Область техники, к которой относится изобретение
Изобретение имеет отношение к новым спиро-аминосоединениям, обладающим антагонистической активностью к орексиновому рецептору 1.
Изобретение также имеет отношение к способу получения таких соединений, фармацевтическим композициям, содержащим одно или более соединений формулы (VI), и их применению в качестве антагонистов орексинового рецептора 1.
Уровень техники
Орексины являются нейропептидами, открытыми в 1998 двумя группами исследователей. Орексин А представляет собой пептид, состоящий из 33 аминокислот, а орексин В - пептид, состоящий из 28 аминокислот. Орексины секретируются отдельной группой нейронов в боковой части гипоталамуса и связываются с сопряженными с G-белком рецепторами, а именно OX1 и OX2. Орексиновый рецептор 1 (OX1) селективно связывает орексин А, а орексиновый рецептор 2 (OX2) способен связывать и орексин А и орексин В.
Известно, что орексины стимулируют потребление корма крысами, таким образом, подтверждая свою модулирующую роль в механизмах, связанных с потреблением пищи.
Кроме того, было показано, что орексины регулируют структуру сна и тем самым создают возможность потенциально новых терапевтических способов лечения нарколепсии, а также инсомнии и других нарушений сна. Недавно было показано, что орексины участвуют в механизмах привыкания, таким образом, применение их в качестве модуляторов может предоставить возможность лечения компульсивных нарушений и привыкания к наркотическим средствам. Орексиновые рецепторы располагаются в мозге млекопитающих и принимают участие в некоторых патологических процессах.
В международной патентной заявке WO 2009/016560 ряд производных транс-3-аза-бицикло[3.1.0]гексана раскрываются как орексиновые антагонисты.
Новые производные пирролидина и пиперидина были раскрыты в WO 2009/040730, производные N-ароил-циклических аминов были раскрыты в международной патентной заявке WO 02/090355, а пиперазиновые соединения в WO 03/051873, все они являются новыми структурами, предложенными в качестве антагонистов орексиновых рецепторов,
Ряд производных N-ароил-циклических аминов раскрывается в международной патентной заявке WO 2004/026866 как непептидные антагонисты орексиновых рецепторов человека. В частности, 43 пиперидиновых соединения, в которых бета положение при атоме азота было замещено водородом или метальными группами, были проверены на активность по отношению к рецепторам OХ1 и OХ2.
В этих документах описываются соединения, обладающие активностью по отношению к двум типам рецепторов.
Целью настоящего изобретения является предоставление соединений, обладающих селективной антагонистической активностью к орексиновому рецептору 1.
Раскрытие изобретения
Цель настоящего изобретения достигается посредством спиро-аминосоединения формулы (VI):
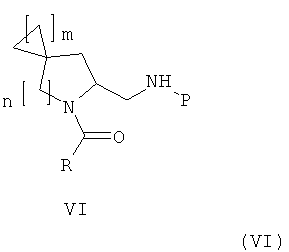
где
m представляет собой 1 или 2 или 3
n представляет собой 1 или 2,
R выбирают из 5- или 6-членного ароматического кольца и 5- или 6-членного гетероароматического кольца, содержащего от 1 до 3 гетероатомов, выбранных из S, О и N, причем такое кольцо является замещенным одним или двумя заместителями, выбранными из группы, состоящей из (С1-С3)алкила, галогена, (С3-С5)циклоалкилоксигруппы, (С1-С3)алкилкарбонила, фенила, необязательно замещенного одним или более атомами галогена, 5- или 6-членного гетероцикла, содержащего, по меньшей мере, один атом азота;
P представляет собой заместитель Q или COQ, где Q представляет собой группу, выбранную из фенила, пиридила, пиримидила, хинолила, изохинолила, хиноксалила, бензофуранила, имидазотриазолила, при этом Q необязательно замещен одним или более заместителями, выбранными из группы, состоящей из (С1-С3)алкила, галогена, трифторметила, карбамидной группы, метилкарбамидной группы, карбоксигруппы; метилкарбоксигруппы,
или его фармацевтически приемлемой соли.
В этом изобретении соединения формулы (VI) могут существовать в виде R и S энантиомеров и в виде рацемической смеси. Это изобретение включает в объем правовой охраны все возможные изомеры и рацемические смеси. Где бы ни находились дополнительные центры симметрии, это изобретение включает все возможные диастереоизомеры, а также соответствующие смеси.
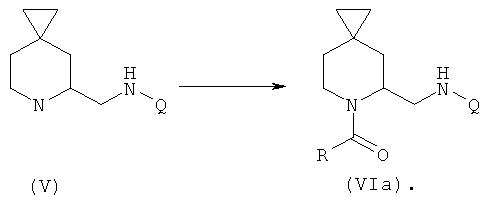
В первом варианте осуществления в спиро-аминосоединении формулы (VI) Р представляет собой Q. Это соединение имеет формулу (VIa):
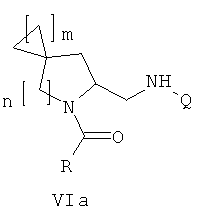
Во втором варианте осуществления в спиро-аминосоединении формулы (VI) Р представляет собой COQ. Это соединение имеет формулу (VIb):
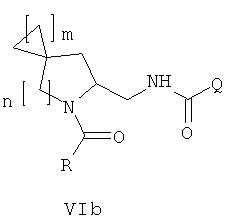
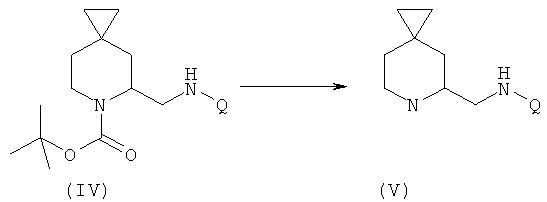
Дополнительный аспект данного изобретения касается способа получения соединения формулы (VIa), включающего следующие стадии, представленные на схеме:
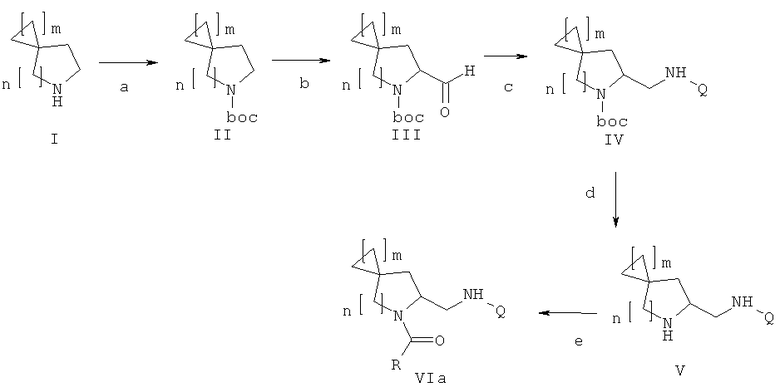
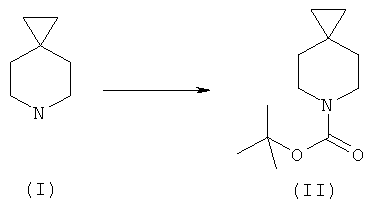
a) защита соединения формулы (I) защитной группой ВОС для получения соединения формулы (II);
b) взаимодействие соединения формулы (II) с сильными основаниями и диметилформамидом, что дает соединение формулы (III);
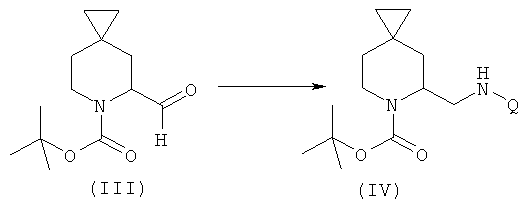
c) добавление амина формулы Q-NH2, где Q выбирают из группы, состоящей из фенила, пиридила, пиримидила, хинолила, изохинолила, хиноксалила, бензофуранила, имидазотриазолила, такой Q необязательно замещен одним или более заместителями, выбранными из группы, состоящей из (C1-C3)алкила, галогена, трифторметила, карбамидной группы, метилкарбамидной группы, карбоксигруппы, метилкарбоксигруппы, в присутствии восстановителя для получения соединения формулы (IV);
d) отщепление BOC-группы от соединения формулы (IV) для получения соединения формулы (V);
e) взаимодействие соединения формулы (V) с RCOOH в присутствии агентов сочетания или с соответствующими хлорангидридами RCOCl в присутствии основания, где R выбирают из 5- или 6-членного ароматического кольца и 5- или 6-членного гетероароматического кольца, содержащего от 1 до 3 гетероатомов, выбранных из S, О и N, причем такое кольцо является замещенным одним или двумя заместителями, выбранными из группы, состоящей из (С1-С3)алкила, галогена, (С3-С5)циклоалкоксигруппы, (С1-С3)алкилкарбонила, фенила, необязательно замещенного одним или более атомами галогена, 5- или 6-членного гетероцикла, содержащего, по меньшей мере, один атом азота.
В дополнительном аспекте данное изобретение касается способа получения соединения формулы (VIb), включающего следующие стадии, представленные на схеме ниже:
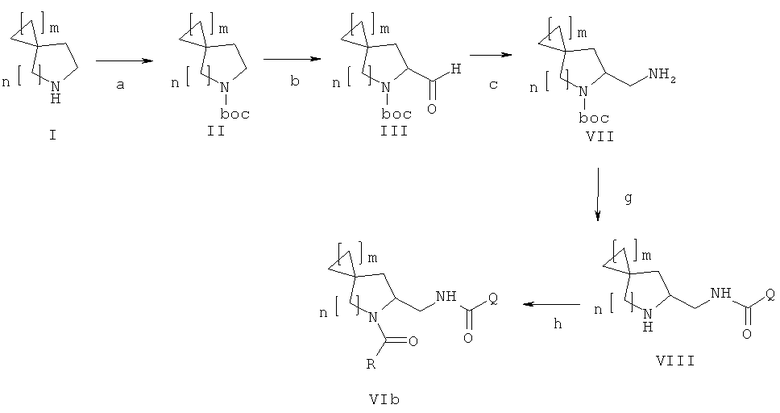
a) защита соединения формулы (I) защитной группой ВОС с получением соединения формулы (II);
b) взаимодействие соединения формулы (II) с сильными основаниями и диметилформамидом, что дает соединение формулы (III);
f) проведение восстановительного аминирования соединения формулы (III) с получением амина формулы (VII);
g) взаимодействие соединения формулы (VII) с QCOOH в присутствии агентов сочетания или с соответствующими хлорангидридами QCOCl в присутствии основания с получением амида формулы (VIII), где Q является группой, выбранной из фенила, пиридила, пиримидила, хинолила, изохинолила, хиноксалила, бензофуранила, имидазотриазолила, такой Q необязательно замещен одним или более заместителями, выбранными из группы, состоящей из (C1-C3)алкила, галогена, трифторметила, карбамидной группы, метилкарбамидной группы, карбоксигруппы, метилкарбоксигруппы;
h) отщепление BOC-группы от соединения формулы (VIII) и взаимодействие с RCOOH в присутствии агентов сочетания или с соответствующими хлорангидридами RCOCl в присутствии основания с получением соединения формулы VIb, где R выбирают из 5- или 6-членного ароматического кольца и 5- или 6-членного гетероароматического кольца, содержащего от 1 до 3 гетероатомов, выбранных из S, О и N, причем такое кольцо является замещенным одним или более заместителями, выбранными из группы, состоящей из (С1-С3)алкила, галогена, (С3-С5)циклоалкилоксигруппы, (С1-С3)алкилкарбонила, фенила, необязательно замещенного одним или более атомами галогена, 5- или 6-членного гетероцикла, содержащего, по меньшей мере, один атом азота.
В другом аспекте изобретение имеет отношение к фармацевтической композиции, содержащей соединение формулы (VI), предпочтительно (VIa), и фармацевтически приемлемый носитель.
В другом аспекте изобретение имеет отношение к соединению формулы (VI) как лекарственному средству, в частности это касается его применения для производства медикамента для лечения патологий, при которых необходим антагонист рецептора OХ1, например, в случае лечения ожирения, нарушений сна, компульсивных расстройств (навязчивых состояний), лекарственной зависимости (наркомании), шизофрении.
Осуществление изобретения
Таким образом, изобретение имеет отношение к спиро-аминосоединению формулы (VI):

где
m является 1 или 2 или 3,
n представляет собой 1 или 2,
R выбирают из 5- или 6-членного ароматического кольца и 5- или 6-членного гетероароматического кольца, содержащего от 1 до 3 гетероатомов, выбранных из S, О и N, причем такое кольцо является замещенным одним или двумя заместителями, выбранными из группы, состоящей из (С1-С3)алкила, галогена, (С3-С5)циклоалкилоксигруппы, (С1-С3)алкилкарбонила, фенила, необязательно замещенного одним или более атомами галогена, 5- или 6-членного гетероцикла, содержащего, по меньшей мере, один атом азота;
Р представляет собой заместитель Q или COQ, где Q представляет собой группу, выбранную из фенила, пиридила, пиримидила, хинолила, изохинолила, хиноксалила, бензофуранила, имидазотриазолила, такой Q необязательно замещен одним или двумя заместителями, выбранными из (С1-С3)алкила, галогена, трифторметила, карбамидной группы, метилкарбамидной группы, карбоксигруппы, метилкарбоксигруппы;
или его фармацевтически приемлемой соли.
В первом варианте осуществления в спиро-аминосоединении формулы (VI) Р представляет собой Q. Это соединение имеет формулу (VIa):
В этом варианте осуществления предпочтительно n представляет собой 2, а m является 1.
Предпочтительно первый вариант осуществления изобретения относится к соединению с пиперидиновым циклом, в котором кольцо из трех спироуглеродных атомов находится в гамма-положении к атому азота. Предпочтительно R представляет собой фенил или гетероциклическое кольцо.
Более предпочтительно, когда R представляет собой гетероциклическое кольцо, оно является тиазольным кольцом, даже более предпочтительно тиазольным кольцом, замещенным, по меньшей мере, одним заместителем, выбранным из группы, состоящей из метила, фенила, фенила, замещенного одним или более галогенами.
Более предпочтительно, когда R представляет собой фенил, этот фенил может замещаться группой, выбранной из циклопропил(С1-С3)алкилоксигруппы, триазолила, пиримидила.
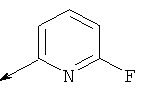
Предпочтительно Q представляет собой пиридильное кольцо, даже более предпочтительно пиридил, замещенный одним или более заместителями, выбранными из группы, состоящей из трифторметила, карбоксигруппы, метилкарбоксигруппы, метила и галогена.
Предпочтительные соединения изобретения выбирают из группы, состоящей из:
метил 5-хлор-2-(((6-(5-(4-фторфенил)-2-метилтиазол-4-карбонил)-6-азаспиро[2.5]октан-5-ил)метил)амино)бензоата
(2-метил-5-фенилтиазол-4-ил)(5-(((5-(трифторметил)пиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)метанона
(2-метил-5-фенилтиазол-4-ил)(5-(((6-метилпиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)метанона
(2-метил-5-фенилтиазол-4-ил)(7-(((6-метилпиридин-2-ил)амино)метил)-8-азаспиро[4.5]декан-8-ил)метанона
(5-(((5-хлорпиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)(2-(циклопропилметокси)фенил)метанона
(5-(((5-хлорпиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)(2-(пиримидин-2-ил)фенил)метанона
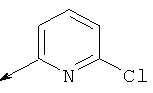
(5-(((5-хлорпиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)(2-метил-5-фенилтиазол-4-ил)метанона
(5-(((5-хлорпиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)(5-(4-фторфенил)-2-метилтиазол-4-ил)метанона
(5-(((5-хлорпиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)(5-метил-2-(2Н-1,2,3-триазол-2-ил)фенил)метанона
(5-(((5-хлорпиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)(5-метил-2-(пиримидин-2-ил)фенил)метанона
(5-(((5-фторпиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)(2-метил-5-фенилтиазол-4-ил)метанона
(7-(((5-хлорпиридин-2-ил)амино)метил)-8-азаспиро[4.5]декан-8-ил)(2-метил-5-фенилтиазол-4-ил)метанона
(R)-(2-метил-5-фенилтиазол-4-ил)(5-(((6-метилпиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)метанона
(R)-(5-(((5-хлорпиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)(2-метил-5-фенилтиазол-4-ил)метанона
(R)-(5-(((5-хлорпиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)(5-фтор-2-(пиримидин-2-ил)фенил)метанона
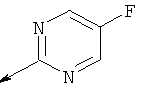
(R)-(5-метил-2-(пиразин-2-ил)фенил)(5-(((6-(трифторметил)пиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)метанона
(S)-(2,5-дихлорфенил)(5-(((4,б-диметилпиримидин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)метанона
(S)-(2-(2Н-1,2,3-триазол-2-ил)фенил)(5-(((5-хлорпиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)метанона
(S)-(2-метил-5-фенилтиазол-4-ил)(5-(((4-метилпиримидин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)метанона
(S)-(2-метил-5-фенилтиазол-4-ил)(5-(((5-(трифторметил)пиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)метанона
(S)-(2-метил-5-фенилтиазол-4-ил)(5-(((5-метилпиримидин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)метанона
(S)-(2-метил-5-фенилтиазол-4-ил)(5-(((6-(трифторметил)пиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)метанона
(S)-(2-метил-5-фенилтиазол-4-ил)(5-(((6-метилпиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)метанона
(S)-(5-(((4,6-дифторпиримидин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)(2-метил-5-фенилтиазол-4-ил)метанона
(S)-(5-(((4,6-диметилпиримидин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)(2-метил-5-фенилтиазол-4-ил)метанона
(S)-(5-(((5-хлорпиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)(2-(пиразин-2-ил)фенил)метанона
(S)-(5-(((5-хлорпиридин-2-ил)амино)метил)-6-азаспиро [2.5]октан-6-ил)(2-(пиримидин-2-ил)фенил)метанона
(S)-(5-(((5-хлорпиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)(2-фтор-6-(2Н-1,2,3-триазол-2-ил)фенил)метанона
(S)-(5-(((5-хлорпиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)(2-фтор-6-(пиридин-2-ил)фенил)метанона
(S)-(5-(((5-хлорпиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)(2-фтор-6-(пиримидин-2-ил)фенил)метанона
(S)-(5-(((5-хлорпиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)(2-метил-5-фенилтиазол-4-ил)метанона
(S)-(5-(((5-хлорпиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)(5-фтор-2-(2Н-1,2,3-триазол-2-ил)фенил)метанона
(S)-(5-(((5-хлорпиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)(5-фтор-2-(пиразин-2-ил)фенил)метанона
(S)-(5-(((5-хлорпиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)(5-фтор-2-(пиридин-2-ил)фенил)метанона
(S)-(5-(((5-хлорпиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)(5-фтор-2-(пиримидин-2-ил)фенил)метанона
(S)-(5-(((5-хлорпиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)(5-метил-2-(пиридин-2-ил)фенил)метанона
(S)-(5-(((5-хлорпиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)(5-метил-2-(пиримидин-2-ил)фенил)метанона
(S)-(5-(((5-фторпиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)(2-метил-5-фенилтиазол-4-ил)метанона
(S)-(5-(((6-хлорпиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)(2-метил-5-фенилтиазол-4-ил)метанона
(S)-(5-(((6-хлорпиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)(5-метил-2-(2Н-1,2,3-триазол-2-ил)фенил)метанона
(S)-(5-(((6-хлорпиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)(5-метил-2-(пиразин-2-ил)фенил)метанона
(S)-(5-(((6-хлорпиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)(5-метил-2-(пиримидин-2-ил)фенил)метанона
(S)-(5-(((6-фторпиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)(2-метил-5-фенилтиазол-4-ил)метанона
(S)-(5-метил-2-(2Н-1,2,3-триазол-2-ил)фенил)(5-(((5-(трифторметил)пиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)метанона
(S)-(5-метил-2-(2Н-1,2,3-триазол-2-ил)фенил)(5-(((6-(трифторметил)пиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)метанона
(S)-(5-метил-2-(пиразин-2-ил)фенил)(5-(((6-(трифторметил)пиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)метанона
(S)-(5-метил-2-(пиримидин-2-ил)фенил)(5-(((5-(трифторметил)пиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)метанона
(S)-(5-метил-2-(пиримидин-2-ил)фенил)(5-(((6-(трифторметил)пиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)метанона
(S)-[1,1'-бифенил]-2-ил(5-(((4,б-диметилпиримидин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)метанона
(7-(((5-хлорпиридин-2-ил)амино)метил)-8-азаспиро[4.5]декан-8-ил)(2-метил-5-фенилтиазол-4-ил)метанона
(2-метил-5-фенилтиазол-4-ил)(7-(((6-метилпиридин-2-ил)амино)метил)-8-азаспиро[4.5]декан-8-ил)метанона
(S)-(6-(((5-хлорпиридин-2-ил)амино)метил)-5-азаспиро[2.4]гептан-5-ил)(5-метил-2-(2Н-1,2,3-триазол-2-ил)фенил)метанона
(S)-(5-метил-2-(2Н-1,2,3-триазол-2-ил)фенил)(6-(((6-метилпиридин-2-ил)амино)метил)-5-азаспиро[2.4]гептан-5-ил)метанона
(S)-(6-(((5-хлорпиридин-2-ил)амино)метил)-5-азаспиро[2.4]гептан-5-ил)(5-метил-2-(пиримидин-2-ил)фенил)метанона
В первом варианте осуществления изобретения, когда m=2, n предпочтительно равно 2.
В этом варианте осуществления:
- Q предпочтительно выбирают из пиридила, пиридила, замещенного одним или более заместителями, выбранными из (С1-С3)алкила, трифторметила, галогена; и
- R выбирают из фенила и 5-членного гетероароматического кольца, содержащего два гетероатома, выбранных из S, О и N, причем такое кольцо является замещенным одним заместителем, выбранным из (С1-С3)алкила, пиримидила, тиазолила, фенила, необязательно замещенного одним или более атомами галогена.
Предпочтительными соединениями в тех случаях, когда m=2, являются:
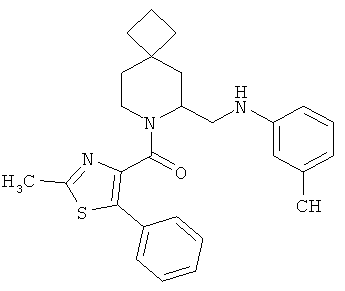
(2-метил-5-фенилтиазол-4-ил)(6-((6-метилпиридин-2-иламино)метил)-7-азаспиро[3.5]нонан-7-ил)метанон
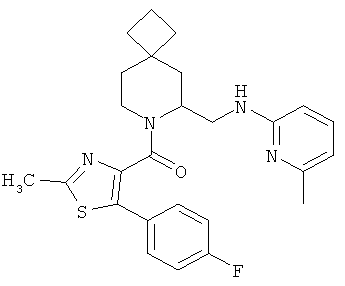
(5-(4-фторфенил)-2-метилтиазол-4-ил)(6-((6-метилпиридин-2-иламино)метил)-7-азаспиро[3.5]нонан-7-ил)метанон

