Изобретение относится к области химии полимеров и нанотехнологиям и может быть использовано для получения полимерных наночастиц низкосиалированного эритропоэтина с высокой степенью сорбции, перпективных для лечения неврологических заболеваний
Уровень техники
Известно, что при ишемии тканей головного мозга, организм включает защитные механизмы. Одним, из таких механизмов является, в частности, ишемическое прекондиционирование (феномен прерывистой ишемии или метаболической адаптации). На прекондиционирование также можно воздействовать лекарственными препаратами, так называемое фармакологическое прекондиционирование. Влияние может осуществляться таким препаратом, как эритропоэтин.
Эритропоэтин вырабатывается астроцитами в зоне пенумбры (ишемической полутени мозга). Число рецепторов ЭПО в нейронах пенумбры возрастает. Эритропоэтин стимулирует синтез белков, угнетает влияние возбуждающих аминокислот на нейроны, угнетает воспаление, ингибирует апоптоз нейронов. Стимулирует нейро- и ангиогенез после повреждения головного мозга. Наряду с этим нейропротективная роль эритропоэтина заключается в стимуляции эритропоэза, увеличении поступления кислорода в мозг. Воздействуя на эндотелий, ЭПО модифицирует синтез факторов, регулирующих кровоток, факторов регулирующих выживание эндотелиальных клеток, их предшественников, факторов нейро - и ангиогенеза [Грицук С.Ф., «Анестезия и механизмы защиты мозга». Вестник интенсивной терапии. - 2011. №1. С. 59-67].
В следующей работе также рассматриваются вопросы прекондиционирования при ишемическом повреждении мозга и влияние на эти процессы эритропоэтина. Основываясь на результатах данной работы, было выявлено, что создание методов и средств нейропротекции, основанных на феномене прекондиционирования и стимуляции эндогенной нейропротекции, может быть весьма перспективным. В этой работе также осуществляли ведение эритропоэтина. Его использовали в дозах 2500 и 5000 Ед/кг за 30 минут до 40-минутной окклюзии средней мозговой артерии у крыс. Было выявлено, что это уменьшает ишемическое повреждение мозга. Наибольшим защитным эффектом обладает введение эритропоэтина в дозе 5000 Ед/кг. Реперфузионное повреждение мозга уменьшается в случае введение эритропоэтина в дозе 5000 Ед/кг через 3 и 12 часов после 30 минутной окклюзии средней мозговой артерии. Полученные результаты делают перспективным применение эритропоэтина в клинической практике для повышения устойчивости головного мозга к ишемии.
Выявленная роль транзиторных ишемических атак как прекондиционирующего фактора дает основание для оптимизации прогноза при ишемическом инсульте [Шмонин А.А. «Эндогенная нейропротекция при ишемических нарушениях мозгового кровообращения (клинико-экспериментальное исследование)», автореферат диссертации на соискание ученой степени кандидата медицинских наук. Санкт-Петербург. 2012. С. 22].
Включение в комплекс интенсивной терапии больных с тяжелой черепно-мозговой травмой препарата рекомбинантного эритропоэтина человека (Эпокрина) способствовало более раннему выходу больных из комы (в 1-й группе к 10 суткам посттравматического периода вышли из комы 70,38% больных, во 2-й группе - 100% больных; р=0,02), снижению частоты возникновения гнойно-септических и нейро-трофических осложнений в 3,3 раза (р<0,001), сокращению длительности интенсивного лечения, требующего пребывания больного в отделении реанимации и интенсивной терапии в 2,4 раза (р=0,02). [Белоусова М.Е., «Оптимизация интенсивной терапии тяжелой черепно-мозговой травмы при использовании рекомбинантного эритропоэтина человека», автореферат диссертации на соискание ученой степени кандидата медицинских наук. Ростов-на-Дону. 2011. 24 с].
Во всех этих работах использовался эритропоэтин, как препарат выбора при повреждения головного мозга, сопровождающихся ишемией.
При этом, последние исследования показывают, что использование особых форм эритропоэтина - низкосиалированных, а также особых форм его доставки через гематоэнцефалический барьер, более перспективно.
Так, в патенте RU 2335296, 10.10.2008, предложена композиция с применением низкосиалированной формы РЭПО, которая используется для профилактики и лечения неврологических заболеваний. Было проведено сравнительное исследование нейрофизиологической активности препаратов РЭПО и низкосиалированного ЭПО при изучении их влияния на регуляцию экспрессии нейротофических факторов NGF (Nerve growth factor - фактор роста нервов) и BDNF (Brain derived Neurotrophic factor - нейротрофический фактор мозга) in vitro и in vivo.
Нейротрофины - семейство регуляторных белков нервной ткани, которые синтезируются нейронами и клетками нейроглии, и способствуют дифференцировке и поддержанию жизнеспособности и функционирования периферических и центральных нейронов. При этом используется аутокринный и паракринный механизмы регуляции. Нейротрофины регулируют нейрональную дифференцировку, индуцируют ветвление дендритов (арборизацию) и рост аксонов (спрутинг) в направлении клеток-мишеней. В зрелой нервной системе нейротрофины регулируют как краткосрочную синаптическую передачу, так и долговременное потенцирование, участвуя таким образом в обеспечении пластичности нервной системы, необходимой для ее нормального функционирования. Регуляция экспрессии нейротрофинов и их рецепторов в мозге в настоящее время рассматривается как один из самых перспективных путей воздействия на ряд патологических состояний ЦНС, связанных с дегенерацией и гибелью нейронов. В то же время известные к настоящему времени соединения, способные регулировать экспрессию нейротрофинов в мозге, например, антидепрессанты, обладают негативными побочными эффектами, ограничивающими их использование.
Для сравнительного исследования влияния препаратов РЭПО и низкосиалированной формы ЭПО на экспрессию NGF и BDNF в коре головного мозга in vivo использовали самцов крыс линии Вистар, массой 200 г. Изучаемые препараты однократно вводили внутрибрюшинно в дозе 100 мкг/кг массы животного. Контрольным животным вводили растворитель. В каждой группе было по четыре животных. Через 1 ч после введения препарата крыс забивали с помощью углекислотной асфиксии и сразу после этого выделяли фронтальную кору мозга. Ткань замораживали в жидком СО2 и хранили при - 80°С. Выделение тотальной РНК, обратную транскрипцию и ПЦР, а также оценку уровня экспрессии нейротрофинов проводили аналогично описанному выше.
В результате проведенного исследования обнаружено, что через час после введения как РЭПО, так и низкосиалированного ЭПО, происходит статистически достоверная стимуляция экспрессии обоих нейротрофинов в 2-3 раза.
Таким образом, в результате проведенных исследований было показано, что при введении РЭПО и его низкосиалированного ЭПО, как in vitro, так и in vivo, обнаружено сильное изменение экспрессии BDNF и NGF, составляющее 2-5 раз по сравнению с контролем. Такое увеличение экспрессии является длительным и наблюдается как через 1 час, так и через 4 часа после введения. Наиболее значительным и длительным эффектом на увеличение экспрессии BDNF обладает препарат низкосиалированного ЭПО.
Поддержание жизнеспособности нейронов является NGF- и BDNF-зависимым процессом, осуществляемым нейроглиальными клетками. Можно предположить, что увеличение экспрессии NGF и BDNF под действием рекомбинантного эритропоэтина и его низкосиалированной формы является одним из основных молекулярных механизмов терапевтического действия гормона на центральную нервную систему. Поскольку BDNF обладает высокой нейропротекторной и нейротрофической активностью, то на основе низкосиалированного ЭПО возможно создание фармпрепарата, проявляющего активность при нейродегенеративных заболеваниях.
Низкосиалированный эритропоэтин в этой работе предполагалось использовать методом отбора из банка клеточных линий эффективных продуцентов ЭПО с повышенным содержанием низкосиалированных форм; методом очистки и фракционирования пула ЭПО с целью более полного извлечения низкосиалированных изоформ, что является сложным и многофакторным процессом. Данное изобретение не подразумевает наноразмерной системы доставки, определение оптимальных параметров НЧ, нагруженных нсЭПО и его сорбции.
Многие работы посвящены наноразмерной системе доставки лекарственных средств в различные органы, в том числе, и в головной мозг.
Так, Тарасов В.В. разработал подходы к стандартизации наноразмерных форм, выявил основные параметры, определяющие качество полученных наноразмерных форм: размер частиц, степень включения/сорбции действующего вещества, агрегационная устойчивость [Тарасов В.В. «Разработка и применение полимерных систем доставки препаратов нейротропного действия», автореферат диссертации на соискание ученой степени кандидата фармацевтических наук. Москва. 2012. с. 25].
Была разработана технология получения наноразмерных форм препаратов нейротропного действия: ПБЦА-НЧ (полибутилцианоакрилата) феназепама со степенью включения 85,7% ±4,5; ПБЦА-НЧ тетрапептида структуры Phe-β-A1a-G1y-Trp-NH2 со степенью сорбции биологически активного вещества 67,2±3,7%; ПБЦА-НЧ эритропоэтина со степенью сорбции - 46±6,3%. Выявлено, что частицы с включенным феназепамом и сорбированным тетрапептидом устойчивы при хранении в течение 27 месяцев при температурах +4°С и +20°С.
В данной работе используется другой полимерный носитель, степень сорбции очень низкая, при этом размер НЧ с эритропоэтином очень большой: 329±26 нм, при этом используется не низкосиалированный эритропоэтин.
Данная работа была принята авторами за прототип.
Задачей изобретения является разработка высоковоспроизводимого способа получения наночастиц низкосиалированного эритропоэтина с высокой степенью сорбции для лечения неврологических заболеваний.
Целью данного изобретения является получение оптимального размера наночастиц сополимера лактида с гликолидом с высокой степенью сорбции для низкосиалированного эритропоэтина.
Технический результат
Способ позволяет получать НЧ, отличающиеся высокой стабильностью, также он высокоэкономичен, так как позволяет выбрать оптимальное соотношение реагентов для получения заданного препарата, с требуемыми характеристиками. Т.е. практически не остается непрореагировавшим раствор низкосиалированного эритропоэтина, который сам по себе является дорогостоящим препаратом. Кроме того, данный способ позволяет расширить арсенал средств для эффективного использования в неврологической практике, так обладает всеми необходимыми параметрами: размером наночастиц меньше 150 нм для прохождения через ГЭБ и наивысшей степенью сорбции препарата на НЧ. Способ прост для воспроизведения, не требует дорогостоящего оборудования, обладает высокой точностью.
Раскрытие изобретения
Предметом настоящего изобретения является способ получения наночастиц низкосиалированного эритропоэтина с высокой степенью сорбции для лечения неврологических заболеваний, согласно которому
растворяют сополимер PDLGA-93, представляющего собой сополимер D,L-лактида и гликолида, при соотношении D,L-лактида и гликолида 75/25 и средневесовой молекулярной массой 70-80 кДа, в ацетоне при температуре 20-25°С для получения 1-2% раствора сополимера;
растворяют поливиниловый спирт молекулярной массой 30-70 кДа в бидистиллированной деионизированной воде при температуре 50-60°С для получения 1-2% раствора стабилизатора;
затем для получения наночастиц сополимера смешивают полученные выше раствор сополимера и раствор стабилизатора при постоянном перемешивании;
смешивают полученные наночастицы с концентрацией в водной суспензии 0,05-0,10 г/мл с водным раствором низкосиалированного эритропоэтина с концентрацией 0,5-1,0 мг/мл в массовом соотношении 100 к 1;
далее для получения наночастиц низкосиалированного эритропоэтина добавляют натрий-ацетатный буфер для достижения диапазона рН 3,6-4,0 и инкубируют в течение 14 часов при температуре 20-25°С.
Более предпочтительным является способ получения наночастиц, отличающий тем, что для получения 1% раствора сополимера PDLGA-93 в ацетоне берут 100 мг сополимера и 10 мл ацетона.
Более предпочтительным является способ получения наночастиц, отличающий тем, что что для получения 1% раствора сополимера PDLGA-93 в ацетоне берут 5 мг сополимера и 0,5 мл ацетона.
Более предпочтительным является способ получения наночастиц, отличающий тем, что для получения 1% раствора стабилизатора берут 100 мг поливинилового спирта и 10 мл деионизированной воды.
Более предпочтительным является способ получения наночастиц, отличающий тем, что для получения 1% раствора стабилизатора берут 5 мг поливинилового спирта и 0,5 мл деионизированной воды.
Более предпочтительным является способ получения наночастиц, отличающий тем, что полученные наночастицы с концентрацией 0,067 г/мл и массой 10 мг смешивают с водным раствором низкосиалированного эритропоэтина с концентрацией 0,67 мг/мл и массой 0,1 мг в одинаковых объемах, равных 0,15 мл.
Более предпочтительным является способ получения наночастиц, отличающий тем, что натрий-ацетатный буфер добавляют в объеме 0,3 мл, равном общему объему образца наночастиц и раствора низкосиалированного эритропоэтина, для достижения значения рН 3,8-4,0.
Более предпочтительным является способ получения наночастиц, отличающий тем, что инкубирование с натрий-ацетатным буфером осуществляют при температуре 22-25°С°.
Изобретение поясняется чертежами.
