ОБЛАСТЬ ТЕХНИКИ
Данное изобретение относится к новым производным N-ацилгидразона и, более конкретно, к новым производным N-ацилгидразона, обладающим ингибиторной активностью в отношении Т-клеток и/или активностью против лимфонеоплазии, их стереоизомерам и фармацевтически приемлемым солям, их использованию для приготовления фармацевтических композиций, фармацевтическим композициям, их содержащим, способам лечения с помощью этих композиций и способам получения новых производных N-ацилгидразона.
УРОВЕНЬ ТЕХНИКИ
Иммунодепрессивные медикаменты
Ключевой задачей трансплантологии является подавление иммунного ответа, ведущего к отторжению трансплантата. Таким образом, разработка иммунодепрессивных медикаментов, способных селективно и эффективно регулировать иммунный ответ по отношению к имплантату, является ключевой целью для повышения успешности трансплантаций. Т-клетки являются основными мишенями для предотвращения отторжения трансплантатов, и большинство клинически применяемых иммунодепрессивных медикаментов ингибируют ответ Т-клеток.
Азатиоприн является первым иммунодепрессивным агентом, применяемым при трансплантации органов. Однако азатиоприн ингибирует синтез ДНК, что приводит к развитию таких побочных эффектов, как лейкопения, макроцитоз и подавление роста костного мозга, по причине чего азатиоприн был переведен в терапию второй линии после введения циклоспорина. Циклоспорин и такролимус, ингибиторы кальциневрина, используются в настоящее время при трансплантации органов в качестве терапии первой линии.
Эти два медикамента блокируют экспрессию интерлейкина-2 (IL-2) путем ингибирования кальциневрина. Далее эти медикаменты могут подавлять активацию Т-клеток на ранней стадии, тем самым сильно подавляя иммунный ответ, приводящий к отторжению трансплантата. Однако, эти медикаменты необходимо принимать в течение длительного времени, и они вызывают такие побочные эффекты, как нефротоксичность, анемия и гипертония, а также инфекционные и онкологические заболевания в результате ослабления иммунной системы. В последние годы, с целью уменьшения побочных эффектов ингибиторов кальциневрина, в комбинации с ингибитором кальциневрина применяли мофетила микофенолат (ингибитор нуклеотидного синтеза), но он вызывает такие побочные эффекты, как болезни пищеварительного тракта, лейкопению и анемию.
Сиролимус и эверолимус, ингибиторы mTOR (мишень рапамицина в клетках млекопитающих), являются медикаментами, ингибирующими пролиферацию Т-клеток путем блокады сигнальных путей рецептора IL-2. Эти медикаменты в основном назначаются в комбинации с ингибитором кальциневрина, потому что эффективность этих медикаментов самих по себе невелика. Однако, эти медикаменты усиливают побочные эффекты ингибитора кальциневрина, такие как гиперлипидемия и тромбоцитопения.
FTY720, антагонист сфингозин-1-фосфата, является медикаментом, уменьшающим иммунные ответы путем блокировки миграции Т-клеток от лимфоидных органов к имплантатам, и в сравнении с другими медикаментами обладает низкой токсичностью. Однако FTY720 при приеме с другими медикаментами (например, анестетиками и бета-блокираторами общего действия) может вызывать инфаркт. В области трансплантации органов, комбинированная терапия с использованием циклоспорина находится в стадии клинических испытаний (см. N Engl J Med. 351, 2715-2729; N Engl J Med. 352, 1371-1373; Nat Med. 11, 605-613; Business Insights. 2010, BI 00022-067).
Как было описано выше, используемые в настоящее время для предотвращения отторжения трансплантатов иммунодепрессанты токсичны и вызывают такие побочные эффекты, как воспаления и онкологические заболевания, вследствие ослабления иммунной системы. Поэтому необходима разработка новых медикаментов, способных селективно разрушать Т-клетки, реагирующие на трансплантационные антигены.
Реакция "трансплантат против хозяина"
Аллогенная трансплантация гематопоэтических стволовых клеток (ТГСК, HSCT) является наиболее эффективным и постоянным способом лечения различных злокачественных заболеваний крови и иммунодефицитных заболеваний и используется для лечения более 20000 пациентов в год во всем мире (согласно отчету Центра международных исследований трансплантации крови и костного мозга, CIBMTR). В последние годы предпринимались попытки применения ТГСК при аутоиммунных заболеваниях, солидном раке и трансплантациях. Несмотря на быстрое развитие аллогенной трансплантации гематопоэтических стволовых клеток (например, разработка технологии идентификации антигенов лейкоцитов человека (HLA) и новых иммунодепрессантов) в последние 20 лет, реакция «трансплантат против хозяина» (РТПХ, GVHD), являющаяся осложнением, вызываемым Т-клетками донора, все еще остается главной причиной пост-трансплантационной смертности. Острая РТПХ (степени II-IV), которая развивается в основном в первые 100 дней после трансплантации, проявляется у 25-60% пациентов в случае идентичности HLA между кровными родственниками и у 45-70% пациентов в случае отсутствия кровной родственной связи, и 70% пациентов с этим заболеванием (III-IV степени) умирает. Острая РТПХ развивается в три стадии. Во время первой стадии, провоспалительные факторы (TNF-α и LPS), образующиеся после высоких доз химиотерапии и системной радиационной терапии перед трансплантацией, активируют дендритные клетки периферических лимфоидных органов. Во второй стадии, аллоантиген-специфичные Т-клетки, порождаемые активированными дендритными клетками, дифференцируют в эффекторные клетки. На последней стадии, аллоантиген-специфичные эффекторные Т-клетки мигрируют в кишечник, печень и кожу, которые являются главными мишенями, и вызывают там воспалительные повреждения тканей.
Доля положительного ответа на действие ингибитора кальциневрина, который применяется в качестве терапии первой линии для предотвращения острой РТПХ, составляет менее 50%, и таким образом доля предотвращения очень низка в сравнении с количеством трансплантаций. Лечение после развития РТПХ зависит от стероидной терапии, но стероидная терапия демонстрирует долю положительного ответа менее 50%, и поэтому классифицируется как терапия высокого риска с высокой смертностью (см. Annu Rev Immunol. 25, 139-170; Blood. 114, 4327-4336; Lancet. 373, 1550-1561, Curr Opin Immunol. 11, 509-515). Таким образом, срочно необходимо развитие новых эффективных медикаментов для предотвращения и лечения острой РТПХ.
Множественный склероз
Множественный склероз является воспалительным заболеванием центральной нервной системы (ЦНС), которое является аутоиммунным заболеванием, вызываемым реакцией Т-клеток на происходящий от миелина антиген. На начальной стадии заболевание развивается по рецидивно-ремиссионной схеме и затем прогрессирует во вторичный прогрессивный множественный склероз вследствие нарастающего накопления повреждений головного и спинного мозга. Нарастающее накопление повреждений головного и спинного мозга ведет к потере зрения, нарушению баланса и движений, нарушению речи и сенсорики, двустороннему параличу, нарушению половой функции и сложностям с мочеотделением и дефекацией; в тяжелых случаях приводит к общему параличу.
Терапия при лечении множественного склероза в основном делится на лечение обострений болезни, терапию, изменяющую течение заболевания, симптоматическую терапию и профилактическую терапию. Лечение обострений болезни проводят для снижения воспаления и создания иммунодепрессивного эффекта, а терапию, изменяющую течение заболевания применяют для задержки прогрессирования заболевания, т.е. для предотвращения развития прогрессирующего множественного склероза.
При лечении обострений болезни, с целью облегчения симптомов и предотвращения получения перманентных повреждений, в течение 3-5 дней внутривенно вводят высокие дозы (500-1000 мг/кг) глюкокортикоидов. Глюкокортикоиды ингибируют миграцию иммунных клеток в мозг и уменьшают отек, тем самым ингибируя воспаление, возникающее на острой стадии (см. Immunology and Cell Biology. 76, 55-64; Annu. Rev. Immunol. 20, 125-163). Хотя глюкокортикоиды оказывают крайне положительный эффект при применении во время острой стадии болезни, они не могут предотвратить прогрессирование заболевания в качестве терапии, изменяющей течение болезни.
В качестве терапии, изменяющей течение болезни, проводят иммунотерапию, призванную излечить заболевание и предотвратить его рецидивы. Медикаментами, одобренными Управлением по контролю продуктов питания и лекарственных средств США (US FDA) для применения в качестве иммунотерапии, являются интерферон-бета (IFN-β), глатирамер ацетат (GA), митоксантрон и натализумаб. Однако, они обладают следующими побочными эффектами (см. N Engl J Med. 362, 456-458):
IFN-β, наиболее популярный в настоящее время препарат, обладает противовоспалительным и антивирусным эффектом, ингибирует экспрессию антигенов и предотвращает активацию Т-клеток. Кроме того, IFN-β активирует костимуляторные молекулы для стимулирования апоптоза аутореактивных клеток. Однако, IFN-β можно вводить только внутримышечным или подкожным путем, и при его введении в течение длительного времени он вызывает эритему и отек в месте введения, а также такие побочные эффекты, как мышечную боль, озноб и аутоиммунные заболевания.
Митоксантрон (новатрон) имеет очень малую молекулярную массу, поэтому он проникает через мягкие мозговые оболочки и ингибирует пролиферацию Т-клеток, В-клеток и макрофагов в мягких мозговых оболочках и антиген-представляющую функцию антиген-представляющих клеток (APCs), тем самым облегчая симптомы множественного склероза. Однако, дозировка митоксантрона должна быть ограничена, так как он оказывает тяжелую нагрузку на сердце.
Глатирамер ацетат (ГА, GA) является аналогом основного миелинового белка (ОМБ, МВР). ГА образует ГА/ОМБ комплекс при связывании ОМБ с молекулой HLA класса II, тем самым ингибируя активацию ОМБ-реактивных Т-клеток путем конкурирования с ОМБ при его связывании с рецептором Т-клеток (TCR). ГА обладает эффектом снижения количества рецидивов во время рецидивно-ремиссионного множественного склероза и облегчает симптомы множественного склероза при его рецидивах, наподобие IFN-β, но также увеличивает частоту развития постоянных «черных дыр» в центральной нервной системе, по сравнению с IFN-β.
Натализумаб является гуманизированным моноклональным антителом, связывающимся напрямую с α4 подгруппой (CD49; адгезионная молекула на поверхности лейкоцитов) интегрина VLA-4 (very late antigen-4) для предотвращения связывания между лейкоцитами и васкулярными эндотелиальными клетками, тем самым предотвращая вхождение активных Т-клеток в центральную нервную систему. Он демонстрирует отличную эффективность в отношении рецидивно-ремиссионного множественного склероза, но обладает побочным эффектом в виде развития прогрессирующей мультифокальной лейкоэнцефалопатии (PML) у 0,3-0,9% пациентов после 2 лет приема.
Финголимод является синтетическим аналогом S1P (сфингозин 1-фосфат) рецептора мириоцина и является первым медикаментом для орального приема, недавно одобренным Управлением по контролю продуктов питания и лекарственных средств США (US FDA).
Финголимод предотвращает перемещение активированных лимфоцитов от вторичных лимфоидных тканей в центральную нервную систему путем связывания S1PR с поверхностью активированных хелперных Т-клеток 1 типа. Однако, сообщалось о таких побочных эффектах, как инфекция, макулярный отек, головная боль, грипп, диарея, люмбаго и повышения уровня печеночных ферментов (см. Nat. Rev. Neurol. 7, 255-262; Nat Rev Drug Discov. 11, 909-925).
Таким образом, существует потребность в разработке новых медикаментов, которые способны блокировать прогрессирование вторичного множественного склероза, демонстрируя при этом высокую терапевтическую эффективность при кратковременном оральном приеме (см. N Engl J Med. 362, 456-458).
Лимфонеоплазия
При употреблении в этом тексте, термин «лимфонеоплазия» относится к раку лимфатических клеток (В-клеток, Т-клеток и NK/T-клеток) в костном мозге и лимфоидной ткани. Лимфонеоплазия в целом подразделяется на лейкемию, лимфому и множественную миелому. Лимфоидная лейкемия является раком крови, при котором незрелые лимфоциты в костном мозге превращаются в раковые клетки, которые аккумулируются в ткани и распространяются системно через кровь. Лимфоидная лейкемия в целом подразделяется на острую лимфоидную лейкемию и хроническую лимфоидную лейкемию. Лимфома является раком крови, при котором лимфоциты в лимфоидной ткани превращаются в раковые клетки, аккумулируются в ткани и распространяются в периферическую кровь и костный мозг. Она подразделяется на лимфому Ходжкина и неходжкинскую лимфому. Множественная миелома является раком крови, при котором клетки плазмы в периферической лимфоидной ткани превращаются в раковые клетки, которые затем аккумулируются в костном мозге (см. see J Clin Invest. 2012; 122:3396-3397; Blood. 1994; 84:1361-1392; Lancet. 2012; 380:848-857).
Лечение лимфонеоплазии проводится с помощью стандартной химиотерапии с применением комбинации циклофосфамида, доксорубицина, винкристина и преднизона. В отношении начальной ответной эффективности этих медикаментов, ремиссия у взрослых пациентов наступает приблизительно в 85% случаев, однако также высока вероятность рецидива: пятилетний срок выживания без рецидива болезни составляет только 30-40%. Кроме того, при приеме этих медикаментов высок риск инфекции из-за их высокой цитотоксичности, также они часто вызывают такие побочные эффекты как нейротоксичность, проблемы с пищеварением и кровотечения (см. N Engl J Med. 1998; 339:21-26; N Engl J Med 2006; 354:166-78; Nat Rev Drug Discov. 2007; 6:149-165).
Согласно недавним сообщениям, медикаменты, обладающие цитотоксичным эффектом в отношении специфических клеток, также обладают противораковым эффектом, если эти клетки превращаются в раковые. Общалось, что иммунодепрессант В-клеток ибрутиниб, ингибирующий тирозинкиназу Брутона (BTK), являющуюся сигнальным медиатором рецептора В-клеток (BCR), обладает противораковым действием в отношении рецидивирующей или устойчивой к лечению лимфомы В-клеток. Бортезомиб, ингибитор NF-kB активности, используется в качестве иммунодепрессанта для ингибирования Т-клеток и В-клеток. Бортезомиб был одобрен Управлением по контролю продуктов питания и лекарственных средств США (US FDA) в качестве терапевтического агента для лечения множественного склероза и также недавно использовался в комбинации с ибрутинибом для лечения лимфомы В-клеток. Субероиланилид гидроксамовой кислоты (SAHA), являющийся ингибитором гистон деацетилазы (HDACi), обладает противораковым эффектом в отношении кожной лимфомы Т-клеток. Ингибиторный эффект SAHA в отношении заболевания «трансплантат против хозяина» (GVHD) был недавно обнаружен на животных моделях, вследствие чего SAHA вызвал интерес в качестве иммунодепрессанта (см. Semin Cancer Biol. 2011 Nov; 21:335-46; Br J Haematol. 2013; 161:43-56; N Engl J Med. 2008; 359:906-917; Nat Rev Drug Discov. 2006; 5:769-784; J. Clin. Invest. 2008; 118:2562-2573). Таким образом, иммунодепрессивные медикаменты, нацеленные на специфические иммунные клетки, можно клинически использовать в качестве новых противораковых агентов, способных специфично воздействовать на специфические формы рака.
ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Техническая проблема
Целью данного изобретения является предоставление новых соединений, способных селективно и эффективно удалять трансплантационные антиген- и аутоантиген-специфичные Т-клетки, которые тем самым можно использовать в качестве агентов для предотвращения и лечения трансплантационного иммунного отторжения и/или лечения аутоиммунных заболеваний. Кроме того, целью данного изобретения является предоставление новых соединений, обладающих прекрасной апоптической эффективностью против лимфолейкозных клеток, и вследствие этого могут использоваться в качестве агентов для лечения лимфонеоплазии.
Таким образом, целью данного изобретения является предоставление новых производных N-ацилгидразона, обладающих селективной ингибиторной активностью в отношении Т-клеток и/или анти-лимфолейкозной активностью, их стереоизомеров, фармацевтически активных солей, фармацевтических композиций, их содержащих, способов лечения с использованием этих композиций, и способов получения новых производных N-ацилгидразона.
Техническое решение
Данное изобретение относится к соединению следующей формулы 1, его стереоизомерам или его фармацевтически приемлемой соли:
[Формула 1]
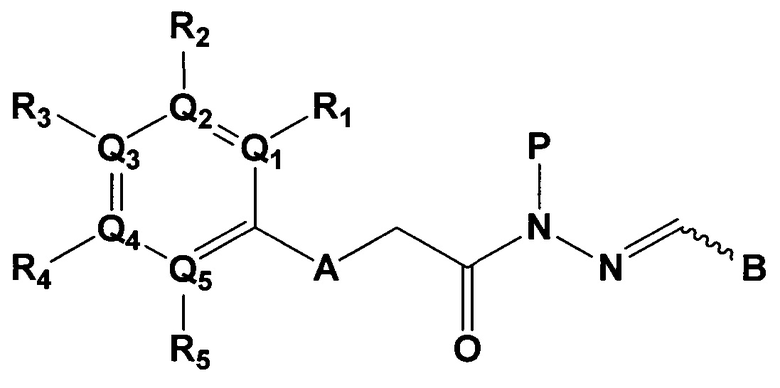
в котором
А является N-H, О, или S;
Q1, Q2, Q3, Q4 и Q5 каждый независимо являются С или N;
R1, R2, R3, R4 и R5 группы каждая независимо являются отсутствующей, или являются -Н, -CF3, -F, -Br, -Cl, цианидом, -СН2ОН группой, -(CO)NH2 группой, -(С1-С6)алкил группой, -(С1-3)алкокси группой, -NH2 группой, -N(CH3)2 группой, или 4, 5 или 6-членным гетероарилом или гетероциклоалкилом, содержащим от 1 до 3 членов выбранных из группы, состоящей из N, О и S (упомянутый гетероарил или гетероциклоалкил содержит по меньшей мере один заместитель выбираемый из -Н, галогена и амина);
Р является -Н, -(С1-С3)ОН группой, -(С1-С6)алкил группой, -(СО)(С1-С6)алкил группой;
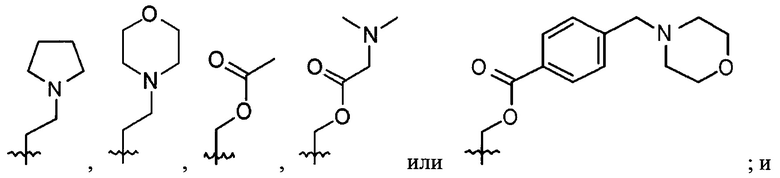
В выбирают из группы, состоящей из:
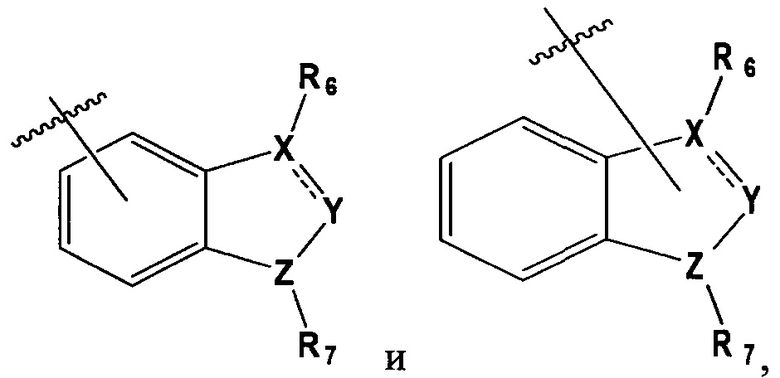
в которой, X, Y и Z каждый независимо являются С, N или S, и
R6 и R7 каждый независимо отсутствует, или является -Н, -Br, -(С1-С6)алкил группой, -(C1-С3)ОН группой,
В одном аспекте данного изобретения, соединения формулы 1 далее описываются следующим образом:
А является N-H, О или S;
Q1, Q2, Q3, Q4 и Q5 являются С;
R2 и R4 являются Н;
R1, R3 и R5 каждый независимо являются -Н, -F, -Br, -Cl, метилгруппой, этилгруппой, -СН2ОН группой, цианидом, -NH2 группой, или 4, 5 или 6-членным гетероарилом или гетероциклоалкилом, содержащим от 1 до 3 членов, выбранных из группы, состоящей из N, О и S;
Р является -Н, метилом, -CH2OH группой, -СН2СН2 ОН группой,
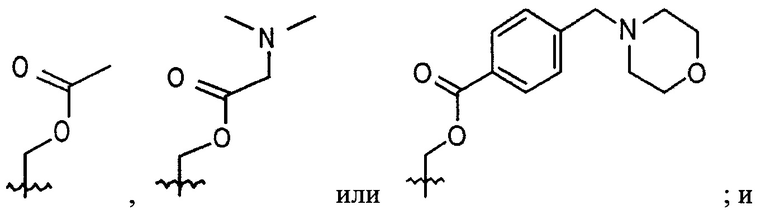
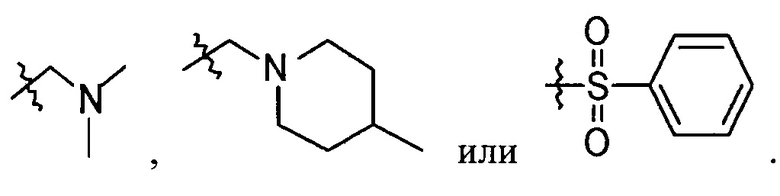
В выбирают из группы, состоящей из:
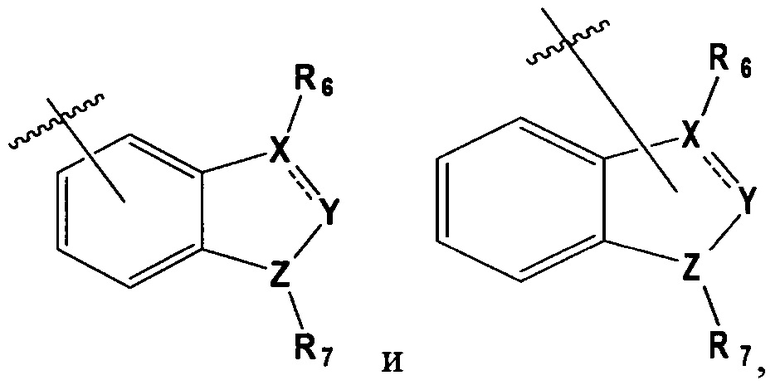
в которой Х и Y является С,
Z является N, и
R6 и R7 каждый независимо являются -Н, метилом или -СН2СН2ОН группой.
Соединения по формуле 1, согласно данному изобретению, могут в целом использоваться в виде фармацевтически приемлемой соли. Фармацевтически приемлемые соли включают фармацевтически приемлемые соли присоединения основания или соли присоединения кислоты, например, соли металлов, такие как соли щелочных и щелочноземельных металлов, соли аммония, соли присоединения органических аминов, соли присоединения аминокислот и сульфонатные соли. Соли присоединения кислоты включают соли присоединения неорганической кислоты, такие как соль хлористого водорода, соль серной кислоты и соль фосфорной кислоты; и соли присоединения органической кислоты, такие как алкил сульфонат, арил сульфонат, ацетат, малат, фумарат, тартрат, цитрат и лактат. Примеры солей металлов включают соли щелочных металлов, такие как соль лития, соль натрия и соль калия; соли щелочноземельных металлов, такие как соль магния, соль кальция, соль алюминия и соль цинка. Примеры солей аммония включают соль аммония и соль тетраметиламмония. Примеры солей присоединения органических аминов включают соли с морфолином и пиперидином. Примеры солей присоединения аминокислот включают соли с глицином, фенилаланином, глутаминовой кислотой и лизином. Примеры сульфонатных солей включают мезилат, тозилат и соли бензолсульфоновой кислоты.
Термин "стереоизомер" означает изомерные молекулы, имеющие одинаковую молекулярную формулу и связи, но отличающиеся своей трехмерной ориентацией.
Специфические примеры предпочтительных соединений формулы 1 по данному изобретению включают:
Соединение 065
(E)-N'-((1Н-индол-4-ил)метилен)-2-(мезитилокси)ацетогидразид;
Соединение 092
(Е)-N'-((1Н-индол-5-ил)метилен)-2-(4-бром-2,6-диметилфенокси)ацетогидразид;
Соединение 108
(E)-N'-((1Н-индол-6-ил)метилен)-2-(мезитилокси)ацетогидразид;
Соединение 109
(E)-N'-((1Н-индол-2-ил)метилен)-2-(мезитилокси)ацетогидразид;
Соединение 112
(Е)-N'-((1Н-индол-4-ил)метилен)-2-(4-бром-2,6-диметилфенокси)ацетогидразид;
Соединение 133
(Е)-N'-((1Н-индол-6-ил)метилен)-2-(4-бром-2,6-диметилфенокси)ацетогидразид;
Соединение 135
(E)-N'-((1Н-индол-4-ил)метилен)-2-(мезитиламино)ацетогидразид;
Соединение 137
(Е)-N'-((1Н-индол-4-ил)метилен)-2-(2,6-диметил-4-(пиридин-3-ил)фенокси)ацетогидразид;
Соединение 139
(Е)-N'-((1Н-индол-6-ил)метилен)-2-(2,6-диметил-4-(пиридин-3-ил)фенокси)ацетогидразид;
Соединение 146
(Е)-N'-((1Н-индол-4-ил)метилен)-2-(2-(пиридин-3-ил)фенокси)ацетогидразид;
Соединение 147
(E)-N'-((1Н-индол-4-ил)метилен)-2-(мезитилтио)ацетогидразид;
Соединение 149
(Е)-N'-((1Н-индол-4-ил)метилен)-2-(2,6-диметил-4-(пиримидин-5-ил)фенокси)ацетогидразид;
Соединение 155
(Е)-N'-((1Н-индол-4-ил)метилен)-2-(4-хлор-2,6-диметилфенокси)ацетогидразид;
Соединение 156
(Е)-N'-((1Н-индол-3-ил)метилен)-2-(4-хлор-2,6-диметилфенокси)ацетогидразид;
Соединение 157
(Е)-N'-((1Н-индол-6-ил)метилен)-2-(4-хлор-2,6-диметилфенокси)ацетогидразид;
Соединение 158
(Е)-N'-((1Н-индол-3-ил)метилен)-2-(мезитилтио)ацетогидразид;
Соединение 159
(E)-N'-((1Н-индол-6-ил)метилен)-2-(мезитилтио)ацетогидразид;
Соединение 164
(Е)-2-(мезитилокси)-N-метил-N'-((1-метил-1Н-индол-4-ил)метилен)ацетогидразид;
Соединение 177
(Е)-N'-((1Н-индол-4-ил)метилен)-N-(2-гидроксиэтил)-2-(мезитилокси)ацетогидразид;
Соединение 180
(E)-N'-((1-(2-гидроксиэтил)-1Н-индол-4-ил)метилен)-2-(мезитилокси)ацетогидразид;
Соединение 182
(E)-N'-((1Н-индол-4-ил)метилен)-2-(2,4-диметил-6-(пиридин-3-ил)фенокси)ацетогидразид;
Соединение 183
(E)-N'-((1Н-индол-6-ил)метилен)-2-(2,4-диметил-6-(пиридин-3-ил)фенокси)ацетогидразид;
Соединение 184
(Е)-N'-((1Н-индол-5-ил)метилен)-2-(2,4-диметил-6-(пиридин-3-ил)фенокси)ацетогидразид;
Соединение 187
(E)-N'-((1Н-индол-4-ил)метилен)-2-((2-метилпиридин-3-ил)окси)ацетогидразид;
Соединение 195
(Е)-N'-((1Н-индол-6-ил)метилен)-N-(2-гидроксиэтил)-2-(мезитилокси)ацетогидразид;
Соединение 217
(E)-N'-((1Н-индол-6-ил)метилен)-2-(2,4-диметил-6-(пиридин-4-ил)фенокси)ацетогидразид;
Соединение 218
(Е)-N'-((1Н-индол-6-ил)метилен)-2-(2-(фуран-3-ил)-4,6-диметилфенокси)ацетогидразид;
Соединение 227
(Е)-N'-((1Н-индол-4-ил)метилен)-2-(4-(гидроксиметил)-2,6-диметилфенокси)ацетогидразид;
Соединение 228
(Е)-N'-((1Н-индол-6-ил)метилен)-2-(4-(гидроксиметил)-2,6-диметилфенокси)ацетогидразид;
Соединение 229
(Е)-N'-((1Н-индол-6-ил)метилен)-2-(2,4-диметил-6-(тетрагидрофуран-3-ил)фенокси)ацетогидразид;
Соединение 232
(Е)-N'-((1Н-индол-4-ил)метилен)-2-(4-амино-2,6-диметилфенокси)ацетогидразид;
Соединение 233
(Е)-N'-((1Н-индол-6-ил)метилен)-N-(гидроксиметил)-2-(мезитилокси)ацетогидразид;
Соединение 236
(Е)-(2-((1Н-индол-6-ил)метилен)-1-(2-(мезитилокси)ацетил)гидразинил)метил 2-(диметиламино)ацетат;
Соединение 237
(Е)-N'-((1Н-индол-6-ил)метилен)-2-(4-циано-2,6-диметилфенокси)ацетогидразид;
Соединение 238
(Е)-(2-((1Н-индол-4-ил)метилен)-1-(2-(мезитилокси)ацетил)гидразинил)метил 2-(диметиламино)ацетат;
Соединение 243
(Е)-N'-((1Н-индол-6-ил)метилен)-N-ацетил-2-(мезитилокси)ацетогидразид;
Соединение 244
(Е)-N'-((1Н-индол-4-ил)метилен)-N-(гидроксиметил)-2-(мезитилокси)ацетогидразид;
Соединение 245
(Е)-(2-((1Н-индол-4-ил)метилен)-1-(2-(мезитилокси)ацетил)гидразинил)метил 4-(морфолинометил)бензоат;
Соединение 252
(Е)-(2-((1Н-индол-6-ил)метилен)-1-(2-(мезитилокси)ацетил)гидразинил)метил ацетат;
Соединение 258
(Е)-N'-((1Н-индол-6-ил)метилен)-2-(2,6-диметилфенокси)ацетогидразид;
Соединение 259
(Е)-N'-((1Н-индол-4-ил)метилен)-2-(2-бром-4,6-диметилфенокси)ацетогидразид;
Соединение 260
(Е)-N'-((1Н-индол-6-ил)метилен)-2-(2-бром-4,6-диметилфенокси)ацетогидразид;
Соединение 272
(Е)-(2-((1Н-индол-6-ил)метилен)-1-(2-(мезитилокси)ацетил)гидразинил)метил 4-(морфолинометил)бензоат;
Соединение 311
(E)-N'-((1Н-индол-6-ил)метилен)-2-(2-метил-6-(тетрагидрофуран-3-ил)фенокси)ацетогидразид;
Соединение 312
(Е)-N'-((1Н-индол-4-ил)метилен)-2-(2-метил-6-(тетрагидрофуран-3-ил)фенокси)ацетогидразид;
Соединение 313
(E)-N'-((1Н-индол-6-ил)метилен)-2-(2-(фуран-3-ил)-6-метилфенокси)ацетогидразид;
Соединение 314
(E)-N'-((1Н-индол-4-ил)метилен)-2-(2-(фуран-3-ил)-6-метилфенокси)ацетогидразид;
Соединение 317
(E)-N'-((1Н-индол-4-ил)метилен)-2-(2-(фуран-2-ил)-4,6-диметилфенокси)ацетогидразид;
Соединение 319
(Е)-N'-((1Н-индол-4-ил)метилен)-2-((2-(фуран-3-ил)пиридин-3-ил)окси)ацетогидразид;
Соединение 320
(Е)-N'-((1Н-индол-4-ил)метилен)-2-(2,4-диметил-6-(тетрагидрофуран-3-ил)фенокси)ацетогидразид;
Соединение 322
(E)-N'-((1Н-индол-6-ил)метилен)-2-(4-(фуран-3-ил)-2,6-диметилфенокси)ацетогидразид;
Соединение 323
(Е)-N'-((1Н-индол-6-ил)метилен)-2-(2,6-диметил-4-(тетрагидрофуран-3-ил)фенокси)ацетогидразид;
Соединение 329
(E)-N'-((1Н-индол-4-ил)метилен)-2-((2-этил-6-метилпиридин-3-ил)окси)ацетогидразид;
Соединение 331
(E)-N'-((1Н-индол-6-ил)метилен)-2-((2-(пирролидин-1-ил)пиридин-3-ил)окси)ацетогидразид;
Соединение 332
(Е)-N'-((1Н-индол-6-ил)метилен)-2-(2,4-диметил-6-(тетрагидрофуран-2-ил)фенокси)ацетогидразид;
Соединение 333
(E)-N'-((1Н-индол-4-ил)метилен)-2-(2-(фуран-3-ил)-4,6-диметилфенокси)ацетогидразид;
Соединение 336
(Е)-N'-((1Н-индол-4-ил)метилен)-2-(4-(фуран-3-ил)-2,6-диметилфенокси)ацетогидразид;
Соединение 337
(Е)-N'-((1Н-индол-4-ил)метилен)-2-(2,6-диметил-4-(тетрагидрофуран-3-ил)фенокси)ацетогидразид;
Соединение 343
(Е)-N'-((1Н-индол-6-ил)метилен)-2-((2-(тиофен-3-ил)пиридин-3-ил)окси)ацетогидразид;
Соединение 344
(E)-N'-((1Н-индол-4-ил)метилен)-2-(2,4-диметил-6-(тиофен-2-ил)фенокси)ацетогидразид;
Соединение 346
(Е)-N'-((1Н-индол-4-ил)метилен)-2-(2-(фуран-2-ил)-4-метоксифенокси)ацетогидразид;
Соединение 375
(Е)-2-(2,4-диметил-6-(тетрагидрофуран-3-ил)фенокси)-N-метил-N'-((1-метил-1Н-индол-6-ил)метилен)ацетогидразид;
Соединение 378
(Е)-N'-((1Н-индол-6-ил)метилен)-2-(4-фтор-2-метил-6-(тетрагидрофуран-3-ил)фенокси)ацетогидразид;
Соединение 379
(Е)-N'-((1Н-индол-4-ил)метилен)-2-(4-фтор-2-метил-6-(тетрагидрофуран-3-ил)фенокси)ацетогидразид;
Соединение 380
(Е)-2-(4-фтор-2-метил-6-(тетрагидрофуран-3-ил)фенокси)-N-метил-N'-((1-метил-1Н-индол-6-ил)метилен)ацетогидразид; и
Соединение 457
(Е)-N'-((1Н-индол-6-ил)метилен)-2-(2,4-диметил-6-(тетрагидро-2Н-пиран-4-ил)фенокси)ацетогидразид.
Специфические примеры более предпочтительных соединений формулы 1 по данному изобретению включают:
Соединение 065
(E)-N'-((1Н-индол-4-ил)метилен)-2-(мезитилокси)ацетогидразид;
Соединение 108
(E)-N'-((1Н-индол-6-ил)метилен)-2-(мезитилокси)ацетогидразид;
Соединение 229
(Е)-N'-((1Н-индол-6-ил)метилен)-2-(2,4-диметил-6-(тетрагидрофуран-3-ил)фенокси)ацетогидразид; и
Соединение 457
(Е)-N'-((1Н-индол-6-ил)метилен)-2-(2,4-диметил-6-(тетрагидро-2Н-пиран-4-ил)фенокси)ацетогидразид.
Данное изобретение также представляет фармацевтические композиции, включающие производные N-ацилгидразона формулы 1, их стереоизомеры или их фармацевтически приемлемые соли; и фармацевтически приемлемый носитель.
Предпочтительным образом, такая композиция используется для предотвращении или лечения заболевания, ассоциирующегося с ингибированием активности Т клеток. Специфические примеры таких заболеваний включают заболевание «трансплантат против хозяина» (GVHD) после трансплантации гематопоэтических стволовых клеток или после трансплантации органов, множественный склероз и ревматоидный артрит.
Предпочтительным образом, такая композиция используется для лечения лимфонеоплазии. Специфические примеры лимфонеоплазии включают Т лимфоидную лейкемию, В лимфоидную лейкемию, NK лейкемию, NKT лейкемию, множественную миелому, Т лимфому и В лимфому.
Полезные эффекты
Данное изобретение представляет новые соединения обладающие N-ацилгидразоновой структурой, которые можно использовать в качестве селективных ингибиторов Т клеток и/или анти-лимфолейкозых медикаментов.
Эти соединения эффективно ингибируют активные Т клетки. А именно, эти соединения селективно и эффективно удаляют Т-клетки, специфичные к трансплантационным антигенам и аутоантигенам, и тем самым могут использоваться для предотвращения и лечения реакции иммунного отторжения после трансплантации и лечения аутоиммунных заболеваний. Такими примерами служат заболевание «трансплантат против хозяина» (GVHD) после трансплантации гематопоэтических стволовых клеток или после трансплантации органов, множественный склероз и ревматоидный артрит, и др.
Кроме того, эти соединения обладают превосходным апоптическим эффектом по отношению к лимфолейкозным клеткам, и таким образом могут использоваться как агенты для лечения лимфонеоплазии. Такие примеры включают Т лимфоидную лейкемию, В лимфоидную лейкемию, NK лейкемию, NKT лейкемию, множественную миелому, Т лимфому и В лимфому, и др.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
На Фиг. 1 и 2 изображены результаты теста ингибиторной активности соединений по данному изобретению против аллоантиген-специфичных Т клеток в условиях клеточной культуры.
На Фиг. 3 изображены результаты теста ингибиторной активности соединений по данному изобретению против аллоантиген-специфичных Т клеток.
На Фиг. 4 изображены результаты теста ингибиторной активности соединений по данному изобретению против острого заболевания «трансплантат против хозяина».
На Фиг. 5 изображены результаты теста терапевтической активности соединений по данному изобретению в отношении множественного склероза.
ЛУЧШИЙ ВАРИАНТ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Получение соединений
Соединения формулы 1 по данному изобретению можно получить способами, известными из различных источников. Способы получения соединений формулы 1 будут далее подробно описаны с нижеследующими схемами и примерами.
[Схема 1]
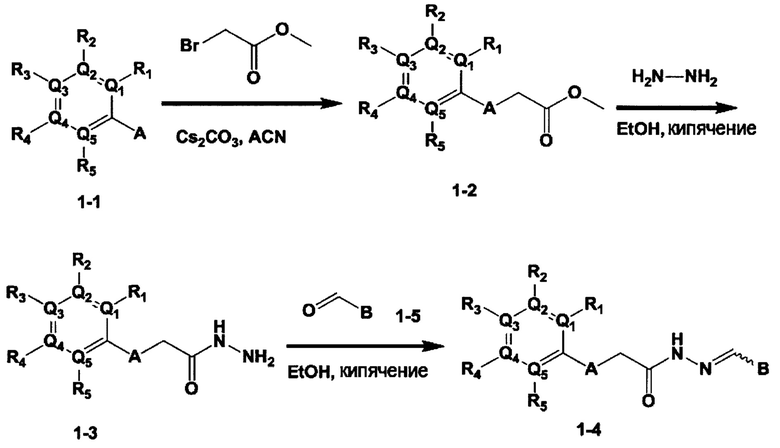
Синтез соединения 1-2 (реакция с метил бромацетатом):
Соединение 1-1 и метил бромацетат растворили в ацетонитриле, и затем добавили карбонат цезия, после чего перемешивали. По окончании реакции, реакционную смесь отфильтровали сквозь цеолиты чтобы удалить карбонат цезия, упарили при пониженном давлении, и очистили с получением соединения 1-2,
Синтез соединения 1-3 (реакция с гидразином):
Соединение 1-2 и гидразин моногидрат растворили в этаноле, после чего перемешивали. По окончании реакции, реакционную смесь упарили при пониженном давлении, и очистили с получением соединения 1-3 в виде твердого вещества белого цвета.
Синтез соединения 1-4 (реакция с альдегидом):
Соединение 1-3 и альдегид растворили в этаноле, после чего перемешивали. По окончании реакции, реакционную смесь упарили при пониженном давлении чтобы удалить этанол, после чего очистили с получением соединения 1-4,
Тем же способом можно синтезировать соединения 013, 014, 034, 065, 083, 092, 100, 108, 109, 112, 118, 121, 127, 133, 135, 136, 137, 138, 139, 146, 147, 149, 152, 155, 156, 157, 158, 159, 180, 182, 183, 184, 187, 190, 191, 192, 193, 194, 196, 205, 206, 211, 217, 218, 227, 228, 229, 237, 256, 258, 259, 260, 279, 280, 286, 288, 291, 293, 301, 302, 303, 304, 310, 311, 312, 313, 314, 317, 318, 319, 320, 322, 323, 326, 327, 329, 330, 331, 332, 333, 336, 337, 343, 344, 345, 346, 347, 356, 358, 359, 378, 379 и 457,
Пример 1. Синтез соединения 013
Стадия 1. Получение метил 2-(о-тозилокси)ацетата: о-Крезол (5 г, 46,24 ммоль) растворили в диметилформамиде. Метил бромацетат (7,1 г, 46,24 ммоль) и карбонат калия (19 г, 138,42 ммоль) добавили к этой смеси, после чего перемешивали при комнатной температуре в течение 1 часа. По окончании реакции, к реакционной смеси добавили воду, и проэкстрагировали с помощью этилацетата. Полученный органический слой промыли насыщенным водным раствором аммоний хлорида, высушили над сульфатом магния, и упарили при пониженном давлении с получением метил 2-(0-тозилокси)ацетата, который использовали в следующей стадии без очистки.
Стадия 2. Получение 2-(о-тозилокси)ацетогидразида: К метил 2-(о-тозилокси)ацетату (8,3 г, 46,24 ммоль) добавили избыточное количество гидразина моногидрата, после чего перемешивали при 90°С в течение 10 минут. По окончании реакции, к этой смеси добавили воду, получив при этом твердый осадок, который отфильтровали и промыли диэтиловым эфиром с получением 2-(о-тозилокси)ацетогидразида в виде твердого вещества белого цвета.
Стадия 3. Получение (E)-N'-((1Н-индол-4-ил)метилен)-2-(о-тозилокси)ацетогидразида: 2-(о-тозилокси)ацетогидразид (0,1 г, 0,55 ммоль) и 1Н-индол-4-карбальдегид (0,08 г, 0,55 ммоль) растворили в этаноле, после чего перемешивали при 90°С в течение 3 часов. По окончании реакции, реакционную смесь охладили до комнатной температуры. Добавили гексан. Образовавшийся осадок отфильтровали, и промыли диэтиловым эфиром с получением соединения 013 в виде твердого вещества белого цвета.
1H NMR (400 MHz, DMSO-d6): δ 11,51-11,37 (m, 2H), 8,51-8,25 (m, 0,7H), 7,50-7,46 (m, 2H), 7,23-6,97 (m, 5H), 6,91-6,79 (m, 2H), 5,22-4,68 (m, 1H), 2,26-2,22 (m, 3H).
Пример 2. Синтез соединения 014
2-(о-тозилокси)ацетогидразид (0,1 г, 0,55 ммоль) и 1Н-индол-5-карбальдегид (0,08 г, 0,55 ммоль) растворили в этаноле, после чего перемешивали при 90°С в течение 3 часов. По окончании реакции, реакционную смесь охладили до комнатной температуры, после чего удалили растворитель. Гексан и этил ацетат добавили к этой смеси. Образовавшийся осадок отфильтровали, и промыли диэтиловым эфиром с получением соединения 014 в виде твердого вещества белого цвета.
