реакционную смесь перемешивали в течение ночи при комнатной температуре. К реакционной смеси добавляли NaHCO3 (насыщенный раствор), затем смесь экстрагировали (3×) при помощи DCM. Объединенные органические слои сушили при помощи MgSO4, фильтровали и концентрировали. Это вещество использовали без дополнительной очистки.
3-(3-((циклопропилметил)(((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)амино)циклобутил)пропановая кислота
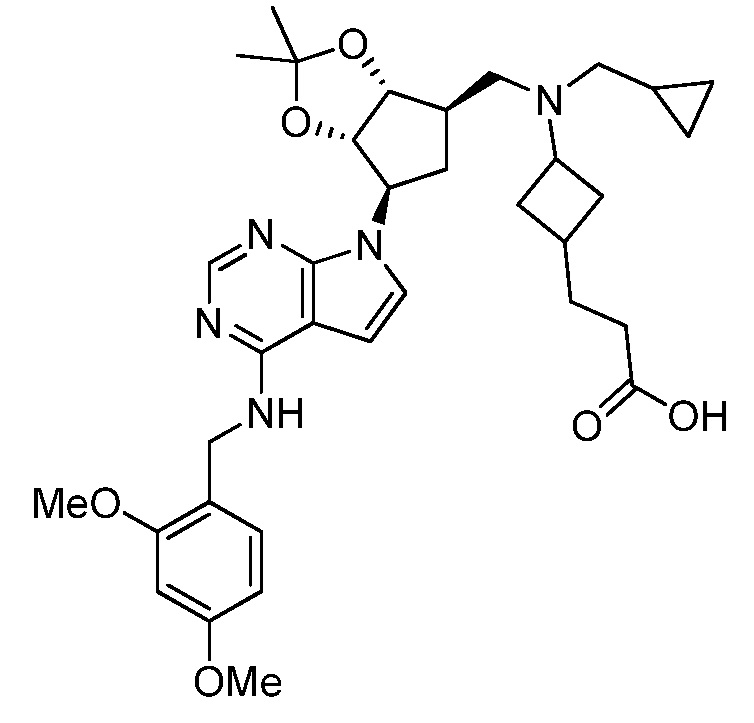
Гидроксид лития, моногидрат (0,62 г, 15 ммоль) добавляли к раствору этил 3-(3-((циклопропилметил)(((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)амино)циклобутил)пропаноата (0,98 г, 1,5 ммоль) в тетрагидрофуране (13 мл) и метаноле (3 мл). Реакционную смесь перемешивали в течение 24 часов при комнатной температуре, подкисляли при помощи 1н раствора HCl до pH=6. Летучие вещества удаляли в вакууме и оставшуюся воду удаляли путем азеотропной перегонки с этанолом с последующей лиофилизацией в течение 18 часов. Полученное не совсем белое твердое вещество использовали без дополнительной очистки.
N-(2-амино-4-(трет-бутил)фенил)-3-(3-((циклопропилметил)(((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)амино)циклобутил)пропанамид
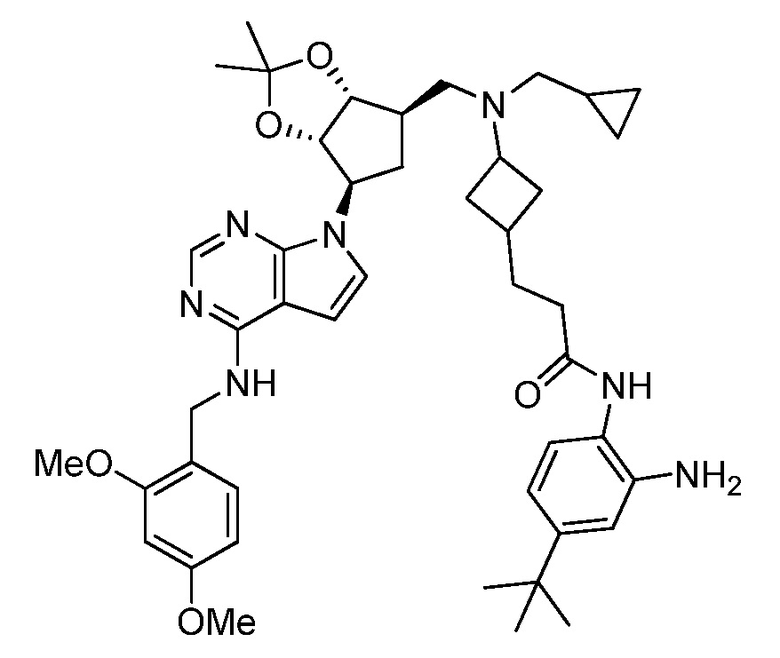
Гексафторфосфат N,N,N’,N’-тетраметил-0-(7-азабензотриазол-1-ил)урония (0,846 г, 2,22 ммоль) добавляли к раствору 3-(3-((циклопропилметил)(((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)амино)циклобутил)пропановой кислоты (0,94 г, 1,5 ммоль) и N,N-диизопропилэтиламина (0,852 мл, 4,89 ммоль) и [8]4-трет-бутилбензол-1,2-диамина (0,292 г, 1,78 ммоль) в N,N-диметилформамиде (9,19 мл, 119 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре, частично концентрировали примерно до 2 мл и добавляли NaHCO3 (насыщенный раствор). Смесь экстрагировали при помощи EtOAc (3×) и объединенные органические слои сушили при помощи MgSO4, фильтровали и концентрировали. Остаток очищали флэш-хроматографией (DCM/7н NH3 в MeOH 95:5) с получением желаемого соединения (0,92 г) в виде твердого вещества.
7-((3aS,4R,6R,6aR)-6-(((3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(циклопропилметил)амино)метил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-N-(2,4-диметоксибензил)-7H-пирроло[2,3-d]пиримидин-4-амин
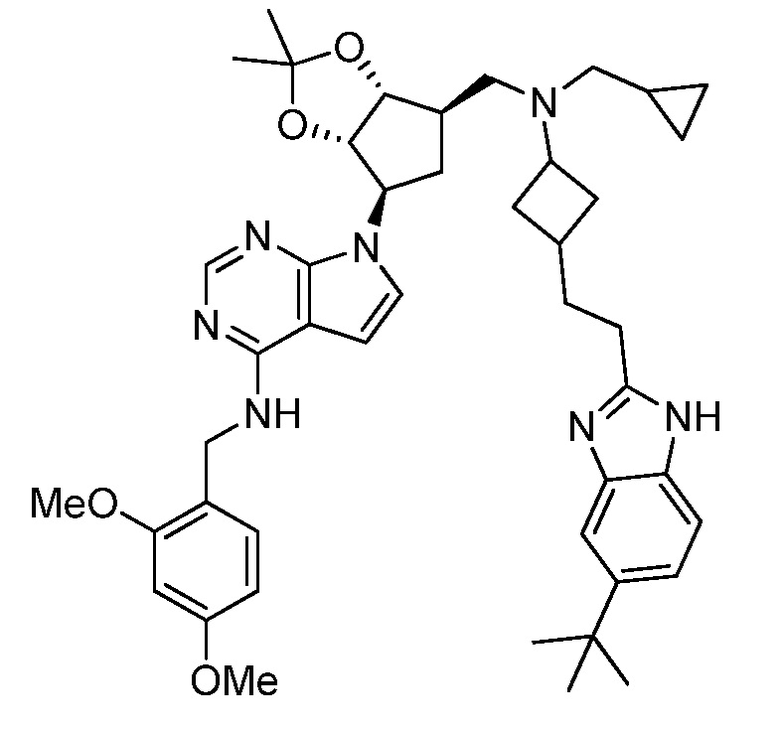
N-(2-амино-4-(трет-бутил)фенил)-3-(3-((циклопропилметил)(((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)амино)циклобутил)пропанамид (1,1 г, 1,4 ммоль) в уксусной кислоте (5 мл) нагревали при 60°C в течение ночи. Раствор концентрировали и очищали флэш-хроматографией (DCM/7н NH3 ион MeOH 93:7) с получением желаемого соединения (0,57 г) в виде бесцветного пенистого вещества.
(1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-(((3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(циклопропилметил)амино)метил)циклопентан-1,2-диол
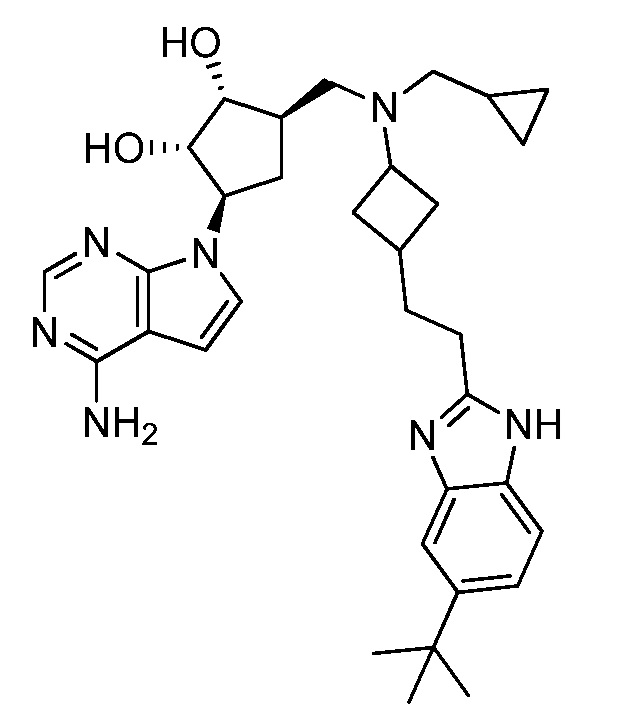
Трифторуксусную кислоту (10 мл) добавляли к смеси воды (1 мл) и 7-((3aS,4R,6R,6aR)-6-(((3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(циклопропилметил)амино)метил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-N-(2,4-диметоксибензил)-7H-пирроло[2,3-d]пиримидин-4-амина (0,52 г, 0,68 ммоль) при комнатной температуре. Реакционную смесь перемешивали в течение ночи при комнатной температуре и добавляли триэтилсилан (0,22 мл, 1,4 ммоль). Летучие вещества удаляли в вакууме и полученный остаток брали для поглощения в MeOH (15 мл), добавляли 500 мг K2CO3 и 8 капель H2O и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Смесь фильтровали и фильтровальную лепешку промывали при помощи 10 мл MeOH. Фильтрат концентрировали и полученный остаток очищали флэш-хроматографией (DCM/7н NH3 в MeOH 90:10) с получением желаемого продукта (0,196 г) в виде не совсем белого пенистого вещества. MS (ESI+) для C33H45N7O2 m/z 572,6 [M+H]+; MS (ESI-) для C33H45N7O2 m/z 570,3 [M-H]-; ВЭЖХ чистота >90% (время удерживания, 2,850 минут).
1H ЯМР (400 МГц, d4-MeOH) δH 7,944 (с, 1H), 7,361 (с, 1H), 7,280-7,259 (м, 1H), 7,172-7,150 (м, 1H), 7,092-7,078 (м, 1H), 6,484-6,475 (д, J=3,6 Гц, 1H), 4,222-4,185 (м, 1H), 3,815-3,779 (м, 1H), 3,329 (м, 0,5H (метин транс изомера)), 2,961 (м, 0,5H (метин цис изомера), 2,745-2,627 (м, 3H), 2,503-2,450 (м, 1H), 2,301-2,187 (м, 5H), 2,036-1,890 (м, 2H), 1,793-1,776 (м, 3H), 1,529-1,385 (м, 2H), 1,246 (с, 9H), 0,808-0,739 (м, 1H), 0,394-0,362 (м, 2H), 0,012-0,013 (м, 2H).
Время удерживания: 2,850 минут. ВЭЖХ Условия: колонка Agilent Zorbax Exlipse XDB-C18, 4,6×50 мм (1,8 мкм наполнителя), Растворитель A- Вода (0,1% TFA), Растворитель B- Ацетонитрил (0,07% TFA) 6 мин. градиент от 5 до 95% B; 1 мин. удерживание; затем повторный цикл.
Соединение 45: этил 3-(3-((((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)(изобутил)амино)циклобутил)пропаноат
Амин этил 3-(3-((((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)амино)циклобутил)пропаноат (1,7 г, 2,8 ммоль) брали для поглощения в метанол (20 мл) и добавляли цианоборогидрид натрия (0,35 г, 5,6 ммоль). pH доводили до приблизительно 6 с использованием 10% раствора AcOH в MeOH, затем добавляли одной порцией изобутиральдегид (0,33 мл, 3,6 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Добавляли еще 1,3 эквивалента изобутиральдегида и перемешивание продолжали в течение ночи. К реакционной смеси добавляли NaHCO3 (насыщенный раствор), затем смесь экстрагировали (3×) при помощи DCM. Объединенные органические слои сушили при помощи MgSO4 и концентрировали. Остаток очищали флэш-хроматографией (DCM/7н NH3 в MeOH 97:3) с получением желаемого соединения (1,75 г) в виде бесцветного пенистого вещества.
3-(3-((((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)(изобутил)амино)циклобутил)пропановая кислота
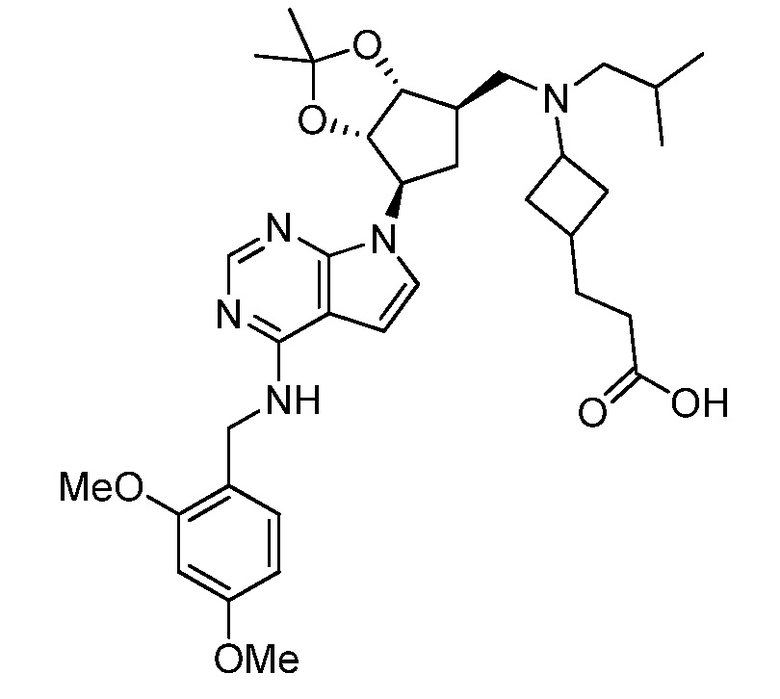
Гидроксид лития моногидрат (1,11 г, 26,4 ммоль) добавляли к раствору этил 3-(3-((((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)(изобутил)амино)циклобутил)пропаноата (1,75 г, 2,64 ммоль) в тетрагидрофуране (13 мл, 160 ммоль) и метаноле (3 мл, 70 ммоль). Реакционную смесь перемешивали в течение 24 часов при комнатной температуре, подкисляли 1н раствором HCl до pH=6, летучие вещества удаляли в вакууме и оставшуюся воду удаляли путем азеотропной перегонки с этанолом с последующей лиофилизацией в течение 18 часов. Полученное не совсем белое твердое вещество использовали без дополнительной очистки.
N-(2-амино-4-(трет-бутил)фенил)-3-(3-((((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)(изобутил)амино)циклобутил)пропанамид
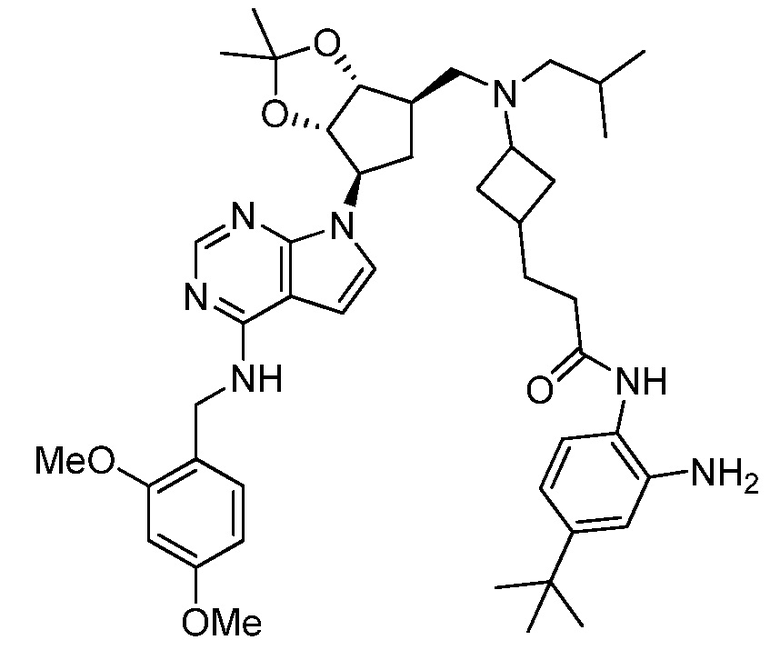
Гексафторфосфат N,N,N’,N’-тетраметил-0-(7-азабензотриазол-1-ил)урония (1,52 г, 4,01 ммоль) добавляли к раствору 3-(3-((((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)(изобутил)амино)циклобутил)пропановой кислоты (1,7 г, 2,7 ммоль) и N,N-диизопропилэтиламина (1,54 мл, 8,82 ммоль) и 4-трет-бутилбензол-1,2-диамина (0,527 г, 3,21 ммоль) в N,N-диметилформамиде (16,6 мл). Реакционную смесь перемешивали в течение ночи при комнатной температуре, частично концентрировали примерно до 2 мл и затем добавляли NaHCO3 (насыщенный раствор). Затем смесь экстрагировали при помощи EtOAc (3×) и объединенные органические слои сушили при помощи MgSO4, фильтровали, концентрировали и очищали флэш-хроматографией (DCM/7н NH3 в MeOH 95:5) с получением желаемого амида (1,71 г) в виде твердого вещества.
7-((3aS,4R,6R,6aR)-6-(((3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(изобутил)амино)метил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-N-(2,4-диметоксибензил)-7H-пирроло[2,3-d]пиримидин-4-амин
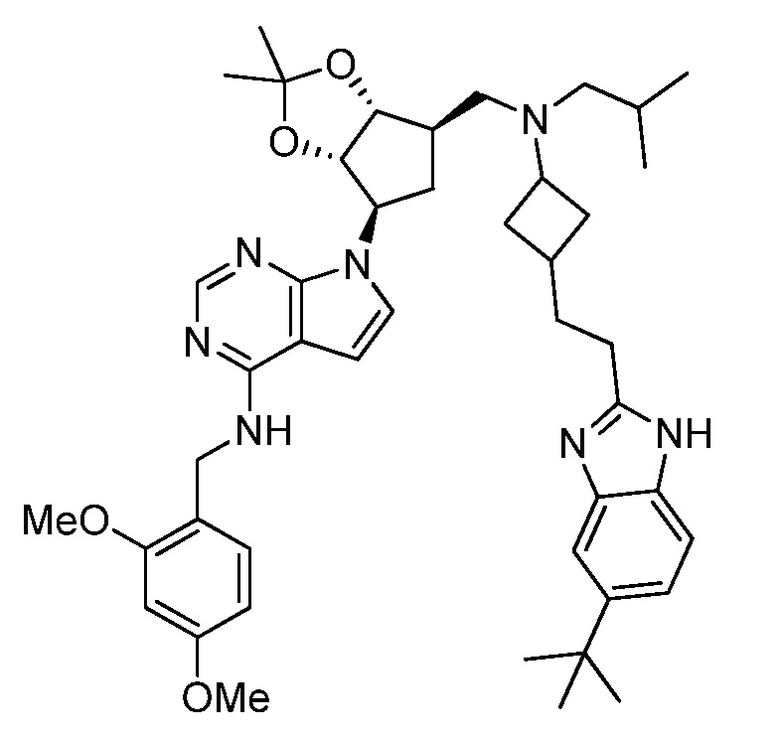
Раствор N-(2-амино-4-(трет-бутил)фенил)-3-(3-((((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)(изобутил)амино)циклобутил)пропанамида (1,71 г, 2,19 ммоль) в уксусной кислоте (6 мл) перемешивали в течение ночи при 60°C, летучие вещества удаляли в вакууме и полученный остаток очищали флэш-хроматографией (SiO2, DCM/7н NH3 в MeOH 94:6) с получением желаемого соединения (0,9 г) в виде пенистого вещества.
(1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-(((3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(изобутил)амино)метил)циклопентан-1,2-диол
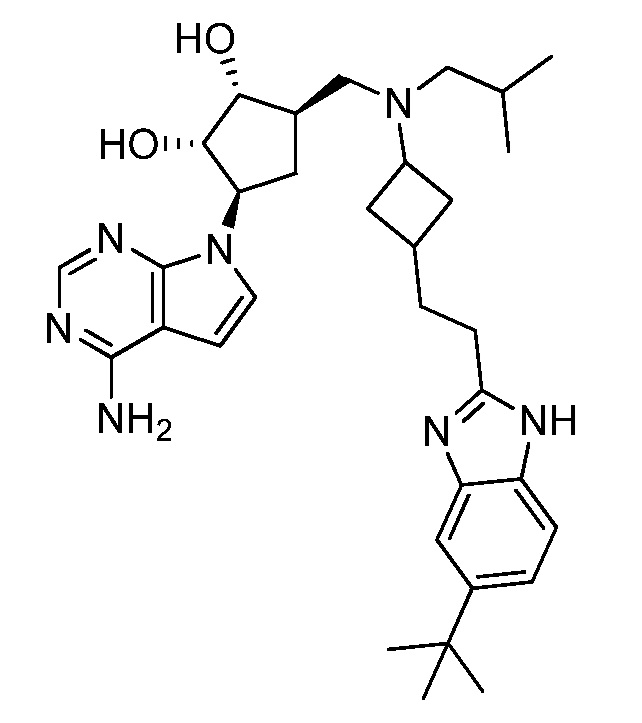
Трифторуксусную кислоту (20 мл, 300 ммоль) добавляли к смеси воды (2 мл, 100 ммоль) и 7-((3aS,4R,6R,6aR)-6-(((3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)-(изобутил)амино)метил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-N-(2,4-диметоксибензил)-7H-пирроло[2,3-d]пиримидин-4-амина (0,9 г, 1 ммоль) при комнатной температуре. Реакционную смесь перемешивали в течение ночи и добавляли триэтилсилан (0,38 мл, 2,4 ммоль). Летучие вещества удаляли в вакууме и полученный остаток брали для поглощения в MeOH (15 мл), добавляли 500 мг K2CO3 и 8 капель H2O и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Смесь фильтровали и фильтровальную лепешку промывали при помощи 10 мл MeOH. Фильтрат концентрировали и полученный остаток очищали флэш-хроматографией (DCM/7н NH3 в MeOH 90:10) с получением желаемого продукта (0,274 г) в виде не совсем белого пенистого вещества. MS (ESI+) для C33H47N7O2 m/z 574,6 [M+H]+; MS (ESI-) для C33H45N7O2 m/z 572,4 [M-H]-; ВЭЖХ чистота >86% (время удерживания, 2,918 минут).
1H ЯМР (400 МГц, d4-MeOH) δH 8,078 (с, 1H), 7,497 (с, 1H), 7,416-7,396 (м, 1H), 7,305-7,284 (м, 1H), 7,216-7,200 (м, 1H), 6,621-6,612 (д, J=3,6 Гц, 1H), 4,368-4,334 (м, 1H), 3,930-3,894 (м, 1H), 2,934-2,918 (м, 1H), 2,866-2,797 (м, 2H), 2,652-2,583 (м, 1H), 2,444-2,361 (м, 2H), 2,287-2,199 (м, 2H), 2,166-2,119 (м, 3,5H (содержит метин транс изомера)), 2,048-2,012 (м, 1H), 1,921-1,748 (м, 3,5H (содержит метин цис изомера)), 1,622-1,494 (м, 2H), 1,380 (с, 9H), 1,269-1,252 (м, 1H), 0,932-0,879 (м, 6H).
Соединение 46: (1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-((((1r,3S)-3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(циклобутил)-амино)метил)циклопентан-1,2-диол
Диастереоизомеры разделяли при помощи SFC. Это вещество брали для поглощения в MeOH/H2O и лиофилизировали с получением белого порошка (23,7 мг).
1H ЯМР (400 МГЦ, MeOD) δH м.д. 8,06 (с, 1H), 7,48 (шир.с, 1H), 7,39 (д, J=8,71 Гц, 1H), 7,28 (дд, J=8,60, 1,76 Гц, 1H), 7,19 (д, J=3,52 Гц, 1H), 6,60 (д, J=3,52 Гц, 1H), 4,84 (м, 1H), 4,28 (дд, J=7,26, 6,22 Гц, 1H), 3,84 (т, J=5,70 Гц, 1H), 3,16 (м, 1H), 2,99 (м,1H), 2,80 (т, J=7,15 Гц, 2H), 2,73 (дд, J=13,68, 6,22 Гц, 1H), 2,49 (дд, J=13,68, 7,67 Гц, 1H), 2,38 (м, 1H), 2,22 (м, 3H), 2,00 (м, 4H), 1,91 (м, 3H), 1,60 (м, 5H), 1,36 (с, 9H).
Соединение 47: (1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-((((1r,3S)-3-(2-(5-бром-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(метил)амино)метил)циклопентан-1,2-диол
Диастереоизомеры разделяли при помощи SFC. Это вещество брали для поглощения в MeOH/H2O и лиофилизировали с получением белого порошка (21 мг).
1H ЯМР (400 МГц, MeOD) δH м.д. 8,06 (с, 1H), 7,63 (шир.с, 1H), 7,39 (м, 1H), 7,30 (дд, J=8,50, 1,66 Гц, 1H), 7,20 (д, J=3,52 Гц, 1H), 6,60 (д, J=3,52 Гц, 1H), 4,32 (дд, J=7,67, 6,01 Гц, 1H), 3,88 (м, 1H), 2,82 (т, J=7,15 Гц, 2H), 2,71 (м, 1H), 2,52 (м, 1H), 2,41 (м, 2H), 2,25 (м, 2H), 2,18 (с, 3H), 2,03 (м, 1H), 1,92 (м, 3H), 1,62 (м, 1H), 1,51 (м, 2H).
Соединение 48: (1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-((((1s,3R)-3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(изобутил)-амино)метил)циклопентан-1,2-диол
Диастереоизомеры разделяли при помощи SFC. После лиофилизации (78 мг) получали бесцветное твердое вещество.
Соединение 49: (1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-((((1s,3R)-3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(этил)амино)метил)циклопентан-1,2-диол
Диастереоизомеры разделяли при помощи SFC.
Lux-3 (2×15 см), 30% этанола (0,2% DEA))/CO2, 100 бар, 65 мл/мин 220 нм. Объем вводимой пробы: 0,4 мл, 6,2 мг/мл метанол.
1H ЯМР (400 МГц, d4-MeOH) δH 8,081 (с, 1H), 7,499 (с, 1H), 7,416-7,395 (м, 1H), 7,308-7,282 (м, 1H), 7,226-7,216 (м, 1H), 6,619-6,610 (м, 1H), 4,344-4,310 (м, 1H), 3,922-3,895 (м, 1H), 3,410-3,329 (м, 1H), 2,875-2,837 (м, 2H), 2,738-2,689 (м, 1H), 2,659-2,607 (м, 2H), 2,535-2,483 (м, 1H), 2,452-2,380 (м, 1H), 2,311-2,224 (м, 1H), 2,158-2,121 (м, 3H), 2,061-2,030 (м, 2H), 1,913-1,863 (м, 2H), 1,674-1,590 (м, 1H), 1,381 (с, 9H), 1,056-1,020 (т, J=7,2 Гц, 3H).
Соединение 50: (1R,2S,3R,5R)-3-(6-амино-9H-пурин-9-ил)-5-(((3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(изопропил)амино)метил)циклопентан-1,2-диол
этил 3-(3-((((1R,2R,3S,4R)-4-(6-((2,4-диметоксибензил)амино)-9H-пурин-9-ил)-2,3-дигидроксициклопентил)метил)(изопропил)амино)циклобутил)- пропаноат
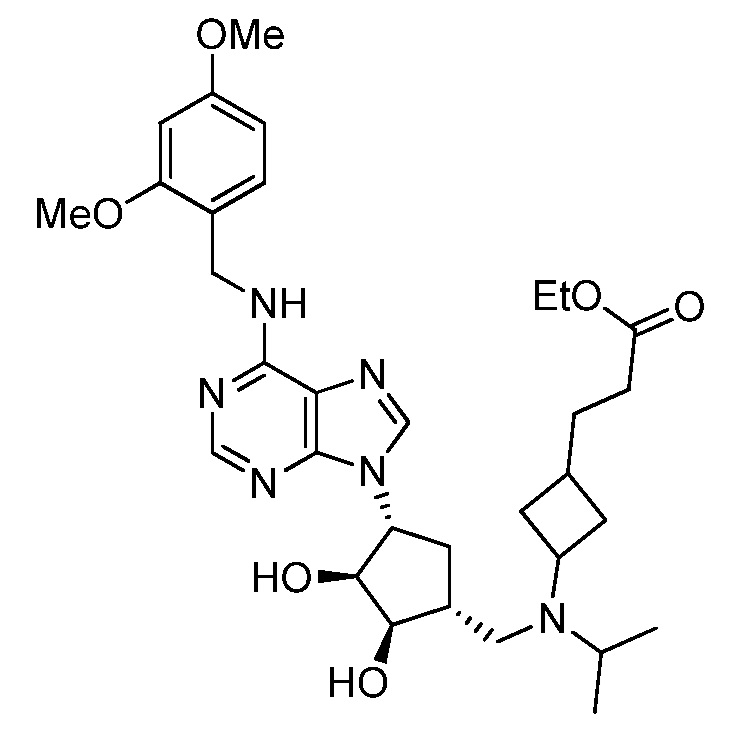
Амин этил 3-(3-((((1R,2R,3S,4R)-4-(6-((2,4-диметоксибензил)амино)-9H-пурин-9-ил)-2,3-дигидроксициклопентил)метил)амино)циклобутил)пропаноат (1,5 г, 2,5 ммоль) брали для поглощения в ацетонитрил (66 мл) и добавляли изопропилиодид (2,5 мл, 25 ммоль) и триэтиламин (5,2 мл, 37 ммоль). Реакционную смесь нагревали до 80°C в течение 12 часов. Добавляли еще 15 эквивалентов TEA и еще 15 эквивалентов iPrI и реакцию продолжали еще в течение 8 часов. Добавляли еще по 15 эквивалентов каждого из iPrI и TEA и нагревание продолжали в течение ночи. Реакционную смесь концентрировали и добавляли насыщенный раствор Na2CO3 (20 мл) и DCM (20 мл). Слои разделяли и водный слой снова экстрагировали 3 раза, объединенные органические слои сушили и очищали флэш-хроматографией (SiO2, DCM/7н NH3 в MeOH 97:3).
Полученный остаток растворяли в 30 мл DCM и промывали при помощи 20 мл насыщенного раствора NaHCO3 и 10 мл 1н раствора NaOH. Водный слой экстрагировали при помощи DCM 3 раза, объединенные органические слои сушили над MgSO4 и растворитель удаляли с получением желаемого продукта (1,3 г) в виде пенистого/твердого вещества.
(3-((((1R,2R,3S,4R)-4-(6-((2,4-диметоксибензил)амино)-9H-пурин-9-ил)-2,3-дигидроксициклопентил)метил)(изопропил)-амино)циклобутил)пропановая кислота
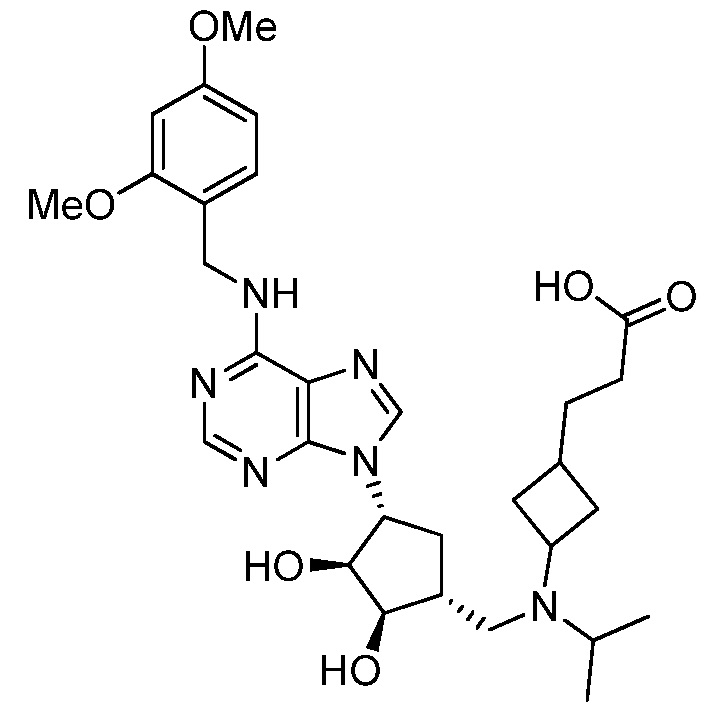
Гидроксид лития моногидрат (0,838 г, 20,0 ммоль) добавляли к раствору этил 3-(3-(3-((((1R,2R,3S,4R)-4-(6-((2,4-диметоксибензил)амино)-9H-пурин-9-ил)-2,3-дигидроксициклопентил)метил)(изопропил)амино)циклобутил)-пропановой кислоты (1,3 г, 2,0 ммоль) в тетрагидрофуране (30 мл, 300 ммоль) и метаноле (6,5 мл, 160 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре, подкисляли при помощи 1н раствора HCl до pH=6. Летучие вещества удаляли в вакууме и оставшуюся воду удаляли путем азеотропной перегонки с этанолом, с последующей лиофилизацией. Полученное твердое вещество использовали без дополнительной очистки.
N-(2-амино-4-(трет-бутил)фенил)-3-(3-((((1R,2R,3S,4R)-4-(6-((2,4-диметоксибензил)амино)-9H-пурин-9-ил)-2,3-дигидроксициклопентил)метил)(изопропил)амино)циклобутил)-пропанамид
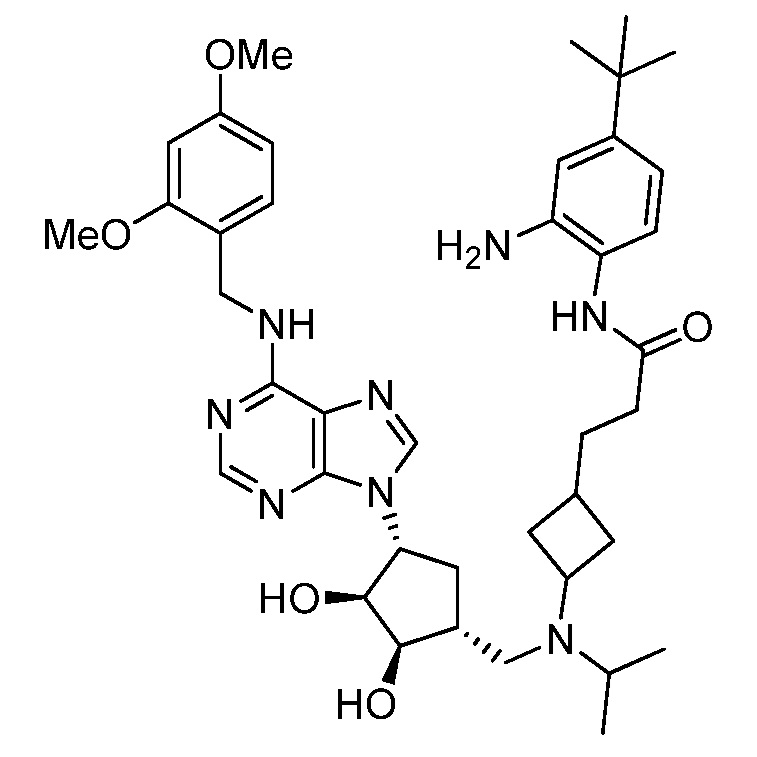
Гексафторфосфат N,N,N’,N’-тетраметил-0-(7-азабензотриазол-1-ил)урония (1,19 г, 3,13 ммоль) добавляли к раствору 3-{3-[{[(3aR,4R,6R,6aS)-6-{6-[(2,4-диметоксибензил)амино]-9H-пурин-9-ил}-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил]метил}{изопропил)амино]циклобутил}пропановой кислоты (1,30 г, 2,09 ммоль) и N,N-диизопропилэтиламина (1,20 мл, 6,89 ммоль) и 4-трет-бутилбензол-1,2-диамина (0,411 г, 2,50 ммоль) в N,N-диметилформамиде (12,9 мл). Реакционную смесь перемешивали в течение 2 часов, добавляли NaHCO3 (насыщенный раствор) и смесь экстрагировали при помощи EtOAc (3×) и объединенные органические слои сушили над MgSO4 фильтровали и концентрировали. Остаток очищали флэш-хроматографией (DCM → DCM/7н NH3 в MeOH 95:5) с получением желаемого амида (1,4 г) в виде твердого вещества.
9-((3aS,4R,6R,6aR)-6-(((3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(изопропил)амино)метил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-N-(2,4-диметоксибензил)-9H-пурин-6-амин
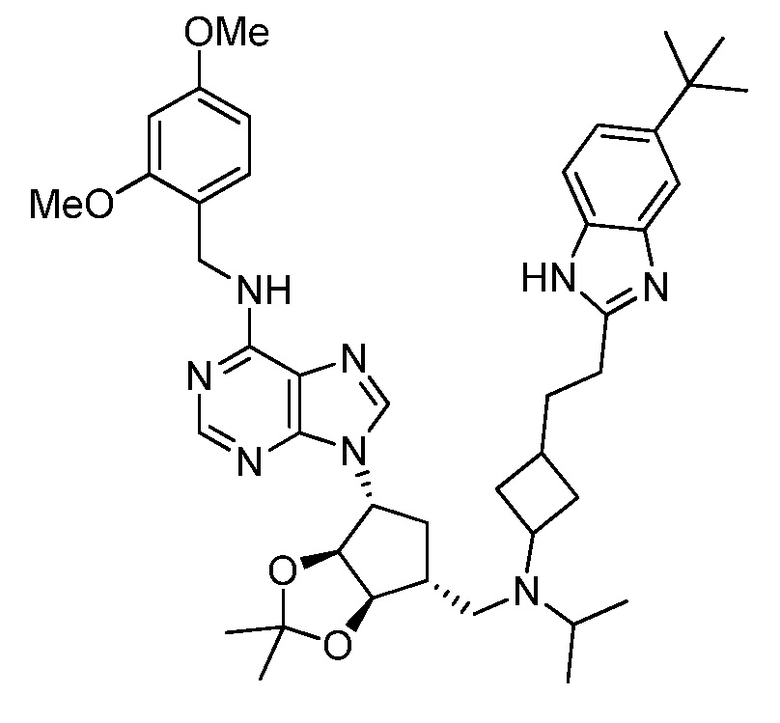
N-(2-амино-4-(трет-бутил)фенил)-3-(3-((((1R,2R,3S,4R)-4-(6-((2,4-диметоксибензил)амино)-9H-пурин-9-ил)-2,3-дигидроксициклопентил)метил)(изопропил)амино)циклобутил)-пропанамид (1,4 г, 1,8 ммоль) в уксусной кислоте (5 мл, 90 ммоль) перемешивали в течение ночи при 60°C. Реакционную смесь концентрировали и очищали флэш-хроматографией (DCM → DCM/7н NH3 в MeOH 94:6) с получением желаемого соединения (0,91 г) в виде пенистого вещества.
(1R,2S,3R,5R)-3-(6-амино-9H-пурин-9-ил)-5-(((3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(изопропил)-амино)метил)циклопентан-1,2-диол
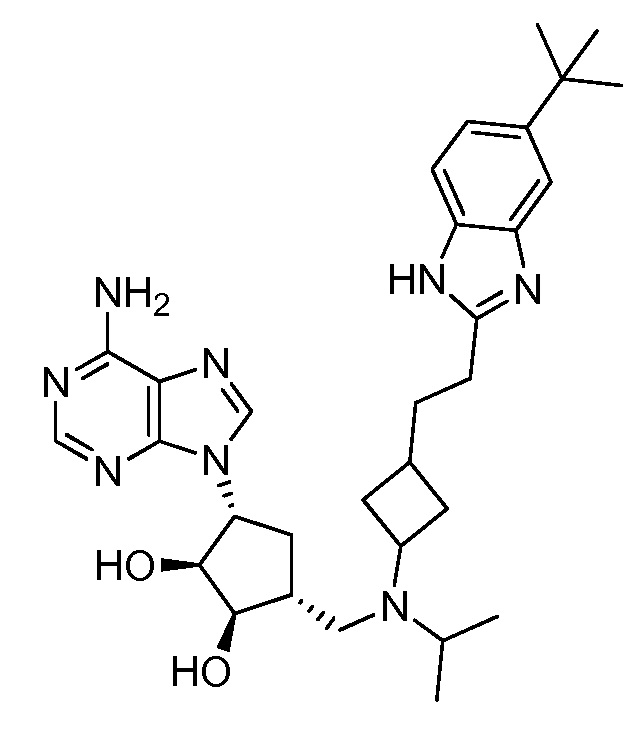
Трифторуксусную кислоту (10 мл, 100 ммоль) добавляли к смеси воды (1 мл, 60 ммоль) и 9-((3aS,4R,6R,6aR)-6-(((3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(изопропил)- амино)метил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]-диоксол-4-ил)-N-(2,4-диметоксибензил)-9H-пурин-6-амина (0,91 г, 1,2 ммоль) при комнатной температуре. Реакционную смесь перемешивали в течение ночи при комнатной температуре. Реакционную смесь затем нагревали до 35°C и добавляли триэтилсилан (0,39 мл, 2,4 ммоль). Реакционную смесь перемешивали при 35°C еще в течение 2 дней. Летучие вещества удаляли в вакууме и полученный остаток брали для поглощения в MeOH (15 мл). Добавляли 500 мг K2CO3 и 8 капель H2O и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Смесь фильтровали и фильтровальную лепешку промывали при помощи 10 мл MeOH. Фильтрат концентрировали и полученный остаток очищали флэш-хроматографией (DCM/7н NH3 в MeOH 90:10) с получением желаемого продукта (0,142 г) в виде бесцветного твердого вещества после нескольких дней лиофилизации.
Соединение 51: (1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-((((1s,3R)-3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(циклобутил)-амино)метил)циклопентан-1,2-диол
Диастереоизомеры разделяли при помощи SFC (25 мг).
1H ЯМР (400 МГц, MeOD) δH м.д. 8,06 (с, 1H), 7,48 (шир.с, 1H), 7,38 (д, J=7,88 Гц, 1H), 7,27 (дд, J=8,60, 1,55 Гц, 1H), 7,19 (д, J=3,52 Гц, 1H), 6,60 (д, J=3,73 Гц, 1H), 4,85 (м, 1H), 4,29 (м, 1H), 3,85 (т, J=5,60 Гц, 1H), 3,41 (м, 1H), 3,17 (м, 1H), 2,83 (т, J=7,36 Гц, 2H), 2,74 (дд, J=13,68, 6,63 Гц, 1H), 2,51 (дд, J=13,79, 7,57 Гц, 1H), 2,38 (м, 1H), 2,18 (м, 3H), 2,09 (м, 1H), 2,02 (м, 5H), 1 83 (м, 2H), 1,60 (м, 3H), 1,36 (с, 9H).
Соединение 52: (1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-((((1r,3S)-3-(2-(5,6-дихлор-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(метил)-амино)метил)циклопентан-1,2-диол
Диастереоизомеры разделяли при помощи SFC. Это вещество брали для поглощения в MeOH/H2O и лиофилизировали с получением твердого вещества кремового цвета (64 мг).
1H ЯМР (400 МГц, MeOD) δH м.д. 8,06 (с, 1H), 7,63 (с, 2H), 7,20 (д, J=3,52 Гц, 1H), 6,59 (д, J=3,73 Гц, 1H), 4,32 (дд, J=7,88, 6,01 Гц, 1H), 3,88 (дд, J=5,60, 4,77 Гц, 1H), 2,82 (т, J=7,26 Гц, 2H), 2,69 (м, 1H), 2,48 (м, 1H), 2,38 (м, 2H), 2,24 (м, 3H), 2,15 (с, 3H),1,91 (м, 3H), 1,61 (м, 1H), 1,50 (м, 2H).
Соединение 53: (1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-((((1r,3S)-3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(изобутил)-амино)метил)циклопентан-1,2-диол
Диастереоизомеры разделяли при помощи SFC. После лиофилизации выделяли бесцветное твердое вещество (85 мг).
Соединение 54: (1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-((((1s,3R)-3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)-(циклопропилметил)амино)метил)циклопентан-1,2-диол
Диастереоизомеры разделяли при помощи SFC. Это вещество брали для поглощения в MeOH/H2O и лиофилизировали с получением белого порошка (53 мг).
1H ЯМР (400 МГц, MeOD) δH м.д. 8,06 (с, 1H), 7,48 (шир.с, 1H), 7,39 (д, J=8,50 Гц, 1H), 7,28 (дд, J=8,60, 1,76 Гц, 1H), 7,21 (д, J=3,52 Гц, 1H), 6,60 (д, J=3,52 Гц, 1H), 4,32 (дд, J=7,67, 5,80 Гц, 1H), 3,91 (м, 1H), 3,44 (м, 1H), 2,84 (т, J=7,57 Гц, 2H), 2,78 (дд, J=13,27, 7,05 Гц, 1H), 2,58 (дд, J=13,06, 7,67 Гц, 1H), 2,40 (м, 3H), 2,11 (т, J=6,22 Гц, 3H), 2,02 (м, 2H), 1,87 (м, 2H), 1,62 (м, 1H), 1,37 (с, 9H), 0,87 (м, 1H), 0,49 (м, 2H), 0,12 (м, 2H).
Соединение 55: (1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-((((1s,3R)-3-(2-(5-бром-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(метил)амино)метил)циклопентан-1,2-диол
Диастереоизомеры разделяли при помощи SFC.
1H ЯМР (400 МГЦ, MeOD) δH м.д. 8,05 (с, 1H), 7,63 (с, 1H), 7,39 (м, 1H), 7,30 (дд, J=8,50, 1,66 Гц, 1H), 7,20 (д, J=3,52 Гц, 1H), 6,59 (д, J=3,73 Гц, 1H), 4,32 (дд, J=7,77, 5,91 Гц, 1H), 3,88 (дд, J=5,70, 4,66 Гц, 1H), 2,99 (м, 1H), 2,84 (т, J=7,57 Гц, 2H), 2,48 (м, 1H), 2,41 (дд, J=7,98, 4,87 Гц, 1H), 2,34 (м, 1H), 2,24 (м, 1H), 2,15 (с, 3H), 2,10 (м, 3H), 2,01 (м, 2H), 1,86 (т, J=8,19 Гц, 2H), 1,61 (м, 1H).
Соединение 56: (1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-((изопропил(3-(2-(5-(трифторметокси)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)амино)метил)циклопентан-1,2-диол
N-(2-амино-4-(трифторметокси)фенил)-3-(3-((((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)(изопропил)амино)циклобутил)пропанамид
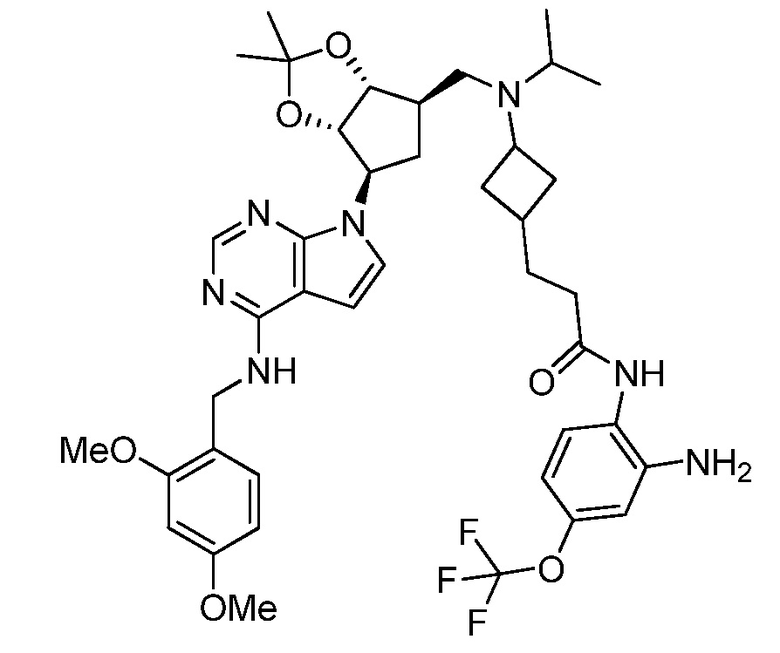
Гексафторфосфат N,N,N’,N’-тетраметил-0-(7-азабензотриазол-1-ил)урония (1,19 г, 3,14 ммоль) добавляли к раствору 3-(3-((((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)(изопропил)амино)циклобутил)пропановой кислоты (1,3 г, 2,1 ммоль) и N,N-диизопропилэтиламина (1,20 мл, 6,90 ммоль) и 4-(трифторметокси)бензол-1,2-диамина (0,482 г, 2,51 ммоль) в N,N-диметилформамиде (13,0 мл, 167 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре и частично концентрировали примерно до 2 мл и затем добавляли NaHCO3 (насыщенный раствор). Смесь экстрагировали при помощи EtOAc (3×) и объединенные органические слои сушили при помощи MgSO4, фильтровали и концентрировали. Остаток очищали флэш-хроматографией (DCM/7н NH3 в MeOH 95:5) с получением желаемого амида (1,4 г) в виде твердого вещества.
N-(2,4-диметоксибензил)-7-((3aS,4R,6R,6aR)-6-((изопропил(3-(2-(5-(трифторметокси)-1H-бензо[d]имидазол-2-ил)этил)-циклобутил)амино)метил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-7H-пирроло[2,3-d]пиримидин-4-амин
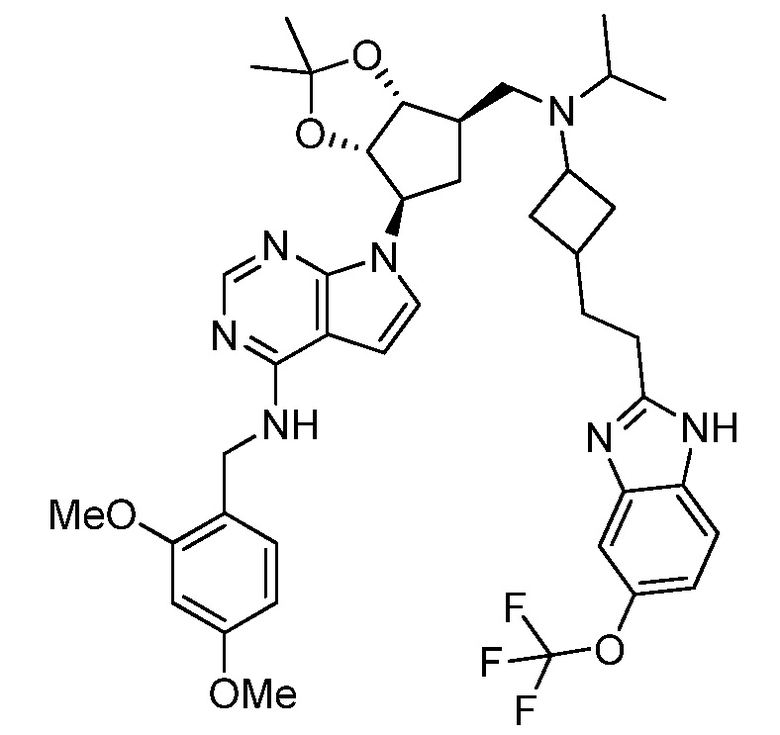
N-(2-амино-4-(трифторметокси)фенил)-3-(3-((((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)(изопропил)амино)циклобутил)пропанамид (1,4 г, 1,8 ммоль) нагревали в AcOH при 60°C в течение ночи. Реакционную смесь концентрировали в вакууме с получением неочищенного продукта.
(1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-((изопропил(3-(2-(5-(трифторметокси)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)амино)метил)циклопентан-1,2-диол
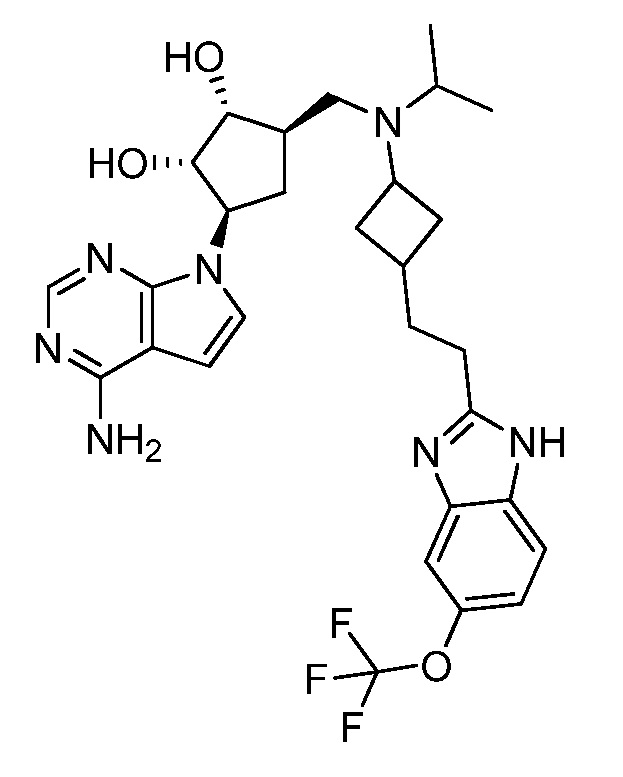
Трифторуксусную кислоту (10 мл, 100 ммоль) добавляли к смеси воды (1 мл, 60 ммоль) и N-(2,4-диметоксибензил)-7-((3aS,4R,6R,6aR)-6-((изопропил(3-(2-(5-(трифторметокси)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)амино)метил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-7H-пирроло[2,3-d]пиримидин-4-амина (0,91 г, 1,2 ммоль) при комнатной температуре. Реакционную смесь перемешивали в течение ночи при комнатной температуре, затем гасили путем добавления триэтилсилана (0,37 мл, 2,3 ммоль). Летучие вещества удаляли в вакууме и полученный остаток брали для поглощения в MeOH (15 мл). Добавляли 500 мг K2CO3 и 8 капель H2O и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Смесь фильтровали и фильтровальную лепешку промывали при помощи 10 мл MeOH. Фильтрат концентрировали и полученный остаток очищали флэш-хроматографией (DCM/7н NH3 в MeOH 90:10) с получением желаемого продукта (0,232 г) в виде не совсем белого пенистого вещества. MS (ESI+) для C29H36F3N7O3 m/z 588,2 [M+H]+; MS (ESI-) для C29H36F3N7O3 m/z 586,2 [M-H]-; ВЭЖХ чистота >90% (время удерживания, 2,570 минут).
1H ЯМР (400 МГц, d4-MeOH) δH 8,082 и 8,079 (с, 1H, перекрывающие пики из-за цис и транс изомеров), 7,554-7,524 (м, 1H), 7,414 (с, 1H), 7,225-7,209 (м, 1H), 7,155-7,127 (м, 1H), 6,618-6,609 (м, 1H), 4,363-4,323 (м, 1H), 3,976-3,932 (м, 1H), 3,606-3,524 (м, 0,5H (метин транс изомера)), 3,156-3,110 (м, 0,5H (метин цис изомера), 3,089-3,006 (м, 1H), 2,731-2,679 (м, 1H), 2,544-2,360 (м, 2H), 2,256-2,239 (м, 3H), 2,093-2,061 (м, 2H), 1,987-1,861 (м, 3H), 1,648-1,568 (м, 2H), 1,072-1,006 (м, 6H).
Соединение 57: (1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-((метил((1s,3R)-3-(2-(5-(трифторметокси)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)амино)метил)циклопентан-1,2-диол
Диастереоизомеры разделяли при помощи SFC. Это вещество лиофилизировали с получением твердого вещества (78 мг).
1H ЯМР (400 МГц, d4-MeOH) δH 8,076 (с, 1H), 7,548-7,527 (м, 1H), 7,414 (с, 1H), 7,227-7,218 (м, 1H), 7,148-7,123 (м, 1H), 6,616-6,607 (м, 1H), 4,361-4,327 (м, 1H), 3,926-3,899 (м, 1H), 3,037-3,000 (м, 1H), 2,907-2,870 (м, 2H), 2,538-2,283 (м, 4H), 2,178-2,013 (м, 8H), 1,913-1,872 (м, 2H), 1,680-1,599 (м, 1H).
Соединение 58: (1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-((((1s,3R)-3-(2-(5,6-дихлор-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(метил)амино)-метил)циклопентан-1,2-диол
Диастереомеры разделяли при помощи SFC. Вещество брали для поглощения в MeOH/H2O и лиофилизировали с получением желто-коричневого порошка (73 мг).
1H ЯМР (400 МГц, MeOD) δH м.д. 8,06 (с, 1H), 7,64 (с, 2H), 7,21 (д, J=3,73 Гц, 1H), 6,59 (д, J=3,52 Гц, 1H), 4,32 (дд, J=7,77, 6,12 Гц, 1H), 3,89 (м, 1H), 3,01 (м, 1H), 2,86 (т, J=7,67 Гц, 2H), 2,51 (м, 1H), 2,40 (м, 2H), 2,27 (м, 1H), 2,18 (с, 3H), 2,11 (м, 3H), 2,02 (кв., J=6,43 Гц, 2H),1,88 (т, J=8,19 Гц, 2H), 1,63 (м, 1H).
Соединение 59: (1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-((((1s,3R)-3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(циклобутилметил)- амино)метил)циклопентан-1,2-диол
Стадия 1: этил 3-((1R,3s)-3-((циклобутилметил)(((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)амино)циклобутил)пропаноат
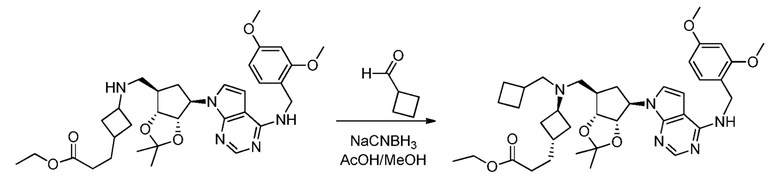
Амин этил 3-[3-({[(3aR,4R,6R,6aS)-6-{4-[(2,4-диметоксибензил)амино]-7H-пирроло[2,3-d]пиримидин-7-ил}-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил]метил}амино)циклобутил]пропаноат (1,8 г, 3,0 ммоль) брали для поглощения в метанол (20 мл, 600 ммоль) и добавляли цианоборогидрид натрия (0,37 г, 5,9 ммоль). pH доводили до приблизительно 6 с использованием 10% раствора AcOH в метаноле, затем добавляли одной порцией циклобутанкарбоксальдегид (0,32 г, 3,8 ммоль). Реакцию осуществляли в течение 5 часов, к этому времени ВЭЖХ анализ показал, что реакция остановилась. Добавляли еще 1,3 эквивалентов циклобутанкарбоксальдегида, и реакция продолжалась в течение ночи. К реакционной смеси добавляли NaHCO3 (насыщенный раствор), затем смесь экстрагировали 3 раза при помощи DCM. Объединенные органические слои сушили при помощи MgSO4 и концентрировали с получением желтого смолистого вещества. Цис и транс изомеры можно было разделить на диоксиде кремния. Осуществляли очистку методом жидкостной хроматографии (DCM/7н NH3 в MeOH 96:4) с получением 2 отдельных партий продукта, каждая из которых была обогащена одним соответствующим изомером до около 90%. Верхний изомер: 0,38 г (5:1 смесь, цис). Нижний изомер: 0,31 г (7:1 смесь, транс). MS (ESI+) для C35H49N5O6 m/z 676,7 [M+H]+; ВЭЖХ чистота >69% (время удерживания, 3,791).
Стадия 2: N-(2-амино-5-(трет-бутил)фенил)-3-((1R,3s)-3-((циклобутилметил)(((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)амино)циклобутил)пропанамид
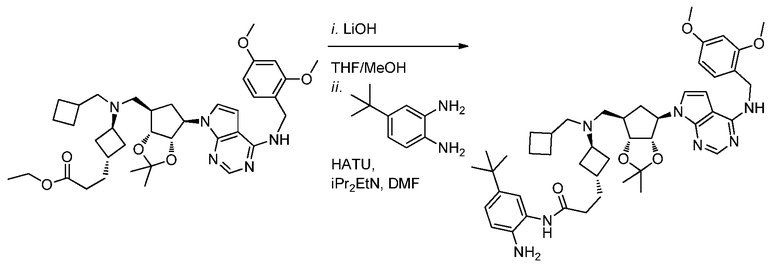
Нижний изомер (транс): Гидроксид лития моногидрат (0,192 г, 4,59 ммоль) добавляли к раствору этил 3-((1R,3s)-3-((циклобутилметил)(((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)амино)циклобутил)пропаноата (0,31 г, 0,46 ммоль) в тетрагидрофуране (6 мл, 70 ммоль) и метаноле (1,5 мл, 37 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре, и к следующему утру исходное вещество было израсходовано и преобразовано в кислоту. Реакционную смесь подкисляли при помощи 1н раствора HCl до pH=6. Летучие вещества удаляли в вакууме и оставшуюся воду удаляли путем азеотропной перегонки с этанолом, с последующей лиофилизацией в течение 24 часов. Полученное не совсем белое твердое вещество использовали без дополнительной очистки. ВЭЖХ чистота >94% (время удерживания 3,344).
Гексафторфосфат N,N,N',N'-тетраметил-O-(7-азабензотриазол-1-ил)урония (0,273 г, 0,718 ммоль) добавляли к раствору 3-{транс-3-[(циклобутилметил){[(3aR,4R,6R,6aS)-6-{4-[(2,4-диметоксибензил)амино]-7H-пирроло[2,3-d]пиримидин-7-ил}-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил]метил}амино]циклобутил}пропановой кислоты (0,31 г, 0,48 ммоль) и N,N-диизопропилэтиламина (0,275 мл, 1,58 ммоль) и 4-трет-бутилбензол-1,2-диамина (0,0943 г, 0,574 ммоль) в N,N-диметилформамиде (2,96 мл, 38,3 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре, и к следующему утру исходное вещество было израсходовано. Реакционную смесь частично концентрировали примерно до 2 мл и затем добавляли NaHCO3 (насыщенный раствор). Смесь экстрагировали при помощи EtOAc 3 раза и объединенные органические слои сушили при помощи MgSO4 и концентрировали. Полученный остаток очищали жидкостной хроматографией (DCM/7н NH3 в MeOH 95:5) с получением N-(2-амино-5-(трет-бутил)фенил)-3-((1R,3s)-3-((циклобутилметил)(((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)амино)циклобутил)пропанамида (0,29 г; 76%) в виде фиолетово-коричневого аморфного твердого вещества. ВЭЖХ чистота >20% (время удерживания 3,650 минут).
Стадия 3: 7-((3aS,4R,6R,6aR)-6-((((1s,3R)-3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(циклобутилметил)амино)метил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-N-(2,4-диметоксибензил)-7H-пирроло[2,3-d]пиримидин-4-амин
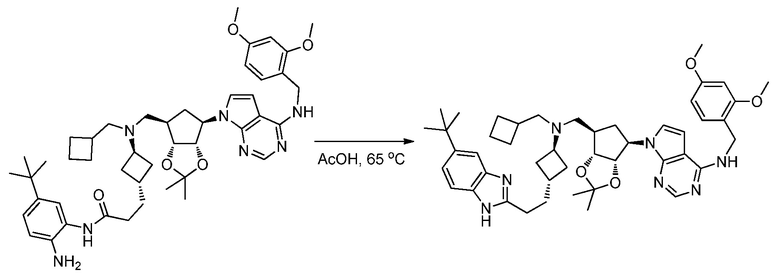
Раствор N-(2-амино-5-(трет-бутил)фенил)-3-((1R,3s)-3-((циклобутилметил)(((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)амино)циклобутил)пропанамида (0,3 г, 0,4 ммоль) в уксусной кислоте (1,0 мл, 20 ммоль) перемешивали в течение ночи при 65oC, и к следующему утру исходное вещество было израсходовано. Летучие вещества удаляли в вакууме и полученный остаток очищали жидкостной хроматографией (DCM/7н NH3 в MeOH 93:7) с получением 7-((3aS,4R,6R,6aR)-6-((((1s,3R)-3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(циклобутилметил)амино)метил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-N-(2,4-диметоксибензил)-7H-пирроло[2,3-d]пиримидин-4-амина в виде не совсем белого твердого вещества. ВЭЖХ чистота >73% (время удерживания 3,709 минут).
Стадия 4: (1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-((((1s,3R)-3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(циклобутилметил)амино)метил)циклопентан-1,2-диол
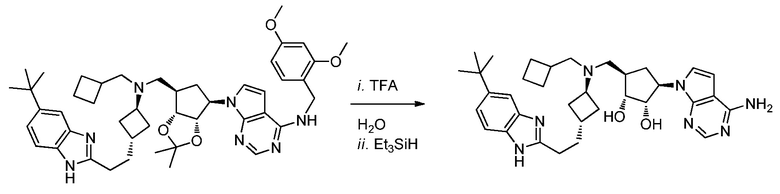
Трифторуксусную кислоту (5 мл, 70 ммоль) добавляли к смеси воды (0,5 мл, 30 ммоль) и 7-((3aS,4R,6R,6aR)-6-((((1s,3R)-3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(циклобутилметил)амино)метил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-N-(2,4-диметоксибензил)-7H-пирроло[2,3-d]пиримидин-4-амина (0,23 г, 0,30 ммоль) при комнатной температуре. Реакцию осуществляли в течение ночи, затем ярко-розовую суспензию гасили триэтилсиланом (0,095 мл, 0,59 ммоль). Летучие вещества удаляли в вакууме и полученный остаток брали для поглощения в MeOH (15 мл). Добавляли 500 мг K2CO3 и 8 капель H2O и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Смесь фильтровали и фильтровальную лепешку промывали при помощи 10 мл метанола. Фильтрат концентрировали и полученный остаток очищали жидкостной хроматографией (DCM/7н NH3 в MeOH 90:10) с получением (1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-((((1s,3R)-3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(циклобутилметил)амино)метил)циклопентан-1,2-диола (0,018 г, 10%) в виде бесцветного твердого вещества. MS (ESI+) для C34H47N7O2 m/z 586,4 [M+H]+; ВЭЖХ чистота >93% (время удерживания, 2,070 минут).
1H ЯМР (400 МГц, d4-MeOH) δH 8,083 (с, 1H), 7,501 (с, 1H), 7,421-7,400 (м, 1H), 7,315-7,290 (м, 1H), 7,218-7,209 (м, 1H), 6,621-6,612 (м, 1H), 4,350-4,317 (м, 1H), 3,930-3,903 (м, 1H), 3,403-3,367 (м, 1H), 2,880-2,843 (м, 2H), 2,722-2,360 (м, 6H), 2,323-2,241 (м, 2H), 2,173-1,606 (м, 13H), 1,387 (с, 9H).
Соединение 60: (1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-((изопропил((1r,3S)-3-(2-(5-(трифторметокси)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)амино)метил)циклопентан-1,2-диол
Диастереоизомеры разделяли при помощи SFC. После лиофилизации выделяли бесцветное твердое вещество (62 мг).
Соединение 61: (1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-((изопропил((1s,3R)-3-(2-(5-(трифторметокси)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)амино)метил)циклопентан-1,2-диол
Диастереоизомеры разделяли при помощи SFC. Вещество брали для поглощения в MeOH/H2O и лиофилизировали с получением не совсем белого порошка (89 мг).
