ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
Данная не являющаяся предварительной заявка, поданная согласно Разделу 37 Кодекса федеральных регламентов (Code of Federal Regulations; CFR), §1.53(b), испрашивает приоритет согласно Разделу 35 Свода законов США (United States Code; USC), §119(e), предварительной заявки США с серийным №61/555398, поданной 3 ноября 2011 года, которая включена посредством ссылки во всей своей полноте.
ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Данное изобретение в целом относится к соединениям для лечения расстройств, опосредованных тирозинкиназой Брутона (Bruton's tyrosine kinase; Btk), включая воспаление, иммунологическое расстройство и рак, и более конкретно к соединениям, которые ингибируют активность Btk. Изобретение также относится к способам применения этих соединений для диагностики или лечения in vitro, in situ и in vivo клеток млекопитающих или ассоциированных патологических состояний.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Протеинкиназы, самое большое семейство ферментов человека, включают в себя существенно больше 500 белков. Тирозинкиназа Брутона (Btk) является членом Tec-семейства тирозинкиназ и представляет собой регулятор развития В-клеток на ранних этапах, а также активации, передачи сигнала и выживаемости зрелых В-клеток.
Передача сигнала В-клетками через В-клеточный рецептор (B-cell receptor; BCR) может приводить к целому ряду биологических последствий, которые в свою очередь зависят от стадии развития В-клетки. Величина и продолжительность BCR-опосредуемых сигналов должна строго регулироваться. Аберрантная BCR-опосредуемая передача сигнала может быть причиной нарушения активации В-клеток и/или образования патогенных аутоантител, что приводит к многочисленным аутоиммунным и/или воспалительным заболеваниям. Результатом мутации Btk у людей является сцепленная с X-хромосомой агаммаглобулинемия (X-linked agammaglobulinaemia; XLA). Это заболевание характеризуется нарушением созревания В-клеток, уменьшением уровня иммуноглобулинов, нарушением не зависимых от Т-клеток иммунных ответов и заметным ослаблением устойчивой кальциевой сигнализации при BCR-стимуляции. Подтверждение участия Btk в аллергических расстройствах и/или аутоиммунном заболевании и/или воспалительном заболевании было получено на Btk-дефицитных мышиных моделях. Например, на стандартных мышиных доклинических моделях системной красной волчанки (systemic lupus erithematosus, SLE), показано, что дефицит Btk приводит к заметному улучшению прогрессирования данного заболевания. Кроме того, Btk-дефицитные мыши также могут быть устойчивы к развитию коллаген-индуцированного артрита и могут быть менее восприимчивы к индуцированному стафилококками артриту. Многочисленные экспериментальные данные подтверждают роль В-клеточной и гуморальной иммунной системы в патогенезе аутоиммунных и/или воспалительных заболеваний. Терапевтические средства на основе белков (такие как ритуксан), разработанные с целью истощения В-клеток, представляют подход к лечению ряда аутоиммунных и/или воспалительных заболеваний. Вследствие участия Btk в активации В-клеток ингибиторы Btk могут быть полезны в качестве ингибиторов опосредуемой В-клетками патогенной активности (такой как продуцирование аутоантител). Btk также экспрессируется в остеокластах, тучных клетках и моноцитах, и показано, что ее присутствие важно для функционирования этих клеток. Например, дефицит Btk у мышей ассоциирован с нарушением опосредуемой IgE (иммуноглобулин Е) активации тучных клеток (заметным уменьшением высвобождения TNF-альфа (tumor necrosis factor - фактор некроза опухоли) и других воспалительных цитокинов), а дефицит Btk у людей ассоциирован с заметным снижением продуцирования TNF-альфа активированными моноцитами.
Таким образом, ингибирование активности Btk может быть полезно для лечения аллергических расстройств и/или аутоиммунных и/или воспалительных заболеваний, таких как: SLE, ревматоидный артрит, многочисленные васкулитоподобные заболевания, идиопатическая тромбоцитопеническая пурпура (idiopathic thrombocytopenic purpura; ITP), тяжелая миастения, аллергический ринит и астма (Di Paolo et al. (2011) Nature Chem. Biol. 7(1): 41-50; Liu et al. (2011) Jour, of Pharm. and Exper. Ther. 338(1): 154-163). Помимо этого, сообщалось, что Btk участвует в апоптозе; таким образом, ингибирование активности Btk может быть полезно в случае рака, а также для лечения В-клеточной лимфомы, лейкоза и других гематологических злокачественных новообразований. Кроме того, с учетом роли Btk в функционировании остеокластов ингибирование активности Btk может быть полезно для лечения поражений костей, таких как остеопороз. Имеются сообщения о конкретных ингибиторах Btk (Liu (2011) Drug Metab. and Disposition 39(10): 1840-1849; US 7884108, WO 2010/056875; US 7405295; US 7393848; WO 2006/053121; US 7947835; US 2008/0139557; US 7838523; US 2008/0125417; US 2011/0118233; заявка PCT/US2011/050034 "PYRIDINONES/PYRAZINONES, METHOD OF MAKING, AND METHOD OF USE THEREOF", поданная 31 августа 2011 года; заявка PCT/US2011/050013 "PYRIDAZINONES, METHOD OF MAKING, AND METHOD OF USE THEREOF", поданная 31 августа 2011 года; заявка США с серийным №13/102720 "PYRIDONE AND AZA-PYRIDONE COMPOUNDS AND METHODS OF USE", поданная 6 мая 2011 года).
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
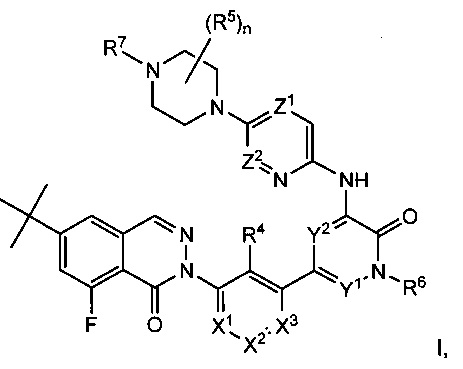
Данное изобретение с целом относится к соединениям формулы I и II, соединениям 8-фторфталазин-1(2h)-онов с активностью, модулирующей тирозинкиназу Брутона (Btk), имеющим структуры:
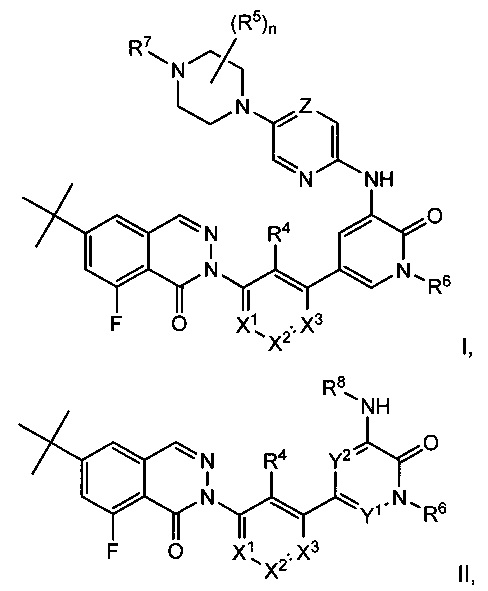
включая стереоизомеры, таутомеры или их фармацевтически приемлемые соли. Различные заместители определены в данном описании ниже.
Один из аспектов изобретения относится к фармацевтической композиции, содержащей соединение формулы I или II и фармацевтически приемлемый носитель, глидант, разбавитель или эксципиент. Фармацевтическая композиция может дополнительно содержать второй терапевтический агент.
Другой аспект изобретения относится к способу изготовления фармацевтической композиции, включающему объединение соединения формулы I или II с фармацевтически приемлемым носителем.
Изобретение включает способ лечения заболевания или расстройства, включающий введение терапевтически эффективного количества соединения формулы I или II пациенту с заболеванием или расстройством, выбранным из иммунных расстройств, рака, сердечно-сосудистого заболевания, вирусной инфекции, воспаления, расстройств метаболизма/функции эндокринной системы и неврологических расстройств, и опосредованным тирозинкиназой Брутона.
Изобретение включает набор для лечения состояния, опосредованного тирозинкиназой Брутона, включающий в себя: а) первую фармацевтическую композицию, содержащую соединение формулы I или II; и b) инструкции к применению.
Изобретение включает соединение формулы I или II для применения в качестве лекарственного средства и для применения в лечении заболевания или расстройства, выбранного из иммунных расстройств, рака, сердечно-сосудистого заболевания, вирусной инфекции, воспаления, расстройств метаболизма/функции эндокринной системы и неврологических расстройств, и опосредованного тирозинкиназой Брутона.
Изобретение включает применение соединения формулы I или II в изготовлении лекарственного средства для лечения иммунных расстройств, рака, сердечно-сосудистого заболевания, вирусной инфекции, воспаления, расстройств метаболизма/функции эндокринной системы и неврологических расстройств, и при этом лекарственное средство опосредованно действует на тирозинкиназу Брутона.
Изобретение включает способы получения соединения формулы I или II.
КРАТКОЕ ОПИСАНИЕ ФИГУР
На Фиг. 1 показано получение 6-трет-бутил-8-фтор-2-(2-(гидроксиметил)-3-(1-метил-5-(5-(4-метилпиперазин-1-ил)пиридин-2-иламино)-6-оксо-1,6-дигидропиридин-3-ил)фенил)фталазин-1(2Н)-она 101 исходя из 4-трет-бутилбензоилхлорида 101а.
На Фиг. 2 показано получение 6-трет-бутил-8-фтор-2-(3-(гидроксиметил)-4-(1-метил-5-(5-(4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-иламино)-6-оксо-1,6-дигидропиридин-3-ил)пиридин-2-ил)фталазин-1(2Н)-она 102 исходя из 2,6-дибром-4-фторбензальдегида 102а.
На Фиг. 3 показано получение 6-трет-бутил-8-фтор-2-(3-(гидроксиметил)-4-(1-метил-5-(5-(4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-иламино)-6-оксо-1,6-дигидропиридин-3-ил)пиридин-2-ил)-фталазин-1(2Н)-она 103 исходя из 2-бром-4-хлорникотинальдегида 103а.
На Фиг. 4 показано получение (S)-6-трет-бутил-8-фтор-2-(3-(гидроксиметил)-4-(1-метил-5-(3-метил-5-(4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-иламино)-6-оксо-1,6-дигидропиридин-3-ил)пиридин-2-ил)фталазин-1(2Н)-она 105 исходя из (3S)-трет-бутил-3-метил-4-(6-нитропиридин-3-ил)пиперазин-1-карбоксилата 105а.
На Фиг. 5 показано получение (R)-6-трет-бутил-8-фтор-2-(3-(гидроксиметил)-4-(1-метил-5-(5-(2-метил-4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-иламино)-6-оксо-1,6-дигидропиридин-3-ил)пиридин-2-ил)фталазин-1(2Н)-она 106 исходя из 1-метил-3-(5-(2-метил-4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-иламино)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2(1Н)-она 106а.
На Фиг. 6 показано получение (S)-6-трет-бутил-8-фтор-2-(4-(гидроксиметил)-5-(1-метил-5-(5-(2-метил-4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-иламино)-6-оксо-1,6-дигидропиридин-3-ил)пиридин-3-ил)фталазин-1(2Н)-она 109 исходя из 3-бром-5-(6-трет-бутил-8-фтор-1-оксо-1,2-дигидрофталазин-2-ил)пиридин-4-карбальдегида 109а.
На Фиг. 7 показано получение 6-трет-бутил-2-(4-(5-(5-((2S,5R)-2,5-диметил-4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-иламино)-1-метил-6-оксо-1,6-дигидропиридин-3-ил)-3-(гидроксиметил)пиридин-2-ил)-8-фторфталазин-1(2Н)-она 110 исходя из 2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-(5-(5-((2S,5R)-2,5-диметил-4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-иламино)-1-метил-6-оксо-1,6-дигидропиридин-3-ил)никотинальдегида 110а.
На Фиг. 8 показано получение (S)-6-трет-бутил-2-(4-(5-(5-(2-этил-4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-иламино)-1-метил-6-оксо-1,6-дигидропиридин-3-ил)-3-(гидроксиметил)пиридин-2-ил)-8-фторфталазин-1(2Н)-она 112 исходя из (8)-2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-(5-(5-(2-этил-4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-иламино)-1-метил-6-оксо-1,6-дигидропиридин-3-ил)никотинальдегида 112а.
ПОДРОБНОЕ ОПИСАНИЕ ТИПИЧНЫХ ВОПЛОЩЕНИЙ
Теперь будет приведена подробная ссылка на некоторые воплощения изобретения, примеры которых иллюстрируются сопровождающими структурами и формулами. Несмотря на то, что изобретение будет описано в сочетании с приведенными воплощениями, очевидно, что изобретение не предполагает ограничения этими воплощениями. Напротив, предполагается, что изобретение охватывает все альтернативные варианты, модификации и эквиваленты, которые могут быть включены в объем настоящего изобретения, определенный формулой изобретения. Специалисту в данной области техники будут известны многие способы и вещества, аналогичные или эквивалентные изложенным в данном описании, которые могут быть использованы при практическом применении настоящего изобретения. Настоящее изобретение никоим образом не ограничено описанными способами и веществами. В том случае, если в одной или более включенных ссылках из литературы, патентов и аналогичных материалов имеется отличие от данной заявки или противоречие с данной заявкой, включая, но не ограничиваясь этим, определенные термины, употребление терминов, описанные методы или тому подобное, то следует руководствоваться раскрытым в данной заявке. Если не указано иное, все технические и научные термины, использованные в данном описании, имеют значение, обычно понимаемое средним специалистом в области техники, к которой данное изобретение относится. Несмотря на то, что при практическом применении или испытании данного изобретения могут быть использованы способы и материалы, аналогичные или эквивалентные изложенным в данном описании, подходящие способы и материалы описаны ниже. Все публикации, патентные заявки, патенты и другие ссылки, упоминаемые в данном описании, включены посредством ссылки во всей своей полноте. Использованная в данной заявке номенклатура основывается на систематической номенклатуре IUPAC (International Union of Pure and Applied Chemistry - Международный союз по теоретической и прикладной химии), если не указано иное.
ОПРЕДЕЛЕНИЯ
При указании числа заместителей термин "один или более" относится к диапазону от одного заместителя до максимально возможного числа замещений, т.е. означает замену заместителями от одного атома водорода до всех атомов водорода включительно. Термин "заместитель" означает атом или группу атомов, заменяющий(ие) атом водорода в исходной молекуле. Термин "замещенный" означает, что конкретная группа уже несет один или более заместителей. Если какая-либо группа может нести несколько заместителей, и предложен ряд возможных заместителей, то такие заместители выбраны независимо и необязательно будут одинаковыми. Термин "незамещенный" означает, что конкретная группа не несет заместителей. Термин "возможно замещенный" означает, что конкретная группа не замещена или замещена одним или более заместителями, независимо выбранными из группы возможных заместителей. При указании числа заместителей термин "один или более" означает от одного заместителя до максимально возможного числа замещений, т.е. означает замену заместителями от одного атома водорода до всех атомов водорода включительно.
Термин "алкил", использованный в данном описании, относится к насыщенному линейному или разветвленному одновалентному углеводородному радикалу из одного-двенадцати атомов углерода (С1-С12), причем этот алкильный радикал возможно может быть независимо замещен одним или более заместителями, описанными ниже. В другом воплощении алкильный радикал содержит одного до восьми атомов углерода (C1-C8) или от одного до шести атомов углерода (С1-С6). Примеры алкильных групп включают, но не ограничиваются этим, метил (Me, -СН3), этил (Et, -СН2СН3), 1-пропил (н-Pr, н-пропил, -СН2СН2СН3), 2-пропил (изо-Pr, изопропил, -СН(СН3)2), 1-бутил (н-Bu, н-бутил, -СН2СН2СН2СН3), 2-метил-1-пропил (изо-Bu, изобутил, -СН2СН(СН3)2), 2-бутил (втор-Bu, втор-бутил, -СН(СН3)СН2СН3), 2-метил-2-пропил (трет-Bu, трет-бутил, -С(СН3)3), 1-пентил (н-пентил, -СН2СН2СН2СН2СН3), 2-пентил (-СН(СН3)СН2СН2СН3), 3-пентил (-СН(СН2СН3)2), 2-метил-2-бутил (-С(СН3)2СН2СН3), 3-метил-2-бутил (-СН(СН3)СН(СН3)2), 3-метил-1-бутил (-СН2СН2СН(СН3)2), 2-метил-1-бутил (-СН2СН(СН3)СН2СН3), 1-гексил (-СН2СН2СН2СН2СН2СН3)), 2-гексил (-СН(СН3)СН2СН2СН2СН3), 3-гексил (-СН(СН2СН3)(СН2СН2СН3)), 2-метил-2-пентил (-С(СН3)2СН2СН2СН3), 3-метил-2-пентил (-СН(СН3)СН(СН3)СН2СН3), 4-метил-2-пентил (-СН(СН3)СН2СН(СН3)2), 3-метил-3-пентил (-С(СН3)(СН2СН3)2), 2-метил-3-пентил (-СН(СН2СН3)СН(СН3)2), 2,3-диметил-2-бутил (-С(СН3)2СН(СН3)2), 3,3-диметил-2-бутил (-СН(СН3)С(СН3)3), 1-гептил, 1-октил и им подобные.
Термин "алкилен", использованный в данном описании, относится к насыщенному линейному или разветвленному двухвалентному углеводородному радикалу из одного-двенадцати атомов углерода (С1-С12), причем этот алкиленовый радикал возможно может быть независимо замещен одним или более заместителями, описанными ниже. В другом воплощении алкиленовый радикал содержит от одного до восьми атомов углерода (C1-C8) или от одного до шести атомов углерода (С1-С6). Примеры алкиленовых групп включают, но не ограничиваются этим, метилен (-СН2-), этилен (-СН2СН2-), пропилен (-СН2СН2СН2-) и им подобные.
Термин "алкенил" относится к линейному или разветвленному одновалентному углеводородному радикалу из двух-восьми атомов углерода (С2-С8), имеющему по меньшей мере один сайт ненасыщенности, т.е. углерод-углеродную sp2-двойную связь, причем этот алкенильный радикал возможно может быть независимо замещен одним или более заместителями, изложенными в данном описании, и включает радикалы, имеющие ориентации "цис" и "транс" или альтернативно, ориентации Έ" и "Z". Примеры включают, но этим не ограничиваются, этиленил или винил (-СН=СН2), аллил (-СН2СН=СН2) и им подобные.
Термин "алкенилен" относится к линейному или разветвленному двухвалентному углеводородному радикалу из двух-восьми атомов углерода (С2-С8), имеющему по меньшей мере один сайт ненасыщенности, т.е. углерод-углеродную sp2-двойную связь, причем этот алкениленовый радикал возможно может быть независимо замещен одним или более заместителями, изложенными в данном описании, и включает в себя радикалы, имеющие ориентации "цис" и "транс" или, альтернативно, ориентации "Е" и "Z". Примеры включают, но не ограничиваются этим, этиленилен или винилен (-СН=СН-), аллил (-СН2СН=СН-) и им подобные.
Термин "алкинил" относится к линейному или разветвленному одновалентному углеводородному радикалу из двух-восьми атомов углерода (С2-С8), имеющему по меньшей мере один сайт ненасыщенности, т.е. углерод-углеродную sp-тройную связь, причем этот алкинильный радикал возможно может быть независимо замещен одним или более заместителями, изложенными в данном описании. Примеры включают, но не ограничиваются, этим этинил (-С≡СН), пропинил (пропаргил, -СН2С≡СН) и им подобные.
Термин "алкинилен" относится к линейному или разветвленному двухвалентному углеводородному радикалу из двух-восьми атомов углерода (С2-С8), имеющему по меньшей мере один сайт ненасыщенности, т.е. углерод-углеродную sp-тройную связь, причем этот алкиниленовый радикал возможно может быть независимо замещен одним или более заместителями, изложенными в данном описании. Примеры включают, но не ограничиваются этим, этинилен (-С≡С-), пропинилен (пропаргилен, -СН2С≡С-) и им подобные.
Термины "карбоцикл", "карбоциклил", "карбоциклическое кольцо" и "циклоалкил" относятся к одновалентному неароматическому, насыщенному или частично ненасыщенному кольцу, имеющему 3-12 атомов углерода (C3-C12) в виде моноциклического кольца или 7-12 атомов углерода в виде бициклического кольца. Бициклические карбоциклы, имеющие 7-12 атомов, могут быть организованы, например, в виде бицикло-[4,5]-, -[5,5]-, -[5,6]- или -[6,6]-системы, а бициклические карбоциклы, имеющие 9 или 10 атомов в кольце, могут быть организованы в виде бицикло-[5,6]- или -[6,6]-системы или в виде соединенных мостиковой связью систем, таких как бицикло[2.2.1]гептан, бицикло[2.2.2]октан и бицикло[3.2.2]нонан. Спиро-группировки также включены в объем этого определения. Примеры моноциклических карбоциклов включают, но не ограничиваются этим, циклопропил, циклобутил, циклопентил, 1-циклопент-1-енил, 1-циклопент-2-енил, 1-циклопент-3-енил, циклогексил, 1-циклогекс-1-енил, 1-циклогекс-2-енил, 1-циклогекс-3-енил, циклогексадиенил, циклогептил, циклооктил, циклононил, циклодецил, циклоундецил, циклододецил и им подобные. Карбоциклильные группы возможно независимо замещены одним или более заместителями, изложенными в данном описании.
"Арил" означает одновалентный ароматический углеводородный радикал из 6-20 атомов углерода (С6-С20), образованный в результате удаления одного атома водорода от одного атома углерода исходной ароматической кольцевой системы. Некоторые арильные группы представлены в типичных структурах как "Ar". Арил включает бициклические радикалы, содержащие ароматическое кольцо, конденсированное с насыщенным, частично ненасыщенным кольцом или ароматическим карбоциклическим кольцом. Типичные арильные группы включают, но не ограничиваются этим, радикалы, образованные из бензола (фенил), замещенных бензолов, нафталина, антрацена, радикалы бифенил, инденил, инданил, радикалы, образованные из 1,2-дигидронафталина, радикал 1,2,3,4-тетрагидронафтил и тому подобное. Арильные группы возможно независимо замещены одним или более заместителями, изложенными в данном описании.
"Арилен" означает двухвалентный ароматический углеводородный радикал из 6-20 атомов углерода (С6-С20), образованный в результате удаления двух атомов водорода от двух атомов углерода исходной ароматической кольцевой системы. Некоторые ариленовые группы представлены в типичных структурах как "Ar". Арилен включает бициклические радикалы, содержащие ароматическое кольцо, сконденсированное с насыщенным, частично ненасыщенным кольцом или ароматическим карбоциклическим кольцом. Типичные ариленовые группы включают, но не ограничиваются этим, радикалы, происходящие из бензола (фенилен), замещенных бензолов, нафталина, антрацена, бифенилена, инденилена, инданилена, 1,2-дигидронафталина, радикал 1,2,3,4-тетрагидронафтил и тому подобное. Ариленовые группы возможно замещены одним или более заместителями, изложенными в данном описании.
Термины "гетероцикл", "гетероциклил" и "гетероциклическое кольцо" используются в данном описании взаимозаменяемо и относятся к насыщенному или частично ненасыщенному (т.е. имеющему одну или более двойных и/или тройных связей в пределах кольца) карбоциклическому радикалу, содержащему от 3 до примерно 20 кольцевых атомов, в котором по меньшей мере один атом в кольце представляет собой гетероатом, выбранный из атомов азота, кислорода, фосфора и серы, а остальные кольцевые атомы представляют собой атомы С, причем один или более чем один кольцевой атом возможно независимо замещен одним или более заместителями, описанными ниже. Гетероцикл может представлять собой моноцикл, имеющий 3-7 кольцевых членов (2-6 атомов углерода и 1-4 гетероатома, выбранных из Ν, О, Ρ и S), или бицикл, имеющий 7-10 кольцевых членов (4-9 атомов углерода и 1-6 гетероатомов, выбранных из Ν, О, Ρ и S), например: бицикло-[4,5]-, -[5,5]-, -[5,6]- или -[6,6]-систему. Гетероциклы описаны в Paquette Leo Α.; "Principles of Modern Heterocyclic Chemistry" (W.A. Benjamin, New York, 1968), в частности в главах 1, 3, 4, 6, 7 и 9; "The Chemistry of Heterocyclic Compounds, A series of Monographs" (John Wiley & Sons, New York, 1950 до настоящего времени), в частности, в томах 13, 14, 16, 19 и 28; и в J. Am. Chem. Soc. (1960) 82: 5566. "Гетероциклил" также включает радикалы, при этом гетероциклические радикалы конденсированы с насыщенным, частично ненасыщенным кольцом или ароматическим карбоциклическим или гетероциклическим кольцом. Примеры гетероциклических колец включают, но не ограничиваются этим, морфолин-4-ил, пиперидин-1-ил, пиперазинил, пиперазин-4-ил-2-он, пиперазин-4-ил-3-он, пирролидин-1-ил, тиоморфолин-4-ил, S-диоксотиоморфолин-4-ил, азокан-1-ил, азетидин-1-ил, октагидропиридо[1,2-а]пиразин-2-ил, [1,4]диазепан-1-ил, пирролидинил, тетрагидрофуранил, дигидрофуранил, тетрагидротиенил, тетрагидропиранил, дигидропиранил, тетрагидротиопиранил, пиперидино, морфолино, тиоморфолино, тиоксанил, пиперазинил, гомопиперазинил, азетидинил, оксетанил, тиетанил, гомопиперидинил, оксепанил, тиепанил, оксазепинил, диазепинил, тиазепинил, 2-пирролинил, 3-пирролинил, индолинил, 2Н-пиранил, 4Н-пиранил, диоксанил, 1,3-диоксоланил, пиразолинил, дитианил, дитиоланил, дигидропиранил, дигидротиенил, дигидрофуранил, пиразолидинилимидазолинил, имидазолидинил, 3-азабицикло[3.1.0]гексанил, 3-азабицикло[4.1.0]гептанил, азабицикло[2.2.2]гексанил, 3Н-индолил, хинолизинил и N-пиридилмочевины. Спиро-группировки также включены в объем этого определения. Примерами гетероциклической группы, где 2 кольцевых атома замещены группировками оксо (=O), являются пиримидинонил и 1,1-диоксо-тиоморфолинил. Приведенные в данном описании гетероциклические группы возможно независимо замещены одним или более заместителями, изложенными в данном описании.
Термин "гетероарил" относится к одновалентному ароматическому радикалу, состоящему из 5-, 6- или 7-членных колец, и включает в себя конденсированные кольцевые системы (по меньшей мере одно из колец в которых является ароматическим) из 5-20 атомов, содержащие один или более гетероатомов, независимо выбранных из атомов азота, кислорода и серы. Примерами гетероарильных групп являются пиридинил (включая, например, 2-гидроксипиридинил), имидазолил, имидазопиридинил, пиримидинил (включая, например, 4-гидроксипиримидинил), пиразолил, триазолил, пиразинил, тетразолил, фурил, тиенил, изоксазолил, тиазолил, оксадиазолил, оксазолил, изотиазолил, пирролил, хинолинил, изохинолинил, тетрагидроизохинолинил, индолил, бензимидазолил, бензофуранил, циннолинил, индазолил, индолизинил, фталазинил, пиридазинил, триазинил, изоиндолил, птеридинил, пуринил, оксадиазолил, триазолил, тиадиазолил, фуразанил, бензофуразанил, бензотиофенил, бензотиазолил, бензоксазолил, хиназолинил, хиноксалинил, нафтиридинил и фуропиридинил. Гетероарильные группы возможно независимо замещены одним или более заместителями, изложенными в данном описании.
Гетероциклические или гетероарильные группы могут быть присоединены по углероду (углерод-связанные) или азоту (азот-связанные), если такое возможно. В качестве примера, а не ограничения, углерод-связанные гетероциклы или гетероарилы связываются по положению 2, 3, 4, 5 или 6 пиридина, по положению 3, 4, 5 или 6 пиридазина, по положению 2, 4, 5 или 6 пиримидина, по положению 2, 3, 5 или 6 пиразина, по положению 2, 3, 4 или 5 фурана, тетрагидрофурана, тиофурана, тиофена, пиррола или тетрагидропиррола, по положению 2, 4 или 5 оксазола, имидазола или тиазола, по положению 3, 4 или 5 изоксазола, пиразола или изотиазола, по положению 2 или 3 азиридина, по положению 2, 3 или 4 азетидина, по положению 2, 3, 4, 5, 6, 7 или 8 хинолина или по положению 1, 3, 4, 5, 6, 7 или 8 изохинолина.
В качестве примера, а не ограничения, азот-связанные гетероциклы или гетероарилы связываются по положению 1 азиридина, азетидина, пиррола, пирролидина, 2-пирролина, 3-пирролина, имидазола, имидазолидина, 2-имидазолина, 3-имидазолина, пиразола, пиразолина, 2-пиразолина, 3-пиразолина, пиперидина, пиперазина, индола, индолина, 1Н-индазола, по положению 2 изоиндола или изоиндолина, по положению 4 морфолина и по положению 9 карбазола или β-карболина.
Термины "лечить" и "лечение" относятся к терапевтическому лечению, при котором у объекта должно быть ослаблено (облегчено) нежелательное физиологическое изменение или расстройство, такое как развитие или распространение артрита или рака. Для задач данного изобретения полезные или желаемые клинические результаты включают, но не ограничиваются этим, ослабление симптомов, снижение степени заболевания, стабильное (т.е. без ухудшения) состояние заболевания, задержку или замедление прогрессирования заболевания, улучшение или временное облегчение болезненного состояния и ремиссию (будь то частичную или полную), как детектируемую, так и недетектируемую. "Лечение" также может означать продление выживаемости по сравнению с ожидаемой выживаемостью в отсутствие получения лечения. Нуждающиеся в таком лечении включают тех, кто уже имеет данное состояние или расстройство.
Фраза "терапевтически эффективное количество" означает количество соединения по настоящему изобретению, которое (1) лечит конкретное заболевание, состояние или расстройство, (2) ослабляет, улучшает или устраняет один или более симптомов конкретного заболевания, состояния или расстройства либо (3) предотвращает или задерживает начало развития одного или более симптомов конкретного заболевания, состояния или расстройства, изложенного в данном описании. В случае рака терапевтически эффективное количество лекарственного средства может уменьшать количество раковых клеток; уменьшать размер опухоли; ингибировать (т.е. замедлять до некоторой степени и предпочтительно останавливать) инфильтрацию раковых клеток в периферические органы; ингибировать (т.е. замедлять до некоторой степени и предпочтительно останавливать) опухолевый метастаз; ингибировать, до некоторой степени, опухолевый рост; и/или ослаблять до некоторой степени один или более симптомов, ассоциированных с раком. В зависимости от возможности лекарственного средства предотвращать рост и/или уничтожать существующие раковые клетки, оно может быть цитостатическим и/или цитотоксическим. В случае терапии рака эффективность можно измерить, например, оценивая время до прогрессирования заболевания (ТТР) и/или определяя коэффициент ответа (RR).
"Воспалительное нарушение", как использовано в данном описании, может относиться к любому заболеванию, расстройству или синдрому, при котором чрезмерный или нерегулируемый воспалительный ответ вызывает гипертрофированные воспалительные симптомы, повреждение ткани реципиента или утрату функции тканей. "Воспалительное нарушение" также относится к патологическому состоянию, опосредованному притоком лейкоцитов и/или хемотаксисом нейтрофилов.
"Воспаление", как использовано в данном описании, относится к локализованному протективному ответу, вызванному повреждением или разрушением тканей, который направлен на разрушение, ослабление или создание барьера (изолирование) как в отношении вредоносного агента, так и поврежденной ткани. Воспаление, в значительной мере ассоциировано с притоком лейкоцитов и/или хемотаксисом нейтрофилов. Воспаление может возникнуть в результате инфекции патогенными микроорганизмами и вирусами и неинфекционными путями, например, в результате травмы или реперфузии после инфаркта миокарда или инсульта, иммунного ответа на чужеродный антиген и аутоиммунных ответов. Соответственно, воспалительные нарушения, поддающиеся лечению соединениями формулы I и II, охватывают расстройства, ассоциированные как с ответами специфической защитной системы, так и с ответами неспецифической защитной системы.
Термин "специфическая защитная система" относится к компоненту иммунной системы, который реагирует на присутствие специфических антигенов. Примеры воспаления, являющегося результатом ответа специфической защитной системы, включают классический ответ на чужеродные антигены, аутоиммунные заболевания и реакцию гиперчувствительности отсроченного типа, опосредованную Т-клетками. Хронические воспалительные заболевания, отторжение твердых трансплантированных тканей и органов, например, трансплантатов почки и костного мозга, и реакция "трансплантат против хозяина" (GVHD), представляют собой другие примеры воспалительных ответов специфической защитной системы.
Термин "неспецифическая защитная система", как он использован в данном описании, относится к воспалительным нарушениям, которые опосредованы лейкоцитами, не обладающими иммунологической памятью (например, гранулоцитами и макрофагами). Примеры воспаления, являющегося результатом, по меньшей мере отчасти, ответа неспецифической защитной системы, включают воспаление, ассоциированное с такими состояниями, как (острый) респираторный дистресс-синдром взрослых (ARDS) или синдромы полиорганной недостаточности; реперфузионная травма; острый гломерулонефрит; реактивный артрит; дерматозы с острыми воспалительными компонентами; острый гнойный менингит или другие воспалительные нарушения центральной нервной системы, такие как инсульт; термическое повреждение; воспалительное заболевание кишечника; синдромы, ассоциированные с переливанием гранулоцитов, и цитокин-индуцируемая токсичность.
"Аутоиммунное заболевание", как использовано в данном описании, относится к любой группе расстройств, при которых повреждение ткани ассоциировано с гуморальными или клеточно-опосредованными ответами на собственные компоненты организма.
"Аллергическое заболевание", как использовано в данном описании, относится к любым симптомам, поражению ткани или потере функции ткани в результате аллергической реакции. "Ассоциированное с артритом заболевание", как использовано в данном описании, относится к любому заболеванию, характеризующемуся воспалительными поражениями суставов различной этиологии. "Дерматит", как использовано в данном описании, относится к любому из большого семейства заболеваний кожи, характеризующихся воспалением кожи различной этиологии. "Отторжение трансплантата", как использовано в данном описании, относится к любой иммунной реакции, направленной против трансплантированной ткани, как например, органов, или клеток (например, костного мозга), характеризующейся потерей функции трансплантированных и окружающих ее тканей, болью, отеком, лейкоцитозом и тромбоцитопенией. Терапевтические способы по настоящему изобретению включают способы лечения расстройств, ассоциированных с активацией воспалительных клеток.
"Активация воспалительных клеток" относится к индукции под действием стимула (в том числе цитокинов, антигенов или аутоантител, но ими не ограничиваясь) пролиферативного клеточного ответа, продуцированию растворимых медиаторов (в том числе цитокинов, кислородных радикалов, ферментов, простаноидов или вазоактивных аминов, но ими не ограничиваясь) или экспрессии на клеточной поверхности новых медиаторов или больших количеств медиаторов (в том числе антигенов главного комплекса гистосовместимости или молекул клеточной адгезии, но ими не ограничиваясь) в воспалительных клетках (в том числе моноцитах, макрофагах, Т-лимфоцитах, В-лимфоцитах, гранулоцитах (т.е. полиморфоядерных лейкоцитах, таких как нейтрофилы, базофилы и эозинофилы), тучных клетках, дендритных клетках, клетках Лангерганса и эндотелиальных клетках, но ими не ограничиваясь). Специалистам в данной области будет очевидно, что активация одного фенотипа или комбинации этих фенотипов в этих клетках может способствовать инициации, сохранению или обострению воспалительного нарушения.
Термин "NSAID" представляет собой аббревиатуру к "нестероидному противовоспалительному лекарственному средству" и относится к терапевтическому агенту, обладающему аналгезирующим, жаропонижающим (снижая повышенную температуру тела и ослабляя боль без нарушения сознания) и, в более высоких дозах, противовоспалительным действиями (уменьшая воспаление). Термин "нестероидный" используется для различения этих лекарственных средств и стероидов, которые (среди большого разнообразия других эффектов) обладают аналогичным эйкозаноид-подавляющим противовоспалительным действием. Особенность NSAID в качестве анальгетиков заключается в том, что они является ненаркотическими. NSAID включают аспирин, ибупрофен и напроксен. Обычно NSAID показаны для лечения острых или хронических состояний, при которых наличествует боль и воспаление. Как правило, NSAID показаны для облечения симптомов следующих состояний: ревматоидного артрита, остеоартрита, воспалительных артропатий (например, анкилозирующего спондилита, псориатического артрита, синдрома Рейтера, острой формы подагры, дисменорреи, метастатической костной боли, головной боли и мигрени, послеоперационной боли, боли от легкой до умеренной степени тяжести вследствие воспаления и тканевого повреждения, лихорадки, кишечной непроходимости и почечной колики. Большинство NSAID действуют как неселективные ингибиторы фермента циклооксигеназа, ингибируя оба изофермента, как циклооксигеназу-1 (СОХ-1), так и циклооксигеназу-2 (СОХ-2). Циклооксигеназа катализирует образование простагландинов и тромбоксана из арахидоновой кислоты (которая высвобождается из клеточного фосфолипидного бислоя под действием фосфолипазы А2). В процессе воспаления простагландины действуют (среди других веществ) в качестве молекул-мессенджеров. Ингибиторы СОХ-2 включают целекоксиб, эторикоксиб, лумиракоксиб, парекоксиб, рофекоксиб и валдекоксиб.
Термин "рак" относится к физиологическому состоянию или описывает физиологическое состояние у млекопитающих, которое обычно характеризуется нерегулируемым клеточным ростом. Термин "опухоль" подразумевает наличие одного или более типов раковых клеток. Примеры рака включают, но не ограничиваются этим, карциному, лимфому, бластому, саркому и лейкоз или лимфолейкозы. Более конкретные примеры таких видов рака включают плоскоклеточный рак (например, плоскоклеточный рак эпителиальной природы), рак легкого, в том числе мелкоклеточный рак легкого, немелкоклеточную карциному легкого ("NSCLC"), аденокарциному легкого и плоскоклеточную карциному легкого, рак брюшины, гепатоклеточный рак, желудочный рак или рак желудка, в том числе рак желудочно-кишечного тракта, рак поджелудочной железы, глиобластому, рак шейки матки, рак яичников, рак печени, рак мочевого пузыря, гепатому, рак молочной железы, рак толстой кишки, рак прямой кишки, колоректальный рак, рак эндометрия или матки, рак слюнных желез, рак почки или почечный рак, рак предстательной железы, рак вульвы, рак щитовидной железы, печеночную карциному, анальный рак, рак пениса, а также рак головы и шеи.
"Гематологические злокачественные новообразования" (британское написание "haematological" отличается от принятого в США написания "hematological") представляют собой типы рака, которые воздействуют на кровь, костный мозг и лимфатические узлы. Поскольку эти три системы тесно связаны через иммунную систему, то заболевание, затрагивающее одну из них, в равной степени часто воздействует и на остальные: несмотря на то, что лимфома является заболеванием лимфатических узлов, зачастую она распространяется в костный мозг, затрагивая кровь. Гематологические злокачественные новообразования представляют собой злокачественные неоплазии ("рак"), и как правило их лечат специалисты в области гематологии и/или онкологии. В некоторых центрах "гематологию/онкологию" относят к одной из узких специализаций терапии внутренних болезней, тогда как в других считают отдельными подразделениями (имеются также специалисты в области хирургической и радиационной онкологии). Не все гематологические расстройства являются злокачественными ("раковыми"); помощь при этих иных заболеваниях крови также может быть оказана гематологом. Гематологические злокачественные новообразования могут возникать из любой из двух основных линий клеток крови: линий миелоидных и лимфоидных клеток. Линия миелоидных клеток обычно продуцирует гранулоциты, эритроциты, тромбоциты, макрофаги и тучные клетки; линия лимфоидных клеток продуцирует Β-, Т-, NK-клетки (natural killer cells - природные киллерные клетки) и плазматические клетки. Лимфомы, лимфоцитарные лейкозы и миелома происходят из лимфоидной линии, в то время как острый и хронический миелогенный лейкоз, миелодиспластические синдромы и миелопролиферативные расстройства имеют миелоидное происхождение. Лейкозы включают острый лимфобластный лейкоз (ALL), острый миелогенный лейкоз (AML), хронический лимфоцитарный лейкоз (CLL), хронический миелогенный лейкоз (CML), острый моноцитарный лейкоз (AMOL) и мелкоклеточную лимфоцитарную лимфому (SLL). Лимфомы включают лимфомы Ходжкина (все четыре подтипа) и неходжкинские лимфомы (все подтипы).
"Химиотерапевтический агент" представляет собой химическое соединение, полезное в лечении рака, независимо от механизма действия. Классы химиотерапевтических агентов включают, но не ограничиваются этим: алкилирующие агенты, антиметаболиты, алкалоиды растительного происхождения, являющиеся "веретенными ядами", цитотоксические/противоопухолевые антибиотики, ингибиторы топоизомеразы, антитела, фотосенсибилизирующие вещества и ингибиторы киназ. Химиотерапевтические агенты включают соединения, используемые в "терапии направленного действия" и традиционной химиотерапии. Примеры химиотерапевтических агентов включают: эрлотиниб (TARCEVA®, Genentech/OSI Pharm.), доцетаксел (TAXOTERE®, Sanofi-Aventis), 5-FU (фторурацил, 5-фторурацил, №CAS (Chemical abstract service - Химическая реферативная служба)): 51-21-8), гемцитабин (GEMZAR®, Lilly), PD-0325901 (№CAS: 391210-10-9, Pfizer), цисплатин (цис-диамин, дихлорплатина(Н), №CAS: 15663-27-1), карбоплатин (№CAS: 41575-94-4), паклитаксел (TAXOL®, Bristol-Myers Squibb Oncology, Princeton, N.J.), трастузумаб (HERCEPTIN®, Genentech), темозоломид (4-метил-5-оксо-2,3,4,6,8-пентазабицикло-[4.3.0]нона-2,7,9-триен-9-карбоксамид, №CAS: 85622-93-1, TEMODAR®, TEMODAL®, Schering Plough), тамоксифен ((Z)-2-[4-(1,2-бут-1-енил)фенокси]-N,N-диметил-этанамин, NOLVADEX®, ISTUBAL®, VALODEX®) и доксорубицин (ADRIAMYCIN®), Akti-1/2, HPPD (оксим 5-(1-(2-гидроксиэтил)-3-(пиридин-4-ил)-1Н-пиразол-4-ил)-2,3-дигидро-1Н-инден-1-она) и рапамицин.
Другие примеры химиотерапевтических агентов включают: оксалиплатин (ELOXATIN®, Sanofi), бортезомиб (VELCADE®, Millennium Pharm.), сутент (SUNITINIB®, SU11248, Pfizer), летрозол (FEMARA®, Novartis), иматиниба мезилат (GLEEVEC®, Novartis), XL-518 (ингибитор Mek (Mitogen-activated protein kinase/Extracellular signal related kinase Kinase - киназа митоген-активируемой протеинкиназы/внеклеточной сигнал-регулируемой киназы), Exelixis, WO 2007/044515), ARRY-886 (ингибитор Mek, AZD6244, Array BioPharma, Astra Zeneca), SF-1126 (ингибитор PI3K (phosphatidyl inositol 3 kinase -фосфатидилинозитол-3-киназа), Semafore Pharmaceuticals), BEZ-235 (ингибитор PI3K, Novartis), XL-147 (ингибитор PI3K, Exelixis), PTK787/ZK 222584 (Novartis), фулвестрант (FASLODEX®, AstraZeneca), лейковорин (фолиновая кислота), рапамицин (сиролимус, RAPAMUNE®, Wyeth), лапатиниб (TYKERB®, GSK572016, Glaxo Smith Kline), лонафарниб (SARASAR™, SCH 66336, Schering Plough), сорафениб (NEXAVAR®, BAY43-9006, Bayer Labs), гефитиниб (IRESSA®, AstraZeneca), иринотекан (CAMPTOSAR®, CPT-11, Pfizer), типифарниб (ZARNESTRA™, Johnson & Johnson), ABRAXANE™ (без кремофора), препараты наночастиц паклитаксела, сконструированных с использованием альбумина (American Pharmaceutical Partners, Schaumberg, II), валдетаниб (rINN, ZD6474, ZACTIMA®, AstraZeneca), хлорамбуцил, AG1478, AG1571 (SU 5271; Sugen), темсиролимус (TORISEL®, Wyeth), пазопаниб (GlaxoSmithKline), канфосфамид (TELCYTA®, Telik), тиотепа и циклофосфамид (CYTOXAN®, NEOSAR®); алкилсульфонаты, такие как бусульфан, импросульфан и пипосульфан; азиридины, такие как бензодопа, карбоквон, метуредопа и уредопа; этиленимины и метиламеламины, в том числе алтретамин, триэтиленмеламин, триэтиленфосфорамид, триэтилентиофосфорамид и триметиломеламин; ацетогенины (особенно буллатацин и буллатацинон); камптотецин (в том числе синтетический аналог топотекан); бриостатин; каллистатин; СС-1065 (в том числе его синтетические аналоги адозелезин, карзелезин и бизелезин); криптофицины (в частности криптофицин 1 и криптофицин 8); доластатин; дуокармицин (в том числе синтетические аналоги, KW-2189 и СВ1-ТМ1); элеутеробин; панкратистатин; саркодиктиин; спонгистатин; азотные аналоги иприта, такие как хлорамбуцил, хлорнафазин, хлорфосфамид, эстрамустин, ифосфамид, мехлоретамин, гидрохлорид мехлоретаминоксида, мелфалан; новембихин, фенестерин, преднимустин, трофосфамид, урациловый иприт; нитрозомочевины, такие как кармустин, хлорозотоцин, фотемустин, ломустин, нимустин и ранимустин; антибиотики, такие как энедииновые антибиотики (например, калихеамицин, калихеамицин гамма 11, калихеамицин омега И (Angew Chem. Intl. Ed. Engl. (1994) 33: 183-186); динемицин, динемицин А; бисфосфонаты, такие как клодронат; эсперамицин; а также неокарциностатиновый хромофор и родственные хромофоры хромопротеиновых энедииновых антибиотиков), аклациномизины, актиномицин, аутрамицин, азасерин, блеомицины, кактиномицин, карабицин, карминомицин, карцинофилин, хромомицины, дактиномицин, даунорубицин, деторубицин, 6-диазо-5-оксо-L-норлейцин, морфолино-доксорубицин, цианоморфолино-доксорубицин, 2-пирролино-доксорубицин и дезоксидоксо-рубицин, эпирубицин, эзорубицин, идарубицин, неморубицин, марселломицин, митомицины, такие как митомицин С, микофеноловую кислоту, ногаламицин, оливомицины, пепломицин, порфиромицин, пуромицин, квеламицин, родорубицин, стрептонигрин, стрептозоцин, туберцидин, убенимекс, зиностатин, зорубицин; антиметаболиты, такие как метотрексат и 5-фторурацил (5-FU); аналоги фолиевой кислоты, такие как деноптерин, метотрексат, птероптерин, триметрексат; аналоги пурина, такие как флударабин, 6-меркаптопурин, тиамиприн, тиогуанин; аналоги пиримидина, такие как анцитабин, азацитидин, 6-азауридин, кармофур, цитарабин, дидезоксиуридин, доксифлуридин, эноцитабин, флоксуридин; андрогены, такие как калустерон, дромостанолона пропионат, эпитиостанол, мепитиостан, тестолактон; антиадренергические средства, такие как аминоглутетимид, митотан, трилостан; средство для восполнения фолиевой кислоты, такое как фролиновая кислота; ацеглатон; альдофосфамидгликозид; аминолевулиновую кислоту; энилурацил; амсакрин; бестрабуцил; бисантрен; эдатраксат; дефофамин; демеколцин; диазиквон; элфорнитин; эллиптиния ацетат; эпотилон; этоглуцид; галлия нитрат; гидроксимочевину; лентинан; лонидаинин; майтансиноиды, такие как майтансин и ансамитоцины; митогуазон; митоксантрон; мопиданмол; нитраэрин; пентостатин; фенамет; пирарубицин; лозоксантрон; подофиллиновую кислоту; 2-этилгидразид; прокарбазин; полисахаридный комплекс PSK® (JHS Natural Products, Eugene, OR); разоксан; ризоксин; сизофиран; спирогерманий; тенуазоновую кислоту; триазиквон; 2,2',2ʺ-трихлортриэтиламин; трихотецены (особенно, токсин Т-2, верракурин А, роридин А и ангуидин); уретан; виндезин; дакарбазин; манномустин; митобронитол; митолактол; пипоброман; гацитозин; арабинозид ("Ara-C"); циклофосфамид; тиотепа; 6-тиогуанин; меркаптопурин; метотрексат; аналоги платины, такие как цисплатин и карбоплатин; винбластин; этопозид (VP-16); ифосфамид; митоксантрон; винкристин; винорелбин (NAVELBINE®); новантрон; тенипозид; эдатрексат; дауномицин; аминоптерин; капецитабин (XELODA®, Roche); ибандронат; СРТ-11; ингибитор топоизомеразы RFS 2000; дифторметилорнитин (DMFO); ретиноиды, такие как ретиноевая кислота; и фармацевтически приемлемые соли, кислоты и производные любого из упомянутого выше.
В определение "химиотерапевтического агента" также включены: (1) антигормональные агенты, действие которых заключается в регуляции или ингибировании воздействия гормонов на опухоли, как например, антиэстрогены и селективные модуляторы рецептора эстрогена (selective estrogen receptor modulators; SERM), в том числе, например, тамоксифен (включая NOLVADEX®; тамоксифена цитрат), ралоксифен, дролоксифен, 4-гидрокситамоксифен, триоксифен, кеоксифен, LY117018, онапристон и FARESTON® (торемифина цитрат); (2) ингибиторы ароматазы, ингибирующие фермент ароматазу, которая регулирует продуцирование эстрогенов в надпочечниках, такие как, например, 4(5)-имидазолы, аминоглутетимид, MEGASE® (мегестрола ацетат), AROMASIN® (эксеместан; Pfizer), форместан, фадрозол, RIVISOR® (ворозол), FEMARA® (летрозол; Novartis) и ARIMIDEX® (анастрозол; AstraZeneca); (3) антиандрогены, такие как флутамид, нилутамид, бикалутамид, лейпролид и гозерелин; а также троксацитабин (1,3-диоксолановый аналог нуклеозида цитозина); (4) ингибиторы протеинкиназ, такие как ингибиторы MEK (WO 2007/044515); (5) ингибиторы липид-киназ; (6) антисмысловые олигонуклеотиды, в частности такие, которые ингибируют экспрессию генов в путях передачи сигналов, участвующих в аберрантной пролиферации клеток, например, РКС-альфа (протеинкиназа С), Raf и H-Ras, такие как облимерсен (GENASENSE®, Genta Inc.); (7) рибозимы, такие как ингибиторы экспрессии VEGF (vascular endothelial growth factor - сосудистый эндотелиальный фактор роста) (например, ANGIOZYME®) и ингибиторы экспрессии HER2; (8) вакцины, как например, генотерапевтические вакцины, например, ALLOVECTIN®, LEUVECTIN® и VAXID®; rIL-2 (рекомбинантный интерлейкин-2) PROLEUKIN®; ингибиторы топоизомеразы 1, такие как LURTOTECAN®; rmRH ABARELIX®; (9) антиангиогенные агенты, такие как бевацизумаб (AVASTIN®, Genentech); и фармацевтически приемлемые соли, кислоты и производные любого из упомянутого выше.
В определение "химиотерапевтического агента" также включены терапевтические антитела, такие как алемтузумаб (Campath), бевацизумаб (AVASTIN®, Genentech); цетуксимаб (ERBITUX®, Imclone); панитумумаб (VECTIBIX®, Amgen), ритуксимаб (RITUXAN®, Genentech/Biogen Idee), пертузумаб (OMNITARG™, 2С4, Genentech), трастузумаб (HERCEPTIN®, Genentech), тозитумомаб (Bexxar, Corixia) и конъюгат антитела и лекарственного средства, гемтузумаб-озогамицин (MYLOTARG®, Wyeth).
Гуманизированные моноклональные антитела с терапевтическим потенциалом в качестве химиотерапевтических агентов в комбинации с ингибиторами Btk по изобретению включают: алемтузумаб, аполизумаб, азелизумаб, атлизумаб, бапинеузумаб, бевацизумаб, биватузумаб-мертанзин, кантузумаб-мертанзин, цеделизумаб, цертолизумаб-пегол, сидфузитузумаб, сидтузумаб, даклизумаб, экулизумаб, эфализумаб, эпратузумаб, эрлизумаб, фелвизумаб, фонтолизумаб, гемтузумаб-озогамицин, инотузумаб-озогамицин, ипилимумаб, лабетузумаб, линтузумаб, матузумаб, меполизумаб, мотавизумаб, мотовизумаб, натализумаб, нимотузумаб, ноловизумаб, нумавизумаб, окрелизумаб, омализумаб, паливизумаб, пасколизумаб, пекфузитузумаб, пектузумаб, пертузумаб, пекселизумаб, раливизумаб, ранибизумаб, ресливизумаб, реслизумаб, ресивизумаб, ровелизумаб, руплизумаб, сибротузумаб, сиплизумаб, сонтузумаб, такатузумаб-тетраксетан, тадоцизумаб, тализумаб, тефибазумаб, тоцилизумаб, торализумаб, трастузумаб, тукотузумаб целмолейкин, тукуситузумаб, умавизумаб, уртоксазумаб и визилизумаб.
Термин "метаболит" относится к продукту, образуемому в процессе метаболизма конкретного соединения или его соли в организме. Метаболиты соединения могут быть идентифицированы с использованием общепринятых методик, известных в данной области техники, а их активности определены с использованием таких тестов, которые изложены в данном описании. Такие продукты могут получаться из вводимого соединения, например, в результате его окисления, восстановления, гидролиза, амидирования, деамидирования, этерификации, деэтерификации, ферментативного расщепления и тому подобного. Соответственно, изобретение включает метаболиты соединений по изобретению, в том числе соединения, получаемые способом, включающим приведение соединения формулы I или II по данному изобретению в контакт с млекопитающим в течение периода времени, достаточного для получения продукта его метаболизма.
Термин "инструкция по применению" обычно относится к инструкциям, традиционно включаемым в промышленные упаковки терапевтических продуктов, которые содержат информацию о показаниях, применении, дозировке, введении, противопоказаниях и/или предупреждениях касательно применения таких терапевтических продуктов.
Термин "хиральный" относится к молекулам, обладающим свойством не совпадать при наложении на зеркально отображаемого партнера, тогда как термин "ахиральный" относится к молекулам, совпадающим при наложении на своего зеркально отображаемого партнера.
Термин "стереоизомеры" относится к соединениям, которые имеют идентичный химический состав, но различаются расположением атомов или групп в пространстве.
"Диастереомер" относится к стереоизомеру с двумя или более центрами хиральности, и такие молекулы не являются зеркальными отображениями друг друга. Диастереомеры имеют разные физические свойства, например точки плавления, точки кипения, спектральные свойства и реакционные способности. Смеси диастереомеров можно разделить с помощью аналитических методик высокого разрешения, таких как электрофорез и хроматография.
Термин "энантиомеры" относится к двум стереоизомерам соединения, которые не совпадают с зеркальными отображениями друг друга.
В данном описании в основном использованы стереохимические определения и правила, представленные в S.P. Parker, Ed., McGraw-Hill Dictionary of Chemical Terms (1984), McGraw-Hill Book Company, New York; и Eliel E. and Wilen S., "Stereochemistry of Organic Compounds", John Wiley & Sons, Inc., New York, 1994. Соединения по изобретению могут содержать асимметрические или хиральные центры и ввиду этого существовать в разных стереоизомерных формах. Предполагается, что все стереоизомерные формы соединений по изобретению, включая, но не ограничиваясь этим, диастереомеры, энантиомеры и атропоизомеры, а также их смеси, такие как рацемические смеси, составляют часть настоящего изобретения. Многие органические соединения существуют в оптически активных формах, т.е. они обладают способностью вращать плоскость плоскополяризованного света. При описании оптически активного соединения для обозначения абсолютной конфигурации молекулы относительно ее хирального(ых) центра(ов) используют префиксы D и L или R и S. Для обозначения знака направления вращения плоскополяризованного света соединением применяют префиксы d и I или (+) и (-), при этом (-) или I означает, что соединение является левовращающим. Соединение с префиксом (+) или d является правовращающим. Для данной химической структуры эти стереоизомеры являются идентичными, за исключением того, что они являются зеркальными отображениями друг друга. Конкретный стереоизомер также может быть определен как энантиомер, и смесь таких изомеров часто называют энантиомерной смесью. Смесь с соотношением энантиомеров 50:50 называется рацемической смесью или рацематом, которая может образовываться, если не соблюдалось никакой стереоселекции или стереоспецифичности в химической(ом) реакции или процессе. Термины "рацемическая смесь" и "рацемат" относятся к эквимолярной смеси двух энантиомерных разновидностей, не обладающей оптической активностью. Энантиомеры можно выделить по отдельности из рацемической смеси методом хирального разделения, таким как сверхкритическая жидкостная хроматография (SFC). Определение конфигурации при хиральных центрах в разделенных энантиомерах может быть предварительным, и изображенные в Таблице 1 структуры приведены в целях иллюстрации, тогда как для стереохимического определения ожидается получение рентгеноструктурных данных.
Термин "таутомер" или "таутомерная форма" относится к структурным изомерам, обладающим разной энергией, взаимопревращение которых протекает через низкий энергетический барьер. Например, протонные таутомеры (также известные как прототропные таутомеры) характеризуются взаимопревращениями в результате миграции протона, как например при кето-енольной и имин-енаминной изомерии. Валентные таутомеры включают взаимопревращения посредством реорганизации некоторых из связывающих электронов.
Термин "фармацевтически приемлемые соли" означает соли, которые не являются нежелательными ни в биологическом, ни в других отношениях. Фармацевтически приемлемые соли включают соли присоединения как кислоты, так и основания. Фраза "фармацевтически приемлемый" указывает на то, что вещество или композиция должны быть совместимы химически и/или токсикологически с другими ингредиентами, входящими в состав композиции, и/или с млекопитающим, подвергаемым лечению ими.
Термин "фармацевтически приемлемая соль присоединения кислоты" означает такие фармацевтически приемлемые соли, которые образуются с неорганическими кислотами, такими как соляная кислота, бромистоводородная кислота, серная кислота, азотная кислота, угольная кислота, фосфорная кислота, и органическими кислотами, выбранными из классов алифатических, циклоалифатических, ароматических, аралифатических, гетероциклических, карбоновых и сульфоновых органических кислот, таких как муравьиная кислота, уксусная кислота, пропионовая кислота, гликолевая кислота, глюконовая кислота, молочная кислота, пировиноградная кислота, щавелевая кислота, яблочная кислота, малеиновая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, винная кислота, лимонная кислота, аспарагиновая кислота, аскорбиновая кислота, глутаминовая кислота, антраниловая кислота, бензойная кислота, коричная кислота, миндальная кислота, эмбоновая кислота, фенилуксусная кислота, метансульфоновая кислота (мезилат), этансульфоновая кислота, п-толуолсульфоновая кислота и салициловая кислота.
Термин "фармацевтически приемлемая соль присоединения основания" означает такие фармацевтически приемлемые соли, которые образуются с органическим или неорганическим основанием. Примеры приемлемых солей с неорганическими основаниями включают соли натрия, калия, аммония, кальция, магния, железа, цинка, меди, марганца и алюминия. Соли, происходящие из фармацевтически приемлемых органических нетоксичных оснований, включают соли первичных, вторичных и третичных аминов, замещенных аминов, в том числе природных замещенных аминов, циклических аминов и основных ионообменных смол, таких как изопропиламин, триметиламин, диэтиламин, триэтиламин, трипропиламин, этаноламин, 2-диэтиламиноэтанол, триметамин, дициклогексиламин, лизин, аргинин, гистидин, кофеин, прокаин, гидрабамин, холин, бетаин, этилендиамин, глюкозамин, метилглюкамин, теобромин, пурины, пиперазин, пиперидин, N-этилпиперидин и полиаминные смолы.
"Сольват" относится к ассоциации или комплексу одной или более молекул растворителя и соединения по изобретению. Примеры растворителей, которые образуют сольваты, включают, но не ограничиваются этим, воду, изопропанол, этанол, метанол, DMSO (диметилсульфоксид), этилацетат, уксусную кислоту и этаноламин.
Термин "ЕС50" представляет собой "полумаксимальную эффективную концентрацию" и означает концентрацию конкретного соединения в плазме крови, необходимую для получения конкретного эффекта in vivo, равного 50% от максимального.
Термин "Ki" относится к константе ингибирования и означает абсолютную аффинность связывания конкретного ингибитора с рецептором. Ее измеряют, используя анализы конкурентного связывания, и она равна концентрации, при которой этот конкретный ингибитор будет занимать 50% рецепторов в случае отсутствия какого-либо конкурентного лиганда (например, радиоактивного лиганда). Величины Ki можно преобразовать логарифмически в величины pKi (-log Ki), при этом более высокие значения указывают на экспоненциально более высокую эффективность.
Термин "IC50" относится к "концентрации, вызывающей половину от максимального ингибирования" и означает концентрацию конкретного соединения, необходимую для получения 50% ингибирования биологического процесса in vitro. Величины IC50 можно преобразовать логарифмически в величины pIC50 (-log IС50), при этом более высокие значения указывают на экспоненциально более высокую эффективность. Величина IC50 не является абсолютной величиной, а зависит от экспериментальных условий, например, от используемых концентраций, и ее можно преобразовать в абсолютную константу ингибирования (Ki), используя уравнение Ченга-Пруссофа (Biochem. Pharmacol. (1973) 22: 3099). Можно рассчитать параметры для другого процента ингибирования, такие как IC70, IC90 и т.д.
Термины "соединение по данному изобретению", и "соединения по настоящему изобретению", и "соединения формулы I" включают в себя соединения формул I и стереоизомеры, геометрические изомеры, таутомеры, сольваты, метаболиты и фармацевтически приемлемые соли и их пролекарства.
Подразумевается, что любая формула или структура, приведенная в данном описании, включая соединения формулы I и II, также представляет гидраты, сольваты и полиморфы таких соединений и их смеси.
Подразумевается, что любая формула или структура, приведенная в данном описании, включая соединения формулы I и II, также представляет меченные изотопом формы этих соединения, равно как и немеченые формы. Меченные изотопом соединения имеют структуры, изображенные формулами, приведенными в данном описании, но с учетом того, что один или более чем один атом заменен атомом, имеющим выбранную(ое) атомную массу или массовое число. Примеры изотопов, которые могут быть инкорпорированы в соединения по изобретению, включают изотопы водорода, углерода, азота, кислорода, фосфора, фтора и хлора, такие как, но не ограничиваясь этим, 2Н (дейтерий, D), 3Н (тритий), 11С, 13С, 14С, 15N, 18F, 31Р, 32Р, 35S, 36Cl и 125I. Разные меченные изотопом соединения по настоящему изобретению, например, соединения, в которые инкорпорированы такие радиоактивные изотопы, как 3Н, 13С и 14С. Такие меченные изотопом соединения могут быть полезны в исследованиях метаболизма, исследованиях кинетики реакций, методах детекции или визуализации, таких как позитронно-эмиссионная томография (PET) или однофотонная эмиссионная компьютерная томография (SPECT), в том числе в анализах распределения лекарственного средства или субстрата в тканях, или в лечении пациентов радиоактивными средствами. Меченные или замещенные дейтерием терапевтические соединения по изобретению могут иметь улучшенные свойства в отношении DMPK (drug metabolism and pharmacokinetics - метаболизм и фармакокинетика лекарственных средств), связанные с всасыванием, распределением, метаболизмом и экскрецией (absorption, distribution, methabolism and excretion; ADME). Замена на более тяжелые изотопы, такие как дейтерий, может давать некоторые терапевтические преимущества, обусловленные большей метаболической стабильностью, например, увеличенный in vivo период полувыведения или снижение дозировки. Для исследований с использованием PET или SPECT может быть полезно 18F-меченное соединение. В общем случае, меченные изотопом соединения по данному изобретению и их пролекарства могут быть получены путем осуществления методик, описанных на схемах или в разделах Примеры и Подготовительные примеры, изложенных ниже, в результате замены не меченного изотопом реагента легко доступным меченным изотопом реагентом. Кроме того, замена на более тяжелые изотопы, в частности дейтерий (т.е. 2Н или D), может давать некоторые терапевтические преимущества, обусловленные большей метаболической стабильностью, например, увеличенный in vivo период полувыведения, или снижение дозировки, или улучшение терапевтического индекса. Очевидно, что в этом случае дейтерий считается заместителем в соединении формулы (I). Концентрация такого более тяжелого изотопа, в частности дейтерия, может быть определена по фактору изотопного обогащения. Подразумевается, что любой атом в соединениях по данному изобретению, специально не обозначенный как конкретный изотоп, представляет собой любой стабильный изотоп этого атома. Если не указано иное, то когда положение конкретно обозначено как "Н" или "водород", понимают, что атом водорода в данном положении имеет свой природный изотопный состав. Соответственно подразумевается, что любой атом в соединениях по данному изобретению, конкретно обозначенный как дейтерий (D), представляет собой дейтерий.
СОЕДИНЕНИЯ 8-ФТОРФТАЛАЗИН-1(2Н)-ОНОВ
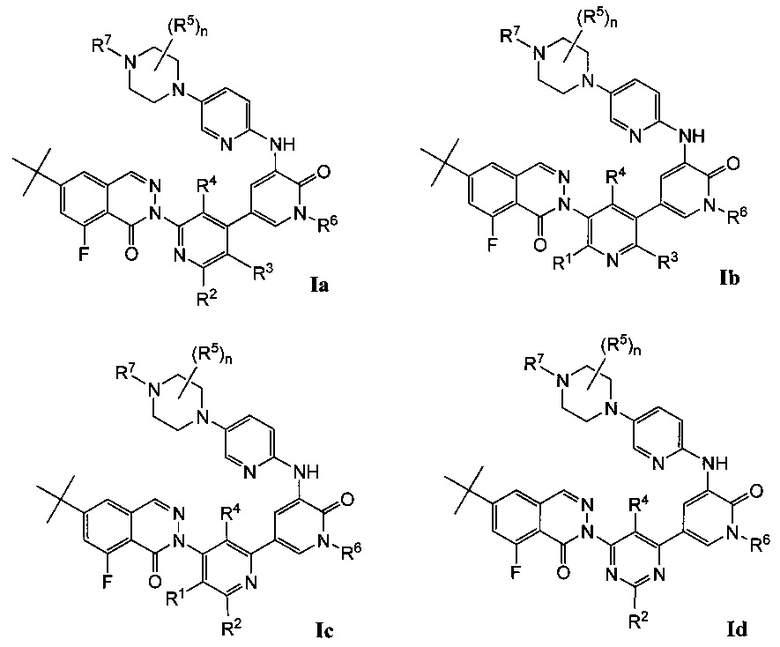
Согласно настоящему изобретению предложены соединения 8-фторфталазин-1(2h)-онов формулы I, в том числе формул Ia-If, и фармацевтические композиции на их основе, которые потенциально полезны в лечении заболеваний, состояний и/или расстройств, модулируемых киназой Btk:
или их стереоизомеры, таутомеры или фармацевтически приемлемые соли, где:
X1 представляет собой CR1 или N;
X2 представляет собой CR2 или N;
X3 представляет собой CR3 или N;
где один или два из Χ1, X2 и X3 представляют собой N;
R1, R2 и R3 независимо выбраны из Н, F, Cl, CN, -СН3, -СН2СН3, -СН2ОН, -CH2F, -CHF2, -CF3, -СН2СН2ОН, -NH2, -NHCH3, -N(CH3)2, -ОН, -OCH3, -OCH2CH3 и -OCH2CH2OH;
R4 выбран из Η, F, Cl, CN, -CH2OH, -CH(CH3)OH, -C(CH3)2OH, -CH(CF3)OH, -CH2F, -CHF2, -CH2CHF2, -CF3, -C(O)NH2, -C(O)NHCH3, -C(O)N(CH3)2, -NH2, -NHCH3, -N(CH3)2, -NHC(O)CH3, -OH, -OCH3, -OCH2CH3, -OCH2CH2OH, циклопропила, циклопропилметила, 1-гидроксициклопропила, имидазолила, пиразолила, 3-гидрокси-оксетан-3-ила, оксетан-3-ила и азетидин-1-ила;
R5 выбран из -СН3, -СН2СН3, -СН2ОН, -CH2F, -CHF2, -CF3, -CN и -CH2CH2OH;
или две группы R5 образуют 3-, 4-, 5- или 6-членное карбоциклическое или гетероциклическое кольцо;
или группа R5 и группа R7 образуют 3-, 4-, 5- или 6-членное карбоциклическое или гетероциклическое кольцо;
n равно 0, 1, 2, 3 или 4;
R6 выбран из Н, -СН3, -СН2СН3, -СН2СН2ОН, -CH2F, -CHF2, -CF3, -NH2, -NHCH3, -N(CH3)2, -OH, -OCH3, -OCH2CH3 и -OCH2CH2OH;
R7 выбран из Η, -СН3, -S(O)2CH3, циклопропила, азетидин-3-ила, оксетан-3-ила и морфолин-4-ила;
Z1 представляет собой CR8 или N, где R8 выбран из Н, F, Cl, -СН3, -СН2СН3, -СН2СН2ОН, -NH2, -NHCH3, -N(CH3)2, -ОН, -ОСН3, -ОСН2СН3 и -ОСН2СН2ОН;
Z2 представляет собой CR9 или N, где R9 выбран из Н, -СН3, -СН2СН3 и -СН2СН2ОН; и
Y1 и Y2 независимо выбраны из СН и N, причем каждый Y1 и Y2 не представляет собой N.
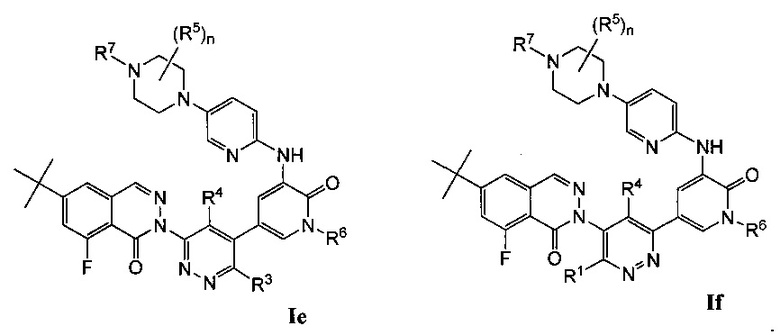
Типичные воплощения соединений формулы I включают соединения формул Ia-If:
Типичные воплощения соединений формулы I включают соединения, где X1 представляет собой Ν, X2 представляет собой CR2, и X3 представляет собой CR3.
Типичные воплощения соединений формулы I включают соединения, где X1 представляет собой CR1, X2 представляет собой N, и X3 представляет собой CR3.
Типичные воплощения соединений формулы I включают соединения, где X1 представляет собой CR1, X2 представляет собой CR2, и X3 представляет собой N.
Типичные воплощения соединений формулы I включают соединения, где X1 и X3 представляют собой Ν, X1 и X2 представляют собой N, или X2 и X3 представляют собой N.
Типичные воплощения соединений формулы I включают соединения, где X2 представляет собой CR2, и R2 представляет собой F.
Типичные воплощения соединений формулы I включают соединения, где X1 и X3 представляют собой СН.
Типичные воплощения соединений формулы I включают соединения, где R4 представляет собой -СН2ОН.
Типичные воплощения соединений формулы I включают соединения, где R5 представляет собой -СН3, и n равно 1 или 2.
Типичные воплощения соединений формулы I включают соединения, где R7 представляет собой оксетан-3-ил.
Типичные воплощения соединений формулы I включают соединения, где Y1 представляет собой СН.
Типичные воплощения соединений формулы I включают соединения, где Y2 представляет собой СН.
Типичные воплощения соединений формулы I включают соединения, где Y1 представляет собой N.
Типичные воплощения соединений формулы I включают соединения, где Y2 представляет собой N.
Типичные воплощения соединений формулы I включают соединения, где Z1 представляет собой СН.
Типичные воплощения соединений формулы I включают соединения, где Z2 представляет собой СН.
Типичные воплощения соединений формулы I включают соединения, где Z1 представляет собой N.
Типичные воплощения соединений формулы I включают соединения, где Z2 представляет собой N.
