Настоящее изобретение относится к замещенным производным пиримидина и к их применению в терапии. Более конкретно, но не исключительно, настоящее изобретение относится к соединениям, способным ингибировать одну или несколько протеинкиназ, а в частности, polo-подобных киназ.
Предшествующий уровень техники
Семейство Polo-подобных киназ состоит из ключевых ферментов, регулирующих клеточный цикл и играющих основную роль в регуляции вступления клетки в фазу митоза и ее прохождения этой фазы. Многие опухолевые клетки экспрессируют высокие уровни PLK1 и являются восприимчивыми к антисмысловым олигонуклеотидам, направленным против этого белка.
Для инициации митоза требуется активация фактора, стимулирующего М-фазу (MPF), то есть комплекса, образующегося между CDKl и циклинами B-типа [Nurse, P. (1990) Nature, 344, 503-508]. Эти циклины аккумулируются в процессе прохождения S-фазы и G2-фазы клеточного цикла и стимулируют ингибирование фосфорилирования MPF-комплекса под действием киназ WEEl, MIKl и MYTl. В конце G2-фазы соответствующее дефосфорилирование под действием фосфатазы с двойной специфичностью, CDC25C, стимулирует активацию MPF [Nigg, E. A. (2001) Nat. Rev. Mol. Cell Biol., 2, 21-32]. В интерфазе циклин В локализуется в цитоплазме, а затем, в процессе прохождения профазы, он фосфорилируется и транслоцируется в ядро. Считается, что аккумуляция активного MPF в ядре во время профазы играет важную роль в инициации прохождения M-фазы [Takizawa, C.G. and Morgan, D.O. (2000) Curr. Opin. Cell Biol., 12, 658-665]. Однако, ядерный MPF не подвергается действию киназы WEEl до тех пор, пока он не вступит во взаимодействие с CDC25C. Сама фосфотаза CDC25C, локализующаяся в цитоплазме во время интерфазы, аккумулируется в ядре на стадии профазы. Проникновение циклина В и CDC25C в ядро стимулируется фосфорилированием под действием PLK1 [Roshak, A.K., Capper, E.A., Imburgia, C., Fornwald, J., Scott, G. and Marshall, L.A. (2000) Cell. Signalling, 12, 405-411]. Таким образом, эта киназа является важным регулятором инициации вступления в М-фазу.
У человека присутствуют три близкородственные polo-подобные киназы (PLK) [Glover, D.M., Hagan, I.M. and Tavares, A.A. (1998) Genes Dev., 12, 3777-3787]. Они содержат в высокой степени гомологичный N-концевой каталитический киназный домен, а их С-концы содержат две или три консервативных области, polo-боксы. Функция polo-боксов пока еще точно не ясна, однако известно, что зависимая от polo-бокса активность PLK1 необходима для «правильного» перехода из метафазы в анафазу и цитокинеза [Seong, Y.-S., Kamijo, K., Lee, J.-S., Fernandez, E., Kuriyama, R., Miki, T. и Lee, K.S. (2002) J. Biol. Chem., 277, 32282-32293]. Из трех киназ PLK лучше всего охарактеризована киназа PLK1, и она регулирует события цикла деления клеток, включая начало митоза, активацию сверочных точек повреждения ДНК, регуляцию образования комплекса, стимулирующего анафазу, фосфорилирование протеосомы, а также удвоение и созревание центросомы. Ранее было также показано, что у млекопитающих киназа PLK2 (также известная как SNK) и киназа PLK3 (также известная как PRK и FNK) представляют собой продукты предраннего гена. Очевидно, что активность киназы PLK3 достигает своего пика в процессе прохождения фазы S и фазы G2. Она также активируется в процессе активации сверочной точки повреждения ДНК и при сильном окислительном стрессе. PLK3 также играет важную роль в регуляции динамики образования микротрубочек и функции центросомы в клетках, а нарушение регуляции экспрессии PLK3 приводит к прекращению клеточного цикла и к апоптозу клеток [Wang, Q., Xie, S., Chen, J., Fukusawa, K., Naik, U., Traganos, F., Darzynkiewicz, Z., Jhanwar-Uniyal, M. and Dai, W. (2002) Mol. Cell Biol., 22, 3450-3459]. Из трех PLK PLK2 представляет собой наименее изученный гомолог. Обе PLK2 и PLK3 могут также выполнять важные постмитотические функции [Kauselmann, G., Weiler, M., Wulff, P., Jessberger, S., Konietzko, U., Scafidi, J., Staubli, U., Bereiter-Hahn, J., Strebhardt, K. and Kuhl, D. (1999) EMBO J., 18, 5528-5539].
Тот факт, что человеческие PLK регулируют некоторые фундаментальные аспекты митоза, был проиллюстрирован путем микроинъекции анти-PLK1 антитела в человеческие опухолевые клетки [Lane, H. A. and Nigg, E. A. (1996) J. Cell. Biol., 135, 1701-1713]. Такая обработка не оказывала влияния на репликацию ДНК, но приводила к нарушению цикла деления клеток. Деление клеток прекращалось на фазе митоза, и эти клетки обнаруживали аномальное распределение конденсированного хроматина и моноастральных микротрубочек, образующихся в дуплицированных, но в неразделенных центросомах. В противоположность этому, неиммортализованные человеческие клетки прекращали свой рост на G2-фазе в стадии одиночных одноядерных клеток. Кроме того, если функция PLK1 блокируется опосредуемой аденовирусом доставкой доминантно-негативного гена, то, в данном случае, во многих опухолевых клеточных линиях наблюдался опухолеспецифический апоптоз, тогда как нормальные эпителиальные клетки, несмотря на прекращения их деления на фазе митоза, не подвергались митотическому «коллапсу», наблюдаемому в опухолевых клетках [Cogswell, J.P., Brown, C.E., Bisi, J.E. and Neill, S.D. (2000) Cell Growth Differ., 11, 615-623]. Таким образом, активность PLK1 необходима для функционального созревания центросом на поздней фазе G2/ранней профазе и для последующего образования биполярного веретена. Кроме того, полученные результаты позволяют сделать предположение о присутствии в нормальных клетках контрольной точки созревания центросом, которая чувствительна к ингибированию PLK1. Истощение клеточной PLK1 под действием короткой интерферирующей РНК (киРНК) также подтверждает, что этот белок необходим для осуществления многих митотических процессов и завершения цитокинеза [Liu, X. and Erikson, R.L. (2003) Proc. Natl. Acad. Sci. USA, 100, 5789-5794]. О возможной терапевтической эффективности ингибирования PLK также позволяют предположить результаты исследования с использованием PLK1-специфических антисмысловых олигонуклеотидов, которые, как было показано, индуцируют ингибирование роста раковых клеток in vitro и in vivo [Spankuch-Schmitt, B., Wolf, G., Solbach, C., Loibl, S., Knecht, R., Stegmuller, M., von Minckwitz, G., Kaufmann, M. and Strebhardt, K. (2002) Oncogene, 21, 3162-3171]. Было показано, что конститутивная экспрессия PLK1 в клетках млекопитающих приводит к их перерождению в злокачественные клетки [Smith, M.R., Wilson, M.L., Hamanaka, R., Chase, D., Kung, H., Longo, D.L. and Ferris, D.K. (1997) Biochem. Biophys. Res. Commun., 234, 397-405]. Кроме того, сверхэкспрессия PLK1 часто наблюдается в человеческих опухолях, а поэтому экспрессия PLK1 является прогностическим фактором у пациентов, страдающих опухолями различных типов [Takahashi, T., Sano, B., Nagata, T., Kato, H., Sugiyama, Y., Kunieda, K., Kimura, M., Okano, Y. and Saji, S. (2003) Cancer Science, 94, 148-152; Tokumitsu, Y., Mori, M., Tanaka, S., Akazawa, K., Nakano, S. and Niho, Y. (1999) Int. J. Oncol., 15, 687-692; Wolf, G., Elez, R., Doermer, A., Holtrich, U., Ackermann, H., Stutte, H.J., Altmannsberger, H.-M., Rϋbsamen-Waigmann, H. and Strebhardt, K. (1997) Oncogene, 14, 543-549].
Хотя совершенно очевидно, что ингибирование киназы PLK, при ее использовании в фармакологии, может оказывать терапевтическое действие [Kraker, A.J. and Booher, R.N. (1999) In Annual Reports in Medicinal Chemistry (Vol. 34) (Doherty, A.M., ed.), pp. 247-256, Academic Press], однако, в настоящее время имеется крайне мало информации, касающейся возможности использования небольших молекул-ингибиторов PLK в качестве лекарственных средств. Одним из немногих биохимических ингибиторов PLK1, охарактеризованных в настоящее время, являются сцитонемин, то есть натуральный морепродукт, имеющий симметричную индольную молекулу [Stevenson, C.S., Capper, E.A., Roshak, A.K., Marquez, B., Eichman, C., Jackson, J.R., Mattern, M., Gerwick, W.H., Jacobs, R.S. and Marshall, L.A. (2002) J. Pharmacol. Exp. Ther., 303, 858-866; Stevenson, C.S., Capper, E.A., Roshak, A.K., Marquez, B., Grace, K., Gerwick, W.H., Jacobs, R.S. and Marshall, L.A. (2002) Inflammation Research, 51, 112-114]. Сцитонемин ингибирует фосфорилирование CDC25C под действием рекомбинантной киназы PLK1 с величиной IC50, составляющей 2 мкМ (при концентрации АТФ 10 мкМ). Такое ингибирование, очевидно, является обратимым, и осуществляется по механизму смешанного конкурентного действия АТФ. Аналогичное действие наблюдалось также и против других белков, таких как серин-треонин-киназы и участвующие в клеточном цикле киназы с двойной специфичностью, включая MYTl, CHK1, CDK1/циклин B и PKC. Сцитонемин обнаруживал явно выраженное антипролиферативное действие на различные человеческие клеточные линии in vitro. Другие небольшие молекулы-ингибиторы PLK и их применение для лечения пролиферативных расстройств описаны в Международной патентной заявке WO2004/067000 под названием «Cyclacel Limited».
Задачей настоящего изобретения является идентификация новых небольших молекул-ингибиторов PLK. Более конкретно, целью настоящего изобретения является получение небольших молекул-ингибиторов PLK, которые могут быть использованы в терапевтических целях для лечения ряда пролиферативных расстройств.
Описание сущности изобретения
В своем первом аспекте, настоящее изобретение относится к соединению формулы VIII или к его фармацевтически приемлемой соли или фармацевтически приемлемому сложному эфиру:

где
X представляет собой NR7;
Y представляет собой O или N-(CH2)nR19;
n равно 1, 2 или 3;
m равно 1 или 2;
каждый из R1 и R2 независимо представляет собой H, алкил или циклоалкил;
каждый из R4 и R4’ независимо представляет собой H или алкил; или
R4 и R4’, взятые вместе, образуют спиро-циклоалкильную группу;
R19 представляет собой H, алкильную, арильную или циклоалкильную группу;
R6 представляет собой OR8 или галоген; и
каждый из R7 и R8 независимо представляет собой H или алкил.
Во втором своем аспекте, настоящее изобретение относится к соединению формулы VI, или к его фармацевтически приемлемой соли или фармацевтически приемлемому сложному эфиру,
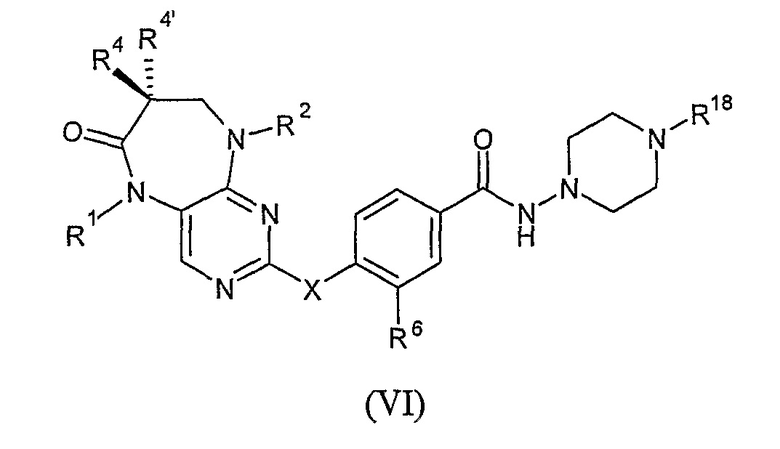
где
X представляет собой NR7;
каждый из R1 и R2 независимо представляет собой H, алкил или циклоалкил;
каждый из R4 и R4’ независимо представляет собой H или алкил; или
R4 и R4’, взятые вместе, образуют спиро-циклоалкильную группу;
R18 представляет собой H или алкил, где указанная алкильная группа необязательно замещена R6;
каждый R6 независимо представляет собой OR8 или галоген; и
каждый из R7 и R8 независимо представляет собой H или алкил.
В своем третьем аспекте, настоящее изобретение относится к соединению формулы VII, или к его фармацевтически приемлемой соли или фармацевтически приемлемому сложному эфиру,
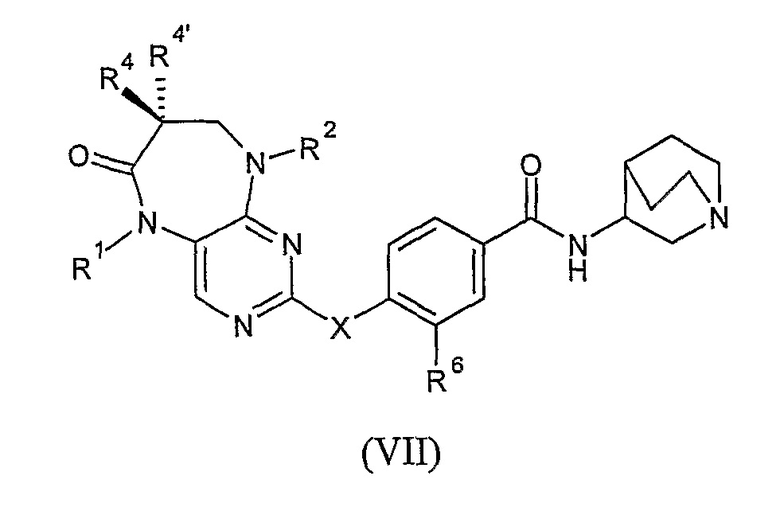
где
X представляет собой NR7;
каждый из R1 и R2 независимо представляет собой H, алкил или циклоалкил;
каждый из R4 и R4’ независимо представляет собой H или алкил; или
R4 и R4’, взятые вместе, образуют спиро-циклоалкильную группу;
R6 представляет собой OR8 или галоген; и
каждый из R7 и R8 независимо представляет собой H или алкил.
В своем четвертом аспекте, настоящее изобретение относится к соединению, выбранному из таких соединений, как:
№
В своем пятом аспекте, настоящее изобретение относится к фармацевтической композиции, содержащей соединение, описанное выше, в смеси с фармацевтически приемлемым разбавителем, наполнителем или носителем.
В своем шестом аспекте, настоящее изобретение относится к применению соединения, описанного выше, в целях приготовления лекарственного препарата для лечения пролиферативного расстройства.
В своем седьмом аспекте, настоящее изобретение относится к способу лечения пролиферативного расстройства, где указанный способ включает введение индивидууму терапевтически эффективного количества соединения, описанного выше.
В своем восьмом аспекте, настоящее изобретение относится к способу лечения PLK-зависимого расстройства, где указанный способ включает введение индивидууму терапевтически эффективного количества соединения, описанного выше.
В своем девятом аспекте, настоящее изобретение относится к способу получения соединений, определенных выше.
Подробное описание изобретения
Используемый в описании термин «алкил» включает насыщенные алкильные группы с прямой и разветвленной цепью, которые могут быть замещенными (моно- или полизамещенными) или незамещенными. Предпочтительной алкильной группой является C1-20-алкильная группа, более предпочтительно, C1-15алкильная группа, еще более предпочтительно, C1-12алкильная группа, еще более предпочтительно, С1-6алкильная группа, а наиболее предпочтительно, С1-3алкильная группа. Особенно предпочтительными алкильными группами являются, например, метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, пентил и гексил. Подходящими заместителями являются, например, одна или более групп R6. Предпочтительной алкильной группой является незамещенная алкильная группа.
Используемый в описании термин «циклоалкил» означает циклическую алкильную группу, которая может быть замещенной (моно- или полизамещенной) или незамещенной. Предпочтительной циклоалкильной группой является C3-12циклоалкильная группа, а более предпочтительно, C3-6циклоалкильная группа. Подходящими заместителями являются, например, одна или более групп R6.
Используемый в описании термин «арил» означает ароматическую C6-12-группу, которая может быть замещенной (моно- или полизамещенной) или незамещенной. Типичными примерами являются фенил и нафтил и т.п. Подходящими заместителями являются, например, одна или более групп R6.
Соединения формулы VIII
Как упоминалось выше, в одном из своих аспектов, настоящее изобретение относится к соединению формулы VIII, или к его фармацевтически приемлемой соли или фармацевтически приемлемому сложному эфиру.
В одном из своих особенно предпочтительных вариантов, настоящее изобретение относится к соединению формулы VIIIa, или к его фармацевтически приемлемой соли или фармацевтически приемлемому сложному эфиру,
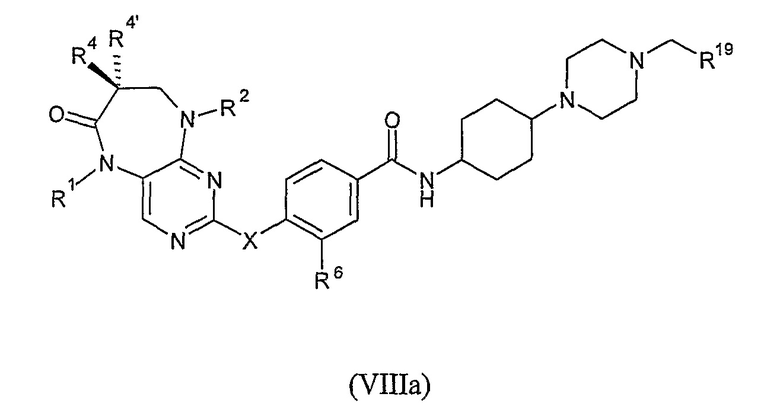
где
X представляет собой NR7;
каждый из R1 и R2 независимо представляет собой H, алкил или циклоалкил;
каждый из R4 и R4’ независимо представляет собой H или алкил; или
R4 и R4’, взятые вместе, образуют спиро-циклоалкильную группу;
R19 представляет собой H, алкильную, арильную или циклоалкильную группу;
R6 представляет собой OR8 или галоген; и
каждый из R7 и R8 независимо представляет собой H или алкил.
В одном из предпочтительных вариантов изобретения, R19 представляет собой H, алкильную или циклоалкильную группу.
Предпочтительно, R1 представляет собой H или алкил, а более предпочтительно, алкил. Еще более предпочтительно, R1 представляет собой метил или этил, а более предпочтительно, Me.
Предпочтительно, R2 представляет собой циклоалкильную группу, а более предпочтительно, C3-6-циклоалкильную группу. Еще более предпочтительно, R2 представляет собой циклопентильную или циклогексильную группу, а еще более предпочтительно, циклопентил.
Предпочтительно, R7 представляет собой H или алкил, более предпочтительно, H или метил, а еще более предпочтительно, H.
В одном из предпочтительных вариантов изобретения, один из R4 и R4’ представляет собой алкил, а другой представляет собой H или алкил.
В другом предпочтительном варианте изобретения, каждый из R4 и R4’ независимо представляет собой алкил. Более предпочтительно, R4 и R4’ оба представляют собой метил.
В другом предпочтительном варианте изобретения, R4 и R4’ оба представляют собой H.
В другом предпочтительном варианте изобретения, R4 и R4’, взятые вместе, образуют спиро-циклоалкильную группу, а более предпочтительно, спиро-C3-6циклоалкильную группу. Еще более предпочтительно, R4 и R4’, взятые вместе, образуют спиро-C3 или C4циклоалкильную группу, а более предпочтительно, C3циклоалкильную группу.
В одном из предпочтительных вариантов изобретения, R6 представляет собой OR8, а еще более предпочтительно, OMe.
Предпочтительно, R19 представляет собой циклопропил.
В другом своем особенно предпочтительном варианте, настоящее изобретение относится к соединению формулы VIIIb, или к его фармацевтически приемлемой соли или фармацевтически приемлемому сложному эфиру,
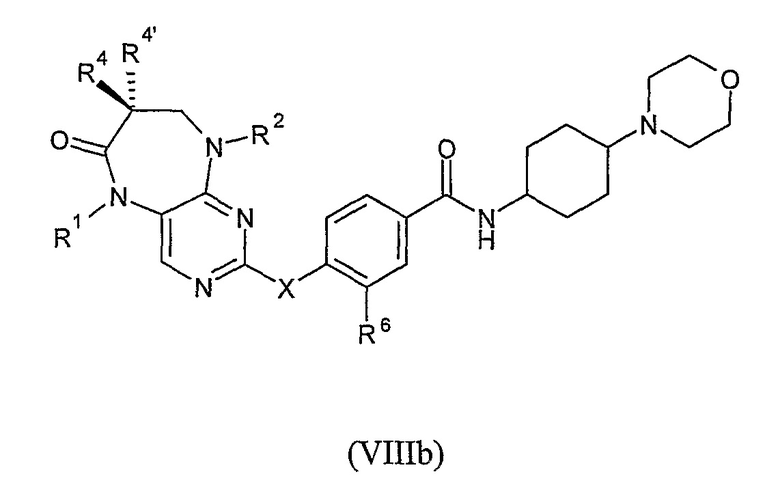
где R1, R2, R6, R4, R4’ и X являются такими, как они были определены выше.
В другом своем особенно предпочтительном варианте, настоящее изобретение относится к соединению формулы VIIIc, или к его фармацевтически приемлемой соли или фармацевтически приемлемому сложному эфиру,

где Х, R1, R2, R6, R4, R4’ и R19 являются такими, как они были определены выше.
В одном особенно предпочтительном варианте изобретения, указанное соединение формулы VIII выбрано из таких соединений, как
№
и их фармацевтически приемлемые соли и сложные эфиры.
Более предпочтительно, соединение формулы VIII выбрано из таких соединений, как
№
и их фармацевтически приемлемые соли или сложные эфиры.
Еще более предпочтительно, соединением согласно изобретению является соединение [371].
Соединения формулы VII
В одном из своих аспектов, настоящее изобретение относится к соединению формулы VII, или к его фармацевтически приемлемой соли или фармацевтически приемлемому сложному эфиру,
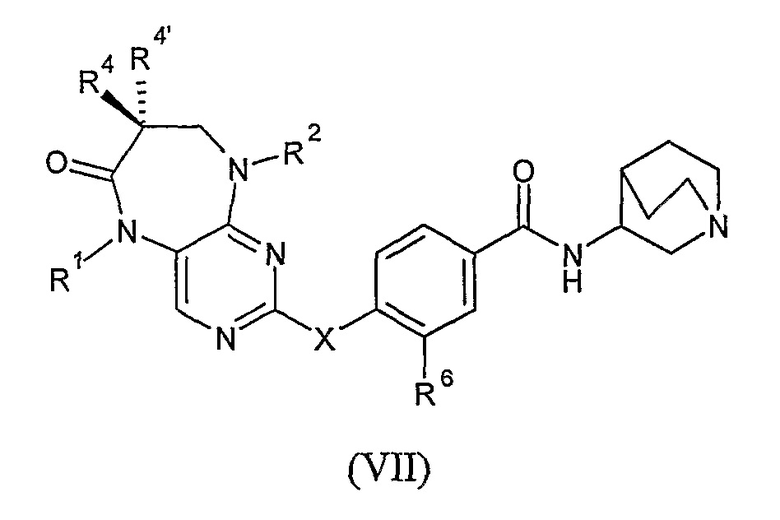
где
X представляет собой NR7;
каждый из R1 и R2 независимо представляет собой H, алкил или циклоалкил;
каждый из R4 и R4’ независимо представляет собой H или алкил; или
R4 и R4’, взятые вместе, образуют спиро-циклоалкильную группу; и
R6 представляет собой OR8 или галоген; и
каждый из R7 и R8 независимо представляет собой H или алкил.
Предпочтительно, R1 представляет собой алкил, а более предпочтительно, метил.
Предпочтительно, R2 представляет собой циклоалкильную группу, а более предпочтительно, C3-6-циклоалкильную группу. Еще более предпочтительно, R2 представляет собой циклопентильную или циклогексильную группу, а еще более предпочтительно, циклопентил.
Предпочтительно, R7 представляет собой H или алкил, более предпочтительно, H или метил, а еще более предпочтительно, H.
В одном из предпочтительных вариантов изобретения, один из R4 и R4’ представляет собой алкил, а другой представляет собой H или алкил.
В другом предпочтительном варианте изобретения, каждый из R4 и R4’ независимо представляет собой алкил. Более предпочтительно, R4 и R4’, оба, представляют собой метил.
В другом предпочтительном варианте изобретения, R4 и R4’, оба, представляют собой H.
В другом предпочтительном варианте изобретения, R4 и R4’, взятые вместе, образуют спиро-циклоалкильную группу, а более предпочтительно, спиро-C3-6циклоалкильную группу. Еще более предпочтительно, R4 и R4’, взятые вместе, образуют спиро-C3 или C4циклоалкильную группу, а более предпочтительно, C3циклоалкильную группу.
Предпочтительно, R6 представляет собой OR8, а еще более предпочтительно, OMe.
В одном из предпочтительных вариантов изобретения, соединение формулы VII выбрано из таких соединений, как
№
и их фармацевтически приемлемые соли или сложные эфиры.
Соединения формулы VI
В одном из своих аспектов, настоящее изобретение относится к соединению формулы VI, или к его фармацевтически приемлемой соли или фармацевтически приемлемому сложному эфиру,

