Изобретение касается производных 3,3а,4,5-тетра-гидро-1Н-оксазоло[3,4-а] хинолин-1-она, их получения, а также терапевтического использования.
Известны (EP 0322263) производные 3,3а,4,5-тетрагидро-1Н-оксазоло[3,4-а] хинолин-1-она, пригодные для использования в качестве антидепрессантов.
Предусмотренные настоящим изобретением соединения отвечают общей формуле I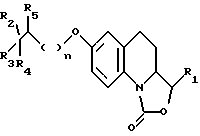
в которой n равно 0 или 1;
R1 представляет собой атом водорода, либо этенильную, метильную, этильную, фенильную, гидроксиметильную или метоксиметильную группу; а также
либо R2 обозначает метильную, трифторметильную или цианогруппу,
R3 соответствует атому водорода, либо гидроксильной или бензилоксигруппе, а
R4 и R5 являются атомами водорода;
либо R2 и R4 вместе образуют группу -(CH2)4-,
R3 представляет собой гидроксильную группу, а
R5 обозначает атом водорода;
либо R2 и R5 вместе образуют группу -O-(CH2)3-, а
R3 и R4 являются атомами водорода;
либо R2 и R5 вместе образуют группу -(CH5)4-,
R3 соответствует гидроксильной группе, а
R4 представляет собой атом водорода.
Предусмотренные настоящим изобретением соединения могут находиться в форме различных изомеров, в том числе в форме энантиомеров и диастереоизомеров. Настоящее изобретение охватывает указанные различные формы, а также их смеси, включая рацемические смеси.
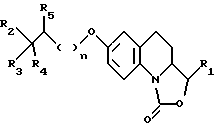
Соединения, отвечающие формуле (I), можно получить в соответствии со способом, представленным в Приложении 1, причем указанный способ предполагает, что соединение, описываемое формулой II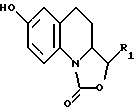
в которой R1 представляет собой атом водорода либо этенильную, метильную, фенильную, гидроксиметильную или метоксиметильную группу,
обрабатывают соединением, соответствующим формуле III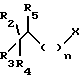
в которой X обозначает атом галогена, либо лабильную группу, такую как мезилокси- или тозилоксигруппа,
с получением соединения, отвечающего формуле I, в которой R1 соответствует приведенным выше обозначениям, после чего восстанавливают указанное соединение, описываемое формулой I, в которой R1 является этенильной группой, с получением соединения, отвечающего формуле I, в которой R1 представляет собой этильную группу. Соединения, предусмотренные формулой II, в которой R1 представляет собой атом водорода, этенильную, метильную или фенильную группу, могут быть получены из этил 2-формил-6-метокси-1,2,3,4-тетрагидрохинолин-1-карбоксилата, известного соединения, получение которого описано в ЕР 0322263.
В том случае, если R1 обозначает атом водорода, указанный способ состоит в том, что:
- этил 2-формил-6-метокси-1,2,3,4-тетрагидрохинолин-1-карбоксилат обрабатывают восстанавливающим агентом, таким как борогидрид натрия или борогидрид калия,
- циклизируют образующееся при этом соединение, приводя его во взаимодействие с основанием, таким как метоксид натрия, и, наконец,
- подвергают деметилированию 7-метокси-3,3а,4,5-тетрагидро-1Н- оксазоло[3,4-а]хинолин-1-он, отвечающий формуле IV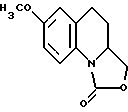
В том случае, если R1 является этенильной, метильной или фенильной группой, указанный способ заключается в том, что:
- этил 2-формил-6-метокси-1,2,3,4-тетрагидрохинолин-1-карбоксилат обрабатывают органомагниевым соединением, описываемым формулой R1MgX, где R1 представляет собой этенильную, метильную или фенильную группу, а X обозначает атом галогена,
- циклизируют образующееся при этом соединение, приводя его во взаимодействие с основанием, таким как метоксид натрия, и, наконец,
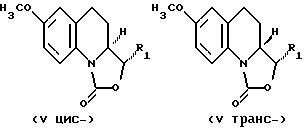
- подвергают деметилированию 7-метокси-3,3а,4,5-тетрагидро-1Н-оксазоло[3,4-а]хинолин-1-он, отвечающий формуле V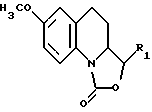
в которой R1 соответствует приведенным выше обозначениям. Соединения, предусмотренные формулой II, в которой R1 представляет собой гидроксиметильную или метоксиметильную группу, могут быть получены из 3-этенил-7-гидрокси-3,3а, 4,5-тетрагидро-1Н-оксазоло[3,4-а] хинолин-1-она, соединения, отвечающего формуле II, в которой R1 является этенильной группой, следующим образом:
- защищают соответствующую гидроксильную группу, в результате чего образуется соединение, описываемое формулой VI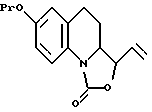
в которой Pr обозначает защитную группу, такую как бензильная группа;
- обрабатывают указанное соединение озоном, а затем - восстанавливающим агентом, таким как борогидрид натрия, с получением соединения, описываемого формулой Vll, которое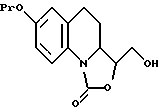
либо лишают защитных групп с получением соединения, предусмотренного формулой II, где R1 является гидроксиметильной группой, либо обрабатывают диметил сульфатом с образованием соединения, соответствующего формуле Vlll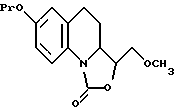
с которого затем снимают защитные группы с получением соединения, соответствующего формуле II, где R1 представляет собой метоксиметильную группу.
Соединения, отвечающие формуле I, в которой R1 обозначает этенильную, метильную, этильную, гидроксиметильную или метоксиметильную группу, могут существовать в форме цис- и транс-изомеров,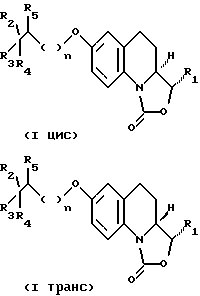
которые в свою очередь получают из цис- и транс-изомеров соответствующих соединений, предусмотренных формулой II, причем последние получают описанным выше способом после разделения с помощью хроматографии цис- и транс-изомеров указанного производного, предусмотренного формулой V.
Соединения, отвечающие формуле I, в которой
R1 представляет собой этенильную группу, а
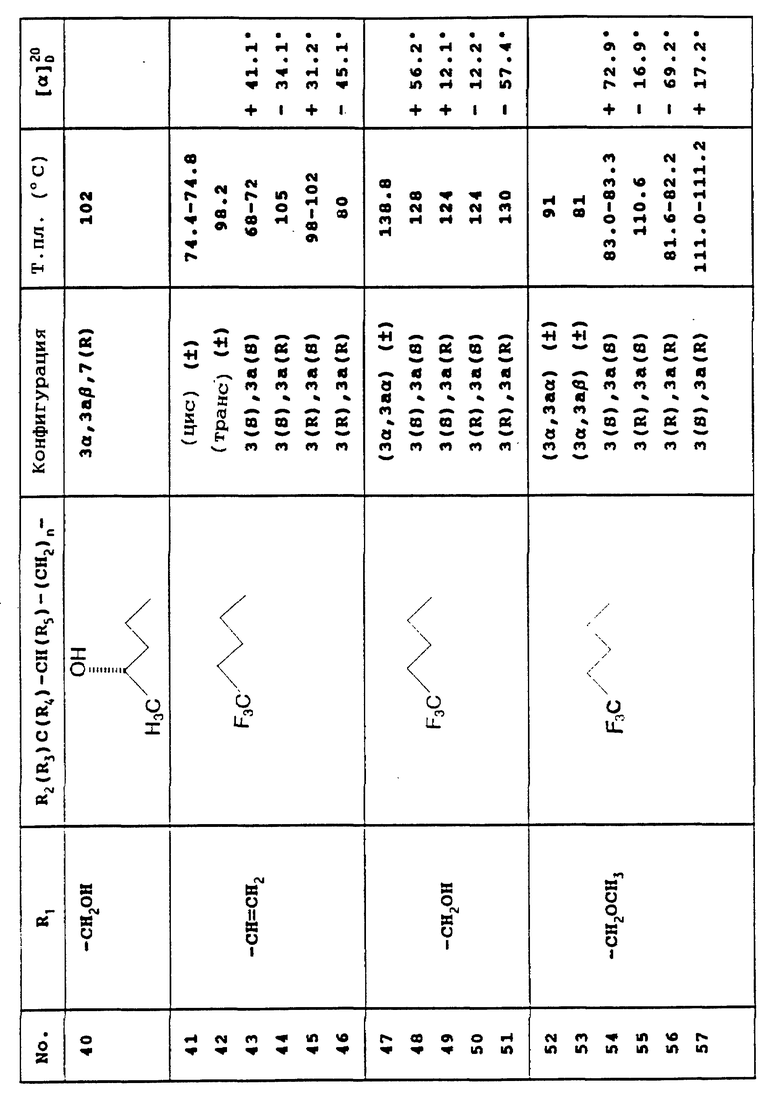
R3 - бензилоксигруппу;
либо в альтернативном случае
R1 является гидроксиметильной или метоксиметильной группой, а
R3 - гидроксильной или бензилоксигруппой; а
радикалы R4 и R5 обозначают атомы водорода,
могут быть получены также в соответствии со способом, представленным в Приложении 2, причем указанный способ предусматривает, что
- 3-этенил-7-гидрокси-3,3а, 4,5-тетрагидро-1Н- оксазоло[3,4-а]хинолин-1-он, соединение, отвечающее формуле II, в которой R1 является этенильной группой, обрабатывают соединением, описываемым формулой R2CH(OH)-(CH2)-(CH2)nX, где X представляет собой атом галогена либо лабильную группу, такую как тозилокси- или мезилоксигруппа,
- обрабатывают полученное таким образом соединение, отвечающее формуле IX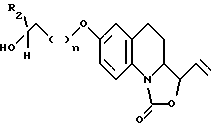
бензил галогенидом,
- приводят образующееся при этом соединение, описываемое формулой X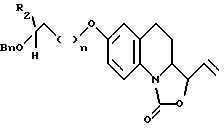
во взаимодействие с озоном, а затем - с восстанавливающим агентом, таким как борогидрид натрия, с получением производного, отвечающего формуле XI, которое затем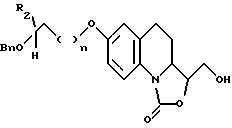
либо лишают защитных групп с образованием соответствующего соединения, предусмотренного формулой (I), где R1 обозначает гидроксиметильную группу, а R3 - гидроксильную группу,
- либо обрабатывают диметил сульфатом с получением соединения, описываемого формулой Xll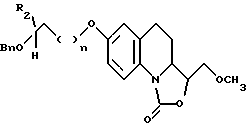
- из которого в дальнейшем удаляют защитную группу с образованием соответствующего соединения, предусмотренного формулой I, в которой R1 представляет собой метоксиметильную группу, а R3 является гидроксильной группой.
Указанные энантиомеры и диастереоизомеры соединений, отвечающих формуле I, получают из энантиомеров и диастереоизомеров соединений, описываемых формулой II, и/или из энантиомеров соединений, соответствующих формуле III.
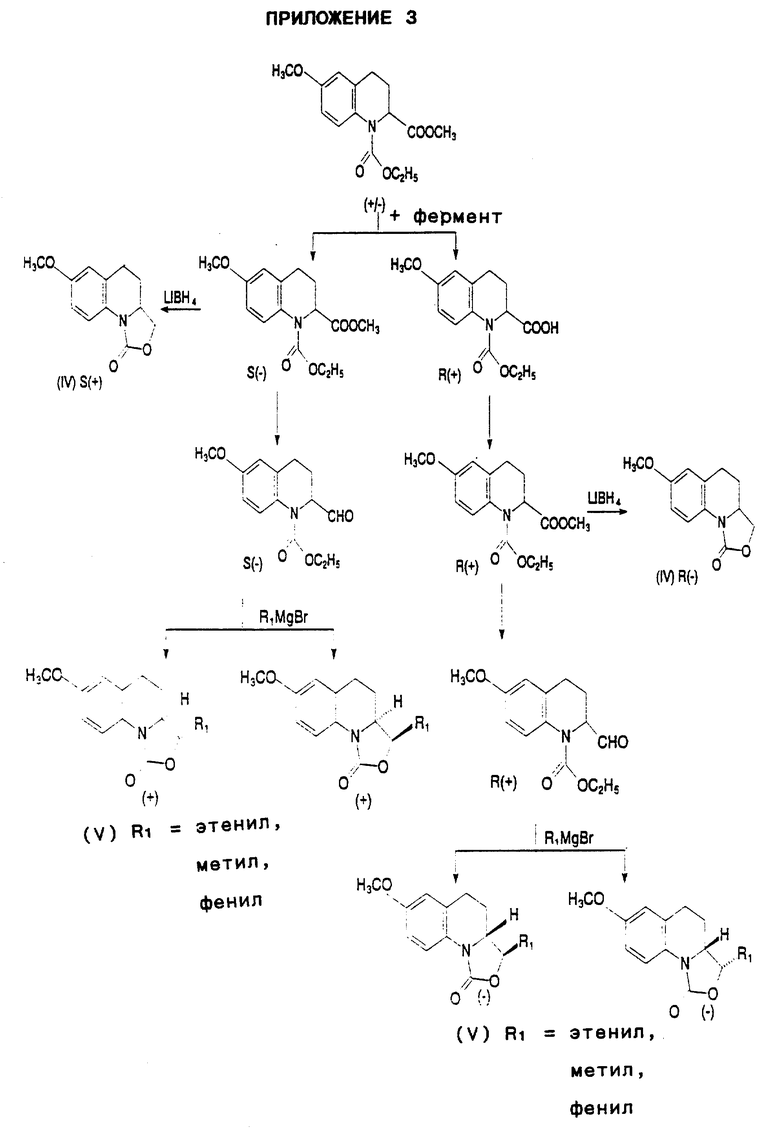
В свою очередь, указанные энантиомеры и диастереоизомеры соединений, предусмотренных формулой II, получают из энантиомеров и диастереоизомеров соединений, отвечающих формуле IV или V, выделяемых из рацемического этил 6-метокси-2-метокси-карбонил-1,2,3,4-тетрагидрохинолин-1-карбоксилата в соответствии со способом, представленным в Приложении 3.
