Область изобретения
Настоящее изобретение относится к использованию новых производных, которые ингибируют киназу Btk и являются полезными для лечения аутоиммунных и воспалительных заболеваний, вызванных аберрантной активацией В-клеток.
Уровень техники
Протеинкиназы являются одним из самых крупных семейств ферментов человека и регулируют различные сигнальные процессы посредством добавления фосфатных групп к белкам (Т. Hunter, Cell 1987 50:823-829). В частности, тирозинкиназы фосфорилируют белки по фенольному остатку тирозина. Семейство тирозинкиназ включает в себя киназы, которые контролируют рост, миграцию и дифференцировку клеток. Аномальная активность киназ наблюдается при различных заболеваниях человека, включая рак, аутоиммунные и воспалительные заболевания. Таким образом, протеинкиназы являются одними из ключевых регуляторов клеточной сигнализации, они являются целью для модуляции клеточных функций посредством небольших молекулярных ингибиторов киназ и, тем самым, являются хорошей целью для разработки лекарственных препаратов. В дополнение к лечению опосредованных киназами процессов заболеваний, селективные и эффективные ингибиторы киназы также полезны при изучении клеточных сигнальных процессов и идентификации других клеточных мишеней в терапевтических целях.
Существуют хорошие доказательства того, что В-клетки играют ключевую роль в патогенезе аутоиммунных и/или воспалительных заболеваний. Терапевтические вещества на основе белков, разрушающие В клетки, такие как Ритуксан, эффективны против вызываемых аутоантителами воспалительных заболеваний, таких как ревматоидный артрит (Rastetter и соавт. Annu Rev Med 2004 55:477). Таким образом, ингибиторы протеинкиназ, которые играют роль в В-клеточной активации, должны быть полезными терапевтическими веществами для патологий, опосредованных В-клетками, таких как производство аутоантител.
Сигналинг, осуществляющийся через рецептор В-клеток (BCR) контролирует ряд ответов В-клетки, включая пролиферацию и дифференцировку в зрелые продуцирующие антитела клетки.
BCR является одним из ключевых регуляторов для В-клеточной активности и аберрантный сигналинг может привести к нерегулируемой В-клеточной пролиферации и образованию патогенных аутоантител, которые вызывают множество аутоиммунных и/или воспалительных заболеваний. Тирозинкиназа Брутона (Btk) не является BCR-связанной киназой, она находится вблизи мембраны и следует сразу за BCR по нисходящему пути сигналинга. Было показано, что отсутствие Btk блокирует BCR сигналинг и, следовательно, ингибирование Btk может быть полезным терапевтическим подходом для блокировки патологических процессов, опосредованных В-клетками.
Киназа Btk является членом Тес семейства тирозинкиназ, и, как было показано, является критическим регулятором раннего развития В-клеток и активации и выживания зрелых В-клеток (Khan и соавт. Immunity 1995 3:283; Ellmeier и соавт. J. Exp. Med. 2000 192:1611). Мутация киназы Btk у человека приводит к заболеванию агаммаглобулинемии, сцепленной с Х-хромосомой (XLA) (обзор у Rosen и соавт. New Eng. J. Med. 1995 333:431 и Lindvall и соавт. Immunol. Rev. 2005 203:200). У этих пациентов нарушена иммунная реакция, нарушено созревание В-клеток, повышенные уровни иммуноглобулинов и периферических В-клеток, уменьшенный Т-клеточнонезависимый иммунный ответ, а также ослабленная мобилизация кальция после стимуляции BCR
Подтверждение роли Btk в аутоиммунных и воспалительных заболеваниях также было получено на Btk-дефицитной модели мышей. В доклинических моделях системной красной волчанки (СКВ) на мышах, Btk-дефицитные мыши показывают заметные улучшения прогрессирования заболевания. Кроме того, Btk-дефицитные мыши устойчивы к коллаген-индуцированному артриту (Jansson и Holmdahl Clin. Exp. Immunol. 1993 94:459). Была продемонстрирована дозозависимая эффективность селективного ингибитора Btk на мышиной модели артрита (Z. Pan и соавт., Chem. Med Chem. 2007 2:58-61).
Btk также экспрессируется в клетках, отличных от В-лимфоцитов, которые могут быть вовлечены в патологический процесс. Например, Btk экспрессируется тучными клетками и Btk-дефицитные тучные клетки, полученный из костного мозга демонстрируют нарушение антиген индуцированной дегрануляции (Iwaki и соавт. J. Biol. Chem. 2005 280:40261). Это подтверждает, что киназа Btk могла бы быть полезна для лечения патологического ответа тучных клеток, такого как аллергия и астма. Также моноциты больных сцепленной с Х-хромосомой агаммаглобулинемией, у которых отсутствует Btk-активность, показали снижение продукции ФНО-альфа после стимуляции (Horwood и соавт. J Exp Med 197:1603, 2003). Таким образом, ФНО-альфа опосредованное воспаление может модулироваться низкомолекулярными ингибиторами Btk. Кроме того, Btk, как сообщается, играют важную роль в апоптозе (Islam и Smith Immunol. Rev. 2000 178:49) и, таким образом Btk ингибиторы были бы полезны для лечения некоторых В-клеточных лимфом и лейкемий (Feldhahn и соавт. J. Exp. Med. 2005 201:1837).
Краткое описание изобретения
В настоящем изобретении предложены Btk-ингибирующие соединения Формулы I, способы их применения, как описано в настоящей заявке далее:
В настоящем изобретении предложено соединение Формулы I,
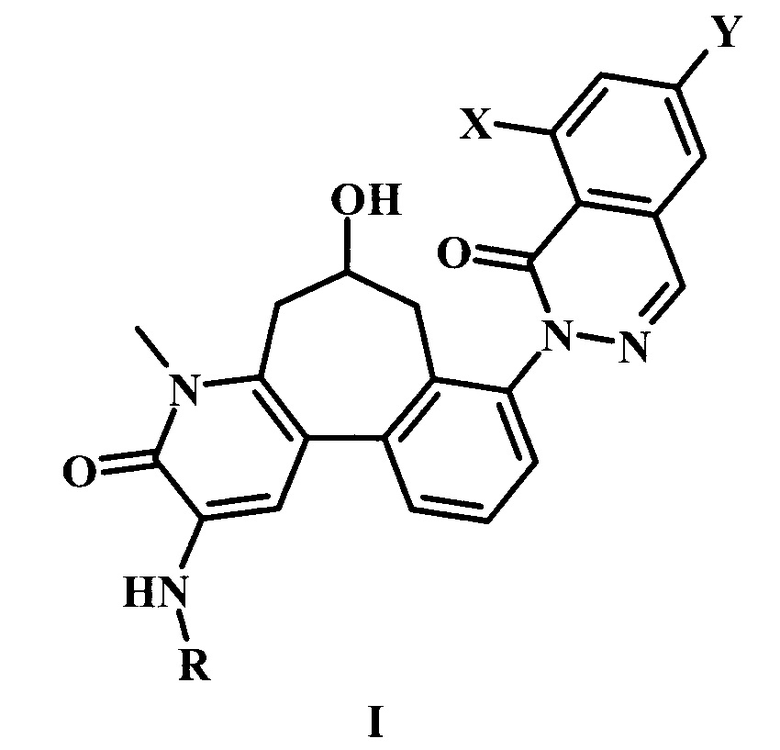
где:
X представляет собой галоген;
Y представляет собой Н или низший алкил;
R представляет собой -R1-R2-R3;
R1 представляет собой гетероарил;
R2 представляет собой -С(=O) или отсутствует;
R3 представляет собой гетероциклоалкил, возможно замещенный одним или более R3'; и
каждый R3' независимо представляет собой низший алкил, галоген, низший алкокси, или низший галоалкил;
или его фармацевтически приемлемая соль.
В настоящем изобретении предложен способ лечения воспалительного или аутоиммунного состояния, содержащий введение нуждающемуся в этом пациенту терапевтически эффективного количества соединения Формулы I.
В настоящем изобретении предложен фармацевтическая композиция, содержащая соединение Формулы I, смешанное с по меньшей мере одним фармацевтически приемлемым носителем, эксципиентом или разбавителем.
Подробное описание изобретения
Определения
Единственное число как используется в настоящем изобретении относится и к единственному и к множественному числу, например, «соединение» относится к одному или более соединений или по меньшей мере одному соединению. Соответственно единственное число, «один или более» и «по меньшей мере один» могут использоваться в настоящем изобретении взаимозаменяемо.
Фраза «как определено в настоящем изобретении выше» относится к наиболее широкому определению для каждой группы, как указано в разделе краткое описание изобретения или наиболее широком пункте формулы изобретения. Во всех остальных воплощениях приведенных ниже, заместители, которые могут быть представлены в каждом воплощении и которые специально не определены, сохраняют наиболее широкое значение из раздела краткое описание изобретения.
Как используется в данном описании, независимо от того, находится ли термин в переходной фразе или в теле пункта формулы изобретения, термины "содержит(-ат)" и "содержащий" следует толковать как имеющие открытое значение. То есть, термины должны быть интерпретированы как синонимы фраз «имеющие по меньшей мере» или «включающие по меньшей мере". При использовании в контексте способа, термин «содержащий» означает, что способ включает по меньшей мере перечисленные стадии, но может включать дополнительные стадии. При использовании в контексте соединений или композиций, термин «содержащий» означает, что соединение или композиция содержит по меньшей мере перечисленные признаки или компоненты, но также могут включать в себя дополнительные признаки и компоненты.
Как используется в настоящем изобретении, если специально не указано иного, союз «или» используется во «включительном» смысле как «или/и», а не в «исключительном» как «тот, или иной».
Термин «независимо» используется в настоящем изобретении для обозначения того, что радикал применяется в отдельно взятом случае независимо от наличия или отсутствия радикала, имеющего то же или другое определение в пределах одного соединения. Таким образом, в соединение, в котором Rʺ встречается дважды и определяется как "независимо углерод или азот", оба радикала Rʺ могут быть углеродом, оба Rʺ могут быть азотом, или один Rʺ может быть углеродом, а другой азотом.
Когда любая переменная встречается более чем один раз в любом остатке или формуле, изображающей или описывающей соединения, используемые или заявленные в настоящем изобретении, их определение в каждом случае является независимым от их значения в каждом другом случае. Также, комбинации заместителей и/или переменных возможны только в случае, если такие комбинации дают стабильные соединения.
Символы 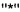 на конце связи или
на конце связи или 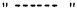 проходящий через связь, каждый относится к точке присоединения функциональной группы или другого химического остатка к остову молекулы, частью которой он является. Так, например:
проходящий через связь, каждый относится к точке присоединения функциональной группы или другого химического остатка к остову молекулы, частью которой он является. Так, например:
 .
.
Связь, нарисованная внутри кольца (в противоположность связанности с отдельной вершиной) означает, что связь может быть присоединена к любому подходящему кольцевому атому.
Термин "необязательный" или "возможно" как используется в настоящем изобретении описывает последующие события или обстоятельства, которые могут, но не обязаны случиться, и что описание включает случаи, при которых события или обстоятельства случаются и случаи, когда их нет. Например, «возможно замещенный» означает, что возможно замещенный остаток может включать атом водорода или заместитель.
Фраза «необязательная связь» означает, что связь может присутствовать или не присутствовать, и что описание включает простую, двойную или тройную связь. Если заместитель определен как «связь» или «отсутствует», атомы, связанные с данным заместителем тогда являются связанными напрямую.
Термин «около» используется в настоящем изобретении для обозначения приблизительно, в области, примерно или вблизи. Когда термин «около» используется вместе с числовым диапазоном, он изменяет этот диапазон расширением верхней и нижней границы указанных числовых значений. В общем, термин «около» используется в настоящем изобретении для изменения числового значения выше и ниже указанного значения на отклонение в 20%
Соединения формулы I могут проявлять таутомеризм. Таутомерные соединения могут существовать в виде двух или более взаимопереходящих видов. Прототропные таутомеры образуются в результате миграции ковалентно связанного атома водорода между двумя атомами. Таутомеры обычно существуют в равновесии, и попытки изолировать отдельные таутомеры обычно приводят к получению смеси, химические и физические свойства которой соответствуют смеси соединений. Положение равновесия зависит от химических свойств внутри молекулы. Например, у многих алифатических альдегидов и кетонов, таких как ацетальдегид, преобладает кето-форма, в то время как у фенолов преобладает енольная форма. Часто встречающиеся прототропные таутомеры включают кето/енол (-С(=O)-СН- ↔ -С(-ОН)=СН-), амид/имидовая кислота (-C(=O)-NH- ↔ -С(-OH)=N-) и амидиновые (-C(=NR)-NH- ↔ -C(-NHR)=N-) таутомеры. Последние два особенно распространены в гетероарильных и гетероциклических кольцах, и настоящее изобретение охватывает все таутомерные формы заявленных соединений.
Технические и научные термины, используемые в настоящем изобретении, имеют значение, обычно понимаемое квалифицированным специалистом в данной области техники, к которой относится настоящее изобретение, если не указано иного. Различные способы и материалы, на которые ссылается настоящее изобретение, известны специалистам в данной области техники. Стандартные справочные издания, в которых излагаются общие принципы фармакологии включают Goodman и Gilman's The Pharmacological Basis of Therapeutics, 10th Ed., McGraw Hill Companies Inc., New York (2001). Любые подходящие материалы и/или способы, известные квалифицированным специалистам могут быть использованы при осуществлении настоящего изобретения. Однако, предпочтительные материалы и способы описаны. Материалы, реагенты и т.п., на которые ссылается следующее описание и примеры могут быть получены из коммерческих источников, если не указано иного.
Определения, описанные в настоящем изобретении, могут соединяться с образованием химически релевантных комбинаций, таких как "гетероалкиларил", "галоалкилгетероарил", "арилалкилгетероциклил", "алкилкарбонил", "алкоксиалкил", "циклоалкилалкил" и т.п. Когда термин "алкил" используется в качестве суффикса после очередного термина, как в "фенилалкиле" или "гидроксиалкиле", это предназначено для обозначения алкильной группы, как определено выше, замещенной одним или двумя заместителями, выбранными из другой специально обозначенной группы. Так, например, "фенилалкил" относится к алкильной группе, содержащей от одного до двух фенильных заместителей, и, таким образом, включает бензил, фенилэтил и бифенил. "Алкиламиноалкил" представляет собой алкильную группу, имеющую от одного до двух алкиламино заместителей. "Гидроксиалкил" включает в себя 2-гидроксиэтил, 2-гидроксипропил, 1-(гидроксиметил)-2-метилпропил, 2-гидроксибутил, 2,3-дигидроксибутил, 2-(гидроксиметил), 3-гидроксипропил и т.д. Соответственно, используемый в настоящем изобретении термин «гидроксиалкил» используется для обозначения подгруппы гетероалкильных групп, определенных ниже. Термин -(ар)алкил относится как к незамещенным алкильным или аралкильным группам. Термин (гетеро)алкил или (гет)арил относится как к арильным, так и к гетероарильным группам.
Термин "спироциклоалкил", как используется в настоящем изобретении, означает спироциклическую циклоалкильную группу, такую как, например, спиро[3.3]гептан. Термин спирогетероциклоалкил, как используется в настоящем изобретении, означает спироциклический гетероциклоалкил, такой как, например, 2,6-диазаспиро[3.3]гептан.
Термин "ацил", используемый в настоящем изобретении, означает группу формулы -C(=O)R, где R представляет собой водород или низший алкил как определено в настоящем изобретении. Термин "алкилкарбонил", используемый в настоящем изобретении, означает группу формулы C(=O)R, где R представляет собой алкил, как определено в настоящем изобретении. Термин С1-6 ацил относится к группе -C(=O)R, содержащей 6 атомов углерода. Термин "арилкарбонил", используемый в настоящем изобретении, означает группу формулы C(=O)R, где R представляет собой арильную группу; термин "бензоил", используемый в настоящем изобретении, означает арилкарбонильную группу, где R представляет собой фенил.
Термин «эфир», как используется в настоящем изобретении, обозначает группу формулы -C(=O)OR, где R представляет собой низший алкил, как определено в настоящем изобретении.
Термин "алкил", используемый в настоящем изобретении, означает насыщенный, моновалентный, углеводородный остаток с неразветвленной или разветвленной цепочкой, содержащий от 1 до 10 атомов углерода. Термин "низший алкил" означает углеводородный остаток с неразветвленной или разветвленной цепочкой, содержащий от 1 до 6 атомов углерода. "С1-10 алкил", используемый в настоящем изобретении, относится к алкилу, состоящему из 1-10 углеродных атомов. Примеры алкильных групп включают, без ограничения, низшие алкильные группы, включая метил, этил, пропил, i-пропил, n-бутил, изобутил, трет-бутил или пентил, изопентил, неопентил, гексил, гептил и октил.
Когда термин "алкил" используется в качестве суффикса со следующим далее термином, как, например, в терминах "фенилалкил" или "гидроксиалкил", он предназначен для обозначения алкильной группы, как определено выше, замещенной от одного до двух заместителей, выбранных из другой специфично названной группы. Так, например, "фенилалкил" обозначает радикал R'Rʺ-, где R' представляет собой фенильный радикал, a Rʺ представляет собой алкиленовый радикал, как определено в настоящем изобретении, с пониманием того, что точка присоединения фенилалкильного остатка будет на алкиленовом радикале. Примеры арилалкильных радикалов включают, без ограничения, бензил, фенилэтил, 3-фенилпропил. Термины "арилалкил" или "аралкил" интерпретируются одинаково, исключая R' представляет собой арильный радикал. Термины "(гет)арилалкил" или "(гет)аралкил" интерпретируются одинаково, исключая R' представляет собой возможно арильный или гетероарильный радикал.
Термин «галоалкил» или «гало-низший алкил» или «низший алкил» относится к углеводородному остатку с прямой или разветвленной цепочкой, содержащему 1-6 атомов углерода, где один или более атомов замещены одним или более атомами галогена.
Термин "алкилен" или «алкиленил», используемый в настоящем изобретении, означает бивалентный насыщенный линейный углеводородный радикал из от 1 до 10 атомов углерода (например, (СН2)n) или разветвленный насыщенный бивалентный углеводородный радикал из 2-10 атомов углерода (например, -СНМе- или -CH2CH(i-Pr)CH2-), если не указано иного. За исключением случая с метиленом, открытые валентности алкиленовой группы не присоединены к одному атому. Примеры алкиленовых радикалов включают, без ограничения, метилен, этилен, пропилен, 2-метил-пропилен, 1,1-диметил-этилен, бутилен, 2-этилбутилен.
Термин "алкокси", используемый в настоящем изобретении, означает -О-алкил группу, где алкил является таким, как определено выше, такую как метокси, этокси, n-пропилокси, i-пропилокси, n-бутилокси, i-бутилокси, t-бутилокси, пентилокси, гексилокси, включая их изомеры. "Низший алкокси", используемый в настоящем изобретении, означает алкокси группу с "низшей алкильной" группой, как определено выше. "С1-10 алкокси", используемый в настоящем изобретении, относится к -О-алкилу, где алкил представляет собой С1-10.
Термин "РСу3" относится к фосфину, трехзамещенному тремя циклическими остатками.
Термин "галоалкокси" или "гало-низший алкокси" или "низший галоалкокси" относится к низшей алкоксигруппе, где один или более атомов углерода замещены одним или более атомами галогена.
Термин "гидроксиалкил" как используется в настоящем изобретении, означает алкильный радикал, как определено в настоящем изобретении, где от одного до трех атомов водорода на разных атомах углерода заменены гидроксильными группами.
Термин "алкилсульфонил" и "арилсульфонил", как используется в настоящем изобретении, относится к группе формулы -S(=O)2R, где R представляет собой алкил или арил соответственно, и алкил и арил являются такими, как определено в настоящем изобретении. Термин "гетероалкилсульфонил", как используется в настоящем изобретении, относится к группе формулы -S(=O)2R, где R представляет собой "гетероалкил", как определено в настоящем изобретении.
Термины «алкилсульфониламино» и "арилсульфониламино", как используется в настоящем изобретении, относится к группе формулы -NR'S(=O)2R, где R представляет собой алкил или арил соответственно, R' представляет собой водород или C1-3 алкил, и алкил и арил являются такими, как определено в настоящем изобретении.
Термин "циклоалкил", используемый в настоящем изобретении, относится к насыщенному карбоциклическому кольцу, содержащему от 3 до 8 атомов углерода, например, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил или циклооктил. "С3-7 циклоалкил", используемый в настоящем изобретении, относится к циклоалкилу, содержащему 3-7 атомов углерода в карбоциклическом кольце.
Термин "карбоксиалкил", как используется в настоящем изобретении, относится к алкильному остатку, в котором один атом водорода заменен карбоксилом, с понимание того, что точка присоединения гетероалкильного радикала находится на атоме углерода. Термин "карбокси" или "карбоксил" относится к остатку -CO2H.