1H NMR (400 MHz, DMSO-d6): δ 11,38-11,28 (m, 2H), 8,33-8,07 (m, 1H), 7,80-7,79 (m, 1H), 7,54-7,52 (m, 1H), 7,44-7,38 (m, 2H), 7,17-7,11 (m, 2H), 6,88-6,83 (m, 2H), 6,49 (m, 1H), 5,15-4,63 (m, 2H), 2,24-2,21 (m, 3Н).
Пример 3. Синтез соединения 034
2-(о-тозилокси)ацетогидразид (0,1 г, 0,55 ммоль) и 1H-индол-6-карбальдегид (0,101 г, 0,67 ммоль) растворили в EtOH, после чего перемешивали при 90°С в течение 18 часов. По окончании реакции, реакционную смесь охладили до комнатной температуры, и упарили при пониженном давлении. Образовавшийся осадок отфильтровали, и промыли диэтиловым эфиром с получением соединения 034,
1H NMR (400 MHz, DMSO-d6): δ 11,41-11,20 (m, 1,6H), 8,33 (s, 0,3H), 8,06 (m, 0,4H), 7,69-7,41 (m, 4H), 7,18-7,06 (m, 2H), 6,90-6,80 (m, 2H), 6,45 (m, 1H), 5,14-4,63 (m, 2H), 2,23-2,16 (m, 3H).
Пример 4. Синтез соединения 065
Стадия 1. Получение метил 2-(мезитилокси)ацетата: 2,4,6-триметилфенол (1 г, 7,3 ммоль) растворили в диметилформамиде. Метил бромацетат (1,1 г, 7,3 ммоль) и карбонат калия (3 г, 22 ммоль) добавили к этой смеси, после чего перемешивали при комнатной температуре в течение 16 часов. По окончании реакции, к реакционной смеси добавили воду, и проэкстрагировали с помощью этилацетата. Полученный органический слой трижды промыли водой, высушили над сульфатом магния, и упарили при пониженном давлении с получением 2-(мезитилокси)ацетата (1,5 г 100%).
Стадия 2. Получение 2-(мезитилокси)ацетогидразида: Метил 2-(мезитилокси)ацетат (1,5 г, 7,3 ммоль) растворили в тетрагидрофуране. Добавили гидразин моногидрат (5,16 г, 103 ммоль), после чего перемешивали при комнатной температуре в течение 1 часа. По окончании реакции, реакционную смесь упарили при пониженном давлении. Концентрат разбавили водой и отфильтровали. Фильтрат промыли водой и диэтиловым эфиром с получением 2-(мезитилокси)ацетогидразида (1,36 г, 89%) в виде твердого вещества белого цвета.
Стадия 3. Получение (E)-N'-((1Н-индол-4-ил)метилен)-2-(мезитилокси)ацетогидразида: 2-(мезитилокси)ацетогидразид (0,1 г, 0,48 ммоль) и 1Н-индол-4-карбальдегид (0,08 г, 0,53 ммоль) растворили в этаноле, после чего перемешивали при 90°С в течение 3 часов. По окончании реакции, реакционную смесь охладили до комнатной температуры. Добавили гексан. Образовавшийся осадок отфильтровали, и промыли этанолом с получением соединения 065 (58 мг, 36%) в виде твердого вещества белого цвета.
1Н NMR (400 MHz, DMSO-d6): δ 11,48-11,35 (m, 2H), 8,70 (s, 0,5H), 8,23 (s, 0,5H), 7,50-7,43 (m, 2H), 7,25-7,10 (m, 2,5H), 6,85-6,76 (m, 2,5H), 4,84 (s, 1H), 4,36 (s, 1H), 2,23 (s, 6H), 2,19 (s, 3H).
Пример 5. Синтез соединения 083
Стадия 1. Получение метил 2-(о-толуидин)ацетата: о-толуидин (1 г, 9,3 ммоль) растворили в диметилформамиде. Метил бромацетат (1,3 г, 9,3 ммоль) и карбонат калия (3,9 г, 28 ммоль) добавили к этой смеси, после чего перемешивали при комнатной температуре в течение 16 часов. По окончании реакции, к реакционной смеси добавили воду, и проэкстрагировали с помощью этилацетата. Полученный органический слой трижды промыли водой, высушили над сульфатом магния, и упарили при пониженном давлении с получением метил 2-(о-толуидин)ацетата, который использовали в следующей стадии.
Стадия 2. Получение 2-(о-толуидин)ацетогидразида: Метил 2-(о-толуидин)ацетат (1,7 г, 9,3 ммоль) растворили в тетрагидрофуране. Добавили гидразин моногидрат (5,16 г, 103 ммоль), после чего перемешивали при комнатной температуре в течение 16 часов. По окончании реакции, реакционную смесь упарили при пониженном давлении. Полученный концентрат очистили с помощью колоночной хроматографии с получением 2-(о-толуидин)ацетогидразида (0,3 г, 18%) в виде жидкого вещества желтого цвета.
Стадия 3. Получение (E)-N'-((1Н-индол-4-ил)метилен)-2-(о-толуидин)ацетогидразида: 2-(о-толуидин)ацетогидразид (0,12 г, 0,67 ммоль) и 1Н-индол-4-карбальдегид (0,12 г, 0,803 ммоль) растворили в этаноле, после чего перемешивали при 90°С в течение 3 часов. По окончании реакции, реакционную смесь охладили до комнатной температуры, и упарили при пониженном давлении. Образовавшийся осадок отфильтровали, и промыли диэтиловым эфиром с получением соединения 083 (58 мг, 5%) коричневого цвета.
1Н NMR (400 MHz, DMSO-d6): δ 11,40-11,20 (m, 2,4H), 8,27-8,07 (m, 1H), 7,79-7,37 (m, 5H), 7,00-6,97 (m, 2H), 6,52-6,47 (m, 3H), 4,26 (d, J=5,44 Hz, 1,4H), 3,82-3,80 (d, J=5,88 Hz, 1H), 2,13-2,11 (m, 3H).
Пример 6. Синтез соединения 100
2-(о-тозилокси)ацетогидразид (0,1 г, 0,56 ммоль) и 1H-индол-6-карбальдегид (0,97 г, 0,67 ммоль) растворили в этаноле, после чего перемешивали при 90°С в течение 3 часов. По окончании реакции, реакционную смесь охладили до комнатной температуры, и упарили при пониженном давлении. Полученный твердый остаток очистили с помощью колоночной хроматографии с получением соединения 100 (0,04 г, 47%).
1Н NMR (400 MHz, DMSO-d6): δ 11,37 (bs, 0,4H), 8,37 (bs, 0,4H), 8,26-8,25 (m, 1H), 8,09 (bs, 0,6H), 7,85 (s, 1H), 7,60 (s, 1H), 7,16-7,08 (m, 2H), 6,88-6,81 (m, 2H), 5,15-4,64 (m, 2H), 2,23-2,20 (m, 3H).
Пример 7. Синтез соединения 108
2-(мезитилокси)ацетогидразид (0,12 г, 0,57 ммоль) и 1H-индол-6-карбальдегид (0,1 г, 0,69 ммоль) растворили в этаноле, после чего перемешивали при 90°С в течение ночи. По окончании реакции, реакционную смесь охладили до комнатной температуры, и упарили при пониженном давлении. Полученный твердый остаток очистили с помощью колоночной хроматографии (этил ацетат:гексан = 1:1) и перекристаллизации (этил ацетат:гексан = 1:1) с получением соединения 108 (0,03 г, 16%).
1Н NMR (400 MHz, DMSO-d6): δ 11,39-11,20 (m, 2H), 8,48 (s, 0,5H), 8,01 (s, 0,3H), 7,68 (s, 0.5Н), 7,57-7,24 (m, 3H), 6,83 (s, 2H), 6,45-6,41 (m, 1H), 4,73-4,30 (m, 2H), 2,21-2,14 (m, 9H).
Пример 8. Синтез соединения 109
2-(мезитилокси)ацетогидразид (0,12 г, 0,57 ммоль) и 1H-индол-2-карбальдегид (0,1 г, 0,69 ммоль) растворили в этаноле, после чего перемешивали при 90°С в течение 16 часов. По окончании реакции, реакционную смесь охладили до комнатной температуры, и упарили при пониженном давлении. Полученный твердый остаток очистили с помощью колоночной хроматографии и перекристаллизации с получением соединения 109 (0,07 г, 36%).
1Н NMR (400 MHz, DMSO-d6): δ 11,35-11,30 (m, 2H), 8,47 (s, 0,6H), 8,01 (s, 0,4H), 7,78-7,70 (m, 1H), 7,53-7,34 (m, 3H), 6,83 (m, 2H), 6,49-6,49 (m, 1H), 4,72-4,29 (m, 2H), 2,21-2,17 (m, 9H).
Пример 9. Синтез соединения 112
Стадия 1. Получение метил 2-(4-бром-2,6-диметилфенокси)ацетата: 4-бром-2,6-диметилфенол (3,0 г, 14,9 ммоль) и метил бромацетат (1,55 мл, 16,4 ммоль) растворили в ацетонитриле. Добавили карбонат цезия (14,6 г, 44,8 ммоль), после чего перемешивали при комнатной температуре в течение 1 дня. По окончании реакции, к реакционной смеси добавили насыщенный водный раствор гидрокарбоната натрия, и проэкстрагировали с помощью этилацетата. Полученный органический слой промыли насыщенным солевым раствором, высушили над безводным сульфатом натрия, отфильтровали, и упарили при пониженном давлении с получением метил 2-(4-бром-2,6-диметилфенокси)ацетата (4,1 г, 100%) без дополнительной очистки.
Стадия 2. Получение 2-(4-бром-2,6-диметилфенокси)ацетогидразида: Метил 2-(4-бром-2,6-диметилфенокси)ацетат (4,1 г, 14,9 ммоль) и гидразин моногидрат (0,87 мл, 17,9 ммоль) растворили в этаноле, после чего перемешивали в течение 1 для при кипячении с обратным холодильником. По окончании реакции, реакционную смесь упарили при пониженном давлении. Концентрированный остаток высушили и очистили с получением 2-(4-бром-2,6-диметилфенокси)ацетогидразида (2,5 г, 51%) в виде твердого вещества белого цвета.
Стадия 3. Получение (Е)-N'-((1Н-индол-4-ил)метилен)-2-(4-бром-2,6-диметилфенокси)ацетогидразида: 2-(4-бром-2,6-диметилфенокси)ацетогидразид (0,18 г, 0,66 ммоль) и индол-4-карбоксальдегид (0,11 г, 0,73 ммоль) растворили в этаноле, после чего перемешивали в течение ночи при кипячении с обратным холодильником. По окончании реакции, реакционную смесь упарили при пониженном давлении. Концентрированный остаток высушили и очистили с получением соединения 112 (52 мг, 20%) в виде твердого вещества белого цвета.
1H NMR (400 MHz, DMSO-d6): δ 11,52, 11,48 (s, 1H), 11,37 (bs, 1H), 8,67, 8,23 (s, 1H), 7,51-7,44 (m, 2H), 7,29-7,24 (m, 2,65H), 7,18-7,12 (m, 2H), 6,77 (s, 0,47H), 4,90, 4,42 (s, 2H), 2,29, 2,28 (m, 6H).
Пример 10. Синтез соединения 118
Стадия 1. Получение метил 2-(2,6-диметилфенокси)ацетата: 2,6-диметилфенол (3,0 г, 24,6 ммоль) и метил бромацетат (2,57 мл, 27,0 ммоль) растворили в ацетонитриле. Добавили карбонат цезия (24,0 г, 73,7 ммоль), после чего перемешивали при комнатной температуре в течение 1 дня. По окончании реакции, к реакционной смеси добавили насыщенный водный раствор гидрокарбоната натрия, и проэкстрагировали с помощью этилацетата. Полученный органический слой промыли насыщенным солевым раствором, высушили над безводным сульфатом натрия, отфильтровали, и упарили при пониженном давлении с получением метил 2-(2,6-диметилфенокси)ацетата (4,77 г, 100%) без дополнительной очистки.
Стадия 2. Получение 2-(2,6-диметилфенокси)ацетогидразида: Метил 2-(2,6-диметилфенокси)ацетат (4,77 г, 24,6 ммоль) и гидразин моногидрат (1,43 мл, 29,4 ммоль) растворили в этаноле, после чего перемешивали в течение 1 дня при кипячении с обратным холодильником. По окончании реакции, реакционную смесь упарили при пониженном давлении. Концентрированный остаток высушили и очистили с получением 2-(2,6-диметилфенокси)ацетогидразида (2,2 г, 46%) в виде твердого вещества белого цвета.
Стадия 3. Получение (Е)-N'-((1Н-индол-4-ил)метилен)-2-(2,6-диметилфенокси)ацетогидразида: 2-(2,6-диметилфенокси)ацетогидразид (100 мг, 0,52 ммоль) и индол-4-карбоксальдегид (82,2 мг, 0,57 ммоль) растворили в этаноле, после чего перемешивали в течение ночи при кипячении с обратным холодильником. По окончании реакции, реакционную смесь упарили при пониженном давлении. Концентрированный остаток высушили и очистили с получением соединения 118 (71,6 мг, 43%) в виде твердого вещества белого цвета.
1H NMR (400 MHz, DMSO-d6): δ 11,51, 11,46 (s, 1H), 11,37 (bs, 1H), 8,70, 8,24 (s, 1H), 7,51-7,43 (m, 2H), 7,26-6,78 (m, 6H), 4,89, 4,41 (s, 2H), 2,29, 2,28 (m, 6H).
Пример 11. Синтез соединения 121
Стадия 1. Получение метил 2-(2,6-диизопропилфенокси)ацетата: 2,6-диизопропилфенол (10 г, 56,1 ммоль) растворили в ацетонитриле. Метил бромацетат (8,5 г, 56,1 ммоль) и карбонат цезия (37 г, 112,4 ммоль) добавили к этой смеси, после чего перемешивали в течение 16 часов при кипячении с обратным холодильником. По окончании реакции, к реакционной смеси добавили воду, и проэкстрагировали с помощью этилацетата. Полученный органический слой трижды промыли водой, высушили над сульфатом магния, и упарили при пониженном давлении с получением метил 2-(2,6-диизопропилфенокси)ацетата (14 г, 100%), который использовали в следующей стадии.
Стадия 2. Получение 2-(2,6-диизопропилфенокси)ацетогидразида:
2-(2,6-диизопропилфенокси)ацетат (14 г, 56,1 ммоль) растворили в EtOH. Добавили гидразин моногидрат (2,5 г, 56,1 ммоль), после чего перемешивали при комнатной температуре в течение 16 часов. По окончании реакции, реакционную смесь упарили при пониженном давлении. Полученный концентрат очистили с помощью колоночной хроматографии с получением 2-(2,6-диизопропилфенокси)ацетогидразида (8,8 г, 62%).
Стадия 3. Получение (Е)-N'-((1Н-индол-4-ил)метилен)-2-(2,6-диизопропилфенокси)ацетогидразида: 2-(2,6-диизопропилфенокси)ацетогидразид (0,1 г, 0,399 ммоль) и 1Н-индол-4-карбальдегид (0,07 г, 0,479 ммоль) растворили в этаноле, после чего перемешивали при 100°С в течение 16 часов. По окончании реакции, реакционную смесь упарили при пониженном давлении. Полученный концентрат очистили с помощью колоночной хроматографии с получением соединения 121 (0,03 г, 20%).
1H NMR (400 MHz, DMSO-d6): δ 11,57-11,22 (m, 2H), 8,62-8,20 (m, 1H), 7,36-6,72 (m, 8H), 4,83-4,33 (m, 2H), 1,19-1,16 (m, 16H).
Пример 12. Синтез соединения 127
Стадия 1. Получение метил 2-(2,6-ди-трет-бутил-4-метилфенокси)ацетата: 2,6-ди-трет-бутил-4-метилфенол (10 г, 45,4 ммоль) растворили в ацетонитриле. Метил бромацетат (7 г, 45,4 ммоль) и карбонат цезия (30 г, 91 ммоль) добавили к этой смеси, после чего перемешивали в течение 16 часов при кипячении с обратным холодильником. По окончании реакции, к реакционной смеси добавили воду, и проэкстрагировали с помощью этилацетата. Полученный органический слой трижды промыли водой, высушили над сульфатом магния, и упарили при пониженном давлении с получением метил 2-(2,6-ди-трет-бутил-4-метилфенокси)ацетата (13,3 г, 100%), который использовали в следующей стадии.
Стадия 2. Получение 2-(2,6-ди-трет-бутил-4-метилфенокси)ацетогидразида:
метил 2-(2,6-ди-трет-бутил-4-метилфенокси)ацетат (13,3 г, 45,4 ммоль) растворили в EtOH. Добавили гидразин моногидрат (2,3 г, 45,4 ммоль), после чего перемешивали при комнатной температуре в течение 16 часов. По окончании реакции, реакционную смесь упарили при пониженном давлении. Полученный концентрат очистили с помощью колоночной хроматографии с получением 2-(2,6-ди-трет-бутил-4-метилфенокси)ацетогидразида (6 г, 45%).
Стадия 3. Получение (E)-N'-((1Н-индол-4-ил)метилен)-2-(2,6-ди-трет-бутил-4-метилфенокси)ацетогидразида: 2-(2,6-ди-трет-бутил-4-метилфенокси)ацетогидразид (0,1 г, 0,342 ммоль) и 1Н-индол-4-карбальдегид (0,07 г, 0,41 ммоль) растворили в этаноле, после чего перемешивали при 100°С в течение 16 часов. По окончании реакции, образовавшийся осадок отфильтровали, и промыли ацетонитрилом с получением соединения 127 (0,025 г, 18%).
1H NMR (400 MHz, DMSO-d6): δ 8,52-8,22 (s, 1H), 7,78 (m, 1,6H), 7,41 (m, 0,8H), 7,14 (m, 3,2H), 6,92 (m, 0,7H), 4,77 (s, 1,3H), 4,23 (s, 0,5H), 2,33 (d, J=4,56 Hz, 3H), 1,38 (s, 18H).
Пример 13. Синтез соединения 133
2-(4-бром-2,6-диметилфенокси)ацетогидразид (0,1 г, 0,37 ммоль) и 1Н-индол-6-карбальдегид (0,11 г, 0,74 ммоль) растворили в этаноле, после чего перемешивали в течение 12 часов при кипячении с обратным холодильником. По окончании реакции, реакционную смесь упарили при пониженном давлении, получив кристаллическое твердое вещество, который отфильтровали, и промыли этанолом, и очистили с помощью колоночной хроматографии с получением соединения 133.
1Н NMR (400 MHz, DMSO-d6): δ 11,42 (d, J=9,72 Hz, 1H), 11,33-11,22 (s, 1H), 8,47 (s, 0,5H), 8,03 (s, 0,5H), 7,70 (s, 0,5H), 7,59-7,37 (m, 3H), 7,29 (m, 2,5H), 6,45(m, 1H), 4,80 (s, 1H), 4,37 (s, 1H), 2,32 (d, J=5,36 Hz, 6H).
Пример 14. Синтез соединения 135
Стадия 1. Получение метил 2-(мезитиламино)ацетата: 2,4,6-триметилбензоламид (10 г, 74 ммоль) растворили в ацетонитриле. Метил бромацетат (11,3 г, 74 ммоль) и карбонат цезия (48 г, 148 ммоль) добавили к этой смеси, после чего перемешивали в течение 16 часов при кипячении с обратным холодильником. По окончании реакции, к реакционной смеси добавили воду, и проэкстрагировали с помощью этилацетата. Полученный органический слой трижды промыли водой, высушили над сульфатом магния, и упарили при пониженном давлении с получением метил 2-(мезитиламино)ацетат (15,3 г, 100%), который использовали в следующей стадии.
Стадия 2. Получение 2-(мезитиламино)ацетогидразида: Метил 2-(мезитиламино)ацетат (15,3 г, 74 ммоль) растворили в EtOH. Добавили гидразин моногидрат (3,7 г, 74 ммоль), после чего перемешивали при 100°С в течение 16 часов при кипячении с обратным холодильником. По окончании реакции, реакционную смесь упарили при пониженном давлении. Полученное твердое вещество отфильтровали получив 2-(мезитиламино)ацетогидразид (3,9 г, 25%).
Стадия 3. Получение (E)-N'-((1Н-индол-4-ил)метилен)-2-(мезитиламино)ацетогидразида: 2-(мезитиламино)ацетогидразид (0,1 г, 0,48 ммоль) и 1Н-индол-4-карбальдегид (0,084 г, 0,58 ммоль) растворили в этаноле, после чего перемешивали при 100°С в течение 16 часов. По окончании реакции, реакционную смесь упарили при пониженном давлении. Полученный концентрат очистили с помощью колоночной хроматографии, после чего упарили при пониженном давлении. Полученное твердое кристаллическое вещество отфильтровали с получением соединения 135 (0,089 г, 55%).
1H NMR (400 MHz, DMSO-d6): δ 11,47-11,34 (m, 2H), 8,41-8,20 (m, 1H), 7,47 (m, 2H), 7,48-7,10 (m, 2H), 6,91 (s, 0,6H), 6,74 (m, 2H), 4,36-3,60 (m, 3H), 2,25-2,12 (m, 9H).
Пример 15. Синтез соединения 136
2-(мезитиламино)ацетогидразид (0,1 г, 0,48 ммоль) и 1H-индол-3-карбальдегид (0,084 г, 0,58 ммоль) растворили в этаноле, после чего перемешивали при 100°С в течение 16 часов. По окончании реакции, реакционную смесь упарили при пониженном давлении. Полученный концентрат очистили с помощью колоночной хроматографии, после чего упарили при пониженном давлении. Полученное твердое кристаллическое вещество отфильтровали с получением соединения 136 (0,06 г, 37%).
1H NMR (400 MHz, DMSO-d6): δ 11,53 (bs, 1H), 11,25-11,06 (m, 1H), 8,33-8,03 (m, 2H), 7,70 (m, 1H), 7,40 (m, 1H), 7,21-7,05 (m, 2H), 6,74-6,71 (m, 2H), 4,36-4,01 (m, 3H), 2,25-2,12 (m, 9H).
Пример 16. Синтез соединения 137
Стадия 1. Получение 2-(2,6-диметил-4-(пиридин-2-ил)фенокси)ацетогидразида: 2-(4-бром-2,6-диметилфенокси)ацетогидразид (0,5 г, 1,8 ммоль) растворили в смеси диметоксиэтан/вода (2:1) и диметилформамиде. Пиридин-2-илборную кислоту (0,27 г, 2,2 ммоль) и Pd(dppf)Cl2 (0,074 г, 0,055 ммоль), карбонат натрия (0,58 г, 5,5 ммоль) добавили к этой смеси, после чего реакцию проводили в микроволновом реакторе при 120°С в течение 15 минут. По окончании реакции, к реакционной смеси добавили воду, и проэкстрагировали с помощью дихлорметана. Полученный органический слой высушили над сульфатом магния, и упарили при пониженном давлении. Концентрат очистили с помощью колоночной хроматографии с получением 2-(2,6-диметил-4-(пиридин-2-ил)фенокси)ацетогидразида (0,34 г, 69%).
Стадия 2. Получение (E)-N'-((1Н-индол-4-ил)метилен)-2-(2,6-диметил-4-(пиридин-2-ил)фенокси)ацетогидразида: 2-(2,6-диметил-4-(пиридин-2-ил)фенокси)ацетогидразид (0,1 г, 0,37 ммоль) и 1H-индол-4-карбальдегид (0,11 г, 0,74 ммоль) растворили в этаноле, после чего перемешивали при 100°С в течение 12 часов. По окончании реакции, полученное твердое вещество отфильтровали с получением соединения 137 (0,09 г, 61%).
1Н NMR (400 MHz, DMSO-d6): δ 11,54-11,35 (m, 2H), 8,84 (s, 1H), 8,68 (s, 0,5H), 8,5 (m, 1H), 8,23 (s, 0,5H), 8,02 (m, 1H), 7,49-6,78(m, 7H), 4,93 (s, 1H), 4,38 (s, 1H), 2,35 (d, J=3,08 Hz, 6H).
Пример 17. Синтез соединения 138
2-(2,6-диметил-4-(пиридин-2-ил)фенокси)ацетогидразид (0,1 г, 0,37 ммоль) и 1H-индол-3-карбальдегид (0,11 г, 0,74 ммоль) растворили в этаноле, после чего перемешивали при 100°С в течение 12 часов. По окончании реакции, полученное твердое вещество отфильтровали с получением соединения 138 (0,08 г, 46%).
1Н NMR (400 MHz, DMSO-d6): δ 11,57-11,22 (m, 2H), 8,84-7,00 (m, 12H), 4,91 (s, 1H), 4,40 (s, 1H), 2,36 (m, 6H).
Пример 18. Синтез соединения 139
2-(2,6-диметил-4-(пиридин-2-ил)фенокси)ацетогидразид (0,1 г, 0,37 ммоль) и 1H-индол-6-карбальдегид (0,11 г, 0,74 ммоль) растворили в этаноле, после чего перемешивали при 100°С в течение 12 часов. По окончании реакции, реакционную смесь упарили при пониженном давлении. Полученный концентрат очистили с помощью колоночной хроматографии с получением соединения 139 (0,1 г, 52%).
1H NMR (400 MHz, DMSO-d6): δ 11,45-11,22 (m, 2H), 8,84-7,23 (m, 11H), 6,45 (m, 1H), 4,88 (s, 0,8H), 4,11 (s, 1H), 2,31 (d, J=5,76 Hz, 6H).
Пример 19. Синтез соединения 146
Стадия 1. Получение метил 2-(2-(пиридин-3-ил)фенокси)ацетата:
2-(пиридин-3-ил)фенол (Соединение 6-6) (0,25 г, 1,4 ммоль) растворили в ацетонитриле. Метил бромацетат (0,15 г, 1,4 мл) и карбонат калия (0,6 г, 0,4 ммоль) добавили к этой смеси, после чего перемешивали при 16 часов. По окончании реакции, реакционную смесь отфильтровали через цеолиты, и упарили при пониженном давлении с получением метил 2-(2-(пиридин-3-ил)фенокси)ацетата (0,35 г, 100%), который использовали в следующей стадии.
Стадия 2. Получение 2-(2-(пиридин-3-ил)фенокси)ацетогидразида:
метил 2-(2-(пиридин-3-ил)фенокси)ацетат (0,32 г, 1,3 ммоль) растворили в EtOH.
Добавили гидразин моногидрат (0,078 г, 1,56 ммоль), после чего перемешивали в течение 16 часов при кипячении с обратным холодильником. По окончании реакции, реакционную смесь упарили при пониженном давлении. Полученный концентрат очистили с помощью колоночной хроматографии с получением 2-(2-(пиридин-3-ил)фенокси)ацетогидразида (0,1 г, 31%).
Стадия 3. Получение (Е)-N'-((1Н-индол-4-ил)метилен)-2-(2-(пиридин-3-ил)фенокси)ацетогидразида: 2-(2-(пиридин-3-ил)фенокси)ацетогидразид (0,1 г, 0,41 ммоль) и 1H-индол-4-карбальдегид (0,072 г, 0,49 ммоль) растворили в этаноле, после чего перемешивали при 100°С в течение 16 часов. По окончании реакции, реакционную смесь упарили при пониженном давлении. Полученный концентрат очистили с помощью колоночной хроматографии, после чего упарили при пониженном давлении. Полученное твердое кристаллическое вещество отфильтровали с получением соединения 146 (0,030 г, 20%).
1H NMR (400 MHz, DMSO-d6): δ 11,54-11,33 (bs, 2H), 8,83 (m, 1H), 8,53-8,49 (m, 1H), 8,42 (s, 0,3H), 8,25 (s, 0,7H), 8,07-8,03 (m, 1H), 7,49-7,32 (m, 6H), 7,23-6,94 (m, 5H), 5,27-4,74 (m, 2H).
Пример 20. Синтез соединения 147
Стадия 1. Получение метил 2-(мезитилтио)ацетата: 2,4,6-триметилбензолтиол (1,5 г, 10 ммоль) растворили в ацетонитриле. Метил бромацетат (1,7 г, 11 ммоль) и карбонат калия (3 г, 2,2 ммоль) добавили к этой смеси, после чего перемешивали в течение 16 часов. По окончании реакции, реакционную смесь отфильтровали через цеолиты. Фильтрат разбавили насыщенным водным раствором аммоний хлорида, и проэкстрагировали с помощью этилацетата. Полученный органический слой высушили над сульфатом магния, и упарили при пониженном давлении с получением метил 2-(мезитилтио)ацетата, который использовали в следующей стадии.
Стадия 2. Получение 2-(мезитилтио)ацетогидразида: Метил 2-(мезитилтио)ацетат (2,5 г, 11 ммоль) растворили в EtOH. Добавили гидразин моногидрат (0,56 г, 11 ммоль), после чего перемешивали при 100°С в течение 16 часов при кипячении с обратным холодильником. По окончании реакции, реакционную смесь упарили при пониженном давлении. Полученный концентрат разбавили насыщенным водным раствором аммоний хлорида, и проэкстрагировали с помощью этилацетата. Полученный органический слой высушили над сульфатом магния, упарили при пониженном давлении, и очистили с помощью колоночной хроматографии с получением 2-(мезитилтио)ацетогидразида (1 г, 41%).
Стадия 3. Получение (E)-N'-((1Н-индол-4-ил)метилен)-2-(мезитилтио)ацетогидразида:
2-(мезитилтио)ацетогидразид (0,1 г, 0,45 ммоль) и 1Н-индол-4-карбальдегид (0,078 г, 0,54 ммоль) растворили в этаноле, после чего перемешивали при 100°С в течение 16 часов. По окончании реакции, реакционную смесь упарили при пониженном давлении. Полученный концентрат очистили с помощью колоночной хроматографии, после чего упарили при пониженном давлении. Полученное твердое кристаллическое вещество отфильтровали с получением соединения 147 (0,028 г, 6,3%).
1Н NMR (400 MHz, DMSO-d6): δ 11,32-11,27 (m, 2H), 8,30-8,17 (m, 1H), 7,47 (m, 2H), 7,47-6,88 (m, 5H), 3,72-3,33 (m, 2H), 2,44-1,96 (m, 9H).
Пример 21. Синтез соединения 149
Стадия 1. Получение метил 2-(2,6-диметил-4-(пиримидин-5-ил)фенокси)ацетата:
2,6-диметил-4-(пиримидин-5-ил)фенол (Соединение 6-3) (0,32 г, 1,6 ммоль) растворили в ацетонитриле. Метил бромацетат (0,24 г, 1,6 ммоль) и карбонат цезия (1 г, 3,2 ммоль) добавили к этой смеси, после чего перемешивали в течение 12 часов при кипячении с обратным холодильником. По окончании реакции, реакционную смесь отфильтровали через цеолиты. Фильтрат разбавили водой, и проэкстрагировали с помощью этилацетата. Полученный органический слой высушили над сульфатом магния, и упарили при пониженном давлении. Концентрат очистили с помощью колоночной хроматографии с получением метил 2-(2,6-диметил-4-(пиримидин-5-ил)фенокси)ацетата (0,44 г, 100%).
Стадия 2. Получение 2-(2,6-диметил-4-(пиримидин-5-ил)фенокси)ацетогидразида:
2-(2,6-диметил-4-(пиримидин-5-ил)фенокси)ацетат (0,44 г, 1,6 ммоль) растворили в EtOH. Добавили гидразин моногидрат (0,12 г, 2,4 ммоль), после чего перемешивали при 100°С в течение 12 часов при кипячении с обратным холодильником. По окончании реакции, реакционную смесь упарили при пониженном давлении, и очистили с помощью колоночной хроматографии с получением 2-(2,6-диметил-4-(пиримидин-5-ил)фенокси)ацетогидразида (0,085 г, 19%).
Стадия 3. Получение (E)-N'-((1Н-индол-4-ил)метилен)-2-(2,6-диметил-4-(пиримидин-5-ил)фенокси)ацетогидразида: 2-(2,6-диметил-4-(пиримидин-5-ил)фенокси)ацетогидразид (0,085 г, 0,31 ммоль) и 1Н-индол-4-карбальдегид (0,068 г, 0,47 ммоль) растворили в этаноле, после чего перемешивали при 100°С в течение 12 часов. По окончании реакции, полученное твердое вещество отфильтровали с получением соединения 149 (0,06 г, 70%).
1H NMR (400 MHz, DMSO-d6): δ 11,55 (bs, 1H), 11,35 (bs, 1H), 9,11 (m, 3,2H), 8,7 (s, 0,5H), 8,24 (s, 0,5H), 7,55-6,8 (m, 7H), 4,95 (s, 1H), 4,47 (s, 1H), 2,38 (d, J=3,2 Hz, 6H).
Пример 22. Синтез соединения 152
Стадия 1. Получение метил 2-(4-(2-аминопиримидин-5-ил)-2,6-диметилфенокси)ацетата: 4-(2-аминопиримидин-5-ил)-2,6-диметилфенол (Соединение 6-4) (0,27 г, 1,24 ммоль) растворили в ацетонитриле. Метил бромацетат (0,12 г, 1,24 ммоль) и карбонат цезия (0,81 г, 2,3 ммоль) добавили к этой смеси, после чего перемешивали в течение 12 часов при кипячении с обратным холодильником. По окончании реакции, реакционную смесь отфильтровали через цеолиты. Фильтрат разбавили водой, и проэкстрагировали с помощью дихлорметана. Полученный органический слой высушили над сульфатом магния, упарили при пониженном давлении, и очистили с помощью колоночной хроматографии с получением метил 2-(4-(2-аминопиримидин-5-ил)-2,6-диметилфенокси)ацетата (0,28 г, 70%).
Стадия 2. Получение 2-(4-(2-аминопиримидин-5-ил)-2,6-диметилфенокси)ацетогидразид: метил 2-(4-(2-аминопиримидин-5-ил)-2,6-диметилфенокси)ацетат (0,28 г, 1 ммоль) растворили в EtOH. Добавили гидразин моногидрат (0,06 г, 1,2 ммоль), после чего перемешивали при 100°С в течение 8 часов при кипячении с обратным холодильником. По окончании реакции, реакционную смесь упарили при пониженном давлении. Образовавшийся осадок отфильтровали с получением 2-(2,6-диметил-4-(пиримидин-5-ил)фенокси)ацетогидразида (0,31 г, 100%).
Стадия 3. Получение (E)-N'-((1Н-индол-6-ил)метилен)-2-(4-(2-аминопиримидин-5-ил)-2,6-диметилфенокси)ацетогидразида: 2-(2,6-диметил-4-(пиримидин-5-ил)фенокси)ацетогидразид (0,080 г, 0,28 ммоль) и 1Н-индол-4-карбальдегид (0,061 г, 0,41 ммоль) растворили в этаноле, после чего перемешивали при 100°С в течение 12 часов. По окончании реакции, образовавшийся осадок отфильтровали, и очистили с помощью колоночной хроматографии с получением соединения 152.
1H NMR (400 MHz, DMSO-d6): δ 9,14-7,01 (m, 10Н), 4,93 (s, 1H), 4,42 (s, 1H), 2,37 (d, J=7,96 Hz, 6H).
Пример 23. Синтез соединения 155
Стадия 1. Получение метил 2-(4-хлор-2,6-диметилфенокси)ацетата: 4-хлор-2,6-диметилфенол (0,5 г, 3,2 ммоль) растворили в ацетонитриле. Метил бромацетат (0,49 г, 3,2 ммоль) и карбонат цезия (1,8 г, 6,4 ммоль) добавили к этой смеси, после чего перемешивали в течение 16 часов. По окончании реакции, реакционную смесь отфильтровали через цеолиты. Фильтрат разбавили водой, и проэкстрагировали с помощью дихлорметана. Полученный органический слой высушили над сульфатом магния, и упарили при пониженном давлении с получением метил 2-(4-хлор-2,6-диметилфенокси)ацетата (0,4 г, 55%), который использовали в следующей стадии.
Стадия 2. Получение 2-(4-хлор-2,6-диметилфенокси)ацетогидразида:
метил 2-(4-хлор-2,6-диметилфенокси)ацетат (0,5 г, 2,2 ммоль) растворили в EtOH.
Добавили гидразин моногидрат (0,13 г, 2,6 ммоль), после чего перемешивали в течение 12 часов при кипячении с обратным холодильником. По окончании реакции, реакционную смесь упарили при пониженном давлении. Концентрат разбавили водой, и проэкстрагировали с помощью дихлорметана. Полученный органический слой высушили над сульфатом магния, и упарили при пониженном давлении с получением 2-(4-хлор-2,6-диметилфенокси)ацетогидразид (0,22 г, 44%).
Стадия 3. Получение (Е)-N'-((1Н-индол-4-ил)метилен)-2-(4-хлор-2,6-диметилфенокси)ацетогидразида: 2-(4-хлор-2,6-диметилфенокси)ацетогидразид (0,08 г, 0,35 ммоль) и 1Н-индол-4-карбальдегид (0,076 г, 0,53 ммоль) растворили в этаноле, после чего перемешивали в течение 12 часов при кипячении с обратным холодильником. По окончании реакции, образовавшийся осадок отфильтровали с получением соединения 155 (0,063 г, 51%).
1H NMR (400 MHz, DMSO-d6): δ 11,48-11,33 (m, 1,5H), 8,64 (s, 0,3H), 8,21 (s, 0,4H), 7,44 (m, 2H), 7,24-6,75 (m, 5H), 4,87 (s, 1H), 4,39 (s, 1H), 2,25 (d, J=3,48 Hz, 6H).
Пример 24. Синтез соединения 156
2-(4-хлор-2,6-диметилфенокси)ацетогидразид (0,08 г, 0,35 ммоль) и 1Н-индол-4-карбальдегид (0,076 г, 0,53 ммоль) растворили в этаноле, после чего перемешивали в течение 12 часов при кипячении с обратным холодильником. По окончании реакции, образовавшийся осадок отфильтровали, и очистили с помощью колоночной хроматографии с получением соединения 156 (0,03 г, 24%).
1Н NMR (400 MHz, DMSO-d6): δ 11,20 (bs, 1H), 11,1 (bs, 1H), 8,54 (s, 0,5H), 8,21 (d, J=7,16 Hz, 5H), 8,14 (s, 0,5H), 7,85 (m, 0,5H), 7,75 (m, 1H), 7,40 (m, 1H), 7,20-7,01 (m, 4H), 4,84 (s, 1H), 4,34 (s, 1H), 2,2 (d, J=3,48 Hz, 6H).
Пример 25. Синтез соединения 157
2-(4-хлор-2,6-диметилфенокси)ацетогидразид (0,08 г, 0,35 ммоль) и 1H-индол-6-карбальдегид (0,076 г, 0,53 ммоль) растворили в этаноле, после чего перемешивали в течение 12 часов при кипячении с обратным холодильником. По окончании реакции, образовавшийся осадок отфильтровали с получением соединения 157 (0,043 г, 34%).
1H NMR (400 MHz, DMSO-d6): δ 11,40-11,2 (m, 1,5H), 8,45 (s, 0,5H), 8,02 (s, 0,4H), 7,69-7,26 (m, 4H), 7,11 (d, J=5,72 Hz, 2H), 6,46 (m, 1H), 4,79 (s, 0,8H), 4,36 (s, 1H), 2,25 (d, J=4,92 Hz, 6H).
Пример 26. Синтез соединения 158
2-(мезитилтио)ацетогидразид (0,2 г, 0,89 ммоль) и 1H-индол-3-карбальдегид (0,155 г, 1,1 ммоль) растворили в этаноле, после чего перемешивали при 100°С в течение 16 часов. По окончании реакции, реакционную смесь упарили при пониженном давлении. Полученное твердое кристаллическое вещество отфильтровали с получением соединения 158 (0,035 г, 11%).
1H NMR (400 MHz, DMSO-d6): δ 11,52 (bs, 1H), 11,04-11,00 (m, 1H), 8,25-7,96 (m, 2H), 7,75-7,71 (m, 1H), 7,40 (m, 1H), 7,18-6,88 (m, 4H), 3,70 (s, 1,4H), 2,45-2,13 (m, 9H).
Пример 27. Синтез соединения 159
2-(мезитилтио)ацетогидразид (0,2 г, 0,89 ммоль) и 1H-индол-6-карбальдегид (0,16 г, 1,1 ммоль) растворили в этаноле, после чего перемешивали при 100°С в течение 16 часов. По окончании реакции, реакционную смесь упарили при пониженном давлении. Концентрат очистили с помощью колоночной хроматографии, после чего упарили при пониженном давлении. Полученное твердое кристаллическое вещество отфильтровали с получением соединения 159 (0,023 г, 7,3%).
1H NMR (400 MHz, DMSO-d6): δ 11,22 (bs, 2H), 8,13-7,94 (m, 1H), 7,65-7,18 (m, 4H), 6,93-6,86 (m, 2H), 6,43 (m, 1H), 3,68-3,30 (m, 2H), 2,49-2,08 (m, 9H).
Пример 28. Синтез соединения 180
2-(мезитиламино)ацетогидразид (0,03 г, 0,14 ммоль) и 1-(2-гидроксиэтил)-1Н-индол-4-карбальдегид (0,03 г, 0,16 ммоль) растворили в диметилсульфоксиде и уксусной кислоте, после чего перемешивали при комнатной температуре в течение 12 часов. По окончании реакции, к реакционной смеси добавили воду, и проэкстрагировали с помощью диэтилового эфира. Полученный органический слой промыли сульфатом натрия, и упарили при пониженном давлении. Концентрат перекристаллизовали с получением соединения 180 (0,002 г, 3,3%).
1Н NMR (400 MHz, DMSO-d6): δ 11,44 (d, J=25 Hz, 1H), 8,67 (s, 0,5H), 8,21 (s, 0,5H), 7,55 (m, 1H), 7,43 (m, 1H), 7,26-7,09 (m, 2H), 6,82-6,73 (m, 3H), 4,86 (m, 1H), 4,81 (s, 1H), 4,33 (s, 1H), 4,23 (m, 2H), 3,69 (m, 2H), 2,2 (d, J=0,6 Hz, 6H), 2,17 (s, 3H).
Пример 29. Синтез соединения 182
Стадия 1. Получение метил 2-(2,4-диметил-6-(пиридин-3-ил)фенокси)ацетата:
2,4-диметил-6-(пиридин-3-ил)фенол (Соединение 6-7) (0,7 г, 3,51 ммоль) растворили в ацетонитриле. Метил бромацетат (0,54 г, 3,51 ммоль) и карбонат цезия (2,3 г, 7,02 ммоль) добавили к этой смеси, после чего перемешивали в течение 16 часов при кипячении с обратным холодильником. По окончании реакции, реакционную смесь отфильтровали через цеолиты. Фильтрат разбавили водой, и проэкстрагировали с помощью этилацетата. Полученный органический слой высушили над сульфатом магния, упарили при пониженном давлении, и очистили с помощью колоночной хроматографии с получением метил 2-(2,4-диметил-6-(пиридин-3-ил)фенокси)ацетат (0,71 г, 75%).
Стадия 2. Получение 2-(2,4-диметил-6-(пиридин-3-ил)фенокси)ацетогидразида:
2-(2,4-диметил-6-(пиридин-3-ил)фенокси)ацетат (0,71 г, 2,63 ммоль) растворили в EtOH. Добавили гидразин моногидрат (0,13 г, 2,63 ммоль), после чего перемешивали при 100°С в течение 16 часов при кипячении с обратным холодильником. По окончании реакции, реакционную смесь упарили при пониженном давлении, и очистили с помощью колоночной хроматографии с получением 2-(2,4-диметил-6-(пиридин-3-ил)фенокси)ацетогидразида (0,71 г, 100%).