Согласно настоящему изобретению также предложены соединения 8-фторфталазин-1(2h)-онов формулы II, которые потенциально полезны в лечении заболеваний, состояний и/или расстройств, модулируемых киназой Btk:
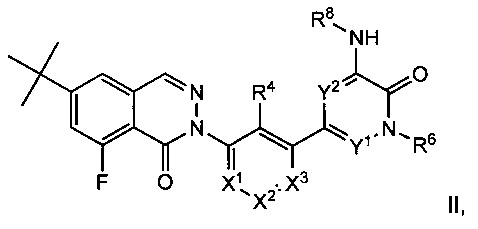
или их стереоизомеры, таутомеры или фармацевтически приемлемые соли, где:
X1 представляет собой CR1 или N;
X2 представляет собой CR2 или N;
X3 представляет собой CR3 или N;
где один или два из Χ1, X2 и X3 представляют собой N;
Y1 и Y2 независимо выбраны из СН и N, причем каждый Y1 и Y2 не представляет собой N;
R1, R2 и R3 независимо выбраны из Н, F, Cl, CN, -СН3, -СН2СН3,-СН2ОН, -CH2F, -CHF2, -CF3, -СН2СН2ОН, -NH2, -NHCH3, -N(CH3)2, -ОН, -OCH3, -OCH2CH3 и -OCH2CH2OH;
R4 выбран из Η, F, Cl, CN, -CH2OH, -CH(CH3)OH, -C(CH3)2OH, -CH(CF3)OH, -CH2F, -CHF2, -CH2CHF2, -CF3, -C(O)NH2, -C(O)NHCH3, -C(O)N(CH3)2, -NH2, -NHCH3, -N(CH3)2, -NHC(O)CH3, -OH, -OCH3, -OCH2CH3, -OCH2CH2OH, -OP(0)(OH)2, циклопропила, циклопропилметила, 1-гидроксициклопропила, имидазолила, пиразолила, 3-гидрокси-оксетан-3-ила, оксетан-3-ила и азетидин-1-ила;
R6 выбран из Η, -СН3, -СН2СН3, -СН2СН2ОН, -CH2F, -CHF2, -CF3, -NH2, -NHCH3, -N(CH3)2, -OH, -OCH3, -OCH2CH3 и -OCH2CH2OH;
R8 выбран из С6-С20арила, С3-С12карбоциклила, С2-С20гетероциклила, C1-С20гетероарила, групп -(С6-С20арил)-(С2-С20гетероциклил), -(C1-С20гетероарил)-(С2-С20гетероциклил), -(С1-С20гетероарил)-(С2-С20гетероциклил)-(С2-С20гетероциклил), -(С1-С20гетероарил)-(С2-С20)гетероциклил)-(С1-С6алкил), -(С1-С20гетероарил)-(С1-С6алкил) и -(С1-С20гетероарил)-С(=О)-(С2-С20гетероциклил); где арил, карбоциклил, гетероциклил и гетероарил возможно замещены одной или более группами, выбранными из F, Cl, Br, I, CN, -СН3, -СН2СН3, СН(СН3)2, -СН2СН(СН3)2, -СН2ОН, -СН2ОСН3, -С(СН3)2ОН, -СН(ОН)СН(СН3)2, -С(СН3)2СН2ОН, -CH2CH2SO2CH3, -СН2ОР(O)(ОН)2, -C(CH3)2CONH2, -СН2ОСН3, -СН2СН2ОН, -СН2СН2ОСН3, -CH2F, -CHF2, -CF3, -CH2CF3, -CH2CHF2, -CH(CH3)CN, -C(CH3)2CN, -CH2CN, -CO2H, -CO2CH3, -CO2C(CH3)3, -COCH(OH)CH3, -C(O)CH3, -C(O)CH2CH3, -C(O)CH(CH3)2, -C(O)NH2, -C(O)NHCH3, -C(O)N(CH3)2, -NH2, -NHCH3, -N(CH3)2, -NHC(O)CH3, -N(CH3)COCH3, -NHS(O)2CH3, -N(CH3)C(CH3)2CONH2, -N(CH3)CH2CH2S(O)2CH3, -NO2, =O, -OCH2CH2N(CH3)2, -OP(O)(OH)2, -OH, -OCH3, -OCH2CH3, -OCH2CH2OCH3, -OCH2CH2OH, -S(O)2N(CH3)2, -SCH3, -S(O)2CH3, -S(O)3H, циклопропила, оксетанила, азетидинила, 1-метилазетидин-3-илокси, N-метил-N-оксетан-3-иламино, азетидин-1-илметила и морфолино.
Типичные воплощения соединений формулы II включают соединения, где R8 представляет собой -(С1-С20гетероарил)-(С2-С20гетероциклил).
Типичные воплощения соединений формулы II включают соединения, где R8 представляет собой пиридинил.
Типичные воплощения соединений формулы II включают соединения, где R8 представляет собой -(пиридинил)-(пиперазинил).
Типичные воплощения соединений формулы II включают соединения, где R8 представляет собой С1-С20гетероарил.
Типичные воплощения соединений формулы II включают соединения, где R8 выбран из:
пиримидинила,
6,7-дигидро-4Н-тиазоло[5,4-с]пиридин-2-ила,
5-(морфолин-4-карбонил)-2-пиридила,
пиразолила,
тиазолила,
6,7-дигидро-4Н-пиразоло[1,5-а]пиразин-2-ила,
оксазолила,
изоксазолила,
имидазолила,
5-(6,7-дигидро-4Н-пиразоло[5,1-с][1,4]оксазин-2-ила,
1,2,3-триазолила,
4,5,6,7-тетрагидропиразоло[1,5-а]пиразинила,
пиразинила и
5,6-дигидро-4Н-пирроло[1,2-b]пиразол-2-ила.
Типичные воплощения соединений формулы II включают соединения, где R8 представляет собой
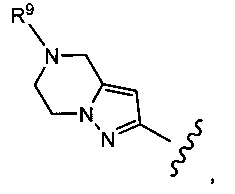
где R9 выбран из Н, -СН3, -СН2ОСН3, -СН2СН3, -СН(СН3)2, -СН2СН2ОН, -СН2СН2ОСН3, -CH2F, -CHF2, -CF3, -CH2CF3, -CH2CHF2, -CH(CH3)CN, -C(CH3)2CN, -CH2CN, -CH2CH2CN, -C(O)CH3, -C(O)CH2CH3, -C(O)CH(CH3)2, -NH2, -NHCH3, -N(CH3)2, -OH, -OCH3, -OCH2CH3>-OCH2CH2OH, циклопропила, циклопропилметила, оксетанила и оксетанилметила.
Типичные воплощения соединений формулы II включают соединения, где X1 представляет собой Ν, X2 представляет собой CR2, и X3 представляет собой CR3.
Типичные воплощения соединений формулы II включают соединения, где X1 представляет собой CR1, X2 представляет собой N, и X3 представляет собой CR3.
Типичные воплощения соединений формулы II включают соединения, где X1 представляет собой CR1, X2 представляет собой CR2, и X3 представляет собой N.
Типичные воплощения соединений формулы II включают соединения, где X1 и X3 представляют собой Ν, X1 и X2 представляют собой N, или X2 и X3 представляют собой N.
Типичные воплощения соединений формулы II включают соединения, где X2 представляет собой CR2, и R2 представляет собой F.
Типичные воплощения соединений формулы II включают соединения, где X1 и X3 представляют собой СН.
Типичные воплощения соединений формулы II включают соединения, где R4 представляет собой -СН2ОН.
Соединения формулы I и II по изобретению могут содержать асимметрические или хиральные центры и ввиду этого существовать в разных стереоизомерных формах. Предполагается, что все стереоизомерные формы соединений по изобретению, включая, но не ограничиваясь этим, диастереомеры, энантиомеры и атропоизомеры, а также их смеси, такие как рацемические смеси, составляют часть настоящего изобретения.
Помимо этого, настоящее изобретение охватывает все диастереомеры, включая цис/транс- (геометрические) и конформационные изомеры. Например, если соединение формулы I содержит двойную связь или конденсированное кольцо, то в объем изобретения включены цис- и транс-формы, а также их смеси.
Если в структурах, приведенных в данном описании, стереохимия какого-либо конкретного хирального атома не конкретизирована, то в качестве соединений по изобретению охватываются и включаются все стереоизомеры. Если стереохимия указана сплошной клиновидной или пунктирной линией, изображающей конкретную конфигурацию, то указан и определен именно этот стереоизомер.
Соединения по настоящему изобретению могут существовать в несольватированных, а также сольватированных формах с фармацевтически приемлемыми растворителями, такими как вода, этанол и им подобные, и предполагается, что данное изобретение охватывает как сольватированные, так и несольватированные формы.
Соединения по настоящему изобретению также могут существовать в разных таутомерных формах, и все такие формы находятся в пределах объема изобретения. Термин "таутомер" или "таутомерная форма" относится к структурным изомерам, обладающим разной энергией, взаимопревращение которых протекает через низкий энергетический барьер. Например, протонные таутомеры (также известные как прототропные таутомеры) характеризуются взаимопревращениями в результате миграции протона, как например при кето-енольной и имин-енаминной изомерии. Валентные таутомеры включают взаимопревращения посредством реорганизации некоторых из связывающих электронов.
БИОЛОГИЧЕСКАЯ ОЦЕНКА
Величины относительной эффективности соединений формулы I в качестве ингибиторов ферментативной активности (или другой биологической активности) можно определить, установив концентрацию, при которой соединение ингибирует активность до предварительно оговоренной степени, и затем сравнив результаты. В типичном случае, предпочтительно определяют концентрацию, при которой активность в биохимическом анализе ингибируется на 50%, т.е. ингибирующую на 50% концентрацию или "IC50". Определение величин IC50 можно провести, используя традиционные методы, известные в данной области техники. В общем случае IС50 можно определить посредством измерения активности заданного фермента в присутствии изучаемого ингибитора в диапазоне концентраций. Затем строят график зависимости экспериментально полученных значений ферментативной активности от используемых концентраций ингибитора. Концентрацию ингибитора, при которой активность фермента составляет 50% (по сравнению с активностью в отсутствие какого-либо ингибитора), принимают за величину IC50. Аналогично, концентрации, вызывающие другой ингибирующий эффект, можно определить посредством соответствующих определений активности. Например, в некоторых случаях постановки задачи может оказаться желательным ввести концентрацию, вызывающую ингибирование на 90%, т.е. IC90, и т.д.
Соединения формулы I тестировали согласно стандартному биохимическому анализу киназы Btk (пример 901).
Общая методика для стандартного анализа киназы Btk с использованием клеток, которую можно использовать для тестирования соединений формулы I, представляет собой анализ Btk с использованием клеток линии Ramos (пример 902).
Стандартный клеточный анализ В-клеточной пролиферации можно использовать для тестирования соединений формулы I с применением В-клеток, выделенных в чистом виде из селезенки мышей Balb/c (пример 903).
Стандартный анализ Т-клеточной пролиферации можно использовать для тестирования соединений формулы I с применением Т-клеток, выделенных в чистом виде из селезенки мышей Balb/c (пример 904).
Анализ ингибирования CD86 можно выполнить для соединений формулы I в отношении ингибирования ими В-клеточной активности, используя общее количество мышиных спленоцитов, выделенных в чистом виде из селезенок мышей Balb/c в возрасте 8-16 недель (пример 905).
Анализ выживаемости клеток при В-клеточном ALL (B-ALL) можно выполнить для соединений формулы I с целью измерения числа жизнеспособных B-ALL-клеток в культуре (пример 906).
Анализ CD69 в цельной крови можно выполнить для соединений формулы I с целью определения способности соединений ингибировать продуцирование CD69 В-лимфоцитами в цельной крови человека, активированными посредством перекрестного сшивания поверхностного IgM с козьими F(ab')2 против IgM человека. CD69 представляет собой лектин С-типа, тип II, вовлеченный в миграцию лимфоцитов и секрецию цитокинов. Экспрессия CD69 представляет собой один из самых ранних существующих индикаторов активации лейкоцитов, и ее быстрая индукция осуществляется на уровне активации транскрипции (Vazquez et al. (2009) Jour, of Immunology, Published October 19, 2009, doi:10.4049/jimmunol.0900839). Зависимое от концентрации ингибирование антигенной стимуляции рецепторов селективными ингибиторами Btk индуцирует экспрессию на поверхности клеток маркера активации лимфоцитов CD69 (Honigberg et al. (2010) Proc. Natl. Acad. Sci. 107(29): 13075-13080). Таким образом, ингибирование CD69 селективными ингибиторами Btk может коррелировать с терапевтической эффективностью некоторых связанных с В-клетками нарушений. Значения IC70, полученные в анализе CD69 в цельной крови человека (от англ. hu blood) с использованием FACS (метод сортировки флуоресцентно-активированных клеток - fluorescence-activated cell sorting), показаны для типичных соединений формулы I в Таблицах 1 и 2.
Цитотоксическую или цитостатическую активность типичных соединений формулы I можно измерить посредством: внесения линии пролиферирующих опухолевых клеток млекопитающих в среду для культивирования клеток, добавления соединения формулы I, культивирования клеток в течение периода времени от примерно 6 часов до примерно 5 суток; и измерения жизнеспособности клеток (пример 908). Анализы in vitro с использованием клеток применяют для измерения жизнеспособности, т.е. пролиферации (IC50), цитотоксичности (ЕС50) и индукции апоптоза (активации каспаз), и они могут быть полезны в прогнозировании клинической эффективности в отношении гематологических злокачественных новообразований и солидных опухолей.
Эффективность in vitro комбинаций соединений формулы I с химиотерапевтическими агентами можно измерить в анализе клеточной пролиферации из примера 908, люминесцентном анализе жизнеспособности клеток CellTiter-Glo® от Promega Corp., Madison, WI. Этот метод гомогенного анализа основан на экспрессии рекомбинантной люциферазы из Coleoptera (US 5583024; US 5674713; US 5700670), и в нем определяют число жизнеспособных клеток в культуре на основании оцененного количества присутствующего АТФ (аденозинтрифосфат), индикатора метаболически активных клеток (Crouch et al. (1993) J. Immunol. Meth. 160: 81-88; US 6602677). Анализ CellTiter-Glo® проводят в 96- или 384-луночном формате, что делает его подходящим для автоматизированного высокопроизводительного скрининга (HTS) (Cree et al. (1995) Anticancer Drugs 6: 398-404). Процедура гомогенного анализа заключается в добавлении одного реагента (реагента CellTiter-Glo®) непосредственно в клетки, культивируемые в среде, дополненной сывороткой. Необходимости в стадиях промывки клеток, удаления среды и многократного пипетирования нет. Данная система позволяет обнаруживать минимально 15 клеток/лунка в 384-луночном формате через 10 минут после добавления реагента и перемешивания.
Гомогенный формат в режиме "добавить-перемешать-измерить" ("add-mix-measure") приводит к лизису клеток и генерации сигнала люминесценции, пропорционального количеству присутствующего АТФ. Количество АТФ прямо пропорционально числу клеток, присутствующих в культуре. В анализе CellTiter-Glo® генерируется люминесцентный сигнал типа свечения ("glow-type"), образуемый в результате люциферазной реакции, который, как правило, имеет время полужизни больше пяти часов в зависимости от используемого типа клеток и используемой среды. Число жизнеспособных клеток выражают в относительных единицах люминесценции (RLU). Субстрат, люциферин жуков-светляков, подвергается окислительному декарбоксилированию под действием рекомбинантной люциферазы светлячков, при этом одновременно происходит превращение АТФ в AMP (аденозинмонофосфат) и образование фотонов. Продолжительное время полужизни снимает необходимость в применении инжекторов для реагентов и обеспечивает оперативность в отношении осуществления непрерывного или периодического режима обработки большого количества планшетов. В этом анализе клеточной пролиферации можно использовать различные многолуночные форматы, например 96- или 384-луночный формат. Данные можно регистрировать с помощью люминометра или визуализирующего устройства - CCD-камеры (charge coupled device - устройство с зарядовой связью). Выходной сигнал люминесценции приведен в виде относительных световых единиц (relative light units; RLU), измеренных в течение некоторого периода времени.
Антипролиферативную эффективность типичных соединений формулы I и их комбинаций с химиотерапевтическими агентами измеряли в анализе CellTiter-Glo® (пример 908) в отношении некоторых клеточных линий гематологических опухолей. Оценивали величины ЕС50 для тестируемых соединений и комбинаций.
Получали типичные соединения формулы I, приведенные в Таблицах 1 и 2, определяли их характеристики и тестировали на предмет ингибирования Btk в соответствии со способами по данному изобретению, и они имеют следующие структуры и соответствующие названия (ChemDraw Ultra, версия 9.0.1, и ChemBioDraw, версия 11.0, CambridgeSoft Corp., Cambridge, ΜΑ). В том случае, когда соединение формулы I или промежуточное соединение соотносится более чем с одним названием, такое соединение будет определяться по химической структуре.
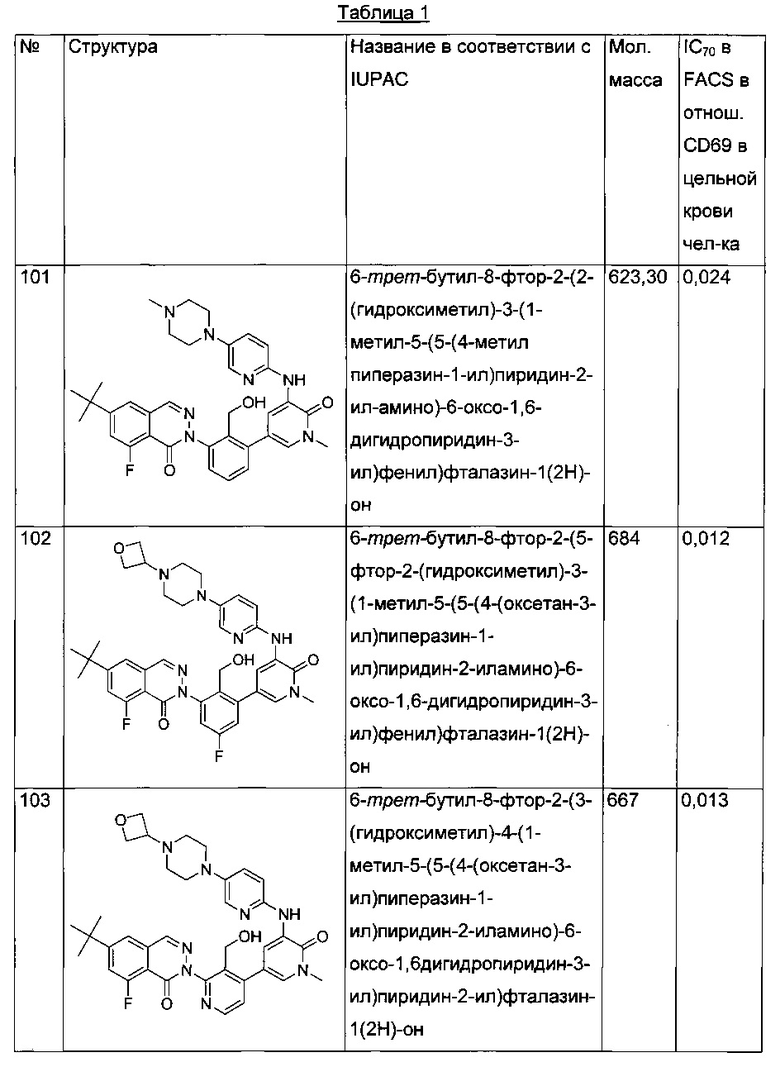
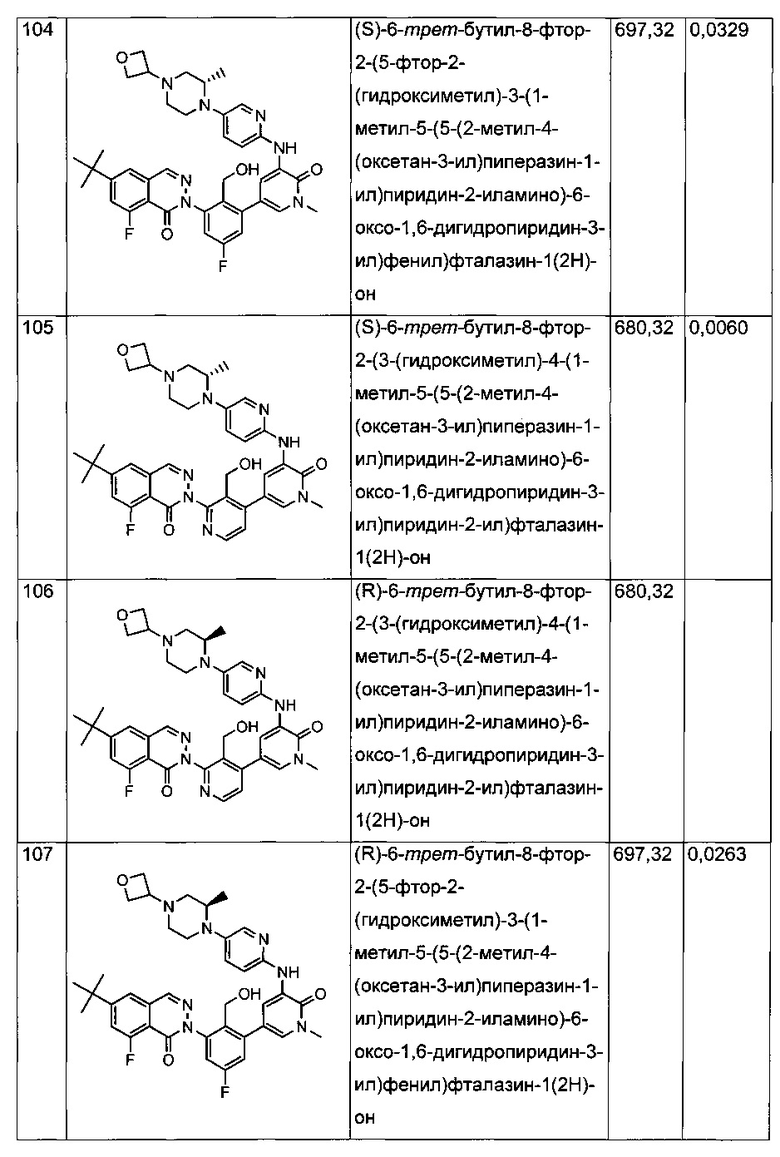

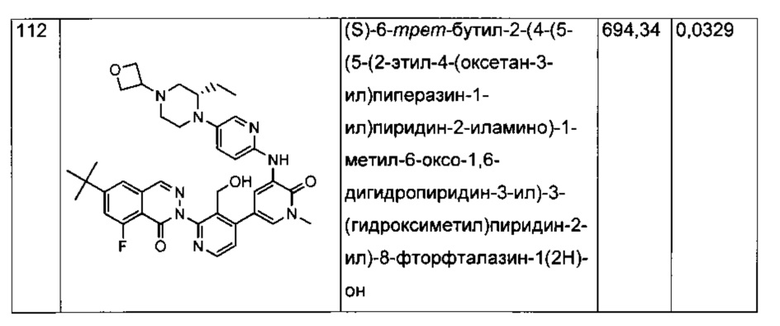
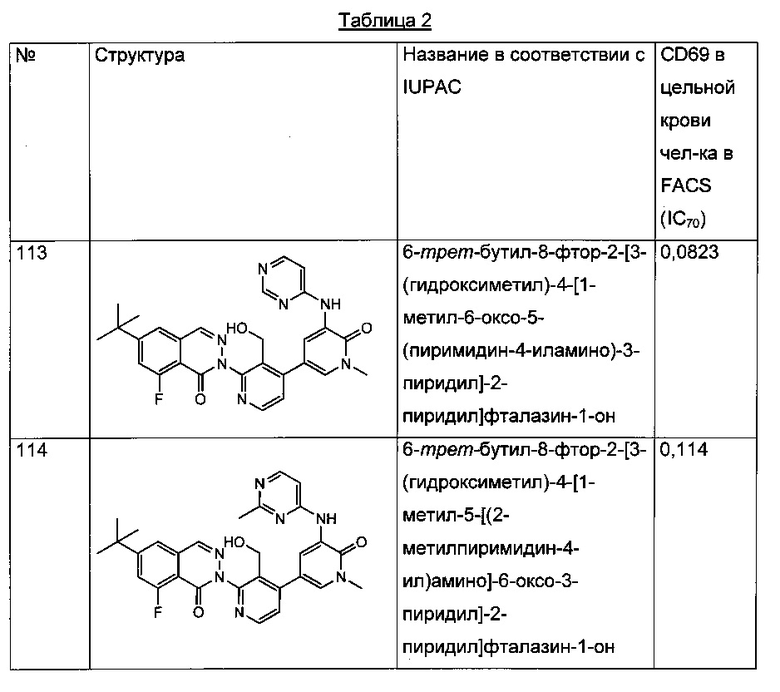
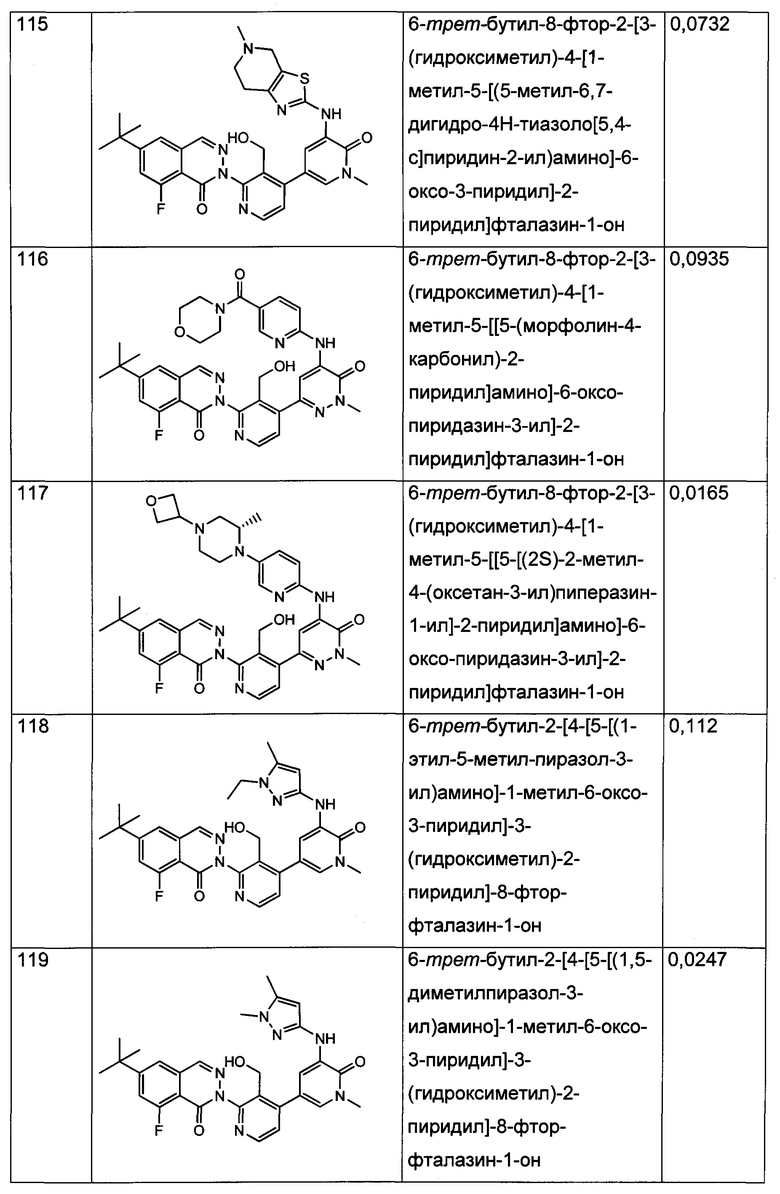
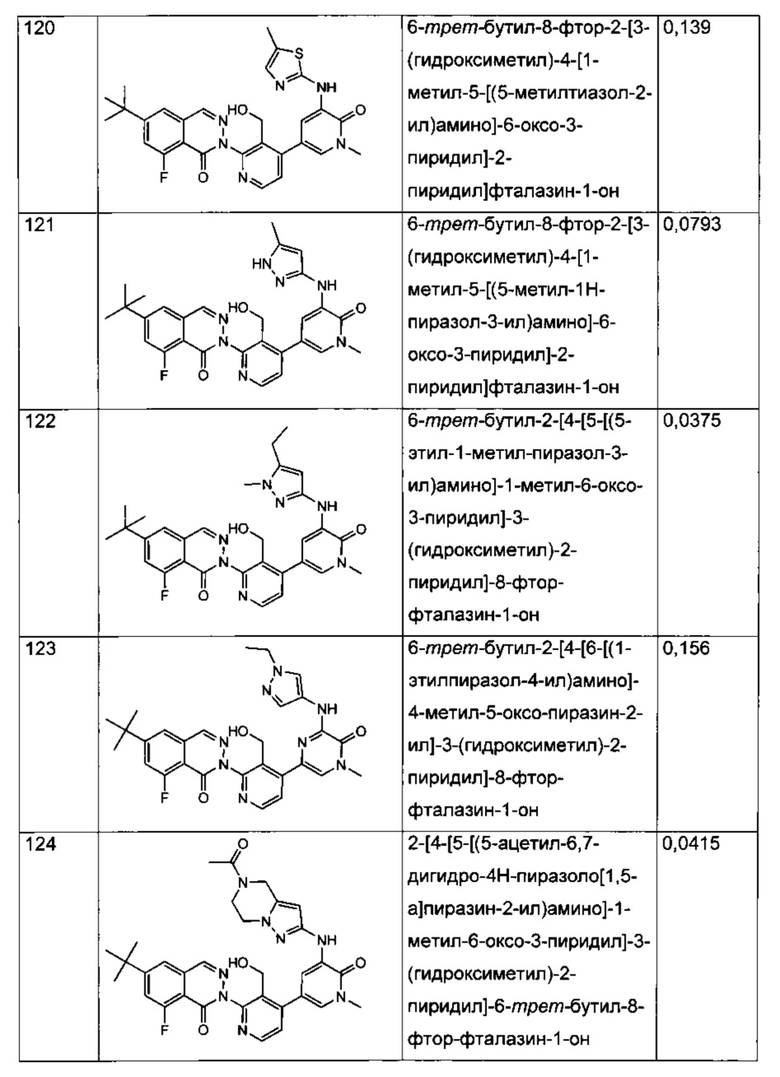
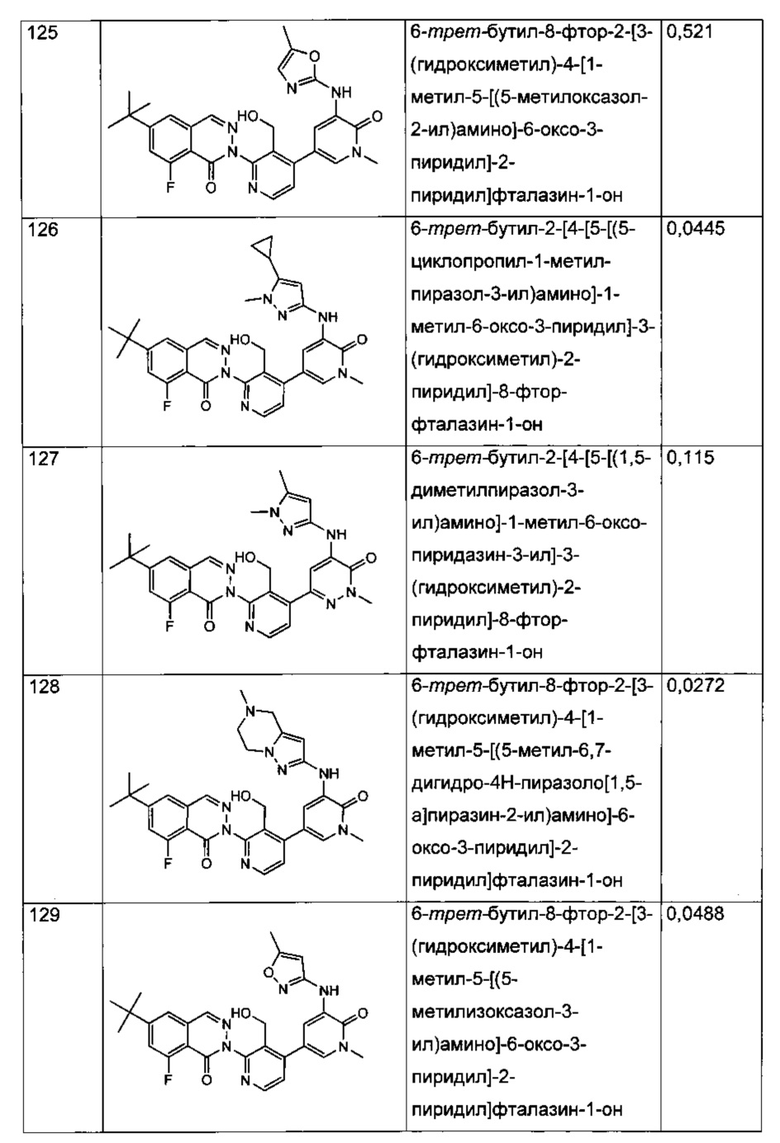
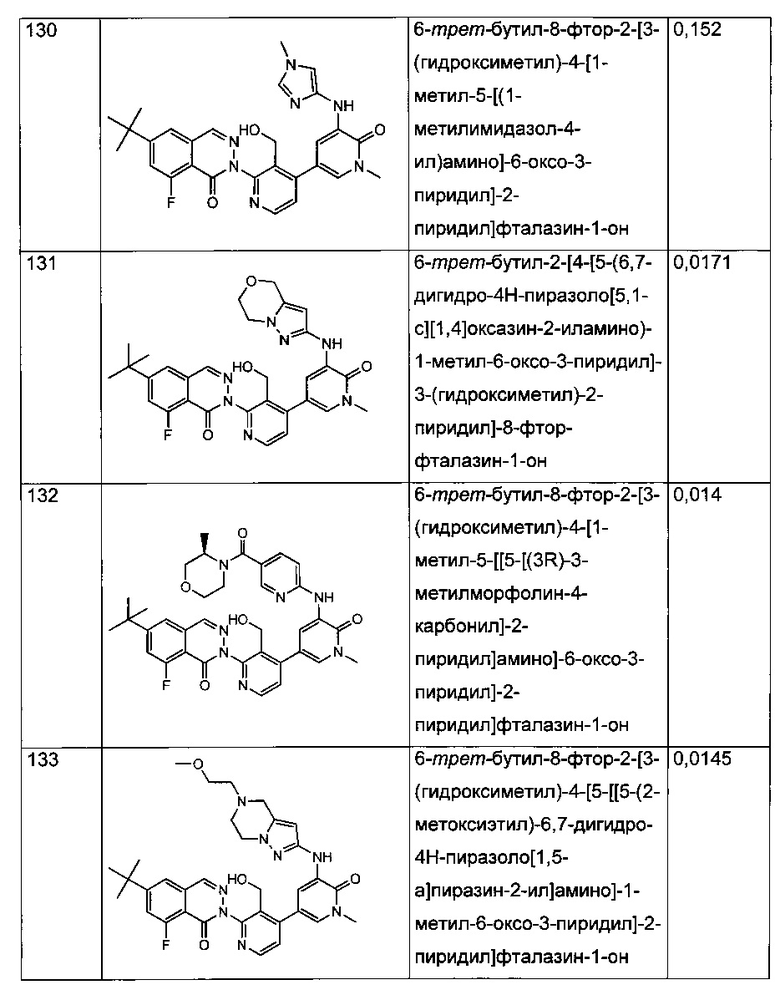
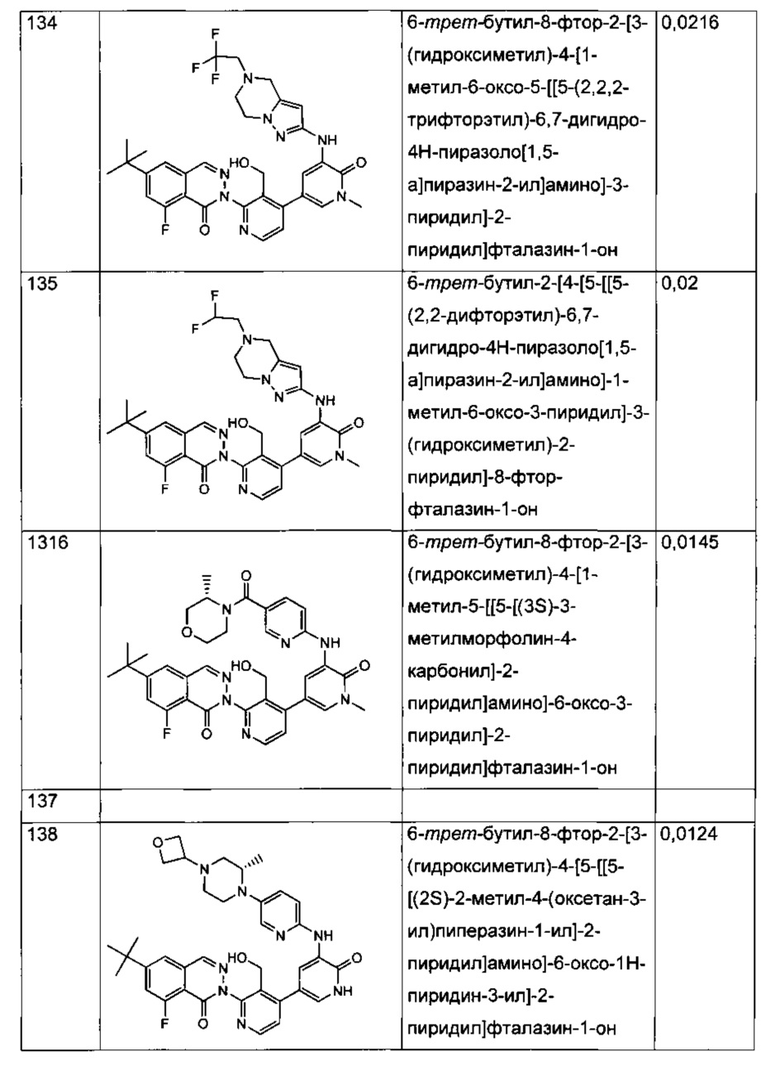
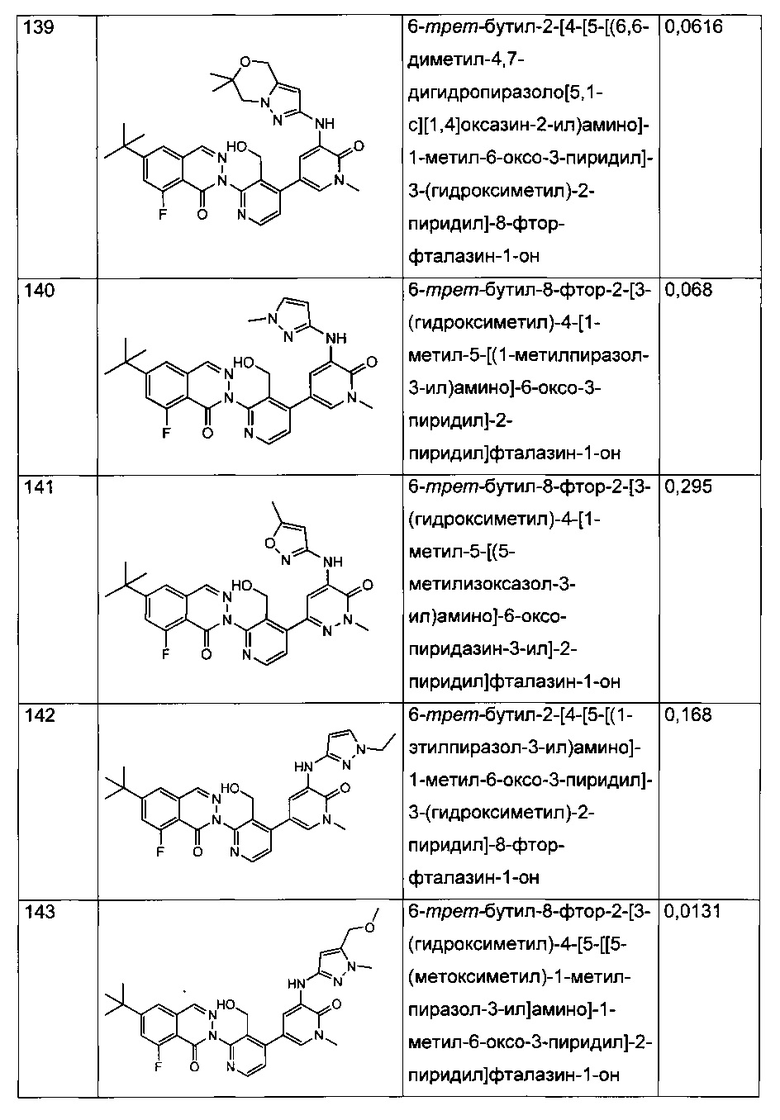

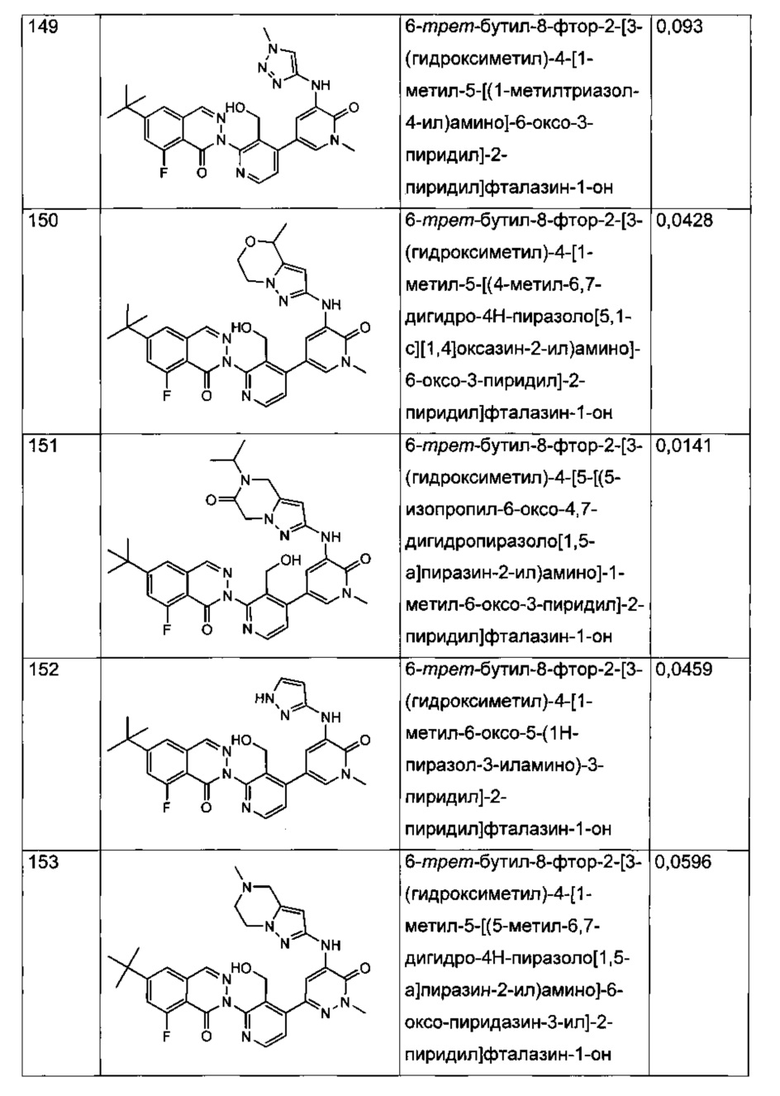
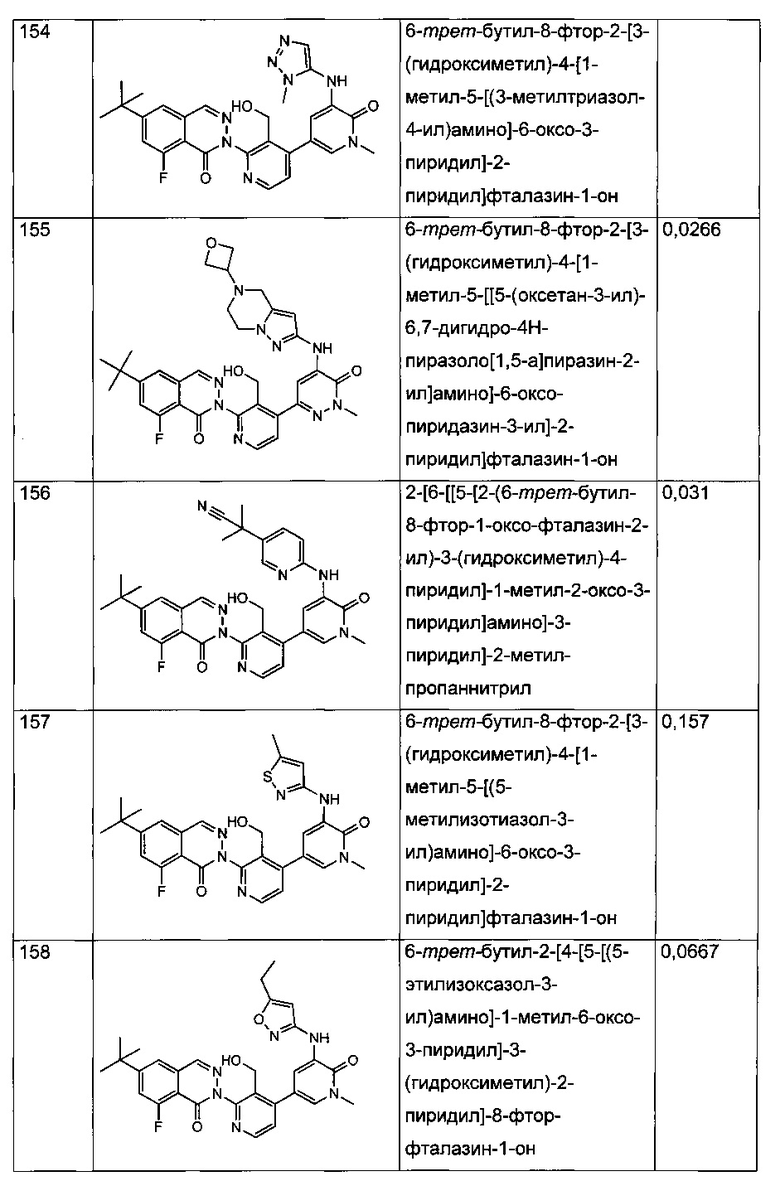
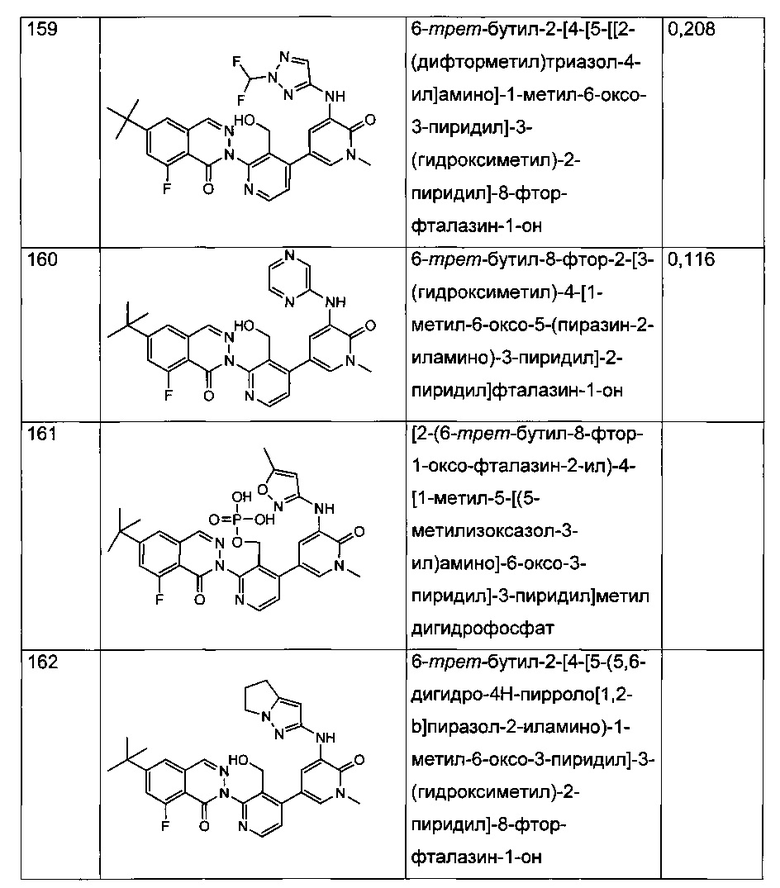
ВВЕДЕНИЕ СОЕДИНЕНИЙ ФОРМУЛЫ I
Соединения по изобретению можно вводить любым путем, соответствующим подвергаемому лечению состоянию. Подходящие пути включают пероральный, парентеральный (в том числе подкожный, внутримышечный, внутривенный, интраартериальный, интрадермальный, интратекальный и эпидуральный), трансдермальный, ректальный, назальный, местный (в том числе трансбуккальный и сублингвальный), вагинальный, внутрибрюшинный, внутрилегочный и интраназальный. Что касается местного иммуносупрессивного лечения, то соединения можно вводить в область поражения, включая перфузию трансплантата ингибитором или иное приведение трансплантата в контакт с ингибитором перед трансплантацией. Очевидно, что предпочтительный путь может варьировать в зависимости, например, от состояния реципиента. Если соединение вводят перорально, оно может быть в составе препарата в виде пилюли, капсулы, таблетки и т.д. вместе с фармацевтически приемлемым носителем или эксципиентом. Если соединение вводят парентерально, оно может быть в составе препарата вместе с фармацевтически приемлемым парентеральным разбавителем и в стандартной инъекционной лекарственной форме, как описано подробно ниже.
Доза для лечения пациентов, являющихся людьми, может изменяться от примерно 10 мг до примерно 1000 мг соединения формулы I или II. Типичная доза может составлять от примерно 100 мг до примерно 300 мг соединения. Дозу можно вводить один раз в сутки (QD), два раза в сутки (BID) или чаще, в зависимости от фармакокинетических и фармакодинамических свойств, включая всасывание, распределение, метаболизм и экскрецию конкретного соединения. К тому же, на дозировку и схему введения могут влиять факторы, определяющие токсичность. При пероральном введении пилюлю, капсулу или таблетку можно принимать путем проглатывания один раз в сутки или с меньшей частотой в течение определенного периода времени. Эта схема может быть повторена для ряда циклов терапии.
СПОСОБЫ ЛЕЧЕНИЯ СОЕДИНЕНИЯМИ ФОРМУЛЫ I И II
Соединения формулы I и II по настоящему изобретению полезны для лечения пациента - человека или животного, страдающего от обусловленных аномальным(ой) клеточным(ой) ростом, функцией или поведением заболеваний или расстройств, ассоциированных с киназой Btk, таких как иммунное расстройство, сердечно-сосудистое заболевание, вирусная инфекция, воспаление, расстройство метаболизма/эндокринной системы или неврологическое расстройство, которые таким образом можно лечить способом, включающим введение для этой цели соединения по настоящему изобретению, определенного выше. Пациента - человека или животного, страдающего от рака, также можно лечить способом, включающим введение для этой цели соединения по настоящему изобретению, определенного выше. Тем самым можно нормализовать или улучшить состояние пациента.
Соединения формулы I и II могут быть полезны для диагностики или лечения in vitro, in situ и in vivo клеток млекопитающих, организмов или ассоциированных патологических состояний, таких как системное и локальное воспаление, иммуновоспалительные заболевания, такие как ревматоидный артрит, подавление иммунитета, отторжение органных трансплантатов, аллергические реакции, язвенный колит, болезнь Крона, дерматит, астма, системная красная волчанка, синдром Шегрена, рассеянный склероз, склеродермия/системный склероз, идиопатическая тромбоцитопеническая пурпура (ITP), связанный с антинейтрофильными цитоплазматическими антителами (ANCA) васкулит, хроническая обструктивная болезнь легких (COPD), псориаз, и для оказания общего защитного действия в отношении суставов.
Способы по изобретению также включают лечение таких заболеваний, как артритоподобные заболевания, такие как ревматоидный артрит, моносуставной артрит, остеоартрит, подагрический артрит, спондилит; болезнь Бехчета; сепсис, септический шок, эндотоксический шок, грамотрицательный сепсис, грамположительный сепсис и синдром токсического шока; вторичный по отношению к септицемии синдром полиорганной недостаточности, травма или геморрагия; офтальмические расстройства, такие как аллергический конъюнктивит, весенний конъюнктивит, увеит и ассоциированная с щитовидной железой офтальмопатия; эозинофильная гранулема; легочные или респираторные заболевания, такие как астма, хронический бронхит, аллергический ринит, ARDS, хроническое легочное воспалительное заболевание (например, хроническая обструктивная болезнь легких), силикоз, легочный саркоидоз, плеврит, альвеолит, васкулит, эмфизема, пневмония, бронхоэктаз и кислородное отравление легких; реперфузионная травма миокарда, головного мозга или конечностей; фиброз, такой как кистозный фиброз; келоидное образование или образование рубцовой ткани; атеросклероз; аутоиммунные заболевания, такие как системная красная волчанка (SLE), аутоиммунный тиреоидит, рассеянный склероз, некоторые формы диабета и синдром Рейно; и ассоциированные с отторжением трансплантатов расстройства, такие как GVHD и отторжение аллотрансплантата; хронический гломерулонефрит; воспалительные заболевания кишечника, такие как хроническое воспалительное заболевание кишечника (CIBD), болезнь Крона, язвенный колит и некротизирующий энтероколит; воспалительные дерматозы, такие как контактный дерматит, атопический дерматит, псориаз или крапивница; лихорадка и миалгии вследствие инфекции; воспалительные нарушения центральной или периферической нервной системы, такие как менингит, энцефалит и повреждение головного или спинного мозга вследствие небольшой травмы; синдром Шегрена; заболевания с вовлечением диапедеза лейкоцитов; алкогольный гепатит; бактериальная пневмония; заболевания, опосредованные комплексом антиген-антитело; гиповолемический шок; сахарный диабет I типа; острая и замедленная гиперчувствительность; болезненные состояния вследствие патологического изменения лейкоцитов и метастаза; термическое повреждение; синдромы, ассоциированные с переливанием гранулоцитов, и цитокин-индуцируемая токсичность.
Способы по изобретению включают лечение рака, выбранного из рака молочной железы, яичника, шейки матки, предстательной железы, семенника, мочеполового тракта, пищевода, гортани, глиобластомы, нейробластомы, рака желудка, кожи, кератоакантомы, рака легкого, эпидермоидной карциномы, крупноклеточной карциномы, немелкоклеточного рака легкого (NSCLC), мелкоклеточной карциномы, аденокарциномы легкого, рака кости, толстой кишки, аденомы, рака поджелудочной железы, аденокарциномы, рака щитовидной железы, фолликулярной карциномы, недифференцированной карциномы, папиллярной карциномы, семиномы, меланомы, саркомы, рака мочевого пузыря, рака печени и желчных протоков, рака почки, панкреатического рака, миелоидных расстройств, лимфомы, волосатоклеточного рака, рака щечной полости, носоглотки, глотки, губы, языка, полости рта, тонкого кишечника, толстой кишки-прямой кишки, толстого кишечника, прямой кишки, головного мозга и центральной нервной системы, болезни Ходжкина, лейкоза, рака бронха, щитовидной железы, печени и внутрипеченочных желчных протоков, гепатоклеточного рака, желудочного рака, глиомы/глиобластомы, эндометриального рака, рака почки и почечной лоханки, мочевого пузыря, тела матки, шейки матки, множественной миеломы, острого миелогенного лейкоза, хронического миелогенного лейкоза, лимфоцитарного лейкоза, хронического лимфоидного лейкоза (CLL), миелоидного лейкоза, рака ротоглотки, неходжкинской лимфомы, и ворсинчатой аденомы толстой кишки.
Способы по изобретению могут найти применение в лечении субъектов, которые подвержены или могут быть подвержены реперфузионной травме, т.е. поражению, являющемуся результатом таких ситуация, при которых ткань или орган претерпевают период ишемии с последующей реперфузией. Термин "ишемия" относится к локализованной тканевой анемии вследствие затруднения притока артериальной крови. Преходящая ишемия с последующей реперфузией обычно приводит к активации нейтрофилов и их миграции через эндотелий кровеносных сосудов в пораженной области. Аккумуляция активированных нейтрофилов в свою очередь приводит к образованию реакционно-способных метаболитов кислорода, которые повреждают компоненты вовлеченных ткани или органа. Это явление "реперфузионной травмы" обычно ассоциируется с такими состояниями, как сосудистый инсульт (включая глобальную и фокальную ишемию), геморрагический шок, миокардиальная ишемия или инфаркт, органная трансплантация и спазм сосудов головного мозга. Для иллюстрации: реперфузионная травма происходит при прекращении процедур шунтирования сердца или во время остановки сердца, когда после прекращения получения сердцем крови начинается реперфузия. Ожидается, что ингибирование активности Btk может привести к снижению количества случаев реперфузионной травмы в таких ситуациях.
ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ
Чтобы применить соединение по данному изобретению для терапевтического лечения млекопитающих, в том числе людей, обычно его изготавливают в соответствии с обычной фармацевтической практикой в виде фармацевтической композиции. Согласно этому аспекту изобретения предложена фармацевтическая композиция, содержащая соединение по данному изобретению вместе с фармацевтически приемлемым разбавителем или носителем.
Типичную композицию изготавливают путем смешивания соединения по настоящему изобретению и носителя, разбавителя или эксципиента. Подходящие носители, разбавители и эксципиенты хорошо известны специалистам в данной области техники и включают такие вещества, как углеводы, воски, растворимые и/или набухаемые в воде полимеры, гидрофильные или гидрофобные вещества, желатин, масла, растворители, вода и им подобные. Выбор конкретного используемого носителя, разбавителя или эксципиента будет зависеть от средства и цели предполагаемого применения соединения по настоящему изобретению. Как правило, растворители выбирают из растворителей, признанных специалистами в данной области безвредными (GRAS) для введения млекопитающему. В общем случае, безвредными растворителями являются нетоксичные водные растворители, такие как вода и другие нетоксичные растворители, которые растворимы в воде или смешиваются с водой. Подходящие водные растворители включают воду, этанол, пропиленгликоль, полиэтиленгликоли (например, ПЭГ400, ПЭГ300) и т.д. и их смеси. Композиции также могут включать в себя одно или более чем одно из следующего: буферы, стабилизирующие агенты, поверхностно-активные вещества, увлажняющие агенты, смазывающие вещества, эмульгаторы, суспендирующие агенты, консерванты, антиоксиданты, придающие непрозрачность агенты, глиданты, технологические добавки, красители, подсластители, отдушки, корригенты и другие известные вспомогательные вещества для обеспечения наилучшей презентации лекарственного средства (т.е. соединения по настоящему изобретению или фармацевтической композиции на его основе) или содействия производству фармацевтического продукта (т.е. лекарственного средства).
Композиции могут быть изготовлены с использованием традиционных методик растворения и смешивания. Например, лекарственную субстанцию (т.е. соединение по настоящему изобретению или стабилизированную форму соединения (например, комплекс с производным циклодекстрина или другим известным агентом комплексообразования)) растворяют в подходящем растворителе в присутствии одного или более эксципиентов, описанных выше. Соединение по настоящему изобретению обычно изготавливают в составе фармацевтических лекарственных форм для обеспечения легко контролируемой дозировки лекарственного средства и облегчения соблюдения больным предписанной схемы лечения.
Фармацевтическая композиция (или препарат) для применения может быть упакована по-разному в зависимости от способа, используемого для введения лекарственного средства. В общем случае, изделие, предназначенное для распространения, содержит контейнер с размещенным в нем фармацевтическим препаратом в соответствующей форме. Подходящие контейнеры хорошо известны специалистам в данной области техники и включают такие материалы, как бутылки (пластиковые и стеклянные), саше, ампулы, пластиковые пакеты, металлические цилиндрические сосуды и им подобное. Контейнер также может включать в себя блок, предохраняющий от неумелого обращения, для предупреждения неосторожного доступа к содержимому упаковки. Помимо этого, контейнер снабжен этикеткой с описанием содержимого контейнера. Этикетка также может содержать соответствующие предостережения.
Фармацевтические композиции на основе соединений по настоящему изобретению могут быть изготовлены для различных путей и типов введения. Например, соединение формулы I или II желаемой степени чистоты возможно может быть смешано с фармацевтически приемлемыми разбавителями, носителями, эксципиентами или стабилизаторами (Remington's Pharmaceutical Sciences (1995) 16th edition, Osol A. Ed.) в форме лиофилизированной композиции, измельченного порошка или водного раствора. Изготовление композиций может быть осуществлено путем смешивания при температуре окружающей среды при соответствующем рН и желаемой степени чистоты с физиологически приемлемыми носителями, т.е. носителями, являющимися нетоксичными для реципиентов в применяемых дозировках и концентрациях. Величина рН композиции зависит главным образом от конкретного применения и концентрации соединения, но может изменяться в диапазоне от примерно 3 до примерно 8. Подходящим воплощением является композиция, изготовленная в ацетатном буфере, при рН 5.
Как правило, соединение можно хранить в виде твердой композиции, лиофилизированной композиции или в виде водного раствора.
Изготовление, дозирование и введение фармацевтических композиций по изобретению будет осуществлено способом, согласующимся с надлежащей медицинской практикой, т.е. в количествах, концентрациях, в соответствии со схемами, порядком, наличием разбавителей и путем введения. Факторы, рассматриваемые в этом контексте, включают конкретное подлежащее лечению расстройство, конкретное подлежащее лечению млекопитающее, клиническое состояние отдельного пациента, причину расстройства, место доставки агента, способ введения, схему введения и другие факторы, известные врачам. "Терапевтически эффективное количество" подлежащего введению соединения будет обусловлено такого рода соображениями и составляет минимальное количество, необходимое для улучшения или лечения гиперпролиферативного расстройства.
Из общих соображений первоначальное фармацевтически эффективное количество ингибитора, вводимого парентерально, из расчета на одну дозу будет находиться в диапазоне примерно 0,01-100 мг/кг, а именно примерно 0,1-20 мг/кг массы тела пациента в сутки, причем типичный начальный диапазон для используемого соединения составляет 0,3-15 мг/кг/сутки.
Приемлемые разбавители, носители, эксципиенты и стабилизаторы нетоксичны для реципиентов в применяемых дозировках и концентрациях и включают буферы, такие как фосфат, цитрат и другие органические кислоты; антиоксиданты, включая аскорбиновую кислоту и метионин; консерванты (такие как октадецилдиметилбензиламмония хлорид; гексаметонийхлорид; бензалконийхлорид, бензетонийхлорид; фенол, бутиловый или бензиловый спирт; алкилпарабены, такие как метил- или пропил-парабен; катехол; резорцин; циклогексанол; 3-пентанол и м-крезол); низкомолекулярные (менее чем примерно 10 остатков) полипептиды; белки, такие как сывороточный альбумин, желатин или иммуноглобулины; гидрофильные полимеры, такие как поливинилпирролидон; аминокислоты, такие как глицин, глутамин, аспарагин, гистидин, аргинин или лизин; моносахариды, дисахариды и другие углеводы, включая глюкозу, маннозу или декстрины; хелатирующие агенты, такие как EDTA (этилендиаминтетрауксусная кислота); сахара, такие как сахароза, маннит, трегалоза или сорбит; солеобразующие противоионы, такие как натрий; комплексы с металлами (например, комплексы Zn-белок); и/или неионные поверхностно-активные вещества, такие как TWEEN™, PLURONICS™ или полиэтиленгликоль (ПЭГ). Кроме того, активные фармацевтические ингредиенты могут быть заключены в микрокапсулы, приготовленные, например, с использованием методов коацервации или путем межфазной полимеризации, например, в микрокапсулы из гидроксиметилцеллюлозы или желатина и микрокапсулы из поли-(метилметакрилата), соответственно, в коллоидные системы лекарственной доставки (например, липосомы, микросферы из альбумина, микроэмульсии, наночастицы и нанокапсулы) или в макроэмульсии. Такие методы описаны в Remington's Pharmaceutical Sciences, 16th edition, Osol A. Ed. (1980).
На основе соединений формулы I или II можно изготовить препараты с длительным высвобождением. Подходящие примеры препаратов с длительным высвобождением включают полупроницаемые матрицы из гидрофобных полимеров в твердой форме, содержащих соединение формулы I или II, причем эти матрицы существуют в форме штампованных изделий, например пленок или микрокапсул. Примеры матриц с длительным высвобождением включают сложные полиэфиры, гидрогели (например, поли(2-гидроксиэтил-метакрилат) или поли(виниловый спирт)), полилактиды (US 3773919), сополимеры L-глутаминовой кислоты и гамма-этил-L-глутамата, неразлагаемый этилен-винилацетат, разлагаемые сополимеры молочной кислоты и гликолевой кислоты, такие как LUPRON DEPOT™ (инъецируемые микросферы, состоящие из сополимера молочной кислоты и гликолевой кислоты и из ацетата лейпролида), и поли-D-(-)-3-гидроксимасляную кислоту.
Данные композиции включают композиции, подходящие для путей введения, подробно изложенных в данном описании. Может быть удобным представление композиций в стандартной лекарственной форме, и они могут быть изготовлены любым из способов, хорошо известных в области фармации. Методы и композиции обычно можно найти в Remington's Pharmaceutical Sciences (Mack Publishing Co., Easton, PA). Такие способы включают стадию приведения активного ингредиента в ассоциацию с носителем, который состоит из одного или более дополнительных ингредиентов. В общем случае композиции изготавливают путем непрерывного и равномерного приведения активного ингредиента в ассоциацию с жидкими носителями или тонкоизмельченными твердыми носителями или ими обоими и затем, если необходимо, придания формы продукту.
Композиции на основе соединения формулы I или II, подходящие для перорального введения, могут быть изготовлены в виде дискретных единиц, таких как пилюли, капсулы, облатки или таблетки, каждая из которых содержит предварительно определенное количество соединения формулы I или II. Прессованные таблетки могут быть изготовлены в подходящей машине путем прессования активного ингредиента в свободно текучей форме, такой как порошок или гранулы, возможно в смеси со связующим веществом, смазывающим веществом, инертным разбавителем, консервантом, поверхностно-активным или диспергирующим агентом. Формованные таблетки могут быть изготовлены в подходящей машине путем формования смеси порошкообразного активного ингредиента, увлажненного инертным жидким разбавителем. Возможно, что таблетки могут иметь покрытие или риску и возможно могут быть изготовлены таким образом, чтобы обеспечить медленное или регулируемое высвобождение из него активного ингредиента. Для перорального применения можно изготовить таблетки, лепешки, пастилки для рассасывания, водные или масляные суспензии, диспергируемые порошки или гранулы, эмульсии, твердые или мягкие капсулы, например желатиновые капсулы, сиропы или эликсиры. Композиции на основе соединения формулы I или II, предназначенные для перорального применения, могут быть изготовлены в соответствии с любым способом, известным в области изготовления фармацевтических композиций, и такие композиции могут содержать один или более агентов, включая подсластители, корригенты, красители и консерванты, с целью придания препарату приятного вкуса. Приемлемыми являются таблетки, содержащие активный ингредиент в смеси с нетоксичным фармацевтически приемлемым эксципиентом, подходящим для изготовления таблеток. Этими эксципиентами могут быть, например, инертные разбавители, такие как карбонат кальция или натрия, лактоза, фосфат кальция или натрия; гранулирующие и разрыхляющие агенты, такие как кукурузный крахмал или альгиновая кислота; связующие вещества, такие как крахмал, желатин или аравийская камедь; и смазывающие вещества, такие как стеарат магния, стеариновая кислота или тальк. Таблетки могут не иметь покрытия или могут быть покрыты с использованием известных методов, включая микроинкапсулирование, для задержки распадаемости и всасывания в желудочно-кишечном тракте и тем самым для обеспечения длительного действия в течение большего периода времени. Например, можно использовать такое вещество-замедлитель, как глицерилмоностеарат или глицерилдистеарат, сами по себе или вместе с воском.
Для лечения глазных или других внешних тканей, например, полости рта и кожи, предпочтительно применяют композиции в виде мази или крема для местного применения, содержащих активный(е) ингредиент(ы) в количестве, например, 0,075-20% масс/масс. При изготовлении в составе мази активные ингредиенты могут быть использованы либо с парафиновой, либо со смешивающейся с водой мазевой основой. Альтернативно, активные ингредиенты могут быть изготовлены в составе крема с использованием основы для крема типа масло-в-воде. При желании водная фаза основы для крема может включать многоатомный спирт, т.е. спирт, имеющий две или более чем две гидроксильные группы, такой как пропиленгликоль, бутан-1,3-диол, маннит, сорбит, глицерин и полиэтиленгликоль (включая ПЭГ400) и их смеси. Композиции для местного применения при желании могут включать в себя соединение, которое усиливает всасывание или проникновение активного ингредиента через кожу или другие зоны воздействия. Примеры таких усилителей проникновения через кожу включают диметилсульфоксид и родственные аналоги. Масляная фаза эмульсий по данному изобретению может быть составлена из известных ингредиентов известным образом. При том, что эта фаза может содержать только эмульгатор, желательно, чтобы она содержала смесь по меньшей мере одного эмульгатора с жиром или маслом либо и с жиром, и с маслом обоими. Предпочтительно, гидрофильный эмульгатор включают вместе с липофильным эмульгатором, который действует как стабилизатор. Также предпочтительно включение как масла, так и жира. Эмульгатор(ы) совместно со стабилизатором(ами) или без него(них) образуют так называемый эмульгирующий воск, и этот воск вместе с маслом и жиром составляет так называемую эмульгирующую мазевую основу, которая образует масляную дисперсионную фазу композиций в форме крема. Эмульгаторы и стабилизаторы эмульсий, подходящие для использования в композиции по изобретению, включают Tween® 60, Span® 80, цетостеариловый спирт, бензиловый спирт, миристиловый спирт, глицерилмоностеарат и лаурилсульфат натрия.
Водные суспензии соединений формул I и II содержат активные вещества в смеси с эксципиентами, подходящими для изготовления водных суспензий. Такие эксципиенты включают суспендирующий агент, такой как карбоксиметилцеллюлозы натриевая соль, кроскармелоза, повидон, метилцеллюлоза, гидроксипропилметилцеллюлоза, альгинат натрия, поливинилпирролидон, трагакантовая камедь и аравийская камедь, и диспергирующие или увлажняющие агенты, такие как природный фосфатид (например, лецитин), продукт конденсации алкиленоксида с жирной кислотой (например, полиоксиэтиленстеарат), продукт конденсации этиленоксида с длинноцепочечным алифатическим спиртом (например, гептадекаэтиленокси-цетанол), продукт конденсации этиленоксида с неполным сложным эфиром, являющимся производным жирной кислоты и ангидрида гексита (например, полиоксиэтиленсорбитан-моноолеат). Водная суспензия также может содержать один или более консервантов, таких как этил- или н-пропил-п-гидроксибензоат, один или более красителей, один или более корригентов и один или более подсластителей, таких как сахароза или сахарин.
Фармацевтические композиции на основе соединений формул I и II могут быть в форме стерильного препарата для инъекций, такого как стерильная водная или масляная суспензия для инъекций. Эта суспензия может быть изготовлена согласно известным в данной области способам с использованием таких подходящих диспергирующих или увлажняющих агентов и суспендирующих агентов, которые упомянуты выше. Стерильный препарат для инъекций также может представлять собой стерильный инъекционный раствор или стерильную инъекционную суспензию в нетоксичном приемлемом для парентерального введения разбавителе или растворителе, как например, раствор в 1,3-бутандиоле, или может быть изготовлен из лиофилизированного порошка. Среди приемлемых разбавителей и растворителей, которые могут быть использованы, находится вода, раствор Рингера и изотонический раствор хлорида натрия. Помимо этого, в качестве растворителя или суспендирующей среды традиционно можно использовать стерильные нелетучие масла. Для этой цели может быть использовано любое маловязкое нелетучее масло, включая синтетические моно- или диглицериды. В дополнение к этому, жирные кислоты, такие как олеиновая кислота, таким же образом могут быть использованы в изготовлении инъекционных средств.
Количество активного ингредиента, которое можно комбинировать с веществом-носителем для изготовления разовой лекарственной формы, будет варьировать в зависимости от подвергаемого лечению реципиента и конкретного способа введения. Например, композиция с замедленным высвобождением, предназначенная для перорального введения людям, может содержать приблизительно 1-1000 мг активного вещества в смеси с соответствующим и удобным количеством вещества-носителя, которое может варьировать от примерно 5 до примерно 95% от массы всей композиции (масса:масса). Фармацевтическая композиция может быть изготовлена таким образом, чтобы беспрепятственно обеспечить введение измеряемого количества. Например, водный раствор, предназначенный для внутривенной инфузии, может содержать от примерно 3 до 500 мкг активного ингредиента на один миллилитр раствора с тем, чтобы можно было осуществлять инфузию подходящего объема со скоростью примерно 30 мл/ч.
Композиции, подходящие для парентерального введения, включают водные и неводные стерильные растворы для инъекций, которые могут содержать антиоксиданты, буферы, бактериостатики и растворенные вещества, делающие композицию изотоничной крови предполагаемого реципиента; и водные и неводные стерильные суспензии, которые могут включать в себя суспендирующие агенты и загустители.
Композиции, подходящие для местного введения в глаз, также включают глазные капли, где активный ингредиент растворен или суспендирован в подходящем носителе, особенно в водном растворителе для активного ингредиента. Активный ингредиент предпочтительно присутствует в таких композициях в концентрации примерно 0,5-20% масс/масс, например примерно 0,5-10% масс/масс, например примерно 1,5% масс/масс.
