ОБЛАСТЬ ТЕХНИКИ
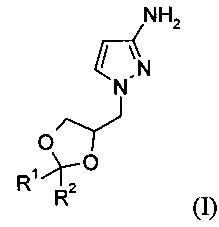
Изобретение относится к способу получения соединения формулы (I)

где R1 и R2 независимо выбраны из атома водорода, С1-6алкила, С3-7циклоалкила, С3-6алкенила или фенила, при этом С1-6алкил, циклоалкил, С3-6алкенил или фенил может быть необязательно замещенным и иметь в качестве заместителей атом галогена, гидроксил, C1-6алкоксикарбонил или фенил, или
R1 и R2 вместе с атомом углерода, к которому они присоединены, образуют С3-7циклоалкил; который может быть использован в синтезе и производстве фармацевтических активных соединений, описанных в патенте США US 7741327 В2.
УРОВЕНЬ ТЕХНИКИ
В патенте US 7741327 В2 описаны различные методики синтеза аминопиразольных производных формулы (I).
Однако найдено, что основное промежуточное соединение 1-нитропиразол представляет собой высокоэнергетическое соединение и является потенциально взрывоопасным в условиях реакции. Кроме того, общий выход вышеупомянутых методик синтеза является низким или средним, что обусловлено наличием реакционных стадий с низким выходом, образованием нескольких побочных продуктов, неселективными реакциями и неполным превращением. Поэтому одна из целей изобретения состоит в том, чтобы найти альтернативную методику синтеза, которая может быть использована в производственном масштабе и которая позволяет получать продукт с более высоким выходом и желаемой чистоты и без необходимости использовать опасные промежуточные соединения.
Данная цель могла бы быть достигнута путем использования способа согласно изобретению, как описано ниже.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
ОПРЕДЕЛЕНИЯ
В контексте данного описания термин "С1-6алкил", один или в комбинации, означает алкильную группу с насыщенной линейной или разветвленной цепью, содержащую от 1 до 6, конкретно от 1 до 4, атомов углерода, например такую, как метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, изобутил, трет-бутил, н-пентил, 3-метилбутил, н-гексил, 2-этилбутил и тому подобное. Конкретные "С1-6алкильные" группы представляют собой метил и этил. Более конкретно "С1-6алкильная" группа представляет собой метил.
Термин "алкоксид", один или в комбинации, означает группу алкил-О-, где "алкил" означает алкильную группу с насыщенной линейной или разветвленной цепью, например такую, как метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, изобутил, трет-бутил, н-пентил, 3-метилбутил, н-гексил, 2-этилбутил и тому подобное; например метоксид, этоксид, пропоксид, изо-пропоксид, н-бутоксид, изо-бутоксид, 2-бутоксид, трет-бутоксид, гексилоксид и тому подобное. Конкретные "алкоксидные" группы представляют собой трет-бутоксид, метоксид и этоксид, и более конкретно "алкоксидная" группа представляет собой трет-бутоксид.
Термин "C1-6алкокси", один или в комбинации, означает группу С1-6алкил-О-, где "C1-6алкил" является таким, как определено выше; например метокси, этокси, пропокси, изопропокси, н-бутокси, изо-бутокси, 2-бутокси, трет-бутокси, гексилокси и тому подобное. Конкретные "С1-6алкокси"-группы представляют собой метокси и этокси, и более конкретно "C1-6алкокси"-группа представляет собой метокси.
Термин "фенил С1-6алкил" относится к C1-6алкильной группе, такой как определено выше, где по меньшей мере один из атомов водорода С1-6алкильной группы заменен фенильной группой. Примерами конкретных фенилС1-6алкильных групп являются бензил, 4-метилбензил, 4-фторбензил, 3-метоксибензил, 4-метоксибензил, 2-фенилэтил и 3-фенилпропил. Более конкретно фенилС1-6алкильная группа представляет собой бензил.
Термин "С3-7циклоалкил", один или в комбинации, относится к насыщенному углеродному кольцу, содержащему от 3 до 7 атомов углерода, конкретно от 3 до 6 атомов углерода, например к циклопропилу, циклобутилу, циклопентилу, циклогексилу, циклогептилу и т.п. Конкретные "С3-7циклоалкильные" группы представляют собой циклопропил, циклопентил и циклогексил.
Термин "карбокси", один или в комбинации, относится к группе -СООН.
Термин "циано", один или в комбинации, относится к группе -CN.
Термин "галоген" означает атом фтора, хлора, брома или иода. Конкретно галоген представляет собой атом фтора, хлора или брома.
Термин "гидрокси", один или в комбинации, относится к группе -ОН.
Термин "карбонил", один или в комбинации, относится к группе -С(О)-.
Соединения общей формулы (I), которые содержат один или несколько хиральных центров, могут присутствовать либо в виде рацематов, диастереоизомерных смесей, либо в виде оптически активных индивидуальных изомеров. Рацематы могут быть разделены на энантиомеры с использованием известных методик. В частности, диастереоизомерные соли, которые могут быть разделены путем кристаллизации, образуются в результате взаимодействия рацемических смесей с оптически активной кислотой, такой как, например, D- или L-винная кислота, миндальная кислота, яблочная кислота, молочная кислота или камфорсульфоновая кислота.
Подробнее, изобретение относится к способу получения соединения формулы (I)
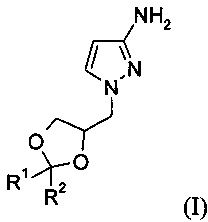
где R1 и R2 независимо выбраны из атома водорода, C1-6алкила, С3-7циклоалкила, С3-6алкенила или фенила, при этом С1-6алкил, циклоалкил, С3-6алкенил или фенил может быть необязательно замещенным и иметь в качестве заместителей атом галогена, гидроксил, C1-6алкоксикарбонил или фенил, или
R1 и R2 вместе с атомом углерода, к которому они присоединены, образуют С3-7циклоалкил,
включающему следующие стадии:
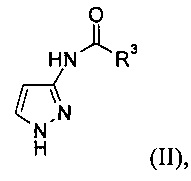
а) защиту 3-аминопиразола с образованием соединения формулы (II)
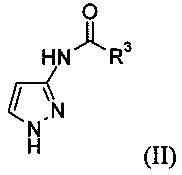
где R3 представляет собой С1-6алкил, циклоалкил или фенил;
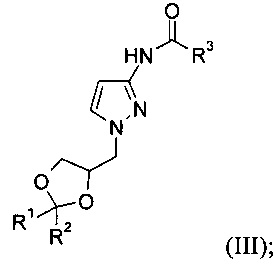
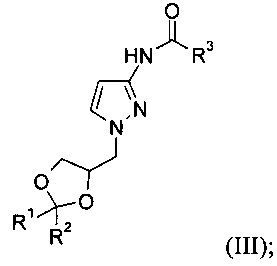
б) 1-замещение защищенного 3-аминопиразола формулы (II) с образованием соединения формулы (III)
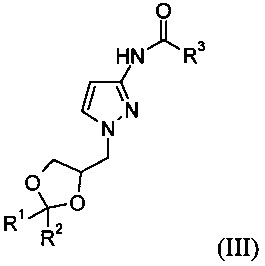
в) гидролиз защищенного 3-аминопиразола формулы (III) в присутствии основания с образованием соединения формулы (I)
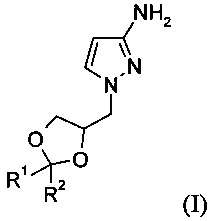
Изобретение относится, в частности, к способу получения соединения формулы (Ia)
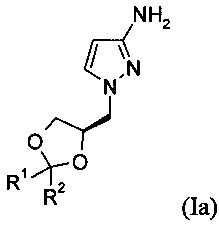 .
