Область техники
Настоящее изобретение относится к области фармацевтической технологии. Более конкретно, настоящее изобретение относится к кристаллической форме соединения, используемого в качестве антагониста минералокортикоидного рецептора, способу ее получения и применению кристаллических форм соединения в изготовлении лекарственного средства для лечения и/или предупреждения повреждения почек или сердечно-сосудистых заболеваний.
Уровень техники
Альдостерон представляет собой минералокортикоидный гормон, синтезируемый в коре надпочечников, и может связываться с минералокортикоидным рецептором и активировать рецептор для содействия сохранению натрия и выведению калия. Это может играть важную роль в поддержании электролитного баланса и изменении структуры и функции эндотелиальных клеток, гладкомышечных клеток сосудов и фибробластов артериальной стенки, а также артериальной адвентициальной оболочки и матрикса в его среде. Высокий уровень альдостерона может привести к аномальной активации минералокортикоидного рецептора, что может привести к электролитному дисбалансу, повреждению кровеносных сосудов, фиброзу и т.п. и в результате к сердечно-сосудистым заболеваниям, таким как гипертония, повреждению органов, таких как почки, сердце и головной мозг, гормональным нарушениям и тому подобному. Препарат, который блокирует связывание альдостерона и минералокортикоидного рецептора посредством конкурентного связывания с минералокортикоидным рецептором, может, следовательно, ингибировать опосредованное альдостероном повреждение и уменьшить распространение вышеуказанного заболевания.
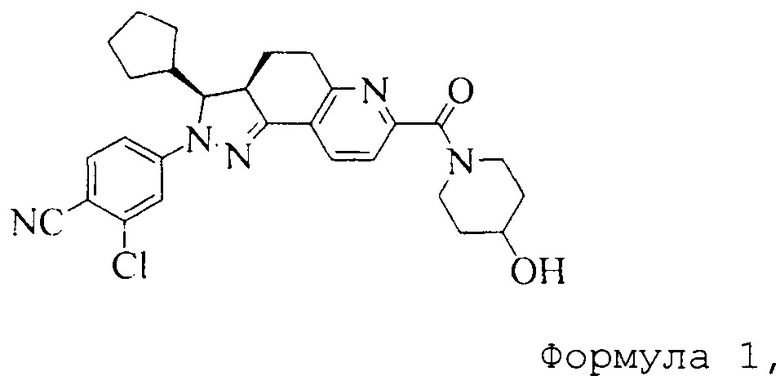
Соединение, представленное формулой (1), 2-хлор-4-[(3S,3aR)-3-циклопентил-7-(4-гидроксипиперидин-1-карбонил)-3,3a,4,5-тетрагидро-2H-пиразоло[3,4-f]хинолин-2-ил]бензонитрил, как раскрыто в заявке WO 2012022121, является антагонистом рецептора альдостерона, который может селективно связываться с минералокортикоидным рецептором, и имеет низкое сродство к глюкокортикоидным и андрогенным рецепторам.
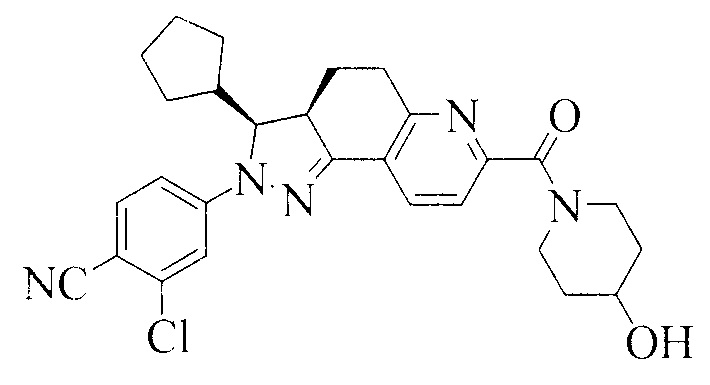
Формула 1
WO 2012022121 раскрывает способ получения соединения, представленного формулой (1), которое получают путем хирального разделения рацемической смеси, содержащей соединение, представленное формулой (1), посредством упаривания досуха на ротационном испарителе. Полученное соединение находится в аморфной форме.
Соединение формулы (1) имеет два хиральных центра. Для того чтобы получить единственный изомер, специалисты в данной области техники будут разделять рацемическую смесь. В соответствии с WO 2012022121, рацемическую смесь, содержащую соединение, представленное формулой (1), сначала получают, а затем разделяют с получением соединения, представленного формулой (1), что делает сложным получение соединения, представленного формулой (1), на предприятиях, действующих в соответствии со стандартом GMP, что приводит к возникновению трудностей в индустриализации и высокой стоимости производства.
Сущность изобретения
Изучение кристаллической формы является очень важным в разработке лекарственного препарата. Различные кристаллические формы соединения будут приводить к различиям в свойствах, таких как стабильность и растворимость. Таким образом, авторы настоящего изобретения провели множество исследований кристаллической формы соединения, представленного формулой (1), и определили и обнаружили некоторые полезные кристаллические формы соединения формулы (1).
При получении соединения формулы (1) авторы настоящего изобретения провели этап разделения заранее так, чтобы можно было легко получить соединение, представленное формулой (1), в производственных условиях в соответствии со стандартом GMP и промышленное масштабирование могло быть благополучно выполнено.
Первой задачей настоящего изобретения является предоставление кристаллических форм соединения формулы (1).
Второй задачей настоящего изобретения является предоставление способа получения соединения формулы (1).
Третьей задачей настоящего изобретения является предоставление способа получения кристаллических форм соединения формулы (1) и способа преобразования любой из кристаллических форм в другую кристаллическую форму.
Другой задачей настоящего изобретения является предоставление применения кристаллических форм соединения формулы (1) для лечения и/или предупреждения повреждения почек или сердечно-сосудистых заболеваний (в том числе повреждения сердца, гипертонии, сердечной недостаточности, инфаркта миокарда, стенокардии, сердечной гипертрофии, миокардита, фиброза сердца и кровеносных сосудов, дисфункции барорецептора или аритмии), а также применения кристаллических форм соединения формулы (1) в производстве лекарственного средства для лечения и/или предупреждения повреждения почек или сердечно-сосудистых заболеваний (в том числе повреждения сердца, гипертонии, сердечной недостаточности, инфаркта миокарда, стенокардии, сердечной гипертрофии, миокардита, фиброза сердца и кровеносных сосудов, дисфункции барорецептора или аритмии).
Техническими решениями в соответствии с настоящим изобретением являются следующие:
1. Кристаллическая форма соединения, представленного формулой (1), 2-хлор-4-[(3S,3aR)-3-циклопентил-7-(4-гидроксипиперидин-1-карбонил)-3,3a,4,5-тетрагидро-2H-пиразоло[3,4-f]хинолин-2-ил]бензонитрил
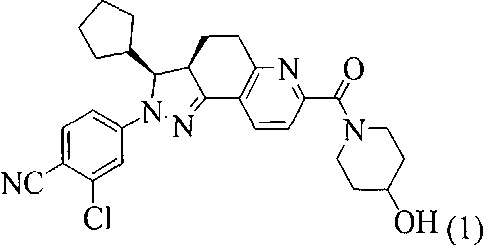 ,
,
которая характеризуется тем, что имеет порошковую рентгеновскую дифрактограмму, содержащую следующие характеристические пики, выраженные в градусах 2θ, при измерении с применением CuКα излучения:
кристаллическая форма I: 14,8°±0,2°, 17,4°±0,2°, 19,4°±0,2°, 19,8°±0,2°;
кристаллическая форма II: 14,6°±0,2°, 19,9°±0,2°, 21,2°±0,2°, 24,6°±0,2°;
кристаллическая форма III: 15,3°±0,2°, 19,5°±0,2°, 20,5°±0,2°, 25,0°±0,2°.
2. Кристаллическая форма соединения согласно решению 1, отличающаяся тем, что имеет порошковую рентгеновскую дифрактограмму, содержащую следующие характеристические пики, выраженные в градусах 2θ, при измерении с применением CuКα излучения:
кристаллическая форма I: 14,8°±0,2°, 16,9°±0,2°, 17,4°±0,2°, 19,4°±0,2°, 19,8°±0,2°, 26,2°±0,2°;
кристаллическая форма II: 14,6°±0,2°, 18,0°±0,2°, 18,7°±0,2°, 19,9°±0,2°, 21,2°±0,2°, 24,6°±0,2°;
кристаллическая форма III: 10,0°±0,2°, 15,3°±0,2°, 15,8°±0,2°, 19,5°±0,2°, 20,5°±0,2°, 25,0°±0,2°.
3. Кристаллическая форма соединения согласно решению 1 или 2, отличающаяся тем, что имеет порошковую рентгеновскую дифрактограмму, содержащую следующие характеристические пики, выраженные в градусах 2θ, при измерении с применением CuКα излучения:
кристаллическая форма I: 9,8°±0,2°, 12,9°±0,2°, 14,8°±0,2°, 15,4°±0,2°, 16,9°±0,2°, 17,4°±0,2°, 19,4°±0,2°, 19,8°±0,2°, 22,6°±0,2°, 26,2°±0,2°;
кристаллическая форма II: 4,5°±0,2°, 9,0°±0,2°, 12,2°±0,2°, 14,0°±0,2°, 14,6°±0,2°, 18,0°±0,2°, 18,7°±0,2°, 19,9°±0,2°, 21,2°±0,2°, 24,6°±0,2°;
кристаллическая форма III: 3,8°±0,2°, 10,0°±0,2°, 15,3°±0,2°, 15,8°±0,2°, 17,9°±0,2°, 19,5°±0,2°, 20,5°±0,2°, 25,0°±0,2°, 26,0°±0,2°, 27,2°±0,2°.
