Объектом изобретения является способ получения адгезионных клеток, в соответствии с которым адгезионные клетки вводят в культуральный сосуд, который содержит микроносители в культуральной среде, и несколько последовательных пассажей клеток проводят в одном сосуде, причем каждый раз используя всю или часть клеточной популяции предшествующего пассажа клеток для проведения следующего пассажа клеток. Изобретение также относится к применению этого способа для получения биологических агентов, в частности, для изготовления вакцин или лекарственных соединений.
В 1980-х годах развитие технологии культивирования клеток на микроносителях упростило крупномасштабное получение адгезионных клеток и, соответственно, получение биологических агентов. Получение адгезионных клеток, предназначенных для получения биологических агентов для фармацевтического применения, тем не менее должно соответствовать некоторому числу нормативных требований, в частности: запрету использования адгезионных клеток после некоторого числа «пассажей клеток» из-за существования риска морфологической и/или биологической трансформации клеток. Это, в частности, относится к случаю клеток линии Vero.
В US 4664912 описан способ, используемый в промышленном масштабе для получения партии адгезионных клеток из клеточной затравки, имеющей свое происхождение из рабочего банка клеток. Он основан на последовательности пассажей клеток, которые каждый раз проходят в различных биореакторах, рабочие объемы которых увеличиваются на протяжении последовательных пассажей клеток. Это дает возможность каждый раз увеличивать количество микроносителей, одновременно сохраняя оптимальную концентрацию микроносителей в культуральной среде, которая обычно составляет от 1 до 5 г/л. Клеточная биомасса поэтому увеличивается на протяжении последовательных пассажей клеток до тех, пор пока не будет достигнута желаемая промышленная партия клеток. Перенос клеток из одного биореактора в другой биореактор проводят после открепления адгезионных клеток от микроносителей посредством обработки трипсином и затем блокирования действия фермента внесением сывороточных белков или сыворотки в среду для того, чтобы сохранить в максимальной степени целостность клеток. Полученную суспензию клеток затем переносят (в присутствии или отсутствии используемых микроносителей) в больший биореактор, который содержит большее число новых микроносителей. Однако этот способ промышленного получения адгезионных клеток требует применения и работы с большим количеством материала, что влияет на стоимость получения биологических объектов.
Для снижения стоимости получения адгезионных клеток, предназначенных для получения биологических объектов, в EP 1060241 предложен более быстрый способ получения, в котором больше не требуется начинать каждый раз с клеточной затравки из рабочего банка при необходимости получения промышленной партии клеток. Способ состоит из переноса после каждого пассажа клеток большинства клеток (от 80 до 90% клеточной биомассы) в один или несколько других биореакторов для продолжения амплификации клеточной биомассы и для промышленного получения партии клеток, в то время как оставшиеся 10-20% клеток сохраняют для поддержания запаса «питающих клеток», из которого можно получить дополнительные партии клеток. Тем не менее, этот способ имеет следующие недостатки:
- полученные партии клеток проявляют некоторую гетерогенность в том, что не все они имеют одинаковое число пассажей клеток;
- поддержание запаса «питающих» клеток в культуре во время каждой операции переноса неизбежно приводит к «старению» клеток, которое прямо связано с числом проведенных пассажей клеток, и поэтому может использоваться только в течение ограниченного периода времени для уже указанных нормативных причин.
Для того чтобы освободиться от использования протеолитических ферментов, таких как трипсин, который опасен для целостности клеток, в Ohlson et al., Cytotechnology (1994), vol. 14, pages 67-80, описана технология переноса адгезионных клеток с микроносителей на микроносители (перенос с шариков на шарики) без ферментативной обработки. Близкий контакт между микроносителями, покрытыми адгезионными клетками и не покрытыми микроносителями, обеспечивает перенос клеток на непокрытые микроносители, на которых клетки можно амплифицировать. Поэтому, для усиления клеточного роста непокрытые микроносители добавляют к культуральной среде, которая содержит микроносители, покрытые адгезионными клетками, и культуральную среду периодически перемешивают для обеспечения контакта между микроносителями. Тем не менее, получаемая клеточная популяция является «несинхронизированной», поскольку клетки находятся на разных стадиях клеточного цикла, что может быть главным недостатком для получения биологических агентов.
Все еще существует необходимость оптимизации способов крупномасштабного получения адгезионных клеток, а также получения биологических агентов из них, таким образом, чтобы снизить стоимость получения.
Для достижения этого объектом настоящего изобретения является:
способ получения адгезионных клеток, в соответствии с которым:
а. адгезионные клетки вносят в культуральный сосуд, который содержит микроносители в культуральной среде;
b. клетки амплифицируют, проведя последовательные пассажи клеток в этом одном культуральном сосуде, причем каждый пассаж клеток, следующий за первым пассажем клеток, проводят:
i. используя всю или часть клеточной популяции, которая получена в течение предшествующего пассажа клеток, после проведения ферментативной обработки клеточной популяции для открепления клеток от микроносителей; и
ii. внося культуральную среду и увеличивая количество микроносителей; и
c. клеточную популяцию, полученную в течение финального пассажа клеток, собирают после необязательного проведения ферментативной обработки указанной клеточной популяции для открепления клеток от микроносителей.
Объектом настоящего изобретения также является:
способ получения биологического агента, продуцируемого адгезионными клетками, в соответствии с которым:
а. адгезионные клетки вносят в культуральный сосуд, который содержит микроносители в культуральной среде;
b. клетки амплифицируют, проводя нескольких последовательных пассажей клеток в этом одном культуральном сосуде, причем каждый пассаж клеток, следующий за первым пассажем клеток, проводят:
i. используя всю или часть клеточной популяции, которая получена в течение предшествующего пассажа клеток, после проведения ферментативной обработки указанной клеточной популяции для открепления клеток от микроносителей; и
ii. внося культуральную среду и увеличивая количество микроносителей;
c. клеточную популяцию, полученную в течение финального клеточного пассажа, обрабатывают таким образом, чтобы она продуцировала биологический агент, причем указанную обработку проводят в том же культуральном сосуде, который использовали для амплификации клеток; и
d. собирают биологический агент.
В соответствии с одним аспектом процесса получения биологического агента им является инфекционный агент, и обработку клеточной популяции проводят ее заражением, осуществляемым в течение финального пассажа клеток, причем указанным инфекционным агентом является инфекционная среда.
В соответствии с одним конкретным аспектом изобретения инфекционным агентом является вирус бешенства, а инфекционной средой является вирусная инфекционная среда, свободная от любого продукта животного происхождения.
В общем, число пассажей клеток, проводимых в одном культуральном сосуде, составляет 2, 3 или 4.
Концентрация микроносителей в культуральной среде в течение первого пассажа клеток составляет обычно <1 г/л и предпочтительно ≤0,5 г/л.
Очень предпочтительно, чтобы для ферментативной обработки использовали раствор, содержащий протеолитический фермент, такой как трипсин.
В соответствии с другим аспектом способа по изобретению каждый пассаж клеток, следующий за первым пассажем клеток, проводят, увеличивая объем культуральной среды.
Предпочтительно, чтобы первый пассаж клеток проводили в объеме культуральной среды, который составляет от 1/5 до половины рабочего объема культурального сосуда.
В другом варианте осуществления способа по изобретению культуральная среда свободна от сыворотки животного происхождения.
Предпочтительно, чтобы культуральная среда была свободна от продуктов животного происхождения.
В соответствии с другим аспектом концентрация белка в культуральной среде составляет ≤15 мг/л.
По еще одному аспекту культуральная среда также содержит клеточный протекторный агент.
Предпочтительно, чтобы клеточным протекторным агентом был поливинилпирролидон или полоксамер.
В соответствии с еще одним вариантом осуществления способа по изобретению культуральный сосуд представляет собой биореактор, который имеет рабочий объем между 3 и 3000 литрами, предпочтительно между 20 и 1000 литрами и особенно предпочтительно между 20 и 500 литрами.
В еще одном варианте осуществления изобретения культуральным сосудом является биореактор для однократного применения.
В одном конкретном аспекте способа по изобретению адгезионными клетками являются клетки Vero.
В общем, собираемая клеточная популяция содержит по меньшей мере 60-кратное количество клеток, исходно внесенных в культуральный сосуд.
В другом аспекте изобретение относится к способу получения адгезионных клеток, в соответствии с которым:
а. размораживают запас адгезионных клеток, затем
b. с размороженными клетками проводят один из вариантов осуществления способа по изобретению.
В еще одном аспекте изобретение относится к способу получения адгезионных клеток, в соответствии с которым после получения адгезионных клеток в первом культуральном сосуде в соответствии со способом по изобретению:
а. клеточную популяцию, собранную после ферментативной обработки указанной клеточной популяции для открепления клеток от микроносителей, переносят во второй культуральный сосуд, который имеет больший рабочий объем и который содержит культуральную среду, содержащую большее количество микроносителей относительно количества микроносителей, присутствующего в течение последнего пассажа клеток, проведенного в первом культуральном сосуде, и
b. один из вариантов осуществления способа по изобретению выполняют в этом втором сосуде.
В одном конкретном аспекте один из вариантов осуществления способа по изобретению повторяют в третьем культуральном сосуде, который имеет больший рабочий объем, чем рабочий объем второго культурального сосуда.
Изобретение также относится к использованию клеток, которые были получены в соответствии со способом по изобретению, для получения биологических агентов.
В итоге, изобретение относится к способу получения адгезионных клеток в культуральном сосуде, который содержит микроносители в культуральной среде, в соответствии с которым количество получаемых клеток увеличивается с кратностью ≥ 60 в результате проведения последовательных пассажей клеток в одном и том же культуральном сосуде.
Подробное описание изобретения
Изобретение относится к способу получения адгезионных клеток, в соответствии с которым для амплификации клеток и для получения промышленных партий клеток проводят несколько последовательных пассажей клеток в одном и том же клеточном культуральном сосуде. Посредством этого способа уменьшается число используемых культуральных сосудов, а получаемые партии клеток являются гомогенными, поскольку они все имеют одинаковое число пассажей клеток. Этот способ также служит для получения биологических агентов.
В рамках настоящего изобретения «пассаж клеток» начинается, когда суспензию адгезионных клеток вносят в контакт с микроносителями в культуральной среде, и обычно заканчивается, когда адгезионные клетки освобождают от микроносителей с помощью ферментативной обработки, и они снова представляют собой суспензию в культуральной среде. Пассаж клеток обычно включает в себя следующие фазы:
- фазу колонизации микроносителей, которая соответствует периоду времени, в течение которого клетки, которые были приведены в контакт с микроносителями в культуральной среде, прикрепляются к микроносителям;
- фазу амплификации клеток, прикрепленных к микроносителям, которая соответствует периоду времени, в течение которого клетки умножаются на микроносителях до тех пор, пока доступная поверхность колонизированных микроносителей более чем на 70% и предпочтительно более чем на 80% покрыта клетками. Когда более 70% доступной поверхности колонизированных микроносителей покрыто клетками, считается, что адгезионные клетки являются «по существу конфлюэнтными» или достигли «стадии конфлюэнтности»; и
- фазу открепления по существу конфлюэнтных клеток от микроносителей посредством ферментативной обработки, так что максимум клеток открепляется от своей подложки (в общем, более 80% и предпочтительно более 90%) в течение короткого интервала времени (в общем, менее 30 минут и часто в течение периода времени меньше 20 минут). Клеточная популяция затем представляет собой по существу суспензию клеток, высвобожденных из своих микроносителей (или открепленных от своих микроносителей).
В случае настоящего изобретения в зависимости от использования получаемых адгезионных клеток финальный пассаж клеток, проводимый в культуральном сосуде, включает в себя или не включает в себя фазу открепления.