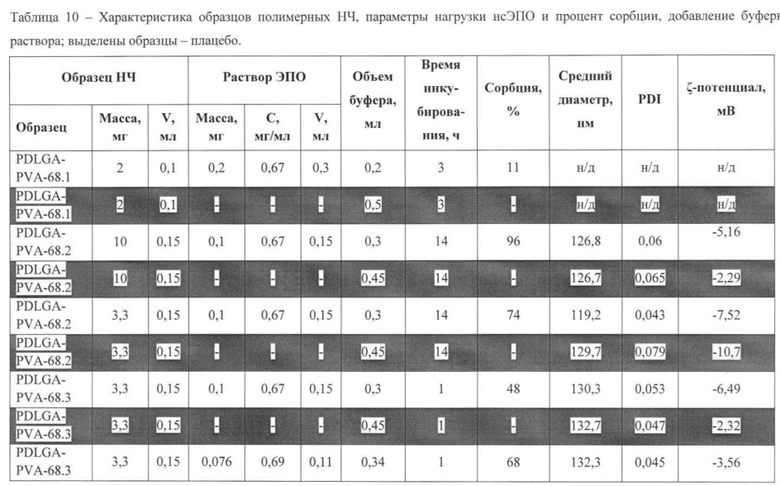
Фиг. 1. ЯМР-спектр образца PDLGA-54 до очистки, содержание мономера 3%.
Фиг. 2. ЯМР-спектр образца PDLGA-54 после очистки, мономер полностью удален.
Фиг. 3. ЯМР-спектр образца PDLGA. 74. Химический состав. 77% лактида и 23% гликолида.
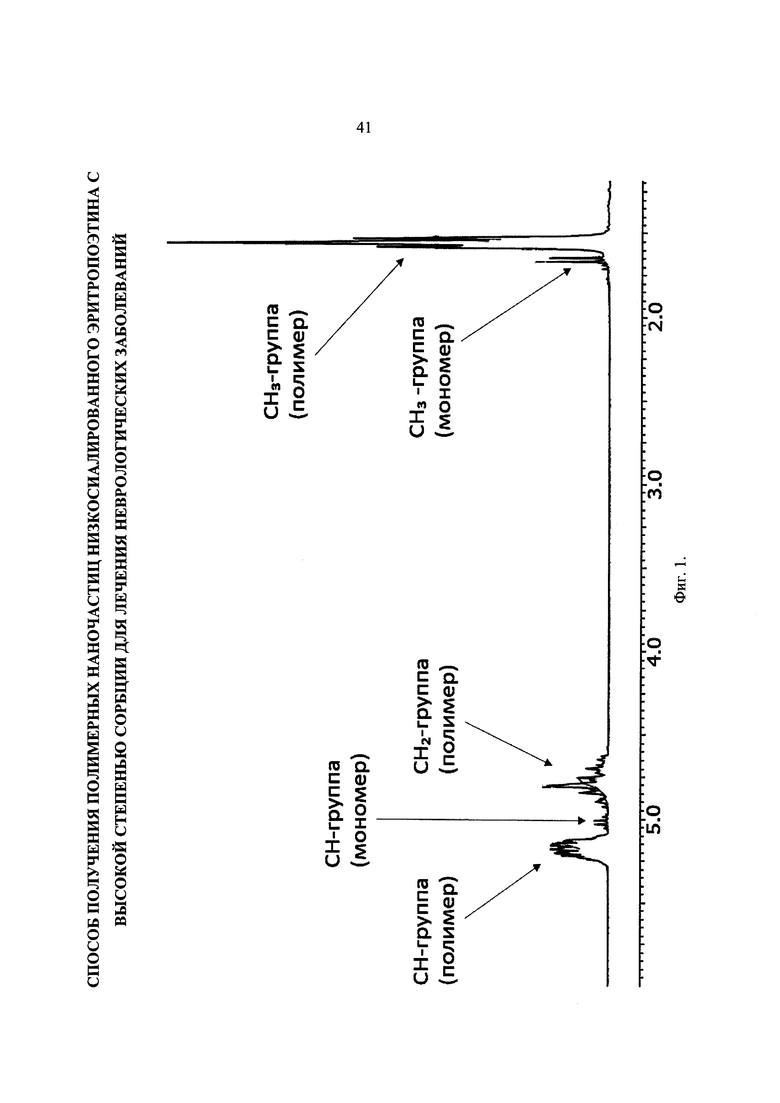
Фиг. 4. ЯМР-спектр образца PDLGA. 93. Химический состав. 74% лактида и 26% гликолида.
Фиг. 5. Хроматограмма образца PDLGA-54. Mw=26 кДа, PDI=1,8.
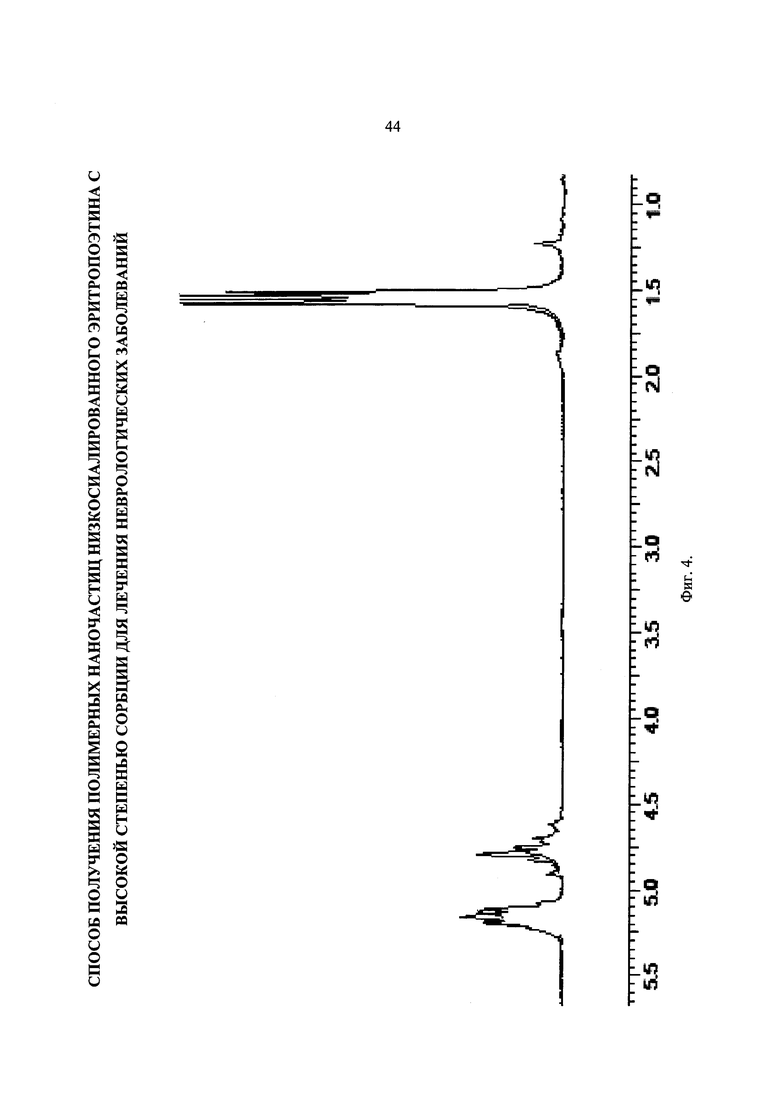
Фиг. 6. Хроматограмма образца PDLGA-74. Mw=130 кДа, PDI=2,3.
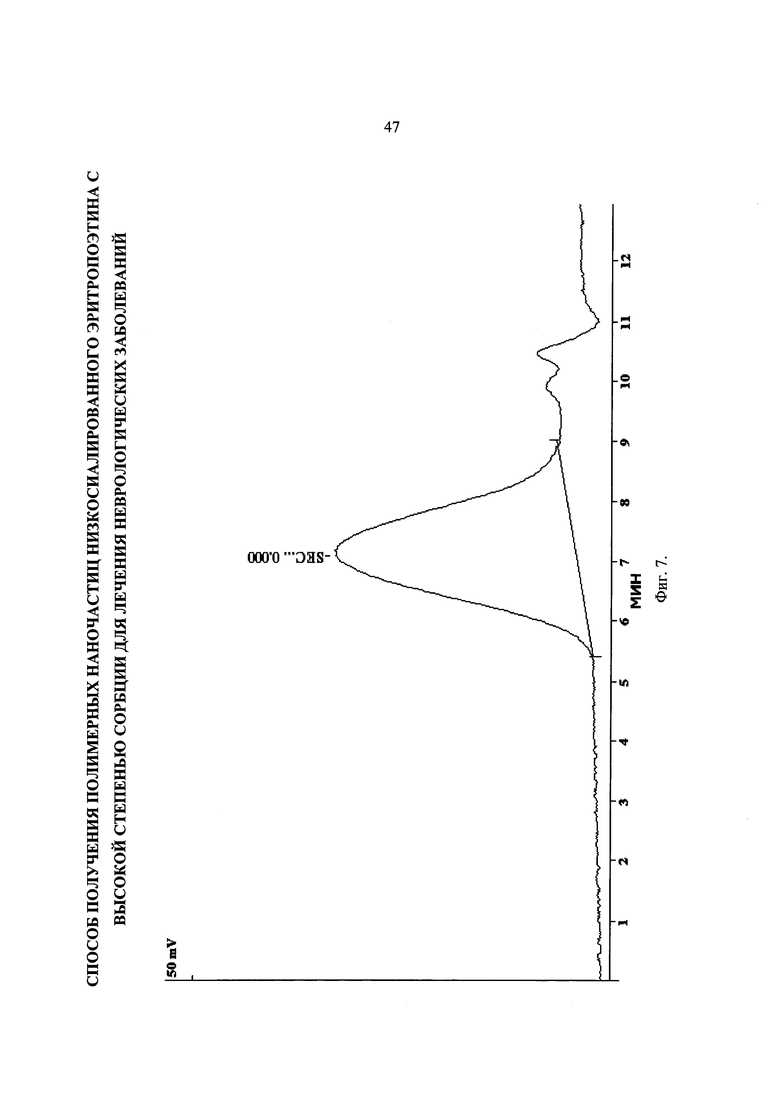
Фиг. 7. Хроматограмма образца PDLGA-93. Mw=75кДа, PDI=1,86.
Фиг. 8. Термограмма образца PDLGA-74, температура стеклования Tg=45 ?С.
Фиг. 9. Термограмма образца PDLGA-54, температура стеклования Tg=42 ?С.
Фиг. 10. Термограмма образца PDLGA-93, температура стеклования Tg=43,5 ?С.
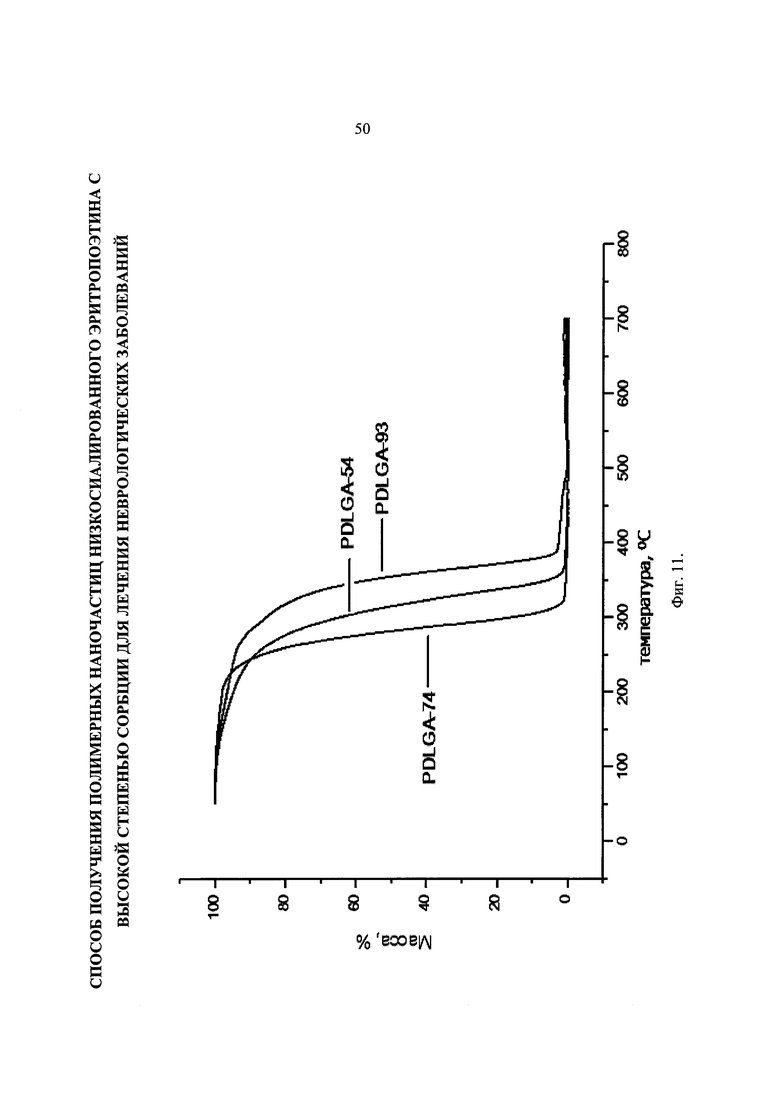
Фиг. 11. ТГА-кривые образцов PDLGA-54, PDLGA-74, PDLGA-93.
Фиг. 12. ИК-спектр образца PDLGA-74.
