Перекрестные ссылки на родственные заявки
Для настоящего изобретения испрашивается приоритет согласно 35 U.S.C. § 119(e) по предварительной заявке США №62/651,512, поданной 2 апреля 2018 года, содержание которой включено в настоящий текст посредством ссылки во всей своей полноте.
Положение о правах на изобретения, сделанные в ходе спонсируемого правительством исследования и разработки
Неприменимо
Ссылка на “список последовательностей”, таблицу или компьютерную программу с перечислением приложений, поданных на компакт-диске
Неприменимо
Предпосылки создания изобретения
Система комплемента играет центральную роль в клиренсе иммунных комплексов и иммунных ответах на возбудители инфекций, чужеродные антигены, зараженные вирусом клетки и опухолевые клетки. Ненадлежащая или избыточная активация системы комплемента может привести к вредоносным и даже потенциально угрожающим жизни последствиям из-за сильного воспаления и вызываемого им разрушения ткани. Эти последствия клинически проявляются в форме различных нарушений, включая септический шок; ишемию миокарда, а также кишечника/реперфузионное повреждение; отторжение трансплантата; отказ органов; нефрит; патологическое воспаление и аутоиммунные заболевания.
Система комплемента состоит из группы белков, которые в норме присутствуют в плазме крови в неактивном состоянии. Активация системы комплемента охватывает главным образом три разных пути, а именно классический, альтернативный и лектиновый пути (V. M. Holers, In Clinical Immunology: Principles и Practice, ed. R. R. Rich, Mosby Press; 1996, 363-391): 1) Классический путь представляет собой кальций/магний-зависимый каскад, который обычно активируется образованием комплексов антиген-антитело. Он также может активироваться независимым от антител образом путем связывания C-реактивного белка, сформировавшего комплекс с лигандом, а также многими патогенами, включая грамм-отрицательные бактерии. 2) Альтернативный путь представляет собой магний-зависимый каскад, который активируется при отложении и активации C3 на определенных восприимчивых поверхностях (например, полисахариды клеточных стенок дрожжей и бактерий, и некоторые биополимерные материалы). 3) Лектиновый путь включает стартовое связывание манноза-зависимого лектина и последующую активацию C2 и C4, которые являются общими с классическим путем (Matsushita, M. et al., J. Exp. Med. 176: 1497-1502 (1992); Suankratay, C. et al., J. Immunol. 160: 3006-3013 (1998)).
Активация системы комплемента генерирует биологически активные фрагменты белков комплемента, например С3а, С4а и С5а анафилотоксинов и C5b-9 мембраноатакующих комплексов (МАК), все из которых вызывают воспалительный ответ путем воздействия на хемотаксис лейкоцитов; активации макрофагов, нейтрофилов, тромбоцитов, тучных клеток и клеток эндотелия; и усиления сосудистой проницаемости, цитолиза и поражения ткани.
Комплемент C5a представляет собой один из наиболее мощных провоспалительных медиаторов в системе комплемента. (Анафилактический C5a пептид в 100 раз активнее, в расчете на мольные количества, в создании воспалительного ответа, чем C3a.) C5a представляет собой активированную форму C5 (молекулярный вес 190 кДа). C5a присутствует в плазме крови человека в количестве примерно 80 мкг/мл (Kohler, P. F. et al., J. Immunol. 99: 1211-1216 (1967)). Он состоит из двух полипептидных цепочек, α и β, с приблизительными молекулярными весами 115 кДа и 75 кДа, соответственно (Tack, B. F. et al., Biochemistry 18: 1490-1497 (1979)). Биосинтезируясь в виде одноцепочечной промолекулы, C5 ферментативно расщепляется на двухцепочечную структуру во время процессинга и секреции. После расщепления две полученные цепочки удерживаются вместе за счет по меньшей мере одной дисульфидной связи, а также нековалентных взаимодействий (Ooi, Y. M. et al., J. Immunol. 124: 2494-2498(1980)).
C5 расщепляется на фрагменты C5a и C5b во время активации комплементного пути. Ферменты конвертазы, отвечающие за активацию C5, представляют собой много-субъединичные комплексы из C4b, C2a и C3b для классического пути, и из (C3b)2, Bb и P для альтернативного пути (Goldlust, M. B. et al., J. Immunol. 113: 998-1007 (1974); Schreiber, R. D. et al, Proc. Natl. Acad. Sci. 75: 3948-3952 (1978)). C5 активируется расщеплением по положению 74-75 (Arg-Leu) в α-цепи. После активации высвобождается 74-аминокислотный пептид C5a весом 11,2 кДа из амино-терминального участка α-цепи. C5a и C3a оба являются сильными стимуляторами нейтрофилов и моноцитов (Schindler, R. et al., Blood 76: 1631-1638 (1990); Haeffner-Cavaillon, N. et al., J. Immunol. 138: 794-700 (1987); Cavaillon, J. M. et al., Eur. J. Immunol. 20: 253-257 (1990)).
В дополнение к своим анафилотоксическим свойствам, C5a вызывает хемотаксичную миграцию нейтрофилов (Ward, P. A. et al., J. Immunol. 102: 93-99 (1969)), эозинофилов (Kay, A. B. et al., Immunol. 24: 969-976 (1973)), базофилов (Lett-Brown, M. A. et al., J. Immunol. 117: 246-252 1976)) и моноцитов (Snyderman, R. et al., Proc. Soc. Exp. Biol. Med. 138: 387-390 1971)). C5a и C5b-9 оба активируют клетки эндотелия к выработке адгезивных молекул, необходимых для секвестрации активированных лейкоцитов, которые участвуют в механизмах воспаления и повреждения тканей (Foreman, K. E. et al., J. Clin. Invest. 94: 1147-1155 (1994); Foreman, K. E. et al., Inflammation 20: 1-9 (1996); Rollins, S. A. et al., Transplantation 69: 1959-1967 (2000)). C5a также опосредует воспалительные реакции, вызывая сокращение гладкой мускулатуры, повышая проницаемость сосудов, инициируя дегрануляцию базофилов и тучных клеток, и индуцируя высвобождение лизосомальных протеаз и окислительных свободных радикалов (Gerard, C. et al., Ann. Rev. Immunol. 12: 775-808 (1994)). Кроме того, C5a модулирует экспрессию генов в печени в острой фазе и усиливает иммунный ответ в целом, повышая выработку ФНО-α, IL-1-β, IL-6, IL-8, простагландинов и лейкотриенов (Lambris, J. D. et al., In: The Human Complement System in Health и Disease, Volanakis, J. E. ed., Marcel Dekker, New York, pp. 83-118).
Считается, что анафилактические и хемотаксические эффекты C5a работают через его взаимодействие с C5a рецептором. Человеческий C5a рецептор (C5aR) представляет собой 52 кДа мембраносвязанный рецептор, связанный с G-белком, который экспрессируется на нейтрофилах, моноцитах, базофилах, эозинофилах, гепатоцитах, гладких мышцах легких и эндотелиальных клетках, а также в тканях почечных клубочков (Van-Epps, D. E. et al., J. Immunol. 132: 2862-2867 (1984); Haviland, D. L. et al., J. Immunol. 154:1861-1869 (1995); Wetsel, R. A., Immunol. Leff. 44: 183-187 (1995); Buchner, R. R. et al., J. Immunol. 155: 308-315 (1995); Chenoweth, D. E. et al., Proc. Natl. Acad. Sci. 75: 3943-3947 (1978); Zwirner, J. et al., Мол. Immunol. 36:877-884 (1999)). Связывающийся с лигандом сайт C5aR является сложным и состоит из по меньшей мере двух физически разделимых связывающихся доменов. Один связывается с амино-концом C5a (аминокислоты 1-20) и дисульфидно-связанным ядром (аминокислоты 21-61), а второй связывается с карбоксильным концом C5a (аминокислоты 62-74) (Wetsel, R. A., Curr. Opin. Immunol. 7: 48-53 (1995)).
C5a играет важную роль в воспалении и повреждении тканей. При искусственном кровообращении и гемодиализе C5a образуется как результат активации альтернативного пути системы комплемента, когда человеческая кровь входит в контакт с искусственной поверхностью аппарата искусственного кровообращения или аппарата для диализа почек (Howard, R. J. et al., Arch. Surg. 123: 1496-1501 (1988); Kirklin, J. K. et al., J. Cardiovasc. Surg. 86: 845-857 (1983); Craddock, P. R. et al., N. Engl. J. Med. 296: 769-774 (1977)). C5a повышает проницаемость капилляров и вызывает эдему, сужение бронхов, легочную вазоконстрикцию, активацию лейкоцитов и тромбоцитов и их инфильтрацию в ткани, в частности в легких (Czermak, B. J. et al., J. Leukoc. Biol. 64: 40-48 (1998)). Было показано, что введение моноклональных C5a-антител снижает дисфункцию эндотелия коронарных сосудов, вызванную экстракорпоральным кровообращением и остановкой сердца (Tofukuji, M. et al., J. Thorac. Cardiovasc. Surg. 116: 1060-1068 (1998)).
C5a также задействован в синдроме острой дыхательной недостаточности (ARDS), хронической обструктивной болезни легких (ХОБЛ) и полиорганной недостаточности (MOF) (Hack, C. E. et al., Am. J. Med. 1989: 86: 20-26; Hammerschmidt DE et al. Lancet 1980; 1: 947-949; Heideman M. et al. J. Trauma 1984; 4: 1038-1043; Marc, MM, et al., Am. J. Respir. Cell и Mol. Biol., 2004: 31: 216-219). C5a усиливает выработку в моноцитах двух важных провоспалительных цитокинов, ФНО-α и IL-1. Было также показано, что C5a играет важную роль в развитии повреждения тканей, и в особенности легочной ткани, в животных моделях септического шока (Smedegard G et al. Am. J. Pathol. 1989; 135: 489-497; Markus, S., et al., FASEB Journal (2001), 15: 568-570). В моделях сепсиса с использованием крыс, свиней и нечеловекоподобных приматов, C5a-антитела, введенные животным перед лечением эндотоксином или E. coli, приводили к уменьшению повреждений ткани, а также уменьшению выработки IL-6 (Smedegard, G. et al., Am. J. Pathol. 135: 489-497 (1989); Hopken, U. et al., Eur. J. Immunol. 26: 1103-1109 (1996); Stevens, J. H. et al., J. Clin. Invest. 77: 1812-1816 (1986)). Более важно, что блокада C5a посредством C5a-поликлональых антител существенно повышает уровень выживаемости в сепсис-моделях лигирования слепой кишки/прокола у крыс (Czermak, B.J. et al., Nat. Med. 5: 788-792 (1999)). Эта модель имеет много общих аспектов с клиническими проявлениями сепсиса у человека. (Parker, S.J. et al., Br. J. Surg. 88: 22-30 (2001)). В той же модели сепсиса было показано, что C5a-антитела подавляют апоптоз тимоцитов (Guo, R.F. et al., J. Clin. Invest. 106: 1271-1280 (2000)) и предотвращают MOF (Huber-Lang, M. et al., J. Immunol. 166: 1193-1199 (2001)). C5a-антитела также выполняли защитную функцию в модели повреждения легких у крыс с фактором из яда кобры, и в повреждении легких, индуцируемом иммунным комплексом (Mulligan, M. S. et al. J. Clin. Invest. 98: 503-512 (1996)). Важность C5a при повреждении легких, индуцируемом иммунным комплексом, была позже подтверждена у мышей (Bozic, C. R. et al., Science 26: 1103-1109 (1996)).
C5a является активным медиатором при ишемии и реперфузии миокарда. Инактивация компонентов комплемента снижала масштаб инфаркта миокарда у мышей (Weisman, H. F. et al., Science 249: 146-151 (1990)), а введение C5a-антител уменьшало повреждения в крысиной модели ишемии-реперфузии на задних лапах (Bless, N. M. et al., Am. J. Physiol. 276: L57-L63 (1999)). Реперфузионные повреждения при инфаркте миокарда также значительно уменьшались у свиней, которым вводили моноклональный анти-C5a иммуноглобулин G (Amsterdam, E. A. et al., Am. J. Physiol. 268:H448-H457 (1995)). Антагонист рекомбинантного человеческого C5aR уменьшал масштаб инфаркта в свиной модели операционной реваскуляризации (Riley, R. D. et al., J. Thorac. Cardiovasc. Surg. 120: 350-358 (2000)).
C5a-активируемые нейтрофилы участвуют также во многих буллезных заболеваниях (например, буллезный пемфигоид, обыкновенная пузырчатка и эксфолиативная пузырчатка). Это хронические и рецидивирующие воспалительные нарушения, клинически характеризуемые стерильными пузырьками, которые возникают в субэпидермальном пространстве кожи и слизистой. Считается, что антитела к кератиноцитам, локализованным в кожных базальных мембранах обеспечивают отсоединение базальных кератиноцитов эпидермиса от подлежащей базальной мембраны; при этом пузырьки характеризуются также накоплением нейтрофилов как в более высоких слоях кожи, так и внутри полости пузырьков. В экспериментальных моделях снижение числа нейтрофилов или отсутствие компонентов комплемента (полное или С5-селективное) может подавлять образование субэпидермальных пузырьков, даже в присутствии высоких титров антител.
Уровень комплемента повышен у пациентов с ревматоидным артритом (Jose, P. J. et al., Ann. Rheum. Dis. 49: 747-752 (1990); Grant, E.P., et al., J. of Exp. Med., 196(11): 1461-1471, (2002)), волчаночным нефритом (Bao, L., et al., Eur. J. of Immunol., 35(8), 2496-2506, (2005)) и системной красной волчанкой (SLE) (Porcel, J. M. et al., Clin. Immunol. Immunopathol. 74: 283-288 (1995)). Концентрация C5a коррелирует со степенью тяжести болезненного состояния. Вызванный коллагеном артрит у мышей и крыс имеет сходство с ревматоидным артритом у людей. Мыши с дефицитом C5a рецепторов демонстрируют полную защиту от артрита, вызванного инъекцией моноклональных антител к коллагену (Banda, N.K., et al., J. of Immunol., 2003, 171: 2109-2115). Поэтому подавление C5a и/или C5a рецептора (C5aR) можно использовать для лечения этих хронических заболеваний.
Считается, что система комплемента активирована у пациентов с воспалительным заболеванием кишечника (ВЗК) и участвует в патогенезе этого заболевания. Компоненты активированного комплемента были обнаружены на поверхности эпителиальных клеток, а также в мышечном слое слизистой оболочки и подслизистых кровеносных сосудов у пациентов, страдающих воспалительным заболеванием кишечника (ВЗК) (Woodruff, T.M., et al., J of Immunol., 2003, 171: 5514-5520).
Экспрессия C5aR повышена в реагирующих астроцитах, микроглии и клетках эндотелия в воспаленной центральной нервной системе человека (Gasque, P. et al., Am. J. Pathol. 150: 31-41 (1997)). Возможно, C5a задействован в развитии нейродегенеративных заболеваний, таких как болезнь Альцгеймера (Mukherjee, P. et al., J. Neuroimmunol. 105: 124-130 (2000); O'Barr, S. et al., J. Neuroimmunol. (2000) 105: 87-94; Farkas, I., et al. J. Immunol. (2003) 170:5764-5771), болезнь Паркинсона, болезнь Пика и трансмиссивная губкообразная энцефалопатия. Активация нейронных C5aR может индуцировать апоптоз (Farkas I et al. J. Physiol. 1998; 507: 679-687). Поэтому подавление C5a и/или C5a рецептора (C5aR) можно использовать для лечения нейродегенеративных заболеваний.
Есть некоторые доказательства того, что выработка C5a ухудшает воспаление, связанное с атопическим дерматитом (Neuber, K., et al., Immunology 73:83-87, (1991)) и хронической аллергической сыпью (Kaplan, A.P., J. Allergy Clin. Immunol. 114; 465-474, (2004).
Теперь известно, что псориаз является болезнью, в которой задействованы Т-клетки (Gottlieb, E. L. et al., Nat. Med. 1: 442-447 (1995)). Однако, нейтрофилы и тучные клетки также могут участвовать в патогенезе этого заболевания (Terui, T. et al., Exp. Dermatol. 9: 1-10; 2000); Werfel, T. et al., Arch. Dermatol. Res. 289: 83-86 (1997)). Накопление нейтрофилов под роговым слоем эпидермиса наблюдается в сильно воспаленных областях псориатических бляшек, и вытяжки из псориазных болячек содержат очень высокие концентрации C5a и демонстрируют потенциальную хемотаксическую активность в отношении нейтрофилов, и этот эффект можно подавить добавлением антител к C5a. C5a являются хемоаттрактантами для T-клеток и нейтрофилов (Nataf, S. et al., J. Immunol. 162: 4018-4023 (1999); Tsuji, R. F. et al., J. Immunol. 165: 1588-1598 (2000); Cavaillon, J. M. et al., Eur. J. Immunol. 20: 253-257 (1990)). Кроме того, было показано экспрессирование C5aR в плазмацитоидных дендритных клетках (pDC), выделенных из болячек кожной красной волчанки, и эти клетки показали хемотаксическое поведение в отношении C5a, что говорит о том, что блокада C5aR на pDC может быть эффективным средством для уменьшения инфильтрации pDC в воспаленную кожу при красной волчанке и псориазе. Таким образом, C5a может служить важной терапевтической мишенью при лечении псориаза.
Иммуноглобулин G-содержащие иммунные комплексы участвуют в патофизиологии при ряде аутоиммунных заболеваний, таких как системная красная волчанка, ревматоидный артрит, синдром Шегрена, синдром Гудпасчера и пневмонит гиперчувствительности (Madaio, M. P., Semin. Nephrol. 19: 48-56 (1999); Korganow, A. S. et al., Immunity 10: 451-459 (1999); Bolten, W. K., Kidney Int. 50: 1754-1760 (1996); Ando, M. et al., Curr. Opin. Pulm. Med. 3: 391-399 (1997)). Эти заболевания очень гетерогенны и обычно поражают один или больше из следующих органов: кожа, кровеносные сосуды, суставы, почки, сердце, легкие, нервная система и печень (включая цирроз и фиброз печени). Классической животной моделью воспалительного ответа при этих заболеваниях иммунного комплекса является феномен Артюса, с инфильтрацией полиморфноядерных клеток, кровоизлиянием и экссудацией плазмы (Arthus, M., C.R. Soc. Biol. 55: 817-824 (1903)). Недавние исследования показали, что C5aR-дефицитные мыши защищены от повреждения тканей, вызванного иммунным комплексом (Kohl, J. et al., Mol. Immunol. 36: 893-903 (1999); Baumann, U. et al., J. Immunol. 164: 1065-1070 (2000)). Эти результаты находятся в согласии с наблюдением, что малый пептидный анти-C5aR антагонист подавляет воспалительный ответ, вызванный отложением иммунного комплекса (Strachan, A. J. et al., J. Immunol. 164: 6560-6565 (2000)). Вместе со своим рецептором, C5a играет важную роль в патогенезе заболеваний, в которых задействован иммунный комплекс. Ингибиторы C5a и C5aR могут быть полезны в лечении этих заболеваний.
Описание уровня техники
Непептидные антагонисты C5a рецептора были описаны в литературе как эффективные для лечения эндотоксического шока у крыс (Stracham, A.J., et al., J. of Immunol. (2000), 164(12): 6560-6565); и для лечения ВЗК (воспалительного заболевания кишечника) в крысиной модели (Woodruff, T.M., et al., J of Immunol., 2003, 171: 5514-5520). Непептидные модуляторы C5a рецептора были также описаны в патентной литературе компаниями Neurogen Corporation, (например, WO 2004/043925, WO 2004/018460, WO 2005/007087, WO 03/082826, WO 03/08828, WO 02/49993, WO 03/084524); Dompe S.P.A. (WO 02/029187); The University of Queenland (WO 2004/100975); и ChemoCentryx (WO 2010/075257).
В литературе есть серьезные экспериментальные доказательства связи повышенного уровня C5a и ряда заболеваний и нарушений, в частности аутоиммунных и воспалительных заболеваний и нарушений. Таким образом, в данной области есть потребность в новых низкомолекулярных органических модуляторах, например агонистах, частичных агонистах и, предпочтительно, антагонистах C5a рецептора (C5aR), которые полезны для подавления патогенных событий, например хемотаксиса, связанного с повышенным уровнем активности анафилатоксина. Настоящее изобретение удовлетворяет эту и другие потребности.
Краткое описание изобретения
В одном аспекте в настоящем изобретении описаны соединения, имеющие формулы (IA), (IB), (IC), (IIA), (IIB) и (IIC):
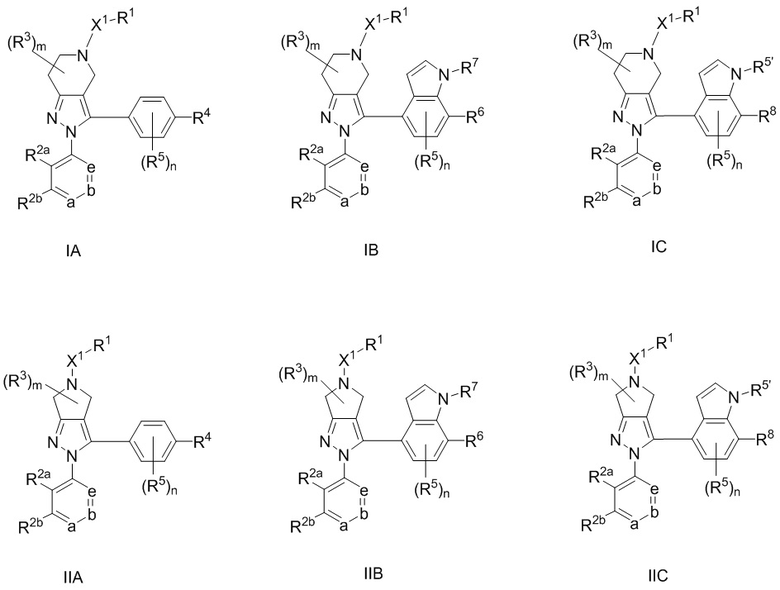
или их фармацевтически приемлемая соль, где символы, буквы и подстрочные индексы n, m, a, b, e, X1, R1, R2a, R2b, R3, R4, R5, R5', R6, R7 и R8 имеют значения, указанные ниже в описании.
Помимо описанных в настоящем тексте соединений, в настоящем изобретении описаны также фармацевтические композиции, содержащие одно или больше из указанных соединений, а также способы применения этих соединений в терапевтических методах, в первую очередь для лечения заболеваний, связанных с C5a сигнальной активностью.
В другом аспекте в настоящем изобретении описаны способы диагностики заболевания у пациента. В этих способах, описанные в настоящем тексте соединения вводят субъекту в меченой форме, затем проводят диагностическую визуализацию для определения присутствия или отсутствия C5aR и/или локализации клеток, экспрессирующих C5aR рецептор. В соответствующем аспекте способ диагностики заболевания осуществляют путем контакта образца ткани или крови с меченым соединением, описанным в настоящем тексте, и определяют присутствие, отсутствие, количество или локализацию C5aR в образце.
Краткое описание чертежей
На фиг. 1 показано высвобождение активного вещества интермедиата 4 из соединения по Примеру 1 (0,5 мг/кг моль-экв., AUC = 4,160 нг⋅час/мл).
На фиг. 2 показано высвобождение активного вещества интермедиата 1 из соединения по Примеру 4 (0,5 мг/кг моль-экв., AUC=753 нг⋅час/мл).
Подробное описание изобретения
Сокращения и определения
Термин "алкил", сам по себе и как часть другого заместителя, означает, если не указано иное, линейный или разветвленный углеводородный радикал, имеющий обозначенное число атомов углерода (например, C1-8 означает 1-8 атомов углерода). Примеры алкильных групп включают метил, этил, н-пропил, изопропил, н-бутил, трет-бутил, изобутил, втор-бутил, н-пентил, н-гексил, н-гептил, н-октил и т.п. Термин "алкенил" означает ненасыщенную алкильную группу, содержащую одну или больше двойных связей. Аналогично, термин «алкинил» означает ненасыщенную алкильную группу, содержащую одну или больше тройных связей. Примеры таких ненасыщенных алкильных групп включают винил, 2-пропенил, кротил, 2-изопентенил, 2-(бутадиенил), изобутенил, 2,4-пентадиенил, 3-(1,4-пентадиенил), этинил, 1- и 3-пропинил, 3-бутинил и их высшие гомологи и изомеры. Термин "циклоалкил" относится также к бициклическим и полициклическим углеводородным кольцам, таким как, например, бицикло[2.2.1]гептан, бицикло[2.2.2]октан и т.д. Термин "гетероциклоалкил" относится к циклоалкильной группе, содержащей 1-5 гетероатомов, выбранных из N, O, и S, где атомы азота и серы необязательно окислены, и атом(ы) азота необязательно кватернизован(ы). Гетероциклоалкил может представлять собой моноциклическую, бициклическую или полициклическую кольцевую систему. Неограничивающие примеры гетероциклоалкильных групп включают пирролидин, имидазолидин, пиразолидин, бутиролактам, валеролактам, имидазолидинон, гидантоин, диоксолан, фталимид, пиперидин, 1,4-диоксан, морфолин, тиоморфолин, тиоморфолин-S-оксид, тиоморфолин-S,S-оксид, пиперазин, пиран, пиридон, 3-пирролин, тиопиран, пирон, тетрагидрофуран, тетрагидротиофен, хинуклидин и т.п. Гетероциклоалкильная группа может быть присоединена к остальной части молекулы через атом углерода в цикле или гетероатом в цикле.
Термин "алкилен", сам по себе или как часть другого заместителя, означает двухвалентный радикал, являющийся производным алкана, в качестве примера можно привести -CH2CH2CH2CH2-. В типичном случае алкильная (или алкиленовая) группа содержит от 1 до 24 атомов углерода, предпочтительными по настоящему изобретению являются группы, содержащие 10 или меньше атомов углерода. «Низший алкил» или «низший алкилен» представляет собой короткоцепочечную алкильную или алкиленовую группу, обычно содержащую четыре или меньше атомов углерода. Аналогично, «алкенилен» или «алкинилен» означает ненасыщенные формы «алкилена», содержащие двойные или тройные связи, соответственно.
Термин “гетероалкил”, сам по себе или в комбинации с другим термином, означает, если не указано иное, устойчивый линейный или разветвленный цепочечный или циклический углеводородный радикал, или их комбинацию, состоящий из указанного числа атомов углерода и 1-3 гетероатомов, выбранных из группы, состоящей из O, N, Si и S, где атомы азота и серы необязательно могут быть окислены, а гетероатом азота необязательно может быть кватернизован. Гетероатом(ы) O, N и S могут располагаться в любом внутреннем положении гетероалкильной группы. Гетероатом Si может располагаться в любом положении гетероалкильной группы, включая положение, по которому алкильная группа присоединена к остальной части молекулы. Примеры включают -CH2-CH2-O-CH3, -CH2-CH2-NH-CH3, -CH2-CH2-N(CH3)-CH3, -CH2-S-CH2-CH3, -CH2-CH2,-S(O)-CH3, -CH2-CH2-S(O)2-CH3, -CH=CH-O-CH3, -Si(CH3)3, -CH2-CH=N-OCH3 и -CH=CH-N(CH3)-CH3. До двух гетероатомов могут располагаться последовательно, как, например, в -CH2-NH-OCH3 и -CH2-O-Si(CH3)3. Сходным образом, термин “гетероалкенил” и “гетероалкинил”, сам по себе или в комбинации с другим термином, означает, если не указано иное, алкенильную группу или алкинильную группу, соответственно, которая содержит указанное число атомов углерода и 1-3 гетероатомов, выбранных из группы, состоящей из O, N, Si и S, где атомы азота и серы необязательно могут быть окислены, а гетероатом азота необязательно может быть кватернизован. Гетероатом(ы) O, N и S могут располагаться во внутреннем положении гетероалкильной группы.
Термин “гетероалкилен”, сам по себе или как часть другого заместителя, означает двухвалентный радикал, насыщенный или ненасыщенный или полиненасыщенный, образованный из гетероалкила, например -CH2-CH2-S-CH2CH2- и -CH2-S-CH2-CH2-NH-CH2-, -O-CH2-CH=CH-, -CH2-CH=C(H)CH2-O-CH2- и -S-CH2-C≡C-. В случае гетероалкиленовых групп гетероатомы могут также занимать одно или оба терминальных положений (например, алкиленокси, алкилендиокси, алкиленамино, алкилендиамино и т.п.).
Термины "алкокси," "алкиламино" и "алкилтио" (или тиоалкокси) применяются в их обычном смысле и относятся к алкильным группам, присоединенным к остальной части молекулы через атом кислорода, аминогруппу или атом серы, соответственно. Кроме того, для диалкиламино-групп алкильные фрагменты могут быть одинаковыми или разными, а также могут объединяться с формированием 3-7-членного цикла с атомом азота, к которому они присоединены. Соответственно, группа, изображаемая как -NRaRb, охватывает пиперидинил, пирролидинил, морфолинил, азетидинил и т.п.
Термин "гидроксиалкил” применяется в своем обычном смысле и относится к разветвленной или линейной алкильной группе, замещенной по меньшей мере одной гидроксильной группой. Гидроксильная группа может находиться в любом положении алкильной группы. Например, термин "C1-4гидроксилалкил" включает гидроксиметил, гидроксиэтил, гидроксипропил, гидроксиизопропил и т.п.
Термин "галоген" сам по себе или как часть другого заместителя означает, если не указано иное, атом фтора, хлора, брома или иода. Кроме того, такие термины как "галогеналкил" включают моногалогеналкил и полигалогеналкил. Например, термин "C1-4 галогеналкил" включает трифторметил, 2,2,2-трифторэтил, 4-хлорбутил, 3-бромпропил и т.п.
Термин "арил" означает, если не указано иное, полиненасыщенную, в типичном случае ароматическую, углеводородную группу, которая может представлять собой один цикл или несколько циклов (до трех циклов), сопряженные или связанные ковалентно. Термин «гетероарил» означает арильные группы (или циклы), содержащие от одного до пяти гетероатомов, выбранных из N, O и S, где атомы азота и серы необязательно окислены, и атом(ы) азота необязательно кватернизован(ы). Гетероарильная группа может быть присоединена к остальной части молекулы через гетероатом. Неограничивающие примеры арильных групп включают фенил, нафтил и бифенил, а неограничивающие примеры гетероарильных групп включают пиридил, пиридазинил, пиразинил, пиримидинил, триазинил, хинолинил, хиноксалинил, хиназолинил, циннолил, фталазинил, бензотриазинил, пуринил, бензоимидазолил, бензопиразолил, бензоксазолил, бензотриазолил, бензизоксазалил, изобензофурил, изоиндолил, индолизинил, бензотриазинил, тиенопиридинил, тиенопиримидинил, пиразолопиримидинил, пирролопиридил, имидазопиридинил, бензотиаксолил, бензофуранил, бензотиенил, индолил, хинолил, изохинолил, изотиазолил, пиразолил, индазолил, птеридинил, имидазолил, триазолил, тетразолил, оксазолил, изоксазолил, тиадиазолил, пирролил, тиазолил, фурил, тиенил и т.п. Заместители в каждой из перечисленных выше арильных или гетероарильных циклических системах выбраны из группы приемлемых заместителей, описанных ниже.
Термин "фармацевтически приемлемые соли" включает соли веществ, полученные с относительно нетоксичными кислотами или основаниями, в зависимости от конкретных заместителей в описанных в настоящем тексте соединениях. Когда соединения по настоящему изобретению содержат относительно кислые функциональные группы, можно получить основно-аддитивные соли путем взаимодействия нейтральной формы таких соединений с достаточным количеством желаемого основания, даже без растворителя или в подходящем инертном растворителе. Примеры солей, являющихся производными фармацевтически приемлемых неорганических оснований, включают соли алюминия, аммония, кальция, меди, железа(II), железа(III), лития, магния, марганца, калия, натрия, цинка и т.д. Соли, являющиеся производными фармацевтически приемлемых органических оснований, включают соли первичных, вторичных и третичных аминов, включая замещенные амины, циклические амины, природные амины и т.д., такие как аргинин, бетаин, кофеин, холин, N,N'-дибензилэтилендиамин, диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этилендиамин, N-этилморфолин, N-этилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лизин, метилглюкамин, морфолин, пиперазин, пиперидин, полиаминовые смолы, прокаин, пурины, теобромин, триэтиламин, триметиламин, трипропиламин, трометамин и т.п. Когда соединения по настоящему изобретению содержат относительно основные функциональные группы, можно получить кислотно-аддитивные соли путем взаимодействия нейтральной формы таких соединений с достаточным количеством желаемой кислоты, без растворителя или в подходящем инертном растворителе. Примеры фармацевтически приемлемых кислотно-аддитивных солей включают соли с неорганическими кислотами, такими как хлористоводородная, бромистоводородная, азотная, угольная, моногидроугольная, фосфорная, моногидрофосфорная, дигидрофосфорная, серная, моногидросерная, иодистоводородная или фосфористая кислота и т.п., а также соли с относительно нетоксичными органическими кислотами, такими как уксусная, пропионовая, изомасляная, малоновая, бензойная, янтарная, субериновая, фумаровая, миндальная, фталевая, бензолсульфоновая, пара-толуолсульфоновая, лимонная, винная, метансульфоновая и т.п. Также охватываются соли с аминокислотами, такие как аргинаты и т.п., и соли таких органических кислот, как глюкуроновая или галактуроновая кислоты и т.п. (см, например, Berge, S.M., et al, “Pharmaceutical Salts”, Journal of Pharmaceutical Science, 1977, 66, 1-19). Некоторые частные соединения по настоящему изобретению содержат и основные, и кислотные функциональные группы, что позволяет таким соединениям образовывать как основно-аддитивные, так и кислотно-аддитивные соли.
Нейтральные формы соединений можно регенерировать путем взаимодействия соли с основанием или кислотой и выделения материнского соединения обычным способом. Материнская форма соединения отличается от различных солевых форм определенными физическими характеристиками, такими как растворимость в полярных растворителях, но во всем остальном соли эквивалентны материнским соединениям, в терминах настоящего изобретения.
Помимо солевых форм в настоящем изобретении описаны соединения, находящиеся в форме пролекарства. Пролекарства соединений, описанных в настоящем тексте, представляют собой соединения, которые легко претерпевают химические изменения в физиологических условиях, давая соединения по настоящему изобретению. Кроме того, пролекарства можно превратить в соединения по настоящему изобретению химическими или биологическими способами ex vivo. Например, пролекарства могут медленно превращаться в соединения по настоящему изобретению, когда они помещены в трансдермальный пластырь с соответствующим ферментом или химическим реагентом.
Некоторые соединения по настоящему изобретению могут существовать в несольватированных формах, а также в сольватированных формах, включая гидратированные формы. В целом, сольватированные формы эквивалентны несольватированным формам, и все они охватываются настоящим изобретением. Некоторые соединения по настоящему изобретению могут существовать в нескольких кристаллических или аморфных формах. В целом, все физические формы эквивалентны для областей применения, охватываемых настоящим изобретением, и входят в объем настоящего изобретения.
Некоторые соединения по настоящему изобретению имеют асимметричные атомы углерода (оптические центры) или двойные связи; все рацематы, диастереомеры, геометрические изомеры, региоизомеры и индивидуальные изомеры (например, отдельные энантиомеры) входят в объем настоящего изобретения. Соединения по настоящему изобретению могут также иметь неприродные соотношения изотопов по одному или больше атомов, составляющих эти соединения. Например, соединения могут быть радиоактивно помечены радиоактивными изотопами, такими как, например, тритий (3H), иод-125 (125I) или углерод-14 (14C). Все изотопные вариации соединений по настоящему изобретению, радиоактивные и нерадиоактивные, входят в объем настоящего изобретения.
1Термин "пролекарственный компонент" означает группу, которая придает желаемые свойства соединению, улучшающие, например, стабильность соединения, время нахождения в кровеносной системе in vivo или растворимость. Соединение, имеющее пролекарственный компонент, метаболизируется (обычно через гидролиз или ферментативно) после введения субъекту, что и дает активное соединение. Соединения с пролекарственным компонентом могут стать активными при расщеплении, но соединения с пролекарственным компонентом могут иметь активность в их непрореагировавших формах. Кроме того, пролекарственный компонент сам может быть активным. Примеры пролекарственных компонентов, охватываемых настоящей заявкой, включают (но не ограничиваются только ими) фосфат, фосфометил, гидроксиметил, аминокислоту, дипептидные и трипептидные фрагменты. Другие охватываемые настоящим изобретением варианты осуществления описаны далее в настоящем тексте.
Термин “аминокислота” относится к природным и неприродным аминокислотам. Неприродные аминокислоты означают соединения, которые имеют такую же базовую химическую структуру, как природные аминокислоты, т.е. α-углерод, который связан с атомом водорода, карбоксильной группой, аминогруппой и R-группой, например (но не ограничиваясь только ими) гомосерин, норлейцин, метионин сульфоксид, метилсульфоний метионин. Такие аналоги имеют модифицированные R-группы (такие как 2,5-диаминопентановая кислота) или модифицированные пептидные подвески, но сохраняют такую же базовую химическую структуру, как у природных аминокислот. Аминокислоты по настоящему изобретению включают N-метилированные и N-ацилированные фрагменты. Когда терминальной группой аминокислоты является атом N, он может быть диметилированным.
При использовании в настоящем тексте волнистая линия " ", которая пересекает простую, двойную или тройную связь в любой изображенной в настоящем тексте химической структуре, обозначает точку присоединения этой простой, двойной или тройной связи к остальной части молекулы.
", которая пересекает простую, двойную или тройную связь в любой изображенной в настоящем тексте химической структуре, обозначает точку присоединения этой простой, двойной или тройной связи к остальной части молекулы.
Описание вариантов осуществления
Соединения
В одном аспекте в настоящем изобретении описаны соединения, имеющие формулы (IA), (IB), (IC), (IIA), (IIB) и (IIC):
или их фармацевтически приемлемая соль, где
член цикла a представляет собой N или C(R2c), член цикла b представляет собой N или C(R2d), и член цикла e представляет собой N или C(R2e), где не более чем один из a, b и e представляет собой N;
X1 выбран из группы, состоящей из связи, C1-8 алкилена, C(O), C(O)-C1-4 алкилена и S(O)2;
R1 выбран из группы, состоящей из следующих:
a) 5-10-членный гетероарил, содержащий в качестве вершин кольца(ец) 1-4 гетероатома, выбранных из N, O и S;
b) C6-10 арил;
c) C3-8 циклоалкил;
d) 4-8-членный гетероциклоалкил, содержащий в качестве вершин кольца(ец) 1-2 гетероатома, выбранных из N, O и S; и
e) C1-8 алкил, C1-8 алкокси, C1-8 галогеналкил, -C(O)NR1aR1b и -CO2R1a; где R1a и R1b каждый независимо выбраны из группы, состоящей из атома водорода, C1-8 алкила, C6-10 арила и -C1-6 алкилен-C6-10 арила;
где группа -X1-R1 необязательно замещена 1-5 заместителями Rx;
R2a и R2e каждый независимо выбраны из группы, состоящей из атома водорода, C1-6 алкила, C1-6 алкокси, C1-6 галогеналкила, -O-C1-6 галогеналкила, -S-C1-6 алкила, -C1-6 алкил-O-C1-6 алкила, -C1-6 алкил-S-C1-6 алкила, CN и галогена, и по меньшей мере один из R2a и R2e отличается от атома водорода;
R2b, R2c и R2d каждый независимо выбраны из группы, состоящей из атома водорода, C1-6 алкила, C1-6 алкокси, C1-6 галогеналкила, -O-C1-6 галогеналкила, -S-C1-6 алкила, -C1-6 алкил-O-C1-6 алкила, -C1-6 алкил-S-C1-6 алкила, циано-группы и галогена;
каждый R3 независимо выбран из группы, состоящей из гидроксила, C1-4 алкила, C1-4 галогеналкила и C1-4 гидроксиалкила, и опционально две R3 группы у одного и того же атома углерода объединены с образованием оксо-группы (=O), и опционально две R3 группы и атомы углерода, к которым они присоединены, образуют 3-6-членное кольцо, содержащее в качестве членов цикла 0-2 гетероатомов, выбранных из O, N и S;
R4 выбран из группы, состоящей из -NHP1, -NHC(O)NHP1, -CH2NHP1 и -CH2NHC(O)NHP1;
каждый R5 независимо выбран из группы, состоящей из C1-8 алкила, C1-8 алкокси, C1-8 галогеналкила, C1-8 галогеналкокси, C1-8 гидроксиалкила, галогена, OH, CN, C(O)R5a и CO2R5a;
R5' выбран из группы, состоящей из атома водорода, C1-8 алкила, C1-8 галогеналкила, C1-8 гидроксиалкила, C(O)R5a и CO2R5a; где каждый R5a независимо выбран из группы, состоящей из атома водорода, C1-4 алкила и C1-4 галогеналкила;
R6 выбран из группы, состоящей из атома водорода, C1-6 алкила, C1-6 алкокси, C1-6 галогеналкила, -O-C1-6 галогеналкила, -S-C1-6 алкила, -C1-6 алкил-O-C1-6 алкила, -C1-6 алкил-S-C1-6 алкила, циано-группы и галогена;
R7 представляет собой P1; и
R8 представляет собой -CH2OP1;
каждый P1 представляет собой пролекарственный компонент;
каждый Rx независимо выбран из группы, состоящей из галогена, CN, C1-4 алкила, C1-4 алкокси, C1-4 галогеналкила, C1-4 галогеналкокси, C1-4 гидроксиалкила, C2-4 алкенила, C3-6 циклоалкила, CO2-C1-4 алкила и CONH2;
подстрочный индекс m равен 0, 1, 2, 3 или 4; и
подстрочный индекс n равен 0, 1, 2 или 3.
В одной группе вариантов осуществления соединения по настоящему изобретению имеют формулу (IA). В другой группе вариантов осуществления соединения по настоящему изобретению имеют формулу (IB). В другой группе вариантов осуществления соединения по настоящему изобретению имеют формулу (IC). В другой группе вариантов осуществления соединения по настоящему изобретению имеют формулу (IIA). В другой группе вариантов осуществления соединения по настоящему изобретению имеют формулу (IIB). В другой группе вариантов осуществления соединения по настоящему изобретению имеют формулу (IIC).
В некоторых вариантах соединений, имеющих формулу (IA), (IB), (IC), (IIA), (IIB), (IIC), или их фармацевтически приемлемых солей, а также любых из перечисленных выше групп вариантов осуществления, P1 выбран из группы, состоящей из:
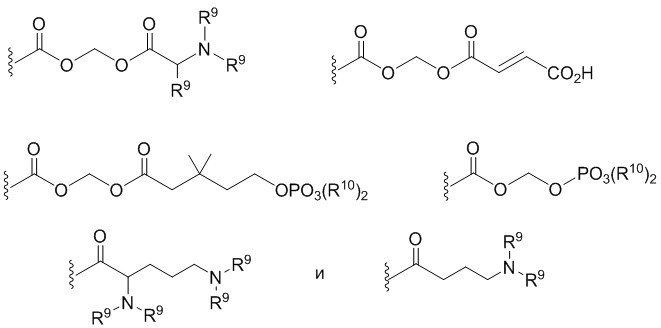
где каждый R9 независимо выбран из группы, состоящей из H и C1-3 алкила; и
каждый R10 независимо выбран из группы, состоящей из H, C1-3 алкила, фенила и бензила.
В некоторых вариантах соединений, имеющих формулу (IA), (IB), (IC), (IIA), (IIB), (IIC), или их фармацевтически приемлемых солей, а также любых из перечисленных выше групп вариантов осуществления, P1 выбран из группы, состоящей из:
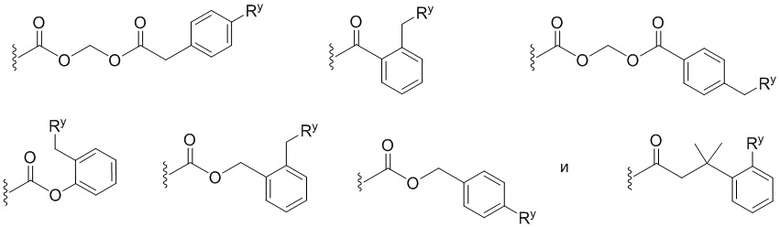 ,
,
где
каждый Ry независимо выбран из группы, состоящей из -OP(O)(ORy1)2, -OC(O)CH2N(Ry2)2, -N(Ry2)2 и пиперазина;
каждый Ry1 независимо выбран из группы, состоящей из H, C1-3 алкила и бензила;
каждый Ry2 независимо представляет собой H или C1-3 алкил; и
каждое фенильное кольцо, содержащее заместитель Ry или -CH2Ry, дополнительно замещено 0-3 заместителями, независимо выбранными из группы, состоящей из нитро-группы, галогена, CN, CF3, C1-4 алкила, C1-4 алкокси, C1-4 галогеналкила, C1-4 галогеналкокси и C1-4 гидроксиалкила.
В некоторых вариантах соединений, имеющих формулу (IA), (IB), (IC), (IIA), (IIB), (IIC), или их фармацевтически приемлемых солей, а также любых из перечисленных выше групп вариантов осуществления, P1 выбран из группы, состоящей из -CH2OH, -P(O)(OR10)2 и -CH2-O-P(O) (OR10)2, где каждый R10 независимо выбран из группы, состоящей из H, C1-3 алкила, фенила и бензила,
В некоторых вариантах соединений, имеющих формулу (IA), (IB), (IC), (IIA), (IIB), (IIC), или их фармацевтически приемлемых солей, а также любых из перечисленных выше групп вариантов осуществления, P1 выбран из группы, состоящей из аминокислоты, дипептида и трипептида. В некоторых вариантах осуществления указанные аминокислота, дипептид или трипептид представляют собой природные аминокислоты. В некоторых вариантах осуществления указанные аминокислотные, дипептидные или трипептидные фрагменты независимо выбраны из группы, состоящей из глицина, аланина, валина, лейцина, изолейцина, лизина, цистеина, аспартата, глутамата, гистидина и фенилаланина, где N-атом каждого аминокислотного фрагмента может быть метилированным или ацилированным.
Аминокислоты по настоящему изобретению могут быть ковалентно связаны с остальной частью молекулы любым подходящим образом. Подходящие линкеры включают (но не ограничиваются только ими) амидные фрагменты между аминной группой и гидроксильной группой, сложноэфирные фрагменты между карбоксильной группой и гидроксильной группой, и сульфонамидный фрагмент (N-S линкер) между тиогруппой и аминогруппой. Обычно аминокислоты связаны с остальной частью молекулы через альфа-аминогруппу или альфа-карбоксильную группу; однако когда R-группа аминокислоты содержит функциональную группу, она тоже может служить точкой связывания. Например, карбоксильная группа в глутамате может служить точкой связывания с остальной частью молекулы. Аналогичным образом, тиол в цистеине также может служить точкой связывания с остальной частью молекулы.
В некоторых вариантах соединений, имеющих формулу (IA), (IB), (IC), (IIA), (IIB), (IIC), или их фармацевтически приемлемых солей, а также любых из перечисленных выше групп вариантов осуществления, P1 выбран из группы, состоящей из:
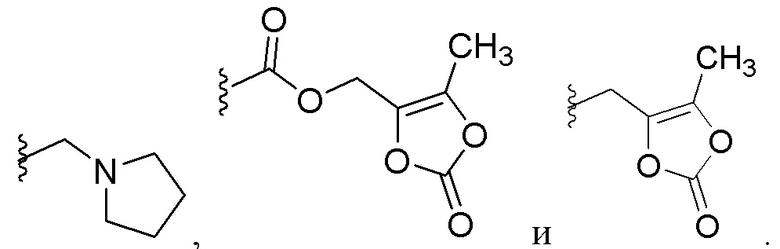
В соединениях, имеющих формулу (IA), (IB), (IC), (IIA), (IIB), (IIC), или их фармацевтически приемлемых солях, а также в любой из указанных выше групп вариантов, в некоторых частных вариантах осуществления X1 представляет собой связь; в других частных вариантах осуществления X1 представляет собой C(O); в других частных вариантах осуществления X1 представляет собой C1-8 алкилен; в других частных вариантах осуществления X1 представляет собой C(O)-C1-4 алкилен или S(O)2.
В соединениях, имеющих формулу (IA), (IB), (IC), (IIA), (IIB), (IIC), или их фармацевтически приемлемых солях, а также в любой из указанных выше групп вариантов или частных вариантов осуществления, в некоторых дополнительных вариантах осуществления R1 представляет собой 5-10-членный гетероарил, содержащий в качестве вершин кольца(ец) 1-4 гетероатома, выбранных из N, O и S; и группа -X1-R1 необязательно замещена 1-4 заместителями Rx. В других дополнительных вариантах осуществления R1 выбран из группы, состоящей из пиразолила, пиридила, пиримидинила, имидазолила, тиазолила, тиадиазолила и пиразинила; и группа -X1-R1 необязательно замещена 1-4 заместителями Rx.
В соединениях, имеющих формулу (IA), (IB), (IC), (IIA), (IIB), (IIC), или их фармацевтически приемлемых солях, а также в любой из указанных выше групп вариантов или частных вариантов осуществления, в некоторых дополнительных вариантах осуществления R1 представляет собой C6-10 арил; и группа -X1-R1 необязательно замещена 1-4 заместителями Rx. В других дополнительных вариантах осуществления R1 представляет собой фенил; и группа -X1-R1 необязательно замещена 1-4 заместителями Rx.
В соединениях, имеющих формулу (IA), (IB), (IC), (IIA), (IIB), (IIC), или их фармацевтически приемлемых солях, а также в любой из указанных выше групп вариантов или частных вариантов осуществления, в некоторых дополнительных вариантах осуществления R1 представляет собой C3-8 циклоалкил; и группа -X1-R1 необязательно замещена 1-4 заместителями Rx. В других дополнительных вариантах осуществления R1 выбран из группы, состоящей из циклобутила, циклопентила и циклогексила; и группа -X1-R1 необязательно замещена 1-4 заместителями Rx.
В соединениях, имеющих формулу (IA), (IB), (IC), (IIA), (IIB), (IIC), или их фармацевтически приемлемых солях, а также в любой из указанных выше групп вариантов или частных вариантов осуществления, в некоторых дополнительных вариантах осуществления R1 представляет собой 4-8-членный гетероциклоалкил, содержащий в качестве вершин кольца(ец) 1-2 гетероатома, выбранных из N, O и S; и где группа -X1-R1 необязательно замещена 1-4 заместителями Rx. В других дополнительных частных вариантах осуществления R1 выбран из группы, состоящей из оксетанила, тетрагидрофуранила, тетрагидропиранила и морфолинила; и группа -X1-R1 необязательно замещена 1-4 заместителями Rx.
В соединениях, имеющих формулу (IA), (IB), (IC), (IIA), (IIB), (IIC), или их фармацевтически приемлемых солях, а также в любой из указанных выше групп вариантов или частных вариантов осуществления, в некоторых дополнительных вариантах осуществления R1 выбран из группы, состоящей из C1-8 алкила, C1-8 алкокси, C1-8 галогеналкила, -C(O)NR1aR1b и -CO2R1a; где R1a и R1b каждый независимо выбраны из группы, состоящей из атома водорода, C1-8 алкила, C6-10 арила и -C1-6 алкилен-C6-10 арила; и группа -X1-R1 необязательно замещена 1-4 заместителями Rx.
В соединениях, имеющих формулу (IA), (IB), (IC), (IIA), (IIB), (IIC), или их фармацевтически приемлемых солях, а также в любой из указанных выше групп вариантов или частных вариантов осуществления, в некоторых дополнительных вариантах осуществления R1 выбран из группы, состоящей из фенила, пиридила, пиримидинила и пиразинила; и группа -X1-R1 необязательно замещена 1-4 заместителями Rx.
В соединениях, имеющих формулу (IA), (IB), (IC), (IIA), (IIB), (IIC), или их фармацевтически приемлемых солях, а также в любом из указанных выше вариантов осуществления, в некоторых дополнительных вариантах осуществления члены цикла a и b представляют собой CH; R2b представляет собой H; член цикла e представляет собой C(R2e), и R2a и R2e независимо выбраны из группы, состоящей из C1-6 алкила, C1-6 алкокси, C1-6 галогеналкила,-O-C1-6 галогеналкила, -S-C1-6 алкила, -C1-6 алкил-O-C1-6 алкила, -C1-6 алкил-S-C1-6 алкила, CN и галогена.
В соединениях, имеющих формулу (IA), (IB), (IC), (IIA), (IIB), (IIC), или их фармацевтически приемлемых солях, а также в любом из указанных выше вариантов осуществления, в некоторых дополнительных вариантах осуществления члены цикла a и b представляют собой CH; R2b представляет собой H; член цикла e представляет собой C(R2e), и R2a и R2e независимо выбраны из группы, состоящей из C1-6 алкила, C1-6 алкокси и галогена.
В соединениях, имеющих формулу (IA), (IB), (IC), (IIA), (IIB), (IIC), или их фармацевтически приемлемых солях, а также в любом из указанных выше вариантов осуществления, в некоторых дополнительных вариантах осуществления подстрочный индекс n равен 0, 1 или 2, и каждый R5, если он присутствует, выбран из группы, состоящей из F, Cl, CN, C1-4 алкила и C1-4 алкокси. В других дополнительных частных вариантах осуществления подстрочный индекс n равен 0, 1 или 2, и каждый R5, если он присутствует, выбран из группы, состоящей из F, Cl, CN, CH3 и OCH3.
В соединениях, имеющих формулу (IA), (IB), (IC), (IIA), (IIB), (IIC), или их фармацевтически приемлемых солях, а также в любом из указанных выше вариантов осуществления, в некоторых дополнительных вариантах осуществления подстрочный индекс m равен 0, 1 или 2, и каждый R3, если он присутствует, представляет собой C1-4 алкил.
В частной группе вариантов соединений, имеющих формулу (IA), (IB), (IC), (IIA), (IIB), (IIC), или их фармацевтически приемлемых солях R1 выбран из группы, состоящей из фенила или пиридила, группа -X1-R1 необязательно замещена 1-4 заместителями Rx; члены цикла a и b представляют собой CH; R2b представляет собой H; член цикла e представляет собой C(R2e), и R2a и R2e независимо выбраны из группы, состоящей из C1-6 алкила, C1-6 алкокси и галогена; m равен 0, 1 или 2, и каждый R3, если он присутствует, представляет собой CH3, n равен 0, 1 или 2, и каждый R5, если он присутствует, выбран из группы, состоящей из F, Cl, CN, CH3 и OCH3.
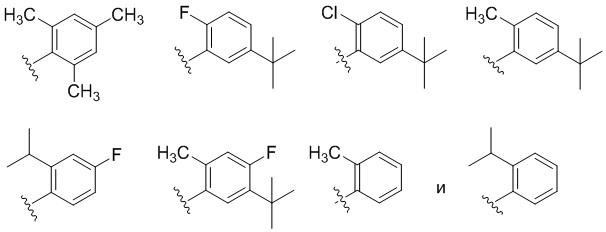
В некоторых вариантах соединений, имеющих формулу (IA), (IB), (IC), (IIA), (IIB), (IIC), или их фармацевтически приемлемых солей, R1 выбран из группы, состоящей из
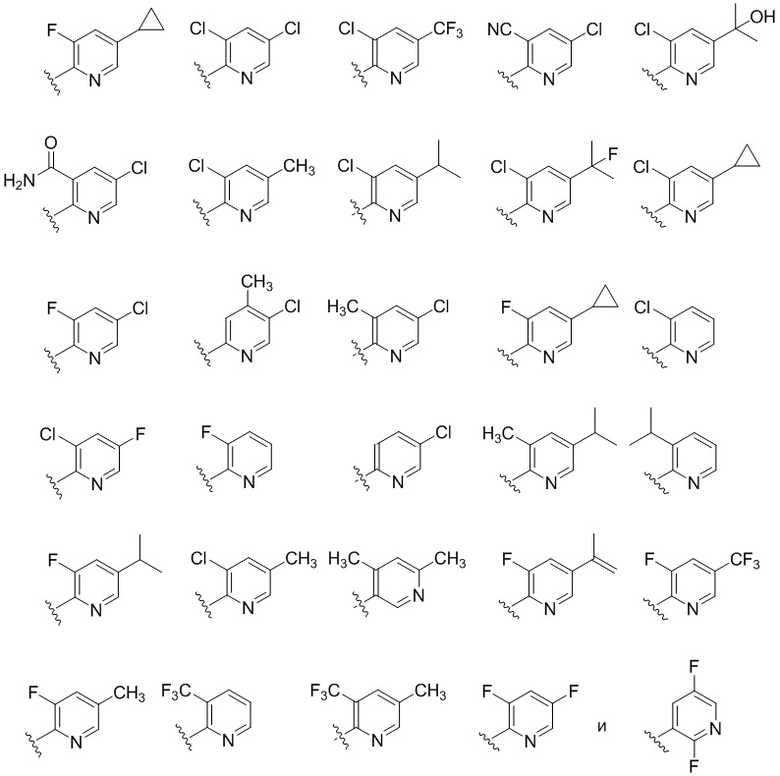
В некоторых вариантах соединений, имеющих формулу (IA), (IB), (IC), (IIA), (IIB), (IIC), или их фармацевтически приемлемых солей, -X1-R1 выбран из группы, состоящей из:
 .
.
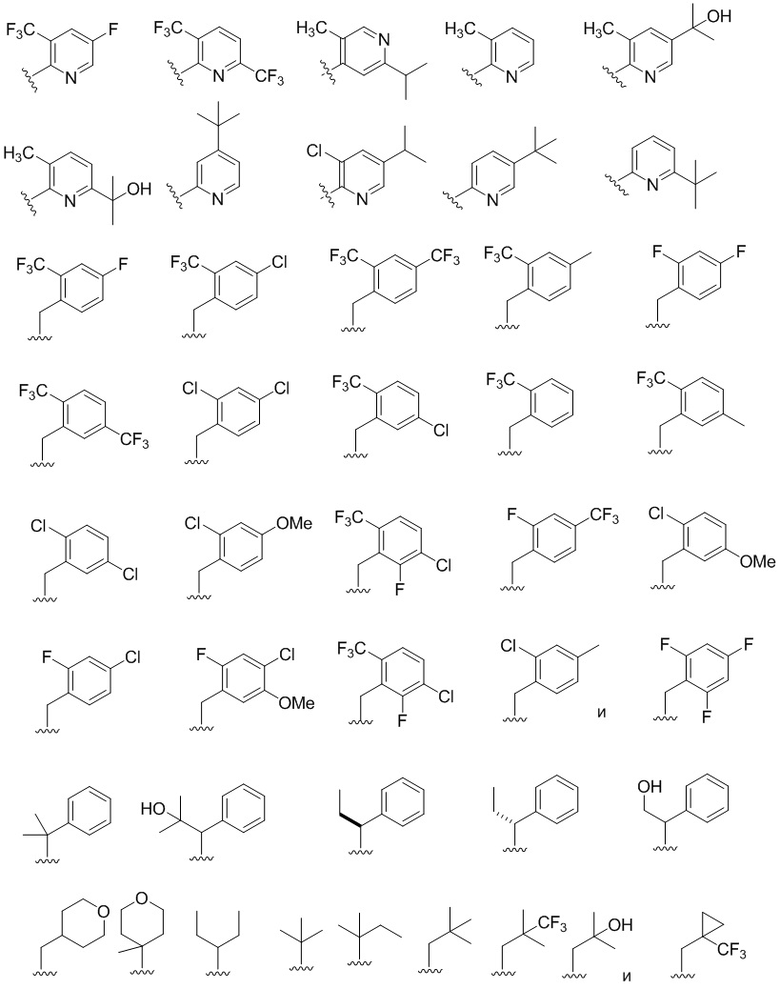
В некоторых вариантах соединений, имеющих формулу (IA), (IB), (IC), (IIA), (IIB), (IIC), или их фармацевтически приемлемых солей, R1 выбран из группы, состоящей из:
 .