1H ЯМР (400 МГц, MeOD) δH м.д. 8,06 (с, 1H), 7,52 (д, J=8,71 Гц, 1H), 7,40 (с, 1H), 7,19 (д, J=3,52 Гц, 1H), 7,12 (м, 1H), 6,59 (д, J=3,52 Гц, 1H), 4,88 (м, 1H), 4,33 (м, 1H), 3,94 (т, J=5,39 Гц, 1H), 3,52 (м, 1H), 3,01 (м, 1H), 2,87 (т, J=7,15 Гц, 2H), 2,68 (дд, J=13,48, 7,88 Гц, 1H), 2,47 (дд, J=13,27, 7,46 Гц, 1H), 2,37 (м, 1H), 2,21 (м, 3H), 2,04 (м, 3H), 1,84 (м, 2H), 1,58 (м, 1H), 1,02 (д, J=6,63 Гц, 3H), 0,98 (д, J=6,43 Гц, 3H).
Соединение 62: (1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-((метил(3-(2-(5-(оксетан-3-ил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)амино)метил)циклопентан-1,2-диол
N-(4-(оксетан-3-ил)фенил)ацетамид
(4-Ацетамидофенил)бороновую кислоту (670 мг, 3,7 ммоль), иодид никеля(II) (35 мг, 0,11 ммоль), транс-2-аминоциклогексанол (17 мг, 0,11 ммоль) и гексаметилдисилазан натрия (690 мг, 3,7 ммоль) отвешивали в микроволновой реакционный сосуд. Сверху помещали мембрану, продували азотом и добавляли изопропиловый спирт (5,7 мл, 75 ммоль). Сосуд продували азотом в течение 10 минут и добавляли 3-иодоксетан (344 мг, 1,87 ммоль) в 0,75 мл изопропилового спирта. Мембрану заменяли крышкой для микроволнового сосуда и смесь нагревали в микроволновом реакторе (условия микроволнового облучения: микроволновой реактор CEM Discovery Explorer; Линейное изменение времени: 10 минут; 80°C в течение 30 минут; мощность: 300 Вт). Неочищенную реакционную смесь разбавляли при помощи 8 мл EtOH и суспензию фильтровали через слой solka floc®. Этот слой промывали при помощи 35 мл EtOH и фильтрат концентрировали. Неочищенное вещество очищали флэш-хроматографией (SiO2 c элюированием при помощи 40-60% EtOAc/CH2Cl2) с получением желаемого продукта в виде масла (200 мг).
N-(2-нитро-4-(оксетан-3-ил)фенил)ацетамид
Серную кислоту (9,4 мл, 180 ммоль) осторожно добавляли к 70% азотной кислоте (7:3, азотная кислота:вода, 11 мл, 170 ммоль), смесь охлаждали при 0°C примерно в течение 5-10 минут. Смесь перемешивали в течение 10 минут при 0°C, затем давали нагреться до комнатной температуры путем удаления ледяной бани. Кислотный раствор переносили в делительную воронку и добавляли метиленхлорид (20 мл, 300 ммоль). Воронку встряхивали в течение 5 минут и фазам давали разделиться.
Органическую фазу (верхнюю фазу) отделяли и способ повторяли с дополнительным количеством 20 мл CH2Cl2. Органические экстракты объединяли, предполагалось, что органическая фаза содержала около 5 г (~80 ммоль) безводного HNO3. При использовании 50-кратного избытка, это требовало около 25 мл раствора. Раствор азотной кислоты охлаждали на ледяной бане. N-(4-Оксетан-3-илфенил)ацетамид (210 мг, 0,70 ммоль) обрабатывали при помощи 25 мл охлажденного раствора HNO3/CH2Cl2 и оставляли для перемешивания примерно в течение 30 минут. Реакционную смесь осторожно выливали в 45 мл 10% раствора NH4OH и осторожно встряхивали. Фазы разделяли и водную фазу промывали при помощи 20 мл CH2Cl2. Объединенную органическую фазу сушили над Na2SO4, фильтровали и концентрировали. Это неочищенное вещество очищали флэш-хроматографией (SiO2 c элюированием при помощи 25-35% EtOAc/CH2Cl2) с получением желаемого продукта в виде твердого вещества (170 мг).
2-нитро-4-(оксетан-3-ил)анилин
Суспензию N-(2-нитро-4-оксетан-3-илфенил)ацетамида (125 мг, 0,529 ммоль) в водном растворе гидразина (8 мл, 160 ммоль) нагревали при 70°C в течение 2 часов, реакционную смесь охлаждали до 45°C и гидразин удаляли в вакууме с получением твердого вещества. Это неочищенное вещество очищали флэш-хроматографией (SiO2, c элюированием при помощи 20% EtOAc/CH2Cl2, с получением желаемого продукта (71 мг).
4-(оксетан-3-ил)бензол-1,2-диамин
Раствор 2-нитро-4-оксетан-3-иланилина (91 мг, 0,47 ммоль) в этаноле (6,1 мл) осторожно обрабатывали при помощи 10% палладия на углероде (10 мг, 0,009 ммоль) в виде суспензии в этаноле. Из реакционной колбы откачивали газ и колбу заполняли газообразным водородом три раза и реакционную смесь оставляли для перемешивания в атмосфере водорода в течение 2 часов. Реакционную смесь фильтровали через слой solka floc® и этот слой промывали при помощи 25 мл MeOH. Фильтрат концентрировали с получением масла, которое отверждалось в условиях высокого вакуума в течение ночи, с получением желаемого соединения (72 мг). Это вещество использовали как таковое на следующей стадии.
1H ЯМР (400 МГЦ, CDCl3) δH м.д. 6,81 (д, J=1,52 Гц, 1H), 6,70 (м, 2H), 5,02 (дд, J=8,34, 5,81 Гц, 2H), 4,74 (м, 2H), 4,09 (м, 1H),3,45 (шир.с, 2H), 3,37 (шир.с, 2H).
N-(2,4-диметоксибензил)-7-((3aS,4R,6R,6aR)-2,2-диметил-6-((метил(3-(2-(5-(оксетан-3-ил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)амино)метил)тетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-7H-пирроло[2,3-d]пиримидин-4-амин
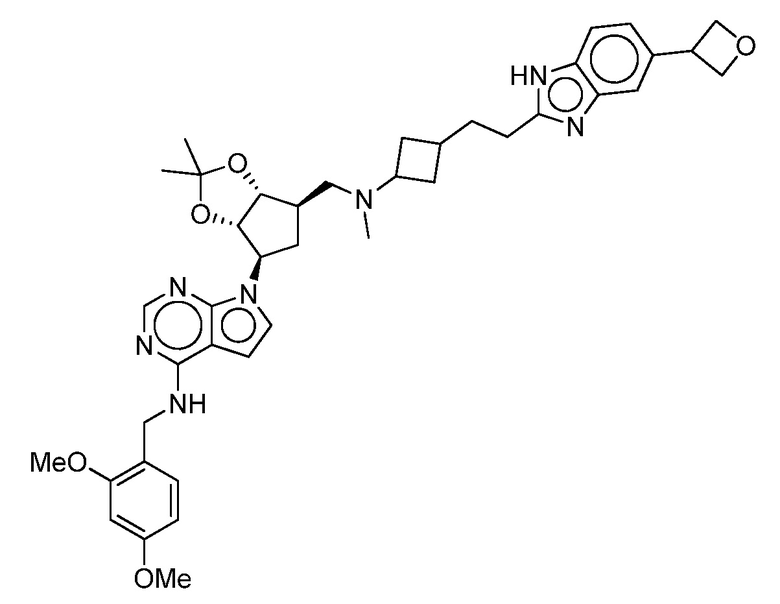
Раствор 3-(3-((((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)(метил)амино)циклобутил)пропановой кислоты (250 мг, 0,42 ммоль) и 4-оксетан-3-илбензол-1,2-диамина (72 мг, 0,44 ммоль) в N,N-диметилформамиде (4,3 мл, 56 ммоль) обрабатывали N,N-диизопропилэтиламином (0,24 мл, 1,4 ммоль) по каплям, с последующим добавлением гексафторфосфата N,N,N',N'-тетраметил-0-(7-азабензотриазол-1-ил)урония (240 мг, 0,632 ммоль) одной порцией. Реакционную смесь перемешивали при комнатной температуре в течение 6 часов, затем реакционную смесь концентрировали в условиях высокого вакуума. Остаток распределяли между 30 мл EtOAc (добавляли некоторое количество MeOH для облегчения солюбилизации продукта) и 30 мл смеси 1/1 H2O/насыщенный раствор NaHCO3. Водную фазу экстрагировали при помощи 30 мл EtOAc и объединенную органическую фазу сушили над Na2SO4, фильтровали и концентрировали с получением стеклообразного вещества/жесткой пены. Неочищенное вещество очищали флэш-хроматографией (SiO2, c элюированием при помощи 6-7% 7н NH3 в CH3OH/CH2Cl2. Было обнаружено два комплекта продуктов, менее полярная пара и более полярная пара, что соответствовало 2 региоизомерам. Каждый региоизомер перерабатывали отдельно на следующей стадии.
Амид (130 мг) брали для поглощения в 5 мл ледяную уксусную кислоту и нагревали при 65°C в течение 2,25 часов, реакционную смесь охлаждали и помещали в холодильник на ночь. Уксусную кислоту удаляли в условиях высокого вакуума с использованием теплой водяной бани. Две партии неочищенного продукта брали для поглощения в 30 мл CH2Cl2 и органическую фазу промывали 10 мл порциями насыщенного раствора NaHCO3 и 2% раствора Na2CO3, сушили над Na2SO4, фильтровали и концентрировали. Неочищенное вещество очищали флэш-хроматографией (SiO2 c элюированием при помощи 5,5-6,5% 7н NH3 в CH3OH/CH2Cl2, с получением желаемого соединения (140 мг).
(1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-((метил(3-(2-(5-(оксетан-3-ил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)амино)метил)циклопентан-1,2-диол
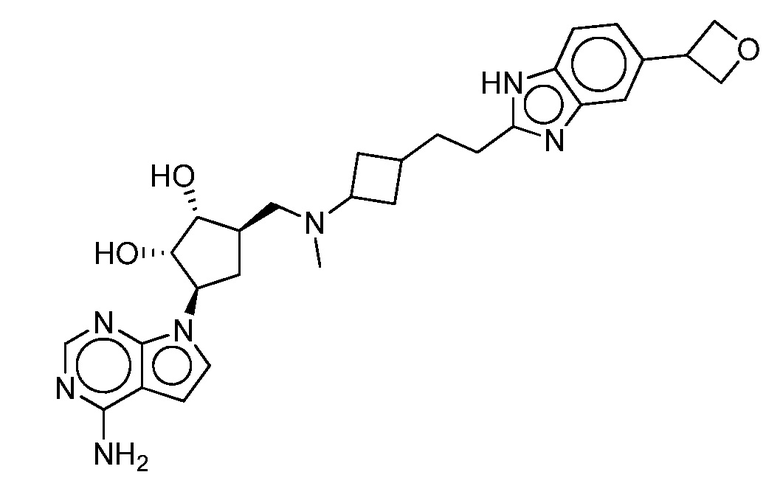
N-(2,4-диметоксибензил)-7-((3aS,4R,6R,6aR)-2,2-диметил-6-((метил(3-(2-(5-(оксетан-3-ил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)амино)метил)тетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-7H-пирроло[2,3-d]пиримидин-4-амин (115 мг, 0,159 ммоль) растворяли в смеси трифторуксусной кислоты (4,00 мл, 51,9 ммоль) и воды (0,40 мл, 22 ммоль), которая была предварительно охлаждена при 0°C на ледяной бане. Раствор перемешивали при 0°C в течение 2 часов, реакционной смеси давали нагреться до комнатной температуры. Через 1 час реакционную смесь концентрировали в вакууме. Остаток брали для поглощения в 6 мл MeOH, концентрировали и этот способ повторяли два раза. Полученный остаток помещали в условия высокого вакуума. Неочищенный остаток разбавляли при помощи 2 мл MeOH, обрабатывали при помощи 140 мг K2CO3 и 10 капель H2O и оставляли для перемешивания при комнатной температуре, пока раствор не становился щелочным, как определяли при помощи pH бумаги. Раствор фильтровали через тонкую фритту и твердые вещества промывали при помощи MeOH. Фильтрат концентрировали с получением твердого вещества, которое помещали в условия высокого вакуума на ночь. Неочищенное вещество очищали методом препаративной ТСХ на двух 20 см × 20 см × 1,0 мм пластинах для препаративной ТСХ, c элюированием при помощи 14% 7н NH3 в CH3OH/CH2Cl2, с получением продукта в виде бесцветного стеклообразного вещества (37 мг).
1H ЯМР (400 МГц, MeOD) δH м.д. 8,06 (с, 1H), 7,52 (шир.с, 1H), 7,48 (д, J=8,29 Гц, 1H), 7,28 (м, 1H), 7,20 (т, J=3,42 Гц, 1H), 6,60 (д, J=3,52 Гц, 1H), 5,12 (м, 2H), 4,80 (м, 2H), 4,38 (м, 1H), 4,32 (м, 1H), 3,89 (кв., J=5,60 Гц, 1H), 3,04 (м, 1H), 2,85 (м, 2H), 2,70 (м, 1H), 2,52 (м, 1H), 2,41 (м, 2H), 2,27 (дд, J=10,99, 6,63 Гц, 2H), 2,19 (с, 3H), 2,17 (с, 3H), 2,14 (м, 1H), 2,03 (д, J=7,88 Гц, 1H), 1,91 (м, 3H), 1,62 (м, 1H), 1,51 (м, 1H).
Соединение 63: (2R,3R,4S,5R)-2-(6-амино-9H-пурин-9-ил)-5-((метил((1r,3S)-3-(2-(5-(оксетан-3-ил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)амино)метил)тетрагидрофуран-3,4-диол
Диастереомеры разделяли при помощи SFC. Вещество брали для поглощения в MeOH/H2O и лиофилизировали с получением желто-коричневого порошка (58 мг).
1H ЯМР (400 МГц, MeOD) δH м.д. 8,26 (с, 1H), 8,19 (с, 1H), 7,51 (с, 1H), 7,47 (д, J=8,29 Гц, 1H), 7,26 (дд, J=8,29, 1,45 Гц, 1H), 5,97 (д, J=3,94 Гц, 1H), 5,11 (дд, J=8,29, 6,01 Гц, 2H), 4,79 (т, J=6,32 Гц, 2H), 4,69 (м, 1H), 4,36 (м, 1H), 4,22 (т, J=5,60 Гц, 1H), 4,15 (м, 1H), 2,79 (т, J=7,15 Гц, 2H), 2,72 (м, 1H), 2,66 (м, 2H), 2,21 (м, 2H), 2,14 (с, 3H), 1,88 (м, 3H), 1,45 (м, 2H).
Соединение 64: (2R,3R,4S,5R)-2-(6-амино-9H-пурин-9-ил)-5-((метил(3-(2-(5-(оксетан-3-ил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)амино)метил)тетрагидрофуран-3,4-диол
Стадия 1: 3-(3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(метил)амино)циклобутил)пропановая кислота
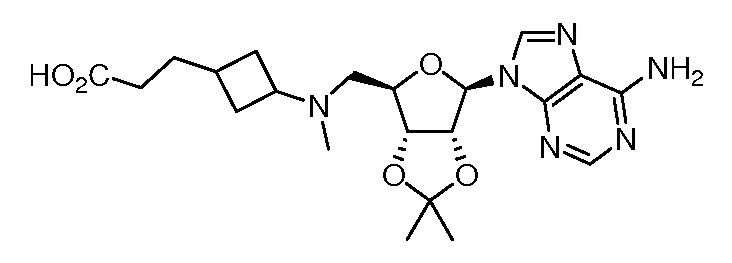
Раствор этил 3-(3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(метил)амино)циклобутил)пропаноата (0,39 г, 0,82 ммоль) в метаноле (14 мл) обрабатывали 1 M водным раствором гидроксида натрия (1,56 мл, 1,56 ммоль) и реакционную смесь нагревали при 50°C при перемешивании в течение 3,5 часов; ВЭЖХ/ЖХ-MS анализ показал преобразование в желаемый продукт. Реакционную смесь концентрировали в вакууме и водный остаток разбавляли водой (10 мл) и экстрагировали при помощи CH2Cl2 (3×5 мл). Водный слой обрабатывали 1 M водным раствором хлористого водорода (1,44 мл, 1,44 ммоль) для доведения до pH 7. Прозрачный бесцветный раствор лиофилизировали с получением неочищенного указанного в заголовке соединения (0,487 г, 110%) в виде слегка беловатого твердого вещества, выход с учетом 1,56 ммоль NaCl (91 мг): MS (ESI+) для C21H30N6O5 m/z 447,1 (M+H)+; MS (ESI-) для C21H30N6O5 m/z 445,2 (M-H)-; ВЭЖХ чистота >95% (время удерживания, 1,949 мин).
Стадия 2: N-(2-амино-5-(оксетан-3-ил)фенил)-3-(3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(метил)амино)циклобутил)пропанамид
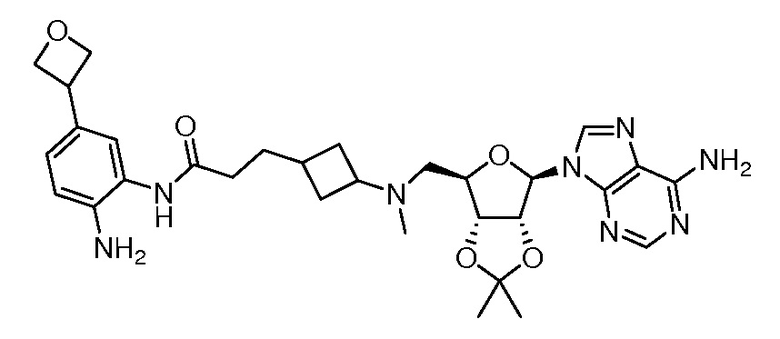
Суспензию полученной на предыдущей стадии неочищенной 3-(3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(метил)амино)циклобутил)пропановой кислоты и 4-(оксетан-3-ил)бензол-1,2-диамина (0,135 г, 0,822 ммоль) в метиленхлориде (8,0 мл) обрабатывали N,N-диизопропилэтиламином (0,716 мл, 4,11 ммоль) и охлаждали до -5°C (лед/насыщенный солевой раствор). Добавляли гексафторфосфат N,N,N',N'-тетраметил-O-(7-азабензотриазол-1-ил)урония [HATU] (0,469 г, 1,23 ммоль) и реакционную смесь перемешивали в течение 5,25 часов с нагреванием до 15°C; ВЭЖХ/ЖХ-MS анализ показал полное преобразование. Реакционную смесь концентрировали в вакууме и разбавляли при помощи CH2Cl2 (15 мл) и воды (7,5 мл). Отделенный водный слой экстрагировали при помощи CH2Cl2 (2×10 мл). Объединенные органические слои сушили (Na2SO4) и концентрировали в вакууме с получением коричневато-фиолетового полупрозрачного маслянистого/пенистого вещества. Очистка колоночной хроматографией (2×8 см диоксид кремния; 0-5% 7н метанольный раствор NH3/CH2Cl2) давала оба амидных региоизомера указанного в заголовке соединения (0,45 г, 82%) в виде полупрозрачного розового пенообразного вещества. MS (ESI+) для C30H40N8O5 m/z 593,3 (M+H)+; MS (ESI-) для C30H40N8O5 m/z 591,3 (M-H)- и 637,4 (M+HCO2)-; ВЭЖХ чистота 90% (время удерживания, 2,097 мин).
Стадия 3: 9-((3aR,4R,6R,6aR)-2,2-диметил-6-((метил(3-(2-(5-(оксетан-3-ил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)амино)метил)тетрагидрофуро[3,4-d][1,3]диоксол-4-ил)-9H-пурин-6-амин
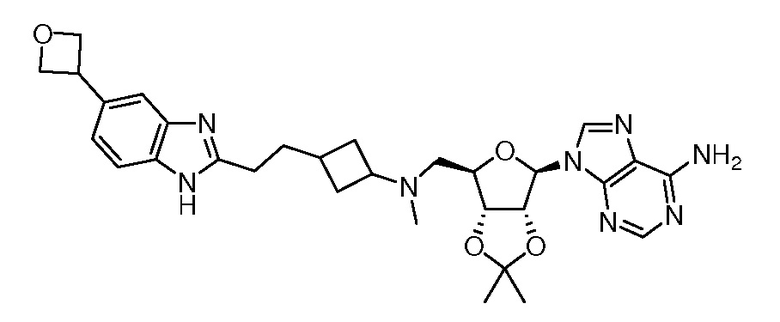
N-(2-амино-5-(оксетан-3-ил)фенил)-3-(3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(метил)амино)циклобутил)пропанамид (0,446 г, 0,752 ммоль) брали для поглощения в уксусную кислоту (7,7 мл, 140 ммоль) и нагревали при 65°C в течение 3,5 часов; ВЭЖХ/ЖХ-MS анализ показал полное преобразование. Через 3,75 часа уксусную кислоту удаляли дистилляцией с минимальным нагревом, с получением оранжевого масла, которое брали для поглощения в CH2Cl2 (45 мл) и промывали насыщенным водным раствором NaHCO3 (2×30 мл). Водный слой обрабатывали при помощи NaCl до насыщения и экстрагировали при помощи CH2Cl2 (2×20 мл). Объединенные органические слои сушили (Na2SO4) и концентрировали в вакууме с получением светло-оранжевого масла. Очистка колоночной хроматографией (2×8 см диоксид кремния; 0-5% 7н метанольный раствор NH3/CH2Cl2) давала указанное в заголовке соединение (0,28 г, 65%) в виде светло-оранжевого пенообразного вещества. MS (ESI+) для C30H38N8O4 m/z 575,3 (M+H)+; MS (ESI-) для C30H38N8O4 m/z 573,3 (M-H)-; ВЭЖХ чистота >95% (время удерживания 2,142 мин).
Стадия 4: (2R,3R,4S,5R)-2-(6-амино-9H-пурин-9-ил)-5-((метил(3-(2-(5-(оксетан-3-ил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)амино)метил)тетрагидрофуран-3,4-диол
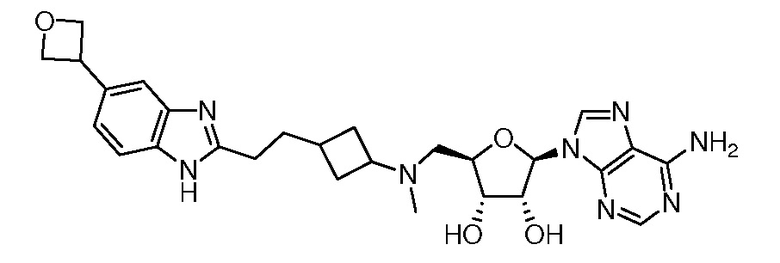
В охлажденную (ледяная баня) колбу, содержащую 9-((3aR,4R,6R,6aR)-2,2-диметил-6-((метил(3-(2-(5-(оксетан-3-ил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)амино)метил)тетрагидрофуро[3,4-d][1,3]диоксол-4-ил)-9H-пурин-6-амин (0,28 г, 0,42 ммоль), добавляли предварительно охлажденный (ледяная баня) раствор трифторуксусной кислоты (6,4 мл, 84 ммоль) в воде (0,75 мл, 42 ммоль). Реакционную смесь перемешивали в течение 5,75 часов при 0°C; ВЭЖХ/ЖХ-MS анализ показал, что исходное вещество почти полностью израсходовано. По прошествии 6 часов колбу удаляли из охлаждающей бани и летучие вещества удаляли дистилляцией при комнатной температуре. Остаток разбавляли при помощи MeOH (15 мл) и обрабатывали карбонатом калия (0,32 г, 2,3 ммоль) и водой (1 мл) и смесь перемешивали в течение 20 минут при комнатной температуре; pH 2. Добавляли дополнительное количество карбоната калия (0,20 г, 1,4 ммоль) и смесь перемешивали в течение 20 минут; pH 8-9. Раствор фильтровали через тонкую фритту, с промывкой при помощи MeOH и фильтрат концентрировали в вакууме с получением желто-коричневого полутвердого вещества. Очистка колоночной хроматографией (3×8 см диоксид кремния; 10-20% 7н метанольный раствор NH3/CH2Cl2) давала указанное в заголовке соединение (123 мг, 55%) в виде почти бесцветного стеклообразного вещества: MS (ESI+) для C27H34N8O4 m/z 535,3 (M+H)+; MS (ESI-) для C27H34N8O4 m/z 533,3 (M-H)-; ВЭЖХ чистота >95% (время удерживания 1,765 мин);
1H ЯМР (400 МГц, d4-MeOH) смесь цис/транс изомеров δH 8,29-8,25 (м, 1H), 8,21-8,17 (м, 1H), 7,51 (с, 1H), 7,47 (д, J=8,3 Гц, 1H), 7,26 (дд, J=1,6, 8,3 Гц, 1H), 6,00-5,96 (м, 1H), 5,11 (дд, J=5,8, 8,3 Гц, 2H), 4,81-4,76 (м, 2H), 4,73-4,68 (м, 1H), 4,40-4,31 (м, 1H), 4,27-4,13 (серия м, 2H), 3,13-3,03 (м, 0,4H), 2,86-2,66 (серия м, 4,6H), 2,30-1,80 (серия м, 8,6H), 1,55-1,40 (м, 1,4H).
Соединение 65: (2R,3R,4S,5R)-2-(6-амино-9H-пурин-9-ил)-5-((метил((1s,3R)-3-(2-(5-(оксетан-3-ил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)амино)метил)тетрагидрофуран-3,4-диол
Диастереомеры разделяли при помощи SFC. Это вещество брали для поглощения в MeOH/H2O и лиофилизировали с получением белого порошка (35 мг).
1H ЯМР (400 МГц, MeOD) δH м.д. 8,28 (с, 1H), 8,19 (с, 1H), 7,51 (с, 1H), 7,48 (д, J=8,29 Гц, 1H), 7,27 (дд, J=8,29, 1,45 Гц, 1H), 5,98 (д, J=4,15 Гц, 1H), 5,12 (дд, J=8,40, 5,91 Гц, 2H), 4,79 (т,J=6,32 Гц, 2H), 4,69 (дд, J=5,39, 4,15 Гц, 1H), 4,36 (м, 1H), 4,23 (т, J=5,60 Гц, 1H), 4,17 (м,1H), 3,05 (м, 1H), 2,83 (т, J=7,46 Гц, 2H), 2,67 (м, 2H), 2,16 (с, 3H), 2,08 (м, 2H), 1,98 (м, 3H), 1,83 (м, 2H).
Соединение 67: (2R,3R,4S,5R)-2-(6-амино-9H-пурин-9-ил)-5-[({3-[2-(5-циклобутил-1H-1,3-бензодиазол-2-ил)этил]циклобутил}(пропан-2-ил)амино)метил]оксолан-3,4-диол
Стадия 1: Бензил 3-[3-({[(3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметил-тетрагидро-2H-фуро[3,4-d][1,3]диоксол-4-ил]метил}амино)циклобутил]пропаноат
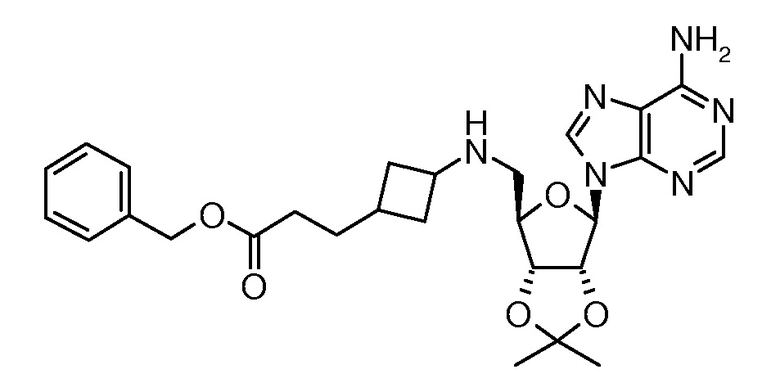
Суспензию 9-[(3aR,4R,6R,6aR)-6-(аминометил)-2,2-диметил-тетрагидро-2H-фуро[3,4-d][1,3]диоксол-4-ил]-9H-пурин-6-амина (1,45 г, 4,736 ммоль), бензил 3-(3-оксоциклобутил)пропаноата (1,21 г, 5,209 ммоль) и уксусной кислоты (246,45 мкл, 4,31 ммоль) в DCE:iPrOH (4:1, 50 мл) перемешивали при комнатной температуре в течение 1 часа. К реакционной смеси добавляли еще одну аликвоту DCE (40 мл) и iPrOH (5 мл) и реакцию продолжали в течение 1 часа. Затем добавляли STAB (1,28 г, 6,03 ммоль) и реакционную смесь перемешивали в течение 18 часов. Реакцию гасили 1н раствором Na2CO3 (10 мл) и продукт экстрагировали при помощи DCM (2×30 мл). Смесь сушили над Na2SO4, фильтровали и упаривали досуха. Очистка колоночной хроматографией на силикагеле, с элюированием смесью 7н NH3 в MeOH:DCM (1:99-3:97), давала желаемый продукт в виде бесцветного масла, 1,51 г (58%); MS (ESI+) для C27H34N6O5 m/z 523,65 [M+H]+; ВЭЖХ чистота 97% (время удерживания, 1,43 мин);
1H ЯМР (500 МГц, ХЛОРОФОРМ-d) δH м.д. 8,35 (д, J=5,3 Гц, 1H), 7,87 (д, J=33,8 Гц, 1H), 7,40-7,29 (м, 5H), 6,08-5,94 (м, 1H), 5,59-5,42 (м, 3H), 5,10 (д, J=4,0 Гц, 2H), 5,03-4,96 (м, 1H), 4,33 (дкв., J=7,3, 3,9 Гц, 1H), 3,12 (ддд, J=23,0, 14,6, 7,5 Гц, 1H), 2,85-2,77 (м, 1H), 2,74 (дд, J=12,5, 6,6 Гц, 1H), 2,39-2,07 (м, 4H), 1,90-1,64 (м, 5H), 1,61 (с, 4H), 1,38 (с, 3H), 1,28-1,06 (м, 1H).
Стадия 2. Бензил 3-[3-({[(3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметил-тетрагидро-2H-фуро[3,4-d][1,3]диоксол-4-ил]метил}(пропан-2-ил)амино)циклобутил]пропаноат
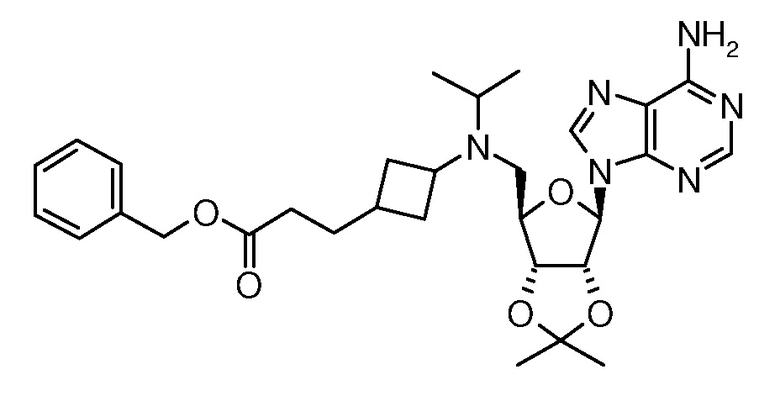
K2CO3 (528,92 мг, 3,83 ммоль) добавляли к раствору бензил 3-[3-({[(3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметил-тетрагидро-2H-фуро[3,4-d][1,3]диоксол-4-ил]метил}амино)циклобутил]пропаноата (1,00 г, 1,91 ммоль) и 2-иодпропана (0,57 мл, 5,74 ммоль) в MeCN и перемешивали при 95oC в герметично закрытой пробирке в течение 18 часов. Реакционную смесь разбавляли при помощи EtOAc (20 мл), фильтровали и упаривали досуха. Очистка хроматографией на силикагеле, c элюированием смесью 7н NH3 в MeOH:DCM (1:99-5:95), давала желаемый продукт в виде бесцветного масла, 700 мг (65%); MS (ESI+) для C30H40N6O5 m/z 565,70 [M+H]+; ВЭЖХ чистота 96% (время удерживания, 1,48 мин);
1H ЯМР (500 МГц, ХЛОРОФОРМ-d) δH м.д. 8,35 (д, J=3,5 Гц, 1H), 7,88 (д, J=3,2 Гц, 1H), 7,44-7,29 (м, 5H), 6,03 (т, J=2,2 Гц, 1H), 5,62-5,42 (м, 3H), 5,10 (д, J=3,3 Гц, 2H), 5,06-4,92 (м, 1H), 4,26 (дт, J=9,9, 3,4 Гц, 1H), 3,46-2,84 (м, 2H), 2,88-2,61 (м, 1H), 2,51 (ддд, J=14,0, 9,1, 7,5 Гц, 1H), 2,33-2,15 (м, 2H), 2,50-2,13 (м, 2H), 2,16-1,74 (м, 4H), 1,60 (с, 3H), 1,43-1,35 (м, 4H), 0,96 (д, J=6,7 Гц, 3H), 0,79 (д, J=6,6 Гц, 3H).
Стадия 3. 3-[3-({[(3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметил-тетрагидро-2H-фуро[3,4-d][1,3]диоксол-4-ил]метил}(пропан-2-ил)амино)циклобутил]пропановая кислота
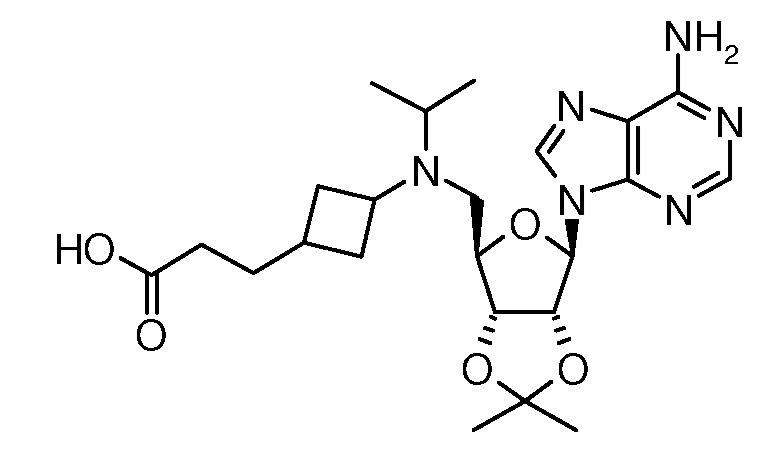
10% Pd-C (70 мг) добавляли к раствору бензил 3-[3-({[(3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметил-тетрагидро-2H-фуро[3,4-d][1,3]диоксол-4-ил]метил}(пропан-2-ил)амино)циклобутил]пропаноата (790 мг, 1,40 ммоль) в EtOH (20 мл) и перемешивали в атмосфере водорода в течение 18 часов при комнатной температуре. Добавляли еще одну аликвоту 10% Pd-C (70 мг) и продолжали перемешивание реакционной смеси в атмосфере водорода в течение 4 часов. Смесь фильтровали и упаривали в вакууме и затем выпаривали из DCM (2×20 мл) с получением 680 мг (количественный выход) белого пенистого твердого вещества; MS (ESI+) для C23H34N6O5 m/z 475,20 [M+H]+; ВЭЖХ чистота 100% (время удерживания, 1,11 мин);
1H ЯМР (500 МГц, ХЛОРОФОРМ-d) δH м.д. 8,29 (д, J=16,3 Гц, 1H), 7,97 (д, J=16,2 Гц, 1H), 6,86 (с, 2H), 6,05 (дд, J=4,3, 1,7 Гц, 1H), 5,66-5,43 (м, 1H), 5,00 (ддд, J=19,3, 6,3, 3,2 Гц, 1H), 4,30 (с, 1H), 3,47-2,85 (м, 2H), 2,60 (ддд, J=38,8, 24,1, 13,5 Гц, 2H), 2,19 (ддд, J=14,7, 11,9, 7,1 Гц, 2H), 2,07-1,94 (м, 2H), 1,81 (дд, J=65,1, 6,9 Гц, 3H), 1,66-1,46 (м, 5H), 1,45-1,22 (м, 4H), 1,00 (д, J=6,4 Гц, 3H), 0,89 (дд, J=12,2, 6,6 Гц, 3H).
Стадия 4. 3-[3-({[(3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметил-тетрагидро-2H-фуро[3,4-d][1,3]диоксол-4-ил]метил}(пропан-2-ил)амино)циклобутил]-N-(2-амино-4/5-циклобутилфенил)пропанамид
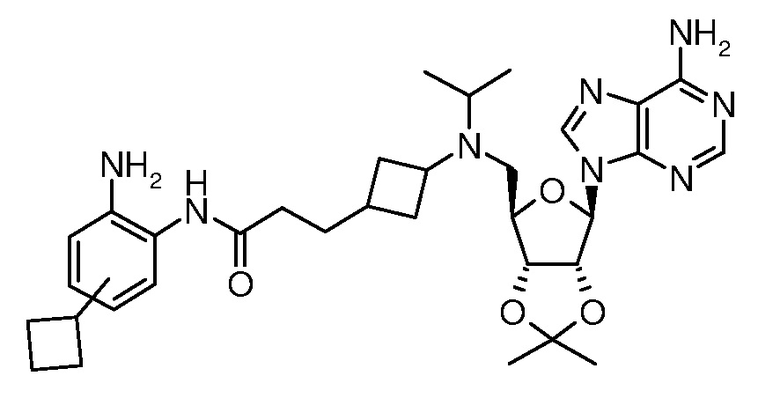
TEA (0,54 мл, 3,90 ммоль) добавляли к раствору 3-[3-({[(3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметил-тетрагидро-2H-фуро[3,4-d][1,3]диоксол-4-ил]метил}(пропан-2-ил)амино)циклобутил]пропановой кислоты (308,46 мг, 0,65 ммоль), 4-циклобутилбензол-1,2-диамина (210,90 мг, 1,30 ммоль), этил (2E)-циано(гидроксиимино)этаноата (184,75 мг, 1,30 ммоль) и EDC.HCl (249,21 мг, 1,30 ммоль) в DCM (15 мл) при комнатной температуре и перемешивали в течение двух часов. Реакционную смесь концентрировали в вакууме, затем добавляли DCM (50 мл). Смесь промывали насыщенным раствором NaHCO3 (2×30 мл). Водный слой экстрагировали при помощи DCM (50 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и упаривали. Продукт очищали колоночной хроматографией на силикагеле, элюировали при помощи EtOAc и затем 7н NH3 в MeOH:DCM (5:95), с получением серого масла, 468 мг (93%); MS (ESI+) для C33H46N8O4 m/z 619,35 [M+H]+; ВЭЖХ чистота 80% (время удерживания, 1,44 мин).
Стадия 5. 9-[(3aR,4R,6R,6aR)-6-[({3-[2-(5-циклобутил-1H-1,3-бензодиазол-2-ил)этил]циклобутил}(пропан-2-ил)амино)метил]-2,2-диметил-тетрагидро-2H-фуро[3,4-d][1,3]диоксол-4-ил]-9H-пурин-6-амин
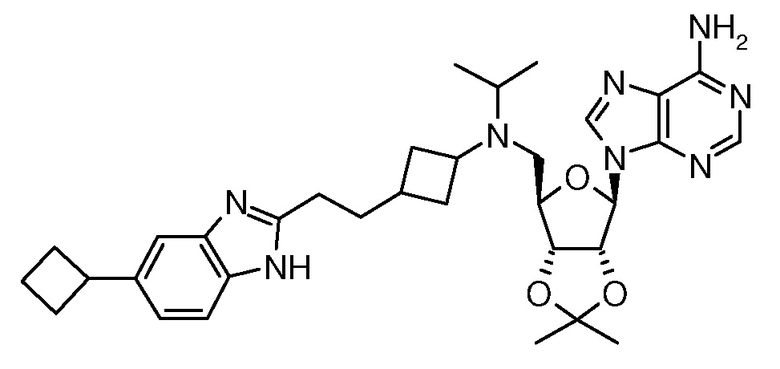
AcOH (10 мл) добавляли к 3-[3-({[(3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметил-тетрагидро-2H-фуро[3,4-d][1,3]диоксол-4-ил]метил}(пропан-2-ил)амино)циклобутил]-N-(2-амино-4/5-циклобутилфенил)пропанамиду (468 мг, 0,61 ммоль) и нагревали до 65oC при перемешивании в течение 4 часов. Реакционную смесь концентрировали в вакууме, затем растворяли в DCM (100 мл) и промывали насыщенным раствором NaHCO3 (2×80 мл), сушили над Na2SO4, фильтровали и упаривали. Очистка флэш-хроматографией на силикагеле (Biotage, Isolera, 25 г картридж), c элюированием смесью 3н раствор аммиака в MeOH:DCM (0-1:9) давала желаемый продукт с чистотой приблизительно 80%. Дополнительная очистка методом препаративной ВЭЖХ давала желаемый продукт в виде серого масла, 120 мг (27%); MS (ESI+) для C33H44N8O3 m/z 601 [M+H]+; ВЭЖХ чистота 100% (время удерживания, 1,43 мин);
1H ЯМР (500 МГц, ХЛОРОФОРМ-d) δH м.д. 8,62 (д, J=53,1 Гц, 6H), 8,57 (с, 2H), 8,25 (д, J=19,3 Гц, 1H), 7,90 (д, J=20,3 Гц, 1H), 7,50 (дд, J=8,3, 3,9 Гц, 1H), 7,41 (д, J=4,5 Гц, 1H), 7,18-7,06 (м, 1H), 6,52 (д, J=96,3 Гц, 2H), 6,07 (дд, J=10,2, 1,3 Гц, 1H), 5,52-5,39 (м, 1H), 5,07 (дд, J=6,2, 3,4 Гц, 1H), 4,44 (тд, J=9,3, 5,1 Гц, 1H), 3,59 (дкв., J=17,4, 8,7 Гц, 1H), 3,36-3,10 (м, 2H), 3,08-2,92 (м, 2H), 2,87-2,72 (м, 2H), 2,43-2,27 (м, 2H), 2,24-1,65 (м, 10H), 1,57 (с, 4H), 1,37 (с, 3H), 1,10 (д, J=6,6 Гц, 3H), 0,91 (дд, J=8,9, 6,8 Гц, 3H).
Стадия 6. (2R,3R,4S,5R)-2-(6-амино-9H-пурин-9-ил)-5-[({3-[2-(5-циклобутил-1H-1,3-бензодиазол-2-ил)этил]циклобутил}(пропан-2-ил)амино)метил]оксолан-3,4-диол
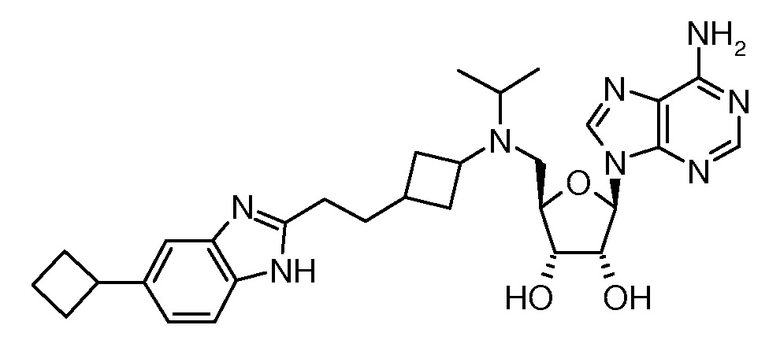
12н раствор HCl (24 ммоль, 2 мл) добавляли по каплям к раствору 9-[(3aR,4R,6R,6aR)-6-[({3-[2-(5-циклобутил-1H-1,3-бензодиазол-2-ил)этил]циклобутил}(пропан-2-ил)амино)метил]-2,2-диметил-тетрагидро-2H-фуро[3,4-d][1,3]диоксол-4-ил]-9H-пурин-6-амина (120 мг, 0,164 ммоль) в MeOH (2 мл) при 0oC при перемешивании. Смеси затем давали нагреться до комнатной температуры и продолжали перемешивание в течение 6 часов. Реакционную смесь охлаждали до 0oC и подщелачивали при помощи 7н раствора NH3 в MeOH (10 мл). Смесь затем упаривали в вакууме. Неочищенный продукт абсорбировали на силикагеле (1 мл), помещали на Si флэш-картридж (10 г) и очищали, c элюированием смесью 7н NH3 в MeOH:DCM (1:9), с получением белого твердого вещества, 36 мг (38%); MS (ESI+) для C30H40N8O3 m/z 561,45 [M+H]+; ВЭЖХ чистота 100% (время удерживания, 1,13 мин);
1H ЯМР (500 МГц, ХЛОРОФОРМ-d) δH м.д. 8,29 (д, J=4,6 Гц, 1H), 8,20 (д, J=1,6 Гц, 1H), 7,50-7,18 (м, 2H), 7,06 (ддд, J=8,3, 4,6, 1,3 Гц, 1H), 6,01-5,90 (м, 1H), 4,73 (дд, J=9,8, 5,1 Гц, 1H), 4,26 (кв., J=5,4 Гц, 1H), 4,14-4,03 (м, 1H), 3,60-3,15 (м, 2H), 3,07-2,86 (м, 2H), 2,84-2,67 (м, 3H), 2,42-2,31 (м, 2H), 2,25-2,11 (м, 4H), 2,10-1,95 (м, 2H), 1,92-1,74 (м, 4H), 1,57 (дд, J=12,2, 6,2 Гц, 1H), 1,02 (дд, J=6,6, 4,0 Гц, 3H), 0,95 (дд, J=6,6, 2,3 Гц, 3H).
Соединение 68: (2R,3R,4S,5R)-2-(6-Амино-9H-пурин-9-ил)-5-{[(3-{2-[5-(1-метокси-2-метилпропан-2-ил)-1H-1,3-бензодиазол-2-ил]этил}циклобутил)(метил)амино]метил}оксолан-3,4-диол
Стадия 1: Бензил 3-[3-({[(3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметил-тетрагидро-2H-фуро[3,4-d][1,3]диоксол-4-ил]метил}амино)циклобутил]пропаноат
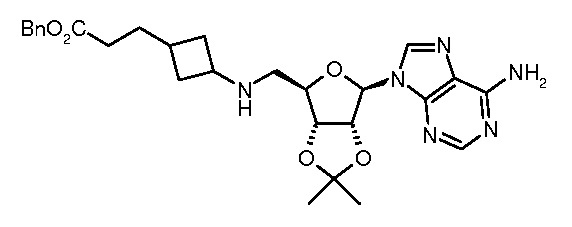
Суспензию 9-[(3aR,4R,6R,6aR)-6-(аминометил)-2,2-диметил-тетрагидро-2H-фуро[3,4-d][1,3]диоксол-4-ил]-9H-пурин-6-амина (5,00 г, 16,3 ммоль), бензил 3-(3-оксоциклобутил)-пропаноата (4,17 г, 18,0 ммоль) и уксусной кислоты (0,85 мл, 14,8 ммоль) в DCE:iPrOH (7:2) (90 мл) перемешивали при комнатной температуре в течение 2 часов. Добавляли по порциям триацетоксиборогидрид натрия (4,40 г, 20,8 ммоль) и смесь оставляли для перемешивания в течение 18 часов при комнатной температуре. Реакцию гасили при помощи раствора 1M Na2CO3 (10 мл) и продукт экстрагировали при помощи DCM (3×30 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и упаривали досуха. Очистка колоночной флэш-хроматографией на силикагеле, с элюированием смесью 1% 7M NH3 в MeOH:99% DCM, давала продукт в виде желтого масла (5,25 г, 55%, чистота 89%): MS (ESI+) для C27H34N6O5 m/z 523,6 [M+H]+; ЖХ чистота 89% (время удерживания, 1,60 мин);
1H ЯМР (500 МГц, CDCl3) δH 8,35 (д, J=6,0 Гц, 1H), 7,91 (с, 1H), 7,30-7,39 (м, 5H), 6,01 (дд, J=3,0 Гц, 1,6, 1H), 5,72 (шир.с, 2H), 5,50 (дт, J=6,4 Гц, 3,3, 1H), 5,10 (д, J=3,8 Гц, 2H), 4,98-5,04 (м, 1H), 4,31-4,38 (м, 1H), 2,97-3,34 (м, 1H), 2,72-2,85 (м, 2H), 2,22-2,35 (м, 3H), 2,14 (тд, J=8,3, 4,3 Гц, 1H), 1,73-1,90 (м, 4H), 1,64-1,71 (м, 1H,), 1,62 (д, J=1,4 Гц, 3H), 1,39 (с, 3H), 1,10-1,27 (м, 1H).
Стадия 2: Бензил 3-[3-({[(3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметил-тетрагидро-2H-фуро[3,4-d][1,3]диоксол-4-ил]метил}(метил)амино)циклобутил]пропаноат
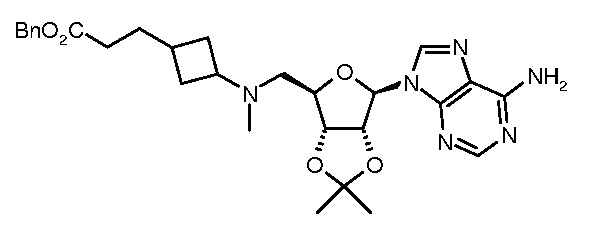
Бензил 3-[3-({[(3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметил-тетрагидро-2H-фуро[3,4-d][1,3]диоксол-4-ил]метил}амино)циклобутил]пропаноат (3,2 г, 6,12 ммоль) растворяли в метаноле (32 мл). Добавляли раствор формальдегида в воде (37%) (0,92 мл, 12,3 ммоль) и перемешивали в течение 45 минут, затем добавляли цианоборогидрид натрия (0,54 г, 8,57 ммоль) по порциям. Реакционную смесь перемешивали в течение 2 часов при комнатной температуре, затем добавляли воду (1 мл) и растворитель выпаривали при комнатной температуре. Остаток очищали хроматографией с использованием смеси 7M аммиака в метаноле/DCM, с получением желаемого соединения в виде желтого масла (смесь диастереомеров) (2,05 г, 62%, чистота 82%): MS (ESI+) для C28H36N6O5 m/z 537,6 [M+H]+; ЖХ чистота 82% (время удерживания, 1,60 мин);
1H ЯМР (500 МГц, CDCl3) δH 8,32-8,38 (м, 1H), 7,91-7,97 (м, 1H), 7,32-7,39 (м, 5H), 6,05-6,10 (м, 1H), 5,61 (шир.с, 2H), 5,53 (ддд, J=16,5, 6,4, 1,8 Гц, 1H), 5,11 (м, 2H), 4,93-5,00 (м, 1H), 4,32-4,40 (м, 1H), 2,50-2,88 (м, 1H), 2,37-2,49 (м, 2H), 2,21-2,30 (м, 2H), 2,10 (м, 3H), 1,91-2,04 (м, 1H), 1,73-1,79 (м, 2H), 1,64-1,72 (м, 2H), 1,56-1,63 (м, 4H), 1,41 (с, 3H), 1,15 (кв., J=9,7 Гц, 1H).
Стадия 3: 3-[3-({[(3aR,4R,6R,6aR)-6-(6-Амино-9H-пурин-9-ил)-2,2-диметил-тетрагидро-2H-фуро[3,4-d][1,3]диоксол-4-ил]метил}(метил)амино)циклобутил]пропановая кислота
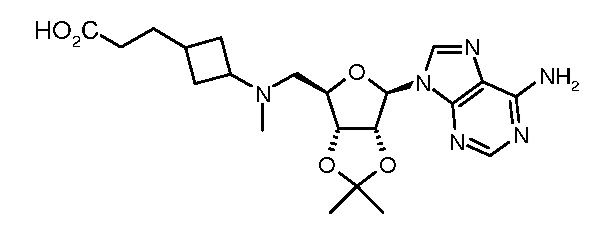
Бензил 3-[3-({[(3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметил-тетрагидро-2H-фуро[3,4-d][1,3]диоксол-4-ил]метил}(метил)амино)циклобутил]пропаноат (1,19 г, 2,22 ммоль) растворяли в этаноле (24 мл) и добавляли 10% палладий на угле (пастообразное вещество, содержание влаги 50%) (0,24 г). Суспензию перемешивали в атмосфере водорода в течение 18 часов. Затем суспензию фильтровали и твердое вещество промывали этанолом (10 мл). Поскольку реакция не была завершена, добавляли дополнительное количество палладия на угле (0,21 г), и реакция продолжалась в атмосфере водорода еще в течение 24 часов. Фильтровали через двойной стекловолоконный фильтр, промывали этанолом и упаривали досуха, с получением желаемого соединения в виде белого пенистого вещества (смесь диастереомеров) (0,85 г, 86%): MS (ESI+) для C21H30N6O5 m/z 447,5 [M+H]+; ЖХ чистота 86% (время удерживания, 1,08 мин);
1H ЯМР (500 МГц, d4-MeOD) δH 8,18-8,35 (м, 2H), 6,14-6,30 (м, 1H), 5,25-5,56 (м, 1H), 5,01-5,11 (м, 1H), 4,35-4,52 (м, 1H), 3,63-3,81 (м, 1H), 3,24-3,30 (м, 1H), 2,98-3,16 (м, 1H), 2,89 (ддд, J=17,2, 13,4, 3,7 Гц, 1H), 2,36 (м, 2H), 2,17-2,26 (м, 1H), 1,98-2,17 (м, 3H), 1,67-1,92 (м, 3H), 1,49-1,64 (м, 4H), 1,39 (с, 3H), 1,20-1,33 (м, 1H).
Стадия 4: Метил 2-(4-фторфенил)-2-метилпропаноат
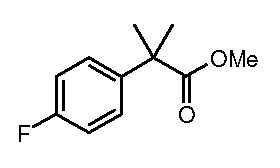
Гидрид натрия (60% суспензия в минеральном масле) (2,64 г, 66 ммоль) промывали гептаном (2×20 мл) и суспендировали в ТГФ (40 мл). Добавляли раствор метил 2-(4-фторфенил)ацетата (5,05 г, 30 ммоль) в ТГФ (10 мл) и перемешивали в течение 30 минут. Добавляли метилиодид (5,6 мл, 90 ммоль) 1 мл порциями в течение 30 минут, сначала при охлаждении до 10°C, затем при осторожном нагревании до 50°C до прекращения газовыделения. Через 4,5 часа добавляли воду (50 мл) и смесь экстрагировали при помощи EtOAc (2×50 мл). Объединенные органические фазы промывали насыщенным солевым раствором (30 мл) и сушили над MgSO4, затем фильтровали и упаривали досуха, при этом оставалось оранжевое масло (5,08 г, 79%, чистота 91% по данным 1Н ЯМР): MS (ESI+) для C11H13FO2 m/z 196,2 [M+H]+; ЖХ чистота 80% (время удерживания, 1,94 мин);
1H ЯМР (500 МГц, CDCl3) δH 7,29-7,34 (м, 2H), 6,98-7,05 (м, 2H), 3,66 (с, 3H), 1,58 (с, 6H).
Стадия 5: 2-(4-Фторфенил)-2-метилпропан-1-ол
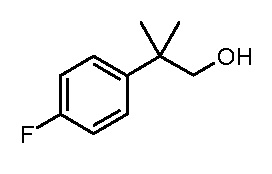
Метил 2-(4-фторфенил)-2-метилпропаноат (5,08 г, 26,9 ммоль) растворяли в ТГФ (51 мл) и охлаждали до 0°C, затем добавляли раствор литийалюминийгидрида (1M в ТГФ) (38,8 мл, 38,8 ммоль) по каплям в течение 30 минут. Когда добавление было завершено, реакционную смесь нагревали до комнатной температуры и перемешивали в течение 3 часов. После повторного охлаждения на льду, осторожно добавляли воду (1,35 мл), с последующим добавлением 15% раствора NaOH в воде (1,35 мл), и снова добавляли воду (4,05 мл). Суспензию перемешивали при комнатной температуре в течение 30 минут, затем твердое вещество отфильтровывали и промывали при помощи ТГФ (2×30 мл). Растворитель выпаривали и продукт очищали хроматографией с использованием смеси EtOAc/гептан с получением прозрачного масла (3,48 г, 80%): MS (ESI+) для C10H13FO m/z 168,2 [M+H]+; ЖХ чистота 94% (время удерживания, 1,76 мин);
1H ЯМР (500 МГц, CDCl3) δH 7,41-7,32 (м, 2H), 7,15-7,00 (м, 2H), 3,62 (д, J=6,4 Гц, 2H), 1,35 (с, 6H).
Стадия 6: 1-Фтор-4-(1-метокси-2-метилпропан-2-ил)бензол
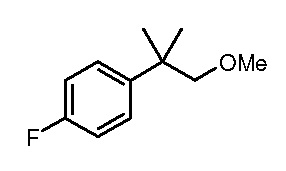
Гидрид натрия (1,664 г, 41,6 ммоль, 60% дисперсия в минеральном масле) суспендировали в безводном ТГФ (18 мл) в атмосфере N2 и к суспензии медленно добавляли 2-(4-фторфенил)-2-метилпропан-1-ол (3,500 г, 20,8 ммоль) в безводном ТГФ (18 мл) при 0°C. После завершения добавления реакционную смесь нагревали до комнатной температуры и оставляли в течение 1 часа. Медленно добавляли иодметан (6,5 мл, 0,104 ммоль) при комнатной температуре и реакционную смесь оставляли для взаимодействия в течение 3 часов. Реакцию гасили медленным добавлением H2O (35 мл). Слои разделяли и водный слой экстрагировали при помощи EtOAc (3×35 мл). Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали в вакууме с получением неочищенного продукта. Продукт очищали колоночной флэш-хроматографией на силикагеле с использованием в качестве элюента от 100% гептана до 10% EtOAc:90% гептан, с получением продукта в виде бесцветного масла (2,991 г, 79%): ЖХ чистота 98% (время удерживания, 2,16 мин);
1H ЯМР (500 МГц, CDCl3) δH 7,40-7,32 (м, 2H), 7,11-6,96 (м, 2H), 3,39 (с, 2H), 3,33 (с, 3H), 1,33 (с, 6H).
Стадия 7: 1-Фтор-4-(1-метокси-2-метилпропан-2-ил)-2-нитробензол
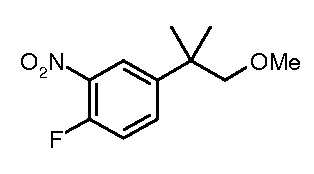
Фтор-4-(1-метокси-2-метилпропан-2-ил)бензол (2,987 г, 16,4 ммоль) охлаждали на соляной бане лед/вода до -20°C и медленно добавляли по каплям серную кислоту (27 мл) при перемешивании. При добавлении серной кислоты раствор становился ярко-оранжевым. Медленно добавляли азотную кислоту (3 мл) по каплям в течение 15-20 минут. При добавлении азотной кислоты раствор становился темно-желтым/коричневым, и осаждалось некоторое количество белого твердого вещества. Реакционную смесь оставляли в течение 30 минут, затем выливали на лед (450 г). Смесь экстрагировали при помощи DCM (2×225 мл) и объединенные органические экстракты сушили над MgSO4, фильтровали и концентрировали в вакууме с получением неочищенного продукта. Продукт очищали колоночной флэш-хроматографией на силикагеле с использованием от 100% гептана до 20% EtOAc:80% гептана, с получением продукта в виде желтого масла (2,239 г, 60%): ЖХ чистота 96% (время удерживания, 2,18 мин);
1H ЯМР (500 МГц, CDCl3) δH 8,08 (дд, J=7,1, 2,5 Гц, 1H), 7,67 (ддд, J=8,7, 4,1, 2,5 Гц, 1H), 7,23 (дд, J=10,6, 8,8 Гц, 1H), 3,41 (с, 2H), 3,33 (с, 3H), 1,37 (с, 6H).
Стадия 8: 1-Азидо-4-(1-метокси-2-метилпропан-2-ил)-2-нитробензол
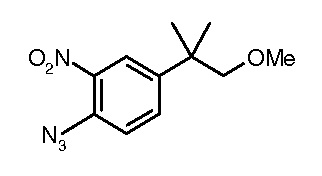
Фтор-4-(1-метокси-2-метилпропан-2-ил)-2-нитробензол (2,227 г, 9,80 ммоль) растворяли в DMF (25 мл) и добавляли азид натрия (1,274 г, 19,6 ммоль) при комнатной температуре и реакционную смесь перемешивали в течение ночи. Реакцию гасили водой (75 мл) и смесь экстрагировали при помощи TBME (3×75 мл). Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали в вакууме с получением неочищенного продукта. Продукт очищали колоночной флэш-хроматографией на силикагеле с использованием в качестве элюента от 100% гептана до 15% EtOAc:85% гептана, с получением продукта в виде желтого масла (2,301 г, 75%, чистота 80%): ЖХ чистота 79% (время удерживания, 2,15 мин);
1H ЯМР (500 МГц, CDCl3) δH 7,97 (д, J=2,2 Гц, 1H), 7,66 (дд, J=8,5, 2,2 Гц, 1H), 7,33-7,22 (м, 1H), 3,40 (с, 2H), 3,33 (с, 3H), 1,36 (с, 6H).
Стадия 9: 4-(1-Метокси-2-метилпропан-2-ил)бензол-1,2-диамин
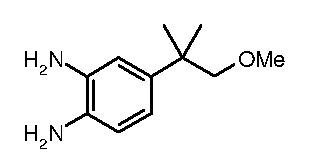
Азидо-4-(1-метокси-2-метилпропан-2-ил)-2-нитробензол (1,102 г, 3,52 ммоль) растворяли в EtOH (30 мл) и добавляли Pd/C (10% масс.) (0,110 г, 10% масс.). Реакционную смесь продували 3 раза при помощи N2, затем 3 раза при помощи H2 и реакционную смесь перемешивали при комнатной температуре в течение ночи. Смесь фильтровали через Целит и фильтрат концентрировали в вакууме с получением неочищенного продукта. Продукт очищали колоночной флэш-хроматографией на силикагеле с использованием в качестве элюента от 100% гептана до 100% EtOAc, с получением продукта в виде бледно-коричневого масла, которое отверждалось до темно-оранжевого твердого вещества (0,481 г, 63%, 90% чистота): MS (ESI+) для C11H18N2O m/z 195,1 [M+H]+; ЖХ чистота 87% (время удерживания, 0,99 мин);
1H ЯМР (500 МГц, CDCl3) δH 6,65 (дд, J=11,2, 1,9 Гц, 2H), 6,58 (д, J=7,9 Гц, 1H), 3,26 (с, 2H), 3,24 (с, 3H), 1,20 (с, 6H).
Стадия 10: 3-[3-({[(3aR,4R,6R,6aR)-6-(6-Амино-9H-пурин-9-ил)-2,2-диметил-тетрагидро-2H-фуро[3,4-d][1,3]диоксол-4-ил]метил}(метил)амино)циклобутил]-N-[2-амино-4-(1-метокси-2-метилпропан-2-ил)фенил]пропанамид
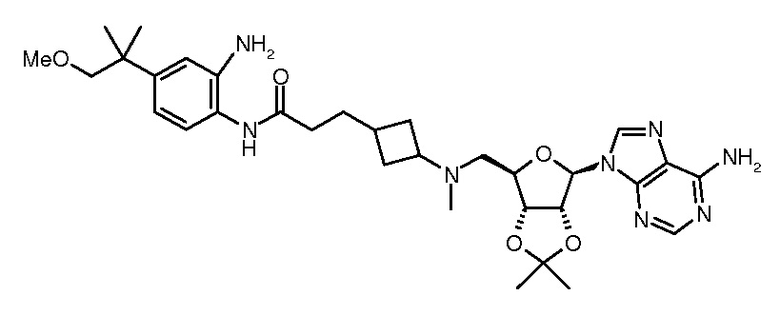
3-[3-({[(3aR,4R,6R,6aR)-6-(6-Амино-9H-пурин-9-ил)-2,2-диметил-тетрагидро-2H-фуро[3,4-d][1,3]диоксол-4-ил]метил}(метил)амино)циклобутил]пропановую кислоту (0,620 г, 1,39 ммоль), 4-(1-метокси-2-метилпропан-2-ил)бензол-1,2-диамин (0,466 г, 2,08 ммоль, чистота 87%), EDC.HCl (0,532 г, 2,78 ммоль) и OXYMA (этил-циано(гидроксиимино)ацетат) (0,395 г, 2,78 ммоль) добавляли в колбу с мешалкой, затем продували при помощи N2. Добавляли безводный DCM (22 мл) и безводный Et3N (1,2 мл, 8,33 ммоль) при комнатной температуре и реакционную смесь оставляли для взаимодействия в течение ночи. Реакцию гасили путем добавления насыщенного раствора NaHCO3 (25 мл) и органический слой отделяли. Водный слой экстрагировали при помощи DCM (2×25 мл) и объединенные органические слои сушили над MgSO4, фильтровали и концентрировали в вакууме с получением неочищенного продукта. Продукт очищали колоночной флэш-хроматографией на силикагеле с использованием сначала 100% EtOAc для элюирования дианилина, затем от 100% DCM до 20% 2M NH3 в MeOH:80% DCM в качестве элюента, с получением продукта в виде коричневого масла (1,081 г, количественный выход): MS (ESI+) для C32H46N8O5 m/z 623,4 [M+H]+; ЖХ чистота 98% (время удерживания, 1,36 мин);
1H ЯМР (500 МГц, CDCl3) δH 8,39 (д, J=7,8 Гц, 1H), 7,99 (с, 1H), 7,42-7,30 (м, 1H), 7,24-7,00 (м, 1H), 6,96-6,81 (м, 1H), 6,12 (д, J=12,6 Гц, 1H), 5,77 (д, J=6,7 Гц, 1H), 5,63 (д, J=5,0 Гц, 1H), 5,14-4,93 (м, 1H), 4,60-4,31 (м, 1H), 4,28-3,69 (м, 2H), 3,48-3,03 (м, 5H), 2,65-2,48 (м, 1H), 2,46-2,33 (м, 1H), 2,34-2,10 (м, 7H), 2,10-1,98 (м, 1H), 2,00-1,66 (м, 4H), 1,62 (д, J=15,7 Гц, 13H), 1,44 (д, J=6,5 Гц, 4H), 1,30 (т, J=3,3 Гц, 7H), 1,12 (д, J=52,3 Гц, 1H).
Стадия 11: 9-[(3aR,4R,6R,6aR)-6-{[(3-{2-[5-(1-Метокси-2-метилпропан-2-ил)-1H-1,3-бензодиазол-2-ил]этил}циклобутил)(метил)амино]метил}-2,2-диметил-тетрагидро-2H-фуро[3,4-d][1,3]диоксол-4-ил]-9H-пурин-6-амин
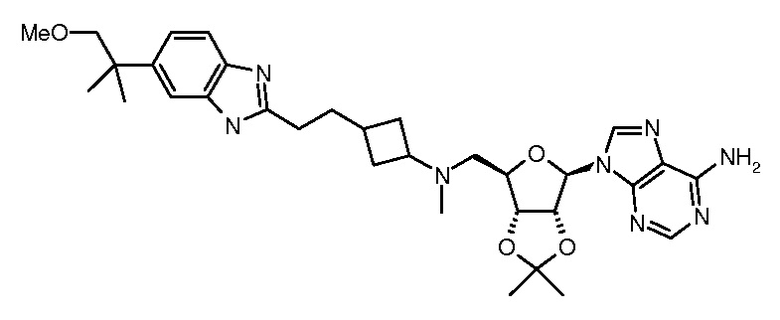
3-[3-({[(3aR,4R,6R,6aR)-6-(6-Амино-9H-пурин-9-ил)-2,2-диметил-тетрагидро-2H-фуро[3,4-d][1,3]диоксол-4-ил]метил}(метил)амино)циклобутил]-N-[2-амино-4-(1-метокси-2-метилпропан-2-ил)фенил]пропанамид (1,036 г, 1,66 ммоль) растворяли в AcOH (17 мл) и нагревали до 50°C в течение 5 часов. Реакционную смесь концентрировали в вакууме и остаток растворяли в DCM (75 мл) и добавляли насыщенный раствор NaHCO3 (75 мл). Органический слой отделяли и водный слой экстрагировали при помощи DCM (2×75 мл). Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали в вакууме с получением неочищенного продукта в виде темно-оранжевого масла (0,850 г, 94%). Продукт очищали с использованием нейтральной препаративной ВЭЖХ, с получением продукта в виде бледно-желтого масла (0,485 г, 47%, 88% чистота): MS (ESI+) для C32H44N8O4 m/z 605,4 [M+H]+; ЖХ чистота 88% (время удерживания, 1,25 мин);
1H ЯМР (500 МГц, CDCl3) δH 10,44 (дд, J=207,7, 22,9 Гц, 1H), 8,41 (д, J=17,1 Гц, 1H), 8,16 (д, J=13,8 Гц, 1H), 7,89-7,58 (м, 1H), 7,54-7,29 (м, 2H), 6,15 (д, J=10,9 Гц, 1H), 5,92-5,56 (м, 3H), 4,98 (д, J=12,5 Гц, 1H), 4,76-4,39 (м, 1H), 3,47 (д, J=6,2 Гц, 2H), 3,33 (д, J=4,8 Гц, 3H), 2,98-2,43 (м, 4H), 2,42-2,00 (м, 6H), 1,99-1,88 (м, 2H), 1,77-1,66 (м, 1H), 1,64 (с, 3H), 1,61-1,48 (м, 1H), 1,44 (д, J=8,1 Гц, 3H), 1,40 (д, J=7,9 Гц, 6H), 1,28 (дд, J=16,0, 9,0 Гц, 1H).
