ПЕРЕКРЕСТНАЯ ССЫЛКА НА СМЕЖНЫЕ ЗАЯВКИ
Настоящая заявка является частичным продолжением заявки на патент США № 13/998,974 (поданной 30 декабря 2013 года), которая истребует приоритет, заявленный в предварительной заявке на патент США № 61/747,799 (поданной 31 декабря 2012 года) и в предварительной заявке на патент США № 61/962,158 (поданной 1 ноября 2013 года), которые полностью включены в настоящий документ путем ссылки.
ОБЛАСТЬ ПРИМЕНЕНИЯ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к области клеточной дифференцировки, включая подготовку эмбриональных стволовых клеток и других плюрипотентных клеток, которые поддерживают плюрипотентность в агрегированном клеточном кластере, с целью их дифференцировки в клетки-предшественники энтодермы, панкреатические эндокринные клетки, клетки мезодермы или клетки эктодермы. В одном аспекте данного изобретения раскрывается способ генерации кластеров плюрипотентных стволовых клеток и поддержания их в суспензионной культуре для дифференциации энтодертмы поджелудочной, панкреатических эндокринных клеток-предшественников и одногормонных панкреатических эндокринных клеток.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Последние достижения в области заместительной клеточной терапии для лечения сахарного диабета 1 типа и нехватка островков Лангерганса с возможностью трансплантации заставили обратить внимание на разработку источников инсулин-продуцирующих клеток, или β-клеток, подходящих для приживления трансплантата. Один подход представляет собой получение функциональных бета-клеток из плюрипотентных стволовых клеток, таких как, например, эмбриональные стволовые клетки.
При эмбриональном развитии позвоночных плюрипотентные клетки дают начало группе клеток, содержащих три зародышевых листка (эктодерму, мезодерму и энтодерму), в ходе процесса, известного как гаструляция. Ткани, такие как ткань щитовидной железы, вилочковой железы, поджелудочной железы, кишечника и печени, будут развиваться из энтодермы через промежуточную стадию. Промежуточная стадия данного процесса представляет собой образование дефинитивной энтодермы.
К концу гаструляции энтодерма разделяется на передний и задний отделы, которые могут быть распознаны по экспрессии ряда факторов, однозначно выделяющих переднюю, среднюю и заднюю области энтодермы. Например, HHEX и SOX2 идентифицируют переднюю область, в то время как CDX1, 2 и 4 идентифицируют заднюю половину энтодермы.
Миграция ткани энтодермы приближает энтодерму к различным мезодермальным тканям, которые способствуют регионализации кишечной пробирки. Это достигается за счет целого ряда секретируемых факторов, таких как FGFs, Wnts, TGF-Bs, ретиноевая кислота (RA), лиганды BMP и их антагонисты. Например, FGF4 и BMP способствуют экспрессии Cdx2 в предполагаемой энтодерме задней кишки и подавляют экспрессию передних генов Hhex и SOX2 (Development 2000 г., 127:1563-1567). Было продемонстрировано, что сигнализация WNT также действует параллельно с сигнализацией FGF, способствуя развитию задней кишки и препятствуя зачаткам передней кишки (Development 2007 г., 134:2207-2217). Наконец, ретиноевая кислота, секретируемая мезенхимой, регулирует границу между передней и задней кишкой (Curr Biol 2002 г., 12:1215-1220).
Уровень экспрессии специфических факторов транскрипции может использоваться для определения типа ткани. Во время преобразования дефинитивной энтодермы в примитивную кишечную пробирку кишечная пробирка становится разделенной на широкие домены, которые можно наблюдать на молекулярном уровне с помощью ограниченных паттернов экспрессии генов. Например, регионализованный домен поджелудочной железы в кишечной пробирке демонстрирует очень высокую экспрессию PDX1 и очень низкую экспрессию CDX2 и SOX2. уровень экспрессии PDX1, NKX6.1, PTF1A и NKX2.2 высок в ткани поджелудочной железы; а уровень экспрессии CDX2 высок в ткани кишечника.
Образование поджелудочной железы происходит при дифференцировке дефинитивной энтодермы в панкреатическую энтодерму. Дорсальный и вентральный домены поджелудочной железы формируются из эпителия передней кишки. Передняя кишка также дает начало формированию пищевода, трахеи, легких, щитовидной железы, желудка, печени, поджелудочной железы и системы желчных протоков.
Клетки панкреатической энтодермы экспрессируют панкрео-дуоденальный, содержащий гомеобокс ген PDX1. В отсутствие PDX1 поджелудочная железа не развивается дальше образования вентрального и дорсального зачатков. Следовательно, экспрессия PDX1 является важной стадией органогенеза поджелудочной железы. Зрелая поджелудочная железа содержит как экзокринную так и эндокринную ткани, которые образуются при дифференцировке панкреатической энтодермы.
D’Amour et al. описывают получение обогащенных культур дефинитивной энтодермы, полученной из эмбриональных стволовых клеток человека в присутствии высокой концентрации активина и низкой концентрации сыворотки (Nature Biotechnol 2005, 23:1534-1541; патент США № 7,704,738). Трансплантация этих клеток под капсулу почки у мышей приводила к дифференцировке в более зрелые клетки с характеристиками ткани энтодермы (патент США № 7,704,738). Клетки дефинитивной энтодермы, полученные из эмбриональных стволовых клеток человека, могут быть далее дифференцированы в PDX1-положительные клетки после добавления FGF10 и ретиноевой кислоты (публикация заявки на патент США № 2005/0266554A1). Последующая трансплантация таких клеток-предшественников панкреатических клеток в жировое тело иммунодефицитных мышей привела к образованию функциональных панкреатических эндокринных клеток с последующей 3-4-месячной стадией созревания (патент США № 7,993,920 и патент США № 7,534,608).
Fisk с соавторами сообщают о системе получения клеток панкреатических островков из эмбриональных стволовых клеток человека (патент США № 7,033,831). Низкомолекулярные ингибиторы также применяли для индуцирования клеток-предшественников панкреатических эндокринных клеток. Например, низкомолекулярные ингибиторы рецептора TGF-ß и рецепторов BMP (Development 2011, 138:861-871; Diabetes 2011, 60:239-247) использовали для значительного увеличения количества панкреатических эндокринных клеток. Кроме того, для генерирования клеток дефинитивной энтодермы или клеток-предшественников панкреатических клеток также применяли низкомолекулярные активаторы (Curr Opin Cell Biol 2009, 21:727-732; Nature Chem Biol 2009, 5:258-265).
В последнее время были намного улучшены протоколы культивирования клеток-предшественников, таких как плюрипотентные стволовые клетки. Публикация РСТ No. WO2007/026353 (Amit с соавторами) раскрывает технологии поддержания человеческих эмбриональных стволовых клеток в недифференцированном состоянии в двухмерной культуре. Ludwig с соавторами, 2006 (Nature Biotechnology, 24: 185-7) раскрывает среду на основе TeSR1 для культивирования человеческих эмбриональных стволовых клеток на матрице. заявкой на патент США на патент США № No. 2007/0155013 (Akaike с соавторами) Описывает способ выращивания плюрипотентных стволовых клеток в суспензии с использованием носителя, который прикрепляется к плюрипотентным стволовым клеткам, и в заявке на патент США на патент США № No. 2009/0029462 (Beardsley с соавторами) раскрывает способы роста плюрипотентных стволовых клеток в суспензии с использованием микроносителей или клеточной инкапсуляции. В публикации РСТ No. WO 2008/015682 (Amit с соавторами) описан способ роста и поддержания человеческих эмбриональных стволовых клеток в суспензионной культуре в особых условиях культивирования при отсутствии субстратной подложки.
заявкой на патент США на патент США № No. 2008/0159994 (Mantalaris др.) описан способ культивирования инкапсулированных в частицы альгината человеческих эмбриональных стволовых клеток в трехмерной культуральной системе.
Несмотря на эти достижения, все еще необходим способ успешного культивирования в трехмерной культуральной системе плюрипотентных стволовых клеток, которые могут дифференцироваться в функциональные эндокринные клетки
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
Предшествующее краткое изложение сущности изобретения, а также последующее подробное описание изобретения, будут более понятными при рассмотрении вместе с прилагаемыми фигурами. В целях иллюстрирования изобретения, на фигурах показаны варианты осуществления настоящего изобретения. Необходимо понимать, однако, что изобретение не ограничивается показанными точными конструкциями, примерами и инструментарием.
На Фиг. 1a представлены микрофотографии обработанных Dispase® эмбриональных стволовых клеток человека (hES) клеточной линии H1 сразу после отделения (левая панель) и спустя 24 часа в не допускающей прикрепления статической культуре (правая панель) в соответствии с примером 1. Клетки после отделения (левая панель) напоминали фрагменты монослоя со средним диаметром фрагмента приблизительно 20-30 мкм, каждый фрагмент состоял из скопления клеток. Спустя 24 часа в не допускающей прикрепления статической культуре клетки сформировали кластерные конфигурации.
На Фиг. 1b представлены результаты проточной цитометрии для CD9, SSEA4, CXCR4, TRA-1-60 и TRA-1-81 для обработанных Dispase® эмбриональных стволовых клеток человека из линии стволовых клеток H1 после культивирования в течение 4 дней в центрифужной пробирке на 125 мл, содержащей 25 мл носителя mTeSR®1 в соответствии с примером 1. Клетки показали высокую экспрессию маркеров плюрипотентности (CD9, SSEA4, TRA-1-60 и TRA-1-81) почти без выражения CXCR4, маркера дифференцировки.
На Фиг. 1c представлены микрофотографии обработанных Dispase® эмбриональных стволовых клеток человека из линии стволовых клеток H1 после 72 и 96 часов дифференцировки в конце стадии 1. На Фиг. 1с заметны неприкрепленные агрегаты клеток после 72 часа при 4-кратном увеличении (левая панель), 96 часов при 4-кратном увеличении (центральная панель) и 96 часов при 10-кратном увеличением (правая панель).
На Фиг. 1d показаны результаты проточной цитометрии обработанных Dispase® эмбриональных стволовых клеток человека из линии стволовых клеток H1 в конце 1 стадии дифференциации для маркеров CD9, CD184 (CXCR4) и CD99 (см. пример 1). Как показано на Фиг. 1d, экспрессия CD9, маркера плюрипотентности, практически исчезла, в то время как экспрессия маркеров окончательной дифференцировки энтодермы CXCR4 (CD184) и CD99 была достаточно высокой.
На Фиг. 1e показаны результаты количественной полимеразной цепной реакции с обратной транскрипцией (qRT-PCR) для экспрессии выбранных генов, ассоциированных с плюрипотентностью и генов, ассоциированных с окончательной дифференцировкой энтодермы обработанных Dispase® эмбриональных стволовых клеток человека из линии стволовых клеток H1 в конце 1 стадии по сравнению с недифференцироваными клетками H1 (WA01) hES (см. пример 1). Клетки в конце стадии 1 показали резкое снижение экспрессии генов плюрипотентности (CD9, NANOG и POU5F1/OCT4) и повышение экспрессии генов, связанных с окончательной дифференцировкой энтодермы (CXCR4, Cerberus (CER1), GSC, Foxa2, GATA4, GATA6, MNX1 и SOX17), по сравнению с недифференцированными клетками линии hES WA01.
На Фиг. 1f показаны микрофотографии обработанных Dispase® эмбриональных стволовых клеток человека из линии стволовых клеток H1, показывающие дальнейшую дифференцировку от окончательно дифференцированной энтодермы в панкреатическую энтодерму (см. Пример 1). По мере прогрессирования дифференцировки явно заметны морфологические изменения в клетках и клеточных кластерах от стадии 2, сутки 1 (верхняя левая панель) до стадии 2, сутки 3 (верхняя правая панель), далее до стадии 3, сутки 4 (нижняя левая панель) и стадии 4, сутки 1 (нижняя правая панель).
На Фиг. 2а изображены данные, полученные в результате проточной цитометрии обработанных EDTA клеток эмбриона человека из линии стволовых клеток H1 после 2 дней культивирования в перемешиваемой культурной суспензии, обработанной EDTA, и перед переходом к дифференцировке культуры, для маркеров, связанных с плюрипотентностью и дифференцировкой, согласно примеру 2. Данные показывают высокую экспрессию маркеров плюрипотентности (CD9, SSEA4, TRA-1-60 и TRA-1-81) почти без выражения маркера дифференцировки (CXCR4).
На Фиг. 2b показаны микрофотографии обработанных EDTA клеток эмбриона человека из линии стволовых клеток H1, дифференцированных на стадии 1, сутки 3 для клеток, выращенных в центрифужной пробирке и стадии 2, сутки 2, стадии 4 сутки 1 и стадии 4 сутки 3 для клеток, выращенных в центрифужной пробирке или колбе Эрленмейера согласно примеру 2. В суспензии дифференцированых культур формируются по существу равномерные и однородные популяции клеток в сферических агрегатах.
На Фиг. 2c показаны данные, полученные в результате проточной цитометрии обработанных EDTA эмбриональных стволовых клеток человека из линии стволовых клеток H1 в конце 1 стадии на маркеры клеточной поверхности, показывающие плюрипотентность и дифференцировку энтодермы. Как показано на Фиг. 2c экспрессия CD9, маркера плюрипотентности, практически исчезла, в то время как экспрессия маркера окончательной дифференцировки энтодермы CXCR4 (CD184) была достаточно высокой.
На Фиг. 2d показаны результаты qRT-PCR для экспрессии выбранных генов, ассоциированных с плюрипотентностью и генов, ассоциированных с окончательной дифференцировкой энтодермы обработанных EDTA эмбриональных стволовых клеток человека из линии стволовых клеток H1 в конце 1 стадии по сравнению с недифференцироваными клетками H1 (WA01) hES (см. пример 2). На Фиг. 2d показано резкое снижение экспрессии генов плюрипотентности (CD9, NANOG и POU5F1/OCT4) и повышение экспрессии генов, связанных с окончательной дифференцировкой энтодермы (CXCR4, Cerberus (CER1), Foxa2, GATA4, GATA6, MNX1 и SOX17).
На Фиг. 2e показаны данные экспрессии маркеров дифференцирования (Nkx6.1, CDX2, Sox2 и Chromagranin), полученные в результате проточной цитометрии обработанных EDTA эмбриональных стволовых клеток человека из линии стволовых клеток H1, которые были дифференцированы с начала стадии 1 в клетки панкреатической энтодермы с помощью суспендирования в центрифужных пробирках или колбах Эрленмейера согласно примеру 2. Данные проточной цитометрии показывают в обоих форматах суспензии высокий уровень Nkx6.1, транскрипционного фактора, необходимого для функциональных β-клеток, и высокие уровни маркеров эндокринной поджелудочной железы, таких как синаптофизин (данные не показаны) и хромогранин.
На Фиг. 2f показаны результаты qRT-PCR экспрессии отдельных генов, ассоциирующихся с дифференцировкой обработанных EDTA эмбриональных стволовых клеток человека из линии стволовых клеток H1, которые были дифференцированы с начала стадии 1 в клетки панкреатической энтодермы с помощью суспендирования в центрифужных пробирках или колбах Эрленмейера согласно Примеру 2. Эти данные сравнивали с данным экспрессии в клеточной линии WA01 hES. Результаты RT-PCR показывают высокие уровни нескольких панкреатических генов-предшественников.
На Фиг. 3a показаны микрофотографии стволовых клеток эмбриона человека линии H1, отделившиеся от статической культуры после обработки Accutase®. Как показано на Фиг. 3, клетки были удалены с поверхности в виде мелких агрегатов.
На Фиг. 3b показаны фазоконтрастные микрофотографии стволовых клеток эмбриона человека линии H1, отделившиеся от статической культуры после обработки Accutase® и после выращенные в суспензионной культуре в течение трех суток. На Фиг. 3b видно формирование по существу равномерных сферических популяций кластеров клеток
На Фиг. 3c показана микрофотография стволовых клеток эмбриона человека линии H1, отделившиеся от статической культуры после обработки Accutase®, выращенные в суспензионной культуре в течение трех суток и подвергнувшиеся серийной пассировке с использованием диссоциации Accutase®.
На Фиг. 4a показаны микрофотографии стволовых клеток эмбриона человека линии H1, приведенных в состояние суспензионной культуры с использованием различных протоколов направленной дифференцировки на различных стадиях дифференцировки. На Фиг. 4 показаны микрофотографии клеток на каждой стадии дифференцировки
На Фиг. 4б приведены результаты проточной цитометрии для маркеров дифференцировки (CXCR4, CD56 и Pdx1) стволовых клеток эмбриона человека линии H1, приведенных в состояние суспензионной культуры с использованием различных протоколов направленной дифференцировки на различных стадиях дифференцировки (часы после начала дифференцировки). В конце процесса дифференцировки на 4-е сутки 4-й стадии высокий процент клеток показал положительную экспрессию Pdx1.
На Фиг. 4c показаны уровни глюкозы у сытых мышей линии SCID-Bg, которым с помощью прибора TheraCyte™ были имплантированы инкапсулированные дифференцированные клетки.
На Фиг. 5a показаны результаты, полученные в результате проточной цитометрии обработанных EDTA эмбриональных стволовых клеток человека из линии стволовых клеток H1 до начала процесса дифференцировки на маркеры клеточной поверхности, показывающие плюрипотентность и дифференцировку энтодермы. Как показано На Фиг. 5a, наблюдалась высокая экспрессия маркеров плюрипотентности CD9, SSEA4, TRA-1-60 и TRA-1-80.
На Фиг. 5b показано фазоконтрастное изображение клеток и результаты проточной цитометрии маркеров дифференцировки CXCR4/CD184 и CD99, а также маркера плюрипотентности CD9 при трех различных настройках подачи во время стадии 1. Тестирование проводили при следующих условиях: (A) замена среды через 24 часа после начала дифференцировки, без замены среды через 48 часов; (B) замена среды через 24 часа после начала дифференцировки и добавление болюса глюкозы через 48 часов; и (C) без замены среды в течение стадии 1 с добавлением болюсов глюкозы и GDF8 через 24 ч после начала дифференцировки и последующего добавления болюса глюкозы через 48 часов после начала.
На Фиг. 5с показаны фазоконтрастные изображения дифференцированных клеток, проявляющих морфологию поджелудочной энтодермы, которые были дифференцированы с использованием следующих настроек подачи во время формирования дефинитивной энтодермы: (A) замена среды через 24 часа после начала дифференцировки, без замены среды через 48 часов; (B) замена среды через 24 часа после начала дифференцировки и добавление болюса глюкозы через 48 часов; и (C) без замены среды в течение стадии 1 с добавлением болюсов глюкозы и GDF8 через 24 ч после начала дифференцировки и последующего добавления болюса глюкозы через 48 часов после начала.
На Фиг. 5d показаны результаты проточной цитометрии для отдельных маркеров экспрессии генов поджелудочной (Nkx6.1 и хромагранин) и выбранных непанкреатических генов (CDX2 и Sox2) дифференцированной клетки в конце стадии 4, которые были дифференцированы с помощью следующих настроек подачи при формировании дефинитивной энтодермы: (A) замена среды через 24 часа после начала дифференцировки, без замены среды через 48 часов; (B) замена среды через 24 часа после начала дифференцировки и добавление болюса глюкозы через 48 часов; и (C) без замены среды в течение стадии 1 с добавлением болюсов глюкозы и GDF8 через 24 ч после начала дифференцировки и последующего добавления болюса глюкозы через 48 часов после начала.
На Фиг. 5e показаны результаты qRT-PCR выбранной экспрессии генов поджелудочной железы и непанкреатических генов для дифференцированных клеток, в конце 4-й стадии, которые были дифференцированы с использованием следующих настроек подачи во время формирования дефинитивной энтодермы: (A) замена среды через 24 часа после начала дифференцировки, без замены среды через 48 часов; (B) замена среды через 24 часа после начала дифференцировки и добавление болюса глюкозы через 48 часов; и (C) без замены среды в течение стадии 1 с добавлением болюсов глюкозы и GDF8 через 24 ч после начала дифференцировки и последующего добавления болюса глюкозы через 48 часов после начала. Данные показаны в виде кратной разницы в экспрессии относительно недифференцированных клеток линии H1 (WA01) (исходная экспрессия 1).
Фиг. 5f показывает экспрессию C-пептида у мышей линии SCID-Bg с имплантированными клетками, дифференцированными в соответствии с условием А (замена среды через 24 часа после начала дифференцировки, без замены среды через 48 часов). Каждой мыши линии SCID-Bg имплантировали 5 млн клеток под почечную капсулу. Как показано на Фиг. 5f, от 12 недель после имплантации человеческий С-пептид был обнаружен на уровнях выше 1 нг/мл, а через 16 недель уровни С-пептида в среднем составляли 2,5 нг/мл.
Фиг. 5g показывает эффект обработки глюкозой выбранных мышей линии SCID-BG до и после введения (например, имплантации) клеток, дифференцированных по условию А (замена среды через 24 часа после начала дифференцировки без замены среды через 48 часов). Как показано на Фиг. 5g, обработка глюкозой вызвала значительное увеличение циркулирующего С-пептида человека в среднем от 0,93 нг/мл в голодном состоянии до 2,39 нг/мл в сытом состоянии.
Фиг. 5h показывает эффект введения стрептозотоцина (STZ) (т.е. STZ-индуцированного диабета) мышам линии SCID-BG, которым вводили клетки, дифференцированные по условию А (замена среды через 24 ч после начала дифференцировки, без замены среды через 48 часов). Как показано на Фиг. 5h, у животных с трансплантатом функциональной GSIS-компетентной ткани (т. е. такой, в которую были введены клетки) поддерживается нормальный уровень глюкозы в крови в отличие от необработанной контрольной группы, у которой развился откровенный диабет.
На Фиг. 6а показаны микрофотографии стволовых клеток эмбриона человека линии H1, выращенных на трех сферах микроносителя Cytodex® до дифференцировки.
На Фиг. 6b показаны микрофотографии стволовых клеток эмбриона человека линии H1, выращенных на трех сферах микроносителя Cytodex® на различных стадиях дифференцировки.
На Фиг. 6с показано количество клеток (клетки/см2) в зависимости от суток дифференцировки для стволовых клеток эмбриона человека линии H1, выращенных и дифференцированных на носителях в среде, содержащей активин A (AA) и WNT3A (носители WTN3A/АА), микроносителях в среде, содержащей активин A и WNT3a (микроносители WTN3A/AA), носителях в среде, содержащей MCX и GDF8 (MCX/GDF8 носители) и микроносителях в среде, содержащей MCX и GDF8 (MCX/GDF8 микроносители).
На Фиг. 6d показано количество клеток (клетки/мл) в зависимости от суток дифференцировки для стволовых клеток эмбриона человека линии H1, выращенных и дифференцированных на носителях в среде, содержащей активин A и WNT3A (носители WTN3A/АА), микроносителях в среде, содержащей активин A и WNT3a (микроносители WTN3A/AA), носителях в среде, содержащей MCX и GDF8 (MCX/GDF8 носители) и микроносителях в среде, содержащей MCX и GDF8 (MCX/GDF8 микроносители).
На Фиг. 6е показаны результаты проточной цитометрии для первой стадии дифференцировки клеток, выращенных на культуре микроносителя или плоской культуре в присутствии: (a) WNT3A и AA; или (2) MCX и GDF8 в виде точечной диаграммы клеточной экспрессии CXCR4/CD184 (по оси Y) и CD9 (по оси Х).
На Фиг. 6f показаны результаты проточной цитометрии для первой стадии дифференцировки клеток, выращенных на культуре микроносителя или плоской культуры в присутствии: (a) WNT3A и AA; или (2) MCX и GDF8 в общей экспрессии каждого из маркеров (CXCR4 и CD9).
На Фиг. 6g показаны результаты количественной PCR экспрессии выбранных генов, ассоциированных с дифференцировкой стволовых клеток эмбриона человека линии H1, которые были дифференцированы выращиванием на плоской культуре или сферах микроносителя в суспензионной культуре в присутствии: (a) WNT3A и AA; или (2) MCX и GDF8.
На Фиг. 7 показано количество клеток на различных стадиях дифференцировки в биореакторе со стадии 1, 1 сутки до 4-й стадии, 3 суток для клеток, дифференцированных в соответствии с протоколом примера 7. Количество клеток показано в млн клеток/мл, что определено посредством цитометра на основе изображений (NucleoCounter®).
На Фиг. 8 показаны среднесуточные уровни рН среды в биореакторе как функцию по времени (сутки дифференцировки) согласно протоколу дифференцировки примера 7. Уровни рН определяли с помощью NOVA BioProfile® FLEX (Nova Biomedical Corporation, г. Уолтем, штат Массачусетс, США).
Фиг. 9 показывает среднесуточный уровень лактата среды в биореакторе как функцию по времени (сутки дифференцировки), согласно протоколу дифференцировки примера 7. Уровни лактата определяли с помощью NOVA BioProfile® FLEX (Nova Biomedical Corporation, г. Уолтем, штат Массачусетс, США).
На Фиг. 10 показана среднесуточный уровень глюкозы в среде в биореакторе как функцию по времени (сутки дифференцировки) согласно протоколу дифференцировки примера 7. Уровни глюкозы определяли с помощью NOVA BioProfile® FLEX (Nova Biomedical Corporation, г. Уолтем, штат Массачусетс, США).
Фиг.11 показана недифференцированная экспрессия гена, определенная способом QRT-PCR, для стадии 0, 1 сутки (т.е. через двадцать четыре часа после посева), клетки дифференцированы в соответствии с протоколом примера 7 для массива плюрипотентности, который содержит выбранные гены, связанные с плюрипотентностью.
На Фиг. 12 показана недифференцированная экспрессия гена, определенная способом QRT-PCR, для стадии 0, 1 сутки (то есть двадцать четыре часа после посева) клеток для дефинитивной энтодермы («DE») массива, который содержит выбранные гены, связанные с дефинитивной энтодермой (пример 7).
Фиг.13 показывает недифференцированную экспрессию гена, определенную способом QRT-PCR, для стадии 0, 3 суток (т.е. семьдесят два часа после посева клеток) для массива плюрипотентности, который содержит выбранные гены, связанные с плюрипотентностью (смотри пример 7).
Фиг. 14 показывает недифференцированную экспрессию гена, определенную способом QRT-PCR, для стадии 0, 3 суток (т.е. семьдесят два часа после посева) клетки для массива DE, который содержит выбранные гены, связанные с DE (смотри пример 7).
На Фиг. 15 показаны результаты флуоресцентной активированный сортировки клеток (FACS) для CD9, CD184/CXCR4, SSEA4, TRA-1-60 и TRA-1-81 для недифференцированной стадии 0, сутки 3 (т.е. семьдесят два часа после посева) клеток (смотри пример 7). Результаты также приведены в Таблице 8.
На Фиг. 16 показана недифференцированная экспрессия гена, определенная способом QRT-PCR, для некоторых генов стадии 0, 1 сутки (то есть двадцать четыре часа после посева) и стадии 0, сутки 3 (т.е. семьдесят два часа после посева), клетки дифференцированы в соответствии с протоколом примера 7. В частности, на фиг. 16 показано небольшое увеличение экспрессии генов для GATA4, РКГ, MIXL1 и Т и увеличение ≥100x экспрессии GATA2 в процессе стадии 0 до направленной дифференцировки.
На Фиг. 17 показана недифференцированная экспрессия гена, определенная способом QRT-PCR, для массива DE, который содержит выбранные гены, связанных с DE, для стадии 0, 1 сутки (то есть двадцать четыре часа после посева) и стадии 0, сутки 3 ( т.е. семьдесят два часа после посева дифференцированных клеток) в соответствии с протоколом примера 7. В частности, Фиг.17 показывает увеличение в ≥100x CER1, FGF17 и экспрессии FGF4 в процессе стадии 0 до направленной дифференцировки.
Фиг. 18 и 19 показывают экспрессию генов для стадии 1, сутки 1 клеток, дифференцированных в соответствии с протоколом примера 7. Фиг. 18 показывает экспрессию генов, определенную способом QRT-PCR, для массива плюрипотентности, который содержит выбранные гены, связанные с плюрипотентностью, для стадии 1, сутки 1 клетки. Фиг. 19 показывает экспрессию генов, определенную способом QRT-PCR, для массива DE, который содержит выбранные гены, связанные с DE, для стадии 1, сутки 1 клетки. Фиг. 18 и 19 иллюстрируют существенные изменения в структуре экспрессии генов, такие как ~700x увеличение экспрессии Foxa2 и 1000x увеличение экспрессии CER1, EOMES, FGF17, FGF4, GATA4, GATA6, РКГ, MIXL1 и T.
Фиг. 20 и 21 показывают экспрессию генов для стадии 1, сутки 3 клеток, дифференцированных в соответствии с протоколом примера 7. Фиг. 20 показывает экспрессию генов, определенную способом QRT-PCR, для массива плюрипотентности, который содержит выбранные гены, связанные с плюрипотентностью, для стадии 1, сутки 3 клетки. Фиг. 21 показывает экспрессию генов, определенную способом QRT-PCR, для массива DE, который содержит выбранные гены, связанные с DE, для стадии 1, сутки 3 клетки.
Фиг. 22 показывает результаты FACS для CD9, CD184 (также известный как CXCR4) и CD99 на стадии 1, сутки 3 клеток, дифференцированных в соответствии с протоколом примера 7. Наблюдался почти полный переход от CD9-экспрессирующей/CXCR4-отрицательной популяции плюрипотентных клеток при инициировании дифференцировки (Фиг. 15) к гомогенной популяции клеток, экспрессирующих CXCR4 (98,3% клеток CXCR4-положительны, ± 1,9SD) в конце стадии 1(Фиг. 22).
На Фиг. 23 показана экспрессия генов, определенная способом qRT-PCR, для массива DE, который содержит выбранные гены, связанные с DE, для стадии 1, сутки 3; стадии 2, сутки 1; и стадии 2, сутки 3 для клеток, дифференцированных в соответствии с протоколом примера 7. Фиг. 23 показывает, что уровни экспрессии HNF4α и GATA6 на стадии 2 сутки 1 и 3, увеличиваются, в то время как гены с высоким уровнем экспрессии на 3 сутки стадии 1 (CXCR4, EOMES, FGF17, FGF4, MNX1, PRDM1, SOX17 и ФВ) показали снижение экспрессии в конце стадии 2.
Фиг. 24 показывает экспрессию генов передней кишки AFP, Pdx1 и Prox1, определенную способом QRT-PCR, для стадии 2, сутки 1 клеток и стадии 2, сутки 3 клеток, дифференцированных в соответствии с протоколом примера 7. Как показано На Фиг. 24, экспрессия этих генов увеличилась.
Фиг. 25 показывает результаты FACS для Pdx1, Foxa2, хромогранина, Nkx2.2 и SOX2 для стадии 3, сутки 3 клеток, выращенных в среде стадии 3 (таблица 7) и дифференцированых в соответствии с протоколом примера 7. Как показано На Фиг. 25, маркеры экспрессии клеток соответствуют энтодерме поджелудочной линии, как определено по экспрессии PDX1 и FOXA2 (90,9% ± 11,9 СО PDX1-положительной и 99,2% ± 0,6 СО FOXA2-положительной).
Фиг. 26 показывает экспрессию генов, определенную способом QRT-PCR, для стадии 4 массива, который содержит выбранные гены, связанные с 4-й стадией, на стадии 3, 1 сутки и 3 стадиисутки 3 клеток, дифференцированных в соответствии с протоколом примера 7, Фиг. 26 показывает, что эти клетки обладают повышенным уровнем генов, обычно экспрессирующих в поджелудочной железе (ARX, GAST, GCG, INS, Isl1, NEUROD1, Ngn3, Nkx2.2, Nkx6.1, Pax4, PAX6, Ptf1a и SST ).
Фиг. 27 показывает результаты FACS для Nkx6.1, хромагранина (CHGA), CDX2, Sox2, Nkx2.2, Pdx1, Foxa2 и NeuroD для стадии 4, сутки 3 клеток, дифференцированных в соответствии с протоколом примера 7. Как показано На Фиг. 27, на стадии 4 сутки 3 у клеток сохраняются высокие уровни экспрессии PDX1 и FOXA2 и далее проявляется паттерн экспрессии, соответствующий сочетанию панкреатических эндокринных клеток (28,1% ± 12,5 СО хромогранин-положительных) и панкреатических клеток-предшественников (58,3% ± 9,7 СО NKX6.1-положительных).
На Фиг. 28 показана экспрессия генов, определенную способом qRT-PCR, для стадии 4 массива, который содержит выбранные гены, связанные с 4-й стадией, на стадии 3, сутки 3; стадии 4 сутки 1 и стадии 4, сутки 3 для клеток, дифференцированных в соответствии с протоколом примера 7. Фиг. 28 показывает повышенный уровень экспрессии генов, обычно выраженных в поджелудочной железе (ARX, GAST, GCG, IAPP, INS, ISL1, MAFB, NEUROD1, NGN3, NKX2.2, NKX6.1, PAX4, PAX6, PTF1A и SST).
Фиг. 29 показывает средние результаты FACS для Nkx6.1, хромагранина (CHGA), CDX2, Sox2, Nkx2.2, Pdx1, Foxa2 и NeuroD для стадии 4, сутки 3 клеток, дифференцированных в соответствии с протоколом примера 7. В частности, Фиг. 29 показывает средний паттерн экспрессии FACS клеток-предшественников панкреатических клеток, генерируемых в 3L шкале из другой партии посевного материала.
Фиг. 30 показывает средние результаты FACS для Nkx6.1, хромагранина (CHGA), CDX2, Sox2, Nkx2.2, Pdx1, Foxa2 и NeuroD для стадии 4, сутки 3 клеток, дифференцированных в соответствии с протоколом примера 7. До дифференцировки в стадии 4, 3 суток клетки, клетки размножили для формирования ISM, а затем вырастили на стадии 0 либо в разработанной в лаборатории среде «IH3», либо в среде Essential8™, в каждую из которых добавили 0,5% BSA. Клетки, выращенные в среде IH3 названы «IH3-P-выращенные клетки», а клетки, выращенные в Essential8™, названы «EZ8-выращенные клетки». Существенной разницы в характере экспрессии у клеток, выращенных в различных среде, не наблюдалось.
Фиг. 31 показывает средние результаты FACS для Nkx6.1, хромагранина (CHGA), CDX2, SOX2, Nkx2.2, Pdx1, Foxa2 и NeuroD для стадии 4, сутки 3 клеток, которые ранее были выращены при разных уровнях рН в стадии 0 (пример 7). Не наблюдалось существенных изменений в профиле клеток стадии 4, сутки 3.
Фиг. 32 сравнивает результаты FACS для Nkx6.1, хромогранина (CHGA), CDX2, SOX2, Nkx2.2, Pdx1, Foxa2 и NeuroD для стадии 4, сутки 3 клеток, которые не были обработаны с пеногасителем-С, и стадии 4, сутки 3 клеток, которые были обработаны эмульсией пеногасителя-С (94 части на миллион) (смотри пример 7). Эмульсия пеногасителя-С(Sigma № по каталогу A8011) не показала эффекта на профиль стадии 4 суток 3 клеток.
Фигуры от 33 до 35 показывают экспрессию гена, определенную способом QRT-PCR, для отдельных генов клеток, дифференцированных в соответствии с протоколом примера 8. Фиг. 33 показывает экспрессию генов, определенную способом QRT-PCR, для некоторых генов в клетках за двадцать четыре часа до начала дифференцировки (смотри пример 8). Как показано На Фиг. 33, клетки из биореактора сохраняют экспрессию генов, характерную для плюрипотентности (POU5F1, NANOG, Sox2 и ZFP42) и показывают минимальное или отсутствующее индуцирование генов, характерных для дифференцировки (AFP и Foxa2: < 50-кратное увеличение; FOXD3, GATA2, GATA4, GSC, HAND2, MIXL1 и T: <10-кратное увеличение экспрессии). Фиг. 34 показывает экспрессию генов, определенную способом QRT-PCR, для отдельных генов клеток через двадцать четыре часа после начала дифференцировки. Фиг. 35 показывает экспрессию генов, определенную способом QRT-PCR, для отдельных генов клеток через семьдесят два часа после начала дифференцировки.
Фигуры 36(а)-36(е) показывают экспрессию гена, определенную способом QRT-PCR, для отдельных генов клеток, дифференцированных на стадиях 2-3 и 4 в соответствии с протоколом примера 8. В частности, эти фигуры показывают экспрессию генов в клетках на стадии 2, сутки 1; стадии 2, сутки 2; стадии 2, сутки 3; стадии 3, сутки 3; и, в зависимости от гена, стадии 4, сутки 1. Фиг. 36(а) показывает экспрессию гена альфа-фетопротеина, Atoh1 и CDX2. Фиг. 36(b) показывает экспрессию генов GAST Hand1, HHEX и HNF4a. Фиг. 36(c) показывает экспрессию гена Nkx2.2, Nkx6.1, OSR1 и Pdx1. Фиг. 36(d) показывает экспрессию генов Prox1, PFT1a, SOX17 и SOX2. Фиг. 36(е) показывает экспрессию гена SOX9. Данные показаны в виде разницы экспрессии относительно недифференцированных клеток линии H1 (WA01) HES (исходная экспрессия 1).
Фиг. 37 показывает экспрессию генов, определенную способом QRT-PCR, для отдельных генов клеток на стадии 4, сутки 3 дифференцировки в соответствии с протоколом в примере 8. Как показано На Фиг. 37, в конце дифференцировки на стадии 3, сутки 3 клеток, дифференцированных в панкреатические клетки-предшественники, характеризующихся высоким уровнем экспрессии Pdx1 (> 1×106кратное индуцирование) и других панкреатических генов (> 1000кратное индуцирование ARX, GCG, GAST, INS, ISL, NEUROD1, Ngn3, Nkx2.2, Nkx6.1, Pax4, Ptf1a и SST) и почти полную потерю экспрессии OCT4/POU5F1 по сравнению с недифференцированными стволовыми клетками эмбриона человека линии H1.
Фиг. 38 показывает подсчет клеток ежедневно в течение протокола дифференцировки по примеру 8. В частности, Фиг.38 показывает плотность клеток в зависимости от суток процесса. Фиг. 38 показывает количество клеток для дифференцировки протоколов в двух пробегах реакторов (PRD1205 и PRD1207), проведенных при рН 6,8 и 7,2. Для сравнения также показано количество клеток клеточного дрейфа.
Фиг. 39 (а) - Фиг. 39 (d) иллюстрируют биологическую активность in vivo стадии 4 сутки 3 клеток, дифференцированных в соответствии с протоколом примера 8 и имплантированных мышам линии SCID-BG. Клетки имплантировали подкожно с помощью устройства TheraCyte™ под почечную капсулу или имплантированные после инкубации на сверхмалых носителях. Мыши проверялись на уровень глюкозы и С-пептида в крови каждые четыре недели после имплантации трансплантата. На Фиг. 39 (а) показаны уровни С-пептида после имплантации 5×106 или 10×106 клеток на стадии 4 сутки 3 в устройстве TheraCyte™ в зависимости от времени. На Фиг. 39 (b) показаны уровни глюкозы у сытых животных после имплантации 5×106 или 10×106 клеток на стадии 4 сутки 3 в устройстве TheraCyte™. Мыши на фиг. 39 (b) были обработаны STZ для абляции функции β-клеток-хозяев перед имплантацией. Фиг. 39 (с) показывает уровень С-пептида, полученный после имплантации ранее криоконсервированных клеток на стадии 4 сутки 3 в устройстве TheraCyte™ в зависимости от времени (недели после имплантации). Фиг. 39 (d) сравнивает уровни C-пептида мышей, получивших почечные трансплантат не криоконсервированных/свежих клеток стадии 4, сутки 3 или криоконсервированных клеток стадии 4, сутки 3, имплантированных сразу после разморозки (D0) или 1 сутки после разморозки ( D1).
Фиг. 40A-40D показывают графики экспрессии CXCR4, CD99 и CD9 клеток, дифференцированных в течение трех суток в соответствии с протоколом примера 9, которые были обработаны в стадии 1, 1 сутки следующим: MCX-соединением и GDF-8 (Фиг. 40A); только MCX (Фиг. 40В); WNT3A и активином А (Фиг. 40C); и только WNT3A (Фиг. 40D). Эти цифры показывают, что в суспензионной культуре добавление 3 мкМоль MCX в отсутствие веществ семейства TGF-β за одни сутки дифференцировки генерируется дефинитивная энтодерма на уровне, сопоставимом с полученным при обработке клеток с 3 мкМоль MCX плюс 100 нг/мл GDF-8 или 20 нг/мл WNT-3A плюс 100 нг/мл активина за одни сутки.
Фиг. 41А-41D показывают графики экспрессии CXCR4, CD99, CD9 и дифференцированных клеток в течение трех суток в соответствии с протоколом примера 10, обработанных различными количествами MCX на стадии 1, 1 сутки. В частности, клетки на стадии 1, сутки 1 были обработаны: 4 мкМоль MCX (Фиг. 41A); 3 мкМоль MCX (Фиг. 41B); 2 мкМоль MCX (Фиг. 41C); и 1,5 мкМоль MCX (Фиг. 41D).
На Фиг. 42A и 42B показаны графики FACS для CXCR4, CD99 и CD9 клеток, дифференцированных в течение трех суток в соответствии с протоколом примера 11. В частности, эти цифры показывают роль частоты замены среды в суспензионной культуре. Фиг. 42A графики экспрессии CXCR4, CD99, CD9 и дифференцированных клеток в течение трех суток в соответствии с протоколом примера10 с полной заменой среды на стадии 1. Фиг. 42B графики экспрессии CXCR4, CD99, CD9 и дифференцированных клеток в течение трех суток в соответствии с протоколом примера 10 без замены среды на сутки 3. Эти данные свидетельствуют, что в системе суспензионной культуры, у тех культур, которые получают замену среды на третьи сутки (фиг.42А) дифференцировки, в результате дефинитивная энтодерма проявляется так же, как у культур, которые не получают замену среды на трое суток (фиг.42B).
Фиг. 43A и 43B показывают графики экспрессии CXCR4, CD99, CD9 и дифференцированных клеток в течение трех суток в соответствии с протоколом примера 12. В частности, на этих фигурах показана роль GlutaMAX™ в суспензионной культуре. Клетки культивировали на стадии 1 в среде с добавлением 1×GlutaMAX™ (Фиг. 43A) или без GlutaMAX™ или без любого глутамина (0 Моль GlutaMAX™) (Фиг. 43B). Эти данные свидетельствуют о том, что в системе суспензионной культуры добавление GlutaMAX™ не влияет на эффективность, с которой образуется дефинитивная энтодерма.
Фигуры 44А до 44D показывают влияние различных количеств бикарбоната натрия на клетки, дифференцированные в соответствии с протоколом примера 13. Фиг. 44A и 44B показывают графики экспрессии CXCR4, CD99, CD9 и дифференцированных клеток в течение трех суток в соответствии с протоколом примера 13 при введении 3,64 г/л (фиг.44A) или 2,49 г/л (фиг.44B) на стадии 1. Фигурки 44С и 44D фиг.показывают фазоконтрастные микрофотографии дифференцированных клеток в течение трех суток в соответствии с протоколом примера 13 при введении 3,64 г/л (Фиг.44C) или 2,49 г/л (Фиг.44D) на стадии 1.
Фиг. 45 показывает ежедневные количество клеток для клеточной плотности в зависимости от дифференцировки клеток, дифференцированных в соответствии с протоколом примера 14. Количество клетки было определено посредством цитометра на основе изображений (NucleoCounter®).
На Фиг. 46 показаны среднесуточные уровни рН среды в биореакторе как функцию по времени (сутки дифференцировки) согласно протоколу дифференцировки примера 14. Уровни рН определяли с помощью NOVA BioProfile® FLEX (Nova Biomedical Corporation, г. Уолтем, штат Массачусетс, США).
На Фиг. 47 показана среднесуточный уровень глюкозы в среде в биореакторе как функцию по времени (сутки дифференцировки) согласно протоколу дифференцировки примера 14. Уровни глюкозы определяли с помощью NOVA BioProfile® FLEX (Nova Biomedical Corporation, г. Уолтем, штат Массачусетс, США).
Фиг. 48 показывает среднесуточный уровень лактата среды в биореакторе как функцию по времени (сутки дифференцировки), согласно протоколу дифференцировки примера 14. Уровни лактата определяли с помощью NOVA BioProfile® FLEX (Nova Biomedical Corporation, г. Уолтем, штат Массачусетс, США).
Фиг. 49 показывает экспрессию генов, определенную способом QRT-PCR, в кратном выражении по сравнению с недифференцированными клетками для массива плюрипотентности, который содержит выбранные гены, связанные с плюрипотентностью, для стадии 0, 1-3 суток и стадии 1, 1-3 суток клеток, дифференцированных в соответствии с протоколом примера 14.
Фиг. 50 показывает экспрессию генов, определенную способом QRT-PCR, в кратном выражении по сравнению с недифференцированными клетками для массива DE, который содержит выбранные гены, связанные с DE, для стадии 0, 1-3 суток, стадии 1, 1-3 суток и стадии 2, сутки 1-3 клеток, дифференцированных в соответствии с протоколом примера 14.
Фиг. 51 показывает результаты FACS для маркеров, связанных с плюрипотентностью (CD184/CXCR4, SSEA4, TRA-1-60 и TRA-1-81) для стадии 0 клеток до дифференцирования в соответствии с протоколом примера 14. В частности, фиг. 51 показывает высокую экспрессию маркеров, связанных с плюрипотентностью.
Фиг. 52 показывает графики экспрессии маркеров дефинитивной энтодермы CXCR4, CD99, CD9 и дифференцированных клеток в конце стадии 1 в соответствии с протоколом примера 14.
На Фиг. 53 показана экспрессия генов, определенная способом qRT-PCR в кратном выражении по сравнению с недифференцированными клетками для GAPDH, AFP, HHEX, HNF4α, PDX1 и PROX1 для стадии 2, сутки 1; стадии 2 сутки 2 и стадии 2, сутки 3 для клеток, дифференцированных в соответствии с протоколом примера 14. Фиг. 53 показывает увеличение экспрессии генов передней кишки (AFP, HHEX, Pdx1 и Prox1).
Фиг. 54 показывает экспрессию генов, определенную способом QRT-PCR, в кратном выражении по сравнению с недифференцированными клетками для GAPDH, AFP, CDX2, GAST, HNF4A, NKX2-2, OSR1, Pdx1 и PFT1A для стадии 2, сутки 1-3 суток и стадия 3, сутки 1-3 клеток, дифференцированых в соответствии с протоколом примера 14. Как показано На Фиг. 54, экспрессия Pdx1 увеличилась 60кратно, от 12 000x относительно контроля в конце стадии 2, 3 суток и до 739 000x относительно контроля над в конце стадии 3, 3 суток
Фиг. 55 показывает экспрессию генов, определенную способом QRT-PCR, в кратном выражении по сравнению с недифференцированными клетками для некоторых генов стадии 3, 1 сутки до 3 и стадии 4, сутки 1-3 клеток, дифференцированных в соответствии с протоколом примера 14. В частности, верхняя панель на Фиг. 55 показывает экспрессию генов GAPDH, AFP, ALB, ARX, CDX2, CHGA, GAST, GCG, IAAP, INS, Isl1 и MAFB. Нижняя панель Фиг. 55 показывает экспрессию генов MAFB, MUCS, NEUROD1, NEUROG3, NKX2-2, NKX6-1, Pax4, Pdx1, POUSF1, Ptf1a, SST и ZlC1.
Фиг. 56 показывает микрофотографии конечной стадии клеток, дифференцированных в соответствии с протоколом примера 14. Показанные на Фиг. 56 репрезентативные микрофотографии (4x) клеточных скоплений на стадии 0 и в конце дифференцировки на стадиях с 1 по 4.
Фиг. от 57 до 80 показывают экспрессию гена, определенную способом QRT-PCR, в кратном выражении по сравнению с недифференцированными клетками, у клеток,дифференцированных в соответствии с различными вариантами осуществления протокола из примера 15 через 0 часов, 6 часов, 24 часа, 30 часов, 48 часов и 72 часа дифференцировки для следующих генов: AFP (Фиг. 57); CD99 (Фиг. 58); CD9 (Фиг. 59); CDH1 (Фиг. 60); CDH2 (Фиг. 61); CDX2 (Фиг. 62); CER1 (Фиг. 63); CXCR4 (Фиг. 64); FGF17 (Фиг. 65); FGF4 (Фиг. 66); FOXA (Фиг. 67); GADPH (Фиг. 68); GATA4 (Фиг. 69); GATA6 (Фиг. 70); GSC (Фиг. 71); KIT (Фиг. 72); MIXL1 (Фиг. 73); MNX1 (Фиг. 74); NANOG (Фиг. 75); OTX2 (Фиг. 76); POUF5F1 (Фиг. 77); SOX17 (Фиг. 78); SOX7 (Фиг. 79) и T (Фиг. 80).
Фиг. 81 показывает процент клеток на стадиях клеточного цикла G0/G1 для клеток через 6 часов, 24 часа, 30 часов, 48 часов и 72 часа дифференцировки в соответствии с различными вариантами осуществления протокола примера 15. В частности, Фиг. 81 показывает результаты для кластеров, которые были обработаны в первые сутки дифференцировки с одним из шести условий: (1) чистая, (2) 3 мкМоль MCX плюс 100 нг/мл GDF-8 (№ по каталогу 120-00, Peprotech), (3) 3 мкМоль только MCX, (4) 100 нг/мл только GDF-8, (5) 20 нг/мл WNT-3A (№ по каталогу 1324-WN-002, R&D Systems, штат Миннесота, США) плюс 100 нг/мл активина А (№ по каталогу 338-AC, R&D Systems, штат Миннесота, США) или (6) 20 нг/мл только WNT-3A.
Фиг. 82 показывает эффекты обработки EDU клеточных скоплений, дифференцированных в соответствии с протоколом примера 15. Левая панель показывает процент клеток на стадиях клеточного цикл G2/M для клеток через 0 часов, 6 часов, 24 часа, 30 часов, 48 часов и 72 часа дифференцировки в соответствии с различными вариантами осуществления протокола примера 15. В частности, левая панель показывает результаты для кластеров, которые лечились в первые сутки дифференцировки по одному из шести условий: (1) чистая, (2) 3 мкМоль MCX плюс 100 нг/мл GDF-8 (№ по каталогу 120-00, Peprotech), (3) 3 мкМоль только MCX, (4) 100 нг/мл только GDF-8, (5) 20 нг/мл WNT-3A (№ по каталогу 1324-WN-002, R&D Systems, штат Миннесота, США) плюс 100 нг/мл активина А (№ по каталогу 338-AC, R&D Systems, штат Миннесота, США) или (6) 20 нг/мл только WNT-3A. В одном наборе данных эти кластеры также обрабатывали EDU. На правой панели Фиг. 82 приведен % EDU-положительных клеток через 0 часов, 6 часов, 24 часа, 30 часов, 48 часов и 72 часа дифференцировки в соответствии с различными вариантами осуществления протокола примера 15.
Фиг. 83 показывает общие эксплуатационные параметры, используемые в протоколах примере 15.
Фиг. 84 показывает количество включений EDU в клетках через 6 часов, 24 часа, 30 часов, 48 часов и 72 часа дифференцировки в соответствии с различными вариантами осуществления протокола примера 15. В частности, на фиг. 84 показаны результаты EDU-инкубированных клеточных кластеров, которые были обработаны в первые сутки дифференцировки по одному из шести условий: (1) чистая, (2) 3 мкМоль MCX плюс 100 нг/мл GDF-8 (№ по каталогу 120-00, Peprotech), (3) 3 мкМоль только MCX, (4) 100 нг/мл только GDF-8, (5) 20 нг/мл WNT-3A (№ по каталогу 1324-WN-002, R&D Systems, штат Миннесота, США) плюс 100 нг/мл активина А (№ по каталогу 338-AC, R&D Systems, штат Миннесота, США) или (6) 20 нг/мл только WNT-3A.
Фиг. 85 показывает процент клеток на стадиях клеточного цикла G0/G1 для клеток через 6 часов, 24 часа, 30 часов, 48 часов и 72 часа дифференцировки в соответствии с различными вариантами осуществления протокола примера 15. В частности, Фиг. 85 показывает результаты для кластеров, которые были обработаны в первые сутки дифференцировки с одним из шести условий: (1) чистая, (2) 3 мкМоль MCX плюс 100 нг/мл GDF-8 (№ по каталогу 120-00, Peprotech), (3) 3 мкМоль только MCX, (4) 100 нг/мл только GDF-8, (5) 20 нг/мл WNT-3A (№ по каталогу 1324-WN-002, R&D Systems, штат Миннесота, США) плюс 100 нг/мл активина А (№ по каталогу 338-AC, R&D Systems, штат Миннесота, США) или (6) 20 нг/мл только WNT-3A.
Фиг. 86 показывает процент клеток в S-фазе клеточного цикла для клеток через 6 часов, 24 часа, 30 часов, 48 часов и 72 часа дифференцировки в соответствии с различными вариантами осуществления протокола примера 15. В частности, Фиг. 86 показывает результаты для кластеров, которые были обработаны в первые сутки дифференцировки с одним из шести условий: (1) чистая, (2) 3 мкМоль MCX плюс 100 нг/мл GDF-8 (№ по каталогу 120-00, Peprotech), (3) 3 мкМоль только MCX, (4) 100 нг/мл только GDF-8, (5) 20 нг/мл WNT-3A (№ по каталогу 1324-WN-002, R&D Systems, штат Миннесота, США) плюс 100 нг/мл активина А (№ по каталогу 338-AC, R&D Systems, штат Миннесота, США) или (6) 20 нг/мл только WNT-3A.
Фиг. 87 показывает процент клеток в S-фазе клеточного цикла для клеток после 0 часов, 6 часов, 24 часа, 30 часов, 48 часов и 72 часа дифференцировки в соответствии с различными вариантами осуществления протокола примера 15. В частности, Фиг. 87 показывает результаты для кластеров, которые были обработаны в первые сутки дифференцировки с одним из шести условий: (1) чистая, (2) 3 мкМоль MCX плюс 100 нг/мл GDF-8 (№ по каталогу 120-00, Peprotech), (3) 3 мкМоль только MCX, (4) 100 нг/мл только GDF-8, (5) 20 нг/мл WNT-3A (№ по каталогу 1324-WN-002, R&D Systems, штат Миннесота, США) плюс 100 нг/мл активина А (№ по каталогу 338-AC, R&D Systems, штат Миннесота, США) или (6) 20 нг/мл только WNT-3A.
Фиг. 88А - 88E показывают экспрессию гена, определенную способом QRT-PCR, в кратном выражении по сравнению с недифференцированными клетками для клеток, дифференцированных в соответствии с различными вариантами осуществления протокола из примера 15 после 0 часов, 6 часов, 24 часа, 30 часов, 48 часов и 72 часа дифференцировки. Фиг. 88A показывает экспрессию генов, определенную способом QRT-PCR, в кратном выражении по сравнению с недифференцированными клетками для CD99, CD9, CDH1 и CDH2. Фиг. 88A показывает экспрессию генов, определенную способом QRT-PCR, в кратном выражении по сравнению с недифференцированными клетками для CXD2, CER1, CXCR4 и FGF17. Фиг. 88C показывает экспрессию генов, определенную способом QRT-PCR, в кратном выражении по сравнению с недифференцированными клетками для FGF4, FOXA, GATA4 и GATA6. Фиг. 88D показывает экспрессию генов, определенную способом QRT-PCR, в кратном выражении по сравнению с недифференцированными клетками для GSC, KIT, MIXL1 и MNX1. Фиг. 88E показывает экспрессию генов, определенную способом QRT-PCR, в кратном выражении по сравнению с недифференцированными клетками для NANOG, Otx2, POUF5F1 и SOX17. На Фиг. 88F показана экспрессия генов, определенная способом qRT-PCR, в кратном выражении по сравнению с недифференцированными клетками для SOX7 и Т. Основные данные для фигур 88A-88F показаны на Фиг. 58-67 и 69-80.
Фиг. 89 показывает определенные способом QRT-PCR паттерны экспрессии генов плюрипотентных клеток, культивируемые в среде для дифференцировки эктодермы в соответствии с протоколом примера 16. Как показано на Фиг. 89, клетки дифференцируют в направлении клеток нейронных линий . В частности, левая панель Фиг. 89 показывает паттерны экспрессии генов для линии индуцированных плюрипотентных стволовых клеток, образованных из клеток тканей пуповины (UTC). На правой панели на фиг 89 показаны паттерны экспрессии генов для суб-клона WB0106 клеточной линии H1 HES.
Фиг. 90 показывает определенные способом QRT-PCR паттерны экспрессии генов плюрипотентных клеток, культивируемых в среде для дифференцировки мезодермы в соответствии с протоколом примера 16. Как показано на Фиг. 90, клетки дифференцируют в направлении клеток сердечной линии. В частности, левая панель Фиг. 90 показывает паттерны экспрессии генов для линии индуцированных плюрипотентных стволовых клеток, образованных из клеток тканей пуповины (UTC). На правой панели на фиг 90 показаны паттерны экспрессии генов для суб-клона WB0106 клеточной линии H1 HES.
Фиг. 91 показывает определенные способом QRT-PCR паттерны экспрессии генов плюрипотентных клеток, культивируемые в среде для дифференцировки эктодермы в соответствии с протоколом примера 16. Как показано на Фиг. 91, клетки дифференцируются в направлении клеток нейронных линий . В частности, левая панель Фиг. 91 показывает паттерны экспрессии генов для линии индуцированных плюрипотентных стволовых клеток, образованных из клеток тканей пуповины (UTC). На правой панели на фиг 91 показаны паттерны экспрессии генов для суб-клона WB0106 клеточной линии H1 HES.
Фиг. 92 показывает паттерны экспрессии белка для PAX6, Sox2 и POU5F1/OCT4, что определено с помощью FACS, плюрипотентных клеток, культивированных в течение трех суток в среде для дифференцировки эктодермы в соответствии с протоколом примера 16. В частности, левые панели Фиг. 92 показывают паттерны экспрессии Pax6, Sox2 и POU5F1/OCT4 для линии индуцированных плюрипотентных стволовых клеток, образованных из клеток тканей пуповины(UTC). Правая панель на Фиг. 92 показывает паттерны экспрессии белка для PAX6, Sox2 и POU5F1/OCT4 для суб-клона WB0106 клеточной линии H1 HES.
Фиг. 93 показывает определенные способом QRT-PCR паттерны экспрессии генов плюрипотентных клеток, культивируемых в среде для дифференцировки мезодермы в соответствии с протоколом примера 16. Как показано на Фиг. 93, клетки дифференцируют в направлении клеток сердечной линии. В частности, левая панель Фиг. 93 показывает паттерны экспрессии генов для линии индуцированных плюрипотентных стволовых клеток, образованных из клеток тканей пуповины (UTC). На правой панели на фиг 93 показаны паттерны экспрессии генов для суб-клона WB0106 клеточной линии H1 HES.
Фиг. 94 показывает микрофотографии клеток, дифференцированных в среде для дифференцировки мезодермы в соответствии с протоколом примера 16. Как показано на Фиг. 94, клетки дифференцируют в направлении клеток сердечной линии. В частности, левые панели фигуры 94 показывают микрофотографии клеток суб-клона WB0106 клеточной линии H1 HES на 3 сутки, 5 сутки и 10 сутки дифференцировки. Правая панель Фиг. 94 показывает микрофотографию линии индуцированных плюрипотентных стволовых клеток, образованных из клеток тканей пуповины (UTC IPSCs) после 10 суток дифференцировки.
Фиг. 95 показывает микрофотографии клеток, дифференцированных в среде для дифференцировки эктодермы в соответствии с протоколом примера 16. Как показано на Фиг. 95, клетки дифференцируют в направлении клеток нейронных линий . В частности, левые панели фигуры 95 показывают микрофотографии клеток суб-клона WB0106 клеточной линии H1 HES на 3 сутки, 5 сутки и 10 сутки дифференцировки. Правая панель Фиг. 95 показывает микрофотографию линии индуцированных плюрипотентных стволовых клеток, образованных из клеток тканей пуповины (UTC iPCS) после 10 суток дифференцировки.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Эта заявка посвящена подготовке эмбриональных стволовых клеток и других плюрипотентных клеток, которые сохраняют плюрипотентность в агрегированном кластере клеток для дифференцировки в клетки-предшественники энтодермы, эндокринные клетки поджелудочной железы, клетки мезодермы или клетки эктодермы. Для ясности описания, а не для ограничения изобретения, подробное описание настоящего изобретения разделено на следующие подразделы, описывающие или иллюстрирующие определенные особенности, варианты осуществления или области применения настоящего изобретения.
Определения
Стволовые клетки представляют собой недифференцированные клетки, определяемые как обладающие способностью на одноклеточном уровне к самообновлению и дифференцировки. Стволовые клетки могут производить клетки-потомки, включая самообновляющиеся прогениторные клетки, необновляющиеся прогениторные клетки и окончательно дифференцированные клетки. Стволовые клетки также характеризуются своей способностью дифференцироваться in vitro в функциональные клетки различных линий дифференцировки из множества зародышевых листков (энтодермы, мезодермы и эктодермы). Стволовые клетки также дают начало тканям множества зародышевых листков после трансплантации и вносят значительный вклад в образование большинства, если не всех тканей после инъекции в бластоцисты.
Стволовые клетки классифицируются по потенциалу развития. «Культура клеток» или «культура» по существу обозначает клетки, взятые из живого организма и выращенные в контролируемых условиях («в культуре» или «культивированные»). Первичная культура клеток обозначает культуру клеток, тканей или органов, взятых непосредственно из организма (-ов) до первого пересева. Клетки размножают в культуре, когда их помещают в ростовую среду в условиях, облегчающих одно или оба из роста и деления клеток, что приводит к большей популяции клеток. При размножении клеток в культуре скорость пролиферации клеток иногда измеряют по количеству времени, которое необходимо клеткам для удвоения численности (также называемое временем удвоения).
Термин «выращивание» или «рост», используемый здесь, описывает процесс увеличения количества плюрипотентных стволовых клеток посредством культивирования по меньшей мере примерно на 5%, 10%, 15%, 20%, 25%, 30%, 35%, 40% , 45%, 50%, 60%, 75%, 90%, 100%, 200%, 500%, 1000% или более, а также на уровни, указанные в процентах. Следует понимать, что количество плюрипотентных стволовых клеток, которые могут быть получены из одной плюрипотентной стволовой клетки, зависит от способности к пролиферации плюрипотентных стволовых клеток. Способность к пролиферации плюрипотентных стволовых клеток может быть рассчитана по времени удвоения клетки, т. е. по времени, которое необходимо клетке, чтобы пройти митотическое деление в культуре, а также времени, в течение которого плюрипотентные стволовые клетки могут поддерживаться в недифференцированном состоянии, что эквивалентно количеству пассажей, умноженному на количество суток между каждым пассажем.
Дифференцировка - это процесс, посредством которого неспециализированная («некоммитированная») или менее специализированная клетка приобретает черты специализированной клетки, такой как нервная клетка или мышечная клетка. Дифференцированная клетка или клетка, индуцированная дифференциацией, представляет собой клетку, занявшую более специализированное («коммитированное») положение в линии дифференцировки клетки. Термин «коммитированная» применительно к процессу дифференцировки относится к клетке, дошедшей в ходе процесса дифференцировки до стадии, с которой в нормальных условиях она продолжит дифференцироваться до конкретного типа клеток или подмножества типов клеток и не сможет в нормальных условиях дифференцироваться в иной тип клеток или вернуться к менее дифференцированному типу. «Дедифференцировка» обозначает процесс, в ходе которого клетка возвращается к менее специализированной (или коммитированной) позиции в клеточной линии дифференцировки. Применяемый в настоящем документе термин «клеточная линия дифференцировки» означает наследственность клетки, т.е. из каких клеток произошла данная клетка и каким клеткам она может дать начало. В клеточной линии дифференцировки клетка помещается в наследственную схему развития и дифференцировки. Маркер, специфичный для линии дифференцировки, относится к характеристике, специфически ассоциированной с фенотипом клеток интересующей линии дифференцировки, и его можно использовать для оценки дифференцировки некоммитированной клетки в клетки интересующей линии дифференцировки.
В настоящем документе термин «маркеры» означает молекулы нуклеиновых кислот или полипептидов с дифференциальной экспрессией в исследуемых клетках. В данном контексте под дифференциальной экспрессией понимают повышенный уровень положительного маркера и пониженный уровень отрицательного маркера по сравнению с недифференцированной клеткой. Обнаруживаемый уровень маркерной нуклеиновой кислоты или полипептида в интересующих клетках оказывается значительно выше или ниже по сравнению с другими клетками, что позволяет идентифицировать интересующую клетку и отличить ее от других клеток с помощью любого из множества способов, известных в данной области.
В настоящем документе клетка «положительна по» заданному маркеру или «положительна», если заданный маркер явно обнаруживается в клетке. Аналогично клетка «отрицательна по» заданному маркеру или «отрицательна», если заданный маркер не обнаруживают в клетке. В частности, положительность по FACS как правило выше, чем 2%, в то время как отрицательный предел по FACS как правило менее 1%. Положительность по PCR, как правило, составляет менее 34 циклов (Cts); в время как отрицательность по PCR составляет, как правило, более 34,5 циклов.
В настоящем документе термины «плотность клеток» и «плотность посева» применяются взаимозаменяемо и относятся к числу клеток, высеваемых на единицу площади поверхности твердого или полутвердого плоского или криволинейного субстрата.
В настоящем документе термин «суспензионная культура» относится к культуре клеток, отдельным клеткам или кластерам, суспендированным в среде, а не прикрепленным к какой-либо поверхности.
В настоящем документе термин «бессывороточный» относится к среде, лишенной человеческой или животной сыворотки. Соответственно, бессывороточная культуральная среда не содержит сыворотку или части сыворотки.
В попытках воспроизвести дифференцировку плюрипотентных стволовых клеток в функциональные панкреатические эндокринные клетки в культуре клеток, процесс дифференцировки часто рассматривается как прогрессирование через несколько последовательных стадий. В настоящем документе различные стадии определяются временем культивирования и реактивами, изложенными в приведенных здесь примерах.
В настоящем документе «дефинитивная энтодерма» относится к клеткам, обладающим характеристиками клеток, происходящих от эпибласта при гаструляции, и формирующим желудочно-кишечный тракт и его производные. Клетки дефинитивной энтодермы экспрессируют по меньшей мере один из следующих маркеров: FOXA2 (также известный как гепатоцитный ядерный фактор 3-β (HNF3β)), GATA4, GATA6, MNX1, SOX17, CXCR4, Cerberus, OTX2, brachyury, goosecoid, C-Kit, CD99 и MIXL1. Маркерные характеристики клеток дефинитивной энтодермы включают CXCR4, FOXA2 и SOX17. Таким образом, клетки дефинитивной энтодермы могут быть охарактеризованы их экспрессией CXCR4, FOXA2 и SOX17. Дополнительно, в зависимости от длительности времени, на протяжении которого клеткам позволяется оставаться на стадии 1, можно наблюдать рост в HNF4α.
Используемый в настоящей заявке термин «панкреатическая эндокринная клетка» относится к клеткам, способным к секреции по меньшей мере одного из следующих гормонов: инсулин, глюкагон, соматостатин или панкреатический полипептид. В дополнение к этим гормонам, маркерные характеристики панкреатических эндокринных клеток включают один или несколько из NGN3, NeuroD1, ISL1, PDX1, NKX6.1, PAX4, ARX, NKX2.2, и PAX6. Панкреатические эндокринные клетки, экспрессирующие маркеры бета-клеток, могут характеризоваться их экспрессией инсулина и по меньшей мере одного из следующих транскрипционных факторов: PDX1, NKX2.2, NKX6.1, NeuroD1, ISL1, HNF3β, MAFAPAX4 и PAX6.
В настоящем документе взаимозаменяемо применяются выражения «d1», «d 1» и «сутки 1»; «d2», «d 2» и «сутки 2»; «d3», «d 3» и «день 3» и так далее. Эти комбинации цифр и букв относятся к конкретным суткам инкубации на различных стадиях в процессе постадийного протокола дифференцировки настоящей заявки.
Термины «глюкоза» и «D-глюкоза» используются в настоящем документе взаимозаменяемо и относятся к декстрозе, сахару, широко встречающемуся в природе.
В настоящем документе термины «NeuroD» и «NeuroD1» используются взаимозаменяемо для обозначения белка, экспрессируемого в клетках-предшественниках панкреатических эндокринных клеток, и кодирующего его гена.
«LDN» и «LDN-193189» относится к ((6-(4-(2-(пиперидин-1-ил)этокси)фенил)-3-(пиридин-4-ил)пиразоло[1,5-a]пиримидин гидрохлориду; DM-3189)) ингибитору рецептора BMP, доступному под торговой маркой STEMOLECULETM от Stemgent, Inc., г. Кембридж, штат Массачусетс, США.
Выделение, размножение и культивирование плюрипотентных стволовых клеток
Плюрипотентные стволовые клетки могут экспрессировать одно или более из указанных TRA-1-60 и TRA-1-81 антител (Thomson и др. 1998, Science 282:1145-1147). Дифференцирование плюрипотентных стволовых клеток in vitro приводит к потере экспрессии TRA-1-60 и TRA-1-81. Недифференцированные плюрипотентные стволовые клетки, как правило, имеют щелочнофосфатазную активность, которую можно обнаружить путем обработки клеток 4% раствором параформальдегида, а затем выращивая с Vector Red в качестве субстрата, как описано производителем (Vector Laboratories, Inc., г. Берлингейм, штат Калифорния, США). Недифференцированные плюрипотентные стволовые клетки также, как правило, экспрессируют OCT4 и TERT, определяемые с помощью RT-PCR.
Другим желательным фенотипическим свойством выращенных плюрипотентных клеток является потенциал дифференцировки в клетки всех трех зародышевых листков: в энтодертмальные, мезодермальные и эктодермальные ткани. Плюрипотентность стволовых клеток можно подтвердить, например, путем инъекции клеток мышам с тяжелым комбинированным иммунодефицитом (SCID), фиксирования образующихся тератом с помощью 4% параформальдегида и их последующего гистологического исследования на наличие клеточных типов, происходящих от трех зародышевых листков. Плюрипотентность можно альтернативно определить по созданию эмбриоидных телец и анализа эмбриоидных телец на наличие маркеров, ассоциирующихся с тремя зародышевыми листками.
Выращенные линии плюрипотентных стволовых клеток можно кариотипировать с использованием стандартного метода G-бэндинга и сравнить с опубликованными кариотипами соответствующих видов приматов. Желательно получить клетки, имеющие «нормальный кариотип», т. е. эуплоидные клетки, в которых все хромосомы человека присутствуют и не имеют видимых изменений. Плюрипотентные клетки можно легко размножить в культуре путем использования различных питательных слоев или с помощью сосудов, покрытых матриксными белками. Альтернативно для стандартного размножения клеток можно использовать химически определенные поверхности в комбинации со средами определенного состава, такими как среды mTeSR®1 (StemCell Technologies, г. Ванкувер, провинции Британская Колумбия, Канада).
Культивирование в суспензионной культуре в соответствии со способом, указанным в некоторых вариантах осуществления настоящего изобретения, осуществляется с помощью посева плюрипотентных стволовых клеток в сосуд для культивирования, при плотности клеток, которая способствует выживанию клеток и пролиферации, но ограничивает дифференцировку. Как правило, применяется плотность посева, которая обеспечивает отсутствие дифференцировки клеток. Следует иметь в виду, что, хотя можно высеивать суспензии отдельных стволовых клеток, предпочтительнее высеивать небольшие скопления клеток.