Указанный способ предполагает разделение с помощью энзиматического гидролиза энантиомеров этил 6-метокси-2-метоксикарбонил-1,2,3,4-тетрагидрохинолин-1-карбоксилата, в ходе которого
- вышеупомянутое рацемическое соединение, находящееся в буферном растворе, таком как смесь однозамещенного фосфата калия и двузамещенного фосфата натрия, либо в двухфазной среде, такой как смесь толуола и буферного раствора, обрабатывают энзимным экстрактом, таким как эстераза печени свиньи, ацетоновые порошки печени лошади, свиньи, быка или кролика, и, в особенности, ацетоновый порошок печени овцы (выпускаемая Sigma), и
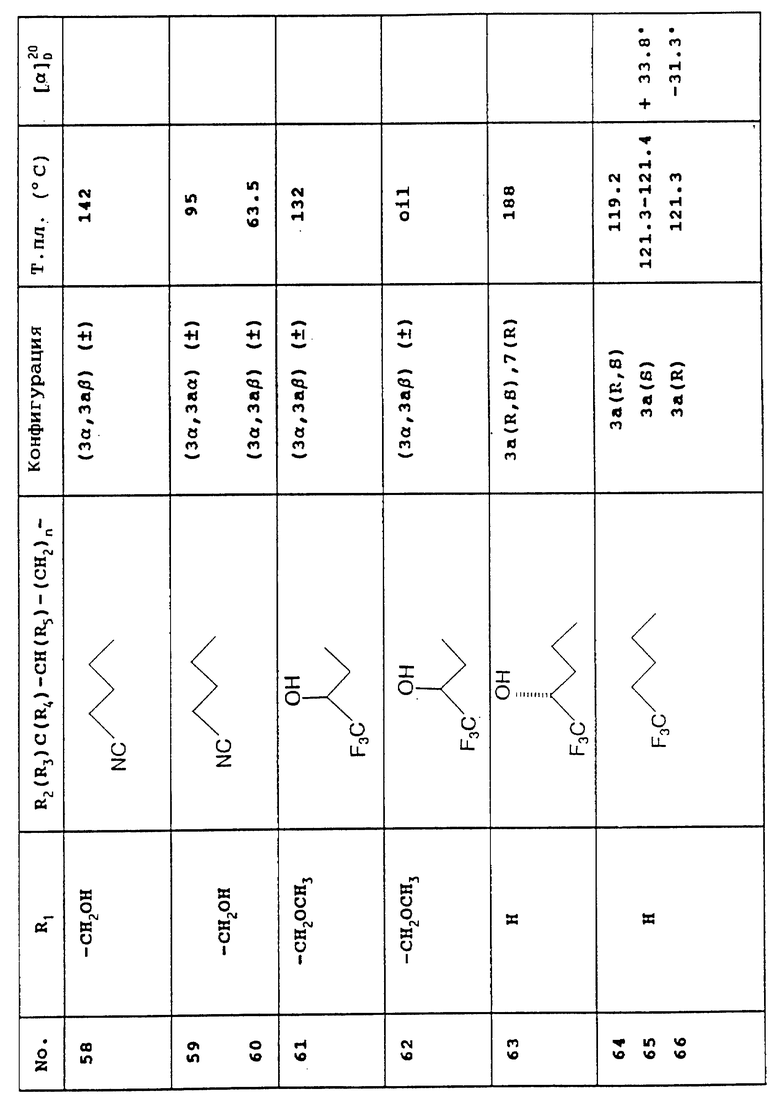
- с помощью экстракции отделяют этил S-(-)-6-метокси-2-метокси- карбонил-1,2,3,4-тетрагидрохинолин-1-карбоксилат от этил R(+)-2-карбокси-6-метокси-1,2,3,4-тетрагидрохинолин-1-карбоксилата.
В том случае, если этил R-(+)-2-карбокси-6-метокси-1,2,3,4-тетрагидрохинолин-1-карбоксилат обрабатывают тионил хлоридом в растворителе, таком как толуол, а затем - метанолом, получают этил R-(+)-6-метокси-2- метоксикарбонил-1,2,3,4-тетрагидрохинолин-1-карбоксилат.
В том случае, если этил R-(+)- и S-(-)-6-метокси-2-метоксикарбонил-1,2,3,4-тетрагидрохинолин-1- карбоксилат обрабатывают борогидридом лития, получают, соответственно, R-(-)- и S-(+)-энантиомеры 7-метокси-3,3а,4,5-тетрагидро-1Н-оксазоло-[3,4-а]- хинолин-1-она (IV).
В том случае, если этил R-(+)- и S-(-)-6-метокси-2-метоксикарбонил-1,2,3,4-тетрагидрохинолин-1- карбоксилат обрабатывают гидридом диизобутилалюминия в растворителе, таком как толуол, получают, соответственно, этил R-(+)- и S-(-)-2-формил-6-метокси-1,2,3,4-тетрагидрохинолин-1- карбоксилат, который при приведении его во взаимодействие с органомагниевым соединением, отвечающим формуле R1MgX, в которой
R1 представляет собой этенильную, метильную или фенильную группу, а
X является атомом водорода, в растворителе, таком как тетрагидрофуран, затем приводят во взаимодействие с метоксидом натрия в растворителе, таком как толуол, после чего подвергают хроматографическому разделению с получением, с одной стороны, (-) диастереоизомеров соединения, отвечающего формуле V, в конфигурации [3(R),3a(R)] и [3(S),3a(R)], а с другой стороны, соответствующих (+) диастереоизомеров в конфигурации [3(S), 3a(S)] и [3(R),3a(S)].
Для того, чтобы получить используемый в качестве исходного вещества рацемический этил 6-метокси-2-метоксикарбонил-1,2,3,4-тетрагидрохинолин-1-карбоксилат:
- 6-метоксихинолин приводят во взаимодействие с цианидом калия или цианидом триметилсилила, а также с бензоил хлоридом в присутствии растворителя, такого как дихлорметан,
- образующийся при этом 1-бензоил-2-циано-6-метокси-1,2-дигидрохинолин приводят во взаимодействие с бромистоводородной кислотой в уксусной кислоте, затем - с водным нашатырным спиртом и, наконец, - с уксусной кислотой,
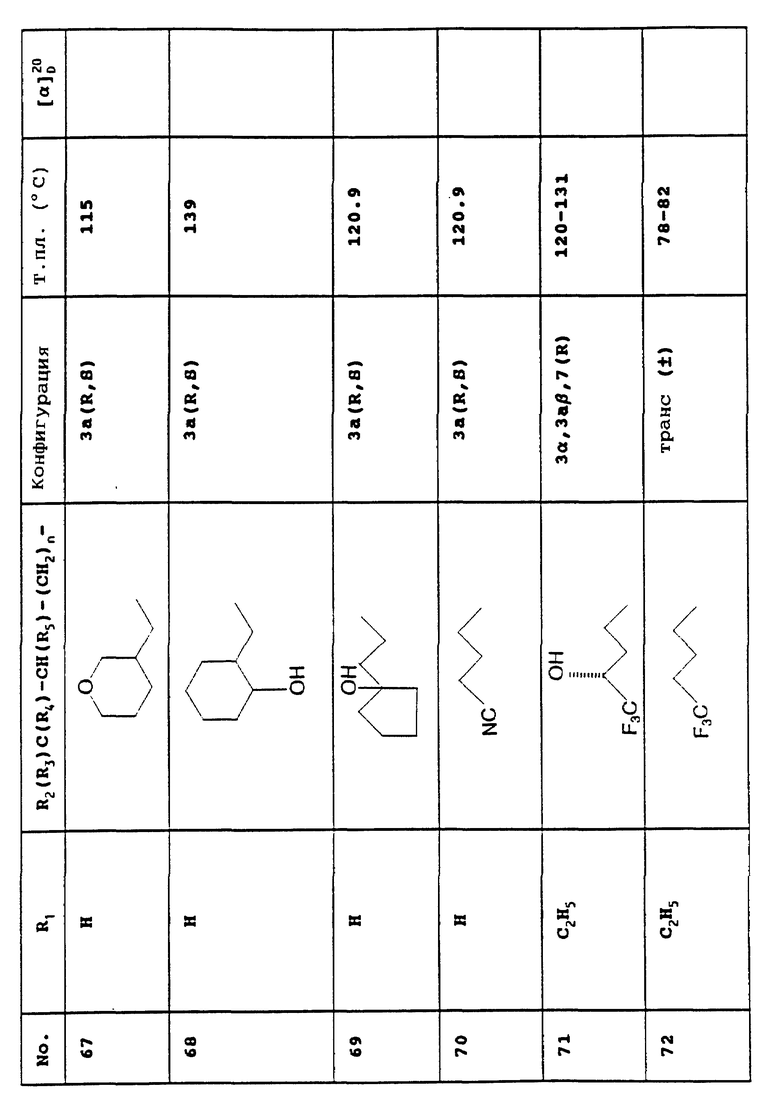
- полученный таким образом 2-карбокси-6-метоксихинолин обрабатывают тионил хлоридом в растворителе, таком как толуол, а затем - метанолом с образованием 6-метокси-2-метоксикарбонил- хинолина,
- указанное соединение восстанавливают в атмосфере водорода в присутствии оксида платины и хлористоводородного спирта в метаноле с получением 6-метокси-2-метоксикарбонил-1,2,3,4-тетрагидохинолина,
- указанное соединение обрабатывают этил хлороформатом в растворителе, таком как дихлорметан, в присутствии карбоната калия.
Приведенные далее Примеры иллюстрируют настоящее изобретение.
Пример 1.
[3α,3aβ,7(R)] -3-Этенил-7-[4.4.4-трифтор-3-гидроксибутокси] -3,3а,4,5-тетрагидро-1 H-оксазоло[3,4-а] хинолин-1-он
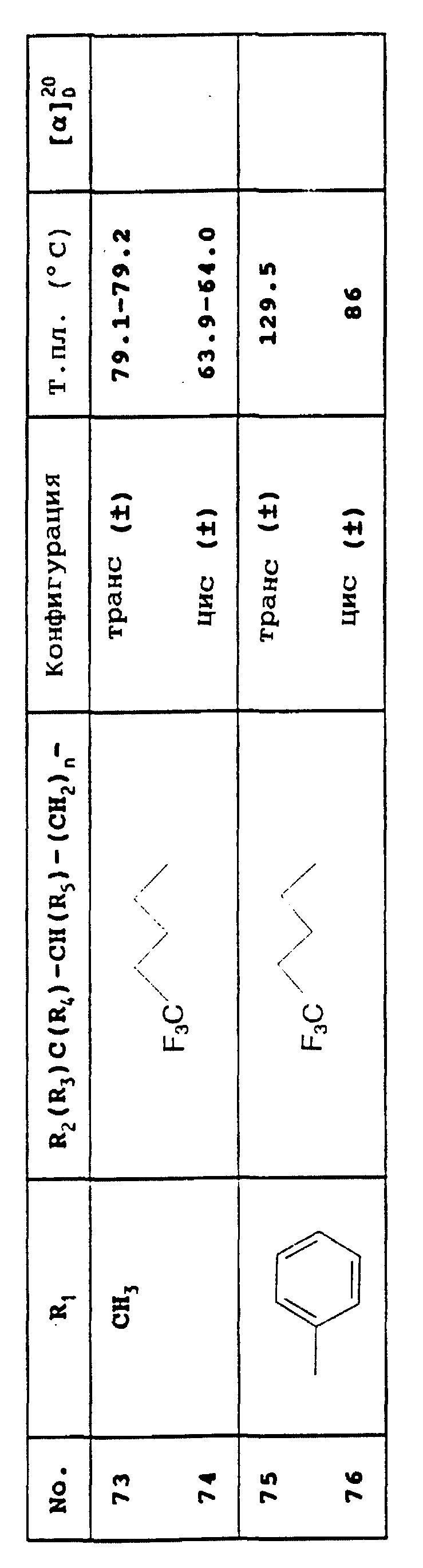
1.1. Цис- и транс-(±)-3-этенил-7-метокси-3,3а,4,5- тетрагидро-1Н-оксазоло[3,4-а] хинолин-1-он
К перемешиваемому раствору 116,3 г (0,442 М) этил 2-формил-6-метокси-1,2,3,4-тетрагидрохинолин-1-карбоксилата в 800 мл тетрагидрофурана, охлажденному до температуры -30oC, в атмосфере аргона в течение 30 мин добавляют 486 мл (0,486 М) 1 М бромида винилмагния. Указанную реакционную среду продолжают перемешивать в течение 2 ч, после чего добавляют холодный насыщенный водный раствор хлорида аммония. Полученную смесь дважды экстрагируют этил ацетатом, соответствующие экстракты промывают водой, высушивают над сульфатом натрия, после чего выпаривают растворитель при пониженном давлении. Образующееся при этом масло вновь растворяют в 340 мл толуола и выдерживают при температуре дефлегмации для удаления следов воды. Затем при 90oC добавляют 1 мл 10%-ного раствора метоксида натрия в метаноле и вновь выдерживают указанную смесь при температуре дефлегмации, отгоняя образующийся этанол. Выпаривают полученную смесь до сухого состояния, выделенный остаток отбирают в этил ацетат и промывают водой, соответствующую органическую фазу высушивают над сульфатом натрия, после чего выпаривают растворитель при пониженном давлении. Выделенный продукт подвергают хроматографии на силикагельной колонке, элюция смесью гептана и этил ацетата в соотношении 4:1, с выходом 32,9 г транс-производного поименованного соединения (т.пл.: 100oC) и 13,4 г соответствующего цис-производного (т.пл.: 134oC).
1.2. Транс-(±)-3-этенил-7-гидрокси-3,3а, 4,5-тетрагидро-1H- оксазоло[3,4-а]хинолин-1-он
К раствору 24,6 г (0,1 М) транс-(±)-3-этенил-7-метокси-3,3а,4,5-тетрагидро-1H- оксазоло[3,4-а]хинолин-1-она в 280 мл дихлорметана по каплям добавляют 19 мл (0,20 М) трибромида бора при температуре 0oC, а затем, через 1 ч, - насыщенный раствор бикарбоната натрия до нейтрального pH. Фильтруют указанную смесь, а соответствующий фильтрат экстрагируют дихлорметаном, содержащим 10% метанола. Полученную органическую фазу промывают водой и высушивают над сульфатом натрия, после чего при пониженном давлении выпаривают растворитель. Образующийся при этом твердый остаток растирают в смеси дихлорметана и метанола в соотношении 1:1. Фильтруют полученную смесь и высушивают ее с конечным выходом 21,4 г продукта.