(6-((5-хлорпиридин-2-иламино)метил)-7-азаспиро[3.5]нонан-7-ил)(2-метил-5-фенилтиазол-4-ил)метанон
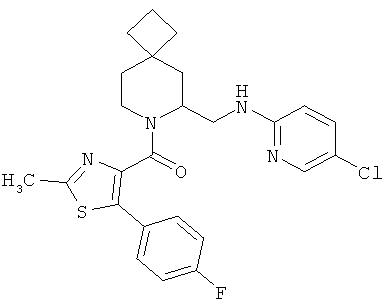
(6-((5-хлорпиридин-2-иламино)метил)-7-азаспиро[3.5]нонан-7-ил)(5-(4-фторфенил)-2-метилтиазол-4-ил)метанон
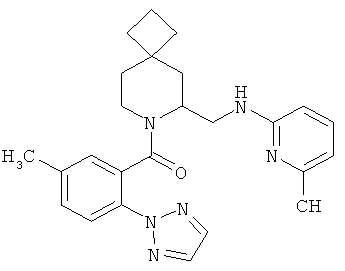
(5-метил-2-(2Н-1,2,3-триазол-2-ил)фенил)(6-((6-метилпиридин-2-иламино)метил)-7-азаспиро[3.5]нонан-7-ил)метанон
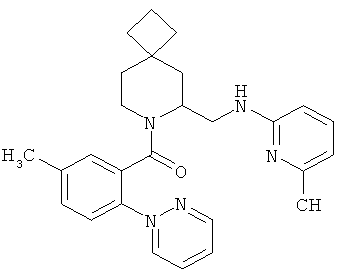
(5-метил-2-(пиримидин-2-ил)фенил)(6-((6-метилпиридин-2-иламино)метил)-7-азаспиро[3.5]нонан-7-ил)метанон
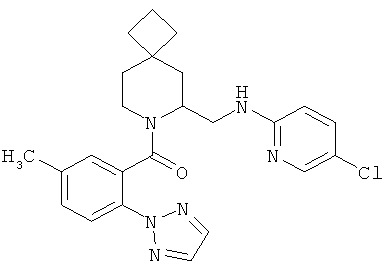
(6-((5-хлорпиридин-2-иламино)метил)-7-азаспиро[3.5]нонан-7-ил)(5-метил-2-(2Н-1,2,3-триазол-2-ил)фенил)метанон
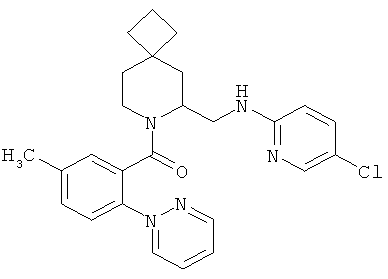
(6-((5-хлорпиридин-2-иламино)метил)-7-азаспиро[3.5]нонан-7-ил)(5-метил-2-(пиримидин-2-ил)фенил)метанон

(2-метил-5-фенилтиазол-4-ил)(6-((5-(трифторметил)пиридин-2-иламино)метил)-7-азаспиро[3.5]нонан-7-ил)метанон
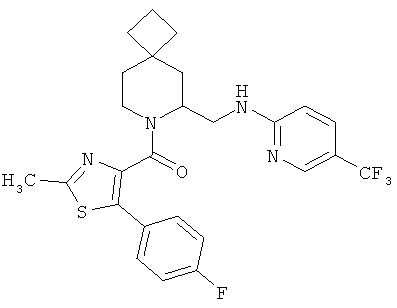
(5-(4-фторфенил)-2-метилтиазол-4-ил)(6-((5-(трифторметил)пиридин-2-иламино)метил)-7-азаспиро[3.5]нонан-7-ил)метанон
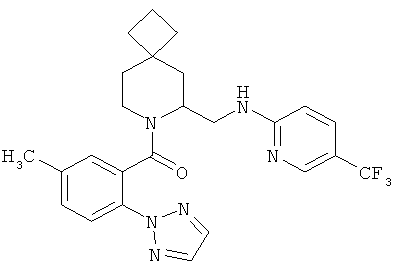
(5-метил-2-(2Н-1,2,3-триазол-2-ил)фенил)(6-((5-(трифторметил)пиридин-2-иламино)метил)-7-азаспиро[3.5]нонан-7-ил)метанон
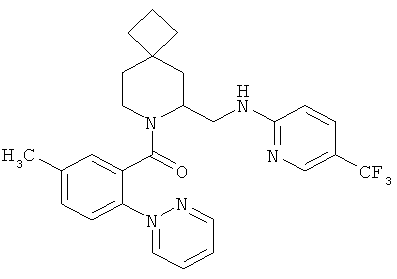
(5-метил-2-(пиримидин-2-ил)фенил)(б-((5-(трифторметил)пиридин-2-иламино)метил)-7-азаспиро[3.5]нонан-7-ил)метанон.
Когда m=3, n предпочтительно равно 2.
В этом варианте осуществления:
- Q предпочтительно выбирают из пиридила, пиридила, замещенного трифторметилом; пиридила, замещенного (С1-С3)алкилом;
- R предпочтительно выбирают из фенила и 5-членного гетероароматического кольца, содержащего два гетероатома, выбранных из S, О и N, причем такой R является замещенным одним или двумя заместителями, выбранными из (С1-С3)алкила, галогена, пиримидила, тиазолила, фенила, необязательно замещенного одним или двумя атомами галогена.
Более предпочтительно, Q представляет собой пиридил, замещенный трифторметилом или пиридил, замещенный (С1-С3)алкилом, а R представляет собой 5-членное гетероароматическое кольцо, содержащее два гетероатома, выбранных из S, О и N, еще более предпочтительно тиазолил, причем такой R является замещенным одним или двумя заместителями, выбранными из (С1-С3)алкила и галогена.
Предпочтительными соединениями в тех случаях, когда n=3, являются:
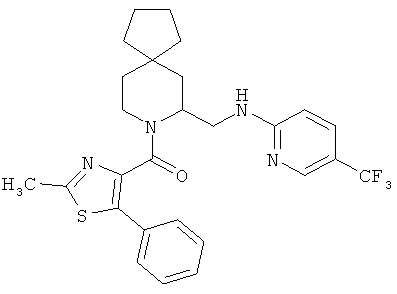
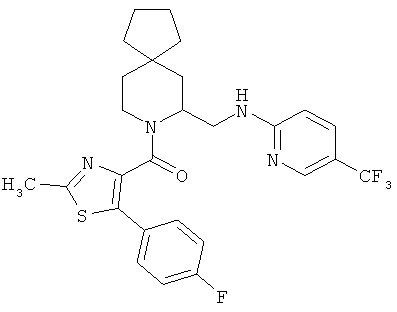
(2-метил-5-р-толилтиазол-4-ил)(7-((5-(трифторметил)пиридин-2-иламино)метил)-8-азаспиро[4.5]декан-8-ил)метанон
(5-(4-фторфенил)-2-метилтиазол-4-ил)(7-((5-(трифторметил)пиридин-2-иламино)метил)-8-азаспиро[4.5]декан-8-ил)метанон