Композиции, подходящие для местного введения в полость рта, включают пастилки для рассасывания, содержащие активный ингредиент в корригирующей основе, обычно сахарозе и аравийской камеди или трагаканте; пастилки, содержащие активный ингредиент в инертной основе, такой как желатин и глицерин или сахароза и аравийская камедь; и жидкости для полоскания рта, содержащие активный ингредиент в подходящем жидком носителе.
Композиции для ректального введения могут быть представлены в виде суппозитория с подходящей основой, содержащей, например масло какао или салицилат.
Композиции, подходящие для внутрилегочного или назального введения, имеют размер частиц, например, в диапазоне 0,1-500 микрон (в том числе размеры частиц в диапазоне от 0,1 до 500 микрон с таким шагом, как 0,5, 1, 30 микрон, 35 микрон и т.д.), которые вводят посредством быстрой ингаляции через носовой проход или посредством ингаляции через полость рта, чтобы достичь альвеолярных мешочков. Подходящие композиции включают водные или масляные растворы активного ингредиента. Композиции, подходящие для введения аэрозоля или сухого порошка, могут быть изготовлены традиционными способами и могут быть доставлены вместе с другими терапевтическими агентами, такими как соединения, до сих пор используемые в лечении или профилактике расстройств, которые описаны ниже.
Композиции, подходящие для вагинального введения, могут быть представлены в виде композиций в форме пессариев, тампонов, кремов, гелей, паст, пенок или спрея, содержащих помимо активного ингредиента такие носители, целесообразность использования которых в данной области техники известна.
Композиции могут быть упакованы в контейнеры для однократного приема или для многократного приема, например, герметично закрытые ампулы и флаконы, и их можно хранить в высушенном сублимационной сушкой (лиофилизированном) состоянии, требующем только добавления стерильного жидкого носителя, например воды, для инъекций непосредственно перед применением. Экстемпоральные растворы и суспензии для инъекций готовят из стерильных порошков, гранул и таблеток описанного ранее вида. Предпочтительными стандартными дозированными лекарственными формами являются формы, содержащие суточную дозу или стандартную суточную субдозу активного ингредиента, как приведено в данном описании выше, или соответствующую ее долю.
Согласно изобретению также предложены ветеринарные композиции, содержащие по меньшей мере один активный ингредиент, который определен выше, вместе с приемлемым в ветеринарии соответствующим носителем. Приемлемыми в ветеринарии носителями являются вещества, полезные для введения композиции, и они могут быть твердыми, жидкими или газообразными веществами, которые в остальном инертны или приемлемы в области ветеринарии и совместимы с активным ингредиентом. Эти ветеринарные композиции можно вводить парентерально, перорально или любым другим желаемым путем.
КОМБИНИРОВАННАЯ ТЕРАПИЯ
Соединения формул I и II могут быть применены по отдельности или в комбинации с другими терапевтическими агентами для лечения заболевания или расстройства, изложенного в данном описании, такого как воспаление или гиперпролиферативное расстройство (например, рак). В некоторых воплощениях соединение формулы I или II объединяют в фармацевтической комбинированной композиции, или схеме введения в виде комбинированной терапии, с дополнительным, вторым терапевтическим соединением, которое обладает противовоспалительными или антигиперпролиферативными свойствами, или таким соединением, которое полезно для лечения воспаления, нарушения иммунного ответа или гиперпролиферативного расстройства (например, рака). Дополнительным терапевтическим средством может быть противовоспалительный агент, иммуномодулирующий агент, химиотерапевтический агент, усилитель апоптоза, нейротропный фактор, агент для лечения сердечно-сосудистого заболевания, агент для лечения заболевания печени, противовирусный агент, агент для лечения заболевания крови, агент для лечения диабета и агент для лечения иммунодефицитных состояний. Вторым терапевтическим агентом может быть противовоспалительный агент NSAID. Вторым терапевтическим агентом может быть химиотерапевтический агент. Второе соединение в такой фармацевтической комбинированной композиции или схеме введения предпочтительно обладает взаимодополняющими активностями по отношению к соединению формулы I или II, такими, что они не оказывают неблагоприятного влияния друг на друга. Такие соединения соответственно присутствуют в комбинации в количествах, эффективных для предполагаемой задачи. В одном из воплощений композиция по данному изобретению содержит соединение формулы I или II либо стереоизомер, таутомер, сольват, метаболит или фармацевтически приемлемую соль или их пролекарство в комбинации с таким терапевтическим агентом, как NSAID.
Введение при комбинированной терапии может быть осуществлено по одновременной или последовательной схеме. В случае последовательного введения комбинация может быть введена за два или более введений. Комбинированное введение включает совместное введение, при котором используются отдельные композиции или единая фармацевтическая композиция, и последовательное введение в любом порядке, при котором, предпочтительно, есть период времени, когда оба активных агента (или все активные агенты) одновременно проявляют свои биологические активности.
Подходящими дозировками для любого из вышеупомянутых вводимых совместно агентов являются дозировки, используемые в настоящее время, и они могут быть снижены вследствие объединенного действия (синергизма) нового идентифицированного агента и других терапевтических агентов или схем лечения.
Комбинированная терапия может обеспечивать "синергизм" и проявлять "синергетический характер", т.е. эффект, достигаемый при совместном использовании активных ингредиентов, больше суммы эффектов, получаемых в результате применения этих соединений по отдельности. Синергетический эффект может быть получен тогда, когда активные ингредиенты: (1) совместно включают в состав композиции при ее изготовлении и вводят или доставляют одновременно в комбинированной, стандартной дозированной лекарственной форме; (2) доставляют путем чередования или параллельно в виде отдельных композиций; или (3) с использованием какой-либо другой схемы (введения). При доставке с использованием чередующейся терапии синергетический эффект может быть получен, когда соединения вводят или доставляют последовательно, например, посредством разных инъекций в отдельных шприцах, отдельных пилюлях или капсулах либо отдельных капельницах. В общем случае, при чередующейся терапии эффективную дозировку каждого активного ингредиента вводят последовательно, т.е. сериями, тогда как при комбинированной терапии эффективные дозировки двух или более активных ингредиентов вводят совместно.
В конкретном воплощении, относящемся к терапии, соединение формулы I или II либо его стереоизомер, таутомер, сольват, метаболит или фармацевтически приемлемая соль или их пролекарство могут быть скомбинированы с другими терапевтическими агентами, гормональными агентами или агентами на основе антител, как например, агенты, изложенные в данном описании, а также скомбинированы с хирургическим лечением и лучевой терапией. Таким образом, комбинированные терапии по настоящему изобретению включают введение по меньшей мере одного соединения формулы I или II либо его стереоизомера, таутомера, сольвата, метаболита или фармацевтически приемлемой соли или их пролекарства и применение по меньшей мере одного другого способа лечения рака. Количества соединения(ий) формулы I или II и другого(их) фармацевтически активного(ых) терапевтического(их) агента(ов) и относительные временные интервалы введения будут выбраны с целью достижения желаемого комбинированного терапевтического эффекта.
МЕТАБОЛИТЫ СОЕДИНЕНИЙ ФОРМУЛ I И II
Кроме того, в объем данного изобретения включены продукты метаболизма in vivo соединений формул I и II, изложенных в данном описании. Такие продукты могут получаться из вводимого соединения, например, в результате его окисления, восстановления, гидролиза, амидирования, деамидирования, этерификации, деэтерификации, ферментативного расщепления и тому подобного. Соответственно, изобретение включает метаболиты соединений формулы I и II, в том числе соединения, получаемые способом, включающим приведение соединения по данному изобретению в контакт с млекопитающим в течение периода времени, достаточного для получения продукта его метаболизма.
Продукты метаболизма обычно идентифицируют посредством получения меченного радиоактивным (например, 14С или 3Н) изотопом соединения по изобретению, введения его парентеральным способом в детектируемой дозе (например, более чем примерно 0,5 мг/кг) животному, такому как крыса, мышь, морская свинка, обезьяна, или человеку, с обеспечением достаточного для осуществления метаболизма времени (обычно примерно от 30 секунд до 30 часов) и выделения продуктов его превращения из мочи, крови или других биологических образцов. Эти продукты легко выделить, поскольку они являются мечеными (другие выделяют посредством применения антител, способных к связыванию с эпитопами, сохранившимися в метаболите). Структуры метаболитов определяют традиционным образом, например, анализом с использованием MS (масс-спектрометрия), LC/MS (жидкостная хроматография в сочетании с MS) или ЯМР (ядерный магнитный резонанс). В общем случае, анализ метаболитов осуществляют так же, как и традиционные исследования метаболизма лекарственных средств, хорошо известные специалистам в данной области. Продукты метаболизма, при условии, что в других случаях они не обнаруживаются in vivo, полезны в диагностических анализах для определения терапевтической дозы соединений по изобретению.
ИЗДЕЛИЯ ПРОИЗВОДСТВА
В другом воплощении изобретения предложены изделие производства или "набор", содержащие вещества, полезные для лечения описанных выше заболеваний и расстройств. В одном из воплощений набор включает в себя контейнер, содержащий соединение формулы I или II либо стереоизомер, таутомер, сольват, метаболит или фармацевтически приемлемую соль или их пролекарство. Кроме того, набор может содержать этикетку или инструкцию по применению, находящуюся на контейнере или вместе с ним. Термин "инструкция по применению" обычно относится к инструкциям, традиционно включаемым в промышленные упаковки терапевтических продуктов, которые содержат информацию о показаниях, применении, дозировке, введении, противопоказаниях и/или предупреждениях касательно применения таких терапевтических продуктов. Подходящие контейнеры включают, например, бутылки, флаконы, шприцы, блистерную упаковку и т.д. Контейнер может быть сделан из разнообразных материалов, таких как стекло или пластик. Контейнер может включать в себя соединение формулы I или II либо композицию на его основе, которые эффективны для лечения указанного состояния, и может иметь стерильное входное отверстие (например, контейнер может представлять собой мешочек или флакон для внутривенных растворов, имеющий пробку, поддающуюся прокалыванию иглой для подкожных инъекций). По меньшей мере одним активным агентом в композиции является соединение формулы I или II. На этикетке или в инструкции по применению указывается, что данную композицию применяют для лечения выбранного, такого как рак. Помимо этого, на этикетке или в инструкции по применению может быть указано, что подлежащим лечению пациентом является пациент с таким расстройством, как гиперпролиферативное расстройство, нейродегенерация, гипертрофия сердца, боль, мигрень или нейротравматическое заболевание или событие. В одном из воплощений на этикетке или в инструкциях по применению указывается, что данную композицию, содержащую соединение формулы I или II, можно применять для лечения расстройства, являющегося результатом аномального клеточного роста. На этикетке или в инструкции по применению также может быть указано, что данную композицию можно применять для лечения других расстройств. Альтернативно или помимо этого данное изделие производства может дополнительно содержать второй контейнер, содержащий фармацевтически приемлемый буфер, такой как бактериостатическая вода для инъекций (BWFI), забуференный фосфатом физиологический раствор, раствор Рингера и раствор декстрозы. Кроме того, оно может включать в себя другие материалы, подходящие с коммерческой точки зрения и точки зрения потребителя, в том числе другие буферы, разбавители, фильтры, иглы и шприцы.
Набор может дополнительно содержать указания относительно введения соединения формулы I или II и, если она присутствует, второй фармацевтической композиции. Например, если набор включает в себя первую композицию, содержащую соединение формулы I или II, и вторую фармацевтическую композицию, то такой набор может дополнительно содержать указания относительно одновременного, последовательного или раздельного введения первой и второй фармацевтических композиций нуждающемуся в этом пациенту.
В другом воплощении наборы подходят для доставки твердых пероральных форм соединения формулы I или II, таких как таблетки или капсулы. Такой набор предпочтительно включает в себя ряд стандартных дозировок. Такие наборы могут включать в себя карточку с дозировками, расположенными в порядке их предполагаемого применения. Примером такого набора является "блистерная упаковка". Блистерные упаковки хорошо известны в упаковочной индустрии и широко используются для упаковки фармацевтических стандартных лекарственных форм. При желании может быть придана памятка, например в виде чисел, букв или других маркировок либо с календарем-вкладышем, где указаны дни в схеме лечения, в которые можно вводить такие дозировки.
Согласно одному из воплощений набор может содержать (а) первый контейнер с содержащимся в нем соединением формулы I или II; и возможно (b) второй контейнер с содержащейся в нем второй фармацевтической композицией, включающей в себя второе соединение, обладающее антигиперпролиферативной активностью. Альтернативно или помимо этого набор может дополнительно включать в себя третий контейнер, содержащий фармацевтически приемлемый буфер, такой как бактериостатическая вода для инъекций (BWFI), забуференный фосфатом физиологический раствор, раствор Рингера и раствор декстрозы. Кроме того, он может включать в себя другие материалы, подходящие с коммерческой точки зрения и точки зрения потребителя, в том числе другие буферы, разбавители, фильтры, иглы и шприцы.
В некоторых других воплощениях, где набор включает в себя композицию на основе соединения формулы I или II и второй терапевтический агент, такой набор может содержать контейнер для помещения в него отдельных композиций, такой как разделенная бутылка или разделенный пакет из фольги, однако отдельные композиции также могут содержаться в одном неразделенном контейнере. Обычно набор содержит указания по введению отдельных компонентов. Форма набора имеет особое преимущество в тех случаях, когда отдельные компоненты предпочтительно вводят в разных лекарственных формах (например, пероральной и парентеральной), вводят с различными интервалами дозирования или в тех случаях, когда согласно предписанию врача желательно провести подбор дозы индивидуальных компонентов комбинации.
ПОЛУЧЕНИЕ СОЕДИНЕНИЙ ФОРМУЛ I И II
Соединения формулы I и II могут быть синтезированы с использованием путей синтеза, включающих способы, аналогичные хорошо известным в области химии, в частности, в свете содержащегося в данном документе описания, и способы, используемые для других гетероциклов, описанные в работах: Comprehensive Heterocyclic Chemistry II, Editors Katritzky and Rees, Elsevier, 1997, например в томе 3; Liebigs Annalen der Chemie, (9): 1910-16, (1985); Helvetica Chimica Acta, 41: 1052-60, (1958); Arzneimittel-Forschung, 40(12): 1328-31, (1990), каждая из которых явным образом включена посредством ссылки. Обычно исходные соединения приобретают в коммерческих источниках, таких как Aldrich Chemicals (Milwaukee, WI), или легко получают, используя способы, хорошо известные специалистам в данной области техники (например, получают способами, в целом описанными в Louis F. Fieser and Mary Fieser, Reagents for Organic Synthesis, v. 1-23, Wiley, N.Y. (1967-2006 ed.) или Beilsteins Handbuch der organischen Chemie, 4, Aufl. ed. Springer-Verlag, Berlin, включая приложения (также доступные через онлайн-базу данных Бейльштейна).
Методологии химических преобразований в ходе синтеза и использования защитных групп (введения и удаления защиты), полезные в синтезе соединений формулы I и II, и необходимые реагенты и промежуточные соединения известны в данной области техники и включают, например, таковые, описанные в R. Larock, Comprehensive Organic Transformations, VCH Publishers (1989); T.W.Greene and P.G.M. Wuts, Protective Groups in Organic Synthesis, 3rd Ed., John Wiley and Sons (1999); and L.Paquette, ed., Encyclopedia of Reagents for Organic Synthesis, John Wiley and Sons (1995) и в их последующих изданиях.
Соединения формулы I и II могут быть получены по отдельности или в виде библиотек соединений, содержащих по меньшей мере 2, например от 5 до 1000 соединений или от 10 до 100 соединений. Библиотеки соединений формулы I и II можно получить, используя комбинаторный подход "расщепляй и смешивай" ("split and mix") или множественные параллельные синтезы с применением методик проведения химических реакций либо в жидкой фазе, либо в твердой фазе, известных специалистам в данной области. Таким образом, согласно следующему аспекту изобретения предложена библиотека соединений, содержащая по меньшей мере 2 соединения или их фармацевтически приемлемые соли.
На фигурах и в разделе Примеры приведены типичные способы получения соединений формулы I и II. Специалистам в данной области техники будет очевидно, что для синтеза соединений формулы I и II можно использовать и другие пути синтеза. Хотя на фигурах и в разделе Примеры изображены и описаны конкретные исходные вещества и реагенты, они могут быть легко заменены на другие исходные вещества и реагенты для получения целого ряда производных и/или реакционных условий. Помимо этого, многие типичные соединения, полученные описанными способами, могут быть дополнительно модифицированы в свете этого описания с использованием общепринятых методов химии, хорошо известных специалистам в данной области техники.
При получении соединений формул I может потребоваться защита функциональной группы (например, первичного или вторичного амина) в промежуточных соединениях. Необходимость в такой защите будет варьировать в зависимости от природы такой функциональной группы и условий способов получения. Подходящие амино-защитные группы включают ацетил, трифторацетил, трет-бутоксикарбонил (ВОС), бензилоксикарбонил (CBz) и 9-флуоренилметиленоксикарбонил (Fmoc). Необходимость в такой защите легко определяется специалистом в данной области техники. Общее описание защитных групп и их применение см. в Т.W. Greene, Protective Groups in Organic Synthesis, John Wiley & Sons, New York, 1999.
Экспериментальные методики, промежуточные соединения и реагенты, полезные для получения соединений формулы I и II, можно найти в заявке US 2012/0010191, "Pyridone and aza-pyridone compounds and methods of use (Соединения пиридонов и азапиридонов и способы применения)", поданной 6 мая 2011 года, которая включена посредством ссылки во всей своей полноте.
На Фиг. 1-8 описан синтез типичных воплощений соединений формулы I или II, более полно описанный в примерах 101-112, который может быть полезен для получения других соединений формулы I и II.
ОБЩИЕ МЕТОДИКИ ПОЛУЧЕНИЯ
Общая методика: сочетание по Сузуки
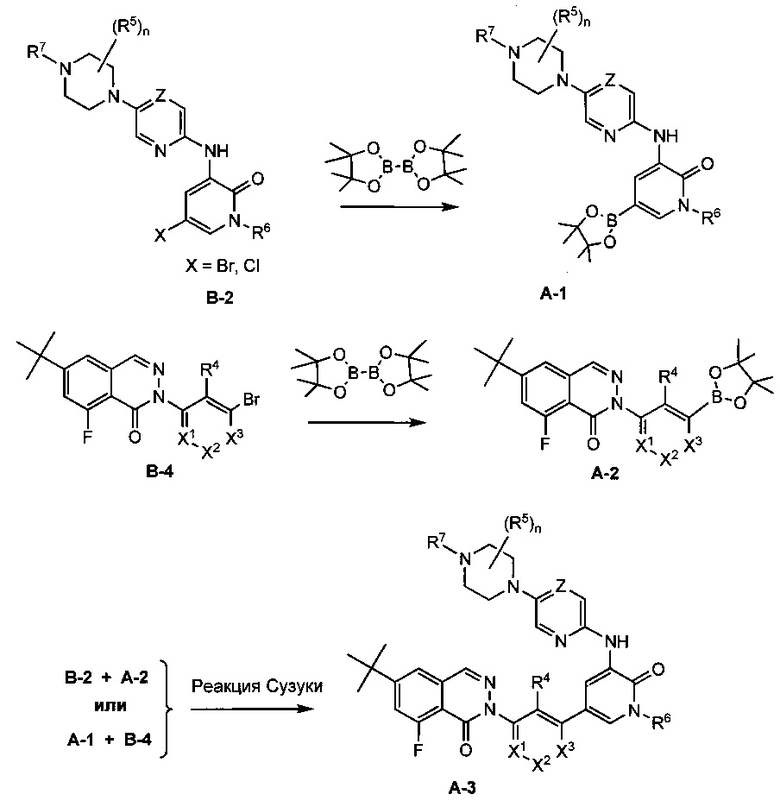
Реакцию сочетания по Сузуки используют для образования углерод-углеродных связей с целью присоединения колец для образования соединений формулы I и II и промежуточных соединений А-3 (Suzuki (1991) Pure Appl. Chem. 63: 419-422; Miyaura and Suzuki (1979) Chem. Reviews 95(7): 2457-2483; Suzuki (1999) J. Organometal. Chem. 576: 147-168). Сочетание по Сузуки представляет собой палладий-опосредуемую реакцию перекрестного сочетания гетероарилгалогенида, такого как В-2 или В-4, с бороновой кислотой, такой как А-1 или А-2. Например, соединение В-2 может быть объединено примерно с 1,5 эквивалента 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолана) и растворено примерно в 3 эквивалентах карбоната натрия в виде 1 молярного раствора в воде и в равном объеме ацетонитрила. Добавляют низковалентный палладиевый реагент в каталитическом или превышающем каталитическое количестве, такой как дихлорид бис(трифенилфосфин)палладия(II). В некоторых случаях для подведения рН водного слоя используют ацетат калия вместо карбоната натрия. Затем реакционную смесь нагревают до примерно 140-150°С под давлением в микроволновом реакторе (Biotage АВ, Uppsala, Sweden) в течение 10-30 минут. Содержимое экстрагируют этилацетатом или другим органическим растворителем. После упаривания органического слоя бор-содержащий сложный эфир А-1 может быть очищен на диоксиде кремния или обращенно-фазовой HPLC (высокоэффективная жидкостная хроматография). Заместители представлены в том виде, как определено, или в виде их защищенных форм или предшественников. Аналогичным образом, промежуточный бромид В-4 можно подвергнуть борилированию с получением соединения А-2.
В результате сочетания по Сузуки соединений В-2 и А-2 или соединений А-1 и В-4 получают соединение формулы I или промежуточное соединение А-3. Эфир бороновой кислоты (или бороновую кислоту) (1,5 экв.) А-1 или А-2 и палладиевый катализатор, такой как хлорид бис(трифенилфосфин)палладия(М) (0,05 экв.), добавляют к смеси галоген-содержащего промежуточного соединения (1 экв.) В-2 или В-4 в ацетонитриле и 1 Μ водном растворе карбоната натрия (в объеме, равном объему ацетонитрила). Реакционную смесь нагревают до примерно 150°С в микроволновом реакторе в течение примерно 15 мин. По LC/MS определяют, когда реакция завершается полностью. К смеси добавляют воду и выпавший в осадок продукт отфильтровывают и очищают посредством HPLC, получая продукт А-3. Заместители представлены в том виде, как определено, или в виде их защищенных форм или предшественников.
На стадии сочетания по Сузуки можно использовать разнообразные палладиевые катализаторы. В реакции сочетание по Сузуки могут быть использованы различные низковалентные катализаторы на основе Pd(II) и Pd(0), в том числе PdCl2(PPh3)2, Pd(t-Bu)3, Pd(dppf)Cl2 CH2Cl2 ([1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий(II) в комплексе с дихлорметаном), Pd(PPh3)4, Pd(OAc)/PPh3, Cl2Pd[(PEt3)]2, Pd(DIPHOS)2 [бис(1,2-бис(дифенилфосфино)этан)палладий(0)], Cl2Pd(bipy) (bipy означает 2,2'-бипиридин), [PdCl(Ph2PCH2PPh2)]2, Cl2Pd[Р(о-толил)3]2, Pd2(dba)3/P(o-толил)3 (dba означает дибензилиденацетон), Pd2(dba)/P(фурил)3, Cl2Pd[Р(фурил)3]2, Cl2Pd(PMePh2)2, Cl2Pd[P(4-F-Ph)3]2, Cl2Pd[P(C6F6)3]2, Cl2Pd[P(2-COOH-Ph)(Ph)2]2, Cl2Pd[P(4-COOH-Ph)(Ph)2]2 и инкапсулированные катализаторы Pd (от англ. encapsulated palladium catalysts) EnCat™ 30, Pd EnCat™ TPP30 и Pd(II)EnCat™ BINAP30 (2,2'-бис(дифенилфосфино)-1,1'-бинафтил; US 2004/0254066).
Общая методика: реакция Бухвальда
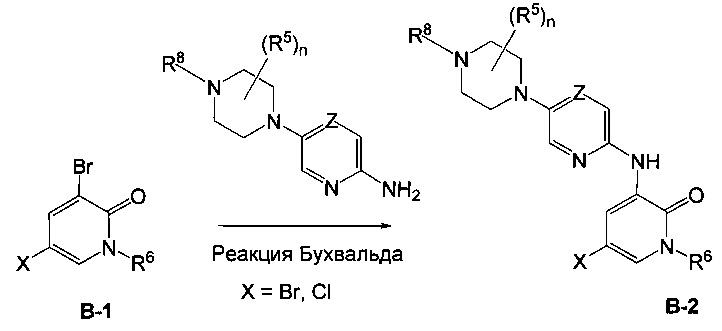
Реакция Бухвальда используется для аминирования 6-бром-содержащих промежуточных соединений В-1 (Wolf and Buchwald (2004) Org. Synth Coll. Vol., 10: 423; Paul et al. (1994) Jour. Amer. Chem. Soc, 116: 5969-5970). К раствору галоген-содержащего промежуточного соединения В-1 в DMF (диметилформамид) добавляют соответствующий пиперазинил-пиридинил- или пиперазинил-пиримидиниламин (200 моль%), Cs2CO3 (50 моль%), Pd2(dba)3 (5 моль%) и 4,5-бис(дифенилфосфино)-9,9-диметилксантен (ксантфос, регистрационный №в CAS 161265-03-8; 10 моль%). Реакционную смесь нагревают до примерно 110°С под давлением в микроволновом реакторе (Biotage АВ, Uppsala, Sweden) в течение примерно 30 мин. Полученный раствор концентрируют в вакууме, получая соединение В-2. Могут быть полезны другие палладиевые катализаторы и фосфиновые лиганды.
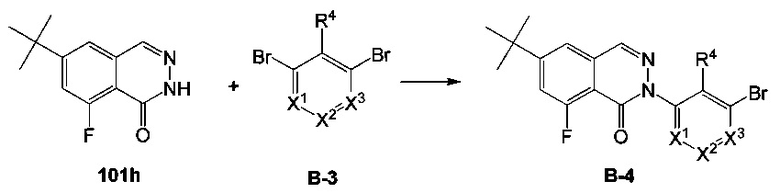
Кроме того промежуточные N-гетероариламиды В-4 можно получить в условиях реакции Бухвальда, используя промежуточные циклические амиды (R7), такие как 6-трет-бутил-8-фторфталазин-1(2Н)-он 101h и гетероарилдибромиды В-3.
МЕТОДЫ РАЗДЕЛЕНИЯ
В способах получения соединений формулы I и II предпочтительным может быть отделение продуктов реакции друг от друга и/или от исходных веществ. Желаемые продукты с каждой стадии или серии стадий разделяют и/или очищают до желаемой степени гомогенности методами, общеизвестными в данной области техники. Обычно такие методы разделения включают в себя многофазную экстракцию, кристаллизацию из растворителя или смеси растворителей, дистилляцию, сублимацию или хроматографию. Хроматография может включать в себя любое количество методов, в том числе, например: обращенно-фазовую и нормально-фазовую; гель-проникающую; ионообменную хроматографию; методы и устройство для жидкостной хроматографии высокого, среднего и низкого давления; мелкомасштабную аналитическую хроматографию; хроматографию с псевдоподвижным слоем (simulated moving bed; SMB) и препаративную тонкослойную и толстослойную хроматографию, а также методы мелкомасштабной тонкослойной и флэш-хроматографии.
Другой класс методов разделения включает обработку смеси реагентом, выбранным для связывания с желаемым продуктом, непрореагировавшим исходным веществом, побочным продуктом реакции или тому подобным либо оказывающим на все вышеперечисленное какое-либо иное действие, способствующее разделению. Такие реагенты включают адсорбенты или абсорбенты, как например, активированный уголь, молекулярные сита, ионообменные среды или им подобные. Альтернативно, такими реагентами могут быть кислоты в случае основного вещества, основания в случае кислотного вещества, связывающие реагенты, такие как антитела, связывающие белки, селективные хелаторы, такие как краун-эфиры, реагенты для жидкость/жидкостной ионной экстракции (liquid/liquid ion (LIX) extraction) или им подобные. Выбор соответствующих методов разделения зависит от природы рассматриваемых веществ, например, от точки кипения и молекулярной массы при дистилляции и сублимации, присутствия или отсутствия полярных функциональных групп при хроматографии, стабильности материалов в кислотных и щелочных средах при многофазной экстракции и тому подобного.
Диастереомерные смеси могут быть разделены на свои индивидуальные диастереомеры на основе различий их физико-химических свойств методами, хорошо известными специалистам в данной области техники, такими как хроматография и/или фракционная кристаллизация. Энантиомеры можно разделить путем превращения энантиомерной смеси в диастереомерную смесь в результате взаимодействия с соответствующим оптически активным соединением (например, хиральным вспомогательным веществом, таким как хиральный спирт или хлорангидрид кислоты Мошера), разделения диастереомеров и превращения (например, посредством гидролиза) индивидуальных диастереоизомеров в соответствующие чистые энантиомеры. Кроме того, некоторые соединения по настоящему изобретению могут быть атропоизомерами (например, замещенные биарилы), и они рассматриваются как часть данного изобретения. Энантиомеры также могут быть разделены с применением колонки для хиральной HPLC.
Индивидуальный стереоизомер, например энантиомер, по существу не содержащий своего стереоизомера, можно получить путем разделения рацемической смеси, используя такой метод, как образование диастереомеров с применением оптически активных разделяющих агентов (Eliel Ε. and Wilen S., "Stereochemistry of Organic Compounds", John Wiley & Sons, Inc., New York, 1994; Lochmuller C.H. (1975) J. Chromatogr., 113(3): 283-302). Рацемические смеси хиральных соединений по изобретению могут быть разделены и соединения выделены любым подходящим методом, включая: (1) образование ионных диастереомерных солей с хиральными соединениями и разделение фракционной кристаллизацией или другими методами, (2) образование диастереомерных соединений с использованием хиральных дериватизирующих реагентов, разделение диастереомеров и превращение в чистые стереоизомеры и (3) разделение по существу чистых или обогащенных стереоизомеров непосредственно в хиральных условиях. См.: "Drug Stereochemistry, Analytical Methods and Pharmacology", Irving W. Wainer, Ed., Marcel Dekker, Inc., New York (1993).
Согласно способу (1) диастереомерные соли могут быть образованы в результате взаимодействия энантиомерно чистых хиральных оснований, таких как бруцин, хинин, эфедрин, стрихнин, α-метил-β-фенилэтиламин (амфетамин) и им подобные, с асимметрическими соединениями, несущими кислотную функциональную группу, такую как группа карбоновой кислоты и сульфоновой кислоты. Разделение диастереомерных солей можно осуществить фракционной кристаллизацией или ионообменной хроматографией. Что касается разделения оптических изомеров аминосоединений, то к образованию диастереомерных солей может приводить добавление хиральных карбоновых или сульфоновых кислот, таких как камфорсульфоновая кислота, винная кислота, миндальная кислота или молочная кислота.
Альтернативно, согласно способу (2) подлежащее разделению вещество приводят во взаимодействие с одним из энантиомеров хирального соединения с образованием диастереомерной пары (Eliel Ε. and Wilen S., "Stereochemistry of Organic Compounds", John Wiley & Sons, Inc., New York, 1994, p. 322). Диастереомерные соединения могут образовываться в результате взаимодействия асимметрических соединений с энантиомерно чистыми хиральными дериватизирующими реагентами, такими как ментильные производные, с последующим разделением диастереомеров и гидролизом с получением чистого или обогащенного энантиомера. Метод определения оптической чистоты включает в себя получение хиральных сложных эфиров, таких как ментиловый эфир, например (-)-ментил-хлорформиат, в присутствии основания, или эфир кислоты Мошера, α-метокси-α-(трифторметил)фенилацетат (Jacob III, J. Org. Chem. (1982) 47: 4165), в рацемической смеси и анализ 1Н ЯМР-спектра на наличие двух атропоизомерных энантиомеров или диастереомеров. Стабильные диастереомеры атропоизомерных соединений можно разделить и выделить нормально-фазовой и обращенно-фазовой хроматографией, следуя способам разделения атропоизомерных нафтил-изохинолинов (WO 96/15111). Согласно способу (3) рацемическую смесь двух энантиомеров можно разделить хроматографией с использованием хиральной неподвижной фазы ("Chiral Liquid Chromatography" (1989), W.J. Lough, Ed., Chapman and Hall, New York; Okamoto, J. Chromatogr. (1990) 513: 375-378). Обогащенные или очищенные энантиомеры можно различить методами, используемыми для различения других хиральных молекул с асимметрическими атомами углерода, такими как оптическое вращение и круговой дихроизм.
ПРИМЕРЫ
Пример 101а. 4-трет-Бутилбензоилхлорид 101а
Смесь 4-трет-бутилбензойной кислоты (1000 г; 5,6 моль) в хлорангидриде сернистой кислоты (sulfurous dichloride) (1,5 л) кипятили с обратным холодильником в течение 3 часов. Затем смесь концентрировали в вакууме и неочищенное соединение 101а использовали на следующей стадии без дополнительной очистки.
Пример 101b. 4-трет-Бутил-N-(2-гидрокси-1,1-диметил-этил)-бензамид 101b
Раствор соединения 101а в DCM (200 мл) по каплям при 0-10°С добавляли к раствору 2-амино-2-метил-1-пропанола (1000 г; 10,5 моль) в CH2Cl2 (2000 мл). Через 5 минут после исходного добавления образовывался белый осадок. Полученную суспензию перемешивали при комнатной температуре в течение ночи. Твердое вещество удаляли фильтрованием и промывали CH2Cl2 (1000 мл). Фильтрат концентрировали путем упаривания на роторном испарителе, получая соединение 101b в виде светло-желтой смолы, которую использовали на следующей стадии без дополнительной очистки.
Пример 101с. 2-(4-трет-Бутил-фенил)-4,4-диметил-4,5-дигидро-оксазол 101с
Смесь соединения 101b (1000 г; неочищенного) в тионилхлориде 1,5 л) кипятили с обратным холодильником в течение 1 часа. Реакционную смесь охлаждали до комнатной температуры и выливали в 500 мл перемешиваемого Et2O, за это время образовывался белый осадок. Осадок собирали фильтрованием и промывали Et2O, растворяли в 500 мл воды и нейтрализовали 25%-ным NaOH. Желтый водный раствор экстрагировали EtOAC (3×500 мл) и объединенную органическую фазу промывали 500 мл рассола, сушили над Na2SO4, фильтровали и концентрировали в вакууме, получая соединение 101с (530 г; 40,8% за 3 стадии) в виде белого твердого вещества. 1Н ЯМР (300 МГц, CDCl3) δ 7.87 (d, J=8,4 Гц, 2Н), 7.41 (d, J=8,4 Гц, 2Н), 4.08 (s, 2Н), 1.37 (s, 6Н), 1.33 (s, 9Н).
Пример 101d. 5-трет-Бутил-2-(4,4-диметил-4,5-дигидро-оксазол-2-ил)-бензальдегид 101d
К раствору соединения 101с (50 г; 0,22 моль) в безводном THF (тетрагидрофуран) (750 мл) добавляли 2,4 Μ раствор н-бутиллития в гексане (225 мл) при -78°С в атмосфере азота. Прозрачный янтарный раствор нагревали до -20°С и перемешивали в течение 4 ч. Реакционная смесь приобретала цвет красного янтаря и становилась мутной. Смесь снова охлаждали до -78°С и быстро перемешивали, после чего по каплям добавляли 72 мл DMF с такой скоростью, чтобы поддерживать температуру ниже -60°С. После добавления реакционную смесь перемешивали при -78°С в течение 15 мин, затем перемешивали при -20°С в течение 1 ч и при комнатной температуре в течение 1 ч. Реакционную смесь гасили, используя 200 мл 0,5 Μ водного KHSO4. Добавляли дополнительное количество раствора KHSO4 для подведения рН до приблизительно 4-5. Водную фазу экстрагировали EtOAC (3×500 мл), объединенные органические фазы промывали 400 мл рассола и сушили над Na2SO4, фильтровали и концентрировали в вакууме, получая соединение 101d (35 г; 62%) в виде желтого твердого вещества, которое использовали на следующей стадии без дополнительной очистки. LCMS, ESI (electrospray ionization - ионизация электрораспылением), m/z 260 (М+1)+.
Пример 101е. 2-(4-трет-Бутил-2-1,3-диоксинан-2-ил-фенил)-4,4-диметил-4,5-дигидрооксазол 101е
Смесь соединения 101d (60 г; 0,23 моль), п-толуолсульфоната пиридиния (4 г; 0,02 моль) и 1,3-пропандиола (60 мл) в толуоле (500 мл) нагревали до температуры дефлегмации в течение ночи и охлаждали до комнатной температуры после определения завершения реакции с помощью LCMS. Реакционную смесь промывали 200 мл 50%-ного водного раствора NaHCO3, 200 мл воды и 200 мл рассола. Органический слой сушили над Na2SO4, фильтровали и концентрировали в вакууме, получая остаток, который очищали хроматографией на силикагеле с использованием смеси 1:5 EtOAc/петролейный эфир в качестве элюента, получая соединение 101е (16 г; 21,7%) в виде прозрачного желтого смолообразного вещества. LCMS (ESI) 318 (М+Н)+. 1Н ЯМР (300 МГц, CDCl3) δ 7.79 (s, 1Η), 7.67 (d, J=8,0 Гц, 1Η), 7.36 (dd, J=1,6; 8,4 Гц, 1Η), 6.32 (s, 1H), 4.23 (dd, J=5,2; 11,2 Гц, 2H), 4.06 (s, 2H), 4.04-3.98 (m, 2H), 2.27-2.21 (m, 1H), 1.48-1.38 (m, 1H), 1.38 (s, 6H), 1.32 (s, 9H).
Пример 101f. 2-(4-трет-Бутил-2-1,3-диоксинан-2-ил-6-фтор-фенил)-4,4-диметил-4,5-дигидро-оксазол 101f
К раствору соединения 101е (20 г; 63 ммоль) в безводном THF (400 мл) добавляли 2,4 Μ раствор н-бутиллития в гексане (65 мл) при -78°С в атмосфере Ν2. Прозрачный желтый раствор перемешивали при -17°С в течение 3 ч, после чего он приобретал густой красно-оранжевый цвет. Реакционный раствор снова охлаждали до -78°С, быстро перемешивали и по каплям в течение 10 мин добавляли раствор N-фторбензолсульфонимида (29 г; 92 ммоль) в безводном THF (100 мл). Реакционную смесь перемешивали при -78°С в течение 5 мин, -20°С в течение 30 мин, затем при комнатной температуре в течение 1 ч. Реакционную смесь выливали в 150 мл 50%-ного водного NH4Cl и экстрагировали 300 мл EtOAc. Отделенную органическую фазу промывали 150 мл воды и 150 мл рассола, сушили над Na2SO4, фильтровали и концентрировали в вакууме, получая остаток, который очищали хроматографией на силикагеле с использованием смеси 1:5 EtOAc/CH2Cl2 в качестве элюента, получая соединение 101f (7 г; 33%) в виде желтого твердого вещества. LCMS (ESI) m/z 336 (М+Н)+. 1Н ЯМР (300 МГц, CDCl3) δ 7.53 (d, J=1,6 Гц, 1Н), 7.08 (dd, J=2,0; 12,0 Гц, 1Н), 5.90 (s, 1Н), 4.24 (dd, J=5,2; 10,8 Гц, 2Н), 4.06 (s, 2Н), 3.98-3.91 (m, 2Н), 2.26-2.19 (m, 1Н), 1.45-1.42 (m, 1Н), 1.42 (s, 6Н), 1.30 (s, 9Н).
Пример 101g. 5-трет-Бутил-7-фтор-3-метоксиизобензофуран-1(3Н)-он 101g
Смесь соединения 101f (68,8 г; 205,4 ммоль), метанола (1340 мл) и 50%-ной водной серной кислоты (881 мл) перемешивали при температуре дефлегмации в течение ночи. Реакционную смесь выливали в 400 мл воды, экстрагировали DCM (3×1000 мл). Объединенные органические экстракты промывали 400 мл рассола, сушили над Na2SO4, фильтровали и концентрировали досуха, получая соединение 101g (43 г) в виде беловатого твердого вещества, которое использовали на следующей стадии без дополнительной очистки LCMS (ESI) m/z 225 (М+Н)+.
Пример 101h. 6-трет-Бутил-8-фторфталазин-1(2Н)-он 101h
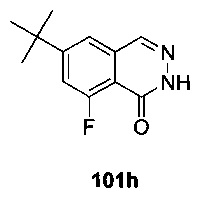
К раствору соединения 101g (40 г; 168 ммоль) в ледяной уксусной кислоте (360 мл) добавляли гидразина моногидрат (240 мл) при 0-10°С под защитой N2. Полученную суспензию перемешивали в атмосфере азота при 50°С в течение 1,5 часов. Реакционную смесь выливали в 300 мл воды при непрерывном перемешивании. Водную фазу экстрагировали DCM (2×500 мл) и объединенную органическую фазу сушили над Na2SO4, фильтровали и концентрировали, получая остаток, который очищали перекристаллизацией в DMC и Et2O, получая соединение 101h (17 г; 37,6% за 2 стадии) в виде беловатого твердого вещества. 1Н ЯМР (300 МГц, CDCl3) δ 8.11 (d, J=2,7 Гц, 1Н), 7.46 (m, 2Н), 1.39 (s, 9Н). LCMS (ESI) m/z 221 (M+H)+.
Пример 101i. 2-(6-трет-Бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-6-хлорбензальдегид 101i
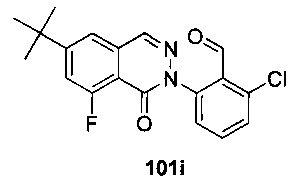
В круглодонную колбу вместимостью 10 мл добавляли 6-трет-бутил-8-фтор-2Н-фталазин-1-он 101h (640 мг; 2,9 ммоль), 2-хлор-6-фторбензальдегид (506 мг; 3,2 ммоль) и карбонат цезия (488 мг; 1,5 ммоль). Колбу вакуумировали и трижды наполняли азотом, затем в реакционную колбу добавляли этокситриметилсилан (684 мг; 5,8 ммоль) и DMF (5 мл). Полученную смесь нагревали до 60°С. Через 4 ч перемешивания раствор оставляли охладиться до температуры окружающей среды и реакцию гасили, по каплям добавляя 2 мл H2O. Желаемый продукт начинал выпадать в осадок из смеси DMF и воды. Твердое вещество собирали фильтрованием после охлаждения до 5°С и промывали смесью DMF/вода (2/1, 2 мл; предварительно охлажденной до 6°С) и H2O (2 мл). Осадок на фильтре сушили в вакуумном сушильном шкафу при 65°С в течение ночи, получая 519 мг (52%) соединения 101i в виде желтого твердого вещества. MS: [М+Н]+ 359.
Пример 101j. 6-трет-Бутил-2-(3-хлор-2-(гидроксиметил)фенил)-8-фторфталазин-1(2Н)-он 101j
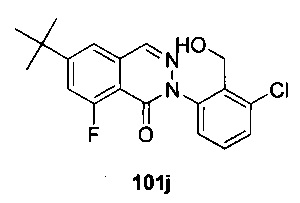
2-(6-трет-Бутил-8-фтор-1-оксо-1Н-фталазин-2-ил)-6-хлор-бензальдегид 101i (519 мг; 1,4 ммоль) растворяли в DCM (2 мл) с перемешиванием при комнатной температуре и затем к раствору добавляли 1 мл iΡΑ. Полученный раствор охлаждали до 4°С и добавляли NaBH4 (27 мг; 0,7 ммоль) в виде одной порции. Через 30 мин перемешивания реакцию гасили добавлением H2O (2 мл). Водный слой экстрагировали CH2Cl2 (2×5 мл), промывали рассолом и сушили над MgSO4. Фильтрат концентрировали, получая остаток, который очищали хроматографией на силикагеле с элюированием смесью 0-20% EtOAc/CH2Cl2, получая соединение 101j в виде белого твердого вещества (385 мг; 72%). MS: [М+Н]+ 361.
Пример 101. 6-трет-Бутил-8-фтор-2-(2-(гидроксиметил)-3-(1-метил-5-(5-(4-метилпиперазин-1-ил)пиридин-2-иламино)-6-оксо-1,6-дигидропиридин-3-ил)фенил)фталазин-1(2Н)-он 101
В колбу емкостью 10 мл добавляли по порядку 6-трет-бутил-2-(3-хлор-2-гидроксиметил-фенил)-8-фтор-2Н-фталазин-1-он 101j (100 мг; 0,27 ммоль), 1-метил-3-[5-(4-метил-пиперазин-1-ил)-пиридин-2-иламино]-5-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-1Н-пиридин-2-он (141 мг; 0,33 ммоль), PCy3 (6 мг), Pd(dba)2 (6 мг) и K2CO3 (112 мг; 0,8 ммоль). Колбу вакуумировали и наполняли азотом. Эту последовательность повторяли три раза. Затем к реакционной смеси добавляли 20%-ный водный 1,4-диоксан. Полученную смесь нагревали до 90°С до слабого кипения с обратным холодильником и перемешивали в течение 1,5 ч в атмосфере азота. Реакционную смесь охлаждали до комнатной температуры, фильтровали через набивку диатомовой земли в качестве фильтрующего агента (CELITE®) и концентрировали с получением остатка, который очищали препаративной TLC (тонкослойная хроматография) (10%-ный МеОН в CH2Cl2), получая соединение 101 (80 мг; 46%) в виде желтого твердого вещества. MS: [M+H]+ 624. 1Н ЯМР (400 МГц, DMSO-d6) δ 8.53 (d, J=2,0 Гц, 1H), 8.50 (d, J=1,6 Гц, 1H), 8.36 (s, 1Н), 7.87 (d, J=3,2 Гц, 2H), 7.73 (d, J=12,8 Гц, 1H), 7.51 (t, J=7,6 Гц, 1Н), 7.41 (d, J=3,2 Гц, 1Н), 7.39 (d, J=3,6 Гц, 1Н), 7.35 (dd, J=2,8; 9,2 Гц, 1Н), 7.28 (d, J=2 Гц, 1Н), 7.20 (d, J=9,2 Гц, 1Н), 4.58 (t, J=4,8 Гц, 1Н), 4.36 (s, 2Н), 3.58 (s, 3Н), 3.04 (m, 4Н), 2.44 (m, 4Н), 2.21 (s, 3Н), 1.38 (s, 9Н).
Пример 102а. 2,6-Дибром-4-фторбензальдегид 102а
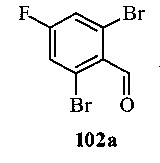
К раствору 1,3-дибром-5-фтор-2-иодбензола (50 г; 132 ммоль) в безводном толуол (300 мл), охлажденному до -35°С, добавляли раствор хлорида изопропилмагния (84 мл; 171 ммоль; 2,0 Μ в диэтиловом эфире) в течение 30 минут, все время поддерживая внутреннюю температуру ниже -25°С. Получали прозрачный коричневый раствор. Перемешивание продолжали в течение 1,5 ч. Затем добавляли безводный DMF (34 мл; 436 ммоль) в течение 30 минут. Температура реакционной смеси повышалась до -19°С. Реакционную смесь нагревали до 10°С (комнатная температура) в течение 1 ч и перемешивали при этой температуре в течение 1,5 ч. Реакцию гасили насыщенным водным NH4Cl (100 мл), фильтровали и упаривали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (с элюированием смесью петролейный эфир/этилацетат от 50:1 до 20:1), получая соединение 102а (20 г; выход 54%) в виде желтого твердого вещества.
Пример 102b. 2-Бром-6-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-фторбензальдегид 102b
К раствору соединения 102а (767 мг; 2,72 ммоль), 6-трет-бутил-8-фторфталазин-1(2Н)-она 101 h (300 мг; 1,36 ммоль) в диоксане (50 мл) добавляли KOAc (267 мг; 2,72 ммоль), CuI (259 мг; 1,36 ммоль) и 4,7-диметокси-1,10-батофенантролин (327 мг; 1,36 ммоль). Барботирования азота через полученный раствор в течение 30 мин смесь перемешивали при 90 градусах в течение 10 ч. Ее оставляли охлаждаться до комнатной температуры и добавляли H2O (100 мл). Водный слой отделяли и экстрагировали этилацетатом (2×200 мл). Объединенные органические слои промывали рассолом (100 мл) и сушили над сульфатом натрия. Сушильный агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении. Остаток очищали на колонке для флэш-хроматографии с элюированием смесью РЕ/ЕА (15:1), получая 102b (172 мг; 30%). LCMS: [М+Н]+ 421. 1Н ЯМР (500 МГц, CDCl3) δ 10.20 (s, 1Н), 8.20 (s, 1Н), 7.49-7.51 (m, 3Н), 7.25 (m, 1Н), 1.36 (s, 9Н).
Пример 102с. 2-(6-трет-Бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-фтор-6-(1-метил-5-(5-(4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-иламино)-6-оксо-1,6-дигидропиридин-3-ил)бензальдегид 102с
В круглодонную колбу загружали 2-бром-6-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-фторбензальдегид 102b (100 мг; 0,24 ммоль), 1-метил-3-(5-(4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-иламино)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2(1Н)-он (131 мг; 0,28 ммоль), PdCl2 (dppf) (25 мг; 0,03 ммоль), K3PO4⋅3H2O (149 мг; 0,56 ммоль), THF (10 мл) и Н2O (5 мл). После трех циклов вакуумирования/продувания аргоном смесь нагревали при 70°С в течение 6 ч. Затем фильтровали и фильтрат упаривали в вакууме. Остаток очищали колоночной флэш-хроматографией с элюированием смесью 1:3 петролейный эфир/этилацетат, получая соединение 102с в виде желтого твердого вещества (98 мг; 60%). LCMS: [М+Н]+ 682.
Пример 102. 6-трет-Бутил-8-фтор-2-(3-(гидроксиметил)-4-(1-метил-5-(5-(4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-иламино)-6-оксо-1,6-дигидропиридин-3-ил)пиридин-2-ил)фталазин-1(2Н)-он 102
Смесь 2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-фтор-6-(1-метил-5-(5-(4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-иламино)-6-оксо-1,6-дигидро-пиридин-3-ил)бензальдегида 102с (98 мг; 0,14 ммоль), NaBH4 (17 мг; 0,43 ммоль) и СН3ОН (10 мл) перемешивали при 25°С в течение 1 ч. Затем концентрировали при пониженном давлении и добавляли воду (10 мл). Полученную смесь экстрагировали, используя CH2Cl2 (10 мл×2). Объединенный CH2Cl2-экстракт концентрировали при пониженном давлении и остаток очищали обращенно-фазовой препаративной HPLC, получая соединение 102 (45 мг; 46%). LCMS: [М+Н]+ 684. 1Н ЯМР (500 МГц, DMSO-d6) δ 8.52-8.56 (m, 2Н), 8.40 (s, 1Н), 7.88 (d, J=6,0 Гц, 2H), 7.74-7.77 (m, 1Н), 7.34-7.40 (m, 3Н), 7.29 (d, J=3,0 Гц, 1H), 7.22 (d, J=9,5 Гц, 1H), 4.54-4.56 (m, 2Н), 4.44-4.47 (m, 2Н), 4.32 (s, 2Н), 3.58 (s, 3Н), 3.43 (s, 1Н), 3.06-3.08 (m, 4Н), 2.36-2.39 (m, 5Н), 1.38 (s, 9Н).
Пример 103а. 2-Бром-4-хлорникотинальдегид 103а
К раствору 2-бром-4-хлорпиридина (1,6 г; 8,0 ммоль) в безводном тетрагидрофуране (40 мл), охлажденному при -70°С, добавляли раствор диизопропиламида лития (5,0 мл; 10,0 ммоль; 2,0 М) в течение 5 минут и перемешивали при -70°С в течение еще 1 ч. В течение 3 минут вводили безводный DMF (1,3 г) и смесь перемешивали в течение еще 30 минут. Затем ее гасили насыщенным NH4Cl (30 мл) и экстрагировали этилацетатом (20 мл×3). Объединенный органический слой сушили над безводным Mg2SO4, фильтровали и упаривали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с элюированием смесью петролейный эфир/этилацетат (20:1), получая соединение 103а в виде желтого твердого вещества (900 мг; 48%). 1Н ЯМР (500 МГц, DMSO-d6) δ 10.21 (s, 1Н), 8.52 (d, J=5,5 Гц, 1Η), 7.79 (d, J=5,0 Гц, 1 Η).
Пример 103b. 2-(6-трет-Бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-хлорникотинальдегид 103b
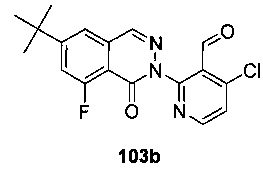
К раствору 2-бром-4-хлорникотинальдегида 103а (446 мг; 2,05 ммоль), 6-трет-бутил-8-фторфталазин-1(2Н)-она 101h (300 мг; 1,36 ммоль) в диоксане (50 мл) добавляли KAcO (267 мг; 2,72 ммоль), CuI (259 мг; 1,36 ммоль) и 4,7-диметокси-1,10-батофенантролин (327 мг; 1,36 ммоль). После барботирования азота через полученный раствор в течение 30 мин смесь перемешивали при 90°С в течение 10 ч. Ее оставляли охлаждаться до комнатной температуры и добавляли H2O (100 мл). Водный слой отделяли и экстрагировали этилацетатом (2×200 мл). Объединенный органический слой промывали рассолом (100 мл) и сушили над сульфатом натрия. Сушильный агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении. Остаток очищали на колонке для флэш-хроматографии с элюированием смесью 10:1 РЕ/ЕА, получая соединение 103b (120 мг; 25%). LCMS: [М+Н]+ 360. 1Н ЯМР (500 МГц, CDCl3) δ 10.36 (s, 1Η), 8.69 (d, J=5,5 Гц, 1Η), 8.28 (d, J=2,0 Гц, 1Η), 7.28-7.56 (m, 3Н), 1.49 (s, 9Н).
Пример 103с. 2-(6-трет-Бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-(1-метил-5-(5-(4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-иламино)-6-оксо-1,6-дигидропиридин-3-ил)никотинальдегид 103с
Смесь соединения 103b (100 мг; 0,28 ммоль), 1-метил-3-(5-(4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-иламино)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2(1Н)-она (131 мг; 0,28 ммоль), PdCl2 (dppf) (25 мг; 0,03 ммоль) и K3PO4⋅3H2O (149 мг; 0,56 ммоль) суспендировали в смеси THF (10 мл)/H2O (5 мл). После трех циклов вакуумирования/продувания аргоном смесь нагревали при 70°С в течение 6 ч. Затем фильтровали и фильтрат упаривали в вакууме. Остаток очищали колоночной флэш-хроматографией с элюированием смесью 1:3 петролейный эфир/этилацетат, получая соединение 103с в виде желтого твердого вещества (150 мг; 60%). LCMS: [М+Н]+ 665.
Пример 103. 6-трет-Бутил-8-фтор-2-(3-(гидроксиметил)-4-(1-метил-5-(5-(4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-иламино)-6-оксо-1,6-дигидропиридин-3-ил)пиридин-2-ил)-фталазин-1(2Н)-он 103
Смесь соединения 103с (120 мг; 0,18 ммоль), NaBH4 (21 мг; 0,54) и СН3ОН (10 мл) перемешивали при 25°С в течение 1 ч. Затем концентрировали при пониженном давлении и добавляли воду (10 мл). Полученную смесь экстрагировали, используя CH2Cl2 (10 мл×2). Объединенный CH2Cl2-экстракт концентрировали при пониженном давлении и остаток очищали обращенно-фазовой препаративной HPLC, получая соединение 103 (45 мг; 38%). LCMS: [М+Н]+ 667. 1Н ЯМР (500 МГц, CDCl3) δ 8.66-8.68 (m, 2Н), 8.35 (s, 1Н), 7.96 (d, J=3,0 Гц, 1H), 7.81 (s, 1Н), 7.65 (d, J=2,5 Гц, 1H), 7.55-7.59 (гл, 3Н), 6.83 (d, J=8,5 Гц, 1H), 4.71-4.74 (m, 4Н), 4.51-4.52 (m, 2Н), 4.07-4.08 (m, 1Н), 3.73 (s, 3Н), 3.56 (s, 1Н), 3.20-3.22 (m, 4Н), 2.56-2.58 (m, 4Н), 1.59 (s, 9Н).
Пример 104а. (S)-2-(6-трет-Бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-фтор-6-(1-метил-5-(5-(2-метил-4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-иламино)-6-оксо-1,6-дигидропиридин-3-ил)бензальдегид 104а
В одногорлую круглодонную колбу емкостью 50 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали 2-бром-6-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-фторбензальдегид 102b (100 мг; 0,24 ммоль), (S)-1-метил-3-(5-(2-метил-4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-иламино)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2(1Н)-он 105f (115 мг; 0,24 ммоль), PdCl2(dppf) (22 мг; 0,03 ммоль), K3PO4 (102 мг; 0,48 ммоль), ацетат натрия (39 мг; 0,48 ммоль), THF (15 мл) и воду (5 мл). После трех циклов вакуумирования/продувания аргоном смесь нагревали при 100°С в течение 2 ч. Затем фильтровали и фильтрат упаривали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 40:1 дихлорметан/метанол, получая соединение 104а в виде желтого твердого вещества (82 мг; 49%). MS: [М+Н]+ 696,3.
Пример 104. (5)-6-трет-Бутил-8-фтор-2-(5-фтор-2-(гидроксиметил)-3-(1-метил-5-(5-(2-метил-4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-иламино)-6-оксо-1,6-дигидропиридин-3-ил)фенил)фталазин-1(2Н)-он 104
Смесь (8)-2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-фтор-6-(1-метил-5-(5-(2-метил-4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-иламино)-6-оксо-1,6-дигидропиридин-3-ил)бензальдегида 104а (80 мг; 0,12 ммоль), NaBH4 (14 мг; 0,36) и метанола (20 мл) перемешивали при 25°С в течение 1 ч. Затем реакционную смесь гасили водой (10 мл) и концентрировали при пониженном давлении. Остаток экстрагировали дихлорметаном (2×30 мл) и объединенный дихлорметановый экстракт концентрировали при пониженном давлении. Остаток очищали, получая соединение 104 (46 мг; 55%) в виде желтого твердого вещества. MS: [М+Н]+ 698,3. 1Н ЯМР (500 МГц, DMSO-d6) δ 8.54 (d, J=2,0 Гц, 1H), 8.52 (d, J=2,0 Гц, 1H), 8.42 (s, 1Н), 7.88 (s, 1Н), 7.86 (d, J=3,0 Гц, 1H), 7.77-7.74 (m, 1Н), 7.40-7.34 (m, 3Н), 7.30 (dd, J=2,5; 9,5 Гц, 1H), 7.23 (d, J=9,0 Гц, 1H), 4.57-4.54 (m, 2Н), 4.49-4.45 (m, 1Н), 4.43-4.40 (m, 1Н), 4.34-4.29 (m, 2Н), 3.70-3.65 (m, 1Н), 3.58 (s, 3Н), 3.40-3.38 (m, 2Н), 3.12-3.06 (m, 1Н), 2.97-2.91 (m, 1Н), 2.36-2.29 (m, 3Н), 2.19-2.15 (m, 1Н), 1.38 (s, 9Н), 0,93 (d, J=6,5 Гц, 3Н).
Пример 105а. (3S)-трет-Бутил-3-метил-4-(6-нитропиридин-3-ил)пиперазин-1-карбоксилат 105а
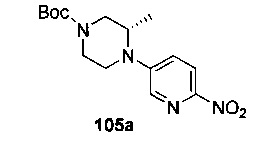
К раствору 5-бром-2-нитропиридина (30 г; 148 ммоль) в DMSO (350 мл) добавляли K2CO3 (14 г; 104 ммоль) и (38)-трет-бутил-3-метилпиперазин-1-карбоксилат (10,0 г; 50 ммоль). Смесь перемешивали при 65°С в течение ночи и затем после охлаждения до комнатной температуры выливали в воду (700 мл). Твердый осадок собирали и сушили под вакуумом. Далее его очищали на колонке для флэш-хроматографии с элюированием смесью 20:1 петролейный эфир/этилацетат и затем CH2Cl2, получая соединение 105а в виде желтого твердого вещества (8,05 г; 50%). LCMS: [М+Н]+ 323.
Пример 105b. (33)-трет-Бутил-4-(6-аминопиридин-3-ил)-3-метил пиперазин-1-карбоксилат 105b
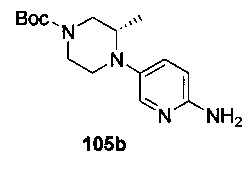
Колбу емкостью 500 мл продували азотом и в нее загружали соединение 105а (5,8 г; 18 ммоль), 10%-ный палладий на угле (50% влаги; 2,0 г) и этанол (200 мл). Колбу вакуумировали, заполняли газообразным водородом и содержимое перемешивали в течение 16 ч при комнатной температуре. Затем водород удаляли в вакууме и заменяли на азот. Катализатор удаляли фильтрованием через набивку CELITE® и фильтрат концентрировали при пониженном давлении, получая соединение 105b в виде коричневого твердого вещества (4,9 г; 96%). LCMS: [М+Н]+ 293.
Пример 105с. (35)-трет-Бутил-4-(6-(5-бром-1-метил-2-оксо-1,2-дигидропиридин-3-иламино)пиридин-3-ил)-3-метилпиперазин-1-карбоксилат 105с
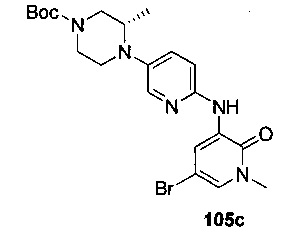
В одногорлую круглодонную колбу емкостью 100 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали 1,4-диоксан (50 мл), соединение 105b (4,0 г; 13,7 ммоль), 3,5-дибром-1-метилпиридин-2(1Н)-он (5,5 г; 20,8 ммоль) и карбонат цезия (11 г; 35 ммоль). После барботирования азота через полученную смесь в течение 30 минут добавляли ксантфос (272 мг; 0,47 ммоль) и трис(дибензилиденацетон)дипалладий(0) (430 мг; 0,47 ммоль) и реакционную смесь нагревали при температуре дефлегмации в течение 3 ч. По прошествии этого времени реакционную смесь охлаждали до комнатной температуры, распределяли между этилацетатом (300 мл) и водой (300 мл) и фильтровали. Водный слой отделяли и экстрагировали этилацетатом (150 мл×2). Органические слои объединяли, промывали рассолом (150 мл) и сушили над сульфатом натрия. Сушильный агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении. Остаток очищали на колонке для флэш-хроматографии с элюированием смесью 50:1 CH2Cl2/метанол, получая соединение 105с в виде желтого твердого вещества (5,4 г; 83%). LCMS: [М+Н]+ 478.
Пример 105d. (3S)-5-Бром-1-метил-3-(5-(2-метилпиперазин-1-ил)пиридин-2-иламино)пиридин-2(1Н)-он 105d
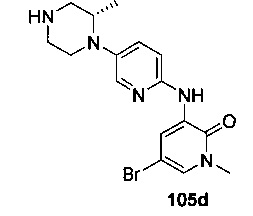
Смесь соединения 105с (3,1 г; 6,5 ммоль) и 4,0 Μ HCl в диоксане (10 мл) перемешивали в течение 5 ч при комнатной температуре. Затем концентрировали в вакууме. Остаток подщелачивали водным 1,0 Μ NaOH и дважды экстрагировали CH2Cl2. Объединенные органические слои промывали водой и концентрировали при пониженном давлении, получая соединение 105d в виде желтого твердого вещества (2,3 г; 95%). LCMS: [М+Н]+ 380.
Пример 105е. (3S)-5-Бром-1-метил-3-(5-(2-метил-4-(оксетан-3-ил)пиперазин-1-ил) пиридин-2-иламино)пиридин-2(1Н)-он 105е
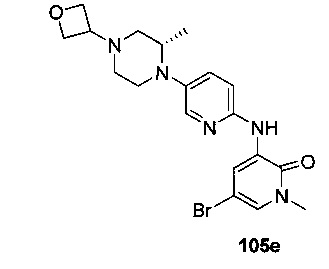
Смесь соединения 105d (2,35 г; 6,2 ммоль), оксетан-3-она (0,49 г; 6,8 ммоль), NaBH3CN (4,75 г; 22,5 ммоль) и хлорида цинка (3 г; 22,7 ммоль) в метаноле (125 мл) перемешивали в течение 5 часов при 50°С. Смесь добавляли к воде и три раза экстрагировали, используя CH2Cl2. Органические слои концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией с элюированием смесью 25:1 CH2Cl2/метанол, получая соединение 105е в виде желтого твердого вещества (2,6 г; 98%). LCMS: [М+Н]+ 434.
Пример 105f. (3S)-1-Метил-3-(5-(2-метил-4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-иламино)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2(1Н)-он 105f
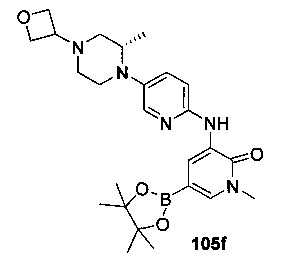
В одногорлую круглодонную колбу емкостью 100 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали соединение 105е (1,0 г; 1,0 экв.; 2,3 ммоль), Pin2B2 (бис(пинаколато)дибор) (1,46 г; 2,50 экв.; 5,75 ммоль), Pd2(dba)3 (105 мг; 0,05 экв.; 0,125 ммоль), X-Phos (2-дициклогексилфосфино-2',4',6'-триизопропилбифенил) (93 мг; 0,1 экв.; 0,23 ммоль), КОАс (676 мг; 3,0 экв.; 6,9 ммоль) и диоксан (50 мл). После трех циклов вакуумирования/продувания аргоном смесь нагревали при 90°С в течение 4 ч. Затем охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток промывали смесью 3:1 петролейный эфир/этилацетат (80 мл), получая соединение 105f в виде желтого твердого вещества (1,0 г; 90%). MS: [М+Н]+ 482.
Пример 105g. (8)-2-(6-трет-Бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-(1-метил-5-(5-(2-метил-4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-иламино)-6-оксо-1,6-дигидропиридин-3-ил)никотинальдегид 105g
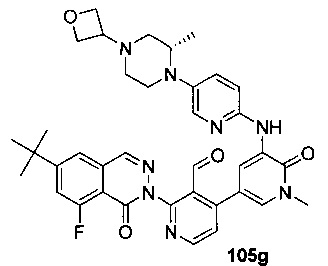
В круглодонную колбу загружали 2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-хлорникотинальдегид 103b (100 мг; 0,28 ммоль), соединение 105f (135 мг; 0,28 ммоль), PdCl2(dppf) (25 мг; 0,03 ммоль), K3PO4⋅3H2O (149 мг; 0,56 ммоль), THF (10 мл) и Н2O (5 мл). После трех циклов вакуумирования/продувания аргоном смесь нагревали при 70°С в течение 6 ч. Затем фильтровали и фильтрат упаривали в вакууме. Остаток очищали колоночной флэш-хроматографией с элюированием смесью 1:3 петролейный эфир/этилацетат, получая соединение 105д в виде желтого твердого вещества (113 мг; 60%). LCMS: [М+Н]+ 679.
Пример 105. (5)-6-трет-Бутил-8-фтор-2-(3-(гидроксиметил)-4-(1-метил-5-(3-метил-5-(4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-иламино)-6-оксо-1,6-дигидропиридин-3-ил)пиридин-2-ил)фталазин-1(2Н)-он 105
Смесь соединения 105g (100 мг; 0,15 ммоль), NaBH4 (17 мг; 0,45 ммоль) и СН3ОН (10 мл) перемешивали при 25°С в течение 1 ч. Смесь экстрагировали, используя CH2Cl2 (10 мл×2). Объединенные CH2Cl2-экстракты концентрировали при пониженном давлении. Остаток очищали препаративной обращенно-фазовой HPLC, получая соединение 105 (65 мг; 65%). LCMS: [М+Н]+ 681. 1Н ЯМР (500 МГц, DMSO-d6) δ 8.62 (d, J=2,0 Гц, 1H), 8.53-8.57 (m, 1Н), 8.42 (s, 2Н), 7.90 (d, J=1,5 Гц, 1H), 7.85 (d, J=2,0 Гц, 1H), 7.76-7.79 (m, 1Н), 7.52 (d, J=5,0 Гц, 1H), 7.46 (d, J=2,0 Гц, 1H), 7.36-7.38 (m, 1Н), 7.24-7.26 (m, 1Н), 4.54-4.57 (m, 2Н), 4.46-4.48 (m, 1Н), 4.40-4.41 (m, 3Н), 3.68-3.70 (m, 1Н), 3.57 (s, 3Н), 3.37-3.40 (m, 1Н), 3.09-3.11 (m, 1Н), 2.93-2.95 (m, 1Н), 2.52-2.54 (m, 2Н), 2.32-2.36 (m, 2Н), 2.18 (s, 1Н), 1.39 (s, 9Н), 0,93 (d, J=6,0 Гц, 3Н).
Пример 106а. (R)-1-Метил-3-(5-(2-метил-4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-иламино)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2(1Н)-он 106а
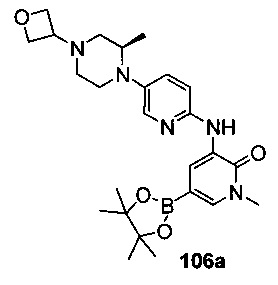
К раствору (R)-5-бром-1-метил-3-(5-(2-метил-4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-иламино)пиридин-2(1Н)-она (2,0 г; 4,60 ммоль), 4,4,4',4',5,5,5',5'-окта-метил-2,2'-би(1,3,2-диоксаборолана) (3,50 г; 13,80 ммоль) в диоксане (50 мл) добавляли PdCl2(dppf) (377,10 мг; 0,46 ммоль) и KOAc (2,70 г; 27,80 ммоль). См. Фиг. 6. Смесь перемешивали при 100°С в течение 12 ч в атмосфере аргона. Смесь фильтровали и фильтрат упаривали в вакууме. Остаток очищали колоночной хроматографией с элюированием смесью CH2Cl2/метанол (15:1), получая соединение 106а (1,10 г; 49%) в виде коричневого твердого вещества. MS: [М+Н]+ 482,3.
Пример 106b. (R)-2-(6-трет-Бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-(1-метил-5-(5-(2-метил-4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-иламино)-6-оксо-1,6-дигидропиридин-3-ил)никотинальдегид 106b
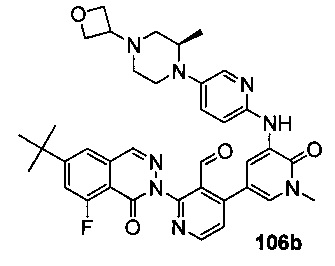
Смесь соединения 106а (260,0 мг; 0,56 ммоль), 2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-хлорникотинальдегида 103b (200,0 мг; 0,56 ммоль), PdCl2(dppf) (50,0 мг; 0,056 ммоль), NaOAc (90,0 мг; 1,1 ммоль), K3PO4 (300 мг; 1,1 ммоль) в ацетонитриле (30 мл) нагревали при 100°С в течение 3 ч. Растворитель выпаривали в вакууме. Остаток очищали колоночной флэш-хроматографией с элюированием смесью CH2Cl2/метанол (10:1), получая соединение 106b (120 мг; 34%) в виде коричневого твердого вещества. LCMS: [М+Н]+ 679,3.
Пример 106. (R)-6-трет-Бутил-8-фтор-2-(3-(гидроксиметил)-4-(1-метил-5-(5-(2-метил-4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-иламино)-6-оксо-1,6-дигидропиридин-3-ил)пиридин-2-ил)фталазин-1(2Н)-он 106
К раствору соединения 106b (90,0 мг; 0,13 ммоль) в МеОН (10 мл) добавляли NaBH4 (15,0 мг; 0,39 ммоль). Смесь перемешивали при 20°С в течение 2 ч. Затем упаривали и остаток очищали препаративной обращенно-фазовой HPLC, получая соединение 106 (7,3 мг; 8%) в виде белого твердого вещества. LCMS: (М+Н)+ 681,4. 1Н ЯМР (500 МГц, DMSO) δ 8.63 (d, J=2,5 Гц, 1Н), 8.57 (d, J=5,0 Гц, 1Н), 8.54 (d, J=2,0 Гц, 1Н), 8.47 (s, 1Н), 7.91 (s, 1Н), 7.86 (d, J=3,0 Гц, 1Н), 7.78 (d, J=12,5 Гц, 1Н), 7.53 (d, J=5,0 Гц, 1Н), 7.47 (d, J=2,0 Гц, 1Н), 7.36-7.39 (m, 1Н), 7.25 (d, J=9,5 Гц, 1Н), 4.90 (s, 1Н), 4.54-4.58 (m, 2Н), 4.47 (t, J=6,0 Гц, 1Н), 4.39-4.43 (m, 3Н), 3.69 (s, 1Н), 3.61 (s, 3Н), 3.37-3.42 (m, 1Н), 3.09-3.11 (m, 1Н), 2.95 (t, J=9,0 Гц, 1Н), 2.54-2.56 (m, 1Н), 2.30-2.37 (m, 2Н), 2.19 (t, J=8,0 Гц, 1Н), 1.39 (s, 9Н), 0,94-0.93 (d, J=6,5 Гц, 3Н).