Для обеспечения плюрипотентных стволовых клеток достаточной и постоянной подачей питательных веществ и факторов роста в суспензионной культуре, культуральная среда могут быть заменена или пополнена на ежедневной основе или по заранее установленному графику, например, каждые 1-5 суток. Большие кластеры плюрипотентных стволовых клеток могут привести к началу клеточной дифференцировки, таким образом, могут быть приняты меры для избежания появления больших агрегатов плюрипотентных стволовых клеток. В соответствии с некоторыми вариантами осуществления изобретения, образовавшиеся кластеры плюрипотентных стволовых клеток диссоциируют, например, каждые 2-7 суток, и отдельные клетки или небольшие скопления клеток, либо разделяют и переносят в другой культуральный сосуд (т. е. пересеивают), или оставляют в том же культуральном сосуде и заменяют либо пополняют культуральную среду.
Большие скопления плюрипотентных стволовых клеток, включая осадок плюрипотентных стволовых клеток, полученный центрифугированием, могут быть подвергнуты ферментативному расщеплению и/или механической диссоциации. Ферментативное расщепление скоплений плюрипотентных стволовых клеток может быть выполнено помещением скопления клеток в фермент, например, в коллагеназу IV типа, Dispase® или Accutase®. Механическая диссоциация больших скоплений плюрипотентных стволовых клеток может быть выполнена с использованием устройства, предназначенного для разделения скоплений до заданного размера. Дополнительно или альтернативно, механическая диссоциация может быть выполнена вручную с помощью иглы или пипетки.
Культуральным сосудом для культивирования плюрипотентных стволовых клеток в суспензии в соответствии со способами, описанными в некоторых вариантах осуществления изобретения, может быть любой сосуд тканевой культуры (например, обладающий достаточным уровнем чистоты для культивирования плюрипотентных стволовых клеток), внутренняя поверхность которого выполнена так, что плюрипотентные стволовые клетки, культивируемые в нем, не могут удерживаться или прикрепляться к такой поверхности (например, сосуд, не обработанный для тканевого культивирования, для предотвращения прикрепления или присоединения к поверхности). Для получения необходимой разрастающейся культуры, культивирование в соответствии с некоторыми вариантами осуществления изобретения осуществляется с помощью управляемой системы культивирования (предпочтительно, системы культивирования с компьютерным управлением), в котором параметры культуры, такие как температура, перемешивание, рН и уровень кислорода автоматически контролируются и регулируются с использованием подходящего устройства. Как только определены нужные параметры культуры, система может быть настроена на автоматическую регулировку параметров культуры по мере необходимости для улучшения роста и дифференцировки плюрипотентных стволовых клеток.
Плюрипотентные стволовые клетки можно культивировать в динамических условиях (т. е. в условиях, в которых плюрипотентные стволовые клетки постоянно движутся в суспензионной культуре, например, в перемешиваемой суспензионной культуральной системе) или в нединамических условиях (например, в статической культуре), сохраняя их способность к пролиферации, плюрипотентности и кариотипическую стабильность в течение нескольких пересевов.
Для нединамического культивирования плюрипотентных стволовых клеток, плюрипотентные стволовые клетки можно культивировать в чашках Петри, Т-колбах, HyperFlasks® (Corning Incorporated, г. Корнинг, штат Нью-Йорк, США), CellStacks® (Corning Incorporated, г. Корнинг, штат Нью-Йорк, США) или Cell Factories (NUNC™ Cell Factory™ Systems (Thermo Fisher Scientific, Inc., г. Питтсбург, штат Пенсильвания, США)) с покрытием или без. Для динамического культивирования плюрипотентных стволовых клеток, плюрипотентные стволовые клетки можно культивировать в подходящем сосуде, например, центрифужных пробирках или колбах Эрленмейера, на нержавеющей стали, стекле, в одноразовом пластиковом шейкере или сосуде-смесителе. Культуральный сосуд может быть соединен с блоком управления и, таким образом, представляют собой управляемую систему культивирования. Перемешивание в культуральном сосуде (например, центрифужная пробирка или колба Эрленмейера) осуществляется непрерывно или периодически. Перемешивание в культуральном сосуде осуществляется в той степени, чтобы поддерживать плюрипотентные стволовые клетки в суспензии.
Плюрипотентные стволовые клетки могут быть выращены в любой среде, которая обеспечивает достаточное количество питательных веществ и внешних стимулов для стимуляции росту и деления. Подходящие среды включают в себя E8™, IH3 и mTeSR®1 или mTeSR®2. Среда можно периодически заменять для обновления запаса питательных веществ и удаления побочных продуктов, выделяемых клетками. В соответствии с некоторыми вариантами осуществления изобретения, культуральную среду меняли ежедневно.
ИСТОЧНИКИ ПЛЮРИПОТЕНТНЫХ СТВОЛОВЫХ КЛЕТОК
Любые плюрипотентные стволовые клетки могут быть использованы в способах по настоящему изобретению. К типам плюрипотентных стволовых клеток, которые можно использовать, относятся устойчивые линии плюрипотентных клеток, получаемые из ткани, формируемой после вынашивания плода, в том числе из преэмбриональной ткани (такой как бластоциста), эмбриональной ткани или ткани плода, взятой в любой момент в период вынашивания, как правило, но не обязательно, до срока приблизительно 10-12 недель беременности. Неограничивающими примерами являются устойчивые линии эмбриональных стволовых клеток человека (hES) или эмбриональные зародышевые клетки человека, такие как, например, эмбриональные стволовые клетки человека линий H1, H7 и H9 (WiCell Research Institute, г. Мэдисон, штат Висконсин, США). Также допустимыми являются клетки, взятые из популяции плюрипотентных стволовых клеток, уже культивированных в отсутствие питающих клеток.
Также соответствуют целям настоящего изобретения индуцибельные плюрипотентные клетки (IPS) или перепрограммированные плюрипотентные клетки, которые могут быть получены из взрослых соматических клеток с помощью принудительной экспрессии ряда факторов, относящихся к плюрипотентным транскрипционным факторам, таким как OCT4, NANOG, Sox2, KLF4 и ZFP42(Annu Rev Genomics Hum Genet 2011, 12:165-185). Эмбриональные стволовые клетки человека, применяемые в способах настоящего изобретения, также могут быть подготовлены, как описано Thomson и др. (патент США № 5,843,780; Science, 1998 г., 282:1145-1147; Curr Top Dev Biol 1998, 38:133-165; Proc Natl Acad Sci U.S.A. 1995, 92:7844-7848). Также подходят мутантные линии эмбриональных стволовых клеток человека, такие как, к примеру, BG01v (BresaGen, Athens, Ga.), или клетки, полученные из зрелых соматических клеток человека, такие как, к примеру, клетки, описанные в Takahashi с соавторами, Cell 131: 1-12 (2007). Плюрипотентные стволовые клетки, пригодные для применения в настоящем изобретении, могут быть получены с помощью методов, описанных у Li с соавторами: (Cell Stem Cell 4: 16-19, 2009); Maherali et al. (Cell Stem Cell 1: 55-70, 2007); Stadtfeld et al. (Cell Stem Cell 2: 230-240); Nakagawa et al. (Nature Biotechnology 26: 101-106, 2008); Takahashi et al. (Cell 131: 861-872, 2007); и публикации заявки на патент США № 2011-0104805. Другие источники плюрипотентных стволовых клеток включают индуцированные плюрипотентные клетки (IPS, Cell, 126(4): 663-676). Другие источники клеток, подходящих для использования в способах по изобретению, включают клетки, полученные из ткани пуповины человека, клетки, полученные из амниотической жидкости, клетки, клетки, полученные из плаценты человека и человеческие партеноты. В одном варианте осуществления, клетки, полученные из ткани пуповины человека могут быть получены с использованием методов, описанных в патенте США №7510873, раскрытие которого включено в качестве ссылки в полном объеме, где они относятся к выделению и характеризации клеток. В другом варианте, клетки, полученные из ткани плаценты, могут быть получены с использованием методов, описанных в заявке на патент США на патент США № 2005/0058631, раскрытие которой включено в качестве ссылки в полном объеме, как она относится к выделению и характеризации клеток. В другом варианте, клетки, полученные из амниотической жидкости могут быть получены с использованием методов, описанных в заявке на патент США на патент США № 2007/0122903, раскрытие которой включено в качестве ссылки в полном объеме, как она относится к выделению и характеризации клеток.
Характеристики плюрипотентных стволовых клеток хорошо известны специалистам в данной области, и продолжается выявление дополнительных характеристик плюрипотентных стволовых клеток. К маркерам плюрипотентных стволовых клеток относится, например, экспрессия одного или более (например 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14 или все) из следующих маркеров: ABCG2, cripto, FOXD3, CONNEXIN43, CONNEXIN45, OCT4, SOX2, NANOG, hTERT, UTF1, ZFP42, SSEA-3, SSEA-4, TRA-1-60 и TRA-1-81. В одном варианте осуществления, плюрипотентные стволовые клетки, пригодные для использования в способах по изобретению, экспрессируют один или более (например, 1, 2, 3 или все) маркеров CD9, SSEA4, TRA-1-60 и TRA-1-81 и лишены экспрессии маркера дифференцировки CXCR4 (также известного как CD184), что было определено с помощью проточной цитометрии. В другом варианте осуществления плюрипотентные стволовые клетки, пригодные для использования в способах по изобретению, экспрессируют один или несколько (например, 1, 2 или все) маркеров CD9, NANOG и POU5F1/OCT4, что было определено с помощью RT-PCR.
Примеры плюрипотентных стволовых клеток включают линию эмбриональных стволовых клеток человека H9 (код NIH: WA09), эмбриональные стволовые клетки человека линии H1 (код NIH: WA01), эмбриональные стволовые клетки человека линии H7 (код NIH: WA07), а также эмбриональные стволовые клетки человека линии SA002 (Cellartis, Швеция). В одном из вариантов осуществления настоящего изобретения указанные плюрипотентные стволовые клетки представляют собой эмбриональные стволовые клетки, например, клетки линии H1 hES. В альтернативных вариантах используются плюрипотентные стволовые клетки не эмбрионального происхождения.
Дифференцировка клеток, экспрессирующих маркеры, характерные для линии панкреатической энтодермы, из плюрипотентных стволовых клеток
Рост плюрипотентных стволовых клеток
Настоящее изобретение, в некоторых вариантах, как описано ниже, относится к изоляции и культивированию стволовых клеток, в частности культивированию кластеров стволовых клеток, которые сохраняют плюрипотентность в динамической суспензионной культуральной системе. Плюрипотентные кластеры клеток могут быть дифференцированы для производства функциональных β клеток.
Плюрипотентные стволовые клетки, используемые в способах по настоящему изобретению, предпочтительно выращиваются в динамическом культуральной суспензии до дифференцировки к желаемой конечной точке. Было обнаружено, что плюрипотентные стволовые клетки могут культивироваться и расти в виде кластеров клеток в суспензии в подходящей среде без потери плюрипотентности. Такое культивирование может произойти в динамической суспензионной культуральной системе, где клетки или клеточные кластеры поддерживаются в движении для предотвращения потери плюрипотентности. Полезные динамические суспензионные культуральные системы включают в себя системы, оснащенные средствами для поддержания движения в культуре, например, методом перемешивания, встряхивания, рециркуляции или пропускания газов через среду. Такое перемешивание может быть прерывистым или непрерывным, оно должно обеспечивать достаточное движение кластеров клеток, чтобы способствовать росту и предотвращать преждевременную дифференцировку. Предпочтительно перемешивание включает непрерывное перемешивание например, посредством лопасти, вращающейся с определенной скоростью. Лопасть может быть закругленной или плоской. Скорость движения лопасти должна быть такой, чтобы кластеры поддерживались в суспендированном состоянии и оседание было минимальным. Кроме того, угол лопасти может быть изменен, чтобы способствовать движению клеток и кластеров клеток вверх для избежания оседания. Кроме того, тип лопастного колеса, угол и скорость вращения могут быть скоординированы таким образом, что клетки и кластеры будут поддерживаться в состоянии однородной коллоидной суспензии.
Суспензионное культивирование и роста кластеров плюрипотентных стволовых клеток может быть достигнуто путем переноса статических культивированных стволовых клеток в соответствующую динамическую культуральную систему, такую как одноразовые пластиковые, многоразовые пластиковые, изготовленные из нержавеющей стали или стекла сосуды, например центрифужную пробирку или колбу Эрленмейера. Например, стволовые клетки, культивированные в адгезивной статической среде, т.е. на поверхности носителей, могут быть сначала удалены с поверхности методом обработки хелатирующим агентом или ферментом. Подходящие ферменты включают, без ограничений, следующие: коллагеназа типа I, Dispase® (Sigma Aldrich LLC, г. Сент-Луис, штат Миссури, США) или коммерчески доступная готовая форма, продаваемая под торговым названием Accutase® (Sigma Aldrich LLC, г. Сент-Луис, штат Миссури, США). Accutase® представляет собой раствор для открепления клеток, содержащий коллагенолитические и протеолитические ферменты (выделенные из ракообразных) и не содержит продуктов, выделенных из млекопитающих или бактерий. Таким образом, в одном варианте осуществления фермент является коллагенолитическим, или протеолитическим ферментом, или раствором для открепления клеток, содержащим коллагенолитические и протеолитические ферменты. Подходящие хелатирующие агенты включают, без ограничений, этилендиаминтетрауксусную кислоту (EDTA). В некоторых вариантах осуществления плюрипотентные стволовые клетки культуры инкубируют с ферментом или хелатирующим агентом, предпочтительно до того момента, когда края колонии начнут сворачиваться и отделяться, но до полного отрыва колоний от поверхности культуры. В одном варианте осуществления клеточные культуры инкубируют при комнатной температуре. В одном варианте осуществления клетки инкубируют при температуре выше 20°С, выше 25°С, выше 30°С или выше 35°С, например, при температуре между примерно 20°С и примерно 40°С, примерно от 25°С до примерно 40°С, примерно от 30°С до около 40°С, например, приблизительно 37°С. В одном варианте осуществления клетки инкубировали в течение по меньшей мере примерно 1, по меньшей мере приблизительно 5, по меньшей мере приблизительно 10, по меньшей мере около 15, по меньшей мере около 20 минут, например, от примерно 1 до примерно 30 минут, от примерно 5 до примерно 30 минут, от примерно 10 до примерно 25 минут, от 15 до 25 минут, например, приблизительно 20 минут. В одном варианте осуществления способ включает в себя стадию удаления фермента или хелатирующего агента из культуры клеток после обработки. В одном варианте осуществления клеточную культуру промывают один раз или два раза или более после удаления фермента или хелатирующего агента. В одном варианте культуру клеток промывают подходящей культуральной средой, такой как mTeSR®1 (Stem Cell Technologies, Ванкувер, Британская Колумбия, Канада). В одном варианте осуществления используется ингибитор Rho-киназы (например, Y-27632, Axxora № по каталогу ALX-270-333, San Diego, CA). Ингибитор Rho-киназы может быть в концентрации от примерно 1 до примерно 100 мкМоль, приблизительно от 1 до 90 мкМоль, от около 1 до около 80 мкМоль, от около 1 до около 70 мкМоль, от приблизительно 1 до приблизительно 60 мкМоль, от около 1 до около 50 мкМоль, от приблизительно 1 до приблизительно 40 мкМоль, от около 1 до около 30 мкМоль, от около 1 до около 20 мкМоль, от около 1 до около 15 мкМоль, от приблизительно 1 до приблизительно 10 мкМоль или 10 мкМоль. В одном варианте осуществления ингибитор Rho-киназы добавляется в количестве по меньшей мере 1 мкМоль, по меньшей мере 5 мкМоль или по меньшей мере 10 мкМоль. Клетки могут быть сняты с поверхности статической системе культивирования с скребком или резиновой палочкой. Среда и клетки могут быть перемещены в динамическую культуральную систему с помощью стеклянной пипетки или других подходящих средств. В предпочтительном варианте, среда в динамической культуральной системе заменяется ежедневно.
Изобретение обеспечивает в одном варианте осуществления способы культивирования и роста плюрипотентных стволовых клеток в трехмерной суспензионной культуре. В частности, предоставлены способы культивирования и роста плюрипотентных стволовых клеток путем формирования агрегированных кластеров клеток этих плюрипотентных стволовых клеток. Клеточные кластеры могут образовывать в результате обработки плюрипотентных стволовых клеток культур ферментом (например, нейтральной протеазой, или Dispase®) или хелатирующим агентом до культивирования клеток. Предпочтительно клетки культивируют в перемешиваемой или встряхиваемой суспензионной культуральной системе. В одном варианте осуществления настоящего изобретения также представлено образование из таких кластеров плюрипотентных стволовых клеток дифференцированных клеток, экспрессирующих маркеры, характерные для линии клеток поджелудочной энтодермы
Предпочтительно, клеточные кластеры состоят из агрегированных плюрипотентных стволовых клеток. Агрегированные стволовые клетки экспрессируют один или несколько маркеров плюрипотентности, например, один или более (например, 1, 2, 3 или все) из маркеров CD9, SSEA4, TRA-1-60, TRA-и 1-81, а также показывают отсутствие экспрессия одного или более маркеров дифференцировки, например, лишены экспрессии CXCR4. В одном варианте осуществления, агрегированные стволовые клетки экспрессируют маркеры плюрипотентности CD9, SSEA4, TRA-1-60, TRA-и 1-81 и лишены экспрессии маркера дифференцировки CXCR4
Один вариант представляет собой способ культивирования плюрипотентных стволовых клеток в виде клеточных кластеров в суспензионной культуре. Клеточные кластеры представляют собой агрегированные плюрипотентные стволовые клетки, культивируемые в динамической перемешиваемой или встряхиваемой суспензионной культуральной системе. Клеточные кластеры могут быть перемещены из плоской прикрепленной культуры путем добавления фермента, такого как нейтральная протеаза, например Dispase, в качестве клеткоотделяющего агента к перемешиваемой или встряхиваемой суспензионной культуральной системе. Примеры подходящих ферментов включают, без ограничений, коллагеназу IV типа, Dispase® или Accutase®. Клетки поддерживают плюрипотентность в перемешиваемой или встряхиваемой культуральной системе, в частности в перемешиваемой суспензионной культуральной системе.
Другой вариант осуществления изобретения относится к способу культивирования плюрипотентных стволовых клеток как клеточных кластеров в суспензионной культуре, в котором кластеры клеток представляют собой агрегированные плюрипотентные стволовые клетки, перемещенные из плоской прикрепленной культуры с помощью хелатирующего агента, например, EDTA, и культивированные в перемешиваемой или встряхиваемой суспензионной культуральной системе. Клеточные кластеры поддерживают плюрипотентность в перемешиваемой или встряхиваемой суспензионной культуральной системе, в частности в перемешиваемой суспензионной культуральной системе.
Другой вариант осуществления изобретения относится к способу культивирования плюрипотентных стволовых клеток как клеточных кластеров в суспензионной культуре, в котором кластеры клеток представляют собой агрегированные плюрипотентные стволовые клетки, перемещенные из плоской прикрепленной культуры с помощью фермента Accutase® и культивированные в перемешиваемой или встряхиваемой суспензионной культуральной системе. Клеточные кластеры поддерживают плюрипотентность в динамичной суспензионной культуральной системе.
Клеточные кластеры по изобретению могут быть дифференцированы в мезодермальные клетки, такие как сердечные клетки, клетки эктодермы, нервные клетки, одногормональные положительные клетки или клетки поджелудочной энтодермы. Способ может дополнительно включать в себя дифференцировку, например, дифференцировку клеток поджелудочной энтодермы в клетки-предшественники панкреатических клеток и клетки, экспрессирующие гормон поджелудочной железы. В другом варианте, клетки-предшественники панкреатических клеток характеризуются экспрессией транскрипционных факторов β-клеток Pdx1 и Nkx6.1.
В одном варианте осуществления, стадия дифференцировки осуществляется через по меньшей мере 12 часов, по меньшей мере 24 часа, по меньшей мере 36 часов, по меньшей мере 48 часов, по меньшей мере 72 часа, по меньшей мере 96 часов, по меньшей мере 120 часов, по меньшей мере 144 ч, по меньшей мере 168 часов, по меньшей мере 196 часов или, более предпочтительно, от около 48 часов до около 72 часа в суспензионной культуральной системе. Дифференцировка может быть осуществлена с использованием постадийного прогрессирования компонентов среды, например, как описано в примерах (например, см. таблицу A и таблицы 1а и 1c).
В предпочтительном варианте, трехмерный кластер клеток получают путем выращивания плюрипотентных стволовых клеток в плоской прикрепленной культуре; выращивания плюрипотентных стволовых клеток в агрегированные кластеры клеток; и переноса кластеров плюрипотентных стволовых клеток из плоской прикрепленной культуры в динамическую культуральную суспензию с использованием фермента или хелатирующего агента. Еще один предпочтительный вариант осуществления представляет собой способ выращивание и дифференцировки плюрипотентных стволовых клеток в динамически перемешиваемой суспензионной культуральной системе путем выращивания плюрипотентных стволовых клеток в плоской прикрепленной культуре; выращивания плюрипотентных стволовых клеток в агрегированные кластеры клеток; и переноса кластеров плюрипотентных стволовых клеток из плоской прикрепленной культуры в динамическую культуральную суспензию с использованием фермента или хелатирующего агента; и дифференцировки кластеров плюрипотентных клеток в динамической перемешиваемой суспензионной культуральной системе для создания популяции клетки-предшественника панкреатических клеток.
Другое вариант осуществления представляет собой выделенный трансплантируемый продукт стволовых клеток, содержащий дифференцированные стволовые клетки, полученные из суспензии разросшихся кластеров плюрипотентных стволовых клеток, которые дифференцируются в клетки-предшественники панкреатических клеток. Более конкретно, выделенный трансплантируемый продукт стволовых клеток получают путем выращивания плюрипотентных стволовых клеток в плоской прикрепленной культуре; выращивания плюрипотентных стволовых клеток в агрегированные кластеры клеток; и переноса кластеров плюрипотентных стволовых клеток из плоской прикрепленной культуры в динамическую культуральную суспензию с использованием фермента или хелатирующего агента; и дифференцировки кластеров плюрипотентных клеток в динамически перемешиваемой суспензионной культуральной системе. Выделенный трансплантируемый продукт стволовых клеток предпочтительно используют для лечения диабета.
В другом варианте осуществления способ включает в себя трансплантацию в животное, больное диабетом для дальнейшего созревания in vivo в функциональные панкреатические эндокринные клетки.
Другой вариант осуществления представляет собой способ выращивания и дифференцировки плюрипотентных стволовых клеток в суспензионной культуральной системе, содержащей растущие плюрипотентные стволовые клетки в плоской прикрепленной культуре; удаления плюрипотентных стволовых клеток с плоской прикрепленной культуры с использованием фермента; прикрепления плюрипотентных стволовых клеток к микроносителям в статической культуре; выращивания плюрипотентных клеток в динамически перемешиваемой суспензионной культуральной системе; и дифференцировки плюрипотентных клеток в динамически перемешиваемой суспензионной культуральной системе для создания популяции клетки-предшественника панкреатических клеток.
Микроносители могут быть любой формы, известной в данной области техники крепления клеток, в частности, микроносители могут быть сферами. Микроноситель может состоять их материалов натурального или синтетического происхождения. Примеры включают микроносители на коллагеновой основе, на основе декстрана или на основе целлюлозы. Например, сферы микроносителя могут быть модифицированными полистирольными сферами с прикрепленным к поверхности катионным триметил аммонием для обеспечения микроносителю положительно заряженной поверхности. Диаметр сферы может изменяться в диапазоне от около 90 до около 200 мкм, в альтернативном варианте от около 100 до около 190 мкм, в альтернативном варианте от около 110 до около 180 мкм, в альтернативном варианте от около 125 до 175 мкм в диаметре. Сферы микроносителей могут состоять из тонкого слоя денатурированного коллагена, химически связанного с матрицей из сшитого декстрана. Сферы микроносителей могут состоять из следующих материалов: стекло, керамика, полимеры (такие как полистирол) или металлы. Кроме того, микроносители могут быть непокрытыми или с покрытием, например, из кремния или белка, такого как коллаген. В дополнительном аспекте микроноситель может состоять или иметь покрытие из соединений, улучшающих связывание клетки с микроносителем и улучшающих отделение клетки от микроносителя, включая, помимо прочего, такие соединения, как натрия гиалуронат, поли(моностеароилглицерид-ко-янтарная кислота), поли-D,L-лактид-ко-гликолид, фибронектин, ламинин, эластин, лизин, n-изопропилакриламид, витронектин и коллаген. Примеры дополнительно включают микроносители, обладающие микротоком, такие как микроносители с частичной гальванической парой цинка и меди, которая производит низкие уровни биологически значимого электричества; или микроносители, являющиеся парамагнетическими, такие как парамагнетические кальций-альгинатные микроносители.
В некоторых вариантах осуществления популяцию клеток панкреатической энтодермы получают путем поэтапной дифференцировки плюрипотентных клеточных кластеров. В некоторых вариантах осуществления плюрипотентные клетки представляют собой эмбриональные плюрипотентные стволовые клетки человека. В одном аспекте настоящего изобретения клетка, экспрессирующая маркеры, характерные для линии дефинитивной энтодермы, представляет собой клетку-предшественник первичной полоски. В альтернативном аспекте настоящего изобретения клетка, экспрессирующая маркеры, характерные для линии дефинитивной энтодермы, представляет собой клетку мезэнтодермы.
В некоторых вариантах осуществления настоящее изобретение относится к способу поэтапной дифференцировки плюрипотентных клеток, содержащему культивирование клеток на стадиях 3-5 в динамической суспензионной культуре. В некоторых вариантах осуществления образованная панкреатическая энтодермальная популяция трансплантируется в животное, больное диабетом, для дальнейшего созревания in vivo до функциональных эндокринных клеток. Настоящее изобретение также предусматривает системы или наборы для использования со способами настоящего изобретения.
Настоящее изобретение также представляет клетку или популяцию клеток, которые можно получить методами настоящего изобретения. Настоящее изобретение также представляет клетку или популяцию клеток, полученную методами настоящего изобретения.
Изобретение представляет методы обработки. В частности, настоящее изобретение представляет способ обработки пациента, страдающего диабетом или подвергающегося риску развития данного заболевания.
Настоящее изобретение также представляет клетку или популяцию клеток, которые можно получить или полученные методом настоящего изобретения для применения в методе обработки. В частности, настоящее изобретение также представляет клетку или популяцию клеток, которые можно получить или полученные методом настоящего изобретения для применения в методе обработки пациента, страдающего диабетом или подвергающегося риску развития данного заболевания. Диабет может быть диабетом типа I или типа II.
В одном из вариантов осуществления метод обработки включает имплантацию клеток, которые получены или могут быть получены методом настоящего изобретения, пациенту.
В одном варианте осуществления способа обработки включает дифференцировку плюрипотентных стволовых клеток в пробирке в стадии 1, стадии 2, стадии 3, стадии4 или стадии 5 клеток, например, как описано здесь, и имплантацию дифференцированных клеток в тело пациента.
В одном из вариантов осуществления метод также содержит стадию культивирования плюрипотентных стволовых клеток, например как описано в настоящем документе, предшествующую стадии дифференцировки плюрипотентных стволовых клеток.
В одном из вариантов осуществления метод также включает стадию дифференцировки клеток in vivo, после стадии имплантации.
В одном из вариантов осуществления пациент является млекопитающим, предпочтительно человеком.
В одном из вариантов осуществления клетки можно имплантировать в форме дисперсных клеток или клеток, образующих кластеры, которые можно вводить в воротную вену печени методом инфузии. Клетки можно альтернативно вводить в биосовместимые разлагающиеся полимерные опорные материалы, пористые неразлагающиеся устройства или в инкапсулированном виде для защиты от иммунного ответа организма-хозяина. Клетки можно имплантировать в подходящее место в организме реципиента. Места имплантации включают в себя, например, печень, естественную поджелудочную железу, пространство под почечной капсулой, сальник, брюшную полость, субсерозное пространство, кишечник, желудок или подкожный карман.
Для стимуляции дальнейшего дифференцировки, выживаемости или активности имплантированных клеток in vivo, до, одновременно с или после введения клеток можно вводить дополнительные факторы, такие как факторы роста, антиоксиданты или противовоспалительные вещества. Данные факторы могут секретироваться эндогенными клетками и воздействовать на внедренные клетки in situ. Дифференцирование имплантированных клеток можно индуцировать любой комбинацией известных в данной области эндогенных факторов роста и экзогенным введением известных в данной области факторов роста.
Количество клеток, используемых при имплантации, зависит от ряда различных факторов, включая состояние пациента и его реакцию на лечение, и может быть определено специалистом в данной области.
В одном из вариантов осуществления метод обработки также включает встраивание клеток перед имплантацией в трехмерный опорный материал. Клетки можно поддерживать in vitro на данном опорном материале перед имплантацией пациенту. Опорный материал, содержащий клетки, альтернативно можно имплантировать непосредственно в организм пациента без дополнительного культивирования in vitro. В опорный материал может быть необязательно включено по меньшей мере одно фармацевтическое вещество, обеспечивающее выживаемость и функционирование трансплантированных клеток.
В некоторых вариантах осуществления настоящего изобретения один или несколько из следующих вариантов может быть использован в способах по настоящему изобретению.
Стадия 3: приблизительно 5 нг/мл, от приблизительно 3 нг/мл до приблизительно 6 нг/мл
Приблизительно 8 мМоль, от приблизительно 1 мМоль до приблизительно 8 мМоль, от приблизительно 3 мМоль до приблизительно 5 мМоль
или
Стадии 3-4:
Приблизительно 25 мМоль, от приблизительно 10 до приблизительно 25 мМоль
или
Стадия 5
Менее чем приблизительно 11 мМоль, от приблизительно 1 мМоль до приблизительно 10 мМоль
или
Стадия 5
Более чем приблизительно 25 мМоль, от приблизительно 25 мМоль до приблизительно 50 мМоль
Публикации, цитируемые в настоящем документе, полностью включаются в настоящий документ посредством ссылки. Настоящее изобретение дополнительно иллюстрируется, среди прочего, следующими примерами.
ПРИМЕРЫ
НАСТОЯЩЕЕ ИЗОБРЕТЕНИЕ ДАЛЕЕ ПРОИЛЛЮСТРИРОВАНО СЛЕДУЮЩИМИ НЕОГРАНИЧИВАЮЩИМИ ПРИМЕРАМИ.
ПРИМЕР 1
СУСПЕНДИРОВАНИЕ И КЛАСТЕРИЗАЦИЯ ЧЕЛОВЕЧЕСКИХ ЭМБРИОНАЛЬНЫХ СТВОЛОВЫХ КЛЕТОК КЛЕТОЧНОЙ ЛИНИИ H1 С ПОМОЩЬЮ DISPASE/НЕЙТРАЛЬНОЙ ПРОТЕАЗЫ
Клетки линии эмбриональных стволовых клеток человека Н1, (клетки WA01, WiCell, г. Мэдисон, штат Висконсин, США) при пассировании 41 раз промывали PBS (№ по каталогу 14190, Invitrogen) и обрабатывали 1 мг/мл раствора Dispase® (Neutral Protease, Sigma Aldrich Co LLC, № по каталогу D4818, г. Сент-Луис, штат Миссури, США) в DMEM/F12 (Invitrogen № по каталогу 11330, г. Гранд Айленд, штат Нью-Йорк, США). Клетки инкубировали при 37°С в течение 15-25 минут, пока края колонии не стали скручиваться и отходить, но перед полным отрывом колоний от поверхности культуры. Dispase® затем удаляли, и чашку Петри дважды промывали средой mTeSR®1 (Stem Cell Technologies, г. Ванкувер, Британская Колумбия, Канада), содержащей 10 мкМоль Y-27632 (Axxora, № по каталогу ALX-270-333, г. Сан-Диего, штат Калифорния, США). Среду mTeSR®1, содержащую 10 мкМоль Y-27632, затем добавляли в чашку для культивирования в кол-ве 5 мл/60 см2, и клетки снимали с поверхности скребком или резиновой палочкой. Среду и клетки переносили в коническую пробирку на 50 мл с помощью стеклянной пипетки и кластеры центрифугировали при 90 g (ОЦС, относительная центробежная сила) в течение 3 минут.
После центрифугирования среду отсасывали и клетки осторожно ресуспендировали и быстро измельчали в 12 мл среды mTeSR®1, содержащей по 10 мкМоль Y-27632 на каждые 225-240 см2 общей плоской культуры (эквивалентно одной колбе T225 или четырем чашкам 10 см, приблизительно 90 миллионов клеток). Суспензию клеток затем переносили (1 мл/лунку) на 6-луночные планшеты Ultra Low Binding Culture (Corning Incorporated, № по каталогу 3471, г. Корнинг, штат Нью-Йорк, США), содержащие 2 мл/лунка свежей среды mTeSR®1 с 10 мкМоль Y-27632. Отделенные таким образом клетки напоминал фрагменты монослоя со средним диаметром поднимаемых фрагментов около 20-30 мкМоль (фиг.1а), каждый из которых состоит из скопления клеток. Эти фрагменты монослоя инкубировали в суспензии в течение 2 часов (время инкубации может лежать в диапазоне от 0,5 до 4 часов), при этом наблюдались точечные агрегаты фрагментов. Агрегаты затем быстро растирают кратко со стеклянной пипеткой 10 мл среды и инкубировали в течение ночи (агрегаты могут переходить непосредственно в суспензию) в планшет слабого связывания (агрегаты также можно инкубировать на необработанной пластике для культивирования клеток и обработанной пластике для культивирования клеток).
После инкубации в течение ночи (18-24 часа), клетки и среду переносили непосредственно в центрифужную пробирку на 125 мл (Corning Incorporated, № по каталогу 4500-125, г. Корнинг, штат Нью-Йорк, США), содержащую 25 мл среды mTeSR®1 и перемешивали со скоростью 50 оборотов в минуту (диапазон может составлять 30-80+ оборотов в минуту) с получением конечного объема приблизительно 75 мл. Среду меняли ежедневно в течение 4 суток. Плюрипотентность определяли после 4 суток в культуре и результаты проточной цитометрии показали высокую экспрессию маркеров плюрипотентности (CD9, SSEA4, TRA-1-60, TRA-и 1-81) почти без экспрессии маркера дифференцировки (CXCR4) , См. рис. 1b. Эти данные показывают, что клетки линии H1 HES могут быть успешно перенесены как кластеры клеток плоской прикрепленной культуры в суспензионную культуру с помощью Dispase® как клеткоотделяющего агента и поддерживать плюрипотентность в динамической суспензионной культуральной системе. Этот пример также может быть осуществлен с помощью встряхивания, а не с перемешиванием суспензионной системы на носителях и в колбах Эрленмейера с сопоставимыми результатами.
После 4 суток в суспензионной культуре (дифференцировка может также начаться через 24-120 часов после формирования агрегатов, предпочтительно культура должна сформироваться за 2-3 суток до начала дифференцировки), плюрипотентные агрегаты клеток дифференцируются с использованием постадийного прогрессирования компонентов среды, чтобы побудить клетки к формированию панкреатического пути развития. Центрифужное перемешивание для дифференцировки агрегатов проходит на скорости 65 оборотов в минуту. Среда и компоненты показаны в таблице 1а.
В конце стадии 1 образцы подвергали проточной цитометрии и PCR. Суспензионные дифференцированные культуры образовали однородную и гомогенную популяцию клеток в виде рыхлых агрегатов в конце стадии 1 (фиг.1c), с практически отсутствующей экспрессией маркера плюрипотентности (CD9), в то время как маркеры окончательного дифференцировки энтодертмы были достаточно высоки, 97,2% CXCR4 (CD184)-положительны и 97,3% CD99-положительны (фиг.1D). Эти результаты коррелируют с результатами Qrt-PCR, которые показали резкое снижение экспрессии генов плюрипотентности (CD9, NANOG и POU5F1/OCT4) и большим увеличением в генах, связанных с дефинитивной энтодермой (CXCR4, Cerberus, РКГ, Foxa2, GATA4, GATA6, MNX1 и SOX17) по сравнению с недифференцированными клетками линии HES WA01 (фиг.1е).
Окончательные кластеры энтодермы были затем дополнительно дифференцированы в направлении примитивной кишки путем удаления веществ семейства TGF-β, GDF8, и добавлением FGF7 в среду. После трех суток культивирования с FGF7, кластеры дифференцировались в поджелудочные PDX1-экспрессирующие клетки добавлением полностью-транс-ретиноевой кислоты либо к среде, содержащей высокий уровень глюкозы (25 мМоль) и низкую концентрацию богатого липидами бычьего сывороточного альбумина (AlbuMAX® (Life Technologies Corporation, г. Карлсбад, штат Калифорния, США)), либо к среде, содержащей относительно низкую концентрацию глюкозы (8 мМоль) и 2% бычьего сывороточного альбумина без жирных кислот. Подробное добавление компонентов в этих средах перечислено в таблице 1а. В конце дифференцировки был проведен анализ образцов на экспрессию маркеров панкреатических клеток-предшественников. Было отмечено, что кластеры, дифференцированные либо с условием низкий уровень глюкозы + 2% FAF-BSA (А), или с высоким содержанием глюкозы + 0,1% AlbuMAX® (B) - что было определено с помощью проточной цитометрии, экспрессировали высокие уровни NKX6.1, необходимого фактора транскрипции функциональных β-клеток, и высокие уровни маркеров эндокринной поджелудочной, таких как синаптофизин и хромогранин (таблица 1b). Эти результаты согласуются с результатами RT-PCR, которые показали высокие уровни нескольких панкреатических генов-предшественников, выраженные в образцах, взятых по условиям А и В (данные не показаны).
Типичные морфологии кластеров клеток в ходе прогрессирования через дифференцировку от дефинитивной энтодермы (DE) (фиг.1c) к клеткам примитивной кишки и панкреатической энтодермы (фиг.1f) показали видимые морфологические изменения в клетках и клеточных кластерах. Как правило, плюрипотентные кластеры плотные и темные по данным фазоконтрастной микроскопии, затем становятся менее плотными по ходу того как клетки прогрессируют до клеток примитивной кишки в стадии 2. Эта морфология реверсирует при последующей обработке полностью-транс-ретиноевой кислотой и кластеры вновь становятся более плотными и равномерными, с гладкой границей кластера.
Клетки, дифференцированные по условию B в стадии 4, были обработаны в течение дополнительных 5 суток в стадии 5 средой, содержащей ингибитор ALK5 (таблица 1c). Этот период дополнительного созревания в культуре приводит к значительному увеличению экспрессии эндокринных маркеров: INS, GCG, SST, PPY и PCSK1. Клеточные кластеры затем имплантировали в почечную капсулу мышей линии SCID-BGв соответствии с утвержденным протоколом исследования IACUC, и далее у мышей в течение 20 недель измеряли уровень С-пептида каждые 2-4 недели в голодном и сытом состоянии. Через 4 недели после имплантации, после 20 часового голодания, а затем стимуляции глюкозой, С-пептид не был обнаружен. К 6 неделе, 2 из 5 мышей показали некоторое положительное (0,087 и 0,137 нг/мл) количество человеческого С-пептида, и к 10 неделе, 5 из 5 мышей были положительными (0,085 - 0,291 нг/мл) на содержание С-пептида. Через 16 недель, после 20 часового голодания и стимуляции глюкозой, все 4 мыши (4/4) показали положительную (0,377 - 3,627 нг/мл) экспрессию С-пептида.
Эти результаты показывают, что агрегаты плюрипотентных клеток могут быть сформированы, а затем дифференцированы в суспензионной культуре для генерирования популяции панкреатических клеток-предшественников, характеризующихся экспрессией факторов транскрипции β-клеток, таких как Pdx1 и Nkx6.1. Таким образом, дифференцированные кластеры клеток, имплантированные и дозревающие in vivo , способны экспрессировать инсулин в ответ на нагрузку глюкозой в физиологически соответствующих уровнях.
Протокол дифференцирования
(конечная концентрация глюкозы)
8мМоль глюкозы
8 мМоль (A) или
25 мМоль глюкозы (B)
или 0,1% Albumax (бычьего сывороточного альбумина) и 2мМоль L-Glutamine (B)
И/ИЛИ
Малые молекулы
0-24 часа
GDF8 (100 нг/мл) на 0-96 часа
ITS-X (1:50 000)
ITS-X (1:50 000)
ITS-X (1:200)
RA (2мкМоль)
SANT (0,25мкМоль)
AA (5 нг/мл)
LDN (100 нМоль)
SANT (0,25мкМоль)
Cypi (100 нМоль)
TppB (500 нМоль)
LDN (100 нМоль)
Результаты проточной цитометрии для выбранных маркеров дифференцировки
NKX6.1
CDX2
Инсулин
Синаптофизин
Хромогранин
BSA
Albumax
Протокол дифференцирования
(конечная концентрация глюкозы)
(25мМоль глюкозы)
и 2мМоль L-глутамина
И/ИЛИ
Малые молекулы
Cypi (100 нМоль)
LDN (100 нМоль)
ALKVi (10мМоль)
ПРИМЕР 2
СУСПЕНДИРОВАНИЕ И КЛАСТЕРИЗАЦИЯ ЧЕЛОВЕЧЕСКИХ ЭМБРИОНАЛЬНЫХ СТВОЛОВЫХ КЛЕТОК КЛЕТОЧНОЙ ЛИНИИ H1 С ПОМОЩЬЮ EDTA
Клетки линии эмбриональных стволовых клеток человека Н1, (клетки WA01, WiCell, г. Мэдисон штат Висконсин, США) при пассировании 41 раз промывали один раз PBS (№ по каталогу 14190, Invitrogen) и обрабатывали EDTA, неферментативным клеткоотделяющим агентом / агентом пассирования (Lonza, № по каталогу 17-7-11 Е). Клетки инкубировали при комнатной температуре в течение 8 минут. Затем удаляли EDTA, и через 1 или 2 минуты (9-10 минут общего воздействия EDTA) планшет промывали средой mTeSR®1, содержащей 10 мкМоль Y-27632 (№ по каталогу Axxora ALX-270-333, г. Сан-Диего, штат Калифорния, США) и отделившиеся клетки собирали в 50 мл коническую пробирку с помощью стеклянной пипетки. После одного дополнительный цикла промывания планшета средой mTeSR®1, содержащей 10 мкМоль Y-27632, смыв собирали и объединяли с отделившимися клетками. Следует отметить, что некоторые клетки оставались на пластине через 9-10 минут после контакта с EDTA при комнатной температуре, и отделившиеся клетки не были полностью измельчены в суспензию отдельных клеток. Вместо этого клетки были удалены с поверхности в виде мелких агрегатов. Затем среду и клетки переносили в 50 мл коническую пробирку с помощью стеклянной пипетки и проводили подсчет клеток (NucleoCounter®-ChemoMetec A/S, № по каталогу YC-Т100, Дания). При необходимости добавляли дополнительную среду mTeSR®1, содержащую 10 мкМоль Y-27632 для того, чтобы довести концентрацию клеток до 1,0-1,5 млн клеток/мл.
Клетки не центрифугировали, так как кластеры свободно агрегированы и могут разделиться на отдельные клетки при центрифугировании в осадок и ресуспендировании с помощью пипетки. Вместо этого, среда и клетки в пробирки осторожно перемешивали до формирования однородной суспензии. При желании, можно также удлинить период обработки EDTA и отделять клетки вблизи суспензии отдельных клеток. Затем суспензию клеток переносили в обработанные двумя нетканевыми культурами 6-луночные планшеты (Becton Dickinson, № по каталогу Falcon 351146, г. Франклин-Лейкс, штат Нью-Джерси, США) при 37°C в увлажненном инкубаторе с 5% CO2, по 3 мл/лунку из стеклянной пипетки. Клетки инкубировали в суспензии в течение 2 ч, после чего наблюдались точечные агрегаты. Агрегаты затем измельчали осторожным пипетированием с помощью стеклянной пипетки для разделения крупных агрегатов и создания суспензии однородных, равномерных кластеров, затем инкубировали в течение ночи без воздействия.
Через 18-24 часа клетки и среды центрифугировали в 50 мл конических пробирках при 90g (ОЦС) в течение 3 минут. Отработанный супернатант среды утилизировали, клеточные агрегаты суспендировали в свежей mTeSR®1, и суспензию переносили в центрифужную пробирку (Corning Incorporated, № по каталогу 4500-125, г. Корнинг, штат Нью-Йорк, США), перемешивали при 55 оборотах в минуту при 37°C в увлажненном 5% СО2-инкубаторе. Среду меняли ежедневно в течение 2 суток. Плюрипотентность была определена после 2 суток в перемешанной суспензии культуры до перехода к дифференцировки культуры. Результаты проточной цитометрии для CD9, SSEA4, TRA-1-60, TRA-1-81 и экспрессии CXCR4 приведены в виде графика разброса на фиг. 2а. Эти данные показывают высокую экспрессию маркеров плюрипотентности (CD9, SSEA4, TRA-1-60, TRA-1-81) и низкую или отсутствующую экспрессию маркера дифференцировки (CXCR4). Эти результаты показывают, что клетки линии H1 HES могут быть перенесены в суспензионную культуру из плоской прикрепленной культуры с использованием неферментативного способа отделения и будут поддерживать плюрипотентность в динамической суспензионной культуральной системе.
Через 2 суток в суспензионной культуре, плюрипотентные агрегаты клеток дифференцируются с постадийным прогрессированием компонентов среды, чтобы побудить клетки к формированию панкреатического пути развития. Счетчик перемешивания поддерживают на скорости 55 оборотов в минуту. Среда и компоненты показаны в таблице 2а.
В конце стадии 1 образцы подвергали проточной цитометрии и PCR. Суспензионные дифференцированные культуры образовали однородную и гомогенную популяцию клеток в виде рыхлых агрегатов в конце стадии 1 (Фиг.2b), с практически отсутствующей экспрессией маркера плюрипотентности (CD9), в то время как экспрессия CXCR4 (CD184), маркера дифференцировки дефинитивной энтодертмы, была достаточно высокой, 95,9% ± 1,8sd (Фиг. 2c) после трех пробегов центрифуги. Эти результаты коррелируют с результатами Qrt-PCR, которые показали резкое снижение экспрессии генов плюрипотентности (CD9, NANOG и POU5F1/OCT4) и большим увеличением в генах, связанных с дефинитивной энтодермой (CXCR4, Cerberus, РКГ, Foxa2, GATA4, GATA6, MNX1 и SOX17) по сравнению с недифференцированными клетками линии HES WA01 (Фиг. 2d).
Кластеры дефинитивной энтодермы из центрифужных пробирок затем объединяют, перемещают либо в другую центрифужную пробирку или колбу Эрленмейера (с системой встряхивания) и направляют на дальнейшую дифференцировку в клетки примитивной кишки путем удаления из среды GDF8 и добавления FGF7. После трех суток культуры с FGF7, кластеры дифференцировались в поджелудочные Pdx1-экспрессирующие клетки добавлением полностью-транс-ретиноевой кислоты к среде, содержащей относительно низкую концентрацию глюкозы (8 мМ) и 2% свободной жирной кислоты бычьего сывороточного альбумина. Подробное добавление компонентов в этих средах перечислено в таблице 2а. В конце дифференцировки был проведен анализ образцов на экспрессию маркеров панкреатических клеток-предшественников. В результате проточной цитометрии в обеих суспензиях наблюдались высокие уровни Nkx6.1, транскрипционного фактора, необходимого для функциональных β-клеток, и высокие уровни маркеров эндокринной поджелудочной, таких как синаптофизин и хромогранин (Табл. 2b и Фиг. 2e). Эти результаты согласуются с результатами RT-PCR, которые показали очень похожие высокие уровни нескольких панкреатических генов-предшественников, выраженных в образцах, полученных во центрифужных пробирках или колбах Эрленмейера (Фиг. 2F).
Эти результаты показывают, что агрегаты плюрипотентныя клеток могут быть сформированы, а затем дифференцированы в суспензионной культуре в различных форматах суспензионной культуры, в том перемешиваемой или встряхиваемой суспензионной системе для генерирования популяции панкреатических клеток-предшественников, характеризующейся экспрессией факторов транскрипции β-клеток, таких как Pdx1 и Nkx6.1.
Компоненты среды и протокол дифференцировки
(8мМоль глюкозы)
3,64 г/л NaCO3
(8мМоль глюкозы)
2,41 г/л NaCO3
1:50 000 ITS-X
1x GlutaMax
1:200 ITS-X
1x GlutaMax
100 нг/мл
50 нг/мл
50 нг/мл
(только сутки 1)
[10 мкМоль]
(0-24 часа)
[2 мкМоль]
SANT [0,25 мкМоль]
TPPB [100 нМоль]
LDN (только сутки 1) [100 нМоль]
Cypi [100 нМоль]
ингибитор ALK5 [1 мкМоль]
TPPB [100 нМоль]
2 дня SF
Сутки 1 и 2,
Без замены на сутки 3
Сутки 1 и 3,
Без замены на сутки 2
Сутки 1 и 2,
Без замены на сутки 3
и сутки 2,
Без замены на сутки 3
Результаты проточной цитометрии для выбранных маркеров дифференцировки
NKX6.1
CDX2
SOX2
NKX2.2
Синаптофизин
Хромогранин
ПРИМЕР 3
СУСПЕНЗИОННАЯ КЛАСТЕРИЗАЦИЯ И СЕРИЙНОЕ СУСПЕНЗИОННОЕ ПАССИРОВАНИЕ ЧЕЛОВЕЧЕСКИХ ЭМБРИОНАЛЬНЫХ СТВОЛОВЫХ КЛЕТОК КЛЕТОЧНОЙ ЛИНИИ H1
Клетки линии эмбриональных стволовых клеток человека Н1, (клетки WA01, WiCell, г. Мэдисон штат Висконсин, США) в пассировании 40, выращенные на тканевой культуре, обработанной полистиролом, покрытым Matrigel® (Corning Incorporated, г. Корнинг, штат Нью-Йорк, США), дважды промывали PBS (№ по каталогу 14190, Invitrogen) и обрабатывали раствором Accutase® половинной эффективности (одна часть PBS на одну часть Accutase®, Sigma-Aldrich, № по каталогу A-6964, г. Сент-Луис, штат Миссури, США). Клетки инкубировали при комнатной температуре в течение 3 с половиной минут. (Accutase® представляет собой раствор для открепления клеток, состоящий из коллагенолитических и протеолитических ферментов (выделенных из ракообразных), и не содержащий продуктов, выделенных из млекопитающих или бактерий). Затем Accutase®удаляли, и через 3 минуты (6 1/2 минут общего воздействия Accutase®) планшет промывали средой mTeSR®1, содержащей 10 мкМоль Y-27632 и отделившиеся клетки собирали в 50 мл коническую пробирку с помощью стеклянной пипетки. После одного дополнительный цикла промывания планшета средой mTeSR®1, содержащей 10 мкМоль Y-27632, смыв собирали и объединяли с отделившимися клетками. Некоторые клетки оставались на планшете после воздействия Accutase® и отделившиеся клетки не были полностью измельчены в суспензию отдельных клеток. Клетки снимали с поверхности в виде мелких агрегатов (Фиг.3А). Среду и клетки переносили в 50 мл коническую пробирку с помощью стеклянной пипетки и проводили подсчет клеток. При необходимости добавляли дополнительную среду mTeSR®1, содержащую 10 мкМоль Y-27632 для того, чтобы довести концентрацию клеток до 1,0-1,5 млн клеток/мл.
Клетки не центрифугировали, так как кластеры свободно агрегированы и могут разделиться на отдельные клетки при центрифугировании в осадок и ресуспендировании с помощью пипетки. Вместо этого, среда и клетки в пробирки осторожно перемешивали до формирования однородной суспензии. Затем суспензию клеток переносили на 6-луночный планшет Ultra Low Binding Culture при 37°C в увлажненный инкубатор с 5% CO2, по 3 мл/лунку с помощью стеклянной пипетки. Клетки инкубировали в суспензии в течение 90 минут, после чего наблюдались точечные агрегаты. Агрегаты затем кратковременно обрабатывали и переносили непосредственно в центрифужную пробирку 125 мл, содержащую 25 мл среды mTeSR®1 перемешанной при 55 оборотах в минуту (всего конечный объем был примерно 75 мл). Среду меняют ежедневно в течение 3 суток, и плюрипотентность в культуре была определена на 3 сутки. Изображения кластеров, полученные с помощью фазоконтрастной микроскопии, показали популяцию кластеров сферической формы, которая сформировалась после 90 минут в статической суспензионной культуре после роста в течение трех суток в культуре (фиг.3b). В конце трех суток в суспензионной культуре, клетки анализируют на плюрипотентность с помощью проточной цитометрии на наличие маркеров CD9, SSEA4, TRA-1-60, TRA-1-81 и CXCR4. Клетки поддерживали высокий уровень экспрессии маркеров плюрипотентности (CD9, SSEA4, TRA-1-60, TRA-1-81), при этом практически отсутствовала экспрессия CXCR4, маркера дифференцировки (таблица 3). Эти результаты показывают, что клетки линии H1 hES могут быть перенесены в суспензионную культуру из плоской прикрепленной культуры с использованием ферментативного способ отделения, например, с помощью Accutase®, и будет поддерживать плюрипотентность в динамической суспензионной культуральной системе.
Плюрипотентные кластеры затем последовательно пассировали с помощью диссоциации Accutase® в течение дополнительных 20 пассажей. На каждом пассаже 50 миллионов клеток осаждали под действием гравитации в течение 2 минут в 50 мл конической пробирке, дважды промывали PBS и обрабатывали раствором Accutase® половинной эффективности на водяной бане при 37°C с аккуратным вращением пробирки на второй и четвертой минуте после добавления Accutase®. После шести минут инкубации из пробирки удаляли Accutase®, не нарушая клеточный осадок. Затем клетки инкубировали еще 3 минуты (9 минут общей экспозиции Accutase®). Пробирку затем промывали средой mTeSR®1, содержащей 10 мкМоль Y-27632, измельчали дважды с помощью стеклянной пипетки, и суспендированные клетки пропускали через 70 мкМоль клеточный фильтр (BD Falcon, № по каталогу 352350, г. Франклин-Лейкс, штат Нью-Джерси, США). Выполняли два дополнительных промывания пробирки со средой mTeSR®1, содержащей 10 мкМоль Y-27632, и пропускали через клеточный фильтр.
Среду и клетки в пробирке осторожно перемешивали до формирования однородной суспензии. Затем суспензию клеток переносили в 6-луночный планшет Ultra Low Binding Culture при 37°C в увлажненный 5% CO2 инкубатор по 3 мл/лунку с помощью стеклянной пипетки и инкубировали в суспензии в течение 2 часов (протестированы временные периоды 0-28 часов), после чего агрегаты переносили в стеклянную центрифужную пробирку с доведением до конечного объема среды 80 мл. Альтернативно, суспензия клеток может быть непосредственно помещена в центрифужную пробирку, перемешиваемую при 55 оборотах в минуту или колбу Эрленмейера, перемешиваемую при 40 оборотах в минуту, и кластеры, образованные в перемешиваемой суспензии (Фиг.3с) доводят до конечного объема среды 80 мл.
С помощью этого метода последовательного пассирования, клетки пассировали 20 раз, с приблизительной кратностью пересева 1: 3 в каждом пассаже. Плюрипотентность измеряли при каждом пассировании с помощью проточной цитометрии, а кариотипирование было проведено с помощью анализа флуоресцентной гибридизации in-situ (FISH) для хромосом 12 и 17; две хромосомы определены как потенциально нестабильные в клетках линии hES. Результаты проточной цитометрии для CD9, SSEA4, TRA-1-60, TRA-1-81 и экспрессии CXCR4 показаны в виде графика и показывают высокий уровень экспрессии маркеров плюрипотентности и низкой или отсутствующей экспрессии маркера дифференцировки ( CXCR4), в то время как FISH анализы для хромосом 12 и 17 показали нормальное количество копий. Эти данные показывают, что клетки линии H1 hES могут быть сохранены в суспензионной культуре после стандартного серийного пассирования с использованием Accutase®, применения ферментативного метод клеточной диссоциации с использованием ферментов, происходящих не от млекопитающих, и будут поддерживать плюрипотентность и стабильный кариотип в динамической суспензионной культуральной системе, при генерировании 1×109 клеток на каждую исходную клетку в течение 20 пассажей. EDTA также может быть использована для этой серии суспензии в 6 пассажах.
Результаты проточной цитометрии на маркеры плюрипотентности клеток, показанные как функция по времени на основе результатов для маркеров CD9, SSEA4, TRA-1-60, TRA-1-81 и CD184 (CXCR4)
(сутки культуры)
ПРИМЕР 4
НАПРАВЛЕННАЯ ДИФФЕРЕНЦИРОВКА СУСПЕНЗИОННОЙ КУЛЬТУРЫ ЧЕЛОВЕЧЕСКИХ ЭМБРИОНАЛЬНЫХ СТВОЛОВЫХ КЛЕТОК КЛЕТОЧНОЙ ЛИНИИ H1
Клетки линии эмбриональных стволовых клеток человека Н1 (клетки WA01, WiCell, г. Мэдисон штат Висконсин, США) при пассировании 40 были сняты с плоской прикрепленной культуры с помощью Accutase® и перенесены в формат суспензионной культуры. Клетки поддерживали в динамической суспензионной культуральной системе в течение 30 пассажей с использованием метода, описанного в примере 3.
Плюрипотентность была подтверждена в течение первых 20 пассажей, как показано в таблице 3, со стабильно высокими уровнями маркеров плюрипотентности, которые сохранялись во всей культуре, что было определено с помощью проточной цитометрии. Чтобы подтвердить плюрипотентность и продемонстрировать способность источника клеток лечить диабет, клетки дифференцировали в панкреатические клетки-предшественники в динамической суспензионной культуральной системе с использованием метода ступенчатой прогрессии в различных средах, содержащих морфогены или факторы роста, предназначенные для нормального развития поджелудочной. Этот процесс приводит к появлению популяции панкреатических клеток-предшественников, которые характеризуются высокой коэкспрессией Pdx1 и Nkx6.1. Когда эти клетки были пересажены, они далее вызревали в ткани, способные к стимулированной глюкозой секреции инсулина, способные секретировать инсулин в ответ на глюкозу и поддерживать нормальный уровень глюкозы в крови при стрептозотоцин-индуцированной модели диабета. Смотрите Фиг. 4c и таблицу 4с.
Для генерирования этих клетки клеток-предшественников панкреатических клеток, человеческие эмбриональные стволовые клетки линии H1, которые были выращены и поддерживались в динамической суспензионной культуральной системе в течение 16 пассажей, дифференцировались по методу, описанному в примере 3. Таким образом, клетки были выращены в течение 30 пассажей, протестированы на плюрипотентность в течение первых 20 пассажей; клетки дифференцировались на 16 пассаже. Плюрипотентные клетки в формате кластера переносили из среды mTeSR®1 в раствор FBC (Таблица 4a) на 3 часа при 4°C. Кластеры клеток затем переносили в суспензию в 3-литровый стеклянный биореактор, регулируемый блоком управления Sartorius Stedim BioStat B-DCU (Goettingen, Германия) и суспендировали в среде для дифференцировки в количестве 0,55 клеток/мл в соответствии с таблицей 4b Клетки поддерживали 14 суток в закрытой стерильной суспензии в биореакторе с регулируемыми показателями температуры, рН и растворенного кислорода (DO) (FermProbe® рН электрод 225 мм, модель № F-635, датчик растворенного кислорода OxyProbe® 12 мм, номер модели D-145 от Broadley-James Corporation, г. Ирвин штат Калифорния, США).
В течение пробега, уровни бикарбоната среды поддерживали в 3,64 г/л, рН поддерживали на уровне рН 7,4 с помощью регулирования потока СО2 в общем объеме среды ≤1,6 л. Свободное пространство биореактора непрерывно обрабатывалось CO2, воздухом и O2 под контролем блока управления Sartorious с установленным уровнем растворенного кислорода 20% на стадии 1 и установленным уровнем растворенного кислорода 30% на стадии 2 с последующим постоянным потоком газа 150cc/мин. Поток кислорода регулируется относительно содержания растворенного кислорода и поток CO2 регулируется относительно уровня рН. Температуру поддерживали на уровне 37°C в течение всего пробега электрическим нагреваемым кожухом. В начале пробега и для каждой замены среды (93% среды удалено за счет замены), лопасти (3”, резьбовое лопастное колеса из нержавеющей стали, работающее с частотой 70 оборотов в минуту) были остановлен, и среда была удалена или добавлена с помощью перистальтического насоса через погружную трубку в биореакторе, подключенную к трубке C-Flex® с использованием трубосварочного стана Terumo™ для поддержания замкнутой системы. Изображения клеток/кластеров были получены в конце каждой стадии дифференцировки, и подвергнутые проточной цитометрии образцы собирали и анализировали на экспрессию CXCR4 на стадии 1 сутки 3 и 3 суток в конце стадии 2 (фиг.4а). Наблюдался почти полное переход популяции от CXCR4-отрицательной плюрипотентной клеточной популяции в начале дифференцировки (таблица 3, канал 16) в популяцию CXCR4-положительную (98,5% клеток CXCR4-положительных, Фиг.4В) клетки. Эти клетки затем перешли к практически CXCR4-отрицательному состоянию через 3 суток в конце стадии 2 (7,0% клеток CXCR4-положительно), а к концу стадии 3 клетки почти полностью перешли в CD56-положительное состояние. В конце процесса дифференцировки на 4-е сутки 4-й стадии, клетки были на 88,5% Pdx1-положительными (4б) и показали паттерны экспрессии, соответствующие сочетанию панкреатических эндокринных клеток (33,5% хромогранин-положительные) и панкреатических клеток-предшественников (65,7% Nkx6.1-положительные). Характерные для данной стадии паттерны экспрессии маркеров указывают на эффективную постадийную дифференцировку от популяции плюрипотентных клеток в клетки поджелудочной железы. В конце процесса дифференцировки было получено 2,77 млн клеток/мл (4,1 миллиардов клеток в 1,5 л), что указывает на полный рост 5 дифференцированных клеток на каждую введенную клетку линии HES.
В конце концов, 500 мл были удалены центрифугированием и промыванием и были испытаны на животной модели приживления, созревания и функционирования. Остальные 1000 мл клеточной суспензии обрабатывали с помощью системы kSep®400 (KBI Biosystems, г. Дарем, штат Северная Каролина, США) для промывания, фильтрации и концентрирования клеточного продукта в полностью замкнутой системе. Клеточный продукт концентрируют от исходного объема 1 л в 50 мл концентрированных клеток до конечной концентрации 41 млн клеток/мл. Эти концентрированные клетки затем разливают в 24 флакона, по 1,2 мл в каждый с использованием автоматизированной машины для разливания во флаконы (Fill-It, TAP, Hertfordshire UK) и замораживают путем помещения в холодильник с жидким азотом.
500 мл дифференцированных клеток, которые были промыты и концентрированы стандартным центрифугированием, трансплантировали в дозе 5 миллионов клеток на мышь линии SCID-Bg непосредственно под почечную капсулу, или размещали внутрь устройств иммунозащитной макроинкапсуляции (TheraCyte™, г. Ирвин штат Калифорния, США ), которые были имплантировали подкожно (6 животных на каждое условие). Через 12 недель после имплантации, имплантированные клетки экспрессировали значительные уровни циркулирующего человеческого C-пептида (> 0,1нг/мл), что было определено с помощью ИФА (ИФА человеческого C-пептида, Mercodia № по каталогу 10-1141-01) в ответ на голодание и последующее кормление и через 16-20 недель животные показывали более 1 нг/мл циркулирующего С-пептида (таблица 4в).
Через 27 недель (190 суток) после имплантации, два животных с устройства инкапсулироваными иммунозащищенными трансплантатами получили одну большую дозу высокой стрептозотицина (STZ)для избирательного уничтожения всех эндогенных мышиных островковых β-клеток и индуцировать диабет (250 мг/кг). После следующих двух недель после STZ-обработки, достаточной, чтобы вызвать явный диабет у контрольных животных, уровни глюкозы в крови животных с тансплантатами оставались в пределах нормы (<150 мг/дл). Через 29 недель после имплантации и две недели после введения STZ два животных были затем проверены на стимулируемую глюкозой секрецию инсулина (GSIS) и показали заметное увеличение циркуляции человеческого c-пептида в ответ на введение глюкозы. Кроме того, когда каждый из трансплантатов были удалены на 209 сутки (29,5 недели) после имплантации, уровень глюкозы в крови животных значительно увеличился до > 500 мг/дл.
Эти результаты показывают, что продукт, выделенный из стволовых клеток эмбриона человека и предназначенный для лечения диабета, может быть получен из суспензии выращенных и дифференцированных стволовых клеток. Продукт может быть генерирован в масштабируемой, перемешивающей системе биореактора с замкнутым циклом и клеточный продукт может быть обработан промыванием в замкнутой системе и концентрирован, как и требуется для коммерческого производства cGMP. Продукт, выделенный из стволовых клеток эмбриона человека, подходит для лечения диабета в широко используемой модели животного диабета,что подтверждено GSIS-компетенций, обладает способностью регулировать уровень глюкозы в крови, и показывает возвращение диабетического состоянии после удаления клеточной терапии.
Состав раствора FBC
b NF = Национальный формуляр
c FCC = Кодекс пищевых химикатов
d Н/Д = Нет данных
e ACS = Химическое общество США
Компоненты среды и протокол дифференцировки
(конечная концентрация глюкозы)
3,64 г/л NaCO3
(8 мМоль глюкозы)
2% не содержащего жирных кислот бычьего сывороточного альбумина (FAF-BSA)
и 2мМоль L-глутамина
И/ИЛИ
Малые молекулы
0-24 часа
GDF8 (100 нг/мл) на 24-72 часа
ITS-X (1:50 000)
ITS-X (1:50 000)
ITS-X (1:200)
RA (2мкМоль)
SANT (0,25 мкМоль)
AA (5 нг/мл)
TppB (200 нМоль)
LDN (100 нМоль) на
0-24 часа стадия 3
SANT (0,25 мкМоль)
Cypi (100 нМоль)
SCIO (2μM)
TppB (100 нМоль)
(Номенклатура Время 0 = первый посев в новой стадии; время 24, 48 или 96 часов = время после замены среды стадии)
Экспрессия C-пептида (нг/мл)
ПРИМЕР 5
НАПРАВЛЕННАЯ ДИФФЕРЕНЦИРОВКА В ФОРМАТ СУСПЕНЗИОННОЙ КУЛЬТУРЫ ПРИКРЕПЛЕННЫХ ЧЕЛОВЕЧЕСКИХ ЭМБРИОНАЛЬНЫХ СТВОЛОВЫХ КЛЕТОК КЛЕТОЧНОЙ ЛИНИИ H1
Клетки линии эмбриональных стволовых клеток человека Н1 (клетки WA01, WiCell, Мэдисон Висконсин) при пассировании 41 были сняты с плоской прикрепленной культуры с помощью EDTA и перенесены в формат суспензионной культуры по методу, описанному в примере 2.
Плюрипотентность клеточных агрегатов измеряли при помощи проточной цитометрии, как показано на Фиг.5а, при этом наблюдалась высокая экспрессия маркеров плюрипотентности CD9, SSEA4, TRA-1-61, TRA-и 1-80, указывающая на высокий уровень плюрипотентности клеток. Эти плюрипотентные клетки затем дифференцируются в панкреатическое клетки-предшественники в динамически перемешиваемой суспензионной культуральной системе с использованием метода ступенчатой прогрессии в различных средах, содержащих малые молекулы и факторы роста, предназначенные для нормального развития поджелудочной. Этот процесс производит популяции панкреатических клеток-предшественников, характеризующихся коэкспрессией факторов транскрипции панкреатических клеток, Pdx1 и Nkx6.1. Когда эти клетки были пересажены, они далее вызревали в ткани, способные к стимулированной глюкозой секреции инсулина, которая может исправить высокий уровень глюкозы в крови в стрептозотоцин-индуцированной модели диабета.
Для генерирования популяции панкреатических клеток-предшественников, плюрипотентные клетки в формате кластера, поддерживаемые в среде mTeSR®1, переносили в 0,2-литровый стеклянный биореактор с перемешиванием суспензии (Dasgip, № по каталогу DS0200 TBSC, г. Шрусбери, штат Массачусетс, США) с регулируемой контроллером температурой, рН и уровнем растворенного кислорода. Плюрипотентные кластеры клеток культивировали в биореакторе в течение двух суток. В это время (стадия 1, сутки 0) среду заменяли и была инициирована дифференцировка, клеточные агрегаты были суспендированы в количестве примерно 0,7 млн клеток/мл в среде для дифференцировки в соответствии с таблицей 5а. Клетки затем выдерживали в этой закрытой стерильной суспензии в биореакторе в течение 14 суток. На протяжении дифференцировки, уровни бикарбоната среды поддерживали в 3,64 г/л, рН поддерживали на уровне 7,4 с помощью регулирования потока СО2 в общем объеме среды 0,3 л. Свободное пространство биореактора непрерывно обрабатывалось CO2 и воздухом под контролем системы управления Dasgip с установленным уровнем растворенного кислорода 30% при постоянном потоке газа 5 л/час. Воздушный поток регулируется относительно содержания растворенного кислорода и поток CO2 регулируется относительно уровня рН.
Компоненты среды и протокол дифференцировки
(конечная концентрация глюкозы)
3,64 г/л NaCO3
(8 мМоль глюкозы)
3,64 г/л NaCO3
(8 мМоль глюкозы)
3,64 г/л NaCO3
(8 мМоль глюкозы)
3,64 г/л NaCO3
(8 мМоль глюкозы)
и 2мМоль L-глутамина
И/ИЛИ
Малые молекулы
Как указано
GDF8 (100 нг/мл)
Как указано
ITS-X (1:50 000)
ITS-X (1:50 000)
ITS-X (1:200)
RA (2 мкМоль)
SANT (0,25 мкМоль)
AA (5 нг/мл)
TppB (200 нМоль)
LDN (100 нМоль) в течение 0-24 часов, стадия 3
SANT (0,25 мкМоль)
Cypi (100 нМоль)
SCIO (2µM)
TppB (100 нМоль)
В этом примере и в описании SCIO представляет собой ингибитор Alk5 с химическим названием 4-{[2-(5-хлор-2-фторфенил)-5-(1-метилэтил)пиримидин-4-ил]амино}-N-(2-гидроксипропил)пиридин-3-карбоксамид и CAS № 674794-97-9. Химическая структура SCIO представлена ниже:

На этой стадии температуру поддерживали на уровне 37°C. В начале пробега и для каждой замены среды (95% среды удалено за счет замены), лопасть была остановлена и среда была удалена, добавлена с помощью перистальтического насоса через погружную трубку в биореакторе, подключенную к трубке C-Flex® с использованием трубосварочного стана Terumo™ для поддержания замкнутой системы.
Несколько различных настроек подачи были испытаны во время стадии 1: (a) замена среды через 24 часа после начала дифференцировки, без замены среды через 48 часов; (b) замена среды через 24 часа после начала дифференцировки и добавление болюса глюкозы через 48 часов; и (c) без замены среды в течение стадии 1 с добавлением болюсов глюкозы и GDF8 через 24 ч после начала дифференцировки и последующего добавления болюса глюкозы через 48 часов после начала.
Количество клеток в момент начала, середины и конца процесса, взятые из для каждого реактора, указано в таблице 5b. В конце стадии 1 клетки отбирали для определения паттернов экспрессии белка с помощью проточной цитометрии. Клетки, дифференцированные по условию A- замена среды через 24 часа после начала дифференцировки к дефинитивную энтодерму, после чего без замены среды в течение следующих 48 часов - показали лучшие результаты при измерении индуцирования маркеров дифференцировки (CD99 и CXCR4) и снижения экспрессии маркера плюрипотентности (CD9) (5б). Более высокая экспрессия CXCR4 и CD99 в сочетании с низкой экспрессией CD9 в конце окончательного формирования энтодермы коррелирует с более высокой экспрессией панкреатических генов и низкой экспрессией генов, показывающих развитие альтернативных органов на поздних стадиях дифференцировки (Фиг. 5d и 5e). В частности, отсутствие замены среды в течение первой стадии дифференцировки или добавление глюкозы к среде на стадии 1 в формате объемной подачи привело к снижению уровней CXCR4 в конце стадии 1, которое коррелировало с отличающейся морфологией агрегатов на конец четвертой стадии дифференцировки (Фиг. 5c). В частности, условия В и С имели более низкую экспрессию гена поджелудочной (Nkx6.1 и CHGA) и более высокую экспрессию не-панкреатических генов (CDX2 и Sox2) в конце стадии 4, что было измерено с помощью проточной цитометрии (Фиг.5d и Таблица 5b). Эти выводы были подтверждены проведением qRT-PCR (Фиг. 5e), так как условие A показало значительно более высокую экспрессию панкреатических генов, чем условие С, условие B дало результаты, средние между А и С. Кроме того, условие С показало значительно более высокие уровни индикативных генов альтернативного непанкреатического развития, например, CDX2, AFP и альбумина (Фиг. 5e). Эти данные показывают, что однородная дефинитивная энтодерма (DE) с высоким уровнем экспрессии CXCR4, которая генерируется без замены среды за последние 48 часов формирования DE, способна позже превратиться в чистую популяцию клеток панкреатической энтодермы.
В конце стадии 4 дифференцировки, клетки, дифференцированные по условию A, были удалены из биореактора, промыты в среде MCDB131, содержащей 0,1% FAF-BSA, и имплантированы мышам линии SCID-BG. Каждой мыши трансплантировали 5 миллионов клеток непосредственно под почечную капсулу. Каждый 4 недели после имплантации брались пробы крови и измерялись уровни глюкозы и С-пептида в крови. Через 12 недель после имплантации человеческий С-пептид был обнаружен на уровнях выше 1 нг/мл, а через 16 недель уровни С-пептида в среднем составляли 2,5 нг/мл. (фиг.5f). На 20 неделе после имплантации уровень С-пептида был измерен у животных в голодном и затем сытом состоянии. Обработка глюкозой вызвала значительное увеличение циркулирующего С-пептида человека от 0,93 нг/мл в голодном состоянии, до 2,39 нг/мл в сытом состоянии (фиг.5 г), что указывает на то, что трансплантированные клетки вызрели в функциональную GSIS-компетентную ткань. Кроме того, когда животным ввели стрептозотоцин (STZ) (β-клетки мыши более чувствительны к STZ и сильнее разрушаются по сравнению с β-клетками человека) для вызова диабетического состояния, животные с трансплантатом функциональной GSIS-компетентной ткани поддерживали нормальный уровень глюкозы в крови в отличие от необработанных контрольных животных, у которых развился явный диабет (Фиг. 5h). Эти результаты показывают, что животные с HES-дифференцированным трансплантатом клеток были защищены от STZ-индуцированного диабета с помощью функционального трансплантата ткани поджелудочной железы .
Количество клеток и данные проточной цитометрии
(Условие)
(Условие)
(Условие)
ПРИМЕР 6
НАПРАВЛЕННАЯ ДИФФЕРЕНЦИРОВКА В ФОРМАТ СУСПЕНЗИОННОЙ КУЛЬТУРЫ МИКРОНОСИТЕЛЕЙ С ПРИКРЕПЛЕННЫМИ ЧЕЛОВЕЧЕСКИМИ ЭМБРИОНАЛЬНЫМИ СТВОЛОВЫМИ КЛЕТКАМИ КЛЕТОЧНОЙ ЛИНИИ H1
Сферы микроносителя Cytodex® 3 (С3) (Sigma-Aldrich Co LLC, г. Сент-Луис, штат Миссури, США, № по каталогу C3275) были подготовлены к культуре путем замачивания 400 мг сфер в стеклянных сцинтилляционных флаконах с кремниевым покрытием объемом 20 мл, содержащих 15 мл PBS Дульбекко (DPBS), в течение 4-24 часов. Cytodex® 3 состоит из тонкого слоя денатурированного коллагена, химически связанного с матриксом из сшитого декстрана. Слой денатурированного коллагена на носителях Cytodex® 3 может разлагаться различными протеазами, включая трипсин и коллагеназу, что дает возможность извлекать клетки из микроносителя, сохраняя максимальную жизнеспособность, функциональность и целостность клеток.
После замачивания сферы автоклавировали, промывали стерильной DPBS и ресуспендировали в кондиционированной среде мышиных эмбриональных фибробластов (MEF-CM) с добавлением 10 мкМоль Y-27632. Сферы затем переносили в 125 мл стеклянные центрифужные пробирки Corning® (Corning Incorporated, г. Корнинг, штат Нью-Йорк, США) при плотности 100 мг сфер/колба. Центрифужные пробирки, содержащие сферы и MEF-CM с Y-27632 уравновешивали в увлажненном инкубаторе с 5% СО2 при 37°C в течение по крайней мере 60 минут.
Клетки линии эмбриональных стволовых клеток человека Н1 (клетки WA01, WiCell, г. Мэдисон, штат Висконсин, США) при пассировании 44 были сняты с плоской прикрепленной культуры с помощью TrypLE™ (Life Technologies Corporation, г. Гранд Айленд, штат Нью-Йорк, США) (8-минутная инкубация при 37°C с образованием суспензии отдельных клеток). Клетки затем промывали и суспендировали в MEF-CM с Y-27632 и 11 млн клеток линии hES крепились к сферам в течение 6 часов во время статического инкубационного периода. Затем в центрифужную пробирку добавляли MEF-CM с Y-27632, доводя конечный объем носителя до 75 мл, и клетки и сферы перемешивали в стеклянной центрифужной пробирке при скорости лопастей 50 оборотов в минуту. Клетки выращивали таким образом в течение 5 суток с ежедневной заменой 50 мл среды MEF-CM. Через 5 суток в культуре, колбы содержали 53×106 клеток (± 12×106 СО). В качестве контроля, один миллион клеток линии Н1 HES также высевали на дно 6-луночной чашки Петри из полистирола, покрытой разведенным 1:30 препаратом Matrigel™ и поддерживали с ежедневной заменой среды MEF-CM.
Через 5 суток в плюрипотентной культуре эти клетки были дифференцированы в панкреатические клетки-предшественники в динамической суспензионной культуральной системе путем постадийного прогрессирования в различных средах, содержащих одно или оба из малых молекул и факторов роста, предназначенных для рекапитуляции морфогенов нормального развития поджелудочной железы. Два состава среды были протестированы в качестве способа поддержания рекапитуляции нормального развития поджелудочной железы; один с использованием активина А и Wnt3a для формирования DE, и другой с использованием соединения MCX с GDF8 для формирования DE (таблицы 6a и 6b, соответственно). Среду меняли ежедневно и образцы характеризовались методами RT-PCR и проточной цитометрии для определения свойств клеток. Фазоконтрастные изображения клеток на микроносителях были получены, и график морфологии клеток в плюрипотентные культуры по времени вплоть до инициирования дифференцировки клеток показан на Фиг. 6а. График по времени, показывающий дифференцировку культур, показан на Фиг. 6b. Также было определено количество клеток, взятое в различные моменты времени по ходу эксперимента, а результаты представлены в зависимости от площади поверхности (клеток/см2 на фиг. 6С) или объема носителя (клеток/мл на Фиг. 6d) для препаратов среды на плоской культуры или суспендированной культуре на микроносителях.
Клетки характеризуются в различных точках в течение всего процесса по результатам проточной цитометрии и RT-PCR. Результаты проточной цитометрии первой стадии дифференцировки и формирование дефинитивной энтодермы показаны в виде точечной диаграммы клеточной экспрессии CXCR4 (по оси Y) и CD9 (по оси Х) на Фиг. 6е, также показана общая экспрессия каждого маркера на Фиг. 6f. Результаты показывают, что при любых условиях значительное большинство клеток образуют дефинитивную энтодерму, что определено коэффициентом усиления экспрессии CXCR4 и потерей экспрессии поверхностного маркера плюрипотентности CD9. Кроме того, наиболее эффективное формирование дефинитивной энтодермы происходит в порядке убывания при обработке следующим образом: микроносители MCX/GDF8> плоская культура MCX/GDF8 > микроносители WNT3A/AA > планарная культура WNT3A/А.А. Так проявляется специфическое действие среды на клетки, так как клетки, обработанные MCX/GDF8 показывают низкую экспрессию CERBERUS (Cer 1), GOOSECOID и FGF17 (Фиг. 6g). Тем не менее, все условия обработки показывают сходные уровни экспрессии генов дефинитивной энтодермы; CD99, CXCR4, FOXA2, KIT и SOX17 (Фиг. 6g и таблица 6 с). Эти процессы порождают популяции панкреатических клеток-предшественников, характеризующихся коэкспрессией факторов транскрипции панкреатических клеток, Pdx1 и Nkx6.1. Когда эти клетки были пересажены, они далее вызревали в ткани, способные к стимулированной глюкозой секреции инсулина, которая может исправить высокий уровень глюкозы в крови в стрептозотоцин-индуцированной модели диабета.
Ниже, в таблице 6a, B27 представляет собой добавку Gibco® B-27® (Life Technologies Corporation, г. Карлсбад, штат Калифорния, США).
Как используется в данном примере, соединение MCX представляет собой 14-проп-2-ен-1-ил-3,5,7,14,17,23,27-гептаазатетрацикло[19.3.1.1 ~ 2,6 ~ 0,1 ~ 8,12. ~] гептакоза-1 (25), 2 (27), 3,5,8 (26), 9,11,21,23-нон-ан-16-он, который имеет следующую формулу (формула 1):
Другие циклические анилин-пиридинотриазины также могут быть использованы вместо описанного выше соединения MCX. Такие соединения включают, но не ограничиваются 14-метил-3,5,7,14,18,24,28-гептаазатетрацикло [20.3.1.1 ~ 2,6 ~ .- 1 ~ 8,12 ~] октакоза-1 (26 ), 2 (28), 3,5,8 (27), 9,11,22,24-нонаен-17-он-е и 5-хлор-1,8,10,12,16,22,26, 32-октаазапентацикло [24.2.2.1 ~ 3,7 ~ -1 ~ 9,13 ~ 0,1 ~ 14,18 ~] тритриаконта-3 (33), 4,6,9 (32), 10, 12,14 ( 31), 15,17-нонаен-23-он. Эти соединения приведены ниже (формула 2 и формула 3):