где
X представляет собой NR7;
каждый из R1 и R2 независимо представляет собой H, алкил или циклоалкил;
один из R4 и R4’ представляет собой алкил, а другой представляет собой H или алкил; или
R4 и R4’, взятые вместе, образуют спиро-циклоалкильную группу;
R18 представляет собой H или алкил, где указанная алкильная группа необязательно замещена R6;
каждый R6 независимо представляет собой OR8 или галоген; и
каждый из R7 и R8 независимо представляет собой H или алкил.
Предпочтительно, R1 представляет собой алкил, а более предпочтительно, метил.
Предпочтительно, R2 представляет собой циклоалкильную группу, а более предпочтительно, C3-6-циклоалкильную группу. Еще более предпочтительно, R2 представляет собой циклопентильную или циклогексильную группу, а еще более предпочтительно, циклопентил.
Предпочтительно, R7 представляет собой H или алкил, более предпочтительно, H или метил, а еще более предпочтительно, H.
В одном из предпочтительных вариантов изобретения, один из R4 и R4’ представляет собой алкил, а другой представляет собой H или алкил.
В другом предпочтительном варианте изобретения, каждый из R4 и R4’ независимо представляет собой алкил. Более предпочтительно, R4 и R4’, оба, представляют собой метил.
В другом предпочтительном варианте изобретения, R4 и R4, оба, представляют собой H.
В другом предпочтительном варианте изобретения, R4 и R4’, взятые вместе, образуют спиро-циклоалкильную группу, а более предпочтительно, спиро-C3-6циклоалкильную группу. Еще более предпочтительно, R4 и R4’, взятые вместе, образуют спиро-C3 или C4циклоалкильную группу, а более предпочтительно, C3циклоалкильную группу.
Предпочтительно, R6 представляет собой OMe или F.
Предпочтительно, R18 представляет собой метил или CH2CH2OH.
В одном из предпочтительных вариантов изобретения, соединение формулы VI выбрано из таких соединений, как
и их фармацевтически приемлемые соли и сложные эфиры.
В одном из особенно предпочтительных вариантов изобретения, указанным соединением является соединение [254], или его фармацевтически приемлемая соль или фармацевтически приемлемый сложный эфир.
В одном из наиболее предпочтительных вариантов изобретения, указанным соединением является HCl-соль соединения [254], то есть 4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-3-метокси-N-(4-метилпиперазин-1-ил)бензамид ⋅ HCl.
В другом своем аспекте, настоящее изобретение относится к соединению, выбранному из таких соединений, как
№
и их фармацевтически приемлемые соли и сложные эфиры.
Предпочтительное соединение выбрано из таких соединений, как
№
и их фармацевтически приемлемые соли и сложные эфиры.
Преимущественно, некоторые соединения согласно изобретению обладают улучшенной селективностью к одной или нескольким киназам по сравнению с уже известными структурно родственными соединениями.
В одном из предпочтительных вариантов изобретения, соединение согласно изобретению обладает большей селективностью к одной или нескольким определенным киназам, по сравнению с одной или более другими киназами.
Так, например, в одном из предпочтительных вариантов изобретения, соединения согласно изобретению способны ингибировать преимущественно киназы семейства PLK, по сравнению с одной или более другими протеинкиназами; а поэтому данное соединение обладает способностью «селективно» ингибировать киназы семейства PLK, по сравнению с одной или более другими протеинкиназами. Используемый в описании термин «селективный» означает, что указанное соединение является селективным к одной или нескольким PLK, по сравнению с одной или более другими протеинкиназами. Предпочтительно, отношение селективности в отношении киназ семейства PLK по сравнению с одной или более другими протеинкиназами превышает примерно 2:1, более предпочтительно, примерно 5:1 или примерно 10:1, а еще более предпочтительно, примерно 20:1, или 50:1 или 100:1. Отношение селективностей может быть определено самим специалистом в данной области.
В одном из особенно предпочтительных вариантов изобретения, соединение согласно изобретению обладает селективностью к PLK1. Таким образом, в одном из предпочтительных вариантов изобретения, соединение согласно изобретению обладает способностью ингибировать преимущественно PLK1, по сравнению с одной или более другими протеинкиназами. Предпочтительно, отношение селективности данного соединения в отношении PLK1 по сравнению с одной или более другими протеинкиназами превышает примерно 2:1, более предпочтительно, примерно 5:1 или примерно 10:1, а еще более предпочтительно, примерно 20:1, или 50:1 или 100:1.
В еще более предпочтительном варианте изобретения, соединение согласно изобретению обладает селективностью к PLK1, по сравнению с PLK2 и/или PLK3. Таким образом, в одном из предпочтительных вариантов изобретения, соединение согласно изобретению обладает способностью ингибировать преимущественно PLK1, по сравнению с PLK2 и/или PLK3. Предпочтительно, отношение селективности данного соединения к PLK1 к селективности этого соединения к PLK2 и/или PLK3 превышает примерно 2:1, более предпочтительно, примерно 5:1 или примерно 10:1, а еще более предпочтительно, примерно 20:1, или 50:1 или 100:1.
В целях иллюстрации можно отметить, что соединение [218], по сравнению с уже известными выбранными соединениями, обладает селективностью к PLK1, по сравнению с PLK2 и PLK3. Так, например, селективность соединения [218] согласно изобретению к PLK1 по сравнению с селективностью к PLK2 в два раза превышает соответствующую селективность соединения [I-64], описанного в WO 07/095188, примерно в 6 раз превышает соответствующую селективность соединения [I-76], и в 7 раз превышает соответствующую селективность соединения [I-4], описанного в WO 07/095188. Аналогичным образом, селективность соединения [218] к PLK1, по сравнению с селективностью к PLK3 в 5 раз превышает селективность соединения [I-64], описанного в WO 07/095188, примерно в 12 раз превышает селективность соединения [I-76], и в 5 раз превышает селективность соединения [I-4], описанного в WO 07/095188. Более подробное описание можно найти в прилагаемых примерах.
В другом предпочтительном варианте изобретения, соединения согласно изобретению, по сравнению с уже известными структурно родственными соединениями, обладают превосходной растворимостью и/или превосходными фармакокинетическими свойствами.
Так, например, соединение [254], по сравнению с соединениями [I-76] и [I-253], описанными в WO 07/095188, обладает превосходной растворимостью и/или превосходными фармакокинетическими свойствами, а поэтому указанное соединение является более эффективным для лечения заболеваний. Более подробное описание сравнительных исследований также можно найти в прилагаемых примерах.
Аналогичным образом, соединение [371], по сравнению с уже известными структурно родственными соединениями, обладает благоприятными фармакокинетическими свойствами с точки зрения их системного действия и биологической доступности при пероральном введении.
В одном из своих предпочтительных вариантов, настоящее изобретение относится к подгруппе соединений, которые отличаются низким уровнем их выведения из клеток под действием переносчиков АТФ-связывающего кластера (ABC), таких как Р-гликопротеин (P-gp), который служит в качестве «насоса» для выведения лекарственного средства. Известно, что субстраты, способствующие выведению P-gp, обычно обнаруживают плохую пероральную всасываемость, что обусловлено действием P-gp, который может переносить абсорбированное лекарственное средство обратно в кишечник [Ambudkar S.V., et al. (2003) Oncogene, 22, 7468-7485]. P-gp может также снижать системное действие субстратов посредством стимуляции их экскреции в желчь и мочу. Переносчики ABC, помимо того, что они служат в качестве насосов для выведения лекарственного средства, могут сообщать опухолевым клеткам резистентность к лекарственному средству в результате повышения уровня экспрессии. Экспрессия P-gp хорошо известна как механизм, посредством которого раковые клетки могут приобретать резистентность ко многим лекарственным средствам (MDR) [Gottesman M.M., et al. (2002) Nat. Rev. Cancer, 2, 48-58]. Некоторые опухоли, происходящие от клеток, которые обычно экспрессируют P-gp, могут обладать природной резистентностью к лекарственным средствам [Ambudkar S.V., et al. (2005) Trends Pharmacol. Sci., 26, 385-387]. Лекарственные средства, которые легко выводятся из клеток, не могут аккумулироваться в больших количествах в клетках, экспрессирующих указанные переносчики, что ограничивает эффективность данной терапии из-за пониженного уровня взаимодействия «лекарственное средство – мишень».
Поэтому соединения с низким уровнем выведения из клеток могут найти свое применение в лечении рака, а также опухолей, происходящих от эпителиальных клеток и злокачественных опухолей, резистентных к лекарственным средствам, путем перорального введения этих соединений, причем вероятность индуцирования резистентности к этим соединениям при повышенном уровне экспрессии «насосов», служащих для их выведения, очень низка. Особенно предпочтительными соединениями согласно изобретению являются соединения, которые обнаруживают низкий уровень выведения из клеток, являются соединения [371], [372] и [377]-[391]. Более подробное описание анализа на биологическую доступность этих соединений и их выведения из клеток можно найти в примере 9 и на фигуре 3.
Кроме того, соединения согласно изобретению, которые имеют низкую степень асимметрии в клетках с резистентностью ко многим лекарственным средствам (MDR)/родительских клетках в анализе на биологическую доступность, также являются более эффективными в отношении ингибирования PLK1 в опухолевых клетках с MDR, чем известные соединения. Таким образом, эти соединения могут оказаться более эффективными в качестве терапевтических ингибиторов PLK. Подробное описание экспериментов по измерению уровней ингибирования PLK1 парах клеточных линий A2780-A2780/ADR приводится в прилагаемых примерах.
Терапевтическое применение
Было обнаружено, что соединения согласно изобретению обладают антипролиферативной активностью, а поэтому предполагается, что они могут быть использованы для лечения пролиферативных заболеваний, таких как рак, лейкозы и другие расстройства, ассоциированные с нерегулируемой пролиферацией клеток, такие как псориаз и рестеноз.
Так, например, в одном из своих аспектов, настоящее изобретение относится к применению соединения согласно изобретению или к его фармацевтически приемлемой соли в целях получения лекарственного препарата для лечения пролиферативного расстройства.
Используемый в описании термин «получение лекарственного препарата» включает применение одного или нескольких вышеописанных соединений непосредственно в качестве лекарственного препарата, а также их применение в программе скрининга других антипролиферативных средств или их применение на любой стадии получения такого лекарственного препарата.
Как определено в настоящей заявке, антипролиферативный эффект согласно изобретению может быть продемонстрирован по способности соединения ингибировать пролиферацию клеток анализе in vitro с использованием целых клеток, например, с использованием любой из клеточных линий AGS, H1299 или SJSA-1. Путем проведения таких анализов можно определить, обладает ли данное соединение антипролиферативным действием согласно изобретению.
В одном из своих предпочтительных вариантов, настоящее изобретение относится к применению одного или нескольких соединений для лечения пролиферативных расстройств. Предпочтительным пролиферативным расстройством является рак или лейкоз. Используемый в описании термин «пролиферативное расстройство» в его широком смысле включает любое расстройство, лечение которого требует регуляции клеточного цикла, например, сердечно-сосудистые расстройства, такие как рестеноз и кардиомиопатия; аутоиммунные расстройства, такие как гломерулонефрит и ревматоидный артрит; кожные болезни, такие как псориаз; воспалительные заболевания; заболевания, вызываемые грибками; заболевания, вызываемые паразитами, такие как малярия; эмфизема и аллопеция. При этих расстройствах, соединения согласно изобретению могут индуцировать апоптоз или, если это необходимо, обеспечивать остановку роста нужных клеток.
В одном из предпочтительных вариантов изобретения, указанным пролиферативным расстройством является рак или лейкоз, а более предпочтительно, рак.
В одном из предпочтительных вариантов изобретения, указанным пролиферативным расстройством является солидная опухоль.
В другом предпочтительном варианте изобретения, указанным пролиферативным расстройством является рак крови. Предпочтительно, таким раком крови является лейкоз, а более предпочтительно, прогрессирующий лейкоз или синдром миелодисплазии (СМД). Другими примерами являются острый миелогенный лейкоз (ОМЛ), острый лимфоцитарный лейкоз (ОЛЛ) или хронический лимфоцитарный лейкоз (ОЛЛ).
В другом предпочтительном варианте изобретения, указанным пролиферативным расстройством является гломерулонефрит.
В другом предпочтительном варианте изобретения, указанным пролиферативным расстройством является ревматоидный артрит.
В другом предпочтительном варианте изобретения, указанным пролиферативным расстройством является псориаз.
В другом предпочтительном варианте изобретения, указанным пролиферативным расстройством является хроническая обструктивная болезнь легких.
Соединения согласно изобретению могут ингибировать любые стадии или фазы клеточного цикла, например, такие как образование ядерной оболочки, выход из фазы покоя клеточного цикла (G0), прохождение G1-фазы, деконденсация хромосомы, разрушение ядерной оболочки, переход через ограничительную точку «Старт» (START), инициация репликации ДНК, события репликации ДНК, терминация репликации ДНК, удвоение центросомы, прохождение G2-фазы, активация митотических или мейотических функций, конденсация хромосомы, отделение центросомы, инициация образования микротрубочек, образование и функции веретена, взаимодействие с белками-переносчиками микротрубочек, отделение и сегрегация хроматида, инактивация митотических функций, образование сократительного кольца и цитокинетические функции. В частности, соединения согласно изобретению могут оказывать влияние на некоторые функции генов, такие как связывание хроматина, образование комплексов репликации, сигнал «разрешения» на репликацию, фосфорилирование или другая активность, направленная на вторичную модификацию, протеолитическое разложение, связывание микротрубочек, связывание актина, связывание септина, активность, направленная на инициацию образования центра организации микротрубочек и связывание с компонентами пути передачи сигнала прохождения клеточного цикла.
В одном из вариантов изобретения, соединение согласно изобретению вводят в количестве, достаточном для ингибирования, по меньшей мере, одного фермента PLK.
В другом своем аспекте, настоящее изобретение относится к способу лечения PLK-зависимого расстройства, где указанный способ включает введение индивидууму, нуждающемуся в этом, соединения согласно изобретению или его фармацевтически приемлемой соли, определенных выше, в количестве, достаточном для ингибирования PLK.
Polo-подобные киназы (PLK) принадлежат к семейству сериновых/треониновых протеинкиназ. Митотические мутанты Drosophila melanogaster в своем polo-локусе имеют аномалии веретена [Sunkel et al., J. Cell Sci., 1988, 89, 25], при этом было обнаружено, что ген polo кодирует митотическую киназу [Llamazares et al., Genes Dev., 1991, 5, 2153]. У человека имеются три близкородственных PLK [Glover et al., Genes Dev., 1998, 12, 3777]. Такие PLK содержат в высокой степени гомологичный аминоконцевой каталитический киназный домен, а у их карбоксиконцов содержатся две или три консервативных области, polo-боксы. Функция polo-боксов пока еще точно не установлена, однако известно, что они участвуют в доставке PLK в субклеточные компартменты [Lee et al., Proc. Natl. Acad. Sci. USA, 1998, 95, 9301; Leung et al., Nat. Struct. Biol., 2002, 9, 719] и в опосредовании взаимодействий с другими белками [Kauselmann et al., EMBO J., 1999, 18, 5528], либо они могут составлять часть ауторегуляторного домена [Nigg, Curr. Opin. Cell Biol., 1998, 10, 776]. Кроме того, зависимая от polo-бокса активность PLK1 необходима для «правильного» перехода из метафазы в анафазу и для цитокинеза [Yuan et al., Cancer Res., 2002, 62, 4186; Seong et al., J. Biol. Chem., 2002, 277, 32282].
Проведенные исследования показали, что человеческие PLK регулируют некоторые фундаментальные аспекты митоза [Lane et al., J. Cell. Biol., 1996, 135, 1701; Cogswell et al., Cell Growth Differ., 2000, 11, 615]. В частности, активность PLK1 необходима для функционального созревания центросом на поздней фазе G2/ранней профазе и последующего образования биполярного веретена. Истощение клеточной PLK1 под действием короткой интерферирующей РНК (киРНК) также подтверждает, что этот белок необходим для осуществления многих митотических процессов и завершения цитокинеза [Liu et al., Proc. Natl. Acad. Sci. USA, 2002, 99, 8672].
В более предпочтительном варианте настоящего изобретения, соединение согласно изобретению вводят в количестве, достаточном для ингибирования PLK1.
Из трех киназ PLK, лучше всего охарактеризована киназа PLK1, и эта киназа регулирует ряд циклов деления клеток, включая начало митоза [Toyoshima-Morimoto et al., Nature, 2001, 410, 215; Roshak et al., Cell. Signalling, 2000, 12, 405], активацию сверочных точек повреждения ДНК [Smits et al., Nat. Cell Biol., 2000, 2, 672; van Vugt et al., J. Biol. Chem., 2001, 276, 41656], регуляцию образования комплекса, стимулирующего анафазу [Sumara et al., Mol. Cell, 2002, 9, 515; Golan et al., J. Biol. Chem., 2002, 277, 15552; Kotani et al., Mol. Cell, 1998, 1, 371], фосфорилирование протеосомы [Feng et al., Cell Growth Differ., 2001, 12, 29], и удвоение и созревание центросомы [Dai et al., Oncogene, 2002, 21, 6195].
В одном из особенно предпочтительных вариантов изобретения, соединениями согласно изобретению являются АТФ-антагонисты, являющиеся ингибиторами PLK1.
В контексте настоящего изобретения, термин «АТФ-антагонизм» означает способность соединения ингибитора ослаблять или предотвращать каталитическую активность PLK, то есть перенос фосфорной группы от АТФ на макромолекулярный субстрат PLK благодаря обратимому или необратимому связыванию в активном центре фермента с последующим нарушением или предотвращением АТФ-связывания.
В другом предпочтительном варианте изобретения, соединение согласно изобретению вводят в количестве, достаточном для ингибирования PLK2 и/или PLK3.
В другом своем аспекте, настоящее изобретение относится к способу ингибирования PLK в клетках, где указанный способ включает контактирование указанных клеток с соединением формулы I или с его фармацевтически приемлемой солью в количестве, достаточном для ингибирования PLK в указанных клетках.
В еще одном своем аспекте, настоящее изобретение относится к способу лечения пролиферативного расстройства, где указанный способ включает ингибирование PLK путем введения индивидууму, нуждающемуся в этом, терапевтически эффективного количества соединения формулы I или его фармацевтически приемлемой соли в целях лечения указанного пролиферативного расстройства.
В другом предпочтительном варианте изобретения, соединение согласно изобретению вводят в количестве, достаточном для ингибирования, по меньшей мере, одной киназы «Аврора». Предпочтительной киназой «Аврора» является киназа «Аврора А», киназа «Аврора В» или киназа «Аврора С».
В другом своем аспекте, настоящее изобретение относится к способу лечения расстройства, ассоциированного с действием киназы «Аврора», где указанный способ включает введение индивидууму, нуждающемуся в этом, соединения согласно изобретению или его фармацевтически приемлемой соли, определенных выше, в количестве, достаточном для ингибирования киназы «Аврора».
В другом своем аспекте, настоящее изобретение относится к применению соединения согласно изобретению в целях ингибирования протеинкиназы.
В другом своем аспекте, настоящее изобретение относится к способу ингибирования протеинкиназы, где указанный способ включает контактирование указанной протеинкиназы с соединением согласно изобретению.
Предпочтительно, указанную протеинкиназу выбирают из киназы «Аврора» и PLK. Более предпочтительно, указанной протеинкиназой является PLK, а наиболее предпочтительно, PLK1.
Фармацевтические композиции
В другом своем аспекте, настоящее изобретение относится к фармацевтической композиции, содержащей соединение согласно изобретению в смеси с одним или несколькими фармацевтически приемлемыми разбавителями, наполнителями или носителями. Хотя соединения согласно изобретению (включая их фармацевтически приемлемые соли, сложные эфиры и фармацевтически приемлемые сольваты) могут быть введены отдельно, однако, обычно, а в частности, при их применении в медицине, их вводят в смеси с фармацевтическим носителем, наполнителем или разбавителем. Фармацевтические композиции могут быть применены для лечения человека или животных, то есть они могут применяться в медицине и в ветеринарии.
Примеры таких наполнителей, подходящих для получения различных других форм фармацевтических композиций, описанных в настоящей заявке, можно найти в руководстве «Handbook of Pharmaceutical Excipients, 2nd Edition, (1994), Edited by A Wade and P.J. Weller».
Носители или разбавители, подходящие для их применения в терапевтических целях, хорошо известны специалистам-фармацевтам, и описаны, например, в руководстве Remington’s Pharmaceutical Sciences, Mack Publishing Co. (A. R. Gennaro edit. 1985).
Примерами подходящих носителей являются лактоза, крахмал, глюкоза, метилцеллюлоза, стеарат магния, маннит, сорбит и т.п. Примерами подходящих разбавителей являются этанол, глицерин и вода.
Выбор фармацевтических носителей, наполнителей или разбавителей может быть осуществлен в зависимости от нужного способа введения и в соответствии со стандартной фармацевтической практикой. В качестве носителей, наполнителей или разбавителей или помимо этих носителей, наполнителей или разбавителей фармацевтические композиции могут содержать любые подходящие связующие вещества, замасливатели, суспендирующие агенты, агенты для нанесения покрытий или солюбилизирующие агенты.
Примерами подходящих связующих веществ являются крахмал, желатин, природные сахара, такие как глюкоза, безводная лактоза, свободно растекающаяся лактоза, бета-лактоза, кукурузные подсластители, природные и синтетические смолы, такие как аравийская камедь, трагакантовая камедь или альгинат натрия, карбоксиметилцеллюлоза и полиэтиленгликоль.
Примерами подходящих замасливателей являются олеат натрия, стеарат натрия, стеарат магния, бензоат натрия, ацетат натрия, хлорид натрия и т.п.
В фармацевтической композиции могут присутствовать консерванты, стабилизаторы, красители и даже ароматизаторы. Примерами консервантов являются бензоат натрия, сорбиновая кислота и сложные эфиры п-гидроксибензойной кислоты. Могут быть также использованы антиоксиданты и суспендирующие агенты.
Соли/сложные эфиры
Соединения согласно изобретению могут присутствовать в виде солей или сложных эфиров, а в частности, фармацевтически приемлемых солей или сложных эфиров.
Фармацевтически приемлемыми солями соединений согласно изобретению являются подходящие кислотно-аддитивные или основно-аддитивные соли данных соединений. Описание подходящих фармацевтически приемлемых солей можно найти в публикации Berge et al., J. Pharm. Sci., 66, 1-19 (1977). Соли могут быть образованы, например, сильными неорганическими кислотами, такими как минеральные кислоты, например, серная кислота, фосфорная кислота или галогенводородная кислота; сильными органическими карбоновыми кислотами, такими как алканкарбоновые кислоты, имеющие 1-4 атомов углерода, которые являются незамещенными или замещенными (например, галогеном), такие как уксусная кислота; насыщенными или ненасыщенными дикарбоновыми кислотами, такими как, например, щавелевая, малоновая, янтарная, малеиновая, фумаровая, фталевая или тетрафталевая кислоты; гидроксикарбоновыми кислотами, такими как, например, аскорбиновая, гликолевая, молочная, яблочная, винная или лимонная кислота; аминокислотами, например, аспарагиновой или глутаминовой кислотой; бензойной кислотой; или органическими сульфоновыми кислотами, такими как (C1-C4)-алкил- или арилсульфоновые кислоты, которые являются незамещенными или замещенными (например, галогеном), такие как метан- или п-толуолсульфоновая кислота. Предпочтительной солью является HCl-соль.
Сложные эфиры могут быть образованы органическими кислотами или спиртами/гидроксидами, в зависимости от этерификации функциональной группы. Органическими кислотами являются карбоновые кислоты, такие как алканкарбоновые кислоты, имеющие 1-12 атомов углерода, которые являются незамещенными или замещенными (например, галогеном), такие как уксусная кислота; насыщенные или ненасыщенные дикарбоновые кислоты, такие как, например, щавелевая, малоновая, янтарная, малеиновая, фумаровая, фталевая или тетрафталевая кислоты; гидроксикарбоновые кислоты, такие как, например, аскорбиновая, гликолевая, молочная, яблочная, винная или лимонная кислота; аминокислоты, например, аспарагиновая или глутаминовая кислота; бензойная кислота; или органические сульфоновые кислоты, такие как (C1-C4)-алкил- или арилсульфоновые кислоты, которые являются незамещенными или замещенными (например, галогеном), такие как метан- или п-толуолсульфоновая кислота. Подходящими гидроксидами являются неорганические гидроксиды, такие как гидроксид натрия, гидроксид калия, гидроксид кальция, гидроксид алюминия. Спиртами являются алкановые спирты, имеющие 1-12 атомов углерода, которые могут быть незамещенными или замещенными (например, галогеном).
Энантиомеры/таутомеры
Во всех аспектах, обсуждаемых выше, настоящее изобретение включает, если это необходимо, все энантиомеры и таутомеры соединений согласно изобретению. Специалистам известны соединения, обладающие оптическими свойствами (то есть имеющие один или несколько хиральных атомов углерода) или таутомерными свойствами. Соответствующие энантиомеры и/или таутомеры могут быть выделены/получены известными методами.
Стереоизомеры и геометрические изомеры
Некоторые соединения согласно изобретению могут существовать в виде стереоизомеров и/или геометрических изомеров, например, они могут иметь один или несколько асимметрических и/или геометрических центров, а поэтому они могут существовать в двух или более и/или геометрических формах. В настоящем изобретении рассматривается применение всех отдельных стереоизомеров и геометрических изомеров указанных агентов-ингибиторов и их смесей. Термины, используемые в настоящей заявке, охватывают все указанные формы, при условии, что эти формы сохраняют соответствующую функциональную активность (хотя необязательно в аналогичной степени).
Настоящее изобретение также включает все подходящие изотопные варианты данного агента или его фармацевтически приемлемой соли. Изотопный вариант агента согласно изобретению или его фармацевтически приемлемой соли определен как вариант, в котором, по меньшей мере, один атом заменен атомом, имеющим тот же самый порядковый номер, но по своей атомной массе отличающийся от атома, обычно встречающегося в природе. Примерами изотопов, которые могут быть включены в данный агент и его фармацевтически приемлемые соли, являются изотопы водорода, углерода, азота, кислорода, фосфора, серы, фтора и хлора, такие как 2Н, 3H, 13C, 14C, 15N, 17О, 18O, 31P, 32P, 35S, 18F и 36Cl, соответственно. Некоторые изотопы агента и его фармацевтически приемлемых солей, например, изотопы, в число которых входит радиоактивный изотоп, такой как 3H или 14C, могут быть использованы в исследованиях по распределению лекарственных средств и/или субстратов в ткани. Соединения, меченные тритием, то есть 3H, и углеродом-14, то есть изотопами 14C, являются особенно предпочтительными из-за легкости их получения и детектирования. Кроме того, замещение изотопами, такими как дейтерий, то есть 2H, может давать некоторые терапевтические преимущества, благодаря их более высокой метаболической стабильности, например, увеличение времени полужизни in vivo или снижение требуемой дозы, а поэтому, в некоторых случаях, такое замещение может оказаться предпочтительным. Изотопы агента согласно изобретению и его фармацевтически приемлемых солей обычно могут быть получены стандартными методами с использованием соответствующих изотопов подходящих реагентов.
Сольваты
Настоящее изобретение также включает сольватные формы соединений согласно изобретению. Термины, используемые в настоящей заявке, охватывают все эти формы.
Полиморфы
Настоящее изобретение также относится к соединениям согласно изобретению, которые имеют различные кристаллические формы, полиморфные формы и (без)водные формы. Специалистам-фармацевтам хорошо известно, что химические соединения могут быть выделены в любых указанных формах путем внесения незначительных изменений методом очистки и/или выделения с использованием растворителей при синтезе таких соединений.
Пролекарства
Настоящее изобретение также включает соединения согласно изобретению в форме пролекарства. Такими пролекарствами обычно являются соединения согласно изобретению, в которых одна или несколько соответствующих групп были модифицированы так, чтобы такую модификацию можно было удалить после введения указанных соединений человеку или млекопитающему. Такое удаление модификаций обычно осуществляют под действием фермента, обычно присутствующего у данного индивидуума, хотя для удаления модификации in vivo может быть введен второй агент вместе с указанным пролекарством. Примерами таких модификаций являются сложный эфир (например, любой из сложных эфиров, описанных выше), который может быть удален под действием эстеразы и т.п. Другие такие системы хорошо известны специалистам.
Введение
Фармацевтические композиции согласно изобретению могут быть адаптированы для перорального, ректального, вагинального, парентерального, внутримышечного, внутрибрюшинного, внутриартериального, интратекального, интрабронхиального, подкожного, интрадермального, внутривенного, интраназального, трансбуккального или подъязычного введения.
Препараты для перорального введения приготавливают в форме спрессованных таблеток, драже, таблеток, гелей, капель и капсул. Эти композиции, предпочтительно, содержат от 1 до 250 мг, а более предпочтительно, от 10 до 100 мг активного ингредиента на дозу.
Другие формы для введения включают растворы или эмульсии, которые могут быть введены внутривенно, внутриартериально, интратекально, подкожно, интрадермально, внутрибрюшинно или внутримышечно, и которые получают из стерильных или стерилизуемых растворов. Фармацевтические композиции согласно изобретению могут быть также приготовлены в форме суппозиториев, пессариев, суспензий, эмульсий, лосьонов, мазей, кремов, гелей, спреев, растворов или тонкодисперсных порошков.
Альтернативным способом чрескожного введения является наложение кожного пластыря. Так, например, активный ингредиент может быть включен в крем, состоящий из водной эмульсии полиэтиленгликолей или жидкого вазелинового масла. Активный ингредиент может быть также включен, в концентрации от 1 до 10% масс., в мазь, состоящую из основы, такой как белый воск или белое мягкое вазелиновое масло в комбинации, если это необходимо, со стабилизаторами и консервантами.
Формы для инъекций могут содержать от 10 до 1000 мг, предпочтительно, от 10 до 250 мг активного ингредиента на дозу.
Композиции могут быть приготовлены в стандартной лекарственной форме, то есть в форме дискретных частей, содержащих одноразовую стандартную дозу или множество дробных доз данной стандартной дозы.
Дозы
Специалист в данной области может легко без излишнего экспериментирования определить дозу одной из композиций согласно изобретению, подходящую для введения индивидууму. Обычно врач определяет фактическую дозу, которая может быть наиболее подходящей для данного пациента и зависит от ряда факторов, включая активность конкретно используемого соединения, метаболическую стабильность и продолжительность действия такого соединения, возраст, массу тела, общее состояние здоровья, пол и режим питания пациента, способ и время введения, скорость выведения, комбинацию лекарственных средств, тяжесть конкретного состояния индивидуума и терапию, которой подвергается данный индивидуум. Указанные в описании дозы представляют собой репрезентативную усредненную дозу, обычно применяемую в данных случаях. Само собой разумеется, что в каждом конкретном случае дозы могут быть выше или ниже указанных интервалов, и эти дозы также входят в объем настоящего изобретения.
В зависимости от ситуации, данный агент может быть введен в дозе, составляющей от 0,01 до 30 мг/кг массы тела, например, от 0,1 до 10 мг/кг, а более предпочтительно, от 0,1 до 1 мг/кг массы тела.
В репрезентативном варианте изобретения, для лечения злокачественно опухоли пациенту может быть введена одна или несколько доз, составляющих 10-150 мг/день.
Комбинации
В особенно предпочтительном варианте изобретения, одно или несколько соединений согласно изобретению вводят в комбинации с с одним или несколькими другими активными агентами, например, с противораковыми лекарственными средствами, имеющимися в продаже. В этих случаях соединения согласно изобретению могут быть введены поочередно, одновременно или последовательно с одним или несколькими другими активными агентами.
Противораковые лекарственные средства обычно являются более эффективными при их использовании в комбинации друг с другом. В частности, комбинированная терапия является предпочтительной с точки зрения устранения сопутствующего сильного токсического действия, а также с точки зрения ее механизма действия и механизма(ов) вырабатывания резистентности. Кроме того, большинство лекарственных средств также желательно вводить в их максимально допустимых дозах с минимальными интервалами времени между введением доз. Основное преимущество комбинаций химиотерапевтических лекарственных средств заключается в том, что они могут стимулировать аддитивный или даже синергический эффект посредством биохимических взаимодействий, а также могут снижать вероятность развития резистентности в опухолевых клетках на ранней стадии, которые должны быть, в той или иной степени, восприимчивыми к начальной химиотерапии, проводимой с использованием одного агента. Примером использования биохимических взаимодействий при выборе комбинаций лекарственных средств является введение лейковорина в целях повышения уровня связывания активного внутриклеточного метаболита 5-фторурацила с его мишенью, а именно, тимидилат-синтазой, с последующим повышением его цитотоксических эффектов.
В современных методах лечения рака и лейкоза используется множество комбинаций. Более широкий обзор методов, применяемых во врачебной практике можно найти в руководстве «Oncologic Therapies», изданном E. E. Vokes и H. M. Golomb, и опубликовано в Springer.
Предпочтительные комбинации могут быть выбраны путем исследования рост-ингибирующей активности тестируемых соединений с использованием агентов, которые, как известно или как предполагается, являются эффективными для лечения конкретного ракового заболевания на начальной стадии или для элиминации клеточных линий, происходящих от данных раковых опухолей. Такой способ может быть также применен для определения порядка введения агентов, то есть до введения данного лекарственного средства, одновременно с его введением или после его введения. Указанная схема введения может иметь ту особенность, что во всех циклах действуют идентифицированные в настоящем описании агенты.
Способы
В другом своем аспекте, настоящее изобретение относится к способу получения соединения согласно изобретению, где указанный способ включает стадии:
(i) превращения соединения формулы (II) в соединение формулы (IV) либо непосредственно, либо путем выделения соединения формулы (III); и
(ii) превращения указанного соединения формулы (IV) в соединение формулы (I)
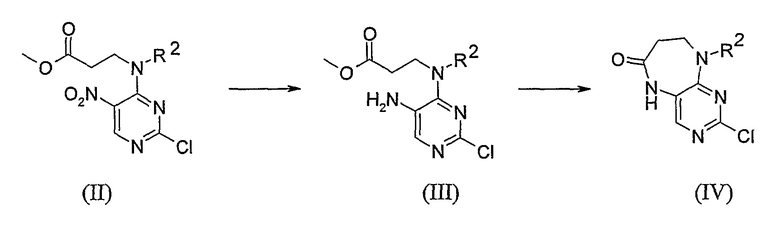
Стадия (i), предпочтительно, включает нагревание указанного соединения формулы (II) с NH4Cl и Fe в EtOH/H2O.
Настоящее изобретение также проиллюстрировано на нижеследующих неограничивающих примерах со ссылкой на описанные ниже фигуры, где
На фигуре 1 проиллюстрирована площадь под кривой (AUC), которая представляет собой зависимость концентрации соединений А-Н в плазме от времени для каждого тестируемого соединения. Во всех проводимых экспериментах, доза носителя остается постоянной (для внутривенного введения = цитратный буфер, pH 3, 1 мл/кг; для перорального введения = DMA/ПЭГ400/10 мМ тартратный буфер, pH 4 (1:3:6), 5 мл/кг);
На фигуре 2 представлены такие же данные, как и на фигуре 1, но в форме биологической доступности (% F) каждого соединения для перорального введения.
На фигуре 3 проиллюстрировано сравнение выбранных соединений с уже известными соединениями A’-J’ (определенными в таблице 8) в родительских опухолевых клетках и в опухолевых клетках, резистентных к многим лекарственным средствам. Отношения асимметрии приводятся для соединения [378].
ПРИМЕРЫ
Общий экспериментальный метод
Химические соединения и растворители были закуплены у коммерческих поставщиков и были использованы, если это не оговорено особо, в том виде, в котором они были получены. В качестве стандартного осушителя для органических растворов, если это не оговорено особо, был использован безводный MgSO4. ЯМР-спектры были зарегистрированы с помощью устройства Varian INOVA-500. Химические сдвиги выражены в миллионных долях по отношению к внутреннему стандарту тетраметилсилану. Константы взаимодействия (J) равны приблизительно 0,1 Гц. В описании использованы следующие сокращения: с (s), синглет; д (d), дублет; т (t), триплет; кв. (q), квартет; квин. (qu), квинтуплет; м (m), мультиплет и шир. (br), широкий. Масс-спектры были получены на одноквадрупольном масс-спектрометре Waters ZQ2000 с ионизацией электрораспылением (ESI). Аналитическую и препаративную ОФ-ВЭЖХ осуществляли на колонке с силикагелем Vydac 218TP54 (250×4,6 мм) или Vydac 218TPl022 (250×22 мм) с элюированием в линейном градиенте системы вода/ацетонитрил (содержащей 0,1% трифторуксусную кислоту) при скорости потока 1 мл/мин (в аналитической хроматографии) и 9 мл/мин (в препаративной хроматографии). При этом использовали следующие аналитические градиенты:
время = 0 мин
время = 20 мин
время = 25 мин
Альтернативно, хроматография может быть осуществлена на колонках Xbridge (100×4,6 мм) и Xbridge (100×19 мм) с элюированием в линейном градиенте системы вода/ацетонитрил (содержащей 0,1% гидроксид аммония) при скорости потока 1 мл/мин (в аналитической хроматографии) и 20 мл/мин (в препаративной хроматографии). При этом использовали следующие аналитические градиенты:
время = 0 мин
время = 5 мин
время=10 мин
время=12 мин
Флэш-хроматографию проводили на колонках, предварительно упакованных силикагелем (EM Kieselgel 60, 0,040-0,063 мм, Merck) или реагентом ISOLUTE (Biotage).
Сокращения:
Пример 1
Соединение [2]: 4-(9-циклопентил-5-метил-6-оксо-6,7,8,9-тетрагидро-5H-пиримидо[4,5-b][1,4]диазепин-2-иламино)-N-(1-метилпиперидин-4-ил)бензамид
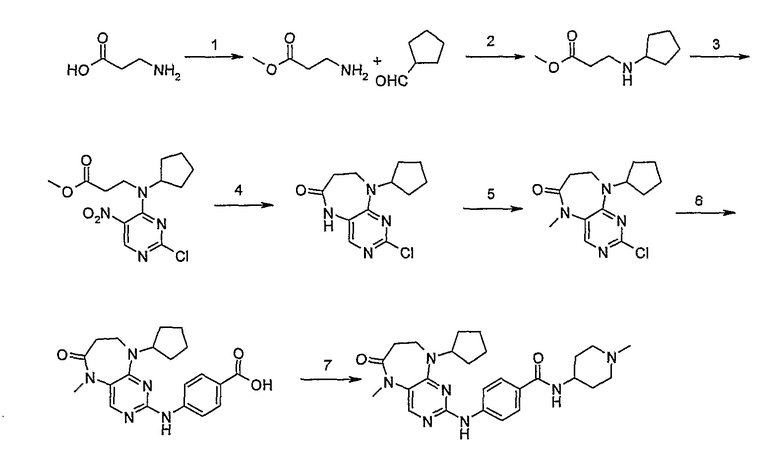
1. Тионилхлорид (2,1 экв.), MeOH, 0°C для добавления, кипячение с обратным холодильником – 2 ч.; 2. циклопентанон (0,77 экв.), ацетат натрия (0,77 экв.), триацетоксиборгидрид натрия (1,11 экв.), ДХМ, к.т., 16 ч.; 3. 2,4-дихлор-5-нитропиримидин (1,1 экв.), K2CO3 (1 экв.), ацетон, 0°C – к.т., 16 ч.; 4, NH4Cl (8,5 экв.), Fe (8 экв.), EtOH/H2O (4:1), кипячение с обратным холодильником – 2,5 ч.; - не выделяли. 5. MeI (1,18 экв.), NaH (1,07 экв.), ДМФ, -10°C – к.т., 3 ч.; 6. 4-амино-3-метоксибензойная кислота (1,5 экв.), концентрированная HCl, H2O/EtOH (4:1), кипячение с обратным холодильником – 48 ч.; 7. DIPEA (2 экв.), TBTU (1,1 экв.), 4-аминометилпиперидин (1,2 экв.), ДХМ, к.т., 16 часов.
Стадия 1: Метил-3-аминопропаноат
β-аланин (9,37 г, 0,105 моль) добавляли к MeOH (50 мл), и смесь охлаждали до 0°C на ледяной бане, а затем по каплям добавляли тионилхлорид [осторожно: при добавлении происходит экзотермическая реакция]. После добавления реакционную смесь нагревали до комнатной температуры, а затем кипятили с обратным холодильником в течение 2 ч. Растворитель выпаривали при пониженном давлении и полученное масло обрабатывали трет-бутилметиловым эфиром, а затем полученные кристаллы отфильтровывали и сушили в вакууме с получением белого кристаллического твердого вещества (11 г, колич.); 1H (ДМСО-d6): δ 2,73 (дд, J=7 Гц, 2H, CH2), 2,98 (дд, J=7 Гц, 2H, CH2), 3,61 (с, 3Η, CH3), 8,28 (шир.с., 2Η, NH2); МС(+ve): 104,1.
Стадия 2: Метил-3-(циклопентиламино)пропаноат
Метил-3-аминопропаноат (9,37 г, 0,09 моль) солюбилизировали в ДХМ (200 мл), а затем добавляли циклопентанон (6,43 мл, 0,07 моль), ацетат натрия (5,96 г, 0,07 моль) и триацетоксиборгидрид натрия (22 г, 0,10 моль). Реакционную смесь перемешивали при комнатной температуре в течение 16 ч. После этого добавляли 20% бикарбонат натрия (100 мл) и 2M гидроксид натрия (50 мл) и продукт экстрагировали ДХМ/Η2O. Органические экстракты объединяли, промывали насыщенным NaCl, сушили MgSO4, фильтровали и фильтрат выпаривали при пониженном давлении, а затем сушили в вакууме с получением продукта в виде бледно-желтого масла (8,90 г, 55%); МС(+ve): 172,4.
Стадия 3: Метил-3-[циклопентил(2-хлор-5-нитропиримидин-4-ил)амино]пропаноат
Метил-3-(циклопентиламино)пропаноат (838 мг, 0,005 моль) и K2CO3 (676 мг, 0,005 моль) добавляли к ацетону (5 мл), и полученную смесь охлаждали до 0°C на ледяной бане, а затем добавляли 2,4-дихлор-5-нитропиримидин (1,044 г, 1,1 экв.). Затем реакционную смесь (RM) нагревали до комнатной температуры и продолжали перемешивать еще 16 часов, после чего добавляли еще 0,12 экв. пиримидина. Затем перемешивание продолжали еще 3 часа. Реакционную смесь выпаривали при пониженном давлении и продукт экстрагировали EtOAc/H2O. Органические экстракты объединяли, промывали насыщенным NaCl, сушили MgSO4, фильтровали и фильтрат выпаривали при пониженном давлении, а затем сушили в вакууме с получением продукта в виде коричневого маслянистого остатка (1,04 г, 65%); Rt = 16 минут (Vydac 1); ME(+ve): 329,1.
Стадия 4: 2-хлор-9-циклопентил-5,7,8,9-тетрагидро-6H-пиримидо[4,5-b][1,4]диазепин-6-он
Метил-3-[циклопентил(2-хлор-5-нитропиримидин-4-ил)амино]пропаноат (1,00 г, 0,003 моль) и NH4Cl (1,38 г, 0,025 моль, 8,5 экв.) добавляли к EtOH/H2O (4:1 мл, 10 мл), и смесь кипятили с обратным холодильником, после чего порциями добавляли порошок железа (1,36 г, 0,024 моль, 8 экв.). Затем реакционную смесь кипятили с обратным холодильником еще 2 часа. За ходом реакции наблюдали с помощью ВЭЖХ, и если не наблюдалось присутствия исходного вещества (SM), то реакционную смесь в горячем виде фильтровали через целит. Целит промывали EtOAc (10 мл) и EtOH (10 мл) [оба горячими], а затем фильтрат выпаривали при пониженном давлении и сушили в вакууме, в результате чего получали продукт в виде коричневого твердого вещества (350 мг, 43%); Rt = 12 минут (Vydac 1); МС(+ve): 267,2.
Стадия 5: 2-хлор-9-циклопентил-5-метил-5,7,8,9-тетрагидро-6H-пиримидо[4,5-b][1,4]диазепин-6-он
2-хлор-9-циклопентил-5,7,8,9-тетрагидро-6H-пиримидо[4,5-b][1,4]диазепин-6-он (318 мг, 0,0012 моль) и MeI (88 мкл, 0,0014 моль, 1,18 экв.) добавляли в ДМФ (5 мл), и раствор охлаждали до -10°C ацетоном/сухим льдом, после чего добавляли NaH (30 мг, 0,0013 моль, 1,07 экв.). Реакционную смесь перемешивали при 0°С в течение 30 минут и при комнатной температуре в течение 30 минут. Затем реакционную смесь концентрировали, и продукт экстрагировали EtOAc/H2O. Органические экстракты объединяли, промывали насыщенным NaCl, сушили MgSO4, фильтровали и фильтрат выпаривали при пониженном давлении, а затем сушили в вакууме с получением продукта в виде пурпурного маслянистого остатка (252 мг, 75%); Rt = 13 минут (Vydac 1); МС(+ve): 281,2.
Стадия 6: (Соединение [1]) 4-(9-циклопентил-5-метил-6-оксо-6,7,8,9-тетрагидро-5H-пиримидо[4,5-b][1,4]диазепин-2-иламино)бензойная кислота.
Метод проведения реакции сочетания А
2-хлор-9-циклопентил-5-метил-5,7,8,9-тетрагидро-6H-пиримидо[4,5-b][1,4]диазепин-6-он (248 мг, 0,0009 моль) и 4-амино-3-метоксибензойную кислоту (221 мг, 0,0013 моль, 1,5 экв.), концентрированную HCl (152 мкл) и H2O/EtOH (8:2 мл) добавляли к RBF, и полученную реакционную смесь кипятили с обратным холодильником в течение 4 часов. За ходом реакции наблюдали с помощью ВЭЖХ, и если не наблюдалось присутствия исходного вещества (время реакции 4 часа), то реакционную смесь концентрировали при пониженном давлении, и продукт экстрагировали ДХМ/H2O. Органические экстракты объединяли, промывали насыщенным NaCl, сушили MgSO4, фильтровали и фильтрат выпаривали при пониженном давлении, а затем сушили в вакууме с получением коричневого маслянистого остатка. К остатку добавляли несколько капель MeOH, и образовавшийся твердый осадок собирали путем вакуумной фильтрации, промывали MeOH, а затем сушили в вакууме с получением продукта в виде пурпурного твердого вещества (51 мг, 14%); Rt = 11,2 мин (0_60_20 мин, чистота 100%); 1H-ЯМР (ДМСО-d6): δ 1,61 (шир.с, 4H, циклопент-H), 1,72 (шир.с, 2Η, циклопент-H), 1,94 (шир.с, 2H, циклопент-H), 2,59 (дд, J=4,5 Гц, 2H, CH2), 3,18 (с, 3Η, CH3), 3,63 (дд, J=4,5 Гц, 2H, CH2), 3,95 (с, 3Η, CH3), 7,51 (с, 1Η), 7,56 (д, J=8 Гц, 1H, phe-H), 7,82 (с, 1H), 8,10 (с, 1H), 8,47 (д, J=8,5 Гц, 1H, phe-H), 12,64 (шир.с, 1Η, NH); MS+ve: 412,2.
Стадия 7: (Соединение [2]): 4-(9-циклопентил-5-метил-6-оксо-6,7,8,9-тетрагидро-5H-пиримидо[4,5-b][1,4]диазепин-2-иламино)-N-(1-метилпиперидин-4-ил)бензамид
4-(9-циклопентил-5-метил-6-оксо-6,7,8,9-тетрагидро-5Η-пиримидо[4,5-b][1,4]диазепин-2-иламино)бензойную кислоту (35 мг, 0,085 ммоль), DIPEA (28 мкл, 0,17 ммоль, 2 экв.) и TBTU (30 мг, 0,093 ммоль, 1,1 экв.) добавляли к 3 мл ДХМ и полученный раствор перемешивали при комнатной температуре в течение 30 минут, а затем добавляли 4-аминометилпиперидин (13 мкл, 0,10 ммоль, 1,2 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Затем реакционную смесь концентрировали, и продукт экстрагировали ДХМ/H2O. Органические экстракты объединяли, промывали насыщенным NaCl, сушили MgSO4, фильтровали и фильтрат выпаривали при пониженном давлении, а затем сушили в вакууме с получением желтого маслянистого остатка. (21 мг, 49%); Rt = 9,86 мин (0_60_20 мин, чистота 100%); 1Н-ЯМР (CD3OD): δ 1,69-1,75 (м, 6H), 1,82 (шир.с, 2H), 1,96-1,99 (м, 2H), 2,03-2,05 (шир.с, 2H), 2,18-2,23 (м, 2H), 2,34 (с, 3H, CH3), 2,68 (дд, J=4,5 Гц, 2H, CH2), 2,95-2,97 (м, 2H), 3,28 (с, 3H, CH3), 3,73 (дд, J=4,5 Гц, 2H, CH2), 3,89-3,94 (м, 1H, CH), 4,01 (с, 3Η, CH3), 4,91-4,96 (м, 1H, CH), 7,49-7,51 (м, 2Η), 8,02 (шир.с, 1H), 8,50 (д, J = 8,5 Гц, 1H, phe-H); МС(+ve): 508,2.
Промежуточные соединения сложных аминоэфиров
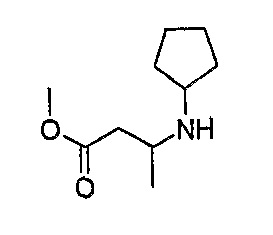
Нижеследующие промежуточные соединения получали методом, описанным в примере 1, стадии 2:
Метиловый эфир 3-циклопентиламиномасляной кислоты
МС(+ve): 186,3
Метиловый эфир 3-циклогексиламиномасляной кислоты
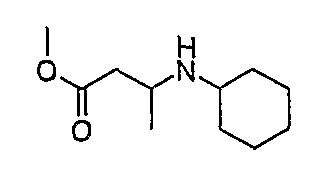
МС(+ve): 200,3
Метиловый эфир 3-циклопентиламино-2-метилпропионовой кислоты
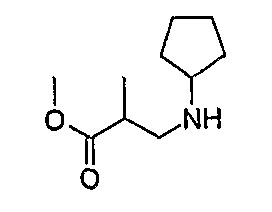
1H-ЯМР (CDCl3): 1,18 (3H, д, J=7 Гц, CH3), 1,33 (2H, м, CH2), 1,53 (2H, м, CH2), 1,67 (2H, м, CH2), 1,82 (2H, м, CH2), 2,63 (2H, м, CH2), 2,87 (1H, м, CH), 3,06 (1H, м, CH), 3,69 (3H, с, OCH3).
Метиловый эфир 3-циклогексиламинопропионовой кислоты
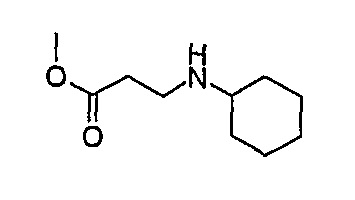
МС(+ve): 186,4.
Метиловый эфир 3-циклопентиламино-4-метилпентановой кислоты
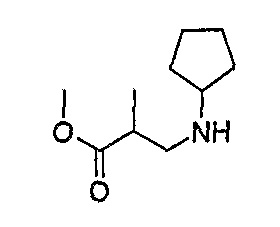
1H-ЯМР (CDCl3): 1,18 (3H, д, J=7 Гц, CH3), 1,33 (2H, м, CH2), 1,53 (2H, м, CH2), 1,67 (2H, м, CH2), 1,82 (2H, м, CH2), 2,63 (2H, м, CH2), 2,87 (1H, м, CH), 3,06 (1H, м, CH), 3,69 (3H, с, OCH3).
Метиловый эфир 3-(1-этилпропиламино)пропионовой кислоты
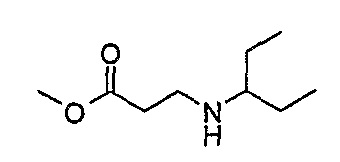
1H-ЯМР (CDCl3): 0,89 (6H, т, J=8 Гц, CH3), 1,44 (4H, м, CH2CH3), 1,77 (1H, шир.с, NH), 2,39 (1H, м, CH), 2,53 (2H, м, CH2), 2,89 (2H, м, CH2), 3,69 (3H, с, OCH3)
Метиловый эфир 3-(тетрагидропиран-4-иламино)пропионовой кислоты
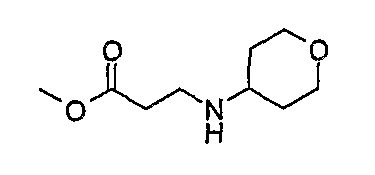
1Н-ЯМР (CDCl3): 1,41 (2H, м, CH2), 1,86 (2H, м, CH2), 2,54 (2H, м, CH2), 2,71 (1H, м, CH), 2,95 (2H, м, CH2), 3,44 (2H, т, J=11,5 Гц, CH2), 3,71 (3H, с, OCH3), 3,99 (2H, д, J=11 Гц, CH2)
Метиловый эфир 1-циклопентиламинометилциклопропанкарбоновой кислоты
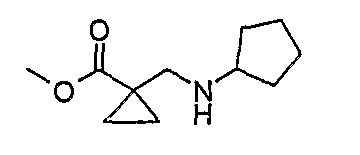
1Н-ЯМР (CDCl3): 0,84 (3H, т, J=7 Гц, CH3), 1,25 (4H, м, CH2), 1,37 (2H, м, CH), 1,54 (2H, м, CH), 1,70 (2H, м, CH), 1,83 (2H, м, CH), 2,71 (2H, с, CH2), 3,10 (1H, м, CH), 4,16 (2H, кв., J=7 Гц, CH2CH3).
Этиловый эфир 3-циклопентиламино-2,2-диметилпропионовой кислоты
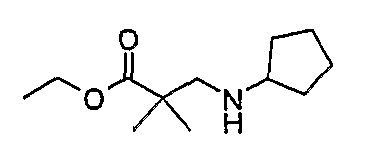
1H-ЯМР (CDCl3): 1,19 (6H, с, CH3), 1,25 (3H, т, J=7 Гц, CH3), 1,28 (2H, м, CH), 1,49 (2H, м, CH), 1,65 (2H, м, CH), 2,64 (1H, с, CH2), 3,01 (1H, м, CH), 4,10 (2H, кв., J=7,5 Гц, CH2).
Альтернативный метод. Стадия 2а: метил-3-аминопропаноат
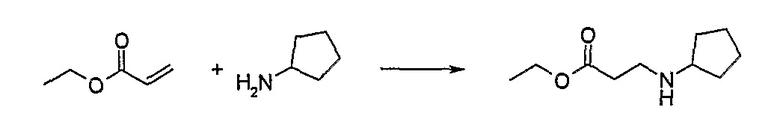
Метилакрилат (4,50 мл, 4,302 г, 49,97 ммоль, 0,99 экв.) по каплям добавляли к раствору циклопентиламина (5,00 мл, 4,315 г, 50,68 ммоль) в метаноле (120 мл) при -60°C (сухой лед, ацетон). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение ночи. Растворитель удаляли (при пониженном давлении, в вакууме) с получением продукта в виде масла (8,236 г, выход неочищенного продукта 96%). Протонный ЯМР показал, что продукт содержал ~25% диалкилированного побочного продукта.
Аналогичным методом получали нижеследующие соединения.
Этиловый эфир 3-фениламинопропионовой кислоты