Стадия 3. Получение (Е)-N'-((1Н-индол-4-ил)метилен)-2-(2,4-диметил-6-(пиридин-3-ил)фенокси)ацетогидразида: 2-(2,4-диметил-6-(пиридин-3-ил)фенокси)ацетогидразид (0,2 г, 0,74 ммоль) и 1Н-индол-4-карбальдегид (0,13 г, 0,88 ммоль) растворили в этаноле, после чего перемешивали при 100°С в течение 16 часов. По окончании реакции, реакционную смесь упарили при пониженном давлении. Полученный концентрат очистили с помощью колоночной хроматографии с получением соединения 182 (0,1 г, 34%).
1H NMR (400 MHz, DMSO-d6): δ 11,32 (bs, 1H), 11,22 (bs, 1H), 8,78-8,75 (m, 1H), 8,57-8,50 (m, 1,5H), 8,04-7,98 (m, 1,5H), 7,48-7,41 (m, 3H), 7,21-7,01 (m, 4,5H), 6,33 (m, 0,5H), 4,46-4,00 (m, 2H), 2,35-2,27 (m, 6H).
Пример 30. Синтез соединения 183
2-(2,4-диметил-6-(пиридин-3-ил)фенокси)ацетогидразид (0,2 г, 0,74 ммоль) и 1H-индол-6-карбальдегид (0,13 г, 0,88 ммоль) растворили в этаноле, после чего перемешивали при 100°С в течение 16 часов. По окончании реакции, реакционную смесь упарили при пониженном давлении. Концентрат очистили с помощью колоночной хроматографии с получением соединения 183 (0,14 г, 47%).
1H NMR (400 MHz, DMSO-d6): δ 11,29-11,14 (m, 2H), 8,77-8,74 (m, 1H), 8,52-8,51 (m, 1H), 8,36 (s, 0,5H), 8,02-7,96 (m, 1H), 7,87 (s, 0,5H), 7,67-7,33 (m, 5H), 7,11-7,05 (m, 2,5H), 6,45-6,41 (m, 1H), 4,41-3,97 (m, 2H), 2,37-2,21 (m, 9H).
Пример 31. Синтез соединения 184
2-(2,4-диметил-6-(пиридин-3-ил)фенокси)ацетогидразид (0,2 г, 0,74 ммоль) и 1H-индол-5-карбальдегид (0,13 г, 0,88 ммоль) растворили в этаноле, после чего перемешивали при 100°С в течение 16 часов. По окончании реакции, реакционную смесь упарили при пониженном давлении. Концентрат очистили с помощью колоночной хроматографии с получением соединения 184 (0,14 г, 48%).
1Н NMR (400 MHz, DMSO-d6): δ 11,38-11,32 (m, 1H), 11,11-11,09 (m, 1H), 8,78-8,75 (m, 1H), 8,51 (m, 1H), 8,37 (s, 0,5H), 8,04-7,97 (m, 1H), 7,86-7,77 (m, 1H), 7,58-7,05 (m, 6,5H), 6,51-6,45 (m, 1H), 4,40-3,96 (m, 2H), 2,32-2,27 (m, 6H).
Пример 32. Синтез соединения 187
Стадия 1. Получение метил 2-(2-метилпиридин-3-илокси)ацетата: 2-метилпиридин-3-ол (0,6 г, 4,4 ммоль) растворили в ацетонитриле. Добавили метил бромацетат (0,41 мл, 4,4 ммоль) и карбонат цезия (1,7 г, 5,3 ммоль), после чего перемешивали при 80°С в течение 12 часов. По окончании реакции, к реакционной смеси добавили воду, и проэкстрагировали с помощью этилацетата. Полученный органический слой высушили над сульфатом магния, упарили при пониженном давлении, и очистили с помощью колоночной хроматографии с получением метил 2-(2-метилпиридин-3-илокси)ацетата (0,34 г, 42%), который использовали в следующей стадии.
Стадия 2. Получение 2-(2-метилпиридин-3-илокси)ацетогидразида: Метил 2-(2-метилпиридин-3-илокси)ацетат (0,34 г, 1,1 ммоль) растворили в EtOH. Добавили гидразин моногидрат (0,14 мл, 1,6 ммоль), после чего перемешивали в течение 12 часов при кипячении с обратным холодильником. По окончании реакции, реакционную смесь упарили при пониженном давлении. Концентрат отфильтровали с получением 2-(2-метилпиридин-3-илокси)ацетогидразида в неочищенном виде.
Стадия 3. Получение (E)-N'-((1Н-индол-4-ил)метилен)-2-(2-метилпиридин-3-илокси)ацетогидразида: 2-(4-хлор-2,6-диметилфенокси)ацетогидразид (0,1 г, 0,55 ммоль) и 1H-индол-4-карбальдегид (0,088 г, 0,61 ммоль) растворили в этаноле, после чего перемешивали в течение 3 часов при кипячении с обратным холодильником. По окончании реакции, образовавшийся осадок отфильтровали с получением соединения 187 (0,14 г, 82%).
1H NMR (400 MHz, DMSO-d6): δ 11,54 (bs, 1H), 11,42 (m, 1H), 8,49 (s, 0,4H), 8,24 (s, 0,6H), 8,0 (m, 1H), 7,47 (m, 2H), 7,29-7,10 (m, 4H), 6,96 (s, 1H), 5,28 (s, 1,3H), 4,74 (s, 0,7H), 2,39 (d, J=18,2 Hz, 3H).
Пример 33. Синтез соединения 190
Стадия 1. Получение метил 2-(2,4-диметилпиридин-3-илокси)ацетата:
2,4-диметилпиридин-3-ол (0,5 г, 3,2 ммоль) растворили в ацетонитриле. Добавили метил бромацетат (0,35 мл, 3,9 ммоль) и карбонат цезия (1,25 г, 3,9 ммоль), после чего перемешивали при 100°С в течение 12 часов. По окончании реакции, к реакционной смеси добавили воду, и проэкстрагировали с помощью этилацетата. Полученный органический слой высушили над сульфатом магния, упарили при пониженном давлении, и очистили с помощью колоночной хроматографии с получением метил 2-(2,4-диметилпиридин-3-илокси)ацетата (0,18 г, 28%), который использовали в следующей стадии.
Стадия 2. Получение 2-(2,4-диметилпиридин-3-илокси)ацетогидразида:
метил 2-(2,4-диметилпиридин-3-илокси)ацетат (0,18 г, 0,9 ммоль) растворили в EtOH. Добавили гидразин моногидрат (0,05 мл, 1,1 ммоль), после чего перемешивали в течение 3 часов при кипячении с обратным холодильником. По окончании реакции, реакционную смесь упарили при пониженном давлении. Концентрат перекристаллизовали из этанола с получением 2-(2,4-диметилпиридин-3-илокси)ацетогидразида.
Стадия 3. Получение (E)-N'-((1Н-индол-4-ил)метилен)-2-(2,4-диметилпиридин-3-илокси)ацетогидразида: 2-(4-хлор-2,6-диметилфенокси)ацетогидразид (0,08 г, 0,41 ммоль) и 1Н-индол-4-карбальдегид (0,065 г, 0,45 ммоль) растворили в этаноле, после чего перемешивали в течение 3 часов при кипячении с обратным холодильником. По окончании реакции, образовавшийся осадок отфильтровали от реакционной смеси и очистили с помощью колоночной хроматографии с получением соединения 190 (0,045 г, 33%).
1H NMR (400 MHz, DMSO-d6): δ 11,52-11,33 (m, 1H), 8,62 (s, 0,5H), 8,2 (s, 0,5H,), 8,08 (m, 1H), 7,49-6,77 (m, 6H), 4,94 (s, 1H), 4,45 (s, 1H), 2,47 (m, 3H), 2,29 (m, 3H).
Пример 34. Синтез соединения 191
2-(4-хлор-2,6-диметилфенокси)ацетогидразид (0,065 г, 0,33 ммоль) и 1H-индол-6-карбальдегид (0,053 г, 0,36 ммоль) растворили в этаноле, после чего перемешивали в течение 12 часов при кипячении с обратным холодильником. По окончании реакции, реакционную смесь упарили при пониженном давлении. Концентрат очистили с помощью колоночной хроматографии с получением соединения 191 (0,088 г, 82%).
1H NMR (400 MHz, DMSO-d6): δ 11,41 (m, 1H), 11,25 (m, 1H), 8,43 (s, 0,5H), 8,06 (m, 1,5H,), 7,7-7,07 (m, 5H), 6,43 (m, 1H), 4,86 (s, 1H), 4,66 (s, 1H), 2,39 (d, J=3,08 Hz, 3H), 2,28 (d, J=3,08 Hz, 3H).
Пример 35. Синтез соединения 192
Стадия 1. Получение метил 2-(2-(трифторметил)фенокси)ацетат:
2-(трифторметил)фенол (1 г, 6,2 ммоль) растворили в ацетонитриле. Метил бромацетат (0,94 г, 6,2 ммоль) и карбонат цезия (4 г, 12 ммоль) добавили к этой смеси, после чего перемешивали в течение 16 часов. По окончании реакции, реакционную смесь отфильтровали через цеолиты. Фильтрат разбавили насыщенным водным раствором гидрокарбоната натрия, и проэкстрагировали с помощью этилацетата. Полученный органический слой высушили над сульфатом магния, и упарили при пониженном давлении с получением метил 2-(2-(трифторметил)фенокси)ацетат (1,4 г, 100%), который использовали в следующей стадии.
Стадия 2. Получение 2-(2-(трифторметил)фенокси)ацетогидразид:
метил 2-(2-(трифторметил)фенокси)ацетат (1,4 г, 6,2 ммоль) растворили в EtOH. Добавили гидразин моногидрат (2 г, 40 ммоль), после чего перемешивали в течение 16 часов при кипячении с обратным холодильником. По окончании реакции, реакционную смесь упарили при пониженном давлении. Концентрат разбавили водой, и проэкстрагировали с помощью дихлорметана. Полученный органический слой высушили над сульфатом магния, и упарили при пониженном давлении с получением 2-(2-(трифторметил)фенокси)ацетогидразида (1,4 г, 96%).
Стадия 3. Получение (Е)-N'-((1Н-индол-4-ил)метилен)-2-(2-(трифторметил)фенокси)ацетогидразида: 2-(2-(трифторметил)фенокси)ацетогидразид (0,2 г, 0,85 ммоль) и 1H-индол-4-карбальдегид (0,13 г, 0,85 ммоль) растворили в этаноле, после чего перемешивали при 100°С в течение 16 часов. По окончании реакции, реакционную смесь упарили при пониженном давлении. Концентрат очистили с помощью колоночной хроматографии с получением соединения 192 (0,27 г, 88%).
1H NMR (400 MHz, DMSO-d6): δ 11,57-11,33 (m, 2H), 8,43-8,25 (m, 1H), 7,64-7,44 (m, 4H), 7,25-6,96 (m, 6H), 5,37-4,84 (m, 2H).
Пример 36. Синтез соединения 193
2-(2-(трифторметил)фенокси)ацетогидразид (0,2 г, 0,85 ммоль) и 1Н-индол-5-карбальдегид (0,13 г, 0,85 ммоль) растворили в этаноле, после чего перемешивали при 100°С в течение 16 часов. По окончании реакции, реакционную смесь упарили при пониженном давлении. Концентрат очистили с помощью колоночной хроматографии с получением соединения 193 (0,18 г, 43%).
1H NMR (400 MHz, DMSO-d6): δ 11,57-11,33 (m, 2H), 8,25-8,06 (m, 1H), 7,77-7,79 (m, 1H), 7,62-7,51 (m, 4H), 7,41-7,36 (m, 2,5H), 7,16-7,07 (m, 2,5H), 7,47 (m, 1H), 5,30-4,79 (m, 2H).
Пример 37. Синтез соединения 194
2-(2-(трифторметил)фенокси)ацетогидразид (0,2 г, 0,85 ммоль) и 1H-индол-6-карбальдегид (0,13 г, 0,85 ммоль) растворили в этаноле, после чего перемешивали при 100°С в течение 16 часов. По окончании реакции, реакционную смесь упарили при пониженном давлении. Концентрат очистили с помощью колоночной хроматографии с получением соединения 194 (0,17 г, 55%).
1H NMR (400 MHz, DMSO-d6): δ 11,49-11,26 (m, 2H), 8,27-8,07 (m, 1H), 7,71-7,54 (m, 4H), 7,44-7,42 (m, 2H), 7,20-7,06 (m, 2H), 6,45 (s, 1H), 5,31-4,80 (m, 2H).
Пример 38. Синтез соединения 196
Стадия 1. Получение метил 2-(2-хлорпиридин-3-илокси)ацетата:
2-хлорпиридин-3-ол (0,6 г, 4,6 ммоль) растворили в ацетонитриле. Метил бромацетат (0,52 мл, 9,26 ммоль) и карбонат цезия (1,8 г, 5,6 ммоль) добавили к этой смеси, после чего перемешивали в течение 3 часов. По окончании реакции, к реакционной смеси добавили воду, и проэкстрагировали с помощью дихлорметана. Полученный органический слой высушили над сульфатом магния, упарили при пониженном давлении, и очистили с помощью колоночной хроматографии с получением метил 2-(2-хлорпиридин-3-илокси)ацетата (0,86 г, 92%), который использовали в следующей стадии.
Стадия 2. Получение 2-(2-хлорпиридин-3-илокси)ацетогидразида:
метил 2-(2-хлорпиридин-3-илокси)ацетат (0,4 г, 1,98 ммоль) растворили в EtOH. Добавили гидразин моногидрат (0,1 мл, 2,2 ммоль), после чего перемешивали в течение 12 часов при кипячении с обратным холодильником. По окончании реакции, реакционную смесь упарили при пониженном давлении с получением 2-(2-хлорпиридин-3-илокси)ацетогидразида.
Стадия 3. Получение (Е)-N'-((1Н-индол-4-ил)метилен)-2-(2-хлорпиридин-3-илокси)ацетогидразида: 2-(2-хлорпиридин-3-илокси)ацетогидразид (0,1 г, 0,49 ммоль) и 1Н-индол-4-карбальдегид (0,086 г, 0,59 ммоль) растворили в этаноле, после чего перемешивали при 100°С в течение 12 часов. По окончании реакции, полученное твердое вещество отфильтровали с получением соединения 196 (0,15 г, 92%).
1H NMR (400 MHz, DMSO-d6): δ 11,61 (bs, 1H), 11,36 (m, 1H), 8,46 (s, 0,2H), 8,25 (s, 0,8H), 7,56-7,09 (m, 7H), 6,97 (m, 1H), 5,42 (s, 1,5H), 4,88 (s, 0,5H), 4,9 (m, 1H), 4,88 (s 2H), 4,08 (t, J=6,28 Hz, 2H), 3,58 (m, 2H), 2,18 (d, J=4,28 Hz, 9H).
Пример 39. Синтез соединения 205
2-(мезитилокси)ацетогидразид (0,10 г, 0,48 ммоль) и 1Н-бензо[d]имидазол-5-карбальдегид (0,079 г, 0,48 ммоль) растворили в EtOH, после чего перемешивали при 100°С в течение 16 часов. По окончании реакции, реакционную смесь охладили до комнатной температуры, и упарили при пониженном давлении. Полученное твердое вещество очистили с помощью колоночной хроматографии с получением соединения 205 (0,058 г, 36%).
1H NMR (400 MHz, DMSO-d6): δ 12,62-12,48 (m, 1H), 11,47-11,39 (m, 1H), 8,52 (s, 0,5H), 8,29-8,23 (m, 1H), 8,05 (s, 0,5H), 7,88-7,43 (m, 3H), 6,83-6,82 (m, 2H), 4,74-4,31 (m, 2H), 2,23-2,14 (m, 9H).
Пример 40. Синтез соединения 206
2-(мезитилокси)ацетогидразид (0,10 г, 0,39 ммоль) и бензо[d][1,2,3]тиадиазол-5-карбальдегид (0,079 г, 0,48 ммоль) растворили в EtOH, после чего перемешивали при 100°С в течение 16 часов. По окончании реакции, реакционную смесь охладили до комнатной температуры, и упарили при пониженном давлении. Полученное твердое вещество очистили с помощью колоночной хроматографии и перекристаллизации с получением соединения 206 (0,11 г, 65%).
1H NMR (400 MHz, DMSO-d6): δ 11,81-11,73 (m, 1H), 8,89-8,85 (m, 1H), 8,69 (s, 0,5H), 8,47-8,36 (m, 1H), 8,23-8,07 (s, 1,5H), 6,84-8,81 (m, 2H), 4,80-4,36 (m, 2H), 2,22-2,16 (m, 9H).
Пример 41. Синтез соединения 211
2-(мезитилокси)ацетогидразид (0,10 г, 0,39 ммоль) и 3-((4-метилпиперидин-1-ил)метил)-1H-индол-4-карбальдегид (0,089 г, 0,42 ммоль) растворили в диметилсульфоксиде, после чего перемешивали при комнатной температуре в течение 12 часов. По окончании реакции, реакционную смесь охладили до комнатной температуры, разбавили водой, и проэкстрагировали с помощью этилацетата. Полученный органический слой высушили над сульфатом магния, и упарили при пониженном давлении. Образовавшийся осадок отфильтровали с получением соединения 211 (0,078 г, 45%).
1Н NMR (400 MHz, DMSO-d6,): δ 11,42-11,17 (m, 1H), 11,1 (m, 1H), 9,08 (s, 0,5H), 8,6 (s, 0,5Н), 7,59 (d, J=7,60 Hz, 5H), 7,43-7,27 (m, 1,5H), 7,13 (t, J=7,68 Hz, 0,5H), 7,0,3 (t, J=7,72 Hz, 0,5H), 6,83 (d, J=10,0 Hz, 2H), 4,74 (s, 1H), 4,34 (s, 1H), 3,59 (s, 1H), 3,50 (m, 1H), 2,86 (m, 2H), 2,23 (s, 3H), 2,18 (m, 6H), 1,86 (m, 2H), 1,47 (m, 2H), 1,28 (m, 1H), 1,01 (m, 2H), 0,81 (m, 3H).
Пример 42. Синтез соединения 217
Стадия 1. Получение метил 2-(2,4-диметил-6-(пиридин-4-ил)фенокси)ацетата: 2,4-диметил-6-(пиридин-4-ил)фенол (Соединение 6-8) (0,12 г, 0,6 ммоль) растворили в ацетонитриле. Метил бромацетат (0,092 г, 0,6 ммоль) и карбонат цезия (0,2 г, 0,6 ммоль) добавили к этой смеси, после чего перемешивали в течение 16 часов. По окончании реакции, реакционную смесь упарили при пониженном давлении, и очистили с помощью колоночной хроматографии с получением метил 2-(2,4-диметил-6-(пиридин-4-ил)фенокси)ацетата (0,1 г, 61%).
Стадия 2. Получение 2-(2,4-диметил-6-(пиридин-4-ил)фенокси)ацетогидразида:
2-(2,4-диметил-6-(пиридин-4-ил)фенокси)ацетат (0,1 г, 0,37 ммоль) растворили в EtOH. Добавили гидразин моногидрат (0,13 г, 2,63 ммоль), после чего перемешивали при 100°С в течение 16 часов при кипячении с обратным холодильником. По окончании реакции, реакционную смесь упарили при пониженном давлении, и очистили с помощью колоночной хроматографии с получением 2-(2,4-диметил-6-(пиридин-3-ил)фенокси)ацетогидразида (0,1 г, 100%).
Стадия 3. Получение (E)-N'-((1Н-индол-6-ил)метилен)-2-(2,4-диметил-6-(пиридин-4-ил)фенокси)ацетогидразида: 2-(2,4-диметил-6-(пиридин-3-ил)фенокси)ацетогидразид (0,06 г, 0,22 ммоль) и 1Н-индол-4-карбальдегид (0,032 г, 0,22 ммоль) растворили в этаноле, после чего перемешивали в течение 16 часов при кипячении с обратным холодильником. По окончании реакции, реакционную смесь упарили при пониженном давлении.
Концентрат очистили с помощью колоночной хроматографии с получением соединения 217 (0,04 г, 45%).
1H NMR (400 MHz, DMSO-d6): δ 11,28-11,12 (m, 2H), 8,61-8,58 (m, 2H), 8,38-7,89 (2s, 1H), 7,68 (s, 0,5H), 7,63-7,34 (m, 5,5H), 7,14-7,09 (m, 2,5H), 6,46-6,42 (m, 1H), 4,44 3,99 (2s, 2H), 2,34-2,29 (m, 6H).
Пример 43. Синтез соединения 218
Стадия 1. Получение метил 2-(2-(фуран-3-ил)-4,6-диметилфенокси)ацетата:
2-(фуран-3-ил)-4,6-диметилфенол (Соединение 6-2) (0,2 г, 1,1 ммоль) растворили в ацетонитриле. Метил бромацетат (0,16 г, 1,1 ммоль) и карбонат цезия (0,35 г, 1,1 ммоль) добавили к этой смеси, после чего перемешивали в течение 16 часов. По окончании реакции, реакционную смесь отфильтровали через цеолиты. Фильтрат упарили при пониженном давлении, и очистили с помощью колоночной хроматографии с получением метил 2-(2-(фуран-3-ил)-4,6-диметилфенокси)ацетата (0,18 г, 64%).
Стадия 2. Получение 2-(2-(фуран-3-ил)-4,6-диметилфенокси)ацетогидразида:
метил 2-(2-(фуран-3-ил)-4,6-диметилфенокси)ацетат (0,1 г, 0,38 ммоль) растворили в EtOH. Добавили гидразин моногидрат (1 г, 20 ммоль), после чего перемешивали в течение 16 часов при кипячении с обратным холодильником. По окончании реакции, реакционную смесь упарили при пониженном давлении с получением 2-(2-(фуран-3-ил)-4,6-диметилфенокси)ацетогидразида (0,03 г, 30%).
Стадия 3. Получение (E)-N'-((1Н-индол-6-ил)метилен)-2-(2-(фуран-3-ил)-4,6-диметилфенокси)ацетогидразида: 2-(2-(фуран-3-ил)-4,6-диметилфенокси)ацетогидразид (0,03 г, 0,12 ммоль) и 1H-индол-6-карбальдегид (0,017 г, 0,12 ммоль) растворили в этаноле, после чего перемешивали в течение 16 часов при кипячении с обратным холодильником. По окончании реакции, реакционную смесь упарили при пониженном давлении. Концентрат очистили с помощью колоночной хроматографии с получением соединения 218 (0,02 г, 45%).
1Н NMR (400 MHz, DMSO-d6): δ 11,38-11,37 (m, 1H), 11,29-11,16 (m, 1H), 8,46, 8,30 (2s, 1H), 8,26 (s, 0,5H), 8,00 (s, 0,5H), 7,73 (m, 1,5H), 7,60-7,20 (m, 4,5H), 7,02-6,98 (m, 2H), 6,51-6,39 (m, 1H), 4,66 (s, 1H), 4,20 (m, 1H), 2,30-2,27 (m, 6H).
Пример 44. Синтез соединения 227
Стадия 1. Получение метил 2-(4-(гидроксиметил)-2,6-диметилфенокси)ацетата:
4-(гидроксиметил)-2,6-диметилфенол (0,034 г, 0,22 ммоль) растворили в метаноле. Метил бромацетат (0,034 г, 0,22 ммоль) и карбонат калия (0,031 г, 0,22 ммоль) добавили к этой смеси, после чего перемешивали в течение 16 часов. По окончании реакции, к реакционной смеси добавили насыщенный водный раствор аммоний хлорида, и проэкстрагировали с помощью этилацетата. Полученный органический слой высушили над сульфатом магния, и упарили при пониженном давлении с получением метил 2-(4-(гидроксиметил)-2,6-диметилфенокси)ацетата (0,04 г, 80%), который использовали в следующей стадии.
Стадия 2. Получение 2-(4-(гидроксиметил)-2,6-диметилфенокси)ацетогидразида:
Метил 2-(4-(гидроксиметил)-2,6-диметилфенокси)ацетат (0,3 г, 1,3 ммоль) растворили в EtOH. Добавили гидразин моногидрат (1 г, 18 ммоль), после чего перемешивали в течение 60°С в течение 16 часов. По окончании реакции, реакционную смесь упарили при пониженном давлении с получением 2-(4-(гидроксиметил)-2,6-диметилфенокси)ацетогидразида (0,25 г, 83%).
Стадия 3. Получение (E)-N'-((1Н-индол-4-ил)метилен)-2-(4-(гидроксиметил)-2,6-диметилфенокси)ацетогидразида: 2-(2-(трифторметил)фенокси)ацетогидразид (0,1 г, 0,45 ммоль) и 1Н-индол-4-карбальдегид (0,065 г, 0,45 ммоль) растворили в этаноле, после чего перемешивали при 100°С в течение 16 часов. По окончании реакции, реакционную смесь упарили при пониженном давлении. Концентрат очистили с помощью колоночной хроматографии с получением соединения 227 (0,05 г, 32%).
1H NMR (400 MHz, DMSO-d6): δ 11,53-11,46 (m, 1H), 11,37 (bs, 1H), 8,69 (s, 1H), 8,22 (s, 1H), 7,50-7,43 (m, 2H), 7,25-7,11 (m, 2,5H), 6,98 (m, 2H), 6,76 (s, 0,5H), 5,10-5,08 (m, 1H), 4,86 (s, 1H), 4,39-4,38 (m, 3H). 2,27-2,26 (m, 6H).
Пример 45. Синтез соединения 228
2-(2-(трифторметил)фенокси)ацетогидразид (0,07 г, 0,31 ммоль) и 1H-индол-6-карбальдегид (0,045 г, 0,31 ммоль) растворили в этаноле, после чего перемешивали в течение 16 часов при кипячении с обратным холодильником. По окончании реакции, реакционную смесь упарили при пониженном давлении. Концентрат очистили с помощью колоночной хроматографии с получением соединения 228 (0,05 г, 46%).
1Н NMR (400 MHz, DMSO-d6): δ 11,44-11,40 (m, 1H), 11,33-11,23 (m, 1H), 8,50-8,03 (2s, 1H), 7,77 (s, 0,5H), 7,59-6,76 (m, 3,5H), 6,98 (s, 2H), 6,47-6,42 (m, 1H), 5,11-5,07 (m, 1H), 4,77 (s, 1H), 4,39-4,34 (m, 3H), 2,26-2,24 (m, 6H).
Пример 46. Синтез соединения 229
Стадия 1. Получение метил 2-(2,4-диметил-6-(тетрагидрофуран-3-ил)фенокси)ацетата:
2,4-диметил-6-(тетрагидрофуран-3-ил)фенол (Соединение 7-2) (0,4 г, 2,1 ммоль) растворили в ацетонитриле. Метил бромацетат (0,32 г, 2,1 ммоль) и карбонат цезия (1,4 г, 4,2 ммоль) добавили к этой смеси, после чего перемешивали в течение 16 часов. По окончании реакции, реакционную смесь отфильтровали через цеолиты. Фильтрат упарили при пониженном давлении, и очистили с помощью колоночной хроматографии с получением метил 2-(2,4-диметил-6-(тетрагидрофуран-3-ил)фенокси)ацетата (0,42 г, 76%).
Стадия 2. Получение 2-(2,4-диметил-6-(тетрагидрофуран-3-ил)фенокси)ацетогидразида:
метил 2-(2,4-диметил-6-(тетрагидрофуран-3-ил)фенокси)ацетат (0,18 г, 0,68 ммоль) растворили в EtOH. Добавили гидразин моногидрат (1 г, 20 ммоль), после чего перемешивали в течение 16 часов при кипячении с обратным холодильником. По окончании реакции, реакционную смесь упарили при пониженном давлении с получением 2-(2,4-диметил-6-(тетрагидрофуран-3-ил)фенокси)ацетогидразида (0,1 г, 56%).
Стадия 3. Получение (Е)-N'-((1Н-индол-6-ил)метилен)-2-(2,4-диметил-6-(тетрагидрофуран-3-ил)фенокси)ацетогидразида: 2-(2,4-диметил-6-(тетрагидрофуран-3-ил)фенокси)ацетогидразид (0,1 г, 0,38 ммоль) и 1H-индол-6-карбальдегид (0,067 г, 0,45 ммоль) растворили в этаноле, после чего перемешивали в течение 16 часов при кипячении с обратным холодильником. По окончании реакции, реакционную смесь упарили при пониженном давлении. Концентрат очистили с помощью колоночной хроматографии (этил ацетат:гексан = 2:1) с получением соединения 229 (0,063 г, 43%).
1Н NMR (400 MHz, DMSO-d6): δ 11,48-11,44 (m, 1H), 11,35-11,23 (m, 1H), 8,48 (s, 0,5H), 8,05 (s, 0,5H), 7,72 (s, 0,5H), 7,59-7,27 (m, 3,5H), 6,96-6,88 (m, 2H), 6,47-6,43 (m, 1H), 4,77 (s, 1H), 4,34 (s, 1H), 4,02-3,92 (m, 2H), 3,77-3,73 (m, 2H), 3,53-3,43 (m, 1H), 2,26-2,20 (m, 7Н),1,89-2,20 (m, 1Н).
Пример 47. Синтез соединения 237
Стадия 1. Получение метил 2-(4-циано-2,6-диметилфенокси)ацетата:
4-гидрокси-3,5-диметилбензонитрил (200 мг, 1,4 ммоль), метил бромацетат (0,15 мл, 1,4 ммоль) и карбонат цезия (500 мг, 1,68 ммоль) растворили в ацетонитриле (4 мл), после чего перемешивали при 80°С в течение 2 часов. По окончании реакции, реакционную смесь проэкстрагировали с помощью этилацетата, высушили над сульфатом магния, и упарили при пониженном давлении с получением метил 2-(4-циано-2,6-диметилфенокси)ацетата (303 мг, 99%).
Стадия 2. Получение 2-(4-циано-2,6-диметилфенокси)ацетогидразида:
метил 2-(4-циано-2,6-диметилфенокси)ацетат (200 мг, 0,91 ммоль) и гидразин (0,05 мл, 1,0 ммоль) растворили в EtOH (2 мл), после чего перемешивали при 80°С в течение 12 часов. По окончании реакции, реакционную смесь упарили при пониженном давлении с получением 2-(4-циано-2,6-диметилфенокси)ацетогидразида (180 мг, 90%).
Стадия 3. Получение (Е)-N'-((1Н-индол-6-ил)метилен)-2-(4-циано-2,6-диметилфенокси)ацетогидразида: 2-(4-циано-2,6-диметилфенокси)ацетогидразид (92 мг, 0,42 ммоль) и 1H-индол-6-карбальдегид (67 мг, 0,46 ммоль) растворили в EtOH 1 мл, после чего перемешивали при 80°С в течение 12 часов. По окончании реакции, реакционную смесь очистили с помощью колоночной хроматографии (этил ацетат:гексан = 1:1), перекристаллизовали из смеси гексан/этил ацетат, и отфильтровали с получением соединения 237 (31 мг, 21%).
1H NMR (400 MHz, DMSO-d6): δ 11,45 (m, 1H), 11,32-11,24 (m, 1H), 8,45-8,04 (2s, 1H), 7,71 (s, 0,5H), 7,59-7,57 (m, 3H), 7,53-7,38 (m, 2H), 7,27 (m, 0,5H), 6,45 (m, 1H), 4,92 (s, 1H), 4,47 (s, 1H), 2,30 (d, J=4,24 Hz, 6H).
Пример 48. Синтез соединения 256
Соединение 108 (100 мг, 0,35 ммоль) и NBS (57 мг, 0,35 ммоль) растворили в CH2Cl2 мл, после чего перемешивали при комнатной температуре в течение 2 часов. По окончании реакции, реакционную смесь отфильтровали. Фильтрат очистили с помощью колоночной хроматографии (этил ацетат:гексан = 2:1) с получением соединения 256 (40 мг, 32%).
1H NMR (400 MHz, DMSO-d6): δ 11,45 (bs, 1H), 8,52 (s, 1H), 8,06 (s, 1H), 7,73 (s, 0,5H), 7,61 (m, 1H), 7,54-7,39 (m, 2H), 6,83 (m, 2H), 2,21 (m, 9H).
Пример 49. Синтез соединения 258
2-(2,6-диметилфенокси)ацетогидразид (100 мг, 0,51 ммоль) и 1H-индол-6-карбальдегид (74 мг, 0,51 ммоль) растворили в EtOH (2 мл), после чего перемешивали при 100°С в течение 12 часов. После удаления растворителя, остаток перекристаллизовали из тетрагидрофурана и гексана с получением соединения 258 (42 мг, 26%).
1H NMR (400 MHz, DMSO-d6): δ 11,38 (m, 1H), 11,29 (m, 1H), 8,49, 8,04 (2s, 1H), 7,71 (s, 0,5H), 7,58 (m, 1H), 7,52-7,27 (m, 2,5H), 7,05 (m, 2H), 6,96 (m, 1H), 6,45 (m, 1H), 4,79 (s, 2H), 4,37 (s, 2H), 2,27 (m, 6H).
Пример 50. Синтез соединения 259
Стадия 1. Получение метил 2-(2-бром-4,6-диметилфенокси)ацетата:
2-бром-4,6-диметилфенол (200 мг, 1,4 ммоль), метил бромацетат (0,15 мл, 1,4 ммоль) и карбонат цезия (500 мг, 1,68 ммоль) растворили в ацетонитриле (4 мл), после чего перемешивали при 100°С в течение 3 часов. По окончании реакции, реакционную смесь проэкстрагировали с помощью этилацетата, высушили над сульфатом магния, и очистили с помощью колоночной хроматографии (этил ацетат:гексан = 1:3) с получением метил 2-(2-бром-4,6-диметилфенокси)ацетата (516 мг, 100%).
Стадия 2. Получение 2-(2-бром-4,6-диметилфенокси)ацетогидразида:
метил 2-(2-бром-4,6-диметилфенокси)ацетат (423 мг, 1,55 ммоль) и гидразин (0,08 мл, 1,57 ммоль) растворили в EtOH (2 мл), после чего перемешивали при 100°С в течение 12 часов. По окончании реакции, реакционную смесь упарили при пониженном давлении с получением 2-(2-бром-4,6-диметилфенокси)ацетогидразида (393 мг, 93%).
Стадия 3. Получение (Е)-N'-((1Н-индол-4-ил)метилен)-2-(2-бром-4,6-диметилфенокси)ацетогидразида: 2-(2-бром-4,6-диметилфенокси)ацетогидразид (85 мг, 0,31 ммоль) и 1Н-индол-4-карбальдегид (45 мг, 0,31 ммоль) растворили в EtOH (2 мл), после чего перемешивали при 100°С в течение 12 часов. По окончании реакции, реакционную смесь упарили при пониженном давлении. Концентрат перекристаллизовали из этанола, и отфильтровали с получением соединения 259 (50 мг, 40%).
1H NMR (400 MHz, DMSO-d6): δ 11,47-11,44 (m, 1H), 11,34 (bs, 1H), 8,68 (s, 1H), 8,23 (s, 1H), 7,47 (m, 2H), 7,25 (m, 2H), 7,17-7,13 (m, 4H), 7,12 (s, 1H), 6,78 (s, 1H), 4,98 (s, 1H), 4,48 (s, 2H), 2,32-2,24 (m, 9H).
Пример 51. Синтез соединения 260
2-(2-бром-4,6-диметилфенокси)ацетогидразид (85 мг, 0,31 ммоль) и 1Н-индол-6-карбальдегид (45 мг, 0,31 ммоль) растворили в EtOH (2 мл), после чего перемешивали при 100°С в течение 12 часов. По окончании реакции, реакционную смесь очистили с помощью колоночной хроматографии (этил ацетат:гексан = 1:1) с получением соединения 260 (60 мг, 48%).
1H NMR (400 MHz, DMSO-d6): δ 11,38 (s, 1H), 11,29-11,20 (m, 1H), 8,48-8,02 (2s, 1H), 7,71 (s, 0,5H), 7,58-7,25 (m, 3H), 7,28-7,25 (m, 0,5H), 7,05 (m, 1H), 6,45 (m, 1H), 4,88, (2s, 2H), 2,28 (m, 6H).
Пример 52. Синтез соединения 279
2-(о-тозилокси)ацетогидразид (100 мг, 0,56 ммоль) и 1H-индол-6-карбальдегид (81 мг, 0,56 ммоль) растворили в EtOH (2 мл), после чего перемешивали при 90°С в течение 6 часов. По окончании реакции, образовавшийся осадок отфильтровали с получением соединения 279 (50 мг, 29%).
1Н NMR (400 MHz, DMSO-d6): δ 11,30 (m, 1H), 11,28 (m, 1H), 8,34 (s, 0,5H), 8,08 (s, 0,5H), 7,67 (d, J=2,8 Hz, 1H), 7,46-7,37 (m, 2H), 7,18-7,10 (m, 2H), 6,89-6,81 (m, 2H), 6,47 (m, 1H), 5,15 (s, 1H), 4,65 (s, 1H), 2,23 (d, J=13,2 Hz, 3H).
Пример 53. Синтез соединения 280
Стадия 1. Получение 1-(фенилсульфонил)-1Н-индол-6-карбальдегид: 1Н-индол-4-карбальдегид (1000 мг, 6,89 ммоль), гидрид натрия (360 мг, 8,26 ммоль) и бензилсульфонил хлорид (1 мл, 6,89 ммоль) растворили в тетрагидрофуране 10 мл, после чего перемешивали при комнатной температуре в течение 12 часов. По окончании реакции, реакционную смесь проэкстрагировали с помощью этилацетата, высушили над сульфатом магния, и очистили с помощью колоночной хроматографии (этил ацетат:гексан = 1:3) с получением 1-(фенилсульфонил)-1Н-индол-6-карбальдегида (500 мг, 25%).
Стадия 2. Получение (Е)-2-(мезитилокси)-N'-((1-(фенилсульфонил)-1Н-индол-4-ил)метилен)ацетогидразида: 2-(мезитилокси)ацетогидразид (30 мг, 0,14 ммоль) и 1Н-индол-4-карбальдегид (44 мг, 0,16 ммоль) растворили в EtOH (2 мл), после чего перемешивали при 100°С в течение 12 часов. По окончании реакции, образовавшийся осадок отфильтровали с получением соединения 280 (30 мг, 39%).
1H NMR (400 MHz, DMSO-d6): δ 11,64 (m, 1H), 8,65-8,18 (2s, 1H), 8,02-7,97 (m, 4H), 7,7-7,2 (m, 6H), 4,78 (s, 1H), 4,36 (s, 1H), 2,20 (m, 9H).
Пример 54. Синтез соединения 286
Стадия 1. Получение метил 2-(2,6-диметилпиридин-4-илокси)ацетата:
2,6-диметилпиридин-4-ол (300 мг, 2,44 ммоль), метил 2-бромацетат (0,27 мл, 2,93 ммоль) и карбонат цезия (2,4 г, 7,32 ммоль) растворили в ацетонитриле 6 мл, после чего перемешивали при 80°С в течение 1 часа. По окончании реакции, реакционную смесь отфильтровали под пониженным давлением. Фильтрат упарили при пониженном давлении, и очистили с помощью колоночной хроматографии (гексан:этил ацетат = 3:1) с получением метил 2-(4-фтор-2-метилфенокси)ацетата (200 мг, 42%).
Стадия 2. Получение 2-(2,6-диметилпиридин-4-илокси)ацетогидразида:
метил 2-(2,6-диметилпиридин-4-илокси)ацетат (200 мг, 1,03 ммоль) и гидразин (0,05 мл, 1,13 ммоль) растворили в EtOH (3 мл), после чего перемешивали при 100°С в течение 12 часов. По окончании реакции, образовавшийся осадок отфильтровали с получением 2-(4-фтор-2-метилфенокси)ацетогидразида (190 мг, 79%).
Стадия 2. Получение (E)-N'-((1Н-индол-4-ил)метилен)-2-(2,6-диметилпиридин-4-илокси)ацетогидразида: 2-(2,6-диметилпиридин-4-илокси)ацетогидразид (30 мг, 0,15 ммоль) и 1Н-индол-4-карбальдегид (22 мг, 0,15 ммоль) растворили в EtOH (2 мл), после чего перемешивали при 90°С в течение 12 часов. По окончании реакции, образовавшийся осадок отфильтровали с получением соединения 286 (20 мг, 41%).
1H NMR (400 MHz, DMSO-d6): δ 11,53 (m, 1H), 11,38 (m, 1H), 8,53-8,26 (s, 1H), 7,49 (m, 2H), 7,19 (m, 2,5H), 7,11 (s, 0,5H), 6,70-6,64 (m, 2H), 5,26 (s, 1H), 4,74 (s, 1H), 2,34 (m, 6H).
Пример 55. Синтез соединения 288
2-(2,6-диметилпиридин-4-илокси)ацетогидразид (100 мг, 0,51 ммоль) и 1Н-индол-6-карбальдегид (74 мг, 0,51 ммоль) растворили в EtOH (2 мл), после чего перемешивали при 110°С в течение 12 часов. К реакционной смеси добавили воду, и проэкстрагировали с помощью этилацетата. Полученный органический слой высушили над сульфатом магния, и очистили с помощью колоночной хроматографии (гексан:этил ацетат = 2:1) с получением соединения 288 (30 мг, 18%).
1H NMR (400 MHz, DMSO-d6): δ 11,43 (m, 1H), 11,28 (m, 1H), 8,35-8,07 (2s, 1H), 7,68 (d, J=18,0 Hz, 1H), 7,57 (d, J=8,4 Hz, 1H), 6,66 (d, J=22,0 Hz, 2H), 6,47 (s, 1H), 5,20 (s, H), 4,69 (s, 1H), 2,34 (m, 6H).
Пример 56. Синтез соединения 291
Стадия 1. Получение метил 2-(4-фтор-2-метилфенокси)ацетата: 4-фтор-2-метилфенол (500 мг, 3,96 ммоль), метил 2-бромацетат (0,36 мл, 3,96 ммоль) и карбонат цезия (1,9 мг, 5,94 ммоль) растворили в ацетонитриле (8 мл), после чего перемешивали при 90°С в течение 5 часов. К реакционной смеси добавили воду, и проэкстрагировали с помощью этилацетата. Полученный органический слой высушили над сульфатом магния, и очистили с помощью колоночной хроматографии (гексан:этил ацетат = 3:1) с получением метил 2-(4-фтор-2-метилфенокси)ацетата (450 мг, 57%).
Стадия 2. Получение 2-(4-фтор-2-метилфенокси)ацетогидразида:
метил 2-(4-фтор-2-метилфенокси)ацетат (400 мг, 2,02 ммоль) и гидразин (0,12 мл, 2,42 ммоль) растворили в EtOH 4 мл, после чего перемешивали при 90°С в течение 3 часов. По окончании реакции, образовавшийся осадок отфильтровали с получением 2-(4-фтор-2-метилфенокси)ацетогидразида (280 мг, 70%).
Стадия 3. Получение (Е)-N'-((1Н-индол-6-ил)метилен)-2-(4-фтор-2-метилфенокси)ацетогидразида: 2-(4-фтор-2-метилфенокси)ацетогидразид (100 мг, 0,50 ммоль) и 1H-индол-6-карбальдегид (73 мг, 0,50 ммоль) растворили в EtOH (2 мл), после чего перемешивали при 90°С в течение 4 часов. К реакционной смеси добавили воду, и проэкстрагировали с помощью этилацетата. Полученный органический слой высушили над сульфатом магния, и очистили с помощью колоночной хроматографии (гексан:этил ацетат = 2:1) с получением соединения 291 (37 мг, 23%).