Примеры подходящих соединений описаны в заявке на патент США на патент США № 2010/0015711, раскрытие которой включено в полном объеме, как она относится к соединениям MCX, связанным с циклическими анилин-приридинотриазинами и их синтезом.
Ингибитор Cyp26, используемый в этом примере на стадии 4 представлял собой N-{4-[2-этил-1-(1H-1,2,4-триазол-1-ил)бутил]фенил}-1,3-бензотиазол-2-амин, имеющий CAS № 201410-53-9 и следующую структуру.
Этот ингибитор Cyp26 также известен как Cypi. Структура и синтез этого ингибитора Cyp26 описана в патенте США № 7,378,433, раскрытие которого включено путем ссылки в полном объеме, как он относится к ингибиторам Cyp26 и их синтезу.
Препараты среды и протокол дифференцировки
11 мМоль глюкозы
17,5 мМоль глюкозы
25 мМоль глюкозы
Показатель
И/или
Малые
молекулы
(100 нг/мл)
Wnt3a
(20 нг/мл)
(100 нг/мл)
(50 нг/мл)
(100 нг/мл)
ПП
(2µM)
SANT1
(250nM)
(100 нг/мл)
ALK5i
(1 мкМоль)
TPB (50 нМоль)
(100 нг/мл)
ALK5i
(1 мкМоль)
Препараты среды и протокол дифференцировки
8 мМоль глюкозы
10,5 мМоль глюкозы
25 мМоль глюкозы
25 мМоль глюкозы
роста
Малые молекулы, агонист/
антагонист
100 нг/мл
MCX (только сутки 1)
3 мкмоль
1:50000 ITS-X
50 нг/мл
1:50000 ITS-X
AA (5 нг/мл)
RA (2 мкМоль)
SANT (250 мкМоль)
LDN 193189
1:200 ITS:X
SANT (250 нМоль)
LDN 193189 (200 нМоль)
ингибитор Cyp26 (100 нМоль)
1:200 ITS:X
Пример 7
Суб-клон клеточной линии H1 (WA01) hES - WB0106 используется для этого примера. Суб-клон WB0106 был получен в Научно-исследовательском институте WiCell (Мэдисон, Висконсин) от посевного материала линии H1, называемого DDL-13. Суб-клон WB0106 H1 линии был получен из флакона DDL-13, размороженного при пассировании 23 в среде mTeSR®1 на подложке Matrigel™, а затем пассированного с EDTA. WB0106 был заморожен при пассировании 28 и был выбран для этих исследований на основе нормального кариотипа (FISH и G-полосы), способности дифференцироваться в панкреатические клетки-предшественники и компетентности для формирования кластеров и роста в суспензионной культуре.
Флакон WB0106 WCB затем размораживали в среде на подложке Matrigel™ в колбе T225 (Corning Incorporated, г. Корнинг, штат Нью-Йорк, США) и при первом пассировании клетки разделили на несколько колб T225. На втором пассировании клетки из нескольких колб T225 были объединены и использованы для заполнения одной двухслойной емкости для клеток Cell Stack™ (CS2). После формирования в CS2 конфлюэнтного слоя на 70%, к портам среды прикрепили систему трубок с крышками C-Flex®, к которым подсоединяли ведущие к насосу трубки для замыкания системы. После того как система была замкнута с помощью трубок C-Flex®, к ней приваривались с помощью аппарата Terumo пакеты или бутыли, куда перистальтическим насосом переносились объемы жидкости (среда, PBS-/-, Accutase® или суспендированные клетки).
Для отделения клеток от CS2, клетки промывали один раз PBS-/-, затем обрабатывали с раствором Accutase® половинной эффективности, разбавленным PBS-/- и инкубировали в течение 4-5 минут. Затем Accutase® удаляли, и через 3 минуты после нанесения раствора фермента, CS2 был использован для стимулирования клеточного отделения. Флакон со средой, дополненной 0,5% BSA, содержащей 10 мкмоль/л ингибитора Rho-киназы, Y-27632, закачивали в CS2 для промывания и инактивации остаточной Accutase®, после чего смыв собирали. Добавляли второй промывочный объем, собирали смыв и объединяли с первым. Затем 2,0-2,5 × 108 клеток в 200 мл переносили на однослойную емкость для клеток CellSTACK® и инкубировали при 37°C в течение 2 часов в увлажненном инкубаторе с 5% CO2. Используя замкнутый контур трубок C-Flex® с трубкой для насоса, прикрепленной между двумя портами среды CellSTACK®, клеточную суспензию измельчали в течение 5 минут при 75 оборотах в минуту с помощью перистальтического насоса для гомогенизации агрегатов. Замкнутый контур трубок был заменен стерильными фильтрами 0,2 микрона для газообмена и CellSTACK® инкубировали в течение ночи при 37°C в увлажненном инкубаторе с 5% CO2. После инкубации в течение ночи (12-22 часов, 18 часов оптимально), клетки в CellSTACK® образовали округлые сферические агрегаты (кластеры) плюрипотентных клеток.
Среду с добавлением 0,5% BSA, содержащую суспендированные кластеры клеток, переносили из CellSTACK® в одноразовую 1-литровую центрифужную пробирку (Corning; г. Корнинг, штат Нью-Йорк, США) вместе с 0,4 л свежей среды, дополненной 0,5% BSA и выдерживали 55-65 оборотах в минуту. Через двадцать четыре часа после переноса, одноразовую 1-литровую центрифужную пробирку удаляли из увлажненного инкубатора с 5% CO2 и кластерам давали отстояться в течение 5-10 минут. Затем среду отсасывали, пока не осталось 200 мл и добавляли в центрифужную пробирку 400 мл дополнительной свежей культуральной среды. Этот процесс повторяли в конце 2 суток (через 48 часов после переноса).
В конце 3 суток (72 часа после перевода в центрифужную пробирку с CS2), клеточные кластеры обрабатывали Accutase® для пассирования и дальнейшего роста. Процесс пассирования был инициирован путем удаления 1 л одноразовой центрифужной пробирки из увлажненного 5% CO2 инкубатора. Пробирку помещали в биологически безопасный бокс центрифуги для поддержания гомогенной суспензии клеток. После этого суспензию клеток удаляли из центрифужной пробирки 100 мл пипеткой, равномерно распределяется между четырьмя 175 мл коническими пробирками из поликарбоната (ThermoFisher-Nalgene; г. Буффало, штат Нью-Йорк, США), и центрифугировали в течение 5 минут при 80-200 ОЦС. Отработанную среду отсасывали, не нарушая клеточный осадок. Затем в каждую пробирку добавляли 25 мл DPBS без кальция или магния (DPBS-/), и клетки объединяли в одну коническую пробирку и центрифугировали в течение 5 минут при 80-200 ОЦС. DPBS-/ отсасывали из конической пробирки и в пробирку добавили 30 мл раствора 50% Accutase®/50% DPBS-/. Клеточные скопления пипетировали 1-3 раза, а затем периодически перемешивали в течение 4 минут, затем центрифугировали в течение 5 минут при 80-200 ОЦС. Accutase® затем отсасывали настолько полно, насколько это возможно, не нарушая клеточный осадок, и коническую пробирку непрерывно и осторожно постукивали в течение 3-5 минут до тех пор, пока суспензия клеток не стала равномерной молочно-белой. 10 мл среды с добавлением 0,5% BSA, содержащей 10 мкмоль/л ингибитора Rho-киназы, Y-27632, добавляли к суспензии клеток и измельчали 2-4 раза, чтобы инактивировать остаточный Accutase®. 90 мл среды, дополненной 0,5% BSA, содержащей 10 мкмоль/л ингибитора Rho-киназы, Y-27632, добавляли к клеткам и суспензию пропускали через клеточное сито 40 мкМоль (BD Falcon; г. Франклин-Лейкс, штат Нью-Джерси, США).
Плотность клеток в объеме 100 мл отфильтрованной клеточной суспензии определяли с помощью аппарата NucleoCounter® NC-100 (ChemoMetec A/S, г. Аллерод, Дания) и добавляли дополнительную среду до конечной концентрации клеток 1×106 клеток/мл в среде, дополненной 0,5% BSA содержащий 10 мкмоль/л ингибитора Rho-киназы, Y-27632. Затем 225 мл (225 миллионов клеток) клеточной суспензии переносили в 1-литровый вращающийся сосуд одноразового и инкубировали в течение 1 часа без перемешивания в увлажненном инкубаторе с 5% CO2. Затем колбу вынимали из инкубатора и перемешивали при 100 оборотах в минуту на вращаемой пластине в биологически безопасном боксе в течение 1-3 минут. В то время как суспензия клеток смешивалась, дополнительно к суспензии клеток добавляли 225 мл среды, дополненной 0,5% BSA, содержащий 10 мкмоль/л ингибитора Rho-киназы, Y-27632. Центрифужную пробирку затем возвращали в увлажненный инкубатор с 5% CO2 на 30 минут. Затем колбу вынимали из инкубатора и перемешивали при 100 оборотах в минуту на вращаемой пластине в биологически безопасном боксе в течение 1-3 минут. В то время как суспензия клеток смешивалась, дополнительно к суспензии клеток добавляли 150 мл среды, дополненной 0,5% BSA, содержащей 10 мкмоль/л в Rho-киназы, Y-27632, чтобы довести окончательный объем до 600 мл, и колбу возвращали к перемешиваемой суспензии в инкубаторе , Через 24 и 48 часов после диссоциации Accutase®, кластерам клеток дали осесть на дно колбы в течение 5-10 минут. Для гарантированного сведения к минимуму потерь кластеров, 400 мл отработанной среды удаляли из колбы аспирацией и заменяли свежей средой. Используя этот процесс, H1 клетки были преобразованы из прикрепленной на подложке культуры в кластеры клеток в суспензионной культуре.
Через 72 часа после первоначальной обработки Accutase® процесс клеточной диссоциации кластеров и посев (пассирование) в центрифужную пробирку повторяли для поддержания клеток в состоянии суспензии для многократного пассирования (диапазон испытаний: 1-10 пассажей). Описанный выше процесс был повторен, с той разницей, что после первых 24 часов среда не была удалена, а было добавлено 200 мл свежей среды. Через 48 часов после диссоциации Accutase®, кластерам клеток дали осесть на дно колбы в течение 5-10 минут, отсасывали 600 мл среды и добавляли в колбу 400 мл свежей среды.
Эти клетки, прошедшие пассажи суспензии и культивирование, можно затем криоконсервировать и хранить для будущего использования. Для того чтобы подготовить суспензию разросшихся клеток для криоконсервации, клеточные кластеры диссоциировали с Accutase®, как описано выше для пассирования суспензии, за исключением того, что клетки не пропускали через клеточный фильтр 40 микронов. Было определено количество клеток в 100 мл клеточной суспензии, созданной из раствора в каждой одноразовой 1-литровой колбе. Клеточные суспензии затем объединяли и центрифугировали в течение 5 минут при 80-200 ОЦС. Среду из центрифужной пробирки затем удаляли настолько полно, насколько это возможно, не нарушая клеточный осадок. Холодный (< 4°C) CryoStor®10 (Stem Cell Technologies, Inc., г. Ванкувер, Британская Колумбия, Канада) затем добавляли по каплям таким образом, чтобы достичь конечной концентрации 150 миллионов клеток на мл, и раствор клеток держали на ледяной бане во время переноса в 1,8 мл флакон для криоконсервирования Corning® (Corning Incorporated, г. Корнинг, штат Нью-Йорк, США) или 15 мл пакет для криоконсервирования Miltenyi (Miltenyi Biotec Inc. г. Оберн, штат Калифорния, США).
Суспензия разросшихся клеток затем была заморожена в ампуле при высокой плотности в морозильнике с контролируемой скоростью следующим образом. Камеру предварительно охлаждали до 4°C и температуру поддерживали до тех пор, пока пробирка с образцом не достигала температуры 6°C. Затем температуру в камере понижали со скоростью 2°C/мин, пока пробирка с образцом не достигала температуры -7°C. После того, как пробирка с образцом достигла температуры -7°C, камеру охлаждали на 20°C/мин до температуры -45°C. После чего температуру в камере поднимали на 10°C/мин до тех пор, пока температура в камере не достигала -25°C, а затем дополнительно охлаждали камеру на 0,8°C/мин до тех пор, пока пробирка с образцом не достигала температуры -45°C. Температуру в камере затем снижали на 35°C/мин до тех пор, пока температура в камере не достигала -160°C. Температура в камере затем удерживалась на уровне -160°C в течение по крайней мере 10 минут, после чего пробирки переносили в хранилище с жидким азотом, в часть с газовой фазой.
Для высевания в биореактор с механическим перемешиванием, криоконсервированные клетки высокой плотности удаляли из хранилища с жидким азотом, размораживали и использовали для посева в замкнутом 3-литровом стеклянном биореакторе (DASGIP; г. Юлих, Германия). Четыре или пять пробирок удаляли из хранилища с жидким азотом, части с газовой фазой, и устанавливали непосредственно в ванну с водой температурой 37°C на 105 секунд. Содержимое размороженных пробирок затем переносили с помощью 2 мл стеклянной пипетки в 50 мл коническую пробирку. Затем в пробирку добавляли по каплям 9 мл среды (IH3 или Essential8™ (E8™)), содержащей 0,5% BSA и дополненной 10 мкмоль/л ингибитора Rho-киназы, Y-27632. Затем клетки центрифугировали при 80-200 ОЦС в течение 5 минут. Супернатант из пробирки отсасывали, добавляли 10 мл свежей среды (IH3 или E8™), содержащей 0,5% BSA и дополненной 10 мкмоль/л ингибитора Rho-киназы, Y-27632, и объем, содержащий клетки, пипеткой переносили во флаконы для переноса среды (Cap2V8®, SaniSure, г. Мурпарк, штат Калифорния, США). Содержимое флаконов затем нагнетали непосредственно в биореактор через стерильные сваренные C-Flex трубки с помощью перистальтического насоса. При подготовке к посеву плюрипотентных стволовых клеток в биореактор, подготовили 1,5 л среды (IH3 или E8™ с добавлением 0,5% BSA, содержащей 10 мкмоль/л ингибитора Rho-киназы, Y-27632), подогретого до 37°С, перемешанной при 70 оборотах в минуту, отрегулированной до 6,8-7,1 рН с помощью СО2, с уровнем растворенного кислорода 30% (содержание СО2, воздуха, О2 и N2 регулируется). Сразу после посева в биореактор, из него отбирали пробы для подсчета клеток, и средний объем доводили по мере необходимости для получения конечной концентрации клеток 0,225×106 клеток/мл.
Клетки, высеянные в биореактор с механическим перемешиванием, сформировали кластеры клеток в непрерывно помешиваемом резервуаре, и эти кластеры выдерживали в среде плюрипотентности (IH3 или E8™, дополненной 0,5% BSA) в реакторе в течение трех суток. Среду меняли ежедневно, частичная замена среды выполнялась через 24 часа после посева, удаляли 1-1,3 литра отработанной среды и добавляли 1,5 л свежей среды. Спустя сорок восемь часов после посева, удаляли 1,5-1,8 л отработанной среды и добавляли 1,5 л свежей среды. Через 72 часа после посева была инициирована дифференцировка плюрипотентных клеток путем удаления > 90% использованной среды и добавления среды для дифференцировки (таблица 7).
После начала процесс дифференцировки, клетки поддерживали в течение 12 или более суток в закрытой стерильной суспензии в биореакторе с регулируемой температурой (37°), рН 7,4 (для дифференцировки) с установленным уровнем растворенного кислорода (10% DO на стадии 1 и 30% DO на прочих стадиях, содержание СО2, О2, N2 и воздуха регулируется). На протяжении всего процесса дифференцировки при каждой замене среды, лопастное колесо было остановлено на 5-20 минут до удаления среды через погружную трубку для стабилизации кластеров. Среду в биореакторе удаляли или добавляли из/в закрытого флакона или пакета с помощью перистальтического насоса через погруженную пробирку, соединенную с трубками C-Flex® с использованием сварщика трубок Terumo™ для поддержания замкнутой системы. Рабочее колесо и нагреватель были вновь запущены после добавления в сосуд достаточного количества среды, чтобы полностью погрузить колесо.
Для того чтобы контролировать процесс биореактора, образцы среды, содержащие кластеры клеток, забирались ежедневно для определения количество клеток и жизнеспособности (NucleoCounter®), как показано на Фиг. 7. Наблюдался общий рост клеток в процессе, высеянное количество 0,225×106 жизнеспособных клеток/мл разрослось до количества в среднем 0,92×106 жизнеспособных клеток/мл на стадии 4 сутки 3. При поддержании определенного уровня кислотности (рН 7,0-6,8) во время посева в биореактор и кластеризации плюрипотентных клеток, средний выход клеток на стадии 4 суток 3 увеличился в среднем на 1,3×106 клеток/мл (фиг.7) ,
В дополнение к ежедневным подсчетам, образцы среды биореактора были проанализированы NOVA BioProfile® FLEX (Nova Biomedical Corporation, г. Уолтем, штат Массачусетс, США). Было отмечено, что, согласно предварительным установкам реактора, рН среды на стадии 0 был кислым по отношению к гомеостатическому стандартному показателю рН 7,4, общему для большинства питательных сред, и рН среды в реакторе снизился в течение стадии 0 в результате клеточного метаболизма (Фиг. 8). Эти результаты коррелируют с тенденцией увеличения концентрации молочной кислоты и снижения уровня глюкозы до конца 6 суток дифференцировки (Фиг.9 и 10). Вместе, эти данные свидетельствуют о том, что клеток в реакторе быстро растут и поглощают глюкозу в течение стадии 0 и первых двух стадий дифференцировки (1-6 сутки). Тем не менее, с начала стадии 3 и далее, клеточной метаболизм (снижение уровней лактата и повышение уровней глюкозы) в реакторе не коррелирует с пиком численности клеток в стадии 3 с последующим несоответствием плотности клеток на протяжении 4-й стадии.
Следовало определить, соответствуют ли специфические для стадии изменения рН и метаболизма изменениям в паттернах экспрессии мРНК. Тестирование образцов клеток из биореактора проводили с использованием четырех массивов Biosystems® Low Density Arrays (Life Technologies Corporation, г. Карлсбад, штат Калифорния, США) обозначенных «плюрипотентность», «дефинитивная энтодерма (DE)», «кишечная трубка» (GT) и «4 стадия (S4)», после чего результаты сравнивали со справочными данными, полученными от образцов недифференцированных клеток линии H1 (WB0106) hES в качестве контроля, чтобы стандартизировать экспрессию всех пробегов и массивов.
Также для каждой стадии дифференциации была определена экспрессия гена с использованием этих же массивов. Также наблюдался тот факт, что посевной клеточный материал, размороженный в биореакторе, показал недифференцированные паттерны экспрессии генов на стадии 0 сутки 1 и стадии 3, 0 сутки (24 и 72 часа после посева в биореактор: Фиг. 11, 12, 13 и 14). Эти результаты хорошо коррелируют с результатами проточной цитометрии, которые показали высокие уровни экспрессии CD9, SSEA4, TRA-1-60, TRA-и 1-81, а также отсутствие CXCR4/CD184 (Фиг. 15 и таблица 8). Хотя проточная цитометрия и анализы qRT-PCR экспрессии генов показали надежные и стабильные паттерны экспрессии генов плюрипотентности (CD9, NANOG, POU5F1, SOX2, TDGF и ZFP42) в соответствии со стабильным плюрипотентным состоянием, также отмечалось умеренное, но нестабильное увеличение экспрессии генов для GATA4, РКГ, MIXL1 и Т; и ≥ 100-кратное увеличение экспрессии CER1, FGF17, FGF4 и GATA2 в некоторых образцах в процессе стадии 0 до направленной дифференцировки (Фиг. 16 и 17).
По завершении стадии 0 (72 часа после посева реактора), клетки переносили в среду для дифференцировки (таблица 7), содержащую MCX и GDF8. Двадцать четыре часа после этой замены среды были отмечены значительные изменения в структуре экспрессии генов (Фиг. 18 и 19), такие как ~700x увеличение экспрессии Foxa2 и 1000x увеличение экспрессии CER1, EOMES, FGF17, FGF4, GATA4, GATA6, РКГ, MIXL1 и T. Эти повышенные уровни экспрессии указывали на прохождение клетками мезодермальной стадии. Было также отмечено, что уровни CDX2 были повышены на стадии 1 сутки 1 по сравнению с недифференцированными клетками (470x увеличение экспрессии по сравнению с контролем), однако это было преходящее повышение уровня экспрессии и экспрессия CDX2 упала на 94% между стадией 1, 1 сутки и стадией 1, сутки 3, вернувшись к уровню, сопоставимому с тем, который наблюдался до начала дифференцировки (Фиг. 14, 19 и 21).
Через 72 часа после воздействия среды дифференцировки DE, клетки экспрессировали профиль в соответствии с спецификациями дефинитивной энтодермы, а пиковые уровни CXCR4 и Foxa2 и SOX17 экспрессировались на уровне > 1000x относительно справочных. В соответствии с параметрами дефинитивной энтодермы было также отмечено, что экспрессия генов CER1, EOMES, FGF17, FGF4, GATA4, GATA6, РКГ MIXL1 и Т снизилась с повышенных уровней, наблюдаемых на стадии 1 1 сутки (Фиг.20 и 21).
Изменения в экспрессии генов, наблюдаемые Qrt-PCR, коррелируют с результатами, полученными с помощью проточной цитометрии. Наблюдался почти полный переход от CD9-экспрессирующей/CXCR4-отрицательной популяции плюрипотентных клеток при инициировании дифференцировки (Фиг. 15) к гомогенной популяции клеток, экспрессирующих CXCR4 (98,3% клеток CXCR4-положительны, ± 1,9SD) в конце стадии 1(Фиг. 22).
После завершения формирования дефинитивной энтодермы (стадия 1) среду заменяли на другую, содержащую FGF7, морфоген, который используется, чтобы вызвать образование примитивный передней кишки (стадия 2). В соответствии с образованием примитивной кишки, уровни экспрессии HNF4α и GATA6 на стадии 2 сутки 1 и 3, увеличиваются, в то время как гены с высоким уровнем экспрессии на 3 сутки стадии 1 (CXCR4, EOMES, FGF17, FGF4, MNX1, PRDM1, SOX17 и ФВ) показали снижение экспрессии в конце стадии 2 (Фиг. 23). Экспрессия генов передней кишки (AFP, Pdx1 и Prox1) повысилась (Фиг. 24).
После того как клетки были культивированы в среде во время стадии 2 в течение 72 часа, культура была перенесена в среду стадии 3 (таблица 7). В данной среде маркеры экспрессии клеток соответствуют энтодерме поджелудочной линии, сто определено по экспрессии PDX1 и FOXA2 (90,9% ± 11,9 СО PDX1-положительной и 99,2% ± 0,6 СО FOXA2-положительной), показано на Фиг. 25. Эти результаты были подтверждены данными из образцов, подвергнутых анализу экспрессии гена с помощью Qrt-PCR . Экспрессия генов PDX1 увеличилась 5-кратно за 24 часа с конца стадии 2 сутки 3 (38 000-кратно относительно H1) до конца стадии 3 сутки 1 (200 000-кратно относительно H1) и снова двукратно через 48 часов на стадии 3 сутки 3 (435 000-кратно относительно H1). Эти данные указывали на формирование панкреатических клеток (Фиг. 26). Это наблюдение было дополнительно подтверждено повышенным уровнем генов, обычно экспрессирующих в поджелудочной железе (ARX, GAST, GCG, INS, Isl1, NEUROD1, Ngn3, Nkx2.2, Nkx6.1, Pax4, PAX6, Ptf1a и SST ), как показано на Фиг. 26. Кроме того, наблюдалась очень низкая или отсутствующая экспрессия OCT4/POU5F1 (2-10% относительно контроля или 32-37 образцов Cts по данным Qrt-PCR) и высоким уровнем экспрессии других маркеров энтодертмальных линий AFP, ALB, и CDX2, что указывает на спецификации и переход клеточной популяции в биореакторе из клеток кишечной трубки в панкреатические клетки
Как показано на Фиг. 27, в конце процесса дифференцировки на стадии 4 сутки 3 у клеток сохраняются высокие уровни экспрессии PDX1 и FOXA2 и далее проявляется паттерн экспрессии, соответствующий сочетанию панкреатических эндокринных клеток (28,1% ± 12,5 СО хромогранин-положительных) и клеток-предшественников панкреатических клеток (58,3% ± 9,7 СО NKX6.1-положительных). Характерные для данной стадии паттерны экспрессии маркеров указывают на эффективную постадийную дифференцировку от популяции плюрипотентных клеток в клетки-предшественники панкреатических клеток. Результаты, полученные с помощью проточной цитометрии, были дополнительно подтверждены данными Qrt-PCR. Все гены, обычно экспрессирующиеся в поджелудочной железе (ARX, GAST, GCG, IAPP, INS, ISL1, MAFB, NEUROD1, NGN3, NKX2.2, NKX6.1, PAX4, PAX6, PTF1A и SST), показали повышенный уровень экспрессии. (Фиг. 28).
Паттерн экспрессии, который наблюдается на Фиг. 27, оставался соответственным во время нескольких пробегов с несколькими изменениями процесса, например, с различным посевным материалом, средой стадии 0, рН среды стадии 0 и использованием пеногасителя. Были протестированы разные источники посевного материала и каждый раз происходила эффективная генерация эндодермальных панкреатических клеток с >90% FOXA2, >75% PDX1, и >50% NKX6.1 (Фиг. 29). Кроме того, не наблюдалось существенного различия в паттернах экспрессии продукта биореактора между клетками, которые были выращены на стадии 0 в разработанной в лаборатории среде под названием «IH3» с добавлением 0,5% BSA или коммерчески доступной среде: Essential8™ с добавлением 0,5% BSA (Фиг. 30). Когда исследовали роль рН в стадии 0 культуры, было отмечено, что клетки, выращенные в стадии 0 при относительно низком рН (6,8) показали увеличение роста в биореактор по отношению к средней перспективе (Фиг.7), но никаких существенных изменений в клеточном профиле на сутки 3 стадии 4 (Фиг. 31). Кроме того, использование пеногасителя C эмульсии (Sigma № по каталогу A8011) в количестве 94 части на миллион явно уменьшило количество пузырей, образующихся при барботировании но не повлияло на профиль клеток с конца стадии 0 до 4-й стадии сутки 3 клеток (таблица 9 и Фиг. 32).
В конце каждой дифференцировки в биореакторе клеточный продукт криоконсервировали. Клетки промывали в MCDB131 с 3,63 г/л бикарбоната натрия или MCDB131 с 3,63 г/л бикарбоната натрия, глюкозой (до 8 мм) и 1 × Glutamax, и затем переносили в холодную (<4°C) среду для криоконсервации, состоящую из 57,5% MCDB131 с 2,43 г/л бикарбоната натрия, 30% свободного от ксенобиотиков KSR, 10% DMSO и 2,5% HEPES (конечная концентрация 25 мМоль). Затем клетки замораживали в морозильной камере с контролируемой скоростью (ХПН) с помощью охлаждающего профиля, который поддерживал клеточные кластеры в среде криоконсервации при температуре окружающей среды в течение максимум 15 минут, с последующим снижением температуры до 4°C в течение 45 мин и дальнейшим снижением температуры на 2°C/мин до -7°C (образец). Затем образец быстро охлаждался, снижение температуры в камере шло со скоростью 25°C/мин до -45°C. Затем было предусмотрено увеличение температуры в камере на 10°C/мин до -25°C (камера). Затем образец охлаждали со скоростью 0,2°C/мин до тех пор, пока температура не достигала -40°C. Затем камеру охлаждали до -160°C со скоростью 35°C/мин и выдерживали при этой температуре в течение 15 минут. Образцы были перемещены в контейнер хранилища с жидким азотом, часть с газовой фазой, для хранения после прекращения пробега CRF.
Клетки могут быть разморожены удалением из газовой фазы хранилища с жидким азотом и перенесены во флакон на водяную баню температурой 37°C. Пробирку осторожно встряхивали на водяной бане в течение менее 2 минут до того момента, пока во флаконе не оставался небольшой кристалл льда. Содержимое сосуда затем переносили в 50 мл коническую колбу и разбавляли по каплям в течение двух минут с использованием среды MCDB131 с 2,43 г/л бикарбоната натрия и 2% BSA, до конечного объема 20 мл. Общее количество клеток определяли с помощью аппарата NucleoCounter® и клеточную суспензию переносили в на сверхмалые носители в чашку для культивирования на 1 час. Затем клетки выделяли из среды в 50 мл коническую колбу, супернатант удаляли и клетки ресуспендировали в среде 4 стадии носитель для анализа или исследования in vivo.
В качестве альтернативы, после разморозки клетки из флакона переносили в пустую стеклянную центрифужную пробирку 125 мл Corning® (Corning, Incorporated, г. Корнинг, штат Нью-Йорк, США) и по каплям добавляли 10 мл среды MCDB131, содержащей 2,43 г/л бикарбоната натрия и 2% BSA. Конечный объем доводили той же средой до 80 мл. Общее количество клеток определяли с помощью NucleoCounter® и клеточную суспензию перемешивали при 40-65 оборотах в минуту в течение ночи (12-28 часов). Затем клетки характеризовали или использовали для исследования in vivo.
Композиция среды IH3 приведена ниже, а также описана в публикации заявки на патент США № 2013/0236973, раскрытие которой включено путем ссылки в полном объеме, как она относится к подходящей культуральной среде. Количество BSA в среде IH3 может изменяться.
0,5% FAF-BSA класса реагентов
1 нг/мл TGF-β1
100 нг/мл bFGF
20 нг/мл IGF-1
0,25 мМ аскорбиновой кислоты.
Сутки/Дата
(3,64 г/л NaCO3)
(3,64 г/л NaCO3)
(3,64 г/л NaCO3)
(3,64 г/л NaCO3)
2,5 мМоль глюкозы
1:50 000 ITS-X
Glutamax 1:100
2,5 мМоль глюкозы
1:50 000 ITS-X
Glutamax 1:100
2,5 мМоль глюкозы
1:200 ITS-X
Glutamax 1:100
2,5 мМоль глюкозы
1:200 ITS-X
Glutamax 1:100
GDF8
100 нг/мл
50 нг/мл
50 нг/мл
MCX
[2 мкМоль]
SANT [0,25 мкМоль]
TPPB [100 нМоль]
Только сутки 1
LDN 100
TPPB [100 нМоль]
Все дни относительно 0 ч.
Сутки 1 и 2,
Без замены на сутки 3
Сутки 1 и 3,
Без замены на сутки 2
Сутки 1 и 2,
Без замены на сутки 3
Материалы:
• Клетки человеческой эмбриональной клеточной линии H1 (hES), (клетки WA01, WiCell, Madison WI)
• PBS (№ по каталогу 14190, Invitrogen)
• Y-27632 (Axxora № по каталогу ALX-270-333, San Diego, CA)
• EDTA, (Lonza, № по каталогу 17-7-11E)
• NucleoCounter®-(ChemoMetec A/S, № по каталогу YC-T100, г. Аллерод, Дания)
• Обработанные двумя нетканевыми культурами 6-луночные планшеты (Becton Dickinson, № по каталогу Falcon 351146, Franklin Lakes, NJ)
• Accutase®, (Sigma, № по каталогу A-6964, г. Сент-Луис, штат Миссури, США)
• Датчики биореактора для определения уровня pH и количества растворенного кислорода (DO) (FermProbe® рН электрод 225 мм, модель № F-635, датчик растворенного кислорода OxyProbe® 12 мм, номер модели D-145 от Broadley-James Corporation, г. Ирвин штат Калифорния, США)
• Устройство иммунозащитной макроинкапсуляции (TheraCyte™, г. Ирвин штат Калифорния, США)
• мМоль ИФА человеческого C-пептида (MERCODIA № по каталогу 10-1141-01)
• GlutaMAX™, MCDB131 и ITS-X Invitrogen
• FAF-BSA (Proliant)
• Ретиноевая кислота, глюкоза 45% (2,5M), SANT (ингибитор Shh) (Sigma)
• GDF8 (Peprotech)
• MCX
• FGF7 (R & D Systems)
• LDN-193189 (антагонист рецептора BMP) (Stemgent)
• TPPB (активатор PKC) (ChemPartner)
• Изготовленная на заказ среда MCDB 131
Пример 8
Созревание и функция полученных в биореакторе и криоконсервированных кластеров панкреатических клеток-предшественников
Для генерирования достаточного количество клеток для каждого биореакторы разморозили по одному флакону клеток линии H1 hES (WB0106) 31 пассирования из главного банка клеток. Клетки были выращены прикрепленными в среде mTeSR®1 в течение нескольких пассажей на Matrigel™ с использованием EDTA-пассирования до получения достаточного количества клеток, чтобы засеять пять покрытых Matrigel™ двухслойных емкостей для клеток CellSTACKs® (CS2). Клетки были выращены прикрепленными и после формирования в CS2 конфлюэнтного слоя на 70%, к портам среды прикрепили систему трубок с крышками C-Flex®, к которым подсоединяли ведущие к насосу трубки для замыкания системы. После того как система была замкнута с помощью трубок C-Flex®, к ней приваривались с помощью аппарата Terumo пакеты или бутыли, куда перистальтическим насосом переносились объемы жидкости (среда, PBS-/-, Accutase® или суспендированные клетки).
Для отделения клеток от CS2, клетки промывали один раз фосфатно-солевым буфером Дульбекко без кальция или магния (PBS-/-), затем обрабатывали с раствором Accutase® половинной эффективности, разбавленным PBS-/ и инкубировали в течение 4-5 минут. Раствор Accutase® затем удаляли, и через 3 минуты после нанесения раствора фермента добавляли CS2 для стимулирования клеточного отделения. Емкость со средой mTeSR®1, содержащей 10 мкмоль/л ингибитора Rho-киназы, Y-27632, закачивали в CS2 для промывания и инактивации остаточной Accutase®, затем смыв собирали. Добавляли второй промывочный объем, собирали смыв и объединяли с первым. 1,6-2,0 × 109 клеток извлекали из CS2s в конечном объеме 2 литра. 2,0-2,5 × 108 клеток на слое переносили в четыре CS2 или восемь однослойных емкостей Cell Stacks™ и инкубировали при 37°C в течение 2 часов в увлажненном инкубаторе с 5% CO2 в объеме 200 мл на слой.
Используя замкнутый контур трубок C-Flex® с прикрепленной трубкой для насоса, прикрепленной между портами среды CellSTACK®, клеточную суспензию измельчали в течение 5 минут при 75 оборотах в минуту с помощью перистальтического насоса для гомогенизации агрегатов. CellSTACKs® затем инкубировали в течение ночи при 37°C в течение 18 часов в увлажненном инкубаторе с 5% CO2. 2 л клеток и сред из CellStack™ затем объединяли и переносили, по 1 литр каждого, в два 3 л DASGIP биореактора вместе с 1,5 л свежей среды mTeSR® на каждый биореактор. Клетки поддерживали в течение двух дополнительных суток в среде mTeSR® до начала дифференцировки, с полной заменой среды через 24 часа после посева биореактора. После этого была инициирована и направлена дифференцировка, как описано в таблице 10. Клетки поддерживали всего в течение 14 или 15 суток (2 суток mTeSR® + 12 или 13 суток в процессе дифференцировки) в закрытой стерильной суспензии в биореакторе регулируемой температуры (37°C), рН (изменяемым или регулируемым СО2 до 6,8 или 7,2 для плюрипотентных клеток и 7,4 для дифференцировки) и уровнем растворенного кислорода (30% DO, уровни СО2/воздуха регулируются). Лопастное колесо было остановлено на 5-20 минут до удаления среды через погружную трубку для стабилизации кластеров. Среду удаляли или добавляли с помощью перистальтического насоса через погруженную пробирку, соединенную с трубками C-Flex® (Cole-Parmer North America, г. Вернон Хилс, штат Иллинойс, США) с использованием сварщика трубок Terumo™ для поддержания замкнутой системы. Рабочее колесо и нагреватель были вновь запущены после добавления достаточного количества среды, чтобы полностью погрузить колесо.
Два производственных пробега были начаты в 3-литровых реакторах с использованием этих методов. В первом пробеге реактора были протестированы два различных показателя рН в течение первых двух суток в плюрипотентной культуральной среде. Реактор 1 был установлен на показатель pH 7,2 с фиксированной скоростью инфузии газа CO2 5%, так что рН снижался по мере окисления среды реактора с течением времени за счет метаболической активности клеток. Реактор 2 был установлен на показатель рН 7,2 с помощью регулируемого уровня газа CO2. Во втором пробеге реактора был установлен показатель рН 6,8 для реактора 1 и 7,2 для реактора 2 в обоих регулировались уровни газа СО2.
Для того чтобы контролировать процессы в биореакторе, в конце каждой стадии дифференцировки были взяты образцы кластеров клеток и проанализированы с помощью проточной цитометрии (таблица 11; таблица 12). Наблюдался почти полный переход от CD9-экспрессирующей/CXCR4-отрицательной популяции плюрипотентных клеток при инициировании дифференцировки к гомогенной популяции клеток, экспрессирующих CXCR4 (96,9-98,1% клеток CXCR4-положительны) по завершению формирования дефинитивной энтодермы.
Результаты, полученные с помощью проточной цитометрии коррелирует с результатами парных образцов, проанализированных методом RT-PCR. Образцы тестировались на протяжении всего процесса для оценки характеристик экспрессии генов в процессе дифференцировки от плюрипотентности к превращению в панкреатические. До начала направленной дифференцировки была протестирована мРНК кластеров клеток в биореакторе на массивах Low Density для панели генов, ассоциированных с плюрипотентностью или ранним началом дифференцировки.
Было отмечено, что клетки из биореактора сохраняют экспрессию генов, характерную для плюрипотентности (POU5F1, NANOG, Sox2 и ZFP42) и показывают минимальное или отсутствующее индуцирование генов, характерных для дифференцировки (AFP и Foxa2: < 50-кратное увеличение; FOXD3, GATA2, GATA4, GSC, HAND2, MIXL1 и T: <10-кратное увеличение экспрессии) по сравнению с контрольными недифференцированными клетками линии H1 (Фиг. 33). Однако, как только клетки вступили в контакт со средой дифференцировки на стадии 1 сутки 1, паттерны экспрессии резко изменились, уровни экспрессии CDX2, CER1, FGF17, FGF4, Foxa2, GATA4, GATA6, РКГ MIXL1, MNX1 и Brachyury (Т) увеличились в 100-1000 раз относительно уровней недифференцированных клеток линии HES Н1 (фиг.34). К концу стадии 1 сутки 3 (формирование дефинитивной энтодермы), экспрессия CD9, CDX2, FGF4, MIXL1, NANOG, POU5F1 и Brachyury (Т) уменьшилась относительно таковой на стадии 1 сутки 1, а экспрессия характерных окончательных генов энтодермы, таких как CD99, CER1, CXCR4, FGF17, GATA4, GATA6, KIT, OTX или SOX17, достигла максимума (Фиг. 35).
В конце стадии 1 клеточная культуральная среда была заменена: вместо среды, содержащей GDF8 добавили среду, содержащую FGF7. Было отмечено несколько различных паттернов экспрессии генов: увеличение экспрессии определенных генов в течение стадии 2 (AFP, Atoh1, HHEX, OSR1, Pdx1, Prox1, Sox2 и SOX9), снижение экспрессии (Hand1 и SOX17), стабильно высокая постоянная экспрессия (HNF4α), или низкая/отсутствующая экспрессия (CDX2, GAST, Nkx2.2, Nkx6.1 и Ptf1a) (фиг.36а-е). Эти модели показали, что клетки в реакторе приобретают характеристики клеток передней кишки (AFP, Atoh1, HHEX, HNF4α, OSR1, Pdx1, Prox1, Sox2 и SOX9), экспрессия маркеров мезодермы (Hand1 и SOX17) снизилась. Тем не менее, к концу стадии 2 клетки еще не были специализированы более конкретно в вызревшие клетки кишки или панкреатические клетки (CDX2, GAST, Nkx2.2, Nkx6.1 и Ptf1a).
К концу стадии 3 клетки были специализированы в панкреатические, что было определено по экспрессии Pdx1, показавшей > 100000-кратное увеличение мРНК относительно недифференцированного контроля (Фиг. 36) и результатам проточной цитометрии, показавшей 76-98% клеток, экспрессирующих Pdx1 (табл 11). Также наблюдалась индукция других генов поджелудочной железы (GAST, NKX2.2, NKX6.1, PROX1, PTF1a и SOX9) и кишечника, таких как AFP и CDX2; что указывало на постепенное созревание и спецификацию клеток.
К концу процесса дифференцировки на сутки 3 или 4 стадии 4, клетки показали паттерны экспрессии, соответствующие сочетанию панкреатических эндокринных клеток (47-54% хромагранин-положительных) и панкреатических клеток-предшественников (33-52% NKX6.1-положительных), как показано в таблицах 11 и 12. Эти стадиеспецифические паттерны экспрессии маркеров конкретно указывают на эффективную постадийную дифференцировку из популяции плюрипотентных клеток в панкреатические клетки-предшественники, характеризующихся высоким уровнем экспрессии Pdx1 (>1×106 кратное индуцирование) и других панкреатических генов (> 1000кратное индуцирование ARX, GCG, GAST, INS, ISL, NEUROD1, Ngn3, Nkx2.2, Nkx6.1, Pax4, Ptf1a и SST) и почти полной потерей экспрессии OCT4/POU5F1 по сравнению с недифференцированным эмбриональными стволовыми клетками человека линии H1 (Фиг. 37).
В конце процесса дифференцировки было получено 0,08-0,45 × 106 клеток/мл (фиг.38: ежедневный подсчет количества клеток). Клетки, полученные в этом процессе, были криоконсервированы или непосредственно имплантированы в животных либо подкожно с помощью устройства TheraCyte™, либо под почечную капсулу. Для криоконсервирирования клеток их переносили в среду криоконсервации, состоящую из 57,5% MCDB131 с 2,43 г/л бикарбоната натрия,30% свободного от ксенобиотиков KSR, 10% DMSO и 2,5% HEPES (конечная концентрация 25 мМоль). После того, как кластеры клеток были суспендированы в среде криоконсервации при комнатной температуре, пробирки для криоконсервации переносили в морозильник с контролируемой скоростью (CRF) в течение 15 минут. Температуру в камере затем снижали до 4°C в течение 45 минут, и далее снижали на 2°C/мин до -7°C (образец). Затем образец быстро охлаждался, снижение температуры в камере шло со скоростью 25°C/мин до -45°C. Затем было предусмотрено увеличение температуры в камере на 10°C/мин до -25°C (камера). Затем образец охлаждали со скоростью 0,2°C/мин до тех пор, пока температура не достигала -40°C. Затем камеру охлаждали до -160°C со скоростью 35°C/мин и выдерживали при этой температуре в течение 15 минут. Образцы были перемещены в контейнер хранилища с жидким азотом, часть с газовой фазой, для хранения после прекращения пробега CRF.
После того как клетки были сохранены в газовой фазе жидкого азота, их размораживали путем извлечения из хранилища и перенесения на водяную баню при 37°C. Пробирку осторожно встряхивали на водяной бане в течение менее 2 минут до того момента, пока во флаконе не оставался небольшой кристалл льда. Содержимое сосуда затем переносили в 50 мл коническую колбу и разбавляли по каплям в течение двух минут с использованием среды MCDB131 с 2,43 г/л бикарбоната натрия и 2% BSA, до конечного объема 20 мл. Общее количество клеток определяли с помощью аппарата NucleoCounter® и клеточную суспензию переносили в на сверхмалые носители в чашку для культивирования на 1 час. Затем клетки выделяли из среды в 50 мл коническую колбу, супернатант удаляли и клетки ресуспендировали в 4 стадии среды. Затем клетки имплантировали в животных либо подкожно с помощью устройства TheraCyte™, или под почечную капсулу, или клетки инкубировали в чашках Петри со сверхнизкой связывающей способностью течение ночи и затем имплантировали животному.
У животных отслеживали уровень глюкозы и С-пептида в крови каждые четыре недели после имплантации трансплантата. Животные, которым были имплантированы не-криоконсервированные панкреатические клетки-предшественники с помощью устройства TheraCyte™ или путем прямого размещения клеток под почечную капсулу, показали вызревание клеток и экспрессию C-пептида 1 нг/мл через 16 недель, позже поднявшуюся до 2 нг/мл C-пептида через 20 недель после имплантации (Фиг. 39а и 39d). Кроме того, при обработке STZ для абляции функции β-клеток, у животных с имплантатами поддерживали нормогликемию до удаления трансплантатов, тем самым указывая, что трансплантаты оказались способны защитить животных от диабета, вызванного одной большой дозой STZ (Фиг. 39b ).
Это картина наблюдалась у животных, которым были имплантированы криоконсервированные клетки. Животные, которым под почечную капсулу были имплантированы криоконсервированные панкреатические клетки-предшественники, которые культивировали в течение 1 часа после разморозки (1207B), показали среднее количество С-пептида 0,56 нг/мл и 1,09 нг/мл через 16 и 20 недель соответственно, в то время как клетки, культивированные в течение ночи после разморозки (1207C), показали в среднем 0,81 нг/мл и 1,35 нг/мл С-пептида через 16 и 20 недель соответственно (Фиг. 39d). Животные, которым были имплантированы криоконсервированные панкреатические клетки-предшественники с помощью устройства TheraCyte™, показали более 1 нг/мл C-пептида через 16 недель, и также как не-криоконсервированные контроли, оказались способны к экспрессии терапевтических уровней С-пептида через неделю после обработки STZ (0,98 нг/мл, Фиг. 39с). Эти результаты показывают, что панкреатические криоконсервированные клеток-предшественников может функционировать с эффективностью, сравнимой с эффективностью не-криоконсервированных контролей при испытании на животных моделях.
(3,64 г/л NaCO3)
(3,64 г/л NaCO3)
(3,64 г/л NaCO3)
2,5 мМоль глюкозы
1:50 000 ITS-X
Glutamax 1:100
2,5 мМоль глюкозы
1:50 000 ITS-X
Glutamax 1:100
2,5 мМоль глюкозы
1:200 ITS-X
Glutamax 1:100
2,5 мМоль глюкозы
1:200 ITS-X
Glutamax 1:100
GDF8
100 нг/мл
50 нг/мл
50 нг/мл
(только сутки 0)
[1:1000; 10 мкМоль]
MCX
[3 мкМоль]
SANT [0,25 мкМоль]
TPPB [100 нМоль]
Только сутки 1
LDN 100
TPPB [100 нМоль]
Сутки 1 и 2,
Без замены на с3
Сутки 1 и 3,
Без замены на сутки 2
Сутки 1 и 2,
Без замены на с3
Болюс глюкозы на сутки 3
Примечание:
• Базовая среда в данной таблице 10 опционально может содержать 5 мМоль глюкозы на стадиях 1-5, при условии, что в качестве добавки не используется Glutamax.
• В приведенной выше таблице 10 также указано, что на стадии 4 может быть добавлено ([100 нМоль]) Cypi.
Расчет напряжения сдвига, испытываемого клеточными агрегатами в биореакторе с механическим перемешиванием
Было определено напряжение сдвига, которое испытывают клеточные агрегаты в 2,7 литра перемешиваемой DASGIP суспензии в биореакторе при скорости перемешивания 70 оборотов в минуту в 3l DASGIP биореакторе. Для того чтобы рассчитать значения напряжения сдвига, были сделаны следующие заявленные допущения.
Предположения:
1. Максимальное напряжение сдвига, испытываемое клеточными агрегатами, не является результатом турбулентных вихрей
2. Максимальное напряжение сдвига, испытываемое клеточными агрегатами, не является результатом трения двух агрегатов друг об друга или агрегата о лопасть.
3. Напряжение. вызванное столкновением с перегородками (т.е. погруженными трубками и зондами) в этих расчетах не рассматриваются
Для целей расчетов в данном описании использовались перечисленные ниже номенклатура и физические параметры.
Номенклатура:

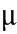


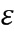
Параметры :

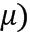

Указанные в списке параметры биореактора и среды использовались в указанных ниже уравнениях
Уравнения :
Числа Рейнольдса:
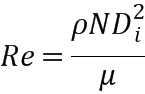
Максимальное напряжение сдвига агрегатов (Cherry and Kwon 1990)

Мощность, рассеиваемая (ε) на единицу массы

Потребленная электороэнергия (P)

Расчет числа мощности был основан на эмпирической корреляции, полученной Нагатой (1975) для емкостей без преград с механическим перемешиванием.

где
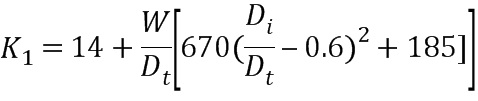

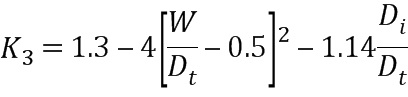
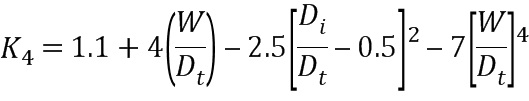
Был рассчитан максимальный сдвиг клеточных агрегатов при скорости перемешивания 70 оборотов в минуту в биореакторе 2,7 л DASGIP, он составил, по меньшей мере, 0,25 паскалей (2,5 дин/см2). Клетки, содержащиеся во внешнем слой кластеров, испытывали самые высокие уровни напряжения сдвига. Данные значения напряжения сдвига сильно зависят от изложенных предположений.
Пример 9
Дифференцировка эмбриональных стволовых клеток клеточной линии WA01 в клетки дефинитивной энтодермы: роль MCX/GDF8 в суспензионной культуре
Кластеры плюрипотентных эмбриональных стволовых клеток человека из линии стволовых клеток H1 (NIH код: WA01) высевали при плотности клеток в пределах от 0,25×106 до 2×106 клеток/мл в колбах Эрленмейера / перемешиваемых колбах, центрифужных пробирках или на непокрытых 6-луночных планшетах низкого связывания или обработанных нетканевой культурой 6-луночных планшетах, посев проводится в среду MCDB-131, содержащую 3,64 г/мл бикарбоната натрия и 5,5 мМоль глюкозы (№ по каталогу A13051 DJ, Invitrogen, CA), дополненной 2% очищенной от жирных кислот BSA (№ по каталогу 68700, Proliant, IA), 1X GlutaMAX™ (№ по каталогу 35050-079 , Invitrogen, Калифорния), дополнительно добавлено 2,5 мМоль глюкозы (№ по каталогу G8769, Sigma) и ITS-х с концентрацией базового раствора 1: 50 000 (№ по каталогу 51500056, Invitrogen, Калифорния). Среда MCDB-131, дополненная таким образом, в настоящем документе будет упоминаться как «базовая среда стадии 1». Кластеры в этой среде в первые сутки дифференцировки были обработаны либо 3 мкМоль MCX (ингибитор GSK3B, 14-проп-2-ен-1-ил-3,5,7,14,17,23,27-гептаазатетрацикло [19.3.1.1~2,6~.1~8,12~]гептакоза-1(25),2(27),3,5,8(26),9,11,21,23-нонен-16-он, заявка на патент США № 12/494,789; включенная в настоящий документ посредством ссылки в полном объеме) и 100 нг/мл GDF-8 (№ по каталогу 120-00, Peprotech), либо только 3 мкМоль MCX, или 20 нг/мл WNT-3A (№ по каталогу 1324-WN-002, R&D Systems, штат Миннесота, США) плюс 100 нг/мл активина А (№ по каталогу 338-AC, R&D Systems, штат Миннесота, США), или 20 нг/мл только WNT-3A. На второй сутки, клетки переносили в свежую базовую среду стадии 1 с добавлением либо 100 нг/мл GDF8, или 100 нг/мл активина A. Образцы собирали для проточной цитометрии, PCR и анализа Вестерн-блот в различные моменты времени, начиная от нулевого времени (непосредственно перед добавление базовой среды плюс добавки) до момента спустя 72 часа после начала дифференцировки.
Эффективность, с которой генерировалась дефинитивная энтодерма, была определена после 3 суток дифференцировки при каждом условии, измерением с использованием проточной цитометрии процента клеток, экспрессирующих маркеры клеточной поверхности CXCR4, CD99 и CD9. Данные (как показано на графиках экспрессии на Фиг. 40a-d и обобщено в таблице 13) показывают, что в суспензионной культуре при добавлении 3 мкМоль MCX в отсутствие веществ семейства TGF-β за первые сутки дифференцировки генерируется дефинитивная энтодерма на уровнях, сравнимых с проявляющимися при обработке клеток 3 мкМоль MCX плюс 100 нг/мл GDF-8 или 20 нг/мл WNT-3A плюс 100 нг/мл активина А за одни сутки .
(Сутки 1 → Сутки 2 и 3)
(% посредством анализа FACS)
(% посредством анализа FACS)
(% от родительских)
Пример 10
Дифференцировка эмбриональных стволовых клеток из клеточной линии WA01 в дефинитивную энтодерму: дозозависимый эффект соединений MCX в суспензионной культуре в зависимости от концентрации
Кластеры плюрипотентных эмбриональных стволовых клеток человека из линии стволовых клеток H1 (NIH код: WA01) высевали при плотности клеток в пределах от 0,25×106 до 2×106 клеток/мл в колбах Эрленмейера/перемешиваемых колбах, центрифужных пробирках на базовую среду стадии 1, как описано в примере 9.Кластеры обрабатывали базовой средой стадии 1, содержащей 1,5, 2, 3 или 4 мкМоль MCX на первые сутки дифференцировки и свежей базовой средой стадии 1, содержащей 100 нг/мл GDF-8, на 2 сутки. На третьи сутки среду на заменяли. Образцы собирали для проточной цитометрии и PCR-анализа по истечению третьих суток дифференцировки.
Эффективность, с которой генерировалась дефинитивная энтодерма, была определена измерением с использованием проточной цитометрии процента клеток, экспрессирующих маркеры клеточной поверхности CXCR4, CD99 и CD9. Данные (как показано на графиках экспрессии на Фиг. 41A-D и обобщено в таблице 14) показывают, что в суспензионных культурах, добавление MCX при концентрациях менее 2 мкМоль приводит в результате к появлению меньшего количества клеток дефинитивной энтодермы (о чем свидетельствует более низкий процент CXCR4-положительных клеток и более высокий процент CD9-положительных клеток). Кроме того, при концентрациях выше 4 мкМоль, MCX проявляет вредное воздействие на клетки, что приводит к снижению жизнеспособности клеток. Тем не менее, за счет увеличения концентрации BSA, эффекты MCX могут быть модулированы таким образом, что концентрация ≥ 4 мкМоль/л может быть применена. С другой стороны, концентрации ≤ 1,5 мкМоль/л может быть применена для создания дефинитивной энтодермы при использовании более низких концентраций BSA.
Пример 11
Дифференцировка эмбриональных стволовых клеток из клеточной линии WA01 в дефинитивную энтодерму: роль частоты замены среды в суспензионной культуре
Кластеры плюрипотентных эмбриональных стволовых клеток человека из линии стволовых клеток H1 (NIH код: WA01) высевали при плотности клеток в пределах от 0,25×106 до 2×106 клеток/мл в колбах Эрленмейера/перемешиваемых колбах, центрифужных пробирках на базовую среду стадии 1, как описано в примере 9.Кластеры обрабатывали базовой средой стадии 1, содержащей 3 мкМоль MCX на первые сутки дифференцировки и свежей базовой средой стадии 1, содержащей 100 нг/мл GDF-8, на 2 сутки. В контрольных культурах выполняли замену среды на третьи сутки; в отдельном сосуде замену среды на третьи сутки не проводили. Образцы собирали для проточной цитометрии и PCR-анализа по истечению третьих суток дифференцировки.
Эффективность, с которой генерировалась дефинитивная энтодерма, была определена при каждом из условий измерением с использованием проточной цитометрии процента клеток, экспрессирующих маркеры клеточной поверхности CXCR4, CD99 и CD9. Результаты показаны на графиках экспрессии На Фиг. 42А и В и приведены в таблице 15.
Пример 12
Дифференцировка эмбриональных стволовых клеток из клеточной линии WA01 в дефинитивную энтодерму: Использование GlutaMAX™ в суспензионной культуре
Кластеры плюрипотентных эмбриональных стволовых клеток человека из линии стволовых клеток H1 (NIH код: WA01) высевали при плотности клеток в пределах от 0,25×106 до 2×106 клеток/мл в колбах Эрленмейера / перемешиваемых колбах, центрифужных пробирках.
Настоящий пример выполняли для определения необходимости добавки Glutamax™ для генерации дефинитивной энтодермы путем суспендирования кластеров в базовой среде стадии 1 (как описано в примере 9) с и без GlutaMAX™, среда была дополнена 3 мкМоль MCX на первые сутки дифференцировки с заменой базовой среды стадии 1, содержащей 100 нг/мл GDF-8, на 2 сутки. На третьи сутки среду на заменяли. Образцы собирали для проточной цитометрии и PCR-анализа по истечению третьих суток дифференцировки.
Эффективность, с которой генерировалась дефинитивная энтодерма, была определена при каждом из условий измерением с использованием проточной цитометрии процента клеток, экспрессирующих маркеры клеточной поверхности CXCR4, CD99 и CD9. Результаты показаны на графиках экспрессии на Фиг. 43А и В и приведены в таблице 16.
Пример 13
Дифференцировка эмбриональных стволовых клеток из клеточной линии WA01 в дефинитивную энтодерму: роль концентраций бикарбоната натрия в суспензионной культуре
Кластеры плюрипотентных эмбриональных стволовых клеток человека из линии стволовых клеток H1 (NIH код: WA01) высевали при плотности клеток в пределах от 0,25×106 до 2×106 клеток/мл в колбах Эрленмейера / перемешиваемых колбах, центрифужных пробирках на базовую среду стадии 1, как описано в примере 9, ( среда содержала 3,64 г/л бикарбонат натрия), или на модифицированную базовую среду стадии 1, которая содержала 2,43 г/л бикарбоната натрия. Кластеры обрабатывали базовой средой стадии 1, содержащей MCX и GDF-8, как описано в примере 12. Образцы собирали для проточной цитометрии по истечению третьих суток дифференцировки. На каждые сутки дифференцировки также делались фазоконтрастные изображения.
Эффективность, с которой генерировалась дефинитивная энтодерма, была определена измерением с использованием проточной цитометрии процента клеток, экспрессирующих маркеры клеточной поверхности CXCR4, CD99 и CD9. Результаты показаны на графиках экспрессии на Фиг. 44а и В и приведены в таблице 17. В суспензионных культурах, низкий уровень бикарбоната натрия, 2,43 г/л, по всей видимости, приводят к менее эффективной генерации дефинитивной энтодермы (в среднем 87,4% клеток экспрессируют CXCR4), в отличие от более высокого уровня 3,64 г/л (в среднем 97.35% клеток экспрессируют CXCR4). Кроме того, было отмечено, что различия в уровнях бикарбоната коррелировали с различиями в морфологии кластера в конце стадии 1, что было отмечено с помощью фазоконтрастной микроскопии (Фиг.44 C и D). Кроме того, клетки, дифференцированные при высоких уровнях бикарбоната, как наблюдалось, формировали менее плотные клеточные кластеры, чем клетки, дифференцированные при 2,43 г/л бикарбоната.
Пример 14
Создание кластеров клеток-предшественников панкреатических клеток из человеческих индуцированных плюрипотентных стволовых клеток в масштабируемом процессе в биореакторе
Клеточная терапия требует большого количества (> 108) клеток в дозе. Этот пример демонстрирует процесс, способный дифференцировать индуцированные массы плюрипотентных стволовых клеток (IPS клетки) в количествах, на 3-5 порядков больших, чем это позволяют нынешние производственные практики клеточной терапии.
В этом примере была использована клеточная линия IPS - UTC (полученная из клеток ткани пуповины, ранее описанных в заявке на патент США 13/330,931 (публикации заявки на патент США 2013/0157365), раскрытие которой включено путем ссылки в полном объеме, как она относится к получению клеточных линий iPS). Клетки были получены из мышиных эмбриональных фидерных клеток с использованием трансфекции плазмид без оставления следов и криоконсервированных при пассировании 15.
Из этих криоконсервированных клеток была создана серия клеточных банков путем добавления содержимого пробирки исходного материала в ходе процесса разморозки флакона в рекомбинантный человеческий ламинин (hrLaminin № по каталогу LN-521 от Biolamina, г. Стокгольм, Швеция) в среде Essential8™ (E8) ™ от Life Technologies Corporation (г. Гранд Айленд, штат Нью-Йорк, США) для генерирования внутрилабораторного посевного материала. Размороженный и выращенный материал называется «предварительно-предварительный главный банк клеток» (Pre-Pre МСВ) и служит в качестве посевного материала для будущих банков. Из трех последовательных клеточных банков Pre-Pre МСВ затем генерируется предварительный главный банк клеток (Pre МСВ), главный банк клеток (MCB), и рабочий банк клеток (WCB). Один флакон WCB затем размораживают, выращивают на hrLaminin с использованием пассирования EDTA в течение трех пассажей в E8™. Клетки были впервые высеяны после разморозки в колбу T225 (Corning; г. Корнинг, штат Нью-Йорк, США), а затем пересеяны на несколько колб T225. Несколько колбы T225 затем пассажировали и объединяли, чтобы засеять одну однослойную емкость для клеток Cell Stack™ (CS1). После того, как клетки в CS1 были объединены, клетки промывали один раз PBS-/-, обрабатывали раствором Accutase® половинной эффективности, разбавляли PBS-/- и инкубировали в течение 4-5 минут. Затем Accutase® удаляли, и через 3 минуты после нанесения раствора фермента, CS1 был использован для стимулирования клеточного отделения. E8™ с добавлением 0,5% BSA, содержащий 10 мкмоль/л ингибитора Rho-киназы, Y-27632, был добавлен к CS1 для промывания и инактивации остаточных Accutase®. Смыв собирали и добавляли второй объем для промывания, смыв с которого также собрали и объединили с первым смывом.
Клетки переносили в среду, дополненную 0,5% BSA, содержащую 10 мкмоль/л ингибитора Rho-киназы, Y-27632, в 1-литровую центрифужную пробирку одноразового использования (Corning г. Корнинг, штат Нью-Йорк, США) в концентрации 1×106 клеток/мл в 225 мл. Клеткам дали возможность кластеризоваться в статической суспензии в течение 60 минут в увлажненном инкубаторе с 5% CO2, затем перемешивали в течение 5 минут при 55-65 оборотах в минуту и добавляли 225 мл дополнительной среды, дополненной 0,5% BSA, содержащей 10 мкмоль/л ингибитора Rho-киназы, Y-27632. Клеткам дали отстояться в статической культуре в течение 30 дополнительных минут, а затем в центрифужную пробирку добавили 150 мл дополнительной среды с добавлением 0,5% BSA, содержащей 10 мкмоль/л ингибитора Rho-киназы, Y-27632. После этого клетки непрерывно перемешивали при скорости 50-70 оборотов в минуту в увлажненном инкубаторе с 5% CO2. Двадцать четыре часа спустя центрифужную пробирку вынимали из инкубатора и кластерам давали отстояться в течение 5-10 минут. Затем среду отсасывали, пока не осталось 200 мл и добавляли в центрифужную пробирку 400 мл дополнительной свежей культуральной среды. Этот процесс повторяли в конце 2 суток (через 48 часов после переноса).
Через 72 часа после первоначальной обработки Accutase® процесс клеточной диссоциации кластеров и посев (пассирование) в центрифужную пробирку повторяли для поддержания клеток в состоянии суспензии для многократного пассирования (диапазон испытаний: 1-10 пассажей).
Используя этот процесс, плюрипотентные клетки UTC были преобразованы из прикрепленной на подложке культуры в суспензионную культуру в виде кластеров клеток и затем выращены в суспензии. Эти суспензии пересевали и культивируемые клетки затем криоконсервировали и сохраняли для последующего использования. Для того чтобы подготовить суспензию разросшихся клеток для криоконсервации, клеточные кластеры диссоциировали с Accutase®, как описано выше, за исключением того, что клетки не пропускали через клеточный фильтр 40 мкМоль. Клетки в каждой одноразовой 1-литровой колбе затем подсчитывали, объединяли по мере необходимости и центрифугировали в течение 5 минут при 80-200 ОЦС. Затем супернатант удаляли настолько полно, насколько это возможно, не нарушая клеточный осадок. Холодный (< 4°C) CryoStor®10 затем добавляли по каплям таким образом, чтобы достичь конечной концентрации 150 миллионов клеток на мл, и раствор клеток держали на ледяной бане во время переноса в 1,8 мл флакон для криоконсервирования Corning (Corning; г. Корнинг, штат Нью-Йорк, США) или 15 мл пакет для криоконсервирования Miltenyi (Miltenyi Biotec Inc. г. Оберн, штат Калифорния, США).
Суспензия разросшихся клеток затем была заморожена в ампуле при высокой плотности в морозильнике с контролируемой скоростью следующим образом. Камеру предварительно охлаждали до 4°C и температуру поддерживали до тех пор, пока пробирка с образцом не достигала температуры 6°C. Затем температуру в камере понижали со скоростью 2°C/мин, пока пробирка с образцом не достигала температуры -7°C. После того, как пробирка с образцом достигла температуры -7°C, камеру охлаждали на 20°C/мин до температуры -45°C. После чего температуру в камере поднимали на 10°C/мин до тех пор, пока температура в камере не достигала -25°C, а затем дополнительно охлаждали камеру на 0,8°C/мин до тех пор, пока пробирка с образцом не достигала температуры -45°C. Температуру в камере затем снижали на 35°C/мин до тех пор, пока температура в камере не достигала -160°C. Температура в камере затем удерживалась на уровне -160°C в течение по крайней мере 10 минут, после чего пробирки переносили в хранилище с жидким азотом, в часть с газовой фазой.
Для высевания в биореактор с механическим перемешиванием, криоконсервированные клетки высокой плотности удаляли из хранилища с жидким азотом, размораживали и использовали для посева в замкнутом 0,2-литровом стеклянном биореакторе (DASGIP; г. Юлих, Германия). Пробирки для криоконсервации были извлечены из хранилища с жидким азотом, части с газовой фазой, и помещены непосредственно на водяную баню при 37°C на 105 секунд. Содержимое размороженных пробирок затем переносили с помощью 2 мл стеклянной пипетки в 50 мл коническую пробирку. Затем в пробирку по каплям добавляли 9 мл E8™, содержащей 0,5% BSA с добавлением 10 мкмоль/л ингибитора Rho-киназы, Y-27632. Затем клетки центрифугировали при 80-200 ОЦС в течение 5 минут. После этого супернатант отсасывали из пробирки и добавляли 10 мл свежей E8, содержащей 0,5% BSA и дополненной 10 мкмоль/л ингибитора Rho-киназы, Y-27632. Этот объем, содержащий клетки, пипеткой переносили во флаконы для переноса среды (Cap2V8®, SaniSure, г. Мурпарк, Калифорния, США) и содержимое флаконов затем нагнетали непосредственно в биореактор через стерильные сваренные C-flex трубки с помощью перистальтического насоса. При подготовке к посеву плюрипотентных стволовых клеток в биореактор, подготовили 0,15 л среды E8™ с добавлением 0,5% BSA, содержащего 10 мкмоль/л ингибитора Rho-киназы, Y-27632, подогретой до 37°С, перемешанной при 70 оборотах в минуту, отрегулированной до 6,8-7,1 рН с помощью СО2, с уровнем растворенного кислорода 30% (содержание СО2, воздуха, О2 и N2 регулируется). Сразу после посева в биореактор, из него отбирали пробы для подсчета клеток, и средний объем доводили по мере необходимости для получения конечной концентрации клеток 0,225×106 клеток/мл.
Клетки, в биореактор с механическим перемешиванием, сформировали кластеры в биореакторе с механическим перемешиванием, После посева, кластеры клеток поддерживали в реакторе в среде E8™ с добавлением 0,5% BSA в течение трех суток. Среду заменяли ежедневно; через 24 часа после посева удалили 90% от использованной среды и добавили 0,15 литра свежей среды. Через 48 после посева удалили 90% от использованной среды и добавили 0,15 л свежей среды. Через 72 часа после посева была инициирована дифференцировка плюрипотентных клеток путем удаления > 90% использованной среды и добавления среды для дифференцировки (таблица 18).
После начала процесс дифференцировки, клетки поддерживали в течение 12 или более суток в закрытой стерильной суспензии в биореакторе с регулируемой температурой (37°C), рН 7,4 (для дифференцировки) с установленным уровнем растворенного кислорода (10% DO на стадии 1 и 30% DO на прочих стадиях, содержание СО2, О2, N2 и воздуха регулируется). На протяжении всего процесса дифференцировки при каждой замене среды, лопастное колесо было остановлено на 5-20 минут до удаления среды через погружную трубку для стабилизации кластеров. Среду в биореакторе удаляли или добавляли из/в закрытого флакона или пакета с помощью перистальтического насоса через погруженную пробирку, соединенную с трубками C-Flex® с использованием сварщика трубок Terumo™ для поддержания замкнутой системы. Рабочее колесо и нагреватель были вновь запущены после добавления в сосуд достаточного количества среды, чтобы полностью погрузить колесо.
Для того чтобы контролировать процесс биореактора, образцы среды, содержащие кластеры клеток, забирались ежедневно для определения количество клеток и жизнеспособности (NucleoCounter®), как показано на Фиг. 45. В процессе наблюдался общий рост клеток, инокулят 0,225×106 жизнеспособных клеток/мл разросся до 0,65×106 жизнеспособных клеток/мл на стадии 4 сутки 3 (Фиг. 45).
В дополнение к ежедневным подсчетам, образцы среды биореактора были проанализированы NOVA BioProfile® FLEX (Nova Biomedical Corporation, г. Уолтем, штат Массачусетс, США). Было отмечено, что, относительно установленной кислотности реактора на начало стадии 0 (рН 6,8), рН среды в течение стадии 0 стал более кислым (рН 6,8) (Фиг.46). Изначальные настройки кислотности на стадии 0, казалось, уменьшали метаболическую активность клеток, при относительно низких уровнях молочной кислоты и высоком содержанием глюкозы, которые наблюдались в среде на стадии 0. Как только клетки начали дифференцировку во второй половине стадии 3, клетки поглотили практически всю глюкозу (Фиг.47) в среде и начали генерировать высокий уровень молочной кислоты (Фиг.48). Кроме того, в течение стадий 1 и 2 наблюдалась увеличивает плотность клеток (Фиг.45).
Для того чтобы определить, соответствуют ли стадиеспецифические изменения рН и метаболизма соответствует постадийным изменениям паттернов экспрессии mRNA, измеренным с помощью Qrt-PCR, было сделано следующее. Были использованы четыре массива Applied Biosystems Low Density (Life™, г. Карлсбад, штат Калифорния, США) обозначенные «плюрипотентность», «дефинитивная энтодерма (DE)», «кишечная трубка»(GT) или «4 стадия (S4)». Результаты представлены в виде кратных разниц относительно недифференцированных клеточных образцов UTCiPS, где недифференцированные клетки выступают в качестве контроля для стандартизации экспрессии всех пробегов и массивов.
С использованием этих массивов была определена экспрессия гена на каждой стадии дифференцировки. Именно тогда стало заметно, что посевной материал размороженных клеток в биореакторе показал недифференцированные паттерны экспрессии генов в стадии 0 сутки 1, 2, и 3 (24, 48 и 72 часа после посева биореактора: Фиг. 49 и 50). Эти результаты хорошо коррелируют с результатами проточной цитометрии, которые показали высокие уровни экспрессии CD9, SSEA4, TRA-1-60, TRA-и 1-81, а также отсутствие CXCR4/CD184 (Фиг.51). Результат проточной цитометрии и данные Qrt-PCR показал надежные и стабильные паттерны экспрессии генов плюрипотентности (CD9, NANOG, POU5F1, SOX2, TDGF и ZFP42) и отсутствие экспрессии генов, которые характерно экспрессируют в ходе дифференцировки (CD99, CDH2, CDX2, CER1, CXCR4, EOMES, FGF17, FGF4, Foxa2, GATA2, GATA4, GATA6, РКГ Hand2, HNF4α KIT, MNX1, MIXL1, PRDM1, PTHR1R, SOX17, SOX7, Т, TMPRSS2 и ФВ), характеризуя стабильное плюрипотентное состояние.
По завершении стадии 0 (72 часа после посева реактора), клетки переносили в среду для дифференцировки (таблица 18), содержащую MCX и GDF8. Через двадцать четыре часа после этой замены среды авторы отметили значительные изменения в структуре экспрессии генов (Фиг. 49 и 50, кратность увеличение экспрессии по сравнению с недифференцированным контролем), например, >10-кратное увеличение экспрессии FOXA2, HAND2, PRDM1, PTH1R и SOX17, > 100-кратное увеличение экспрессии CER1, FGF4, GATA4, GATA6, РКГ и MNX1 и > 1000-кратное увеличение экспрессии EOMES, FGF17, MIXL1 и T. Эти повышенные уровни экспрессии указывали на прохождение клетками мезодермальной стадии. Было также отмечено, что уровни CDX2 повысились на стадии 1 сутки 1 по сравнению с недифференцированными клетками (2700x увеличение экспрессии по сравнению с контролем), однако это было преходящее повышение уровня экспрессии и экспрессия CDX2 упала на 97% в течение стадии 1 сутки 3, до уровней, сопоставимых с наблюдаемым до индукции дифференцировки (фиг.49 и 50, кратное увеличение экспрессии по сравнению с недифференцированным контролем).
Через 72 часа после контакта со средой дифференцировки стадии 1, клетки экспрессировали профиль в соответствии с спецификацией к дефинитивной энтодерме, а уровни CXCR4 достигли ~ 400x увеличения по сравнению со справочными данными контроля, экспрессия Foxa2 достигла 136x увеличения по сравнению со справочными данными контроля и экспрессия SOX17 достигла 470 000x увеличения по сравнению со справочными данными контроля. В соответствии с характеристиками дефинитивной энтодермы, было также отмечено, что экспрессия генов CER1, EOMES, FGF4, GSC, MIXL1 и T в конце стадии 1 (сутки 3) снизилась с повышенных уровней, наблюдаемых на стадии 1 сутки 1 (Фиг. 49 и 50, кратность увеличения экспрессии по сравнению с недифференцированным контролем).
Эти изменения в экспрессии генов, наблюдаемые при Qrt-PCR, коррелируют с результатами, полученными с помощью проточной цитометрии. Наблюдался почти полный переход от CD9-экспрессирующей/CXCR4-отрицательной популяции плюрипотентных клеток при инициировании дифференцировки (Фиг. 51) к гомогенной популяции клеток, экспрессирующих CXCR4 (98,3% клеток CXCR4-положительны) в конце стадии 1 (Фиг. 52).
После завершения формирования дефинитивной энтодермы (стадия 1) среду заменяли другой, содержащей FGF7, морфогена, который используется, чтобы вызвать образование примитивный передней кишки. В соответствии с образованием примитивной кишки уровни экспрессии HNF4α и GATA6 на стадии 2 сутки 1 и 3 были увеличены, в то время как гены с высоким уровнем экспрессии на стадии 1 сутки 3 (CXCR4, EOMES, FGF17, FGF4, MNX1, PRDM1, SOX17 и VWF) показали снижение экспрессии к концу стадии 2 (Фиг. 50 и 53, кратность увеличения экспрессии по сравнению с недифференцированным контролем). Экспрессия генов передней кишки (AFP, HHEX, Pdx1 и Prox1) была увеличена (Фиг. 53, кратное увеличение экспрессии по сравнению с недифференцированным контролем).
После того как клетки были культивированы в среде во время стадии 2 в течение 72 часа, культура была перенесена в среду стадии 3 (таблица 18). После внесения в эту среду маркеры экспрессии клеток показали результаты, характерные для энтодертмы поджелудочной линии что было определено анализом экспрессии гена методом Qrt-PCR. Экспрессия Pdx1 увеличилась 60кратно, от 12 000x относительно контроля в конце стадии 2, 3 сутки и до 739 000x относительно контроля над в конце стадии 3, 3 сутки. Эти данные показывают, что клетки дифференцируются в панкреатические (Фиг.54). Это наблюдение также было подтверждено увеличенными уровни экспрессии, в сравнении с недифференцированным контролем, определенных генов, обычно выраженных в поджелудочной железе (ARX, GAST, GCG, INS, Isl1, NEUROD1, Ngn3, Nkx2.2, Nkx6.1, Pax4, PAX6, Ptf1a и SST ), что показано на Фиг. 54 и 55. Интересно, что не наблюдалась экспрессия OCT4/POU5F1 (37 образцов Cts проверено способом qRT-PCR), но наблюдались высокие уровни экспрессии прочих маркеров энтодермальных линий AFP, ALB, и CDX2. Это указывает на то, что популяции клеток в биореакторе дифференцировали из популяции плюрипотентных клеток сначала в клетки относительно пластичной кишечной трубки, а потом в панкреатические клетки (Фиг. 54 и 55).
В конце стадии 4 процесса дифференцировки клетки сохраняли высокий уровень экспрессии Pdx1 (95,6% Pdx1-положительны, по FACS, ~ 1000000кратное индуцирование по сравнению с контролем по данным Qrt-PCR) и Foxa2 (99,5% Foxa2-положительны по FACS). Клетки показали паттерны экспрессии, соответствующие таковым у панкреатических клеток-предшественников (39,2% Nkx6.1-положительных по FACS) и популяции панкреатических эндокринных клеток (9,4% PAX6-положительных, 12,4% хромагранин-положительных, 15,2% NKX2.2-положительных; все по FACS). Характерные для данной стадии паттерны экспрессии маркеров указывают на эффективную постадийную дифференцировку от популяции плюрипотентных клеток в клетки-предшественники панкреатических клеток. Эти результаты, наблюдаемые в проточной цитометрии, были подтверждены Qrt-PCR. Было также отмечено, что все гены, обычно экспрессирующиеся в поджелудочной железе (ARX, GAST, GCG, IAPP, INS, ISL1, MAFB, NEUROD1, NGN3, NKX2.2, NKX6.1, PAX4, PAX6, PTF1A и SST), показали повышенный уровень экспрессии на стадии 4 суток 3. (Фиг. 55). Для справки, на Фиг. 56 показана репрезентативная микрофотография (4x) клеточных кластеров в конце каждой стадии.
(3,64 г/л NaCO3)
(3,64 г/л NaCO3)
(3,64 г/л NaCO3)
(3,64 г/л NaCO3)
2,5 мМоль глюкозы
1:50 000 ITS-X
Glutamax 1:100
2,5 мМоль глюкозы
1:50 000 ITS-X
Glutamax 1:100
2,5 мМоль глюкозы
1:200 ITS-X
Glutamax 1:100
2,5 мМоль глюкозы
1:200 ITS-X
Glutamax 1:100
GDF8
100 нг/мл
50 нг/мл
50 нг/мл
MCX
[2 мкМоль]
SANT [0,25 мкМоль]
TPPB [100 нМоль]
Только сутки 1
LDN 100
TPPB [100 нМоль]
Все дни относительно 0 ч.
Сутки 1 и 2,
Без замены на сутки 3
Сутки 1 и 3,
Без замены на сутки 2
Сутки 1 и 2,
Без замены на сутки 3
(млн клеток / мл)
(млн клеток / мл)
Материалы:
• Клетки человеческой эмбриональной клеточной линии H1 (hES), (клетки WA01, WiCell, Madison WI)
• PBS (№ по каталогу 14190, Invitrogen)
• Y-27632 (Axxora № по каталогу ALX-270-333, San Diego, CA)
• EDTA, (Lonza, № по каталогу 17-7-11E)
• NucleoCounter®-(ChemoMetec A/S, № по каталогу YC-T100, г. Аллерод, Дания)
• Обработанные двумя нетканевыми культурами 6-луночные планшеты (Becton Dickinson, № по каталогу Falcon 351146, Franklin Lakes, NJ)
• Accutase®, (Sigma-Aldrich, № по каталогу A-6964, г. Сент-Луис, штат Миссури, США)
• Датчики биореактора для определения уровня pH и количества растворенного кислорода (DO) (FermProbe® рН электрод 225 мм, модель № F-635, датчик растворенного кислорода OxyProbe® 12 мм, номер модели D-145 от Broadley-James Corporation, г. Ирвин штат Калифорния, США)
• Устройство иммунозащитной макроинкапсуляции (TheraCyte™, г. Ирвин штат Калифорния, США)
• ИФА человеческого C-пептида (MERCODIA № по каталогу 10-1141-01)
• GlutaMAX™, MCDB131 и ITS-X Invitrogen
• FAF-BSA (Proliant)
• Ретиноевая кислота, глюкоза 45% (2,5M), SANT (ингибитор Shh) (Sigma)
• GDF8 (Peprotech)
• MCX
• FGF7 (R & D Systems)
• LDN-193189 (антагонист рецептора BMP) (Stemgent)
• TPPB (активатор PKC) (ChemPartner)
Пример 15
Дифференцировка эмбриональных стволовых клеток из клеточной линии WA01 в дефинитивную энтодерму: роль MCX/GDF8 в качестве регулятора клеточного цикла в суспензионной культуре
Кластеры плюрипотентных эмбриональных стволовых клеток человека из линии стволовых клеток H1 (NIH код: WA01) высевали по 0,5×106 клеток/мл в колбах Эрленмейера с перемешиванием в среду MCDB-131, содержащую 3,64 г/мл бикарбоната натрия и 5,5 мМоль глюкозы (№ по каталогу A13051 DJ, Invitrogen, CA), которая была дополнена 2% 2% BSA без жирных кислот (№ по каталогу 68700, Proliant, И. А.), 1X GlutaMAX™ (№ по каталогу 35050-079, Invitrogen, Калифорния), дополнительно 2,5 мМоль глюкозы (№ по каталогу G8769, Sigma) и ITS-х в исходной концентрации 1: 50 000 ( № по каталогу 51500056, Invitrogen, CA). Среда MCDB-131, дополненная таким образом, будут упоминаться как базовая среда стадии 1 или «Чистая» среда для целей данного примера. Ингибитор GSK3B, 14-проп-2-ен-1-ил-3,5,7,14,17,23,27-гептаазатетрацикло [19.3.1.1~2,6~.1~8,12~]гептакоза-1(25),2(27),3,5,8(26),9,11,21,23-нонен-16-он, заявка на патент США № 12/494,789; включенная в настоящий документ посредством ссылки в полном объеме, называется MCX.
Кластеры обрабатывали в первые сутки дифференцировки с одним из шести условий: (1) чистая, (2) 3 мкМоль MCX плюс 100 нг/мл GDF-8 (№ по каталогу 120-00, Peprotech), (3) 3 мкМоль только MCX, (4) 100 нг/мл только GDF-8, (5) 20 нг/мл WNT-3A (№ по каталогу 1324-WN-002, R&D Systems, штат Миннесота, США) плюс 100 нг/мл активина А (№ по каталогу 338-AC, R&D Systems, штат Миннесота, США) или (6) 20 нг/мл только WNT-3A.
Среда в каждом из условий была заменена через 24 и 48 ч после начала дифференцировки. В это время, у клеток по условиям 1, 2, 3 и 4 среда была заменена на свежую базовую среду стадии 1 с добавлением 100 нг/мл GDF8, а у клеток по условиям 5 и 6 среда была заменена на свежую базовую среду стадии 1 с добавлением 100 нг/мл активина A.
За час до начала дифференцировки и на 5, 23, 29, 47 или 71 час после начала дифференцировки (упоминается как «время 0»), образцы суспензии были перенесены на обработанные двумя нетканевыми культурами шестилуночные планшеты и инкубированы с EdU (Click-iT® EdU Kit, Life Technologies Corporation, г. Карлсбад, штат Калифорния, США) в течение одного часа. Инкубированные с EdU клетки затем оценивали с помощью проточной цитометрии на 0, 6, 24, 30, 48, или 72 ч после начала дифференцировки для измерения процента клеток на стадиях G0/G1, S, или G2/M клеточного цикла (фиг.81-87).
По данному протоколу наблюдались существенные различия в процентном отношении клеток на стадиях G0/G1, S, или G2/M клеточного цикла (Фиг. 82-87), и было отмечено, что клетки, обработанные MCX и MCX + GDF8, показали почти 40% уменьшение включений EdU по сравнению с другими четырьмя условиями обработки (Фиг. 81). Это уменьшение включений EDU сопровождалось увеличением на 38% количества G0/G1 клеток в образце, обработанном MCX + GDF8 и увеличением на 54% количества G0/G1 клеток в образце, обработанном только MCX. Эти изменения включений EDU и увеличение перехода к G0/G1 через 6 часов после начала дифференцировки не наблюдалось в клетках, обработанных GDF8, WNT3A, WNT-3A+ активин А или чистой средой. Скорее, клетки, обработанные GDF8, WNT-3A, WNT-3A + активин А или чистой средой, продемонстрировали минимальное снижение процента клеток с включениями EDU (в среднем 48,1%, SD ± 1,2) и в средне 13% -ное снижение количество клеток в G0/G1 через шесть часов после начала дифференцировки (стандартное отклонение, ± 5%), как показано на Фиг.81 и 82.
Подобные различия наблюдались позднее в процессе распределения между значениями G0/G1 для клеток, обработанных MCX или MCX + GDF8 по сравнению с другими условиями обработки. Через 30 часов после времени 0 обнаружили, что обработанные MCX или MCX + GDF8 клетки показали на 43-45% меньшее количество клеток в G0/G1, по сравнению с клетками, обработанными WNT-3A + активин А, GDF8, WNT-3A, или чистой средой. Этот разрыв между процентом G0/G1 клеток был не изменился через 48 ч после начала дифференцировки, на стадиях клеточного цикла G0/G1 наблюдалось 71,9-75,5% клеток, обработанных MCX или MCX + GDF8 , при соответственных значениях 48,5% для GDF8, 55,8 % для WNT3A, 57,7% для Wnt-3A + активин А и 49% для чистой среды. В дополнение к наблюдаемым различиям включений EDU и G0/G1 профилей, клетки, обработанные MCX или MCX + GDF8 показали в результате на 15-33% больше клеток в фазе S клеточного цикла через 30 и 48 часов после времени 0, по сравнению с данными по клеткам, обработанным WNT3a + активин, GDF8, WNT-3A или чистой средой(фиг.84 и 85).
Данные (экспрессии гена CD99, CD9, CDH1, CDH2, CDX2, CER1, CXCR4, FGF17, FGF4, Foxa2, GATA4, GATA6, РКГ, KIT, MIXL1, MNX1, NANOG, Otx2, POU5F1, SOX17, SOX7 и Т показаны на Фиг. 57-80 и 88a-f) показали, что в суспензионной культуре, добавление MCX с или без вещества семейства TGF-β, GDF8, в первые сутки дифференцировки вызывает генерирование дефинитивной энтодермы, сравнимое с таковой при обработке клеток 20 нг/мл WNT-3A +100 нг/мл активина А на первые сутки по результатам измерения экспрессии генов в конце формирования дефинитивной энтодермы. Тем не менее, в соответствии с различиями в клеточного цикла, наблюдаемыми в процессе формирования дефинитивной энтодермы, были замечены промежуточные различия в экспрессии генов. В образцах, обработанных MCX или MCX + GDF8, гены Т (Brachyury), GATA4 и CDX2 индуцировались на уровне, существенно более высоком, чем у клеток, обработанных Wnt-3A + активин А или трех других протестированных условий в течение первых 24 часов дифференцировки ( Фигуры 88 B, C, и D). С другой стороны, экспрессия генов плюрипотентности (NANOG и POU5F1/OCT4) значительно уменьшилась через 24 часа в образцах, обработанных MCX или MCX + GDF8, по сравнению с исходной клеточной популяцией или другими четырьмя тестируемыми условиями (фиг.88E). Величина индукции экспрессии генов, таких как FGF4, Foxa2 и SOX17, была значительно ниже в образцах MCX или MCX + GDF8, по сравнению с другими четырьмя тестируемыми условиями, в течение 24 часов после начала дифференцировки, однако через 48 часов экспрессия FGF4, Foxa2 и SOX17 у всех образцов была на сопоставимых уровнях. (Фиг. 88c и e).
Пример 16
Генерирование эктодермальной и мезодермальной ткани с использованием процесса дифференцировки в масштабируемой суспензии.
Этот пример демонстрирует процесс, во время которого происходит как рост, так и дифференциация плюрипотентных стволовых клеток (PSC) и тем самым достигается масштабируемый производственный процесс генерирования эктодермальной или мезодермальной ткани.
Две клеточные линии были выращены в суспензии для обеспечения посевного материала для этих исследований: суб-клон H1 (WA01) клеточной линии hES WB0106 и линия индуцированных плюрипотентных стволовых клеток (iPSC), сгенерированная из клеток ткани пуповины (UTC). Как описано в предыдущих примерах, выращенные в суспензии клетки замораживали при высокой плотности в морозильной камере с контролируемой скоростью, затем размораживали для высевания в закрытом 3-литровом стеклянном биореакторе (DASGIP; г. Юлих, Германия) или одноразовом 3-литровом биореакторе (Mobius®, EMD Millipore Corporation, г. Биллерика, штат Массачусетс, США) в конечной концентрации клеток 0,225×106 клеток/мл. Клетки, которые высевали в биореактор с механическим перемешиванием, образовывали кластеры клеток в резервуаре непрерывного перемешивания и выдерживали в среде плюрипотентности (E8™ с добавлением 0,5% BSA) в реакторе в течение трех суток. Через 72 часа после посева была инициирована дифференцировка плюрипотентных клеток путем перенесения клеточных кластеров в пластиковые одноразовые колбы Эрленмейера (125 мл колбы PETG, № по каталогу 4112, Thermo Scientific Рочестер Нью-Йорк) в соответствующую среду для дифференцировки (таблица 19), для формирования мезодермы/сердечной ткани ( 1) или эктодермы/нервной ткани (2).
После начала процесса дифференцировки клетки поддерживали в течение десяти (10) суток при 100 оборотах в минуту в увлажненной инкубаторе с 5% СО2 на движущейся платформе (MAXQ 416hp, Thermo Scientific, Рочестер Нью-Йорк). На сутки 1, сутки 3, сутки 5 и сутки 7 после начала дифференцировки, среды в колбе заменяли на свежие среде, как показано в таблице 19. До начала дифференцировки образцы были подвергнуты анализу qRT-PCR для получения эталонных величин, а затем процедуру повторяли на 3, 5, 7 и 10 сутки после начала дифференцировки.
Для проверки того, возможно ли определить наличие эктодермальных или мезодермальных специфических изменений в паттернах экспрессии мРНК с помощью QRT-PCR, были использованы три массива Applied Biosystems Low Density (Life™, Карлсбад, Калифорния), обозначенные «Плюрипотентность», «Дефинитивная энтодерма (DE)» и «стадия 6 (S6)», и результаты сравнивали с данными образцов соответствующих недифференцированных плюрипотентных стволовых клеток в качестве контроля для стандартизации экспрессии.
С помощью этих массивов определяются паттерны экспрессии гена плюрипотентных клеток, культивируемых в эктодермальной (фиг.89) или мезодермальной (фиг.90) среде дифференцировки. Было отмечено, что клетки, дифференцированные в перемешиваемых колбах, обработанные по любому из условий, демонстрируют снижение экспрессии генов плюрипотентности, таких как NANOG, POU5F1/OCT4, TDGF1 и ZFP42, по мере роста культуры с 3 суток по 10 сутки, что было измерено на массиве плюрипотентности. Экспрессия CXCR4 повысилась в образцах клеток HES или плюрипотентных клеток, дифференцированных в эктодерму или мезодерму. Эти результаты коррелируют с данными Qrt-PCR, показывающими высокую экспрессию генов, характерных для дифференцировки. Клетки, обработанные средой для эктодермальной дифференцировки, показали повышенный уровень экспрессии ARX, NeuroD, Nkx6.1, PAX6 (> 100 раз), и ZIC1 (> 1000 раз) по данным Qrt-PCR с 3 по10 сутки после начала дифференцировки (Фиг. 91). Эти данные были подтверждены FACS массива, который показал, что на третьи (3) сутки после начала дифференцирования в клетки эктодерм, клетки IPSC и hES поддерживали высокий уровень экспрессии SOX2 (гена плюрипотентности и нейронных стволовых клеток), но лишились экспрессии POU5F1/OCT4 (генов плюрипотентности), но при этом проявилась экспрессия PAX6 (ген нервной и эндокринной дифференцировки) (Фиг.92).
Схожая кинетика дифференцировки наблюдалась в клетках, обработанных средой для мезодермальной дифференцировки. В то время как экспрессия плюрипотентных генов снизилась в течение 10-дневной дифференцировки (Фиг. 90), на 3 сутки наблюдалась ранняя индукция генов, характерных для ранней переходной стадии дифференцировки мезодерму (CER1, EOMES, CKIT и VWF), уровень экспрессии данных генов на десятые сутки опустился практически до исходного (Фиг. 93). Было также отмечено, что экспрессия генов, характерных для мезодермы на 3, 5, 7 и 10 сутки после начала дифференцировки показала раннюю высокую экспрессию генов (CDH2, CDX2, GATA6, HNF4α, MNX1, PRDM1 и SOX17 на Фиг. 93). Та же картина индукции экспрессии генов наблюдалась в обоих образцах клеток IPS и HES, указывая на то, что процесс дифференцировки был направленным, а не спонтанным по своей природе.
Эти изменения в экспрессии генов, наблюдаемые с помощью, Qrt-PCR коррелируют с результатами, полученными с помощью фазоконтрастной микроскопии и иммунного окрашивания кластеров, подвергнутых криосекции. На 10 сутки в мезодермальной дифференцированной суспензионной культуре, примерно 1 из 10 кластеров начал спонтанно «сокращаться», что позволило предположить, что клетки дифференцировались в миокардиальную ткань (фиг.94, левая панель, 10 сутки, белые полосы). Окрашенное поперечное сечение некоторых кластеров показало слоистую непрерывную картину с окрашенным β-тубулином,что свидетельствует о формировании мышечной ткани (Фиг. 94, справа).
Разительно отличается морфологическая картина, наблюдаемая для кластеров, дифференцированных в эктодермальные клетки (Фиг. 95, слева) по сравнению с кластерами, дифференцированными в мезодермальные клетки (Фиг.94). Кластеры во всем объеме эктодермальной дифференцировки были крупнее и плотнее, чем клетки, дифференцированные в мезодермальные клетки, и у клеток, дифференцированных в эктодермальные клетки, экспрессия β-тубулина менее выражена. Клетки, показывающие экспрессию β-тубулина, показали более дендритную картину при окрашивании (Фиг. 95, правая панель, белые стрелки), характерную для нейронов.
Эти результаты в сочетании с данными Qrt-PCR и FACS, показывают, что клетки депонированные в банк и выращенные в суспензии, могут быть дифференцированы в суспензионной культуре в мезодермальные или эктодермальные клетки направленным и воспроизводимым образом.
Сутки/Дата
Сутки 0-4
Сутки 5-10
Сутки 0-6
Сутки 7-10
(2,5 г/л NaCO3 final)
(2,5 г/л NaCO3 final)
(2,5 г/л NaCO3 final)
(2,5 г/л NaCO3)
2,5 мМоль глюкозы
Glutamax 1:100
1:100 ITS-X
2,5 мМоль глюкозы
Glutamax 1:100
1:100 ITS-X или
1X B-27
2,5 мМоль глюкозы
Glutamax 1:100
2,5 мМоль глюкозы
Glutamax 1:100
1X B-27
ALKVi [7,5 мкМоль]
MCX [2 мкМоль]
Только сутки 3 и 4:
IWP-4 [8 мкМоль]
Каждые сутки отсчитываются от времени начала
Сутки 0, 1 и 3
Сутки 5 и 7
Сутки 0, 1, 3 и 5
Сутки 7
В то время как изобретение было описано и проиллюстрировано со ссылкой на различные конкретные материалы, процедур и примеров, следует понимать, что изобретение не ограничено конкретными комбинациями материала и процедур, выбранных для этой цели. Многочисленные изменения, касающиеся таких деталей изобретения, будут очевидными для специалистов в данной области техники. Подразумевается, что все описания и примеры представлены только в качестве иллюстрации и находятся в пределах сущности и объема настоящего изобретения, что отражено в нижеприведенной формуле изобретения. Все ссылки, патенты и заявки на патенты, приведенные в этой заявке, полностью включены в этот документ путем отсылки.