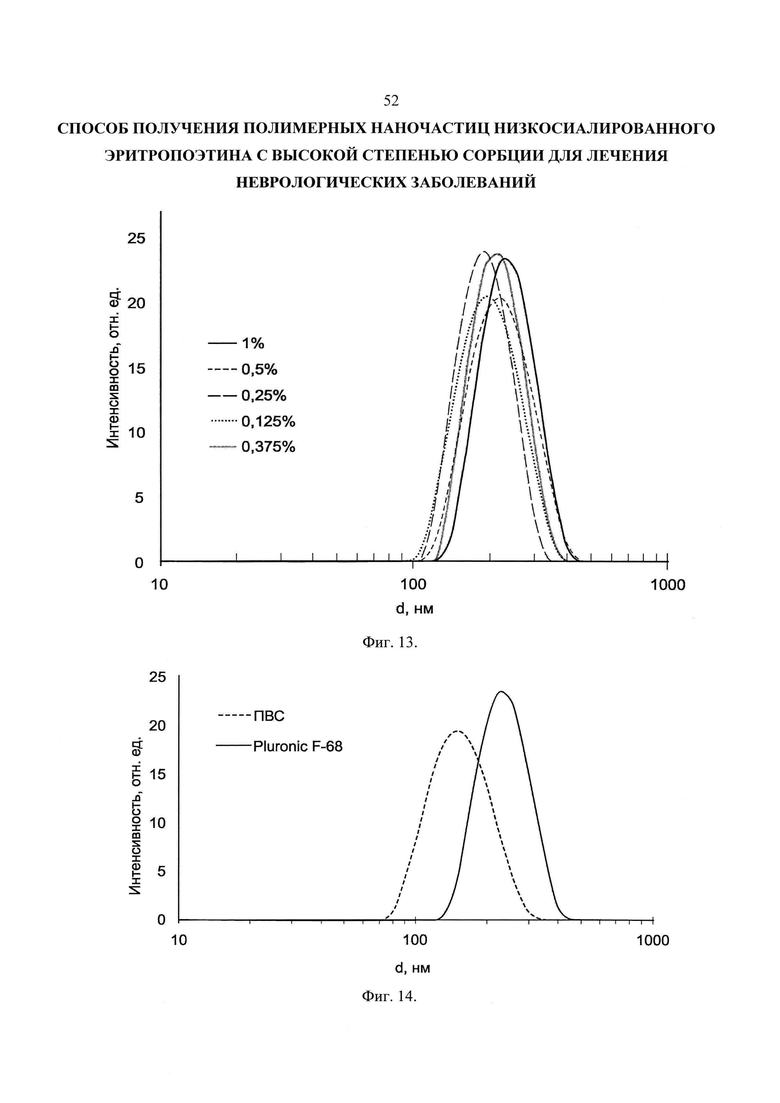
Фиг. 13. Влияние концентрации PluronicF-68 в водной фазе на распределение наночастиц по размерам (образцы PDLGA-PLU-19, 20, 21, 22, 23).
Фиг. 14. Распределение наночастиц по размерам в суспензии PDLGA-PVA-17 (стабилизатор. ПВС) и PDLGA-PLU-19.
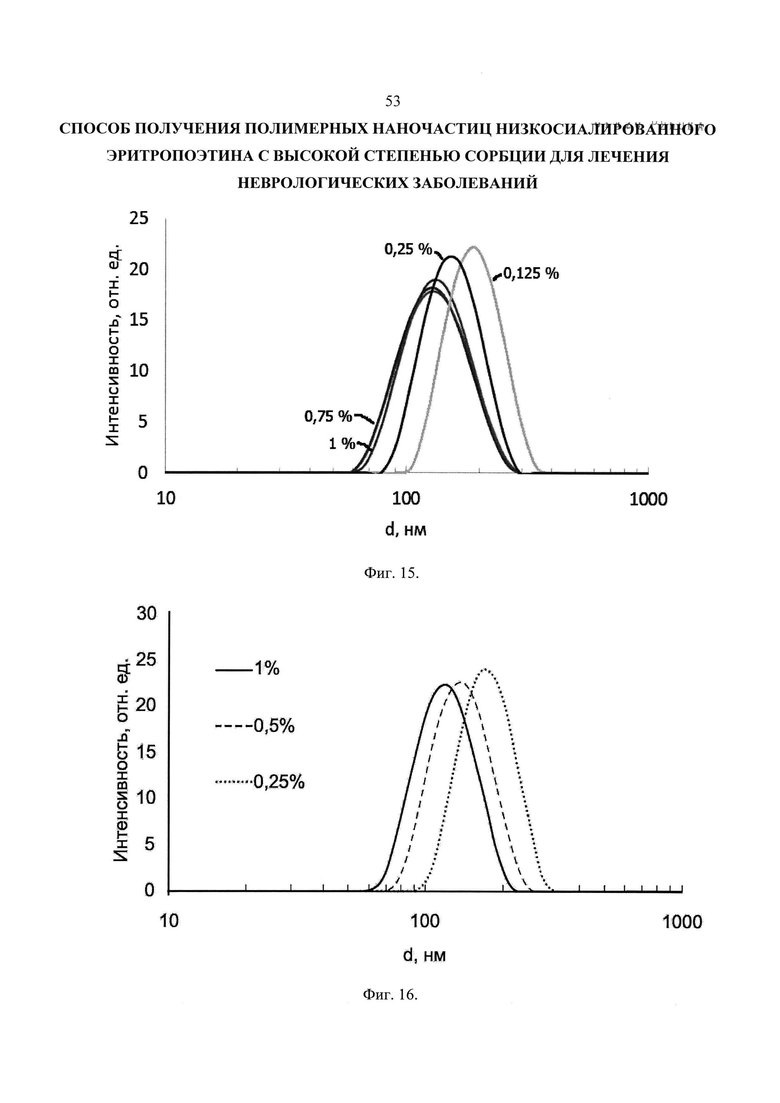
Фиг. 15. Распределение наночастиц на основе PDLGA-74 по размерам в зависимости от концентрации ПВС в водной фазе. Концентрация органической фазы 1%.
Фиг. 16. Распределение наночастиц на основе PDLGA-93 по размерам в зависимости от концентрации ПВС в водной фазе. Концентрация органической фазы 1%.
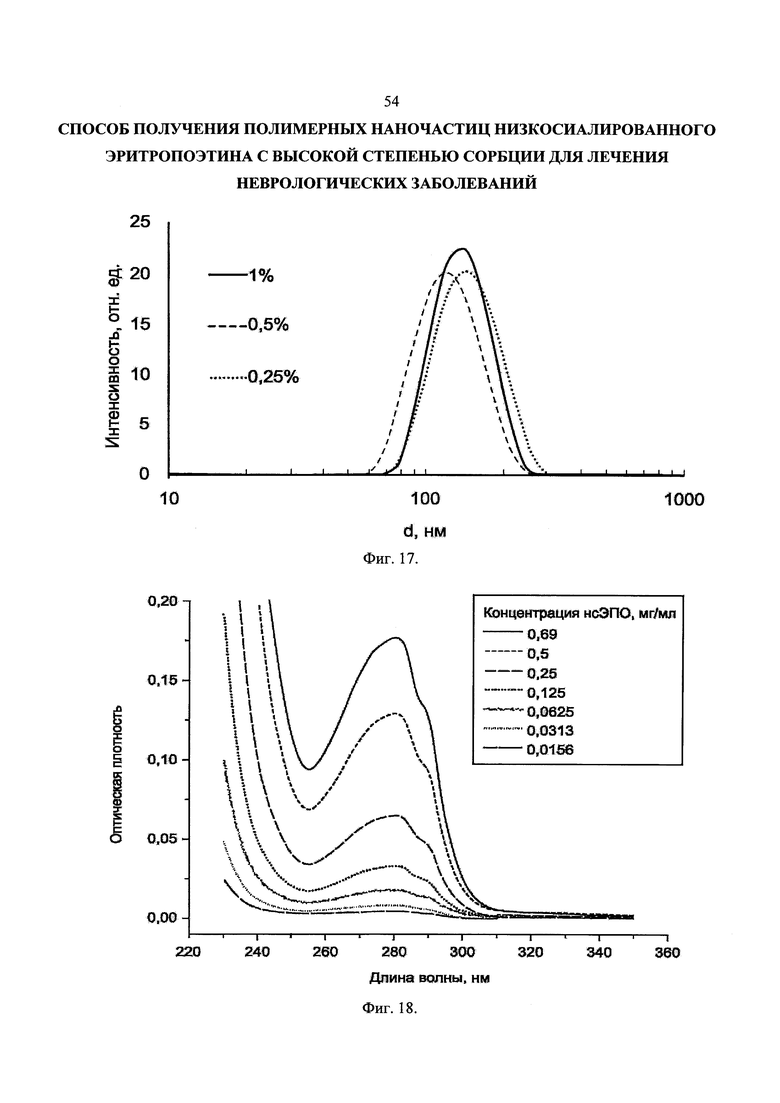
Фиг. 17. Распределение наночастиц на основе PDLGA-93 по размерам в зависимости от концентрации PDLGA-93 в органической фазе. Концентрация водной вазы 0,5%.
Фиг. 18. Спектры водных растворов нсЭПО различной концентрации.
Фиг. 19. Калибровочная кривая для водных растворов нсЭПО.
Фиг. 20. Вид типичных спектров супернатанта суспензии образца, нагруженного ЭПО (красная кривая) в сравнении с контролем (черная кривая).
Получение полимерных наночастиц.
Известно, что разработка наносомальных форм белков и пептидов ограничена в связи с неустойчивостью их молекул в условиях химического синтеза. Гомогенизация, а также контакт с органическими растворителем могут привести к денатурации белка и, как следствие, потери его активности.
Наночастицы на основе полилактидов получали методом замены растворителя.
Метод замены растворителя (нанопреципитации) - одностадийная процедура, позволяющая получить водную суспензию полимерных наночастиц, стабилизированных поверхностно-активными веществами (ПАВ). Для проведения процесса необходимы следующие компоненты: полимер, растворитель для полимера, стабилизатор (ПАВ) и фаза, в которой полимер нерастворим, но растворим ПАВ, обычно вода. Причем растворитель полимера должен смешиваться с водой и легко выпариваться, такими свойствами обладает ацетон. Раствор полимера в ацетоне (органическая фаза) приливают к перемешиваемому раствору стабилизатора в воде (водная фаза). После чего органический растворитель выпаривают при определенной температуре. При смешивании двух фаз органический растворитель смешивается с водой, в результате чего "хороший" растворитель для полимера заменяется на "плохой" - воду и полимер коллапсирует в частицу, которая тут же покрывается стабилизатором, присутствующим в водной фазе.
Приготовление органической фазы
Необходимую навеску сополимера D,L-лактида и гликолида 75/25 растворяют в 10 мл ацетона для получения раствора с концентрацией от 0,25 до 1%, от 2,5 до 10 мг/мл. Раствор перемешивают на магнитной мешалке при комнатной температуре до полного растворения полимера и гомогенизации раствора.
Приготовление водной фазы
Необходимую навеску PluronicF-68 или поливинилового спирта растворяют в 10 мл бидистилированной деионизованной воды для получения раствора с концентрацией от 0,125 до 1%, от 1,25 до 10 мг/мл. Раствор готовят в стеклянном стакане объемом 50 мл. В случае PluronicF-68 раствор перемешивают при комнатной температуре на магнитной мешалке до визуального растворения вещества. Раствор поливинилового спирта перемешивают на магнитной мешалке при температуре 50°С до визуального растворения вещества.
Смешивание фаз и выпаривание органического растворителя Стакан, содержащий водную фазу нагревают до 55°С. При постоянном перемешивании (700 rpm) медленно добавляют органическую фазу к водной в пропорции 1:1 с помощью автоматического дозатора. Скорость перемешивания увеличивают до 1000 rpm и оставляют систему перемешиваться для выпаривания ацетона при температуре 55°С на 1 час в вытяжном шкафу.
Фильтрация водной суспензии Полученную на предыдущем этапе суспензию фильтруют через предварительно смоченный деионизованной водой стеклянный фильтр с размером пор 3 мкм для удаления механических примесей и возможного осадка. Объем суспензии доводят до 10 мл, доливая деионизованную воду через тот же стеклянный фильтр с целью вымыть находящийся в нем объем суспензии.
Удаление не включенного в состав частиц стабилизатора Суспензии центрифугируют при ускорении 20 000 g в течение 30 мин. Супернатант осторожно отбирают, а осадок ресуспендируют в исходном объеме воды. Образованный осадок перемешивают механически и затем подвергают суспензии озвучиванию в ультразвуковой ванне в течение 30 мин. При наблюдении агломератов процедуру озвучивания повторяют до полного ресуспендирования осадка. В случае неполного ресуспендирования осадка после трехктратной процедуры озвучивания суспензию повторно пропускают через стеклянный фильтр с размером пор 3 мкм.
Финальную концентрацию суспензии определяют следующим образом. 1 мл суспензии наливают в предварительно взвешенную тару. Воду выпаривают на плите при 150°С. По разнице между начальным и конечным весом тары рассчитывают концентрацию суспензии. Осадок в таре ресуспендированию не подлежит. Потери при этом незначительны.