1H NMR (400 MHz, DMSO-d6): δ 11,43-11,41 (m, 1H), 11,34-11,30 (m, 1H), 8,34-8,07 (2s, 1H), 7,72 (d, J=27,7 Hz, 1H), 7,64 (m, 1H), 7,44 (m, 2H), 6,95 (m, 3H), 5,15 (s, H), 4,63 (s, H), 2,22 (s, 3H)
Пример 57. Синтез соединения 293
Стадия 1. Получение метил 2-(2-этил-6-метилпиридин-3-илокси)ацетата: 2-этил-6-метилпиридин-3-ол (500 мг, 3,62 ммоль), метил 2-бромацетат (0,33 мл, 3,62 ммоль) и карбонат цезия (1,78 г, 5,43 ммоль) растворили в ацетонитриле 5 мл, после чего перемешивали при 100°С в течение 5 часов. По окончании реакции, к реакционной смеси добавили воду, и проэкстрагировали с помощью этилацетата. Полученный органический слой высушили над сульфатом магния, и очистили с помощью колоночной хроматографии (гексан:этил ацетат = 3:1) с получением метил 2-(2-этил-6-метилпиридин-3-илокси)ацетата (565 мг, 70%).
Стадия 2. Получение 2-(2-этил-6-метилпиридин-3-илокси)ацетогидразида:
метил 2-(2-этил-6-метилпиридин-3-илокси)ацетат (400 мг, 1,79 ммоль) и гидразин (0,1 мл, 2,15 ммоль) растворили в EtOH 4 мл, после чего перемешивали при 90°С в течение 5 часов. По окончании реакции, реакционную смесь отфильтровали под пониженным давлением с получением 2-(2-этил-6-метилпиридин-3-илокси)ацетогидразида (242 мг, 65%).
Стадия 3. Получение (E)-N'-((1Н-индол-6-ил)метилен)-2-(2-этил-6-метилпиридин-3-илокси)ацетогидразида: 2-(2-этил-6-метилпиридин-3-илокси)ацетогидразид (100 мг, 0,50 ммоль) и 1H-индол-6-карбальдегид (73 мг, 0,50 ммоль) растворили в EtOH (3 мл), после чего перемешивали при 90°С в течение 4 часов. По окончании реакции, образовавшийся осадок отфильтровали с получением соединения 293 (46 мг, 29%).
1Н NMR (400 MHz, DMSO-d6): δ 11,43 (bs, 1H), 11,30-11,27 (s, 1H), 8,32-8,06 (2s, 1H), 7,64 (m, 1H), 7,42 (m, 2H), 7,36 (m, 1H), 7,02 (m, 1H), 6,45 (s, 1H), 5,17 (s, 1H), 4,66 (s, 1H), 2,75 (m, 2H), 2,45 (s, 3H), 1,21 (m, 3H).
Пример 58. Синтез соединения 301
Стадия 1. Получение метил 2-(2,6-диметилпиримидин-4-илокси)ацетата:
2,6-диметилпиримидин-4-ол (300 мг, 2,42 ммоль), метил бромацетат (0,22 мл, 2,90 ммоль) и карбонат цезия (945 мг, 2,90 ммоль) растворили в ацетонитриле (4 мл), после чего перемешивали при 80°С в течение 2 часов. По окончании реакции, реакционную смесь отфильтровали под пониженным давлением. Фильтрат упарили при пониженном давлении, и очистили с помощью колоночной хроматографии (этил ацетат:гексан = 1:3) с получением метил 2-(2,6-диметилпиримидин-4-илокси)ацетата (170 мг, 36%).
Стадия 2. Получение 2-(2,6-диметилпиримидин-4-илокси)ацетогидразида:
метил 2-(2,6-диметилпиримидин-4-илокси)ацетат (170 мг, 0,87 ммоль) и гидразин (0,05 мл, 0,95 ммоль) растворили в EtOH (2 мл), после чего перемешивали при 100°С в течение 12 часов. По окончании реакции, реакционную смесь упарили при пониженном давлении с получением 2-(2,6-диметилпиримидин-4-илокси)ацетогидразида (149 мг, 88%).
Стадия 3. Получение (E)-N'-((1Н-индол-4-ил)метилен)-2-(2,6-диметилпиримидин-4-илокси)ацетогидразида: 2-(2,6-диметилпиримидин-4-илокси)ацетогидразид (57 мг, 0,29 ммоль) и 1Н-индол-4-карбальдегид (42 мг, 0,30 ммоль) растворили в EtOH 1 мл, после чего перемешивали при 100°С в течение 12 часов. По окончании реакции, образовавшийся осадок отфильтровали с получением соединения 301 (20 мг, 21%).
1Н NMR (400 MHz, DMSO-d6): δ 11,51-11,33 (m, 2H), 8,47-8,24 (m, 1H), 7,49 (m, 2H), 7,19 (m, 2H), 7,10 (m, 1H), 6,71 (m, 1H), 5,47 (s, 1H), 4,94 (s, 1H).
Пример 59. Синтез соединения 302
Стадия 1. Получение метил 2-(2-трет-бутил-4-метоксифенокси)ацетата:
2-трет-бутил-4-метоксифенол (300 мг, 1,67 ммоль), метил бромацетат (0,18 мл, 2,00 ммоль) и карбонат цезия (653 мг, 2,00 ммоль) растворили в ацетонитриле (4 мл), после чего перемешивали при 80°С в течение 1 часа. По окончании реакции, реакционную смесь отфильтровали под пониженным давлением. Фильтрат упарили при пониженном давлении, и очистили с помощью колоночной хроматографии (этил ацетат:гексан = 1:3) с получением метил 2-(2-трет-бутил-4-метоксифенокси)ацетата (259 мг, 62%).
Стадия 2. Получение 2-(2-трет-бутил-4-метоксифенокси)ацетогидразида:
метил 2-(2-трет-бутил-4-метоксифенокси)ацетат (259 мг, 1,03 ммоль) и гидразин (0,06 мл, 1,13 ммоль) растворили в EtOH (2 мл), после чего перемешивали при 80°С в течение 12 часов. По окончании реакции, реакционную смесь упарили при пониженном давлении с получением 2-(2-трет-бутил-4-метоксифенокси)ацетогидразида (231 мг, 89%).
Стадия 3. Получение (Е)-N'-((1Н-индол-4-ил)метилен)-2-(2-трет-бутил-4-метоксифенокси)ацетогидразида: 2-(2-трет-бутил-4-метоксифенокси)ацетогидразид (90 мг, 0,36 ммоль) и 1H-индол-4-карбальдегид (57 мг, 0,39 ммоль) растворили в EtOH (2 мл), после чего перемешивали при 90°С в течение 12 часов. По окончании реакции, образовавшийся осадок отфильтровали с получением соединения 302 (60 мг, 44%).
1Н NMR (400 MHz, DMSO-d6): δ 11,50 (m, 1H), 11,37 (m, 1H), 8,47-8,27 (2s, 1H), 7,49 (m, 2H), 7,16 (m, 2H), 6,95 (m, 1H), 6,80 (m, 1H), 6,74 (m, 2H), 5,13 (s, 1H), 4,12 (s, 1H), 3,69 (s, 3H), 1,38 (s, 9H).
Пример 60. Синтез соединения 303
2-(2-трет-бутил-4-метоксифенокси)ацетогидразид (90 мг, 0,36 ммоль) и 1H-индол-6-карбальдегид (57 мг, 0,39 ммоль) растворили в EtOH (2 мл), после чего перемешивали при 90°С в течение 12 часов. По окончании реакции, к реакционной смеси добавили воду, и проэкстрагировали с помощью этилацетата. Полученный органический слой высушили над сульфатом магния, и очистили с помощью колоночной хроматографии (этил ацетат:гексан = 1:2) с получением соединения 303 (66 мг, 48%).
1H NMR (400 MHz, DMSO-d6): δ 11,41 (m, 1H), 11,26 (m, 1H), 8,31-8,08 (2s, 1H), 7,71 (m, 1H), 7,56 (m, 1H), 7,38 (m, 2H), 6,85 (m, 1H), 6,74 (m, 2H), 6,46 (m, 2H), 5,06 (s, 1H), 4,58 (s, 1H), 3,69 (s, 3H), 1,36 (s, 9H).
Пример 61. Синтез соединения 304
Стадия 1. Получение метил 2-((2-метилпиридин-3-ил)окси)ацетата:
3-гидрокси-2-метилпиридин (500 мг, 4,58 ммоль) и метил бромацетат (0,48 мл, 5,04 ммоль) растворили в ацетонитриле. Добавили карбонат цезия (2,24 г, 6,82 ммоль), после чего перемешивали при комнатной температуре в течение 1 дня. По окончании реакции, к реакционной смеси добавили насыщенный водный раствор гидрокарбоната натрия, и проэкстрагировали с помощью этилацетата. Полученный органический слой промыли насыщенным солевым раствором, высушили над безводным сульфатом натрия, отфильтровали, и упарили при пониженном давлении. Концентрированный остаток очистили с помощью колоночной хроматографии (силикагель; гексан/этил ацетат, 6/4) с получением метил 2-((2-метилпиридин-3-ил)окси)ацетата (356 мг, 43%).
Стадия 2. Получение 2-((2-метилпиридин-3-ил)окси)ацетогидразида:
Соединение 1-2 (метил 2-((2-метилпиридин-3-ил)окси)ацетат, 356 мг, 1,94 ммоль) и гидразин моногидрат (0,19 мл, 3,93 ммоль) растворили в этаноле, после чего перемешивали в течение 1 дня при кипячении с обратным холодильником. По окончании реакции, реакционную смесь упарили при пониженном давлении. Концентрированный остаток высушили и очистили с получением 2-((2-метилпиридин-3-ил)окси)ацетогидразида (240 мг, 67%) в виде твердого вещества белого цвета.
Стадия 3. Получение (E)-N'-((1Н-индол-6-ил)метилен)-2-((2-метилпиридин-3-ил)окси)ацетогидразида: 2-((2-метилпиридин-3-ил)окси)ацетогидразид (240 мг, 1,32 ммоль) и индол-6-карбоксальдегид (211,6 мг, 1,46 ммоль) растворили в этаноле, после чего перемешивали в течение ночи при кипячении с обратным холодильником. По окончании реакции, реакционную смесь упарили при пониженном давлении. Концентрированный остаток очистили с помощью колоночной хроматографии (силикагель; метилен хлорид/метанол, 10/1) с получением соединения 304 (46 мг, 11%) в виде твердого вещества коричневого цвета.
1Н NMR (400 MHz, DMSO-d6): δ 11,47 (m, 1H), 11,29 (m, 1H). 8,34-8,00 (m, 2H), 7,69 (d, 1H, J=23,4 Hz), 7,58 (d, 1H, J=8,2 Hz), 7,47-7,32 (m, 2H), 7,29-7,23 (m, 1H), 7,21-7,13 (m, 1H), 6,47 (s, 1H), 5,24-4,73 (m, 2H), 2,43 (s, 3H).
Пример 62. Синтез соединения 310
Стадия 1. Получение метил 2-(2-(фуран-3-ил)пиридин-3-илокси)ацетата:
2-(фуран-3-ил)пиридин-3-ол (Соединение 6-10) (100 мг, 0,62 ммоль), метил бромацетат (0,07 мл, 0,75 ммоль) и карбонат цезия (244 мг, 0,75 ммоль) растворили в ацетонитриле (4 мл), после чего перемешивали при 80°С в течение 12 часов. По окончании реакции, реакционную смесь отфильтровали под пониженным давлением, и упарили при пониженном давлении. Концентрат очистили с помощью колоночной хроматографии (этил ацетат:гексан = 1:3) с получением метил 2-(2-(фуран-3-ил)пиридин-3-илокси)ацетата (128 мг, 89%).
Стадия 2. Получение 2-(2-(фуран-3-ил)пиридин-3-илокси)ацетогидразида:
метил 2-(2-(фуран-3-ил)пиридин-3-илокси)ацетат (128 мг, 0,54 ммоль) и гидразин (0,03 мл, 0,6 ммоль) растворили в EtOH (2 мл), после чего перемешивали при 90°С в течение 6 часов. По окончании реакции, реакционную смесь упарили при пониженном давлении с получением 2-(2-(фуран-3-ил)пиридин-3-илокси)ацетогидразида (108 мг, 84%).
Стадия 3. Получение (E)-N'-((1Н-индол-6-ил)метилен)-2-(2-(фуран-3-ил)пиридин-3-илокси)ацетогидразида: 2-(2-(фуран-3-ил)пиридин-3-илокси)ацетогидразид (100 мг, 0,43 ммоль) и 1H-индол-6-карбальдегид (70 мг, 0,44 ммоль) растворили в EtOH (2 мл), после чего перемешивали при 100°С в течение 12 часов. По окончании реакции, к реакционной смеси добавили воду, и проэкстрагировали с помощью этилацетата. Полученный органический слой высушили над сульфатом магния, и очистили с помощью колоночной хроматографии (этил ацетат:гексан = 3:7) с получением соединения 310 (60 мг, 39%).
1H NMR (400 MHz, DMSO-d6): δ 11,57 (bs, 1H), 11,30 (bs, 1H), 8,78-8,57 (2s, 1H), 8,30 (s, 0,2H), 8,21 (m, 1H), 8,11 (s, 1H), 7,75 (m, 1H), 7,72 (s, 0,9H), 7,59-7,40 (m, 5H), 7,26 (m, 1H), 7,16 (m, 1H), 6,47 (m, 1H), 5,36 (s, 1H), 4,87 (s, 1H).
Пример 63. Синтез соединения 311
Стадия 1. Получение метил 2-(2-метил-6-(тетрагидрофуран-3-ил)фенокси)ацетата:
2-метил-6-(тетрагидрофуран-3-ил)фенол (Соединение 7-3) (117 мг, 0,66 ммоль), метил бромацетат (0,07 мл, 0,79 ммоль) и карбонат цезия (258 мг, 0,79 ммоль) растворили в ацетонитриле (2 мл), после чего перемешивали при 100°С в течение 2 часов. По окончании реакции, реакционную смесь отфильтровали под пониженным давлением, и упарили при пониженном давлении. Концентрат очистили с помощью колоночной хроматографии (этил ацетат:гексан = 1:3) с получением метил 2-(2-метил-6-(тетрагидрофуран-3-ил)фенокси)ацетата (141 мг, 86%).
Стадия 2. Получение 2-(2-метил-6-(тетрагидрофуран-3-ил)фенокси)ацетогидразида:
метил 2-(2-метил-6-(тетрагидрофуран-3-ил)фенокси)ацетат (141 мг, 0,56 ммоль) и гидразин (0,03 мл, 0,6 ммоль) растворили в EtOH (2 мл), после чего перемешивали при 100°С в течение 12 часов. По окончании реакции, реакционную смесь упарили при пониженном давлении с получением 2-(2-(фуран-3-ил)-6-метилфенокси)ацетогидразида (127 мг, 90%).
Стадия 3. Получение (E)-N'-((1Н-индол-6-ил)метилен)-2-(2-метил-6-(тетрагидрофуран-3-ил)фенокси)ацетогидразида: 2-(2-(фуран-3-ил)-6-метилфенокси)ацетогидразид (70 мг, 0,28 ммоль) и 1H-индол-6-карбальдегид (45 мг, 0,31 ммоль) растворили в EtOH (2 мл), после чего перемешивали при 100°С в течение 12 часов. По окончании реакции, к реакционной смеси добавили воду, и проэкстрагировали с помощью этилацетата. Полученный органический слой высушили над сульфатом магния, и очистили с помощью колоночной хроматографии (этил ацетат:гексан = 1:1) с получением соединения 311 (70 мг, 67%).
1H NMR (400 MHz, DMSO-d6): δ 11,31 (m, 1H), 11,20 (m, 1H), 8,47-8,04 (2s, 1H), 7,71 (s, 0,5H), 7,58 (m, 1H), 7,58-7,38 (m, 3H), 7,27 (m, 0,5H), 7,17 (m, 1H), 7,06 (m, 2H), 6,45 (m, 1H), 4,81 (s, 1H), 4,04 (m, 1H), 3,97 (m, 2H), 3,78 (m, 2H), 3,52 (m, 1H), 2,29 (m, 3H), 2,29 (m, 1H), 1,19 (m, 1H).
Пример 64. Синтез соединения 312
2-(2-(фуран-3-ил)-6-метилфенокси)ацетогидразид (70 мг, 0,28 ммоль) и 1Н-индол-4-карбальдегид (45 мг, 0,31 ммоль) растворили в EtOH (2 мл), после чего перемешивали при 100°С в течение 12 часов. По окончании реакции, к реакционной смеси добавили воду, и проэкстрагировали с помощью этилацетата. Полученный органический слой высушили над сульфатом магния, и очистили с помощью колоночной хроматографии (этил ацетат:гексан = 1:1) с получением соединения 312 (70 мг, 67%).
1H NMR (400 MHz, DMSO-d6): δ 11,35 (m, 1H), 11,3 (m, 1H), 8,66-8,23 (2s, 1H), 7,46 (m, 2H), 7,25 (m, 0,5H), 7,16 (m, 5H), 6,77 (m, 0,5H), 4,90 (s, 1H), 4,40 (s, 1H), 4,03 (m, 1H), 3,77 (m, 2H), 3,54 (m, 1H), 3,34 (m, 1H), 2,29 (s, 3H), 2,29 (m, 1H), 1,89 (m, 1H).
Пример 65. Синтез соединения 313
Стадия 1. Получение метил 2-(2-(фуран-3-ил)-6-метилфенокси)ацетата:
2-(фуран-3-ил)-6-метилфенол (Соединение 6-9) (100 мг, 0,57 ммоль), метил бромацетат (0,04 мл, 0,68 ммоль) и карбонат цезия (160 мг, 0,68 ммоль) растворили в ацетонитриле (4 мл), после чего перемешивали при комнатной температуре в течение 12 часов. По окончании реакции, реакционную смесь отфильтровали под пониженным давлением. Фильтрат упарили при пониженном давлении, и очистили с помощью колоночной хроматографии (этил ацетат:гексан = 1:3) с получением метил 2-(2-(фуран-3-ил)-6-метилфенокси)ацетата (146 мг, 100%).
Стадия 2. Получение 2-(2-(фуран-3-ил)-6-метилфенокси)ацетогидразида:
метил 2-(2-(фуран-3-ил)-6-метилфенокси)ацетат (146 мг, 0,57 ммоль) и гидразин (0,03 мл, 0,6 ммоль) растворили в EtOH (2 мл), после чего перемешивали при 100°С в течение 6 часов. По окончании реакции, реакционную смесь упарили при пониженном давлении с получением 2-(2-(фуран-3-ил)-6-метилфенокси)ацето-гидразида (121 мг, 83%).
Стадия 3. Получение (E)-N'-((1Н-индол-6-ил)метилен)-2-(2-(фуран-3-ил)-6-метилфенокси)ацетогидразида: 2-(2-(фуран-3-ил)-6-метилфенокси)ацетогидразид (70 мг, 0,28 ммоль) и 1H-индол-6-карбальдегид (45 мг, 0,31 ммоль) растворили в EtOH (2 мл), после чего перемешивали при 100°С в течение 12 часов. По окончании реакции, к реакционной смеси добавили воду, и проэкстрагировали с помощью этилацетата. Полученный органический слой высушили над сульфатом магния, и очистили с помощью колоночной хроматографии (этил ацетат:гексан = 1:1) с получением соединения 313 (20 мг, 19%).
1Н NMR (400 MHz, DMSO-d6): δ 11,41 (m, 2H), 8,45-8,01 (2s, 1H), 8,30 (m, 1H), 7,72 (m, 1H), 7,46 (m, 4H), 7,10 (m, 4H), 6,44 (m, 1H), 4,70 (s, 1H), 4,25 (s, 1H), 2,30 (m, 3H).
Пример 66. Синтез соединения 314
2-(2-(фуран-3-ил)-6-метилфенокси)ацетогидразид (70 мг, 0,28 ммоль) и 1Н-индол-4-карбальдегид (45 мг, 0,31 ммоль) растворили в EtOH (2 мл), после чего перемешивали при 100°С в течение 12 часов. По окончании реакции, к реакционной смеси добавили воду, и проэкстрагировали с помощью этилацетата. Полученный органический слой высушили над сульфатом магния, и очистили с помощью колоночной хроматографии (этил ацетат:гексан = 1:1) с получением соединения 314 (28 мг, 26%).
1H NMR (400 MHz, DMSO-d6): δ 11,44 (m, 1H), 11,34 (m, 1H), 8,64 (s, 1H), 8,18 (s, 1H), 8,32 (m, 1H), 7,75 (m, 1H), 7,47 (m, 3H), 7,27-7,00 (m, 5,5H), 6,57 (s, 0,5H), 4,78 (s, 1H), 4,29 (s, 1H), 2,37 (m, 3H).
Пример 67. Синтез соединения 317
Стадия 1. Получение метил 2-(2-(фуран-2-ил)-4,6-диметилфенокси)ацетата: 2-(фуран-2-ил)-4,6-диметилфенол (Соединение 6-18) (133 мг, 0,71 ммоль), метил 2-бромацетат (0,08 мл, 0,85 ммоль) и карбонат цезия (276 мг, 0,85 ммоль) растворили в ацетонитриле (1 мл), после чего перемешивали при 100°С в течение 1 часа. По окончании реакции, к реакционной смеси добавили воду, и проэкстрагировали с помощью этилацетата. Полученный органический слой высушили над сульфатом магния, и очистили с помощью колоночной хроматографии (этил ацетат:гексан = 1:4) с получением метил 2-(2-(фуран-2-ил)-4,6-диметилфенокси)ацетата (168 мг, 100%).
Стадия 2. Получение 2-(2-(фуран-2-ил)-4,6-диметилфенокси)ацетогидразида: Метил 2-(2-(фуран-2-ил)-4,6-диметилфенокси)ацетат (168 мг, 0,65 ммоль) и гидразин (0,03 мл, 0,72 ммоль) растворили в EtOH (2 мл), после чего перемешивали при 100°С в течение 12 часов. По окончании реакции, реакционную смесь упарили при пониженном давлении с получением 2-(2-(фуран-2-ил)-4,6-диметилфенокси)ацетогидразида (169 мг, 100%).
Стадия 3. Получение (E)-N'-((1Н-индол-4-ил)метилен)-2-(2-(фуран-2-ил)-4,6-диметилфенокси)ацетогидразида: 2-(2-(фуран-2-ил)-4,6-диметилфенокси)ацетогидразид (85 мг, 0,33 ммоль) и 1Н-индол-4-карбальдегид (52 мг, 0,36 ммоль) растворили в EtOH (2 мл), после чего перемешивали при 100°С в течение 24 часов. По окончании реакции, образовавшийся осадок отфильтровали с получением соединения 317 (140 мг, 31%).
1H NMR (400 MHz, DMSO-d6): δ 11,57 (m, 1H), 11,46 (m, 1H), 8,64-8,24 (2s, 1H), 7,75 (m, 1H), 7,47 (m, 3H), 7,11 (m, 4,5H), 6,62 (m, 1,5H), 4,80 (s, 1H), 4,30 (s, 1H), 2,32 (m, 6H).
Пример 68. Синтез соединения 318
2-(2-(фуран-2-ил)-4,6-диметилфенокси)ацетогидразид (85 мг, 0,33 ммоль) и 1H-индол-6-карбальдегид (52 мг, 0,36 ммоль) растворили в EtOH (2 мл), после чего перемешивали при 90°С в течение 24 часов. По окончании реакции, к реакционной смеси добавили воду, и проэкстрагировали с помощью этилацетата. Полученный органический слой высушили над сульфатом магния, и очистили с помощью колоночной хроматографии (этил ацетат:гексан = 45:55) с получением соединения 318 (30 мг, 23%).
1H NMR (400 MHz, DMSO-d6): δ 11,44 (m, 1H), 11,20 (m, 1H), 8,47-8,02 (2s, 1H), 7,73 (m, 1H), 7,44 (m, 4H), 7,23 (m, 0,5H), 7,14 (m, 1H), 7,02 (m, 1H), 6,60 (m, 1H), 6,43 (m, 1H), 4,72 (s, 1H), 4,44 (s, 1H), 2,32 (m, 6H).
Пример 69. Синтез соединения 319
2-(2-(фуран-3-ил)пиридин-3-илокси)ацетогидразид (70 мг, 0,30 ммоль) и 1Н-индол-4-карбальдегид (48 мг, 0,33 ммоль) растворили в EtOH (2 мл), после чего перемешивали при 80°С в течение 12 часов. По окончании реакции, к реакционной смеси добавили воду, и проэкстрагировали с помощью этилацетата. Полученный органический слой высушили над сульфатом магния, и очистили с помощью колоночной хроматографии (этил ацетат:гексан = 1:1). Полученное вещество перекристаллизовали из смеси тетрагидрофуран/гексан, и отфильтровали с получением соединения 319 (50 мг, 46%).
1Н NMR (400 MHz, DMSO-d6): δ 11,62 (m, 1H), 11,39 (m, 1H), 8,74-8,46 (m, 1H), 8,30 (s, 1H), 8,21 (m, 1H), 7,74 (m, 1H), 7,52 (m, 3H), 7,20 (m, 4H), 7,01 (m, 1H), 5,43 (s, 1H), 4,90 (s, 1H).
Пример 70. Синтез соединения 320
2-(2,4-диметил-6-(тетрагидрофуран-3-ил)фенокси)ацетогидразид (40 мг, 0,15 ммоль) и 1Н-индол-4-карбальдегид (22 мг, 0,15 ммоль) растворили в EtOH (2 мл), после чего перемешивали при 90°С в течение 4 часов. По окончании реакции, реакционную смесь упарили при пониженном давлении, и очистили с помощью колоночной хроматографии (этил ацетат:гексан = 1:1) с получением соединения 320 (17 мг, 29%).
1H NMR (400 MHz, DMSO-d6): δ 11,45 (bs, 1H), 11,35 (bs, 1H), 8,45 (s, 1H), 8,25-8,16 (2s, 1H), 7,45 (m, 2H), 7,25 (m, 3H), 7,17 (m, 1H), 7,11 (m, 1H), 6,96 (m, 1H), 5,49-4,94 (s, 2H), 4,01 (m, 2H), 3,77 (m, 2H), 3,51 (m, 1H), 2,28 (s, 3H), 2,22 (s, 3H).
Пример 71. Синтез соединения 322
Стадия 1. Получение метил 2-(4-(фуран-3-ил)-2,6-диметилфенокси)ацетата: 4-(фуран-3-ил)-2,6-диметилфенол (Соединение 6-5) (120 мг, 0,46 ммоль), метил 2-бромацетат (0,05 мл, 0,55 ммоль) и карбонат цезия (448 мг, 1,39 ммоль) растворили в ацетонитриле (2 мл), после чего перемешивали при 90°С в течение 3 часов. По окончании реакции, к реакционной смеси добавили воду, и проэкстрагировали с помощью этилацетата. Полученный органический слой высушили над сульфатом магния, и очистили с помощью колоночной хроматографии (гексан:этил ацетат = 9:1) с получением 2-(4-(фуран-3-ил)-2,6-диметилфенокси)ацетата (130 мг, 79%).
Стадия 2. Получение 2-(4-(фуран-3-ил)-2,6-диметилфенокси)ацетогидразида:
2-(4-(фуран-3-ил)-2,6-диметилфенокси)ацетат (125 мг, 0,48 ммоль) и гидразин (0,024 мл, 0,48 ммоль) растворили в EtOH (2 мл), после чего перемешивали при 90°С в течение 2 часов. По окончании реакции, реакционную смесь отфильтровали под пониженным давлением, и разбавили диэтиловым эфиром (10 мл), после чего перемешивали в течение 10 минут. Образовавшийся осадок отфильтровали с получением 2-(4-(фуран-3-ил)-2,6-диметилфенокси)ацето-гидразида (102 мг, 82%).
Стадия 3. Получение (Е)-N'-((1Н-индол-6-ил)метилен)-2-(4-(фуран-3-ил)-2,6-диметилфенокси)ацетогидразида: 2-(4-(фуран-3-ил)-2,6-диметилфенокси)ацетогидразид (40 мг, 0,15 ммоль) и 1Н-индол-6-карбальдегид (22 мг, 0,15 ммоль) растворили в EtOH (2 мл), после чего перемешивали при 90°С в течение 3 часов. По окончании реакции, реакционную смесь упарили при пониженном давлении, и очистили с помощью колоночной хроматографии (гексан:этил ацетат = 1:1) с получением соединения 322 (19 мг, 32%).
1Н NMR (400 MHz, DMSO-d6): δ 11,41-11,38 (m, 1H), 11,30-11,20 (m, 1H), 8,50-8,04 (2s, 1H), 8,08 (m, 1H), 7,88 (m, 1H), 7,61 (m, 1H), 7,48-7,37 (m, 2H), 7,38 (m, 1H), 7,27 (m, 3H), 6,89 (m, 1H), 6,47 (m, 1H), 4,81 (s, 1H), 4,34 (s, 1H), 2,07 (m, 6H).
Пример 72. Синтез соединения 323
Стадия 1. Получение метил 2-(2,6-диметил-4-(тетрагидрофуран-3-ил)фенокси)ацетата:
2,6-диметил-4-(тетрагидрофуран-3-ил)фенол (Соединение 7-6) (68 мг, 0,35 ммоль), метил 2-бромацетат (0,04 мл, 0,45 ммоль) и карбонат цезия (341 мг, 1,05 ммоль) растворили в ацетонитриле (2 мл), после чего перемешивали при 100°С в течение 4 часов. По окончании реакции, реакционную смесь отфильтровали под пониженным давлением. Фильтрат упарили при пониженном давлении, и очистили с помощью колоночной хроматографии (гексан:этил ацетат = 9:1) с получением метил 2-(2,6-диметил-4-(тетрагидро-фуран-3-ил)фенокси)ацетата (74 мг, 79%).
Стадия 2. Получение 2-(2,6-диметил-4-(тетрагидрофуран-3-ил)фенокси)ацетогидразида:
метил 2-(2,6-диметил-4-(тетрагидрофуран-3-ил)фенокси)ацетат (67 мг, 0,25 ммоль) и гидразин (0,012 мл, 0,25 ммоль) растворили в EtOH (2 мл), после чего перемешивали при 90°С в течение 2 часов. По окончании реакции, реакционную смесь упарили при пониженном давлении, разбавили диэтиловым эфиром 10 мл, и перемешивали в течение 10 минут. Образовавшийся осадок отфильтровали с получением 2-(2,6-диметил-4-(тетрагидрофуран-3-ил)фенокси)ацетогидразида (45 мг, 66%).
Стадия 3. Получение (Е)-N'-((1Н-индол-6-ил)метилен)-2-(2,6-диметил-4-(тетрагидрофуран-3-ил)фенокси)ацетогидразида: 2-(2,6-диметил-4-(тетрагидрофуран-3-ил)фенокси)ацетогидразид (30 мг, 0,11 ммоль) и 1H-индол-6-карбальдегид (17 мг, 0,11 ммоль) растворили в EtOH 1 мл, после чего перемешивали при 90°С в течение 3 часов. По окончании реакции, реакционную смесь упарили при пониженном давлении, и очистили с помощью колоночной хроматографии (гексан:этил ацетат = 1:1) с получением соединения 323 (15 мг, 34%).
1H NMR (400 MHz, DMSO-d6): δ 11,42-11,39 (m, 1H), 11,37-11,32 (m, 1H), 8,47-8,03 (s, 1H), 7,57-7,48 (m, 2H), 7,40-7,37 (m, 2H), 6,95 (m, 2H), 6,47 (m, 1H), 4,74 (s, 1H), 4,31 (s, 1H), 3,96 (m, 3H), 3,75 (m, 2H), 2,32 (s, 3H), 2,21 (s, 3H).
Пример 73. Синтез соединения 326
Стадия 1. Получение метил 2-(2,3-диметилфенокси)ацетата:
2,3-диметилфенол (500 мг, 4,09 ммоль), метил 2-бромацетат (0,45 мл, 4,90 ммоль) и карбонат цезия (4,0 г, 12,3 ммоль) растворили в ацетонитриле (8 мл), после чего перемешивали при 100°С в течение 3 часов. По окончании реакции, реакционную смесь отфильтровали под пониженным давлением. Фильтрат упарили при пониженном давлении, и очистили с помощью колоночной хроматографии (гексан:этил ацетат = 9:1) с получением метил 2-(2,3-диметилфенокси)ацетата (628 мг, 83%).
Стадия 2. Получение 2-(2,3-диметилфенокси)ацетогидразида:
метил 2-(2,3-диметилфенокси)ацетат (600 мг, 3,09 ммоль) и гидразин (0,15 мл, 3,09 ммоль) растворили в EtOH 8 мл, после чего перемешивали при 90°С в течение 2 часов. По окончании реакции, реакционную смесь упарили при пониженном давлении, разбавили диэтиловым эфиром 10 мл, и перемешивали в течение 10 минут. Образовавшийся осадок отфильтровали с получением 2-(2,3-диметилфенокси)ацетогидразида (572 мг, 95%). Стадия 3. Получение (Е)-N'-((1Н-индол-4-ил)метилен)-2-(2,3-диметилфенокси)ацетогидразида: 2-(2,3-диметилфенокси)ацетогидразид (100 мг, 0,55 ммоль) и 1H-индол-4-карбальдегид (75 мг, 0,52 ммоль) растворили в EtOH (3 мл), после чего перемешивали при 90°С в течение 2 часов. По окончании реакции, реакционную смесь упарили при пониженном давлении, и очистили с помощью колоночной хроматографии (гексан:этил ацетат = 1:1) с получением соединения 326 (93 мг, 56%).
1Н NMR (400 MHz, DMSO-d6): δ 11,50-11,44 (m, 1H), 11,37-11,32 (m, 1H), 8,52-8,25 (2s, 1H), 7,50 (m, 2H), 7,23 (m, 2H), 7,06 (m, 2H), 6,81 (m, 1H), 6,75 (m, 1H), 5,18 (s, 1H), 4,65 (s, 1H), 2,23 (s, 3H), 2,21 (s, 3H).
Пример 74. Синтез соединения 327
2-(2,3-диметилфенокси)ацетогидразид (100 мг, 0,55 ммоль) и 1Н-индол-4-карбальдегид (75 мг, 0,52 ммоль) растворили в EtOH (3 мл), после чего перемешивали при 90°С в течение 2 часов. По окончании реакции, реакционную смесь упарили при пониженном давлении, и очистили с помощью колоночной хроматографии (гексан:этил ацетат = 1:1) с получением соединения 327 (104 мг, 62%).
1Н NMR (400 MHz, DMSO-d6): δ 11,41 (bs, 1H), 11,27 (bs, 1H), 8,34-8,07, (2s, 1H), 7,69 (d, J=22,9 Hz, 1H), 7,56 (m, 1H), 7,41 (m, 2H), 6,98 (m, 1H), 6,70 (m, 2H), 6,46 (m, 1H), 4,82 (d, 2H), 2,49 (s, 3H), 2,23 (s, 3H).
Пример 75. Синтез соединения 329
2-(2-этил-6-метилпиридин-3-илокси)ацетогидразид (30 мг, 0,14 ммоль)и 1Н-индол-4-карбальдегид (21 мг, 0,37 ммоль) растворили в EtOH 1 мл, после чего перемешивали при 90°С в течение 4 часов. По окончании реакции, образовавшийся осадок отфильтровали с получением соединения 329 (19 мг, 40%).
1Н NMR (400 MHz, DMSO-d6): δ 11,52 (bs, 1H), 11,37 (bs, 1H), 8,49-8,25 (2s, 1H), 7,48 (m, 2H), 7,17 (m, 3H), 6,98 (m, 2H), 5,23 (s, 1H), 4,70 (s, 1H), 2,78 (m, 2H), 2,35 (s, 3H), 1,18 (m, 3H).
Пример 76. Синтез соединения 330
Стадия 1. Получение метил 2-(4-бром-2-цианофенокси)ацетата:
5-бром-2-гидроксибензонитрил (400 мг, 2,01 ммоль), метил 2-бромацетат (0,2 мл, 2,2 ммоль) и карбонат цезия (716 мг, 2,2 ммоль) растворили в ацетонитриле (4 мл), после чего перемешивали при 80°С в течение 2 часов. По окончании реакции, реакционную смесь отфильтровали под пониженным давлением. Фильтрат упарили при пониженном давлении, и очистили с помощью колоночной хроматографии (этил ацетат:гексан = 1:3) с получением метил 2-(4-бром-2-цианофенокси)ацетата (451 мг, 83%).
Стадия 2. Получение 2-(4-бром-2-цианофенокси)ацетогидразида:
метил 2-(4-бром-2-цианофенокси)ацетат (451 мг, 1,67 ммоль) и гидразин (0,09 мл, 1,84 ммоль) растворили в EtOH (2 мл), после чего перемешивали при 100°С в течение 12 часов. По окончании реакции, образовавшийся осадок отфильтровали с получением 2-(4-бром-2-цианофенокси)ацетогидразида (398 мг, 88%).
Стадия 3. Получение (Е)-N'-((1Н-индол-4-ил)метилен)-2-(4-бром-2-цианофенокси)ацетогидразида: 2-(4-бром-2-цианофенокси)ацетогидразид (100 мг, 0,37 ммоль) и 1Н-индол-4-карбальдегид (53 мг, 0,39 ммоль) растворили в EtOH (2 мл), после чего перемешивали при 90°С в течение 12 часов. По окончании реакции, к реакционной смеси добавили воду, и проэкстрагировали с помощью этилацетата. Полученный органический слой высушили над сульфатом магния, и очистили с помощью колоночной хроматографии (этил ацетат:гексан = 2:1) с получением соединения 330 (21 мг, 0,05 ммоль, 14%).
1H NMR (400 MHz, DMSO-d6): δ 11,6 (bs, 1H), 11,37 (m, 1H), 8,45-8,26 (2s, 1H), 8,03 (m, 1H), 7,78 (m, 1H), 7,47 (m, 2H), 7,16 (m, 3H), 6,98 (m, 1H), 5,47 (s, 1H), 4,93 (s, 1H).
Пример 77. Синтез соединения 331
Стадия 1. Получение метил 2-(2-(пирролидин-1-ил)пиридин-3-илокси)ацетата:
2-(пирролидин-1-ил)пиридин-3-ол (Соединение 7-9) (78 мг, 0,41 ммоль), метил бромацетат (0,05 мл, 0,49 ммоль) и карбонат цезия (159 мг, 0,49 ммоль) растворили в ацетонитриле (2 мл), после чего перемешивали при 80°С в течение 3 часов. По окончании реакции, реакционную смесь отфильтровали под пониженным давлением. Фильтрат упарили при пониженном давлении, и очистили с помощью колоночной хроматографии (этил ацетат:гексан = 1:5) с получением метил 2-(2-(пирролидин-1-ил)пиридин-3-илокси)ацетата (86 мг, 88%).
Стадия 2. Получение 2-(2-(пирролидин-1-ил)пиридин-3-илокси)ацетогидразида:
метил 2-(2-(пирролидин-1-ил)пиридин-3-илокси)ацетат (86 мг, 0,36 ммоль) и гидразин (0,02 мл, 0,39 ммоль) растворили в EtOH (2 мл), после чего перемешивали при 100°С в течение 12 часов. По окончании реакции, реакционную смесь упарили при пониженном давлении с получением 2-(2-(пирролидин-1-ил)пиридин-3-илокси)ацето-гидразида (85 мг, 99%).
Стадия 3. Получение (Е)-N'-((1Н-индол-6-ил)метилен)-2-(2-(пирролидин-1-ил)пиридин-3-илокси)ацетогидразида: 2-(2-(пирролидин-1-ил)пиридин-3-илокси)ацетогидразид (85 мг, 0,36 ммоль) и 1H-индол-6-карбальдегид (57 мг, 0,39 ммоль) растворили в EtOH (2 мл), после чего перемешивали при 100°С в течение 12 часов. По окончании реакции, реакционную смесь проэкстрагировали с помощью этилацетата, высушили над сульфатом магния, и очистили с помощью колоночной хроматографии (этил ацетат:гексан = 1:1) с получением соединения 331 (64 мг, 48%).
1Н NMR (400 MHz, DMSO-d6): δ 11,44 (m, 1H), 11,27 (m, 1H), 8,32-8,07 (2s, 1H), 7,69 (m, 2H), 7,56 (m, 1H), 7,43 (m, 2H), 7,06 (m, 1H), 6,55 (m, 1H), 6,46 (m, 1H), 5,09 (s, 1H), 4,59 (s, 1H), 3,59 (m, 4H), 1,82 (m, 4H).
Пример 78. Синтез соединения 332
Стадия 1. Получение метил 2-(2,4-диметил-6-(тетрагидрофуран-2-ил)фенокси)ацетата:
2,4-диметил-6-(тетрагидрофуран-2-ил)фенол (Соединение 7-5) (200 мг, 1,04 ммоль), метил 2-бромацетат (0,095 мл, 1,14 ммоль) и карбонат цезия (327 мг, 1,14 ммоль) растворили в ацетонитриле (2 мл), после чего перемешивали при 100°С в течение 3 часов. По окончании реакции, реакционную смесь отфильтровали под пониженным давлением. Фильтрат упарили при пониженном давлении, и очистили с помощью колоночной хроматографии (этил ацетат:гексан = 1:3) с получением метил 2-(2,4-диметил-6-(тетрагидрофуран-2-ил)фенокси)ацетата (145 мг, 0,73 ммоль, 30%).
Стадия 2. Получение 2-(2,4-диметил-6-(тетрагидрофуран-2-ил)фенокси)ацетогидразида:
метил 2-(2,4-диметил-6-(тетрагидрофуран-2-ил)фенокси)ацетат (80 мг, 0,30 ммоль) и гидразин (0,02 мл, 0,33 ммоль) растворили в EtOH (2 мл), после чего перемешивали при 100°С в течение 6 часов. По окончании реакции, реакционную смесь упарили при пониженном давлении с получением 2-(2,4-диметил-6-(тетрагидро-фуран-2-ил)фенокси)ацетогидразида (80 мг, 100%).
Стадия 3. Получение (Е)-N'-((1Н-индол-6-ил)метилен)-2-(2,4-диметил-6-(тетрагидрофуран-2-ил)фенокси)ацетогидразида: 2-(2,4-диметил-6-(тетрагидрофуран-2-ил)фенокси)ацетогидразид (80 мг, 0,30 ммоль) и 1H-индол-6-карбальдегид (43 мг, 0,32 ммоль) растворили в EtOH (2 мл), после чего перемешивали при 100°С в течение 12 часов. По окончании реакции, к реакционной смеси добавили воду, и проэкстрагировали с помощью этилацетата. Полученный органический слой высушили над сульфатом магния, и очистили с помощью колоночной хроматографии (этил ацетат:гексан = 1:1) с получением соединения 332 (60 мг, 51%).
1H NMR (400 MHz, DMSO-d6): δ 11,36 (m, 1H), 11,2 (m, 1H), 8,48-8,03 (2s, 1H), 7,70 (s, 0,5H), 7,50 (m, 4H), 7,39 (m, 0,5H), 6,85 (m, 2H), 6,47 (m, 1H), 4,74-4,31 (2s, 1H), 4,31 (m, 1H), 3,38 (m, 2H), 2,22 (m, 6H), 1,58 (m, 2H), 1,45 (m, 2H).