(±)(2-метил-5-фенилтиазол-4-ил)(7-(((6-метилпиридин-2-ил)амино)метил)-8-азаспиро[4.5]декан-8-ил)метанон
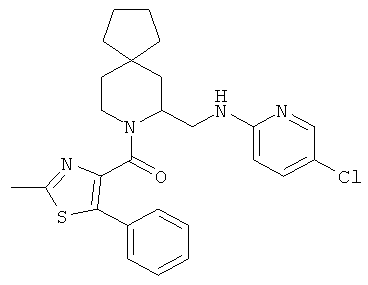
(±)(7-(((5-хлорпиридин-2-ил)амино)метил)-8-азаспиро[4.5]декан-8-ил)(2-метил-5-фенилтиазол-4-ил)метанон
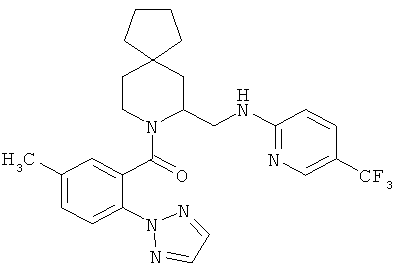
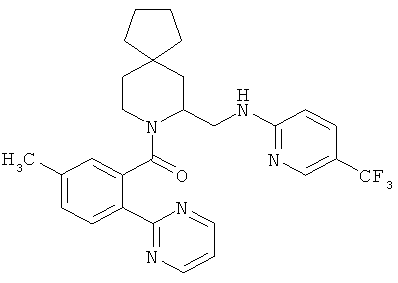
(5-метил-2-(2Н-1,2,3-триазол-2-ил)фенил)(7-((5-(трифторметил)пиридин-2-иламино)метил)-8-азаспиро[4.5]декан-8-ил)метанон
(5-метил-2-(пиримидин-2-ил)фенил)(7-((5-(трифторметил)пиридин-2-иламино)метил)-8-азаспиро[4.5]декан-8-ил)метанон.
Когда n=1, m предпочтительно равно 1.
В этом варианте осуществления:
- Q предпочтительно выбирают из пиридила, пиридила, замещенного одним или более галогенами; пиридила, замещенного (С1-С3)алкилом;
- R предпочтительно выбирают из фенила и 5-членного гетероароматического кольца, содержащего два или три гетероатома, выбранных из S, О и N, причем такой R является замещенным одним или двумя заместителями, выбранными из (С1-С3)алкила, галогена, пиримидила, тиазолила, фенила, необязательно замещенного одним или более атомами галогена, 5- или 6-членного гетероцикла, содержащего, по меньшей мере, один атом азота.
- Более предпочтительно, Q представляет собой пиридил, замещенный одним или более галогенами; пиридил, замещенный (С1-С3)алкилом, а R представляет собой фенил, замещенный одним или двумя заместителями, выбранными из (С1-С3)алкила и 5- или 6-членного гетероцикла, содержащего, по меньшей мере, один атом азота.
Предпочтительными соединениями в тех случаях, когда m=1, являются:
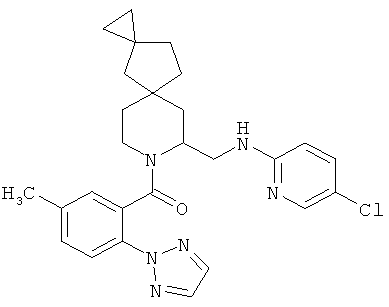
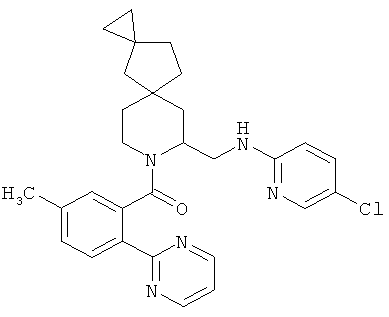
(6-((5-хлорпиридин-2-иламино)метил)-5-азаспиро[2.4]гептан-5-ил)(5-метил-2-(2Н-1,2,3-триазол-2-ил)фенил)метанон
(6-((5-хлорпиридин-2-иламино)метил)-5-азаспиро[2.4]гептан-5-ил)(5-метил-2-(пиримидин-2-ил)фенил)метанон;
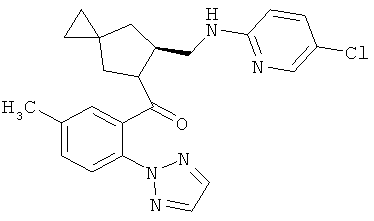
S)-(6-(((5-хлорпиридин-2-ил)амино)метил)-5-азаспиро[2.4]гептан-5-ил)(5-метил-2-(2Н-1,2,3 -триазол-2-ил)фенил)метанон
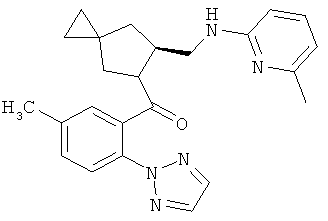
8)-(5-метил-2-(2Н-1,2,3-триазол-2-ил)фенил)(6-(((6-метилпиридин-2-ил)амино)метил)-5-азаспиро[2.4]гептан-5-ил)метанон
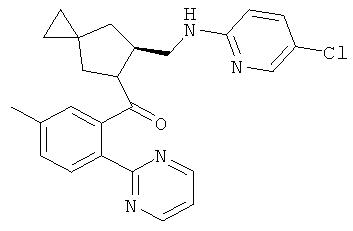
8)-(б-(((5-хлорпиридин-2-ил)амино)метил)-5-азаспиро[2.4]гептан-5-ил)(5-метил-2-(пиримидин-2-ил)фенил)метанон
Во втором варианте осуществления в спиро-аминосоединении формулы (VI) Р представляет собой COQ. Это соединение имеет формулу (VIb):
Изобретение также имеет отношение к способу получения соединения формулы (Via), включающему следующие стадии, представленные в схеме ниже:
a) защита соединения формулы (I) защитной группой BOC с получением соединения формулы (II);
b) взаимодействие соединения формулы (II) с сильными основаниями и диметилформамидом, что дает соединение формулы (III);
c) добавление амина формулы Q-NH2, где Q выбирают из группы, состоящей из фенила, пиридила, пиримидила, хинолила, изохинолила, хиноксалила, бензофуранила, имидазотриазолила, причем такой Q необязательно замещен одним или более заместителями, выбранными из группы, состоящей из (С1-С3)алкила, галогена, трифторметила, карбамидной группы, метилкарбамидной группы, карбоксигруппы, метилкарбоксигруппы, в присутствии восстановителя для получения соединения формулы (IV);
d) отщепление BOC-группы от соединения формулы (IV) для получения соединения формулы (V);
e) взаимодействие соединения формулы (V) с RCOOH в присутствии агентов сочетания или с соответствующими хлорангидридами RCOCl в присутствии основания, где R выбирают между 5- или 6-членным ароматическим кольцом и 5- или 6-членным гетероароматическим кольцом, содержащим от 1 до 3 гетероатомов, выбранных из S, О и N, причем такое кольцо является замещенным одним или двумя заместителями, выбранными из группы, состоящей из (С1-С3)алкила, галогена, (С3-С5)циклоалкилоксигруппы, (С1-С3)алкилкарбонила, фенила, необязательно замещенного одним или более атомами галогена, 5- или 6-членного гетероцикла, содержащего, по меньшей мере, один атом азота.
Кроме того, изобретение имеет отношение к способу получения соединения формулы (VIb), включающему следующие стадии, представленные в схеме ниже:
a) защита соединения формулы (I) защитной группой ВОС для получения соединения формулы (II);
b) взаимодействие соединения формулы (II) с сильными основаниями и диметилформамидом, что дает соединение формулы (III);
f) проведение восстановительного аминирования соединения формулы (III) для получения амина формулы (VII);
g) взаимодействие соединения формулы (VII) с QCOOH в присутствии агентов сочетания или с соответствующими хлорангидридами RCOCl в присутствии основания для получения амида формулы (VIII), где Q выбирают из группы, состоящей из фенила, пиридила, пиримидила, хинолила, изохинолила, хиноксалила, бензофуранила, имидазотриазолила, причем такой Q необязательно замещен одним или более заместителями, выбранными из группы, состоящей из (С1-С3)алкила, галогена, трифторметила, карбамидной группы, метилкарбамидной группы, карбоксигруппы, метилкарбоксигруппы;
h) отщепление ВОС-группы от соединения формулы (VIII) и взаимодействие с RCOOH в присутствии агентов сочетания или с соответствующими хлорангидридами RCOCl в присутствии основания, чтобы получить соединение формулы VIb, где R выбирают между 5- или 6-членным ароматическим кольцом и 5- или 6-членным гетероароматическим кольцом, содержащим от 1 до 3 гетероатомов, выбранных из S, О и N, причем такое кольцо является замещенным одним или двумя заместителями, выбранными из группы, состоящей из (С1-С3)алкила, галогена, (С3-С5)циклоалкилоксигруппы, (С1-С3)алкилкарбонила, фенила, необязательно замещенного одним или более атомами галогена, 5- или 6-членного гетероцикла, содержащего, по меньшей мере, один атом азота.
Соединения формулы (I) стадии а) являются или коммерчески доступными и/или описаны в литературе. На стадии b) в числе сильных оснований может использоваться втор-бутиллитий (втор-BuLi).
В наиболее предпочтительном варианте осуществления изобретение имеет отношение к пиперидиновым соединениям, содержащим спирокольцо из трех атомов углерода.
Кроме того, дополнительной целью изобретения является получение соединений формулы VIa, где m=1 и n=2, включающее следующие стадии:
а) взаимодействие соединения формулы (I) с трет-бутил дикарбонатом в органическом растворителе с получением соединения формулы (II);
b) взаимодействие соединений формулы (II) с сильным основанием и диметилформамидом с получением соединения формулы (III)
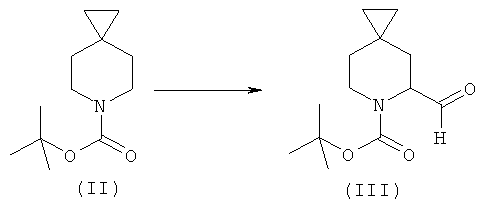
или взаимодействие соединений формулы (II) с основанием и N,N,N'N'-тетраметил этилендиамином при -60°C в органическом растворителе и затем с диметилформамидом при -78°C;
с) взаимодействие соединений формулы (III) с соединениями формулы Q-NH2 при использовании восстановителя в органическом растворителе в течение примерно 18 часов при комнатной температуре с получением соединений формулы (IV), где Q выбирают из группы, состоящей из фенила, пиридила, пиримидила, хинолила, изохинолила, хиноксалила, бензофуранила, имидазотриазолила, причем такой Q необязательно замещен одним или более заместителями, выбранными из группы, состоящей из (С1-С3)алкила, галогена, трифторметила, карбамидной группы, метилкарбамидной группы, карбоксигруппы, метилкарбоксигруппы;
d) взаимодействие соединений формулы (IV) с трифторуксусной кислотой в подходящем органическом растворителе в течение примерно 4 часов с получением соединений формулы (V);
е) взаимодействие соединений формулы (V) с соединениями формулы RCOOH с использованием подходящего конденсирующего агента и основания в органическом растворителе в течение примерно 18 часов с получением соединений формулы (Via), где R выбирают из 5- или 6-членного ароматического кольца и 5- или 6-членного гетероароматического кольца, содержащего от 1 до 3 гетероатомов, выбранных из S, О и N, причем такое кольцо является замещенным одним или более заместителями, выбранными из группы, состоящей из (С1-С3)алкила, галогена, (С3-С5)циклоалкилоксигруппы, (С1-С3)алкилкарбонила, фенила, необязательно замещенного одним или более атомами галогена, 5- или 6-членного гетероцикла, содержащего, по меньшей мере, один атом азота.
Способ получения соединений формулы VIa, где m=1 и n=2, предусматривает предпочтительное применение дихлорметана в качестве органического растворителя на стадии d) и NаВН(ОАс)3 в дихлорэтане на стадии с).
Предпочтительным агентом сочетания на стадии е) является O-(бензотриазол-1-ил) N,N,N',N'-тетраметилуронийгексафторфосфат, диизопропилэтиламин в качестве основания, диметилформамид/дихлорметан в качестве растворителей.
Согласно изобретению соединения получают, используя простой способ, удобный для увеличения в масштабе и не имеющий слишком длинных и дорогостоящих стадий получения, дающий высокий выход стабильного соединения фармацевтического качества.
Соединения изобретения как таковые или их фармацевтически приемлемую соль можно использовать в медицине, в частности в качестве антагонистов орексинового рецептора 1.
Их можно использовать в комбинации с фармацевтически приемлемым носителем и необязательно с подходящими эксципиентами для получения фармацевтических композиций. Термин "фармацевтически приемлемый носитель" означает растворители, носители, разбавители и т.п., которые используются при введении соединений изобретения.
Такие фармацевтические композиции могут быть введены парентеральным, пероральным, защечным, подъязычным, ректальным, местным или чрескожным путем.
Композиции данного изобретения, пригодные для перорального введения, можно удобно разделить на отдельные единицы дозирования, такие как таблетки, капсулы, облатки, порошки или пилюли, или использовать как жидкие суспензии.
Кроме того, таблетки могут содержать подходящие и обычно используемые в фармацевтической области эксципиенты, например предварительно желатинизированный крахмал, микрокристаллическую целлюлозу, натрия гликолят крахмала, тальк, лактозу, стеарат магния, сахарозу, стеариновую кислоту, маннитол.
Композиции для удобного парентерального введения включают стерильные препараты.
Композиции для удобного местного введения могут быть разработаны в виде кремов, паст, масел, мазей, эмульсий, пен, гелей, капель, растворов для опрыскивания и трансдермальных (чрескожных) пластырей.
Соединения изобретения могут использоваться в производстве медикамента для лечения патологий, при которых необходимо применение антагониста рецептора OX1, например, для лечения ожирения и нарушений сна, компульсивных нарушений, лекарственной и алкогольной зависимости, шизофрении.
Далее изобретение будет подробно описано с помощью следующих примеров, имеющих отношение к получению некоторых изобретательских соединений и оценке их активности в отношении рецептора OX1 и рецептора OX2.
В нижеприведенных методах после исходных материалов, как правило, дается ссылка на описание. Исходный материал необязательно должен быть приготовлен по упомянутому описанию. Стереохимию примеров определяли, исходя из предположения, что абсолютная конфигурация центров сохраняется.
Использованные в следующих примерах реагенты были коммерчески доступны от разных поставщиков (например, Sigma-Aldrich, Acros или Apollo scientific) и использовались без дополнительной очистки. Были использованы растворители в сухом виде. Реакции в безводном окружении проводили под избыточным давлением сухого N2.
Микроволновые реакции проводили на приборе Biotage Initiator 2.5.
Спектры протонного ядерного магнитного резонанса (1Н ЯМР) регистрировали с помощью прибора Bruker Avance 400 МГц. Химические сдвиги даются в м.д. (5) с использованием полосы остаточного растворителя в качестве внутреннего стандарта. Для параметров расщепления использовали следующие сокращения: s, синглет; d, дублет; t, триплет; q, квартет; m, мультиплет; b, широкий сигнал. Когда регистрировали более чем один конформер, то обычно приводили химические сдвиги наиболее распространенного конформера.
Масс-спектры (MS) регистрировали с помощью классического спектрометра Ion Trap Thermo LCQ, применяя положительный ES(+) и отрицательный ES(-) режим ионизации.
ВЭЖХ спектры получали с помощью прибора Waters Alliance 2965 и детектора UV-Vis Waters 2996. Использовали следующий метод хроматографии (с использованием Phenomenex Luna C18, 150*4.6, 5 мкм): 35 мин элюирование при 30°C, подвижная фаза состояла из разных смесей ацетонитрил/метанол/KH2PO4 (20 мМ pH 2,5), скорость потока 0,6 мл/мин.
ВЭЖХ-спектры для определения хиральной чистоты получали с помощью прибора Agilent 1200 и УФ-детектора DAD G1315D. Применяли следующий метод хроматографии (с использованием колонок Phenomenex LUX 5u cellulose-1, 250*4,6 мм): 30 мин элюирование при 30°C, подвижная фаза 90% н-гексан 10% этанол +0,1% DEA, скорость потока 0,5 мл/мин.
UPLC-спектры (спектры сверхпроизводительной жидкостной хроматографии) получали на приборе Waters Acquity UPLC-SQD, используя колонку Acquity UPLC-BEH C18 (1,7 мкМ; 50х2,1 мм).
Очистку методом препаративной хиральной ВЭЖХ проводили с помощью препаративного жидкостного хроматографа Shimadzu LC-8A и УФ-детектора SPD-20A. Использовали следующие методы хроматографии (с использованием Phenomenex LUX 5u cellulose-1, AXIA 250*21,20 мм):
А: подвижная фаза 90% н-гексан 10% этанол +0,1% DEA, скорость потока 10 мл/мин.
В: подвижная фаза 60% н-гексан 40% этанол+0,1% DEA, скорость потока 10 мл/мин.
С: подвижная фаза 93% н-гексан 7% изопропанол+0,1% DEA, скорость потока 10 мл/мин.
D: подвижная фаза 95% н-гексан 5% изопропанол+0,1% DEA, скорость потока 10 мл/мин.
Е: подвижная фаза 80% н-гексан 20% этанол+0,1% DEA, скорость потока 10 мл/мин.
Флэш-хроматографию с силикагелем проводили на силикагеле 230-400 меш (поставляемом компанией Merck AG Darmstadt, Германия); в ряде способов применяли автоматические флэш-хроматографические системы Biotage (системы Spl и Isolera), используя силикагелевые картриджи Biotage.
Тонкослойную хроматографию проводили с использованием пластин Kieselgel 60F-254 (Merck TLC), проявляли УФ светом, водным раствором перманганата, парами йода.
Пример 1. Получение промежуточного соединения 1: 6-азаспиро [2.5]октан гидробромида 5
Бензил 6-азаспиро [2-5]октан-6-карбоксилат (9 г; 36 ммоль), получение которого уже было описано, например, в WO 2008084300, растворили в 35% HB2 в AcOH (10 мл) при 0°C и перемешивали в течение 3 часов. Раствор обработали гексаном (200 мл). После декантирования растворителя добавили 80 мл Et2O. Полученное твердое вещество отфильтровали, промыли эфиром и гексаном (50 мл), затем высушили под вакуумом с получением 6,3 г промежуточного соединения 1 (светло-кремовое твердое вещество).
MS (ESI) m/z: 112 [M+H]+
1H-ЯМР (DMSO-d6) δ ppm 8.35 (m, 2H) 3.07 (m, 4H) 1.50-1.54 (m, 4H) 0.38 (s, 4H).
Пример 2. Получение промежуточного соединения 2: трет-бутил 6-азаспиро [2.5]октан-6-карбоксилата
К суспензии 6-азаспиро [2.5]октан гидробромида (промежуточное соединение 1;
3,4 г; 17,6 ммоль) в дихлорметане (50 мл), охлажденной до 0°С, добавили триэтиламин (5,14 мл; 3,9 ммоль), затем добавили раствор трет-бутил дикарбоната (4,24 г; 19,4 ммоль) в дихлорметане (20 мл) за 20 минут. Прозрачный раствор перемешивали при комнатной температуре в течение 18 часов, затем добавили дихлорметан (50 мл), органический раствор промыли водой (2×20 мл), HCl 0.5 N (20 мл) затем опять водой (2×20 мл). Органический растворитель обезвоживали (Na2SO4) и выпаривали с получением 3,7 г промежуточного соединения 2 (светло-желтое твердое вещество).
1Н-ЯМР(CDCl3) δ ppm 3.43 (m, 4H) 1.46 (s, 9H) 1.32 (m, 4H) 0.32 (s, 4H).
Пример 3. Получение промежуточного соединения 3: (±) трет-бутил 5-формил-6-азаспиро [2.5]октан-6-карбоксилата
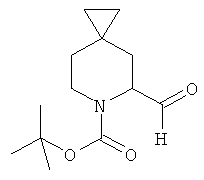
Растворенный в 20 мл Et2O трет-бутил-6-азаспиро [2.5]октан-6-карбоксилат (промежуточное соединение 2; 1 г; 4,73 ммоль) охладили до -60°C, затем добавили N,N,N',N'-тетраметилэтилендиамин (0,71 мл; 4,73 ммоль) и втор-BuLi 1,4М в гексане (4,05 мл; 5,68 ммоль). После 10 минут при -60°C температуру поднимали в течение 30 минут до -20°C, затем реакционную смесь охладили до -78°C и добавили диметилформамид (0,55 мл; 7,09 ммоль, растворенный в 5 мл Et2O). Через 30 минут медленно добавили насыщенный водный раствор NH4Cl (8 мл), затем реакционной смеси позволили достичь комнатной температуры. Реакционную смесь экстрагировали Et2O (3×50 мл), органический растворитель высушили (Na2SO4) и выпарили с получением сырого продукта, который очистили с помощью хроматографии с силикагелем (петролейный эфир/этилацетат от 95/5 до 85/15). (±) трет-бутил 5-формил-6-азаспиро [2.5]октан-6-карбоксилат получили в виде светло-желтого твердого вещества (400 мг).
MS (ESI) m/z 262 [M+Na]+.
1Н-ЯМР (СDСl3) δ ppm 9.65 (s, 1H) 4.61 (m, 1H) 4.02 (m, 1H) 3.10 (m, 1H) 2.08 (m, 1H) 1.80 (m, 1H) 1.51 (m, 1H) 1.49 (s, 9H) 0.88 (m, 1H) 0.35-0.42 (m, 4H).
Пример 4. Получение промежуточного соединения 4: (±)трет-бутил-5-((6-метилпиридин-2-иламино)метил)-6-азаспиро[2.5]октан-6-карбоксилата
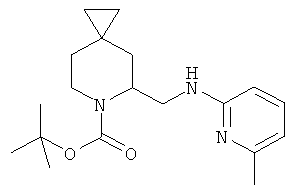
(±) трет-бутил-5-формил-6-азаспиро [2.5]октан-6-карбоксилат (промежуточное соединение 3; 400 мг; 1,67 ммоль) растворили в дихлорэтане (5 мл), затем добавили уксусную кислоту (5 экв.) и 2-амино-6-пиколин (217 мг; 2,0 ммоль). Через 3 часа при комнатной температуре добавили NaBH(OAc)3 (560 мг; 2,63 ммоль) и держали реакционную смесь при комнатной температуре, постоянно перемешивая, в течение 18 часов. Реакционную смесь выливали в водный NaHCO3 и экстрагировали этилацетатом. Органические слои объединили, высушили (Na2SO4) и концентрировали под вакуумом; полученное вещество очистили с помощью колоночной хроматографии с силикагелем (петролейный эфир/этилацетат=8/2 до 1/1). Получили 360 мг промежуточного соединения 4 в виде бесцветного масла.
MS (ESI) m/z: 354 [M+Na]+. 1Н-ЯМР (CDCl3) δ ppm 7.32 (m, 1H) 6.44 (d, 1H) 6.24 (d, 1H) 4.65 (m, 1H) 4.55 (m, 1H) 4.10 (m, 1H) 3.37 (m, 1H) 3.05 (m, 1H) 2.37 (s, 3H) 1.8-2.1 (m, 2H) 1.49 (s, 9H) 1.15 (m, 1H) 0.85 (m, H) 0.25-0.48 (m, 4H).
Пример 5. Получение промежуточного соединения 5: (±)трет-бутил-5-((5-хлорпиридин-2-иламино)метил)-6-азаспиро[2.5]октан-6-карбоксилата
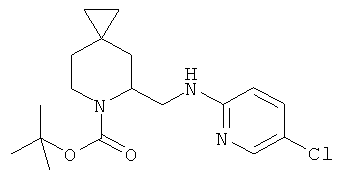
(±) терет-бутил-5-формил-6-азаспиро [2.5]октан-6-карбоксилат (промежуточное соединение 3; 450 мг; 1,88 ммоль) растворили в дихлорэтане (5 мл), затем добавили уксусную кислоту (5 экв.) и 2-амино-5-хлорпиридин (290 мг; 2,25 ммоль). Через 3 часа при комнатной температуре добавили NaBH(OAc)3 (636 мг; 3,0 ммоль) и держали реакционную смесь, постоянно перемешивая, при комнатной температуре в течение 18 часов. Реакционную смесь вылили в водный NaHCO3 и экстрагировали этилацетатом. Органические слои объединили, высушили (Na2SO4) и концентрировали под вакуумом; сырой продукт очистили колоночной хроматографией с силикагелем, используя петролейный эфир/этилацетат=8/2. Получили 240 мг промежуточного соединения 5 в виде бесцветного масла.
1Н-ЯМР (CDCl3) δ ppm 8.03 (d, 1H) 7.33 (dd, 1H) 6.33 (d, 1H) 4.7 (m, 1H) 4.57 (m, 1H) 4.13 (m, 1H) 3.85 (m, 1H) 3.36 (m, 1H) 2.99 (m, 1H) 2.16 (m, 1H) 1.88-1.95 (m, 1H) 1.42 (s, 9H) 0.99 (m, 1H) 0.82 (m, H) 0.45-0.50 (m, 2H) 0.28-0.32 (m, 2H).
Пример 6. Получение промежуточного соединения 6: (±) трет-бутил-5-((5-трифторметилпиридин-2-иламино)метил)-6-азаспиро[2.5]октан-6-карбоксилата
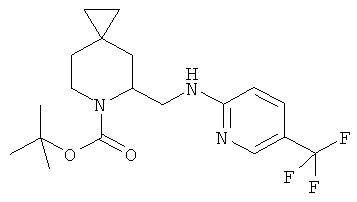
(±) трет-бутил-3-формил-6-азаспиро [2.5]октан-6-карбоксилат (промежуточное соединение 3; 450 мг; 1,88 ммоль) растворили в дихлорэтане (5 мл), затем добавили уксусную кислоту (5 экв.) и 2-амино-5-трифторметилпиридин (365 мг; 2,25 ммоль). Через 3 часа при комнатной температуре добавили NaBH(OAc)3 (636 мг; 3,0 ммоль) и держали реакционную смесь, постоянно перемешивая, при комнатной температуре в течение 18 часов. Реакционную смесь выливали в водный NaHCO3 и экстрагировали этилацетатом. Органические слои объединили, высушили (Na2SO4) и концентрировали под вакуумом; сырой продукт очистили с помощью колоночной хроматографии с силикагелем, используя градиент дихлорметан до дихлорметан/этилацетат=95/5). Получили 200 мг промежуточного соединения 6 в виде желтого масла.
MS (ESI) m/z: 408 [M+Na]+
1Н-ЯМР (CDCl3) δ ppm 8.35 (s, 1H) 7.6 (d, 1H) 6.5 (d, 1H) 5.85 (m, 1H) 5.40 (d, 1H) 5.15 (m, 1H) 3.75 (m, 1H) 3.51 (m, 1H) 2.25-2.45 (m, 2H) 1.40-1.75 (m, 10H) 0.3-0.55 (m, 4H).
Пример 7. Получение промежуточного соединения 7: (±) N-(6-азаспиро[2.5]октан-5-илметил)-6-метилпиридин-2-амина
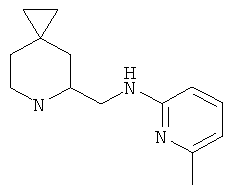
(±) трете-бутил-5-((6-метилпиридин-2-иламино)метил)-6-азаспиро[2.5]октан-6-карбоксилат (промежуточное соединение 4; 350 мг; 1,05 ммоль) растворили в дихлорметане (10 мл) и охладили до 0°C, затем добавили трифторуксусную кислоту (2 мл) Через 1 час при 0°C и еще 2 часа при комнатной температуре раствор выпарили, вновь растворенный в дихлорметане остаток промыли насыщенным водным раствором NaHCO3. Органические слои высушили (Na2SO4) и концентрировали под вакуумом. Сырой продукт очистили с помощью колоночной хроматографии с силикагелем (CHCl3/MeOH=8/2). Получили 65 мг промежуточного соединения 7 в виде светло-желтого масла.
MS (ESI) m/z: 232 [М+Н]+
1Н-ЯМР (CDCl3) δ ppm 7.32 (m, 1H) 6.46 (d, 1H) 6.32 (d, 1H) 5.75 (m, 1H) 3.45-3.65 (m, 4H) 3.05 (m, 1H) 2.42 (s, 3Н) 2.06-2.18 (m, 1H) 1.0-1.29 (m, 2H) 0.36-0.52 (m, 4H).
Пример 8. Получение промежуточного соединения 8: (±) (6-азаспиро[2.5]октан-5-илметил)-5-хлорпиридин-2-амина
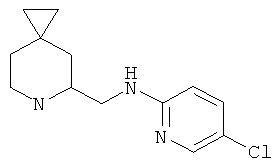
(±) трет-бутил-5-((5-хлорпиридин-2-иламино)метил)-6-азаспиро[2.5]октан-6-карбоксилат (промежуточное соединение 5; 240 мг; 0,68 ммоль) растворили в дихлорметане (10 мл) и охладили до 0°C, затем добавили трифторуксусную кислоту (2 мл). Через 1 час при 0°C и еще 2 часа при комнатной температуре раствор выпарили, вновь растворенный в дихлорметане остаток промыли насыщенным водным раствором NaHCO3. Объединенные органические слои высушили (Na2SO4) и концентрировали под вакуумом; сырой продукт очистили с помощью колоночной хроматографии с силикагелем (градиент дихлорметана до дихлорметан/MeOH=9/1). Получили 150 мг промежуточного соединения 8 в виде светло-желтого масла.
1Н-ЯМР (CDCl3) δ ppm 7.96 (d, 1H) 7.30 (m, 1H) 6.42 (d, 1H) 6.25 (m, 1H) 3.74 (m, 1H) 3.37-3.55 (m, 3H) 2.93 (m, 1H) 2.20 (m, 1H) 2.0-2.66 (m, 1H) 1.20 (m, 1H) 1.04 (m, 1H) 0.42-0.58 (m, 4H).
Пример 9. Получение промежуточного соединения 9: (±) N-(6-азаспиро[2.5]октан-5-илметил)-5-(трифторметил)пиридин-2-амина
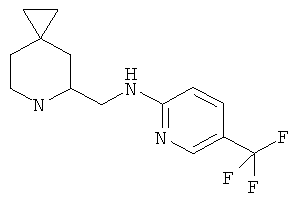
(±) трет-бутил-5-((5-трифторметилпиридин-2-иламино)метил)-6-азаспиро[2.5]октан-6-карбоксилат (промежуточное соединение 5; 200 мг; 0,520 ммоль) растворили в дихлорметане (10 мл) и охладили до 0°C, затем добавили трифторуксусную кислоту (2 мл). Через 1 час при 0°C и 18 часов при комнатной температуре раствор выпарили, остаток, растворенный в дихлорметане, промыли насыщенным водным раствором NaHCO3. Объединенные органические слои высушили (Na2SO4) и концентрировали под вакуумом; полученный сырой продукт очистили колоночной хроматографией с силикагелем (градиент от дихлорметан/этилацетат=9/1 до дихлорметан/МеОН=9/1). Получили 60 мг промежуточного соединения 9 в виде светло-желтого масла.
MS (ESI) m/z: 286 [М+Н]+.
Пример 10. Получение промежуточного соединения 10: метил 2-(6-азаспиро[2.5]октан-5-илметиламино)-5-хлорбензоата
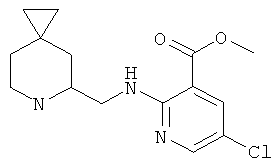
К раствору (±) трет-бутил-5-формип-6-азаспиро [2.5]октан-6-карбоксилата (промежуточное соединение 3; 160 мг; 0,669 ммоль) в дихлорэтане (10 мл) добавили метил-2-амино-5-хлорбензоат (149 мг; 0,802 ммоль) и ледяной AcОН (0,191 мл; 3,343 ммоль). Реакционной смеси давали дойти до комнатной температуры в течение 3 часов, затем добавили натрий триацетоксиборогидрид (227 мг; 1,070 ммоль) и перемешивали реакционную смесь при той же самой температуре в течение ночи. Смесь разбавили дихлорметаном и промыли насыщенным раствором NAHСО3 (2х). Органическую фазу отделили на разделительном картридже и выпарили. Полученный остаток (273 мг; 0,669 ммоль) растворили в смеси трифторуксусная кислота:дихлорметан 3:1 (1,2:0,4 мл), охлаждая на ледяной бане. Затем реакционную смесь перемешивали в течение 2 часов при комнатной температуре. Растворители выпарили, остаток растворили в дихлорметане и промыли раствор насыщенным раствором NaHCO3 (2х). После выпаривания органической фазы остаток очистили с помощью SPE-Si картриджа (5 г), элюируя смесью дихлорметан:
МеОН (от дихлорметана до дихлорметан:МеОН 95:5). Получили 78 мг промежуточного соединения 10.
1Н ЯМР: (CDCl3) δ ррт 7.87-7.86 (d, 2Н), 7.3-7.27 (dd, 1Н) 6.68-6-66 (d, 1H) 3.87 (s, 3H) 3.25-3.21 (т, 2Н) 3.15-3.11 (т, 1Н) 3.06-3.0 (т, 1Н) 2.86-2.79 (dt, 1Н) 2.74 (bs, 1Н) 1.98-1.91 (dt, 1Н) 1.78-1.72 (t, 1Н) 0.97-0.93 (т, 1Н) 0.88-0.83 (т, 1Н) 0.41-0.35 (т, 2Н) 0.32-0.28 (m, 2H)
Пример 11. Получение промежуточного соединения 12: 4-метилового эфира (S)-2-[трет-бутоксикарбонил-(2-метоксикарбонилэтил)амино]янтарной кислоты
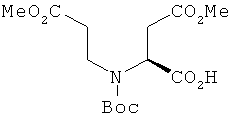
К раствору хлорангидрида 4-метилового эфира (8)-2-аминоянтарной кислоты (промежуточное соединение 11; 250 г; 1,36 моль) в воде (600 мл), охлажденного до 0°C, добавили триэтиламин (474 мл; 3,4 моль) и метилакрилат (368 мл; 4,09 моль). Смесь сильно перемешали и нагрели до комнатной температуры. После промывки петролейным эфиром (2×2 л) добавили трет-бутанол (200 мл) и Boc2O (370 г; 1,70 моль) и сильно перемешивали в течение 16 часов. Смесь промыли петролейным эфиром (2×2 л). Водный раствор охладили до 0°С, а значение pH довели до 3,0 с помощью концентрированной НСl. Продукт экстрагировали с этилацетатом (3×1 л), органические экстракты объединили, промыли насыщенным NaCl (1 л), высушили (Na2SO4) и выпарили с получением 420 г (выход 92%) 4-метилового эфира (S)-2-[трет-бутоксикарбонил-(2-метоксикарбонил-этил)-амино]-янтарной кислоты.
1H-ЯМР (CDCl3) δ ppm 5.45 (s, 1Н), 4.44 (m, 1Н), 3.80-3.87 (m, 1Н), 3.71 (s, 3H), 3.68 (s, 3H), 3.45-3.53 (m, 1H), 3.20 (m, 1H), 2.57-2.90(m, 3H), 1.43 (s, 9H)
Пример 12. Получение промежуточного соединения 13: трет-бутиламиновой соли 1-трет-бутилового эфира (S)-4-оксо-пиперидин-1,2-дикарбоновой кислоты
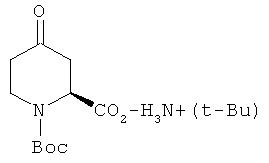
К раствору 4-метилового эфира (S)-2-[трет-бутоксикарбонил-(2-метоксикарбонил-этил)-амино]янтарной кислоты (промежуточное соединение 12; 420 г; 1,35 моль) в THF (2л) в атмосфере азота, охлажденного до 0°C, добавили NaOMe/MeOH (1,5 л; 2,7М) за полчаса. Полученный желтый раствор перемешивали при 85°C в течение 3 часов (через 1 час была получена суспензия). THF (1,5 л) отогнали при пониженном давлении и добавили воду (2 л). Полученную смесь перемешивали при 110°С в течение 20 часов. Смесь промыли этилацетатом (2×1 л). Водный раствор охладили до 0°С, а значение pH довели до 2,5 с помощью концентрированной НСl. Продукт экстрагировали этилацетатом (3×1 л), органические экстракты объединили, промыли насыщенным NaCl (1 л), высушили (Na2SO4) и профильтровали. Фильтрат охладили до 0°C и добавили при перемешивании трет-бутиламин (145 мл; 1,35 моль). Желтое твердое вещество собрали фильтрованием и высушили, затем кипятили в изопропиловом спирте (1,5 л). Суспензию охладили до 5°C и собрали фильтрованием, что дало 180 г промежуточного соединения 3 (легкое белое твердое вещество, выход:45%).
1Н-ЯМР (D2O) δ ppm 8.94 (s, 1H), 5.10 (m, 1H), 4.06 (m, 1H), 3.70 (m, 1H), 2.86(m, 2H),2.55(m,2H),1.47(s,9H)
[α]D 25 - 14.4(c1.0, H2O)
Пример 13. Получение промежуточного соединения 14: 1-трет-бутилового эфира 2-бензил- (8)-4-оксо-пиперидин-1,2-дикарбоксилата
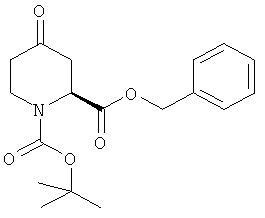
К суспензии трет-бутиламиновой соли 1-трет-бутилового эфира (S)-4-оксо-пиперидин-1,2-дикарбоновой кислоты (промежуточное соединение 13; 300 г; 0,95 моль) в этилацетате (2,5 л) перемешивали при 0°С медленно добавили водную НСl 0.5N (3 л), насыщенную NaCl. После полного растворения органический слой декантировали, промыли насыщенным NaCl, высушили (Na2SO4) и выпарили с выходом 200 г кислотного производного. К раствору кислотного производного (200 г; 0,82 моль) в безводном дихлорметане (1,5 л) добавили бензиловый спирт (88,9 г; 0,82 моль), N,N'-дициклогексилкарбодиимид (186 г; 0,91 моль), 4-диметиламинпиридин (11 г; 0,09 моль) при 0°C, затем нагрели до комнатной температуры и перемешивали при комнатной температуре в течение 10 часов. Смесь профильтровали, а жидкую фазу выпарили с получением сырого продукта, который очистили хроматографией с силикагелем (петролейный эфир/этилацетат от 10/1 до 5/1). Получили 1-трет-бутиловый эфир 2-бензил-(S)-4-оксо-пиперидин-1,2-дикарбоксилата в виде бесцветного масла (210 г), выход:73%.
1Н-ЯМР (CDCl3) δ ppm 7.34-7.41 (m, 5H), 5.20 (s, 2H), 4.91 (m, 1H), 4.08 (m, 1H), 3.70 (m, 1H), 2.82 (m, 2H), 2.51 (m, 2H), 1.45 (d, 9H)
Пример 14. Получение промежуточного соединения 15: 1-трет-бутиловый эфир 2-бензил-(8)-4-метилен-пиперидин-1,2-дикарбоксилата