Термины "гетероарил" или «гетероароматический», используемые в настоящем изобретении, означают моноциклический или бициклический радикал из 5-12 кольцевых атомов, имеющий по меньшей мере одно ароматическое или частично ненасыщенное кольцо, содержащее от четырех до восьми атомов на кольцо, включающих один или более гетероатомов N, О или S, остальные кольцевые атомы представляют собой углерод, принимая во внимание, что точка присоединения гетероарильного радикала будет находиться на ароматическом или частично ненасыщенном кольце. Как хорошо известно специалистам в области техники, гетероарильные кольца обладают меньшим ароматическим характером, чем их полностью углеродные аналоги. Таким образом, для целей изобретения, гетероарильная группа должна обладать только некоторой степенью ароматического характера. Примеры гетероарильных остатков включают моноциклические ароматические гетероциклы с 5-6 кольцевыми атомами и 1-3 гетероатомами, включая, без ограничения, пиридинил, пиримидинил, пиразинил, оксазинил, пирролил, пиразолил, имидазолил, оксазолил, 4,5-дигидро-оксазолил, 5,6-дигидро-4Н-[1,3]оксазолил, изоксазол, тиазол, изотиазола, триазолин, тиадиазол и оксадиаксолин, которые могут быть возможно замещены одним или более, предпочтительно одним или двумя заместителями, выбранными из гидрокси, циано, алкил, алкокси, тио, низший галогеналкокси, алкилтио, гало, низший галогеналкил, алкилсульфинил, алкилсульфонил, галоген, амино, алкиламино, диалкиламино, аминоалкил, алкиламиноалкил и диалкиламиноалкил, нитро, алкоксикарбонил и карбамоил, алкилкарбамоил, диалкилкарбамоил, арилкарбамоил, алкилкарбониламино и арикарбониламино. Примеры бициклических остатков включают, без ограничения, хинолинил, изохинолинил, бензофурил, бензотиофенил, бензоксазол, бензизоксазол, бензотиазол, нафтиридинил, 5,6,7,8-тетрагидро-[1,6]нафтиридинил и бензизотиазол. Бициклические остатки могут быть возможно замещены в любом кольце, однако точки присоединения находятся на кольце, содержащем гетероатом.
Термины «гетероциклил», «гетероциклоалкил» или «гетероцикл», как используется в настоящем изобретении обозначает моновалентный насыщенный циклический радикал, состоящий из одного или более колец, предпочтительно одного или двух колец, включая спироциклические кольцевые системы, из 3-8 атомов на кольцо, включающих один или более кольцевых гетероатомов (выбранных из N, O или S(O)0-2), и которые возможно могут быть независимо замещены одним или более, предпочтительно одним или двумя заместителями, выбранными из гидрокси, оксо, циано, низший алкил, низший алкокси, низший галогеналкокси, алкилтио, галоген, низший галогеналкил, гидроксиалкил, нитро, алкоксикарбонил, амино, алкиламино, алкилсульфонил, арилсульфонил, алкиламиносульфонил, ариламиносульфонил, алкилсульфониламино, арилсульфониламино, алкиламинокарбонил, ариламинокарбонил, алкилкарбониламино, арилкарбониламино и их ионных форм, если не указано иное. Примеры гетероциклических радикалов включают, без ограничения, морфолинил, пиперазинил, пиперидинил, азетидинил, пирролидинил, гексагидроазепинил, оксетанил, тетрагидрофуранил, тетрагидротиофенил, оксазолидинил, тиазолидинил, изоксазолидинил, тетрагидропиранил, тиоморфолинил, хинуклидинил и имидазолинил и их ионные формы. Примерами могут быть также бициклические, такие как, например, 3,8-диазабицикло[3.2.1]октан, 2,5-диазабицикло[2.2.2]октан или октагидро-пиразино[2,1-с][1,4]оксазин.
Ингибиторы Btk
В настоящем изобретении предложено соединение Формулы I,
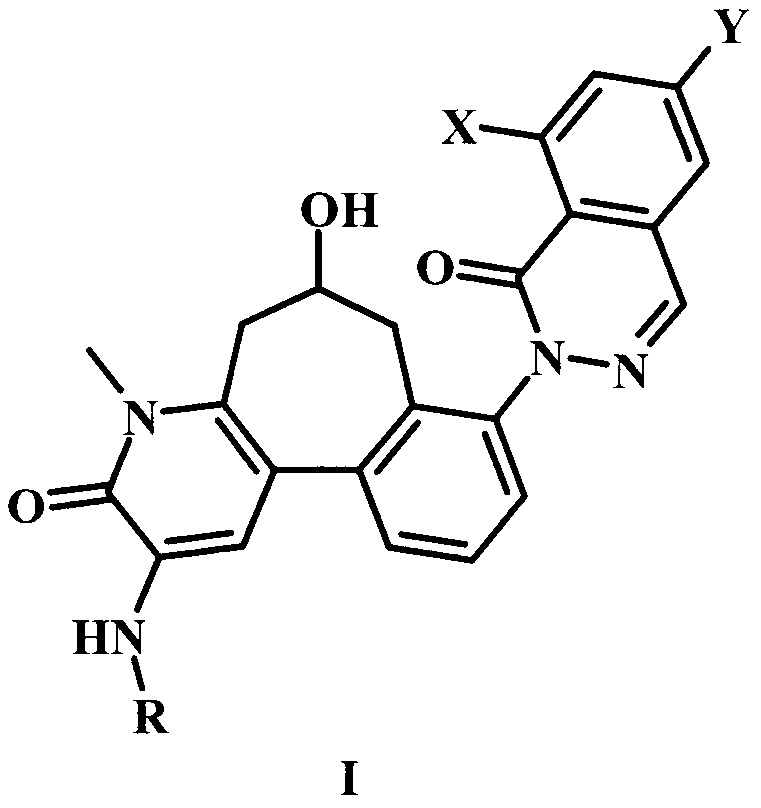
где:
X представляет собой галоген;
Y представляет собой Н или низший алкил;
R представляет собой -R1-R2-R3;
R1 представляет собой гетероарил;
R2 представляет собой -С(=O) или отсутствует;
R3 представляет собой гетероциклоалкил, возможно замещенный одним или более R3'; и
каждый R3' независимо представляет собой низший алкил, галоген, низший алкокси, или низший галоалкил;
или его фармацевтически приемлемая соль.
В настоящем изобретении предложено соединение Формулы I, где X представляет собой F.
В настоящем изобретении предложено вышеуказанное соединение Формулы I, где R1 представляет собой пиридил.
В настоящем изобретении предложено вышеуказанное соединение Формулы I, где R2 представляет собой -С(=O).
В настоящем изобретении предложено вышеуказанное соединение Формулы I, где R3 представляет собой морфолинил.
В настоящем изобретении предложено вышеуказанное соединение Формулы I, где Y представляет собой Н.
Альтернативно, в настоящем изобретении предложено соединение Формулы I, где Y представляет собой трет-бутил.
В настоящем изобретении предложено соединение Формулы I, где R2 отсутствует.
В настоящем изобретении предложено соединение Формулы I, где X представляет собой F, R2 отсутствует и R1 представляет собой пиридил.
В настоящем изобретении предложено вышеуказанное соединение Формулы I, где R3 представляет собой пирролидинил, возможно замещенный одним или более R3'.
В настоящем изобретении предложено вышеуказанное соединение Формулы I, где R3' представляет собой метил.
В настоящем изобретении предложено вышеуказанное соединение Формулы I, где X представляет собой F и Y представляет собой Н.
Альтернативно, в настоящем изобретении предложено соединение Формулы I, где X представляет собой F и Y представляет собой трет-бутил.
В настоящем изобретении предложено вышеуказанное соединение Формулы I, где X представляет собой F, R1 представляет собой пиридил, R2 отсутствует, R3 представляет собой пирролидинил, R3' представляет собой метил, и Y представляет собой циклопропил или диалкиламино.
В настоящем изобретении предложено соединение Формулы I, выбранное из группы, состоящей из:
8-(6-трет-бутил-8-фтор-1-оксофталазин-2-ил)-6-гидрокси-4-метил-2-[[5-(морфолин-4-карбонил)пиридин-2-ил]амино]-6,7-дигидро-5H-бензо[1,2]циклогепта[6,7-d]пиридин-3-он;
8-(6-трет-бутил-8-фтор-1-оксофталазин-2-ил)-6-гидрокси-4-метил-2-[[5-[(2S)-1-метилпирролидин-2-ил]пиридин-2-ил]амино]-6,7-дигидро-5Н-бензо[1,2]циклогепта[6,7-d]пиридин-3-он;
(6S)-8-(6-трет-бутил-8-фтор-1-оксофталазин-2-ил)-6-гидрокси-4-метил-2-[[5-(морфолин-4-карбонил)пиридин-2-ил]амино]-6,7-дигидро-5H-бензо[1,2]циклогепта[6,7-d]пиридин-3-он;
(6R)-8-(6-трет-бутил-8-фтор-1-оксофталазин-2-ил)-6-гидрокси-4-метил-2-[[5-(морфолин-4-карбонил)пиридин-2-ил]амино]-6,7-дигидро-5H-бензо[1,2]циклогепта[6,7-d]пиридин-3-он;
8-(8-фтор-1-оксофталазин-2-ил)-6-гидрокси-4-метил-2-[[5-(морфолин-4-карбонил)пиридин-2-ил]амино]-6,7-дигидро-5Н-бензо[1,2]циклогепта[6,7-d]пиридин-3-он;
(6S)-8-(8-фтор-1-оксофталазин-2-ил)-6-гидрокси-4-метил-2-[[5-(морфолин-4-карбонил)пиридин-2-ил]амино]-6,7-дигидро-5H-бензо[1,2]циклогепта[6,7-d]пиридин-3-он; и
(6R)-8-(8-фтор-1-оксофталазин-2-ил)-6-гидрокси-4-метил-2-[[5-(морфолин-4-карбонил)пиридин-2-ил]амино]-6,7-дигидро-5Н-бензо[1,2]циклогепта[6,7-d]пиридин-3-он.
В настоящем изобретении предложен способ лечения воспалительного и/или аутоиммунного состояния, содержащий введение нуждающемуся в этом пациенту терапевтически эффективного количества соединения Формулы I.
В настоящем изобретении предложен способ лечения ревматоидного артрита, содержащий введение нуждающемуся в этом пациенту терапевтически эффективного количества соединения Формулы I.
В настоящем изобретении предложен способ лечения астмы, содержащий введение нуждающемуся в этом пациенту терапевтически эффективного количества соединения Формулы I.
В настоящем изобретении предложена фармацевтическая композиция содержащая соединение Формулы I.
В настоящем изобретении предложена фармацевтическая композиция содержащая соединение Формулы I, смешанное по меньшей мере с одним фармацевтически приемлемым носителем, эксципиентом или разбавителем.
В настоящем изобретении предложено применение соединения Формулы I в качестве терапевтически активного вещества.
В настоящем изобретении предложено применение соединения Формулы I для производства лекарственного средства для лечения воспалительного нарушения.
В настоящем изобретении предложено применение соединения Формулы I для производства лекарственного средства для лечения аутоиммунного заболевания.
В настоящем изобретении предложено применение соединения Формулы I для производства лекарственного средства для лечения ревматоидного артрита.
В настоящем изобретении предложено применение соединения Формулы I для производства лекарственного средства для лечения астмы.
В настоящем изобретении предложено применение соединения, как описано выше, для лечения воспалительного и/или аутоиммунного состояния.
В настоящем изобретении предложено применение соединения, как описано выше, для лечения ревматоидного артрита.
В настоящем изобретении предложено применение соединения, как описано выше, для лечения астмы.
В настоящем изобретении предложено соединение, способ или композиция, как здесь описано.
Соединения и Получение
Примеры репрезентативных соединений, охватываемых настоящим изобретением и находящихся в объеме настоящего изобретения, приведены в следующей таблице. Эти примеры и методики, следующие далее, приведены для более ясного понимания квалифицированными специалистами настоящего изобретения и для облегчения его осуществления. Они не должны рассматриваться в качестве ограничивающих объем настоящего изобретения, но только в качестве его иллюстрации и отображения.
В основном, номенклатура, использованная в настоящем изобретении основана на AUTONOMTM v.4.0, компьютеризованной системе генерации названий химических соединений по ИЮПАК института Бельштейн. Если наблюдается несоответствие между изображенной структурой и названием, данным для этой структуры, изображенная структура обладает более высоким весом. Кроме того, если стереохимия структуры или части структуры не обозначена, например, связью или пунктирной линией, структура или часть структуры должны интерпретироваться как охватывающие все их стереоизомеры.
В ТАБЛИЦЕ I показаны примеры соединений в соответствии с общей Формулой I.:

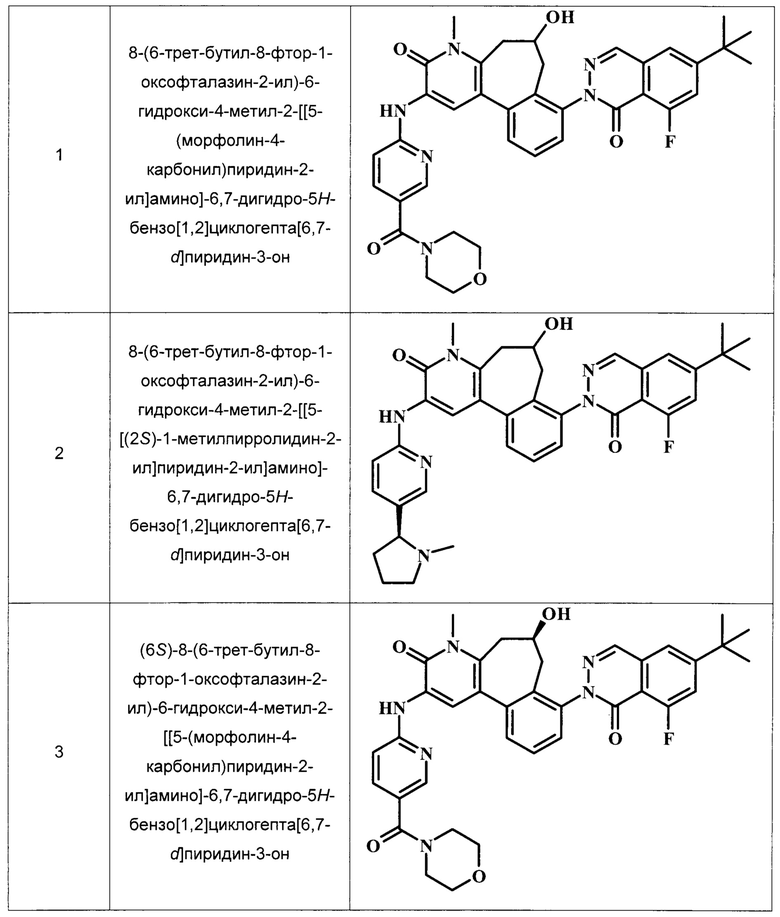
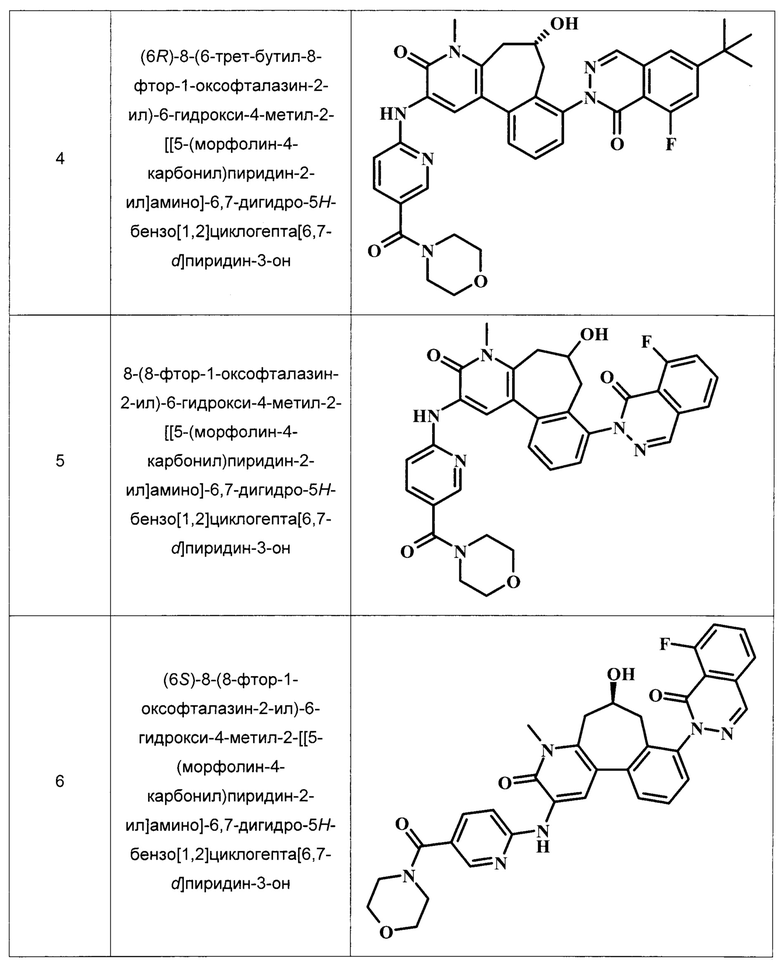
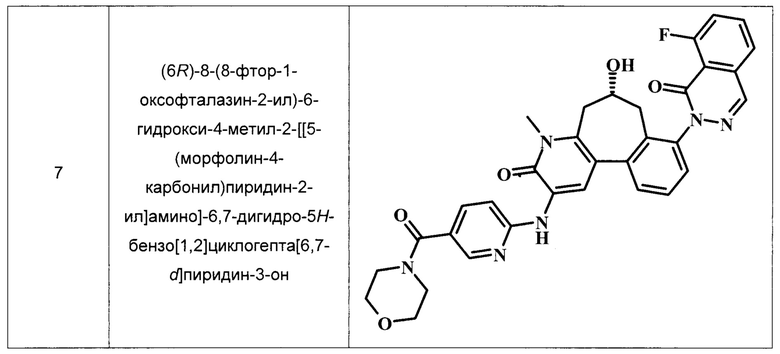
Общие Схемы Синтеза
Соединения настоящего изобретения могут быть получены любыми обычными средствами. Подходящие методики для синтеза этих соединений предложены в примерах. В основном, соединения настоящего изобретения могут быть получены в соответствии со схемами, данными ниже.
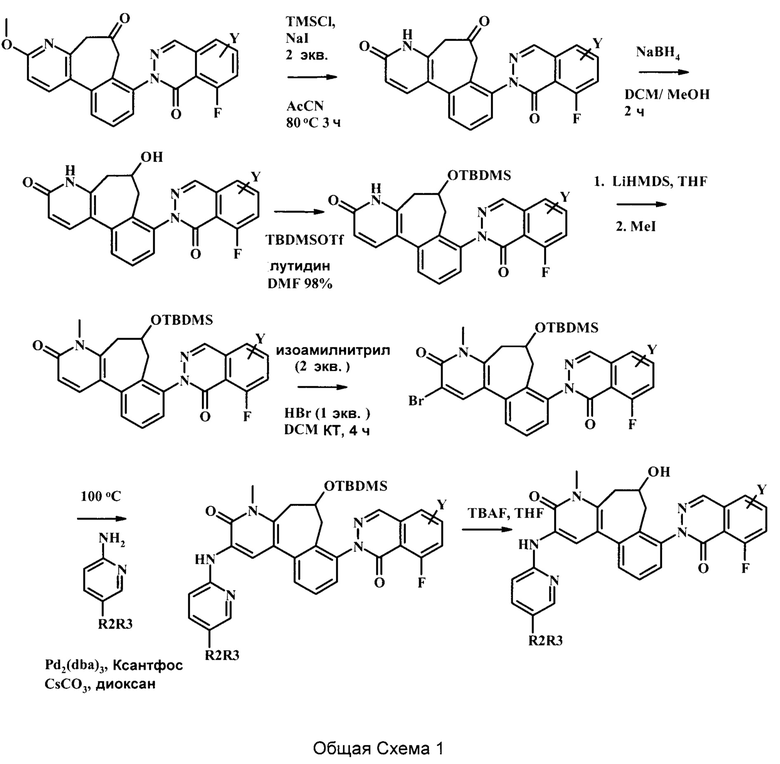
На вышеуказанной Общей Схеме 1, R2 может быть -С(=O) или отсутствует, R3 может представлять собой гетероциклоалкил, возможно замещенный одним или более R3', каждый R3' может независимо представлять собой низший алкил, галоген, низший алкокси, или низший галоалкил, и Y может представлять собой Н или низший алкил.
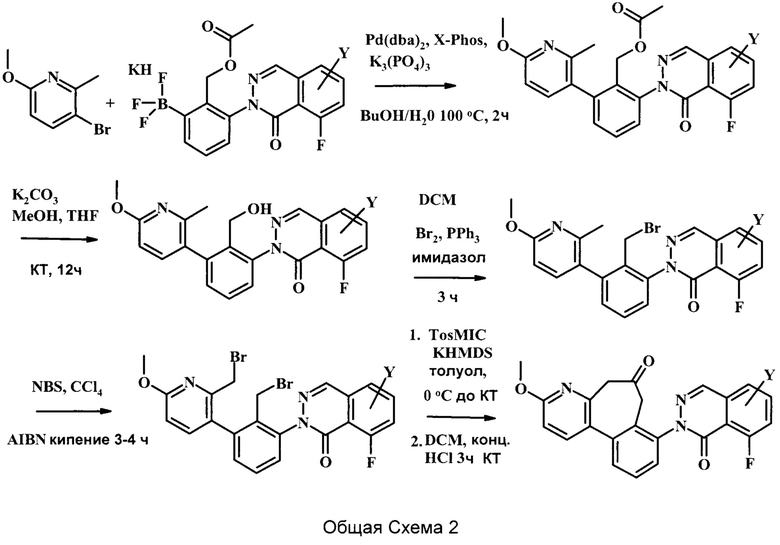
На вышеуказанной Общей Схеме 2, R2 может представлять собой -С(=O) или отсутствует, R3 может представлять собой гетероциклоалкил, возможно замещенный одним или более R3', каждый R3' может независимо представлять собой низший алкил, галоген, низший алкокси, или низший галоалкил, и Y может представлять собой Н, низший алкил,
Фармацевтические композиции и введение
Соединения по настоящему изобретению могут быть приготовлены в виде различных лекарственных форм и носителей для перорального введения. Пероральный прием может быть проведен в форме таблеток, покрытых оболочкой таблеток, драже, твердых и мягких желатиновых капсул, растворов, эмульсий, сиропов или суспензий. Соединения настоящего изобретения являются эффективными при введении другими путями, включая непрерывное (внутривенно-капельное) местное, парентеральное, внутримышечное, внутривенное, подкожное, трансдермальное (которое может включать агент для улучшения проникновения), буккальное, носовое, ингаляцию и ведение суппозиториев среди других путей введения. Предпочтительным способом введения является, как правило, пероральное с использованием удобного ежедневного режима дозирования, который может быть скорректирован в зависимости от степени заболевания и реакции пациента на активный ингредиент.