Пример 79. Синтез соединения 333
2-(2-(фуран-3-ил)-4,6-диметилфенокси)ацетогидразид (30 мг, 0,12 ммоль) и 1H-индол-4-карбальдегид (17 мг, 0,12 ммоль) растворили в EtOH (2 мл), после чего перемешивали при 90°С в течение 6 часов. По окончании реакции, образовавшийся осадок отфильтровали с получением соединения 333 (14 мг, 31%).
1Н NMR (400 MHz, DMSO-d6): δ 11,48-11,43 (m, 1H), 11,34 (s, 1H), 8,43 (d, J=10,6 Hz, 1H), 8,64-8,27 (2s, 1H), 7,72 (m, 1H), 7,48 (m, 2H), 7,39 (m, 1H), 7,24 (m, 2H), 7,01 (m, 1H), 6,98 (m, 1H), 4,74 (s, 1H), 4,23 (s, 1H), 2,27 (s, 3H), 2,24 (s, 3H).
Пример 80. Синтез соединения 336
2-(4-(фуран-3-ил)-2,6-диметилфенокси)ацетогидразид (30 мг, 0,11 ммоль) и 1Н-индол-4-карбальдегид (17 мг, 0,15 ммоль) растворили в EtOH 1 мл, после чего перемешивали при 90°С в течение 10 часов. По окончании реакции, реакционную смесь упарили при пониженном давлении, и очистили с помощью колоночной хроматографии (гексан:этил ацетат=1:1) с получением соединения 336 (18 мг, 41%).
1H NMR (400 MHz, DMSO-d6): δ 11,51 (bs, 1H), 11,35 (s, 1H), 8,35-8,27 (2s, 1H), 8,08 (m, 1H), 7,50 (m, 1H), 7,46 (m, 1H), 7,32 (m, 1H), 7,24 (m, 2H), 7,05 (m, 1H), 6,90 (m, 1H), 5,04 (s, 1H), 4,77 (s, 1H), 2,31 (d, J=16,0 Hz, 3H), 2,16 (s, 3H).
Пример 81. Синтез соединения 337
2-(2,6-диметил-4-(тетрагидрофуран-3-ил)фенокси)ацетогидразид (50 мг, 0,19 ммоль) и 1Н-индол-4-карбальдегид (100 мг, 0,55 ммоль) растворили в EtOH 1 мл, после чего перемешивали при 90°С в течение 3 часов. По окончании реакции, к реакционной смеси добавили воду, и проэкстрагировали с помощью этилацетата. Полученный органический слой высушили над сульфатом магния, и очистили с помощью колоночной хроматографии (гексан:этил ацетат = 1:1) с получением соединения 337 (31 мг, 50%).
1H NMR (400 MHz, DMSO-d6): δ 11,42 (bs, 1H), 11,29 (m, 1H), 8,55-8,16 (2s, 1H), 7,58 (m, 2H), 7,40 (m, 2H), 6,95 (m, 2H), 6,47 (m, 1H), 5,26 (s, 1H), 4,73 (s, 1H), 3,96 (m, 3H), 3,75 (m, 2H), 2,32 (s, 3H), 2,21 (s, 3H).
Пример 82. Синтез соединения 343
Стадия 1. Получение метил 2-(2-(тиофен-3-ил)пиридин-3-илокси)ацетата: 2-(тиофен-3-ил)пиридин-3-ол (Соединение 6-12) (50 мг, 0,28 ммоль), метил 2-бромацетат (0,03 мл, 0,31 ммоль) и карбонат цезия (101 мг, 0,31 ммоль) растворили в ацетонитриле (1 мл), после чего перемешивали при 80°С в течение 3 часов. По окончании реакции, к реакционной смеси добавили воду, и проэкстрагировали с помощью этилацетата. Полученный органический слой высушили над сульфатом магния, и очистили с помощью колоночной хроматографии (этил ацетат:гексан = 1:1) с получением метил 2-(2-(тиофен-3-ил)пиридин-3-илокси)ацетата (48 мг, 67%).
Стадия 2. Получение 2-(2-(тиофен-3-ил)пиридин-3-илокси)ацетогидразида:
метил 2-(2-(тиофен-3-ил)пиридин-3-илокси)ацетат (48 мг, 0,19 ммоль) и гидразин (0,01 мл, 0,21 ммоль) растворили в EtOH (2 мл), после чего перемешивали при 90°С в течение 12 часов. По окончании реакции, реакционную смесь упарили при пониженном давлении с получением 2-(2-(тиофен-3-ил)пиридин-3-илокси)-ацетогидразида (48 мг, 100%).
Стадия 3. Получение (E)-N'-((1Н-индол-6-ил)метилен)-2-(2-(тиофен-3-ил)пиридин-3-илокси)ацетогидразида: 2-(2-(тиофен-3-ил)пиридин-3-илокси)ацетогидразид (48 мг, 0,19 ммоль) и 1H-индол-6-карбальдегид (30 мг, 0,21 ммоль) растворили в EtOH (2 мл), после чего перемешивали при 85°С в течение 12 часов. По окончании реакции, реакционную смесь упарили при пониженном давлении, и очистили с помощью колоночной хроматографии (этил ацетат:гексан = 1:1) с получением соединения 343 (31 мг, 42%).
1H NMR (400 MHz, DMSO-d6): δ 11,50 (m, 1H), 11,30 (m, 1H), 8,62-8,50 (m, 1H), 8,29-8,21 (m, 1H), 8,11 (s, 1H), 7,98 (m, 1H), 7,67 (m, 1H), 7,57 (m, 3H), 7,45 (m, 2H), 7,29 (m, 1H), 6,47 (m, 1H), 5,37 (s, 1H), 4,86 (s, 1H).
Пример 83. Синтез соединения 344
Стадия 1. Получение метил 2-(2,4-диметил-6-(тиофен-2-ил)фенокси)ацетата:
2,4-диметил-6-(тиофен-2-ил)фенол (Соединение 6-13) (100 мг, 0,49 ммоль), метил 2-бромацетат (0,05 мл, 0,53 ммоль) и карбонат цезия (190 мг, 0,53 ммоль) растворили в ацетонитриле (1 мл), после чего перемешивали при 100°С в течение 1 часа. По окончании реакции, реакционную смесь отфильтровали под пониженным давлением. Фильтрат упарили при пониженном давлении, и очистили с помощью колоночной хроматографии (этил ацетат:гексан = 1:4) с получением метил 2-(2,4-диметил-6-(тиофен-2-ил)фенокси)ацетата (141 мг, 100%).
Стадия 2. Получение 2-(2,4-диметил-6-(тиофен-2-ил)фенокси)ацетогидразида:
Метил 2-(2,4-диметил-6-(тиофен-2-ил)фенокси)ацетат (141 мг, 0,51 ммоль) и гидразин (0,03 мл, 0,56 ммоль) растворили в EtOH (2 мл), после чего перемешивали при 85°С в течение 12 часов. По окончании реакции, реакционную смесь упарили при пониженном давлении с получением 2-(2,4-диметил-6-(тиофен-2-ил)-фенокси)ацетогидразида (128 мг, 91%).
Стадия 3. Получение (E)-N'-((1Н-индол-4-ил)метилен)-2-(2,4-диметил-6-(тиофен-2-ил)фенокси)ацетогидразида: 2-(2,4-диметил-6-(тиофен-2-ил)фенокси)ацетогидразид (70 мг, 0,25 ммоль) и 1Н-индол-4-карбальдегид (40 мг, 0,26 ммоль) растворили в EtOH (2 мл), после чего перемешивали при 100°С в течение 12 часов. По окончании реакции, реакционную смесь упарили при пониженном давлении, и очистили с помощью колоночной хроматографии (этил ацетат:гексан = 1:1) с получением соединения 344 (31 мг,31%).
1H NMR (400 MHz, DMSO-d6): δ 11,42 (bs, 0,5H), 11,35 (m, 1,5H), 8,68 (s, 1H), 8,15 (s, 0,5H), 7,61-7,59 (m, 2H), 7,35-7,05 (m, 7H), 6,49 (m, 0,5H), 4,72 (s, 1H), 4,25 (s, 1H), 2,28 (m, 6H).
Пример 84. Синтез соединения 345
2-(2,4-диметил-6-(тиофен-2-ил)фенокси)ацетогидразид (70 мг, 0,25 ммоль) и 1Н-индол-6-карбальдегид (40 мг, 0,26 ммоль) растворили в EtOH (2 мл), после чего перемешивали при 100°С в течение 12 часов. По окончании реакции, реакционную смесь упарили при пониженном давлении, и очистили с помощью колоночной хроматографии (этил ацетат:гексан = 1:1) с получением соединения 345 (20 мг, 20%).
1H NMR (400 MHz, DMSO-d6): δ 11,35 (m, 1H), 11,20 (m, 1H), 8,49-7,97 (2s, 1H), 7,71 (s, 1H), 7,61-7,34 (m, 6,5H), 7,16-7,09 (m, 1H), 7,02 (m, 1H), 6,44 (m, 1H), 4,65-4,20 (2s, 1H), 2,19 (m, 6H).
Пример 85. Синтез соединения 346
Стадия 1. Получение метил 2-(2-(фуран-2-ил)-4-метоксифенокси)ацетата:
2-(фуран-2-ил)-4-метоксифенол (Соединение 6-14) (205 мг, 1,09 ммоль), метил 2-бромацетат (0,1 мл, 1,08 ммоль) и карбонат цезия (1,05 г, 3,24 ммоль) растворили в ацетонитриле (4 мл), после чего перемешивали при 90°С в течение 9 часов. По окончании реакции, реакционную смесь отфильтровали под пониженным давлением. Фильтрат упарили при пониженном давлении, и очистили с помощью колоночной хроматографии (гексан:этил ацетат = 9:1) с получением метил 2-(2-(фуран-2-ил)-4-метоксифенокси)ацетата (194 мг, 68%).
Стадия 2. Получение 2-(2-(фуран-2-ил)-4-метоксифенокси)ацетогидразида:
метил 2-(2-(фуран-2-ил)-4-метоксифенокси)ацетат (189 мг, 0,72 ммоль) и гидразин (0,035 мл, 0,72 ммоль) растворили в EtOH (2 мл), после чего перемешивали при 90°С в течение 3 часов. По окончании реакции, реакционную смесь упарили при пониженном давлении, разбавили диэтиловым эфиром 10 мл, и перемешивали в течение 10 минут.
Образовавшийся осадок отфильтровали с получением 2-(2-(фуран-2-ил)-4-метоксифенокси)ацето-гидразида (146 мг, 80%).
Стадия 3. Получение (E)-N'-((1Н-индол-4-ил)метилен)-2-(2-(фуран-2-ил)-4-метоксифенокси)ацетогидразида: 2-(2-(фуран-2-ил)-4-метоксифенокси)ацетогидразид (50 мг, 0,19 ммоль) и 1Н-индол-4-карбальдегид (28 мг, 0,19 ммоль) растворили в EtOH 1 мл, после чего перемешивали при 90°С в течение 4 часов. По окончании реакции, реакционную смесь упарили при пониженном давлении, и очистили с помощью колоночной хроматографии (гексан:этил ацетат = 1:1) с получением соединения 346 (21 мг, 28%).
1Н NMR (400 MHz, DMSO-d6): δ 11,57-11,54 (m, 1H), 11,34-11,28 (m, 1H), 8,27-7,75 (2s, 1H), 7,74 (m, 1H), 7,30 (m, 3H), 7,17 (m, 5H), 6,98 (m, 1H), 6,61 (m, 1H), 5,26 (s, 1H), 4,73 (s, 1H), 3,76 (s, 3H).
Пример 86. Синтез соединения 347
2-(2-(фуран-2-ил)-4-метоксифенокси)ацетогидразид (50 мг, 0,19 ммоль) и 1H-индол-6-карбальдегид (28 мг, 0,19 ммоль) растворили в EtOH 1 мл, после чего перемешивали при 90°С в течение 4 часов. По окончании реакции, реакционную смесь упарили при пониженном давлении, и очистили с помощью колоночной хроматографии (гексан:этил ацетат = 1:1) с получением соединения 347 (39 мг, 53%).
1H NMR (400 MHz, DMSO-d6): δ 11,50-11,47 (m, 1H), 11,32-11,28 (m, 1H), 8,30-8,09 (2s, 1H), 7,75 (m, 2H), 7,65 (m, 1H), 7,31 (m, 4H), 7,06 (m, 1H), 6,87 (m, 1H), 6,60 (s, 1H), 6,46 (s, 1H), 5,20 (s, 1H), 4,70 (s, 1H), 3,76 (s, 3H).
Пример 87. Синтез соединения 356
Стадия 1. Получение метил 2-(3-(фуран-2-ил)фенокси)ацетата:
3-(фуран-2-ил)фенол (Соединение 6-15) (183 мг, 1,14 ммоль), метил 2-бромацетат (0,11 мл, 1,14 ммоль) и карбонат цезия (556 мг, 1,71 ммоль) растворили в ацетонитриле (2 мл), после чего перемешивали при 90°С в течение 4 часов. По окончании реакции, реакционную смесь отфильтровали под пониженным давлением. Фильтрат упарили при пониженном давлении, и очистили с помощью колоночной хроматографии (гексан:этил ацетат = 9:1) с получением метил 2-(3-(фуран-2-ил)фенокси)ацетата (228 мг, 86%).
Стадия 2. Получение 2-(3-(фуран-2-ил)фенокси)ацетогидразида:
метил 2-(3-(фуран-2-ил)фенокси)ацетат (220 мг, 0,95 ммоль) и гидразин (0,05 мл, 0,95 ммоль) растворили в EtOH (2 мл), после чего перемешивали при 90°С в течение 2 часов. По окончании реакции, реакционную смесь упарили при пониженном давлении, разбавили диэтиловым эфиром 10 мл, и перемешивали в течение 10 минут.
Образовавшийся осадок отфильтровали с получением 2-(3-(фуран-2-ил)фенокси)ацетогидразида (185 мг, 84%).
Стадия 3. Получение (Е)-N'-((1Н-индол-4-ил)метилен)-2-(3-(фуран-2-ил)фенокси)ацетогидразида: 2-(3-(фуран-2-ил)фенокси)ацетогидразид (50 мг, 0,21 ммоль) и 1Н-индол-4-карбальдегид (32 мг, 0,21 ммоль) растворили в EtOH 1 мл, после чего перемешивали при 90°С в течение 17 часов. По окончании реакции, реакционную смесь упарили при пониженном давлении, и очистили с помощью колоночной хроматографии (гексан:этил ацетат = 1:1) с получением соединения 356 (47 мг, 61%).
1H NMR (400 MHz, DMSO-d6): δ 11,54-11,49 (m, 1H), 11,41-11,38 (m, 1H), 8,58-8,27 (2s, 1H), 7,75 (m, 1H), 7,49 (m, 2H), 7,46 (m, 3H), 7,30 (m, 3H), 7,23 (m, 1H), 6,98 (m, 1H), 6,94 (m, 1H), 6,57 (m, 1H), 5,26 (s, 1H), 4,74 (s, 1H).
Пример 88. Синтез соединения 358
Стадия 1. Получение метил 2-(3-(пиридин-3-ил)фенокси)ацетата:
3-(пиридин-3-ил)фенол (Соединение 6-16) (379 мг, 2,22 ммоль), метил 2-бромацетат (0,2 мл, 2,22 ммоль) и карбонат цезия (1086 мг, 3,33 ммоль) растворили в ацетонитриле 5 мл, после чего перемешивали при 100°С в течение 4 часов. По окончании реакции, реакционную смесь отфильтровали под пониженным давлением. Фильтрат упарили при пониженном давлении, и очистили с помощью колоночной хроматографии (гексан:этил ацетат = 7:3) с получением метил 2-(3-(пиридин-3-ил)фенокси)ацетата (420 мг, 78%).
Стадия 2. Получение 2-(3-(пиридин-3-ил)фенокси)ацетогидразида:
метил 2-(3-(пиридин-3-ил)фенокси)ацетат (410 мг, 1,69 ммоль) и гидразин (0,08 мл, 1,69 ммоль) растворили в EtOH 4 мл, после чего перемешивали при 90°С в течение 2 часов. По окончании реакции, реакционную смесь упарили при пониженном давлении, разбавили диэтиловым эфиром 10 мл, и перемешивали в течение 10 минут. Образовавшийся осадок отфильтровали с получением 2-(3-(пиридин-3-ил)фенокси)ацетогидразида (365 мг, 89%).
Стадия 3. Получение (Е)-N'-((1Н-индол-4-ил)метилен)-2-(3-(пиридин-3-ил)фенокси)ацетогидразида: 2-(3-(пиридин-3-ил)фенокси)ацетогидразид (50 мг, 0,20 ммоль) и 1H-индол-4-карбальдегид (30 мг, 0,20 ммоль) растворили в EtOH 1 мл, после чего перемешивали при 90°С в течение 17 часов. По окончании реакции, образовавшийся осадок отфильтровали с получением соединения 358 (39 мг, 53%).
1H NMR (400 MHz, DMSO-d6): δ 11,53 (bs, 1H), 11,39-11,34 (m, 1H), 8,90 (m, 1H), 8,58 (m, 1H), 8,57-8,26 (2s, 1H), 8,05 (m, 1H), 7,39 (m, 6H), 7,13 (m, 3H), 6,99 (m, 1H), 5,32 (s, 1H), 4,80 (s, 1H).
Пример 89. Синтез соединения 359
2-(3-(пиридин-3-ил)фенокси)ацетогидразид (50 мг, 0,21 ммоль) и 1H-индол-6-карбальдегид (32 мг, 0,21 ммоль) растворили в EtOH 1 мл, после чего перемешивали при 90°С в течение 17 часов. По окончании реакции, реакционную смесь упарили при пониженном давлении, и очистили с помощью колоночной хроматографии (гексан:этил ацетат = 1:1) с получением соединения 359 (28 мг, 38%).
1Н NMR (400 MHz, DMSO-d6): δ 11,45-11,42 (m, 1H), 11,33-11,28 (m, 1H), 8,91 (m, 1H), 8,88 (m, 1H), 8,56-8,23 (s, 1H), 8,09 (m, 1H), 7,66 (m, 1H), 7,56 (m, 1H), 7,39 (m, 6H), 7,05 (m, 1H), 6,46 (m, 1H), 5,26 (s, 1H), 4,76 (s, 1H).
Пример 90. Синтез соединения 378
Стадия 1. Получение метил 2-(4-фтор-2-метил-6-(тетрагидрофуран-3-ил)фенокси)ацетата:
4-фтор-2-метил-6-(тетрагидрофуран-3-ил)фенол (Соединение 7-4) (368 мг, 1,87 ммоль) и метил бромацетат (0,21 мл, 2,25 ммоль) растворили в ацетонитриле. Добавили карбонат цезия (915 мг, 2,81 ммоль), после чего перемешивали при комнатной температуре в течение 1 дня. По окончании реакции, к реакционной смеси добавили насыщенный водный раствор гидрокарбоната натрия, и проэкстрагировали с помощью этилацетата. Полученный органический слой промыли насыщенным солевым раствором, высушили над безводным сульфатом натрия, отфильтровали, и упарили при пониженном давлении. Остаток очистили с помощью колоночной хроматографии (силикагель; гексан/этил ацетат, 9/1) с получением метил 2-(4-фтор-2-метил-6-(тетрагидрофуран-3-ил)фенокси)ацетата (415 мг, 83%).
Стадия 2. Получение 2-(4-фтор-2-метил-6-(тетрагидрофуран-3-ил)фенокси)ацетогидразида: метил 2-(4-фтор-2-метил-6-(тетрагидрофуран-3-ил)фенокси)ацетат (415 мг, 1,55 ммоль) и гидразин моногидрат (91,0 мкл, 3,93 ммоль) растворили в этаноле, после чего перемешивали в течение 1 дня при кипячении с обратным холодильником. По окончании реакции, реакционную смесь упарили при пониженном давлении. Концентрированный остаток очистили с помощью колоночной хроматографии (силикагель; дихлорметан/метанол, 9/1) с получением 2-(4-фтор-2-метил-6-(тетрагидрофуран-3-ил)фенокси)ацетогидразида (316 мг, 76%) в виде бесцветного масла.
Стадия 3. Получение (E)-N'-((1Н-индол-6-ил)метилен)-2-(4-фтор-2-метил-6-(тетрагидрофуран-3-ил)фенокси)ацетогидразида: 2-(4-фтор-2-метил-6-(тетрагидрофуран-3-ил)фенокси)ацетогидразид (230 мг, 0,86 ммоль) и индол-6-карбоксальдегид (137 мг, 0,94 ммоль) растворили в этаноле, после чего перемешивали в течение ночи при кипячении с обратным холодильником. По окончании реакции, реакционную смесь упарили при пониженном давлении, и очистили с помощью колоночной хроматографии (силикагель; этил ацетат:гексан, 3/7) с получением соединения 378 (145 мг, 45%) в виде твердого вещества белого цвета.
1H NMR (400 MHz, DMSO-d6): δ 11,48-11,44 (m, 1H), 11,35-11,23 (m, 1H), 8,48, 8,05 (2s, 1H), 7,72 (s, 0,5H), 7,59-7,27 (m, 3,5H), 6,96-6,88 (m, 2H), 6,47-6,43 (m, 1H), 4,77 (s, 1H), 4,34 (s, 3H), 4,02-3,92 (m, 2H), 3,77-3,73 (m, 2H), 3,53-3,43 (m, 2H), 2,26-2,20 (m, 7H), 1,89-2,20 (m, 1H).
Пример 91. Синтез соединения 379
2-(4-фтор-2-метил-6-(тетрагидрофуран-3-ил)фенокси)ацетогидразид (80,5 мг, 0,30 ммоль) и индол-4-карбоксальдегид (52,3 мг, 0,36 ммоль) растворили в этаноле, после чего перемешивали в течение 1 дня при кипячении с обратным холодильником. По окончании реакции, реакционную смесь упарили при пониженном давлении, и очистили с помощью колоночной хроматографии (силикагель; этил ацетат:гексан, 3/7) с получением соединения 379 (42 мг, 36%) в виде твердого вещества белого цвета.
1H NMR (400 MHz, DMSO-d6): δ 11,48-11,44 (m, 1H), 11,35-11,23 (m, 1H), 8,48, 8,05 (2s, 1H), 7,72 (s, 0,5H), 7,59-7,27 (m, 3,5H), 6,96-6,88 (m, 2H), 6,47-6,41 (m, 1H), 4,75 (s, 1H), 4,31 (s, 3H), 4,02-3,92 (m, 2H), 3,75-3,71(m, 2H), 3,53-3,43 (m, 2H), 2,26-2,21 (m, 7H), 1,88-2,22 (m, 1H).
Пример 92. Синтез соединения 457
Стадия 1. Получение метил 2-(2,4-диметил-6-(тетрагидро-2Н-пиран-4-ил)фенокси)ацетата:
2,4-диметил-6-(тетрагидро-2Н-пиран-4-ил)фенол (Соединение 7-7) (156 мг, 0,76 ммоль) и метил бромацетат (0,089 мл, 0,91 ммоль) растворили в ацетонитриле. Добавили карбонат цезия (370 мг, 1,14 ммоль), после чего перемешивали при комнатной температуре в течение 1 дня. По окончании реакции, к реакционной смеси добавили насыщенный водный раствор гидрокарбоната натрия, и проэкстрагировали с помощью этилацетата. Полученный органический слой промыли насыщенным солевым раствором, высушили над безводным сульфатом натрия, отфильтровали, и упарили при пониженном давлении. Остаток очистили с помощью колоночной хроматографии (силикагель; гексан/этил ацетат, 9/1) с получением метил 2-(2,4-диметил-6-(тетрагидро-2Н-пиран-4-ил)фенокси)ацетата (168 мг, 80%).
Стадия 2. Получение 2-(2,4-диметил-6-(тетрагидро-2Н-пиран-4-ил)фенокси)ацетогидразида:
Метил 2-(2,4-диметил-6-(тетрагидро-2Н-пиран-4-ил)фенокси)ацетат (168 мг, 0,60 ммоль) и гидразин моногидрат (36,0 мкл, 0,72 ммоль) растворили в метаноле, после чего перемешивали в течение 1 дня при кипячении с обратным холодильником. По окончании реакции, реакционную смесь упарили при пониженном давлении. К концентрату добавили гексан. Полученный белый осадок отфильтровали, и высушили с получением 2-(2,4-диметил-6-(тетрагидро-2Н-пиран-4-ил)фенокси)ацетогидразида (114 мг, 68%) в виде твердого вещества белого цвета.
Стадия 3. Получение (E)-N'-((1Н-индол-6-ил)метилен)-2-(2,4-диметил-6-(тетрагидро-2Н-пиран-4-ил)фенокси)ацетогидразида: 2-(2,4-диметил-6-(тетрагидро-2Н-пиран-4-ил)фенокси)ацетогидразид (114 мг, 0,41 ммоль) и индол-6-карбоксальдегид (65,3 мг, 0,45 ммоль) растворили в этаноле, после чего перемешивали в течение ночи при кипячении с обратным холодильником. По окончании реакции, реакционную смесь упарили при пониженном давлении, и очистили с помощью колоночной хроматографии (силикагель; этил ацетат:гексан = 7:3) с получением соединения 457 (78 мг, 47%) в виде твердого вещества коричневого цвета.
1Н NMR (400 MHz, DMSO-d6): δ 11,44-11,40 (m, 1H), 11,32-11,23 (m, 1H), 8,49 (s, 0,5H), 8,04 (2s, 0,45H), 7,72 (s, 0,53H), 7,60-7,26 (m, 3,5H), 6,93-6,88 (m, 2H), 6,48-6,43 (m, 1H), 4,77 (s, 0,9H), 4,33 (s, 1,1H), 3,91 (bs, 2H), 3,22-3,18 (m, 2H), 2,25-2,23(m, 6H), 1,67-1,61 (m, 4H).
На следующей схеме 2 показаны способы получения соединений 233, 272, 236, 252, 244, 245, 238 и 289 из соединений 108, 065 и 187 полученных по схеме 1.
[Схема 2]
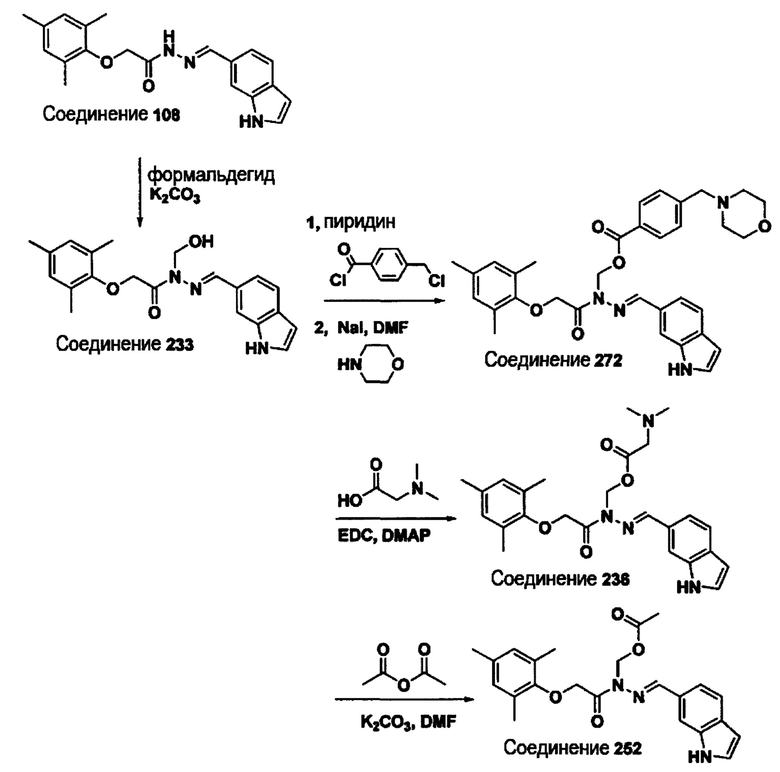
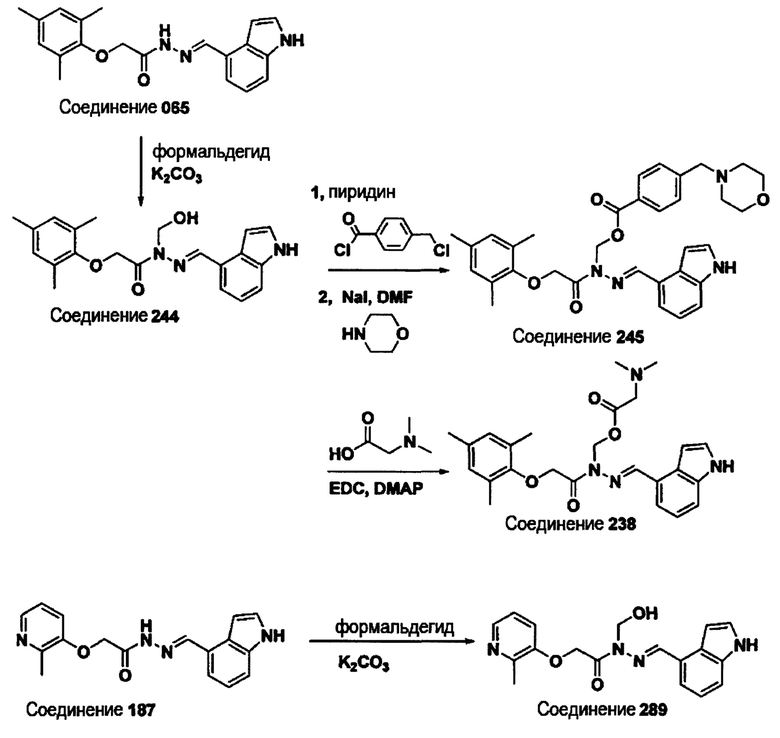
Пример 93. Синтез соединения 233
Соединение 108 (510 мг, 1,52 ммоль), формальдегид 35% (0,6 мл, 15,2 ммоль), карбонат калия (210 мг, 1,52 ммоль) растворили в диметилформамиде (1 мл), после чего перемешивали при 40°С в течение 48 часов. По окончании реакции, к этой смеси добавили воду, после чего отфильтровали полученный белый осадок. Фильтрат упарили при пониженном давлении, и очистили с помощью колоночной хроматографии с получением соединения 233 (110 мг, 20%).
1Н NMR (400 MHz, DMSO-d6): δ 11,43-11,40 (m, 1H), 8,52 (s, 1H), 8,07-7,37 (m, 5H), 6,83 (d, J=4,44 Hz, 2H), 6,45 (m, 1H), 6,45 (m, 2H), 5,52 (d, J=15,5 Hz, 2H), 4,89 (s, 1H), 4,03 (s, 1H), 2,22 (m, 9H).
Пример 94. Синтез соединения 244
Соединение 065 (510 мг, 1,52 ммоль), формальдегид 35% (0,6 мл, 15,2 ммоль) и карбонат калия (210 мг, 1,52 ммоль) растворили в диметилформамиде (1 мл), после чего повысили температуру до 40°С и перемешивали в течение 2 дней. По окончании реакции, к этой смеси добавили воду, после чего отфильтровали полученный белый осадок. Фильтрат упарили при пониженном давлении, и очистили с помощью колоночной хроматографии (этил ацетат:гексан = 1:1) получив Соединение 244 (110 мг, 20%).
1Н NMR (400 MHz, DMSO-d6): δ 11,4 (m, 1H), 8,69-8,24 (2s, 1H), 7,67 (m, 1H), 7,50 (m, 1H), 7,22 (m, 3H), 6,83 (m, 2H), 6,45 (m, 1H), 5,54 (m, 2H), 4,83 (s, 1H), 4,36 (s, 1H), 2,31 (m, 9H).
Пример 95. Синтез соединения 289
Соединение 187 (110 мг, 0,36 ммоль) растворили в диметилформамиде (1,5 мл).
Формальдегид (0,09 мл, 1,08 ммоль) и карбонат калия (41 мг, 0,36 ммоль) добавили к этой смеси, после чего перемешивали при 45°С в течение 16 часов. По окончании реакции, образовавшийся осадок отфильтровали с получением соединения 289 (50 мг, 41%).
1H NMR (400 MHz, DMSO-d6): δ 11,56 (m, 1H), 8,51-8,27 (m, 1H), 8,02 (m, 1H), 7,66 (d, J=8,0 Hz, 1H), 7,51 (m, 1H), 7,31-6,97 (m, 4H), 5,75 (m, 2H), 5,42-4,76 (m, 2H), 2,43 (m, 3H).
Пример 96. Синтез соединения 272
Соединение 233 (200 мг, 0,55 ммоль) и 3-(хлорметил)бензил хлорид (113 мг, 0,60 ммоль) растворили в пиридине (2 мл), после чего перемешивали в течение 3 часов при комнатной температуре. По окончании реакции, к реакционной смеси добавили воду, и проэкстрагировали с помощью этилацетата. Полученный органический слой высушили над сульфатом магния, и упарили при пониженном давлении. Концентрат растворили в диметилформамиде, после чего морфолин (0,04 мл, 0,44 ммоль) и йодид натрия (cat.) добавили к этой смеси, после чего перемешивали в течение 4 часов при комнатной температуре. По окончании реакции, к этой смеси добавили воду, тем самым получив белый осадок, который отфильтровали с получением соединения 272 (20 мг, 0,03 ммоль, 32%).
1Н NMR (400 MHz, DMSO-d6): δ 11,49 (m, 1H), 8,55 (s, 0,5H), 7,88 (s, 0,5H), 7,81 (m, 3H), 7,69-7,63 (m, 1,5H), 7,57-7,51 (m, 1,5H), 7,41 (m, 3H), 6,83 (d, J=12,0 Hz, 2H), 6,59 (m, 1H), 6,50 (d, J=1,80 Hz, 2H), 4,77 (s, 1H), 4,34 (s, 1H), 3,52 (m, 6H), 2,30 (m, 4H), 2,20 (m, 9H)
Пример 97. Синтез соединения 245
Соединение 244 (33 мг, 0,09 ммоль) и 3-(хлорметил)бензил хлорид (17 мг, 0,12 ммоль) растворили в пиридине (0,2 мл), после чего перемешивали в течение 3 часов при комнатной температуре. По окончании реакции, к реакционной смеси добавили воду, и проэкстрагировали с помощью этилацетата. Полученный органический слой высушили над сульфатом магния, и упарили при пониженном давлении. Концентрат растворили в диметилформамиде, после чего морфолин (0,02 мл, 0,24 ммоль) и йодид натрия (cat.) добавили к этой смеси, после чего перемешивали в течение 4 часов при комнатной температуре. К реакционной смеси добавили воду, и проэкстрагировали с помощью этилацетата. Полученный органический слой высушили над сульфатом магния, и очистили с помощью колоночной хроматографии (этил ацетат:гексан = 1:1~2:1)с получением соединения 245 (18 мг, 58%).
1H NMR (400 MHz, DMSO-d6): δ 11,52-46 (m, 1H), 8,71-8,24 (2s, 1H), 7,89-7,76 (m, 3H), 7,70-7,65 (m, 1H), 7,44-7,23 (m, 5H), 6,84 (m, 2H), 6,50 (d, J=14,7 Hz, 2H), 4,83 (s, 1H), 4,36 (s, 1H), 3,53-3,49 (m, 6H), 2,31 (m, 4H), 2,22 (m, 9H).
Пример 98. Синтез соединения 238
Соединение 244 (20 мг, 0,05 ммоль), N,N-диметилглицин (6 мг, 0,05 ммоль), EDC (24 мг, 0,13 ммоль) и 4-диметиламинопиридин (3 мг, 0,03 ммоль) растворили в метилен хлориде: диметилформамид = 1 мл:1 мл, после чего перемешивали в течение 2 часов при комнатной температуре. К реакционной смеси добавили воду, и проэкстрагировали с помощью метиленхлорида. Полученный органический слой высушили над сульфатом магния, и перекристаллизовали из гексана с получением соединения 238 (10 мг, 44%).
1Н NMR (400 MHz, DMSO-d6: δ 11,4 (m, 1H), 8,70 (s, 0,5H), 8,23 (s, 0,5H), 7,74-7,56 (m, 2H), 7,34-7,21 (m, 3H), 6,84 (d, J=5,92 Hz, 2H), 6,27 (d, J=15,2 Hz, 2H), 4,83 (s, 1H), 4,36 (s, 1H),2,19 (m, 15H).
Пример 99. Синтез соединения 236
Соединение 233 (20 мг, 0,06 ммоль), N,N-диметилглицин (16 мг, 0,15 ммоль), EDC (28 мг, 0,15 ммоль) и 4-диметиламинопиридин (4 мг, 0,03 ммоль) растворили в диметилацетамиде (1 мл), после чего перемешивали в течение 1,5 часов при 40°С.Реакционную смесь охладили до 0°С, после чего перемешивали в течение 30 минут медленно добавляя 1,5 мл воды. После дополнительного перемешивания в течение 1 часа, к реакционной смеси добавили воду, и проэкстрагировали с помощью метиленхлорида. Полученный органический слой упарили при пониженном давлении. Концентрат очистили с помощью колоночной хроматографии (этил ацетат:метилен хлорид = 1:4) с получением соединения 236 (10 мг, 37%).
1H NMR (400 MHz, DMSO-d6): δ 11,52 (m, 1H), 8,54, 8,06 (s, 1H), 7,89 (s, 0,5H), 7,59-7,56 (m, 3H), 6,85 (d, J=7,48 Hz, 2H), 6,5 (m, 1H), 6,45 (d, J=18,0 Hz, 1H), 4,76 (s, 1H), 4,34 (s, 1Н), 3,15 (d, J=18,9 Hz, 2H), 2,19 (m, 15H).
Пример 100. Синтез соединения 252
Соединение 233 (30 мг, 0,08 ммоль), безводную уксусную кислоту (8 мг, 0,08 ммоль) и триэтиламин (8 мг, 0,08 ммоль) растворили в диметилформамиде (1 мл), после чего перемешивали при комнатной температуре в течение 12 часов. К реакционной смеси добавили воду, тем самым получив белый осадок, который отфильтровали после чего перекристаллизовали из гексана с получением соединения 252 (20 мг, 0,04 ммоль, 60%).
1H NMR (400 MHz, DMSO-d6): δ 11,47 (m, 1Н), 8,69-8,24 (2s, 1Н), 7,67-7,62 (m, 1Н), 7,50 (m, 1Н), 7,31-7,09 (m, 2,5H), 6,84 (d, J=7,4 Hz, 2H), 6,77 (d, J=3,2 Hz, 0,5H), 6,45 (m, 1Н), 5,54 (m, 2H), 4,84 (s, 1Н), 4,36 (s, 1Н), 2,24 (m, 9H).
На следующей схеме 3 показаны способы получения соединений 164, 177, 243, 195, 198, 201, 375 и 380 из соединений 065, 108, 229 и 378 полученных по схеме 1,
[Схема 3]
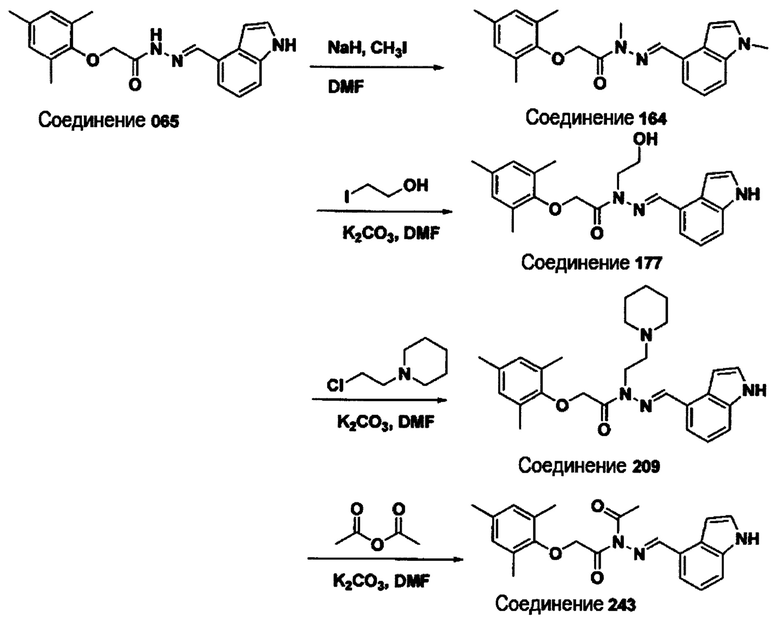
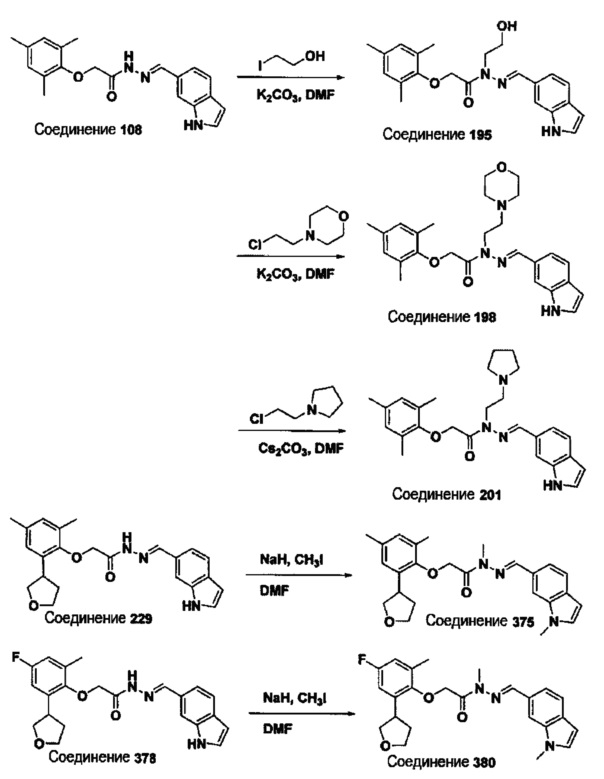
Пример 101. Синтез соединения 164
Соединение 065 (265 мг, 0,79 ммоль), 2-йодометан (336 мг, 2,37 ммоль), гидрид натрия (38 мг, 1,58 ммоль) растворили в диметилформамиде (2 мл), после чего перемешивали при 90°С в течение 12 часов. К реакционной смеси добавили воду, и проэкстрагировали с помощью этилацетата. Полученный органический слой высушили над сульфатом магния, и упарили при пониженном давлении. Концентрат очистили с помощью колоночной хроматографии с получением соединения 164 (152 мг, 0,42 ммоль, 53%).
1Н NMR (400 MHz, DMSO-d6: δ 8,18 (s, 0,5H), 7,48 (s, 0,5H), 7,36 (d, J=3,0 Hz, 1H), 7,32 (m, 1H), 7,18 (m, 0,5H), 6,81 (s, 2H), 6,77 (d, J=3,0 Hz, 1H), 4,96 (s, 2H), 3,77 (s, 3H), 3,38 (s, 3H), 2,18 (s, 9H).
Пример 102. Синтез соединения 375
Соединение 229 (50 мг, 0,13 ммоль) растворили в диметилформамиде. Гидрид натрия (16,7 мг, 0,38 ммоль) и метил йодид (54,5 мг, 0,38 ммоль) добавили к этой смеси. После реакции при комнатной температуре в течение 2 часов, реакционную смесь упарили при пониженном давлении. Остаток очистили с помощью колоночной хроматографии (силикагель; метилен хлорид/метанол, 10/1) с получением соединения 375 (8,5 мг, 16%) в виде твердого вещества коричневого цвета.