.
Дополнительно изобретение относится к способу получения 1-((R)2,2-диметил-[1,3]диоксолан-4-илметил)-1H-пиразол-3-иламина
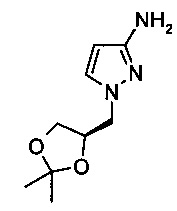
Стадия а)
Стадия а) относится к защите 3-аминопиразола с образованием соединения формулы (II)
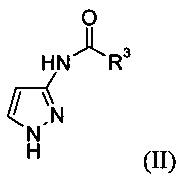
и в основном данную реакцию проводят с использованием карбоксилирующего агента при температуре реакции от 20 до 100°С.
В реакционный сосуд вносят подходящую систему растворителей и 3-аминопиразол. Порядок добавления может определяться удобством или другими особенностями процесса, известными специалисту в данной области техники.
Несмотря на то, что реакцию можно проводить во многих неспиртовых растворителях, в основном данную реакцию выполняют в растворителе, выбранном из тетрагидрофурана, уксусной кислоты, воды, изо-пропилацетата или этилацетата. Более конкретно растворитель представляет собой этилацетат.
Затем к 3-аминопиразолу и подходящему растворителю добавляют карбоксилирующий агент. Согласно конкретному варианту осуществления изобретения карбоксилирующий агент, используемый на стадии а), представляет собой уксусный ангидрид, ацетилхлорид, бензойный ангидрид, бензоилхлорид или пивалоилхлорид. Более конкретно карбоксилирующий агент представляет собой уксусный ангидрид.
Количество карбоксилирующего агента обычно определяется количеством молярных эквивалентов 3-аминопиразола и в основном составляет 1,0-2,0 молярных эквивалента.
В основном температура реакции находится в диапазоне от 40 до 80°С. Более конкретно температура реакции равна 60°С.
Через определенное время, обычно через 1-6 ч, ход реакции контролируют в основном путем ВЭЖХ (высокоэффективной жидкостной хроматографии). Полученный амид формулы (II) может быть выделен с использованием методик, известных специалисту в данной области техники, таких как фильтрование. Полученный продукт сушат под вакуумом, в основном при температуре от 30 до 60°С, до постоянной массы.
В основном для защиты 3-аминопиразола используют защитную группу, которая может быть удалена в некислотных условиях, более конкретно в присутствии основания. Такие защитные группы известны специалисту в данной области техники.
Стадия б)
Стадия б) включает алкилирование соединения формулы (II) с образованием соединения формулы (III), данную реакцию выполняют с использованием алкилирующего агента в органическом растворителе с добавлением основания и литиевой соли при температуре от 70 до 150°С.
В реакционный сосуд вносят соединение формулы (II) и подходящую систему растворителей. Порядок добавления может определяться удобством или другими особенностями процесса, известными специалисту в области химии.
Данную реакцию можно проводить во многих органических растворителях. Согласно конкретному варианту осуществления изобретения растворитель, используемый на стадии б), представляет собой диметилформамид, диметилацетамид, N-метилпирролидинон или диметилсульфоксид. Более конкретно растворитель представляет собой диметилформамид.
Затем к соединению формулы (II) и подходящему растворителю добавляют основание. Конкретное основание, используемое на стадии б), представляет собой натриевую, литиевую или калиевую соль алкоксида. Более конкретно основание представляет собой трет-бутоксид натрия. Количество основания обычно определяется количеством молярных эквивалентов соединения формулы (II) и в основном составляет 1,0-3,0 молярных эквивалента.
После добавления основания добавляют литиевую соль. Конкретная литиевая соль, используемая на стадии б), представляет собой хлорид лития, бромид лития или иодид лития. Конкретная литиевая соль представляет собой хлорид лития. Количество литиевой соли обычно определяется количеством молярных эквивалентов соединения формулы (II) и в основном составляет 0,5-3,0 молярных эквивалента, и более конкретно 1,0-1,5 эквивалента.
К полученной смеси добавляют алкилирующий агент, который представляет собой оксанановое производное. Согласно конкретному варианту осуществления изобретения алкилирующий агент представляет собой
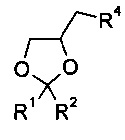
где R1 и R2 являются такими, как определено выше, и
R4 представляет собой атом хлора, брома, иода или -O-SO2-R5, где R5 представляет собой C1-6алкил, фенил или фенил, содержащий от 1 до 3 заместителей, независимо выбранных из С1-6алкила, атома галогена или нитро. Более конкретно алкилирующий агент представляет собой
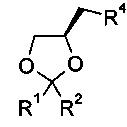
где R1, R2 и R4 являются такими, как определено выше.
Дополнительный конкретный алкилирующий агент представляет собой

где R1 и R2 являются такими, как определено выше.
Синтез соединения формулы (III) выполняют в основном при температуре от 70 до 150°С. Более конкретно температура реакции на стадии б) находится в диапазоне от 90 до 110°С.
Ход реакции можно контролировать путем ВЭЖХ. В зависимости от исходных растворителей и температуры реакция обычно завершается через 3-24 ч, и может потребоваться добавление еще одной порции основания и оксананового производного. После удаления органического реакционного растворителя путем перегонки реакция может быть остановлена путем добавления воды. Полученный продукт, соединение формулы (III), может быть экстрагирован с использованием органического растворителя, такого как этилацетат, изо-пропилацетат, 2-метил-тетрагидрофуран или дихлорметан. Конкретный экстракционный растворитель представляет собой дихлорметан. Полученный продукт может быть кристаллизован и выделен путем фильтрования, или после удаления экстракционного растворителя полученный неочищенный продукт формулы (III) может быть использован на стадии в) без дополнительной очистки.