Соединение или соединения по настоящему изобретению, а также их фармацевтически используемые соли, вместе с одним или более традиционными эксципиентами, носителями или разбавителями, могут быть сделаны в форме фармацевтических композиций и единичных дозированных форм. Фармацевтические композиции и единичные дозированные формы могут состоять из обычных ингредиентов в обычных пропорциях, с или без дополнительных активных соединений или основ, и единичные дозированные формы могут содержать любое подходящее эффективное количество активного ингредиента, соразмерное с предполагаемым используемым дневным диапазоном дозировки. Фармацевтические композиции могут быть использованы в качестве твердых веществ, таких как таблетки или наполненные капсулы, полутвердых веществ, порошков, препаратов с замедленным высвобождением или жидкостей, таких как растворы, суспензии, эмульсии, эликсиры или заполненные капсулы для перорального применения, или в форме суппозиториев для ректального или вагинального введения, или в виде стерильных растворов для инъекций для парентерального применения. Типичный препарат будет содержать примерно от 5% до примерно 95% активного соединения или соединений (масс/масс). Термин "препарат" или "дозированная форма" используется для обозначения твердых и жидких лекарственных форм активного соединения и квалифицированному специалисту в данной области будет понятно, что активный ингредиент может существовать в форме различных препаратов, в зависимости от органа-мишени или ткани и желаемой дозы и фармакокинетических параметров.
Термин "эксципиент", как используется в настоящем изобретении, относится к соединению, которое полезно при получении фармацевтической композиции, оно, как правило, безопасно, нетоксично и ни биологически, ни еще каким-либо образом не является нежелательным, и включает эксципиенты, которые приемлемы для применения в ветеринарии, а также для фармацевтического использования для людей. Соединения по настоящему изобретению могут быть вводиться отдельно, однако обычно их вводят в смеси с одним или несколькими подходящими фармацевтическими эксципиентами, разбавителями или носителями, выбранными с учетом предполагаемого способа введения и обычной фармацевтической практикой.
"Фармацевтически приемлемый" означает такой, который полезен при приготовлении фармацевтической композиции, и как правило, безопасный, нетоксичный и ни биологически, ни по иному нежелательный и включает в себя такой, который приемлем для ветеринарии а также для фармацевтического использования для людей.
Форма активного ингредиента "фармацевтически приемлемая соль" может также изначально придать желательные фармакокинетические свойства активному ингредиенту, которые отсутствовали в не-солевой форме, и даже может положительно влиять на фармакодинамику активного ингредиента в отношении его терапевтической активности в организме. Фраза «фармацевтически приемлемая соль" соединения означает, соль, которая является фармацевтически приемлемой и которая обладает требуемой фармакологической активностью исходного соединения. Такие соли включают: (1) кислотно-аддитивные соли, образованные с неорганическими кислотами, такими как соляная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и т.п., или образованные с органическими кислотами, такими как уксусная кислота, пропионовая, капроновая кислоты, циклопентанпропионовая кислота, гликолевая кислота, пировиноградная кислота, молочная кислота, малоновая кислота, янтарная кислота, яблочная кислота, малеиновая кислота, фумаровая кислота, винная кислота, лимонная кислота, бензойная кислота, 3-(4-гидроксибензоил)бензойная кислота, коричная кислота, миндальная кислота, метансульфоновая кислота, этансульфоновая кислота, 1,2-этан-дисульфоновая кислота, 2-гидроксиэтансульфоновая кислота, бензолсульфоновая кислота, 4-хлорбензолсульфоновая кислота, 2-нафталинсульфоновая кислота, 4-толуол сульфоновая кислота, камфор сульфоновая кислота, 4-метилбицикло[2,2,2]-окт-2-ен-1-карбоновая кислота, глюкогептоновая кислота, 3-фенилпропионовая кислота, триметилуксусная кислота, третичная капроновая кислота, лаурилсерная кислота, глюконовая кислота, глутаминовая кислота, гидроксинафтоевая кислота, салициловая кислота, стеариновая кислота, муконовая кислота и т.п., или (2) соли, образованные в случае, когда протон кислоты присутствует в исходном соединении или он заменен ионом металла, например, ионом щелочного металла, ионом щелочноземельного металла или ионом алюминия, или связан с органическим основанием, таким как этаноламин, диэтаноламин, триэтаноламин, трометамин, N-метилглюкамин и т.п.
Препараты в твердой форме включают порошки, таблетки, пилюли, капсулы, саше, суппозитории и диспергируемые гранулы. Твердый носитель может быть одним или несколькими веществами, которые также могут действовать как разбавители, вкусовые добавки, растворители, ароматизаторы, солюбилизаторы, любриканты, суспендирующие агенты, связующие вещества, консерванты, разрыхлители для таблеток или инкапсулирующий материал. В порошках носитель обычно представляет собой тонко измельченное твердое вещество, которое является смесью с мелко измельченным активным компонентом. В таблетках активный компонент обычно смешивают с носителем, имеющим необходимую связывающую способность в подходящих пропорциях и спрессовывают до нужной формы и размера. Подходящие носители включают, без ограничения, карбонат магния, стеарат магния, тальк, сахар, лактозу, пектин, декстрин, крахмал, желатин, трагакант, метилцеллюлозу, натрий-карбоксиметилцеллюлозу, легкоплавкий воск, масло какао, и т.п. Твердые лекарственные формы могут содержать, в дополнение к активному компоненту, красители, ароматизаторы, стабилизаторы, буферы, искусственные и натуральные подсластители, диспергаторы, загустители, солюбилизирующие агенты и т.п.
Жидкие препараты, также подходящие для перорального введения, включают жидкие препараты, включая эмульсии, сиропы, эликсиры, водные растворы, водные суспензии. Они включают в себя твердые формы препаратов, которые предназначены для превращения в жидкую форму незадолго до использования. Эмульсии могут быть получены в растворах, например, в водном растворе пропиленгликоля, или могут содержать эмульгаторы, такие как лецитин, сорбитанмоноолеат или камедь. Водные растворы могут быть получены путем растворения активного компонента в воде и добавления подходящих красителей, ароматизаторов, стабилизаторов и загустителей. Водные суспензии могут быть получены диспергированием тонкоизмельченного активного компонента в воде с вязким материалом, таким как природные или синтетические камеди, смолы, метилцеллюлоза, натрий-карбоксиметилцеллюлоза и другие хорошо известные суспендирующие агенты.
Соединения по настоящему изобретению могут быть приготовлены для парентерального введения (например, путем инъекции, например болюсной инъекции или инфузии) и могут быть представлены в единичной дозированной форме в ампулах, предварительно заполненных шприцах, растворов для инфузий небольшого объема или в многодозовых контейнерах с добавлением консерванта. Композиции могут быть в таких формах, как суспензии, растворы или эмульсии в масляных или водных носителях, например, растворы в водном растворе полиэтиленгликоля. Примеры масляных или неводных носителей, разбавителей, растворителей или носителей включают пропиленгликоль, полиэтиленгликоль, растительные масла (например, оливковое масло) и инъекционные органические сложные эфиры (например, этилолеат), и могут содержать вспомогательные вещества, такие как консерванты, увлажняющие, эмульгирующие или суспендирующие, стабилизирующие и/или диспергирующие агенты. Альтернативно, активный ингредиент может быть в форме порошка, полученного путем асептического выделения стерильного твердого вещества или путем лиофилизации из раствора для разбавления перед использованием подходящим носителем, например, стерильной, апирогенной водой.
Соединения по настоящему изобретению могут быть приготовлены для местного введения в эпидермис в виде мазей, кремов или лосьонов или в виде трансдермального пластыря. Мази и кремы могут, например, быть приготовлены на водной или масляной основе с добавлением подходящих загустителей и/или желирующих агентов. Лосьоны могут быть приготовлены на водной или масляной основе и обычно также содержат один или несколько эмульгаторов, стабилизирующих агентов, диспергирующих агентов, суспендирующих агентов, загустителей или красителей. Препараты подходящие для местного применения в полости рта включают пастилки, содержащие активные вещества в ароматизированной основе, как правило, сахарозе и аравийской камеди или трагаканте; пастилки, содержащие активный ингредиент в инертной основе, такой как желатин и глицерин или сахароза и гуммиарабик, и жидкости для полоскания рта, содержащие активный ингредиент в подходящем жидком носителе.
Соединения по настоящему изобретению могут быть приготовлены для введения в виде суппозиториев. Легкоплавкий воск, такой как смесь глицеридов жирных кислот или масло какао, расплавляются сначала и активный компонент равномерно диспергируют, например, путем перемешивания. Расплавленную гомогенную смесь затем выливают в формы подходящего размера, дают остыть и затвердеть.
Соединения по настоящему изобретению могут быть приготовлены для вагинального применения. Подходящими являются вагинальные суппозитории, тампоны, кремы, гели, пасты, пены или спреи, содержащие в дополнение к активному ингредиенту такие носители, которые известны в уровне техники.
Соединения по настоящему изобретению могут быть приготовлены для назального введения. Растворы или суспензии вводят непосредственно в полость носа с помощью обычных средств, например, капельницей, пипеткой или распылителем. Композиции могут быть представлены в одной или многодозовой формах. В последнем случае для капельницы или пипетки, это может быть достигнуто путем введения пациенту необходимого, заданного объема раствора или суспензии. В случае распыления, это может быть достигнуто, например, путем регулировки распылительной помпы.
Соединения по настоящему изобретению могут быть приготовлены для аэрозольного введения, в частности, в дыхательные пути и в том числе для интраназального введения. Соединение обычно имеет небольшой размер частиц, например, порядка пяти (5) микрон или менее. Такой размер частиц может быть получен с помощью известных в данной области способов, например, путем измельчения. Активный ингредиент содержится в герметичной упаковке с подходящим пропеллентом, таким как хлорфторуглерод (ХФУ), например, дихлордифторметан, трихлорфторметан или дихлортетрафторэтан или диоксид углерода или другой подходящий газ. Аэрозоль для удобства может содержать поверхностно-активное вещество, такое как лецитин. Доза препарата может контролироваться дозирующим клапаном. Кроме того, активные ингредиенты могут быть представлены в виде сухого порошка, например порошковой смеси соединения в подходящей порошковой основе, такой как лактоза, крахмал, производные крахмала, такие как гидроксипропилметилцеллюлоза и поливинилпирролидон (ПВП). Порошок-носитель образует гель в носовой полости. Порошковая композиция может быть представлена в виде единичной дозированной формы, например, в капсулах или картриджах, например, желатиновых или блистерной упаковке, из которой порошок может быть введен с помощью ингалятора.
При желании, препараты могут быть приготовлены с кишечными покрытиями, адаптированными для замедленного или контролируемого высвобождения активного ингредиента. Например, соединения по настоящему изобретению могут быть загружены в трансдермальные или подкожные устройства доставки лекарственных средств. Эти системы доставки желательны, когда необходимо длительное высвобождение соединения и когда соблюдение пациентом режима лечения имеет решающее значение. Соединения в трансдермальных системах доставки часто присоединяются к твердой подложке, адгезивной в отношении кожи. Соединение настоящего изобретения также может быть комбинировано с усилителем проникновения, например, Азон (1-додецилаза-циклогептан-2-оном). Системы доставки с непрерывным высвобождением вводятся подкожно в субдермальный слой хирургическим путем или инъекций. Подкожные имплантаты инкапсулируют соединения в жирорастворимых мембранах, например, силиконовом каучуке или биоразлагаемом полимере, например, полимолочной кислоте.
Подходящие препараты наряду с фармацевтическими носителями, разбавителями и эксципиентами описаны в Remington: The Science and Practice of Pharmacy 1995, под редакцией E.W. Martin, Mack Publishing Company, 19th edition, Easton, Pennsylvania. Квалифицированный специалист может изменить композиции в рамках раскрытого в настоящем изобретении для получения множества препаратов для конкретного пути введения, не делая композиции настоящего изобретения нестабильными или снижая их терапевтическую активность.
Изменение соединений по настоящему изобретению с целью сделать их более растворимыми в воде или другом носителе, например, может быть легко достигнуто путем незначительных изменений (образование соли, этерификация и т.д.), которые находятся в пределах знаний квалифицированного специалиста в данной области. Также вполне укладывается в обычные для данной области техники знания изменения способа введения и режима дозирования конкретного соединения для того, чтобы управлять фармакокинетикой соединений настоящего изобретения для максимально положительного эффекта у больных.
Термин "терапевтически эффективное количество", как используется в настоящем изобретении, означает количество, необходимое для уменьшения симптомов заболевания у человека. Доза будет скорректирована с учетом индивидуальных требований в каждом конкретном случае. Эта дозировка может варьироваться в широких пределах в зависимости от многих факторов, таких как тяжесть заболевания, подлежащего лечению, возраста и общего состояния здоровья пациента, других лекарственных средств, которые принимает пациент, пути и формы введения и предпочтения и опыт лечащего врача. Для перорального введения подходящей для монотерапии и/или в комбинированной терапии является суточная доза примерно от 0,01 до примерно 1000 мг/кг веса тела в день. Предпочтительная суточная доза составляет от приблизительно 0,1 до приблизительно 500 мг/кг массы тела, более предпочтительно от 0,1 до 100 мг/кг массы тела и наиболее предпочтительно 1,0 и около 10 мг/кг массы тела в сутки. Таким образом, для введения пациенту весом 70 кг, диапазон доз будет от около 7 мг до 0,7 г в день. Суточная доза может вводиться в виде одной дозы или в виде разделенных доз, как правило, от 1 до 5 доз в день. Как правило, лечение начинают с меньших доз, которые меньше, чем оптимальная доза соединения. После этого доза увеличивается на малое количество, пока не будет достигнут оптимальный эффект для конкретного пациента. Специалист в лечении заболеваний, описанных в настоящем изобретении, сможет без дополнительных экспериментов и в зависимости от личных знаний, опыта и раскрытия настоящего изобретения, установить терапевтически эффективное количество соединения по настоящему изобретению для конкретного заболевания и пациента.
Фармацевтические препараты предпочтительно находятся в единичных дозированных формах. В такой форме препарат разделен на стандартные дозы, содержащие соответствующие количества активного компонента. Единичная дозированная форма может быть упакованным препаратом, упаковкой, содержащей дискретные количества препарата, такие как упакованные таблетки, капсулы и порошки во флаконах или ампулах. Кроме того, единичная дозированная форма может быть капсулой, таблеткой, саше или пастилкой или она может быть соответствующим числом любой из них в упакованном виде.
Показания и способы лечения
Соединения общей Формулы I ингибируют тирозинкиназу Брутона (Btk). Активация Btk вышестоящими киназами приводит к активации фосфолипазы-Сγ, которая, в свою очередь, стимулирует высвобождение провоспалительных медиаторов. Соединения Формулы I полезны при лечении артрита и других воспалительных и аутоиммунных заболеваний. Соединения в соответствии с Формулой I являются, соответственно, полезными для лечения артритов. Соединения Формулы I полезны для ингибирования киназы Btk в клетках и для модуляции развития В-клеток. Настоящее изобретение также включает фармацевтические композиции, содержащие соединение Формулы I, смешанное с фармацевтически приемлемым носителем, эксципиентами или разбавителями.
Соединения, описанные в настоящем изобретении, являются ингибиторами киназ, в частности ингибиторами киназы Btk Эти ингибиторы могут быть полезны для лечения одного или более заболеваний, чувствительных к ингибированию киназ, в том числе заболеваний чувствительных к ингибированию Btk киназы и/или ингибированию В-клеточной пролиферации у млекопитающих. Не желая быть связанным с какой-либо конкретной теорией, полагают, что взаимодействие соединения по изобретению с Btk приводит к ингибированию активности Btk и, следовательно, к фармацевтической полезности этих соединений. Таким образом, настоящее изобретение включает в себя способ лечения млекопитающих, например, человека, с заболеванием, чувствительным к ингибированию активности Btk и/или ингибированию В-клеточной пролиферации, содержащий введение млекопитающим с таким заболеванием эффективного количества по крайней мере одного химического соединения, заявленного в настоящем изобретении. Эффективная концентрация может быть установлена экспериментально, например, путем анализа концентрации соединения в крови или теоретически, путем расчета биодоступности. Другие киназы, которые могут быть затронуты в дополнение к Btk включают, без ограничения, другие тирозинкиназы и серин/треонин киназы.
Киназ играют существенную роль в сигнальных путях, контролирующих основные клеточные процессы, таких как пролиферация, дифференцировка и смерть (апоптоз) клеток. Аномальная киназная активность участвует в широком круге заболеваний, в том числе различных видах рака, аутоиммунных и/или воспалительных заболеваниях и острой воспалительной реакции. Разнообразная роль киназ в ключевых сигнальных путях клетки предоставляет значительные возможности для выявления новых лекарственных препаратов, нацеленных на киназы и сигнальные пути.
Один из вариантов настоящего изобретения включает в себя способ лечения пациента, имеющего аутоиммунное и/или воспалительное заболевание или острую воспалительную реакцию, чувствительные к ингибированию активности Btk и/или В-клеточной пролиферации.
Аутоиммунные и/или воспалительные заболевания, на которые можно воздействовать соединениями и композициями в соответствии с настоящим изобретением, включают, без ограничения: псориаз, аллергию, болезнь Крона, синдром раздраженного кишечника, болезнь Шегрена, отторжение трансплантата ткани и сверхострое отторжение трансплантированных органов, бронхиальную астму, системную красную волчанку (и связанный с ней гломерулонефрит), дерматомиозит, рассеянный склероз, склеродермию, васкулиты (ANCA-ассоциированные и другие васкулиты), аутоиммунное гемолитическое состояние и тромбоцитопению, синдром Гудпасчера (и связанный с ним гломерулонефрит и легочное кровотечение), атеросклероз, ревматоидный артрит, хроническую идиопатическую тромбоцитопеническую пурпуру (ИТП), болезнь Аддисона, болезнь Паркинсона, болезнь Альцгеймера, диабет, септический шок и миастению.
В настоящее изобретение включены способы лечения, при которых по крайней мере одно химическое соединение, предусмотренное настоящим изобретением, вводят в комбинации с противовоспалительным средством. Противовоспалительные средства включают, без ограничения, НПВП, неспецифические и СОХ-2 специфические ингибиторы циклооксигеназ, соединения золота, кортикостероиды, метотрексат, антагонисты рецепторов фактора некроза опухоли (TNF), иммунодепрессанты и метотрексат.
Примеры НПВП включают, без ограничения, ибупрофен, флурбипрофен, напроксен и напроксен натрия, диклофенак, комбинации диклофенака натрия и мизопростола, сулиндак, оксапрозин, дифлунизал, пироксикам, индометацин, этодолак, фенопрофен кальция, кетопрофен, натрия набуметон, сульфасалазин, толметин натрия и гидроксихлорохин. Примеры НПВП также включают СОХ-2 специфичные ингибиторы, такие как целекоксиб, вальдекоксиб, лумиракоксиб и/или эторикоксиб.
В некоторых воплощениях противовоспалительным средством является салицилат. Салицилаты включают, без ограничения, ацетилсалициловую кислоту или аспирин, салицилат натрия, а также холин и салицилат магния.
Противовоспалительное средство также может быть кортикостероидом. Например, кортикостероидом может быть кортизон, дексаметазон, метилпреднизолон, преднизолон, преднизолон фосфат натрия или преднизон.
В дополнительных воплощениях противовоспалительным агентом является соединение золота, такое как ауротиомалат натрия или ауранофин.
Изобретение также включает воплощения, в которых противовоспалительным средством является метаболический ингибитор, такой как ингибитор дигидрофолатредуктазы, такой как метотрексат или ингибитор дигидрооротатдегидрогеназы, такой как лефлуномид.
Другие воплощения настоящего изобретения относятся к комбинациям, в которых по крайней мере одно противовоспалительное соединения является анти-С5 моноклональным антителом (например, экулизумаб или пекселизумаб), антагонистом ФНО, таким как инфликсимаб или энтанерцепт, который является анти-ФНО-альфа моноклональным антителом.
Кроме того, другие воплощения изобретения относятся к комбинациям, в которых по крайней мере один активный агент является иммунодепрессантом, таким как иммунодепрессант, выбранный из метотрексата, лефлуномида, циклоспорина, такролимуса, азатиоприна и микофенолятмофетила.