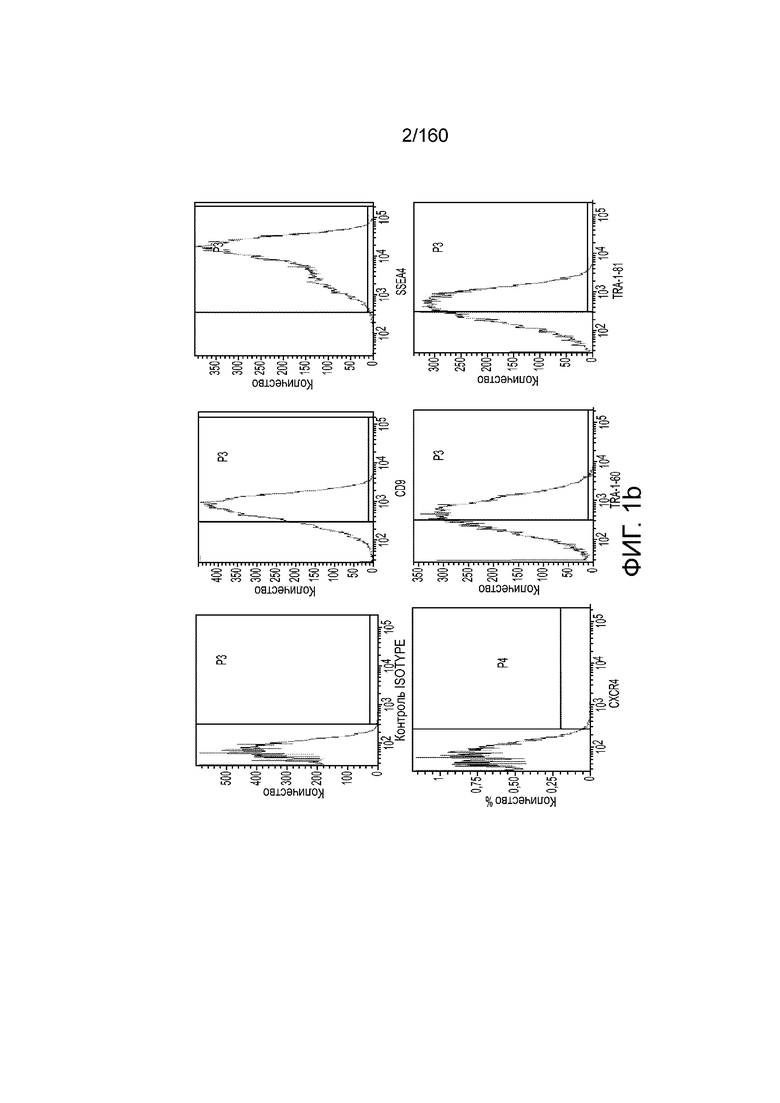

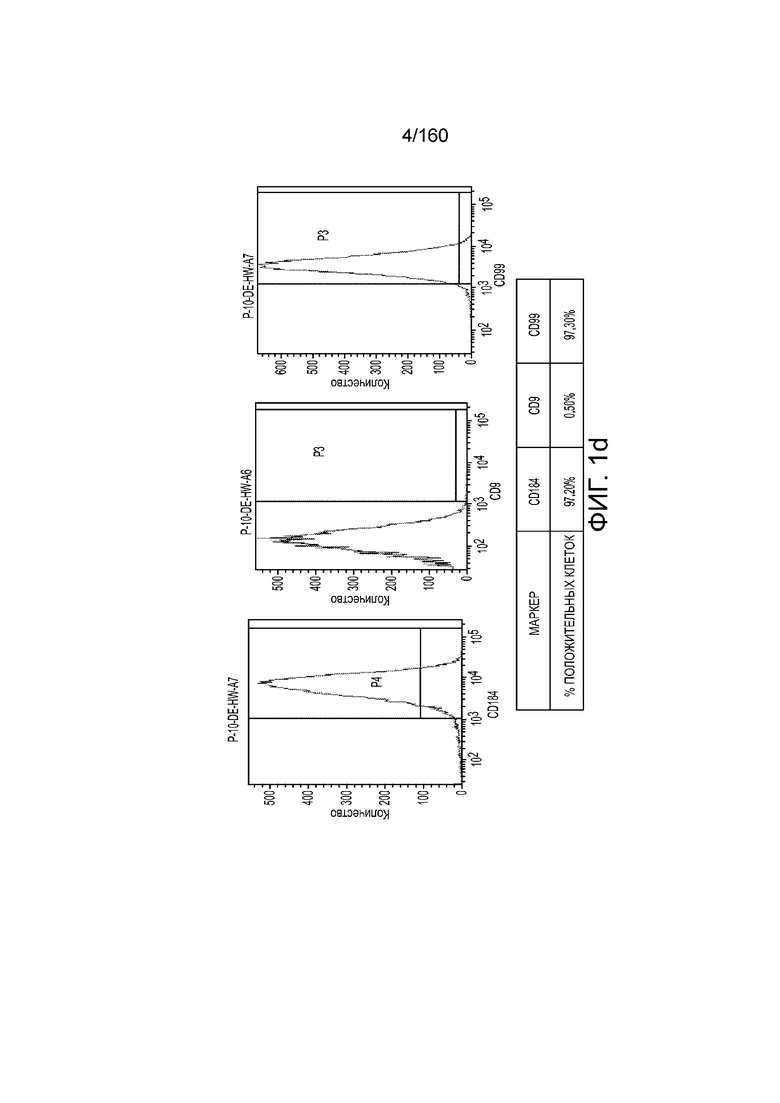


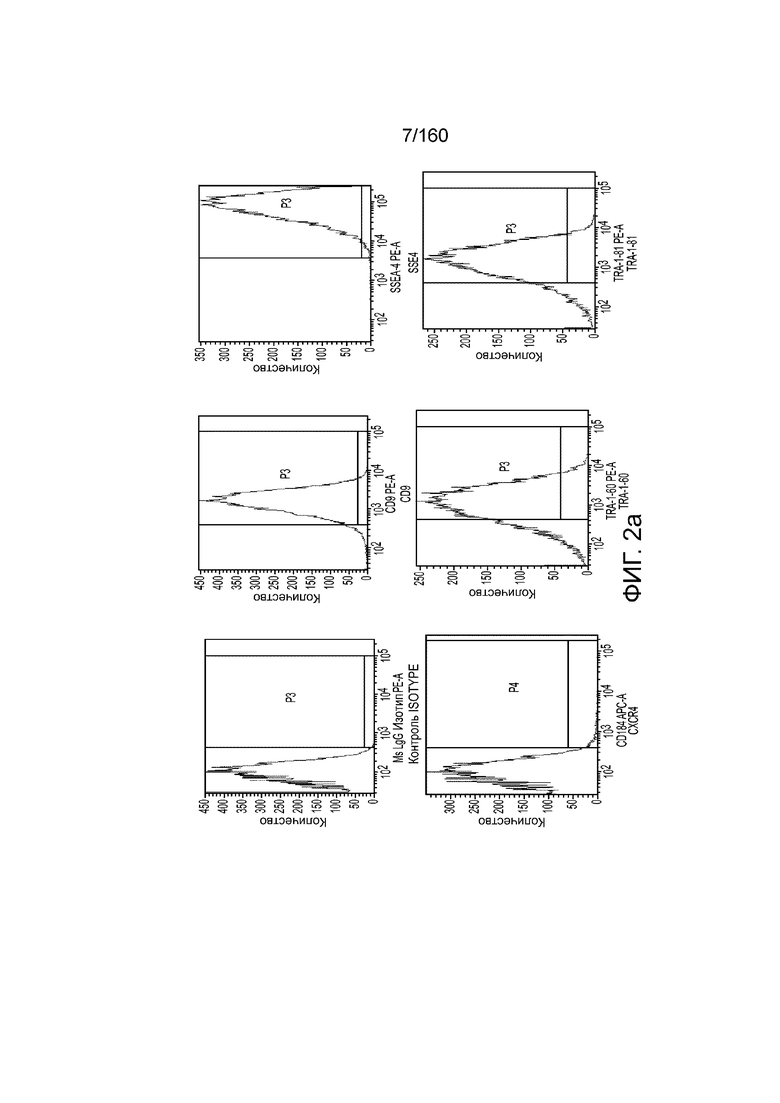

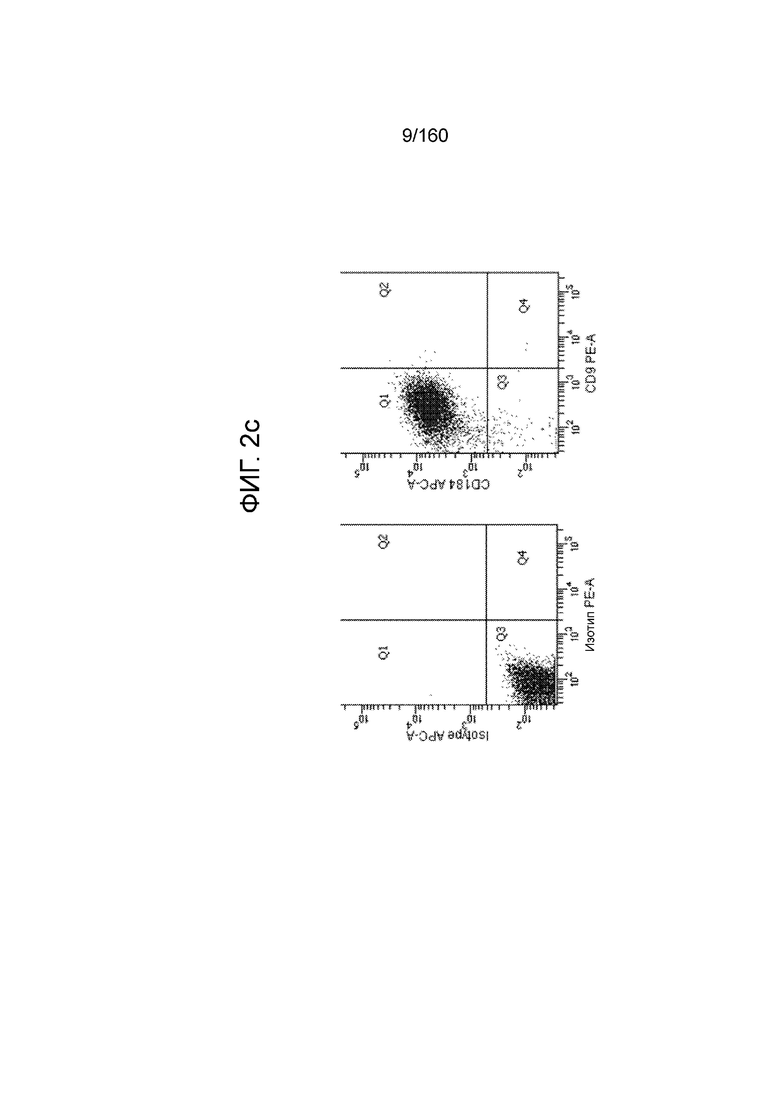
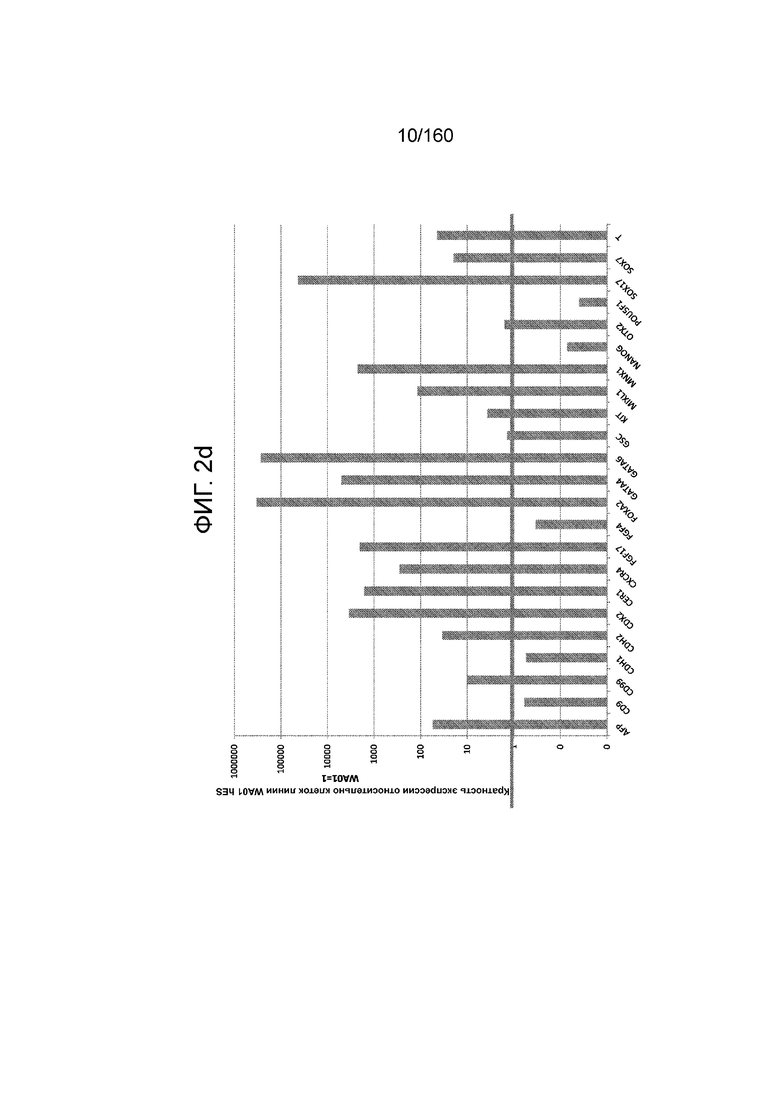














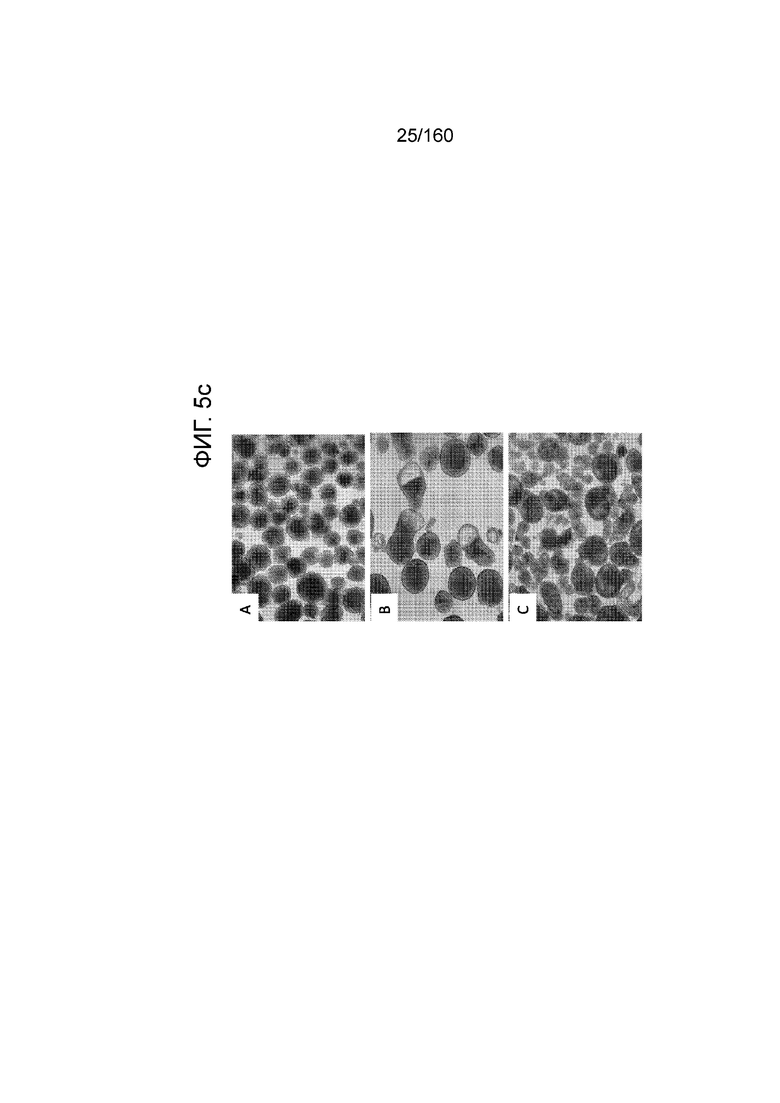





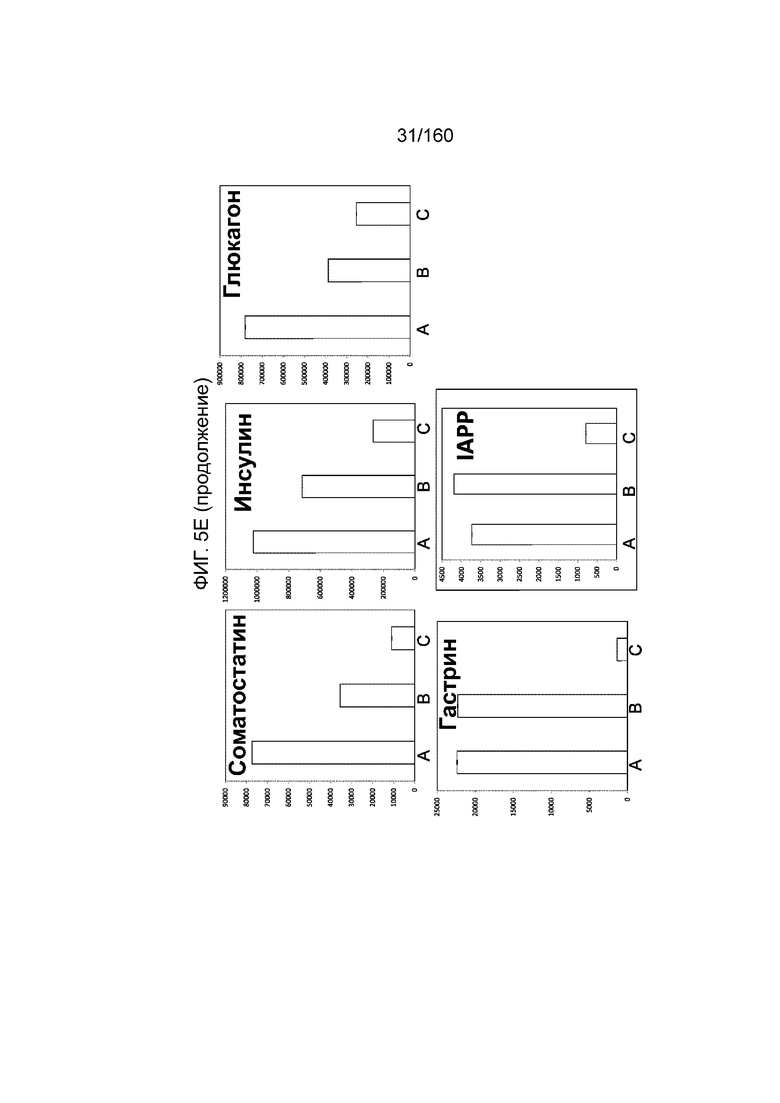

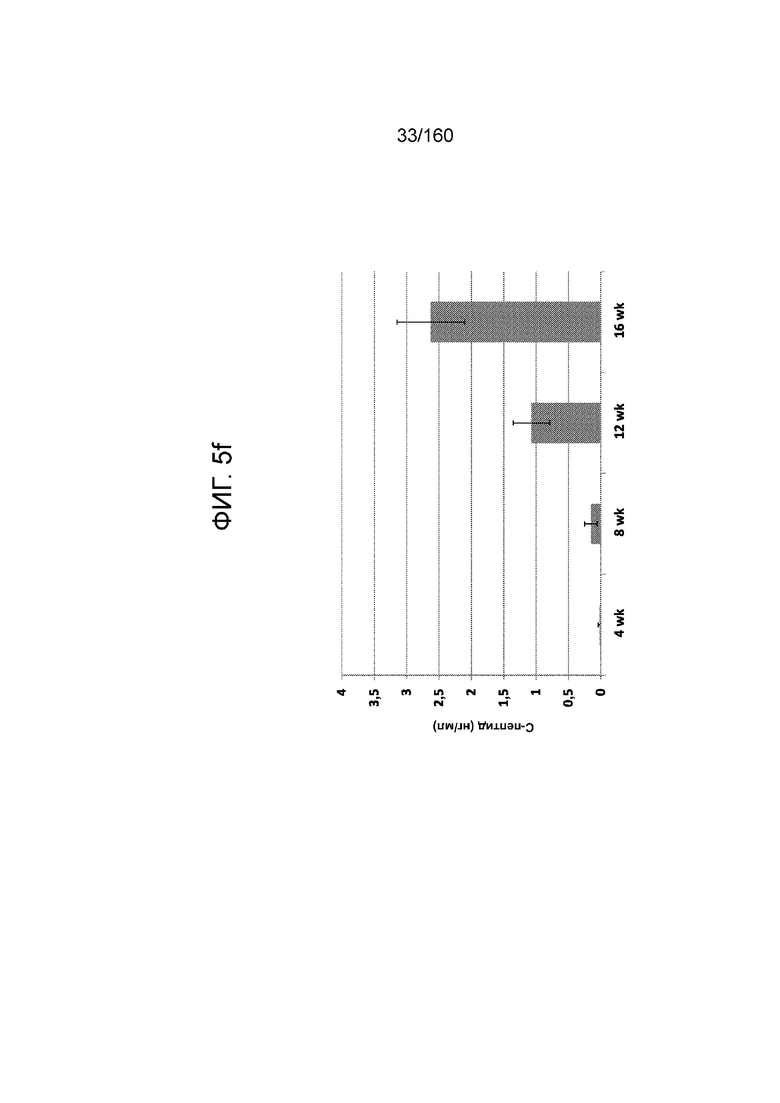



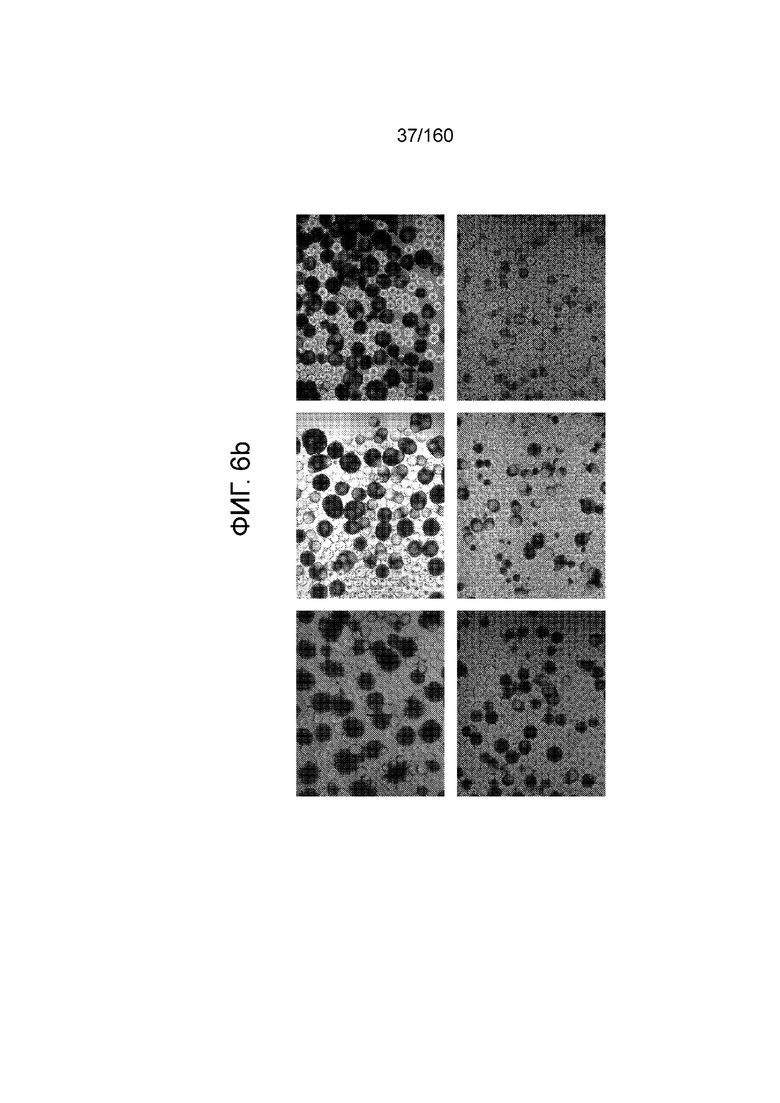


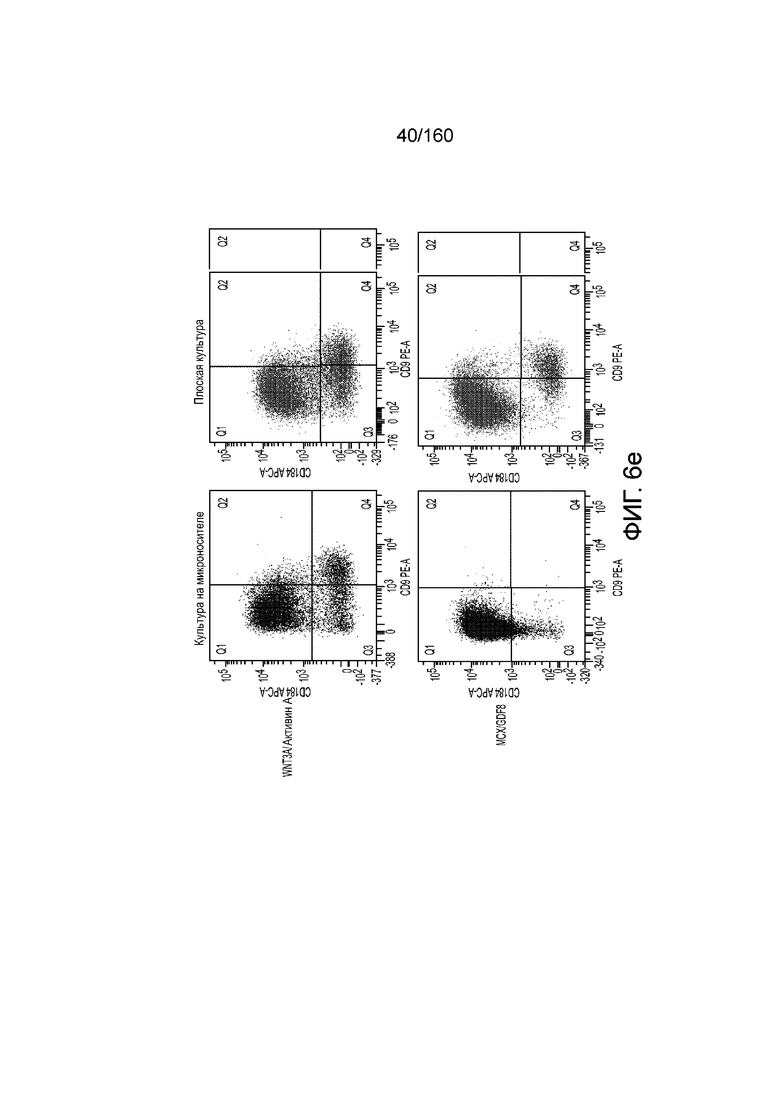

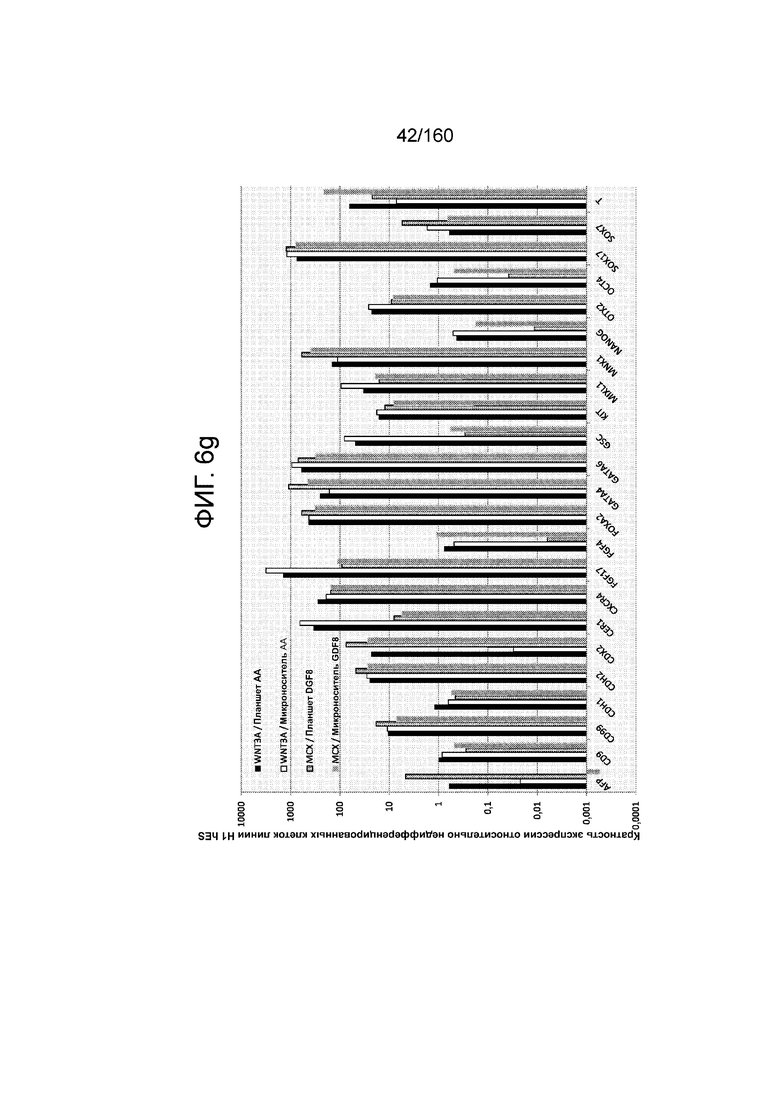
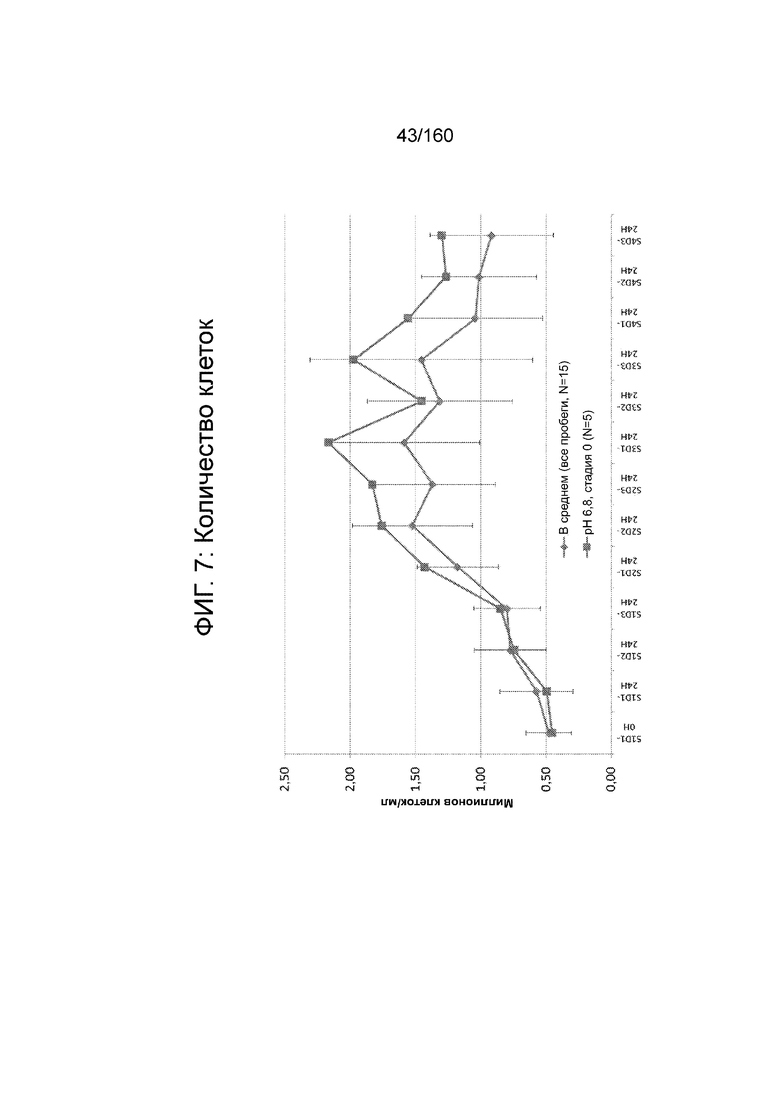


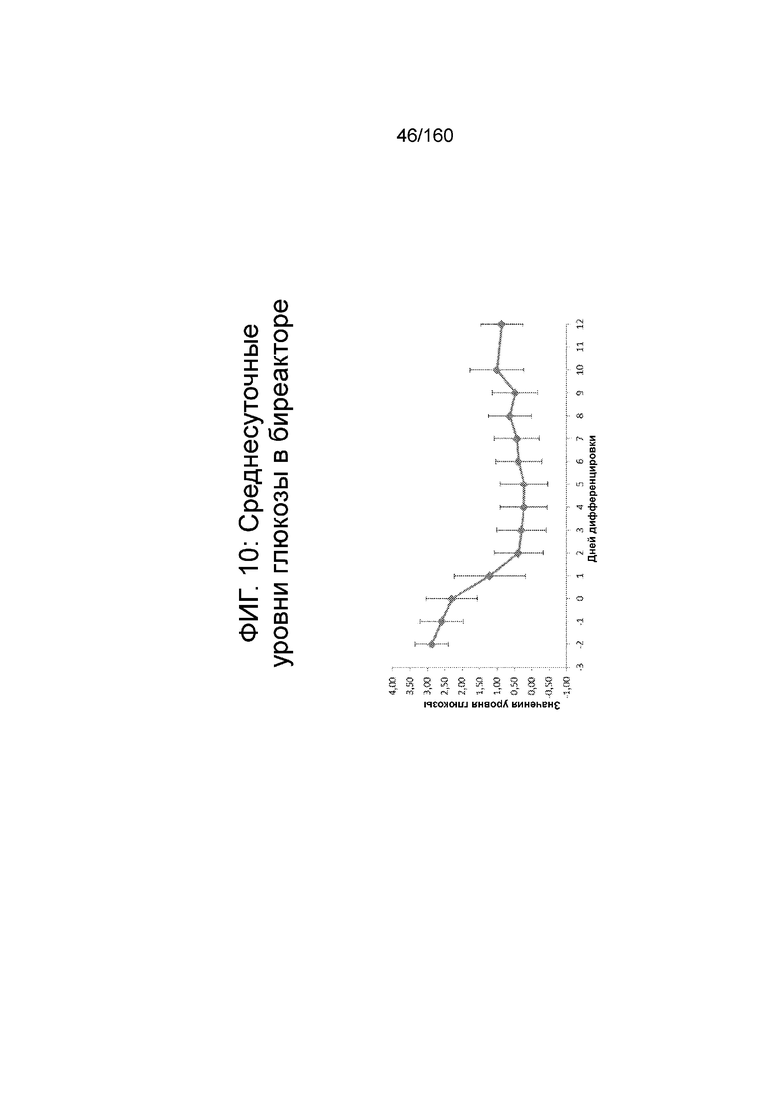
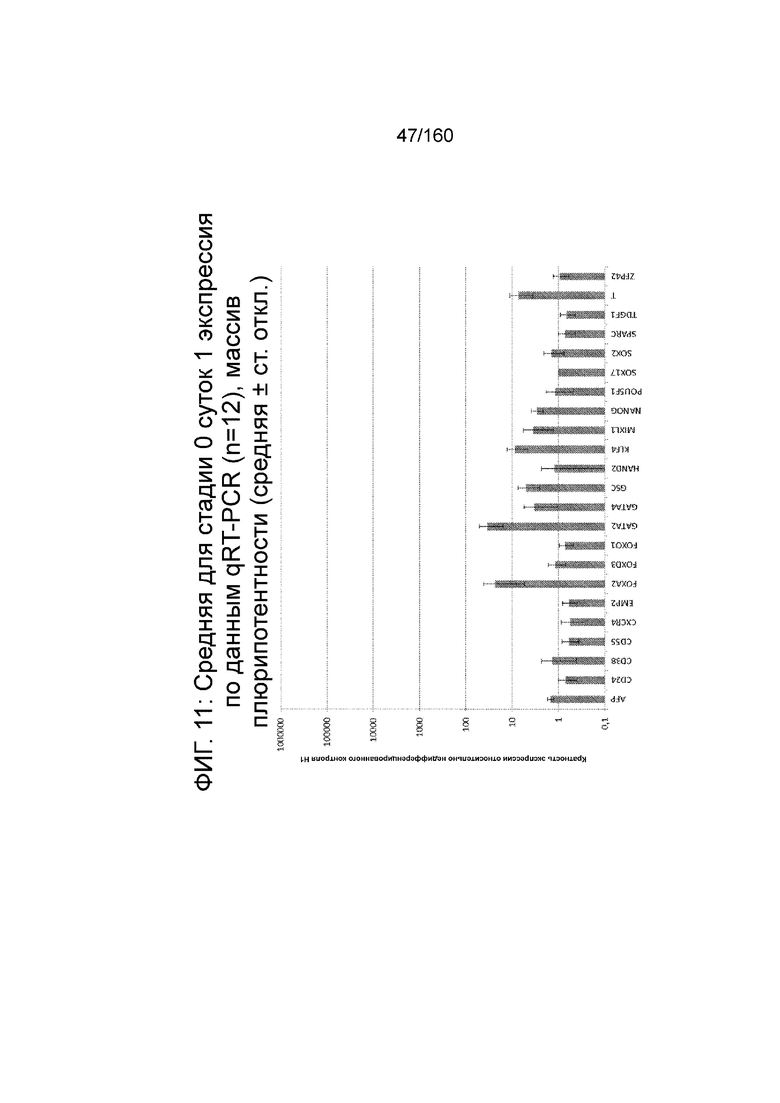
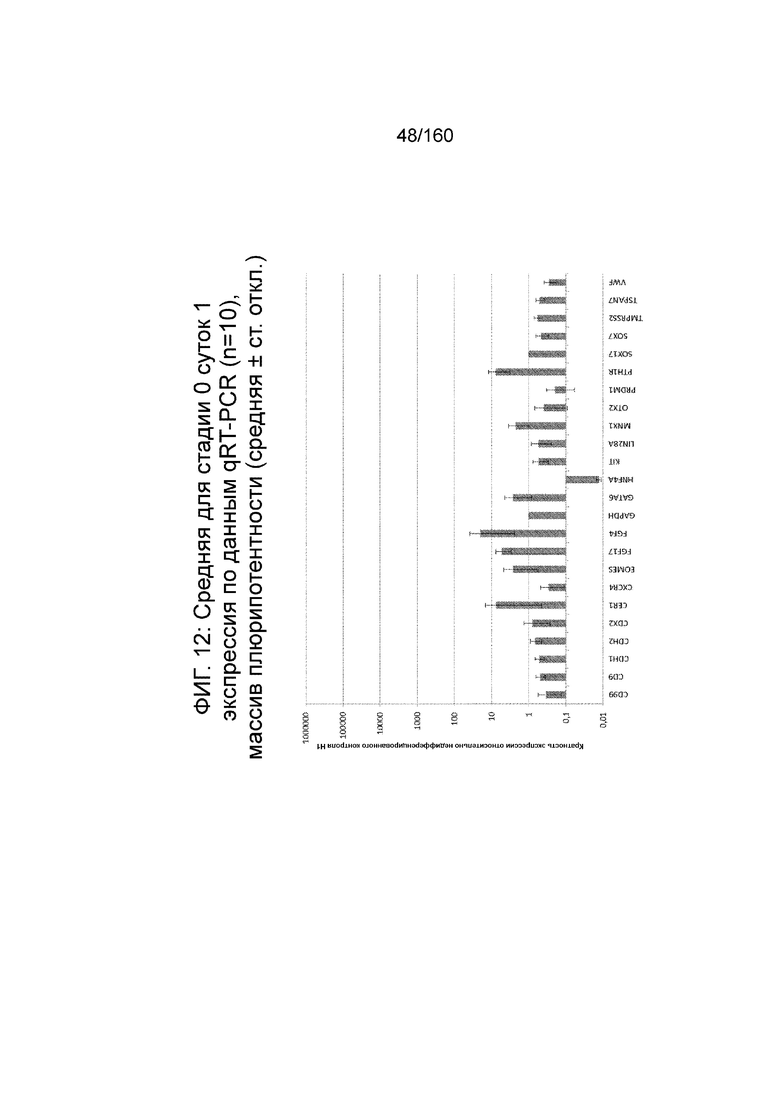
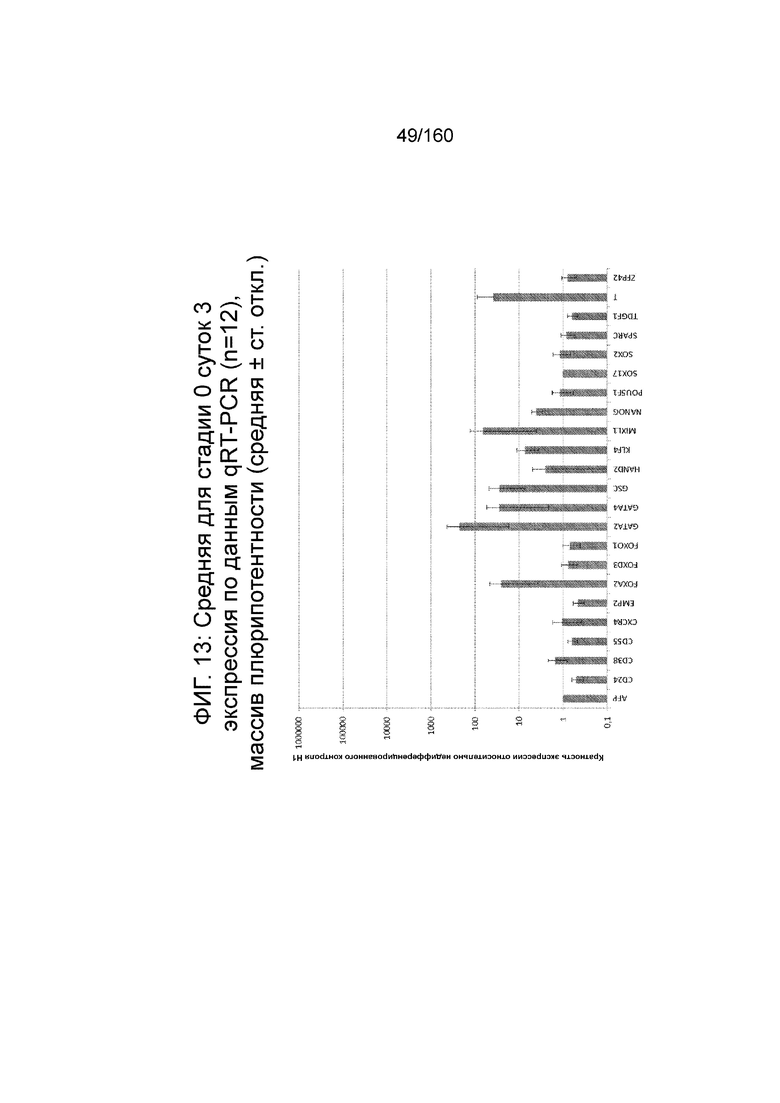
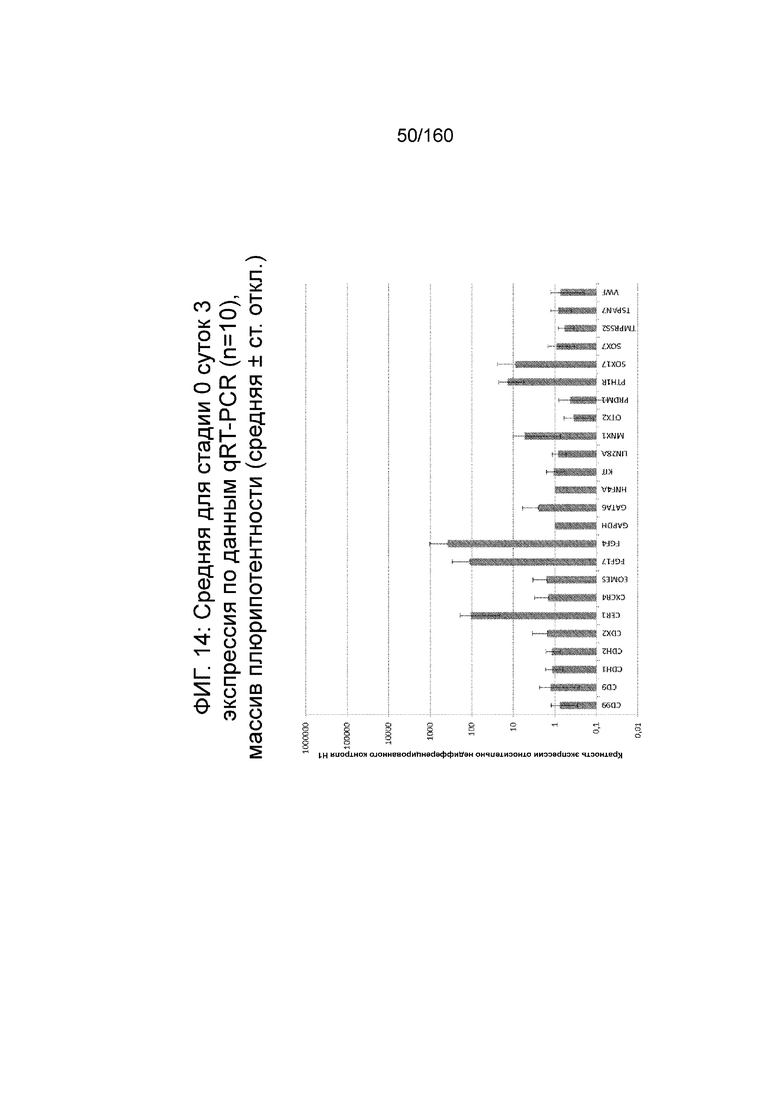