Соединение формулы (III)
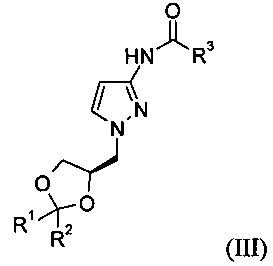
где R1 представляет собой метил, R2 представляет собой метил и R3 представляет собой метил.
Стадия в)
Стадия в) включает гидролиз 1-алкилированного 3-аминопиразол амида формулы (III) с образованием соединения формулы (I). Стадию в) выполняют в растворителе с использованием основания при температуре от 40 до 100°С.
В реакционный сосуд вносят соединение формулы (III) и подходящую систему растворителей. Порядок добавления может определяться удобством или другими особенностями процесса, известными специалисту в данной области техники.
Несмотря на то, что данную реакцию можно проводить во многих органических растворителях, конкретный растворитель, используемый на стадии в), представляет собой метанол, этанол или воду или их смесь. Более конкретно растворитель представляет собой воду.
Затем к соединению формулы (II) и подходящему растворителю добавляют основание. Согласно конкретному варианту осуществления изобретения основание, используемое на стадии в), представляет собой гидроксид натрия, гидроксид лития или гидроксид калия, согласно более конкретному варианту осуществления изобретения основание представляет собой гидроксид натрия.
Количество основания обычно определяется количеством молярных эквивалентов соединения формулы (III) и в основном составляет 3-6 молярных эквивалентов. Синтез соединения формулы (I) выполняют в основном при температуре от 40 до 100°С. Согласно более конкретному варианту осуществления изобретения температура реакции на стадии в) находится в диапазоне от 60 до 80°С.
Ход реакции можно контролировать путем ВЭЖХ. В зависимости от исходных растворителей и температуры реакция обычно завершается через 8-48 ч.
Изобретение также включает способ, такой как определено выше, включающий стадии а), б) и в) и дополнительно включающий следующие стадии:
г) взаимодействие соединения формулы (Ia) с соединением формулы (IV) с образованием соединения формулы (V)
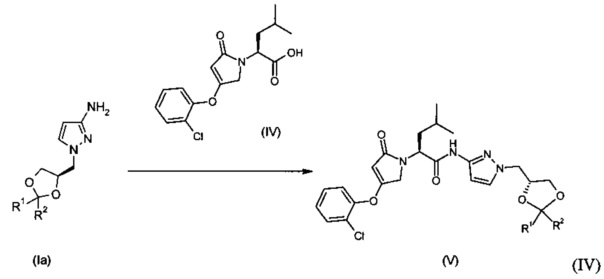
и
д) снятие защитной группы у соединения формулы (V) с получением (S)-2-[4-(2-хлор-фенокси)-2-оксо-2,5-дигидро-пиррол-1-ил]-4-метил-пентановой кислоты [1-((R)-2,3-дигидрокси-пропил)-1H-пиразол-3-ил]-амида
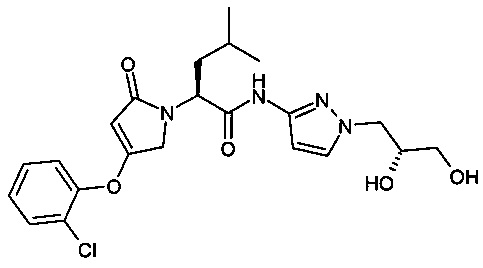
Стадия г)
Стадия г) включает синтез амида из соединения формулы (Ia) и соединения формулы (IV) с образованием соединения формулы (V). Стадию г) выполняют в растворителе с использованием реагента для образования амидной связи и катализатора при температуре от -10 до 25°С.
В реакционный сосуд вносят соединение формулы (IV), соединение формулы (Ia) и подходящую систему растворителей. Порядок добавления может определяться удобством или другими особенностями процесса, известными специалисту в данной области техники.
Несмотря на то, что данную реакцию можно проводить во многих органических растворителях, конкретный растворитель, используемый на стадии г), представляет собой дихлорметан.
К полученной смеси добавляют реагент для образования амидной связи и катализатор. Согласно конкретному варианту осуществления изобретения реагент для образования амидной связи, используемый на стадии г), представляет собой 1-(3-диметиламинопропил)-3-этилкарбодиимид и катализатор, используемый на стадии г), представляет собой 1-гидроксибензотриазол.
Количество соединения формулы (Ia) обычно определяется количеством молярных эквивалентов соединения формулы (IV) и в основном составляет 1,0-2,0 молярных эквивалента. Количество реагента для образования амидной связи обычно определяется количеством молярных эквивалентов соединения формулы (IV) и в основном составляет 1,0-3,0 молярных эквивалента. Количество катализатора обычно определяется количеством молярных эквивалентов соединения формулы (IV) и в основном составляет 0,05-1,1 молярного эквивалента.
Синтез соединения формулы (V) выполняют в основном при температуре от -10 до 25°С. Согласно более конкретному варианту осуществления изобретения температура реакции на стадии г) находится в диапазоне от 0 до 15°С.
Ход реакции можно контролировать путем ВЭЖХ. В зависимости от температуры реакции реакция обычно завершается через 1-24 ч. Реакция может быть остановлена путем добавления воды. После удаления водной фазы органический реакционный растворитель удаляют путем перегонки. Полученный амид формулы (V) может быть выделен с использованием методик, известных специалисту в данной области техники, таких как фильтрование. Полученный продукт сушат под вакуумом, в основном при температуре в диапазоне от 30 до 60°С, до постоянной массы.
Стадия д)
Стадия д) включает удаление кетальной защитной группы у соединения формулы (V) с получением (S)-2-[4-(2-хлор-фенокси)-2-оксо-2,5-дигидро-пиррол-1-ил]-4-метил-пентановой кислоты [1-((R)-2,3-дигидрокси-пропил)-1H-пиразол-3-ил]-амида. Стадию д) выполняют в растворителе с использованием кислоты при температуре от 0 до 40°С.
В реакционный сосуд вносят соединение формулы (V) и подходящую систему растворителей. Порядок добавления может определяться удобством или другими особенностями процесса, известными специалисту в данной области техники.
Несмотря на то, что данную реакцию можно проводить во многих органических растворителях, конкретные растворители, используемые на стадии д) представляют собой этанол, 2-пропанол, тетрагидрофуран и 2-метилтетрагидрофуран. Более конкретно растворитель представляет собой 2-пропанол.
К полученной смеси добавляют кислоту. Согласно конкретному варианту осуществления изобретения кислота, используемая на стадии д), представляет собой водную HCl в концентрации 1,0-6,0 н.
Количество кислоты обычно определяется количеством молярных эквивалентов соединения формулы (V) и в основном составляет 1-10 молярных эквивалентов.
Синтез (S)-2-[4-(2-хлор-фенокси)-2-оксо-2,5-дигидро-пиррол-1-ил]-4-метил-пентановой кислоты [1-((R)-2,3-дигидрокси-пропил)-1H-пиразол-3-ил]-амида выполняют в основном при температуре от 0 до 40°С. Согласно более конкретному варианту осуществления изобретения температура реакции на стадии д) находится в диапазоне от 15 до 25°С.