4. Способ получения соединения, представленного формулой (1), 2-хлор-4-[(3S,3aR)-3-циклопентил-7-(4-гидроксипиперидин-1-карбонил)-3,3a,4,5-тетрагидро-2Н-пиразоло[3,4-f]хинолин-2-ил]бензонитрил,
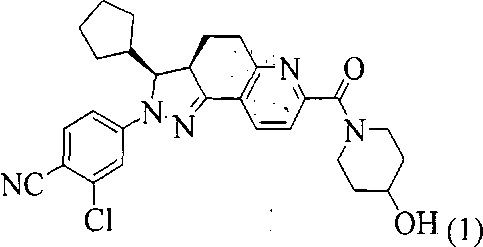 ,
,
который характеризуется тем, что содержит стадии:
(9) хирального разделения этил 2-(3-хлор-4-цианофенил)-3-циклопентил-3,3а,4,5-тетрагидро-2Н-пиразоло[3,4-f]хинолин-7-карбоксилата для получения (3S,3aR)-этил 2-(3-хлор-4-цианофенил)-3-циклопентил-3,3а,4,5-тетрагидро-2Н-пиразоло[3,4-f]хинолин-7-карбоксилата;
(10) гидролиза (3S,3aR)-этил 2-(3-хлор-4-цианофенил)-3-циклопентил-3,3а,4,5-тетрагидро-2Н-пиразоло[3,4-f]хинолин-7-карбоксилата для получения (3S,3aR)-2-(3-хлор-4-цианофенил)-3-циклопентил-3,3а,4,5-тетрагидро-2Н-пиразоло[3,4-f]хинолин-7-карбоновой кислоты;
(11) проведения реакции конденсации (3S,3aR)-2-(3-хлор-4-цианофенил)-3-циклопентил-3,3а,4,5-тетрагидро-2Н-пиразоло[3,4-f]хинолин-7-карбоновой кислоты и 4-гидроксипиперидина для получения соединения, представленного формулой (1).
5. Способ согласно решению 4, дополнительно включающий одну из следующих стадий: помещение соединения, представленного формулой (1), полученного на стадии (11), в безводный низший спирт, ацетонитрил, смешанный растворитель из этилацетата и этанола, смешанный растворитель из метанола и тетрагидрофурана или смешанный растворитель из ацетонитрила и ацетона, нагревание полученного раствора до тех пор, пока он не станет прозрачным, последующее охлаждение полученного раствора для выпадения твердого вещества и фильтрование и сушка выпавшего твердого вещества; или
помещение соединения, представленного формулой (1), полученного на стадии (11), в низший спирт, чтобы растворить его, а затем добавление полученного раствора по каплям к воде, фильтрование полученной смеси и, необязательно, сушка отфильтрованного вещества под вакуумом; или
промывание соединения, представленного формулой (1), полученного на стадии (11), смешанным раствором из воды и ацетонитрила, фильтрование полученной смеси и, необязательно, сушка отфильтрованного вещества под вакуумом; или
растворение соединения, представленного формулой (1), полученного на стадии (11), в ацетоне, добавление полученного раствора по каплям в н-гептан и фильтрование полученной смеси.
5-1. Способ согласно решению 4 или 5, дополнительно включающий стадию (8) непосредственно перед стадией (9):
(8) проведение реакции конденсации (Е)-этил 6-циклопентилметилен-5-оксо-5,6,7,8-тетрагидрохинолин-2-карбоксилата и гидрохлорида 2-хлор-4-гидразинобензонитрила для получения этил 2-(3-хлор-4-цианофенил)-3-циклопентил-3,3а,4,5-тетрагидро-2Н-пиразоло[3,4-f]хинолин-7-карбоксилата.
5-2. Способ согласно решению 5-1, дополнительно включающий стадию (7) непосредственно перед стадией (8):
(7) проведение реакции образования соли 2-хлор-4-гидразинобензонитрила и соляной кислоты для получения гидрохлорида 2-хлор-4-гидразинобензонитрила.
5-3. Способ согласно решению 5-2, дополнительно включающий стадию (6) непосредственно перед стадией (7):
(6) проведение реакции замещения 2-хлор-4-фторбензонитрила и гидразингидрата для получения 2-хлор-4-гидразинбензонитрила.
5-4. Способ согласно решению 5-3, дополнительно включающий стадию (5) непосредственно перед стадией (6):
(5) проведение реакции конденсации этил 5-оксо-5,6,7,8-тетрагидрохинолин-2-карбоксилата и циклопентанкарбальдегида для получения (E)-этил 6-циклопентилметилен-5-оксо-5,6,7,8-тетрагидрохинолин-2-карбоксилата.
5-5. Способ согласно решению 5-4, дополнительно включающий стадию (4) непосредственно перед стадией (5):
(4) проведение реакций гидролиза и этерификации 5-оксо-5,6,7,8-тетрагидрохинолин-2-карбонитрила для получения этил 5-оксо-5,6,7,8-тетрагидрохинолин-2-карбоксилата.
5-6. Способ согласно решению 5-5, дополнительно включающий стадию (3) непосредственно перед стадией (4):
(3) проведение реакции замещения 5-оксо-5,6,7,8-тетрагидрохинолин-N-оксида, хлорида N,N-диметилкарбаминовой кислоты и триметилсилилцианида для получения 5-оксо-5,6,7,8-тетрагидрохинолин-2-карбонитрила.
5-7. Способ согласно решению 5-6, дополнительно включающий стадию (2) непосредственно перед стадией (3):
(2) проведение реакции окисления 5-оксо-5,6,7,8-тетрагидрохинолина для получения 5-оксо-5,6,7,8-тетрагидрохинолин-N-оксида.
5-8. Способ согласно решению 5-7, дополнительно включающий стадию (1) непосредственно перед стадией (2):
(1) проведение реакций конденсации и присоединения 1,3-циклогександиона, ацетата аммония и акролеина для получения 5-оксо-5,6,7,8-тетрагидрохинолина.
6. Способ получения кристаллической формы I соединения, представленного формулой (1), в соответствии с решением 1, 2 или 3, который характеризуется помещением соединения 2-хлор-4-[(3S,3aR)-3-циклопентил-7-(4-гидроксипиперидин-1-карбонил)-3,3a,4,5-тетрагидро-2Н-пиразоло[3,4-f]хинолин-2-ил]бензонитрила в безводный низший спирт, ацетонитрил, смесь этилацетата и этанола, смесь метанола и тетрагидрофурана или смесь ацетонитрила и ацетона, нагреванием полученного раствора до тех пор, пока он не станет прозрачным, последующим охлаждением полученного раствора для выпадения твердого вещества и фильтрованием и сушкой выпавшего твердого вещества для получения кристаллической формы I; или растворением соединения 2-хлор-4-[(3S,3aR)-3-циклопентил-7-(4-гидроксипиперидин-1-карбонил)-3,3a,4,5-тетрагидро-2H-пиразоло[3,4-f]хинолин-2-ил]бензонитрила в ацетоне, добавлением полученного раствора по каплям в н-гептан и фильтрованием полученной смеси для получения кристаллической формы I.
7. Способ получения кристаллической формы III соединения, представленного формулой (1), в соответствии с решением 1, 2 или 3, который характеризуется помещением соединения 2-хлор-4-[(3S,3aR)-3-циклопентил-7-(4-гидроксипиперидин-1-карбонил)-3,3a,4,5-тетрагидро-2H-пиразоло[3,4-f]хинолин-2-ил]бензонитрила в низший спирт, чтобы растворить его, а затем добавлением полученного раствора по каплям к воде и фильтрованием полученной смеси для получения в результате кристаллической формы III; или промыванием соединения 2-хлор-4-[(3S,3aR)-3-циклопентил-7-(4-гидроксипиперидин-1-карбонил)-3,3a,4,5-тетрагидро-2H-пиразоло[3,4-f]хинолин-2-ил]бензонитрила смесью воды и ацетонитрила и фильтрованием полученной смеси для получения в результате кристаллической формы III.
8. Способ получения кристаллической формы II соединения, представленного формулой (1), в соответствии с решением 1, 2 или 3, который характеризуется помещением соединения 2-хлор-4-[(3S,3aR)-3-циклопентил-7-(4-гидроксипиперидин-1-карбонил)-3,3a,4,5-тетрагидро-2H-пиразоло[3,4-f]хинолин-2-ил]бензонитрила в низший спирт, чтобы растворить его, последующим добавлением полученного раствора по каплям к воде и фильтрованием полученной смеси для получения в результате кристаллической формы III; или промыванием соединения 2-хлор-4-[(3S,3aR)-3-циклопентил-7-(4-гидроксипиперидин-1-карбонил)-3,3a,4,5-тетрагидро-2H-пиразоло[3,4-f]хинолин-2-ил]бензонитрила смесью воды и ацетонитрила и фильтрованием полученной смеси для получения в результате кристаллической формы III; затем сушкой полученной кристаллической формы III под вакуумом с получением кристаллической формы II.
В соответствии с настоящим изобретением термин "низший спирт" относится к метанолу, этанолу, н-пропанолу и тому подобному.
9. Фармацевтическая композиция, отличающаяся тем, что указанная фармацевтическая композиция содержит кристаллическую форму соединения, представленного формулой (1), согласно решению 1, 2 или 3 и фармацевтически приемлемый носитель, где указанная кристаллическая форма содержит кристаллические формы I, II и III или их комбинацию.
9-1. Настоящее изобретение также предоставляет фармацевтическую композицию, содержащую кристаллические формы I, II и III соединения, представленного формулой (1), или их комбинацию. Указанная фармацевтическая композиция может также содержать фармацевтически приемлемый носитель, такой как эксципиент, связующее вещество, увлажнитель, разрыхлитель, загуститель и тому подобное.
10. Применение кристаллической формы соединения, представленного формулой (1), в соответствии с решением 1, 2 или 3 в изготовлении лекарственного средства для лечения и/или предупреждения повреждения почек или сердечно-сосудистых заболеваний, где указанная кристаллическая форма содержит кристаллические формы I, II, III или их комбинацию.
11. Применение согласно решению 10, отличающееся тем, что сердечно-сосудистые заболевания включают в себя повреждение сердца, гипертонию, сердечную недостаточность, инфаркт миокарда, стенокардию, сердечную гипертрофию, миокардит, фиброз сердца и кровеносных сосудов, дисфункцию барорецептора или аритмию.
12. Способ лечения и/или предупреждения повреждения почек или сердечно-сосудистых заболеваний, где указанный способ включает введение нуждающемуся в этом субъекту терапевтически эффективного количества кристаллической формы соединения, представленного формулой (1), в соответствии с решением 1, 2 или 3, где кристаллическая форма содержит кристаллические формы I, II, III или их комбинацию.
13. Способ согласно решению 12, отличающийся тем, что указанные сердечно-сосудистые заболевания включают в себя повреждение сердца, гипертонию, сердечную недостаточность, инфаркт миокарда, стенокардию, сердечную гипертрофию, миокардит, фиброз сердца и кровеносных сосудов, дисфункцию барорецептора или аритмию.