В контексте настоящего изобретения последовательные пассажи клеток проводят в дном и том же культуральном сосуде, используя всю или часть клеточной популяции, полученной в течение предшествующего пассажа клеток, для проведения следующего пассажа клеток. Обычно для проведения следующего пассажа клеток используют по меньшей мере 80% клеточной популяции, полученной в течение предшествующего пассажа клеток. Для получения максимального количества клеток предпочтительно проводить пассажи клеток, каждый раз используя всю клеточную популяцию, полученную в течение предшествующего пассажа клеток, для проведения следующего пассажа клеток. Хотя в конце каждого пассажа клетки освобождают (открепляют) от их микроносителей посредством ферментативной обработки, отсутствует рекомендованный в прототипе перенос клеточной биомассы в один или несколько других клеточных культуральных сосудов для продолжения клеточной амплификации. Амплификацию клеточной биомассы проводят в настоящем изобретении в одном и том же культуральном сосуде. Этот способ очень предпочтителен, поскольку аналогичное количество клеток получают за такой же период времени без необходимости использования и работы с несколькими культуральными сосудами, что уменьшает пространство, требуемое для получения промышленного количества клеток, и, соответственно, по существу снижает стоимость производства. Удивительно то, что хотя амплификация клеточной биомассы требует ферментативной обработки на каждом клеточном пассаже, количество клеток, которое получается в конце осуществления способа по изобретению, значительно превышает количество клеток, полученное с использованием стандартной технологии «перенос с микроносителей на микроносители» (смотрите пример 2).
Каждый новый пассаж клеток, который соответствует пассажу клеток, следующему за первым пассажем клеток, проводят в одном культуральном сосуде. Для начала пассажа, следующего за первым пассажем клеток, культуральную среду и объем микроносителей, превышающий количество микроносителей, внесенное в течение предшествующего пассажа клеток, вносят для увеличения доступной поверхности клеточной подложки. Следует понимать, что термин «новый пассаж клеток» или «пассаж, следующий за первым пассажем клеток», означает пассаж клеток, следующий за пассажем клеток, который проводили в культуральном сосуде. Следует также понимать, что внесение или добавление микроносителей в культуральный сосуд соответствует внесению непокрытых микроносителей. Для облегчения прикрепления адгезионных клеток предпочтительно использовать новые микроносители. Хотя обычно объем культуральной среды соответственно увеличивается с каждым пассажем, следующим за первым пассажем клеток, увеличение количества микроносителей обычно пропорционально выше увеличения объема среды, что, в общем, приводит к постепенному увеличению концентрации микроносителей в культуральной среде в течение последовательных пассажей клеток. В течение финального пассажа клеток при использовании, например, в качестве микроносителей декстрановых микрошариков, продаваемых под наименованием CytodexTM (CytodexTM 1, 2 или 3), концентрация микроносителей в культуральной среде обычно составляет от 1 до 7 г/л, но может достигать 10-15 г/л. Способ по изобретению, в соответствии с которым клеточная биомасса увеличивается в результате последовательных пассажей клеток в одном и том же культуральном сосуде, называют «способ все-в-одном» (смотрите фиг.1b).
Обычно в одном культуральном сосуде проводят 2 клеточных пассажа, 3 клеточных пассажа или 4 клеточных пассажа. В зависимости от дальнейшего использования полученного продукта клеточная популяция, которую собирают в конце выполнения способа по изобретению, представляет собой либо суспензию клеток, высвобожденных из своих микроносителей (в этом случае финальный пассаж клеток проводят, включая стадию открепления клеток), либо суспензию клеток, прикрепленных к микроносителям (в этом случае финальный пассаж клеток проводят, исключая стадию открепления клеток). Когда способ получения адгезионных клеток включает в себя два последовательных пассажа клеток, проводимых в одном и том же культуральном сосуде, способ по изобретению означает выполнение следующих стадий:
а. культуральную среду, микроносители и адгезионные клетки вносят в культуральный сосуд,
b. клеткам обеспечивают условия культивирования, которые позволяют им прикрепляться к микроносителям и пролиферировать на них,
с. клетки открепляют от микроносителей посредством ферментативной обработки и часть клеток (необязательно) удаляют из культурального сосуда,
d. культуральную среду и микроносители вносят повторно, так чтобы количество вносимых микроносителей превышало количество ранее внесенных микроносителей,
е. клеткам повторно обеспечивают условия культивирования, которые позволяют им прикрепляться к микроносителям и пролиферировать на них, и
f. полученную популяцию клеток собирают после необязательного открепления клеток от их микроносителей посредством ферментативной обработки, причем стадии (а)-(е) проводят в одном и том же культуральном сосуде.
Стадии (а)-(с) соответствуют первому клеточному пассажу, а стадии (d)-(f) соответствуют второму клеточному пассажу, который заканчивается со сбором клеток.
Когда в одном сосуде проводят более двух последовательных пассажей клеток, это означает повтор стадий (c), (d) и (e) после стадии (е) в одном культуральном сосуде перед выполнением стадии (f) сбора клеток. Обычно стадии (c), (d) и (e) повторяют один раз после стадии (е), что соответствует проведению трех последовательных пассажей клеток, или стадии (c), (d) и (e) повторяют два раза после стадии (е), что соответствует проведению 4 последовательных пассажей клеток. Предпочтительно выполнять стадию (с) (которая соответствует откреплению клеток от их микроносителей), когда клетки по существу являются конфлюэнтными. Обычно объем культуральной среды также увеличивают при каждом добавлении микроносителей (стадия d).
Когда популяцию клеток, собираемую в конце осуществления способа по изобретению, используют для создания запаса клеток, финальный пассаж клеток обычно включает в себя стадию открепления клеток посредством ферментативной обработки, которую обычно проводят в том же культуральном сосуде. Клеточная популяция затем представляет собой по существу суспензию клеток, высвобожденных из своих микроносителей.
Когда популяцию клеток используют для получения биологического агента, финальный пассаж клеток часто проводят, не включая стадию открепления клеток. Популяцию клеток, полученных в форме суспензии клеток, прикрепленных к микроносителям, затем напрямую обрабатывают в том же культуральном сосуде таким образом, что она продуцирует целевой биологический агент. Предполагается, что термин «биологический агент» означает любое вещество или организм, который может продуцироваться адгезионными клетками. В частности, это могут быть вирусы или белки (антитела, антигены, ферменты и т.п.). Когда способ получения биологического агента, продуцируемого адгезионными клетками, включает в себя два последовательных пассажа клеток, проводимых в одном культуральном сосуде, способ по изобретению, таким образом, означает выполнение следующих стадий:
а. культуральную среду, микроносители и адгезионные клетки вносят в культуральный сосуд,
b. клеткам обеспечивают условия культивирования, которые позволяют им прикрепляться к микроносителям и пролиферировать на них,
с. клетки открепляют от микроносителей посредством ферментативной обработки и часть клеток (необязательно) удаляют из культурального сосуда,
d. культуральную среду и микроносители вносят повторно, так чтобы количество вносимых микроносителей превышало количество ранее внесенных микроносителей,
е. клеткам повторно обеспечивают условия культивирования, которые позволяют им прикрепляться к микроносителям и пролиферировать на них,
f. полученную клеточную популяцию обрабатывают таким образом, чтобы она продуцировала биологический агент, и
g. собирают биологический агент, причем стадии (a)-(f) проводят в одном и том же культуральном сосуде.
Когда больше двух последовательных пассажей клеток проводят в одном сосуде, это означает повторение стадий (c), (d) и (e) после стадии (e) в одном культуральном сосуде перед выполнением стадии (f) получения биологического агента. Обычно стадии (c), (d) и (e) повторяют один раз после стадии (е), что соответствует проведению трех последовательных пассажей клеток, или стадии (c), (d) и (e) повторяют дважды после стадии (е), что соответствует проведению 4 последовательных пассажей клеток. Предпочтительно выполнять стадию (с) (которая соответствует откреплению клеток от своих микроносителей), когда клетки по существу конфлюэнтны. Обычно культуральный объем также увеличивают каждый раз при добавлении микроносителей (стадия d).
Когда задачей является получение рекомбинантного белка, такого как, например, цитокин, антитело или вакцинный белок, клеточную суспензию помещают в условия культивирования, которые обеспечивают продукцию этого белка с использованием подходящей среды для получения белка. Например, можно упомянуть среду, описанную в EP 0354129 для получения рекомбинантных белков в клетках СНО и Vero.
Когда биологическим агентом является инфекционный агент, суспензию клеток, прикрепленных к микроносителям, инфицируют внесением инфекционного агента (бактерии, вирусы, паразиты и т.п.) в культуральный сосуд, обычно после замены культуральной среды инфекционной средой. Инфекционным биологическим агентом, в частности, может быть рекомбинантный вирус (рекомбинантные поксвирусы, рекомбинантные аденовирусы) или могут быть вирусы, такие как вирус бешенства, вирус гриппа, полиовирус и т.п. Биологический агент обычно собирают, удаляя культуральный супернатант в одну или несколько стадий - смотрите пример 7. Если биологический агент, наоборот, является внутриклеточным, как в случае нелитических вирусов, часто предпочтительно собрать супернатант и клетки, которые впоследствии обрабатывают литическими агентами.
Используемая для получения биологических агентов среда, в частности, инфекционная среда, которую используют для получения вирусов, таких как вирус бешенства, предпочтительно может быть свободна от сыворотки животного происхождения, от белка животного происхождения или даже от любого продукта животного происхождения.
Микроносители, подходящие для объекта изобретения, обычно имеют форму микрошариков, которые предпочтительно являются непористыми, для того чтобы облегчить действие ферментов. Они имеют диаметр обычно между 90 и 250 мкм. Их плотность немного выше плотности культуральной среды, для того чтобы обеспечить их выделение простым отстаиванием, но одновременно она не должна быть слишком высокой, чтобы получить полное ресуспендирование микрошариков при умеренной интенсивности перемешивания среды. При стандартных условиях культивирования плотность микроносителей обычно находится между 1,020 и 1,050 г/мл. Поверхность микрошариков выбирают так, чтобы облегчить прикрепление шариков. Матрикс микроносителей предпочтительно является нежестким, для того чтобы обеспечить лучшее сохранение клеток при столкновении между шариками. Средняя доступная поверхность для прикрепления клеток обычно составляет между 4000 и 5000 см2/г микрошариков. Эти характеристики, в частности, относятся к микрошарикам с перекрестно-сшитым декстрановым матриксом, продаваемым под наименованием CytodexTM (Cytodex 1, Cytodex 2, Cytodex 3), но также они относятся к другим микрошарикам, основой матрикса которых является перекрестно-сшитый полистирол (Biosilon, Solohill), или стеклянным микрошарикам (Sigma Aldrich).
В контексте настоящего изобретения концентрация микроносителей в течение первого пассажа клеток, в частности, при использовании в качестве микроносителей микрошариков CytodexTM, таких как микрошарики CytodexTM 1, обычно снижена до концентрации <1 г/л, причем в прототипе микроносители используют в концентрации между 1 и 5 г/л. Обычно она составляет ≤0,5 г/л; более определенно она составляет 0,1-0,4 г/л, и конкретнее она составляет 0,1-0,3 г/л. Эта концентрация фактически соответствует исходной концентрации микроносителей в культуральной среде после внесения клеток в культуральный сосуд. Таким образом, она составляет <1 г/л, предпочтительно ≤0,5 г/л; в частности, она составляет 0,1-0,4 г/л и более конкретно она составляет 0,1-0,3 г/л.
Исходное количество клеток, которое вносят в культуральный сосуд, выбирают так, чтобы более 80% микроносителей были колонизированы клетками. Для получения данной степени колонизации в культуральный сосуд обычно вносят исходное количество клеток, которое по меньшей мере в 5-10 раз выше количества микроносителей, присутствующих в культуральной среде. Например, в случае получения клеток Vero исходное количество клеток, которое вносят в культуральный сосуд, обычно находится между 5×103 и 5×104 клеток/см2 микрошариков CytodexTM, что представляет собой приблизительно от 5 до 50 клеток на микрошарик. Фактически, поскольку концентрация микроносителей в культуральной среде в течение первого пассажа клеток ниже той, которую обычно используют в прототипе, соответственно следует, что исходная концентрация клеток также будет ниже.