Ход реакции можно контролировать путем ВЭЖХ. В зависимости от температуры реакции реакция обычно завершается через 1-24 ч. Реакция может быть остановлена путем добавления воды. Полученный продукт может быть экстрагирован с использованием органического растворителя, такого как этилацетат, изо-пропилацетат, 2-метил-тетрагидрофуран или трет-бутилметиловый эфир. Конкретный экстракционный растворитель представляет собой трет-бутилметиловый эфир. После удаления органического растворителя путем перегонки продукт может быть разбавлен этанолом и использован без дополнительной очистки для изготовления лекарственных препаратов.
Далее изобретение проиллюстрировано примерами, которые никоим образом не должны быть истолкованы как ограничение объема изобретения конкретными методиками, описанными в этих примерах.
ПРИМЕРЫ
Пример 1
Получение N-(1H-пиразол-3-ил)-ацетамида
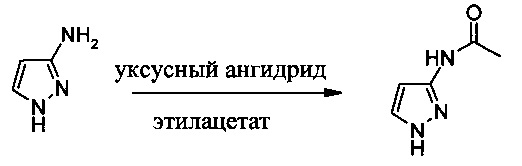
В стеклянную колбу объемом 2 л вносили 200 г (2,36 моль) 3-аминопиразола и 900 г этилацетата. Данную смесь перемешивали в течение 30 мин при 45°С с получением гомогенного раствора. К смеси добавляли 245 г (2,36 моль) уксусного ангидрида в течение 1,2 ч. Полученную смесь перемешивали в течение 3 ч при 60°С. Согласно ВЭЖХ-анализу в реакционной смеси присутствовал 3-аминопиразол. В течение 15 мин при температуре приблизительно 60°С в смесь добавляли еще одну порцию (9,7 г, 0,09 моль) уксусного ангидрида. Полученную суспензию перемешивали дополнительно в течение 2 ч при температуре приблизительно 60°С. Затем смесь охлаждали до 20-25°С и перемешивали при этой же температуре в течение 15 ч. Твердое вещество собирали путем фильтрования и промывали этилацетатом (630 г). Затем твердое вещество сушили в вакуумной печи (45-50°С/Р≤-0,1 мПа) в течение приблизительно 24 ч с получением N-(1H-пиразол-3-ил)-ацетамида (295,2 г; выход: 94%) в виде желтоватого твердого вещества. 1Н ЯМР (400 МГц, d6-ДМСО) δ: 12.26 (s, 1H), 10.33 (s, 1Н), 7.57 (s, 1H), 6.47 (s, 1H), 1.99 (s, 3Н).
Пример 2
Получение N-[1-((R)-2,2-диметил-[1,3]диоксолан-4-илметил)-1H-пиразол-3-ил]-ацетамида
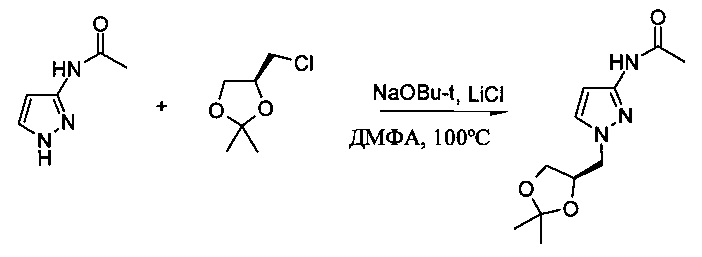
В колбу объемом 3 л вносили 200,0 г (1,60 моль) N-(1H-пиразол-3-ил)-ацетамида и 1 л безводного ДМФА (N,N-диметилформамида). К перемешиваемой суспензий добавляли за один прием 173,8 г (1,75 моль) трет-бутоксида натрия. К данной смеси добавляли за один прием 82,0 г (1,91 моль) хлорида лития. И затем к смеси добавляли за один прием 267 г (1,75 моль) (S)-(-)-4-(хлорметил)-2,2-диметил-1,3-диметил-1,3-диоксолана. Данную смесь перемешивали в течение 5 ч при 100°С. К полученной смеси добавляли дополнительно 31,6 г (0,32 моль) трет-бутоксида натрия и затем 48,5 г (0,32 моль) (S)-(-)-4-(хлорметил)-2,2-диметил-1,3-диметил-1,3-диоксолана. Данную смесь перемешивали при 100°С дополнительно в течение 5 ч. К полученной смеси добавляли еще 31,6 г (0,32 моль) трет-бутоксида натрия и затем 48,5 г (0,32 моль) (S)-(-)-4-(хлорметил)-2,2-диметил-1,3-диметил-1,3-диоксолана. Смесь перемешивали в течение 16 ч при 100°С. Затем смесь охлаждали до 60°С и концентрировали при пониженном давлении (60-25 мбар [(6,0-2,5)⋅103 Па]; 60°С) с удалением 720 г растворителей. К остатку добавляли 1,6 л воды. Полученный раствор экстрагировали дихлорметаном (ДХМ) (3,2 л). Объединенные органические фазы промывали 20% (по массе) раствором хлорида натри (1,6 л) и концентрировали путем перегонки под вакуумом (30-40°С (порциями)/Р<-100 мбар [104 Па]) с получением 585 г (316 г после коррекции с учетом % по массе; выход: 82,8%) N-[1-((R)-2,2-диметил-[1,3]диоксолан-4-илметил)-1H-пиразол-3-ил]-ацетамида в виде красно-коричневого масла, который использовали на следующей стадии без дополнительной очистки. 1Н ЯМР (400 МГц, d6-ДМСО) δ: 10.37 (s, 1H), 7.57 (s, 1Н), 6.44 (s, 1Н), 4.33-4.36 (m, 1Н), 4.08-4.11 (m, 2Н), 3.98-4.02 (m, 1Н), 3.71-3.75 (m, 1Н), 1.98 (s, 3Н), 1.3l (s, 3Н), 1,26 (s, 3Н).