Стадия 12: (2R,3R,4S,5R)-2-(6-Амино-9H-пурин-9-ил)-5-{[(3-{2-[5-(1-метокси-2-этилпропан-2-ил)-1H-1,3-бензодиазол-2-ил]этил}циклобутил)(метил)амино]метил}оксолан-3,4-диол
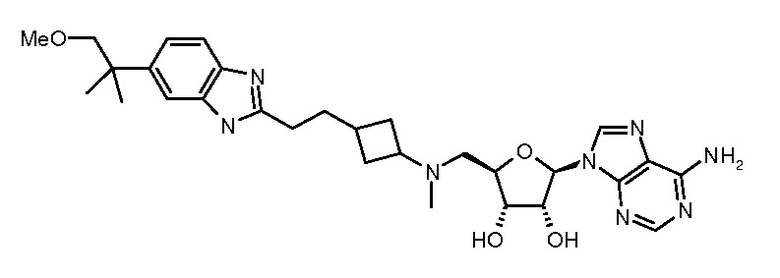
9-[(3aR,4R,6R,6aR)-6-{[(3-{2-[5-(1-Метокси-2-метилпропан-2-ил)-1H-1,3-бензодиазол-2-ил]этил}циклобутил)(метил)амино]метил}-2,2-диметил-тетрагидро-2H-фуро[3,4-d][1,3]диоксол-4-ил]-9H-пурин-6-амин (0,485 г, 0,706 ммоль, чистота 88%) растворяли в MeOH (20 мл) и добавляли концентрированную HCl (4,9 мл, 10 об.) при комнатной температуре и реакционную смесь оставляли для взаимодействия в течение 2,5 часов. Реакционную смесь концентрировали в вакууме и остаток растворяли в минимальном количестве MeOH (~2 мл). Реакцию гасили путем добавления насыщенного раствора NaHCO3 (10 мл) и EtOAc (30 мл). Слои разделяли и водный слой экстрагировали при помощи EtOAc (2×30 мл). Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали в вакууме с получением неочищенного продукта. Продукт очищали колоночной флэш-хроматографией на силикагеле с использованием в качестве элюента от 100% DCM до 20% 2M NH3 в MeOH:80% DCM, с получением продукта в виде белого пенистого вещества (0,213 г, 53%): MS (ESI+) для C29H40N8O4 m/z 565,4 [M+H]+; ЖХ чистота 100% (время удерживания, 2,11 мин) (7 мин);
1H ЯМР (500 МГц, d4-MeOD) δH 8,28 (д, J=4,1 Гц, 1H), 8,22 (д, J=3,2 Гц, 1H), 7,51 (с, 1H), 7,42 (дд, J=8,5, 2,1 Гц, 1H), 7,29 (дд, J=8,5, 1,5 Гц, 1H), 6,01 (т, J=3,9 Гц, 1H), 4,84-4,74 (м, 1H), 4,41-4,29 (м, 1H), 4,29-4,19 (м, 1H), 3,48 (с, 2H), 3,30 (с, 3H), 3,14-2,98 (м, 1H), 2,99-2,89 (м, 1H), 2,90-2,73 (м, 2H), 2,39 (с, 3H), 2,37-2,30 (м, 1H), 2,28-2,10 (м, 2H), 2,08-1,86 (м, 4H), 1,70-1,51 (м, 1H), 1,38 (с, 6H).
Соединение 69: (1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-((((1r,3S)-3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(метил)амино)метил)циклопентан-1,2-диол.
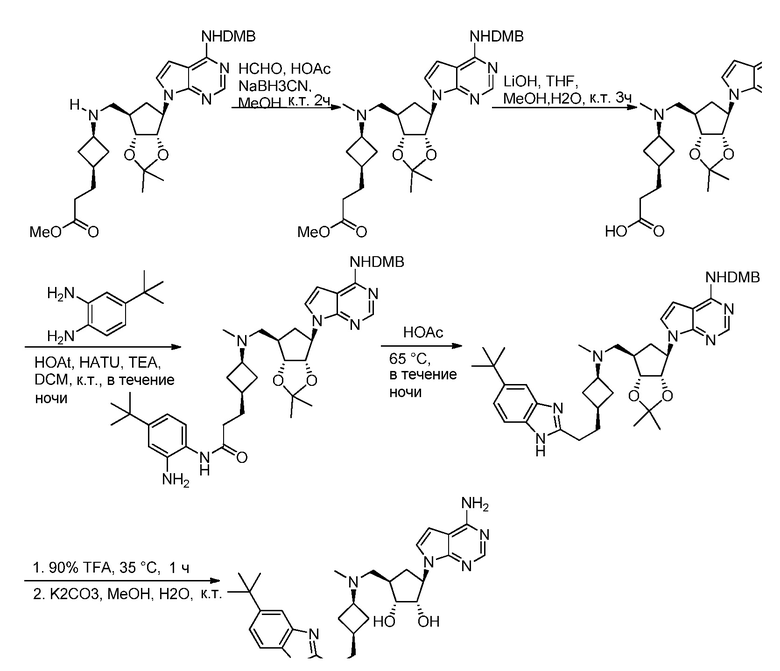
(1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-((((1r,3S)-3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(метил)амино)метил)циклопентан-1,2-диол.
1H ЯМР (500 МГц, MeOD): δH 8,06 (с, 1H), 7,47 (шир.с, 1H), 7,38 (д, J=8,0 ГЦ, 1H), 7,27 (д, J=8,0 Гц, 1H), 7,19 (с, 1H), 6,60 (с, 1H), 4,32 (с, 1H), 3,88 (с, 1H), 2,80 (шир.с, 2H), 2,68 (шир.с, 1H), 2,49-2,10 (м, 9H), 1,90 (шир.с, 2H), 1,62-1,49 (м, 3H), 1,35 (с, 9H) м.д.; ESI-MS (m/z): 532,3 [M+1]+.
Бензил 3-(3-((((3aR,4R,6R,6aR)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)амино)циклобутил)пропаноат
Раствор 7-((3aR,4R,6R,6aR)-6-(аминометил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)-N-(2,4-диметоксибензил)-7H-пирроло[2,3-d]пиримидин-4-амина (20 г, 43,91 ммоль), бензил 3-(3-оксоциклобутил)пропаноата (12,2 г, 52,69 ммоль) и HOAc (15 мл) в DCE (200 мл) перемешивали при 30°C в течение 3 часов. Добавляли NaBH(OAc)3 (18,6 г, 87,81 ммоль) и реакционную смесь перемешивали при 30°C еще в течение 1 часа. Смесь промывали водой (100 мл ×2) и насыщенным солевым раствором (100 мл), сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали колоночной хроматографией на силикагеле с использованием EA:DCM:MeOH=10:10:1 в качестве элюента, с получением желаемого соединения (21 г, выход: 65%, цис/транс=52/47) в виде желтого твердого вещества.
1H ЯМР (500 МГц, MeOD): δH 8,16 (шир.с, 1H), 7,36-7,29 (м, 5H), 7,22 (д, J=3,5 Гц, 1H), 7,15 (д, J=8,5 Гц, 1H), 6,66 (шир.с, 1H), 6,54 (д, J=2,0 Гц, 1H), 6,43 (дд, J=8,5 и 2,0 Гц, 1H), 6,20 (шир.с, 1H), 5,41-5,39 (м, 1H), 5,08 (д, J=3,0 Гц, 1H), 4,99-4,95 (м, 1H), 4,66 (с, 2H), 4,30-4,25 (м, 1H), 3,83 (с, 3H), 3,76 (с, 3H), 3,38-3,35 (м, 0,5H), 3,11-3,06 (м, 0,5H), 2,92-2,82 (м, 2H), 2,29-2,18 (м, 3H), 2,12-2,05 (м, 0,5H), 1,90-1,68 (м, 4H), 1,63-1,61 (м, 1H), 1,60 (с, 3H), 1,38 (с, 3H), 1,35-1,28 (м, 0,5H), 1,25 (т, J=6,5 Гц, 2H), 1,15-1,12 (м, 0,5H) м.д.; ESI-MS (отрицательный режим, m/z): 670,3 [M-1]+.
Бензил 3-(3-((((3aR,4R,6R,6aR)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(изопропил)амино)циклобутил)пропаноат
Смесь бензил 3-(3-((((3aR,4R,6R,6aR)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)амино)циклобутил)пропаноата (4 г, 5,95 ммоль), 2-иодпропана (6 г, 35,72 ммоль) и K2CO3 (2,5 г, 17,86 ммоль) в CH3CN (50 мл) перемешивали при температуре кипения с обратным холодильником в течение 2 дней. Смесь охлаждали до комнатной температуры, фильтровали и фильтровальную лепешку промывали при помощи CH3CN (20 мл). Фильтрат концентрировали и остаток очищали при помощи Combi-Flash (80 г силикагеля, с использованием градиента EA:DCM:MeOH = от 10:10:0 до 10:10:1, 60 мл/мин 40 минут, общий объем растворителя 2,4 л) с получением желаемого соединения (3 г, выход: 71 %) в виде желтого твердого вещества.
1H ЯМР (500 МГц, MeOD): δH 8,15 (с, 1H), 7,38-7,29 (м, 5H), 7,19 (д, J=4,0 Гц, 1H), 7,14 (д, J=8,0 Гц, 1H), 6,65 (д, J=3,0 Гц, 1H), 6,53 (д, J=2,0 Гц, 1H), 6,41 (дд, J=8,5 и 2,0 Гц, 1H), 6,20 (д, J=2,5 Гц, 1H), 5,36-5,32 (м, 1H), 5,09 (с, 1H), 5,07 (с, 1H), 4,94-4,89 (м, 3H), 4,65 (с, 2H), 4,17-4,14 (м, 1H), 3,82 (с, 3H), 3,75 (с, 3H), 3,41-3,35 (м, 0,5H), 3,04-2,95 (м, 0,5H), 2,94-2,86 (м, 1H), 2,72-2,52 (м, 2H), 2,25 (т, J=7,5 Гц, 1H), 2,21 (т, J=8,0 Гц, 1H), 2,05-1,90 (м, 2H), 1,90-1,82 (м, 0,5H), 1,76-1,70 (м, 0,5H), 1,65-1,55 (м, 5H), 1,42-1,34 (м, 4H), 0,95 (д, J=6,5 Гц, 3H), 0,83-0,80 (м, 3H) м.д.; ESI-MS (m/z): 714,4 [M+1]+.
3-(3-((((3aR,4R,6R,6aR)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(изопропил)амино)циклобутил)пропановая кислота
К раствору бензил 3-(3-((((3aR,4R,6R,6aR)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(изопропил)-амино)циклобутил)пропаноата (2,7 г, 3,78 ммоль) в ТГФ/MeOH (15 мл/15 мл) добавляли раствор LiOH.H2O (1,6 г, 37,82 ммоль) в воде (5 мл). Смесь перемешивали при 30°C в течение 2 часов. Летучие вещества удаляли при пониженном давлении. Остаток разбавляли водой (10 мл) и экстрагировали при помощи EA (15 мл ×2). Водные слои суспензии доводили до pH=3-4 при помощи 1н раствора HCl и экстрагировали при помощи EA (30 мл ×3). Объединенные органические слои промывали насыщенным солевым раствором (50 мл). Органическую фазу сушили над Na2SO4, фильтровали и концентрировали с получением желаемого соединения в виде желтого твердого вещества (2,9 г).
1H ЯМР (500 МГц, MeOD): δH 8,20 (с, 1H), 7,92 (с, 0,5H), 7,38-7,32 (м, 3H), 7,28-7,23 (м, 1,5H), 7,14 (д, J=8,5 Гц, 1H), 6,68 (шир.с, 1H), 6,56 (д, J=2,0 Гц, 1H), 6,44 (д, J=8,5 Гц, 1H), 6,25 (с, 1H), 5,51-5,47 (м, 1H), 5,18-5,13 (м, 1H), 4,66 (с, 2H), 4,61 (с, 1H), 4,43-4,40 (м, 1H), 3,96-3,90 (м, 0,5H), 3,85 (с, 3H), 3,77 (с, 3H), 3,65-3,58 (м, 0,5H), 3,50-3,40 (м, 2H), 2,46-2,36 (м, 1H), 2,20-2,00 (м, 4H), 1,98-1,70 (м, 2,5H), 1,70-1,58 (м, 4,5H), 1,40 (с, 3H), 1,18 (д, J=5,0 Гц, 3H), 0,90 (т, J=6,0 Гц, 3H) м.д.; ESI-MS (m/z): 624,3 [M+1]+.
N-(2-амино-4-(трет-бутил)фенил)-3-(3-((((3aR,4R,6R,6aR)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(изопропил)амино)циклобутил)пропанамид
К раствору 3-(3-((((3aR,4R,6R,6aR)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(изопропил)амино)циклобутил)пропановой кислоты (2,7 г, 4,33 ммоль), 4-(трифторметокси)бензол-1,2-диамины (1,23 г, 6,49 ммоль), HATU (2,5 г, 6,49 ммоль) и HOAT (0,88 г, 6,49 ммоль) в DCM (30 мл) добавляли TEA (1,8 мл, 12,99 ммоль). Смесь перемешивали при комнатной температуре в течение 2 часов. К смеси добавляли DCM (70 мл) и промывали водой (20 мл ×2) и насыщенным солевым раствором (50 мл). Органическую фазу сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали при помощи Combi-Flash (80 г силикагеля, с использованием градиента EA:DCM:MeOH = от 10:10:0 до 10:10:2, 60 мл/мин 40 минут общий объем растворителя 2,4 л) с получением желаемого соединения (2,2 г, выход: 67% (две стадии) в виде коричневого твердого вещества.
1H ЯМР (500 МГц, MeOD): δH 8,21 (с, 1H), 7,28-7,12 (м, 3H), 6,73 (шир.с, 1H), 6,69 (шир.с, 1H), 6,56-6,52 (м, 2H), 6,42 (дд, J=8,0 и 2,5 Гц, 1H), 6,26 (д, J=1,5 Гц, 1H), 5,48 (д, J=6,0 Гц, 1H), 5,20-5,16 (м, 1H), 4,66 (с, 2H), 4,46-4,42 (м, 1H), 4,08-3,98 (м, 0,5H), 3,84 (с, 3H), 3,76 (с, 3H), 3,75-3,69 (м, 0,5H), 3,60-3,38 (м, 2H), 2,60-2,30 (м, 3H), 1,92-1,86 (м, 1H), 1,80-1,70 (м, 1H), 1,59 (д, J=3,5 Гц, 3H), 1,40 (с, 3H), 1,25 (т, J=7,5 Гц, 3H), 1,00-0,80 (м, 2H) м.д.; ESI-MS (m/z): 798,3 [M+1]+.
N-(2,4-диметоксибензил)-7-((3aR,4R,6R,6aR)-6-((изопропил(3-(2-(5-(трифторметокси)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)амино)метил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)-7H-пирроло[2,3-d]пиримидин-4-амин
Раствор N-(2-амино-4-(трет-бутил)фенил)-3-(3-((((3aR,4R,6R,6aR)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(изопропил)-амино)циклобутил)пропанамида
(2,2 г, 2,5 ммоль) в HOAc (20 мл) перемешивали при 65°C в течение 5 часов. Смесь охлаждали до комнатной температуры и концентрировали. Остаток растворяли в DCM (50 мл), промывали 15% раствором Na2CO3 (20 мл ×2), водой (20 мл) и насыщенным солевым раствором (30 мл). Органическую фазу сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали при помощи Combi-Flash (80 г силикагеля, с использованием градиента EA:DCM:MeOH = от 10:10:0 до 10:10:2, 60 мл/мин 35 минут, общий объем растворителя 2,1 л) с получением желаемого соединения (1,4 г) в виде коричневого твердого вещества, которое разделяли при помощи хиральной ВЭЖХ, с получением цис-изомера (600 мг, выход: 28%) и транс изомера (480 мг, выход: 22%).
цис-изомер: 1H ЯМР (500 МГц, MeOD): δH 8,17 (с, 1H), 7,52 (д, J=9,0 Гц, 1H), 7,39 (с, 1H), 7,20 (д, J=4,0 Гц, 1H), 7,12 (д, J=8,0 Гц, 1H), 6,68 (шир.с, 1H), 6,49 (д, J=2,5 Гц, 1H), 6,39 (дд, J=8,5 и 2,0 Гц, 1H), 6,19 (д, J=2,0 Гц, 1H), 5,35 (дд, J=6,0 и 2,0 Гц, 1H), 4,68-4,60 (м, 2H), 4,18-4,13 (м, 1H), 3,78 (с, 3H), 3,73 (с, 3H), 3,10-3,02 (м, 1H), 2,95-2,88 (м, 1H), 2,78-2,72 (м, 2H), 2,69-2,57 (м, 2H), 2,12-2,02 (м, 2H), 1,86-1,76 (м, 2H), 1,57 (с, 3H), 1,52-1,40 (м, 2H), 1,37 (с, 3H), 0,96 (д, J=6,5 Гц, 3H), 0,82 (д, J=6,5 Гц, 3H) м.д.; ESI-MS (m/z): 780,4 [M+1]+.
транс-изомер: 1H ЯМР (500 МГц, MeOD): δH 8,15 (с, 1H), 7,52 (д, J=9,0 Гц, 1H), 7,40 (с, 1H), 7,20 (д, J=3,5 Гц, 1H), 7,12 (д, J=8,0 Гц, 1H), 6,68 (шир.с, 1H), 6,51 (д, J=2,0 Гц, 1H), 6,40 (дд, J=8,0 и 2,5 Гц, 1H), 6,20 (д, J=2,5 Гц, 1H), 5,35 (дд, J=6,0 и 2,0 Гц, 1H), 4,64 (с, 2H), 4,20-4,16 (м, 1H), 3,82 (с, 3H), 3,73 (с, 3H), 3,50-3,42 (м, 1H), 2,95-2,90 (м, 1H), 2,80 (т, J=6,0 Гц, 2H), 2,75-2,58 (м, 2H), 2,12-1,95 (м, 4H), 1,75-1,65 (м, 2H), 1,58 (с, 3H), 1,38 (с, 3H), 0,97 (д, J=6,5 Гц, 3H), 0,83 (д, J=6,5 Гц, 3H) м.д.; ESI-MS (m/z): 780,4 [M+1]+.
Соединение 70: (1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-((((1s,3R)-3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(метил)амино)метил)циклопентан-1,2-диол
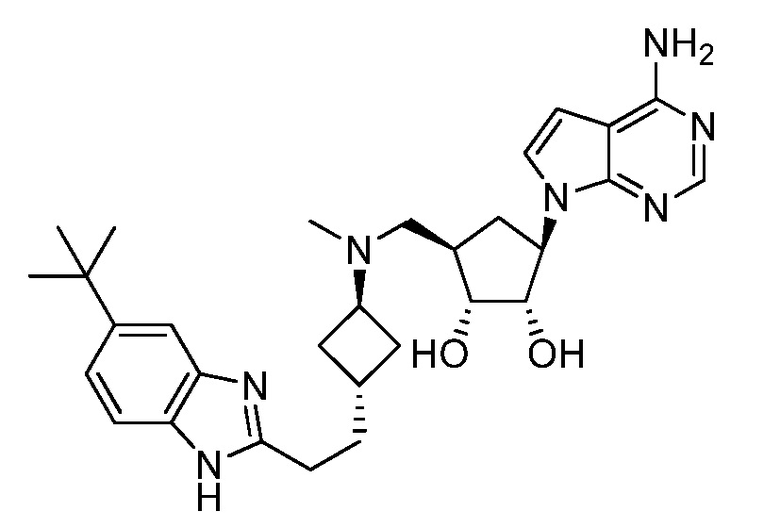
метил 3-((1R,3s)-3-((((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)(метил)амино)циклобутил)пропаноат
Раствор метил 3-((1R,3s)-3-((((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)амино)циклобутил)пропаноата (1,85 г, 3,12 ммоль) и NaBH3CN (590 мг, 9,36 ммоль) в MeOH (25 мл) доводили до pH=6 при помощи AcOH, затем добавляли формальдегид (936 мг, 31,2 ммоль). Реакционную смесь перемешивали при 25°C в течение ночи. Реакцию гасили насыщенным раствором NaHCO3 (5 мл), упаривали, добавляли воду (10 мл), экстрагировали при помощи DCM (150 мл ×3), промывали насыщенным солевым раствором (80 мл), сушили и концентрировали. Остаток очищали при помощи SGC с получением желаемого соединения (1,85 г, выход 97%) в виде белого твердого вещества.
1H ЯМР (500 МГц, MeOD): δH 8,12 (с, 1H), 7,22 (д, J=3,5 Гц, 1H), 7,14 (д, J=8,5 Гц, 1H), 6,64 (д, J=3,0 Гц, 1H), 6,55 (д, J=2,5 Гц, 1H), 6,44 (дд, J=8,5 и 2,5 Гц, 1H), 5,01-4,98 (м, 2H), 4,65 (с, 2H), 4,60-4,58 (м, 1H), 4,30 (с, 1H), 3,84 (с, 3H), 3,76 (с, 3H), 3,66 (с, 3H), 3,44-3,38 (м, 1H), 2,80-2,73 (м, 2H), 2,48-2,43 (м, 4H), 2,32 (т, J=7,5 Гц, 2H), 2,20-2,16 (м, 4H), 1,95-1,94 (м, 2H), 1,82-1,81 (м, 2H), 1,56 (с, 3H), 1,31 (с, 3H) м.д.; ESI-MS (m/z): 608,3 [M+1]+.
3-((1R,3s)-3-((((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)(метил)амино)циклобутил)пропановая кислота
Раствор метил 3-((1R,3s)-3-((((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)(метил)амино)циклобутил)пропаноата
(1,85 г, 3,04 ммоль) и LiOH (382 мг, 15,24 ммоль) в ТГФ/MeOH/H2O (1:1:1, 30 мл) перемешивали при 50°C в течение 2 часов. Реакционную смесь концентрировали с получением желаемого соединения (2,25 г, соль, чистота 85%) в виде белого твердого вещества. Это неочищенное вещество использовали непосредственно на следующей стадии без дополнительной очистки.
1H ЯМР (500 МГц, MeOD): δH 8,11 (с, 1H), 7,92 (с, 1H), 7,30 (д, J=3,0 Гц, 1H), 7,14 (д, J=8,0 Гц, 1H), 6,67 (д, J=3,0 Гц, 1H), 6,54 (д, J=2,0 Гц, 1H), 6,44 (дд, J=10,0 и 2,5 Гц, 1H), 5,05-5,03 (м, 2H), 4,73 (д, J=6,0 Гц, 1H), 4,65 (с, 2H), 3,90-3,85 (м, 1H), 3,85 (с, 3H), 3,77 (с, 3H), 3,30-3,18 (м, 2H), 2,82 (с, 3H), 2,65-2,55 (м, 1H), 2,53-2,46 (м, 3H), 2,27-2,20 (м, 3H), 2,12-2,11 (м, 2H), 1,83-1,82 (м, 2H), 1,57 (с, 3H), 1,32 (с, 3H) м.д.; ESI-MS (m/z): 594,3 [M+1]+.
N-(2-амино-4-(трет-бутил)фенил)-3-((1R,3s)-3-((((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)(метил)амино)-циклобутил)пропанамид
Раствор 3-((1R,3s)-3-((((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)(метил)амино)циклобутил)пропановой кислоты (2,25 г, 3,8 ммоль), HOAt (680 мг, 5 ммоль) и HATU (1,9 г, 5 ммоль) в DCM (60 мл) перемешивали при комнатной температуре в течение 1 часа, затем добавляли по каплям 4-трет-бутилбензолдиамин (656 мг, 4 ммоль) и TEA (1,21 г, 12 ммоль) в DCM (3 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. К реакционной смеси добавляли воду (20 мл) и DCM (60 мл), экстрагировали при помощи DCM (60 мл ×2), промывали насыщенным солевым раствором (10 мл), сушили и концентрировали. Остаток очищали при помощи SGC, с получением желаемого соединения (1,1 г, выход 46%) в виде желтоватого твердого вещества.
1H ЯМР (500 МГц, MeOD): δH 8,11 (с, 1H), 7,21 (д, J=4,0 Гц, 1H), 7,13 (д, J=8,5 Гц, 1H), 6,98 (д, J=8,5 Гц, 1H), 6,92 (д, J=2,0 Гц, 1H), 6,77 (дд, J=8,0 и 1,5 Гц, 1H), 6,64 (д, J=3,5 Гц, 1H), 6,54 (д, J=2,0 Гц, 1H), 6,43 (дд, J=8,0 и 1,5 Гц, 1H), 5,02-4,99 (м, 2H), 4,65-4,61 (м, 3H), 3,83 (с, 3H), 3,75 (с, 3H), 3,60-3,52 (м, 1H), 2,97-2,83 (м, 2H), 2,58 (с, 3H), 2,54-2,38 (м, 4H), 2,33-2,29 (м, 2H), 2,18-2,04 (м, 3H), 1,94-1,91 (м, 2H), 1,55 (с, 3H), 1,30 (с, 3H), 1,25 (с, 9H) м.д.; ESI-MS (m/z): 740,5 [M+1]+.
7-((3aS,4R,6R,6aR)-6-((((1s,3R)-3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(метил)амино)метил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-N-(2,4-диметоксибензил)-7H-пирроло[2,3-d]пиримидин-4-амин
Раствор N-(2-амино-4-(трет-бутил)фенил)-3-((1R,3s)-3-((((3aR,4R,6R,6aS)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)метил)(метил)амино)-циклобутил)пропанамида (1,1 г, 1,49 ммоль) в AcOH (8 мл) нагревали до 65°C в течение 3 часов. Реакционную смесь упаривали, растворяли в MeOH (5 мл) доводили до pH=8 при помощи насыщенного раствора NaHCO3, концентрировали и очищали методом препаративной ТСХ, с получением желаемого соединения (620 мг, 58%) в виде белого твердого вещества.
1H ЯМР (500 МГц, MeOD): δH 8,10 (с, 1H), 7,48 (шир.с, 1H), 7,38 (шир.с, 1H), 7,28 (д, J=8,5 Гц, 1H), 7,21 (д, J=3,0 Гц, 1H), 7,13 (д, J=8,5 Гц, 1H), 6,62 (шир.с, 1H), 6,53 (с, 1H), 6,42 (д, J=8,5 Гц, 1H), 4,98-4,90 (м, 2H), 4,64 (с, 2H), 4,50 (шир.с, 1H), 3,83 (с, 3H), 3,75 (с, 3H), 3,01-2,98 (м, 1H), 2,83 (д, J=7,5 Гц, 1H), 2,44-2,34 (м, 4H), 2,16 (с, 3H), 2,12-1,96 (м, 6H), 1,84 (т, J=7,5 Гц, 2H), 1,53 (с, 3H), 1,36 (с, 9H), 1,28 (с, 3H) м.д.; ESI-MS (m/z): 722,4 [M+1]+.
(1R,2S,3R,5R)-3-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-((((1s,3R)-3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(метил)амино)метил)циклопентан-1,2-диол
Раствор 7-((3aS,4R,6R,6aR)-6-((((1s,3R)-3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)-(метил)амино)метил)-2,2-диметилтетрагидро-3aH-циклопента[d][1,3]диоксол-4-ил)-N-(2,4-диметоксибензил)-7H-пирроло[2,3-d]пиримидин-4-амина (620 мг, 0,86 ммоль) в TFA (5 мл, 90%) перемешивали при 25°C в течение 1 часа. Реакционную смесь концентрировали досуха, растворяли в MeOH (5 мл) и доводили до pH=8 при помощи насыщенного раствора K2CO3. Смесь перемешивали при комнатной температуре в течение 0,5 часа. Затем реакционную смесь концентрировали с получением остатка. Остаток очищали методом препаративной ВЭЖХ, с получением желаемого соединения (330 мг, выход 73%) в виде белого твердого вещества.
1H ЯМР (500 МГц, MeOD): δH 8,07 (с, 1H), 7,49 (д, J=1,5 Гц, 1H), 7,40 (д, J=11,0 Гц, 1H), 7,29 (дд, J=11,0 и 2,5 Гц, 1H), 7,21 (д, J=5,0 Гц, 1H), 6,60 (д, J=4,0 Гц, 1H), 4,92-4,89 (м, 1H), 4,33 (дд, J=9,5 и 8,5 Гц, 1H), 3,90 (д, J=5,5 Гц, 1H), 3,03-2,99 (м, 1H), 2,85 (д, J=9,5 Гц, 1H), 2,50-2,22 (м, 4H), 2,16 (с, 3H), 2,15-1,98 (м, 5H), 1,88 (т, J=10,0 Гц, 2H), 1,68-1,59 (м, 1H), 1,37 (с, 9H) м.д.; ESI-MS (m/z): 532,3 [M+1]+.
Соединение 71: (2R,3R,4S,5R)-2-(6-амино-9H-пурин-9-ил)-5-((((1r,3S)-3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(метил)амино)метил)тетрагидрофуран-3,4-диол
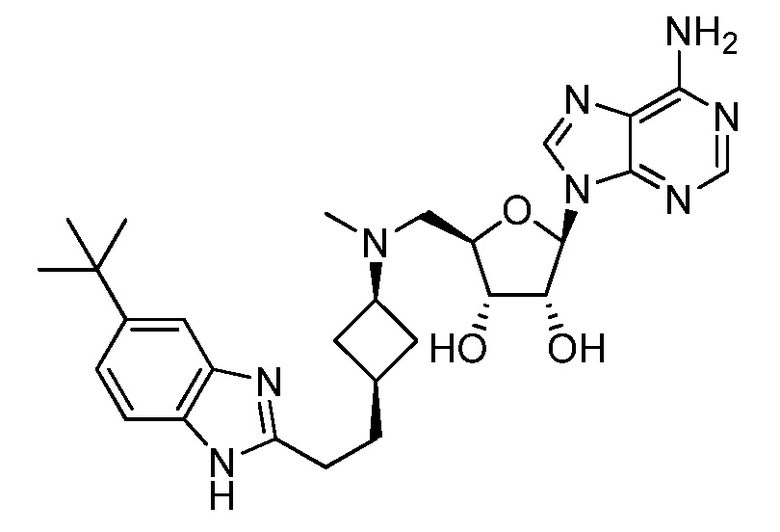
Смесь цис-9-((3aR,4R,6R,6aR)-6-((((1r,3S)-3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(метил)-амино)метил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)-9H-пурин-6-амина (920 мг, 1,60 ммоль) в 3 M HCl/MeOH (20 мл) перемешивали при 35°C в течение 2 часов и упаривали досуха. Остаток растворяли в MeOH (15 мл) и добавляли насыщенный раствор K2CO3 для доведения раствора до pH 8. Затем смесь перемешивали в течение 5 минут и фильтровали. Фильтрат концентрировали и неочищенное вещество очищали методом препаративной ВЭЖХ, с получением целевого соединения (400 мг, выход: 47%) в виде белого твердого вещества.
1H ЯМР (500 МГц, MeOD): δH 8,27 (с, 1H), 8,20 (с, 1H), 7,47 (с, 1H), 7,47 (с, 1H), 7,40-7,37 (м, 1H), 7,29-7,26 (м, 1H), 5,98 (д, J=4,5 Гц, 1H), 4,69 (т, J=4,5 Гц, 1H), 4,24-4,20 (м, 1H), 4,18-4,15 (м, 1H), 2,81-2,76 (м, 2H), 2,75-2,69 (м, 1H), 2,67-2,62 (м, 2H), 2,25-2,18 (м, 1H), 2,14 (с, 3H), 1,90-1,85 (м, 3H), 1,49-1,41 (м, 2H), 1,36 (с, 9H) м.д.; ESI-MS (m/z): 535,3 [M+1]+.
Соединение 72: (2R,3R,4S,5R)-2-(6-амино-9H-пурин-9-ил)-5-((((1s,3R)-3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(метил)амино)метил)тетрагидрофуран-3,4-диол
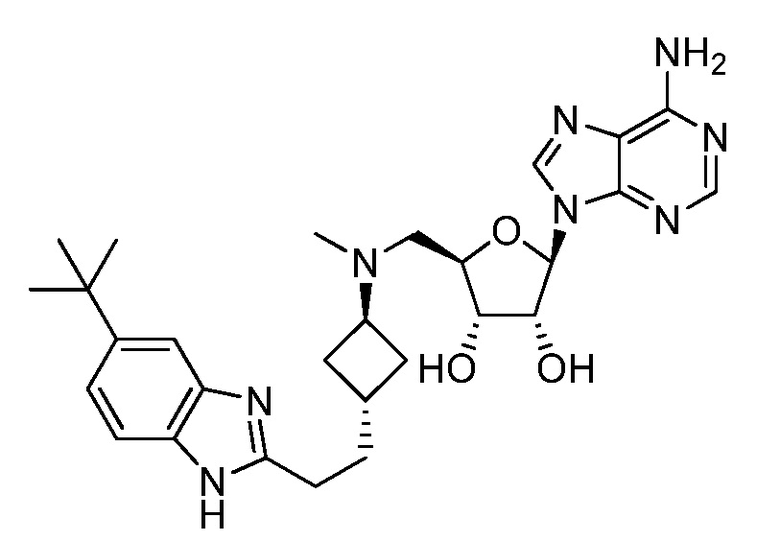
Смесь транс-9-((3aR,4R,6R,6aR)-6-((((1r,3S)-3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(метил)-амино)метил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)-9H-пурин-6-амина (420 мг, 0,73 ммоль) в 3 M HCl/MeOH (20 мл) перемешивали при 35°C в течение 2 часов и упаривали досуха. Остаток растворяли в MeOH (15 мл) и добавляли насыщенный раствор K2CO3 для доведения раствора до pH 8. Затем смесь перемешивали в течение 5 минут и фильтровали. Фильтрат концентрировали и неочищенное вещество очищали методом препаративной ВЭЖХ, с получением целевого соединения (198 мг, выход: 51%) в виде белого твердого вещества.
1H ЯМР (500 МГц, MeOD): δH 8,28 (с, 1H), 8,19 (с, 1H), 7,48 (с, 1H), 7,38 (д, J=8,0 Гц, 1H), 7,28-7,26 (м, 1H), 5,98 (д, J=4,0 Гц, 1H), 4,69 (т, J=5,5 Гц, 1H), 4,23 (т, J=5,0 Гц, 1H), 4,19-4,16 (м, 1H), 3,06-3,03 (м, 1H), 2,80 (т, J=7,5 Гц, 2H), 2,68-2,64 (м, 2H), 2,16 (с, 3H), 2,09-1,95 (м, 5H), 1,85-1,80 (м, 2H), 1,36 (с, 9H) м.д.; ESI-MS (m/z): 535,3 [M+1]+.
Бензил 3-(3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(этил)амино)циклобутил)пропаноат
К смеси бензил 3-(3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)амино)циклобутил)пропаноата (3,76 г, 7,2 ммоль) и NaBH3CN (5,9 г, 93,6 ммоль) в MeOH (40 мл) добавляли AcOH для доведения до pH=6. Затем добавляли 40% MeCHO (8,7 мл, 122,5 ммоль) и смесь перемешивали при 30°C в течение 1,5 часов. Добавляли воду (15 мл) и смесь концентрировали в вакууме. Затем смесь экстрагировали при помощи DCM (30 мл ×3). Объединенную органическую фазу промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали. Неочищенное вещество очищали при помощи SGC (DCM: MeOH=100:1-20:1), с получением целевого соединения (2,6 г, выход: 66%) в виде белого твердого вещества.
1H ЯМР (500 МГц, MeOD): δH 8,26 (с, 1H), 8,23 (м, 1H), 7,37-7,28 (м, 5H), 6,24 (с, 1H), 5,52-5,49 (м, 1H), 5,10-5,06 (м, 3H), 4,41-4,40 (м, 1H), 3,20-2,90 (м, 2H), 2,80-2,60 (м, 2H), 2,12-1,77 (м, 4H), 1,74-1,60 (м, 3H), 1,39 (с, 3H), 1,26-1,22 (м, 3H), 0,94-0,89 (м, 3H) м.д.; ESI-MS (m/z): 551,3 [M+1]+.
3-(3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(этил)-амино)циклобутил)пропановая кислота
К раствору бензил 3-(3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(этил)амино)циклобутил)пропаноата (2,6 г, 4,73 ммоль) в MeOH (40 мл) добавляли 10% Pd/C (2,3 г) и смесь перемешивали в атмосфере H2 при 50°C в течение 20 часов. Смесь фильтровали и фильтрат концентрировали с получением целевого соединения (2 г, выход: 92%) в виде белого твердого вещества.
1H ЯМР (500 МГц, MeOD): δH 8,27 (с, 1H), 8,25 (с, 1H), 6,30 (с, 1H), 5,52-5,49 (м, 1H), 5,14-5,12 (м, 1H), 4,51-4,47 (м, 1H), 3,43-3,36 (м, 2H), 3,22-3,15 (м, 1H), 2,94-2,89 (м, 2H), 2,28-2,05 (м, 4H), 1,96-1,80 (м, 2H), 1,74-1,69 (м, 1H), 1,65-1,59 (м, 5H), 1,41-1,36 (м, 4H), 1,03-0,99 (м, 3H) м.д.; ESI-MS (m/z): 461,3 [M+1]+.
N-(2-амино-4-(трет-бутил)фенил)-3-(3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(этил)амино)циклобутил)пропанамид
К раствору 3-(3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(этил)амино)циклобутил)пропановой кислоты (2 г, 4,35 ммоль), HOAT (768 мг, 5,65 ммоль), HATU (2,2 г, 5,65 ммоль) и TEA (3 мл, 21,3 ммоль) в DCM (40 мл) добавляли 4-трет-бутилбензол-1,2-диамин (785 мг, 4,79 ммоль) и смесь перемешивали при комнатной температуре в течение 2 часов. Добавляли воду (15 мл) и смесь экстрагировали при помощи DCM (30 мл ×2). Объединенную органическую фазу промывали при помощи H2O (20 мл ×2). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали. Неочищенное вещество очищали при помощи SGC (DCM: MeOH=70:1-20:1), с получением целевого соединения (1,6 г, выход: 61%) в виде белого твердого вещества.
1H ЯМР (500 МГц, MeOD): δH 8,28-8,27 (м, 1H), 8,24-8,23 (м, 1H), 7,10-6,92 (м, 2H), 6,81-6,76 (м, 1H), 6,22 (с, 1H), 5,54-5,52 (м, 1H), 5,03 (с, 1H), 4,36 (с, 1H), 3,03-2,50 (м, 5H), 2,32-2,26 (м, 2H), 2,16-1,80 (м, 4H), 1,73-1,69 (м, 2H), 1,59 (с, 3H), 1,39 (с, 3H), 1,28-1,24 (м, 9H), 0,94-0,85 (м, 3H) м.д.; ESI-MS (m/z): 607,3 [M+1]+.
9-((3aR,4R,6R,6aR)-6-(((3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(этил)амино)метил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)-9H-пурин-6-амин
Раствор N-(2-амино-4-(трет-бутил)фенил)-3-(3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(этил)-амино)циклобутил)пропанамида (1,6 г, 2,64 ммоль) в AcOH (20 мл) перемешивали при 65°C в течение 15 часов. Раствор концентрировали в вакууме и разбавляли при помощи DCM (30 мл). Смесь промывали насыщенным раствором NaHCO3 (20 мл ×2) и насыщенным солевым раствором (20 мл ×1). Объединенную органическую фазу сушили над Na2SO4, фильтровали и концентрировали с получением целевого соединения 1,5 г (выход: 97%) в виде белого твердого вещества.
1H ЯМР (500 МГц, MeOD): δH 8,28-8,26 (м, 1H), 8,22 (с, 1H), 7,48 (с, 1H), 7,40-7,38 (м, 1H), 7,30-7,27 (м, 1H), 6,20-6,18 (м, 1H), 5,52-5,49 (м, 1H), 5,02-4,98 (м, 1H), 4,34-4,31 (м, 1H), 2,95-2,92 (м, 1H), 2,79-2,68 (м, 4H), 2,56-2,50 (м, 2H), 2,09-1,81 (м, 5H), 1,71-1,63 (м, 1H), 1,58 (с, 3H), 1,38 (с, 3H), 1,36 (с, 9H), 1,35-1,28 (м, 1H), 0,89-0,85 (м, 3H) м.д.; ESI-MS (m/z): 589,3 [M+1]+.
Смесь 9-((3aR,4R,6R,6aR)-6-(((3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(этил)амино)метил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)-9H-пурин-6-амина (1,35 г, 2,30 ммоль) в 3 M HCl/MeOH (20 мл) перемешивали при 35°C в течение 2 часов и упаривали досуха. Остаток растворяли в MeOH (15 мл) и добавляли насыщенный раствор K2CO3 для доведения раствора до pH 8. Затем смесь перемешивали в течение 5 минут и фильтровали. Фильтрат концентрировали и неочищенное вещество разделяли при помощи хиральной ВЭЖХ и очищали методом препаративной ВЭЖХ, с получением цис продукта (280 мг, общий выход: 22%) и транс продукта (150 мг, общий выход: 12%) в виде белых твердых вещества.
Соединение 73: (2R,3R,4S,5R)-2-(6-амино-9H-пурин-9-ил)-5-((((1r,3S)-3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(этил)амино)метил)тетрагидрофуран-3,4-диол
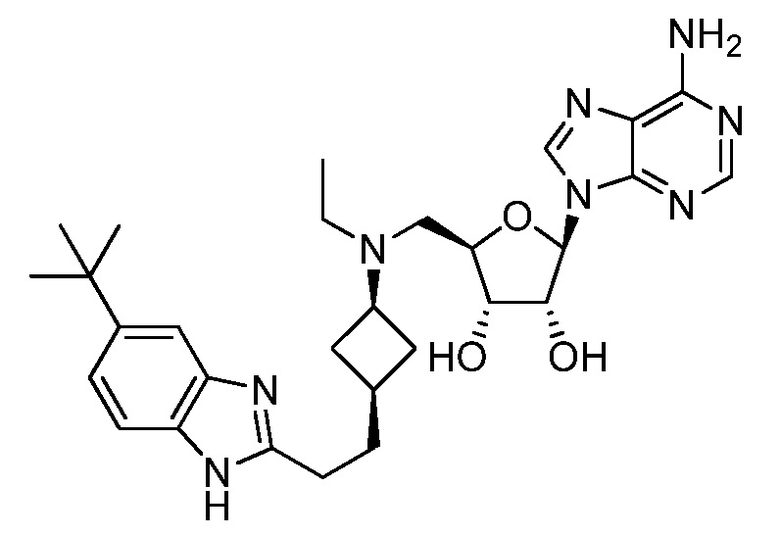
Цис-изомер: 1H ЯМР (500 МГц, MeOD): δH 8,27 (с, 1H), 8,20 (с, 1H), 7,47 (с, 1H), 7,39-7,37 (м, 1H), 7,29-7,26 (м, 1H), 5,97 (д, J=4,5 Гц, 1H), 4,67 (т, J=5,0 Гц, 1H), 4,24 (т, J=5,5 Гц, 1H), 4,18-4,14 (м, 1H), 3,06-3,03 (м, 1H), 2,91-2,81 (м, 2H), 2,79-2,75 (м, 2H), 2,64-2,58 (м, 2H), 2,25-2,19 (м, 1H), 1,89-1,86 (м, 3H), 1,52-1,47 (м, 2H), 1,36 (с, 9H), 0,98 (т, J=7,0 Гц, 3H) м.д.; ESI-MS (m/z): 549,3 [M+1]+.
Соединение 74: (2R,3R,4S,5R)-2-(6-амино-9H-пурин-9-ил)-5-((((1s,3R)-3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(этил)амино)метил)тетрагидрофуран-3,4-диол
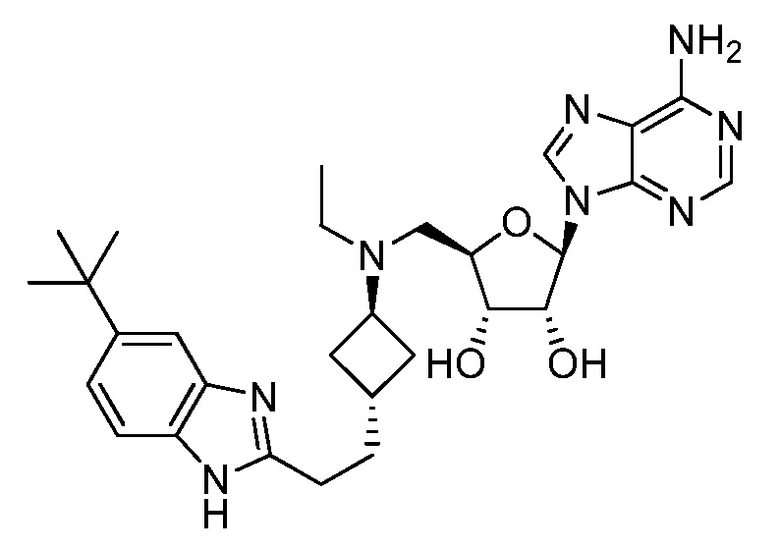
Транс-изомер: 1H ЯМР (500 МГц, MeOD): δH 8,28 (с, 1H), 8,20 (с, 1H), 7,48 (с, 1H), 7,39-7,37 (м, 1H), 7,29-7,26 (м, 1H), 5,97 (д, J=4,0 Гц, 1H), 4,67 (т, J=5,0 Гц, 1H), 4,24 (т, J=6,0 Гц, 1H), 4,18-4,14 (м, 1H), 3,42-3,35 (м, 1H), 2,91-2,78 (м, 4H), 2,64-2,58 (м, 2H), 2,10-2,07 (м, 3H), 1,99-1,96 (м, 2H), 1,85-1,79 (м, 2H), 1,35 (м, 9H), 0,98 (т, J=6,5 Гц, 3H) м.д.; ESI-MS (m/z): 549,3 [M+1]+.
Соединение 76: (2R,3R,4S,5R)-2-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-((изопропил((1s,3R)-3-(2-(5-(трифторметокси)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)амино)метил)тетрагидрофуран-3,4-диол
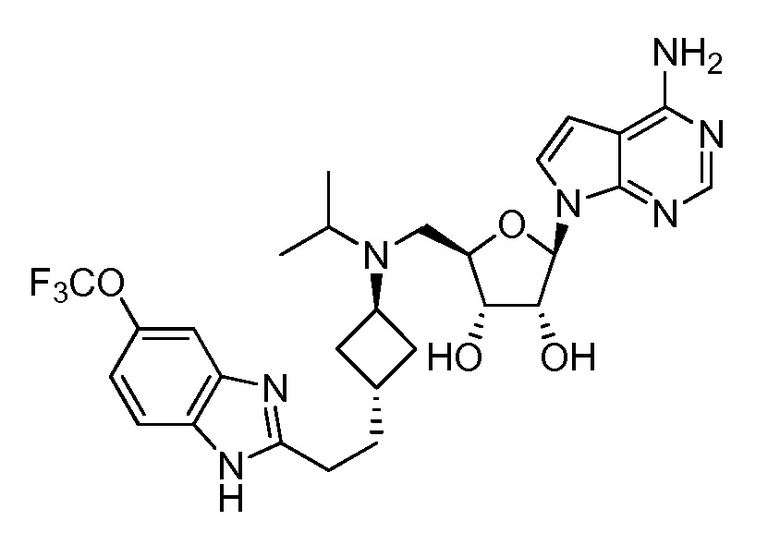
Раствор транс-N-(2,4-диметоксибензил)-7-((3aR,4R,6R,6aR)-6-((изопропил(3-(2-(5-(трифторметокси)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)амино)метил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)-7H-пирроло[2,3-d]пиримидин-4-амина (480 мг, 0,62 ммоль) в 90% TFA (5 мл) перемешивали при 30°C в течение 2 часов. Летучие вещества удаляли при пониженном давлении. К остатку добавляли MeOH (6 мл) и доводили до pH=9~10 при помощи NH3·H2O. Смесь перемешивали при комнатной температуре в течение 30 минут и концентрировали. Остаток очищали методом препаративной ВЭЖХ, с получением желаемого соединения (182 мг, выход: 50%) в виде белого твердого вещества.
1H ЯМР (400 МГц, MeOD): δH 8,09 (с, 1H), 7,52 (д, J=8,8 Гц, 1H), 7,39 (с, 1H), 7,27 (д, J=3,6 Гц, 1H), 7,12 (д, J=8,8 Гц, 1H), 6,64 (д, J=3,2 Гц, 1H), 6,11 (д, J=4,0 Гц, 1H), 4,44 (т, J=4,8 Гц, 1H), 4,12 (т, J=6,0 Гц, 1H), 4,06-4,01 (м, 1H), 3,62-3,53 (м, 1H), 3,10-3,00 (м, 1H), 2,92-2,65 (м, 4H), 2,25-2,15 (м, 2H), 2,10-1,98 (м, 3H), 1,85-1,76 (м, 2H), 1,03 (д, J=6,4 Гц, 3H), 0,98 (д, J=6,4 Гц, 3H) м.д.; 19F ЯМР (400 МГц, MeOD): δ -59,80 м.д.; ESI-MS (m/z): 590,3 [M+1]+.
Соединения 77 и 78
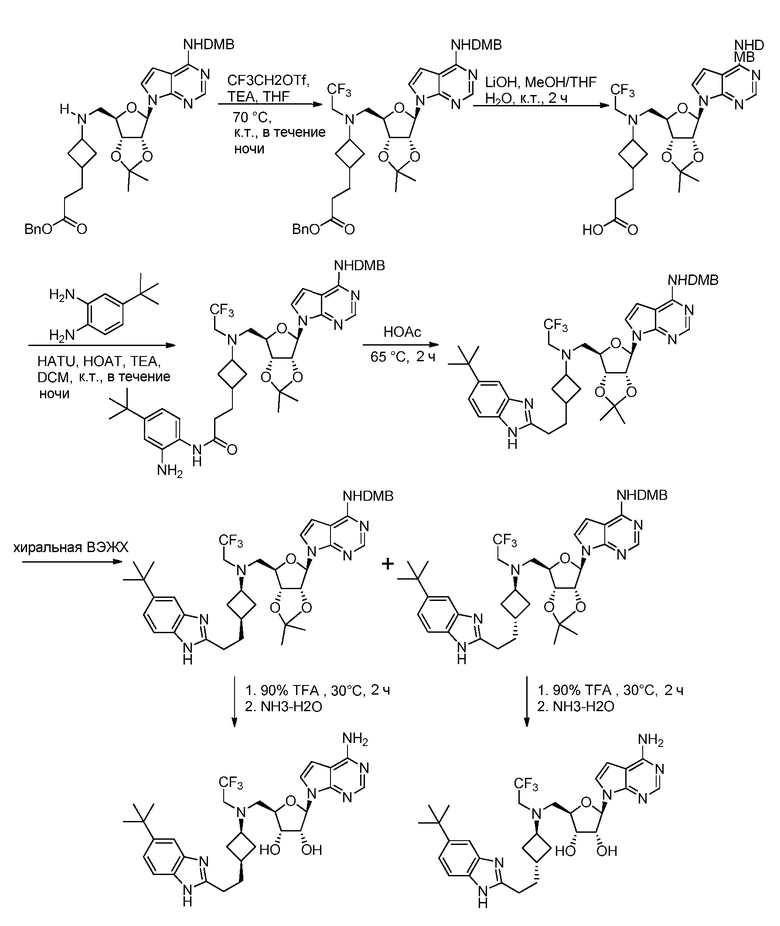
Соединение 78: (2R,3R,4S,5R)-2-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-((((1r,3S)-3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(2,2,2-трифторэтил)амино)метил)тетрагидрофуран-3,4-диол:
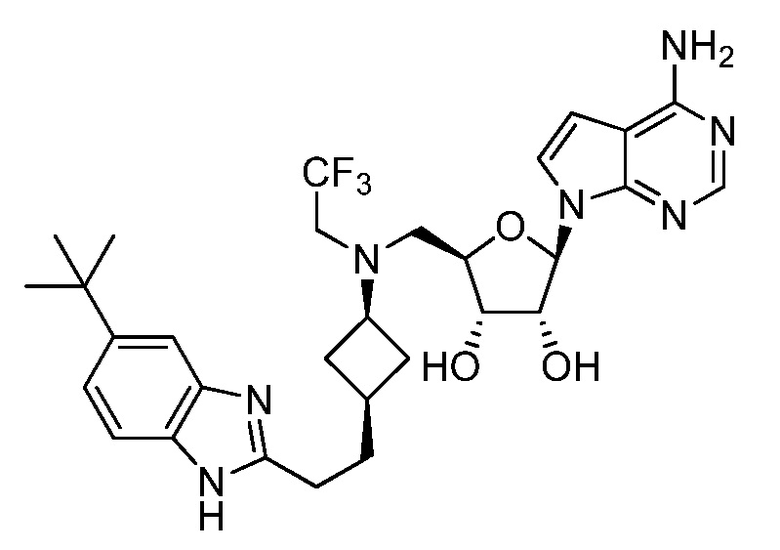
1H ЯМР (500 МГц, MeOD): δH 8,09 (с, 1H), 7,48 (шир.с, 1H), 7,40-7,37 (м, 1H), 7,28 (дд, J=10,5 и 1,5 Гц, 1H), 7,21 (д, J=4,5 Гц, 1H), 6,65 (д, J=4,5 Гц, 1H), 6,11 (д, J=5,0 Гц, 1H), 4,44 (т, J=6,0 Гц, 1H), 4,13-4,08 (м, 2H), 3,30-3,15 (м, 3H), 3,09-3,03 (м, 1H), 2,97-2,90 (м, 1H), 2,78 (т, J=9,0 Гц, 2H), 2,28-2,20 (м, 2H), 1,92-1,78 (м, 3H), 1,55-1,45 (м, 2H), 1,37 (с, 9H) м.д.; LC-MS (m/z): 602,3 [M+1]+.
Соединение 77: (2R,3R,4S,5R)-2-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-((((1s,3R)-3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(2,2,2-трифторэтил)амино)метил)тетрагидрофуран-3,4-диол:
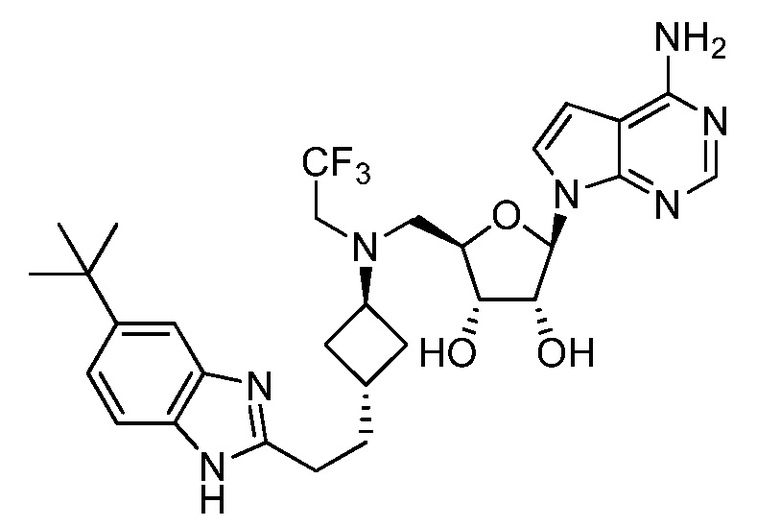
1H ЯМР (500 МГц, MeOD): δH 8,09 (с, 1H), 7,48 (шир.с, 1H), 7,39 (д, J=10,5 Гц, 1H), 7,28 (дд, J=10,5 и 2,0 Гц, 1H), 7,21 (д, J=5,0 Гц, 1H), 6,64 (д, J=4,5 Гц, 1H), 6,12 (д, J=6,0 Гц, 1H), 4,45 (т, J=6,0 Гц, 1H), 4,14-4,09 (м, 2H), 3,68-3,60 (м, 1H), 3,30-3,15 (м, 2H), 3,11-3,04 (м, 1H), 2,97-2,90 (м, 1H), 2,80 (т, J=9,5 Гц, 2H), 2,12-2,03 (м, 3H), 2,00-1,90 (м, 2H), 1,90-1,80 (м, 2H), 1,36 (с, 9H) м.д.; LC-MS (m/z): 602,3 [M+1]+.
Бензил 3-(3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(метил)амино)циклобутил)пропаноат
К смеси бензил 3-(3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)амино)циклобутил)пропаноата (3,76 г, 7,2 ммоль) и NaBH3CN (5,9 г, 93,6 ммоль) в MeOH (40 мл) добавляли AcOH для доведения до pH=6. Затем добавляли 37% HCHO (8,7 мл, 122,4 ммоль) и смесь перемешивали при 30°C в течение 1,5 часов. Добавляли воду (15 мл) и смесь концентрировали в вакууме. Затем смесь экстрагировали при помощи DCM (30 мл ×3). Объединенную органическую фазу промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали. Неочищенное вещество очищали при помощи SGC (DCM: MeOH=100:1-20:1), с получением целевого соединения (2,4 г, выход: 67%) в виде белого твердого вещества.
1H ЯМР (500 МГц, MeOD): δH 8,27 (с, 1H), 8,22 (д, J=2,5 Гц, 1H), 7,35-7,30 (м, 5H), 6,22 (с, 1H), 5,55-5,52 (м, 1H), 5,09 (с, 2H), 5,04-5,01 (м, 1H), 4,40-4,38 (м, 1H), 2,76-2,65 (м, 3H), 2,29-2,22 (м, 2H), 2,18 (с, 3H), 2,11-1,95 (м, 2H), 1,78-1,71 (м, 2H), 1,64-1,61 (м, 2H), 1,59 (с, 3H), 1,41-1,39 (м, 1H), 1,38 (с, 3H) м.д.; ESI-MS (m/z): 537,3 [M+1]+.
3-(3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(метил)-амино)циклобутил)пропановая кислота
К раствору бензил 3-(3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(метил)амино)циклобутил)пропаноата (2,4 г, 4,48 ммоль) в MeOH (40 мл) добавляли 10% Pd/C (2,3 г) и смесь перемешивали в атмосфере H2 при 50°C в течение 15 часов. Смесь фильтровали и фильтрат концентрировали с получением целевого соединения (1,9 г, выход: 95%) в виде белого твердого вещества.
1H ЯМР (500 МГц, MeOD): δH 8,27 (с, 1H), 8,24 (с, 1H), 6,27 (с, 1H), 5,54-5,52 (м, 1H), 5,09-5,07 (м, 1H), 4,50-4,47 (м, 1H), 3,17-3,07 (м, 2H), 3,02-2,90 (м, 1H), 2,39-2,35 (м, 3H), 2,31-2,05 (м, 4H), 1,91-1,70 (м, 2H), 1,64-1,51 (м, 5H), 1,39 (с, 3H), 1,23-1,15 (м, 1H) м.д.; ESI-MS (m/z): 447,2 [M+1]+.
N-(2-амино-4-(трет-бутил)фенил)-3-(3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(метил)амино)циклобутил)пропанамид
К раствору 3-(3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(метил)амино)циклобутил)пропановой кислоты (1,9 г, 4,26 ммоль), HOAT (753 мг, 5,54 ммоль), HATU (2,1 г, 5,54 ммоль) и TEA (3 мл, 21,3 ммоль) в DCM (40 мл) добавляли 4-трет-бутилбензол-1,2-диамин (769 мг, 4,69 ммоль) и смесь перемешивали при комнатной температуре в течение 2 часов. Добавляли воду (15 мл) и смесь экстрагировали при помощи DCM (30 мл ×2). Объединенную органическую фазу промывали при помощи H2O (20 мл ×2). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали. Неочищенное вещество очищали при помощи SGC (DCM: MeOH=70:1-20:1), с получением целевого соединения (1,8 г, выход: 72%) в виде белого твердого вещества.
1H ЯМР (500 МГц, MeOD): δH 8,28 (с, 1H), 8,23 (д, J=3,0 Гц, 1H), 7,10-6,92 (м, 2H), 6,81-6,76 (м, 1H), 6,21 (с, 1H), 5,56 (с, 1H), 5,49 (д, J=3,0 Гц, 1H), 5,01 (с, 1H), 4,36 (с, 1H), 2,69-2,49 (м, 3H), 2,31-2,27 (м, 2H), 2,13-2,00 (м, 5H), 1,88-1,81 (м, 2H), 1,75-1,65 (м, 2H), 1,60 (с, 3H), 1,41-1,35 (м, 4H), 1,28-1,25 (м, 9H) м.д.; ESI-MS (m/z): 593,4 [M+1]+.
9-((3aR,4R,6R,6aR)-6-((((1r,3S)-3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(метил)амино)метил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)-9H-пурин-6-амин
Раствор N-(2-амино-4-(трет-бутил)фенил)-3-(3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(метил)амино)циклобутил)пропанамида (1,8 г, 3,04 ммоль) в AcOH (20 мл) перемешивали при 65°C в течение 15 часов. Раствор концентрировали в вакууме и разбавляли при помощи DCM (30 мл). Смесь промывали насыщенным раствором NaHCO3 (20 мл ×2) и насыщенным солевым раствором (20 мл ×1). Объединенную органическую фазу сушили над Na2SO4, фильтровали и концентрировали с получением 1,7 г (выход: 97%) желаемого продукта.
Продукт разделяли при помощи хиральной ВЭЖХ, с получением 920 мг цис-изомера и 420 мг транс-изомера.
Цис-Изомер: 1H ЯМР (500 МГц, MeOD): δH 8,27 (с, 1H), 8,21 (с, 1H), 7,48 (с, 1H), 7,40-7,38 (м, 1H), 7,29-7,26 (м, 1H), 6,19 (д, J=2,5 Гц, 1H), 5,55-5,52 (м, 1H), 4,99-4,97 (м, 1H), 4,35-4,31 (м, 1H), 2,77-2,73 (м, 2H), 2,62-2,46 (м, 3H), 2,10-2,01 (м, 4H), 1,84-1,81 (м, 3H), 1,58 (с, 3H), 1,38-1,36 (м, 12H), 1,19-1,14 (м, 3H) м.д.; ESI-MS (m/z): 575,3 [M+1]+.
Транс-изомер: 1H ЯМР (500 МГц, MeOD): δH 8,28 (с, 1H), 8,21 (с, 1H), 7,48 (с, 1H), 7,40-7,38 (м, 1H), 7,30-7,27 (м, 1H), 6,19 (д, J=1,5 Гц, 1H), 5,55-5,52 (м, 1H), 5,01-4,98 (м, 1H), 4,36-4,34 (м, 1H), 2,96-2,92 (м, 2H), 2,77 (т, J=7,5 Гц, 2H), 2,58-2,50 (м, 2H), 2,09 (с, 3H), 2,04-1,90 (м, 4H), 1,82-1,79 (м, 1H), 1,70-1,66 (м, 2H), 1,59 (с, 3H), 1,38 (с, 3H), 1,36 (с, 9H) м.д.; ESI-MS (m/z): 575,3 [M+1]+.
Соединение 87: (2R,3R,4S,5R)-2-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-((изопропил((1r,3S)-3-(2-(5-(трифторметокси)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)амино)метил)тетрагидрофуран-3,4-диол
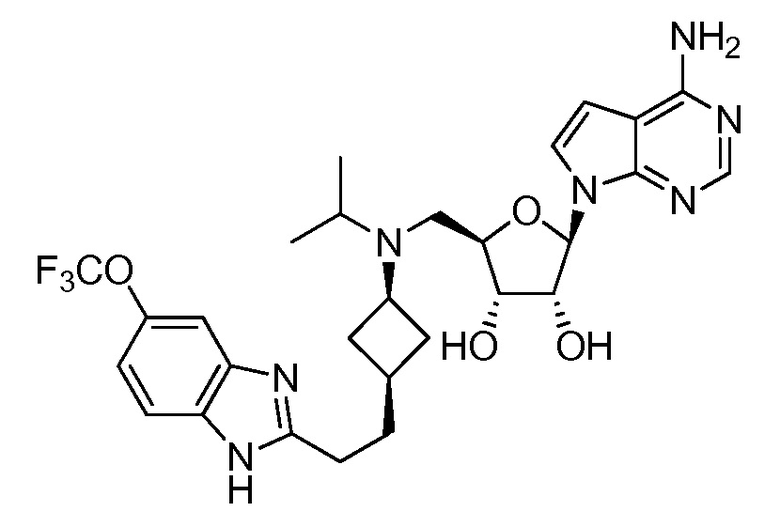
Раствор цис-N-(2,4-диметоксибензил)-7-((3aR,4R,6R,6aR)-6-((изопропил(3-(2-(5-(трифторметокси)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)амино)метил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)-7H-пирроло[2,3-d]пиримидин-4-амина (600 мг, 0,77 ммоль) в 90% TFA (5 мл) перемешивали при 30°C в течение 2 часов. Летучие вещества удаляли при пониженном давлении. К остатку добавляли MeOH (6 мл) и доводили до pH=9~10 при помощи NH3·H2O. Смесь перемешивали при комнатной температуре в течение 30 минут и концентрировали. Остаток очищали при помощи препаративной ВЭЖХ, с получением желаемого соединения (260 мг, выход: 57%) в виде белого твердого вещества.
1H ЯМР (400 МГц, MeOD): δH 8,09 (с, 1H), 7,52 (д, J=8,8 Гц, 1H), 7,39 (с, 1H), 7,27 (д, J=4,0 Гц, 1H), 7,12 (дд, J=8,4 и 0,8 Гц, 1H), 6,64 (д, J=4,0 Гц, 1H), 6,12 (д, J=4,4 Гц, 1H), 4,43 (т, J=5,2 Гц, 1H), 4,11 (т, J=5,2 Гц, 1H), 4,05-4,01 (м, 1H), 3,20-3,10 (м, 1H), 3,08-3,00 (м, 1H), 2,88-2,65 (м, 4H), 2,25-2,15 (м, 2H), 1,92-1,82 (м, 3H), 1,65-1,55 (м, 2H), 1,02 (д, J=6,4 Гц, 3H), 0,98 (д, J=6,4 Гц, 3H) м.д.; 19F ЯМР (400 МГц, MeOD): δ -59,80 м.д.; ESI-MS (m/z): 590,3 [M+1]+.
Соединения 90 и 75:
Бензил 3-(3-((((3aR,4R,6R,6aR)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)амино)циклобутил)пропаноат
К раствору 7-((3aR,4R,6R,6aR)-6-(аминометил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)-N-(2,4-диметоксибензил)-7H-пирроло[2,3-d]пиримидин-4-амина (2,5 г, 5,49 ммоль), бензил 3-(3-оксоциклобутил)пропаноата (1,66 г, 7,14 ммоль) и HOAc (329 мг, 5,49 ммоль) в DCE (40 мл) добавляли NaB(OAc)3H (2,33 г, 11 ммоль) одной порцией. Затем полученную реакционную смесь перемешивали при комнатной температуре в течение ночи. Добавляли насыщенный водный раствор NaHCO3 (40 мл) для гашения реакции, затем смесь экстрагировали при помощи DCM (50 мл ×3), сушили над безводным Na2SO4 и концентрировали. Неочищенное вещество очищали при помощи SGC (DCM:MeOH=100:1 до 50:1), с получением желаемого соединения (2,3 г, выход: 64%) в виде белого твердого вещества.