В конце каждого пассажа клетки открепляют от микроносителей в течение короткого периода времени (в общем менее 30 минут и предпочтительно в течение менее 15 минут) путем обработки клеток раствором фермента, имеющего протеолитическую активность (протеазы). В контексте изобретения клетки обычно открепляют от микроносителей в культуральном сосуде, который использовали для проведения последовательных пассажей клеток, что означает, что все фазы культивирования клеток и все обработки клеток, которые проводят в течение последовательных пассажей, осуществляют в одном и том же культуральном сосуде. Также, необязательно, можно открепить клетки от микроносителей после переноса их во второй сосуд, где проходит ферментативная обработка, и затем повторно внести полученную клеточную суспензию в первый культуральный сосуд, в котором проходят последовательные клеточные пассажи. Этот способ не является предпочтительным, поскольку он приводит к потере клеток в ходе проведения переноса.
Раствор протеолитического фермента обычно содержит сериновую протеазу, такую как трипсин, проназа® или диспаза®. Когда микроносителями являются микрошарики Cytodex 3, также можно использовать папаин, фицин или коллагеназу. Обычно для открепления адгезионных клеток от микрошариков CytodexTM используют раствор трипсина. Предпочтительно, чтобы протеаза была неживотного происхождения, то есть ее получают, используя способ, в котором не используется материал животного происхождения. Ее получают, например, с использованием растительного материала, химическим синтезом или генетической рекомбинацией с использованием бактерий, дрожжей, грибов или растений. Например, можно использовать раствор фермента, свободный от какого-либо продукта животного происхождения, продаваемый Invitrogen под торговым наименованием TrypLETM Select или TrypLETM Express. Эту протеазу, белковая последовательность которой описана в WO 94/25583, получают ферментацией штамма DSM 2672 Fusarium oxysporum или с помощью генетической рекомбинации. Она имеет ферментативную активность, аналогичную трипсину. Для облегчения открепления клеток к раствору фермента можно добавить хелатирующий агент, который связывает ионы кальция, такой как, например, EDTA, EGTA или цитрат, или, необязательно, адгезионные клетки можно обработать хелатирующим агентом перед проведением ферментативной обработки. Концентрация протеазы и, необязательно, хелатирующего агента а среде, а также температура, при которой проводят ферментативную обработку клеток (обычно между 20 и 38°C), задают таким образом, чтобы более 80% клеток открепились от своей подложки за короткий период времени (≤30 минут). Перед проведением самой ферментативной обработки обычно удаляют по меньшей мере половину объема культуральной среды. Обычно удаляют приблизительно 2/3 объема культуральной среды. Затем протеолитическую активность нейтрализуют добавлением в среду ингибитора обычно пептидной или белковой природы, который нейтрализует действие протеаз. Предпочтительно, чтобы композиция ингибитора была свободной от любого загрязнения животного происхождения. Этим ингибитором являются, например, рекомбинантный апротинин или экстракты очищенных фракций, содержащих трапсиновый ингибитор из соевых бобов или лимской фасоли (Worthington Biochemical). Среду обычно перемешивают на протяжении фазы открепления клеток от микроносителей, за исключением периода, в течение которого удаляют культуральную среду.
Количественную оценку полученной клеточной суспензии обычно проводят, используя обычные системы подсчета, с помощью которых также можно определить жизнеспособность клеток. Хотя часть клеточной популяции можно удалить из культурального сосуда при избыточном росте клеток, часто используют всю клеточную популяцию для инициации пассажа клеток, следующего за первым пассажем клеток, который проводят в том же сосуде. Для увеличения клеточной биомассы в течение последовательных пассажей необходимо ввести в культуральный сосуд в начале каждого пассажа клеток, следующего за первым пассажем клеток (который начинается после стадии открепления клеток с использованием ферментативной обработки), большее количество микроносителей, чем количество ранее внесенных микроносителей. Если в течение последовательных пассажей клеток поддерживают постоянный объем культуры, то это означает увеличение концентрации микроносителей на каждом клеточном пассаже, следующем за первым пассажем клеток. С другой стороны, в течение последовательных пассажей клеток можно поддерживать постоянную концентрацию микроносителей, если объем культуральной среды увеличивается в таком же соотношении на каждом новом клеточном пассаже. Весьма предпочтительно в начале каждого пассажа клеток, следующего за первым пассажем клеток, увеличивать как объем культуральной среды, так и концентрацию микроносителей, для того чтобы получить максимальную амплификацию клеток. Ориентировочно, на каждом новом клеточном пассаже клетки культивируют в объеме культуральной среды, который в 1,2-3 раза превышает объем, в котором клетки содержали в течение предшествующего пассажа. Аналогичным образом, на каждом новом клеточном пассаже концентрация микроносителей в культуральной среде в 2-10 раз превышает концентрацию, которая была в течение предшествующего пассажа клеток. В контексте настоящего изобретения обычно нецелесообразно удалять использованные микроносители в конце каждого пассажа клеток (то есть в конце стадии открепления клеток). Даже хотя указанные микроносители могут быть повторно колонизированы клетками, количество использованных микроносителей из предшествующих пассажей клеток обычно не учитывают при вычислении количества носителей, которые вносят в начале каждого нового пассажа клеток. Фаза прикрепления клеток на микроносители обычно длится от 1 до 10 часов в зависимости от типа клеток. После фазы прикрепления может быть предпочтительно удалить всю или часть культуральной среды после осаждения микроносителей, и заменить ее новой средой, для того чтобы ускорить пролиферацию клеток, прикрепленных к микроносителям.
Культуральной средой, подходящей для объекта изобретения, может быть стандартная среда для культивирования клеток с добавленной сывороткой животного происхождения. Предпочтительно, чтобы культуральная среда не содержала ни сыворотку, ни сывороточный белок. Культуральная среда, в частности, может быть свободна от любого белка животного происхождения или даже от любого продукта животного происхождения. Предполагается, что термин «белок или продукт животного происхождения» означает белок или продукт, способ получения которых включает в себя по меньшей мере одну стадию, в которой используют материал животного происхождения или человеческий материал. Особенно предпочтительно, чтобы среда, используемая для культивирования клеток, могла быть свободна от любого белка или могла содержать очень маленькое количество белков в виде рекомбинантных белков или белков, выделенных из растений (сои, риса и т.п.) или из дрожжей. Они наиболее часто включают низкомолекулярные белки (≤10 кДа) (также называемые полипептиды) в очень низкой концентрации. Общая концентрация белка в данной культуральной среде обычно составляет ≤15 мг/л по методу Бредфорд. В частности, это так для среды VP SFM, продаваемой InVitrogen, которая подходит для способа по изобретению, в частности, для культивирования клеток Vero. Также можно упомянуть бессывороточную среду Opti ProTM (InVitrogen), Episerf (InVitrogen), Ex-cell® MDCK (Sigma-Aldrich), Ex-CellTM Vero (SAFC biosciences), бессывороточную MP-BHK® (MP Biomedicals), бессывороточную SFC-10 BHK express (Promo cell), безбелковую SFC-20 BHK express (Promo cell), HyQ PF Vero (Hyclone ref. SH30352.02), Hyclone SFM4 Megavir, среду MDSS2 (Axcell biotechnology), модифицированную по Искову среду DMEM (Hyclone), питательную среду Хэма (Ham-F10, Ham-F12), среду Лейбовитца L-15 (Hyclone), среду Pro Vero (Lonza) и среду Power MDCK (Lonza), которые свободны от любого продукта животного происхождения и которые содержат мало белков или совсем не содержат их.
Когда культуральная среда свободна от животной сыворотки или сывороточного белка или имеет общую концентрацию белка <15 мг/л (по Брэдфорд), обычно добавляют клеточный протекторный агент, который защищает клетки от усилий сдвига, возникающих при перемешивании среды. Наиболее часто используемые клеточные протекторные агенты обычно имеют поверхностно-активные свойства. Ими, в частности, являются полимеры винилового спирта, также известные как поливиниловые спирты (ПВС), полимеры этиленгликоля, также известные как полиэтиленгликоли (ПЭГ), полимеры 1-винил-2-пирролидона, также известные как поливинилпирролидон (ПВП) или полоксамеры, которые представляют собой «блок-сополимеры» этиленоксида и пропиленоксида, имеющие формулу HO(C2H4O)a(C3H6O)b(C2H4O)aH, в которой а обозначает число этиленоксидных звеньев, а b обозначает число пропиленоксидных звеньев. Эти клеточные протекторные агенты обычно используют в диапазоне концентраций от 0,001% до 2% (вес/объем) в культуральной среде. Среди особенно предпочтительных клеточных протекторных агентов можно упомянуть полоксамер 188 и ПВР. Полоксамер 188 имеет средний молекулярный вес приблизительно 8400 дальтон и используется в культуральной среде в концентрации обычно между 0,05 и 0,2% (вес/объем). ПВП также является рекомендуемым агентом, поскольку он стимулирует рост клеток, как описано в WO 01/40443. ПВП обычно используют в диапазоне среднего молекулярного веса от 20 кДа до 360 кДа, предпочтительно в диапазоне среднего молекулярного веса от 20 кДа до 40 кДа, с концентрацией в культуральной среде, которая обычно составляет от 0,01% до 2%, и предпочтительно с концентрацией 0,05%-0,5% (вес/объем). ПВР также можно охарактеризовать не только посредством его молекулярного веса, но также по его К-значению, в котором учитывается средний молекулярный вес ПВП, а также вариации в молекулярном весе в любую сторону от среднего значения. Для вычисления К-значения дана ссылка на уравнение, описанное в статье Cryobiology, 8, 453-464 (1971): К-значение вычисляют на основе относительной вязкости 1%-го раствора ПВП по формуле:
Log η отн./C=75K02/(1+1,5K0C)+K0 ,
где K=1000K0 ,
C представляет концентрацию ПВП в граммах на 100 мл среды.
η отн. является вязкостью раствора по сравнению с вязкостью растворителя.
ПВП, подходящий для объекта изобретения, имеет К-значение, которое обычно находится между 18 и 60 предпочтительно между 26 и 35. Ориентировочно для получения запаса клеток Vero в соответствии со способом по изобретению культуральную среду на основе VPSFM, продаваемой Invitrogen, содержащую в качестве клеточного протекторного агента ПВП, имеющий К-значение примерно 30, в концентрации 0,1% (вес/объем) или полоксамер 188 в концентрации 0,1% (вес/объем), можно использовать в качестве культуральной среды, свободной от любого продукта животного происхождения и имеющей очень низкое содержание белка (<15 мг/л по методу Брэдфорд).
Обычно композиция культуральной среды остается постоянной в течение последовательных пассажей клеток, но при необходимости может оказаться полезным добавление питательных добавок, таких как глюкоза и/или глутамин. При последовательных пассажах клеток также может быть полезно в фазах пролиферации клеток обновлять всю или часть клеточной среды, если это необходимо для клеток. Это оценивают стандартными способами тестирования, имеющимися в распоряжении опытных специалистов в данной области, такими как измерение содержания глюкозы, глутамина, лактата, ионов аммония. В контексте настоящего изобретения культуральную среду обычно непрерывно перемешивают, прилагая силу, достаточную для постоянного поддержания микроносителей в суспензии в культуральной среде, за исключением времени, когда удаляют всю или часть культуральной среды.
Объем культуральной среды в течение первого пассажа клеток обычно представляет собой от половины до 1/5 рабочего объема культурального сосуда. В случае культуральных сосудов с большой емкостью (биореакторов более чем на 100 литров), определенная конфигурация которых позволяет культивировать клетки, прикрепленные к микроносителям, в малом объеме (таких как, например, биореактор, оборудованный нижней конической зоной седиментации), объем культуральной среды может быть меньше (1/6-1/10 от рабочего объема биореактора) или даже меньше и может представлять 1/20 рабочего объема биореактора. Как было указано ранее, объемы культуральной среды обычно увеличиваются по мере прохождения курса последовательных пассажей клеток, причем финальный пассаж клеток часто проводят в объеме культуральной среды, который соответствует по меньшей мере 70% рабочего объема сосуда.