Пример 3
Получение 1-((R)-2,2-диметил-[1,3]диоксолан-4-илметил)-1H-пиразол-3-иламина
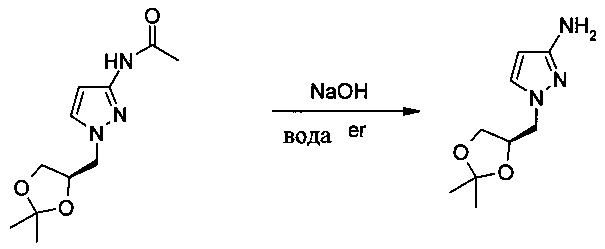
В стеклянную колбу объемом 5 л, оснащенную механической мешалкой и термометром, вносили 584 г (315 г после коррекции с учетом % по массе; 1,28 моль) N-[1-((R)-2,2-диметил-[1,3]диоксолан-4-илметил)-1H-пиразол-3-ил]-ацетамида и 1,2 л очищенной воды. Данную смесь перемешивали в течение 3 ч при 60-65°C с получением гомогенного раствора. После охлаждения смеси до 40-45°С добавляли порциями 214 г (5,14 моль) NaOH (в виде твердого вещества). Данную смесь перемешивали в течение 24 ч при 90°С. Затем смесь охлаждали до 20-25°С и экстрагировали изопропилацетатом (2 кг). Объединенные органические фазы концентрировали путем перегонки под вакуумом (35-45°С (порциями)/Р<-0,1 мПа) с получением желтого масла. Данный остаток разбавляли путем добавления 414 г трет-бутилметилового эфира и затем 750 г гептана. Полученную суспензию перемешивали в течение 15 ч при 20-25°С. Твердое вещество собирали путем фильтрования и промывали смесью трет-бутилметиловый эфир/гептан (1100 г; 1/2 об./об.). Затем твердое вещество сушили в вакуумной печи (30-35°С/Р≤-0,1 мПа) в течение приблизительно 24 ч с получением 215 г (выход после 2 стадий: 68%) 1-((R)-2,2-диметил-[1,3]диоксолан-4-илметил)-1Р-пиразол-3-иламина в виде желтоватого твердого вещества. 1Н ЯМР (400 МГц, d6-ДМСО) δ: 7.31 (s, 1Н), 5.38 (s, 1Н), 4.55 (s, 2Н), 4.26-4.32 (m, 1Н), 3.92-3.98 (m, 3Н), 3.7-3.73 (m, 1H), 1.31 (s, 3Н), 1.25 (s, 3Н).
Пример 4
Получение N-(1H-пиразол-3-ил)-бензамида
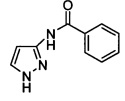
3-Аминопиразол (9 г, 108 ммоль) растворяли в ДХМ (250 мл). Добавляли за один прием N-метилморфолин (26,5 г, 262 ммоль). Медленно добавляли бензоилхлорид (34,86 г, 248 ммоль) при комнатной температуре. После перемешивания в течение 16 ч при комнатной температуре смесь концентрировали при пониженном давлении с получением твердого вещества. Полученное твердое вещество растворяли в метаноле (200 мл). Медленно добавляли водный раствор NaOH (2,5 М, 120 мл, 300 ммоль), и добавляли ТГФ (50 мл) для получения гомогенного раствора. После перемешивания в течение 20 мин при комнатной температуре смесь концентрировали при пониженном давлении и вливали в воду (300 мл). Осадок собирали путем фильтрования и сушили в печи с получением 17,55 г светло-желтого твердого вещества (выход: 87,7%). 1Н ЯМР (400 МГц, d6-ДМСО) δ: 12.45 (s, 1Н), 10.79 (s, 1Н), 7.99-8.02 (m, 2Н), 7.67 (s, 1Н), 7.48-7.59 (m, 3Н), 6.65 (s, 1H).
Пример 5
Получение N-[1-((R)-2,2-диметил-[1,3]диоксолан-4-илметил)-1H-пиразол-3-ил]-бензамида
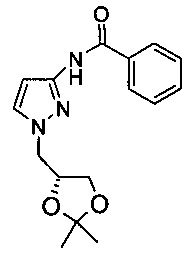
N-(1H-Пиразол-3-ил)-бензамид (2,5 г, 20 ммоль) и 4-хлор-бензолсульфоновой кислоты 2,2-диметил-[1,3]диоксолан-4-илметиловый эфир (6,75 г, 22 ммоль) растворяли в безводном 1,4-диоксане (30 мл). Добавляли за один прием трет-бутоксид натрия (2,28 г, 23,8 ммоль) при комнатной температуре. Полученную желтую суспензию нагревали при перемешивании до температуры дефлегмации в течение 23 ч. Смесь охлаждали до 10°С, добавляли воду (80 мл). Данную смесь экстрагировали этилацетатом (30 мл × 2), органический слой промывали 1 н. раствором NaOH (20 мл), 10% водным NaCl (40 мл × 2). Органический слой сушили над Na2SO4, концентрировали при пониженном давлении с получением неочищенного продукта в виде масла. После очистки на силикагелевой (silicon) колонке (гексан / ЕА (этилацетат) = 5:1 - гексан / ЕА = 3:1) получали желтоватое твердое вещество (3 г; выход: 50%). 1Н ЯМР (400 МГц, d6-ДМСО) δ: 10.86 (s, 1Н), 7.99-8.01 (d, 2Н), 7.67 (s, 1Н), 7.47-7.58 (m, 3Н), 6.63 (s, 1H), 4.4-4.42 (m, 1Н), 4.16-4.38 (m, 2Н), 4.01-4.05 (m, 1H), 3.75-3.78 (m, 1Н), 1.33 (s, 3Н), 1.25 (s, 3Н).
Пример 6
Получение 1-((R)-2,2-диметил-[1,3]диоксолан-4-илметил)-1H-пиразол-3-иламина
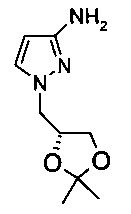
N-[1-((R)-2,2-Диметил-[1,3]диоксолан-4-илметил)-1H-пиразол-3-ил]-бензамид (150 мг, 0,5 ммоль) растворяли в метаноле (2 мл), и добавляли за один прием воду (1 мл). Данную смесь перемешивали в течение 30 мин при комнатной температуре. Добавляли NaOH (120 мг, 3 ммоль). Данную смесь перемешивали при температуре дефлегмации в течение 18 ч. Согласно анализу с использованием UPLC (сверхэффективной жидкостной хроматографии) превращение составляло ~20%, поэтому смесь перемешивали дополнительно в течение 20 ч. Превращение составляло только ~40%. В смесь добавляли еще одну порцию NaOH (120 мг, 3 ммоль). Смесь перемешивали при температуре дефлегмации еще в течение 20 ч. Превращение составляло ~70%. Далее реакцию не продолжали.
Пример 7
Получение 2,2-диметил-N-(1H-пиразол-3-ил)-пропионамида

3-Аминопиразол (9 г, 108 ммоль) растворяли в ДХМ (250 мл). Добавляли за один прием триэтиламин (27 г, 262 ммоль). Медленно добавляли пивалоилхлорид (30 г, 248 ммоль) при комнатной температуре. После перемешивания в течение 6 ч при комнатной температуре растворитель частично выпаривали при пониженном давлении с получением твердого вещества. Полученное твердое вещество растворяли в метаноле (200 мл). Медленно добавляли водный NaOH (2,5М, 120 мл) и данную смесь перемешивали в течение 30 мин при комнатной температуре. Растворитель выпаривали при пониженном давлении. Полученную суспензию переносили в воду (300 мл). Твердое вещество собирали путем фильтрования и сушили с получением указанного в заголовке соединения в виде желтого твердого вещества (13,5 г; выход: 76%). 1Н ЯМР (400 МГц, d6-ДМСО) δ: 12.3 (s, 1H), 9.77 (s, 1H), 7.58 (s, 1Н), 6.49 (s, 1Н), 1.2 (s, 9H).