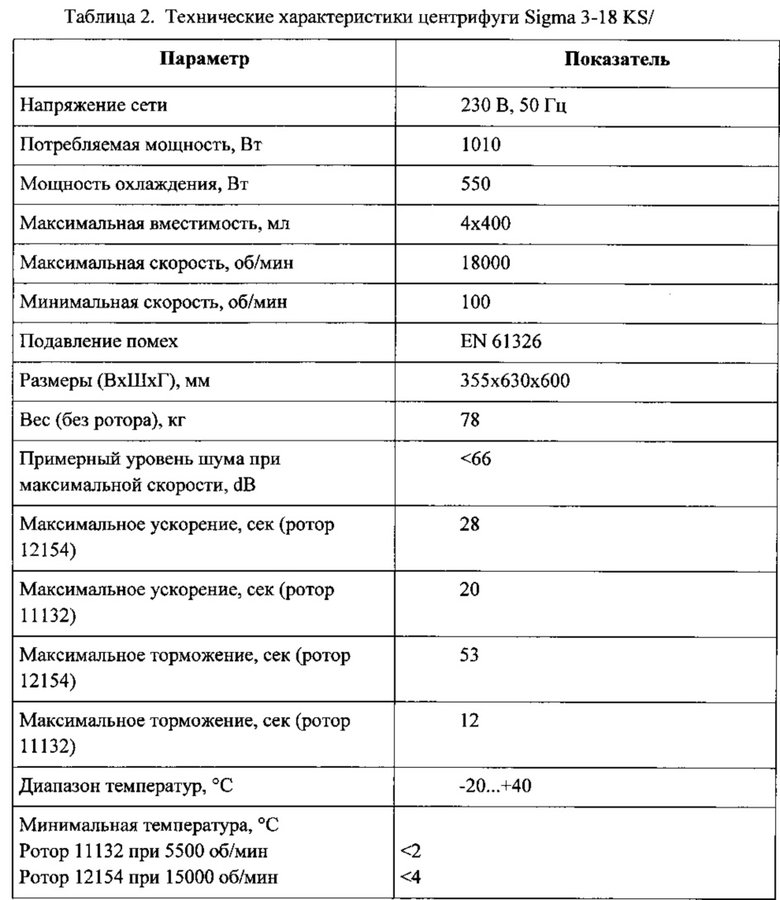
В эксперименте использовалась лабораторная магнитная мешалка HeidolphMRHei-Tec с подогревом и датчиком контроля температуры HeidolphEKTHei-Соп.Технические характеристики приведены в таблице 1. Для получения частиц использовали цилиндрические тефлоновые мешальники длиной 1,5 см.

Центрифуга SIGMA 3-18KS с угловым ротором с герметичной алюминиевой крышкой 30×1,5/2 мл для пробирок 15008, 15040. Технические характеристики центрифуги приведены в таблице 2.
Наночастицы, полученные по такой методике, способны при определенных условиях сорбировать эритропоэтин. Сорбция обусловлена слабыми водородными связями, возникающими между молекулами стабилизатора и белка. Для проведения нагрузки наночастиц плацебо необходимо инкубировать их совместно с раствором эритропоэтина при определенных условиях: соотношение массы наночастицы/белок, рН среды и др. После инкубирования не сорбировавшийся на наночастицы белок удаляют центрифугированием суспензии и их последующим ресупендированием.
Для лиофилизации полученной суспензии (плацебо или нагруженной эритропоэтином) необходимо довести суспензию до определенной концентрации, после чего добавить криопротектор, препятствующий слипанию наночастиц при заморозке. Заморозку водной суспензии проводят при температуре -35°С, после чего проводят лиофилизацию в лиофильной сушке, принципы работы которой позволяют сохранить структуру и стабильность полученного препарата.
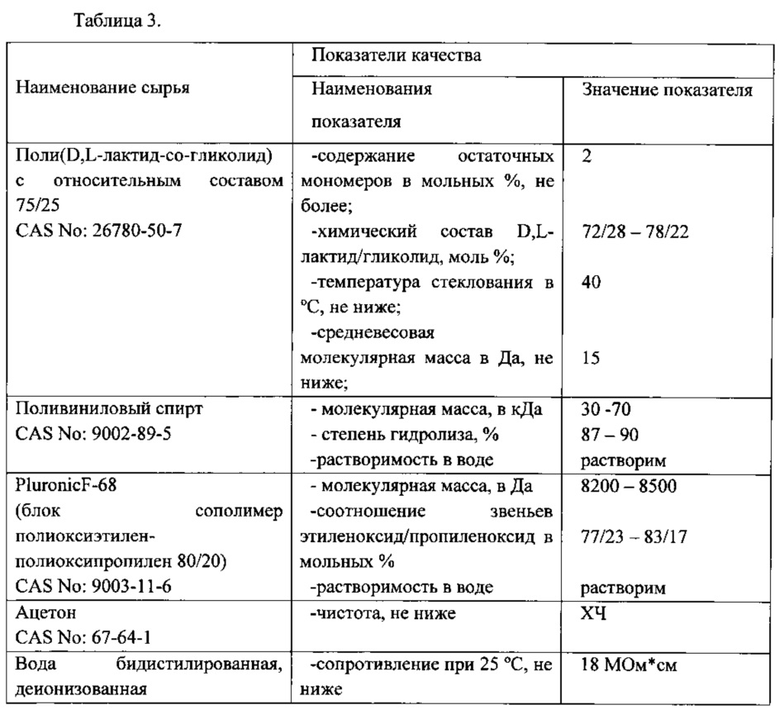
Характеристики сырья, используемого в эксперименте, представлены в Таблице 3.
Были изучены основные технологические факторы, влияющие на получение наноразмерных форм препарата нейротропного действия. Выявлено, что среди факторов, оказывающих влияние на получение наноразмерных форм существенно влияют: значение рН полимеризационной среды, соотношение полимер/растворитель, скорость и продолжительность перемешивания в процессе полимеризации, продолжительность инкубации и концентрация низкосиалированного эритропоэтина).
Очевидно, что на эффективность сорбции соединений различной природы на НЧ влияет целый ряд факторов, определяющих сродство этих соединений к поверхности, среди которых важную роль играет возможность электростатического взаимодействия между сорбируемым соединением и поверхностью наночастиц, с образованием псевдополиэлектролитных комплексов. Именно это взаимодействие может быть ключевым в случае эритропоэтина, обладающего выраженными кислотными свойствами (величина изоэлектрической точки эритропоэтина составляет рН 3,5-4,0, что обусловлено наличием сиаловых кислот в терминальных положениях углеводных цепочек эритропоэтина).
Кроме того, стабилизировать связывание целевых молекул с поверхностью НЧ могут и гидрофобные взаимодействия фрагментов молекул с наиболее гидрофобными участками поверхности НЧ. Для изучения возможности сорбции эритропоэтина были синтезированы НЧ на основе различных полимеров, полученные в присутствии стабилизаторов (поливиниловый спирт-ПВС, PluronicF-68) различной природы и не только определены размеры НЧ, но и оценены их поверхностные заряды.
Важное значение на степень сорбции имеет также соотношение лактида и гликолида в получаемом полимере. Было отдано предпочтение соотношению 75/25, так как в экспериментальном исследовании было показано, что в аналогичных условиях, при соотношении, например, 50:50 степень сорбции была значительно меньше.
Выяснилось, что размер частиц, правда, с другим носителем-прогестероном, а не с эритропоэтином, наблюдался средним при соотношении 75/25, чем при соотношениях 50:50, 65:35, 85:15 и 100:0 [Wu XS. Synthesis, characterization, biodegradation, and drug delivery application of biodegradable lactic/glycolic acid polymers: Part III. Drug delivery application. Artif Cells Blood Substit Immobil Biotechnol. 2004; 32(4):575-91].
Молекулярная масса (MM) полимера один из факторов, влияющих на размер получаемых наночастиц и сроки их деградации. Для исследования влияния ММ сополимера на размер частиц синтезировали 3 образца сополимера лактида и гликолида с различными молекулярными массами:
- PDLGA-54 - сополимер лактида с гликолидом 75/25 (средневесовая молекулярная масса Mw=26 кДа);
- PDLGA-74 - сополимер лактида с гликолидом 75/25 (средневесовая молекулярная масса Mw=130 кДа):
- PDLGA-93 - сополимер лактида с гликолидом 75/25 (средневесовая молекулярная масса Mw=75 кДа):
Методом гель-проникающей хроматографии получено молекулярно-массовое распределение полимеров. Синтез полимеров проводили в реакторе периодического действия с добавлением оловосодержащего катализатора. При этом в концентрация олова в реакционной смеси не превышала 100 ppm, в то время как стандарт ASTMF-2579 допускает содержание олова не более 150 ppm в сополимерах лактида и гликолида, предназначенных для медицинских изделий [Standard Specification for Amorphous Poly(lactide) and Poly(lactide-co-glycolide) Resins for Surgical Implants. ASTM-F2579 - 10, 2010]. В качестве мономеров использовали D,L-лактид с чистотой более 99% (Purac, Голландия) и гликолид (SigmaAldrich, США) с чистотой более 99%, дополнительную очистку не проводили. Концентрация кислотных групп в мономерах определенная спектрофотометрически не превышала 3,5×10-7 моль/г. Синтезированные полимеры очищали от непрореагировавшего мономера и других примесей методом переосаждения из раствора в спирт.
Синтезированные полимеры изучены с применением ряда физико-химических методов исследования, которые позволили комплексно охарактеризовать синтезированные образцы. А также подтвердить их чистоту и соответствие заданным параметрам.
Ядерный магнитный резонанс Исследование полимеров методом ядерного магнитного резонанса (ЯМР) позволило установить химический состав и чистоту синтезированных образцов. Спектры получали на приборе фирмы Bruker (Германия) модель WP_250 SY при рабочей частоте 250,13 МГц, внутренний стандарт - Me4Si Для исследования образцы полилактида растворяли в дейтерированном хлороформе CDCl3. ЯМР-спектр образца PDLGA-54 до очистки с расшифровкой всех пиков приведен на Фиг. 1. Для расчета здесь и далее будут использованы сигналы СН-групп лактида (мономера и полимера) и CH2-группы гликолида, т.к. они лучше разнесены в сравнении с сигналами СН3 - групп. Расчет состава по приведенным на спектре интегралам дает следующий результат: 71% звеньев лактида, 26% звеньев гликолида и 3% мономера лактида. Остатки мономера, не вступившего в реакцию полимеризации могут негативно повлиять на характеристики полимера и неконтролируемо ускорить сроки его деградации, поэтому концентрацию остаточного мономера необходимо минимизировать (стандарт ASTM-F2579 допускает мольное содержание мономера не более 2% [Standard Specification for Amorphous Poly(lactide) and Poly(lactide-co-glycolide) Resins for Surgical Implants. ASTM-F2579 - 10, 2010]).
После очистки полимера, как видно из спектра (Фиг. 2), мономер полностью удаляется, конечный состав продукта: 72% лактида и 28% гликолида. Расхождение в 3% с заданным составом допускается стандартами [Standard Specification for Amorphous Poly(lactide) and Poly(lactide-co-glycolide) Resins for Surgical Implants. ASTM-F2579 - 10, 2010].
На Фиг. 3 и 4 представлены ЯМР-спектры образцов PDLGA-74 и PDLGA-93 после очистки. Видно, что мономер удален из обоих материалов. Состав сополимера PDLGA-74: 77% лактида и 23% гликолида, состав PDLGA - 93: 74% лактида и 26% гликолида
Таким образом, исследование методом ЯМР показало, что состав синтезированных образцов отличается от заданного не более, чем на 3%, при этом после очистки материалы не содержат непрореагировавшего мономера. Синтезированные образцы по всем исследованным параметрам удовлетворяют требованиям стандартов. Для получения наночастиц использовали все три синтезированных образца.
Гель-проникающая хроматография
Исследование полимеров методом гель-проникающей хроматографии (ГПХ) позволяет получить информацию о молекулярно-массовом распределении полимеров, определить ширину этого распределения (индекс полидисперсности) и среднестатистические значения молекулярной массы (среднечисленная ММ - Mn, средневесовая ММ - Mw). Исследования проводили на жидкостном хроматографе Laboratorniprisroje (Чехословакия) при 40°С на колонках Phenogel500 (размер пор 104 Å) и Phenogel1000 (размер пор 105 Å), в качестве растворителя использовали тетрагидрофуран, скорость подачи элюента 1 мл/мин. Образцы полимеров растворяли в тетрагидрофуране(концентрация около 20 мг/мл). Значение Mw и индекса полидисперсности определяли с помощью программного комплекса МультиХром.
Хроматограмма образца PDLGA-54 представлена на Фиг. 5. Пик с началом при 6 мин и окончанием в 10 мин характеризует молекулярно-массовое распределение полимера. В результате обработки найденные значения средневесовой молекулярной массы и индекса полидисперсности составили Mw=26 кДа и 1,8 соответственно.
Хроматограмма образца PDLGA-74 представлена на Фиг. 6. Это самый высокомолекулярный образец среди синтезированных. Получили значение Mw=130 кДа и PDI=2,3.
На Фиг. 7 представлена хроматограмма образца PDLGA-93. Mw=75кДа и PDI=1.86
Таким образом, синтезированные образцы PDLGA-54, PDLGA-74, PDLGA-93 имеют средневесовую молекулярную массу Mw равную 26, 130 и 75 кДа соответственно.
Дифференциальная сканирующая калориметрия Теплофизические свойства синтезированных образцов были исследованы методом дифференциальной сканирующей калориметрии (ДСК). С помощью этого метода можно определить такие характеристики полимеров, как: температура стеклования, температура кристаллизации и плавления, величины тепловых эффектов соответствующих переходов и др. Теплофизические характеристики образцов влияют на скорость деградации материалов, а также на стабильность полученных на их основе полимерных наночастиц.
ДСК кривые сняты на приборе Mettler Toledo DSC30 (температурный диапазон прибора -150-600°С) в динамическом режиме при скорости нагревания 20°С/мин. Измерения производились в потоке азота с расходом 50 мл/мин. Температуру стеклования определяли с помощью стандартного программного обеспечения, поставляемого с прибором.