В-клетки и предшественники В-клеток, экспрессирующие Btk, вовлечены в патологию В-клеточных злокачественных новообразований, в том числе, без ограничения, В-клеточной лимфомы, лимфомы (в том числе лимфомы Ходжкина и неходжкинскую лимфому), волосатоклеточной лимфомы, множественной миеломы, хронического и острого миелоцитарного лейкоза и острого и хронического лимфоцитарного лейкоза.
Было показано, что Btk является ингибитором сигнального комплекса Fas/APO-1 (CD-95) индуцирующего гибель (DISC) в линиях лимфоидных В-клеток. Судьба лейкемических/лимфомных клеток может постоянно находиться в равновесии между противостоянием проапоптотических эффектов каспаз, активируемых DISC и вышестоящего антиапоптотического регуляторного механизма с участием Btk и/или ее субстратов. (Vassilev и соавт., J. Biol. Chem. 1998, 274, 1646-1656).
Кроме того, было обнаружено, что Btk ингибиторы являются полезными в качестве хемосенсорных агентов, и, таким образом, могут быть использованы в комбинации с другими химиотерапевтическими препаратами, в частности, препаратами, которые индуцируют апоптоз. Примеры других химиотерапевтических препаратов, которые могут быть использован в комбинации с хемосенсорными Btk ингибиторами включают ингибиторы топоизомеразы I (камптотецин или топотекан), ингибиторы топоизомеразы II (например, дауномицин и этопозид), алкилирующие агенты (например, циклофосфамид, мелфалан и BCNU), агенты, направленные на тубулин (например, таксол и винбластин) и биологические агенты (например, антитела, такие как антитела к CD20, IDEC 8, иммунотоксины и цитокины).
Btk активность также может быть связана с некоторыми экспрессирующимися при лейкемии слитыми в результате транслокации частей хромосом 9 и 22 генами bcr-abl. Это нарушение обычно наблюдается при хронической миелоидной лейкемии. Btk конститутивно фосфорилируется bcr-abl киназой, которая инициирует нисходящие сигналы выживания, который предотвращает апоптоз в bcr-abl клетках. (N. Feldhahn и соавт. J. Exp. Med. 2005 201(11):1837-1852).
Способы лечения
В настоящем изобретении предложен способ лечения воспалительного и/или аутоиммунного состояния, включающий введение нуждающемуся в этом пациенту терапевтически эффективного количества соединения Формулы I.
В настоящем изобретении предложен способ лечения воспалительного состояния, включающий введение нуждающемуся в этом пациенту терапевтически эффективного количества соединения Формулы I.
В настоящем изобретении предложен способ лечения ревматоидного артрита, включающий введение нуждающемуся в этом пациенту терапевтически эффективного количества соединения Формулы I.
В настоящем изобретении предложен способ лечения астмы, включающий введение нуждающемуся в этом пациенту терапевтически эффективного количества соединения Формулы I.
В настоящем изобретении предложен способ лечения воспалительного и/или аутоиммунного состояния, включающий введение нуждающемуся в этом пациенту терапевтически эффективного количества Btk ингибирующего соединения Формулы I.
В настоящем изобретении предложен способ лечения артрита, включающий введение нуждающемуся в этом пациенту терапевтически эффективного количества Btk ингибирующего соединения Формулы I.
В настоящем изобретении предложен способ лечения астмы, включающий введение нуждающемуся в этом пациенту терапевтически эффективного количества Btk ингибирующего соединения Формулы I.
В настоящем изобретении предложен способ ингибирования пролиферации В-клеток, включающий введение нуждающемуся в этом пациенту терапевтически эффективного количества Btk ингибирующего соединения Формулы I.
В настоящем изобретении предложено способ ингибирования активности Btk, включающий введение Btk ингибирующего соединения Формулы I, где соединение Btk ингибитора показывает IC50 50 мкмоль или менее в in vitro биохимическом анализе активности Btk.
В одном варианте вышеуказанного способа, соединение Btk ингибитора показывает IC50 100 нмоль или менее в in vitro биохимическом анализе активности Btk.
В другом варианте вышеуказанного способа, соединение Btk ингибитора показывает IC50 10 нмоль или менее в in vitro биохимическом анализе активности Btk.
В настоящем изобретении предложен способ лечения воспалительного состояния, включающий совместное введение нуждающемуся в этом пациенту терапевтически эффективного количества противовоспалительного соединения в комбинации с соединением Btk ингибитора Формулы I.
В настоящем изобретении предложен способ лечения артрита, включающий совместное введение нуждающемуся в этом пациенту терапевтически эффективного количества противовоспалительного соединения в комбинации с соединением Btk ингибитора Формулы I.
В настоящем изобретении предложен способ лечения лимфомы или BCR-ABL1+ лейкозных клеток посредством введения нуждающемуся в этом пациенту терапевтически эффективного количества Btk ингибирующего соединения Формулы I.
В настоящем изобретении предложено применение соединения, как описано выше, для лечения воспалительного и/или аутоиммунного состояния.
В настоящем изобретении предложено применение соединения, как описано выше, для получения лекарственного средства для лечения воспалительного и/или аутоиммунного состояния.
В настоящем изобретении предложено соединение, как описано выше, для применения для лечения воспалительного и/или аутоиммунного состояния.
В настоящем изобретении предложено соединение, как описано выше, для применения при лечении ревматоидного артрита.
В настоящем изобретении предложено соединение, как описано выше, для применения при лечении астмы.
В настоящем изобретении предложено изобретение, как здесь описано.
Примеры
Общие Аббревиатуры
Обычно используемые сокращения включают: ацетил (Ас), азо-бис-изобутирилнитрил (AIBN), атмосферы (атм), 9-борабицикло[3.3.1]нонан (9-BBN или BBN), 2,2'-бис(дифенилфосфино)-1,1'-бинафтил (BINAP), трет-бутоксикарбонил (Boc), ди-трет-бутилпирокарбонат или boc-ангидрид (BOC2O), бензил (Bn), бутил (Bu), регистрационный номер в системе кодирования в реферативном журнале "Chemical Abstracts" (CASRN), бензилоксикарбонил (CBZ или Z), карбонилдиимидазол (CDI), 1,4-диазабицикло[2.2.2]октан (DABCO), диэтиламиносеры трифторид (DAST), дибензилиденацетон (DBA), 1,5-диазабицикло[4.3.0]нон-5-ен, (DBN), 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU), N,N'-дициклогексилкарбодиимид (DCC), 1,2-дихлорэтан (DCE), дихлорметан (ДХМ), 2,3-дихлор-5,6-дициано-1,4-бензохинон (DDQ), диэтилазодикарбоксилат (DEAD), ди-изо-пропилазодикарбоксилат (DIAD), ди-изо-бутилалюминия гидрид (DIBAL или DIBAL-H), ди-изо-пропилэтиламин (DIPEA), N,N-диметилацетамид (DMA), 4-N,N-диметиламинопиридин (DMAP), N,N-диметилформамид (ДМФ), диметилсульфоксид (ДМСО), 1,1'-бис-(дифенилфосфино)этан (DPPE), 1,1'-бис-(дифенилфосфино)ферроцен (DPPF), 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид (EDCl), 2-этокси-1-этоксикарбонил-1,2-дигидрохинолин (EEDQ), этил (Et), этилацетат (EtOAc), этанол, (EtOH), 2-этокси-2Н-хинолин-1-карбоновой кислоты этиловый эфир (EEDQ), диэтиловый эфир (Et2O), этилизопропиловый эфир (EtOiPr), O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфатуксусная кислота (HATU), уксусная кислота (НОАс), 1-N-гидроксибензотриазол (HOBt), высокоэффективная жидкостная хроматография (ВЭЖХ), изопропанол (IPA), изопропилмагнийхлорид (iPrMgCl), гексаметилдисилазан (HMDS), гексан (hex), жидкостная хроматография с масс-спектрометрией (ЖХ-МС), лития гексаметилдисилазан (LiHMDS), мета-хлорпербензойная кислота (m-СРВА), метанол (МеОН), температура плавления (т.пл. или MP), MeSO2-(мезил или Ms), метил (Me), ацетонитрил (MeCN), м-хлорпербензойная кислота (МСРВА), масс-спектр (ms или MS), метил-трет-бутиловый эфир (МТВЕ), метил-тетрагидрофуран (МеТГФ), N-бромсукцинимид (NBS), н-бутиллитий (nBuLi), N-карбоксиангидрид (NCA), N-хлорсукцинимид (NCS), N-метилморфолин (НММ), N-метилпирролидон (NMP), хлорхромат (РСС), дихлор-((бис-дифенилфосфино)ферроценил)палладий(II) (Pd(dppf)Cl2), палладия(II)ацетат (Pd(OAc)2), трис(дибензилиденацетон)дипалладий (0) (Pd2(DBA)3), дихромат пиридиния (PDC), фенил (Ph), пропил (Pr), изо-пропил (i-Pr), фунт на квадратный дюйм (PSI), пиридин (pyr), 1,2,3,4,5-пентафенил-1-(ди-трет-бутилфосфино)ферроцен (Q-Phos), комнатная температура (собственная температура, кт или КТ), втор-бутиллитий (sBuLi), трет-бутилдиметилсилил или Трет-BuMe2Si (TBDMS), тетра-н-бутиламмония фторид (TBAF), триэтиламин (TEA или Et3N), 2,2,6,6-тетраметилпиперидин-1-оксил (TEMPO), триметилсилилэтоксиметил (SEM), трифлат или CF3SO2-(Tf), трифторуксусная кислота (ТФУ), 1,1'-бис-2,2,6,6-тетраметилгептан-2,6-дион (TMHD), О-бензотриазол-1-ил-N,N,N',N'-тетраметилурония тетрафторборат (TBTU), тонкослойная хроматография (ТСХ), тетрагидрофуран (ТГФ), триметилсилил или Me3Si (TMS), р-толуолсульфокислота (TsOH или pTsOH), 4-Me-C6H4SO2 или тозил (Ts) и N-уретан-N-карбоксиангидрид (UNCA). Обычные номенклатуры, включая префиксы нормальный (N), изо (I-), вторичный (втор-), третичный (трет-) и нео имеют свои обычные значения при использовании их с алкильной частью. (J. Rigaudy и D.P. Klesney, Nomenclature in Organic Chemistry, IUPAC 1979 Pergamon Press, Oxford).
Общие условия
Соединения настоящего изобретения могут быть получены с использованием коммерчески доступных исходных соединений посредством обычных способов и методик синтеза, известных квалифицированным специалистам. Ниже показаны реакционные схемы, подходящие для получения таких соединений. Дополнительные иллюстрации можно найти в конкретных примерах.


Подробное описание общих экспериментов
Реагенты покупали у компаний Aldrich, Oakwood, Matrix или других поставщиков и использовали без дополнительной очистки. Реакции, использующие микроволновое излучение для нагревания, проводились с использованием или системы Personal Chemistry Emrys Optimizer или системы СЕМ Discovery. Очистка в милиграммовом или граммовом масштабе проводилась способами, известными квалифицированным специалистам, такими как элюция с силикагелевой флеш колонки; препаративная очистка на флеш колонках также в некоторых случаях проводилась не предварительно заполненных мультиграммовых силикагелевых колонках (RediSep) элюируемых с помощью системы CombiFlash. Biotage™ и ISCO™ также представляют собой флеш-колоночные средства, которые могут применяться в настоящем изобретении для очистки промежуточных соединений.
С целью оценки идентичности и чистоты соединения, проводили запись LC/MS (жидкостная хроматография/масс-спектроскопия) спектра с использованием следующей системы. Для измерения масс-спектра - система, состоящая из спектрометра Micromass Platform II: Электроспрей ионизация в режиме детекции положительно заряженных ионов (диапазон масс: 150-1200). Одновременное хроматографическое разделение проводилось с помощью следующей ВЭЖХ системы: ES Industries Chromegabond WR С-18 3u 120  (3.2×30 мм) катридж для колонки; Подвижная фаза А: вода (0.02% ТФУ) и Фаза В: ацетонитрил (0.02% ТФУ); градиент от 10% В до 90% В за 3 минуты; время уравновешивания - 1 минута; скорость потока 2 мл/минуту.
(3.2×30 мм) катридж для колонки; Подвижная фаза А: вода (0.02% ТФУ) и Фаза В: ацетонитрил (0.02% ТФУ); градиент от 10% В до 90% В за 3 минуты; время уравновешивания - 1 минута; скорость потока 2 мл/минуту.
Многие соединения формулы 1 также очищали обратно-фазовой ВЭЖХ, с использованием способов, хорошо известных квалифицированным специалистам. В некоторых случаях, препаративная ВЭЖХ очистка проводилась с использованием РЕ Sciex 150 EX Mass Spec, управляющим сборником фракций Gilson 215, соединенным с системой препаративной ВЭЖХ Shimadzu и автоинжектором Leap.Соединения собирались из элюирующего потока с использованием LC/MS детекции в режиме детекции положительно заряженных ионов: Элюция соединений с колонок С-18 (2.0×10 см, элюируемых при 20 мл/мин) проводилась с использованием подходящего линейного режима градаций в течение 10 минут Растворителя (А) 0.05% ТФУ/H2O и Растворителя (В) 0.035% ТФУ/ацетонитрил. Для внесения в систему ВЭЖХ, неочищенные образцы растворили в смеси метанола, ацетонитрила и ДМСО.
Полученные соединения охарактеризовались посредством 1H-NMR с использованием Bruker 400 МГЦ ЯМР спектрометра.
Соединения настоящего изобретения могут быть синтезированы в соответствии с известными методиками. Следующие примеры и ссылки приведены с целью понимания настоящего изобретения. Примеры, однако, не предназначены для ограничения изобретения, действительный объем прав которого изложен в прилагаемой формуле изобретения. Название реагирующих веществ и конечных продуктов в примерах были созданы с использованием программного обеспечения AutoNom 2000.
Примеры Получения
Абсолютная стереохимия для примеров 3, 4, 6 и 7 основана на сравнении предполагаемой биологической эффективности и/или времени задержки на хиральной сверхкритической жидкостной хроматографии и не является подтвержденной.
Получение Промежуточного соединения А
Стадия 1. Получение 2-диметоксиметил-6-фтор-бензойной кислоты

Раствор 1-диметоксиметил-3-фтор-бензола (100 г, 588 ммоль) в тетрагидрофуране (1 л) охладили до -60°С в атмосфере N2. s-BuLi (1.4 М, 664 ммоль, 475 мл) добавили при -60°С. Получившийся красный раствор перемешивали при -60°С в течение 1 ч. В 2 л колбу в атмосфере N2 добавили чистый сухой лед (355 г, 5.88 моль) и тетрагидрофуран (300 мл) с последующим n-BuLi (5 мл) для удаления остаточной влажности. Вышеуказанный красный анионный раствор добавили в смесь сухого льда в тетрагидрофуране в течение 2 ч. Получившийся светло-коричневый раствор перемешивали в течение дополнительных 20 мин. После завершения реакции, добавили воду (1 л), и затем реакционную смесь нейтрализовали концентрированной водной HCl (70 мл) до рН=2. Органический слой отделили и сохранили и водный слой экстрагировали этилацетатом (500 мл). Объединенные органические слои промыли водой (2×300 мл). Растворитель удалили и затем получившийся неочищенный продукт кристаллизовали с получением 2-диметоксиметил-6-фторбензойной кислоты в виде светло-коричневого осадка (84 г, 66% выход).
Стадия 2. Получение 8-фтор-2Н-фталазин-1-он

Раствор 2-(диметоксиметил)-6-фторбензойной кислоты (60 г, 280 ммоль), уксусной кислоты (39.8 г, 38 мл) и гидразина (16.8 г, 16.3 мл, 420 ммоль) в изопропиловом спирте (150 мл) кипятили с обратным холодильником при 100°С в атмосфере N2. Через 2 ч, реакция завершилась. Добавили этилацетат (200 мл), и затем воду (400 мл) для образования двух фаз. Водную фазу экстрагировали этилацетатом (6×200 мл). Органические слои объединили. Растворитель удалили с получением 8-фтор-2Н-фталазин-1-она в виде желтого осадка (32 г, 68% выход).
Стадия 3. Получение 2-хлор-6-(8-фтор-1-оксо-1Н-фталазин-2-ил)-бензальдегид
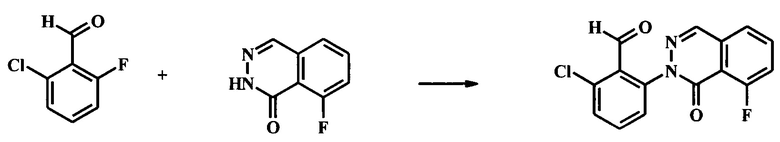
Раствор 8-фтор-2Н-фталазин-1-она (32 г, 195 ммоль), 2-хлор-6-фторбензальдегида (40.1 г, 254 ммоль), и карбоната цезия (63.7 г, 195 ммоль) в N,N-диметилацетамиде (200 мл) нагревали при 55°С в течение 24 ч. Воду (100 мл) добавили в эту реакционную смесь. Суспензию перемешивали в течение 1 ч. Осадок отфильтровали и промыли с помощью IPA/вода (1:2, 300 мл), с последующей водой (2×200 мл) с получением 2-хлор-6-(8-фтор-1-оксо-1Н-фталазин-2-ил)-бензальдегида в виде желтого осадка (50 г, 84% выход).
Стадия 4. Получение 2-(3-хлор-2-гидроксиметил-фенил)-8-фтор-2Н-фталазин-1-он

В суспензию борогидрида натрия (1.19 г, 29.7 ммоль) в изопропиловом спирте (130 мл) медленно добавили раствор 2-хлор-6-(8-фтор-1-оксо-1Н-фталазин-2-ил)-бензальдегида (30 г, 99.1 ммоль) в N,N-диметилацетамиде (220 мл). Смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь охладили на ледяной бане, и медленно добавили насыщенный раствор NH4Cl (220 мл). Суспензию отфильтровали и промыли с помощью IPA/вода (1:2, 200 мл) с получением 2-(3-хлор-2-гидроксиметил-фенил)-8-фтор-2Н-фталазин-1-она в виде желтого осадка (27 г, 88%).
Стадия 5. Получение уксусной кислоты 2-хлор-6-(8-фтор-1-оксо-1Н-фталазин-2-ил)-бензилового эфира

К раствору 2-(3-хлор-2-гидроксиметил-фенил)-8-фтор-2Н-фталазин-1-она (26 г, 85.3 ммоль) в CH2Cl2 (300 мл) добавили триэтиламин (11.2 г, 111 ммоль), уксусный ангидрид (11.0 г, 111 ммоль) и затем DMAP (5.21 г, 4.26 ммоль). Получившуюся смесь перемешивали при комнатной температуре до завершения реакции. Реакцию погасили водой (200 мл). Органический слой промыли насыщенным водным NaHCO3 (200 мл) и затем солевым раствором (200 мл). Растворитель удалили, и добавили гептаны/этилацетат (7:1, 240 мл). Суспензию нагревали при 60°С в течение 2 ч. Медленно охладили до комнатной температуры в течение ночи. Осадок собрали посредством фильтрации и промыли гептаном с получением уксусной кислоты 2-хлор-6-(8-фтор-1-оксо-1Н-фталазин-2-ил)-бензилового эфира в виде желтого осадка (28.5 г, 96%).
Промежуточное соединение А
Стадия 6. Получение калия (2-(ацетоксиметил)-3-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)фенил)трифторборат
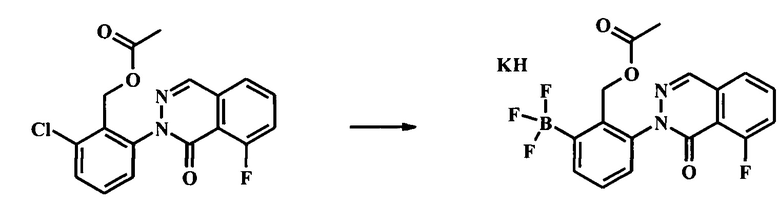
Смесь уксусной кислоты 2-хлор-6-(8-фтор-1-оксо-1Н-фталазин-2-ил)-бензилового эфира (27 г, 77.9 ммоль), бис(пинаколато)дибора (29.7 г, 117 ммоль), Pd(OAc)2 (719 мг, 3.36 ммоль), X-PHOS (3.19 г, 6.72 ммоль), и ацетата калия (16.6 г, 169 ммоль) в метилтетрагидрофуране (200 мл) дегазировали. Получившуюся смесь нагревали при 75°С в течение ночи. После этого смесь охладили, добавили 2 н HCl (100 мл) и смесь перемешивали в течение 1 ч и отфильтровали через слой целита. Органический слой из фильтрата разделили, промыли водой (180 мл) и сконцентрировали с получением густого масла. Масло растворили в метаноле (300 мл) и обработали раствором фтороводорода калия (3 М, 77.9 мл, 234 ммоль). Получившуюся суспензию нагревали при 45°С в течение 3 ч и затем перемешивали при комнатной температуре в течение ночи. Осадок собрали посредством фильтрации и промыли метанолом с получением калия (2-(ацетоксиметил)-3-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)фенил)трифторбората в виде желтого осадка (26 г, 92% выход).