Пример 107а. (R)-2-(6-трет-Бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-фтор-6-(1-метил-5-(5-(2-метил-4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-иламино)-6-оксо-1,6-дигидро-пиридин-3-ил)бензальдегид 107а
В одногорлую круглодонную колбу емкостью 50 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали (R)-1-метил-3-(5-(2-метил-4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-иламино)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2(1Н)-он (173 мг; 1,0 экв.; 0,36 ммоль), 2-бром-6-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-фторбензальдегид 101i (152 мг; 1,0 экв.; 0,36 ммоль), K3PO4 (152 мг; 2,0 экв.; 0,72 ммоль), PdCl2(dppf) (26 мг; 0,1 экв.; 0,036 ммоль), NaOAc (59 мг; 2,0 экв.; 0,72 ммоль), CH3CN (20 мл) и Н2O (0,8 мл). После трех циклов вакуумирования/продувания аргоном смесь нагревали при 80°С в течение 2 ч. Затем охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 40:1 DCM/EtOH, получая соединение 107а в виде желтого твердого вещества (77 мг; 31%). MS: [М+Н]+ 696,3.
Пример 107. (R)-6-трет-Бутил-8-фтор-2-(5-фтор-2-(гидроксиметил)-3-(1-метил-5-(5-(2-метил-4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-иламино)-6-оксо-1,6-дигидропиридин-3-ил)фенил)фталазин-1(2Н)-он 107
В одногорлую круглодонную колбу емкостью 50 мл, оборудованную магнитной мешалкой, загружали соединение 107а (77 мг; 1,0 экв.; 0,11 ммоль), NaBH4 (21 мг; 5,0 экв.; 0,55 ммоль) и МеОН (10 мл). Смесь перемешивали при комнатной температуре в течение 1 ч. Затем фильтровали и фильтрат концентрировали. Остаток очищали препаративной обращенно-фазовой HPLC, получая соединение 107 (38 мг; 49%). MS: [М+Н]+ 698,3. 1Н ЯМР (500 МГц, CDCl3) δ 8.59 (d, J=2,0 Гц, 1Η), 8.29 (d, J=2,5 Гц, 1Η), 7.98(d, J=2,5 Гц, 1Η), 7.81 (s, 1Η), 7.56-7.54 (m, 2Η), 7.40 (s, 1Н), 7.31-7.27 (m, 2Н), 7.11-7.10 (m, 1Н), 6.81 (d, J=9,0 Гц, 1H), 4.71-4.62 (m, 4Н), 4.35 (s, 2Н), 3.73-3.70 (m, 4Н), 3.53-3.46 (m, 2Н), 3.08 (t, J=5,0 Гц, 2H), 2.56-2.46 (m, 3Н), 2.22-2.18 (m, 1Н), 1.43 (s, 9Н), 0,98 (d, J=6,5 Гц, 3Н).
Пример 108а. 2-(6-трет-Бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-6-(5-(5-((2S,5R)-2,5-диметил-4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-иламино)-1-метил-6-оксо-1,6-дигидропиридин-3-ил)-4-фторбензальдегид 108а
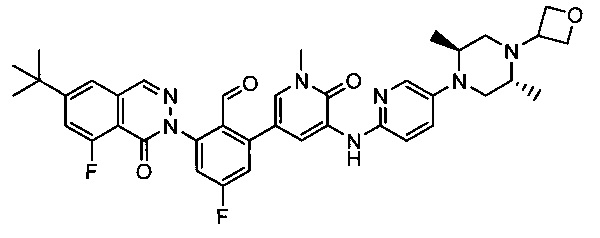
В одногорлую круглодонную колбу емкостью 25 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали 2-бром-6-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-фторбензальдегид 102b (84 мг; 0,20 ммоль), 3-(5-((2S,5R)-2,5-диметил-4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-иламино)-1-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2(1Н)-он (99 мг; 0,20 ммоль), Pd(dppf)Cl2 (15 мг; 0,02 ммоль), K3PO4 (127 мг; 0,6 ммоль), ацетат натрия (49 мг; 0,6 ммоль), ацетонитрил (5 мл) и воду (0,5 мл). После трех циклов вакуумирования/продувания аргоном смесь нагревали при температуре дефлегмации в течение 2 ч. Затем охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 30:1 дихлорметан/метанол, получая соединение 108а в виде белого твердого вещества (54 мг; 38%).
MS: [М+Н]+ 710,3.
Пример 108. 6-трет-Бутил-2-(3-(5-(5-((25,5Я)-2,5-диметил-4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-иламино)-1-метил-6-оксо-1,6-дигидропиридин-3-ил)-5-фтор-2-(гидроксиметил)фенил)-8-фторфталазин-1(2Н)-он 108
При 0°С к раствору 2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-6-(5-(5-((2S,5R)-2,5-диметил-4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-иламино)-1-метил-6-оксо-1,6-дигидропиридин-3-ил)-4-фторбензальдегида 108а (54 мг; 0,076 ммоль) в метаноле (5 мл) добавляли боргидрид натрия (9,0 мг; 0,23 ммоль). Смесь перемешивали в течение 60 минут. Затем гасили водой (1,0 мл) и концентрировали при пониженном давлении. Остаток очищали препаративной обращенно-фазовой HPLC, получая соединение 108 (12 мг; 22%). MS: [М+Н]+ 712,3. 1Н ЯМР (500 МГц, CDCl3) δ 8.63 (d, J=2,5 Гц, 1H), 8.31 (d, J=2,0 Гц, 1H), 8.05 (d, J=2,5 Гц, 1H), 7.87 (s, 1Н), 7.58-7.55 (m, 2Н), 7.44 (d, J=2,0 Гц, 1H), 7.36 (s, 1Н), 7.30 (d, J=2,0 Гц, 1H), 7.12 (dd, J=2,5; 8,5 Гц, 1H), 6.82 (d, J=8,5 Гц, 1H), 4.76-4.73 (m, 2Н), 4.68-4.61 (m, 2Н), 4.38-4.36 (m, 2Н), 3.74-3.72 (m, 2Н), 3.72 (s, 3Н), 3.22-3.20 (m, 1Н), 2.94-2.92 (m, 1Н), 2.74-2.72 (m, 2Н), 2.50-2.48 (m, 1Н), 1.98-1.96 (m, 1Η), 1.45 (s, 9Η), 0,93-0.91 (m, 6Н).
Пример 109а. 3-Бром-5-(6-трет-бутил-8-фтор-1-оксо-1,2-дигидро-фталазин-2-ил)пиридин-4-карбальдегид 109а

В герметично закрываемую пробирку, оборудованную магнитной мешалкой, загружали 6-трет-бутил-8-фтор-1,2-дигидрофталазин-1-он 101 h (220 мг; 1,0 ммоль), 3,5-дибромпиридин-4-карбальдегид (530 мг; 2,0 ммоль), CuI (190 мг; 1,0 ммоль), 4,7-диметокси-1,10-батофенантролин (244 мг; 1,0 ммоль), Cs2CO3 (652 мг; 2,0 ммоль) и диоксан (8 мл). После трех циклов вакуумирования/продувания аргоном смесь нагревали при 110°С в течение 5 ч. Затем фильтровали и фильтрат упаривали в вакууме. Остаток очищали колоночной хроматографией на силикагеле с элюированием смесью этилацетат/петролейный эфир (1:3; об./об.), получая соединение 109а (118 мг; 29,3%) в виде твердого вещества. LCMS: [М+Н]+ 406.
Пример 109b. 3-(6-трет-Бутил-8-фтор-1-оксо-1,2-дигидрофталазин-2-ил)-5-[1-метил-5- ({5-[(2S)-2-метил-4-(оксетан-3-ил)пиперазин-1-ил]пиридин-2-ил}амино)-6-оксо-1,6-дигидро-пиридин-3-ил]пиридин-4-карбальдегид 109b
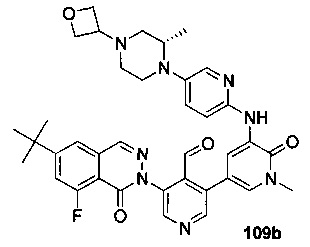
В одногорлую круглодонную колбу емкостью 25 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали соединение 109а (118 мг; 0,29 ммоль), 1-метил-3-({5-[(28)-2-метил-4-(оксетан-3-ил)пиперазин-1-ил]пиридин-2-ил}амино)-5-(тетраметил-1,3,2-диоксаборолан-2-ил)-1,2-дигидропиридин-2-он 105f (140 мг; 0,29 ммоль), Pd(dppf)Cl2 (24,8 мг; 0,03 ммоль), KOAc (58,9 мг; 0,60 ммоль), K3PO4⋅3H2O (159,8 мг; 0,60 ммоль), ацетонитрил (6 мл) и воду (3 капли). После трех циклов вакуумирования/продувания аргоном смесь нагревали при 110°С в течение 2 ч. Затем реакционную смесь охлаждали до комнатной температуры, фильтровали и фильтрат упаривали в вакууме. Остаток очищали препаративной TLC с использованием в качестве подвижной фазы смеси дихлорметан/метанол (20:1; об./об.), получая соединение 109b (95 мг; 36%) в виде красного твердого вещества. MS: [М+Н]+ 679.
Пример 109. (S)-6-трет-Бутил-8-фтор-2-(4-(гидроксиметил)-5-(1-метил-5-(5-(2-метил-4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-иламино)-6-оксо-1,6-дигидропиридин-3-ил)пиридин-3-ил)фталазин-1(2Н)-он 109
К суспензии соединения 109b (95 мг; 0,106 ммоль) при 0°С в метаноле (5 мл) добавляли боргидрид натрия (24 мг; 0,636 ммоль) и смесь перемешивали в течение 30 минут. Затем реакционную смесь гасили водой (1,0 мл) и концентрировали. Остаток очищали препаративной обращенно-фазовой HPLC, получая соединение 109 (13,5 мг; 18,7%). LCMS: [М+Н]+ 681. 1Н ЯМР (500 МГц, CDCl3) δ 8.76 (s, 1Н), 8.65 (d, J=2,0 Гц, 1Н), 8.61 (s, 1Н), 8.32 (d, J=2,5 Гц, 1Н), 7.96 (d, J=3,0 Гц, 1Н), 7.82 (s, 1Н), 7.56-7.58 (m, 2Н), 7.40 (d, J=2,0 Гц, 1Н), 7.28-7.31 (m, 1Н), 6.81 (d, J=9,0 Гц, 1Н), 4.61-4.70 (m, 4Н), 4.43 (s, 2Н), 3.95 (s, 1Н), 3.72 (s, 3Н), 3.45-3.53 (m, 2Н), 3.07 (t, J=5,25 Гц, 2Н), 2.43-2.56 (m, 3Н), 2.18-2.22 (m, 1Н), 1.43 (s, 9Н), 0,99 (d, J=6,5 Гц, 3Н).
Пример 110а. 2-(6-трет-Бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-(5-(5-((2S,5R)-2,5-диметил-4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-иламино)-1-метил-6-оксо-1,6-дигидропиридин-3-ил)никотинальдегид 110а
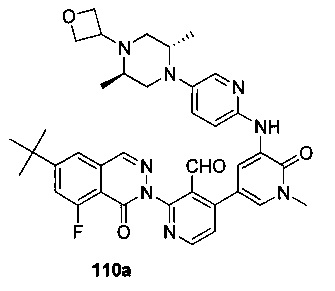
В одногорлую круглодонную колбу емкостью 100 мл загружали 2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-хлорникотинальдегид 10b (216 мг; 0,6 ммоль), 3-(5-((2S,5R)-2,5-диметил-4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-иламино)-1-метил-5-(4,4,5-триметил-1,3,2-диоксаборолан-2-ил)пиридин-2(1Н)-он (360 мг; 0,72 ммоль), Pd(dppf)Cl2 (30 мг, 0,03 ммоль), K3PO4 (270 мг; 1,2 ммоль) и NaOAc-3H2O (180 мг; 1,2 ммоль) в CH3CN (80 мл). Систему вакуумировали и заполняли N2. Реакционную смесь нагревали при 100°С в течение 2 ч. Затем охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали колоночной флэш-хроматографией с элюированием смесью 25:1 CH2Cl2/MeOH, получая соединение 110а (160 мг; 42%) в виде желто-коричневого твердого вещества. MS: [М+Н]+693,3.
Пример 110. 6-трет-Бутил-2-(4-(5-(5-((2S,5R)-2,5-диметил-4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-иламино)-1-метил-6-оксо-1,6-дигидропиридин-3-ил)-3-(гидроксиметил)пиридин-2-ил)-8-фторфталазин-1(2Н)-он 110
Смесь соединения 110а (100 мг; 0,15 ммоль) и NaBH4 (20 мг; 0,45 ммоль) в МеОН (30 мл) перемешивали при 30°С в течение 2 ч. Смесь гасили водой и экстрагировали EtOAC (10 мл×3). Объединенный EtOAc-экстракт концентрировали при пониженном давлении и остаток очищали препаративной обращенно-фазовой HPLC, получая соединение 110 (80 мг; 80%). MS: [М+Н]+ 695,3. 1Н ЯМР (500 МГц, CDCl3) δ 8.72 (d, J=2 Гц, 1H), 8.67 (d, J=5 Гц, 1H), 8.35 (d, J=2,5 Гц, 1H), 8.05 (d, J=3 Гц, 1H), 7.88 (s, 1Н), 7.68 (d, J=2,5 Гц, 1H),7.58-7.55 (m, 3Н), 7.38-7.36 (m, 1Н), 6.82 (d, J=8,5 Гц, 1H), 4.78-4.71 (m, 2Н), 4.67-4.61 (m, 2Н), 4.50 (s, 2Н), 4.07 (t, J=6 Гц, 1H), 3.78-3.74 (m, 4Н), 3.22-3.20 (m, 1Н), 2.92 (d, J=3 Гц, 1H), 2.77-2.71 (m, 2Н), 2.51-2.48 (m, 1Н), 1.20-1.95 (m, 1Н), 1.45 (s, 9Н), 0,93-0.90 (m, 6Н).
Пример 111а. (S)-трет-Бутил-3-этил-4-(6-нитропиридин-3-ил)пиперазин-1-карбоксилат 111а
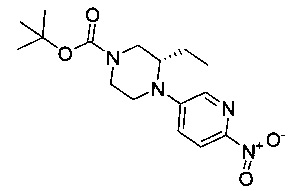
В одногорлую круглодонную колбу емкостью 250 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали 1,4-диоксан (50 мл), 5-бром-2-нитропиридин (2,02 г; 10 ммоль), (5)-трет-бутил-3-этилпиперазин-1-карбоксилат (2,14 г; 10,0 ммоль), Pd2(dba)3 (458 мг; 0,50 ммоль), ксантфос (576 мг; 1,0 ммоль) и карбонат цезия (6,52 г; 20 ммоль). После трех циклов вакуумирования/продувания аргоном смесь нагревали при 100°С в течение ночи. По прошествии этого времени реакционную смесь охлаждали до комнатной температуры. Затем фильтровали и фильтрат упаривали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 3:1 петролейный эфир/этилацетат, получая соединение 111а (700 мг; 22%) в виде желтого твердого вещества. MS: [М+Н]+ 336.
Пример 111b. (S)-трет-Бутил-4-(6-аминопиридин-3-ил)-3-этилпиперазин-1-карбоксилат 111b
Одногорлую круглодонную колбу емкостью 100 мл продували азотом и в нее загружали соединение 111а (0,7 г; 2,08 ммоль), 10%-ный палладий на угле (50% влаги, 208 мг) и метанол (40 мл). Колбу со смесью вакуумировали, заполняли газообразным водородом и содержимое перемешивали при комнатной температуре в течение 6 ч. Затем водород удаляли и колбу заполняли азотом. Катализатор удаляли фильтрованием через набивку CELITE® и фильтрат концентрировали при пониженном давлении, получая соединение 111b (568 мг; 89%). MS: [М+Н]+ 306.
Пример 111с. (S)-трет-Бутил-4-(6-(5-бром-1-метил-2-оксо-1,2-дигидро-пиридин-3-иламино) пиридин-3-ил)-3-этилпиперазин-1-карбоксилат 111с
В одногорлую круглодонную колбу емкостью 100 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали 1,4-диоксан (50 мл), соединение 111b (568 мг; 1,86 ммоль), 3,5-дибром-1-метилпиридин-2(1Н)-он (498 мг; 1,86 ммоль), Pd2(dba)3 (85 мг; 0,093 ммоль), ксантфос (107 мг; 0,186 ммоль) и карбонат цезия (1,198 г; 3,72 ммоль). После трех циклов вакуумирования/продувания аргоном смесь нагревали при 100°С в течение 6 ч. Затем фильтровали и фильтрат упаривали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 100:1 дихлорметан/метанол, получая соединение 111с (502 мг; 55%) в виде желтого твердого вещества. MS: [М+Н]+ 492.
Пример 111d. (3)-5-Бром-3-(5-(2-этилпиперазин-1-ил)пиридин-2-иламино)-1-метил пиридин-2(1Н)-он 111d
Смесь соединения 111с (502 мг; 1,02 ммоль), дихлорметана (2 мл) и 4,0 Μ HCl в диоксане (4 мл) перемешивали при комнатной температуре в течение 5 ч. Затем концентрировали при пониженном давлении, получая неочищенное соединение 111d в виде желтого твердого вещества (263 мг; 66%), которое использовали на следующей стадии без дополнительной очистки. MS: [М+Н]+ 392.
Пример 111е. (S)-5-Бром-3-(5-(2-этил-4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-иламино)-1-метилпиридин-2(1Н)-он 111е
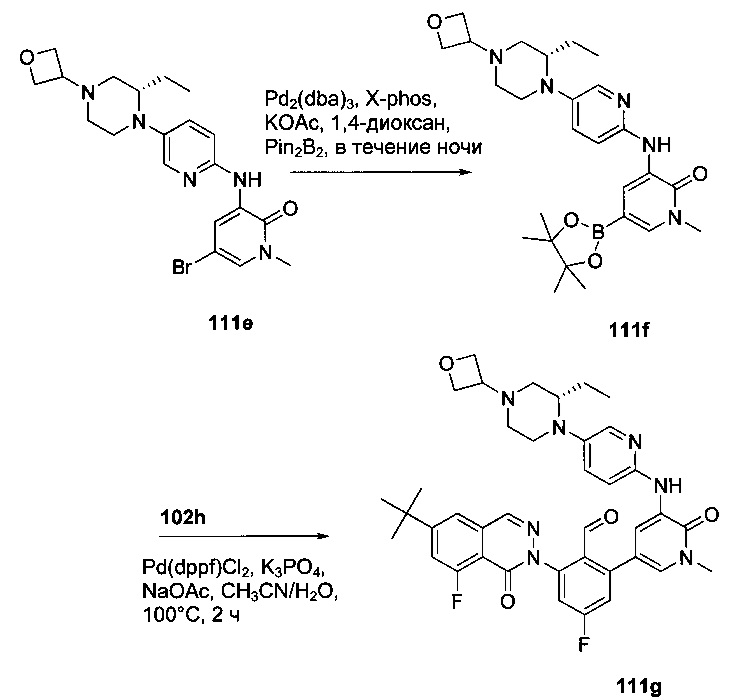
Смесь соединения 111d (263 мг; 0,67 ммоль), оксетан-3-она (96 мг; 1,34 ммоль), NaBH3CN (104 мг; 1,68 ммоль) и хлорида цинка (227 мг; 1,68 ммоль) в метаноле (10 мл) перемешивали при 50°С в течение 5 часов. Затем к реакционной смеси добавляли воду (10 мл). Полученную смесь концентрировали при пониженном давлении. Остаток три раза экстрагировали дихлорметаном. Объединенный органический слой концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 50:1 дихлорметан/метанол, получая соединение 111е (203 мг; 68%). MS: [М+Н]+ 448.
Пример 111f. (S)-3-(5-(2-Этил-4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-иламино)-1-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2(1Н)-он 111f
В одногорлую круглодонную колбу емкостью 100 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали соединение 111е (3219 мг; 7,20 ммоль), Pin2B2 (9072 мг; 36,0 ммоль), Pd2(dba)3 (329 мг; 0,36 ммоль), X-phos (302 мг; 0,72 ммоль), ацетат калия (2117 мг; 21,6 ммоль) и диоксан (50 мл). После трех циклов вакуумирования/продувания аргоном смесь нагревали при 60°С в течение 16 ч. Затем охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток промывали смесью 8:1 петролейный эфир/этилацетат (80 мл), получая соединение 111f в виде желтого твердого вещества (3,0 г; 84%).
Пример 111g. (S)-2-(6-трет-Бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-6-(5-(5-(2-этил-4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-иламино)-1-метил-6-оксо-1,6-дигидропиридин-3-ил)-4-фторбензальдегид 111g
В одногорлую круглодонную колбу емкостью 25 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали 2-бром-6-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-фторбензальдегид 102h (168 мг; 0,40 ммоль), соединение 111f (198 мг; 0,40 ммоль), Pd(dppf)Cl2 (15 мг; 0,02 ммоль), K3PO4 (170 мг; 0,8 ммоль), ацетат натрия (66 мг; 0,8 ммоль), ацетонитрил (5 мл) и воду (0,8 мл). После трех циклов вакуумирования/продувания аргоном смесь нагревали при температуре дефлегмации в течение 2 ч. Затем охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 30:1 дихлорметан/метанол, получая соединение 111g в виде белого твердого вещества (100 мг; 35%). MS: [М+Н]+ 710,3.
Пример 111. (S)-6-трет-Бутил-2-(3-(5-(5-(2-этил-4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-ил-амино)-1-метил-6-оксо-1,6-дигидропиридин-3-ил)-5-фтор-2-(гидроксиметил)фенил)-8-фторфталазин-1(2Н)-он 111
К раствору соединения 111g (100 мг; 0,14 ммоль) при 0°С в метаноле (5 мл) добавляли боргидрид натрия (16,0 мг; 0,42 ммоль). Смесь перемешивали в течение 60 минут. Затем ее гасили водой (1,0 мл) и концентрировали. Остаток очищали препаративной обращенно-фазовой HPLC, получая соединение 111 (32 мг; 32%). MS: [М+Н]+ 712,3. 1Н ЯМР (500 МГц, CDCl3) δ 8.57 (d, J=2,0 Гц, 1Н), 8.30 (d, J=2,0 Гц, 1Н), 7.94 (s, 1Н), 7.80 (s, 1Н), 7.58 (s, 2Н), 7.41 (d, J=2,0 Гц, 1Н), 7.31-7.28 (m, 2Н), 7.12 (dd, J=2,5; 8,5 Гц, 1Н), 6.82 (d, J=9,0 Гц, 1Н), 4.73-4.70 (m, 4Н), 4.37 (s, 2Н), 3.74-3.72 (m, 1Н), 3.71 (s, 3Н), 3.56-3.54 (m, 1Н), 3.34-3.32 (m, 1Н), 3.15-3.13 (m, 2Н), 2.58-2.36 (m, 4Н), 1.45 (s, 9Н), 1.44-1.43 (m, 2Н), 0,83 (t, J=7,5 Гц, 3Н).
Пример 112а. (S)-2-(6-трет-Бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-(5-(5-(2-этил-4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-иламино)-1-метил-6-оксо-1,6-дигидропиридин-3-ил)никотинальдегид 112а
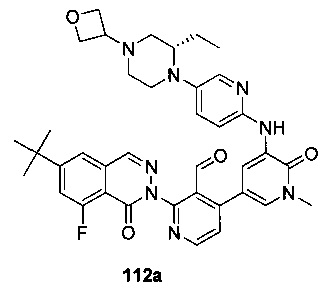
В круглодонную колбу емкостью 50 мл загружали 2-(6-трет-бутил-8-фтор-1-оксо-фталазин-2(1Н)-ил)-4-хлорникотинальдегид 103b (150 мг; 0,43 ммоль), (S)-3-(5-(2-этил-4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-иламино)-1-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2(1Н)-он (206 мг; 0,43 ммоль), PdCl2(dppf) (33 мг; 0,04 ммоль), K3PO4 (202 мг; 0,86 ммоль), NaOAc (71 мг; 0,86 ммоль), CH3CN (10 мл) и Н2O (2 мл). После трех циклов вакуумирования/продувания аргоном смесь нагревали при 100°С в течение 3 ч. Затем фильтровали и фильтрат упаривали в вакууме. Остаток очищали колоночной флэш-хроматографией с элюированием смесью 1:3 петролейный эфир/этилацетат, получая соединение 112а в виде желтого твердого вещества (146 мг; 49%). LCMS: [М+Н]+ 693.
Пример 112. (S)-6-трет-Бутил-2-(4-(5-(5-(2-этил-4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-иламино)-1-метил-6-оксо-1,6-дигидропиридин-3-ил)-3-(гидроксиметил)пиридин-2-ил)-8-фторфталазин-1(2Н)-он 112
Смесь соединения 112а (140 мг; 0,20 ммоль), NaBH4 (21 мг; 0,60) и СН3ОН (8 мл) перемешивали при 25°С в течение 1 ч. Затем реакционную смесь гасили водой (10 мл), смесь экстрагировали, используя CH2Cl2 (15 мл×2). Объединенный CH2Cl2-экстракт концентрировали при пониженном давлении. Остаток очищали обращенно-фазовой препаративной HPLC, получая соединение 112 (70 мг; 50%). LCMS: [М+Н]+ 695. 1Н ЯМР (500 МГц, DMSO) δ 8.50-8.60 (m, 2Н), 8.44 (s, 1Н), 7.90 (d, J=1,5 Гц, 1Н), 7.82 (d, J=3,0 Гц, 1Н), 7.75-7.78 (m, 1Н), 7.51-7.53 (m, 1Н), 7.45 (d, J=2,5 Гц, 1Н), 7.33-7.35 (m, 1Н), 7.22-7.24 (m, 1Н), 4.47-4.59 (m, 2Н), 4.36-4.45 (m, 4Н), 3.60 (s, 3Н), 3.46-3.50 (m, 1Н), 3.38-3.41 (m, 3Н), 3.14-3.17 (m, 1Н), 2.96-3.00 (m, 1Н), 2.60-2.64 (m, 1Н), 2.50-2.55 (m, 1Н), 2.14-2.17 (m, 1Н), 2.06-2.10 (m, 1Н), 1.66-1.69 (m, 1Н), 1.39 (s, 9Н), 1.21-1.28 (m, 1Н), 0,77-0.80 (m, 3Н).
Пример 113а. 5-Бром-1-метил-3-(пиримидин-4-иламино)пиридин-2(1Н)-он 113а
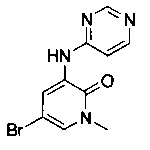
В одногорлую круглодонную колбу емкостью 100 мл, оборудованную магнитной мешалкой и входным отверстием для азота, загружали 3,5-дибром-1-метилпиридин-2(1Н)-он (2,00 г; 21,0 ммоль), 2-аминопиримидин (5,61 г; 21,0 ммоль), карбонат цезия (13,7 г; 42,1 ммоль), DMF (5 мл) и 1,4-диоксан (70 мл). После барботирования азота через полученную суспензию в течение 30 мин добавляли ксантфос (1,10 г; 1,89 ммоль) и трис(дибензилиденацетон)-дипалладий(0) (963 мг; 1,05 ммоль). К колбе подсоединяли обратный холодильник и реакционную смесь нагревали при 100°С в течение 4 ч. По прошествии этого времени смесь охлаждали до комнатной температуры и разбавляли смесью 90:10 метиленхлорид/метанол (150 мл) и водой (100 мл) и проводили разделение слоев. Водный слой экстрагировали смесью 90:10 метиленхлорид/метанол (50 мл) и объединенные органические слои промывали рассолом и сушили над сульфатом натрия. Сушильный агент удаляли фильтрованием. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали колоночной флэш-хроматографией (диоксид кремния, 90:10 метиленхлорид/метанол), получая соединение 113а с выходом 58% (3,42 г) в виде аморфного светло-зеленого твердого вещества: т.пл. 217-219°С; 1Н ЯМР (500 МГц, CDCl3) δ 9.29 (s, 1Н), 8.77 (s, 1Н), 8.72 (d, J=2,5 Гц, 1H), 8.36 (d, J=6,0 Гц, 1Η), 7.69 (d, J=2,5 Гц, 1Η), 7.37 (dd, J=5,5; 1,0 Гц, 1Η), 3.53 (s, ЗН); MS (ESI+) m/z 281,0 (M+H).
Пример 113b. 1-Метил-3-(пиримидин-4-иламино)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2(1Н)-он 113b
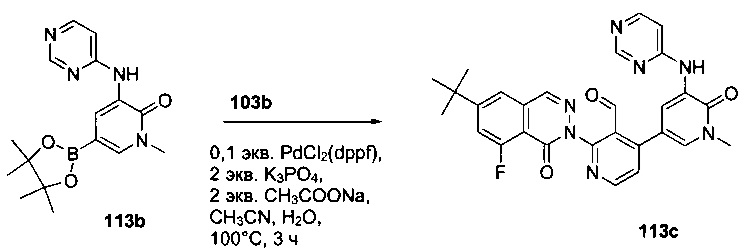
В одногорлую круглодонную колбу емкостью 250 мл, оборудованную магнитной мешалкой и холодильником, загружали соединение 113а (4,0 г; 14 ммоль), X-phos (400 мг; 0,7 ммоль), Pd2(dba)3 (635 мг; 0,7 ммоль), ацетат калия (7,3 мг; 28 ммоль), 4,4,4',4',5,5,5',5,-октаметил-2,2'-би(1,3,2-диоксаборолан) (10,6 г; 42 ммоль) и 1,4-диоксан (100 мл). После трех циклов вакуумирования/продувания аргоном реакционную смесь нагревали при 60°С в течение 8 ч. Затем охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали колоночной флэш-хроматографией с элюированием смесью 5:1 петролейный эфир/этилацетат, получая соединение 113b в виде бледно-желтого твердого вещества (3,8 мг; 82%). MS: [М+Н]+ 329,5.
Пример 113с. 2-(6-трет-Бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-(1-метил-6-оксо-5-(пиримидин-4-иламино)-1,6-дигидропиридин-3-ил)никотинальдегид 113
В круглодонную колбу емкостью 100 мл, оборудованную обратным холодильником, загружали 2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-хлорникотинальдегид 103b (110 мг; 0,31 ммоль), соединение 113b (100 мг; 0,31 ммоль), PdCl2 (dppf) (25 мг; 0,030 ммоль), K3PO4 (241 мг; 0,93 ммоль), NaOAc (76 мг; 0,93 ммоль), ацетонитрил (15 мл) и воду (0,5 мл). После трех циклов вакуумирования/продувания аргоном смесь нагревали при 100°С в течение 3 ч. Затем фильтровали и фильтрат упаривали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 1:20 метанол/дихлорметан, получая соединение 113с в виде красного твердого вещества (80 мг; 51%). MS-ESI: [М+Н]+ 526,3.
Пример 113. 6-трет-Бутил-8-фтор-2-[3-(гидроксиметил)-4-[1-метил-6-оксо-5-(пиримидин-4-иламино)-3-пиридил]-2-пиридил]фталазин-1-он 113
Смесь соединения 113с (80 мг; 0,14 ммоль), NaBH4 (27 мг; 0,70 ммоль) и метанола (10 мл) перемешивали при комнатной температуре в течение 0,5 ч. Смесь гасили водой (2 мл) и концентрировали при пониженном давлении. Остаток очищали обращенно-фазовой препаративной HPLC, получая соединение 113 (57 мг; 71%). MS-ESI: [М+Н]+ 528,3. 1Н ЯМР (500 МГц, DMSO-d6) δ 9.26 (s, 1Н), 8.76 (d, J=2,5 Гц, 1Η), 8.67 (s, 1Η), 8.59 (d, J=5,0 Гц, 1Η), 8.54 (d, J=2,5 Гц, 1Η), 8.31 (d, J=6,0 Гц, 1Η), 7.90 (d, J=1,5 Гц, 1Η), 7.79-7.76 (m, 1Η), 7.69 (d, J=2,0 Гц, 1H), 7.54 (d, J=5,0 Гц, 1H), 7.34-7.32 (m, 1Н), 4.94 (t, J=5,0 Гц, 1H), 4.42-4.40 (m, 2Н), 3.62 (s, 3Н), 1.39 (s, 9Н).
Пример 114а. 5-Бром-1-метил-3-(2-метилпиримидин-4-иламино)пиридин-2(1Н)-он 114а
Следуя методике из примера 113а и исходя из 2-метилпиримидин-4-амина (2,0 г; 18,3 ммоль) и 3,5-дибром-1-метилпиридин-2(1Н)-она (9,6 г; 36 ммоль), получали соединение 114а в виде желтого твердого вещества (2,3 г; 43,4%). MS: [М+Н]+ 295. 1Н ЯМР (500 МГц, DMSO-d6) δ 9.20 (s, 1Н), 8.78 (s, 1Н), 8.26 (d, J=4,5 Гц, 1H), 7.68 (s, 1Н), 7.18 (d, J=4,5 Гц, 1H), 3.59 (s, 3Н), 2.52 (s, 3Н).
Пример 114b. 1-Метил-3-(2-метилпиримидин-4-иламино)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2(1Н)-он 114b
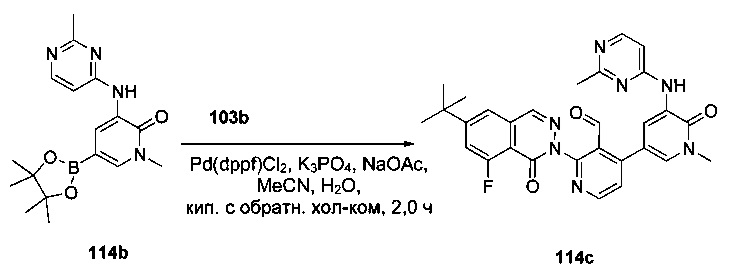
В одногорлую круглодонную колбу емкостью 100 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали бис(пинаколато)дибор (689 мг; 2,61 ммоль), 1,4-диоксан (30 мл), соединение 114а (307 мг; 1,04 ммоль), Pd2(dba)3 (47 мг; 0,050 ммоль), X-phos (48 мг; 0,10 ммоль) и ацетат калия (305 мг; 3,12 ммоль). Смесь нагревали при 65°С в течение 6 ч. Затем фильтровали и фильтрат упаривали в вакууме, получая соединение 114b (300 мг; 84%) в виде коричневого твердого вещества. MS: [М+Н]+ 342,2.
Пример 114с. 2-(6-трет-Бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-(1-метил-5-(2-метилпиримидин-4-иламино)-6-оксо-1,6-дигидропиридин-3-ил)никотинальдегид 114с
В одногорлую круглодонную колбу емкостью 50 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали соединение 114b (236 мг; 0,69 ммоль), 2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-хлорникотинальдегид 103b (250 мг; 0,69 ммоль), PdCl2(dppf) (29 мг; 0,035 ммоль), K3PO4 (296 мг; 1,39 ммоль), ацетат натрия (114 мг; 1,39 ммоль), ацетонитрил (15 мл) и воду (1 мл). Систему вакуумировали и заполняли N2. Реакционную смесь нагревали при 100°С в течение 2 ч. Затем охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 30:1 дихлорметан/метанол, получая соединение 114с (134 мг; 36%) в виде желтого твердого вещества. MS-ESI: [М+Н]+ 540,2.
Пример 114. 6-трет-Бутил-8-фтор-2-[3-(гидроксиметил)-4-[1-метил-5-[(2-метилпиримидин-4-ил)амино]-6-оксо-3-пиридил]-2-пиридил]фталазин-1-он 114
При 0°С к суспензии 2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-(1-метил-5-(2-метилпиримидин-4-иламино)-6-оксо-1,6-дигидропиридин-3-ил)никотинальдегида 114с (130 мг; 0,24 ммоль) в метаноле (20 мл) добавляли боргидрид натрия (27 мг; 0,72 ммоль). Реакционную смесь перемешивали в течение 20 минут и затем гасили водой (10 мл). Затем концентрировали при пониженном давлении и остаток экстрагировали дихлорметаном (3×20 мл). Объединенный органический слой сушили и концентрировали при пониженном давлении. Остаток очищали препаративной обращенно-фазовой HPLC, получая соединение 114 (20 мг; 15%) в виде белого твердого вещества. MS-ESI: [М+Н]+ 542,3. 1Н ЯМР (500 МГц, CDCl3) 8.97 (d, J=2,5 Гц, 1Η), 8.71 (d, J=5,0 Гц, 1Η), 8.36 (d, J=2,0 Гц, 1Η), 8.28 (d, J=5,5 Гц, 1Η), 8.09-8.06 (m, 1Η), 7.86 (d, J=2,5 Гц, 1Η), 7.60-7.56 (m, 3Н), 6.61 (d, J=6,0 Гц, 1H), 4.54-4.43 (m, 2H), 4.16-4.13 (m, 1H), 3.75 (s, 3Н), 2.62 (s, 3Н), 1.46 (s, 9H).
Пример 115а. 5-Метил-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-амин 115а
Раствор 1-метил-4-пиперидона (11,3 г; 100 ммоль) в 2-пропаноле (80 мл) нагревали до 50°С. К раствору последовательно добавляли раствор цианамида (4,2 г; 100 ммоль) в 2-пропаноле (25 мл) и порошок серы (3,2 г; 100 ммоль). После добавления каталитического количества пирролидина (1,3 мл) полученную смесь перемешивали при 50°С в течение 2 часов. Реакционную смесь оставляли охлаждаться до комнатной температуры и перемешивали в течение ночи. Затем охлаждали до или ниже 10°С в бане с ледяной водой и перемешивали в течение 1 часа при этой же температуре. Выпавшие в осадок кристаллы собирали фильтрованием и промывали 2-пропанолом (20 мл). Влажные кристаллы сушили в вакууме, получая соединение 115а (10 г; 59%). MS: [М+Н]+ 170. 1Н ЯМР (500 МГц, DMSO-de) δ 6.70 (s, 2Н), 3.31 (s, 2Н), 2.61 (t, J=5,5 Гц, 2H), 2.45 (m, 2Н), 2.33 (s, 3Н).
Пример 115b. 5-Бром-1-метил-3-(5-метил-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-иламино)пиридин-2(1Н)-он 115b
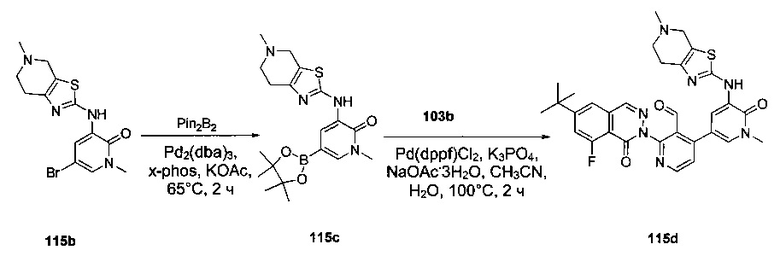
Следуя методикам, описанным для соединения 113а, и исходя из соединения 115а (4,0 г; 23,5 ммоль) и 3,5-дибром-1-метилпиридин-2(1Н)-она (3,0 г; 17,8 ммоль), получали соединение 115b в виде желтого твердого вещества (2,8 г; 44%). MS: [М+Н]+ 357.
Пример 115с. 1-Метил-3-(5-метил-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-иламино)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2(1Н)-он 115с
Соединение 115b (997 мг; 2,8 ммоль) растворяли в диоксане (50 мл), затем добавляли бис(пинаколато)дибор (3,0 г; 12,0 ммоль), Pd2(dba)3 (128 мг; 0,14 ммоль), X-phos (134 мг; 0,28 ммоль) и ацетат калия (823 мг; 8,4 ммоль). После трех циклов вакуумирования/продувания аргоном смесь нагревали при 65°С в течение 2 ч. Смесь охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали под давлением и остаток промывали петролейным эфиром (2×10 мл), получая соединение 115с в виде желтого твердого вещества (968 мг; 86%), которое использовали на следующей стадии без дополнительной очистки. MS-ESI: [М+Н]+ 403,2.
Пример 115d. 2-(6-трет-Бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-(1-метил-5-(5-метил-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-иламино)-6-оксо-1,6-дигидропиридин-3-ил)никотинальдегид 115d
В круглодонную колбу, оборудованную обратным холодильником, загружали 2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-хлорникотинальдегид 103b (144 мг; 0,40 ммоль), соединение 115с (240 мг; 0,60 ммоль), PdCl2(dppf) (20 мг; 0,020 ммоль), K3PO4 (180 мг; 0,80 ммоль), тригидрат ацетата натрия (120 мг; 0,80 ммоль) и смесь ацетонитрил/вода (15 мл/1 мл). После трех циклов вакуумирования/продувания аргоном смесь нагревали при 100°С в течение 2 ч. Затем охлаждали до комнатной температуры и фильтровали. Фильтрат упаривали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 25:1 дихлорметан/метанол, получая соединение 115d в виде желтого твердого вещества (100 мг; 42%). MS-ESI: [М+Н]+ 600,3.
Пример 115. 6-трет-Бутил-8-фтор-2-[3-(гидроксиметил)-4-[1-метил-5-[(5-метил-6,7-дигидро-4Н-тиазоло[5,4-с]пиридин-2-ил)амино]-6-оксо-3-пиридил]-2-пиридил]фталазин-1-он 115
К смеси соединения 115d (100 мг; 0,15 ммоль) в метаноле (6 мл) добавляли NaBH4 (18 мг; 0,45 ммоль). Смесь перемешивали при 30°С в течение 1 ч и затем гасили водой (10 мл). Смесь экстрагировали дихлорметаном (3×30 мл) и объединенный органический слой концентрировали при пониженном давлении. Остаток очищали препаративной обращенно-фазовой HPLC, получая соединение 115 (35 мг; 36%) в виде белого твердого вещества. MS-ESI: [М+Н]+ 602,2. 1Н ЯМР (500 МГц, CDCl3) δ 8.68 (d, J=5,0 Гц, 1H), 8.42 (d, J=2,5 Гц, 1H), 8.35 (d, J=2,5 Гц, 1Η), 8.30 (s, 1Η), 7.77 (d, J=2,0 Гц, 1Η), 7.58-7.53 (m, 3Н), 4.49-4.45 (m, 2Н), 4.10 (bs, 1Н), 3.73 (s, 3Н), 3.58 (s, 2Н), 2.83-2.79 (m, 4Н), 2.52 (s, 3Н), 1.45 (s, 9H).
Пример 116а. 6-трет-Бутил-2-(4-хлор-3-(гидроксиметил)пиридин-2-ил)-8-фторфталазин-1(2Н)-он 116а
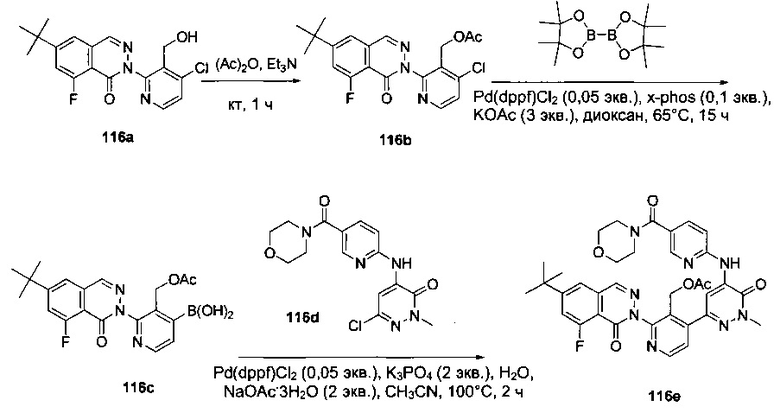
К раствору 2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-хлорникотинальдегида 103b (2,0 г; 5,5 ммоль) в метаноле (30 мл) добавляли NaBH4 (700 мг; 16,5 ммоль) при комнатной температуре. Реакционную смесь перемешивали в течение 1 ч и гасили водой (30 мл). Затем смесь концентрировали при пониженном давлении и остаток экстрагировали дихлорметаном (3×30 мл). Объединенную органическую фазу сушили над безводным Na2SO4, фильтровали и упаривали при пониженном давлении, получая соединение 116а в виде белого твердого вещества (1,8 г; 90%). MS: [М+Н]+ 362,3.
Пример 116b. (2-(6-трет-Бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-хлорпиридин-3-ил)метилацетат 116b
В одногорлую круглодонную колбу емкостью 50 мл, оборудованную магнитной мешалкой, загружали соединение 116а (1,6 г; 4,4 ммоль), уксусный ангидрид (10 мл) и триэтиламин (1 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 30:1 дихлорметан/метанол, получая соединение 116b в виде коричневого твердого вещества (1,4 г; 82%). MS: [М+Н]+ 404,3.
Пример 116с. 3-(Ацетоксиметил)-2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)пиридин-4-илбороновая кислота 116с
В одногорлую круглодонную колбу емкостью 100 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали соединение 116b (1,0 г; 2,5 ммоль), Pin2B2 (3,2 г; 12,5 ммоль), Pd(dppf)Cl2 (75 мг; 0,125 ммоль), X-phos (75 мг; 0,25 ммоль), ацетат калия (750 мг; 7,5 ммоль) и диоксан (60 мл). После трех циклов вакуумирования/продувания аргоном смесь нагревали при 65°С в течение 15 ч. Затем охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток промывали смесью 3:1 петролейный эфир/этилацетат (10 мл), получая соединение 116с в виде желтого твердого вещества (1,0 г; LCMS чистота: 75%). MS: [М+Н]+ 414,2.
Пример 116d. 6-Хлор-2-метил-4-({5-[(морфолин-4-ил)карбонил]пиридин-2-ил}амино)-2,3-дигидропиридазин-3-он 116d
В одногорлую круглодонную колбу емкостью 100 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали 1,4-диоксан (40 мл), (6-аминопиридин-3-ил)(морфолино)метанон (2,07 г; 10,0 ммоль), 4-бром-6-хлор-2-метилпиридазин-3(2Н)-он (3,35 г; 15,0 ммоль), Pd2(dba)3 (915 мг; 1,0 ммоль), ксантфос (578 мг; 1,0 ммоль) и карбонат цезия (6,52 г; 20 ммоль). После трех циклов вакуумирования/продувания аргоном смесь нагревали при 100°С в течение 8 ч. Затем охлаждали до комнатной температуры и фильтровали. Твердое вещество промывали дихлорметаном (2×20 мл). Объединенный фильтрат сушили над безводным Na2SO4 и концентрировали при пониженном давлении, получая соединение 116d (2,45 г; 51%) в виде желтого твердого вещества. MS: [М+Н]+ 350,1.
Пример 116е. (2-(6-трет-Бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-(1-метил-5-(5-(морфолин-4-карбонил)пиридин-2-иламино)-6-оксо-1,6-дигидропиридазин-3-ил)пиридин-3-ил)метилацетат 116е
В круглодонную колбу емкостью 50 мл, оборудованную обратным холодильником, загружали соединение 116d (175 мг; 0,50 ммоль), 3-(ацетоксиметил)-2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)пиридин-4-илбороновую кислоту 116с (300 мг; 0,75 ммоль), Pd(dppf)Cl2 (25 мг; 0,025 ммоль), K3PO4 (220 мг; 1,0 ммоль), тригидрат ацетата натрия (150 мг; 1,0 ммоль) и смесь ацетонитрил/вода (20/1 мл). После трех циклов вакуумирования/продувания аргоном смесь нагревали при 100°С в течение 2 ч. Затем фильтровали и фильтрат упаривали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 25:1 дихлорметан/метанол, получая соединение 116е в виде желтого твердого вещества (150 мг; 45%). MS-ESI: [М+Н]+ 683,3.
Пример 116. 6-трет-Бутил-8-фтор-2-[3-(гидроксиметил)-4-[1-метил-5-[[5-(морфолин-4-карбонил)-2-пиридил]амино]-6-оксо-пиридазин-3-ил]-2-пиридил]фталазин-1-он 116
Смесь соединения 116е (150 мг; 0,20 ммоль) и гидроксида лития (85 мг; 2,0 ммоль) в смеси THF/изопропанол (5/3 мл) и воде (2 мл) перемешивали при 30°С в течение 1 ч. Смесь упаривали при пониженном давлении и остаток экстрагировали этилацетатом (3×20 мл). Объединенный этилацетатный экстракт концентрировали при пониженном давлении и остаток очищали препаративной обращенно-фазовой HPLC, получая соединение 116 (80 мг; 80%) в виде белого твердого вещества. MS-ESI: [М+Н]+ 641,3. 1Н ЯМР (500 МГц, CDCl3) δ 8.78 (s, 1Н), 8.75 (d, J=5,0 Гц, 1Η), 8.47 (s, 1Η), 8.45 (d, J=2,0 Гц, 1Η), 8.34 (d, J=3,0 Гц, 1Η), 7.79 (dd, J=2,0; 8,0 Гц, 1H), 7.64 (d, J=4,5 Гц, 1H), 7.57 (s, 1H), 7.52 (dd, J=1,5; 12,5 Гц, 1H), 7.02 (d, J=8,5 Гц, 1H), 4.57-4.55 (m, 2H), 3.94 (s, 3Н), 3.81-3.75 (m, 6H), 1.64-1.62 (m, 2H), 1.44 (s, 9H).
Пример 117а. (S)-трет-Бутил-4-(6-(6-хлор-2-метил-3-оксо-2,3-дигидропиридазин-4-иламино)пиридин-3-ил)-3-метилпиперазин-1-карбоксилат 117а

В одногорлую круглодонную колбу емкостью 250 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали (8)-трет-бутил-4-(6-аминопиридин-3-ил)-3-метилпиперазин-1-карбоксилат 105b (2,5 г; 8,5 ммоль), 4-бром-6-хлор-2-метилпиридазин-3(2Н)-он (2,2 г; 10,0 ммоль), ксантфос (240 мг; 0,40 ммоль), трис(дибензилиденацетон)дипалладий(0) (360 мг; 0,40 ммоль), Cs2CO3 (5,5 г; 17 ммоль) и 1,4-диоксан (100 мл). После трех циклов вакуумирования/продувания аргоном смесь нагревали при 100°С в течение 2,5 ч. Затем охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали колоночной хроматографией на силикагеле с элюированием смесью дихлорметан/метанол (от 40:1 до 30:1), получая соединение 117а в виде бледно-желтого твердого вещества (3,2 г; 86%). MS-ESI: [М+Н]+ 435,1.
Пример 117b. (S)-6-Хлор-2-метил-4-(5-(2-метилпиперазин-1-ил)пиридин-2-иламино)пиридазин-3(2Н)-он 117b
Смесь соединения 117а (3,0 г; 6,9 ммоль) и 4,0 Μ HCl в этаноле (20 мл) перемешивали при комнатной температуре в течение 2 ч. Затем смесь концентрировали при пониженном давлении, получая неочищенное соединение 117b в виде желтого твердого вещества (2,5 г; 98%), которое использовали на следующей стадии без дополнительной очистки. MS-ESI: [М+Н]+ 335,1.
Пример 117с. (S)-6-Хлор-2-метил-4-(5-(2-метил-4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-иламино)пиридазин-3(2Н)-он 117с
Смесь соединения 117b (2,3 г; 6,8 ммоль), оксетан-3-она (1,4 г; 20,0 ммоль), NaBH3CN (620 мг; 10 ммоль) и хлорида цинка (1,36 г; 10 ммоль) в метаноле (20 мл) перемешивали при 50°С в течение 3 часов. Смесь добавляли к воде (40 мл) и концентрировали при пониженном давлении. Остаток три раза экстрагировали дихлорметаном. Объединенный органический слой сушили и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 50:1 дихлорметан/метанол, получая соединение 117с (2,0 г; 75%). MS-ESI: [М+Н]+ 391,2.
Пример 117d. (S)-(2-(6-трет-Бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-(1-метил-5-(5-(2-метил-4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-иламино)-6-оксо-1,6-дигидропиридазин-3-ил)пиридин-3-ил)метилацетат 117d
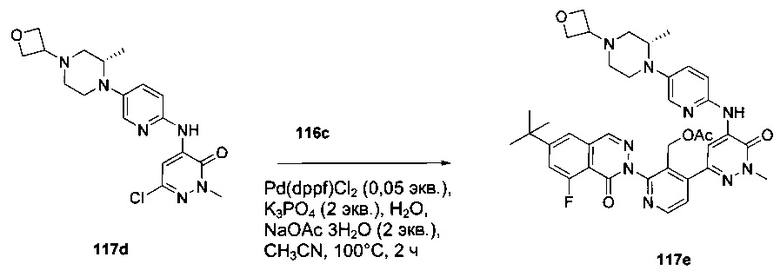
В круглодонную колбу, оборудованную обратным холодильником, загружали соединение 117с (200 мг; 0,50 ммоль), 3-(ацетоксиметил)-2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)пиридин-4-ил-бороновую кислоту 116с (300 мг; 0,75 ммоль), Pd(dppf)Cl2 (25 мг; 0,025 ммоль), K3PO4 (220 мг; 1,0 ммоль), тригидрат ацетата натрия (150 мг; 1,0 ммоль) и смесь ацетонитрил/вода (20/1 мл). После трех циклов вакуумирования/продувания аргоном смесь нагревали при 100°С в течение 2 ч. Затем охлаждали до комнатной температуры и фильтровали. Фильтрат упаривали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 25:1 дихлорметан/метанол, получая соединение 117d в виде желтого твердого вещества (110 мг; 42%). MS-ESI: [М+Н]+ 724,4.
Пример 117. 6-трет-Бутил-8-фтор-2-[3-(гидроксиметил)-4-[1-метил-5-[[5-[(25)-2-метил-4-(оксетан-3-ил)пиперазин-1-ил]-2-пиридил]амино]-6-оксо-пиридазин-3-ил]-2-пиридил]фталазин-1-он 117
Смесь соединения 117d (110 мг; 0,15 ммоль) и гидроксида лития (65 мг; 1,5 ммоль) в смеси THF/изопропанол (5/3 мл) и воде (2 мл) перемешивали при 30°С в течение 1 ч. Смесь упаривали при пониженном давлении и остаток экстрагировали этилацетатом (2×20 мл). Объединенный этилацетатный экстракт концентрировали при пониженном давлении и остаток очищали препаративной обращенно-фазовой HPLC, получая соединение 117 (100 мг; 95%) в виде желтого твердого вещества. MS-ESI: [М+Н]+ 682,4. 1Н ЯМР (500 МГц, CDCl3) δ 8.73 (d, J=5,0 Гц, 1Η), 8.60 (s, 1Η), 8.33 (d, J=2,5 Гц, 1Η), 8.24 (s, 1Η), 8.07 (s, 1Η), 7.65 (d, J=5,0 Гц, 1H), 7.56-7.51 (m, 2Н), 7.34 (s, 1Н), 6.96 (d, J=9,0 Гц, 1Н), 4.73-4.55 (m, 5H), 3.92 (m, 4H), 3.73-3.74 (m, 1H), 3.55-3.53 (m, 1H), 3.16-3.15 (m, 2H), 2.64-2.39 (m, 4H), 1.44 (s, 9H), 1.09-1.07 (m, 3Н).
Пример 118а. 5-Бром-1-метил-3-(5-метил-1Н-пиразол-3-иламино)пиридин-2(1Н)-он 118а
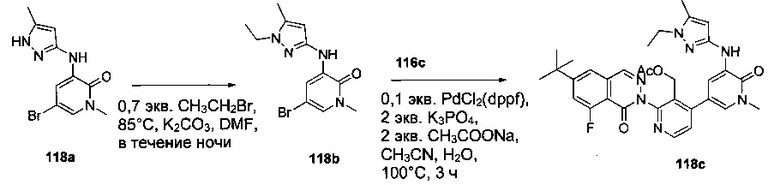
В одногорлую круглодонную колбу емкостью 100 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали 1,4-диоксан (15 мл), 5-метил-1Н-пиразол-3-амин (1 г; 10 ммоль) (1), 3,5-дибром-1-метилпиридин-2(1Н)-он (4 г; 15 ммоль) (2) и карбонат цезия (6,4 г; 20 ммоль). Добавляли ксантфос (400 мг; 0,8 ммоль) и Pd2(dba)3 (700 мг; 0,8 ммоль) и реакционную смесь нагревали при 100°С в течение 5 ч. По прошествии этого времени реакционную смесь охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении и остаток очищали на колонке для флэш-хроматографии с элюированием смесью дихлорметан:метанол (20:1), получая соединение 118а (1,0 г; 35%). MS: [М+Н]+ 283.
Пример 118b. 5-Бром-3-(1-этил-5-метил-1Н-пиразол-3-иламино)-1-метил-пиридин-2(1Н)-он 118b
В круглодонную колбу емкостью 100 мл загружали соединение 118а (800 мг; 2,83 ммоль), бромэтан (216 мг; 1,98 ммоль), K2CO3 (780 мг; 5,66 ммоль) и DMF (20 мл). Смесь нагревали при 85°С в течение ночи. Затем фильтровали и фильтрат упаривали в вакууме. Остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 1:20 метанол/дихлорметан, получая соединение 118b в виде красного твердого вещества (298 мг; 37%). MS-ESI: [М+Н]+ 311,0. 1Н ЯМР (500 МГц, DMSO-d6) δ 8.28 (s, 1Н), 7.99 (d, J=2,5 Гц, 1H), 7.35 (d, J=2,5 Гц, 1H), 5.85 (s, 1Н), 3.98-3.94 (m, 2Н), 3.48 (s, 3Н), 2.19 (s, 3Н), 1.27 (t, J=7,0 Гц, 3Н).
Пример 118с. (2-(6-трет-Бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-(5-(1-этил-5-метил-1Н-пиразол-3-иламино)-1-метил-6-оксо-1,6-дигидропиридин-3-ил)пиридин-3-ил)метилацетат 118с
В круглодонную колбу емкостью 50 мл, оборудованную обратным холодильником, загружали соединение 118b (103 мг; 0,33 ммоль), 3-(ацетоксиметил)-2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)пиридин-4-илбороновую кислоту 116с (136 мг; 0,33 ммоль), PdCl2(dppf) (27 мг; 0,033 ммоль), K3PO4 (171 мг; 0,66 ммоль), ацетат натрия (54 мг; 0,66 ммоль), ацетонитрил (10 мл) и воду (0,5 мл). После трех циклов вакуумирования/продувания аргоном смесь нагревали при 100°С в течение 3 ч. Затем фильтровали и фильтрат упаривали в вакууме. Остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 1:20 метанол/дихлорметан, получая соединение 118с в виде желтого твердого вещества (80 мг; 41%). MS-ESI: [М+Н]+ 600,2.
Пример 118. 6-трет-Бутил-2-[4-[5-[(1-этил-5-метил-пиразол-3-ил)амино]-1-метил-6-оксо-3-пиридил]-3-(гидроксиметил)-2-пиридил]-8-фтор-фталазин-1-он 118
Смесь соединения 118с (80 мг; 0,13 ммоль), гидроксида лития (13 мг; 0,53 ммоль), THF (6 мл), изопропанола (4 мл) и воды (2 мл) перемешивали при комнатной температуре в течение 0,5 ч. Смесь концентрировали при пониженном давлении и разбавляли водой (5 мл). Затем экстрагировали дихлорметаном (2×10 мл). Объединенный дихлорметановый экстракт концентрировали при пониженном давлении и остаток очищали обращенно-фазовой препаративной HPLC, получая соединение 118 (20 мг; 26%) в виде белого твердого вещества. MS-ESI: [М+Н]+ 558,3. 1Н ЯМР (500 МГц, DMSO-d6) δ 8.56 (d, J=5,0 Гц, 1H), 8.53 (d, J=2,5 Гц, 1H), 8.10 (s, 1Н), 8.05 (d, J=2,5 Гц, 1H), 7.90 (d, J=2,0 Гц, 1H), 7.79-7.76 (m, 1Η), 7.50 (d, J=5,0 Гц, 1Η), 7.40 (d, J=2,5 Гц, 1Η), 5.88 (s, 1Η), 4.92 (t, J=5,0 Гц 1H), 4.46-4.45 (m, 2Н), 3.91 (q, J=7,5 Гц, 2H), 3.59 (s, 3Н), 2.19 (s, 3Н), 1.40 (s, 9Н), 1.27 (t, J=7,0 Гц, 3Н).
Пример 119а. 5-Бром-3-(1,5-диметил-1Н-пиразол-3-иламино)-1-метил-пиридин-2(1Н)-он 119а
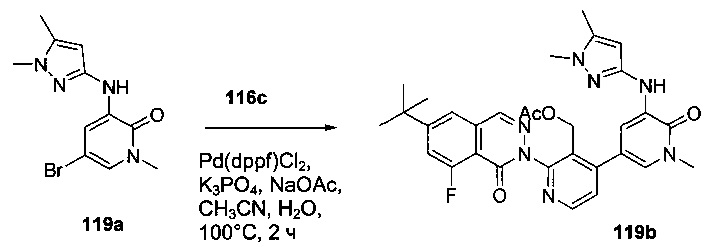
Раствор 5-бром-1-метил-3-(5-метил-1Н-пиразол-3-иламино)пиридин-2(1Н)-она 118а (2,8 г; 9,9 ммоль) в безводном DMF (10 мл) обрабатывали 60%-ной дисперсией NaH в минеральном масле (0,51 г; 13 ммоль) с перемешиванием в атмосфере азота. После прекращения выделения газа реакционную смесь перемешивали в течение еще 30 минут. Через этот промежуток времени реакционную смесь обрабатывали иодметаном (0,98 г; 7,0 ммоль), продолжая перемешивание в атмосфере азота в течение 2 часов. Медленно добавляли воду (50 мл) и смесь фильтровали. Фильтрат экстрагировали этилацетатом (3×30 мл). Объединенный экстракт концентрировали при пониженном давлении и остаток очищали колоночной флэш-хроматографией с элюированием смесью 3:1 петролейный эфир/этилацетат, получая соединение 119а (0,70 г; 24%). MS: [М+Н]+ 297.
Пример 119b. (2-(6-трет-Бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-(5-(1,5-диметил-1Н-пиразол-3-иламино)-1-метил-6-оксо-1,6-дигидропиридин-3-ил)пиридин-3-ил)метилацетат 119b
В круглодонную колбу емкостью 50 мл, оборудованную обратным холодильником, загружали соединение 119а (180 мг; 0,50 ммоль), 3-(ацетоксиметил)-2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)пиридин-4-илбороновую кислоту 116с (207 мг; 0,50 ммоль), Pd(dppf)Cl2 (41 мг; 0,050 ммоль), K3PO4 (212 мг; 1,0 ммоль), ацетат натрия (82 мг; 1,0 ммоль), воду (0,5 мл) и ацетонитрил (10 мл). После трех циклов вакуумирования/продувания аргоном смесь нагревали при температуре дефлегмации в течение 2 ч. Затем охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 30:1 дихлорметан/метанол, получая соединение 119b в виде белого твердого вещества (175 мг; 60%). MS-ESI: [М+Н]+ 586,4.
Пример 119. 6-трет-Бутил-2-[4-[5-[(1,5-диметилпиразол-3-ил)амино]-1-метил-6-оксо-3-пиридил]-3-(гидроксиметил)-2-пиридил]-8-фтор-фталазин-1-он 119
Смесь соединения 119b (150 мг; 0,26 ммоль) и гидроксида лития (61,4 мг; 2,6 ммоль) в смеси изопропанол/THF (1:1; 4 мл) и воде (1 мл) перемешивали при 30°С в течение 1 ч. Смесь упаривали в вакууме и остаток разбавляли водой (5 мл). Затем экстрагировали этилацетатом (2×10 мл). Объединенный этилацетатный экстракт концентрировали при пониженном давлении и остаток очищали препаративной обращенно-фазовой HPLC, получая соединение 119 (75,0 мг; 54%) в виде белого твердого вещества. MS-ESI: [М+Н]+ 544,3. 1Н ЯМР (500 МГц, CDCl3) δ 8.66 (d, J=5,0 Гц, 1Н), 8.35 (d, J=2,0 Гц, 1Н), 7.97 (d, J=2,0 Гц, 1Н), 7.59-7.53 (m, 4Н), 7.37 (s, 1Н), 5.74 (s, 1Н), 4.51 (s, 2Н), 4.07 (s, 1Н), 3.72 (s, 3Н), 3.70 (s, 3Н), 2.25 (s, 3Н), 1.46 (s, 9Н).
Пример 120а. 5-Бром-1-метил-3-(5-метилтиазол-2-иламино)пиридин-2(1Н)-он 120а
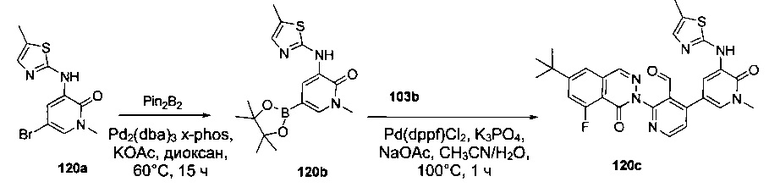
В одногорлую круглодонную колбу емкостью 100 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали 1,4-диоксан (50 мл), 5-метилтиазол-2-амин (2,28 г; 20,0 ммоль), 3,5-дибром-1-метилпиридин-2(1Н)-он (5,34 г; 20,0 ммоль), карбонат цезия (13,0 г; 40,0 ммоль), ксантфос (1,16 г; 2,0 ммоль) и трис(дибензилиденацетон)дипалладий(0) (916 мг; 1,0 ммоль). Систему подвергали трем циклам вакуумирования/продувания аргоном и нагревали при температуре дефлегмации в течение 5 ч. Затем охлаждали до комнатной температуры и фильтровали. Твердое вещество промывали дихлорметаном (3×50 мл). Объединенный фильтрат концентрировали при пониженном давлении и остаток промывали ацетонитрилом (30 мл), получая соединение 120а (5 г; неочищенное; 83%) в виде желтого твердого вещества. MS-ESI: [М+Н]+ 300,1.
Пример 120b. 1-Метил-3-(5-метилтиазол-2-иламино)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2(1Н)-он 120b
В круглодонную колбу емкостью 250 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали соединение 120а (5,0 г; 16,6 ммоль), Pin2B2 (21,1 г; 83,0 ммоль), трис(дибензилиденацетон)дипалладий(0) (760 мг; 0,83 ммоль), x-phos (810 мг; 1,7 ммоль), ацетат калия (4,9 г; 50 ммоль) и 1,4-диоксан (80 мл). Реакционную смесь подвергали трем циклам вакуумирования/продувания аргоном и нагревали при 65°С в течение 3 ч. Затем охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток промывали петролейным эфиром, получая соединение 120b (20,0 г; неочищенное) в виде коричневого твердого вещества, которое использовали на следующей стадии без дополнительной очистки. MS-ESI: [М+Н]+ 348,2.
Пример 120с. 2-(6-трет-Бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-(1-метил-5-(5-метилтиазол-2-иламино)-6-оксо-1,6-дигидропиридин-3-ил)никотинальдегид 120с
В одногорлую круглодонную колбу емкостью 250 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали соединение 120b (174 мг; 0,50 ммоль), 2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-хлорникотинальдегид 103b (180 мг; 0,50 ммоль), K3PO4 (212 мг; 1,0 ммоль), ацетат натрия (82 мг; 1,0 ммоль), 1,1'-бис(дифенилфосфино)ферроцендихлор-палладий(II) (18 мг; 0,025 ммоль), ацетонитрил (8 мл) и воду (0,5 мл). Реакционную смесь подвергали трем циклам вакуумирования/продувания аргоном и нагревали при 100°С в течение 1 ч. Анализ реакционной смеси с использованием LCMS показал полное превращение в желаемый продукт. Реакционную смесь охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток разбавляли дихлорметаном (50 мл) и водой (30 мл). Водный слой экстрагировали дихлорметаном (2×30 мл). Объединенный органический экстракт сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Темный остаток очищали колоночной хроматографией на силикагеле с элюированием смесью дихлорметан/метанол (от 80:1 до 30:1), получая соединение 120с (150 мг; 55%) в виде желтого твердого вещества. MS-ESI: [М+Н]+ 545,3.
Пример 120. 6-трет-Бутил-8-фтор-2-[3-(гидроксиметил)-4-[1-метил-5-[(5-метилтиазол-2-ил)амино]-6-оксо-3-пиридил]-2-пиридил]фталазин-1-он 120
К раствору соединения 120с (98 мг; 0,18 ммоль) в смеси метанол/дихлорметан (3/3 мл) добавляли NaBH4 (21 мг; 0,55 ммоль) при комнатной температуре. После перемешивания реакционной смеси в течение 1 ч LCMS показала полное завершение реакции. Смесь гасили водой (5 мл) и концентрировали при пониженном давлении. Остаток экстрагировали дихлорметаном (3×15 мл). Объединенный органический слой промывали рассолом (20 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали препаративной обращенно-фазовой HPLC, получая соединение 120 (30 мг; 31%) в виде белого твердого вещества. MS-ESI: [М+Н]+ 546,7. 1Н ЯМР (500 МГц, DMSO-d6) δ 9.92 (s, 1Н), 8.58 (d, J=5,5 Гц, 1H), 8.56 (d, J=2,5 Гц, 1H), 8.54 (d, J=2,0 Гц, 1H), 7.91 (d, J=2,0 Гц, 1H), 7.78 (d, J=13,0 Гц, 1Η), 7.56 (d, J=2,0 Гц, 1Η), 7.51 (d, J=5,0 Гц, 1Η), 6.96 (d, J=1,0 Гц, 1H), 4.91 (bs, 1Н), 4.42-4.41 (m, 2Н), 3.60 (s, 3Н), 2.28 (d, J=1,0 Гц, 3Н), 1.40 (s, 9Н).
Пример 121а. (2-(6-трет-Бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-(1-метил-5-(5-метил-1Н-пиразол-3-иламино)-6-оксо-1,6-дигидропиридин-3-ил)пиридин-3-ил)метилацетат 121а
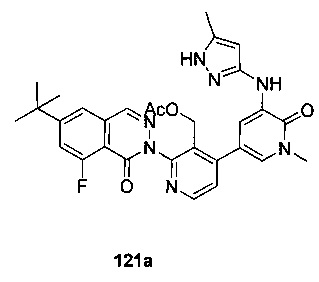
В круглодонную колбу емкостью 50 мл, оборудованную обратным холодильником, загружали 5-бром-1-метил-3-(5-метил-1Н-пиразол-3-иламино)пиридин-2(1Н)-он 118а (142 мг; 0,50 ммоль), 3-(ацетоксиметил)-2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1H)-ил)пиридин-4-илбороновую кислоту 116с (310 мг; 0,75 ммоль), Pd(dppf)Cl2 (18 мг; 0,025 ммоль), K3PO4 (212 мг; 1,0 ммоль), ацетат натрия (82 мг; 1,0 ммоль), ацетонитрил (10 мл) и воду (0,2 мл). После трех циклов вакуумирования/продувания аргоном смесь нагревали при 100°С в течение 2 ч. Затем фильтровали и фильтрат упаривали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 25:1 дихлорметан/метанол, получая соединение 121а в виде коричневого твердого вещества (100 мг; 35%). MS-ESI: [М+Н]+ 572,3.
Пример 121. 6-трет-Бутил-8-фтор-2-[3-(гидроксиметил)-4-[1-метил-5-[(5-метил-1Н-пиразол-3-ил)амино]-6-оксо-3-пиридил]-2-пиридил]фталазин-1-он 121
Смесь соединения 121а (86 мг; 0,15 ммоль) и гидроксида лития (36 мг; 1,5 ммоль) в смеси THF/изопропанол (5:3; 8 мл) и воде (2 мл) перемешивали при 30°С в течение 1 ч. Смесь упаривали при пониженном давлении и остаток разбавляли водой (5 мл). Затем экстрагировали этилацетатом (2×10 мл). Объединенный этилацетатный экстракт концентрировали при пониженном давлении и остаток очищали препаративной обращенно-фазовой HPLC, получая соединение 121 (24 мг; 30%) в виде белого твердого вещества. MS-ESI: [М+Н]+ 530,3. 1Н ЯМР (500 МГц, CDCl3) δ 8.65 (d, J=5,0 Гц, 1H), 8.35 (d, J=2,0 Гц, 1H), 8.04 (d, J=2,5 Гц, 1H), 7.58-7.53 (m, 4Н), 7.44 (s, 1Н), 5.78 (s, 1Н), 4.51-4.49 (m, 2Н), 3.73 (s, 3Н), 2.03 (s, 3Н), 1.45 (s, 9Н).
Пример 122а. 5-Бром-3-(5-этил-1Н-пиразол-3-иламино)-1-метилпиридин-2(1Н)-он 122а
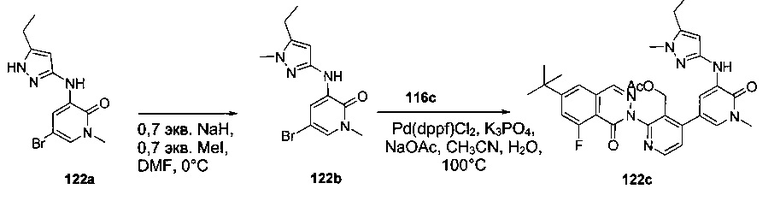
В одногорлую круглодонную колбу емкостью 100 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали 1,4-диоксан (80 мл), 6-этил-1Н-пиразол-3-амин (3,33 г; 30,0 ммоль), 3,5-дибром-1-метилпиридин-2(1Н)-он (9,6 г; 36 ммоль) и карбонат цезия (19,5 г; 60 ммоль). После барботирования азота через суспензию в течение 10 минут добавляли ксантфос (1,73 мг; 3,0 ммоль) и трис(дибензилиденацетон)дипалладий(0) (1,36 мг; 1,5 ммоль). Систему подвергали трем циклам вакуумирования/продувания аргоном и нагревали при температуре дефлегмации в течение 2 ч. Затем незамедлительно фильтровали. Твердое вещество промывали диоксаном (3×30 мл) и объединенный фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с элюированием смесью петролейный эфир/этилацетат (от 2:1 до 1:2), получая соединение 122а (3,8 г; 43%) в виде красного твердого вещества. MS-ESI: [М+Н]+ 297,0.