14. Кристаллическая форма соединения, представленного формулой (1), в соответствии с решением 1, 2 или 3 для лечения и/или предупреждения повреждения почек или сердечно-сосудистых заболеваний, где кристаллическая форма содержит кристаллические формы I, II, III или их комбинацию.
15. Кристаллическая форма в соответствии с решением 14, отличающаяся тем, что указанные сердечно-сосудистые заболевания включают в себя повреждение сердца, гипертонию, сердечную недостаточность, инфаркт миокарда, стенокардию, сердечную гипертрофию, миокардит, фиброз сердца и кровеносных сосудов, дисфункцию барорецептора или аритмию.
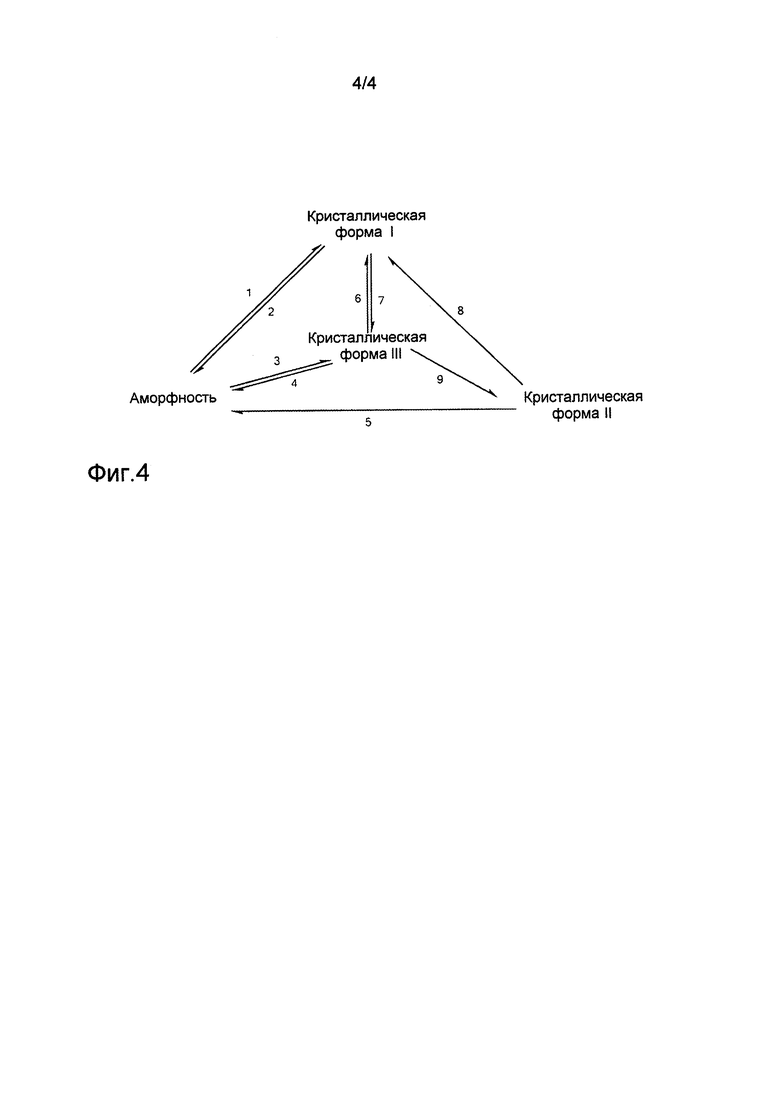
16. Кристаллические формы I, II и III, а также аморфная форма соединения, представленного формулой (1), могут быть преобразованы друг в друга при определенном условии. Настоящее изобретение также предоставляет преобразование между кристаллической формой I, кристаллической формой II, кристаллической формой III и аморфной формой.
Аморфная форма может быть перекристаллизована из безводного этанола с получением кристаллической формы I; кристаллические формы I, II и III или их комбинация могут быть растворены в низшем спирте в качестве растворителя и затем упарены досуха на ротационном испарителе с получением аморфной формы;
кристаллическая форма II может быть перекристаллизована из безводного этанола для получения кристаллической формы I; аморфная форма может быть растворена в метаноле, и полученный в результате раствор затем добавляют по каплям к воде для получения кристаллической формы III;
кристаллическая форма III может быть высушена при комнатной температуре с получением кристаллической формы II;
кристаллическая форма I может быть промыта системой из ацетонитрила и воды с получением кристаллической формы III; и
кристаллическая форма III может быть перекристаллизована из безводного этанола с получением кристаллической формы I.
Описание чертежей
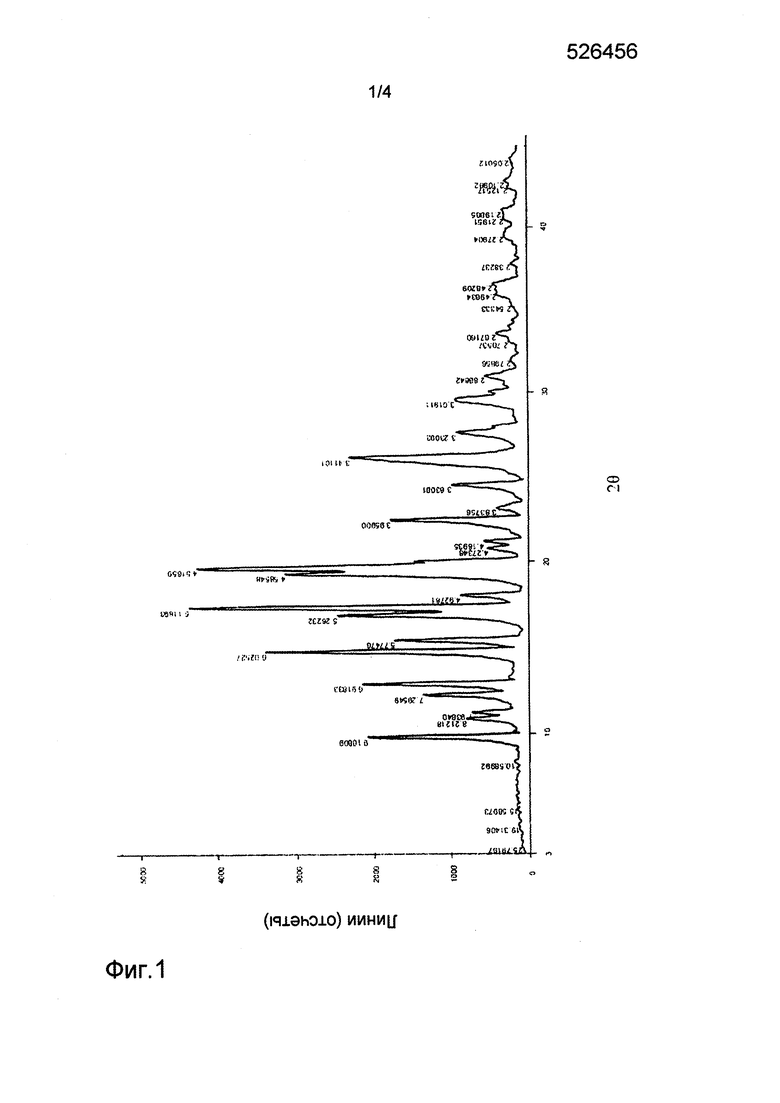
На фиг.1 представлен спектр рентгеновской дифракции для кристаллической формы I соединения формулы (1);
на фиг.2 представлен спектр рентгеновской дифракции для кристаллической формы II соединения формулы (1);
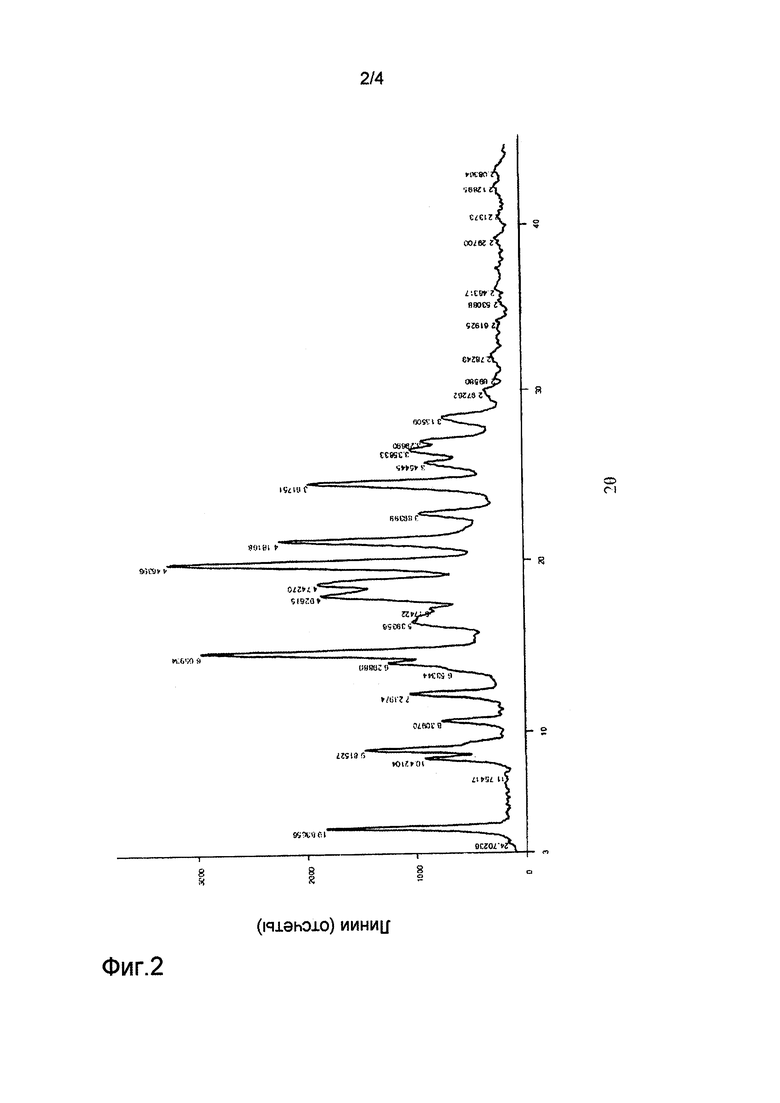
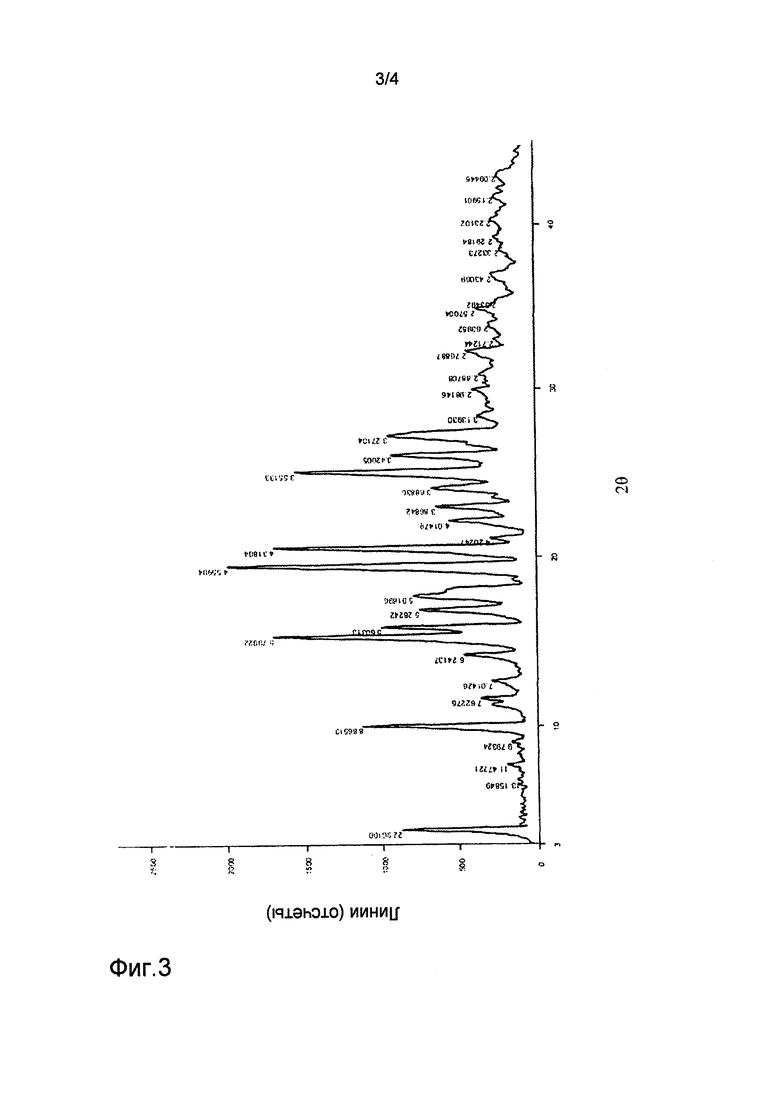
на фиг.3 представлен спектр рентгеновской дифракции для кристаллической формы III соединения формулы (1);
на фиг.4 представлена взаимосвязь преобразований между кристаллической формой I, кристаллической формой II, кристаллической формой III и аморфной формой соединения формулы (1), в которой:
1. перекристаллизация из этанола;
2. растворение в низшем спирте, а затем упаривание досуха на ротационном испарителе;
3. растворение в метаноле, а затем отделение осаждением при добавлении воды;
4. растворение в низшем спирте, а затем упаривание досуха на ротационном испарителе;
5. растворение в низшем спирте, а затем упаривание досуха на ротационном испарителе;
6. перекристаллизация из этанола;
7. промывание системой ацетонитрил/вода;
8. перекристаллизация из этанола;
9. высушивание при комнатной температуре.
Способы осуществления изобретения
Настоящее изобретение будет проиллюстрировано подробно с помощью следующих способов осуществления в виде примеров. Тем не менее, следует понимать, что объем настоящего изобретения не ограничивается следующими примерами.
Пример 1: Получение 2-хлор-4-[(3S,3aR)-3-циклопентил-7-(4-гидроксипиперидин-1-карбонил)-3,3a,4,5-тетрагидро-2H-пиразоло[3,4-f]хинолин-2-ил]бензонитрила
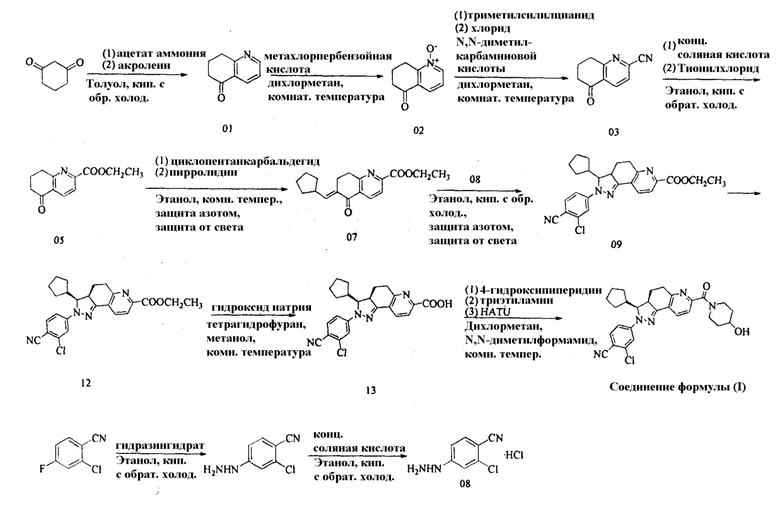
1. Получение 5-оксо-5,6,7,8-тетрагидрохинолина (01)
Уравнение реакции:

Две реакции проводили параллельно:
В 100-литровый реакционный сосуд вносили толуол (45 л), а затем добавляли 1,3-циклогександион (15 кг) при перемешивании. Полученную смесь нагревали до тех пор, пока твердое вещество не растворится. В полученный раствор добавляли ацетат аммония (24 кг). Полученную смесь нагревали до кипения с обратным холодильником в течение 12 часов и охлаждали до 0°С. К полученной смеси медленно добавляли в общей сложности 15 кг акролеина отдельными порциями. Смесь медленно нагревали до кипения с обратным холодильником, подвергали взаимодействию в течение 12 часов, охлаждали и разделяли на слои. Нижний слой дважды промывали толуолом (5 л × 2). Органические слои объединяли и концентрировали досуха с получением в общей сложности 7,4 кг сырого 5-оксо-5,6,7,8-тетрагидрохинолина в виде черной жидкости, выход: 18,8%.
2. Получение 5-оксо-5,6,7,8-тетрагидрохинолин-N-оксида (02) Уравнение реакции:

Сырой 5-оксо-5,6,7,8-тетрагидрохинолин (7,4 кг) загружали в 100-литровый реакционный сосуд. Дихлорметан добавляли в реакционный сосуд до общего объема 50 л. Полученную смесь охлаждали до -10°С. Метахлорпербензойную кислоту (13 кг) добавляли отдельными порциями. Затем смесь перемешивали в течение 20 часов при комнатной температуре. Реакционную смесь затем фильтровали при помощи вакуумной системы. Осадок на фильтре дважды промывали дихлорметаном и объединяли с фильтратом. Органический раствор промывали насыщенным раствором тиосульфата натрия до такой степени, пока йодид калия на крахмальной индикаторной бумаге не окажется синим, и сушили над безводным сульфатом натрия с получением раствора (50 л), который не подвергали дополнительной обработке и непосредственно использовали на следующей стадии.
3. Получение 5-оксо-5,6,7,8-тетрагидрохинолин-2-карбонитрил (03)
Уравнение реакции:

В 100-литровый реакционный сосуд вносили вышеуказанный раствор 5-оксо-5,6,7,8-тетрагидро-хинолин-N-оксида (50 л), затем добавляли триметилсилилцианид (10 кг), а затем медленно добавляли хлорид N,N-диметилкарбаминовой кислоты (11 кг). Реакционную смесь перемешивали при комнатной температуре в течение 48 часов. Насыщенный водный раствор гидроксида натрия медленно добавляли отдельными порциями, чтобы довести рН до 8-9. Полученную смесь разделяли на слои и экстрагировали. Водную фазу экстрагировали дихлорметаном три раза (8 л × 3). Органические фазы объединяли, промывали водой один раз (20 л). Полученную органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением красно-черной жидкости (около 8 л). Жидкость охлаждали и кристаллизовали этанолом с получением 5-оксо-5,6,7,8-тетрагидрохинолин-2-карбонитрила (930 г), выход: 10,7% (вычислено на основе исходного вещества 5-оксо-5,6,7,8-тетрагидрохинолина).
4. Получение этил 5-оксо-5,6,7,8-тетрагидрохинолин-2-карбоксилата (05)
Уравнение реакции:
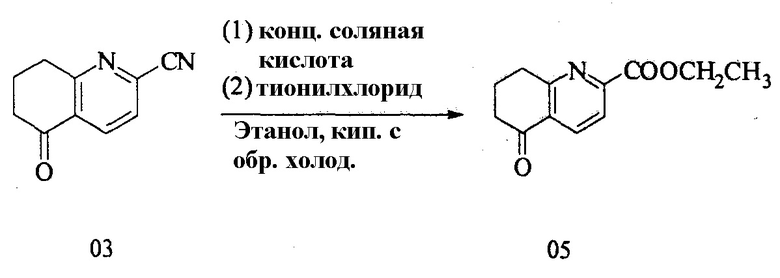
Три реакции проводили параллельно:
В 2-литровую круглодонную колбу вносили безводный этанол (800 мл) и 5-оксо-5,6,7,8-тетрагидрохинолин-2-карбонитрил (280 г). Затем концентрированную соляную кислоту (400 мл) добавляли в ледяной бане. Смесь нагревали и перемешивали в течение 16 ч с обратным холодильником. Реакционную смесь затем охлаждали и концентрировали. После добавления безводного этанола (1 л) полученную смесь охлаждали до 0°С. После добавления по каплям тионилхлорида (200 мл) полученную смесь нагревали и перемешивали в течение 10 ч с обратным холодильником. Реакционную смесь концентрировали и остаток растворяли в дихлорметане. Полученный раствор доводили раствором бикарбоната натрия до рН>7 и разделяли на слои. Водную фазу экстрагировали дихлорметаном три раза. Органические фазы объединяли, сушили, концентрировали с получением этил 5-оксо-5,6,7,8-тетрагидрохинолин-2-карбоксилата (875 г) в общей сложности, выход: 8,18%.