Термограмму образца PDLGA-74 снимали в диапазоне температур от - 20 до 200°С (Фиг. 8). В диапазоне температур 45-50°С виден переход материала в высокоэластичное состояние, который характеризуется температурой стеклования Tg=45°С.
При наличии кристалличности в образце на термограмме при 160-180°С проявился бы переход, соответствующий плавлению кристаллической решетки, но такого эффекта не наблюдается, что говорит об аморфной структуре образца. Отсутствие кристалличности объясняется тем, что стеорегулярность полимерной цепи лактида нарушается статистическими включениями гликолида и все полимеры состава 75/25 будут полностью аморфны. Поэтому остальные термограммы снимали в диапазоне температур от -20 до 120°С.
Термограмма образца PDLGA-54 представлена на Фиг. 9. Температура стеклования Tg составила 42°С, более низкое значение в сравнении с образцом PDLGA-74 объясняется более низким значением молекулярной массы образца. Из термограммы образца (Фиг. 10) PDLGA-93 видно, что для него Tg=43,5°С.
Таким образом, в результате исследования образцов методом ДСК показано, что они имеют аморфную структуру, а значение температуры стеклования находится в диапазоне 42-45°С.
Термогравиметрический анализ
Термогравиметрический анализ (ТГА) представляет собой исследование потери массы образца при его нагревании. На основании полученных данных можно определить температуру деструкции материала, а также оценить его состав, поскольку разные компоненты сгорают при различной температуре. ТГА-кривые получены на приборе Mettler Toledo TG50 (весы М3 microbalance - диапазон измерения 0-150 мг, точность 1 мкг, температурный диапазон 30-900°С) в динамическом режиме от 30 до 700°С со скоростью нагрева 10°С/мин. Измерения производились в потоке воздуха с расходом 200 мл/мин. Температуру деструкции определяли с помощью стандартного программного обеспечения, поставляемого с прибором.
ТГА-кривые образцов представлены на Фиг. 11. Из кривых видно, что температура деструкции образца PDLGA-54 составляет 250°С, образца PDLGA-74 около 230°С, для образца PDLGA-93 эта температура самая высокая - 280°С. Небольшая потеря массы образцов в диапазоне температур до 200°С обусловлена испарением содержащейся в материалах воды и низкомолекулярных фракций полимера. Вид кривой в области 200-300°С определяется преимущественно молекулярно-массовым распределением полимера. Для самого узкого распределения характерно резко начало деструкции (PLGA-74, PDLGA-93), тогда как для PDLGA-54 оно более плавное, что указывает на содержание фракций с небольшой молекулярной массой.
ИК-спектроскопия
Спектроскопия в инфракрасной области (ИК-спектроскопия)-метод исследования, позволяющий по инфракрасным спектрам поглощение установить строение молекул различных веществ.
Спектры синтезированных полимеров снимали на ИК-Фурье спектрометре Thermo Scientific Nicolet IR200e в области 500-4000 см-1. Образцы исследовали в твердом виде, помещая их в виде плоских пластинок на кристалл.
ИК-спектр образца сополимера PDLGA-74 приведен на Фиг. 12. Спектры двух других образцов - PDLGA-54 и PDLGA-93 выглядели аналогично, с небольшой разницей в высоте пиков, поэтому здесь они не приведены. Небольшие пики в области 3500 см-1 соответствуют ОН-группам воды содержащейся в образце и концевым группам полимера. Практически незаметные пики указывают на то, что в образцах воды практически нет. Дублет при 2995 и 2946 см-1 относится к ассиметричным колебания СН-группы. Высокий пик при 1751 см-1 соответствует колебаниям С=О связи в карбоксильной группе. Пики при 1452 и 1382 см-1 отвечают колебаниям -СН3 группы, содержащихся в звене лактида. Пики в области 1050-1269 см-1 отвечают эфирной связи -С-O-. Поглощение при 1250-1500 см-1 соответствуют деформационным колебаниям -СН3 и -CH2 групп [D'Avila Carvalho Erbetta С. Synthesis and Characterization of Poly(D,L-Lactide-co-Glycolide) Copolymer // Journal of Biomaterials and Nanobiotechnology. 2012. Vol. 03. №02. P. 208-225].
Таким образом, методом ИК-спектроскопии подтвержден химический состав синтезированных полимеров. Показано, что содержание воды в синтезированных образцах пренебрежимо мало. Данный метод среди прочих выделяется простотой и скоростью получения результатов, поэтому может быть использован как экспресс-метод детектирования сополимера лактида и гликолида в смеси других твердых веществ.
Исследование влияния условий процесса на размер и стабильность наночастиц плацебо
Исследовали влияние следующих параметров на размер и стабильность суспензии наночастиц:
- молекулярная масса сополимера лактида и гликолида 75/25;
- концентрация органической фазы;
- тип стабилизатора и его концентрация в водной фазе.
Наработка образцов наночастиц плацебо при варьировании условий эксперимента Процесс получения полимерных наночастиц (НЧ) методом замены растворителя нестационарный, поэтому каждый образец получали в трех разных экспериментах для исследования воспроизводимости характеристик. Параметры приготовления суспензии НЧ приведены в таблице 4.

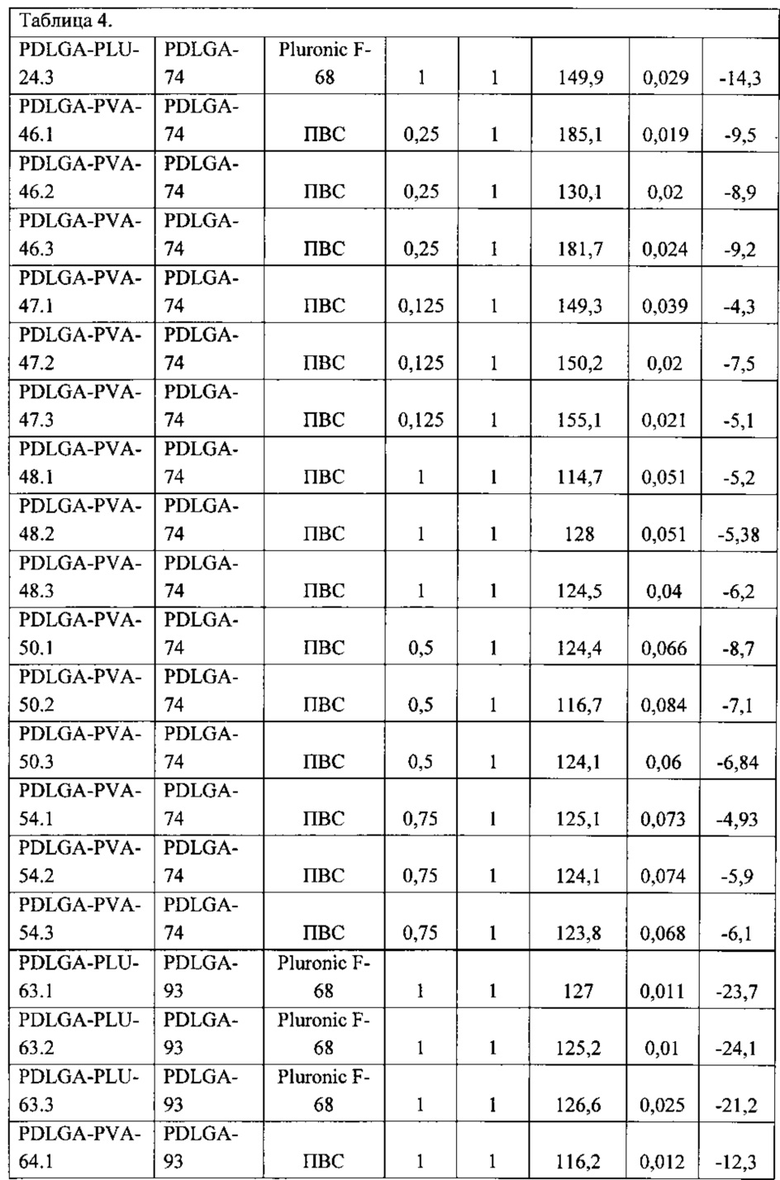
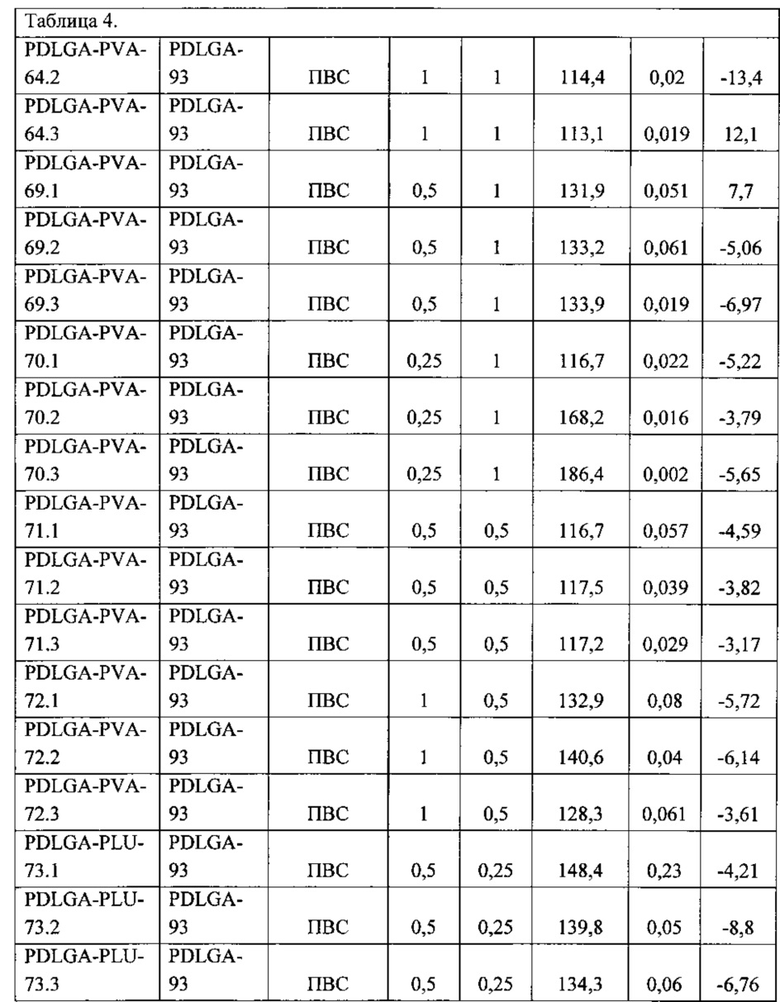
Влияние концентрации PluronicF-68 в водной фазе на распределение наночастиц по размерам представлено на Фиг. 13, использовали полимер PDLGA-54. Поскольку каждый эксперимент повторяли 3 раза, на Фиг. 13 и далее представлено усредненное по группе из 3-х образцов распределение наночастиц. Следует отметить что размер пика в распределении и средний размер, приведенный в таблице могут отличаться, т.к. средний размер - статистическая величина, при ее расчете учитывается ширина распределения, его симметричность и др.
Из Фиг. 13 и Таблицы 4 видно, что максимальный размер 239 нм наблюдается для частиц, полученных при концентрации водной фазы 1%, при снижении этой величины размер частиц падает и достигает минимума при концентрации 0,25 и 0,125% - 185 нм и 190 нм соответственно.
Сравнивая образцы PDLGA-PVA-17 и PDLGA-PLU-19, полученные при одинаковой концентрации водной фазы, но разном типе стабилизатора можно увидеть, что в случае поливинилового спирта размер меньше на 100 нм: 147 нм против 239 нм (Фиг. 14).
Стоит отметить, что наночастицы стабилизированные PluronicF-68 не всегда успешно ресуспендировались после центрифугирования, существенная часть частиц оставалась в виде агломератов, что по-видимому связано с недостаточно хорошим стабилизирующим эффектом этого полимера. В случае же поливинилового спирта осадок после центрифугирования ресуспендировался хорошо, при этом размер полученных частиц, как видно из Фиг. 14, был существенно меньше, поэтому для дальнейших экспериментов было решено использовать в качестве стабилизатора именно ПВС.
Влияние концентрации поливинилового спирта в водной фазе на распределение наночастиц по размерам представлено на Фиг. 15. Суспензии получены с использованием PDLGA-74 - образцы: PDLGA-PVA-45, 46, 47, 48 и 54.
Анализ распределений, приведенных на Фиг. 15 показывает, что уменьшение концентрации ПВС с 1 до 0,5% практически не оказывает влияния на размер частиц и ширину распределения. По-видимому, это связано с тем, что стабилизатор присутствует в избытке. Дальнейшее снижение концентрации приводит к увеличению размеров частиц.
Такой эффект можно объяснить тем, что ПВС становится недостаточно для того, чтобы эффективно стабилизировать всю площадь поверхности наночастиц, которые увеличиваются в размерах тем самым уменьшая суммарную площадь поверхности.
Следует отметить, что при концентрациях от 1 до 0,25% масса образующегося осадка в процессе получения осадка не превышает 5 мг. В случае концентрации 0,125% масса осадка резко возросла - до 62 мг, это можно объяснить тем, что часть сополимера лактида и гликолида коллапсирует в массу из-за критического недостатка стабилизатора.
Меньший размер частиц в случае концентрации 0,125%, чем в случае 0,25% можно объяснить тем, что в результате выпадения сополимера в осадок, фактическая концентрация органической фазы уменьшается, стабилизатора становится достаточно для стабилизации наночастиц меньших размеров.