1H NMR (400 MHz, DMSO-d6): δ 11,48-11,44 (m, 1H), 11,35-11,23 (m, 1H), 8,48, 8,05 (2s, 1H), 7,72 (s, 0,5H), 7,59-7,27 (m, 3,5H), 6,96-6,88 (m, 2H), 6,47-6,43 (m, 1H), 4,77 (s, 1H), 4,34 (s, 3H), 4,02-3,92 (m, 2H), 3,77-3,73 (m, 2H), 3,56 (s, 3H), 3,53-3,43 (m, 2H), 2,26-2,20 (m, 7H), 1,89-2,20 (m, 1H).
Пример 103. Синтез соединения 380
Соединение 378 (115 мг, 0,28 ммоль) растворили в диметилформамиде. Гидрид натрия (36,9 мг, 0,85 ммоль) и метил йодид (0,53 мл, 0,85 ммоль) добавили к этой смеси. После реакции при комнатной температуре в течение 1 дня, к реакционной смеси добавили воду, и проэкстрагировали с помощью этилацетата. Полученный органический слой упарили при пониженном давлении. Концентрат очистили с помощью колоночной хроматографии (силикагель; гексан/этил ацетат, 3/7) с получением соединения 380 (26 мг, 22%) в виде твердого вещества белого цвета.
1Н NMR (400 MHz, CDCl3): δ 7,88 (s, 1H), 7,59 (d, 1H, J=8,3 Hz), 7,47 (s, 1H), 7,43 (dd, 1H, J=8,3, 1,3 Hz), 7,13 (d, 1H, J=3,0 Hz), 6,88 (dd, 1H, J=9,5, 3,1 Hz), 6,79 (dd, 1H, J=8,7, 3,0 Hz), 6,49 (d, 1H, J=3,0 Hz), 5,03, 5,01 (ABq, 2H, J=10,3, 15,8 Hz), 4,14-4,04 (m, 2H), 3,94-3,84 (m, 2H), 3,81 (s, 3H), 3,74-3,71 (m, 1H), 3,49 (s, 3H), 2,44-2,38 (m, 1H), 2,36 (s, 3H), 1,98-1,89 (m, 1H).
Пример 104. Синтез соединения 177
Соединение 065 (265 мг, 0,79 ммоль), 2-йодоэтанол (0,12 мл, 1,58 ммоль) и карбонат калия (436 мг, 3,16 ммоль) растворили в диметилформамиде (2 мл), после чего перемешивали при 50°С в течение 12 часов. По окончании реакции, к реакционной смеси добавили воду, и проэкстрагировали с помощью этилацетата. Полученный органический слой промыли насыщенным солевым раствором, высушили над сульфатом магния, и очистили с помощью колоночной хроматографии с получением соединения 177 (121 мг, 0,32 ммоль, 41%).
1Н NMR (400 MHz, DMSO-d6: δ 11,33 (bs, 1H), 8,37 (s, 1H), 7,44 (d, J=7,92 Hz, 1H), 7,39 (t, J=2,88 Hz, 1H), 7,26 (d, J=7,08 Hz, 1H), 7,12 (t, J=7,8 Hz, 1H), 6,82 (s, 2H), 6,79 (m, 1H), 4,99 (m, 2H), 4,96 (s, 2H), 4,14 (t, J=5,76 Hz, 2H), 3,3 (m, 2H), 2,20 (s, 6H), 2,16 (s, 3H).
Пример 105. Синтез соединения 195
Соединение 108 (265 мг, 0,79 ммоль), 2-йодоэтанол (0,12 мл, 1,58 ммоль) и карбонат калия (436 мг, 3,16 ммоль) растворили в диметилформамиде (2 мл), после чего перемешивали при 50°С в течение 12 часов. По окончании реакции, к реакционной смеси добавили воду, и проэкстрагировали с помощью этилацетата. Полученный органический слой промыли насыщенным солевым раствором, высушили над сульфатом магния, и очистили с помощью колоночной хроматографии с получением соединения 195 (50 мг, 17%).
1Н NMR (400 MHz, DMSO-d6: δ 11,27 (bs, 1H), 8,19 (s, 1H), 7,64 (s, 1H), 7,50 (d, J=8,2 Hz, 1H), 7,39 (t, J=2,96 Hz, 1H), 7,30 (m, 1H), 6,83 (s, 2H), 6,41 (m, 1H), 4,9 (m, 1H), 4,88 (s 2H), 4,08 (t, J=6,28 Hz, 2H), 3,58 (m, 2H), 2,18 (d, J=4,28 Hz, 9H).
Пример 106. Синтез соединения 198
Соединение 108 (100 мг, 0,29 ммоль), 4-(2-хлорэтил)морфолин (0,11 г, 0,58 ммоль) и карбонат калия (164 мг, 1,2 ммоль) растворили в диметилформамиде (2 мл), после чего перемешивали при 90°С в течение 12 часов. По окончании реакции, к реакционной смеси добавили воду, и проэкстрагировали с помощью этилацетата. Полученный органический слой высушили над сульфатом магния, и очистили с помощью колоночной хроматографии с получением соединения 198,
1Н NMR (400 MHz, DMSO-d6: δ 11,26 (bs, 1H), 8,13 (s, 1H), 7,66 (s, 1H), 7,52 (d, J=8,2 Hz, 1H), 7,39 (t, J=2,96 Hz, 1H), 7,30 (m, 1H), 6,82 (s, 2H), 6,41 (m, 1H), 4,87 (s, 2H), 4,16 (m 2H), 3,46 (m, 4H), 2,22 (d, J=4,28 Hz, 9H).
Пример 107. Синтез соединения 201
Соединение 108 (40 мг, 0,12 ммоль), 1-(2-хлорэтил)пирролидин (40 мг, 0,24 ммоль) и карбонат цезия (116 мг, 0,36 ммоль) растворили в диметилформамиде, после чего перемешивали при 100°С в течение 12 часов. По окончании реакции, к реакционной смеси добавили воду, и проэкстрагировали с помощью дихлорметана. Полученный органический слой высушили над сульфатом магния, и очистили с помощью колоночной хроматографии с получением соединения 201 (17 мг, 33%).
1Н NMR (400 MHz, DMSO-d6: δ 11,26 (bs, 1H), 8,11 (s, 1H), 7,65 (s, 1H), 7,52 (d, J=8,2 Hz, 1H), 7,39 (t, J=2,96 Hz, 1H), 7,31 (m, 1H), 6,82 (s, 2H), 6,4 (m, 1H), 4,87 (s, 2H), 4,15 (s, 2H), 2,53 (m, 6H), 2,18 (d, J=3,2 Hz, 9H), 1,68 (m, 4H).
Пример 108. Синтез соединения 243
Соединение 065 (30 мг, 0,08 ммоль), безводную уксусную кислоту (9 мг, 0,08 ммоль) и карбонат калия (12 мг, 0,08 ммоль) растворили в диметилформамиде 2 мл, после чего перемешивали при комнатной температуре в течение 12 часов. По окончании реакции, к реакционной смеси добавили воду, и проэкстрагировали с помощью этилацетата.
Полученный органический слой высушили над сульфатом магния, и упарили при пониженном давлении. Концентрат очистили с помощью колоночной хроматографии с получением соединения 243 (517 мг, 64%).
1Н NMR (400 MHz, DMSO-d6): δ 11,62 (m, 1H), 8,44-8,37 (m, 1H), 8,25 (s, 0,5H), 8,80 (m, 1H), 7,51-7,33 (m, 2,5Н), 7,02 (d, J=3,48 Hz, 0,5H), 6,85 (d, J=5,8 Hz, 1H), 4,83 (s, 1H), 4,37 (s, 1H), 2,66 (m, 3H), 2,10 (m, 9H).
На следующей схеме 4 показаны способы получения соединений 072, 232, 237 и 239.
[Схема 4]
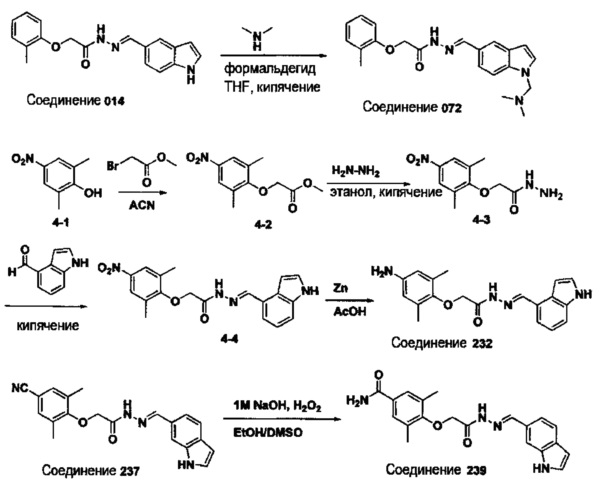
Пример 109. Синтез соединения 072
Соединение 014 (100 мг, 1,52 ммоль), формальдегид (48,6 мг, 1,62 ммоль), и 2М раствор диметиламина в тетрагидрофуране (1,62 мл, 3,24 ммоль) перемешивали в течение 6 часов при кипячении с обратным холодильником. По окончании реакции, к реакционной смеси добавили воду, и проэкстрагировали с помощью этилацетата.
Полученный органический слой высушили над сульфатом магния, и упарили при пониженном давлении. Концентрат очистили с помощью колоночной хроматографии с получением соединения 072 (6 мг, 5%).
1Н NMR (400MHz, DMSO-d6 δ 11,4 (bs, 0,8H), 8,32-8,06 (m, 0,7H), 7,80-7,79 (m, 1H), 7,63-7,61 (m, 1H), 7,57-7,55 (m, 1H), 7,41-7,40 (m, 1H), 7,14-7,10 (m, 2H), 6,87-6,79 (m, 2H), 6,51-6,50 (m, 1H), 5,14-4,63 (m, 4H), 2,23-2,18 (m, 9H).
Пример 110. Синтез соединения 232
Стадия 1. Получение метил 2-(2,6-диметил-4-нитрофенокси)ацетата (Соединение 4-2):
2,6-диметил-4-нитрофенол (1000 мг, 5,98 ммоль) и метил бромацетат (0,6 мл, 5,98 ммоль) и карбонат цезия (2,9 г, 8,97 ммоль) растворили в ацетонитриле 10 мл, после чего перемешивали при 80°С в течение 12 часов. К реакционной смеси добавили воду, и проэкстрагировали с помощью этилацетата. Полученный органический слой высушили над сульфатом магния, и очистили с помощью колоночной хроматографии (этил ацетат:гексан = 1:1) с получением метил 2-(2,6-диметил-4-нитрофенокси)ацетат (682 мг, 48%).
Стадия 2. Получение 2-(2,6-диметил-4-нитрофенокси)ацетогидразида (Соединение 4-3):
метил 2-(2,6-диметил-4-нитрофенокси)ацетат (300 мг, 1,25 ммоль), гидразин моногидрат (0,07 мл, 1,40 ммоль) растворили в EtOH (3 мл), после чего перемешивали при 90°С в течение 12 часов. Реакционную смесь очистили с помощью колоночной хроматографии (этил ацетат:гексан = 1:1) с получением 2-(2,6-диметил-4-нитро-фенокси)ацетогидразида (280 мг, 96%).
Стадия 3. Получение (Е)-N'-((1Н-индол-4-ил)метилен)-2-(2,6-диметил-4-нитрофенокси)ацетогидразида (Соединение 4-4):
2-(2,6-диметил-4-нитрофенокси)ацетогидразид (289 мг, 1,21 ммоль), 1H-индол-4-карбальдегид (175 мг, 1,21 ммоль) растворили в EtOH (3 мл), после чего перемешивали при 90°С в течение 12 часов. Реакционную смесь очистили с помощью колоночной хроматографии (этил ацетат:гексан = 1:1) с получением (Е)-N'-((1Н-индол-4-ил)метилен)-2-(2,6-диметил-4-нитрофенокси)ацетогидразида (127 мг, 29%).
Стадия 4. Получение (Е)-N'-((1Н-индол-4-ил)метилен)-2-(4-амино-2,6-диметилфенокси)ацетогидразида (Соединение 232):
(E)-N'-((1Н-индол-4-ил)метилен)-2-(2,6-диметил-4-нитрофенокси)ацетогидразид (20 мг, 0,05 ммоль) и цинк (74 мг, 1 ммоль) растворили в уксусной кислоте (1 мл), после чего перемешивали в течение 6 часов при комнатной температуре. Реакционную смесь очистили с помощью колоночной хроматографии (этил ацетат:гексан = 2:1) с получением соединения 232 (2 мг, 11%).
1H NMR (400 MHz, CD3OD): δ 8,68 (s, 0,6H), 8,20 (s, 0,4H), 7,51-7,30 (m, 3H), 7,21-7,15 (m, 2H), 6,83 (m, 0,5H), 6,45 (d, J=8,04H, 1,5H), 4,62 (s, 1H), 4,40 (s, 1H), 2,25 (s, 6H).
Пример 111. Синтез соединения 239
Соединение 237 (16 мг, 0,05 ммоль) растворили в EtOH:диметилсульфоксид = 4 мл:1 мл. Реакционную смесь охладили до 0°С, и добавили 1М раствор гидроксида натрия и 30% H2O2, Температуру реакционной смеси повысили до комнатной, после чего перемешивали в течение 2 часов. По окончании реакции, к реакционной смеси добавили воду, и проэкстрагировали с помощью этилацетата. Полученный органический слой высушили над сульфатом магния, и упарили при пониженном давлении. Концентрат очистили с помощью колоночной хроматографии с получением соединения 239 (8 мг, 44 %).
1Н NMR (400 MHz, DMSO-d6): δ 11,43 (d, J=15,4 Hz, 1H), 11,32-11,21 (d, J=40,9 Hz, 1H), 8,48-8,04 (2s, 1H), 7,84 (m, 1H), 7,71 (s, 0,5H), 7,29-7,22 (m, 1,5H), 6,45 (m, 1H), 4,85 (s, 1H), 4,42 (s, 1H), 2,30 (d, J=5,16 Hz, 6H).
На следующей схеме 5 показаны способы получения альдегидных соединений (Соединения 1-5) из схемы 1,
[Схема 5]
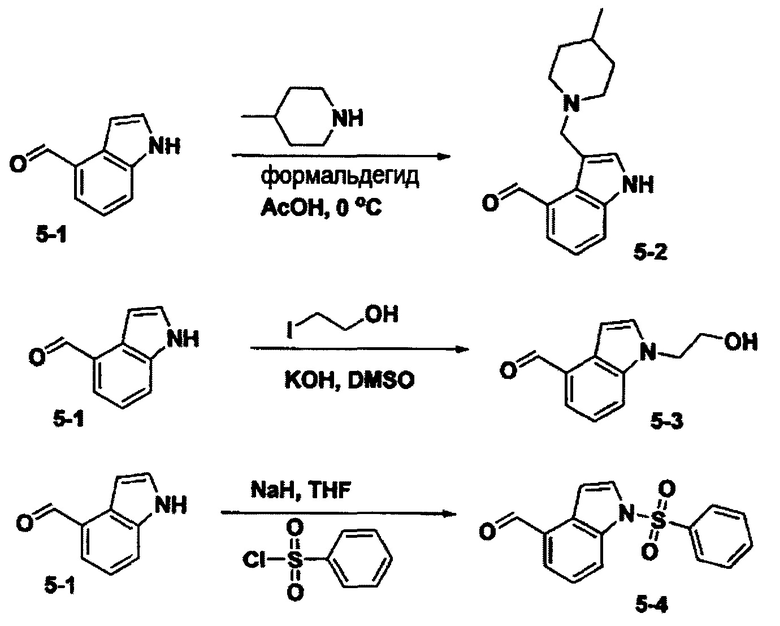
Пример получения 1. Синтез соединения 5-2 (Полупродукт соединения 211):
Соединение 5-1 (200 мг, 1,38 ммоль) и формальдегид 37% (1,1 мл, 1,66 ммоль) растворили в уксусной кислоте. Реакционную смесь охладили до 0°С, и добавили 4-метилпиперидин (154 мл, 1,38 ммоль), после чего перемешивали при комнатной температуре в течение 3 часов. По окончании реакции, к реакционной смеси добавили 1M NaOH, и проэкстрагировали с помощью этилацетата. Полученный органический слой промыли насыщенным солевым раствором, высушили над сульфатом магния, и упарили при пониженном давлении с получением соединения 5-2 (311 мг, 87%).
Пример получения 2. Синтез соединения 5-3 (Полупродукт соединения 180):
Соединение 5-1 (125 мг, 1,06 ммоль), 2-йодоэтанол (0,1 мл, 1,59 ммоль) и KOH (96 мг, 2,12 ммоль) растворили в диметилсульфоксиде (2 мл), после чего перемешивали при комнатной температуре в течение 12 часов. По окончании реакции, к реакционной смеси добавили воду, и проэкстрагировали с помощью этилацетата. Полученный органический слой очистили с помощью колоночной хроматографии с получением соединения 5-3 (120 мг, 59,8%).
Пример получения 3. Синтез соединения 5-4 (Полупродукт соединения 280):
Соединение 5-1 (1 г, 6,89 ммоль) растворили в тетрагидрофуране, после чего добавили гидрид натрия (360 мг, 8,27 ммоль). Добавили бензолсульфонил хлорид (1,2 г, 6,89 ммоль), после чего перемешивали при комнатной температуре в течение 12 часов. По окончании реакции, к реакционной смеси добавили воду, и проэкстрагировали с помощью этилацетата. Полученный органический слой очистили с помощью колоночной хроматографии с получением соединения 5-4 (500 мг, 25%).
На следующей схеме 6 показаны способы получения исходных соединений (Соединения 1-1) из схем 1 и 7.
[Схема 6]
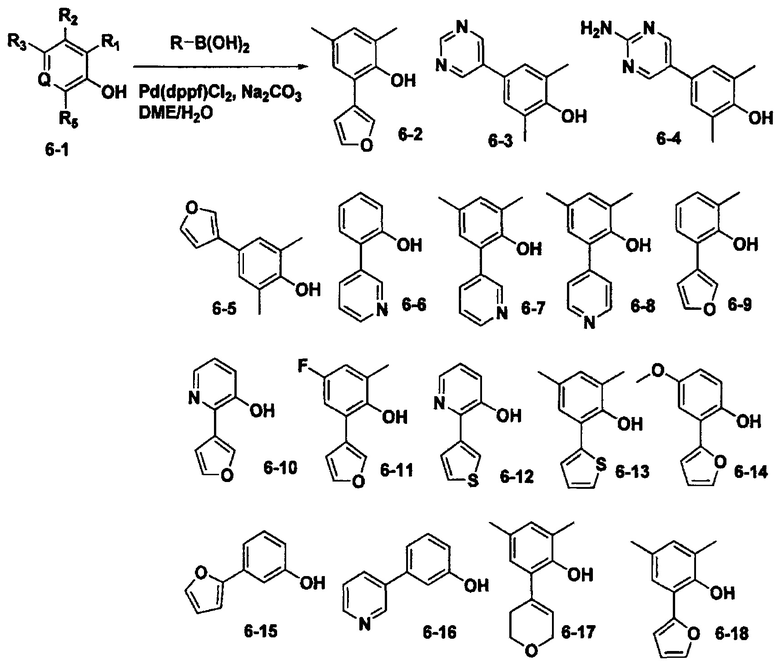
Пример получения 4. Получение 2-(фуран-3-ил)-4,6-диметилфенола (Соединение 6-2):
2-Бром-4,6-диметилфенол (1,8 г, 8,9 ммоль) растворили в смеси диметоксиэтан/вода (2:1). Фуран-3-илборную кислоту (1 г, 8,9 ммоль), Pd(dppf)Cl2 (0,36 г, 0,45 ммоль) и карбонат натрия (3,7 г, 45 ммоль) добавили к этой смеси, после чего перемешивали в течение 1 часа при кипячении с обратным холодильником. По окончании реакции, к реакционной смеси добавили насыщенный водный раствор гидрокарбоната натрия, и проэкстрагировали с помощью этилацетата. Полученный органический слой высушили над сульфатом магния, упарили при пониженном давлении, и очистили с помощью колоночной хроматографии с получением 2-(фуран-3-ил)-4,6-диметилфенола (0,9 г, 54%).
Пример получения 5. Получение 2,6-диметил-4-(пиримидин-5-ил)фенола (Соединение 6-3): 4-Бром-2,6-диметилфенол (0,5 г, 2,5 ммоль) растворили в смеси диметоксиэтан/вода (2:1). Пиримидин-5-илборную кислоту (0,37 г, 2,8 ммоль), Pd(dppf)Cl2 (0,02 г, 0,13 ммоль) и карбонат натрия (0,79 г, 7,5 ммоль) добавили к этой смеси, после чего реакцию проводили в микроволновом реакторе при 120°С в течение 15 минут. По окончании реакции, к реакционной смеси добавили воду, и проэкстрагировали с помощью этилацетата. Полученный органический слой высушили над сульфатом магния, упарили при пониженном давлении, и очистили с помощью колоночной хроматографии с получением 2,6-диметил-4-(пиримидин-5-ил)фенола (0,1 г, 20%).
Пример получения 6. Получение 4-(2-аминопиримидин-5-ил)-2,6-диметилфенола (Соединение 6-4); 2-Аминопиримидин-5-илборную кислоту (0,76 г, 5,4 ммоль) растворили в смеси диметоксиэтан/вода (2:1). 4-Бром-2,6-диметилфенол (0,91 г, 4,52 ммоль) и Pd(dppf)Cl2 (0,19 г, 0,14 ммоль), карбонат натрия (1,4 г, 14 ммоль) добавили к этой смеси, после чего реакцию проводили в микроволновом реакторе при 120°С в течение 15 минут. По окончании реакции, к реакционной смеси добавили воду, и проэкстрагировали с помощью дихлорметана. Полученный органический слой высушили над сульфатом магния, упарили при пониженном давлении, и очистили с помощью колоночной хроматографии с получением 4-(2-аминопиримидин-5-ил)-2,6-диметилфенола (0,27 г, 27%).
Пример получения 7. Получение 4-(фуран-3-ил)-2,6-диметилфенола (Соединение 6-5): 4-Бром-2,6-диметилфенол (350 мг, 1,74 ммоль), 3-фуранилборную кислоту (269 мг, 2,26 ммоль), Pd(dppf)Cl2 (71 мг, 0,087 ммоль) и карбонат натрия (553 мг, 5,22 ммоль) растворили в смеси DME: вода = 4 мл: 2 мл, после чего перемешивали при микроволновом облучении при 120°С в течение 20 минут. По окончании реакции, реакционную смесь отфильтровали через цеолиты. Полученный фильтрат проэкстрагировали с помощью этилацетата, высушили над сульфатом магния, и очистили с помощью колоночной хроматографии (гексан:этил ацетат = 9:1) с получением 4-(фуран-3-ил)-2,6-диметилфенола (225 мг, 50%).
Пример получения 8. Получение 2-(пиридин-3-ил)фенола (Соединение 6-6):
2-Бромфенол (0,5 г, 2,9 ммоль) растворили в смеси диметоксиэтан/вода (2:1). Пиридин-3-илборную кислоту (0,71 г, 5,8 ммоль) и Pd(dppf)Cl2 (0,12 г, 0,14 ммоль), карбонат натрия (1,2 г, 14 ммоль) добавили к этой смеси, после чего перемешивали в течение 1 часа при кипячении с обратным холодильником. По окончании реакции, к реакционной смеси добавили воду, и проэкстрагировали с помощью этилацетата. Полученный органический слой высушили над сульфатом магния, упарили при пониженном давлении, и очистили с помощью колоночной хроматографии с получением 2-(пиридин-3-ил)фенола (0,25 г, 20%).
Пример получения 9. Получение 2,4-диметил-6-(пиридин-3-ил)фенола (Соединение 6-7): 2-бром-4,6-диметилфенол (1 г, 5 ммоль) растворили в смеси диметоксиэтан/вода (2:1). Пиридин-3-илборную кислоту (0,62 г, 5 ммоль) и Pd(dppf)Cl2 (0,20 г, 0,25 ммоль), карбонат натрия (2,1 г, 25 ммоль) добавили к этой смеси, после чего перемешивали в течение 1 часа при кипячении с обратным холодильником. По окончании реакции, к реакционной смеси добавили воду, и проэкстрагировали с помощью этилацетата. Полученный органический слой высушили над сульфатом магния, упарили при пониженном давлении, и очистили с помощью колоночной хроматографии с получением 2,4-диметил-6-(пиридин-3-ил)фенола (0,7 г, 70%).
Пример получения 10. Получение 2,4-диметил-6-(пиридин-4-ил)фенол (Соединение 6-8): 2-Бром-4,6-диметилфенол (0,5 г, 2,5 ммоль) растворили в смеси диметоксиэтан/вода (2:1). Пиридин-4-илборную кислоту (0,32 г, 2,5 ммоль) и Pd(dppf)Cl2 (0,10 г, 0,13 ммоль), карбонат натрия (1 г, 13 ммоль) добавили к этой смеси, после чего перемешивали в течение 1 часа при кипячении с обратным холодильником. По окончании реакции, к реакционной смеси добавили воду, и проэкстрагировали с помощью этилацетата. Полученный органический слой высушили над сульфатом магния, упарили при пониженном давлении, и очистили с помощью колоночной хроматографии с получением 2,4-диметил-6-(пиридин-4-ил)фенола (0,15 г, 30%).
Пример получения 11. Получение 2-(фуран-3-ил)-6-метилфенола (Соединение 6-9): 2-Бром-6-метилфенол (800 мг, 4,28 ммоль), 3-фуран борную кислоту (574 мг, 5,13 ммоль), карбонат натрия (1400 мг, 12,8 ммоль) и Pd2(dppf)2Cl2 (699 мг, 0,82 ммоль) растворили в смеси диметоксиэтан/вода 10 мл/10 мл, после чего перемешивали с микроволновым облучением при 120°С в течение 20 минут. По окончании реакции, реакционную смесь отфильтровали через цеолиты. Фильтрат разбавили водой, и проэкстрагировали с помощью этилацетата. Полученный органический слой высушили над сульфатом магния, и очистили с помощью колоночной хроматографии (этил ацетат:гексан = 1:3) с получением 2-(фуран-3-ил)-6-метилфенола (273 мг, 26%).
Пример получения 12. Получение 2-(фуран-3-ил)пиридин-3-ола (Соединение 6-10): 2-Бромпиридин-3-ол (200 мг, 1,15 ммоль), 3-фуран борную кислоту (574 мг, 1,38 ммоль), карбонат натрия (365 мг, 12,8 ммоль) и Pd2(dppf)2Cl2 (187 мг, 0,23 ммоль) растворили в смеси диметоксиэтан/вода 2 мл/1 мл, после чего перемешивали с микроволновым облучением при 120°С в течение 20 минут. По окончании реакции, реакционную смесь отфильтровали через цеолиты. Фильтрат разбавили водой, и проэкстрагировали с помощью этилацетата. Полученный органический слой высушили над сульфатом магния, и очистили с помощью колоночной хроматографии (этил ацетат:гексан = 1:3) с получением 2-(фуран-3-ил)пиридин-3-ола (50 мг, 27%).
Пример получения 13. Получение 4-фтор-2-(фуран-3-ил)-6-метилфенола (Соединение 6-11): 2-бром-4-фтор-6-метилфенол (337 мг, 1,20 ммоль), 3-фуранилборную кислоту (162 мг, 1,45 ммоль), Pd(dppf)Cl2 (98,3 мг, 0,12 ммоль) и карбонат натрия (383 мг, 3,61 ммоль) растворили в смеси DME: вода = 20 мл: 10 мл, после чего перемешивали с микроволновым облучением при 120°С в течение 20 минут. Реакционную смесь отфильтровали через цеолиты. Фильтрат упарили при пониженном давлении, и очистили с помощью колоночной хроматографии (силикагель; гексан/этил ацетат, 9/1) с получением 4-фтор-2-(фуран-3-ил)-6-метилфенола (96 мг, 30%) в виде желтого масла.
Пример получения 14. Получение 2-(тиофен-3-ил)пиридин-3-ола (Соединение 6-12): 2-Бромпиридин-3-ол (100 мг, 0,58 ммоль), 3-тиофенилборную кислоту (88 мг, 0,69 ммоль), карбонат натрия (184 мг, 2,07 ммоль) и Pd2(dppf)2Cl2 (94 мг, 0,12 ммоль) растворили в смеси диметоксиэтан/вода 1 мл/1 мл, после чего перемешивали с микроволновым облучением при 120°С в течение 20 минут. По окончании реакции, реакционную смесь отфильтровали через цеолиты. Фильтрат разбавили водой, и проэкстрагировали с помощью этилацетата. Полученный органический слой высушили над сульфатом магния, и очистили с помощью колоночной хроматографии (этил ацетат:гексан = 1:3) с получением 2-(тиофен-3-ил)пиридин-3-ола (50 мг, 48%).
Пример получения 15. Получение 2,4-диметил-6-(тиофен-2-ил)фенола (Соединение 6-13): 2-Бром-4,6-диметилфенол (500 мг, 2,49 ммоль), 2-тиофенилборную кислоту (88 мг, 2,99 ммоль), карбонат натрия (791 мг, 7,47 ммоль) и Pd2(dppf)2Cl2 (406 мг, 0,49 ммоль) растворили в смеси диметоксиэтан/вода 6 мл/3 мл, после чего перемешивали с микроволновым облучением при 120°С в течение 20 минут. По окончании реакции, реакционную смесь отфильтровали через цеолиты. Фильтрат разбавили водой, и проэкстрагировали с помощью этилацетата. Полученный органический слой высушили над сульфатом магния, и очистили с помощью колоночной хроматографии (этил ацетат:гексан = 1:3) с получением 2,4-диметил-6-(тиофен-2-ил)фенола (231 мг, 45%).
Пример получения 16. Получение 2-(фуран-2-ил)-4-метоксифенола (Соединение 6-14): 2-Бром-4-метоксифенол (400 мг, 1,97 ммоль), 2-фуранилборную кислоту (287 мг, 2,56 ммоль), Pd(dppf)Cl2 (81 мг, 0,1 ммоль) и карбонат натрия (626 мг, 5,91 ммоль) растворили в смеси DME: вода = 4 мл: 2 мл, после чего перемешивали с микроволновым облучением при 120°С в течение 20 минут. По окончании реакции, реакционную смесь отфильтровали через цеолиты. Фильтрат разбавили водой, и проэкстрагировали с помощью этилацетата. Полученный органический слой высушили над сульфатом магния, и очистили с помощью колоночной хроматографии (гексан:этил ацетат = 9:1) с получением 2-(фуран-2-ил)-4-метоксифенола (212 мг, 57%).
Пример получения 17. Получение 3-(фуран-2-ил)фенола (Соединение 6-15):
3-Бромфенол (500 мг, 2,86 ммоль) и фуран-2-борную кислоту (420 мг, 3,76 ммоль) растворили в смеси диметоксиэтан/вода = 5 мл/2,5 мл. Pd(dppf)Cl2 (118 мг, 0,14 ммоль) и карбонат натрия (919 мг, 8,67 ммоль) добавили к этой смеси, после чего перемешивали с микроволновым облучением при 120°С в течение 20 минут. По окончании реакции, реакционную смесь отфильтровали через цеолиты. Фильтрат разбавили водой, и проэкстрагировали с помощью этилацетата. Полученный органический слой высушили над сульфатом магния, и очистили с помощью колоночной хроматографии(гексан:этил ацетат = 9:1) с получением 3-(фуран-2-ил)фенола (191 мг, 41%).
Пример получения 18. Получение 3-(пиридин-3-ил)фенола (Соединение 6-16):
3-Бромфенол (500 мг, 2,86 ммоль) и пиридин-3-илборную кислоту (422 мг, 3,76 ммоль) растворили в смеси диметоксиэтан/вода = 5 мл/2,5 мл. Pd(dppf)Cl2 (118 мг, 0,14 ммоль) и карбонат натрия (919 мг, 8,67 ммоль) добавили к этой смеси, после чего перемешивали с микроволновым облучением при 120°С в течение 20 минут. По окончании реакции, реакционную смесь отфильтровали через цеолиты. Фильтрат разбавили водой, и проэкстрагировали с помощью этилацетата. Полученный органический слой высушили над сульфатом магния, и очистили с помощью колоночной хроматографии (гексан:этил ацетат = 9:1) с получением 3-(пиридин-3-ил)фенола (386 мг, 78%).
Пример получения 19. Получение 2-(3,6-дигидро-2Н-пиран-4-ил)-4,6-диметилфенола (Соединение 6-17): 2-Бром-4,6-диметилфенол (430 мг, 2,14 ммоль), 2-(3,6-дигидро-2Н-пиран-4-ил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (494 мг, 2,35 ммоль), Pd(dbpf)Cl2 (69,7 мг, 0,11 ммоль) и карбонат натрия (680 мг, 6,45 ммоль) растворили в смеси диметоксиэтан: вода = 3 мл: 1 мл, после чего перемешивали с микроволновым облучением при 120°С в течение 30 минут. Реакционную смесь отфильтровали через цеолиты. Фильтрат упарили при пониженном давлении, и очистили с помощью колоночной хроматографии (силикагель; гексан/этил ацетат, 9/1) с получением 2-(3,6-дигидро-2Н-пиран-4-ил)-4,6-диметилфенола (160 мг, 37%) в виде бесцветного масла.
Пример получения 20. Получение 2-(фуран-2-ил)-4,6-диметилфенола (Соединение 6-18): 2-Бром-4,6-диметилфенол (500 мг, 2,49 ммоль), фуран-2-илборную кислоту (88 мг, 2,99 ммоль), карбонат натрия (791 мг, 7,47 ммоль) и Pd2(dppf)2Cl2 (406 мг, 0,49 ммоль) растворили в смеси диметоксиэтан/вода 6 мл/3 мл, после чего перемешивали с микроволновым облучением при 120°С в течение 20 минут. По окончании реакции, реакционную смесь отфильтровали через цеолиты. Фильтрат разбавили водой, и проэкстрагировали с помощью этилацетата. Полученный органический слой высушили над сульфатом магния, и очистили с помощью колоночной хроматографии (этил ацетат:гексан = 1:3) с получением 2-(фуран-2-ил)-4,6-диметилфенола (231 мг, 45%).
На следующей схеме 7 показаны способы получения исходных соединений (Соединения 1-1) из схемы 1,
[Схема 7]
Пример получения 21. Получение 2,4-диметил-6-(тетрагидрофуран-3-ил)фенола (Соединение 7-2): 2-(фуран-3-ил)-4,6-диметилфенол (Соединение 6-2) (0,6 г, 3,2 ммоль) растворили в метаноле. Добавили палладий на угле (0,06 г), после чего перемешивали под шариком с водородом при комнатной температуре в течение 6 часов. По окончании реакции, реакционную смесь отфильтровали через цеолиты. Фильтрат упарили при пониженном давлении, и очистили с помощью колоночной хроматографии с получением 2,4-диметил-6-(тетрагидрофуран-3-ил)фенола (0,45 г, 73%).
Пример получения 22. Получение 2-метил-6-(тетрагидрофуран-3-ил)фенола (Соединение 7-3): 2-(фуран-3-ил)-6-метилфенол (Соединение 6-9) (173 мг, 0,70 ммоль) и палладий на угле (25 мг, 15%) растворили в вмеси метанол: тетрагидрофуран = 1 мл: 1 мл, после чего перемешивали под шариком с водородом в течение 48 часов. По окончании реакции, реакционную смесь отфильтровали через цеолиты. Фильтрат упарили при пониженном давлении, и очистили с помощью колоночной хроматографии (гексан:этил ацетат = 3:1) с получением 2-метил-6-(тетрагидрофуран-3-ил)фенола (117 мг, 93%).
Пример получения 23. Получение 4-фтор-2-метил-6-(тетрагидрофуран-3-ил)фенола (Соединение 7-4): 4-фтор-2-(фуран-3-ил)-6-метилфенол (Соединение 6-11) (471 мг, 2,45 ммоль) растворили в метаноле. Добавили палладий на угле (64,3 мг, 5 весовых процентов), после чего перемешивали под шариком с водородом в течение 24 часов. По окончании реакции, реакционную смесь отфильтровали через цеолиты. Фильтрат упарили при пониженном давлении. Концентрат очистили с помощью колоночной хроматографии (силикагель; гексан/этил ацетат, 9/1) с получением 4-фтор-2-метил-6-(тетрагидрофуран-3-ил)фенола (368 мг, 76%) в виде твердого вещества белого цвета.
Пример получения 24. Получение 2,4-диметил-6-(тетрагидрофуран-2-ил)фенола (Соединение 7-5): 2-(фуран-2-ил)-4,6-диметилфенол (Соединение 6-18) (200 мг, 1,06 ммоль) растворили в метаноле (3 мл). Добавили палладий на угле (30 мг), после чего перемешивали под шариком с водородом в течение 12 часов. По окончании реакции, реакционную смесь отфильтровали через цеолиты. Фильтрат упарили при пониженном давлении с получением 2,4-диметил-6-(тетрагидрофуран-2-ил)фенола (200 мг, 98%).
Пример получения 25. Получение 2,6-диметил-4-(тетрагидрофуран-3-ил)фенола (Соединение 7-6): 4-(фуран-3-ил)-2,6-диметилфенол (Соединение 6-5) (110 мг, 0,74 ммоль) растворили в смеси тетрагидрофуран: метанол = 1 мл: 1 мл. Добавили палладий на угле (15 мг, 15%), после чего перемешивали под шариком с водородом в течение 18 часов. По окончании реакции, реакционную смесь отфильтровали через цеолиты. Фильтрат упарили при пониженном давлении с получением 2,6-диметил-4-(тетрагидрофуран-3-ил)фенола (74 мг, 72%).
Пример получения 26. Получение 2,4-диметил-6-(тетрагидро-2Н-пиран-4-ил)фенола (Соединение 7-7): 2-(3,6-дигидро-2Н-пиран-4-ил)-4,6-диметилфенол (Соединение 6-17) (234 мг, 1,14 ммоль) растворили в метаноле. Добавили палладий на угле (92 mg, 40 весовых процентов), после чего перемешивали под шариком с водородом в течение 24 часов. По окончании реакции, реакционную смесь отфильтровали через цеолиты. Фильтрат упарили при пониженном давлении. Остаток очистили с помощью колоночной хроматографии (силикагель; гексан/этил ацетат, 9/1) с получением 2,4-диметил-6-(тетрагидро-2Н-пиран-4-ил)фенола (156 мг, 66%) в виде твердого вещества белого цвета.
Пример получения 27. Получение 2-(пирролидин-1-ил)пиридин-3-ола (Соединение 7-9): 2-бромпиридин-3-ол (200 мг, 1,15 ммоль) и пирролидин (184 мг, 2,58 ммоль) поместили в реакционный сосуд, после чего проводили реакцию с одновременным микроволновым облучением при 120°С в течение 20 минут. К реакционной смеси добавили раствор 1 М HCl для нейтрализации, и проэкстрагировали с помощью этилацетата и дихлорметана. Полученный органический слой высушили над сульфатом магния, и упарили при пониженном давлении. Концентрат очистили с помощью колоночной хроматографии с получением 2-(пирролидин-1-ил)пиридин-3-ола (78 мг, 41%).
Структурные формулы соответствуют изображенным в следующих Таблицах 1-9.