Пример 122b. 5-Бром-3-(5-этил-1-метил-1Н-пиразол-3-иламино)-1-метилпиридин-2(1Н)-он 122b
К смеси соединения 122а (598 мг; 2,0 ммоль) в DMF (10 мл) добавляли NaH (64 мг; 1,6 ммоль) при 0°С и смесь перемешивали в течение 0,5 ч. К смеси по каплям при 0°С добавляли подметан (227 мг; 1,6 ммоль) в DMF. Смесь перемешивали в течение 0,5 ч и гасили водой (20 мл). Затем экстрагировали дихлорметаном (3×30 мл). Объединенный экстракт концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле с элюированием смесью петролейный эфир/этилацетат (от 5:1 до 2:1), получая соединение 122b (360 мг; 58%) в виде светло-желтого твердого вещества. MS-ESI: [М+Н]+ 311,1.
Пример 122с. (2-(6-трет-Бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-(5-(5-этил-1-метил-1Н-пиразол-3-иламино)-1-метил-6-оксо-1,6-дигидропиридин-3-ил)пиридин-3-ил)метилацетат 122с
В круглодонную колбу емкостью 50 мл, оборудованную обратным холодильником, загружали соединение 122b (310 мг; 1,0 ммоль), 3-(ацетоксиметил)-2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)пиридин-4-илбороновую кислоту 116с (1,24 г; 3,0 ммоль), K3PO4 (424 мг; 2,0 ммоль), ацетат натрия (164 мг; 2,0 ммоль), 1,1'-бис(дифенилфосфино)ферроцендихлор-палладий(II) (73 мг; 0,10 ммоль), ацетонитрил (10 мл) и воду (0,5 мл). Систему подвергали трем циклам вакуумирования/продувания азотом и нагревали при 100°С под защитой N2 в течение 2,5 ч. Реакционную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. Остаток разбавляли дихлорметаном (50 мл) и водой (50 мл). Водный слой отделяли и экстрагировали дихлорметаном (3×20 мл). Объединенный органический слой сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Темный остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 60:1 дихлорметан/метанол, получая соединение 122с (150 мг; 25%) в виде желтого масла. MS-ESI: [М+Н]+ 600,0.
Пример 122. 6-трет-Бутил-2-[4-[5-[(5-этил-1-метил-пиразол-3-ил)амино]-1-метил-6-оксо-3-пиридил]-3-(гидроксиметил)-2-пиридил]-8-фтор-фталазин-1-он 122
К раствору соединения 122с (150 мг; 0,25 ммоль) в смеси THF/изопропанол/вода (2,5/1/0,5 мл) добавляли гидроксид лития (70 мг; 2,5 ммоль) при 35°С. После перемешивания реакционной смеси в течение 3 ч LCMS показала полное завершение реакции. Смесь выливали в воду (15 мл) и экстрагировали дихлорметаном (3×20 мл). Объединенный органический слой промывали рассолом (30 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Оставшееся твердое вещество очищали препаративной обращенно-фазовой HPLC, получая соединение 122 (49 мг; 35%) в виде белого твердого вещества. MS-ESI: [М+Н]+ 557,8. 1Н ЯМР (500 МГц, CDCl3) δ 8.59 (d, J=5,0 Гц,1Н), 8.51 (d, J=2,5 Гц, 1H), 7.92 (d, J=2,0 Гц, 1H), 7.88 (d, J=1,5 Гц, 1H), 7.73 (m, 1Н), 7.64 (d, J=5,0 Гц,1Н), 7.41 (d, J=1,5 Гц, 1H), 5.92 (s, 1H), 4.59 (s, 2H), 3.72 (s, 3Н), 3.68 (s, 3Н), 2.64 (m, 2H), 1.47 (s, 9H), 1.27 (t, J=2,5, 3Н).
Пример 123а. 1-Этил-4-нитро-1Н-пиразол 123а
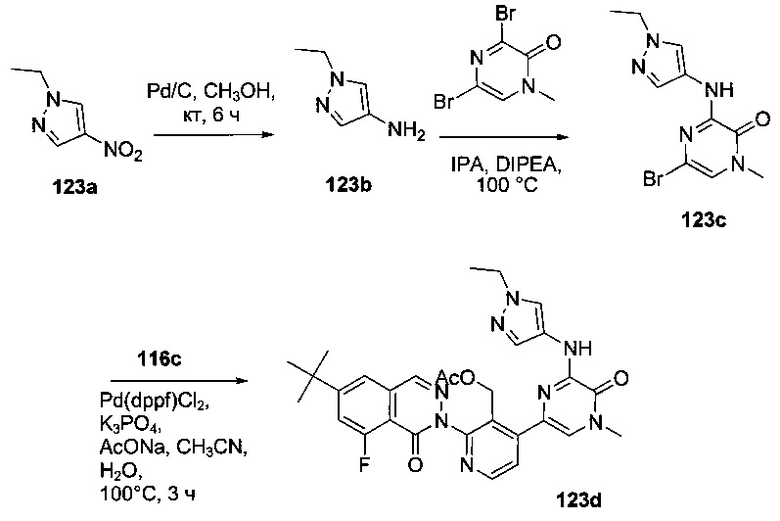
К раствору 4-нитро-1Н-пиразола (5,0 г; 44,2 ммоль) в безводном DMF (100 мл) добавляли NaH (60%-ный в масле) (1,94 г; 48,6 ммоль), перемешивая при -25°С в атмосфере азота. Смесь перемешивали в течение 30 мин, затем добавляли бромэтан (5,30 г; 48,6 ммоль). Смесь продолжали перемешивать в атмосфере азота при -25°С в течение 6 ч. Раствор разбавляли этилацетатом (100 мл), промывали водой (2×50 мл) и рассолом (50 мл), сушили над сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении, получая неочищенное соединение 123а (5,0 г; 80%), которое использовали на следующей стадии без дополнительной очистки. MS-ESI: [М+Н]+ 142,0.
Пример 123b. 1-Этил-1Н-пиразол-4-амин 123b
Одногорлую круглодонную колбу емкостью 100 мл продували водородом и в нее загружали соединение 123а (4,5 г; 31,9 ммоль), 10%-ный палладий на угле (10% влаги; 2,0 г) и метанол (50 мл). Смесь перемешивали при комнатной температуре в течение 6 ч. Катализатор удаляли фильтрованием через набивку CELITE® и фильтрат концентрировали при пониженном давлении, получая соединение 123b (3,3 г; 93%). MS-ESI: [М+Н]+ 112,0.
Пример 123с. 5-Бром-3-(1-этил-1Н-пиразол-4-иламино)-1-метилпиразин-2(1Н)-он 123с
В одногорлую круглодонную колбу емкостью 100 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали соединение 123b (500 мг; 4,5 ммоль), 3,5-дибром-1-метилпиразин-2(1Н)-он (2,40 г; 9,0 ммоль), диизопропилэтилацетат (DIPEA) (3 мл) и изопропанол (IPA) (50 мл). Смесь нагревали при 100°С в течение 2 ч. Затем охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении, получая соединение 123с (802 мг; 60%) в виде белого твердого вещества. MS-ESI: [М+Н]+ 298,0.
Пример 123d. (2-(6-трет-Бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-(6-(1-этил-1Н-пиразол-4-иламино)-4-метил-5-оксо-4,5-дигидропиразин-2-ил)пиридин-3-ил)метил ацетат 123d
В круглодонную колбу емкостью 50 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали соединение 123с (151 мг; 0,51 ммоль), 3-(ацетоксиметил)-2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)пиридин-4-илбороновую кислоту 116с (206 мг; 0,50 ммоль), PdCl2(dppf) (22 мг; 0,030 ммоль), K3PO4 (216 мг; 1,02 ммоль), ацетат натрия (84 мг; 1,02 ммоль), ацетонитрил (10 мл) и воду (0,5 мл). После трех циклов вакуумирования/продувания аргоном смесь нагревали при 100°С в течение 3 ч. Затем фильтровали и фильтрат упаривали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 1:3 петролейный эфир/этилацетат, получая соединение 123d в виде желтого твердого вещества (158 мг; 53%). MS-ESI: [М+Н]+ 587,2.
Пример 123. 6-трет-Бутил-2-[4-[6-[(1-этилпиразол-4-ил)амино]-4-метил-5-оксо-пиразин-2-ил]-3-(гидроксиметил)-2-пиридил]-8-фтор-фталазин-1-он 123
Смесь соединения 123d (152 мг; 0,26 ммоль) и гидроксида лития (61 мг; 2,56 ммоль) в смеси изопропанол/THF (1:1; 10 мл) и воде (3 мл) перемешивали при комнатной температуре в течение 1 ч. Смесь упаривали при пониженном давлении и остаток экстрагировали этилацетатом (2×10 мл). Объединенный этилацетатный экстракт концентрировали при пониженном давлении и остаток очищали препаративной обращенно-фазовой HPLC, получая соединение 123 (55 мг; 39%) в виде желтого твердого вещества. MS-ESI: [М+Н]+ 545,2. 1Н ЯМР (500 МГц, DMSO-d6) δ 9.72 (s, 1Н), 8.58 (d, J=5,0 Гц, 1H), 8.54 (d, J=2,5 Гц, 1H), 8.18 (s, 1Н), 7.90 (d, J=2,0 Гц, 1H), 7.80-7.75 (m, 1Н), 7.74 (s, 1Н), 7.71 (d, J=5,0 Гц, 1H), 7.51 (s, 1Н), 4.90 (t, J=5,0 Гц, 1H), 4.61-4.53 (m, 2Н), 4.09 (q, J=7,5 Гц, 2H), 3.54 (s, 3Н), 1.41 (s, 9Н), 1.35 (t, J=6,0 Гц, 3Н).
Пример 124а. (3-Нитро-1Н-пиразол-5-ил)метанол 124а
Трехгорлую круглодонную колбу емкостью 3 л, оборудованную механической мешалкой, капельной воронкой и входным отверстием для азота, продували азотом, в нее загружали 3-нитропиразол-5-карбоновую кислоту (28,0 г; 178 ммоль) и THF (420 мл) и охлаждали до -5°С, используя баню со смесью лед/ацетон. Добавляли раствор комплекса боран-THF (1,0 М; 535 мл; 535 ммоль) с такой скоростью, чтобы поддерживать внутреннюю температуру реакции ниже 5°С. По завершении добавления охлаждающую баню удаляли и реакционную смесь перемешивали при комнатной температуре в течение 18 ч. По прошествии этого времени реакционную смесь охлаждали до -5°С, используя баню со смесью лед/ацетон, добавляли воду (70 мл) и 4 н. соляную кислоту (70 мл) и реакционную смесь перемешивали при температуре дефлегмации в течение 1 ч для разрушения комплекса борана с пиразолом. Реакционную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении до объема приблизительно 30 мл. Добавляли этилацетат (175 мл) и смесь перемешивали в течение 15 мин. Водный слой отделяли и экстрагировали этилацетатом (4x200 мл). Объединенные органические слои промывали насыщенным водным бикарбонатом натрия (2×50 мл), рассолом (50 мл) и сушили над сульфатом натрия, сушильный агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении, получая соединение 124а с выходом 94% (24,0 г) в виде светло-желтого твердого вещества: 1Н ЯМР (300 МГц, DMSO-d6) δ 13.90 (br s, 1H), 6.87 (s, 1H), 5.58 (t, 1H, J=5,4 Гц), 4.53 (d, 2H, J=5,1 Гц); MS (ESI+) m/z 144,0 (M+H).
Пример 124b. (1-(2-Бромэтил)-3-нитро-1Н-пиразол-5-ил)метанол 124b
Трехгорлую круглодонную колбу емкостью 1 л, оборудованную механической мешалкой и терморегулятором, продували азотом, в нее загружали соединение 124а (25,0 г; 175 ммоль), DMF (250 мл) и карбонат цезия (70,0 г; 215 ммоль) и нагревали при 104°С в течение 5 мин. Затем реакционную смесь охлаждали до 0°С, используя баню со смесью лед/ацетон, и порциями добавляли дибромэтан (329 г; 1,75 моль) (никакого экзотермического эффекта не наблюдали). Реакционную смесь перемешивали при 0°С в течение 1 ч, затем при комнатной температуре в течение 4 ч. По прошествии этого времени медленно добавляли раствор KH2PO4 (40 г) в воде (400 мл). Реакционную смесь перемешивали при комнатной температуре в течение 30 мин. Добавляли этилацетат (450 мл), водный слой отделяли и экстрагировали этилацетатом (2×100 мл). Объединенные органические слои промывали водой (200 мл), рассолом (200 мл), сушили над сульфатом натрия и сушильный агент удаляли фильтрованием. Фильтрат концентрировали при пониженном давлении, получая с выходом 86% 37,5 г неочищенного соединения 124b в виде оранжевого масла: 1Н ЯМР (300 МГц, CDCl3) δ 6.85 (s, 1Н), 4.82 (d, 2Н, J=5,4 Гц), 4.66 (t, 2Н, J=6,3 Гц), 3.83 (t, 2Н, J=6,3 Гц); MS (ESI+) m/z 249,9 (M+H).
Пример 124с. 1-(2-Бромэтил)-5-(бромметил)-3-нитро-1Н-пиразол 124с
Трехгорлую круглодонную колбу емкостью 500 л, оборудованную механической мешалкой, входным отверстием для азота и обратным холодильником, продували азотом и в нее загружали соединение 124b (37,0 г; 148 ммоль) и хлороформ (160 мл). Реакционную смесь охлаждали до -5°С, используя баню со смесью лед/ацетон, и порциями добавляли трибромид фосфора (40,0 г; 148 ммоль). Охлаждающую баню удаляли и реакционную смесь перемешивали при температуре дефлегмации в течение 2 ч. По прошествии этого времени реакционную смесь охлаждали до -5°С и добавляли насыщенный водный бикарбонат натрия (250 мл) до достижения рН 8,5. Смесь экстрагировали этилацетатом (3×150 мл) и объединенные органические слои промывали насыщенным водным карбонатом натрия (2×50 мл), рассолом (75 мл), сушили над сульфатом натрия и сушильный агент удаляли фильтрованием. Фильтрат концентрировали при пониженном давлении, получая желтый остаток, который растворяли с осторожным нагреванием в метиленхлориде (60 мл). Добавляли гексаны (приблизительно 20 мл), и раствор становился мутным. Смесь нагревали, пока не образовывался твердый осадок, добавляли метиленхлорид (9 мл), и раствор становился прозрачным. Раствор оставляли охлаждаться до комнатной температуры и через 4 ч образовавшиеся кристаллы собирали, используя вакуумную фильтрацию. Осадок на фильтре промывали охлажденной во льду смесью 1:2 метиленхлоридтексаны (2×20 мл), получая 1-(2-бромэтил)-5-(бромметил)-3-нитро-1Н-пиразол (19,7 г). Объединенные фильтраты упаривали и процедуру проводили еще раз, получая дополнительно 9,70 г 1-(2-бромэтил)-5-(бром-метил)-3-нитро-1H-пиразола. Твердые вещества объединяли и сушили под высоким вакуумом в течение 18 ч, получая с выходом 57% 26,0 г соединения 124с в виде белых кристаллов: т.пл. 95-97°С; 1Н ЯМР (300 МГц, CDCl3) δ 6.93 (s, 1Η), 4.63 (t, 2Η, J=6,0 Гц), 4.54 (s, 2Н), 3.86 (t, 2Н, J=6,0 Гц).
Пример 124d. 2-Нитро-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин 124d
В одногорлую круглодонную колбу емкостью 250 мл, оборудованную магнитной мешалкой, загружали соединение 124с (3,0 г; 9,64 ммоль) в THF (35 мл) и водный аммиак (135 мл; 25-28%-ный). Смесь перемешивали при комнатной температуре в течение 72 ч в атмосфере азота. Затем реакционную смесь концентрировали при пониженном давлении и полученный остаток распределяли между этилацетатом (100 мл) и водой (100 мл). Водный слой экстрагировали этилацетатом (2×50 мл). Объединенный органический слой промывали 10%-ным карбонатом калия (2×100 мл), рассолом (200 мл) и сушили над сульфатом натрия. Сушильный агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении, получая соединение 124d в виде желтого твердого вещества (1,23 г; 76%). MS: [М+Н]+ 169.
Пример 124е. 1-(2-Нитро-6,7-дигидропиразоло[1,5-а]пиразин-5(4Н)-ил)этанон 124е
К раствору соединения 124d (672 мг; 4,0 ммоль) в дихлорметане (20 мл) добавляли ацетилхлорид (936 мг; 12,0 ммоль) и K2CO3 (1104 мг; 8,0 ммоль). Смесь перемешивали в течение ночи. Затем фильтровали и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 100:1 дихлорметан/метанол, получая соединение 124е в виде белого твердого вещества (500 мг; 60%). MS: [М+Н]+ 211,2.
Пример 124f. 1-(2-Амино-6,7-дигидропиразоло[1,5-а]пиразин-5(4Н)-ил)этанон 124f
Одногорлую круглодонную колбу емкостью 50 мл продували азотом и в нее загружали соединение 124е (492 мг; 2,34 ммоль), 10%-ный палладий на угле (50% влаги; 234 мг) и метанол (20 мл). Колбу со смесью вакуумировали, заполняли газообразным водородом и содержимое перемешивали при комнатной температуре в течение 2 ч. Затем откачивали водород и колбу заполняли азотом. Катализатор удаляли фильтрованием через набивку CELITE® и фильтрат концентрировали при пониженном давлении, получая соединение 124f (380 мг; 80%). MS: [М+Н]+ 181,1.
Пример 124g. 3-(5-Ацетил-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин-2-иламино)-5-бром-1-метилпиридин-2(1Н)-он 124g
В одногорлую круглодонную колбу емкостью 50 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали соединение 124f (270 мг; 1,5 ммоль), 1,4-диоксан (20 мл), Pd2(dba)3 (137 мг; 0,15 ммоль), ксантфос (173 мг; 0,30 ммоль) и карбонат цезия (978 мг; 3,0 ммоль). После трех циклов вакуумирования/продувания аргоном смесь нагревали при 100°С в течение 6 ч. По прошествии этого времени реакционную смесь охлаждали до комнатной температуры. Затем фильтровали и фильтрат упаривали в вакууме. Остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 50:1 дихлорметан/метанол, получая соединение 124g (540 мг; 89%) в виде желтого твердого вещества. MS: [М+Н]+ 368,0.
Пример 124h. 3-(5-Ацетил-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин-2-иламино)-1-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пириди,н-2(1Н)-он 124h
В одногорлую круглодонную колбу емкостью 50 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали соединение 124g (365 мг; 1,0 ммоль), Pin2B2 (1,26 г; 5,0 ммоль), Pd2(dba)3 (91 мг; 0,10 ммоль), X-phos (92 мг; 0,20 ммоль), ацетат калия (294 мг; 3,0 ммоль) и диоксан (10 мл). После трех циклов вакуумирования/продувания аргоном смесь нагревали при 60°С в течение 16 ч. Затем охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 50:1 метиленхлорид/метанол, получая соединение 124h в виде коричневого твердого вещества (330 мг; 80%). MS: [М+Н]+ 414,2.
Пример 124i. 4-(5-(5-Ацетил-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин-2-иламино)-1-метил-6-оксо-1,6-дигидропиридин-3-ил)-2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)никотинальдегид 124i

Следуя методике, описанной в примере 123d и исходя из 2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-хлорникотинальдегида 103b (200 мг; 0,57 ммоль) и соединения 124h (343 мг; 0,83 ммоль), получали соединение 124i в виде желтого твердого вещества (300 мг; 86%). MS-ESI: [М+Н]+ 611,3.
Пример 124. 2-[4-[5-[(5-Ацетил-6,7-дигидро-4Н-пиразоло[1,5-а]пиразин-2-ил)амино]-1-метил-6-оксо-3-пиридил]-3-(гидроксиметил)-2-пиридил]-6-трет-бутил-8-фтор-фталазин-1-он 124
Следуя методике из примера 120 и исходя из соединения 124i (200 мг; 0,33 ммоль), получали соединение 124 в виде белого твердого вещества (54 мг; 27%). MS-ESI: [М+Н]+ 613,3. 1Н ЯМР (500 МГц, DMSO-d6, Τ=80°С) δ 8.53 (d, J=8,0 Гц, 1H), 8.47 (d, J=5,0 Гц, 1H), 7.94-7.92 (m, 2Н), 7.84 (d, J=2,0 Гц, 1H), 7.67 (dd, J=2,5; 22,0 Гц, 1H), 7.46 (d, J=8,5 Гц, 1H), 7.34 (d, J=4,0 Гц, 1H), 5.98 (s, 1Н), 4.63-4.57 (m, 3Н), 4.44 (d, J=8,0 Гц, 2H), 3.98 (bs, 2Н), 3.89-3.86 (m, 2Н), 3.58 (s, 3Н), 2.08 (s, 3Н), 1.41 (s, 9Н).
Пример 125а. 5-Бром-1-метил-3-(5-метилоксазол-2-иламино)пиридин-2(1Н)-он 125а
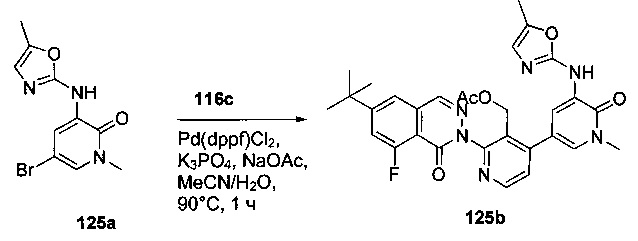
В одногорлую круглодонную колбу емкостью 100 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали 5-метилоксазол-2-амин (276 мг; 2,82 ммоль), 3,5-дибром-1-метилпиридин-2(1Н)-он (753 мг; 2,82 ммоль), трис-(дибензилиденацетон)дипалладий(0) (256 мг; 0,28 ммоль), ксантфос (324 мг; 0,56 ммоль), Cs2CO3 (1,8 г; 5,64 ммоль) и 1,4-диоксан (30 мл). После трех циклов вакуумирования/продувания аргоном смесь нагревали при 92°С в течение 3 ч. Затем охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 100:1 дихлорметан/метанол, получая соединение 125а в виде белого твердого вещества (702 мг; 88%). MS-ESI: [М+Н]+ 284,1.
Пример 125b. (2-(6-трет-Бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-(1-метил-5-(5-метилоксазол-2-иламино)-6-оксо-1,6-дигидропиридин-3-ил)пиридин-3-ил)метилацетат 125b
В одногорлую круглодонную колбу емкостью 50 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали соединение 125а (150 мг; 0,53 ммоль), 3-(ацетоксиметил)-2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)пиридин-4-илбороновую кислоту 116с (438 мг; 1,06 ммоль), Pd(dppf)Cl2 (39 мг; 0,053 ммоль), K3PO4 (225 мг; 1,06 ммоль), ацетат натрия (87 мг; 1,06 ммоль), воду (0,5 мл) и ацетонитрил (10 мл). После трех циклов вакуумирования/продувания аргоном смесь нагревали при 90°С в течение 1 ч. Затем охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали колоночной хроматографией на силикагеле с элюированием смесью дихлорметан/метанол (от 100:1 до 50:1), получая соединение 125b в виде желтого твердого вещества (120 мг; 40%). MS-ESI: [М+Н]+ 573,3.
Пример 125. 6-трет-Бутил-8-фтор-2-[3-(гидроксиметил)-4-[1-метил-5-[(5-метилоксазол-2-ил)амино]-6-оксо-3-пиридил]-2-пиридил]фталазин-1-он 125
Смесь соединения 125b (114 мг; 0,20 ммоль) и гидроксида лития (120 мг; 5,0 ммоль) в смеси изопропанол/ТНР/вода (2:2:1;10 мл) перемешивали при 35°С в течение 30 мин. Смесь концентрировали при пониженном давлении и остаток разбавляли водой (5 мл). Полученную смесь три раза экстрагировали дихлорметаном. Объединенный органический слой затем концентрировали при пониженном давлении и полученный остаток очищали препаративной обращенно-фазовой HPLC, получая соединение 125 (52 мг; 49%). MS-ESI: [М+Н]+ 530,9. 1Н ЯМР (500 МГц, DMSO-d6) δ 9.25 (s, 1Н), 8.57 (d, J=5,0 Гц, 1H), 8.53 (d, J=3,0 Гц, 1H), 8.25 (d, J=2,5 Гц, 1H), 7.90 (d, J=1,0 Гц, 1H), 7.78 (dd, J=1,0; 13,0 Гц, 1H), 7.61 (d, J=2,0 Гц, 1H), 7.50 (d, J=5,0 Гц, 1H), 6.64 (d, J=1,5 Гц, 1H), 4.92 (bs, 1Η), 4.40 (d, J=7,0 Гц, 2H), 3.60 (s, 3Н), 2.22 (s, 3Н), 1.39 (s, 9Н).
Пример 126а. 3-Циклопропил-3-оксопропаннитрил 126а
К раствору ацетонитрила (0,34 мл; 6,58 ммоль) в THF (3 мл) при -78°С под защитой N2 по каплям добавляли диизопропиламид лития (3,3 мл; 2Mb THF; 6,58 ммоль). Реакционную смесь перемешивали при -78°С в течение 3 ч. Затем добавляли этилциклопропанкарбоксилат (0,50 г; 4,38 ммоль) в THF (2 мл) и смесь оставляли нагреваться до комнатной температуры в течение 1 ч. Добавляли воду (2 мл) и растворитель удаляли при пониженном давлении. Добавляли дихлорметан (2 мл) и рН смеси подводили до 5, используя 2 н. HCl. Затем экстрагировали дихлорметаном (5 мл×2). Объединенный органический слой сушили над Na2SO4 и концентрировали, получая соединение 126а в виде желтого масла, которое использовали на следующей стадии без дополнительной очистки.
Пример 126b. 3-Циклопропил-1Н-пиразол-5-амин 126b
К раствору соединения 126а (477 мг; 4,38 ммоль) в метаноле (5 мл) добавляли гидразина гидрат (5 мл; 80%). Реакционную смесь нагревали при 75°С в течение 15 ч. Метанол удаляли при пониженном давлении и остаток экстрагировали дихлорметаном (2×8 мл). Объединенный экстракт сушили над Na2SO4 и концентрировали. Остаток очищали на колонке для флэш-хроматографии с элюированием смесью 100:1 дихлорметан/метанол, получая соединение 126b в виде желтого масла (250 мг; 46% за две стадии). MS: [М+Н]+ 124.
Пример 126с. трет-Бутил-5-амино-3-циклопропил-1Н-пиразол-1-карбоксилат 126с
К смеси соединения 126b (0,25 г; 2,0 ммоль) и K2CO3 (0,828 г; 6,0 ммоль) в THF (5 мл) добавляли (Вос)2O (0,436 г; 2,0 ммоль) в THF (5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 15 ч. Затем фильтровали и фильтрат концентрировали при пониженном давлении. Остаток очищали на колонке для флэш-хроматографии с элюированием смесью 6:1 петролейный эфир/этилацетат, получая соединение 126с в виде белого твердого вещества (240 мг; 54%). MS: [М-Вос]+ 124.
Пример 126d. 5-Бром-3-(3-циклопропил-1Н-пиразол-5-иламино)-1-метилпиридин-2(1Н)-он 126d
В одногорлую круглодонную колбу емкостью 100 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали 1,4-диоксан (15 мл), соединение 126с (455 мг; 1,95 ммоль), 3,5-дибром-1-метилпиридин-2(1Н)-он (0,40 г; 1,5 ммоль) и карбонат цезия (1,22 г; 3,75 ммоль). После барботирования азота через полученную смесь в течение 30 минут добавляли ксантфос (87 мг; 0,15 ммоль) и трис(дибензилиденацетон)дипалладий(0) (70 мг; 0,075 ммоль). Реакционную смесь кипятили с обратным холодильником в течение 15 ч. По прошествии этого времени реакционную смесь охлаждали до комнатной температуры и фильтровали. Фильтрат распределяли между этилацетатом (30 мл) и водой (30 мл). Водный слой отделяли и экстрагировали этилацетатом (2×50 мл). Объединенный органический слой промывали рассолом (50 мл) и сушили над сульфатом натрия. Сушильный агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении. Остаток очищали на колонке с силикагелем с элюированием смесью 50:1 дихлорметан/метанол, получая соединение 126d в виде желтого твердого вещества (320 мг; 70%). MS: [М+Н]+ 309. 1Н ЯМР (500 МГц, DMSO-d6) δ 11.85 (s, 1Н), 8.23 (s, 1Н), 8.02 (d, J=2,5 Гц, 1H), 7.35 (d, J=2,5 Гц, 1H), 5.77 (d, J=2,0 Гц, 1H), 3.46 (s, 3Н), 1.84-1.82 (m, 1Н), 0,92-0.90 (m, 2Н), 0,65-0.64 (m, 2Н).
Пример 126е. 5-(3-Циклопропил-1Н-пиразол-5-иламино)-1-метил-6-оксо-1,6-дигидропиридин-3-илбороновая кислота 126е
В одногорлую круглодонную колбу емкостью 50 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали соединение 126d (205 мг; 0,665 ммоль), 4,4,4',4',5,5,5',5,-октаметил-2,2,-би(1,3,2-диоксаборолан) (1,0 г; 4,0 ммоль), диоксан (16 мл), PdCl2(dppf) (54,3 мг; 0,066 ммоль) и ацетат калия (0,39 мг; 4,0 ммоль). После барботирования аргона через полученную смесь в течение 30 минут ее перемешивали при 105°С в течение 4 ч в атмосфере аргона. Затем охлаждали до комнатной температуры и фильтровали. Фильтрат упаривали при пониженном давлении, получая неочищенное сединение 126е, которое использовали без дополнительной очистки. MS: [М+Н]+ 275.
Пример 126f. 5-Бром-3-(5-циклопропил-1-метил-1Н-пиразол-3-иламино)-1-метилпиридин-2(1Н)-он 126f
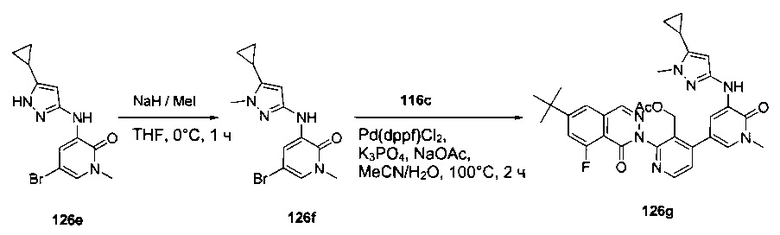
При 0°С к раствору соединения 126е (500 мг; 1,6 ммоль) в DMF (6 мл) добавляли NaH (60%-ный в масле) (80 мг; 2,0 ммоль). Реакционную смесь перемешивали при 0°С в течение 1 ч. Добавляли подметан (213 мг; 1,5 ммоль) и полученную смесь перемешивали при комнатной температуре в течение еще 2 ч. Затем к смеси добавляли воду (10 мл). Полученную суспензию фильтровали, промывали водой и сушили в вакууме, получая соединение 126f в виде белого твердого вещества (350 мг; 68%). MS-ESI: [М+Н]+ 323,1.
Пример 126g. (2-(6-трет-Бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-(5-(5-циклопропил-1-метил-1Н-пиразол-3-иламино)-1-метил-6-оксо-1,6-дигидропиридин-3-ил)пиридин-3-ил)метилацетат 126g
В герметично закрываемую колбу, оборудованную магнитной мешалкой, загружали соединение 126f (200 мг; 0,62 ммоль), (2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-3-ил)метилацетат 116с (268 мг; 0,65 ммоль), Pd(dppf)Cl2 (18 мг; 0,025 ммоль), ацетат натрия (74 мг; 0,90 ммоль), K3PO4 (191 мг; 0,90 ммоль) и смесь ацетонитрил/вода (5 мл/0,5 мл). После трех циклов вакуумирования/продувания аргоном смесь нагревали при 100°С в течение 2,0 ч. Затем охлаждали до комнатной температуры и фильтровали. Фильтрат упаривали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 10:1 дихлорметан/метанол, получая соединение 126g (100 мг; 26%) в виде коричневого твердого вещества. MS-ESI: [М+Н]+ 612,3.
Пример 126. 6-трет-Бутил-2-[4-[5-[(5-циклопропил-1-метил-пиразол-3-ил)амино]-1-метил-6-оксо-3-пиридил]-3-(гидроксиметил)-2-пиридил]-8-фтор-фталазин-1-он 126
Смесь соединения 126g (100 мг; 0,16 ммоль) и гидроксида лития (72 мг; 3,0 ммоль) в смеси изопропанол/THF (1:1; 4 мл) и воде (1 мл) перемешивали при 50°С в течение 2 ч. Смесь упаривали при пониженном давлении. Остаток очищали обращенно-фазовой комбифлэш-хроматографией, получая соединение 126 (32 мг; 35%) в виде белого твердого вещества. MS-ESI: [М+Н]+ 570,3. 1Н ЯМР (500 МГц, DMSO-d6) δ 8.63 (d, J=5,0 Гц, 1H), 8.33 (d, J=2,0 Гц, 1H), 7.94 (d, J=1,5 Гц, 1Н), 7.56-7.55 (m, 2Н), 7.53-7.51 (m, 2Н), 7.33 (s, 1Н), 5.53 (s, 1Н) 4.49-4.48 (m, 2Н), 4.04-4.02 (m, 1Н), 3.79 (s, 3Н), 3.69 (s, 3Н), 1.69-1.65 (m, 1Н), 1.43 (s, 9Н), 0,97-0.94 (m, 2Н), 0,68-0.65 (m, 2Н).
Пример 127а. 6-Хлор-2-метил-4-(5-метил-1Н-пиразол-3-иламино)-пиридазин-3(2Н)-он 127а
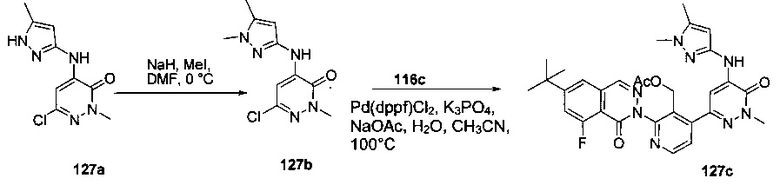
В одногорлую круглодонную колбу емкостью 100 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали 1,4-диоксан (20 мл), 5-этил-1Н-пиразол-3-амин (971 мг; 10,0 ммоль), 4-бром-6-хлор-2-метилпиридазин-3(2H)-он (2,46 г; 11,0 ммоль) и карбонат цезия (6,52 г; 20,0 ммоль). После барботирования азота через суспензию в течение 10 минут добавляли ксантфос (1,74 г; 3,0 ммоль) и трис(дибензилиденацетон)дипалладий(0) (1,37 г; 1,5 ммоль). Систему подвергали трем циклам вакуумирования/продувания аргоном и нагревали при температуре дефлегмации в течение 2 ч. Затем незамедлительно фильтровали, пока реакционная смесь оставалась горячей. Твердое вещество промывали диоксаном (3×30 мл) и объединенный фильтрат концентрировали при пониженном давлении. Остаток очищали хроматографией на силикагеле с элюированием смесью 6:1 петролейный эфир/этилацетат, получая соединение 127а (1,8 г; 75%) в виде желтого твердого вещества. MS-ESI: [М+Н]+ 239,9.
Пример 127b. 6-Хлор-4-(1,5-диметил-1Н-пиразол-3-иламино)-2-метил-пиридазин-3(2Н)-он 127b
К смеси соединения 127а (480 мг; 2,0 ммоль) в безводном DMF (10 мл) добавляли NaH (чистота 60%) (64 мг; 1,6 ммоль) при 0°С и полученную смесь перемешивали в течение 0,5 ч. К смеси по каплям при 0°С добавляли подметан (227 мг; 1,6 ммоль) в DMF (5 мл). Реакционную смесь перемешивали в течение еще 1,5 ч и гасили водой (20 мл). Затем экстрагировали дихлорметаном (3×20 мл) и объединенный органический слой упаривали при пониженном давлении. Остаток очищали обращенно-фазовой комбифлэш-хроматографией (А: 2%-ный водный NH4HCO3, В: ацетонитрил), получая соединение 127b (120 мг; 24%) в виде светло-желтого твердого вещества. MS-ESI: [М+Н]+ 254,3.
Пример 127с. (2-(6-трет-Бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-(5-(1,5-диметил-1Н-пиразол-3-иламино)-1-метил-6-оксо-1,6-дигидропиридазин-3-ил)пиридин-3-ил)метилацетат 127с
В одногорлую круглодонную колбу емкостью 50 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали соединение 127b (120 мг; 0,47 ммоль), (2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-3-ил)метилацетат 116с (536 мг; 1,3 ммоль), Pd(dppf)Cl2 (34 мг; 0,047 ммоль), K3PO4 (199 мг; 0,94 ммоль), ацетат натрия (77 мг; 0,94 ммоль), ацетонитрил (10 мл) и воду (0,2 мл). После трех циклов вакуумирования/продувания аргоном смесь нагревали при 100°С в течение 2 ч. Затем охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали колоночной хроматографией на силикагеле с элюированием смесью дихлорметан/метанол (от 100:1 до 30:1), получая соединение 127с (180 мг; 65%) в виде черного масла. MS-ESI: [М+Н]+ 587,1.
Пример 127. 6-трет-Бутил-2-[4-[5-[(1,5-диметилпиразол-3-ил)амино]-1-метил-6-оксо-пиридазин-3-ил]-3-(гидроксиметил)-2-пиридил]-8-фтор-фталазин-1-он 127
К раствору соединения 127с (176 мг; 0,30 ммоль) в пропан-2-оле (4 мл), тетрагидрофуране (4 мл) и воде (1,0 мл) добавляли гидроксид лития (72 мг; 3,0 ммоль). Смесь перемешивали при 30°С в течение 2 ч. Затем упаривали при пониженном давлении и остаток очищали препаративной обращенно-фазовой HPLC, получая соединение 127 (30 мг; 18%) в виде белого твердого вещества. MS-ESI: [M+H]+ 544,8. 1Н ЯМР (500 МГц, MeOD) δ 8.66 (d, J=5,0 Гц, 1Н), 8.51 (d, J=2,5 Гц, 1H), 7.88 (s, 1H), 7.88 (s, 1H), 7.76-7.72 (m, 2H), 5.97 (s, 1H), 4.68 (s, 2H), 3.89 (S, 3Н), 3.75 (s, 3Н), 2.29 (s, 3Н), 1.47 (s, 9H).
Пример 128а. 5-Метил-2-нитро-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин 128а
В одногорлую круглодонную колбу емкостью 1 л, оборудованную магнитной мешалкой и входным отверстием для азота, загружали THF (350 мл), соединение 124с (10,0 г; 32,2 ммоль), 2 Μ раствор метиламина в THF (113 мл; 225 ммоль) и перемешивали при комнатной температуре в течение 72 ч. По прошествии этого времени реакционную смесь концентрировали досуха при пониженном давлении и полученное твердое вещество перемешивали со смесью этилацетата (75 мл) и 10%-ного водного карбоната калия (75 мл). Водный слой отделяли и экстрагировали этилацетатом (2×75 мл). Объединенные органические экстракты промывали 10%-ным водным карбонатом калия (75 мл), затем рассолом (50 мл) и сушили над сульфатом натрия. Сушильный агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении, получая соединение 128а с выходом 97% (5,70 г) в виде желтого твердого вещества. 1Н ЯМР (300 МГц, CDCl3) δ 6.62 (s, 1Н), 4.28 (t, 2Н, J=5,4 Гц), 3.67 (s, 2Н), 2.95 (t, 2Н, J=5,4 Гц), 2.52 (s, 3Н); MS (ESI+) m/z 183,0 (M+H).
Пример 128b. 5-Метил-4,5,6,7-тетрагидропиразоло[1,5-a]пиразин-2-амин 128b
Реакционный сосуд Парра емкостью 500 мл продували азотом и в него загружали 10%-ный палладий на угле (50% влаги; 800 мг сухого веса) и раствор соединения 128а (4,00 г; 2,20 ммоль) в этаноле (160 мл). Сосуд подсоединяли к гидрогенизатору Парра, вакуумировали, заполняли газообразным водородом до давления 45 ф/кв. дюйм (310 кПа) и встряхивали в течение 2 ч. По прошествии этого времени водород откачивали и сосуд заполняли азотом. Добавляли CELITE® 521 (1,0 г) и смесь фильтровали через набивку CELITE® 521. Осадок на фильтре промывали этанолом (2×75 мл) и объединенные фильтраты концентрировали досуха при пониженном давлении, получая с выходом 99% соединение 128b (3,31 г) в виде оранжевого твердого вещества. 1Н ЯМР (300 МГц, CDCl3) δ 5.34 (s, 1Н), 3.98 (t, 2Н, J=5,4 Гц), 3.52 (s, 3Н), 2.84 (t, 2Н, J=5,7 Гц), 2.45 (s, ЗН); MS (ESI+) m/z 153,1 (M+H).
Пример 128с. 5-Бром-1-метил-3-(5-метил-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин-2-иламино) пиридин-2(1Н)-он 128с
В герметично закрываемую колбу, оборудованную магнитной мешалкой, загружали соединение 128b (1,02 г; 6,7 ммоль), 3,5-дибром-1-метилпиридин-2(1Н)-он (2,15 г; 8,1 ммоль), Pd2(dba)3 (610 мг; 0,67 ммоль), 2,2-бис(дифенилфосфино)-1,1-бинафтил (775 мг; 1,34 ммоль), карбонат цезия (4,37 г; 13,6 ммоль) и 1,4-диоксан (30 мл). После трех циклов вакуумирования/продувания аргоном смесь нагревали при 110°С в течение 2 ч. Затем фильтровали и фильтрат упаривали в вакууме. Остаток очищали колоночной хроматографией на силикагеле с элюированием смесью дихлорметан/метанол (15:1, об./об.), получая соединение 128с (380 мг; 14%) в виде белого твердого вещества. LCMS: [М+Н]+ 338.
Пример 128d. 1-Метил-3-(5-метил-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин-2-иламино)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2(1Н)-он 128d
В одногорлую круглодонную колбу емкостью 100 мл, оборудованную магнитной мешалкой и холодильником, загружали соединение 128с (1,0 г; 3 ммоль), Pin2B2 (3,8 г; 15 ммоль), Pd(dppf)Cl2 (137 мг; 0,15 ммоль), X-phos (143 мг; 0,3 ммоль), ацетат калия (88 мг; 9 ммоль) и 1,4-диоксан (50 мл). После трех циклов вакуумирования/продувания аргоном реакционную смесь нагревали при 60°С в течение 15 ч. Затем охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток промывали петролейным эфиром, получая соединение 128d в виде желтого твердого вещества (0,87 г; 75%). MS: [М+Н]+ 386.
Пример 128е. 2-(6-трет-Бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-(1-метил-5-(5-метил-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин-2-иламино)-6-оксо-1,6-дигидропиридин-3-ил)никотинальдегид 128е

В круглодонную колбу емкостью 25 мл, оборудованную обратным холодильником, загружали соединение 128d (193 мг; 0,50 ммоль), 2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-хлорникотинальдегид 103b (180 мг; 0,50 ммоль), 1,1'-бис(дифенилфосфино)ферроцендихлорпалладий(II) (17 мг; 0,025 ммоль), K3PO4 (212 мг; 1,0 ммоль), ацетат натрия (82 мг; 1,0 ммоль), ацетонитрил (6 мл) и воду (0,5 мл). После трех циклов вакуумирования/продувания аргоном смесь нагревали при 100°C в течение 1 ч. Анализ реакционной смеси с использованием LCMS показал полное превращение в желаемый продукт. Реакционную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. Остаток разбавляли дихлорметаном (20 мл) и водой (10 мл). Водный слой отделяли и экстрагировали дихлорметаном (3×10 мл). Объединенный органический слой сушили над Na2SO4 и фильтровали. Фильтрат концентрировали при пониженном давлении. Темный остаток очищали колоночной хроматографией на силикагеле с элюированием смесью дихлорметан/метанол (от 80:1 до 30:1), получая соединение 128е (190 мг; 65%) в виде желтого твердого вещества. MS-ESI: [М+Н]+ 583,3.
Пример 128. 6-трет-Бутил-8-фтор-2-[3-(гидроксиметил)-4-[1-метил-5-[(5-метил-6,7-дигидро-4Н-пиразоло[1,5-а]пиразин-2-ил)амино]-6-оксо-3-пиридил]-2-пиридил]фталазин-1-он 128
К раствору соединения 128е (158 мг; 0,27 ммоль) в смеси метанол/дихлорметан (5/5 мл) добавляли NaBH4 (31 мг; 0,82 ммоль) при комнатной температуре. После перемешивания реакционной смеси в течение 1 ч LCMS показала полное завершение реакции. Реакцию гасили водой (10 мл) и реакционную смесь концентрировали при пониженном давлении. Остаток экстрагировали дихлорметаном (3×20 мл). Объединенный органический слой промывали рассолом (30 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали препаративной обращенно-фазовой HPLC, получая соединение 128 (95 мг; 60%) в виде белого твердого вещества. MS-ESI: [М+Н]+ 585,3. 1Н ЯМР (500 МГц, CDCl3) δ 8.66 (d, J=5,0 Гц, 1H), 8.35 (d, J=2,0 Гц, 1H), 8.00 (d, J=2,5 Гц, 1H), 7.58-7.54 (m, перекрывание, 4Н), 7.44 (s, 1Н), 5.70 (s, 1Н), 4.52-4.39 (m, 2Н), 4.10-4.09 (m, 3Н), 3.70 (s, 3Н), 3.59 (s, 2Н), 2.88-2.86 (m, 2Н), 2.47 (s, 3Н), 1.43 (s, 9Н).
Пример 129а. 5-Бром-1-метил-3-(5-метилизоксазол-3-иламино)пиридин-2(1Н)-он 129а
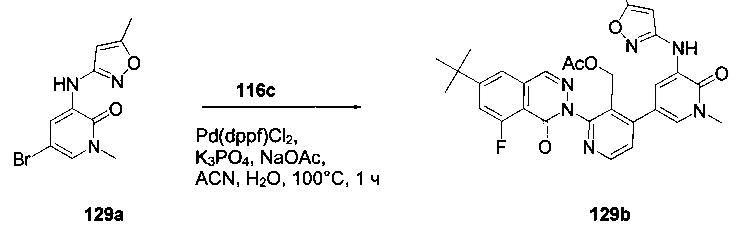
В одногорлую круглодонную колбу емкостью 100 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали 5-метилизоксазол-3-амин (1,0 г; 10,2 ммоль), 3,5-дибром-1-метилпиридин-2(1Н)-он (4,09 г; 15,3 ммоль), Pd2(dba)3 (467 мг; 0,51 ммоль), ксантфос (598 мг; 1,02 ммоль), Cs2CO3 (6,65 г; 20,4 ммоль) и диоксан (50 мл). После трех циклов вакуумирования/продувания аргоном реакционную смесь нагревали при 100°С в течение 3 ч. Анализ реакционной смеси с использованием LCMS показал полное превращение в желаемый продукт. Смесь фильтровали, когда она была еще горячей. Фильтрат охлаждали до комнатной температуры и полученный осадок собирали фильтрованием, получая соединение 129а (1,6 г; 55%) в виде желтого твердого вещества. MS-ESI: [М+Н]+ 284,1.
Пример 129b. (2-(6-трет-Бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-(1-метил-5-(5-метилизоксазол-3-иламино)-6-оксо-1,6-дигидропиридин-3-ил)пиридин-3-ил)метилацетат 129b
В одногорлую круглодонную колбу емкостью 50 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали 3-(ацетоксиметил)-2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)пиридин-4-илбороновую кислоту 116с (438 мг; 1,06 ммоль), соединение 129а (150 мг; 0,60 ммоль), Pd(dppf)Cl2 (19 мг; 0,026 ммоль), K3PO4 (224 мг; 1,06 ммоль), ацетат натрия (87 мг; 1,06 ммоль), воду (5 капель) и ацетонитрил (10 мл). После трех циклов вакуумирования/продувания аргоном реакционную смесь нагревали при 100°С в течение 1 ч. Анализ реакционной смеси с использованием LCMS показал полное превращение в желаемый продукт. Смесь охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении, получая соединение 129b (300 мг; 87%) в виде темного масла, которое использовали на следующей стадии без дополнительной очистки. MS-ESI: [М+Н]+ 573,3.
Пример 129. 6-трет-Бутил-8-фтор-2-[3-(гидроксиметил)-4-[1-метил-5-[(5-метилизоксазол-3-ил)амино]-6-оксо-3-пиридил]-2-пиридил]фталазин-1-он 129
К раствору соединения 129b (280 мг; 0,49 ммоль) в THF (4 мл), изопропаноле (4 мл) и воде (2 мл) добавляли гидроксид лития (24 мг; 0,98 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Смесь концентрировали при пониженном давлении и остаток очищали препаративной обращенно-фазовой HPLC, получая соединение 129 в виде белого твердого вещества (85 мг; 33%). MS-ESI: [М+Н]+ 531,3. 1Н ЯМР (500 МГц, DMSO-d6) δ 9.03 (s, 1Н), 8.58 (d, J=5,0 Гц, 1H), 8.54 (d, J=3,0 Гц, 1H), 7.99 (d, J=2,0 Гц, 1H), 7.91 (d, J=1,0 Гц, 1H), 7.78 (dd, J=1,0; 13,0 Гц, 1H), 7.56 (d, J=2,0 Гц, 1H), 7.51 (d, J=5,0 Гц, 1H), 6.26 (s, 1Н), 4.92 (s, 1Н), 4.43 (d, J=6,0 Гц, 2H), 3.61 (s, 3Н), 2.32 (s, 3Н), 1.40 (s, 9Н).
Пример 130а. 5-Бром-1-метил-3-(1-метил-1Н-имидазол-4-иламино)-пиридин-2(1Н)-он 130а
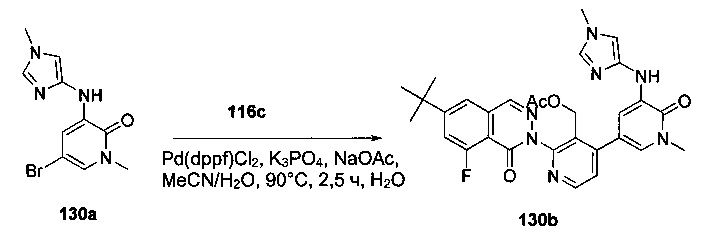
В одногорлую круглодонную колбу емкостью 100 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали 1,4-диоксан (50 мл), 1-метил-1Н-имидазол-4-амин (1,1 г; 11,3 ммоль), 3,5-дибром-1-метилпиридин-2(1Н)-он (3,0 г; 11,3 ммоль), Pd2(dba)3 (1,0 г; 1,13 ммоль), ксантфос (1,3 г; 2,26 ммоль) и карбонат цезия (7,3 г; 22,6 ммоль). После трех циклов вакуумирования/продувания аргоном смесь нагревали при 92°С в течение 4,5 ч. Затем охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали колоночной хроматографией на силикагеле с элюированием смесью дихлорметан/метанол (от 100:1 до 50:1), получая соединение 130а (2,4 г; 76%) в виде желтого твердого вещества. MS-ESI: [М+Н]+ 283,1.
Пример 130b. (2-(6-трет-Бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-(1-метил-5-(1-метил-1Н-имидазол-4-иламино)-6-оксо-1,6-дигидропиридин-3-ил)пиридин-3-ил)метилацетат 130b
В круглодонную колбу емкостью 50 мл, оборудованную магнитной мешалкой, загружали соединение 130а (150 мг; 0,53 ммоль), 3-(ацетоксиметил)-2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)пиридин-4-илбороновую кислоту 116с (438 мг; 1,06 ммоль), PdCl2(dppf) (43 мг; 0,053 ммоль), K3PO4 (225 мг; 1,06 ммоль), ацетат натрия (87 мг; 1,06 ммоль), ацетонитрил (10 мл) и воду (0,2 мл). После барботирования азота через смесь в течение 10 минут к колбе подсоединяли обратный холодильник и реакционную смесь нагревали при 90°С в течение 2,5 ч. Затем охлаждали до комнатной температуры и фильтровали. Фильтрат упаривали при пониженном давлении и полученный остаток очищали колоночной хроматографией на силикагеле с элюированием смесью дихлорметан/метанол (от 50:1 до 20:1), получая соединение 130b в виде желтого твердого вещества (120 мг; 40%). MS-ESI: [М+Н]+ 572,3.
Пример 130. 6-трет-Бутил-8-фтор-2-[3-(гидроксиметил)-4-[1-метил-5-[(1-метилимидазол-4-ил)амино]-6-оксо-3-пиридил]-2-пиридил]фталазин-1-он 130
Смесь соединения 130b (100 мг; 0,18 ммоль) и гидрата гидроксида лития (189 мг; 4,5 ммоль) в смеси изопропанол/ТНР/вода (2:2:1; 10 мл) перемешивали при 35°С в течение 30 мин. Смесь упаривали при пониженном давлении и к остатку добавляли воду (5 мл). Затем экстрагировали дихлорметаном (3×10 мл). Объединенный экстракт концентрировали при пониженном давлении и остаток очищали препаративной обращенно-фазовой HPLC, получая соединение 130 (41,1 мг; 44,4%) в виде белого твердого вещества. MS-ESI: [М+Н]+ 530,3. 1Н ЯМР (500 МГц, DMSO-d6) δ 8.56-8.53 (m, 2Н), 7.90 (d, J=2,0 Гц, 1H), 7.78 (dd, J=2,0; 12,5 Гц, 1H), 7.61 (s, 1Н), 7.51 (d, J=5,0 Гц, 1H), 7.41-7.39 (m, 2Н), 7.37 (d, J=2,0 Гц, 1H), 6.97 (d, J=1,5 Гц, 1H), 5.04-5.02 (m, 1Н), 4.41-4.39 (m, 2Н), 3.59 (s, 3Н), 3.58 (s, 3Н), 1.39 (s, 9Н).
Пример 131а. 2-Нитро-6,7-дигидро-4Н-пиразоло[5,1-с][1,4]оксазин 131а
В одногорлую круглодонную колбу емкостью 250 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали 1-(2-бромэтил)-5-(бромметил)-3-нитро-1Н-пиразол 124с (3,00 г; 9,59 ммоль) и 4 Μ водную бромистоводородную кислоту (120 мл) и полученную смесь нагревали при температуре дефлегмации в течение 24 ч. По прошествии этого времени реакционную смесь концентрировали при пониженном давлении до объема приблизительно 6 мл и остаток перемешивали в 2 Μ водном гидроксиде натрия (40 мл) в течение 2 ч. По прошествии этого времени добавляли метиленхлорид (40 мл) и смесь перемешивали в течение 15 мин. Водный слой отделяли и экстрагировали метиленхлоридом (2×50 мл). Объединенные органические экстракты промывали рассолом (100 мл) и сушили над сульфатом натрия. Сушильный агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении, получая с выходом 62% 1,01 г соединения 131а в виде белого твердого вещества: т.пл. 110-112°С; 1Н ЯМР (300 МГц, CDCl3) 6.68 (s, 1Н), 4.87 (s, 2Н), 4.28 (t, 2Н, J=5,4 Гц), 4.20 (t, 2Н, J=5,1 Гц); MS (ESI+) m/z 170,0 (M+H).
Пример 131b. 6,7-Дигидро-4Н-пиразоло[5,1-с][1,4]оксазин-2-амин 131b
Сосуд гидрогенизатора Парра емкостью 500 мл продували азотом и в него загружали соединение 131а (1,01 г; 5,92 ммоль), 10%-ный палладий на угле (50% влаги; 125 мг сухого веса) и этанол (50 мл). Сосуд вакуумировали, заполняли газообразным водородом до давления 25 ф/кв. дюйм (172,4 кПа) и встряхивали в течение 2 ч в аппарате Парра для гидрирования. Затем из сосуда откачивали водород и заполняли азотом. Катализатор удаляли фильтрованием через набивку CELITE® 521 и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией, используя силикагель 400 сс и элюируя 3%-ным метанолом в метиленхлориде. Собирали фракции, получая после концентрирования при пониженном давлении соединение 131b с выходом 73% (601 мг) в виде желтого твердого вещества: т.пл. 74-76°С; 1Н ЯМР (300 МГц, CDCl3) δ 5.37 (s, 1Н), 4.72 (s, 2Н), 4.07 (t, 2Н, J=5,1 Гц), 3.98 (t, 2Н, J=5,1 Гц), 3.57 (br s, 2H); MS (ESI+) m/z 140,4 (M+H).
Пример 131c. 5-Бром-3-(6,7-дигидро-4Н-пиразоло[5,1-c][1,4]оксазин-2-иламино)-1-метил пиридин-2(1Н)-он 131с
В трехгорлую круглодонную колбу емкостью 50 мл, оборудованную магнитной мешалкой, обратным холодильником и входным отверстием для азота, загружали 1,4-диоксан (20 мл), соединение 131b (600 мг; 4,31 ммоль), 3,5-дибром-1-метил-пиридин-2(1Н)-он (1,44 г; 5,40 ммоль) и карбонат цезия (3,08 г; 9,48 ммоль). После барботирования азота через полученный раствор в течение 30 мин добавляли ксантфос (300 мг; 0,52 ммоль) и трис(дибензилиденацетон)-дипалладий(0) (320 мг; 0,35 ммоль) и реакционную смесь нагревали при температуре дефлегмации в течение 2 ч. По прошествии этого времени реакционную смесь охлаждали до комнатной температуры, распределяли между этилацетатом (75 мл) и водой (75 мл) и фильтровали. Водный слой отделяли и экстрагировали этилацетатом (2×25 мл). Органические слои объединяли, промывали рассолом (50 мл) и сушили над сульфатом натрия. Сушильный агент удаляли фильтрованием и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией, используя силикагель 400 сс и элюируя 1%-ным метанолом в метиленхлориде. Собирали фракции, получая после концентрирования при пониженном давлении соединение 131с с выходом 31% (433 мг) в виде зеленого твердого вещества: т.пл. 195-197°С; 1Н ЯМР (300 МГц, CDCl3) 7.92 (d, 1Н, J=2,4 Гц), 7.44 (s, 1Н), 6.90 (d, 1Н, J=2,4 Гц), 5.65 (s, 1Н), 4.80 (s, 2Н), 4.13 (s, 2Н), 3.61 (s, 5Н); MS (ESI+) m/z 324,9 (M+H).
Пример 131d. 3-(6,7-Дигидро-4Н-пиразоло[5,1-с][1,4]оксазин-2-иламино)-1-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2(1Н)-он 131d
В круглодонную колбу емкостью 250 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали смесь соединения 131с (1,3 г; 4,0 ммоль), бис(пинаколато)дибора (2,03 г; 8,0 ммоль), PdCl2(dppf) (439 мг; 0,60 ммоль), ацетата калия (784 мг; 8,0 ммоль) и 1,4-диоксана (60 мл). После барботирования азота через эту смесь в течение 30 мин ее нагревали при температуре дефлегмации в течение 15 ч. По завершении реакции смесь охлаждали до комнатной температуры и фильтровали. Твердое вещество промывали этилацетатом (100 мл). Объединенный фильтрат упаривали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 30:1 дихлорметан/метанол, получая соединение 131d (446 мг; 30%). MS: [М+Н]+ 373.
Пример 131е. 2-(6-трет-Бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-(5-(6,7-дигидро-4Н-пиразоло[5,1-с][1,4]оксазин-2-иламино)-1-метил-6-оксо-1,6-дигидропиридин-3-ил)никотинальдегид 131е
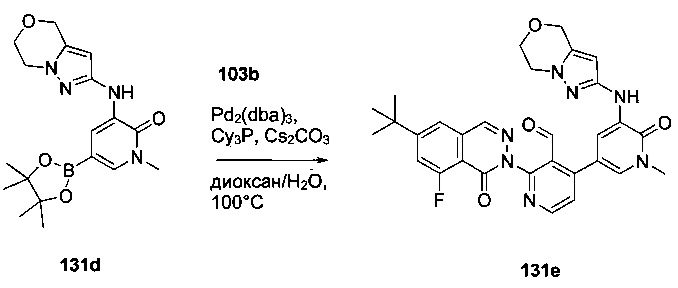
В круглодонную колбу емкостью 50 мл, оборудованную обратным холодильником, загружали 2-(6-трет)-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-хлорникотинальдегид 103b (216 мг; 1 экв.; 0,60 ммоль), соединение 131d (446 мг; 2 экв.; 1,2 ммоль), Pd2(dba)3 (55 мг; 0,1 экв.; 0,060 ммоль), Су3Р (трициклогексилфосфин) (67 мг; 0,4 экв.; 0,24 ммоль), Cs2CO3 (395 мг; 2 экв.; 1,2 ммоль), воду (0,5 мл) и диоксан (10 мл). После трех циклов вакуумирования/продувания N2 реакционную смесь перемешивали при 100°С в течение 2 ч. Затем охлаждали до комнатной температуры и фильтровали. Фильтрат упаривали при пониженном давлении и полученный остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 4:1 петролейный эфир/этилацетат, получая соединение 131е (272 мг; 80%) в виде коричневого твердого вещества. MS-ESI: [М+Н]+ 569,8.
Пример 131. 6-трет-Бутил-2-[4-[5-(6,7-дигидро-4Н-пиразоло[5,1-с][1,4]оксазин-2-иламино)-1-метил-6-оксо-3-пиридил]-3-(гидроксиметил)-2-пиридил]-8-фтор-фталазин-1-он 131
В одногорлую круглодонную колбу емкостью 50 мл, оборудованную магнитной мешалкой, загружали соединение 131е (220 мг; 1,0 экв.; 0,39 ммоль), NaBH4 (73 мг; 5,0 экв.; 1,90 ммоль) и метанол (10 мл). Смесь перемешивали при комнатной температуре в течение 1 ч и гасили водой (5 мл). Затем концентрировали при пониженном давлении и полученный остаток экстрагировали дихлорметаном (3×10 мл). Объединенный органический слой концентрировали при пониженном давлении и остаток очищали препаративной обращенно-фазовой HPLC, получая соединение 131 (105 мг; 47%). MS-ESI: [М+Н]+ 571,8. 1Н ЯМР (500 МГц, CDCl3) δ 8.66 (d, J=5,0 Гц, 1H), 8.35 (d, J=2,5 Гц, 1H), 8.03 (d, J=2,5 Гц, 1H), 7.59-7.53 (m, 4Н), 7.48 (s, 1Н), 5.73 (s, 1Н), 4.81 (s, 2Н), 4.51 (bs, 2Н), 4.12-4.05 (m, перекрывание, 5Н), 3.73 (s, 3Н), 1.46 (s, 9Н).
Пример 132а. (R)-(6-Аминопиридин-3-ил)(3-метилморфолино)метанон 132а
К раствору (R)-3-метилморфолина (2,02 г; 20 ммоль) в этаноле (25 мл) добавляли 1-этил-3-(3-диметиламинопропил)карбодиимид (EDCl) (3,33 г; 17,4 ммоль), гидроксибензотриазол (HOBt) (2,35 г; 17,4 ммоль) и 6-амино-никотиновую кислоту (2,0 г; 14,5 ммоль). После перемешивания в течение 18 ч при комнатной температуре реакционную суспензию фильтровали и фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 3:1 этилацетат/петролейный эфир, получая соединение 132а в виде белого твердого вещества (1,6 г; 36%). MS-ESI: [М+Н]+ 222,3.
Пример 132b. (R)-5-Бром-1-метил-3-(5-(3-метилморфолин-4-карбонил)-пиридин-2-иламино)пиридин-2(1Н)-он 132b
В одногорлую круглодонную колбу емкостью 100 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали соединение 132а (332 мг; 1,5 ммоль), 3,5-дибром-1-метилпиридин-2(1Н)-он (Н-001) (480 мг; 1,8 ммоль) и карбонат цезия (978 мг; 3,0 ммоль). После барботирования азота через суспензию в течение 3 минут добавляли ксантфос (87 мг; 0,15 ммоль) и трис(дибензилиденацетон)дипалладий(0) (69 мг; 0,075 ммоль). Систему подвергали трем циклам вакуумирования/продувания аргоном и нагревали при температуре дефлегмации в течение 2,5 ч. Затем охлаждали до комнатной температуры и фильтровали. Твердое вещество промывали дихлорметаном (2×50 мл) и объединенный фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с элюированием смесью петролейный эфир/этилацетат (от 2:1 до 1:2), получая соединение 132b (430 мг; 70%) в виде желтого твердого вещества. MS-ESI: [М+Н]+ 407,3.
Пример 132с. (R)-1-Метил-5-(5-(3-метилморфолин-4-карбонил)пиридин-2-иламино)-6-оксо-1,6-дигидропиридин-3-илбороновая кислота 132с

В круглодонную колбу емкостью 100 мл, оборудованную обратным холодильником, загружали соединение 132b (580 мг; 1,42 ммоль), бис(пинаколато)дибор (1,2 г; 4,5 ммоль), Pd2(dba)3 (137 мг; 0,15 ммоль), X-phos (71 мг; 0,15 ммоль), ацетат калия (294 мг; 3,0 ммоль) и 1,4-диоксан (20 мл). После трех циклов вакуумирования/продувания аргоном смесь нагревали при 70°С в течение 2 ч. Затем фильтровали и фильтрат упаривали при пониженном давлении. Остаток промывали петролейным эфиром, получая соединение 132с (500 мг; 94%) в виде белого твердого вещества, которое использовали на следующей стадии без дополнительной очистки. MS-ESI: [М+Н]+ 373,1.
Пример 132d. (R)-2-(6-трет-Бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-(1-метил-5-(5-(3-метилморфолин-4-карбонил)пиридин-2-иламино)-6-оксо-1,6-дигидропиридин-3-ил)никотинальдегид 132d
В одногорлую круглодонную колбу емкостью 25 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали соединение 132с (260 мг; 0,70 ммоль), 2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-хлорникотин-альдегид 103b (180 мг; 0,50 ммоль), K3PO4 (297 мг; 1,4 ммоль), ацетат натрия (114 мг; 1,4 ммоль), 1,1'-бис(дифенилфосфино)ферроцендихлор-палладий(II) (57 мг; 0,070 ммоль) и смесь ацетонитрил/вода (10/0,2 мл). После трех циклов вакуумирования/продувания N2 смесь нагревали при 100°С в течение 1,5 ч. Анализ реакционной смеси с использованием LCMS показал полное превращение в желаемый продукт. Реакционную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. Остаток разбавляли дихлорметаном (20 мл) и водой (20 мл). Водный слой отделяли и экстрагировали дихлорметаном (3×20 мл). Объединенный органический слой сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Темный остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 60:1 дихлорметан/метанол, получая соединение 132d (120 мг; 37%) в виде желтого твердого вещества. MS-ESI: [М+Н]+ 652,3.
Пример 132. 6-трет-Бутил-8-фтор-2-[3-(гидроксиметил)-4-[1-метил-5-[[5-[(3R)-3-метилморфолин-4-карбонил]-2-пиридил]амино]-6-оксо-3-пиридил]-2-пиридил]фталазин-1-он 132
К раствору соединения 132d (120 мг; 0,18 ммоль) в смеси метанол/дихлорметан (4/4 мл) добавляли NaBH4 (15 мг; 0,60 ммоль). Смесь перемешивали при комнатной температуре в течение 1 ч и гасили водой (2 мл). Затем упаривали при пониженном давлении и остаток очищали препаративной обращенно-фазовой HPLC, получая соединение 132 (60 мг; 50%) в виде белого твердого вещества. MS-ESI: [М+Н]+ 654,3. 1Н ЯМР (500 МГц, MeOD-d4) δ 8.94 (d, J=2,5 Гц, 1Η), 8.61 (d, J=5,0 Гц, 1Η), 8.51 (d, J=2,5 Гц, 1Η), 8.35 (d, J=2,0 Гц, 1Η), 7.88 (d, J=1,5 Гц, 1H), 7.75 (d, J=1,5 Гц, 1H), 7.72-7.69 (m, 1Н), 7.65 (d, J=5,0 Гц, 1H), 7.61 (d, J=2,5 Гц, 1H), 7.13 (d, J=7,5 Гц, 1H), 4.62-4.58 (m, 2Н), 4.36-4.31 (m, 1Н), 3.61-3.84 (m, 2Н), 3.71-3.67 (m, перекрывание, 5Н), 3.56-3.47 (m, 2Н), 1.47 (s, 9Н), 1.39 (d, J=7,0 Гц, 3Н).
Пример 133а. 5-(2-Метоксиэтил)-2-нитро-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин 133а
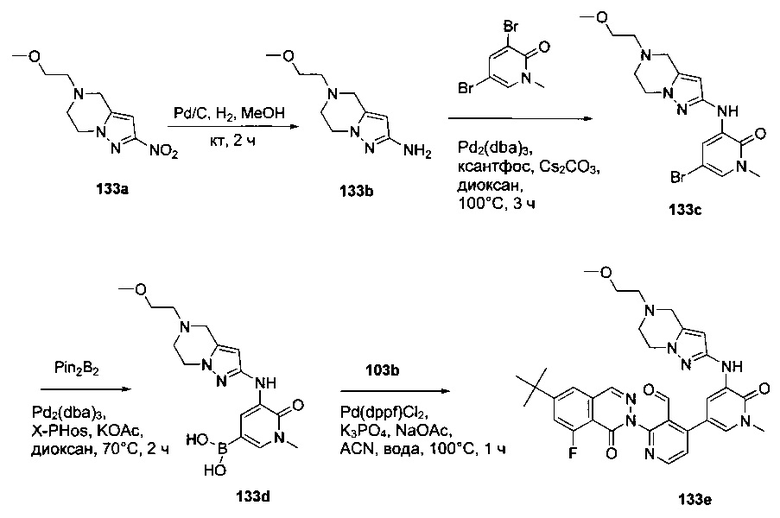
К раствору 2-нитро-4,5,6,7-тетрагидропиразоло[1,5-а]пиразина 124d (190 мг; 1,13 ммоль) в ацетонитриле (10 мл) добавляли K2CO3 (311,9 мг; 2,26 ммоль) и 1-бром-2-метоксиэтан (188,3 мг; 1,36 ммоль). Реакционную смесь нагревали при 80°С в течение 17 ч в условиях облучения микроволнами. Анализ реакционной смеси с использованием LCMS показал полное превращение в желаемый продукт. Смесь охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении, получая соединение 133а в виде белого твердого вещества (230 мг; 90%), которое использовали на следующей стадии без дополнительной очистки. MS-ESI: [М+Н]+ 227,0.
Пример 133b. 5-(2-Метоксиэтил)-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин-2-амин 133b
К раствору соединения 133а (286 мг; 1,26 ммоль) в метаноле (10 мл) добавляли Pd/C (28,6 мг). Систему вакуумировали и затем заполняли Н2. После перемешивания при комнатной температуре в течение 2 ч смесь фильтровали. Фильтрат концентрировали при пониженном давлении, получая соединение 133b в виде желтого твердого вещества (240 мг; 97%), которое использовали на следующей стадии без дополнительной очистки. MS-ESI: [М+Н]+ 197,0.
Пример 133с. 5-Бром-3-(5-(2-метоксиэтил)-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин-2-иламино)-1-метилпиридин-2(1Н)-он 133с
В круглодонную колбу емкостью 100 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали соединение 133b (230 мг; 1,17 ммоль), 3,5-дибром-1-метилпиридин-2(1Н)-он (468,4 мг; 1,76 ммоль), Pd2(dba)3 (53,5 мг; 0,0585 ммоль), ксантфос (67,6 мг; 0,117 ммоль), Cs2CO3 (762,8 мг; 2,34 ммоль) и диоксан (20 мл). После трех циклов вакуумирования/продувания N2 смесь нагревали при 100°С в течение 3 ч. Анализ реакционной смеси с использованием LCMS показал полное превращение в желаемый продукт. Смесь охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 40:1 дихлорметан/метанол, получая соединение 133с в виде темного твердого вещества (380 мг; 85%). MS-ESI: [М+Н]+ 382,2.
Пример 133d. 3-(5-(2-Метоксиэтил)-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин-2-иламино)-1-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2(1Н)-он 133d
В одногорлую круглодонную колбу емкостью 50 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали соединение 133с (330 мг; 0,86 ммоль), Pin2B2 (329 мг; 1,30 ммоль), Pd2(dba)3 (40 мг; 0,043 ммоль), X-phos (41 мг; 0,086 ммоль), ацетат калия (169 мг; 1,726 ммоль) и диоксан (10 мл). После трех циклов вакуумирования/продувания N2 смесь нагревали при 70°С в течение 2 ч. Анализ реакционной смеси с использованием LCMS показал полное превращение в желаемый продукт. Смесь охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток промывали петролейным эфиром, получая соединение 133d в виде темного масла (240 мг; 80%), которое использовали на следующей стадии без дополнительной очистки. MS-ESI: [М+Н]+ 348,3.