Культуральный сосуд оборудован системой для перемешивания (механической, посредством тока воздуха и т.п.) для поддержания микроносителей в суспензии в клеточной культуральной среде и имеет средство для обновления среды в соответствии с потребностями клеточной культуры и/или средства для тестирования и регуляции температуры, рН, давления кислорода, насыщения газом, необязательно, азотом или воздухом, и метаболитов или питательных веществ (лактазы, глюкозы, глутамина, ионов аммония и т.п.). Эти устройства хорошо известны опытным специалистам в данной области, которые знают, как их использовать в соответствии с размером и конфигурацией используемого сосуда. Например, культуральный сосуд по изобретению может иметь форму центрифуги или биореактора. Когда рабочий объем сосуда превышает 2 литра, обычно используют биореактор, который обычно может представлять собой многоразовый стеклянный или металлический контейнер, либо, когда биореакторами являются одноразовые биореакторы, сосуды могут представлять собой одноразовые мешки, продаваемые, в частности, P. Guerin под названием Nucleo PG-ATMI™. Также можно использовать, например, систему Biowave (Wave BioreactorTM), продаваемую General Electrics, одноразовую STR-систему (биореактор с перемешиванием в резервуаре) BioreactorTM (Sartorius), систему SUBTM (одноразовый биореактор) (Hyclone) или готовую систему для клеток (Millipore). В контексте способа по изобретению, главной задачей которого является получение партий клеток в промышленном масштабе, используют биореактор, рабочий объем которого находится между 3 литрами и 1000 литрами, но чаще используют биореактор с рабочим объемом 20-500 литров.
В изобретении «адгезионными клетками» являются клетки, устоявшиеся как линии, или клетки, полученные непосредственно в результате выделения из животного или человека, здоровых или опухолевых тканей, которым при используемых условиях культивирования необходима твердая подложка для размножения или нормального развития. Они обычно образуют монослой клеток на своей подложке из-за контактного ингибирования. Клетки, которые при используемых условиях культивирования не нуждаются в твердой подложке для размножения и которые могут расти в суспензии в культуральной среде, поэтому, фактически исключены. Линии адгезионных клеток можно получить из первичных культур здоровых и опухолевых клеток, но также их можно получить в результате трансформации клеток с использованием иммортализующих агентов, как в случае клеточной линии PER.C6.
В качестве примера линий адгезионных клеток, подходящих для объекта изобретения, следует упомянуть мышиные клеточные линии, такие как 3T3, NTCT или линия WEHI, линии клеток хомячка, такие как линия BHK (в частности, линии ВНК21) или линия СНО, линии клеток собаки, такие как линия MDCK, линии клеток свиньи, такие как линия PK15, бычьи клеточные линии, такие как линия MDBK, линии клеток обезьяны, такие как Vero, LLC-MK2, FRHL2 или линия MA104, и линии клеток человека, такие как MRC5, 293, PER.C6, Hela, ECV или линия A 431. Эти адгезионные клеточные линии также могут представлять собой линии, трансфецированные рекомбинантным вектором (плазмидой, вирусом и т.п.), если они предназначены для продукции рекомбинантных белков.
Посредством способа по изобретению популяция адгезионных клеток увеличивается по меньшей мере в 40 раз, предпочтительно по меньшей мере в 60 раз и особенно предпочтительно по меньшей мере в 100 раз в результате проведения последовательных пассажей клеток в одном и том же культуральном сосуде. Это можно осуществить, поскольку способ по изобретению дает возможность увеличивать в 5-40 раз, предпочтительно в 10-30 раз, площадь поверхности клеточной подложки в течение последовательных пассажей клеток, которые проводят в данном одном сосуде. В способах предшествующего уровня техники данный порядок амплификации клеток достигался с использованием по меньшей мере двух сосудов, но чаще трех культуральных сосудов разного размера (смотрите пример 5). Способ по изобретению очень предпочтителен, поскольку такое же промышленное количество клеток, как получаемое с помощью способов предшествующего уровня техники, получают за такое же время, одновременно снижая расходы, связанные с использованием и содержанием культуральных сосудов и пространства, необходимых для получения данных партий клеток.
Способ получения адгезионных клеток по изобретению можно предпочтительно проводить прямым внесением в культуральный сосуд клеток сразу после разморозки, не осуществляя стандартно рекомендуемый период адаптации, в течение которого проводят один или несколько «адаптационных» пассажей клеток для «адаптации» клеток к более сложным условиям культивирования, таким как культивирование в присутствии микрошариков CytodexTM в концентрации ≤0,5 г/л в культуральной среде и/или культивирование в среде, которая не содержит сыворотки или которая содержит очень мало белка (≤15 мг/л). В данном случае запас адгезионных клеток размораживают в соответствии с процедурами, хорошо известными опытным специалистам в данной области, и затем суспензию размороженных клеток вносят напрямую в культуральный сосуд, который содержит микроносители в культуральной среде, для проведения способа, описанного в изобретении. Как было указано ранее, в частности при использовании микрошариков CytodexTM в качестве микроносителей, концентрация микроносителей в культуральной среде в течение первого пассажа клеток обычно снижена до концентрации ≤0,5 г/л; она обычно составляет от 0,1 до 0,4 г/л и более конкретно составляет от 0,1 до 0,3 г/л. Культуральная среда также не должна содержать сыворотку или сывороточные белки. Культуральная среда даже может быть полностью свободна от белков или может иметь очень низкое содержание белка (≤15 мг/л). Запас замороженных клеток может находиться в пробирке (в этом случае количество клеток обычно низкое, от 107 до 5×108 клеток) или предпочтительно может находиться в мешке, который содержит в 100 раз больше клеток. Крупномасштабный способ получения, в частности, ускоряется, поскольку содержание размороженных мешков можно высевать непосредственно в культуральный сосуд большой емкости.
Когда запас адгезионных клеток, который был получен с использованием способа по изобретению, недостаточен, то клеточную биомассу, которая была получена из одного и того же клеточного культурального сосуда, можно дополнительно увеличивать, перенося клеточную популяцию:
- либо в один или несколько культуральных сосудов, которые можно использовать для проведения последовательных пассажей клеток обычным образом, то есть перенося после каждого пассажа клеток полученную клеточную биомассу в другой культуральный сосуд с большей емкостью; и более предпочтительно
- во второй культуральный сосуд, который имеет гораздо больший рабочий объем (обычно по меньшей мере в 10 раз больше, наиболее часто в 10-50 раз больше первого сосуда) и повторно применяя способ по изобретению к клеткам, которые были перенесены в этот второй сосуд. Таким образом, в результате проведения способа еще более значительно сокращается число культуральных сосудов, используемых для получения промышленных партий клеток, а также требуемое пространство.
Для оценки экономического преимущества осуществления способа по изобретению в контексте получения адгезионных клеток в промышленном масштабе можно указать общепринятую схему промышленного получения клеток Vero, предназначенных для продукции полиовируса, описанную в Reviews of Infectious Diseases, vol 6, supplement 2, S341-S344 (1984). Общепринятая схема включает в себя пять последовательных пассажей клеток, первый из которых проводят в 1-литровом биореакторе, второй - в 5-литровом биореакторе, третий - в 20-литровом биореакторе, четвертый - в 150-литровом биореакторе, и в конце пятый пассаж проводят в 1000-литровом биореакторе. Посредством способа по изобретению можно провести первые три клеточных пассажа в одном 20-литровом биореакторе и затем провести последние два пассажа, обычно перенося клетки в 150-литровый биореактор и затем в 1000-литровый биореактор. Также можно повторять способ по изобретению дважды, проводя первые три пассажа клеток в одном 20-литровом биореакторе и затем перенося клетки напрямую в один 500-литровый или 1000-литровый биореактор, в котором проводят последние два пассажа клеток. В обоих случаях количество клеток того же порядка, что и полученное с применением общепринятой схемы, получают за такой же период времени, но в первом случае - сохраняя 2 биореактора (1-литровый и 5-литровый), а во втором случая - сохраняя 3 биореактора (1-л, 5-л и 150-л (смотрите пример 5).
Способ получения адгезионных клеток, особенно предпочтительный с экономической точки зрения, состоит из повтора способа по изобретению в двух культуральных сосудах, размер которых сильно отличается. В этом отношении объектом изобретения поэтому является:
способ получения адгезионных клеток, в соответствии с которым:
а. адгезионные клетки вносят в первый культуральный сосуд, который содержит микроносители в культуральной среде;
b. клетки амплифицируют, проводя нескольких последовательных пассажей клеток в этом первом культуральном сосуде, причем каждый пассаж клеток, следующий за первым пассажем клеток, проводят, каждый раз используя всю или часть клеточной популяции, которая получена в течение предшествующего пассажа клеток и клетки которой были откреплены от микроносителей посредством ферментативной обработки, и каждый раз добавляя культуральную среду и увеличивая количество микроносителей;
c. клеточную популяцию, полученную в течение финального пассажа клеток, проводимого в этом первом культуральном сосуде, собирают после открепления клеток от микроносителей посредством ферментативной обработки;
d. собранную клеточную популяцию переносят во второй культуральный сосуд, который имеет больший рабочий объем и который содержит культуральную среду, содержащую большее количество микроносителей, чем количество микроносителей, которое присутствовало в течение финального клеточного пассажа, проведенного в первом культуральном сосуде;
е. клетки амплифицируют, проводя нескольких последовательных пассажей клеток в этом втором культуральном сосуде, причем каждый пассаж клеток, следующий за первым пассажем клеток, проводят, каждый раз используя всю или часть клеточной популяции, которая получена в течение предшествующего пассажа клеток и клетки которой были откреплены от микроносителей посредством ферментативной обработки, и каждый раз добавляя культуральную среду и увеличивая количество микроносителей;
f. клеточную популяцию, полученную в течение финального пассажа клеток, проводимого в этом втором культуральном сосуде, собирают после необязательного открепления клеток от микроносителей посредством ферментативной обработки, и, необязательно,
g. стадии (d)-(f) повторяют снова в третьем культуральном сосуде, имеющем еще больший рабочий объем.
В общем, рабочий объем второго культурального сосуда в 20-50 раз превышает объем первого культурального сосуда.
Предпочтительно, чтобы адгезионные клетки, которые вносят на стадии (а) способа, были взяты из запаса замороженных клеток, которые были разморожены непосредственно перед внесением в первый культуральный сосуд.
Повтор способа по изобретению также можно выполнять для получения биологического агента. В этом отношении объектом изобретения поэтому является:
способ получения биологического агента, продуцируемого клетками, прикрепленными к микроносителям, в соответствии с которым:
а. адгезионные клетки вносят в первый культуральный сосуд, который содержит микроносители в культуральной среде;
b. клетки амплифицируют, проводя нескольких последовательных пассажей клеток в этом первом культуральном сосуде, причем каждый пассаж клеток, следующий за первым пассажем клеток, проводят, каждый раз используя всю или часть клеточной популяции, которая получена в течение предшествующего пассажа клеток и клетки которой были откреплены от микроносителей посредством ферментативной обработки, и каждый раз добавляя культуральную среду и увеличивая количество микроносителей;
c. клеточную популяцию, полученную в течение финального пассажа клеток, проводимого в этом первом культуральном сосуде, собирают после открепления клеток от микроносителей посредством ферментативной обработки;
d. собранную клеточную популяцию переносят во второй культуральный сосуд, который имеет больший рабочий объем и который содержит культуральную среду, содержащую большее количество микроносителей, чем количество микроносителей, которое присутствовало в течение финального клеточного пассажа, проведенного в первом культуральном сосуде;
е. клетки амплифицируют, проводя нескольких последовательных пассажей клеток в этом втором культуральном сосуде, причем каждый пассаж клеток, следующий за первым пассажем клеток, проводят, каждый раз используя всю или часть клеточной популяции, которая получена в течение предшествующего пассажа клеток и клетки которой были откреплены от микроносителей посредством ферментативной обработки, и каждый раз добавляя культуральную среду и увеличивая количество микроносителей;
f. клеточную популяцию, полученную в течение финального клеточного пассажа, проведенного во втором культуральном сосуде, обрабатывают таким образом, что она продуцирует биологический агент, причем указанную стадию (f) проводят в том же культуральном сосуде, который использовали для проведения стадий (d) и (e); и
g. собирают биологический агент.