.
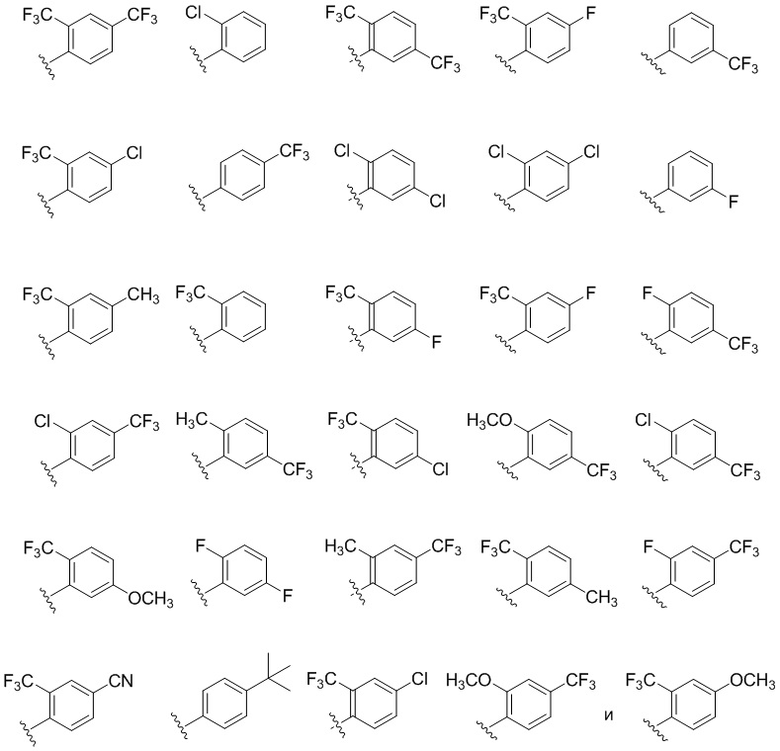
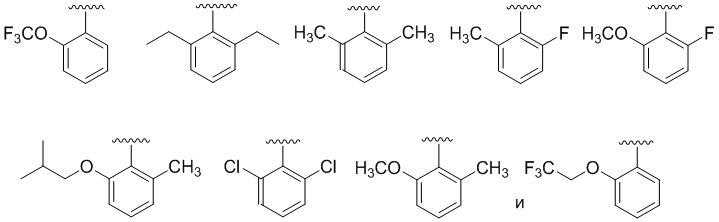
В некоторых вариантах соединений, имеющих формулу (IA), (IB), (IC), (IIA), (IIB), (IIC), или их фармацевтически приемлемых солей, R1 выбран из группы, состоящей из:
 .
.
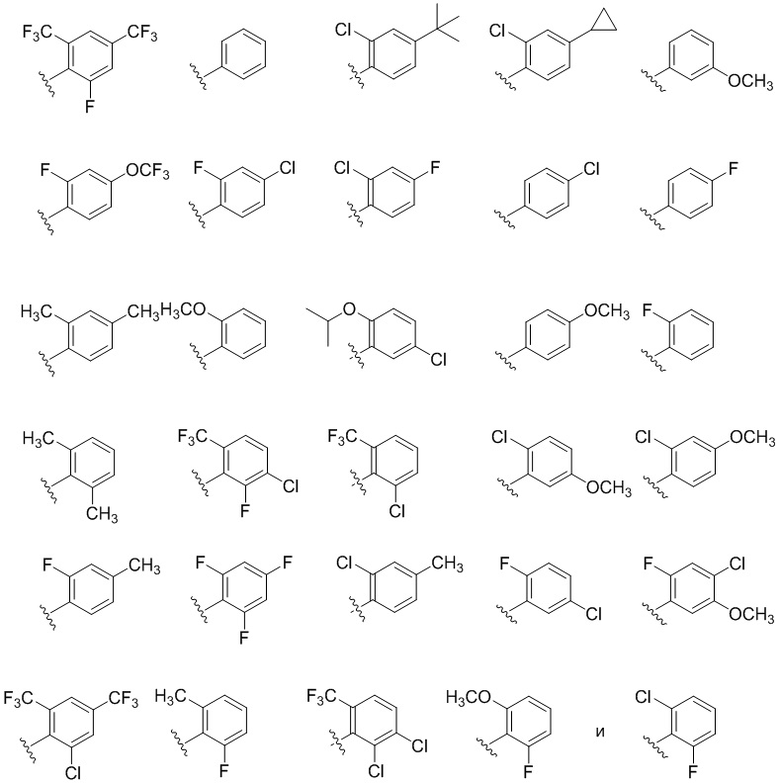
В некоторых вариантах соединений, имеющих формулу (IA), (IB), (IC), (IIA), (IIB), (IIC), или их фармацевтически приемлемых солей, R1 выбран из группы, состоящей из:
 .
.
В соединениях, имеющих формулу (IA), (IB), (IC), (IIA), (IIB), (IIC), или их фармацевтически приемлемых солях, а также в любом из указанных выше вариантов осуществления, в некоторых дополнительных вариантах осуществления группа 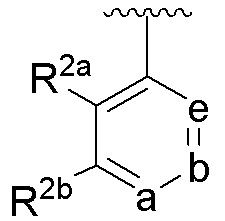 выбрана из группы, состоящей из
выбрана из группы, состоящей из
 .
.
В соединениях, имеющих формулу (IA), (IB), (IC), (IIA), (IIB), (IIC), или их фармацевтически приемлемых солях, а также в любом из указанных выше вариантов осуществления, в некоторых дополнительных вариантах осуществления n равен 0.
В соединениях, имеющих формулу (IA), (IB), (IC), (IIA), (IIB), (IIC), или их фармацевтически приемлемых солях, а также в любом из указанных выше вариантов осуществления, в некоторых дополнительных вариантах осуществления подстрочный индекс n равен 2, и две группы R3 присоединены к одному и тому же атому углерода и объединены с образованием оксо-группы (=O).
В некоторых вариантах осуществления, соединение по настоящему изобретению представляет собой соединение, описанное в разделе Примеры и таблицах.
Получение соединений
Некоторые соединения по настоящему изобретению можно получить по методикам, описанным в настоящей заявке в разделе Примеры. Кроме того, описаны также синтезы некоторых промежуточных соединений, которые могут применяться для синтеза соединений по настоящему изобретению.
Фармацевтические композиции
Помимо описанных выше соединений, композиции для модулирования C5a активности у людей и животных обычно содержат фармацевтически приемлемый носитель или разбавитель.
Термин “композиция” при использовании в настоящем тексте охватывает продукт, содержащий указанные ингредиенты в указанных количествах, а также любой продукт, который получается, напрямую или опосредованно, при комбинировании указанных ингредиентов в указанных количествах. Под термином “фармацевтически приемлемый” понимается носитель, разбавитель или вспомогательное вещество, которое должно быть совместимо с другими ингредиентами препарата и не наносить вреда принимающему его пациенту.
Фармацевтические композиции для введения соединений по настоящему изобретению можно выпускать в виде дозированных лекарственных форм, и их можно готовить любым из способов, известных в фармакологии и в области введения лекарственных препаратов. Все способы включают стадию объединения действующего вещества с носителем, который содержит один или несколько вспомогательных ингредиентов. В целом, фармацевтические композиции получают путем однородного и тщательного смешивания действующего вещества с жидким носителем или тонко измельченным твердым носителем, или с обоими, и затем, при необходимости, придания продукту формы желаемого препарата. В фармацевтической композиции действующее вещество присутствует в количестве, достаточном для оказания целевого эффекта на болезнь или на патологическое состояние.
Фармацевтические композиции, содержащие действующее вещество, могут иметь форму, подходящую для перорального применения, например они могут быть в виде таблеток, саше, пастилок, водных или масляных суспензий, диспергируемых порошков или гранул, эмульсий и самоэмульгирующихся препаратов, как описано в Заявке на Патент США 2002-0012680, твердых или мягких капсул, сиропов, эликсиров, растворов, буккальных пластырей, геля для полости рта, жевательной резинки, жевательных таблеток, шипучих порошков и шипучих таблеток. Композиции, предназначенные для перорального приема, можно готовить любыми способами, известными в области производства фармацевтических композиций. Такие композиции могут содержать один или больше агентов, выбранных из группы, состоящей из подсластителей, ароматизаторов, красителей, антиоксидантов и консервантов, для создания фармацевтически привлекательных и приятных на вкус препаратов. Таблетки содержат действующее вещество в смеси с нетоксичными фармацевтически приемлемыми вспомогательными веществами, подходящими для производства таблеток. Такими вспомогательными веществами могут быть, например, инертные разбавители, такие как целлюлоза, диоксид углерода, оксид алюминия, карбонат кальция, карбонат натрия, глюкоза, маннит, сорбит, лактоза, фосфат кальция или фосфат натрия; гранулирующие агенты и агенты, ускоряющие распад таблеток, например кукурузный крахмал или альгиновую кислоту; связующие агенты, например поливинилпирролидон, целлюлозу, крахмал, желатин или смолу акации, и лубриканты, например стеарат магния, стеариновую кислоту или тальк. Таблетки могут не иметь покрытия или они могут быть покрыты кишечнорастворимой оболочкой, или иметь покрытие, нанесенное каким-либо другим известным способом, с целью замедления распада и всасывания в желудочно-кишечном тракте, тем самым обеспечивая пролонгированное действие в течение более длительного времени. Например, можно применять такие замедляющие распад материалы как глицерил моностеарат или глицерил дистеарат. На таблетки можно также наносить покрытие способами, описанными в Патентах США № 4,256,108; 4,166,452 и 4,265,874, с образованием осмотических таблеток с замедленным высвобождением.
Препараты, предназначенные для перорального приема, могут также иметь вид твердых желатиновых капсул, где действующее вещество смешано с инертным твердым разбавителем, например карбонатом кальция, фосфатом кальция или каолином, или в виде мягких желатиновых капсул, где действующее вещество смешано с водной или масляной средой, например арахисовым маслом, жидким парафином или оливковым маслом. Кроме того, можно готовить эмульсии с не растворяющимися в воде ингредиентами, такими как масла, и стабилизировать их поверхностно-активными веществами, такими как моно- или диглицериды, сложные эфиры ПЭГ и т.п.
Водные суспензии содержат действующие вещества в смеси со вспомогательными веществами, подходящими для производства водных суспензий. Такие вспомогательные вещества представляют собой суспендирующие агенты, например натрия карбоксиметилцеллюлозу, метилцеллюлозу, гидроксипропилметилцеллюлозу, альгинат натрия, поливинилпирролидон, трагакантовую камедь, и смолу акации; диспергирующие или увлажняющие агенты могут представлять собой природные фосфатиды, например лецитин, или продукты конденсации алкиленоксида с жирными кислотами, например полиоксиэтилен стеарат, или продукты конденсации этиленоксида с длинноцепочечными алифатическими спиртами, например гептадекаэтиленоксицетанол, или продукты конденсации этиленоксида с неполными сложными эфирами, полученными из жирных кислот и гекситола, такие как полиоксиэтилен сорбитмоноолеат, или продукты конденсации этиленоксида с неполными сложными эфирами, полученными из жирных кислот и ангидридами гекситола, например полиэтилен сорбитанмоноолеат. Водные суспензии могут также содержать один или больше консервантов, например этил или н-пропил пара-гидроксибензоат, один или больше красителей, один или больше ароматизаторов, и один или больше подсластителей, таких как сахароза или сахарин.
Масляные суспензии можно получать путем суспендирования действующего вещества в растительном масле, например в арахисовом масле, оливковом масле, сезамовом масле или кокосовом масле, или в минеральном масле, таком как жидкий парафин. Масляные суспензии могут содержать загустители, например пчелиный воск, твердый парафин или цетиловые спирты. Можно добавлять подсластители, такие как перечисленные выше, и красители, для получения приятной на вкус композиции для перорального приема. Такие композиции можно стабилизировать добавлением антиоксиданта, такого как аскорбиновая кислота.
Диспергируемые порошки и гранулы, подходящие для приготовления водной суспензии при добавлении воды, содержат действующее вещество в смеси с диспергирующим или увлажняющим агентом, суспендирующим агентом и одним или больше консервантами. Примеры диспергирующих или увлажняющих агентов, суспендирующих агентов приведены выше. Могут также присутствовать дополнительные вспомогательные вещества, например подсластители, ароматизаторы и красители.
Фармацевтические композиции по настоящему изобретению могут также иметь форму эмульсий типа масло-в-воде. Масляной фазой может служить растительное масло, например оливковое масло или арахисовое масло, или минеральное масло, например жидкий парафин, или их смесь. Подходящие эмульгаторы могут представлять собой природные смолы, например смолу акации или трагакантовую камедь, природные фосфатиды, например соевое масло, лецитин, и сложные эфиры или неполные сложные эфиры, полученные из жирных кислот и ангидридов гекситола, например сорбитан моноолеат, и продукты конденсации указанных неполных сложных эфиров с этиленоксидом, например полиоксиэтилен сорбитанмоноолеат. Эмульсия может также содержать подсластители и ароматизаторы.
В состав сиропов и эликсиров могут входить подсластители, например глицерин, пропиленгликоль, сорбит или сахароза. Такие препараты могут также содержать средства, уменьшающие раздражение, консервант, ароматизаторы и красители. Растворы для перорального приема можно готовить в комбинации с, например, циклодекстрином, ПЭГ и поверхностно-активными веществами.
Фармацевтические композиции могут иметь форму стерильных инъецируемых водных или масляных суспензий. Такую суспензию можно готовить по известным в данной области методикам, с применением подходящих диспергаторов или увлажняющих агентов и суспендирующих агентов, которые были указаны выше. Стерильные инъецируемые препараты могут также представлять собой стерильные инъецируемые растворы или суспензии в нетоксичном парентерально приемлемом разбавителе или растворителе, например могут иметь форму раствора в 1,3-бутандиоле. Среди подходящих носителей и растворителей, которые можно использовать, находятся вода, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, стерильные масла часто применяют в качестве растворителя или суспендирующей среды. Для этой цели можно использовать любое безвкусное нелетучее масло, включая синтетические моно- и диглицериды. Кроме того, в приготовлении инъецируемых препаратов находят применение жирные кислоты, такие как олеиновая кислота.
Описанные в настоящем тексте соединения можно также вводить в форме суппозиториев для ректального введения лекарственного средства. Такие композиции можно готовить путем смешивания лекарственного средства с подходящим нераздражающим вспомогательным веществом, которое является твердым при комнатной температуре, но переходит в жидкое состояние при температуре тела, и поэтому плавится в прямой кишке с высвобождением лекарственного средства. Такие материалы включают масло какао и полиэтиленгликоли. Кроме того, соединения можно вводить в виде глазных препаратов как капли или мази. Кроме того, можно осуществлять чрескожное введение рассматриваемых соединений посредством ионофорезных пластырей и т.п. Для местного нанесения применяют кремы, мази, гели, растворы или суспензии, содержащие соединения по настоящему изобретению. В контексте настоящего изобретения, местное нанесение включает также применение полосканий и растворов для рта.
Соединения по настоящему изобретению можно также соединять с носителем, представляющем собой подходящие полимеры, в качестве направленных носителей лекарственного средства. Такие полимеры могут включать поливинилпирролидон, пирановый сополимер, полигидрокси-пропил-метакриламид-фенол, полигидроксиэтил-аспартамид-фенол или полиэтиленоксид-полилизин, замещенный пальмитоильными остатками. Кроме того, соединения по настоящему изобретению можно соединять с носителем, относящимся к классу биоразлагаемых полимеров, которые можно применять для достижения контролируемого высвобождения лекарственного средства, например: полимолочная кислота, полигликолевая кислота, сополимеры полимолочной и полигликолевой кислоты, поли-эпсилон-капролактон, полигидроксимасляная кислота, полиортоэфиры, полиацетали, полидигидропираны, полицианоакрилаты и сшитые или амфипатические блок-сополимеры гидрогелей. Полимеры и полупроницаемые полимерные матриксы можно формовать в изделия, такие как клапаны, стенты, трубки, протезы и т.п. В одном варианте осуществления настоящего изобретения соединение по настоящему изобретению соединяют с полимером или полупроницаемым полимерным матриксом, сформованным в виде стента или стент-графта.
Фармацевтические композиции по настоящему изобретению могут содержать одно или больше дополнительных терапевтических средств. Эти одно или больше дополнительных терапевтических средств выбраны из группы, состоящей из кортикостероидов, стероидов, иммунодепрессантов, агонистов иммуноглобулина G, ингибиторов дипептидилпептидазы IV, антагонистов лимфоцитарного функционально-ассоциированного антигена-3, лигандов интерлейкина-2, ингибиторов лигандов интерлейкина-1 бета, ингибиторов альфа-субъединицы рецептора IL-2, стимуляторов гена HGF, антагонистов IL-6, антагонистов IL-5, стимуляторов альфа-1 антитрипсина, антагонистов каннабиноидного рецептора, ингибиторов гистондеацетилазы, ингибиторов AKT протеинкиназ, ингибиторов CD20, ингибиторов Abl тирозинкиназы, ингибиторов JAK тирозинкиназ, ингибиторов лигандов ФНО-альфа, модуляторов гемоглобина, антагонистов ФНО, ингибиторов протеасом, модуляторов CD3, ингибиторов Hsp 70 семейства, агонистов иммуноглобулина, антагонистов CD30, антагонистов тубулина, агонистов сфингозин-1-фосфатного рецептора, ингибиторов лиганда фактора роста соединительной ткани, ингибиторов каспазы, лигандов адренокортикотропного гормона, ингибиторов Btk тирозинкиназы, ингибиторов компонента системы комплемента C1s, агонистов рецептора эритропоэтина, ингибиторов стимуляторов B-лимфоцитов, ингибиторов циклинзависимой киназы 2, стимуляторов гликопротеинового лиганда P-селектина 1, ингибиторов mTOR, ингибиторов фактора элонгации 2, ингибиторов молекулы клеточной адгезии, агонистов фактора XIII, ингибиторов кальциневрина, агонистов иммуноглобулина G1, ингибиторов инозин-монофосфат-дегидрогеназы, ингибиторов компонента системы комплемента C1s, модуляторов тимидинкиназ, модуляторов CTLA-4, антагонистов рецептора ангиотензина II, модуляторов рецептора ангиотензина II, антагонистов рецептора 12А из суперсемейства ФНО, антагонистов CD52, ингибиторов аденозиндеаминазы, ингибиторов антиген дифференцировки Т-клеток CD6, лигандов FGF-7, ингибиторов дигидрооротат-дегидрогеназы, ингибиторов Syk тирозинкиназы, антагонистов рецептора интерферона I типа, ингибиторов интерферона альфа, ингибиторов фактора ингибирования миграции макрофагов, антагонистов интегрина альфа-V/бета-6, стимуляторов цистеинпротеазы, ингибиторов p38 MAP киназы, ингибиторов гена TP53, ингибиторов шига-подобного токсина I, стимуляторов фукозилтрансферазы 6, лигандов интерлейкина 22, ингибиторов гена IRS1, стимуляторов протеинкиназ C, ингибиторов протеинкиназы C альфа, антагонистов CD74, антагонистов иммуноглобулин гамма Fc рецептора 2B, ингибиторов антигена T-клеток CD7, антагонистов CD95, стимуляторов N-ацетилманнозамин-киназы, лигандов кардиотропина-1, ингибиторов эластазы лейкоцитов, антагонистов лиганда CD40 рецептора, модуляторов лиганда CD40 рецептора, антагонистов IL-17, антагонистов TLR-2, ингибиторов маннан-связывающей лектин-зависимой серинпротеазы-2 (MASP-2), ингибиторов фактора B, ингибиторов фактора D, модуляторов C3aR, модуляторов C5aR2, антагонистов T-клеточного рецептора, ингибиторов PD-1, ингибиторов PD-L1, ингибиторов TIGIT, ингибиторов TIM-3, ингибиторов LAG-3, ингибиторов VISTA, агонистов STING, ингибиторов IDO, модуляторов аденозинового рецептора, ингибиторов CD39, ингибиторов CD73, антагонистов хемокиновых рецепторов, в особенности CXCR1, CXCR2, CXCR3, CXCR4, CXCR7, CCR1, CCR2, CCR3, CCR4, CCR5, CCR7, CCR7, CCR9, CX3CR1 и CXCR6, и их комбинаций.
В некоторых вариантах осуществления одно или больше дополнительных терапевтических средств выбраны из группы, состоящей из следующих: обинутузумаб, ритуксимаб, окрелизумаб, циклофосфамид, преднизон, гидрокортизон, гидрокортизона ацетат, кортизона ацетат, тиксокортол пивалат, преднизолон, метилпреднизолон, триамцинолона ацетонид, триамцинолоновый спирт, мометазон, амцинонид, будесонид, десонид, флуоцинонид, флуоцинолона ацетонид, галцинонид, бетаметазон, бетаметазон натрия фосфат, дексаметазон, дексаметазон натрия фосфат, флуокортолон, гидрокортизон-17-валерат, галометазон, аклометазона дипропионат, беклометазон, бетаметазона валерат, бетаметазона дипропионат, предникарбат, клобетазон-17-бутират, клобетазол-17-пропионат, флуокортолона капроат, флуокортолона пивалат, флупреднидена ацетат, гидрокортизон-17-бутират, гидрокортизон-17-ацепонат, гидрокортизон-17-бутепрат, циклесонид и предникарбат, GB-0998, иммугло, бегеломаб, алефацепт, альдеслейкин, гевокизумаб, даклизумаб, базиликсумаб, инолимомаб, беперминоген перплазмид, сирукумаб, тоцилизумаб, клазакизумаб, меполизумаб, финголимод, панобиностат, трицирибин, нилотиниб, иматиниб, тофацитиниб, момелотиниб, пефицитиниб, итацитиниб, инфликсимаб, PEG-bHb-CO, этанерцепт, иксазомиб, бортезомиб, муромонаб, отеликсизумаб, гусперимус, брентуксимаб ведотин, понезимод, KRP-203, FG-3019, эмрикасан, кортикотропин, ибрутиниб, синрайз, конестат, метоксиполиэтиленгликоль-эпоэтин бета, белимумаб, блисибимод, атацицепт, селициклиб, нейхулизумаб, эверолимус, сиролимус, денилейкин дифитокс, LMB-2, натализумаб, катридекаког, циклоспорин, такролимус, воклоспорин, канакинумаб, микофенолят, мизорибин, CE-1145, TK-DLI, абатацепт, белатацепт, олмесартан медоксомил, спарсентан, TXA-127, BIIB-023, алемтузумаб, пентостатин, итолизумаб, палифермин, лефлуномид, PRO-140, ценикривирок, фостаматиниб, анифролумаб, сифалимумаб, BAX-069, BG-00011, лосмапимод, QPI-1002, ShigamAbs, TZ-101, F-652, репариксин, ладариксин, PTX-9908, аганирсен, APH-703, сотрастаурин, милатузумаб, SM-101, T-Guard, APG-101, DEX-M74, кардиотрофин-1, типрелестат, ASKP-1240, BMS-986004, HPH-116, KD-025, OPN-305, TOL-101, дефибротид, помалидомид, тимоглобулин, лаквинимод, реместемцел-L, лошадиный анти-тимоцитарный иммуноглобулин, стемпейцел, LIV-гамма, Октагам 10%, t2c-001, 99mTc-сестамиби, Clairyg, Просорба, помалидомид, лаквинимод, теплизумаб, FCRx, солнатид, форалумаб, ATIR-101, BPX-501, ACP-01, ALLO-ASC-DFU, ирбесартан + пропагерманий, ApoCell, каннабидиол, RGI-2001, саратин, конъюгат анти-CD3 бивалентное антитело-дифтерийный токсин, NOX-100, LT-1951, OMS721, ALN-CC5, ACH-4471, AMY-101, Acthar гель и CD4+CD25+ регуляторные T-клетки, MEDI7814, P32, P59, пембролизумаб, ниволумаб, атезолизумаб, авелумаб, дурвалумаб, CCX354, CCX721, CCX9588, CCX140, CCX872, CCX598, CCX6239, CCX587, CCX624, CCX282, CCX025, CCX507, CCX430, CCX765, CCX758, CCX771, CCX662, CCX650 и их комбинации. Дополнительное обсуждение комбинированной терапии включено в настоящую заявку в разделе “Способы применения”.
Способы применения
Соединения по настоящему изобретению можно применять в качестве агонистов, (предпочтительно) антагонистов, частичных агонистов, обратных агонистов рецепторов C5a в различном контексте, как in vitro, так и in vivo. В одном варианте осуществления соединения по настоящему изобретению представляют собой антагонист C5aR, который можно применять для подавления связывания лиганда C5a рецептора (например, C5a) с C5a рецептором in vitro или in vivo. В целом, такие способы включают стадию контактирования C5a рецептора с достаточным количеством одного или более модуляторов C5a рецептора, описанных в настоящем тексте, в присутствии лиганда C5a рецептора в водном растворе и в условиях, подходящих для связывания данного лиганда с C5a рецептором. C5a рецептор может присутствовать в суспензии (например, в виде изолированной мембраны или препарата клеток), в выращенной или выделенной клетке, или в ткани или органе.
Предпочтительно количество модулятора C5a рецептора, контактирующего с рецептором, должно быть достаточно для ингибирования связывания C5a с C5a рецептором in vitro, и оно измеряется, например, с применением радиолигандного анализа, анализа мобилизации кальция или анализа хемотаксиса, как описано в настоящем тексте.
В одном варианте осуществления настоящего изобретения C5a модуляторы по настоящему изобретению используются для модулирования, предпочтительно ингибирования, активности передачи сигнала C5a рецептора, например посредством контактирования одного или более соединений по настоящему изобретению с C5a рецептором (in vitro или in vivo) в условиях, подходящих для связывания модуляторов с рецептором. Рецептор может находиться в растворе или суспензии, в препаратах выращенных или выделенных клеток, или в пациенте. Любое модулирование активности передачи сигнала можно оценить путем детектирования влияния на мобилизацию ионов кальция или путем детектирования влияния на C5a рецептор-опосредуемый клеточный хемотаксис. В целом, эффективное количество C5a модулятора(ов) - это количество, достаточное для модулирования активности передачи сигнала C5a рецептора in vitro в анализе мобилизации кальция, или C5a рецептор-опосредуемого клеточного хемотаксиса в анализе миграции.
Когда соединения по настоящему изобретению используются для ингибирования C5a рецептор-опосредуемого клеточного хемотаксиса, предпочтительно хемотаксиса лейкоцитов (например, нейтрофилов), в in vitro анализе хемотаксиса, такие методы включают контактирование белых кровяных телец (в частности, белых кровяных телец приматов, в особенности белых кровяных телец человека) с одним или более соединениями по настоящему изобретению. Предпочтительно концентрация достаточна для ингибирования хемотаксиса белых кровяных телец в in vitro анализе хемотаксиса, так что уровень хемотаксиса, наблюдаемый в контрольном опыте, значительно выше, чем уровень, наблюдаемый в анализе с добавлением соединения по настоящему изобретению.
В другом варианте осуществления соединения по настоящему изобретению могут также применяться для лечения пациентов, страдающих от патологических состояний, чувствительных к модулированию рецептора C5a. При использовании в настоящем тексте термин "лечение" охватывает как лечение, модифицирующее заболевание, так и симптоматическое лечение, любое из которых может быть профилактическим (т.е. до появления симптомов, для предотвращения, задержки или уменьшения степени тяжести симптомов) или терапевтическим (т.е. после появления симптомов, для уменьшения степени тяжести и/или длительности симптомов). При использовании в настоящем тексте состояние считается «чувствительным к модулированию рецептора C5a», если модулирование активности рецептора C5a приводит к снижению ненормальной активности рецептора C5a. При использовании в настоящем тексте термин "пациенты" включает приматов (в особенности людей), домашних животных-компаньонов (таких как собаки, кошки, лошади и т.п.) и сельскохозяйственных животных (таких как крупный рогатый скот, свиньи, овцы и т.п.), где дозировки соответствуют описанным в настоящем тексте.
Состояния, которые можно лечить модулированием C5a:
Аутоиммунные заболевания - например, ревматоидный артрит, системная красная волчанка, синдром Гийена-Барре, панкреатит, волчаночный нефрит, волчаночный гломерулонефрит, псориаз, болезнь Крона, васкулит, синдром раздраженного кишечника, дерматомиозит, рассеянный склероз, бронхиальная астма, болезнь плотного осадка, пемфигус, пемфигоид, склеродерма, миастения гравис, аутоиммунные гемолитические и тромбоцитопенические состояния, синдром Гудпасчера (и связанные с ним гломерулонефрит и легочное кровоточение), C3-гломерулопатия, C3-гломерулонефрит, мезангиопролиферативный гломерулонефрит, болезнь Кавасаки, ИГ нефропатия, иммуноваскулит, отторжение тканевого трансплантата, реакция «трансплантат против хозяина», сверхострое отторжение пересаженных органов; и т.п.
Воспалительные расстройства и родственные патологические состояния - например, нейтропения, сепсис, септический шок, болезнь Альцгеймера, рассеянный склероз, нейтрофилия, инсульт, воспалительное заболевание кишечника (ВЗК), воспаление вследствие сильных ожогов, повреждение легких и ишемически-реперфузионное повреждение, остеоартрит, а также острый синдром расстройства дыхания (у взрослых) (ARDS), хроническая обструктивная болезнь легких (ХОБЛ), синдром системной воспалительной реакции (SIRS), атопический дерматит, псориаз, хроническая уртикария и синдром полиорганной недостаточности (MODS), гемолитико-уремический синдром, атипичный гемолитико-уремический синдром (aHUS). Также включены патологические осложнения, связанные с инсулинозависимым сахарным диабетом (включая диабетическую ретинопатию), волчаночная нефропатия, нефрит Хеймана, мембранный нефрит и другие формы гломерулонефрита, контактный сензитивный ответ, и воспаление вследствие контакта крови с искусственными поверхностями, что может вызывать активацию комплемента, например во время экстракорпорального кровообращения (например, во время гемодиализа или через аппарат для сердечно-легочной реанимации, например, в связи с сосудистой хирургией, такой как аорто-коронарное шунтирование или замена сердечного клапана), или в связи с контактом с поверхностью других искусственных сосудов или контейнеров (например, устройство поддержки желудочка, аппарат «искусственное сердце», трубки для переливания, мешки для хранения крови, плазмафарез, тромбофарез и т.п.). Также включены заболевания, родственные ишемически-реперфузионному повреждению, такие как возникающие вследствие пересадки трансплантатов, включая пересадку солидных органов, и синдромы, такие как ишемически-реперфузионное повреждение, ишемический колит и ишемия сердца. Соединения по настоящему изобретению могут также применяться в лечении возрастной дегенерации желтого пятна (Hageman et al, P.N.A.S,102: 7227-7232, 2005).
Сердечно-сосудистые и церебро-сосудистые нарушения - например, инфаркт миокарда, тромбоз коронарных артерий, закупорка сосудов, постоперационная реокклюзия сосудов, атеросклероз, травматическое повреждение центральной нервной системы и ишемическая болезнь сердца. В одном варианте осуществления эффективное количество соединения по настоящему изобретению можно вводить пациенту, у которого есть риск развития инфаркта миокарда или тромбоза (например, у которого присутствуют один или больше признанных фактора риска для инфаркта миокарда или тромбоза, такие как (но не ограничиваясь только ими) ожирение, курение, высокое кровяное давление, гиперхолестеринемия, более ранние случаи или наследственные случаи инфаркта миокарда или тромбоза для снижения риска инфаркта миокарда или тромбоза.
Онкологические заболевания или нарушения - например, меланома, рак легких, лимфома, саркома, карцинома, фибросаркома, липосаркома, хондросаркома, остеогенная саркома, ангиосаркома, лимфангиосаркома, синовиома, мезотелиома, менингиома, лейкемия, лимфома, лейомиосаркома, рабдомиосаркома, плоскоклеточная карцинома, базальноклеточная карцинома, аденокарцинома, папиллярная карцинома, цистаденокарцинома, бронхогенная карцинома, почечноклеточный рак, гепатоцеллюлярная карцинома, переходно-клеточный рак, хориокарцинома, семинома, эмбриональная карцинома, опухоль Вильма, плеоморфная аденома, папиллома клеток печени, аденома канальцев почек, цистаденома, папиллома, аденома, лейомиома, рабдомиома, гемангиома, лимфангиома, остеома, хондрома, липома и фиброма.
Васкулитные заболевания - Васкулитные заболевания характеризуются воспалением сосудов. Инфильтрация лейкоцитов приводит к разрушению стенки сосудов, и считается, что активность комплемента играет важную роль в инициировании миграции лейкоцитов, а также в образующемся повреждении в сайте воспаления (Vasculitis, Second Edition, Edited by Ball и Bridges, Oxford University Press, pp 47-53, 2008). Описанные в настоящей заявке соединения можно применять для лечения лейкокластического васкулита, АНЦА-ассоциированного васкулита (васкулита, при котором в крови определяются антинейтрофильные цитоплазматические антитела), иммунного васкулита, гранулематоза Вегенера, микроскопического полиангиита, синдрома Черджа-Стросса, пурпуры Шенлейна-Геноха, узелкового периартериита, быстропрогрессирующего гломерулонефрита (RPGN), криоглобулинемии, гигантоклеточного артериита (ГКА), болезни Бехчета и синдрома дуги аорты.
ВИЧ-инфекция и СПИД - описанные в настоящей заявке модуляторы рецептора C5a можно применять для подавления ВИЧ-инфекции, замедления развития СПИД или снижения степени тяжести симптомов ВИЧ-инфекции и СПИД.
Нейродегенеративные нарушения и связанные с ними заболевания - В других аспектах описанные в настоящей заявке антагонисты C5a можно применять для лечения болезни Альцгеймера, рассеянного склероза и угасания когнитивной функции, связанных с операциями в условиях искусственного кровообращения и похожими процедурами.
В одном варианте осуществления настоящего изобретения соединения по настоящему изобретению можно применять для лечения заболеваний, выбранных из группы, состоящей из сепсиса (и связанных с ним нарушений), ХОБЛ, ревматоидного артрита, волчаночного нефрита и рассеянного склероза.
Методы лечения, описанные в настоящем изобретении, включают введение пациенту эффективного количества одного или более соединений, описанных в настоящем тексте. Подходящие пациенты включают пациентов, страдающих заболеванием или расстройством или подверженных (т.е. профилактическое лечение) заболеванию или расстройству, указанным в настоящем тексте. Типичные пациенты для описанного в настоящем изобретении лечения включают млекопитающих, в частности приматов, в особенности людей. Другие подходящие пациенты включают домашних животных-компаньонов, таких как собаки, кошки, лошади и т.п., или сельскохозяйственных животных, таких как крупный рогатый скот, свиньи, овцы и т.п.
В целом, описанные в настоящем изобретении способы лечения включают введение пациенту эффективного количества одного или больше соединений, описанных в настоящем тексте. В предпочтительном варианте осуществления соединение (соединения) по настоящему изобретению предпочтительно вводят пациенту (например, человеку) перорально или наружно. Эффективное количество может представлять собой количество, достаточное для модулирования активности рецептора C5a, и/или количество, достаточное для уменьшения или ослабления симптомов у пациента. Предпочтительно вводимое количество достаточно для создания в плазме крови концентрации соединения (или его активного метаболита, если соединение представляет собой пролекарство) достаточно высокой для детектируемого ингибирования хемотаксиса белых кровяных телец (например, нейтрофилов) in vitro. Режимы лечения могут варьироваться в зависимости от применяемого соединения и конкретного состояния, подвергающегося лечению; для лечения большинства нарушений, предпочтительная частота введения составляет 4 раза в сутки или меньше. В целом, прием 2 раза в сутки более предпочтителен, и особенно предпочтительно введение 1 раз в сутки. Следует понимать, однако, что конкретный уровень дозировки и режим введения для каждого конкретного пациента зависит от ряда факторов, включая активность конкретного соединения, возраст, вес тела, общее состояние здоровья, пол, диета, время введения, способ введения, скорость выведения, комбинация лекарств (например, другие лекарственные средства, которые вводят пациенту) и степень тяжести конкретного заболевания, подвергающегося лечению, а также мнение лечащего врача. В целом, предпочтительно применение минимальной дозировки, достаточной для обеспечения эффективной терапии. За терапевтической эффективностью для пациентов можно следить с помощью медицинских или ветеринарных критериев, подходящих для конкретного состояния, подвергающегося лечению.
Дозировки порядка от примерно 0,1 мг до примерно 140 мг на килограмм веса тела в сутки могут применяться в лечении или профилактики состояний, включающих патогенную активность C5a (от примерно 0,5 мг до примерно 7 г на человека в сутки). Количество действующего вещества, которое можно комбинировать с носителями для производства однократной дозированной формы, варьируется в зависимости от конкретного пациента, подвергающегося лечению, и от применяемого способа введения. Дозированные готовые формы обычно содержат от примерно 1 мг до примерно 500 мг действующего вещества. Для соединений, которые вводят перорально, чрескожно, внутривенно или подкожно, предпочтительно, чтобы вводилось достаточное количество соединения для достижения концентрации в плазме крови от 5 нг (нанограмм)/мл до 10 мкг (микрограмм)/мл плазмы крови, более предпочтительно - достаточное количество соединения для достижения концентрации в плазме крови от 20 нг/мл до 1 мкг/мл плазмы крови, наиболее предпочтительно - достаточное количество соединения для достижения концентрации в плазме крови от 50 нг/мл до 200 нг/мл плазмы крови. Для прямого введения в синовиальную оболочку (для лечения артрита), следует вводить достаточное количество соединения для локального достижения примерно 1 микромолярной концентрации.
Частота введения также может варьироваться в зависимости от применяемого соединения и конкретного состояния, подвергающегося лечению. Однако, для лечения большинства заболеваний предпочтителен режим введения 4 раза в сутки, три раза в сутки или меньше, при этом особенно предпочтителен режим введения один раз в сутки или 2 раза в сутки. Следует понимать, однако, что конкретный уровень дозировки для каждого конкретного пациента зависит от ряда факторов, включая активность конкретного соединения, возраст, вес тела, общее состояние здоровья, пол, диета, время введения, способ введения, скорость выведения, комбинация лекарств (например, другие лекарственные средства, которые вводят пациенту), степень тяжести конкретного заболевания, подвергающегося лечению, и другие факторы, включая мнение лечащего врача.
Комбинированная терапия
Описанные в настоящем изобретении соединения могут применяться в комбинации с одним или более дополнительными терапевтическими средствами, которые используются в лечении, профилактике, подавлении или облегчении заболеваний или патологических состояний, для которых могут применяться соединения и композиции по настоящему изобретению. Такие одно или больше дополнительных терапевтических средств можно вводить способом и в количествах, в норме применяемых для них, одновременно или последовательно с соединением или композицией по настоящему изобретению. Когда соединение или композиция по настоящему изобретению применяется одновременно с одним или больше другими лекарственными средствами, предпочтительной является фармацевтическая композиция, содержащая такие другие лекарственные средства в дополнение к соединению или композиции по настоящему изобретению. Соответственно, фармацевтические композиции по настоящему изобретению включают такие, которые содержат также одно или больше других действующих веществ или терапевтических средств, в дополнение к соединению или композиции по настоящему изобретению.
Примерами одного или более дополнительных терапевтических средств являются кортикостероиды, стероиды, иммунодепрессанты, агонисты иммуноглобулина G, ингибиторы дипептидилпептидазы IV, антагонисты лимфоцитарного функционально-ассоциированного антигена-3, лиганды IL-2, ингибиторы лигандов IL-1 бета, ингибиторы IL2RA, стимуляторы гена HGF, антагонисты IL-6, антагонисты IL-5, стимуляторы альфа-1 антитрипсина, антагонисты каннабиноидного рецептора, ингибиторы гистондеацетилазы, ингибиторы AKT протеинкиназ, ингибиторы CD20, ингибиторы Abl тирозинкиназы, ингибиторы JAK тирозинкиназ, ингибиторы лигандов ФНО-альфа, модуляторы гемоглобина, антагонисты ФНО, ингибиторы протеасом, модуляторы CD3, ингибиторы Hsp 70 семейства, агонисты иммуноглобулина, антагонисты CD30, антагонисты тубулина, агонисты сфингозин-1-фосфатного рецептора, ингибиторы лиганда фактора роста соединительной ткани, ингибиторы каспазы, лиганды адренокортикотропного гормона, ингибиторы Btk тирозинкиназы, ингибиторы компонента системы комплемента C1s, агонисты рецептора эритропоэтина, ингибиторы стимуляторов B-лимфоцитов, ингибиторы циклинзависимой киназы 2, стимуляторы гликопротеинового лиганда P-селектина 1, ингибиторы mTOR, ингибиторы фактора элонгации 2, ингибиторы молекулы клеточной адгезии, агонисты фактора XIII, ингибиторы кальциневрина, агонисты иммуноглобулина G1, ингибиторы инозин-монофосфат-дегидрогеназы, ингибиторы компонента системы комплемента C1s, модуляторы тимидинкиназ, модуляторы CTLA-4, антагонисты рецептора ангиотензина II, модуляторы рецептора ангиотензина II, антагонисты рецептора 12А из суперсемейства ФНО, антагонисты CD52, ингибиторы аденозиндеаминазы, ингибиторы антиген дифференцировки Т-клеток CD6, лиганды FGF-7, ингибиторы дигидрооротат-дегидрогеназы, ингибиторы Syk тирозинкиназы, антагонисты рецептора интерферона I типа, ингибиторы интерферона альфа, ингибиторы фактора ингибирования миграции макрофагов, антагонисты интегрина альфа-V/бета-6, стимуляторы цистеинпротеазы, ингибиторы p38 MAP киназы, ингибиторы гена TP53, ингибиторы шига-подобного токсина I, стимуляторы фукозилтрансферазы 6, лиганды интерлейкина 22, ингибиторы гена IRS1, стимуляторы протеинкиназ C, ингибиторы протеинкиназы C альфа, антагонисты CD74, антагонисты FCGR2B, ингибиторы антигена T-клеток CD7, антагонисты CD95, стимуляторы N-ацетилманнозамин-киназы, лиганды кардиотропина-1, ингибиторы эластазы лейкоцитов, антагонисты лиганда CD40 рецептора, модуляторы лиганда CD40 рецептора, антагонисты IL-17, антагонисты TLR-2, ингибиторы маннан-связывающей лектин-зависимой серинпротеазы-2 (MASP-2), ингибиторы фактора B, ингибиторы фактора D, модуляторы C3aR, модуляторы C5aR2, антагонисты T-клеточного рецептора, ингибиторы PD-1, ингибиторы PD-L1, ингибиторы TIGIT, ингибиторы TIM-3, ингибиторы LAG-3, ингибиторы VISTA, агонисты STING, ингибиторы IDO, модуляторы аденозинового рецептора, ингибиторы CD39, ингибиторы CD73, антагонисты хемокиновых рецепторов, в особенности CXCR1, CXCR2, CXCR3, CXCR4, CXCR7, CCR1, CCR2, CCR3, CCR4, CCR5, CCR7, CCR7, CCR9, CX3CR1 и CXCR6, и их комбинации.
В некоторых вариантах осуществления дополнительное терапевтическое средство, применяющееся в описанных в настоящем тексте терапевтических методах, выбрано из группы, состоящей из следующих: обинутузумаб, ритуксимаб, окрелизумаб, циклофосфамид, преднизон, гидрокортизон, гидрокортизона ацетат, кортизона ацетат, тиксокортол пивалат, преднизолон, метилпреднизолон, триамцинолона ацетонид, триамцинолоновый спирт, мометазон, амцинонид, будесонид, десонид, флуоцинонид, флуоцинолона ацетонид, галцинонид, бетаметазон, бетаметазон натрия фосфат, дексаметазон, дексаметазон натрия фосфат, флуокортолон, гидрокортизон-17-валерат, галометазон, аклометазона дипропионат, беклометазон, бетаметазона валерат, бетаметазона дипропионат, предникарбат, клобетазон-17-бутират, клобетазол-17-пропионат, флуокортолона капроат, флуокортолона пивалат, флупреднидена ацетат, гидрокортизон-17-бутират, гидрокортизон-17-ацепонат, гидрокортизон-17-бутепрат, циклесонид и предникарбат, GB-0998, иммугло, бегеломаб, алефацепт, альдеслейкин, гевокизумаб, даклизумаб, базиликсумаб, инолимомаб, беперминоген перплазмид, сирукумаб, тоцилизумаб, клазакизумаб, меполизумаб, финголимод, панобиностат, трицирибин, нилотиниб, иматиниб, тофацитиниб, момелотиниб, пефицитиниб, итацитиниб, инфликсимаб, PEG-bHb-CO, этанерцепт, иксазомиб, бортезомиб, муромонаб, отеликсизумаб, гусперимус, брентуксимаб ведотин, понезимод, KRP-203, FG-3019, эмрикасан, кортикотропин, ибрутиниб, синрайз, конестат, метоксиполиэтиленгликоль-эпоэтин бета, белимумаб, блисибимод, атацицепт, селициклиб, нейхулизумаб, эверолимус, сиролимус, денилейкин дифитокс, LMB-2, натализумаб, катридекаког, циклоспорин, такролимус, воклоспорин, канакинумаб, микофенолят, мизорибин, CE-1145, TK-DLI, абатацепт, белатацепт, олмесартан медоксомил, спарсентан, TXA-127, BIIB-023, алемтузумаб, пентостатин, итолизумаб, палифермин, лефлуномид, PRO-140, ценикривирок, фостаматиниб, анифролумаб, сифалимумаб, BAX-069, BG-00011, лосмапимод, QPI-1002, ShigamAbs, TZ-101, F-652, репариксин, ладариксин, PTX-9908, аганирсен, APH-703, сотрастаурин, милатузумаб, SM-101, T-Guard, APG-101, DEX-M74, кардиотрофин-1, типрелестат, ASKP-1240, BMS-986004, HPH-116, KD-025, OPN-305, TOL-101, дефибротид, помалидомид, тимоглобулин, лаквинимод, реместемцел-L, лошадиный анти-тимоцитарный иммуноглобулин, стемпейцел, LIV-гамма, Октагам 10%, t2c-001, 99mTc-сестамиби, Clairyg, Просорба, помалидомид, лаквинимод, теплизумаб, FCRx, солнатид, форалумаб, ATIR-101, BPX-501, ACP-01, ALLO-ASC-DFU, ирбесартан + пропагерманий, ApoCell, каннабидиол, RGI-2001, саратин, конъюгат анти-CD3 бивалентное антитело-дифтерийный токсин, NOX-100, LT-1951, OMS721, ALN-CC5, ACH-4471, AMY-101, Acthar гель и CD4+CD25+ регуляторные T-клетки, MEDI7814, P32, P59, пембролизумаб, ниволумаб, атезолизумаб, авелумаб, дурвалумаб, CCX354, CCX721, CCX9588, CCX140, CCX872, CCX598, CCX6239, CCX587, CCX624, CCX282, CCX025, CCX507, CCX430, CCX765, CCX758, CCX771, CCX662, CCX650 и их комбинации.
Заболевание или нарушение, подлежащее лечению, определяет, какое дополнительное терапевтическое средство или терапевтические средства более всего подходят для введения в комбинации с соединениями по настоящему изобретению - это может определить квалифицированный специалист в данной области.
Весовое соотношение соединения по настоящему изобретению и второго действующего вещества может варьироваться и зависит от эффективной дозировки каждого ингредиента. Обычно применяют эффективную дозировку каждого ингредиента. Так, например, когда соединение по настоящему изобретению комбинируют с НПВП, весовое соотношение соединения по настоящему изобретению и НПВП обычно находится в диапазоне от примерно 1000:1 до примерно 1:1000, предпочтительно от примерно 200:1 до примерно 1:200. Комбинации соединения по настоящему изобретению и других действующих веществ обычно также находятся в указанных диапазонах, но в каждом случае следует применять эффективную дозировку каждого действующего вещества.
Нефармацевтическое применение
В другом аспекте настоящего изобретения соединения по настоящему изобретению можно применять в различных нефармацевтических in vitro и in vivo областях применения. Например, соединения по настоящему изобретению могут быть помечены и использованы в качестве зонда для детектирования и локализации C5a рецептора (препараты клеток или образцы срезов тканей). Соединения по настоящему изобретению можно также применять в качестве положительного контроля в тестах активности C5a рецептора, т.е. в качестве стандартов для определения способности кандидата связываться с C5a рецептором, или в качестве радиоактивно меченых трейсеров для позитрон-эмиссионной томографии (ПЭТ) или для однофотонной эмиссионной компьютерной томографии (SPECT). Такие методы можно применять для характеристики C5a рецепторов в живых субъектах. Например, модулятор C5a рецептора может быть помечен с помощью ряда хорошо известных методик (например, введение радионуклидной метки, такой как тритий) и инкубирован с образцом в течение надлежащего времени инкубирования (например, сначала определенного в ходе отслеживания динамики связывания). После инкубирования несвязанное соединение удаляют (например, промывкой), а связанное соединение детектируют с помощью любого метода, подходящего для применяемой метки (например, радиоавтография или измерение активности сцинтилляционным методом для радиоактивно-меченых соединений; для детектирования люминесцентных групп и флуоресцентных групп можно применять спектроскопические методы). Для контроля можно аналогичным образом тестировать соответствующий образец, содержащий меченое соединение и большее (например, в 10 раз большее) количество немеченого соединения. Большее количество детектируемой метки, оставшейся в тестируемом образце, чем в контрольном образце, указывает на присутствие C5a рецептора в образце. Детектирование, включая авторадиографию рецептора (картографирование рецептора) для C5a рецептора в культурах клеток или в образцах тканей можно проводить как описано Kuhar в разделах 8,1,1-8,1,9 в книге Current Protocols in Pharmacology (1998) John Wiley & Sons, New York.
Описанные в настоящем тексте соединения можно также применять в различных хорошо известных методах разделения клеток. Например, модуляторы можно связать с внутренней поверхностью планшета для выращивания клеток или с другой подложкой, для применения в качестве аффинных лигандов для иммобилизации и соответственно выделения C5a рецепторов (например, для выделения рецептор-экспрессирующих клеток) in vitro. В одной предпочтительной области применения модулятор, связанный с флуоресцентным маркером, таким как флуоресцеин, контактирует с клетками, которые затем анализируют (или выделяют) методом сортировки флуоресцентно-активированных клеток (FACS).
- Примеры
Следующие далее примеры приведены для иллюстрации, а не для ограничения объема настоящего изобретения.
Описанные ниже реагенты и растворители могут быть получены из коммерческих источников, таких как Aldrich Chemical Co. (Milwaukee, Wisconsin, USA). 1H-ЯМР спектры записывали на Varian Mercury 400 МГц ЯМР-спектрометре. Значения хим.сдвигов приведены относительно тетраметилсилана (ТМС) и описаны в порядке: мультиплетность (с, синглет; д, дублет; т, триплет; кв, квартет; м, мультиплет) и число протонов. Результаты масс-спектрометрии приведены в виде значения массы, деленной на заряд, и далее относительная интенсивность каждого иона (в скобках). В примерах приведено одно значение m/e для каждого M+H (или, если указано, M-H) иона, содержащего наиболее распространенные изотопы атомов. Во всех случаях изотопное распределение соответствует ожидаемой формуле. Масс-спектрометрию с ионизацией электрораспылением (ESI) проводили на масс-спектрометре Hewlett-Packard MSD с ионизацией электрораспылением, оснащенном ВЭЖХ HP1100 для ввода образца. Обычно аналит растворяли в метаноле в концентрации 0,1 мг/мл, и 1 микролитр полученного раствора вводили в масс-спектрометр, сканирующий ионы в диапазоне от 100 до 1500 дальтон. Все соединения анализировали в режиме ESI с регистрацией положительных ионов, используя смесь ацетонитрил/вода с 1% муравьиной кислоты в качестве растворителя. Описанные ниже соединения можно анализировать в режиме ESI с регистрацией отрицательных ионов, используя 2 мМ раствор NH4OAc в смеси ацетонитриле/вода в качестве растворителя.
В Примерах и в остальном тексте заявки применяются следующие сокращения:
EtOH: Этанол
EtONa: Этоксид натрия
ТГФ: Тетрагидрофуран
ТСХ: Тонкослойная хроматография
MeOH: Метанол
Соединения по настоящему изобретению можно синтезировать как описано ниже, применяя разнообразные реакции, известные квалифицированному специалисту в данной области техники. Квалифицированному специалисту в данной области будет также понятно, что могут применяться и альтернативные методы синтеза целевых соединений, и что подходы, используемые в тексте настоящего документа, не являются исчерпывающими, но дают работающие и практичные способы получения соединений по настоящему изобретению.
Некоторые молекулы, заявленные в настоящем патенте, могут существовать в различных энантиомерных и диастереомерных формах, и все такие варианты соединений входят в объем настоящего изобретения.
Подробное описание экспериментальных методик, используемых для синтеза ключевых соединений в настоящем тексте, ведет к молекулам, которые описаны идентифицирующими их физическими данными, а также описывающими их изображениями химической структуры.
Квалифицированным специалистам в данной области техники будет также понятно, что во время стандартных методик обработки в органической химии часто применяются кислоты и основания. Иногда во время описанных в настоящем тексте экспериментальных процедур образуются соли родительских соединений, если они обладают необходимой собственной кислотностью или основностью.
Синтез интермедиата 1: 3-(6-хлор-7-метокси-1H-индол-4-ил)-2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин 
Осторожно: Формирование диазониевых соединений потенциально опасно, пожалуйста, соблюдайте осторожность и используйте надлежащие средства персональной защиты!
Стадия a: В 250-миллилитровую колбу, содержащую 90 мл концентрированной соляной кислоты, при перемешивании на магнитной мешалке добавляли 2,6-диэтиланилин (10,0 г, 67,0 ммоль). Реакционную смесь перемешивали 30 минут и охлаждали на бане лед/соль до внутренней температуры в колбе -5°C. Медленно добавляли в полученную смесь раствор нитрита натрия (5,5 г, 80,0 ммоль) в воде (60 мл), поддерживая внутреннюю температуру ниже 5°C.
Отдельно при механическом перемешивании добавляли дигидрат хлорид олова(II) (31,6 г, 140 ммоль) в 500-миллилитровую 3-горлую круглодонную колбу, в которую предварительно наливали концентрированную соляную кислоту (60 мл). Полученный раствор затем охлаждали в ледяной бане.
Суспензию диазониевой соли затем фильтровали в 500-миллилитровую колбу, содержащую охлажденный раствор хлорида олова, при интенсивном перемешивании. Через 90 минут реакционную смесь переносили в 500-миллилитровую колбу Эрленмейера, и колбу промывали водой (20 мл) и хлороформом (8 мл). Объединенную смесь перемешивали в течение ночи при комнатной температуре. Весь жидкий слой декантировали, получая влажное твердое вещество. Полученное вещество сушили в вакууме один день и затем переносили в 500-миллилитровую трехгорлую круглодонную колбу, оснащенную верхнеприводной механической мешалкой, и перемешивали с эфиром (180 мл). Полученную смесь охлаждали в ледяной бане и медленно добавляли раствор NaOH (10н., 30 мл) в полученную смесь, поддерживая внутреннюю температуру ниже 12°C. После добавления смесь оставляли на 2 часа в ледяной бане. Эфирный слой декантировали в 500-миллилитровую колбу и пропускали через этот эфирный раствор поток хлороводорода при перемешивании. Выпавший осадок отделяли фильтрованием, получая (2,6-диэтилфенил)гидразин гидрохлорид. МС: (ES) m/z вычислено для C10H17N2 [M+H]+ 165,1, найдено 165,1.
Стадия b: N,N-диизопропилэтиламин (8 мл, 46,0 ммоль) добавляли в смесь (2,6-диэтилфенил)гидразина гидрохлорида (8 г, 39,9 ммоль), трет-бутил 3-циано-4-оксопиперидин-1-карбоксилата (5 г, 22,3 ммоль) и EtOH (60 мл) в 250-миллилитровой круглодонной колбе при перемешивании на магнитной мешалке. Полученную смесь перемешивали при кипячении в течение 3 часов. Добавляли ледяную уксусную кислоту (12 мл, 208 ммоль), и смесь перемешивали при кипячении еще 2 часа. Растворитель удаляли при пониженном давлении, остаток растворяли в EtOAc и промывали раствором NaOH (2н.), насыщенным раствором хлорида натрия и сушили над MgSO4. Растворитель удаляли при пониженном давлении, и остаток очищали методом флэш-хроматографии на силикагеле (5-55% EtOAc в гексане), получая трет-бутил 3-амино-2-(2,6-диэтилфенил)-6,7-дигидро-2H-пиразоло[4,3-c]пиридин-5(4H)-карбоксилат. МС: (ES) m/z вычислено для C21H31 N4O2 [M+H]+ 371,2, найдено 371,2.
Осторожно: Формирование диазониевых соединений потенциально опасно, пожалуйста, соблюдайте осторожность и используйте надлежащие средства персональной защиты!