1H ЯМР (500 МГц, MeOD): δH 8,14 (д, J=2,5 Гц, 1H), 7,35-7,28 (м, 5H), 7,20 (д, J=4,0 Гц, 1H), 7,13 (д, J=8,0 Гц, 1H), 6,64 (д, J=3,0 Гц, 1H), 6,53 (д, J=2,0 Гц, 1H), 6,42 (д, J=8,5 Гц, 1H), 6,18 (д, J=2,5 Гц, 1H), 5,40-5,39 (м, 1H), 5,08-5,07 (м, 2H), 4,96-4,94 (м, 1H), 4,64 (с, 2H), 4,26-4,24 (м, 1H), 3,82 (с, 3H), 3,75 (с, 3H), 3,10-3,05 (м, 0,55H), 2,86-2,82 (м, 2H), 2,26-2,20 (м, 3H), 2,10-1,59 (м, 5H), 1,58 (с, 3H), 1,37 (с, 3H), 1,35-1,25 (м, 0,6H), 1,15-1,08 (м, 0,5H) м.д.; LC-MS (m/z): 672,4 [M+1]+.
Бензил 3-(3-((((3aR,4R,6R,6aR)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(изопропил)амино)циклобутил)пропаноат
Бензил 3-(3-((((3aR,4R,6R,6aR)-6-(4-((2,4-диметоксибензил)-амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)амино)циклобутил)пропаноат (2,3 г, 3,43 ммоль) смешивали с K2CO3 (3,3 г, 24 ммоль) и 2-иодпропаном (5,8 г, 34,3 ммоль) в MeCN (25 мл) в герметично закрытой пробирке, затем нагревали до 95°C при перемешивании в течение 20 часов. Реакционную смесь фильтровали и промывали при помощи MeCN (30 мл), фильтрат упаривали в вакууме с получением желаемого соединения (2,1 г, выход: 88%) в виде белого твердого вещества, которое использовали на следующей стадии без дополнительной очистки.
1H ЯМР (500 МГц, MeOD): δH 8,13 (с, 1H), 7,35-7,29 (м, 5H), 7,18 (д, J=3,0 Гц, 1H), 7,13 (д, J=8,5 Гц, 1H), 6,64 (д, J=3,0 Гц, 1H), 6,53 (д, J=2,5 Гц, 1H), 6,41 (дд, J=8,5 и 2,5 Гц, 1H), 6,18 (д, J=2,5 Гц, 1H), 5,33-5,32 (м, 1H), 5,08-5,06 (м, 2H), 4,90-4,89 (м, 1H), 4,64 (с, 2H), 4,15-4,14 (м, 1H), 3,83 (с, 3H), 3,75 (с, 3H), 3,37-3,36 (м, 0,43H), 3,01-2,98 (м, 0,59H), 2,92-2,88 (м, 1H), 2,70-2,40 (м, 3H), 2,24-2,18 (м, 2H), 2,10-1,80 (м, 3H), 1,75-1,69 (м, 2H), 1,61-1,52 (м, 5H), 1,38 (с, 3H), 0,94 (д, J=6,5 Гц, 3H), 0,81-0,79 (м, 3H) м.д.; LC-MS (m/z): 714,0 [M+1]+.
3-(3-((((3aR,4R,6R,6aR)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(изопропил)амино)циклобутил)пропановая кислота
Бензил 3-(3-((((3aR,4R,6R,6aR)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(изопропил)амино)циклобутил)пропаноат
(1,5 г, 2,1 ммоль) растворяли в MeOH (25 мл), добавляли Pd/C (10% на углероде, 70% воды, 742 мг) и полученную смесь перемешивали при 35°C под давлением 1 атм H2 в течение ночи. Смесь затем фильтровали и промывали при помощи MeOH (15 мл ×3), фильтрат упаривали в вакууме с получением желаемого соединения (1,12 г, выход: 85%) в виде белого твердого вещества, которое использовали на следующей стадии без дополнительной очистки.
1H ЯМР (500 МГц, MeOD): δH 8,18 (с, 1H), 7,90 (с, 1H), 7,35-7,32 (м, 1H), 7,23-7,22 (м, 1H), 7,12 (д, J=8,5 Гц, 1H), 6,66 (шир.с, 1H), 6,54 (д, J=2,0 Гц, 1H), 6,43-6,41 (м, 1H), 6,24-6,23 (м, 1H), 5,47-5,46 (м, 1H), 5,13-5,12 (м, 2H), 4,64 (с, 2H), 4,41-4,37 (м, 1H), 3,84 (с, 3H), 3,75 (с, 3H), 3,64-3,58 (0,6H), 3,46-3,40 (м, 1,7H), 2,45-1,60 (м, 9H), 1,57 (с, 3H), 1,38 (с, 3H), 1,11 (д, J=7,0 Гц, 3H), 0,87 (д, J=6,5 Гц, 3H) м.д.; LC-MS (m/z): 624,0 [M+1]+.
N-(2-амино-4-(трет-бутил)фенил)-3-(3-((((3aR,4R,6R,6aR)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(изопропил)амино)циклобутил)пропанамид
К раствору 3-(3-((((3aR,4R,6R,6aR)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(изопропил)амино)циклобутил)пропановой кислоты (1,1 г, 1,76 ммоль), HATU (1 г, 2,64 ммоль), HOAT (359 мг, 2,64 ммоль) в DCM (30 мл) добавляли раствор 4-трет-бутилбензол-1,2-диамина (433 мг, 2,64 ммоль) и TEA (533 мг, 5,28 ммоль) в DCM (10 мл) по каплям, затем полученную реакционную смесь перемешивали при комнатной температуре в течение ночи. После разбавления при помощи DCM (50 мл), смесь промывали водой (30 мл ×3), сушили и концентрировали. Неочищенное вещество очищали при помощи SGC (DCM:MeOH=100:1 до 40:1), с получением желаемого соединения (680 мг, выход: 50%) в виде белого твердого вещества.
1H ЯМР (500 МГц, MeOD): δH 8,16 (с, 1H), 7,19 (д, J=3,5 Гц, 1H), 7,14-7,10 (м, 1,7H),6,99-6,97 (м, 0,6H), 6,92 (с, 0,6H), 6,77 (дд, J=8,0, 2,0 Гц, 1H), 6,66-6,65 (м, 1H), 6,54-6,53 (м, 1H), 6,42 (д, J=8,0 Гц, 1H), 6,20 (д, J=2,0 Гц, 1H), 5,36-5,35 (м, 1H), 4,96-4,95 (м, 1H), 4,65 (с, 2H), 3,83 (с, 3H), 3,75 (с, 3H), 3,17-2,73 (м, 4H), 2,33-1,71 (м, 8H), 1,58 (с, 3H), 1,53-1,50 (м, 1H), 1,38 (с, 3H), 1,28 (с, 9H), 1,0 (д, J=5,5 Гц, 3H), 0,84 (д, J=5,0 Гц, 3H) м.д.; LC-MS (m/z): 770,0 [M+1]+.
7-((3aR,4R,6R,6aR)-6-(((3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(изопропил)амино)метил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)-N-(2,4-диметоксибензил)-7H-пирроло[2,3-d]пиримидин-4-амин
N-(2-Амино-4-(трет-бутил)фенил)-3-(3-((((3aR,4R,6R,6aR)-6-(4-((2,4-диметоксибензил)амино)-7H-пирроло[2,3-d]пиримидин-7-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(изопропил)амино)циклобутил)пропанамид (670 мг, 0,87 ммоль) растворяли в HOAc (8 мл) и затем нагревали до 65°C при перемешивании в течение ночи. Растворитель удаляли в вакууме. Остаток растворяли в DCM (60 мл), затем промывали NaHCO3 (насыщенный раствор, 20 мл) и водой (20 мл), органическую фазу сушили и концентрировали. Неочищенное вещество очищали методом препаративной ТСХ (DCM:MeOH=10:1), с получением желаемого соединения (470 мг, выход: 73%) в виде белого твердого вещества, которое затем разделяли при помощи хиральной ВЭЖХ, с получением цис (243 мг) и транс изомеров (180 мг) в виде белых твердых веществ.
Цис-изомер: 1H ЯМР (500 МГц, MeOD): δH 8,15 (с, 1H), 7,47 (с, 1H), 7,38-7,37 (м, 1H), 7,27 (дд, J=8,5 и 1,5 Гц, 1H), 7,17 (д, J=4,0 Гц, 1H), 7,10 (д, J=8,5 Гц, 1H), 6,65 (д, J=3,0 Гц, 1H), 6,49 (д, J=2,5 Гц, 1H), 6,38 (дд, J=8,0 и 2,5 Гц, 1H), 6,18 (д, J=2,5 Гц, 1H), 5,33 (дд, J=6,5 и 2,5 Гц, 1H), 4,92-4,91 (м, 1H), 4,63 (с, 2H), 4,16-4,15 (м, 1H), 3,78 (с, 3H), 3,72 (с, 3H), 3,08-3,07 (м, 1H), 2,97-2,96 (м, 1H), 2,72-2,66 (м, 4H), 2,09-2,03 (м, 2H), 1,82-1,78 (м, 3H), 1,56 (с, 3H), 1,48-1,40 (м, 2H), 1,38 (с, 3H), 1,36 (с, 9H), 0,95 (д, J=7,0 Гц, 3H), 0,81 (д, J=6,5 Гц, 3H) м.д.; LC-MS (m/z): 752,0 [M+1]+.
Транс-изомер: 1H ЯМР (500 МГц, MeOD): δH 8,14 (с, 1H), 7,47 (с, 1H), 7,38-7,37 (м, 1H), 7,27 (дд, J=8,5 и 1,5 Гц, 1H), 7,18 (д, J=3,5 Гц, 1H), 7,10 (д, J=8,0 Гц, 1H), 6,64 (д, J=4,0 Гц, 1H), 6,50 (д, J=2,0 Гц, 1H), 6,39-6,37 (м, 1H), 6,18 (д, J=2,0 Гц, 1H), 5,33 (дд, J=6,0 и 2,5 Гц, 1H), 4,90 (дд, J=6,5 и 3,5 Гц, 1H), 4,62 (с, 2H), 4,17-4,15 (м, 1H), 3,80 (с, 3H), 3,71 (с, 3H), 3,44-3,43 (м, 1H), 2,93-2,90 (м, 1H), 2,77-2,68 (м, 3H), 2,62-2,60 (м, 1H), 2,05-1,93 (м, 5H), 1,68-1,67 (м, 2H), 1,56 (с, 3H), 1,36 (с, 12H), 0,95 (д, J=7,0 Гц, 3H), 0,81 (д, J=6,5 Гц, 3H) м.д.; LC-MS (m/z): 752,0 [M+1]+.
Соединение 90: (2R,3R,4S,5R)-2-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-((((1r,3S)-3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(изопропил)-амино)метил)тетрагидрофуран-3,4-диол
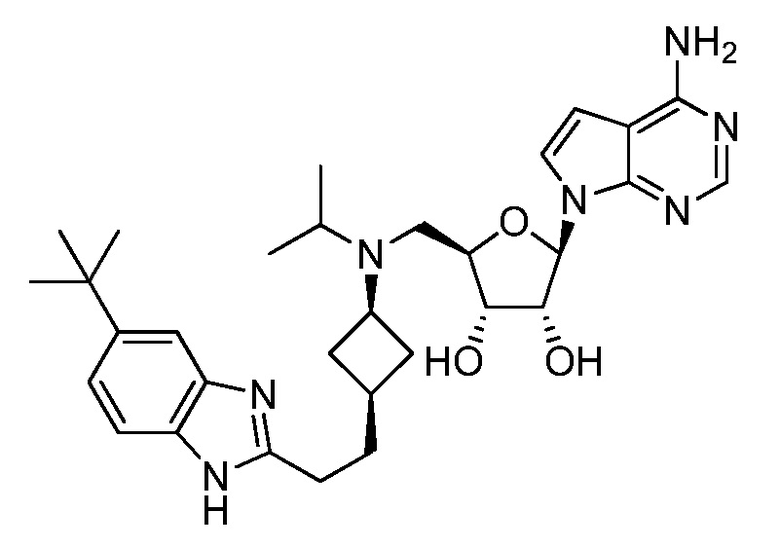
К смеси TFA (2,7 мл) и воды (0,3 мл) добавляли цис изомер-7-((3aR,4R,6R,6aR)-6-(((3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(изопропил)амино)метил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)-N-(2,4-диметоксибензил)-7H-пирроло[2,3-d]пиримидин-4-амин (235 мг, 0,31 ммоль). Раствору давали выстояться при 35°C в течение 2 часов и упаривали досуха. Остаток совместно упаривали с метанолом два раза. Затем остаток растворяли в MeOH (20 мл). Раствор найтрализовали при помощи K2CO3 (124 мг, растворенный в 1 мл H2O) при перемешивании при комнатной температуре в течение 1 часа. Растворитель удаляли в вакууме, затем неочищенное вещество очищали методом препаративной ВЭЖХ, с получением желаемого соединения (90 мг, выход: 51%) в виде белого твердого вещества.
1H ЯМР (500 МГц, MeOD): δH 8,08 (с, 1H), 7,47 (с, 1H), 7,39-7,37 (м, 1H), 7,28 (д, J=2,5 Гц, 1H), 7,26 (д, J=4,5 Гц, 1H), 6,62 (д, J=4,5 Гц, 1H), 6,10 (д, J=5,5 Гц, 1H), 4,43-4,41 (м, 1H), 4,12-4,09 (м, 1H), 4,04-4,01 (м, 1H), 3,15-3,13 (м, 1H), 3,04-3,01 (м, 1H),2,86-2,68 (м, 4H), 2,21-2,17 (м, 2H), 1,88-1,85 (м, 3H), 1,60-1,57 (м, 2H), 1,36 (с, 9H), 1,01 (д, J=8,0 Гц, 3H), 0,97 (д, J=8,0 Гц, 3H) м.д.; LC-MS (m/z): 562,5 [M+1]+.
Соединение 75: (2R,3R,4S,5R)-2-(4-амино-7H-пирроло[2,3-d]пиримидин-7-ил)-5-((((1s,3R)-3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(изопропил)амино)-метил)тетрагидрофуран-3,4-диол
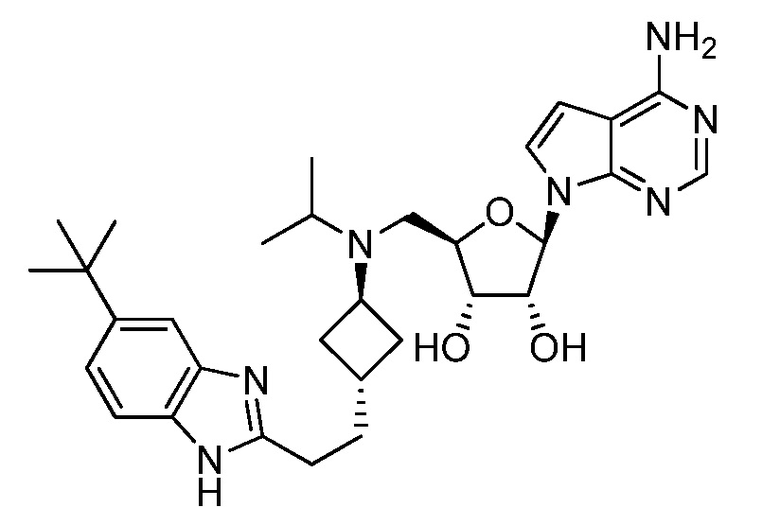
К смеси TFA (2,7 мл) и воды (0,3 мл) добавляли транс изомер 7-((3aR,4R,6R,6aR)-6-(((3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(изопропил)амино)метил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)-N-(2,4-диметоксибензил)-7H-пирроло[2,3-d]пиримидин-4-амина (175 мг, 0,23 ммоль). Раствору давали выстояться при 35°C в течение 2 часов и упаривали досуха. Остаток упаривали вместе с метанолом два раза. Затем остаток растворяли в MeOH (20 мл). Раствор найтрализовали при помощи K2CO3 (97 мг, растворенный в 1 мл H2O) при перемешивании при комнатной температуре в течение 1 часа. Растворитель удаляли в вакууме, затем неочищенное вещество очищали методом препаративной ВЭЖХ, с получением желаемого соединения (95 мг, выход: 71%) в виде белого твердого вещества.
1H ЯМР (500 МГц, MeOD): δH 8,09 (с, 1H), 7,49 (с, 1H), 7,41-7,39 (м, 1H), 7,30-7,27 (м, 2H), 6,64 (д, J=4,0 Гц, 1H), 6,12 (д, J=5,5 Гц, 1H), 4,45-4,42 (м, 1H), 4,13-4,11 (м, 1H), 4,05-4,03 (м, 1H), 3,58-3,54 (м, 1H), 3,06-3,02 (м, 1H), 2,89-2,80 (м, 3H), 2,74-2,70 (м, 1H), 2,20-2,17 (м, 2H), 2,03-1,99 (м, 3H), 1,84-1,81 (м, 3H), 1,38 (с, 9H), 1,03 (д, J=8,5 Гц, 3H), 0,98 (д, J=8,0 Гц, 3H) м.д.; LC-MS (m/z): 562,5 [M+1]+.
Соединение 96: (2R,3R,4S,5R)-2-(6-амино-9H-пурин-9-ил)-5-((метил((1r,3S)-3-(2-(5-(трифторметокси)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)амино)метил)тетрагидрофуран-3,4-диол
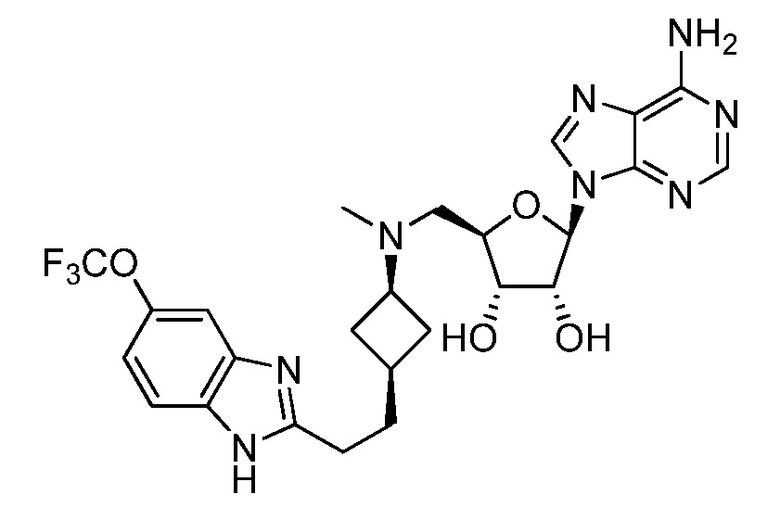
N-(2-амино-4-(трифторметокси)фенил)-3-(3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(метил)амино)циклобутил)пропанамид
К раствору 3-(3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(метил)амино)циклобутил)пропановой кислоты (1,2 г, 2,91 ммоль), HATU (2,17 г, 5,83 ммоль) и HOAT (0,91 г, 5,83 ммоль) в DCM (17 мл) добавляли 4-(трифторметокси)бензол-1,2-диамин (1,1 г, 5,83 ммоль) и TEA (2,05 мл, 14,56 ммоль). Смесь перемешивали при комнатной температуре в течение ночи. Смесь разбавляли при помощи DCM (50 мл) и промывали водой (15 мл ×3) и насыщенным солевым раствором (30 мл). Органическую фазу сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали при помощи Combi-Flash (40 г силикагеля, с использованием градиента EA:DCM:MeOH = от 10:10:0 до 10:10:8, 40 мл/мин, 50 минут, общий объем растворителя 2,0 л), с получением желаемого соединения (1,0 г, выход: 60 %) в виде желтого твердого вещества.
1H ЯМР (500 МГц, MeOD): δH 8,29 (с, 1H), 8,25-8,24 (м, 1H), 7,20-7,13 (м, 1H), 6,95-6,85 (м, 0,4H), 6,73 (шир.с, 0,8H), 6,54 (д, J=7,5 Гц, 0,8H), 6,22 (д, J=2,0 Гц, 1H), 5,60-5,55 (м, 1H), 5,03-5,00 (м, 1H), 4,40-4,35 (м, 1H), 3,40-3,35 (м, 0,3H), 3,00-2,92 (м, 0,7H), 2,70-2,47 (м, 3H), 2,36-2,28 (м, 2H), 2,20-1,80 (м, 6H), 1,76-1,66 (м, 2H), 1,60 (с, 3H), 1,40 (с, 3H), 1,26-1,16 (м, 1H) м.д.; ESI-MS (m/z): 621,3 [M+1]+.
9-((3aR,4R,6R,6aR)-2,2-диметил-6-((метил(3-(2-(5-(трифторметокси)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)-амино)метил)тетрагидрофуро[3,4-d][1,3]диоксол-4-ил)-9H-пурин-6-амин
Раствор N-(2-амино-4-(трифторметокси)фенил)-3-(3-((((3aR,4R,6R,6aR)-6-(6-амино-9H-пурин-9-ил)-2,2-диметилтетрагидрофуро[3,4-d][1,3]диоксол-4-ил)метил)(метил)амино)циклобутил)пропанамида (1,0 г, 1,61 ммоль) в HOAc (10 мл) перемешивали при 65°C в течение ночи. Смесь охлаждали до комнатной температуры и концентрировали. Остаток растворяли в DCM (50 мл), промывали насыщенным раствором NaHCO3 (10 мл ×2), водой (20 мл) и насыщенным солевым раствором (30 мл). Остаток разделяли при помощи хиральной ВЭЖХ, с получением цис изомера(460 мг, выход: 47%) и транс изомера (220 мг, выход: 23%).