Т.пл.: 216oC.
Используя в качестве исходного вещества цис-(±)-3-этенил-7-метокси-3,3а, 4,5- тетрагидро-1H-оксазоло[3,4-а] хинолин-1-он, получают цис-(±)-3- этенил-7-гидрокси-3,3а,4,5-тетрагидро-1H-оксазоло[3,4-а]хинолин-1-он.
Т.пл.: 250oC
1.3. [3α,3aβ,7(R)] 3-Этенил-7-[4,4,4-трифтор-3- гидроксибутокси]-3,3а, 4,5-тетрагидро-1Н-оксазоло[3,4-а]хинолин-1-он
К раствору 21,2 г (0,092 М) транс-(±)-3-этенил-7-гирокси- 3,3а,4,5-тетрагидро-1Н-оксазоло[3,4-а] хинолин-1-она в 150 мл ацетонитрила в атмосфере аргона при комнатной температуре добавляют 25,4 г (0,183 М) карбоната калия, а затем - 34,9 г (0,138 М) 1-иод-3(R)-гидрокси-4,4,4-трифторбутана. Через 4 ч указанную смесь разбавляют дихлорметаном и промывают водой. Соответствующую органическую фазу высушивают над сульфатом натрия и при пониженном давлении выпаривают растворитель. Образующееся при этом масло очищают с помощью хроматографии на силикагельной колонке, элюция хлороформом, содержащим 0-3% метанола. После кристаллизации из смеси ацетона и диизопропилового эфира получают 30 г продукта.
Т.пл.: 135,8oC.
Пример 2.
[3α,3aβ,7(R)] -3-Гидроксиметил-7-[4.4.4-трифтор-3-бензилоксибутокси] -3,3а,4,5- тетрагидро-1Н-оксазоло[3,4-а]хинолин-1-он
2.1. [3α,3aβ,7(R)] -3-Этенил-7-[4,4,4-трифтор-3-бензилоксибутокси]-3,3а, 4,5-тетрагидро- 1Н-оксазоло[3,4-а]хинолин-1-он
К раствору 30,0 г (840 мМ) [3α,3aβ,7(R)] -3-этенил-7-[4,4,4-трифтор-3-гидроксибутокси] -3,3а,4,5-тетрагидро-1H- оксазоло[3,4-а]хинолин-1-она в 300 мл толуола добавляют раствор 13,4 г гидроксида натрия в 13,4 мл воды, 2,7 г (8,4 мМ) бромида тетрабутиламмония, а затем - 43,1 г (0,252 М) бензил бромида. Указанную смесь перемешивают в течение 2 ч при комнатной температуре, после чего экстрагируют этил ацетатом, промывают водой и высушивают над сульфатом натрия, а растворитель выпаривают при пониженном давлении. Образующийся при этом масляный остаток очищают с помощью хроматографии на силикагельной колонке, элюция смесью гептана и хлороформома в соотношении 1:1. Получают 35 г продукта в форме масла.
2.2. [3α,3aβ,7(R)] -3-Гидроксиметил-7-[4,4,4-трифтор-3-бензилоксибутокси]- 3,3а,4,5-тетрагидро-1H-оксазоло[3,4-а]хинолин-1-он)
Через раствор 31,5 г (70,4 мМ) [3α,3aβ, (R)]-3-этенил-7- [4,4,4-трифтор-3-бензилоксибутокси] -3,3а,4,5-тетрагидро-1Н- оксазоло[3,4-а]хинолин-1-она в 515 мл дихлорметана и 780 мл метанола в течение 3 ч продувают озон при температуре -30oC. Затем с помощью струи азота удаляют использованный озон и, поддерживая температуру -30oC, добавляют 26,8 г (0,704 М) борогидрида натрия. Через 5 мин добавляют 21,8 г (0,352 М) диметил сульфида и позволяют указанной смеси нагреться до комнатной температуры. Затем образовавшуюся смесь промывают водой, соответствующую органическую фазу высушивают над сульфатом натрия и концентрируют полученный раствор при пониженном давлении. Выделенный продукт очищают с помощью хроматографии на силикагельной колонке, элюция дихлорметаном, содержащим 0-5% метанола. В результате перекристаллизации из диэтилового эфира получают 26,5 г продукта.
Т.пл.: 111,6oC.
Пример [3α,3aβ,7(R)] -3-Метоксиметил-7-[4,4,4-трифтор-3-бензилоксибутокси]-3,3а,4,5- тетрагидро-1H-оксазоло[3,4-а]хинолин-1-он
К раствору 7,8 г (17 мМ) [3α,3aβ,7(R)] -3-гидроксиметил-7-[4,4,4- трифтор-3-бензилоксибутокси]-3,3а,4,5-тетрагидро-1H- оксазоло[3,4-а]хинолин-1-она в 85 мл толуола добавляют 0,54 г (1,7 мМ) бромида тетрабутиламмония, а затем - 6,4 г (51 мМ) диметил сульфата. После этого в течение 30 мин по каплям добавляют раствор 2,7 г (67 мМ) гидроксида натрия в 2,7 мл воды. Указанную реакционную среду перемешивают в течение 30 мин, после чего разбавляют этил ацетатом, экстрагируют соответствующую органическую фазу, промывают ее водой и высушивают над сульфатом натрия, а растворитель выпаривают при пониженном давлении. С помощью хроматографии на силикагельной колонке, элюция смесью гептана и этил ацетата в соотношении 1:1, выделяют 6,2 г продукта в форме масла.
Пример 4. [3α,3aβ,7(R)] -3-Метоксиметил-7- [4.4.4-трифтор-3- гидроксибутокси]-3,3а,4,5-тетрагидро-1H-оксазоло[3,4-а]хинолин-1-он
Раствор 2,8 г (6 мМ) [3α,3aβ,7(R)] 3-метоксиметил-7-[4,4,4-трифтор-3-бензилоксибутокси] -3,3а,4,5- тетрагидро-1Н-оксазоло[3,4-а]хинолин-1-она подвергают гидрогенированию в течение 16 ч в 30 мл этанола, в присутствии следов хлористоводородной кислоты и 0,6 г 10%-ного палладиево-угольного катализатора, содержащего 50% воды. Указанную смесь фильтруют через силикагель, а растворитель выпаривают при пониженном давлении. Выделенный продукт очищают с помощью хроматографии на силикагельной колонке, элюция смесью гептана и этил ацетата 1:1. В результате перекристаллизации из диэтилового эфира получают 1,4 г продукта.
Т.пл.: 94,8oC.
Пример 5. [3α,3aβ,7(R)] -7-3-гидроксибутокси-3-гидроксиметил-3,3а,4,5-тетрагидро-1-H- оксазоло[3,4-а]хинолин-1-он
5.1. Транс-7-бензилокси-3-этенил-3,3а,4,5-тетрагидро-1H-оксазоло [3,4-а] хинолин-1-он
К раствору 10 г (0,043 М) транс-(±)-3-этенил-7-гидрокси- 3,3а,4,5-тетрагидро-1Н-оксазоло[3,4-а] хинолин-1-она (полученного на Стадии 2 Примера 1) и 12 г (0,086 М) карбоната калия в 250 мл ацетонитрила и 50 мл диметилформамида добавляют 8,9 г (0,052 М) бензил бромида. Указанную смесь перемешивают при температуре 80oC в течение 1 ч, затем, пока она остается горячей, фильтруют и промывают ацетонитрилом, после чего при пониженном давлении выпаривают досуха. Образующийся при этом остаток отбирают в этил ацетат и несколько раз промывают водой, соответствующую органическую фазу высушивают над сульфатом натрия и концентрируют при пониженном давлении. В результате перекристаллизации из диизопропилового эфира получают 12,8 г продукта.
Т.пл.: 96oC.
5.2. Транс-7-бензилокси-3-гидроксиметил-3,3а, 4,5- тетрагидро-1Н-оксазоло[3,4-а]хинолин-1-он
Через раствор 12,5 г (0,039 М) транс-7-бензилокси-3-этенил-3,3а,4,5-тетрагидро-1Н- оксазоло[3,4-а] хинолин-1-она в 450 мл дихлорметана и 350 мл метанола, охлажденный до температуры -40oC, в течение 3 ч 30 мин продувают озон. После этого с помощью струи азота удаляют избыток озона и маленькими порциями добавляют 14,8 г (0,39 М) борогидрида натрия, а затем - 12,6 г (0,195 М) диметил сульфида и оставляют указанную смесь на ночь при комнатной температуре. Эту смесь дважды экстрагируют дихлорметаном, соответствующую органическую фазу промывают водой, насыщенным раствором хлорида натрия и высушивают, а растворитель выпаривают при пониженном давлении. Посте очистки с помощью хроматографии на силикагельной колонке, элюция смесью дихлорметана и метанола в соотношении 95:5, получают 10,9 г продукта.
Т.пл.: 144oC.
5.3. Транс-7-гидрокси-3-гидроксиметил-3,3а, 4,5- тетрагидро-1H-оксазоло[3,4-а]хинолин-1-он
3,0 г (9,2 мМ) транс-7-бензилокси-3-гидроксиметил-3,3а,4,5-тетрагидро-1Н-оксазоло [3,4-а] хинолин-1-она, растворенного в 20 мл этанола и 40 мл тетрагидрофурана, подвергают гидрогенированию в течение 1 ч в присутствии 1 г 10%-ного палладиево-угольного катализатора, содержащего 50% воды. Указанную смесь фильтруют через силикагель, а растворитель выпаривают при пониженном давлении. Получают 1,1 г продукта.
Т.пл.: 250oC.
5.4. [3α,3aβ,7(R)] 7-(3-гидроксибутокси)-3-гидроксиметил-3,3а,4,5-тетрагидро-1Н- оксазоло[3,4-а]хинолин-1-он
К раствору 500 мг (2,13 мМ) транс-7-гидрокси-3-гидроксиметил- 3,3а, 4,5-тетрагидро-1Н-оксазоло[3,4-а] хинолин-1-она в 3 мл ацетонитрила и 2 мл диметилформамида добавляют 590 мг (4,26 мМ) карбоната калия, а затем - раствор 623 мг (2,55 мМ) 3(S)-гидроксибутил п-толуолсульфоната в 5 мл ацетонитрила. Указанную смесь перемешивают при температуре 80oC в течение 2 ч, после чего добавляют 20 мл воды и дважды экстрагируют этил ацетатом. Соответствующую органическую фазу высушивают над сульфатом натрия и концентрируют при пониженном давлении. Получаемое при этом твердое вещество подвергают хроматографии на силикагельной колонке, элюция смесью этил ацетата и циклогексана в соотношении 1:1, после чего растирают в диизопропиловом эфире. Получают 420 мг продукта.
Т.пл.: 102oC.
Пример 6. [3(S), 3а(S), 7(R)1-(+)-3-Метоксиметил-7-[4,4,4- трифтор-3-гидроксибутокси]-3,3а,4,5-тетрагидро-1Н-оксазоло[3.4-а]- хинолин-1-он
6.1. 1-Бензоил-2-циано-6-метокси-1,2-дигидрохинолин
Раствор 10 г (63 мМ) 6-метоксихинолина в 80 мл дихлорметана смешивают с раствором 12,4 г (188 мМ) цианида калия, после чего к образовавшейся смеси медленно добавляют 14,5 мл (125 мМ) бензоил хлорида. Указанную смесь перемешивают в течение 18 ч, отделяют органическую фазу, а соответствующую водную фазу экстрагируют дихлорметаном. Полученные органические фракции промывают водной 5%-ной соляной кислотой, затем - водой, водным раствором гидроксида натрия и снова водой, высушивают их над сульфатом натрия, а растворитель выпаривают при пониженном давлении. Образующееся при этом масло кристаллизуют из 95%-ного этанола. Получают 13,4 г продукта.
Т.пл.: 124oC.
6.2. 2-Карбокси-6-метоксихинолин
К раствору 217 г (0,747 М) 1-бензоил-2-циано-6-метокси 1,2-дигидрохинолина в 270 мл уксусной кислоты добавляют 270 мл 48%-ной бромистоводородной кислоты и выдерживают образовавшуюся смесь при дефлегмации в течение 30 мин. Фильтруют указанную смесь, ополаскивают ее диэтиловым эфиром, полученное при этом твердое вещество суспендируют в 2 л воды и нагревают до температуры 90oC. Затем добавляют водный нашатырный спирт до pH 8-9 и фильтруют указанную смесь, пока она еще остается горячей. При температуре 50oC с помощью уксусной кислоты фильтрат подкисляют до pH 4-5, затем охлаждают. Отфильтровывают кристаллизованный продукт, ополаскивают его водой, после чего перекристаллизовывают из 250 мл уксусной кислоты и промывают диэтиловым эфиром. Получают 129 г продукта.
Т.пл.: 187oC.
6.3. 6-Метокси-2-метоксикарбонилхинолин
К суспензии 129 г (0,635 М) 2-карбокси-6-меоксихинолина в 1200 мл толуола по каплям добавляют 230 мл (3,17 М) тионил хлорида и выдерживают образовавшуюся смесь при повышенной температуре в течение 3 ч 30 мин. Концентрируют указанный раствор при пониженном давлении, а полученное при этом твердое вещество растворяют в 300 мл метанола. Указанную смесь перемешивают в течение 30 мин, после чего при пониженном давлении выпаривают растворитель, а образующийся при этом продукт отбирают в диэтиловый эфир и собирают с помощью фильтрации. Выделенное твердое вещество отбирают в этил ацетат и разбавляют водным нашатырным спиртом, отделяют соответствующую органическую фазу, обрабатывают ее животным углем, фильтруют и концентрируют при пониженном давлении. Получают 90 г продукта.
Т.пл.: 129oC.
6.4. (±)-6-Метокси-2-метоксикарбонил-1,2,3,4- тетрагидрохинолин
К раствору 50 г (0,23 М) 6-метокси-2-метоксикарбонилхинолина в 1000 мл метанола добавляют раствор 6н хлористоводородного этанола до pH 1, после чего указанную смесь подвергают гидрогенизации в течение 18 ч в присутствии 2,6 г гидратированного оксида платины. Затем с помощью фильтрации удаляют использованный катализатор, а растворитель выпаривают при пониженном давлении. Растирают полученный продукт в смеси диизопропилового эфира и петролейного эфира, в результате чего получают 56 г продукта в форме гидрохлорида.
Т.пл.: 129oC.