Изопентил нитрит (4 мл, 28,6 ммоль) медленно добавляли при комнатной температуре в смесь трет-бутил 3-амино-2-(2,6-диэтилфенил)-6,7-дигидро-2H-пиразоло[4,3-c]пиридин-5(4H)-карбоксилата (3 г, 8,1 ммоль), CuBr (4 г, 27,9 ммоль) и MeCN (50 мл) в 250-миллилитровой круглодонной колбе при перемешивании на магнитной мешалке. Полученную смесь перемешивали при комнатной температуре 1 час, разбавляли этилацетатом, фильтровали через целит, промывали насыщенным раствором NH4Cl и сушили над MgSO4. Растворитель удаляли при пониженном давлении, и остаток очищали методом флэш-хроматографии на силикагеле (2-25% EtOAc в гексане), получая трет-бутил 3-бром-2-(2,6-диэтилфенил)-6,7-дигидро-2H-пиразоло[4,3-c]пиридин-5(4H)-карбоксилат. МС: (ES) m/z вычислено для C21H29BrN3O2 [M+H]+ 434,1, найдено 434,2.
Стадия c: Иодметан (1,5 мл, 24 ммоль) добавляли в суспензию 4-бром-2-хлор-6-нитрофенола (3,2 г, 12,7 ммоль) и K2CO3 (3 г, 21,7 ммоль) в ДМФА (40 мл) в 250-миллилитровой круглодонной колбе при перемешивании на магнитной мешалке. Полученную смесь перемешивали при 45°C в течение 4 часов, разбавляли этилацетатом, промывали насыщенным раствором хлорида натрия и сушили над MgSO4. Растворитель удаляли при пониженном давлении, и остаток очищали методом флэш-хроматографии на силикагеле (2-25% EtOAc в гексане), получая 5-бром-1-хлор-2-метокси-3-нитробензол. МС: (ES) m/z вычислено для C7H6BrClNO3 [M+H]+ 265,9, найдено 265,9.
Раствор винилмагний бромида в ТГФ (1 M, 40 мл, 40 ммоль) быстро добавляли в раствор 5-бром-1-хлор-2-метокси-3-нитробензола (3,2 г, 12 ммоль) в безводном ТГФ (40 мл) в атмосфере азота и при интенсивном перемешивании при -60°C. Реакционную смесь оставляли нагреваться до -30°C в течение 1,5 часов. Реакцию гасили насыщенным водным раствором NH4Cl, и смесь оставляли нагреваться до комнатной температуры в течение 1 часа. Реакционную смесь разбавляли этилацетатом, промывали насыщенным раствором хлорида натрия и сушили над MgSO4. Растворитель удаляли при пониженном давлении, и остаток очищали методом флэш-хроматографии на силикагеле (2-20% EtOAc в гексане), получая 4-бром-6-хлор-7-метокси-1H-индол. МС: (ES) m/z вычислено для C9H8BrClNO [M+H]+ 259,9, найдено 259,9.
В суспензию 4-бром-6-хлор-7-метокси-1H-индола (1,2 г, 4,6 ммоль), 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолана) (2,4 г, 9,5 ммоль) и KOAc (2,3 г, 23,4 ммоль) в ДМСО (10 мл) добавляли комплекс Pd(dppf)Cl2 с дихлорметаном (600 мг, 0,73 ммоль). Реакционную смесь дегазировали (N2) 2 минуты и перемешивали при 120°C в течение 2 часов. Реакционную смесь разбавляли этилацетатом, фильтровали через целит, промывали насыщенным раствором хлорида натрия и сушили над MgSO4. Растворитель удаляли при пониженном давлении, и остаток очищали методом флэш-хроматографии на силикагеле (5-20% EtOAc в гексане), получая 6-хлор-7-метокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-индол. МС: (ES) m/z вычислено для C15H20BClNO3 [M+H]+ 308,1, найдено 308,1.
В суспензию трет-бутил 3-бром-2-(2,6-диэтилфенил)-6,7-дигидро-2H-пиразоло[4,3-c]пиридин-5(4H)-карбоксилата (600 мг, 1,4 ммоль), 6-хлор-7-метокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-индола (550 мг, 1,8 ммоль) и K2CO3 (500 мг, 3,6 ммоль) в п-диоксане (6 мл) и H2O (1 мл) добавляли комплекс Pd(dppf)Cl2 с дихлорметаном (300 мг, 0,37 ммоль). Реакционную смесь дегазировали (N2) 2 минуты и перемешивали в атмосфере азота при 100°C в течение 2 часов. Реакционную смесь разбавляли этилацетатом, фильтровали через целит, промывали насыщенным раствором хлорида натрия и сушили над MgSO4. Растворитель удаляли при пониженном давлении, и остаток очищали методом флэш-хроматографии на силикагеле (5-20% EtOAc в гексане), получая трет бутил 3-(6-хлор-7-метокси-1H-индол-4-ил)-2-(2,6-диэтилфенил)-6,7-дигидро-2H-пиразоло[4,3-c]пиридин-5(4H)-карбоксилат. МС: (ES) m/z вычислено для C30H36ClN4O3 [M+H]+ 535,2,1, найдено 535,2.
Описанный выше трет-бутил 3-(6-хлор-7-метокси-1H-индол-4-ил)-2-(2,6-диэтилфенил)-6,7-дигидро-2H-пиразоло[4,3-c]пиридин-5(4H)-карбоксилат растворяли в дихлорметане (5 мл) и добавляли 4н. раствор HCl в диоксане (5 мл, 20 ммоль). Полученную смесь перемешивали при комнатной температуре 2 часа. После окончания реакции, растворитель упаривали в вакууме, получая 3-(6-хлор-7-метокси-1H-индол-4-ил)-2-(2,6-диэтилфенил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин гидрохлорид. МС: (ES) m/z вычислено для C25H29ClN4O [M+H]+ 435,2, найдено 435,2.
Стадия d: N,N-диизопропилэтиламин (0,2 мл, 1,15 ммоль) добавляли в суспензию 3-(6-хлор-7-метокси-1H-индол-4-ил)-2-(2,6-диэтилфенил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридина гидрохлорида (100 мг, 0,21 ммоль), 2-хлор-5-(трифторметил)пиримидина (45 мг, 0,25 ммоль) и Li2CO3 (20 мг, 0,27 ммоль) в MeCN (10 мл) при перемешивании на магнитной мешалке. Полученную смесь перемешивали при 75°C в течение 30 минут. После охлаждения до комнатной температуры реакционную смесь разбавляли этилацетатом, промывали насыщенным раствором хлорида натрия и сушили над MgSO4. Растворитель удаляли при пониженном давлении, и остаток очищали методом препаративной ТСХ (40% EtOAc в гексане), затем растирали в MeOH, получая 3-(6-хлор-7-метокси-1H-индол-4-ил)-2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин. 1H ЯМР (400 МГц, CDCl3) δ 8,48 (с, 2H), 8,46 (с, 1H), 7,06-7,27 (м, 4H), 6,62 (д, J=1,0 Гц, 1H), 6,42-6,49 (м, 1H), 4,83 (с, 2H), 4,36 (т, J=5,7 Гц, 2H), 4,00 (с, 3H), 3,03 (т, J=5,7 Гц, 2H), 2,10-2,40 (м, 4H), 0,80-1,08 (м, 6H). МС: (ES) m/z вычислено для C30H29ClF3N6O [M+H]+ 581,2, найдено 581,2.
Синтез интермедиата 2: [4-[2-(2,6-диэтилфенил)-5-[5-(трифторметил)пиримидин-2-ил]-6,7-дигидро-4H-пиразоло[4,3-c]пиридин-3-ил]-5-фтор-1H-индол-7-ил]метанол
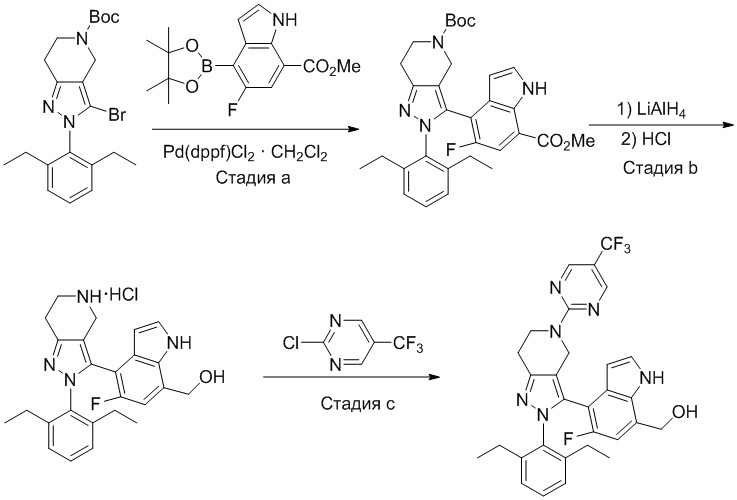
Стадия a: Раствор винилмагний бромида в ТГФ (1M, 341 мл, 341 ммоль) добавляли в раствор 4-бром-5-фтор-2-нитробензойной кислоты (15,0 г, 56,8 ммоль) в безводном ТГФ (200 мл) в атмосфере азота при -50°C. Реакционную смесь перемешивали при той же температуре и оставляли нагреваться до -40°C в течение 1,5 часов. Реакционную смесь гасили насыщенным водным раствором NH4Cl и оставляли нагреваться до комнатной температуры в течение 1 часа. Реакционную смесь подкисляли 1н. водным раствором HCl, разбавляли этилацетатом, промывали насыщенным раствором хлорида натрия и сушили над Na2SO4. Растворитель удаляли при пониженном давлении, получая неочищенный остаток.
Описанный выше неочищенный остаток перемешивали в смеси H2SO4 (25 мл) и MeOH (250 мл) при кипячении 5 часов. Затем охлаждали до комнатной температуры и упаривали при пониженном давлении. Полученный остаток разбавляли этилацетатом и насыщенным раствором хлорида натрия. Органический слой отделяли, сушили над Na2SO4, упаривали при пониженном давлении и очищали методом флэш-хроматографии на силикагеле (0-50% EtOAc в гексане), получая метил 4-бром-5-фтор-1H-индол-7-карбоксилат. МС: (ES) m/z вычислено для C10H8BrFNO2 [M+H]+ 271,9, найдено 271,9.
В суспензию метил 4-бром-5-фтор-1H-индол-7-карбоксилата (0,900 г, 3,3 ммоль), 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолана) (1,51 г, 5,94 ммоль) и KOAc (1,62 г, 16,5 ммоль) в ДМСО (19 мл) добавляли комплекс Pd(dppf)Cl2 с дихлорметаном (400 мг, 0,49 ммоль). Реакционную смесь дегазировали (N2) 2 минуты и перемешивали при 115°C в течение 1,5 часов. Реакционную смесь разбавляли этилацетатом, фильтровали через целит, промывали насыщенным раствором хлорида натрия и сушили над Na2SO4. Растворитель удаляли при пониженном давлении, и остаток очищали методом флэш-хроматографии на силикагеле (0-100% CH2Cl2/гексан), получая метил 5-фтор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-индол-7-карбоксилат. МС: (ES) m/z вычислено для C16H20BFNO4 [M+H]+ 320,1, найдено 320,1.
В суспензию трет-бутил 3-бром-2-(2,6-диэтилфенил)-6,7-дигидро-2H-пиразоло[4,3-c]пиридин-5(4H)-карбоксилата (1,00 г, 2,31 ммоль), метил 5-фтор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-индол-7-карбоксилата (740 мг, 2,31 ммоль) и K2CO3 (1,28 г, 9,24 ммоль) в п-диоксане (14 мл) и воде (2,5 мл) добавляли комплекс Pd(dppf)Cl2 с дихлорметаном (400 мг, 0,49 ммоль). Реакционную смесь дегазировали (N2) 2 минуты и перемешивали в атмосфере азота при 100°C в течение 2,5 часов. Реакционную смесь разбавляли этилацетатом, промывали водным раствором NaHCO3 и сушили над Na2SO4. Растворитель удаляли при пониженном давлении, и остаток очищали методом флэш-хроматографии на силикагеле (0-70% EtOAc в гексане), получая трет-бутил 2-(2,6-диэтилфенил)-3-(5-фтор-7-метоксикарбонил-1H-индол-4-ил)-6,7-дигидро-4H-пиразоло[4,3-c]пиридин-5-карбоксилат. МС: (ES) m/z вычислено для C31H36FN4O4 [M+H]+ 547,2, найдено 547,2.
Стадия b: Описанный выше трет-бутил 2-(2,6-диэтилфенил)-3-(5-фтор-7-метоксикарбонил-1H-индол-4-ил)-6,7-дигидро-4H-пиразоло[4,3-c]пиридин-5-карбоксилат (1,00 г, 1,83 ммоль) растворяли в ТГФ (35 мл) и добавляли раствор LiAlH4 в эфире (1M, 2,7 мл) при 0°C. Полученную смесь перемешивали при 0°C в течение 40 минут. Затем гасили водой, разбавляли смесью IPA/CHCl3 (1:3), промывали насыщенным раствором хлорида натрия и сушили над Na2SO4. Растворитель удаляли при пониженном давлении, и остаток очищали методом флэш-хроматографии на силикагеле (0-90% EtOAc в гексане), получая трет-бутил 2-(2,6-диэтилфенил)-3-[5-фтор-7-(гидроксиметил)-1H-индол-4-ил]-6,7-дигидро-4H-пиразоло[4,3-c]пиридин-5-карбоксилат. МС: (ES) m/z вычислено для C30H36FN4O3 [M+H]+ 519,2, найдено 519,2.
Описанный выше трет-бутил 2-(2,6-диэтилфенил)-3-(5-фтор-7-(гидроксиметил)-1H-индол-4-ил]-6,7-дигидро-4H-пиразоло[4,3-c]пиридин-5-карбоксилат (650 мг, 1,25 ммоль) растворяли в дихлорметане (13 мл) и добавляли 4н. раствор HCl в диоксане (35 мл, 140 ммоль). Полученную смесь перемешивали при комнатной температуре 1,5 часа. После окончания реакции растворитель упаривали в вакууме, получая [4-[2-(2,6-диэтилфенил)-4,5,6,7-тетрагидропиразоло[4,3-c]пиридин-3-ил]-5-фтор-1H-индол-7-ил]метанол гидрохлорид. МС: (ES) m/z вычислено для C25H28FN4O [M+H]+ 419,2, найдено 419,2.
Стадия c: Триэтиламин (1,50 мл, 10,7 ммоль) добавляли в суспензию [4-[2-(2,6-диэтилфенил)-4,5,6,7-тетрагидропиразоло[4,3-c]пиридин-3-ил]-5-фтор-1H-индол-7-ил] метанола гидрохлорида (600 мг, 1,32 ммоль) и 2-хлор-5-(трифторметил)пиримидина (350 мг, 1,9 ммоль) в MeCN (70 мл). Полученную смесь перемешивали при 80°C в течение 30 минут. После охлаждения до комнатной температуры реакционную смесь разбавляли этилацетатом, промывали водным раствором NaHCO3 и сушили над Na2SO4. Растворитель удаляли при пониженном давлении, и остаток очищали методом флэш-хроматографии на силикагеле (0-90% EtOAc в гексане), получая [4-[2-(2,6-диэтилфенил)-5-[5-(трифторметил)пиримидин-2-ил]-6,7-дигидро-4H-пиразоло[4,3-c]пиридин-3-ил]-5-фтор-1H-индол-7-ил]метанол. 1H ЯМР (400 МГц, CDCl3) δ 9,05 (ушир.с, 1H), 8,47 (ушир.с, 2 H), 7,27 (м, 1H), 7,16 (м, 2H), 6,86 (д, J=7,26 Гц, 1H), 6,56 (д, J=10,0 Гц, 1H), 6,37 (т, J=2,6 Гц, 1H), 4,88 (м, 3H), 4,68 (д, J=16,4 Гц, 1H), 4,43 (м, 1H), 4,29 (м, 1H), 3,04 (т, J=6,0 Гц, 2H), 2,38-2,58 (м, 3H), 2,17 (секстет, J=7,3 Гц, 1H), 1,94 (секстет, J=7,3 Гц, 1H), 1,21 (т, J=7,4 Гц, 3H), 0,75 (т, J=7,4 Гц, 3H). МС: (ES) m/z вычислено для C30H29F4N6O [M+H]+ 565,2, найдено 565,2.
Синтез интермедиата 3: 1-(4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-2,5-дифторфенил)мочевина
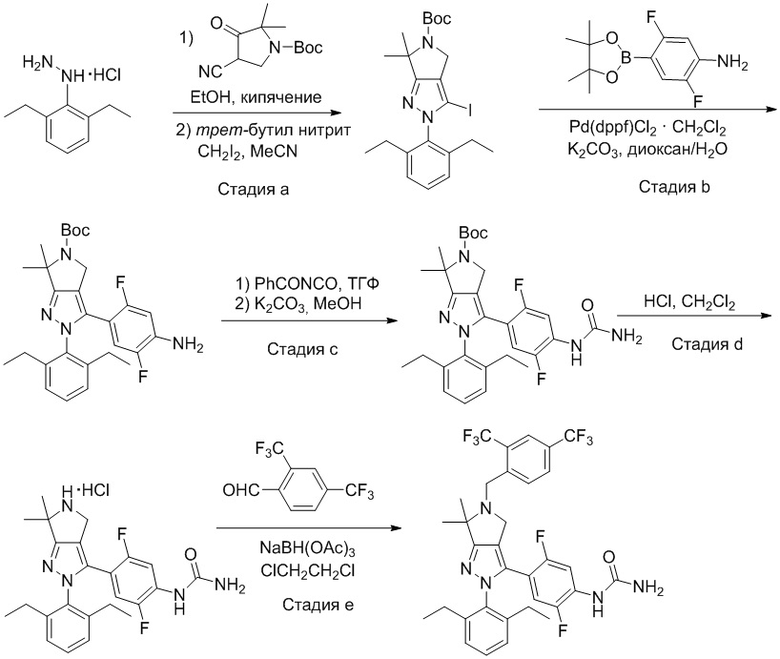
Стадия a: Пиридин (4,0 мл, 49,5 ммоль) добавляли в смесь (2,6-диэтилфенил) гидразина гидрохлорида (5,0 г, 24,9 ммоль), трет-бутил 4-циано-2,2-диметил-3-оксопирролидин-1-карбоксилата (5,0 г, 21,0 ммоль) и EtOH (60 мл) в 250-миллилитровой круглодонной колбе при перемешивании на магнитной мешалке. Полученную смесь перемешивали при 70°C в течение 24 часов. Растворитель удаляли при пониженном давлении, остаток разбавляли этилацетатом и промывали водным раствором лимонной кислоты, насыщенным водным раствором NaHCO3, насыщенным водным раствором хлорида натрия и сушили над MgSO4. Растворитель удаляли при пониженном давлении, и остаток кристаллизовали из циклогексана, получая трет-бутил 3-амино-2-(2,6-диэтилфенил)-6,6-диметил-2,6-дигидропирроло[3,4-c]пиразол-5(4H)-карбоксилат. МС: (ES) m/z вычислено для C22H33N4O2 [M+H]+ 385,2, найдено 385,2.
Осторожно: Формирование диазониевых соединений потенциально опасно, пожалуйста, соблюдайте осторожность и используйте надлежащие средства персональной защиты!
Трет-бутилнитрит (0,5 мл, 3,8 ммоль) медленно добавляли при комнатной температуре в смесь трет-бутил 3-амино-2-(2,6-диэтилфенил)-6,6-диметил-2,6-дигидропирроло[3,4-c]пиразол-5(4H)-карбоксилата (1 г, 2,6 ммоль), дииодметана (1,5 мл, 18,6 ммоль) и MeCN (15 мл) в 100-миллилитровой круглодонной колбе при перемешивании на магнитной мешалке. Полученную смесь перемешивали при 45°C в течение 3 часов, затем разбавляли толуолом, промывали насыщенным раствором NH4Cl/NH4OH (3:1), насыщенным водным раствором хлорида натрия и сушили над MgSO4. Растворитель удаляли при пониженном давлении, и остаток очищали методом флэш-хроматографии на силикагеле (от 2 до 25% EtOAc в гексане), получая трет-бутил 2-(2,6-диэтилфенил)-3-иод-6,6-диметил-2,6-дигидропирроло[3,4-c]пиразол-5(4H)-карбоксилат. МС: (ES) m/z вычислено для C22H31IN3O2 [M+H]+ 496,1, найдено 496,2.
Стадия b: Смесь 4-бром-2,5-дифторанилина (1,5 г, 7,2 ммоль), 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолана) (2,2 г, 8,7 ммоль), KOAc (1,8 г, 18,3 ммоль) и комплекса Pd(dppf)Cl2 с дихлорметаном (580,0 мг, 0,7 ммоль) в диоксане (12 мл) перемешивали при 95°C в течение 2 часов в атмосфере азота. Полученную смесь затем охлаждали до комнатной температуры и фильтровали через целит. Фильтрат собирали, упаривали при пониженном давлении и очищали методом флэш-хроматографии на силикагеле (от 0 до 50% EtOAc в гексане), получая 2,5-дифтор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилин. МС: (ES) m/z вычислено для C12H17BF2NO2 [M+H]+ 256,1, найдено 256,2.
В суспензию трет-бутил 2-(2,6-диэтилфенил)-3-иод-6,6-диметил-2,6-дигидропирроло[3,4-c]пиразол-5(4H)-карбоксилата (0,7 г, 1,4 ммоль), 2,5-дифтор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилина (0,7 г, 2,7 ммоль), K2CO3 (1,3 г, 7,2 ммоль) в диоксане (10 мл) и воде (2 мл) добавляли комплекс Pd(dppf)Cl2 с дихлорметаном (300,0 мг, 0,37 ммоль). Реакционную смесь дегазировали (N2) в течение 2 минут и перемешивали в атмосфере N2 при 100°C в течение 2 часов. Реакционную смесь разбавляли этилацетатом, фильтровали через целит, промывали насыщенным раствором хлорида натрия, сушили над MgSO4 и фильтровали. Растворитель упаривали при пониженном давлении, и остаток очищали методом флэш-хроматографии на силикагеле (от 2 до 10% EtOAc в гексане), получая трет-бутил 3-(4-амино-2,5-дифторфенил)-2-(2,6-диэтилфенил)-6,6-диметил-2,6-дигидропирроло[3,4-c]пиразол-5(4H)-карбоксилат. МС: (ES) m/z вычислено для C28H35F2N4O2 [M+H]+ 497,3, найдено 497,5.
Стадия c: Смесь трет-бутил 3-(4-амино-2,5-дифторфенил)-2-(2,6-диэтилфенил)-6,6-диметил-2,6-дигидропирроло[3,4-c]пиразол-5(4H)-карбоксилата (0,5 г, 1,0 ммоль) и бензоилизоцианата (0,5 г, 3,4 ммоль) в ТГФ (10 мл) перемешивали 3 часа при комнатной температуре. Полученную смесь упаривали при пониженном давлении, получая трет-бутил 3-(4-(3-бензоилуреидо)-2,5-дифторфенил)-2-(2,6-диэтилфенил)-6,6-диметил-2,6-дигидропирроло[3,4-c]пиразол-5(4H)-карбоксилат.
Смесь трет-бутил 3-(4-(3-бензоилуреидо)-2,5-дифторфенил)-2-(2,6-диэтилфенил)-6,6-диметил-2,6-дигидропирроло[3,4-c]пиразол-5(4H)-карбоксилата (~1,0 ммоль, см. выше) и K2CO3 (1,3 г, 7,2 ммоль) в MeOH (15 мл) перемешивали 2 часа при комнатной температуре и затем 20 минут при 50°C. Полученную смесь экстрагировали этилацетатом. Органический слой отделяли, сушили над MgSO4, упаривали при пониженном давлении и очищали методом флэш-хроматографии на силикагеле (от 10 до 50% EtOAc в гексане), получая трет-бутил 2-(2,6-диэтилфенил)-3-(2,5-дифтор-4-уреидофенил)-6,6-диметил-2,6-дигидропирроло[3,4-c] пиразол-5(4H)-карбоксилат. МС: (ES) m/z вычислено для C29H36F2N5O3 [M+H]+ 540,3, найдено 540,3.
Стадия d: Описанный выше трет-бутил 2-(2,6-диэтилфенил)-3-иод-6,6-диметил-2,6-дигидропирроло[3,4-c]пиразол-5(4H)-карбоксилат растворяли в дихлорметане (10 мл) и добавляли раствор HCl в диоксане (4н., 5 мл). Полученную смесь перемешивали при комнатной температуре 12 часов. После окончания реакции растворитель упаривали в вакууме, получая 1-(4-(2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c] пиразол-3-ил)-2,5-дифторфенил)мочевины гидрохлорид. МС: (ES) m/z вычислено для C24H28F2N5O [M+H]+ 440,2, найдено 440,3.
Стадия e: N,N-диизопропилэтиламин (0,2 мл, 1,2 ммоль) добавляли в суспензию 1-(4-(2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-2,5-дифторфенил)мочевины гидрохлорида (0,1 г, 0,2 ммоль) и 2,4-бис(трифторметил) бензальдегида (0,2 г, 0,8 ммоль) в 1,2-дихлорэтане (10 мл) при перемешивании на магнитной мешалке. После перемешивания при комнатной температуре в течение 10 минут, порциями добавляли NaBH(OAc)3 (0,3 г, 1,4 ммоль). Полученную смесь перемешивали при 45°C в течение 2 часов. После охлаждения до комнатной температуры, реакционную смесь разбавляли этилацетатом, промывали водным раствором NaHCO3, насыщенным водным раствором хлорида натрия и сушили над MgSO4. Растворитель удаляли при пониженном давлении, и остаток очищали методом препаративной ТСХ (50% EtOAc в гексане), затем методом ВЭЖХ (MeCN/H2O, с 1% ТФУК), получая 1-(4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-2,5-дифторфенил)мочевину. 1H ЯМР (400 МГц, CDCl3) δ 8,18 (д, J=8,3 Гц, 1H), 7,88-7,98 (м, 2H), 7,75-7,83 (м, 1H), 7,31 (т, J=7,7 Гц, 1H), 7,14 (д, J=7,7 Гц, 2H), 6,79-6,85 (ушир, 1H), 6,40 (дд, J=6,5, 12,1 Гц, 1H), 4,79 (с, 2H), 4,13 (с, 2H), 3,74 (с, 2H), 2,20-2,34 (м, 4H), 1,51 (с, 6H), 1,06 (т, J=7,6 Гц, 6H). МС: (ES) m/z вычислено для C33H32F8N5O [M+H]+ 666,2, найдено 666,2.
Синтез интермедиата 4: 4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индол
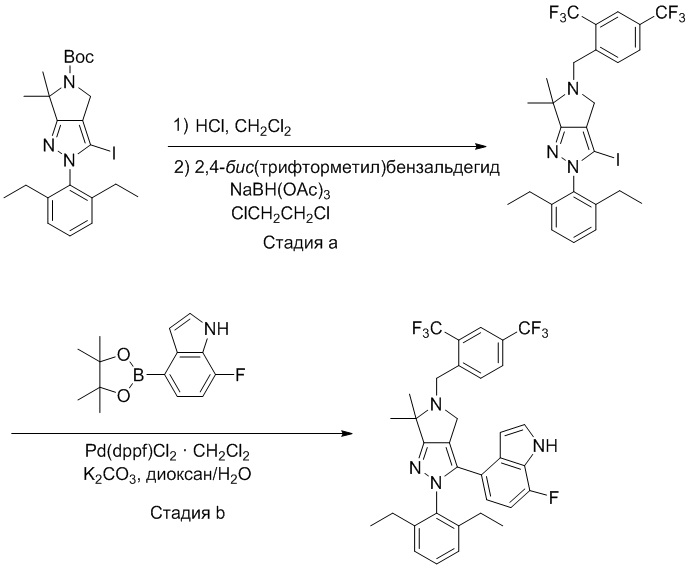
Стадия a: Полученный как описано выше трет-бутил 2-(2,6-диэтилфенил)-3-иод-6,6-диметил-2,6-дигидропирроло[3,4-c]пиразол-5(4H)-карбоксилат растворяли в дихлорметане (10 мл) и добавляли 4н. раствор HCl в диоксане (5 мл, 20 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 12 часов. После окончания реакции растворитель упаривали в вакууме, получая 2-(2,6-диэтилфенил)-3-иод-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол гидрохлорид. МС: (ES) m/z вычислено для C17H23IN3 [M+H]+ 396,1, найдено 396,2.
N,N-диизопропилэтиламин (0,3 мл, 1,73 ммоль) добавляли в суспензию 2-(2,6-диэтилфенил)-3-иод-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразола гидрохлорида (680,0 мг, 1,57 ммоль) и 2,4-бис(трифторметил)бензальдегида (800 мг, 3,3 ммоль) в 1,2-дихлорэтане (10 мл) при перемешивании на магнитной мешалке. После перемешивания при комнатной температуре в течение 10 минут, порциями добавляли NaBH(OAc)3 (800 мг, 3,77 ммоль). Полученную смесь перемешивали при 45°C в течение 2 часов. После охлаждения до комнатной температуры, реакционную смесь разбавляли этилацетатом, промывали водным раствором NaHCO3, насыщенным водным раствором хлорида натрия и сушили над MgSO4. Растворитель удаляли при пониженном давлении, и остаток очищали методом флэш-хроматографии на силикагеле (от 2 до 25% EtOAc в гексане), получая 5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-3-иод-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол. МС: (ES) m/z вычислено для C26H27F6IN3 [M+H]+ 622,1, найдено 622,1.
Стадия b: В суспензию 4-бром-7-фтор-1H-индола (1,00 г, 4,67 ммоль), бис(пинаколато)диборона (1,31 г, 5,14 ммоль) и KOAc (1,15 г, 11,7 ммоль) в диоксане (15 мл) добавляли комплекс Pd(dppf)Cl2 с дихлорметаном (416 мг, 0,51 ммоль). Реакционную смесь дегазировали азотом 2 минуты и перемешивали при 100°C в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры, разбавляли этилацетатом и фильтровали через целит. Растворитель удаляли при пониженном давлении, и остаток очищали методом флэш-хроматографии на силикагеле (0-30% EtOAc в гексане), получая 7-фтор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-индол. МС: (ES) m/z вычислено для C14H18BFNO2 [M+H]+ 262,1, найдено 262,1.
Смесь 5-(2,4-бис(трифторметил)бензил)-3-иод-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразола (200 мг, 0,32 ммоль), 7-фтор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-индола (150 мг, 0,57 ммоль), K2CO3 (276 мг, 2,0 ммоль) и комплекса Pd(dppf)Cl2 с дихлорметаном (60 мг, 0,07 ммоль) в диоксане (6 мл) и воде (1 мл) перемешивали при 100°C в течение 5 часов в атмосфере азота. Смесь охлаждали до комнатной температуры, разбавляли этилацетатом и фильтровали через слой целита. Фильтрат собирали, упаривали в вакууме, и остаток очищали методом флэш-хроматографии на силикагеле (0-50% EtOAc в гексане), получая 4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индол. 1H ЯМР (400 МГц, CDCl3) δ 8,44 (с, 1H), 8,19 (д, J=8,0 Гц, 1H), 7,86 (с, 1H), 7,75 (д, J=8,4 Гц, 1H), 7,22 (м, 2H), 7,07 (д, J=7,6 Гц, 2H), 6,61 (м, 1H), 6,47 (м, 2H), 4,15 (с, 2H), 3,71 (с, 2H), 2,37 (м, 2H), 2,22 (м, 2H), 1,56 (с, 6H), 1,00 (т, J=7,6 Гц, 6H). МС: (ES) m/z вычислено для C34H32F7N4 [M+H]+ 629,2, найдено 629,2.
Пример 1. Синтез (фосфоноокси)метил 4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-карбоксилата
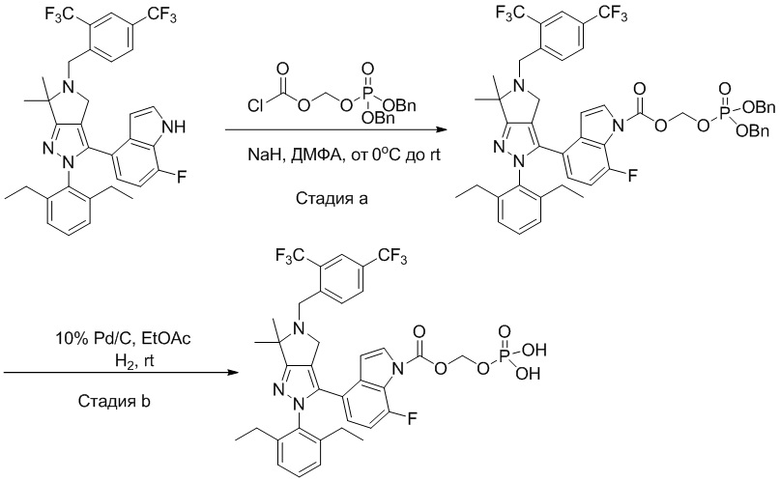
Стадия a: В перемешиваемый раствор 4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индола (240 мг, 0,35 ммоль) в ДМФА добавляли NaH (60%, 60 мг, 1,5 ммоль) при 0°C. Смесь перемешивали при 0°C в течение 10 минут, затем добавляли дибензилоксифосфорилокси-метил хлорформиат (263 мг, 0,71 ммоль) при 0°C. Полученную смесь перемешивали и оставляли нагреваться до комнатной температуры в течение 30 минут. После окончания реакции, реакцию гасили водой, экстрагировали этилацетатом, сушили над Mg2SO4 и упаривали в вакууме. Сырой продукт очищали методом хроматографии на силикагеле (10-50% EtOAc в гексане), получая ((бис(бензилокси)фосфорил)окси)метил 4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-карбоксилат. МС: (ES) m/z вычислено для C50H47F7N4O6P [M+H]+ 963,3, найдено 963,3.
Стадия b: В раствор ((бис(бензилокси)фосфорил)окси)метил 4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-карбоксилата (79 мг, 0,08 ммоль) в этилацетате (40 мл) добавляли 10% Pd/C (100 мг), и гидрировали под давлением 45 фунт/кв.дюйм в течение 20 минут. Реакционную смесь фильтровали через целит, промывали осадок смесью 1:1 EtOAc/MeOH (15 мл) и упаривали досуха. Остаток очищали методом ВЭЖХ (MeCN/H2O, с 0,1% ТФУК), получая (фосфоноокси)метил 4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-карбоксилат. 1H ЯМР (400 МГц, d6-ДМСО) δ 8,13 (д, J=8,2 Гц, 1H), 8,01 (д, J=8,2 Гц, 1H), 7,95 (с, 1H), 7,80 (д, J=4,8 Гц, 1H), 7,30 (т, J=7,8 Гц, 1H), 7,12 (д, J=7,8 Гц, 2H), 6,95 (дд, J=8,6, 12,1 Гц, 1H), 6,56-6,66 (м, 2H), 5,70 (д, J=14,4 Гц, 2H), 4,15 (с, 2H), 3,60 (с, 2H), 3,35 (ушир, 2H), 2,14-2,22 (м, 4H), 1,47 (с, 6H), 0,90 (т, J=7,6 Гц, 6H). МС: (ES) m/z вычислено для C36H35F7N4O6P [M+H]+ 783,2, найдено 783,2.
- Пример 2. Синтез (S)-(4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-ил)-L-цистеина
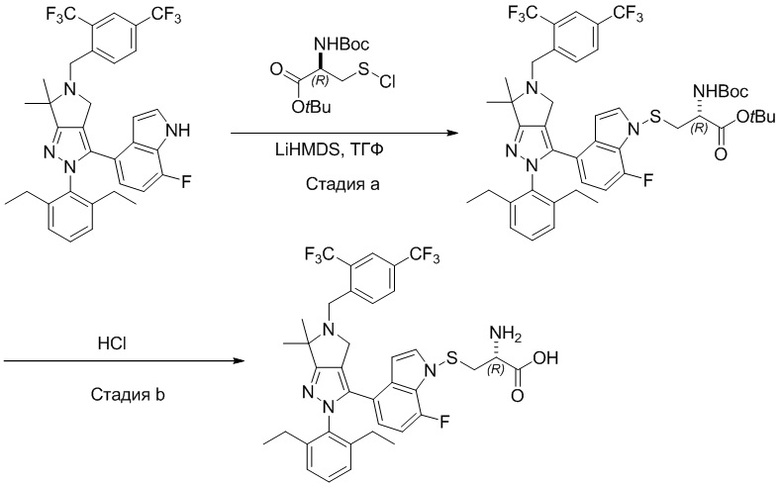
Стадия a: В перемешиваемый раствор ди-трет бутил 3,3'-дисульфандиил(2R,2'R)-бис(2-((трет-бутоксикарбонил)амино)пропаноата) (830 мг, 1,5 ммоль) в дихлорэтане (6 мл) при 0°C добавляли тионилхлорид (0,14 мл, 1,9 ммоль). Смесь перемешивали при 0°C в течение 15 минут, формируя трет-бутил N-(трет-бутоксикарбонил)-S-хлор-L-цистеинат, который использовали напрямую в следующей стадии.
В перемешиваемый раствор 4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индола (240 мг, 0,35 ммоль) в ТГФ (10 мл) в атмосфере азота при -45°C добавляли 1M раствор LiHMDS в ТГФ (0,8 мл, 0,8 ммоль). После перемешивания в течение 15 минут при -45°C, добавляли трет-бутил N-(трет-бутоксикарбонил)-S-хлор-L-цистеинат, полученный как описано выше (~1,5 ммоль). Полученную смесь перемешивали и оставляли нагреваться до комнатной температуры в течение 30 минут. После окончания реакцию гасили водой, экстрагировали этилацетатом, сушили над MgSO4 и упаривали в вакууме. Сырой продукт очищали методом хроматографии на силикагеле (10-50% EtOAc в гексане), получая трет-бутил (S)-(4-(5-(2,4- (трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-ил)-N-(трет-бутоксикарбонил)-L-цистеинат. МС: (ES) m/z вычислено для C46H53F7N5O4S [M+H]+ 904,4, найдено 904,5.
Стадия b: В раствор трет-бутил (S)-(4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-ил)-N-(трет-бутоксикарбонил)-L-цистеината (220 мг, 0,24 ммоль) в дихлорметане (6 мл) добавляли 4н. раствор HCl в диоксане (5 мл, 20 ммоль). Полученную смесь перемешивали при комнатной температуре в течение ночи. После окончания реакции смесь упаривали в вакууме. Остаток очищали методом ВЭЖХ (MeCN/H2O, с 0,1% ТФУК), получая (S)-(4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-ил)-L-цистеин. 1H ЯМР (400 МГц, CD3OD) δ 8,24 (д, J=7,6 Гц, 1H), 7,98-8,10 (м, 2H), 7,63-7,66 (м, 2H), 7,22-7,37 (м, 2H), 6,97-7,03 (м, 1H), 6,63-6,68 (м, 1H), 6,43-6,47 (м, 1H), 4,71-4,86 (м, 5H), 4,07-4,27 (м, 3H), 3,14-3,33 (м, 2H), 2,37-2,49 (м, 2H), 2,02-2,26 (м, 2H), 1,85-1,94 (м, 6H), 1,25-1,42 (м, 3H), 0,77-0,84 (м, 3H). МС: (ES) m/z вычислено для C37H37F7N5O2S [M+H]+ 748,3, найдено 748,2.
Пример 3. Синтез (4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-ил)фосфоновой кислоты
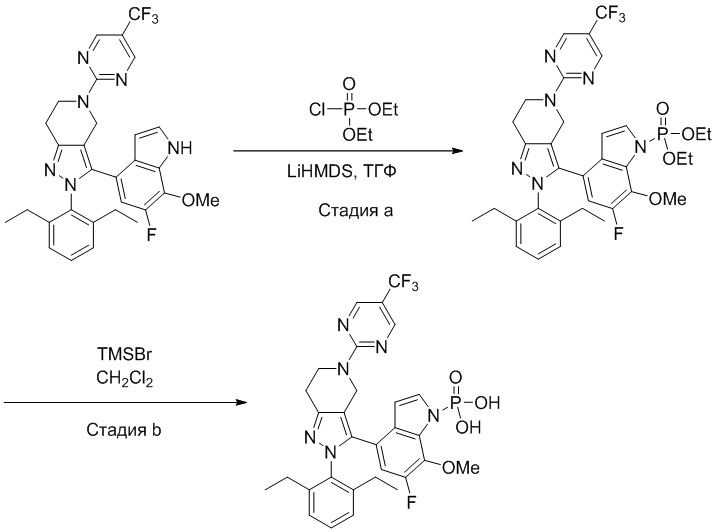
Стадия a: В перемешиваемый раствор 2-(2,6-диэтилфенил)-3-(6-фтор-7-метокси-1H-индол-4-ил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридина (230 мг, 0,4 ммоль) в ТГФ (10 мл) в атмосфере азота при -70°C добавляли 1M раствор LiHMDS в ТГФ (0,6 мл, 0,6 ммоль). После перемешивания в течение 15 минут при -70°C, добавляли диэтилхлорфосфат (0,1 мл, 0,8 ммоль). Полученную смесь оставляли нагреваться до комнатной температуры в течение 1 часа. После окончания реакции смесь гасили водой, экстрагировали этилацетатом, промывали насыщенным водным раствором NaHCO3, сушили над MgSO4 и упаривали в вакууме. Сырой продукт очищали методом хроматографии на силикагеле (5-25% EtOAc в гексане), получая диэтил (4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-ил)фосфонат. МС: (ES) m/z вычислено для C34H38F4N6O4P [M+H]+ 701,3, найдено 701,3.
Стадия b: В раствор диэтил (4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-ил)фосфоната (75 мг, 0,11 ммоль) в дихлорметане (6 мл) при 0°C добавляли TMSBr (0,2 мл, 1,5 ммоль). Полученную смесь оставляли нагреваться до комнатной температуры в течение 1 часа и затем перемешивали при 40°C в течение ночи. После окончания реакции реакционную смесь упаривали в вакууме. Остаток очищали методом ВЭЖХ (MeCN/H2O, с 0,1% ТФУК), получая (4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-ил)фосфоновую кислоту. 1H ЯМР (400 МГц, CD3OD) δ 8,58 (с, 2H), 7,68 (дд, J=2,5, 3,5 Гц, 1H), 7,32 (т, J=7,7 Гц, 1H), 7,16 (ушир, 2H), 6,49-6,57 (м, 2H), 4,82-4,90 (м, 4H), 4,39 (с, 2H), 3,99 (с, 3H), 2,98 (т, J=5,9 Гц, 2H), 2,05-2,42 (м, 4H), 0,78-1,25 (м, 6H). МС: (ES) m/z вычислено для C30H30F4N6O4P [M+H]+ 645,2, найдено 645,4.
Пример 4. Синтез (фосфоноокси)метил 4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил) пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбоксилата
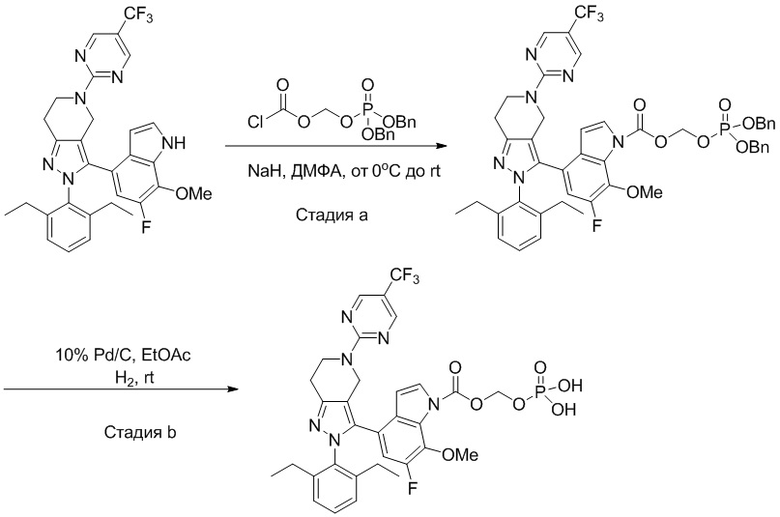
Стадия a: В перемешиваемый раствор 2-(2,6-диэтилфенил)-3-(6-фтор-7-метокси-1H-индол-4-ил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридина (570 мг, 1,0 ммоль) в ДМФА добавляли NaH (60%, 60 мг, 1,5 ммоль) при 0°C. Смесь перемешивали при 0°C в течение 10 минут, затем добавляли дибензилокси-фосфорилоксиметил хлорформиат (526 мг, 1,42 ммоль). Полученную смесь перемешивали и оставляли нагреваться до комнатной температуры в течение 30 минут. После окончания реакции смесь гасили водой, экстрагировали этилацетатом, сушили над MgSO4 и упаривали в вакууме. Сырой продукт очищали методом хроматографии на силикагеле (10-100% EtOAc в гексане), получая ((бис(бензилокси)фосфорил)окси)метил 4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбоксилат. МС: (ES) m/z вычислено для C46H44F4N6O7P [M+H]+ 899,3, найдено 899,3.
Стадия b: В раствор ((бис(бензилокси)фосфорил)окси)метил 4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбоксилата (120 мг, 0,13 ммоль) в этилацетате (45 мл) добавляли 10% Pd/C (200 мг) и гидрировали под давлением 50 фунт/кв.дюйм в течение 20 минут. Реакционную смесь фильтровали через целит, промывали осадок смесью 1:1 EtOAc/MeOH (15 мл), упаривали досуха. Остаток очищали методом ВЭЖХ (MeCN/H2O, с 0,1% ТФУК), получая (фосфоноокси)метил 4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил) пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбоксилат. 1H ЯМР (400 МГц, CD3OD) δ 8,58 (с, 2H), 7,87 (д, J=3,9 Гц, 1H), 7,33 (т, J=7,6 Гц, 1H), 7,16 (ушир.с, 2H), 6,59-6,64 (м, 2H), 5,87 (д, J=14,4 Гц, 2H), 4,74-4,86 (м, 4H), 4,39 (с, 2H), 3,97 (с, 3H), 2,98 (т, J=5,7 Гц, 2H), 2,05-2,42 (м, 4H), 0,76-1,28 (м, 6H). МС: (ES) m/z вычислено для C32H32F4N6O7P [M+H]+ 719,2, найдено 719,2.
Пример 5. Синтез (E)-4-(((4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбонил)окси)метокси)-4-оксобут-2-еновой кислоты
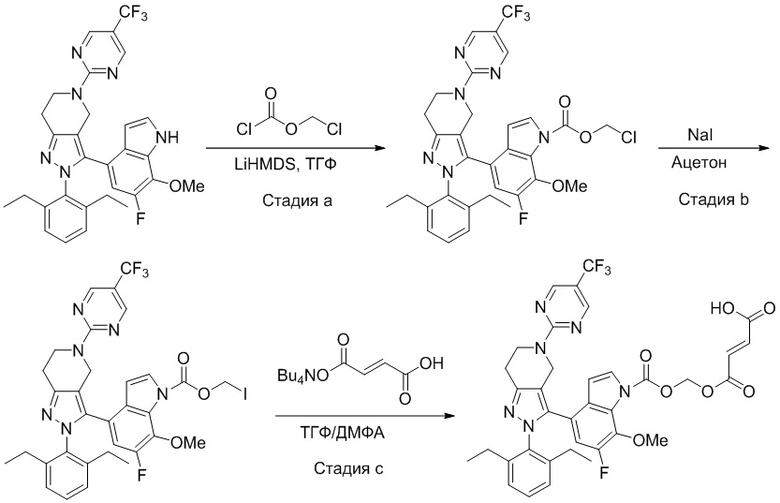
Стадия a: В перемешиваемый раствор 2-(2,6-диэтилфенил)-3-(6-фтор-7-метокси-1H-индол-4-ил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридина (280 мг, 0,5 ммоль) в ТГФ (10 мл) при -78°C добавляли 1M раствор LiHMDS в ТГФ (0,8 мл, 0,8 ммоль). Смесь перемешивали при -78°C в течение 15 минут, затем добавляли хлорметил хлорформиат (80 мкл, 0,9 ммоль). Полученную смесь перемешивали и оставляли нагреваться до комнатной температуры в течение 30 минут. После окончания реакции смесь гасили насыщенным водным раствором NH4Cl, экстрагировали этилацетатом, сушили над Mg2SO4 и упаривали в вакууме, получая хлорметил 4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбоксилат, который напрямую использовали в следующей стадии. МС: (ES) m/z вычислено для C32H30ClF4N6O3 [M+H]+ 657,2, найдено 657,2.
Стадия a: NaI (350 мг, 2,33 ммоль) добавляли в перемешиваемый раствор хлорметил 4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбоксилата (300 мг, 0,46 ммоль) в ацетоне (10 мл) при комнатной температуре. Полученную смесь перемешивали при 45°C в течение ночи. После окончания реакции смесь гасили насыщенным раствором хлорида натрия, экстрагировали этилацетатом, сушили над MgSO4 и упаривали в вакууме. Сырой продукт очищали методом хроматографии на силикагеле (5-25% EtOAc в гексане), получая иодметил 4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбоксилат. МС: (ES) m/z вычислено для C32H30F4IN6O3 [M+H]+ 749,1, найдено 749,2.
Стадия c: В раствор иодметил 4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил) пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбоксилата (50 мг, 0,07 ммоль) в ТГФ (5 мл) при 0°C добавляли тетрабутиламмониевую соль фумаровой кислоты (25 мг, 0,07 ммоль) в ДМФА (1 мл). Полученную смесь оставляли нагреваться до комнатной температуры в течение 1 часа. После окончания реакции смесь упаривали в вакууме. Остаток очищали методом ВЭЖХ (MeCN/H2O, с 0,1% ТФУК), получая (E)-4-(((4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил) пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбонил)окси)метокси)-4-оксобут-2-еновую кислоту. 1H ЯМР (400 МГц, CD3OD) δ 8,58 (с, 2H), 7,87 (д, J=3,9 Гц, 1H), 7,33 (т, J=7,6 Гц, 1H), 7,16 (ушир.с, 2H), 6,59-6,64 (м, 2H), 5,87 (д, J=14,4 Гц, 2H), 4,74-4,86 (м, 4H), 4,39 (с, 2H), 3,97 (с, 3H), 2,98 (т, J=5,7 Гц, 2H), 2,05-2,42 (м, 4H), 0,76-1,28 (м, 6H). МС: (ES) m/z вычислено для C36H33F4N6O7 [M+H]+ 737,2, найдено 737,2.
Пример 6. Синтез ((диметилглицил)окси)метил 4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбоксилата
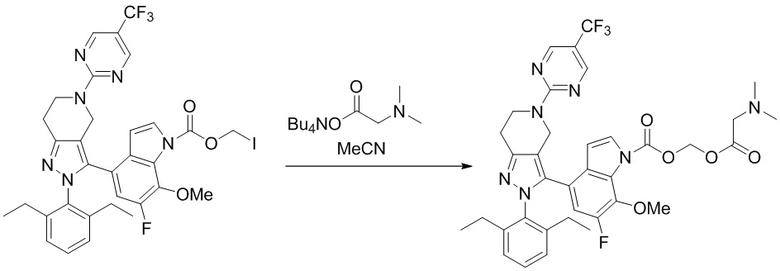
В раствор иодметил 4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбоксилата (40 мг, 0,06 ммоль) в MeCN (5 мл) при 0°C добавляли тетрабутиламмония диметилглицинат (20 мг, 0,06 ммоль) в ДМФА (1 мл). Полученную смесь оставляли нагреваться до комнатной температуры в течение 1 часа. После окончания реакции смесь гасили 1н. раствором HCl (0,1 мл, 0,1 ммоль) и очищали методом ВЭЖХ (MeCN/H2O, с 0,1% ТФУК), получая ((диметилглицил)окси)метил 4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбоксилат в виде соли с ТФУК. 1H ЯМР (400 МГц, CD3OD) δ 8,59 (с, 2H), 7,92 (д, J=3,9 Гц, 1H), 7,34 (т, J=7,6 Гц, 1H), 7,17 (ушир.с, 2H), 6,63-6,74 (м, 2H), 5,83 (с, 2H), 4,81-4,88 (ушир.с, 1H), 4,46 (с, 2H), 4,39 (с, 2H), 3,97 (с, 3H), 3,45 (с, 5H), 2,95-3,03 (м, 5H), 2,05-2,42 (м, 4H), 0,76-1,28 (м, 6H). МС: (ES) m/z вычислено для C36H38F4N7O5 [M+H]+ 724,2, найдено 724,2.
Пример 7. Синтез ((4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-2,5-дифторфенил)карбамоил) фосфамидной кислоты
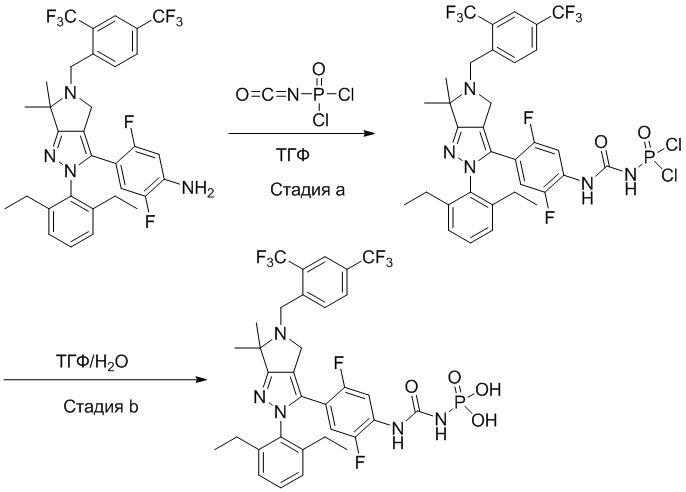
Стадия a: В раствор 4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-2,5-дифторанилина (100 мг, 0,16 ммоль) в ТГФ (6 мл) при -50°C добавляли фосфоизоцианатодихлорид (0,04 мл, 0,41 ммоль). Смесь оставляли нагреваться до комнатной температуры в течение 1 часа и затем упаривали в вакууме. Остаток растирали в гексане, получая ((4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-2,5-дифторфенил)карбамоил)фосфамидодихлорид, который использовали напрямую в следующей стадии.
Стадия b: В раствор описанного выше ((4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-2,5-дифторфенил) карбамоил)фосфамидодихлорида (~0,16 ммоль) в ТГФ (6 мл) при комнатной температуре добавляли воду (3 мл). Смесь перемешивали при комнатной температуре 2 часа, затем добавляли 1н. раствор NaOH (0,5 мл, 0,5 ммоль). Смесь перемешивали еще 2 часа. После окончания реакции смесь очищали методом ВЭЖХ (MeCN/H2O, с 0,1% ТФУК), получая ((4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-2,5-дифторфенил)карбамоил)фосфамидную кислоту. 1H ЯМР (400 МГц, ДМСО-d6) δ 9,60 (ушир, 1H), 8,29 (с, 1H), 8,16 (д, J=8,2 Гц, 2H), 7,92-8,08 (м, 2H), 7,38 (д, J=7,9 Гц, 1H), 7,21 (д, J=7,7 Гц, 2H), 6,45 (дд, J=6,7, 11,9 Гц, 1H), 4,13 (с, 2H), 3,64 (с, 2H), 3,24-3,48 (ушир, 2H), 2,16 (кв, J=7,6 Гц, 4H), 1,42 (с, 6H), 0,95 (т, J=7,6 Гц, 6H). МС: (ES) m/z вычислено для C33H33F8N5O4P [M+H]+ 746,2, найдено 746,2.
Пример 8. Синтез ((3,3-диметил-5-(фосфоноокси)пентаноил)окси)метил 4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбоксилата
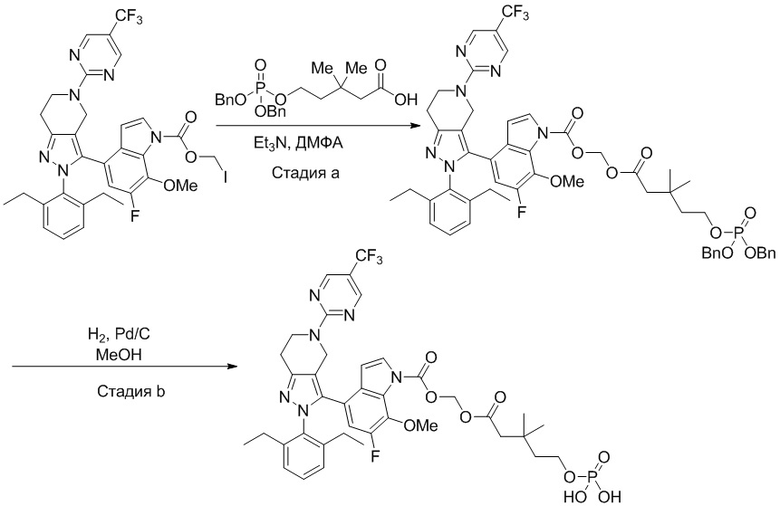
Стадия a: В колбу, содержащую 4,4-диметилдигидро-2H-пиран-2,6(3H)-дион (5 г, 35 ммоль) в ТГФ (140 мл), добавляли по каплям 1M раствор LiAlH4 в ТГФ (35 мл, 35 ммоль). Смесь нагревали при 75°C в течение 2 часов. После окончания реакцию гасили водой, и смесь фильтровали. Фильтрат упаривали, получая 3,3-диметилпентан-1,5-диол.
В раствор 3,3-диметилпентан-1,5-диола (1 г, 7,6 ммоль) в ТГФ (15,6 мл) добавляли по каплям 1M раствор tBuOK (8,3 мл, 8,3 ммоль), затем тетрабензилдифосфат (4,2 г, 7,8 ммоль). После нагревания при 70°C в течение 16 часов, смесь упаривали и очищали методом колоночной хроматографии на силикагеле (0-100% EtOAc в гексане), получая дибензил (5-гидрокси-3,3-диметилпентил)фосфат. МС: (ES) m/z вычислено для C21H29O5P [M+H]+ 393,2, найдено 393,1.
В раствор дибензил (5-гидрокси-3,3-диметилпентил)фосфата (400 мг, 1 ммоль) в ДМФА (10 мл) добавляли дихромат пиридиния (2,3 г, 6 ммоль). Смесь перемешивали при комнатной температуре 2 часа, затем упаривали и очищали методом колоночной хроматографии на силикагеле (0-100% EtOAc в гексане). Очищенный остаток растворяли в 10 мл смеси 1:1 tBuOH и H2O. В раствор добавляли NaH2PO4 (0,61 г, 5 ммоль), затем хлорит натрия (0,46 г, 5 ммоль) и 2M раствор 2-метил-2-бутена (5 мл, 10 ммоль). Смесь перемешивали при комнатной температуре 16 часов. После окончания реакции, смесь упаривали в вакууме, и сырой продукт очищали методом колоночной хроматографии на силикагеле (0-100% EtOAc в гексане), получая 5-((бис(бензилокси)фосфорил)окси)-3,3-диметилпентановую кислоту. МС: (ES) m/z вычислено для C21H27O6P [M+H]+ 407,2, найдено 407,1.