К суспензии метил(трифенил)фосфонийбромида (248 г; 0,69 моль) в безводном толуоле (1 л) добавили гексаметилдисилазид натрия (258 мл; 0,69 моль) при 0°С, после завершения добавления реакционную смесь нагрели до комнатной температуры и перемешивали при комнатной температуре в течение 1 часа, затем охладили до 0°С и добавили раствор 1-трет-бутилового эфира 2-бензил-(S)-4-оксо-пиперидин-1,2-дикарбоксилата (промежуточное соединение 14; 210 г; 0,63 моль) в безводном толуоле (0,5 л). Полученную смесь нагрели до комнатной температуры и перемешивали при комнатной температуре в течение 1 часа, добавили воду (500 мл), экстрагировали этилацетатом. Объединенные органические экстракты высушили (Na2SO4), профильтровали и выпарили с получением сырого продукта, который очистили с помощью хроматографии с силикагелем (петролейный эфир/этилацетат от 10/1 до 5/1). Получили 1-трет-бутиловый эфир 2-бензил-(S)-4-метилен-пиперидин-1,2-дикарбоксилата в виде бесцветного масла (120 г), выход 57,4%.
1Н-ЯМР (CDCl3) δ ppm 7.29-7.41 (m, 5H), 4.73-5.25 (m, 5H), 4.14 (m, 1H), 3.06 (m, 1H), 3.80(m. 1H), 2.47(m, 1H), 2.13 (m, 2H), 1.35 (d, 9H)
Пример 15. Получение промежуточного соединения 16: 6-трет-бутиловый эфир 5-бензил-(S)-6-аза-спиро[2.5]октан-5,6-дикарбоксилата
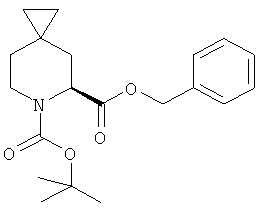
К раствору 1-трет-бутилового эфира 2-бензил-(8)-4-метилен-пиперидин-1,2-дикарбоксилата (промежуточное соединение 15, полученное методом А, 120 г; 0.36 моль) в безводном THF (1 л) медленно добавили диазометан в эфире (500 мл) [который был приготовлен из метил-3-нитро-1-нитроксогуанидина (213 г) в 40% КОН (1 л)] при температуре от -25°С до -35°С в атмосфере азота и медленно нагрели до комнатной температуры (около 4 часов) и перемешивали при комнатной температуре в течение 10 часов. Смесь профильтровали, жидкую фазу выпарили и получили сырой продукт, который очистили с помощью хроматографии с силикагелем (петролейный эфир/этилацетат от 10/1 до 5/1). Получили 6-трет-бутиловый эфир 5-бензил-(S)-6-Аза-спиро[2.5]октан-5,6-дикарбоксилата в виде бесцветного масла (110 г), выход:88%.
1Н-ЯМР (CDCl3) δ ppm 7.36 (m, 5Н), 5.14-5.33 (m, 2Н), 4.90 (m, 1Н), 3.98 (m, 1Н), 3.17 (m, 1Н), 2.19 (m, 1Н), 1.92 (m, 1Н), 1.42 (d, 9Н), 0.80 (m, 1Н), 0.10-0.30 (m, 4Н)
Пример 16. Получение промежуточного соединения 17: 6-трет-бутилового эфира (S)-6-аза-спиро[2.5]октан-5,6-дикарбоновой кислоты
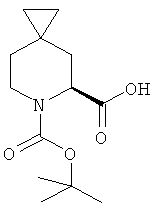
К раствору 6-трет-бутилового эфира 5-бензил-(S)-6-аза-спиро[2.5]октан-5,6-дикарбоксилата (промежуточное соединение 16; 110 г; 0,32 мол, ее:66%) в безводном метаноле (1 л) добавили Pd/BaSO4 (50 г) в атмосфере водорода, реакционную смесь перемешивали при комнатной температуре в течение ночи. Полученную смесь профильтровали, а жидкую фазу выпарили, чтобы получить 68 г 6-трет-бутилового эфира (S)-6-аза-спиро[2.5]октан-5,6-дикарбоновой кислоты (легкое белое твердое вещество, выход: 85%) которую перекристаллизовывали с РЕ/EtOAc (4:1) с получением 38 г 6-трет-бутилового эфира (S)-6-аза-спиро[2.5]октан-5,б-дикарбоновой кислоты.
1Н-ЯМР (CDCl3) δ ppm 12.67 (s, 1Н), 4.60 (m, 2Н), 3.85 (m, 1Н), 3.10 (m, 1Н), 2.08 (m, 1H), 1.79 (m, 1H), 1.40 (d, 9H), 0.84 (m, 1H), 0.28-0.32 (m, 4H)
Пример 17. Получение промежуточного соединения 18: трет-бутилового эфира (S)-5-гидроксиметил-6-аза-спиро[2.5]октан-6-карбоновой кислоты
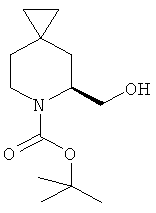
К раствору 6-трет-бутилового эфира (S)-6-аза-спиро[2.5]октан-5,6-дикарбоновой кислоты (промежуточное соединение 17; 35 г; 0,122 моль) в безводном THF (300 мл) добавили BH3/THF (1М; 360 мл; 0,3б5 моль) в атмосфере азота при 0°С.Когда добавление было сделано, реакционную смесь нагревали до комнатной температуры и перемешивали при комнатной температуре в течение ночи. Полученную смесь выпаривали, чтобы получить сырой продукт, который очищали с помощью хроматографии с силикагелем (петролейный эфир/этилацетат от 10/1 до 5/1), трет-бутиловый эфир (S)-5-гидроксиметил-6-аза-спиро[2.5]октан-6-карбоновой кислоты был получен в виде бесцветного масла (31 г, выход 86%).
1Н-ЯМР (CDCl3) δ ppm 4.40 (m, 1H), 4.01 (m, 1H), 3.66 (m, 1H), 3.06 (m, 1H), 2.03 (m, 1H), 1.87 (m, 1H), 1.50 (s, 9H), 1.02 (d, 1H), 0.85 (d, 1H), 0.27-0.43 (m, 4H);
MS Calcd.: 241; MS Found: 142 ([M-100+1]+).
Альтернативно промежуточное соединение 18 получали из промежуточного соединения 16. К суспензии алюмогидрида лития (0,57 г; 15 ммоль) в безводном THF (30 мл) добавили раствор 6-трет-бутилового эфира 5-бензил-(S)-6-аза-спиро[2.5]октан-5,6-дикарбоксилата (промежуточное соединение 16; 3,45 г; 10 ммоль) в THF (20 мл) при 0°С, после того как добавление было закончено, реакционную смесь перемешивали при 0°С в течение 2 часов. Полученную смесь гасили Na2SO4·10H2O, фильтровали, а фильтрат высушивали (Na2SO4) и выпаривали с получением сырого продукта, который очищали с помощью хроматографии с силикагелем (петролейный эфир/этилацетат от 10/1 до 5/1). Получили трет-бутиловый эфир (S)-5-гидроксиметил-6-аза-спиро[2.5]октан-6-карбоновой кислоты в виде бесцветного масла (2,0 г; выход 83%).
Пример 18. Получение промежуточного соединения 19: (S) трет-бутил 5-формил-6-азаспиро [2.5]октан-6-карбоксилата
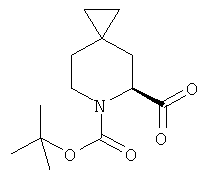
К растворенному в 80 мл DCM (S)-трет-бутил-5-(гидроксиметил)-6-азаспиро[2.5]октан-6-карбоксилату (промежуточное соединение 18; 20 г; 82,0 ммоль) добавили TEMPO (2,6 г; 16 ммоль) и BAIB (29,3 г; 90 ммоль). Через 2 часа при 25°C реакционную смесь разбавили DCM (150 мл), промыли водным раствором Na2S2O3 затем водой, высушили (Na2SO4) и выпарили, чтобы получить сырой продукт, который очистили с помощью хроматографии с силикагелем (петролейный эфир до петролейный эфир/этилацетат 95/5). Получили (S) трет-бутил-5-формил-6-азаспиро [2.5]октан-6-карбоксилат в виде светло-желтого твердого вещества (7 г).
1Н-ЯМР (CDCl3) δ ppm 9.66 (s, 1H), 4.68 (m, 1H), 3.97 (m, 1H), 3.13 (m, 1H), 2.07 (m, 1H), 1.83 (m, 1H), 1.50 (m, 10H), 0.88 (m, 1H), 0.35-0.50 (m, 2H), 0.20-0.30 (m, 2H)
Пример 19. Получение промежуточных соединений 20-24
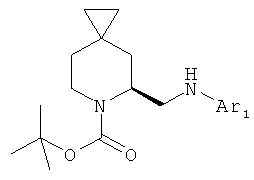
Общая методика 1:
(S) трет-бутил 5-формил-6-азаспиро [2.5]октан-6-карбоксилат (промежуточное соединение 19:1 экв.) растворили в дихлорэтане (2,5-5 мл/ммоль), затем добавили уксусную кислоту (5 экв.) и соответствующий 2-аминопиридин (1,2 экв.). Через 1-3 часа при комнатной температуре добавили NaBH(OAc)3 (1,6 экв.), реакция продолжалась при комнатной температуре и при перемешивании в течение 18 часов. Реакционную смесь выливали в водный NaHCO3 и экстрагировали этилацетатом. Органические слои объединили, высушили (Na2SO4) и концентрировали под вакуумом; сырой продукт очистили с помощью колоночной хроматографии с силикагелем, используя петролейный эфир/этилацетат=8/2 до 1/1 или DCM/MeOH=98/2 до 9/1. Промежуточные соединения 20-24 получали в виде масел.
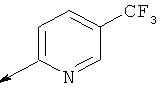
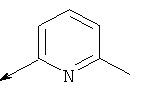
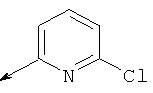
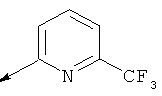
Следующие промежуточные соединения были получены согласно Общей методике 1:
Характеристика промежуточных соединений 20-24:

Пример 20. Получение промежуточного соединения 25: трет-бутилового эфира (S)-5-(1,3-диоксо-1,3-дигидро-изоиндол-2-илметил)-6-аза-спиро[2.5]октан-6-карбоновой кислоты
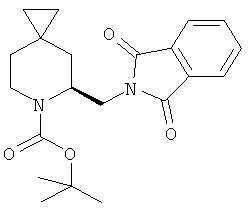
К раствору трет-бутилового эфира (S)-5-гидроксиметил-6-аза-спиро[2.5]октан-6-карбоновой кислоты (промежуточное соединение 18; 10 г; 42 ммоль) в THF (300 мл) добавили трифенилфосфин (13 г; 50 ммоль), фталимид (6,2 г; 42 ммоль) и диэтил-азодикарбоксилат (21,75 г; 50 ммоль) при 0°С в атмосфере азота. После того как добавление было закончено, реакционную смесь нагревали до комнатной температуры и перемешивали при комнатной температуре 10 часов. Полученную смесь выпарили и получили сырой продукт, который очистили хроматографией с силикагелем (петролейный эфир/этилацетат от 10/1 до 5/1). Получили трет-бутиловый эфир (S)-5-(1,3-Диоксо-1,3-дигидро-изоиндол-2-илметил)-6-аза-спиро[2.5]октан-6-карбоновой кислоты в виде легкого белого твердого вещества (13 г; выход 83,6%).
1Н-ЯМР (400 MHz, CDCl3) δ ppm 7.73-7.92 (m, 5H), 4.79 (m, 1H), 4.5 l(m, 1H), 4.08 (m, 1H), 3.48(m, 1H), 3.33(m, 1H), 2.21(m, 1H), 1.93(m, 1H), 1.25(m, 1H), 1.09 (s, 9H), 0.88 (m,lH), 0.33-0.6 l(m,4H)
Пример 21. Получение промежуточного соединения 26: трет-бутилового эфира (S)-5-аминометил-6-аза-спиро[2.5]октан-6-карбоновой кислоты

К раствору трет-бутилового эфира (S)-5-(1,3-диоксо-1,3-дигидро-изоиндол-2-илметил)-6-аза-спиро [2.5]октан-6-карбоновой кислоты (промежуточное соединение 25; 12 г; 32,4 ммоль) в этаноле (150 мл) добавили гидрат гидразина (8,1 г; 162 ммоль). После того как добавление было закончено, реакционную смесь перемешивали при 80°С в течение 5 часов. Полученную смесь профильтровали, фильтрат выпарили с получением сырого продукта, который очистили с помощью хроматографии с силикагелем (петролейный эфир/этилацетат от 10/1 до 1/1). Получили трет-бутиловый эфир (S)-5-аминометил-6-аза-спиро[2.5]октан- 6-карбоновой кислоты в виде бесцветного масла (6,88 г; выход 86%).
1Н-ЯМР (400 МН2, СDСl3) δ ppm 4.28 (m, 1H), 4.06 (m, 1H), 3.20 (m, 1H), 2.96 (m, 1H), 1.79 (m, 1H), 2.10(m, 1H), 1.89(m, 1H), 1.49(s, 9H), 0.95(d, 1H), 0.80 (d, 1H), 0.27-0.44 (m, 4H);
Пример 22. Получение промежуточных соединений 27-33
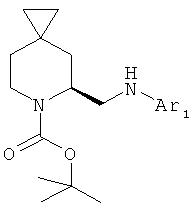
Общая методика 2
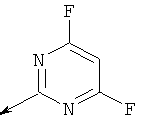
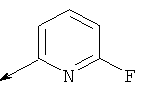
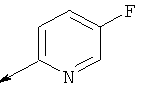
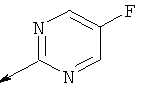
К раствору трет-бутилового эфира (S)-5-аминометил-6-аза-спиро[2.5]октан-6-карбоновой кислоты (промежуточное соединение 26; 2 ммоль) в изопропаноле (50 мл) и N,N-диизопропилэтиламина (3 ммоль) добавили Ar1-X (где Х=2 Cl, 2F или 2 Вr; 1,1 ммоль). После добавления реакционную смесь перемешивали до превращения исходного вещества и (в зависимости от Ar1-X) в температурном диапазоне от -20° до 90°С. Полученную смесь выпарили и получили сырой продукт, который очистили с помощью хроматографии с силикагелем (петролейный эфир/этилацетат от 10/1 до 3/1).
Следующие промежуточные соединения были получены согласно Общей методике 2:

Пример 23. Получение промежуточных соединений 34-45

Общая методика 3:
Одно из промежуточных соединений 20-24 и 27-33 (1 экв.) растворили в дихлорметане (10 мл/ммоль) и охладили до 0°С, затем добавили трифторуксусную кислоту (2 мл/ммоль). Через 1 час при 0°C и 2 часа при комнатной температуре раствор выпарили, остаток, вновь растворенный в дихлорметане, промыли насыщенным водным раствором NaHCO3. Органические слои высушили (Na2SO4) и концентрировали под вакуумом. Сырой продукт очистили с помощью колоночной хроматографии с силикагелем (СНСl3/МеОН=8/2). Промежуточные соединения были получены в виде светло-желтых масел.
Общая методика 4:
К раствору одного из промежуточных соединений 20-24 и 27-33 в этилацетате добавили HCl(g)/EtOAc (4,0 M) при 0°С. После добавления реакционную смесь перемешивали при 0°С в течение 1 часа. Полученные твердые вещества собрали фильтрованием, промыли петролейным эфиром и высушили, получив промежуточные соединения в виде гидрохлоридов.
Следующие промежуточные соединения были получены по Общей методике 3 или 4:
Характеристика промежуточных соединений 34-45:
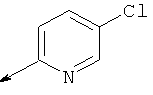
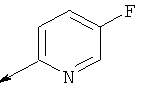
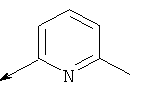
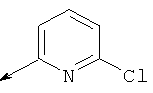

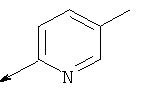
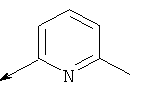
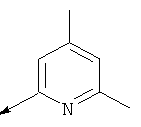
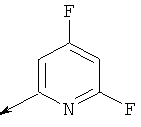
Пример 24. Получение промежуточных соединений 46-50
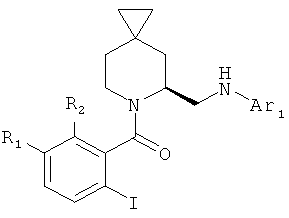
Общая методика 5
Промежуточные соединения 34-45 (1 экв.) растворили в безводном дихлорметане (10 мл/ммоль) при 0°C с TEA (3 экв.), добавили соответствующий 2-иодобензоилхлорид, растворенный в безводном дихлорметане. Через 2 часа смесь выливали в водный NaHCO3 и экстрагировали дихлорметаном. Органические слои объединили, высушили (Na2SO4) и концентрировали под вакуумом; сырой продукт очистили с помощью колоночной хроматографии с силикагелем (Гексан/AcOEt 9/1).
Следующие промежуточные соединения были получены согласно Общей методике 5:
Характеристика промежуточных соединений 46-50:
Пример 25. Получение промежуточного соединения 52: 2-метилового эфира 1-трет-бутил-(2S,4R)-4-(трет-бутил-диметил-силанилокси)-пирролидин-1,2-дикарбоксилата
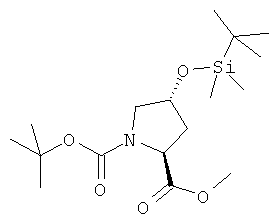
К раствору 2-метилового эфира 1-трет-бутил-(2S,4R)-4-гидрокси-пирролидин-1,2-дикарбоксилата (промежуточное соединение 51; 38,7 г; 160 ммоль) в дихлорметане (700 мл) при 0°С добавили имидазол (21,7 г; 320 ммоль) и трет-бутилдиметилсилил хлорид (26,2 г; 170 моль). Смесь перемешивали при комнатной температуре в течение 4 часов. Смесь профильтровали, затем жидкую фазу промыли HCl 0,5N (500 мл), насыщенным водным NaHCO3 (500 мл), затем рассолом (2×500 мл). Органический растворитель осушили (Na2SO4) и выпарили с получением 58 г промежуточного соединения 52 (желтое твердое вещество, выход 100%).
1Н-ЯМР (400 MHz, CDCl3) δ ppm 4.34-4.46 (m, 2H), 3.75 (m, 3H), 3.58-3.65 (m, 1H), 3.32-3.44 (m, 1H), 2.20 (m, 1H), 2.04 (m, 1H), 1.48-1.43 (d, 9H), 0.92 (s, 9H), 0.05 (s, 6H).
Пример 26. Получение промежуточного соединения 53: трет-бутилового эфира (2S,4R)-4-(трет-бутил-диметил-силанилокси)-2- гидроксиметил-пирролидин-1-карбоновой кислоты

К суспендированному раствору алюмогидрида лития (7,2 г; 190 ммоль) в THF (200 мл) в атмосфере азота по каплям добавили 2-метиловый эфир 1-трет-бутил-(2S,4R)-4-(трет-бутил-диметил-силанилокси)-пирролидин-1,2-дикарбоксилата (промежуточное соединение 52, 58 г; 160 ммоль) в THF (300 мл) при 0°С. Раствор перемешивали при 0°С 2 часа, затем добавили Na2SO4·10H2O, а смесь профильтровали, фильтрат концентрировали, чтобы получить 51,4 г промежуточного соединения 53 (желтое масло, выход 96%).
1Н-ЯМР (400 MHz, CDCl3) δ ppm 4.95 (m, 1H), 4.30 (m, 1H), 4.15 (m, 1H), 3.71 (m, 1H), 3.57 (m, 1H), 3.45 (m, 1H), 3.36 (m, 1H), 1.98 (m, 1H), 1.49 (s, 9H), 0.89 (s, 9H), 0.06 (s, 6H)
Пример 27. Получение промежуточного соединения 54: трет-бутилового эфира (2S,4R)-4-(трет-бутил-диметил-силанилокси)-2-(4-Нитро-бензоилоксиметил)-пирролидин-1-карбоновой кислоты