Для выделения чистого основания указанный продукт разбавляют водным нашатырным спиртом, экстрагируют этил ацетатом, высушивают над сульфатом натрия, а также выпаривают растворитель при пониженном давлении.
Т.пл.: < 50oC.
6.5. Этил (±)-6-метокси-2-метоксикарбонил-1,2,3,4-тетрагидро-хинолин-1 -карбоксилат
К раствору 69,3 г (0,313 М) (±)-6- метокси-2-метоксикарбонил-1,2,3,4-тетрагидрохинолина и 39 мл (0,407 мл) этил хлороформата в 1400 мл дихлорметана добавляют 85 г (0,63 М) карбоната калия и выдерживают образовавшуюся смесь при дефлегмации в течение 18 ч. Фильтруют указанную смесь, соответствующий фильтрат концентрируют при пониженном давлении, а образующийся при этом остаток подвергают хроматографии на силикагельной колонке, элюция смесью этил ацетата и циклогексана в соотношении 3:7. Получают 80,4 г продукта в форме масла.
6.6. Этил S-(-)-6-метокси-2-метоксикарбонил-1,2,3,4- тетрагидрохинолин-1-карбоксилат и
этил R-(+)-2-карбокси-6- метокси-1,2,3,4-тетрагидрохинолин-1-карбоксилат
3г(10,2мМ)этил(±)-6-метокси-2-метоксикарбонил-1,2,3,4-тетрагидрохинолин-1- карбоксилата суспендируют в 80 мл 0,01 М фосфатного буфера (однозамещеный фосфат калия + двузамещенный фосфат калия), pH 7. Затем pH указанной смеси с помощью водного раствора 1 М гидроксида натрия доводят до 7,3 и добавляют 7,5 г ацетонового порошка печени овцы. Образовавшуюся смесь перемешивают в течение 14 ч при комнатной температуре, поддерживая постоянное значение pH с помощью водного раствора 1 М гидроксида натрия, затем фильтруют указанную смесь через целит и промывают использованный целит приблизительно 400 мл диэтилового эфира. Соответствующую водную фазу трижды экстрагируют 400 мл диэтилового эфира, объединяют полученные органические фракции, высушивают их над сульфатом магния, фильтруют и выпаривают в вакууме. Получают 1,5 г маслянистого продукта, соответствующего этил S-(-)-6-метокси-2- метоксикарбонил-1,2,3,4-тетрагидрохинолин-1-карбоксилату.
ее: 99% с помощью хиральной HPLC
[α]
Соответствующую водную фазу с помощью 10%-ной соляной кислоты подкисляют до pH 4,5, затем трижды экстрагируют 100 мл диэтилового эфира. Объединяют полученные эфирные фазы, высушивают их над сульфатом магния и фильтруют, после чего растворитель выпаривают в вакууме. Получают 1,04 г этил R-(+)-2- карбокси-6-метокси-1,2,3,4-тетрагидрохинолин-1-карбоксилата.
ее: 88% с помощью хиральной HPLC
[α]
6.7. Этил S-(-)-2-формил-6-метокси-1,2,3,4-тетрагидрохинолин-1- карбоксилат
К раствору 34,6 г (0,118 М) этил S-(-)-6-метокси-2- метоксикарбонил-1,2,3,4-тетрагидрохинолин-1-карбоксилата в 700 мл толуола при температуре -70oC по каплям добавляют 235 мл (0,234 М) 1,5 М раствора гидрида диизобутилалюминия в толуоле. Образовавшуюся смесь перемешивают в течение 15 мин при температуре -70oC, затем при перемешивании медленно добавляют 17 мл метанола, позволяя при этом указанной смеси нагреться до комнатной температуры. Добавляют 1,5 л 1,5 М раствора соляной кислоты, отделяют органическую фазу, а соответствующую водную фазу экстрагируют диэтиловым эфиром. Полученные органические фракции несколько раз промывают водой, а также выпаривают растворитель при пониженном давлении. Получают 23,8 г продукта.
[α]
6.8. [3(S),3a(S)]-(+)- и [3(R),3а(S)]-3-этенил-7-метокси- 1,2,3,4,-тетрагидро-1Н-оксазоло[3,4-а]хинолин-1-он
К раствору 23,8 г (90 мМ) этил S-(-)-2-формил-6-метокси- 1,2,3,4-тетрагидрохинолин-1-карбоксилата в 260 мл тетрагидрофурана, охлажденному до температуры -40oC, в атмосфере аргона в течение 1 ч 30 мин добавляют 99 мл (99 мМ) 1 М бромида винилмагния. Указанную реакционную среду перемешивают в течение 1 ч, после чего добавляют холодный насыщенный водный раствор хлорида аммония. Полученную смесь дважды экстрагируют диэтиловым эфиром, промывают водой, высушивают над сульфатом натрия, после чего выпаривают растворитель при пониженном давлении. Образующееся при этом масло вновь растворяют в 172 мл толуола и нагревают с дефлегмацией для удаления следов воды. Затем при температуре 90oC добавляют 0,8 мл 10%-ного раствора метоксида натрия в метаноле. Указанную смесь вновь нагревают с дефлегмацией, от гоняя образующийся этанол, после чего позволяют ей остыть до комнатной температуры, а полученный раствор подвергают хроматографии на силикагельной колонке, элюция смесью циклогексана и этил ацетата в соотношении 4:1. Получают 5,3 г [3(S), 3a(S)]-соединения.
Т.пл.: 80-83oC
[α]
а также 3 г соответствующего [3(R),3а(S)]-соединения,
Т.пл.: 137-138oC
[α]
6.9. [3(S), 3а(S)] -(+)-3-этенил-7-гидрокси-3,3а,4,5- тетрагидро-1Н-оксазоло[3,4-а]хинолин-1-он
К раствору 5,3 г (22 мМ) [3(S),3а(S)]-(+)-3-этенил-7-метокси- -3,3а,4,5, -тетрагидро-1Н-оксазоло[3,4-а] хинолин-1-она в 13 мл дихлорметана при температуре 0oC по каплям добавляют 43 мл (43 мМ) 1 М раствора трибромида бора в дихлорметане. Образовавшуюся смесь перемешивают в течение 30 мин, после чего добавляют разбавленный водный нашатырный спирт до нейтрального pH. Затем добавляют 5-10 мл метанола, в 2 раза концентрируют указанную реакционную среду при пониженном давлении и фильтруют ее. Полученный при этом осадок ополаскивают водой, затем - диэтиловым эфиром, после чего высушивают. Выделяют 4,7 г продукта.
Т.пл.: 215oC
[α]
6.10. [3(S),3а(S),7(R)]-(+)-3-Этенил-7-[4,4,4-трифтор-3- гидроксибутокси]-3,3а,4,5-тетрагидро-1Н-оксазоло[3,4-а]хинолин-1-он
К раствору 4,7 г (20 мМ) [3(S),3а(S)]-(+)-3-этенил-7-гидрокси- 3,3а, 4,5-тетрагидро-1Н-оксазоло[3,4-а]хинолин-1-она в 55 мл ацетонитрила добавляют 5,6 г (41 мМ) карбоната калия, а затем - 9,1 г (31 мМ) 3(R)-гидрокси-4,4,4-трифторбутил п-толуолсульфоната. Указанную смесь нагревают с дефлегмацией в течение 16 ч, после чего разбавляют дихлорметаном и промывают водой. В дальнейшем полученную органическую фазу высушивают над сульфатом натрия, а затем при пониженном давлении выпаривают растворитель. Выделенное при этом масло очищают с помощью хроматографии на колонке силикагеля. После кристаллизации из диизопропилового эфира получают 6,0 г продукта.
Т.пл. 145-146oC.
[α]
6.11. [3(S), 3а(S),7(R)]-(+)-3-Этенил-7-[4,4,4-трифтор-3- бензилоксибутокси]-3,3а,4,5-тетрагидро-1Н-оксазоло[3,4-а]хинолин-1-он
К раствору 6,0 г (17 мМ) [3(S),3а(S),7(R)]-(+)-3- Этенил-7-[4,4,4-трифтор-3-гидроксибутокси] -3,3а,4,5-тетрагидро-1Н- оксазоло[3,4-а]хинолин-1-она в 60 мл толуола добавляют раствор 2,7 г (67 мМ) гидроксида натрия в 2,7 мл воды, 0,53 г (1,7 мМ) бромида тетрабутиламмония, а затем - 6,0 мл (50 мМ) бензил бромида. Указанную смесь перемешивают при комнатной температуре в течение 16 ч, после чего экстрагируют этил ацетатом, промывают водой, высушивают над сульфатом натрия и при пониженном давлении выпаривают растворитель. Выделенное при этом масло очищают с помощью хроматографии на силикагельной колонке, элюция смесью циклогексана и этил ацетата в соотношении 8:2. Получают 7,1 г продукта в форме медленно кристаллизующегося масла.
Т.пл.: 84oC.
[α]
6.12. [3(S), 3a(S),7(R)]-(+)-3-Гидроксиметил-7-[4,4,4-трифтор- 3-бензилоксибутокси]-3,3а,4,5-тетрагидро-1Н-оксазоло-[3.4-а]хинолин- 1-он
В раствор 7,0 г (16 мМ) [3(S),3a(S),7(R)]-(+)-3-этенил-7-[4,4,4-трифтор-3-бензилоксибутокси] - 3,3а,4,5-тетрагидро-1Н-оксазоло[3,4-а]хинолин-1-она в 170 мл дихлорметана и 240 мл метанола в течение 2 ч подают озон при температуре -40oC. Затем удаляют озон струей азота, после чего добавляют 5,9 г (160 мМ) борогидрида натрия при той же температуре. Через 5 мин добавляют 5,7 мл (78 мМ) диметил сульфида, позволяют смеси нагреться до комнатной температуры, затем промывают водой, а полученную при этом органическую фазу высушивают над сульфатом натрия и концентрируют при пониженном давлении. Выделенный продукт растирают с диэтиловым эфиром, после чего фильтруют. Получают 4,7 г продукта.
Т.пл.: 118oC
[α]
6.13. [3(S), 3a(S), 7(R)1-(+)-3-Метоксиметил-7-[4,4,4-трифтор- 3-бензилоксибутокси]-3,3а,4,5-тетрагидро-1Н-оксазоло-[3,4-а] хинолин-1-он
К раствору 4,6 г (10 мМ) [3(S),3а(S),7(R)]-(+)-3- гироксиметил-7-[4,4,4-трифтор-3-гидроксибутокси]-3,3а,4,5- тетрагидро-1Н-оксазоло[3,4-а]хинолин-1-она в 60 мл толуола добавляют 0,32 г (1 мМ) бромида тетрабутил аммония, а затем - 7,7 г (61 мМ) диметил сульфата и раствор 3,2 г (82 мМ) гидроксида натрия в 3,2 мл воды. Указанную смесь перемешивают в течение 1 ч, разбавляют этил ацетатом, после чего экстрагируют полученную органическую фазу, промывают ее водой, высушивают над сульфатом натрия и при пониженном давлении выпаривают растворитель. С помощью хроматографии на силикагельной колонке, элюция смесью циклогексана и этил ацетата в соотношении 7:3, получают 6,2 г продукта в форме масла.
[α]
6.14. [3(S),3a(S),7(R)]-(+)-3-Метоксиметил-7-[4,4,4-трифтор- 3-гидроксибутокси]-3,3а,4,5-тетрагидро-1Н-оксазоло[3,4-а]- хинолин-1-он
Раствор 4,1 г (8,8 мМ) [3(S),3a(S),7(R)]-(+)-3- метоксиметил-7-[4,4,4-трифтор-3-бензилоксибутокси]-3,3а,4,5- тетрагидро-1Н-оксазоло[3,4-а]хинолин-1-она в 80 мл этанола подвергают гидрогенизированию в течение 1 ч в присутствии 0,8 г 10%-ного палладиевоугольного катализатора, содержащего 50% воды, а также следов солянокислого этанола. Указанную смесь фильтруют через силикагель и при пониженном давлении выпаривают растворитель. Поименованный продукт кристаллизуют из смеси ацетона и диизопропилового эфира, а затем - из смеси диизопропилового эфира и изопропанола в соотношении 97:3.
Получают 1,0 г продукта.
Т.пл..: 120,7-120,9oC.
[α]
Пример 7.
[3(R), 3a(S), 7(R)] -(+)-3-Метоксиметил-7-[4,4,4-трифтор-3- гидроксибутокси]-3,3а,4,5-тетрагидро-1Н-оксазоло[3,4-а]- хинолин-1-он
7.1. [3(R),3а(S)]-(+)-3-Этенил-7-гидрокси-3,3а,4,5-тетрагидро- 1Н-оксазоло[3,4-а]хинолин-1-он
2,0 г (8,0 мМ)[3(R),3а(S)]-(+)-3-этенил-7-метокси- 3,3а,4,5-тетрагидро-1Н-оксазоло[3,4-а]хинолин-1-она (полученного на Стадии 8 Примера 6) обрабатывают при условиях, описанных в Стадии 9 Примера 6.
Получают 1,9 г продукта.
[α]
7.2. [3(R),3а(S),7(R)]-(+)-3-Этенил-7-[4,4,4-трифтор-3- гидроксибутокси] -3,3а,4,5-тетрагидро-1Н-оксазоло[3,4-а]хинолин-1-он
Раствор 1,9 г (8,0 мМ) [3(R),3а(S)]-(+)-3-этенил-7-гидрокси- 3,3а,4,5-тетрагидро-1Н-оксазоло[3,4-а] хинолин-1-она в 26 мл ацетонитрила и 10 мл диметилформамида обрабатывают при условиях, описанных в Стадии 10 Примера 6.
Получают 2,6 г продукта.
Т.пл.: 143-145oC.
[α]
7.3. [3(R), 3a(S), 7(R)]-(+)-3-Этенил-7-[4,4,4-трифтор-3- бензилоксибутокси]-3,3а,4,5-тетрагидро-1Н-оксазоло[3,4-а]хинолин- 1-он
2,5 г (7,0 мМ) [3(R),3а(S),7(R)]-(+)-3-этенил-7-[4,4,4- трифтор-3-гидроксибутокси] -3,3а, 4,5-тетрагидро-1Н-оксазоло- [3,4-а]хинолин-1-она обрабатывают при условиях, описанных в Стадии 11 Примера 6.
Получают 3,0 г продукта.
Т.пл.: 73-74oC.