Необязательно, стадии (d) и (е) можно повторить в третьем культуральном сосуде перед обработкой клеточной популяции таким образом, чтобы она продуцировала биологический агент.
Как было указано ранее, продуцируемым биологическим агентом может быть, например, рекомбинантный белок или вирус, такой как вирус бешенства.
Объектом изобретения также является применение клеток, которые были получены посредством одного из способов по изобретению, для продукции биологических агентов.
В итоге, изобретение относится к способу получения адгезионных клеток в культуральной среде, которая содержит микроносители, в соответствии с которым последовательные пассажи клеток проводят в одном и том же культуральном сосуде с целью увеличения популяции клеток в 40 и более раз, предпочтительно в 60 и более раз и в особенности предпочтительно в 120 и более раз. Используемым культуральным сосудом предпочтительно является биореактор, который имеет рабочий объем по меньшей мере 20 литров.
Культуральной средой, как было указано в контексте изобретения, может быть стандартная среда с добавленной сывороткой, но предпочтительно является средой, свободной от сыворотки животного происхождения. Особенно предпочтительно использовать среду, свободную от любого продукта животного происхождения и концентрация белка которой составляет ≤15 мг/л.
На фиг.1 представлены два способа амплификации клеток, прикрепленных к микроносителям, посредством последовательных пассажей клеток: а) в соответствии с общепринятым способом последовательные пассажи клеток проводят в разных культуральных сосудах с увеличивающимся рабочим объемом и (b) в соответствии со способом по изобретению (способ все-в-одном) последовательные пассажи клеток проводят в одном и том же культуральном сосуде. Клетки находятся в замороженной форме на стадии 0. Стадия 0→1 соответствует переносу размороженных клеток в биореактор. Стадия 1 соответствует первому пассажу клеток. Стадия 1→2 соответствует переносу популяции клеток, полученной в конце первого пассажа клеток после обработки протеолитическим ферментом с целью открепления клеток от микроносителей, либо в случае способа (а) во второй биореактор, который имеет больший рабочий объем, либо в случае способа (b) - в тот же культуральный сосуд. Стадия 2 соответствует второму клеточному пассажу. В случае способа (b) второй клеточный пассаж обычно проводят в большем объеме культуральной среды и в присутствии большей концентрации микроносителей. Стадия 2→3 соответствует переносу популяции клеток, полученной в конце второго пассажа клеток после обработки протеолитическим ферментом с целью открепления клеток от микроносителей, либо в случае способа (а) в третий биореактор, который имеет больший рабочий объем, либо в случае способа (b) - в тот же культуральный сосуд. Стадия 3 соответствует третьему клеточному пассажу. В случае способа (b) третий клеточный пассаж обычно проводят в большем объеме культуральной среды и в присутствии большей концентрации микроносителей, чем в течение второго пассажа клеток.
На фиг.2 представлен вид микрошариков под микроскопом (20-кратное увеличение) после 8-дневного периода культивирования клеток Vero, осуществляемого (а) способом все-в-одном или (b) способом переноса с шариков на шарики (смотрите пример 2 для условий культивирования).
Настоящее изобретение будет легко понятно в свете следующих примеров, которые служат для иллюстрации изобретения, однако не ограничивая его содержание.
Пример 1: амплификация клеток Vero путем проведения 3 последовательных пассажей клеток в одном и том же 2-литровом биореакторе
В этом примере исследовали влияние различных параметров, таких как исходная концентрация микроносителей, присутствие или отсутствие сыворотки в культуральной среде и природа клеточного протекторного агента, на рост клеток.
1.1) Используемые материалы
Биореактор:
Эксперименты проводили в одноразовых биореакторах с емкостью 2,4 литра, продаваемых Millipore под наименованием Cellready (Mobius). Они оборудованы датчиком рН, датчиком рО2 и температурным датчиком, а также винтовым перемешивающим устройством.
Микроносители:
Использовали микрошарики Cytodex 1, поставляемые GE Healthcare. Микрошарики оставляли набухать в течение 24 часов в фосфатном буфере (1×C PBS, pH~7,4) после удаления количества, необходимого для проведения каждого пассажа клеток. Затем их промывали три раза тем же буфером и затем стерилизовали автоклавированием. Непосредственно перед внесением в биореактор буфер для стерилизации замещали равным объемом культуральной среды после осаждения микрошариков. 1 г микрошариков представляет адгезионную поверхность приблизительно 4400 см2.
Протестированная культуральная среда:
VPSFM/K30: бессывороточная среда VPSFM (Invitrogen), свободная от продуктов животного происхождения, с добавленным 0,1% (вес/объем) поливинилпирролидона (ПВП) К30, поставляемого ISP.
VPSFM/F68: бессывороточная среда VPSFM (Invitrogen), свободная от продуктов животного происхождения, с добавленным 0,1% (вес/объем) полоксамера 180, поставляемого BASF.
VPSFM/K30/SVF: среда VPSFM/K30 с добавлением 4 % фетальной телячьей сыворотки с инактивированной системой системой комплемента.
Клетки:
Клетки Vero, источником которых является банк клеток, содержащийся в замороженной форме в концентрации 50×106 клеток/мл в бессывороточной среде, содержащей 10% диметилсульфоксида, в пробирках для замораживания клеток (Nunc, кат. номер: 430663, 5 мл).
1.2) Рабочий протокол, используемый для оценки параметров в отношении исходной концентрации микроносителей и клеточного протекторного агента в способе « все-в-одном»
Один и тот же рабочий протокол использовали для исследования этих двух параметров.
Концентрации 0,1 г/л и 0,3 г/л были протестированы, для того чтобы оценить эффект очень низкой концентрации микроносителей в течение первого пассажа клеток на рост клеток Vero.
66×106 клеток в 2 литрах среды VPSFM/K30, содержащей 0,6 г микроносителей, вносили в биореактор 1 (bio 1) (что равно количеству 25000 клеток/см2 адгезионной поверхности и представляет исходную концентрацию микроносителей 0,3 г/л) после подведения регулируемых параметров в биореакторе, таких как рН - до 7,2-7,5, температура - до 37°C и pO2 - до ~25%.
22×106 клеток в 2 литрах среды VPSFM/K30, содержащей 0,2 г микроносителей, вносили в биореактор 2 (bio 2) (что равно количеству 25000 клеток/см2 адгезионной поверхности и представляет исходную концентрацию микроносителей 0,1 г/л).
На всем протяжении культивирования клеток культуральную среду постоянно перемешивали, за исключением фаз осаждения микрошариков, которые использовали для обновления культуральной среды или для уменьшения объема культуральной среды.
В день D3 клетки обрабатывали трипсином по следующему протоколу.
После осаждения микрошариков ~300 мл культуральной среды VPSFM/K30 оставляли в биореакторе и затем добавляли ~300 мл 0,025 М раствора цитрата натрия, содержащего 600 мг рекомбинантного трипсина (Roche, кат. номер 04618734), в фосфатном буфере без кальция и магния. Среду перемешивали с небольшой скоростью. После проверки фактического открепления клеток с использованием тестового образца (в общем, данное открепление занимало от 15 до 30 минут) действие трипсина останавливали, добавляя ~300 мл раствора VPSFM/K30, содержащего 1 мг/мл ингибитора трипсина (Sigma, кат. номер T6522). После определения количества клеток часть клеточной суспензии удаляли, для того чтобы оставшееся количество клеток соответствовало приблизительно 25×103 клеток/см2 адгезионной поверхности после внесения либо 2,4 г микроносителей (bio 1), либо 0,6 г микроносителей (bio 2) в общем объеме культуральной среды 2 литра, для проведения второго пассажа клеток. Регулируемые параметры снова подводили, так же как и в первом пассаже клеток. Через 4-6 часов среду замещали свежей культуральной средой и затем, необязательно, обновляли второй раз после 24-48 часов культивирования. В день D7 клетки снова трипсинизировали по такому же протоколу, которые использовали в D3. После подсчета клеток часть клеточной суспензии также удаляли, для того чтобы оставшееся количество клеток соответствовало приблизительно 25×103 клеток/см2 адгезионной поверхности после внесения либо 6 г микроносителей (bio 1), либо 2,4 г микроносителей (bio 2) в общем объеме культуральной среды 2 литра для проведения третьего пассажа клеток. Регулируемые параметры биореактора затем снова подводили. Через 4-6 часов среду замещали свежей культуральной средой и затем, необязательно, обновляли второй раз после 24-48 часов культивирования. В D10 клетки собирали для оценки степени амплификации клеток в соответствии с исходной концентрацией микроносителей в культуральной среде.
Для оценки роли клеточных протекторных агентов были протестированы полоксамер 188 и поливинилпирролидон (ПВП) К30 с использованием такого же рабочего протокола и с исходной концентрацией микроносителей 0,3 г/л.
1.3) Рабочий протокол, используемый для оценки роли сы воротки в способе «все-в-одном»
Рабочий протокол, такой же как описанный в разделе 1.2, использовали со следующими отличиями:
- тестировали среду VPSFM/K30/SVF,
- исходная концентрация микроносителей составляла 0,3 г/л,
- обработку трипсином проводили в дни D5 и D8. Перед каждой обработкой трипсина суспензию микрошариков промывали 3 раза 600 мл промывочного буфера (фосфатный буфер (1×C PBS)), используя для удаления сыворотки последовательность стадий, включающую в себя промывание, осаждение и удаление промывочного буфера.
1.4) Результаты
Для оценки эффекта различных исследуемых параметров на рост клеток концентрацию клеток измеряли через равные промежутки времени, используя систему определения количества клеток Nucleocounter (Chemometec®), и вычисляли число суммарных поколений клеток. Каждый раз отбирали два образца суспензии микрошариков. Указанные значения представляют средние значения для двух образцов, взятых для каждой проанализированной временной точки.
Количество клеток и число суммарных поколений клеток, наблюдаемое в ходе культивирования, представлено для каждого исследуемого параметра в 3 таблицах, приведенных ниже.
1.4.1) параметр «исходная концентрация микроносителей»
**: после обработки трипсином и подведения концентрации клеток до 25×103 клеток/см2 адгезионной поверхности.
***: для вычисления числа суммарных поколений, учитывали возможное подведение концентрации клеток, которые проводили в течение последовательных пассажей клеток, и использовали общую формулу, приведенную ниже: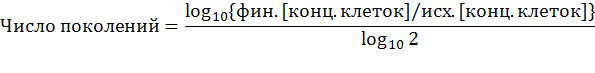
фин.[конц.клеток]: соответствует концентрации клеток на рассматриваемый день
исх.[конц.клеток]: соответствует исходной концентрации клеток в D0
Хотя количество клеток, полученное после 3 последовательных пассажей клеток меньше в биореакторе, в котором исходная концентрация микроносителей составляла 0,1 г/л, время удвоения клеточной популяции, с другой стороны, было немного короче в этом биореакторе, поскольку наблюдаемое число суммарных поколений клеток было выше на всем протяжении испытаний.
Удивительно, но очень низкая концентрация микроносителей в течение первого пассажа клеток (~0,1 г/л) не оказывает отрицательного влияния на рост клеток. Напротив, клетки имеют тенденцию делиться еще более активно по сравнению с тем, когда они находятся в биореакторе, который содержит более высокую концентрацию микроносителей (0,3 г/л).
1.4.2) параметр «клеточного протекторного агента»
**: после обработки трипсином и подведения концентрации клеток до 25×103 клеток/см2 адгезионной поверхности.
Результаты показывают, что добавление полоксамера 188 или ПВП(К30) к бессывороточной культуральной среде имеет в общем одинаковый эффект на амплификацию клеток, проводимую по способу «все-в-одном».