Пример 133е. 2-(6-трет-Бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-(5-(5-(2-метоксиэтил)-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин-2-иламино)-1-метил-6-оксо-1,6-дигидропиридин-3-ил)никотинальдегид 133е
В круглодонную колбу емкостью 25 мл, оборудованную обратным холодильником, загружали 2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-хлорникотинальдегид 103b (100 мг; 0,28 ммоль), соединение 133d (144,4 мг; 0,42 ммоль), Pd(dppf)Cl2 (11,3 мг; 0,0139 ммоль), K3PO4 (117,8 мг; 0,556 ммоль), ацетат натрия (45,6 мг; 0,556 ммоль), ацетонитрил (10 мл) и воду (3 капли). После трех циклов вакуумирования/продувания N2 смесь нагревали при 100°С в течение 1 ч под защитой N2. Анализ реакционной смеси с использованием LCMS показал полное превращение в желаемый продукт. Смесь охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 40:1 дихлорметан/метанол, получая соединение 133е в виде желтого твердого вещества (140 мг; 80%). MS-ESI: [М+Н]+ 627,3.
Пример 133. 6-трет-Бутил-8-фтор-2-[3-(гидроксиметил)-4-[5-[[5-(2-метоксиэтил)-6,7-дигидро-4Н-пиразоло[1,5-а]пиразин-2-ил]амино]-1-метил-6-оксо-3-пиридил]-2-пиридил]фталазин-1-он 133
К раствору соединения 133е (180 мг; 0,287 ммоль) в дихлорметане (5 мл) и метаноле (5 мл) добавляли NaBH4 (21,7 мг; 0,574 ммоль). После перемешивания при комнатной температуре в течение 1 ч смесь гасили водным NH4Cl (5 мл) и концентрировали при пониженном давлении. Остаток экстрагировали дихлорметаном (3×20 мл). Объединенный органический слой промывали рассолом, сушили над Na2SO4, концентрировали при пониженном давлении и очищали препаративной обращенно-фазовой HPLC, получая соединение 133 (50 мг; 27%) в виде белого твердого вещества. MS-ESI: [М+Н]+ 629,3. 1Н ЯМР (500 МГц, DMSO-d6) δ 8.57 (d, J=5,5 Гц, 1H), 8.54 (d, J=2,5 Гц, 1H), 8.22 (s, 1Н), 8.04 (d, J=2,5 Гц, 1H), 7.90 (d, J=1,5 Гц, 1H), 7.78 (dd, J=1,5; 13,5 Гц, 1H), 7.51 (d, J=5,5 Гц, 1H), 7.39 (d, J=2,0 Гц, 1H), 5.89 (s, 1Н), 4.90-4.88 (m, 1Н), 4.42 (s, 2Н), 3.92 (t, J=5,5 Гц, 2H), 3.61 (s, 2Н), 3.59 (s, 3Н), 3.50 (t, J=5,0 Гц, 2H), 3.25 (s, 3Н), 2.90 (t, J=5,5 Гц, 2H), 2.67 (t, J=5,0 Гц, 2H), 1.40 (s, 9Н).
Пример 134а. 2-Нитро-5-(2,2,2-трифторэтил)-4,5,6,7-тетрагидро-пиразоло[1,5-а]пиразин 134а
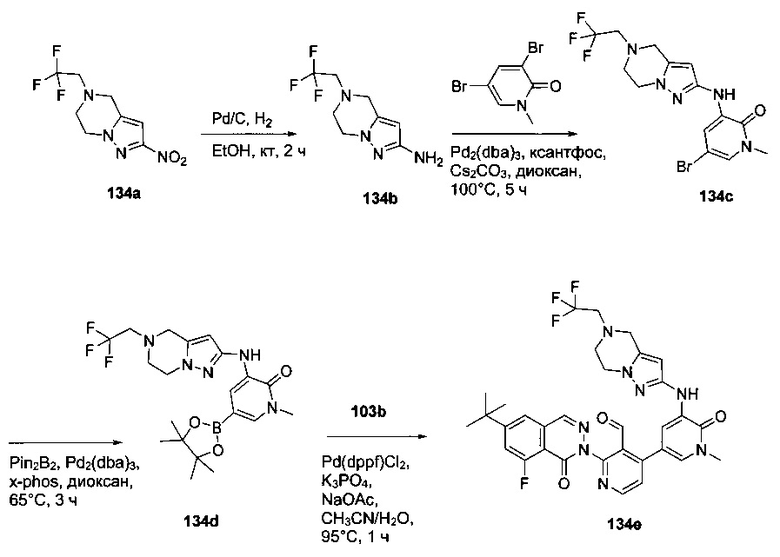
В герметично закрываемую колбу, оборудованную магнитной мешалкой, загружали 1-(2-бромэтил)-5-(бромметил)-3-нитро-1Н-пиразол 124с (632 мг; 2,0 ммоль), 2,2,2-трифторэтанамин (594 мг; 6,0 ммоль) и DMSO (5 мл) и нагревали при 120°С в течение 2 ч. Смесь охлаждали до комнатной температуры и разбавляли водой (20 мл). Полученную смесь экстрагировали этилацетатом (3×20 мл). Объединенный органический слой сушили и фильтровали. Фильтрат концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 50:1 дихлорметан/метанол, получая соединение 134а (392 мг; 78%) в виде желтого твердого вещества. MS-ESI: [М+Н]+ 250,9.
Пример 134b. 5-(2,2,2-Трифторэтил)-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин-2-амин 134b
К раствору соединения 134а (390 мг; 1,56 ммоль) в этаноле (20 мл) добавляли Pd/C (примерно 200 мг). Реакционный сосуд заполняли газообразным водородом (с использованием баллона) и содержимое перемешивали при комнатной температуре в течение 2 ч. После завершения реакции смесь фильтровали через набивку CELITE®. Фильтрат концентрировали при пониженном давлении, получая соединение 134b в виде желтого твердого вещества (308 мг; 90%), которое использовали на следующей стадии без дополнительной очистки. MS-ESI: [М+Н]+ 221,1.
Пример 134с. 5-Бром-1-метил-3-(5-(2,2,2-трифторэтил)-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин-2-иламино)пиридин-2(1Н)-он 134с
В круглодонную колбу емкостью 100 мл, оборудованную обратным холодильником, загружали соединение 134b (300 мг; 1,36 ммоль), 3,5-дибром-1-метилпиридин-2(1Н)-он (364 мг; 1,36 ммоль), карбонат цезия (887 мг; 2,7 ммоль) и 1,4-диоксан (20 мл). После барботирования азота через суспензию в течение 10 минут добавляли ксантфос (78 мг; 0,136 ммоль) и Pd2(dba)3 (62 мг; 0,068 ммоль). Систему подвергали трем циклам вакуумирования/продувания аргоном и нагревали при температуре дефлегмации в течение 5 ч. Затем охлаждали до комнатной температуры и фильтровали. Твердое вещество промывали дихлорметаном (30 мл). Объединенный фильтрат концентрировали при пониженном давлении. Твердый остаток очищали колоночной хроматографией на силикагеле с элюированием смесью дихлорметан/метанол (от 80/1 до 30/1), получая соединение 134с (420 мг; 76%) в виде желтого твердого вещества. MS-ESI: [M+H]+ 406,0.
Пример 134d. 1-Метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3-(5-(2,2,2-трифтор-этил)-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин-2-иламино)пиридин-2(1Н)-он 134d
В одногорлую круглодонную колбу емкостью 50 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали соединение 134с (400 мг; 0,98 ммоль), Pin2B2 (750 мг; 2,96 ммоль), Pd2(dba)3 (45 мг; 0,05 ммоль), x-phos (48 мг; 0,1 ммоль), ацетат калия (294 мг; 3,0 ммоль) и 1,4-диоксан (15 мл). Реакционную смесь подвергали трем циклам вакуумирования/продувания аргоном и нагревали при 65°С в течение 3 ч. Затем охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении. Полученный остаток промывали петролейным эфиром, получая соединение 134d (400 мг; 90%) в виде коричневого твердого вещества, которое использовали на следующей стадии без дополнительной очистки. MS-ESI: [М+Н]+ 453,9.
Пример 134е. 2-(6-трет-Бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-(1-метил-6-оксо-5-(5-(2,2,2-трифторэтил)-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин-2-иламино)-1,6-дигидропиридин-3-ил)никотинальдегид 134е
В круглодонную колбу емкостью 25 мл, оборудованную обратным холодильником, загружали соединение 134d (200 мг; 0,44 ммоль), 2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-хлорникотинальдегид 103b (159 мг; 0,44 ммоль), K3PO4 (186 мг; 0,88 ммоль), ацетат натрия (72 мг; 0,88 ммоль), 1,1'-бис(дифенилфосфино)ферроцендихлорпалладий(II) (16 мг; 0,022 ммоль) и смесь ацетонитрил/вода (8/0,2 мл). После трех циклов вакуумирования/продувания N2 смесь нагревали при 100°С в течение 1 ч под защитой N2. Анализ реакционной смеси с использованием LCMS показал полное превращение в желаемый продукт. Реакционную смесь охлаждали до комнатной температуры и разбавляли дихлорметаном (30 мл) и водой (15 мл). Водный слой отделяли и экстрагировали дихлорметаном (2×15 мл). Объединенный органический экстракт сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Темный остаток очищали колоночной хроматографией на силикагеле с элюированием смесью дихлорметан/метанол (от 80/1 до 30/1), получая соединение 134е (128 мг; 45%) в виде желтого твердого вещества. MS-ESI: [М+Н]+ 651,3.
Пример 134. 6-трет-Бутил-8-фтор-2-[3-(гидроксиметил)-4-[1-метил-6-оксо-5-[[5-(2,2,2-трифторэтил)-6,7-дигидро-4Н-пиразоло[1,5-а]пиразин-2-ил]амино]-3-пиридил]-2-пиридил]фталазин-1-он 134
К раствору соединения 134е (110 мг; 0,169 ммоль) в смеси метанол/дихлорметан (4/4 мл) добавляли NaBH4 (19 мг; 0,51 ммоль) при комнатной температуре. После перемешивания реакционной смеси в течение 1 ч LCMS показала полное завершение реакции. Смесь гасили водой (20 мл) и экстрагировали дихлорметаном (3×30 мл). Объединенный органический слой промывали рассолом (20 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали препаративной обращенно-фазовой HPLC, получая соединение 134 (50 мг; 45%) в виде белого твердого вещества. MS-ESI: [М+Н]+ 653,3. 1Н ЯМР (500 МГц, CDCl3) δ 8.66 (d, J=5,0 Гц, 1H), 8.35 (d, J=2,5 Гц, 1H), 8.00 (d, J=2,5 Гц, 1H), 7.59-7.58 (m, 1Н), 7.56-7.55 (m, 1Н), 7.53-7.52 (m, 2Н), 7.45 (s, 1Н), 5.73 (s, 1Н), 4.51-4.49 (m, 2Н), 4.11-4.08 (m, 2Н), 4.05-4.03 (m, 1Н), 3.94 (s, 2Н), 3.73 (s, 3Н), 3.24-3.18 (m, 4Н), 1.45 (s, 9Н).
Пример 135а. 5-(2,2-Дифторэтил)-2-нитро-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин 135а
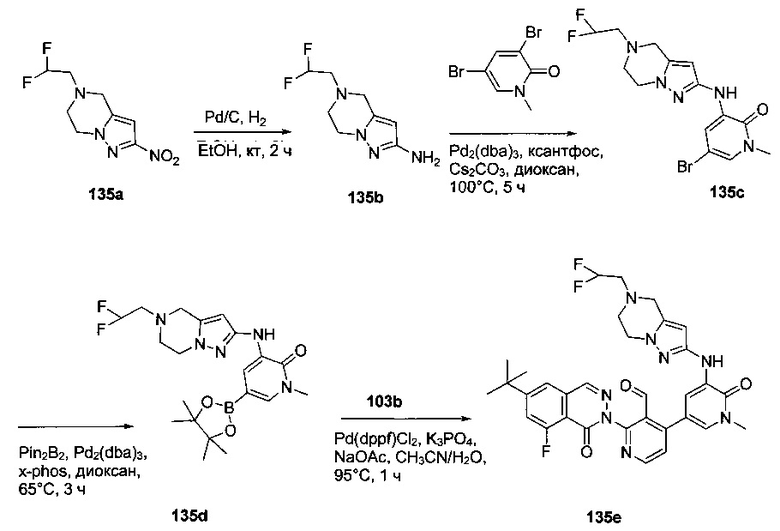
В герметично закрываемую колбу, оборудованную магнитной мешалкой, загружали 1-(2-бромэтил)-5-(бромметил)-3-нитро-1Н-пиразол 124с (632 мг; 2,0 ммоль), 2,2-дифторэтанамин (486 мг; 6,0 ммоль) и DMSO (5 мл). Все это нагревали при 120°С в течение 2 ч. Смесь охлаждали до комнатной температуры и разбавляли водой (20 мл). Полученную смесь экстрагировали этилацетатом (3×20 мл). Объединенный органический слой сушили и фильтровали. Фильтрат концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 50:1 дихлорметан/метанол, получая соединение 135а (371 мг; 80%) в виде желтого твердого вещества. MS-ESI: [М+Н]+ 233,2.
Пример 135b. 5-(2,2-Дифторэтил)-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин-2-амин 135b
К раствору соединения 135а (370 мг; 1,59 ммоль) в этаноле (20 мл) добавляли Pd/C (примерно 200 мг). Реакционный сосуд заполняли газообразным водородом (с использованием баллона) и содержимое перемешивали при комнатной температуре в течение 2 ч. После завершения реакции смесь фильтровали через набивку CELITE®. Фильтрат концентрировали при пониженном давлении, получая соединение 135b в виде желтого твердого вещества (293 мг; 91%), которое использовали на следующей стадии без дополнительной очистки. MS-ESI: [М+Н]+ 203,2.
Пример 135с. 5-Бром-3-(5-(2,2-дифторэтил)-4,5,6,7-тетрагидро-пиразоло[1,5-а]пиразин-2-иламино)-1-метилпиридин-2(1Н)-он 135с
В круглодонную колбу емкостью 50 мл, оборудованную обратным холодильником, загружали соединение 135b (290 мг; 1,43 ммоль), 3,5-дибром-1-метилпиридин-2(1Н)-он (383 мг; 1,43 ммоль), карбонат цезия (932 мг; 2,86 ммоль) и 1,4-диоксан (20 мл). После барботирования азота через суспензию в течение 10 минут добавляли ксантфос (82 мг; 0,143 ммоль) и Pd2(dba)3 (65 мг; 0,072 ммоль). Систему подвергали трем циклам вакуумирования/продувания аргоном и нагревали при температуре дефлегмации в течение 5 ч. Затем охлаждали до комнатной температуры и фильтровали. Твердое вещество промывали дихлорметаном (30 мл). Объединенный органический фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с элюированием смесью дихлорметан/метанол (от 80/1 до 30/1), получая соединение 135с (400 мг; 72%) в виде желтого твердого вещества. MS-ESI: [М+Н]+ 387,8.
Пример 135d. 3-(5-(2,2-Дифторэтил)-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин-2-иламино)-1-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2(1Н)-он 135d
В одногорлую круглодонную колбу емкостью 50 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали соединение 135с (400 мг; 1,03 ммоль), Pin2B2 (785 мг; 3,1 ммоль), Pd2(dba)3 (45 мг; 0,050 ммоль), X-phos (48 мг; 0,10 ммоль), ацетат калия (294 мг; 3,0 ммоль) и 1,4-диоксан (20 мл). Реакционную смесь подвергали трем циклам вакуумирования/продувания аргоном и нагревали при 65°С в течение 3 ч. Затем охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении. Полученный остаток промывали петролейным эфиром, получая соединение 135d (412 мг; 92%) в виде коричневого твердого вещества, которое использовали на следующей стадии без дополнительной очистки. MS-ESI: [М+Н]+ 436,0.
Пример 135е. 2-(6-трет-Бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-(5-(5-(2,2-дифторэтил)-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин-2-иламино)-1-метил-6-оксо-1,6-дигидропиридин-3-ил)никотинальдегид 135е
В круглодонную колбу емкостью 25 мл, оборудованную обратным холодильником, загружали соединение 135d (200 мг; 0,46 ммоль), 2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-хлорникотинальдегид 103b (166 мг; 0,46 ммоль), K3PO4 (195 мг; 0,92 ммоль), ацетат натрия (75 мг; 0,92 ммоль), 1,1'-бис(дифенилфосфино)ферроцендихлорпалладий(II) (17 мг; 0,023 ммоль) и смесь ацетонитрил/вода (8/0,2 мл). После трех циклов вакуумирования/продувания N2 смесь нагревали при 100°С в течение 1 ч. Анализ реакционной смеси с использованием LCMS показал полное превращение в желаемый продукт.Реакционную смесь охлаждали до комнатной температуры и разбавляли дихлорметаном (30 мл) и водой (15 мл). Водный слой экстрагировали дихлорметаном (2×15 мл). Объединенный органический экстракт сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Темный остаток очищали колоночной хроматографией на силикагеле с элюированием смесью дихлорметан/метанол (от 80/1 до 30/1), получая соединение 135е (110 мг; 38%) в виде желтого твердого вещества. MS-ESI: [М+Н]+ 633,3.
Пример 135. 6-трет-Бутил-2-[4-[5-[[5-(2,2-дифторэтил)-6,7-дигидро-4Н-пиразоло[1,5-а]пиразин-2-ил]амино]-1-метил-6-оксо-3-пиридил]-3-(гидроксиметил)-2-пиридил]-8-фтор-фталазин-1-он 135
К раствору соединения 135е (100 мг; 0,158 ммоль) в смеси метанол/дихлорметан (4/4 мл) добавляли NaBH4 (18 мг; 0,475 ммоль) при комнатной температуре. После перемешивания реакционной смеси в течение 1 ч LCMS показала полное завершение реакции. Реакционную смесь гасили водой (20 мл) и экстрагировали дихлорметаном (3×20 мл). Объединенный органический слой промывали рассолом (20 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали препаративной обращенно-фазовой HPLC, получая соединение 135 (60 мг; 60%) в виде белого твердого вещества. MS-ESI: [М+Н]+ 635,3. 1Н ЯМР (500 МГц, CDCl3) δ 8.66 (d, J=5,0 Гц, 1H), 8.35 (d, J=2,5 Гц, 1H), 8.00 (d, J=2,5 Гц, 1H), 7.59-7.58 (m, 1Н), 7.56-7.55 (m, 1Н), 7.53-7.52 (m, 2Н), 7.45 (s, 1Н), 6.07-5.83 (m, 1Н), 5.72 (s, 1Н), 4.51-4.49 (m, 2H), 4.11-4.08 (m, 2H), 4.05-4.03 (m, 1H), 3.94 (s, 2H), 3.73 (s, 3H), 3.13-3.10 (m, 2H), 3.00-2.94 (m, 2H), 1.45 (s, 9H).
Пример 136a. (S)-(6-Аминопиридин-3-ил)(3-метилморфолино)метанон 136a
К раствору (S)-3-метилморфолина (1,5 г; 15,0 ммоль) в этаноле (20 мл) добавляли EDCI (3,33 г; 17,4 ммоль), HOBt (2,35 г; 17,4 ммоль) и 6-амино-никотиновую кислоту (2,07 г; 15,0 ммоль) при комнатной температуре. После перемешивания в течение 18 ч полученную суспензию фильтровали. Твердое вещество очищали колоночной хроматографией на силикагеле с элюированием от смеси 2:1 петролейный эфир/этилацетат до этилацетата, получая соединение 136а (1,0 г; 30%) в виде белого твердого вещества. MS-ESI: 222,3 (М+Н)+.
Пример 136b. (S)-5-Бром-1-метил-3-(5-(3-метилморфолин-4-карбонил)пиридин-2-иламино)пиридин-2(1Н)-он 136b
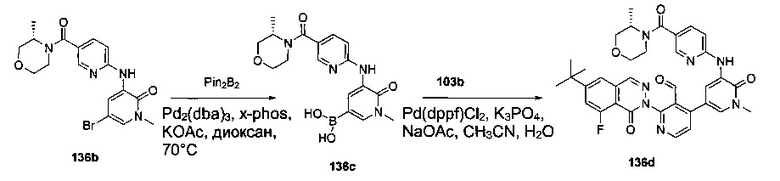
В одногорлую круглодонную колбу емкостью 50 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали соединение 136а (222 мг; 1,0 ммоль), 3,5-дибром-1-метилпиридин-2(1Н)-он (320 мг; 1,2 ммоль), карбонат цезия (652 мг; 2 ммоль) и 1,4-диоксан (10 мл). После барботирования азота через суспензию в течение 10 минут добавляли ксантфос (58 мг; 0,10 ммоль) и трис(дибензилиденацетон)дипалладий(0) (46 мг; 0,050 ммоль). Систему подвергали трем циклам вакуумирования/продувания аргоном и нагревали при температуре дефлегмации в течение 2,5 ч. Затем охлаждали до комнатной температуры и фильтровали. Твердое вещество промывали дихлорметаном (2×30 мл) и объединенный фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с элюированием смесью петролейный эфир/этилацетат (от 2:1 до 1:2), получая соединение 136b (280 мг; 69%) в виде желтого твердого вещества. MS-ESI: [М+Н]+ 407,3.
Пример 136с. (S)-1-Метил-5-(5-(3-метилморфолин-4-карбонил)пиридин-2-иламино)-6-оксо-1,6-дигидропиридин-3-илбороновая кислота 136с
В круглодонную колбу емкостью 100 мл, оборудованную обратным холодильником, загружали соединение 136b (600 мг; 1,5 ммоль), бис(пинаколато)дибор (1,2 г; 4,5 ммоль), Pd2(dba)3 (137 мг; 0,15 ммоль), X-phos (71 мг; 0,15 ммоль), ацетат калия (294 мг; 3,0 ммоль) и 1,4-диоксан (20 мл). После трех циклов вакуумирования/продувания аргоном смесь нагревали при 70°С в течение 2 ч. Затем фильтровали и фильтрат упаривали при пониженном давлении. Остаток промывали петролейным эфиром, получая соединение 136с (520 мг; 93%) в виде белого твердого вещества, которое использовали непосредственно без дополнительной очистки. MS-ESI: [М+Н]+ 373,1.
Пример 136d. (S)-2-(6-трет-Бутил-в-фторН -оксофталазин-2(1Н)-ил)-4-(1-метил-5-(5-(3-метилморфолин-4-карбонил)пиридин-2-иламино)-6-оксо-1,6-дигидропиридин-3-ил)никотинальдегид 136d
В одногорлую круглодонную колбу емкостью 100 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали соединение 136с (260 мг; 0,70 ммоль), 2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-хлорникотинальдегид 103b (180 мг; 0,50 ммоль), K3PO4 (297 мг; 1,4 ммоль), ацетат натрия (114 мг; 1,4 ммоль), 1,1'-бис(дифенилфосфино) ферроцендихлорпалладий(II) (57 мг; 0,07 ммоль) и смесь ацетонитрил/вода (10/0,2 мл). После трех циклов вакуумирования/продувания N2 смесь нагревали при 100°С в течение 1,5 ч. Анализ реакционной смеси с использованием LCMS показал полное превращение в желаемый продукт. Реакционную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. Остаток разбавляли дихлорметаном (20 мл) и водой (10 мл). Водный слой отделяли и экстрагировали дихлорметаном (3×20 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Темный остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 60:1 дихлорметан/метанол, получая соединение 136d (140 мг; 43%) в виде желтого твердого вещества. MS-ESI: [М+Н]+ 652,3.
Пример 136. 6-трет-Бутил-8-фтор-2-[3-(гидроксиметил)-4-[1-метил-5-[[5-[(38)-3-метилморфолин-4-карбонил]-2-пиридил]амино]-6-оксо-3-пиридил]-2-пиридил]фталазин-1-он 136
К раствору соединения 136d (140 мг; 0,21 ммоль) в смеси метанол/дихлорметан (4/4 мл) добавляли NaBH4 (16 мг; 0,63 ммоль). Смесь перемешивали при комнатной температуре в течение 1 ч и гасили водой (2 мл). Затем упаривали при пониженном давлении и остаток очищали препаративной обращенно-фазовой HPLC, получая соединение 136 (40 мг; 28%) в виде белого твердого вещества. MS-ESI: [М+Н]+ 654,3. 1Н ЯМР (500 МГц, DMSO-d6) δ 9.00 (s, 1Н), 8.77 (d, J=8,0 Гц, 1H), 8.57 (d, J=5,0 Гц, 1H), 8.53 (d, J=2,5 Гц, 1H), 8.25 (d, J=2,5 Гц, 1H), 7.90 (d, J=1,5 Гц, 1H), 7.78 (d, J=1,0 Гц, 1H), 7.65-7.63 (m, 1Н), 7.59 (d, J=2,5 Гц, 1H), 7.53 (d, J=5,0 Гц, 1H), 7.37 (d, J=8,5 Гц, 1H), 4.91-4.89 (m, 1Н), 4.45-4.39 (m, 2Н), 4.17-4.13 (m, 1Н), 3.79-3.67 (m, 2Н), 3.58-3.53 (m, перекрывание, 5Н), 3.41-3.36 (m, 1Н), 3.29-3.25 (m, 1Н), 1.39 (s, 9Н), 1.22 (d, J=7.5 Гц, 3Н).
Пример 138а. (S)-трет-Бутил-4-(6-(5-хлор-2-метоксипиридин-3-иламино)пиридин-3-ил)-3-метилпиперазин-1-карбоксилат 138а
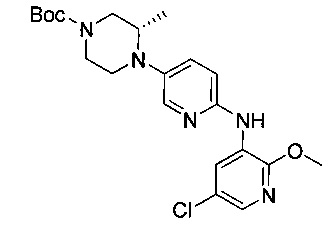
В одногорлую круглодонную колбу емкостью 100 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали 1,4-диоксан (40 мл), (S)-трет-бутил-4-(6-аминопиридин-3-ил)-3-метилпиперазин-1-карбоксилат 105b (2,04 г; 7,0 ммоль), 3-бром-5-хлор-2-метоксипиридин (2,8 г; 12,6 ммоль), Pd2(dba)3 (640 мг; 0,70 ммоль), ксантфос (404,6 мг; 0,70 ммоль) и карбонат цезия (4,56 г; 14,0 ммоль). После трех циклов вакуумирования/продувания аргоном смесь нагревали при 100°С в течение 4 ч. По прошествии этого времени реакционную смесь охлаждали до комнатной температуры. Затем фильтровали и фильтрат упаривали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 1:3 этилацетат/петролейный эфир, получая соединение 138а (1,7 г; 57%) в виде желтого твердого вещества. MS-ESI: [М+Н]+ 434,2.
Пример 138b. (8)-5-Хлор-3-(5-(2-метилпиперазин-1-ил)пиридин-2-иламино)пиридин-2(1Н)-он 138b
Раствор соединения 138а (1,0 г; 2,3 ммоль) в смеси диоксан/HCl (30 мл) перемешивали при 100°С в течение 2 ч. Затем смесь упаривали при пониженном давлении, получая соединение 138b (1,48 г; неочищенное) в виде желтого твердого вещества. MS-ESI: [М+Н]+ 320,3.
Пример 138с. (5)-5-Хлор-3-(5-(2-метил-4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-иламино) пиридин-2(1Н)-он 138с
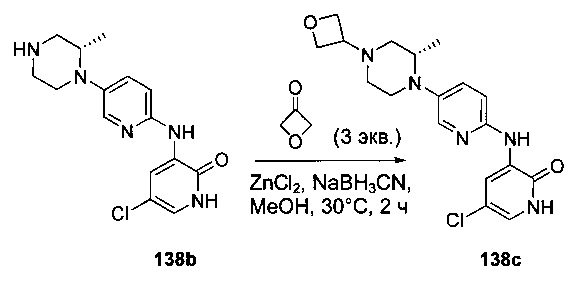
К раствору соединения 138b (1,0 г; 3,1 ммоль) в метаноле (35 мл) добавляли оксетан-3-он (669,6 мг; 9,3 ммоль), NaBH3CN (585,9 мг; 9,3 ммоль) и ZnCl2 (1,26 г; 9,3 ммоль). Реакционную смесь перемешивали при 30°С в течение 2 ч. Смесь упаривали при пониженном давлении и остаток разбавляли водой (20 мл). Затем экстрагировали дихлорметаном (3×30 мл). Объединенный дихлорметановый экстракт концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 40:1 этилацетат/метанол, получая соединение 138с (291 мг; 25%) в виде красного твердого вещества. MS-ESI: [М+Н]+ 376,3.
Пример 138. (S)-6-трет-Бутил-8-фтор-2-(3-(гидроксиметил)-4-(5-(5-(2-метил-4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-иламино)-6-оксо-1,6-дигидропиридин-3-ил)пиридин-2-ил)фталазин-1(2Н)-он 138
В герметично закрываемую колбу, оборудованную магнитной мешалкой, загружали соединение 138с (150 мг; 0,40 ммоль), 3-(ацетоксиметил)-2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)пиридин-4-илбороновую кислоту 116с (495,6 мг; 1,2 ммоль), Pd2(dba)3 (36,6 мг; 0,040 ммоль), Р(Cy)3 (44,6 мг; 0,16 ммоль), Cs2CO3 (391,2 мг; 1,2 ммоль), диоксан (8 мл) и воду (0,2 мл). После трех циклов вакуумирования/продувания аргоном смесь нагревали при 120°С в течение 4 ч. По прошествии этого времени реакционную смесь охлаждали до комнатной температуры. Затем фильтровали и фильтрат упаривали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 40:1 этилацетат/метанол и далее очищали препаративной обращенно-фазовой HPLC, получая соединение 138 (48 мг; 18%) в виде желтого твердого вещества. MS-ESI: [М+Н]+ 667,3. 1Н ЯМР (500 МГц, DMSO-d6) δ 12.05 (s, 1Н), 8.65 (d, J=2,0 Гц, 1H), 8.57 (d, J=5,0 Гц, 1H), 8.55 (d, J=3,0 Гц, 1Η), 8.44 (s, 1Η), 7.91 (d, J=2,0 Гц, 1Η), 7.87 (d, J=2,5 Гц, 1Η), 7.79 (dd, J=1,0 Гц; 13,0 Гц, 1Н), 7.54 (d, J=5,0 Гц, 1Н), 7.39 (dd, J=2,5 Гц; 9,0 Гц, 1Н), 7.26-7.23 (m, 2Н), 4.97-4.95 (m, 1Н), 4.58-4.54 (m, 2Н), 4.48-4.46 (m, 1Н), 4.43-4.41 (m, 1Н), 4.38-4.37 (m, 2Н), 3.69-3.68 (m, 1Н), 3.41-3.39 (m, 1Н), 3.11-3.09 (m, 1Н), 2.97-2.93 (m, 1H), 2.56-2.54 (m, 1H), 2.35-2.32 (m, 2H), 2.21-2.17 (m, 1H), 1.40 (s, 9H), 0,94 (d, J=6,5 Гц, 3Н).
Пример 139а. (3-Нитро-1Н-пиразол-5-ил)метанол 139а
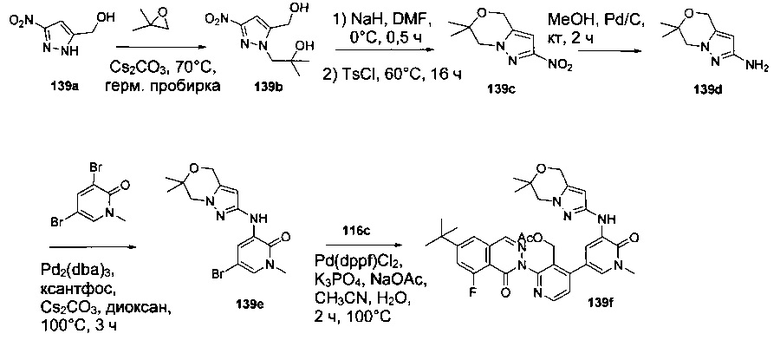
Смесь 3-нитро-1Н-пиразол-5-карбоновой кислоты (4,71 г; 30 ммоль), BH3/THF (75 мл; 1 моль/л; 75 ммоль) перемешивали при 60°С в течение 2 ч. Смесь охлаждали до комнатной температуры и добавляли 4 Μ HCl (19 мл; 75 ммоль). Смесь перемешивали при 70°С в течение 2 ч. После охлаждения до комнатной температуры смесь концентрировали при пониженном давлении. Остаток распределяли между этилацетатом и рассолом (100:100 мл). Водную фазу экстрагировали этилацетатом (4×50 мл). Объединенный органический слой сушили над Na2SO4 и упаривали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с элюированием смесью петролейный эфир/этилацетат (от 5:1 до 1:1), получая соединение 139а (3,5 г; 79%) в виде белого твердого вещества. MS-ESI: [М+Н]+ 144,2.
Пример 139b. 1-(5-(Гидроксиметил)-3-нитро-1Н-пиразол-1-ил)-2-метилпропан-2-ол 139b
В герметично закрываемую пробирку загружали соединение 139а (2,145 г; 15 ммоль), Cs2CO3 (978 мг; 3,0 ммоль) и 2,2-диметилоксиран (15 мл). Смесь перемешивали при 70°С в течение 3 ч. После охлаждения до комнатной температуры смесь концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с элюированием смесью петролейный эфир/этилацетат (от 5:1 до 1:1), получая соединение 139b (1,2 г; 38%) в виде белого твердого вещества. MS-ESI: [М+Н]+ 216,2.
Пример 139с. 6,6-Диметил-2-нитро-6,7-дигидро-4Н-пиразоло[5,1-с][1,4]оксазин 139с
К раствору соединения 139b (1,1 г; 5,1 ммоль) в DMF (10 мл) добавляли NaH (60%-ную дисперсию в минеральном масле; 246 мг; 6,14 ммоль) при 0°С. Полученную суспензию перемешивали в течение 30 мин, затем добавляли п-толуолсульфонилхлорид (1169 мг; 6,14 ммоль). Смесь перемешивали при 60°С в течение ночи. После охлаждения до комнатной температуры добавляли насыщенный раствор хлорида аммония и смесь экстрагировали дихлорметаном. Объединенный органический слой промывали рассолом, сушили над Na2SO4 и упаривали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с элюированием градиентом смеси петролейный эфир/этилацетат (от 9:1 до 2:1), получая соединение 139с (228 мг; 22%). MS-ESI: [М+Н]+ 198,3.
Пример 139d. 6,6-Диметил-6,7-дигидро-4Н-пиразоло[5,1-с][1,4]оксазин-2-амин 139d
Одногорлую круглодонную колбу емкостью 50 мл продували азотом и в нее загружали соединение 139с (0,21 г; 1,25 ммоль), 10%-ный палладий на угле (50% влаги; 125 мг) и метанол (10 мл). Колбу вакуумировали, заполняли газообразным водородом и содержимое перемешивали при комнатной температуре в течение 2 ч. Затем откачивали водород и колбу заполняли азотом. Катализатор удаляли фильтрованием через набивку CELITE® и фильтрат концентрировали при пониженном давлении, получая соединение 139d (167 мг; 93%). MS-ESI: [М+Н]+ 168,1.
Пример 139е. 5-Бром-3-(6,6-диметил-6,7-дигидро-4Н-пиразоло[5,1-с][1,4]оксазин-2-иламино)-1-метилпиридин-2(1Н)-он 139е
В круглодонную колбу емкостью 100 мл, оборудованную обратным холодильником, загружали 1,4-диоксан (10 мл), соединение 139d (167 мг; 1,0 ммоль), 3,5-дибром-1-метилпиридин-2(1Н)-он (320 мг; 1,2 ммоль), Pd2(dba)3 (91 мг; 0,10 ммоль), ксантфос (116 мг; 0,20 ммоль) и карбонат цезия (652 мг; 2,0 ммоль). После трех циклов вакуумирования/продувания аргоном смесь нагревали при 100°С в течение 3 ч. Затем фильтровали и фильтрат упаривали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 100:1 дихлорметан/метанол, получая соединение 139е (210 мг; 60%) в виде желтого твердого вещества. MS-ESI: [М+Н]+ 352,9.
Пример 139f. (2-(6-трет-Бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-(5-(6,6-диметил-6,7-дигидро-4Н-пиразоло[5,1-с][1,4]оксазин-2-иламино)-1-метил-6-оксо-1,6-дигидропиридин-3-ил)пиридин-3-ил)метилацетат 139f
В одногорлую круглодонную колбу емкостью 25 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали соединение 139е (177 мг; 0,50 ммоль), 3-(ацетоксиметил)-2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)пиридин-4-илбороновую кислоту 116с (207 мг; 0,50 ммоль), Pd(dppf)Cl2 (41 мг; 0,050 ммоль), ацетат натрия (82 мг; 1,0 ммоль), тригидрат K3PO4 (266 мг; 1,0 ммоль), воду (6 капли) и ацетонитрил (5 мл). После трех циклов вакуумирования/продувания аргоном смесь нагревали при 100°С в течение 2 ч. Затем фильтровали и фильтрат упаривали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 50:1 дихлорметан/метанол, получая соединение 139f (161 мг; 50%) в виде коричневого твердого вещества. MS-ESI: [М+Н]+ 642,3.
Пример 139. 6-трет-Бутил-2-[4-[5-[(6,6-диметил-4,7-дигидропиразоло[5,1-с][1,4]оксазин-2-ил)амино]-1-метил-6-оксо-3-пиридил]-3-(гидроксиметил)-2-пиридил]-8-фтор-фталазин-1-он 139
Смесь соединения 139f (160 мг; 0,25 ммоль) и гидроксида лития (60 мг; 2,5 ммоль) в смеси изопропанол/TIHF (1:1; 4 мл) и воде (1 мл) перемешивали при 30°С в течение 1 ч. Смесь упаривали при пониженном давлении и остаток разбавляли водой (5 мл). Затем экстрагировали этилацетатом (3×10 мл). Объединенный этилацетатный экстракт концентрировали при пониженном давлении и остаток очищали препаративной обращенно-фазовой HPLC, получая соединение 139 (60 мг; 40%) в виде светло-желтого твердого вещества. MS-ESI: [М+Н]+ 599,8. 1Н ЯМР (500 МГц, DMSO-d6) δ 8.57 (d, J=5,0 Гц, 1H), 8.54 (d, J=2,5 Гц, 1H), 8.34 (s, 1H), 8.08 (d, J=2,5 Гц, 1H), 7.90 (d, J=1,5 Гц, 1H), 7.79-7.76 (m, 1H), 7.51 (d, J=5,0 Гц, 1H), 7.39 (d, J=2,0 Гц, 1H), 5.95 (s, 1H), 4.92 (bs, 1H), 4.73 (s, 2H), 4.42 (s, 2H), 3.80 (s, 2H), 3.60 (s, 3Н), 1.40 (s, 9H), 1.26 (s, 6H).
Пример 140а. 5-Бром-1-метил-3-(1-метил-1Н-пиразол-3-иламино)пиридин-2(1Н)-он 140а
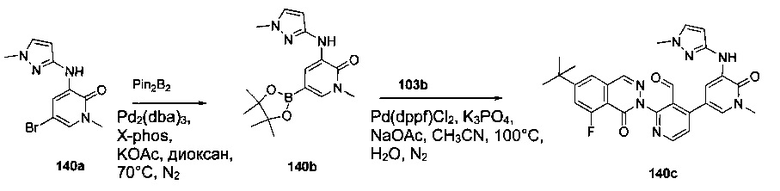
В одногорлую круглодонную колбу емкостью 250 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали 1,4-диоксан (100 мл), 1-метил-1Н-пиразол-3-амин (970 мг; 10,0 ммоль), 3,5-дибром-1-метилпиридин-2-(1Н)-он (2,9 г; 11 ммоль) и карбонат цезия (6,5 г; 20,0 ммоль). После барботирования азота через суспензию в течение 10 минут добавляли трис(дибензилиденацетон)дипалладий(0) (457 мг; 0,50 ммоль) и ксантфос (587 мг; 1,0 ммоль). Систему подвергали трем циклам вакуумирования/продувания аргоном и нагревали при температуре дефлегмации в течение 2 ч. Затем охлаждали до комнатной температуры и фильтровали. Твердое вещество промывали дихлорметаном (2×50 мл) и объединенный органический фильтрат концентрировали. Остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 30:1 дихлорметан/метанол, получая соединение 140а в виде желтого твердого вещества (900 мг; 32%). MS-ESI: [М+Н]+ 283,1.
Пример 140b. 1-Метил-3-(1-метил-1Н-пиразол-3-иламино)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2(1Н)-он 140b
В круглодонную колбу емкостью 100 мл, оборудованную обратным холодильником, загружали соединение 140а (564 мг; 2,0 ммоль), бис(пинаколато)дибор (1,5 г; 6,0 ммоль), Pd2(dba)3 (183 мг; 0,20 ммоль), X-phos (95 мг; 0,20 ммоль), ацетат калия (392 мг; 4,0 ммоль) и 1,4-диоксан (20 мл). После трех циклов вакуумирования и продувания аргоном смесь нагревали при 70°С в течение 2 ч. Затем охлаждали до комнатной температуры и фильтровали. Фильтрат упаривали при пониженном давлении. Остаток промывали петролейным эфиром, получая соединение 140b (600 мг; 91%) в виде коричневого твердого вещества, которое использовали без дополнительной очистки. MS-ESI: [М+Н]+ 331,0.
Пример 140с. 2-(6-трет-Бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-(1-метил-5-(1-метил-1Н-пиразол-3-иламино)-6-оксо-1,6-дигидропиридин-3-ил)никотинальдегид 140с
В круглодонную колбу емкостью 50 мл, оборудованную обратным холодильником, загружали 2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-хлорникотинальдегид 103b (359 мг; 1,0 ммоль), соединение 140b (660 мг; 2,0 ммоль), K3PO4 (424 мг; 2,0 ммоль), 1,1'-бис(дифенилфосфино)ферроцен-дихлорпалладий(II) (82 мг; 0,10 ммоль), ацетат натрия (164 мг; 2,0 ммоль) и смесь ацетонитрил/вода (15/0,2 мл). После трех циклов вакуумирования/продувания N2 смесь нагревали при 100°С в течение 1,5 ч. Смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. Остаток разбавляли дихлорметаном (30 мл) и водой (30 мл). Органический слой отделяли и водный слой экстрагировали дихлорметаном (3×20 мл). Объединенный органический слой сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Темный остаток очищали колоночной хроматографией на силикагеле с элюированием смесью дихлорметан/метанол (от 80/1 до 50/1), получая соединение 140с (280 мг; 53%) в виде желтого твердого вещества. MS-ESI: [М+Н]+ 528,1.
Пример 140. 6-трет-Бутил-8-фтор-2-[3-(гидроксиметил)-4-[1-метил-5-[(1-метилпиразол-3-ил)амино]-6-оксо-3-пиридил]-2-пиридил]фталазин-1-он 140
К раствору соединения 140с (270 мг; 0,50 ммоль) в THF (5 мл), пропан-2-оле (5 мл) и воде (2 мл) добавляли гидроксид лития (36 мг; 1,5 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч и концентрировали при пониженном давлении. Остаток разбавляли дихлорметаном (20 мл) и водой (10 мл). Органический слой отделяли и водный слой экстрагировали дихлорметаном (3×15 мл). Объединенный органический слой промывали рассолом и концентрировали при пониженном давлении. Остаток очищали препаративной обращенно-фазовой HPLC, получая соединение 140 (53 мг; 20%) в виде белого твердого вещества. MS-ESI: [М+Н]+ 530,3. 1Н ЯМР (500 МГц, DMSO-d6) δ 8.56 (d, J=5,0 Гц, 1H), 8.53 (d, J=2,0 Гц, 1H), 8.21 (s, 1Н), 8.03 (d, J=2,5 Гц, 1H), 7.89 (s, 1Н), 7.78-7.5 (m, 1Н), 7.51-7.48 (m, 2Н), 7.39 (d, J=2,5 Гц, 1H), 6.07 (d, J=2,0 Гц, 1H), 4.91 (s, 1Н), 4.42 (s, 2Н), 3.71 (s, 3Н), 3.58 (s, 3Н), 1.39 (s, 9Н).
Пример 141а. 6-Хлор-2-метил-4-(5-метилизоксазол-3-иламино)пиридазин-3(2Н)-он 141а
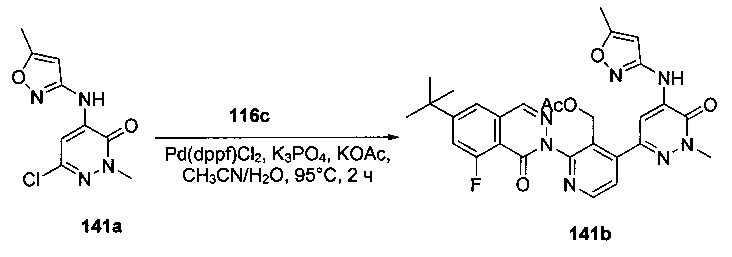
В одногорлую круглодонную колбу емкостью 100 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали 1,4-диоксан (40 мл), 5-метилизоксазол-3-амин (559 мг; 5,7 ммоль), 4-бром-6-хлор-2-метилпиридазин-3(2Н)-он (1,89 г; 8,55 ммоль), Pd2(dba)3 (521 мг; 0,57 ммоль), ксантфос (329 мг; 0,57 ммоль) и карбонат цезия (3,72 г; 11,4 ммоль). После трех циклов вакуумирования/продувания аргоном смесь нагревали при 100°С в течение 3 ч. Затем смесь охлаждали до 80°С и фильтровали. Фильтрат охлаждали до комнатной температуры. Затем фильтровали, получая соединение 141а (600 мг; 44%) в виде желтого твердого вещества. MS-ESI: [М+Н]+ 241,0.
Пример 141b. (2-(6-трет-Бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-(1-метил-5-(5-метилизоксазол-3-иламино)-6-оксо-1,6-дигидропиридазин-3-ил)пиридин-3-ил)метилацетат 141b
В одногорлую круглодонную колбу емкостью 100 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали соединение 141а (288 мг; 1,2 ммоль), 3-(ацетоксиметил)-2-(6-трет-бутил-8-фтор-1-оксо-фталазин-2(1Н)-ил)пиридин-4-илбороновую кислоту 116с (1,49 г, 3,6 ммоль), Pd(dppf)Cl2 (99 мг; 0,12 ммоль), ацетат калия (235 мг; 2,4 ммоль), K3PO4 (523 мг; 2,4 ммоль), ацетонитрил (20 мл) и воду (10 капель). После трех циклов вакуумирования/продувания аргоном смесь нагревали при 95°С в течение 2 ч. Затем фильтровали и фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 1:1 этилацетат/петролейный эфир, получая соединение 141b (220 мг; 32%) в виде желтого твердого вещества. MS-ESI: [М+Н]+ 574,2.
Пример 141. 6-трет-Бутил-8-фтор-2-[3-(гидроксиметил)-4-[1-метил-5-[(5-метилизоксазол-3-ил)амино]-6-оксо-пиридазин-3-ил]-2-пиридил]фталазин-1-он 141
Смесь соединения 141а (117,3 мг; 0,20 ммоль) и моногидрата гидроксида лития (84 мг; 2,0 ммоль) в смеси изопропанол/THF (1:1; 8 мл) и воде (1 мл) перемешивали при 35°С в течение 0,5 ч. Смесь концентрировали при пониженном давлении. Остаток распределяли между водой (10 мл) и дихлорметаном (10 мл). Водную фазу экстрагировали дихлорметаном (3×10 мл). Объединенный органический слой концентрировали при пониженном давлении и остаток очищали препаративной обращенно-фазовой HPLC, получая соединение 141 (70 мг; 66%) в виде бледно-желтого твердого вещества. MS-ESI: [М+Н]+ 532,2. 1Н ЯМР (500 МГц, DMSO-d6) δ 9.92 (s, 1Н), 8.65 (d, J=5,5 Гц, 1H), 8.55 (d, J=2,5 Гц, 1H), 7.90 (d, J=1,5 Гц, 1H), 7.85 (s, 1Н), 7.80-7.77 (m, 1Н), 7.62 (d, J=5,0 Гц, 1H), 6.33 (s, 1Н), 4.85 (s, 1Н), 4.56-4.53 (m, 1Н), 4.45-4.42 (m, 1Н), 3.80 (s, 3Н), 2.36 (s, 3Н), 1.40 (s, 9Н).
Пример 142а. 3-Бром-5-иодпиридин-2-ол 142а
В одногорлую круглодонную колбу емкостью 100 мл, оборудованную магнитной мешалкой, загружали ацетонитрил (50 мл), TFA (трифторуксусная кислота) (10 мл), 3-бромпиридин-2-ол (4,0 г; 11,56 ммоль), N-иодсукцинимид (5,2 г; 11,56 ммоль). Смесь перемешивали при комнатной температуре в течение 15 ч. Смесь разбавляли водой (100 мл) и образовавшееся белое твердое вещество собирали фильтрованием, получая соединение 142а (6,6 г; 96%) в виде белого твердого вещества. MS-ESI: [М+Н]+ 300.
Пример 142b. 3-Бром-5-иод-1-метилпиридин-2(1Н)-он 142b
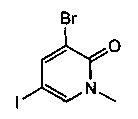
В одногорлую круглодонную колбу емкостью 100 мл, оборудованную магнитной мешалкой, загружали DMF (50 мл), соединение 142а (6,0 г; 20,0 ммоль), CH3I (4,26 г; 30,0 ммоль) и K2CO3 (5,52 г; 40,0 ммоль). Смесь перемешивали при комнатной температуре в течение 2 ч и разбавляли водой (200 мл). Образовавшееся белое твердое вещество собирали фильтрованием, получая соединение 142b (5,97 г; 95%) в виде белого твердого вещества. MS-ESI: [М+Н]+ 314.
Пример 142с. (4-(5-Бром-1-метил-6-оксо-1,6-дигидропиридин-3-ил)-2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)пиридин-3-ил)метилацетат 142с
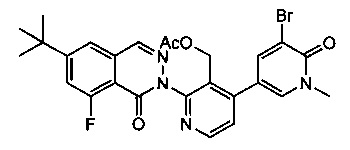
В одногорлую круглодонную колбу емкостью 50 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали соединение 142b (1,37 г; 4,38 ммоль; 1,0 экв.), 3-(ацетоксиметил)-2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)пиридин-4-илбороновую кислоту 116с (2,07 мг; 5,0 ммоль), Pd(dppf)Cl2 (179 мг; 0,219 ммоль; 0,050 экв.), ацетат натрия (718 мг; 8,76 ммоль; 2,0 экв.), K3PO4 (1,86 г; 8,76 ммоль; 2,0 экв.), ацетонитрил (20 мл) и воду (1 мл). После трех циклов вакуумирования/продувания аргоном смесь нагревали при 30°С в течение 2 ч. Затем охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 100:1 дихлорметан/этанол, получая соединение 142с (800 мг; 32%) в виде желтого твердого вещества. MS-ESI: [М+Н]+ 555,2. 1Н ЯМР (500 МГц, DMSO-d6) δ 8.66 (d, J=5,0 Гц, 1H), 8.56 (d, J=2,5 Гц, 1H), 8.14 (d, J=2,5 Гц, 1H), 8.08 (d, J=2,0 Гц, 1H), 7.91 (d, J=1,5 Гц, 1H), 7.82-7.79 (m, 1Н), 7.62 (d, J=6,5 Гц, 1Η), 5.02 (s, 2Η), 3.59 (s, 3Н), 1.78 (s, 3Н), 1.40 (s, 9Н).
Пример 142d. (2-(6-трет-Бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-(5-(1-этил-1Н-пиразол-3-иламино)-1-метил-6-оксо-1,6-дигидропиридин-3-ил)пиридин-3-ил)метилацетат 142d
Во флакон емкостью 1 драхма (3,7 мл) добавляли соединение 142с (40 мг; 0,074 ммоль), 1-этил-1Н-пиразол-3-амин (1,2 экв), карбонат цезия (1,5 экв), ксантфос (10 моль%) и трис(дибензилиденацетон)дипалладий(0) (5 моль%) в безводном 1,4-диоксане (0,2 М). Реакционную смесь затем перемешивали при 80°С в течение 3 часов. После охлаждения до комнатной температуры реакционную смесь далее разбавляли дихлорметаном (3 мл) и промывали водой (2×3 мл). Органический слой сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Неочищенный продукт 142d затем переносили на следующую стадию без очистки.
Пример 142. 6-трет-Бутил-2-[4-[5-[(1-этилпиразол-3-ил)амино]-1-метил-6-оксо-3-пиридил]-3-(гидроксиметил)-2-пиридил]-8-фтор-фталазин-1-он 142
Во флакон емкостью 1 драхма (3,7 мл) добавляли соединение 142d (1 экв.) в смеси 4:1 THF и воды (1 мл). Затем к этой смеси добавляли гидроксид лития (1,5 экв.) и реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Далее реакционную смесь разбавляли дихлорметаном (3 мл) и промывали водой (2×3 мл). Органический слой собирали, сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Неочищенное вещество очищали обращенно-фазовой хроматографией, получая соединение 142 (7,6 мг; выход 19%). 1Н ЯМР (400 МГц, DMSO-d6) δ 8.55 (s, 1Н), 8.52 (d, J=2,5 Гц, 1H), 8.16 (s, 1Н), 8.06 (d, J=2,5 Гц, 1H), 7.89 (m, 1Н), 7.75 (dd, J=13,1; 1,9 Гц, 1H), 7.53 (d, J=2,2 Гц, 1H), 7.51 (d, J=12,6 Гц, 1H), 7.40 (d, J=2,1 Гц, 1H), 6.07 (s, 1Н), 4.46 (br s 1H), 4.00 (q, J=7,2 Гц, 2H), 3.60 (s, 3Н), 3.17 (s, 2H), 1.39 (s, 9H), 1.34 (t, J=7,2 Гц, ЗН). ES-MS m/z 544,3 [М+1].
Пример 143. 6-трет-Бутил-8-фтор-2-[3-(гидроксиметил)-4-[5-[[5-(метоксиметил)-1-метил-пиразол-3-ил]амино]-1-метил-6-оксо-3-пиридил]-2-пиридил]фталазин-1-он 143
Следуя методикам из примера 142 и заменяя 1-этил-1Н-пиразол-3-амин на 5-(метоксиметил)-1-метил-1Н-пиразол-3-амин, получали соединение 143 (11,2 мг; выход 54%). 1Н ЯМР (400 МГц, DMSO-d6) δ 8.56 (d, J=5,0 Гц, 1H), 8.53 (d, J=2,5 Гц, 1H), 8.19 (s, 1Н), 8.03 (d, J=2,4 Гц, 1H), 7.89 (d, J=1,8 Гц, 1H), 7.76 (dd, J=13,2; 1,7 Гц, 1H), 7.50 (d, J=5,0 Гц, 1H), 7.39 (d, J=2,4 Гц, 1H), 6.11 (s, 1Н), 4.43 (d, J=5,4 Гц, 2H), 4.38 (s, 2H), 3.66 (s, 3Н), 3.59 (s, 3Н), 3.26 (s, 3Н), 1.39 (s, 9H). ES-MS m/z 574,3 [М+1].
Пример 144. 6-трет-Бутил-8-фтор-2-[3-(гидроксиметил)-4-[1-метил-5-[[1-метил-5-(пирролидин-1-карбонил)пиразол-3-ил]амино]-6-оксо-3-пиридил]-2-пиридил]фталазин-1-он 144
Следуя методикам из примера 142 и заменяя 1-этил-1Н-пиразол-3-амин на (3-амино-1-метил-1Н-пиразол-5-ил)(пирролидин-1-ил)метанон, получали соединение 144 (14,6 мг; выход 65%). 1Н ЯМР (400 МГц, DMSO-d6) δ 8.59-8.50 (m, 2Н), 8.28 (s, 1Н), 8.04 (d, J=2,3 Гц, 1H), 7.89 (d, J=1,8 Гц, 1H), 7.76 (dd, J=13,1; 1,7 Гц, 1H), 7.51 (d, J=5,1 Гц, 1H), 7.42 (d, J=2,3 Гц, 1H), 6.46 (s, 1Н), 4.87 (t, J=5,1 Гц, 1H), 4.47-4.40 (m, 2Н), 3.79 (s, 3Н), 3.60 (s, 3Н), 3.48 (dd, J=19,0; 6,7 Гц, 4H), 1.91-1.82 (m, 4Н), 1.39 (s, 9Н). ES-MS m/z 627,4 [М+1].
Пример 145. 6-трет-Бутил-2-[4-[5-[[1-(2,2-дифторэтил)-5-метил-пиразол-3-ил]амино]-1-метил-6-оксо-3-пиридил]-3-(гидроксиметил)-2-пиридил]-8-фтор-фталазин-1-он 145
Следуя методикам из примера 142 и заменяя 1-этил-1Н-пиразол-3-амин на 1-(2,2-дифторэтил)-5-метил-1Н-пиразол-3-амин, получали соединение 145 (11 мг; выход 26%). 1Н ЯМР (400 МГц, DMSO-d6) δ 8.55 (d, J=5,0 Гц, 1H), 8.52 (d, J=2,7 Гц, 1Η), 8.20 (s, 1Η), 8.11 (d, J=2,4 Гц, 1Η), 7.90 (d, J=1,6 Гц, 1Η), 7.75 (dd, J=13,1; 1,8 Гц, 1Η), 7.50 (d, J=5,0 Гц, 1Η), 7.42 (d, J=2,4 Гц, 1H), 5.97 (s, 1Η), 4.88 (t, J=5,0 Гц, 1H), 4.46-4.33 (m, 5Н), 3.59 (s, 3Н), 2.21 (s, 3Н), 1.39 (s, 9Н). ES-MS m/z 594,3 [М+1].
Пример 146а. Этил-2-(5-(гидроксиметил)-3-нитро-1Н-пиразол-1-ил)ацетат 146а
В одногорлую круглодонную колбу емкостью 100 мл, оборудованную магнитной мешалкой, загружали ацетонитрил (30 мл), (3-нитро-1Н-пиразол-5-ил)метанол (1,43 г; 10,0 ммоль), Cs2CO3 (490 мг; 1,5 ммоль) и этил-2-бромацетат (2,00 г; 12 ммоль). Смесь перемешивали при 40°С в течение 5 ч. Затем охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 30:1 дихлорметан/метанол, получая соединение 146а (1,65 г; 72%) в виде желтого твердого вещества. MS-ESI: [М+Н]+ 229,9.
Пример 146b. Этил-2-(5-(хлорметил)-3-нитро-1Н-пиразол-1-ил)ацетат 146b
К смеси соединения 146а (1,50 г; 6,55 ммоль) в CHCl3 (60 мл), охлажденной при 0°С, медленно добавляли SOCl2 (2,34 г; 19,6 ммоль), все время поддерживая внутреннюю температуру ниже 5°С. Эту реакционную смесь нагревали до 50°С и перемешивали при этой температуре в течение 3 ч. Затем охлаждали до 0°С и гасили водой. Органический слой отделяли и упаривали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 30:1 дихлорметан/метанол, получая соединение 146b (1,1 г; 68%) в виде желтого твердого вещества. MS-ESI: [М+Н]+ 247,9.
Пример 146с. 5-Метил-2-нитро-4,5-дигидропиразоло[1,5-а]пиразин-6(7Н)-он 146с
К раствору соединения 146b (1,0 г; 4,0 ммоль) в дихлорметане (30 мл) добавляли раствор CH3NH2 (1,07 г; 12,0 ммоль; 35%-ный в метаноле). Эту реакционную смесь перемешивали при комнатной температуре в течение 3 ч и разбавляли водой (30 мл). Органический слой отделяли, сушили над Na2SO4 и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 30:1 дихлорметан/метанол, получая соединение 146с (450 мг; 57%) в виде желтого твердого вещества. MS-ESI: [М+Н]+ 196,9.
Пример 146d. 2-Амино-5-метил-4,5-дигидропиразоло[1,5-а]пиразин-6(7Н)-он 146d
К раствору соединения 146с (450 мг; 2,3 ммоль) в этаноле (30 мл) добавляли Pd/C (10%-ный; 400 мг). Реакционный сосуд заполняли газообразным водородом (с использованием баллона) и содержимое перемешивали при комнатной температуре в течение 2 ч. После завершения реакции смесь фильтровали через набивку CELITE® и фильтрат концентрировали при пониженном давлении, получая соединение 146d в виде желтого твердого вещества (320 мг; 84%), которое использовали без дополнительной очистки на следующей стадии. MS-ESI: [М+Н]+167,1.
Пример 146е. 2-(5-Бром-1-метил-2-оксо-1,2-дигидропиридин-3-иламино)-5-метил-4,5-дигидропиразоло[1,5-а]пиразин-6(7Н)-он 146е
В круглодонную колбу емкостью 100 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали соединение 146d (300 мг; 1,8 ммоль), 3,5-дибром-1-метилпиридин-2(1Н)-он (482 мг; 1,8 ммоль), карбонат цезия (1,17 г; 3,6 ммоль) и 1,4-диоксан (20 мл). После барботирования азота через суспензию в течение 10 минут добавляли ксантфос (104 мг; 0,18 ммоль) и трис(дибензилиденацетон)дипалладий(0) (82 мг; 0,090 ммоль). Систему подвергали трем циклам вакуумирования/продувания аргоном и нагревали при температуре дефлегмации в течение 5 ч. Затем охлаждали до комнатной температуры и фильтровали. Твердое вещество промывали дихлорметаном (2×30 мл). Объединенный фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с элюированием смесью дихлорметан/метанол (от 80/1 до 30/1), получая соединение 146е (390 мг; 61%) в виде желтого твердого вещества.
Пример 146f. (2-(6-трет-Бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-(1-метил-5-(5-метил-6-оксо-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин-2-иламино)-6-оксо-1,6-дигидропиридин-3-ил)пиридин-3-ил)метилацетат 146f
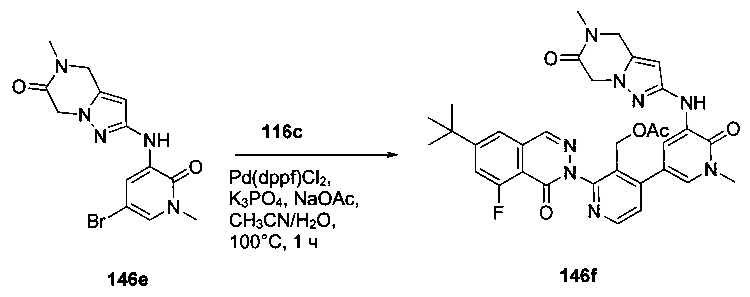
В круглодонную колбу емкостью 25 мл, оборудованную обратным холодильником, загружали 3-(ацетоксиметил)-2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)пиридин-4-илбороновую кислоту 116с (165 мг; 0,40 ммоль), соединение 146е (141 мг; 0,40 ммоль), K3PO4 (170 мг; 0,80 ммоль), ацетат натрия (66 мг; 0,80 ммоль), Pd(dppf)Cl2 (15 мг; 0,020 ммоль) и смесь ацетонитрил/вода (8/0,2 мл). Смесь подвергали трем циклам вакуумирования/продувания азотом и нагревали при 100°С под защитой N2 в течение 1 ч. Анализ реакционной смеси с использованием LCMS показал полное превращение в желаемый продукт. Реакционную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. Остаток разбавляли дихлорметаном (30 мл) и водой (30 мл). Органический слой отделяли и водный слой экстрагировали дихлорметаном (2×30 мл). Объединенный органический экстракт сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Темный остаток очищали колоночной хроматографией на силикагеле с элюированием смесью дихлорметан/метанол (от 80/1 до 30/1), получая соединение 146f (115 мг; 45%) в виде желтого твердого вещества. MS-ESI: [М+Н]+ 641,4.
Пример 146. 6-трет-Бутил-8-фтор-2-[3-(гидроксиметил)-4-[1-метил-5-[(5-метил-6-оксо-4,7-дигидропиразоло[1,5-а]пиразин-2-ил)амино]-6-оксо-3-пиридил]-2-пиридил]фталазин-1-он 146
К раствору соединения 146f (110 мг; 0,172 ммоль) в смеси THF/изопропанол/вода (4/2/2 мл) добавляли гидроксид лития (21 мг; 0,86 ммоль). Смесь перемешивали при 30°С в течение 1 ч. После завершения реакции смесь концентрировали при пониженном давлении. Остаток разбавляли водой (10 мл) и экстрагировали этилацетатом (3×20 мл). Объединенный органический слой сушили и концентрировали при пониженном давлении. Остаток очищали препаративной обращенно-фазовой HPLC, получая соединение 146 в виде белого твердого вещества (45 мг; 44%). MS-ESI: [М+Н]+ 599,2. 1Н ЯМР (500 МГц, DMSO-d6) δ 8.57 (d, J=5,0 Гц, 1Η), 8.54 (d, J=2,5 Гц, 1Η), 8.41 (s, 1Η), 8.08 (d, J=2,5 Гц, 1Η), 7.91 (d, J=1,5 Гц, 1Η), 7.78 (d, J=13,0 Гц, 1Η), 7.51 (d, J=5,0 Гц, 1Η), 7.42 (d, J=2,5 Гц, 1H), 6.08 (s, 1Н), 4.91-4.89 (m, 1Н), 4.62 (s, 2Н), 4.57 (s, 2Н), 4.45-4.43 (m, 2Н), 3.61 (s, 3Н), 2.98 (s, 3Н), 1.40 (s, 9Н).
Пример 147а. 1-(6-Нитропиридин-3-ил)азетидин-3-ол 147а
В одногорлую круглодонную колбу емкостью 100 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали ацетонитрил (50 мл), 5-фтор-2-нитропиридин (1,2 г; 7,9 ммоль), K2CO3 (2,1 г; 15,8 ммоль) и гидрохлорид азетидин-3-ола (1,3 г; 11,9 ммоль). Смесь нагревали при 60°С в течение 1 ч. По прошествии этого времени реакционную смесь охлаждали до комнатной температуры. Затем фильтровали и фильтрат упаривали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с элюированием смесью дихлорметан/метанол (от 50:1 до 20:1), получая соединение 147а (1,1 г; 73%) в виде желтого твердого вещества. MS-ESI: [М+Н]+ 196,0.
Пример 147b. 1-(6-Аминопиридин-3-ил)азетидин-3-ол 147b
Одногорлую круглодонную колбу емкостью 100 мл продували азотом и в нее загружали соединение 147а (1,0 г; 5,1 ммоль), 10%-ный палладий на угле (10% влаги; 100 мг) и этанол (40 мл). Колбу вакуумировали, заполняли газообразным водородом и содержимое перемешивали при комнатной температуре в течение 5 ч. Затем откачивали водород и колбу заполняли азотом. Катализатор удаляли фильтрованием через набивку CELITE® и фильтрат концентрировали при пониженном давлении, получая соединение 147b в виде желтого твердого вещества (792 мг; 85%). MS-ESI: [М+Н]+ 166,1.
Пример 147с. 5-Бром-3-(5-(3-гидроксиазетидин-1-ил)пиридин-2-иламино)-1-метил пиридин-2(1Н)-он 147с
В одногорлую круглодонную колбу емкостью 100 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали соединение 147b (792 мг; 4,8 ммоль), 3,5-дибром-1-метилпиридин-2(1Н)-он (1,9 г; 7,2 ммоль), трис-(дибензилиденацетон)дипалладий(0) (440 мг; 0,48 ммоль), ксантфос (555 мг; 0,96 ммоль), Cs2CO3 (3,1 г; 9,6 ммоль) и 1,4-диоксан (40 мл). После трех циклов вакуумирования/продувания аргоном смесь нагревали при 90°С в течение 3,0 ч. Затем охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали колоночной хроматографией на силикагеле с элюированием смесью дихлорметан/метанол (от 50:1 до 20:1), получая соединение 147с в виде желтого твердого вещества (1,5 г; 89%). MS-ESI: [М+Н]+ 351,1.
Пример 147d. (2-(6-трет-Бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-(5-(5-(3-гидроксиазетидин-1-ил)пиридин-2-иламино)-1-метил-6-оксо-1,6-дигидропиридин-3-ил)пиридин-3-ил)метилацетат 147d

В колбу емкостью 100 мл, оборудованную обратным холодильником, загружали соединение 147с (285 мг; 0,81 ммоль), 3-(ацетоксиметил)-2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)пиридин-4-илбороновую кислоту 116с (669 мг; 1,62 ммоль), Pd(dppf)Cl2 (66,2 мг; 0,081 ммоль), K3PO4 (343,4 мг; 1,62 ммоль), ацетат натрия (132,8 мг; 1,62 ммоль), воду (0,5 мл) и ацетонитрил (15 мл). После трех циклов вакуумирования/продувания аргоном смесь нагревали при 100°С в течение 3 ч. Затем охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали колоночной хроматографией на силикагеле с элюированием смесью дихлорметан/метанол (от 50:1 до 30:1), получая соединение 147d в виде коричневого твердого вещества (140,0 мг; 27%). MS-ESI: [М+Н]+ 640,3.
Пример 147. 6-трет-Бутил-8-фтор-2-[4-[5-[[5-(3-гидроксиазетидин-1-ил)-2-пиридил]амино]-1-метил-6-оксо-3-пиридил]-3-(гидроксиметил)-2-пиридил]фталазин-1-он 147
Смесь соединения 147d (140 мг; 0,22 ммоль) и гидроксида лития (132 мг; 5,5 ммоль) в смеси изопропанол/ТНР/вода (2:2:1,10 мл) перемешивали при комнатной температуре в течение 1 часа. Смесь концентрировали при пониженном давлении и разбавляли водой (10 мл). Полученную смесь три раза экстрагировали дихлорметаном. Объединенный органический слой концентрировали при пониженном давлении и полученный остаток очищали препаративной обращенно-фазовой HPLC, получая соединение 147 (40 мг; 31%) в виде желтого твердого вещества. MS-ESI: [М+Н]+ 598,2. 1Н ЯМР (500 МГц, DMSO-d6) δ 8.57-8.53 (m, 3Н), 8.32 (s, 1Н), 7.90 (s, 1Н), 7.77 (d, J=13,0 Гц, 1Н), 7.51-7.49 (m, 2Н), 7.43 (s, 1Н), 7.19 (d, J=8,5 Гц, 1Н), 6.88 (dd, J=3,0; 8,5 Гц, 1Н), 5.57-5.56 (m, 1Н), 4.89-4.87 (m, 1Н), 4.55-4.52 (m, 1Н), 4.43-4.41 (m, 2Н), 4.05-4.02 (m, 2Н), 3.60 (s, 3Н), 3.45-3.42 (m, 2Н), 1.39 (s, 9Н).
Пример 148а. 2-Метил-1-(2-нитро-6,7-дигидропиразоло[1,5-а]пиразин-5(4Н)-ил)пропан-1-он 148а

К раствору 2-нитро-4,5,6,7-тетрагидропиразоло[1,5-а]пиразина 124d (160 мг; 0,952 ммоль) в дихлорметане (10 мл) добавляли триэтиламин (197 мг; 1,90 ммоль). После перемешивания при комнатной температуре в течение 5 минут добавляли раствор изобутирилхлорида (111,0 мг; 1,047 ммоль) в дихлорметане (2 мл) и смесь перемешивали в течение 1 ч. Анализ реакционной смеси с использованием LCMS показал полное превращение в желаемый продукт. Затем смесь концентрировали при пониженном давлении, получая соединение 148а в виде белого твердого вещества (220 мг; 97%), которое использовали на следующей стадии без дополнительной очистки. MS-ESI: [М+Н]+ 239,0.
Пример 148b. 1-(2-Амино-6,7-дигидропиразоло[1,5-а]пиразин-5(4Н)-ил)-2-метилпропан-1-он 148b
К раствору соединения 148а (220 мг; 0,924 ммоль) в метаноле (20 мл) добавляли Pd/C (22 мг). Систему вакуумировали и повторно заполняли Н2. После перемешивания при комнатной температуре в течение 2 ч смесь фильтровали. Фильтрат концентрировали при пониженном давлении, получая соединение 148b в виде желтого твердого вещества (190 мг; 98%), которое использовали на следующей стадии без дополнительной очистки. MS-ESI: [М+Н]+ 209,1.
Пример 148с. 5-Бром-3-(5-изобутирил-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин-2-иламино-1-метилпиридин-2(1Н)-он 148с
В круглодонную колбу емкостью 100 мл, оборудованную обратным холодильником, загружали соединение 148b (190 мг; 0,913 ммоль), 3,5-дибром-1-метилпиридин-2(1Н)-он (365,6 мг; 1,37 ммоль), Pd2(dba)3 (41,7 мг; 0,0456 ммоль), ксантфос (52,8 мг; 0,0913 ммоль), Cs2CO3 (595,3 мг; 1,826 ммоль) и диоксан (20 мл). Систему подвергали трем циклам вакуумирования/продувания азотом и нагревали при 100°С под защитой N2 в течение 3 ч. Анализ реакционной смеси с использованием LCMS показал полное превращение в желаемый продукт. Смесь охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток промывали ацетонитрилом (2 мл), получая соединение 148с в виде темного твердого вещества (240 мг; 67%), которое использовали на следующей стадии без дополнительной очистки. MS-ESI: [М+Н]+ 394,2.