Пример 8
Получение N-[1-((R)-2,2-диметил-[1,3]диоксолан-4-илметил)-1H-пиразол-3-ил]-2,2-диметил-пропионамида
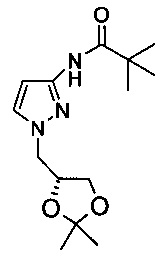
2,2-Диметил-N-(1H-пиразол-3-ил)-пропионамид (1,67 г, 10 ммоль) растворяли в ДМФА (17 мл). В данную смесь добавляли (S)-4-хлорметил-2,2-диметил-[1,3]диоксолан (1,52 г, 10 ммоль). И в конце добавляли трет-бутоксид натрия (0,98 г, 10 ммоль) при комнатной температуре. Полученную смесь нагревали при перемешивании до 100°С в течение 23 ч в атмосфере N2. Согласно UPLC-анализу в смеси оставалось 26% 2,2-диметил-N-(1H-пиразол-3-ил)-пропионамида. Добавляли еще одну порцию 2,2-диметил-N-(1H-пиразол-3-ил)-пропионамида (0,76 г, 5 ммоль) и трет-бутоксида натрия (0,49 г, 5 ммоль). Смесь перемешивали при 100°С дополнительно в течение 24 ч. Согласно ВЭЖХ-анализу в смеси оставалось 9,4% 2,2-диметил-N-(1H-пиразол-3-ил)-пропионамида, селективность N1/N2-алкилированных продуктов составляла 2,8:1. ДМФА частично выпаривали при пониженном давлении. Добавляли воду (10 мл). Данную смесь экстрагировали ДХМ (2×12 мл), органический слой промывали 10% водным раствором NaCl (2×20 мл). Органический слой сушили над Na2SO4, концентрировали при пониженном давлении с получением масла, которое очищали на силикагелевой (silicon) колонке (гексан / ЕА = 5:1 - гексан / ЕА = 3:1) с выходом желтоватого твердого вещества (0,84 г; выход: 30%). 1Н ЯМР (400 МГц, d6-ДМСО) δ: 9.85 (s, 1Н), 7.59 (s, 1Н), 6.47 (s, 1H), 4.36-4.39 (m, 1Н), 4.08-4.12 (m, 2Н), 4.0-4.02 (m, 1Н), 3.73-3.75 (m, 1Н), 1.33 (s, 3Н), 1.26 (s, 3Н), 1.19 (s, 9H).
Получение 1-((R)-2,2-диметал-[1,3]диоксолан-4-илметил)-1H-пиразол-3-иламина
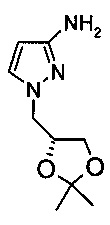
N-[1-((R)-2,2-Диметил-[1,3]диоксолан-4-илметил)-1H-пиразол-3-ил]-2,2-диметил-пропионамид (1,42 г, 5 ммоль) растворяли в метаноле (15 мл), и добавляли за один прием воду (3 мл). Добавляли за один прием NaOH (1,67 г, 40 ммоль). Данную смесь перемешивали в течение 40 ч при 70°С. Согласно ВЭЖХ-анализу превращение составляло 2,8%. Далее реакцию не продолжали.
Пример 9
Получение (S)-2-[4-(2-хлор-фенокси)-2-оксо-2,5-дигидро-пиррол-1-ил]-4-метил-пентановой кислоты [1-((R)-2,2-диметил-[1,3]диоксолан-4-илметил)-1H-пиразол-3-ил]-амида
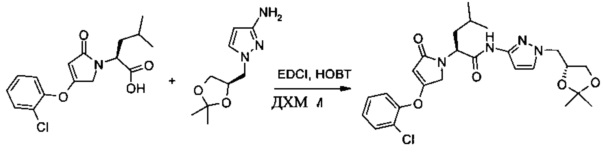
В эмалированный химический реактор объемом 200 л вносили дихлорметан (ДХМ) (46,3 кг). В реактор добавляли еще одну порцию дихлорметана (112,0 кг) и затем (S)-2-[4-(2-хлор-фенокси)-2-оксо-2,5-дигидро-пиррол-1-ил]-4-метилпентановую кислоту (11,9 кг, 36,8 моль) и 1-((R)-2,2-диметил-[1,3]диоксолан-4-илметил)-1H-пиразол-3-иламин (8,0 кг, 40,6 моль). К данной смеси добавляли при перемешивании 1-гидроксибензотриазол (0,44 кг). После охлаждения до 4°С добавляли в течение 3 ч в 4 приема 14,0 кг 1-(3-диметиламинопропил)-3-этилкарбодиимида (5,0 кг + 3,1 кг + 3,8 кг + 2,1 кг), поддерживая температуру смеси в диапазоне 4-10°С. Смесь перемешивали при 4-12°С дополнительно в течение 5 ч. Согласно ВЭЖХ-анализу через 2,5 ч реакция была завершена. Смесь переносили в эмалированный химический реактор объемом 500 л. Температуру смеси устанавливали ниже 10°С и затем реакцию останавливали путем добавления воды (119,0 кг). Водную фазу отделяли и органический растворитель удаляли путем перегонки под вакуумом до конечного объема приблизительно 30 л. К остатку добавляли этилацетат (106,0 кг). Данную смесь перемешивали в течение 30 мин, охлаждали до 0-10°С и промывали 5% (по массе) раствором лимонной кислоты, 10% (по массе) раствором карбоната натрия и 2,5% (по массе) раствором хлорида натрия. Растворитель удаляли путем перегонки под вакуумом при 18-30°С до конечного объема приблизительно 30 л. К оставшемуся раствору добавляли н-гептан (64,7 кг) в течение 2,5 ч. Температуру смеси доводили до 0-5°С и затем смесь перемешивали в течение 4,5 ч. Твердое вещество собирали нутч-фильтром, промывали н-гептаном (16,2 кг), сушили в потоке азота в течение 20 ч при 40-45°С с получением ((S)-2-[4-(2-хлорфенокси)-2-оксо-2,5-дигидро-пиррол-1-ил]-4-метил-пентановой кислоты [1-((R)-2,2-диметил-[1,3]диоксолан-4-илметил)-1Н-пиразол-3-ил]-амида (17,2 кг, выход: 88,1%). 1Н ЯМР (300 МГц, ДМСО-d6, этанол удаляли) δ м.д.: 0.90 (d, J=6.4 Гц, 3 Н), 0.94 (d, J=6.4 Гц, 3 Н), 1.05 (br. s., 3 Н), 1.06 (br. s., 3 Н), 1.36-1.64 (m, 2 Н), 1.68-1.84 (m, 1 Н), 3.89 (s, 2 Н), 4.20 (d, J=18.4 Гц, 1 Н), 4.62 (d, J=18.4 Гц, 1 Н), 4.68 (s, 1 Н), 4.78 (s, 1 Н), 4.90 (dd, J=10.7, 4.7 Гц, 1 Н), 6.44 (d, J=2.1 Гц, 1 Н), 7.37 (td, J=7.8, 1.8 Гц, 1 Н), 7.46 (td, J=7.8, 1.2 Гц, 1 Н), 7.50-7.56 (m, 2 Н), 7.66 (dd, J=7.8, 1.2 Гц, 1 Н), 10.81 (s, 1 Н).