1Н-ЯМР (CDCl3): 1,29 (3H, т, J=7,5 Гц, CH3), 2,64 (2H, т, J=6,5 Гц, CH2), 3,48 (2H, т, J=6,5 Гц, CH2), 4,18 (2H, кв., J=7 Гц, CH2), 6,65-6,74 (5H, м, Ar-H).
Промежуточные соединения нитропиримидина
Нижеследующие промежуточные соединения также получали методом, описанным в примере 1, стадии 3:
Метиловый эфир 3-[(2-хлор-5-нитропиримидин-4-ил)циклопентиламино]масляной кислоты
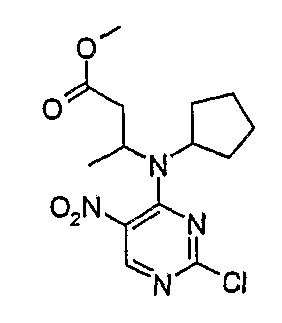
Rt = 18,0 мин (0_60_20 мин); МС(+ve): 343,20,
Метиловый эфир 3-[(2-хлор-5-нитропиримидин-4-ил)циклогексиламино]масляной кислоты
МС(+ve): 357,2.
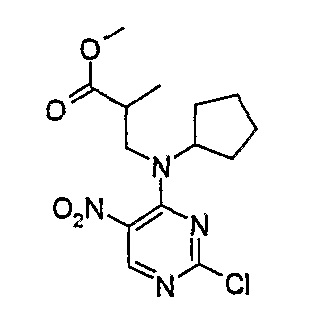
Метиловый эфир 3-[(2-хлор-5-нитропиримидин-4-ил)циклопентиламино]-2-метилпропионовой кислоты
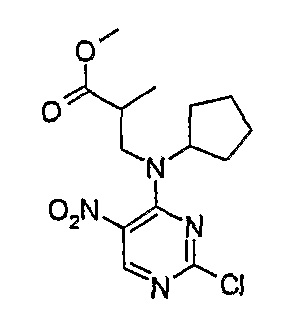
1Н-ЯМР (CDCl3): 1,22 (3H, д, J=12 Гц, CH3), 1,57 (2H, м, CH2), 1,76 (2H, м, CH2), 1,93 (2H, м, CH2), 1,98 (2H, м, CH2), 3,18 (1H, с, CH), 3,61 (3H, м, CH + CH2), 3,68 (3H, с, OCH3), 8,74 (1H, с, Pyr-H).
Метиловый эфир 3-[(2-хлор-5-нитропиримидин-4-ил)циклогексиламино]пропионовой кислоты
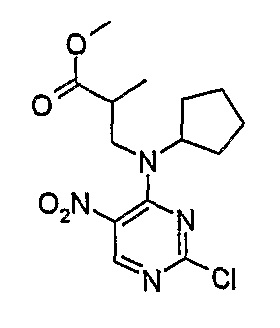
Rt = 17,6 мин (Vydac 1); МС(+ve): 343,1.
Метиловый эфир 3-[(2-хлор-5-нитропиримидин-4-ил)циклопентиламино]-4-метилпентановой кислоты
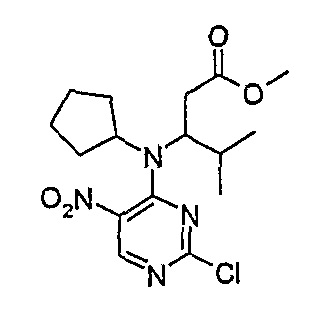
1Н-ЯМР (CDCl3): 0,94 (6H, т, J=7 Гц), 1,45-1,82 (7H, м, 3 × CH2 + CH), 2,17 (1H, м, CH), 2,69 (2H, м, 2 × CH), 3,35 (2H, м, CH2), 2,58 (1H, м, CH), 3,73 (3H, с, OCH3), 8,64 (1H, с, Pyr-H).
Метиловый эфир 3-[(2-хлор-5-нитропиримидин-4-ил)-(1-этилпропил)амино]пропионовой кислоты

МС(+ve) 331,1, 333,1.
Метиловый эфир 3-[(2-хлор-5-нитропиримидин-4-ил)-(тетрагидропиран-4-ил)амино]пропионовой кислоты
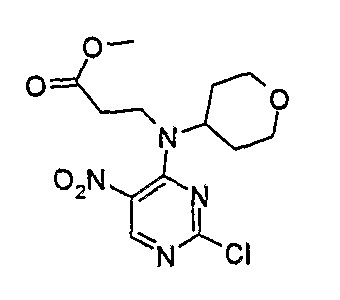
МС(+ve) 345,1, 347,0; Rt = 12,62 мин (Vydac 1).
Этиловый эфир 3-[бензил-(2-хлор-5-нитропиримидин-4-ил)амино]пропионовой кислоты
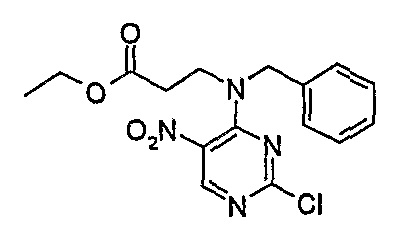
1Н-ЯМР (CDCl3): 1,26 (3H, т, J=7 Гц, CH3), 2,73 (2H, т, J=7 Гц, CH2), 3,85 (2H, м, CH2), 4,16 (2H, кв., J=7 Гц, CH2), 4,71 (2H, с, CH2), 7,20 (2H, м, Ar-H), 7,37 (3H, м, Ar-H), 8,69 (1H, с, Pyr-H).
Этиловый эфир 3-[(2-хлор-5-нитропиримидин-4-ил)фениламино]пропионовой кислоты
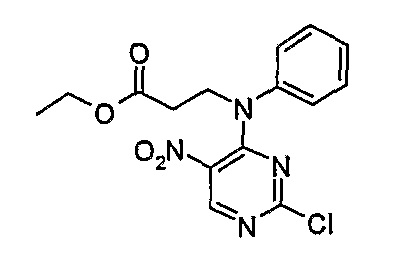
1H-ЯМР (CDCl3): 1,22 (3H, т, J=7,5 Гц, CH3), 2,73 (2H, т, J=7 Гц, CH2), 4,09 (2H, кв., J=7 Гц, CH2), 4,43 (2H, т, J=7 Гц, CH2), 7,14-7,48 (5H, м, Ar-H), 8,57 (1H, с, Pyr-H).
Этиловый эфир 1-{[(2-хлор-5-нитропиримидин-4-ил)циклопентиламино]метил}циклопропанкарбоновой кислоты
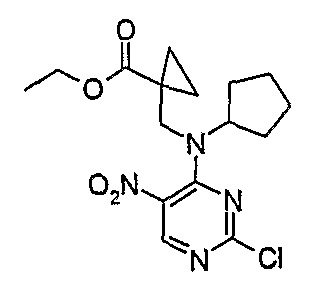
МС(+ve) 381,1, 383,3; Rt = 4,80 мин (XBridge 1).
Этиловый эфир 3-[(2-хлор-5-нитропиримидин-4-ил)циклопентиламино]-2,2-диметилпропионовой кислоты
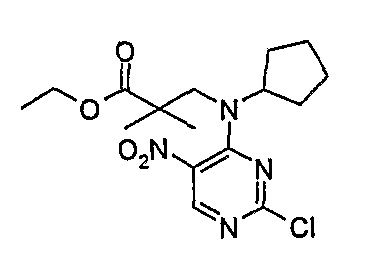
МС(+ve) 371,1, 373,1; Rt = 4,67 мин (XBridge 1).
Сложные эфиры аминопиримидина, образующие хлорпиримидиндиазепины
Нижеследующие соединения выделяли путем реакции восстановления, описанной в примере 1, стадии 4, в виде нециклизованных промежуточных соединений.
Этиловый эфир 3-[(5-амино-2-хлорпиримидин-4-ил)бензиламино]пропионовой кислоты
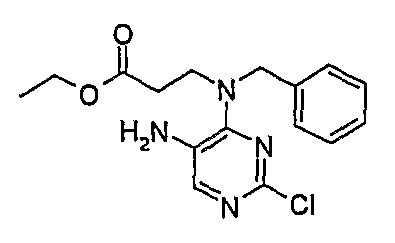
1Н-ЯМР (CDCl3): 1,26 (3H, т, J=7,5 Гц, CH3), 2,73 (2H, т, J=7 Гц, CH2), 3,86 (2H, т, J=7 Гц, CH2), 4,14 (2H, кв., J=7 Гц, CH2), 4,86 (2H, с, CH2), 7,26-7,39 (5H, м, Ar-H), 7,94 (1H, с, Pyr-H).
Этиловый эфир 3-[(5-амино-2-хлорпиримидин-4-ил)фениламино]пропионовой кислоты
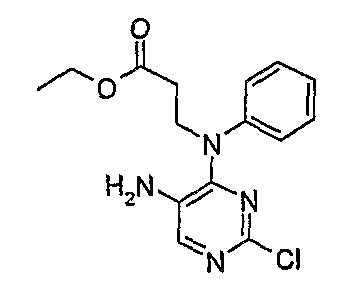
МС(+ve) 321,2, 323,2; Rt = 3,54 мин (XBridge 1).
Этиловый эфир 1-{[(5-амино-2-хлорпиримидин-4-ил)циклопентиламино]метил}циклопропанкарбоновой кислоты
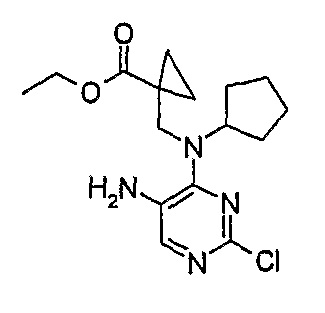
МС(+ve) 339,2, 341,2; Rt = 4,15 мин (XBridge 1).
Этиловый эфир 3-[(5-амино-2-хлорпиримидин-4-ил)циклопентиламино]-2,2-диметилпропионовой кислоты
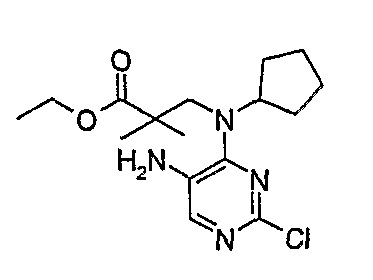
МС(+ve) 341,2, 343,2; Rt= 4,25 мин (XBridge 1).
Затем вышеуказанные соединения подвергали реакции циклизации нижеследующим методом.
Соответствующее соединение (5 ммоль) в ДМФ (10 мл) нагревали до 140°C в течение 2 часов. Растворитель выпаривали в вакууме, добавляли этилацетат (10 мл), и полученное твердое вещество фильтровали и сушили в вакууме с получением соединений, перечисленных ниже.
Примечание: Такая реакция может быть также проведена с использованием ДМСО/NaH.
9-бензил-2-хлор-5,7,8,9-тетрагидропиримидо[4,5-b][1,4]диазепин-6-он
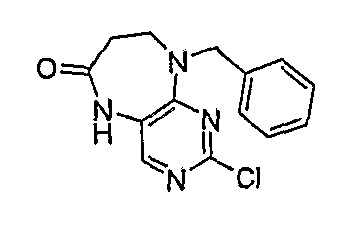
1Н-ЯМР (ДМСО): 2,72 (2H, м, CH2), 3,67 (2H, м, CH2), 4,86 (2H, с, CH2), 7,26-7,37 (5H, м, Ar-H), 7,88 (1H, с, Pyr-H), 9,77 (1H, с, NH).
2-хлор-9-фенил-5,7,8,9-тетрагидропиримидо[4,5-b][1,4]диазепин-6-он
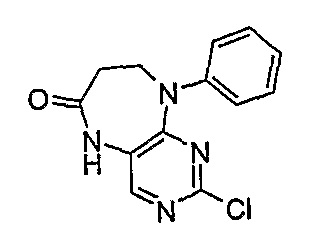
МС(+ve) 275,2, 277,2; Rt = 2,80 мин (XBridge 1).
2-хлор-9-циклопентил-5,7,8,9-тетрагидропиримидо[4,5-b][1,4]диазепин-7,1’-циклопропан]-6-он
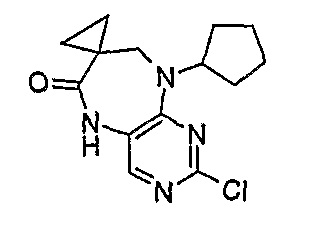
МС(+ve) 293,2, 295,2; Rt = 3,48 мин (XBridge 1).
2-хлор-9-циклопентил-7,7-диметил-5,7,8,9-тетрагидропиримидо[4,5-b][1,4]диазепин-6-он
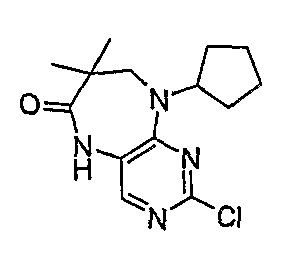
МС(+ve) 295,1, 297,2; Rt = 3,58 мин (XBridge 1).
Алкилированные диазепиноны
Нижеследующие промежуточные соединения были также получены методом, описанным в примере 1, стадии 5:
2-хлор-9-циклопентил-5,8-диметил-5,7,8,9-тетрагидропиримидо[4,5-b][1,4]диазепин-6-он
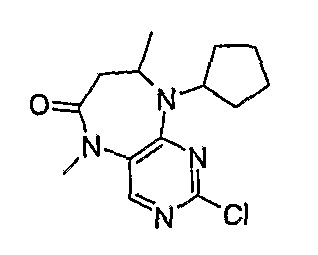
Rt = 14,6 мин (Vydac 1); МС(+ve): 295,2.
2-хлор-9-циклопентил-5,7-диметил-5,7,8,9-тетрагидропиримидо[4,5-b][1,4]диазепин-6-он
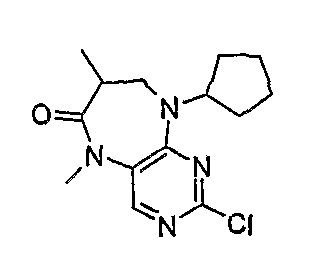
МС(+ve) 295,1, 297,2; Rt = 9,11 мин (XBridge 2).
2-хлор-9-циклопентил-8-изопропил-5-метил-5,7,8,9-тетрагидропиримидо[4,5-b][1,4]диазепин-6-он
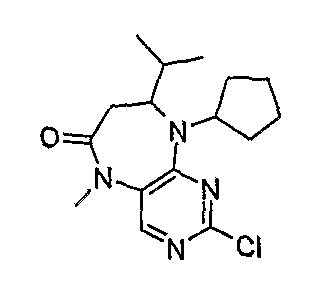
МС(+ve) 323,1, 325,1; Rt = 10,17 мин (XBridge 2).
2-хлор-9-циклопентил-5-этил-7-метил-5,7,8,9-тетрагидропиримидо[4,5-b][1,4]диазепин-6-он
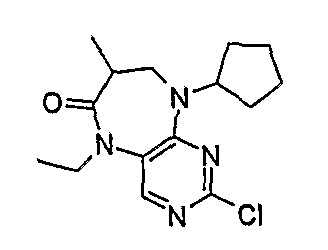
1H-ЯМР (CDCl3): 1,08 (6H, м, 2 × CH3), 1,38 (1H, м, CH), 1,55-1,70 (5H, м, CH + CH2), 1,70 (1H, м, CH), 2,10 (1H, м, CH), 2,73 (1H, м, CH), 3,48 (2H, м, CH2), 4,03 (1H, м, CH), 4,72 (1H, м, CH), 7,98 (1H, с, Pyr-H).
2-хлор-9-(1-этилпропил)-5-метил-5,7,8,9-тетрагидропиримидо[4,5-b][1,4]диазепин-6-он
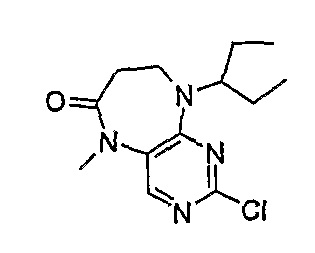
МС(+ve) 283,2, 285,1; Rt = 8,44 мин (XBridge 2).
2-хлор-9-(1-этилпропил)-5-этил-5,7,8,9-тетрагидропиримидо[4,5-b][1,4]диазепин-6-он
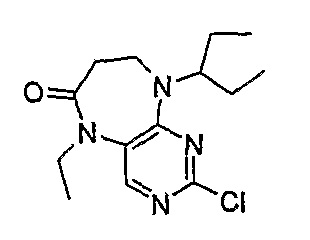
МС(+ve) 297,12, 299,14; Rt = 3,67 мин (XBridge 2).
2-хлор-5-метил-9-(тетрагидропиран-4-ил)-5,7,8,9-тетрагидропиримидо[4,5-b][1,4]диазепин-6-он