Влияние концентрации ПВС в водной фазе на распределение полученных наночастиц по размерам при использовании PDLGA-93 представлено на Фиг. 16. Видно, что распределение смешается в сторону больших размеров при уменьшении концентрации.
На Фиг. 17 приведен анализ влияния концентрации PDLGA-93 в органической фазе на распределение частиц по размерам. Концентрация водной фазе оставалась постоянной - 0,5%. При уменьшении концентрации органической фазы размер сначала падет, это можно объяснить тем, что в малом объеме смеси ацетона и воды становится меньше полимера и частица, образованная при его коллапсе приобретает меньший размер. При дальнейшем уменьшении концентрации водной фазы распределение смещается вправо, чтобы объяснить этот эффект требуется провести дополнительные исследования.
Анализ зависимости среднего размера наночастиц от молекулярной массы используемого сополимера PDLGA показывает, что она практически не оказывает влияния на размер и дзета-потенциал частиц. Воспроизводимость трех одинаковых экспериментов тем лучше, чем выше концентрация стабилизатора в водной фазе, это связано с нестационарностью процесса получения наночастиц. Также можно заметить, что в целом образцы, стабилизированные PluronicF-68 имели больший размер, чем в случае стабилизации поливиниловым спиртом. Однако по величине дзета-потенциала последние уступали частицам с PluronicF-68. Вместе с тем, несмотря на более высокий дзета-потенциал частицы с PluronicF-68 не полностью ресуспендировались после центрифугирования, это является критическим недостатком.
Таким образом, на основании проведенного исследования выявлены оптимальные параметры получения наночастиц, перспективных для нагрузки низкосиалированным эпритропоэтином. Концентрация полимера в органической фазе: 0,5-1%, оптимальным стабилизатором является поливиниловый спирт с концентрацией в водной фазе 0,5-1%.
При использовании полимера PDLGA-54 - сополимера лактида с гликолидом 75/25 со средневесовой молекулярной массой Mw=26 кДа и стабилизатора Pluronic F-68 в различных концентрациях органической фазы, от 0,125 до 0,5% для получения образцов наночастиц, размер частиц был значительно больше, в сравнении с применением стабилизатора ПВС с концентрацией органической фазы 1%.
Так, размер получаемых частиц с применением 1% ПВС не превышал ожидаемой и рекомендуемой величины, а именно:<150 нм.
Тогда как размер наночастиц, получаемых при применении стабилизатора Pluronic F - 68 стабильно превышал эту величину даже в самой низкой концентрации. Средний размер частиц при концентрации органической фазы 0,125% составлял 190,4 нм. Средний размер частиц при концентрации органической фазы 0,25% составлял 184,9 нм; при концентрации органической фазы 0,5% и 1% - 207,9 и 232 нм, соответственно. Концентрация водной фазы во всех случаях составляла 1%.
Так как отчетливо наблюдалась тенденция к преимуществу использования ПВС, то для следующего образца PDLGA-74 - сополимера лактида с гликолидом 75/25 со средневесовой молекулярной массы Mw=130 кДа он был применен в большинстве своем. При концентрации ПВС 0,125% средним размером частиц являлся размер 151,5 нм. При концентрации ПВС 0,25% средним размером частиц являлся размер 165,6 нм. Средний размер частиц при концентрации стабилизатора 0,5% - 121, 7 нм, 1% - 122,4 нм. Размер наночастиц, получаемых при применении стабилизатора Pluronic F - 68 при концентрации 1% составляет 150,3 нм. Концентрация водной фазы также во всех случаях составляла 1%.
Наиболее оптимальный размер частиц получен при концентрации 1% ПВС и 1% водной фазы при использовании образца PDLGA-93 - сополимер лактида с гликолидом 75/25 со средневесовой молекулярной массой Mw=75 кДа и составлял 114,5 нм.
В результате исследований определены оптимальные параметры получения полимерных наночастиц плацебо: концентрация полимера в органической фазе: 0,5 - 1%, тип стабилизатора - ПВС, концентрация стабилизатора в водной фазе - 0,5 - 1%.
Определен приемлемый, наилучший, полимер для оптимального размера наночастиц <150 нм
Поэтому для дальнейших исследований был отобран полимер PDLGA-93 сополимер лактида с гликолидом 75/25 со средневесовой молекулярной массой Mw=75 кДа и стабилизатор: 1% ПВС (PVA(30-70)) в частности, с усредненным диаметром наночастиц 114,5 нм., среднее PDI составляет 0,017.
Нагрузка водной суспензии полимерных наночастиц плацебо низкосиалированным эритропоэтином (нсЭПО)
Процесс нагрузки полученных ранее наночастиц плацебо низкосиалированным эритропоэтином состоит из следующих шагов:
а) Суспензию полимерных наночастиц плацебо известной концентрации центрифугируют при ускорении 50 000 g в течение 30 мин для того, чтобы получить осадок наночастиц определенной массы. Для этой процедуры используют ультрацентрифугу, способную поддерживать вакуум для обеспечения высоких ускорений. После центрифугирования, супернатант удаляют;
б) К осадку, полученному на предыдущем шаге, добавляют раствор низкосиалированного эритропоэтина известной концентрации. При этом соотношение массы наночастиц в осадке и массы добавленного нсЭПО выбирают в диапазоне от 100:1 до 100:5.
в) К полученной смеси добавляют буферный раствор, имеющий рН в диапазон 3,6-4,0. Тип буферного раствора и его объем выбирают так, чтобы буферной емкости раствора было достаточно для поддержания необходимого рН инкубационной среды. Предпочтительно использовать натрий-ацетатный буфер, причем вводить его объем не превышающий более, чем в 3 раза объем раствора введенного ранее нсЭПО;
г) Осадок полимерных наночастиц ресупендируют в полученной среде сначала механически с помощью стерильной металлической иглы, затем путем озвучивания в ультразвуковой ванне в течение 5 мин. При этом мощность озвучивания необходимо контролировать так, чтобы температура воды в ванне в процессе озвучивания не превышала 37°С. При наблюдении агломератов в ресуспендированной суспензии процедуру озвучивания повторяют до полного ресуспендирования осадка наночастиц:
д) Полученную суспензию инкубируют при комнатной температуре в течение 3 часов, перемешивая с помощью шейкера при оборотах 700-800 rpm;
е) После окончания инкубирования суспензию центрифугируют при ускорении 50 000 g и температуре 4-8°С в течение 30 мин для того, чтобы удалить не сорбировавшийся на поверхность наночастиц нсЭПО.
ж) Супернатант отбирают и анализируют на предмет определения концентрации не сорбировавшегося нсЭПО методом спектрофотометрии, а осадок ресуспендируют в дистилированной воде аналогично процедуре, описанной выше.
Исследование и получение образцов полимерных наночастиц, нагруженных нсЭПО.
Отработка условий получения и характеризация суспензионных образцов наночастиц, нагруженных низкосиалированным эритропоэтином (нсЭПО)
Образцы полимерных наночастиц плацебо были получены способом, описанным выше, методом замены растворителя. Условия приготовления суспензии полимерных НЧ и характеристики реагентов приведены в таблице 5. Все суспензии (за исключением образца PDLGA-PLU-63) были приготовлены в 3 экземплярах с одинаковыми параметрами приготовления, это продиктовано необходимостью контроля повторяемости результатов.
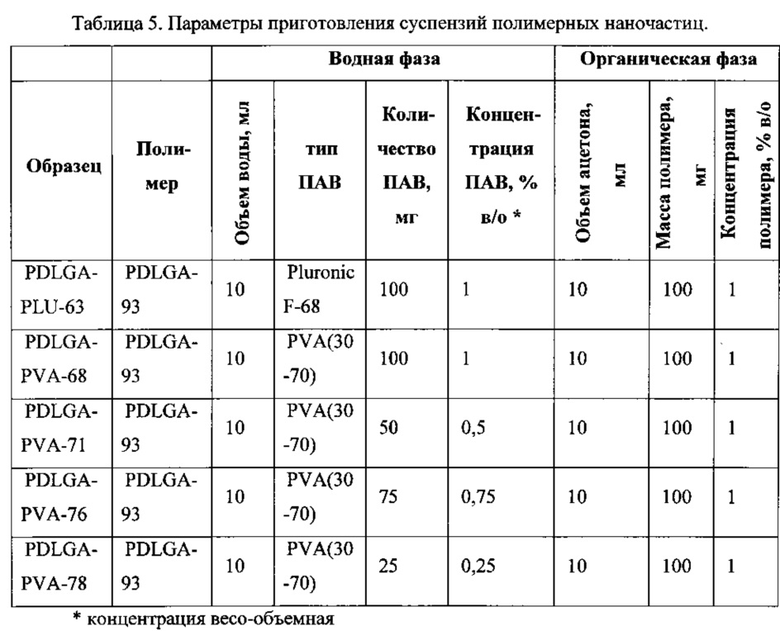
С(ПАВ в/о)=(0,1 г ПАВ*100%)/10 мл=1% ПАВ (в/о)
Полученные суспензии были отфильтрованы через предварительно смоченный деионизованной водой стеклянный фильтр (размер пор - 3 мкм) для удаления механических примесей и возможного осадка. Затем фильтр промывали деионизованной водой для вымывания осевших в нем (фильтре) наночастиц и доводили объем суспензии до 10 мл. Для удаления невключенного в состав частиц стабилизатора (ПАВа) суспензии центрифугировали при ускорении 20 000 g в течение 30 мин, отбирали супернатант и ресуспендировали осадок в исходном объеме воды. Осадок диспергировали как механически, так и с помощью ультразвуковой ванны.
Концентрацию полученной суспензии определяли методом выпаривания. Для этого, 1 мл суспензии наливали в предварительно взвешенную тару, затем воду выпаривали досуха на нагревательной плитке при температуре 150°С. Изменение в разности массы и соответственно концентрация полученной суспензии приведены в таблице 6.

Так же, полученные суспензии были охарактеризованы методом динамического светорассеяния. Исследование проводили на приборе MalvernZetasizerNanoZS в рабочем диапазоне детектируемых размеров частиц от 60 нм до 6 мкм. Фотодетектор был расположен под углом 173° к исходному направлению лазерного луча (длина волны лазерного излучения - 633 нм). Для исключения многократного рассеяния исходные суспензии разбавляли до концентрации около 1 мг/мл. Использовали кварцевую кювету с геометрической длиной пути света 10 мм.
Результаты исследования среднего диаметра, индекса полидисперсности (PDI) и дзета-потенциала образцов представлены в таблице 7.
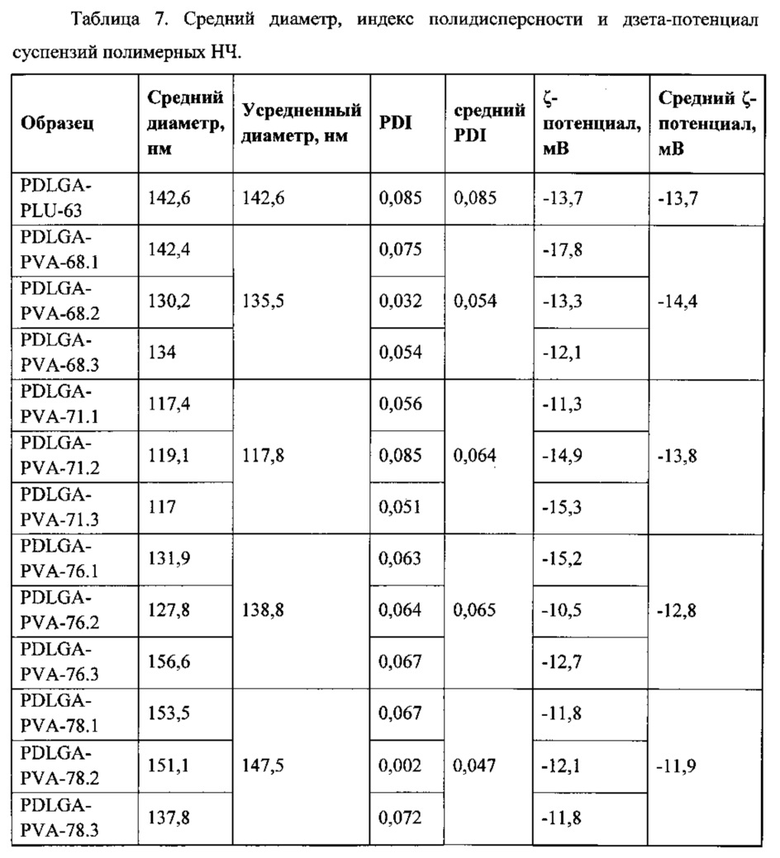
Как видно из таблицы 7, все суспензии удовлетворяют характеристикам, требующимся для успешного прохождения ГЭБ (средний диаметр наночастиц плацебо в суспензии ≤150 нм; Z-потенциал (по модулю) >10 мВ; индекс полидисперсности ≤0.2).
Исследования влияния условий нагрузки наночастиц нсЭПО на процент сорбции и стабильность полученных образцов [Известно, что на процент сорбции белков на наночастицы влияет время и температура инкубирования [Gene S. et al. Intranasal erythropoietin therapy in nervous system disorders. // Expert opinion on drug delivery. 2011. Vol. 8, №1. P. 19-32. Kreuter J. et al. Passage of peptides through the blood-brain barrier with colloidal polymer particles (nanoparticles). // Brain research. 1995. Vol. 674, №1. P. 171-174], но основное влияние оказывает значение водородного показателя раствора [Shi Y. et al. Improvement of in vivo efficacy of recombinant human erythropoietin by encapsulation in PEG-PLA micelle. // International journal of nanomedicine. 2013. Vol. 8. P. 1-11]. Из таблицы 7 следует, что поверхность НЧ является отрицательно заряженной и может экранироваться свободными положительными ионами раствора, а сам эритропоэтин является гликопротеином и способен к поляризации в заряженных средах в присутствии других ионов.