В раствор иодметил 4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбоксилат (70 мг, 0,09 ммоль) в 0,2 мл ДМФА добавляли 5-((бис(бензилокси)фосфорил) окси)-3,3-диметилпентановую кислоту (54 мг, 0,13 ммоль), затем Et3N (0,02 мл, 0,14 ммоль). Смесь перемешивали при комнатной температуре 2 часа, затем упаривали в вакууме. Полученный остаток очищали методом колоночной хроматографии на силикагеле (0-100% EtOAc в гексане) получая (((5-((бис(бензилокси)фосфорил)окси)-3,3-диметилпентаноил)окси) метил 4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбоксилат.
Стадия b: В раствор (((5-((бис(бензилокси)фосфорил)окси)-3,3-диметилпентаноил) окси)метил 4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбоксилата (50 мг, 0,05 ммоль) в MeOH (1 мл) добавляли 10% Pd/C (6 мг, 0,005 ммоль). Смесь перемешивали под давлением водорода из шарика 1 час, затем фильтровали через целит, упаривали и очищали методом ВЭЖХ (MeCN/H2O, с 0,1% ТФУК), получая ((3,3-диметил-5-(фосфоноокси) пентаноил)окси)метил 4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбоксилат. 1H ЯМР (400 МГц, CDCl3) δ 8,54 (с, 2H), 7,63 (д, J=3,9 Гц, 1H), 7,28 (т, J=7,7 Гц, 1H), 7,08 (д, J=7,6 Гц, 2H), 6,59 (д, J=12,1 Гц, 1H), 6,47 (д, J=3,8 Гц, 1H), 6,02 (с, 2H), 5,60 (ушир.с, 2H), 4,76 (ушир.с, 2H), 4,38 (т, J=5,9 Гц, 2H), 4,03 (дд, J=14,1, 7,1 Гц, 2H), 3,99 (с, 3H), 3,10 (т, J=3,1 Гц, 2H), 2,35 (с, 2H), 2,20 (ушир.с, 4H), 1,74 (т, J=7,0 Гц, 2H), 1,05 (ушир.с, 12H). МС: (ES) m/z вычислено для C39H43FN6O9P [M+H]+ 847,3, найдено 847,2.
Пример 9: Синтез (3-фтор-4-фосфонооксифенил)метил N-((4-(5-((2,4-бис(трифторметил) фенил)метил)-2-(2,6-диэтилфенил)-6,6-диметил-4H-пирроло[3,4-c]пиразол-3-ил)-2,5-дифторфенил)карбамоил]карбамата
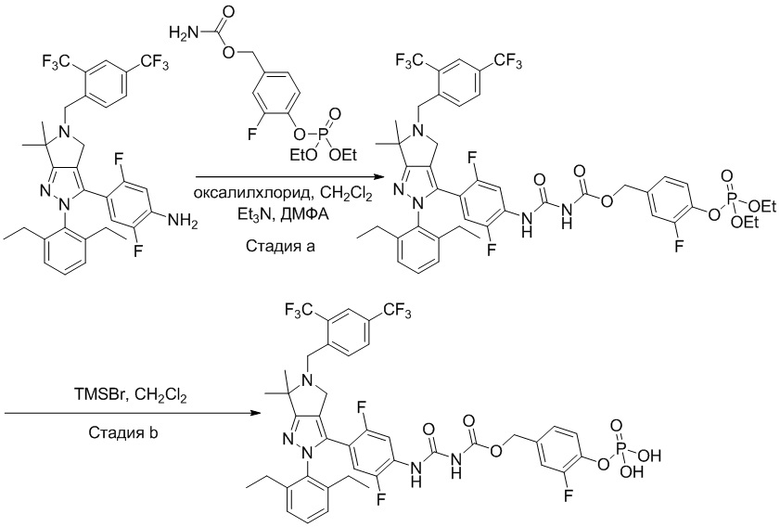
Стадия a: В раствор 3-фтор-4-гидроксибензальдегида (5 г, 35,7 ммоль) в дихлорметане (36 мл) добавляли Et3N (7,5 мл, 53,8 ммоль), затем диэтилхлорфосфат (5,7 мл, 39,4 ммоль). Смесь перемешивали при комнатной температуре 3 часа, затем гасили водой. Органический и водный слой разделяли, и водный слой экстрагировали этилацетатом. Органические слои объединяли, сушили над сульфатом натрия, фильтровали и упаривали в вакууме. Полученный остаток очищали методом колоночной хроматографии на силикагеле, получая диэтил (2-фтор-4-формилфенил)фосфат. МС: (ES) m/z вычислено для C11H14FO5P [M+H]+ 277,1, найдено 277,0.
В раствор диэтил (2-фтор-4-формилфенил)фосфата (8,99 г, 32,5 ммоль) в ТГФ (32,5 мл) при -78°C добавляли NaBH4 (3,6 г, 97,3 ммоль). После перемешивания при -78°C в течение 1 часа реакцию гасили водой. Органический и водный слой разделяли, и водный слой экстрагировали этилацетатом. Органические слои объединяли, сушили над сульфатом натрия, фильтровали и упаривали в вакууме. Остаток очищали методом колоночной хроматографии на силикагеле (85-100% EtOAc в гексане), получая диэтил (2-фтор-4-(гидроксиметил)фенил)фосфат. МС: (ES) m/z вычислено для C11H16FO5P [M+H]+ 279,1, найдено 279,0.
В раствор диэтил (2-фтор-4-(гидроксиметил)фенил)фосфата (1 г, 3,6 ммоль) в ТГФ (8,7 мл) при 0°C добавляли диизопропилэтиламин (0,76 мл, 4,4 ммоль) и трифосген (0,53 г, 1,8 ммоль). После перемешивания при 0°C в течение 1 часа, добавляли NH4OH (1,6 мл, 41 ммоль). Смесь перемешивали при комнатной температуре в течение 16 часов, затем упаривали в вакууме, и остаток очищали методом колоночной хроматографии на силикагеле (0-100% EtOAc в гексане), получая 4-((диэтоксифосфорил)окси)-3-фторбензил карбамат. МС: (ES) m/z вычислено для C12H17FNO6P [M+H]+ 322,1, найдено 322,0.
В раствор 4-((диэтоксифосфорил)окси)-3-фторбензил карбамата (155 мг, 0,48 ммоль) в дихлорметане (4,8 мл) при 0°C добавляли оксалилхлорид (0,06 мл, 0,71 ммоль). Смесь нагревали при 40°C в течение 16 часов, затем упаривали в вакууме. Остаток растворяли в ТГФ (2 мл) и добавляли в раствор 4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-2,5-дифторанилина (100 мг, 0,16 ммоль) в ТГФ (2 мл). Смесь перемешивали при комнатной температуре в течение 5 часов, затем упаривали в вакууме. Остаток очищали методом колоночной хроматографии на силикагеле (0-100% EtOAc в гексане), получая (4-диэтоксифосфорилокси-3-фторфенил)метил N-((4-(5-((2,4-бис(трифторметил)фенил)метил)-2-(2,6-диэтилфенил)-6,6-диметил-4H-пирроло[3,4-c]пиразол-3-ил)-2,5-дифторфенил)карбамоил)карбамат. МС: (ES) m/z вычислено для C45H45F9N5O7P [M+H]+ 970,3, найдено 970,0.
Стадия b: В раствор (4-диэтоксифосфорилокси-3-фторфенил)метил N-((4-(5-((2,4-бис(трифторметил)фенил)метил)-2-(2,6-диэтилфенил)-6,6-диметил-4H-пирроло[3,4-c] пиразол-3-ил)-2,5-дифторфенил)карбамоил)карбамата (77 мг, 0,08 ммоль) в дихлорметане (1 мл) добавляли по каплям TMSBr (0,13 мл, 0,10 ммоль). Смесь перемешивали при комнатной температуре в течение 16 часов, затем упаривали в вакууме. Остаток очищали методом ВЭЖХ (MeCN/H2O, с 0,1% ТФУК), получая (3-фтор-4-фосфонооксифенил)метил N-((4-(5-((2,4-бис(трифторметил)фенил)метил)-2-(2,6-диэтилфенил)-6,6-диметил-4H-пирроло[3,4-c] пиразол-3-ил)-2,5-дифторфенил)карбамоил)карбамат. 1H ЯМР (400 МГц, CD3OD) δ 8,22 (д, J=8,2 Гц, 1H), 8,15 (с, 1H), 8,08-8,15 (м, 2H), 7,46 (дд, J=7,6, 7,9 Гц, 1H), 7,38 (дд, J=8,2, 8,2 Гц, 1H), 7,28 (д, J=7,6 Гц, 2H), 7,17 (д, J=11,0 Гц, 1H), 7,10 (д, J=8,6 Гц, 1H), 6,42 (дд, J=6,5, 11,5 Гц, 1H), 5,14 (с, 2H), 4,77 (с, 2H), 4,57 (с, 2H), 3,34 (с, 2H), 2,24 (кв, J=7,7 Гц, 4H), 1,89 (с, 6H), 1,05 (т, J=7,2 Гц, 6H). МС: (ES) m/z вычислено для C41H37F9N5O7P [M+H]+ 914,2, найдено 914,1.
- Пример 10. Синтез 3-нитро-4-(фосфоноокси)бензил 4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбоксилата
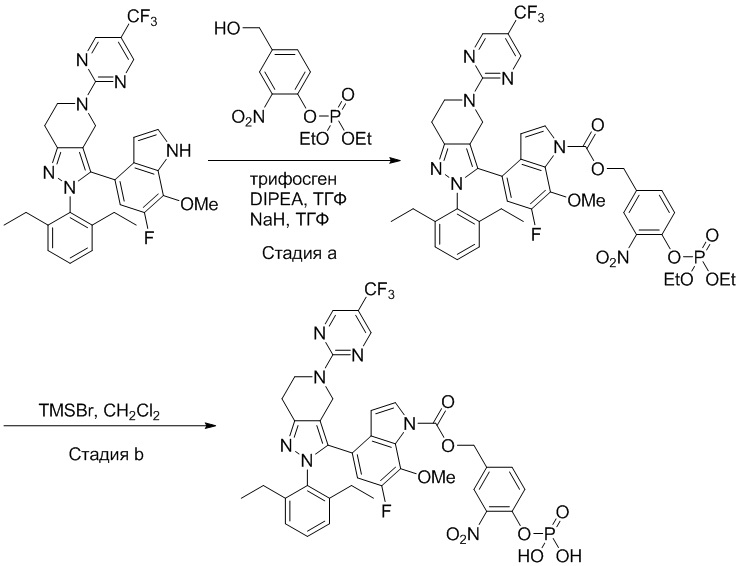
Стадия a: В раствор 4-гидрокси-3-нитробензальдегида (1 г, 6,0 ммоль) в дихлорметане (6 мл) добавляли Et3N (1,25 мл, 9,0 ммоль) и диэтилхлорфосфат (0,95 мл, 6,6 ммоль). Смесь перемешивали при комнатной температуре в течение 16 часов, затем гасили водой. Органический и водный слой разделяли, и водный слой экстрагировали этилацетатом. Органические слои объединяли, сушили над сульфатом натрия, фильтровали и упаривали в вакууме. Остаток очищали методом колоночной хроматографии на силикагеле (0-100% EtOAc в гексане), получая диэтил (4-формил-2-нитрофенил)фосфат. МС: (ES) m/z вычислено для C11H14NO7P [M+H]+ 304,1, найдено 304,0.
В раствор диэтил (4-формил-2-нитрофенил)фосфата (1,36 г, 4,5 ммоль) в ТГФ (4,5 мл) при -78°C добавляли NaBH4 (500 мг, 13,5 ммоль). После перемешивания при -78°C в течение 1 часа, реакцию гасили водой. Органический и водный слой разделяли, и водный слой экстрагировали этилацетатом. Органические слои объединяли, сушили над сульфатом натрия, фильтровали и упаривали. Полученный остаток очищали методом колоночной хроматографии на силикагеле (0-100% EtOAc в гексане), получая диэтил (4-(гидроксиметил)-2-нитрофенил)фосфат. МС: (ES) m/z вычислено для C11H16NO7P [M+H]+ 306,1, найдено 306,0.
В раствор диэтил (4-(гидроксиметил)-2-нитрофенил)фосфата (100 мг, 0,33 ммоль) в ТГФ (1,6 мл) при 0°C добавляли диизопропилэтиламин (0,07 мл, 0,40 ммоль) и трифосген (50 мг, 0,17 ммоль). После перемешивания при 0°C в течение 1 часа, реакцию гасили водой. Органический и водный слой разделяли, и водный слой экстрагировали этилацетатом. Органические слои объединяли, сушили над сульфатом натрия, фильтровали и упаривали, получая сырой хлорформиатный интермедиат.
В раствор 2-(2,6-диэтилфенил)-3-(6-фтор-7-метокси-1H-индол-4-ил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридина (93 мг, 0,16 ммоль) в ТГФ (1 мл) при 0°C добавляли NaH (13 мг, 0,32 ммоль). После перемешивания при 0°C в течение 30 минут, добавляли в смесь раствор сырого хлорформиата (получен как описано выше) в ТГФ (0,5 мл). Раствор перемешивали при комнатной температуре в течение 16 часов. Реакцию гасили водой. Органический и водный слой разделяли, и водный слой экстрагировали этилацетатом. Органические слои объединяли, сушили над сульфатом натрия, фильтровали и упаривали в вакууме. Остаток очищали методом колоночной хроматографии на силикагеле (0-100% EtOAc в гексане), получая 4-((диэтоксифосфорил) окси)-3-нитробензил 4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбоксилат.
Стадия b: В раствор 4-((диэтоксифосфорил)окси)-3-нитробензил 4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбоксилата (128 мг, 0,14 ммоль) в дихлорметане (1,4 мл) добавляли по каплям TMSBr (0,11 мл, 0,86 ммоль). После перемешивания при комнатной температуре в течение 5 часов, добавляли в смесь дополнительное количество TMSBr (0,11 мл, 0,86 ммоль). Смесь перемешивали при комнатной температуре в течение 16 часов, упаривали в вакууме и очищали методом ВЭЖХ (MeCN/H2O, с 0,1% ТФУК), получая 3-нитро-4-(фосфоноокси)бензил 4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбоксилат. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,68 (с, 2H), 8,12 (дд, J=0,9, 2,3 Гц, 1H), 7,85 (д, J=3,8 Гц, 1H), 7,85 (дд, J=2,2, 8,6 Гц, 1H), 7,60 (дд, J=1,1, 8,5 Гц, 1H), 7,29 (т, J=7,6 Гц, 1H), 7,12 (ушир.с, 2H), 6,62 (д, J=3,8 Гц, 1H), 6,55 (д, J=12,4 Гц, 1H), 5,48 (с, 2H), 4,76 (с, 2H), 4,30 (ушир.с, 2H), 3,83 (д, J=1,2 Гц, 3H), 2,91 (т, J=6,0 Гц, 2H), 2,16 (ушир.с, 4H), 2,04 (с, 2H), 0,91 (ушир.с, 6H). МС: (ES) m/z вычислено для C38H34FN7O9P [M+H]+ 840,2, найдено 840,0.
Пример 11. Синтез 3-фтор-4-(фосфоноокси)бензил 4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбоксилата
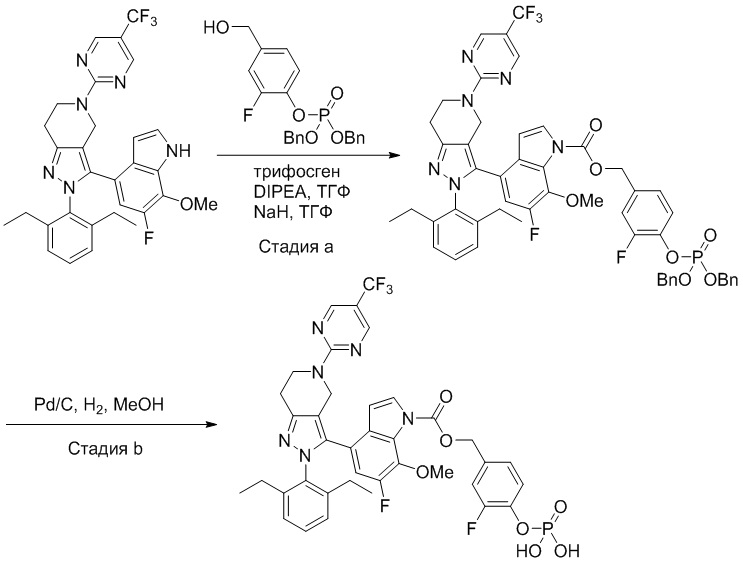
Стадия a: В раствор 3-фтор-4-гидроксибензальдегида (1 г, 7,1 ммоль) в ТГФ (32 мл) добавляли 1M раствор tBuOK в ТГФ (7,6 мл, 7,6 ммоль). Смесь нагревали до 70°C и добавляли тетрабензилфосфат (4,0 г, 7,4 ммоль). После 1 часа при 70°C, добавляли в смесь гексан и смесь фильтровали. Фильтрат упаривали в вакууме, и полученный остаток очищали методом колоночной хроматографии на силикагеле (0-100% EtOAc в гексане), получая дибензил (2-фтор-4-формилфенил)фосфат. МС: (ES) m/z вычислено для C21H18FO5P [M+H]+ 401,1, найдено 401,1.
В раствор дибензил (2-фтор-4-формилфенил)фосфата (2,68 г, 6,7 ммоль) в ТГФ (6,7 мл) при -78°C добавляли NaBH4 (0,76 г, 20,5 ммоль). После перемешивания при -78°C в течение 1 часа, смесь гасили водой. Органический и водный слой разделяли, и водный слой экстрагировали этилацетатом. Объединенные органические слои сушили над сульфатом натрия, фильтровали и упаривали в вакууме. Полученный остаток очищали методом колоночной хроматографии на силикагеле (0-100% EtOAc в гексане), получая дибензил (2-фтор-4-(гидроксиметил)фенил)фосфат. МС: (ES) m/z вычислено для C21H20FO5P [M+H]+ 403,1, найдено 403,0.
В раствор дибензил (2-фтор-4-(гидроксиметил)фенил)фосфата (200 мг, 0,50 ммоль) в ТГФ (2,4 мл) при 0°C добавляли диизопропилэтиламин (0,1 мл, 0,57 ммоль) и трифосген (72 мг, 0,24 ммоль). После перемешивания при 0°C в течение 1 часа, реакцию гасили водой. Органический и водный слой разделяли, и водный слой экстрагировали этилацетатом. Органические слои объединяли, сушили над сульфатом натрия, фильтровали и упаривали, получая сырой хлорформиатный интермедиат.
В раствор 2-(2,6-диэтилфенил)-3-(6-фтор-7-метокси-1H-индол-4-ил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридина (70 мг, 0,12 ммоль) в ТГФ (1,2 мл) при 0°C добавляли NaH (10 мг, 0,24 ммоль). После перемешивания при 0°C в течение 30 минут, добавляли в смесь раствор сырого хлорформиата (получен как описано выше) в ТГФ (1,2 мл). Раствор перемешивали при 0°C в течение 1 часа, затем гасили водой. Органический и водный слой разделяли, и водный слой экстрагировали этилацетатом. Органические слои объединяли, сушили над сульфатом натрия, фильтровали и упаривали в вакууме. Остаток очищали методом колоночной хроматографии на силикагеле (0-100% EtOAc в гексане), получая 4-((бис(бензилокси)фосфорил)окси)-3-фторбензил 4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c] пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбоксилат.
Стадия b: В раствор 4-((бис(бензилокси)фосфорил)окси)-3-фторбензил 4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c] пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбоксилата (100 мг, 0,10 ммоль) в MeOH (1 мл) добавляли 10% Pd/C (10 мг, 0,01 ммоль). Смесь перемешивали под давлением водорода из шарика 1 час, затем фильтровали через целит, упаривали и очищали методом ВЭЖХ (MeCN/H2O, с 0,1% ТФУК), получая 3-фтор-4-(фосфоноокси)бензил 4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбоксилат. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,68 (с, 2H), 7,84 (с, 1H), 7,52-7,38 (м, 2H), 7,37-7,25 (м, 2H), 7,11 (ушир.с, 2H), 6,62 (с, 1H), 6,54 (д, J=12,3 Гц, 1H), 5,40 (с, 2H), 4,76 (с, 2H), 4,30 (с, 2H), 3,83 (с, 3H), 2,90 (ушир.с, 2H), 2,16 (ушир.с, 4H), 2,04 (ушир.с, 2H), 0,90 (ушир.с, 6H). МС: (ES) m/z вычислено для C38H34F5N6O7P [M+H]+ 813,2, найдено 813,2.
Пример 12. Синтез (2-(4-(фосфоноокси)фенил)ацетокси)метил 4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбоксилата
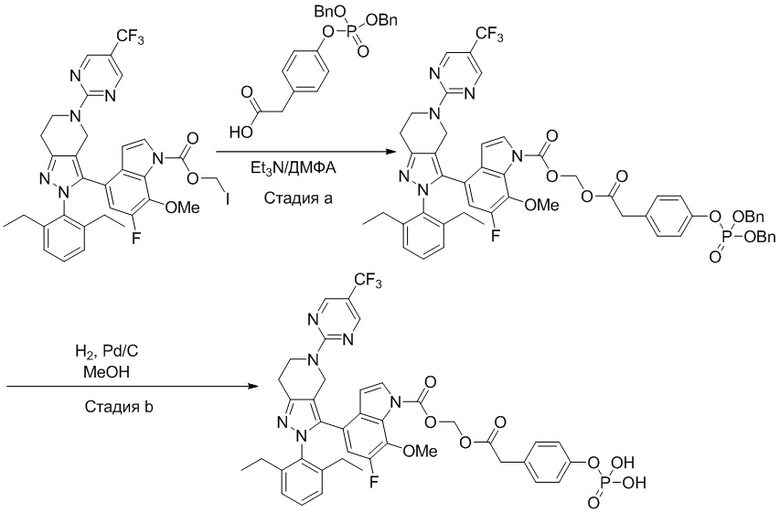
Стадия a: В колбу, содержащую этил 2-(4-гидроксифенил)ацетат (1 г, 5,6 ммоль) в ТГФ (11 мл), добавляли по каплям 1M раствор tBuOK в ТГФ (5,9 мл, 5,9 ммоль) и тетрабензилдифосфат (3 г, 5,6 ммоль). Смесь нагревали при 70°С в течение 2 часов. Добавляли дополнительное количество 1,0 M tBuOK (1,2 мл, 1,2 ммоль) и тетрабензилдифосфата (0,6 г, 1,1 ммоль). Смесь нагревали еще 3 часа. После завершения реакции добавляли гексан и смесь фильтровали. Фильтрат упаривали, и остаток очищали методом колоночной хроматографии на силикагеле (0-100% EtOAc в гексане), получая этил 2-(4-((бис(бензилокси)фосфорил)окси)фенил)ацетат. МС: (ES) m/z вычислено для C24H25O6P [M+H]+ 441,1, найдено 441,1.
В раствор этил 2-(4-((бис(бензилокси)фосфорил)окси)фенил)ацетата (1,54 г, 3,5 ммоль) в ТГФ (9 мл) добавляли по каплям раствор LiOH (0,32 г, 7,6 ммоль) в H2O (9 мл). Смесь перемешивали при комнатной температуре в течение 1 часа. Реакцию гасили 1н. раствором HCl. Водный слой экстрагировали этилацетатом. Органические слои объединяли, сушили над сульфатом натрия, фильтровали и упаривали. Остаток очищали методом колоночной хроматографии на силикагеле (0-100% EtOAc в гексане), получая 2-(4-((бис(бензилокси)фосфорил)окси)фенил)уксусную кислоту. МС: (ES) m/z вычислено для C22H21O6P [M+H]+ 413,1, найдено 413,1.
В раствор иодметил 4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбоксилата (100 мг, 13 ммоль) в ДМФА (0,5 мл) добавляли 2-(4-((бис(бензилокси) фосфорил)окси)фенил)уксусную кислоту (82 мг, 20 ммоль) и триэтиламин (0,03 мл, 0,20 ммоль). Смесь перемешивали при комнатной температуре в течение 16 часов, затем упаривали. Остаток очищали методом колоночной хроматографии на силикагеле (0-100% EtOAc в гексане), получая (2-(4-((бис(бензилокси)фосфорил)окси)фенил)ацетокси)метил 4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбоксилат.
Стадия b: В раствор (2-(4-((бис(бензилокси)фосфорил)окси)фенил)ацетокси)метил 4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло [4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбоксилата (66 мг, 0,06 ммоль) в MeOH (1 мл) добавляли 10% Pd/C (7 мг, 0,006 ммоль). Смесь перемешивали под давлением водорода из шарика 1 час, затем фильтровали через целит, упаривали и очищали методом ВЭЖХ (MeCN/H2O, с 0,1% ТФУК), получая (2-(4-(фосфоноокси)фенил)ацетокси)метил 4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбоксилат. 1H ЯМР (400 МГц, CDCl3) δ 8,47 (с, 2H), 7,38 (д, J=3,8 Гц, 1H), 7,22-7,26 (м, 2H), 7,10 (д, J=7,7 Гц, 2H), 7,01-7,06 (м, 4H), 6,57 (д, J=12,1 Гц, 1H), 6,45 (д, J=3,8 Гц, 1H), 5,97 (с, 2H), 5,02 (ушир.с, 2H), 4,77 (с, 2H), 4,36 (ушир.с, 2H), 3,95 (с, 3H), 3,60 (с, 2H), 3,07 (ушир.с, 2H), 2,05-2,35 (м, 4H), 0,99 (ушир.с, 6H). МС: (ES) m/z вычислено для C40H37F4N6O9P [M+H]+ 853,2, найдено 853,0.
Пример 13. Синтез (4-фосфоноокси-3-(трифторметил)фенил)метил N-((4-(5-((2,4-бис(трифторметил)фенил)метил)-2-(2,6-диэтилфенил)-6,6-диметил-4H-пирроло[3,4-c] пиразол-3-ил)-2,5-дифторфенил)карбамоил)карбамата
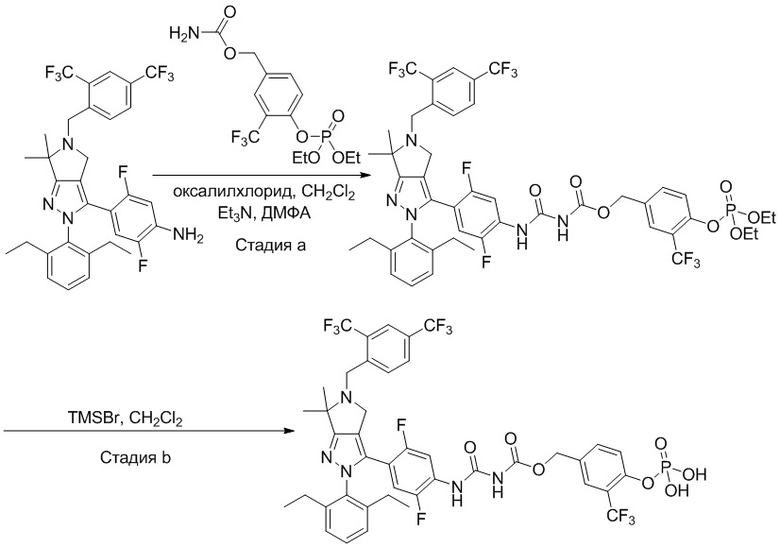
Стадия a: В раствор 4-гидрокси-3-(трифторметил)бензальдегида (1 г, 0,53 ммоль) в дихлорметане (10 мл) добавляли триэтиламин (1,1 мл, 0,79 ммоль), затем диэтилхлорфосфат (0,84 мл, 0,58 ммоль). Смесь перемешивали при комнатной температуре 1 час, затем упаривали в вакууме. Остаток очищали методом колоночной хроматографии на силикагеле (0-100% EtOAc в гексане), получая диэтил (4-формил-2-(трифторметил)фенил)фосфат. МС: (ES) m/z вычислено для C12H14F3O5P [M+H]+ 327,1, найдено 327,0.
В раствор диэтил (4-формил-2-(трифторметил)фенил)фосфата (1,43 г, 4,4 ммоль) в ТГФ (4,5 мл) при -78°С добавляли NaBH4 (0,49 г, 13,2 ммоль). Реакционную смесь нагревали до комнатной температуры и перемешивали 16 часов. После завершения, реакцию гасили водой. Органический и водный слой разделяли, и водный слой экстрагировали этилацетатом. Органические слои объединяли, сушили над сульфатом натрия, фильтровали и упаривали в вакууме. Остаток очищали методом колоночной хроматографии на силикагеле (85-100% EtOAc в гексане), получая диэтил (4-(гидроксиметил)-2-(трифторметил)фенил)фосфат. МС: (ES) m/z вычислено для C12H16F3O5P [M+H]+ 329,1, найдено 329,1.
В раствор диэтил (4-(гидроксиметил)-2-(трифторметил)фенил)фосфата (0,6 г, 1,8 ммоль) в ТГФ (4,4 мл) при 0°С добавляли диизопропилэтиламин (0,39 мл, 2,2 ммоль) и трифосген (0,27 мл, 0,9 ммоль). После перемешивания при 0°С в течение 1 часа, добавляли NH4OH (0,8 мл, 21 ммоль). Смесь перемешивали при комнатной температуре 1 час, затем упаривали в вакууме, и остаток очищали методом колоночной хроматографии на силикагеле (0-100% EtOAc в гексане), получая 4-((диэтоксифосфорил)окси)-3-(трифторметил)бензил карбамат. МС: (ES) m/z вычислено для C13H17F3NO6P [M+H]+ 372,1, найдено 372,0.
В раствор 4-((диэтоксифосфорил)окси)-3-(трифторметил)бензил карбамата (240 мг, 0,65 ммоль) в дихлорметане (2 мл) при 0°С добавляли оксалилхлорид (0,09 мл, 1,0 ммоль). Смесь нагревали при 40°С в течение 16 часов, затем упаривали в вакууме. Остаток растворяли в 2 мл ТГФ и добавляли в раствор 4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-2,5-дифторанилина (200 мг, 0,32 ммоль) в ТГФ (4 мл). Смесь перемешивали при комнатной температуре в течение 5 часов, затем упаривали в вакууме. Остаток очищали методом колоночной хроматографии на силикагеле (0-100% EtOAc в гексане), получая (4-диэтоксифосфорилокси-3-(трифторметил)фенил)метил N-((4-(5-((2,4-бис(трифторметил) фенил)метил)-2-(2,6-диэтилфенил)-6,6-диметил-4H-пирроло[3,4-c]пиразол-3-ил)-2,5-дифторфенил)карбамоил)карбамат.
Стадия b: В раствор (4-диэтоксифосфорилокси-3-(трифторметил)фенил)метил N-((4-(5-((2,4-бис(трифторметил)фенил)метил)-2-(2,6-диэтилфенил)-6,6-диметил-4H-пирроло[3,4-c]пиразол-3-ил)-2,5-дифторфенил)карбамоил)карбамата (176 мг, 0,17 ммоль) в дихлорметане (1,7 мл) добавляли по каплям TMSBr (0,29 мл, 2 ммоль). Смесь перемешивали при комнатной температуре в течение 16 часов, затем упаривали в вакууме. Остаток очищали методом ВЭЖХ (MeCN/H2O, с 0,1% ТФУК), получая (4-фосфоноокси-3-(трифторметил)фенил)метил N-((4-(5-((2,4-бис(трифторметил)фенил)метил)-2-(2,6-диэтилфенил)-6,6-диметил-4H-пирроло[3,4-c]пиразол-3-ил)-2,5-дифторфенил)карбамоил)карбамат. 1H ЯМР (400 МГц, ДМСО-d6) δ: 10,80 (с, 1H), 10,11 (с, 1H), 8,15 (с, 1H), 7,90-8,15 (м, 3H), 7,73 (ушир.с, 1H), 7,63-7,69 (м, 1H), 7,54-7,61 (м, 1H), 7,35-7,42 (м, 1H), 7,18-7,24 (м, 2H), 6,49-6,58 (м, 1H), 5,19 (с, 2H), 4,13 (с, 2H), 3,65 (с, 2H), 2,07-2,23 (м, 4H), 1,30-1,40 (м, 6H), 0,88-1,03 (м, 6H). МС: (ES) m/z вычислено для C42H37F11N5O7P [M+H]+ 964,2, найдено 964,0.
Пример 14. Синтез (3-нитро-4-фосфонооксифенил)метил N-((4-(5-((2,4-бис(трифторметил)фенил)метил)-2-(2,6-диэтилфенил)-6,6-диметил-4H-пирроло[3,4-c] пиразол-3-ил)-2,5-дифторфенил)карбамоил)карбамата
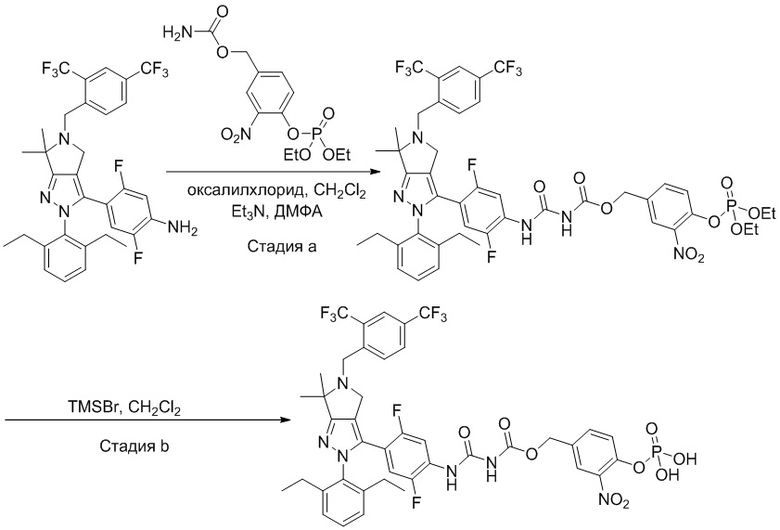
Стадия a: В раствор 4-гидрокси-3-нитробензальдегида (1 г, 6,0 ммоль) в дихлорметане (6 мл) добавляли Et3N (1,25 мл, 9,0 ммоль) и диэтилхлорфосфат (0,95 мл, 6,6 ммоль). Смесь перемешивали при комнатной температуре в течение 16 часов, затем гасили водой. Органический и водный слой разделяли, и водный слой экстрагировали этилацетатом. Органические слои объединяли, сушили над сульфатом натрия, фильтровали и упаривали в вакууме. Остаток очищали методом колоночной хроматографии на силикагеле (0-100% EtOAc в гексане), получая диэтил (4-формил-2-нитрофенил)фосфат. МС: (ES) m/z вычислено для C11H14NO7P [M+H]+ 304,1, найдено 304,0.
В раствор диэтил (4-формил-2-нитрофенил)фосфата (1,36 г, 4,5 ммоль) в ТГФ (4,5 мл) при -78°С добавляли NaBH4 (500 мг, 13,5 ммоль). После перемешивания при -78°С в течение 1 часа, реакцию гасили водой. Органический и водный слой разделяли, и водный слой экстрагировали этилацетатом. Органические слои объединяли, сушили над сульфатом натрия, фильтровали и упаривали. Остаток очищали методом колоночной хроматографии на силикагеле (0-100% EtOAc в гексане), получая диэтил (4-(гидроксиметил)-2-нитрофенил) фосфат. МС: (ES) m/z вычислено для C11H16NO7P [M+H]+ 306,1, найдено 306,1.
В раствор диэтил (4-(гидроксиметил)-2-нитрофенил)фосфата (200 мг, 0,66 ммоль) в ТГФ (1,6 мл) при 0°С добавляли диизопропилэтиламин (0,14 мл, 0,80 ммоль) и трифосген (100 мг, 0,34 ммоль). После перемешивания при 0°С в течение 1 часа, добавляли NH4OH (0,32 мл, 8,2 ммоль). Смесь перемешивали при комнатной температуре 1 час, затем упаривали в вакууме, и остаток очищали методом колоночной хроматографии на силикагеле (0-100% EtOAc в гексане), получая 4-((диэтоксифосфорил)окси)-3-нитробензил карбамат. МС: (ES) m/z вычислено для C12H17N2O8P [M+H]+ 349,1, найдено 349,0.
В раствор 4-((диэтоксифосфорил)окси)-3-(трифторметил)бензил карбамата (0,4 г, 1,2 ммоль) в дихлорметане (11,5 мл) при 0°С добавляли оксалилхлорид (0,15 мл, 1,8 ммоль). Смесь нагревали при 40°С в течение 16 часов, затем упаривали в вакууме. Остаток растворяли в ТГФ (1 мл) и добавляли в раствор 4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-2,5-дифторанилина (200 мг, 0,32 ммоль) в ТГФ (5 мл). Смесь перемешивали при комнатной температуре 3 часа, затем упаривали в вакууме. Остаток очищали методом колоночной хроматографии на силикагеле (0-100% EtOAc в гексане), получая (4-диэтоксифосфорилокси-3-нитрофенил)метил N-((4-(5-((2,4-бис(трифторметил)фенил)метил)-2-(2,6-диэтилфенил)-6,6-диметил-4H-пирроло[3,4-c]пиразол-3-ил)-2,5-дифторфенил)карбамоил)карбамат.
Стадия b: В раствор (4-диэтоксифосфорилокси-3-нитрофенил)метил N-((4-(5-((2,4-бис(трифторметил)фенил)метил)-2-(2,6-диэтилфенил)-6,6-диметил-4H-пирроло[3,4-c] пиразол-3-ил)-2,5-дифторфенил)карбамоил)карбамата (100 мг, 0,10 ммоль) в дихлорметане (1 мл) добавляли по каплям TMSBr (0,09 мл, 0,6 ммоль). После перемешивания при комнатной температуре 3 часа, добавляли в смесь дополнительное количество TMSBr (0,09 мл, 0,6 ммоль). Смесь перемешивали при комнатной температуре в течение 16 часов, упаривали в вакууме и очищали методом ВЭЖХ (MeCN/H2O, с 0,1% ТФУК), получая (3-нитро-4-фосфонооксифенил)метил N-((4-(5-((2,4-бис(трифторметил)фенил)метил)-2-(2,6-диэтилфенил)-6,6-диметил-4H-пирроло[3,4-c]пиразол-3-ил)-2,5-дифторфенил)карбамоил) карбамат. 1H ЯМР (400 МГц, CD3OD) δ: 8,25 (д, J=8,0 Гц, 1H), 8,16 (с, 2H), 8,01-8,12 (м, 1H), 7,82 (с, 1H), 7,54 (с, 2H), 7,46 (т, J=7,7 Гц, 1H), 7,28 (д, J=7,8 Гц, 2H), 6,38 (дд, J=7,6, 11,2 Гц, 1H), 5,20 (с, 2H), 4,81 (с, 2H), 4,66 (с, 2H), 2,25 (кв, J=7,7 Гц, 4H), 1,92 (с, 6H), 1,05 (т, J=7,7 Гц, 6H). МС: (ES) m/z вычислено для C41H37F8N6O9P [M+H]+ 941,2, найдено 941,1
- Пример 15. Синтез 2-(4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбонил)-5-фторбензил дигидрофосфат
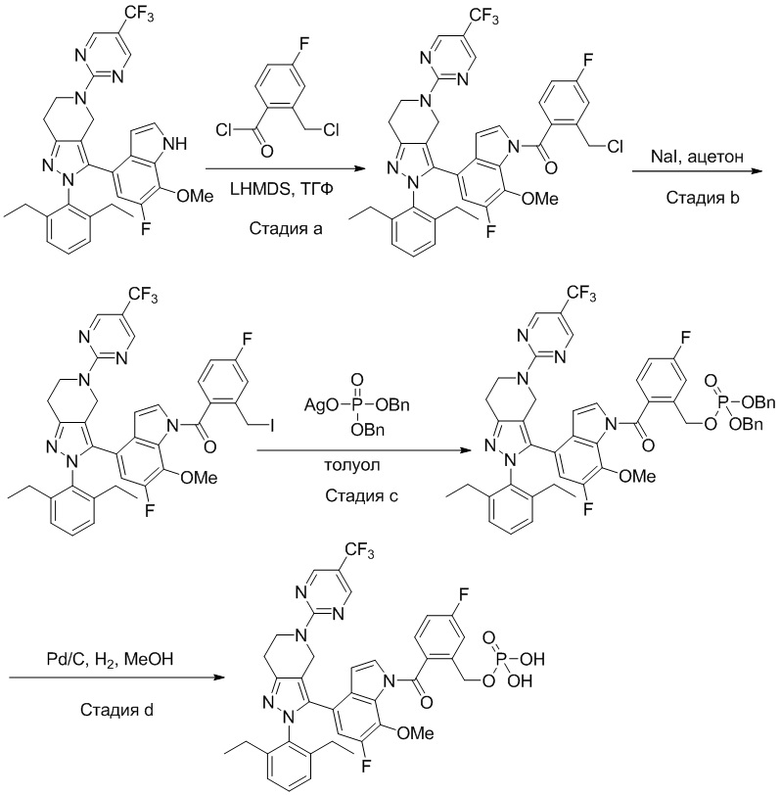
Стадия a: Пробирку, содержащую 5-фторизобензофуран-1(3H)-он (50 мг, 0,33 ммоль), борную кислоту (2 мг, 0,03 ммоль) и триэтилбензиламмония хлорид (6 мг, 0,03 ммоль), нагревали при 110°С. В смесь добавляли тионилхлорид (0,05 мл, 0,69 ммоль). После перемешивания при 110°С в течение 16 часов, смесь упаривали, получая 2-(хлорметил)-4-фторбензоилхлорид.
В раствор 2-(2,6-диэтилфенил)-3-(6-фтор-7-метокси-1H-индол-4-ил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридина (46 мг, 0,08 ммоль) в ТГФ (0,5 мл) при -78°С добавляли 1M раствор LHMDS в ТГФ (0,12 мл, 0,12 ммоль). Смесь перемешивали при -78°С в течение 30 минут, нагревали до 0°С и перемешивали 15 минут, затем снова охлаждали до -78°С. Добавляли по каплям в смесь раствор 2-(хлорметил)-4-фторбензоилхлорида (0,33 ммоль) в ТГФ (0,5 мл). После перемешивания при комнатной температуре в течение 16 часов, реакцию гасили водой. Водный и органический слой разделяли, и водный слой экстрагировали этилацетатом. Органические слои объединяли, сушили над сульфатом натрия, фильтровали и упаривали. Остаток очищали методом колоночной хроматографии на силикагеле (0-100% EtOAc в гексане), получая (2-(хлорметил)-4-фторфенил)(4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-ил)метанон. МС: (ES) m/z вычислено для C38H32ClF5N6O2 [M+H]+ 735,2, найдено 735,1.
Стадия b: В раствор (2-(хлорметил)-4-фторфенил)(4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-ил)метанона (48 мг, 0,065 ммоль) в ацетоне (1 мл) добавляли иодид натрия (32 мг, 0,21 ммоль). Смесь нагревали при 70°С в течение 1 часа, затем упаривали в вакууме. Остаток очищали методом колоночной хроматографии на силикагеле (0-100% EtOAc в гексане), получая (4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-ил)(4-фтор-2-(иодметил)фенил)метанон. МС: (ES) m/z вычислено для C38H32F5IN6O2 [M+H]+ 827,2, найдено 827,0.
Стадия c: В раствор (4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-ил)(4-фтор-2-(иодметил)фенил)метанона (26 мг, 0,031 ммоль) в толуоле ( 1 мл) добавляли дибензилфосфат серебра (24 мг, 0,062 ммоль). Смесь нагревали при 110°С в течение 16 часов, затем упаривали в вакууме. Остаток очищали методом колоночной хроматографии на силикагеле (0-100% EtOAc в гексане), получая дибензил (2-(4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбонил)-5-фторбензил)фосфат.
Стадия d: В раствор дибензил (2-(4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил) пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбонил)-5-фторбензил)фосфата (17 мг, 0,017 ммоль) в MeOH (1 мл) добавляли 10% Pd/C (2 мг). Смесь перемешивали под давлением водорода из шарика 1 час, затем фильтровали через целит, упаривали и очищали методом ВЭЖХ (MeCN/H2O, с 0,1% ТФУК), получая 2-(4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбонил)-5-фторбензил дигидрофосфат. 1H ЯМР (400 МГц, CD3OD) δ 8,58 (с, 2H), 7,60 (д, J=10,0 Гц, 1H), 7,49 (ушир.с, 2H), 7,33 (т, J=7,6 Гц, 1H), 7,11-7,24 (м, 3H), 6,65 (с, 1H), 6,59 (д, J=13,0 Гц, 1H), 5,35 (д, J=7,3 Гц, 2H), 4,87 (с, 2H), 4,38 (с, 2H), 3,75 (с, 3H), 2,98 (с, 2H), 2,25 (ушир.с, 4H), 1,02 (ушир.с, 6H). МС: (ES) m/z вычислено для C38H34F5N6O6P [M+H]+ 797,2, найдено 797,1.
Пример 16. Синтез 2-(4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбонил)бензил диметилглицината
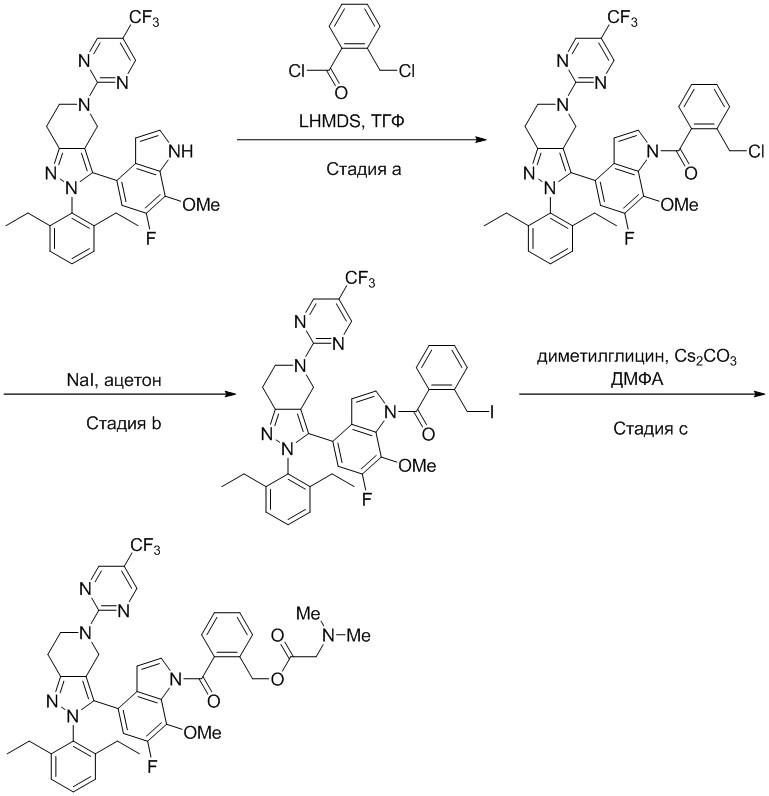
Стадия a: В раствор 2-(2,6-диэтилфенил)-3-(6-фтор-7-метокси-1H-индол-4-ил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридина (500 мг, 0,9 ммоль) в ТГФ (8,8 мл) при -78°С добавляли 1M раствор LHMDS в ТГФ (0,98 мл, 0,98 ммоль). После перемешивания при -78°С в течение 30 минут, добавляли в смесь 2-(хлорметил)бензоилхлорид (0,25 мл, 1,78 ммоль). После перемешивания при комнатной температуре в течение 16 часов, реакцию гасили водой. Водный и органический слой разделяли, и водный слой экстрагировали этилацетатом. Органические слои объединяли, сушили над сульфатом натрия, фильтровали и упаривали. Остаток очищали методом колоночной хроматографии на силикагеле (0-100% EtOAc в гексане), получая (2-(хлорметил)фенил)(4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-ил)метанон. МС: (ES) m/z вычислено для C38H33ClF4N6O2 [M+H]+ 717,2, найдено 717,0.
Стадия b: В раствор (2-(хлорметил)фенил)(4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-ил)метанона (156 мг, 0,22 ммоль) в ацетоне (2,2 мл) добавляли иодид натрия (129 мг, 0,9 ммоль). После нагревания при 70°С в течение 1 часа, реакцию гасили водой. Водный и органический слой разделяли, и водный слой экстрагировали этилацетатом. Органические слои объединяли, сушили над сульфатом натрия, фильтровали и упаривали. Остаток очищали методом колоночной хроматографии на силикагеле (0-100% EtOAc в гексане), получая (4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-ил)(2-(иодметил)фенил)метанон.
Стадия c: В раствор (4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-ил)(2-(иодметил)фенил)метанона (76 мг, 0,094 ммоль) в ДМФА (1 мл) при 0°С добавляли Cs2CO3 (31 мг, 0,1 ммоль) и диметилглицин (10 мг, 0,1 ммоль). После перемешивания при комнатной температуре в течение 16 часов, реакцию гасили водой. Водный и органический слой разделяли, и водный слой экстрагировали этилацетатом. Органические слои объединяли, сушили над сульфатом натрия, фильтровали и упаривали. Остаток очищали методом ВЭЖХ (MeCN/H2O, с 0,1% ТФУК), получая 2-(4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил) пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбонил)бензил диметилглицинат. 1H ЯМР (400 МГц, CDCl3) δ 8,54 (с, 2H), 7,65-7,81 (м, 4H), 7,31 (т, J=7,7 Гц, 1H), 7,22 (д, J=3,7 Гц, 1H), 7,11 (ушир.с, 2H), 6,64 (д, J=12,7 Гц, 1H), 6,52 (д, J=3,7 Гц, 1H), 5,08 (с, 2H), 4,76 (с, 2H), 4,37 (с, 2H), 4,14 (с, 2H), 3,85 (с, 3H), 3,32 (с, 6H), 3,09 (т, J=5,9 Гц, 2H), 2,08 (ушир.с, 4H), 1,04 (ушир.с, 6H). МС: (ES) m/z вычислено для C42H41F4N7O4 [M+H]+ 784,3, найдено 784,2.
Пример 17. Синтез 3-хлор-4-(фосфоноокси)бензил 4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-карбоксилата
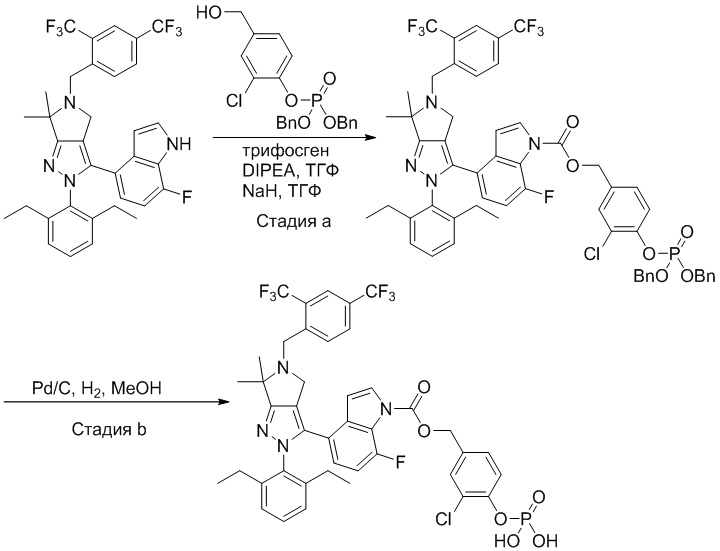
Стадия a: В колбу, содержащую 3-хлор-4-гидроксибензойную кислоту (5 г, 29 ммоль) в MeOH (100 мл) добавляли по каплям тионилхлорид (7,4 мл, 102 ммоль). Смесь нагревали при 60°С в течение 1 часа, затем упаривали в вакууме. Полученный остаток растворяли в ТГФ (100 мл). В раствор добавляли LiAlH4 (4,4 г, 110 ммоль). После нагревания при 65°С в течение 1 часа, реакцию гасили 1н. раствором HCl. Водный и органический слой разделяли, и водный слой экстрагировали этилацетатом. Органические слои объединяли, сушили над сульфатом натрия, фильтровали и упаривали. Остаток очищали методом колоночной хроматографии на силикагеле (0-100% EtOAc в гексане), получая 2-хлор-4-(гидроксиметил)фенол.
В раствор 2-хлор-4-(гидроксиметил)фенола (2 г, 12,7 ммоль) в диоксане (25 мл) добавляли DDQ (2,87 г, 12,7 ммоль). Смесь перемешивали при комнатной температуре в течение 4 часов, затем фильтровали. Фильтрат промывали водой, затем сушили над сульфатом натрия, фильтровали и упаривали. Остаток очищали методом колоночной хроматографии на силикагеле (0-100% EtOAc в гексане), получая 3-хлор-4-гидроксибензальдегид. МС: (ES) m/z вычислено для C7H5ClO2 [M+H]+ 157,0, найдено 157,0.
В колбу, содержащую 3-хлор-4-гидроксибензальдегид (1,4 г, 9,0 ммоль) в ТГФ (14,6 мл) добавляли по каплям 1,0 M раствор tBuOK в ТГФ (9,5 мл, 9,5 ммоль) и тетрабензилдифосфат (4,8 г, 8,9 ммоль). Смесь нагревали при 70°С в течение 2 часов. После завершения добавляли гексан и фильтровали смесь. Фильтрат упаривали, и полученный остаток очищали методом колоночной хроматографии на силикагеле (0-100% EtOAc в гексане), получая дибензил (2-хлор-4-формилфенил)фосфат. МС: (ES) m/z вычислено для C21H18ClO5P [M+H]+ 417,1, найдено 417,0.
В раствор дибензил (2-хлор-4-формилфенил)фосфата (2,75 г, 6,6 ммоль) в ТГФ (6,6 мл) при -78°С добавляли NaBH4 (0,73 г, 19,7 ммоль). После перемешивания при -78°С в течение 1 часа, реакцию гасили водой. Органический и водный слой разделяли, и водный слой экстрагировали этилацетатом. Органические слои объединяли, сушили над сульфатом натрия, фильтровали и упаривали в вакууме. Полученный остаток очищали методом колоночной хроматографии на силикагеле (0-100% EtOAc в гексане), получая дибензил (2-хлор-4-(гидроксиметил)фенил)фосфат. МС: (ES) m/z вычислено для C21H20ClO5P [M+H]+ 419,1, найдено 419,0.
В раствор дибензил (2-хлор-4-(гидроксиметил)фенил)фосфата (250 мг, 0,6 ммоль) в ТГФ (3 мл) при 0°С добавляли диизопропилэтиламин (0,12 мл, 0,69 ммоль) и трифосген (92 мг, 0,3 ммоль). После перемешивания при 0°С в течение 1 часа, реакцию гасили водой. Органический и водный слой разделяли, и водный слой экстрагировали этилацетатом. Органические слои объединяли, сушили над сульфатом натрия, фильтровали и упаривали, получая сырой хлорформиат.
В раствор 4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индола (187 мг, 0,30 ммоль) в ТГФ (1,5 мл) при 0°С добавляли NaH (24 мг, 0,60 ммоль). После перемешивания при 0°С в течение 30 минут, добавляли в смесь раствор сырого хлорформиата (получен как описано выше) в ТГФ (1,5 мл). После перемешивания при 0°С в течение 1 часа, реакцию гасили водой. Органический и водный слой разделяли, и водный слой экстрагировали этилацетатом. Органические слои объединяли, сушили над сульфатом натрия, фильтровали и упаривали в вакууме. Остаток очищали методом колоночной хроматографии на силикагеле (0-100% EtOAc в гексане), получая 4-((бис(бензилокси)фосфорил)окси)-3-хлорбензил 4-(5-(2,4-бис (трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c] пиразол-3-ил)-7-фтор-1H-индол-1-карбоксилат.
Стадия b: В раствор 4-((бис(бензилокси)фосфорил)окси)-3-хлорбензил 4-(5-(2,4-бис (трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c] пиразол-3-ил)-7-фтор-1H-индол-1-карбоксилата (298 мг, 0,28 ммоль) в MeOH (4 мл) добавляли 10% Pd/C (30 мг, 0,03 ммоль). Смесь перемешивали под давлением водорода из шарика 1 час, затем фильтровали через целит, упаривали и очищали методом ВЭЖХ (MeCN/H2O, с 0,1% ТФУК), получая 3-хлор-4-(фосфоноокси)бензил 4-(5-(2,4-бис (трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c] пиразол-3-ил)-7-фтор-1H-индол-1-карбоксилат. 1H ЯМР (400 МГц, CD3OD) δ 8,12 (с, 1H), 8,04-8,12 (м, 2H), 7,83 (с, 1H), 7,55 (с, 1H), 7,51 (д, J=8,4 Гц, 1H), 7,33-7,37 (м, 2H), 7,17 (д, J=7,9 Гц, 2H), 6,82 (дд, J=10,2, 10,2 Гц, 1H), 6,67 (ушир.с, 2H), 5,36 (с, 2H), 4,77 (с, 2H), 4,50 (с, 2H), 2,16-2,29 (м, 4H), 1,93 (ушир.с, 6H), 0,99 (ушир.с, 6H). МС: (ES) m/z вычислено для C42H37ClF7N4O6P [M+H]+ 893,2, найдено 893,1.
Пример 18. Синтез 3-фтор-4-(фосфоноокси)бензил 4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-карбоксилата
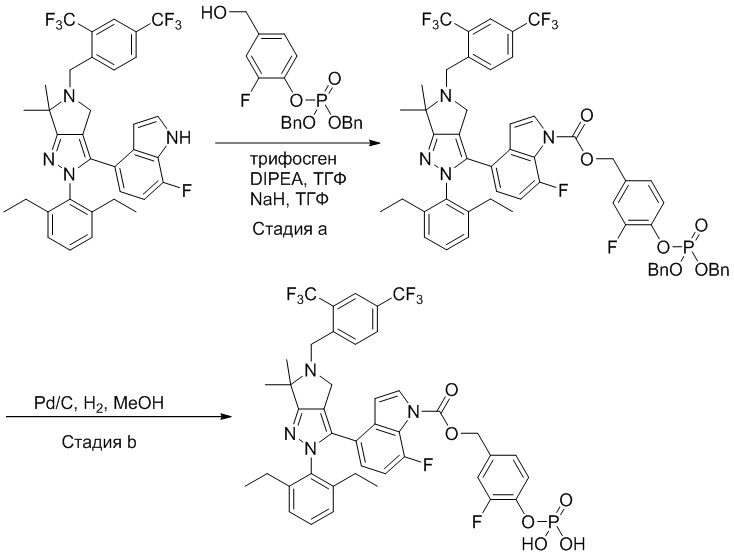
Стадия a: В колбу, содержащую 3-фтор-4-гидроксибензальдегид (1 г, 7,1 ммоль) в ТГФ (32 мл), добавляли по каплям 1,0 M раствор tBuOK в ТГФ (7,6 мл, 7,6 ммоль) и тетрабензилдифосфат (4,0 г, 7,4 ммоль). Смесь нагревали при 70°С в течение 1 часа. После завершения добавляли гексан и смесь фильтровали. Фильтрат упаривали, и остаток очищали методом колоночной хроматографии на силикагеле (0-100% EtOAc в гексане), получая дибензил (2-фтор-4-формилфенил)фосфат. МС: (ES) m/z вычислено для C21H18FO5P [M+H]+ 401,1, найдено 401,1.