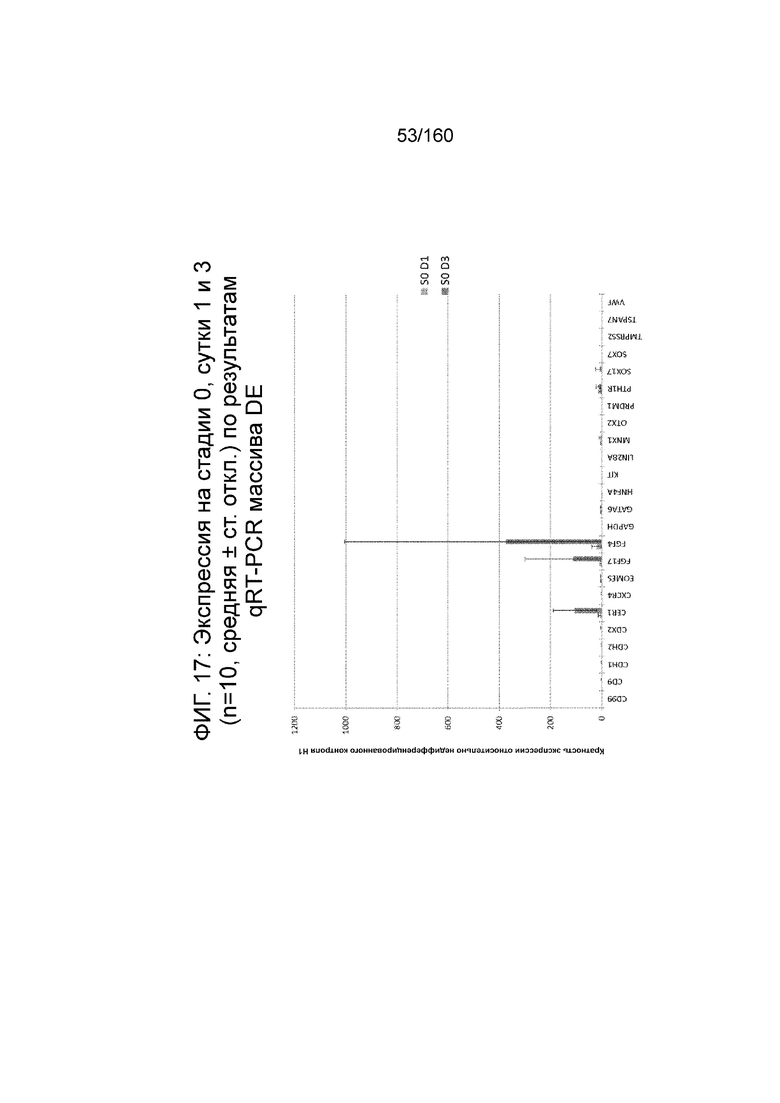

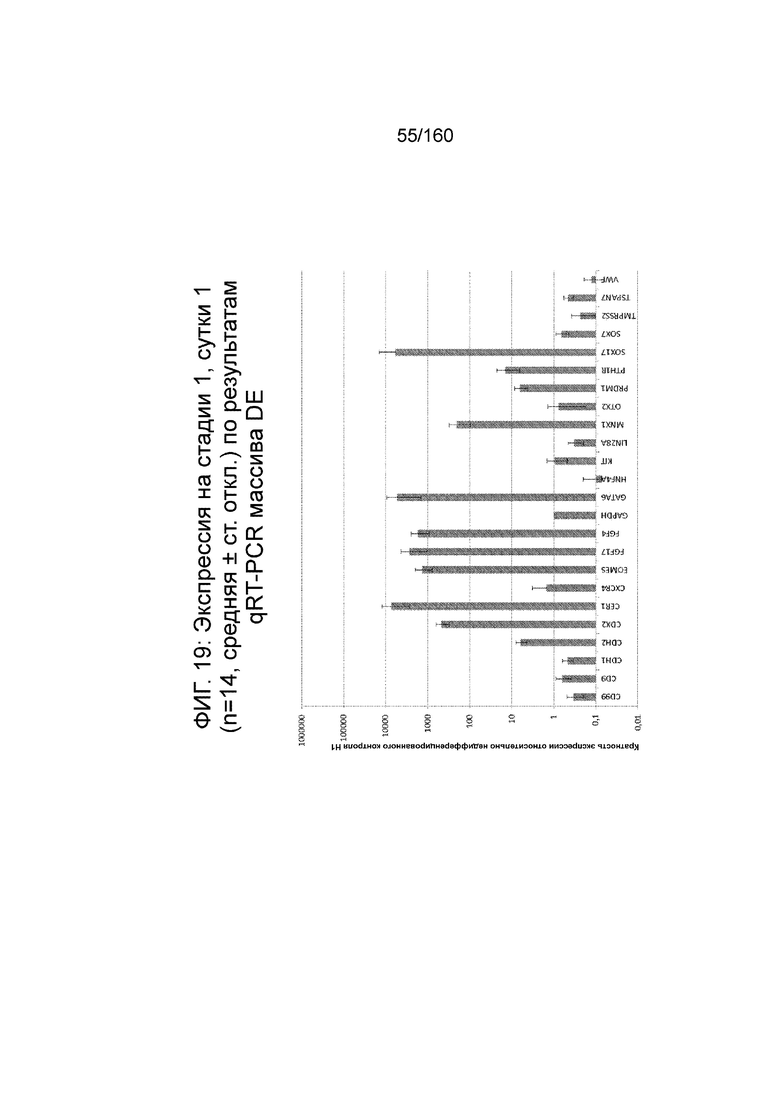



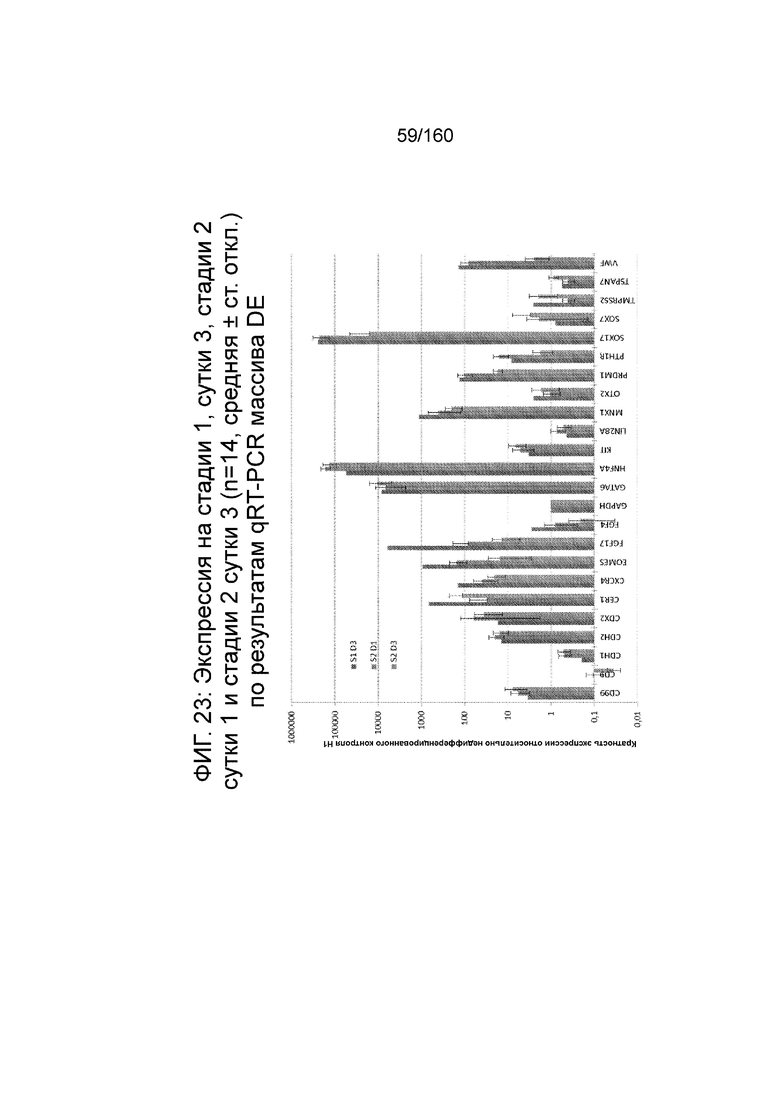
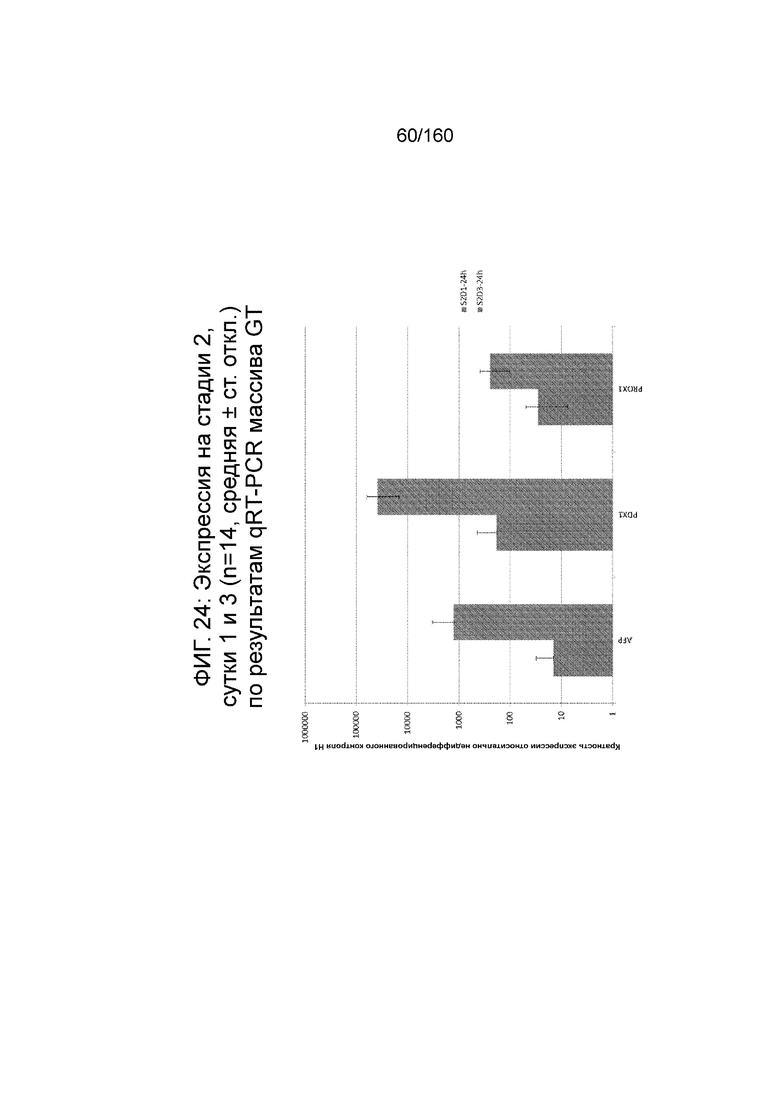
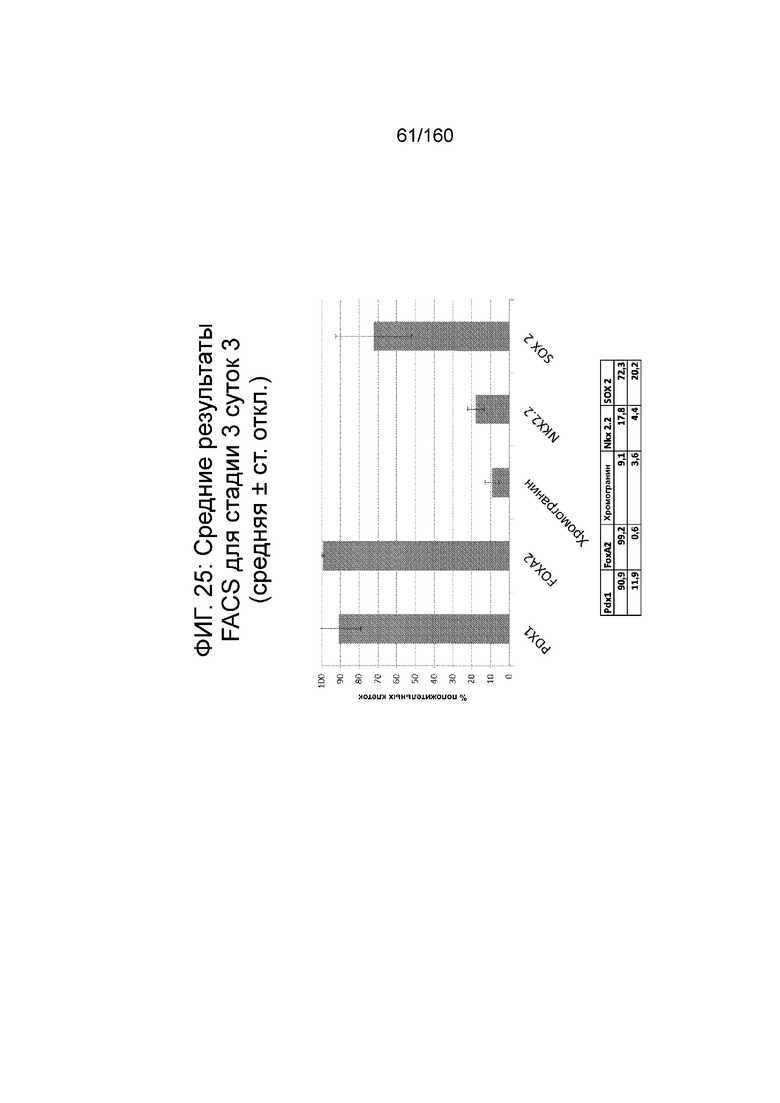



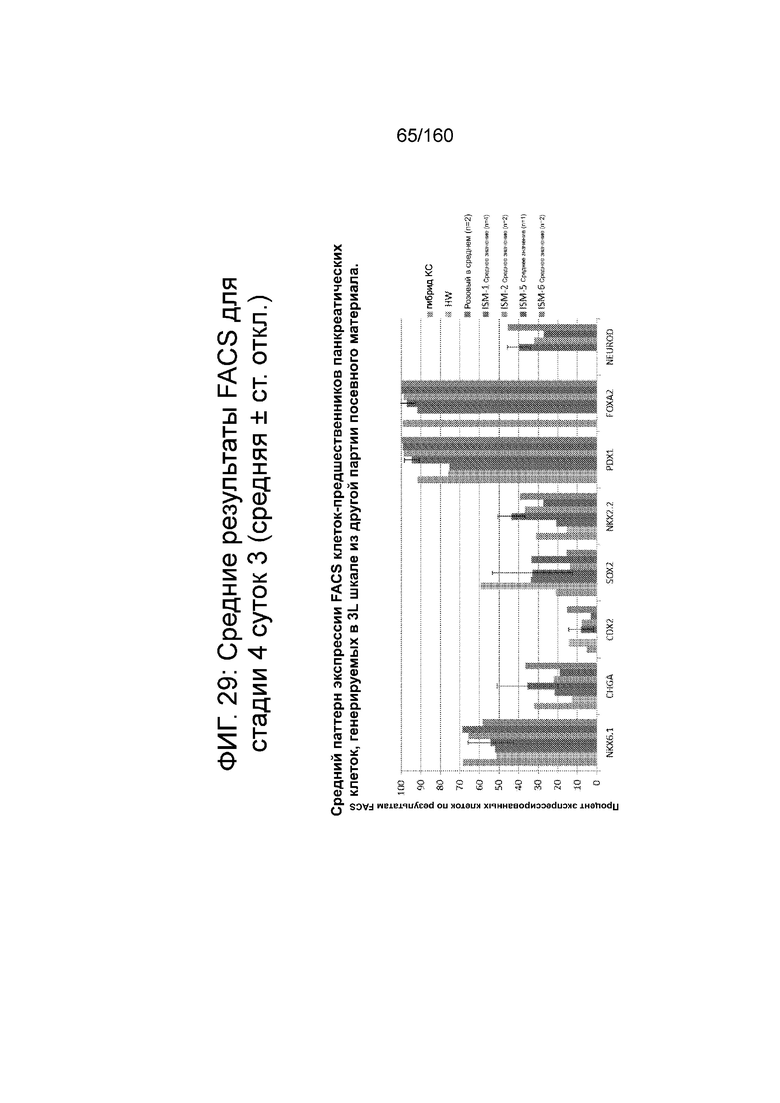

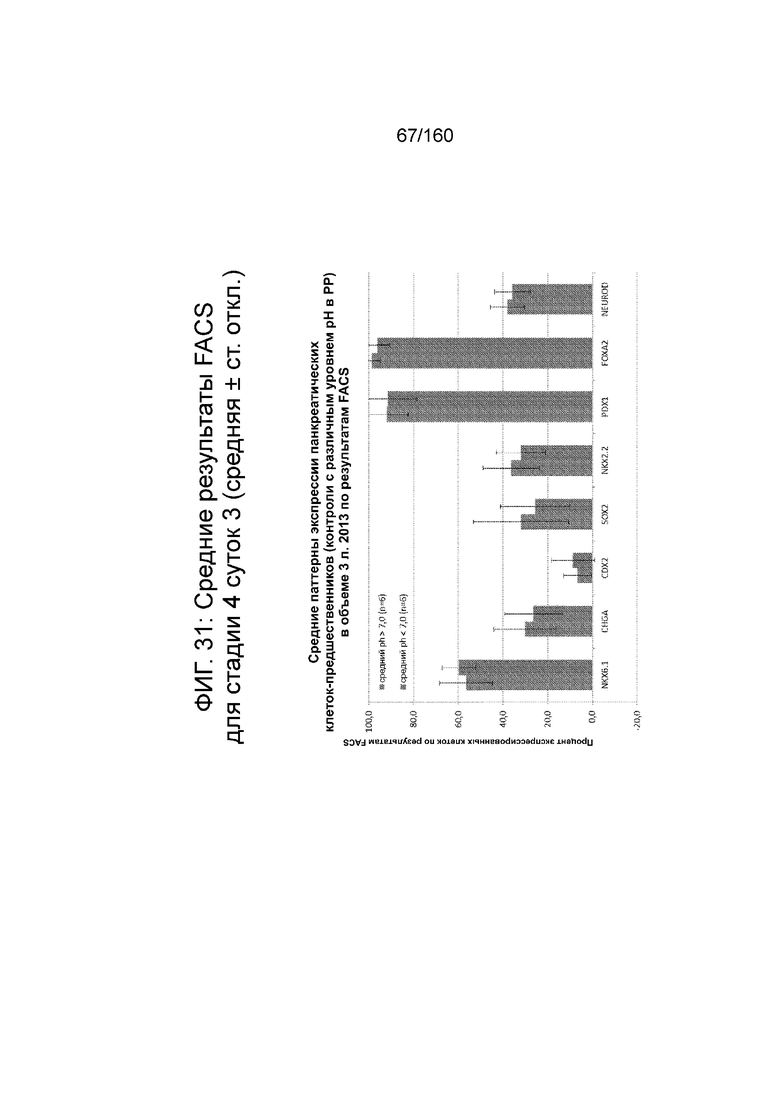


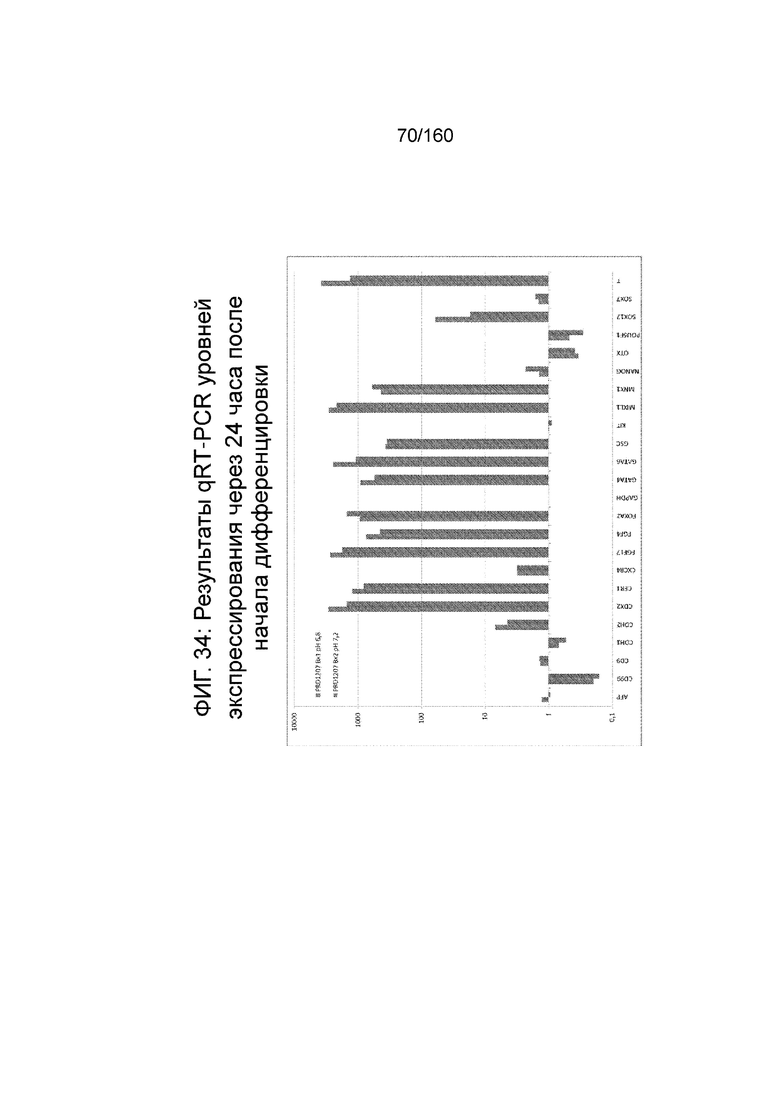








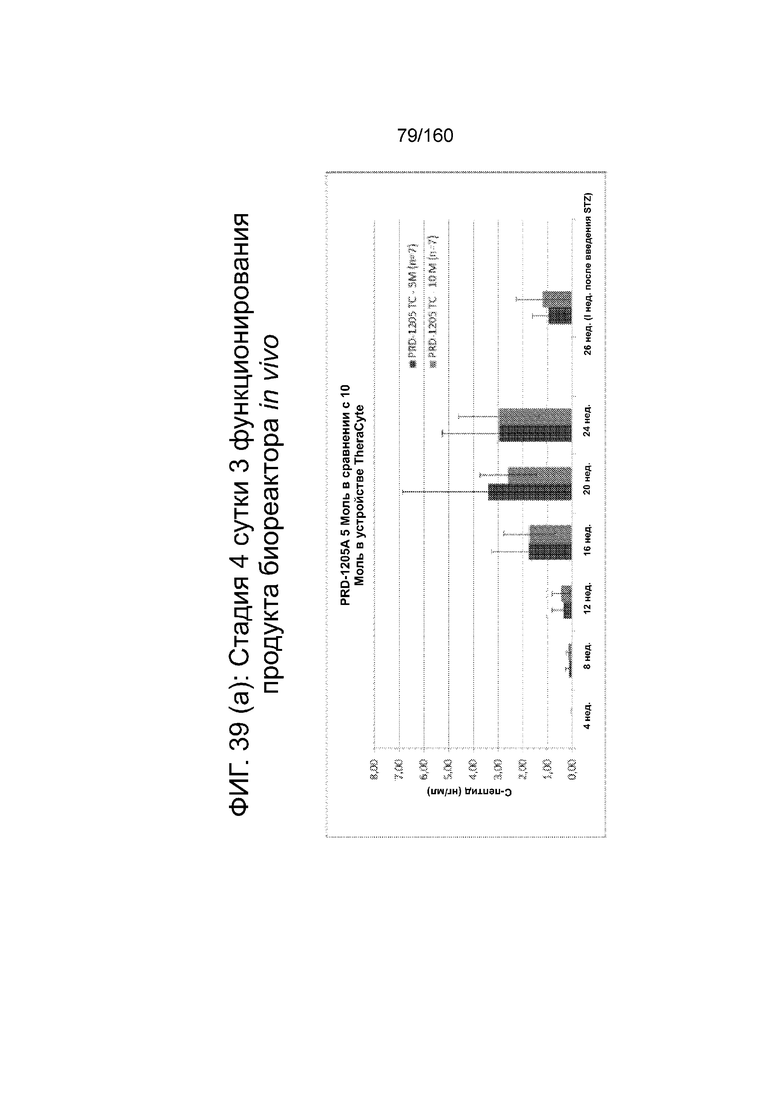
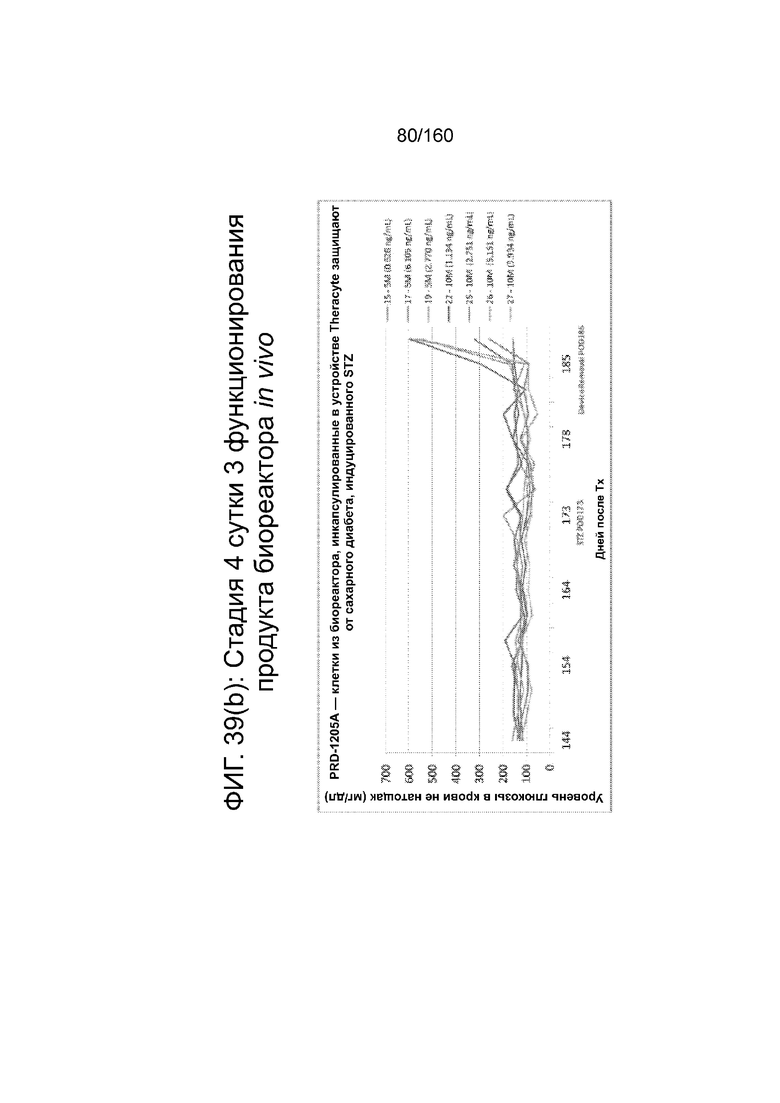


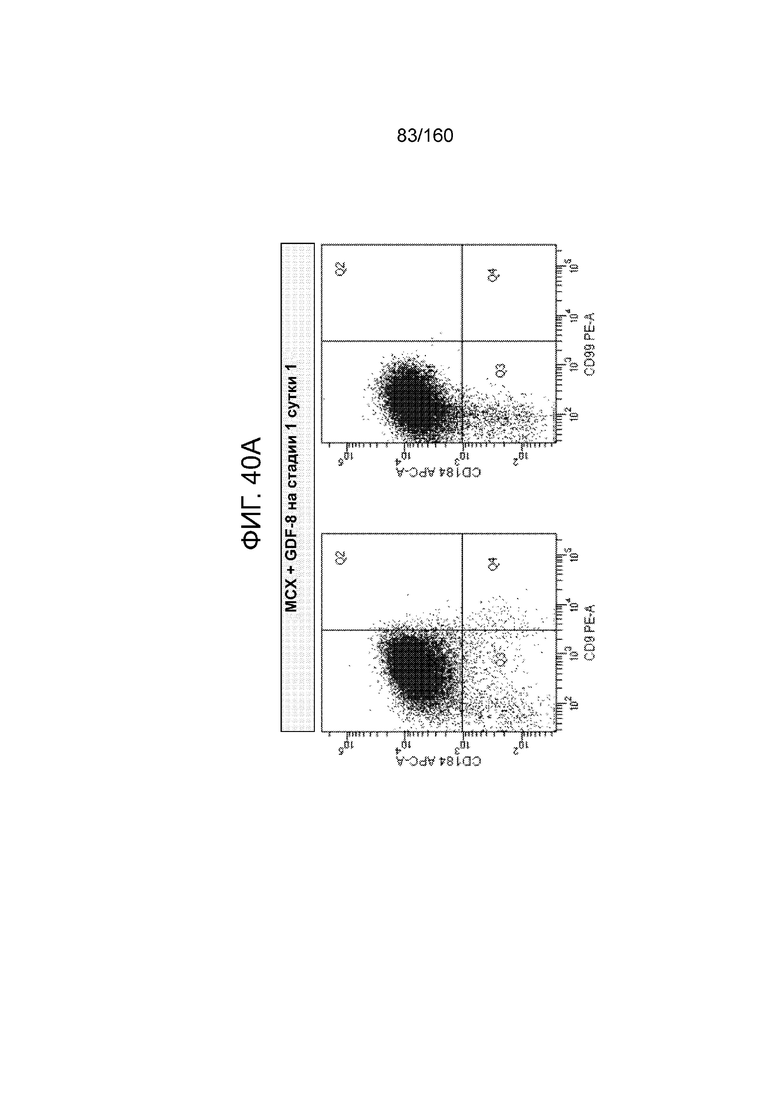

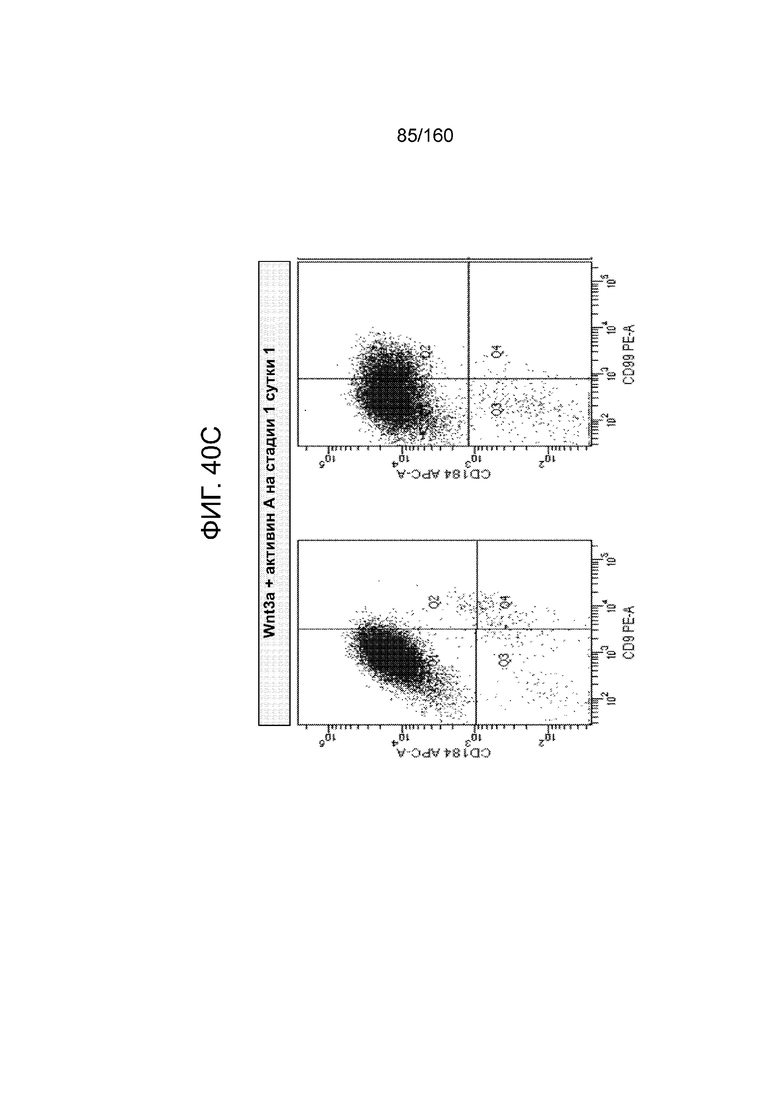





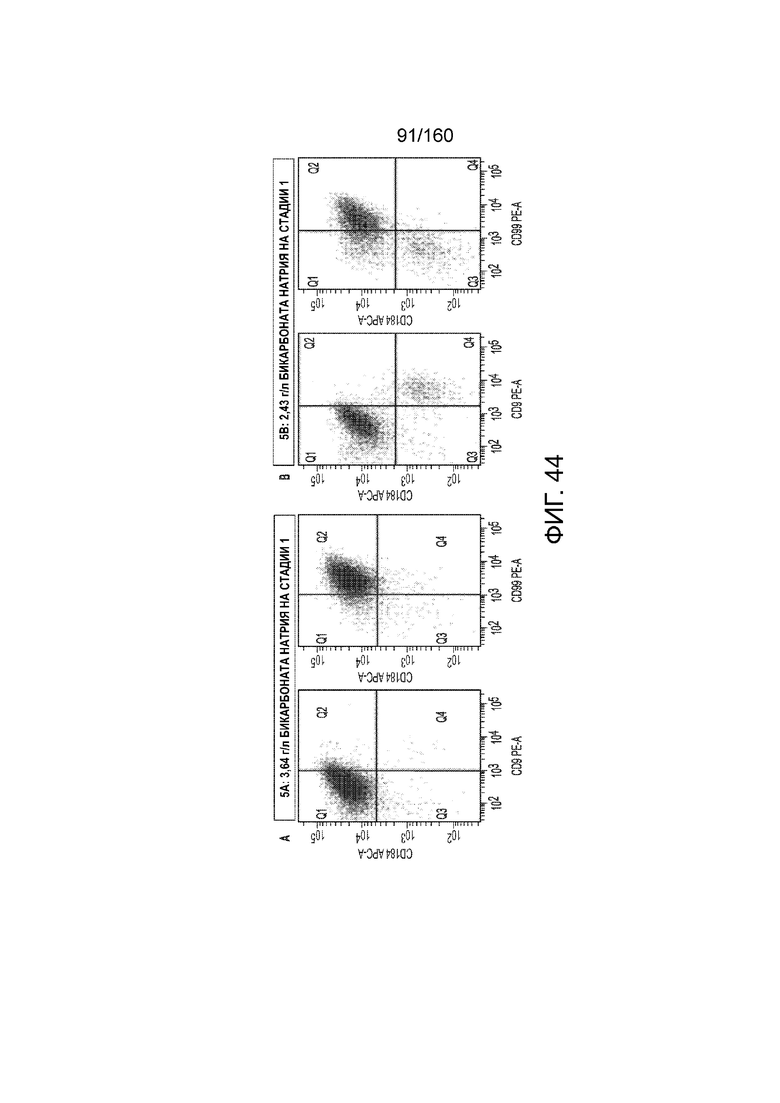
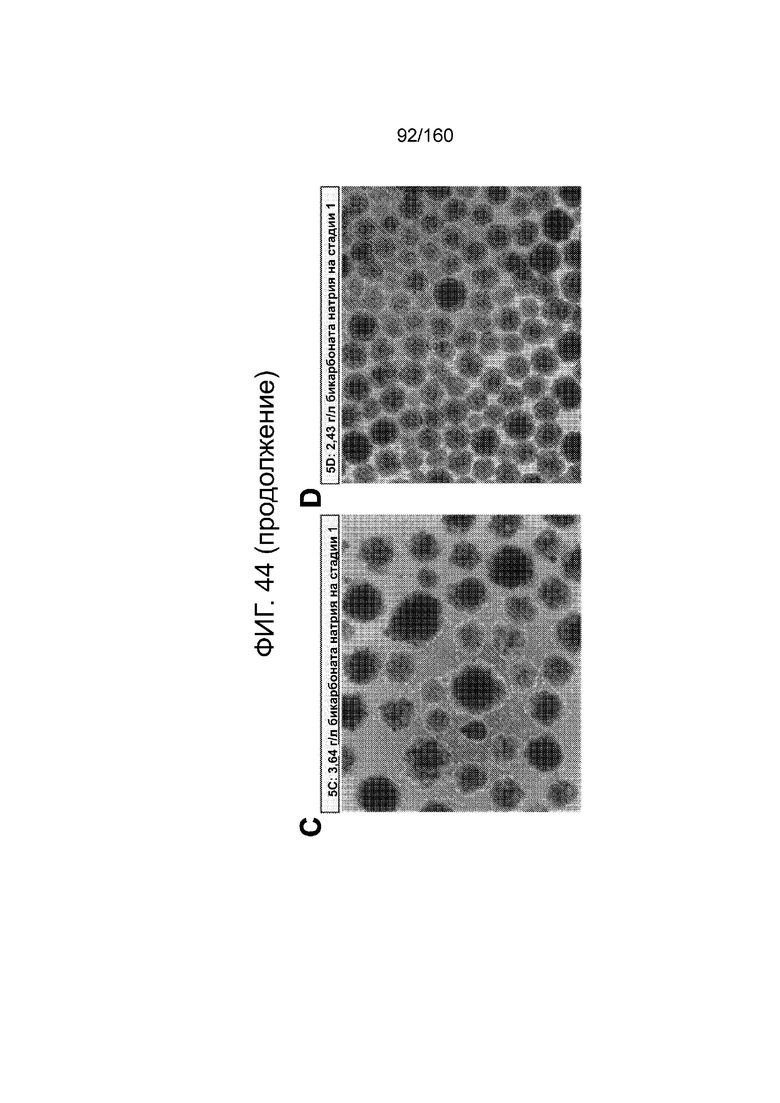
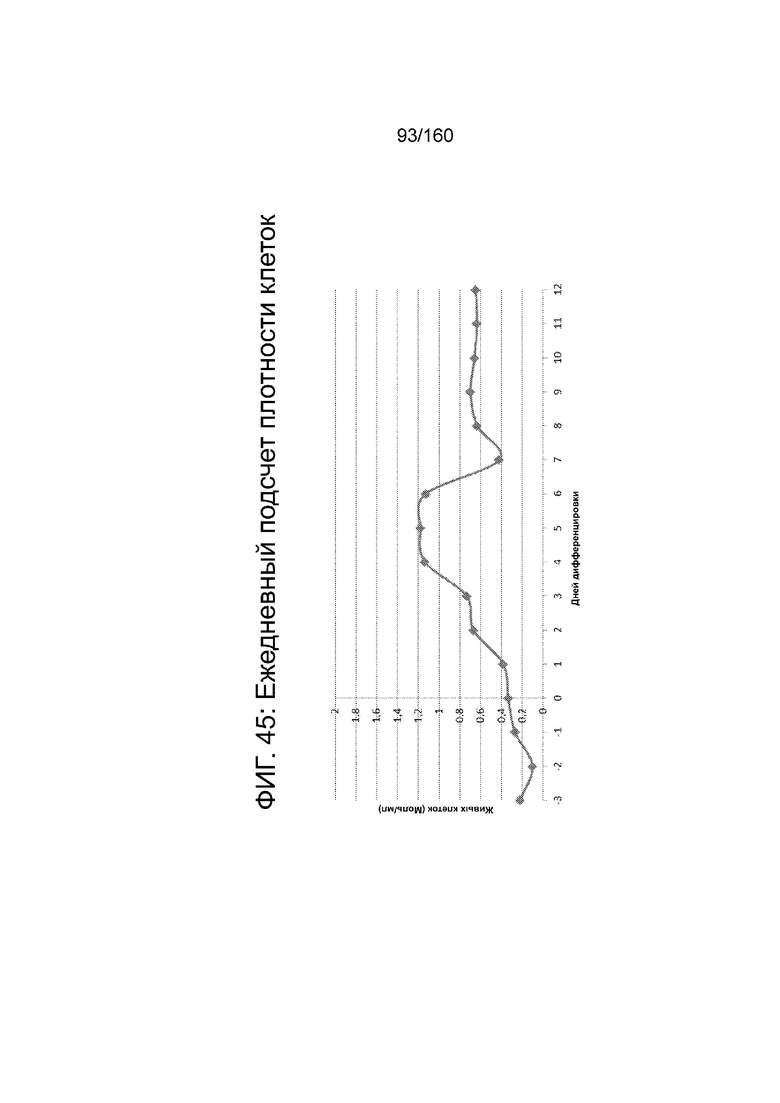
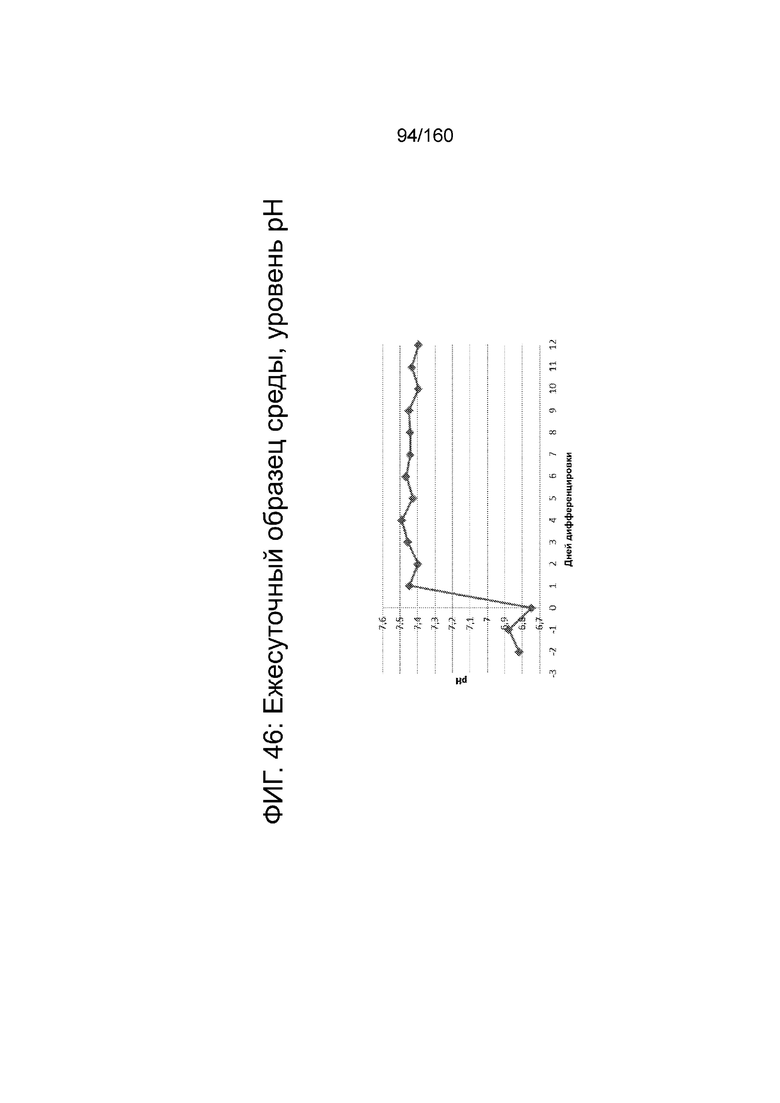