Пример 148d. 3-(5-Изобутирил-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин-2-иламино)-1-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2(1Н)-он 148d
В круглодонную колбу емкостью 100 мл, оборудованную обратным холодильником, загружали соединение 148с (220 мг; 0,558 ммоль), Pin2B2 (212,6 мг; 0,837 ммоль), Pd2(dba)3 (25,5 мг; 0,0279 ммоль), X-Phos (26,6 мг; 0,0558 ммоль), ацетат калия (109,4 мг; 1,12 ммоль) и диоксан (20 мл). Систему подвергали трем циклам вакуумирования/продувания азотом и нагревали при 70°С под защитой N2 в течение 2 ч. Анализ реакционной смеси с использованием LCMS показал полное превращение в желаемый продукт. Затем смесь охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток промывали петролейным эфиром, получая соединение 148d в виде темного масла (200 мг; 81%), которое использовали на следующей стадии без дополнительной очистки. MS-ESI: [М+Н]+ 442,4.
Пример 148е. 2-(6-трет-Бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-(5-(5-изобутирил-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин-2-иламино)-1-метил-6-оксо-1,6-дигидропиридин-3-ил)никотинальдегид 148е
В круглодонную колбу емкостью 100 мл, оборудованную обратным холодильником, загружали 2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-хлорникотинальдегид 103b (150 мг; 0,42 ммоль), соединение 148d (278 мг; 0,63 ммоль), Pd(dppf)Cl2 (17,1 мг; 0,021 ммоль), K3PO4 (178,1 мг; 0,84 ммоль), ацетат натрия (68,9 мг; 0,84 ммоль), ацетонитрил (15 мл) и воду (5 капель). Систему подвергали трем циклам вакуумирования/продувания азотом и нагревали при 100°С под защитой N2 в течение 1 ч. Анализ реакционной смеси с использованием LCMS показал полное превращение в желаемый продукт. Смесь охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток промывали ацетонитрилом (0,5 мл), получая соединение 148е в виде белого твердого вещества (100 мг; 37%), которое использовали на следующей стадии без дополнительной очистки. MS-ESI: [М+Н]+ 638,8.
Пример 148. 6-трет-Бутил-8-фтор-2-[3-(гидроксиметил)-4-[1-метил-5-[[5-(2-метилпропаноил)-6,7-дигидро-4Н-пиразоло[1,5-а]пиразин-2-ил]амино]-6-оксо-3-пиридил]-2-пиридил]фталазин-1-он 148
К раствору соединения 148е (90 мг; 0,141 ммоль) в дихлорметане (5 мл) и метаноле (5 мл) добавляли NaBH4 (10,7 мг; 0,282 ммоль). После перемешивания при комнатной температуре в течение 1 ч реакционную смесь гасили водным NH4Cl и концентрировали при пониженном давлении. Остаток экстрагировали дихлорметаном (3×15 мл). Объединенный органический слой промывали рассолом, сушили над Na2SO4 и концентрировали при пониженном давлении. Остаток очищали препаративной обращенно-фазовой HPLC, получая соединение 148 (40 мг; 44%) в виде белого твердого вещества. MS-ESI: [М+Н]+640,8. 1Н ЯМР (500 МГц, DMSO-d6) δ 8.57 (d, J=5,0 Гц, 1H), 8.54 (d, J=2,5 Гц, 1H), 8.35-8.28 (m, 1Η), 8.06 (s, 1Η), 7.90 (d, J=1,5 Гц, 1Η), 7.78 (dd, J=1,0; 13,0 Гц, 1Η), 7.51 (d, J=5,0 Гц, 1Η), 7.40 (d, J=2,5 Гц, 1Η), 6.01 (s, 1Η), 4.90-4.88 (m, 1Η), 4.78-4.76 (m, 1Η), 4.64-4.62 (m, 1Η), 4.42-4.41 (m, 2Н), 4.03-3.92 (m, 4Н), 3.59 (s, 3Н), 3.00-2.96 (m, 1Н), 1.40 (s, 9Н), 1.04-1.01 (m, 6Н).
Пример 149а. 1-Метил-4-нитро-1Н-1,2,3-триазол 149а
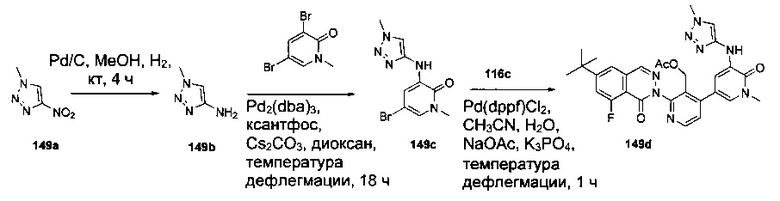
В одногорлую круглодонную колбу емкостью 100 мл, содержащую 4-нитро-2Н-1,2,3-триазол (2,0 г; 17,5 ммоль) и THF (10 мл), при 0°С добавляли NaH (1,7 г; 35,0 ммоль; 2,0 экв.). Смесь перемешивали при 0°С в течение 15 мин. Добавляли раствор CH3I (3,68 г; 26,3 ммоль; 1,5 экв.) в ацетоне (40 мл) и полученную реакционную смесь перемешивали при комнатной температуре в течение 2 ч. По прошествии этого времени реакцию гасили водой (20 мл) при 0°С и реакционную смесь концентрировали при пониженном давлении. Остаток разбавляли дихлорметаном (100 мл). Затем промывали рассолом, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 6:1 петролейный эфир/этилацетат, получая 1-метил-5-нитро-1Н-1,2,3-триазол (1,34 г; 60%) в виде белого твердого вещества и соединение 149а (800 мг; 35%) в виде светло-желтого твердого вещества. 1Н ЯМР (500 МГц, CDCl3) δ 8.34 (s, 1Н), 4.26 (s, 3Н). 1-Метил-5-нитро-1Н-1,2,3-триазол: 1Н ЯМР (500 МГц, CDCl3) δ 8.16 (s, 1Н), 4.31 (s, 3Н).
Пример 149b. 1-Метил-1Н-1,2,3-триазол-4-амин 149b
Следуя методике из примера 148b и исходя из соединения 149а (800 мг; 6,25 ммоль) и 10%-ного палладия на угле (50% влаги, 160 мг), получали соединение 149b в виде желтого твердого вещества (600 мг; 98%). 1Н ЯМР (500 МГц, CDCl3) δ 6.91 (s, 1Н), 3.97 (s, 3Н), 3.65 (brs, 2Н).
Пример 149с. 5-Бром-1-метил-3-(1-метил-1Н-1,2,3-триазол-4-иламино)пиридин-2(1Н)-он 149с
Следуя методике из примера 148с и исходя из соединения 149b (500 мг; 5,10 ммоль; 1,0 экв.) и 3,5-дибром-1-метилпиридин-2(1Н)-она (2,04 г; 7,65 ммоль; 1,5 экв.), получали соединение 149с в виде желтого твердого вещества (760 мг; 52%). MS-ESI: [М+Н]+ 283,9.
Пример 149d. (2-(6-трет-Бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-(1-метил-5-(1-метил-1Н-1,2,3-триазол-4-иламино)-6-оксо-1,6-дигидропиридин-3-ил)пиридин-3-ил)метилацетат 149d
Следуя методике из примера 147d и исходя из соединения 149с (120 мг; 0,42 ммоль; 1 экв.) и 3-(ацетоксиметил)-2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)пиридин-4-илбороновой кислоты (116с) (485 мг; 1,18 ммоль; 2,8 экв.), получали соединение 149d в виде желтого твердого вещества (120 мг; 50%). MS-ESI: [М+Н]+ 573,3.
Пример 149. 6-трет-Бутил-8-фтор-2-[3-(гидроксиметил)-4-[1-метил-5-[(1-метилтриазол-4-ил)амино]-6-оксо-3-пиридил]-2-пиридил]фталазин-1-он 149
Следуя методике из примера 147 и исходя из соединения 149с (90 мг; 0,16 ммоль), получали соединение 149 в виде белого твердого вещества (32 мг; 38%). MS-ESI: [М+Н]+ 530,8. 1Н ЯМР (500 МГц, CDCl3) δ 8.65 (d, J=5,0 Гц, 1H), 8.37 (d, J=1,5 Гц, 1H), 7.69-7.67 (m, 3Н), 7.60-7.57 (m, 2Н), 7.50 (d, J=5,0 Гц, 1H), 7.34 (d, J=1,5 Гц, 1H), 4.53 (s, 2Н), 4.27 (s, 1Н), 4.10 (s, 3Н), 3.74 (s, 3Н), 1.46 (s, 9Н).
Пример 150а. N-Метокси-N-метил-3-нитро-1Н-пиразол-5-карбоксамид 150а
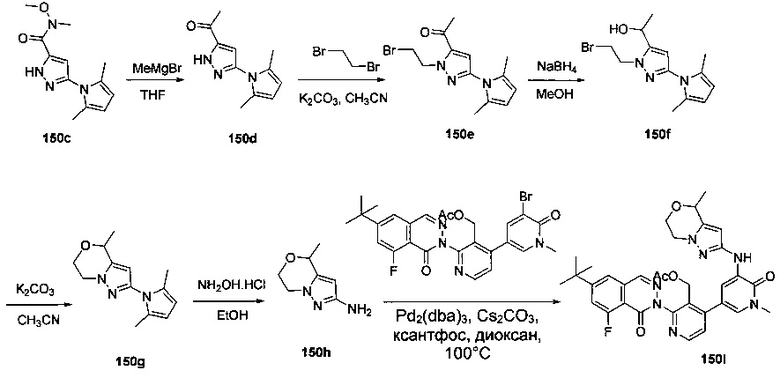
В одногорлую круглодонную колбу емкостью 500 мл, оборудованную магнитной мешалкой, загружали 3-нитро-1Н-пиразол-5-карбоновую кислоту (15,7 г; 1,0 экв.; 100 ммоль), гидрохлорид Ν,Ο-диметилгидроксиламина (19,5 г; 2,0 экв.; 200 ммоль), HATU (гексафторфосфат O-(7-азабензотриазол-1-ил)-1,1,3,3-тетраметилурония) (76,0 г; 2,0 экв.; 200 ммоль), триэтиламин (40,4 г; 4,0 экв.; 400 ммоль) и дихлорметан (300 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Растворитель удаляли при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 100:1 дихлорметан/метанол, получая соединение 150а (16,0 г; 80%) в виде белого твердого вещества. MS-ESI: [М+Н]+ 201,1.
Пример 150b. 3-Амино-N-метокси-N-метил-1Н-пиразол-5-карбоксамид 150b
Одногорлую круглодонную колбу емкостью 250 мл продували азотом и в нее загружали соединение 150а (16,0 г; 1,0 экв.; 80,0 ммоль), 10%-ный палладий на угле (50% влаги; 800 мг) и метанол (100 мл). Колбу вакуумировали, заполняли газообразным водородом и содержимое перемешивали в атмосфере водорода при комнатной температуре в течение ночи. Затем откачивали водород и колбу заполняли азотом. Катализатор удаляли фильтрованием через набивку CELITE®. Фильтрат концентрировали при пониженном давлении, получая соединение 150b (11,0 г; 81%) в виде белого твердого вещества. MS-ESI: [М+Н]+ 171,1.
Пример 150с. 3-(2,5-Диметил-1Н-пиррол-1-ил)-N-метокси-N-метил-1Н-пиразол-5-карбоксамид 150с
В круглодонную колбу емкостью 250 мл, оборудованную магнитной мешалкой и ловушкой Дина-Старка, загружали соединение 150b (11,0 г; 1,0 экв.; 64,7 ммоль), гексан-2,5-дион (11,1 г; 1,5 экв.; 97,2 ммоль), моногидрат п-толуолсульфоновой кислоты (558 мг; 0,05 экв.; 3,24 ммоль) и толуол (100 мл). Реакционную смесь кипятили с обратным холодильником в течение ночи. Полученную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 1:2 петролейный эфир/этилацетат, получая соединение 150с (10,4 г; 65%) в виде белого твердого вещества. MS-ESI: [М+Н]+ 249,0.
Пример 150d. 1-(3-(2,5-Диметил-1Н-пиррол-1-ил)-1Н-пиразол-5-ил)этанон 150d
В круглодонную колбу емкостью 250 мл, оборудованную магнитной мешалкой, загружали соединение 150с (7,44 г; 1,0 экв.; 30,0 ммоль) и THF (100 мл). Под защитой N2 при 0°С добавляли раствор MeMgBr (3,0 Μ в эфире) (25 мл; 2,5 экв.; 75,0 ммоль). Смесь перемешивали при комнатной температуре в течение 5 ч и гасили насыщенным раствором NH4Cl (30 мл). Смесь концентрировали при пониженном давлении и остаток экстрагировали этилацетатом (3×50 мл). Объединенный органический слой упаривали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 4:1 петролейный эфир/этилацетат, получая соединение 150d в виде белого твердого вещества (5,40 г; 89%). MS-ESI: [М+Н]+ 204,0. 1Н ЯМР (500 МГц, CDCl3) δ 6.74 (s, 1Н), 5.92 (s, 2Н), 2.61 (s, 3Н), 2.15 (s, 6Н).
Пример 150е. 1-(1-(2-Бромэтил)-3-(2,5-диметил-1Н-пиррол-1-ил)-1Н-пиразол-5-ил)этанон 150е
В круглодонную колбу емкостью 250 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали соединение 150d (5,4 г; 1,0 экв.; 26,6 ммоль), 1,2-дибромэтан (20,0 г; 4,0 экв.; 106,4 ммоль), K2CO3 (7,34 г; 2,0 экв.; 53,2 ммоль) и ацетонитрил (100 мл). Реакционную смесь кипятили с обратным холодильником в течение 5 ч. Смесь охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 6:1 петролейный эфир/этилацетат, получая соединение 150е (7,5 г; 91%) в виде бесцветного масла. MS-ESI: [М+Н]+ 309,9.
Пример 150f. 1-(1-(2-Бромэтил)-3-(2,5-диметил-1Н-пиррол-1-ил)-1Н-пиразол-5-ил)этанол 150f
В круглодонную колбу емкостью 100 мл, оборудованную магнитной мешалкой, загружали соединение 150е (2,1 г; 1,0 экв.; 6,8 ммоль), NaBH4 (1,29 г; 5,0 экв.; 34,0 ммоль) и метанол (50 мл). Смесь перемешивали при комнатной температуре в течение 2 ч и гасили водой (30 мл). Затем концентрировали при пониженном давлении и остаток экстрагировали дихлорметаном (3×50 мл). Объединенный органический слой концентрировали при пониженном давлении, получая неочищенное соединение 150f, которое использовали на следующей стадии без дополнительной очистки. MS-ESI: [М+Н]+ 311,9.
Пример 150g. 2-(2,5-Диметил-1Н-пиррол-1-ил)-4-метил-6,7-дигидро-4Н-пиразоло[5,1-с][1,4]оксазин 150g
В круглодонную колбу емкостью 100 мл, оборудованную обратным холодильником, загружали соединение 150f (2,0 г; 1,0 экв.; 6,4 ммоль), K2CO3 (1,77 г; 2,0 экв.; 12,8 ммоль), ацетонитрил (50 мл). Реакционную смесь кипятили с обратным холодильником в течение ночи. Затем охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 4:1 петролейный эфир/этилацетат, получая соединение 150g (550 мг; 37%, за две стадии) в виде бесцветного масла. MS-ESI: [М+Н]+ 232,3.
Пример 150h. 4-Метил-6,7-дигидро-4Н-пиразоло[5,1-с][1,4]оксазин-2-амин 150h
В круглодонную колбу емкостью 100 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали соединение 150g (550 мг; 1,0 экв.; 2,38 ммоль), гидроксиламина гидрохлорид (827 мг; 5,0 экв.; 11,9 ммоль) и этанол (30 мл). Смесь кипятили с обратным холодильником в течение 2 суток. Затем охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали препаративной обращенно-фазовой HPLC, получая соединение 150h (30 мг; 8%) в виде желтого масла. MS-ESI: [М+Н]+ 154,1. 1Н ЯМР (500 МГц, CDCl3) δ 5.37 (s, 1Н), 4.73 (q, J=7,0 Гц, 1H), 4.26-4.23 (m, 1Н), 4.08-4.03 (m, 1Н), 3.91-3.90 (m, 1Н), 3.90-3.87 (m, 1Н), 1.65 (d, J=7,0 Гц, 3Н).
Пример 150i. (2-(6-трет-Бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-(1-метил-5-(4-метил-6,7-дигидро-4Н-пиразоло[5,1-с][1,4]оксазин-2-иламино)-6-оксо-1,6-дигидропиридин-3-ил)пиридин-3-ил)метилацетат 150i
В одногорлую круглодонную колбу емкостью 25 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали соединение 150η (25 мг; 1,0 экв.; 0,16 ммоль), (4-(5-бром-1-метил-6-оксо-1,6-дигидропиридин-3-ил)-2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)пиридин-3-ил)метилацетат 142с (181 мг; 2,0 экв.; 0,32 ммоль), Pd2(dba)3 (15 мг; 0,1 экв.; 0,016 ммоль), ксантфос (19 мг; 0,2 экв.; 0,032 ммоль), Cs2CO3 (105 мг; 2,0 экв.; 0,32 ммоль) и диоксан (10 мл). Смесь подвергали трем циклам вакуумирования/продувания аргоном и перемешивали при 100°С в течение 2 ч. Затем охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 20:1 дихлорметан/метанол, получая соединение 150i в виде коричневого твердого вещества (48 мг; 48%).
Пример 150. 6-трет-Бутил-8-фтор-2-[3-(гидроксиметил)-4-[1-метил-5-[(4-метил-6,7-дигидро-4Н-пиразоло[5,1-с][1,4]оксазин-2-ил)амино]-6-оксо-3-пиридил]-2-пиридил]фталазин-1-он 150
В круглодонную колбу емкостью 25 мл, оборудованную магнитной мешалкой, загружали соединение 150i (48 мг; 1,0 экв.; 0,077 ммоль), гидроксид лития (9,2 мг; 5,0 экв.; 0,38 ммоль), смесь изопропанол/THF (4/4 мл) и воду (1 мл). Смесь перемешивали при комнатной температуре в течение 1 ч и концентрировали при пониженном давлении. Остаток очищали препаративной обращенно-фазовой HPLC, получая соединение 150 в виде желтого твердого вещества (6,8 мг; 15%). MS-ESI: [М+Н]+ 585,8. 1Н ЯМР (500 МГц, CDCl3) δ 8.66 (d, J=4,5 Гц, 1H), 8.35 (d, J=2,0 Гц, 1H), 8.03 (d, J=1,5 Гц, 1H), 7.59-7.53 (m, 4Н), 7.47 (s, 1Н), 5.72 (s, 1Н), 4.82-4.78 (m, 1Н), 4.52-4.49 (m, 2Н), 4.33-4.30 (m, 1Н), 4.30-4.27 (m, 1Н), 4.02-3.98 (m, 3Н), 3.73 (s, 3Н), 1.54 (d, J=7,0 Гц, 3Н), 1.48 (s, 9Н).
Пример 151а. Этил-2-(5-(гидроксиметил)-3-нитро-1Н-пиразол-1-ил)ацетат 151а
В одногорлую круглодонную колбу емкостью 100 мл, оборудованную магнитной мешалкой, загружали ацетонитрил (30 мл), (3-нитро-1Н-пиразол-5-ил)метанол (1,43 г; 10,0 ммоль), Cs2CO3 (490 мг; 1,5 ммоль) и этил-2-бромацетат (2,00 г; 12 ммоль). Смесь перемешивали при 40°С в течение 5 ч. Затем охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 30:1 дихлорметан/метанол, получая соединение 151а (1,65 г; 72%) в виде желтого твердого вещества. MS-ESI: [М+Н]+ 229,9.
Пример 151b. Этил-2-(5-(хлорметил)-3-нитро-1Н-пиразол-1-ил)ацетат 151b
К смеси соединения 151а (1,50 г; 6,55 ммоль) в CHCl3 (60 мл), охлажденной при 0°С, медленно добавляли SOCl2 (2,34 г; 19,6 ммоль), все время поддерживая внутреннюю температуру ниже 5°С. Эту реакционную смесь нагревали до 50°С и перемешивали при этой температуре в течение 3 ч. Затем охлаждали до 0°С и гасили водой. Органический слой отделяли и упаривали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 30:1 дихлорметан/метанол, получая соединение 151b (1,1 г; 68%) в виде желтого твердого вещества. MS-ESI: [М+Н]+ 247,9.
Пример 151с. Этил-2-(5-((изопропиламино)метил)-3-нитро-1Н-пиразол-1-ил)ацетат 151с
Смесь соединения 151b (500 мг; 2,04 ммоль), пропан-2-амина (180 мг; 3,06 ммоль) и дихлорметана (20 мл) перемешивали при комнатной температуре в течение 2 ч. Смесь концентрировали при пониженном давлении, получая соединение 151с (400 мг; 74%) в виде желтого твердого вещества, которое использовали на следующей стадии без дополнительной очистки. MS-ESI: [М+Н]+ 271,1.
Пример 151d. 5-Изопропил-2-нитро-4,5-дигидропиразоло[1,5-а]пиразин-6(7Н)-он 151d
Смесь соединения 151с (400 мг; 1,48 ммоль) и метанола (30 мл) перемешивали при 50°С в течение 16 ч. Смесь концентрировали при пониженном давлении. Остаток разбавляли водой (80 мл) и экстрагировали этилацетатом (3×50 мл). Объединенный органический слой сушили над Na2SO4 и концентрировали при пониженном давлении, получая соединение 151d (300 мг; 90%) в виде желтого твердого вещества. MS-ESI: [М+Н]+ 225,3.
Пример 151е. 2-Амино-5-изопропил-4,5-дигидропиразоло[1,5-а]пиразин-6(7Н)-он 151е
К раствору соединения 151d (220 мг; 1,0 ммоль) в этаноле (20 мл) добавляли Pd/C (10%-ный; 22 мг). Сосуд с реакционной смесью заполняли газообразным водородом (с использованием баллона) и содержимое перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь фильтровали через набивку CELITE® и фильтрат концентрировали при пониженном давлении, получая соединение 151е в виде желтого твердого вещества (180 мг; 92%), которое использовали без дополнительной очистки. MS-ESI: [М+Н]+ 195,3.
Пример 151f. 2-(5-Бром-1-метил-2-оксо-1,2-дигидропиридин-3-иламино)-5-изопропил-4,5-дигидропиразоло[1,5-а]пиразин-6(7Н)-он 151f
В круглодонную колбу емкостью 100 мл, оборудованную обратным холодильником, загружали соединение 151е (200 мг; 1,0 ммоль), 3,5-дибром-1-метилпиридин-2(1Н)-он (268 мг; 1,0 ммоль), Pd2(dba)3 (50 мг; 0,050 ммоль), ксантфос (58 мг; 0,10 ммоль), Cs2CO3 (652 мг; 2,0 ммоль) и 1,4-диоксан (20 мл). Систему подвергали трем циклам вакуумирования/продувания аргоном и нагревали при 100°С в течение 2 ч. Затем охлаждали до комнатной температуры и фильтровали. Твердое вещество промывали дихлорметаном (2×10 мл). Объединенный фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с элюированием смесью дихлорметан/метанол (от 80:1 до 30:1), получая соединение 151f (300 мг; 79%) в виде желтого твердого вещества. MS-ESI: [М+Н]+ 380,2.
Пример 151g. 5-Изопропил-2-(1-метил-2-оксо-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,2-дигидропиридин-3-иламино)-4,5-дигидропиразоло[1,5-а]пиразин-6(7Н)-он 151g
В одногорлую круглодонную колбу емкостью 50 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали соединение 151f (200 мг; 0,52 ммоль), Pin2B2 (330 мг; 1,3 ммоль), Pd2(dba)3 (25 мг; 0,026 ммоль), X-phos (25 мг; 0,052 ммоль), ацетат калия (150 мг; 1,5 ммоль) и 1,4-диоксан (10 мл). Смесь подвергали трем циклам вакуумирования/продувания аргоном и нагревали при 65°С в течение 4 ч. Затем фильтровали и фильтрат упаривали при пониженном давлении. Остаток промывали петролейным эфиром, получая неочищенное соединение 151g (300 мг; чистота: 50%) в виде коричневого твердого вещества. MS-ESI: [М+Н]+ 428,2.
Пример 151h. 2-(6-трет-Бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-(5-(5-изопропил-6-оксо-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин-2-иламино)-1-метил-6-оксо-1,6-дигидропиридин-3-ил)никотинальдегид 151h
В круглодонную колбу емкостью 25 мл, оборудованную обратным холодильником, загружали 2-(6-трет-бутил-8-фтор-1-оксоизохинолин-2(1Н)-ил)-4-хлорникотинальдегид 103b (108 мг; 0,30 ммоль), соединение 151g (256 мг; 0,60 ммоль), K3PO4 (127 мг; 0,60 ммоль), моногидрат ацетата натрия (82 мг; 0,60 ммоль), Pd(dppf)Cl2 (12 мг; 0,015 ммоль), воду (0,5 мл) и ацетонитрил (8 мл). Реакционную смесь подвергали трем циклам вакуумирования/продувания азотом и нагревали при 80°С под защитой N2 в течение 2 ч. Затем охлаждали до комнатной температуры и концентрировали при пониженном давлении. Остаток разбавляли дихлорметаном (20 мл) и водой (10 мл). Органический слой отделяли и водный слой экстрагировали дихлорметаном (2×10 мл). Объединенный органический экстракт сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Темный остаток очищали колоночной хроматографией на силикагеле с элюированием смесью дихлорметан/метанол (от 80:1 до 30:1), получая соединение 151h (90 мг; 48%) в виде желтого твердого вещества. MS-ESI: [М+Н]+ 625,2.
Пример 151. 6-трет-Бутил-8-фтор-2-(3-(гидроксиметил)-4-(5-(5-изопропил-6-оксо-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин-2-иламино)-1-метил-6-оксо-1,6-дигидропиридин-3-ил)пиридин-2-ил)фталазин-1(2Н)-он 151
В одногорлую круглодонную колбу емкостью 25 мл загружали соединение 151h (90 мг; 0,14 ммоль), NaBH4 (18 мг; 0,42 ммоль) и метанол (5 мл). Смесь перемешивали при комнатной температуре в течение 1 ч и концентрировали при пониженном давлении. К полученному остатку добавляли воду (10 мл) и смесь экстрагировали дихлорметаном (3×15 мл). Объединенный органический слой концентрировали при пониженном давлении. Остаток очищали препаративной обращенно-фазовой HPLC, получая соединение 151 (50 мг; 55%). MS-ESI: [М+Н]+ 626,8. 1Н ЯМР (500 МГц, CDCl3) δ 8.67 (d, J=5,0 Гц, 1H), 8.36 (d, J=2,5 Гц, 1H), 8.02 (d, J=2,0 Гц, 1H), 7.59-7.58 (m, 3Н), 7.55-7.52 (m, 2Н), 5.90 (s, 1Н), 5.07-5.05 (m, 1Н), 4.74 (s, 2Н), 4.50-4.47 (m, 4Н), 4.09-4.06 (m, 1Н), 3.73 (s, 3Н), 1.45 (s, 9Н), 1.26 (d, J=7,0 Гц, 6H).
Пример 152а. трет-Бутил-3-амино-1Н-пиразол-1-карбоксилат 152а
К смеси 3-аминопиразола (3,0 г; 36 ммоль) и триэтиламина (7,6 г; 75 ммоль) в 1,4-диоксане (35 мл) добавляли (Вос)2O (7,8 г; 36 ммоль). Реакционную смесь перемешивали при 25°С в течение 2 ч. Затем концентрировали при пониженном давлении. Остаток очищали на колонке с силикагелем с элюированием смесью 3:1 петролейный эфир/этилацетат, получая соединение 152а в виде белого твердого вещества (3,4 г; 52%). MS-ESI: [М+Н]+ 184,1.
Пример 152b. 3-(1Н-Пиразол-3-иламино)-5-бром-1-метилпиридин-2(1Н)-он 152а
В одногорлую круглодонную колбу емкостью 250 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали соединение 152а (2,2 г; 12 ммоль), ксантфос (0,69 г; 1,2 ммоль), Pd2(dba)3 (1,1 г; 1,2 ммоль), 3,5-дибром-1-метилпиридин-2(1Н)-он (6,4 г; 24 ммоль), Cs2CO3 (15,6 г; 48 ммоль) и 1,4-диоксан (50 мл). После барботирования азота через полученную смесь в течение 10 минут ее нагревали при 105°С в течение 15 ч. Смесь охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении и оставшуюся смесь промывали метанолом (8 мл), получая соединение 152b в виде бледно-желтого твердого вещества (1,2 г; 37%). MS-ESI: [М+Н]+ 269,1.
Пример 152с. (4-(5-(1 Н-Пиразол-3-иламино)-1-метил-6-оксо-1,6-дигидропиридин-3-ил)-2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)пиридин-3-ил)метилацетат 152с
В герметично закрываемую колбу, оборудованную магнитной мешалкой, загружали соединение 152b (200 мг; 0,74 ммоль), (2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-3-ил)метилацетат 116с (370 мг; 0,74 ммоль), Pd(dppf)Cl2 (30 мг; 0,035 ммоль), ацетат натрия (74 мг; 0,90 ммоль), K3PO4 (191 мг; 0,90 ммоль) и смесь ацетонитрил/вода (5 мл/0,5 мл). После трех циклов вакуумирования/продувания аргоном смесь нагревали при 100°С в течение 2 ч. Затем фильтровали и фильтрат упаривали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 10:1 дихлорметан/метанол, получая соединение 152с (100 мг; 25%) в виде коричневого твердого вещества. MS-ESI: [М+Н]+ 558,3.
Пример 152. 6-трет-Бутил-8-фтор-2-[3-(гидроксиметил)-4-[1-метил-6-оксо-5-(1Н-пиразол-3-иламино)-3-пиридил]-2-пиридил]фталазин-1-он 152
Смесь соединения 152с (100 мг; 0,18 ммоль) и гидрата гидроксида лития (84 мг; 2,0 ммоль) в THF (8 мл), изопропаноле (8 мл) и воде (2 мл) перемешивали при 40°С в течение 0,5 ч. Смесь упаривали при пониженном давлении и остаток разбавляли водой (5 мл). Затем экстрагировали этилацетатом (2×10 мл). Объединенный экстракт концентрировали при пониженном давлении и остаток очищали препаративной обращенно-фазовой HPLC, получая соединение 152 (6,5 мг; 7%) в виде бледно-желтого твердого вещества. MS-ESI: [М+Н]+ 515,8. 1Н ЯМР (500 МГц, CDCl3) δ 8.62-8.61 (m, 1Н), 8.32 (s, 1Н), 8.04 (m, 1Н), 7.55-7.53 (m, 4Н), 7.50-7.49 (m, 1Н), 7.44 (s, 1Н), 6.01 (s, 1Н), 4.49-4.48 (m, 2Н), 3.71 (s, 3Н), 1.45 (s, 9Н).
Пример 153а. 6-Хлор-2-метил-4-(5-метил-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин-2-иламино)пиридазин-3(2Н)-он 153а
В одногорлую круглодонную колбу емкостью 100 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали 1,4-диоксан (30 мл), 5-метил-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин-2-амин 128b (1,70 г; 11,2 ммоль), 4-бром-6-хлор-2-метилпиридазин-3(2Н)-он (2,68 г; 12,0 ммоль) и карбонат цезия (7,30 г; 22,4 ммоль). После барботирования азота через суспензию в течение 30 минут добавляли ксантфос (0,59 г; 1,02 ммоль) и трис(дибензилиденацетон)дипалладий(0) (467 мг; 0,51 ммоль). Систему подвергали трем циклам вакуумирования/продувания аргоном и нагревали при 90°С в течение 2 ч. Затем охлаждали до комнатной температуры и фильтровали. Фильтрат упаривали в вакууме. Остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 30:1 дихлорметан/метанол, получая соединение 153а (1,9 г; 60%) в виде коричневого твердого вещества. LCMS: [М+Н]+ 295,1.
Пример 153b. 1-Метил-5-(5-метил-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин-2-иламино)-6-оксо-1,6-дигидропиридазин-3-илбороновая кислота 153b
В круглодонную колбу емкостью 50 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали соединение 153а (200 мг; 0,68 ммоль), бис(пинаколато)дибор (Pin2B2; 863 мг; 3,40 ммоль), Pd2(dba)3 (55 мг; 0,060 ммоль), X-Phos (60 мг; 0,12 ммоль), ацетат калия (60 мг; 1,36 ммоль) и 1,4-диоксан (10 мл). Колбу вакуумировали и затем заполняли N2. Далее смесь нагревали при 50°С в течение 2 ч. По завершении реакции смесь фильтровали и твердое вещество промывали этилацетатом (10 мл). Объединенный фильтрат упаривали при пониженном давлении, получая соединение 153b в виде бледно-желтого твердого вещества, которое использовали на следующей стадии. MS-ESI: [М+Н]+ 305,1.
Пример 153с. 2-(6-трет-Бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-(1-метил-5-(5-метил-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин-2-иламино)-6-оксо-1,6-дигидропиридазин-3-ил)никотинальдегид 153с
В круглодонную колбу емкостью 25 мл, оборудованную обратным холодильником, загружали соединение 153b (200 мг; 0,66 ммоль), 2-(в-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-хлорникотинальдегид 103b (240 мг; 0,66 ммоль), PdCl2(dppf) (54 мг; 0,066 ммоль), K3PO4 (250 мг; 1,2 ммоль), ацетат натрия (100 мг; 1,20 ммоль), ацетонитрил (10 мл) и воду (0,5 мл). Сосуд вакуумировали и затем заполняли N2. Далее смесь нагревали при 100°С в течение 1 ч. Смесь охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении и остаток очищали на колонке с силикагелем с элюированием смесью 20:1 дихлорметан/метанол, получая соединение 153с в виде бледно-желтого твердого вещества (170 мг; 42%). MS-ESI: [М+Н]+ 584,3.
Пример 153. 6-трет-Бутил-8-фтор-2-[3-(гидроксиметил)-4-[1-метил-5-[(5-метил-6,7-дигидро-4Н-пиразоло[1,5-а]пиразин-2-ил)амино]-6-оксо-пиридазин-3-ил]-2-пиридил]фталазин-1-он 153
Смесь соединения 153с (170 мг; 0,29 ммоль) и NaBH4 (20 мг; 0,50 ммоль) в метаноле (5 мл) перемешивали при комнатной температуре в течение 0,5 ч. Затем реакционную смесь гасили водой (7 мл) и концентрировали при пониженном давлении. Остаток экстрагировали дихлорметаном (2×10 мл). Объединенный дихлорметановый экстракт концентрировали при пониженном давлении и остаток очищали обращенно-фазовой препаративной HPLC, получая соединение 153 в виде бледно-желтого твердого вещества (35 мг; 21%). MS-ESI: [М+Н]+ 586,3. 1Н ЯМР (500 МГц, DMSO-d6) δ 9.39 (s, 1Н), 8.63 (d, J=5,0 Гц, 1H), 8.54 (d, J=2,0 Гц 1H), 7.93 (s, 1Н), 7.90 (d, J=2,0 Гц, 1H), 7.77 (dd, J=1,5; 12,5 Гц, 1H), 7.60-7.59 (m, 1Н), 6.03 (s, 1Н), 4.82-4.81 (m, 1Н), 4.51-4.45 (m, 2Н), 4.01-3.98 (m, 2Н), 3.77 (s, 3Н), 3.62-3.60 (m, 2Н), 2.90-2.86 (m, 2Н), 2.42-2.40 (m, 3Н), 1.34 (s, 9Н).
Пример 154а. 1-Метил-1Н-1,2,3-триазол-5-амин 154а
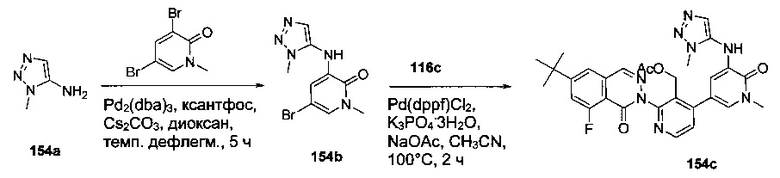
Круглодонную колбу емкостью 50 мл продували азотом и в нее загружали 1-метил-5-нитро-1Н-1,2,3-триазол из примера 149а (0,78 г; 6,09 ммоль), 10%-ный палладий на угле (50% влаги; 160 мг) и метанол (20 мл). Колбу вакуумировали, заполняли газообразным водородом и содержимое перемешивали в течение 4 ч при комнатной температуре. Затем откачивали водород и колбу заполняли азотом. Катализатор удаляли фильтрованием через набивку CELITE® и фильтрат концентрировали при пониженном давлении, получая соединение 154а (418 мг; 70%) в виде желтого твердого вещества. MS: [М+Н]+ 99,3.
Пример 154b. 5-Бром-1-метил-3-(1-метил-1Н-1,2,3-триазол-5-иламино)-пиридин-2(1Н)-он 154b
В одногорлую круглодонную колбу емкостью 100 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали 1,4-диоксан (20 мл), соединение 154а (500 мг; 5,10 ммоль), 3,5-дибром-1-метилпиридин-2(1Н)-он (1362 мг; 5,10 ммоль) и карбонат цезия (3,325 г; 10,2 ммоль). После барботирования азота через суспензию в течение 20 минут добавляли ксантфос (0,59 г; 1,02 ммоль) и трис(дибензилиденацетон)дипалладий(0) (467 мг; 0,51 ммоль). Систему подвергали трем циклам вакуумирования/продувания азотом и нагревали при температуре дефлегмации в течение 5 ч. Затем охлаждали до комнатной температуры и фильтровали. Фильтрат упаривали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 50:1 дихлорметан/метанол, получая соединение 154b (220 мг; 15%) в виде коричневого твердого вещества. LCMS: [М+Н]+ 284,1.
Пример 154с. (2-(6-трет-Бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-(1-метил-5-(1-метил-1Н-1,2,3-триазол-5-иламино)-6-оксо-1,6-дигидропиридин-3-ил)пиридин-3-ил)метилацетат 154с
В круглодонную колбу емкостью 25 мл, оборудованную обратным холодильником, загружали соединение 154b (100 мг; 0,35 ммоль), 3-(ацетоксиметил)-2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)пиридин-4-илбороновую кислоту 116с (174 мг; 0,42 ммоль), Pd(dppf)Cl2 (29 мг; 0,035 ммоль), ацетат натрия (57 мг; 0,70 ммоль), тригидрат K3PO4 (186 мг; 0,70 ммоль), воду (6 капель) и ацетонитрил (6 мл). После трех циклов вакуумирования/продувания аргоном смесь нагревали при 100°С в течение 2 ч. Затем фильтровали и фильтрат упаривали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 25:1 дихлорметан/метанол, получая соединение 154с (85 мг; 42%) в виде коричневого твердого вещества. MS-ESI: [М+Н]+ 573,4.
Пример 154. 6-трет-Бутил-8-фтор-2-[3-(гидроксиметил)-4-[1-метил-5-[(3-метилтриазол-4-ил)амино]-6-оксо-3-пиридил]-2-пиридил]фталазин-1-он 154
Смесь соединения 154с (85 мг; 0,15 ммоль) и гидроксида лития (36 мг; 1,5 ммоль) в смеси изопропанол/THF (1:1; 4 мл) и воде (1 мл) перемешивали при 30°С в течение 1 ч. Смесь упаривали при пониженном давлении и остаток разбавляли водой (5 мл). Затем экстрагировали этилацетатом (2×10 мл). Объединенный этилацетатный экстракт концентрировали при пониженном давлении и остаток очищали препаративной обращенно-фазовой HPLC, получая указанное в заголовке соединение (11 мг; 16%) в виде белого твердого вещества. MS-ESI: [М+Н]+ 531,4. 1Н ЯМР (500 МГц, DMSO-d6) δ 8.54-8.53 (m, 2Н), 8.03 (s, 1Н), 7.91 (d, J=1,5 Гц, 1H), 7.80-7.76 (m, 1Н), 7.75 (s, 1Н), 7.55 (d, J=2,5 Гц, 1H), 7.52 (d, J=5,0 Гц, 1Η), 6.89 (d, J=2,5 Гц, 1Η), 5.04 (t, J=5,0 Гц, 1Η), 4.34 (d, J=5,0 Гц, 2H), 3.88 (s, 3Н), 3.63 (s, 3Н), 1.40 (s, 9Н).
Пример 155а. 2-Нитро-5-(оксетан-3-ил)-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин 155а
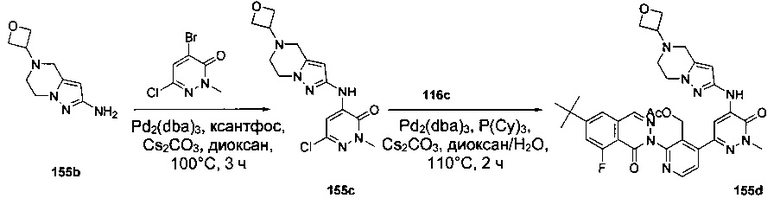
Смесь 2-нитро-4,5,6,7-тетрагидропиразоло[1,5-а]пиразина 124d (238 мг; 1,40 ммоль), оксетан-3-она (252 мг; 3,5 ммоль), NaBH3CN (260 мг; 4,2 ммоль) и хлорида цинка (567 мг; 4,2 ммоль) в метаноле (10 мл) перемешивали при 50°С в течение 6 часов. К смеси добавляли воду и три раза экстрагировали дихлорметаном. Объединенный органический слой концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 50:1 дихлорметан/метанол, получая соединение 155а (200 мг; 64%). MS: [М+Н]+ 225,3.
Пример 155b. 5-(Оксетан-3-ил)-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин-2-амин 155b
Одногорлую круглодонную колбу емкостью 50 мл продували азотом и в нее загружали соединение 155а (0,20 г; 0,89 ммоль), 10%-ный палладий на угле (50% влаги; 89 мг) и метанол (10 мл). Колбу вакуумировали, заполняли газообразным водородом и содержимое перемешивали в течение 2 ч при комнатной температуре. Затем откачивали водород и колбу заполняли азотом. Катализатор удаляли фильтрованием через набивку CELITE® и фильтрат концентрировали при пониженном давлении, получая соединение 155b (140 мг; 81%). MS: [М+Н]+ 195,1.
Пример 155с. 6-Хлор-2-метил-4-(5-(оксетан-3-ил)-4,5,6,7-тетрагидро-пиразоло[1,5-а]пиразин-2-иламино)пиридазин-3(2Н)-он 155с
В одногорлую круглодонную колбу емкостью 100 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали 1,4-диоксан (25 мл), соединение 155b (640 мг; 3,3 ммоль), 4-бром-6-хлор-2-метилпиридазин-3(2Н)-он (1,1 г; 4,95 ммоль), Pd2(dba)3 (302 мг; 0,33 ммоль), ксантфос (381 мг; 0,66 ммоль) и карбонат цезия (2,15 г; 6,6 ммоль). После трех циклов вакуумирования/продувания аргоном смесь нагревали при 100°С в течение 3 ч. По прошествии этого времени реакционную смесь охлаждали до комнатной температуры. Затем фильтровали и фильтрат упаривали при пониженном давлении. Остаток промывали этилацетатом, получая соединение 155с (754 мг; 68%) в виде желтого твердого вещества. MS-ESI: [М+Н]+ 337,3.
Пример 155d. (2-(6-трет-Бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-(1-метил-5-(5-(оксетан-3-ил)-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин-2-иламино)-6-оксо-1,6-дигидропиридазин-3-ил)пиридин-3-ил)метилацетат 155d
В одногорлую круглодонную колбу емкостью 100 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали соединение 155с (367 мг; 1,1 ммоль), 3-(ацетоксиметил)-2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)пиридин-4-илбороновую кислоту 116с (908,6 мг; 2,2 ммоль), Pd2(dba)3 (100,6 мг; 0,11 ммоль), Р(Cy)3 (122,8 мг; 0,44 ммоль), Cs2CO3 (717 мг; 2,2 ммоль), диоксан (20 мл) и воду (0,5 мл). После трех циклов вакуумирования/продувания аргоном смесь нагревали при 110°С в течение 2 ч. Затем фильтровали и фильтрат упаривали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 35:1 дихлорметан/метанол, получая соединение 155d (120 мг; 16%) в виде желтого твердого вещества. MS-ESI: [М+Н]+ 669,8.
Пример 155. 6-трет-Бутил-8-фтор-2-[3-(гидроксиметил)-4-[1-метил-5-[[5-(оксетан-3-ил)-6,7-дигидро-4Н-пиразоло[1,5-а]пиразин-2-ил]амино]-6-оксо-пиридазин-3-ил]-2-пиридил]фталазин-1-он 155
Смесь соединения 155d (117 мг; 0,175 ммоль) и моногидрата гидроксида лития (73,5 мг; 1,75 ммоль) в смеси изопропанол/THF (1:1; 8 мл) и воде (1 мл) перемешивали при 35°С в течение 0,5 ч. Смесь упаривали при пониженном давлении и остаток разбавляли водой (6 мл). Затем смесь экстрагировали дихлорметаном (3×50 мл). Объединенный дихлорметановый экстракт концентрировали при пониженном давлении и остаток очищали препаративной обращенно-фазовой HPLC, получая соединение 155 (30 мг; 27%) в виде желтого твердого вещества. MS-ESI: [М+Н]+ 628,3. 1Н ЯМР (500 МГц, DMSO-d6) δ 9.40 (s, 1Н), 8.64 (d, J=5,5 Гц, 1H), 8.55 (d, J=2,0 Гц, 1H), 7.94 (s, 1Н), 7.90 (d, J=1,5 Гц, 1H), 7.79-7.77 (m, 1Н), 7.61 (d, J=4,5 Гц, 1H), 6.04 (s, 1Н), 7.61-7.60 (m, 1Н), 4.61-4.59 (m, 2Н), 4.50-4.45 (m, 4Н), 4.01-3.99 (m, 2Н), 3.78 (s, 3Н), 3.69-3.65 (m, 1Н), 3.54 (s, 2Н), 2.79-2.76 (m, 2Н), 1.39 (s, 9Н).
Пример 156а. 5-Фтор-N,N-бис(4-метоксибензил)пиридин-2-амин 156
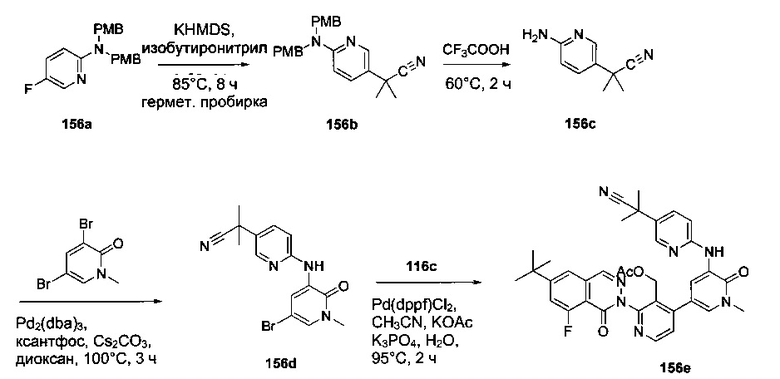
В круглодонную колбу емкостью 100 мл загружали 5-фторпиридин-2-амин (1,12 г; 10,0 ммоль), NaH (288 мг; 12,0 ммоль) и THF (20 мл) при 25°С. Добавляли 4-метоксибензилхлорид (1,87 г; 12,0 ммоль) и перемешивали при 25°С в течение 2 ч. Затем все это концентрировали при пониженном давлении. К остатку добавляли воду (30 мл) и полученную смесь экстрагировали дихлорметаном (3×80 мл). Объединенный экстракт сушили над Na2SO4, фильтровали и упаривали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 1:9 этилацетат/петролейный эфир, получая соединение 156а (620 мг; 18%) в виде желтой жидкости. MS-ESI: [М+Н]+ 353,0.
Пример 156b. 2-(6-(бис(4-Метоксибензил)амино)пиридин-3-ил)-2-метилпропаннитрил 156b
В герметично закрываемую колбу емкостью 25 мл, оборудованную магнитной мешалкой, загружали соединение 156а (528 мг; 1,5 ммоль), KHMDS (гексаметилдисилазид калия) (15 ммоль; 15 мл; 1 моль/л в THF) и изобутиронитрил (1,03 г; 15 ммоль). После трех циклов вакуумирования/продувания аргоном смесь нагревали при 85°С в течение 8 ч. Затем охлаждали до комнатной температуры и гасили, добавляя воду. Смесь концентрировали при пониженном давлении и остаток экстрагировали этилацетатом (3×20 мл). Объединенный экстракт сушили над Na2SO4, фильтровали и упаривали при пониженном давлении, получая соединение 156b (514 мг; 85%) в виде желтой жидкости. MS-ESI: [М+Н]+ 402,0.
Пример 156с. 2-(6-Аминопиридин-3-ил)-2-метилпропаннитрил 156с
Раствор соединения 156b (514 мг; 1,28 ммоль) в CF3COOH (15 мл) перемешивали при 60°С в течение 2 ч. По прошествии этого времени реакционную смесь охлаждали до комнатной температуры. Затем упаривали при пониженном давлении и остаток промывали петролейным эфиром и этилацетатом, получая соединение 156с (600 мг; неочищенное) в виде желтого твердого вещества. MS-ESI: [М+Н]+ 162,3.
Пример 156d. 2-(6-(5-Бром-1-метил-2-оксо-1,2-дигидропиридин-3-иламино)пиридин-3-ил)-2-метилпропаннитрил 156d
В одногорлую круглодонную колбу емкостью 100 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали 1,4-диоксан (35 мл), соединение 156с (483 мг; 3,0 ммоль), 3,5-дибром-1-метил-пиридин-2(1Н)-он (1,6 г; 6,0 ммоль), Pd2(dba)3 (274,5 мг; 0,30 ммоль), ксантфос (346,8 мг; 0,60 ммоль) и карбонат цезия (4,59 г; 15 ммоль). После трех циклов вакуумирования/продувания аргоном смесь нагревали при 100°С в течение 3 ч. По прошествии этого времени реакционную смесь охлаждали до комнатной температуры и фильтровали. Фильтрат упаривали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 1:1 этилацетат/петролейный эфир, получая соединение 156d (400 мг; 38%) в виде желтого твердого вещества. MS-ESI: [М+Н]+ 347,0.
Пример 156е. (2-(6-трет-Бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-(5-(5-(2-цианопропан-2-ил)пиридин-2-иламино)-1-метил-6-оксо-1,6-дигидропиридин-3-ил)пиридин-3-ил)метилацетат 156е
В одногорлую круглодонную колбу емкостью 100 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали соединение 156d (346 мг; 1,0 ммоль), 3-(ацетоксиметил)-2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)пиридин-4-илбороновую кислоту 116с (1,65 г; 4,0 ммоль), Pd(dppf)Cl2 (82,5 мг; 0,10 ммоль), ацетат калия (196 мг; 2,0 ммоль), K3PO4 (424 мг; 2,0 ммоль), ацетонитрил (15 мл) и воду (0,5 мл). После трех циклов вакуумирования/продувания аргоном смесь нагревали при 95°С в течение 2 ч. Затем фильтровали и фильтрат упаривали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 10:1 этилацетат/петролейный эфир, получая соединение 156е (210 мг; 33%) в виде желтого твердого вещества. MS-ESI: [М+Н]+ 635,8.
Пример 156. 2-[6-[[5-[2-(6-трет-Бутил-8-фтор-1-оксо-фталазин-2-ил)-3-(гидроксиметил)-4-пиридил]-1-метил-2-оксо-3-пиридил]амино]-3-пиридил]-2-метил-пропаннитрил 156
Смесь соединения 156е (191 мг; 0,30 ммоль) и моногидрата гидроксида лития (126 мг; 3,0 ммоль) в смеси изопропанол/THF (1:1; 8 мл) и воде (2 мл) перемешивали при 35°С в течение 0,5 ч. Смесь упаривали при пониженном давлении и разбавляли водой (10 мл). Затем экстрагировали дихлорметаном (3×20 мл). Объединенный дихлорметановый экстракт концентрировали при пониженном давлении и остаток очищали препаративной обращенно-фазовой HPLC, получая соединение 156 (30 мг; 17%) в виде бледно-желтого твердого вещества. MS-ESI: [М+Н]+ 594,3. 1Н ЯМР (500 МГц, DMSO-d6) δ 8.83 (s, 1Н), 8.77 (d, J=2,0 Гц, 1Η), 8.59 (d, J=5,0 Гц, 1Η), 8.54 (d, J=2,5 Гц, 1Η), 8.34 (d, J=2,5 Гц, 1H), 7.91 (s, 1Н), 7.79-7.74 (m, 2Н), 7.57-7.53 (m, 2Н), 7.40 (d, J=9,0 Гц, 1H), 4.92 (bs, 1Η), 4.43 (s, 2Η), 3.62 (s, 3Н), 1.67 (s, 6Н), 1.39 (s, 9Н).
Пример 157а. (2-(6-трет-Бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-(1-метил-5-(5-метилизотиазол-3-иламино)-6-оксо-1,6-дигидропиридин-3-ил)пиридин-3-ил)метилацетат 157а
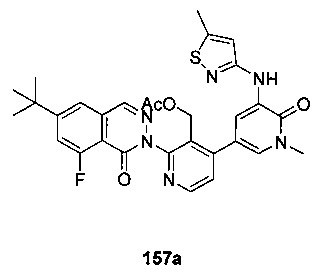
В герметично закрываемую пробирку емкостью 25 мл загружали (4-(5-бром-1-метил-6-оксо-1,6-дигидропиридин-3-ил)-2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)пиридин-3-ил)метилацетат 142с (155 мг; 0,28 ммоль), гидрохлорид 4-фтор-2-(1-оксо-5-метилизотиазол-3-амина (55 мг; 0,33 ммоль), Cs2CO3 (183 мг; 0,56 ммоль), Pd2(dba)3 (27 мг; 0,030 ммоль), ксантфос (35 мг; 0,060 ммоль) и DMF (10 мл). После трех циклов вакуумирования/продувания аргоном смесь нагревали при 110°С в условиях облучения микроволнами в течение 1 часа. Затем охлаждали до комнатной температуры и упаривали при пониженном давлении. Остаток очищали на колонке с силикагелем с элюированием смесью 20:1 метиленхлорид/метанол, получая соединение 157а в виде желтого твердого вещества (64 мг; 39%). MS-ESI: [М+Н]+ 589,2.
Пример 157. 6-трет-Бутил-8-фтор-2-[3-(гидроксиметил)-4-[1-метил-5-[(5-метилизотиазол-3-ил)амино]-6-оксо-3-пиридил]-2-пиридил]фталазин-1-он 157
К раствору соединения 157а (60 мг; 0,10 ммоль) в смеси THF/изопропанол/вода (4 мл/4 мл/1 мл) добавляли гидроксид лития (24 мг; 1,0 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 0,5 ч и концентрировали при пониженном давлении. Остаток разбавляли водой (10 мл) и экстрагировали этилацетатом (3×15 мл). Объединенный органический слой сушили, используя Na2SO4, и концентрировали с получением желтого твердого вещества, которое очищали препаративной обращенно-фазовой HPLC, получая соединение 157 в виде желтого твердого вещества (27 мг; 50%). MS-ESI: [М+Н]+ 546,7. 1Н ЯМР (500 МГц, DMSO-d6) δ 9.28 (s, 1Н), 8.60-8.54 (m, 3Н), 7.90 (s, 1H), 7.78 (d, J=16,0 Гц, 1H), 7.55-7.50 (m, 2H), 7.04 (s, 1H), 4.89 (t, J=6,5 Гц, 1H), 4.43-4.41 (m, 2H), 3.61 (s, 3Н), 2.48 (s, 3Н), 1.40 (s, 9H).
Пример 158а. 5-Бром-3-(5-этилизоксазол-3-иламино)-1-метилпиридин-2(1Н)-он 158а
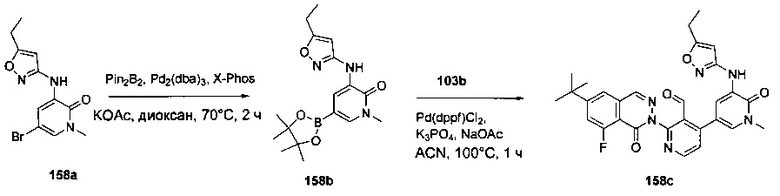
В одногорлую круглодонную колбу емкостью 50 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали 5-этилизоксазол-3-амин (250 мг; 2,23 ммоль), 3,5-дибром-1-метилпиридин-2(1Н)-он (893 мг; 3,35 ммоль), Pd2(dba)3 (102 мг; 0,112 ммоль), ксантфос (129 мг; 0,223 ммоль), Cs2CO3 (1,45 г; 4,46 ммоль) и диоксан (20 мл). Систему подвергали трем циклам вакуумирования/продувания азотом и нагревали при 100°С под защитой N2 в течение 3 ч. Анализ реакционной смеси с использованием LCMS показал полное превращение в желаемый продукт. Затем смесь охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении и остаток очищали препаративной обращенно-фазовой HPLC, получая соединение 158а (300 мг; 45%) в виде белого твердого вещества. MS-ESI: [М+Н]+ 297,8.
Пример 158b. 3-(5-Этилизоксазол-3-иламино)-1-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2(1Н)-он 158b
В круглодонную колбу емкостью 50 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали соединение 158а (250 мг; 0,839 ммоль), Pin2B2 (320 мг; 1,26 ммоль), Pd2(dba)3 (38,4 мг; 0,042 ммоль), X-Phos (48,5 мг; 0,0839 ммоль), ацетат калия (164,4 мг; 1,678 ммоль) и диоксан (20 мл). Систему подвергали трем циклам вакуумирования/продувания азотом и нагревали при 70°С под защитой N2 в течение 2 ч. Анализ реакционной смеси с использованием LCMS показал полное превращение в желаемый продукт. Затем смесь охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток промывали петролейным эфиром, получая соединение 158b (330 мг; неочищенное) в виде темного твердого вещества, которое использовали на следующей стадии без дополнительной очистки. MS-ESI: [М+Н]+ 346,0.
Пример 158с. 2-(6-трет-Бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-(5-(5-этилизоксазол-3-иламино)-1-метил-6-оксо-1,6-дигидропиридин-3-ил)никотинальдегид 158с
В круглодонную колбу емкостью 50 мл, оборудованную обратным холодильником, загружали 2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-хлорникотинальдегид 103b (160 мг; 0,44 ммоль), соединение 158b (228 мг; 0,66 ммоль), Pd(dppf)Cl2 (18 мг; 0,022 моль), K3PO4 (187 мг; 0,88 ммоль), ацетат натрия (72,2 мг; 0,88 ммоль), ацетонитрил (15 мл) и воду (5 капель). Систему подвергали трем циклам вакуумирования/продувания азотом и нагревали при 100°С под защитой N2 в течение 1 ч. Анализ реакционной смеси с использованием LCMS показал полное превращение в желаемый продукт. Затем смесь охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 40:1 дихлорметан/метанол, получая соединение 158с (210 мг; 88%) в виде желтого масла. MS-ESI: [М+Н]+ 543,3.
Пример 158. 6-трет-Бутил-2-[4-[5-[(5-этилизоксазол-3-ил)амино]-1-метил-6-оксо-3-пиридил]-3-(гидроксиметил)-2-пиридил]-8-фтор-фталазин-1-он 158
К раствору соединения 158с (190 мг; 0,35 ммоль) в дихлорметане (5 мл) и метаноле (5 мл) добавляли NaBH4 (26,5 мг; 0,70 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч и гасили водным NH4Cl. Затем концентрировали при пониженном давлении и остаток экстрагировали дихлорметаном. Объединенный экстракт промывали рассолом, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали препаративной обращенно-фазовой HPLC, получая соединение 158 (70 мг; 37%) в виде белого твердого вещества. MS-ESI: [М+Н]+ 545,3. 1Н ЯМР (500 МГц, DMSO-d6) δ 9.01 (s, 1Н), 8.58 (d, J=5,0 Гц, 1H), 8.54 (d, J=2,0 Гц, 1H), 8.01 (d, J=2,0 Гц, 1H), 7.90 (s, 1Н), 7.79 (d, J=13 Гц, 1H), 7.57 (d, J=2,5 Гц, 1H), 7.51 (d, J=5,0 Гц, 1H), 6.26 (s, 1Н), 4.90 (s, 1Н), 4.44-4.43 (m, 2Н), 3.61 (s, 3Н), 2.69-2.65 (m, 2Н), 1.40 (s, 9Н), 1.19 (t, J=8,0 Гц, 3Н).
Пример 159а. 2-(Дифторметил)-4-нитро-2Н-1,2,3-триазол 159а
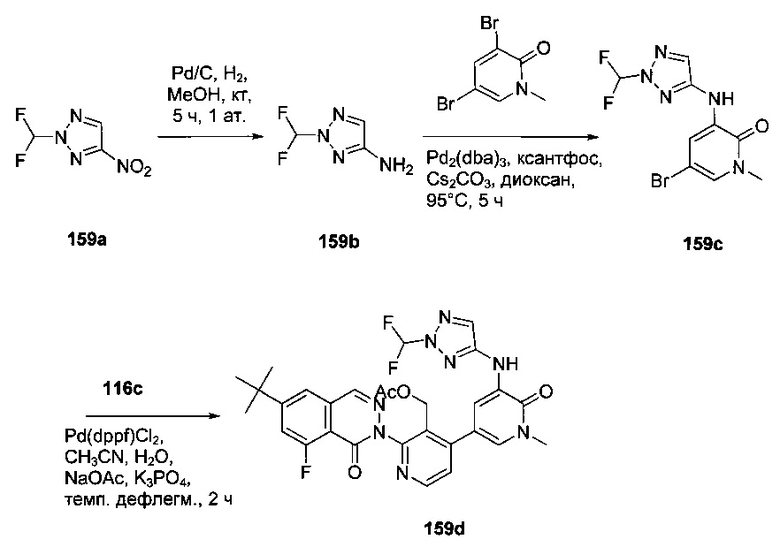
Содержимое одногорлой круглодонной колбы емкостью 100 мл, содержащей 4-нитро-2Н-1,2,3-триазол (500 мг; 4,38 ммоль; 1,0 экв.), 2-хлор-2,2-дифторацетат натрия (1310 мг; 8,76 ммоль; 2,0 экв.), K2CO3 (1140 мг; 8,76 ммоль; 2,0 экв.) и ацетонитрил (20 мл), перемешивали при температуре дефлегмации в течение 5 ч. После охлаждения до комнатной температуры реакционную смесь фильтровали и фильтрат упаривали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 4:1 петролейный эфир/этилацетат, получая соединение 159а (350 мг; 48%) в виде коричневой жидкости. 1Н ЯМР (500 МГц, CDCl3) δ 8.38 (s, 1Н), 7.39 (t, J=57,5 Гц, 1H).
Пример 159b. 2-(Дифторметил)-2Н-1,2,3-триазол-4-амин 159b
Следуя методике из примера 151е и исходя из соединения 159а (300 мг; 1,83 ммоль) и 10%-ного палладия на угле (50% влаги; 60 мг), получали соединение 159b в виде желтой жидкости (200 мг; 81%). 1Н ЯМР (500 МГц, CDCl3) δ 7.24 (s, 1Н), 7.14 (t, J=58,5 Гц, 1Η), 4.08 (brs, 2Η).
Пример 159с. 5-Бром-3-(2-(дифторметил)-2Н-1,2,3-триазол-4-иламино)-1-метилпиридин-2(1Н)-он 159с
Следуя методике из примера 151f и исходя из соединения 159b (170 мг; 1,25 ммоль; 1,0 экв.) и 3,5-дибром-1-метилпиридин-2(1Н)-она (504 г; 1,89 ммоль; 1,5 экв.), получали соединение 159с в виде светло-желтого твердого вещества (280 мг; 70%). MS-ESI: [М+Н]+ 320,1.
Пример 159d. (2-(6-трет-Бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-(5-(2-(дифторметил)-2Н-1,2,3-триазол-4-иламино)-1-метил-6-оксо-1,6-дигидропиридин-3-ил)пиридин-3-ил)метилацетат 159d
Следуя методике из примера 152с и исходя из соединения 159с (150 мг; 0,46 ммоль; 1,0 экв.) и 3-(ацетоксиметил)-2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)пиридин-4-илбороновой кислоты (116с) (285 мг; 0,69 ммоль; 1,5 экв.), получали соединение 159d в виде желтого твердого вещества (90 мг; 32%). MS-ESI: [М+Н]+ 609,3.
Пример 159. 6-трет-Бутил-2-[4-[5-[[2-(дифторметил)триазол-4-ил]амино]-1-метил-6-оксо-3-пиридил]-3-(гидроксиметил)-2-пиридил]-8-фтор-фталазин-1-он 159
Следуя методике из примера 152 и исходя из соединения 159d (80 мг; 0,13 ммоль), получали соединение 159 в виде белого твердого вещества (29 мг; 40%). MS-ESI: [М+Н]+ 567,3. 1Н ЯМР (500 МГц, CDCl3) δ 8.70 (d, J=5,0 Гц, 1H), 8.36 (d, J=2,0 Гц, 1Η), 8.15 (d, J=2,0 Гц, 1Η), 7.84 (s, 1Η), 7.74 (d, J=2,0 Гц, 1Η), 7.62-7.53 (m, 4Η), 7.25 (t, J=59,0 Гц, 1H), 4.50 (s, 2Н), 4.09 (t, J=6,5 Гц, 1H), 3.75 (s, 3Н), 1.45 (s, 9Н).
Пример 160а. 5-Бром-1-метил-3-(пиразин-2-иламино)пиридин-2(1Н)-он 160а
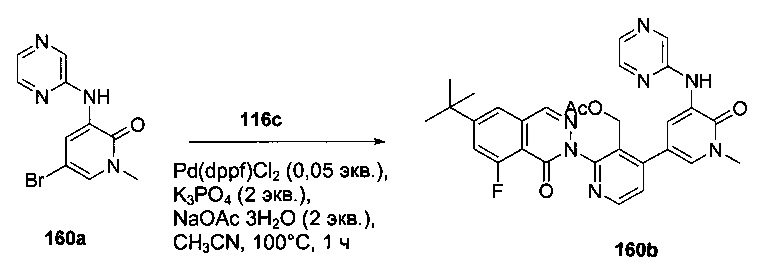
В одногорлую круглодонную колбу емкостью 250 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали пиразин-2-амин (1,0 г; 10 ммоль), 3,5-дибром-1-метилпиридин-2(1Н)-он (2,7 г; 10 ммоль), Pd2(dba)3 (460 мг; 0,50 ммоль), ксантфос (600 мг; 1,0 ммоль), карбонат цезия (6,52 г; 20 ммоль) и 1,4-диоксан (150 мл). После трех циклов вакуумирования/продувания аргоном смесь нагревали при 100°С в течение 2 ч. Затем фильтровали и фильтрат упаривали в вакууме. Остаток очищали перекристаллизацией с метанолом, получая соединение 160а (1,3 г; 47%) в виде светло-зеленого твердого вещества. MS-ESI: [М+Н]+ 281,0.
Пример 160b. (2-(6-трет-Бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-(1-метил-6-оксо-5-(пиразин-2-иламино)-1,6-дигидропиридин-3-ил)пиридин-3-ил)метилацетат 160b
В круглодонную колбу емкостью 50 мл, оборудованную обратным холодильником, загружали соединение 160а (140 мг; 0,50 ммоль), 3-(ацетоксиметил)-2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)пиридин-4-илбороновую кислоту 116с (410 мг; 1,0 ммоль), Pd(dppf)Cl2 (25 мг; 0,025 ммоль), K3PO4 (220 мг; 1,0 ммоль), тригидрат ацетата натрия (136 мг; 1,0 ммоль), ацетонитрил (15 мл) и воду (0,5 мл). Колбу вакуумировали и заполняли N2. Реакционную смесь нагревали при 100°С в течение 1 ч. Затем охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали колоночной хроматографией на силикагеле с элюированием смесью дихлорметан/метанол (от 100:1 до 25:1), получая соединение 160b (150 мг; 52%) в виде желтого твердого вещества. MS-ESI: [М+Н]+ 570,2.
Пример 160. 6-трет-Бутил-8-фтор-2-[3-(гидроксиметил)-4-[1-метил-6-оксо-5-(пиразин-2-иламино)-3-пиридил]-2-пиридил]фталазин-1-он 160
Смесь соединения 160b (120 мг; 0,21 ммоль) и моногидрата гидроксида лития (88 мг; 2,1 ммоль) в смеси THF/изопропанол (4:2; 6 мл) и воде (2 мл) перемешивали при 30°С в течение 1 ч. Смесь упаривали при пониженном давлении и остаток разбавляли водой (10 мл). Затем экстрагировали этилацетатом (2×20 мл). Объединенный этилацетатный экстракт концентрировали при пониженном давлении и остаток очищали препаративной обращенно-фазовой HPLC, получая соединение 160 (50 мг; 45%) в виде белого твердого вещества. MS-ESI: [М+Н]+ 527,8. 1Н ЯМР (500 МГц, CHCl3) δ 8.78 (d, J=2,5 Гц, 1Н), 8.71 (d, J=5,0 Гц, 1Н), 8.36 (d, J=2,0 Гц, 1Н), 8.31 (s, 1Н), 8.19 (s, 1Н), 8.14 (s, 1Н), 8.05 (d, J=2,0 Гц, 1Н), 7.81 (d, J=2,5 Гц, 1Н), 7.59 (s, 1Н), 7.56-7.55 (m, 2Н).
Пример 161а. 1-Метил-3-(5-метилизоксазол-3-иламино)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2(1Н)-он 161а
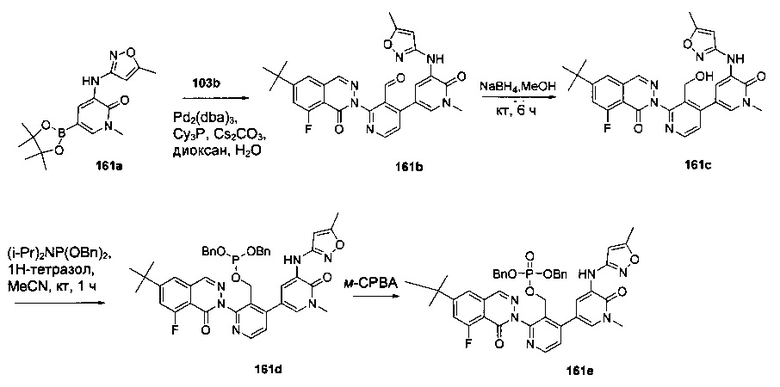
В круглодонную колбу емкостью 50 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали 5-бром-1-метил-3-(5-метилизоксазол-3-иламино)пиридин-2(1Н)-он 129а (330 мг; 1,16 ммоль), Pin2B2 (442 мг; 1,74 ммоль), Pd2(dba)3 (53 мг; 0,058 ммоль), X-Phos (55 мг; 0,116 ммоль), ацетат калия (227 мг; 2,32 ммоль) и диоксан (20 мл). После трех циклов вакуумирования/продувания N2 смесь нагревали при 70°С в течение 2 ч. Анализ реакционной смеси с использованием LCMS показал полное превращение в желаемый продукт. Смесь охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении и остаток промывали петролейным эфиром, получая соединение 161а (300 мг; 78%) в виде желтого твердого вещества. MS-ESI: [М+Н]+ 332,3.
Пример 161b. 2-(6-трет-Бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-(1-метил-5-(5-метилизоксазол-3-иламино)-6-оксо-1,6-дигидропиридин-3-ил)никотинальдегид 161b
В круглодонную колбу емкостью 100 мл, оборудованную обратным холодильником, загружали 2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-хлорникотинальдегид 103b (390 мг; 1,08 ммоль), соединение 161а (537 мг; 1,62 ммоль), карбонат цезия (704 мг; 2,16 ммоль), 1,4-диоксан (30 мл) и воду (3 мл). После барботирования азота через суспензию в течение 10 минут добавляли Cy3P (121 мг; 0,43 ммоль) и Pd2(dba)3 (99 мг; 0,11 ммоль). Систему подвергали трем циклам вакуумирования/продувания азотом и нагревали при температуре дефлегмации в течение 3 ч. Затем смесь охлаждали до комнатной температуры и фильтровали. Твердое вещество промывали дихлорметаном (3×20 мл). Объединенный фильтрат концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле с элюированием смесью 20:1 этилацетат/петролейный эфир, получая соединение 161b (300 мг; 52%). MS-ESI: [М+Н]+ 528,8.