Получение I-1
Стадия 1. Получение 2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-6-(6-метокси-2-метилпиридин-3-ил)бензилацетата
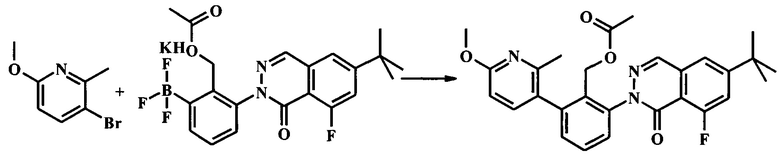
К раствору 3-бромо-6-метокси-2-метилпиридина (830 мг, 4.11 ммоль) в н-бутаноле (56.0 мл) добавили калия (2-(ацетоксиметил)-3-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)фенил)трифторборат (который может быть получен, как описано в Berthel, S.J. et al. US 2011-497093 P, промежуточное соединение в примере I-30; 1.95 г, 4.11 ммоль), воду (14.0 мл), трехосновный фосфат калия (1.74 г, 8.22 ммоль), X-PHOS (196 мг, 411 мкмоль) и бис(дибензилиденацетон)палладий (118 мг, 205 мкмоль) в атмосфере аргона. Реакционную смесь нагревали на масляной бане при 100°С в течение 2 ч до тех пор, пока не осталось исходного вещества по данным ЖХМС и TLC (7:3 гексан/этилацетат). Реакционной смеси дали остыть до комнатной температуры; добавили воду и смесь, экстрагировали этилацетатом (2Х). Органический экстракт промыли солевым раствором, высушили (Na2SO4) и сконцентрировали досуха. Очистка посредством колонки для флеш-хроматографии (Analogix IntelliFlash 280, колонка Analogix SF25-80 г, гексан/этилацетат 0-60% градиент в течение 45 мин) дала 2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-6-(6-метокси-2-метилпиридин-3-ил)бензилацетат (1.63 г, 3.33 ммоль, 81.1% выход) в виде белой пены. LC/MS наблюдаемый [М+Н]+ 490. 1Н NMR (300 MHz, ХЛОРОФОРМ-d) δ ppm 1.42 (s, 9Н) 1.77 (s, 3Н) 2.28 (s, 3Н) 3.97 (s, 3Н) 4.68-5.00 (m, 2Н) 6.63 (d, J=8.31 Hz, 1H) 7.29 (dd, J=7.55, 1.51 Hz, 1H) 7.37-7.62 (m, 5H) 8.21 (d, J=2.64 Hz, 1H).
Стадия 2. Получение 6-трет-бутил-8-фтор-2-[2-гидроксиметил-3-(6-метокси-2-метил-пиридин-3-ил)-фенил]-2Н-фталазин-1-она
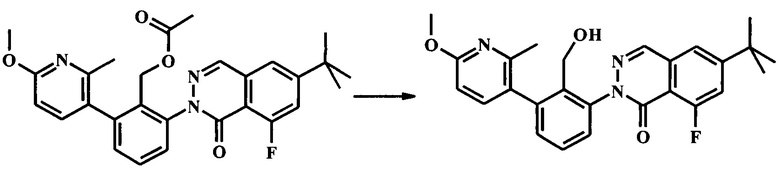
К раствору 2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-6-(6-метокси-2-метилпиридин-3-ил)бензилацетата (2.54 г, 5.19 ммоль) в метаноле (50 мл) и тетрагидрофуране (10 мл) добавили карбонат калия (143 мг, 1.04 ммоль). Реакционную смесь перемешивали в течение ночи. ЖХМС показала полную конверсию. В реакционную смесь добавили рН 2 буфер (10% водный KHSO4/Na2SO4) с последующей водой и смесь экстрагировали дихлорметаном (2Х). Объединенный органический экстракт промыли водой, высушили (Na2SO4) и сконцентрировали досуха с получением количественного выхода неочищенного 6-трет-бутил-8-фтор-2-(2-(гидроксиметил)-3-(6-метокси-2-метилпиридин-3-ил)фенил)фталазин-1(2Н)-она (2.45 г) в виде светло-желтой пены, которую использовали как есть на следующей стадии. LC/MS наблюдаемый [М+Н]+ 448.
Стадия 3. Получение 2-[2-бромметил-3-(6-метокси-2-метил-пиридин-3-ил)-фенил]-6-трет-бутил-8-фтор-2Н-фталазин-1-она

К раствору 6-трет-бутил-8-фтор-2-(2-(гидроксиметил)-3-(6-метокси-2-метилпиридин-3-ил)фенил)фталазин-1(2Н)-она (2.18 г, 4.87 ммоль) в дихлорметане (42.5 мл) добавили имидазол (348 мг, 5.12 ммоль). Реакционную смесь охладили до 0°С. В реакционную смесь добавили трифенилфосфин (1.41 г, 5.36 ммоль) и бром (817 мг, 264 мкл, 5.12 ммоль) в атмосфере аргона. Реакционной смеси дали нагреться до комнатной температуры и перемешивали при этой температуре в течение 3 ч. ТСХ (7:3 гексан/EtOAc) показала отсутствие исходного спирта. Добавили воду и смесь экстрагировали дихлорметаном (2Х). Объединенный органический экстракт высушили (Na2SO4) и сконцентрировали досуха. Очистка посредством колонки для флеш-хроматографии (Analogix IntelliFlash 280, колонка Analogix SF25-80 г, гексан/этилацетат 0-30% градиент в течение 10 мин, затем поддерживали при 30% этилацетате в течение 10 мин) дала чистый 2-(2-(бромметил)-3-(6-метокси-2-метилпиридин-3-ил)фенил)-6-трет-бутил-8-фторфталазин-1(2Н)-он (2.05 г, 4.02 ммоль, 82.4% выход). LC/MS наблюдаемый [М+Н]+ 510, 512. 1Н NMR (300 MHz, ХЛОРОФОРМ-d) δ ppm 1.43 (s, 9Н) 2.31 (s, 3Н) 3.99 (s, 3Н) 4.17 (d, J=10.58 Hz, 1H) 4.36 (d, J=10.58 Hz, 1H) 6.67 (d, J=8.31 Hz, 1H) 7.21-7.27 (m, 1H) 7.37-7.45 (m, 1H) 7.46-7.59 (m, 4H) 8.29 (d, J=2.64 Hz, 1H).
Стадия 4. Получение 2-[2-бромметил-3-(2-бромметил-6-метокси-пиридин-3-ил)-фенил]-6-трет-бутил-8-фтор-2Н-фталазин-1-она
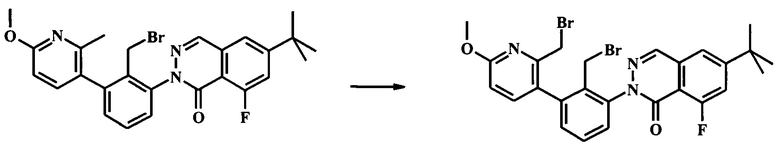
Суспензию 2-(2-(бромметил)-3-(6-метокси-2-метилпиридин-3-ил)фенил)-6-трет-бутил-8-фторфталазин-1(2Н)-она (2.05 г, 4.02 ммоль) в тетрахлориде углерода (363 мл) нагрели до 50°С до растворения большей части и затем добавили AIBN (33.0 мг, 201 мкмоль) и N-бромсукцинимид (751 мг, 4.22 ммоль) в атмосфере азота. Смесь нагревали при кипении с обратным холодильником в течение 3.5 ч. После охлаждения до комнатной температуры, смесь отфильтровали. Осадок промыли тетрахлоридом углерода. Объединенный фильтрат и промывочные жидкости сконцентрировали до небольшого объема. Очистка посредством колонки для флеш-хроматографии (Analogix IntelliFlash 280, колонка Thompson SF25-80 г, гексаны/(1:1 этилацетат/дихлорметан) 10-30% градиент в течение 10 мин, затем поддерживали при 30% этилацетат/дихлорметан в течение 10 мин) дала чистые и смешанные фракции. Смешанные фракции снова подвергли хроматографии и чистый продукт объединили с получением 2-(2-(бромметил)-3-(2-(бромметил)-6-метоксипиридин-3-ил)фенил)-6-трет-бутил-8-фторфталазин-1(2Н)-она (1.79 г, 3.04 ммоль, 75.6% выход) в виде белой пены вместе с 211 мг (10%) восстановленного исходного вещества. Наблюдаемый ЖХ/МС продукта [М+Н]+ 590. 1Н NMR (300 MHz, ХЛОРОФОРМ-d) δ ppm 1.43 (s, 9Н) 4.02 (s, 3Н) 4.08-4.45 (m, 4Н) 6.78 (d, J=8.31 Hz, 1H) 7.39-7.64 (m, 6H) 8.29 (d, J=2.27 Hz, 1H).
Стадия 5. Получение 8-(6-трет-бутил-8-фтор-1-оксо-1Н-фталазин-2-ил)-3-метокси-5Н,7Н-бензо[3,4]циклогепта[1,2-b]пиридин-6-он

К смеси гидроксида натрия (1.3 г, 32.4 ммоль) и йодида тетрабутиламмония (239 мг, 648 мкмоль) в воде (12 мл) и дихлорметане (30 мл) добавили при 0°С по каплям раствор толуолсульфонилметил изоцианата (1.27 г, 6.48 ммоль) в дихлорметане (18.0 мл), с последующим раствором 2-(2-(бромметил)-3-(2-(бромметил)-6-метоксипиридин-3-ил)фенил)-6-трет-бутил-8-фторфталазин-1(2Н)-она (1.91 г, 3.24 ммоль) в дихлорметане (18.0 мл). Ледяную ванну убрали, и смесь интенсивно перемешивали при комнатной температуре в течение 24 ч. ТСХ (силикагель, 7:3 гексаны/этилацетат) показала только следовые количества оставшегося исходного вещества. Добавили воду и смесь экстрагировали дихлорметаном (2Х). Объединенный органический экстракт промыли водой, высушили (Na2SO4) и сконцентрировали досуха с получением неочищенного 6-трет-бутил-8-фтор-2-[6-изоциано-3-метокси-6-(толуол-4-сульфонил)-6,7-дигидро-5Н-бензо[3,4]циклогепта[1,2-b]пиридин-8-ил]-2Н-фталазин-1-она (2.97 г), который сразу взяли на следующую стадию на следующей стадии. К неочищенному 6-трет-бутил-8-фтор-2-[6-изоциано-3-метокси-6-(толуол-4-сульфонил)-6,7-дигидро-5Н-бензо[3,4]циклогепта[1,2-b]пиридин-8-ил]-2Н-фталазин-1-ону (2.97 г) в дихлорметане (100 мл) добавили концентрированную соляную кислоту (15.9 мл, 191 ммоль). Получившуюся смесь перемешивали при комнатной температуре в течение 3 ч, до полного расходования исходного вещества по данным ТСХ (7:3 гексан/этилацетат). Раствор 10% бикарбоната натрия осторожно добавляли до прекращения выделения газа. Добавили дополнительное количество воды, и смесь экстрагировали этилацетатом (2Х). Объединенный органический экстракт промыли солевым раствором, высушили (Na2SO4) и сконцентрировали досуха. Очистка посредством колонки для флеш-хроматографии (Analogix IntelliFlash 280, колонка Analogix SF25-40 г, гексан/этилацетат 0-25% градиент в течение 15 мин, затем поддерживали при 25% гексаны/этилацетат в течение 15 мин.) дала 795 мг 8-(6-трет-бутил-8-фтор-1-оксо-1Н-фталазин-2-ил)-3-метокси-5Н,7Н-бензо[3,4]циклогепта[1,2-b]пиридин-6-она (54% выход). Наблюдаемый ЖХ/МС продукта [М+Н]+ 458. 1Н NMR (300 MHz, ХЛОРОФОРМ-d) δ ppm 1.43 (s, 9Н) 3.38 (br. s., 2H) 3.80 (br. s., 2H) 3.98 (s, 3H) 6.83 (d, J=8.31 Hz, 1H) 7.37-7.66 (m, 5H) 7.84 (d, J=8.31 Hz, 1H) 8.23 (d, J=2.27 Hz, 1H).
Стадия 6. Получение 8-(6-трет-бутил-8-фтор-1-оксо-1Н-фталазин-2-ил)-5,7-дигидро-4Н-бензо[3,4]циклогепта[1,2-b]пиридин-3,6-dione

К раствору 8-(6-трет-бутил-8-фтор-1-оксо-1Н-фталазин-2-ил)-3-метокси-5Н,7Н-6ензо[3,4]циклогепта[1,2-b]пиридин-6-она (795 мг, 1.74 ммоль) в ацетонитриле (16.1 мл) добавили йодид натрия (521 мг, 3.48 ммоль) и хлортриметилсилан (378 мг, 440 мкл, 3.48 ммоль) в атмосфере аргона. Смесь нагревали при 82-83°С в течение 3 ч. ТСХ (6:4 гексан/этилацетат) показала отсутствие исходного вещества, и ЖХМС показала только один пик. После охлаждения, добавили холодный 10% водный тиосульфат натрия. Получившаяся суспензия превратилась в раствор, затем выпал осадок. Добавили некоторое количество этилацетата и твердый продукт собрали посредством фильтрации и промыли несколько раз водой, с последующим этилацетатом и затем эфиром с получением чистого продукта (приблизительно 600 мг) как подтвердили с помощью ЖХМС. Фильтрат и промывочные жидкости поместили на разделительную воронку. Смесь экстрагировали этилацетатом (2Х). Объединенные органические слои высушили над MgSO4, отфильтровали и фильтрат сконцентрировали до небольшого объема. Осадок собрали посредством фильтрации, промыли небольшим объемом этилацетата и эфира. ЖХМС показала наличие чистого продукта. Этот осадок объединили с первым осадком с получением 688 мг 8-(6-трет-бутил-8-фтор-1-оксо-1Н-фталазин-2-ил)-5,7-дигидро-4Н-бензо[3,4]циклогепта[1,2-b]пиридин-3,6-диона (90% выход) в виде осадка кремового цвета. Наблюдаемый ЖХ/МС продукта [М+Н]+ 444. 1Н NMR (300 MHz, ХЛОРОФОРМ-d) δ ppm 1.43 (s, 9Н) 3.01-3.96 (m, 4Н) 6.71 (d, J=9.44 Hz, 1H) 7.35-7.67 (m, 5H) 7.80 (d, J=9.44 Hz, 1H) 8.23 (d, J=2.64 Hz, 1H).
Стадия 7. Получение 8-(6-трет-бутил-8-фтор-1-оксо-1Н-фталазин-2-ил)-6-гидрокси-4,5,6,7-тетрагидро-бензо[3,4]циклогепта[1,2-b]пиридин-3-он
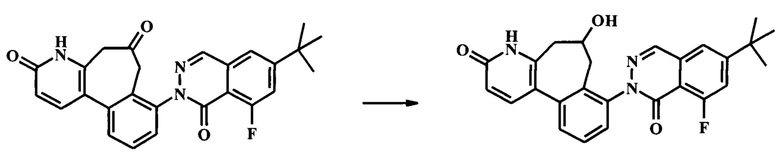
В 250 мл круглодонной колбе, 8-(6-трет-бутил-8-фтор-1-оксо-1Н-фталазин-2-ил)-5,7-дигидро-4Н-бензо[3,4]циклогепта[1,2-b]пиридин-3,6-дион (824 мг, 1.86 ммоль) объединили с дихлорметаном (50 мл) и метанолом (2 мл) с получением бесцветного раствора. Добавили борогидрид натрия (105 мг, 2.79 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч до завершения реакции, как определили по данным ЖХМС. Добавили насыщенный раствор f NH4Cl (10 мл) добавили. Реакционную смесь перемешивали при комнатной температуре в течение 10 мин, затем разбавили водой и дихлорметаном и получившийся осадок собрали посредством фильтрации. Осадок промыли несколько раз водой, затем дихлорметаном/эфиром и затем эфиром. Получившийся кремообразный осадок olid высушили с помощью помпы с получением 762 мг чистого продукта, 8-(6-трет-бутил-8-фтор-1-оксо-1Н-фталазин-2-ил)-6-гидрокси-4,5,6,7-тетрагидро-бензо[3,4]циклогепта[1,2-b]пиридин-3-он. Фильтрат и промывочные жидкости поместили на разделительную воронку и экстрагировали дихлорметаном (3Х). Органические слои высушили над Na2SO4 и сконцентрировали под вакуумом. Неочищенный материал очистили с помощью флеш-хроматографии (силикагель, 24 г, 1.5%-4% МеОН в ДХМ) с получением дополнительных 39 мг чистого продукта, 8-(6-трет-бутил-8-фтор-1-оксо-1Н-фталазин-2-ил)-6-гидрокси-4,5,6,7-тетрагидро-бензо[3,4]циклогепта[1,2-b]пиридин-3-она. Обе порции объединили с получением 801 мг (97% выход) чистого продукта в виде осадка кремового цвета. Наблюдаемый ЖХ/МС продукта [М+Н]+ 445.9. 1Н NMR (300 MHz, DMSO-d6) δ ppm 1.37 (s, 9H) 2.07 (d, J=11.33 Hz, 1H) 2.30-2.45 (m, 1H) 2.61 (br. s., 2H) 4.18-4.51 (m, 1H) 6.34 (d, J=9.06 Hz, 1H) 7.29 (br. s., 1H) 7.43 (d, J=2.27 Hz, 2H) 7.63 (t, J=7.93 Hz, 1H) 7.76 (d, J=13.60 Hz, 1H) 7.88 (br. s., 1H) 8.52 (d, J=4.91 Hz, 1H) 11.89 (br. s., 1H).
Стадия 8. Получение 6-(трет-6утил-диметил-силанилокси)-8-(6-трет-бутил-8-фтор-1-оксо-1Н-фталазин-2-ил)-4,5,6,7-тетрагидро-бензо[3,4]циклогепта[1,2-b]пиридин-3-она
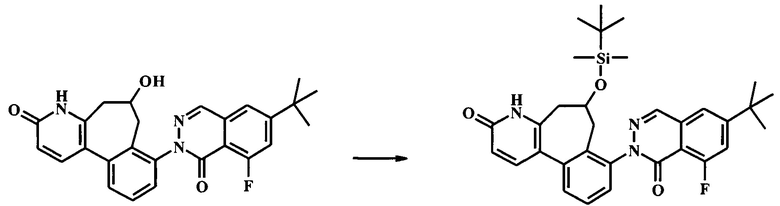
В 25 мл круглодонной колбе, 8-(6-трет-бутил-8-фтор-1-оксо-1Н-фталазин-2-ил)-6-гидрокси-4,5,6,7-тетрагидро-бензо[3,4]циклогепта[1,2-b]пиридин-3-он (636 мг, 1.43 ммоль) объединили с ДМФ (18 мл) с получением бесцветного раствора. Реакционную смесь охладили в ванне со льдом. В реакционную смесь добавили 2,6-лутидин (337 мг, 366 мкл, 3.14 ммоль) и затем покапельно добавили трет-бутилдиметилсилил трифторметансульфонат (830 мг, 721 мкл, 3.14 ммоль). Реакционной смеси дали нагреться до комнатной температуры в течение 40 минут. Реакция завершилась как определили по данным ЖХМС. Добавили метанол, с последующим насыщенным раствором NH4Cl. Перемешивание продолжили в течение 5 мин. Реакционную смесь влили в 150 мл H2O и экстрагировали дихлорметаном (3×75 мл). Органические слои высушили над Na2SO4 и сконцентрировали под вакуумом с получением 0.79 г 6-(трет-бутил-диметил-силанилокси)-8-(6-трет-бутил-8-фтор-1-оксо-1Н-фталазин-2-ил)-4,5,6,7-тетрагидро-бензо[3,4]циклогепта[1,2-b]пиридин-3-она (98% выход). Этот продукт объединили с продуктом из предшествующей порции (0.17 г) с получением 0.96 г соединения, указанного в заголовке. Наблюдаемый ЖХ/МС продукта [М+Н]+ 560.1. 1Н NMR (300 MHz, ХЛОРОФОРМ-d) δ ppm -0.11 (s, 3Н) -0.01 (br. s., 3H) 0.69 (s, 9H) 1.43 (s, 9H) 2.23 (dd, J=13.22, 9.44 Hz, 1H) 2.44-2.88 (m, 3H) 4.54-4.97 (m, 1H) 6.60 (d, J=9.44 Hz, 1H) 7.28-7.56 (m, 6H) 7.65 (br. s., 1H) 8.22 (d, J=2.64 Hz, 1H).