5. Получение (Е)-этил 6-циклопентилметилен-5-оксо-5,6,7,8-тетрагидрохинолин-2-карбоксилата (07)
Уравнение реакции:

Три реакции проводили параллельно:
В 2-литровую одногорлую бутыль вносили этил 5-оксо-5,6,7,8-тетрагидрохинолин-2-карбоксилат (291 г) и этанол (450 мл). Дополнительно добавляли циклопентанкарбальдегид (213 мл) при -20°С и полученную смесь перемешивали в течение 10 минут, затем медленно добавляли пирролидин (110 мл). При защите азотом и защите от света реакционную смесь перемешивали в течение 8 часов при комнатной температуре. Раствор выдерживали при помощи штатива при -20°С в течение 2 часов и фильтровали. Полученное твердое вещество промывали охлажденным этанолом и сушили с получением (Е)-этил 6-циклопентилметилен-5-оксо-5,6,7,8-тетрагидрохинолин-2-карбоксилата (862 г), выход: 72,3%.
6. Получение гидрохлорида 2-хлор-4-гидразинобензонитрила (08)
Уравнение реакции:
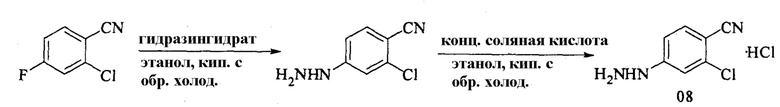
В 100-литровый реакционный сосуд вносили этанол (40 л) и 2-хлор-4-фторбензонитрил (7 кг). Затем добавляли гидразингидрат (4 л). Полученную смесь нагревали с обратным холодильником в течение 5 часов, затем охлаждали и подвергали фильтрованию на центрифуге. Полученное твердое вещество помещали в 100-литровый реакционный сосуд. Добавляли безводный этанол (40 л), а затем медленно добавляли концентрированную соляную кислоту (7,5 л). Полученную смесь нагревали с обратным холодильником в течение 2 часов, подвергали фильтрованию на центрифуге и сушили с получением гидрохлорида 2-хлор-4-гидразинобензонитрила (7 кг), выход: 76,2%.
7. Получение этил 2-(3-хлор-4-цианофенил)-3-циклопентил-3,3а,4,5-тетрагидро-2Н-пиразоло[3,4-f]хинолин-7-карбоксилата (09)
Уравнение реакции:

Четыре реакции проводили параллельно:
В 2-литровую одногорлую бутыль вносили (Е)-этил 6-циклопентилметилен-5-оксо-5,6,7,8-тетрагидрохинолин-2-карбоксилат (215,5 г), гидрохлорид 2-хлор-4-гидразинобензонитрила (191 г) и этанол (900 мл). При защите азотом и защите от света реакционную смесь нагревали с обратным холодильником в течение 9 часов при 80°С, охлаждали до комнатной температуры, выдерживали при помощи штатива при -20°C в течение 2 часов и фильтровали. Полученное твердое вещество промывали охлажденным этанолом и диэтиловым эфиром соответственно и сушили с получением этил 2-(3-хлор-4-цианофенил)-3-циклопентил-3,3а,4,5-тетрагидро-2Н-пиразоло[3,4-f]хинолин-7-карбоксилата (1026 г), выход: 79,3%.
8. Получение (3S,3aR)-этил 2-(3-хлор-4-цианофенил)-3-циклопентил-3,3а,4,5-тетрагидро-2Н-пиразоло[3,4-f]хинолин-7-карбоксилата (12)
Уравнение реакции:
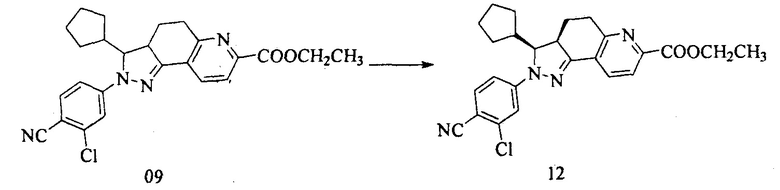
Этил 2-(3-хлор-4-цианофенил)-3-циклопентил-3,3а,4,5-тетрагидро-2Н-пиразоло[3,4-f]хинолин-7-карбоксилат разделяли с помощью SFC (сверхкритической жидкостной хроматографии) с получением двух изомеров. Первый компонент, полученный разделением и сбором, представлял собой (3S,3aR)-этил 2-(3-хлор-4-цианофенил)-3-циклопентил-3,3a,4,5-тетрагидро-2Н-пиразоло[3,4-f]хинолин-7-карбоксилат.
Условия разделения:
Прибор: SFC (Novasep 30-50)
Колонка для выполнения: Chiralpak IA, 20 мкм, 5×25 см
Подвижная фаза: фаза A представляла собой сверхкритический СО2, фаза В представляла собой дихлорметан:тетрагидрофуран:диэтаноламин = 50:50:0,1 (об.:об.:об.), А:В=50:50 (об.:об.)
Скорость потока: 150 г/мин
Длина волны детектирования: 465 нм.
Подготовка образца: этил 2-(3-хлор-4-цианофенил)-3-циклопентил-3,3а,4,5-тетрагидро-2Н-пиразоло[3,4-f]хинолин-7-карбоксилат (1025 г) подвергали ультразвуковому растворению в дихлорметане. Полученную смесь фильтровали с получением раствора образца (приблизительно 50 мг/мл).
Раствор образца разделяли с помощью SFC и первый изомер с появлением пика собирали, т.е. (3S,3aR)-этил 2-(3-хлор-4-цианофенил)-3-циклопентил-3,3а,4,5-тетрагидро-2Н-пиразоло[3,4-f]хинолин-7-карбоксилат (601,58 г).
9. Получение (3S,3aR)-2-(3-хлор-4-цианофенил)-3-циклопентил-3,3a,4,5-тетрагидро-2Н-пиразоло[3,4-f]хинолин-7-карбоновой кислоты (13)
Уравнение реакции:

В 20-литровый реакционный сосуд вносили (3S,3aR)-этил 2-(3-хлор-4-цианофенил)-3-циклопентил-3,3а,4,5-тетрагидро-2Н-пиразоло[3,4-f]хинолин-7-карбоксилат (1066 г), тетрагидрофуран (4 л) и метанол (2 л). Смесь перемешивали при -10°С в течение 10 минут и раствор (1,2 л) гидроксида натрия (192 г) в воде медленно добавляли к ней. Полученную смесь перемешивали при комнатной температуре в течение 4 часов и половину растворителя удаляли в вакууме. Реакционную смесь доводили разбавленной соляной кислотой до рН 3-4 и фильтровали при помощи вакуумной системы. Твердое вещество промывали охлажденным метанолом и диэтиловым эфиром соответственно и сушили с получением (3S,3aR)-2-(3-хлор-4-цианофенил)-3-циклопентил-3,3а,4,5-тетрагидро-2H-пиразоло[3,4-f]хинолин-7-карбоновой кислоты (867 г), выход: 86,7%.
10. Получение 2-хлор-4-[(3S,3aR)-3-циклопентил-7-(4-гидроксипиперидин-1-карбонил)-3,3a,4,5-тетрагидро-2Н-пиразоло[3,4-f]хинолин-2-ил]бензонитрила (соединение формулы (1))
Уравнение реакции:

В 5-литровый реакционный сосуд вносили (3S,3aR)-2-(3-хлор-4-цианофенил)-3-циклопентил-3,3a,4,5-тетрагидро-2Н-пиразоло[3,4-f]хинолин-7-карбоновую кислоту (380 г), дихлорметан (900 мл) и N,N-диметилформамид (360 мл). Триэтиламин (380 мл) дополнительно добавляли при перемешивании. Смесь охлаждали до -10°С и дополнительно перемешивали в течение 10 минут. Добавляли раствор (700 мл) 4-гидроксипиперидина (137 г) в дихлорметане. Полученную смесь перемешивали в течение 5 минут. Добавляли 2-(7-аза-1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилуроний гексафторфосфат (HATU) (380 г) и реакцию проводили при комнатной температуре в течение 3 часов. Затем дополнительно добавляли 2-(7-аза-1H-бензотриазол-1-ил)-1,1,3,3-тетраметилуроний гексафторфосфат (50 г) и реакцию проводили в течение 1 часа. Растворитель удаляли в вакууме. Остаток по каплям добавляли к 10-кратному количеству воды и твердое вещество отделяли. Твердое вещество растворяли в дихлорметане (2 л) и один раз промывали водой (2 л). Водную фазу один раз экстрагировали дихлорметаном (500 мл). Органические фазы объединяли, сушили и концентрировали досуха с получением твердого вещества формулы (1) (290 г), выход: 63,7%.
Молекулярная формула: C28H30ClN5O2; MS (М+Н): 504
1Н-ЯМР (CDCl3, 400 МГц): δ 8,375-8,395 (1Н, д), 7,423-7,474 (2H, м), 7,264-7,276 (1Н, д), 6,968-6,995 (1H, дд), 4,641-4,678 (1Н, дд), 4,17-4,21 (1H, м), 3,99 (1H, с), 3,76-3,79 (1Н, м), 3,515 (1H, м), 3,41-3,44 (1Н, м), 3,230-3,322 (2H, м), 2,995 (1H, м), 2,321-2,352 (1H, м), 2,10-2,15 (2Н, м), 1,978-2,089 (2H, м), 1,863-1,895 (1H, м), 1,758-1,777 (1H, м), 1,433-1,663 (7Н, м), 1,221-1,352 (2H, м).