цис-изомер: 1H ЯМР (500 МГц, MeOD): δH 8,28 (с, 1H), 8,23 (с, 1H), 7,53 (д, J=9,0 Гц, 1H), 7,40 (с, 1H), 7,13 (дд, J=8,5 и 1,0 Гц, 1H), 6,21 (д, J=2,0 Гц, 1H), 5,55 (дд, J=6,5, 2,5 Гц, 1H), 5,01 (кв., J=3,5 Гц, 1H), 4,33-4,37 (м, 1H), 2,81-2,78 (м, 2H), 2,66-2,58 (м, 2H), 2,53-2,49 (м, 1H), 2,13-2,03 (м, 5H), 1,86-1,84 (м, 3H), 1,59 (с, 3H), 1,43-1,39 (м, 4H), 1,22-1,17 (м, 1H) м.д.; LC-MS (m/z): 603,3 [M+1]+.
транс-изомер: 1H ЯМР (500 МГц, MeOD): δH 8,29 (с, 1H), 8,22 (с, 1H), 7,52 (шир.с, 1H), 7,40 (с, 1H), 7,13 (д, J=9,0 Гц, 1H), 6,21 (д, J=2,0 Гц, 1H), 5,55 (дд, J=6,5 и 2,0 Гц, 1H), 5,01 (кв., J=3,0 Гц, 1H), 4,38-4,34 (м, 1H), 2,96-2,92 (м, 1H), 2,84-2,81 (м, 2H), 2,59-2,49 (м, 2H), 2,10 (с, 3H), 2,05-1,91 (м, 4H), 1,85-1,79 (м, 1H), 1,73-1,66 (м, 2H), 1,60 (с, 3H), 1,39 (с, 3H) м.д.; LC-MS (m/z): 603,3 [M+1]+.
Соединение 96
Раствор цис-9-((3aR,4R,6R,6aR)-2,2-диметил-6-((метил(3-(2-(5-(трифторметокси)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)амино)метил)тетрагидрофуро[3,4-d][1,3]диоксол-4-ил)-9H-пурин-6-амина (460 мг, 0,77 ммоль) в 1н HCl/MeOH (10 мл) перемешивали при 30°C в течение 4 часов. Летучие вещества удаляли при пониженном давлении. К остатку добавляли MeOH (10 мл) и доводили до pH=10~11 при помощи NH3·H2O. Смесь перемешивали при комнатной температуре в течение 30 минут и концентрировали. Остаток очищали методом препаративной ВЭЖХ, с получением желаемого соединения (215 мг, выход: 43%) в виде белого твердого вещества.
1H ЯМР (500 МГц, MeOD): δH 8,28 (с, 1H), 8,21 (с, 1H), 7,52 (д, J=8,0 Гц, 1H), 7,40 (с, 1H), 7,12 (дд, J=8,5 и 0,5 Гц, 1H), 6,00 (д, J=4,0 Гц, 1H), 4,71 (т, J=4,5 Гц, 1H), 4,24 (т, J=5,5 Гц, 1H), 4,18 (т, J=6,0 Гц, 1H), 2,84-2,81 (м, 2H), 2,77-2,68 (м, 3H), 2,25-2,22 (м, 2H), 2,17 (с, 3H), 1,91-1,89 (м, 3H), 1,51-1,45 (м, 2H) м.д.; LC-MS (m/z): 563,3 [M+1]+.
Соединение 97: (2R,3R,4S,5R)-2-(6-амино-9H-пурин-9-ил)-5-((метил((1s,3R)-3-(2-(5-(трифторметокси)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)амино)метил)тетрагидрофуран-3,4-диол
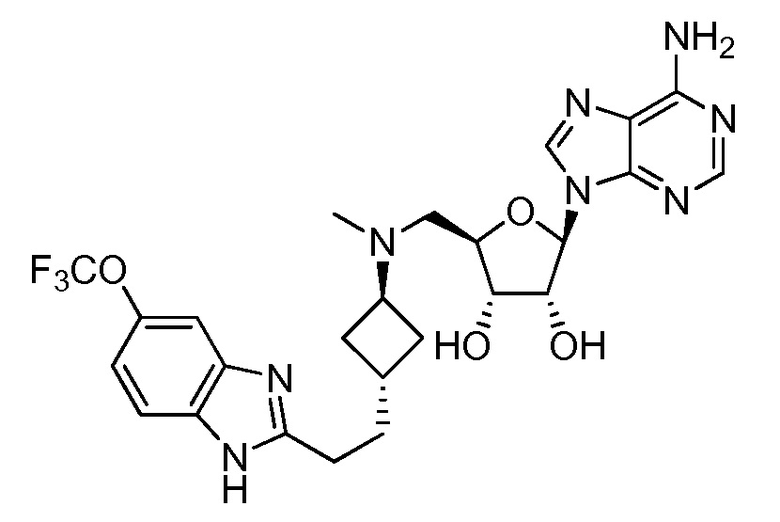
Раствор транс-9-((3aR,4R,6R,6aR)-2,2-диметил-6-((метил(3-(2-(5-(трифторметокси)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)-амино)метил)тетрагидрофуро[3,4-d][1,3]диоксол-4-ил)-9H-пурин-6-амина (220 мг, 0,37 ммоль) в 1н HCl/MeOH (5 мл) перемешивали при 30°C в течение 4 часов. Летучие вещества удаляли при пониженном давлении. К остатку добавляли MeOH (10 мл) и доводили до pH=10~11 при помощи NH3·H2O. Смесь перемешивали при комнатной температуре в течение 30 минут и концентрировали. Остаток очищали методом препаративной ВЭЖХ, с получением желаемого соединения (80 мг, выход: 39%) в виде белого твердого вещества.
1H ЯМР (500 МГц, MeOD): δH 8,29 (с, 1H), 8,20 (с, 1H), 7,52 (д, J=8,5 Гц, 1H), 7,40 (с, 1H), 7,11 (д, J=8,5 Гц, 1H), 6,00 (д, J=4,0 Гц, 1H), 4,72 (т, J=4,5 Гц, 1H), 4,25 (т, J=5,5 Гц, 1H), 4,18-4,20 (м, 1H), 3,04-3,08 (м, 1H), 2,83-2,86 (м, 2H), 2,64-2,72 (м, 2H), 2,17 (с, 3H), 1,97-2,10 (м, 5H), 1,82-1,85 (м, 2H) м.д.; LC-MS (m/z): 563,3 [M+1]+.
Соединение 106
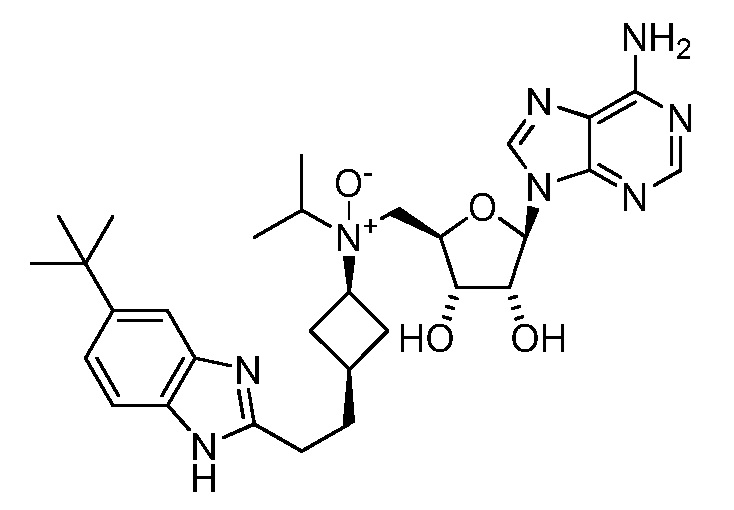
К раствору (2R,3R,4S,5R)-2-(6-амино-9H-пурин-9-ил)-5-((((1r,3S)-3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(изопропил)амино)метил)тетрагидрофуран-3,4-диола (300 мг, 0,53 ммоль) в 30% водном растворе диоксана (20 мл) добавляли MCPBA (91 мг, 0,53 ммоль). Смесь перемешивали при комнатной температуре в течение 3 часов и затем концентрировали. Неочищенное вещество очищали методом препаративной ВЭЖХ, с получением желаемого продукта (160 мг, выход: 45%) в виде белого твердого вещества.
1H ЯМР (500 МГц, MeOD): δH 8,24 (с, 1H), 8,22 (с, 1H), 7,49 (шир.с, 1H), 7,42-7,38 (м, 1H), 7,31-7,29 (м, 1H), 6,00-5,96 (м, 1H), 4,68-4,65 (м, 2H), 4,43-4,36 (м, 1H), 4,05-4,00 (м, 1H), 3,88-3,76 (м, 1H), 3,68-3,49 (м, 1H), 3,46-3,37 (м, 1H), 2,84-2,81 (м, 2H), 2,42-2,15 (м, 4H), 1,95-1,89 (м, 3H), 1,39 (с, 1H), 1,35-1,27 (м, 3H), 1,25-1,21 (м, 3H) м.д.; ESI-MS (m/z): 579,4 [M+1]+.
Соединение 107
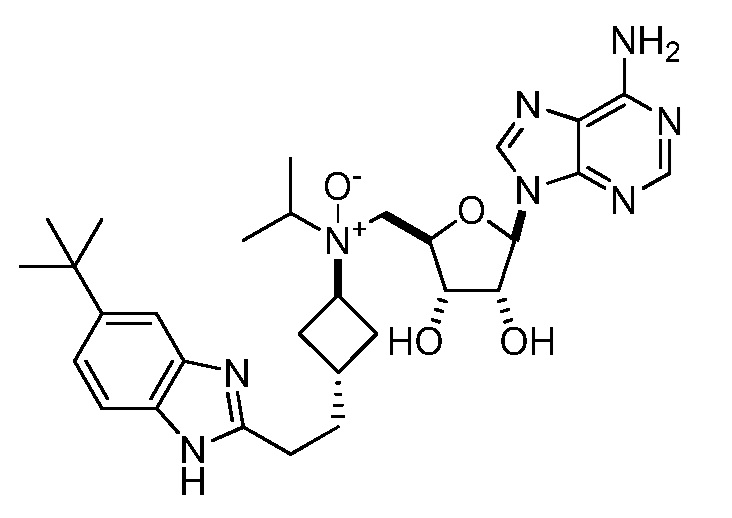
К раствору (2R,3R,4S,5R)-2-(6-амино-9H-пурин-9-ил)-5-((((1s,3R)-3-(2-(5-(трет-бутил)-1H-бензо[d]имидазол-2-ил)этил)циклобутил)(изопропил)амино)метил)тетрагидрофуран-3,4-диола (300 мг, 0,53 ммоль) в 30% водном растворе диоксана (20 мл) добавляли MCPBA (91 мг, 0,53 ммоль). Смесь перемешивали при комнатной температуре в течение 3 часов и затем концентрировали. Неочищенное вещество очищали методом препаративной ВЭЖХ, с получением желаемого продукта (80 мг, выход: 26%) в виде белого твердого вещества.
1H ЯМР (500 МГц, MeOD): δH 8,25-8,22 (м,2H), 7,49 (шир.с, 1H), 7,42-7,38 (м, 1H), 7,31-7,28 (м, 1H), 6,02-5,97 (м, 1H), 4,69-4,60 (м, 2H), 4,47-4,32 (м, 2H), 3,86-3,74 (м, 1H), 3,70-3,54 (м, 1H), 3,45-3,35 (м, 1H), 2,97-2,72 (м, 4H), 2,15-1,75 (м, 5H), 1,39 (с, 1H), 1,34-1,28 (м, 3H), 1,26-1,22 (м, 3H) м.д.; ESI-MS (m/z): 579,7 [M+1]+.
Пример 9: Сверхкритическая Жидкостная Хроматография
Соединения очищали методом Сверхкритической Жидкостной Хроматографии (SFC) с использованием известных процедур. Например, способов, используемых Lotus Separations, LLC, Princeton, NJ. См., например, http://www.lotussep.com. См. также, http://www.greenchemistrygroup.org/Program2009.html.
Условия SFC разделения для некоторых примеров указаны ниже. Другие соединения, описанные в настоящей заявке, можно разделить аналогичными способами.
Пример 10: Протокол биоанализа и общие способы
Клеточная культура. Клеточные линии человеческих гематологических опухолей THP-1, RS4;11 и MV4-11 были получены от ATCC, MOLM-13 клетки были получены от DSMZ. Все линии выращивали в RPMI 1640, содержащей 10% FBS, и поддерживали с использованием рекомендаций поставщика, что касается плотности клеток и условий окружающей среды. Среду дополняли заменимыми аминокислотами и L-Глутамином. THP-1 клетки также дополняли 0,05 мМ β-Меркаптоэтанола.
Анализ метилирования. Клетки высевали при плотности 5Х105 клеток/мл в 12 луночный планшет в конечном объеме 2 мл. К клеткам добавляли соединения до подходящей концентрации из 50 мМ DMSO исходного раствора. Соединение и среду обновляли через каждые два дня в течение семидневного курса инкубации путем подсчета клеток методом вытеснения с использованием трипанового синего (Vicell), осаждения центрифугированием при 200 g в течение 5 минут и ресуспендирования в свежей среде, содержащей соединение при конечной концентрации 5X105 клеток/мл. После инкубации с соединением гистоны экстрагировали из 1Х106 клеток с использованием коммерческого набора для экстракции гистонов (Active Motif). Определяли количество очищенных гистонов с использованием анализа белка BCA (Pierce) с BSA стандартной кривой. 400 нг выделенных гистонов фракционировали методом SDS-PAGE на 4-20% геле и переносили на нитроцеллюлозные мембраны. Мембраны инкубировали с различными первичными и вторичными антителами и получали изображение на системе визуализации Licor (Odyssey). H3K79-Me2 кроличье поликлональное антитело закупали у Abcam. Другие кроличьи поликлональные антитела, включая H3K4-Me3, H3K9-Me3, H3K27-Me2 и H3K27-Me3, закупали у Cell Signalling Technologies (CST). Мышиное моноклональное полное H3 антитело использовали в качестве контроля нагрузки (CST). Флуоресцентно-меченные вторичные антитела закупали у Odyssey.
Анализ роста и жизнеспособности клеток. Клетки собирали из экспоненциально растущих клеточных культур и высевали при плотности 3×104 клеток на лунку. Образцы поддерживали в 96-луночном планшете с черными стенками и прозрачным дном (Corning). Конечную концентрацию 50 мкМ соединения в 0,2% DMSO добавляли в соответствующие лунки в День 0. Обработку MV4-11 и MOLM-13 продолжали 14 дней, тогда как THP-1 клетки обрабатывали в течение 18 дней. Соединение и среду заменяли через каждые два дня в процессе инкубации путем переноса образцов в V-донный планшет (Corning), центрифугирования при 200 g в течение 5 минут в роторе при комнатной температуре, ресуспендирования в свежей среде, содержащей соединение, и переноса обратно в аналитический планшет. Клетки подсчитывали периодически с использованием Guava Viacount анализа и считывали на устройстве EasyCyte Plus (Millipore). Клетки разделяли в аналитических планшетах, когда это было необходимо, для поддержания рекомендованной плотности клеток. Конечные данные подсчета клеток корректировали с учетом разделений клеток и представляли как общее количество жизнеспособных клеток/лунка.
HOKCA9 (qPCR). Клетки обрабатывали соединением в течение 7 дней, подобно тому, как в анализе метилирования. Клетки осаждали центрифугированием при 200 g в роторе при комнатной температуре и РНК полностью выделяли с использованием набора Qiagen RNeasy. Концентрацию и качество РНК определяли с использованием Nanovue (GE Healthcare). Осуществляли обратную транскрипцию всей РНК с использованием набора для высокопроизводительной обратной транскрипции кДНК (Applied Biosystems). Специально сконструированный набор меченных праймеров для HOXA9 закупали у Applied Biosystems. qPCR реакции содержали 50 нг кДНК, 1X меченный праймер и 1X Taqman универсальную смесь для PCR (Applied Biosystems). Образцы исследовали на устройстве 7900 HT Fast Real Time PCR (Applied Biosystems) с PCR условиями 2 мин. 50°C, 10 мин. 95°C, 40 циклов при 15 сек 95°C и 1 мин 60°C. Количество циклов HOXA9 нормализовали к контролирующему гену B2 микроглобулину (B2M специально запланированный контроль от Applied Biosystems). Процент DMSO контроля рассчитывали при помощи уравнения, процент контроля = (2^-ΔΔCT)*100, где ΔΔCT представляет собой разницу между нормализованным HOXA9 образцом и контролем (ΔCT образец - ΔCT контроль = ΔΔCT).
Определение ИК50. Осуществляли 3-кратные разведения испытываемых соединений в DMSO для 10 точек и 1 мкл вносили в 384-луночный микротитровальный планшет. Положительный контроль (100% ингибирующий стандарт) представлял собой 2,5 мкМ конечную концентрацию S-аденозил-L-гомоцистеина, и отрицательный контроль (0% ингибирующий стандарт) содержал 1 мкл DMSO. Соединение затем инкубировали в течение 30 минут с 40 мкл на лунку DOT1L(1-416) (0,25 нМ конечная концентрация в буфере для анализа: 20 мМ TRIS, pH 8,0, 10 мМ NaCl, 0,002% Tween20, 0,005% Бычьего Кожного Желатина, 100 мМ KCl и 0,5 мМ DTT). 10 мкл на лунку смешанного субстрата (тотже буфер для анализа с 200 нМ S-[метил-3H]-аденозил-L метионином, 600 нМ немеченного S-[метил-3H]-аденозил-L метионина и 20 нМ олигонуклеосомы) добавляли для инициирования реакции. Реакцию инкубировали в течение 120 минут при комнатной температуре и гасили при помощи 10 мкл на лунку 100 мкМ S-метил-аденозил-L метионина. Для детекции субстрат из 50 мкл реакционной смеси иммобилизировали на 384-луночном покрытом стрептавидином планшете Flasplate (Perkin Elmer) (также покрытом 0,2% полиэтиленимином) и считывали при помощи сцинтилляционного счетчика Top Count (Perkin Elmer). Значения ИК50 представлены в таблице ниже. В этой таблице, “A” указывает значения ИК50 <0,1 мкМ; “B” указывает значения ИК50 >0,1 мкМ и <1 мкМ; “C” указывает значения ИК50 >1 мкМ и <10 мкМ; и “D” указывает значения ИК50 >10 мкМ и <50 мкМ.
ИК50
Пример 11: Анти-пролиферативные анализы опухолей