Стадия 9. Получение 6-(трет-бутил-диметил-силанилокси)-8-(6-трет-бутил-8-фтор-1-оксо-1Н-фталазин-2-ил)-4-метил-4,5,6,7-тетрагидро-бензо[3,4]циклогепта[1,2-b]пиридин-3-он
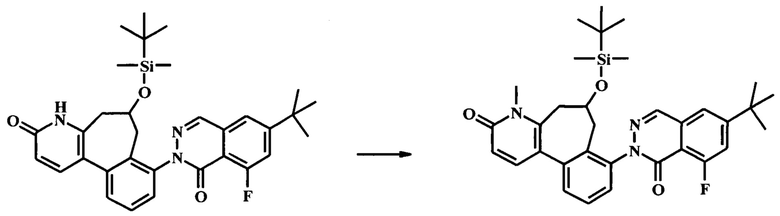
К суспензии 6-(трет-бутил-диметил-силанилокси)-8-(6-трет-бутил-8-фтор-1-оксо-1Н-фталазин-2-ил)-4,5,б,7-тетрагидро-бензо[3,4]циклогепта[1,2-b]пиридин-3-она (0.96 г, 1.72 ммоль) в тетрагидрофуране (25 мл) добавили при 25°С 1М гексаметилдисилазан лития в тетрагидрофуране (1.8 мл, 1.8 ммоль) в атмосфере аргона. Смесь перемешивали в течение 10 мин при комнатной температуре. Добавили метилйодид (487 мг, 214 мкл, 3.43 ммоль). Через 90 мин, реакция была завершена на 90%. Добавили дополнительно 2 капли метилйодида (2 капли) и реакционную смесь перемешивали при комнатной температуре в течение 30 мин. Дополнительного продукта не образовалось. Добавили дополнительное количество 1М гексаметилдисилазан лития в тетрагидрофуране (0.2 мл) и реакционную смесь перемешивали при комнатной температуре в течение 30 мин. Дополнительного продукта не образовалось. Реакционную смесь погасили насыщенным раствором NH4Cl. Реакционную смесь влили в 150 мл H2O и экстрагировали этилацетатом (3×100 мл) и затем дихлорметаном (1Х). Объединенную органическую фазу промыли солевым раствором, высушили над MgSO4 и сконцентрировали под вакуумом. Неочищенный материал очистили с помощью флеш-хроматографии (силикагель, 40 г, 1.5%-3% метанол в дихлорметане) с получением 759 мг 6-(трет-бутил-диметил-силанилокси)-8-(6-трет-бутил-8-фтор-1-оксо-1Н-фталазин-2-ил)-4-метил-4,5,6,7-тетрагидро-бензо[3,4]циклогепта[1,2-b]пиридин-3-она (77% выход) в виде осадка кремового цвета. Наблюдаемый ЖХ/МС продукта [М+Н]+ 574.1. 1Н NMR (300 MHz, ХЛОРОФОРМ-d) δ ppm -0.13 (s, 3Н) -0.02 (s, 3Н) 0.76 (br. s., 9Н) 1.44 (s, 9H) 2.22 (br. s., 1H) 2.65 (d, J=6.80 Hz, 2H) 3.04 (d, J=14.73 Hz, 1H) 3.76 (s, 3H) 4.60-5.08 (m, 1H) 6.63 (d, J=9.44 Hz, 1H) 7.28-7.60 (m, 6H) 8.23 (d, J=2.27 Hz, 1H).
Стадия 10. Получение 2-бромо-6-(трет-бутил-диметил-силанилокси)-8-(6-трет-бутил-8-фтор-1-оксо-1Н-фталазин-2-ил)-4-метил-4,5,6,7-тетрагидро-бензо[3,4]циклогепта[1,2-b]пиридин-3-он
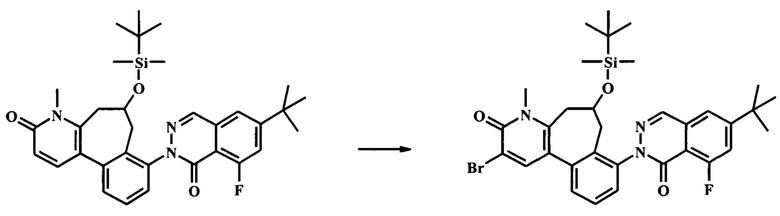
В 10 мл круглодонной колбе, 6-(трет-бутил-диметил-силанилокси)-8-(6-трет-бутил-8-фтор-1-оксо-1Н-фталазин-2-ил)-4-метил-4,5,6,7-тетрагидро-бензо[3,4]циклогепта[1,2-b]пиридин-3-он (260 мг, 453 мкмоль) объединили дихлорметаном (10 мл) с получением бесцветного раствора. Добавили гидробромистую кислоту (80.2 мг, 53.8 мкл, 476 мкмоль) и сразу изоамилнитрил (111 мг, 128 мкл, 952 мкмоль). Реакционную смесь перемешивали при комнатной температуре в течение 4 ч. Реакция завершилась, как определили по данным ЖХМС. Реакционную смесь влили в 75 мл насыщенного NaHCO3/H2O и экстрагировали дихлорметаном (3×50 мл). Объединенные органические слои высушили над Na2SO4 и сконцентрировали под вакуумом. Неочищенный материал очистили с помощью флеш-хроматографии (силикагель, 24 г, 15%-50% этилацетат в гексанах) с получением 278 мг 2-бромо-6-(трет-бутил-диметил-силанилокси)-8-(6-трет-бутил-8-фтор-1-оксо-1Н-фталазин-2-ил)-4-метил-4,5,6,7-тетрагидро-бензо[3,4]циклогепта[1,2-b]пиридин-3-она в виде белого осадка. Наблюдаемый ЖХ/МС продукта [М+Н]+ 652, 654. 1Н NMR (300 MHz, ХЛОРОФОРМ-d) δ ppm -0.13 (s, 3Н) -0.02 (s, 3Н) 0.77 (s, 9Н) 1.44 (s, 9Н) 2.19 (br. s., 1H) 2.66 (br. s., 2H) 3.02 (d, J=13.97 Hz, 1H) 3.84 (s, 3H) 4.60-5.06 (m, 1H) 7.27-7.61 (m, 5H) 7.90 (s, 1H) 8.23 (br. s., 1H).
Стадия 11. Получение 6-(трет-бутил-диметилсиланилокси)-8-(6-трет-бутил-8-фтор-1-оксо-1Н-фталазин-2-ил)-4-метил-2-[5-(морфолин-4-карбонил)-пиридин-2-иламино]-4,5,6,7-тетрагидробензо[3,4]циклогепта[1,2-b]пиридин-3-он
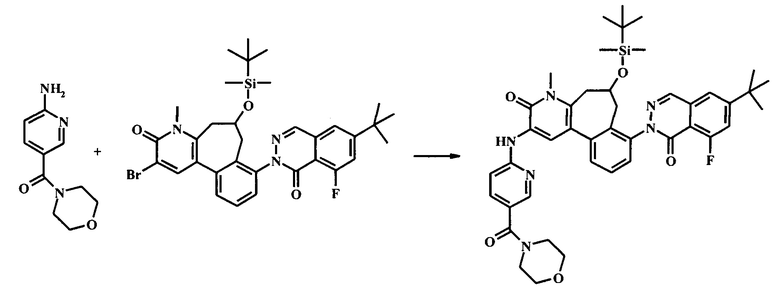
К раствору (6-аминопиридин-3-ил)(морфолино)метанона (87.0 мг, 420 мкмоль) и 2-бромо-6-(трет-бутил-диметил-силанилокси)-8-(6-трет-бутил-8-фтор-1-оксо-1Н-фталазин-2-ил)-4-метил-4,5,6,7-тетрагидро-бензо[3,4]циклогепта[1,2-b]пиридин-3-она (274 мг, 420 мкмоль) в диоксане (7 мл) добавили в атмосфере аргона: карбонат цезия (410 мг, 1.26 ммоль), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (36.4 мг, 63.0 мкмоль) и трис(дибензилиденацетон)дипалладий(0) (19.2 мг, 21.0 мкмоль). Реакционную смесь нагревали при 100°С в течение 14 ч, затем дали остыть до комнатной температуры. Реакционную смесь разбавили дихлорметаном, высушили над Na2SO4 и отфильтровали через целит. Слой целита промыли дихлорметаном. Объединили фильтрат и промывочные жидкости сконцентрировали и неочищенный материал очистили с помощью флеш-хроматографии (силикагель, 40 г, 1% метанол в дихлорметане; затем 1%-4% градиент метанола в дихлорметане) с получением 270 мг 6-(трет-бутил-диметилсиланилокси)-8-(6-трет-бутил-8-фтор-1-оксо-1Н-фталазин-2-ил)-4-метил-2-[5-(морфолин-4-карбонил)-пиридин-2-иламино]-4,5,6,7тетрагидробензо[3,4]циклогепта[1,2-b]пиридин-3-она (83% выход). Наблюдаемый ЖХ/МС продукта [М+Н]+ 779. 1Н NMR (300 MHz, ХЛОРОФОРМ-d) δ ppm -0.14 (s, 3Н) -0.02 (s, 3Н) 0.76 (s, 9Н) 1.44 (s, 9Н) 2.25 (br. s., 1H) 2.64 (br. s., 2H) 3.00 (d, J=14.35 Hz, 1H) 3.71 (d, J=8.69 Hz, 8H) 3.86 (s, 3H) 4.58-5.08 (m, 1H) 6.84 (d, J=8.31 Hz, 1H) 7.28-7.75 (m, 6H) 8.23 (d, J=2.27 Hz и наложение br. S., 2H) 8.35 (br. s., 1H) 8.77 (br. s., 1H).
Пример 1
Стадия 12. Получение 8-(6-трет-бутил-8-фтор-1-оксо-1Н-фталазин-2-ил)-6-гидрокси-4-метил-2-[5-(морфолин-4-карбонил)-пиридин-2-иламино]-4,5,6,7-тетрагидробензо[3,4]циклогепта[1,2-b]пиридин-3-она
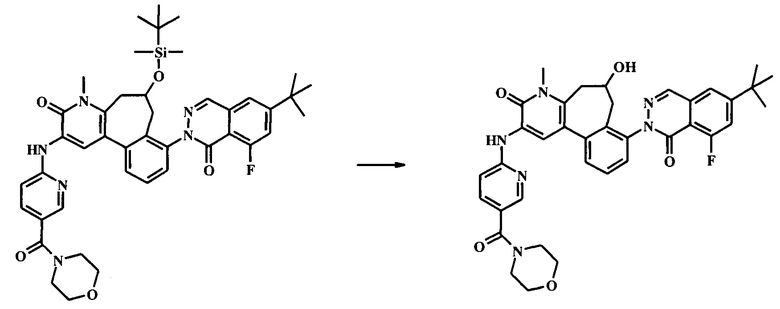
В 100 мл круглодонной колбе, 6-(трет-бутил-диметилсиланилокси)-8-(6-трет-бутил-8-фтор-1-оксо-1Н-фталазин-2-ил)-4-метил-2-[5-(морфолин-4-карбонил)-пиридин-2-иламино]-4,5,6,7-тетрагидробензо[3,4]циклогепта[1,2-b]пиридин-3-он объединили с тетрагидрофураном (6.0 мл) с получением желтого раствора. Добавили раствор фторида тетрабутиламмония (514 мкл 1М в тетрагидрофуране, 514 мкмоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч, влили в 75 мл H2O и экстрагировали этилацетатом (2×75 мл) и дихлорметаном (1Х). Объединенные органические слои высушили над MgSO4 и сконцентрировали под вакуумом. Неочищенный материал очистили с помощью флеш-хроматографии (силикагель, 40 г, 5%-30% (60:10:1 дихлорметан: метанол: NH4OH) в дихлорметане) с получением стекловидного вещества. Это вещество растворили в небольшом объеме дихлорметана и добавили эфир. Получившийся преципитировавшийся осадок собрали посредством фильтрации и тритурировали с эфиром с получением 135 мг чистого продукта, 8-(6-трет-бутил-8-фтор-1-оксо-1Н-фталазин-2-ил)-6-гидрокси-4-метил-2-[5-(морфолин-4-карбонил)-пиридин-2-иламино]-4,5,6,7-тетрагидробензо[3,4]циклогепта[1,2-b]пиридин-3-он в виде белого порошка. Наблюдаемый ЖХ/МС продукта [М+Н]+ 665. 1Н NMR (300 MHz, ХЛОРОФОРМ-d) δ ppm 1.43 (s, 9Н) 2.14-2.88 (m, 3Н) 3.17 (d, J=12.46 Hz, 1H) 3.71 (d, J=9.06 Hz, 8H) 3.83 (s, 3H) 3.95-4.20 (m, 1H) 4.39 (br. s., 1H) 6.83 (d, J=8.31 Hz, 1H) 7.32 (d, J=7.18 Hz, 1H) 7.45-7.73 (m, 5H) 8.03-8.43 (m, 3H) 8.77 (s, 1H). Фильтрат сконцентрировали с получением дополнительных 84 мг чистого продукта в виде белого осадка. Общий выход составил 219 мг (96% выход).
Пример 2
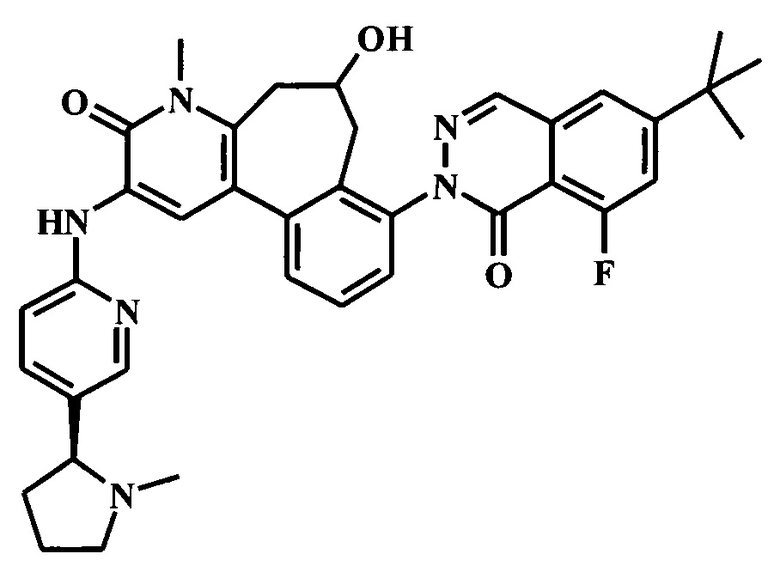
8-(6-трет-бутил-8-фтор-1-оксо-1Н-фталазин-2-ил)-6-гидрокси-4-метил-2-[5-((S)-1-метил-пирролидин-2-ил)-пиридин-2-иламино]-4,5,6,7-тетрагидро-бензо[3,4]циклогепта[1,2-b]пиридин-3-он
Получение способом, аналогичным Примеру 1 за исключением замены на 5-((S)-1-метил-пирролидин-2-ил)-пиридин-2-иламин ((который может быть получен, как описано у Berthel, S.J. et al. US 2011-497093 P, промежуточное соединение в примере 1-18) (6-аминопиридин-3-ил)(морфолино)метанона на стадии 11 дало 55 мг соединения, указанного в заголовке, в виде белого осадка. Наблюдаемый ЖХ/МС продукта [М+Н]+ 635.
Пример 3
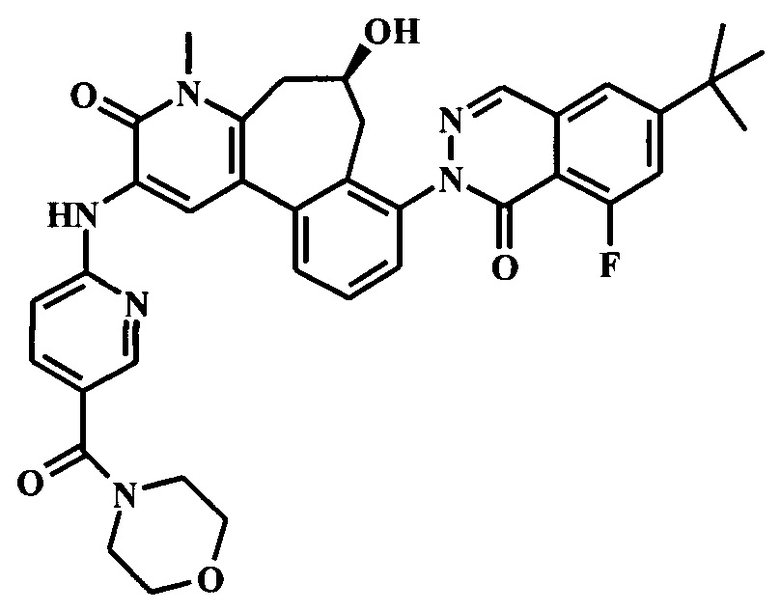
(S)-8-(6-трет-бутил-8-фтор-1-оксо-1Н-фталазин-2-ил)-6-гидрокси-4-метил-2-[5-(морфолин-4-карбонил)-пиридин-2-иламино]-4,5,6,7-тетрагидро-бензо[3,4]циклогепта[1,2-b]пиридин-3-он
В 10 мл круглодонной колбе, 6-(трет-бутил-диметил-силанилокси)-8-(6-трет-бутил-8-фтор-1-оксо-1Н-фталазин-2-ил)-4-метил-4,5,6,7-тетрагидро-бензо[3,4]циклогепта[1,2-b]пиридин-3-он (490 мг, 854 мкмоль) объединили с дихлорметаном (18.8 мл) с получением бесцветного раствора. HBr (151 мг, 101 мкл, 897 мкмоль) добавили и затем сразу изоамилнитрит (210 мг, 241 мкл, 1.79 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 4 ч до завершения по данным ТСХ. Реакционную смесь влили в 75 мл насыщенного NaHCO3/H2O и экстрагировали дихлорметаном (3×50 мл). Органические слои высушили над Na2SO4, отфильтровали и фильтрат сконцентрировали под вакуумом. Материал растворили в этилацетате и сконцентрировали снова с получением 560 мг (100%) 2-бромо-6-(трет-бутил-диметил-силанилокси)-8-(6-трет-бутил-8-фтор-1-оксо-1Н-фталазин-2-ил)-4-метил-4,5,6,7-тетрагидро-бензо[3,4]циклогепта[1,2-b]пиридин-3-она в виде бледно-желтого осадка. Этот рацемический 2-бромо-6-(трет-бутил-диметил-силанилокси)-8-(6-трет-бутил-8-фтор-1-оксо-1Н-фталазин-2-ил)-4-метил-4,5,6,7-тетрагидро-бензо[3,4]циклогепта[1,2-b]пиридин-3-он (360 мг) подвергли хиральному разделению.
Хиральное разделение с использованием сверхкритической жидкостной хроматографии на колонке KROMASIL OD (элюент 30% метанол в CO2) дало 170 мг (S)-2-бромо-6-(трет-бутил-диметил-силанилокси)-8-(6-трет-бутил-8-фтор-1-оксо-1Н-фталазин-2-ил)-4-метил-4,5,6,7-тетрагидро-бензо[3,4]циклогепта[1,2-b]пиридин-3-она в виде элюируемого первым пика и 173 мг (R)-2-бромо-6-(трет-бутил-диметил-силанилокси)-8-(6-трет-бутил-8-фтор-1-оксо-1Н-фталазин-2-ил)-4-метил-4,5,6,7-тетрагидро-бензо[3,4]циклогепта[1,2-b]пиридин-3-она в виде элюируемого вторым пика.
К раствору (6-аминопиридин-3-ил)(морфолино)метанона (8.89 мг, 42.9 мкмоль) и (S)-2-бромо-6-(трет-бутил-диметил-силанилокси)-8-(6-трет-бутил-8-фтор-1-оксо-1Н-фталазин-2-ил)-4-метил-4,5,6,7-тетрагидро-бензо[3,4]циклогепта[1,2-b]пиридин-3-она (28 мг, 42.9 мкмоль) в диоксане (715 мкл) добавили в атмосфере аргона карбонат цезия (41.9 мг, 129 мкмоль), 4,5бис(дифенилфосфино)-9,9-диметилксантен (3.72 мг, 6.44 мкмоль) и трис(дибензилиденацетон)дипалладий(0) (1.96 мг, 2.15 мкмоль). Смесь нагревали при 100°С в течение 14 ч перед тем, как дали остыть до комнатной температуры. Неочищенную реакционную смесь разбавили дихлорметаном (15 мл) и высушили над Na2SO4. Осадки отфильтровали и промыли дихлорметаном. Объединенный фильтрат и промывочные жидкости сконцентрировали под вакуумом. Неочищенный продукт очистили с помощью флеш-хроматографии (силикагель, 12 г, 1%, затем 1%-4% метанола в дихлорметане) с получением 24.5 мг (S)-6-(трет-бутил-диметилсиланилокси)-8-(6-трет-бутил-8-фтор-1-оксо-1Н-фталазин-2-ил)-4-метил-2-[5-(морфолин-4-карбонил)-пиридин-2-иламино]-4,5,6,7-тетрагидробензо[3,4]циклогепта[1,2-b]пиридин-3-она, который использовали как есть на следующей стадии. Наблюдаемый ЖХ/МС продукта [М+Н]+ 779. В 50 мл круглодонной колбе, (S)-6-(трет-бутил-диметилсиланилокси)-8-(6-трет-бутил-8-фтор-1-оксо-1Н-фталазин-2-ил)-4-метил-2-[5-(морфолин-4-карбонил)-пиридин-2-иламино]-4,5,6,7 тетрагидробензо[3,4]циклогепта[1,2-b]пиридин-3-он (24.5 мг, 31.5 мкмоль) объединили с ТГФ (1.5 мл) с получением желтого раствора. Добавили раствор фторида тетрабутиламмония (47.2 мкл 1М в тетрагидрофуране, 47.2 мкмоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч до завершения по данным ЖХМС. Реакционную смесь влили в 75 мл H2O и экстрагировали этилацетатом (2×75 мл) и 1Х дихлорметан. Органические слои высушили над MgSO4 и сконцентрировали под вакуумом. Неочищенный материал очистили с помощью флеш-хроматографии (силикагель, 40 г, 5%-30% (60:10:1 дихлорметан: метанол: NH4OH) в дихлорметане) с получением чистого продукта в виде стеклообразного вещества. Продукт растворили в небольшом объеме дихлорметана и добавили эфир. Белый осадок преципитировался с получением 18 мг (63%) (S)-8-(6-трет-бутил-8-фтор-1-оксо-1Н-фталазин-2-ил)-6-гидрокси-4-метил-2-[5-(морфолин-4-карбонил)-пиридин-2-иламино]-4,5,6,7-тетрагидробензо[3,4]циклогепта[1,2-b]пиридин-3-она в виде белого осадка. Наблюдаемый ЖХ/МС продукта [М+Н]+ 665.