трет-Бутиловый эфир (2S,4R)-4-(трет-бутил-диметил-силанилокси)-2-гидроксиметил-пирролидин-1-карбоновой кислоты (промежуточное соединение 53; 51,4 г; 155 ммоль), растворенный в 500 мл дихлорметана, охладили до 0°С, затем добавили p-нитробензойную кислоту (28,5 г; 171 ммоль), N,N'-дициклогексилкарбодиимид (35,2 г; 171 ммоль) и 4-диметиламинопиридин (1,90 г; 1,б0 ммоль). Смесь перемешивали при комнатной температуре в течение 18 часов, затем профильтровали; фильтрат концентрировали с получением сырого продукта, который очистили с помощью хроматографии с силикагелем (петролейный эфир/этилацетат от 15/1 до 10/1). Получили трет-бутиловый эфир (2S,4R)-4-(трет-бутил-диметил-силанилокси)-2-(4-нитро-бензоилоксиметил)пирролидин-1-карбоновой кислоты в виде белого твердого вещества (70 г; выход 94%).
1Н-ЯМР (400 MHz, CDCl3) δ ppm 8.32 (d, 2H), 8.23 (d, 2H), 4.44-4.56 (m, 4H), 3.42-3.58 (m, 2H), 2.08 (m, 1H), 1.96 (m, 1H), 1.48 (s, 9H), 0.89 (s, 9H), 0.08 (s, 6H)
Пример 28. Получение промежуточного соединения 55: трет-бутилового эфира (2S,4R)-4-гидрокси-2-(4-нитро-бензоил оксиметил)-пирролидин-1-карбоновой кислоты

К раствору трет-бутилового эфира (2S,4R)-4-(трет-бутил-диметил-силанилокси)-2-(4-нитро-бензоилоксиметил)-пирролидин-1-карбоновой кислоты (промежуточное соединение 54; 35 г; 72,9 ммоль) в 300 мл пиридина добавили по каплям 70% пиридин гидрофторид (200 мл) при 0°С. 2 часа держали при комнатной температуре, затем температуру понизили до 0°С и добавили 1500 мл воды, затем позволили реакционной смеси достичь комнатной температуры. Реакционную смесь экстрагировали этилацетатом (2×1,5 л), органический растворитель промыли 1 N HCl (2,0 л), насыщенным водным NaHCO3 (2,0 л) и рассолом (2,0 л), высушили (Na2SO4) и выпарили, чтобы получить 23 г промежуточного соединения 55 (желтое твердое вещество, выход 92%).
1Н-ЯМР (400 MHz, CDCl3) δ ppm 8.32 (d, 2H), 8.20 (d, 2H), 4.34-4.59 (m, 4H), 3.48-3.70 (m, 2H), 1.99-2.20 (m, 3H), 1.46 (s, 9H)
Пример 29. Получение промежуточного соединения 56: трет-бутилового эфира (S)-2-(4-нитро-бензоилоксиметил)-4-оксо-пирролидин-1-карбоновой кислоты
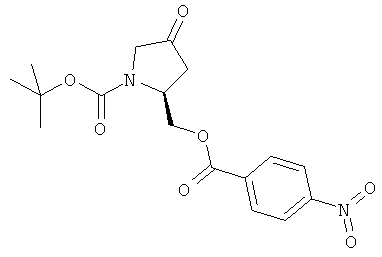
К раствору оксалил хлорида (23,5 мл; 24бммоль) в дихлорметане (400 мл), который был охлажден до -75°С, за 20 минут добавили диметилсульфоксид (35 мл; 492 ммоль) в дихлорметане (60 мл). Через 30 минут при -70°С добавили по каплям раствор трет-бутилового эфира (2S,4R)-4-гидрокси-2-(4-нитро-бензоилоксиметил)-пирролидин-1-карбоновой кислоты (промежуточное соединение 55, 45 г; 123 ммоль) в дихлорметане (300 мл). Через 2 часа при -70°С добавили за 10 минут N,N'-диизопропилэтиламин (60 мл). Через 25 минут при -60°С и 30 минут при комнатной температуре добавили дихлорметан (200 мл). Органический растворитель промыли 0,5N HCl (500 мл), водой (500 мл) и рассолом (500 мл), осушили (Na2SO4) и выпарили, чтобы получить сырой продукт, который очистили с помощью хроматографии с силикагелем (петролейный эфир/этилацетат от 5/1 до 3/1). Получили трет-бутиловый эфир (S)-2-(4-нитро-бензоилоксиметил)-4-оксо-пирролидин-1-карбоновой кислоты в виде белого твердого вещества (28,5 г; выход 70%).
1Н-ЯМР (400 MHz, CDCl2) δ ppm 8.32 (d, 2H), 8.12 (d, 2H). 4.69-4.84 (m, 2H), 4.38 (m, 1H), 4.01 (m, 1H), 3.71 (m, 1H), 2.94 (m, 1H), 2.56 (m, 1H), 1.51 (s, 9H)
Пример 30. Получение промежуточного соединения 57: трет-бутилового эфира (S)-4-метилен-2-(4-нитро-бензоилоксиметил)-пирролидин-1-карбоновой кислоты

Метилтрифенилфосфоний бромид (38 г; 106 ммоль), растворенный в 300 мл толуола охладили до 0°С, затем по каплям добавили натрий гексаметилдисилазид (48,3 мл; 93,7 ммоль). После 2 часов при комнатной температуре охладили до 0°С и добавили раствор трет-бутилового эфира (S)-2-(4-нитро-бензоилоксиметил)-4-оксо-пирролидин-1-карбоновой кислоты (промежуточное соединение 56; 15,5 г; 42,6 ммоль) в 300 мл толуола. Через 10 минут медленно добавили 500 мл воды, затем позволили реакционной смеси достичь комнатной температуры. Органический растворитель промыли рассолом (500 мл), осушили (Na2SO4) и выпарили, чтобы получить сырой продукт, который очистили с помощью хроматографии с силикагелем (петролейный эфир/этилацетат от 15/7 до 7/1). Получили трет-бутиловый эфир (S)-4-Метилен-2-(4-нитро-бензоилоксиметил)-пирролидин-1-карбоновой кислоты в виде желтого твердого вещества (6,82 г; выход 46%).
1Н-ЯМР (400 MHz, CDCl2) δ ppm 8.29 (d, 2H), 8.23 (d, 2H), 5.07 (s, 2H), 4.35-4.50 (m, 3H), 4.16 (m, 1H), 3.93 (m, 1H), 2.89 (m, 1H), 2.52 (m, 1H), 1.48 (s, 9H)
Пример 31. Получение промежуточного соединения 58: трет-бутилового эфира (S)-6-(4-нитро-бензоилоксиметил)-5-аза-спиро[2.4]гептан-5-карбоновой кислоты
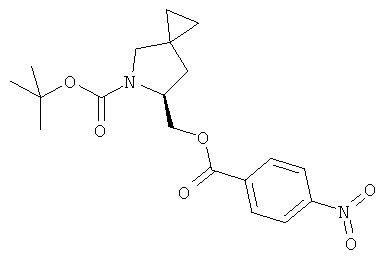
К раствору КОН (66 г) в воде (130 мл) и Et2O (250 мл) при 0°С медленно добавили метил-3-нитро-1-нитроксогуанид (90 г). Через 30 минут при 0°С органическую фазу отделили, чтобы получить раствор диазометана.
трет-Бутиловый эфир (S)-4-метилен-2-(4-нитро-бензоилоксиметил)-пирролидин-1-карбоновой кислоты (промежуточное соединение 57; 11,3 г; 31,2 ммоль), растворенный в 120 мл THF, добавили к палладийдиацетату (3,0 г). Через 2 часа при комнатной температуре охладили до -60°С; медленно добавили раствор диазометана. Смесь перемешивали при температуре от -60°С до 0°С в течение 3 часов и нагревали до комнатной температуры в течение ночи. Смесь профильтровали, затем жидкую фазу концентрировали с получением 9,0 г промежуточного соединения 58 (желтое масло, выход 79%).
1Н-ЯМР (400 MHz, CDCl3) δ ppm 8.30 (d, 2H), 8.25 (d, 2H), 4.30-4.60 (m, 3H), 3.55 (m, 1H), 3.11 (m, 1H), 2.29 (m, 1H), 1.47 (s, 9H), 0.88 (m, 1H), 0.66 (m, 4H)
Пример 32. Получение промежуточного соединения 59: трет-бутилового эфира (S)-6-гидроксиметил-5-аза-спиро[2.4]гептан-5-карбоновой кислоты
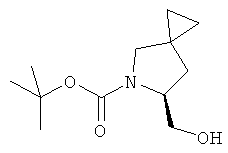
трет-Бутиловый эфир (S)-6-(4-нитро-бензоилоксиметил)-5-аза-спиро[2.4]гептан-5-карбоновой кислоты (промежуточное соединение 58; 8,9 г; 23,7 ммоль), растворенный в 200 мл метанола, охладили до 0°С, затем добавили водный 2N NaOH (100 мл). Смесь перемешивали при комнатной температуре в течение 1 часа, а затем концентрировали. Добавили этилацетат (200 мл), органический растворитель промыли водой (200 мл) и рассолом (200 мл), высушили (Na2O4) и выпарили, чтобы получить сырой продукт, который очистили с помощью хроматографии с силикагелем (петролейный эфир/этилацетат 5/1) и получили 5,0 г сырого продукта, который затем вновь очистили с помощью препаративной ВЭЖХ и получили 3,25 г промежуточного соединения 59 (бесцветное масло, выход 62%).
1Н-ЯМР (400 MHz, CDCl3) δ ppm 4.73 (m, 1H), 3.78 (m, 1H), 3.55 (m, 1H), 3.33 (m, 1H), 2.95 (m, 1H), 2.11 (m, 1H), 1.64 (m, 1H), 1.40 (s, 9H), 0.53 (m, 4H);
MS Calcd.: 227; MS найдено: 228 ([M+1]+).
Пример 33. Получение промежуточного соединения 60: (S)-трет-бутил 6-формил-5-азаспиро[2.4]гептан-5-карбоксилата
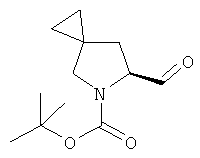
К (S)-трет-бутил-6-(гидроксиметил)-5-азаспиро[2.4]гептан-5-карбоксилату (промежуточное соединение 59; 2,2 г; 9,7 ммоль), растворенному в 10 мл DCM, добавили TEMPO (306 мг; 1,9 ммоль) и BAIB (3,43 мг; 10,6 ммоль). Через 2 часа при 25°С реакционную смесь разбавили DCM (150 мл), промыли водным раствором Na2S2O3, затем водой, высушили (Na2SO4) и выпарили с получением сырого продукта, который очистили с помощью хроматографии с силикагелем (петролейный эфир до петролейный эфир/этилацетат 95/5). Получили (S)-трет-бутил-6-формил-5-азаспиро[2.4]гептан-5-карбоксилат в виде светло-желтого твердого вещества (1,8 г).
1H-ЯМР (DMSO-d6) δ ppm 9.52 (s, 1H) 4.21(m, 1H) 3.20-3.40 (m, 2H) 2.11 (m, 1H) 1.50-1.80 (m, 1H) 1.36-1.41 (m, 9H) 0.50-0.65 (m, 4H)
Пример 34. Получение промежуточного соединения 61: (S)-трет-бутил-6-(((5-хлорпиридин-2-ил)амино)метил)-5-азаспиро[2.4]гептан-5-карбоксилата
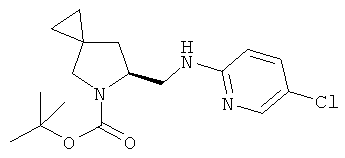
(A)-трет-бутил-6-формил-5-азаспиро[2.4]гептан-5-карбоксилат (промежуточное соединение 60; 700 мг; 3.1 ммоль) растворили в дихлорэтане (15 мл), затем добавили уксусную кислоту (0,9 мл; 15,5 ммоль) и 5-хлорпиридин (400 мг; 3,1 ммоль). Через 2 часа при комнатной температуре добавили NaBH(OAc)3 (1,05 г; 4,97 ммоль), а реакционную смесь перемешивали при комнатной температуре в течение 18 часов. Реакционную смесь выливали в водный NaHCO3 и экстрагировали DCM. Органические слои объединили, высушили (Na2SO4) и концентрировали под вакуумом; сырой продукт очистили с помощью колоночной хроматографии с силикагелем, используя петролейный эфир/этилацетат=95/5 до 85/15. Получили (S)-трет-бутил 6-(((5-хлорпиридин-2-ил)амино)метил)-5-азаспиро[2.4]гептан-5-карбоксилат в виде бесцветного масла (600 мг). MS (ESI) m/z: 338 [M+H]+
Пример 35. Получение промежуточного соединения 62: (S)-трет-бутил-6-(((6-метилпиридин-2-ил)амино)метил)-5-азаспиро[2.4]гептан-5-карбоксилата
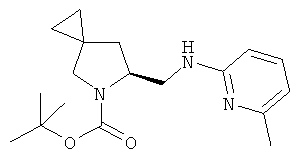
(S)-трет-бутил-6-формил-5-азаспиро[2.4]гептан-5-карбоксилат (промежуточное соединение 60, 700 мг; 3,1 ммоль) растворили в дихлорметане (15 мл), затем добавили уксусную кислоту (0,9 мл; 15,5 ммоль) и 6-метилпиридин (336 мг; 3,1 ммоль). Через 2 часа при комнатной температуре добавили NaВН(OАс)3 (1,05 г; 4,97 ммоль), реакционную смесь перемешивали при комнатной температуре в течение 18 часов. Реакционную смесь выливали в водный NaHCO3 и экстрагировали DCM. Органические слои объединили, высушили (Na2SO4) и концентрировали под вакуумом; сырой продукт очистили с помощью колоночной хроматографии с силикагелем, используя петролейный эфир/этилацетат=95/5 до 85/15. Получили (S)-трет-бути-6-(((6-метилпиридин-2-ил)амино)метил)-5-азаспиро[2.4]гептан-5-карбоксилат в виде бесцветного масла (600 мг).
Пример 36. Получение промежуточного соединения 63: (S)-N-(5-азаспиро[2.4]гептан-6-илметил)-5-хлорпиридин-2-амина
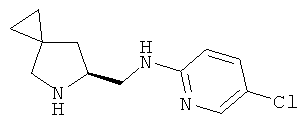
(S)-трет-бутил-6-(((5-хлорпиридин-2-ил)амино)метил)-5-азаспиро[2.4]гептан-5-карбоксилат (промежуточное соединение 60; 600 мг; 1,77 ммоль) растворили в дихлорметане (10 мл) и охладили до 0°С, затем добавили трифторуксусную кислоту (4 мл). Через 1 час при 0°С и 3 часа при комнатной температуре раствор выпарили, остаток, повторно растворенный в дихлорметане, промыли насыщенным водным раствором NaНСО3. Органические слои высушили (Na2SO4) и концентрировали под вакуумом. Сырой продукт очистили с помощью колоночной хроматографии с силикагелем (СНСl3/МеОН=8/2). Получили (S)-R-(5-азаспиро[2.4]гептан-6-илметил)-5-хлорпиридин-2-амин в виде светло-желтого масла (400 мг).
MS (ESI) m/z: 238 [М+Н]+
1Н-ЯМР (CDCl3) δ ppm 8.02 (d, J=3 Hz, 1H) 7.35 (dd, J=8 Hz, 3 Hz, 1H) 6.39 (d, J=8 Hz, 1H) 5.05 (m, 1H) 3.60-3.63 (m, 1H) 3.40-3.48(m, 1H) 3.20-3.30 (m, 1H) 2.85-2.90 (m, 1H) 1.85-1.95 (m, 1H) 1.52-1.61 (m, 1H) 0.50-0.60 (m, 4H)
Пример 37. Получение промежуточного соединения 64: (S)-N-(5-азаспиро[2.4]гептан-6-илметил)-6-метилпиридин-2-амина
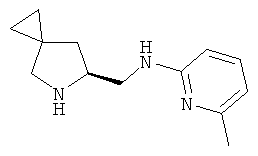
S)-трет-бутил-6-(((6-метилпиридин-2-ил)амино)метил)-5-азаспиро[2.4]гептан-5-карбоксилат (промежуточное соединение 60; 600 мг; 1,89 ммоль) растворили в дихлорметане (10 мл) и охладили до 0°С, затем добавили трифторуксусную кислоту (4 мл). Через 1 час при 0°С и 3 часа при комнатной температуре раствор выпарили, остаток, повторно растворенный в дихлорметане, промыли насыщенным водным раствором NaHCO3. Органические слои высушили (Na2SO4) и концентрировали под вакуумом. Сырой продукт очистили с помощью колоночной хроматографии с силикагелем (СНСl3/МеОН=8/2). Получили (S)-N-(5-азаспиро[2.4]гептан-6-илметил)-6-метилпиридин-2-амин в виде светло-желтого масла (225 мг).
MS(ESI) m/z: 218 [M+H]+.
1H-ЯМР (CDCl3) δ ррm 7.37-7.41 (m, 1Н) 6.55 (d, J=8 Hz, 1H) 6.45 (d, J=8 Hz, 1H) 5.62 (s, 1H) 4.25-4.31 (m, 1H) 3.76-3.80 (m, 1H) 3.60-3.66 (m, 1H) 3.28 (d, J=12 Hz, 1H) 3.07 (d, J=12Hz, 1H) 1.89-2.01 (m, 2H) 0.59-0.81 (m, 4Н)
Пример 38. Получение промежуточного соединения 65: (S)-трет-бутил 6-((1,3-диоксоизоиндолин-2-ил)метил)-5-азаспиро[2.4]гептан-5-карбоксилата
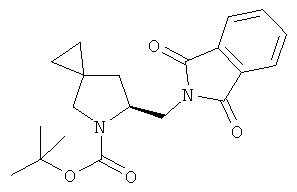
В круглодонную колбу в атмосфере азота к раствору (S)-трет-бутил 6-(гидроксиметил)-5-азаспиро[2.4]гептан-5-карбоксилата (промежуточное соединение 59; 498 мг; 2,19 ммоль) в 10 мл безводного THF добавили трифенилфосфин (947 мг; 3,61 ммоль) и фталимид (541 мг; 3,67 ммоль). Смесь охладили на ледяной бане и добавили по каплям раствор DEAD 40 вес.% в толуоле (1,6 мл; 3,51 ммоль).
Позволили реакционной смеси нагреться до комнатной температуры в течение ночи. На следующее утро смесь потушили небольшим количеством МеОН; затем растворитель удалили, а полученный остаток очистили с помощью флэш-хроматографии на 50 г силикагелевом картридже, элюируя ступенчатым градиентом: циклогексан 100% в двух объемах колонки, циклогексан/AcOEt 95/5 в двух объемах колонки, линейным градиентом до 85/15 в 10 объемах колонки и затем изократически 85/15 в 4 объемах колонки.
Затем собранные фракции выпарили с получением 758 мг (S)-трет-бутил 6-((1,3-диоксоизоиндолин-2-ил)метил)-5-азаспиро[2.4]гептан-5-карбоксилата в виде прозрачного масла (выход 97%).
MS (ESI) m/z 357 [M+H]+; 379 [M+Na]+
1Н-ЯМР (CDCl3) δ ppm 7.83-7.91 (m, 2H) 7.69-7.78 (m, 1H) 4.29-4.52 (m, 1H) 4.0-4.13 (m, 1H) 3.77-3.87 (m, 1H) 3.46-3.61 (m, 1H) 3.09 (m, 1H) 2.21-2.31(m, 1H) 1.26-1.40 (m, 9H) 0.55-0.89 (m, 4H)
Пример 39. Получение промежуточного соединения 66: (S)-трет-бутил 6-(аминометил)-5-азаспиро[2.4]гептан-5-карбоксилата
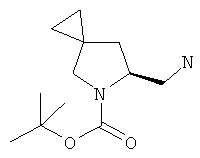
(S)-трет-бутил-6-((1,3-диоксоизоиндолин-2-ил)метил)-5-азаспиро[2.4]гептан-5-карбоксилат (промежуточное соединение 65; 752 мг; 2,11 ммоль) растворили в 20 мл этанола, добавили гидразинмоногидрат (550 мкл; 11,32 ммоль) и перемешивали смесь при комнатной температуре в течение ночи.
Образовавшееся большое количество белого осадка отфильтровали и тщательно промыли диэтиловым эфиром. Жидкую фазу выпарили до сухости, а остаток опять перенесли в диэтиловый эфир. Затем полученную суспензию вновь профильтровали, дополнительно промыв все твердые вещества эфиром. Все собранные жидкие фазы выпарили и получили 420 мг (S)-трет-бутил-6-(аминометил)-5-азаспиро[2.4]гептан-5-карбоксилата в виде светлого вязкого масла (выход 88%).
MS (ESI) m/z 227 [M+H]+ 249 [M+Na]+
1Н-ЯМР (CDCl3) δ ppm 4.38-4.26 (m, 1H) 3.42-3.66 (m, 1H) 2.87-3.09 (m, 3H) 2.14-2.19 (m, 1H) 1.48 (s, 9H) 1.24-1.32 (m, 1H) 0.58-0.75 (m, 4H)
Пример 40. Получение промежуточного соединения 67: (S)-трет-бутил-6-(((4,6-диметилпиримидин-2-ил)амино)метил)-5-азаспиро[2.4]гептан-5-карбоксилата
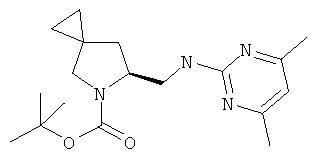
Во флаконе для микроволновой печи в 1 мл изопропилового спирта растворили (S)-трет-бутил-6-(аминометил)-5-азаспиро[2.4]гептан-5-карбоксилат (промежуточное соединение 66; 61 мг; 0,27 ммоль) и 2-хлор-4,6-диметилпиримидин (61 мг; 0,43 ммоль). Добавили DIPEA (0,1 мл; 0,57 ммоль), флакон закупорили и нагрели с помощью микроволнового облучения до 120°С за два цикла по 30 минут каждый.
Затем растворитель удалили, а остаток очистили с помощью флэш-хроматографии на силикагелевом картридже 10 г, элюируя ступенчатым градиентом: циклогексан/AcOEt в двух объемах колонки, затем линейным градиентом до 65/35 в 16 объемах колонки и в конце изократически 65/35 в 3 объемах колонки.
Удаление растворителя давало 37 мг (S)-трет-бутил-6-(((4,6-диметилпиримидин-2-ил)амино)метил)-5-азаспиро[2.4]гептан-5-карбоксилата в виде чистой корки (выход 41%).
MS (ESI) m/z 333 [M+H]+; 355 [M+Na]+
1Н-ЯМР (CDCl3) δ ppm 6.30-6.32 (m, 1H) 5.37-5.72 (m, 1H) 4.11-4.20 (m, 1H) 3.60-3.85 (m, 2H) 3.44-3.58 (m, 1H) 3.09-3.12 (m, 1H) 2.30 (s, 6H) 2.14-2.20 (m, 1H) 1.50 (s, 9H) 0.85-0.95 (m, 1H) 0.50-0.76 (m, 4H)
Пример 41. Получение промежуточного соединения 68: (S)-трет-бутил-6-(((5-хлорпиримидин-2-ил)амино)метил)-5-азаспиро[2.4]гептан-5-карбоксилата
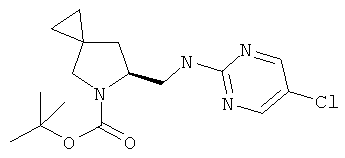
Во флаконе для микроволновой печи в 1 мл изопропилового спирта растворили (S)-трет-бутил-6-(аминометил)-5-азаспиро[2.4]гептан-5-карбоксилат (промежуточное соединение 66; 66 мг; 0,29 ммоль) и 2,5-дихлорпиримидин (65 мг; 0,44). Добавили DIPEA (100 мкл; 0,58 ммоль); флакон закупорили и нагревали до 120°С в течение 20 минут с помощью микроволнового облучения.
Затем растворитель удалили, а остаток очистили с помощью флэш-хроматографии на силикагелевом картридже 10 г, элюируя ступенчатым градиентом: циклогексан/AcOEt в двух объемах колонки, затем линейным градиентом до 80/20 в 10 объемах колонки и в конце изократически 80/20 в 2 объемах колонки. Удаление растворителя давало 60 мг (S)-трет-бутил-6-(((5-хлорпиримидин-2-ил)амино)метил)-5-азаспиро[2.4]гептан-5-карбоксилата в виде твердого белого вещества (выход 61%).
MS (ESI) m/z 339-341 (Cl pattern) [M+H]+ 361-363 [M+Na]+
1Н-ЯМР (CDCl3) δ ppm 8.21 (s,2H) 5.62-6.5 (m, 1H) 4.20-4.30 (m, 1H) 3.53-3.62 (m, 2H) 3.07-3.49 (m, 2H) 2.24-2.29 (m, 1H) 1.61 (m, 1H) 1.49 (m. 9H) 0.56-0.70 (m, 4H)
Пример 42. Получение промежуточного соединения 69: (S)-N-(5-азаспиро(2.4]гептан-6-илметил)-4,6-диметилпиримидин-2-амина