Нагрузку суспензионных образцов НЧ осуществляли следующим образом. Суспензия НЧ нужного объема с известной концентрацией центрифугировали при 50000 g в течение 30 минут, далее удаляли супернатант и получали осадок необходимой массы. Осадок ресуспендировали объемом водного раствора нсЭПО известной концентрации, либо для получения суспензии плацебо разводили осадок деионизованной водой того же объема. К полученной суспензии добавляли ацетатный буфер для варьирования рН, затем инкубировали заданное время. После инкубирования разделяли НЧ с включенным нсЭПО и невключенный нсЭПО в растворе на ультрацентрифуге при 50000 g, температуре 4-8°С, на протяжении 30 мин. Супернатант с невключенным ЛС отбирали для дальнейшего контроля концентрации по описанной ниже методике. Все образцы готовили парно, нагруженные ЛС и плацебо, спектр супернатанта плацебо вычитался из спектра образца.
Определение концентрации нсЭПО спектрофотометрическим методом Известно, что эритропоэтин человека, как и многие другие белки, обладает характерным пиком поглощения в районе 280 нм в ближней ультрафиолетовой области [Lai P., Everett R., Wang F. Structural characterization of human erythropoietin. // Journal of Biological Chemistry. 1986. Vol. 261, №7. P. 3116-3121.].
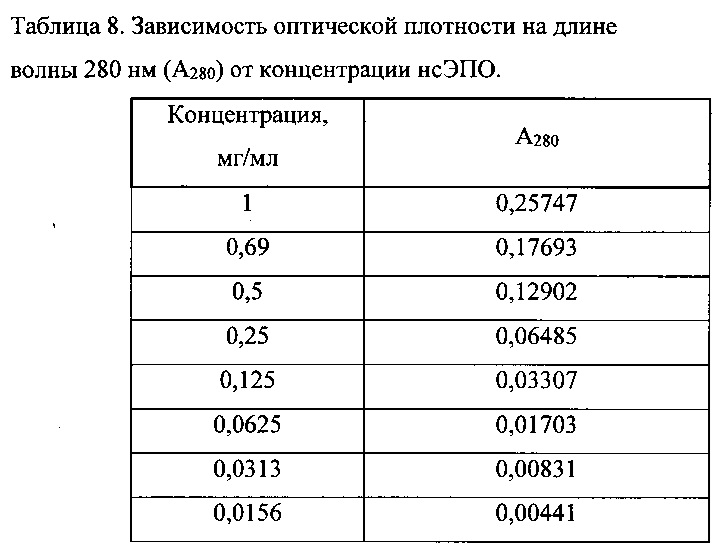
Кроме того, в данной области нет выраженных пиков поглощения других компонентов наноразмерной системы доставки (полимера, ПАВа, стабилизаторов и др.). Ввиду малых количеств образца было решено использовать микрокюветы с геометрической длиной прохождения света 2 мм. Измерения были проведены на приборе ShimadzuUV-3600 (Shimadzucorp., Япония) в диапазоне длин волн 230-350 нм, скорость сканирования - средняя. Спектры водных растворов нсЭПО различной концентрации в буфере представлены на Фиг. 18. Спектр водного раствора с концентрацией 1 мг/мл на Фиг. 18 не представлен из-за его высокой интенсивности. Зависимость оптической плотности на длине волны 280 нм (А280) от концентрации приведена в таблице 8. Измерения для более низких концентраций нсЭПО затруднительны в виду собственного шума прибора (около 0,0001 Abs), отклонения же величины А280 при концентрации 0,0156 мг/мл не превышают 5%. На основании данных Таблицы 8 была построена калибровочная кривая для водных растворов нсЭПО (Фиг. 19) и получено уравнение пересчета величины А280 в концентрацию нсЭПО:
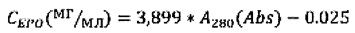
Подставляя в данное уравнение оптическую плотность водного раствора с неизвестной концентрацией нсЭПО, можно определить последнюю.
Так же на основании полученных данных можно определить коэффициент экстинкции для нсЭПО в растворе по формуле:
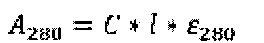
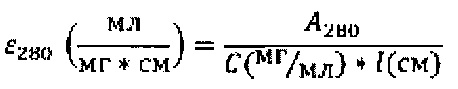
В процессе измерений использовались кюветы с геометрической длиной пути светового луча 2 мм, поэтому l=0,2 см, и, следовательно, коэффициент экстинкции:
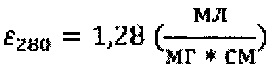
С помощью данной величины при необходимости можно выполнить определение концентрации нсЭПО в водном растворе на другом оборудовании используя другие (например, односантиметровые) кюветы.
В литературе, посвященной сорбции ЛВ на наночастицы, присутствует путаница и неоднозначность в вопросе терминологии, разные группы исследователей по-разному называют и обозначают одни и те же величины. При обсуждением вопросов нагрузки нсЭПО на НЧ мы использовали следующую терминологию. Если у нас есть суспензия НЧ в воде с общей массой наночастиц (полимер + ПАВ)=М, и мы добавляли в суспензию m лекарственного соединения, а после процесса нагрузки на частицы сорбировалось msorbлекарственного соединения, то тогда можно определить следующие обозначения:
Степень сорбции
α=msorb/m
показывает сколько ЛС от введенного в реакцию сорбировалось на НЧ, максимальное значение αmax=1. Данное значение показывает экономическую эффективность процесса нагрузки.
Процент сорбции
α(%)=(msorb/m)*100%
Степень нагрузки (degreeofdownloading, DOD) DOD - это отношение msorb/M, оно показывает терапевтическую эффективность лекарственной формы.
DOD=msorb/M
Процент нагрузки
DOD(%)=(msorb/M)*100%
Степень сорбции так же можно определить через концентрацию лекарственного соединения в растворе до и после нагрузки, действительно:
α=msorb/m=(m-msolv)/m
где msolv - масса ЛС, оставшаяся после нагрузки в растворе, далее:
α=(m-msolv)/m=1-msolv/m=1-Csolv/C
или в процентах (процент сорбции нсЭПО):
α(%)=(1-Csolv/C)*100%
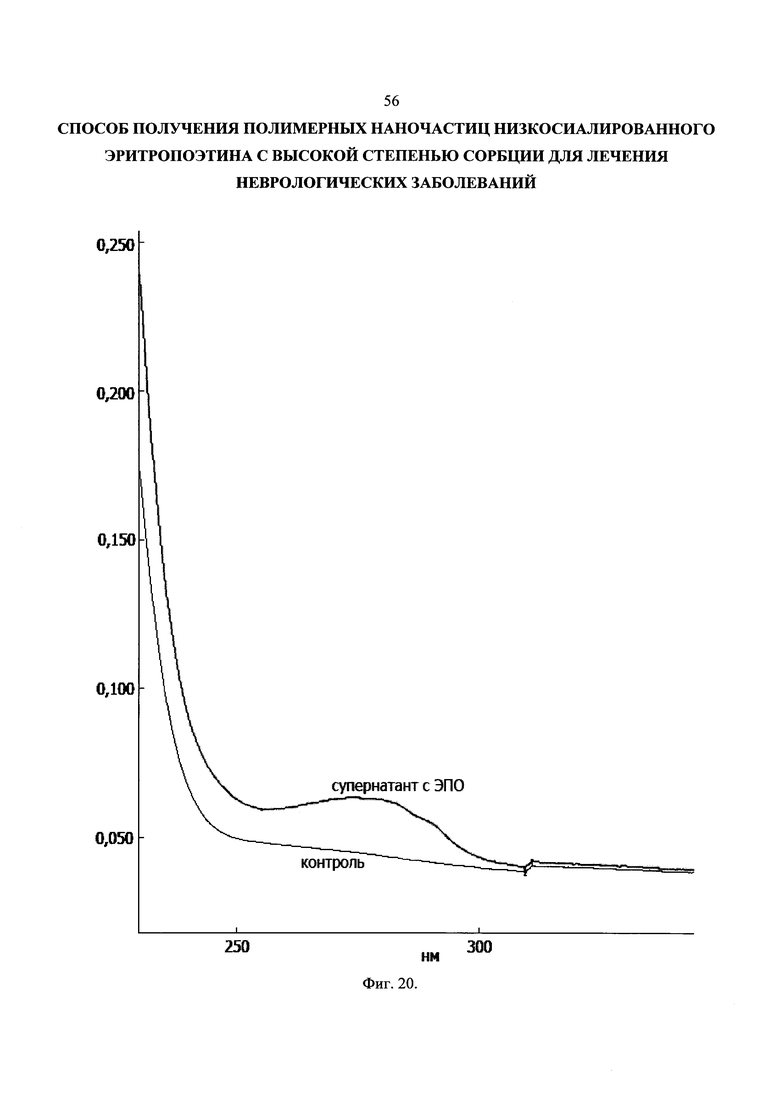
Следовательно, для определения степени сорбции достаточно измерить концентрацию ЛС после нагрузки в растворе (супернатанте), концентрация введенного ЛС известна. Типичный спектр супернатанта суспензии с ЭПО в сравнении с супернатантом суспензии плацебо (контроль) представлен на Фиг. 20.
Для исследования влияние времени инкубирования были приготовлены образцы суспензий, параметры которых отражены в Таблице 9.
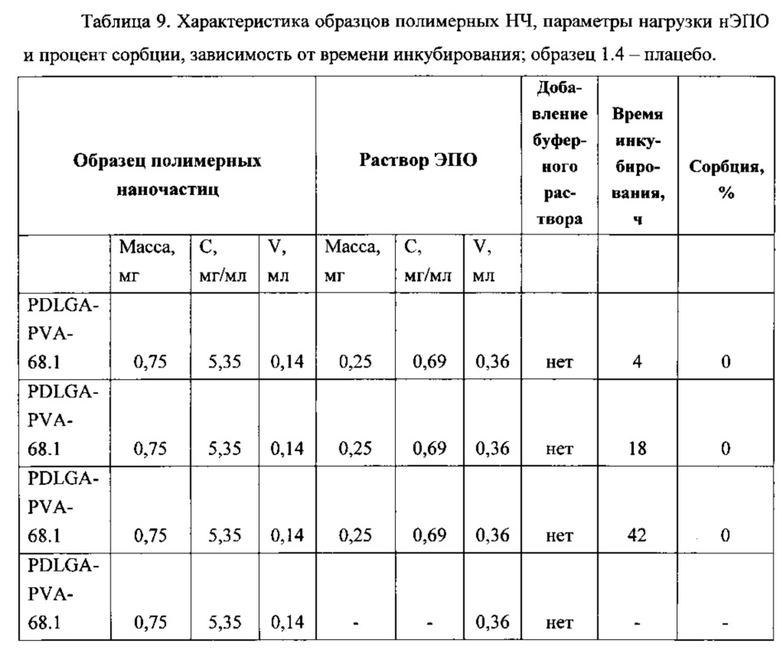
Из Таблицы 9 видно, что при исходном рН вне зависимости от времени инкубирования нсЭПО не сорбируется на поверхность наночастиц и остается в растворе. Мы считаем, что это вызвано нейтральной кислотностью буфера (рН=6.8), в котором растворен нсЭПО. Было предположение, что возможно достичь высокого процента сорбции при ресуспензировании раствором нсЭПО лиофилизата. Образец с2.1 представлял собой лиофилизат полимерных НЧ массой 2 мг, который ресуспенизировали раствором нсЭПО в буфере объемом 0,3 мл, время инкубирования составляло 20 часов. Процент сорбции оказался не более 1%. Были проведены эксперименты с другим типом полимера - блок-сополимер PDLA-PEG-PDLA (образец с3.1), но и в этом случае сорбции нсЭПО на наночастицы практически не наблюдалось.
Выше указывалось, что процент сорбции сильно зависит значения водородного показателя раствора, в котором происходит инкубирование, и при снижении рН до 4 и ниже степень сорбции значительно увеличивается. Для увеличения кислотности раствора был использован ацетатный буфер с рН=3.8, который добавляли в суспензию перед инкубированием.
В результате исследований отобрали образец PDLGA-PVA-68 (образцы PDLGA-PVA-68.1, PDLGA-PVA-68.2 PDLGA-PVA-68.3) с оптимальными параметрами, в частности, с усредненым диаметром наночастиц 135,5 нм, со средним PDI=0,054, Средним ζ-потенциалом, мВ=-14,4 мВ. Этот образец получали из сополимера лактида с гликолидом (D,L-лактида и гликолида) при соотношении лактида и гликолида как: 75/25 и средневесовой молекулярной массой Mw=75 кДа = полимера PDLGA-93. Брали навеску полимера в количестве 100 мг, растворяли в 10 мл ацетона и получали 1% концентрацию этого полимера, органическую фазу. Для получения водной фазы брали навеску поливинилового спирта PVA (30-70) с Mr 30-70 kDa в количестве также 100 мг, растворяли в 10 мл воды, получали 1% раствор водной фазы. При этом раствор поливинилового спирта перемешивают на магнитной мешалке HeidolphMRHei-Tec с подогревом и датчиком контроля температуры HeidolphEKTHei-Con с цилиндрическими тефлоновыми мешальниками длиной 1,5 см, при температуре 50°С до визуального растворения вещества, емкость, содержащую водную фазу нагревают до 55°С, смешивают эти растворы при постоянном перемешивании 700 rpm медленно добавляют органическую фазу к водной в пропорции 1:1 с помощью автоматического дозатора, скорость перемешивания увеличивают до 1000 rpm и оставляют систему перемешиваться для выпаривания ацетона на 1 час в вытяжном шкафу.