1.4.3) параметр «сыворотка»
**: после обработки трипсином.
Эти результаты показывают, что способ «все-в-одном» также применим для клеток, прикрепленных к микроносителям, культивируемым в культуральной среде, содержащей сыворотку.
Пример 2: сравнение вариантов получения клеток Vero либо с использованием способа по изобретению (способ «все-в-одном»), либо с использованием способа простого добавления микроносителей (методика «переноса с шариков на шарики»)
Варианты получения клеток сравнивали после 2 последовательных пассажей клеток, проводимых в соответствии со способом «все-в-одном» или с использованием методики «переноса с шариков на шарики».
Исследования проводили в стеклянных биореакторах емкостью 4 литра, продаваемых компанией Sartoriu под названием Quattro. Они оборудованы датчиком рН, датчиком рО2 и температурным датчиком, а также винтовым перемешивающим устройством.
2.1) рабочий протокол, используемый для выполнения «способа все-в-одном»
Для «способа все-в-одном» применяли такой же принцип, как описанный в разделе 1.2, со следующими отличиями:
- Используемой культуральной средой была среда VPSFM/K30.
- Объем культуральной среды оставался постоянным в течение 2 последовательных пассажей и составлял 4 литра.
- Первый пассаж клеток проводили, используя концентрацию микрошариков 0,3 г/л и внося такое количество клеток, чтобы их концентрация составляла 50000 клеток/см2 адгезионной поверхности.
- Культуральную среду обновляли в D3.
- В день D4 клетки обрабатывали трипсином в соответствии с такими же процедурами, как описанные в разделе 1.2. После трипсинизации и подсчета клеток 2/3 объема клеточной суспензии удаляли для того, чтобы оставшееся количество клеток соответствовало приблизительно 25×103 клеток/см2 адгезионной поверхности после внесения 4,8 г микрошариков (то есть концентрации 1,2 г/л) в общем объеме культуральной среды 4 литра для проведения второго пассажа клеток.
- Культуральную среду обновляли дважды: первый раз - через 4-6 часов после внесения шариков и второй раз - в день D6.
- В день D8 клетки собирали и подсчитывали для оценки степени амплификации клеток. Аликвоту суспензии микрошариков также анализировали под микроскопом для оценки степени покрытия микрошариков клетками.
2.2) рабочий протокол, используемый для выполнения способа «переноса с шариков на шарики»
Применяли такой же рабочий протокол, как описанный в разделе 2.1, за исключением того, что клетки не обрабатывали трипсином в D4. В день D4 2/3 объема суспензии микрошариков удаляли, так чтобы удалить такую же часть клеток и микрошариков, чтобы соответствовать таким же условиям культивирования, как используемые в способе «все-в-одном». Затем вносили суспензию микрошариков, содержащую 4,8 г Cytodex 1 (то есть с концентрацией 1,2 г/л) в общем объеме клеточной культуральной среды 4 литра. Рабочий протокол далее был таким же, как описанный в разделе 2.1.
2.3) Результаты
Для сравнения роста клеток в течение двух тестируемых способов концентрацию клеток измеряли через одинаковые интервалы времени в соответствии с такими же процедурами, как описанные в разделе 1.4.
Количество клеток и число суммарных поколений клеток, наблюдаемые в течение культивирования, приведены ниже в таблице.
**: после подведения концентрации клеток и, необязательно, обработки трипсином (в случае способа «все-в-одном»)
В течение первого пассажа клеток (D0-D3) рост клеток примерно одинаков в двух биореакторах. Напротив, в течение второго пассажа клеток (D3-D8) амплификация клеток значительно слабее в биореакторе, в котором проводили технологию «переноса с шариков на шарики». В D8 количество собранных клеток приблизительно составляет половину от количества, собранного в биореакторе, в котором проводили способ «все-в-одном». Результаты, касающиеся изменения числа суммарных поколений клеток, имеют такую же тенденцию. В день D8 число суммарных поколений клеток превышает 6 в случае клеток, которые амплифицировали по способу «все-в-одном». Это число составляет 5 для клеток, которые амплифицировали по технологии «переноса с шариков на шарики». Клетки, которые амплифицировали по способу «все-в-одном», таким образом, размножались более активно. Эти результаты являются неожиданными, поскольку для проведения последовательных пассажей клеток способ «все-в-одном» требует применения трипсина на клетках, который, как известно, наносит ущерб целостности клеток.
Эти результаты были подтверждены в 2 независимых экспериментах, проведенных при одинаковых рабочих условиях. Вариационный анализ числа суммарных поколений клеток в день D8 показывает достоверную разницу (p=0,0137).
Анализ микрошариков под микроскопом в день D8 показывает, что подавляющее большинство микрошариков покрыты клетками при проведении способа все-в-одном (смотрите фиг.2а). Напротив, только часть микрошариков покрыты клетками при проведении технологии «переноса с шариков на шарики». Постоянное перемешивание среды или периодическое перемешивание среды (в результате повторения, например, 2-часовых циклов, включающих перемешивание в течение 5 минут с последующим периодом «отдыха» в течение 20 минут), после добавления микрошариков Cytodex 1 в D4 по существу не меняло результаты (смотрите фиг.2b).
Пример 3: амплификация клеток Vero путем проведения 2 последовательных пассажей клеток в одном и том же 20-литровом биореакторе
3.1) Используемые материалы
Биореактор:
Использовали 20-литровый биореактор в виде одноразового мешка, продаваемый ATMI под названием Nucleo-20. Датчики pH, pO2 и температуры после калибровки и последующей стерилизации автоклавированием, посредством мешков-футляров для датчиков устанавливали на мешок в соответствии со стандартным протоколом ATMI: соединения Kleenpack®, расположенные, с одной стороны, на мешке и, с другой стороны, на футляре датчика, соединяли и затем датчик вносили в биореактор через созданное таким образом соединение.
Микроносители: микрошарики Cytodex 1, поставляемые GE Healthcare (смотрите раздел 1.1).
Культуральная среда: среда VPSFM/K30 (смотрите раздел 1.1).
Клетки: клетки Vero (смотрите раздел 1.1).
3.2) Рабочий протокол
Шесть литров культуральной среды VPSFM/K30 вносили в Nucleo-20, и затем добавляли 1 литр суспензии микрошариков, содержащей 4 г Cytodex 1 (которая представляет исходную концентрацию микрошариков 0,5 г/л после добавления клеток). После подведения регулируемых параметров внутри Nucleo-20, таких как температура - до 37°C, рН - до 7,2-7,4 и pO2 - до ~25%, и начала умеренного перемешивания среды, чтобы ресуспендировать шарики в культуральной среде, в Nucleo вносили 500×106 клеток в 1 литре культуральной среды VPSFM/K30 после размораживания. На всем протяжении рабочего протокола культуральную среду непрерывно перемешивали, за исключением фаз осаждения микрошариков, которые проводили для обновления культуральной среды или для уменьшения объема культуральной среды.
В день D2 (2 дня после начала культивирования) среду заменяли новой средой VPSFM/K30.
В день D5 клетки трипсинизировали по следующему протоколу.
Прекращали перемешивание, регулирование рН и рО2. Оставляли только температурную регуляцию. После осаждения микрошариков в биореакторе оставляли ~3 литра культуральной среды VPSFM/K30 и затем добавляли ~3 литра 0,025 М раствора цитрата натрия, содержащего 600 мг рекомбинантного трипсина (Roche, кат. номер 04618734) в фосфатном буфере без кальция и магния. Среду затем снова умеренно перемешивали. После проверки фактического открепления клеток путем забора тестового образца (в общем, данное открепление занимало от 15 до 30 минут) действие трипсина останавливали, добавляя ~3 литра раствора VPSFM/K30, содержащего 1 мг/мл ингибитора трипсина (Sigma, кат. номер T6522). Затем добавляли суспензию микрошариков, содержащую 28 г Cytodex 1, что соответствует концентрации микрошариков приблизительно 1,4 г/л в среде после доведения объема культуральной среды до 20 литров культуральной средой VPSFM/K30. Регулируемые параметры внутри Nucleo-20 затем снова подводили до таких же значений, как в первом пассаже клеток. Через 4-6 часов после внесения микрошариков среду заменяли новой культуральной средой. Вторую замену культуральной среды проводили в день D7. В день D9 клетки были по существу конфлюэнтными. Их затем трипсинизировали по такому же протоколу, какой использовали в D5. Уровень амплификации клеток, полученный после 2 пассажей клеток, проведенных в одном биореакторе, определяли из полученной суспензии клеток.
3.3) Результаты
Для измерения уровня полученной амплификации клеток концентрацию клеток измеряли через равные интервалы в соответствии с такими же процедурами, как описанные в разделе 1.4.
Количество и концентрацию клеток, наблюдаемые в течение культивирования, представлены в таблице ниже:
**: после обработки трипсином и адгезии клеток
***: представляет исходное количество посеянных живых клеток
После двух последовательных пассажей клеток, проведенных в одном 20-литровом биореакторе, популяция клеток увеличилась в 68 раз после 9 дней культивирования, тогда как поверхность клеточной подложки увеличилась в 7 раз.
Пример 4: амплификация клеток Vero путем проведения 3 последовательных пассажей в одном и том же 20-литровом биореакторе
4.1) Используемые материалы
Используемые материалы идентичны материалам, описанным в методике примера 3.
4.2) Рабочий протокол
Пассажи клеток проводили по такому же протоколу, как описанный в разделе 3.2, со следующими изменениями:
- первый пассаж проводили, внося 250×106 клеток в 8 литров среды VPSFM/K30, содержащей 2 г микрошариков Cytodex 1 (что представляет концентрацию микрошариков 0,25 г/л);
- в день D5 после первой обработки трипсином для проведения второго пассажа клеток добавляли суспензию микрошариков, содержащую 14 г Cytodex 1, что соответствует концентрации микрошариков приблизительно 1,07 г/л после доведения объема культуральной среды до 13 литров культуральной средой VPSFM/K30;
- в день D9 после второй обработки трипсином для проведения третьего пассажа клеток добавляли суспензию микрошариков, содержащую 60 г Cytodex 1, что представляет концентрацию микрошариков приблизительно 3 г/л после доведения объема культуральной среды до 20 литров культуральной средой VPSFM/K30.
В день D12 клетки были по существу конфлюэнтными. Их затем трипсинизировали по такому же протоколу, какой использовали в D5.
Уровень амплификации клеток, полученный после 3 пассажей клеток, проведенных в одном биореакторе, определяли с использованием полученной финальной суспензии клеток.
4.3) Результаты
Количество и концентрацию клеток, наблюдаемые на протяжении культивирования, представлены в таблице ниже:
**: после обработки трипсином и адгезии клеток
После трех последовательных пассажей клеток Vero, проведенных в одном 20-литровом биореакторе, популяция клеток увеличилась в 126 раз после 12 дней культивирования, тогда как поверхность клеточной подложки увеличилась в 30 раз.
Пример 5: сравнение получения клеток Vero с использованием либо способа по изобретению (последовательные пассажи клеток проводят в одном и том же биореакторе), либо стандартного способа размножения клеток (последовательные пассажи клеток каждый раз проводят в разных биореакторах большего размера)
5.1) Рабочий протокол
Получение клеток Vero исследовали и сравнивали, проводя либо 3 последовательных пассажа клеток в одном 20-литровом биореакторе по протоколу, описанному в примере 4, и используя исходное количество клеток 250×106, полученных непосредственно из банка замороженных клеток, либо проводя 3 последовательных пассажа клеток в разных биореакторах из нержавеющей стали, первый - в 2-литровом биореакторе, второй - в 7-литровом биореакторе и третий - в 28-литровом биореакторе. Используемые условия эксперимента стандартного способа были следующими.