МС(+ve) 297,1, 299,2; Rt = 5,74 мин (XBridge 2).
9-бензил-2-хлор-5-метил-5,7,8,9-тетрагидропиримидо[4,5-b][1,4]диазепин-6-он
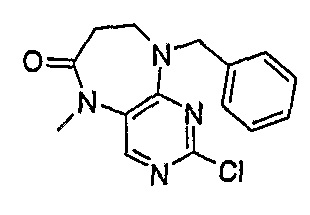
МС(+ve) 303,1, 305,2; Rt = 13,57 мин (Vydac 1).
2-хлор-5-метил-9-фенил-5,7,8,9-тетрагидропиримидо[4,5-b][1,4]диазепин-6-он
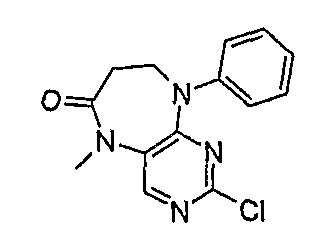
МС(+ve) 289,1, 291,1; Rt = 2,93 мин (XBridge 2).
2-хлор-9-циклопентил-5-метил-5,7,8,9-тетрагидропиримидо[4,5-][1,4]диазепин-7,1’-циклопропан]-6-он
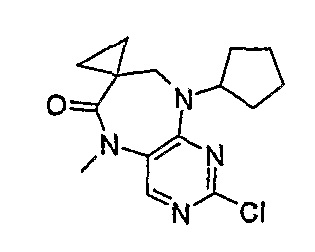
МС(+ve) 307,1, 309,2; Rt = 3,67 мин (XBridge 2).
2-хлор-9-циклопентил-5,7,7-триметил-5,7,8,9-тетрагидропиримидо[4,5-b]{1,4]диазепин-6-он
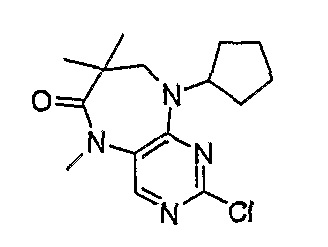
МС(+ve) 309,1, 311,2; Rt = 3,90 мин (XBridge 2).
Пиримидиндиазепиноны, полученные посредством реакции сочетания
Нижеследующие соединения были также получены методом сочетания А, описанным в примере 1, стадии 6:
Соединение [3]: 4-(9-циклопентил-8-метил-6-оксо-6,7,8,9-тетрагидро-5H-пиримидо[4,5-b][1,4]диазепин-2-иламино)-3-метоксибензойная кислота

Твердое вещество пурпурного цвета (29%): lH-ЯМР (ДМСО-d6): δ 1,33 (д, J=Гц, 3H, CH3), 1,52-1,58 (м, 3H, циклопент-H), 1,70-1,78 (м, 3Η, циклопент-H), 1,87-1,93 (м, 2Η, циклопент-H), 2,97-3,00 (м, 2Η, CH2), 3,94 (с, 3Η, CH3), 4,15-4,17 (м, 1Η, CH), 4,93-4,97 (м, 1H, CH), 7,59-7,60 (м, 2H), 7,82 (1, 1H), 8,01 (д, J=9 Гц, 1H, phe-H), 9,55 (шир.с, 1H, OH), 9,96 (с, 1H); МС(+ve): 426,2, Rt = 11,08 мин (чистота 100%, (Vydac 1)).
Соединение [4]: 4-(9-циклопентил-5,8-диметил-6-оксо-6,7,8,9-тетрагидро-5H-пиримидо[4,5-b][1,4]диазепин-2-иламино)-3-метоксибензойная кислота

Твердое вещество пурпурного цвета (67%). 1Н-ЯМР (ДМСО-d6): δ 1,26 (д, J=Гц, 3H, CH3), 1,43-1,93 (м, 8H, циклопент-H), 2,52 (с, 3H, CH3), 2,99-3,02 (м, 2H, CH2), 3,96 (с, 3H, CH3), 4,13-4,16 (м, 1H, CH), 4,65-4,69 (м, 1H, CH), 7,60-7,61 (м, 2H), 8,06 (д, J=9 Гц, 1H, phe-H), 8,17 (с, 1H), 9,35 (шир.с, 1H); МС(+ve): 426,2, Rt = 11,59 мин (чистота 96%, (Vydac 1)).
Соединение [5]: 4-(9-циклогексил-8-метил-6-оксо-6,7,8,9-тетрагидро-5H-пиримидо[4,5-b][1,4]диазепин-2-иламино)-3-метоксибензойная кислота
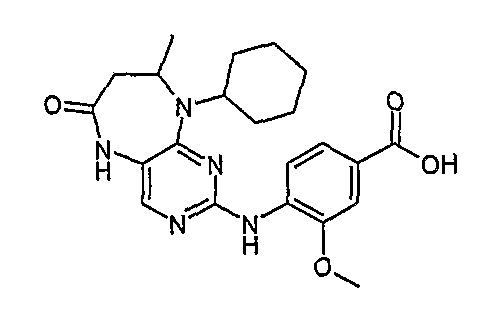
Твердое вещество коричневого цвета (12%); 1Н-ЯМР (ДМСО-d6): δ 1,27 (д, J=6,5 Гц, 3H, CH3), 1,58-1,87 (м, 10H, циклогекс-H), 2,85-2,88 (м, 2H, CH2), 3,94 (с, 3H, CH3), 4,21-4,23 (м, 1H, CH), 4,77 (шир.с, 1H, CH), 7,03 (с, 1H), 7,13 (с, 1H), 7,23 (с, 1H), 7,57-7,58 (м, 2H), 7,086 (с, 1H), 8,07-8,08 (м, 1H), 9,89 (шир.с, 1H, NH); МС(+ve): 426,2, Rt = 11,58 мин (чистота 90%, (Vydac 1)).
Соединение [6]: 4-(9-циклопентил-6-оксо-6,7,8,9-тетрагидро-5H-пиримидо[4,5-b][1,4]диазепин-2-иламино)-3-метоксибензойная кислота
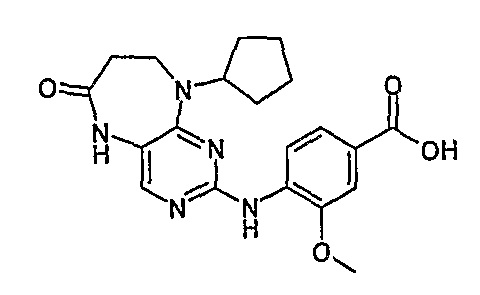
Твердое вещество пурпурного цвета (13%); 1Н-ЯМР (ДМСО-d6): δ 1,55-1,86 (м, 8H, циклопент-H), 2,70-2,73 (м, 2H, CH2), 3,70-3,72 (м, 2H, CH2), 5,02-5,09 (м, 1H, CH), 7,57-7,59 (м, 2H), 7,77 (с, 1H, пиримид-H), 8,06 (д, J=8,5 Гц, 1H, phe-H), 9,54 (шир.с, 1Η, OH), 9,75 (с, 1Η, NH); МС(+ve): 398,2, Rt = 10,55 мин (чистота 96%, (Vydac 1)).
Соединение [7]: 4-(9-циклопентил-8-метил-6-оксо-6,7,8,9-тетрагидро-5H-пиримидо[4,5-b][1,4]диазепин-2-иламино)бензойная кислота

Не совсем белое твердое вещество (10%); 1Н-ЯМР (ДМСО-d6): δ 1,27 (д, J=6,5 Гц, 3H, CH3), 1,46-1,90 (м, 8Η, циклопент-H), 2,83-2,86 (м, 2Η, CH2), 4,07-4,09 (м, 1Η, CH), 5,04-5,09 (м, 1Η, CH), 7,20 (дд, J=3,5 и 8,5 Гц, 2H, phe-H), 7,79 (д, J=2,0 Гц, 1H, пиримид-H), 7,86 (д, J=8,5 Гц, 2H, phe-H), 9,78 (шир.с, 1H, NH); МС(+ve): 382,2, Rt = 10,46 мин (чистота 90%, (Vydac 1)).
Соединение [8]: 4-(9-Циклопентил-5,8-диметил-6-оксо-6,7,8,9-тетрагидро-5H-пиримидо[4,5-b][1,4]диазепин-2-иламино)бензойная кислота
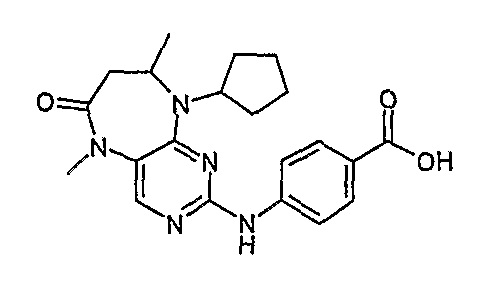
Не совсем белое твердое вещество (25%); 1Н-ЯМР (ДМСО-d6): δ 1,19 (д, J=6,0 Гц, 3H, CH3), 1,32-2,10 (м, 8Η, циклопент-H), 2,73-2,76 (м, 2Η, CH2), 3,22 (с, 3Η, CH3), 4,04 (м, 1Η, CH), 4,70-4,75 (м, 1Η, CH), 7,78-7,85 (м, 4Η, phe-H), 8,12 (с, 1Η, пиримид-H), 9,68 (шир.с, 1Η, NH); МС(+ve): 394,0, Rt = 11,05 мин (чистота 96%, (Vydac 1)).
Соединение [9]: 4-(9-циклогексил-6-оксо-6,7,8,9-тетрагидро-5H-пиримидо[4,5-b][1,4]диазепин-2-иламино)-3-метоксибензойная кислота
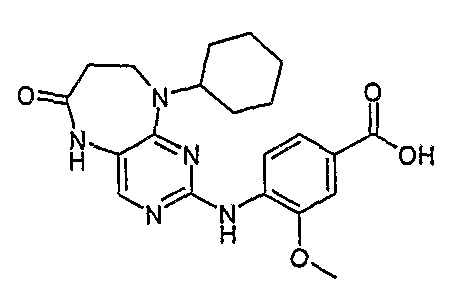
Твердое вещество коричневого цвета (15%); 1Н-ЯМР (ДМСО-d6): δ 1,15-1,86 (м, 10H, циклогекс-H), 2,72 (дд, J=4,5 Гц, 2H, CH2), 3,75 (дд, J=4,5 Гц, 2H, CH2), 3,96 (с, 3H, CH3), 4,58-4,63 (м, 1H, CH), 7,58-7,60 (м, 2Η), 7,79 (с, 1H, пиримид-H), 8,16 (д, J=8,5 Гц, 1H, phe-H), 9,74 (шир.с, 1Η, NH); МС(+ve): 412,2, Rt = 11,19 мин (чистота 100%, (Vydac 1)).
Соединение [10]: 9-циклогексил-2-(4-гидроксифениламино)-5,7,8,9-тетрагидропиримидо[4,5-b][1,4]диазепин-6-он
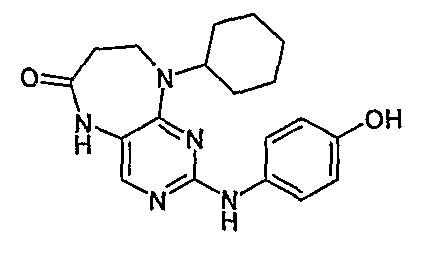
Твердое вещество пурпурного цвета (7%); 1Н-ЯМР (ДМСО-d6): δ 1,17-1,87 (м, 10H, циклогекс-H), 2,72 (шир.с, 2H, CH2), 3,72 (шир.с, 2H, CH2), 4,62-4,66 (м, 1Η, CH), 6,82 (д, J=8,5 Гц, 2H, phe-H), 7,13 (с, 1H), 7,23 (с, 1H), 7,34-7,37 (м, 3H, 2 × phe-H и NH); МС(+ve): 354,2, Rt = 10,63 мин (чистота 100%, (Vydac 1)).
Соединение [11]: 9-циклопентил-2-(4-гидроксифениламино)-7-метил-5,7,8,9-тетрагидропиримидо[4,5-b][1,4]диазепин-6-он

1Н-ЯМР (CD3OD): 1,18 (3H, д, J=7 Гц, CH3), 1,52 (1H, м, CH), 1,61-1,84 (6H, м, CH), 1,99 (1H, м, CH), 2,85 (1H, м, CH), 3,53 (2H, м, CH), 5,14 (1H3 м, CH), 6,74 (2H, м, Ar-H), 7,34 (2H, м, Ar-H), 7,64 (1H, с, Pyr-H) ; МС(+ve): 354,2; Rt = 10,85 мин (Vydac 1).
Соединение [12]: 9-циклопентил-2-(4-гидроксифениламино)-8-изопропил-5-метил-5,7,8,9-тетрагидропиримидо[4,5-b][1,4]диазепин-6-он
1Н-ЯМР (ДМСО): 0,72 (3H, д, J=7 Гц, CHCH3), 0,85 (2H, д, J=7 Гц, CHCH3), 1,29 (1H, м, CH), 1,51 (4H, м, CH), 1,60 (2H, м, CH), 1,93 (1H, м, CH), 2,08 (1H, м, CH), 2,29 (2H, м, 2 × CH), 3,13 (3H, с, N-CH3), 3,82 (1H, м, CH), 4,15 (1H, м, CH), 6,65 (2H, д, J=6 Гц, Ar-H), 7,47 (2H, д, J=6 Гц, Ar-H), 8,14 (1H, с, Pyr-H), 8,99 (1H, с, NH); МС(+ve): 396,1; Rt = 12,54 мин (Vydac 1).
Соединение [13]: 9-Циклопентил-2-(4-гидроксифениламино)-5,8-диметил-5,7,8,9-тетрагидропиримидо[4,5-b][1,4]диазепин-6-он
Белое твердое вещество (27%); 1Н-ЯМР (ДМСО-d6): δ 1,22 (д, J=6,5 Гц, 3H, CH3), 1,38-1,85 (м, 8H, циклопент-H), 2,49-2,53 (м, 2H, CH2), 3,18 (с, 3H, CH3), 4,05-4,10 (м, 1H, CH), 4,51-4,60 (м, 1H, CH), 6,80 (д, J=9,0 Гц, 2H, phe-H), 7,22 (д, J=9,0 Гц, 2H, phe-H), 7,98 (с, 1H, пиримид-H), 9,53 (шир.с, 1H, OH), 10,05 (шир.с, 1H, NH); МС(+ve): 368,3, Rt = 10,81 мин (чистота 93%, (Vydac 1)).
Соединение [14]: 4-(9-Бензил-5-метил-6-оксо-6,7,8,9-тетрагидро-5H-пиримидо[4,5-b][1,4]диазепин-2-иламино)-3-метоксибензойная кислота
1Н-ЯМР (ДМСО): 2,72 (2H, м, CH2), 3,68 (2H, м, CH2), 3,90 (3H, с, OCH3), 4,88 (2H, с, CH2), 7,22-7,44 (6H, м, Ar-H), 7,44 (1H, с, Ar-H), 7,77 (1H, с, Pyr-H), 8,10 (1H, д, J=7 Гц), 8,18 (1H, с, NH); МС(+ve): 434,2; Rt= 10,97 мин (Vydac 1).
Соединение [15}: 4-(5-метил-6-оксо-9-фенил-6,7,8,9-тетрагидро-5H-пиримидо[4,5-b][1,4]диазепин-2-иламино)бензойная кислота
1Н-ЯМР (ДМСО): 2,84 (2H, м, CH2), 3,28 (3H, с, CH3), 4,08 (2H, м, CH2), 7,24 (2H, д, J=8,5 Гц, Ar-H), 7,35-7,37 (3H, м, Ar-H), 7,47-7,52 (4H, м, Ar-H), 8,29 (1H, с, Pyr-H), 9,77 (1H, с, NH); МС(+ve): 390,2; Rt = 1,93 мин (XBridge 2).
Соединение [16]: 4-(9-циклопентил-5,7,7-триметил-6-оксо-6,7,8,9-тетрагидро-5H-пиримидо[4,5-b][1,4]диазепин-2-иламино)бензойная кислота

1H-ЯМР (ДМСО): 1,09 (6H, с, CH3), 1,60 (4H, м, CH), 1,74 (2H, м, CH), 1,88 (2H, м, CH), 3,19 (3H, с, N-CH3), 3,37 (2H, с, CH2), 5,23 (1H, с, CH), 7,82 (4H, м, Ar-H), 7,99 (1H, с, Pyr-H), 9,57 (1H, с, NH); МС(+ve): 410,25; Rt = 2,25 мин (XBridge 2).
Получение и реакция взаимодействия аминобензамидов
4-амино-3-метокси-N-(1-метилпиперидин-4-ил)бензамид
К 4-амино-3-метоксибензойной кислоте (1,064 г мг, 6,37 ммоль) в дихлорметане (50 мл) добавляли DIPEA (2,22 мкл, 12,74 ммоль) и TBTU (2,25 г, 7,0 ммоль), и смесь перемешивали в течение 10 минут. Затем добавляли 4-амино-1-метилпиперидин (0,872 г, 7,65 ммоль), и перемешивание продолжали в течение 16 часов. Полученное твердое вещество фильтровали и сушили в вакууме (1,04 г, 3,95 ммоль, 62%).
1H-ЯМР (ДМСО): 1,54 (2H, м, CH), 1,68 (2H, м, CH), 1,90 (2H, м, CH), 2,15 (3H, с, N-CH3), 2,76 (2H, д, J=11,5 Гц, CH), 3,66 (1H, м, CH), 3,77 (3H, с, OCH3), 6,56 (1H, м, Ar-H), 7,26 (1H, м, Ar-H), 7,75 (1H, д, J=7,5 Гц); МС(+ve): 264,4; Rt = 7,45 мин (Vydac 2).
Аналогичным методом получали нижеследующие аминобензамиды:
4-амино-N-(1-метилпиперидин-4-ил)бензамид, полученный посредством трет-бутилового эфира [4-(1-метилпиперидин-4-илкарбамоил)фенил]карбаминовой кислоты
Трет-бутиловый эфир [4-(1-метилпиперидин-4-илкарбамоил)фенил]карбаминовой кислоты: 1H-ЯМР (ДМСО): 1,44 (9H, с, C(CH3)3), 1,52 (2H, м, CH), 1,71 (2H, м, CH), 1,92 (2H, м, CH), 1,92 (3H, с, N-CH3), 2,74 (2H, м, CH), 3,68 (1H, м, N-CH), 7,47 (2H, д, J=8,5 Гц, Ar-H), 7,72 (2H, д, J=8,5 Гц, Ar-H), 8,00 (1H, д, J=7,5 Гц, NH), 9,54 (1H, с, NH). При проведении метода сочетания В данное соединение использовали без предварительного проведения реакции снятия защиты (см. ниже).
Метод сочетания B
Соединение [17]: 4-(9-циклопентил-5,7-диметил-6-оксо-6,7,8,9-тетрагидро-5H-пиримидо[4,5-b][1,4]диазепин-2-иламино)-3-метокси-N-(1-метилпиперидин-4-ил)бензамид
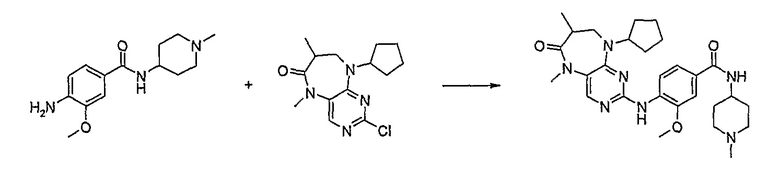
Анилин (149 мг, 0,57 ммоль), пиримидин (53 мг, 0,19 ммоль) и TFA (109 мкл, 0,95 ммоль) в 2,2,2-трифторэтаноле (5 мл) кипятили с обратным холодильником в течение 18 часов. Растворитель выпаривали, а затем добавляли этилацетат (10 мл), промывали насыщенным NaHCO3 и раствором соли, после чего растворитель выпаривали. Затем добавляли этилацетат (10 мл), и полученное твердое вещество фильтровали и сушили в вакууме (30 мг, 0,06 ммоль, 30%).
1H-ЯМР (ДМСО): 1,06 (3H, д, J=7 Гц, CHCH3), 1,52-1,78 (10H, м, алкил CH + CH2), 2,01 (2H, м, CH), 2,36 (3H, с, N-CH3), 2,73 (1H, м, CH), 2,85 (2H, м, CH), 3,45 (1H, м, CH), 3,71 (1H, м, CH), 3,94 (3H, с, OCH3), 5,06 (1H, м, CH), 7,46 (2H, м, Ar_h), 7,61 (1H, с, Ar-H), 7,77 (1H, с, NH), 8,10 (1H, д, J=8 Гц, Ar-H), 8,36 (1H, д, J=8 Гц), Ar-H), 9,44 (1H, с, NH); МС(+ve): 508,2; Rt = 7,30 мин (XBridge 2).
Нижеследующие соединения получали способом, аналогичным способу, описанному в вышеуказанном примере:
Соединение [18]: 4-(9-циклопентил-7-метил-6-оксо-6,7,8,9-тетрагидро-5H-пиримидо[4,5-b][1,4]диазепин-2-иламино)-N-(1-метилпиперидин-4-ил)бензамид
1H-ЯМР (ДМСО): 1,08 (3H, д, J=7 Гц, CHCH3), 1,53-1,80 (10H, м, алкил CH + CH2), 2,01 (2H, м, CH), 2,12 (2H, м, CH), 2,29 (3H, с, N-CH3), 2,75 (1H, м, CH), 2,90 (2H, м, CH), 3,48 (1H, м, CH), 3,77 (1H, м, CH), 5,11 (1H, м, CH), 7,78 (4H, м, Ar-H), 8,04 (1H, д, J=7,5 Гц, NH), 9,35 (1H, с, CONH), 9,43 (1H, с, CONH); МС(+ve): 478,2; Rt = 6,74 мин (XBridge 2).
Соединение [19]: 4-(9-циклопентил-8-изопропил-6-оксо-6,7,8,9-тетрагидро-5H-пиримидо[4,5-b][1,4]диазепин-2-иламино)-3-метокси-N-(1-метилпиперидин-4-ил)бензамид