[α]
7.4. [3(R), 3a(S), 7(R)]-(+)-3-Гидроксиметил-7-[4,4,4-трифтор-3- бензилоксибутокси]-3,3а,4,5-тетрагидро-1Н-оксазоло [3,4-а] хинолин-1-он
3,0 г (6,7 мM) [3(R),3а(S),7(R)]-(+)-3-этенил-7- [4,4,4-трифтор-3-бензилоксибутокси] -3,3а, 4,5-тетрагидро-1Н- оксазоло-[3,4-а]хинолин-1-она обрабатывают при условиях, описанных в Стадии 12 Примера 6.
Получают 2,1 г продукта.
Т.пл. 107oC.
[α]
7.5. [3(R), 3a(S), 7(R)] -(+)-3-Метоксиметил-7-[4,4,4-трифтор-3- бензилоксибутокси]-3,3а,4,5-тетрагидро-1Н-оксазоло[3,4-а]-хинолин-1-он
Раствор 2,1 г (4,7 мМ) [3(R), 3а(S), 7(R)] -(+)-3-гидроксиметил-7- [4,4,4-трифтор-3-гидроксибутокси] -3,3а, 4,5-тетрагидро-1Н-оксазоло [3,4-а] хинолин-1-она в 30 мл толуола и 3 мл дихлорметана обрабатывают при условиях, описанных в Стадии 13 Примера 6.
Получают 1,8 г продукта в форме масла.
[α]
7.6. [3(R), 3а(S), 7(R)]-(+)-3-Метоксиметил-7-[4,4,4- трифтор-3-гидроксибутокси]-3,3а,4,5-тетрагидро-1Н-оксазоло[3,4- а]-хинолин-1-он
1,8 г (3,9 мМ) [3(R),3а(S),7(R)]-(+)-3- метоксиметил-7-[4,4,4-трифтор-3-бензилоксибутокси] -3,3а, 4,5- тетрагидро-1Н-оксазоло[3,4-а]хинолин-1-она обрабатывают при условиях, описанных в Стадии 14 Примера 6.
Получают 0,9 г продукта.
Т.пл.: 165,3-165,5oC.
[α]
Пример 8.
[3(R), 3а(S), 7(R)] -(-)-3-Метоксиметил-7-[4,4,4- трифтор-3-гидроксибутокси]-3,3а,4,5-тетрагидро-1Н- оксазоло[3.4-а]-хинолин-1-он
8.1. Этил R-(+)-6-метокси-2-метоксикарбонил-1,2,3,4- тетрагидрохинолин-1-карбоксилат
К 170 мл метанола по каплям добавляют 21,8 мл (302 мМ) тионил хлорида при температуре -40oC. Через 10 мин добавляют 16,9 г (60,4 мМ) этил R-(+)-2-карбокси-6-метокси-1,2,3,4-тетрагидрохинолин-1-карбоксилата (полученного на Стадии 6 Примера 6), перемешивают указанную смесь в течение 3 ч, позволяя ей при этом нагреться до температуры 0oC, после чего выливают ее в смесь воды и льда и добавляют нашатырный спирт до нейтрального pH. Указанную смесь экстрагируют этил ацетатом, соответствующую органическую фазу промывают водой, высушивают над сульфатом натрия и выпаривают растворитель. Получают 18 г продукта в форме масла.
[α]
8.2. Этил R-(+)-2-формил-6-метокси-1,2,3,4-тетрагидрохинолин- 1-карбоксилат
18 г (0,061 М) этил R-(+)-6-метокси-2-метоксикарбонил- 1,2,3,4-тетрагидрохинолин-1-карбоксилата обрабатывают при условиях, описанных в Стадии 7 Примера 6. Получают 11,6 г продукта.
[α]
8.3. [3(R), 3a(R)] -(-)- и [3(S),3а(R)]-(-)-3-Этенил-7-метокси- 3,3а, 4,5-тетрагидро-1Н-оксазол[3,4-а]хинолин-1-он
11,5 г (43,6 мМ) этил R-(+)-2-формил-6-метокси-1,2,3,4-тетрагидрохинолин-1-карбоксилата обрабатывают при условиях, описанных в Стадии 8 Примера 6.
Получают 4,0 г [3(R),3а(R)]-соединения
Т.пл.: 80oC.
[α]
и 2,2 г [3(S),3а(R)]- соединения
Т.пл.: 140oC.
[α]
8.4. [3(R), 3а(R)] -(-)-3-Этенил-7-гидрокси-3,3а,4,5- тетрагидро-1Н-оксазол[3,4-а]хинолин-1-он
4,0 г (16 мМ) [3(R),3а(R)]-(-)-3-этенил-7-метокси-3,3а,4,5- тетрагидро-1Н-оксазол 3,4-а] хинолин-1-она обрабатывают при условиях, описанных в Стадии 9 Примера 6. Получают 2,5 г продукта.
[α]
8.5. [3(R), 3a(R),7R]-(-)-3-Этенил-7-[4,4,4-трифтор-3- гидроксибутокси] -3,3а,4,5-тетрагидро-1Н-оксазол[3,4-а]хинолин-1-он
Раствор 2,35 г (10 мМ) [3(R),3а(R)]-(-)-3-этенил- 7-гидрокси-3,3а,4,5-тетрагидро-1Н-оксазол[3,4-а] хинолин-1-она в 25 мл ацетонитрила и 10 мл диметилформамида обрабатывают при условиях, описанных в Стадии 10 Примера 6.
Получают 2,5 г продукта.
Т.пл: 92oC
[α]
8.6. [3(R), 3a(R), 7(R)]-(+)-3-Этенил-7-[4,4,4-трифтор-3- бензилоксибутокси]-3,3а,4,5-тетрагидро-1Н-оксазол[3,4-а]хинолин- 1-он
1,82 г (5,09 мМ) [3(R),3а(R),7(R)]-(-)-3-этенил-7-[4,4,4- трифтор-3-гидроксибутокси] -3,3а, 4,5-тетрагидро-1Н-оксазол-[3,4- а]хинолин-1-она обрабатывают при условиях, описанных в Стадии 11 Примера 6.
Получают 2,1 г продукта в форме масла.
[α]
8.7. [3(R), 3a(R), 7(R)]-(+)-3-Гидроксиметил-7-[4,4,4- трифтор-3-бензилоксибутокси]-3,3а,4,5-тетрагидро-1Н-оксазол [3,4-а]- хинолин-1-он
2,1 г (4,7 мМ) [3(R),3а(R),7(R)]-(+)-3-этенил-7-[4,4,4- тpифтop-3-бензилoкcибутoкcи] -3,3a, 4,5-тетpaгидpo-1H-oкcaзoл- [3,4-а]хинолин-1-она обрабатывают при условиях, описанных в Стадии 12 Примера 6.
Получают 1,4 г продукта.
[α]
8.8. [3(R), 3а(R), 7(R)] -(+)-3-Метоксиметил-7-[4,4,4-трифтор-3- бензилоксибутокси]-3,3а,4,5-тетрагидро-1Н-оксазол[3,4-а]- хинолин-1-он
1,4 г (3,1 мМ) [3(R),3а(R),7(R)]-(+)-3-гидроксиметил-7- [4,4,4-трифтор-3-бензилоксибутокси]-3,3а,4,5-тетрагидро-1Н- оксазол[3,4-а]хинолин-1-она обрабатывают при условиях, описанных в Стадии 13 Примера 6. Получают 1,0 г продукта в виде масла.
[α]
8.9. [3(R),3a(R),7(R)]-(-)-3-Метоксиметил-7-[4,4,4-трифтор- 3-гидроксибутокси]-3,3а,4,5-тетрагидро-1Н-оксазол[3,4-а]хинолин-1-он
1,0 г (2,2 мМ) [3(R),3а(R),7(R)]-(+)-3-метоксиметил- 7-[4,4,4-трифтор-3-бензилоксибутокси] -3,3а, 4,5-тетрагидро-1Н-оксазол [3,4-а] хинолин-1-она обрабатывают при условиях, описанных в Стадии 14 Примера 6. Выпарив растворитель при пониженном давлении и проведя хроматографию на силикагельной колонке, элюция смесью циклогексана и этил ацетата в соотношении 1:1, получают 0,65 г продукта.
Т.пл.: 90oC.
[α]
Пример 9.
[3(S), 3а(R), 7(R)] -(+)-3-Метоксиметил-7-[4,4,4- трифтор-3-гидроксибутокси]-3,3а,4,5-тетрагидро-1Н-оксазоло [3,4-а]-хинолин-1-он
9.1. [3(S), 3а(R)] -(-)-3-Этенил-7-гидрокси-3,3а,4,5- тетрагидро-1Н-оксазол[3,4-а]хинолин-1-он
2,2 г (9,0 мМ) [3(S),3а(R)]-3-этенил-7-метокси-3,3а,4,5-тетрагидро-1Н- оксазоло[3,4-а] хинолин-1-она (полученного на Стадии 3 Примера 8) обрабатывают при условиях, описанных в Стадии 9 Примера 6. Получают 1,8 г продукта.
[α]
9.2. [3(S),3а(R),7(R)]-(-)-3-Этенил-7-[4,4,4-трифтор-3-гидроксибутокси]- 3,3а,4,5-тетрагидро-1Н-оксазол[3,4-а]хинолин-1-он
1,7 г (7,5 мМ) [3(S),3а(R)]-(-)-3-этенил-7-гидрокси-3,3а,4,5- тетрагидро-1Н-оксазол[3,4-а] хинолин-1-она обрабатывают при условиях, описанных в Стадии 10 Примера 6. После кристаллизации из диэтилового эфира получают 1,8 г продукта.
Т.пл.: 145oC.
[α]
9.3. [3(S), 3a(R), 7(R)]-(+)-3-Этенил-7-[4,4,4-трифтор-3- бензилоксибутокси]-3,3а,4,5-тетрагидро-1Н-оксазол[3,4-а]хинолин-1-он
1,77 г (4,95 мМ) [3(R),3а(R),7(R)]-(-)-3-этенил-7-[4,4,4- трифтор-3-гидроксибутокси] -3,3а, 4,5-тетрагидро-1Н-оксазол-[3,4- а]хинолин-1-она обрабатывают при условиях, описанных в Стадии 11 Примера 6. После очистки с помощью хроматографии на силикагельной колонке, элюция смесью циклогексана и хлороформа в соотношении 1:9, получают 2,0 г продукта.
[α]
9.4. [3(S), 3a(R), 7(R)]-(+)-3-Гидроксиметил-7-[4,4,4-трифтор-3- бензилоксибутокси]-3,3а,4,5-тетрагидро-1Н-оксазол[3,4-а]- хинолин-1-он
1,9 г (4,2 мМ) [3(S),3а(R),7(R)]-(+)-3- этенил-7-[4,4,4-трифтор-3-бензилоксибутокси] -3,3а, 4,5- тетрагидро-1Н-оксазол-[3,4-а]хинолин-1-она обрабатывают при условиях, описанных в Стадии 12 Примера 6. Сконцентрировав соответствующую органическую фазу при пониженном давлении, получают 1,7 г продукта.
Т.пл.: 98oC.
[α]
9.5. [3(S), 3a(R), 7(R)] -(+)-3-Метоксиметил-7-[4,4,4-трифтор-3- бензилоксибутокси]-3,3а,4,5-тетрагидро-1Н-оксазол[3,4-а]-хинолин- 1-он
1,68 г (3,72 мМ) [3(S),3а(R),7(R)]-(+)-3-гидроксиметил-7- [4,4,4-трифтор-3-бензилоксибутокси]-3,3а,4,5-тетрагидро-1Н-оксазол [3,4-а]хинолин-1-она обрабатывают при условиях, описанных в Стадии 13 Примера 6. С помощью хроматографии на силикагельной колонке, элюция смесью циклогексана и этил ацетата в соотношении 6:4, получают 1,35 г продукта в форме масла.
[α]
9.6. [3(S),3a(R),7(R)]-(+)-3-Метоксиметил-7-[4,4,4-трифтор- 3-гидроксибутокси]-3,3а,4,5-тетрагидро-1Н-оксазол[3,4-а]-хинолин- 1-он
1,29 г (2,77 мМ) [3(S),3a(R),7(R)]-(+)-3- метоксиметил-7-[4,4,4-трифтор-3-бензилоксибутокси]-3,3а,4,5- тетрагидро-1Н-оксазол[3,4-а]хинолин-1-она обрабатывают при условиях, описанных в Стадии 14 Примера 6. Получают 0,33 г продукта.
Т.пл.: 103,6-103,8oC.
[α]
Пример 10.
7(R)-(4,4,4-трифтор-3-гидроксибутокси)-3,3а, 4,5-тетрагидро-1Н- оксазоло[3,4-а]хинолин-1-он
10.1. 7-Метокси-3,3а,4,5-тетрагидро-1Н-оксазоло[3,4-а]хинолин-1-он
К раствору 10,9 г (41,4 мМ) этил 2-формил-6-метокси-1,2,3,4-тетрагидрохинолин-1-карбоксилата в 100 мл метанола, охлажденному до температуры 0oC, по частям добавляют 2,2 г (41,4 мМ) борогидрида калия. Указанную среду перемешивают в течение 1 ч, после чего подвергают гидролизу, разбавляют водой и экстрагируют диэтиловым эфиром. Затем соответствующую органическую фазу промывают водой и высушивают над сульфатом натрия, а растворитель выпаривают при пониженном давлении. Выделенное при этом масло вновь растворяют в 90 мл толуола, нагревают образовавшийся раствор до температуры дефлегмации, чтобы удалить следы воды, и добавляют каталитическое количество 10%-ного метоксида натрия в метаноле при температуре 90oC. Для удаления образовавшегося этанола указанную смесь вновь нагревают до температуры дефлегмации, после чего выпаривают растворитель. Полученный остаток отбирают в этил ацетат, промывают водой, после чего соответствующую органическую фазу высушивают над сульфатом натрия и концентрируют при пониженном давлении. После хроматографии на силикагельной колонке, элюция смесью гептана и этил ацетата в соотношении 4:1, получают 5,0 г продукта.
Т.пл.: 99oC.
10.2. 7-Гидрокси-3,3а,4,5-тетрагидро-1Н-оксазоло[3,4-а] хинолин-1-он
К раствору 4,6 г (21 мМ) этил 7-метокси-3,3а,4,5- тетрагидро-1Н-оксазол[3,4-а] хинолин-1-она в 50 мл дихлорметана по каплям добавляют 4 мл (42 мМ) трибромида бора при температуре 0oC. Через 1 ч указанную среду подвергают гидролизу, добавляя водный нашатырный спирт до нейтрального pH. После этого отфильтровывают образовавшийся осадок и высушивают его в вакууме. Получают 3,1 г продукта.