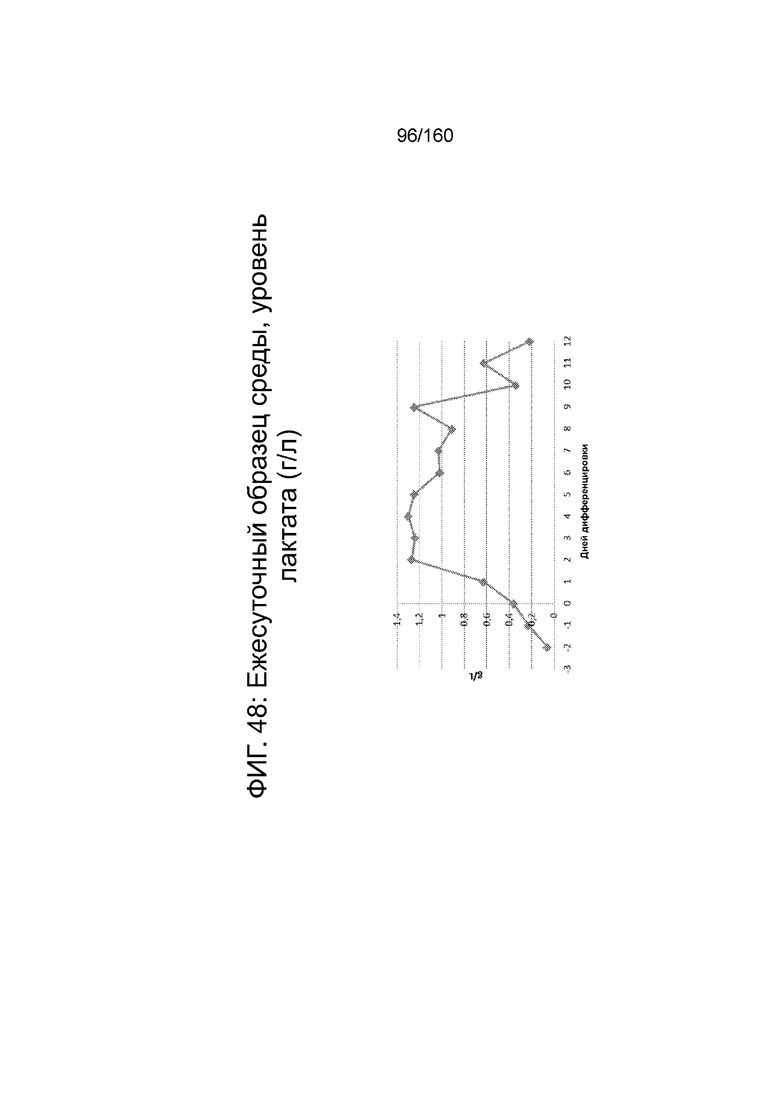
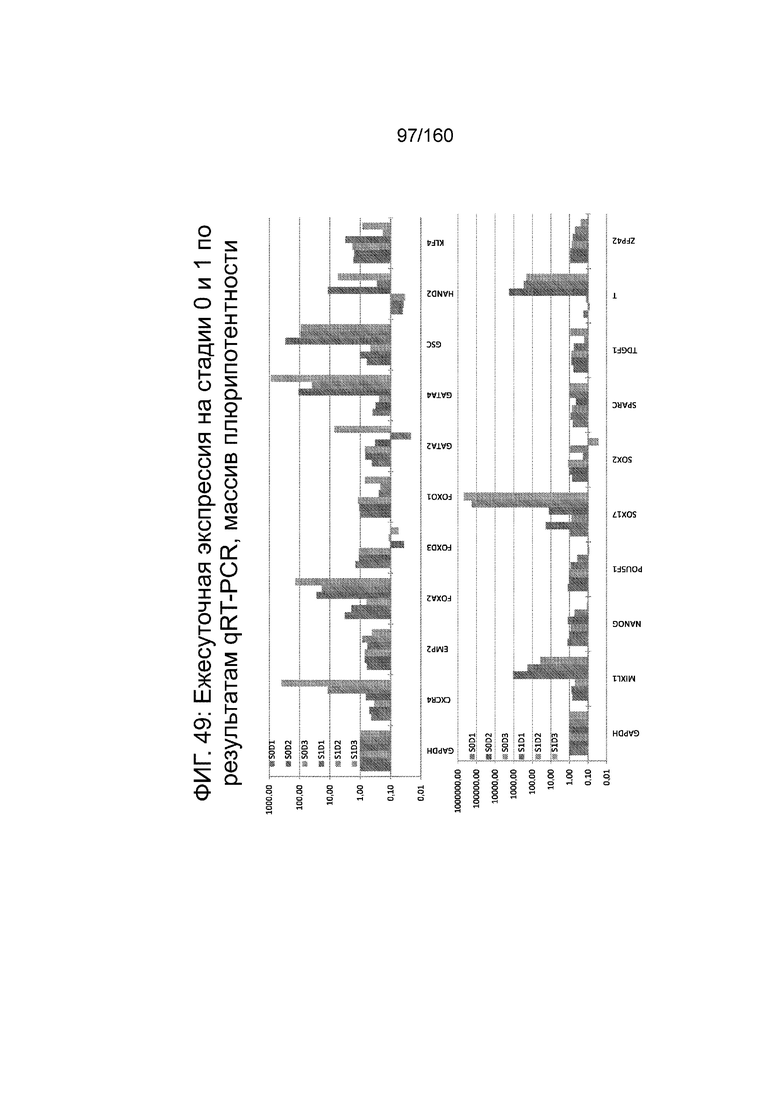



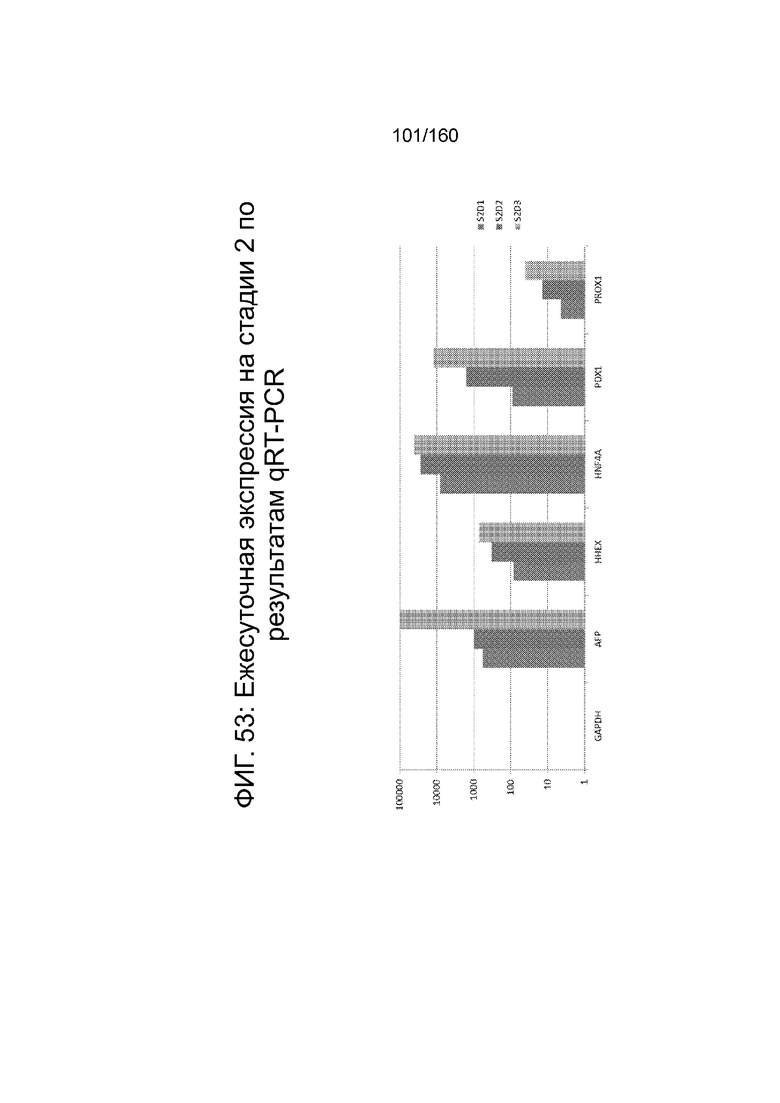

















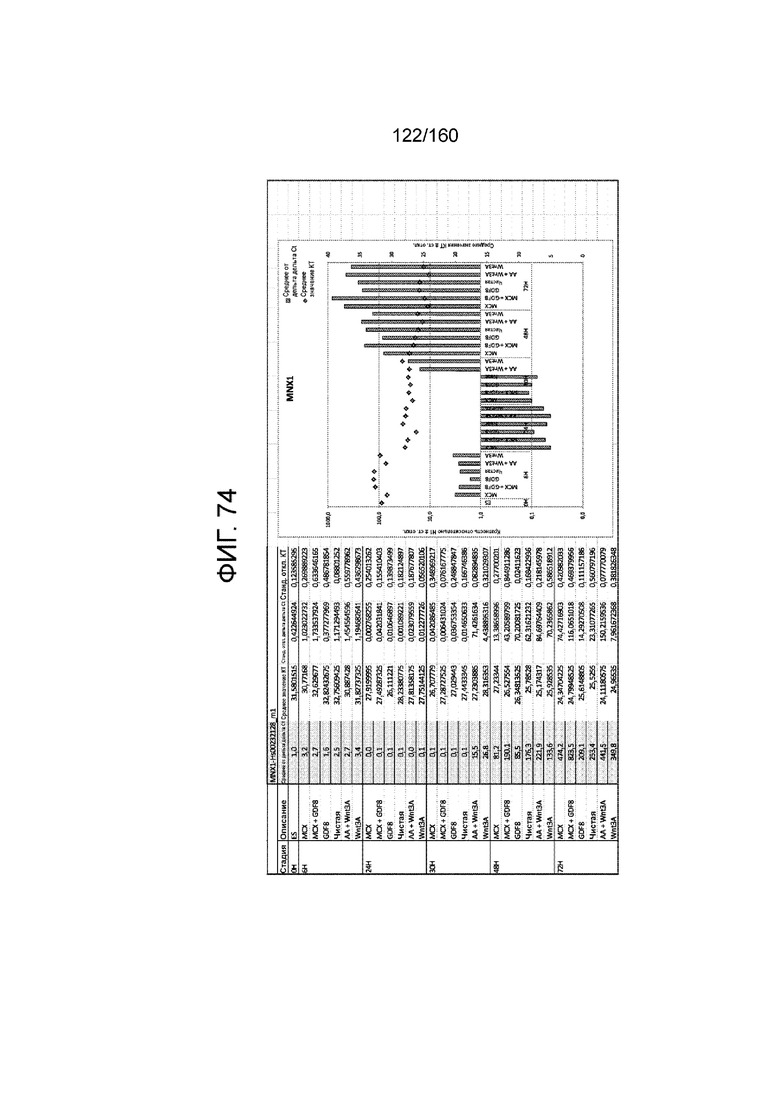




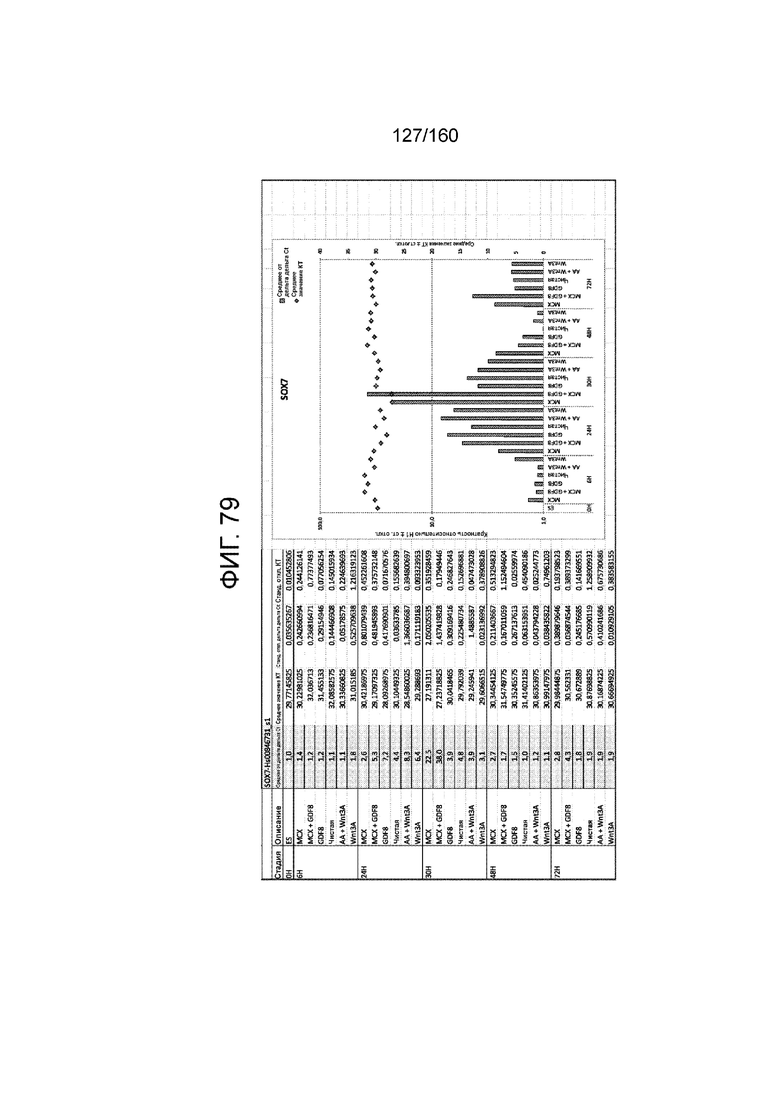

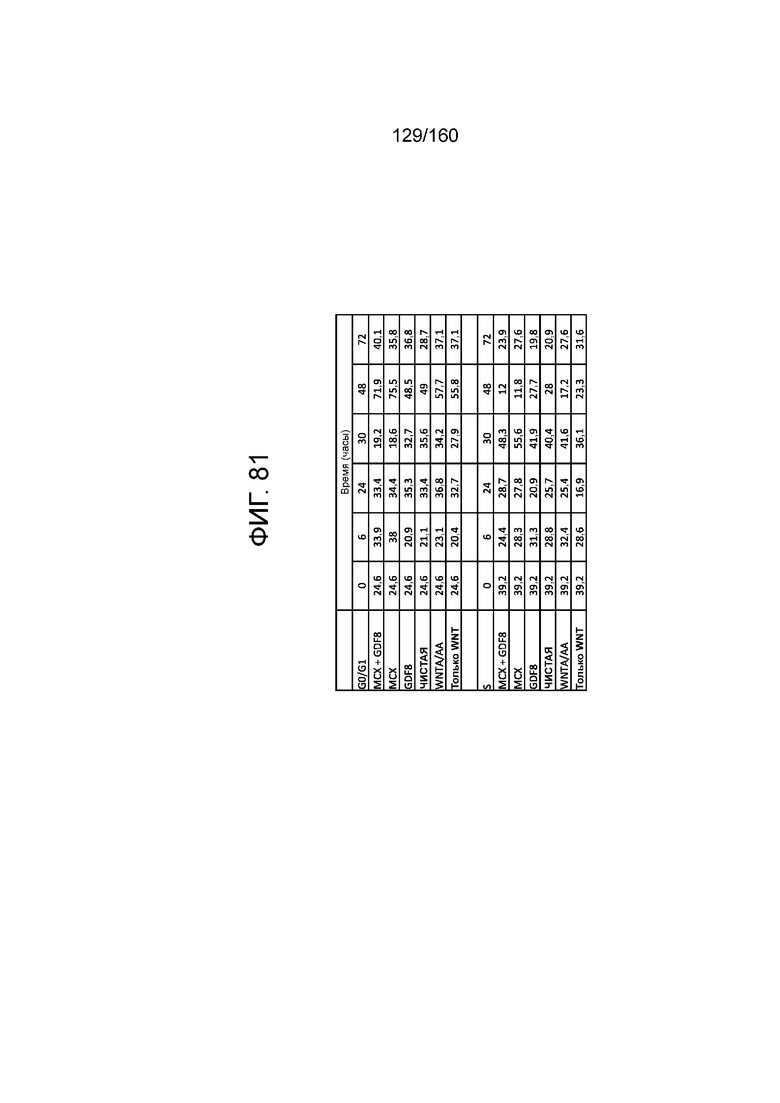



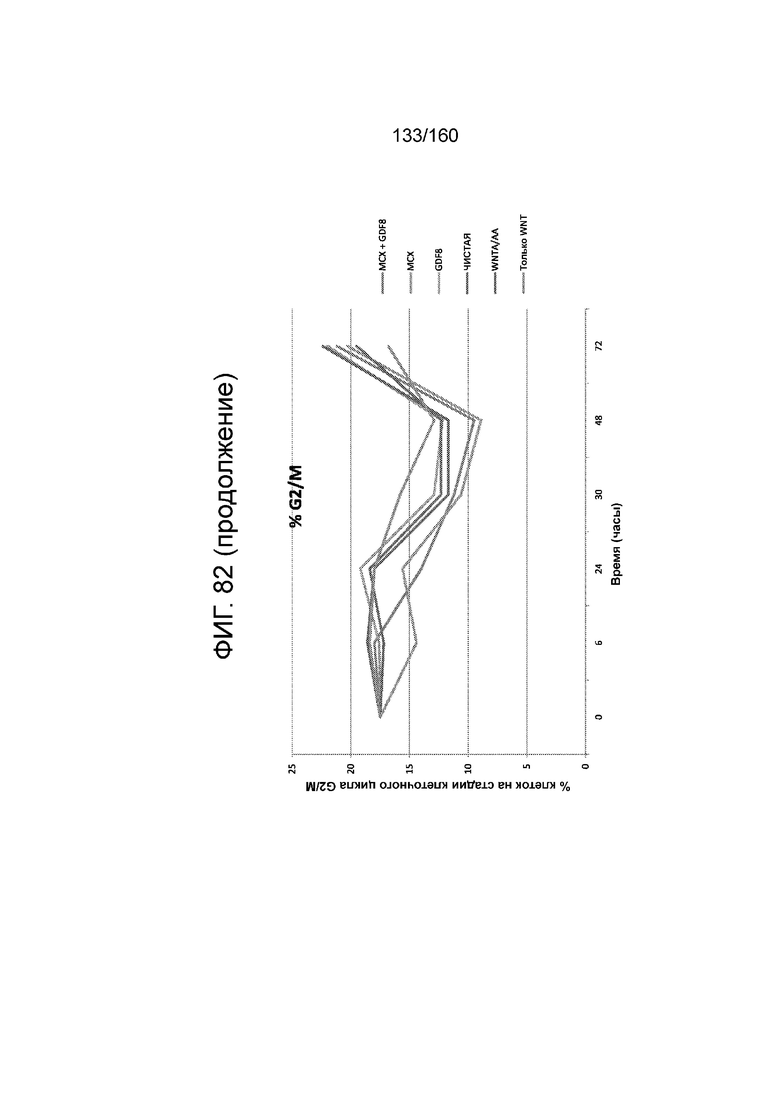




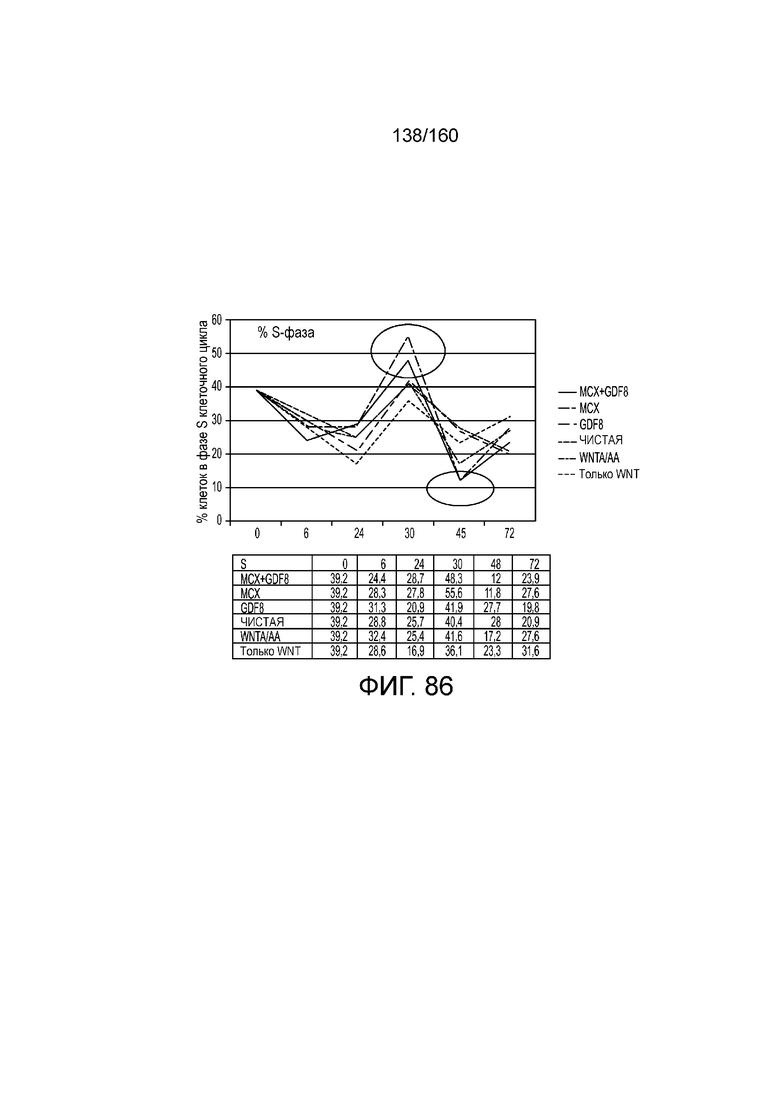
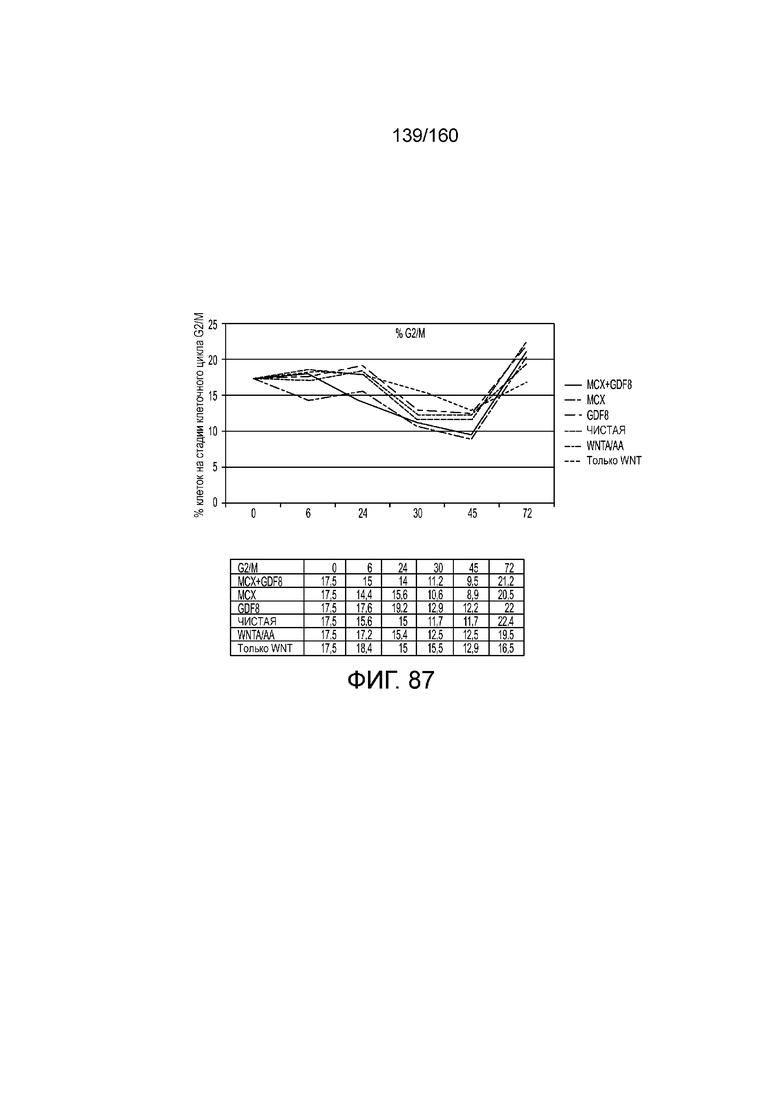












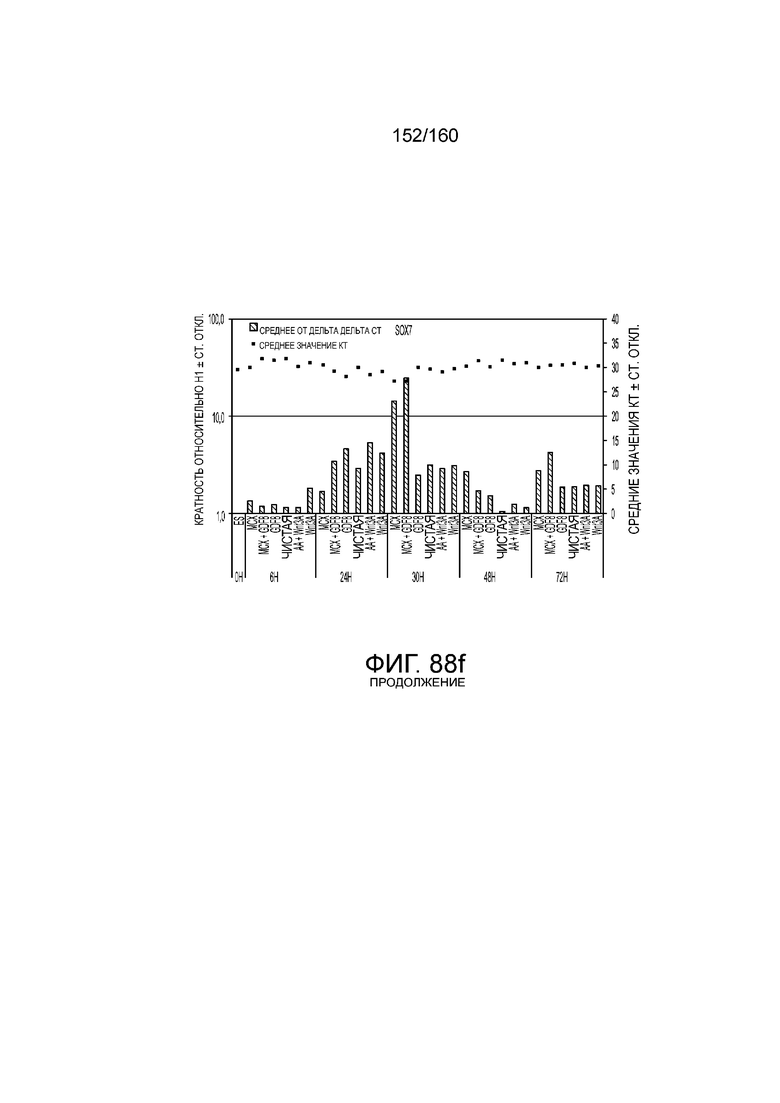

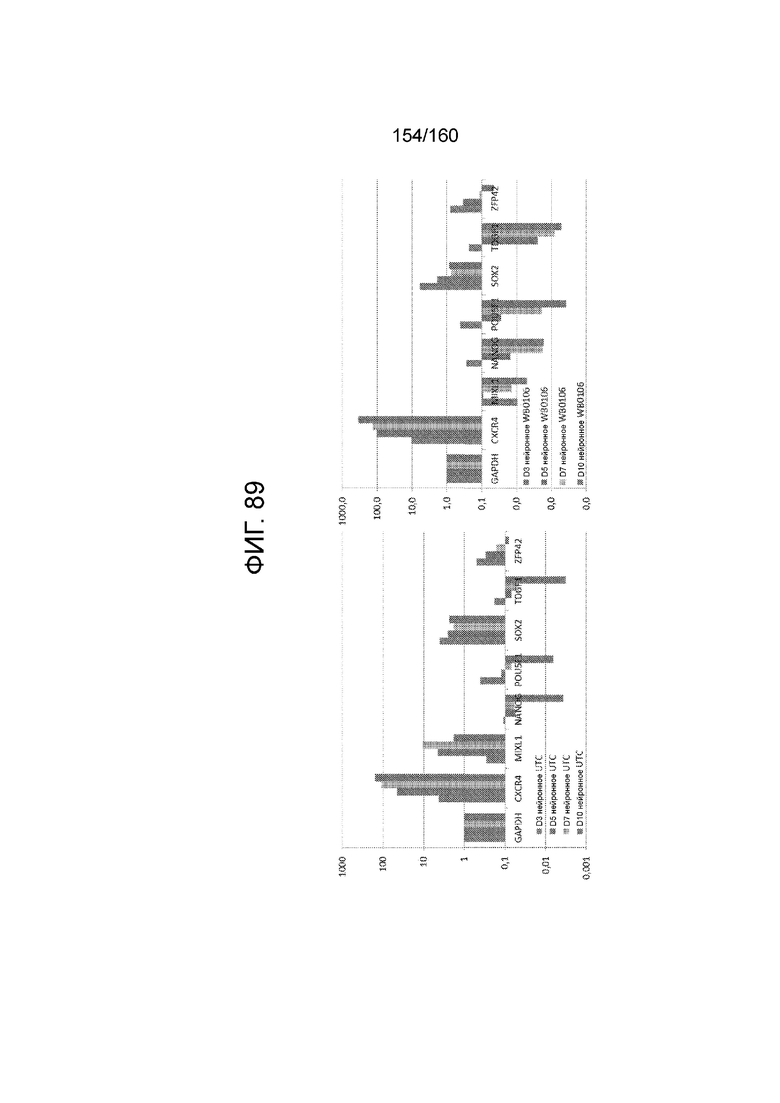


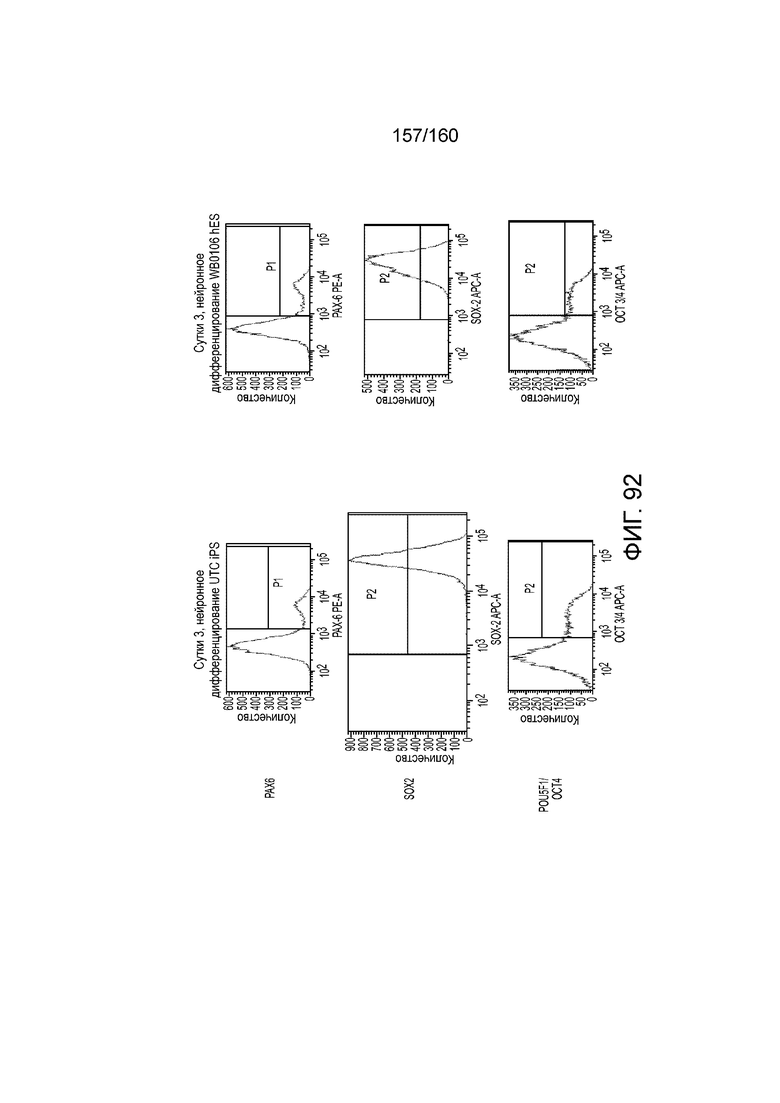


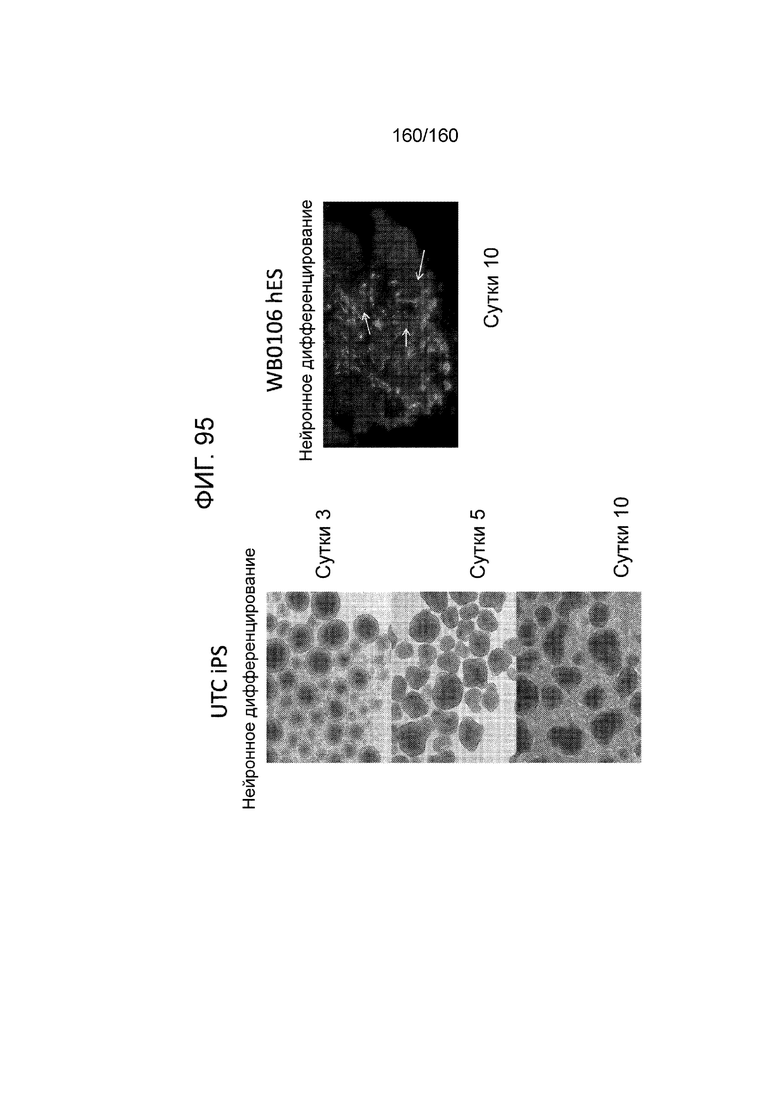
Изобретение относится к области биотехнологии, а именно к получению трехмерных клеточных кластеров и их дифференцировке. Способ включает обработку плюрипотентных стволовых клеток, которые являются индуцированными плюрипотентными стволовыми клетками, клетками, полученными из ткани пуповины человека, партенотами, клетками, полученными из амниотической жидкости, или эмбриональными стволовыми клетками человека линии H1, H7, H9, SA002 или BG01v, культивированных в плоской адгезивной культуре вместе с хелатирующим агентом или ферментом, с высвобождением клеточных агрегатов из плоской адгезивной культуры. Далее осуществляют суспендирование клеточных агрегатов из плоской адгезивной культуры в культурную среду в присутствии ингибитора Rho-киназы без диссоциации клеточных агрегатов до единичных клеток. Затем осуществляют перенос суспензии клеточных агрегатов в динамическую суспензионную культуру и увеличение объема суспензии клеточных агрегатов в динамической суспензионной культуре с получением трехмерных клеточных кластеров, при этом трехмерные клеточные кластеры сохраняют плюрипотентность. Далее возможна дифференциация плюрипотентных клеточных кластеров в динамической суспензионной культуральной системе с получением популяции клеток-предшественников панкреатических клеток, популяции клеток-предшественников нейронов или популяции клеток-предшественников кардиомиоцитов. Изобретение позволяет расширить арсенал средств. 3 н. и 31 з.п. ф-лы, 20 табл., 95 ил., 16 пр.
1. In vitro способ получения трехмерных клеточных кластеров из плюрипотентных стволовых клеток, включающий:
a. обработку плюрипотентных стволовых клеток, культивированных в плоской адгезивной культуре вместе с хелатирующим агентом или ферментом, с высвобождением клеточных агрегатов из плоской адгезивной культуры;
b. суспендирование клеточных агрегатов из плоской адгезивной культуры в культурную среду в присутствии ингибитора Rho-киназы без диссоциации клеточных агрегатов до единичных клеток;
c. перенос суспензии клеточных агрегатов в динамическую суспензионную культуру; и
d. увеличение объема суспензии клеточных агрегатов в динамической суспензионной культуре с получением трехмерных клеточных кластеров, при этом трехмерные клеточные кластеры сохраняют плюрипотентность,
причем плюрипотентные стволовые клетки являются индуцированными плюрипотентными стволовыми клетками, клетками, полученными из ткани пуповины человека, партенотами, клетками, полученными из амниотической жидкости, или эмбриональными стволовыми клетками человека линии H1, H7, H9, SA002 или BG01v.
2. Способ по п. 1, в котором плюрипотентные стволовые клетки обрабатывают ферментом, выбранным из нейтральной протеазы или Accutase®.
3. Способ по п. 2, в котором ферментом является нейтральная протеаза.
4. Способ по п. 3, в котором плюрипотентные стволовые клетки представляют собой H1 hES.
5. Способы по п. 1, в котором клетки в клеточных кластерах экспрессируют CD9, SSEA4, TRA-1-60 и TRA-1-81 и не экспрессируют CXCR4.
6. Способ по п. 1, в котором плюрипотентные стволовые клетки обрабатывают хелатирующим агентом.
7. Способ по п. 6, в котором хелатирующим агентом является этилендиаминтетрауксусная кислота (EDTA).
8. Способ по п. 1, в котором динамически перемешиваемая суспензионная культура содержит микроносители.
9. Способ дифференцировки клеточных кластеров из плюрипотентных стволовых клеток в динамически перемешиваемой суспензионной культуральной системе, включающий:
а. обработку плюрипотентных стволовых клеток, культивированных в плоской адгезивной культуре вместе с хелатирующим агентом или ферментом, с высвобождением клеточных агрегатов из плоской адгезивной культуры;
b. суспендирование клеточных агрегатов из плоской адгезивной культуры в присутствии ингибитора Rho-киназы без диссоциации клеточных агрегатов до единичных клеток;
с. перенос суспензии клеточных агрегатов в динамически перемешиваемую суспензионную культуру;
d. увеличение объема суспензии клеточных агрегатов в динамически перемешиваемой суспензионной культуре с получением плюрипотентных клеточных кластеров, в которых клеточные кластеры экспрессируют CD9, SSEA4, TRA-1-60 и TRA-1-81 и не экспрессируют CXCR4; и
e. дифференциацию плюрипотентных клеточных кластеров в динамической суспензионной культуральной системе с получением популяции клеток-предшественников панкреатических клеток, популяции клеток-предшественников нейронов или популяции клеток-предшественников кардиомиоцитов,
причем плюрипотентные стволовые клетки являются индуцированными плюрипотентными стволовыми клетками, клетками, полученными из ткани пуповины человека, партенотами, клетками, полученными из амниотической жидкости, или эмбриональными стволовыми клетками человека линии H1, H7, H9, SA002 или BG01v.
10. Способ по п. 9, при котором продуцируется популяция клеток-предшественников панкреатических клеток, которые экспрессируют транскрипционные факторы β-клеток.
11. Способ по п. 10, в котором транскрипционные факторы β-клеток представляют собой PDX1 и/или NKX6.1.
12. Способ по п. 9, который включает дифференцировку плюрипотентных клеточных кластеров в динамически перемешиваемой суспензионной культуральной системе с получением популяции клеток-предшественников поджелудочной железы.
13. Cпособ по п. 9, который включает дифференцировку плюрипотентных клеточных кластеров в динамически перемешиваемой суспензионной культуральной системе с получением популяции клеток-предшественников нейронов.
14. Способ по п. 9, который включает дифференцировку плюрипотентных клеточных кластеров в динамически перемешиваемой суспензионной культуральной системе с получением популяции клеток-предшественников кардиомиоцитов.
15. Cпособ по п. 9, в котором плюрипотентные стволовые клетки обрабатывают хелатирующим агентом.
16. Cпособ по п. 15, в котором хелатирующим агентом является EDTA.
17. Cпособ по п. 9, в котором плюрипотентные стволовые клетки обрабатывают ферментом.
18. Способ по п. 17, в котором ферментом является нейтральная протеаза или Accutase.
19. Способ по п. 9, в котором стадия дифференцировки включает культивирование при уровне кислорода от примерно гипоксии до примерно 30% окружающего воздуха, липидов в диапазоне от 0,1 % до примерно 2% или при комбинации этих условий.
20. Способ по п. 12, который включает дифференцировку трехмерных плюрипотентных кластеров стволовых клеток в окончательные клетки эндодермы в культуральной среде, дополненной либо (i) циклическим анилинпиридинотриазином и GDF8, либо (ii) WNT3A и активином A.
21. Способ по п. 20, в котором циклический анилинпиридинотриазин представляет собой 14-проп-2-ен-1-ил-1-3,5,7,14,17,23,27-гептаазатетрацикло[19.3.1.1~2,6~.1~8,12~]-[гептакоза-1(25),2(27),3,5,8(26),9,11,21,23-нонаен-16-он.
22. Способ по п. 9, в котором динамически перемешиваемая суспензионная культуральная система содержит микроносители.
23. In vitro cпособ получения плюрипотентных кластеров стволовых клеток, включающий:
а. обработку плюрипотентных стволовых клеток, культивированных в плоской адгезивной культуре, вместе с ферментативным или хелатирующим агентом при комнатной температуре в течение периода времени, достаточного для высвобождения плюрипотентных стволовых клеток в виде клеточных агрегатов;
b. удаление указанного ферментативного или хелатирующего агента;
с. добавление культуральной среды с добавлением ингибитора Rho-киназы в клеточные агрегаты и суспендирование клеточных агрегатов в культуру без диссоциации клеточных агрегатов в единичные клетки;
d. перенос суспензии клеточных агрегатов в динамическую суспензионную культуральную систему; и
e. увеличение объема суспензии клеточных агрегатов в динамической суспензионной культуре с получением клеточных кластеров, при этом клетки в клеточных кластерах являются плюрипотентными,
причем плюрипотентные стволовые клетки являются индуцированными плюрипотентными стволовыми клетками, клетками, полученными из ткани пуповины человека, партенотами, клетками, полученными из амниотической жидкости, или эмбриональными стволовыми клетками человека линии H1, H7, H9, SA002 или BG01v.
24. Способ по п. 23, в котором способ включает применение хелатирующих агентов.
25. Cпособ по п. 24, в котором хелатирующим агентом является EDTA.
26. Способ по п. 25, в котором способ включает применение ферментативных агентов.
27. Способ по п. 23, в котором ферментативным агентом является нейтральная протеаза или Accutase.
28. Способ по п. 23, который дополнительно включает дифференциацию плюрипотентных кластеров стволовых клеток в динамической суспензионной культуральной системе с получением популяции клеток-предшественников панкреатических клеток, популяции клеток-предшественников нейронов или популяции клеток-предшественников кардиомиоцитов.
29. Способ по п. 1, в котором ингибитор Rho-киназы является Y-27632.
30. Способ по п. 1, в котором культуральная среда содержит ингибитор Rho-киназы при концентрации от 1 до около 100 мкМ.
31. Способ по п. 9, в котором ингибитор Rho-киназы является Y-27632.
32. Способ по п. 9, в котором клеточные агрегаты из плоской адгезивной культуры суспендируют в присутствии от 1 до примерно 100 мкМ ингибитора Rho-киназы.
33. Способ по п. 23, в котором ингибитор Rho-киназы является Y-27632.
34. Способ по п. 23, в котором культуральная среда содержит ингибитор Rho-киназы при концентрации от 1 до около 100 мкМ.
| СПОСОБ УДАЛЕНИЯ НОВООБРАЗОВАНИЙ ИРИДОЦИЛИАРНОЙ ЗОНЫ | 2014 |
|
RU2559756C1 |
| Устройство для снятия жира с кишек | 1929 |
|
SU18625A1 |
| БЛАЖЕВИЧ О.В | |||
| Культивирование клеток | |||
| Курс лекций | |||
| - Мн.: БГУ, 2004 | |||
| Парный автоматический сцепной прибор для железнодорожных вагонов | 0 |
|
SU78A1 |
| EGUIZABAL C | |||
| et al | |||
| "Complete Meiosis from Human Induced Pluripotent Stem Cells", Stem Cells | |||
| Способ приготовления лака | 1924 |
|
SU2011A1 |
| STACPOOL S.R.L | |||
| et al | |||
| "Efficient derivation of neural precursor cells, spinal motor neurons and midbrain dopaminergic neurons from human ES cells at 3% oxygen", Nat Protoc | |||
| Изложница с суживающимся книзу сечением и с вертикально перемещающимся днищем | 1924 |
|
SU2012A1 |
| УСТРОЙСТВО ДЛЯ ПУЛИРОВАНИЯ ДЛЯ ОДНОГО ИЛИ МНОЖЕСТВА МЕДИЦИНСКИХ КОНТЕЙНЕРОВ | 2016 |
|
RU2809303C2 |
| ДИФФЕРЕНЦИРОВКА СТРОМАЛЬНЫХ КЛЕТОК, ПОЛУЧЕННЫХ ИЗ ЖИРОВОЙ ТКАНИ, В ЭНДОКРИННЫЕ КЛЕТКИ ПОДЖЕЛУДОЧНОЙ ЖЕЛЕЗЫ И ИХ ИСПОЛЬЗОВАНИЕ | 2002 |
|
RU2351648C2 |
| ВЕРХОВСКАЯ Л.З | |||
| и др | |||
| "Действие алкоксизамещенных глицерина на морфофункциональные свойства перевиваемой культуры клеток", Криобиология | |||
| Способ приготовления консистентных мазей | 1919 |
|
SU1990A1 |