1H-ЯМР (CD3OD): 0,92 (3H, д, J=6,5 Гц, CHCH3), 1,04 (3H, д, J=6 Гц, CHCH3), 1,65-1,97 (14H, м, алкил CH + CH2), 2,25 (2H, дд, J=11,5 Гц, 11,5 Гц, CH), 2,36 (3H, с, N-CH3), 2,67 (1H, м, CH), 3,03 (3H, м, CH + CH2), 3,51 (1H, м, CH), 3,95 (1H, м, CH), 4,02 (3H, с, OCH3), 5,09 (1H, м, CH), 7,59 (2H, м, Ar-H), 7,87 (1H, с, pyr-H-H), 8,46 (1H, д, J=7 Гц, Ar-H); МС(+ve): 536,3; Rt = 7,68 мин (XBridge 2).
Соединение [20]: 4-(9-циклопентил-5,7-диметил-6-оксо-6,7,8,9-тетрагидро-5H-пиримидо[4,5-b][1,4]диазепин-2-иламино)-3-метокси-N-(1-метилпиперидин-4-ил)бензамид
1H-ЯМР (ДМСО): 1,02 (3H, д, J=6,5 Гц, CHCH3), 1,54-1,80 (12H, м, алкил CH + CH2), 2,10 (3H, м, (CH + CH2), 2,27 (3H, с, N-CH3), 2,84 (3H, м, CH3), 3,15 (3H, с, CH3), 3,46 (1H, м, CH), 3,80 (1H, м, N-CH), 3,95 (3H, с, OCH3), 4,76 (1H, м, CH), 7,50 (2H, м, Ar-H), 7,75 (1H, с, Ar-H), 8,10 (1H, с, NH), 8,14 (1H, д, J=7,5 Гц, Ar-H), 8,40 (1H, д, J=8 Гц, Ar-H); МС(+ve): 522,3; Rt = 7,76 мин (XBridge 2).
Соединение [21]: 4-[9-(1-этилпропил)-6-оксо-6,7,8,9-тетрагидро-5H-пиримидо[4,5-b][1,4]диазепин-2-иламино]-3-метокси-N-(1-метилпиперидин-4-ил)бензамид
1H-ЯМР (ДМСО): 0,85 (6H, д, J=7 Гц, CH2CH3), 1,59 (4H, м, CH2CH3), 1,66 (2H, м, CH), 1,83 (2H, м, CH), 2,37 (3H, с, N-CH3), 2,65 (2H, м, CH2), 2,98 (2H, м, CH), 3,50 (2H, м, CH2), 3,83 (1H, м, N-CH), 3,94 (3H, с, OCH3), 4,93 (1H, с, CH), 4,;49 (2H, м, Ar-H + NH), 7,57 (1H, с, NH), 7,79 (1H, с, Ar-H), 8,15 (1H, д, J=6,5 Гц, Ar-H), 8,37 (1H, д, J=8,5 Гц, Ar-H), 9,41 (1H, с, NH); МС(+ve): 496,2; Rt = 6,73 мин (XBridge 2).
Соединение [22]: 4-[9-(1-этилпропил)-6-оксо-6,7,8,9-тетрагидро-5H-пиримидо[4,5-b][1,4]диазепин-2-иламино]-N-(1-метилпиперидин-4-ил)бензамид
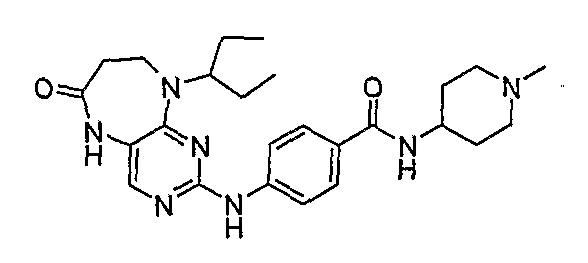
1H-ЯМР (ДМСО): 0,86 (6H, д, J=7 Гц, CH2CH3), 1,56 (4H, м, CH2CH3), 1,62 (2H, м, CH), 1,76 (2H, м, CH), 2,52 (3H, с, N-CH3), 2,63 (2H, м, CH2), 3,22 (2H, м, CH), 3,49 (2H, м, CH2), 3,92 (1H, м, CH), 4,97 (1H, м, CH), 7,78 (4H, м, Ar-H), 8,15 (1H, с, Pyr-H), 9,31 (1H, с, NH), 9,39 (1H, с, NH); МС(+ve): 466,3; Rt = 6,26 мин (XBridge 2).
Соединение [23]: 4-(9-циклопентил-8-изопропил-5-метил-6-оксо-6,7,8,9-тетрагидро-5H-пиримидо[4,5-b][1,4]диазепин-2-иламино)-3-метокси-N-(1-метилпиперидин-4-ил)бензамид
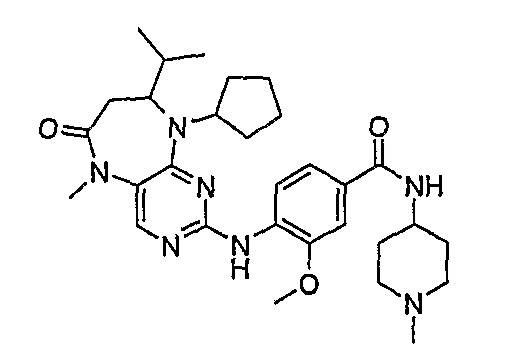
1Н-ЯМР (CD3OD): 0,83 (3H, д, J=7 Гц, CHCH3), 0,93 (3H, д, J=7 Гц, CHCH3), 1,41 (2H, м, CH), 1,65 (4H, м, CH), 1,79 (2H, м, CH), 1,91 (2H, м, CH), 2,17 (2H, м, CH), 2,41 (2H, м, 2 × CH), 2,71 (4H, м, CH3 + CH), 2,91 (2H, м, CH), 3,35 (3H, с, CH3), 3,90 (1H, м, CH), 3,94 (3H, с, OCH3), 4,21 (1H, м, CH), 4,27 (1H, м, CH), 7,18 (2H, м, Ar-H), 8,23 (1H, с, Pyr-H), 8,56 (1H, д, J=8,5 Гц, Ar-H); МС(+ve): 550,3; Rt = 8,40 мин (XBridge 2).
Соединение [24]: 4-[9-(1-этилпропил)-5-метил-6-оксо-6,7,8,9-тетрагидро-5Н-пиримидо[4,5-b][1,4]диазепин-2-иламино]-3-метокси-N-(1-метилпиперидин-4-ил)бензамид
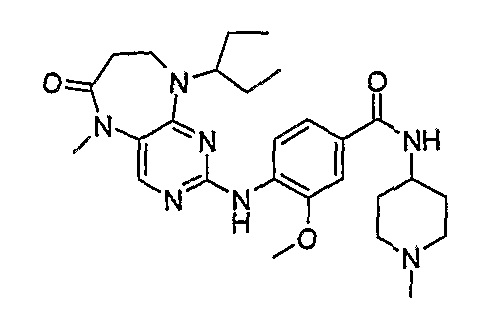
1Н-ЯМР (ДМСО): 0,85 (6H, т, J=7,5 Гц, CH2CH3), 1,62 (8H, м, CH + CH2), 1,76 (2H, д, J=7 Гц, CH), 1,94 (2H, дд, J=10 Гц, 10 Гц, CH), 2,17 (3H, с, N-CH3), 2,64 (2H, м, CH2), 2,80 (2H, д, J=11 Гц, CH), 3,18 (3H, с, N-CH3), 3,49 (2H, м, CH2), 3,75 (1H, м, CH), 3,95 (3H, с, OCH3), 4,65 (1H, м, CH), 7,49 (2H, м, Ar-H), 7,69 (1H, с, Pyr-H), 8,07 (1H, с, NH), 8,10 (1H, д, J=9 Гц, Ar-H), 8,37 (1H, д, J=9 Гц, Ar-H); МС(+ve): 510,3; Rt = 8,35 мин (XBridge 2).
Соединение [25]: 3-метокси-N-(1-метилпиперидин-4-ил)-4-[6-оксо-9-(тетрагидропиран-4-ил)-6,7,8,9-тетрагидро-5H-пиримидо[4,5-b][1,4]диазепин-2-иламино]бензамид
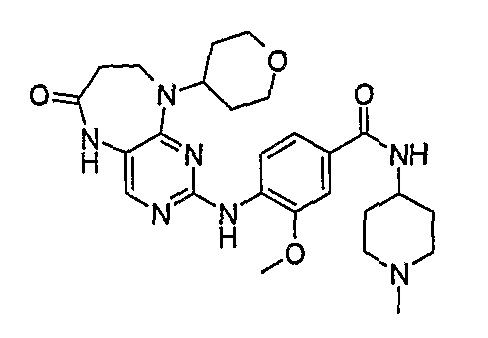
1H-ЯМР (CD3OD): 1,60 (2H, м, CH), 1,69 (2H, д, J=11 Гц, CH), 1,93 (2H, д, J=12 Гц, CH), 1,97 (2H, дд, J=12 Гц, 12 Гц, CH), 2,18 (3H, с, N-CH3), 2,61 (2H, м, CH2), 2,80 (2H, д, J=10,5 Гц, CH), 3,48 (2H, дд, J=11 Гц, CH), 3,62 (2H, м, CH2), 3,75 (1H, м, CH), 3,95 (3H, с, OCH3), 4,04 (2H, д, J=8 Гц, CH), 4,85 (1H, с, CH), 7,49 (2H, м, Ar-H), 7,66 (1H, с, NH), 7,81 (1H, с, Pyr-H), 8,11 (1H, д, J=7,5 Гц, Ar-H), 8,33 (1H, д, J=8 Гц, Ar-H), 9,45 (1H, с, NH); МС(+ve): 510,1; Rt = 5,34 мин (XBridge 2).
Соединение [26]: 3-Метокси-4-[5~метил-6-оксо-9-(тетрагидропиран-4-ил)-6,7,8,9-тетрагидро-5H-пиримидо[4,5-b][1,4]диазепин-2-иламино]-N-(1-метилпиперидин-4-ил)бензамид
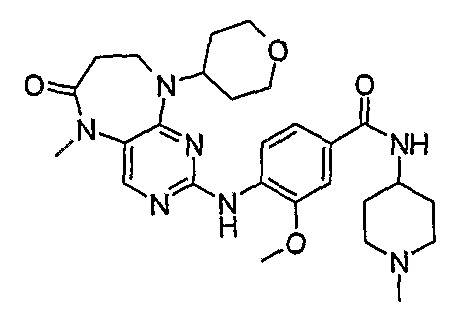
1Н-ЯМР (CD3OD): 1,73 (2H, ддд, J=3 Гц, 12 Гц, 18 Гц, CH), 1,84 (2H, д, J=11 Гц, CH), 1,98 (4H, м, алкил CH), 2,19 (2H, дд, J=10,5 Гц, 10,5 Гц, CH), 2,33 (3H, с, N-CH3), 2,71 (2H, м, CH2), 2,97 (2H, д, J=12 Гц, CH), 3,65 (2H, дд, J=11 Гц, CH), 3,75 (2H, м, CH2), 3,92 (1H, м, CH), 4,02 (3H, с, OCH3), 4,12 (1H, дд, J=4 Гц, 11 Гц, CH), 4,78 (1H, м, CH), 7,53 (2H, м, Ar-H), 8,04 (1H, с, Pyr-H), 8,45 (1H, д, J=9 Гц, Ar-H); МС(+ve): 524,3; Rt= 5,71 мин (XBridge 2).
Соединение [27]: 4-[5-Этил-9-(1-этилпропил)-6-оксо-6,7,8,9-тетрагидро-5H-пиримидо[4,5-b][1,4]диазепин-2-иламино]-3-метокси-N-(1-метилпиперидин-4-ил)бензамид
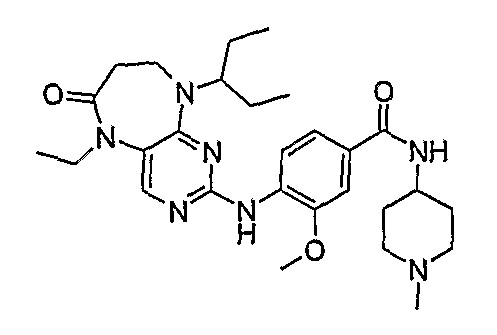
1Н-ЯМР (ДМСО): 0,86 (6H, т, J=7,5 Гц, CH3), 0,94 (3H, д, J=7 Гц, CH3), 1,63 (6H, м, алкил CH + CH2), 1,78 (2H, д, J=11 Гц, CH), 1,94 (2H, дд, J=11,5 Гц, CH), 2,18 (3H, с, N-CH3), 2,57 (2H, м, CH2), 2,80 (2H, м, CH2), 3,47 (2H, м, CH2), 3,77 (3H, м, CH + CH2), 3,96 (3H, с, OCH3), 4,59 (1H, м, CH), 7,50 (2H, м, Ar-H), 7,24 (1H, с, NH), 8,01 (2H, м, Ar-H + Pyr-H), 8,39 (1H, д, J=9 Гц, Ar-H); МС(+ve): 524,3; Rt = 7,56 мин (XBridge 2).
Соединение [29]: 4-(9-Бензил-5-метил-6-оксо-6,7,8,9-тетрагидро-5H-пиримидо[4,5-b][1,4]диазепин-2-иламино)-3-метокси-N-(1-метилпиперидин-4-ил)бензамид
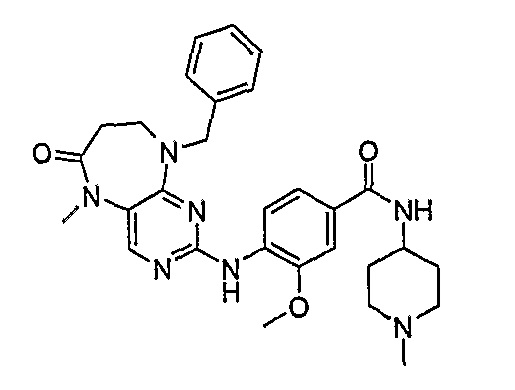
1Н-ЯМР (ДМСО): 1,58 (2H, ддд, J=4 Гц, 12,5 Гц, 24,5 Гц, CH), 1,73 (2H, д, J=1,5 Гц, CH), 1,93 (2H, дд, J=15 Гц, CH), 2,17 (3H, с, N-CH3), 2,69 (2H, м, CH2), 2,78 (2H, д, J=11,5 Гц, CH), 3,22 (3H, с, N-CH3), 3,63 (2H, м, CH2), 3,72 (1H, м, CH), 3,91 (3H, с, OCH3), 4,87 (2H, с, CH2), 7,23-7,32 (4H, м, Ar-H), 7,35-7,43 (2H, м, Ar-H), 7,44 (1H, д, J=2 Гц, Ar-H), 7,72 (1H, с, Pyr-H), 8,04 (1H, д, J=7,5 Гц, Ar-H), 8,12 (1H, д, J=8,5 Гц), 8,16 (1H, с, NH); МС(+ve): 530,4; Rt = 4,55 мин (XBridge 2).
Соединение [30]: 3-Метокси-4-(5-метил-6-оксо-9-фенил-6,7,8,9-тетрагидро-5H-пиримидо[4,5-b][1,4]диазепин-2-иламино)-N-(1-метилпиперидин-4-ил)бензамид

1Н-ЯМР (ДМСО): 1,56 (2H, дд, J=11,5 Гц, 20,5 Гц, CH), 1,75 (2H, д, J=10 Гц, CH), 1,95 (2H, м, CH), 2,18 (3H, с, N-CH3), 2,83 (3H, м, CH + CH2), 3,16 (1H, с, CH), 3,27 (3H, с, N-CH3), 3,71 (1H, м, N-CH), 3,88 (3H, с, OCH3), 4,06 (2H, м, CH2); Mc(+ve): 516,3; Rt = 2,72 мин (XBridge 2).
Соединение [31]: 4-(9-циклопентил-5-метил-6-оксо-6,7,8,9-тетрагидро-5H-спиро[пиримидо[4,5-b][1,4]диазепин-3,1’-циклопропан]-2-иламино)-N-(1-метилпиперидин-4-ил)бензамид
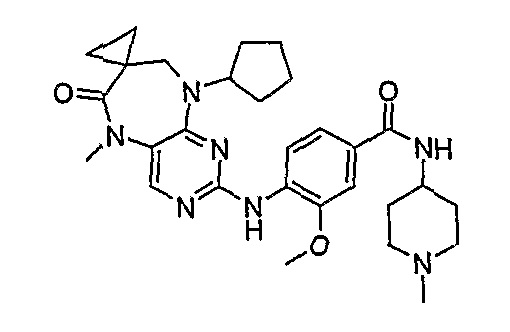
1Н-ЯМР (ДМСО): 0,66 (2H, д, J=6 Гц, циклопропил-CH), 0,91 (2H, д, J=6 Гц, циклопропил-CH), 1,50 (2H, м, CH), 1,58 (4H, м, CH), 1,69 (2H, м, CH), 1,77 (2H, м, CH), 1,95 (4H, м, CH), 2,16 (3H, с, N-CH3), 2,73 (1H, с, CH), 2,79 (2H, д, J=11 Гц, CH), 2,89 (1H, с, CH), 3,16 (3H, с, N-CH3), 3,47 (2H, с, CH2), 3,72 (1H, м, CH), 3,94 (3H, с, OCH3), 4,86 (1H, м, CH), 7,46 (2H, м, Ar-H), 7,67 (1H, с, NH), 7,95 (1H, с, Pyr-H), 8,07 (1H, д, J=8 Гц, Ar-H), 8,38 (1H, д, J=8 Гц, Ar-H); МС(+ve): 534,3; Rt = 3,05 мин (XBridge 2).
Соединение [32]: 4-(9-циклопентил-5,7,7-триметил-6-оксо-6,7,8,9-тетрагидро-5H-пиримидо[4,5-b][1,4]диазепин-2-иламино)-3-метокси-N-(1-метилпиперидин-4-ил)бензамид
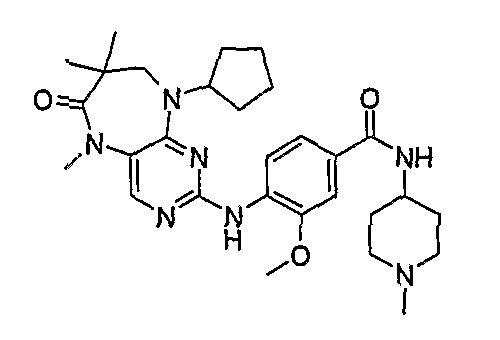
1H-ЯМР (ДМСО): 1,09 (6H, с, CH3), 1,58 (6H, м, CH2), 1,74 (4H, м, CJ), 1,95 (4H, м, CH), 2,16 (3H, с, NCH3), 2,73 (2H, д, J=11,5 Гц, CH), 3,18 (3H, с, NCH3), 3,37 (2H, с, CH2), 3,74 (1H, м, CH), 3,94 (3H, с, OCH3), 5,19 (1H, м, CH), 7,46 (2H, м, Ar-H), 7,67 (1H, с, NH), 7,98 (1H, с, Pyr-H), 8,07 (1H, д, J=8 Гц, Ar-H), 8,36 (1H, д, J=8 Гц); МС(+ve): 536,40; Rt=3,32 мин (XBridge 2).
Реакции сочетания с получением амидов
Нижеследующие соединения также получали методом, описанным в примере 1, стадии 7:
Соединение [33]: 4-(9-циклопентил-5,8-диметил-6-оксо-6,7,8,9-тетрагидро-5H-пиримидо[4,5-b][1,4]диазепин-2-иламино)-3-метокси-N-(1-метилпиперидин-4-ил)бензамид

Желтое твердое вещество (31%). 1Н-ЯМР (CD3OD): δ 1,24 (д, J=6,5 Гц, 3H, CH3), 1,36-2,17 (м, 16H, 8 × циклопент-H и 4 × пиперид-H), 2,51 (с, 3H, CH3), 3,14-3,16 (м, 2H, CH2), 3,28 (с, 3H, CH3), 4,00 (с, 3Η, CH3), 4,04-4,06 (м, 1Η, CH), 4,71-4,74 (м, 1Η, CH), 7,49-7,52 (м, 2Η), 7,99 (с, 1H), 8,41 (д, J=8,5 Гц, 1H, phe-H); МС(+ve): 522,4, Rt = 10,03 мин (чистота 97%, Vydac 1).
Соединение [34]: 4-(9-циклопентил-8-метил-6-оксо-6,7,8,9-тетрагидро-5H-пиримидо[4,5-b][1,4]диазепин-2-иламино)-3-метокси-N-(1-метилпиперидин-4-ил)бензамид

Желтое твердое вещество (38%); МС(+ve): 508,3, Rt = 9,53 мин (чистота 97%, Vydac 1).
Соединение [35]: 4-(9-циклогексил-8-метил-6-оксо-6,7,8,9-тетрагидро-5H-пиримидо[4,5-b][1,4]диазепин-2-иламино)-3-метокси-N-(1-метилпиперидин-4-ил)бензамид

Не совсем белое твердое вещество (33%); 1Н-ЯМР (CD3OD): δ 1,34-1,41 (м, 2H), 1,44 (д, J=6,5 Гц, 3H, CH3), 1,60-1,99 (м, 8H), 2,22-2,26 (м, 2H), 2,73-2,77 (м, 1H), 2,91 (с, 3H, CH3), 2,96-3,25 (м, 5H), 3,61-3,63 (м, 2H, CH2), 4,01 (с, 3Η, CH3), 4,17-4,21 (м, 1Η, CH), 4,33-4,36 (м, 1Η, CH), 7,59 (дд, J=2,0 и 8,5 Гц, 1H, phe-H), 7,63 (д, J=2,0 Гц, 1H, phe-H), 7,69 (с, 1Η, пиримид-H), 8,00 (д, J=8,5 Гц, 1H, phe-H); МС(+ve): 522,2, Rt = 10,17 мин (чистота 100%, Vydac 1).
Соединение [36]: 4-(9-циклопентил-6-оксо-6,7,8,9-тетрагидро-5H-пиримидо[4,5-b][1,4}диазепин-2-иламино)-3-метокси-N-(1-метилпиперидин-4-ил)бензамид