Пример 10
Получение (S)-2-[4-(2-хлор-фенокси)-2-оксо-2,5-дигидро-пиррол-1-ил]-4-метил-пентановой кислоты [1-((R)-2,3-дигидрокси-пропил-1H-пиразол-3-ил]-амида
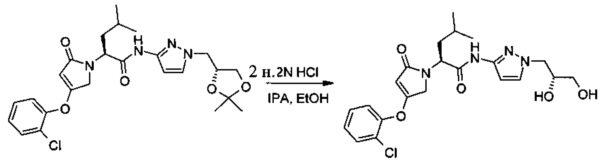
В эмалированный химический реактор объемом 200 л вносили 2-пропанол (42,7 кг) и ((S)-2-[4-(2-хлорфенокси)-2-оксо-2,5-дигидро-пиррол-1-ил]-4-метил-пентановой кислоты [1-((R)-2,2-диметил-[1,3]диоксолан-4-илметил)-1H-пиразол-3-ил]-амид (13,6 кг, 27,0 моль). Данную суспензию перемешивали при 25-30°С до растворения твердого вещества. Температуру смеси доводили до 15°С и затем добавляли в течение 3,5 ч 2,0 н. соляную кислоту (59,5 кг), поддерживая температуру смеси в диапазоне 9-17°С. Данную смесь нагревали до 18-23°С и перемешивали в течение 5,5 ч. Согласно ВЭЖХ-анализу через 2 ч реакция была завершена. Реакционную смесь переносили в эмалированный химический реактор объемом 500 л и разбавляли очищенной водой (28,6 кг) и трет-бутилметиловым эфиром (166,1 кг). Водную фазу отделяли и органическую фазу промывали последовательно 1,0 н. раствором гидроксида натрия, 10,7% (по массе) раствором хлорида натрия и 1,0% (по массе) раствором хлорида натрия. Органическую фазу переносили в эмалированный химический реактор объемом 200 л. Растворители удаляли путем перегонки под вакуумом при 10-26°С до объема приблизительно 27 л. Полученное масло разбавляли этанолом (107,6 кг) и растворители удаляли путем перегонки под вакуумом при 12-30°С с получением 19,85 кг (10,04 кг после коррекции с учетом % по массе; выход: 80,4%) (S)-2-[4-(2-хлор-фенокси)-2-оксо-2,5-дигидро-пиррол-1-ил]-4-метил-пентановой кислоты [1-((R)-2,3-дигидрокси-пропил-1H-пиразол-3-ил]-амида в этаноле. 1Н ЯМР (300 МГц, ДМСО-d6) δ м.д.: 0.90 (d, J=6.3 Гц, 3 Н), 0.94 (d, J=6.3 Гц, 3 Н), 1.33-1.50 (m, 1 Н), 1.49-1.67 (m, 1 Н), 1.68-1.85 (m, 1 Н), 3.16-3.32 (m, 2 Н), 3.70-3.93 (m, 2 Н), 4.09 (m, J=13.6, 3.6 Гц, 1 Н), 4.21 (d, J=18.4 Гц, 1 Н), 4.61 (d, J=18.4 Гц, 1 Н), 4.71 (t, J=5.6 Гц, 1 Н), 4.79 (s, 1 Н), 4.88 (dd, J=10.6, 4.8 Гц, 1 Н), 4.94 (d, J=5.1 Гц, 1 Н), 6.41 (d, J=1.5 Гц, 1 Н), 7.37 (t, J=7.5 Гц, 1 Н), 7.46 (t, J=7.5 Гц, 1 Н), 7.50-7.56 (m, 2 Н), 7.65 (d, J=7.5 Гц, 1 Н), 10.78 (s, 1 Н).
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА, СОДЕРЖАЩИЕ ИХ КОМПОЗИЦИИ, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2004 |
|
RU2346938C2 |
| ПРОИЗВОДНЫЕ ГЕТЕРОАРИЛПИРРОЛИДИНИЛ- И ПИПЕРИДИНИЛКЕТОНА | 2007 |
|
RU2479575C2 |
| СОЕДИНЕНИЕ АМИНОПИРАЗОЛОПИРИМИДИНА, ИСПОЛЬЗУЕМОЕ В КАЧЕСТВЕ ИНГИБИТОРА ТИРОЗИНКИНАЗНОГО РЕЦЕПТОРА НЕЙРОТРОФИЧЕСКОГО ФАКТОРА | 2017 |
|
RU2764523C2 |
| ЗАМЕЩЕННОЕ ГЕТЕРОЦИКЛИЧЕСКОЕ КОНДЕНСИРОВАННОЕ ЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2020 |
|
RU2815814C1 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛАМИНОПИРИМИДИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ ОБОГАЩЕННОЙ ЛЕЙЦИНОВЫМИ ПОВТОРАМИ КИНАЗЫ 2 (LRRK2) ДЛЯ ПРИМЕНЕНИЯ ПРИ ЛЕЧЕНИИ БОЛЕЗНИ ПАРКИНСОНА | 2013 |
|
RU2637948C2 |
| ПИРАЗОЛО[3,4-b]ПИРИДИНОВЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ФОСФОДИЭСТЕРАЗ | 2003 |
|
RU2348633C2 |
| ЗАМЕЩЕННЫЕ АРИЛПИРАЗОЛЫ В КАЧЕСТВЕ АГЕНТОВ ДЛЯ УНИЧТОЖЕНИЯ ПАРАЗИТОВ | 2006 |
|
RU2381218C2 |
| НОВЫЕ ПИРРОЛЬНЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2014 |
|
RU2607788C2 |
| НОВЫЕ ИНДОЛИЗИНОВЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2013 |
|
RU2646223C2 |
| ФТОР-18 И УГЛЕРОД-11 МЕЧЕНЫЕ РАДИОЛИГАНДЫ ДЛЯ ТОМОГРАФИИ С ПОЗИТРОННОЙ ЭМИССИЕЙ (РЕТ), ВИЗУАЛИЗИРУЮЩЕЙ LRRK2 | 2012 |
|
RU2650641C2 |
Изобретение относится к новому способу получения соединения формулы (I), где R1 и R2 независимо выбраны из атома водорода, C1-6алкила, С3-7циклоалкила, С3-6алкенила или фенила, при этом C1-6алкил, циклоалкил, С3-6 алкенил или фенил может быть необязательно замещенным и иметь в качестве заместителей атом галогена, гидроксил, С1-6алкоксикарбонил или фенил, или R1 и R2 вместе с атомом углерода, к которому они присоединены, образуют С3-7циклоалкил, включающему следующие стадии: а) защиту 3-аминопиразола с образованием соединения формулы (II), где R3 представляет собой С1-6алкил, циклоалкил или фенил; б) 1-замещение защищенного 3-аминопиразола формулы (II) с образованием соединения формулы (III) и в) гидролиз защищенного 3-аминопиразола формулы (III) в присутствии основания с образованием соединения формулы (I). Изобретение относится также к новому промежуточному соединению формулы II, где R1, R2 и R3 представляют собой метил. Соединения, полученные согласно изобретению, могут быть использованы в синтезе и производстве соединений, подходящих для лечения заболеваний или патологических состояний, ассоциированных с ингибированием полимеризации актина. 2 н. и 17 з.п. ф-лы, 10 пр.