In Vitro Анти-пролиферативной анализ. Эффективность и селективность анти-пролиферативной активности соединений по настоящему изобретению оценивали с использованием панели MLL-реаранжированных и не-MLL-реаранжированных клеточных линий лейкоза человека. Клеточные линии, используемые в испытании, указаны на Фиг. 1A. MLL-реаранжированная панель включала клеточные линии, происходящие из ALL, AML и бифенотипических лейкозов со слияниями MLL-AF4, MLL-AF9 или MLL-ENL. Эти клеточные линии рекрутируют DOT1L. Панель также включала пять клеточных линий, которые не имели MLL-реаранжировки, и одну клеточную линию, которая содержала частичную тандемную дупликацию MLL гена (MLL-PTD).
Экспоненциально растущие клетки высевали, в трех повторах, в 96-луночные планшеты при плотности 3×104 клеток/лунка в конечном объеме 150 мкл. Клетки инкубировали в присутствии увеличивающихся концентраций Соединения 2. Анти-пролиферативную активность определяли путем измерения жизнеспособности клеток через каждые 3-4 дня в течение 14 дней. В дни, когда осуществляли подсчет клеток, питательную среду и Соединение 2 заменяли и клетки снова разделяли до плотности 5×104 клеток/лунка.
На Фиг. 1 представлены результаты полумаксимальной ингибирующей концентрации (ИК50), которые показывают, что Соединение 2 демонстрирует сильную наномолярную анти-пролиферативную активность против трех из четырех испытанных MLL-реаранжированных клеточных линий (MV4;11 (MLL-AF4), MOLM-13 (MLL-AF9) и KOPN-8 (MLL-ENL). EOL-1 клетки, которые экспрессируют MLL-PTD, также показали высокую чувствительность к Соединению 2 (ИК50=11 нМ). RS4;11 клетки и два типа не-MLL-реаранжированных клеток (Reh и Kasumi-1) были на 1-3 порядка менее чувствительны, и два типа не-MLL-реаранжированных клеток (Jurkat и HL-60) показали отсутствие активности. В целом, результаты показывают, что Соединение 2 эффективно и селективно ингибирует пролиферацию MLL-реаранжированных лейкозных клеточных линий и подгруппу не-MLL-реаранжированных лейкозных клеточных линий.
In Vivo анти-пролиферативной анализ. Противоопухолевую in vivo активность соединений по настоящему изобретению оценивали в мышиной модели ксенотрансплантата MLL-реаранжированного лейкоза.
Четырем группам из 20 (Группы 1, 3, 4 и 5) и одной группе из 8 (Группа 2) самок “голых” мышей (средняя масса тела 0,023 кг) с MV4-11 ксенотрансплантатом опухолей с размерами 80-120 мм3 имплантировали подкожно мининасосы (Alzet Model 2001). Группа 1 получала только носитель из насоса. Группа 2 получала только носитель из насоса плюс интраперитонеальные инъекции три раза в день (с 8-часовыми интервалами) носителя. Группа 3 получала 112 мг/кг/день из насоса плюс интраперитонеальные инъекции три раза в день (с 8-часовыми интервалами) 20 мг/кг соединения 2 для общей суточной дозы 172 мг/кг/день. Группы 4 и 5 получали 112 и 56 мг/кг/день соединения 2 из насосов, соответственно. Насосы были рассчитаны на действие в течение 7 дней, и их заменяли два раза для получения общей продолжительности инфузии 21 день.
По одному образцу крови брали у всех животных в Группах 4 и 5 в дни 7, 14 и 21 и анализировали для определения уровней соединения 2 в плазме. Образцы крови брали у Группы 3 в дни 7 и 14 в следующие точки времени (по 3 животных на точку времени): за 5 минут до введения интраперитонеальной дозы, и через 15 минут, 30 минут, 1, 2 и 4 часа после введения интраперитонеальной дозы. В день 21 через три часа после последней интраперитонеальной инъекции один образец крови брали из Группы 3. Размер опухоли измеряли через каждые 4 дня. Через 21 день испытание прекращали и рассчитывали средний TGI.
Фиг. 2 показывает рост опухоли в течение 21-дневного введения доз. Не было никакой разницы в размере опухоли между двумя контрольными группами, которым вводили носитель. Группа введения высокой дозы посредством мининасоса, дополненной интраперитонеальным введением, показала статистически значимый TGI >70% по сравнению с контролями. Группы введения 56 и 112 мг/кг/день показали на являющиеся статистически значимыми TGI значения 43 и 38%, соответственно, по сравнению с контролями. Соединение 2 указано как Пр. 2 на Фиг. 2.
Фиг. 3A показывает установившееся, согласно оценке, состояние концентрации в плазме соединения 2 в Группе 4 и 5, как было определено при помощи усредненных образцов крови, взятых в дни 7, 14 и 21. Эти данные говорят о том, что средние установившиеся уровни Соединения 2 в плазме находились в пределах от 99 до 152 нг/мл для Группы 4 и от 52 до 238 нг/мл для Группы 5. Средний уровень в плазме в образцах, взятых в день 21, составил 99 нг/мл для Группы 4 и 52 нг/мл для Группы 5.
Фиг. 3B показывает график концентраций соединения 2 в плазме против времени после интраперитонеальной инъекции. Интраперитонеальные инъекции обеспечивали значимое увеличение воздействия Соединения 2 на уровни в плазме, что касается как Cмax (от 4200 до 5000 нг/мл) после каждых трех инъекций в день, так и ежедневных AUC, по сравнению с теми, которые были получены при установившехся уровнях в плазме в результате непрерывной инфузии. В целом, результаты показывают, что Соединение 2 демонстрирует значимую противоопухолевую активность в модели мышиного ксенотрансплантата MLL реаранжированного лейкоза.
ВКЛЮЧЕНИЕ ПОСРЕДСТВОМ ССЫЛКИ
Полное раскрытие каждого из патентных документов и научных статей, на которые имеются ссылки в настоящей заявке, включено посредством ссылки для всех целей.
ЭКВИВАЛЕНТЫ
Настоящее изобретение можно осуществить в других специфических формах, без отступления от сути или существенных характеристик изобретения. Описанные выше варианты воплощения поэтому следует рассматривать во всех отношениях иллюстративными, а не ограничивающими изобретение, описанное в настоящей заявке. Объем настоящего изобретения, таким образом, определяется прилагаемой формулой изобретения, а не представленным выше описанием, и предполагается, что все изменения, которые подпадают под суть и объем эквивалентности формулы изобретения, охватываются настоящим изобретением.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ RMT5 | 2019 |
|
RU2814198C2 |
| НОВОЕ ПИРРОЛОПИРИМИДИНОВОЕ СОЕДИНЕНИЕ ИЛИ ЕГО СОЛЬ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ЕЕ, В ЧАСТНОСТИ, АГЕНТ ДЛЯ ПРЕДОТВРАЩЕНИЯ И/ИЛИ ЛЕЧЕНИЯ ОПУХОЛЕЙ, И ТОМУ ПОДОБНОЕ, НА ОСНОВЕ ИНГИБИТОРНОГО ВОЗДЕЙСТВИЯ НА NAE | 2015 |
|
RU2658008C2 |
| УСИЛИТЕЛЬ ПРОТИВООПУХОЛЕВОГО ВОЗДЕЙСТВИЯ, СОДЕРЖАЩИЙ СОЕДИНЕНИЕ ПИРРОЛОПИРИМИДИНА | 2016 |
|
RU2710380C1 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ КАРБОНУКЛЕОЗИДА, ПРИМЕНЯЕМЫЕ В КАЧЕСТВЕ ПРОТИВОРАКОВЫХ АГЕНТОВ | 2017 |
|
RU2712944C1 |
| КОМПОЗИЦИИ И СПОСОБЫ ИНГИБИРОВАНИЯ ВИРУСНОЙ ПОЛИМЕРАЗЫ | 2013 |
|
RU2654482C2 |
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ, ПРИМЕНЯЕМЫЕ ДЛЯ ЛЕЧЕНИЯ РАССТРОЙСТВ, СВЯЗАННЫХ С NTRK | 2016 |
|
RU2744974C2 |
| НОВЫЙ СПОСОБ ИНГИБИРОВАНИЯ РОСТА ВИРУСОВ И/ИЛИ ВИРУЛИЦИДНЫЙ СПОСОБ И НОВЫЙ АНАЛОГ ПИРАЗИННУКЛЕОТИДА ИЛИ ПИРАЗИННУКЛЕОЗИДА | 2002 |
|
RU2292894C2 |
| АМИДЫ КОНДЕНСИРОВАННОГО ПИПЕРИДИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ ИОННЫХ КАНАЛОВ | 2014 |
|
RU2741810C2 |
| ПРОИЗВОДНЫЕ ПИРРОЛОПИРИМИДИНА, ПОЛЕЗНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ JAK-КИНАЗЫ | 2012 |
|
RU2618673C2 |
| КОНДЕНСИРОВАННОЕ ХИНОЛИНОВОЕ ПРОИЗВОДНОЕ И ЕГО ПРИМЕНЕНИЕ | 2005 |
|
RU2384571C2 |
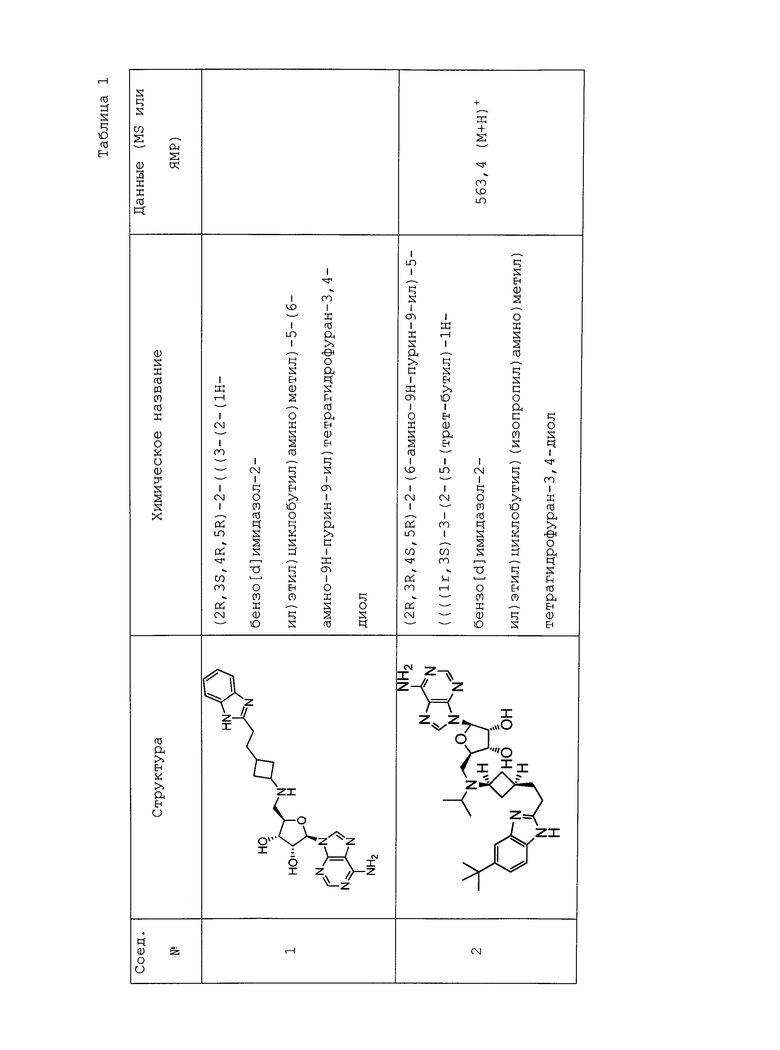
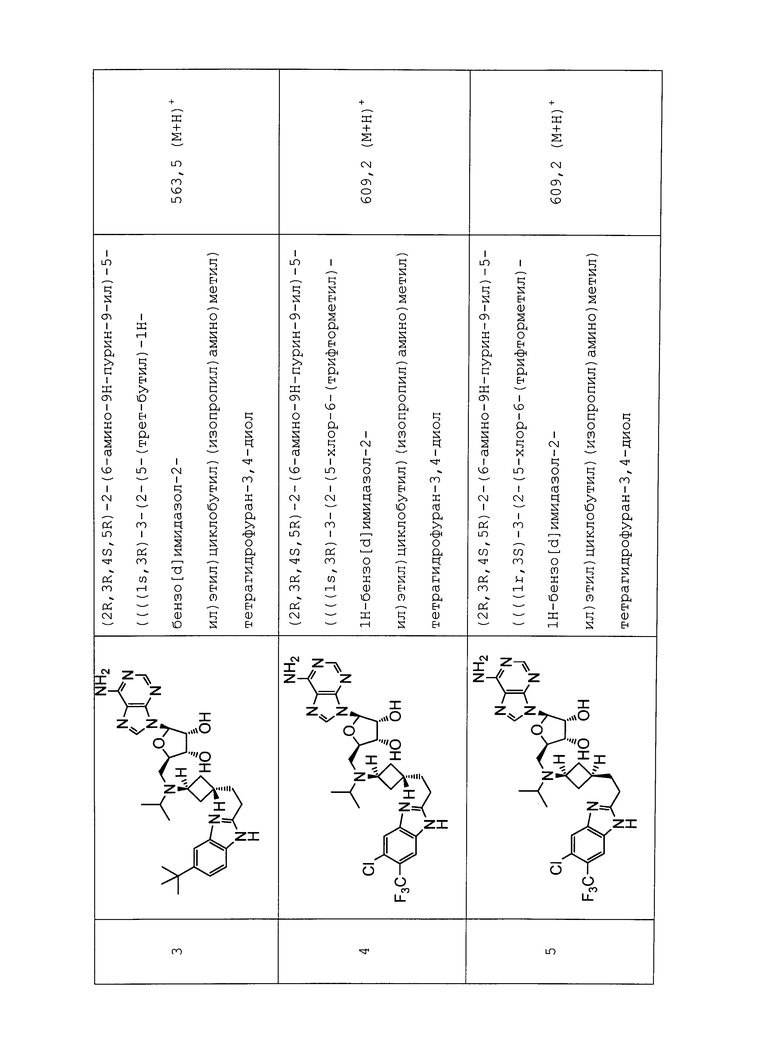
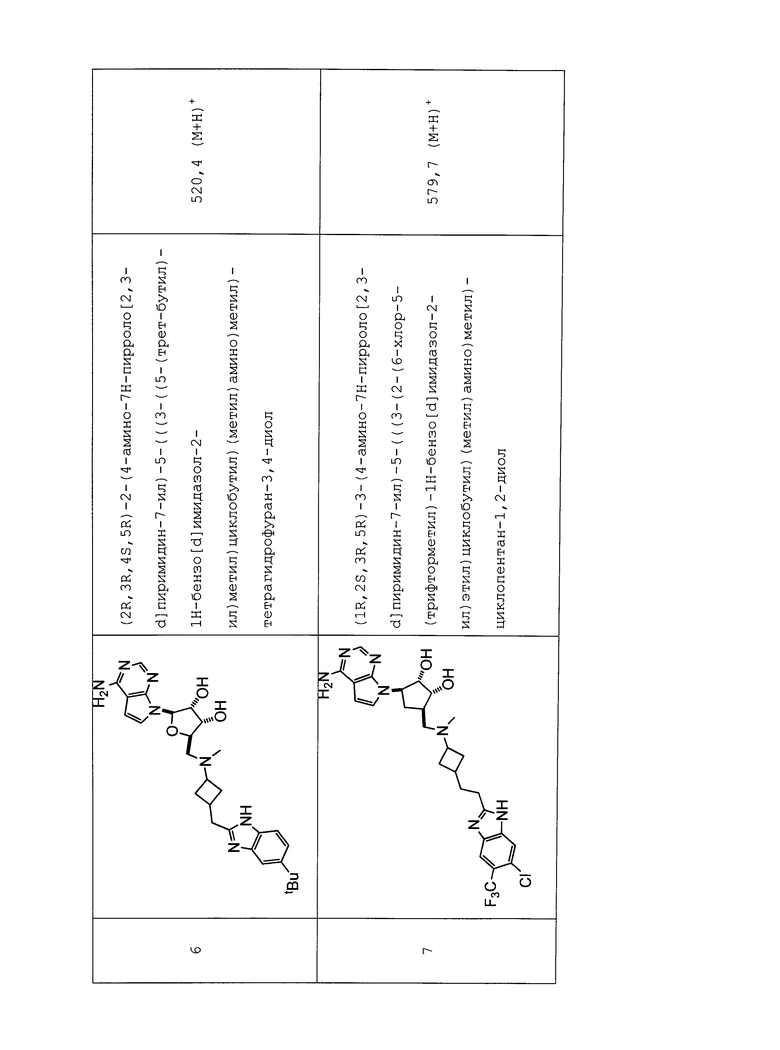
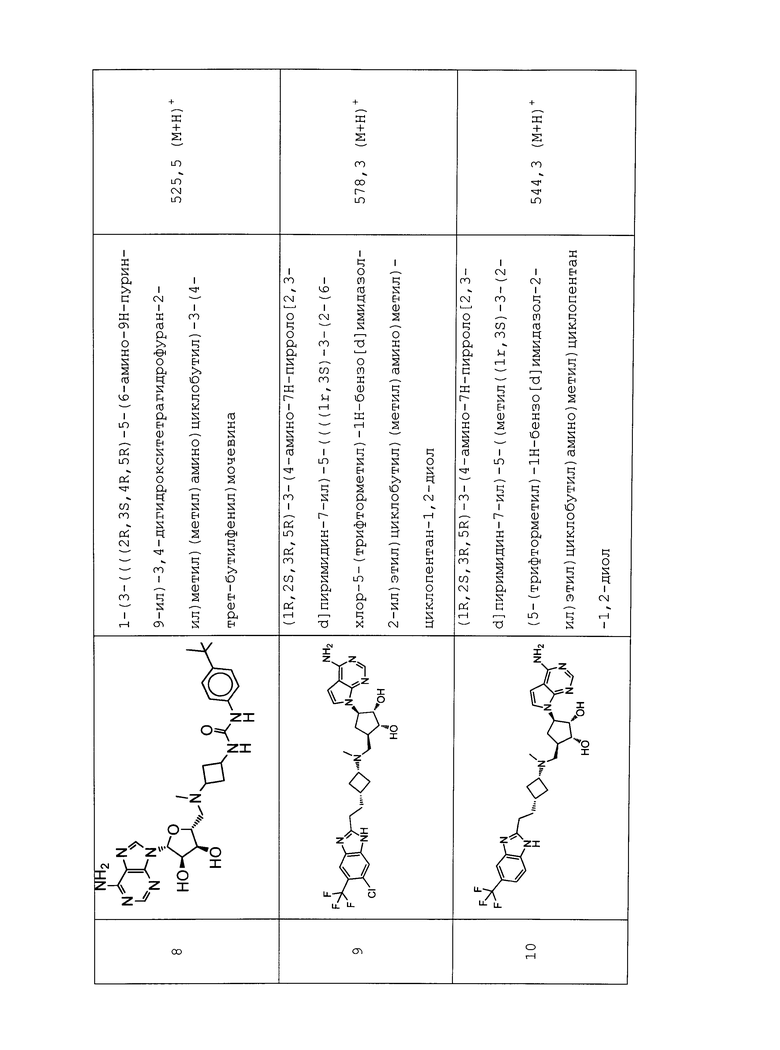
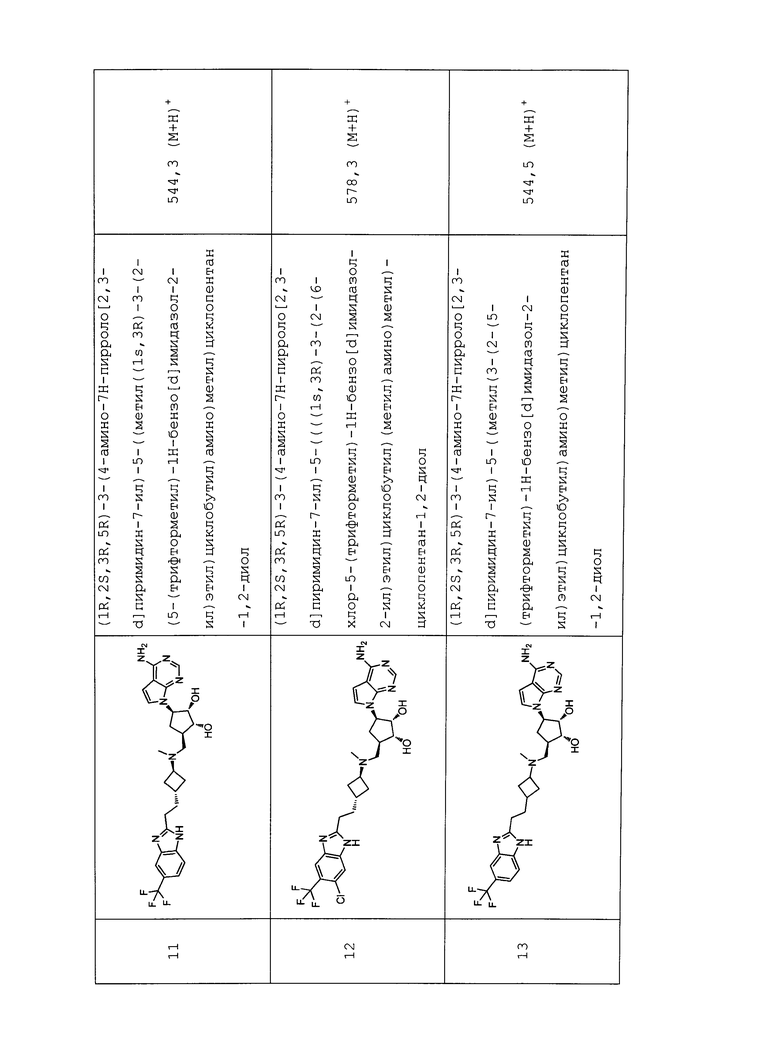
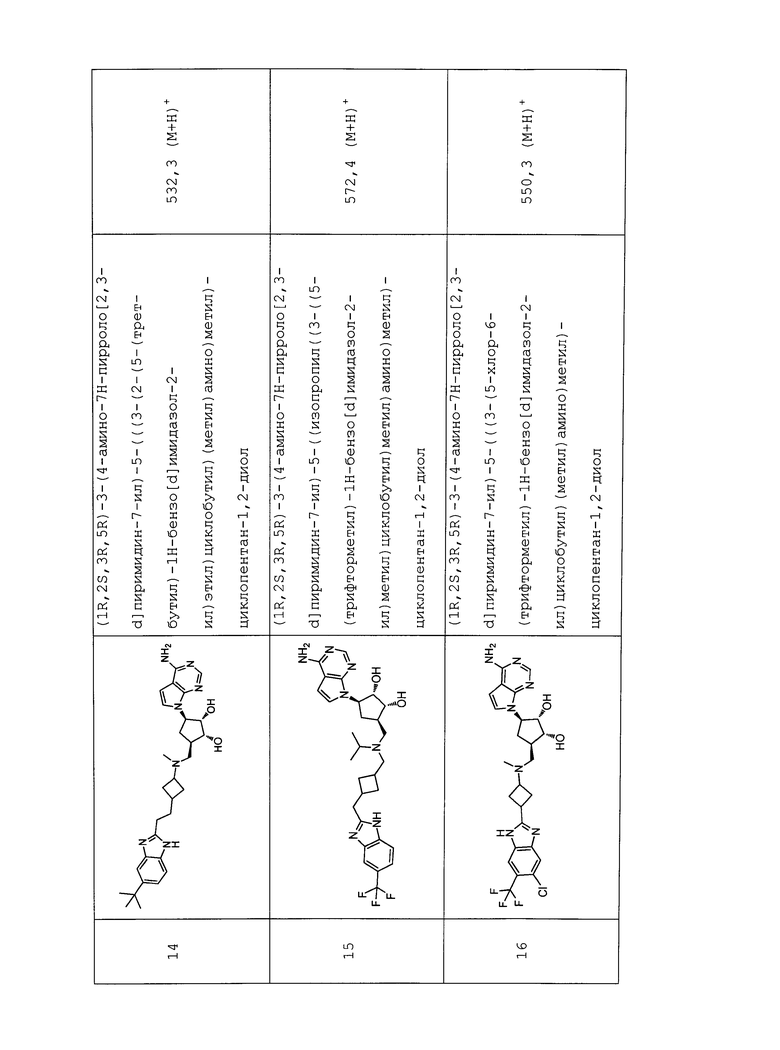
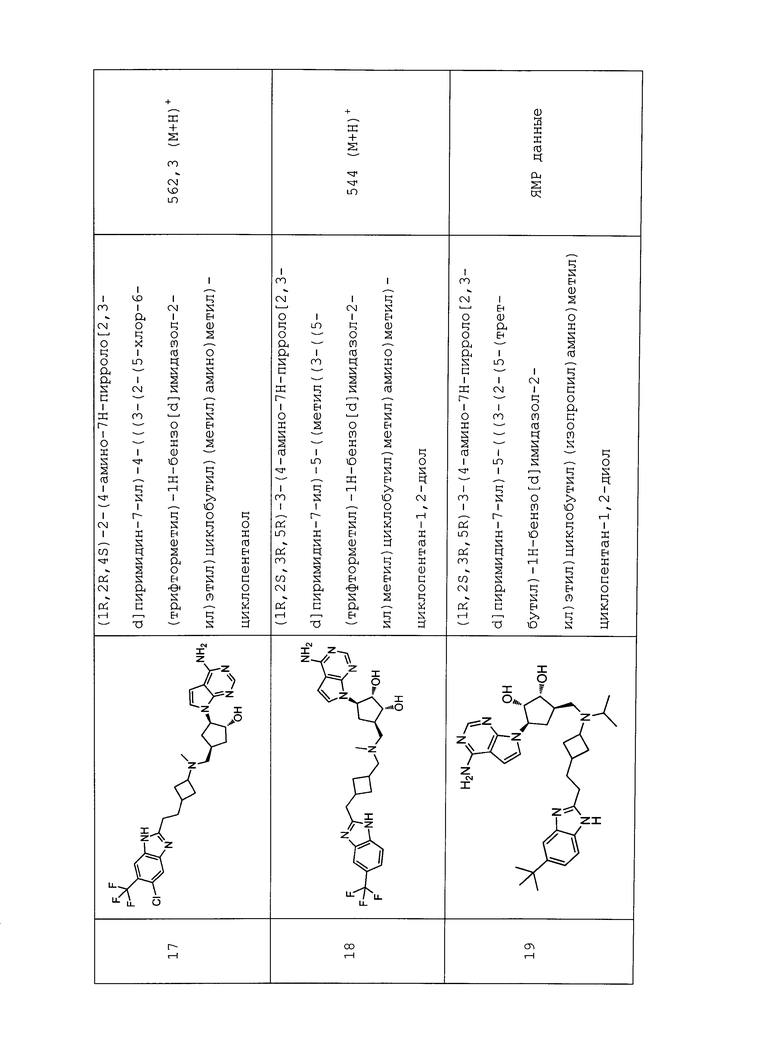
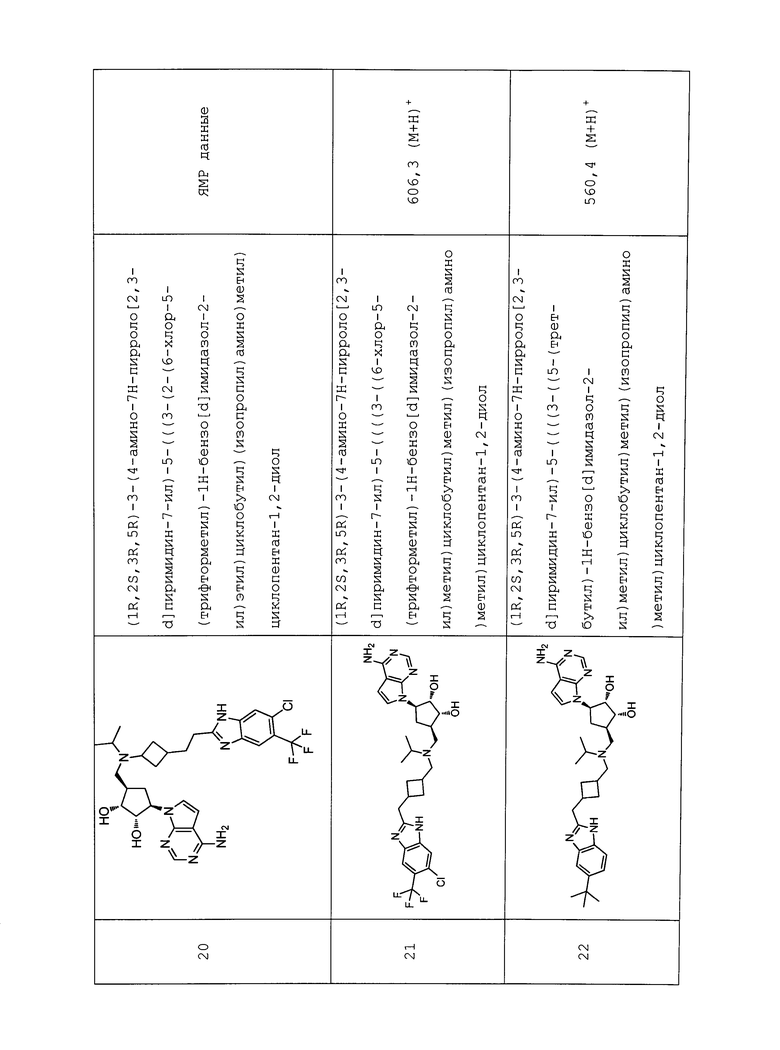
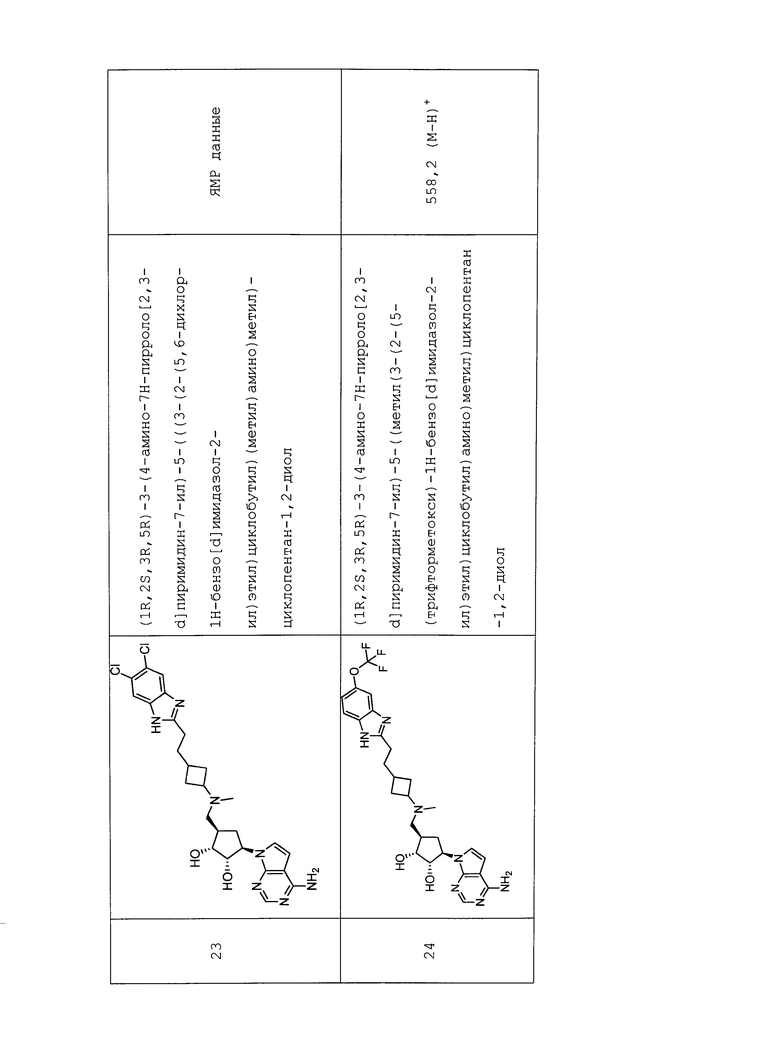
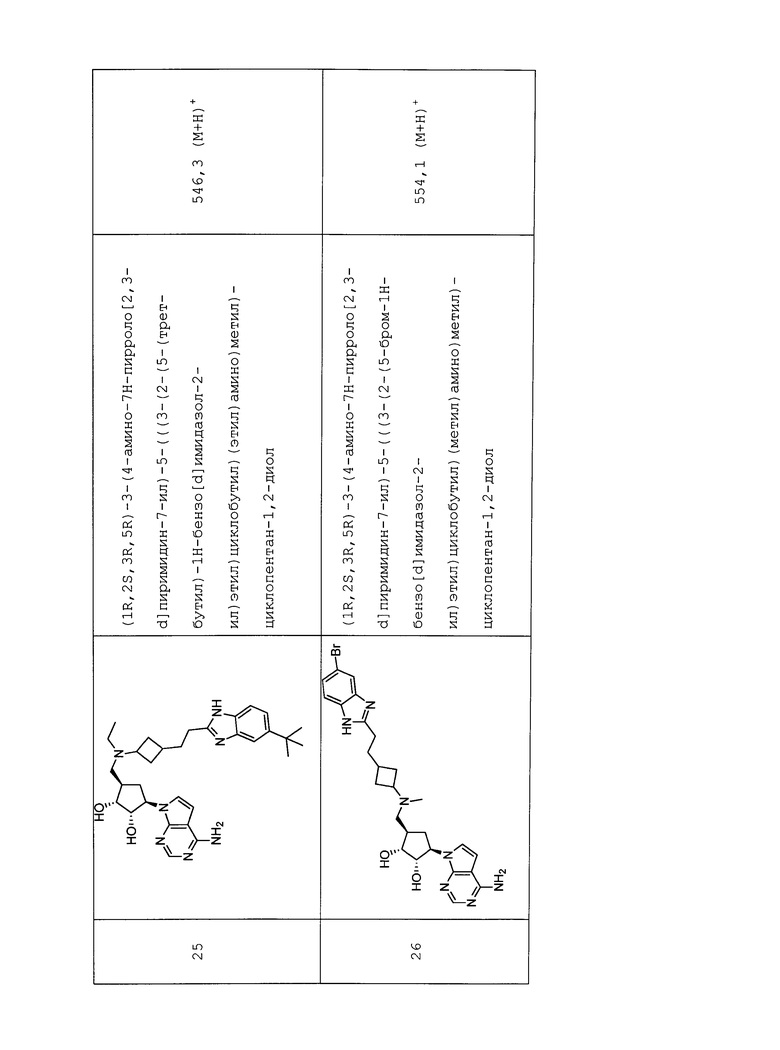
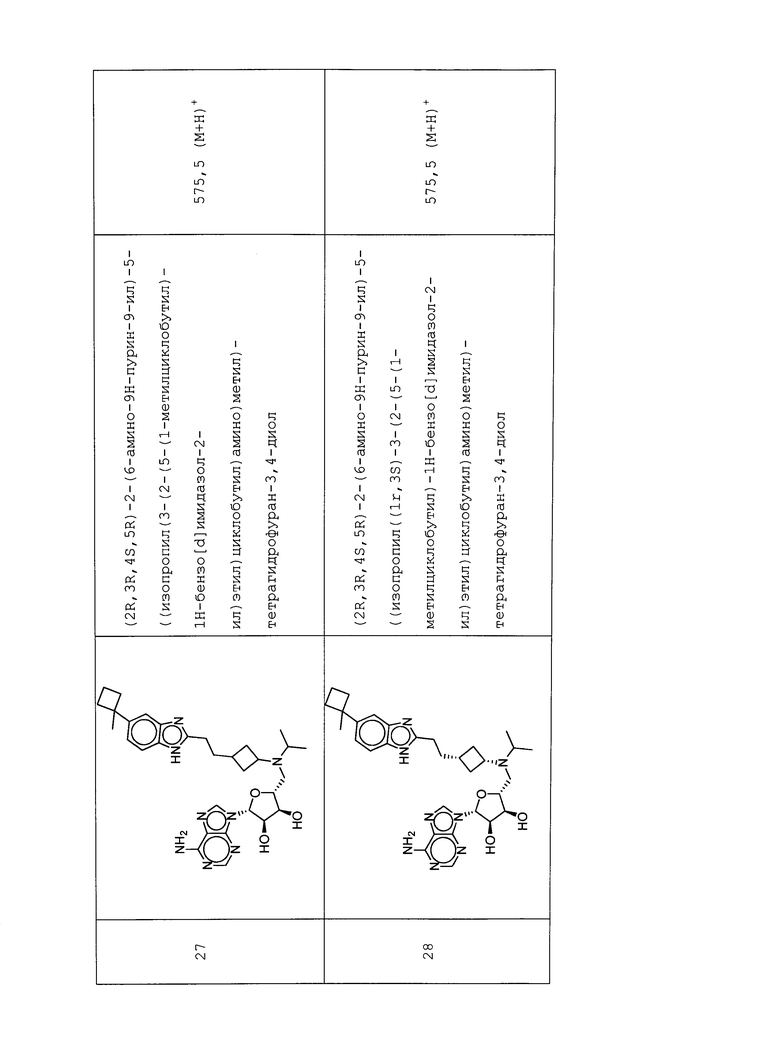
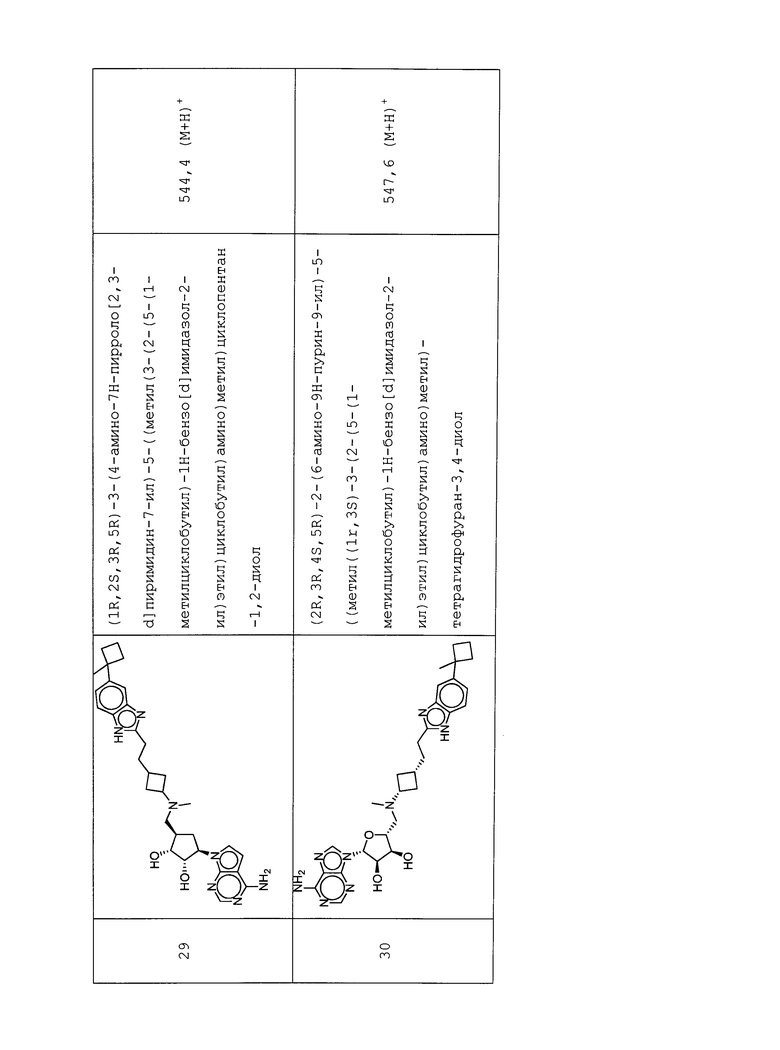
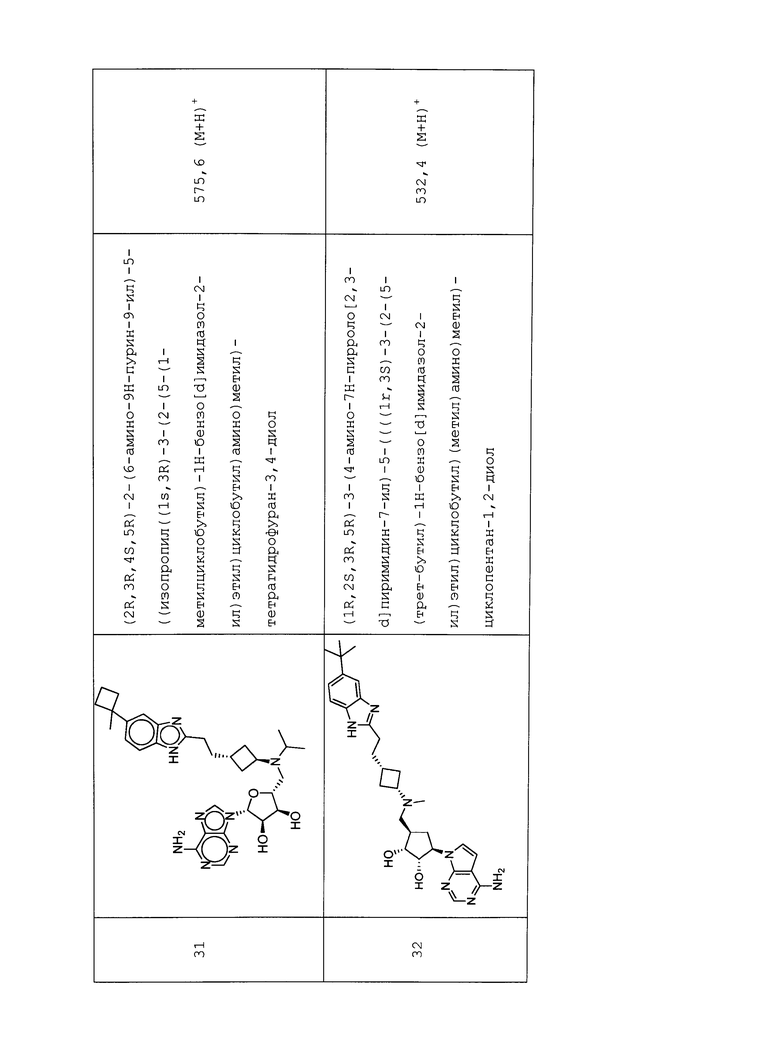
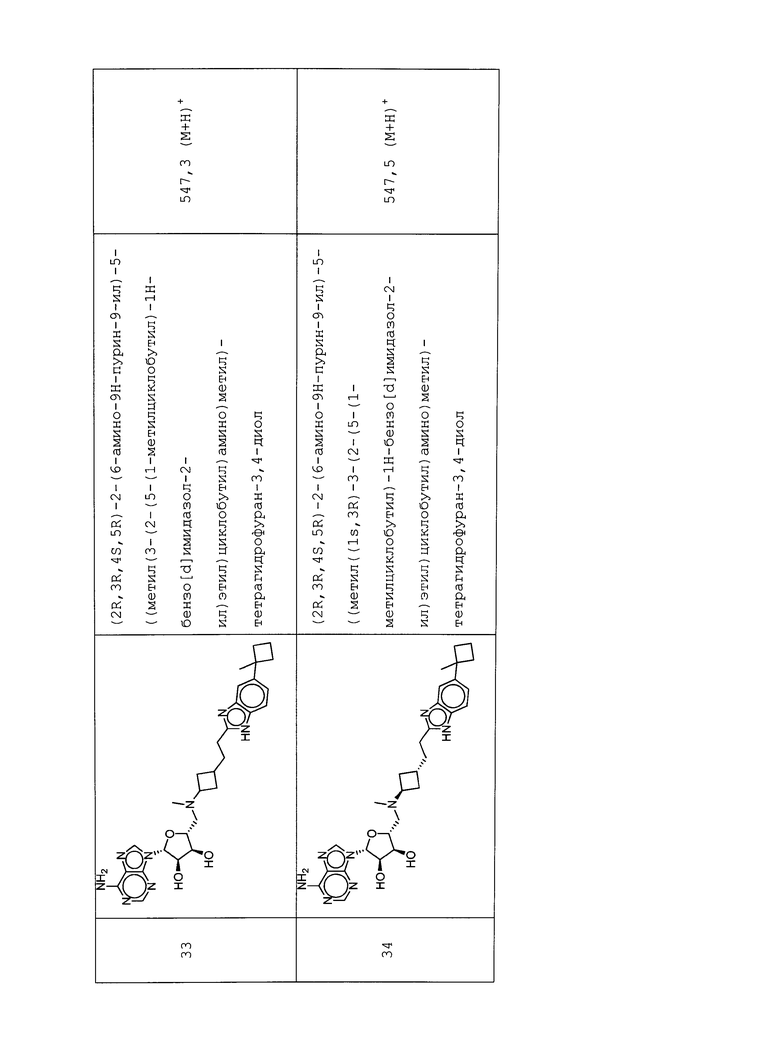
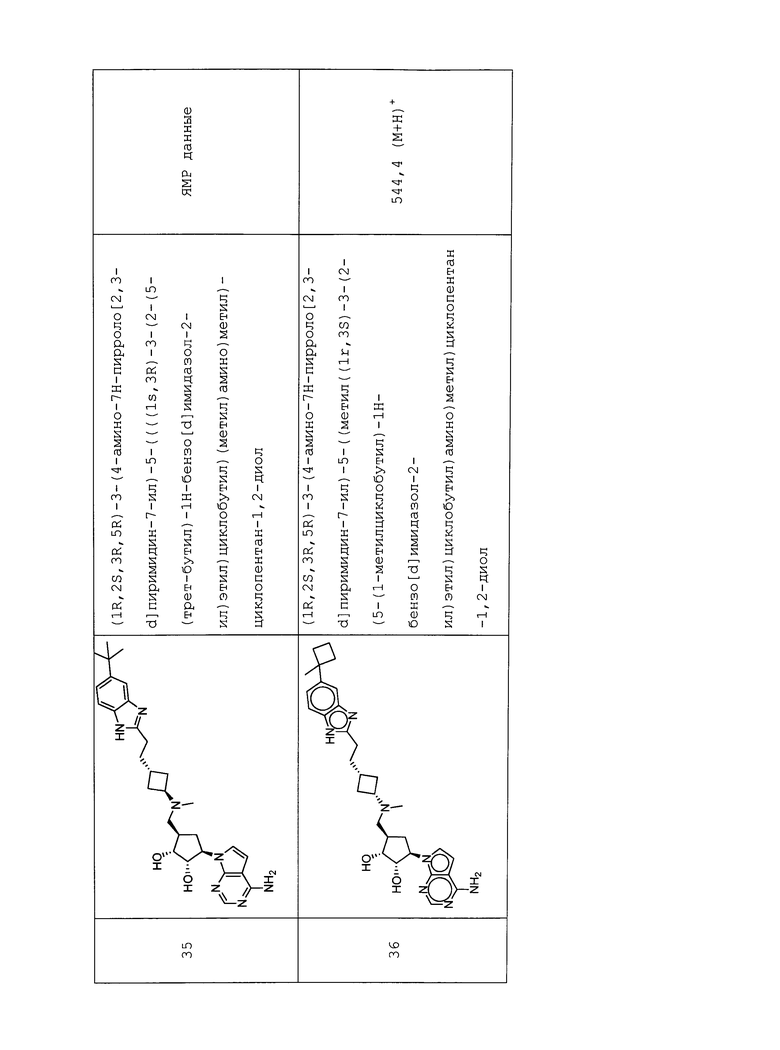
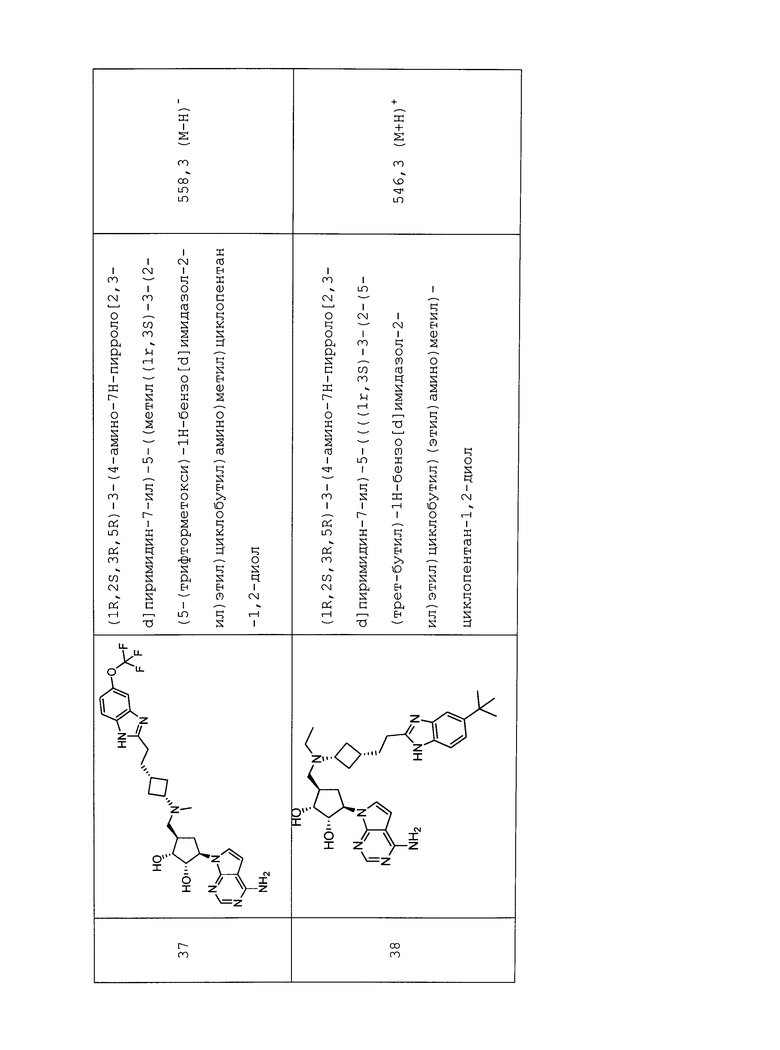
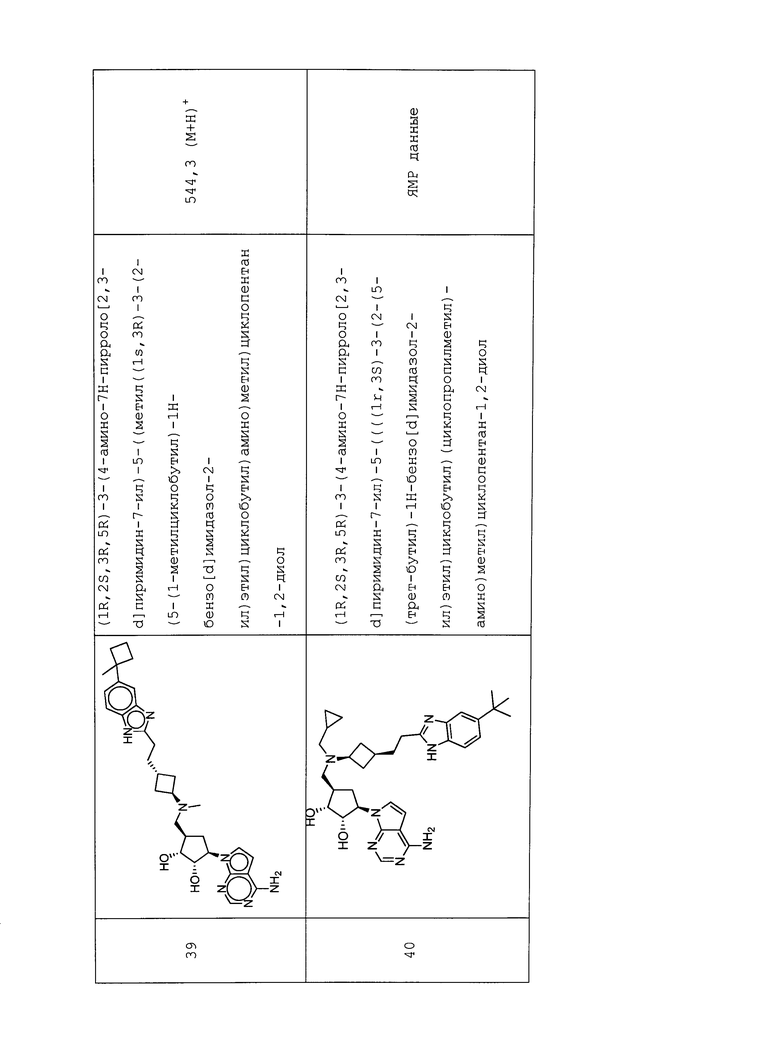
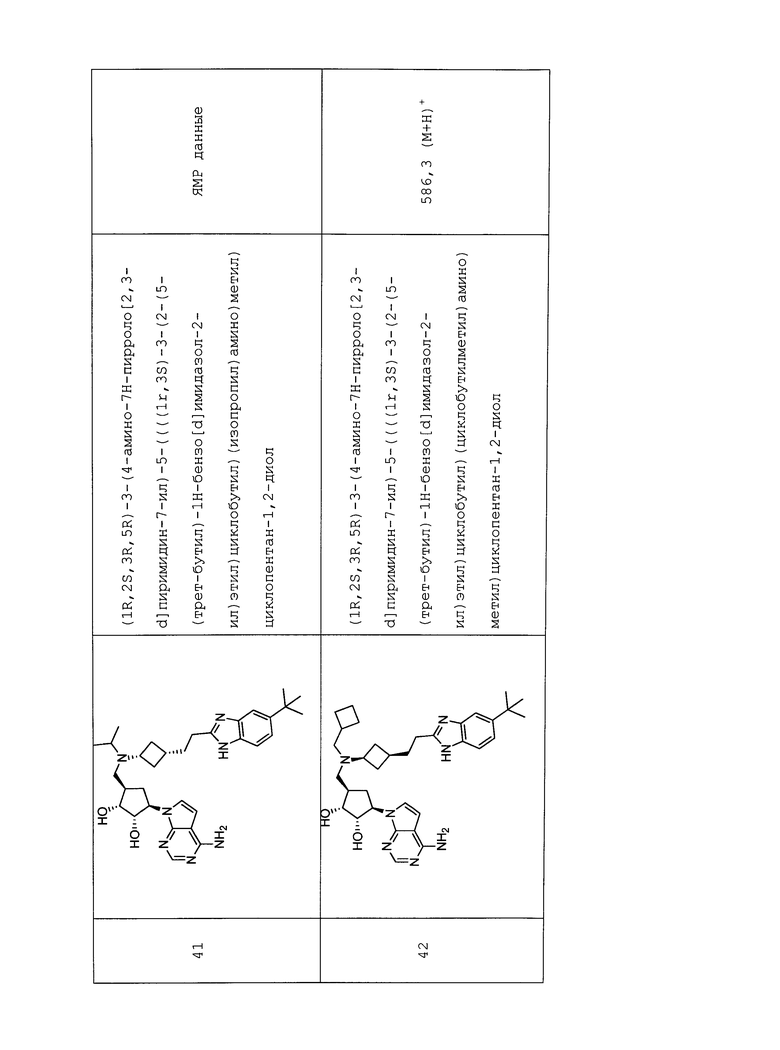
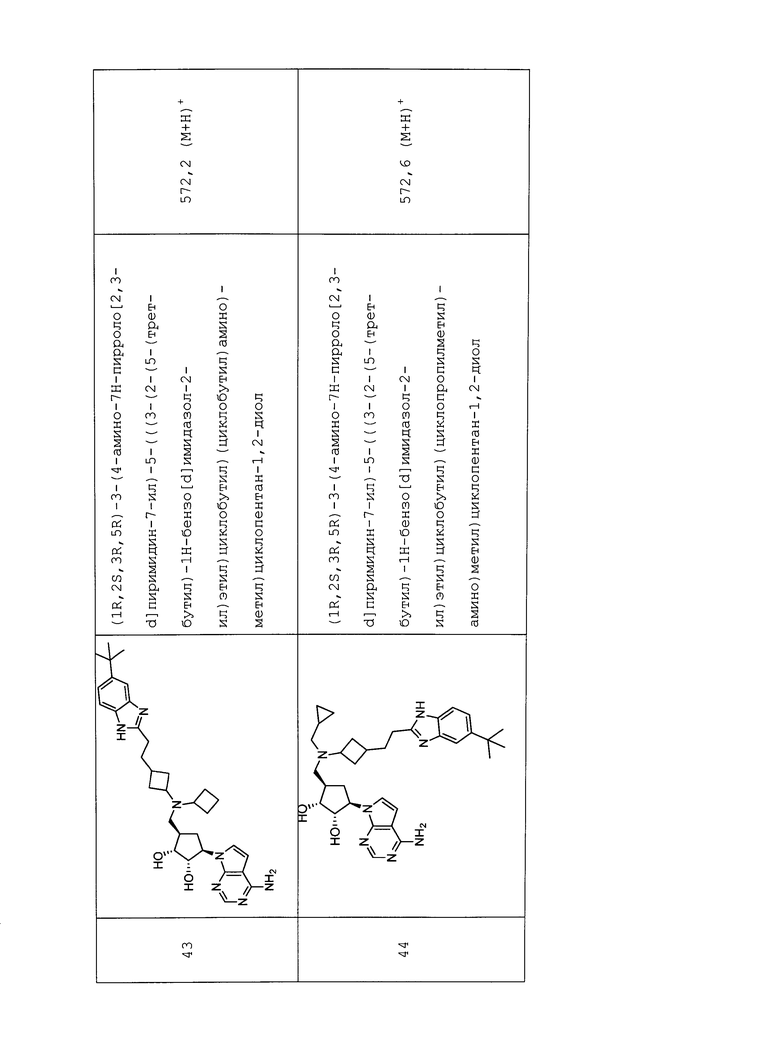
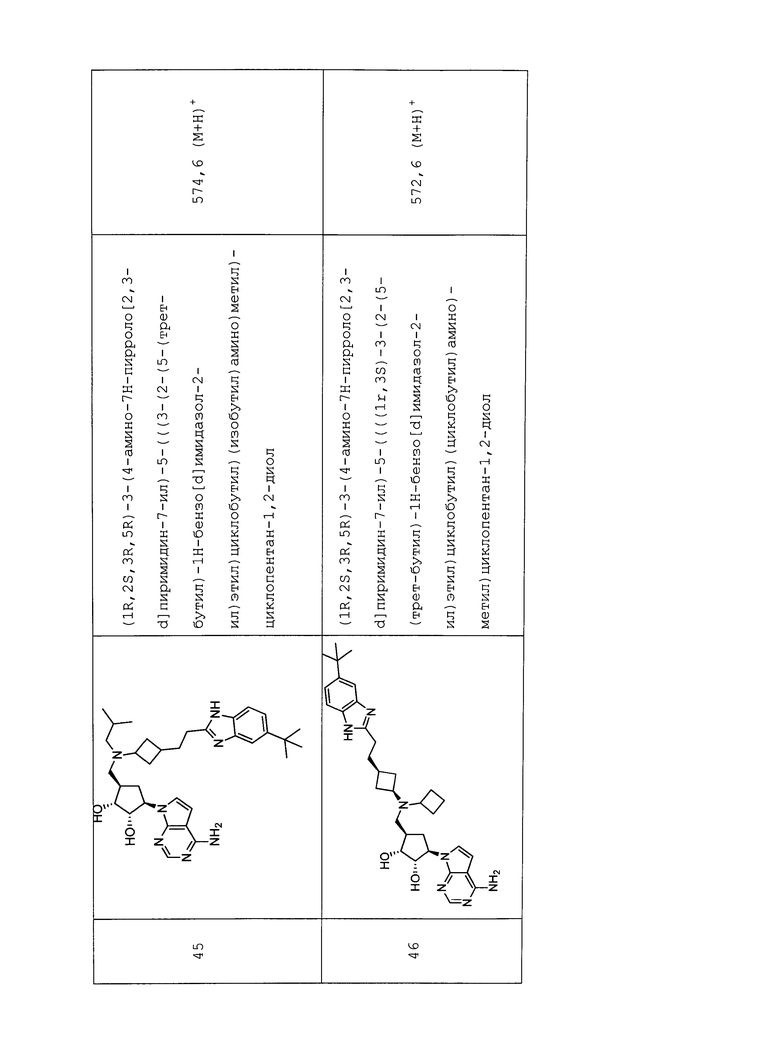
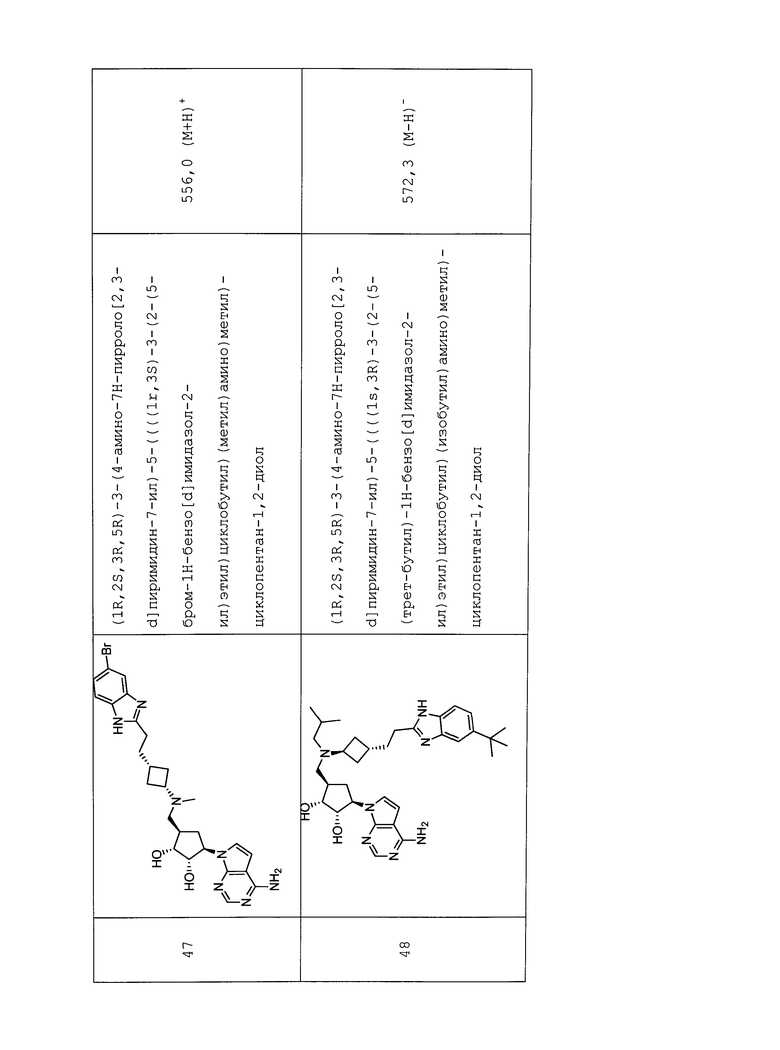
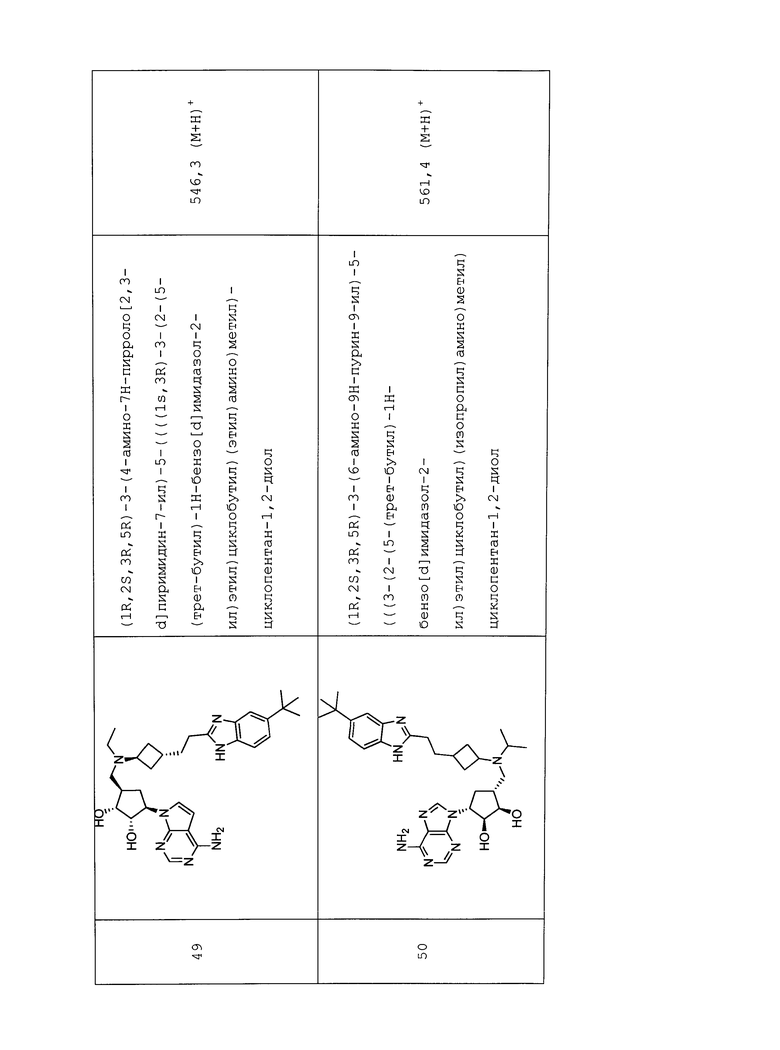
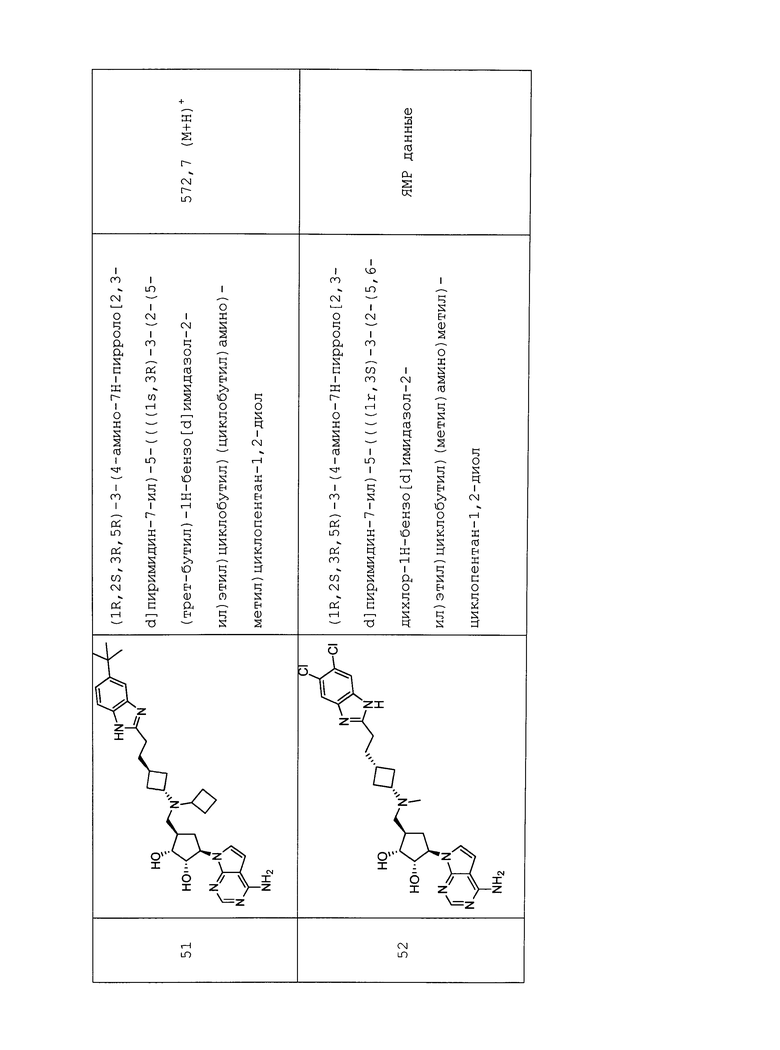
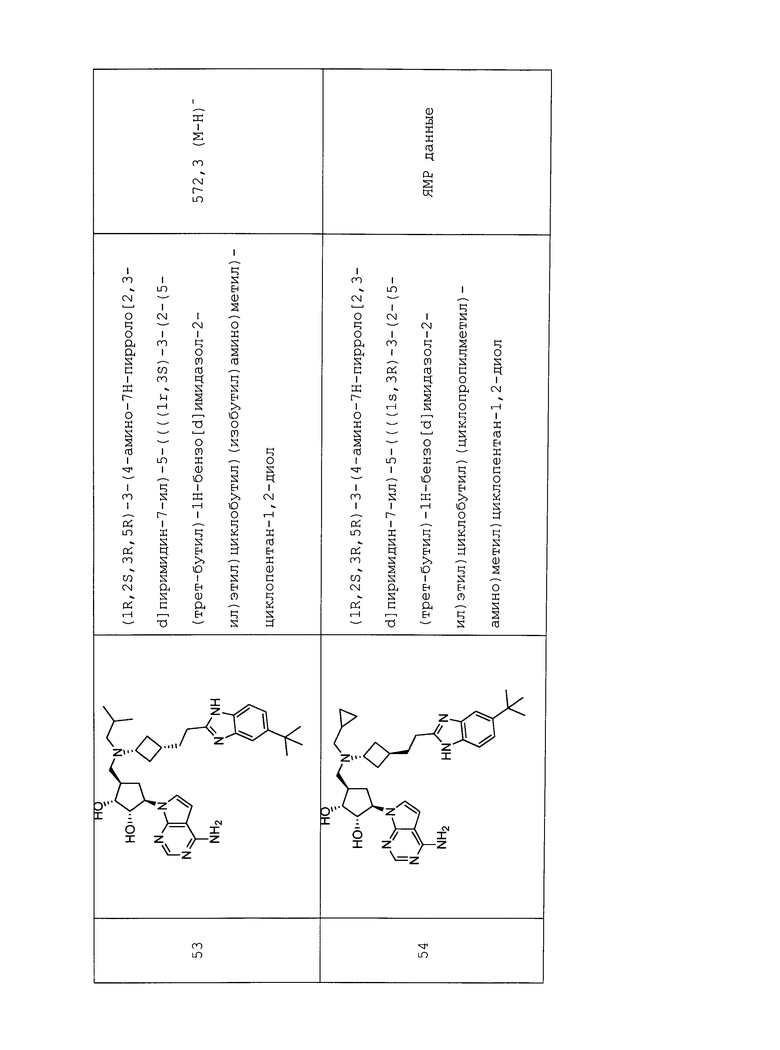
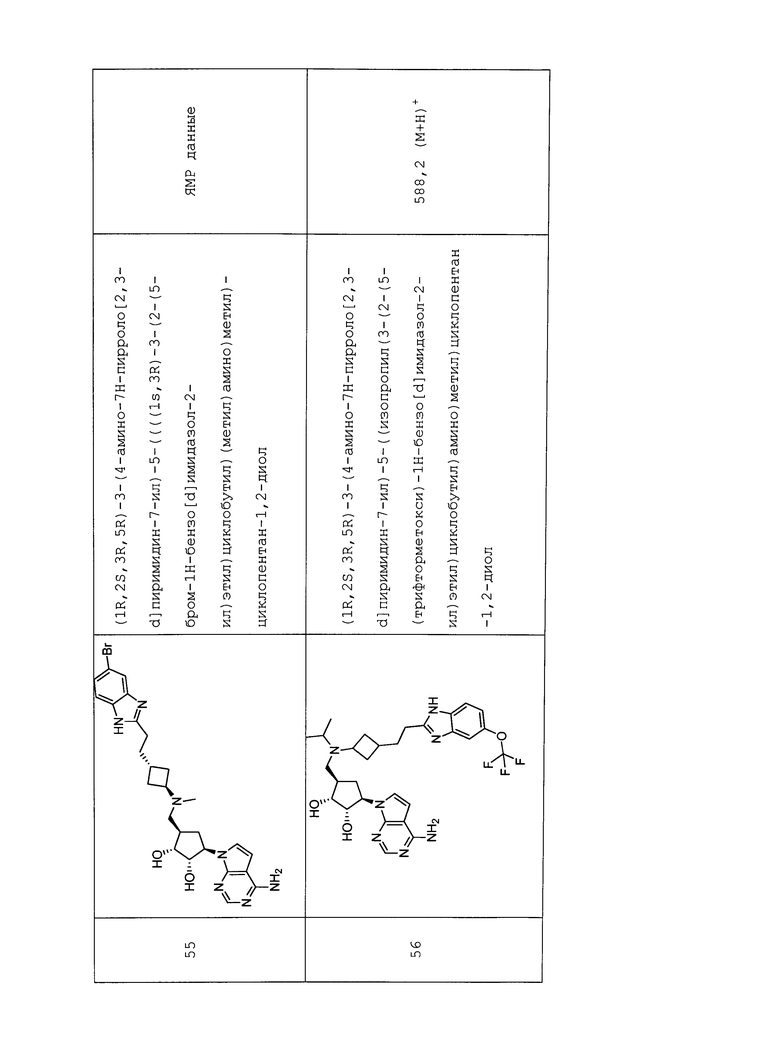
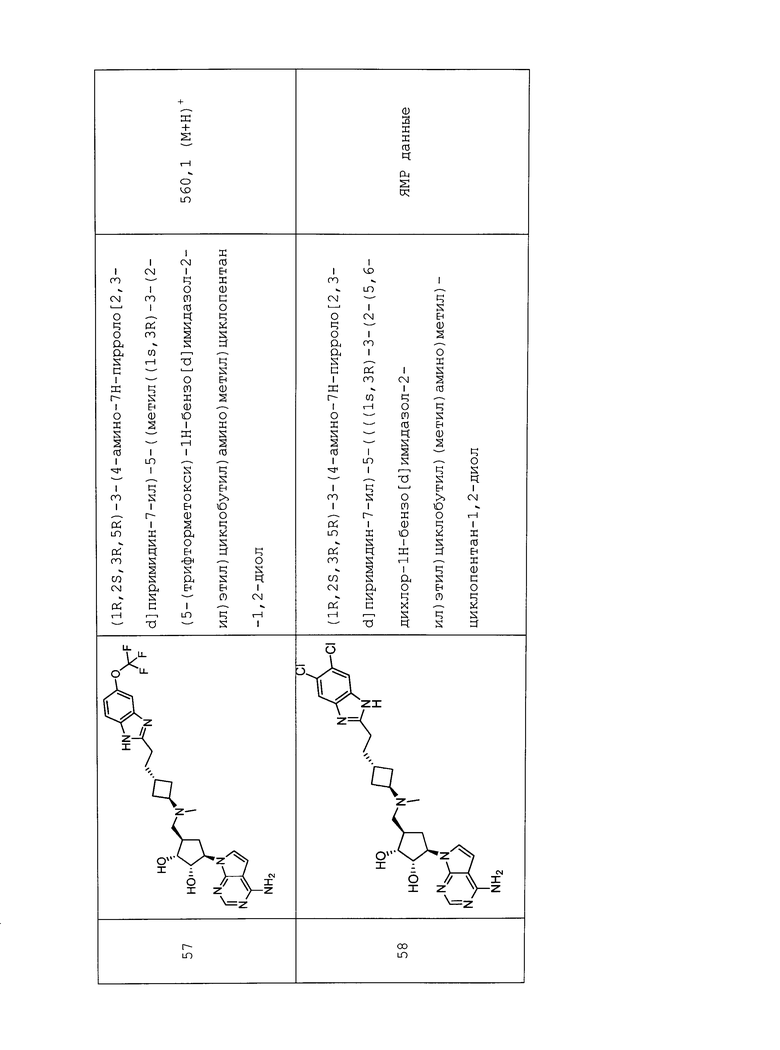
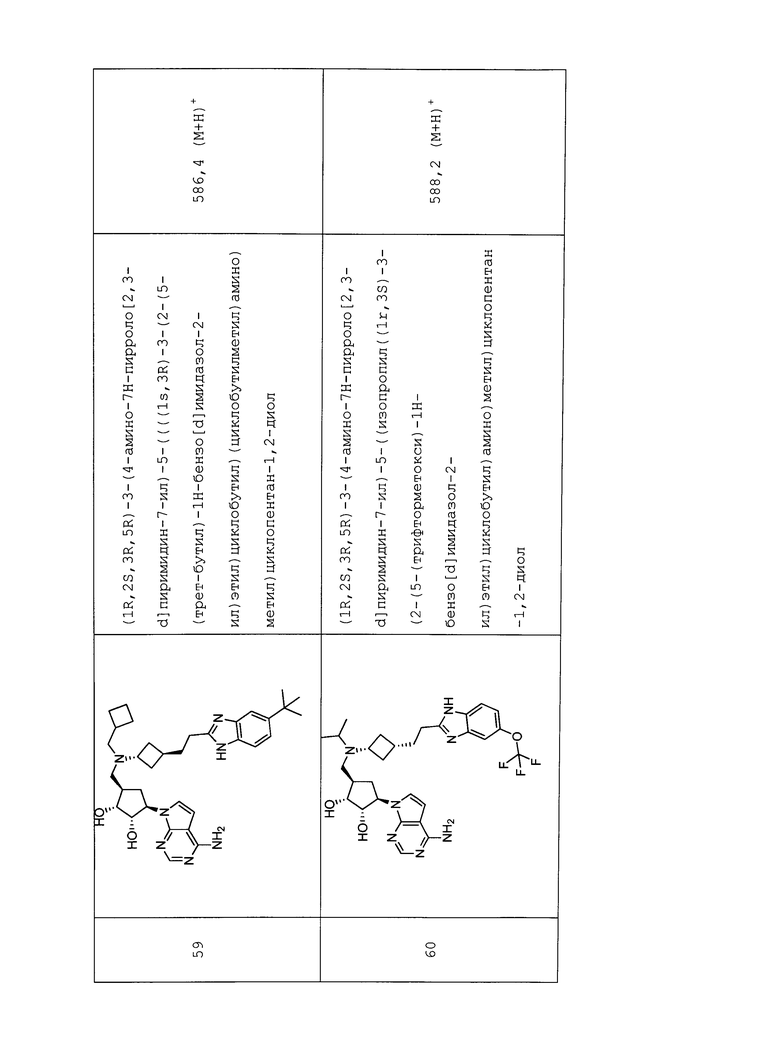
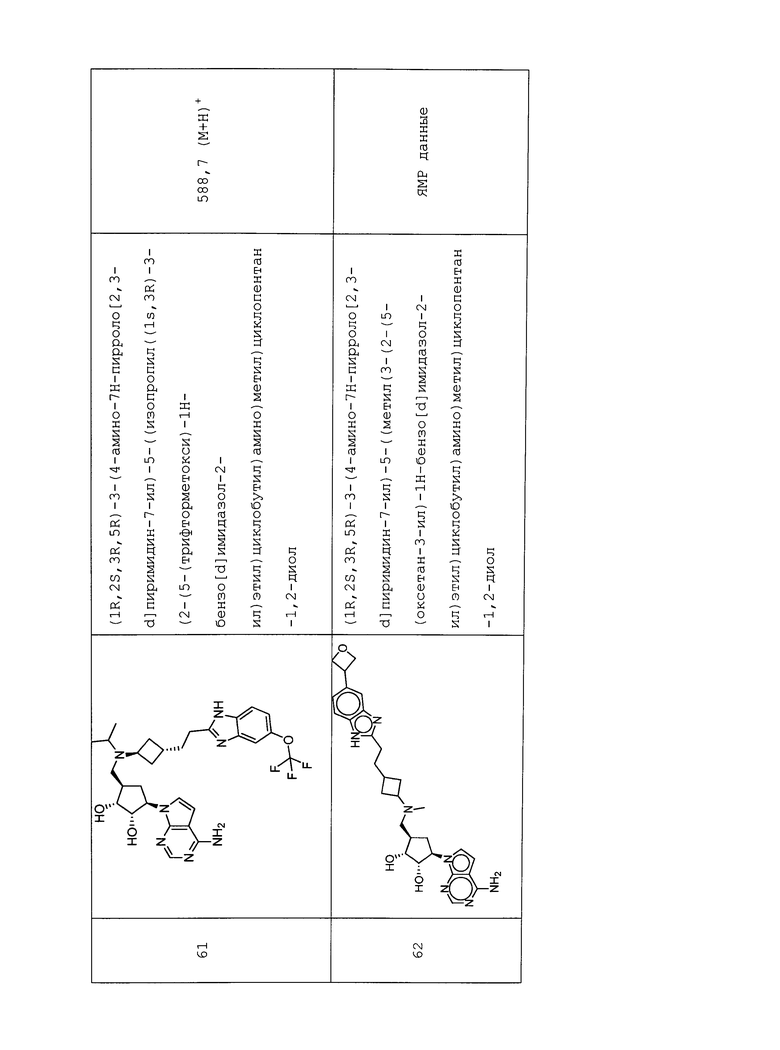
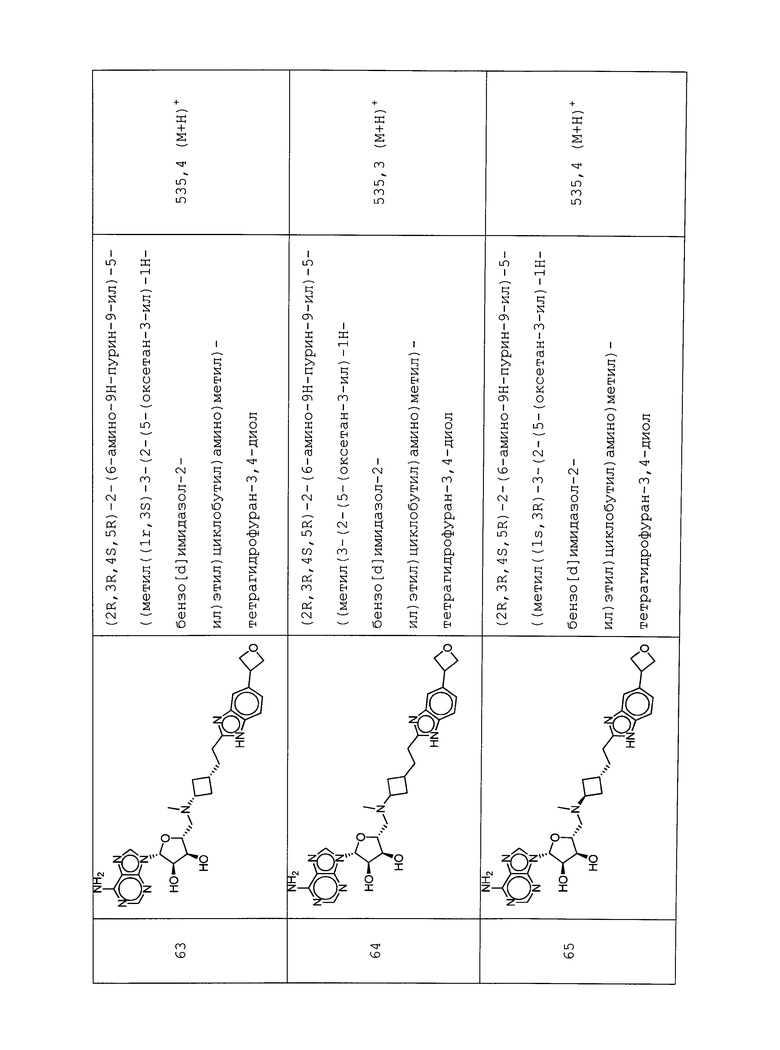
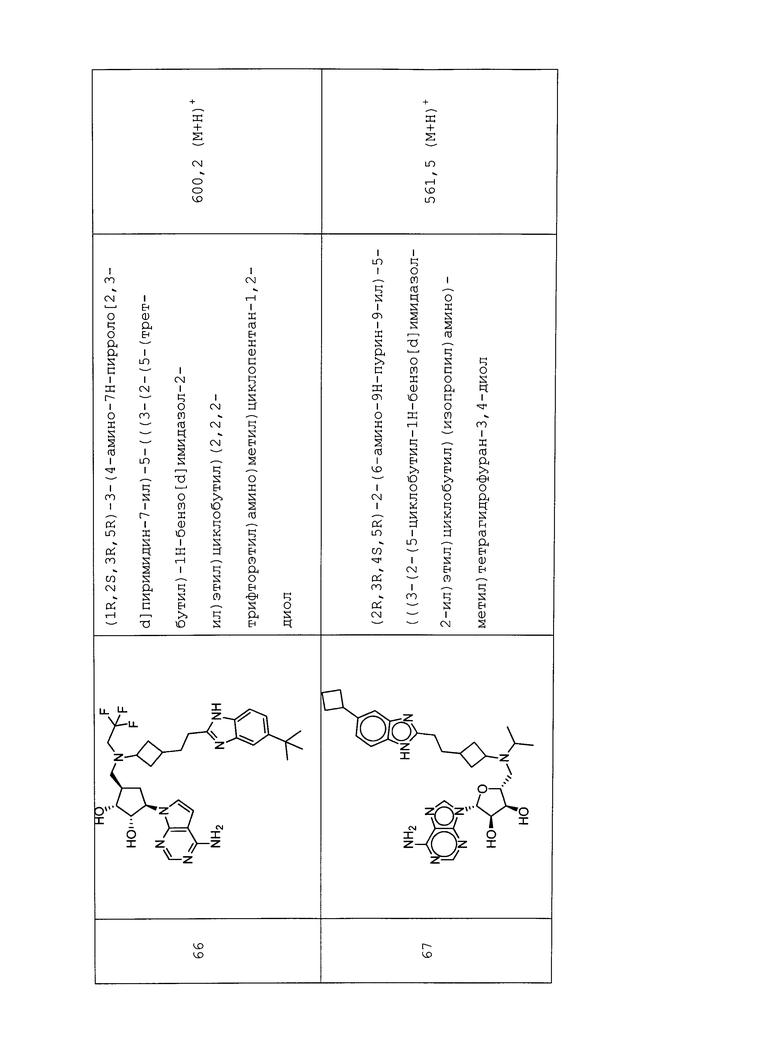
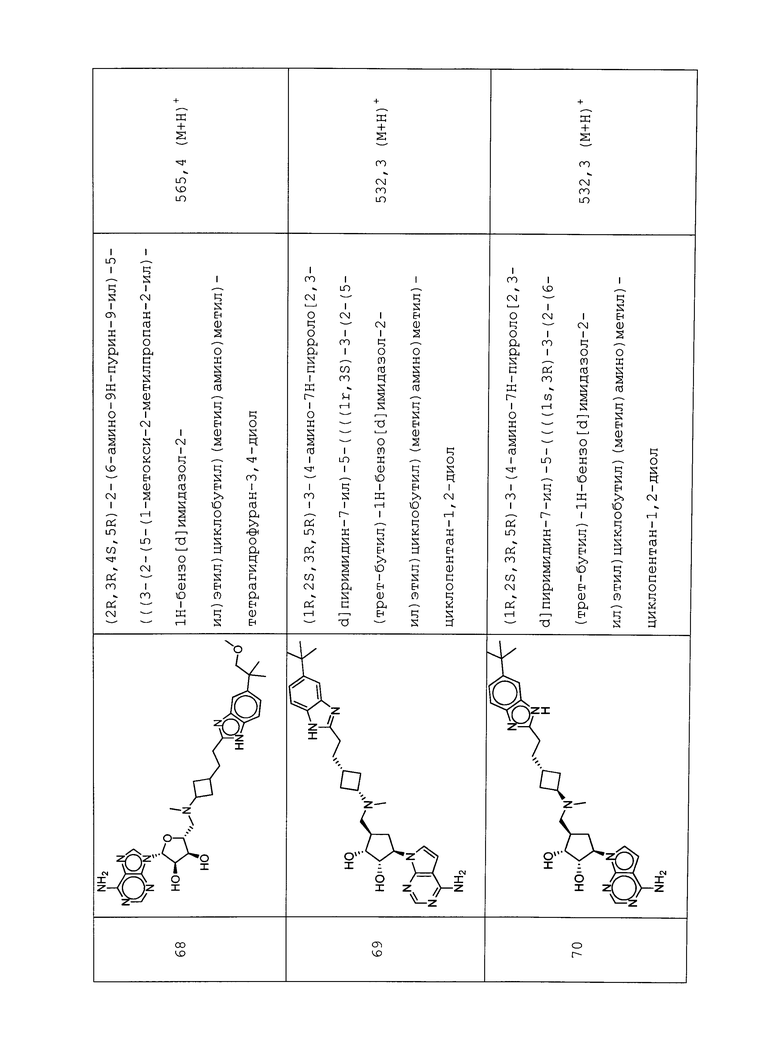
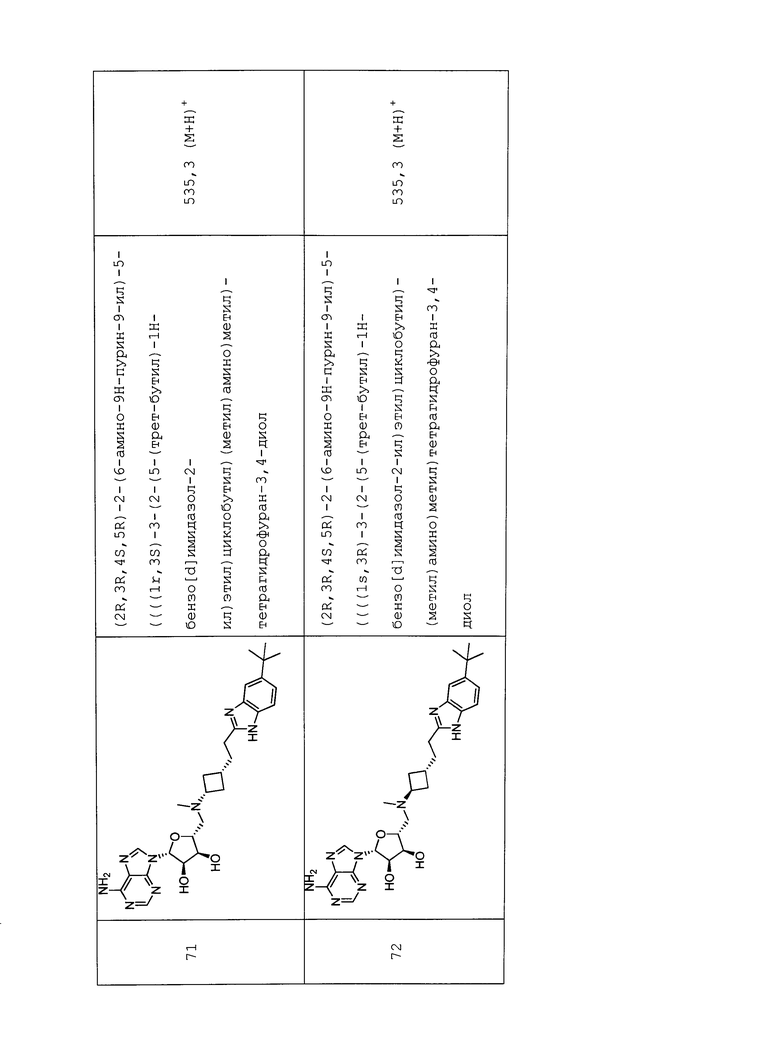
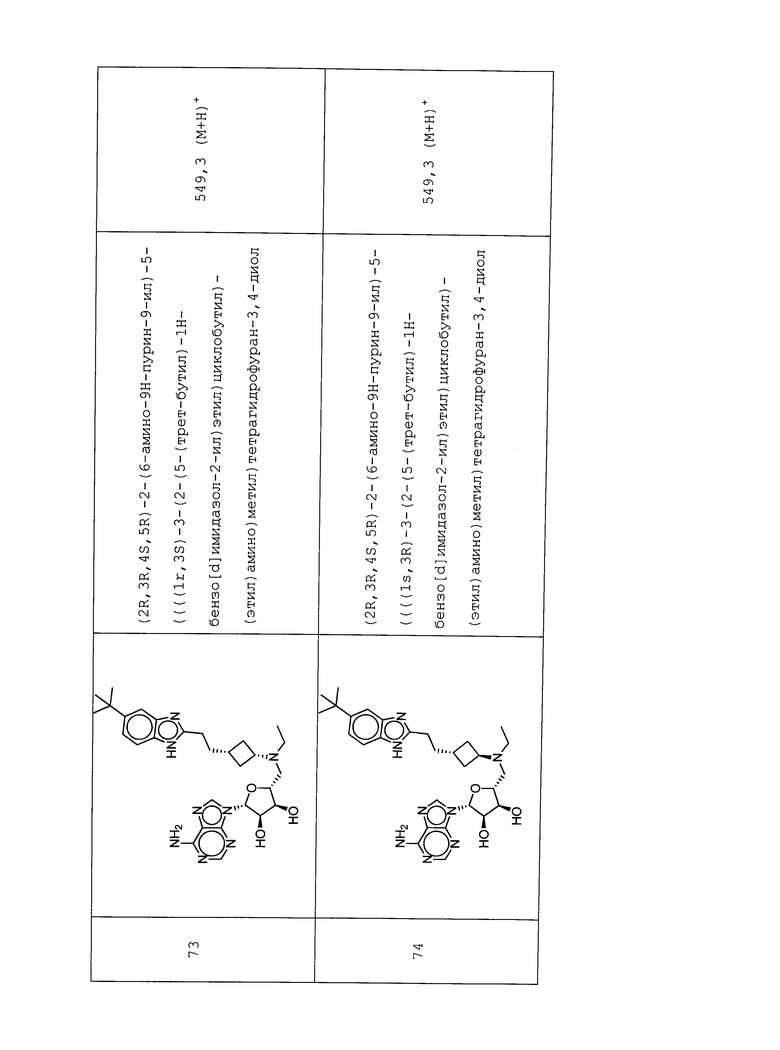
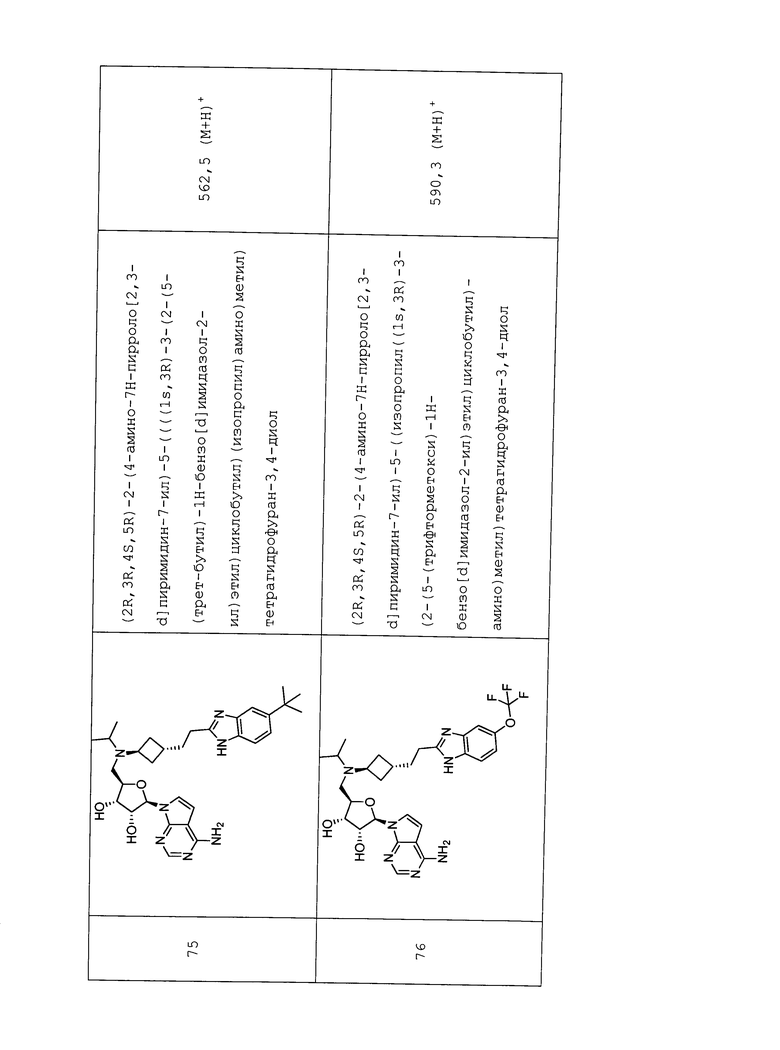
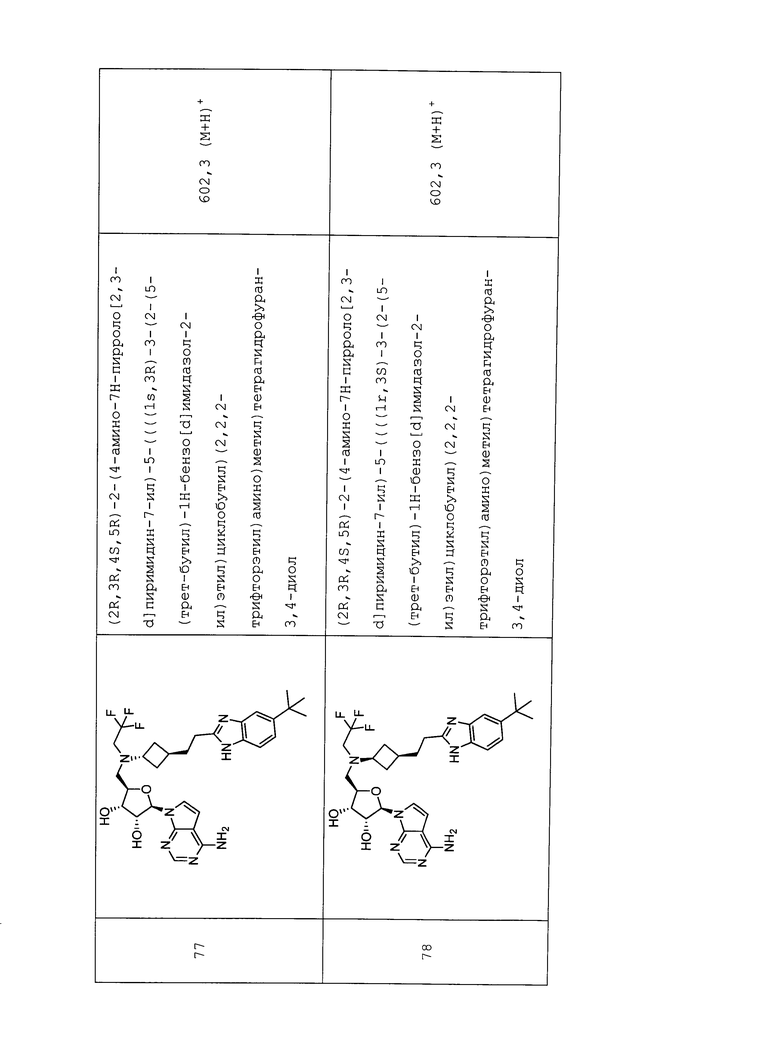
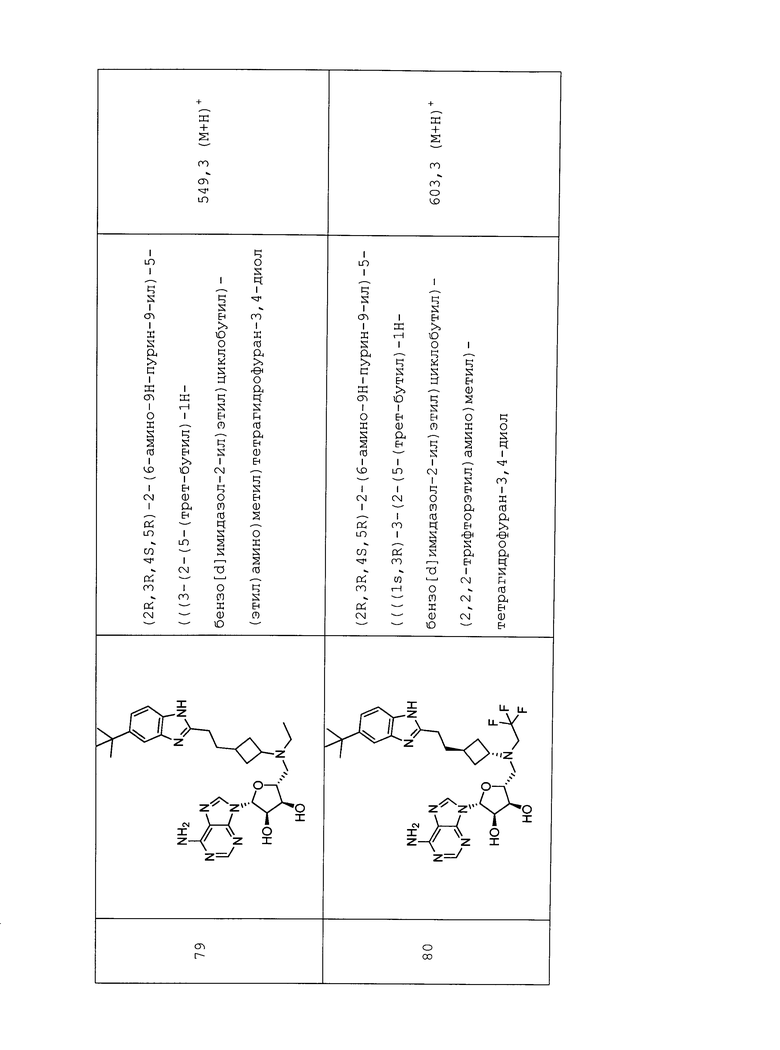
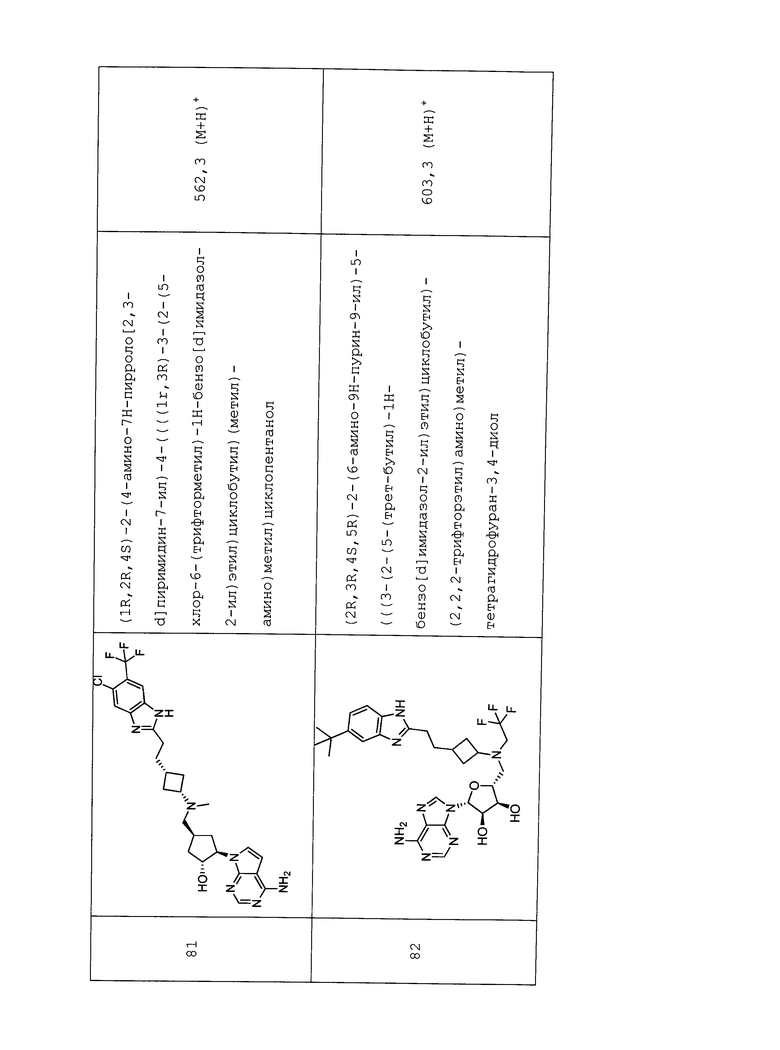
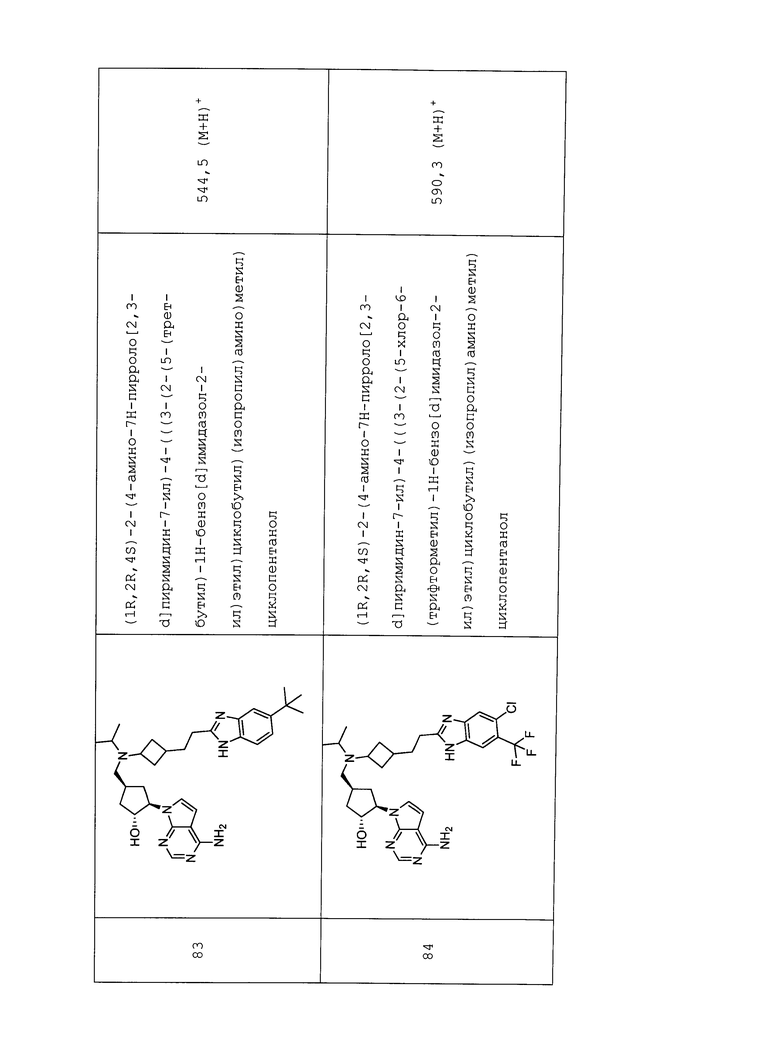
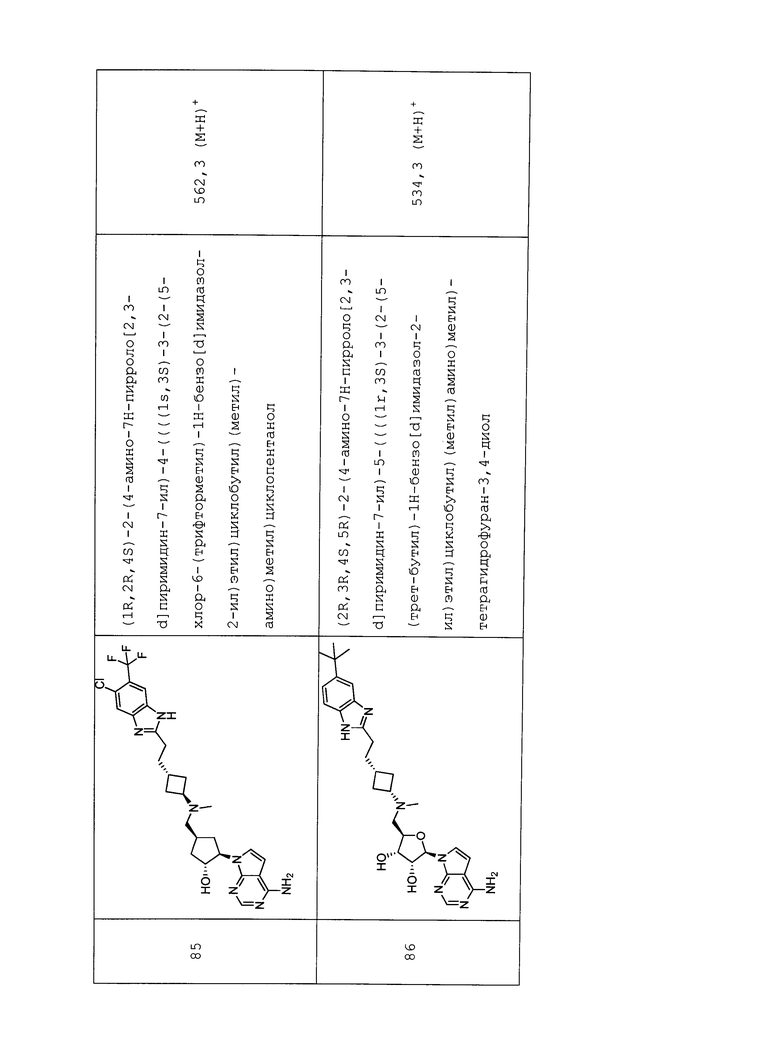
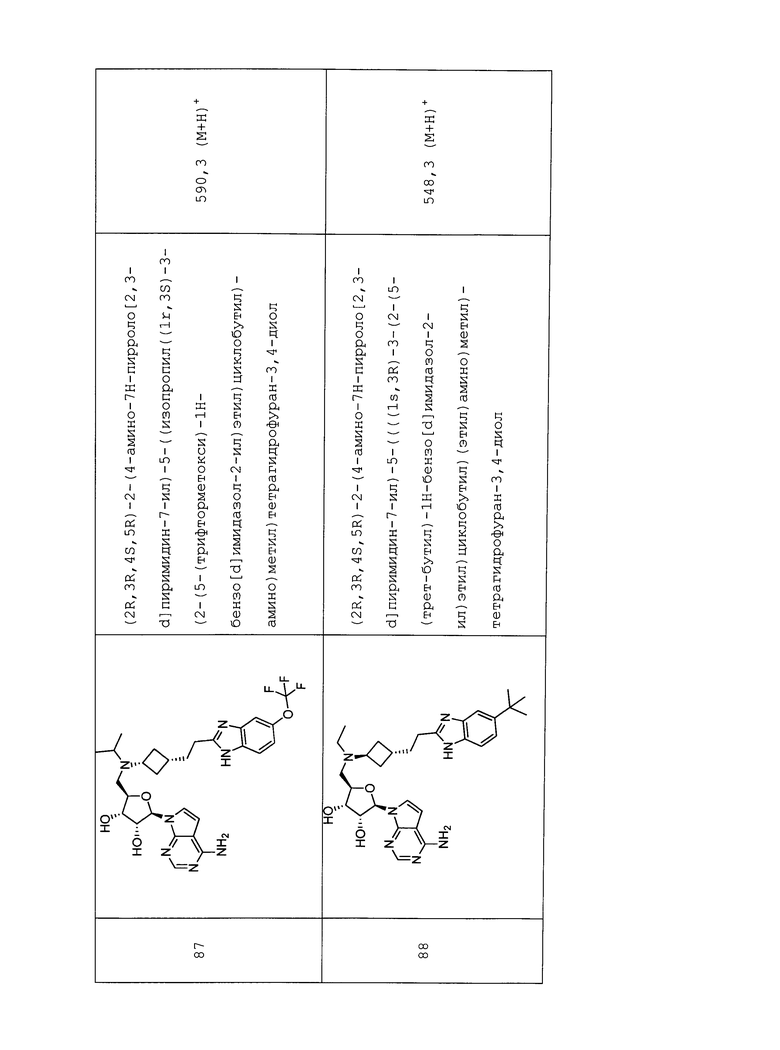
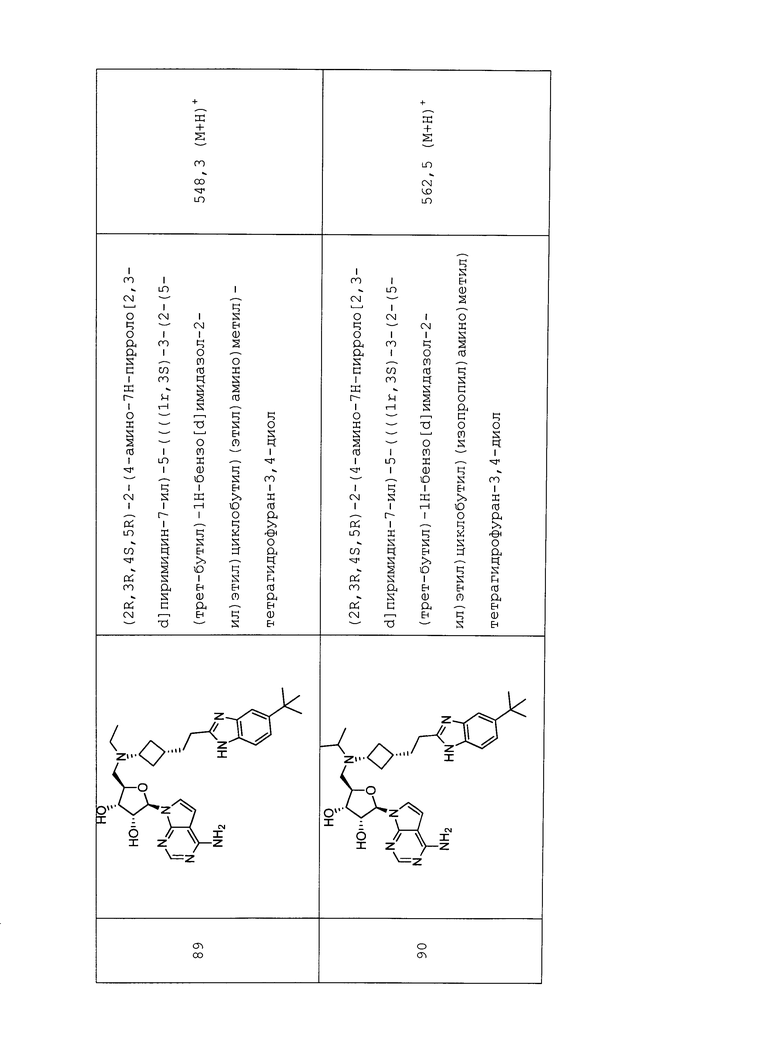
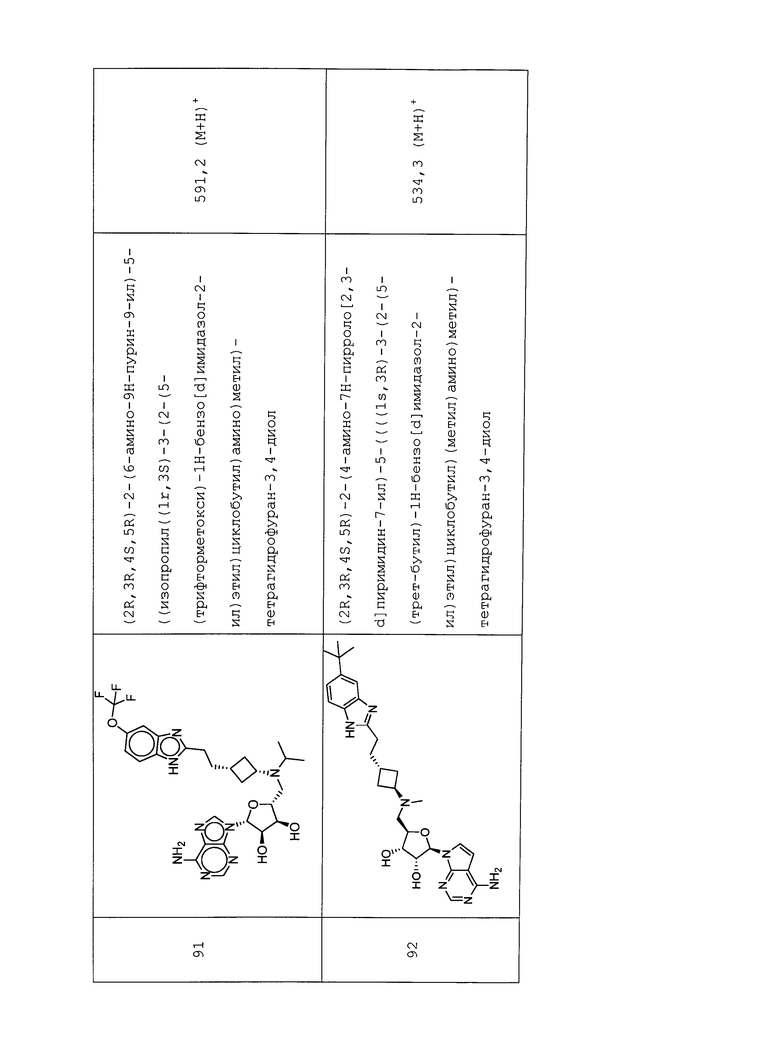
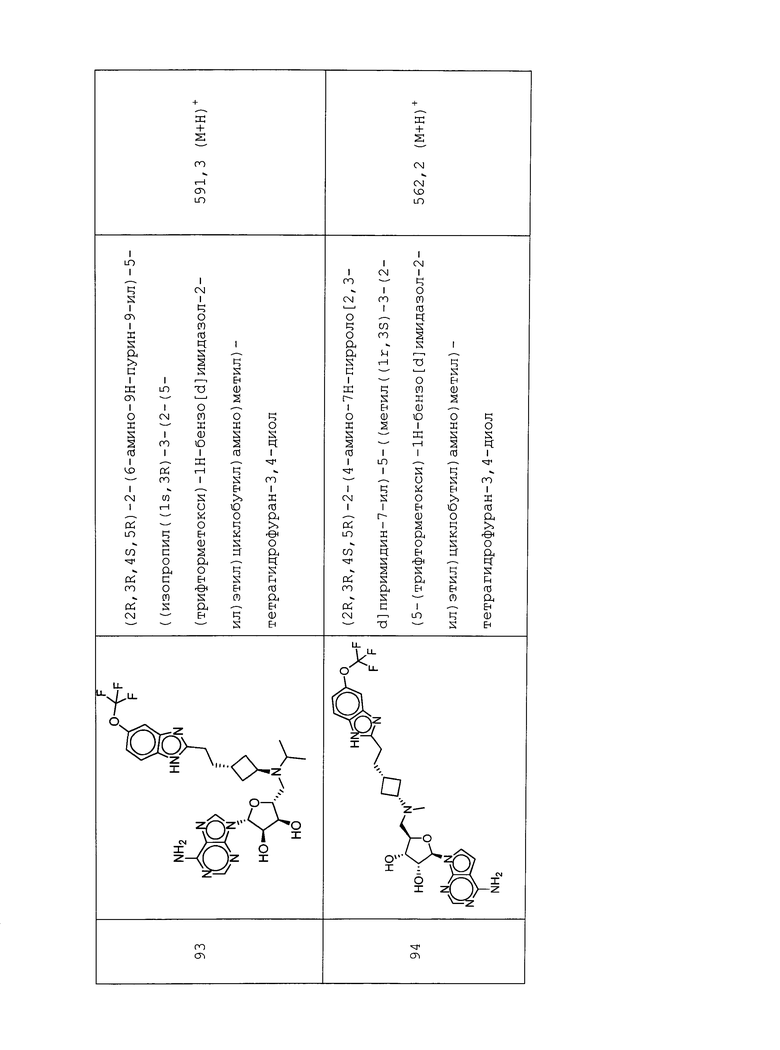
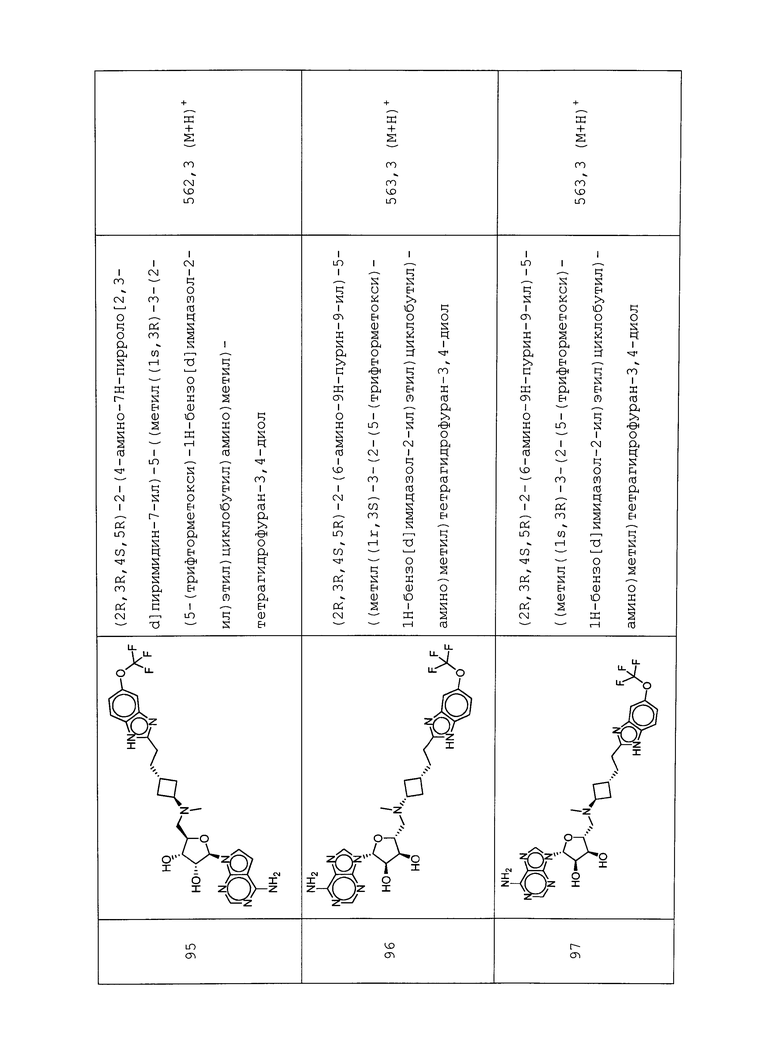
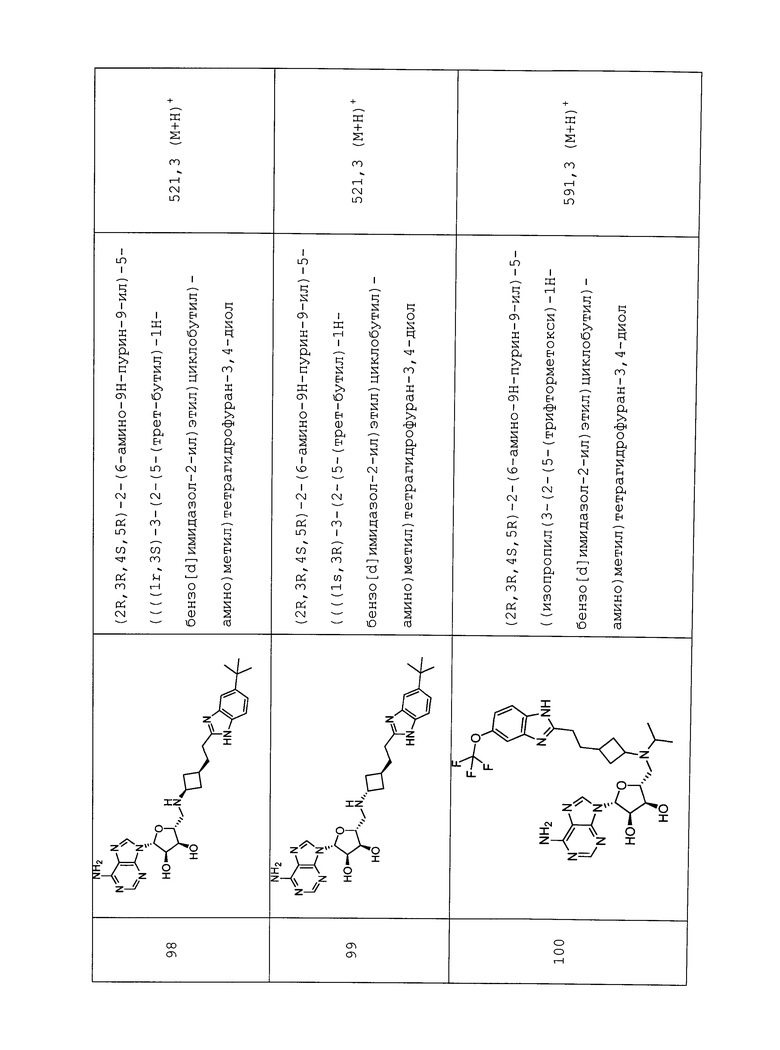
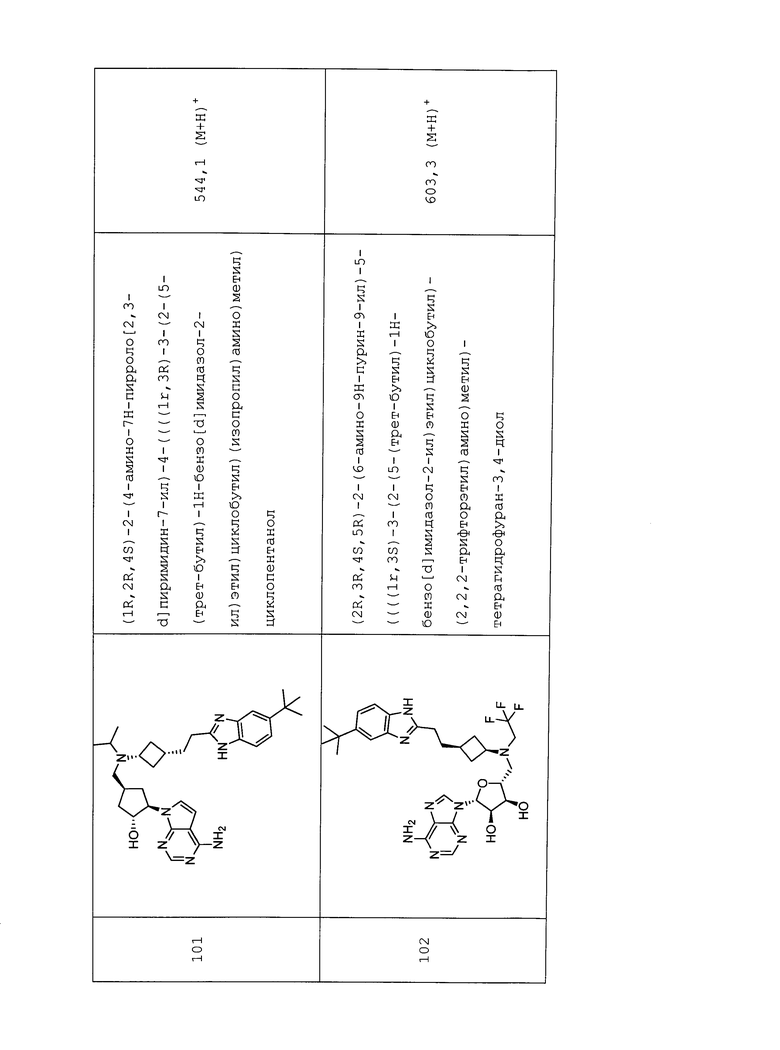
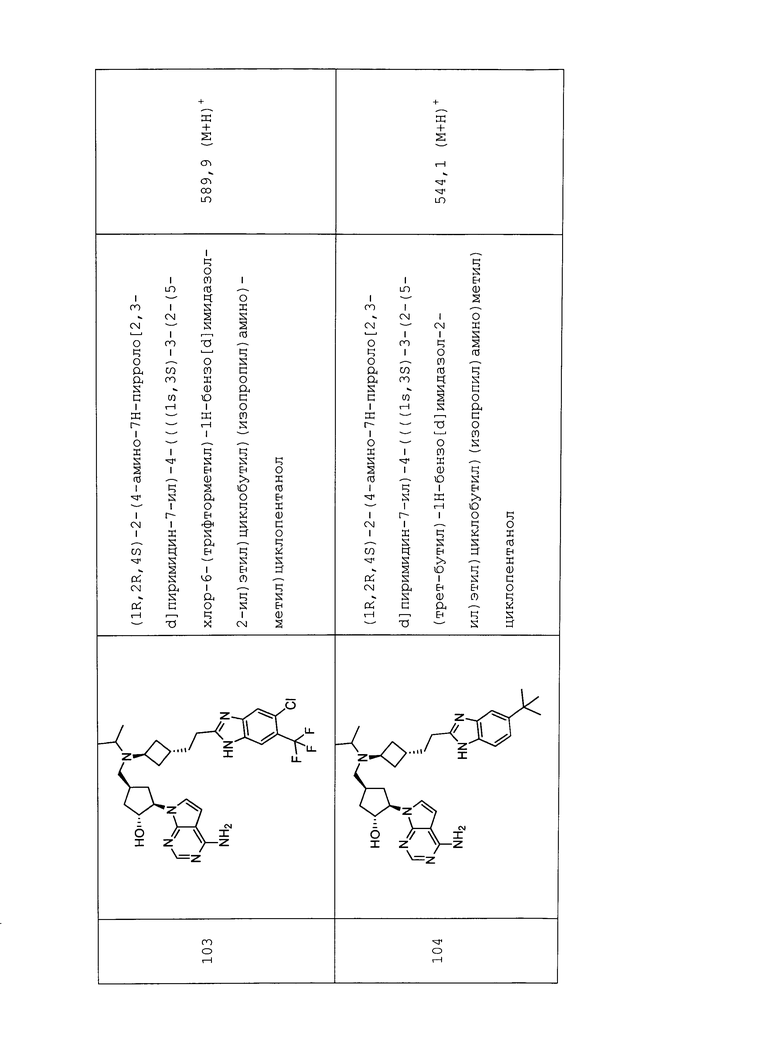
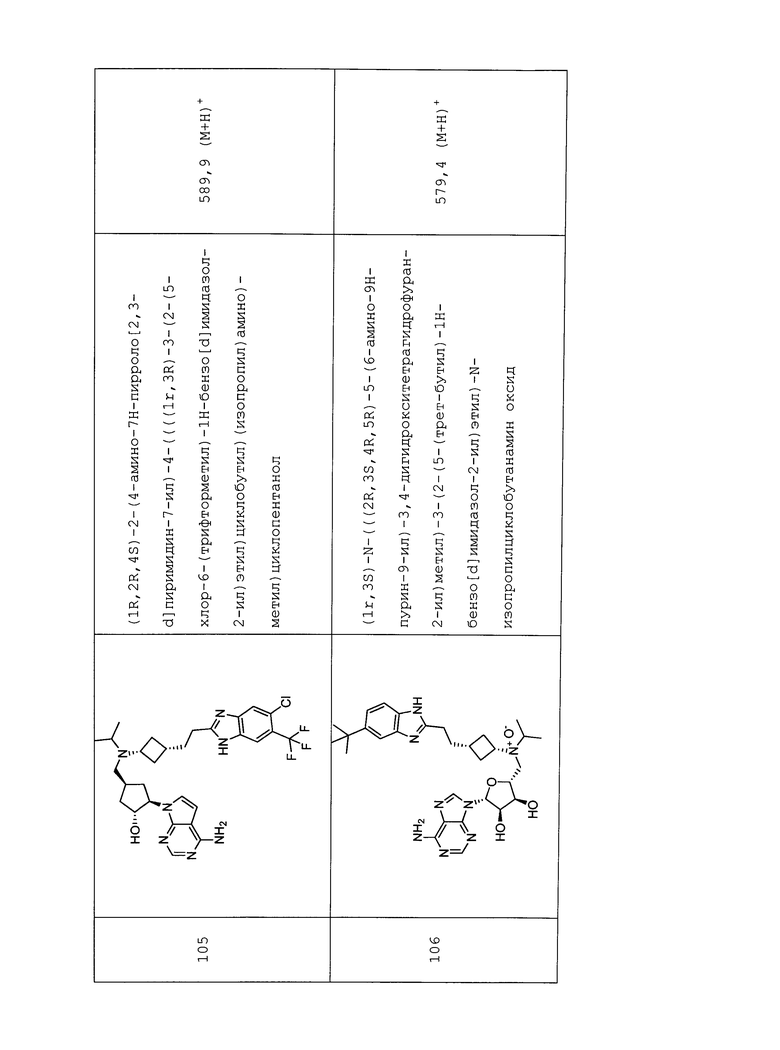
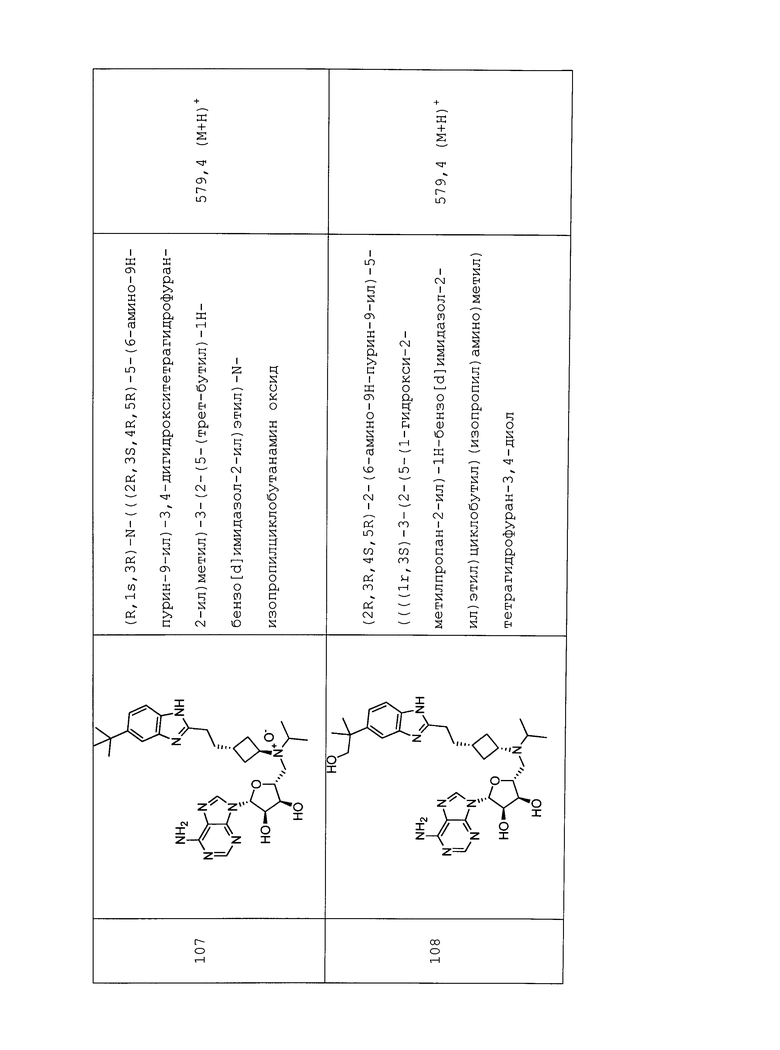
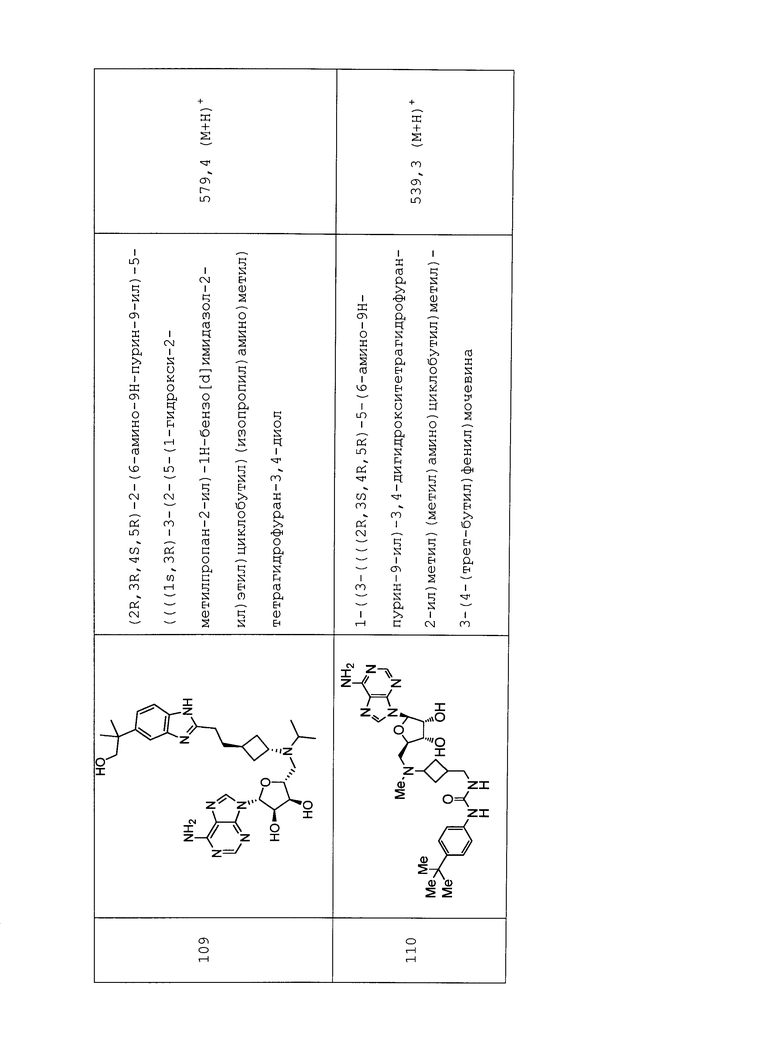
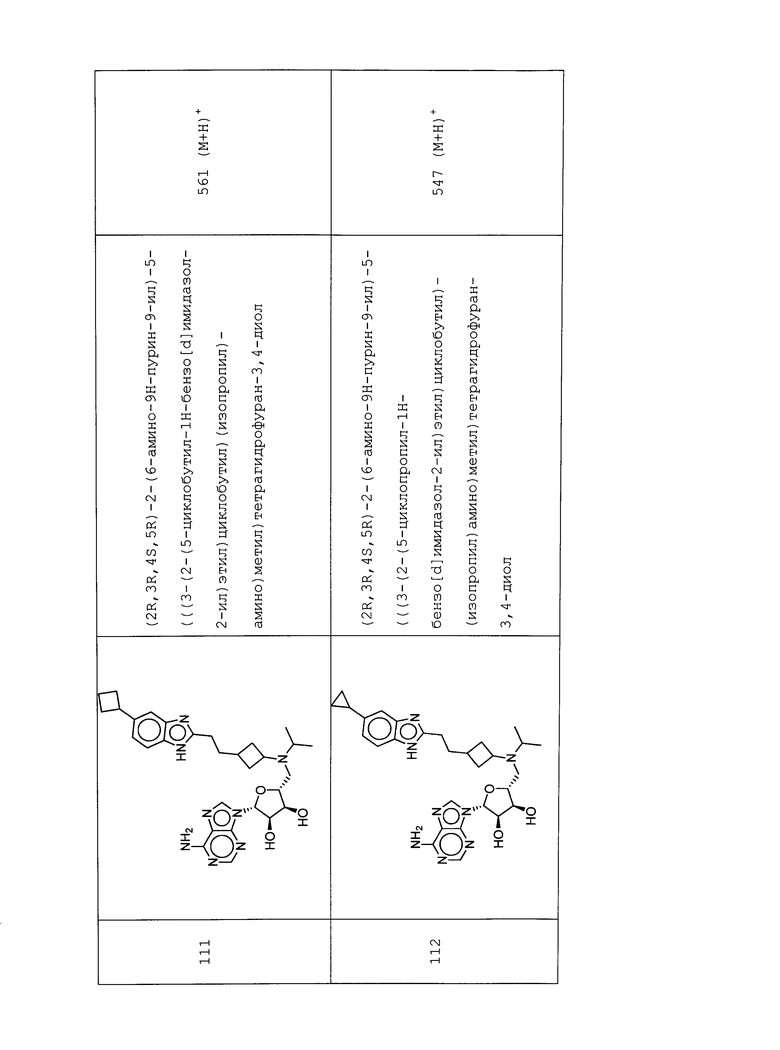
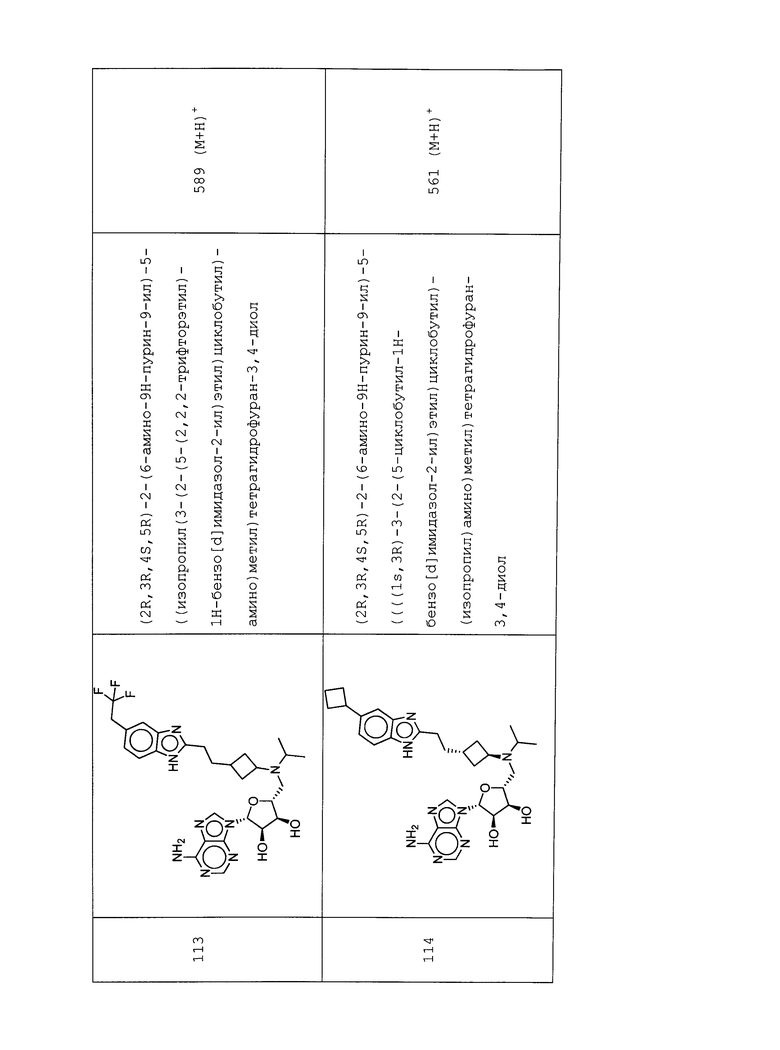
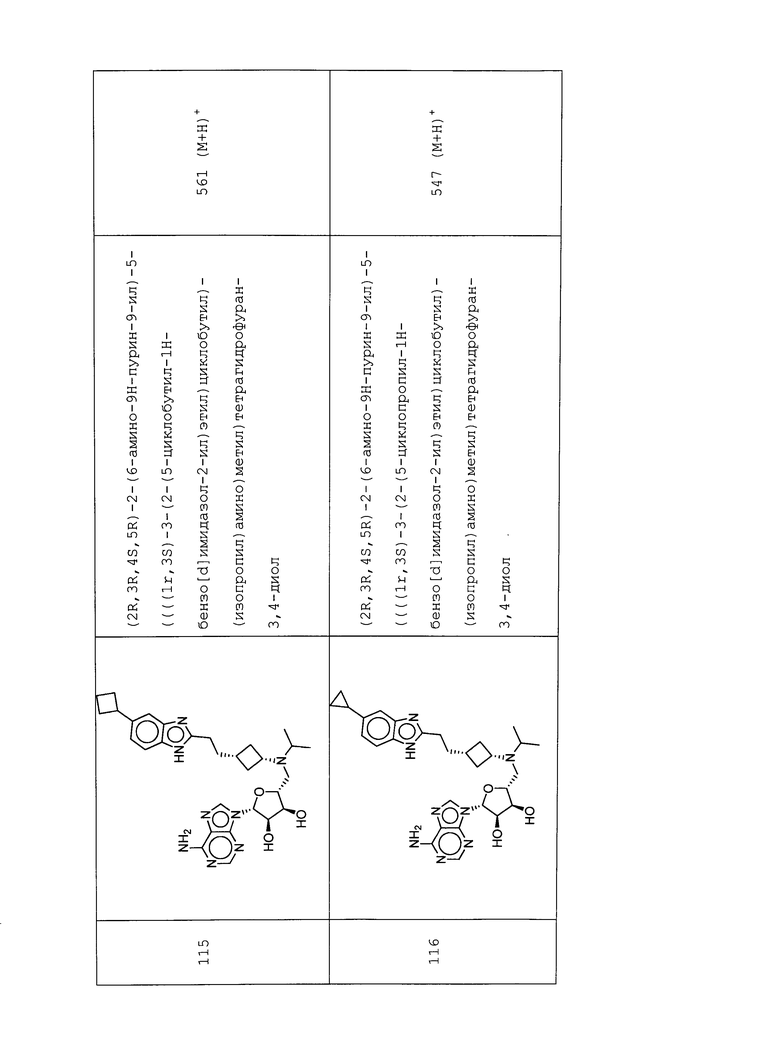
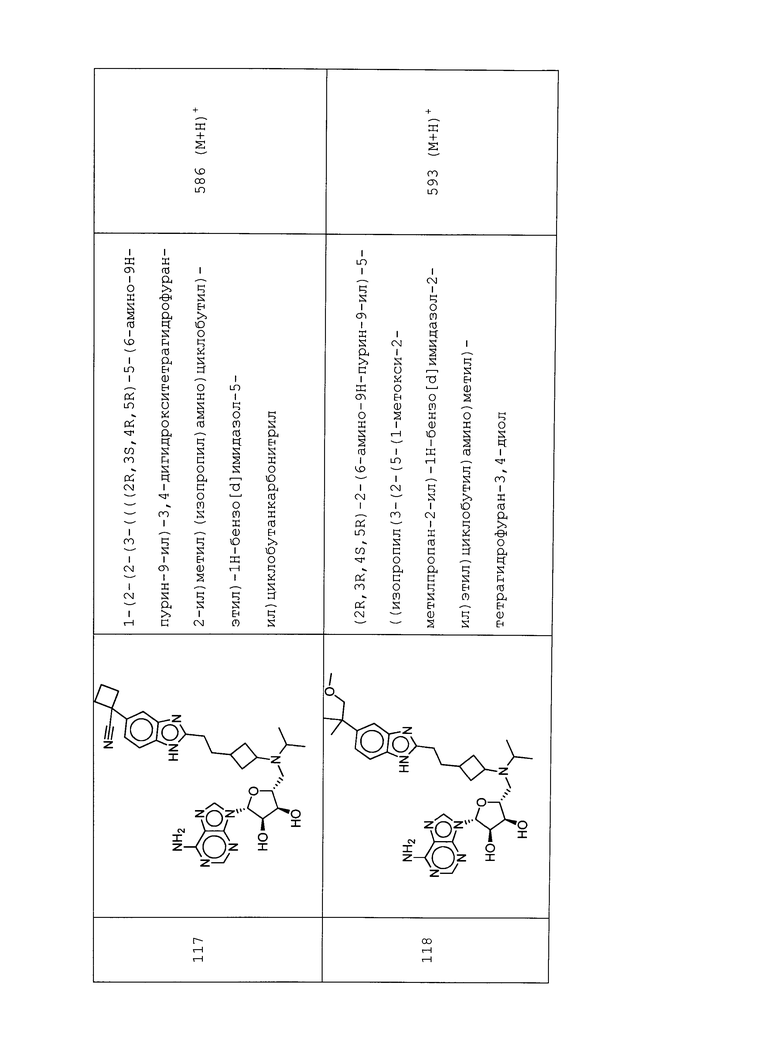
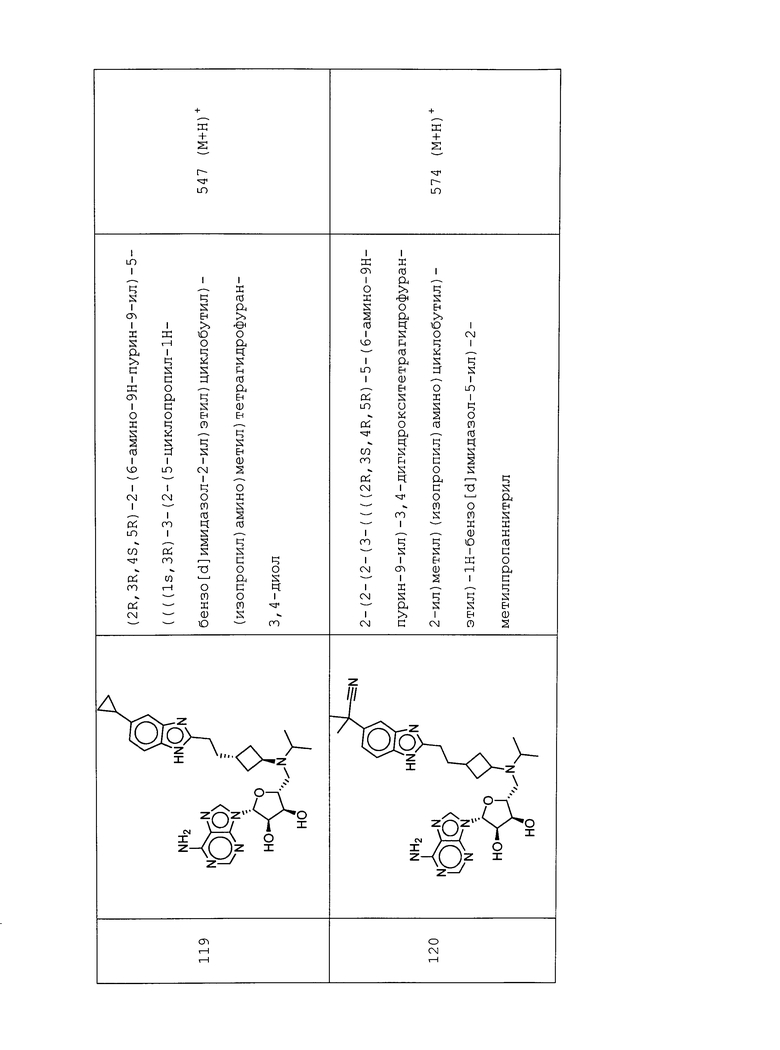
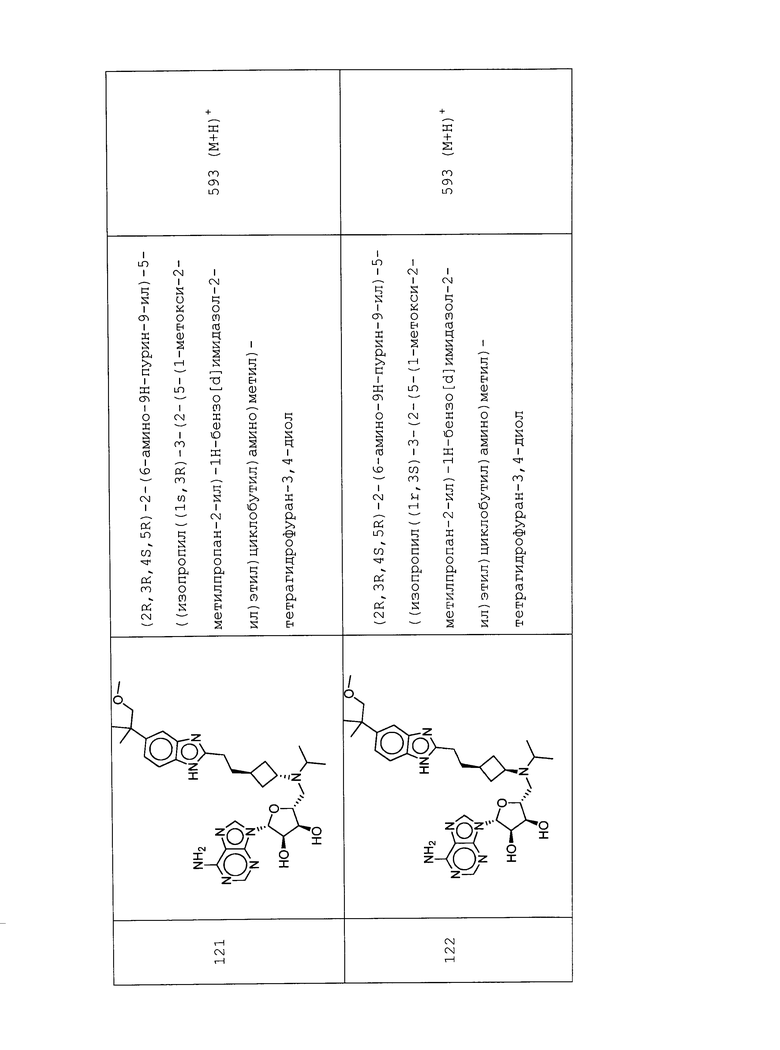
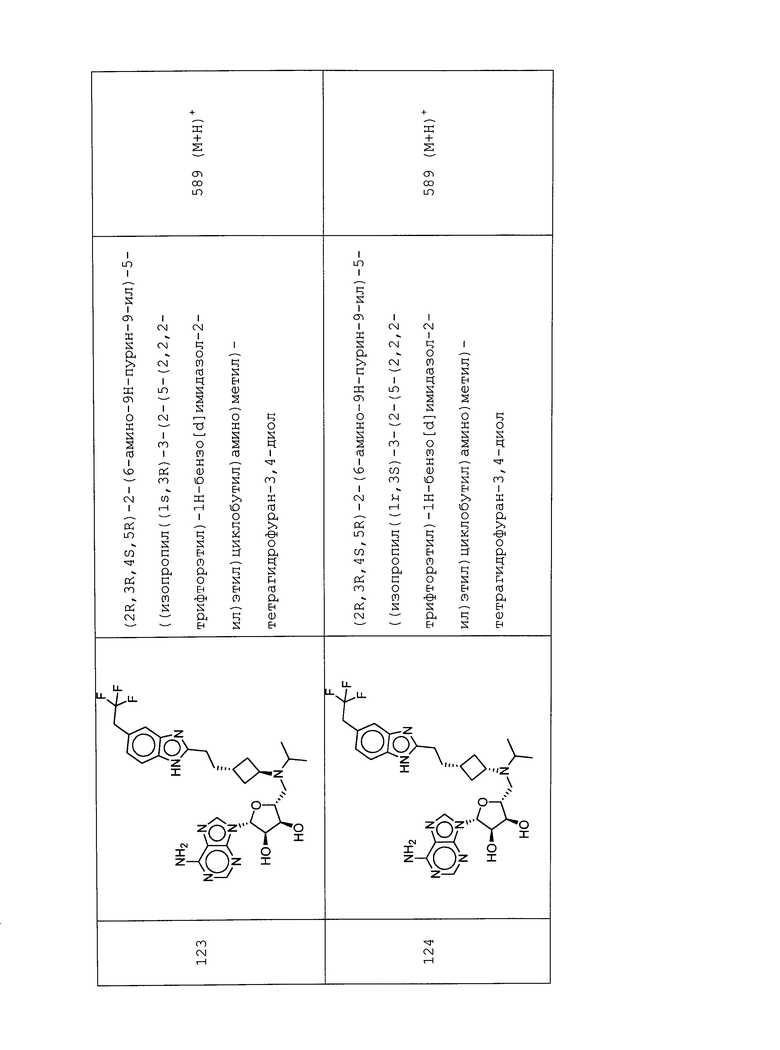
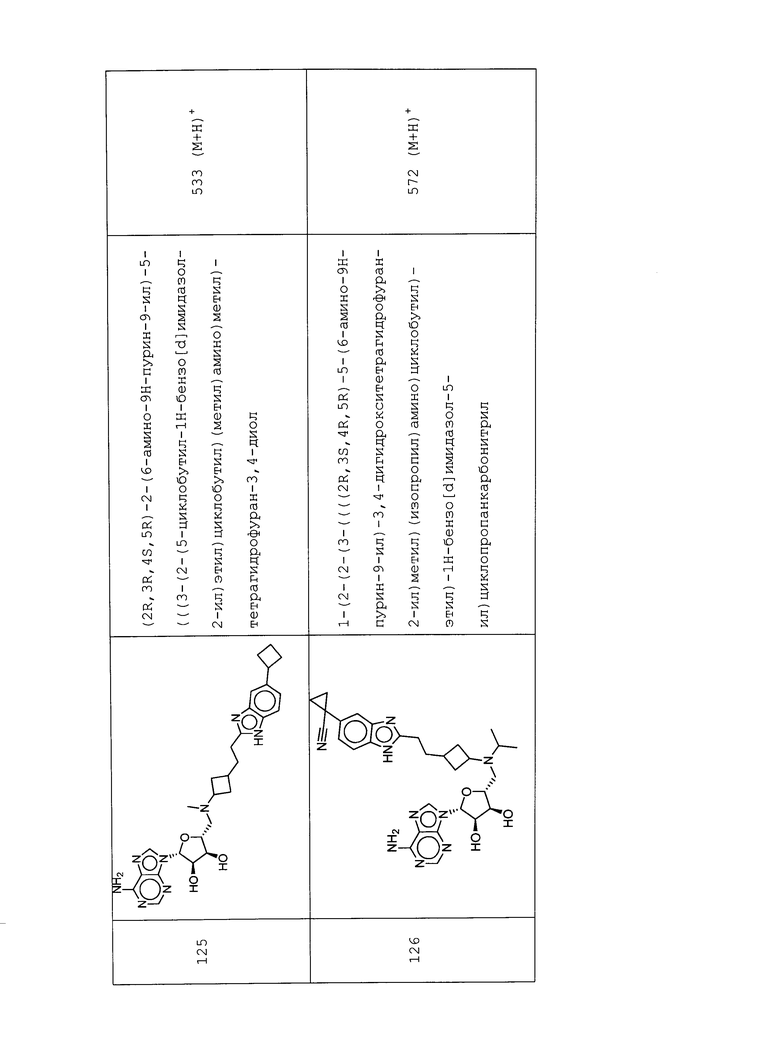
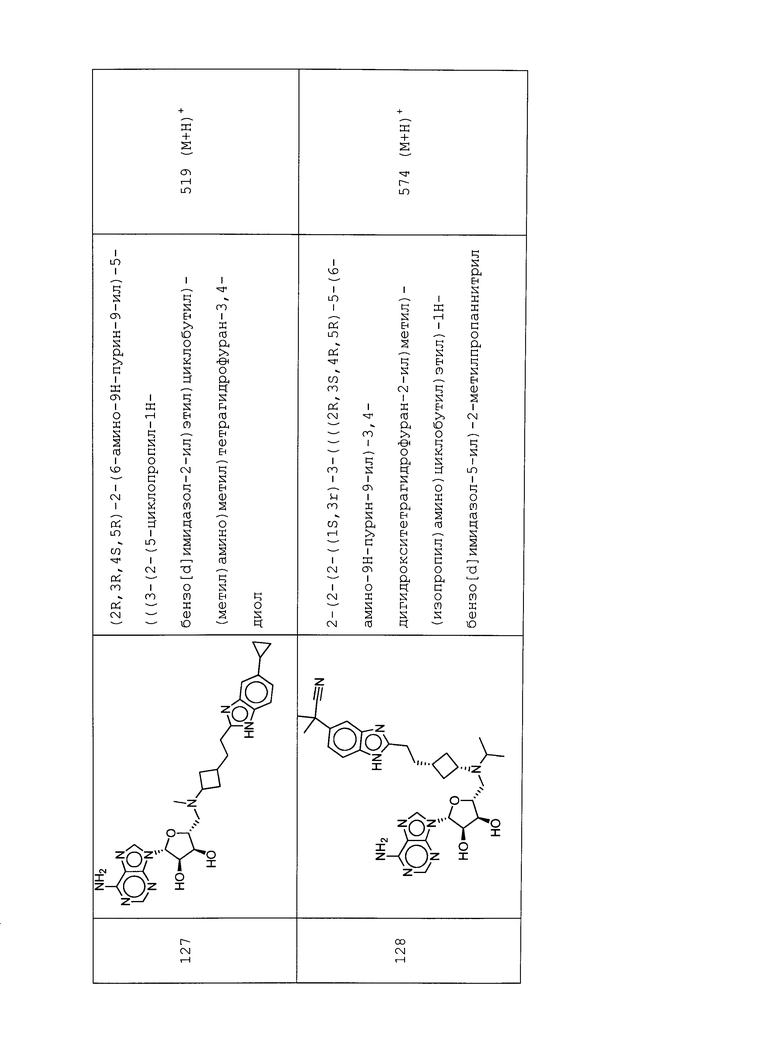
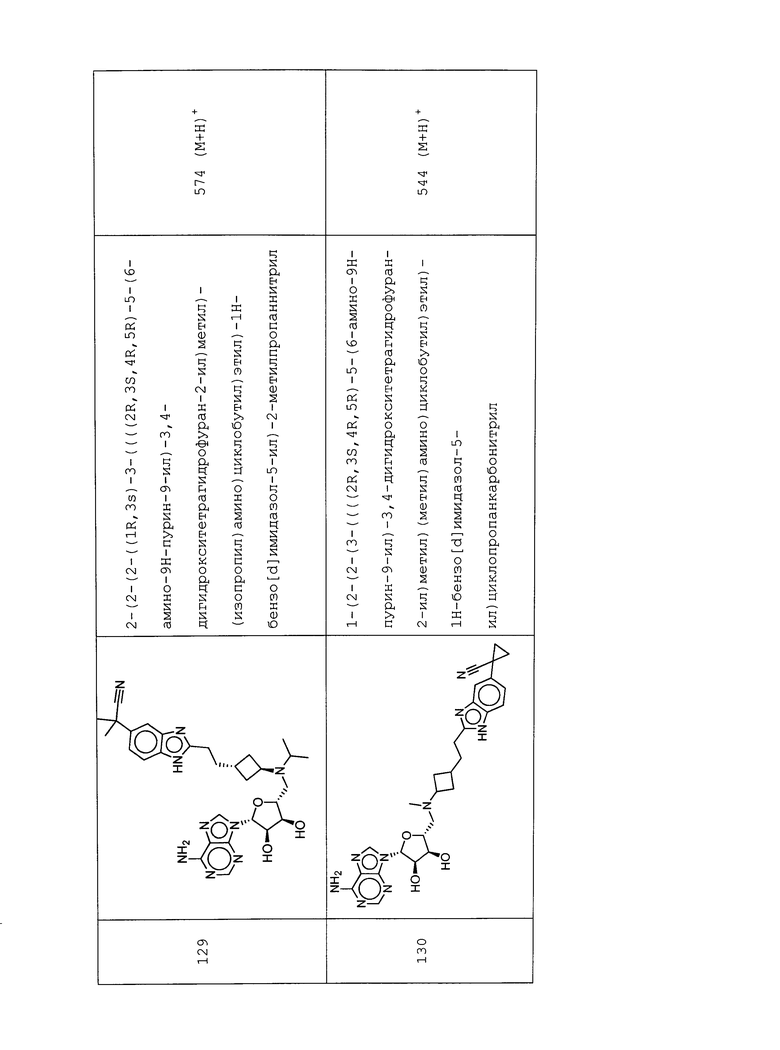
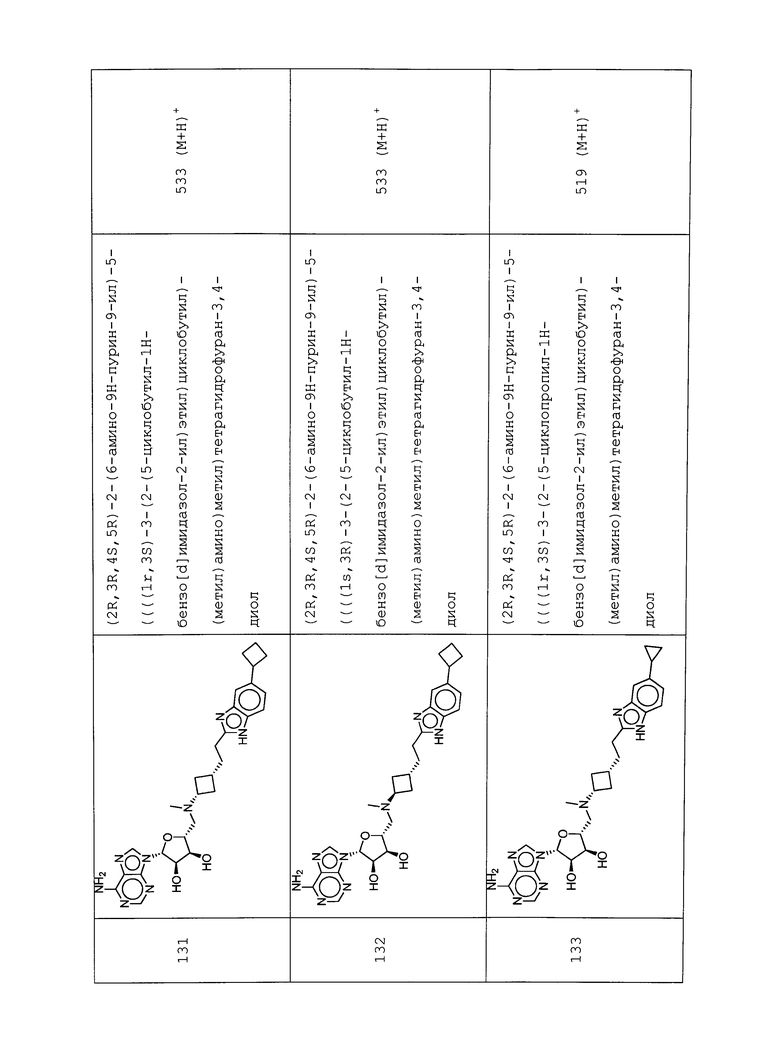
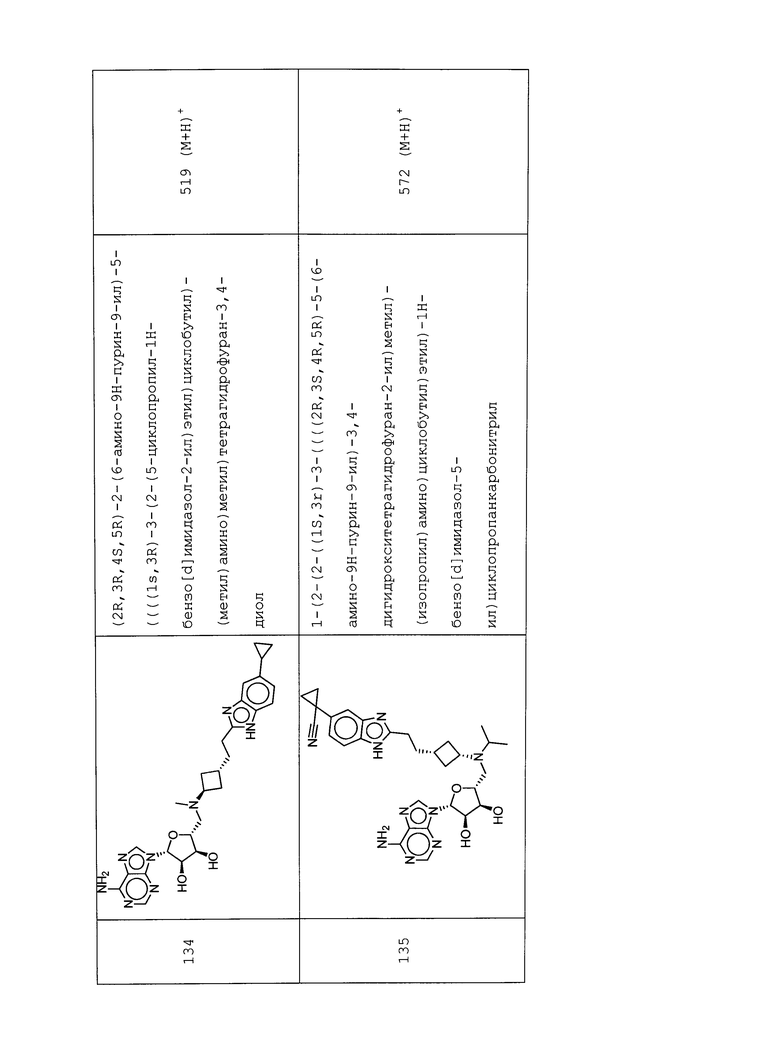
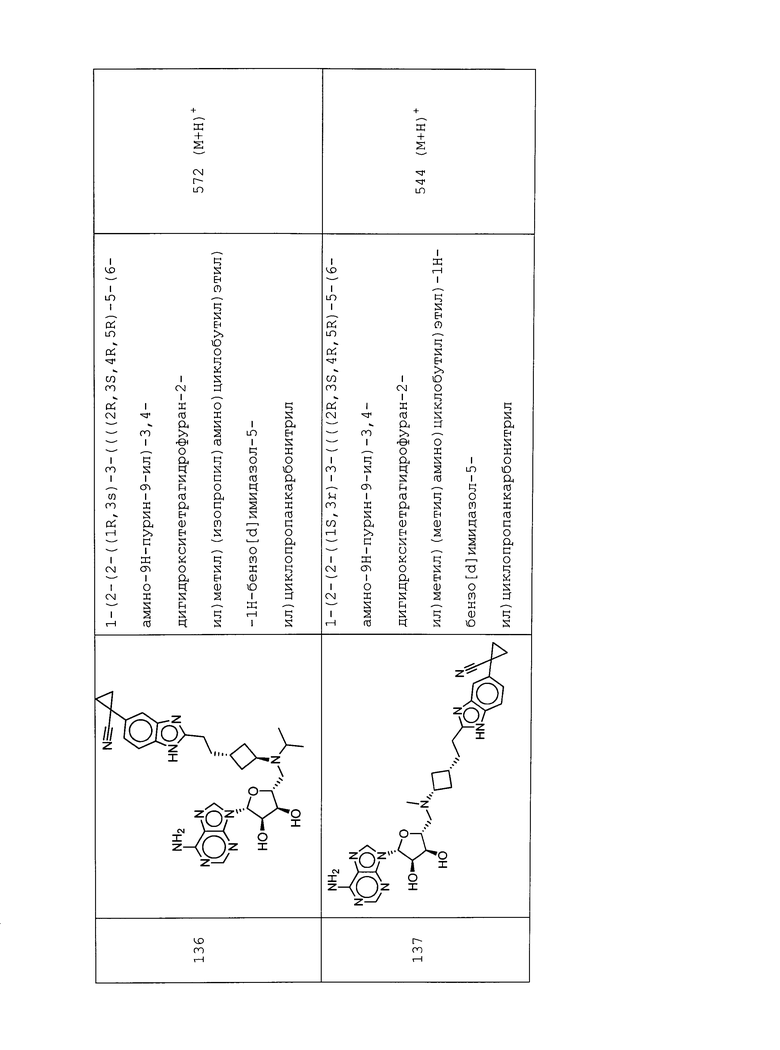
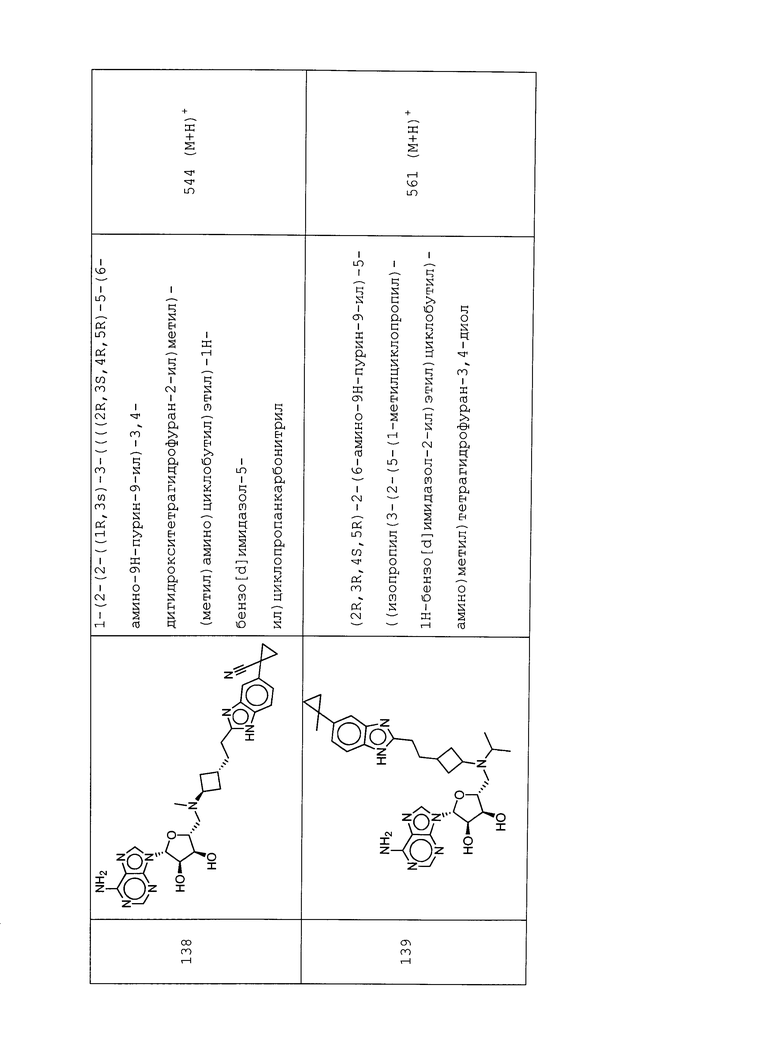
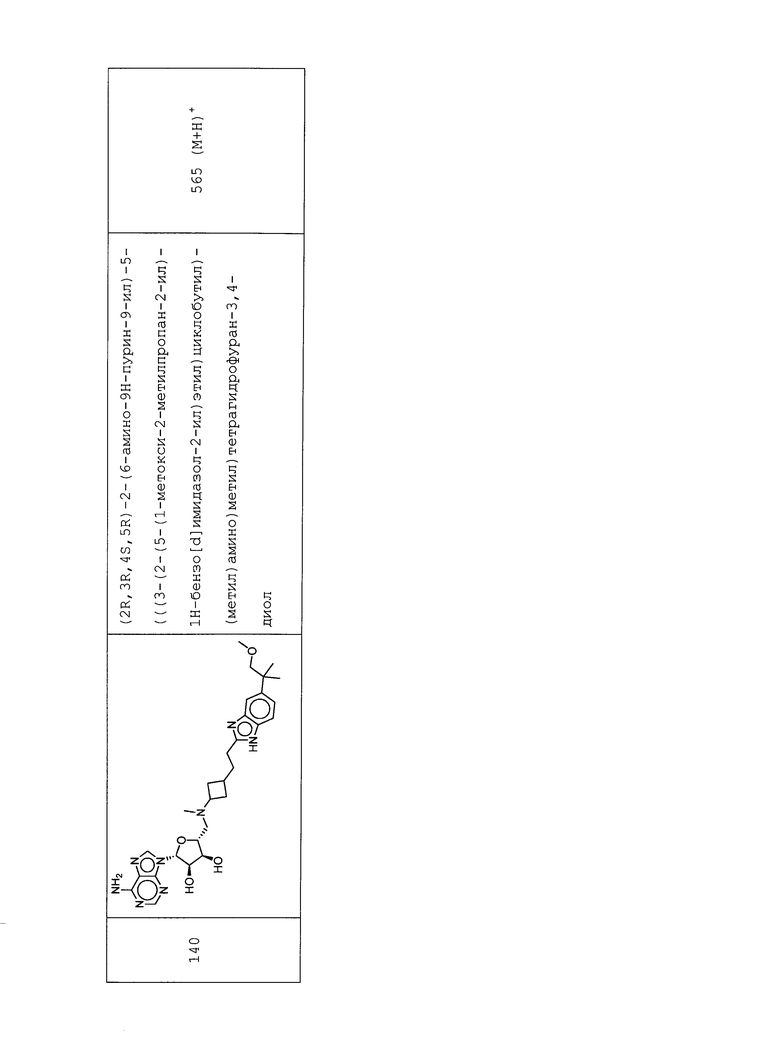

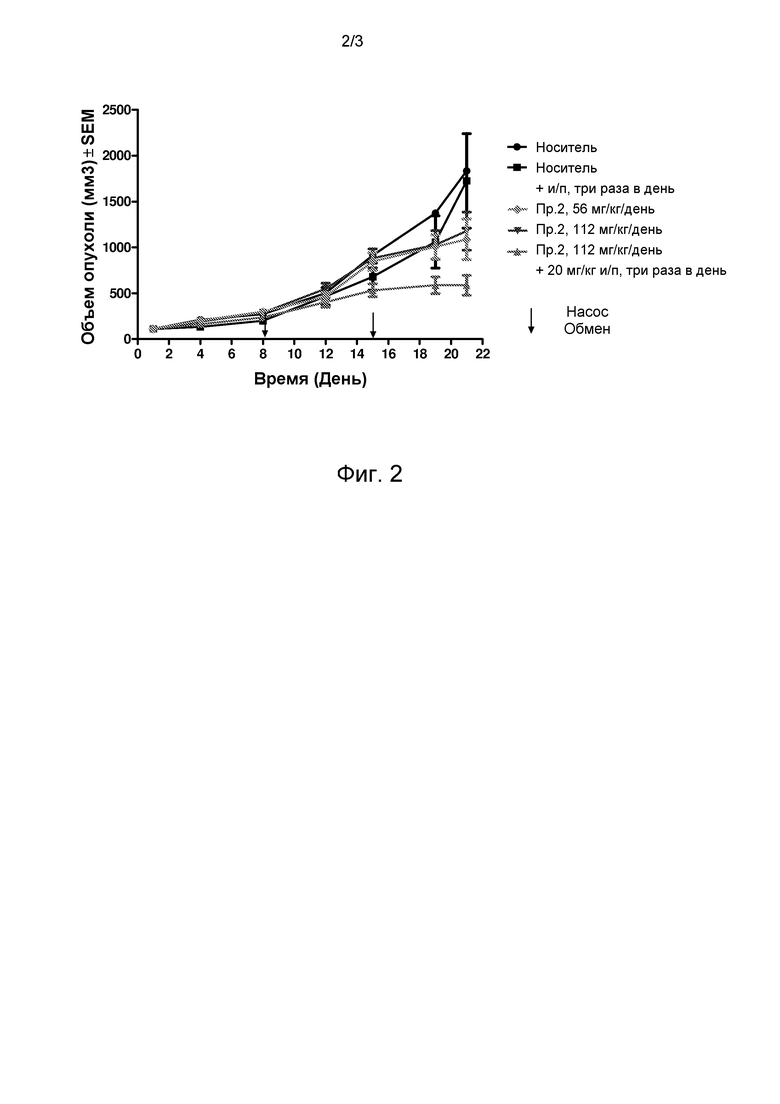
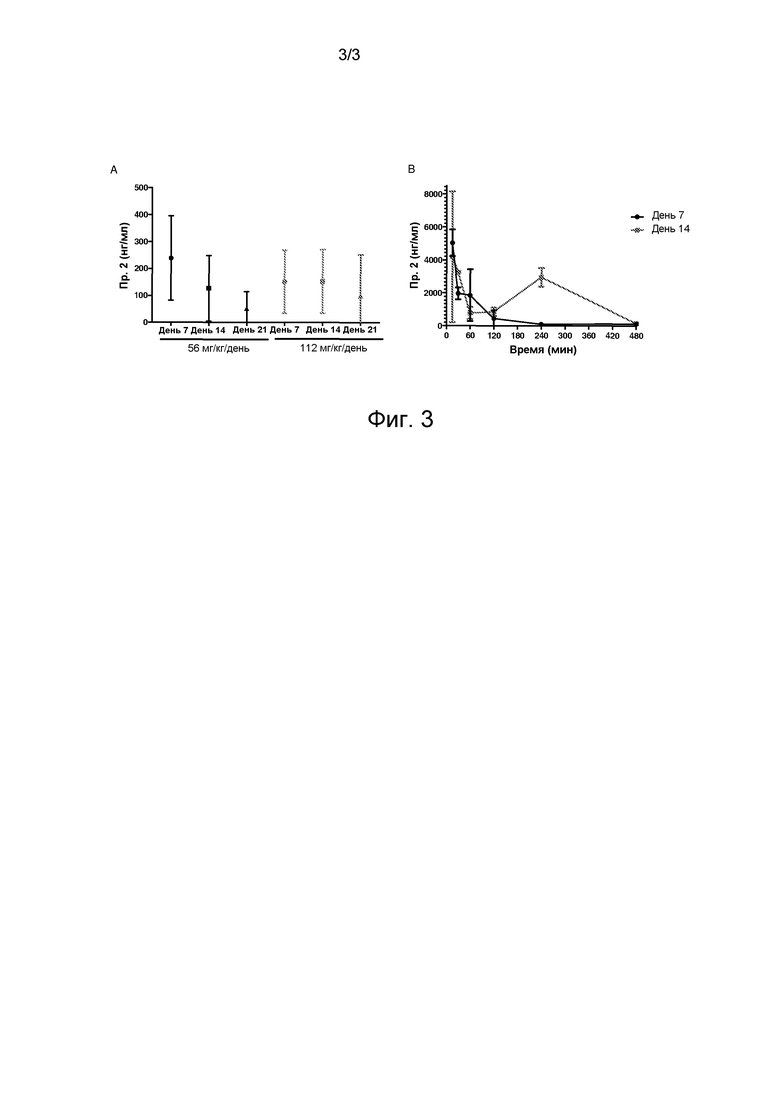
Изобретение относится к соединению формулы (IV) или его N-оксиду, или фармацевтически приемлемой соли такого соединения, где А представляет собой О или СН2; Q представляет собой NH2, NHRb, NRbRc или ОН, где каждый из Rb и Rc независимо представляет собой C1-C6 алкил; X представляет собой N или группу CRx, в которой Rx представляет собой Н; Y представляет собой Н или Rd, где Rd представляет собой C1-С6 алкил, С2-С6 алкенил, С2-С6 алкинил или С3-С8 циклоалкил, и Rd необязательно замещен одним или несколькими заместителями, выбранными из группы, состоящей из галогена и С3-С8 циклоалкила; каждый из R1 и R2 представляет собой Н; каждый из Re, Rf, Rg и Rh независимо представляет собой группу -М2-Т2, в которой М2 представляет собой связь или О-С1-С4 алкильный линкер, и Т2 представляет собой Н, галоген или RS4, где RS4 представляет собой C1-C6 алкил, С2-С6 алкенил, С2-С6 алкинил, С3-С8 циклоалкил или оксетанил, и каждый из О-С1-С4 алкильного линкера и RS4 необязательно замещен одним или несколькими заместителями, выбранными из группы, состоящей из галогена, гидроксила, циано, C1-C6 алкила, С2-С6 алкенила, С2-С6 алкинила и C1-С6 алкоксила; и m имеет значение 0, 1 или 2. Изобретение относится к конкретным соединениям. Соединения по изобретению предназначены для изготовления фармацевтической композиции, обладающей антипролиферативной активностью, включающей терапевтически эффективное количество соединения и фармацевтически приемлемый носитель. Кроме того, соединения применяют для лечения лейкоза и ингибирования DOT1L-опосредованного метилирования белка. Технический результат – замещенное пуриновое или 7-деазапуриновое соединение эффективное для применения в лечении лейкоза и ингибирования DOT1L-опосредованного метилирования белка. 9 н. и 15 з.п. ф-лы, 1 табл., 3 ил., 11 пр.