Т.пл.: > 260oC.
10.3. 7-(R)-(4,4,4-Трифтор-3-гидроксибутокси)-3,3а,4,5- тетрагидро-1Н-оксазоло[3,4-а]хинолин-1-он
К раствору 2,0 г (9,7 мМ) 7-гидрокси-3,3а,4,5-тетрагидро-1H- оксазоло[3,4-а] хинолин-1-она в 10 мл ацетонитрила и 10 мл диметилформамида добавляют 4,3 г (15 мМ) 3-(R)-гидрокси-4,4,4- трифторбутил тозилата и 4,0 г (29 мМ) карбоната калия. Указанную смесь перемешивают в течение 4 ч при температуре 90oC, после чего разбавляют этил ацетатом и промывают водой. Соответствующую органическую фазу высушивают над сульфатом натрия и выпаривают при пониженном давлении. Выделенное при этом масло очищают с помощью хроматографии на силикагельной колонке, элюция смесью гептана и этил ацетата в соотношении 4:1, в результате чего получают 1,9 г продукта.
Т.пл.: 188oC.
Пример 11.
S-(+)-7-(4,4,4-трифторбутокси)-3,3а, 4,5-тетрагидро-1Н- оксазоло[3,4-а] хинолин-1-он
11.1. S-(+)-7-Метокси-3,3а, 4,5-тетрагидро-1Н-оксазоло [3,4-а]хинолин-1-он
К раствору 8,0 г (27 мМ) этил S-(-)-6-метокси-2-метоксикарбонил- 1,2,3,4-тетрагидрохинолин-1-карбоксилата (полученного на Стадии 6 Примера 6) в 80 мл диглима по частям добавляют 0,90 г (41 мМ) борогидрида лития. Указанную среду перемешивают в течение 3 ч при температуре 50oC, после чего выливают в воду и экстрагируют этил ацетатом образовавшийся продукт. Соответствующую органическую фазу высушивают над сульфатом натрия и концентрируют при пониженном давлении, а выделенный остаток растирают в петролейном эфире, содержащем небольшое количество изопропилового спирта. Получают 4,4 г продукта.
Т.пл.: 112oC.
[α]
В соответствии с указанным способом, используя в качестве исходного вещества этил R-(+)-6-метокси-2-метоксикарбонил- 1,2,3,4-тетрагидрохинолин-1-карбоксилат, получают R-(-)-7-метокси- -3,3а, 4,5-тетрагидро-1Н-оксазоло[3,4-а]хинолин-1-он.
Т.пл.: 110oC.
[α]
11.2. S-(+)-7-Гидрокси-3,3а, 4,5-тетрагидро-1Н-оксазоло[3,4-а] - хинолин-1-он
К раствору 4,3 г (20 мМ) этил S-(+)-7-метокси-3,3а,4,5- тетрагидро-1Н-оксазол[3,4-а] хинолин-1-она в 40 мл дихлорметана, охлажденному до температуры 0oC, по каплям добавляют 39 мл (39 мМ) 1М раствора трибромида бора в дихлорметане при температуре 0oC. Указанную среду перемешивают в течение 1 ч, позволяя при этом подняться температуре, после чего добавляют водный раствор аммиака до нейтрального pH, отфильтровывают образовавшийся при этом осадок и высушивают его. Получают 3,0 г продукта.
Т.пл.: > 250oC.
[α]
В соответствии с указанным способом, используя в качестве исходного вещества R-(-)-7-метокси-3,3а, 4,5- тетрагидро-1Н-оксазоло[3,4-а]хинолин-1-он, получают R-(-)-7- гидрокси-3,3а,4,5-тетрагидро-1Н-оксазоло[3,4-а]хинолин-1-он.
[α]
11.3. S-(+)-7-(4,4,4-Трифторбутокси)-3,3а, 4,5-тетрагидро-1Н- оксазоло[3,4-а]хинолин-1-он
К раствору 1,9 г (9,3 мМ) 5-(+)-7-гидрокси-3,3а,4,5-тетрагидро- 1Н-оксазоло[3,4-а] хинолин-1-она в смеси 10 мл диметилформамида и 40 мл ацетонитрила добавляют 2,7 г (13 мМ) 1-бром-4,4,4-трифторбутана и 2,6 г (18 мМ) карбоната калия. Указанную смесь выдерживают в течение 3 ч при температуре 90oC, после чего разбавляют этил ацетатом и промывают водой. Соответствующую органическую фазу высушивают над сульфатом натрия и концентрируют при пониженном давлении, а выделенный остаток подвергают хроматографии на силикагельной колонке, элюция дихлорметаном, содержащим 0,5% метанола. После перекристаллизации из изопропилового спирта получают 2,1 г продукта.
Т.пл.: 121,3-121,4oC.
[α]
Пример 12.
Цис-(+)-3-фенил-7-(4,4,4-трифторбутокси)-3,3а, 4,5-тетрагидро-1Н- оксазоло[3,4-а]хинолин-1-он
12.1. Цис- и транс-(±)-7-метокси-3-фенил-3,3а,4,5-тетрагидро-1Н-оксазоло [3,4-а]хинолин-1-он
Используя в качестве исходных веществ раствор этил 2-формил-6-метокси-1,2,3,4-тетрагидрохинолин-1-карбоксилата в метаноле, охлажденный до температуры 0oC, а также бромид фенилмагния, обрабатывают указанные реагенты при условиях, сходных с описанными в Стадии 1 Примера 1, с получением соответствующих цис-производного,
т.пл.: 99oC; и
транс-производного,
т.пл.: 126oC.
12.2. Цис-(±)-7-гидрокси-3-фенил-3,3а,4,5-тетрагидро-1Н- оксазоло[3,4-а] хинолин-1-он
Используя в качестве исходного вещества цис-(±)-7-метокси-3-фенил-3,3а, 4,5-тетрагидро-1Н-оксазоло [3,4-а] хинолин-1-он, который обрабатывают при условиях, описанных в Стадии 2 Примера 1, получают цис-(±)-7-гидрокси-3- фенил-3,3а,4,5-тетрагидро-1Н-оксазоло[3,4-а]хинолин-1-он.
Т.пл.: 242oC.
Используя в качестве исходного вещества транс-(±)-7-метокси- 3-фенил-3,3а, 4,5-тетрагидро-1Н-оксазоло[3,4-а] хинолин-1-он, получают транс-(±)-7-гидрокси-3-фенил-3,3а,4,5-тетрагидро-1Н- оксазоло[3,4-а]хинолин-1-он.
Т.пл.: 216oC.
12.3. Цис-(±)-3-фенил-7-(4,4,4-трифторбутокси)-3,3а, 4,5- тетрагидро-1Н-оксазоло[3,4-а]хинолин-1-он
К раствору 0,65 г (2,3 мМ) цис-(±)-7-гидрокси-3-фенил-3,3а,4,5-тетрагидро-1Н- оксазоло[3,4-а] хинолин-1-она в 15 мл диметилформамида добавляют 0,66 г (3,5 мМ) 1-бром-4,4,4-трифторбутана и 0,64 г (4,6 мМ) карбоната калия. Указанную смесь выдерживают в течение 4 ч при температуре 90oC, после чего разбавляют этил ацетатом и промывают водой. Затем соответствующую органическую фазу высушивают над сульфатом натрия и концентрируют при пониженном давлении, а выделенный остаток подвергают хроматографии на силикагельной колонке, элюция циклогексаном, содержащим 20% этил ацетата. После растирания в диизопропиловом эфире получают 0,60 г продукта.
Т.пл.: 129,5oC.
Пример 13.
Цис-(+)-3-метил-7-(4,4,4-трифторбутокси)-3,3а, 4,5-тетрагидро-1Н- оксазоло[3,4-а]хинолин-1-он
13.1. Цис-(±)-7-метокси-3-метил-3,3а,4,5-тетрагидро-1Н-оксазоло[3,4-а] хинолин-1-он
Используя в качестве исходных веществ раствор этил 2-формил-6-метокси-1,2,3,4-тетрагидрохинолин-1-карбоксилата в диэтиловом эфире, охлажденный до температуры 0oC, а также бромид метилмагния, обрабатывают указанные реагенты при условиях, сходных с описанными в Стадии 1 Примера 1, с получением соответствующего цис-производного,
т.пл.: 138-139oC; и
транс-производного,
т.пл.: 122-123oC.
13.2. Цис-(±)-7-гидрокси-3-метил-3,3а,4,5-тетрагидро-1Н- оксазоло[3,4-а] хинолин-1-он
Используя в качестве исходного вещества цис-(±)-7-метокси-3-метил-3,3а, 4,5-тетрагидро-1Н-оксазоло [3,4-а] хинолин-1-он, который обрабатывают при условиях, описанных в Стадии 2 Примера 1, получают цис-(±)-7-гидрокси-3- метил-3,3а,4,5-тетрагидро-1Н-оксазоло[3,4-а]хинолин-1-он.
Т.пл.: 258-259oC.
Используя в качестве исходного вещества транс-(±)-7- метокси-3-метил-3,3а, 4,5-тетpaгидро-1Н-оксазоло[3,4-а] хинолин-1-он, получают транс-(±)-7-гидрокси-3-метил-3,3а,4,5- тетрагидро-1Н-оксазоло[3,4-а]хинолин-1-он.
Т.пл.: 240-241oC.
13.3. Цис-(±)-3-метил-7-(4,4,4-трифторбутокси)-3,3а, 4,5- тетрагидро-1Н-оксазоло[3,4-а]хинолин-1-он
К раствору 0,70 г( 3,2 мМ) цис-(±)-7-гидрокси-3-метил-3,3а,4,5-тетрагидро-1Н- оксазоло[3,4-а] хинолин-1-она в 15 мл диметилформамида добавляют 0,90 г (4,8 мМ) 1-бром-4,4,4-трифтор-бутана и 0,90 г (6,4 мМ) карбоната калия. Указанную смесь выдерживают при температуре 90oC в течение 4 ч, после чего разбавляют этил ацетатом и промывают водой. Затем соответствующую органическую фазу высушивают над сульфатом натрия и концентрируют при пониженном давлении, а выделенный остаток подвергают хроматографии на силикагельной колонке, элюция циклогексаном, содержащим 20% этил ацетата. После растирания в диизопропиловом эфире получают 0,80 г продукта.
Т.пл.: 79,1-79,2oC.
Пример 14.
[3α,3aβ,7(R)] 3-Этил-7-[4,4,4-трифторр-3-гидроксибутокси] 3,3а,4,5- -тетрагидро-1Н-оксазоло[3,4-а]хинолин-1-он
0,86 г (2,4 мМ) [3α,3aβ,7(R)] 3-этенил-7-[4,4,4-трифтор-3-гидроксибутокси] -3,3а, 4,5-тетрагидро-1Н- оксазоло[3,4-а]хинолин-1-она (полученного в Примере 1) подвергают гидрогенированию в течение 3 ч в 20 мл метанола в присутствии 0,2 г 5%-ного палладиево-угольного катализатора, содержащего 50% воды. Затем указанную смесь фильтруют и концентрируют при пониженном давлении до сухого состояния. Образовавшееся при этом масло очищают с помощью хроматографии на силикагельной колонке, элюция смесью дихлорметана и метанола в соотношении 95:5, а также с помощью кристаллизации из диизопропилового эфира, в результате чего получают 0,47 г продукта.
Т.пл.: 120-131oC.
Пример 15.
[3(S), 3a(S),7(S)]-3-Метоксиметил-7-[4,4,4-трифтор-3- гидроксибутокси]-3,3а,4,5-тетрагидро-1Н-оксазоло[3,4-а]хинолин-1-он
15.1. [3(S), 3а(S)]-(+)-7-Бензилокси-3-этенил- 3,3а,4,5-тетрагидро-1Н-оксазол[3,4-а]хинолин-1-он
Используя в качестве исходного вещества [3(S),3a(S)]-(+)- З-этенил-7-гидрокси-3,3а, 4,5-тетрагидро-1Н-оксазоло[3,4-а] хинолин-1-он (полученный на Стадии 9 Примера 6), который обрабатывают при условиях, описанных в Стадии 1 Примера 5, получают [3(S),3а(S)]- (+)-7-бензилокси-З-этенил-3,3а,4,5-тетрагидро-1Н-оксазол[3,4-а] хинолин-1-он.
Т.пл.: 86-90oC.
[α]
В соответствии с указанным способом, получают следующие соединения:
- [3(S), 3а(R)]-(-)-7-Бензилокси-3-этенил-3,3а,4,5- тетрагидро-1Н-оксазол[3,4-а]хинолин-1-он.
Т.пл.: 102oC
[α]
- [3(R), 3а(S)]-(+)-7-Бензилокси-3-этенил-3,3а,4,5- тетрагидро-1Н-оксазол[3,4-а]хинолин-1-он.
Т.пл.: 98oC.
[α]
- [3(R),3а(R)]-(-)-7-Бензилокси-3-этенил-3,3а,4,5-тетрагидро-1Н- оксазол[3,4-а]хинолин-1-он.
Т.пл.: 84oC.
[α]
15.2. [3(S),3а(S)]-(+)-7-Бензилокси-3-гидроксиметил-3,3а,4,5- тетрагидро-1Н-оксазол[3,4-а]хинолин-1-он
Используя в качестве исходного вещества соединение, полученное на предыдущей стадии, обрабатывают его при условиях, описанных в Стадии 2 Примера 5, с получением [3(S),3a(S)]-(+)-7-бензилокси-3-гидроксиметил-3,3а,4,5-тетрагидро-1Н- оксазол-[3,4-а]хинолин-1-она.
Т.пл.: 120-140oC.
[α]
Используя тот же способ, получают следующие соединения: -
- [3(S), 3а(R)] -(-)-7-Бензилокси-3-гидроксиметил-3,3а, 4,5-тетрагидро- 1Н-оксазол[3,4-а]хинолин-1-он в форме смолы.
[α]
- [3(R),3а(S)]-(+)-7-Бензилокси-3-гидроксиметил-3,3а,4,5-тетрагидро-1H- -оксазол[3,4-а]хинолин-1-он.
Т.пл.: 138-140oC.
[α]
- [3(R), 3а(R)]-(-)-7-Бензилокси-3-гидроксиметил-3,3а,4,5- тетрагидро-1Н-оксазол[3,4-а]хинолин-1-он в форме смолы.