1. Способ получения соединения формулы (I)
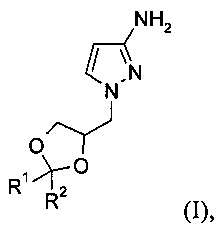
где R1 и R2 независимо выбраны из атома водорода, C1-6алкила, С3-7циклоалкила, С3-6алкенила или фенила, при этом C1-6алкил, циклоалкил, С3-6алкенил или фенил может быть необязательно замещенным и иметь в качестве заместителей атом галогена, гидроксил, С1-6алкоксикарбонил или фенил, или
R1 и R2 вместе с атомом углерода, к которому они присоединены, образуют С3-7циклоалкил,
включающий следующие стадии:
а) защиту 3-аминопиразола с образованием соединения формулы (II)
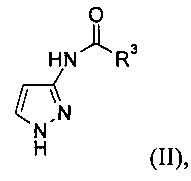
где R3 представляет собой С1-6алкил, циклоалкил или фенил;
б) 1-замещение защищенного 3-аминопиразола формулы (II) с образованием соединения формулы (III)
в) гидролиз защищенного 3-аминопиразола формулы (III) в присутствии основания с образованием соединения формулы (I)

2. Способ по п. 1 для получения соединения формулы (Ia)

3. Способ по п. 1 или 2, отличающийся тем, что соединение формулы (I) или (Ia) представляет собой 1-((R)2,2-диметил-[1,3]диоксолан-4-илметил)-1H-пиразол-3-иламин
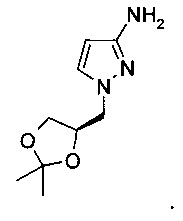
4. Способ по п. 1, отличающийся тем, что стадию а) выполняют с использованием карбоксилирующего агента при температуре реакции от 20 до 100°C.
5. Способ по п. 4, отличающийся тем, что реакцию проводят в растворителе, выбранном из тетрагидрофурана, уксусной кислоты, воды, изо-пропилацетата или этилацетата.
6. Способ по п. 4, отличающийся тем, что карбоксилирующий агент представляет собой уксусный ангидрид, ацетилхлорид, бензойный ангидрид, бензоилхлорид или пивалоилхлорид.
7. Способ по п. 4, отличающийся тем, что температура реакции находится в диапазоне от 40 до 80°C.
8. Способ по п. 1, отличающийся тем, что стадию б) выполняют с использованием алкилирующего агента в органическом растворителе с добавлением основания и литиевой соли при температуре от 70 до 150°C.
9. Способ по п. 8, отличающийся тем, что растворитель, используемый на стадии б), представляет собой диметилформамид, диметилацетамид, N-метилпирролидинон или диметилсульфоксид.
10. Способ по п. 8, отличающийся тем, что основание на стадии б) представляет собой натриевую, литиевую или калиевую соль алкоксида.
11. Способ по п. 8, отличающийся тем, что литиевая соль, используемая на стадии б), представляет собой хлорид лития, бромид лития или иодид лития.
12. Способ по п. 8, отличающийся тем, что алкилирующий агент на стадии б) представляет собой
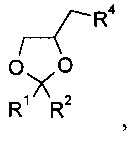
где R1 и R2 являются такими, как определено в п. 1;
R4 представляет собой атом хлора, брома, иода или -O-SO2-R5, где R5 представляет собой С1-6алкил, фенил или фенил, содержащий от 1 до 3 заместителей, независимо выбранных из C1-6алкила, атома галогена или нитро.
13. Способ по п. 12, отличающийся тем, что алкилирующий агент на стадии б) представляет собой
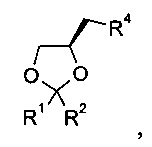
где R1, R2 и R4 являются такими, как определено в п. 12.
14. Способ по п. 8, отличающийся тем, что температура реакции на стадии б) находится в диапазоне от 90 до 110°C.
15. Способ по п. 1, отличающийся тем, что стадию в) выполняют в растворителе с использованием основания при температуре от 40 до 100°C.
16. Способ по п. 15, отличающийся тем, что растворитель, используемый на стадии в), представляет собой метанол, этанол или воду или их смесь.
17. Способ по п. 15, отличающийся тем, что основание, используемое на стадии в), представляет собой гидроксид натрия, гидроксид лития или гидроксид калия.
18. Способ по п. 15, отличающийся тем, что реакцию на стадии в) выполняют при температуре от 60 до 80°C.
19. Соединение формулы (III)
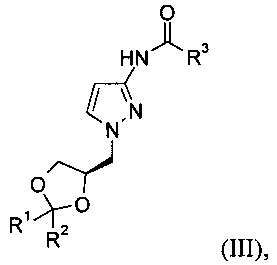
где R1 представляет собой метил, R2 представляет собой метил и R3 представляет собой метил.
| АРОЧНЫЙ ЗАСЫПНОЙ МОСТ | 2003 |
|
RU2236498C1 |
| US 20090264445 A1, 22.10.2009 | |||
| WO 2009127546 A1, 22/10.2009 | |||
| Способ получения гетероциклических производных 1-(1,3-диоксолан-2-илме-тил)-1н-имидазолов или -1н-1,2,4-триазолов или их кислотно-аддитивных солей,в виде смеси или отдельных стереоизомеров | 1978 |
|
SU703020A3 |