Твердое вещество пурпурного цвета (40%); 1Н-ЯМР (CD3OD): δ 1,65-2,25 (м, 8H, циклопент-H), 2,34 (с, 3Η, CH3), 2,73-2,75 (м, 2Η, CH2), 3,69-3,71 (м, 2Η, CH2), 3,90-3,94 (м, 1Η, CH), 4,01 (с, 3Η, CH3), 5,22-5,25 (м, 1Η, CH), 7,48-7,51 (м, 2Η, 2 × phe-H), 7,76 (с, 1Η, пиримид-H), 8,47 (с, 1Η, NH); МС(+ve): 493,4, Rt = 9,21 мин (чистота 92%, Vydac 1).
Соединение [37]: 4-(9-циклопентил-5,8-диметил-6-оксо-6,7,8,9-тетрагидро-5H-пиримидо[4,5-b][1,4]диазепин-2-иламино)-N-(1-метилпиперидин-4-ил)бензамид
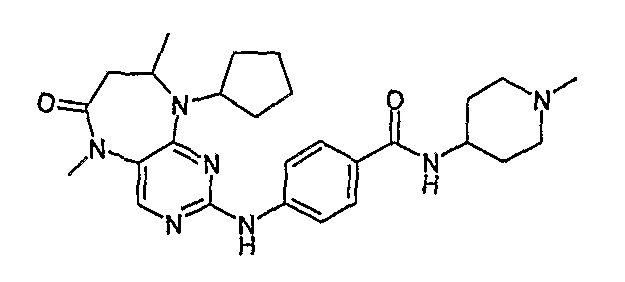
Не совсем белое твердое вещество (45%); 1Н-ЯМР (ДМСО-d6): δ 1,17 (д, J=6,0 Гц, 3H, CH3), 1,29-2,10 (м, 16Η, 8 × циклопент-H и 8 × пиперид-H), 2,21 (шир.с, 3Η, CH3), 2,81-2,83 (м, 2H, CH2), 3,21 (с, 3H, CH3), 3,73-3,76 (м, 1H, CH), 4,03-4,05 (м, 1H, CH), 4,68-4,72 (м, 1H, CH), 7,74-7,79 (м, 4H, phe-H), 8,11 (с, 1H, пиримид-H), 9,47 (шир.с, 1H, NH); МС(+ve): 492,3, Rt = 9,70 мин (чистота 92%, Vydac 1).
Соединение [38]: 4-(9-циклопентил-8-метил-6-оксо-6,7,8,9-тетрагидро-5H-пиримидо[4,5-b][1,4]диазепин-2-иламино)-N-(1-метилпиперидин-4-ил)бензамид
Не совсем белое твердое вещество (15%); 1Н-ЯМР (ДМСО-d6): δ 1,25 (д, J=7,0 Гц, 3H, CH3), 1,51-2,04 (м, 10H), 2,09 (с, 3H, CH3), 2,55-2,66 (м, 6H), 2,78-2,79 (м, 2H, CH2), 3,05-3,15 (м, 1H, CH), 3,95-4,04 (м, 1H, CH), 5,13-5,15 (м, 1H, CH), 7,75 (дд, J=8,5 Гц, 4H, phe-Н), 7,81 (с, 1H, пиримид-Н), 8,24 (д, J=7 Гц, 1H, NH), 9,48 (с, 1H, NH), 9,66 (шир.с, 1H, NH); МС(+ve): 478,3, Rt = 9,28 мин (чистота 100%, Vydac 1).
Соединение [39]: 4-(9-циклогексил-6-оксо-6,7,8,9-тетрагидро-5H-пиримидо[4,5-b][1,4]диазепин-2-иламино)-3-метокси-N-(1-метилпиперидин-4-ил)бензамид
Не совсем белое твердое вещество (19%); 1Н-ЯМР (ДМСО-d6): δ 1,16-1,98 (м, 18H, 10 × циклогекс-Н и 8 × пиперид-Н), 2,18 (с, 3H, CH3), 2,60 (дд, J=5 Гц, 2H, CH2), 3,60 (дд, J=5 Гц, 2H, CH2), 3,73-3,76 (м, 1Η, CH), 3,95 (с, 3Η, CH3), 4,58-4,63 (м, 1H, CH), 7,46 (дд, J=2,0 и 8,0 Гц, 1H, phe-Н), 7,49 (д, J=2 Гц, 1H, phe-Н), 7,60 (с, 1H), 7,78 (с, 1H), 8,10 (д, J=8,0 Гц, 1H, NH), 8,37 (д, J=8,0 Гц, 1H, phe-H), 9,41 (шир.с, 1Η, NH); МС(+ve): 508,4, R4 = 9,70 мин (чистота 95%, Vydac 1).
Пример 2
Общий экспериментальный метод
Аналитическую и препаративную ОФ-ВЭЖХ-МС осуществляли на колонках Waters XBridge (50×4,6 мм C18, 3,5 мкм или 100×4,6 мм C18, 3,5 мкм) и XBridge (100×19 мм C18, 5 мкм) с использованием линейного градиента системы растворителя A (воды, содержащей 0,1% гидроксида аммония и 5% ацетонитрила)/растворителя B (ацетонитрила). Препаративную ОФ-ВЭЖХ осуществляли на колонке Apex Prepsil ODS 10 микрон (22×250 мм) с использованием линейного градиента растворителя A (воды, содержащей 0,1% TFA)/растворителя B (ацетонитрила). Масс-спектры получали на одноквадрупольном масс-спектрометре Waters ZQ2000 с ионизацией электрораспылением (ESI).
Были использованы следующие градиенты:
мл/мин
Промежуточное соединение 1: 2-хлор-9-циклопентил-5-метил-8,9-дигидро-5H-пиримидо[4,5-b][1,4]диазепин-6(7H)-он
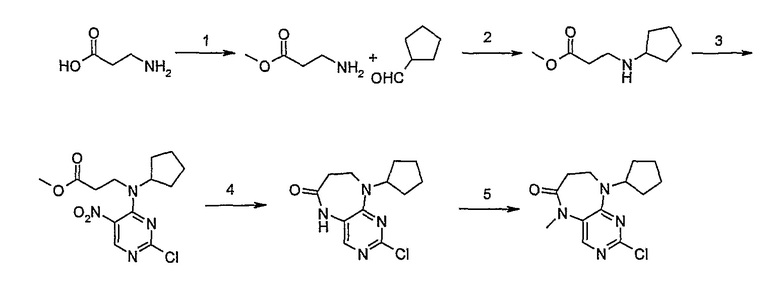
1. Тионилхлорид (2,1 экв.), MeOH, 0°C для добавления, кипячение с обратным холодильником в течение 2 ч.; 2. Циклопентанон (0,77 экв.), ацетат натрия (0,77 экв.), триацетоксиборгидрид натрия (1,11 экв.), ДХМ, к.т., 16 ч.; 3. 2,4-дихлор-5-нитропиримидин (1,1 экв.), K2CO3 (1 экв.), ацетон, 0°C – к.т., 16 ч.; 4. NH4Cl (8,5 экв.), Fe (8 экв.), EtOH/H2O (4:1), кипячение с обратным холодильником - 2,5 ч.; 5. MeI (1,18 экв.), NaH (1,07 экв.), ДМФ, -10°C – к.т., 3 ч.
Стадия 1: Метил 3-аминопропаноат
β-аланин (9,37 г, 0,105 моль, 1,0 экв.) добавляли к MeOH (50 мл), и смесь охлаждали до 0°C, а затем по каплям добавляли тионилхлорид (16 мл, 0,221 моль, 2,1 экв.) [осторожно: при добавлении происходит экзотермическая реакция]. Реакционную смесь оставляли для нагревания до комнатной температуры, а затем кипятили с обратным холодильником в течение 2 ч. Раствор концентрировали в вакууме, обрабатывали трет-бутилметиловым эфиром, и полученные кристаллы удаляли фильтрацией. Продукт представлял собой белое кристаллическое твердое вещество (11 г, 100%); 1H (ДМСО-d6): δ 2,73 (2H, дд, J=7 Гц, CH2), 2,98 (2H, дд, J=7 Гц, CH2), 3,61 (3Η, с, CH3), 8,28 (2Η, шир.с., NH2); МС(+ve): 104,1.
Стадия 2: Метил 3-(циклопентиламино)пропаноат
Метил 3-аминопропаноат (9,37 г, 0,09 моль) растворяли в ДХМ (200 мл). Затем добавляли циклопентанон (6,43 мл, 0,07 моль, 0,77 экв.), ацетат натрия (5,96 г, 0,07 моль, 0,77 экв.) и триацетоксиборгидрид натрия (22 г, 0,10 моль, 1,11 экв.) и реакционную смесь перемешивали при комнатной температуре в течение 16 ч. После этого добавляли 20% бикарбонат натрия (100 мл) и 2M гидроксид натрия (50 мл), и продукт экстрагировали ДХМ/Η2O. Органические экстракты объединяли, промывали насыщенным NaCl, сушили (MgSO4) и выпаривали при пониженном давлении с получением продукта в виде бледно-желтого масла (8,90 г, 55%); МС(+ve): 172,4.
Стадия 3: Метил 3-[циклопентил(2-хлор-5-нитропиримидин-4-ил)амино]пропаноат
Метил-3-(циклопентиламино)пропаноат (838 мг, 0,005 моль, 1 экв.) и K2CO3 (0,676 г, 0,005 моль, 1 экв.) добавляли к ацетону (5 мл), и полученную смесь охлаждали до 0°C, а затем по каплям добавляли 2,4-дихлор-5-нитропиримидин (1,04 г, 0,0055 моль, 1,1 экв.). Затем реакционную смесь нагревали до комнатной температуры и продолжали перемешивать еще 16 часов, после чего добавляли еще 0,12 экв. пиримидина. Затем перемешивание продолжали еще 3 часа. Реакционную смесь выпаривали при пониженном давлении и продукт экстрагировали EtOAc/H2O. Органические экстракты промывали насыщенным NaCl, сушили (MgSO4) и выпаривали при пониженном давлении с получением продукта в виде коричневого маслянистого остатка (1,04 г, 65%); МС+ve: 329,2; Rt = 3,78 мин (Аналитический_1).
Стадия 4: 2-хлор-9-циклопентил-5,7,8,9-тетрагидро-6H-пиримидо[4,5-b][1,4]диазепин-6-он
Метил-3-[циклопентил(2-хлор-5-нитропиримидин-4-ил)амино]пропаноат (1,0 г, 0,003 моль, 1 экв.) и NH4Cl (1,38 г, 0,025 моль, 8,5 экв.) добавляли к EtOH/H2O (4:1, 10 мл), и смесь кипятили с обратным холодильником. После этого порциями добавляли порошок железа (1,36 г, 0,024 моль, 8 экв.), и через 2 часа реакционную смесь подвергали горячей фильтрации через целит, тщательно промывая EtOAc (10 мл) и EtOH (10 мл) [оба раза горячими]. Растворитель выпаривали при пониженном давлении и получали продукт в виде коричневого твердого вещества (0,35 г, 43%); 1H (ДМСО-d6): δ 1,55 (4H, м, 2CH2), 1,7 (2H, м, CH2), 1,83 (2Η, м, CH2), 2,66 (2Η, т, J=5 Гц, CH2), 3,57 (2Η, т, J=5 Гц, CH2), 5,01 (1Η, м, CH), 7,83 (1Η, с, CH), 9,71 (1Η, с, NH); МС(+ve): 267,2; Rt = 2,87 мин (Аналитический_1).
Стадия 5: 2-хлор-9-циклопентил-5-метил-8,9-дигидро-5H-пиримидо[4,5-b][1,4]диазепин-6(7Н)-он
2-хлор-9-циклопентил-5,7,8,9-тетрагидро-6H-пиримидо[4,5-δ][1,4]диазепин-6-он (0,318 г, 0,0012 моль) и MeI (0,088 мл, 0,0014 моль, 1,18 экв.) добавляли к ДМФ (5 мл), и раствор охлаждали до -10°C. Затем добавляли NaH (0,03 г, 0,0013 моль, 1,07 экв.), и реакционную смесь перемешивали при 0°С в течение 30 минут и при комнатной температуре в течение 30 минут. Растворитель выпаривали при пониженном давлении и продукт экстрагировали EtOAc/H2O. Органические экстракты объединяли, промывали насыщенным NaCl, сушили (MgSO4) и выпаривали при пониженном давлении с получением продукта в виде пурпурного маслянистого остатка (0,25 г, 75%); 1H (ДМСО-d6): δ 1,55 (4H, м, 2CH2), 1,7 (2Η, м, CH2), 1,9 (2Η, м, CH2), 2,63 (2Η, т, J=5 Гц, CH2), 3,17 (3Η, с, CH3), 3,65 (2Η, т, J=5 Гц, CH2), 4,74 (1Η, м, CH), 8,14 (1Η, с, CH); МС(+ve): 281,2; Rt = 3,11 мин (Аналитический_1).
Методами, аналогичными методам, описанным выше для получения промежуточного соединения 1, были получены нижеследующие промежуточные соединения 2-4:
Промежуточное соединение 2: 2’-хлор-9’-циклопентил-5’-метил-8’,9’-дигидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-6’(5’Η)-он
В результате реакции взаимодействия этилового эфира 1-аминометилциклопропанкарбоновой кислоты с циклопентаноном, ацетатом натрия и триацетоксиборгидридом натрия в ДХМ получали этиловый эфир 1-циклопентиламинометилциклопропанкарбоновой кислоты
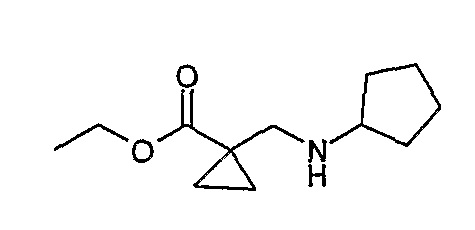
1Н-ЯМР (CDCl3): 0,84 (3H, т, J=7 Гц, CH3), 1,25 (4H, м, CH2), 1,37 (2H, м, CH), 1,54 (2H, м, CH), 1,70 (2H, м, CH), 1,83 (2H, м, CH), 2,71 (2H, с, CH2), 3,10 (1H, м, CH), 4,16 (2H, кв., J=7 Гц, CH2CH3).
В результате реакции взаимодействия метилового эфира 1-циклопентиламинометилциклопропанкарбоновой кислоты с 2,4-дихлор-5-нитропиримидином и K2CO3 в ацетоне получали этиловый эфир 1-{[(2-хлор-5-нитропиримидин-4-ил)циклопентиламино]метил}циклопропанкарбоновой кислоты. После обработки NH4Cl и порошком железа в этаноле получали этиловый эфир 1-{[(5-амино-2-хлорпиримидин-4-ил)циклопентиламино]метил}циклопропанкарбоновой кислоты.
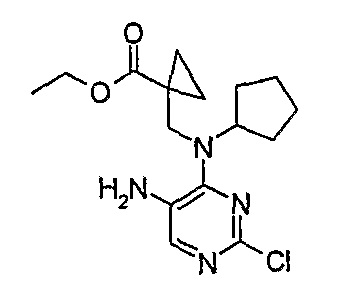
МС(+ve): 339,2, 341,2; Rt = 4,15 мин (Аналитический_2).
Вышеуказанное соединение в ДМФ нагревали до 140°C в течение 2 часов. Растворитель выпаривали в вакууме, добавляли этилацетат, и полученное твердое вещество фильтровали и сушили в вакууме с получением 2’-хлор-9’-циклопентил-8’,9’-дигидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-6’(5’H)-она
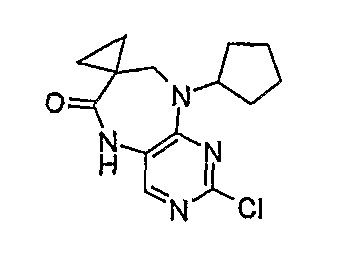
МС(+ve): 293,2, 295,2; Rt = 3,48 мин (Аналитический_2).
В результате реакции взаимодействия 2-хлор-9-циклопентил-5,7,8,9-тетрагидропиримидо[4,5-b][1,4]диазепин-7,1’-циклопропан]-6-она с MeI и NaH в ДМФ получали 2’-хлор-9’-циклопентил-5’-метил-8’,9’-дигидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-6’(5’H)-он
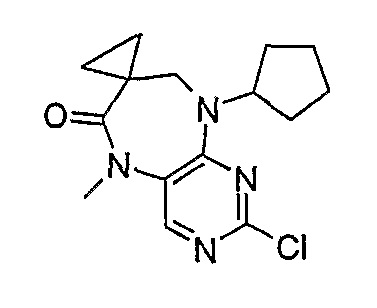
МС(+ve): 307,1, 309,2; Rt = 3,67 мин (Аналитический_3).
Промежуточное соединение 3: 2-хлор-9-циклопентил-5,7,7-триметил-8,9-дигидро-5H-пиримидо[4,5-b][1,4]диазепин-6(7H)-он
В результате реакции взаимодействия этилового эфира 3-амино-2,2-диметилпропионовой кислоты с циклопентаноном, ацетатом натрия и триацетоксиборгидридом натрия в ДХМ получали этиловый эфир 3-циклопентиламино-2,2-диметилпропионовой кислоты.
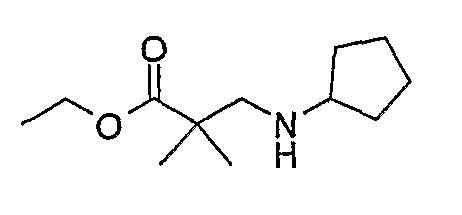
1H-ЯМР (CDCl3): 1,19 (6H, с, CH3), 1,25 (3H, т, J=7 Гц, CH3), 1,28 (2H, м, CH), 1,49 (2H, м, CH), 1,65 (2H, м, CH), 2,64 (1H, с, CH2), 3,01 (1H, м, CH), 4,10 (2H, кв., J=7,5 Гц, CH2).
В результате реакции взаимодействия этилового эфира 3-циклопентиламино-2,2-диметилпропионовой кислоты с 2,4-дихлор-5-нитропиримидином и K2CO3 в ацетоне получали этиловый эфир 3-[(2-хлор-5-нитропиримидин-4-ил)циклопентиламино]-2,2-диметилпропионовой кислоты
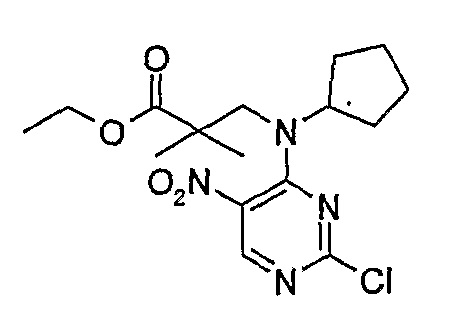
МС(+ve): 371,1, 373,1; Rt = 4,67 мин (Аналитический_2).
В результате реакции взаимодействия этилового эфира 3-[(2-хлор-5-нитропиримидин-4-ил)циклопентиламино]-2,2-диметилпропионовой кислоты с NH4Cl и порошком железа в этаноле получали этиловый эфир 3-[(5-амино-2-хлорпиримидин-4-ил)циклопентиламино]-2,2-диэтилпропионовой кислоты.
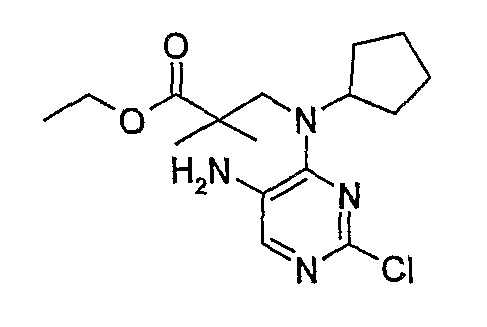
МС(+ve): 341,2, 343,2; Rt = 4,25 мин (Аналитический_2).
Этиловый эфир 3-[(5-амино-2-хлорпиримидин-4-ил)-циклопентиламино]-2,2-диметилпропионовой кислоты в ДМФ нагревали до 140°C в течение 2 часов. Растворитель выпаривали в вакууме, добавляли этилацетат и полученное твердое вещество фильтровали и сушили в вакууме с получением 2-хлор-9-циклопентил-7,7-диметил-5,7,8,9-тетрагидропиримидо[4,5-b][1,4]диазепин-6-она.
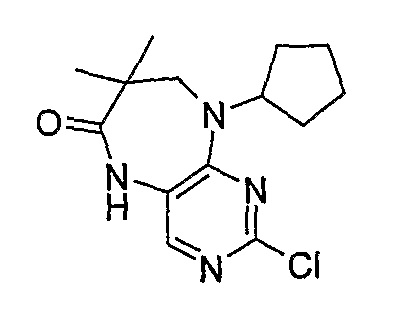
МС(+ve): 295,1, 297,2; Rt= 3,58 мин (Аналитический_2).
В результате реакции взаимодействия 2-хлор-9-циклопентил-7,7-диметил-5,7,8,9-тетрагидропиримидо[4,5-b][1,4]диазепин-6-она с MeI и NaH в ДМФ получали 2-хлор-9-циклопентил-5,7,7-триметил-8,9-дигидро-5H-пиримидо[4,5-b][1,4]диазепин-6(7H)-он.
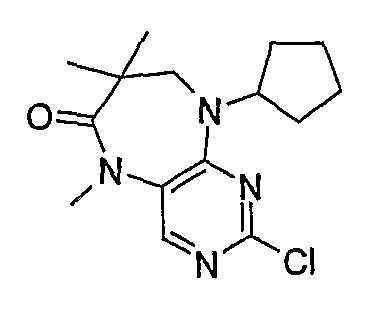
МС(+ve): 309,1, 311,2; Rt = 3,90 мин (Аналитический_3).
Промежуточное соединение 4: 2’-хлор-9’-циклопентил-5’-метил-8’,9’-дигидроспиро[циклобутан-1,7’-пиримидо[4,5-b][1,4]диазепин]-6’(5’H)-он
В результате реакции взаимодействия метилового эфира 1-аминометилциклобутанкарбоновой кислоты с циклопентаноном, ацетатом натрия и триацетоксиборгидридом натрия в ДХМ получали метиловый эфир 1-циклопентиламинометил-циклобутанкарбоновой кислоты.
1H-ЯМР (CDCl3): 1,36 (2H, м, CH2), 1,51 (2H, м, CH2), 1,66 (2H, м, CH2), 1,95 (4H, м, 2 × CH2), 2,15 (2H, м, CH2), 2,43 (4H, м, 2 × CH2), 2,93 (2H, с, CH2), 3,21 (1H, м, CH), 3,76 (3H, с, CH3).
В результате реакции взаимодействия метилового эфира 1-циклопентиламинометилциклобутанкарбоновой кислоты с 2,4-дихлор-5-нитропиримидином и K2CO3 в ацетоне получали метиловый эфир 1-{[(2-хлор-5-нитропиримидин-4-ил)циклопентиламино]метил}циклобутанкарбоновой кислоты
МС(+ve): 369,2, 371,2; Rt = 3,48 мин (Аналитический_1).
В результате реакции взаимодействия метилового эфира 1-{[(2-хлор-5-нитропиримидин-4-ил)циклопентиламино]метил}циклобутанкарбоновой кислоты с NH4Cl и порошком железа в этаноле получали непосредственно выделенный 2’-хлор-9’-циклопентил-8’,9’-дигидроспиро[циклобутан-1,7’-пиримидо[4,5-b] [1,4]диазепин]-6’(5’H)-он.
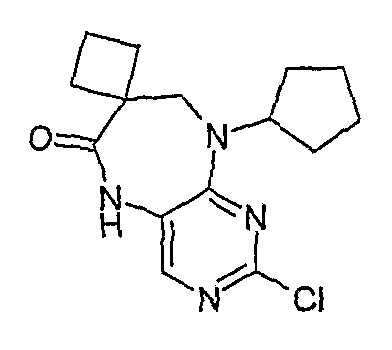
МС(+ve): 307,2, 309,2; Rt = 3,48 мин (Аналитический_1).
В результате реакции взаимодействия 2-хлор-9-циклопентил-5,7,8,9-тетрагидропиримидо[4,5-b][1,4]диазепин-7,l’-циклобутан]-6-она с MeI и NaH в ДМФ получали 2’-хлор-9’-циклопентил-5’-метил-8’,9’-дигидроспиро[циклобутан-1,7’-пиримидо[4,5-b][1,4]диазепин]-6’(5’H)-он
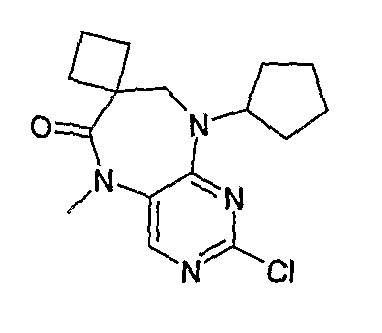
МС(+ve): 321,2, 323,3; Rt = 3,77 мин (Аналитический_1).
Промежуточное соединение 5: 4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-3-метоксибензойная кислота
2’-хлор-9’-циклопентил-5’-метил-8’,9’-дигидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-6’(5’H)-он (промежуточное соединение 2) (0,70 г, 2,5 ммоль, 1 экв.), 4-амино-3-метоксибензойную кислоту (1,25 г, 7,5 ммоль, 3 экв.) и TFA (1,43 г, 12,5 ммоль, 5 экв.) кипятили с обратным холодильником в TFE (25 мл) в течение 18 часов. Растворитель выпаривали в вакууме и полученный остаток растирали с MeOH, в результате чего получали 4-(9’-циклопентил-5’-метил-6’-оксо- 5’,6’,8’,9’-тетрагидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-3-метоксибензойную кислоту (0,81 г, 74%).
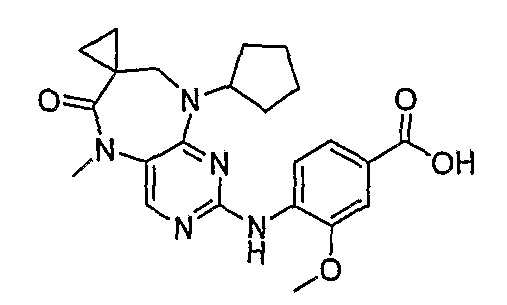
Rt = 1,88 мин (Аналитический_1). МС(+ve): 438,4, МС (-ve): 436,5.
Промежуточное соединение 6: 4-(9-циклопентил-5,7,7-триметил-6-оксо-6,7,8,9-тетрагидро-5H-пиримидо[4,5-b][1,4]диазепин-2-иламино)-3-метоксибензойная кислота
2-хлор-9-циклопентил-5,7,7-триметил-8,9-дигидро-5H-пиримидо[4,5-b][1,4]диазепин-6(7H)-он (промежуточное соединение 3) (0,35 г, 1,14 ммоль, 1 экв.), 4-амино-3-метоксибензойную кислоту (0,57 г, 3,42 ммоль, 3 экв.) и TFA (0,42 мл, 5,7 ммоль, 5 экв.) кипятили с обратным холодильником в TFE (10 мл) в течение 36 часов. Растворитель выпаривали в вакууме и полученный остаток растирали с EtOAc с получением 4-(9-циклопентил-5,7,7-триметил-6-оксо-6,7,8,9-тетрагидро-5H-пиримидо[4,5-b][1,4]диазепин-2-иламино)-3-метоксибензойной кислоты (0,38 г, 76%).
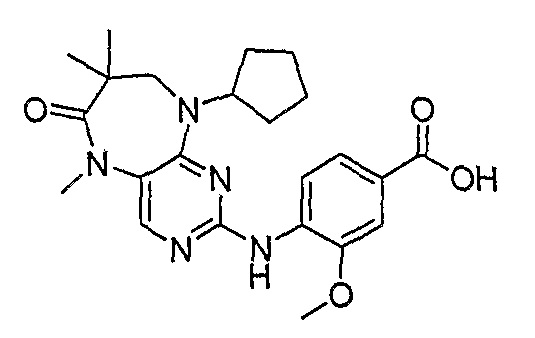
МС(+ve): 440,3, МС(-ve): 438,5.
Промежуточное соединение 7: 4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклобутан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-3-метоксибензойная кислота
2’-хлор-9’-циклопентил-5’-метил-8’,9’-дигидроспиро[циклобутан-1,7’-пиримидо[4,5-b][1,4]диазепин]-6’(5’H)-он (промежуточное соединение 4) (1,02 г, 3,18 ммоль, 1 экв.), 4-амино-3-метоксибензойную кислоту (1,6 г, 9,5 ммоль, 3 экв.) и TFA (1,2 мл, 15,9 ммоль, 5 экв.) кипятили с обратным холодильником в TFE (40 мл) в течение 24 часов. Растворитель выпаривали в вакууме и полученный остаток растирали с EtOAc с получением 4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклобутан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-3-метоксибензойной кислоты (1,08 г, 75%).
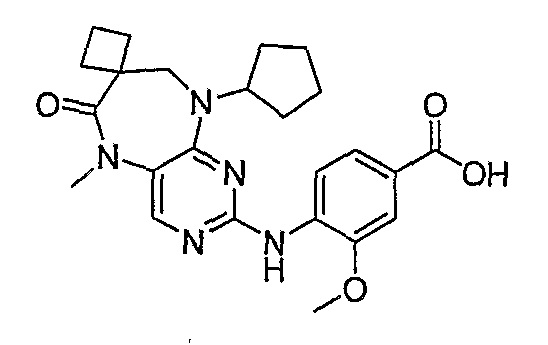
Rt = 2,10 мин (Аналитический_1); МС(+ve): 452,3; МС(-ve): 450,4.
Промежуточное соединение 8: 4-(9-циклопентил-5-метил-6-оксо-6,7,8,9-тетрагидро-5H-пиримидо[4,5-b][1,4]диазепин-2-иламино)-3-метоксибензойная кислота
2-хлор-9-циклопентил-5-метил-8,9-дигидро-5H-пиримидо[4,5-b][1,4]диазепин-6(7H)-он (промежуточное соединение 1) (0,70 г, 2,5 ммоль, 1 экв.), 4-амино-3-метоксибензойную кислоту (1,25 г, 7,5 ммоль, 3 экв.) и TFA (0,93 мл, 12,5 ммоль, 5 экв.) кипятили с обратным холодильником в TFE (25 мл) в течение 20 часов. Растворитель выпаривали в вакууме и полученный остаток растирали с MeOH с получением 4-(9-циклопентил-5-метил-6-оксо-6,7,8,9-тетрагидро-5H-пиримидо[4,5-b][1,4]диазепин-2-иламино)-3-метоксибензойной кислоты (0,66 г, 64%).
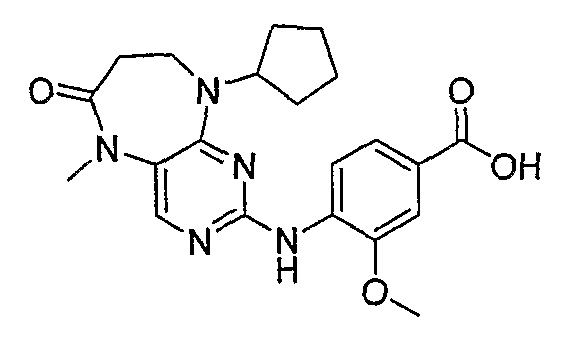
Rt = 1,88 мин (Аналитический_1); МС(+ve): 412,4, МС(-ve): 410,5.
Промежуточное соединение 9: 4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-3-фторбензойная кислота
2’-хлор-9’-циклопентил-5’-метил-8’,9’-дигидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-6’(5’Η)-он (промежуточное соединение 2) (0,42 г, 1,36 ммоль, 1 экв.), 4-амино-3-фторбензойную кислоту (0,63 г, 4,1 ммоль, 3 экв.) и TFA (0,51 мл, 6,8 ммоль, 5 экв.) кипятили с обратным холодильником в TFE (10 мл) в течение 36 часов. Затем добавляли дополнительное количество 4-амино-3-фторбензойной кислоты (0,63 г, 4,1 ммоль, 3 экв.) и TFA (0,51 мл, 6,8 ммоль, 5 экв.), и реакционную смесь снова кипятили с обратным холодильником в течение 4 дней. Растворитель выпаривали в вакууме и полученный остаток растирали с EtOAc с получением 4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-3-фторбензойной кислоты (0,41 г, 71%).
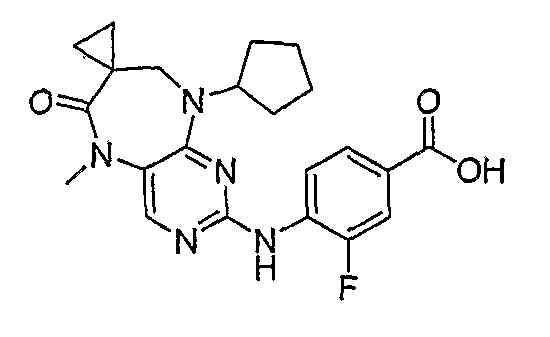
Rt = 1,93 мин (Аналитический_1); МС(+ve): 426,3.
Промежуточное соединение 10: 4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклобутан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-3-фторбензойная кислота
2’-хлор-9’-циклопентил-5’-метил-8’,9’-дигидроспиро[циклобутан-1,7’-пиримидо[4,5-b][1,4]диазепин]-6’(5’H)-он (Промежуточное соединение 4) (0,20 г, 0,63 ммоль, 1 экв.), 4-амино-3-фторбензойную кислоту (0,30 г, 1,9 ммоль, 3 экв.) и TFA (0,24 мл, 3,2 ммоль, 5 экв.) кипятили с обратным холодильником в TFE в течение 18 часов. Затем добавляли 4-амино-3-фторбензойную кислоту (0,20 г) и TFA (0,1 мл) и реакционную смесь снова кипятили с обратным холодильником в течение 2 дней. Затем добавляли TFA (0,24 мл), и реакционную смесь снова кипятили с обратным холодильником в течение 3 дней. Растворитель выпаривали в вакууме, и полученный остаток растирали с EtOAc, в результате чего получали 4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклобутан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-3-фторбензойную кислоту (0,15 г, 55%).
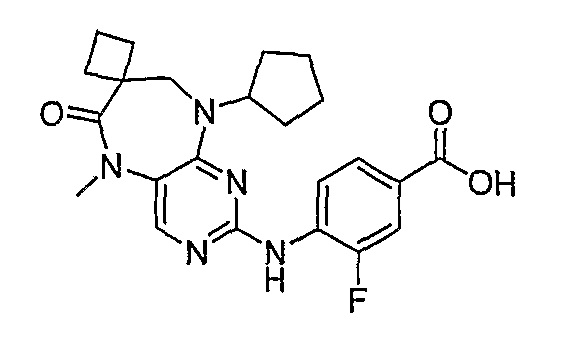
Rt = 2,02 мин (Аналитический_1); МС(+ve): 440,3; МС(-ve): 438,4.
Промежуточное соединение 11: 4-амино-N-(транс-4-(4- (циклопропилметил)пиперазин-1-ил)циклогексил)-3-метоксибензамид
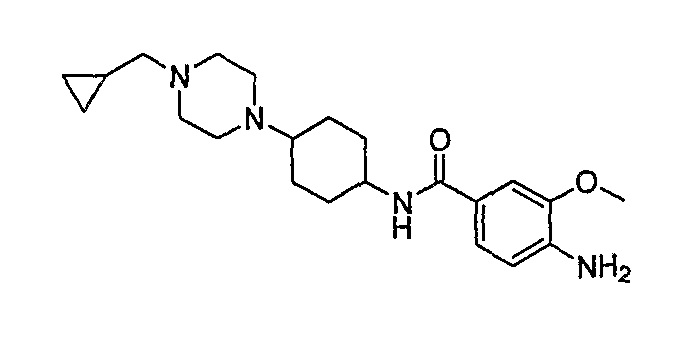
4-амино-N-(транс-4-(4-(циклопропилметил)пиперазин-1-ил)циклогексил)-3-метоксибензамид представляет собой известное соединение и было получено методами, описанными в WO 2007/090844 A1.
Промежуточное соединение 12: 4-(9-циклопентил-5-метил-6-оксо-6, 7,8,9-тетрагидро-5H-пиримидо[4,5-b][1,4]диазепин-2-иламино)-3-метокси-N-(пиперидин-4-ил)бензамид
4-(9-циклопентил-5-метил-6-оксо-6,7,8,9-тетрагидро-5H-пиримидо[4,5-b][1,4]диазепин-2-иламино)-3-метоксибензойную кислоту (промежуточное соединение 8) (0,49 г, 1,2 ммоль, 1 экв.), TBTU (0,42 г, 1,3 ммоль, 1,1 экв.) и DIPEA (0,40 мл, 2,4 ммоль, 2 экв.) перемешивали вместе в ДХМ (25 мл) в течение 30 минут, а затем добавляли 4-амино-1-BOC-пиперидин (0,29 г, 1,4 ммоль, 1,2 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 20 часов, а затем разбавляли ДХМ (30 мл) и последовательно промывали водой (2×15 мл), 50% насыщенным водным бикарбонатом натрия (1×20 мл), насыщенным водным карбонатом калия (1×20 мл) и насыщенным раствором соли (1×20 мл). После концентрирования в вакууме выделяли 4-(9-циклопентил-5-метил-6-оксо-6,7,8,9-тетрагидро-5H-пиримидо[4,5-b][1,4]диазепин-2-иламино)-3-метокси-N-(1-BOC-пиперидин-4-ил)бензамид, который использовали без дополнительной очистки (0,71 г). Rt = 3,58 мин (Аналитический_1).
Затем добавляли метаноловый раствор соляной кислоты и полученный раствор перемешивали при комнатной температуре в течение 4 часов. После концентрирования в вакууме получали гидрохлоридную соль 4-(9-циклопентил-5-метил-6-оксо-6,7,8,9-тетрагидро-5H-пиримидо[4,5-b][1,4]диазепин-2-иламино)-3-метокси-N-(пиперидин-4-ил)бензамида. Эту соль распределяли между ДХМ и водным раствором карбоната калия при pH 8. ДХМ-слой отделяли, а водный слой экстрагировали ДХМ и EtOAc. Объединенные органические слои сушили (MgSO4) и концентрировали в вакууме с получением 4-(9-циклопентил-5-метил-6-оксо-6,7,8,9-тетрагидро-5H-пиримидо[4,5-b][1,4]диазепин-2-иламино)-3-метокси-N-(пиперидин-4-ил)бензамид в виде свободного основания (0,51 г).
Rt = 2,78 мин (Аналитический_1); МС(+ve): 494; МС(-ve): 492,
Соединение [254]: 4-(9’-Циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-3-метокси-N-(4-метилпиперазин-1-ил)бензамид
4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-3-метоксибензойную кислоту (промежуточное соединение 5) (22 мг, 0,05 ммоль, 1 экв.), DIPEA (17 мкл, 0,10 ммоль, 2 экв.) и TBTU (18 мг, 0,055 ммоль, 1,1 экв.) добавляли к 0,5 мл ДМФ, и полученный раствор перемешивали при комнатной температуре в течение 30 минут, а затем добавляли 1-амино-4-метилпиперазин (12 мкл, 0,1 ммоль, 2 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 4 часов, а затем очищали с помощью препаративной ОФ-ВЭЖХ-МС (Препаративный_1) с получением 4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-3-метокси-N-(4-метилпиперазин-1-ил)бензамида (белого твердого вещества, 8 мг, 30%).
Rt = 2,63 мин (Аналитический_1) МС(+ve): 535,5; МС(-ve): 533,6; 1Н-ЯМР (ДМСО-d6) δ м.д.: 0,63-0,70 (2H, м), 0,87-0,94 (2H, м), 1,43-1,54 (2H, м), 1,55-1,63 (2H, м), 1,64-1,73 (2H, м), 1,83-1,94 (2H, м), 2,18 (3H, с), 2,31-2,48 (4H, м), 2,92 (4H, м), 3,16 (3H, с), 3,47 (2H, с), 3,94 (3H, с), 4,78-4,91 (1H, м), 7,35-7,48 (2H, м), 7,68 (1H, с), 7,98 (1H, с), 8,39 (1H, д, J=8,3 Гц), 9,31 (1H, с).
Соединение [218]: (±)-4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклобутан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-3-метокси-N-(хинуклидин-3-ил)бензамид
4-(9’-цклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклобутан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-3-метоксибензойную кислоту (промежуточное соединение 7) (15 мг, 0,033 ммоль), DIPEA (12 мкл, 0,066 ммоль, 2 экв.) и TBTU (12 мг, 0,036 ммоль, 1,1 экв.) добавляли к 0,5 мл ДМФ, и полученный раствор перемешивали при комнатной температуре в течение 5 минут, а затем добавляли дигидрохлорид 3-аминохинуклидина (8 мг, 0,04 ммоль, 1,2 экв.) и DIPEA (13 мкл). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов, а затем очищали с помощью препаративной ОФ-ВЭЖХ-МС (Препаративный_2) с получением (±)-4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклобутан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-3-метокси-N-(хинуклидин-3-ил)бензамида.
Rt = 3,26 мин (Аналитический_1); МС(+ve): 560,4; 1Н-ЯМР (ДМСО-d6) δ м.д.: 8,37 (д, J=8,3 Гц, 1H), 8,08 (д, J=6,8 Гц, 1H), 8,05 (с, 1H), 7,73 (с, 1H), 7,45-7,53 (м, 2H), 4,82 (квин., J=8,3 Гц, 1H), 3,84-4,01 (м, 4H), 3,64 (с, 2H), 3,18 (с, 3H), 3,04-3,14 (м, 1H), 2,88 (т, J=9,8 Гц, 1H), 2,58-2,75 (м, 4H), 2,21-2,33 (м, 2H), 2,07 (с, 2H), 1,97 (шир.с, 2H), 1,87 (шир.с, 2H), 1,76 (д, J=4,9 Гц, 2H), 1,56-1,70 (м, 8H), 1,30 (шир.с, 1H).
Соединение [195]: 4-(9-циклопентил-5,7,7-триметил-6-оксо-6,7,8,9-тетрагидро-5H-пиримидо[4,5-b][1,4]диазепин-2-иламино)-3-метокси-N-(4-метилпиперазин-1-ил)бензамид
4-(9-циклопентил-5,7,7-триметил-6-оксо-6,7,8,9-тетрагидро-5H-пиримидо[4,5-b][1,4]диазепин-2-иламино)-3-метоксибензойную кислоту (промежуточное соединение 6) (20 мг, 0,046 ммоль, 1 экв.), DIPEA (16 мкл, 0,091 ммоль, 2 экв.) и TBTU (16 мг, 0,05 ммоль, 1,1 экв.) добавляли к 0,5 мл ДМФ, и полученный раствор перемешивали при комнатной температуре в течение 5 минут, а затем добавляли 1-амино-4-метилпиперазин (7 мг). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов, а затем очищали с помощью препаративной ОФ-ВЭЖХ-МС (Препаративный_1) с получением 4-(9-циклопентил-5,7,7-триметил-6-оксо-6,7,8,9-тетрагидро-5H-пиримидо[4,5-b][1,4]диазепин-2-иламино)-3-метокси-N-(4-метил-пиперазин-1-ил)бензамида.
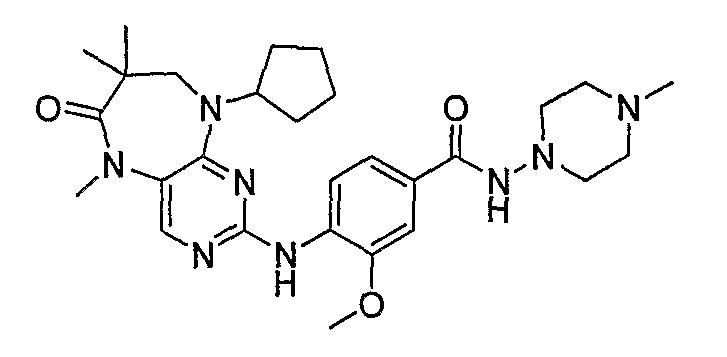
Rt = 2,81 мин (Аналитический_1); МС(+ve): 537,5; 1Н-ЯМР (ДМСО-d6) δ м.д.: 1,09 (6H, с), 1,61 (4H, м), 1,73 (2H, м), 1,87 (2H, м), 2,19 (3H, с), 2,42 (2H, м), 2,92 (4H, м), 3,29 (3H, с), 3,37 (3H, м), 3,93 (3H, с), 3,17 (1H, м), 7,40 (2H, м), 7,68 (1H, с), 7,98 (1H, с), 8,37 (1H, д, J=7 Гц), 9,30 (1H, с).
Соединение [221]: 4-(9’-Циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклобутан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-3-метокси-N-(4-метилпиперазин-1-ил)бензамид
4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклобутан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-3-метоксибензойную кислоту (промежуточное соединение 7) (15 мг, 0,033 ммоль, 1 экв.), DIPEA (12 мкл, 0,066 ммоль, 2 экв.) и TBTU (12 мг, 0,036 ммоль, 1,1 экв.) добавляли к 0,5 мл ДМФ, и полученный раствор перемешивали при комнатной температуре в течение 5 минут, а затем добавляли 1-амино-4-метилпиперазин (5 мг) и DIPEA (13 мкл). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов, а затем очищали с помощью препаративной ОФ-ВЭЖХ-МС (Препаративный_2) с получением 4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклобутан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-3-метокси-N-(4-метилпиперазин-1-ил)бензамида.
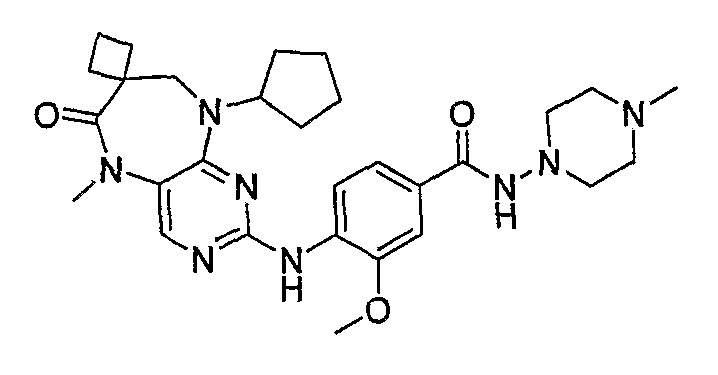
Rt = 2,79 мин (Аналитический_1); МС(+ve): 549,5; 1H-ЯМР (ДМСО-d6) δ м.д.: 9,31 (с, 1H), 8,37 (д, J=8,3 Гц, 1H), 8,05 (с, 1H), 7,72 (с, 1H), 7,37-7,47 (м, 2H), 4,73-4,86 (м, 1H), 3,93 (с, 3H), 3,64 (с, 2H), 3,18 (с, 3H), 2,92 (шир.с, 4H), 2,36 (шир.с, 4H), 2,27 (д, J=9,3 Гц, 2H), 2,18 (с, 3H), 1,97 (шир.с, 2H), 1,79-1,91 (м, 1H), 1,76 (шир.с, 2H), 1,54-1,72 (м, 7H).
Соединение [371]: (9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-N-((транс)-4-(4-(циклопропилметил)пиперазин-1-ил)циклогексил)-3-метоксибензамид
2’-хлор-9’-циклопентил-5’-метил-8’,9’-дигидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-6’(5’H)-он (промежуточное соединение 2) (0,14 г, 0,45 ммоль, 1 экв.), 4-амино-N-(транс-4-(4-(циклопропилметил)пиперазин-1-ил)циклогексил)-3-метоксибензамид (промежуточное соединение 11) (0,26 г, 0,67 ммоль, 1,5 экв.) и TFA (0,17 мл, 2,2 ммоль, 5 экв.) в TFE (3 мл) нагревали при 80°C в течение 18 часов. Затем добавляли дополнительное количество промежуточного соединения 2 (0,14 г) и реакционную смесь снова нагревали в течение 48 часов. Реакционную смесь концентрировали в вакууме и остаток очищали колоночной флэш-хроматографией на двуокиси кремния, элюируя 0 → 20% аммиаком/MeOH в градиенте ДХМ, а затем препаративной ВЭЖХ (Препаративный_4) и наконец, препаративной ОФ-ВЭЖХ-МС (Препаративный_1) с получением 4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-N-((транс)-4-(4-(циклопропилметил)пиперазин-1-ил)циклогексил)-3-метоксибензамида (0,11 г, 25%).
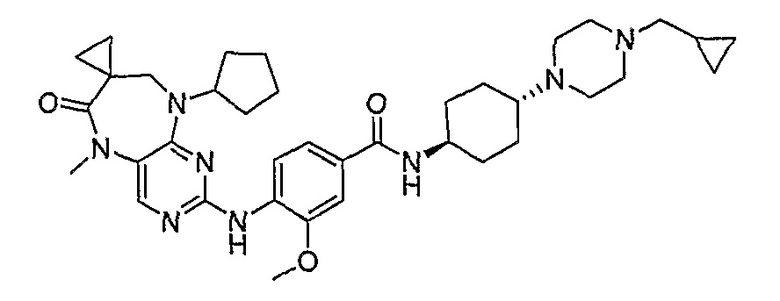
Rt = 3,18 мин (Аналитический_1); ES(+ve): 657,6; ES(-ve): 655,7; 1H-ЯМР (ДМСО-d6) δ м.д.: 0,05 (д, J=4,20 Гц, 3H), 0,44 (д, J=7,74 Гц, 2H), 0,67 (д, J=1,61 Гц, 2H), 0,74-0,85 (м, 1H), 0,90 (с, 2H), 1,23-1,44 (м, 3H), 1,44-1,55 (м, 2H), 1,54-1,64 (м, 2H), 1,64-1,74 (м, 2H), 1,88 (шир.с, 5H), 2,08 (с, 3H), 2,13 (д, J=6,45 Гц, 2H), 2,17-2,28 (м, 1H), 2,29-2,47 (м, 3H), 2,60-2,67 (м, 1H), 3,06-3,23 (м, 5H), 3,47 (с, 2H), 3,65-3,79 (м, 1H), 3,94 (с, 2H), 4,05-4,13 (м, 1H), 4,78-4,91 (м, 1H), 7,48 (с, 2H), 7,67 (с, 1H), 7,98 (с, 1H), 8,00-8,07 (м, 1H), 8,31-8,45 (м, 1H).