Пример 2: Получение кристаллической формы I 2-хлор-4-[(3S,3aR)-3-циклопентил-7-(4-гидроксипиперидин-1-карбонил)-3,3a,4,5-тетрагидро-2Н-пиразоло[3,4-f]хинолин-2-ил]бензонитрила (1)
Соединение, представленное формулой (1) (1 г), полученное в примере 1, добавляли в безводный этанол (3 мл). Смесь нагревали до 80°С, пока раствор не становился прозрачным. Затем смесь медленно охлаждали до комнатной температуры. Твердое вещество отфильтровывали и три раза промывали безводным этанолом. Полученное твердое вещество сушили при 60°С под вакуумом в течение 12 часов с получением кристаллической формы I. Спектр рентгеновской дифракции (XRD) кристаллической формы I показан на фиг. 1, и его основные параметры были следующими:
Пример 3: Получение кристаллической формы I 2-хлор-4-[(3S,3aR)-3-циклопентил-7-(4-гидроксипиперидин-1-карбонил)-3,3a,4,5-тетрагидро-2Н-пиразоло[3,4-f]хинолин-2-ил]бензонитрила (2)
Соединение, представленное формулой (1) (146 мг), полученное в примере 1, растворяли в ацетонитриле (50 мл) при 80°С. Полученную смесь затем медленно охлаждали до комнатной температуры, перемешивали в течение ночи и фильтровали с получением кристаллической формы I.
Пример 4: Получение кристаллической формы I 2-хлор-4-[(3S,3aR)-3-циклопентил-7-(4-гидроксипиперидин-1-карбонил)-3,3a,4,5-тетрагидро-2Н-пиразоло[3,4-f]хинолин-2-ил]бензонитрила (3)
Соединение, представленное формулой (1) (200 мг), полученное в примере 1, вносили в 100 мл круглодонную колбу. Добавляли этилацетат (10 мл). Смесь нагревали до 78°С с обратным холодильником. Затем добавляли этанол (0,5 мл) и смесь перемешивали при 80°С. Полученный раствор медленно охлаждали до комнатной температуры. После 2 дней полученную смесь фильтровали с получением кристаллической формы I.
Пример 5: Получение кристаллической формы I 2-хлор-4-[(3S,3aR)-3-циклопентил-7-(4-гидроксипиперидин-1-карбонил)-3,3a,4,5-тетрагидро-2Н-пиразоло[3,4-f]хинолин-2-ил]бензонитрила (4)
Соединение, представленное формулой (1) (100 мг), полученное в примере 1, помещали в 100 мл круглодонную колбу. Добавляли ацетон (3 мл) и соединение растворяли. К полученной смеси добавляли по каплям н-гептан (20 мл), и твердое вещество выпадало. Полученную смесь фильтровали с получением кристаллической формы I.
Пример 6: Получение кристаллической формы I 2-хлор-4-[(3S,3aR)-3-циклопентил-7-(4-гидроксипиперидин-1-карбонил)-3,3a,4,5-тетрагидро-2Н-пиразоло[3,4-f]хинолин-2-ил]бензонитрила (5)
Соединение, представленное формулой (1) (100 мг), полученное в примере 1, вносили в 100 мл круглодонную колбу. Добавляли смешанный растворитель (0,5 мл, метанол:тетрагидрофуран = 1:1). Полученную смесь нагревали до 60°С. Затем смесь медленно охлаждали до комнатной температуры, и твердое вещество выпадало. Полученную смесь фильтровали с получением кристаллической формы I.
Пример 7: Получение кристаллической формы I 2-хлор-4-[(3S,3aR)-3-циклопентил-7-(4-гидроксипиперидин-1-карбонил)-3,3a,4,5-тетрагидро-2Н-пиразоло[3,4-f]хинолин-2-ил]бензонитрила (6)
Соединение, представленное формулой (1) (100 мг), полученное в примере 1, вносили в 100 мл круглодонную колбу. Добавляли в круглодонную колбу смешанный растворитель (3,5 мл, ацетонитрил:ацетон = 1:1). Смесь растворяли при нагревании при 60°С и перемешивании, затем медленно охлаждали до комнатной температуры, чтобы выпало твердое вещество. Полученную смесь фильтровали с получением кристаллической формы I.
Пример 8: Получение кристаллической формы III 2-хлор-4-[(3S,3aR)-3-циклопентил-7-(4-гидроксипиперидин-1-карбонил)-3,3a,4,5-тетрагидро-2Н-пиразоло[3,4-f]хинолин-2-ил]бензонитрила (1)
Соединение, представленное формулой (1) (200 мг), полученное в примере 1, вносили в метанол (4 мл). Соединение растворяли при 80°С. Полученный раствор добавляли по каплям к воде (40 мл). Полученную смесь фильтровали с получением кристаллической формы III. Спектр рентгеновской дифракции (XRD) кристаллической формы III показан на фиг.3, и его основные параметры были следующими:
Пример 9: Получение кристаллической формы III 2-хлор-4-[(3S,3aR)-3-циклопентил-7-(4-гидроксипиперидин-1-карбонил)-3,3a,4,5-тетрагидро-2Н-пиразоло[3,4-f]хинолин-2-ил]бензонитрила (2)
Соединение, представленное формулой (1) (100 мг), полученное в примере 1, вносили в 10 мл пробирку для центрифуги. Добавляли в пробирку для центрифуги смешанный раствор (8 мл, вода:ацетонитрил = 10:1) и перемешивали. Полученную смесь фильтровали с получением кристаллической формы III.
Пример 10: Получение кристаллической формы II 2-хлор-4-[(3S,3aR)-3-циклопентил-7-(4-гидроксипиперидин-1-карбонил)-3,3a,4,5-тетрагидро-2Н-пиразоло[3,4-f]хинолин-2-ил]бензонитрила
Кристаллическую форму III, полученную в примере 8, сушили в течение 12 ч в вакууме при комнатной температуре с получением кристаллической формы II. Спектр рентгеновской дифракции (XRD) кристаллической формы II показан на фиг. 2, и его основные параметры были следующими:
Анализ 1: Стабильность кристаллической формы I настоящего соединения
Образец:
Кристаллическая форма I соединения, представленного формулой (1): кристаллическую форму I получали в соответствии с примером 2.
Условия испытаний в исследовании влияющих факторов:
Высокотемпературные испытания:
(1) кристаллическую форму I соединения, представленного формулой (1), клали на сухое и чистое часовое стекло и выдерживали при 60°С в течение 10 дней. Образцы брали на 5-й день и 10-й день. Измеряли содержания сопутствующего вещества и соединения, представленного формулой (1), в образце и сравнивали с содержаниями в образце, взятом в день 0;
(2) кристаллическую форму I соединения, представленного формулой (1), упаковывали и герметизировали при помощи полиэтиленового мешка с низкой плотностью для фармацевтического применения во внутреннем слое и полиэфир/алюминий/полиэтиленовой композитной пленки для фармацевтической упаковки во внешнем слое и выдерживали при 60°С в течение 10 дней. Образцы брали на 5-й день и 10-й день. Измеряли содержания сопутствующего вещества и соединения, представленного формулой (1), в образце и сравнивали с содержаниями в образце, взятом в день 0.
Испытание при высокой влажности: кристаллическую форму I соединения, представленного формулой (1), клали на сухое и чистое часовое стекло и выдерживали при 25°С при относительной влажности 90%±5% в течение 10 дней. Образцы брали на 5-й день и 10-й день. Измеряли содержания сопутствующего вещества и соединения, представленного формулой (1), в образце и сравнивали с содержаниями в образце, взятом в день 0.
Испытание освещением: кристаллическую форму I соединения, представленного формулой (1), клали на сухое и чистое часовое стекло и выдерживали при освещенности 4500 лк ± 500 лк в освещенной коробке в течение 10 дней. Образцы брали на 5-й день и 10-й день. Измеряли содержания сопутствующего вещества и соединения, представленного формулой (1), в образце и сравнивали с содержаниями в образце, взятом в день 0.
Измерение содержания соединения, представленного формулой (1)
Содержание соединения, представленного формулой (1), измеряли с помощью метода внешнего стандарта в соответствии с Высокоэффективной Жидкостной Хроматографией в Китайской Фармакопее, Приложение V D, издание 2010 года.
Измерение содержания сопутствующего вещества
Содержание сопутствующего вещества измеряли с помощью метода нормализации площадей в соответствии с Высокоэффективной Жидкостной Хроматографией в Китайской Фармакопее, Приложение V D, издание 2010 года.
Результаты испытаний приведены в таблице 1.
Результаты испытаний исследования влияющих факторов на кристаллическую форму I соединения, представленного формулой (1)
10
98,1
0,97
10
99,0
0,60
90%±5%
10
99,0
0,56
500 лк
10
97,0
2,0
Авторы настоящего изобретения исследовали стабильность кристаллической формы I соединения, представленного формулой (1). Из результатов исследования очевидно, что содержания сопутствующего вещества и соединения, представленного формулой (1), в кристаллической форме I соединения, представленного формулой (1), по существу не изменяются при высокой температуре, при высокой влажности и в условиях освещенности. Кристаллическая форма I превосходила аморфную форму в стабильности, что показало, что кристаллическая форма I соединения, представленного формулой (1), имела относительно высокую стабильность, что являлось подходящим для изготовления, хранения и транспортировки лекарственного препарата и являлось благоприятным для обеспечения достоверности и безопасности в использовании лекарственного препарата.
Анализ 2
Стабильность кристаллической формы II настоящего соединения Образец:
Кристаллическая форма II соединения, представленного формулой (1): кристаллическую форму II получали в соответствии с примером 10.
Условия испытаний в исследовании влияющих факторов:
Высокотемпературные испытания:
(1) кристаллическую форму II соединения, представленного формулой (1), клали на сухое и чистое часовое стекло и выдерживали при 60°С в течение 10 дней. Образец брали на 10-й день. Измеряли содержания сопутствующего вещества и соединения, представленного формулой (1), в образце и сравнивали с содержаниями в образце, взятом в день 0;
(2) кристаллическую форму II соединения, представленного формулой (1), упаковывали и герметизировали при помощи полиэтиленового мешка с низкой плотностью для фармацевтического применения во внутреннем слое и полиэфир/алюминий/полиэтиленовой композитной пленки для фармацевтической упаковки во внешнем слое и выдерживали при 60°С в течение 10 дней. Образец брали на 10-й день. Измеряли содержание сопутствующего вещества и соединения, представленного формулой (1), в образце и сравнивали с содержаниями в образце, взятом в день 0.
Испытание при высокой влажности: кристаллическую форму II соединения, представленного формулой (1), клали на сухое и чистое часовое стекло и выдерживали при 25°С при относительной влажности 90%±5% в течение 10 дней. Образец брали соответственно на 10-й день. Измеряли содержания сопутствующего вещества и соединения, представленного формулой (1), в образце и сравнивали с содержаниями в образце, взятом в день 0.