В раствор дибензил (2-фтор-4-формилфенил)фосфата (2,68 г, 6,7 ммоль) в ТГФ (6,7 мл) при -78°С добавляли NaBH4 (0,76 г, 20,5 ммоль). После перемешивания при -78°С в течение 1 часа, реакцию гасили водой. Органический и водный слой разделяли, и водный слой экстрагировали этилацетатом. Органические слои объединяли, сушили над сульфатом натрия, фильтровали и упаривали в вакууме. Остаток очищали методом колоночной хроматографии на силикагеле (0-100% EtOAc в гексане), получая дибензил (2-фтор-4-(гидроксиметил)фенил)фосфат. МС: (ES) m/z вычислено для C21H20FO5P [M+H]+ 403,1, найдено 403,0.
В раствор дибензил (2-фтор-4-(гидроксиметил)фенил)фосфата (250 мг, 0,62 ммоль) в ТГФ (3 мл) при 0°С добавляли диизопропилэтиламин (0,12 мл, 0,69 ммоль) и трифосген (92 мг, 0,3 ммоль). После перемешивания при комнатной температуре в течение 30 минут, реакцию гасили водой. Органический и водный слой разделяли, и водный слой экстрагировали этилацетатом. Органические слои объединяли, сушили над сульфатом натрия, фильтровали и упаривали, получая сырой хлорформиат.
В раствор 4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индола (195 мг, 0,31 ммоль) в ТГФ (1,5 мл) при 0°С добавляли NaH (24 мг, 0,60 ммоль). После перемешивания при комнатной температуре в течение 30 минут, добавляли в смесь раствор сырого хлорформиата (получен как описано выше) в ТГФ (1,5 мл). После перемешивания при комнатной температуре в течение 16 часов, реакцию гасили водой. Органический и водный слой разделяли, и водный слой экстрагировали этилацетатом. Органические слои объединяли, сушили над сульфатом натрия, фильтровали и упаривали в вакууме. Остаток очищали методом колоночной хроматографии на силикагеле (0-100% EtOAc в гексане), получая 4-((бис(бензилокси) фосфорил)окси)-3-фторбензил 4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-карбоксилат.
Стадия b: В раствор 4-((бис(бензилокси)фосфорил)окси)-3-фторбензил 4-(5-(2,4-бис (трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c] пиразол-3-ил)-7-фтор-1H-индол-1-карбоксилата (307 мг, 0,29 ммоль) в EtOAc (1,5 мл) добавляли 10% Pd/C (30 мг, 0,03 ммоль). Смесь перемешивали под давлением водорода из шарика 3 часа, затем фильтровали через целит, упаривали и очищали методом ВЭЖХ (MeCN/H2O, с 0,1% ТФУК), получая 3-фтор-4-(фосфоноокси)бензил 4-(5-(2,4-бис (трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c] пиразол-3-ил)-7-фтор-1H-индол-1-карбоксилат. 1H ЯМР (400 МГц, CD3OD) δ: 8,13 (с, 1H), 8,05-8,11 (м, 2H), 7,83 (м, 1H), 7,44 (дд, J=8,4, 8,4 Гц, 1H), 7,30-7,37 (м, 2H), 7,23 (д, J=8,4 Гц, 1H), 7,17 (д, J=7,8 Гц, 2H), 6,83 (дд, J=10,4, 10,4 Гц, 1H), 6,67 (ушир.с, 2H), 5,38 (с, 2H), 4,80 (с, 2H), 4,54 (с, 2H), 2,15-2,29 (м, 4H), 1,96 (ушир.с, 6H), 0,99 (ушир.с, 6H). МС: (ES) m/z вычислено для C42H37F8N4O6P [M+H]+ 877,2, найдено 877,1.
Пример 19. Синтез 2-((фосфоноокси)метил)фенил 4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-карбоксилата
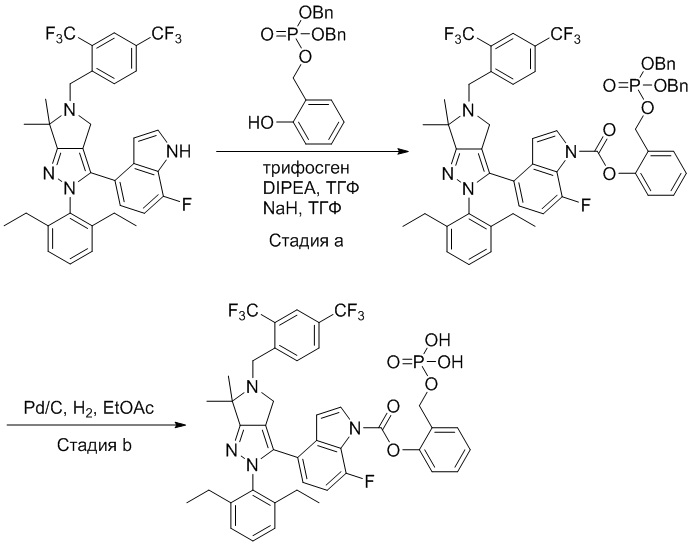
Стадия a: В колбу, содержащую 2-гидроксибензальдегид (10 г, 82 ммоль) в ДМФА (82 мл) добавляли по каплям имидазол (6,1 г, 90 ммоль) и TBDMSCl (13,6 г, 90 ммоль). После перемешивания при комнатной температуре 3 часа, реакцию гасили водой. Смесь экстрагировали этилацетатом. Объединенные органические слои сушили над сульфатом натрия, фильтровали и упаривали. Остаток очищали методом колоночной хроматографии на силикагеле (0-100% EtOAc в гексане), получая 2-((трет-бутилдиметилсилил)окси)бензальдегид. МС: (ES) m/z вычислено для C13H20O2Si [M+H]+ 237,1, найдено 237,1.
В раствор 2-((трет-бутилдиметилсилил)окси)бензальдегида (16,4 г, 69 ммоль) в MeOH (126 мл) добавляли NaBH4 (2,55 г, 67,4 ммоль). После перемешивания при комнатной температуре в течение 2 часов, реакцию гасили водой. Смесь упаривали в вакууме, затем экстрагировали гексаном. Органические слои объединяли, сушили над сульфатом натрия, фильтровали и упаривали в вакууме. Остаток очищали методом колоночной хроматографии на силикагеле (0-10% EtOAc в гексане), получая (2-((трет-бутилдиметилсилил)окси)фенил) метанол.
В колбу, содержащую (2-((трет-бутилдиметилсилил)окси)фенил)метанол (1 г, 4,2 ммоль) в ТГФ (42 мл), добавляли по каплям 1,0 M раствор tBuOK в ТГФ (4,6 мл, 4,6 ммоль) и тетрабензилдифосфат (2,5 г, 4,6 ммоль). Смесь нагревали при 60°С в течение 1 часа. После завершения добавляли гексан и смесь фильтровали. Фильтрат упаривали, и остаток использовали далее без дополнительной очистки.
В раствор сырого остатка в MeCN (42 мл) добавляли по каплям HF-пиридин (4,2 мл). После перемешивания при комнатной температуре в течение 1 часа, смесь упаривали в вакууме. Остаток очищали методом колоночной хроматографии на силикагеле (0-100% EtOAc в гексане), получая дибензил (2-гидроксибензил)фосфат. МС: (ES) m/z вычислено для C21H21O5P [M+H]+ 385,1, найдено 385,0.
В раствор дибензил (2-гидроксибензил)фосфата (366 мг, 0,95 ммоль) в ТГФ (4,8 мл) добавляли диизопропилэтиламин (0,18 мл, 1,0 ммоль) и трифосген (139 мг, 0,47 ммоль). После перемешивания при комнатной температуре в течение 30 минут, реакцию гасили водой. Органический и водный слой разделяли, и водный слой экстрагировали этилацетатом. Органические слои объединяли, сушили над сульфатом натрия, фильтровали и упаривали.
В раствор 4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индола (300 мг, 0,48 ммоль) в ТГФ (4,8 мл) добавляли NaH (37 мг, 0,97 ммоль). После перемешивания при комнатной температуре в течение 20 минут, добавляли в смесь раствор сырого хлорформиата (получен как описано выше) в 1 мл ТГФ. После перемешивания при комнатной температуре в течение 16 часов, реакцию гасили водой. Органический и водный слой разделяли, и водный слой экстрагировали этилацетатом. Органические слои объединяли, сушили над сульфатом натрия, фильтровали и упаривали в вакууме. Остаток очищали методом колоночной хроматографии на силикагеле (0-100% EtOAc в гексане), получая 2-(((бис(бензилокси) фосфорил)окси)метил)фенил 4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-карбоксилат.
Стадия b: В раствор 2-(((бис(бензилокси)фосфорил)окси)метил)фенил 4-(5-(2,4-бис (трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c] пиразол-3-ил)-7-фтор-1H-индол-1-карбоксилата (500 мг, 0,48 ммоль) в EtOAc (2,4 мл) добавляли 10% Pd/C (51 мг). Смесь перемешивали под давлением водорода из шарика 2 часа, затем фильтровали через целит, упаривали и очищали методом ВЭЖХ (MeCN/H2O, с 0,1% ТФУК), получая 2-((фосфоноокси)метил)фенил 4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-карбоксилат. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,12 (д, J=8,1 Гц, 1H), 7,94-8,06 (м, 2H), 7,85 (с, 1H), 7,51 (д, J=7,6 Гц, 1H), 7,35-7,39 (м, 1H), 7,28-7,33 (м, 2H), 7,07-7,20 (м, 3H), 6,90-6,95 (м, 1H), 6,60 (с, 1H), 6,56 (дд, J=8,4, 3,7 Гц, 1H), 5,42 (с, 2H), 4,15 (ушир.с, 2H), 3,59 (ушир.с, 2H), 2,16 (кв, J=7,8 Гц, 4H), 1,48 (ушир.с, 6H), 0,90 (т, J=7,5 Гц, 6H). МС: (ES) m/z вычислено для C42H38F7N4O6P [M+H]+ 859,2, найдено 859,2.
- Пример 20. Синтез 2-(4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-карбонил)бензил дигидрофосфата
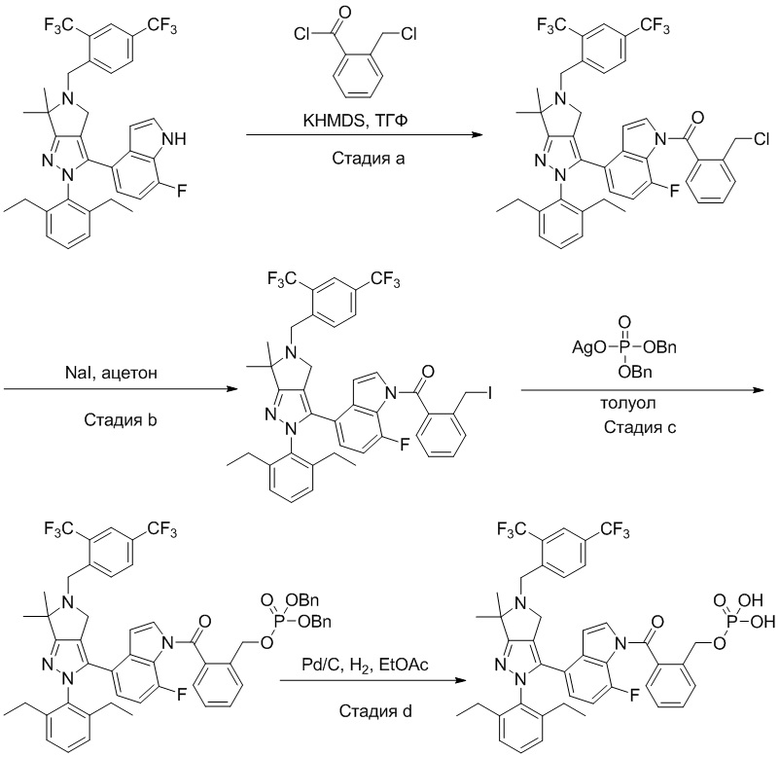
Стадия a: В раствор 4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индола (500 мг, 0,80 ммоль) в ТГФ (8 мл) при -78°С добавляли 0,5 M раствор KHMDS в ТГФ (2,6 мл, 1,3 ммоль). После перемешивания при -78°С в течение 30 минут, добавляли в смесь 2-(хлорметил) бензоилхлорид (0,28 мл, 1,9 ммоль). После перемешивания при комнатной температуре в течение 16 часов, реакционную смесь разбавляли этилацетатом и промывали насыщенным водным раствором NaHCO3. Водный и органический слой разделяли, и водный слой экстрагировали этилацетатом. Органические слои объединяли, сушили над сульфатом натрия, фильтровали и упаривали. Остаток очищали методом колоночной хроматографии на силикагеле (0-100% EtOAc в гексане), получая (4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-ил)(2-(хлорметил)фенил)метанон. МС: (ES) m/z вычислено для C42H36ClF7N4O [M+H]+ 781,3, найдено 781,0.
Стадия b: В раствор (4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-ил)(2-(хлорметил)фенил)метанона (194 мг, 0,25 ммоль) в ацетоне (3,1 мл) добавляли иодид натрия (149 мг, 0,99 ммоль). После нагревания при 70°С в течение 2 часов, реакционную смесь разбавляли этилацетатом и промывали водой. Водный и органический слой разделяли, и водный слой экстрагировали этилацетатом. Органические слои объединяли, сушили над сульфатом натрия, фильтровали и упаривали, получая (4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-ил)(2-(иодметил)фенил)метанон, который использовали в последующей стадии без дополнительной очистки.
Стадия c: В раствор сырого (4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-ил)(2-(иодметил)фенил)метанона (0. 25 ммоль) в толуоле ( 1 мл) добавляли дибензилфосфат серебра (190 мг, 0,49 ммоль). После нагревания смеси при 110°С в течение 3 часов, смесь фильтровали через целит. Фильтрат упаривали, и остаток очищали методом колоночной хроматографии на силикагеле (0-100% EtOAc в гексане), получая дибензил (2-(4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-карбонил)бензил)фосфат.
Стадия d: В раствор дибензил (2-(4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-карбонил)бензил)фосфата (96 мг) в EtOAc (1 мл) добавляли 10% Pd/C (10 мг, 0,009 ммоль). Смесь перемешивали под давлением водорода из шарика 4 часа, затем фильтровали через целит, упаривали и очищали методом ВЭЖХ (MeCN/H2O, с 0,1% ТФУК), получая 2-(4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло [3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-карбонил)бензил дигидрофосфат. 1H ЯМР (400 МГц, CD3OD) δ: 8,12 (с, 1H), 8,09 (ушир.с, 2H), 7,74 (д, J=7,7 Гц, 1H), 7,67 (ддд, J=1,4, 7,3, 7,3 Гц, 1H), 7,57 (дд, J=1,4, 7,7 Гц, 1H), 7,49 (ддд, J=1,4, 7,2, 7,2 Гц, 1H), 7,40 (д, J=3,7 Гц, 1H), 7,36 (д, J=7,7 Гц, 1H), 7,20 (д, J=7,7 Гц, 1H), 6,84 (дд, J=8,3, 11,3 Гц, 1H), 6,72 (дд, J=3,8, 8,4 Гц, 1H), 6,67 (дд, J=1,8, 3,8 Гц, 1H), 5,19 (д, J=7,4 Гц, 2H), 4,75 (с, 2H), 4,55 (с, 2H), 2,26 (кв, J=7,5 Гц, 4H), 1,93 (с, 6H), 1,03 (т, J=7,5 Гц, 6H). МС: (ES) m/z вычислено для C42H38F7N4O5P [M+H]+ 843,3, найдено 843,2.
Пример 21. Синтез ((4-(пиперазин-1-илметил)бензоил)окси)метил 4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбоксилата

Стадия a: Смесь иодметил 4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбоксилата (840 мг, 1,12 ммоль), 4-формилбензойной кислоты (252 мг, 1,68 ммоль) и диизопропилэтиламина (0,74 мл, 4,48 ммоль) в дихлорметане (20 мл) нагревали до 50°C в течение 3 часов. Смесь охлаждали до комнатной температуры, выливали в насыщенный водный раствор NaHCO3 и экстрагировали дихлорметаном. Органический слой отделяли, сушили над Na2SO4, упаривали при пониженном давлении и очищали методом флэш-хроматографии на силикагеле (0-60% EtOAc в гексане), получая ((4-формилбензоил)окси) метил 4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбоксилат. МС: (ES) m/z вычислено для C40H35F4N6O6[M+H]+ 771,2, найдено 771,2.
Стадия b: В 200-миллилитровую колбу, содержащую пиперазин (1,0 г, 11,6 ммоль) в дихлорметане (25 мл), при 0°C добавляли последовательно уксусную кислоту (40 мл), NaBH(OAc)3 (1,5 г, 7,0 ммоль) и ((4-формилбензоил)окси)метил 4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбоксилат (0,5 г, 0,65 ммоль). Смесь оставляли нагреваться до комнатной температуры и перемешивали в течение ночи. Смесь охлаждали до 0°C и добавляли 2M раствор HCl в эфире (20 мл, 40 ммоль). Смесь очищали методом флэш-хроматографии на силикагеле (0-100% EtOAc в гексане, затем 0-60 % MeOH в CH2Cl2). Чистые фракции объединяли, охлаждали до 0°C, добавляли 2M раствор HCl в эфире (10 мл, 20 ммоль) и упаривали при пониженном давлении, получая HCl-соль ((4-(пиперазин-1-илметил)бензоил)окси)метил 4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбоксилата. 1H ЯМР (400 МГц, CD3OD) δ 8,58 (с, 2H), 8,21 (д, J=8,4 Гц, 2H), 7,80 (д, J=3,6 Гц, 1H), 7,75 (д, J=8,0 Гц, 2H), 7,33 (т, J=7,8 Гц, 1H), 7,16 (ушир.с, 2H), 6,59-6,66 (м, 2H), 6,30 (с, 2H), 4,32-4,50 (м, 4H), 3,96 (д, J=1,2 Гц, 3H), 3,40-3,60 (м, 9H), 3,28-3,34 (м, 3H), 2,98 (дд, J=5,8, 5,8 Гц, 2H), 2,25 (ушир.с, 4H), 1,00 (ушир.с, 6H); МС (свободная форма): (ES) m/z вычислено для C44H45F4N8O5[M+H]+ 841,3, найдено 841,7.
Пример 22. Синтез ((4-((фосфоноокси)метил)бензоил)окси)метил 4-(5-(2,4-бис (трифторметил)-бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло [3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-карбоксилата

Стадия a: В раствор 4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индола (3,0 г, 4,76 ммоль) в ТГФ (30 мл) при -78°C добавляли 1M раствор LiHMDS в толуоле (7,61 мл, 7,61 ммоль) по каплям. Смесь перемешивали при той же температуре еще 15 минут. Добавляли в смесь хлорметил хлорформиат (0,83 мл, 9,52 ммоль). Полученную смесь оставляли нагреваться до комнатной температуры и перемешивали 0,5 часа. Смесь гасили насыщенным водным раствором NH4Cl и экстрагировали этилацетатом. Органический слой отделяли, промывали насыщенным водным раствором NaHCO3, сушили над Na2SO4, упаривали при пониженном давлении и очищали методом флэш-хроматографии на силикагеле (0 t-o 40% EtOAc в гексане), получая хлорметил 4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-карбоксилат. МС: (ES) m/z вычислено для C36H33ClF7N4O2[M+H]+ 721,1, найдено 721,0.
Стадия b: Смесь хлорметил 4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-карбоксилата (2,5 г, 3,46 ммоль) и NaI (6,0 г, 40,0 ммоль) в ацетоне (50 мл) нагревали при 45°C в течение 7 часов. Смесь охлаждали до комнатной температуры, выливали в насыщенный водный раствор NaHCO3 и экстрагировали этилацетатом. Органический слой отделяли, сушили над Na2SO4, упаривали при пониженном давлении и очищали методом флэш-хроматографии на силикагеле (0-40% EtOAc в гексане), получая иодметил 4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-карбоксилат. МС: (ES) m/z вычислено для C36H33F7IN4O2[M+H]+ 813,2, найдено 813,2.
Стадия c: Смесь иодметил 4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-карбоксилата (156 мг, 0,19 ммоль) и ((4-(((бис(бензилокси)фосфорил)окси)метил)бензоил)окси)серебра (100 мг, 0,19 ммоль) в толуоле (10 мл) нагревали при 110°C в течение 1 часа. Смесь охлаждали до комнатной температуры, выливали в насыщенный водный раствор NaHCO3 и экстрагировали этилацетатом. Органический слой отделяли, сушили над Na2SO4, упаривали при пониженном давлении и очищали методом флэш-хроматографии на силикагеле (0-60% EtOAc в гексане, затем 0-30% EtOAc в CH2Cl2), получая ((4-(((бис(бензилокси)фосфорил) окси)метил)-бензоил)окси)метил 4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-карбоксилат. МС: (ES) m/z вычислено для C58H53F7N4O8P[M+H]+ 1097,3, наблюдались только фрагменты молекулярного пика.
Стадия d: Смесь((4-(((бис(бензилокси)фосфорил)окси)метил)бензоил)окси)метил 4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-карбоксилата (0,10 г, 0,09 ммоль), ТФУК (1 мл), дихлорметана (1 мл) и воды (1 мл) нагревали при 45°C в течение 6 часов. Смесь охлаждали до комнатной температуры, упаривали при пониженном давлении и очищали методом ВЭЖХ (MeCN/H2O, с 0,1% ТФУК), получая ((4-((фосфоноокси)метил)бензоил)окси) метил 4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-карбоксилат в виде соли с ТФУК. 1H ЯМР (400 МГц, CD3OD) δ 8,01-8,13 (м, 6H), 7,85 (д, J=3,6 Гц, 1H), 7,54 (д, J=8,4 Гц, 2H), 7,35 (дд, J=7,8, 7,8 Гц, 1H), 7,16 (д, J=8,0 Гц, 2H), 6,81-6,88 (м, 1H), 6,66-6,71 (м, 2H), 6,26 (с, 2H), 5,07 (д, J=7,2 Гц, 2H), 4,70 (ушир.с, 2H), 4,40 (ушир.с, 2H), 3,30 (ушир.с, 2H), 2,16-2,30 (м, 4H), 1,88 (с, 6H), 1,00 (т, J=7,6 Гц, 6H); МС: (ES) m/z вычислено для C44H41F7N4O8P[M+H]+ 917,3, найдено 917,1.
Пример 23. Синтез ((4-(пиперазин-1-илметил)бензоил)окси)метил 4-(5-(2,4-бис (трифторметил)-бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло [3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-карбоксилата
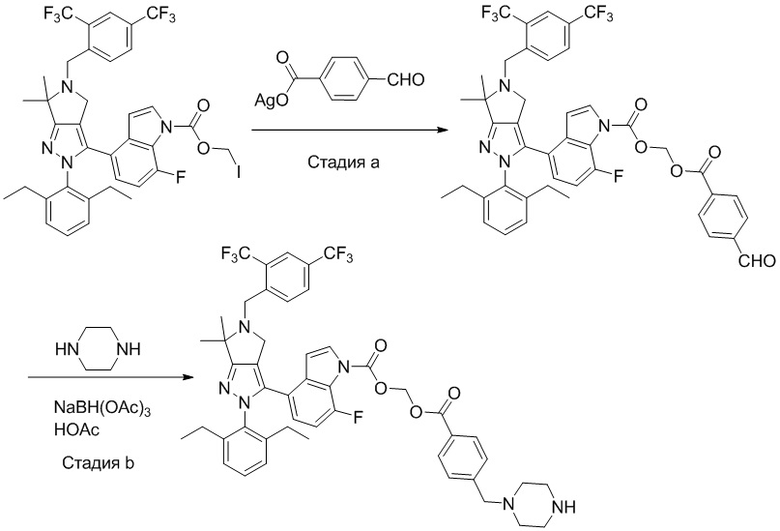
Стадия a: Смесь 4-формилбензойной кислоты (0,5 г, 3,3 ммоль) и LiOH моногидрата (0,15 г, 3,7 ммоль) в ТГФ (7,5 мл), MeOH (1 мл) и воде (0,5 мл) перемешивали при комнатной температуре в течение 15 минут. В смесь добавляли AgNO3 (0,65 г, 3,8 ммоль). Смесь перемешивали еще 15 минут и упаривали досуха при пониженном давлении, получая ((4-формилбензоил)окси)серебро.
Смесь иодметил 4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-карбоксилата (0,30 г, 0,37 ммоль) и описанного выше ((4-формилбензоил)окси)серебра (0,14 г, 0,55 ммоль) в толуоле (5 мл) нагревали при 100°C в течение 1 часа. Смесь охлаждали до комнатной температуры, выливали в насыщенный водный раствор NaHCO3 и экстрагировали этилацетатом. Органический слой отделяли, сушили над Na2SO4, упаривали при пониженном давлении и очищали методом флэш-хроматографии на силикагеле (0-60% EtOAc в гексане), получая ((4-формилбензоил)окси)метил 4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-карбоксилат. МС: (ES) m/z вычислено для C44H38F7N4O5[M+H]+ 835,3, найдено 835,3.
Стадия c: В пробирку, содержащую пиперазин (0,150 г, 1,74 ммоль) в дихлорметане (4 мл), при 0°C добавляли уксусную кислоту (3 мл), NaBH(OAc)3 (0,400 г, 1,88 ммоль) и ((4-формилбензоил)окси)метил 4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-карбоксилат (0,060 г, 0,071 ммоль) последовательно. Смесь оставляли нагреваться до комнатной температуры и перемешивали в течение ночи. Смесь охлаждали до 0°C и гасили реакцию 2M раствором HCl в эфире (2 мл, 4 ммоль). Смесь очищали методом флэш-хроматографии на силикагеле (0-100% MeOH в CH2Cl2). Чистые фракции объединяли, охлаждали до 0°C, добавляли 2M раствор HCl в эфире (2 мл, 4 ммоль) и упаривали при пониженном давлении, получая HCl-соль ((4-(пиперазин-1-илметил)бензоил)окси)метил 4-(5-(2,4-бис(трифторметил) бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-карбоксилата. 1H ЯМР (400 МГц, CD3OD) δ 8,04-8,26 (м, 6H), 7,74-7,90 (м, 3H), 7,35 (дд, J=7,2, 7,2 Гц, 1H), 7,16 (д, J=7,2 Гц, 2H), 6,80-6,90 (м, 2H), 6,66-6,74 (м, 1H), 6,27 (с, 2H), 4,80-5,00 (м, 2H), 4,58 (с, 2H), 4,36 (ушир.с, 1H), 3,63 (ушир.с, 10H), 2,23 (ушир.с, 4H), 2,03 (ушир.с, 6H), 0,99 (т, J=6,4 Гц, 6H); МС: (ES) m/z вычислено для C48H48F7N6O4 [M+H]+ 905,3, найдено 905,3.
Пример 24. Синтез ((4-((фосфоноокси)метил)бензоил)окси)метил 4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбоксилата

Стадия a: Смесь иодметил 4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбоксилата (0,15 г, 0,20 ммоль) и ((4-(((бис(бензилокси)фосфорил)окси)метил)бензоил) окси)серебра (0,11 г, 0,21 ммоль) в толуоле (4 мл) нагревали при 110°C в течение 1 часа. Смесь охлаждали до комнатной температуры, выливали в насыщенный водный раствор NaHCO3 и экстрагировали этилацетатом. Органический слой отделяли, сушили над Na2SO4, упаривали при пониженном давлении и очищали методом флэш-хроматографии на силикагеле (0-40% EtOAc в CH2Cl2), получая ((4-(((бис(бензилокси)фосфорил)окси)метил) бензоил)окси)метил 4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбоксилат. МС: (ES) m/z вычислено для C54H50F4N6O9P[M+H]+ 1033,3, наблюдались только фрагменты молекулярного пика.
Стадия b: Смесь ((4-(((бис(бензилокси)фосфорил)окси)метил)бензоил)окси)метил 4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло [4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбоксилата (0,08 г, 0,08 ммоль), ТФУК (1 мл), дихлорметана (1 мл) и воды (1 мл) нагревали при 45°C в течение 7 часов. Смесь охлаждали до комнатной температуры, упаривали при пониженном давлении и очищали методом ВЭЖХ (MeCN/H2O, с 0,1% ТФУК), получая ((4-((фосфоноокси)метил)бензоил)окси) метил 4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбоксилат. 1H ЯМР (400 МГц, CD3OD) δ 8,55 (с, 2H), 8,10 (д, J=7,6 Гц, 2H), 7,78 (с, 1H), 7,54 (д, J=8,0 Гц, 2H), 7,32 (дд, J=7,2 Гц, 1H), 7,15 (ушир.с, 2H), 6,57-6,64 (м, 2H), 6,28 (с, 2H), 5,08 (д, J=7,6 Гц, 2H), 4,81 (ушир.с, 2H), 4,36 (ушир.с, 2H), 3,95 (с, 3H), 3,25-3,34 (м, 2H), 2,92-3,00 (м, 2H), 2,20 (ушир.с, 4H), 1,00 (ушир.с, 6H); МС: (ES) m/z вычислено для C40H38F4N6O9P[M+H]+ 853,2, найдено 853,0.
- Пример 25. Синтез (глицилокси)метил 4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил) пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбоксилата
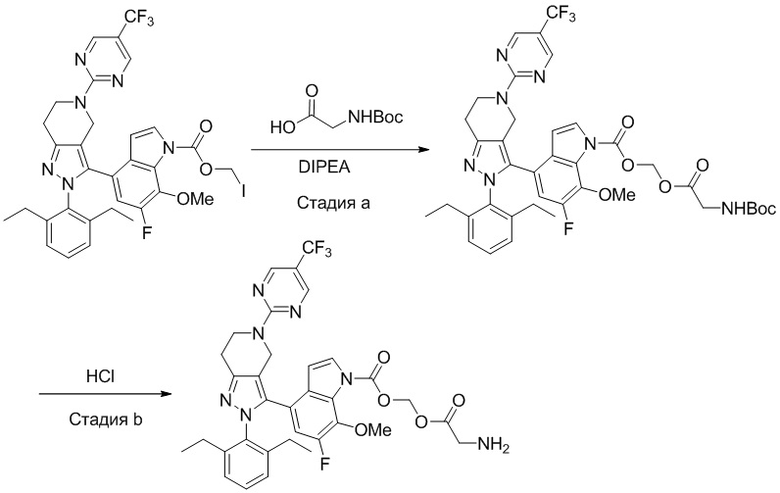
Стадия a: Смесь иодметил 4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбоксилата (0,080 г, 0,10 ммоль), (трет-бутоксикарбонил)глицина (0,056 г, 0,32 ммоль) и диизопропилэтиламина (0,088 мл, 0,53 ммоль) в дихлорметане (3 мл) нагревали при 45°C в течение 1,5 часов. Смесь охлаждали до комнатной температуры, упаривали при пониженном давлении, получая (((трет-бутоксикарбонил)глицил)окси)метил 4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбоксилат. МС: (ES) m/z вычислено для C39H42F4N7O7[M+H]+ 796,3, найдено 796,3.
Стадия b: Смесь(((трет-бутоксикарбонил)глицил)окси)метил 4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c] пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбоксилата (~ 0,10 ммоль) и 4M раствора HCl в диоксане (1,5 мл, 6 ммоль) в дихлорметане (3 мл) перемешивали при комнатной температуре в течение 1,5 часов. Смесь упаривали при пониженном давлении и очищали методом флэш-хроматографии на силикагеле (0-100% EtOAc в гексане, затем 0-80% MeOH в EtOAc). Чистые фракции объединяли, охлаждали до 0°C, добавляли 2M раствор HCl в эфире (1 мл, 2 ммоль) и упаривали при пониженном давлении, получая (глицилокси)метил 4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло [4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбоксилат в виде HCl-соли. 1H ЯМР (400 МГц, CD3OD) δ 8,59 (с, 2H), 7,78-7,83 (м, 1H), 7,34 (дд, J=7,2, 7,2 Гц, 1H), 7,18 (ушир.с, 2H), 6,60-6,68 (м, 2H), 6,17 (с, 2H), 4,83 (ушир.с, 2H), 4,39 (ушир.с, 2H), 4,01 (ушир.с, 2H), 3,97 (с, 3H), 2,96-3,03 (м, 2H), 2,26 (ушир.с, 4H), 1,96-2,03 (м, 2H), 1,20-1,26 (м, 1H), 1,00 (ушир.с, 6H); МС: (ES) m/z вычислено для C34H34F4N7O5[M+H]+ 696,2, найдено 696,2.
Пример 26. Синтез ((L-валил)окси)метил 4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил) пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбоксилата
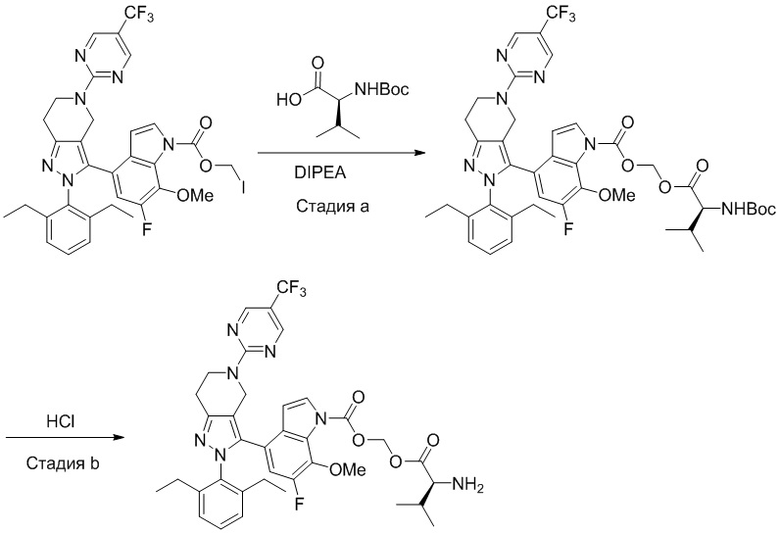
Стадия a: Смесь иодметил 4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбоксилата (0,080 г, 0,10 ммоль), (трет-бутоксикарбонил)-L-валина (0,07 г, 0,32 ммоль) и диизопропилэтиламина (0,09 мл, 0,53 ммоль) в дихлорметане (3 мл) перемешивали при комнатной температуре в течение 2 часов. Смесь упаривали на роторном испарителе при пониженном давлении и очищали методом флэш-хроматографии на силикагеле (0-60% EtOAc в гексане), получая (((трет-бутоксикарбонил)-L-валил)окси)метил 4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c] пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбоксилат; МС: (ES) m/z вычислено для C42H48F4N7O7[M+H]+ 838,4, найдено 838,8.
Стадия b: Смесь (((трет-бутоксикарбонил)-L-валил)окси)метил 4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c] пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбоксилата (31 мг, 0,037 ммоль) и 4M раствора HCl в диоксане (0,5 мл, 2 ммоль) в дихлорметане (0,5 мл) перемешивали при комнатной температуре в течение 1 часа. Смесь упаривали при пониженном давлении и очищали методом флэш-хроматографии на силикагеле (0-30% MeOH в CH2Cl2). Чистые фракции объединяли, охлаждали до 0°C, добавляли 2M раствор HCl в эфире (0,5 мл, 2 ммоль) и упаривали при пониженном давлении, получая ((L-валил)окси)метил 4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c] пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбоксилат в виде HCl-соли. 1H ЯМР (400 МГц, CD3OD) δ 8,60 (с, 2H), 7,81 (с, 1H), 7,36 (дд, J=7,2, 7,2 Гц, 1H), 7,19 (ушир.с, 2H), 6,62-6,72 (м, 2H), 6,24 (д, J=5,6 Гц, 1H), 6,13 (д, J=5,2 Гц, 1H), 5,49 (с, 1H), 4,82 (ушир.с, 2H), 4,41 (ушир.с, 2H), 4,10 (с, 1H), 3,98 (с, 3H), 3,01 (с, 2H), 1,80-2,60 (м, 5H), 0,60-1,40 (м, 14H); МС: (ES) m/z вычислено для C37H40F4N7O5[M+H]+ 738,3, найдено 738,2.
Пример 27. Синтез (глицилокси)метил 4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-карбоксилата

Стадия a: Смесь иодметил 4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-карбоксилата (0,06 г, 0,07 ммоль), (трет-бутоксикарбонил)глицина (0,04 г, 0,18 ммоль) и диизопропилэтиламина (0,08 мл, 0,48 ммоль) в дихлорметане (1 мл) перемешивали при 45°C в течение 1,5 часов. Смесь охлаждали до комнатной температуры и упаривали при пониженном давлении, получая (((трет-бутоксикарбонил)глицил)окси)метил 4-(5-(2,4-бис (трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-карбоксилат. МС: (ES) m/z вычислено для C43H45F7N5O6[M+H]+ 860,3, найдено 860,3.
Стадия b: Смесь (((трет-бутоксикарбонил)глицил)окси)метил 4-(5-(2,4-бис (трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-карбоксилата (~0,07 ммоль) и 4M раствора HCl в диоксане (1 мл, 4 ммоль) в диоксане (2 мл) перемешивали при комнатной температуре в течение 2 часов. Смесь упаривали при пониженном давлении и очищали методом ВЭЖХ (MeCN/H2O, с 0,1% ТФУК), получая (глицилокси)метил 4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-карбоксилат в виде соли с ТФУК. 1H ЯМР (400 МГц, CDCl3) δ 8,37 (д, J=7,6 Гц, 1H), 7,88-7,94 (м, 2H), 7,59 (д, J=4,0 Гц, 1H), 7,20-7,30 (м, 2H), 7,07 (д, J=7,6 Гц, 2H), 6,58-6,70 (м, 3H), 5,96 (с, 2H), 4,50-4,70 (м, 2H), 4,30 (ушир.с, 2H), 3,79 (с, 2H), 2,19 (ушир.с, 4H), 1,83-2,03 (м, 8H), 0,98 (т, J=7,2 Гц, 6H); МС: (ES) m/z вычислено для C38H37F7N5O4[M+H]+ 760,3, найдено 760,1.
Пример 28. Синтез ((L-валил)окси)метил 4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-карбоксилата
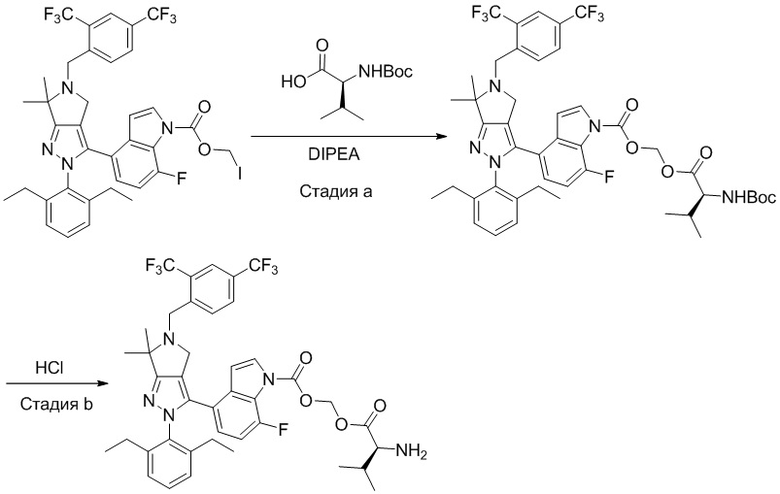
Стадия a: Смесь иодметил 4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-карбоксилата (0,10 г, 0,12 ммоль), (трет-бутоксикарбонил)-L-валина (0,08 г, 0,37 ммоль) и диизопропилэтиламина (0,10 мл, 0,61 ммоль) в дихлорметане (3 мл) перемешивали при 45°C. Через 2 часа смесь охлаждали до комнатной температуры, упаривали при пониженном давлении и очищали методом флэш-хроматографии на силикагеле (0-50% EtOAc в гексане), получая (((трет-бутоксикарбонил)-L-валил)окси)метил 4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-карбоксилат. МС: (ES) m/z вычислено для C46H51F7N5O6[M+H]+ 902,3, найдено 902,3.
Стадия b: В раствор (((трет-бутоксикарбонил)-L-валил)окси)метил 4-(5-(2,4-бис (трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c] пиразол-3-ил)-7-фтор-1H-индол-1-карбоксилата (25 мг, 0,028 ммоль) в дихлорметане (0,3 мл) добавляли 4M раствор HCl в диоксане (0,1 мл, 0,4 ммоль). Смесь перемешивали при комнатной температуре 3 часа и затем упаривали при пониженном давлении, получая ((L-валил)окси)метил 4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-карбоксилат в виде HCl-соли. 1H ЯМР (400 МГц, CDCl3) δ 9,33 (с, 1H), 8,99 (с, 2H), 7,99-8,06 (м, 1H), 7,93 (с, 1H), 7,72 (с, 1H), 7,10-7,30 (м, 3H), 6,97 (ушир.с, 1H), 6,68-6,78 (м, 1H), 6,59-6,66 (м, 1H), 6,16-6,22 (м, 1H), 5,90-5,98 (м, 1H), 4,69 (ушир.с, 2H), 4,47 (ушир.с, 1H), 4,03 (с, 1H), 3,60-3,90 (м, 3H), 1,70-2,60 (м, 10H), 0,75-1,34 (м, 12H); МС: (ES) m/z вычислено для C41H43F7N5O4[M+H]+ 802,3, найдено 802,2.
Пример 29. Синтез ((4-((диметиламино)метил)бензоил)окси)метил 4-(5-(2,4-бис (трифторметил)-бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло [3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-карбоксилата
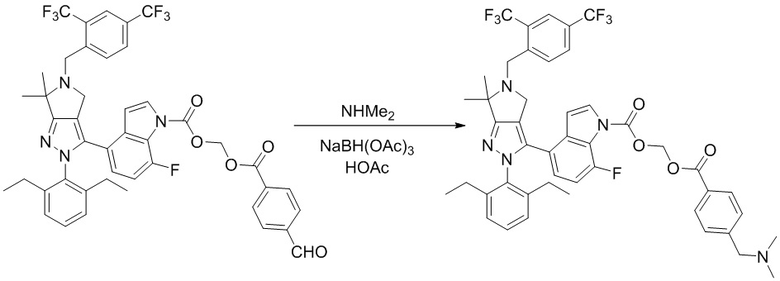
Смесь диметиламина (0,40 мл, насыщенный раствор в дихлорметане), HOAc (0,80 мл), ((4-формилбензоил)окси)метил 4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-карбоксилата (0,080 г, 0,096 ммоль) и NaBH(OAc)3 (0,200 г, 0,94 ммоль) в дихлорметане перемешивали при комнатной температуре в течение 1 часа. Смесь выливали в насыщенный водный раствор NaHCO3 и экстрагировали этилацетатом. Органический слой отделяли, сушили над Na2SO4, упаривали при пониженном давлении и очищали методом флэш-хроматографии на силикагеле (0-100% EtOAc в гексане, затем 0-30% MeOH в EtOAc), получая ((4-((диметиламино)метил)бензоил)окси)метил 4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-карбоксилат. 1H ЯМР (400 МГц, CDCl3) δ 8,15 (д, J=8,0 Гц, 1H), 8,03 (д, J=7,2 Гц, 2H), 7,86 (с, 1H), 7,75 (д, J=8,0 Гц, 1H), 7,66 (м, 1H), 7,40 (д, J=7,6 Гц, 2H), 7,25 (м, 1H), 7,07 (д, J=7,6 Гц, 2H), 6,77 (м, 1H), 6,62 (м, 1H), 6,56 (м, 1H), 6,23 (с, 2H), 4,12 (с, 2H), 3,63 (с, 2H), 3,47 (с, 2H), 2,23 (с, 6H), 2,10-2,40 (м, 4H), 1,55 (с, 6H), 1,01 (т, J=7,4 Гц, 6H); МС: (ES) m/z вычислено для C46H45F7N5O4[M+H]+ 864,3, найдено 864,2.
- Пример 30. Синтез ((диметилглицил)окси)метил 4-(5-(2,4-бис(трифторметил)-бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-карбоксилата
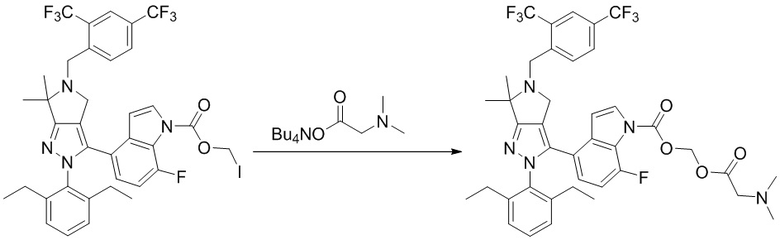
Смесь иодметил 4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-карбоксилата (0,03 г, 0,04 ммоль) и тетрабутиламмония диметилглицината (20 мг, 0,06 ммоль) в ТГФ (0,6 мл) перемешивали при комнатной температуре в течение 0,5 часа. Смесь упаривали при пониженном давлении и очищали методом ВЭЖХ (MeCN/H2O, с 1% HOAc), получая ((диметилглицил)окси)метил 4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-карбоксилат. 1H ЯМР (400 МГц, CD3OD) δ 8,21 (д, J=8,4 Гц, 1H), 7,91-7,96 (м, 2H), 7,80 (д, J=4,0 Гц, 1H), 7,34 (дд, J=7,6, 7,6 Гц, 1H), 7,17 (д, J=7,6 Гц, 2H), 6,81-6,87 (м, 1H), 6,65-6,72 (м, 2H), 6,12 (с, 2H), 4,23 (с, 2H), 4,17 (с, 2H), 3,69 (с, 2H), 2,90 (с, 6H), 2,17-2,36 (м, 4H), 1,57 (с, 6H), 1,02 (т, J=7,4 Гц, 6H); МС: (ES) m/z вычислено для C40H41F7N5O4[M+H]+ 788,3, найдено 788,2.
Пример 31. Синтез ((диметоксифосфорил)окси)метил 4-(5-(2,4-бис(трифторметил) бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-карбоксилата
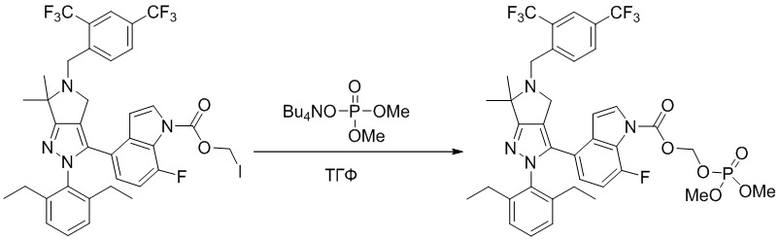
Смесь иодметил 4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-карбоксилата (40 мг, 0,037 ммоль) и тетрабутиламмония диметилфосфата (30 мг, 0,08) ммоль) в ТГФ (6 мл) перемешивали при комнатной температуре в течение 1,5 часов. После окончания реакции смесь разбавляли этилацетатом, промывали насыщенным раствором хлорида натрия и сушили над MgSO4. Растворитель удаляли при пониженном давлении, и остаток очищали методом флэш-хроматографии на силикагеле (5-20% EtOAc в гексане), получая ((диметоксифосфорил)окси)метил 4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-карбоксилат. 1H ЯМР (400 МГц, CD3OD) δ 8,13 (д, J=8,2 Гц, 1H), 7,75-7,88 (м, 2H), 7,66-7,68 (м, 1H), 7,23-7,27 (м, 1H), 7,07-7,09 (м, 2H), 6,77 (дд, J=8,4, 11,8 Гц, 1H), 6,59-6,66 (м, 2H), 5,81 (д, J=14,1 Гц, 2H), 4,12 (с, 2H), 3,79 (с, 3H), 3,76 (с, 3H), 3,70 (с, 2H), 2,14-2,38 (м, 4H), 1,56 (с, 6H), 1,02 (т, J=7,6 Гц, 6H). МС: (ES) m/z вычислено для C38H39F7N4O6P [M+H]+ 811,2, найдено 811,2.
Пример 32. Синтез (4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-ил)метанола
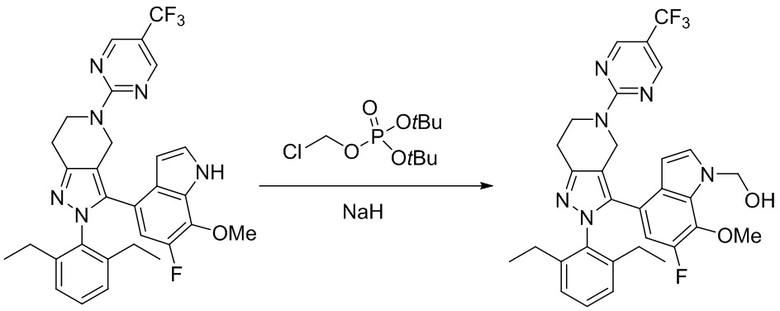
Смесь2-(2,6-диэтилфенил)-3-(6-фтор-7-метокси-1H-индол-4-ил)-5-(5-(трифторметил) пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридина (200 мг, 0,35 ммоль), ди-трет-бутил(хлорметил)фосфата (183 мг, 0,35 ммоль) и NaH (100 мг, 60% в минеральном масле, 2,5 ммоль) в ДМФА (5 мл) перемешивали при комнатной температуре в течение 2 часов. Смесь выливали в воду и экстрагировали этилацетатом. Органический слой отделяли, сушили над Na2SO4, упаривали при пониженном давлении и очищали методом флэш-хроматографии на силикагеле (0-80% EtOAc в гексане), получая (4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-ил)метанол. 1H ЯМР (400 МГц, CDCl3) δ 8,48 (с, 2H), 7,24 (дд, J=7,2, 7,2 Гц, 1H), 7,15 (д, J=3,2 Гц, 1H), 7,00-7,10 (м, 2H), 6,44 (д, J=12,8 Гц, 1H), 6,33 (д, J=3,2 Гц, 1H), 5,59 (д, J=8,0 Гц, 2H), 4,80 (с, 2H), 4,35 (с, 2H), 4,11 (д, J=2,4 Гц, 3H), 3,71 (т, J=8,2 Гц, 1H), 3,02 (т, J=5,8 Гц, 2H), 2,30 (ушир.с, 4H), 1,02 (ушир.с, 6H). МС: (ES) m/z вычислено для C31H31F4N6O2[M+H]+ 595,2, найдено 595,5.
Пример 33. Синтез (4-(2-(2,6-диэтилфенил)-5-(4-(трифторметил)фенил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-5-фтор-1H-индол-7-ил)метил глицината гидрохлорида
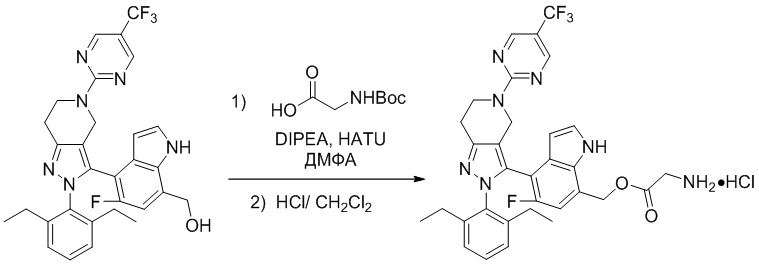
В виалу с ДМФА (3,0 мл) загружали (4-(2-(2,6-диэтилфенил)-5-(4-(трифторметил) фенил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-5-фтор-1H-индол-7-ил)метанол (100 мг, 0,18 ммоль), (трет-бутоксикарбонил)глицин (61 мг, 0,35 ммоль), HATU (134 мг, 0,35 ммоль) и диизопропилэтиламин (68 мг, 0,52 ммоль). Смесь перемешивали при комнатной температуре в течение 24 часов. После окончания реакцию гасили водой и очищали полученный сырой продукт методом хроматографии на силикагеле (10-50% EtOAc в гексане) и упаривали в вакууме. Остаток затем растворяли в дихлорметане (3,0 мл) и добавляли 4н. раствор HCl в диоксане (2,0 мл) при комнатной температуре в течение 2 часов. После окончания реакции удаляли растворитель и растирали остаток в дихлорметане, получая (4-(2-(2,6-диэтилфенил)-5-(4-(трифторметил)фенил)-4,5,6,7-тетрагидро-2H-пиразоло [4,3-c]пиридин-3-ил)-5-фтор-1H-индол-7-ил)метилглицината гидрохлорид. 1H ЯМР (400 МГц, CD3OD) δ 11,07 (ушир.с, 1H), 8,56 (ушир.с, 2H), 7,47-7,50 (м, 1H), 7,19-7,26 (м, 2H), 6,91 (дд, J=1,9, 3,1 Гц, 1H), 6,85 (д, J=10,9 Гц 1H), 6,40-6,43 (м, 1H), 5,53 (с, 2H), 4,90 (д, J=16,1 Гц, 1H), 4,62 (д, J=16,0 Гц, 1H), 4,42-4,50 (м, 1H), 4,25-4,35 (м, 1H), 3,90 (с, 2H), 3,76 (с, 2H), 3,00 (т, J=5,9 Гц, 2H), 2,43-2,50 (м, 2H), 2,12-2,20 (м, 1H), 1,90 -1,98 (м, 1H), 1,22 (т, J=7,8 Гц, 3H), 0,73 (т, J=7,4 Гц, 3H) МС: (ES) m/z вычислено для C32H31F4N7O2[M+H]+ 622,3, найдено 622,2
Пример 34. Синтез (4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-5-фтор-1H-индол-7-ил)метил-L-валината гидрохлорида
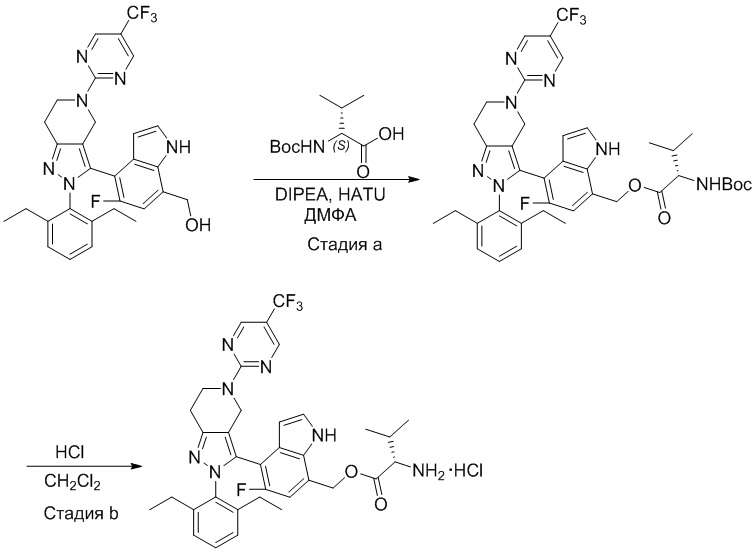
Стадия a: В виалу с ДМФА (3,0 мл) загружали (4-(2-(2,6-диэтилфенил)-5-(4-(трифторметил)фенил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-5-фтор-1H-индол-7-ил)метанол (100 мг, 0,18 ммоль), (трет-бутоксикарбонил)валин (77 мг, 0,35 ммоль), HATU (134 мг, 0,35 ммоль) и диизопропилэтиламин (68 мг, 0,53 ммоль). Реакционную смесь перемешивали при 50°C в течение 24 часов. После окончания реакции смесь гасили водой, и полученный сырой продукт очищали методом хроматографии на силикагеле (10-50% EtOAc в гексане) и упаривали в вакууме, получая (4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил) пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-5-фтор-1H-индол-7-ил)метил(трет-бутоксикарбонил)-L-валинат. МС: (ES) m/z вычислено для C40H45F4N7O4[M+H]+ 764,4, найдено 764,3.
Стадия b: В перемешиваемый раствор (4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил) пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-5-фтор-1H-индол-7-ил)метил (трет-бутоксикарбонил)-L-валината (60 мг, 0,08 ммоль) в дихлорметане (5 мл) добавляли 4н. раствор HCl в диоксане (0,2 мл, 0,8 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 2 часов. После окончания реакции смесь разбавляли водой и насыщенным водным раствором NaHCO3, экстрагировали дихлорметаном, промывали насыщенным раствором хлорида натрия и сушили над MgSO4. Растворитель удаляли при пониженном давлении, и остаток очищали методом ВЭЖХ (MeCN/H2O, с 0,1% ТФУК), получая целевой продукт. Это вещество превращали в HCl-соль, получая (4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c] пиридин-3-ил)-5-фтор-1H-индол-7-ил)метил-L-валината гидрохлорид. 1H ЯМР (400 МГц, ДМСО-d6) δ 11,65 (ушир.с, 1H), 8,61-8,72 (м, 2H), 8,42 (ушир.с, 2H), 7,56 (ушир.с, 1H), 7,15-7,25 (м, 2H), 6,88-6,98 (м, 2H), 6,33 (с, 1H), 5,42-5,55 (м, 2H), 4,74 (дд, J=6,2, 15,2 Гц, 1H), 4,57 (д, J=15,7 Гц, 1H), 4,35-4,45 (м, 1H), 4,15-4,20 (м, 1H), 3,92-4,05 (м, 1H), 2,85-2,95 (м, 2H), 2,28-2,40 (м, 2H), 2,00-2,20 (м, 2H), 1,85-1,92 (м, 1H), 1,12 (т, J=7,4 Гц, 3H), 0,83-0,90 (м, 6H), 0,61-0,63 (м, 3H). МС: (ES) m/z вычислено для C35H37F4N7O2 [M+H]+ 664,3, найдено 664,2.