Соединение [372]: 4-(9-циклопентил-5,7,7-триметил-6-оксо-6,7,8,9-тетрагидро-5H-пиримидо[4,5-b][1,4]диазепин-2-иламино)-N-((транс)-4-(4-(циклопропилметил)пиперазин-1-ил)циклогексил)-3-метоксибензамид
2-хлор-9-циклопентил-5,7,7-триметил-8,9-дигидро-5H-пиримидо[4,5-b][1,4]диазепин-6(7H)-он (промежуточное соединение 3) (0,13 г, 0,43 ммоль, 1 экв.), 4-амино-N-(транс-4-(4-(циклопропилметил)пиперазин-1-ил)циклогексил)-3-метоксибензамид (промежуточное соединение 11) (0,26 г, 0,67 ммоль, 1,5 экв.) и TFA (0,17 мл, 2,2 ммоль, 5 экв.) в TFE (3 мл) нагревали при 80°C в течение 18 часов. Затем добавляли дополнительное количество промежуточного соединения 6 (0,13 г), и реакционную смесь снова нагревали в течение 48 часов. Реакционную смесь концентрировали в вакууме и остаток очищали колоночной флэш-хроматографией на диоксиде кремния, элюируя 0 → 20% аммиаком/MeOH в градиенте ДХМ, а затем препаративной ВЭЖХ (Препаративный_4) и наконец, препаративной ОФ-ВЭЖХ-МС (Препаративный_1) с получением 4-(9-циклопентил-5,7,7-триметил-6-оксо-6,7,8,9-тетрагидро-5H-пиримидо[4,5-b][1,4]диазепин-2-иламино)-N-((транс)-4-(4-(циклопропилметил)пиперазин-1-ил)циклогексил)-3-метоксибензамида (0,03 г, 7%).
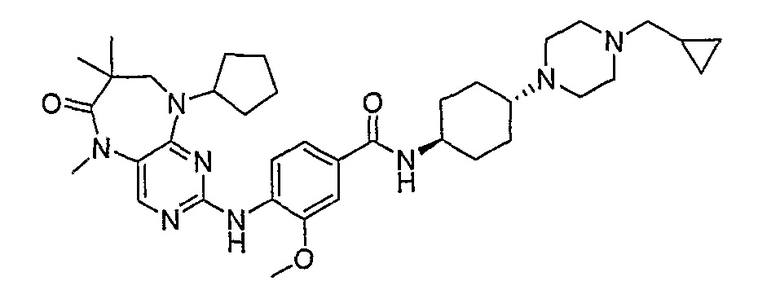
Rt = 3,38 мин (Аналитический_1); ES(+ve): 659,6; ES(-ve): 657,7; 1H-ЯМР (ДМСО-d6) δ м.д.: 0,05 (д, J=3,86 Гц, 3H), 0,40-0,48 (м, 3H), 0,73-0,84 (м, 2H), 1,03-1,13 (м, 10H), 1,18 (с, 1H), 1,24-1,43 (м, 6H), 1,61 (шир.с, 3H), 1,74 (д, J=9,65 Гц, 2H), 1,88 (шир.с, 5H), 2,08 (с, 5H), 2,13 (д, J=6,75 Гц, 2H), 2,17-2,25 (м, 1H), 2,36 (шир.с, 3H), 2,60-2,67 (м, 1H), 3,14-3,22 (м, 2H), 3,37 (с, 1H), 3,51 (с, 1H), 3,94 (с, 2H), 5,18 (т, J=8,52 Гц, 1H), 7,43-7,50 (м, 1H), 7,67 (с, 1H), 7,98 (с, 1H), 8,03 (д, J=7,72 Гц, 1H), 8,35 (д, J=8,36 Гц, 1H).
Альтернативно, для каждого специалиста в данной области очевидно, что соединения примеров 5, 6 могут быть синтезированы посредством реакции взаимодействия промежуточных соединений 5 или 6 с известным соединением транс-4-(4-циклопропилметилпиперазин-1-ил)циклогексиламином (описанным в заявке US 6861422 B2) в стандартных условиях образования амидной связи, таких как условия, описанные в примере 1.
Соединение [345]: 4-(9-циклопентил-5-метил-6-оксо-6,7,8,9-тетрагидро-5H-пиримидо[4,5-b][1,4]диазепин-2-иламино)-3-метокси-N-[1-(тетрагидропиран-4-ил)пиперидин-4-ил]бензамид
4-(9-циклопентил-5-метил-6-оксо-6,7,8,9-тетрагидро-5H-пиримидо[4,5-b][1,4]диазепин-2-иламино)-3-метокси-N-(пиперидин-4-ил)бензамид (промежуточное соединение 12) (35 мг, 0,07 ммоль, 1 экв.) и триацетоксиборгидрид натрия (18 мг, 0,084 ммоль, 1,2 экв.) в ДХМ (0,5 мл) подвергали реакции взаимодействия с тетрагидропиран-4-оном (6,5 мкл, 0,07 ммоль, 1 экв.) при комнатной температуре в течение 2 дней. Затем снова добавляли триацетоксиборгидрид натрия (18 мг, 1,2 экв.), тетрагидропиран-4-он (6,5 мкл, 1 экв.) и уксусную кислоту (4 мкл, 1 экв.). Через два дня реакционную смесь разбавляли ДХМ, промывали водой и органический слой концентрировали в вакууме. Полученный остаток очищали препаративной ОФ-ВЭЖХ-МС (Препаративный_1) с получением 4-(9-циклопентил-5-метил-6-оксо-6,7,8,9-тетрагидро-5H-пиримидо[4,5-b][1,4]диазепин-2-иламино)-3-метокси-N-[1-(тетрагидропиран-4-ил)пиперидин-4-ил]бензамида (10 мг, 25%).
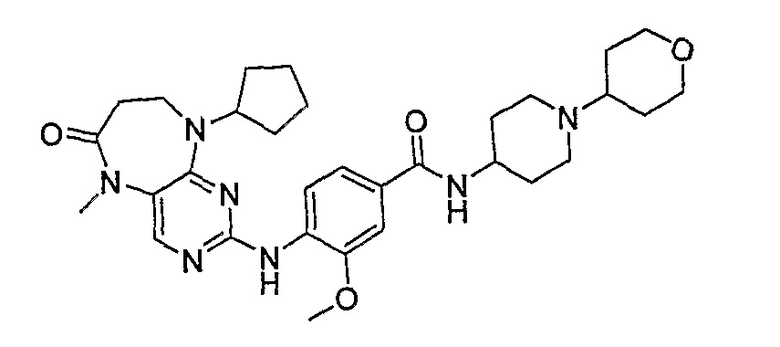
Rt = 2,74 мин (Аналитический_1); ES(+ve): 578,5; ES(-ve): 576,7.
Соединение [373]: 4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклобутан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-3-фтор-N-(4-метилпиперазин-1-ил)бензамид
4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклобутан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-3-фторбензойную кислоту (промежуточное соединение 10) (100 мг, 0,23 ммоль, 1 экв.), DIPEA (80 мкл, 0,46 ммоль, 2 экв.) и TBTU (80 мг, 0,25 ммоль, 1,1 экв.) добавляли к 1 мл ДМФ, и полученный раствор перемешивали при комнатной температуре в течение 10 минут, а затем добавляли 1-амино-4-метилпиперазин (33 мкл, 0,27 ммоль, 1,2 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, а затем разделяли на две равные партии и очищали препаративной ОФ-ВЭЖХ-МС (Препаративный_3) с получением 4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклобутан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-3-фтор-N-(4-метилпиперазин-1-ил)бензамид (0,05 г, 41%).
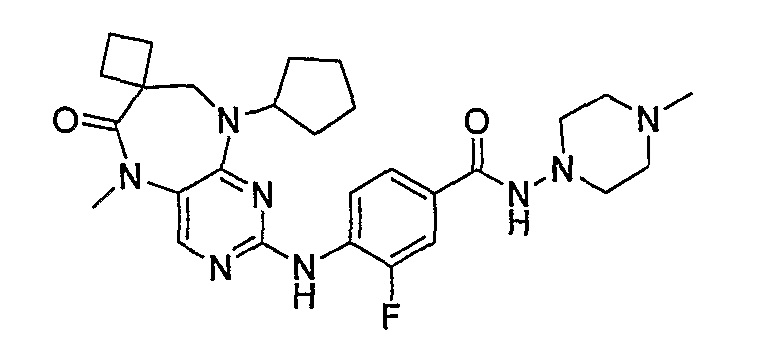
Rt = 2,74 мин (Аналитический_1); МС(+ve): 537,5; 1H-ЯМР (ДМСО-d6) δ м.д.: 1,55-1,65 (8H, м), 1,71 (2H, м), 1,88 (2H, м), 2,18 (3H, с), 2,28 (2H, кв., J=10,5 Гц), 2,41-2,52 (4H, м), 2,89 (4H, м), 3,18 (3H, с), 3,61 (2H, с), 4,76 (1H, квинтет, 8,5 Гц), 7,61 (2H, м), 8,03 (1H, с), 8,12 (1H, дд, J=8 Гц, 8,5 Гц), 8,76 (1H, с), 9,36 (1H, с).
Соединение [374]: 4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-3-фтор-N-(4-метилпиперазин-1-ил)бензамид
4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-3-фторбензойную кислоту (промежуточное соединение 9) (0,27 г, 0,63 ммоль, 1 экв.), DIPEA (0,21 мл, 1,26 ммоль, 2 экв.) и TBTU (0,22 г, 0,69 ммоль, 1,1 экв.) добавляли к 5 мл ДХМ и полученный раствор перемешивали при комнатной температуре в течение 30 минут, а затем добавляли 1-амино-4-метилпиперазин (91 мкл, 0,75 ммоль, 1,2 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 4 часов, а затем промывали водой, концентрировали в вакууме и остаток очищали с помощью препаративной ОФ-ВЭЖХ-МС (Препаративный_1) с получением 4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-3-фтор-N-(4-метилпиперазин-1-ил)бензамида (0,14 г, 44%).
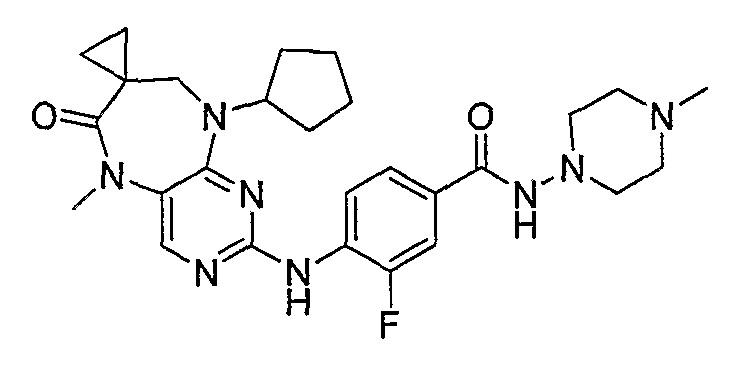
Rt = 2,58 мин (Аналитический_1); МС(+ve): 523,4; 1H-ЯМР (ДМСО-d6) δ м.д.: 0,65-0,67 (2H, с, CH2), 0,85-0,95 (2H, м, CH2), 1,23-1,82 (8H, м, алкил-H), 2,18 (3H, с, CH3), 2,36-2,41 (4H, м, алкил-H), 2,88 (4H, с, алкил-H), 3,16 (3H, с, CH3), 3,44 (2H, с, алкил-H), 4,77-4,80 (1H, м, CH), 7,6 (2H, м, арил-H), 7,96 (1H, с, арил-H), 8,16 (1H, т, J=8 Гц, арил-H), 8,70 (1H, с, NH), 9,35 (1H, с, NH).
Соединение [194]: (±)-4-(9-циклопентил-5,7,7-триметил-6-оксо-6,7,8,9-тетрагидро-5H-пиримидо[4,5-b][1,4]диазепин-2-иламино)-3-метокси-N-(хинуклидин-3-ил)бензамид
4-(9-циклопентил-5,7,7-триметил-6-оксо-6,7,8,9-тетрагидро-5H-пиримидо[4,5-b][1,4]диазепин-2-иламино)-3-метоксибензойную кислоту (промежуточное соединение 6) (20 мг, 0,046 ммоль, 1 экв.), DIPEA (16 мкл, 0,091 ммоль, 2 экв.) и TBTU (16 мг, 0,05 ммоль, 1,1 экв.) добавляли к 0,5 мл ДМФ, и полученный раствор перемешивали при комнатной температуре в течение 5 минут, а затем добавляли дигидрохлорид 3-аминохинуклидина (11 мг, 0,055 ммоль, 1 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов, а затем очищали с помощью препаративной ОФ-ВЭЖХ-МС (Препаративный_1) с получением (±)-4-(9-циклопентил-5,7,7-триметил-6-оксо-6,7,8,9-тетрагидро-5H-пиримидо[4,5-b][1,4]диазепин-2-иламино)-3-метокси-N-(хинуклидин-3-ил)бензамида (8 мг, 32%).
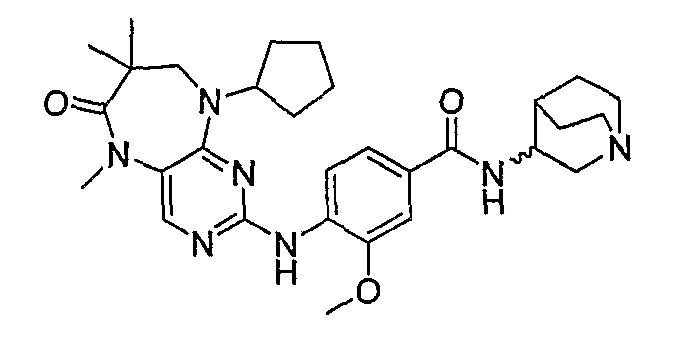
Rt = 3,28 мин (Аналитический_1); МС(+ve): 548,5; 1Н-ЯМР (ДМСО-d6) δ м.д.: 8,37 (1H, д, J=8 Гц), 8,07 (1H, д, J=6,5 Гц), 7,98 (1H, с), 7,69 (1H, с), 7,47-7,50 (2H, м), 5,19 (1H, квин., J=8 Гц), 3,95 (4H, м), 3,37 (2H, с), 3,18 (3H, с), 3,11 (1H, м), 2,89 (1H, м), 2,65-2,72 (4H, м), 1,88 (2H, м), 1,73-1,87 (4H, м), 1,57-1,61 (6H, м), 1,31 (1H, м), 1,09 (6H, с).
Соединение [186]: (±)-4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-3-метокси-N-(хинуклидин-3-ил)бензамид
4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-3-метоксибензойную кислоту (промежуточное соединение 5) (25 мг, 0,057 ммоль, 1 экв.), DIPEA (20 мкл, 0,11 ммоль, 2 экв.) и TBTU (20 мг, 0,063 ммоль, 1,1 экв.) добавляли к 0,5 мл ДМФ, и полученный раствор перемешивали при комнатной температуре в течение 15 минут, а затем добавляли дигидрохлорид 3-аминохинуклидина (11 мг, 0,055 ммоль, 1,1 экв.) и DIPEA (40 мкл). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов, а затем очищали с помощью препаративной ОФ-ВЭЖХ-МС (Препаративный_1) с получением (±)-4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-3-метокси-N-(хинуклидин-3-ил)бензамида.
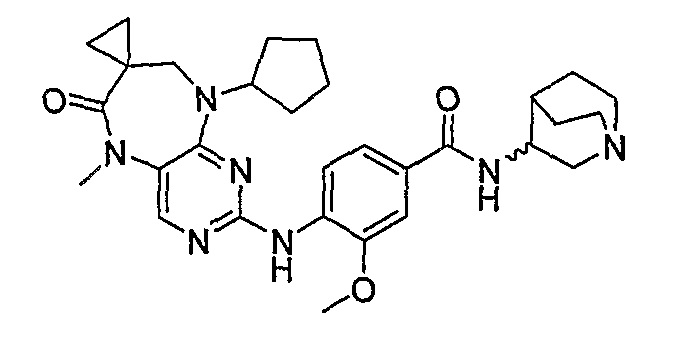
Rt = 3,02 мин (Аналитический_1); МС(+ve): 546,4; 1Н-ЯМР (ДМСО-d6) δ м.д.: 8,40 (1H, д, J=8 Гц), 8,08 (1H, д, J=7 Гц), 7,98 (1H, с), 7,69 (1H, с), 7,47-7,50 (2H, м), 4,87 (1H, квинт., J=9 Гц), 4,10 (1H, кв., J=5,5 Гц), 3,95 (4H, м), 3,47 (2H, с), 3,27 (3H, с), 3,07 (1H, м), 2,08 (1H, м), 2,63-2,86 (4H, м), 1,88 (3H, м), 1,78 (1H, м), 1,68 (2H, м), 1,50-1,62 (5H, м), 1,32 (1H, м), 0,90 (2H, м), 0,66 (2H, м).
Соединение [375]: 4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-N-(4-(2-гидроксиэтил)пиперазин-1-ил)-3-метоксибензамид
4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-3-метоксибензойную кислоту (промежуточное соединение 5) (48 мг, 0,11 ммоль, 1 экв.), DIPEA (36 мкл, 0,22 ммоль, 2 экв.) и TBTU (39 мг, 0,12 ммоль, 1,1 экв.) добавляли к 0,5 мл ДМФ, и полученный раствор перемешивали при комнатной температуре в течение 15 минут, а затем добавляли 2-(4-аминопиперазин-1-ил)этанол (19 мг, 0,13 ммоль, 1,2 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 15 часов, а затем концентрировали в вакууме и очищали с помощью препаративной ОФ-ВЭЖХ-МС (Препаративный_2) с получением 4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-N-(4-(2-гидроксиэтил)пиперазин-1-ил)-3-метоксибензамида (24 мг, 39%).
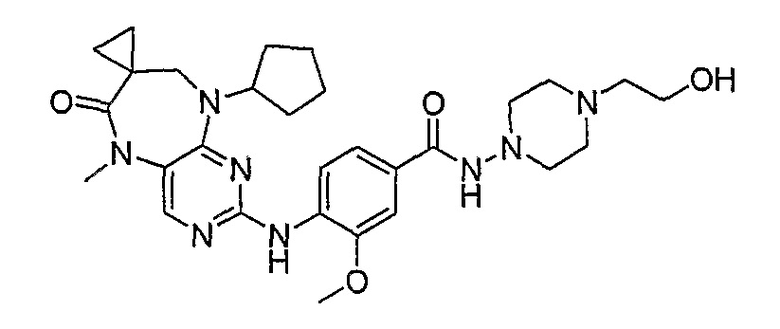
Rt = 2,48 мин (Аналитический_1); МС(+ve): 565,4; 1Н-ЯМР (ДМСО) δ м.д.: 0,66-0,67 (2H, м, CH2), 0,88-0,90 (2H, м, CH2), 1,23-1,90 (8H, м, алкил-H), 2,34-2,41 (4H, м, алкил-H), 2,91 (4H, т, J=4 Гц, алкил-H), 3,16 (3H, с, CH3), 3,46-3,51 (4H, м), 3,93 (3H, с, CH3), 4,38-4,45 (1H, м), 4,81-4,87 (1H, м, CH), 7,40 (2H, д, J=10,5 Гц, арил-H), 7,67 (1H, с), 7,98 (1H, с), 8,39 (1H, д, J=8 Гц, арил-H), 9,30 (1H, с, NH).
Соединение [376]: 4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-3-метокси-N-морфолинобензамид
4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-3-метоксибензойную кислоту (промежуточное соединение 5) (50 мг, 0,11 ммоль, 1 экв.), DIPEA (38 мкл, 0,23 ммоль, 2 экв.) и TBTU (40 мг, 0,12 ммоль, 1,1 экв.) добавляли к 5 мл ДХМ, и полученный раствор перемешивали при комнатной температуре в течение 15 минут, а затем добавляли 4-аминоморфолин (12 мг, 0,14 ммоль, 1,2 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов, а затем концентрировали в вакууме и очищали с помощью препаративной ОФ-ВЭЖХ-МС (Препаративный_1) с получением 4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-3-метокси-N-морфолинобензамида (26 мг, 44%).
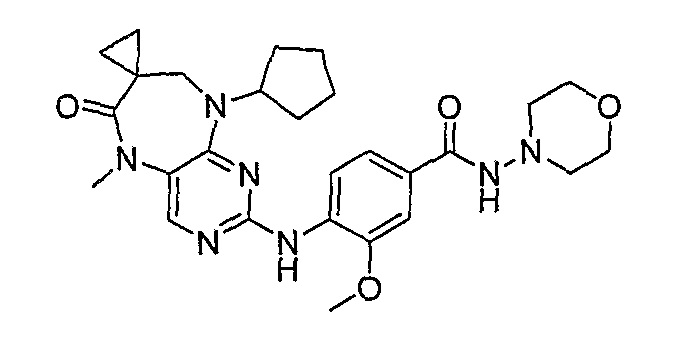
Rt = 2,72 мин (Аналитический_1); МС(+ve): 522,4; 1Н-ЯМР (ДМСО) δ м.д.: 0,65-0,71 (2H, м, CH2), 0,85-0,91 (2H, м, CH2), 1,23-1,88 (8H, м, алкил-H), 2,92 (4H, т, J=4 Гц, алкил-H), 3,16 (3H, с, CH3), 3,47 (2H, с, алкил-H), 3,66 (4H, т, J=4,5 Гц), 3,94 (3H, с, CH3), 4,81-4,87 (1H, м, CH), 7,41 (2H, д, J=9,5 Гц ), 7,69 (1H, с), 7,98 (1H, с), 8,40 (1H, д, J=8 Гц), 9,41 (1H, с, NH).
Соединение [347]: (R)-4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-3-метокси-N-(хинуклидин-3-ил)бензамид
4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-3-метоксибензойную кислоту (Промежуточное соединение 5) (0,11 г, 0,25 ммоль, 1 экв.), DIPEA (0,17 мл) и TBTU (88 мг, 0,28 ммоль, 1,1 экв.) добавляли к 5 мл ДХМ, и полученный раствор перемешивали при комнатной температуре в течение 30 минут, а затем добавляли дигидрохлорид (R)-(+)-3-аминохинуклидина (60 мг, 0,30 ммоль, 1,2 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов, а затем разбавляли ДХМ и последовательно промывали водой (×2), 50% насыщенным водным бикарбонатом натрия, насыщенным водным бикарбонатом натрия и насыщенным раствором соли. Органический слой сушили (MgSO4) и концентрировали в вакууме. Остаток растворяли в минимальном количестве EtOAc, а затем добавляли н-гептан для осаждения (R)-4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-3-метокси-N-(хинуклидин-3-ил)бензамида (белое твердое вещество, 0,13 г, 96%).
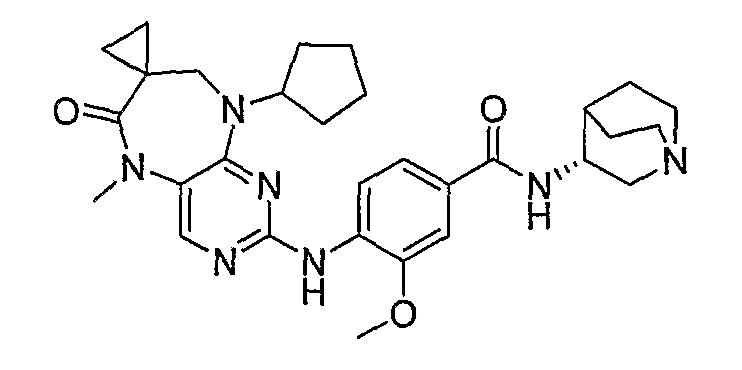
Rt = 3,07 мин (Аналитический_1); МС(+ve): 546,5; МС(-ve): 544,6; 1Н-ЯМР (ДМСО-d6) δ м.д. 0,62-0,72 (2H, м), 0,87-0,94 (2H, м), 1,20-1,38 (1H, м), 1,44-1,54 (2H, м), 1,54-1,64 (4H, м), 1,64-1,74 (2H, м), 1,75-1,85 (1H, м), 1,85-1,95 (3H, м), 2,65-2,79 (4H, м), 2,83-3,01 (1H, м), 3,11-3,21 (4H, м), 3,47 (2H, с), 3,95 (4H, с), 4,77-4,93 (1H, м), 7,42-7,54 (2H, м), 7,69 (1H, с), 7,98 (1H, с), 8,10 (1H, д, J=6,3 Гц), 8,39 (1H, д, J=8,3 Гц).
Соединение [348]: (S)-4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-3-метокси-N-(хинуклидин-3-ил)бензамид
4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-3-метоксибензойную кислоту (промежуточное соединение 5) (0,11 г, 0,25 ммоль, 1 экв.), DIPEA (0,17 мл) и TBTU (88 мг, 0,28 ммоль, 1,1 экв.) добавляли к 5 мл ДХМ, и полученный раствор перемешивали при комнатной температуре в течение 30 минут, а затем добавляли дигидрохлорид (S)-(-)-3-аминохинуклидина (60 мг, 0,30 ммоль, 1,2 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов, а затем разбавляли ДХМ и последовательно промывали водой (×2), 50% насыщенным водным бикарбонатом натрия, насыщенным водным бикарбонатом натрия и насыщенным раствором соли. Органический слой сушили (MgSO4) и концентрировали в вакууме. Остаток растворяли в минимальном количестве EtOAc, а затем добавляли н-гептан для осаждения (S)-4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-3-метокси-N-(хинуклидин-3-ил)бензамида (белое твердое вещество, 0,075 г, 55%).
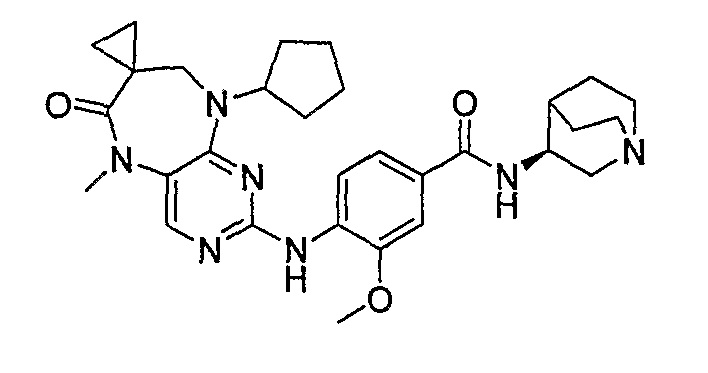
Rt = 3,04 мин (Аналитический_1); МС(+ve): 546,5; МС(-ve): 544,6; 1Н-ЯМР (ДМСО-d6) δ м.д.: 0,59-0,74 (2H, м), 0,86-0,97 (2H, м), 1,20-1,43 (1H, м), 1,45-1,75 (7H, м), 1,76-1,98 (4H, м), 2,66-2,86 (3H, м), 2,89-3,07 (1H, м), 3,17 (3H, с), 3,47 (3H, с), 3,89-4,02 (4H, м), 4,76-4,96 (1H, м), 7,42-7,59 (2H, м), 7,70 (1H, с), 7,98 (1H, с), 8,14 (1H, д, J=6,3 Гц), 8,40 (1H, д, J=7,8 Гц).
Соединение [377]: 4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклобутан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-N-((транс)-4-(4-(циклопропилметил)пиперазин-1-ил)циклогексил)-3-метоксибензамид
2’-хлор-9’-циклопентил-5’-метил-8’,9’-дигидроспиро[циклобутан-1,7’-пиримидо[4,5-b][1,4]диазепин]-6’(5Η)-он (промежуточное соединение 4) (0,065 г, 0,2 ммоль, 1 экв.), 4-амино-N-(транс-4-(4-(циклопропилметил)пиперазин-1-ил)циклогексил)-3–метоксибензамид (промежуточное соединение 11) (0,24 г, 0,6 ммоль, 3 экв.) и TFA (75 мкл, 1 ммоль, 5 экв.) в TFE (6 мл) кипятили с обратным холодильником в течение 40 часов. Реакционную смесь концентрировали в вакууме и остаток распределяли между EtOAc и насыщенным бикарбонатом натрия. Органический слой отделяли, промывали насыщенным раствором соли и концентрировали в вакууме с получением остатка, который очищали препаративной ОФ-ВЭЖХ-МС (Препаративный_1) с получением 4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклобутан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-N-((транс)-4-(4-(циклопропилметил)пиперазин-1-ил)циклогексил)-3-метоксибензамида.
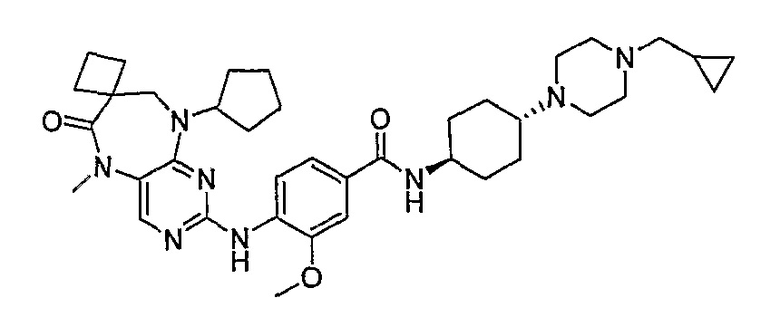
Rt = 3,41 мин (Аналитический_1); ES(+ve): 671,6; ES(-ve): 669,7; 1H-ЯМР (ДМСО-d6) δ м.д.: 0,03 (2H, дд, J=9,5 Гц, 5 Гц), 0,41 (2H, дд, J=10 Гц, 5 Гц), 0,79 (1H, м), 1,20-1,37 (4H, м), 1,55-1,94 (18H, м), 2,04 (2H, д, J=6,5 Гц), 2,16-2,46 (8H, м), 3,15 (3H, с), 3,32 (2H, с), 3,69 (1H, м), 3,90 (3H, с), 4,79 (1H, м), 7,44 (2H, м), 7,68 (1H, с), 8,01 (2H, м), 8,34 (1H, д, J=8,5 Гц).
Соединение [378]: 4-(9-циклопентил-5-метил-6-оксо-6,7,8,9-тетрагидро-5H-пиримидо[4,5-b][1,4]диазепин-2-иламино)-N-((транс)-4-(4-(циклопропилметил)пиперазин-1-ил)циклогексил)-3-метоксибензамид
2-хлор-9-циклопентил-5-метил-8,9-дигидро-5H-пиримидо[4,5-b][1,4]диазепин-6(7H)-он (промежуточное соединение 1) (0,13 г, 0,43 ммоль, 1 экв.), 4-амино-N-(транс-4-(4- (циклопропилметил)пиперазин-1-ил)циклогексил)-3-метоксибензамид (промежуточное соединение 11) (0,26 г, 0,67 ммоль, 1,5 экв.) и TFA (0,17 мл, 2,2 ммоль, 5 экв.) в TFE (3 мл) нагревали при 80°C в течение 18 часов. Затем добавляли дополнительное количество промежуточного соединения 1 (0,10 г) и реакционную смесь снова нагревали в течение 48 часов. Реакционную смесь концентрировали в вакууме и остаток очищали колоночной флэш-хроматографией на диоксиде кремния, элюируя 0 → 20% аммиаком/MeOH в градиенте ДХМ, а затем препаративной ВЭЖХ (Препаративный_4), и наконец, препаративной ОФ-ВЭЖХ-МС (Препаративный_1) с получением 4-(9-циклопентил-5-метил-6-оксо-6,7,8,9-тетрагидро-5H-пиримидо[4,5-b][1,4]диазепин-2-иламино)-N-((транс)-4-(4-(циклопропилметил)пиперазин-1-ил)циклогексил)-3-метоксибензамида (0,11 г, 26%).
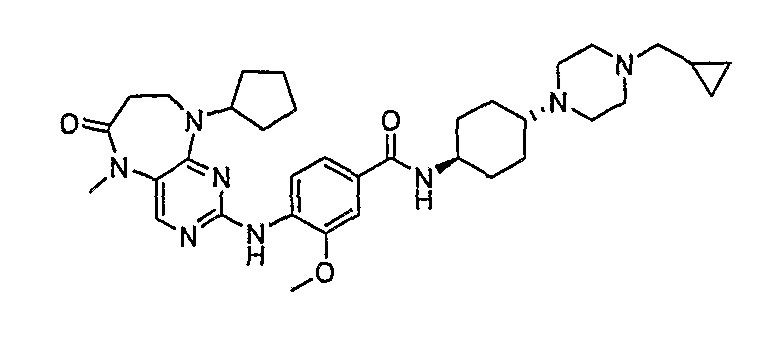
Rt = 2,88 мин (Аналитический_1); ES(+ve): 631,5; ES(-ve): 629,6; 1H-ЯМР (ДМСО-d6) δ м.д.: 0,15-0,11 (м, 2H), 0,40 (д, J=8,06 Гц, 2H), 0,63-0,85 (м, 1H), 1,11-1,41 (м, 3H), 1,57 (шир.с, 2H), 1,69 (д, J=17,73 Гц, 2H), 1,74-1,95 (м, 6H), 2,04 (с, 2H), 2,09 (д, J=6,45 Гц, 2H), 2,16 (д, J=9,67 Гц, 1H), 2,32 (шир.с, 2H), 2,49 (шир.с, 1H), 2,51-2,59 (м, 2H), 2,59 (шир.с, 1H), 3,07-3,17 (м, 3H), 3,27 (с, 5H), 3,53-3,62 (м, 2H), 3,67 (дд, J=7,41, 3,55 Гц, 1H), 3,89 (с, 2H), 4,04 (кв., J=5,37 Гц, 1H), 4,76 (квин., J=8,22 Гц, 1H), 7,38-7,48 (м, 2H), 7,68 (с, 1H), 7,99 (д, J=7,74 Гц, 1H), 8,03 (с, 1H), 8,33 (д, J=8,06 Гц, 1H).
Промежуточные соединения 13-19: 4-(аминозамещенные)циклогексанамины
1. [Соединение получали методом, аналогичным методу III, описанному в публикации Abdel-Magid A.F. et al. (1996) J. Org. Chem., 61, 3849-3862] триацетоксиборгидрид натрия (1,5 экв.), уксусная кислота (1 экв.), амин (1,1 экв.), ТГФ, к.т., 20 ч.; 2. H2, 10% Pd/C, 60°C, метанол, гидрирование в проточном реакторе; 3. иодид натрия (4 экв.), хлортриметилсилан (4 экв.), ацетонитрил, 0°C –к.т., 18 ч.
Стадия 1:
N-бензилоксикарбонил-4-аминоциклогексанон, соединение, известное специалистам [WO2007/002181 A2] (обычно в масштабе 1 ммоль), добавляли в реакционную пробирку, содержащую ТГФ (5 мл), соответствующий замещенный пиперазин, гомопиперазин или морфолин вместе с уксусной кислотой и триацетоксиборгидридом натрия. Реакционную смесь перемешивали при температуре окружающей среды в течение 20 часов. Реакцию гасили раствором NaHCO3 (2 мл), а затем смесь подкисляли до pH2 раствором 1н HCl. Смесь промывали EtOAc, а затем водный слой отделяли и подщелачивали 2н раствором NaOH до pH10. Продукт экстрагировали EtOAc, а затем промывали насыщенным NaCl, сушили (MgSO4) и выпаривали при пониженном давлении с получением нужного бензил-4-(аминозамещенного)циклогексилкарбамата.
Стадия 2:
Каждый бензил-4-(аминозамещенный)циклогексилкарбамат растворяли в метаноле до концентрации 0,05 М. Гидрирование осуществляли в проточном реакторе H-CubeТМ (ThalesNano Inc.) при скорости потока 1 мл/мин над катализатором 10% Pd/C, нагретым до 60°С при полном давлении водорода. После концентрирования при пониженном давлении получали нужный 4-(аминозамещенный)циклогексанаминовый продукт в виде масла. Твердые гидрохлоридные соли могут быть получены путем перемешивания с эфирным раствором HCl.
Если необходимо селективное снятие защиты бензилкарбаматов в присутствии N-бензильной группы (промежуточные соединения 16 и 18), то вместо стадии 2 проводили стадию 3.
Стадия 3:
Иодид натрия (1,5 ммоль, 4 экв.) растворяли в безводном ацетонитриле (5 мл) и раствор перемешивали при добавлении хлортриметилсилана (1,5 ммоль, 4 экв.). Через 5 минут, смесь медленно добавляли к раствору 4-(аминозамещенного)циклогексанамина, растворенного в безводном ацетонитриле (2 мл), охлажденном на бане со льдом-водой. Через 1 час охлаждающую баню удаляли и реакционную смесь перемешивали при температуре окружающей среды в течение 18 часов. Реакционную смесь концентрировали при пониженном давлении и снова растворяли в 1% растворе воды в метаноле. Смесь абсорбировали на колонке SCX II, промывали метанолом и элюировали аммиаком в метаноле. После концентрирования в вакууме получали нужный 4-(аминозамещенный)циклогексанамин.
Соединение [379]: 4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-N-((транс)-4-(4-этилпиперазин-1-ил)циклогексил)-3-метоксибензамид
4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-3-метоксибензойную кислоту (промежуточное соединение 5) (66 мг, 0,15 ммоль, 1 экв.), DIPEA (52 мкл, 0,3 ммоль, 2 экв.) и TBTU (54 мг, 0,17 ммоль, 1,1 экв.) добавляли к 1,5 мл ДМФ и полученный раствор перемешивали при комнатной температуре в течение 20 минут, а затем добавляли 4-(4-этилпиперазин-1-ил)циклогексанамин (промежуточное соединение 13) в виде тригидрохлоридной соли (58 мг, 0,18 ммоль, 1,2 экв.) и DIPEA (78 мкл, 0,45 ммоль, 3 экв.). Затем реакционную смесь перемешивали при комнатной температуре в течение 2 часов, после чего ее очищали препаративной ОФ-ВЭЖХ-МС (Препаративный_2) с получением 4-(9’-циклопентил-5’~метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро [циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-N-((транс)-4-(4-этилпиперазин-1-ил)циклогексил)-3-метоксибензамида (бесцветное стекловидное вещество, 26 мг, 28%).
Rt = 3,00 мин (Аналитический_1) МС(+ve) 631,6; МС(-ve) 629,7; Rt = 3,00 мин (Аналитический_1) МС(+ve) 631,6; МС(-ve) 629,7; 1H-ЯМР (ДМСО-d6) δ м.д.: 0,59-0,72 (2H, м), 0,81-0,92 (2H, м), 0,97 (3H, т, J=7,1 Гц), 1,20-1,42 (4H, м), 1,42-1,55 (2H, м), 1,54-1,64 (2H, м), 1,64-1,73 (2H, м), 1,83 (2H, д, J=11,7 Гц), 1,89 (4H, д, J=7,8 Гц), 2,14-2,42 (7H, м), 3,16 (3H, с), 3,47 (2H, с), 3,64-3,79 (1H, м), 3,94 (3H, с), 4,84 (1H, квин., J=8,7 Гц), 7,40-7,54 (2H, м), 7,67 (1H, с), 7,98 (1H, с), 8,03 (1H, д, J=7,8 Гц), 8,38 (1H, д, J=8,3 Гц).
Соединение [380]: 4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-N-((цис)-4-(4-этилпиперазин-1-ил)циклогексил)-3-метоксибензамид
После синтеза и очистки, соединение [379] выделяли как отдельное соединение 4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-N-((цис-(4-этилпиперазин-1-ил)циклогексил)-3-метоксибензамид (бесцветное стекловидное вещество, 27 мг, 29%).
Rt = 3,13 мин (Аналитический_1) МС(+ve) 631,6; МС(-ve) 629,7; 1Н-ЯМР (ДМСО-d6) δ м.д.: 0,60-0,72 (2H, м), 0,83-0,93 (2H, м), 0,98 (3H, т, J=7,1 Гц), 1,38-1,55 (6H, м), 1,55-1,63 (2H, м), 1,63-1,80 (4H, м), 1,80-1,96 (4H, м), 2,02-2,17 (1H, м), 2,16-2,44 (7H, м), 3,16 (3H, с), 3,47 (2H, с), 3,79-3,92 (1H, м), 3,94 (3H, с), 4,84 (1H, квин., J=8,5 Гц), 7,44-7,58 (2H, м), 7,67 (1H, с), 7,98 (1H, с), 8,03 (1H, д, J=7,3 Гц), 8,37 (2H, д, J=8,8 Гц).
Соединение [381]: 4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-N-(4-(4-(циклопропилметил)-1,4-диазепан-1-ил)циклогексил)-3-метоксибензамид
4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-3-метоксибензойную кислоту (промежуточное соединение 5) (88 мг, 0,2 ммоль, 1 экв.), DIPEA (70 мкл, 0,4 ммоль, 2 экв.) и TBTU (72 мг, 0,24 ммоль, 1,2 экв.) добавляли к 2 мл ДМФ, и полученный раствор перемешивали при комнатной температуре в течение 20 мин, а затем добавляли 4-(4-(циклопропилметил)-1,4-диазепан-1-ил)циклогексанамин (промежуточное соединение 14) (60 мг, 0,24 ммоль, 1,2 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, а затем очищали с помощью препаративной ОФ-ВЭЖХ-МС (Препаративный_2) с получением 4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-N-(4-(4-(циклопропилметил)-1,4-диазепан-1-ил)циклогексил)-3-метоксибензамида в виде смеси цис- и транс-изомеров (не совсем белое твердое вещество, 67 мг, 50%).
Rt = 4,12 мин (широкий пик) (Аналитический_1) МС(+ve) 671,6; МС(-ve) 669,7; 1Н-ЯМР (ДМСО-d6) δ м.д.: 0,01 (2H, д, J=4,4 Гц), 0,40 (2H, д, J=7,3 Гц), 0,63 (2H, шир.с), 0,71-0,83 (1H, м), 0,87 (2H, шир.с), 1,02 (1H, т, J=7,1 Гц), 1,25-1,38 (2,5H, м), 1,39-1,52 (3,5H, м), 1,52-1,61 (2H, м), 1,65 (4H, д, J=5,9 Гц), 1,70-1,80 (3H, м), 1,85 (3H, д, J=4,4 Гц), 2,21-2,31 (1H, м), 2,55-2,83 (8H, м), 3,13 (3,5H, с), 3,35-3,55 (3H, м), 3,69 (0,5H, шир.с), 3,91 (3,5H, с), 4,31 (0,5H, т, J=5,1 Гц), 4,81 (1H, 1, J=8,5 Гц), 7,25-7,54 (2H, м), 7,64 (1H, с), 7,84-8,16 (2H, м), 8,35 (1H, д, J=8,3 Гц).
Соединение [382]: 4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’, 8’,9’-тетрагидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-N-(4-(4-этил-1,4-диазепан-1-ил)циклогексил)-3-метоксибензамид
4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-3-метоксибензойную кислоту (промежуточное соединение 5) (44 мг, 0,1 ммоль, 1 экв.), DIPEA (35 мкл, 0,2 ммоль, 2 экв.) и TBTU (36 мг, 0,12 ммоль, 1,2 экв.) добавляли к 1 мл ДМФ, и полученный раствор перемешивали при комнатной температуре в течение 20 мин, а затем добавляли 4-(4-этил-1,4-диазепан-1-ил)циклогексанамин (промежуточное соединение 15) в виде тригидрохлоридной соли (32 мг, 0,1 ммоль, 1 экв.) и DIPEA (53 мкл, 0,3 ммоль, 3 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, а затем очищали с помощью препаративной ОФ-ВЭЖХ-МС (Препаративный_2) с получением 4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-N-(4-(4-этил-1,4-диазепан-1-ил)циклогексил)-3-метоксибензармида в виде смеси цис- и транс-изомеров (белое твердое вещество, 10 мг, 16%).
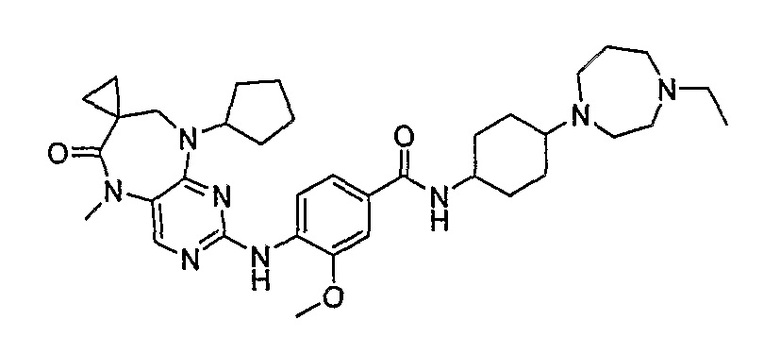
Rt = 3,98 мин, 4,18 мин (широкие пики) (Аналитический_1) МС(+ve) 645,6; МС(-ve) 643,7; 1Н-ЯМР (ДМСО-d6) δ м.д.: 0,57-0,74 (2H, м), 0,82-0,93 (2H, м), 0,95-1,09 (3H, м), 1,30-1,42 (2H, м), 1,42-1,54 (3,5H, м), 1,58 (2H, д, J=4,4 Гц), 1,63-1,75 (3,5H, м), 1,75-1,83 (1,5H, м), 1,88 (3,5H, шир.с), 1,98-2,16 (1,5H, м), 2,59-2,86 (4H, м), 3,17 (4H, с), 3,41-3,53 (3,5H, м), 3,78 (2,5H, м), 3,88-4,02 (3,5H, м), 4,06-4,38 (1H, м), 4,76-4,91 (1H, м), 7,39-7,57 (2H, м), 7,59-7,78 (1H, м), 7,98 (2H, с), 8,31-8,45 (1H, м).
Соединение [383]: N-(4-(4-бензил-1,4-диазепан-1-ил)циклогексил)-4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-3-метоксибензамид
4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-3-метоксибензойную кислоту (промежуточное соединение 5) (66 мг, 0,15 ммоль, 1 экв.), DIPEA (52 мкл, 0,3 ммоль, 2 экв.) и TBTU (54 мг, 0,17 ммоль, 1,1 экв.) добавляли к 1 мл ДМФ, и полученный раствор перемешивали при комнатной температуре в течение 20 мин, а затем добавляли 4-(4-бензил-1,4-диазепан-1-ил)циклогексанамин (промежуточное соединение 16) (54 мг, 0,19 ммоль, 1,27 экв.), растворенный в ДМФ (0,5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, а затем очищали с помощью препаративной ОФ-ВЭЖХ-МС (Препаративный_5) с получением N-(4-(4-бензил-1,4-диазепан-1-ил)циклогексил)-4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-3-метоксибензамида в виде смеси цис- и транс-изомеров (не совсем белое твердое вещество, 33 мг, 31%).
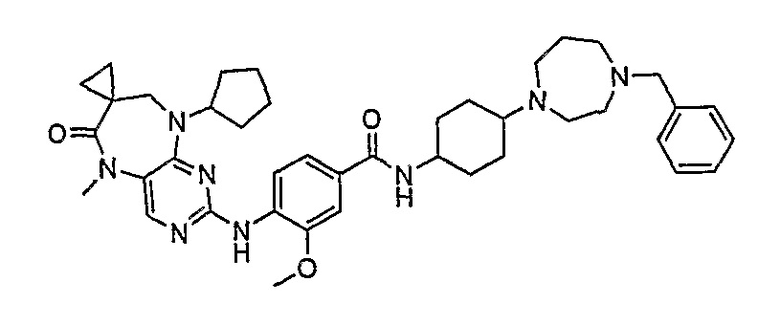
Rt = 4,40 мин (Аналитический_1) МС(+ve) 707,6; МС(-ve) 705,7; 1Н-ЯМР (ДМСО-d6) δ м.д.: 0,73 (2H, шир.с), 0,96 (2H, шир.с), 1,22-2,22 (19H, м), 2,64-2,91 (4H, м), 3,23 (5H, с), 3,53 (8H, с), 3,89-4,24 (4H, м). 4,91 (1H, т, J=8,5 Гц), 5,82 (0H, с), 7,23-8,54 (10H, м).
Соединение [384]: 4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’- тетрагидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-N-((транс)-4-(4-метилпиперазин-1-ил)циклогексил)-3-метоксибензамид
4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-3-метоксибензойную кислоту (промежуточное соединение 5) (66 мг, 0,15 ммоль, 1 экв.), DIPEA (52 мкл, 0,3 ммоль, 2 экв.) и TBTU (54 мг, 0,17 ммоль, 1,1 экв.) добавляли к 1 мл ДМФ, и полученный раствор перемешивали при комнатной температуре в течение 20 мин, а затем добавляли 4-(4-метилпиперазин-1-ил)циклогексанамин (промежуточное соединение 17) (35 мг, 0,18 ммоль, 1,2 экв.), растворенный в ДМФ (0,5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, а затем очищали с помощью препаративной ОФ-ВЭЖХ-МС (Препаративный_2 и Препаративный_3) с получением 4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-N-((транс)-4-(4-метилпиперазин-1-ил)циклогексил)-3-метоксибензамида (белое твердое вещество, 17 мг, 18%).