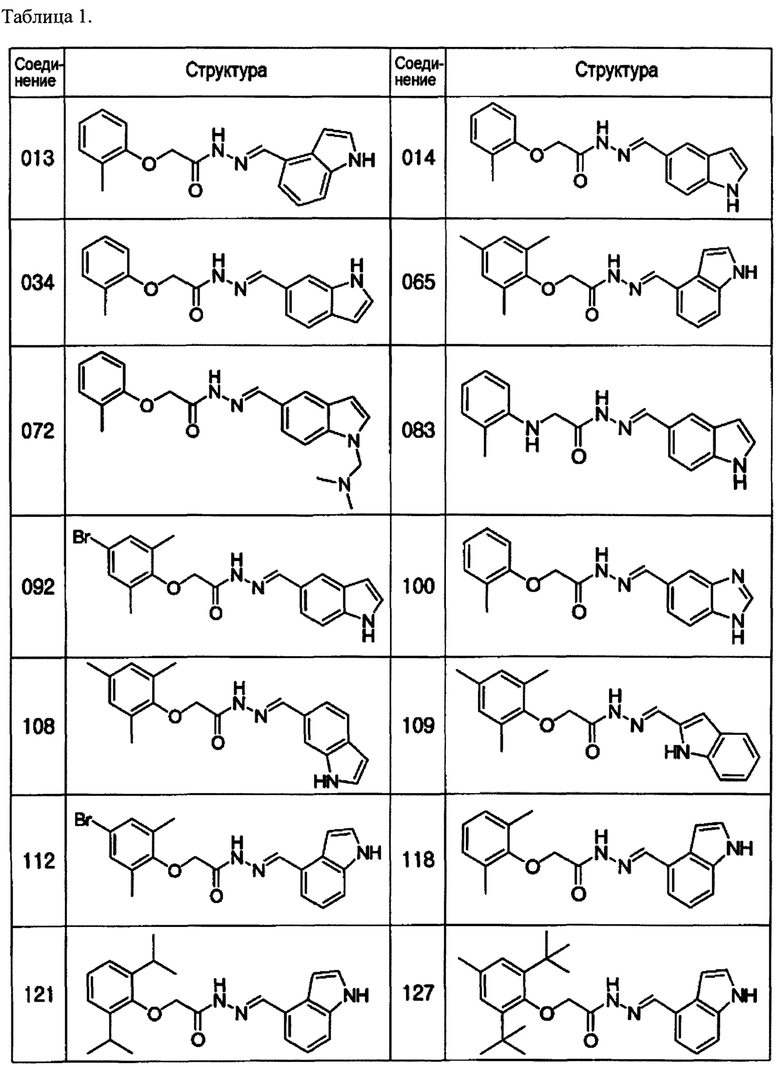
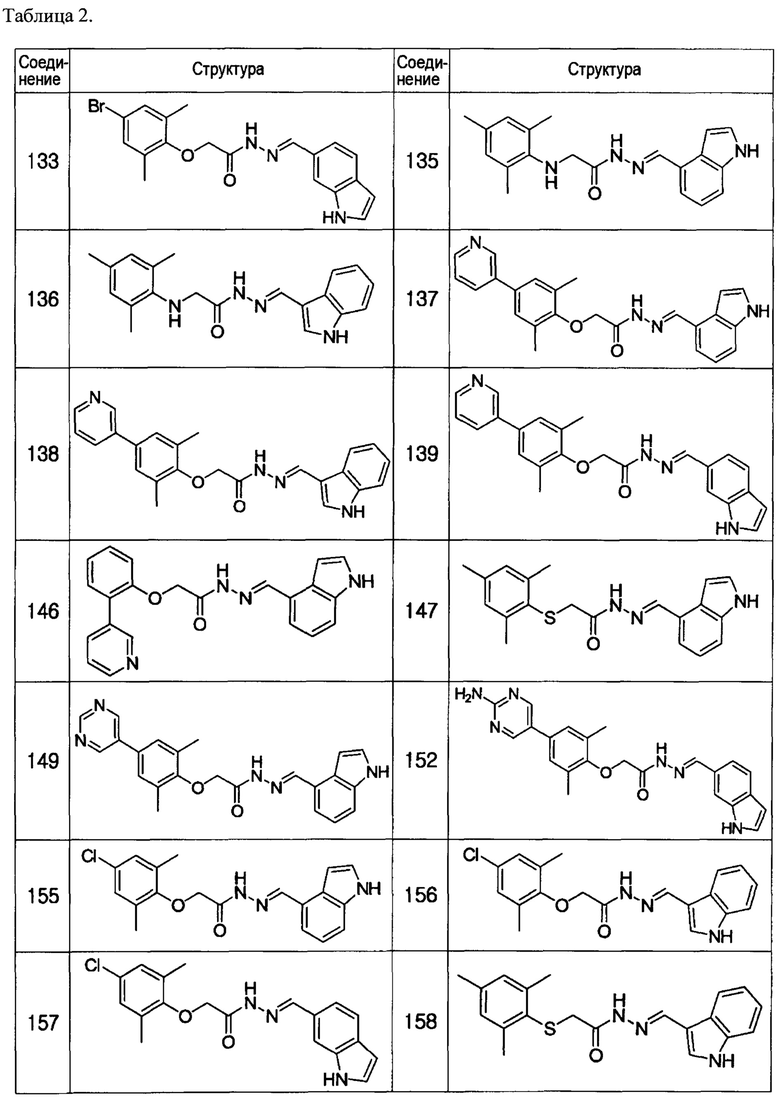
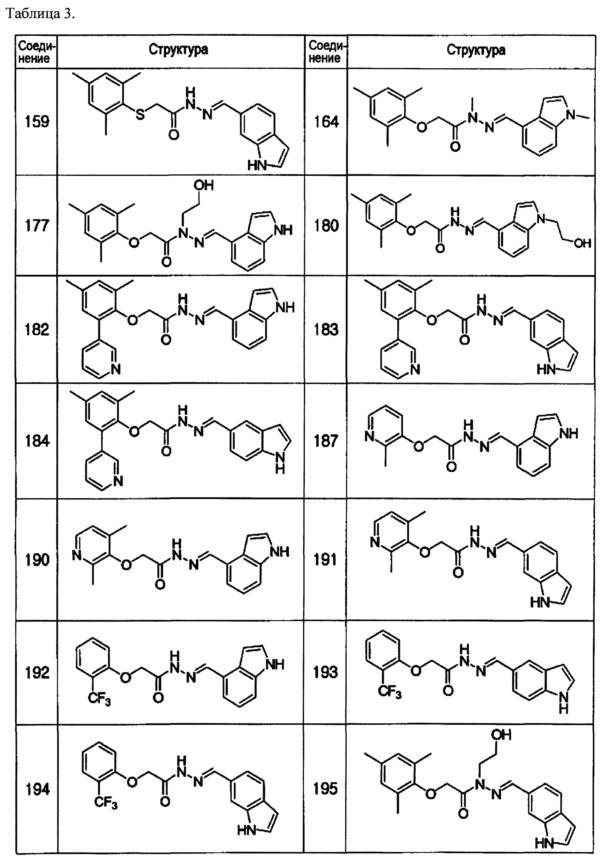
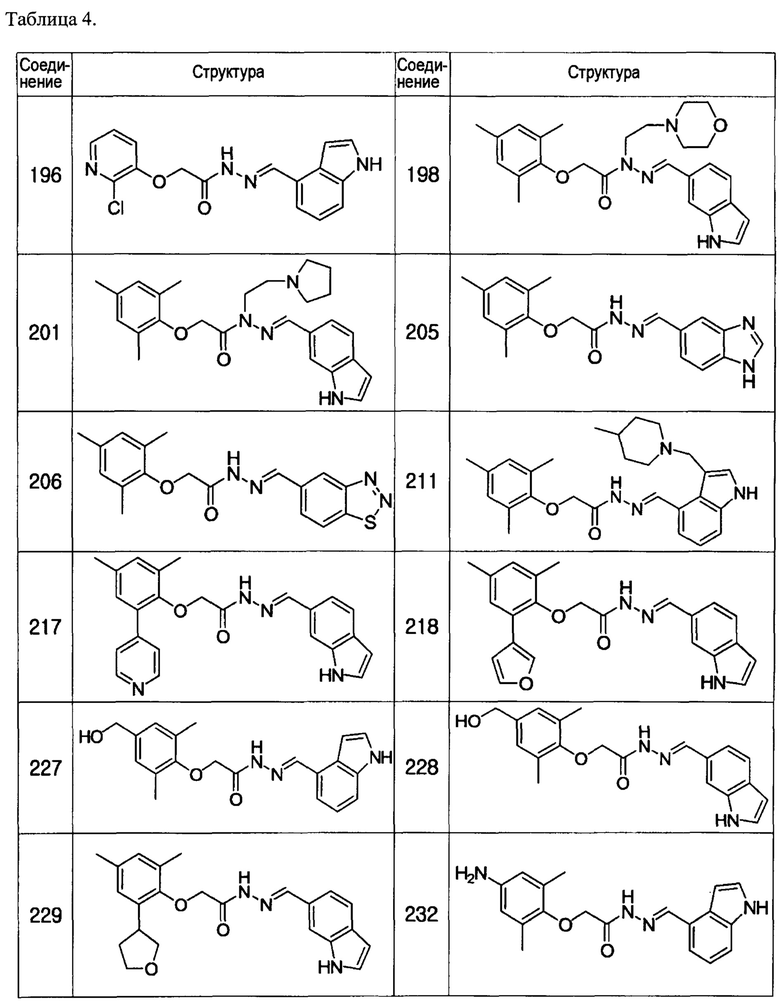
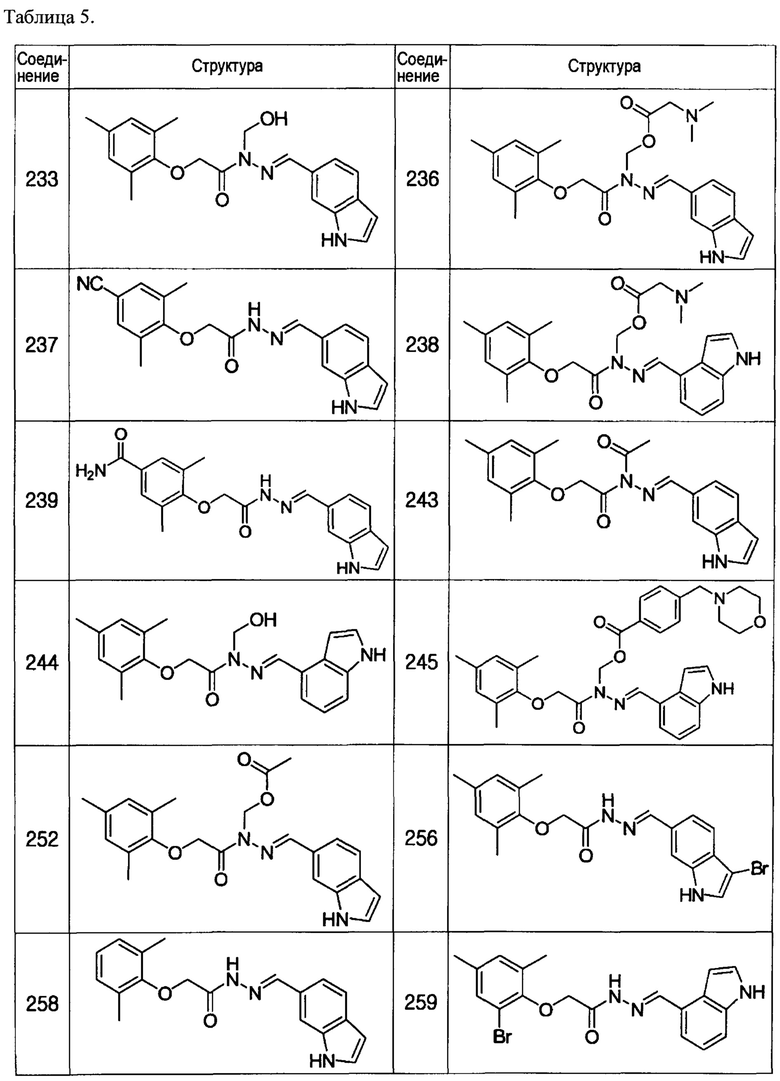
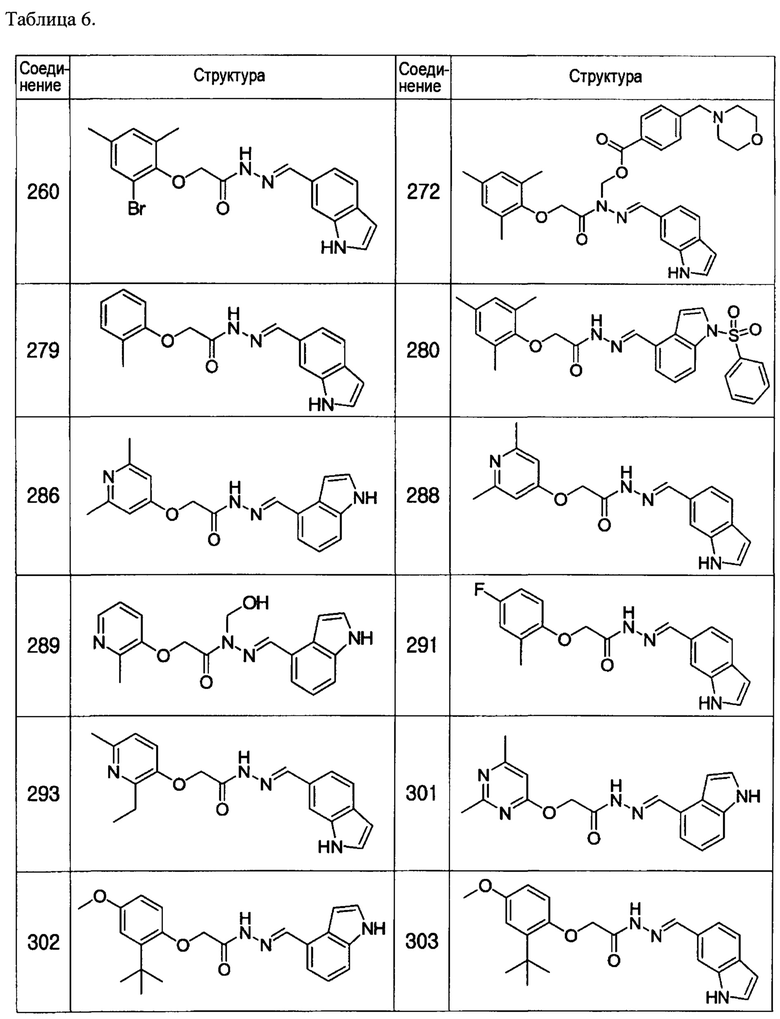
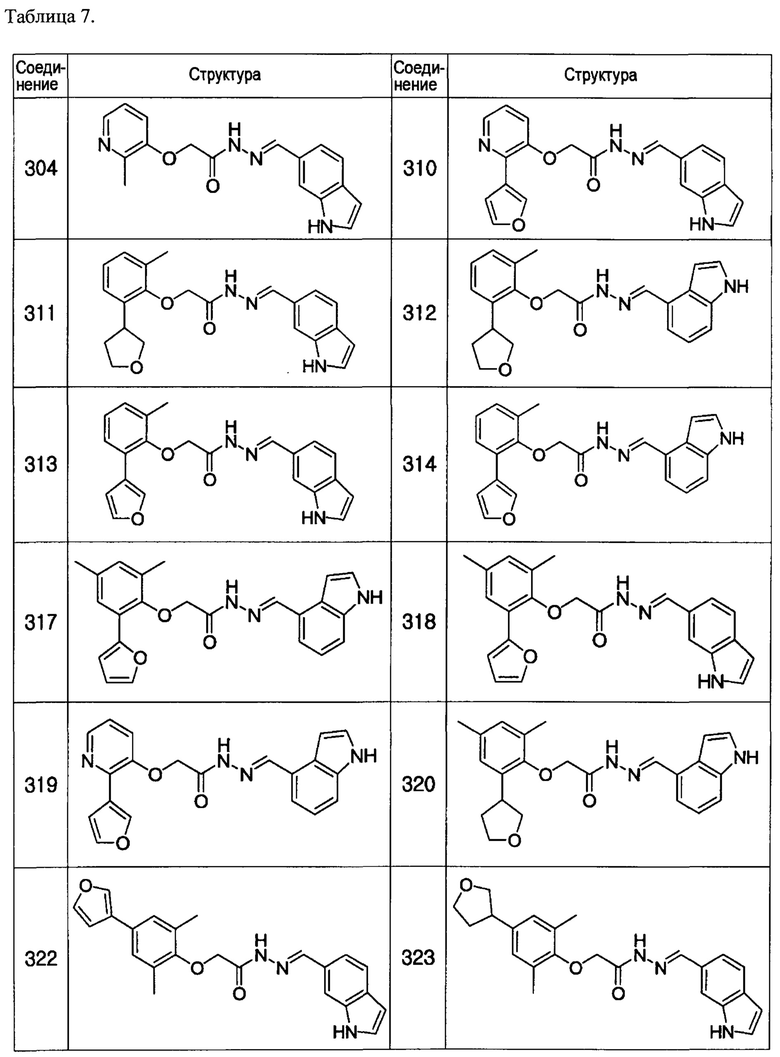
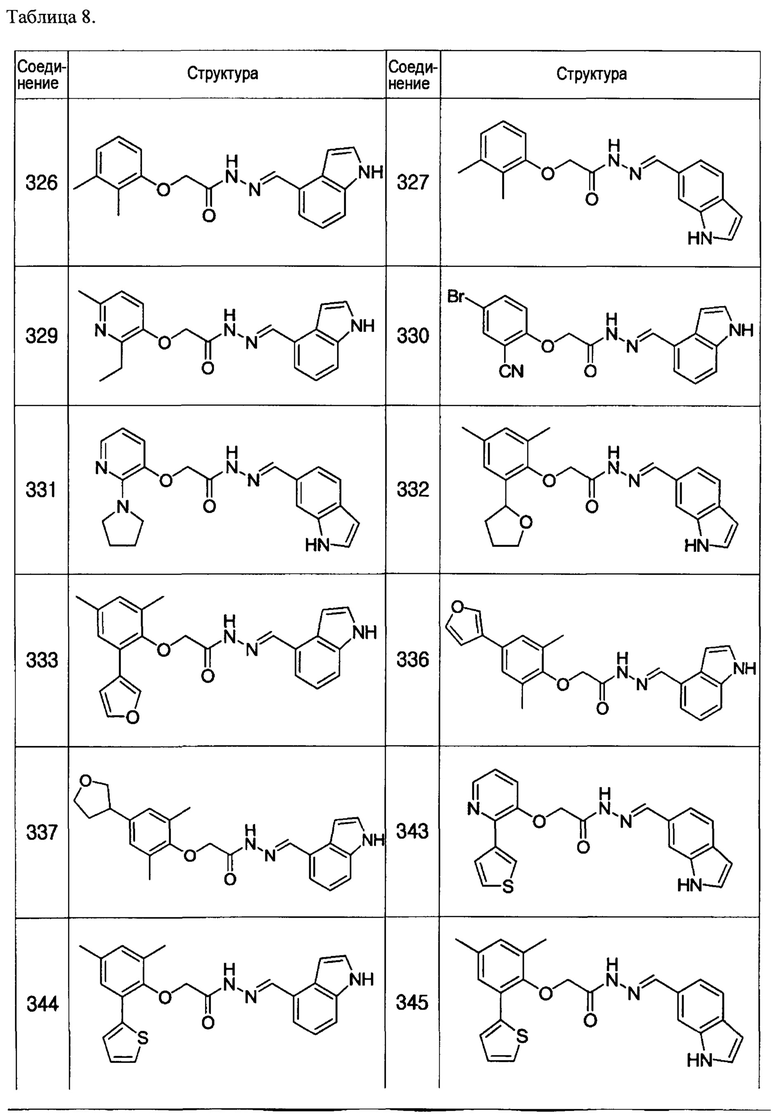
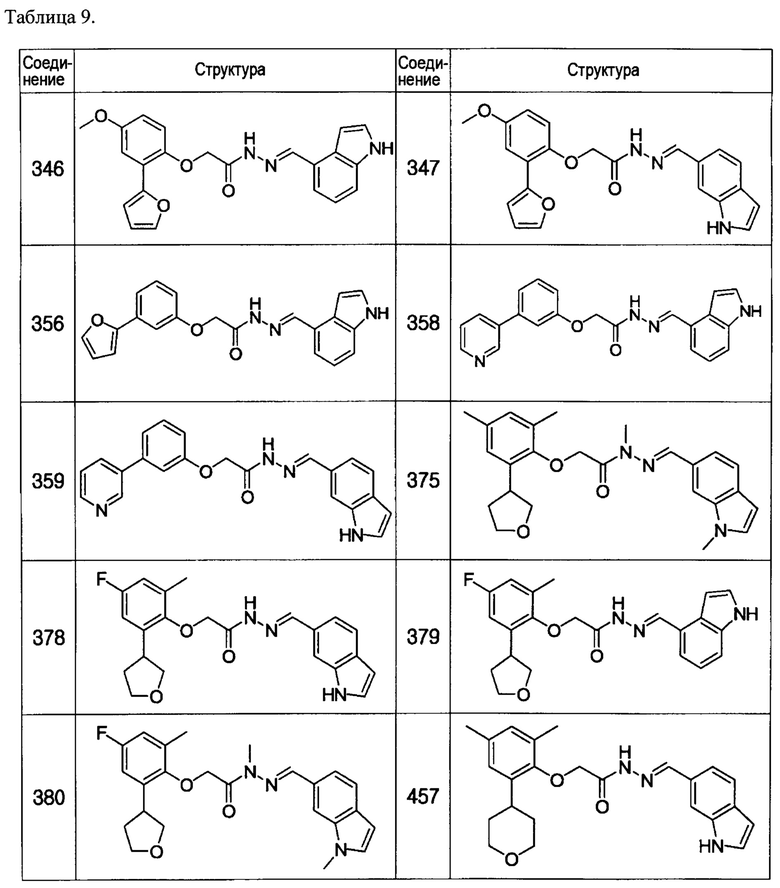
Измерение активности соединений - экспериментальный протокол
Экспериментальный Пример 1: Измерение про-апоптической активности соединений
С целью исследования эффективности полученных соединений, апоптический эффект соединений в отношении клеточной линии человеческих Т-клеток острой лимфобластической лейкемии, Т-клеток Jurkat, оценивали следующим образом.
Т-клетки Jurkat, культивированные в RPMI среде с 10% содержанием эмбриональной бычьей сыворотки (FBS) собрали и окрасили трипановым синим (Sigma). Результат окрашивания показал жизнеспособность клеток 97% или выше. Затем клетки центрифугировали на 1200 об/мин при комнатной температуре в течение 5 минут и повторно суспендировали в 10% FBS-содержащей RPMI среде с концентрацией 1,5×105 клеток/мл. 200 мкл клеточной суспензии поместили в каждую лунку плоскодонного 96-луночного планшета (Costar). Затем, клетки обработали 10 мкл разбавленных соединений в RPMI среде с конечными концентрациями 1 мкмоль/л или 5 мкмоль/л, и инкубировали в 5% CO2 инкубаторе при 37°С в течение 72 часов. В контрольной группе с носителем, клетки обработали 10 мкл RPMI среды с содержанием 1% диметилсульфоксида (DMSO, Sigma). Инкубированные клетки собрали, поместили в 5-мл FACS пробирки (BD Falcon), и промыли 1 мл фосфатного буферного раствора (PBS), после чего повторно суспендировали в 0,1 мл связывающего буферного раствора (10 ммоль/л Hepes-NaOH, pH 7,4, 140 ммоль/л NaCl, 2,5 мл CaCl2). Суспендированные клетки окрасили флуоресцеин изотиоцианатом (FITC)-Аннексином V (BD Biosciences) и 7-амино-актиномицином D (7-AAD, e-Biosciences) при комнатной температуре в течение 15 минут. Окрашенные клетки суспендировали в 0,3 мл связывающего буферного раствора и проанализировали с помощью проточного цитометра. Апоптоз определяли по доле (%) клеток, окрашенных FITC-Аннексином V и 7-AAD от общего количества клеток (см. Таблицы 10 и 11).
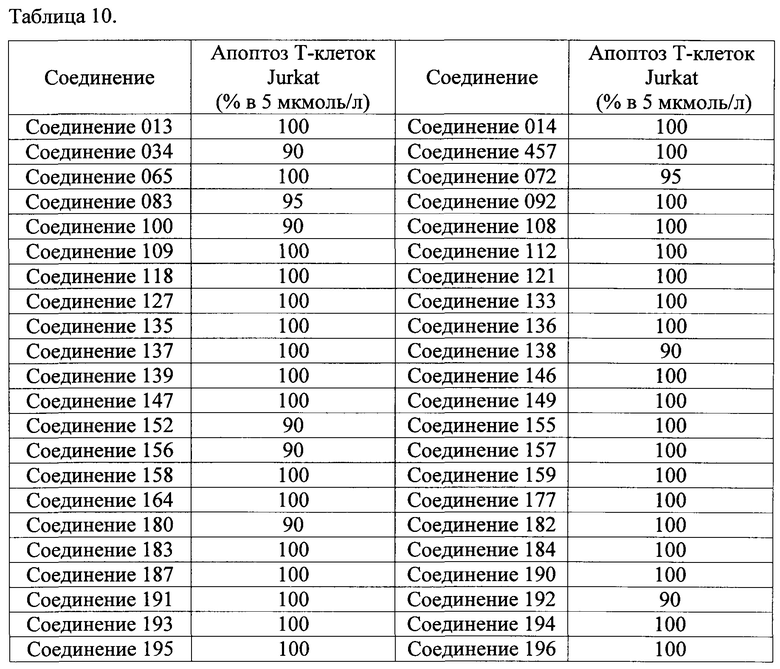
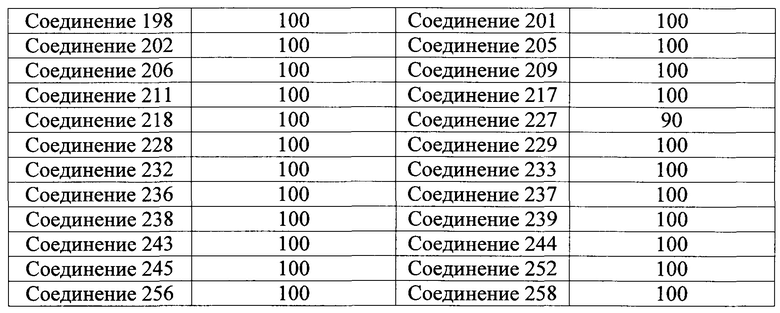
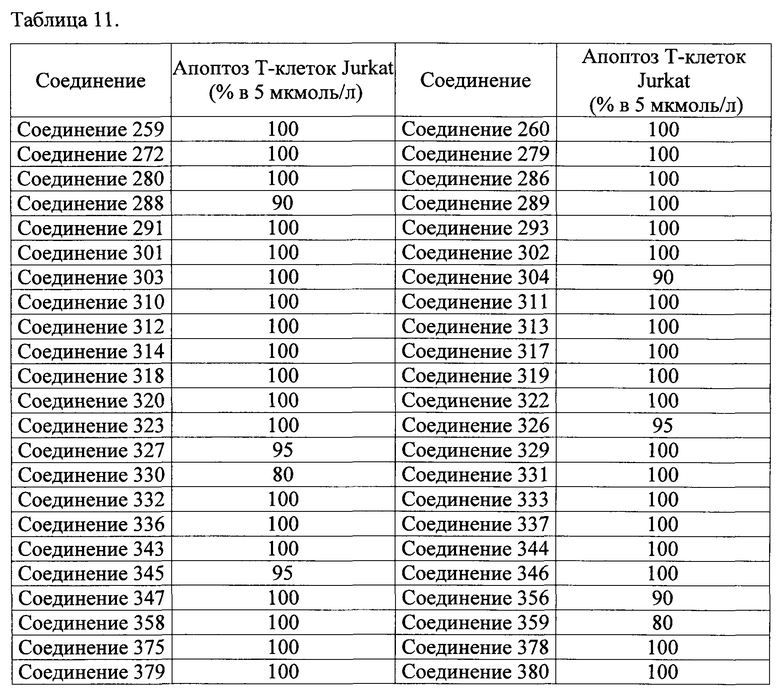
Экспериментальный Пример 2: Ингибиторный эффект в отношении аллоантиген-специфических Т клеток in vitro
С целью оценки ингибиторного эффекта соединения 229 по данному изобретению в отношении аллоантиген-специфичных Т клеток, провели следующий эксперимент.
Лимфатические узлы и селезенки асептически собрали у нормальных C57BL/6 мышей (возраст 8-12 недель, женского пола, 20±2 г) и измельчили с добавлением RPMI среды. После этого, измельченные ткани пропустили через клеточное сито (BD Falcon) для получения клеточных суспензий. Клеточную суспензию селезенки центрифугировали с 1200 об/мин в течение 5 минут. Надосадочную жидкость удалили, после чего к клеткам добавили 1 мл аммоний хлорид/бикарбонат калия (ACK) лизисного буфера (0,15 М NH4Cl, 1 ммоль/л KHCO3, 0,1 ммоль/л Na2EDTA), после чего встряхивали в течение 1 минуты и промыли RPMI средой. Эритроцит-лизированные клетки селезенки центрифугировали вместе с клетками лимфатических узлов, после чего повторно суспендировали в 1 мл буфера мангитно-активированной клеточной сортировки (MACS) (0,5% BSA, 2 ммоль/л EDTA в PBS), и добавили 100 мкл мышиных CD90,2 микрогранул (Miltenyi Biotec) после чего инкубировали при 4°С в течение 15 минут. К инкубированной клеточной суспензии добавили 10 мл MACS буфера, центрифугировали в течение 5 минут, промыли, после чего повторно суспендировали в 2 мл MACS буфера. Суспендированные клетки поместили в autoMACS Pro Separator (Miltenyi Biotec), и отделили Т клетки связанные с CD90,2 микрогранулами. Выделенные Т-клетки промыли фосфатным буферным раствором (PBS), после чего повторно суспендировали в PBS с концентрацией 1×108 клеток/мл. Нормальных BALB/c мышей (возраст 10-12 недель, женского пола, 20-23 г) облучали 950 рад γ радиации с помощью IBL 437С облучателя (CIS bio international), после чего 100 мкл приготовленной ранее суспензии Т-клеток C57BL/6 мышей ввели в хвостовую вену BALB/c мышей. Через 3 дня после инъекции клеток мышей умертвили и извлекли селезенки. Из извлеченных селезенок были получены свободные от эритроцитов клеточные суспензии описанным выше способом, и донорные Т-клетки отделили от селезеночной клеточной суспензии с помощью autoMACS Pro Separator, тем самым получив вторичные реактивные клетки. В то же время, Т-клетки выделили из нормальных C57BL/6 мышей таким же способом, как было описано выше, тем самым получив первичные реактивные клетки. И первичные, и вторичные реактивные клетки повторно суспендировали в 1 мл 1% FBS-содержащей PBS, и обработали 3 мкмоль/л карбоксифлуоресцеин сукцинимидиловым эфиром (CFSE, Invitrogen), после чего оставили реагировать при комнатной температуре в течение 4 минут. Чтобы остановить реакцию к клеточной суспензии добавили холодный 5% FBS-содержащей PBS, после чего промыли. Промытые клетки повторно суспендировали в 10% FBS-содержащей RPMI среды с концентрацией 2×106/мл. Селезенки нормальных BALB/c мышей (возраст 8-12 недель, женского пола, 20±2 г) собрали асептическим образом. Свободные от эритроцитов клетки селезенки были получены таким же способом, как было описано выше, после чего их повторно суспендировали в 1 мл of PBS. 0,5 мг/мл Mitomycin-C (Sigma) добавили к суспензии с концентрацией 0,5 мкг/мл, после чего инкубировали при 37°С в течение 20 минут, после чего трижды промыли избыточным количеством PBS. Промытые клетки повторно суспендировали в 10% FBS-содержащей RPMI среды с концентрацией 4×106 клеток/мл, тем сеемым получив клетки-стимуляторы. Первичные и вторичные реактивные клетки смешали со стимуляторными клетками в соотношении 1:1 отдельно, и 200 мкл каждой клеточной смеси поместили в каждую лунку круглодонного 96-луночного планшета (Costar). После этого, клетки обработали 10 мкл соединения 229 разбавленного в RPMI среде в различных концентрациях, и инкубировали в 5% СO2 инкубаторе при 37°С в течение 72 часов. В контрольной группе с носителем, к клеткам добавили 10 мкл 0,2% DMSO-содержащей RPMI среды. Инкубированные клетки поместили в 5-мл FACS пробирки и промыли 1 мл буфера для клеточной сортировки с возбуждением флуоресценции (FACS) (1% FBS и 0,1% азаид натрия в PBS). Промытые клетки повторно суспендировали в 0,1 мл FACS буферного раствора, после чего обработали 1 мкг мышиного иммуноглобулина G (IgG, e-Biosciences) с целью предотвращения неспецифического связывания антител, после чего инкубировали при 4°С в течение 15 минут. После этого, клетки окрасили 0,5 мкг фикоэритрин-конъюгированного антимышиного Н-2Kb mAb (e-Biosciences) и окрашивали при 4°С в течение 30 минут. После окрашивания, клетки дважды промыли 1 мл FACS буферного раствора, повторно суспендировали в 0,1 мл FACS буферного раствора, окрасили 7-AAD при комнатной температуре в течение 5 минут. Окрашенные клетки суспендировали в 0,3 мл FACS буферного раствора после чего проанализировали с помощью проточного цитометра. Ингибирование пролиферации Т-клеток определили путем измерения отношения (%) клеточной фракции с низкой интенсивностью флуоресценцией CFSE к общему количеству живых реактивных клеток, и апоптоз Т-клеток определили путем измерения отношения (%) клеток, окрашенных 7-амино-актиномицином D (7-AAD) к общему количеству реактивных клеток.
Результаты показали, что доля Т-клеток (CFSElow) размножившихся в ответ на аллоантиген в контрольной группе носителя составила 50% от общего количества Т-клеток, но уменьшилась до 3% или менее в группе, обработанной 0,5 мкмоль/л или больше соединением 229, что позволяет предположить, что пролиферация Т-клеток, реагирующих на аллоантиген, была существенно ингибирована. Кроме того, в условиях вторичной реакции, в которых Т-клетки активированные главным образом аллоантигеном in vivo реагировали с тем же антигеном in vitro, соединение 229 эффективно ингибировало пролиферацию аллоантиген-специфичных Т-клеток (см. Фиг. 1.). С целью проверки того, может ли ингибиторный эффект соединения 229 по отношению к аллоантиген-специфичным Т-клеткам также вызывать апоптоз, клетки окрасили 7-AAD и проанализировали с помощью проточной цитометрии. В результате оказалось, что при первичной реакции, апоптоз в проверочной группе, обработанной 0,5 мкмоль/л или более соединения 229 повысился на 14-19% по сравнению с контрольной группой, и при вторичной реакции, апоптоз в проверочной группе, обработанной 0,25 мкмоль/л или более соединения 229 значительно увеличился (21-42%) % по сравнению с контрольной группой (см. ФИГ. 2 и таблицу 12).
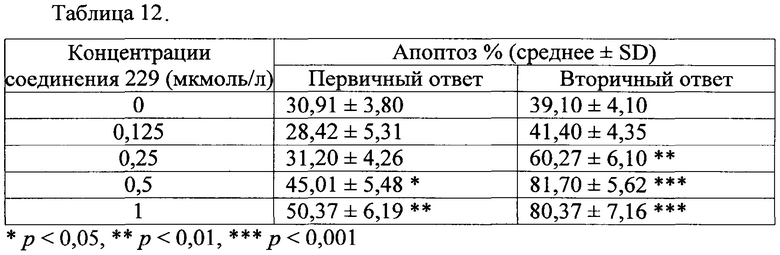
Таким образом, соединение 229 по данному изобретению обладает ингибиторным эффектом в отношении пролиферации всех Т-клеток, которые первично и вторично реагируют на аллоантиген, и этот ингибиторный эффект является следствием вызванного апоптоза. Кроме того, пролиферация вторично активированных Т-клеток ингибируется даже при более низких концентрациях соединения 229, и поэтому соединение 229 можно эффективно использовать в качестве иммунодепрессивного агента не только для предотвращения, но и для лечения отторжения трансплантата.
Экспериментальный Пример 3: Ингибиторный эффект в отношении аллоантиген-специфичных Т-клеток in vivo
С целью оценки ингибиторного эффекта соединения 229 по данному изобретению в отношении Т-клеток, реагирующих на аллоантиген in vivo, провели следующий эксперимент на животных моделях.
Лимфатические узлы и селезенки асептически собрали у нормальных C57BL/6-Ly5,1 мышей (возраст 8-12 недель, женского пола, 20±2 г), и выделили Т-клетки таким же образом, как было описано в экспериментальном примере 2, и затем повторно суспендировали в PBS с концентрацией 1×108 клеток/мл. Нормальных BDF1 мышей (возраст 10-12 недель, женского пола, 20-23 г) облучили 950 рад γ радиацией, после чего 100 мкл ранее приготовленных Т-клеток C57BL/6-Ly5,1 мышей ввели в хвостовую вену BDF1 мышей. Соединение 229 полностью растворили в Cremophor EL (Sigma)/этанол смеси (1:1, v/v) в количестве, соответствующем 15% (объемных) вводимого раствора для внутрибрюшинного введения и в количестве, соответствующем 7,5% (объемных) для орального введения, и растворы соединений разбавили PBS. Раствор вводили внутрибрюшинно (200 мкл/мышь) или орально (500 мкл/мышь) каждый день в течение 5 дней начиная с дня введения клеток. Контрольной группе вводили Cremophor EL-этанол-PBS смесь в таком же соотношении и объеме, как и экспериментальной группе. Через 6 дней после введения клеток мышей умертвили, извлекли селезенки, и свободные от эритроцитов клетки были получены таким же способом, как описано в экспериментальном примере 2. Клетки селезенки окрасили трипановым синим и подсчитали количество живых клеток на орган каждого животного. После этого, клетки селезенки поместили в 5 мл FACS пробирки и промыли 1 мл FACS буферного раствора. Клетки повторно суспендировали в 0,1 мл FACS буферного раствора, после чего обработали 1 мкг of мышиного иммуноглобулина G с целью предотвращения неспецифического связывания антитела, и инкубировали при 4°С в течение 15 минут. После этого, клетки окрасили 0,5 мкг фикоэритрин-конъюгированным антимышиным Ly5,1 mAb (моноклональное антитело) при 4°С в течение 30 минут. Окрашенные клетки дважды промыли 1 мл FACS буферного раствора, повторно суспендировали в 0,3 мл FACS буферного раствора и проанализировали с помощью проточного цитометра. Количество донорных Т-клеток в селезенке определили путем измерения доли (%) донорной фракции Т-клеток (Ly5,1+ клеток) относительно живых клеток на селезенку.
Результаты показали, что количество аллоантиген-специфичных Т-клеток в селезенке мышей, получавших внутрибрюшинно (Фиг. 3А) или орально (Фиг. 3В) соединение 229 по данному изобретению существенно ниже в сравнении с контрольной группой в зависимости от концентрации, и когда соединение 229 вводили в дозировке 50 мг/кг, количество активированных Т-клеток было существенно ниже, что позволяет предположить, что пролиферация Т-клеток, отвечающих на аллоантиген, была полностью ингибирована (см. Фиг. 3А и 3В).
Таким образом, соединение 229 по данному изобретению обладает ингибирующим эффектом в отношении аллоантиген-специфичных Т-клеток in vivo, и может использоваться в качестве орального иммунодепрессивного агента.
Экспериментальный Пример 4: Ингибиторный эффект в отношении острого заболевания «трансплантат против хозяина»
С целью проверки ингибиторного эффекта соединения 229 по данному изобретению в отношении острого заболевания «трансплантат против хозяина», острое заболевание «трансплантат против хозяина» было стимулировано в мышиной модели алогенной трансплантации костного мозга согласно описанию, приведенному ниже, мыши получали соединение 229, и оценивалась эффективность действия соединения.
Лимфатические узлы, селезенки, обе бедренные кости и голени асептически отбирали у нормальных C57BL/6 мышей (возраст 8-12 недель, женского пола, 20-23 г), и Т-клетки изолировали из лимфатических узлов и селезенок таким же образом, как было описано в экспериментальном примере 2 и повторно суспендировали в PBS с концентрацией 8×107 клеток/мл. Бедренные кости и голени обрезали с концов и вымывали костный мозг RPMI средой с помощью шприца (21G для бедренной кости и 26G для голени), который затем суспендировали и пропускали через клеточное сито для создания клеточной суспензии. Суспензию клеток костного мозга центрифугировали на 1200 об/мин в течение 5 минут, удаляли надосадочную жидкость, затем добавляли 1 мл ACK лизисный буфер и встряхивали 1 минуту, после чего промывали RPMI средой. Отцентрифугированные клетки костного мозга повторно суспендировали в 300 мкл MACS буфера, и к этой смесит добавляли 30 мкл CD90,2 микрогранул, после чего инкубировали при 4°С в течение 15 минут. К инкубированной клеточной суспензии добавляли 5 мл MACS буфера, центрифугировали в течение 5 минут, промывали, после чего повторно суспендировали в 2 мл MACS буфера. Суспендированные клетки поместили в autoMACS Pro Separator, и удалили Т-клетки, связанные с CD90,2 микрогранулами. Отделенные клетки костного мозга, лишенные Т-клеток (TCD-BM) промыли PBS и повторно суспендировали в PBS с концентрацией 1×108 клеток/мл. Нормальных BDF1 мышей (возраст 10-12 недель, женского пола, 20-23 г) облучили 850 рад γ радиации с использованием излучателя, после чего 100 мкл трансплантата, полученного смешиванием C57BL/6 мышиных Т-клеток и TCD-BM в соотношении 1:1 вводили в хвостовую вену BDF1 мышей. В группе, получившей соединение 229 по данному изобретению, соединение 229 было полностью растворено в Cremophor EL-этанол смеси (1:1 по объему) в количестве, соответствующем 15% (объемных) вводимого объема, после чего раствор с соединением разбавили PBS, и этот раствор вводили внутрибрюшинно каждой мыши в дозировке 25 мг/кг ежедневно в течение 10 дней со дня введения трансплантата. Тем временем, такролимус, который в данный момент является клинически используемым иммунодепрессивным агентом, вводили мышам из другой группы в том же количестве, как и соединение 229, с целью сравнения ингибиторного эффекта в отношении острого заболевания «трансплантат против хозяина», а мышам из контрольной группы вводили 200 мкл смеси Cremophor EL-этанол-PBS (7,5:7,5:85 по объему) таким же образом, как и экспериментальным группам. После этого, ежедневно контролировали выживание мышей, и тяжесть GVHD оценивали дважды в неделю с помощью клинической системы оценки GVHD, включающей уменьшение веса, сутулость, пониженную активность, нарушение мехового покрова и поражения кожи. Каждый параметр оценивался по трехбалльной шкале (от 0 до 2).
Результаты показали, что мыши в контрольной группе начали умирать начиная с 29 дня после трансплантации клеток и на 70 день продемонстрировали уровень выживания в 10%, а мыши из группы, получавшей лечение такролимусом, начали умирать на 23 день и продемонстрировали финальный уровень выживания в 50%. Между тем, в группах, получавшей лечение соединением 229 по данному изобретению, мышиные смерти начались на 35 и 46 день после трансплантации клеток, и финальный уровень выживания оказался 80%, что существенно выше, чем в контрольной группе. Также, уровень выживания в группе, получавшей лечение соединением 229 оказался выше, чем в группе, получавшей лечение такролимусом, но это повышение было не очень значительным (см. Фиг. 4А). По результатам оценки тяжести GVHD с помощью клинической оценочной системы, у мышей, получавших лечение такролимусом, наблюдалось повреждение мехового покрова и кожи, тогда как у мышей, получавших лечение соединением 229, наблюдались легкие симптомы GVHD, включая легкую потерю веса. Это уменьшение симптомов GVHD при лечении соединением 229 было статистически значительным (см. Фиг. 4В).
Соответственно, соединение 229 по данному изобретению обладает ингибиторным эффектом в мышиных моделях GVHD, при этом ингибиторное действие соединения 229 превосходит таковое у традиционного иммунодепрессивного агента такролимуса. Так, соединение 229 по данному изобретению может эффективно использоваться в качестве нового иммунодепрессивного агента.
Экспериментальный Пример 5: Эффективность при лечении множественного склероза
С целью проверки терапевтической эффективности соединения 229 по данному изобретению в отношении множественного склероза, экспериментальный аутоиммунный энцефаломиелит (ЕАЕ) стимулировали в мышиной модели согласно приведенному ниже описанию, мыши получали соединение 229, и оценивалась эффективность действия соединения.
В первый день стимулирования, миелин дендроцит гликопротеин35-55 (MOG35-55, Peptron) (200 мкг), миобактерия туберкулеза (Difco, Cat No. 231141) (500 μg) и полный адъювант Фрейнда (Sigma Aldrich, Cat No. F5506) смешали и оставили погруженным на 5 минут. 0,1 мл иммерсионного пептида подкожно вводили в оба бока каждой C57BL/6 мыши (возраст 9 недель, женского пола, 18±2 г), после чего 0,1 мг коклюшного токсина (Sigma Aldrich, Cat No. P2980) (200 нг) вводили внутривенно. Через два дня, внутривенно вводили такое же количество токсина. Проверяли утечку иммерсии и факт наступления ЕАЕ посредством визуального наблюдения в течение 7 дней.
Соединение 229 и финголимод, являющийся в данное время клинически используемым препаратом для лечения множественного склероза, полностью растворили в смеси Cremophor EL-этанол (1:1 по объему) в количестве, соответствующем 7,5% (объемных) вводимого объема, раствор соединений разбавили PBS. Две группы мышей получали орально 200 мкл разбавленного раствора соединения 229 в дозировке 25 и 50 мг/кг, и третья группа мышей получала орально 200 мкл разбавленного раствора финголимода в дозировке 1 мг/кг ежедневно с 17 по 37 день (всего 21 раз) после начала эксперимента. В контрольной группе с носителем, 200 мкл смеси Cremophor EL-этанол-PBS (3,75: 3,75: 92,5 по объему) вводили каждой мыши таким же образом, как в экспериментальных группах. Индекс ЕАЕ записывали ежедневно на основе визуальных наблюдений с помощью системы оценки тяжести заболевания по шестизначной шкале от нуля до пяти начиная с седьмого дня эксперимента. Симптомы множественного склероза оценивали по следующим критериям:
0: без патологии;
1: обмякший хвост;
2: обмякший хвост и слабость задних конечностей;
3: паралич задних конечностей;
4: паралич задних конечностей и слабость передних конечностей; и
5: смерть.