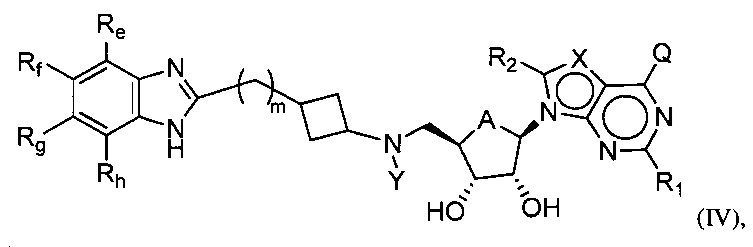
1.Соединение формулы (IV) или его N-оксид, или фармацевтически приемлемая соль такого соединения:
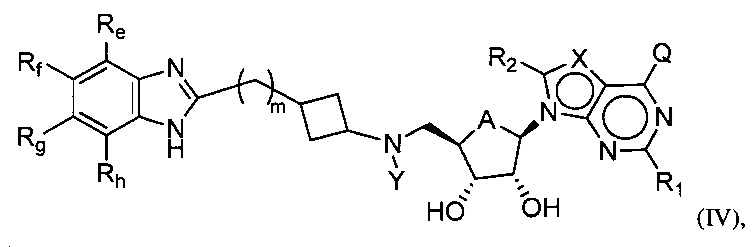
где
А представляет собой О или СН2;
Q представляет собой NH2, NHRb, NRbRc или ОН, где каждый из Rb и Rc независимо представляет собой C1-C6 алкил;
X представляет собой N или группу CRx, в которой Rx представляет собой Н;
Y представляет собой Н или Rd, где Rd представляет собой C1-С6 алкил, С2-С6 алкенил, С2-С6 алкинил или С3-С8 циклоалкил, и Rd необязательно замещен одним или несколькими заместителями, выбранными из группы, состоящей из галогена и С3-С8 циклоалкила;
каждый из R1 и R2 представляет собой Н;
каждый из Re, Rf, Rg и Rh независимо представляет собой группу -М2-Т2, в которой М2 представляет собой связь или О-С1-С4 алкильный линкер, и Т2 представляет собой Н, галоген или RS4, где RS4 представляет собой C1-C6 алкил, С2-С6 алкенил, С2-С6 алкинил, С3-С8 циклоалкил или оксетанил, и каждый из О-С1-С4 алкильного линкера и RS4 необязательно замещен одним или несколькими заместителями, выбранными из группы, состоящей из галогена, гидроксила, циано, C1-C6 алкила, С2-С6 алкенила, С2-С6 алкинила и C1-С6 алкоксила; и
m имеет значение 0, 1 или 2.
2. Соединение по п. 1, где А представляет собой О.
3. Соединение по п. 1, где А представляет собой О и m имеет значение 2.
4. Соединение по п. 1, где X представляет собой N.
5. Соединение по п. 1, где Q представляет собой NH2 или NHRb.
6. Соединение по п. 1, где Y представляет собой Rd.
7. Соединение по п. 6, где Rd представляет собой С1-С6 алкил, необязательно замещенный циклопропилом, циклобутилом или галогеном.
8. Соединение по п. 6, где Rd представляет собой циклопропил или циклобутил, каждый необязательно замещенный галогеном.
9. Соединение по п. 1, где по меньшей мере один из Re, Rf, Rg и Rh представляет собой галоген; C1-C6 алкоксил, необязательно замещенный одним или несколькими атомами галогена; C1-C6 алкил, необязательно замещенный одним или несколькими заместителями, выбранными из циано, галогена, гидрокси и C1-C6 алкоксила; циклопропил, необязательно замещенный одним или несколькими заместителями, выбранными из C1-C6 алкила и циано; или оксетанил, необязательно замещенный одним или несколькими заместителями, выбранными из циано, галогена, гидрокси, С1-С6 алкила и C1-C6 алкоксила.
10. Соединение по п. 9, где по меньшей мере один из Re, Rf, Rg и Rh выбран из группы, состоящей из F; Cl; Br; CF3; OCF3; оксетанила, необязательно замещенного одним или несколькими заместителями, выбранными из циано, галогена, гидрокси, C1-C6 алкила и C1-C6 алкоксила; C1-C4 алкоксила; циклопропила, необязательно замещенного одним или несколькими заместителями, выбранными из C1-C4 алкила и циано; циклобутила, необязательно замещенного одним или несколькими заместителями, выбранными из C1-C4 алкила и циано; и C1-C4 алкила, необязательно замещенного одним или несколькими заместителями, выбранными из циано, галогена, циклопропила, циклобутила, гидроксила и C1-C6 алкоксила.
11. Соединение, выбранное из
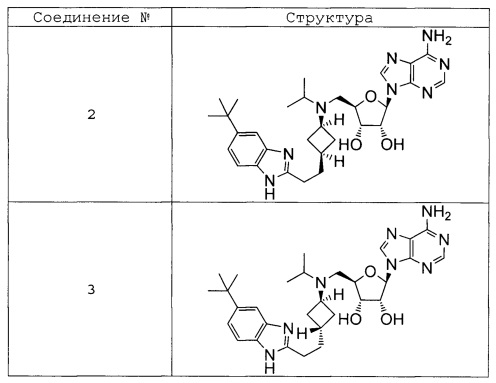
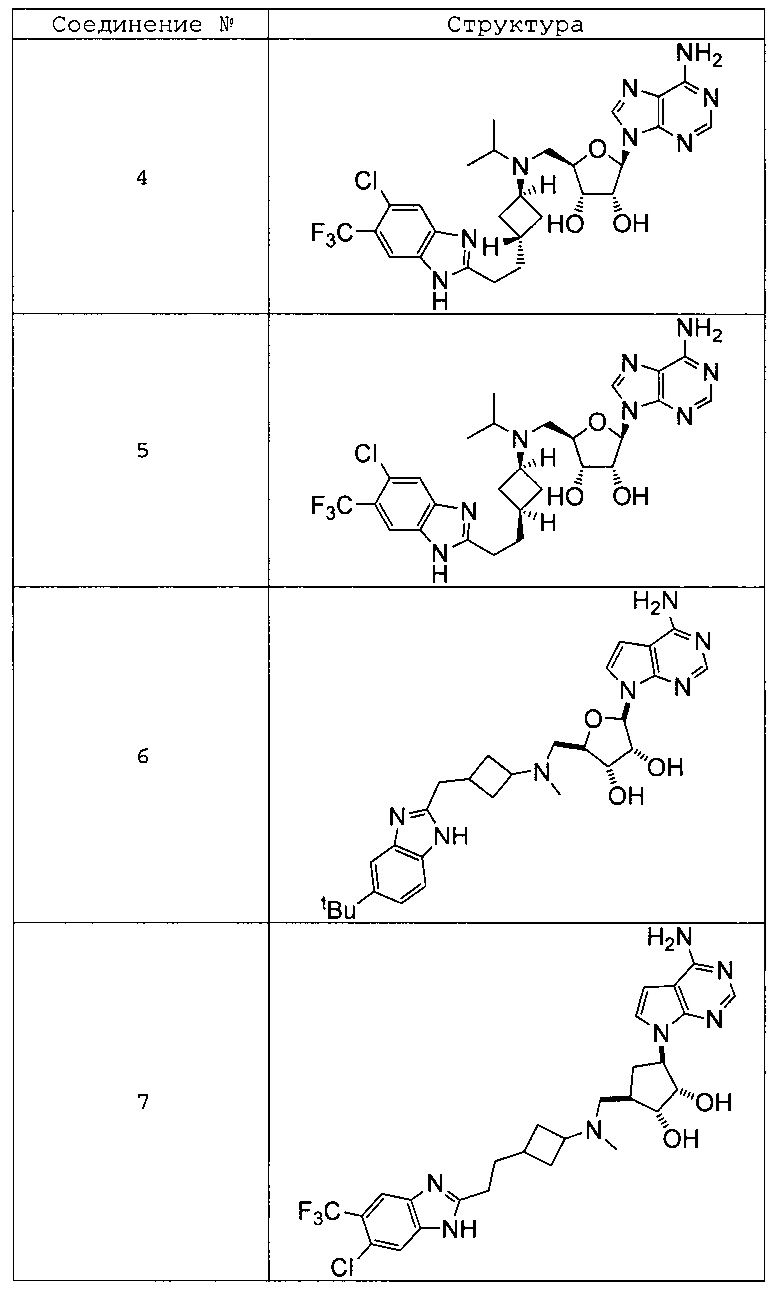
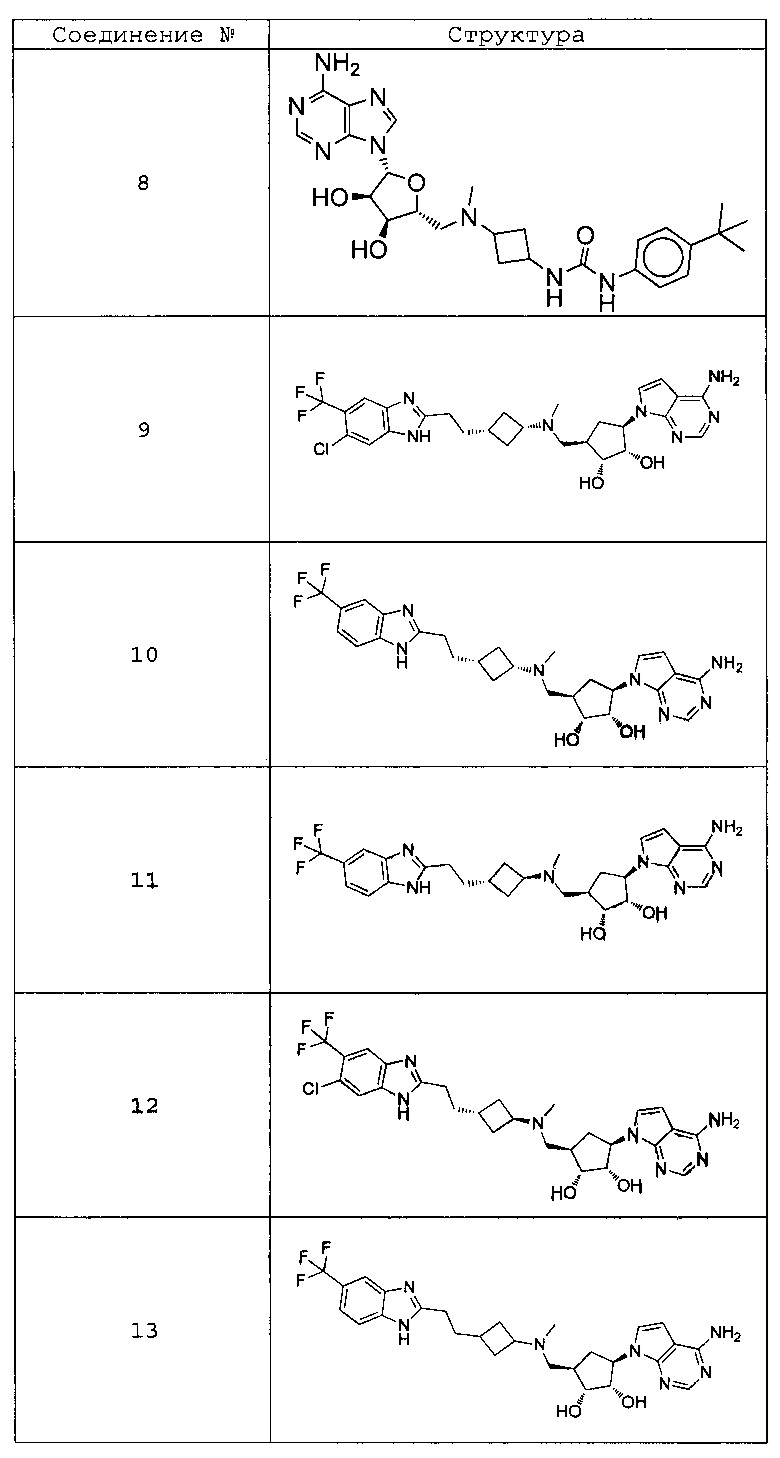
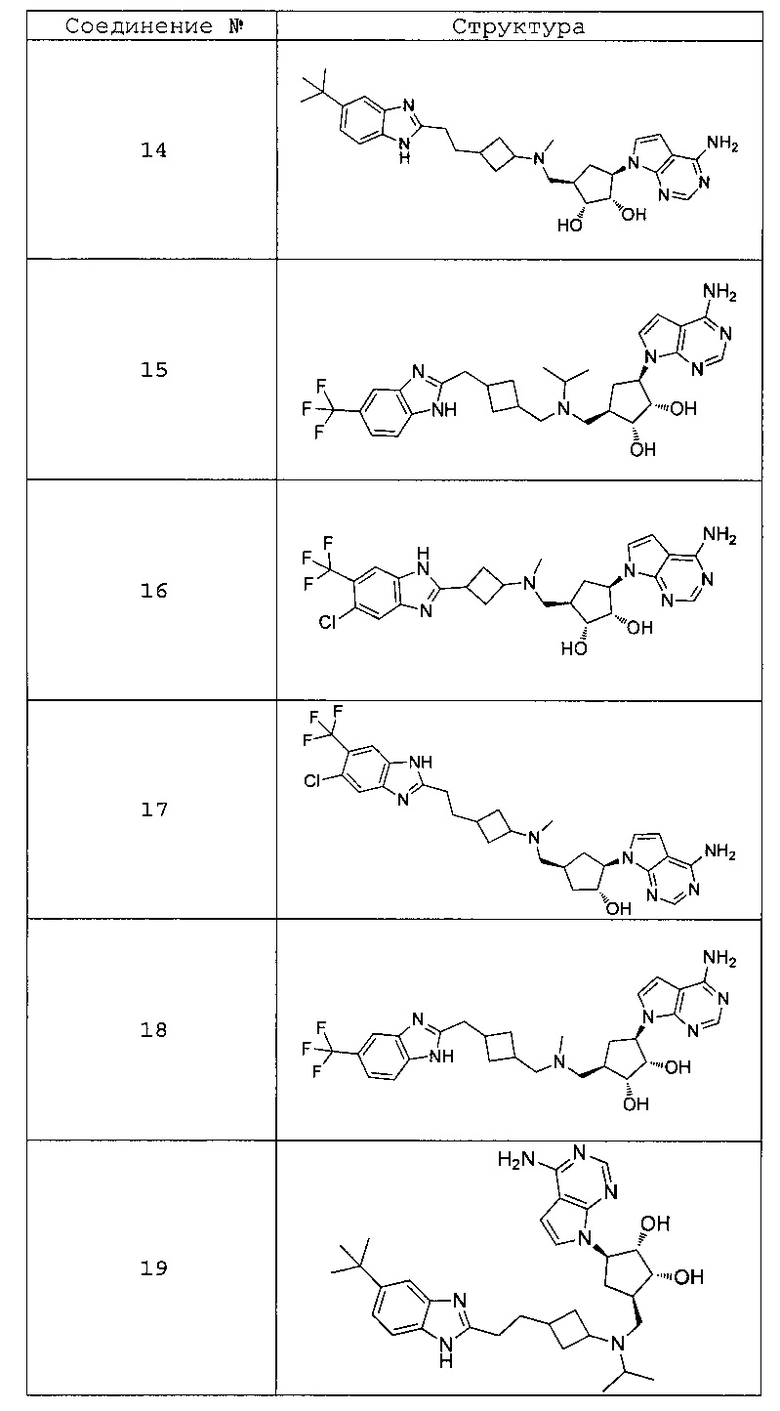
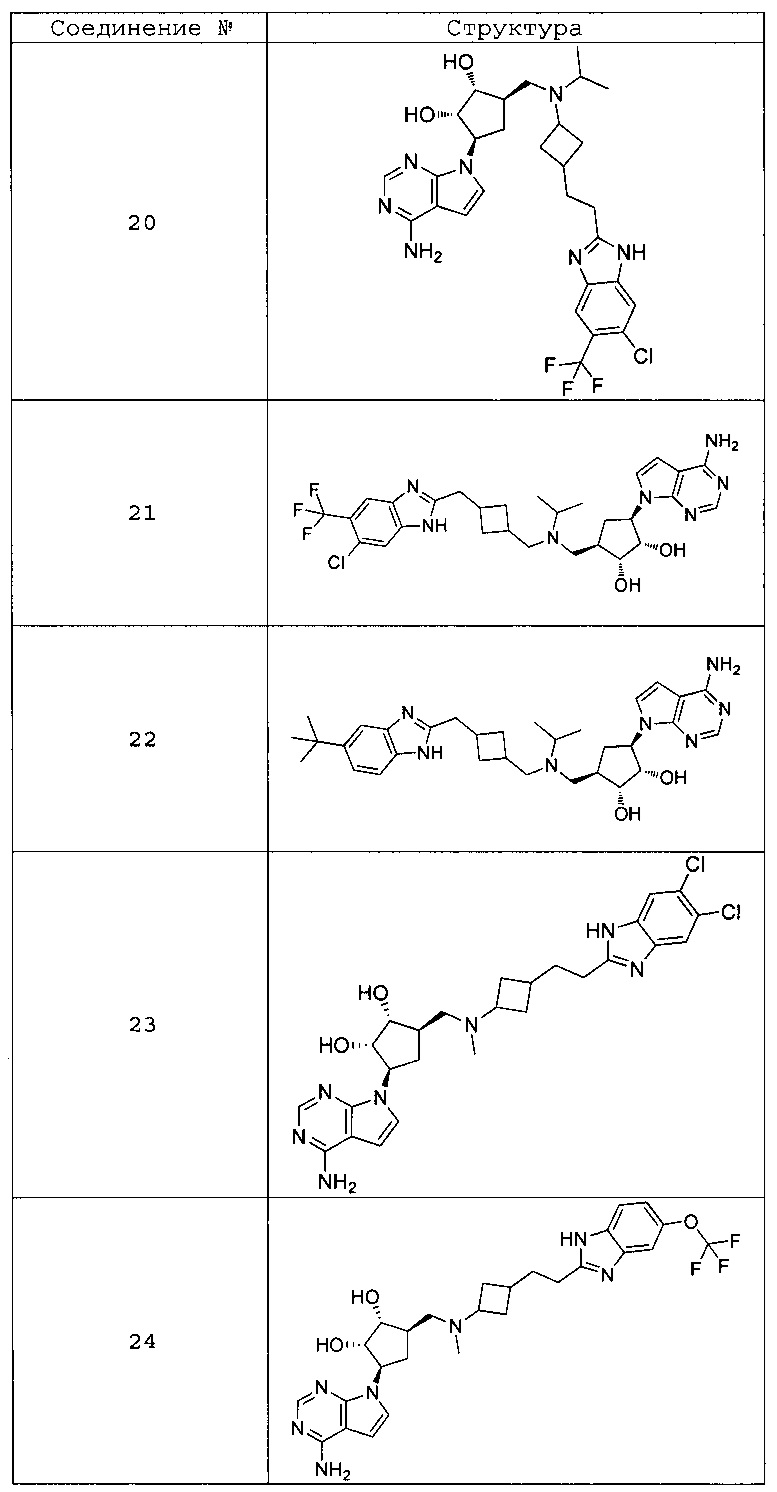
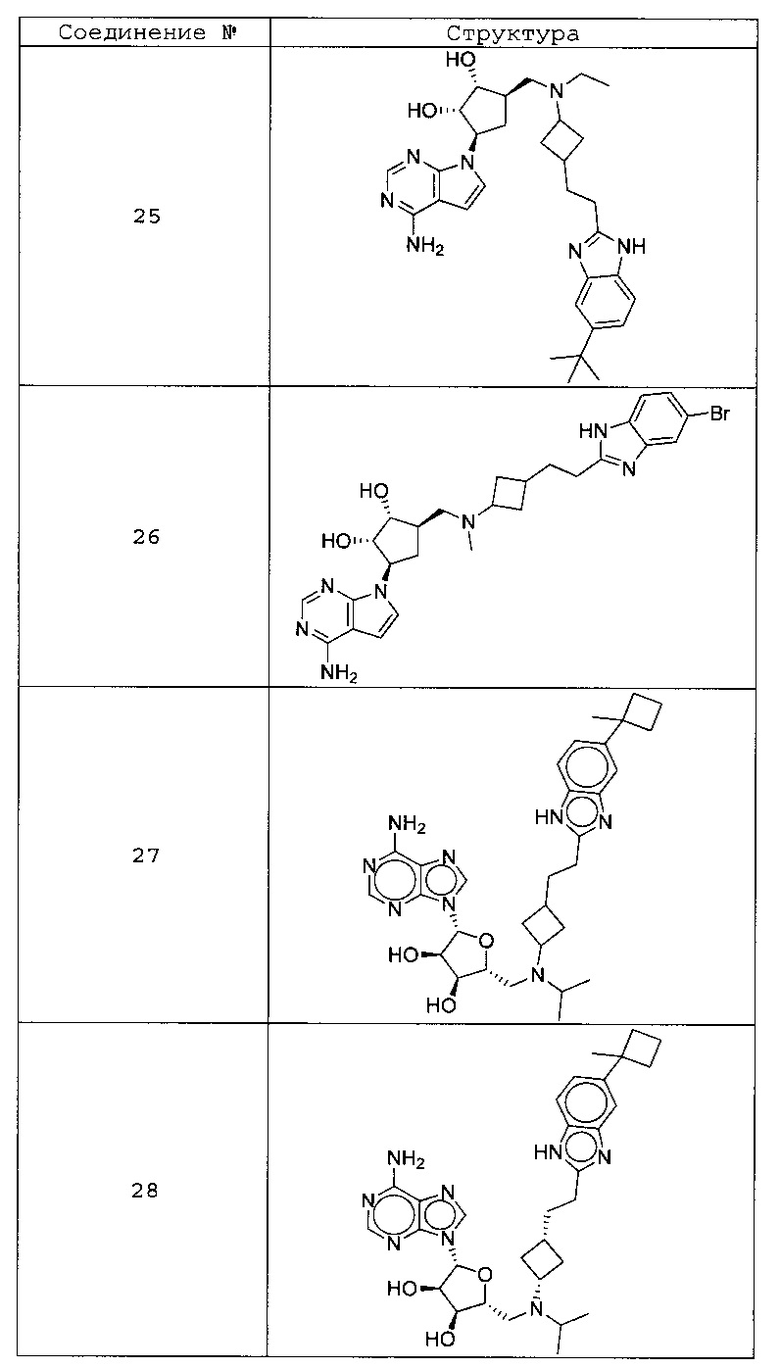
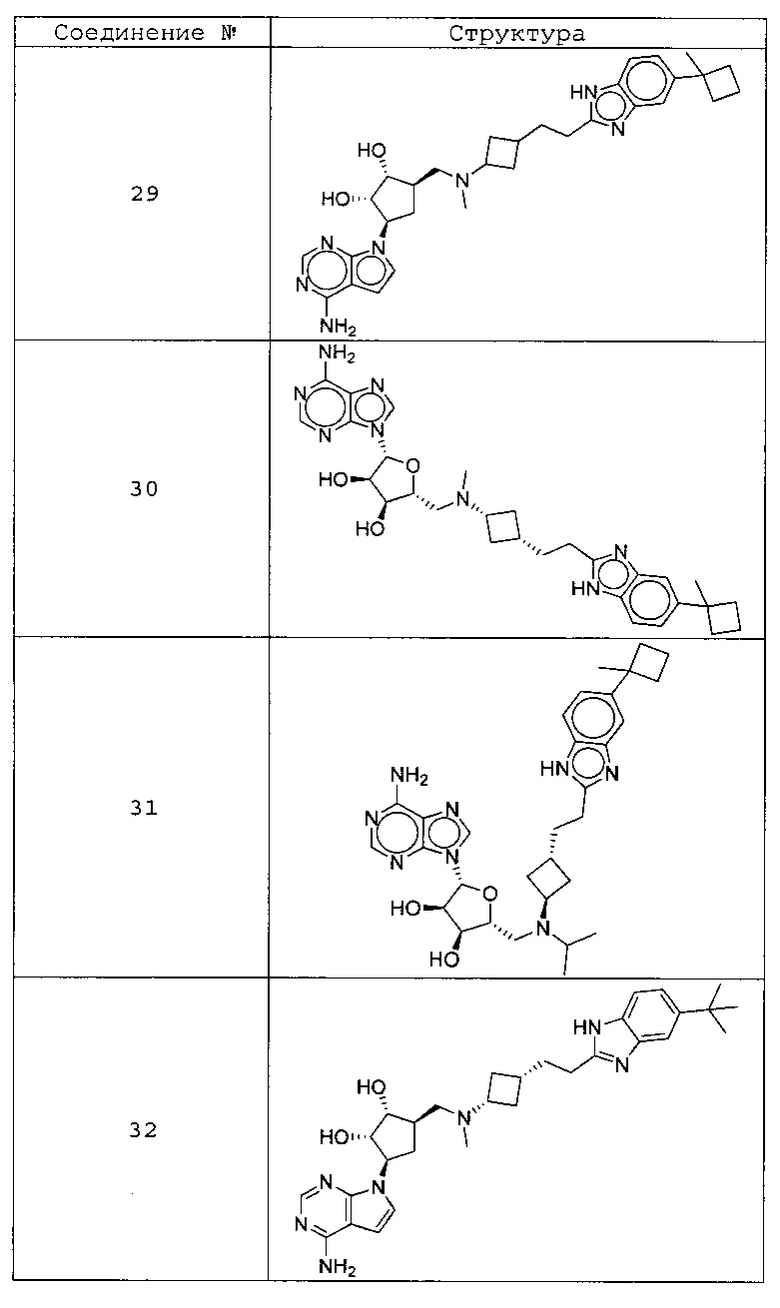
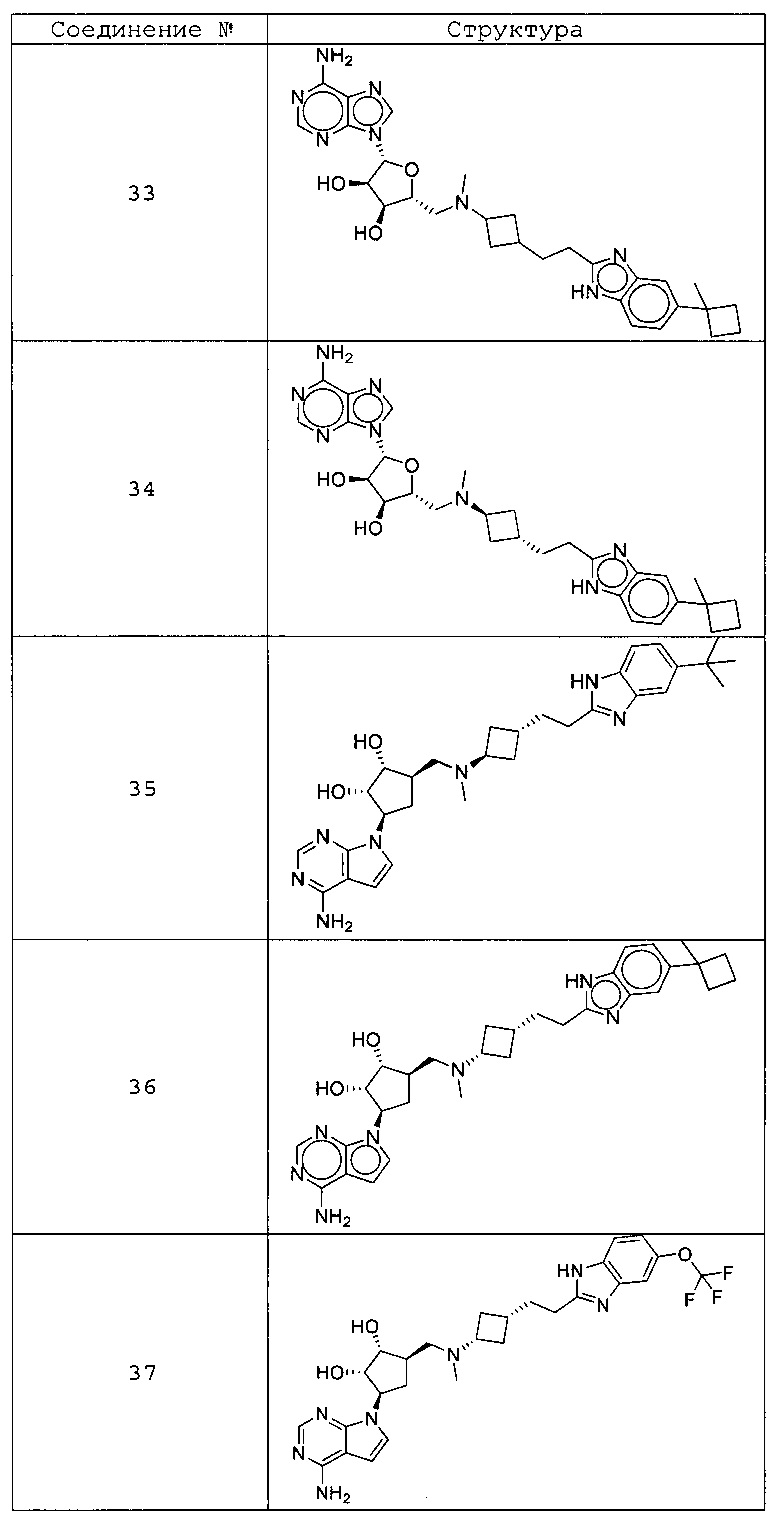