- Пример 35. Синтез 4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-5-фтор-1H-индол-7-ил)метил (S)-2,5-диаминопентаноата гидрохлорида
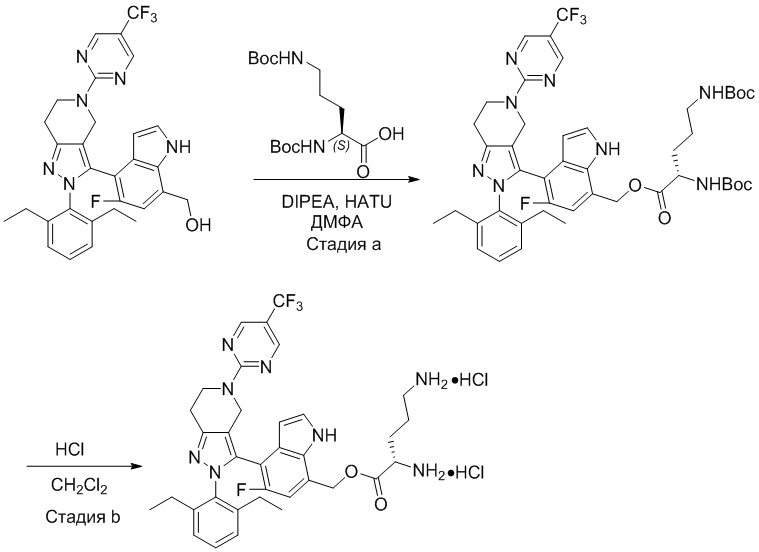
Стадия a: В виалу с ДМФА (3,0 мл) загружали (4-(2-(2,6-диэтилфенил)-5-(4-(трифторметил)фенил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-5-фтор-1H-индол-7-ил)метанол (110 мг, 0,19 ммоль), 2,5-бис(трет-бутоксикарбониламино)пентановую кислоту (96 мг, 0,29 ммоль), HATU (149 мг, 0,39 ммоль) и диизопропилэтиламин (75 мг, 1,17 ммоль). Реакцию перемешивали при комнатной температуре в течение 24 часов. После окончания реакции смесь гасили водой, и полученный сырой продукт очищали методом хроматографии на силикагеле (10-100% EtOAc в гексане) и сушили в вакууме, получая 4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-5-фтор-1H-индол-7-ил)метил (S)-2,5-бис((трет-бутоксикарбонил)амино) пентаноат. МС: (ES) m/z вычислено для C45H54F4N8O6[M+H]+ 879,41, найдено 879,5
Стадия b: В перемешиваемый раствор 4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил) пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-5-фтор-1H-индол-7-ил)метил-(S)-2,5-бис((трет-бутоксикарбонил)-амино)пентаноата (75 мг, 3,6 ммоль) в дихлорметане (5 мл) добавляли 4н. раствор HCl в диоксане (0,18 мл, 0,68 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 2 часов. После окончания реакции разбавляли смесь водой и насыщенным водным раствором NaHCO3, экстрагировали дихлорметаном, промывали насыщенным раствором хлорида натрия и сушили над MgSO4. Растворитель удаляли при пониженном давлении, и остаток очищали методом ВЭЖХ (MeCN/H2O, с 0,1% ТФУК), получая целевой продукт. Это вещество превращали в HCl-соль, получая (4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-5-фтор-1H-индол-7-ил)метил (S)-2,5-диаминопентаноата гидрохлорид. 1H ЯМР (400 МГц, ДМСО-d6) δ 11,77 (ушир.с, 1H), 8,50-8,65 (м, 4H), 7,90-8,25 (м, 2H), 7,55 (ушир.с, 1H), 7,15-7,25 (м, 2H), 6,97 (дд, J=1,9, 10,9 Гц, 1H), 6,91 (д, J=7,4 Гц 1H), 6,34 (ушир.с, 1H), 5,35-5,54 (м, 2H), 4,73 (д, J=15,7 Гц, 1H), 4,56 (д, J=16,0 Гц, 1H), 4,38-4,45 (м, 2H), 4,05-4,25 (м, 2H), 2,85-3,00 (м, 2H), 2,75-2,80 (м, 2H), 2,31-2,40 (м, 2H), 1,95-2,10 (м, 1H), 1,65-1,78 (м, 4H), 1,12 (т, J=7,4 Гц, 3H), 0,65 (т, J=7,4 Гц, 3H), МС: (ES) m/z вычислено для C35H38F4N8O2 [M+H]+ 679,3, найдено 679,2.
Пример 36. Синтез (4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-5-фтор-1H-индол-7-ил)метил L-лизината гидрохлорида
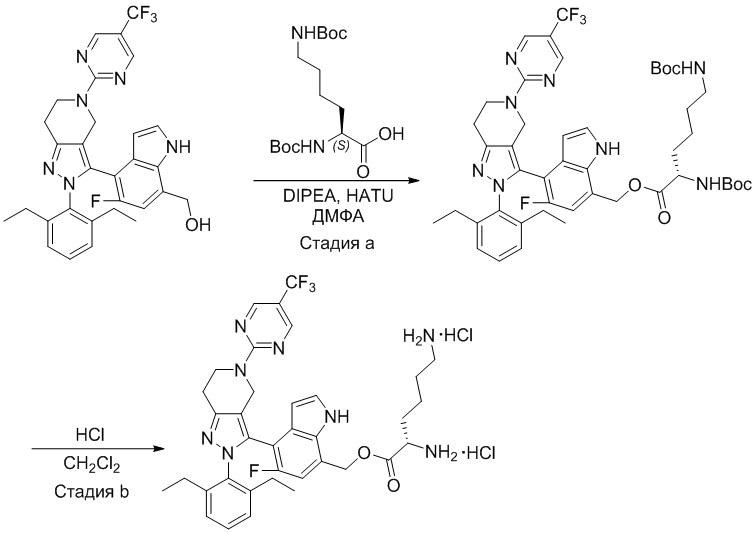
Пример 36 получали по методике, аналогичной описанной для Примера 35, используя N2,N6-бис(трет-бутоксикарбонил)-L-лизин и (4-(2-(2,6-диэтилфенил)-5-(4-(трифторметил)фенил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-5-фтор-1H-индол-7-ил)метанол. 1H ЯМР (400 МГц, ДМСО-d6) δ 11,71 (ушир.с, 1H), 8,50-8,75 (м, 4H), 7,89 (ушир.с, 2H), 7,56 (т, J=2,8 Гц 1H), 7,15-7,25 (м, 2H), 6,94 (д, J=11,0 Гц, 1H), 6,91 (д, J=7,1 Гц 1H), 6,30-6,35 (м, 1H), 5,38-5,54 (м, 2H), 4,73 (д, J=15,6 Гц, 1H), 4,57 (д, J=15,7 Гц, 1H), 4,35-4,45 (м, 1H), 4,15-4,30 (м, 1H), 4,05-4,15 (м, 2H), 2,85-3,00 (м, 2H), 2,65-2,75 (м, 2H), 2,30-2,42 (м, 2H), 2,00-2,15 (м, 1H), 1,75-1,90 (м, 3H), 1,25-1,60 (м, 3H), 1,12 (т, J=7,4 Гц, 3H), 0,65 (т, J=7,4 Гц, 3H),МС: (ES) m/z вычислено для C36H40F4N8O2[M+H]+ 693,3, найдено 693,3.
Пример 37. Синтез (4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-5-фтор-1H-индол-7-ил)метил 4-аминобутаноата гидрохлорида
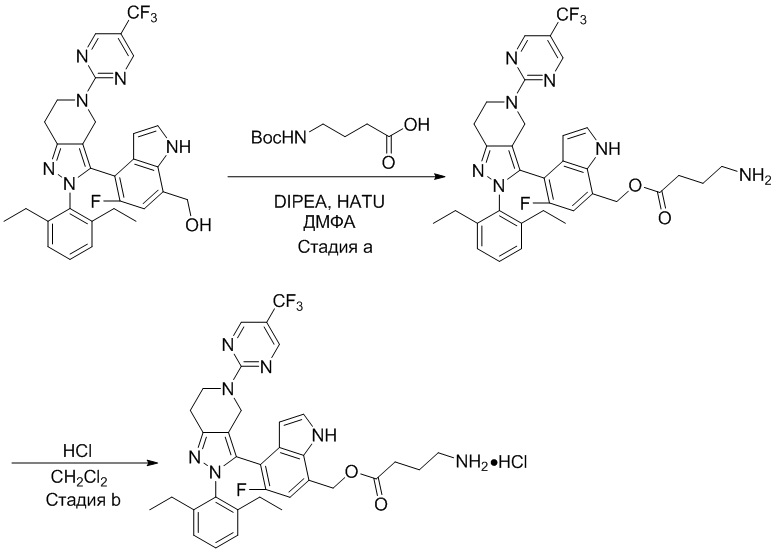
Пример 37 получали по методике, аналогичной описанной для Примера 35, используя 4-((трет-бутоксикарбонил)амино)бутановую кислоту и (4-(2-(2,6-диэтилфенил)-5-(4-(трифторметил)фенил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-5-фтор-1H-индол-7-ил)метанол. 1H ЯМР (400 МГц, ДМСО-d6) δ 11,98 (с, 1H), 8,52-8,72 (м, 2H), 7,79 (ушир.с, 2H), 7,52 (т, J=3,1 Гц 1H), 7,14-7,30 (м, 2H), 6,91 (дд, J=1,6, 7,4 Гц, 1H), 6,84 (д, J=10,9 Гц, 1H), 6,31 (дд, J=1,9, 3,1 Гц, 1H), 5,27 (д, J=3,1 Гц, 2H), 4,74 (д, J=15,6 Гц, 1H), 4,57 (д, J=15,6 Гц, 1H), 4,35-4,50 (м, 2H), 4,15-4,25 (м, 2H), 2,85-3,00 (м, 2H), 2,76-2,84 (м, 2H), 2,31-2,40 (м, 2H), 2,0-2,15 (м, 1H), 1,76-1,90 (м, 3H), 1,12 (т, J=7,4 Гц, 3H), 0,65 (т, J=7,5 Гц, 3H), МС: (ES) m/z вычислено для C34H35F4N7O2[M+H]+ 650,3, найдено 650,3.
Пример 38. Синтез (4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-5-фтор-1H-индол-7-ил)метил-L-гистидината гидрохлорида
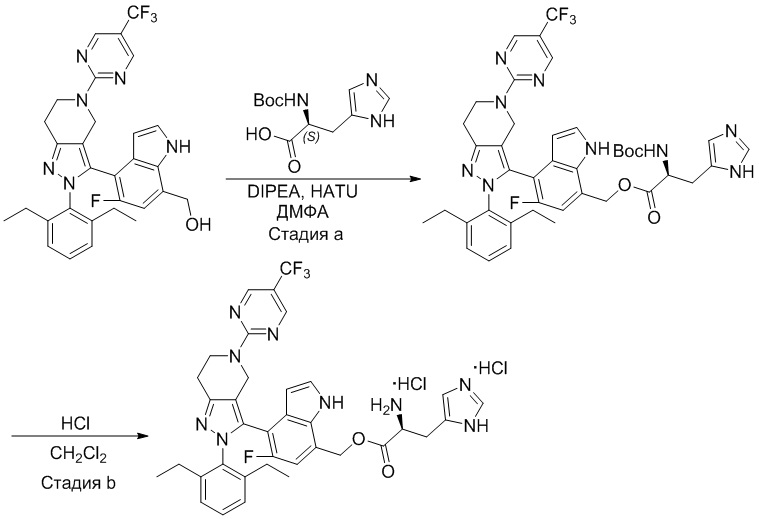
Пример 38 получали по методике, аналогичной описанной для Примера 35, используя (трет-бутоксикарбонил)-L-гистидин и (4-(2-(2,6-диэтилфенил)-5-(4-(трифторметил)фенил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-5-фтор-1H-индол-7-ил)метанол. 1H ЯМР (400 МГц, ДМСО-d6) δ 11,76 (ушир.с, 1H), 9,01 (д, J=3,9 Гц 1H), 8,55-8,75 (м, 4H), 7,55 (д, J=2,8 Гц 1H), 7,48 (с, 1H), 7,15-7,25 (м, 2H), 6,90-6,98 (м, 2H), 6,34 (ушир.с, 1H), 5,37-5,54 (м, 2H), 4,75 (дд, J=2,7, 15,6 Гц, 1H), 4,56 (д, J=15,6 Гц, 1H), 4,35-4,45 (м, 2H), 4,15-4,25 (м, 1H), 3,25-3,35 (м, 2H), 2,85-2,95 (м, 3H), 2,30-2,42 (м, 2H), 2,00-2,10 (м, 1H), 1,82-1,90 (м, 1H), 1,13 (т, J=7,8 Гц, 3H), 0,65 (т, J=7,8 Гц, 3H). МС: (ES) m/z вычислено для C36H35F4N9O2[M+H]+ 702,3, найдено 701,9.
Пример 39. Синтез (S)-3-амино-4-((4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил) пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-5-фтор-1H-индол-7-ил)метокси)-4-оксобутановой кислоты гидрохлорида
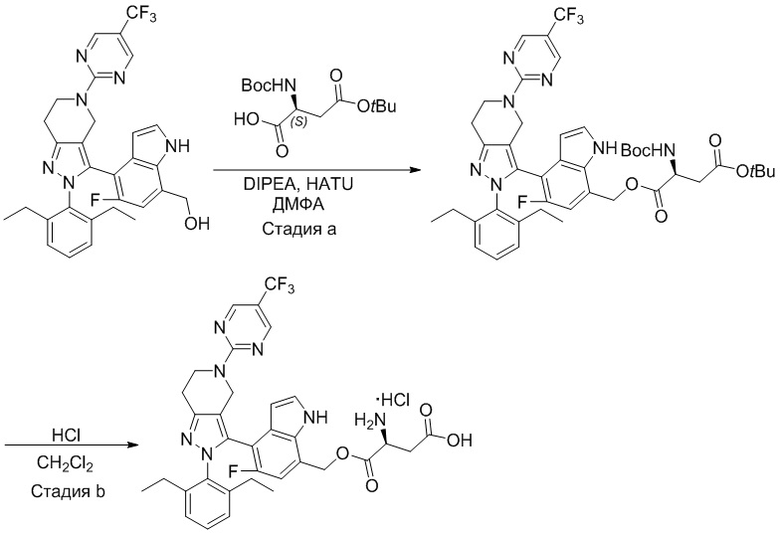
Пример 39 получали по методике, аналогичной описанной для Примера 35, используя (S)-4-(трет-бутокси)-2-((трет-бутоксикарбонил)амино)-4-оксобутановую кислоту и (4-(2-(2,6-диэтилфенил)-5-(4-(трифторметил)фенил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-5-фтор-1H-индол-7-ил)метанол. 1H ЯМР (400 МГц, ДМСО-d6) δ 11,65 (ушир.с, 1H), 8,60-8,75 (м, 2H), 8,46 (ушир.с, 3H), 7,52 (т, J=2,9 Гц 1H), 7,15-7,25 (м, 2H), 6,90 (д, J=7,8 Гц, 1H), 6,87 (ушир.с, 1H), 6,31 (ушир.с, 1H), 5,25-5,40 (м, 2H), 4,57 (д, J=15,7 Гц, 1H), 4,35-4,45 (м, 1H), 4,15-4,30 (м, 2H), 3,53 (с, 2H), 2,99 (д, J=5,5 Гц, 1H), 2,85-2,92 (м, 2H), 2,30-2,38 (м, 2H), 2,00-2,15 (м, 1H), 1,82-1,90 (м, 1H), 1,11 (т, J=7,8 Гц, 3H), 0,65 (т, J=7,8 Гц, 3H). МС: (ES) m/z вычислено для C34H33F4N7O2[M+H]+ 680,3, найдено 680,1.
- Пример 40. Синтез (4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-5-фтор-1H-индол-7-ил)метил L-валилглицината гидрохлорида
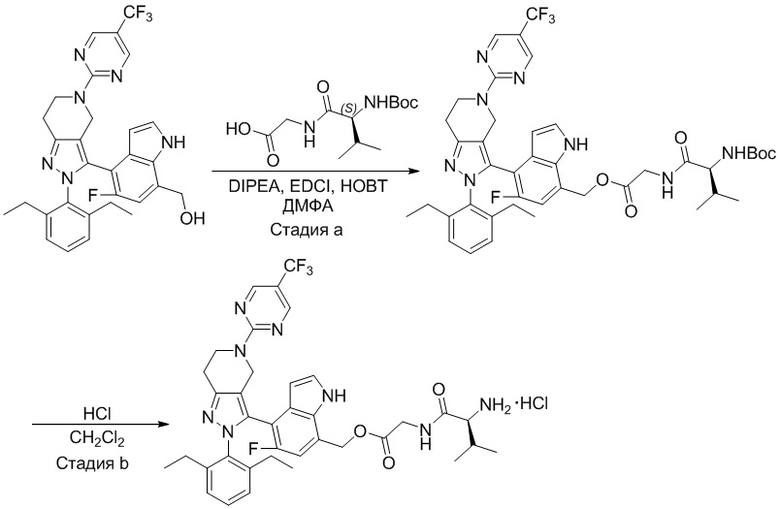
Стадия a: В виалу с ДМФА (3,0 мл) загружали (4-(2-(2,6-диэтилфенил)-5-(4-(трифторметил)фенил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-5-фтор-1H-индол-7-ил)метанол (150 мг, 0,27 ммоль), (трет-бутоксикарбонил)-L-валилглицин (145 мг, 0,53 ммоль), EDCI (101 мг, 0,53 ммоль), HOBT (61 мг, 0,39 ммоль) и DIPEA (102 мг, 0,77 ммоль). Смесь перемешивали при 50°С в течение 24 часов. После окончания реакции смесь обрабатывали, и полученный сырой продукт очищали методом хроматографии на силикагеле (10-60% EtOAc в гексане), получая (4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-5-фтор-1H-индол-7-ил)метил (трет-бутоксикарбонил)-L-валилглицинат.
Стадия b: В перемешиваемый раствор (4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил) пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-5-фтор-1H-индол-7-ил)метил-(трет-бутоксикарбонил)-L-валилглицината (180 мг, 0,02 ммоль) в дихлорметане (5 мл) добавляли 4н. раствор HCl в диоксане (0,25 мл, 0,1 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 5 часов. После окончания реакции растворитель разбавляли водой и насыщенным водным раствором NaHCO3, экстрагировали дихлорметаном, промывали насыщенным раствором хлорида натрия и сушили над Na2SO4. Растворитель удаляли при пониженном давлении, и полученный сырой продукт очищали методом колоночной хроматографии (20-100% EtOAc/гексан), получая целевой продукт, который превращали в HCl-соль, получая (4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил) пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-5-фтор-1H-индол-7-ил)метил-L-валилглицината гидрохлорид. 1H ЯМР (400 МГц, ДМСО-d6) δ 11,58 (ушир.с, 1H), 8,85 (ушир.с, 1H), 8,60-8,75 (м, 2H), 8,09 (ушир.с, 2H), 7,53 (т, J=3,1 Гц 1H), 7,12-7,25 (м, 2H), 6,90 (д, J=6,3 Гц, 1H), 6,86 (д, J=10,9 Гц, 1H), 6,31 (т, J=2,8 Гц, 1H), 5,25-5,35 (м, 2H), 4,72 (д, J=15,9 Гц, 1H), 4,57 (д, J=16,3 Гц, 1H), 4,40-4,45 (м, 1H), 4,10-4,25(м, 2H), 3,91 (дд, J=5,1, 17,2 Гц, 1H), 3,59 (т, J=0,8 Гц, 1H), 2,85-2,92 (м, 2H), 2,31-2,40 (м, 2H), 1,90-2,08 (м, 2H), 1,80-1,90 (м, 1H), 1,12 (т, J=7,8 Гц, 3H), 0,80-0,85 (м, 6H), 0,65 (т, J=7,8 Гц, 3H), МС: (ES) m/z вычислено для C37H40F4N8O3[M+H]+ 721,3, найдено 721,3.
Пример 41. Синтез (4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-5-фтор-1H-индол-7-ил)метил L-изолейцината гидрохлорида
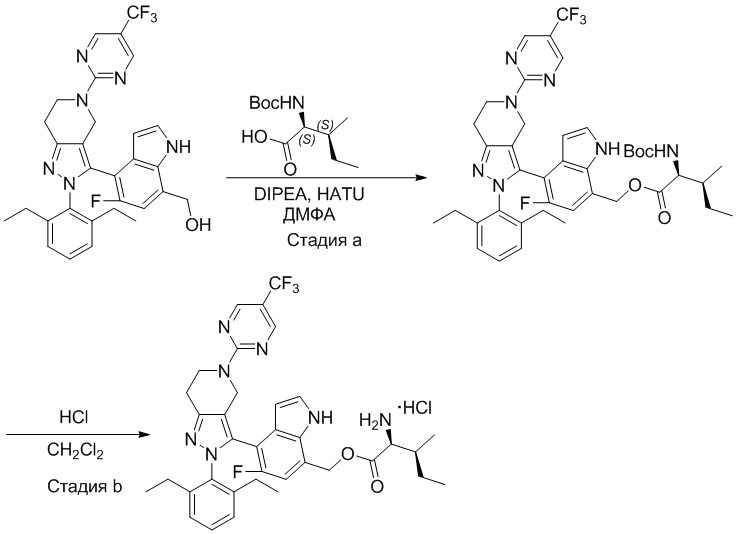
Пример 41 получали по методике, аналогичной описанной для Примера 35, используя (трет-бутоксикарбонил)-L-изолейцин и (4-(2-(2,6-диэтилфенил)-5-(4-(трифторметил)фенил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-5-фтор-1H-индол-7-ил)метанол. 1H ЯМР (400 МГц, ДМСО-d6) δ 11,63 (ушир.с, 1H), 8,60-8,75 (м, 2H), 8,42 (ушир.с, 2H), 7,43-7,60 (м, 1H), 7,14-7,25 (м, 2H), 6,85-6,95 (м, 2H), 6,30-6,35 (м, 1H), 5,38-5,45 (м, 2H), 4,76 (дд, J=5,8, 16,0 Гц, 1H), 4,56 (д, J=15,7 Гц, 1H), 4,35-4,45 (м, 1H), 4,15-4,20 (м, 1H), 3,98-4,05 (м, 1H), 2,82-2,97 (м, 2H), 2,30-2,42 (м, 3H), 2,00-2,10 (м, 1H), 1,80-1,90 (м, 2H), 1,25-1,35 (м, 1H), 1,12 (т, J=7,4 Гц, 3H), 0,72-0,85 (м, 6H), 0,65 (т, J=7,4 Гц, 3H). МС: (ES) m/z вычислено для C36H39F4N7O2[M+H]+ 678,3, найдено 678,3.
Пример 42. Синтез (4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-5-фтор-1H-индол-7-ил)метил L-аланината гидрохлорида

Пример 42 получали по методике, аналогичной описанной для Примера 35, используя (трет-бутоксикарбонил)-L-аланин и (4-(2-(2,6-диэтилфенил)-5-(4-(трифторметил) фенил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-5-фтор-1H-индол-7-ил)метанол. 1H ЯМР (400 МГц, ДМСО-d6) δ 11,62 (ушир.с, 1H), 8,60-8,75 (м, 2H), 8,35 (ушир.с, 2H), 7,56 (т, J=2,7 Гц, 1H), 7,12-7,25 (м, 2H), 6,85-6,95 (м, 2H), 6,30-6,35 (м, 1H), 5,35-5,50 (м, 2H), 4,73 (д, J=17,0 Гц, 1H), 4,56 (д, J=15,76 Гц, 1H), 4,35-4,45 (м, 1H), 4,15-4,22 (м, 2H), 2,85-2,95 (м, 2H), 2,25-2,40 (м, 2H), 2,00-2,10 (м, 1H), 1,82 -1,90 (м, 1H), 1,38 (дд, J=2,4, 7,5 Гц, 3H), 1,12 (т, J=7,8 Гц, 3H), 0,65 (дд, J=7,2, 15,6 Гц, 3H). МС: (ES) m/z вычислено для C33H33F4N7O2[M+H]+ 636,3, найдено 636,2.
Пример 43. Синтез 4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-5-фтор-1H-индол-7-ил)метил-L-фенилаланината гидрохлорида
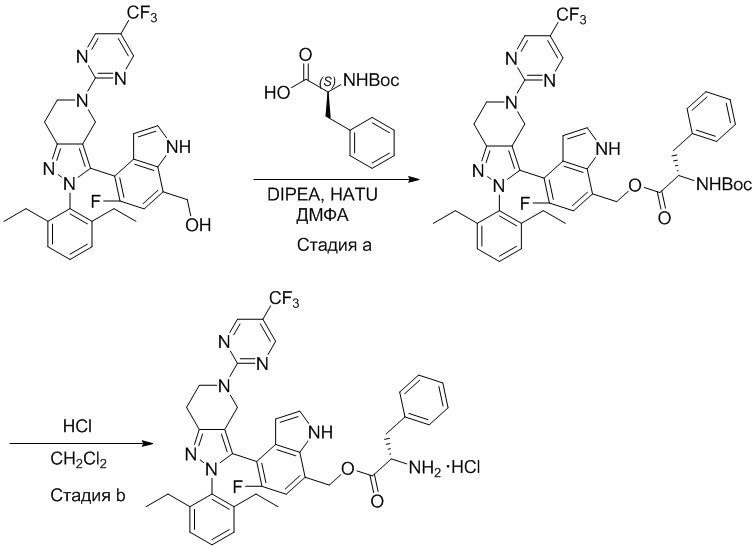
Пример 43 получали по методике, аналогичной описанной для Примера 35, используя (трет-бутоксикарбонил)-L-фенилаланин и (4-(2-(2,6-диэтилфенил)-5-(4-(трифторметил)фенил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-5-фтор-1H-индол-7-ил)метанол. 1H ЯМР (400 МГц, ДМСО-d6) δ 11,55 (ушир.с, 1H), 8,60-8,75 (м, 2H), 8,45-8,52 (м, 2H), 7,52-7,58 (м, 1H), 7,10-7,25 (м, 7H), 6,88-6,95 (м, 1H), 6,71 (дд, J=11,4, 24,3 Гц, 1H), 6,30-6,38 (м, 1H), 5,35-5,45 (м, 2H), 4,73 (д, J=15,9 Гц, 1H), 4,57 (д, J=16,0 Гц, 1H), 4,35-4,45 (м, 2H), 4,15-4,22 (м, 1H), 3,05-3,15 (м, 2H), 2,85-2,95 (м, 2H), 2,30-2,45 (м, 2H), 2,00-2,15 (м, 1H), 1,82-1,90 (м, 1H), 1,13 (т, J=7,4 Гц, 3H), 0,64 (т, J=7,8 Гц, 3H). МС: (ES) m/z вычислено для C39H37F4N7O2[M+H]+ 712,3, найдено 712,2.
Пример 44. Синтез (S)-2-амино-5-((4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил) пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-5-фтор-1H-индол-7-ил)метокси)-5-оксопентановой кислоты гидрохлорида
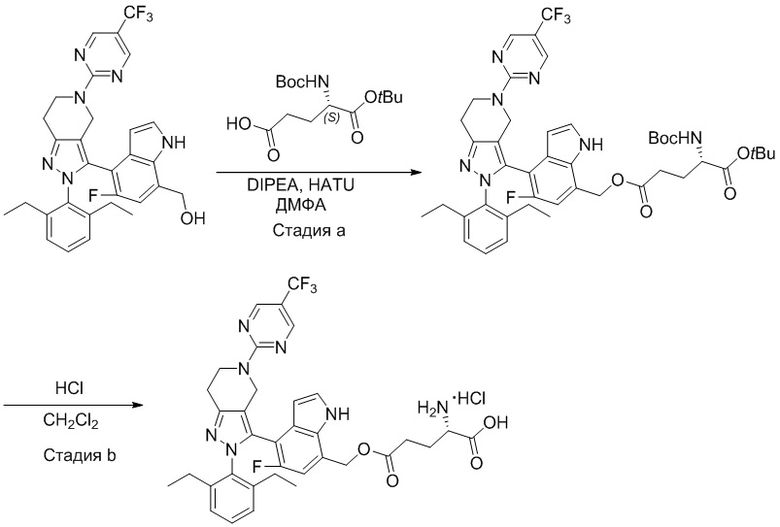
Пример 44 получали по методике, аналогичной описанной для Примера 35, используя (S)-5-(трет-бутокси)-4-((трет-бутоксикарбонил)амино)-5-оксопентановую кислоту и (4-(2-(2,6-диэтилфенил)-5-(4-(трифторметил)фенил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-5-фтор-1H-индол-7-ил)метанол. 1H ЯМР (400 МГц, ДМСО-d6) δ 11,61 (ушир.с, 1H), 8,62-8,75 (м, 2H), 8,35-8,45 (м, 3H), 7,52 (т, J=2,7 Гц 1H), 7,12-7,25 (м, 2H), 6,90 (дд, J=7,8, 11,6 Гц, 1H), 6,84 (д, J=11,0 Гц, 1H), 6,31 (кв, J=2,0 Гц, 1H), 5,23-5,31 (м, 2H), 4,74 (д, J=16,0 Гц, 1H), 4,57 (д, J=16,0 Гц, 1H), 4,35-4,45 (м, 1H), 3,85-4,00 (м, 2H), 2,85-2,95 (м, 2H), 2,45-2,65 (м, 2H), 2,25-2,40 (м, 2H), 1,85-2,10 (м, 3H), 1,80-1,95 (м, 1H), 1,12 (т, J=7,8 Гц, 3H), 0,64 (т, J=7,8 Гц, 3H). МС: (ES) m/z вычислено для C35H35F4N7O4[M+H]+ 694,3, найдено 694,2.
- Пример 45. Синтез (S)-2-ацетамидо-5-((4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил) пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-5-фтор-1H-индол-7-ил)метокси)-5-оксопентановой кислоты
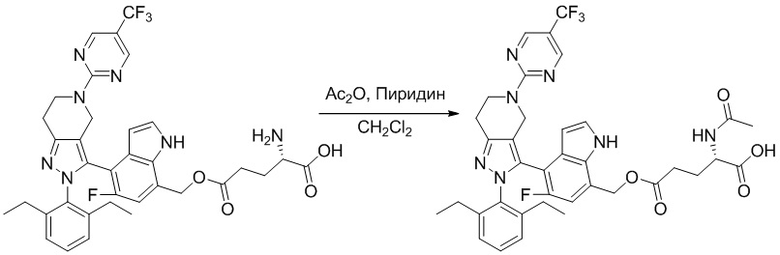
В перемешиваемый раствор (S)-2-амино-5-((4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-5-фтор-1H-индол-7-ил)метокси)-5-оксопентановой кислоты (70 мг, 0,113 ммоль) в дихлорметане (3,0 мл) при комнатной температуре добавляли пиридин (27 мг, 0,4 ммоль) и уксусный ангидрид (30 мг, 0,34 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. После окончания реакции удаляли растворитель, и полученный сырой продукт очищали методом ВЭЖХ (MeCN/H2O, с 0,1% ТФУК), получая (S)-2-ацетамидо-5-((4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-5-фтор-1H-индол-7-ил)метокси)-5-оксопентановую кислоту. 1H ЯМР (400 МГц, ДМСО-d6) δ 11,53 (ушир.с, 1H), 8,55-8,80 (м, 2H), 8,12 (д, J=7,8 Гц, 1H), 7,53 (т, J=2,7 Гц, 1H), 7,15-7,25 (м, 2H), 6,91 (д, J=7,5 Гц, 1H), 6,85 (д, J=10,2 Гц, 1H), 6,31 (т, J=10,2 Гц, 1H), 5,25 (д, J=2,7 Гц, 2H), 4,75 (д, J=16,0 Гц, 1H), 4,58 (д, J=16,0 Гц, 1H), 4,37-4,45 (м, 1H), 4,18-4,25 (м, 2H), 3,45-3,85 (м, 1H), 2,85-2,98 (м, 2H), 2,30-2,45 (м, 4H), 1,95-2,10 (м, 2H), 1,85-1,92 (м, 1H), 1,81 (с, 3H), 1,70-1,78 (м, 1H), 1,12 (т, J=7,8 Гц, 3H), 0,65 (т, J=7,8 Гц, 3H), МС: (ES) m/z вычислено для C37H37F4N7O5[M+H]+ 736,3, найдено 736,2.
Пример 46. Синтез (S)-4-амино-5-((4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил) пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-5-фтор-1H-индол-7-ил)метокси)-5-оксопентановой кислоты гидрохлорида
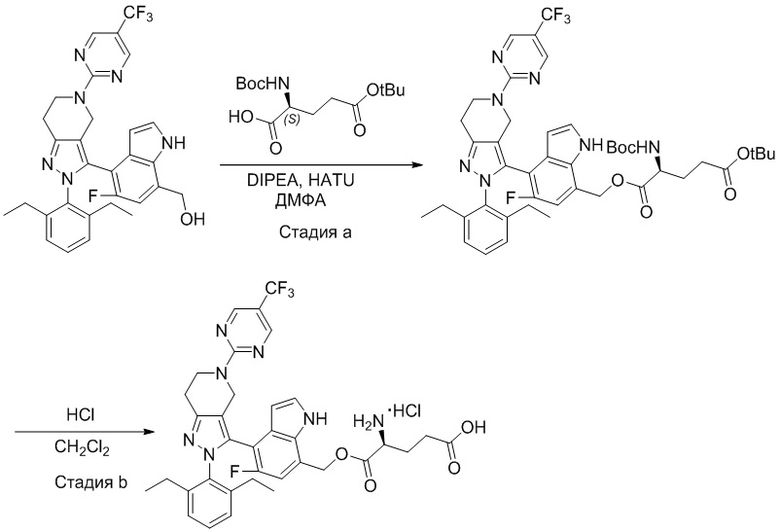
Пример 46 получали по методике, аналогичной описанной для Примера 35, используя (S)-5-(трет-бутокси)-2-((трет-бутоксикарбонил)амино)-5-оксопентановую кислоту и (4-(2-(2,6-диэтилфенил)-5-(4-(трифторметил)фенил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c] пиридин-3-ил)-5-фтор-1H-индол-7-ил)метанол. 1H ЯМР (400 МГц, ДМСО-d6) δ 11,56 (ушир.с, 1H), 8,60-8,75 (м, 2H), 8,30-8,45 (м, 2H), 7,56 (т, J=2,7 Гц 1H), 7,15-7,25 (м, 2H), 6,90-6,98 (м, 2H), 6,30-6,38 (м, 1H), 5,36-5,52 (м, 2H), 4,74 (дд, J=5,6, 16,0 Гц, 1H), 4,57 (д, J=15,7 Гц, 1H), 4,35-4,45 (м, 1H), 4,05-4,25 (м, 2H), 3,35-4,00 (м, 1H), 2,85-2,95 (м, 2H), 2,30-2,45 (м, 4H), 1,95-2,10 (м, 3H), 1,83-1,95 (м, 1H), 1,12 (т, J=7,8 Гц, 3H), 0,65 (дд, J=7,5, 14,9 Гц, 3H), МС: (ES) m/z вычислено для C35H35F4N7O4[M+H]+ 694,3, найдено 694,2.
Пример 47. Синтез (4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-5-фтор-1H-индол-7-ил)метил глицил-L-валината гидрохлорида
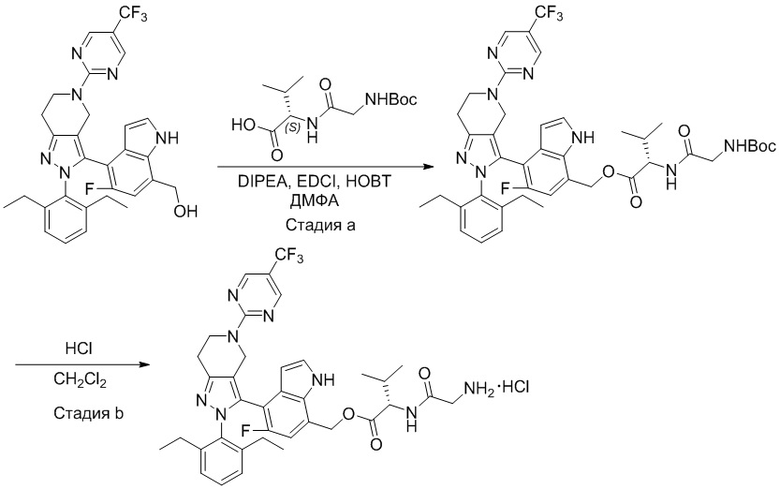
Пример 47 получали по методике, аналогичной описанной для Примера 40, используя (трет-бутоксикарбонил)глицил-L-валин и (4-(2-(2,6-диэтилфенил)-5-(4-(трифторметил)фенил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-5-фтор-1H-индол-7-ил)метанол. 1H ЯМР (400 МГц, ДМСО-d6) δ 11,60 (ушир.с, 1H), 8,60- 8,75 (м, 2H), 8,05 (ушир.с, 2H), 7,55 (ушир.с, 1H), 7,14-7,25 (м, 2H), 6,90-6,95 (м, 1H), 6,86 (дд, J=2,0, 11,0 Гц, 1H), 6,32 (т, J=2,3 Гц, 1H), 5,30-5,45 (м, 2H), 4,78 (д, J=3,5 Гц, 1H), 4,74 (д, J=3,1 Гц, 1H), 4,58 (д, J=16,0 Гц, 2H), 4,35-4,45 (м, 2H), 4,15-4,25 (м, 1H), 3,55-3,70 (м, 2H), 2,85-2,96 (м, 2H), 2,30-2,45 (м, 2H), 2,00-2,10 (м, 2H), 1,83-1,90 (м, 1H), 1,12 (т, J=7,5 Гц, 3H), 0,75-0,85 (м, 6H), 0,63 (т, J=7,4 Гц, 3H). МС: (ES) m/z вычислено для C37H40F4N8O3[M+H]+ 721,3, найдено 721,2.
Пример 48. Синтез (4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-5-фтор-1H-индол-7-ил)метил L-аланил-L-аланината гидрохлорида
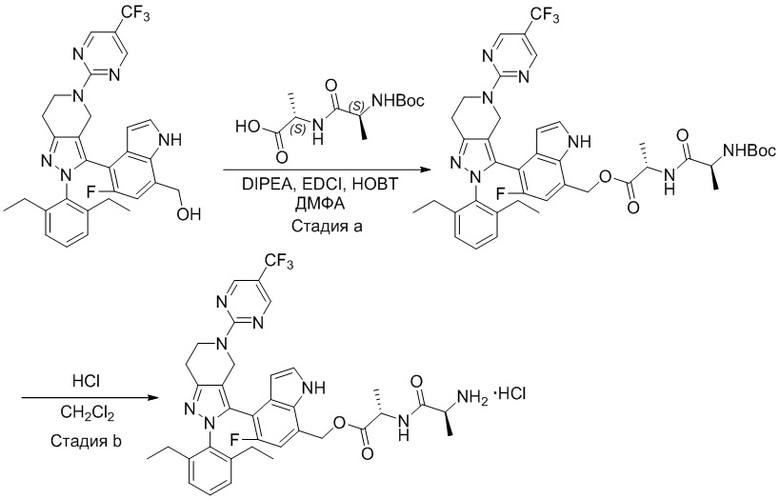
Пример 48 получали по методике, аналогичной описанной для Примера 40, используя (трет-бутоксикарбонил)-L-аланил-L-аланин и (4-(2-(2,6-диэтилфенил)-5-(4-(трифторметил)фенил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-5-фтор-1H-индол-7-ил)метанол. 1H ЯМР (400 МГц, ДМСО-d6) δ 11,58 (ушир.с, 1H), 8,50-8,95 (м, 3H), 8,10 (ушир.с, 2H), 7,53 (ушир.с, 1H), 7,12-7,25 (м, 2H), 6,90 (д, J=7,5 Гц, 1H), 6,84 (дд, J=6,0, 10,6 Гц, 1H), 6,31 (ушир.с, 1H), 5,25-5,38 (м, 2H), 4,73 (д, J=16,5 Гц, 1H), 4,57 (д, J=16,0 Гц, 1H), 4,35-4,45 (м, 2H), 4,15-4,25 (м, 1H), 3,70-3,92 (м, 1H), 2,85-2,95 (м, 2H), 2,30-2,38 (м, 2H), 2,00-2,10 (м, 1H), 1,80-1,87 (м, 1H), 1,30 (т, J=5,6 Гц, 3H), 1,10-1,18 (м, 6H), 0,60-0,66 (м, 3H). МС: (ES) m/z вычислено для C36H38F4N8O3[M+H]+ 707,3, найдено 707,2.
Пример 49. Синтез 4-(((4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-5-фтор-1H-индол-7-ил)метокси)метил)-5-метил-1,3-диоксол-2-она
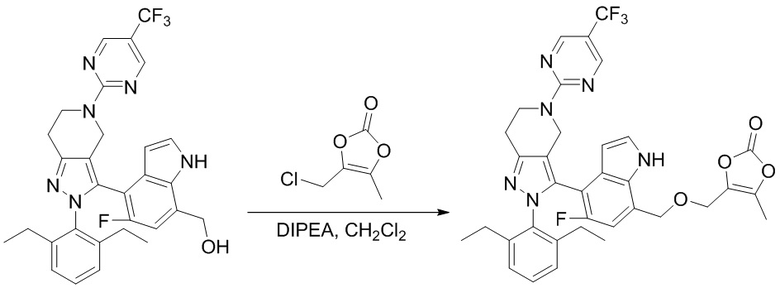
В перемешиваемый раствор (4-(2-(2,6-диэтилфенил)-5-(4-(трифторметил)фенил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-5-фтор-1H-индол-7-ил)метанола (100 мг, 0,18 ммоль) в дихлорметане (4,0 мл) при комнатной температуре добавляли диизопропилэтиламин (151 мг, 1,2 ммоль) и 4-(хлорметил)-5-метил-1,3-диоксол-2-он (150 мг, 0,78 ммоль). Реакцию перемешивали при комнатной температуре в течение 24 часов. После окончания реакции смесь гасили водой, и полученный сырой продукт очищали методом ВЭЖХ (MeCN/H2O, с 0,1% ТФУК), получая 4-(((4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил) пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-5-фтор-1H-индол-7-ил)метокси)метил)-5-метил-1,3-диоксол-2-он. 1H ЯМР (400 МГц, CDCl3) δ 9,17 (ушир.с, 1H), 8,47 (ушир.с, 2H), 7,32-7,40 (м,1H), 7,10-7,35 (м, 2H), 6,85 (д, J=7,4 Гц, 1H), 6,63 (д, J=10,5 Гц, 1H), 6,35-6,45 (м, 1H), 5,49 (д, J=15,1 Гц, 1H), 5,29 (д, J=15,5 Гц, 1H), 4,87 (дд, J=4,3, 16,1 Гц, 1H), 4,66 (дд, J=6,0, 16,5 Гц, 1H), 4,38-4,45 (м, 1H), 4,25-4,35 (м, 1H), 3,04 (т, J=5,8 Гц, 1H), 2,38-2,55 (м, 2H), 2,35 (с, 3H), 2,11-2,25 (м, 2H), 1,85-2,05 (м, 2H), 0,91-1,12 (м, 3H), 0,70-0,89 (м, 3H). МС: (ES) m/z вычислено для C35H32F4N6O4[M+H]+ 677,2, найдено 677,3.
- Пример 50. Синтез (4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-5-фтор-1H-индол-7-ил)метил дигидрофосфата
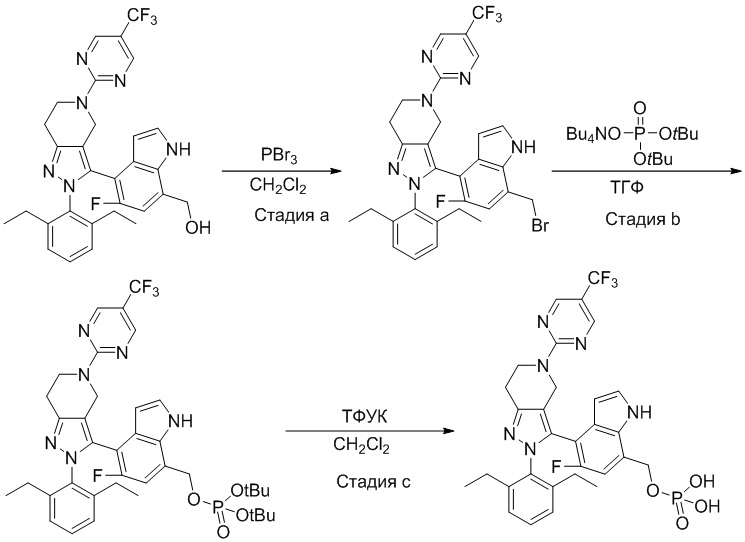
Стадия a: В перемешиваемый раствор (4-(2-(2,6-диэтилфенил)-5-(4-(трифторметил) фенил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-5-фтор-1H-индол-7-ил)метанола (100 мг, 0,16 ммоль) в дихлорметане (30 мл) при -20°C добавляли PBr3 (95 мг, 0,32 ммоль). Реакционную смесь оставляли нагреваться до 0°С и перемешивали 2 часа. После окончания реакции удаляли растворитель в вакууме, получая 3-(7-(бромметил)-5-фтор-1H-индол-4-ил)-2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин, который напрямую использовали в следующей стадии без дополнительной очистки.
Стадия b: В перемешиваемый раствор 3-(7-(бромметил)-5-фтор-1H-индол-4-ил)-2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридина (85 мг, 1,24 ммоль) в ТГФ (4 мл) добавляли тетрабутиламмония ди-трет-бутилфосфат (91 мг, 0,2 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение в течение ночи. Реакционную смесь упаривали, и полученный сырой продукт очищали методом хроматографии на силикагеле (10-100% EtOAc в гексане), получая ди-трет-бутил-((4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-5-фтор-1H-индол-7-ил)метил)фосфат. МС: (ES) m/z вычислено для C38H45F4N6O4P [M+H]+ 757,32, найдено 757,3.
Стадия c: В перемешиваемый раствор ди-трет-бутил-((4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-5-фтор-1H-индол-7-ил)метил)фосфата (70 мг, 0,92 ммоль) в безводном дихлорметане (15 мл) при 0°С добавляли ТФУК (52 мг, 0,46 ммоль) по каплям в течение 5 минут. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. После окончания реакции удаляли растворитель в вакууме, и остаток очищали методом ВЭЖХ (MeCN/H2O, с 0,1% ТФУК), получая (4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-5-фтор-1H-индол-7-ил)метил дигидрофосфат. 1H ЯМР (400 МГц, CD3OD) δ 8,56 (ушир.с, 2H), 7,43 (д, J=3,2 Гц, 1H), 7,15-7,25 (м, 2H), 6,90 (д, J=5,9 Гц, 1H), 6,81 (д, J=11,3 Гц, 1H), 6,37 (д, J=3,1 Гц, 1H), 5,20-5,30 (м, 2H), 4,91 (д, J=16,1 Гц, 1H), 4,64 (д, J=15,6 Гц, 1H), 4,42-4,50 (м, 1H), 4,30-4,37 (м, 1H), 2,98 (т, J=5,1 Гц, 2H), 2,44 (кв, J=7,8 Гц, 2H), 2,12-2,19 (м, 1H), 1,90-1,98 (м, 2H), 1,22 (т, J=7,4 Гц, 3H), 0,72 (т, J=7,4 Гц, 3H), МС: (ES) m/z вычислено для C30H29F4N6O4P[M+H]+ 645,2, найдено 645,6.
Пример 51. Синтез ((4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-5-фтор-1H-индол-7-ил)метокси) метил дигидрофосфата
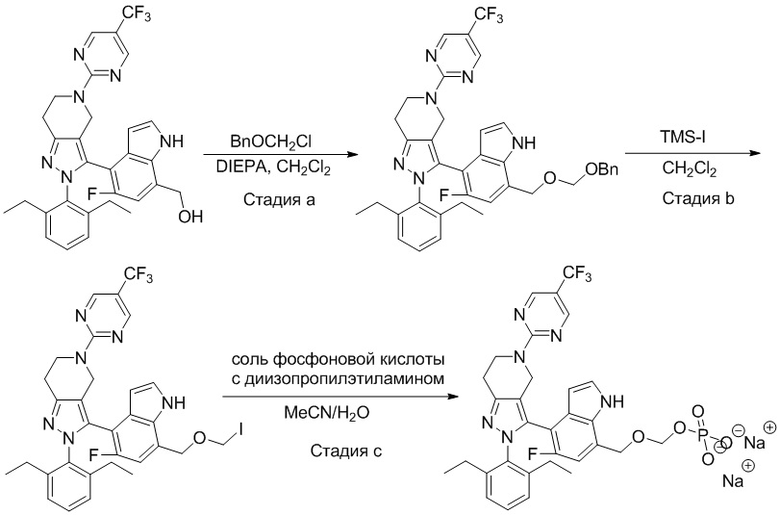
Стадия a: В перемешиваемый раствор (4-(2-(2,6-диэтилфенил)-5-(4-(трифторметил) фенил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-5-фтор-1H-индол-7-ил)метанола (140 мг, 0,25 ммоль) в дихлорметане (4 мл) при комнатной температуре добавляли диизопропилэтиламин (77 мг, 0,74 ммоль) и ((хлорметокси)метил)бензол (77 мг, 0,49 ммоль). Реакционную смесь нагревали до 50°C и перемешивали 4 часа. После окончания реакции смесь охлаждали до комнатной температуры. Смесь разбавляли водой и насыщенным водным раствором NaHCO3, экстрагировали дихлорметаном, промывали насыщенным раствором хлорида натрия и сушили над Na2SO4. Растворитель удаляли при пониженном давлении, и остаток очищали методом хроматографии на силикагеле (10-100% EtOAc в гексане), получая 3-(7-(((бензилокси)метокси)метил)-5-фтор-1H-индол-4-ил)-2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c] пиридин. 1H ЯМР (400 МГц, CDCl3) δ 8,82 (ушир.с, 1H), 8,47 (ушир.с, 2H), 7,32-7,40 (м, 4H), 7,12-7,25 (м, 3H), 6,85 (д, J=7,4 Гц, 1H), 6,63 (д, J=10,5 Гц, 1H), 6,35 (т, J=3,1 Гц, 1H), 4,85-4,98 (м, 7H), 4,69 (д, J=16,8 Гц, 1H), 4,65 (с, 2H), 4,40-4,50 (м, 1H), 4,25-4,35 (м, 1H), 3,0-3,12 (м, 1H), 2,45-2,60 (м, 2H), 2,15-2,25 (м, 1H), 1,85-2,05 (м, 1H), 1,12 (т, J=7,4 Гц, 3H), 0,73 (т, J=7,4 Гц, 3H). МС: (ES) m/z вычислено для C38H36F4N6O2 [M+H]+ 685,74, найдено 685,5.
Стадия b: В перемешиваемый раствор 3-(7-(((бензилокси)метокси)метил)-5-фтор-1H-индол-4-ил)-2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридина (140 мг, 0,20 ммоль) в дихлорметане (3 мл) добавляли триметилсилилиодид (0,5 мл, 0,51 ммоль). Реакционную смесь перемешивали 2 часа при комнатной температуре. После окончания реакции удаляли растворитель в вакууме, получая 2-(2,6-диэтилфенил)-3-(5-фтор-7-((иодметокси)метил)-1H-индол-4-ил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин, который напрямую использовали в следующей стадии без дополнительной очистки.
Стадия c: В перемешиваемую суспензию соли фосфоновой кислоты с диизопропилэтиламином (482 мг, 1,0 ммоль) в MeCN (5 мл) и H2O (5 капель) добавляли 2-(2,6-диэтилфенил)-3-(5-фтор-7-((иодметокси)метил)-1H-индол-4-ил)-5-(5-(трифторметил) пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин (140 мг, 0,19 ммоль). Реакционную смесь перемешивали 2 часа при комнатной температуре. После окончания реакции удаляли растворитель в вакууме, и смесь разбавляли водой и экстрагировали этилацетатом. Растворитель удаляли при пониженном давлении, остаток очищали методом ВЭЖХ (MeCN/H2O, с 0,1% NH4CO3) и лиофилизовывали, получая аммониевую соль ((4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-5-фтор-1H-индол-7-ил)метокси)метил дигидрофосфата. Это вещество превращали в натриевую соль путем разбавления смесью ацетонитрил (0,6 мл)/H2O (0,4 мл) и добавления 0,1M раствора NaOH (213 мкл, 2 экв.) и лиофилизации досуха, получая динатриевую соль ((4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-5-фтор-1H-индол-7-ил)метокси)метил дигидрофосфата. 1H ЯМР (400 МГц, D2O) δ 8,07 (ушир.с, 2H), 7,37 (ушир.с, 1H), 7,07-7,15 (м, 2H), 6,79 (д, J=6,6 Гц, 1H), 6,21 (ушир.с, 2H), 4,87-4,95 (м, 2H), 4,76 (д, J=11,3 Гц, 1H), 4,15-4,25 (м, 2H), 3,60-3,85 (м, 2H), 2,79 (ушир.с, 2H), 2,00-2,25 (м, 4H), 1,65-1,85 (м, 2H), 0,93 (т, J=7,4 Гц, 3H), 0,35-0,50 (м, 3H), МС: (ES) m/z вычислено для C31H31F4N6O5P[M+H]+ 675,2, найдено 675,7.
Пример 52. Синтез 4-((4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-ил)метил)-5-метил-1,3-диоксол-2-она
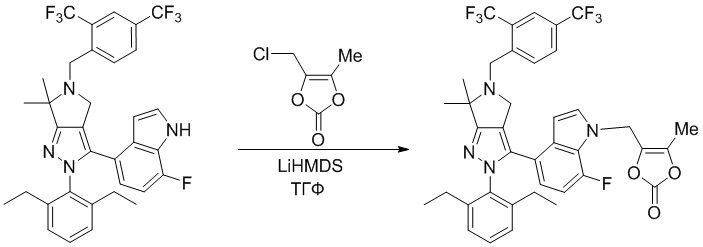
Стадия a: В перемешиваемый раствор 4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индола (400 мг, 0,64 ммоль) в ТГФ (10 мл) добавляли 1M раствор LiHMDS в ТГФ (1 мл, 1 ммоль) при -78°C. После перемешивания в течение 30 минут, добавляли раствор 4-(хлорметил)-5-метил-1,3-диоксол-2-она (113 мг, 0,76 ммоль) в ТГФ (3 мл) при -78°C. Смесь нагревали до комнатной температуры и перемешивали 3 часа. Затем добавляли насыщенный раствор NH4Cl, и смесь экстрагировали этилацетатом. Органические слои объединяли, сушили над Na2SO4, фильтровали и упаривали. Остаток очищали методом колоночной хроматографии на силикагеле (10-30% EtOAc в гексане), получая 4-((4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-ил)метил)-5-метил-1,3-диоксол-2-он. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,16 (д, J=8,2 Гц, 1H), 8,01 (д, J=8,2 Гц, 1H), 7,97 (ушир.с, 1H), 7,82 (д, J=3,5 Гц, 1H), 7,28 (т, J=7,8 Гц, 1H), 7,13 (д, J=8,1 Гц, 1H), 7,08 (д, J=7,0 Гц, 1H), 6,84 (дд, J=8,06, 13,7 Гц, 1H), 6,54 (дд, J=2,8, 3,6 Гц, 1H), 6,45 (дд, J=4,3, 8,2 Гц, 1H), 4,99 (д, J=4,7 Гц, 1H), 4,45 (д, J=4,7 Гц, 1H), 4,15 (ушир.с, 2H), 3,61 (кв, J=3,5 Гц, 1H), 2,33 (с, 3H), 2,20-2,25 (м, 2H), 2,05-2,15 (м, 2H), 1,85-2,05 (м, 1H), 1,47 (с, 6H), 0,90 (т, J=7,4 Гц, 3H), 0,85 (т, J=7,4 Гц, 3H). МС: (ES) m/z вычислено для C39H35F7N4O3 [M+H]+ 741,3, найдено 741,2.
Пример 53. Синтез (5-метил-2-оксо-1,3-диоксол-4-ил)метил 4-(5-(2,4-бис(трифторметил) бензил)-2-(2,6-диэтилфенил)-2,4,5,6-тетрагидро-6,6-диметилпирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-карбоксилата
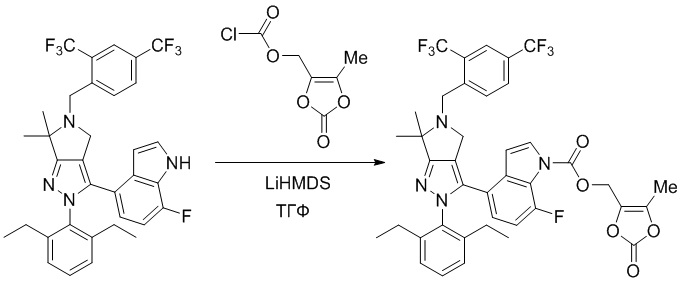
В 50-миллилитровую круглодонную колбу, в которую помещали 4-(гидроксиметил)-5-метил-1,3-диоксол-2-он (0,5 г, 3,84 ммоль) в безводном дихлорметане (6 мл), при -40°C добавляли триэтиламин (0,77 г, 7,62 ммоль) и затем трифосген (0,88 г, 4,58 ммоль) по каплям в течение 5 минут. Реакционную смесь перемешивали при -40°C 1 час, затем нагревали до комнатной температуры на 1 час. После окончания реакции смесь разбавляли водой и экстрагировали дихлорметаном. Объединенные органические слои сушили над Na2SO4, фильтровали и упаривали в вакууме, получая (5-метил-2-оксо-1,3-диоксол-4-ил)метил хлорформиат, который напрямую использовали в следующей стадии без дополнительной очистки.
Пример 53 получали по методике, аналогичной описанной для Примера 52, используя (5-метил-2-оксо-1,3-диоксол-4-ил)метил-хлорформиат и 4-(5-(2,4-бис (трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c] пиразол-3-ил)-7-фтор-1H-индол. 1H ЯМР (400 МГц, CD3OD) δ 8,22 (д, J=6,6 Гц, 1H), 7,90-7,97 (м, 2H), 7,81 (ушир.с, 1H), 7,34 (т, J=7,4 Гц, 1H), 7,16 (д, J=7,4 Гц, 2H), 6,83 (т, J=8,6 Гц, 1H), 6,60-6,75 (м, 2H), 5,24 (с, 2H), 4,23 (ушир.с, 2H), 3,71 (с, 2H), 2,25-2,38 (м, 4H), 2,24 (с, 3H), 1,57 (с, 6H), 1,03 (т, J=7,4 Гц, 6H). МС: (ES) m/z вычислено для C40H35F7N4O5[M+H]+ 785,2, найдено 785,1.