[α]
15.3. [3(S), 3а(S)]-(+)-7-Бензилокси-3-метоксиметил-3,3а,4,5- тетрагидро-1Н-оксазоло[3,4-а]хинолин-1-он
К раствору 1,7 г (0,052 М) [3(S),3а(S)]-(+)-7-бензилокси-3-гидроксиметил-3,3а, 4,5- тетрагидро-1Н-оксазоло[3,4-а]хинолин-1-она в 100 мл толуола и 100 мл дихлорметана добавляют 0,17 г (0,005 М) тетраметиламмоний бромида, затем - 3,0 мл диметил сульфата и наконец - раствор 1,7 г (0,041 М) гидроксида натрия в 1,7 мл воды. Указанную смесь перемешивают в течение 1 ч 30 мин, после чего разбавляют этил ацетатом. Затем отделяют соответствующую органическую фазу, промывают ее водой, высушивают над сульфатом натрия и выпаривают растворитель при пониженном давлении. С помощью хроматографии на силикагельной колонке, элюция смесью циклогексана и этил ацетата в соотношении 3:2, получают 1,2 г продукта.
Т.пл.: 118-120oC.
[α]
В соответствии с указанным способом, получают следующие соединения:
- [3(S), 3а(R)] -(-)-7-Бензилокси-3-метоксиметил-3,3а,4,5- тетрагидро-1Н-оксазол[3,4-а]хинолин-1-он.
Т.пл.: 99oC.
[α]
- [3(R), 3а(S)] -(+)-7-Бензилокси-3-метоксиметил-3,3а,4,5- тетрагидро-1Н-оксазол[3,4-а]хинолин-1-он.
Т.пл.: 96-98oC.
[α]
- [3(R), 3а(R)] -(-)-7-Бензилокси-3-метоксиметил-3,3а,4,5- тетрагидро-1Н-оксазол[3,4-а]хинолин-1-он.
Т.пл.: 122oC.
[α]
15.4. [3(S),3а(S)]-(+)-7-Гидрокси-3-метоксиметил-3,3а,4,5- тетрагидро-1Н-оксазоло[3,4-а]хинолин-1-он
Раствор 1,2 г (0,0035 М) [3(S),3а(S)]-(+)-7-бензилокси-3-метоксиметил-3,3а, 4,5- тетрагидро-1Н-оксазоло-[3,4-а] -хинолин-1-она в течение 5 ч подвергают гидрогенизированию в 50 мл метанола и 40 мл тетрагидрофурана при повышенном давлении и окружающей температуре, в присутствии 0,25 г 10%-ного палладиево-угольного катализатора, содержащего 50% воды. Затем указанную смесь фильтруют через силикагель и выпаривают растворитель при пониженном давлении. Получают 0,9 г продукта.
Т.пл.: 172-176oC.
[α]
В соответствии с указанным способом, получают следующие соединения:
- [3(S),3а(R)]-(+)-7-Гидрокси-3-метоксиметил-3,3а,4,5- тетрагидро-1Н-оксазол[3,4-а]хинолин-1-он.
Т.пл.: 170oC.
[α]
[3(R), 3a(S)] -(-)-7-Гидрокси-3-метоксиметил-3,3а,4,5- тетрагидро-1Н-оксазол[3,4-а]хинолин-1-он.
Т.пл.: 166oC.
[α]
- [3(R),3а(R)]-(-)-7-Гидрокси-3-метоксиметил-3,3а,4,5- тетрагидро-1Н-оксазол[3,4-а]хинолин-1-он.
Т.пл.: 180oC.
[α]
15.5. [3(S), 3a(S),7(S)]-(+)-3-метоксиметил-7-[4,4,4-трифтор- 3-гидроксибутокси]-3,3а,4,5-тетрагидро-1Н-оксазоло[3,4-а]хинолин-1-он
Используя в качестве исходных веществ 0,86 г (3,45 мМ) [3(S),3а(S)]-(+)-7-гидрокси-3-метоксиметил-3,3а,4,5- тетрагидро-1Н-оксазоло[3,4-а]хинолин-1-она и 1,54 г (5,18 мМ) 4,4,4-трифтор-3(S)-гидробутил п-толуолсульфоната, обрабатывают указанные реагенты при условиях, описанных в Стадии 4 Примера 5, с получением 0,38 г продукта в форме масла.
[α]
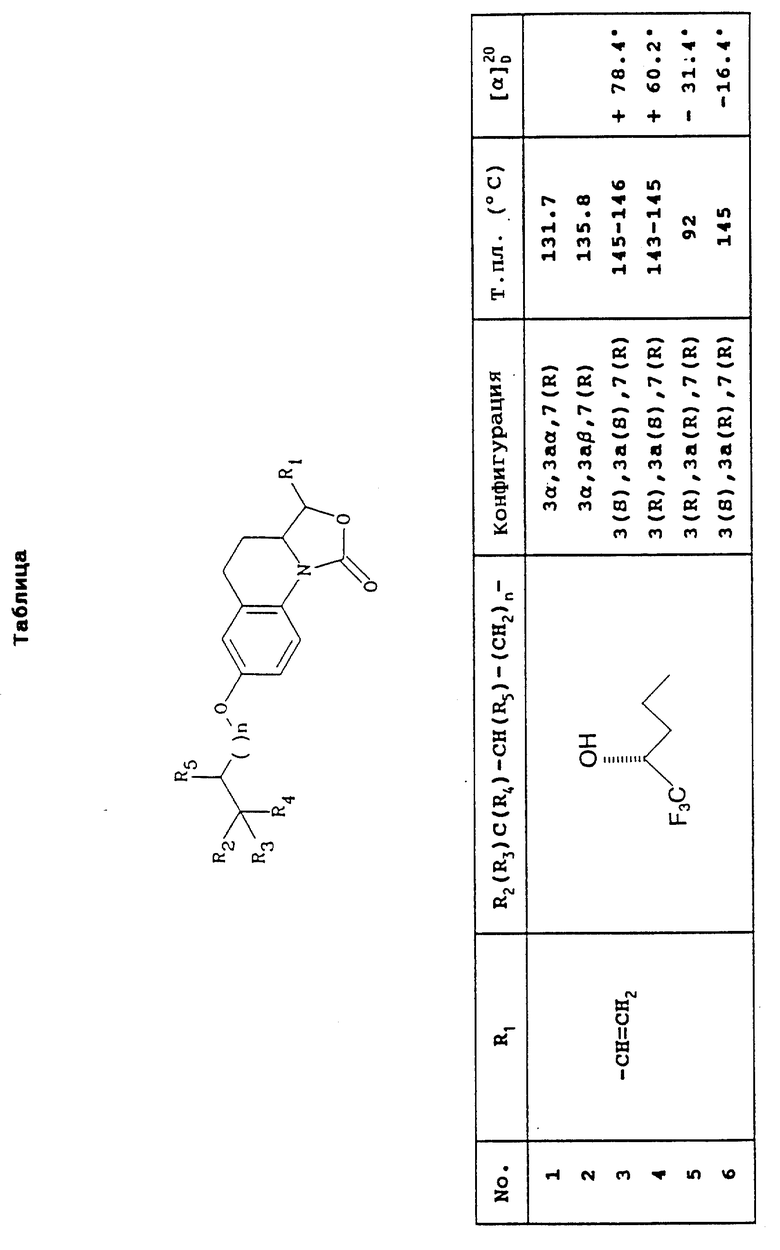
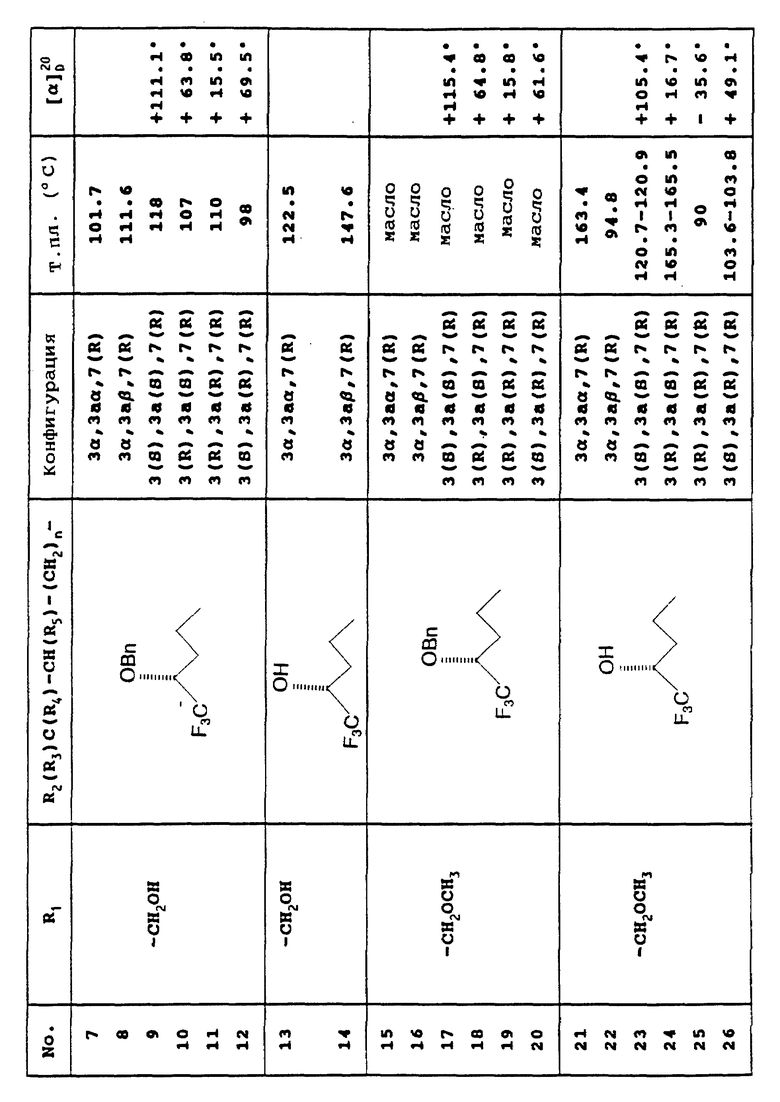
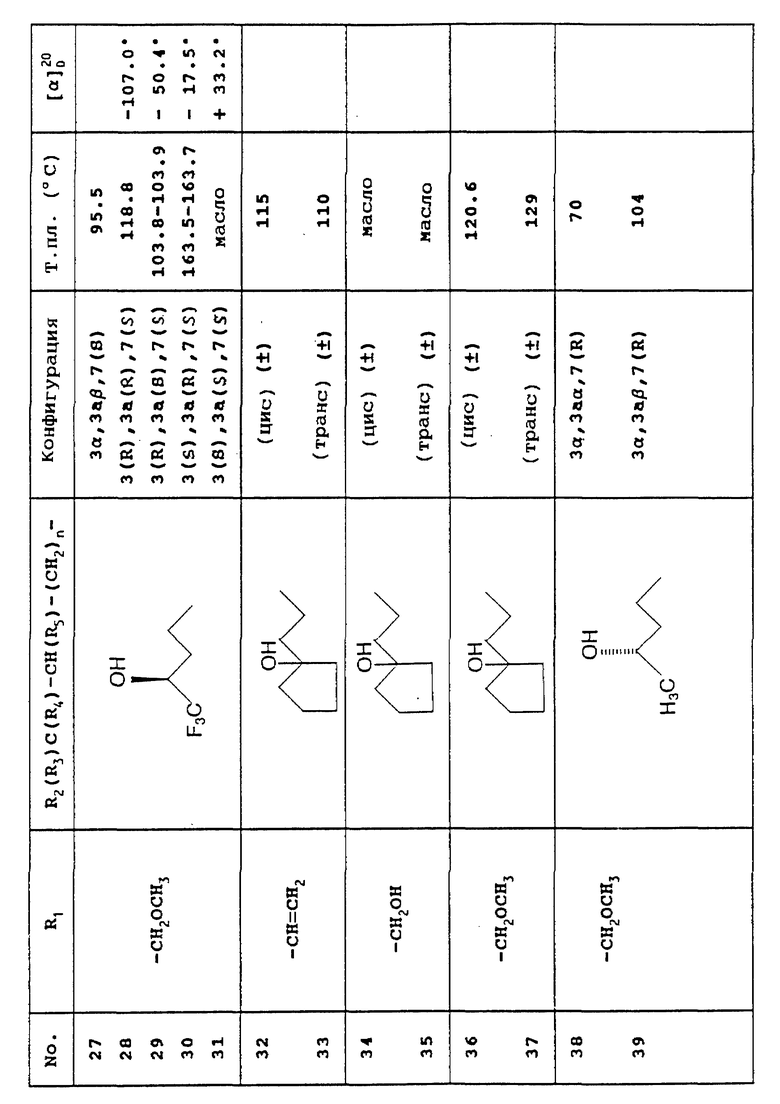
Предусмотренные настоящим изобретением соединения вместе с их физическими свойствами представлены в приведенной ниже Таблице.
Предусмотренные настоящим изобретением соединения явились объектом фармакологических исследований, подтвердивших способность указанных соединений подавлять активность моноамин оксидазы А и моноамин оксидазы В.
Измерение активностей МАО-А и МАО-В проводили в системе in vitro, используя гомогенат мозга крыс в качестве источника фермента (Fowler C. and Strolin-Benedetti M. in J.Neurochem. 40: 1534-1541 (1983)).
Стандартный тест состоит в следующем: мозг крысы гомогенизируют в 20 объемах 0,1 М фосфатного буфера (pH 7,4) и преинкубируют 100 мл гомогената (5 мг ткани) при температуре 37oC в течение 20 мин в отсутствие или в присутствии различных концентраций исследуемого ингибитора. Соответствующую реакцию запускают, добавляя [14C] серотонин ([14C]5HT, конечная концентрация 125 мкм) для измерения активности МАО-А, либо [14C] фенил-этиламин ([14C]PEA, конечная концентрация 8 мкм) для измерения активности МАО-В, в конечном объеме 500 мкл. После 5 мин инкубации в случае [14C]5НT, либо после 1 мин в случае [14C] PEA, указанную реакцию останавливали, добавляя 200 мкл 4н соляной кислоты. Затем соответствующие радиоактивные метаболиты, образовавшиеся в результате окислительного дезаминирования, отделяли от неиспользованного субстрата с помощью экстракции в органическую фазу, а также проводили количественный учет указанных метаболитов посредством измерения радиоактивности.