Клетки Vero после разморозки сначала адаптировали к их условиям культивирования, проводя исходный пассаж в Cell Factories (CF10), внося 40×103 клеток на см2 адгезионной поверхности в 2 литрах культуральной среды VPSFM/K30. После приблизительно 5 дней культивирования полученную популяцию клеток собирали после стадии трипсинизирования. Собранную популяцию клеток использовали для посева в биореактор с рабочим объемом 2 литра, содержащий суспензию 2 г микрошариков Cytodex 1 в двух литрах культуральной среды VPSFM/K30 (концентрация 1 г/л). После тестирования и подведения регулируемых параметров устанавливали температуру на 37°C, pH на 7,2-7,4 и pO2 на ~25% и начинали умеренное перемешивание среды, в 2-литровый биореактор высевали в среднем 220×106 клеток. В день D3 культуральную среду заменяли новой культуральной средой. В день D4 по существу конфлюэнтные клетки трипсинизировали и затем переносили с использованными микроносителями в 7-литровый биореактор, содержащий 7 литров культуральной среды VPSFM/K30, к которой заблаговременно добавили 14 г микрошариков Cytodex 1, что представляет концентрацию микрошариков приблизительно 2 г/л. В день D6 культуральную среду заменяли новой культуральной средой. В день D8 по существу конфлюэнтные клетки трипсинизировали и затем переносили таким же образом в 28-литровый биореактор, содержащий 28 литров культуральной среды, к которой заблаговременно добавили 70 г микрошариков Cytodex 1, что представляет концентрацию микрошариков приблизительно 2,5 г/л. В день D10 культуральную среду заменяли новой культуральной средой. В день D11 по существу конфлюэнтные клетки трипсинизировали и затем собирали и определяли их количество.
5.2) Результаты
Для мониторинга роста клеток при проведении двух тестируемых способов концентрацию клеток измеряли через равные интервалы времени по таким же методикам, как описанные в разделе 1.4.
Количество клеток и уровень амплификации, наблюдаемые в течение культивирования, приведены в таблице ниже:
**: приведенные количества являются средними значениями, полученными в проведенных 3 различных экспериментах.
С использованием способа «все-в-одном» более 29 миллиардов клеток были получены в среднем за 12 дней культивирования после внесения в среднем 253 миллионов непосредственно размороженных клеток в 20-литровый биореактор, то есть со среднем уровнем амплификации 115. С использованием стандартного способа было получено 32 миллиарда клеток в среднем через 11 дней культивирования после исходного внесения в среднем 220 миллионов клеток в 2-литровый биореактор, то есть со средним уровнем амплификации 145. Средняя поверхность клеточной подложки была увеличена в 30 раз в двух способах, но требовала использования 3 биореакторов в случае стандартного способа. Количество клеток, полученное с помощью стандартного способа, было немного выше. Это является следствием того, что клетки, которые использовали для выполнения способа «все-в-одном» и стандартного способа, не находились в одинаковом физиологическом состоянии. Клетки, которые использовали для посева в 20-литровый биореактор в случае способа «все-в-одном», были только что разморожены, в то время как клетки, используемые для посева в 2-литровый биореактор, были гораздо более жизнеспособными, поскольку они уже культивировались в Cell Factory. Результаты в D1 показывают это очень явно; в способе все-в-одном снижение числа клеток примерно на 40% в D1 является результатом стандартного явления «задержки после размораживания». В этот же период времени клетки, которые заблаговременно культивировались в Cell Factory, размножились в стандартном способе (клеточная популяция удвоилась). Несмотря на исходные условия культивирования, которые были явно неблагоприятными в способе «все-в-одном», следует отметить, что в конце культивирования наблюдалась очень маленькая разница между количеством клеток, собранных в двух способах. Можно заключить, что если бы в тестируемых двух способах были одинаковые исходные условия культивирования, то были бы получены одинаковое количество клеток и одинаковый уровень амплификации клеток. Поэтому способ «все-в-одном» является весьма предпочтительными по сравнению со стандартным способом, поскольку такое же количество клеток получают в течение такого же периода времени, экономя при этом средства.
Пример 6: амплификация клеток Vero путем проведения 3 последовательных пассажей клеток в одном и том же 200-литровом биореакторе
6.1) Используемые материалы
За исключением биореактора, которым в данном случае является 200-литровый одноразовый мешок, продаваемый ATMI под названием Nucleo-200, используемые материалы были такими же, как описанные в примере 3.
6.2) Рабочий протокол
Используемый рабочий протокол аналогичен описанному в примере 3со следующими модификациями.
Клетки Vero после размораживания помещали заблаговременно в культивирование в 2 устройства Cell Factory (CF10) в соотношении 40×103 клеток на см2 адгезионной поверхности и 2 литра культуральной среды на CF10. После 5 дней культивирования популяцию клеток собирали после стадии обработки трипсином и далее использовали для посева в Nucleo-200.
Первый пассаж клеток в Nucleo-200 проводили, внося 2,2×109 клеток в 50 литров культуральной среды VPSFM/K30, содержащей 25 г микрошариков Cytodex 1 (что представляет концентрацию микрошариков 0,5 г/л). В день D3 культуральную среду заменяли новой культуральной средой. В день D4 клетки трипсинизировли по протоколу из примера 3, оставляя ~20 литров среды в Nucleo и затем добавляя ~20 литров 0,025 М раствора цитрата натрия, содержащего 3000 мг рекомбинантного трипсина в фосфатном буфере, не содержащем ни кальция, ни магния. После открепления клеток действие трипсина останавливали добавлением 20 литров раствора VPSFM/K30, содержащего 1 мг/мл ингибитора трипсина.
Второй пассаж клеток проводили в том же Nucleo-200, добавляя ко всей полученной клеточной популяции суспензию микрошариков, содержащую 130 г Cytodex 1, что представляет концентрацию микрошариков приблизительно 1 г/л после доведения общего объем среды до 130 литров культуральной средой VPSFM/K30. Культуральную среду обновляли дважды: первый раз сразу после прикрепления клеток на микрошарики, второй раз - в день D6. В день D7 клетки снова трипсинизировали по такому же протоколу, который использовали в D4.
Третий пассаж клеток в том же Nucleo-200 проводили, регулируя популяцию клеток таким образом, чтобы получить концентрацию 20000 клеток/см2 адгезионной поверхности после внесения 450 г микрошариков Cytodex 1 в общем объеме среды, доведенном до 180 литров культуральной средой VPSFM/K30. Аналогичным образом культуральную среду обновляли дважды: сразу после адгезии клеток на микрошариках, а затем - в D10. Затем оценивали число клеток в популяции, полученной после 3 последовательных пассажей клеток, проведенных в одном биореакторе. Таким образом, можно измерить уровень амплификации клеток.
6.3) Результаты
Количество и концентрацию клеток, наблюдаемые в течение культивирования, представлены в таблице ниже:
**: после обработки трипсином и адгезии клеток
***: после обработки трипсином и подведения популяции клеток до концентрации 20000 клеток/смI адгезионной поверхности
После трех последовательных пассажей клеток Vero, проведенных в одном 200-литровом биореакторе, популяция клеток увеличилась в 140 раз через 11 дней культивирования, в то время как поверхность клеточной подложки увеличилась в 18 раз.
Тесты на амплификацию клеток Vero по способу «все-в-одном» также проводили, осуществляя, в частности, два последовательных пассажа клеток в одном 500-литровом одноразовом биореакторе, продаваемом ATMI под названием Nucleo-500. Полученные уровни амплификации клеток были аналогичны уровням, полученным с использованием 200-литрового биореактора, что ясно показывает, что способ по изобретению подходит для очень крупного масштаба.
Пример 7: получение вируса бешенства из партии клеток, полученных проведением 3 последовательных пассажей в одном и том же 200-литровом биореакторе
Партию клеток получали, используя такой же протокол, как описанный в примере 6.
В день D11 культуральную среду заменяли вирусной инфекционной средой и затем клетки инфицировали штаммом Pitman Moore вируса из института Wistar, с множественностью заражения 0,01. Такие же регулируемые параметры относительно температуры, рН, рО2 и умеренного перемешивания среды, как используемые для культивирования клеток, оставляли и подводили для продукции вируса. Вирусную инфекционную среду обновляли в день D3 после вирусной инфекции и культуральные супернатанты собирали для измерения инфекционных титров в дни D7, D10 и D14 после вирусной инфекции. После каждого сбора вируса снова добавляли вирусную инфекционную среду. Инфекционные титры в культуральных супернатантах измеряли посредством стандартного иммунофлуоресцентного теста на клетках ВНК21. Для каждого тестируемого культурального супернатанта приготавливали серию разведений, и затем каждое разведение распределяли по 10 лункам 96-луночного микропланшета. Проводили две серии тестов параллельно. Затем в каждую лунку добавляли суспензию клеток ВНК21. Клетки инкубировали 48 часов при 37°C в 5%-ном CO2. Через 48 часов лунки были покрыты слоем клеток, который затем фиксировали ацетоном. После удаления ацетона и высушивания микропланшета добавляли 50 мкл моноклонального антитела к вирусу бешенства (FDI Fujirebio Diagnostics, кат. № 800092) в разведении 1/70. После инкубирования в течение одного часа с последующими несколькими промывками микропланшеты анализировали под флуоресцентным микроскопом. Лунку считали положительной, если специфичная флуоресценция наблюдалась по меньшей мере в одной клетке. В качестве отрицательного контроля использовали клетки ВНК21, культивированные в отсутствие вируса бешенства, а в качестве положительного контроля использовали клетки ВНК21, культивированные в присутствии референсного штамма вируса бешенства. Инфекционные титры вируса бешенства, содержащегося в тестируемых культуральных супернатантах, определяли по способу Спермана-Карбера и выражали в log10 единицах доз для 50%-ной инфекции клеточной культуры (CCID 50). Наблюдаемые инфекционные титры, полученные в культуральных супернатантах, составляли примерно 7,0 log10 CCID 50.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ КЛЕТОК МЛЕКОПИТАЮЩИХ ДЛЯ ИСПОЛЬЗОВАНИЯ В ПРОИЗВОДСТВЕ ВИРУСОВ | 1998 |