(S)-трет-бутил-6-(((4,6-диметилпиримидин-2-ил)амино)метил)-5-азаспиро[2.4]гептан-5-карбоксилат (промежуточное соединение 68; 18 мг; 0,054 ммоль) растворили в 0,5 мл безводного DCM в атмосфере азота. Добавили TFA (0,2 мл) и встряхивали раствор при комнатной температуре в течение 2 часов. Реакционную смесь загрузили на картридж с 1 г SCX, который затем промыли МеОН, а потом раствором аммиака 2,0 М в МеОН. Основные фракции собрали и выпарили с получением 9 мг (S)-N-(5-азаспиро[2.4]гептан-6-илметил)-4,6-диметилпиримидин-2-амина в виде бесцветного остатка (выход 71%).
MS (ESI) m/z 233 [M+H]+
1Н-ЯМР (CDCl3) δ ppm 6.32 (s, 1H) 5.36 (m, 1H) 3.61-3.67 (m, 2H) 3.41-3.47 (m, 1H) 2.89 (m, 2H) 2.30 (s, 6H) 1.83-1.87 (m, 1H) 1.61-1.65 (m, 1H) 0.52-0.63 (m, 4H)
Пример 43. Получение промежуточного соединения 70: (S)-N-(5-азаспиро[2.4]гептан-6-илметил)-5-хлорпиримидин-2-амина
(S)-трет-бутил-6-(((5-хлорпиримидин-2-ил)амино)метил)-5-азаспиро[2.4]гептан-5-карбоксилат (промежуточное соединение 68; 59 мг; 0,17 ммоль) растворили в 1 мл безводного DCM в атмосфере азота. Добавили TFA (0,3 мл) и встряхивали раствор при комнатной температуре в течение 40 минут.
Реакционную смесь развели в небольшом количестве МеОН и загрузили на картридж с 1 г SCX, который затем промыли МеОН, а потом раствором аммиака 2,0 М в МеОН. Основные фракции собрали и выпарили с получением 37 мг (S)-N-(5-азаспиро[2.4]гептан-6-илметил)-5-хлорпиримидин-2-амина в виде твердого белого вещества (выход 91%).
MS (ESI) m/z 239-241 (Cl pattern) [M+H]+
1Н-ЯМР (CDCl3) δ ppm 8.22 (s,2H) 5.74 (m, 1H) 3.55-3.67 (m, 2H) 3.35-3.41 (m, 1H) 2.86-2.92 (m, 2H) 1.84-1.89 (m, 1H) 1.56-1.61 (m, 1H) 0.45-0.70 (m, 4H).
Пример 44. Получение промежуточного соединения 71: трет-бутил-8-азаспиро[4.5]декан-8-карбоксилата
К раствору 8-азаспиро[4.5]декана (который был описан в WO 9800404 А1 6,2 г; 44,5 ммоль) в дихлорметане (130 мл), охлажденному до 0°С, добавили триэтиламин (7,5 мл; 53,4 ммоль), а затем в течение 20 минут добавили раствор трет-бутилдикарбоната (10,68 г; 48,9 ммоль) в дихлорметане (50 мл). Прозрачный раствор перемешивали при комнатной температуре в течение 18 часов, затем добавили дихлорметан (50 мл), органический раствор промыли водой (2×20 мл), НСl 0,1 N (20 мл), затем вновь водой (2×20 мл). Органический растворитель обезвоживали (Na2SO4) и выпаривали с получением сырого продукта, который очистили на колонке с силикагелем (гексан/AcOEt 95/5) и получили 6 г титульного соединения (светло-желтое твердое вещество).
Пример 45. Получение промежуточного соединения 72: (±)трет-бутил 7-формил-8-азаспиро[4.5]декан-8-карбоксилата
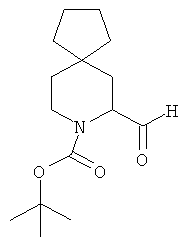
трет-Бутил-8-азаспиро[4.5]декан-8-карбоксилат (промежуточное соединение 71; 6 г; 25,07 ммоль), растворенный в 150 мл Et2O, охладили до -78°С, затем добавили N,N,N',N'-тетраметилэтилендиамин (9,08 мл; 60,16 ммоль) и втор-бутиллитий 1,4 М в гексане (42,95 мл; 60,16 ммоль). Через 10 минут при -60°С температуру подняли до -20°С за 60 минут, затем реакционную смесь охладили до -78°С и добавили диметилформамид (4,47 мл; 60,16 ммоль, растворенный в 5 мл Et2O). Через 30 минут медленно добавили насыщенный водный раствор NH4Cl (30 мл), затем позволили реакционной смеси достичь комнатной температуры. Реакционную смесь экстрагировали Et2O (3×100 мл), органический растворитель осушили (Na2SO4) и выпаривали, чтобы получить 4 г титульного соединения в виде желтого масла.
Пример 46. Получение промежуточного соединения 73: (±)трет-бутил-7-(((6-метилпиридин-2-ил)амино)метил)-8-азаспиро[4.5]декан-8-карбоксилата
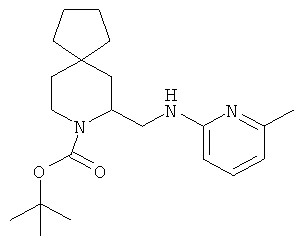
(±)трет-бутил-7-формил-8-азаспиро[4.5]декан-8-карбоксилат (промежуточное соединение 72; 2 г; 7,48 ммоль) растворили в дихлорэтане (25 мл), затем добавили уксусную кислоту (5 экв.) и 2-амино-6-пиколин (810 мг; 7,48 ммоль). Через 3 часа при комнатной температуре добавили NaВН(OАс)3 (2,53 г; 11,9бммоль) и перемешивали реакционную смесь при комнатной температуре в течение 18 часов. Реакционную смесь вылили в водный NaHCO3 и экстрагировали этилацетатом. Органические слои объединили, высушили (Na2SO4) и концентрировали под вакуумом; сырой продукт очистили с помощью колоночной хроматографии на силикагеле (петролейный эфир/этилацетат=9/1) с получением 1,3 г промежуточного соединения в виде бесцветного масла.
Пример 47. Получение промежуточного соединения 74: (±)трет-бутил-7-(((5-хлорпиридин-2-ил)амино)метил)-8-азаспиро[4.5]декан-8-карбоксилата
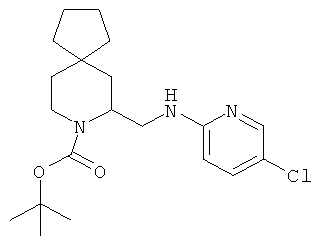
(±)трет-бутил-7-формил-8-азаспиро[4.5]декан-8-карбоксилат (промежуточное соединение 72; 2 г; 7,48 ммоль) растворили в дихлорэтане (25 мл), затем добавили уксусную кислоту (5 экв.) и 2-амино-5-хлорпиридин (961 мг; 7,48 ммоль). Через 3 часа при комнатной температуре добавили NaВН(OАс)3 (2,53 г; 11,96 ммоль) и перемешивали реакционную смесь при комнатной температуре в течение 18 часов. Реакционную смесь вылили в водный NaHCO3 и экстрагировали этилацетатом. Органические слои объединили, высушили (Na2SO4) и концентрировали под вакуумом; сырой продукт очистили с помощью колоночной хроматографии на силикагеле (петролейный эфир/этилацетат=9/1). Получили 1 г промежуточного соединения 74 в виде бесцветного масла.
Пример 48. Получение промежуточного соединения 75: (±)N-(8-азаспиро[4.5]декан-7-илметил)-6-метилпиридин-2-амина
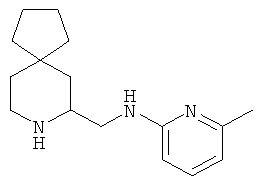
(±)трет-бутил-7-(((6-метилпиридин-2-ил)амино)метил)-8-азаспиро[4.5]декан-8-карбоксилат (промежуточное соединение 73; 1,3 г; 3,62 ммоль) растворили в дихлорметане (10 мл) и охладили до 0°С, затем добавили трифторуксусную кислоту (30 мл). Через 1 час при 0°С и 2 часа при комнатной температуре раствор выпарили, остаток, вновь растворенный в дихлорметане, промыли насыщенным водным раствором NaHCO3. Органические слои высушили (Na2SO4) и концентрировали под вакуумом, чтобы получить 900 мг титульного соединения.
MS (ESI) m/z 260[M+H]+
Пример 49. Получение промежуточного соединения 76: (±)N-(8-азаспиро[4.5]декан-7-илметил)-5-хлорпиридин-2-амина
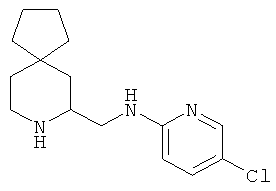
(±)трет-бутил-7-(((5-хлорпиридин-2-ил)амино)метил)-8-азаспиро[4.5]декан-8-карбоксилат (промежуточное соединение 74; 1 г; 2,63 ммоль) растворили в дихлорметане (10 мл) и охладили до 0°С, затем добавили трифторуксусную кислоту (30 мл). Через 1 час при 0°С и 2 часа при комнатной температуре раствор выпарили, остаток, вновь растворенный в дихлорметане, промыли насыщенным водным раствором NaHCO3. Органические слои высушили (Na2SO4) и концентрировали под вакуумом, чтобы получить 600 мг титульного соединения.
MS (ESI) m/z 281 [M+H]+
Получение соединений изобретения:
Пример 50
Соединение 1: (±) (2-метил-5-фенилтиазол-4-ил)(5-((6-метилпиридин-2-иламино)метил)-6-азаспиро[2.5]октан-6-ил)метанон
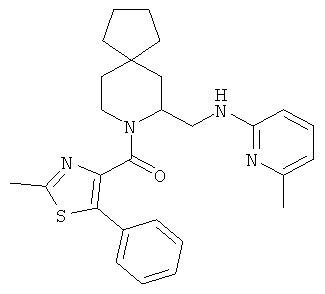
Промежуточное соединение 7 (30 мг; 0,129 ммоль) растворили в дихлорметане (2 мл), затем добавили диизопропилэтиламин (75 мкл, 0,427 ммоль) и O-(бензотриазол-1-ил)-N,N,N'N'-тетраметилуронийгексафторфосфат (69 мг; 0,18 ммоль). Через 30 минут при комнатной температуре 2-метил-5-фенилтиазол-4-карбоновую кислоту, получение которой было описано в США 3282927 (40 мг; 0,18 ммоль), растворили в дихлорметане (2 мл) и диметилформамиде (1 мл) и добавили к реакционной смеси. Через 18 часов при комнатной температуре смесь выливали в водный NaНСО3 и экстрагировали AcOEt. Органические слои объединили, высушили (Na2SO4) и концентрировали под вакуумом; полученный сырой продукт очистили с помощью колоночной хроматографии с силикагелем (петролейный эфир/этилацетат=7/3), получив 12 мг соединения 1 в виде белого твердого вещества.
MS (ESI) m/z 433.1 [М+Н]+; 1Н-ЯМР [Продукт присутствует в виде смеси конформеров. Отнесение проводили к основному компоненту] (CDCl3) δ ppm 7.84 (d, 1H) 7.25-7.64 (m, 5H) 6.48 (d, 1H) 6.11 (d, 1H) 4.95-4.98 (m, 1H) 3.70-3.85 (m, 1H) 3.44-3.55 (m, 1H) 2.95-3.15 (m, 1H) 2.74 (s, 3H) 2.50 (s, 3H) 1.80-1.95 (m, 1H) 1.32-1.15 (m, 2H) 0.89-0.80 (m, 2H) 0.57-0.15 (m,4H).
Энантиомерную смесь (±) (2-метил-5-фенилтиазол-4-ил)(5-((6-метилпиридин-2-иламино)метил)-6-азаспиро[2.5]октан-6-ил)метанона (соединение 1; 30 мг) разделили с помощью хиральной препаративной ВЭЖХ (препаративные хроматографические условия: метод С) с получением двух энантиомеров:
Пример 51
Соединение 2: (2-метил-5-фенилтиазол-4-ил)(5-((6-метилпиридин-2-иламино)метил)-6-азаспиро[2.5]октан-6-ил)метанон (изомер А)
ВЭЖХ время удерживания 36,3 мин (10 мг)
Пример 52
Соединение 3: (2-метил-5-фенилтиазол-4-ил)(5-((6-метилпиридин-2-иламино)метил)-6-азаспиро[2.5]октан-6-ил)метанон (изомер В)
ВЭЖХ время удерживания 38,6 мин (6 мг)
Пример 53
Соединение 4: (±) (5-((5-хлорпиридин-2-иламино)метил)-6-азаспиро[2.5]октан-6-ил)(2-метил-5-фенилтиазол-4-ил)метанон
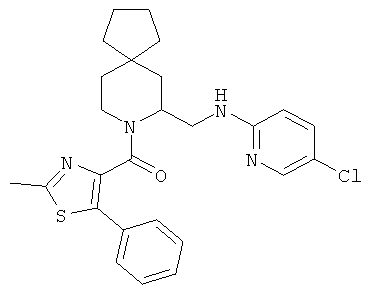
Промежуточное соединение 8 (60 мг; 0,238 ммоль) растворили в дихлорметане (2 мл), затем добавили диизопропилэтиламин (1,37 мкл; 0,786 ммоль) и O-(бензотриазол-1-ил)-N,N,N'N'-тетраметилуронийгексафторфосфат (126 мг; 0,33 ммоль). Через 30 минут при комнатной температуре 2-метил-5-фенилтиазол-4-карбоновую кислоту (73 мг; 0,33 ммоль) растворили в дихлорметане (2 мл) и диметилформамиде (1 мл) и добавили к реакционной смеси. Через 18 часов при комнатной температуре смесь выливали в водный насыщенный раствор NaНСО3 и экстрагировали этилацетатом. Органические слои объединили, высушили (Na2SO4) и концентрировали под вакуумом; сырой продукт очистили с помощью колоночной хроматографии с силикагелем (петролейный эфир/этилацетат=1/1 до этилацетат), получив 37 мг соединения 4 в виде белого твердого вещества.
MS (ESI) m/z 453.1 [М+Н]+; 1Н-ЯМР [продукт присутствует в виде смеси конформеров. Отнесение проводили к основному компоненту] (CDCl3) δ ppm 7.92 (s, 1H) 7.28-7.51 (m, 5H) 6.33 (d, 1H) 5.57 (m, 1H) 4.68-4.71 (m, 1H) 3.89-3.95 (m, 1H) 3.41-3.46 (m, 1H) 3.10-3.25(m, 1H) 2.90-3.05 (m, 1H) 2.81 (s, 3H) 1.80-1.90 (m, 1 H) 1.20-1.5 0(m, 1H) 0.87 (d, 1H) 0.70 (d. 2H) 0.35-0.50 (m, 2H) 0.15-0.30 (m, 2H).
Энантиомерную смесь (±) (5-((5-хлорпиридин-2-иламино)метил)-6-азаспиро[2.5]октан-6-ил)(2-метил-5-фенилтиазол-4-ил)метанона (соединение 4; 75 мг) разделили с помощью хиральной препаративной ВЭЖХ (препаративные хроматографические условия: метод А) с получением двух энантиомеров:
Пример 54
Соединение 5: (5-((5-хлорпиридин-2-иламино)метил)-6-азаспиро[2.5]октан-6-ил)(2-метил-5-фенилтиазол-4-ил)метанон (изомер А)
ВЭЖХ время удерживания 16 мин (23 мг)
Пример 55
Соединение 6; (5-((5-хлорпиридин-2-иламино)метил)-6-азаспиро[2.5]октан-6-ил)(2-метил-5-фенилтиазол-4-ил)метанон (изомер В)
ВЭЖХ время удерживания 18 мин (26 мг)
Пример 56
Соединение 7: (±) (2-метил-5-фенилтиазол-4-ил)(5-((5-(трифторметил)пиридин-2-иламино)метил)-6-азаспиро[2.5]октан-6-ил)метанон
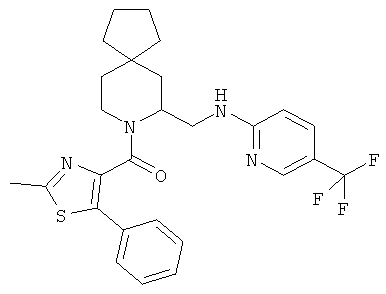
Промежуточное соединение 9 (60 мг; 0,210 ммоль) растворили в дихлорметане (2 мл), добавили диизопропилэтиламин (121 мкл; 0,б93 ммоль) и O-(бензотриазол-1-ил)-N,N,N'N'-тeтpaмeтилypoнийгeкcaфтopфocфaт (112 мг; 0,294 ммоль). Через 30 минут при комнатной температуре 2-метил-5-фенилтиазоле-4-карбоновую кислоту (65 мг; 0,294 ммоль) растворили в дихлорметане (2 мл) диметилформамиде (1 мл) и добавили к реакционной смеси. Через 18 часов при комнатной температуре смесь выливали в водный насыщенный раствор NaHCO3 и экстрагировали этилацетатом. Органические слои объединили, высушили (Na2SO4) и концентрировали под вакуумом; сырой продукт очистили с помощью колоночной хроматографии с силикагелем (градиент от дихлорметан/этилацетат=1/1 до этилацетат), получили 15 мг соединения 7 в виде белого твердого вещества.
MS (ESI) m/z 509 [M+Na]+. 1Н-ЯМР [продукт присутствует в виде смеси конформеров. Отнесение проводили к основному компоненту] (CD3OD) δ ppm 8.05 (s, 1H) 7.39-7.60 (m, 5H) 6.41 (d, 1H) 4.65 (m, 1H) 4.20 (m, 1H) 3.69-3.76 (m, 1H) 3.28-3.35 (m, 1H) 3.10-3.15(m, 1H) 2.43 (s, 3H) 1.82 (m, 1H) 1.30-1.45 (m, 1H) 0.90-1.05 (m, 2H) 0.6-0.68 (m, 1H) 0.30-0.55 (m, 2H) 0.15-0.25 (m, 2H).
Энантиомерную смесь (±) (2-метил-5-фенилтиазол-4-ил)(5-((5-(трифторметил)пиридин-2-иламино)метил)-6-азаспиро[2.5]октан-6-ил)метанон (соединение 7; 8 мг) разделили с помощью хиральной препаративной ВЭЖХ (препаративные хроматографические условия: метод А) с получением чистого энантиомера:
Пример 57
Соединение 8: (2-метил-5-фенилтиазол-4-ил)(5-((5-(трифторметил)пиридин-2-иламино)метил)-6-азаспиро[2.5]октан-6-ил)метанон (один энантиомер)
ВЭЖХ время удерживания 17,9 мин (4 мг).
Пример 58
Соединение 9; (±) (5-((5-хлорпиридин-2-иламино)метил)-6-азаспиро[2.5]октан-6-ил)(2-(циклопропилметокси)фенил)метанон
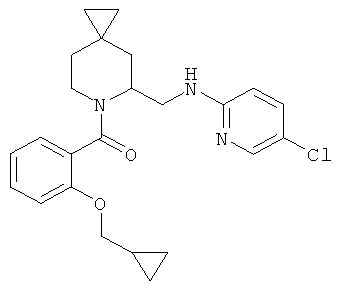
Промежуточное соединение 8 (50 мг; 0,198 ммоль) растворили в дихлорметане (2 мл), затем добавили диизопропилэтиламин (115 мкл; 0,65 ммоль) и O-(бензотриазол-1-ил)-N,N,N'N'-тетраметилуронийгексафторфосфат (105 мг; 0,277 ммоль). Через 30 минут при комнатной температуре 2-(циклопропилметокси)бензойную кислоту, получение которой было описано в Journal of Medicinal Chemistry (1993), 36(10), 1387-92 (промежуточное соединение 10; 53 мг; 0,277 ммоль) растворили в дихлорметане (2 мл) и диметилформамиде (1 мл) и добавили в реакционную смесь. Через 18 часов при комнатной температуре смесь выливали в водный насыщенный раствор NaНСО3 и экстрагировали этилацетатом. Органические слои объединили, высушили (Na2SO4) и концентрировали под вакуумом, полученный сырой продукт очистили колоночной хроматографией с силикагелем (градиент петролейный эфир/этилацетат=8/2 до 1/1), получив 12 мг соединения 9 в виде белого твердого вещества.
MS (ESI) m/z: 426 [М+Н]+. 1Н-ЯМР [продукт присутствует в виде смеси конформеров. Отнесение проводили к основному компоненту] (CDCl3) δ ppm 8.02 (s, 1H) 6.75-7.29 (m, 6H) 6.45 (d, 1H) 5.0-5.32 (m, 2H) 3.75-4.05 (m, 4H) 3.20-3.50 (m, 1H) 2-2.40 (m, 2H) 1.1-1.45 (m, 2H) 0.73-0.76 (m, 1H) 0.2-0.7 (m, 8H).
Пример 59
Соединение 10: (±) (5-((5-хлорпиридин-2-иламино)метил)-6-азаспиро[2.5]октан-6-ил)(5-(4-фторфенил)-2-метилтиазол-4-ил)метанон