Ингибиторную активность, проявляемую по отношению к МАО-А и МАО-В, представляли в виде констант ингибирования КИ (МАО-А) и КИ (МАО-В).
В случае соединений, предусмотренных настоящим изобретением, величина КИ (МАО-А) находится в пределах от 0,4 до 28 нМ, в то время как значение КИ (МАО-В) варьирует от 0,7 до 1000 нМ.
Некоторые соединения, предусмотренные настоящим изобретением, являются избирательными ингибиторами МАО-А, что оказывается возможным в том случае, если величина отношения КИ(МАО-А) / КИ (МАО-В) находится в пределах от 10 до 1000. Между тем, прочие из указанных соединений представляют собой ингибиторы как МАО-А, так и МАО-В, что имеет место при величине отношения КИ (МАО-А) / КИ (МАО-В) в пределах от 0,1 до 10.
Способность подавлять активность МАО была продемонстрирована также in vivo в тесте на усиление действия L-5-гидрокситриптофана (L-5HTP) (Jalfre M. , et al., Arch. Int. Pharmacodyn 259: 194-221 (1982)).
Указанный тест проводили следующим образом: партиям крыс (по 10 особей на каждую дозу) перорально вводили различные дозы исследуемого продукта или носителя, а через 60 мин осуществляли внутрибрюшинное введение L-5HPT в дозе 100 мг/кг, которая сама по себе не вызывает серотонинэргический синдром (общие судороги) у контрольных животных.
Наличие общих судорог оценивали по принципу "все или ничего" через 30 мин после применения L-5HTP. Результаты, полученные для каждой дозы исследуемого продукта, выражали в виде процента животных, у которых наблюдались общие судороги. Затем, исходя из зависимости между эффектом (процентом, характеризующим каждую дозу) и логарифмом соответствующей дозы, с помощью прямолинейной регрессии определяли дозу, при которой общие судороги наблюдаются у 50% животных (ED50), а также ее 95%-ный доверительный интервал.
В случае соединений, предусмотренных настоящим изобретением, величина ED50 находится в пределах от 0,2 до 1,1 мг/кг, что подтверждает способность указанных соединений подавлять активность MAO in vitro.
Кроме того, судя по оценке их токсичности после in vitro по отношению к гепатоцитам крыс и отличных от человека приматов однократного применения, предусмотренные настоящим изобретением соединения очень хорошо переносятся в пределах исследованного спектра доз (вплоть до 100 мкМ).
Полученные результаты свидетельствуют о том, что предусмотренные настоящим изобретением соединения могут быть использованы для производства лекарственных препаратов, проявляющих избирательную ингибирующую способность по отношению к МАО-А, либо ингибирующую способность по отношению как к МАО-А, так и к МАО-В, причем указанные лекарственные препараты могут найти применение особенно при лечении депрессивных состояний, панических припадков, фобий, тревоги, мигрени, возрастных умственных расстройств или слабоумия, а также при предупреждении или лечении нейродегенеративных заболеваний, таких как болезнь Паркинсона и болезнь Альцгеймера, и инсультов.
Предусмотренные настоящим изобретением соединения могут использоваться в комбинации с эксципиентами в форме составов, предназначенных для перорального, парэнтерального или ректального применения, например, в форме таблеток, покрытых оболочкой таблеток, капсул, растворов, суспензий или суппозиториев. В случае перорального применения дневная доза вводимого активного ингредиента может варьировать в пределах от 1 до 100 мг/кг в виде одной дозы или множественных доз. При парэнтеральном и ректальном применении указанная доза может находиться в пределах от 1 до 100 мг/кг.
Заявитель представляет следующие примеры фармацевтического состава по п. 12, в которых в качестве активного вещества используют соединения по изобретению.
Раствор для инъекций
Активное вещество - 5 мг
Глюкоза - 250 мг
Вода для инъекций - ск. треб. до 5 мл на одну ампулу на 5 мл
Активное вещество и глюкоза растворяют в воде для инъекций, полученный раствор фильтруют, разливают в ампулы, запаивают и стерилизуют.
Желатиновая капсула
Активное вещество - 100 мг
Тальк - 24 мг
Силикaгель на одну капсулу на 125 мг - 1 мг
Тонкоизмельченное активное вещество тщательно смешивают с тальком и силикагелем и полученной смесью заполняют желатиновые капсулы.
Таблетка
Активное вещество - 400 мг
Силикагель - 10 мг
Стеариновая кислота - 20 мг
Кукурузный крахмал на одну таблетку на 475 мг - 45 мг
Активное вещество тщательно смешивают с остальными ингредиентами, полученную смесь прессуют в таблетки необходимой формы и размера.
Сироп
Активное вещество - 5 г
Метиловый эфир 4-гидроксибензойной кислоты - 150 мг
Сахароза - 50 мг
Дистиллированная вода - ск. треб. до 100 мг на один флакон 100 мл
Сахарозу растворяют в воде, добавляют активное вещество и метиловый эфир 4-гидроксибензойной кислоты и полученный сироп разливают по флаконам.
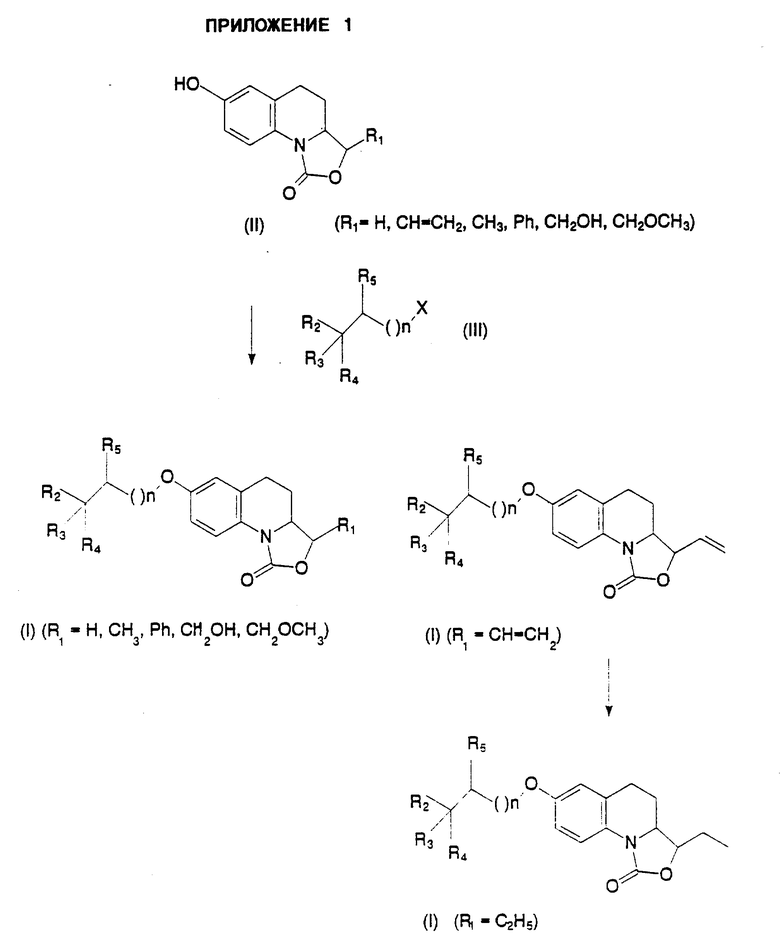
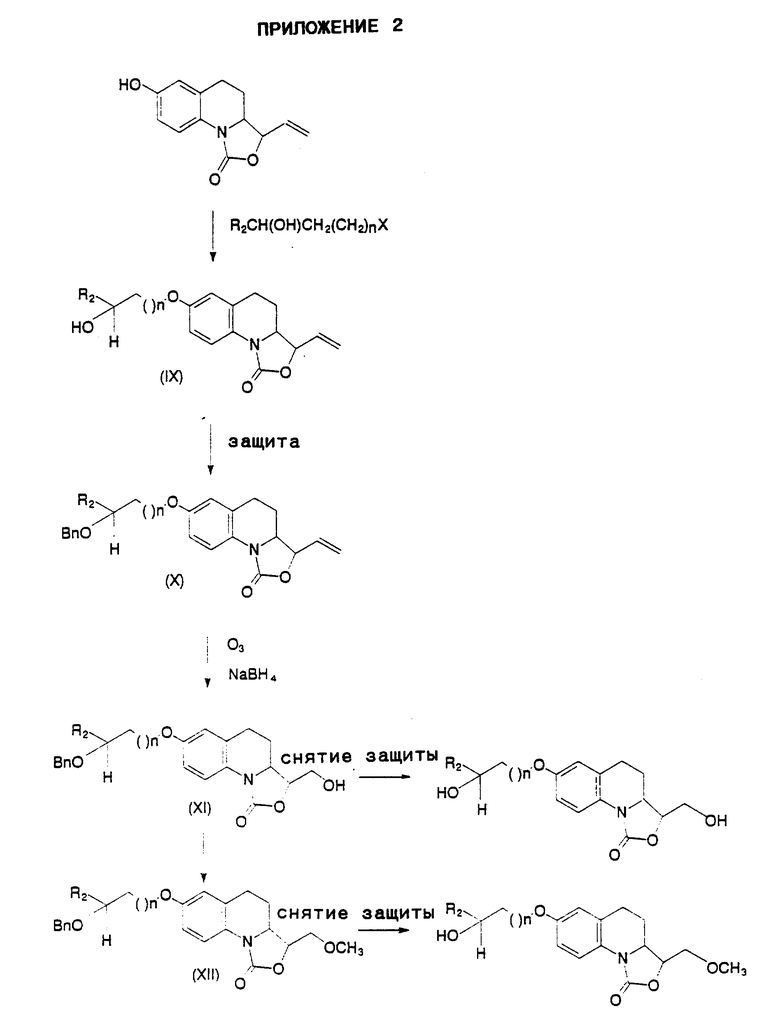
Описываются новые производные оксазолохинолинона общей формулы I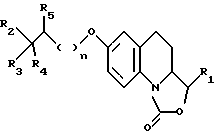
где значения R1 - R5 указаны в п.1 формулы. Соединения проявляют избирательную ингибирующую способность по отношению к МаО-А, либо ингибирующую способность по отношению как к МАО-А, так и к МАО-В. Описывается способ их получения. Описываются также лекарственные препараты, которые могут найти применение при лечении депрессивных состояний, панических припадков, фобий, тревоги, мигрени, возрастных умственных расстройств или слабоумия, а также при предупреждении или лечении нейродегенеративных заболеваний. 4 с. и 8 з.п.ф-лы, 3 ил., 1 табл.
в которой n равно 0 или 1;
R1 представляет собой атом водорода, либо этенильную, метильную, этильную, фенильную, гидроксиметильную или метоксиметильную группу, а также
либо R2 обозначает метильную, трифторметильную или цианогруппу,
R3 соответствует атому водорода, либо гидроксильной или бензилоксигруппе, а
R4 и R5 являются атомами водорода, при условии, что если R2 является метилом, R3 не является водородом, либо R2 и R4 вместе образуют группу -(CH2)4-, R3 представляет собой гидроксильную группу, а R5 обозначает атом водорода, либо R2 и R5 вместе образуют группу -O-(CH2)3-, а R3 и R4 являются атомами водорода; либо R2 и R5 вместе образуют группу -(CH2)4-, R3 соответствует гидроксильной группе, а R4 представляет собой атом водорода,
в форме изомеров, в том числе в форме энантиомеров и диастереоизомеров, а также смесей указанных различных форм, включая рацемические смеси.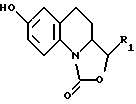
в которой R1 представляет собой атом водорода либо этенильную, фенильную, гидроксиметильную или метоксиметильную группу,
обрабатывают соединением, соответствующим формуле III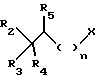
в которой X обозначает атом галогена либо лабильную группу, такую как мезилокси- или тозилоксигруппа,
с получением соединения, отвечающего формуле I, в которой R1 соответствует приведенным выше обозначениям, после чего восстанавливают указанное соединение, описываемой формулой I, в которой R1 является этенильной группой, с получением соединения, отвечающего формуле I, в которой R1 представляет собой этильную группу.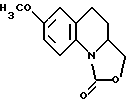
или V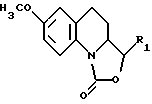
где R1 обозначает этенильную, метильную или фенильную группы, для получения которых осуществляют ферментативный гидролиз этил 2-метоксикарбонил-6-метокси-1,2,3,4-тетрагидрохинолин-1-карбоксилата, с помощью экстракции разделяют S(-) энантиомер этил 2-метоксикарбонил-6-метокси-1,2,3,4-тетрагидрохинолин-1-карбоксилата и R(+) энантиомер этил 2-карбокси-6-метокси-1,2,3,4-тетрагидрохинолин-1-карбоксилата, обрабатывают R(+) энантиомер этил 2-карбокси-6-метокси-1,2,3,4-тетрагидрохинолин-1-карбоксилата тионил хлоридом и метанолом с получением соответствующего 2-метоксикарбонильного производного и S(-) и R(+) энантиомеры указанного 2-метоксикарбонильного производного приводят во взаимодействие либо с борогидридом лития, в результате чего получают соответственно S(+) и R(-) энантиомеры соединения, описываемого формулой IV, либо с гидридом диизобутилалюминия, в результате чего получают соответственно S(-) и R(+) энантиомеры соответствующего 2-фомильного производного, указанные энантиомеры 2-формильного производного обрабатывают органомагниевым соединением, описываемым формулой R1MgX, где R1 такое как определено выше, а X является атомом галогена, а затем - метоксидом натрия, после чего проводят разделение с помощью хроматографии, в результате чего получают указанные дистереоизомеры соединений, отвечающих формуле V.
Приоритет по признакам
04.09.95 R1 - атом водорода, метильная, этильная или фенильная группа; R2 и R3 вместе образуют группу -O-(CH2)3-, а R3 и R4 являются атомами водорода либо R2 и R5 вместе образуют группу -(CH2)4-, R3 соответствует гидроксильной группе, а R4 представляет собой атом водорода;
05.09.94 по другим признакам формулы изобретения
| SU 1284214 A, 1985 | |||
| ВИНТОВОЕ ЗАЖИМНОЕ УСТРОЙСТВО С ПЛАВАЮЩИМИ ГУБКАМИ | 0 |
|
SU322263A1 |
| Устройство для отливки металла под давлением | 1959 |
|
SU128120A1 |