|
RU2230784C2 |
| ВАКЦИНА IPV-DPT | 2007 |
|
RU2447898C2 |
| КУЛЬТУРА ПЛЮРИПОТЕНТНЫХ СТВОЛОВЫХ КЛЕТОК НА МИКРОНОСИТЕЛЯХ | 2009 |
|
RU2555538C2 |
| СПОСОБ КРУПНОМАСШТАБНОГО ПРОИЗВОДСТВА ВИРУСНОГО АНТИГЕНА | 2002 |
|
RU2314344C2 |
| ОБОГАЩЕННАЯ ПИТАТЕЛЬНАЯ СРЕДА ДЛЯ ВЫРАЩИВАНИЯ КЛЕТОК, ИЗВЛЕЧЕННЫХ ИЗ ТКАНИ ПУПОВИНЫ ЧЕЛОВЕКА | 2013 |
|
RU2653449C2 |
| ОБОГАЩЕННАЯ ПИТАТЕЛЬНАЯ СРЕДА ДЛЯ ВЫРАЩИВАНИЯ КЛЕТОК, ИЗВЛЕЧЕННЫХ ИЗ ТКАНИ ПУПОВИНЫ ЧЕЛОВЕКА | 2013 |
|
RU2684306C2 |
| СУСПЕНДИРОВАНИЕ И КЛАСТЕРИЗАЦИЯ ПЛЮРИПОТЕНТНЫХ КЛЕТОК ЧЕЛОВЕКА С ЦЕЛЬЮ ИХ ДИФФЕРЕНЦИРОВКИ В ПАНКРЕАТИЧЕСКИЕ ЭНДОКРИННЫЕ КЛЕТКИ | 2013 |
|
RU2687379C2 |
| СПОСОБ ПОЛУЧЕНИЯ АНТИРАБИЧЕСКОЙ ВАКЦИНЫ | 2005 |
|
RU2287343C1 |
| СПОСОБ ПОЛУЧЕНИЯ ЖИВОЙ КУЛЬТУРАЛЬНОЙ ВАКЦИНЫ ПРОТИВ ВИРУСА ГРИППА | 2009 |
|
RU2420314C1 |
| СУСПЕНДИРОВАНИЕ И КЛАСТЕРИЗАЦИЯ ПЛЮРИПОТЕНТНЫХ СТВОЛОВЫХ КЛЕТОК ЧЕЛОВЕКА С ЦЕЛЬЮ ИХ ДИФФЕРЕНЦИРОВКИ В ПАНКРЕАТИЧЕСКИЕ ЭНДОКРИННЫЕ КЛЕТКИ | 2014 |
|
RU2689710C2 |
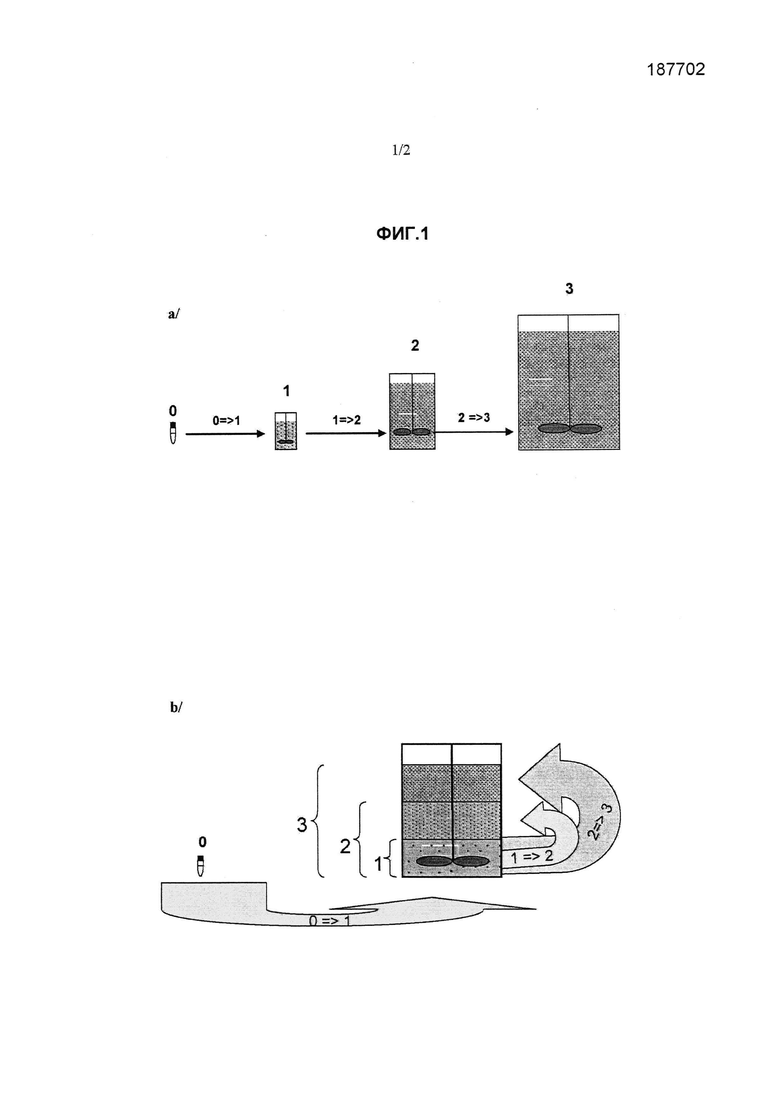

Изобретение относится к клеточной технологии. Описан способ получения адгезионных клеток в соответствии с которым: a. адгезионные клетки вносят в культуральный сосуд, который содержит микроносители в культуральной среде; b. клетки амплифицируют, проводя несколько последовательных пассажей клеток в том же самом культуральном сосуде, причем каждый пассаж клеток, следующий за первым пассажем клеток, проводят: i. используя всю или часть популяции клеток, которая была получена в течение предшествующего пассажа клеток, после проведения ферментативной обработки популяции клеток для открепления клеток от микроносителей; и ii. внося культуральную среду и увеличивая количество микроносителей; и c. популяцию адгезионных клеток, полученную в течение последнего пассажа клеток, собирают после необязательного проведения ферментативной обработки указанной популяции клеток для открепления клеток от микроносителей. Изобретение расширяет арсенал средств для получения адгезионных клеток. 3 н. и 37 з.п. ф-лы, 2 ил., 8 табл., 7 пр.
1. Способ амплификации адгезионных клеток, в соответствии с которым:
a. адгезионные клетки вносят в культуральный сосуд, который содержит микроносители в культуральной среде;
b. клетки амплифицируют, проводя несколько последовательных пассажей клеток в том же самом культуральном сосуде, причем каждый пассаж клеток, следующий за первым пассажем клеток, проводят:
i. используя всю или часть популяции клеток, которая была получена в течение предшествующего пассажа клеток, после проведения ферментативной обработки популяции клеток для открепления клеток от микроносителей; и
ii. внося культуральную среду и увеличивая количество микроносителей; и
c. популяцию адгезионных клеток, полученную в течение последнего пассажа клеток, собирают после необязательного проведения ферментативной обработки указанной популяции клеток для открепления клеток от микроносителей.
2. Способ по п. 1, где число пассажей клеток, проводимых в одном культуральном сосуде, составляет 2, 3 или 4.
3. Способ по п. 1, где концентрация микроносителей в культуральной среде в течение первого пассажа клеток составляет <1 г/л и предпочтительно ≤0,5 г/л.
4. Способ по п. 1, где при проведении ферментативной обработки используют раствор, содержащий протеолитический фермент.
5. Способ по п. 4, где протеолитический фермент представляет собой трипсин.
6. Способ по п. 1, где каждый последующий пассаж клеток проводят, увеличивая объем культуральной среды.
7. Способ по п. 1, где первый пассаж клеток проводят в объеме культуральной среды, который составляет от 1/5 до половины рабочего объема культурального сосуда.
8. Способ по п. 1, где культуральная среда свободна от сыворотки животного происхождения.
9. Способ по п. 1, где культуральная среда свободна от любого продукта животного происхождения.
10. Способ по п. 1, где концентрация белка в культуральной среде составляет ≤15 мг/л.
11. Способ по п. 1, где культуральная среда также содержит клеточный протекторный агент.
12. Способ по п. 11, где клеточным протекторным агентом является поливинилпирролидон или полоксамер.
13. Способ по п. 1, где культуральный сосуд представляет собой биореактор, который имеет рабочий объем между 3 и 3000 литрами, предпочтительно между 20 и 1000 литрами и особенно предпочтительно между 20 и 500 литрами.
14. Способ по п. 13, где культуральным сосудом является биореактор для однократного применения.
15. Способ по п. 1, где адгезионные клетки представляют собой клетки Vero.
16. Способ по любому из пп. 1-15, где популяция клеток, которую собирают на стадии (с), содержит, по меньшей мере, 60-кратное количество клеток, исходно внесенных на стадии (а) способа.
17. Способ по п. 1, дополнительно включающий:
a. перенос популяции клеток, собранной после ферментативной обработки указанной популяции клеток для открепления клеток от микроносителей, во второй культуральный сосуд, который имеет больший рабочий объем и который содержит культуральную среду, содержащую большее количество микропор относительно количества микропор, присутствующего в течение последнего пассажа клеток, проведенного в первом культуральном сосуде, и
b. проведение стадий (b) и (с) способа по п. 1 во втором сосуде.
18. Способ по п. 17, в соответствии с которым стадии (а) и (b) по п. 17 повторяют снова в третьем культуральном сосуде, который имеет больший рабочий объем, чем рабочий объем второго культурального сосуда.
19. Способ получения вируса, продуцируемого адгезионными клетками, в соответствии с которым:
a. адгезионные клетки вносят в культуральный сосуд, который содержит микроносители в культуральной среде;
b. клетки амплифицируют, проводя несколько последовательных пассажей клеток в этом одном культуральном сосуде, причем каждый пассаж клеток, следующий за первым пассажем клеток, проводят:
i. используя всю или часть популяции клеток, которая получена в течение предшествующего пассажа клеток, после проведения ферментативной обработки указанной популяции клеток для открепления клеток от микроносителей; и
ii. внося культуральную среду и увеличивая количество микроносителей;
c. популяцию клеток, получаемую в течение последнего клеточного пассажа, обрабатывают таким образом, чтобы она продуцировала вирус, причем указанную обработку проводят в том же культуральном сосуде, который использовали для амплификации клеток; и
d. собирают вирус.
20. Способ по п. 19, где обработку популяции клеток на стадии (с) проводят, заражая популяцию клеток вирусом в инфекционной среде.
21. Способ по п. 20, в котором вирусом является вирус бешенства.
22. Способ по п. 20 или 21, где инфекционной средой является вирусная инфекционная среда, свободная от любого продукта животного происхождения.
23. Способ по п. 19, где число пассажей клеток, проводимых в одном культуральном сосуде, составляет 2, 3 или 4.
24. Способ по п. 19, где концентрация микроносителей в культуральной среде в течение первого пассажа клеток составляет <1 г/л и предпочтительно ≤0,5 г/л.
25. Способ по п. 24, где при проведении ферментативной обработки используют раствор, содержащий протеолитический фермент.
26. Способ по п. 25, где протеолитический фермент представляет собой трипсин.
27. Способ по п. 19, где каждый последующий пассаж клеток проводят, увеличивая объем культуральной среды.
28. Способ по п. 19, где первый пассаж клеток проводят в объеме культуральной среды, который составляет от 1/5 до половины рабочего объема культурального сосуда.
29. Способ по п. 19, где культуральная среда свободна от сыворотки животного происхождения.
30. Способ по п. 19, где культуральная среда свободна от любого продукта животного происхождения.
31. Способ по п. 19, где концентрация белка в культуральной среде составляет ≤15 мг/л.
32. Способ по п. 19, где культуральная среда также содержит клеточный протекторный агент.
33. Способ по п. 32, где клеточным протекторным агентом является поливинилпирролидон или полоксамер.
34. Способ по п. 19, где культуральный сосуд представляет собой биореактор, который имеет рабочий объем между 3 и 3000 литрами, предпочтительно между 20 и 1000 литрами и особенно предпочтительно между 20 и 500 литрами.
35. Способ по п. 34, где культуральным сосудом является биореактор для однократного применения.
36. Способ по п. 19, где адгезионные клетки представляют собой клетки Vero.
37. Способ по любому из пп. 23-36, где популяция клеток, которую собирают на стадии (с), содержит, по меньшей мере, 60-кратное количество клеток, исходно внесенных на стадии (а) способа.
38. Способ по любому из пп. 23-36, дополнительно включающий:
a. перенос популяции клеток, собранной после ферментативной обработки указанной популяции клеток для открепления клеток от микроносителей, во второй культуральный сосуд, который имеет больший рабочий объем и который содержит культуральную среду, содержащую большее количество микропор относительно количества микропор, присутствующего в течение последнего пассажа клеток, проведенного в первом культуральном сосуде, и
b. проведение стадий (b) и (с) способа по любому из пп. 23-36 во втором сосуде.
39. Способ по п. 38, в соответствии с которым стадии (а) и (b) по п. 38 повторяют снова в третьем культуральном сосуде, который имеет больший рабочий объем, чем рабочий объем второго культурального сосуда.
40. Способ амплификации адгезионных клеток в культуральном сосуде, который содержит микроносители в культуральной среде, в соответствии с которым количество получаемых клеток увеличивается с кратностью ≥60 в результате проведения способа по любому из пп. 1-18.
| XIAO C | |||
| High density cultivation of genetically-engineered CHO cell lines with microcarrier culture systems | |||
| Chin Med Sci J | |||
| Прибор для охлаждения жидкостей в зимнее время | 1921 |
|
SU1994A1 |
| СПОСОБ КРУПНОМАСШТАБНОГО ПРОИЗВОДСТВА ВИРУСНОГО АНТИГЕНА | 2002 |
|
RU2314344C2 |