К раствору гидроксибензотриазола (12 мг; 0,09 ммоль) и 1-этил-3-(3-диметиламинопропил)карбодиимид HCl (23 мг; 0,12 ммоль) в безводном дихлорметане (1,5 мл) добавили 5-(4-фторфенил)-2-метилтиазол-4-карбоновую кислоту (20 мг; 0,08 ммоль) и полученный раствор перемешивали 15 минут. Добавили (±) N-(6 азаспиро[2.5]октан-5-илметил)-5-хлорпиридин-2-амин (промежуточное соединение 8; 20 мг; 0,08 ммоль) и перемешивали смесь в течение дополнительных 3 часов при комнатной температуре. Реакционную смесь разбавили дихлорметаном (2 мл) и промыли насыщенным раствором NaHCO3 (3×5 мл). После высущивания над Na2SO4 и фильтрования органическую фазу выпарили под вакуумом, а остаток очистили с помощью картриджа SPE-Si (5 г), элюируя смесью дихлорметан:МеОН (от дихлорметан до дихлорметан: МеОН 95:5).
Было выделено 39 мг искомого соединения в виде смеси конформеров 1:0,57.
MS(ESI); m/z 471 [МН]+
1Н-ЯМР [продукт присутствует в виде смеси конформеров. Отнесение проводили к основному компоненту] (CDCl3) δ ppm 7.9 (d, 1H) 7.50-7.42 (m, 2H) 7.28-7.25 (dd, 1H) 7.12-7.09 (t, 1H) 6.34-6.31 (d, 1H) 5.65 (m, 1H) 4.66-4.65 (m, 1H) 3.96-3.90 (m, 1H) 3.21-3.17 (m, 1H) 3.01-2.94 (dt, 1H) 2.60 (s, 3H) 2.15-2.05 (dd, 1H) 1.9-1.77 (dt, 1H) 1.46-1.42 (dd, 1H) 0.77-0.74 (d, 1H) 0.49-0.39 (m, 2H) 0.25-0.15 (m, 2H).
Пример 60
Соединение 11; (±)(5-((5-хлорпиридин-2-иламино)метил)-6-азаспиро[2.5]октан-6-ил)(5-метил-2-(2Н-1,2,3-триазол-2-ил)фенил)метанон
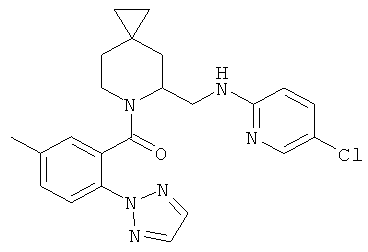
К раствору гидроксибензотриазола (12,9 мг; 0,095 ммоль) и 0-(бензотриазол-1-ил)-N,N,N'N'-тетраметилуронийгексафторфосфат НСl (23 мг; 0,12 ммоль) в безводном дихлорметане (2 мл) добавили 5-метил-2-(2Н-1,2,3-триазол-2-ил)бензойную кислоту (19,4 мг; 0,095 ммоль) и перемешивали полученный раствор в течение 1 часа при комнатной температуре. Добавили (±) N-(6-азаспиро[2.5]октан-5-илметил)-5-хлорпиридин-2-амин (промежуточное соединение 8; 20 мг; 0,08 ммоль), а полученную смесь перемешивали при той же самой температуре в течение ночи. Смесь промыли насыщенным раствором NaНСО3 (3×5 мл).
После высушивания над Na2SO4 и фильтрования органическую фазу выпарили под вакуумом, а остаток очистили с помощью картриджа SPE-Si (2 г), элюируя смесью дихлорметан:МеОН (от дихлорметан до дихлорметан:МеОН 98:2).
Было выделено 29 мг искомого соединения в виде смеси конформеров.
MS (ESI); m/z 436 [МН]+
1Н-ЯМР [продукт присутствует в виде смеси конформеров. Отнесение проводили к основному компоненту] (CDCl3) δ ppm 8.08-8.07 (d, 1H) 7.95-7.92 (m, 1H) 7.85-7.79 (m, 2H) 7.7 (s, 2H) 7.35-7.33 (d, 1H) 6.40-6.38 (d, 1H) 5.61-5.60 (d, 1H) 5.21-5.14 (m, 1H) 3.90-3.84 (m, 1H) 3.66-3.59 (m, 1H) 3.41-3.32 (m, 1H) 3.11-3.01 (m, 1H) 2.45 (s, 3H) 2.29-2.21 (dd, 1H) 1.99-1.85 (m, 1H) 1.19-1.16 (d, 1H) 0.75-0.68 (d, 1H) 0.58-0.27 (m, 4H)
Пример 61
Соединение 12: (±)(5-((5-хлорпиридин-2-иламино)метил)-6-азаспиро[2.5]октан-6-ил)(5-метил-2-(пиримидин-2-ил)фенил)метанон
К раствору гидроксибензотриазола (13 мг; 0,095 ммоль) и 0-(бензотриазол-1-ил)-N,N,N'N'-тетраметилуронийгексафторфосфат HCl (23 мг; 0,. 12 ммоль) в безводном дихлорметане (2 мл) добавили 5-метил-2-(пиримидин-2-ил)бензойную кислоту (20,4 мг; 0,095 ммоль) и полученную смесь перемешивали 1 час при комнатной температуре. Добавили (±) N-(6 азаспиро[2.5]октан-5-илметил)-5-хлорпиридин-2-амин (промежуточное соединение 8; 20 мг; 0,08 ммоль) и полученную смесь перемешивали при той же самой температуре в течение ночи. Реакционную смесь промыли насыщенным раствором NaHCO3 (3×5 мл). После высушивания над Na2S04 и фильтрования органическую фазу выпарили под вакуумом, а остаток очистили с помощью картриджа SPE-Si (2 г), элюируя смесью дихлорметан:МеОН (от дихлорметан до дихлорметан:МеОН 98:2).
Было выделено 10 мг искомого соединения в виде смеси конформеров.
MS (ESI); m/z 446 [МН]+
1Н-ЯМР [продукт присутствует в виде смеси конформеров. Отнесение проводили к основному компоненту] (CDCl3) δ ppm 8.80-8.77 (m, 1H) 8.64-8.6 (d, 1H) 8.36-8.31 (d, 1H) 8.08-8.04 (m, 1H) 7.43-7.17 (m, 3H) 7.08-7.03 (t, 1H) 6.36-6.31 (d, 1H) 5.79 (bs, 1H) 5.19-5.11 (m, 1H) 4.00-3.89 (m. 1H) 3.71-3.62 (m, 1H) 3.50-3.39 (m, 1H) 3.37-3.21 (m, 1H) 2.45 (s, 3H) 2.31-2.24 (dd, 1H) 1.99-1.88 (dt, 1H) 1.25-1.19 (d, 1H) 0.75-0.68 (d, 1H) 0.60-0.13 (m, 4H).
Энантиомерную смесь (±)(5-((5-хлорпиридин-2-иламино)метил)-6-азаспиро[2.5]октан-6-ил)(5-метил-2-(пиримидин-2-ил)фенил)метанона (соединение 12; 550 мг) разделили с помощью хиральной препаративной ВЭЖХ (препаративные хроматографические условия: метод В) с получением одного энантиомера (соединения 13):
Пример 62
Соединение 13: (S)-(5-(((5-хлорпиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)(5-метил-2-(пиримидин-2-ил)фенил)метанон (изомер А)
ВЭЖХ время удерживания 9 мин (150 мг)

Альтернативно соединение 13 было получено с помощью следующей методики: 5-метил-2-(пиримидин-2-ил)бензойную кислоту (428 мг; 2 ммоль; полученную согласно WO 2008147518), промежуточное соединение 34 (500 мг; 2 ммоль) и DIPEA (0,65 мл) растворили в DCM (5 мл) при 0°С, затем добавили ТЗР (50% в DCM; 1,5 г). Смесь перемешивали с обратным холодильником в течение 8 часов, затем при RT в течение ночи. Реакционную смесь промыли 1 М NaOH и водой, высушили (Na2SO4) и выпарили. Сырой продукт очистили с помощью колоночной хроматографии с силикагелем (DCM до DCM/MeOH=95/05). Было выделено 180 мг искомого соединения.
MS (ESI); m/z 446 [МН]+
1Н-ЯМР [продукт присутствует в виде смеси конформеров. Отнесение проводили к основному компоненту] (CDCl3) δ ppm 8.80-8.77 (m, 1H) 8.64-8.6 (d, 1H) 8.36-8.31 (d, 1H) 8.08-8.04 (m, 1H) 7.43-7.17 (m, 3H) 7.08-7.03 (t, 1H) 6.36-6.31 (d, 1H) 5.79 (bs, 1H) 5.19-5.11 (m, 1H) 4.00-3.89 (m, 1H) 3.71-3.62 (m, 1H) 3.50-3.39 (m, 1H) 3.37-3.21 (m. 1H) 2.45 (s, 3H) 2.31-2.24 (dd, 1H) 1.99-1.88 (dt, 1H) 1.25-1.19 (d, 1H) 0.75-0.68 (d, 1H) 0.60-0.13 (m, 4H).
Пример 63
Соединение 15: (±)метил-5-хлор-2-((6-(5-(4-фторфенил)-2-метилтиазол-4-карбонил)-6-азаспиро[2.5]октан-5-ил)метиламино)бензоат

К раствору гидроксибензотриазола (13 мг; 0,097 ммоль) и 0-(бензотриазол-1-ил)-N,N,N'N'-тетраметилуронийгексафторфосфат HCl (23 мг; 0,12 ммоль) в безводном дихлорметане (2 мл) добавили 5-(4-фторфенил)-2-метилтиазол-4-карбоновую кислоту (23 мг; 0,097 ммоль) и полученный раствор перемешивали 1 час при комнатной температуре. Добавили (±) метил-2-(6-азаспиро[2.5]октан-5-илметиламино)-5-хлорбензоат (промежуточное соединение 10) (20 мг; 0,08 ммоль) и полученную смесь перемешивали при той же самой температуре в течение ночи. Смесь промыли насыщенным раствором МаНСОз (3×5 мл).
После высушивания над Na2SO4 и фильтрования органическую фазу выпарили под вакуумом и очистили с помощью картриджа SPE-Si (2 г), элюируя смесью дихлорметан:МеОН (от дихлорметан до дихлорметан:МеОН 98:2).
Было выделено 35 мг искомого соединения.
MS (ESI); m/z 528 [МН]+
Примеры 64-87. Получение соединений 16-35,37-39
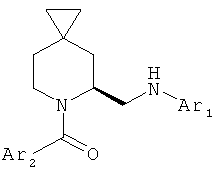
В вышеприведенной формуле Ar1 представляет собой Q, а Аr2 представляет собой R в формуле (VIa).
Общая методика 6
Аr2-CООН (1 экв.; получено согласно WO 2008147518 для соединений 19, 20, 26-35, 37-39 и согласно США 3282927 для соединений 16, 17, 18, 21-25), НОВТ (1 экв.) и EDCI.HCl (1,5 экв.), растворенный в дихлорметане (5 мл/ммоль), перемешивали при 25°С в течение 0,5-2 часов, затем добавили промежуточные соединения 34-45, растворенные в дихлорметане. Через 18 часов смесь выливали в водный насыщенный раствор NaHCO3 и экстрагировали дихлорметаном. Сырой продукт очистили с помощью колоночной хроматографии с силикагелем (DCM до DCM/MeOH=9/1)
Общая методика 7
Ar2-COOH (1 экв.; получено согласно WO 2008147518 для соединений 19, 20, 26-39 и согласно США 3282927 для соединений 16, 17, 18, 21-25), промежуточные соединения 34-45 (1 экв.) и DIPEA (2 экв.) растворили в дихлорметане (5 мл/ммоль) при 0°С, затем добавили Т3Р (50% в дихлорметане, 1,2 экв.). Смесь перемешивали с обратным холодильником в течение 3-5 часов, затем при RT в течение ночи. Реакционную смесь промыли NaOH 1 M и водой, высушили (Na2SO4) и выпарили. Сырой продукт очистили с помощью колоночной хроматографии с силикагелем (DCM до DCM/MeOH=9/1). Энантиомерную чистоту вычисляли как энантиомерный избыток (ее%) с помощью методов хиральной ВЭЖХ.
Следующие соединения были получены согласно Общей методике 6 или 7:
Характеристика соединений 16-39:
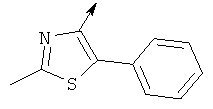
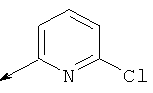
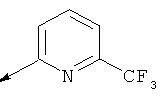
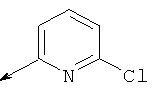
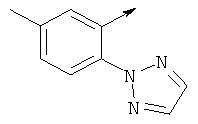
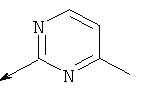
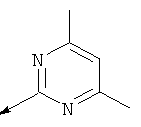
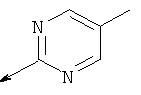
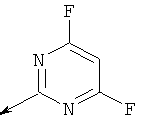
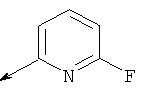
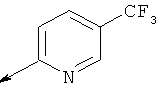
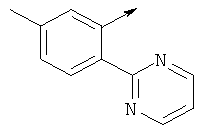
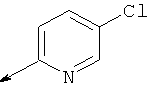
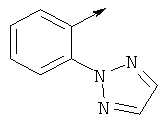
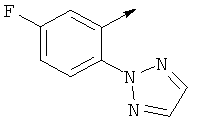
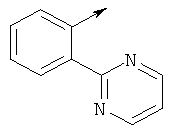
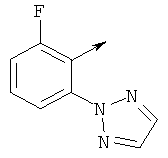

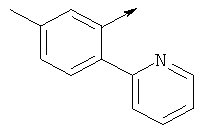
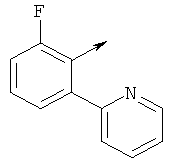