Результаты анализа свидетельствуют о том, что во всех экспериментальных группах ЕАЕ развивался начиная с 7 дня эксперимента и индекс тяжести на 17 день эксперимента составлял 3±1,25, означая 100% развитие острой реакции. В случае мышей из контрольной группы, индекс тяжести составил 3±1 на 17 день эксперимента, 2,5±0,75 на 39 день (период хронической реакции) и 2,33±0,25 на 101 день эксперимента. Контрольная группа продемонстрировала ремиссионно-рецидивирующий характер реакции и высокий индекс тяжести на протяжении всего эксперимента. В финголимодной группе индекс тяжести составил 3±1 на 17 день эксперимента, но только 1,5±0,5 на 39 день эксперимента, что означает что группа, получавшая финголимод, продемонстрировала облегченную острую реакцию и мягкую хроническую реакцию, по сравнению с контрольной группой. Однако, финголимодная группа продемонстрировала увеличение индекса тяжести заболевания с 39 дня эксперимента по окончании введения медикамента и показала высокий индекс тяжести 2,7±0,25 на 101 день эксперимента. Также, группа, получавшая 25 мг/кг соединения 229 по данному изобретению продемонстрировала индекс тяжести заболевания равный 3±1 на 17 день эксперимента, что соответствует индексу тяжести контрольной группы, но продемонстрировала индекс тяжести заболевания равный 1,25±0,75 на 39 день эксперимента и 1,17±0,75 на 55 день эксперимента, что позволяет предположить статистически существенный терапевтический эффект по сравнению с контрольной группой. Однако, в контрольной группе, получавшей 25 мг/кг соединения 229, индекс тяжести заболевания начал расти на 57 день эксперимента и составил 1,83±0,5 на 101 день эксперимента, что меньше чем в контрольной группе, но немного больше, чем на начальной стадии эксперимента (#, р<0,05; ##, р<0,01; и ###, р<0,001, в сравнении с контрольной группой; см. Фиг. 5). Более того, группа, получавшая 50 мг/кг соединения 229 по данному изобретению, продемонстрировала индекс тяжести заболевания равный 3±0,75 на 17 день эксперимента, что соответствует контрольной группе, но продемонстрировала индекс тяжести заболевания равный 0,9±0,25 на 39 день эксперимента и 0,63±0,25 на 101 день эксперимента, что позволяет предположить наличие статистически существенно терапевтического эффекта по сравнению с контрольной группой (*, р<0,05; **, р<0,01; и ***, р<0,001, в сравнении с контрольной группой; см. Фиг. 5). В группе, получавшей 50 мг/кг соединения 229, терапевтический эффект постоянно поддерживался после орального ведения соединения, и этот эффект статистически значительно отличался от такового в группе, получавшей финголимод, на 49 день эксперимента (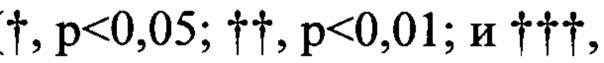 р<0,001, в сравнении с финголимодной группой; см. Фиг. 5). Когда соединение 229 по данному изобретению использовали в дозировке 25 мг/кг, оно продемонстрировало прекрасный терапевтический эффект на начальной стадии, но слегка незначительный эффект при предотвращении рецидива, а когда соединение 229 по данному изобретению использовали в дозировке 50 мг/кг, оно продемонстрировало терапевтический эффект на начальной стадии и непрерывный эффект по предотвращению рецидива. Следовательно, соединение 229 по данному изобретению обладает терапевтическим эффектом против ЕАЕ в мышиной модели, и этот терапевтический эффект более значителен и продолжается в течении более длительного времени по сравнению с финголимодом, который является традиционным терапевтическим агентом против множественного склероза. Таким образом, соединение 229 по данному изобретению является новым терапевтическим агентом для орального применения против множественного склероза, который можно использовать для более эффективной терапевтической стратегии. Экспериментальный Пример 6: Эффективность в отношении апоптоза лимфолейкозных клеточных линий
р<0,001, в сравнении с финголимодной группой; см. Фиг. 5). Когда соединение 229 по данному изобретению использовали в дозировке 25 мг/кг, оно продемонстрировало прекрасный терапевтический эффект на начальной стадии, но слегка незначительный эффект при предотвращении рецидива, а когда соединение 229 по данному изобретению использовали в дозировке 50 мг/кг, оно продемонстрировало терапевтический эффект на начальной стадии и непрерывный эффект по предотвращению рецидива. Следовательно, соединение 229 по данному изобретению обладает терапевтическим эффектом против ЕАЕ в мышиной модели, и этот терапевтический эффект более значителен и продолжается в течении более длительного времени по сравнению с финголимодом, который является традиционным терапевтическим агентом против множественного склероза. Таким образом, соединение 229 по данному изобретению является новым терапевтическим агентом для орального применения против множественного склероза, который можно использовать для более эффективной терапевтической стратегии. Экспериментальный Пример 6: Эффективность в отношении апоптоза лимфолейкозных клеточных линий
С целью проверки ингибиторной эффективности соединения 457 в отношении лимфолейкозных клеточных линий, провели следующий эксперимент.
Каждую из клеточных линий острой Т-клеточная лейкемии Jurkat, Molt4 и CCRF-СЕМ, клеточную линию острой В-клеточной лейкемии Raji и клеточные линии множественного склероза KMS11, KMS12BM, KMS26, KMS28BM, и IM9 повторно суспендировали в 10% FBS-содержащей RPMI среде с концентрацией 1,5×105 клеток/мл, и 200 мкл каждой клеточной суспензии культивировали на плоскодонном 96-луночном планшете (Costar). Клеточную линии острой NK-клеточной лимфомы NK-92MI повторно суспендировали в α-МЕМ среде (с добавками 20% FBS, 1X MEM витаминного раствора, 50 мкмоль/л 2-меркптоэтанола и 1X пенициллин/страптомицин) с концентрацией 1,5×105 клеток/мл, и 200 мкл клеточной суспензии культивировали на плоскодонном 96-луночном планшете. Клетки в каждой лунке обработали 10 мкл каждой из концентраций 0,1, 0,25, 0,5, 0,75 и 1 мкмоль/л соединения, разбавленного RPMI средой, после чего клетки инкубировали в 5% СО2, инкубаторе при 37°С в течение 24 часов. В контрольной группе с носителем, клетки обработали 10 мкл среды с содержанием 0,05% диметилсульфоксида (DMSO, Sigma).
Клетки собрали, после чего апоптоз проанализировали с помощью проточного цитометра после окрашивания с Аннексии V-7-AAD. Инкубированные клетки собрали, поместили в 5-мл FACS пробирку (BD Falcon) и промыли 1 мл FACS буфера (1%. FBS и 0,1% азаида натрия в PBS). Клетки повторно суспендировали в 0,1 мл FACS буферного раствора, после чего эти клетки обработали и окрасили фикоэритрин-Аннексин V при комнатной температуре в течение 10 минут. Через 10 минут окрашивания, клетки обработали и окрасили 7-AAD при комнатной температуре в течение 5 минут. Окрашенные клетки суспендировали в 0,3 мл FACS буферного раствора, после чего проанализировали с помощью проточного цитометра. Как показано в таблицах с 12 по 15, соединения 065, 108, 229 и 457 по данному изобретению продемонстрировали эффект апоптоза по отношению к различным лимфолейкозным клеткам. Таким образом, соединения 065, 108, 229 и 457 по данному изобретению можно использовать в качестве новых медикаментов против лимфонеоплазии.
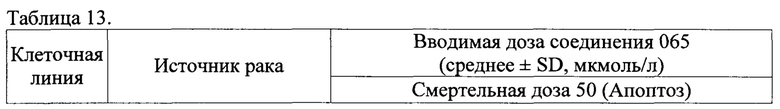
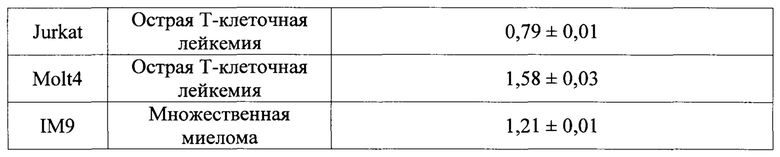

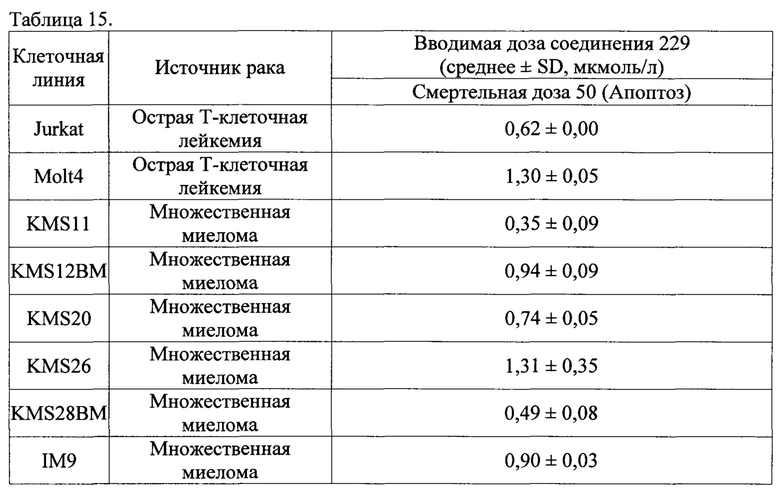

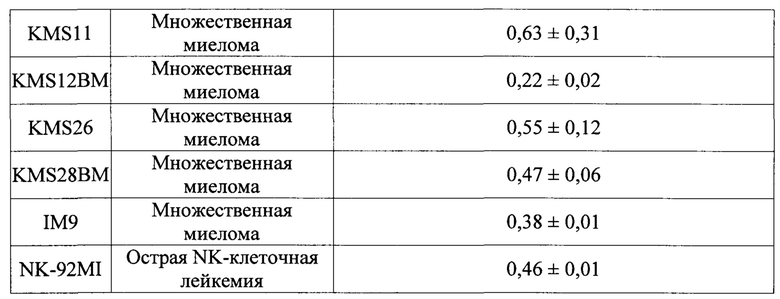
Экспериментальный Пример 7: проверка токсичности
Мышам ICR мужского пола орально вводили суспензию соединения 229 в 0,5% растворе метилцеллюлозы в дозах 10 мг/кг, 50 мг/кг и 100 мг/кг, и в течение 7 дней исследовали коэффициент выживания и массу тела. После каждого введения проверяли наличие жизни, клиническое состояние и изменение веса тела. Проводили гематологические тесты и биохимическую проверку крови. После констатации апоптоза, наличие или отсутствие патологии органов брюшной и грудной полости оценивалось невооруженным глазом.
Результаты показали, что мертвых мышей, равно как и заметных клинических заболеваний, обнаружено не было. При исследовании изменения веса, гематологических проверках, биохимическом исследовании крови и аутопсии также не было обнаружено никаких изменений, связанных с токсичностью. Таким образом, соединение 229 не продемонстрировало токсических изменений в дозировке до 100 мг/кг.
Таким образом, соединение 229 обладает смертельной дозой 50 (LD50) в 100 мг/кг, и является безопасным.
| название | год | авторы | номер документа |
|---|---|---|---|
| Гибридные производные (1Н-1,2,4) триазола и серосодержащих гетероциклов: производных тиазолидин-2,4-диона, тиоморфолин-3-она и 1,4-тиазепан-3-она, обладающих антимикробной активностью | 2020 |
|
RU2771027C1 |
| ТИАЗОЛОПИРИМИДИНОНЫ В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ РЕЦЕПТОРОВ NMDA | 2014 |
|
RU2703273C2 |
| ИНГИБИТОРЫ ТИРОЗИНКИНАЗЫ БРУТОНА | 2014 |
|
RU2648236C2 |
| ПРОИЗВОДНЫЕ ПИПЕРИДИНА В КАЧЕСТВЕ АГОНИСТОВ GPR119 | 2013 |
|
RU2603346C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ БЕНЗОДИАЗЕПИНА | 2010 |
|
RU2683325C2 |
| ПИРАЗОЛ-4-ИЛ-ГЕТЕРОЦИКЛИЛ-КАРБОКСАМИДНЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ПРИМЕНЕНИЯ | 2012 |
|
RU2638552C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ БЕНЗОДИАЗЕПИНА | 2010 |
|
RU2545080C2 |
| БЕНЗОПРОИЗВОДНЫЕ С ШЕСТИЧЛЕННЫМ КОЛЬЦОМ В КАЧЕСТВЕ ИНГИБИТОРА DPP-4 И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2702644C2 |
| ПИПЕРИДИНИЛ НАФТИЛУКСУСНЫЕ КИСЛОТЫ | 2012 |
|
RU2628083C2 |
| АРИЛСУЛЬФОНИЛМЕТИЛЬНЫЕ ИЛИ АРИЛСУЛЬФОНАМИДНЫЕ ПРОИЗВОДНЫЕ АРОМАТИЧЕСКИХ СОЕДИНЕНИЙ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ЛЕЧЕНИЯ РАССТРОЙСТВ, ВОСПРИИМЧИВЫХ К ЛЕЧЕНИЮ ЛИГАНДАМИ ДОФАМИНОВЫХ D РЕЦЕПТОРОВ, С ИХ ПОМОЩЬЮ | 2005 |
|
RU2442781C2 |
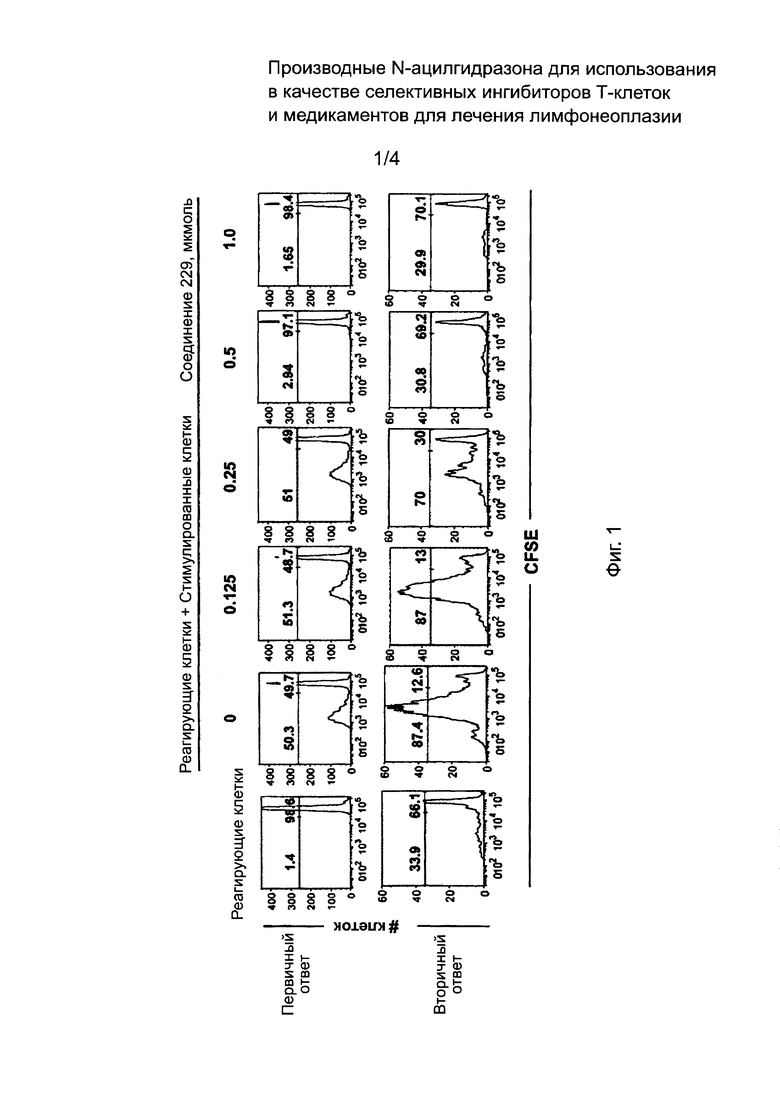
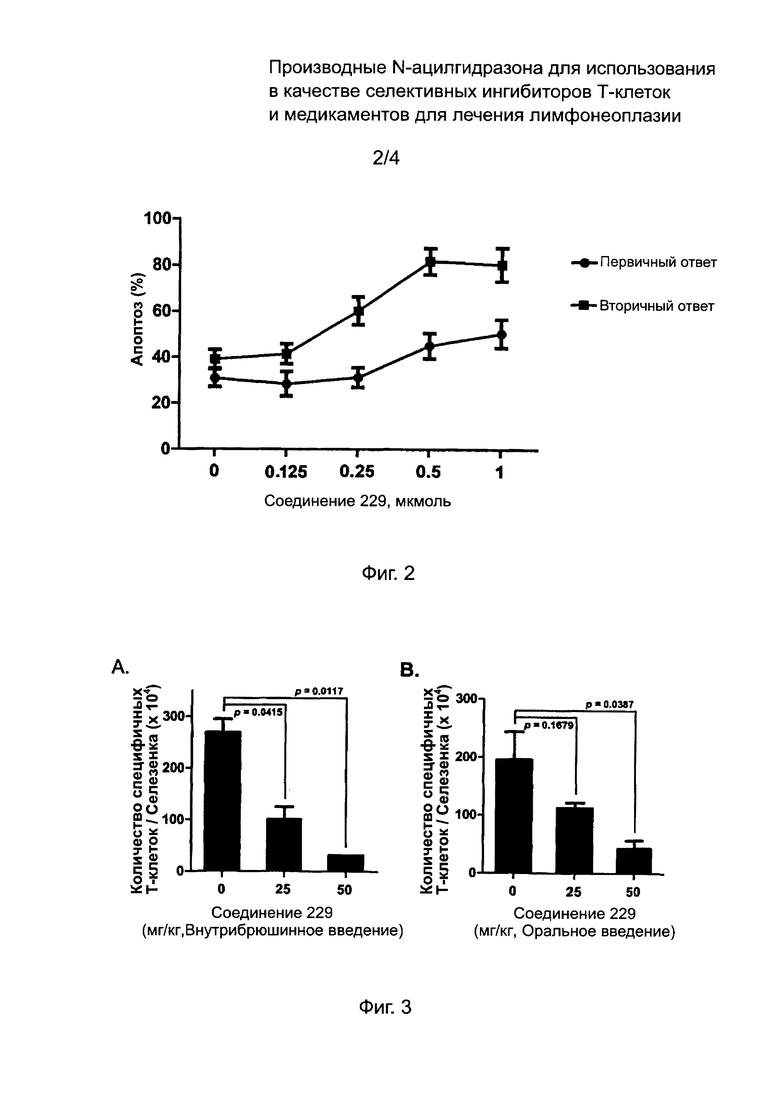
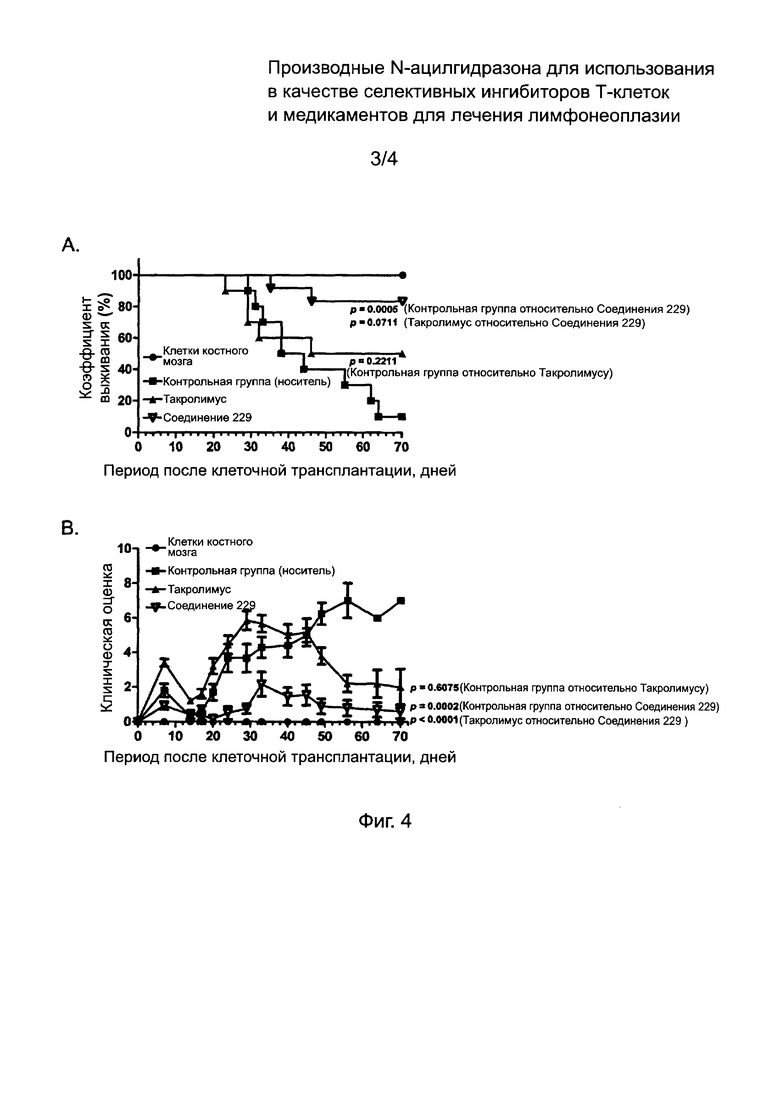
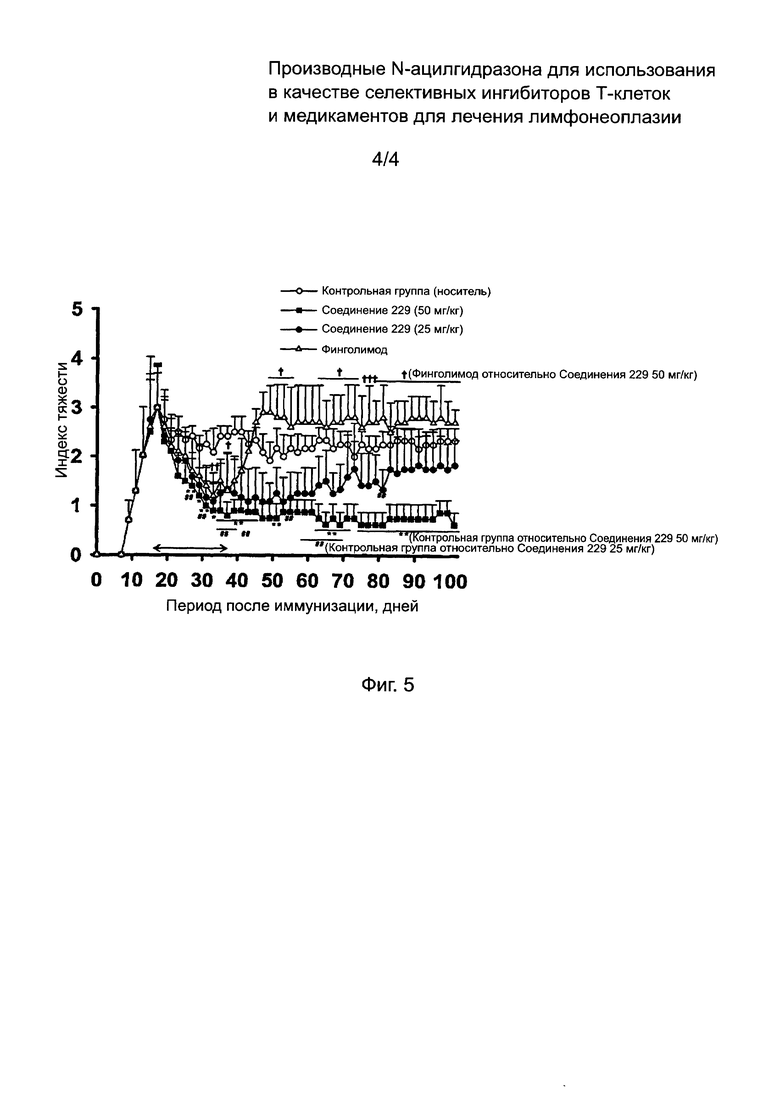
Изобретение относится к новым соединениям формулы 1, его (Е)-стереоизомеру или фармацевтически приемлемой соли:
[Формула 1]
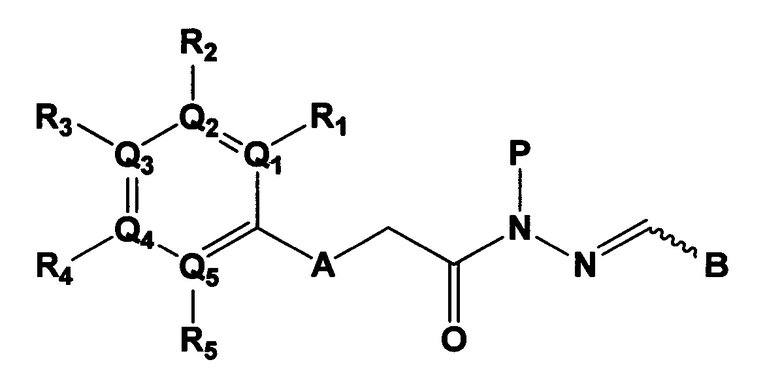 . Технический результат: получены новые соединения, обладающие ингибиторной активностью в отношении Т-клеток, которые могут быть использованы для приготовления фармацевтических композиций для предотвращения или лечения заболеваний, где заболевание является реакцией «трансплантат против хозяина» (РТПХ) после трансплантации органов или гематопоэтических стволовых клеток, множественным склерозом, ревматоидным артритом или лимфонеоплазией. 2 н. и 6 з.п. ф-лы, 16 табл., 143 пр., 5 ил.
. Технический результат: получены новые соединения, обладающие ингибиторной активностью в отношении Т-клеток, которые могут быть использованы для приготовления фармацевтических композиций для предотвращения или лечения заболеваний, где заболевание является реакцией «трансплантат против хозяина» (РТПХ) после трансплантации органов или гематопоэтических стволовых клеток, множественным склерозом, ревматоидным артритом или лимфонеоплазией. 2 н. и 6 з.п. ф-лы, 16 табл., 143 пр., 5 ил.
1. Соединение, соответствующее формуле 1, его (Е)-стереоизомер или фармацевтически приемлемая соль:
[Формула 1]
в котором,
А является N-H, О или S;
Q1 является С;
Q2, Q3, Q4 и Q5 каждый независимо являются С или N;
R1, R2, R4 и R5 каждый независимо отсутствует или является -Н, -CF3, -F, -Br, -Cl, цианидом, -СН2ОН группой, -(CO)NH2 группой, -(С1-С6)алкилгруппой, -(C1-3)алкоксигруппой, -NH2 группой, или 5-, или 6-членным гетероарилом, или гетероциклоалкилом, содержащим от 1 до 2 членов, выбранных из группы, состоящей из N, О и S;
R3 отсутствует или является -Н, -CF3, -F, -Br, цианидом, -СН2ОН группой, -(CO)NH2 группой -(С1-С6)алкилгруппой, -(С1-3)алкоксигруппой, -NH2 группой, или 5-, или 6-членным гетероарилом, или гетероциклоалкилом, содержащим от 1 до 2 членов, выбранных из группы, состоящей из N, О и S;
Р является -Н, метильной группой или -(C1-C3)OH;
 или
или 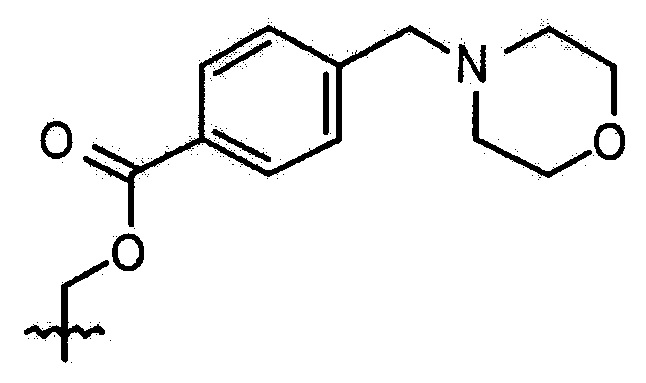 группой; и
группой; и
В является 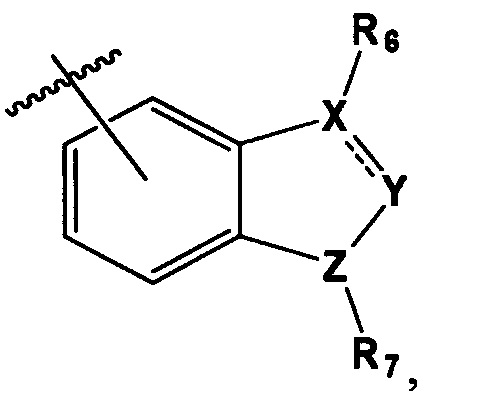
в которой X, Y и Z каждый независимо являются С, N или S, и
R6 и R7 каждый независимо отсутствует или является -Н, -Br, -(С1-С6)алкилгруппой, -CH2CH2OH группой,
 или
или  группой.
группой.
2. Соединение по п. 1, его (Е)-стереоизомер или фармацевтически приемлемая соль, в котором
А является N-H, О или S
Q1, Q2, Q3, Q4 и Q5 являются С;
R2 и R4 являются Н;
R1 и R5 каждый независимо являются -Н, -F, -Br, -Cl, метилгруппой, этилгруппой, -СН2ОН группой, цианидом, -NH2 группой, или 5-, или 6-членным гетероарилом, или гетероциклоалкилом, содержащим от 1 до 2 членов, выбранных из группы, состоящей из N, О и S;
R3 является -Н, -F, -Br, метилгруппой, этилгруппой, -СН2ОН группой, цианидом, -NH2 группой, или 5-, или 6-членным гетероарилом, или гетероциклоалкилом, содержащим от 1 до 2 членов, выбранных из группы, состоящей из N, О и S;
Р является -Н, метилгруппой, -CH2OH группой, -СН2СН2ОН группой,
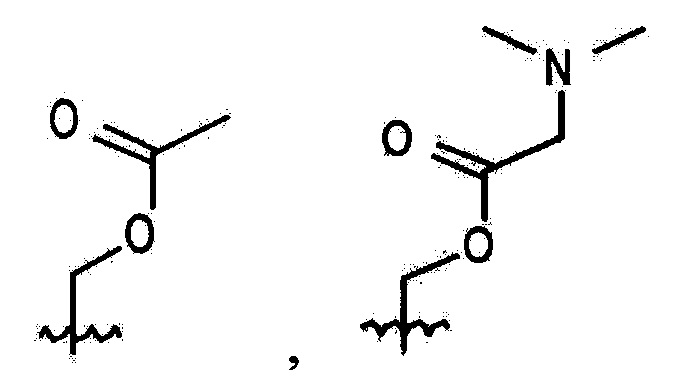 или
или 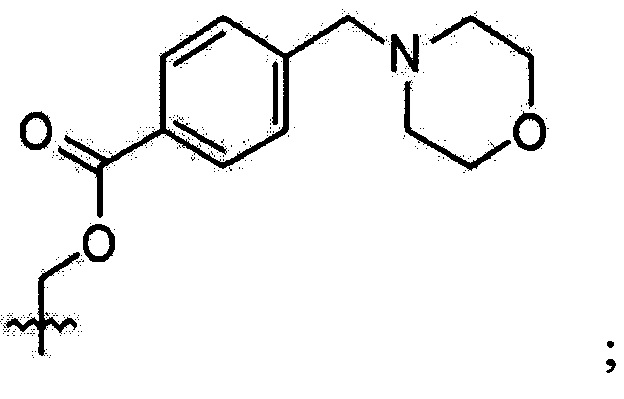 и
и
В является 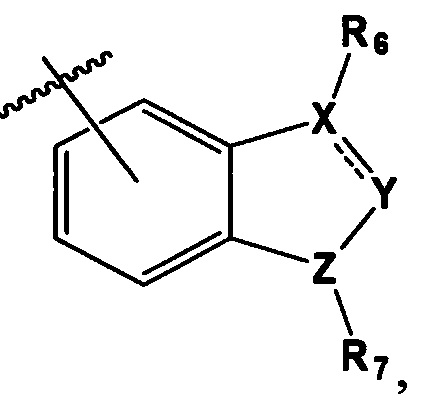
в которой X и Y является С,
Z является N, и
R6 и R7 каждый независимо являются -Н, метилгруппой или -СН2СН2ОН группой.
3. Соединение по п. 1, его (Е)-стереоизомер или фармацевтически приемлемая соль, выбранное из группы, состоящей из:
(E)-N'-((1Н-индол-4-ил)метилен)-2-(мезитилокси)ацетогидразида;
(Е)-N'-((1Н-индол-5-ил)метилен)-2-(4-бром-2,6-диметилфенокси)ацетогидразида;
(E)-N'-((1Н-индол-6-ил)метилен)-2-(мезитилокси)ацетогидразида;
(E)-N'-((1Н-индол-4-ил)метилен)-2-(4-бром-2,6-диметилфенокси)ацетогидразида;
(Е)-N'-((1Н-индол-6-ил)метилен)-2-(4-бром-2,6-диметилфенокси)ацетогидразида;
(E)-N'-((1Н-индол-4-ил)метилен)-2-(мезитиламино)ацетогидразида;
(E)-N'-((1Н-индол-4-ил)метилен)-2-(2,6-диметил-4-(пиридин-3-ил)фенокси)ацетогидразида;
(E)-N'-((1Н-индол-6-ил)метилен)-2-(2,6-диметил-4-(пиридин-3-ил)фенокси)ацетогидразида;
(E)-N'-((1Н-индол-4-ил)метилен)-2-(2-(пиридин-3-ил)фенокси)ацетогидразида;
(E)-N'-((1Н-индол-4-ил)метилен)-2-(мезитилтио)ацетогидразида;
(E)-N'-((1Н-индол-4-ил)метилен)-2-(2,6-диметил-4-(пиримидин-5-ил)фенокси)ацетогидразида;
(E)-N'-((1Н-индол-6-ил)метилен)-2-(мезитилтио)ацетогидразида;
(Е)-2-(мезитилокси)-N-метил-N'-((1-метил-1Н-индол-4-ил)метилен)ацетогидразида;
(E)-N'-((1Н-индол-4-ил)метилен)-N-(2-гидроксиэтил)-2-(мезитилокси)ацетогидразида;
(E)-N'-((1-(2-гидроксиэтил)-1Н-индол-4-ил)метилен)-2-(мезитилокси)ацетогидразида;
(E)-N'-((1Н-индол-4-ил)метилен)-2-(2,4-диметил-6-(пиридин-3-ил)фенокси)ацетогидразида;
(Е)-N'-((1Н-индол-6-ил)метилен)-2-(2,4-диметил-6-(пиридин-3-ил)фенокси)ацетогидразида;
(Е)-N'-((1Н-индол-5-ил)метилен)-2-(2,4-диметил-6-(пиридин-3-ил)фенокси)ацетогидразида;
(Е)-N'-((1Н-индол-4-ил)метилен)-2-((2-метилпиридин-3-ил)окси)ацетогидразида;
(E)-N'-((1Н-индол-6-ил)метилен)-N-(2-гидроксиэтил)-2-(мезитилокси)ацетогидразида;
(Е)-N'-((1Н-индол-6-ил)метилен)-2-(2,4-диметил-6-(пиридин-4-ил)фенокси)ацетогидразида;
(Е)-N'-((1Н-индол-6-ил)метилен)-2-(2-(фуран-3-ил)-4,6-диметилфенокси)ацетогидразида;
(E)-N'-((1Н-индол-4-ил)метилен)-2-(4-(гидроксиметил)-2,6-диметилфенокси)ацетогидразида;
(Е)-N'-((1Н-индол-6-ил)метилен)-2-(4-(гидроксиметил)-2,6-диметилфенокси)ацетогидразида;
(E)-N'-((1Н-индол-6-ил)метилен)-2-(2,4-диметил-6-(тетрагидрофуран-3-ил)фенокси)ацетогидразида;
(Е)-N'-((1Н-индол-4-ил)метилен)-2-(4-амино-2,6-диметилфенокси)ацетогидразида;
(E)-N'-((1Н-индол-6-ил)метилен)-N-(гидроксиметил)-2-(мезитилокси)ацетогидразида;
(Е)-(2-((1Н-индол-6-ил)метилен)-1-(2-(мезитилокси)ацетил)гидразинил)метил 2-(диметиламино)ацетата;
(Е)-N'-((1Н-индол-6-ил)метилен)-2-(4-циано-2,6-диметилфенокси)ацетогидразида;
(Е)-(2-((1Н-индол-4-ил)метилен)-1-(2-(мезитилокси)ацетил)гидразинил)метил 2-(диметиламино)ацетата;
(Е)-N'-((1Н-индол-6-ил)метилен)-N-ацетил-2-(мезитилокси)ацетогидразида;
(E)-N'-((1Н-индол-4-ил)метилен)-N-(гидроксиметил)-2-(мезитилокси)ацетогидразида;
(Е)-(2-((1Н-индол-4-ил)метилен)-1-(2-(мезитилокси)ацетил)гидразинил)метил 4-(морфолинометил)бензоата;
(Е)-(2-((1Н-индол-6-ил)метилен)-1-(2-(мезитилокси)ацетил)гидразинил)метил ацетата;
(E)-N'-((1Н-индол-6-ил)метилен)-2-(2,6-диметилфенокси)ацетогидразида;
(Е)-N'-((1Н-индол-4-ил)метилен)-2-(2-бром-4,6-диметилфенокси)ацетогидразида;
(Е)-N'-((1Н-индол-6-ил)метилен)-2-(2-бром-4,6-диметилфенокси)ацетогидразида;
(Е)-(2-((1Н-индол-6-ил)метилен)-1-(2-(мезитилокси)ацетил)гидразинил)метил 4-(морфолинометил)бензоата;
(E)-N'-((1Н-индол-6-ил)метилен)-2-(2-метил-6-(тетрагидрофуран-3-ил)фенокси)ацетогидразида;
(E)-N'-((1Н-индол-4-ил)метилен)-2-(2-метил-6-(тетрагидрофуран-3-ил)фенокси)ацетогидразида;
(E)-N'-((1Н-индол-6-ил)метилен)-2-(2-(фуран-3-ил)-6-метилфенокси)ацетогидразида;
(E)-N'-((1Н-индол-4-ил)метилен)-2-(2-(фуран-3-ил)-6-метилфенокси)ацетогидразида;
(Е)-N'-((1Н-индол-4-ил)метилен)-2-(2-(фуран-2-ил)-4,6-диметилфенокси)ацетогидразида;
(E)-N'-((1Н-индол-4-ил)метилен)-2-((2-(фуран-3-ил)пиридин-3-ил)окси)ацетогидразида;
(E)-N'-((1Н-индол-4-ил)метилен)-2-(2,4-диметил-6-(тетрагидрофуран-3-ил)фенокси)ацетогидразида;
(E)-N'-((1Н-индол-6-ил)метилен)-2-(4-(фуран-3-ил)-2,6-диметилфенокси)ацетогидразида;
(E)-N'-((1Н-индол-6-ил)метилен)-2-(2,6-диметил-4-(тетрагидрофуран-3-ил)фенокси)ацетогидразида;
(E)-N'-((1Н-индол-4-ил)метилен)-2-((2-этил-6-метилпиридин-3-ил)окси)ацетогидразида;
(E)-N'-((1Н-индол-6-ил)метилен)-2-((2-(пирролидин-1-ил)пиридин-3-ил)окси)ацетогидразида;
(E)-N'-((1Н-индол-6-ил)метилен)-2-(2,4-диметил-6-(тетрагидрофуран-2-ил)фенокси)ацетогидразида;
(E)-N'-((1Н-индол-4-ил)метилен)-2-(2-(фуран-3-ил)-4,6-диметилфенокси)ацетогидразида;
(E)-N'-((1Н-индол-4-ил)метилен)-2-(4-(фуран-3-ил)-2,6-диметилфенокси)ацетогидразида;
(Е)-N'-((1Н-индол-4-ил)метилен)-2-(2,6-диметил-4-(тетрагидрофуран-3-ил)фенокси)ацетогидразида;
(E)-N'-((1Н-индол-6-ил)метилен)-2-((2-(тиофен-3-ил)пиридин-3-ил)окси)ацетогидразида;
(E)-N'-((1Н-индол-4-ил)метилен)-2-(2,4-диметил-6-(тиофен-2-ил)фенокси)ацетогидразида;
(E)-N'-((1Н-индол-4-ил)метилен)-2-(2-(фуран-2-ил)-4-метоксифенокси)ацетогидразида;
(Е)-2-(2,4-диметил-6-(тетрагидрофуран-3-ил)фенокси)-N-метил-N'-((1-метил-1Н-индол-6-ил)метилен)ацетогидразида;
(E)-N'-((1Н-индол-6-ил)метилен)-2-(4-фтор-2-метил-6-(тетрагидрофуран-3-ил)фенокси)ацетогидразида;
(E)-N'-((1Н-индол-4-ил)метилен)-2-(4-фтор-2-метил-6-(тетрагидрофуран-3-ил)фенокси)ацетогидразида;
(Е)-2-(4-фтор-2-метал-6-(тетрагадрофуран-3-ил)фенокси)-N-метил-N'-((1-метил-1Н-индол-6-ил)метилен)ацетогидразида; и
(E)-N'-((1Н-индол-6-ил)метилен)-2-(2,4-диметил-6-(тетрагидро-2Н-пиран-4-ил)фенокси)ацетогидразида.
4. Соединение по п. 3, его (Е)-стереоизомер или фармацевтически приемлемая соль, выбранное из группы, состоящей из:
(E)-N'-((1Н-индол-4-ил)метилен)-2-(мезитилокси)ацетогидразида;
(E)-N'-((1Н-индол-6-ил)метилен)-2-(мезитилокси)ацетогидразида;
(E)-N'-((1Н-индол-6-ил)метилен)-2-(2,4-диметил-6-(тетрагидрофуран-3-ил)фенокси)ацетогидразида; и
(E)-N'-((1Н-индол-6-ил)метилен)-2-(2,4-диметил-6-(тетрагидро-2Н-пиран-4-ил)фенокси)ацетогидразида.
5. Фармацевтическая композиция для предотвращения или лечения заболевания, ассоциирующегося с ингибированием активности Т-клеток, включающая эффективное количество соединения по п. 1 или 2, его (Е)-стереоизомер или фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
6. Фармацевтическая композиция по п. 5, в которой заболевание является реакцией «трансплантат против хозяина» (РТПХ) после трансплантации органов или гематопоэтических стволовых клеток, множественным склерозом или ревматоидным артритом.
7. Фармацевтическая композиция по п. 5, используемая для лечения лимфонеоплазии.
8. Фармацевтическая композиция по п. 7, в которой лимфонеоплазия является Т-лимфоидной лейкемией, В-лимфоидной лейкемией, NK лейкемией, NKT лейкемией, множественной миеломой, Т-лимфомой или В-лимфомой.
| СТАНОК ДЛЯ ОКОРКИ ДРЕВЕСИНЫ | 1949 |
|
SU81111A1 |
| Устройство для электрической сигнализации | 1918 |
|
SU16A1 |
| Походная разборная печь для варки пищи и печения хлеба | 1920 |
|
SU11A1 |
| Кобраков К.И | |||
| и др | |||
| Синтез и спектральные характеристики гидразонов и N-ацилгидразонов, содержащих дихлопиридильные фрагменты | |||
| Химия и химическая технология, том | |||
| Приспособление для автоматической односторонней разгрузки железнодорожных платформ | 1921 |
|
SU48A1 |
| Приспособление для точного наложения листов бумаги при снятии оттисков | 1922 |
|
SU6A1 |
| Andrew R Germain et al, Identification of a selective small molecule inhibitor of breast cancer stem cells, Bioorganic & Medicinal Chemistry Letters, 22, 3571-3574, 25.01.2012 | |||
| WO 2008121877 A2, 09.10.2008 | |||
| WO 2009155362 A1, 23.12.2009 | |||
| ГИДРАЗИДЫ 1Н-ИНДОЛ-3-УКСУСНОЙ КИСЛОТЫ КАК ИНГИБИТОРЫ SPLA | 1994 |
|
RU2127725C1 |