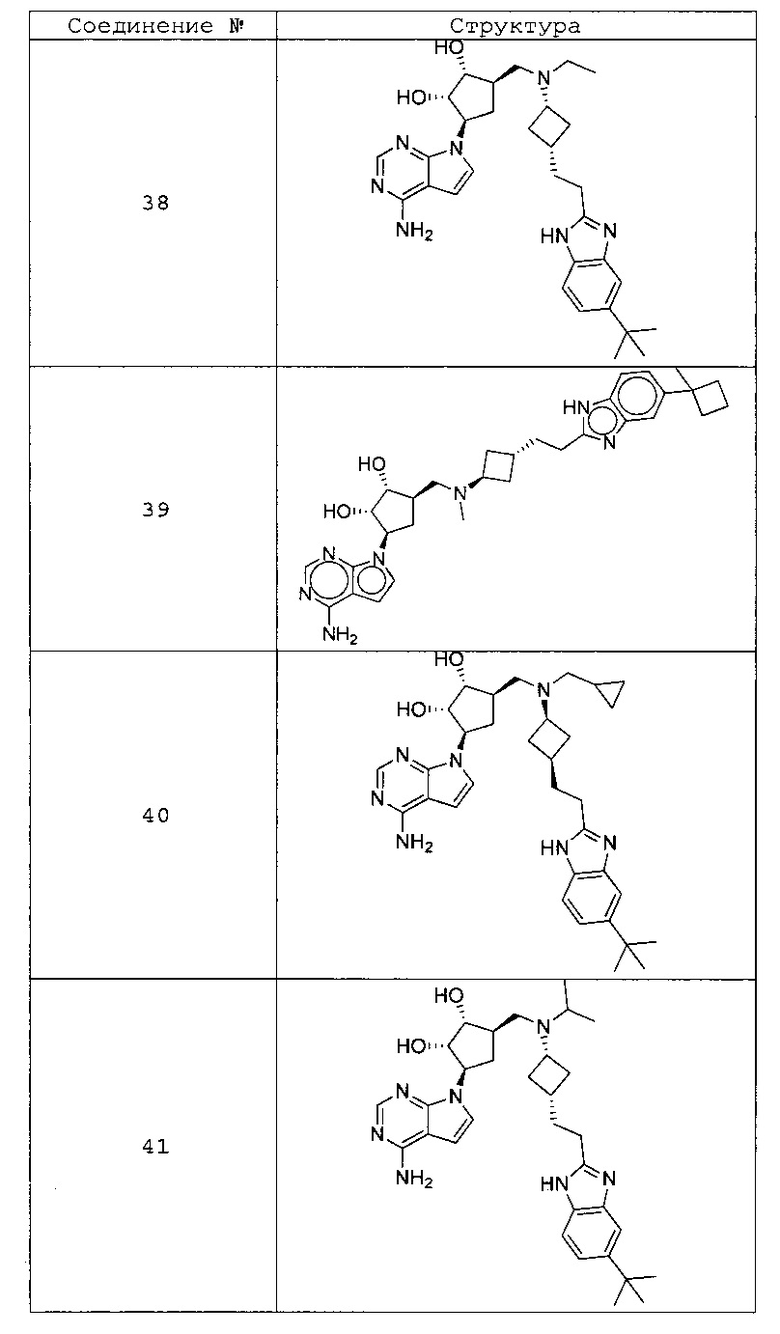
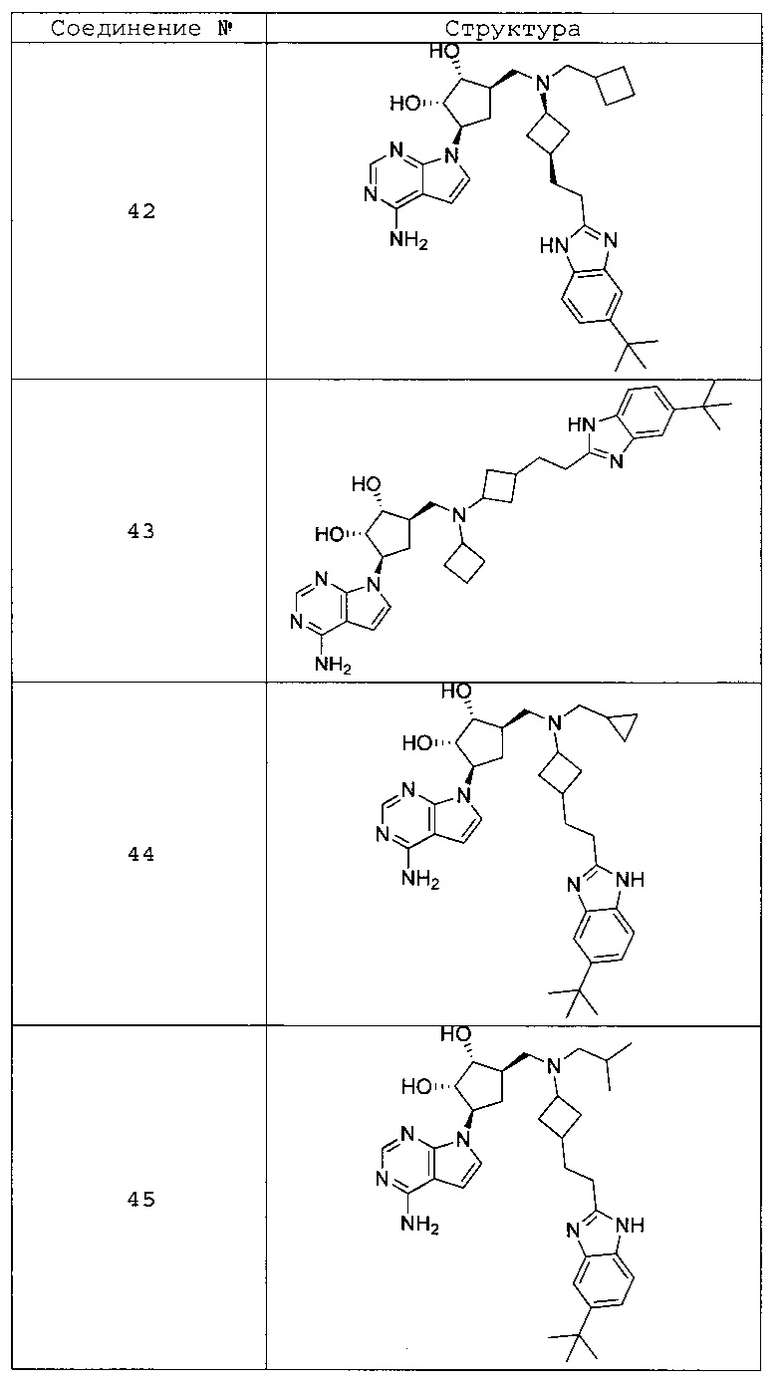
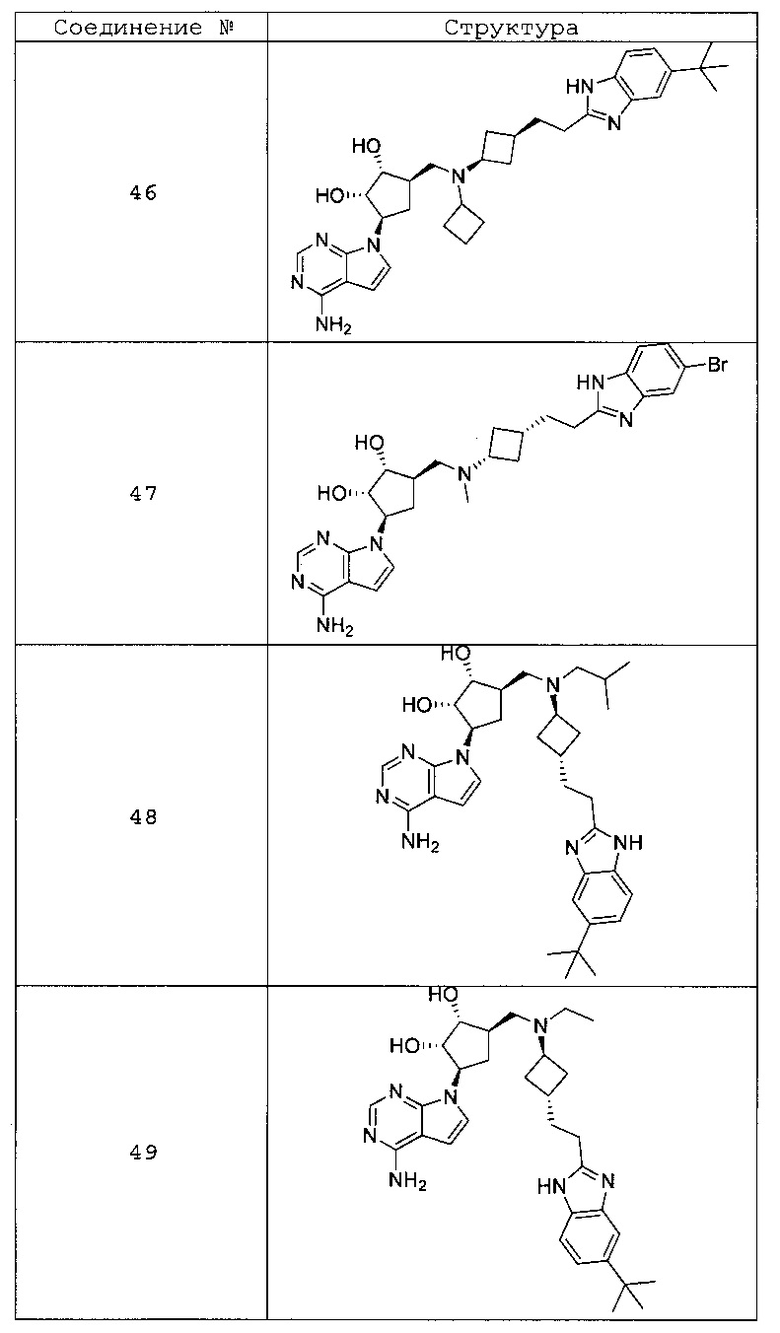
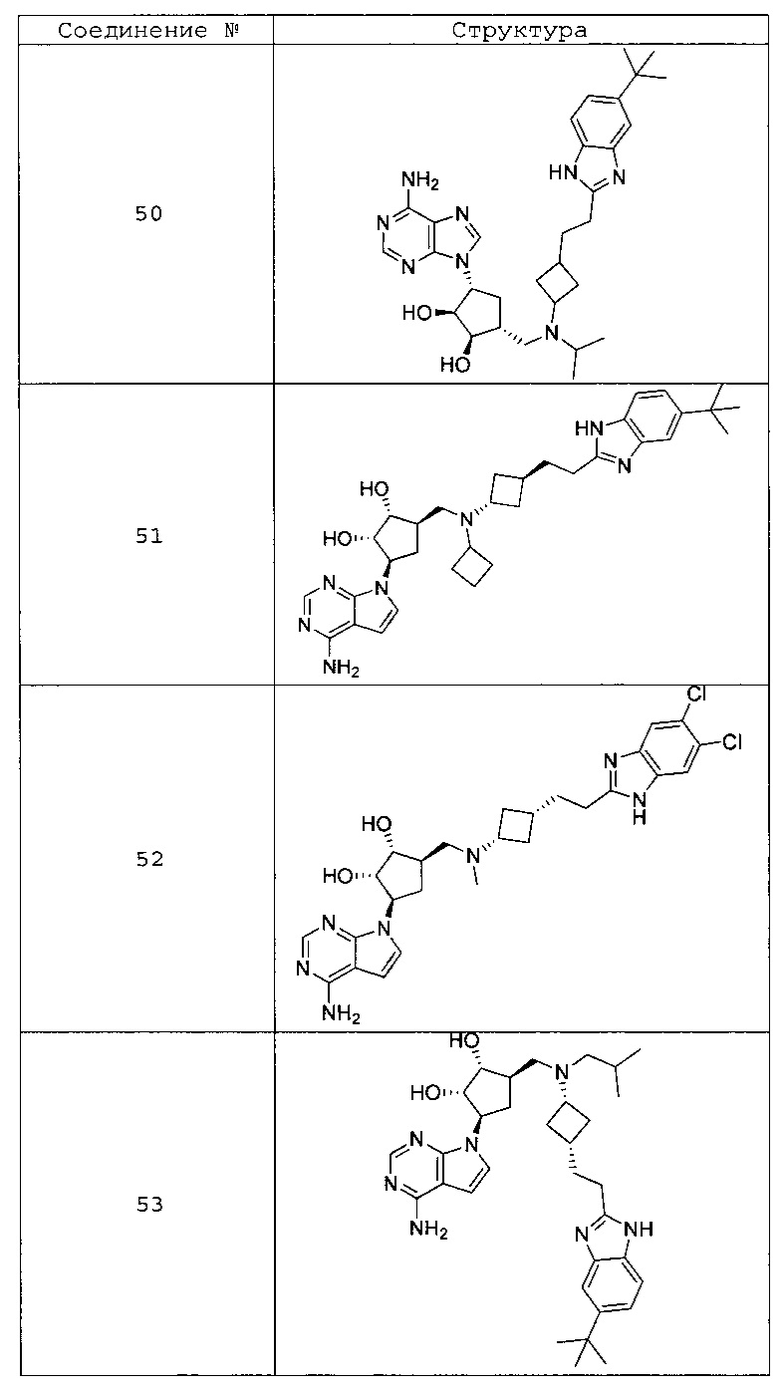
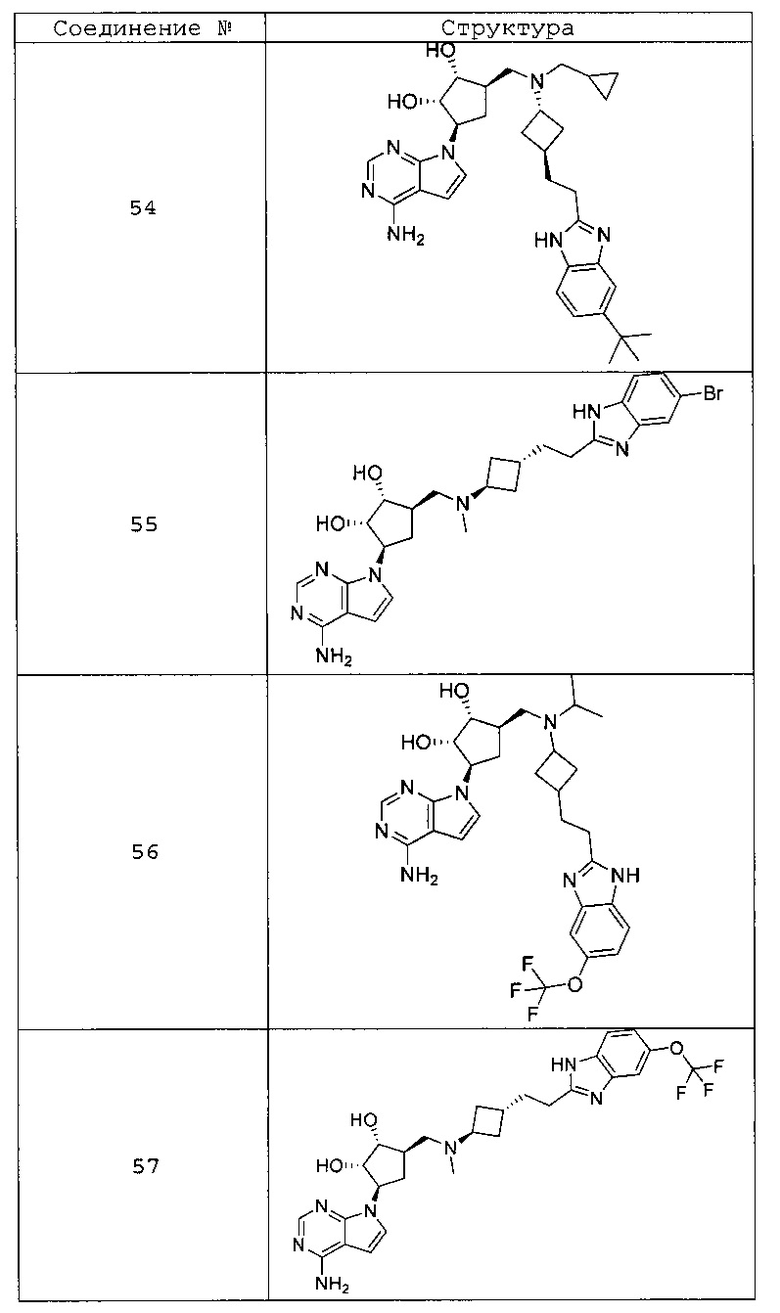
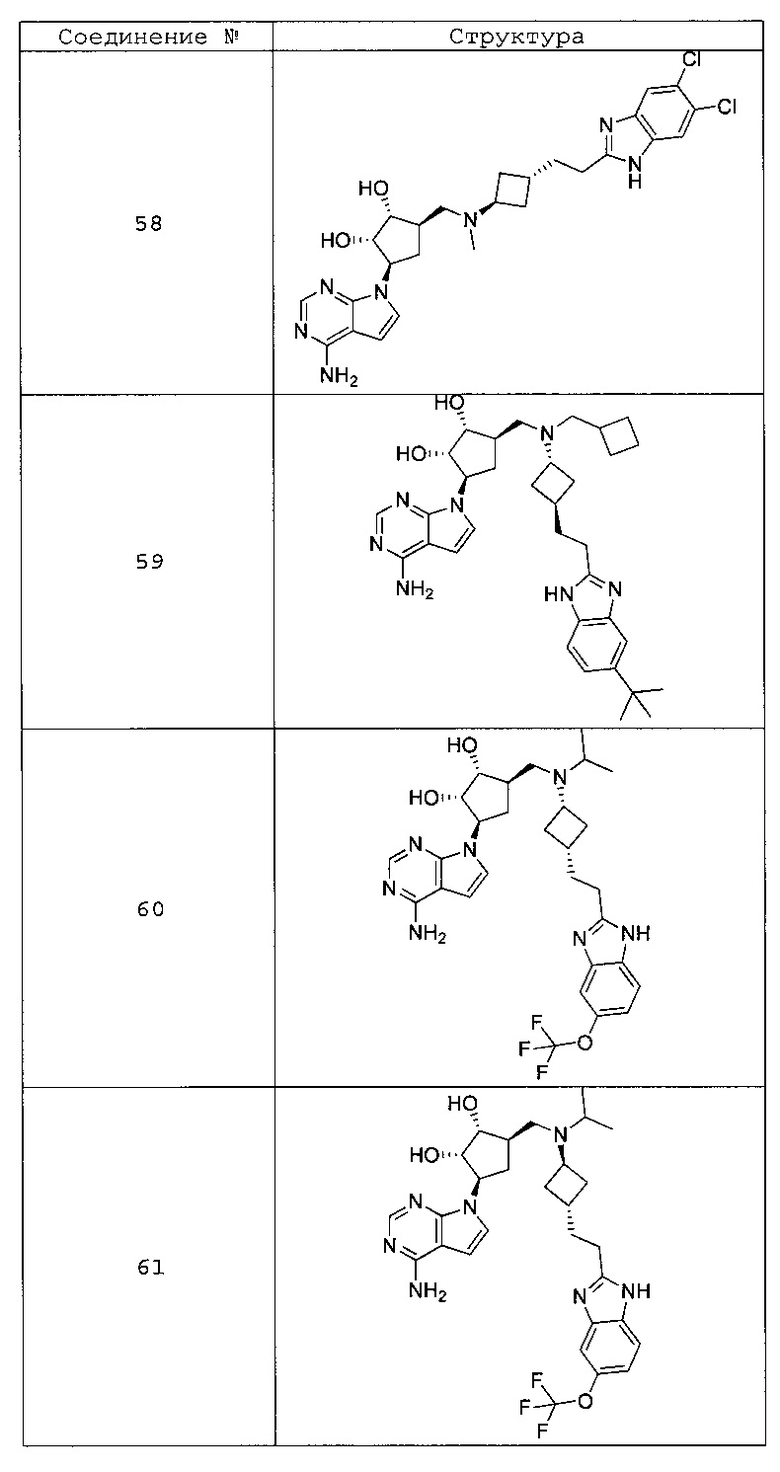
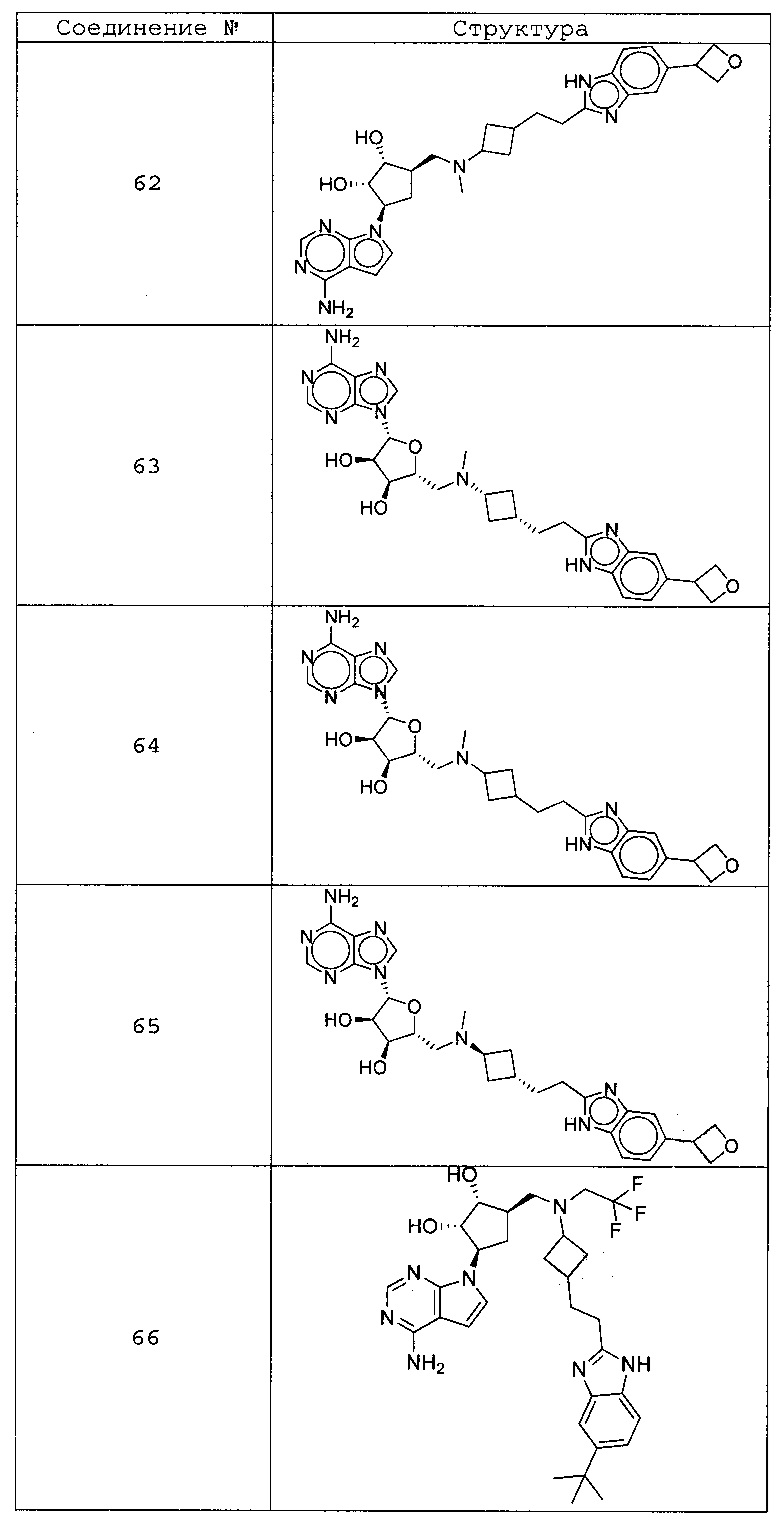
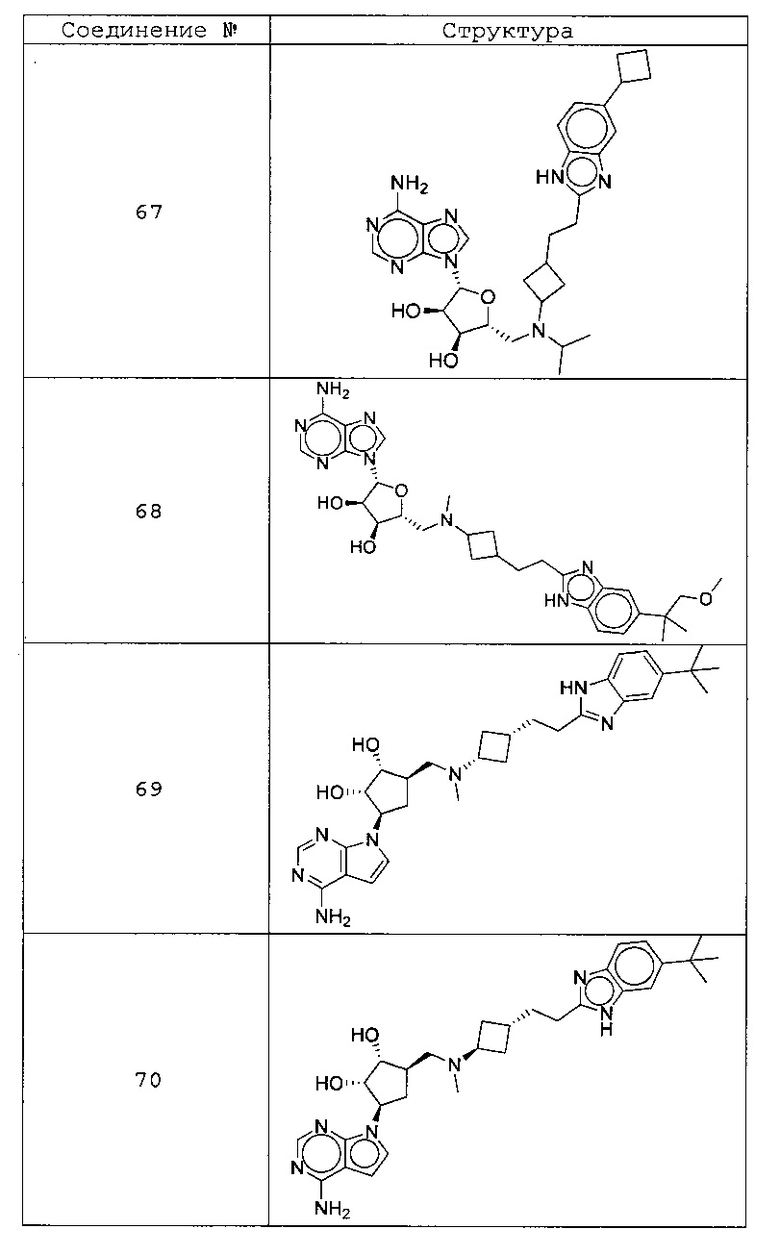
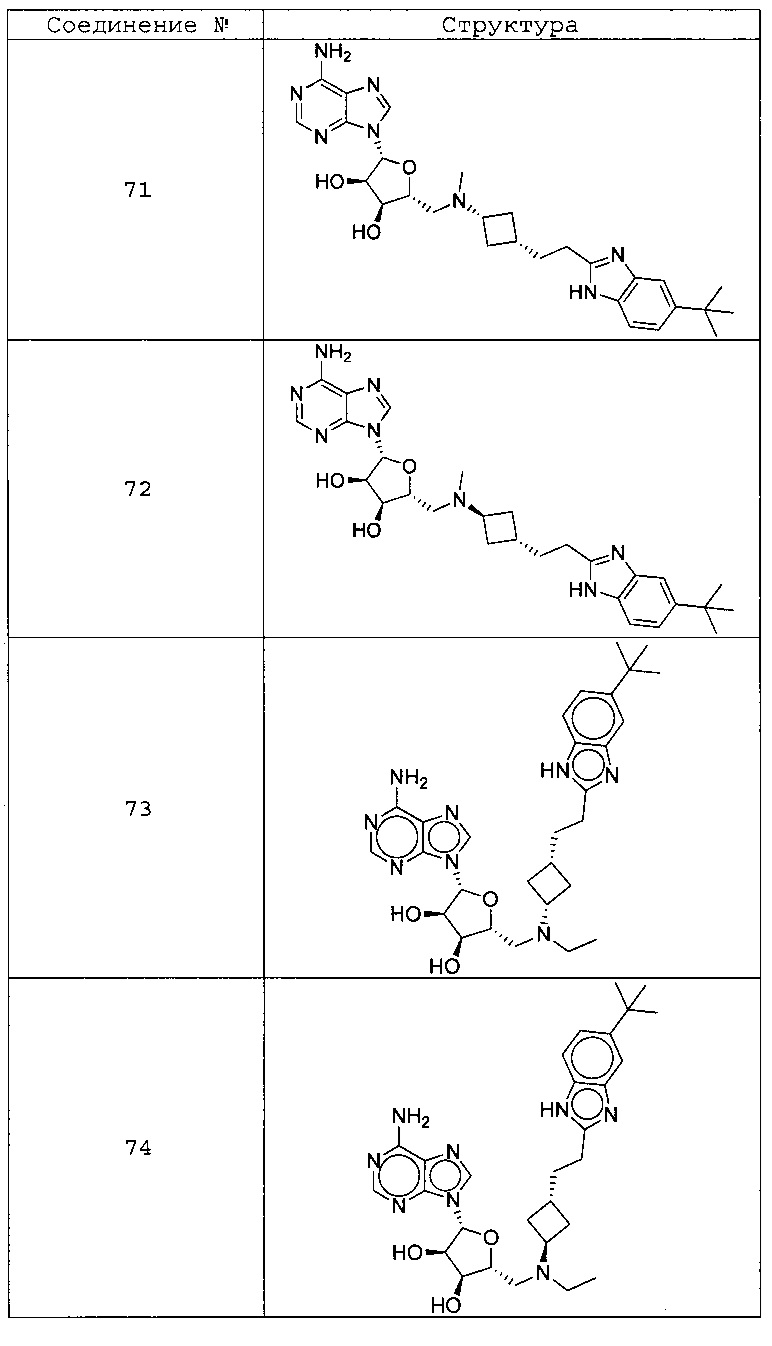
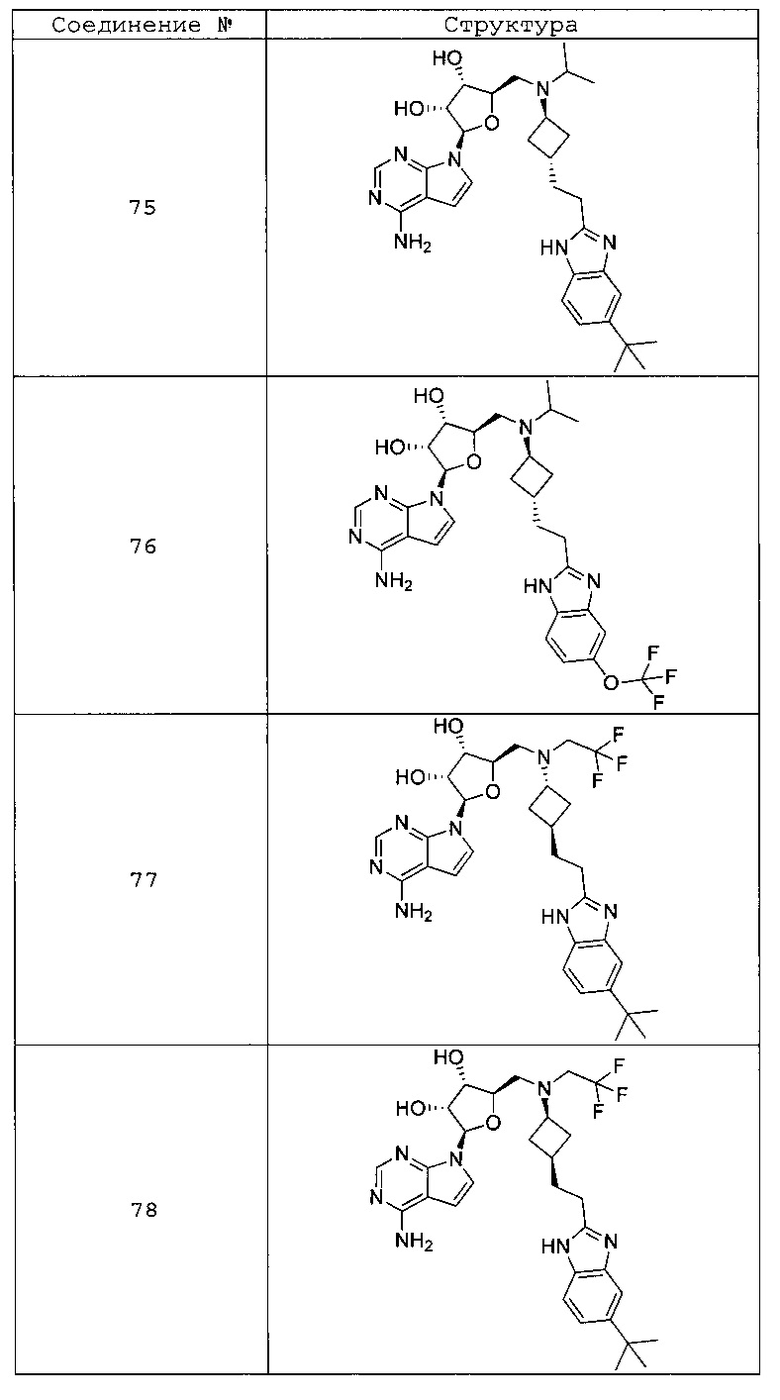
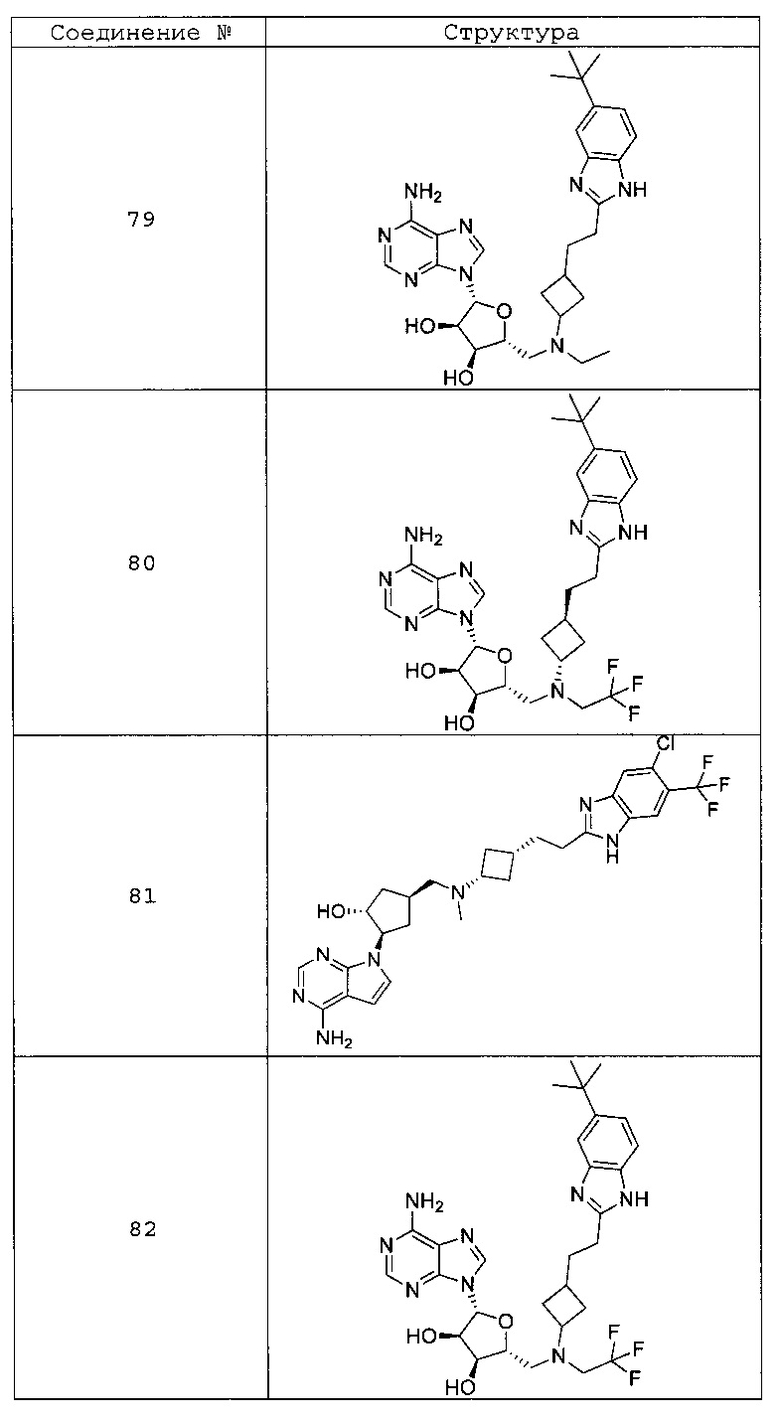
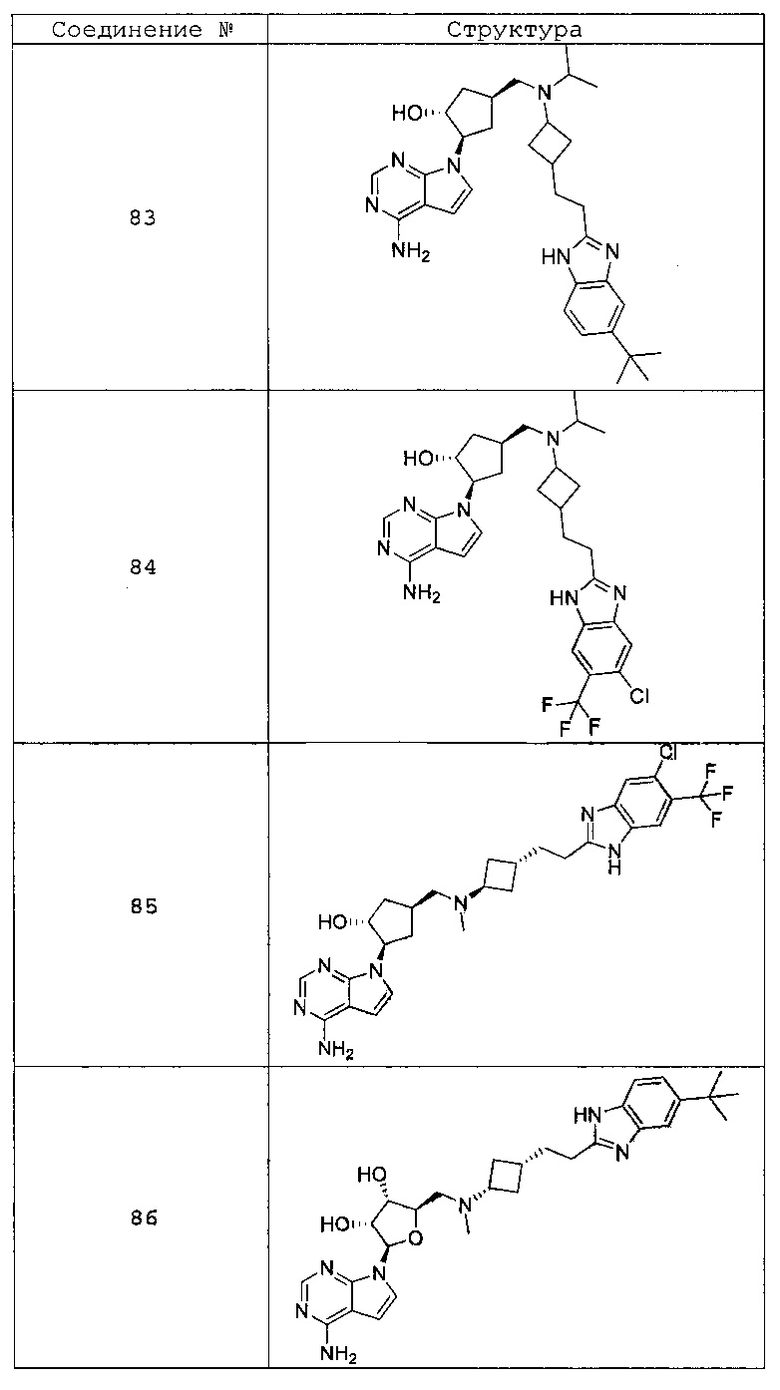
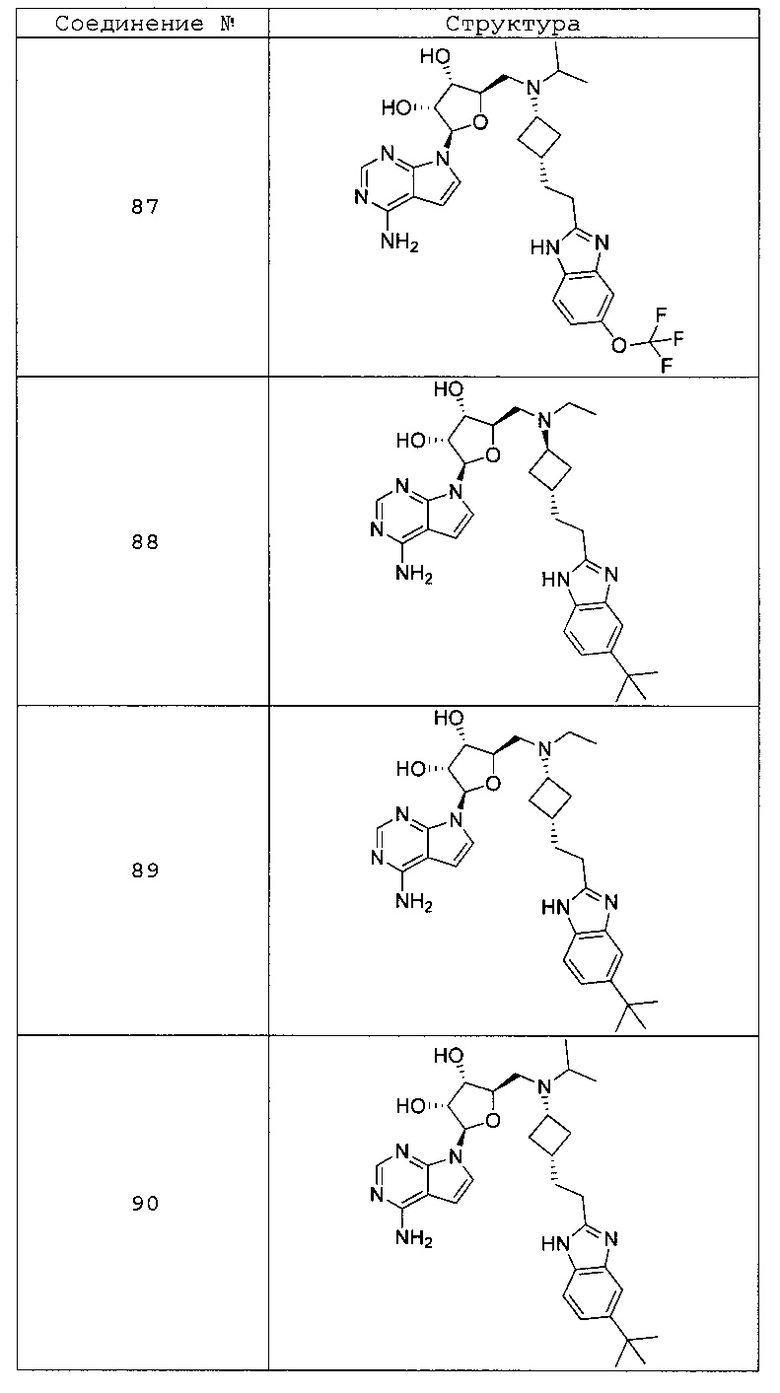
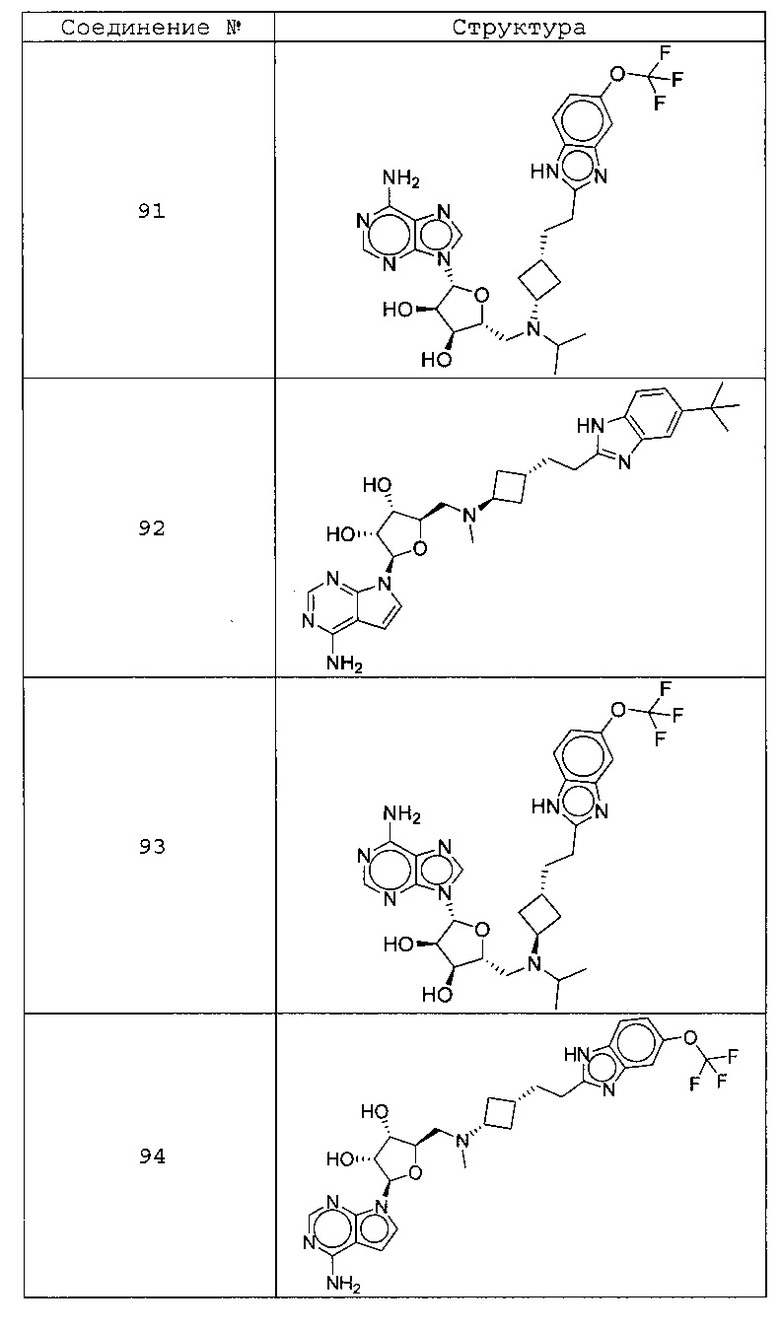
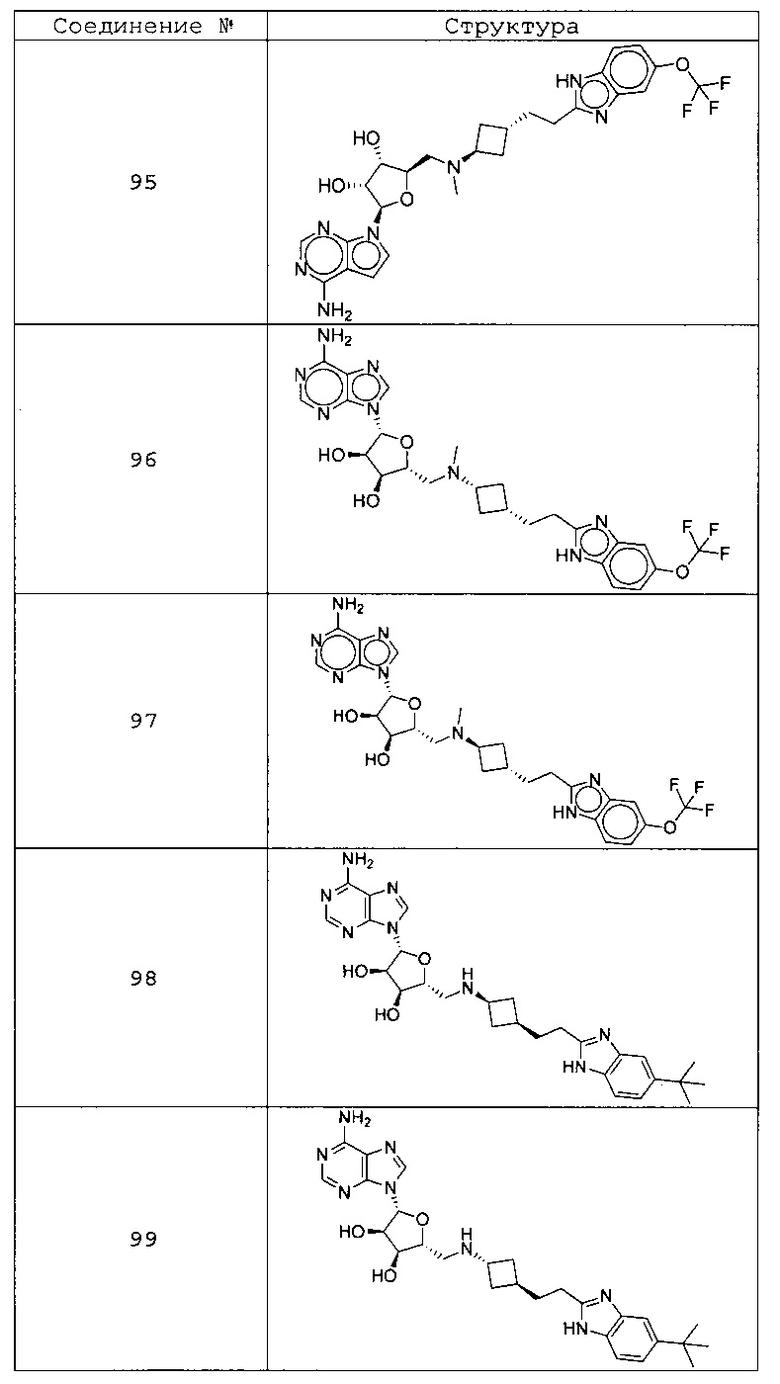
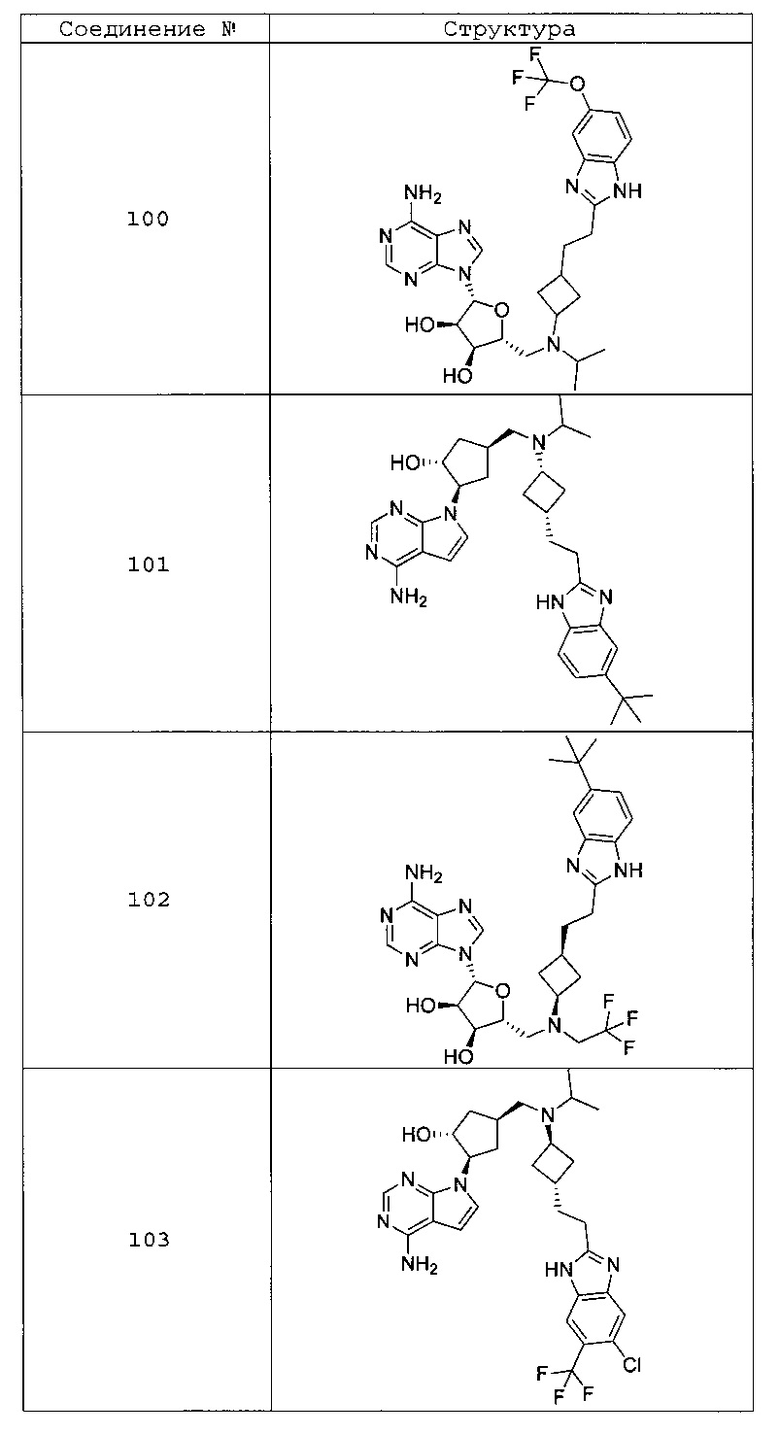
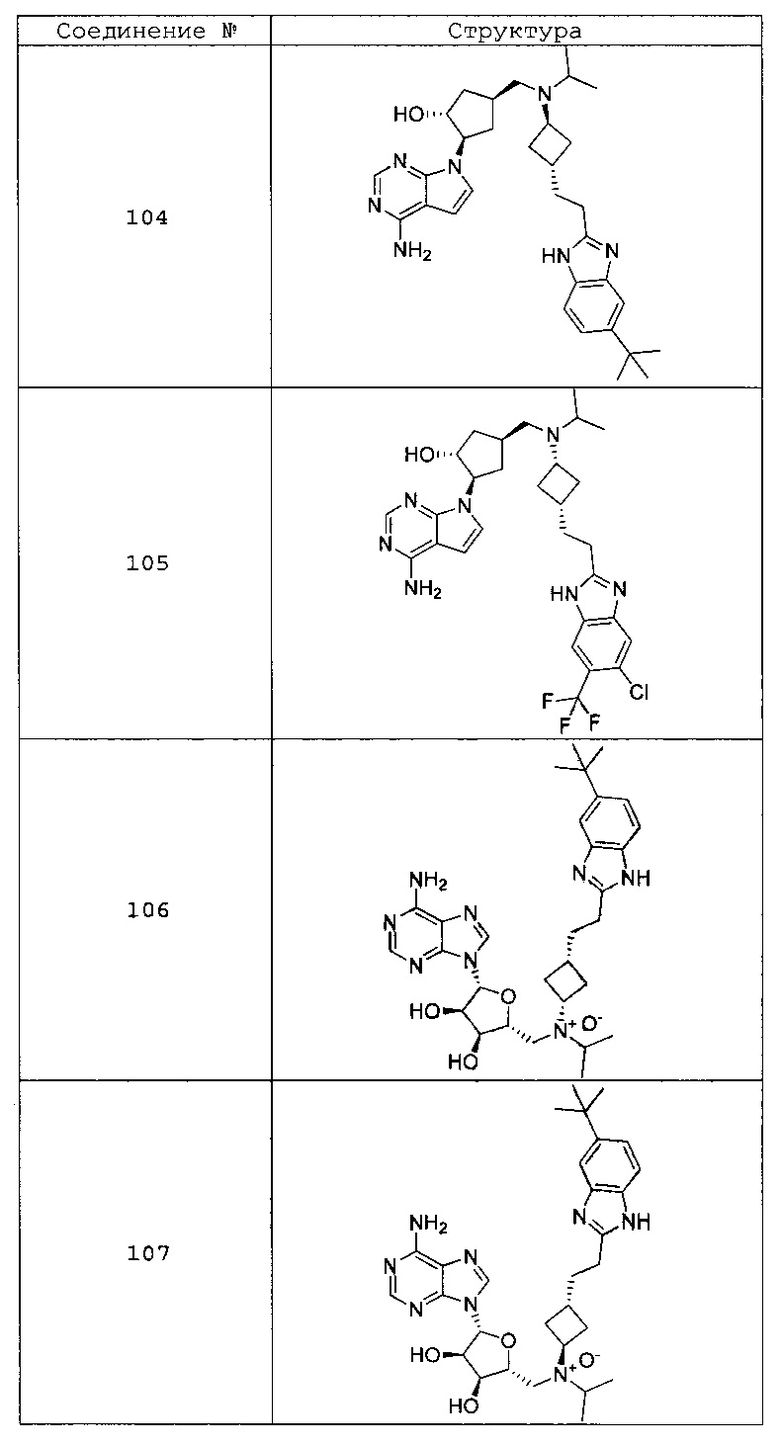
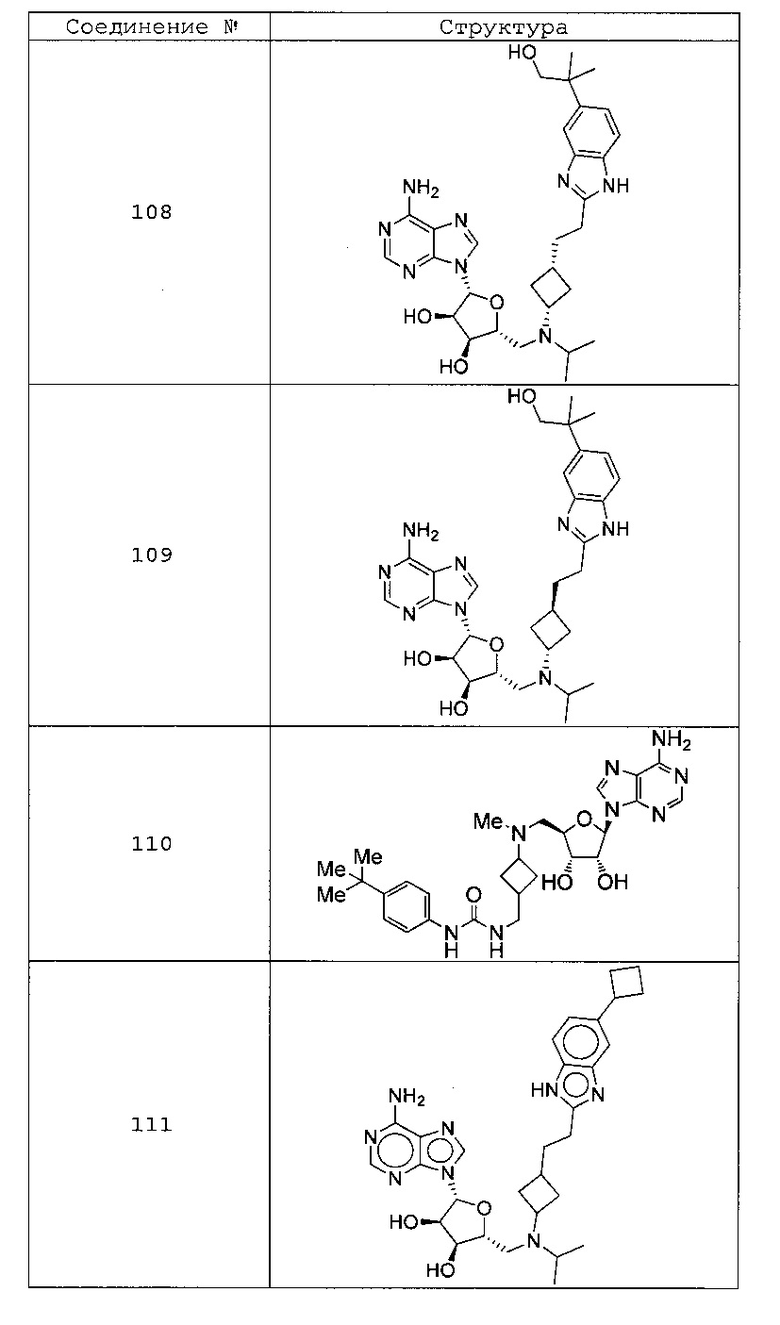
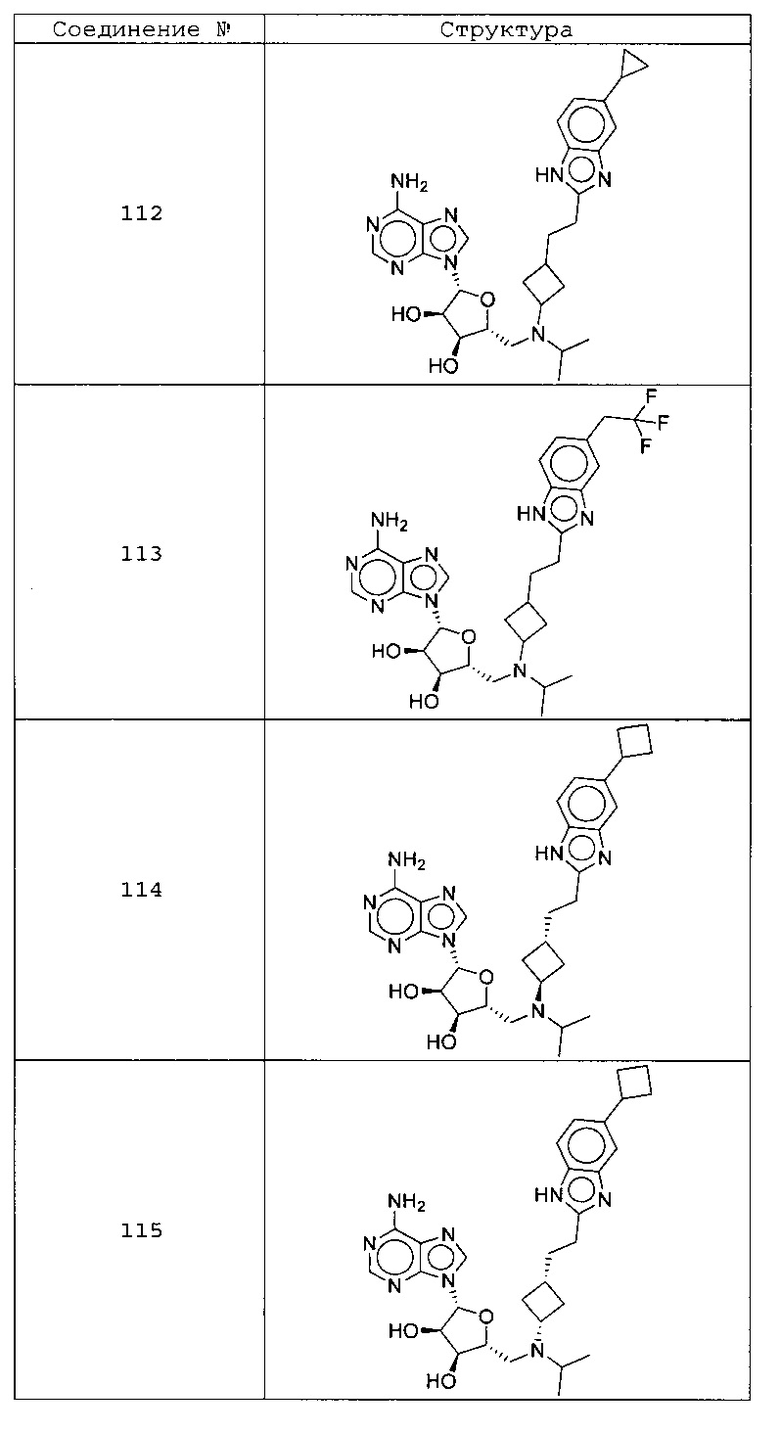
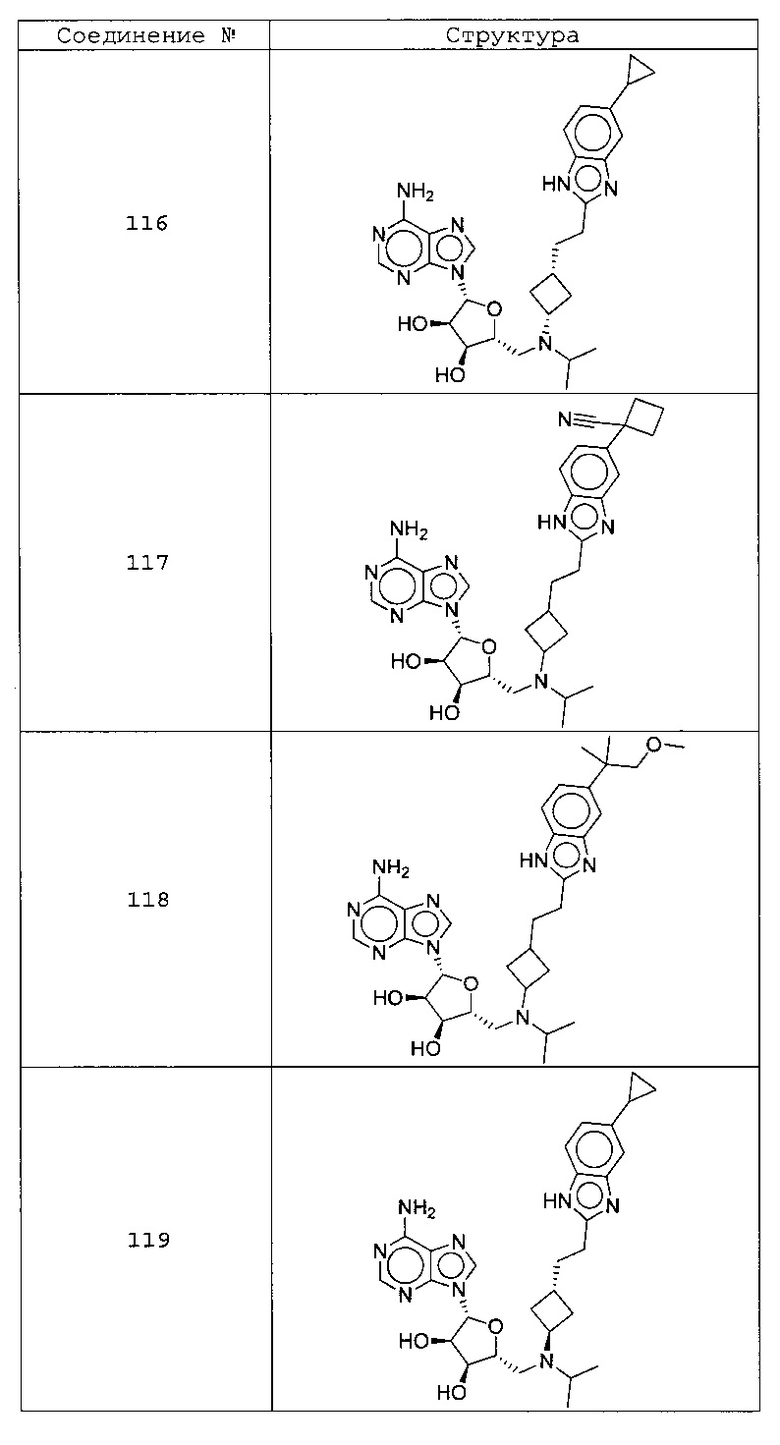
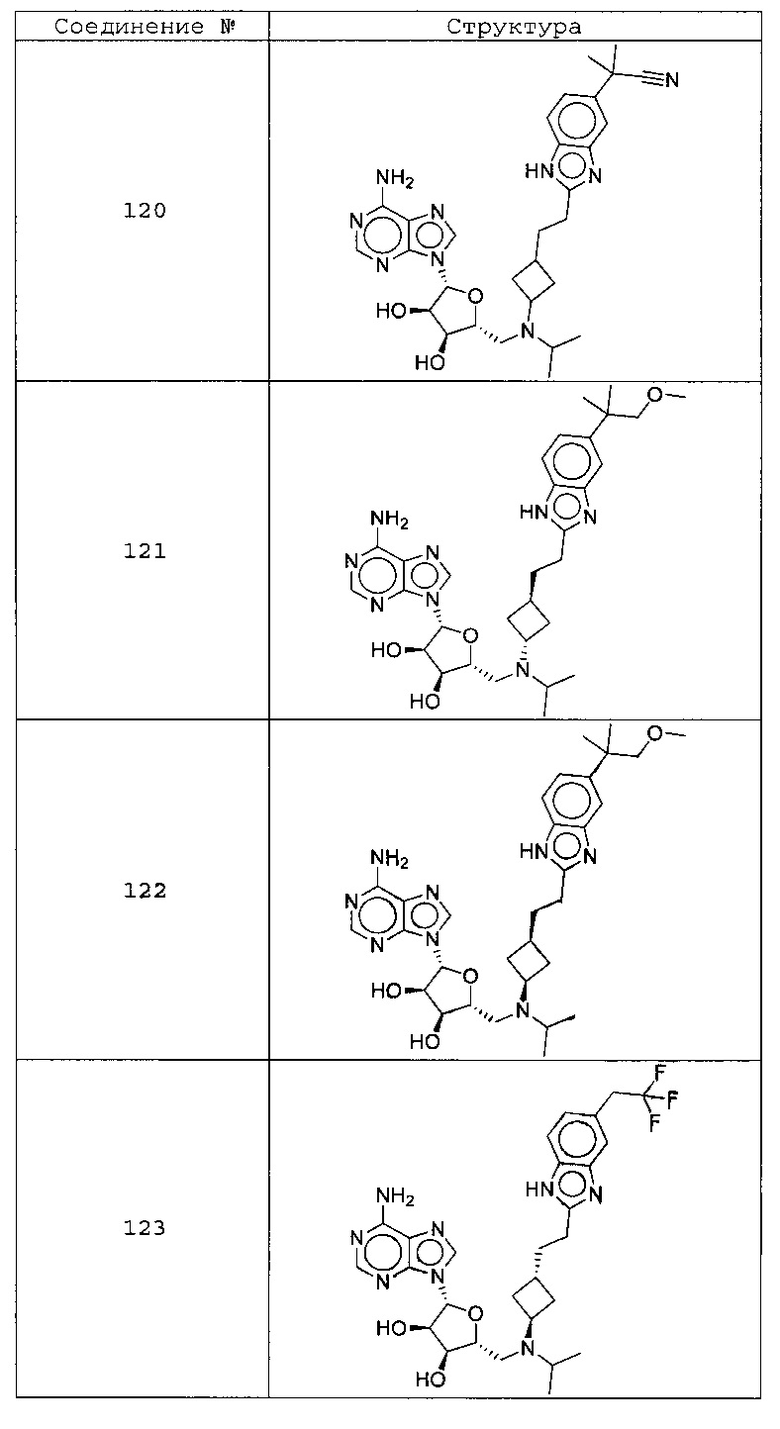
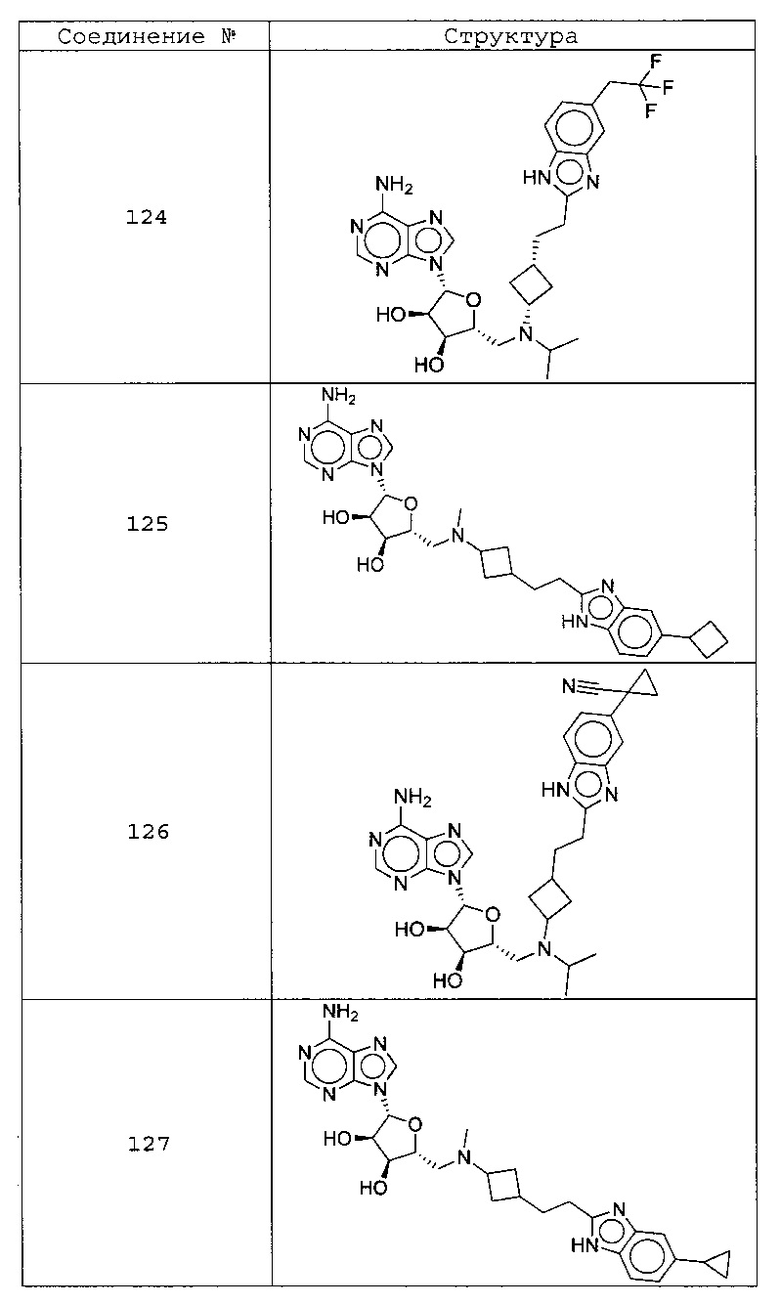
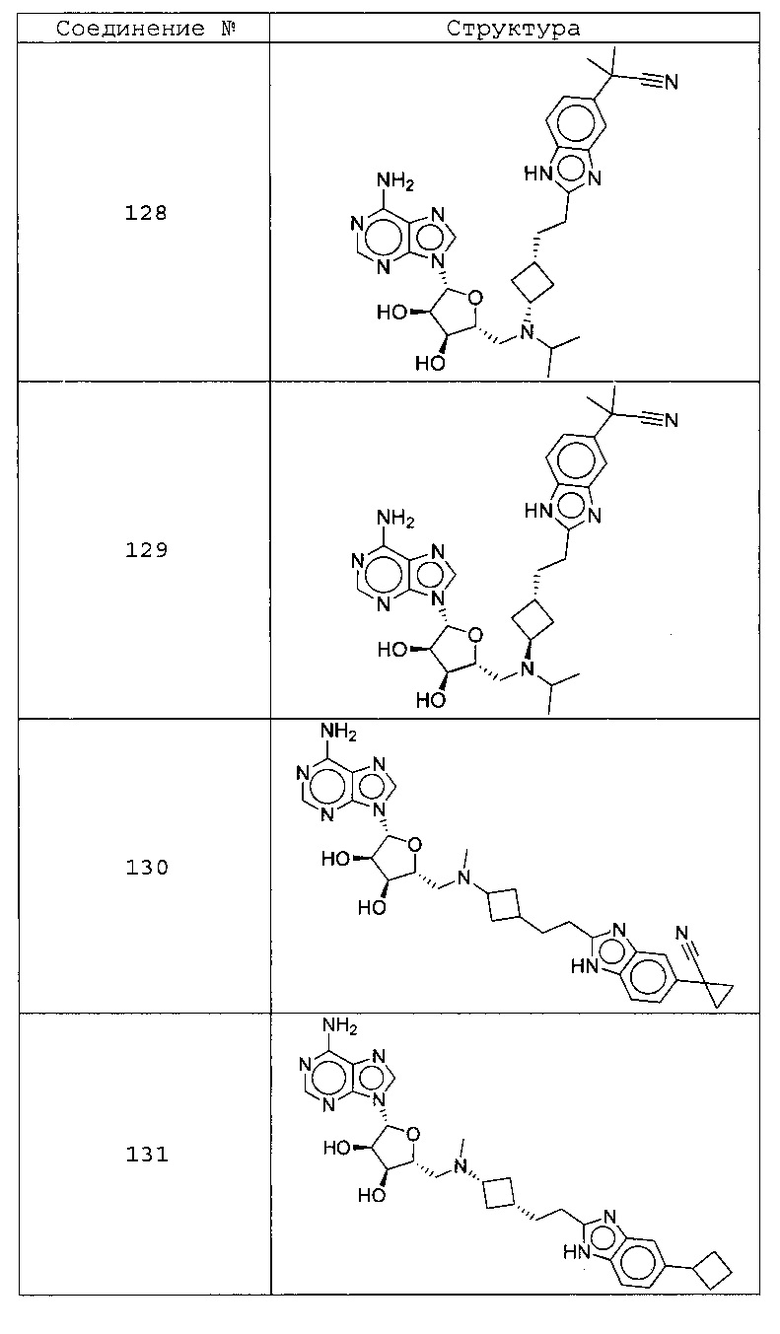
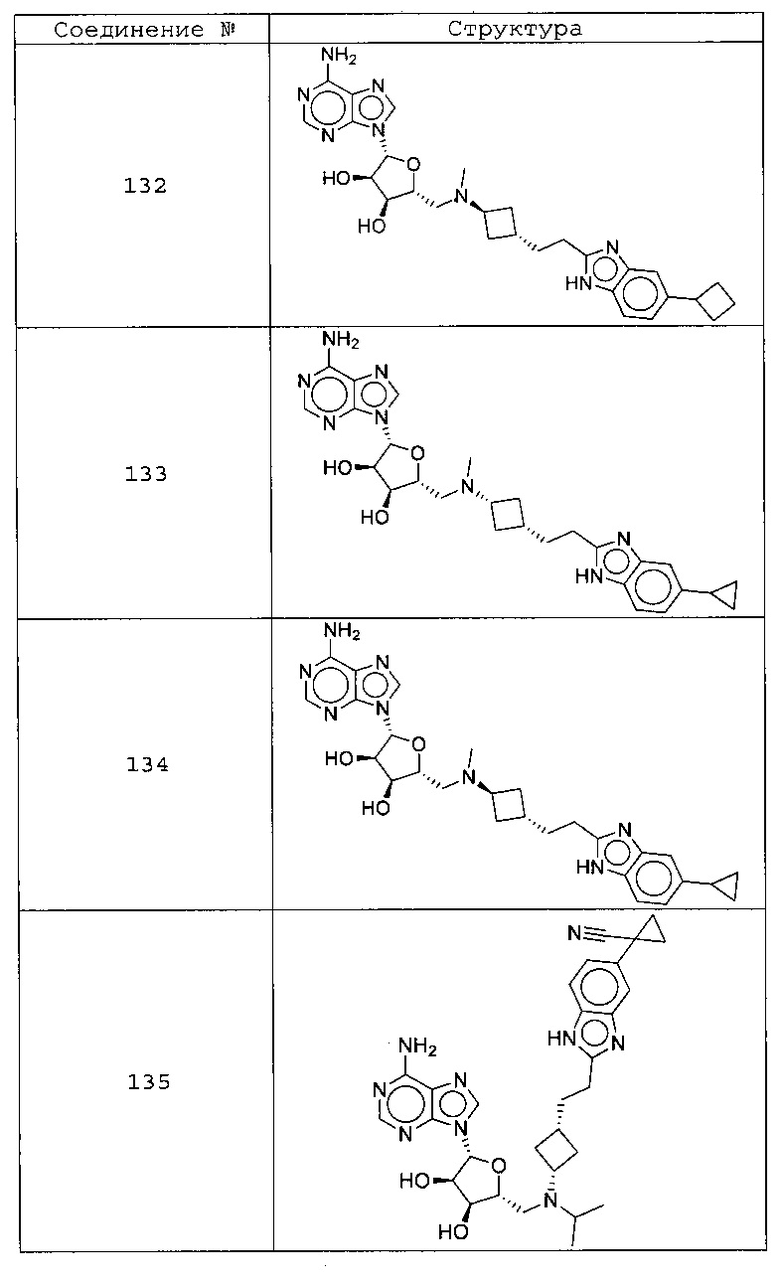
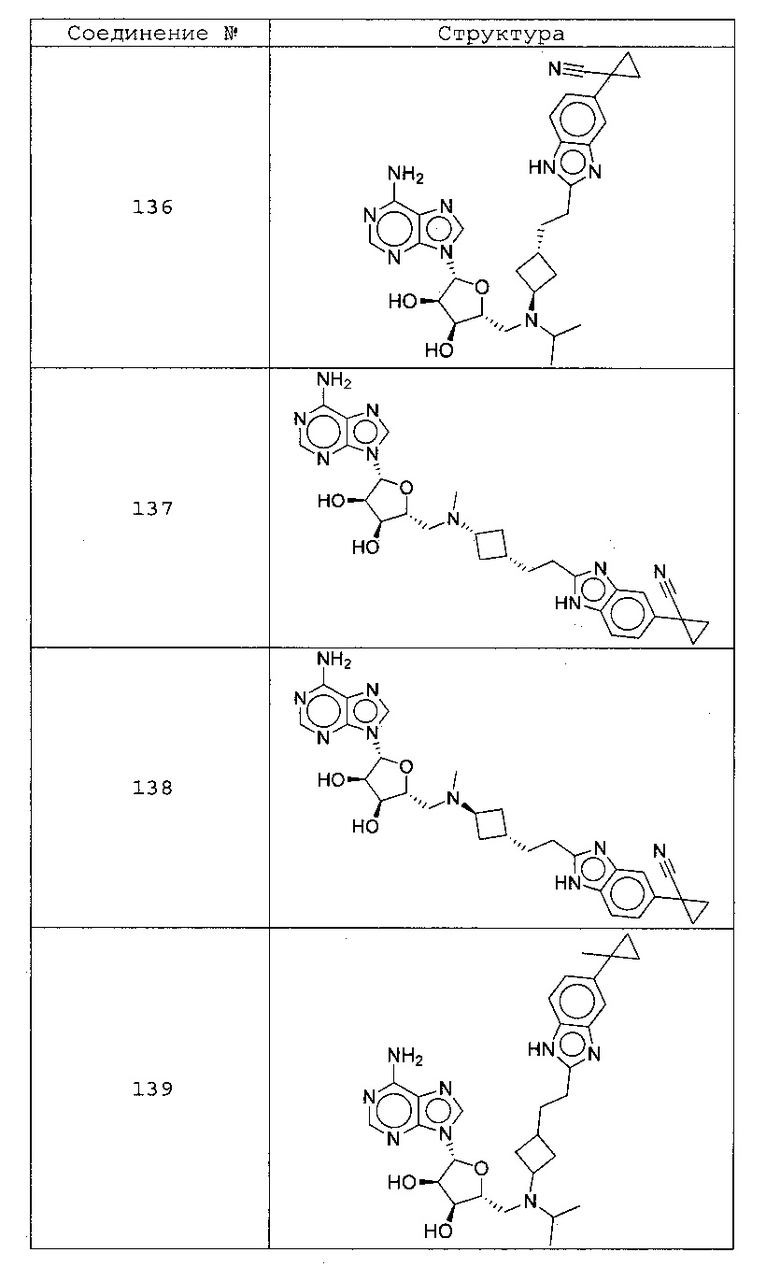
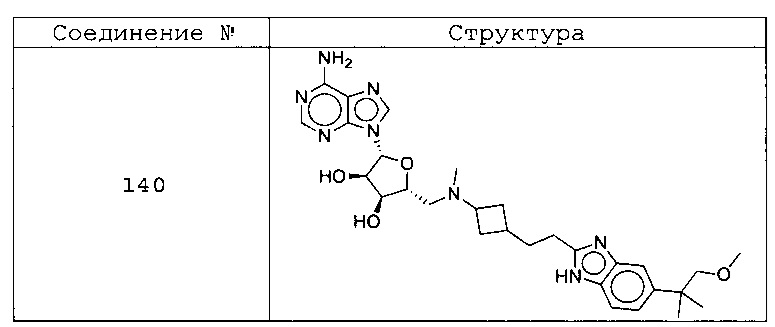
и их фармацевтически приемлемых солей.
12. Соединение по п. 11, где соединение выбрано из
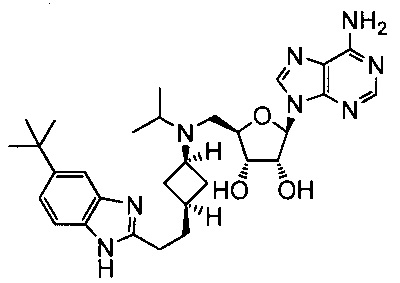 и его фармацевтически приемлемой соли.
и его фармацевтически приемлемой соли.
13. Соединение по п. 12, где соединение представляет собой
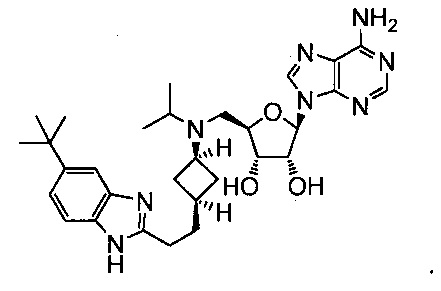
14. Соединение по п. 12, где соединение представляет собой фармацевтически приемлемую соль соединения
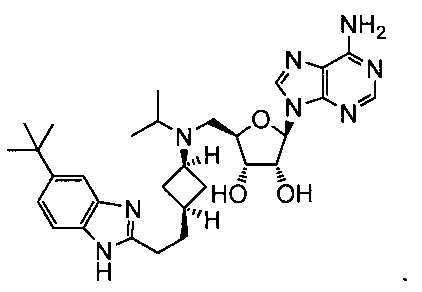
15. Фармацевтическая композиция, обладающая антипролиферативной активностью, включающая терапевтически эффективное количество соединения по любому из пп. 1-10 и фармацевтически приемлемый носитель.
16. Фармацевтическая композиция, обладающая антипролиферативной активностью, включающая терапевтически эффективное количество соединения по п. 11 и фармацевтически приемлемый носитель.
17. Фармацевтическая композиция, обладающая антипролиферативной активностью, включающая терапевтически эффективное количество соединения по п. 12 и фармацевтически приемлемый носитель.
18. Фармацевтическая композиция, обладающая антипролиферативной активностью, включающая терапевтически эффективное количество соединения по п. 13 и фармацевтически приемлемый носитель.
19. Фармацевтическая композиция, обладающая антипролиферативной активностью, включающая терапевтически эффективное количество соединения по п. 14 и фармацевтически приемлемый носитель.
20. Способ лечения лейкоза, включающий введение субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения по любому из пп. 1-14.
21. Способ по п. 20, где лейкоз представляет собой острый миелогенный лейкоз, острый лимфоцитарный лейкоз или лейкоз смешанного происхождения.
22. Способ по п. 20, где лейкоз представляет собой MLL-реаранжированный лейкоз или лейкоз, характеризующийся частичной тандемной дупликацией MLL гена (MLL-PTD).
23. Способ по любому из пп. 20-22, где соединение выбрано из
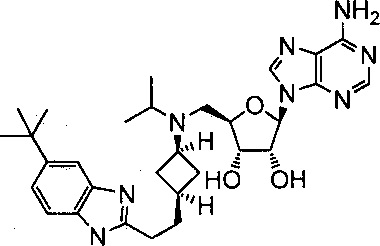 и его фармацевтически приемлемой соли.
и его фармацевтически приемлемой соли.
24. Способ ингибирования DOT1L-опосредованного метилирования белка, включающий введение субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения по любому из пп. 1-14.
| WO 2004022572 А1, 18.03.2004 | |||
| WO 2004007512 A2, 22.01.2004 | |||
| ИНГИБИТОРЫ СОМТ | 2004 |
|
RU2354655C2 |