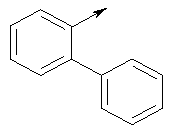
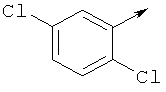
Примеры 87-93. Получение соединений 40-46
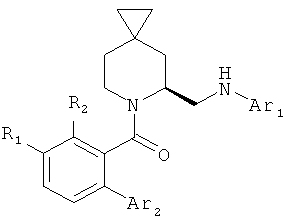
В вышеприведенной формуле Ar1 представляет собой Q и фенил, замещенный R1, R2, Аr2 представляет собой R в формуле (Via).
Общая методика 8
Промежуточные соединения 46-50 (1 экв.) растворили безводным DMF (20 мл/ммоль), затем добавили CsF (2 экв.), Cul (0,2 экв.), [Ph3P]4Pd (0,1 экв.) и соответствующий Аr2-трибутилстаннан (1,5 экв.; полученный согласно Eur. J. Org. Chem. 2003, 1711-1721). Смесь нагревали при 130°С в течение 10 минут (микроволнами), затем выливали в водный насыщенный раствор NH4Cl и экстрагировали AcOEt. Органические слои объединили, высушили (Na2SO4) и концентрировали под вакуумом; сырой продукт очистили с помощью колоночной хроматографии с силикагелем (DCM до DCM/MeOH 9/1). Энантиомерную чистоту вычисляли как энантиомерный избыток (ее%) с помощью методов хиральной ВЭЖХ.
Следующие соединения были получены согласно Общей методике 8:
Характеристика соединений 40-46:
Пример 89-95. Получение соединений 47-53
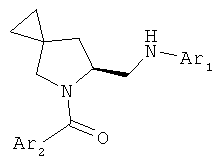
В вышеприведенной формуле Ar1 представляет собой Q, а Аr2 представляет собой R в формуле (VIa).
Общая методика 9
Аr2-CООН (1 экв.; получено согласно WO 2008147518 для соединений 49-51), НОBТ (1 экв.) и EDCI.HCl (1,5 экв.), растворенные в дихлорметане (5 мл/ммоль), перемешивали при 25°С в течение 0,5-2 часов, затем добавили промежуточные соединения 67-68, растворенные в дихлорметане. Через 18 часов смесь выливали в водный насыщенный раствор NaНСО3 и экстрагировали дихлорметаном. Сырой продукт очистили колоночной хроматографией с силикагелем (DCM до DCM/MeOH=9/1)
Общая методика 10
Аr2-CООН (1 экв.; получено согласно WO 2008147518 для соединений 49-51 и согласно США 3282927 для соединений 47-48), промежуточные соединения 67-68 (1 экв.) и DIPEA (2 экв.) растворили в дихлорметане (5 мл/ммоль) при 0°С, затем добавили ТЗР (50% в DCM; 1,2 экв.). Смесь перемешивали с обратным холодильником в течение 3-5 часов, затем при RT в течение ночи. Реакционную смесь промыли 1 М NaOH и водой, высушили (Na2SO4) и выпарили. Сырой продукт очистили колоночной хроматографией с силикагелем (DCM до DCM/MeOH=9/1).
Энантиомерную чистоту всех соединений вычисляли как энантиомерный избыток (ее%) с помощью методов хиральной ВЭЖХ.
Следующие продукты были получены по Общей методике 9 или 10:
Характеристика соединений 47-53:
Пример 96
Соединение 54: (±)(2-метил-5-фенилтиазол-4-ил)(7-(((6-метилпиридин-2-ил)амино)метил)-8-азаспиро[4.5]декан-8-ил)метанон
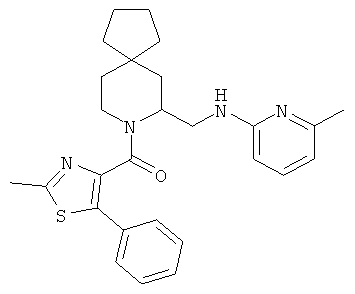
2-метил-5-фенилтиазол-4-карбоновую кислоту, получение которой описано в US 3282927 (85 мг; 0,38 ммоль), НОВТ (52 мг; 0,38 ммоль) и EDCI.HCl (110 мг; 0,578 ммоль) растворенный в дихлорметане (5 мл), перемешивали при 25°С в течение 0,5-2 часов, затем добавили промежуточное соединение 75 (100 мг; 0,38 ммоль), растворенное в дихлорметане (5 мл). Через 18 часов смесь выливали в водный насыщенный раствор NaHCO3 и экстрагировали дихлорметаном. Сырой продукт очистили колоночной хроматографией с силикагелем (DCM до DCM/MeOH=9/1). Выход титульного соединения 36 мг.
MS (ESI) m/z 461 [М+Н]+; 1Н-ЯМР (CDCl3) δ ppm 7.20-7.51 (m, 6H) 6.02-6.46 (m, 2H) 4.63-5.07 (m, 1H) 3.60-3.97 (m, 1H) 3.25-3.51 (m, 2H) 2.83-3.07 (m, 1H) 2.61-2.74 (m, 3H) 2.34-2.39 (m, 3H) 1.76-1.11 (m, 12H) 0.71-0.92 (m, 1H).
Пример 97
Соединение 55: (±)(7-(((5-хлорпиридин-2-ил)амино)метил)-8-азаспиро[4.5]декан-8-ил)(2-метил-5-фенилтиазол-4-ил)метанон
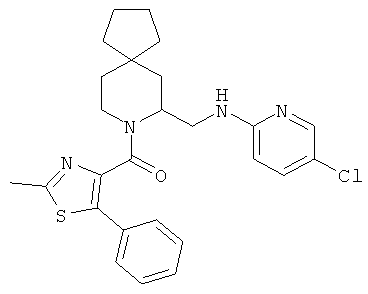
2-метил-5-фенилтиазол-4-карбоновую кислоту, получение которой описано в US 3282927 (78 мг; 0,35 ммоль), НОВТ (48 мг; 0,35 ммоль) и EDCI.HCl (102 мг; 0,53 ммоль), растворенный в дихлорметане (5 мл), перемешивали при 25°С в течение 0,5-2 часов, затем добавили промежуточное соединение 76 (100 мг; 0,38 ммоль), растворенное в дихлорметане (5 мл). Через 18 часов смесь выливали в водный насыщенный раствор NаHСО3 и экстрагировали дихлорметаном. Сырой продукт очистили колоночной хроматографией с силикагелем (DCM до DCM/MeOH=9/1). Выход титульного соединения 52 мг.
MS (ESI) m/z 503 [M+Na]+; 1H-ЯMP (СDCl3) δ ppm 7.91-8.03(m, 1H) 7.23-7.49 (m, 6Н) 6.29-6.41 (m, 1H) 5.32-5.44 (m, 1H) 4.62-5.01 (m, 1H) 3.75-3.95 (m, 1H) 3.36-3.63 (m, 1H) 3.32-3.25 (m, 1H) 2.82-3.20 (m, 1H) 2.61-2.74 (m, 3H). 1.11-1.89 (m, 12H), 0.76-0.95 (m, 1H).
Пример 98
Оценка эффектов соединений изобретения
Полезность соединений согласно настоящему изобретению как антагонистов орексинового рецептора 1 (OX1) определяли методами, хорошо известными специалистам в данной области техники, включая измерение внутриклеточных уровней кальция, [Ca2+]i, методом "FLIPR" (D. Smart, J.C. Jerman, S.J. Brough, S.L. Rushton, P.R. Murdock, F. Jewitt, N.A. Elshourbagy, C.E. Ellis, D.N. Middlemiss & F. Brown; British Journal of Pharmacology (1999) 128, 1-3).
В типичном эксперименте антагонистическая активность в отношении человеческих рецепторов OX1 и OX2 определяется с использованием клеток СНО и НЕК-293, трансфицированных рекомбинантными человеческими рецепторами OX1 и OX2, соответственно, которые высевают с плотностью 2 и 3×104 клеток/лунку, соответственно, в 96-луночные планшеты для флюорометрии. При этом планшет «нагружали» кальциевым красителем (Fluo-4NW/пробенецид в HBSS, Hepes 20 мМ, pH 7,4; Invitrogen) при 37°С в течение 60 минут. После того как температура сохранялась на уровне 22°С в течение 15 мин, измеряли [Ca2+]i прямо в планшете с помощью флуоресцентного планшетного ридера (CellLux Perkin Elmer).
Соединения по изобретению 1-11 растворили в DMSO, разведенном в HBSS (DMSO, 0,3% окончательная), и добавили к лункам. В этом исследовании для сравнения было проверено 5 других соединений с замещенной структурой, подобной соединениям по изобретению, но без спирокольца. Через 5 мин клетки СНО активировали орексином-A, 3 нМ, а клетки НЕК-293 активировали орексином-B, 10 нМ.
Соединения, растворенные в DMSO и разведенные в среде (DMSO; окончат.0,3%), проанализировали в диапазоне концентраций 1нМ-1 мкМ (каждую концентрацию дважды). Антагонистическая активность была выражена как pKb (ко-логарифм эффективной константы диссоциации рассчитывали по модифицированному уравнению Ченга-Прусоффа).
Соединения следующего примера, исследованные согласно этому примеру, дали следующие величины pKb:
Структура сравнительных соединений:
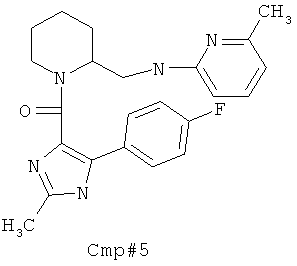
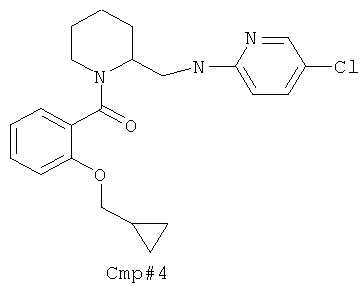
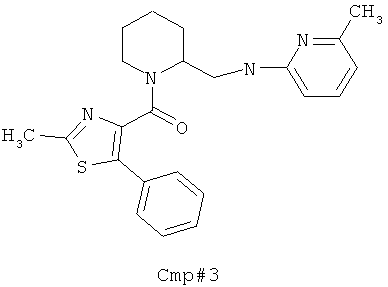
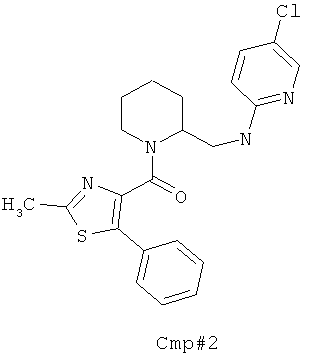
NA: - значение IC50 не могло быть вычислено. Кривая зависимости концентрация - ответ показала менее чем 25% эффект при самых высоких проверенных концентрациях.
<5.0 о <6.0: значение IC50 выше самой высокой проверенной концентрации. Кривая зависимости концентрация - ответ показала менее чем 50% эффект при самых высоких проверенных концентрациях.
Как показано в таблице соединения изобретения неожиданно проявили селективность к рецептору OX1, не так, как сравнительные соединения, которые продемонстрировали антагонистическую активность против двух видов рецепторов и OX1 и OX2.
В частности, соединение 2, имеющее почти одинаковую структуру со сравнительным соединением 2, за исключением спирокольца, показало высокую селективность к рецептору OX1. То же самое наблюдается между соединением 1 и сравнительным соединением 3, соединением 7 и сравнительным соединением 4 и соединением 8 и сравнительным соединением 5.
Пример 99
Фармакокинетику соединения 4 исследовали на самцах крыс Han Wistar. Крысам вводили внутривенно и пероарально (n=3 для каждого способа введения) раствор соединения 2 в дозе 1 мг/кг, приготовленный в молочной кислоте 150 мМ pH 4,5 в воде, 5% DMSO, 10% TWEEN 80. В яремную вену крыс была введена канюля для взятия серии проб, при этом для каждой крысы был получен полный профиль. Другую группу крыс (n=3) обрабатывали внутривенно и забивали через 1 час с целью получения образцов артериальной крови и мозга для оценки проникновения в мозг. Экстракты плазмы и мозга количественно анализировали с помощью специального и чувствительного биоаналитического метода LC-MS/MS. Изменения между индивидуумами в каждой группе из трех крыс были ограниченными (CV для фармакокинетических параметров было ниже 30%).
После внутривенной инъекции соединение находилось в кровообращении со средним значением AUC примерно 740 нг·час/мл. Среднее значение клиренса (выведения) составляло около 320 мл/час, представляя 40% потока крови через печень крысы, что говорит об умеренном выведении и печеночной экстракции. Средний объем распределения (Vss) составлял 330 мл, что представляет собой два объема всей воды в теле крысы и говорит об умеренном распределении соединения вне кровяного компартмента.
После перорального введения наблюдалась довольно быстрая абсорбция, при этом явная максимальная концентрация достигалась за 30 минут для всех трех животных. Среднее значение AUC составляло около 360 нг·час/мл, представляя около 50% AUC после внутривенного введения.
Через один час после внутривенного введения средний уровень в артериальной плазме составлял 126 нг/мл; средний уровень во всем мозге составлял 83 нг/г. Отношение концентраций мозг/плазма давало значение 0,66, что показывает значительное проникновение соединения в мозг.
В заключение, соединение 2, введенное крысам в дозе 1 мг/кг, продемонстрировало характеристики умеренного выведения лекарства с умеренным объемом распределения и хорошее проникновение в мозг. При введении в виде раствора для перорального введения соединение 2 продемонстрировало хорошую биодоступность 50%.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРНЫЕ СОЕДИНЕНИЯ | 2013 |
|
RU2673079C2 |
| СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ РАКА | 2011 |
|
RU2609018C2 |
| СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ РАКА | 2009 |
|
RU2514427C2 |
| ПРОИЗВОДНЫЕ АЗАСПИРОАЛКАНОВ В КАЧЕСТВЕ ИНГИБИТОРОВ МЕТАЛЛОПРОТЕАЗ | 2004 |
|
RU2379303C2 |
| Имидазопиридиновые соединения в качестве ингибиторов PAD | 2018 |
|
RU2782743C2 |
| ИНГИБИТОРЫ ФАКТОРА XIa | 2016 |
|
RU2728783C2 |
| ПРОИЗВОДНЫЕ 5-(7Н-ПИРРОЛО[2,3-d]ПИРИМИДИН-4-ИЛ)-5-АЗАСПИРО[2.5]ОКТАН-8-КАРБОНОВОЙ КИСЛОТЫ В КАЧЕСТВЕ НОВЫХ ИНГИБИТОРОВ JAK-КИНАЗЫ | 2018 |
|
RU2761626C2 |
| ИНГИБИТОРЫ ИОННОГО КАНАЛА, ФАРМАЦЕВТИЧЕСКИЕ СОСТАВЫ И ПРИМЕНЕНИЕ | 2016 |
|
RU2746188C2 |
| ИНДАЗОЛИЛИЗОКСАЗОЛЬНЫЕ ПРОИЗВОДНЫЕ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, ТАКИХ КАК ЗЛОКАЧЕСТВЕННОЕ НОВООБРАЗОВАНИЕ | 2020 |
|
RU2836662C2 |
| БИЦИКЛИЧЕСКИЕ КЕТОНЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2018 |
|
RU2797922C2 |
Изобретение имеет отношение к спиро-аминосоединению формулы (VI), где m представляет собой 1 или 2 или 3, n представляет собой 1 или 2, R выбирают из 6-членного ароматического кольца и 5-членного гетероароматического кольца, содержащего от 1 до 2 гетероатомов, выбранных из S и N, причем такое кольцо является замещенным одним или двумя заместителями, выбранными из группы, состоящей из (С1-С3)алкила, атома галогена, (С3-С5)циклоалкилоксигруппы, фенила, необязательно замещенного одним или более атомами галогена, 5- или 6- членного гетероцикла, содержащего, по меньшей мере, один атом азота, выбираемого из 1,2,3-триазола, пиримидина, пиридина и пиразина; Р представляет собой заместитель Q, где Q выбирают из группы, состоящей из фенила, пиридила, пиримидила, при этом Q необязательно замещен одним или более заместителями, выбранными из группы, состоящей из (С1-С3)алкила, галогена, трифторметила, метилкарбоксигруппы. Изобретение также относится к спиро-аминосоединению формулы (VI), где m представляет собой 1 или 3, n представляет собой 1 или 2, R представляет собой фенил, замещенный заместителем циклопропил (С1-С3)алкокси, Р представляет собой заместитель Q, где Q выбирают из группы, состоящей из фенила, пиридила, пиримидила, при этом Q необязательно замещен одним или более заместителями, выбранными из группы, состоящей из (С1-С3)алкила, галогена, трифторметила, метилкарбоксигруппы. Соединения формулы IV предназначены для производства медикамента для лечения патологий, при которых необходимо применение антагониста орексинового рецептора 1. Технический результат - спиро-аминосоединения, обладающие антагонистической активностью к орексиновому рецептору. 3 н. и 16 з.п. ф-лы, 99 пр.
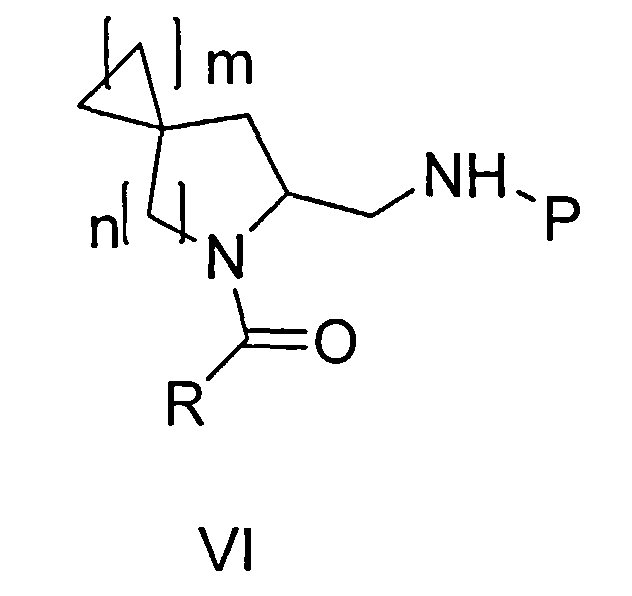
1. Спиро-аминосоединение формулы (VI):
где
m представляет собой 1 или 2 или 3
n представляет собой 1 или 2,
R выбирают из 6-членного ароматического кольца и 5-членного гетероароматического кольца, содержащего от 1 до 2 гетероатомов, выбранных из S и N, причем такое кольцо является замещенным одним или двумя заместителями, выбранными из группы, состоящей из (С1-С3)алкила, атома галогена, (С3-С5)циклоалкилоксигруппы, фенила, необязательно замещенного одним или более атомами галогена, 5- или 6- членного гетероцикла, содержащего, по меньшей мере, один атом азота, выбираемого из 1,2,3-триазола, пиримидина, пиридина и пиразина;
Р представляет собой заместитель Q, где Q выбирают из группы, состоящей из фенила, пиридила, пиримидила, при этом Q необязательно замещен одним или более заместителями, выбранными из группы, состоящей из (С1-С3)алкила, галогена, трифторметила, метилкарбоксигруппы.
2. Соединение по п.1, имеющее структуру формулы (VIa), в котором Р представляет собой Q:
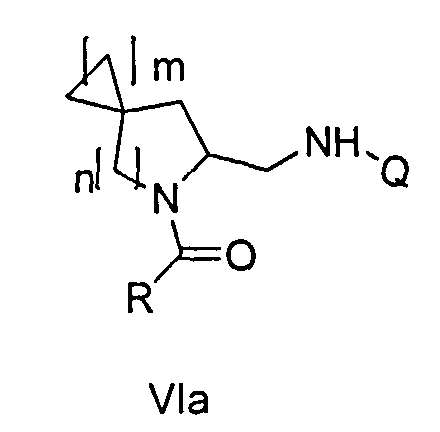
3. Соединение по п.2, в котором n представляет собой 2, а m представляет собой 1.
4. Соединение по п.1, в котором R представляет собой фенил или 5-членное гетероароматическое кольцо.
5. Соединение по п.4, в котором когда R представляет собой гетероароматическое кольцо, оно является тиазольным кольцом.
6. Соединение по п.4, в котором тиазольное кольцо является замещенным одним или двумя заместителем(ми), выбранным(и) из группы, состоящей из метила, фенила, фенила, замещенного одним или более галогенами.
7. Соединение по п.4, в котором R представляет собой фенил, замещенный заместителем, выбранным из триазолила и пиримидила.
8. Соединение по любому из пп.1-7, в котором Q представляет собой пиридильное кольцо, необязательно замещенное одним или более заместителями, выбранными из группы, состоящей из трифторметила, метила и галогена.
9. Соединение по п.2, в котором m=2, а n=2.
10. Соединение по п.2, в котором m=3, а n=2.
11. Соединение по п.10, в котором Q представляет собой пиридил, замещенный трифторметилом, или пиридил, замещенный (С1-С3)алкилом, а R представляет собой 5-членное гетероароматическое кольцо, содержащее два гетероатома, выбранных из S и N, причем такой R является замещенным одним или двумя заместителями, выбранными из (С1-С3)алкила и галогена.
12. Соединение по п.2, в котором m=1, а n=1.
13. Соединение по п.12, в котором Q представляет собой пиридил, замещенный одним или более галогенами, или пиридил, замещенный (С1-С3)алкилом, а R представляет собой фенил, замещенный одним или двумя заместителями, выбранными из (С1-С3)алкила и 5- или 6-членного гетероцикла, содержащего, по меньшей мере, один атом азота.
14. Соединение по п.1, которое выбирают из группы, состоящей из:
(±)(2-метил-5-фенилтиазол-4-ил)(5-((6-метилпиридин-2-иламино)метил)-6-азаспиро[2.5]октан-6-ил)метанона
(R)-(2-метил-5-фенилтиазол-4-ил)(5-(((6-метилпиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)метанона
(S)-(2-метил-5-фенилтиазол-4-ил)(5-(((6-метилпиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)метанона
(±)(5-((5-хлорпиридин-2-иламино)метил)-6-азаспиро[2.5]октан-6-ил)(2-метил-5-фенилтиазол-4-ил)метанона
(R)-(5-(((5-хлорпиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)(2-метил-5-фенилтиазол-4-ил)метанона
(S)-(5-(((5-хлорпиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)(2-метил-5-фенилтиазол-4-ил)метанона
(±)(2-метил-5-фенилтиазол-4-ил)(5-((5-(трифторметил)пиридин-2-иламино)метил)-6-азаспиро[2.5]октан-6-ил)метанона
(S)-(2-метил-5-фенилтиазол-4-ил)(5-(((5-(трифторметил)пиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)метанона
(±)(5-((5-хлорпиридин-2-иламино)метил)-6-азаспиро[2.5]октан-6-ил)(5-(4-фторфенил)-2-метилтиазол-4-ил)метанона
(±)(5-((5-хлорпиридин-2-иламино)метил)-6-азаспиро[2.5]октан-6-ил)(5-метил-2-(2Н-1,2,3-триазол-2-ил)фенил)метанона
(±)(5-((5-хлорпиридин-2-иламино)метил)-6-азаспиро[2.5]октан-6-ил)(5-метил-2-(пиримидин-2-ил)фенил)метанона
(S)-(5-(((5-хлорпиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)(5-метил-2-(пиримидин-2-ил)фенил)метанона
(±)метил 5-хлор-2-((6-(5-(4-фторфенил)-2-метилтиазол-4-карбонил)-6-азаспиро[2.5]октан-5-ил)метиламино)бензоата
(S)-(5-(((5-фторпиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)(2-метил-5-фенилтиазол-4-ил)метанона
(S)-(5-(((6-хлорпиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)(2-метил-5-фенилтиазол-4-ил)метанона
(S)-(2-метил-5-фенилтиазол-4-ил)(5-(((6-(трифторметил)пиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)метанона
(S)-(5-(((6-хлорпиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)(5-метил-2-(2Н-1,2,3-триазол-2-ил)фенил)метанона
(S)-(5-метил-2-(2Н-1,2,3-триазол-2-ил)фенил)(5-(((6-(трифторметил)пиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)метанона
(S)-(2-метил-5-фенилтиазол-4-ил)(5-(((4-метилпиримидин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)метанона
(S)-(5-(((4,6-диметилпиримидин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)(2-метил-5-фенилтиазол-4-ил)метанона
(S)-(2-метил-5-фенилтиазол-4-ил)(5-(((5-метилпиримидин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)метанона
(S)-(5-(((4,б-дифторпиримидин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)(2-метил-5-фенилтиазол-4-ил)метанона
(S)-(5-(((6-фторпиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)(2-метил-5-фенилтиазол-4-ил)метанона
(S)-(5-метил-2-(2Н-1,2,3-триазол-2-ил)фенил)(5-(((5-(трифторметил)пиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)метанона
(S)-(5-метил-2-(пиримидин-2-ил)фенил)(5-(((5-(трифторметил)пиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)метанона
(S)-(2-(2Н-1,2,3-триазол-2-ил)фенил)(5-(((5-хлорпиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)метанона
(S)-(5-(((5-хлорпиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)(5-фтор-2-(2Н-1,2,3-триазол-2-ил)фенил)метанона
(S)-(5-(((5-хлорпиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)(2-(пиразин-2-ил)фенил)метанона
(S)-(5-(((5-хлорпиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)(2-фтор-6-(2Н-1,2,3-триазол-2-ил)фенил)метанона
(S)-(5-(((5-хлорпиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)(5-фтор-2-(пиридин-2-ил)фенил)метанона
(S)-(5-(((5-хлорпиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)(5-метил-2-(пиридин-2-ил)фенил)метанона
(S)-(5-(((5-хлорпиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)(2-фтор-6-(пиридин-2-ил)фенил)метанона
(S)-[1,1'-бифенил]-2-ил(5-(((4,6-диметилпиримидин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)метанона
(S)-(2,5-дихлорфенил)(5-(((4,6-диметилпиримидин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)метанона
(S)-[1,1'-бифенил]-2-ил(5-(((5-хлорпиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)метанона
(S)-[1,1'-бифенил]-2-ил(5-(((4-метилпиримидин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)метанона
(S)-(5-(((5-хлорпиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)(2-(пиримидин-2-ил)фенил)метанона
(S)-(5-(((6-хлорпиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)(5-метил-2-(пиримидин-2-ил)фенил)метанона
(S)-(5-(((6-хлорпиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)(5-метил-2-(пиразин-2-ил)фенил)метанона
(S)-(5-метил-2-(пиразин-2-ил)фенил)(5-(((6-(трифторметил)пиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)метанона
(S)-(5-(((5-хлорпиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)(5-фтор-2-(пиримидин-2-ил)фенил)метанона
(S)-(5-(((5-хлорпиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)(2-фтор-6-(пиримидин-2-ил)фенил)метанона
(S)-(5-(((5-хлорпиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)(5-фтор-2-(пиразин-2-ил)фенил)метанона
(S)-(6-(((5-хлорпиридин-2-ил)амино)метил)-5-азаспиро[2.4]гептан-5-ил)(5-метил-2-(2Н-1,2,3-триазол-2-ил)фенил)метанона
(S)-(5-метил-2-(2Н-1,2,3-триазол-2-ил)фенил)(6-(((6-метилпиридин-2-ил)амино)метил)-5-азаспиро[2.4]гептан-5-ил)метанона
(S)-(6-(((5-хлорпиридин-2-ил)амино)метил)-5-азаспиро[2.4]гептан-5-ил)(5-метил-2-(пиримидин-2-ил)фенил)метанона
(±)(2-метил-5-фенилтиазол-4-ил)(7-(((6-метилпиридин-2-ил)амино)метил)-8-азаспиро[4.5]декан-8-ил)метанона
(±)(7-(((5-хлорпиридин-2-ил)амино)метил)-8-азаспиро[4.5]декан-8-ил)(2-метил-5-фенилтиазол-4-ил)метанона.
15. Соединение по п.14, которое представляет собой
(S)-(5-(((5-хлорпиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)(5-метил-2-(пиримидин-2-ил)фенил)метанон или
(S)-(5-(((5-хлорпиридин-2-ил)амино)метил)-6-азаспиро[2.5]октан-6-ил)(5-фтор-2-(пиримидин-2-ил)фенил)метанон.
16. Спиро-аминосоединение формулы (VI):
где
m представляет собой 1 или 3,
n представляет собой 1 или 2,
R представляет собой фенил, замещенный заместителем циклопропил(С1-С3)алкокси;
Р представляет собой заместитель Q, где Q выбирают из группы, состоящей из фенила, пиридила, пиримидила, при этом Q необязательно замещен одним или более заместителями, выбранными из группы, состоящей из (С1-С3)алкила, галогена, трифторметила, метилкарбоксигруппы.
17. Соединение по п.16, где указанное соединение представляет собой (±)(5-((5-хлорпиридин-2-иламино)метил)-6-азаспиро[2.5]октан-6-ил)(2-(циклопропилметокси)фенил)метанон).
18. Применение соединения по любому из пп.1-17 для производства медикамента для лечения патологий, при которых необходимо применение антагониста орексинового рецептора 1.
19. Применение по п.18, где патологию выбирают из ожирения, нарушений сна, компульсивных нарушений, лекарственной и алкогольной зависимости, шизофрении.
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| СПИРОПИПЕРИДИНЫ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ) | 1993 |
|
RU2168512C2 |
| US 5756504 A, 26.05.1998 | |||