Пример 4
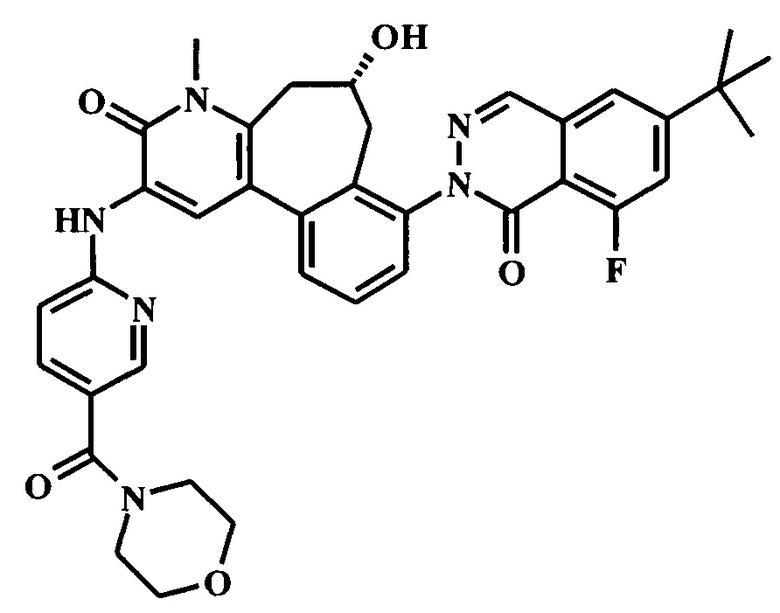
(R)-8-(6-трет-бутил-8-фтор-1-оксо-1Н-фталазин-2-ил)-6-гидрокси-4-метил-2-[5-(морфолин-4-карбонил)-пиридин-2-иламино]-4,5,6,7-тетрагидро-бензо[3,4]циклогепта[1,2-b]пиридин-3-он
К раствору (6-аминопиридин-3-ил)(морфолино)метанона (7.94 мг, 38.3 мкмоль) и (R)-2-бромо-6-(трет-бутил-диметил-силанилокси)-8-(6-трет-бутил-8-фтор-1-оксо-1Н-фталазин-2-ил)-4-метил-4,5,6,7-тетрагидро-бензо[3,4]циклогепта[1,2-b]пиридин-3-она (полученного в примере 3, 25 мг, 38.3 мкмоль) в диоксане (638 мкл) добавили в атмосфере аргона карбонат цезия (37.4 мг, 115 мкмоль), 4,5бис(дифенилфосфино)-9,9-диметилксантен (3.32 мг, 5.75 мкмоль) и трис(дибензилиденацетон)дипалладий(0) (1.75 мг, 1.92 мкмоль). Смесь нагревали при 100°С в течение 14 ч перед тем как ей дали остыть до комнатной температуры. Неочищенную реакционную смесь разбавили дихлорметаном (15 мл) и высушили над Na2SO4. Осадки отфильтровали и промыли дихлорметаном. Объединенный фильтрат и промывочные жидкости сконцентрировали под вакуумом. Неочищенный продукт очистили с помощью флеш-хроматографии (силикагель, 12 г, 1%, затем 1%-4% метанол в дихлорметане) с получением 21.3 мг (R)-6-(трет-бутил-диметилсиланилокси)-8-(6-трет-бутил-8-фтор-1-оксо-1Н-фталазин-2-ил)-4-метил-2-[5-(морфолин-4-карбонил)-пиридин-2-иламино]-4,5,6,7тетрагидробензо[3,4]циклогепта[1,2-b]пиридин-3-она, который использовали как есть на следующей стадии. Наблюдаемый ЖХ/МС продукта [М+Н]+ 779.
В 50 мл круглодонной колбе, (R)-6-(трет-бутил-диметилсиланилокси)-8-(6-трет-бутил-8-фтор-1-оксо-1Н-фталазин-2-ил)-4-метил-2-[5-(морфолин-4-карбонил)-пиридин-2-иламино]-4,5,6,7тетрагидробензо[3,4]циклогепта[1,2-b]пиридин-3-он (21.3 мг, 27.3 мкмоль) объединили с ТГФ (1.5 мл) с получением желтого раствора. Добавили раствор фторида тетрабутиламмония (41.0 мкл 1M в тетрагидрофуране, 41.0 мкмоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч до завершения реакции по данным ЖХМС. Реакционную смесь влили в 75 мл H2O и экстрагировали этилацетатом (2×75 мл) и 1Х дихлорметан. Органические слои высушили над MgSO4 и сконцентрировали под вакуумом. Неочищенный материал очистили с помощью флеш-хроматографии (силикагель, 40 г, 5%-30% (60:10:1 дихлорметан: метанол: NH4OH) в дихлорметане) с получением чистого продукта в виде стеклообразного вещества. Продукт растворили в небольшом объеме дихлорметана и добавили эфир. Белый осадок преципитировался с получением 15 мг (59%) (R)-8-(6-трет-бутил-8-фтор-1-оксо-1Н-фталазин-2-ил)-6-гидрокси-4-метил-2-[5-(морфолин-4-карбонил)-пиридин-2-иламино]-4,5,6,7-тетрагидробензо[3,4]циклогепта[1,2-b]пиридин-3-она в виде белого осадка. Наблюдаемый ЖХ/МС продукта [М+Н]+ 665.
Пример 5
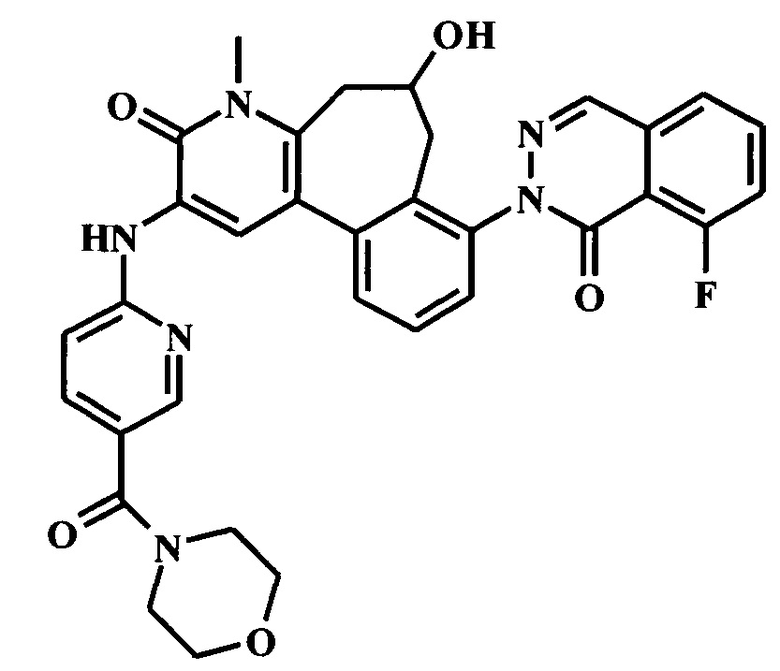
8-(8-фтор-1-оксо-1Н-фталазин-2-ил)-6-гидрокси-4-метил-2-[5-(морфолин-4-карбонил)-пиридин-2-иламино]-4,5,6,7-тетрагидро-бензо[3,4]циклогепта[1,2-b]пиридин-3-он
Получение способом, аналогичным Примеру 1, за исключением замены на калия (2-(ацетоксиметил)-3-(8-фтор-1-оксофталазин-2(1Н)-ил)фенил)трифторборат калия (2-(ацетоксиметил)-3-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)фенил)трифторбората на стадии 1 дало 60 мг соединения, указанного в заголовке, в виде светло-коричневого осадка. Наблюдаемый ЖХ/МС продукта [М+Н]+ 609.
Пример 6
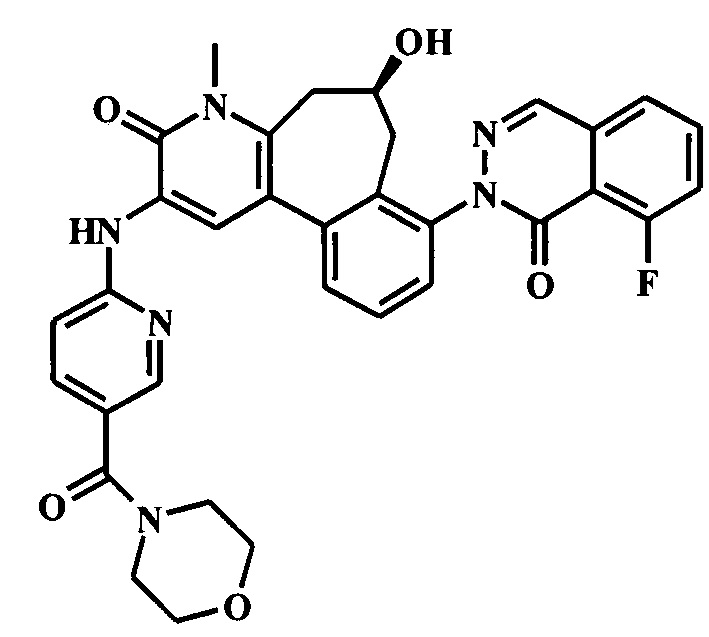
(8)-8-(8-фтор-1-оксо-1Н-фталазин-2-ил)-6-гидрокси-4-метил-2-[5-(морфолин-4-карбонил)-пиридин-2-иламино]-4,5,6,7-тетрагидро-бензо[3,4]циклогепта[1,2-b]пиридин-3-он
Получение способом, аналогичным Примеру 3 за исключением замены на 6-(трет-бутил-диметил-силанилокси)-8-(8-фтор-1-оксо-1Н-фталазин-2-ил)-4-метил-4,5,6,7-тетрагидро-бензо[3,4]циклогепта[1,2-b]пиридин-3-он 6-(трет-бутил-диметил-силанилокси)-8-(6-трет-бутил-8-фтор-1-оксо-1Н-фталазин-2-ил)-4-метил-4,5,6,7-тетрагидро-бензо[3,4]циклогепта[1,2-b]пиридин-3-она дало 87 мг соединения, указанного в заголовке, в виде светло-коричневого осадка. Наблюдаемый ЖХ/МС продукта [М+Н]+ 609.
Пример 7
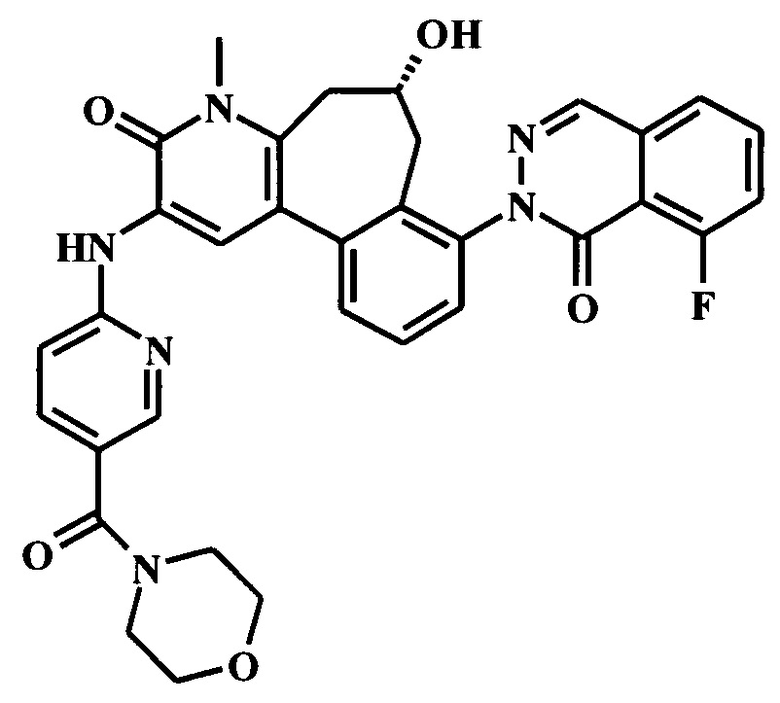
(R)-8-(8-фтор-1-оксо-1Н-фталазин-2-ил)-6-гидрокси-4-метил-2-[5-(морфолин-4-карбонил)-пиридин-2-иламино]-4,5,6,7-тетрагидро-бензо[3,4]циклогепта[1,2-b]пиридин-3-он
Получение способом, аналогичным Примеру 4 за исключением замены на (R)-6-(трет-бутил-диметил-силанилокси)-8-(8-фтор-1-оксо-1Н-фталазин-2-ил)-4-метил-4,5,6,7-тетрагидро-бензо[3,4]циклогепта[1,2-b]пиридин-3-он (R)-6-(трет-бутил-диметил-силанилокси)-8-(6-трет-бутил-8-фтор-1-оксо-1Н-фталазин-2-ил)-4-метил-4,5,6,7-тетрагидро-бензо[3,4]циклогепта[1,2-b]пиридин-3-она дало 11 мг соединения, указанного в заголовке, в виде светло-коричневого осадка. Наблюдаемый ЖХ/МС продукта [М+Н]+ 609.
Биологические примеры
Анализ ингибирования тирозинкиназы Брутона
Анализ представляет собой захват радиоактивного 33Р фосфорилированного продукта посредством фильтрации. Взаимодействие Btk, биотинилированного SH2 пептидного субстрата (Src гомология) и АТФ приводит к фосфорилированию пептидного субстрата. Биотинилированный продукт связан с шариками стрептавидинсефарозы. Все связи, меченные продукты обнаруживались сцинтилляционным счетчиком.
Планшеты для анализа представляли собой 96-луночные полипропиленовые (Greiner) и 96-луночные 1.2 мкм гидрофильные планшеты с PVDF фильтром (Millipore). Приведенные здесь концентрации являются конечными концентрациями при анализе: 10-100 мкмоль соединений в ДМСО (Burdick и Jackson), 5-10 нмоль фермент Btk (His-меченый, полноцепочечный), 30 мкмоль пептидынй субстрат (Biotin-Aca-AAAEEIYGEI-NH2), 100 мкмоль АТФ (Sigma), 8 ммоль имидазол (Sigma, рН 7.2), 8 ммоль глицерол-2-фосфат (Sigma), 200 мкмоль EGTA (Roche Diagnostics), 1 ммоль MnCl2 (Sigma), 20 ммоль MgCl2 (Sigma), 0.1 мг/мл БСА (Sigma), 2 ммоль DTT (Sigma), 1 μCi 33Р ATP (Amersham), 20% стрептавидинсефарозные шарики (Amersham), 50 ммоль EDTA (Gibco), 2 М NaCl (Gibco), 2 М NaCl мас./1% фосфорная кислота (Gibco), microscint-20 (Perkin Elmer).
Значения IC50 рассчитывались на основании 10 точек данных на одно соединение, используя данные полученные на стандартном шаблоне анализа 96-луночных планшет. Одно контрольное соединение и семь неизвестных ингибиторов тестировались на одном планшете в двух повторах. Обычно, соединения разводились полулогарифмично, начиная со 100 мкмоль и заканчивая 3 нмоль. Контрольным соединением выступал стауроспорин. Фон подсчитывали в отсутствии пептидного субстрата. Общую активность определяли в присутствии пептидного субстрата. Следующий протокол использовали для определения ингибирования Btk.
1) Подготовка проб: тест-соединения разводили с полулогарифмическим шагом в буфере для анализа (имидазол, глицерин-2-фосфат, EGTA, MnCl2, MgCl2, БСА).
2) Подготовка шариков
a.) шарики промывали при центрифугировании 500 g
b.) ресуспендировали шарики с PBS и EDTA с получением 20% суспензии шариков
3) Преинкубационная реакционная смесь без субстрата (буфер для анализа, DTT, АТФ, 33Р АТФ) и смесь с субстратом (буфер для анализа, DTT, АТФ, 33Р АТФ, пептидный субстрат) 30°С в течение 15 мин.
4) Для начала анализа, преинкубировали 10 мкл Btk в ферментном буфере (имидазол, глицерин-2-фосфат, БСА) и 10 мкл тестовых соединений в течение 10 мин при комнатной температуре.
5) Добавили 30 мкл реакционной смеси без или с субстратом к Btk и соединениям.
6) Инкубировали 50 мкл общей реакционной смеси в течение 30 мин при 30°С.
7) Перенесли 40 мкл анализируемой смеси к 150 мкл суспензии шариков в фильтр-планшет для остановки реакции.
8) Промыли фильтр-планшеты через 30 мин, со следующими стадиями
a. 3×250 мкл NaCl
b. 3×250 мкл NaCl, содержащий 1% фосфорной кислоты
c. 1×250 мкл H2O
9) Сушили планшеты в течение 1 ч при 65°С или в течение ночи при комнатной температуре
10) Добавили 50 мкл жидкости microscint-20 и посчитали 33Р ед/мин на сцинтиляционном счетчике.
Посчитали процент активности из полученных данных в ед/мин процент активности = (образец - фон) / (общая активность - фон) × 100
Посчитали IC50 из процентной активности, используя сигмоидальную модель односайтового дозового ответа
у=А+((В-А)/(1+((х/С)D))))
х = конц. соед., у = % активности, А = мин, В = макс, С = IC50, D = 1 (наклон кривой)
Анализ TR-FRET (FRET с разрешением по времени) ингибирования тирозинкиназы Брутона (BTK)
Этот анализ конкурирования BTK измеряет эффективность соединения (IC50) для инактивирования состояния тирозинкиназы Брутона с использованием FRET (резонансный перенос энергии флуоресценции) технологии. Комплекс BTK-Eu инкубировали на льду в течение одного часа перед использованием при исходной концентрации 50 нМ BTK-BioeaseTm: 10 нМ Eu-стрептавидин (Perkin-Elmer Catalog # AD0062). Буфер для анализа состоял из 20 мМ HEPES (рН 7.15), 0.1 мМ DTT, 10 мМ MgCl2, 0.5 мг/мл БСА с 3% стабилизатором киназ (Fremont Biosolutions, Catalog # STB-K02). Через 1 ч, вышеуказанную реакционную смесь разбавили в 10 раз буфером для анализа для получения 5 нМ BTK: 1 нМ Eu-стрептавидинового комплекса (донор флуорофора). 18 мкл смеси 0.11 нМ BTK-Eu и 0.11 нМ Kinase Tracer 178 (Invitrogen, Catalog # PV5593,) с одним BTK-Eu в качестве не отрицательного контроля, затем разлили в 384-луночные планшеты с плоским дном (Greiner, 784076). Соединения для тестирования в данном анализе получили в виде 10х концентраций и провели серийное полулогарифмическое разведение в ДМСО, таким образом, чтобы получить 10-точененые кривые. Для активации FRET реакции, соединения получили в виде 10х стоков в ДМСО, добавили в планшеты и планшеты инкубировали 18-24 ч при 14°С.
После инкубирования планшеты считывали на планшетном ридере BMG Pherastar Fluorescent (или эквивалентном) и использовали для измерения энергии эмиссии от европия донора флуорофора (эмиссия при 620 нм) и FRET (эмиссия при 665 нм). Значения лунок отрицательного контроля усреднялись для получения среднего минимума. Положительные значения контролей "без ингибитора" усреднялись для получения среднего максимума. Процент максимальных значений FRET рассчитывался с использованием следующего уравнения:
% макс FRET = 100 × [(FSRсоед - FSRсреди мин) / (FSRсреди макс - FSRсреди мин)]
где FSR = соотношение сигнала FRET. Кривые % макс FRET строились с помощью Activity Base (Excel) и определялись IC50 (%), наклон кривой, z' и %CV. Значения IC50 и стандартное отклонение получены из дублированных кривых (синглетные кривые ингибирования от двух независимых разведений) с использованием Microsoft Excel.
Ингибирование В-клеточной активации в цельной крови, измеренное по экспрессии CD69
Процедура проверки способности ингибиторов Btk подавлять В-клеточный рецептор-опосредованной активацию В-клеток в крови человека выглядит следующим образом:
Цельную кровь человека (HWB) получили от здоровых добровольцев, со следующими ограничениями: 24 ч без приема лекарств, некурящие.
Кровь брали из вены в вакутайнеры, обработанные антикоагулянтом - гепарином натрия. Тестируемые соединения разводили в десять раз от желаемой начальной концентрации препарата в PBS (20х), с последующими трехкратными серийными разведениями в 10% ДМСО в PBS, чтобы получить девятиточечную кривую доза-реакция. 5.5 мкл каждого разведения соединения добавляют в двух повторах в 2 мл 96-луночной планшеты с V-образным дном (Analytical Sales и Services, # 59623-23); 5.5 мкл 10% ДМСО в PBS добавили в контрольные и лунки без соединений. В каждую лунку добавили HWB (100 мкл), и после смешивания планшеты инкубировали при 37°С, 5% CO2, 100% влажности в течение 30 минут. F(ab')2 противо-человеческий козий IgM (Southern Biotech, # 2022-14) (10 мкл 500 мкг/мл раствора, 50 мкг/мл конечная концентрация) добавили в каждую лунку (кроме лунок без соединений) с перемешиванием и планшеты инкубировали дополнительно 20 часов.