Пример 54. Синтез 4-((4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-ил) метил)-5-метил-1,3-диоксол-2-она
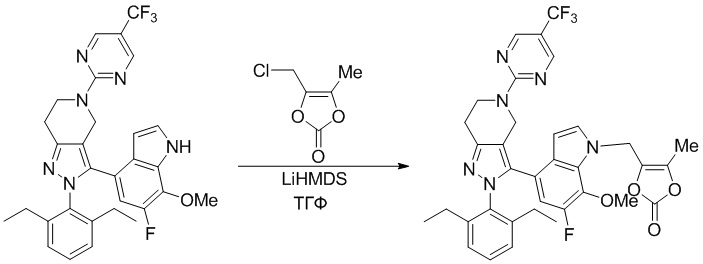
Пример 54 получали по методике, аналогичной описанной для Примера 52, используя 4-(хлорметил)-5-метил-1,3-диоксол-2-он и 2-(2,6-диэтилфенил)-3-(6-фтор-7-метокси-1H-индол-4-ил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин. 1H ЯМР (400 МГц, CD3OD) δ 8,70 (ушир.с, 2H), 7,86 (с, 1H), 7,28 (т, J=6,9 Гц, 1H), 6,59 (ушир.с, 1H), 6,40 (д, J=13,3 Гц, 1H), 5,02 (ушир.с, 2H), 4,78 (ушир.с, 2H), 4,25-4,60 (м, 2H), 3,79 (с, 3H), 3,30 (ушир.с, 2H), 2,89 (ушир.с, 2H), 2,47 (с, 3H), 2,36 (ушир.с, 4H), 0,67-1,12 (м, 6H). МС: (ES) m/z вычислено для C35H32F4N6O4[M+H]+ 677,24, найдено 677,2.
- Пример 55. Синтез (5-метил-2-оксо-1,3-диоксол-4-ил)метил 4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбоксилата
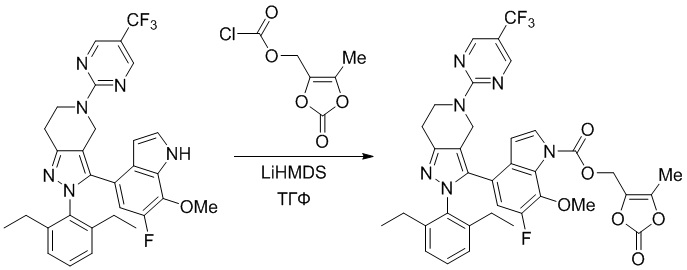
Пример 55 получали по методике, аналогичной описанной для Примера 52, используя (5-метил-2-оксо-1,3-диоксол-4-ил)метил хлорформиат и 2-(2,6-диэтилфенил)-3-(6-фтор-7-метокси-1H-индол-4-ил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин. 1H ЯМР (400 МГц, CD3OD) δ 8,58 (ушир.с, 2H), 7,78 (д, J=1,2 Гц, 1H), 7,32 (т, J=1,2 Гц, 1H), 7,15-7,22 (м, 2H), 6,50-6,65 (м, 2H), 5,28 (с, 2H), 4,38 (ушир.с, 2H), 3,95 (с, 3H), 2,97 (ушир.с, 2H), 2,30-2,40 (м, 6H), 2,26 (с, 3H), 0,98-1,25 (м, 6H). МС: (ES) m/z вычислено для C36H32F4N6O6[M+H]+ 721,23, найдено 721,2.
Пример 56. Синтез 4-((4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-2,4,5,6-тетрагидро-6,6-диметилпирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-карбоксилоилокси)метил)фенил дигидрофосфата
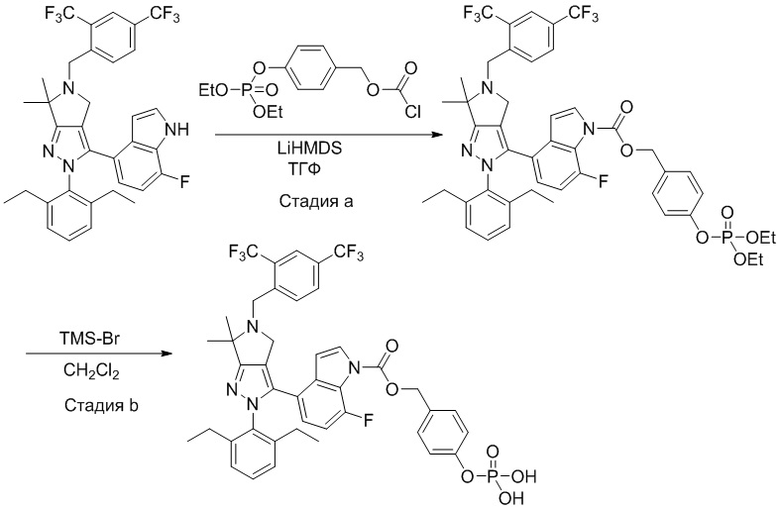
Стадия a: В 50-миллилитровую круглодонную колбу, в которую помещали 4-(гидроксиметил)фенол (0,27 г, 1,73 ммоль), триэтиламин (0,27 г, 1,73 ммоль) и безводный дихлорметан (6 мл), при 0°C добавляли диэтилфосфорилхлорид (0,54 г, 4,32 ммоль) по каплям в течение 5 минут. Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. После окончания реакции смесь экстрагировали этилацетатом. Органические слои объединяли, сушили над Na2SO4, фильтровали и упаривали. Остаток очищали методом колоночной хроматографии на силикагеле (10-80% EtOAc в гексане), получая диэтил 4-(гидроксиметил)-фенилфосфат.
В перемешиваемый раствор диэтил 4-(гидроксиметил)фенилфосфата (0,27 г, 1,73 ммоль) и триэтиламина в безводном дихлорметане (10 мл) при 0°C медленно добавляли трифосген (0,54 г, 4,32 ммоль) в течение 5 минут. Реакционную смесь перемешивали при комнатной температуре в течение 1 часов. После окончания реакции смесь разбавляли водой и экстрагировали дихлорметаном. Объединенные органические слои сушили над Na2SO4, фильтровали и упаривали в вакууме, получая 4-((диэтоксифосфорил)окси)бензил хлорформиат, который напрямую использовали в следующей стадии без дополнительной очистки.
В раствор 4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-2,4,5,6-тетрагидро-6,6-диметилпирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индола (150 мг, 0,23 ммоль) в ТГФ (4 мл) добавляли 1M раствор LiHMDS в ТГФ (0,38 мл, 0,38 ммоль) при -78°C. После перемешивания в течение 30 минут, добавляли раствор 4-((диэтоксифосфорил)окси)бензил хлорформиата (153 мг, 0,47 ммоль) в ТГФ (2 мл) при -78°C. Реакционную смесь нагревали до комнатной температуры и перемешивали 2 часа. После окончания реакции добавляли насыщенный водный раствор NH4Cl, экстрагировали этилацетатом. Органические слои объединяли, сушили над Na2SO4, фильтровали и упаривали. Остаток очищали методом колоночной хроматографии на силикагеле (10-60% EtOAc в гексане), получая 4-((диэтоксифосфорил)окси)бензил-4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-карбоксилат. МС: (ES) m/z вычислено для C46H46F7N4O6P [M+H]+ 915,3, найдено 915,3.
Стадия b: В перемешиваемый раствор 4-((диэтоксифосфорил)окси)бензил-4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-карбоксилата (100 мг, 0,11 ммоль) в дихлорметане (2,5 мл) добавляли бромтриметилсилан (83 мг, 0,55 ммоль) при комнатной температуре в течение 8 часов. После окончания реакции смесь упаривали досуха и очищали методом ВЭЖХ (MeCN/H2O, с 0,1% ТФУК), получая 4-(фосфоноокси)бензил-4-(5-(2,4-бис(трифторметил) бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-карбоксилат. 1H ЯМР (400 МГц, CD3OD) δ 8,13 (с, 1H), 8,06 (с, 2H), 7,82 (д, J=3,5 Гц, 1H), 7,46 (д, J=8,6 Гц, 2H), 7,35 (т, J=7,8 Гц, 1H), 7,22(д, J=7,9 Гц, 2H), 7,16 (д, J=7,8 Гц, 2H), 6,83 (дд, J=8,2, 12,1 Гц, 1H), 6,65-6,75 (м, 2H), 5,40 (с, 2H), 4,72 (ушир.с, 2H), 4,50 (ушир.с, 2H), 2,15-2,30 (м, 4H), 1,93 (с, 6H), 0,99 (т, J=7,4 Гц, 6H) МС: (ES) m/z вычислено для C42H38F7N4O6P [M+H]+ 859,24, найдено 859,2.
Пример 57. Синтез 4-(фосфоноокси)бензил-4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)-пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбоксилата
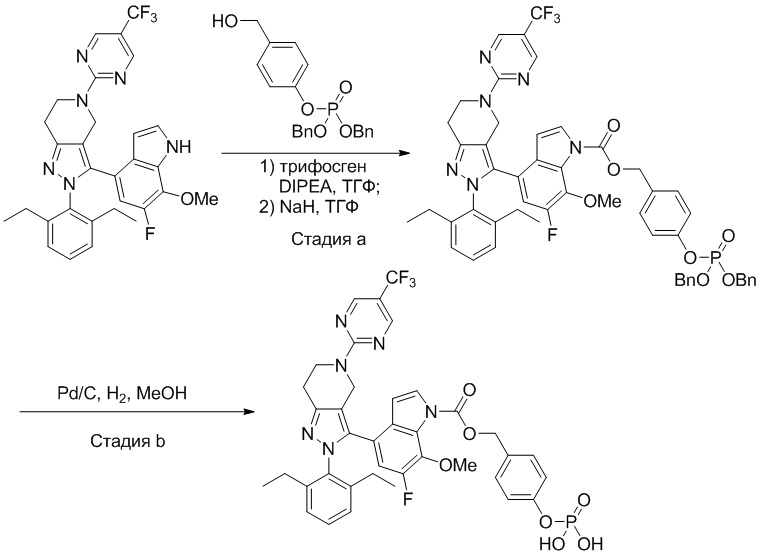
Стадия a: В раствор 4-гидроксибензальдегида (3,0 г, 24,5 ммоль) в 100 мл ТГФ добавляли 1,0 M раствор tBuOK в ТГФ (27,04 мл, 27,04 ммоль). Смесь нагревали до 70°C и добавляли тетрабензилфосфат (14,5 г, 26,95 ммоль). После 1 часа при 70°C, добавляли гексан и смесь фильтровали. Фильтрат упаривали в вакууме, и полученный остаток очищали методом колоночной хроматографии на силикагеле (0-100% EtOAc в гексане), получая дибензил (4-формилфенил)фосфат. МС: (ES) m/z вычислено для C21H19O5P [M+H]+ 383,1, найдено 383,2.
В раствор дибензил (4-формилфенил)фосфата (3,5 г, 9,16 ммоль) в 50 мл ТГФ при -78°C добавляли NaBH4 (0,65 г, 18,3 ммоль). После перемешивания при комнатной температуре в течение 2 часов, смесь гасили водой. Органический и водный слой разделяли, и водный слой экстрагировали этилацетатом. Объединенные органические слои сушили над сульфатом натрия, фильтровали и упаривали в вакууме. Остаток очищали методом колоночной хроматографии на силикагеле (0-100% EtOAc в гексане), получая дибензил-4-(гидроксиметил)фенил)фосфат. МС: (ES) m/z вычислено для C21H21O5P [M+H]+ 385, найдено 385,1.
В раствор дибензил-4-(гидроксиметил)фенил)фосфата (3,56 г, 9,24 ммоль) в 30 мл ТГФ при 0°C добавляли диизопропилэтиламин (2,98, 23,1 ммоль) и трифосген (3,0 г, 10,1 ммоль). После перемешивания при 0°C в течение 1 часа, реакцию гасили водой. Органический и водный слой разделяли, и водный слой экстрагировали этилацетатом. Органические слои объединяли, сушили над сульфатом натрия, фильтровали и упаривали, получая сырой хлорформиат.
В раствор 2-(2,6-диэтилфенил)-3-(6-фтор-7-метокси-1H-индол-4-ил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридина (250 мг, 0,442 ммоль) в 1,2 мл ТГФ при 0°С добавляли NaH (26 мг, 1,05 ммоль). После перемешивания при 0°C в течение 30 минут, добавляли в смесь раствор сырого хлорформиата, полученного как описано выше (247 мг, 0,553 ммоль), в ТГФ (2 мл). Раствор перемешивали при 0°C 1 час, затем гасили водой. Органический и водный слой разделяли, и водный слой экстрагировали этилацетатом. Органические слои объединяли, сушили над сульфатом натрия, фильтровали и упаривали в вакууме. Остаток очищали методом колоночной хроматографии на силикагеле (0-50% EtOAc в гексане), получая 4-((бис(бензилокси)фосфорил)окси)бензил 4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил) пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбоксилат.
Стадия b: В раствор 4-((бис(бензилокси)фосфорил)окси)бензил 4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c] пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбоксилата (150 мг, 0,15 ммоль) в 7 мл ТГФ добавляли 10% Pd/C (50 мг). Смесь перемешивали под давлением водорода из шарика 1 час, затем фильтровали через целит, упаривали и очищали методом ВЭЖХ (MeCN/H2O, с 0,1% ТФУК), получая 4-(фосфоноокси)бензил 4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил) пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-карбоксилат. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,90 (ушир.с, 1H), 6,99 (д, J=3,5 Гц, 1H), 6,20-6,60 (м, 10H), 5,80 (д, J=3,5 Гц, 1H), 5,69 (д, J=12,5 Гц, 1H), 4,53 (с, 2H), 3,94 (ушир.с, 2H), 3,48 (ушир.с, 2H), 3,0 (с, 3H), 2,08 (ушир.с, 2H), 1,68-1,95 (м, 2H), 0,98-1,45 (м, 6H), МС: (ES) m/z вычислено для C38H35F4N6O7P [M+H]+ 795,22, найдено 795,2.
Пример 58. Синтез 2-(4-(4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-2,4,5,6-тетрагидро-6,6-диметилпирроло-[3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-ил)-2-метил-4-оксобутан-2-ил)-3,5-диметилфенил дигидрофосфата
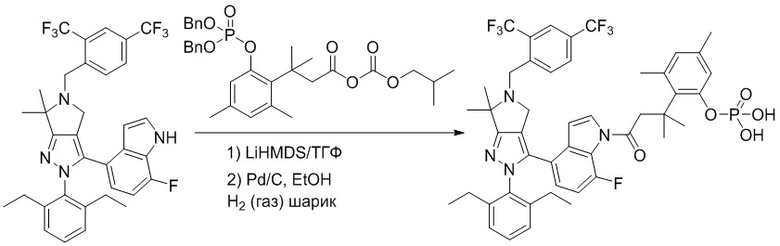
В раствор 3-(2-((бис(бензилокси)фосфорил)окси)-4,6-диметилфенил)-3-метилбутановой кислоты (1,0 г, 2,07 ммоль) в безводном дихлорметане (10 мл) добавляли диизопропилэтиламин и затем изобутилхлорформиат (298 мг, 2,49 ммоль) в дихлорметане (10 мл) при 0°C по каплям в течение 5 минут. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. После окончания реакции смесь разбавляли водой и экстрагировали дихлорметан. Объединенные органические слои сушили над Na2SO4, фильтровали и упаривали в вакууме, получая 3-(2-((бис(бензилокси)фосфорил)окси)-4,6-диметилфенил)-3-метилбутановый (изобутилкарбоновый) ангидрид, который напрямую использовали в следующей стадии без дополнительной очистки.
В раствор 4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индола (400 мг, 0,63 ммоль) в ТГФ (10 мл) добавляли 1M раствор LiHMDS в ТГФ (1,01 мл, 1,01 ммоль) при -78°C. После перемешивания в течение 30 минут, добавляли раствор 3-(2-((бис(бензилокси)фосфорил) окси)-4,6-диметилфенил)-3-метилбутанового (изобутилкарбонового) ангидрида (0,46 г, 0,8 ммоль) в ТГФ (5 мл) при -78°C и перемешивали 1 час. Смесь нагревали до комнатной температуры и перемешивали 2 часа. После окончания реакции добавляли насыщенный водный раствор NH4Cl, и смесь экстрагировали этилацетатом. Органические слои объединяли, сушили над Na2SO4, фильтровали и упаривали. Остаток очищали методом колоночной хроматографии на силикагеле (10-60% EtOAc в гексане), получая дибензил (2-(4-(4-(5-(2,4-бис(трифторметил) бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-ил)-2-метил-4-оксобутан-2-ил)-3,5-диметилфенил)фосфат.
В раствор полученного фосфата (270 мг, 0,25 ммоль) в этаноле (10 мл) добавляли 10% Pd/C (200 мг) при комнатной температуре. Полученную смесь перемешивали в атмосфере водорода (из шарика) 1 час при комнатной температуре. Реакционную смесь фильтровали через целит, и фильтрат упаривали при пониженном давлении, получая 2-(4-(4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-7-фтор-1H-индол-1-ил)-2-метил-4-оксобутан-2-ил)-3,5-диметилфенил дигидрофосфат. 1H ЯМР (400 МГц, ДМСО-d6) δ 12,22 (ушир.с, 1H), 8,47 (д, J=8,2 Гц, 1H), 8,35 (д, J=7,4 Гц, 1H), 8,30 (с, 1H), 8,23 (с, 1H), 7,62 (т, J=8,2 Гц, 1H), 7,44 (д, J=7,0 Гц, 2H), 7,32 (ушир.с, 1H), 7,15 (т, J=10,9 Гц, 1H), 6,82-6,95 (м, 3H), 4,49 (ушир.с, 2H), 3,85-4,05 (м, 4H), 3,66 (ушир.с, 1H), 2,72 (с, 3H), 2,42-2,60 (м, 4H), 2,38 (с, 3H), 1,90 (с, 4H), 1,80 (с, 6H), 1,23 (т, J=7,4 Гц, 6H). МС: (ES) m/z вычислено для C47H48F7N4O5P [M+H]+ 913,3, найдено 913,3.
Пример 59. Синтез 2-(4-(4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло-[4,3-c]пиридин-3-ил)-7-фтор-1H-индол-1-ил)-2-метил-4-оксобутан-2-ил)-3,5-диметилфенил дигидрофосфата
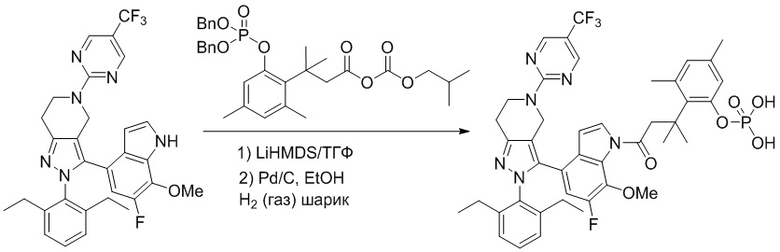
В раствор 2-(2,6-диэтилфенил)-3-(6-фтор-7-метокси-1H-индол-4-ил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридина (150 мг, 0,27 ммоль) в ТГФ (5 мл) добавляли 1M раствор LiHMDS в ТГФ (0,43 мл, 0,43 ммоль) при -78°C. После перемешивания в течение 30 минут, добавляли раствор 3-(2-((бис(бензилокси) фосфорил)окси)-4,6-диметилфенил)-3-метилбутанового ангидрида (193 мг, 0,33 ммоль) в ТГФ (2,5 мл) при -78°C и перемешивали 1 час. Смесь нагревали до комнатной температуры и перемешивали 2 часа. После окончания реакции добавляли насыщенный водный раствор NH4Cl, и смесь экстрагировали этилацетатом. Органические слои объединяли, сушили над Na2SO4, фильтровали и упаривали. Остаток очищали методом колоночной хроматографии на силикагеле (10-40% EtOAc в гексане), получая дибензил (2-(4-(4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c]пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-ил)-2-метил-4-оксобутан-2-ил)-3,5-диметилфенил)фосфат.
В раствор полученного фосфата (120 мг, 0,12 ммоль) в этаноле (5 мл) добавляли 10% Pd/C (100 мг) при комнатной температуре. Полученную смесь перемешивали в атмосфере водорода (из шарика) 1 час при комнатной температуре. Реакционную смесь фильтровали через целит, и фильтрат упаривали при пониженном давлении, получая 2-(4-(4-(2-(2,6-диэтилфенил)-5-(5-(трифторметил)пиримидин-2-ил)-4,5,6,7-тетрагидро-2H-пиразоло[4,3-c] пиридин-3-ил)-6-фтор-7-метокси-1H-индол-1-ил)-2-метил-4-оксобутан-2-ил)-3,5-диметилфенил дигидрофосфат. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,74 (ушир.с, 2H), 7,89 (д, J=1,2 Гц, 1H), 7,30-7,38 (м, 1H), 7,21 (ушир.с, 2H), 7,07 (д, J=6,2 Гц, 2H), 6,96 (с, 1H), 6,64 (с, 1H), 6,54 (с, 1H), 6,46-6,50 (м, 2H), 4,76 (ушир.с, 2H), 4,25-4,45 (м, 2H), 3,72 (с, 6H), 2,91 (ушир.с, 2H), 2,46 (ушир.с, 5H), 2,14 (с, 3H), 1,65-2,05 (м, 4H), 0,80-1,20 (м, 6H). МС: (ES) m/z вычислено для C43H45F4N6O6P [M+H]+ 849,31, найдено 849,2.
Пример 60. Синтез (4-фосфонооксифенил)метил N-((4-(5-((2,4-бис(трифторметил)-фенил)метил)-2-(2,6-диэтилфенил)-6,6-диметил-4H-пирроло[3,4-c]пиразол-3-ил)-2,5-дифтор-фенил)карбамоил)карбамата
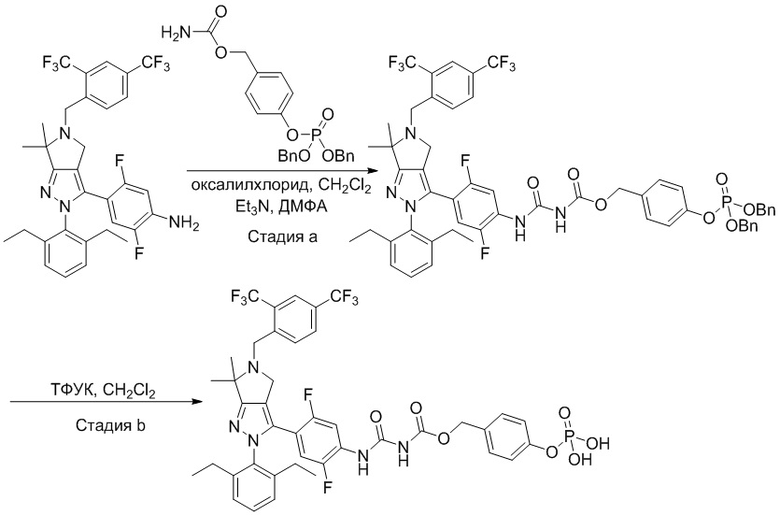
Стадия a: В раствор дибензил-4-(гидроксиметил)фенил)фосфата (3,56 г, 9,24 ммоль) в 2,4 мл ТГФ при 0°C добавляли диизопропилэтиламин (2,98, 23,1 ммоль) и трифосген (3,0 г, 10,1 ммоль). После перемешивания при 0°С в течение 1 часа, добавляли NH4OH (5,0 мл, 41 ммоль). Смесь перемешивали при комнатной температуре в течение 16 часов, затем упаривали в вакууме, и полученный остаток очищали методом колоночной хроматографии на силикагеле (20-80% EtOAc в гексане), получая 4-((бис(бензилокси)фосфорил)окси)бензил карбамат. МС: (ES) m/z вычислено для C22H22NO6P [M+H]+ 428,1, найдено 428,1.
В раствор 4-((бис(бензилокси)фосфорил)окси)бензил карбамата (700 мг, 1,63 ммоль) в дихлорметане (12 мл) при 0°C добавляли оксалилхлорид (0,3 г, 2,45 ммоль). После перемешивания при 0°C в течение 1 часа, смесь нагревали при 50°C в течение 16 часов, затем упаривали в вакууме. Остаток растворяли в ТГФ (2 мл) и добавляли в раствор 4-(5-(2,4-бис (трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c] пиразол-3-ил)-2,5-дифторанилин (150 мг, 0,16 ммоль) в ТГФ (2 мл). Смесь перемешивали при комнатной температуре в течение 5 часов, затем упаривали в вакууме. Остаток очищали методом колоночной хроматографии на силикагеле (0-100% EtOAc в гексане), получая (4-дибензилоксифосфорилокси-3-фенил)метил N-((4-(5-((2,4-бис(трифторметил)фенил)метил)-2-(2,6-диэтилфенил)-6,6-диметил-4H-пирроло[3,4-c]пиразол-3-ил)-2,5-дифторфенил) карбамоил)карбамат.
Стадия b: В раствор 4-дибензилоксифосфорилокси-3-фенил)метил N-((4-(5-((2,4-бис (трифторметил)фенил)метил)-2-(2,6-диэтилфенил)-6,6-диметил-4H-пирроло[3,4-c]пиразол-3-ил)-2,5-дифторфенил)карбамоил)карбамата (150 мг, 0,14 ммоль) в дихлорметане (2 мл) добавляли по каплям 1:1 смесь ТФУК:CH2Cl2 (2 мл) и H2O (0,4 мл). Реакционную смесь нагревали при 50°C в течение 48 часов, затем упаривали в вакууме. Остаток очищали методом ВЭЖХ (MeCN/H2O, с 0,1% ТФУК), получая (4-фосфонооксифенил)метил N-((4-(5-((2,4-бис(трифторметил)фенил)метил)-2-(2,6-диэтилфенил)-6,6-диметил-4H-пирроло[3,4-c] пиразол-3-ил)-2,5-дифтор-фенил)карбамоил)карбамат. 1H ЯМР (400 МГц, ДМСО-d6) δ 10,78 (с, 1H), 10,15 (с, 1H), 8,14 (д, J=8,4 Гц, 1H), 7,95-8,08 (м, 3H), 7,35-7,45 (м, 3H), 7,08-7,25 (м, 4H), 6,53 (дд, J=6,7, 11,7 Гц, 1H), 5,11 (с, 2H), 4,12 (ушир.с, 2H), 3,65 (ушир.с, 2H), 2,05-2,25 (м, 4H), 1,43 (с, 6H), 0,96 (т, J=7,4 Гц, 6H). МС: (ES) m/z вычислено для C41H38F8N5O7P [M+H]+ 896,2, найдено 896,2.
Пример 61. 4-(бензилокси(гидрокси)фосфорил)оксифенил)метил N-((4-(5-((2,4-бис (трифторметил)фенил)метил)-2-(2,6-диэтилфенил)-6,6-диметил-4H-пирроло[3,4-c] пиразол-3-ил)-2,5-дифтор-фенил)карбамоил)карбамат
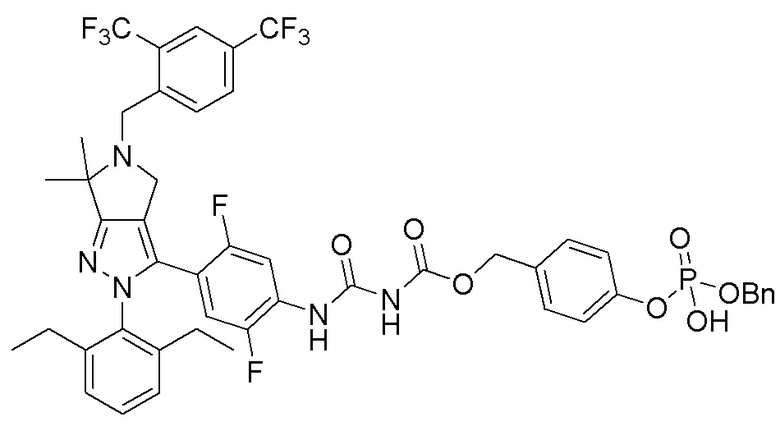
Стадия b в Примере 60 дала также Пример 61. 1H ЯМР (400 МГц, ДМСО-d6) δ 10,79 (с, 1H), 10,14 (ушир.с, 1H), 8,14 (д, J=7,0 Гц, 1H), 7,85- 8,10 (м, 3H), 7,25-7,40 (м, 7H), ), 7,20 (д, J=22,2 Гц, 2H), 7,14 (д, J=9,0 Гц, 2H), 6,53 (дд, J=6,7, 11,4 Гц, 1H), 5,13 (с, 2H), 4,99 (д, J=7,9 Гц, 2H), 4,12 (ушир.с, 2H), 3,65 (ушир.с, 2H), 2,10-2,25 (м, 4H), 1,43 (с, 6H), 0,96 (т, J=7,4 Гц, 6H). МС: (ES) m/z вычислено для C48H44F8N5O7P [M+H]+ 986,3, найдено 986,3.
Пример 62. Синтез N-((4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-2,5-дифторфенил)карбамоил)-2-(диметиламино)ацетамида.
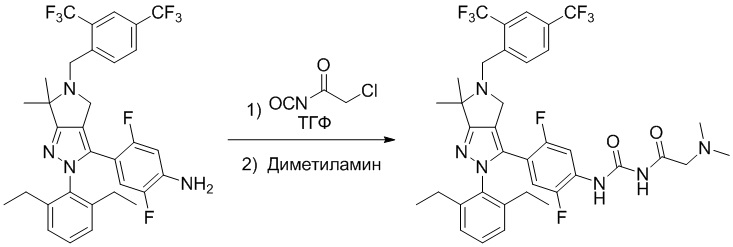
В перемешиваемый раствор 4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-2,5-дифторанилина (400 мг, 0,64 ммоль) в ТГФ (5 мл) добавляли 2-хлорацетил изоцианат при комнатной температуре. После 16 часов перемешивания смесь гасили водой и экстрагировали этилацетатом. Объединенные органические слои сушили над сульфатом натрия, фильтровали и упаривали в вакууме, получая N-((4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-2,5-дифторфенил)карбамоил)-2-хлорацетамид.
В раствор N-((4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-2,5-дифторфенил)карбамоил)-2-хлорацетамида, получение которого описано выше (125 мг, 0,17 ммоль) в ТГФ (5 мл) добавляли диметиламин (0,35 мл, 0,34 ммоль) при комнатной температуре. Реакционную смесь перемешивали 16 часов, затем упаривали в вакууме. Остаток очищали методом ВЭЖХ (H2O/ацетонитрил, 0,1% ТФУК), получая N-((4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-2,5-дифторфенил) карбамоил)-2-(диметиламино)ацетамид. 1H ЯМР (400 МГц, ДМСО-d6) δ 11,3 (ушир.с, 1H), 10,3 (ушир.с, 1H), 9,85 (ушир.с, 1H), 7,90-8,25 (м, 4H), 7,40-7,45 (м, 1H), 7,20-7,28 (м, 2H), 6,55-6,65 (м, 1H), 4,10-4,35 (м, 3H), 3,70 (ушир.с, 2H), 2,83 (с, 6H), 2,17 (кв, J=8,2 Гц, 4H), 1,45 (с, 6H), 0,95 (т, J=7,4 Гц, 6H). МС: (ES) m/z вычислено для C37H38F8N6O2P [M+H]+ 751,3, найдено 751,2.
Пример 63. Синтез 1-(4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-2,5-дифторфенил)-3-(пирролидин-1-илметил)мочевины
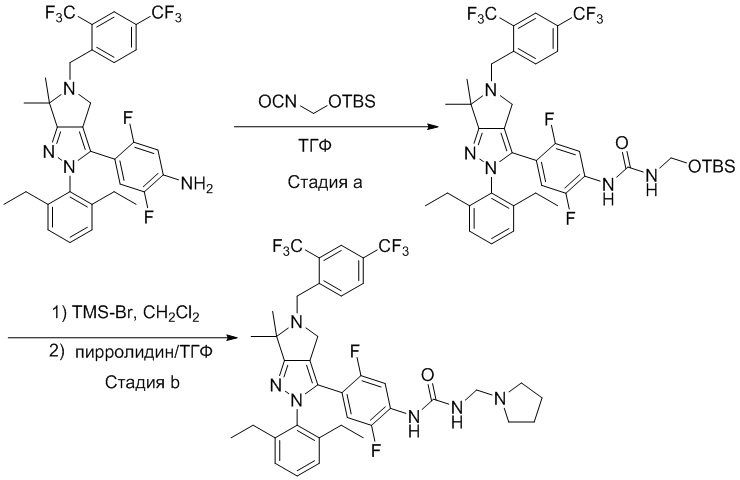
Стадия a: В перемешиваемый раствор 2-гидроксиуксусной кислоты (5,0 г, 65,7 ммоль) в дихлорметане (100 мл) при 0°C добавляли имидазол (11,7 г, 164,3 ммоль) и TBSCl (19,7 г, 131,4 ммоль). Реакционную смесь перемешивали 16 часов при комнатной температуре. Реакционную смесь гасили, добавляя по каплям насыщенный водный раствор NaHCO3, и экстрагировали дихлорметаном. Органический слой промывали насыщенным раствором хлорида натрия, сушили над Na2SO4, фильтровали и упаривали. Полученный сырой продукт напрямую использовали в последующей стадии без дополнительной очистки.
В раствор 2-трет-бутилдиметилсилилоксиуксусной кислоты (10,0 г, 32,6 ммоль) в 50 мл дихлорметана при 0°C добавляли оксалилхлорид (8,23 г, 65,3 ммоль). Реакционную смесь перемешивали 3 часа при комнатной температуре. После окончания реакции удаляли растворитель при пониженном давлении и сушили остаток в вакууме. Полученный сырой продукт напрямую использовали в последующей стадии.
В перемешиваемый раствор 2-трет-бутилдиметилсилилоксиацетил хлорида (5,0 г, 15,4 ммоль) в 1:1 смеси ацетон:H2O (30 мл) добавляли NaN3 (2,5 г, 38,5 ммоль) при комнатной температуре и перемешивали 2 часа. После окончания реакции растворитель удаляли при пониженном давлении, и смесь экстрагировали этилацетатом. Объединенные органические слои сушили над сульфатом натрия, фильтровали и упаривали в вакууме, получая 2-трет-бутилдиметилсилилоксиацетил азид. Полученный азид (3,2 г, 1,63 ммоль) растворяли в 20 мл хлороформа при комнатной температуре, и смесь нагревали при 80°C в течение 2 часов. Растворитель удаляли в вакууме и напрямую использовали в последующей стадии.
Трет-бутил(изоцианатометокси)диметилсилан (0,56 г) растворяли в ТГФ (5 мл) и добавляли в раствор 4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-2,5-дифторанилин (1,25 г, 2,0 ммоль) в ТГФ (10 мл). Смесь перемешивали при комнатной температуре в течение 5 часов, затем упаривали в вакууме. Остаток очищали методом колоночной хроматографии на силикагеле (0-100% EtOAc в гексане, с 1% Et3N), получая 1-(4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-2,5-дифторфенил)-3-(((трет-бутилдиметилсилил)окси)метил)мочевину. МС: (ES) m/z вычислено для C40H47F8N5O2Si [M+H]+ 810,3, найдено 810,1.
Стадия b: В раствор 1-(4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-2,5-дифторфенил)-3-(((трет-бутилдиметилсилил)окси)метил)мочевины (150 мг, 0,19 ммоль) в дихлорметане (3 мл) добавляли по каплям TMSBr (84 мг, 0,55 ммоль) при 0°C. Смесь перемешивали при комнатной температуре 1 час, упаривали в вакууме. Остаток растворяли в дихлорметане (4,0 мл) и добавляли пирролидин (65 мг, 0,92 ммоль). Смесь перемешивали при комнатной температуре в течение 4 часов, затем упаривали в вакууме. Остаток очищали методом ВЭЖХ (MeCN/H2O, с 0,1% ТФУК), получая 1-(4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-2,5-дифторфенил)-3-(пирролидин-1-илметил)мочевину. 1H ЯМР (400 МГц, CD3OD) δ 8,19 (д, J=5,9, Гц, 1H), 8,00-8,10 (м, 3H), 7,42-7,48 (м, 1H), 7,20-7,35 (м, 2H), 6,45 (дд, J=6,6, 11,7 Гц, 1H), 4,61 (с, 2H), 4,35-4,55 (м, 2H), 3,85-4,20 (м, 2H), 3,45-3,55 (м, 2H), 3,10-3,35 (м, 3H), 2,25 (кв, J=7,4 Гц, 4H), 1,90-2,18 (м, 5H), 1,65 (с, 6H), 1,06 (т, J=7,8 Гц, 6H). МС: (ES) m/z вычислено для C38H40F8N6O [M+H]+ 749,3, найдено 749,2.
Пример 64. Синтез 1-(4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-2,5-дифторфенил)-3-(гидроксиметил)мочевины
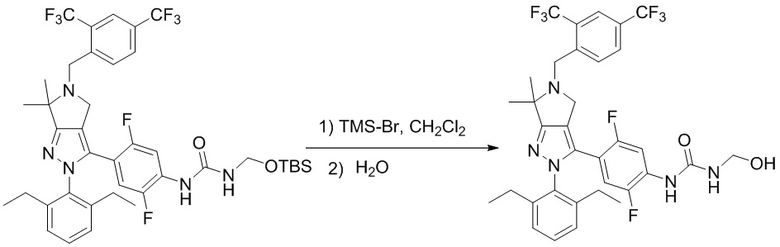
Стадия a: В раствор 1-(4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-2,5-дифторфенил)-3-(((трет-бутилдиметилсилил)окси)метил)мочевины (200 мг, 0,24 ммоль) в дихлорметане (1,4 мл) добавляли по каплям TMSBr (0,11 мл, 0,86 ммоль). Смесь перемешивали при комнатной температуре 1 час, затем упаривали в вакууме и очищали методом ВЭЖХ (MeCN/H2O, с 0,1% ТФУК), получая 1-(4-(5-(2,4-бис(трифторметил)бензил)-2-(2,6-диэтилфенил)-6,6-диметил-2,4,5,6-тетрагидропирроло[3,4-c]пиразол-3-ил)-2,5-дифторфенил)-3-(гидроксиметил) мочевину. 1H ЯМР (400 МГц, CD3OD) δ 8,00-8,25 (м, 4H), 7,42-7,50 (м, 1H), 7,25-7,30 (м, 2H), 6,36-6,45 (м, 1H), 4,75 (ушир.с, 1H), 4,40-4,70 (м, 5H), 2,25 (кв, J=7,4 Гц, 4H), 1,80-1,95 (м, 6H), 1,04 (т, J=3,5 Гц, 6H). МС: (ES) m/z вычислено для C34H33F8N5O2 [M+H]+ 696,3, найдено 696,3.
Пример 65
Этот пример иллюстрирует оценку биологической активности частных вариантов соединений по настоящему изобретению.
Материалы и методы
Клетки
Клетки, экспрессирующие C5a рецепторы
Клетки U937
Клетки U937 представляют собой линию моноцитарных лейкоцитов, которые экспрессируют C5aR и доступны от Американской коллекции типовых культур (ATCC (VA)). Эти клетки выращивали в суспензии в среде RPMI-1640 с добавлением 2 мМ L-глутамина, 1,5 г/л бикарбоната натрия, 4,5 г/л глюкозы, 10 мМ HEPES, 1 мМ пирувата натрия и 10% ФБС (фетальная бычья сыворотка). Клетки выращивали в атмосфере 5% CO2/95% воздух, при 100% влажности при 37°C, пересеивали два раза в неделю в соотношении 1:6 (клетки выращивали при плотности в диапазоне от 1×105 до 2×106 клеток/мл) и собирали при концентрации 1×106 клеток/мл. Перед анализом к клеткам добавляли на ночь 0,5 мМ циклического АМФ (Sigma, OH) и один раз промывали перед использованием. Обработанные цАМФ клетки U937 использовали в тестах связывания с C5aR лигандом и в функциональных тестах.
Выделенные нейтрофилы человека
Опционально можно использовать нейтрофилы человека или грызунов для тестирования активности соединений. Нейтрофилы можно выделить из свежей человеческой крови методами разделения по плотности и центрифугирования. Вкратце, цельную кровь инкубируют с равными частями 3%-ного декстрана и оставляют разделяться на 45 минут. После разделения слоев верхний слой помещают поверх 15 мл Ficoll (15 мл Ficoll на каждые 30 мл суспензии крови) и центрифугируют в течение 30 минут при 400 × g без остановок. Пеллету на дне пробирки отделяют и повторно суспендируют в лизирующем буфере PharmLyse RBC (BD Biosciences, San Jose, CA), после чего образец снова центрифугируют 10 минут при 400 x g без остановок. Полученную пеллету клеток заново суспендируют необходимым образом, она состоит из выделенных нейтрофилов.
Тесты
Подавление связывания лиганда с C5aR
Обработанные цАМФ клетки U937, экспрессирующие C5aR, центрифугировали и заново суспендировали в буфере для проведения анализа (20 мМ HEPES pH 7,1, 140 мМ NaCl, 1 мМ CaCl2, 5 мМ MgCl2, с 0,1% альбумина бычьей сыворотки) в концентрации 3×106 клеток/мл. Анализ связывания проводили следующим образом. 0,1 мл суспензии клеток добавляли в планшеты для проведения анализа, содержащие 5 мкл соединений, что давало финальную концентрацию ~2-10 мкМ каждого скринируемого соединения (или часть зависимости доза-ответ при определении значения IC50 для данного соединения). Затем добавляли 0,1 мл 125I-меченого C5a (получено от Perkin Elmer Life Sciences, Boston, MA), разбавленного в буфере для проведения анализа до финальной концентрации ~50 пкМ, что дает ~30000 импульсов в минуту на лунку, планшеты герметично закрывали и инкубировали примерно 3 часа при 4°C на платформе-шейкере. Реакционные растворы отсасывали на стеклянных фильтрах GF/B, смоченных 0,3%-ным раствором полиэтиленимина (PEI), в вакуумный коллектор клеток (Packard Instruments; Meriden, CT). Добавляли в каждую лунку сцинтилляционную жидкость (40 мкл; Microscint 20, Packard Instruments), планшеты герметично закрывали и замеряли уровень радиоактивности в сцинтиляционном счетчике Topcount (Packard Instruments). Контрольные лунки, содержащие либо только разбавитель (для оценки общего уровня импульсов), либо избыток C5a (1 мкг/мл, для неспецифического связывания), использовали для вычисления процента общего подавления для каждого соединения. Использовали компьютерную программу Prism от GraphPad, Inc. (San Diego, Ca) для вычисления значений IC50. Значения IC50 - это концентрации, необходимые для уменьшения связывания радиоактивно-меченого C5a с рецептором на 50%. (Для дополнительной информации по анализу связывания с лигандом и другим функциональным анализам, см. работы Dairaghi, et al., J. Biol. Chem. 274:21569-21574 (1999), Penfold, et al., Proc. Natl. Acad. Sci. USA. 96:9839-9844 (1999), и Dairaghi, et al,. J. Biol. Chem. 272:28206-28209 (1997)).
Мобилизация кальция
Опционально, соединения можно протестировать на их способность подавлять поток кальция в клетках. Для детектирования высвобождения кальция из внутриклеточных депо, клетки (например, цАМФ-стимулированные U937 или нейтрофилы) инкубируют с 3 мкМ красителем INDO-1AM (Molecular Probes; Eugene, OR) в среде для выращивания клеток в течение 45 минут при комнатной температуре и промывали фосфатно-солевым буфером (PBS). После загрузки INDO-1AM, клетки повторно суспендировали в буфере для анализа потока (сбалансированный солевой раствор Хенкса (HBSS) и 1% FBS). Мобилизацию кальция замеряли с помощью спектрофотометра Photon Technology International (Photon Technology International; New Jersey) с длиной волны возбуждающего излучения 350 нм и двойной одновременной регистрацией испускаемой флуоресценции с длиной волны 400 нм и 490 нм. Относительные концентрации внутриклеточного кальция выражают в виде соотношения испускания 400нм/490нм. Эксперименты проводят при 37°C при непрерывном перемешивании в кюветах, каждая из которых содержит 106 клеток в 2 мл буфера для анализа потока. Хемокиновые лиганды можно применять в диапазоне концентраций от 1 до 100 нМ. Соотношение испускания откладывают на диаграмме относительно времени (обычно 2-3 минуты). Соединения-кандидаты, блокирующие связывание лиганда (до 10 мкМ), добавляют в момент времени 10 секунд, затем добавляют хемокины в момент времени 60 секунд (C5a; R&D Systems; Minneapolis, MN) и контрольный хемокин (SDF-1α; R&D Systems; Minneapolis, MN) в момент времени 150 секунд.
Анализ хемотаксиса
Опционально, соединения можно протестировать на их способность подавлять хемотаксис в клетках. Анализ хемотаксиса проводят с поликарбонатными фильтрами, покрытыми поливинилпирролидоном, с размером пор 5 мкм, в 96-луночных камерах для анализа хемотаксиса (Neuroprobe; Gaithersburg, MD), используя буфер для анализа хемотаксиса (сбалансированный солевой раствор Хенкса (HBSS) и 1% FBS). Используют C5aR лиганды (C5a, R&D Systems; Minneapolis, MN) для оценки подавления соединением C5aR-опосредуемой миграции. Другие хемокины (SDF-1α; R&D Systems; Minneapolis, MN) используют для контроля специфичности. В нижнюю камеру помещают 29 мкл хемокина (0,03 нМ C5a) и различные количества соединения; верхняя камера содержит 100000 клеток U937 или 20 мкл нейтрофилов. Камеры инкубируют 1,5 часа при 37°C и количественно оценивают число клеток в нижней камере либо прямым подсчетом клеток в пяти полях зрения под большим увеличением, либо методом CyQuant (Molecular Probes) - метод флуоресцентного окрашивания, который определяет содержание нуклеиновой кислоты и дает микроскопическую картину.
Идентифицирование ингибиторов C5aR
Анализ
Для тестирования малых органических молекул, которые нарушают связывание C5a рецептора с лигандом, применяли анализ, который детектирует радиоактивный лиганд (например, C5a), связанный с клетками, экспрессирующими C5aR на поверхности (например, цАМФ-стимулированные клетки U937 или выделенные нейтрофилы человека). Для соединений, ингибирующих связывание, конкурентно или неконкурентно, наблюдаются более низкие значения радиоактивности по сравнению с неингибированными контрольными образцами.
Равные количества клеток добавляли в каждую лунку в планшете. Затем клетки инкубировали с радиоактивно-меченым C5a. Несвязанный лиганд удаляли путем промывания клеток, а связанный лиганд определяли количественным подсчетом числа импульсов. Клетки, которые инкубировали без добавления какого-либо органического соединения, давали общее число импульсов (имп/мин общее); неспецифическое связывание определяли путем инкубирования клеток с немеченым лигандом и меченым лигандом. Процент ингибирования определяли по уравнению:
% ингибирования = (1 - [(имп/мин в образце) - (неспецифич. имп/мин)]/[(имп/мин общее) - (неспецифич. имп/мин)]) × 100.
Кривые зависимости доза-ответ
Для выяснения сродства соединений-кандидатов к C5aR, а также подтверждения их способности ингибировать связывание лиганда, определяли их ингибирующую активность в диапазоне концентраций от 1×10-10 до 1×10-4 M. В этом исследовании варьировали количество соединения; число белых кровяных телец и концентрация лиганда оставались постоянными.
Модель эффективности in vivo
Испытуемые соединения оценивали по их потенциальной эффективности при лечении C5a-опосредуемых состояний путем определения эффективности соединения в животной модели. Помимо описанных ниже моделей, другие подходящие животные модели для испытаний этих соединений можно найти в работе Mizuno, M. et al., Expert Opin. Investig. Drugs (2005), 14(7), 807-821, которая включена в настоящий текст в полном объеме посредством ссылки.
Модели C5a-индуцированной лейкопении
C5a-индуцированная лейкопения в мышиной модели с knock-in человеческим геном C5aR
Для исследования эффективности соединений по настоящему изобретению в животной модели, по стандартным методикам создавали рекомбинантных мышей, у которых генетическая последовательность, кодирующая мышиный C5aR, заменена на последовательность, кодирующую человеческий C5aR, что давало hC5aR-KI мышей. У этих мышей введение hC5a приводит к повышенной выработке адгезионных молекул на стенках кровеносных сосудов, которые связывают лейкоциты крови, удаляя их из кровотока. Животным вводили 20 мкг/кг hC5a и через 1 минуту определяли количество лейкоцитов в периферической крови по стандартным методикам. Предварительное введение мышам различных дозировок соединений по настоящему изобретению может практически полностью блокировать hC5a-индуцированную лейкопению.
C5a-индуцированная лейкопения в модели яванских макак (Cynomolgus)
Для исследования эффективности соединений по настоящему изобретению в модели не-человекообразных обезьян, исследовали C5a-индуцированную лейкопению в модели яванских макак (cynomolgus). В этой модели введение hC5a приводит к повышенной выработке адгезионных молекул на стенках кровеносных сосудов, которые связывают лейкоциты крови, удаляя их из кровотока. Животным вводили 10 мкг/кг hC5a и через 1 минуту определяли количество лейкоцитов в периферической крови.
Мышиная модель АНЦА-индуцированного васкулита
В День 0 внутривенно вводили hC5aR-KI мышам 50 мг/кг очищенных антител к миелопероксидазе (Xiao et al, J. Clin. Invest. 110: 955-963 (2002)). Затем мышам раз в день перорально вводили определенные дозы соединений по настоящему изобретению или плацебо в течение семи дней, затем мышей забивали и брали почки для гистологического исследования. Анализ срезов почек показывает значительное снижение числа и степени выраженности серповидных и некротических повреждений в клубочках, по сравнению с животными, которым вводили плацебо.
Мышиная модель неоваскуляризации хориоидеи
Для исследования эффективности соединений по настоящему изобретению в лечении возрастной дегенерации желтого пятна (AMD), мембрану Бруха в глазах hC5aR-KI мышей разрывали методом лазерной коагуляции (Nozika et al, PNAS 103: 2328-2333 (2006). Мышам раз в день перорально или в стекловидное тело в соответствующих дозировках вводили плацебо или соединение по настоящему изобретению в течение 1-2 недель. Заживление вызванных лазером повреждений и неоваскуляризацию определяли методами гистологии и ангиографии.
Модели ревматоидного артрита
Кроличья модель деструктивного воспаления суставов
Для исследования эффективности соединений по настоящему изобретению в ингибировании воспалительного ответа у кроликов на внутрисуставное инъецирование липосахарида LPS (компонента бактериальной мембраны), использовали кроличью модель деструктивного воспаления суставов. В этом исследовании моделируется деструктивное воспаление суставов, наблюдаемое при артрите. Внутрисуставное инъецирование LPS вызывает острый воспалительный ответ, характеризуемый высвобождением цитокинов и хемокинов, многие из которых наблюдаются в суставах при ревматоидном артрите. Наблюдается резкий рост концентрации лейкоцитов в синовиальной жидкости и в синовиальной оболочке в ответ на появление этих хемотаксических медиаторов. Селективные антагонисты хемокиновых рецепторов показали эффективность в этой модели (см. Podolin, et al., J. Immunol. 169(11):6435-6444 (2002)).
LPS исследование на кроликах проводили по методикам, описанным в указанной выше работе Podolin с соавторами, самкам новозеландских кроликов (вес около 2 килограммов) вводили внутрисуставно в одно колено LPS (10 нг) либо вместе с плацебо (фосфатно-солевой буфер с 1% ДМСО), либо с добавлением соединения-кандидата (дозировка 1 = 50 мкМ, или дозировка 2 = 100 мкМ), в общем объеме 1,0 мл. Через 16 часов после инъекции LPS колени промывали и проводили подсчет клеток. Положительный эффект от введения соединений подтверждался гистопатологическим исследованием синовиального воспаления. При гистопатологическом исследовании использовали балльную систему оценки воспаления: 1 - минимальное, 2 - слабое, 3 - умеренное, 4 - среднее.
Тестирование соединений в крысиной модели коллаген-индуцированного артрита
Проводили 17-дневное исследование коллаген-индуцированного артрита II типа для оценки влияния соединения-кандидата на вызванное артритом клиническое распухание голеностопного сустава. Коллаген-индуцированный артрит у крыс является экспериментальной моделью полиартрита, которая широко применяется для доклинических испытаний многочисленных противоревматических препаратов (см. Trentham et al., J. Exp. Med. 146(3):857-868 (1977), Bendele et al., Toxicologic Pathol. 27:134-142 (1999), Bendele et al., Arthritis. Rheum. 42:498-506 (1999)). Отличительными чертами данной модели являются надежное возникновение и развитие устойчивого, легко измеримого многосуставного воспаления, выраженное разрушение хряща вкупе с образованием паннуса, резорбция костной ткани (от слабой до умеренной) и разрастание околохрящевой кости.
Самок крыс линии Льюис (вес примерно 0,2 килограмма) усыпляли изофлураном и инъекционно вводили им неполный адъювант Фрейнда, содержащий 2 мг/мл бычьего коллагена II типа, в основание хвоста и в два сайта на спине в дни 0 и 6 данного 17-дневного исследования. Соединение-кандидат вводили ежедневно посредством подкожной инъекции со дня 0 по день 17 в эффективной дозировке. Проводили измерение диаметра голеностопного сустава штангенциркулем, и уменьшение распухания голеностопного сустава принимали за критерий эффективности.
Крысиная модель сепсиса
Для исследования эффективности соединений по настоящему изобретению в ингибировании генерализованного воспалительного ответа, связанного с сепсисоподобными заболеваниями, использовали крысиную модель сепсиса с лигированием и пункцией слепой кишки (CLP). Исследование крыс с CLP проводили как описано в работе Fujimura N, et al. (American Journal Respiratory Critical Care Medicine 2000; 161: 440-446). Вкратце, крыс-альбиносов линии Wistar обоих полов весом 200-250 грамм не кормили 12 часов до эксперимента. Животных содержали в нормальном режиме с чередованием 12 часовых периодов света и темноты и кормили стандартным крысиным кормом в период до 12 часов перед началом эксперимента. Затем животных разбивали на четыре группы; (i) две группы с имитацией операции и (ii) две группы с CLP. Каждую из этих двух групп (т.е. (i) и (ii)) разбивали на контрольную группу (вводили плацебо) и группу, которой вводили испытуемое соединение. Сепсис вызывался методом CLP. Под короткой анестезией проводили срединную лапаротомию с минимальным рассечением, и слепую кишку лигировали сразу под илеоцекальным клапаном шелковой нитью 3-0, так чтобы сохранить кишечную непрерывность. Противобрыжеечную поверхность слепой кишки перфорировали иглой калибра 18 в двух местах, находящихся на расстоянии 1 см друг от друга, и осторожно сдавливали слепую кишку до выдавливания фекальной массы. Затем кишечник возвращали в брюшную полость и зашивали разрез. В конце операции всех крыс реанимировали физраствором, 3 мл/100 грамм веса тела, подкожно. После операции крысам не давали еды, но они имели неограниченный доступ к воде в течение 16 часов, по истечении которых крыс забивали. Группам с имитацией операции делали лапаротомию и проводили манипуляции со слепой кишкой, но без лигирования или перфорирования. Положительный эффект от введения тестируемых соединений определяли по гистопатологической балльной оценке тканей и органов, а также по замерам нескольких ключевых индикаторов работы печени, почек и перекисного окисления липидов. Для оценки работы печени замеряли уровень аспартаттрансаминазы (AST) и аланинтрансаминазы (ALT). Для оценки работы почек исследовали концентрации в крови азота мочевины и креатинина. Также проводили анализ содержания в крови провоспалительных цитокинов, таких как ФНО-альфа и IL-1бета, методом ELISA.
Мышиная SLE модель экспериментального волчаночного нефрита.
Для изучения влияния соединений по настоящему изобретению на системную красную волчанку (SLE) использовали MRL/lpr мышиную SLE модель. Линия MRL/Mp-Tmfrsf6lpr/lpr (MRL/lpr) представляет собой широко используемую модель человеческого SLE. Для тестирования эффективности соединений в этой модели, самцов мышей MRL/lpr равномерно разделяют между контрольной группой и группой на антагонистах C5aR в возрасте 13 недель. Затем в течение следующих 6 недель вводят соединение или плацебо через осмотические насосы для минимизации стрессовых эффектов у животных. Каждые 2 недели берут образцы крови и мочи в течение 6 недель наступления и развития заболевания. У меньшей части этих мышей развивается гломерулосклероз, приводящий к смерти животного из-за отказа почек. Смертность как индикатор отказа почек является одним из замеряемых критериев, и успешное лечение обычно дает снижение числа внезапных смертей в тест-группах. Кроме того, наличие и степень тяжести поражения почек также можно непрерывно отслеживать по замерам содержания азота мочевины в крови и белка в моче. Ткани и органы также собирали в возрасте 19 недель, проводили гистопатологическое и иммуногистохимическое исследование и оценивали по балльной шкале повреждение тканей и инфильтрацию клеток.
Крысиная модель ХОБЛ
Вызванное дымом воспаление дыхательных путей в крысиной модели можно использовать для оценки эффективности соединений при хронической обструктивной болезни легких (ХОБЛ). Селективные антагонисты хемокинов показали эффективность в этой модели (см. Stevenson, et al., Am. J. Physiol Lung Cell Mol Physiol. 288 L514-L522, (2005)). Крысиную модель острой ХОБЛ создавали как описано в работе Stevenson с соавторами. Испытуемое соединение вводили либо системно перорально или внутривенно, либо применяли местно через небулайзер. Самцов крыс линии Sprague-Dawley (350-400 г) помещали в камеры Perspex и подвергали воздействию сигаретного дыма через насос (50 мл каждые 30 секунд, в паузах свежий воздух). Общее время воздействия на крыс составляло 32 минуты. Крыс забивали через 7 дней после изначального воздействия. Положительный эффект от введения соединений оценивали по уменьшению инфильтрации воспалительных клеток, снижению уровней хемокинов и цитокинов.
В хронической модели, мышей или крыс подвергали ежедневному воздействию табачного дыма в течение периода до 12 месяцев. Соединение вводили системно один раз в сутки перорально или местно через небулайзер. Помимо воспаления, наблюдающегося в острой модели (согласно Stevensen et al.), у животных могут наблюдаться также другие патологии, сходные с наблюдающимися у людей с ХОБЛ, такие как эмфизема (на неё указывает среднелинейный интерсепт) и изменение химии легких (см. работу Martorana et al, Am. J. Respir. Crit Care Med. 172(7): 848-53.
Мышиная EAE модель рассеянного склероза
Экспериментальный аутоиммунный энцефаломиелит (EAE) представляет собой модель рассеянного склероза человека. Вариации этой модели были опубликованы и хорошо известны в данной области. В типичной методике используют мышей C57BL/6 (Charles River Laboratories) для создания EAE модели. Мышей иммунизируют введением 200 мкг миелинового олигодендроцитарного гликопротеина (MOG) 35-55 (Peptide International), эмульгированного в полном адъюванте Фрейнда (CFA), содержащем 4 мг/мл Mycobacterium tuberculosis (Sigma-Aldrich), подкожно в день 0. Кроме того, в день 0 и день 2 животным вводят 200 нг коклюшного токсина (Calbiochem) внутривенно. Клинические симптомы оценивают по шкале от 0 до 5 баллов: 0, нет признаков заболевания; 1, вялый хвост; 2, слабость задних конечностей; 3, паралич задних конечностей; 4, слабость или паралич передних конечностей; 5, умирающее животное. Введение испытуемого соединения начинают в день 0 (профилактическое) или в день 7 (терапевтическое, когда имеется гистологическое доказательство наличия заболевания, но клинические симптомы наблюдаются у малого числа животных), и вводят один или больше раз в сутки в концентрациях, подходящих для оценки их активности и фармакокинетических параметров, например 100 мг/кг подкожно. Эффективность соединений можно оценить путем сравнения степени тяжести (максимальный средний балл в присутствии соединения, в сравнении с плацебо), или путем измерения уменьшения числа макрофагов (F4/80 положительные), выделяемых из спинного мозга. Мононуклеары спинного мозга можно выделить при прерывистом градиенте перколла. Клетки можно окрашивать с применением крысиных анти-мышиных F4/80-PE или крысиных IgG2b-PE (Caltag Laboratories) и количественно подсчитывать методом FACS анализа с использованием 10 мкл Polybeads на образец (Polysciences).