При дальнейшем исследовании образцов PDLGA-PVA-68.1, PDLGA-PVA-68.2 PDLGA-PVA-68.3 было установлено, что наибольший процент сорбции достигается у образца PDLGA-PVA-68.2, а именно 96%.
Не вызывает сомнений (Таблица 10), что снижение рН способствует значительному росту процента сорбции данного ЛС, вплоть до его полной сорбции на поверхности НЧ при низких значениях теоретического процента нагрузки (1%).
Примеры осуществления изобретения иллюстрируют, но не ограничивают настоящее изобретение.
Пример №1.
Приготовление органической фазы.
Навеску сополимера D,L-лактида и гликолида 75/25 со средневесовой молекулярной массой Mw=75 кДа - PDLGA - 93 массой 1,65 мг растворяют в 0,165 мл ацетона для получения раствора с концентрацией 0,01 г/мл. Раствор перемешивают на магнитной мешалке при комнатной температуре до полного растворения полимера и гомогенизации раствора.
Приготовление водной фазы.
Навеску ПВС поливинилового спирта массой 1,65 мг растворяют в 0,165 мл бидистилированной деионизованной воды для получения раствора с концентрацией 0,01 г/мл. Раствор готовят в стеклянном стакане объемом 50 мл.
Смешивание фаз и выпаривание органического растворителя.
Стакан, содержащий водную фазу нагревают до 55°С. При постоянном перемешивании (700 rpm) медленно добавляют органическую фазу к водной в пропорции 1:1 с помощью автоматического дозатора. Скорость перемешивания увеличивают до 1000 rpm и оставляют систему перемешиваться для выпаривания ацетона при температуре 55°С на 1 час в вытяжном шкафу.
Выделяют осадок наночастиц способом указанным выше.
К полученным наночастицам массой 3,3 г при их объеме в водной суспензии 0,15 мл добавляют водный раствор низкосиалированного ЭПО таким же объемом в количестве 0,15 мл с концентрацией 0,67 мг/мл и массой ЭПО 0,1 мг, добавляют двойной объем 0,3 мл натрий-ацетатного буфера при рН 3,8 и инкубируют 1 час при комнатной температуре 25 С°, степень (% сорбции) при этом составляет всего 48%.
Пример №2.
Приготовление органической фазы.
Навеску сополимера D,L-лактида и гликолида 75/25 со средневесовой молекулярной массой Mw=75 кДа - PDLGA - 93 массой 1,65 мг растворяют в 0,165 мл ацетона для получения раствора с концентрацией 0,01 г/мл. Раствор перемешивают на магнитной мешалке при комнатной температуре до полного растворения полимера и гомогенизации раствора.
Приготовление водной фазы.
Навеску ПВС поливинилового спирта массой 1,65 мг растворяют в 0,165 мл бидистилированной деионизованной воды для получения раствора с концентрацией 0,01 г/мл. Раствор готовят в стеклянном стакане объемом 50 мл.
Смешивание фаз и выпаривание органического растворителя.
Стакан, содержащий водную фазу нагревают до 55°С. При постоянном перемешивании (700 rpm) медленно добавляют органическую фазу к водной в пропорции 1:1 с помощью автоматического дозатора. Скорость перемешивания увеличивают до 1000 rpm и оставляют систему перемешиваться для выпаривания ацетона при температуре 55°С на 1 час в вытяжном шкафу.
Выделяют осадок наночастиц способом указанным выше.
К полученным наночастицам массой 3,3 г при их объеме в водной суспензии 0,15 мл добавляют раствор ЭПО таким же объемом в количестве 0,15 мл с концентрацией 0,67 мг/мл и массой низкосиалированного ЭПО 0,1 мг, добавляют двойной объем 0,3 мл натрий-ацетатного буфера при рН 3,8 и инкубируют 14 часов при комнатной температуре 25°С, степень (% сорбции) при этом составляет уже 74%.
Пример №3.
Приготовление органической фазы.
Навеску сополимера D,L-лактида и гликолида 75/25 со средневесовой молекулярной массой Mw=75 кДа - PDLGA - 93 массой 5 мг растворяют в 0,5 мл ацетона для получения раствора с концентрацией 0,01 г/мл. Раствор перемешивают на магнитной мешалке при комнатной температуре до полного растворения полимера и гомогенизации раствора.
Приготовление водной фазы.
Навеску ПВС поливинилового спирта массой 5 мг растворяют в 0,5 мл бидистилированной деионизованной воды для получения раствора с концентрацией 0,01 г/мл. Раствор готовят в стеклянном стакане объемом 50 мл.
Смешивание фаз и выпаривание органического растворителя.
Стакан, содержащий водную фазу нагревают до 55°С. При постоянном перемешивании (700 rpm) медленно добавляют органическую фазу к водной в пропорции 1:1 с помощью автоматического дозатора. Скорость перемешивания увеличивают до 1000 rpm и оставляют систему перемешиваться для выпаривания ацетона при температуре 55°С на 1 час в вытяжном шкафу.
Выделяют осадок наночастиц способом указанным выше.
К полученным наночастицам массой 10 г при их объеме в водной суспензии 0,15 мл добавляют раствор низкосиалированного ЭПО таким же объемом в количестве 0,15 мл с концентрацией 0,67 мг/мл и массой ЭПО 0,1 мг, добавляют двойной объем 0,3 мл натрий-ацетатного буфера при рН 3,8 и инкубируют 14 часов при комнатной температуре 25°С, степень (% сорбции) при этом составляет 96%.
Таким образом, установлено оптимальное соотношение массы НЧ в водной суспензии и низкосиалированного ЭПО, их концентрация, время инкубирования, температура инкубирования, определенный буфер с определенным объемом для достижения оптимальной % сорбции водного раствора низкосиалированного ЭПО на НЧ. При этом диаметр полученных частиц является оптимальным для прохождения через ГЭБ.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ФАРМАЦЕВТИЧЕСКОЙ КОМПОЗИЦИИ, СОДЕРЖАЩЕЙ ЦИННАРИЗИН | 2019 |
|
RU2727964C1 |
| АНТИБАКТЕРИАЛЬНОЕ СРЕДСТВО ДЛЯ ЛЕЧЕНИЯ ВНУТРИКЛЕТОЧНЫХ ИНФЕКЦИЙ | 2006 |
|
RU2308970C1 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ОКТРЕОТИДНЫЕ МИКРОЧАСТИЦЫ | 2003 |
|
RU2404748C2 |
| СИСТЕМА ДОСТАВКИ ВЕЩЕСТВА БЕЛКОВОЙ ПРИРОДЫ В ВИДЕ НАНОЧАСТИЦ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2013 |
|
RU2566069C2 |
| СПОСОБ ПОЛУЧЕНИЯ ФАРМАЦЕВТИЧЕСКОЙ КОМПОЗИЦИИ, СОДЕРЖАЩЕЙ ПЕНТОКСИФИЛЛИН | 2017 |
|
RU2702012C2 |
| НАНОЧАСТИЦЫ, ЛЕГЧЕ ПРОНИКАЮЩИЕ В СЛИЗИСТУЮ ОБОЛОЧКУ ИЛИ ВЫЗЫВАЮЩИЕ МЕНЬШЕ ВОСПАЛЕНИЯ | 2012 |
|
RU2631599C2 |
| ПОЛИЛАКТИДНЫЕ НАНОЧАСТИЦЫ | 2007 |
|
RU2423104C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПОЛИМЕРНЫХ ПРОТИВООПУХОЛЕВЫХ ЧАСТИЦ В ПРОТОЧНОМ МИКРОРЕАКТОРЕ И ЛИОФИЛИЗАТА НА ИХ ОСНОВЕ | 2018 |
|
RU2681933C1 |
| КОМПОЗИЦИИ С ЗАМЕДЛЕННЫМ ВЫСВОБОЖДЕНИЕМ, ВКЛЮЧАЮЩИЕ ОКТРЕОТИД И ДВА ИЛИ БОЛЕЕ СОПОЛИМЕРА ПОЛИЛАКТИДА И ГЛИКОЛИДА | 2006 |
|
RU2464972C2 |
| ПРЕПАРАТИВНАЯ ФОРМА С ЗАМЕДЛЕННЫМ ВЫСВОБОЖДЕНИЕМ, СОДЕРЖАЩАЯ ОКТРЕОТИД И ТРИ ЛИНЕЙНЫХ ПОЛИМЕРА ПОЛИ(ЛАКТИД-СО-ГЛИКОЛИДА) | 2009 |
|
RU2541104C2 |







Изобретение относится к области фармакологии, химии полимеров и нанотехнологиям и может быть использовано для получения полимерных наночастиц низкосиалированного эритропоэтина с высокой степенью сорбции, перпективных для лечения неврологических заболеваний. Способ получения наночастиц заключается в использовании 1% раствора сополимера PDLGA-93, представляющего собой сополимер D,L-лактида и гликолида при соотношении D,L-лактида и гликолида 75/25 и средневесовой молекулярной массой 70-80 кДа, в ацетоне и 1% раствора стабилизатора, представляющего собой раствор поливинилового спирта молекулярной массой 30-70 кДа в бидистиллированной деионизированной воде. Способ позволяет получать наночастицы, отличающиеся высокой стабильностью и наивысшей степенью сорбции препарата на наночастицах для прохождения через гематоэнцефалический барьер. 7 з.п. ф-лы, 20 ил., 4 пр.
1. Способ получения наночастиц низкосиалированного эритропоэтина с высокой степенью сорбции, способных к прохождению через гематоэнцефалический барьер, для лечения неврологических заболеваний, согласно которому
растворяют сополимер PDLGA-93, представляющий собой сополимер D,L-лактида и гликолида при соотношении D,L-лактида и гликолида 75/25 и средневесовой молекулярной массой 70-80 кДа, в ацетоне при температуре 20-25°С для получения 1-2% раствора сополимера;
растворяют поливиниловый спирт молекулярной массой 30-70 кДа в бидистиллированной деионизированной воде при температуре 50-60°С для получения 1-2% раствора стабилизатора;
затем для получения наночастиц сополимера смешивают полученные выше раствор сополимера и раствор стабилизатора при постоянном перемешивании;
смешивают полученные наночастицы с концентрацией в водной суспензии 0,05-0,10 г/мл с водным раствором низкосиалированного эритропоэтина с концентрацией 0,5-1,0 мг/мл в массовом соотношении 100:1;
далее для получения наночастиц низкосиалированного эритропоэтина добавляют натрий-ацетатный буфер для достижения диапазона рН 3,6-4,0 и инкубируют в течение 14 часов при температуре 20-25°С.
2. Способ получения наночастиц по п. 1, отличающий тем, что для получения 1% раствора сополимера PDLGA-93 в ацетоне берут 100 мг сополимера и 10 мл ацетона.
3. Способ получения наночастиц по п. 1, отличающий тем, что что для получения 1% раствора сополимера PDLGA-93 в ацетоне берут 5 мг сополимера и 0,5 мл ацетона.
4. Способ получения наночастиц по п. 1, отличающий тем, что для получения 1% раствора стабилизатора берут 100 мг поливинилового спирта и 10 мл деионизированной воды.
5. Способ получения наночастиц по п. 1, отличающий тем, что для получения 1% раствора стабилизатора берут 5 мг поливинилового спирта и 0,5 мл деионизированной воды.
6. Способ получения наночастиц по п. 1, отличающий тем, что полученные наночастицы с концентрацией 0,067 г/мл и массой 10 мг смешивают с водным раствором низкосиалированного эритропоэтина с концентрацией 0,67 мг/мл и массой 0,1 мг в одинаковых объемах, равных 0,15 мл.
7. Способ получения наночастиц по п. 1, отличающий тем, что натрий-ацетатный буфер добавляют в объеме 0,3 мл для достижения значения рН 3,8-4,0.
8. Способ получения наночастиц по п. 1, отличающий тем, что инкубирование с натрий-ацетатным буфером осуществляют при температуре 22-25°С.
| ЕЛИЗАРОВА О.С | |||
| и др.Нейропротекторный эффект низкосиалированного рекомбинантного эритропоэтина, включенного в наночастицы на основе сополимера молочной и гликолевой кислот у крыс с интрацеребральной посттравматической гематомой.Ж.Экспериментальная и клиническая фармакология, 2012,т.75, N.8, с.7-10 | |||
| C.PINTO REIS et al, Nanoencapsulation I | |||
| Methods for preparation of drug-loaded polymeric nanoparticles | |||
| Nanomedicine: Nanotechnology, Biology, and Medicine, 2006,2, p.12,13,14 | |||
| ОФТАЛЬМИЧЕСКИЕ КОМПОЗИЦИИ В ФОРМЕ ДЕПО ДЛЯ ПЕРИОКУЛЯРНОГО ИЛИ СУБКОНЪЮНКТИВАЛЬНОГО ВВЕДЕНИЯ | 2002 |
|
RU2316315C2 |
| Колосоуборка | 1923 |
|
SU2009A1 |