В конце 20-ти часовой инкубации образцы инкубировали с мечеными флуоресцентной пробой антителами (15 мкл РЕ Mouse anti-Human CD20, BD Pharmingen, # 555623, и/или 20 мкл АРС Mouse anti-Human CD69, BD Pharmingen # 555533) в течение 30 минут, при 37С, 5% CO2, 100% влажности. Включая индуцированный контроль, неокрашенные и одиночные пятна для компенсации корректировки и начальной настройки напряжения. Затем образцы лизировали 1 мл 1Х лизирующего буфера Pharmingen (BD Pharmingen # 555899), и планшеты центрифугировали при 1800 об. в течение 5 минут. Супернатанты удалили отсасыванием и оставшиеся гранулы лизировали еще раз с другим 1 мл 1Х лизирующего буфера Pharmingen, и планшеты развернули вниз, как раньше. Супернатанты аспирировали и оставшиеся гранулы промыли FACs буфером (PBS + 1% FBS). После окончательного центрифугирования, супернатант удалили и гранулы суспендировали в 180 мкл буфера FACs. Образцы перенесли в 96-луночный планшет подходящий для работы на HTS 96 луночной системе на проточном цитофлюориметре BD LSR II.
Используя соответствующие длины волн возбуждения и излучения для использующихся флуорофоров, получили данные и процент положительных клеточных значений получили с использованием программного обеспечения Cell Quest. Результаты первоначально анализировали с помощью программного обеспечения для анализа FACS (Flow Jo). IC50 для тестируемых соединений определяли как концентрацию, которая уменьшала на 50% количество CD69-позитивных клеток, которые также являлись CD20-положительными после стимуляции анти-IgM (среднее по 8 лункам контроля, после вычитания среднего по 8-ми фоновым лункам без соединений). IC50 значения рассчитывались с использованием XLfit версии 3, уравнение 201.
Данные для соединения согласно данному анализу показаны ниже в Таблице II.
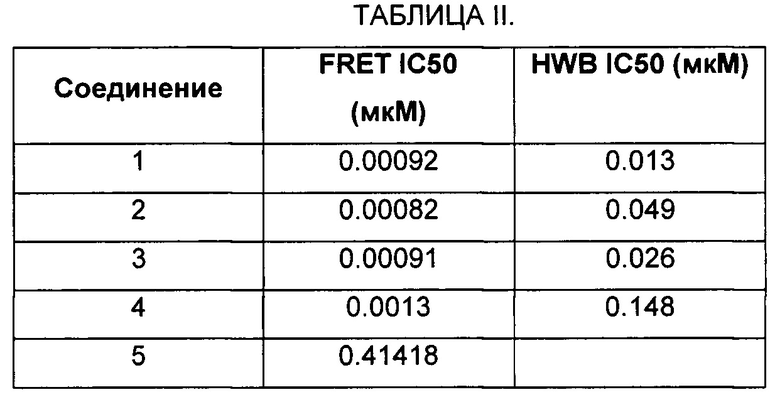

Ингибирование активации В-клеток - В клеточный FLIPR анализ на клетках Ramos
Ингибирование активации В-клеток соединениями настоящего изобретения продемонстрировали посредством определения эффекта тестовых соединений на стимулированные антителами против IgM ответы В-клеток.
В-клеточный FLIPR анализ это основанный на клетках функциональный метод определения эффекта потенциальных ингибиторов на повышение уровня внутриклеточного кальция в ответ на стимуляцию анти IgM антителами. Клетки Ramos (клеточная линия лимфомы Беркитта человека. ATCC-No. CRL-1596) культивировали в Питательной Среде (описана ниже). За один день до анализа клетки Ramos ресуспендировали в свежей питательной среде (такой же как выше) и поместили при концентрации 0.5×106/мл в колбы для культивирования тканей. В день анализа, клетки подсчитывали и устанавливали концентрацию 1×106/мл в питательной среде с добавлением 1 мкМ FLUO-3AM (TefLabs Кат номер. 0116, подготовленный в безводном ДМСО и 10% плюрониловой кислоте) колбе для культивирования тканей, и инкубировали при 37°С (4% CO2) в течение одного часа. Для удаления внеклеточного красителя, клетки собрали центрифугированием (5 мин, 1000 оборотов в минуту), ресуспендировали в FLIPR буфере (см. ниже) при концентрации 1×106 клеток/мл и затем распределили в 96-и луночные покрытые поли-D-лизином черные/прозрачные планшеты (BD-Cat No. 356692) при концентрации 1×105 клеток на лунку. Тестируемые соединения добавили в различных концентрациях в диапазоне от 100 мкМ до 0,03 мкМ (7 концентраций, подробности ниже) и инкубировали с клетками в течение 30 мин при комнатной температуре. Са2+ сигналинг в клетках Ramos был вызван добавлением 10 мг/мл анти-IgM (Southern Biotech, Кат номер. 2020-01) и измерялся на FLIPR (Molecular Devices, захватывает изображения из 96-луночных планшет с помощью ПЗС-камеры с аргоновым лазером при возбуждении 480 нм).
Среда/Буферы:
Питательная среда: RPMI 1640 с L-глутамином (Invitrogen, Кат номер 61870-010.), 10% эмбриональная телячья сыворотка (FBS, Summit Biotechnology Кат номер. FP-100-05), 1 мМ пируват натрия (Invitrogen кат. номер 11360-070).
FLIPR буфер: HBSS (Invitrogen, Кат номер 141175-079), 2 мМ CaCl2 (Sigma Кат номер С-4901), HEPES (Invitrogen, Кат номер 15630-080), 2,5 мм пробенецид (Sigma, Кат номер Р-8761), 0,1% БСА (Sigma, Кат номер А-7906), 11 мМ глюкоза (Sigma, Кат номер G-7528)
Подробности разведения соединений:
С целью получить самую высокую концентрацию для конечного анализа 100 мкМ, 24 мкл 10 мМ стокового раствора соединений (приготовленного в ДМСО) добавляли непосредственно в 576 мкл FLIPR буфера. Тестовые соединения разводили в FLIPR буфере (используя роботизированный пипеттор Biomek 2000) с получением следующей схемы разведения: носитель, 1.00×10-4 М, 1.00×10-5, 3.16×10-6, 1.00×10-6, 3.16×10-7, 1.00×10-7, 3.16×10-8.
Образцы и анализ:
Внутриклеточное увеличение кальция было зарегистрировано с использованием мин-макс статистики (за вычетом спокойной базовой линии из пика, вызванного добавлением стимулирующих антител с использованием программного обеспечения экспорта управления и статистики Molecular Devices FLIPR. IC50 определяли с помощью нелинейного подбора кривой (программное обеспечение GraphPad Prism).
Мышиный коллаген-индуцированный артрит (rnCIA)
В день 0 мышам вводили в основание хвоста или несколько точек на спине с эмульсию коллагена типа II (ID) в полном адъюванте Фрейнда (CFA). После коллагеновой иммунизации у животных будет развиваться артрит в пределах от 21 до 35 дней. Начало артрита является синхронизированным (вторичным) из-за системного введения коллагена в неполном адъюванте Фрейнда (IFA, ID) на 21 день. Животные осматривались каждый день после 20-го дня на предмет любого начала мягкого артрита (оценка 1 или 2, см. описание оценок ниже), который является сигналом для увеличения. После повышения, мышей считали и вводили им терапевтические агенты-кандидаты на указанный период времени (обычно 2-3 недели) и дозировочной частотой, ежедневно (QD) или дважды в день (BID).
Крысиный коллаген-индуцированный артрит (rCIA)
В день 0 крысам вводили эмульсию бычьего коллагена типа II в неполном адъюванте Фрейнда (IFA) внутрикожно (ID) в нескольких местах на спине. Бустер-инъекции коллагеновой эмульсии дается примерно на 7 день (ID) в основание хвоста или альтернативные места на спине. Артрит, как правило, наблюдается на 12-14 дней после первой инъекции коллагена. Животные могут быть оценены на развития артрита, как описано ниже (оценка артрита) с 14-го дня. Животным вводили терапевтические агенты-кандидаты в профилактической манере, начиная с момента вторичного изменения и в течение предписанного времени (обычно 2-3 недели) и частоты дозирования, ежедневно (QT) или дважды в день (BID).
Оценка артрита:
В обеих моделях развивающееся воспаление лап суставов и конечностей оценивали количественно с помощью балльной системы, что предполагает оценку 4 лап в соответствии с критериями, описанными ниже:
Оценка: 1 = отек и/или покраснение лапы или одного места.
2 = отек двух или нескольких суставов.
3 = общий отек лапы с более чем двумя вовлеченными суставами.
4 = тяжелый артрит всей лапы и мест.
Оценка производится в день 0 для базовых измерений и снова начинается при первых признаках или отечности до трех раз в неделю до конца эксперимента. Артритный индекс для каждой мыши получают путем добавления четырех оценок отдельных лап, давая максимальный балл 16 за животное.
Модель in vivo астмы у крыс
Самцов серой крысы сенсибилизировали интраперитонеально 100 мкг О.А. (овальбумин) в 0,2 мл квасцов один раз в неделю в течение трех недель (дни 0, 7 и 14). В день 21 (через неделю после последней сенсибилизации), крысы получали один раз в день либо носитель либо композицию соединения подкожно за 0,5 часа до заражения О.А. аэрозолем (1% О.А. в течение 45 минут) и прекращали через 4 или 24 часа после заражения. Во время умерщвления, сыворотку и плазму собрали из всех животных для серологии и PK, соответственно. Трахейные канюли вставляли и легкие промывали 3 раза PBS. Жидкость бронхоальвеолярного лаважа (БАЛ) анализировалась на общее число лейкоцитов и дифференциальные лейкоциты подсчитывались. Общее число лейкоцитов в аликвоте клеток (20-100 мкл) определяли с помощью Coulter Counter. Для дифференциальной лейкоцитарной формулы, 50-200 мкл пробы центрифугировали в Cytospin и слайд окрашивали Diff-Quik. Пропорции моноцитов, эозинофилов, нейтрофилов и лимфоцитов подсчитывали под световым микроскопом с использованием стандартных морфологических критериев и выразили в процентном виде. Репрезентативные ингибиторы Btk показали снижение общего количества лейкоцитов в БАЛ сенсибилизированных и нагруженных О.А. крыс по сравнению с контрольным уровнем.
Приведенное выше изобретение было описано в некоторых деталях в качестве иллюстрации и, например, с целью понимания и ясности. Для специалиста в данной области очевидно, что изменения и модификации могут быть осуществлены в рамках формулы изобретения. Поэтому следует понимать, что приведенное выше описание предназначено для иллюстрации и не является ограничивающим. Объем изобретения должен быть, следовательно, определен не по отношению к приведенному выше описанию, а с учетом следующей прилагаемой формулы изобретения, наряду с полным объемом эквивалентов, к которым такие требования применимы.
Все патенты, патентные заявки и публикации, цитируемые в данной заявке, включены в качестве ссылки в полном объеме для всех целей в той же степени, как если бы каждый отдельный патент, заявка на патент или публикации были отдельно индивидуально указаны.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЯ 8-ФТОРФТАЛАЗИН-1(2Н)-ОНА В КАЧЕСТВЕ ИНГИБИТОРОВ ТИРОЗИНКИНАЗЫ БРУТОНА | 2012 |
|
RU2622391C2 |
| 3-ОКСО-3,9-ДИГИДРО-1Н-ХРОМЕНО[2,3-c]ПИРРОЛЫ В КАЧЕСТВЕ АКТИВАТОРОВ ГЛЮКОКИНАЗЫ | 2011 |
|
RU2603191C2 |
| ГЕТЕРОАРИЛ ПИРИДОНЫ И АЗАПИРИДОНЫ С ЭЛЕКТРОФИЛЬНОЙ ФУНКЦИОНАЛЬНОСТЬЮ | 2014 |
|
RU2646758C2 |
| ИНГИБИТОРЫ ТИРОЗИНКИНАЗЫ БРУТОНА | 2010 |
|
RU2542585C2 |
| ЗАМЕЩЕННЫЕ АМИДЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ, СПОСОБ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЯ, ЧУВСТВИТЕЛЬНОГО К Btk, СПОСОБ ПОВЫШЕНИЯ ЧУВСТВИТЕЛЬНОСТИ РАКОВЫХ КЛЕТОК К ХИМИОТЕРАПИИ, СПОСОБ УМЕНЬШЕНИЯ ОШИБОК ПРИ ПРИЕМЕ ЛЕКАРСТВА И УЛУЧШЕНИЯ СОБЛЮДЕНИЯ СХЕМЫ ЛЕЧЕНИЯ, СПОСОБ ИНГИБИРОВАНИЯ ГИДРОЛИЗА АТФ, СПОСОБ ОПРЕДЕЛЕНИЯ ПРИСУТСТВИЯ Btk В ОБРАЗЦЕ И СПОСОБ ИНГИБИРОВАНИЯ АКТИВНОСТИ В-КЛЕТОК | 2008 |
|
RU2470923C2 |
| НОВЫЕ ФТАЛАЗИНОВЫЕ ПРОИЗВОДНЫЕ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2014 |
|
RU2636585C2 |
| ПИРИДОНОВЫЕ И АЗАПИРИДОНОВЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ПРИМЕНЕНИЯ | 2011 |
|
RU2617405C2 |
| ПИРАЗОЛ-4-ИЛ-ГЕТЕРОЦИКЛИЛ-КАРБОКСАМИДНЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ПРИМЕНЕНИЯ | 2012 |
|
RU2638552C2 |
| ИНГИБИТОРЫ ТИРОЗИНКИНАЗЫ БРУТОНА | 2013 |
|
RU2619465C2 |
| ПРОЛЕКАРСТВА СОПРЯЖЕННО-БИЦИКЛИЧЕСКИХ АНТАГОНИСТОВ C5aR | 2019 |
|
RU2794327C2 |
Настоящее изобретение относится к соединению общей формулы I, приведенной ниже, или к его фармацевтически приемлемой соли. В соединении формулы I X представляет собой галоген; Y представляет собой Н или низший алкил; R представляет собой -R1-R2-R3; R1 представляет собой пиридил; R2 представляет собой -С(=O) или отсутствует; R3 представляет собой морфолинил или пирролидинил, возможно замещенный низшим алкилом. Описанные выше соединения применяются для модулирования активности ВТК и лечения заболеваний, ассоциированных с избыточной активностью ВТК. Кроме того, настоящие соединения применяются для лечения воспалительных и аутоиммунных заболеваний, ассоциированных с аберрантной пролиферацией B-клеток, таких как ревматоидный артрит. Изобретение также относится к фармацевтической композиции, содержащей соединения формулы I и, по меньшей мере,один носитель, разбавитель или эксципиент. 8 н. и 12 з.п. ф-лы, 2 табл., 7 пр.
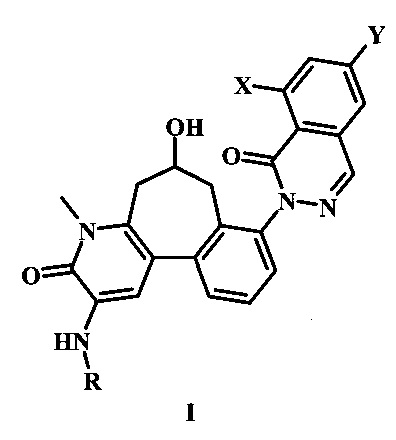
1. Соединение Формулы I,
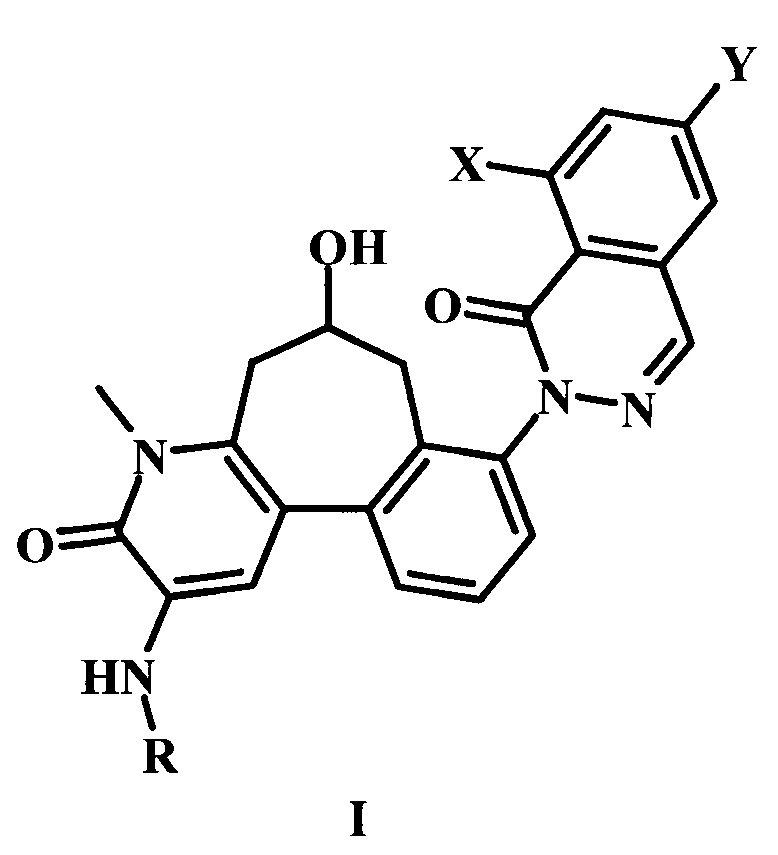
где:
X представляет собой галоген;
Y представляет собой Н или низший алкил;
R представляет собой -R1-R2-R3;
R1 представляет собой пиридил;
R2 представляет собой -С(=O) или отсутствует;
R3 представляет собой морфолинил или пирролидинил, возможно замещенный низшим алкилом,
или его фармацевтически приемлемая соль.
2. Соединение по п. 1, где X представляет собой F.
3. Соединение по п. 1, где R2 представляет собой -С(=O).
4. Соединение по п. 1, где R3 морфолинил.
5. Соединение по п. 1, где Y представляет собой Н.
6. Соединение по п. 1, где Y представляет собой трет-бутил.
7. Соединение по п. 1, где R2 отсутствует.
8. Соединение по п. 7, где R3 представляет собой пирролидинил, возможно замещенный низшим алкилом.
9. Соединение по п. 8, где низший алкил представляет собой метил.
10. Соединение по п. 7, где Y представляет собой Н.
11. Соединение по п. 7, где Y представляет собой трет-бутил.
12. Соединение по п. 1, выбранное из группы, состоящей из:
8-(6-трет-бутил-8-фтор-1-оксофталазин-2-ил)-6-гидрокси-4-метил-2-[[5-(морфолин-4-карбонил)пиридин-2-ил]амино]-6,7-дигидро-5Н-бензо[1,2]циклогепта[6,7-d]пиридин-3-он;
8-(6-трет-бутил-8-фтор-1-оксофталазин-2-ил)-6-гидрокси-4-метил-2-[[5-[(2S)-1-метилпирролидин-2-ил]пиридин-2-ил]амино]-6,7-дигидро-5H-бензо[1,2]циклогепта[6,7-d]пиридин-3-он;
(6S)-8-(6-трет-бутил-8-фтор-1-оксофталазин-2-ил)-6-гидрокси-4-метил-2-[[5-(морфолин-4-карбонил)пиридин-2-ил]амино]-6,7-дигидро-5H-бензо[1,2]циклогепта[6,7-d]пиридин-3-он;
(6R)-8-(6-трет-бутил-8-фтор-1-оксофталазин-2-ил)-6-гидрокси-4-метил-2-[[5-(морфолин-4-карбонил)пиридин-2-ил]амино]-6,7-дигидро-5Н-бензо[1,2]циклогепта[6,7-d]пиридин-3-он;
8-(8-фтор-1-оксофталазин-2-ил)-6-гидрокси-4-метил-2-[[5-(морфолин-4-карбонил)пиридин-2-ил]амино]-6,7-дигидро-5H-бензо[1,2]циклогепта[6,7-d]пиридин-3-он;
(6S)-8-(8-фтор-1-оксофталазин-2-ил)-6-гидрокси-4-метил-2-[[5-(морфолин-4-карбонил)пиридин-2-ил]амино]-6,7-дигидро-5Н-бензо[1,2]циклогепта[6,7-d]пиридин-3-он; и
(6R)-8-(8-фтор-1-оксофталазин-2-ил)-6-гидрокси-4-метил-2-[[5-(морфолин-4-карбонил)пиридин-2-ил]амино]-6,7-дигидро-5H-бензо[1,2]циклогепта[6,7-d]пиридин-3-он.
13. Способ лечения воспалительного или аутоиммунного состояния, содержащий введение нуждающемуся в этом пациенту терапевтически эффективного количества соединения по любому из пп. 1-12.
14. Способ лечения воспалительного состояния, содержащий введение нуждающемуся в этом пациенту терапевтически эффективного количества соединения по любому из пп. 1-12.
15. Способ лечения ревматоидного артрита, содержащий введение нуждающемуся в этом пациенту терапевтически эффективного количества соединения по любому из пп. 1-12.
16. Способ лечения астмы, содержащий введение нуждающемуся в этом пациенту терапевтически эффективного количества соединения по любому из пп. 1-12.
17. Фармацевтическая композиция, ингибирующая киназу Брутона (Btk), содержащая соединение по любому из пп. 1-12, смешанное с по меньшей мере одним фармацевтически приемлемым носителем, эксципиентом или разбавителем.
18. Применение соединения по любому из пп. 1-12 для лечения воспалительного и/или аутоиммунного состояния.
19. Применение соединения по любому из пп. 1-12 для получения лекарственного средства для лечения воспалительного и/или аутоиммунного состояния.
20. Соединение по любому из пп. 1-12 для применения при лечении воспалительного и/или аутоиммунного состояния.
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| Многоступенчатая активно-реактивная турбина | 1924 |
|
SU2013A1 |
| RU 2010136665 A, 20.03.2012. | |||