Мышиная модель трансплантации почки
У мышей можно создать модели пересадки, например, модель аллогенного трансплантата почки от C57BL/6 мышам BALB/c описана в работе Faikah Gueler et al, JASN Express, Aug 27th, 2008. Вкратце, мышей подвергают анестезии и удаляют в блоке левую почку, присоединенную к манжете аорты и почечной вене с небольшой полой манжетой, с мочеточниками. После удаления левой почки у реципиента, манжеты сосудов соединяют с помощью анастомоза с брюшной аортой реципиента и полой веной, соответственно, ниже уровня нативных почечных сосудов. Мочеточник напрямую соединяют с помощью анастомоза с мочевым пузырем. Критическое время хранения трансплантата на холоде составляет 60 минут, а критическое время хранения трансплантата в тепле составляет 30 минут. Правую нативную почку можно удалить во время трансплантации аллографта или в день 4 после трансплантации для исследования долговременной выживаемости. Отслеживают общее физическое состояние мышей как индикатор отторжения. Введение испытуемого соединения животным можно начинать до операции или сразу после трансплантации, например посредством подкожной инъекции один раз в сутки. Отслеживают функционирование почек и животных и их выживаемость. Уровень креатинина в плазме крови отслеживают автоматическим методом (Beckman Analyzer, Krefeld, Germany).
Мышиная модель ишемии/реперфузии
Мышиную модель ишемически-реперфузионного повреждения можно создать как описано в работе Xiufen Zheng et al, Am. J. Pathol, Vol 173:4, Oct, 2008. Вкратце, мышей CD1 возрастом 6-8 недель подвергают анестезии и помещают на теплый коврик для поддержания температуры в ходе операции. После разреза брюшной стенки иссекают почечные ножки и помещают микрососудистую клипсу на левую почечную ножку на 25-30 минут. После ишемии удаляют клипсы вместе с правой почкой, зашивают разрез и оставляют животное восстанавливаться. Берут кровь на анализ концентрации креатинина и азота мочевины как индикаторов здоровья почек. Альтернативно мониторят выживаемость животных с течением времени. Соединение можно вводить животным до и/или после операции; и влияние на концентрации креатинина и азота мочевины, а также на выживаемость животных используют как индикатор эффективности соединения.
Мышиная модель роста опухоли
Мышам C57BL/6 возрастом 6-16 недель подкожно инъецируют 1×105 клеток TC-1 (ATCC, VA) в левый или правый бок. Через 2 недели после инъекции клеток начинают измерять размеры опухоли штангенциркулем каждые 2-4 дня до тех пор, пока опухоль не достигнет размера, требующего забоя животного. При забое животных вскрывают и отбирают селезенку и опухоли. Вырезанные опухоли измеряют и взвешивают. Соединения можно вводить до и/или после инъекции опухолевых клеток; и замедление или подавления роста опухоли используют для оценки эффективности соединения.
Интермедиаты 1-4 являются сильными антагонистами C5aR со значениями IC50 ≤ 5нМ в анализе хемотаксиса с использованием клеток U937, описанном в Примере 65. Характеристичные данные для Примеров 1-64 приведены в таблице 1 (ниже).
[M+H]+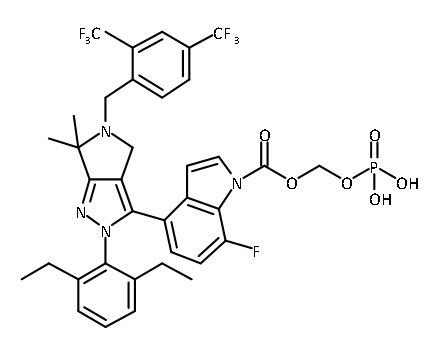
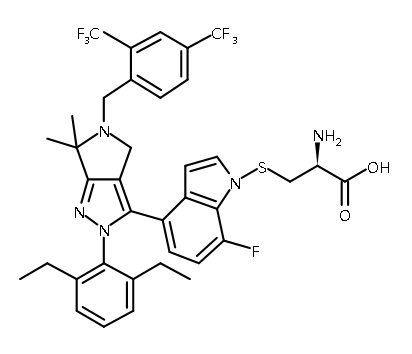
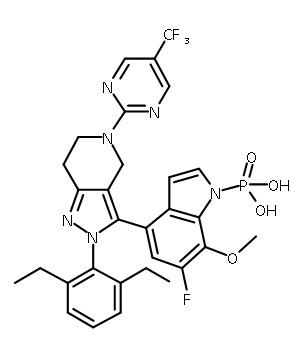
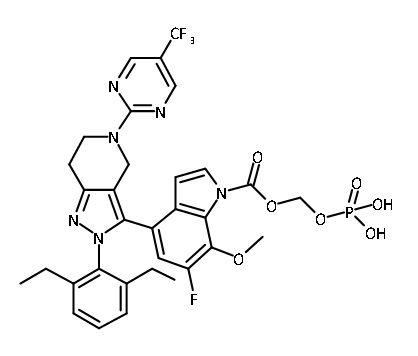
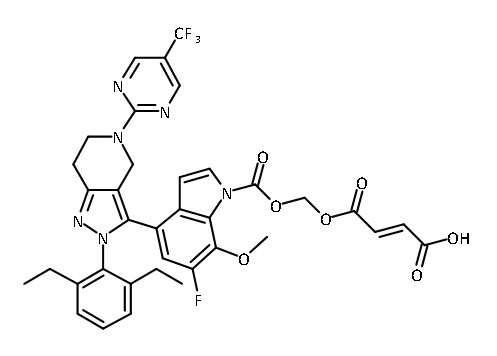
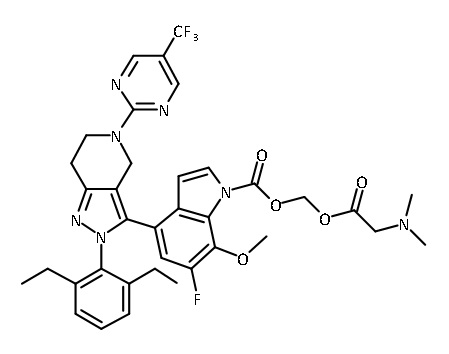
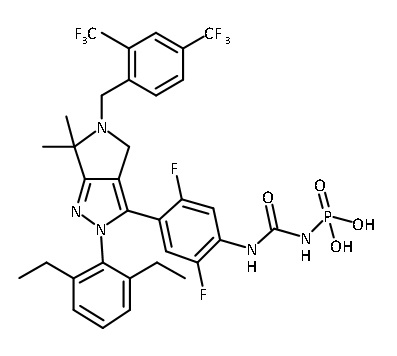
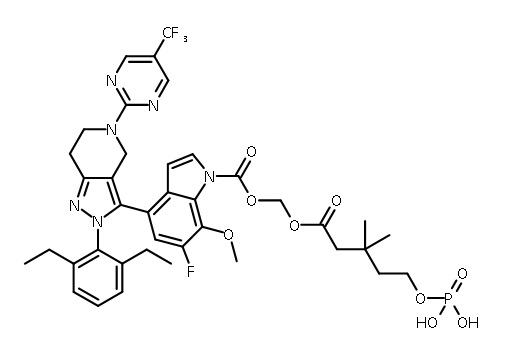

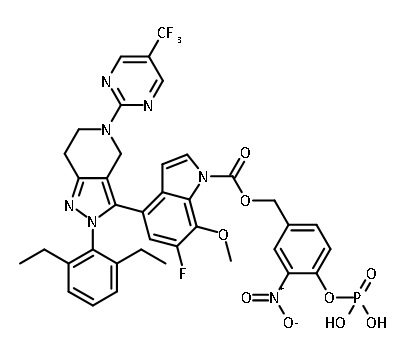
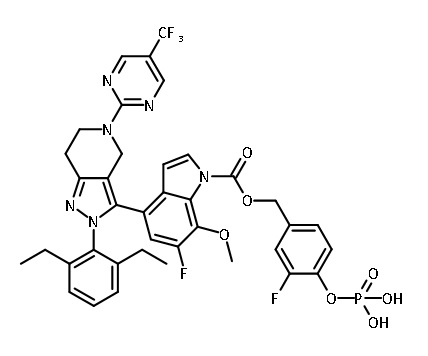
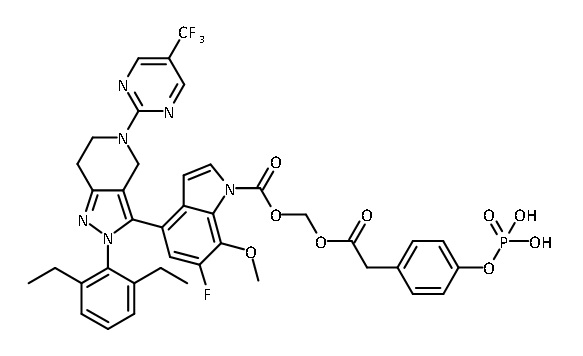

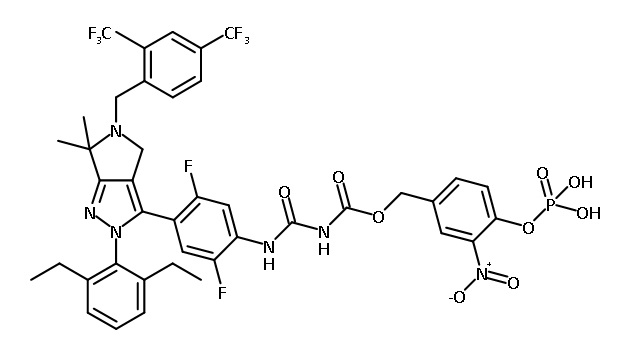
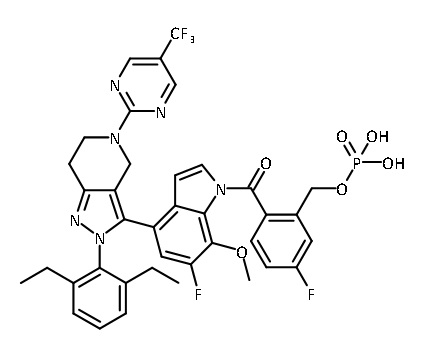
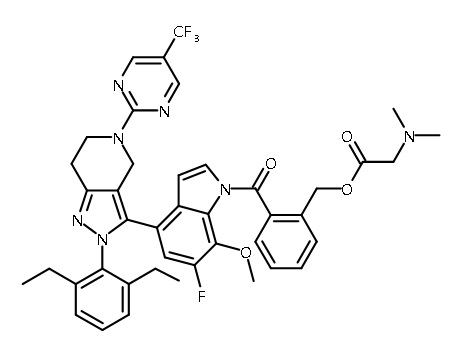
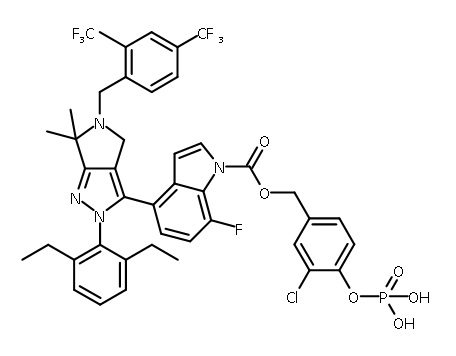
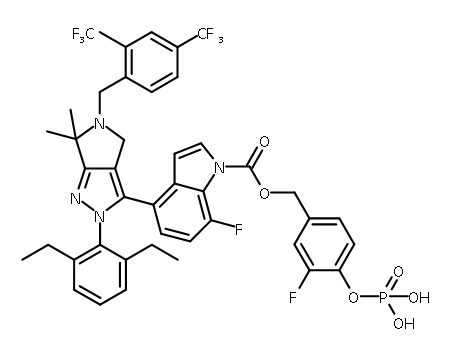
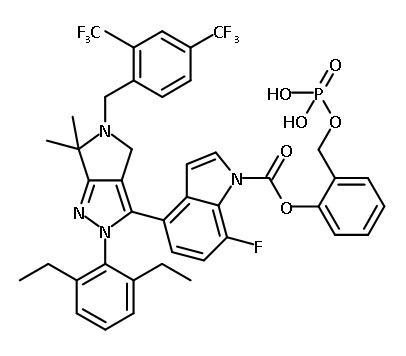
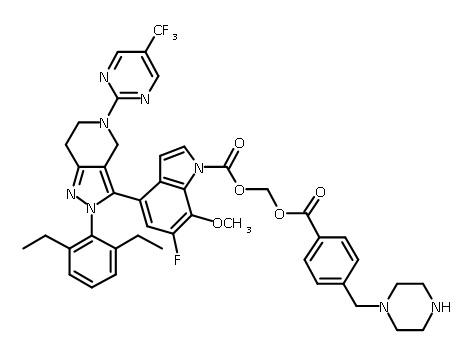
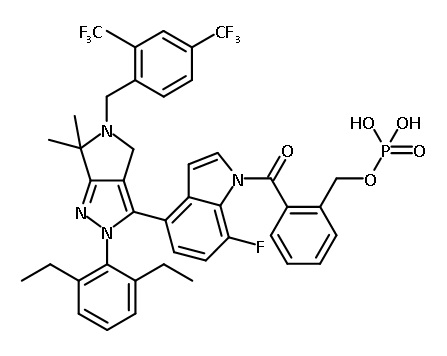
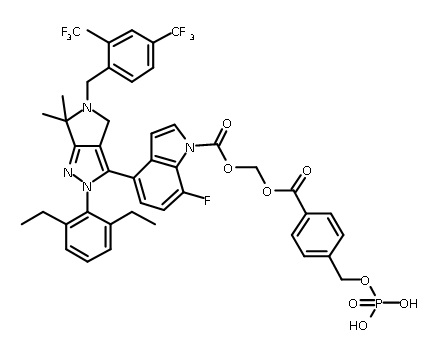

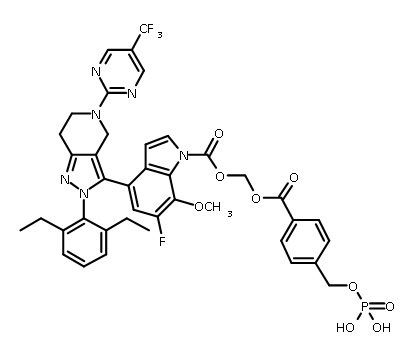
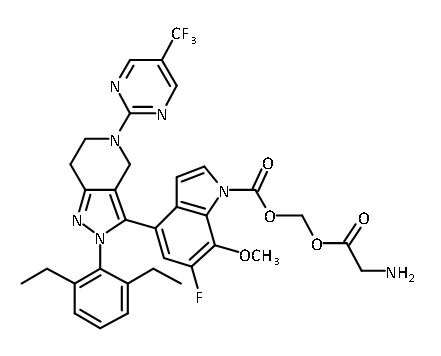
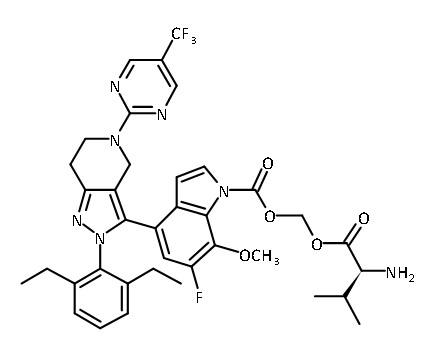
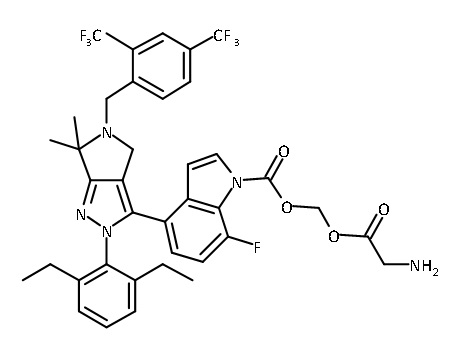
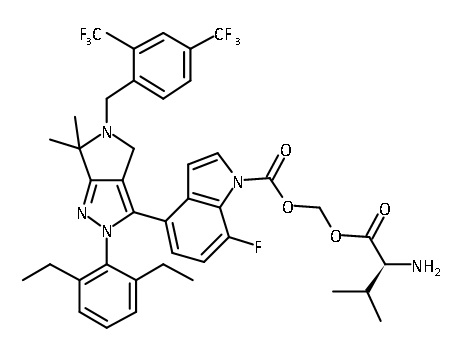

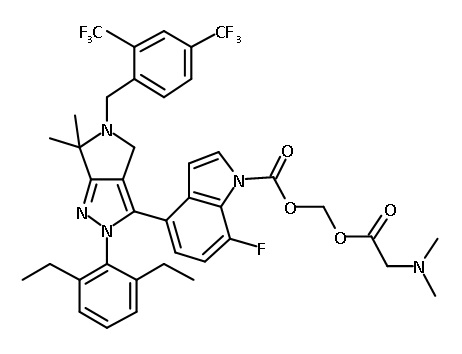
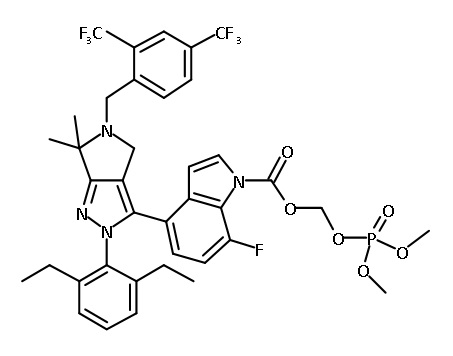

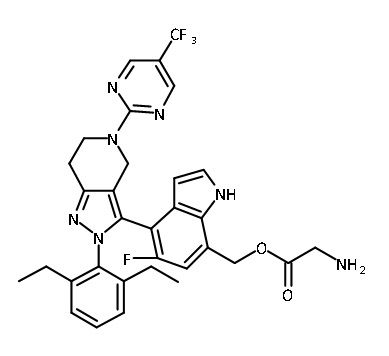
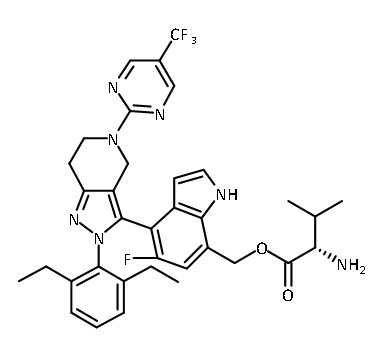
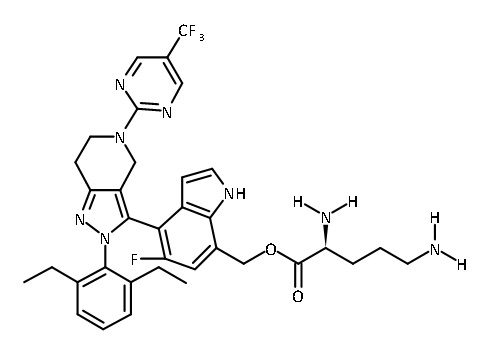
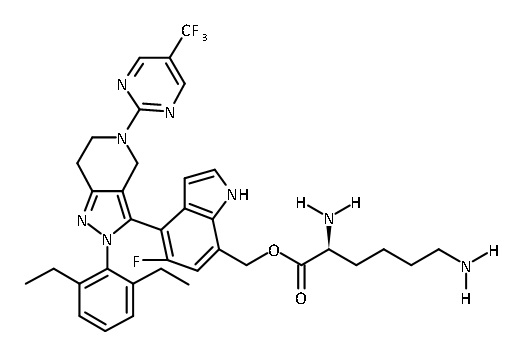
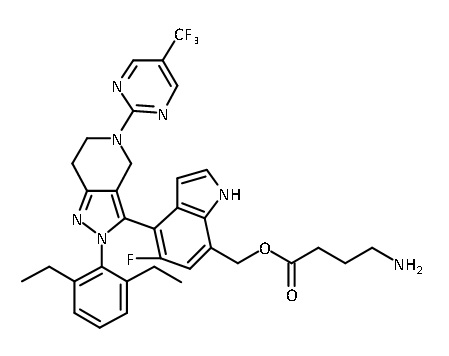
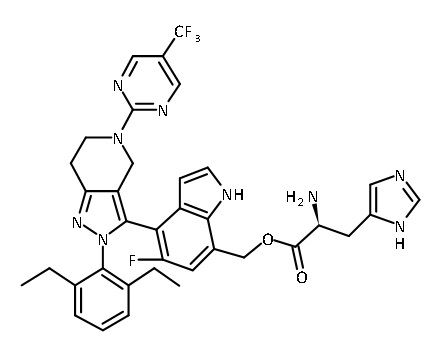
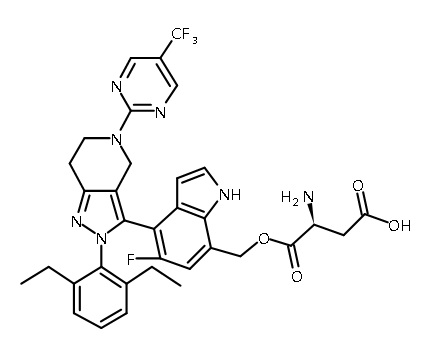
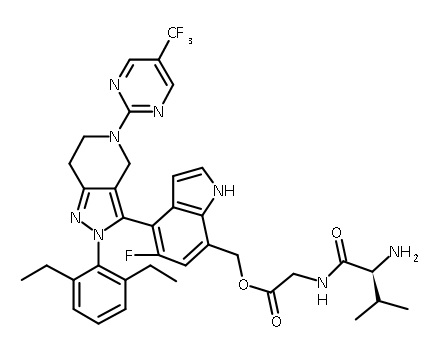

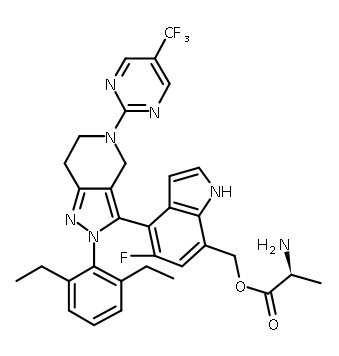
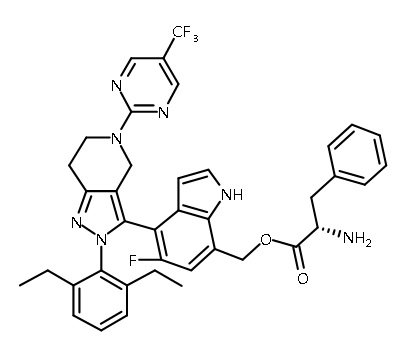
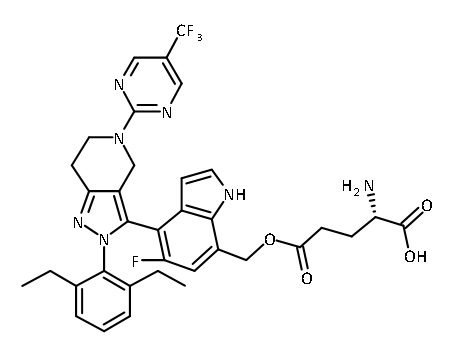
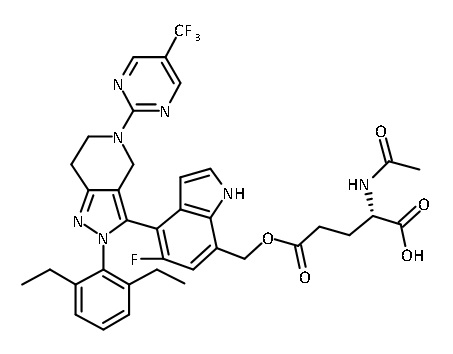
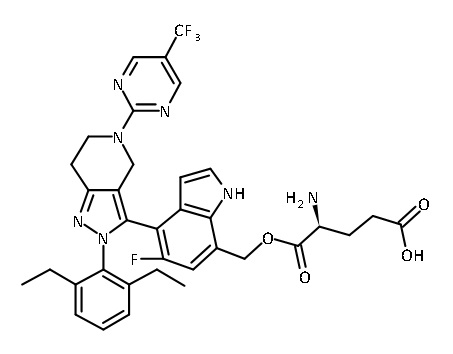
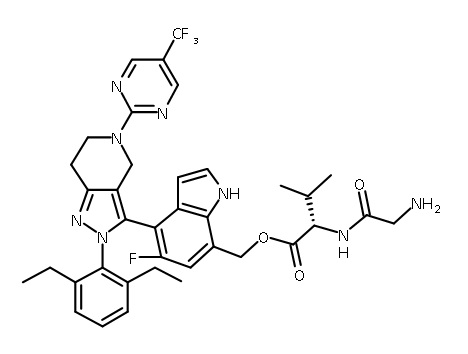
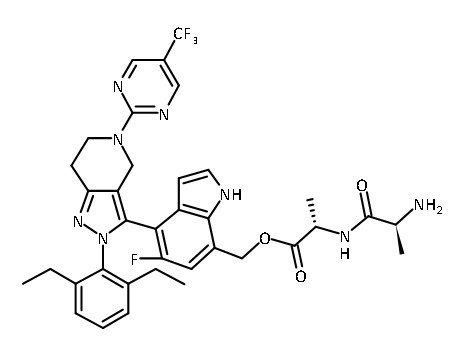
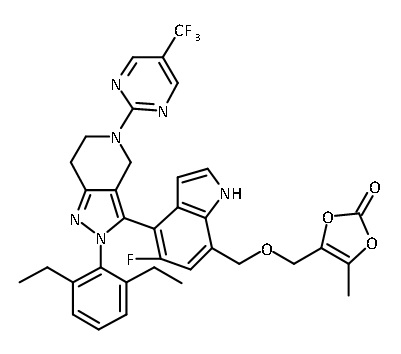
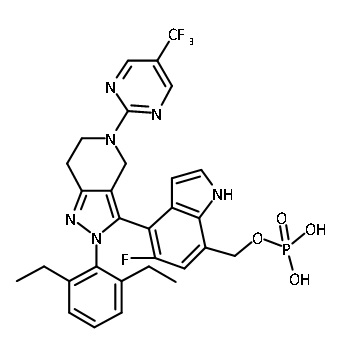


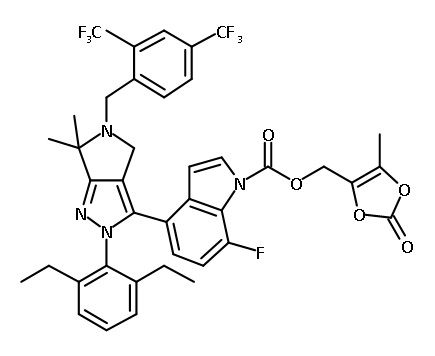
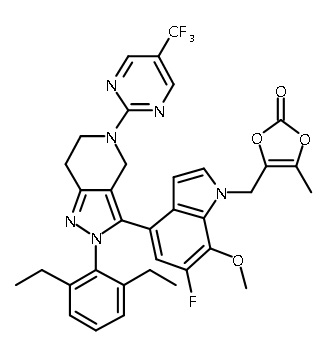

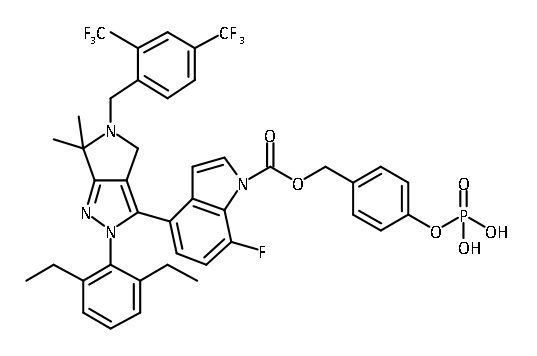
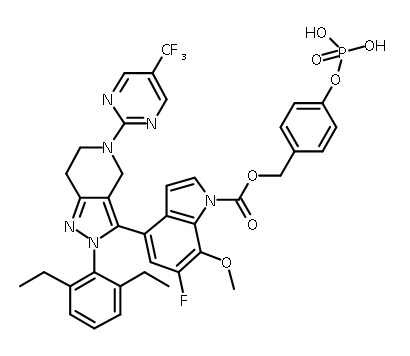
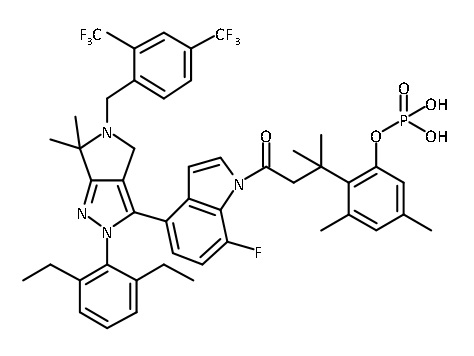
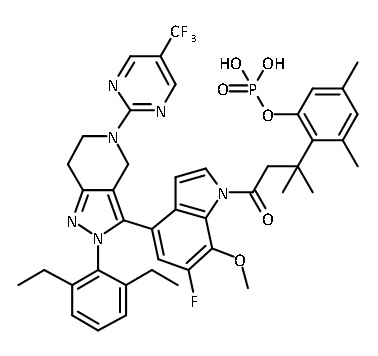
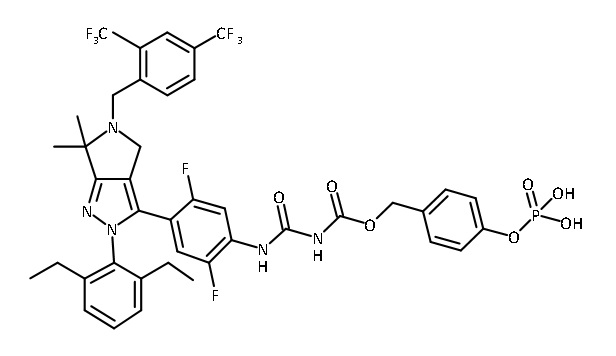
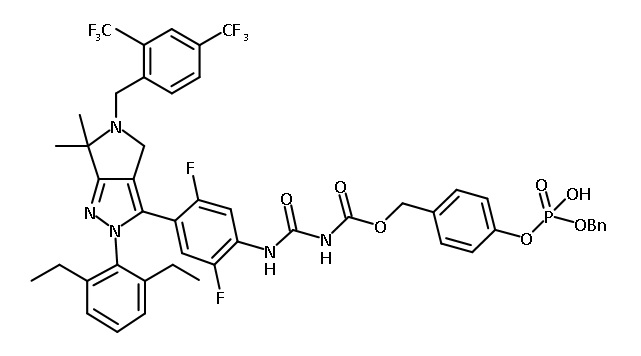
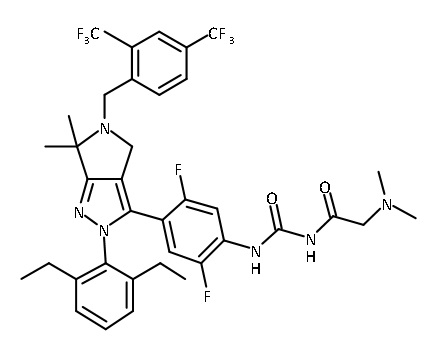
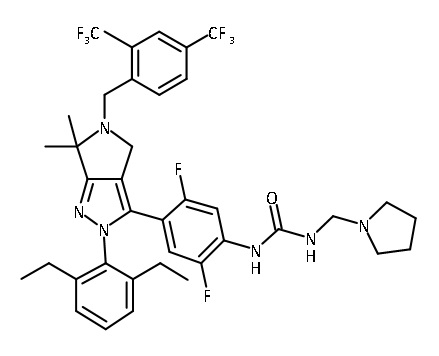
Условия обращенно-фазовой ВЭЖХ, использующиеся при определении времени удерживания в таблице 1:
Колонка: ZORBAX (SB-C18 2,1 x 50 мм, 5 мкм)
Подвижная фаза A: 95% H2O, 5% MeCN (с 0,1% муравьиной кислоты)
Подвижная фаза B: 5% H2O, 95% MeCN (с 0,1% муравьиной кислоты)
Скорость потока: 1,0 мл/мин
Градиент: 20-100% B за 5,5 мин (Метод A)
0-100% B за 4,5 мин (Метод B)
0-100% B за 5,5 мин (Метод C)
Внутривенное введение крысам
Самцов крыс весом от 0,22 до 0,25 кг покупали в Charles River Laboratories (Hollister, Calif.) и подвергали акклиматизации перед экспериментами. Все соединения готовили в форме растворов и вводили животным внутривенно. Соединения из Примера 1 и Примера 4 готовили в смеси 31,6% ДМА/36,8% EtOH/31,6% ПГ, и каждому животному вводили 1 мл/кг. Образцы крови (0,2 мл) брали через яремную вену или в ходе пункции сердца (только в терминальной точке) перед введением дозы, через 2, 5, 10, 15 и 30 минут, 1, 2, 4, 6 и 8 часов после внутривенного введения. Образцы крови брали в охлажденные полипропиленовые пробирки, содержащие калиевую соль ЭДТА в качестве антикоагулянта, и отделяли плазму центрифугированием (Eppendorf Centrifuge 5417R) при 10000 об/мин и 4°С в течение 6 минут, затем хранили при -20°С до проведения анализа.
Образцы плазмы (50 мкл) экстрагировали 200 мкл ацетонитрила, содержащего внутренний стандарт, на линейном шейкере в течение 10 минут и затем центрифугировали при 4450 об/мин в течение 10 минут при 4°С (центрифуга Allegra X-15R, Beckman Coulter, Inc., Fullerton, Calif.). Сто микролитров полученного надосадочного раствора переносили в новый планшет и смешивали с 100 мкл 0,1% муравьиной кислоты в воде для проведения LC-MS/MS анализа. Было обнаружено, что значительные количества действующих веществ высвобождаются после внутривенных инъекций самцам крыс в случае обоих соединений, как показано на фиг. 1 и 2.
Несмотря на то, что в настоящем документе описаны частные варианты осуществления настоящего изобретения, при прочтении описания квалифицированному специалисту в данной области могут стать очевидны вариации раскрытых вариантов осуществления, и квалифицированные специалисты могут, при необходимости, осуществить такие вариации. Соответственно, настоящее изобретение может осуществляться на практике способами, отличными от тех, которые конкретно раскрыты в настоящей заявке, и настоящее изобретение включает все модификации и эквиваленты изобретения, описанного в прилагаемой Формуле изобретения, в соответствии с действующим законодательством. Кроме того, любые комбинации описанных выше элементов во всех их возможных вариациях входят в объем настоящего изобретения, если иное не указано особо или не следует явным образом из контекста.
Все публикации, заявки на патенты, учетные номера и другие ссылки, указанные в настоящем тексте, включены в настоящий текст посредством ссылки, как если бы каждая отдельная публикация или заявка на патент была отдельно и специально указана как включенная в текст посредством ссылки.
| название | год | авторы | номер документа |
|---|---|---|---|
| ДИАРИЛ-ЗАМЕЩЕННЫЕ 6,5-СОПРЯЖЕННЫЕ ЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ КАК ИНГИБИТОРЫ C5aR | 2018 |
|
RU2796983C2 |
| 6-5 КОНДЕНСИРОВАННЫЕ КОЛЬЦА КАК ИНГИБИТОРЫ С5а | 2018 |
|
RU2780338C2 |
| 5-5 КОНДЕНСИРОВАННЫЕ КОЛЬЦА КАК ИНГИБИТОРЫ С5а | 2018 |
|
RU2780322C2 |
| СПОСОБ ЛЕЧЕНИЯ АЛЛЕРГИИ С ИСПОЛЬЗОВАНИЕМ ЗАМЕЩЕННЫХ ПИРАЗОЛОВ | 2001 |
|
RU2259202C2 |
| ЗАМЕЩЕННЫЕ ПИРАЗОЛЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ИНГИБИРОВАНИЯ АКТИВНОСТИ КАТЕПСИНА S | 2001 |
|
RU2278863C2 |
| АНТАГОНИСТЫ ХЕМОКИНОВЫХ РЕЦЕПТОРОВ | 2013 |
|
RU2646762C2 |
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ В КАЧЕСТВЕ ИНГИБИТОРОВ ЭНДОСОМАЛЬНЫХ TOLL-ПОДОБНЫХ РЕЦЕПТОРОВ | 2017 |
|
RU2759678C2 |
| ЗАМЕЩЕННЫЕ ПИРАЗОЛЫ | 2001 |
|
RU2286343C2 |
| ПРОИЗВОДНЫЕ СЕРИНА В КАЧЕСТВЕ АГОНИСТОВ ГРЕЛИНОВЫХ РЕЦЕПТОРОВ | 2015 |
|
RU2695649C2 |
| ПРОИЗВОДНЫЕ ПИРИДИНОНА И ПИРИДАЗИНОНА В КАЧЕСТВЕ ИНГИБИТОРОВ ПОЛИ(ADP-РИБОЗА) ПОЛИМЕРАЗЫ (PARP) | 2007 |
|
RU2472782C2 |
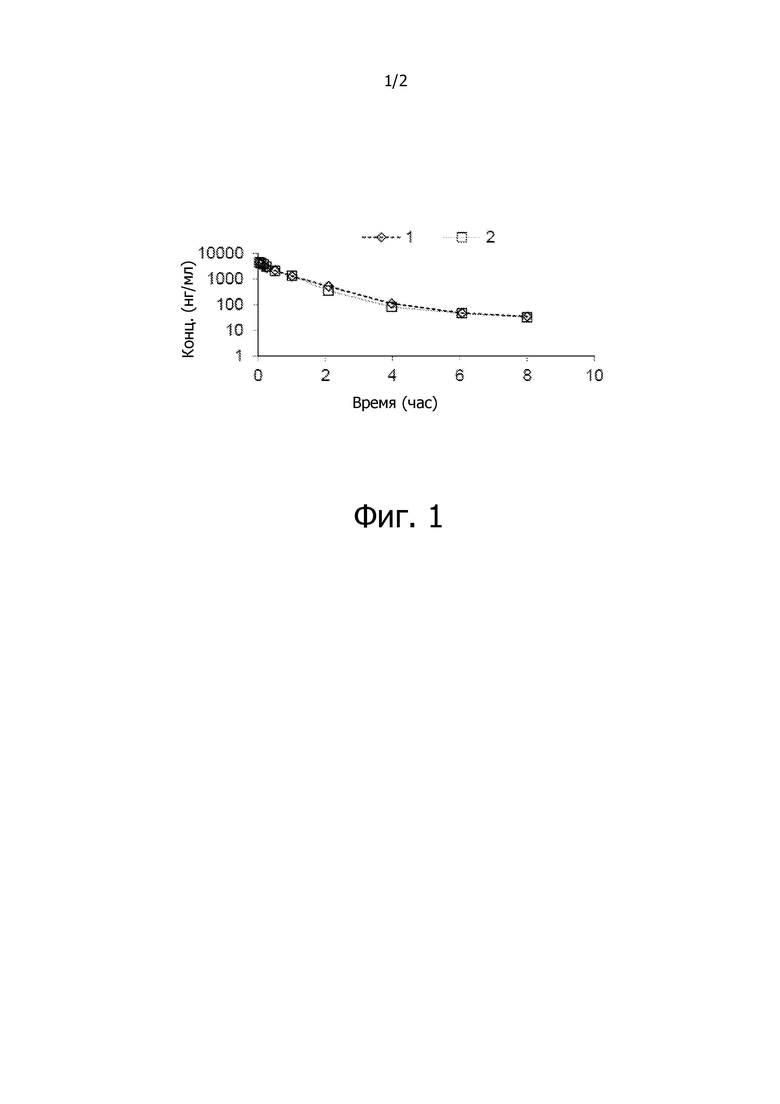
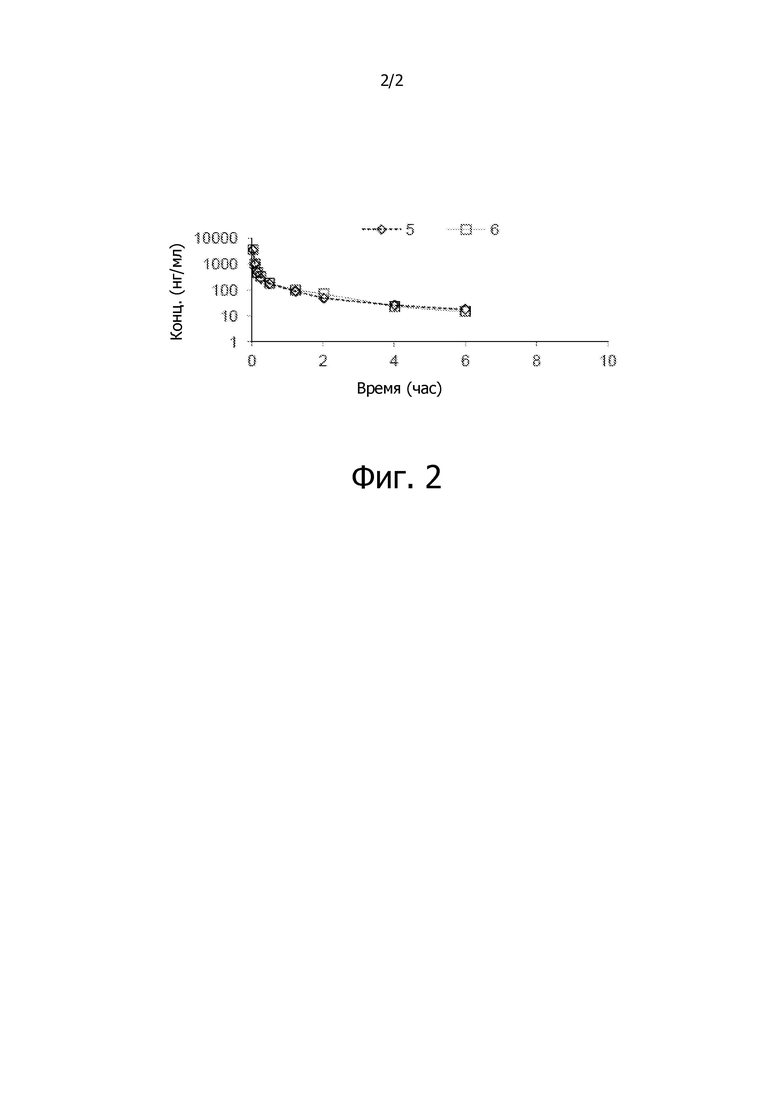
Группа изобретений относится к фармацевтической химии и включает соединения формул (IB), (IC), (IIA) или (IIB) или их фармацевтически приемлемые соли, фармацевтическую композицию и способ лечения на их основе. В формулах (IB), (IC), (IIA) или (IIB) член цикла a представляет собой C(R2c), член цикла b представляет собой C(R2d) и член цикла e представляет собой C(R2e); X1 выбран из группы, состоящей из связи и C1-8 алкилена; R1 выбран из: a) 6-членного гетероарила, содержащего в качестве вершин кольца(ец) 1-4 гетероатома, выбранных из N, O и S; b) C6-10 арила; группа -X1-R1 является незамещенной или замещена 1-5 заместителями Rx; R2a и R2e каждый независимо представляет собой C1-6 алкил; R2b, R2c и R2d каждый независимо представляет собой атом водорода; каждый R3 представляет собой C1-4 алкил; R4 представляет собой -NHC(O)NHP1; каждый R5 представляет собой галоген; R5’ представляет собой атом водорода; R6 выбран из группы, состоящей из C1-6 алкокси и галогена; R7 представляет собой P1; и R8 представляет собой -CH2OP1; каждый P1 представляет собой пролекарственный компонент; каждый Rx независимо выбран из группы, состоящей из галогена, CN, C1-4 алкила, C1-4 алкокси, C1-4 галогеналкила, C1-4 галогеналкокси, C1-4 гидроксиалкила, C2-4 алкенила, C3-6 циклоалкила, CO2-C1-4 алкила и CONH2; подстрочный индекс m равен 0, 1, 2, 3 или 4; и подстрочный индекс n равен 0, 1, 2 или 3; где P1 выбран из групп, указанных в формуле изобретения. Технический результат: соединения формул (IB), (IC), (IIA) или (IIB), обладающие ингибирующей активностью в отношении C5aR. 3 н. и 11 з.п. ф-лы, 2 ил., 1 табл., 65 пр.
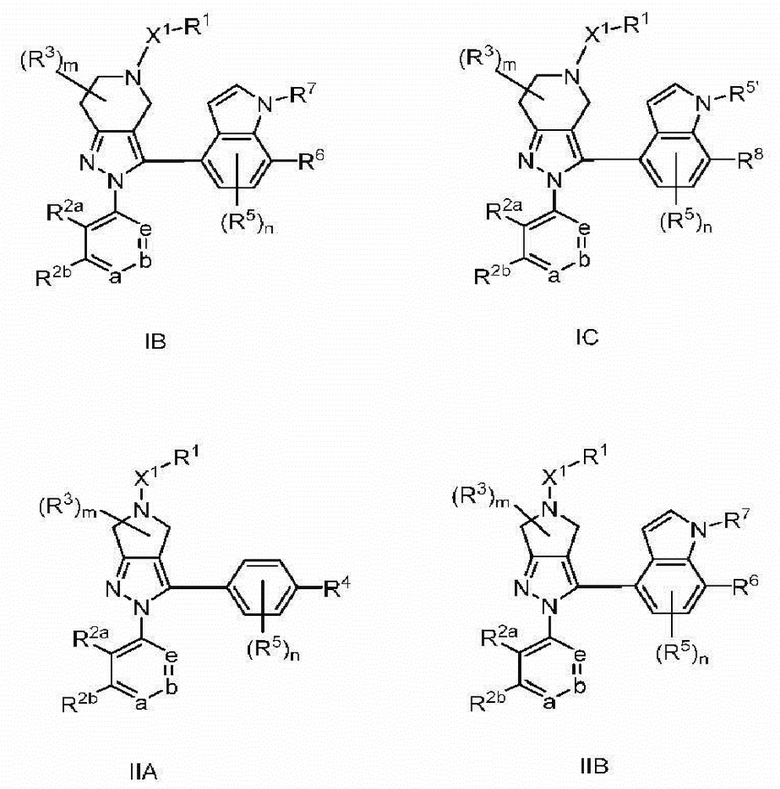
1. Соединение, имеющее любую из формул (IB), (IC), (IIA) или (IIB):
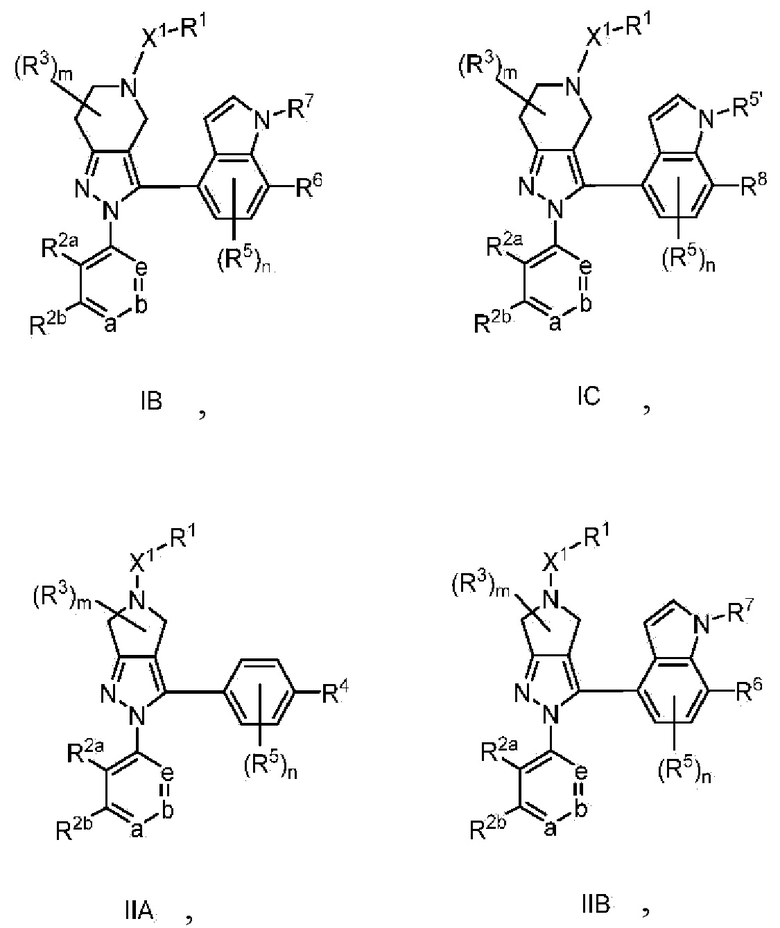
или его фармацевтически приемлемая соль, где
член цикла a представляет собой C(R2c), член цикла b представляет собой C(R2d) и член цикла e представляет собой C(R2e);
X1 выбран из группы, состоящей из связи и C1-8 алкилена;
R1 выбран из группы, состоящей из следующих:
a) 6-членный гетероарил, содержащий в качестве вершин кольца(ец) 1-4 гетероатома, выбранных из N, O и S;
b) C6-10 арил;
где группа -X1-R1 является незамещенной или замещена 1-5 заместителями Rx;
R2a и R2e каждый независимо представляет собой C1-6 алкил;
R2b, R2c и R2d каждый независимо представляет собой атом водорода;
каждый R3 представляет собой C1-4 алкил;
R4 представляет собой -NHC(O)NHP1;
каждый R5 представляет собой галоген;
R5’ представляет собой атом водорода;
R6 выбран из группы, состоящей из C1-6 алкокси и галогена;
R7 представляет собой P1; и
R8 представляет собой -CH2OP1;
каждый P1 представляет собой пролекарственный компонент;
каждый Rx независимо выбран из группы, состоящей из галогена, CN, C1-4 алкила, C1-4 алкокси, C1-4 галогеналкила, C1-4 галогеналкокси, C1-4 гидроксиалкила, C2-4 алкенила, C3-6 циклоалкила, CO2-C1-4 алкила и CONH2;
подстрочный индекс m равен 0, 1, 2, 3 или 4; и
подстрочный индекс n равен 0, 1, 2 или 3;
где (i) P1 выбран из группы, состоящей из:
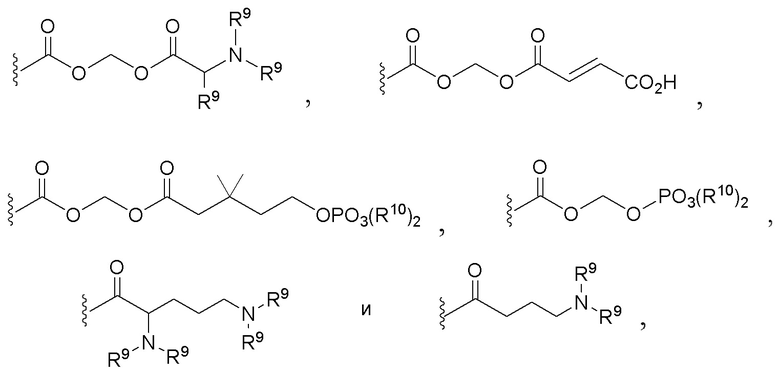
где каждый R9 независимо выбран из группы, состоящей из H и C1-3 алкила; и
каждый R10 независимо выбран из группы, состоящей из H, C1-3 алкила, фенила и бензила; или
(ii) где P1 выбран из группы, состоящей из:
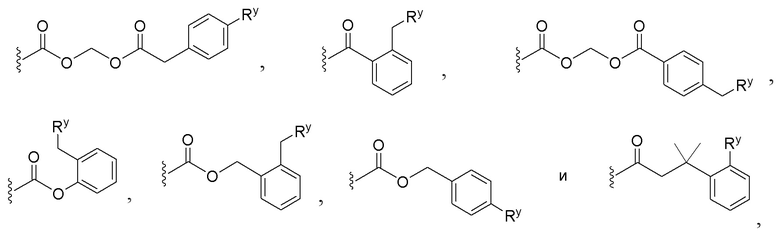
где каждый Ry независимо выбран из группы, состоящей из -OP(O)(ORy1)2, -OC(O)CH2N(Ry2)2, -N(Ry2)2 и пиперазина;
каждый Ry1 независимо выбран из группы, состоящей из H, C1-3 алкила и бензила;
каждый Ry2 независимо представляет собой H или C1-3 алкил; и
каждое фенильное кольцо, содержащее заместитель Ry или -CH2Ry, дополнительно замещено 0-3 заместителями, независимо выбранными из группы, состоящей из нитрогруппы, галогена, CF3 и C1-4 алкила; или
(iii) где P1 выбран из группы, состоящей из -CH2OH, -P(O)(OR10)2 и -CH2-O-P(O)(OR10)2, где каждый R10 независимо выбран из группы, состоящей из H, C1-3 алкила, фенила и бензила; или
(iv) где P1 выбран из группы, состоящей из аминокислоты, дипептида и трипептида, где указанные аминокислотные, дипептидные или трипептидные фрагменты независимо выбраны из группы, состоящей из глицина, аланина, валина, лейцина, изолейцина, лизина, цистеина, аспартата, глутамата, гистидина и фенилаланина, где N-атом каждого аминокислотного фрагмента может быть метилированным или ацилированным; или
(v) где P1 выбран из группы, состоящей из:
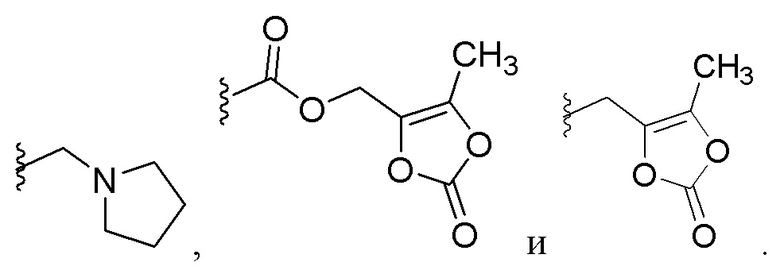
2. Соединение по п. 1 или его фармацевтически приемлемая соль, где R1 представляет собой 6-членный гетероарил, содержащий в качестве вершин кольца(ец) 1-4 гетероатома, выбранных из N, O и S; и где группа -X1-R1 необязательно замещена 1-4 заместителями Rx.
3. Соединение по п. 2 или его фармацевтически приемлемая соль, где R1 выбран из группы, состоящей из пиридила, пиримидинила и пиразинила; и где группа -X1-R1 необязательно замещена 1-4 заместителями Rx.
4. Соединение по любому из пп. 1 - 3 или его фармацевтически приемлемая соль, где R1 представляет собой C6-10 арил; и где группа -X1-R1 необязательно замещена 1-4 заместителями Rx.
5. Соединение по п. 4 или его фармацевтически приемлемая соль, где R1 представляет собой фенил; и где группа -X1-R1 необязательно замещена 1-4 заместителями Rx.
6. Соединение по любому из пп. 1-4 или его фармацевтически приемлемая соль, где R1 выбран из группы, состоящей из фенила, пиридила, пиримидинила и пиразинила; и где группа -X1-R1 необязательно замещена 1-4 заместителями Rx.
7. Соединение по любому из пп. 1-6 или его фармацевтически приемлемая соль, где n равен 0, 1 или 2 и каждый R5, если он присутствует, выбран из группы, состоящей из F и Cl.
8. Соединение по п. 1 или его фармацевтически приемлемая соль, где R1 выбран из группы, состоящей из фенила или пиридила, где группа -X1-R1 необязательно замещена 1-4 заместителями Rx; m равен 0, 1 или 2 и каждый R3, если он присутствует, представляет собой CH3; n равен 0, 1 или 2 и каждый R5, если он присутствует, выбран из группы, состоящей из F и Cl.
9. Соединение по п. 1 или его фармацевтически приемлемая соль, где -X1-R1 выбран из группы, состоящей из:
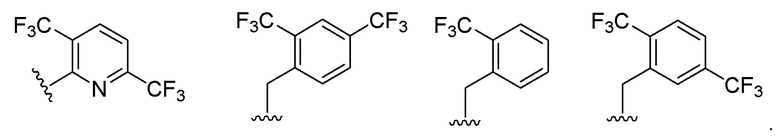
10. Соединение по п. 1 или его фармацевтически приемлемая соль, где R1 выбран из группы, состоящей из:
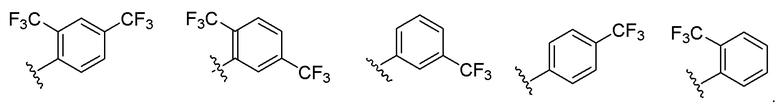
11. Соединение по любому из пп. 1-10 или его фармацевтически приемлемая соль, где 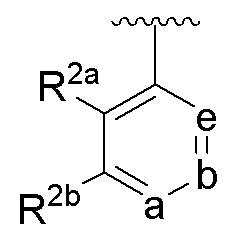 выбран из группы, состоящей из
выбран из группы, состоящей из
12. Соединение по любому из пп. 1-11 или его фармацевтически приемлемая соль, где n равен 0.
13. Фармацевтическая композиция, обладающая ингибирующей активностью в отношении C5aR, содержащая эффективное количество соединения по любому из пп. 1-12 или его фармацевтически приемлемой соли и фармацевтически приемлемый носитель.
14. Способ лечения человека, страдающего от заболевания или расстройства или подверженного заболеванию или расстройству, вовлекающему патологическую активацию рецепторов C5a, включающий введение млекопитающему терапевтически эффективного количества соединения по любому из пп. 1-11 или его фармацевтически приемлемой соли или фармацевтической композиции по п. 13, где заболевание или расстройство выбрано из группы, состоящей из гранулематоза Вегенера, микроскопического полиангиита, C3-гломерулопатии, C3-гломерулонефрита, болезни плотного осадка, атипичного гемолитико-уремического синдрома (aHUS), АНЦА-васкулитов, гнойного гидраденита, системной красной волчанки, волчаночного нефрита, волчаночного гломерулонефрита.
| EA 201490320 A1, 30.06.2014 | |||
| Автомобиль-сани, движущиеся на полозьях посредством устанавливающихся по высоте колес с шинами | 1924 |
|
SU2017A1 |
| US 6893659 B2, 17.05.2005 | |||
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| US 4925846 A1, 15.05.1990. | |||