Испытание освещением: кристаллическую форму II соединения, представленного формулой (1), клали на сухое и чистое часовое стекло и выдерживали при освещенности 5000 лк ± 500 лк в освещенной коробке в течение 10 дней. Образец брали на 10-й день. Измеряли содержания сопутствующего вещества и соединения, представленного формулой (1), в образце и сравнивали с содержаниями в образце, взятом в день 0.
Измерение содержания соединения, представленного формулой (1)
Содержание соединения, представленного формулой (1), измеряли с помощью метода внешнего стандарта в соответствии с Высокоэффективной Жидкостной Хроматографией в Китайской Фармакопее, Приложение V D, издание 2010 года.
Измерение содержания сопутствующего вещества
Содержание сопутствующего вещества измеряли с помощью метода нормализации площадей в соответствии с Высокоэффективной Жидкостной Хроматографией в Китайской Фармакопее, Приложение V D, издание 2010 года.
Результаты испытаний приведены в таблице 2.
Результаты испытаний исследования влияющих факторов на кристаллическую форму II соединения, представленного формулой (1)
Авторы настоящего изобретения исследовали стабильность кристаллической формы II соединения, представленного формулой (1). Из результатов исследования очевидно, что содержания сопутствующего вещества и соединения, представленного формулой (1), в кристаллической форме II соединения, представленного формулой (1), по существу не изменяются при высокой температуре, при высокой влажности и в условиях освещенности. Кристаллическая форма II превосходила аморфную форму в стабильности, что показало, что кристаллическая форма II соединения, представленного формулой (1), имела относительно высокую стабильность, что являлось подходящим для изготовления, хранения и транспортировки лекарственного препарата и являлось благоприятным для обеспечения достоверности и безопасности в использовании лекарственного препарата.
Анализ 3
Свойство защиты органов и понижения давления кристаллической формы I по настоящему изобретению при индуцированном высокосолевом состоянии у крыс линии Dahl, чувствительных к развитию гипертензии при употреблении солевой диеты (Dahl/SS).
Образец и подготовка образца:
Твердая дисперсия кристаллической формы I по настоящему изобретению (кристаллическая форма I по изобретению:PVPK30=1:8 (масс./масс.)): твердую дисперсию кристаллической формы I по настоящему изобретению формулировали с подходящим количеством стерильной воды для инъекций в суспензию, имеющую концентрации 0,03, 0,10, 0,30 и 1,00 мг/мл. Суспензию готовили перед применением каждый день.
Группа животных и модель:
Эксперимент на животных: крысы Dahl/SS мужского пола, свободные от особых патогенов (SPF), 8-9-недельные, приобретенные через Vital River Laboratories у HARLAN LABORATORIES, INC. После одной недели стандартного карантина крыс с хорошими признаками и состоянием использовали в эксперименте.
Крыс Dahl/SS случайным образом разделяли на шесть групп в соответствии с измерением артериального давления до введения: Здоровая контрольная группа (n=10),
Модельная группа (4% NaCl, n=12),
Группы лечения кристаллической формой I по настоящему изобретению, четыре группы:
Группа лечения 0,3 мг/кг/день (n=11),
Группа лечения 1 мг/кг/день (n=11),
Группа лечения 3 мг/кг/день (n=11),
Группа лечения 10 мг/кг/день (n=11),
n представляет собой число крыс.
Способ эксперимента:
Фармакодинамическую активность кристаллической формы I по настоящему изобретению in vivo оценивали на модели крыс Dahl/SS с гипертонией и повреждением почек.
За одну неделю до эксперимента давление крови крыс наблюдали дважды при помощи хвостовой манжеты для измерения артериального давления так, что крысы могли выдержать операцию мониторинга артериального давления. Давление крови крыс наблюдали один раз перед началом эксперимента и использовали в качестве основного артериального давления перед введением. Крыс случайным образом разделяли на группы в соответствии с измеренным давлением крови перед введением. Модель была создана путем кормления крыс кормом с высоким содержанием соли (AIN-93G экспериментальный корм для животного, содержащий 4% NaCl) в течение 42 дней, где крысы имели свободный доступ к пище и воде. И крысы в здоровой контрольной группе питались кормом с низким содержанием натрия.
Крысам в группах лечения кристаллической формой I по настоящему изобретению соответственно вводили кристаллическую форму I по настоящему изобретению в дозировке 0,3, 1, 3 и 10 мг/кг/день. Крысам в группах лечения давали дозы перорально через желудочный зонд два раза в день по 5 мл/кг. Крысам в модельной группе и в здоровой контрольной группе вводили в том же объеме стерильную воду для инъекций.
Измерение артериального давления (систолического артериального давления, сокращенно как SBP): кровяное давление измеряли один раз в неделю в течение шести недель. Изменения артериального давления анализировали для каждой группы.
Функциональное исследование почек и сердца: после эксперимента крыс убивали безболезненным способом. Сердце и билатеральные почки забирали для патоморфологического анализа. Повреждение почек оценивали и проводили полуколичественный анализ на основе гематоксилин-эозинового (HE) контрастного вещества. Повреждение сердца анализировали путем измерения толщины стенки левого желудочка.
Результат эксперимента:
Эффект понижения давления: очевидно, что кристаллическая форма I по изобретению показала значительный эффект снижения давления в модели и показала определенное дозозависимое соотношение (таблица 3).
SBP (мм рт.ст., значение ± стандартное отклонение)
(на 41 день)
Защита почек и сердца: в соответствии с оценкой повреждения почек, группы лечения кристаллической формой I по настоящему изобретению продемонстрировали значительную защиту от повышения степени выраженности повреждения почек (таблица 4). По сравнению с модельной группой, кристаллическая форма I по изобретению значительно уменьшает толщину стенки левого желудочка (таблица 4).
Защита от повреждений почек и повреждения сердца
В моделях гипертензии и нефроза, индуцированных высокосолевым состоянием у крыс Dahl/SS, кристаллическая форма I по изобретению показала значительный эффект снижения давления и защиту от повреждения почек.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ КОНДЕНСИРОВАННОГО ТРИЦИКЛИЧЕСКОГО СОЕДИНЕНИЯ И СООТВЕТСТВУЮЩЕГО ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ | 2020 |
|
RU2785963C1 |
| ПРОИЗВОДНЫЕ ОКСАЗОЛОХИНОЛИНОНА, ИХ ПОЛУЧЕНИЕ И СОДЕРЖАЩИЕ ИХ ЛЕКАРСТВО И ФАРМАЦЕВТИЧЕСКИЙ СОСТАВ | 1995 |
|
RU2141482C1 |
| ИНГИБИТОРЫ RMT5 | 2019 |
|
RU2814198C2 |
| ПРОТИВОВИРУСНЫЕ СРЕДСТВА ДЛЯ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ ВИЧ ИНФЕКЦИИ | 2021 |
|
RU2780103C1 |
| ПРОТИВОВИРУСНЫЕ СРЕДСТВА ДЛЯ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ ВИЧ-ИНФЕКЦИИ | 2020 |
|
RU2780101C2 |
| Бициклические производные пиридина, полезные в качестве ингибитора белков, связывающих жирные кислоты (FABP) 4 и/или 5 | 2013 |
|
RU2648247C2 |
| БИЦИКЛИЧЕСКИЕ ЛАКТАМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2827714C1 |
| НОВЫЕ ИНДОЛИЗИНОВЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2013 |
|
RU2646223C2 |
| АМИДЫ КОНДЕНСИРОВАННОГО ПИПЕРИДИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ ИОННЫХ КАНАЛОВ | 2014 |
|
RU2741810C2 |
| НОВЫЕ ЗАМЕЩЕННЫЕ ТЕТРАЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ТЕТРАГИДРОФУРАНА, ПИРРОЛИДИНА И ТЕТРАГИДРОТИОФЕНА | 2005 |
|
RU2401257C2 |
Настоящее изобретение относится к кристаллическим формам I и II соединения 2-хлор-4-[(3S,3aR)-3-циклопентил-7-(4-гидроксипиперидин-1-карбонил)-3,3a,4,5-тетрагидро-2H-пиразоло[3,4-f]хинолин-2-ил]бензонитрила формулы (I), используемого в качестве антагониста минералокортикоидного рецептора. Кристаллические формы имеют характеристические пики порошковой рентгеновской дифрактограммы, указанные в формуле изобретения. Изобретение также относится к способам получения кристаллических форм I и II и применению полученных кристаллических форм в изготовлении лекарственных средств для лечения и/или предупреждения повреждения почек или сердечно-сосудистых заболеваний. Технический результат – полученные кристаллические формы соединения формулы (I) превосходят его аморфную форму по стабильности не только при высокой температуре и влажности, но и в условиях освещенности. 9 н. и 7 з.п. ф-лы, 4 ил., 4 табл., 10 пр.
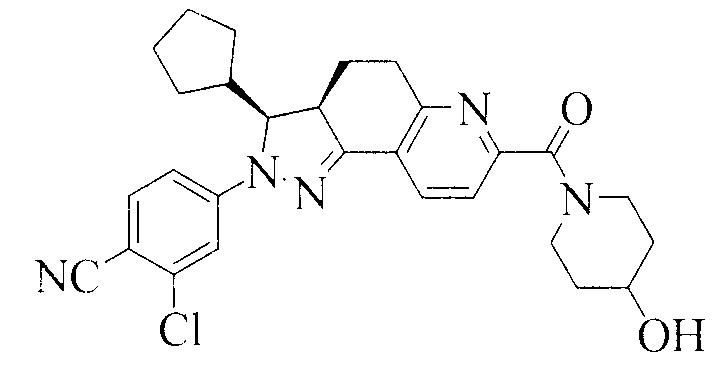
Формула 1
1. Кристаллическая форма соединения, представленного формулой (1), 2-хлор-4-[(3S,3aR)-3-циклопентил-7-(4-гидроксипиперидин-1-карбонил)-3,3а,4,5-тетрагидро-2Н-пиразоло[3,4-f]хинолин-2-ил]бензонитрила,
которая характеризуется тем, что имеет порошковую рентгеновскую дифрактограмму, содержащую следующие характеристические пики, выраженные в градусах 2θ, при измерении с применением СuКα излучения:
кристаллическая форма I: 14,8°±0,2°, 17,4°±0,2°, 19,4°±0,2°, 19,8°±0,2°;
кристаллическая форма II: 14,6°±0,2°, 19,9°±0,2°, 21,2°+0,2°, 24,6°±0,2°.