Пример 161с. 6-трет-Бутил-8-фтор-2-(3-(гидроксиметил)-4-(1-метил-5-(5-метилизоксазол-3-иламино)-6-оксо-1,6-дигидропиридин-3-ил)пиридин-2-ил)фталазин-1(2Н)-он 161с
К раствору соединения 161b (300 мг; 0,57 ммоль) в метаноле (10 мл) добавляли NaBH4 (108 мг; 2,85 ммоль) при комнатной температуре. После перемешивания реакционной смеси в течение 6 ч LCMS показала полное завершение реакции. Затем смесь гасили водой (10 мл) и концентрировали при пониженном давлении. Полученный остаток экстрагировали дихлорметаном (3×20 мл). Объединенный органический слой промывали рассолом (20 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Оставшееся твердое вещество очищали колоночной хроматографией на силикагеле с элюированием смесью петролейный эфир/этилацетат (от 1:20 до 100% этилацетата), получая соединение 161с (164 мг; 54%) в виде белого твердого вещества. MS-ESI: [М+Н]+ 530,8.
Пример 161d. Дибензил-(2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-(1-метил-5-(5-метилизоксазол-3-иламино)-6-оксо-1,6-дигидропиридин-3-ил)пиридин-3-ил)метила фосфит 161d
Смесь соединения 161с (100 мг; 0,19 ммоль), дибензил-диизопропилфосфорамидита (85 мг; 0,25 ммоль), 1Н-тетразола (27 мг; 0,38 ммоль) в ацетонитриле перемешивали при комнатной температуре в течение 1 ч. LCMS показала, что реакция больше не протекает. Смесь, содержащую соединение 161d, использовали на следующей стадии без дополнительной очистки. MS-ESI: [М+Н]+ 775,3.
Пример 161е. Дибензил-(2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-(1-метил-5-(5-метилизоксазол-3-иламино)-6-оксо-1,6-дигидропиридин-3-ил)пиридин-3-ил)метила фосфат 161е)
К смеси соединения 161d в ацетонитриле добавляли м-хлорпербензойную кислоту (49 мг; 0,28 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 5 минут и фильтровали. Фильтрат концентрировали при пониженном давлении и остаток очищали препаративной обращенно-фазовой HPLC, получая соединение 161е (15 мг; 10% за две стадии) в виде белого твердого вещества. MS-ESI: [М+Н]+ 791,3.
Пример 161. [2-(6-трет-Бутил-8-фтор-1-оксо-фталазин-2-ил)-4-[1-метил-5-[(5-метилизоксазол-3-ил)амино]-6-оксо-3-пиридил]-3-пиридил]метила дигидрофосфат 161
Смесь соединения 161е (10 мг; 0,013 ммоль) в трифторуксусной кислоте (297 мг; 2,60 ммоль) перемешивали при комнатной температуре в течение 1 ч. Смесь концентрировали при пониженном давлении и остаток очищали препаративной обращенно-фазовой HPLC, получая соединение 161 (6,7 мг; 84%) в виде белого твердого вещества. MS-ESI: [М+Н]+ 611,3. 1Н ЯМР (500 МГц, DMSO-d6) δ 9.00 (s, 1Η), 8.61 (m, 1Η), 8.50 (bs, 1Η), 8.02 (s, 1Η), 7.87-7.72 (m, 3Н), 7.53 (m, 1Н), 6.27 (s, 1Н), 4.61-4.56 (m, 2Н), 3.46 (s, 3Н), 2.33 (s, 3Н), 1.31 (s, 9Н).
Пример 162а. 5,6-Дигидро-4Н-пирроло[1,2-b]пиразол-2-карбоновая кислота 162а
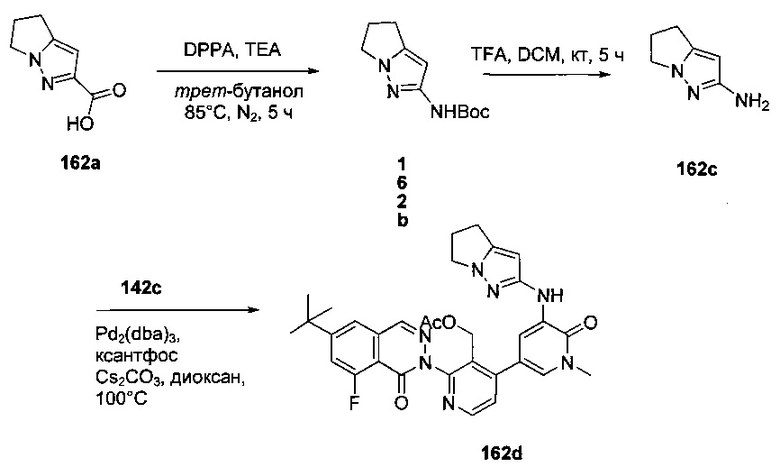
В круглодонную колбу емкостью 25 мл, оборудованную обратным холодильником, загружали этил-5,6-дигидро-4Н-пирроло[1,2-b]пиразол-2-карбоксилат (540 мг; 3,0 ммоль), 2 н. водный раствор гидроксида натрия (3,5 мл) и 1,4-диоксан (3,0 мл). Систему нагревали при 65°С в течение 2,5 ч. Затем охлаждали до комнатной температуры и рН подводили до 2-3, используя концентрированную HCl. Твердое вещество собирали фильтрованием, получая соединение 162а (260 мг; 57%) в виде желтого твердого вещества. MS-ESI: [М+Н]+ 153,3.
Пример 162b. трет-Бутил-5,6-дигидро-4Н-пирроло[1,2-b]пиразол-2-илкарбамат 162b
В одногорлую круглодонную колбу емкостью 25 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали соединение 162а (197,6 мг; 1,3 ммоль), трет-бутанол (5,0 мл), триэтиламин (262,6 мг; 2,6 ммоль) и DPPA (дифенилфосфорилазид) (550 мг; 2,0 ммоль). Систему подвергали трем циклам вакуумирования/продувания азотом и нагревали при 85°С в течение 5 ч. Затем охлаждали до комнатной температуры и очищали с использованием комбифлэш-хроматографии (А: 1% NH4HCO3/вода, В: ацетонитрил), получая соединение 162b (45 мг; 15,5%) в виде желтого твердого вещества. MS-ESI: [М+Н-56]+ 168,3.
Пример 162с. 5,6-Дигидро-4Н-пирроло[1,2-b]пиразол-2-амин 162с
К раствору соединения 162b (45 мг; 0,20 ммоль) в дихлорметане (1,5 мл) добавляли трифторуксусную кислоту (1 мл) при комнатной температуре. Раствор перемешивали в течение 5 ч. Затем концентрировали при пониженном давлении, получая соединение 162с, которое использовали на следующей стадии без дополнительной очистки. MS-ESI: [М+Н]+ 124,3.
Пример 162d. (2-(6-трет-Бутил-8-фтор-1-оксофталазин-2(1Н)-ил)-4-(5-(5,6-дигидро-4Н-пирроло[1,2-Ь]пиразол-2-иламино)-1-метил-6-оксо-1,6-дигидропиридин-3-ил)пиридин-3-ил)метилацетат 162d
В одногорлую круглодонную колбу емкостью 25 мл, оборудованную магнитной мешалкой и обратным холодильником, загружали (4-(5-бром-1-метил-6-оксо-1,6-дигидропиридин-3-ил)-2-(6-трет-бутил-8-фтор-1-оксофталазин-2(1Н)-ил)пиридин-3-ил)метилацетат 142с (166,2 мг; 0,30 ммоль), соединение 162с (24,6 мг; 0,20 ммоль), карбонат цезия (130 мг; 0,40 ммоль) и 1,4-диоксан (4,0 мл). После барботирования азота через суспензию в течение 10 минут добавляли ксантфос (23 мг; 0,040 ммоль) и трис(дибензилиденацетон)дипалладий(0) (14 мг; 0,020 ммоль). Систему подвергали трем циклам вакуумирования/продувания азотом и нагревали при температуре дефлегмации в течение 2,5 ч. Затем охлаждали до комнатной температуры и фильтровали. Твердое вещество промывали дихлорметаном (3×10 мл). Объединенный органический слой концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с элюированием смесью дихлорметан/метанол (от 100:1 до 50:1), получая соединение 162d (50 мг; 42%) в виде желтого твердого вещества. MS-ESI: [М+Н]+ 598,3.
Пример 162. 6-трет-Бутил-2-[4-[5-(5,6-дигидро-4Н-пирроло[1,2-b]пиразол-2-иламино)-1-метил-6-оксо-3-пиридил]-3-(гидроксиметил)-2-пиридил]-8-фтор-фталазин-1-он 162
К раствору соединения 162d (50 мг; 0,080 ммоль) в смеси THF/изопропанол/вода (1/1/0,5 мл) добавляли гидроксид лития (19 мг; 0,80 ммоль) при комнатной температуре. После перемешивания реакционной смеси в течение 2,5 ч LCMS показала полное завершение реакции. Затем смесь выливали в воду (15 мл) и экстрагировали дихлорметаном (3×20 мл). Объединенный органический слой промывали рассолом (30 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Оставшееся твердое вещество очищали препаративной обращенно-фазовой HPLC (А: 1% NH4HCO3/вода, В: ацетонитрил), получая соединение 162 (15 мг; 33,3%) в виде белого твердого вещества. MS-ESI: [М+Н]+ 556,3. 1Н ЯМР (500 МГц, CDCl3) δ 8.62 (d, J=5,0 Гц, 1H), 8.32 (s, 1Н), 7.93 (d, J=2,0 Гц,1Н), 7.55-7.47 (m, 4H), 7.26 (s, 1H), 5.74 (s, 1H), 4.47 (d, J=4,0 Гц, 2H), 4.07 (t, J=2,0 Гц, 2H), 3.70 (s, 3Н), 2.87 (t, J=6,5 Гц, 2H), 2.55-2.52 (m, 2H), 1.45 (s, 9H).
Пример 901. Биохимический анализ Btk
Общая методика стандартного биохимического анализа киназы Btk, которую можно использовать для тестирования соединений формулы I и II, приведена ниже. Готовят основную смесь (master mix) без фермента Btk, содержащую 1×буфер для киназ клеточного сигнального пути (cell signaling kinase buffer) (25 мМ Трис-HCl, рН 7,5; 5 мМ бета-глицерофосфат, 2 мМ дитиотреит; 0,1 мМ Na3VO4, 10 мМ MgCl2), 0,5 мкМ биотинилированный пептидный субстрат 2 для протеинкиназ (РТК) (Promega) и 0,01% BSA (бычий сывороточный альбумин). Готовят основную смесь с ферментом Btk, содержащую 1×буфер для киназ клеточного сигнального пути, 0,5 мкМ биотинилированный пептидный субстрат 2 для РТК, 0,01% BSA и фермент Btk в количестве 100 нг/лунка (0,06 миллиединиц/лунка). Фермент Btk получают следующим образом: полноразмерный ген человеческой Btk дикого типа (номер доступа NM-000061) с С-концевым V5 и меткой 6xHis субклонируют в вектор pFastBac для получения бакуловируса, несущего эту меченную эпитопом Btk. Наработку бакуловируса осуществляют, основываясь на инструкциях Invitrogen, подробно изложенных в опубликованном протоколе этой фирмы "Bac-to-Bac Baculovirus Expression Systems" (Бакуловирусные экспрессирующие системы Bac-to-Bac) (№№ по каталогу 10359-016 и 10608-016). Для инфицирования клеток Sf9 с целью сверхэкспрессии рекомбинантного белка Btk используют 3-й пассаж вируса. Затем белок Btk очищают до гомогенного состояния, используя колонку Ni-NTA (Ni-нитрилацетат). Чистота конечного препарата белка превышает 95% по данным чувствительного окрашивания с использованием Sypro-Ruby. Готовят 200 мкМ раствор АТФ в воде и рН подводят до 7,4 с помощью 1 н. NaOH. Соединения в 5%-ном DMSO в количестве 1,25 мкл переносят в 96-луночный полистироловый планшет Costar 1/2 площади. Соединения тестируют по отдельности и с использованием кривой зависимости дозы от ответа по 11 точкам (исходная концентрация составляет 10 мкМ; разведение 1:2). Основную смесь без фермента (в качестве отрицательного контроля) и основную смесь с ферментом, каждую в объеме по 18,75 мкл, переносят в соответствующие лунки 96-луночного полистиролового планшета Costar 1/2 площади. По 5 мкл 200 мкМ АТФ добавляют к данной смеси в 96-луночном полистироловом планшете Costar 1/2 площади для получения конечной концентрации АТФ 40 мкМ. Реакционную смесь оставляют инкубироваться в течение 1 часа при комнатной температуре. Реакцию останавливают, используя 1×буфер для детекции от Perkin Elmer, содержащий 30 мМ EDTA, 20 нМ SA-APC (стрептавидин-аллофикоцианин) и 1 нМ Ab (антитело) РТ66. Планшет прочитывают, используя флуоресценцию с разрешением во времени, на устройстве Perkin Elmer Envision, в котором применяется фильтр возбуждения при 330 нм, эмиссионный фильтр при 665 нм и 2-й эмиссионный фильтр при 615 нм. Далее рассчитывают значения IC50. Альтернативно, для оценки активности Btk посредством количественного определения фосфорилированного ею пептидного продукта можно использовать анализ LanthaScreen. Уровень FRET (резонансного переноса энергии флуоресценции), происходящего между флуоресцеином на пептидном продукте и тербием на детектирующем антителе, снижается при добавлении ингибиторов Btk, которые ослабляют фосфорилирование пептида. В конечном реакционном объеме 25 мкл человеческой Btk(h) (0,1 нг/25 мкл реакционной смеси) инкубируют с 50 мМ Hepes (N-2-гидроксиэтил-пиперазин-N-2-этансульфоновая кислота) рН 7,5, 10 мМ MgCl2, 2 мМ MnCl2, 2 мМ DTT (дитиотреит), 0,2 мМ NaVO4, 0,01% BSA и 0,4 мкМ флуоресцеин-поли-GAT (Glu, Ala, Tyr). Реакцию инициируют добавлением АТФ до концентрации 25 мкМ (Km для АТФ). После инкубирования в течение 60 минут при комнатной температуре реакцию останавливают, добавляя детектирующее антитело Tb-PY20 в конечной концентрации 2 нМ в 60 мМ EDTA на 30 минут при комнатной температуре. Детекцию осуществляют на устройстве Perkin Elmer Envision с возбуждением при 340 нм и эмиссией при 495 нм и 520 нм. Типичные значения IC50 в отношении ингибирования Btk приведены в Таблицах 1, 2 и 3.
Пример 902. Анализ Btk с использованием клеток линии Ramos
Другая общая методика стандартного анализа киназы Btk с применением клеток, которую можно использовать для тестирования соединений формулы I и II, приведена ниже. Клетки линии Ramos инкубируют при плотности 0,5×107 клеток/мл в присутствии тестируемого соединения в течение 1 ч при 37°С. Затем клетки стимулируют посредством инкубирования с F(ab)2 к человеческому IgM в концентрации 10 мкг/мл в течение 5 минут при 37°С. Клетки собирают центрифугированием, лизируют, анализ белков проводят на осветленном лизате. Равные количества белка от каждого образца подвергают SDS-PAGE (электрофорез в полиакриламидном геле в присутствии додецилсульфата натрия) и вестерн-блоттингу как с антителом к фосфо-Btk(Tyr223) (Cell Signaling Technology, №3531; Epitomics, № по каталогу 2207-1) или с антителом к фосфо-Btk(Tyr551) (BD Transduction Labs, №558034) для оценки аутофосфорилирования Btk, так и с антителом к Btk (BD Transduction Labs, №611116) для контроля общего количества Btk в каждом лизате.
Пример 903. Анализ В-клеточной пролиферации
Общая методика стандартного анализа В-клеточной пролиферации, которую можно использовать для тестирования соединений формулы I и II, приведена ниже. В-клетки выделяют в очищенном виде из селезенок мышей Balb/c в возрасте 8-16 недель, используя набор для выделения В-клеток (Miltenyi Biotech, №по каталогу 130-090-862). Тестируемые соединения разбавляют в 0,25%-ном DMSO и инкубируют с очищенными В-клетками селезенки мышей в количестве 2,5×105 клеток в течение 30 минут, после чего добавляют антитела к IgM мыши (10 мкг/мл; Southern Biotechnology Associates, №каталогу 1022-01) в конечном объеме 100 мкл. После инкубирования в течение 24 ч добавляют 1 мкКи 3Н-тимидина и планшеты инкубируют в течение еще 36 ч, после чего отбирают пробы для анализа с использованием протокола от производителя для SPA-системы (Scintillation Proximity Assay - сцинтилляционный анализ сближения) анализа поглощения [3Н]-тимидина (Amersham Biosciences, №RPNQ0130). Сигнал флуоресценции от SPA-гранул измеряют на счетчике MicroBeta (Wallace Triplex 1450, Perkin Elmer).
Пример 904. Анализ Т-клеточной пролиферации
Общая методика стандартного анализа Т-клеточной пролиферации, которую можно использовать для тестирования соединений формулы I и II, приведена ниже. Т-клетки выделяют в очищенном виде из селезенок мышей Balb/c в возрасте 8-16 недель, используя набор для выделения пан-Т-клеток (Miltenyi Biotech, №по каталогу 130-090-861). Тестируемые соединения разбавляют в 0,25%-ном DMSO и инкубируют с очищенными Т-клетками селезенки мышей в количестве 2,5×105 клеток в планшетах с плоским прозрачным дном, предварительно покрытых в течение 90 минут при 37°С антителами к CD3 (BD, №553057) и к CD28 (BD, №553294), каждым в концентрации 10 мкг/мл. После инкубирования в течение 24 ч добавляют 1 мкКи 3Н-тимидина и планшеты инкубируют в течение еще 36 ч, после чего отбирают пробы для анализа с использованием протокола от производителя для SPA-системы анализа поглощения [3Н]-тимидина (Amersham Biosciences, №RPNQ0130). Сигнал флуоресценции от SPA-гранул измеряют на счетчике MicroBeta (Wallace Triplex 1450, Perkin Elmer).
Пример 905. Анализ ингибирования CD86
Общая методика стандартного анализа ингибирования активности В-клеток, которую можно использовать для тестирования соединений формулы I и II, приведена ниже. Суммарный пул спленоцитов мышей выделяют в очищенном виде из селезенок мышей Balb/c в возрасте 8-16 недель путем лизиса эритроцитов (BD Pharmingen, №555899). Тестируемые соединения разбавляют в 0,5%-ном DMSO и инкубируют со спленоцитами в количестве 1,25×106 клеток в конечном объеме 200 мкл в планшетах с плоским прозрачным дном (Falcon 353072) в течение 60 минут при 37°С. Затем клетки стимулируют, добавляя IgM (15 мкг/мл; Jackson ImmunoResearch, 115-006-020) и инкубируют в течение 24 ч при 37°С, 5% CO2. После инкубирования в течение 24 ч клетки переносят в прозрачные 96-луночные планшеты с коническим дном и собирают центрифугированием при 1200×g в течение 5 мин. Предварительно клетки блокируют, используя CD16/CD32 (BD Pharmingen, №553142), после чего выполняют тройное окрашивание с применением CD19-FITC (флуоресцеинизотиоцианат) (BD Pharmingen, №553785), CD86-PE (phycoerythrin - фикоэритрин) (BD Pharmingen, №553692) и 7AAD (7-амино-актиномицин D) (BD Pharmingen, №51-6898 IE). Клетки подвергают сортингу на устройстве FACSCalibur от BD и производят отбор популяции CD19+/7AAD-. Уровни поверхностной экспрессии CD86 в отобранной популяции измеряют относительно концентрации тестируемого соединения.
Пример 906. Анализ выживаемости клеток при B-ALL
Далее приводится методика стандартного исследования выживаемости клеток при B-ALL (В-клеточный острый лимфобластный лейкоз) с использованием данных по ХТТ (2,3-бис-(2-метокси-4-нитро-5-сульфофенил)-2H-тетразолий-5-карбоксанилид) для измерения числа жизнеспособных клеток. Этот анализ можно использовать для тестирования соединений формулы I и II по их способности ингибировать выживаемость клеток в культуре при B-ALL. Одной из линий В-клеточного острого лимфобластного лейкоза человека, которую можно использовать, является линия SUP-B 15, линия пре-В-клеток ALL человека, предоставляемая АТСС (Американская коллекция типовых культур).
ПреВ-клетки ALL линии SUP-B15 размещают в нескольких 96-луночных титрационных микропланшетах в 100 мкл среды Исков (Iscove), содержащей 20% FBS (фетальная телячья сыворотка), в концентрации 5×105 клеток/мл. Затем добавляют тестируемые соединения в конечной концентрации DMSO 0,4%. Клетки инкубируют при 37°С с 5% CO2 в течение до 3 суток. Через 3 суток клетки распределяют в соотношении 1:3 в свежие 96-луночные планшеты, содержащие тестируемое соединение, и оставляют подрастать в течение еще 3 суток. Через каждые 24 часа в один из дубликатов 96-луночных планшетов добавляют по 50 мкл раствора ХТТ и выполняют измерения поглощения через 2, 4 и 20 часов, следуя указаниям производителя. Далее учитывают результат измерения OD (оптическая плотность) в линейном диапазоне данного анализа (0,5-1,5), проведенного для клеток, обработанных только DMSO, и процентное содержание жизнеспособных клеток в лунках, обработанных соединениями, определяют относительно клеток, обработанных только DMSO.
Пример 907. Анализ CD69 в цельной крови
Человеческую кровь получают от здоровых добровольцев со следующими ограничениями: в течение 1 недели обходящихся без приема лекарственных средств, некурящих. Кровь (приблизительно 20 мл для тестирования 8 соединений) собирают методом венопункции в пробирки Vacutainer® (Becton, Dickinson и Со.) с гепарином натрия.
Растворы соединений формулы I и II в концентрации 10 мМ в DMSO разбавляют 1:10 в 100%-ном DMSO, затем разбавляют путем трехкратных серийных разведений в 100%-ном DMSO для получения кривой зависимости ответа от дозы по десяти точкам. Далее соединения разбавляют 1:10 в PBS (забуференный фосфатом физиологический раствор) и затем аликвоты по 5,5 мкл раствора каждого соединения добавляют в двух повторах в 96-луночный планшет с емкостью лунок 2 мл; в контрольные лунки и в лунки без стимуляции добавляют по 5,5 мкл 10%-ного DMSO в PBS. В каждую лунку добавляют цельную кровь человека (human whole blood, HWB; 100 мкл). После перемешивания планшеты инкубируют при 37°С, 5% CO2, 100%-ной влажности в течение 30 минут. В каждую лунку (за исключением лунок без стимуляции) добавляют при перемешивании козьи F(ab')2 против IgM человека (10 мкл раствора с концентрацией 500 мкг/мл, конечная концентрация 50 мкг/мл) и планшеты инкубируют в течение еще 20 часов. По окончании 20 часов инкубации образцы инкубируют с флуоресцентно меченными антителами в течение 30 минут при 37°С, 5% CO2, 100%-ной влажности. Необходимо включить индуцированный контроль, неокрашенные пробы и взятые отдельно красители для корректировки компенсационных и начальных установок напряжения. Затем осуществляют лизис образцов, используя PharM Lyse™ (BD Biosciences Pharmingen) в соответствии с инструкциями производителя. Далее образцы переносят в 96-луночный планшет, подходящий для работы на 96-луночной HTS-системе от BD Biosciences на установке LSRII. Осуществляют сбор данных и величины средних интенсивностей флуоресценции получают с использованием программного обеспечения DIVA от BD Biosciences. Сначала результаты анализируют с использованием программного обеспечения для FACS-анализа (Flow Jo). Величины IC50 для тестируемых соединений определяют как концентрацию, при которой на 50% уменьшается доля CD69-положительных клеток, которые также являются CD20-положительными, стимулированных антителами против IgM (среднее значение для 8 контрольных лунок после вычитания в качестве фона среднего значения для 8 лунок без стимуляции). Значения ингибирующей концентрации (IC50, IC70, IC90) рассчитывают с использованием 5-й версии Prism, применяя аппроксимацию кривых методом нелинейной регрессии.
Пример 908. Анализ клеточной пролиферации in vitro
Эффективность соединений формулы I или II измеряют в анализе клеточной пролиферации, применяя приведенный далее протокол (Mendoza et al. (2002) Cancer Res. 62: 5485-5488). Люминесцентный анализ жизнеспособности клеток CellTiter-Glo®, в том числе реагенты и протокол, имеется в продаже (Promega Corp., Madison, WI, Технический бюллетень ТВ288). В этом анализе оценивается способность соединений проникать в клетки и ингибировать клеточную пролиферацию. Принцип анализа заключается в определении числа присутствующих жизнеспособных клеток посредством количественного определения АТФ, присутствующего в гомогенном анализе, в котором добавление CellTiter-Glo приводит к лизису клеток и генерации сигнала люминесценции в результате люциферазной реакции. Сигнал люминесценции пропорционален количеству присутствующего АТФ.
Панель линий клеток В-клеточной лимфомы (BJAB, SUDHL-4, TMD8, OCI-Ly10, OCI-Ly3, WSU-DLCL2) размещают в 384-луночном планшете в обычной ростовой среде и в каждую лунку добавляют серийные разведения ингибиторов Btk или только DMSO. Жизнеспособность клеток оценивают после инкубирования в течение 96 часов, используя CellTiter-Glo® (Promega). Данные могут быть представлены в виде относительной клеточной жизнеспособности для обработанных ингибитором ВТК клеток относительно контрольных клеток, обработанных DMSO. Точки на графиках представляют собой средние значения для 4-х повторов для каждого уровня доз. "Усы" представляют SD (standard deviation - стандартное отклонение) от среднего значения.
Методика
1-е сутки - планшеты для рассевания клеток (384-луночные черные ТС-планшеты (планшеты для тканевых культур) с прозрачным дном, для улучшенной микрофотосъемки (microclear), с крышкой от Falcon №353962), собрать клетки, провести посев клеток в количестве 1000 клеток в 54 мкл на одну лунку в 384-луночные планшеты для клеток для выполнения анализа через 3 суток. Среда для культивирования клеток: RPMI (Roswell Park Memorial Institute) или DMEM (модифицированная Дульбекко среда Игла) с высоким содержанием глюкозы, 10% фетальной телячьей сыворотки, 2 мМ L-глутамин, P/S (пенициллин/стрептомицин). Провести инкубацию в течение ночи (over night; Ο/Ν) при 37°С, 5% CO2.
2-е сутки - выполнить добавление лекарственного средства к клеткам с использованием разведения соединений в планшетах для DMSO (серийные разведения 1:2 для 9 точек): добавить по 20 мкл растворов соединений в концентрации 10 мМ во 2-ую колонку 96-луночного планшета. Выполнить серийные разведения 1:2 в поперечном направлении планшета (10 мкл + 20 мкл 100%-ного DMSO) в общей сложности для 9 точек, используя Precision. В планшеты для сред - 96-луночные полипропиленовые планшеты с коническим дном от Nunc (№ по каталогу 249946) (для получения разведения 1:50) добавить по 147 мкл среды во все лунки. Перенести по 3 мкл смеси DMSO+соединение из каждой лунки планшета для DMSO в каждую соответствующую лунку на планшете для сред, используя Rapidplate.
Добавление лекарственного средства к клеткам в планшете для клеток (разведение 1:10): добавить по 6 мкл смеси среды + соединение непосредственно к клеткам (клетки уже находятся в 54 мкл среды). Провести инкубацию в течение 3 суток при 37°С, 5% CO2 в инкубаторе, который не должен часто открываться.
5-е сутки - окраска содержимого планшетов: разморозить буфер CellTiter-Glo при комнатной температуре. Извлечь планшеты для клеток с 37°С и уравновесить до комнатной температуры в течение примерно 30 минут. Добавить буфер CellTiter-Glo к субстрату CellTiter-Glo (бутылка к бутылке). Добавить по 30 мкл реагента CellTiter-Glo (Promega, № по каталогу G7572) в каждую лунку с клетками. Поместить на планшетный шейкер примерно на 30 минут. Снять показания люминесценции, используя планшетный ридер Analyst НТ (в течение полусекунды на одну лунку).
Анализы жизнеспособности клеток и анализы действия комбинаций: клетки высевают в количестве 1000-2000 клеток/лунка в 384-луночные планшеты на 16 ч. На вторые сутки делают девять серийных (1:2) разведений соединений в DMSO в 96-луночном планшете. Далее соединения разбавляют в ростовых средах, используя робот Rapidplate (Zymark Corp., Hopkinton, MA). Разбавленные соединения затем добавляют в четырех повторах в лунки 384-луночных планшетов для клеток и инкубируют при 37°С и 5% CO2. Через 4 суток измеряют относительное число жизнеспособных клеток по люминесценции, используя CellTiter-Glo (Promega) в соответствии с инструкциями производителя, и снимают показания на ридере Wallac Multilabel (PerkinElmer, Foster City). Значения EC50 рассчитывают, используя программное обеспечение Prism® 4.0 (GraphPad, San Diego). Во всех анализах соединения формулы I или II и химиотерапевтические агенты добавляют одновременно или по отдельности с интервалом 4 часа (одно вслед за другим).
Дополнительный типичный анализ in vitro клеточной пролиферации включает следующие стадии:
1) аликвоту 100 мкл клеточной культуры, содержащую примерно 104 клеток в среде, помещают в каждую лунку 384-луночного планшета с непрозрачными стенками;
2) готовят контрольные лунки, содержащие среду и не содержащие клеток;
3) в экспериментальные лунки добавляют соединение и инкубируют в течение 3-5 суток;
4) планшеты уравновешивают до комнатной температуры в течение приблизительно 30 минут;
5) к объему среды для культивирования клеток, присутствующей в каждой лунке, добавляют равный объем реагента CellTiter-Glo;
6) содержимое перемешивают в течение 2 минут на орбитальном шейкере для индукции лизиса клеток;
7) планшет инкубируют при комнатной температуре в течение 10 минут для стабилизации сигнала люминесценции;
8) регистрируют люминесценцию и представляют графические изображения в виде RLU - относительных единиц люминесценции.
Несмотря на то что изложенное выше изобретение для ясности понимания описано довольно подробно посредством иллюстрации и примеров, описания и примеры не следует истолковывать как ограничивающие объем данного изобретения. Соответственно, все подходящие модификации и эквиваленты будут считаться попадающими в объем изобретения, который определен следующей далее формулой изобретения. Описания всей патентной и научной литературы, приведенные в данном описании, явным образом включены во всей своей полноте посредством ссылки.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ ТИРОЗИНКИНАЗЫ БРУТОНА | 2010 |
|
RU2542585C2 |
| ПИРИДОНОВЫЕ И АЗАПИРИДОНОВЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ПРИМЕНЕНИЯ | 2011 |
|
RU2617405C2 |
| Новые соединения пиридопиримидинона для модулирования каталитической активности гистонлизиндеметилаз (KDMS) | 2015 |
|
RU2684396C2 |
| ИНГИБИТОРЫ ТИРОЗИНКИНАЗЫ БРУТОНА | 2014 |
|
RU2618529C2 |
| АМИНОТРИАЗОЛОПИРИДИНЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ | 2009 |
|
RU2552642C2 |
| ГЕТЕРОАРИЛ ПИРИДОНЫ И АЗАПИРИДОНЫ С ЭЛЕКТРОФИЛЬНОЙ ФУНКЦИОНАЛЬНОСТЬЮ | 2014 |
|
RU2646758C2 |
| СПИРО-АМИНОСОЕДИНЕНИЯ, ПРИГОДНЫЕ ДЛЯ ЛЕЧЕНИЯ НАРУШЕНИЙ СНА И ЛЕКАРСТВЕННОГО ПРИВЫКАНИЯ | 2010 |
|
RU2562609C2 |
| ДИГИДРОПИРИДОФТАЛАЗИНОНОВЫЕ ИНГИБИТОРЫ ПОЛИ(АДФ-РИБОЗА)ПОЛИМЕРАЗЫ | 2009 |
|
RU2514937C2 |
| ПОЛИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И СПОСОБЫ ЦЕЛЕНАПРАВЛЕННОЙ ДЕГРАДАЦИИ ПОЛИПЕПТИДОВ БЫСТРО УСКОРЕННОЙ ФИБРОСАРКОМЫ | 2019 |
|
RU2830173C2 |
| КЛАСС КОНДЕНСИРОВАННЫХ КОЛЬЦЕВЫХ СОЕДИНЕНИЙ И ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2021 |
|
RU2831125C1 |

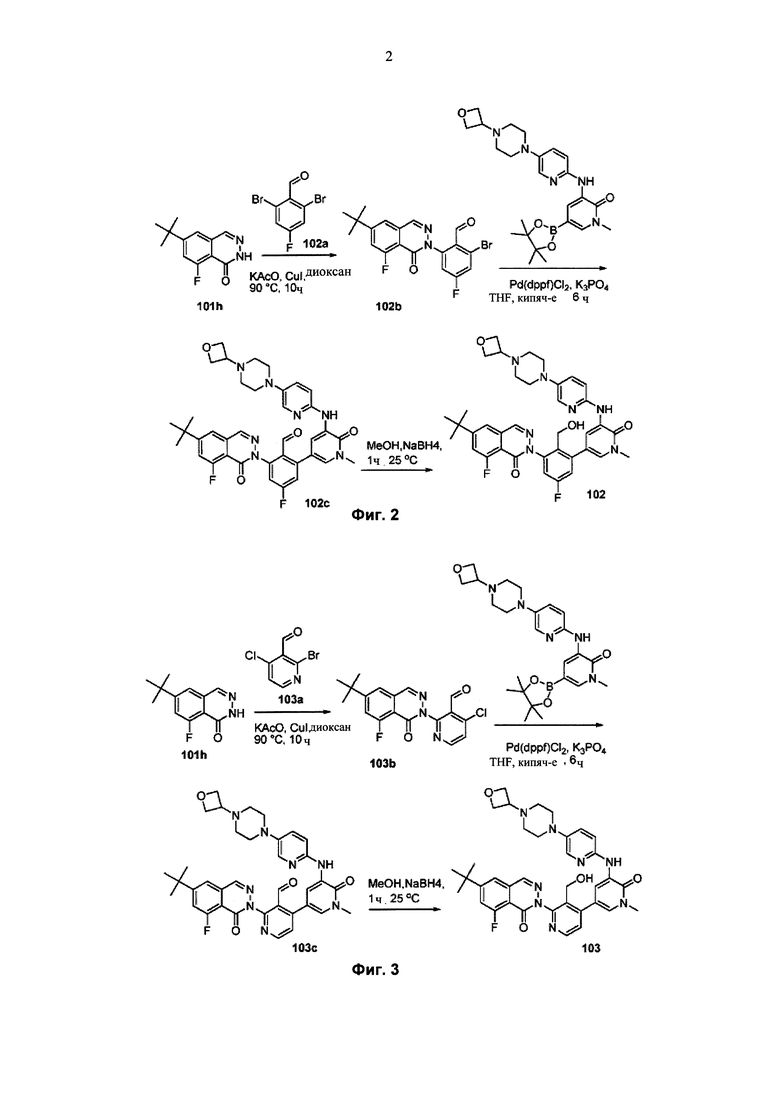
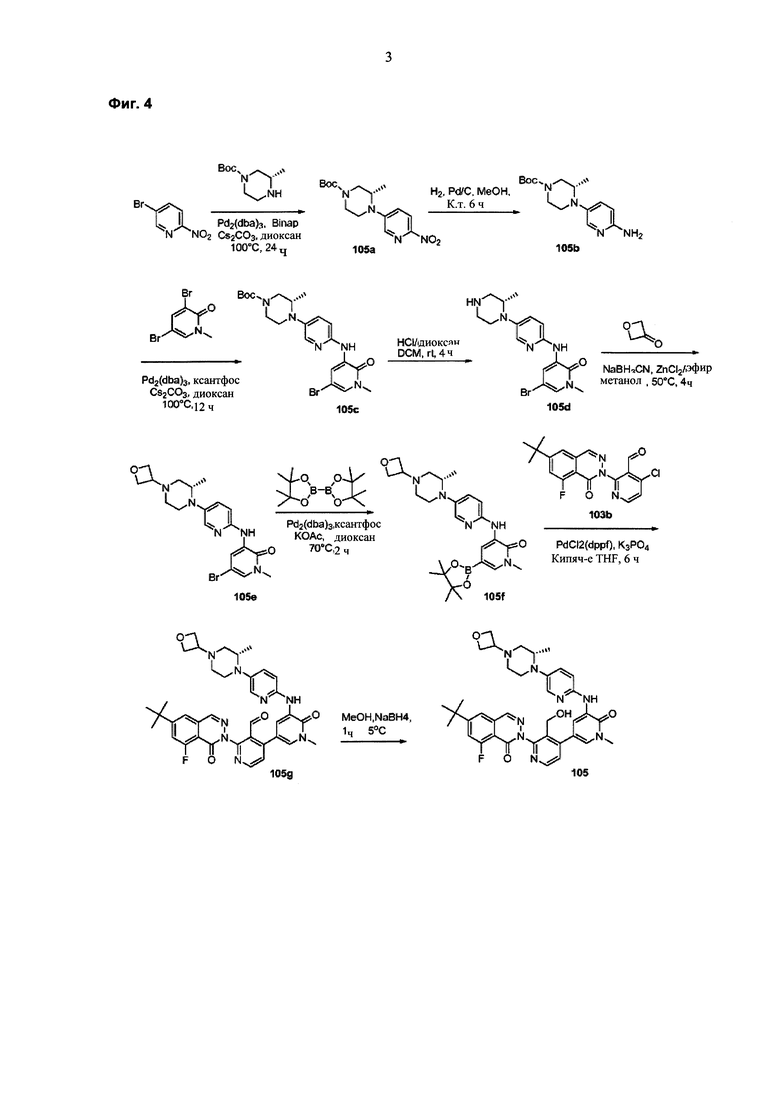
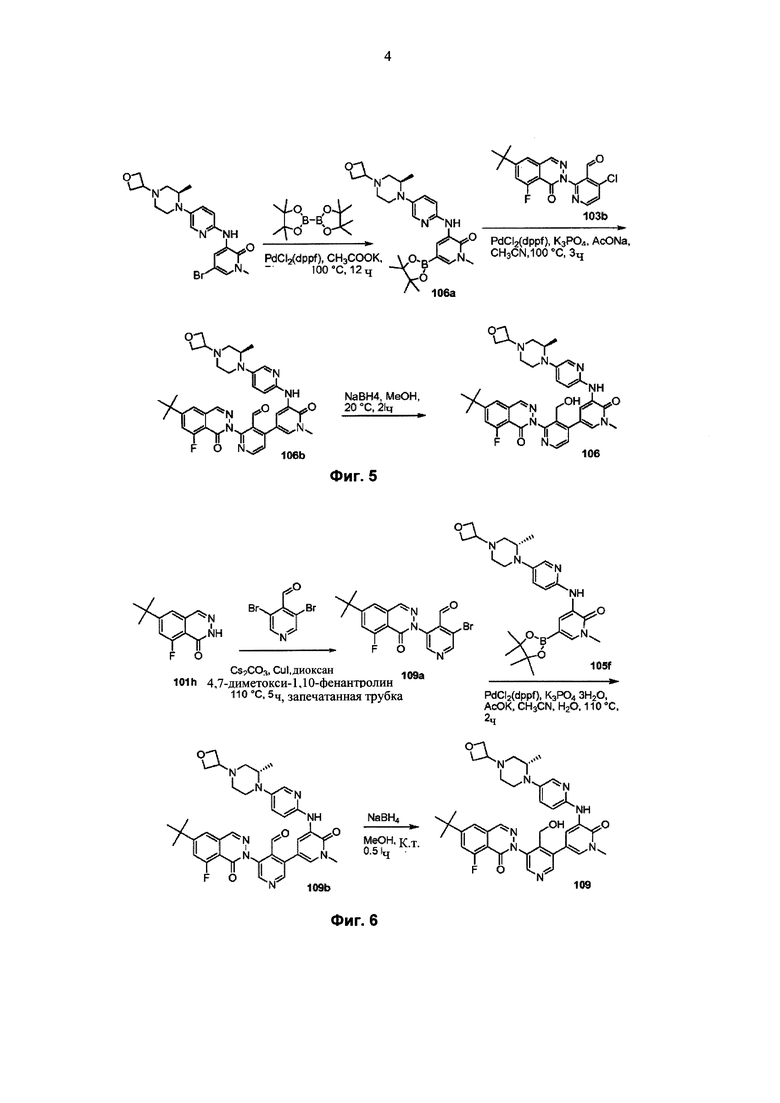
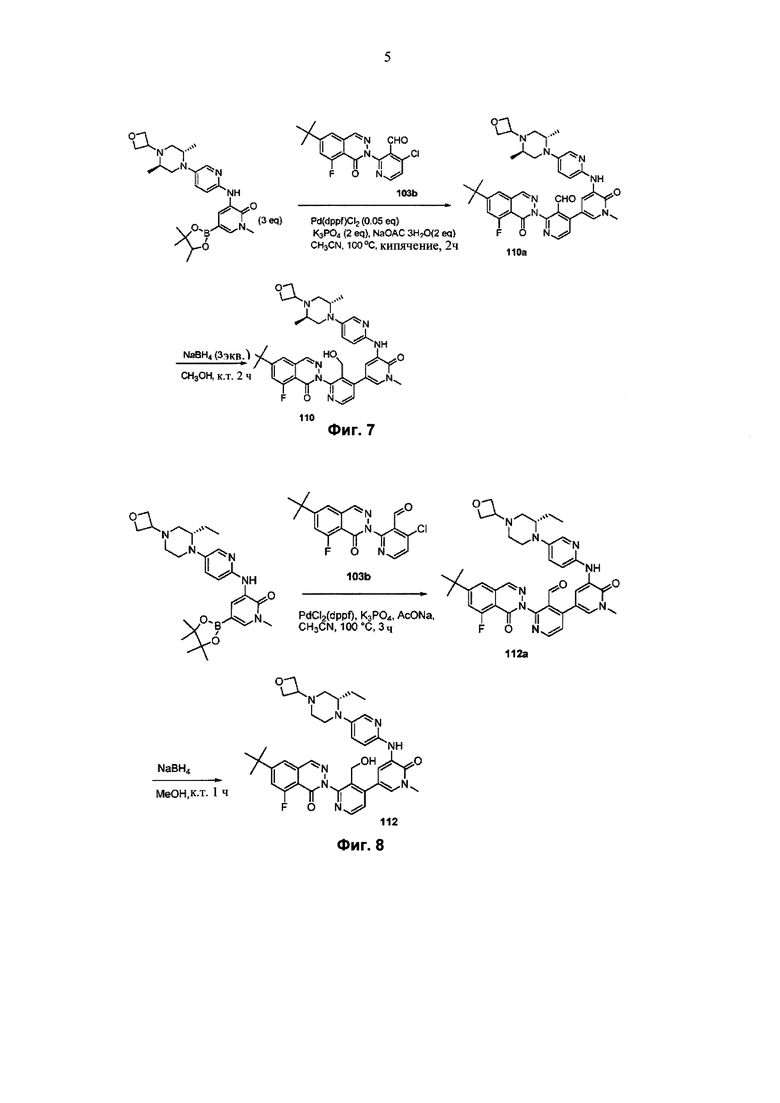
Предложены соединения 8-фторфталазин-1(2H)-онов формулы II, где один из X1, X2 и X3 представляют собой N и остальные символы имеют значения, указанные в п.1 формулы изобретения, или их стереоизомеры, гаутомеры и фармацевтически приемлемые соли. Предложенные соединения ингибируют киназу Btk (тирозинкиназа Брутона) и могут быть использованы для лечения иммунных расстройств, таких как воспаление, опосредованное киназой Btk. Описаны способы применения соединений формулы II для диагностики и лечения in vitro, in situ и in vivo таких расстройств в клетках млекопитающих или ассоциированных патологических состояний. 6 н. и 19 з.п. ф-лы, 8 ил., 2 табл., 69 пр.
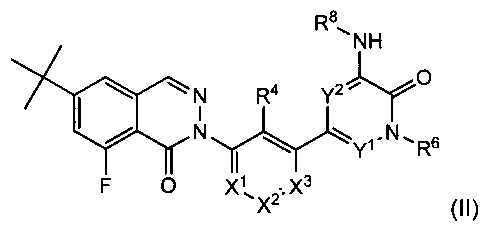
1. Соединение формулы II:
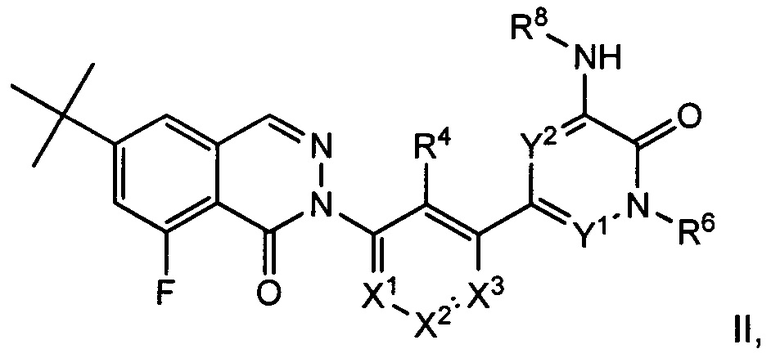
или его стереоизомеры, таутомеры или фармацевтически приемлемые соли, где:
X1 представляет собой CR1 или N;
X2 представляет собой CR2 или N;
X3 представляет собой CR3 или N;
где один из X1, X2 и X3 представляет собой N;
Y1 и Y2 независимо выбраны из СН и N, причем Y1 и Y2 каждый не представляет собой N;
R1, R2 и R3 независимо выбраны из Н и F;
R4 выбран из -СН2ОН и -СН2ОР(O)(ОН)2;
R6 выбран из Н и -СН3;
R8 выбран из пиримидинила, 6,7-дигидро-4Н-тиазоло[5,4-с]пиридин-2-ила, 5-(морфолин-4-карбонил)-2-пиридила, пиразолила, тиазолила, 6,7-дигидро-4Н-пиразоло[1,5-а]пиразин-2-ила, оксазолила, изоксазолила, имидазолила, 6,7-дигидро-4Н-пиразоло[5,1-с][1,4]оксазин-2-ила, 1,2,3-триазолила, пиразинила, 5,6-дигидро-4Н-пирроло[1,2-b]пиразол-2-ила, пиперазин-1-илпиридин-2-ила, 4,7-дигидропиразоло[5,1-с][1,4]оксазин-2-ила, 5-(пирролидин-1-карбонил)пиразол-3-ила, 6-оксо-4,7-дигидропиразоло[1,5-а]пиразин-2-ила, 5-(3-гидроксиазетидин-1-ил)-2-пиридинила, пиридинила, изотиазолила, возможно замещенных одной или более группами, независимо выбранными из -СН3, -СН2СН3, -СН(СН3)2, -СН2ОН, -СН2ОСН3, -СН2СН2ОН, -СН2СН2ОСН3, -CH2CF3, -CH2CHF2, -C(CH3)2CN, -С(O)СН3, -C(O)CH(CH3)2, циклопропила и оксетанила.
2. Соединение по п. 1, где R8 представляет собой
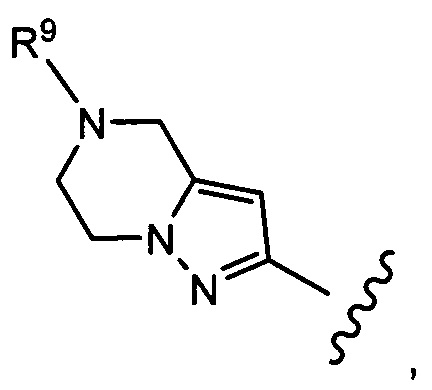
где R9 выбран из -СН3, -СН2СН2ОСН3, -CH2CF3, -CH2CHF2, -С(O)СН3, -С(O)СН(СН3)2 и оксетанила.
3. Соединение по п. 1, где X1 представляет собой N, X2 представляет собой CR2, и X3 представляет собой CR3.
4. Соединение по п. 1, где X1 представляет собой CR1, X2 представляет собой N, и X3 представляет собой CR3.
5. Соединение по п. 1, где X1 представляет собой CR1, X2 представляет собой CR2, и X3 представляет собой N.
6. Соединение по п. 1, где X2 представляет собой CR2, и R2 представляет собой F.
7. Соединение по п. 6, где X1 и X3 представляют собой СН.
8. Соединение по п. 1, где R4 представляет собой -СН2ОН.
9. Соединение по п. 1, выбранное из соединения формулы I:
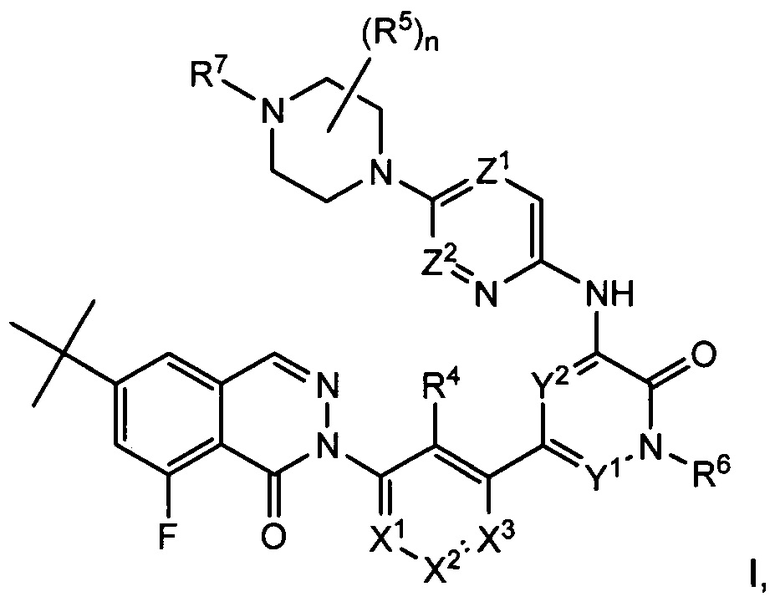
где:
R5 выбран из -СН3 и -СН2СН3;
n равно 0, 1 или 2;
R7 выбран из -СН3 и оксетан-3-ила;
Z1 представляет собой СН;
Z2 представляет собой СН;
Y1 выбран из СН и N, и
Y2 представляет собой СН.
10. Соединение по п. 9, выбранное из соединений формул Ia-Ib, имеющих структуры:
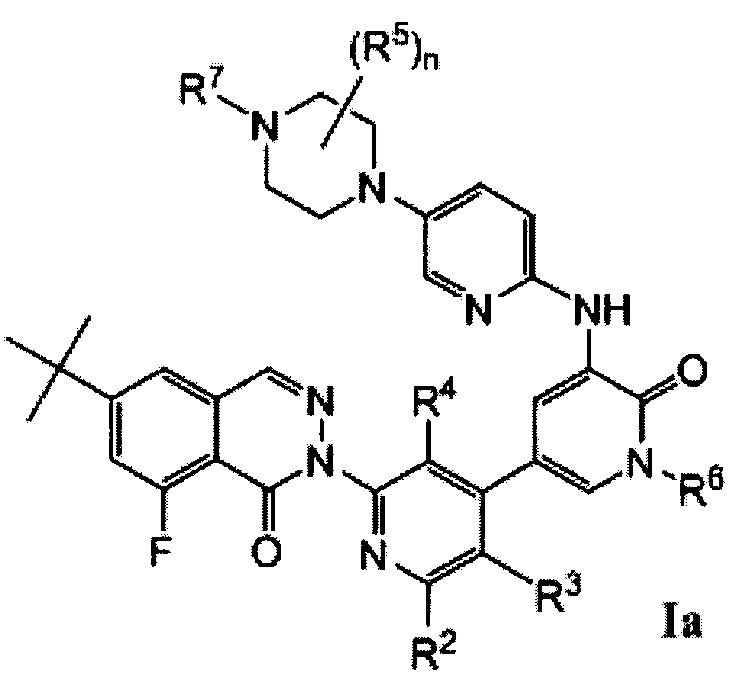
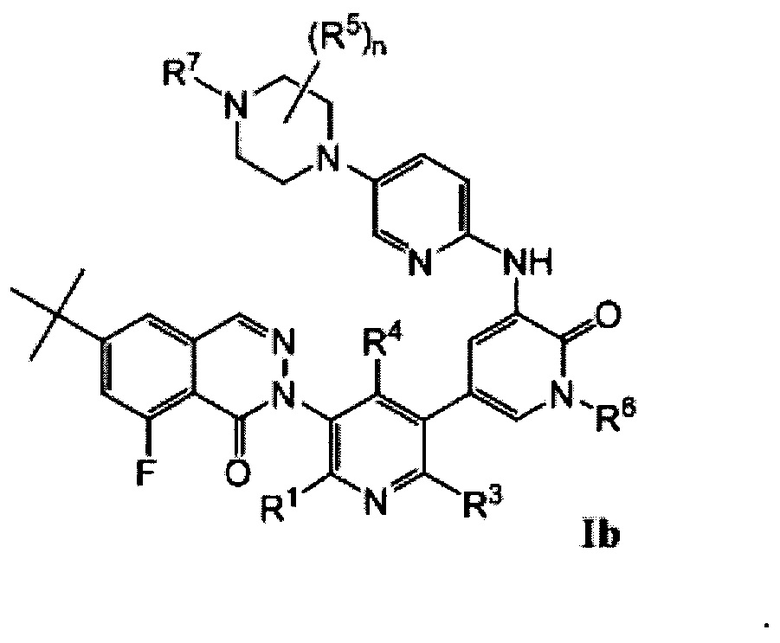
11. Соединение по п. 9, где R5 представляет собой -СН3, и n равно 1 или 2.
12. Соединение по п. 9, где R7 представляет собой оксетан-3-ил.
13. Соединение по п. 9, где Y1 представляет собой СН.
14. Соединение по п. 1, выбранное из следующих соединений:
6-трет-бутил-8-фтор-2-(2-(гидроксиметил)-3-(1-метил-5-(5-(4-метил-пиперазин-1-ил)пиридин-2-ил-амино)-6-оксо-1,6-дигидропиридин-3-ил)фенил)-фталазин-1(2Н)-он,
6-трет-бутил-8-фтор-2-(5-фтор-2-(гидроксиметил)-3-(1-метил-5-(5-(4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-иламино)-6-оксо-1,6-дигидропиридин-3-ил)фенил)фталазин-1(2Н)-он,
6-трет-бутил-8-фтор-2-(3-(гидроксиметил)-4-(1-метил-5-(5-(4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-иламино)-6-оксо-1,6дигидропиридин-3-ил)пиридин-2-ил)фталазин-1(2Н)-он,
(S)-6-трет-бутил-8-фтор-2-(5-фтор-2-(гидроксиметил)-3-(1-метил-5-(5-(2-метил-4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-иламино)-6-оксо-1,6-дигидропиридин-3-ил)фенил)фталазин-1(2Н)-он,
(S)-6-трет-бутил-8-фтор-2-(3-(гидроксиметил)-4-(1-метил-5-(5-(2-метил-4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-иламино)-6-оксо-1,6-дигидропиридин-3-ил)пиридин-2-ил)фталазин-1(2Н)-он,
(R)-6-трет-бутил-8-фтор-2-(3-(гидроксиметил)-4-(1-метил-5-(5-(2-метил-4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-иламино)-6-оксо-1,6-дигидропиридин-3-ил)пиридин-2-ил)фталазин-1(2Н)-он,
(R)-6-трет-бутил-8-фтор-2-(5-фтор-2-(гидроксиметил)-3-(1-метил-5-(5-(2-метил-4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-иламино)-6-оксо-1,6-дигидропиридин-3-ил)фенил)фталазин-1(2Н)-он,
6-трет-бутил-2-(3-(5-(5-((2S,5R)-2,5-диметил-4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-иламино)-1-метил-6-оксо-1,6-дигидропиридин-3-ил)-5-фтор-2-(гидроксиметил)фенил)-8-фторфталазин-1(2Н)-он,
(S)-6-трет-бутил-8-фтор-2-(4-(гидроксиметил)-5-(1-метил-5-(5-(2-метил-4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-иламино)-6-оксо-1,6-дигидропиридин-3-ил)пиридин-3-ил)фталазин-1(2Н)-он,
6-трет-бутил-2-(4-(5-(5-((2S,5R)-2,5-диметил-4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-иламино)-1-метил-6-оксо-1,6-дигидропиридин-3-ил)-3-(гидроксиметил)пиридин-2-ил)-8-фторфталазин-1(2Н)-он,
(S)-6-трет-бутил-2-(3-(5-(5-(2-этил-4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-иламино)-1-метил-6-оксо-1,6-дигидропиридин-3-ил)-5-фтор-2-(гидроксиметил)фенил)-8-фторфталазин-1(2Н)-он,
(S)-6-трет-бутил-2-(4-(5-(5-(2-этил-4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-иламино)-1-метил-6-оксо-1,6-дигидропиридин-3-ил)-3-(гидроксиметил)пиридин-2-ил)-8-фторфталазин-1(2Н)-он.
15. Соединение по п. 1, выбранное из следующих соединений:
6-трет-бутил-8-фтор-2-[3-(гидроксиметил)-4-[1-метил-6-оксо-5-(пиримидин-4-иламино)-3-пиридил]-2-пиридил]фталазин-1-он,
6-трет-бутил-8-фтор-2-[3-(гидроксиметил)-4-[1-метил-5-[(2-метилпиримидин-4-ил)амино]-6-оксо-3-пиридил]-2-пиридил]фталазин-1-он,
6-трет-бутил-8-фтор-2-[3-(гидроксиметил)-4-[1-метил-5-[(5-метил-6,7-дигидро-4Н-тиазоло[5,4-с]пиридин-2-ил)амино]-6-оксо-3-пиридил]-2-пиридил]фталазин-1-он,
6-трет-бутил-8-фтор-2-[3-(гидроксиметил)-4-[1-метил-5-[[5-(морфолин-4-карбонил)-2-пиридил]амино]-6-оксо-пиридазин-3-ил]-2-пиридил]фталазин-1-он,
6-трет-бутил-8-фтор-2-[3-(гидроксиметил)-4-[1-метил-5-[[5-[(2S)-2-метил-4-(оксетан-3-ил)пиперазин-1-ил]-2-пиридил]амино]-6-оксо-пиридазин-3-ил]-2-пиридил]фталазин-1-он,
6-трет-бутил-2-[4-[5-[(1-этил-5-метил-пиразол-3-ил)амино]-1-метил-6-оксо-3-пиридил]-3-(гидроксиметил)-2-пиридил]-8-фтор-фталазин-1-он,
6-трет-бутил-2-[4-[5-[(1,5-диметилпиразол-3-ил)амино]-1-метил-6-оксо-3-пиридил]-3-(гидроксиметил)-2-пиридил]-8-фтор-фталазин-1-он,
6-трет-бутил-8-фтор-2-[3-(гидроксиметил)-4-[1-метил-5-[(5-метилтиазол-2-ил)амино]-6-оксо-3-пиридил]-2-пиридил]фталазин-1-он,
6-трет-бутил-8-фтор-2-[3-(гидроксиметил)-4-[1-метил-5-[(5-метил-1Н-пиразол-3-ил)амино]-6-оксо-3-пиридил]-2-пиридил]фталазин-1-он,
6-трет-бутил-2-[4-[5-[(5-этил-1-метил-пиразол-3-ил)амино]-1-метил-6-оксо-3-пиридил]-3-(гидроксиметил)-2-пиридил]-8-фтор-фталазин-1-он,
6-трет-бутил-2-[4-[6-[(1-этилпиразол-4-ил)амино]-4-метил-5-оксо-пиразин-2-ил]-3-(гидроксиметил)-2-пиридил]-8-фтор-фталазин-1-он,
2-[4-[5-[(5-ацетил-6,7-дигидро-4Н-пиразоло[1,5-а]пиразин-2-ил)амино]-1-метил-6-оксо-3-пиридил]-3-(гидроксиметил)-2-пиридил]-6-трет-бутил-8-фтор-фталазин-1-он,
6-трет-бутил-8-фтор-2-[3-(гидроксиметил)-4-[1-метил-5-[(5-метилоксазол-2-ил)амино]-6-оксо-3-пиридил]-2-пиридил]фталазин-1-он,
6-трет-бутил-2-[4-[5-[(5-циклопропил-1-метил-пиразол-3-ил)амино]-1-метил-6-оксо-3-пиридил]-3-(гидроксиметил)-2-пиридил]-8-фтор-фталазин-1-он,
6-трет-бутил-2-[4-[5-[(1,5-диметилпиразол-3-ил)амино]-1-метил-6-оксо-пиридазин-3-ил]-3-(гидроксиметил)-2-пиридил]-8-фтор-фталазин-1-он,
6-трет-бутил-8-фтор-2-[3-(гидроксиметил)-4-[1-метил-5-[(5-метил-6,7-дигидро-4Н-пиразоло[1,5-а]пиразин-2-ил)амино]-6-оксо-3-пиридил]-2-пиридил]фталазин-1-он,
6-трет-бутил-8-фтор-2-[3-(гидроксиметил)-4-[1-метил-5-[(5-метилизоксазол-3-ил)амино]-6-оксо-3-пиридил]-2-пиридил]фталазин-1-он,
6-трет-бутил-8-фтор-2-[3-(гидроксиметил)-4-[1-метил-5-[(1-метилимидазол-4-ил)амино]-6-оксо-3-пиридил]-2-пиридил]фталазин-1-он,
6-трет-бутил-2-[4-[5-(6,7-дигидро-4Н-пиразоло[5,1-с][1,4]оксазин-2-иламино)-1-метил-6-оксо-3-пиридил]-3-(гидроксиметил)-2-пиридил]-8-фтор-фталазин-1-он,
6-трет-бутил-8-фтор-2-[3-(гидроксиметил)-4-[1-метил-5-[[5-[(3R)-3-метилморфолин-4-карбонил]-2-пиридил]амино]-6-оксо-3-пиридил]-2-пиридил]фталазин-1-он,
6-трет-бутил-8-фтор-2-[3-(гидроксиметил)-4-[5-[[5-(2-метоксиэтил)-6,7-дигидро-4Н-пиразоло[1,5-а]пиразин-2-ил]амино]-1-метил-6-оксо-3-пиридил]-2-пиридил]фталазин-1-он,
6-трет-бутил-8-фтор-2-[3-(гидроксиметил)-4-[1-метил-6-оксо-5-[[5-(2,2,2-трифторэтил)-6,7-дигидро-4Н-пиразоло[1,5-а]пиразин-2-ил]амино]-3-пиридил]-2-пиридил]фталазин-1-он,
6-трет-бутил-2-[4-[5-[[5-(2,2-дифторэтил)-6,7-дигидро-4Н-пиразоло[1,5-а)пиразин-2-ил]амино]-1-метил-6-оксо-3-пиридил]-3-(гидроксиметил)-2-пиридил]-8-фтор-фталазин-1-он,
6-трет-бутил-8-фтор-2-[3-(гидроксиметил)-4-[1-метил-5-[[5-[(3S)-3-метилморфолин-4-карбонил]-2-пиридил]амино]-6-оксо-3-пиридил]-2-пиридил]фталазин-1-он,
6-трет-бутил-8-фтор-2-[3-(гидроксиметил)-4-[5-[[5-[(2S)-2-метил-4-(оксетан-3-ил)пиперазин-1-ил]-2-пиридил]амино]-6-оксо-1Н-пиридин-3-ил]-2-пиридил]фталазин-1-он,
6-трет-бутил-2-[4-[5-[(6,6-диметил-4,7-дигидропиразоло[5,1-с][1,4]оксазин-2-ил)амино]-1-метил-6-оксо-3-пиридил]-3-(гидроксиметил)-2-пиридил]-8-фтор-фталазин-1-он,
6-трет-бутил-8-фтор-2-[3-(гидроксиметил)-4-[1-метил-5-[(1-метилпиразол-3-ил)амино]-6-оксо-3-пиридил]-2-пиридил]фталазин-1-он,
6-трет-бутил-8-фтор-2-[3-(гидроксиметил)-4-[1-метил-5-[(5-метилизоксазол-3-ил)амино]-6-оксо-пиридазин-3-ил]-2-пиридил]фталазин-1-он,
6-трет-бутил-2-[4-[5-[(1-этилпиразол-3-ил)амино]-1-метил-6-оксо-3-пиридил]-3-(гидроксиметил)-2-пиридил]-8-фтор-фталазин-1-он,
6-трет-бутил-8-фтор-2-[3-(гидроксиметил)-4-[5-[[5-(метоксиметил)-1-метил-пиразол-3-ил]амино]-1-метил-6-оксо-3-пиридил]-2-пиридил]фталазин-1-он,
6-трет-бутил-8-фтор-2-[3-(гидроксиметил)-4-[1-метил-5-[[1-метил-5-(пирролидин-1-карбонил)пиразол-3-ил]амино]-6-оксо-3-пиридил]-2-пиридил]фталазин-1-он,
6-трет-бутил-2-[4-[5-[[1-(2,2-дифторэтил)-5-метил-пиразол-3-ил]амино]-1-метил-6-оксо-3-пиридил]-3-(гидроксиметил)-2-пиридил]-8-фтор-фталазин-1-он,
6-трет-бутил-8-фтор-2-[3-(гидроксиметил)-4-[1-метил-5-[(5-метил-6-оксо-4,7-дигидропиразоло[1,5-а]пиразин-2-ил)амино]-6-оксо-3-пиридил]-2-пиридил]фталазин-1-он,
6-трет-бутил-8-фтор-2-[4-[5-[[5-(3-гидроксиазетидин-1-ил)-2-пиридил]амино]-1-метил-6-оксо-3-пиридил]-3-(гидроксиметил)-2-пиридил]фталазин-1-он,
6-трет-бутил-8-фтор-2-[3-(гидроксиметил)-4-[1-метил-5-[[5-(2-метилпропаноил)-6,7-дигидро-4Н-пиразоло[1,5-а]пиразин-2-ил]амино]-6-оксо-3-пиридил]-2-пиридил]фталазин-1-он,
6-трет-бутил-8-фтор-2-[3-(гидроксиметил)-4-[1-метил-5-[(1-метилтриазол-4-ил)амино]-6-оксо-3-пиридил]-2-пиридил]фталазин-1-он,
6-трет-бутил-8-фтор-2-[3-(гидроксиметил)-4-[1-метил-5-[(4-метил-6,7-дигидро-4Н-пиразоло[5,1-с][1,4]оксазин-2-ил)амино]-6-оксо-3-пиридил]-2-пиридил]фталазин-1-он,
6-трет-бутил-8-фтор-2-[3-(гидроксиметил)-4-[5-[(5-изопропил-6-оксо-4,7-дигидропиразоло[1,5-а]пиразин-2-ил)амино]-1-метил-6-оксо-3-пиридил]-2-пиридил]фталазин-1-он,
6-трет-бутил-8-фтор-2-[3-(гидроксиметил)-4-[1-метил-6-оксо-5-(1Н-пиразол-3-иламино)-3-пиридил]-2-пиридил]фталазин-1-он,
6-трет-бутил-8-фтор-2-[3-(гидроксиметил)-4-[1-метил-5-[(5-метил-6,7-дигидро-4Н-пиразоло[1,5-а]пиразин-2-ил)амино]-6-оксо-пиридазин-3-ил]-2-пиридил]фталазин-1-он,
6-трет-бутил-8-фтор-2-[3-(гидроксиметил)-4-[1-метил-5-[(3-метилтриазол-4-ил)амино]-6-оксо-3-пиридил]-2-пиридил]фталазин-1-он,
6-трет-бутил-8-фтор-2-[3-(гидроксиметил)-4-[1-метил-5-[[5-(оксетан-3-ил)-6,7-дигидро-4Н-пиразоло[1,5-а]пиразин-2-ил]амино]-6-оксо-пиридазин-3-ил]-2-пиридил]фталазин-1-он,
2-[6-[[5-[2-(6-трет-бутил-8-фтор-1-оксо-фталазин-2-ил)-3-(гидроксиметил)-4-пиридил]-1-метил-2-оксо-3-пиридил]амино]-3-пиридил]-2-метилпропаннитрил,
6-трет-бутил-8-фтор-2-[3-(гидроксиметил)-4-[1-метил-5-[(5-метилизотиазол-3-ил)амино]-6-оксо-3-пиридил]-2-пиридил]фталазин-1-он,
6-трет-бутил-2-[4-[5-[(5-этилизоксазол-3-ил)амино]-1-метил-6-оксо-3-пиридил]-3-(гидроксиметил)-2-пиридил]-8-фтор-фталазин-1-он,
6-трет-бутил-2-[4-[5-[[2-(дифторметил)триазол-4-ил]амино]-1-метил-6-оксо-3-пиридил]-3-(гидроксиметил)-2-пиридил]-8-фтор-фталазин-1-он,
6-трет-бутил-8-фтор-2-[3-(гидроксиметил)-4-[1-метил-6-оксо-5-(пиразин-2-иламино)-3-пиридил]-2-пиридил]фталазин-1-он,
[2-(6-трет-бутил-8-фтор-1-оксо-фталазин-2-ил)-4-[1-метил-5-[(5-метилизоксазол-3-ил)амино]-6-оксо-3-пиридил]-3-пиридил]метилдигидрофосфат,
6-трет-бутил-2-[4-[5-(5,6-дигидро-4Н-пирроло[1,2-b]пиразол-2-иламино)-1-метил-6-оксо-3-пиридил]-3-(гидроксиметил)-2-пиридил]-8-фтор-фталазин-1-он.
16. Фармацевтическая композиция, ингибирующая активность тирозинкиназы Брутона (Btk), содержащая соединение по любому из пп. 1-15 и фармацевтически приемлемый носитель, глидант, разбавитель или эксципиент.
17. Способ изготовления фармацевтической композиции, ингибирующей активность тирозинкиназы Брутона (Btk), включающий комбинирование соединения по любому из пп. 1-15 с фармацевтически приемлемым носителем.
18. Способ лечения заболевания или расстройства, включающий введение терапевтически эффективного количества фармацевтической композиции по п. 16 пациенту с заболеванием или расстройством, выбранным из иммунных расстройств, рака, сердечно-сосудистого заболевания, вирусной инфекции, воспаления, расстройств метаболизма/функции эндокринной системы и неврологических расстройств, и опосредованным тирозинкиназой Брутона.
19. Способ по п. 18, где заболевание или расстройство представляет собой иммунное расстройство.
20. Способ по п. 19, где иммунное расстройство представляет собой ревматоидный артрит.
21. Способ по п. 18, где заболевание или расстройство представляет собой системное и локальное воспаление, артрит, воспаление, связанное с подавлением иммунитета, отторжение органных трансплантатов, аллергические реакции, язвенный колит, болезнь Крона, дерматит, астму, системную красную волчанку, синдром Шегрена, рассеянный склероз, склеродермию/системный склероз, идиопатическую тромбоцитопеническую пурпуру (ITP), связанный с антинейтрофильными цитоплазматическими антителами (ANCA) васкулит, хроническую обструктивную болезнь легких (COPD), псориаз.
22. Способ по п. 18, где заболевание или расстройство представляет собой рак, выбранный из рака молочной железы, яичника, шейки матки, предстательной железы, семенника, мочеполового тракта, пищевода, гортани, глиобластомы, нейробластомы, рака желудка, кожи, кератоакантомы, рака легкого, эпидермоидной карциномы, крупноклеточной карциномы, немелкоклеточной карциномы легкого (NSCLC), мелкоклеточной карциномы, аденокарциномы легкого, рака кости, толстой кишки, аденомы, рака поджелудочной железы, аденокарциномы, рака щитовидной железы, фолликулярной карциномы, недифференцированной карциномы, папиллярной карциномы, семиномы, меланомы, саркомы, рака мочевого пузыря, рака печени и желчных протоков, рака почки, панкреатического рака, миелоидных расстройств, лимфомы, волосатоклеточного рака, рака щечной полости, носоглотки, глотки, губы, языка, полости рта, тонкого кишечника, толстой кишки-прямой кишки, толстого кишечника, прямой кишки, головного мозга и центральной нервной системы, болезни Ходжкина, лейкоза, рака бронха, щитовидной железы, печени и внутрипеченочных желчных протоков, гепатоклеточного рака, желудочного рака, глиомы/глиобластомы, эндометриального рака, рака почки и почечной лоханки, мочевого пузыря, тела матки, шейки матки, множественной миеломы, острого миелогенного лейкоза, хронического миелогенного лейкоза, лимфоцитарного лейкоза, хронического лимфоидного лейкоза (CLL), миелоидного лейкоза, рака ротоглотки, неходжкинской лимфомы и ворсинчатой аденомы толстой кишки.
23. Набор для лечения состояния, опосредованного тирозинкиназой Брутона, содержащий:
a) фармацевтическую композицию по п. 16; и
b) инструкции к применению.
24. Фармацевтическая композиция по п. 16 для применения в качестве лекарственного средства для лечения заболевания или расстройства, выбранного из иммунных расстройств, рака, сердечно-сосудистого заболевания, вирусной инфекции, воспаления, расстройств метаболизма/функции эндокринной системы и неврологических расстройств, и опосредованного тирозинкиназой Брутона.
25. Применение фармацевтической композиции по п. 16 в изготовлении лекарственного средства для лечения иммунных расстройств, рака, сердечно-сосудистого заболевания, вирусной инфекции, воспаления, расстройств метаболизма/функции эндокринной системы и неврологических расстройств; и при этом лекарственное средство опосредованно действует на тирозинкиназу Брутона.
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| Колосоуборка | 1923 |
|
SU2009A1 |
| Колосоуборка | 1923 |
|
SU2009A1 |
| ЕА 200901313 А1, 30.04.2010. | |||