2. Кристаллическая форма соединения по п. 1, отличающаяся тем, что имеет порошковую рентгеновскую дифрактограмму, содержащую следующие характеристические пики, выраженные в градусах 2θ, при измерении с применением СuКα излучения:
кристаллическая форма I: 14,8°±0,2°, 16,9°±0,2°, 17,4°±0,2°, 19,4°±0,2°, 19,8°±0,2°, 26,2°±0,2°;
кристаллическая форма II: 14,6°±0,2°, 18,0°±0,2°, 18,7°±0,2°, 19,9°±0,2°, 21,2°±0,2°, 24,6°±0,2°.
3. Кристаллическая форма соединения по п. 1 или 2, отличающаяся тем, что имеет порошковую рентгеновскую дифрактограмму, содержащую следующие характеристические пики, выраженные в градусах 2θ, при измерении с применением CuКα излучения:
кристаллическая форма I: 9,8°±0,2°, 12,9°±0,2°, 14,8°±0,2°, 15,4°±0,2°, 16,9°±0,2°, 17,4°±0,2°, 19,4°±0,2°, 19,8°±0,2°, 22,6°±0(2°, 26,2°±0,2°;
кристаллическая форма II: 4,5°±0,2°, 9,0°±0,2°, 12,2°±0,2°, 14,0°±0,2°, 14,6°±0;2°, 18,0°±0,2°, 18,7°±0,2°, 19,9°±0,2°, 21,2°±0,2°, 24,6°±0,2°.
4. Способ получения кристаллической формы соединения по пп. 1-3, где кристаллическая форма представляет собой кристаллическую форму I и указанный способ включает следующие стадии:
добавление соединения, представленного формулой (1) в безводный низший спирт, ацетонитрил, смешанный растворитель из этилацетата и этанола, смешанный растворитель из метанола и тетрагидрофурана или смешанный растворитель из ацетонитрила и ацетона, нагревание полученного раствора до тех пор, пока он не станет прозрачным, последующее охлаждение полученного раствора для осаждения твердого вещества и фильтрование и сушку образовавшегося твердого вещества.
5. Способ получения кристаллической формы соединения по пп. 1-3, где кристаллическая форма представляет собой кристаллическую форму I и указанный способ включает следующие стадии:
растворение соединения, представленного формулой (1) в ацетоне, добавление полученного раствора по каплям в н-гептан и фильтрование полученной смеси.
6. Способ получения кристаллической формы соединения по пп. 1-3, где кристаллическая форма представляет собой кристаллическую форму II и указанный способ включает следующие стадии:
помещение соединения, представленного формулой (1), в низший спирт для его растворения, а затем добавление полученного раствора по каплям к воде, фильтрование полученной смеси и сушка отфильтрованного вещества под вакуумом.
7. Способ получения кристаллической формы соединения по пп. 1-3, где кристаллическая форма представляет собой кристаллическую форму II и указанный способ включает следующие стадии:
промывание соединения, представленного формулой (1), смешанным раствором из воды и ацетонитрила, фильтрование полученной смеси и сушка отфильтрованного вещества под вакуумом.
8. Способ по любому из пп. 4-7, где соединение, представленное формулой (I), получают способом, содержащим стадии:
(9) хирального разделения этил 2-(3-хлор-4-цианофенил)-3-циклопентил-3,3а,4,5-тетрагидро-2Н-пиразоло[3,4-f]хинолин-7-карбоксилата для получения (3S,3aR)-этил 2-(3-хлор-4-цианофенил)-3-циклопентил-3,3а,4,5-тетрагидро-2Н-пиразоло[3,4-f]хинолин-7-карбоксилата;
(10) гидролиза (3S,3aR)-этил 2-(3-хлор-4-цианофенил)-3-циклопентил-3,3а,4,5-тетрагидро-2Н-пиразоло[3,4-f]хинолин-7-карбоксилата для получения (3S,3aR)-2-(3-хлор-4-цианофенил)-3-циклопентил-3,3а,4,5-тетрагидро-2Н-пиразоло[3,4-f]хинолин-7-карбоновой кислоты;
(11) проведения реакции конденсации (3S,3aR)-2-(3-хлор-4-цианофенил)-3-циклопентил-3,3а,4,5-тетрагидро-2Н-пиразоло[3,4-f]хинолин-7-карбоновой кислоты и 4-гидроксипиперидина для получения соединения, представленного формулой (1).
9. Способ по любому из пп. 4-7, где соединение, представленное формулой (I), получают способом, содержащим стадии:
(1) проведения реакции конденсации и реакции присоединения 1,3-циклогександиона, ацетата аммония и акролеина для получения 5-оксо-5,6,7,8-тетрагидрохинолина;
(2) проведения реакции окисления 5-оксо-5,6,7,8-тетрагидрохинолина для получения 5-оксо-5,6,7,8-тетрагидрохинолин-N-оксида;
(3) проведения реакции замещения 5-оксо-5,6,7,8-тетрагидрохинолин-N-оксида в присутствии хлорида N,N-диметилкарбаминовой кислоты и триметилсилилцианида для получения 5-оксо-5,6,7,8-тетрагидрохинолин-2-карбонитрила;
(4) гидролиза и этерификации 5-оксо-5,6,7,8-тетрагидрохинолин-2-карбонитрила для получения этил 5-оксо-5,6,7,8-тетрагидрохинолин-2-карбоксилата;
(5) проведения реакции конденсации этил 5-оксо-5,6,7,8-тетрагидрохинолин-2-карбоксилата и циклопентанкарбальдегида для получения (Е)-этил 6-циклопентилметилен-5-оксо-5,6,7,8-тетрагидрохинолин-2-карбоксилата;
(6) проведения реакции замещения 2-хлор-4-фторбензонитрила и гидразингидрата для получения 2-хлор-4-гидразинбензонитрила;
(7) проведения реакции образования соли 2-хлор-4-гидразинбензонитрила и соляной кислоты для получения гидрохлорида 2-хлор-4-гидразинбензонитрила;
(8) проведения реакции конденсации (Е)-этил 6-циклопентилметилен-5-оксо-5,6,7,8-тетрагидрохинолин-2-карбоксилата и гидрохлорида 2-хлор-4-гидразинбензонитрила для получения этил 2-(3-хлор-4-цианофенил)-3-циклопентил-3,3а,4,5-тетрагидро-2Н-пиразоло[3,4-f]хинолин-7-карбоксилата;
(9) хирального разделения этил 2-(3-хлор-4-цианофенил)-3-циклопентил-3,3а,4,5-тетрагидро-2Н-пиразоло[3,4-f]хинолин-7-карбоксилата для получения (3S,3aR)-этил 2-(3-хлор-4-цианофенил)-3-циклопентил-3,3а,4,5-тетрагидро-2Н-пиразоло[3,4-f]хинолин-7-карбоксилата;
(10) гидролиза (3S,3aR)-этил 2-(3-хлор-4-цианофенил)-3-циклопентил-3,3а,4,5-тетрагидро-2Н-пиразоло[3,4-f]хинолин-7-карбоксилата для получения (3S,3aR)-2-(3-хлор-4-цианофенил)-3-циклопентил-3,3а,4,5-тетрагидро-2Н-пиразоло[3,4-f]хинолин-7-карбоновой кислоты;
(11) проведения реакции конденсации (3S,3aR)-2-(3-хлор-4-цианофенил)-3-циклопентил-3,3а,4,5-тетрагидро-2Н-пиразоло[3,4-f]хинолин-7-карбоновой кислоты и 4-гидроксипиперидина для получения соединения, представленного формулой (1).
10. Фармацевтическая композиция, отличающаяся тем, что содержит кристаллическую форму соединения, представленного формулой (1) по пп. 1, 2 или 3, и фармацевтически приемлемый носитель, где указанная кристаллическая форма представляет собой кристаллические формы I, II или их комбинацию.
11. Применение кристаллической формы соединения, представленного формулой (1) по пп. 1, 2 или 3, для изготовления лекарственного средства для лечения и/или предупреждения повреждения почек или сердечно-сосудистых заболеваний, где указанная кристаллическая форма представляет собой кристаллические формы I, II или их комбинацию.
12. Применение по п. 11, отличающееся тем, что сердечно-сосудистые заболевания включают повреждение сердца, гипертонию, сердечную недостаточность, инфаркт миокарда, стенокардию, сердечную гипертрофию, миокардит, фиброз сердца и кровеносных сосудов, дисфункцию барорецептора или аритмию.
13. Способ лечения и/или предупреждения повреждения почек или сердечно-сосудистых заболеваний, где указанный способ включает введение нуждающемуся в этом субъекту терапевтически эффективного количества кристаллической формы соединения, представленного формулой (1) по пп. 1, 2 или 3, где кристаллическая форма представляет собой кристаллические формы I, II или их комбинацию.
14. Способ по п. 13, отличающийся тем, что указанные сердечнососудистые заболевания включают повреждение сердца, гипертонию, сердечную недостаточность, инфаркт миокарда, стенокардию, сердечную гипертрофию, миокардит, фиброз сердца и кровеносных сосудов, дисфункцию барорецептора или аритмию.
15. Кристаллическая форма соединения, представленного формулой (1) по пп. 1, 2 или 3, предназначенная для лечения и/или предупреждения повреждения почек или сердечно-сосудистых заболеваний, где кристаллическая форма представляет собой кристаллические формы I, II или их комбинацию.
16. Кристаллическая форма по п. 15, отличающаяся тем, что указанные сердечно-сосудистые заболевания включают повреждение сердца, гипертонию, сердечную недостаточность, инфаркт миокарда, стенокардию, сердечную гипертрофию, миокардит, фиброз сердца и кровеносных сосудов, дисфункцию барорецептора или аритмию.
| СПОСОБ, АППАРАТУРА И ЭЛЕКТРОННОЕ УСТРОЙСТВО ДЛЯ ОБРАБОТКИ НАЛОЖЕНИЯ ОКНА ОТОБРАЖЕНИЯ | 2021 |
|
RU2808678C1 |
| Дорожная спиртовая кухня | 1918 |
|
SU98A1 |
| ПИРАЗОЛХИНОЛИНЫ С ИММУНОМОДУЛИРУЮЩЕЙ АКТИВНОСТЬЮ | 2003 |
|
RU2328496C2 |