Rt = 2,85 мин (Аналитический_1) МС(+ve) 617,5; МС(-ve) 615,6; 1Н-ЯМР (ДМСО-d6) δ м.д.: 0,61-0,76 (2H, м), 0,89 (1H, т, J=6,8 Гц), 0,92-0,98 (2H, м), 1,22-1,47 (5H, м), 1,47-1,79 (6H, м), 1,81-2,02 (5H, м), 2,07-2,20 (3H, м), 2,20-2,42 (4H, м), 3,20 (4H, с), 3,34 (3H, с), 3,51 (2H, с), 3,67-3,82 (1H, м), 3,98 (3H, с), 4,88 (1H, квин., J=8,5 Гц), 7,42-7,59 (2H, м), 7,71 (1H, с), 8,02 (1H, с), 8,08 (1H, д, J=7,8 Гц), 8,42 (1H, д, J=8,3 Гц).
Соединение [385]: 4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-N-((цис)-4-(4-метилпиперазин-1-ил)циклогексил)-3-метоксибензамид
После синтеза и очистки было выделено отдельное соединение [384], а именно, 4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-N-((цис)-4-(4-метилпиперазин-1-ил)циклогексил)-3-метоксибензамид (белое твердое вещество, 34 мг, 37%).

Rt = 2,93 мин (Аналитический_1) МС(+ve) 617,5; МС(-ve) 615,6; 1Н-ЯМР (ДМСО-d6) δ м.д.: 0,62-0,80 (2H, м), 0,84-1,02 (4H, м), 1,18-1,36 (4H, м), 1,43-1,84 (12H, м), 1,84-2,03 (4H, м), 2,09-2,22 (4H, м), 2,41 (4H, шир.с), 3,21 (3H, с), 3,52 (2H, с), 3,89-3,97 (1H, м), 3,99 (3H, с), 4,89 (1H, квин., J=8,5 Гц), 7,47-7,61 (2H, м), 7,72 (1H, с), 8,03 (1H, с), 8,08 (1H, д, J=7,3 Гц), 8,43 (1H, д, J=9,3 Гц).
Соединение [386]: N-((транс)-4-(4-бензилпиперазин-1-ил)циклогексил)-4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-3-метоксибензамид
4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-3-метоксибензойную кислоту (промежуточное соединение 5) (66 мг, 0,15 ммоль, 1 экв.), DIPEA (52 мкл, 0,3 ммоль, 2 экв.) и TBTU (54 мг, 0,17 ммоль, 1,1 экв.) добавляли к 1 мл ДМФ, и полученный раствор перемешивали при комнатной температуре в течение 20 минут, а затем добавляли 4-(4-бензилпиперазин-1-ил)циклогексанамин (промежуточное соединение 18) (35 мг, 0,18 ммоль, 1,2 экв.), растворенный в ДМФ (0,5 мл). Затем реакционную смесь перемешивали при комнатной температуре в течение 2 часов, после чего ее очищали препаративной ОФ-ВЭЖХ-МС (Препаративный_2) с получением N-((транс)-4-(4-бензилпиперазин-1-ил)циклогексил)-4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-3-метоксибензамида (белое твердое вещество, 13 мг, 13%).
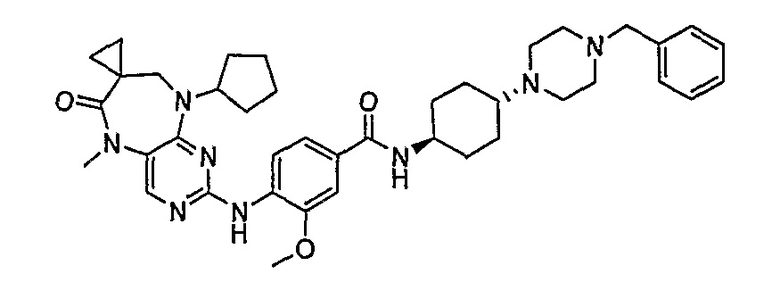
Rt = 3,64 мин (Аналитический_1) МС(+ve) 693,6; МС(-ve) 691,7; 1H-ЯМР (ДМСО-d6) δ м.д.: 0,62-0,79 (2H, м), 0,85-1,02 (2H, м), 1,20-1,46 (4H, м), 1,46-1,58 (2H, м), 1,59-1,67 (2H, м), 1,67-1,78 (2H, м), 1,80-2,02 (6H, м), 2,12 (1H, с), 2,18-2,46 (6H, м), 3,20 (3H, с), 3,43-3,56 (5H, м), 3,67-3,84 (1H, м), 3,97 (3H, с), 4,88 (1H, квин., J=8,4 Гц), 7,22-7,42 (6H, м), 7,44-7,59 (2H, м), 7,71 (1H, с), 8,02 (1H, с), 8,08 (1H, д, J=7,8 Гц), 8,42 (1H, д, J=7,8 Гц).
Соединение [387]: N-((цис)-4-(4-бензилпиперазин-1-ил)циклогексил)-4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-3-метоксибензамид
В результате синтеза и очистки выделяли отдельное соединение [386], а именно, N-((цис)-4-(4-бензилпиперазин-1-ил)циклогексил)-4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-3-метоксибензамид (белое твердое вещество, 20 мг, 19%).
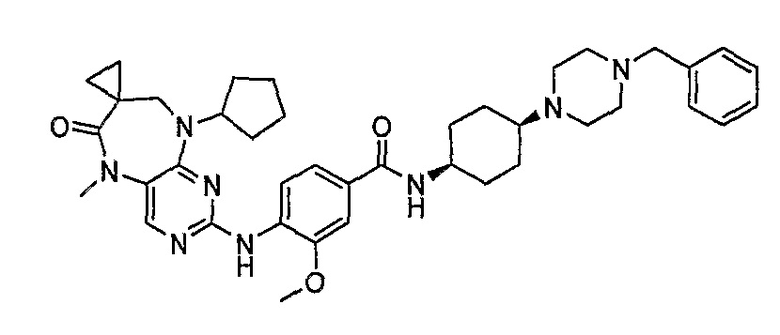
Rt = 3,85 мин (Аналитический_1) МС(+ve) 693,6; МС(-ve) 691,7; 1H-ЯМР (ДМСО-d6) δ м.д.: 0,62-0,79 (2H, м), 0,87-1,01 (2H, м), 1,43-1,59 (5H, м), 1,59-1,67 (2H, м), 1,66-1,74 (2H, м), 1,75-1,84 (2H, м), 1,92 (4H, шир.с), 2,07-2,29 (2H, м), 2,41 (4H, шир.с), 3,20 (3H, с), 3,51 (4H, с), 3,87-4,05 (4H, м), 4,74-4,99 (1H, м), 7,24-7,39 (5H, м), 7,50-7,56 (2H, м), 7,71 (1H, с), 8,02 (1H, с), 8,07 (1H, д, J=6,8 Гц), 8,42 (1H, д, J=8,8 Гц).
Соединение [388]: 4-(9-циклопентил-5,7,7-триметил-6-оксо-6, 7,8,9-тетрагидро-5H-пиримидо[4,5-b][1,4]диазепин-2-иламино)-3-метокси-N-((транс)-4-морфолинциклогексил)бензамид
4-(9-циклопентил-5,7,7-триметил-6-оксо-6,7,8,9-тетрагидро-5H-пиримидо[4,5-b][1,4]диазепин-2-иламино)-3-метоксибензойную кислоту (промежуточное соединение 6) (66 мг, 0,15 ммоль, 1 экв.), DIPEA (52 мкл, 0,3 ммоль, 2 экв.) и TBTU (54 мг, 0,17 ммоль, 1,1 экв.) добавляли к 1,5 мл ДМФ, и полученный раствор перемешивали при комнатной температуре в течение 20 минут, а затем добавляли 4-морфолиноциклогексанамин (промежуточное соединение 16) (33 мг, 0,18 ммоль, 1,2 экв.). Затем реакционную смесь перемешивали при комнатной температуре в течение 2 часов и очищали препаративной ОФ-ВЭЖХ-МС (Препаративный_2) с получением 4-(9-циклопентил-5,7,7-триметил-6-оксо-6,7,8,9-тетрагидро-5H-пиримидо[4,5-b][1,4]диазепин-2-иламино)-3-метокси-N-((транс)-4-морфолиноциклогексил)бензамида (не совсем белое твердое вещество, 10 мг, 11%).

Rt = 3,16 мин (Аналитический_1) МС(+ve) 606,4; МС(-ve) 604,5; 1H-ЯМР (ДМСО-d6) δ м.д.: 1,07-1,18 (7H, м), 1,24-1,49 (4H, м), 1,58-1,71 (4H, м), 1,71-1,84 (2H, м), 1,83-2,03 (6H, м), 2,23 (1H, т, J=10,5 Гц), 2,43-2,51 (3H, м), 3,17-3,28 (4H, м), 3,42 (3H, шир.с), 3,60 (4H, шир.с), 3,64 (1H, шир.с), 3,71-3,83 (1H, м), 3,98 (3H, с), 5,23 (1H, т, J=8,3 Гц), 7,44-7,60 (2H, м), 7,72 (1H, с), 8,02 (1H, с), 8,09 (1H, д, J=7,8 Гц), 8,40 (1H, д, J=8,3 Гц).
Соединение [389]: 4-(9-циклопентил-5,7,7-триметил-6-оксо-6,7,8,9-тетрагидро-5H-пиримидо[4,5-b][1,4]диазепин-2-иламино)-3-метокси-N-((цис)-4-морфолиноциклогексил)бензамид
После синтеза и очистки было выделено отдельное соединение [388], а именно, 4-(9-циклопентил-5,7,7-триметил-6-оксо-6,7,8,9-тетрагидро-5H-пиримидо[4,5-b][1,4]диазепин-2~иламино)-3-метокси-N-((цис)-4-морфолиноциклогексил)бензамид (белое твердое вещество, 22 мг, 24%).
Rt = 3,31 мин (Аналитический_1) MC(+ve) 606,4; МС(-ve) 604,5; 1Н-ЯМР (ДМСО-d6) δ м.д.: 1,13 (6H, с), 1,39-1,60 (4H, м), 1,60-1,71 (4H, м), 1,71-1,85 (4H, м), 1,85-2,01 (4H, м), 2,07-2,22 (2H, м), 2,42-2,48 (4H, м), 3,22 (4H, с), 3,64 (4H, шир.с), 3,87-4,05 (4H, м), 5,14-5,31 (1H, м), 7,45-7,62 (2H, м), 7,71 (1H, с), 8,02 (1H, с), 8,09 (1H, д, J=7,3 Гц), 8,39 (1H, д, J=8,3 Гц).
Соединение [390]: 4-(9’-циклопентил-5’-метил-6’-оксо-5’, 6’,8’,9’-тетрагидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-3-метокси-N-((транс)-4-морфолиноциклогексил)бензамид
4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-3-метоксибензойную кислоту (промежуточное соединение 5) (66 мг, 0,15 ммоль, 1 экв.), DIPEA (52 мкл, 0,3 ммоль, 2 экв.) и TBTU (54 мг, 0,17 ммоль, 1,1 экв.) добавляли к 1,5 мл ДМФ, и полученный раствор перемешивали при комнатной температуре в течение 20 мин, а затем добавляли 4-морфолиноциклогексанамин (помежуточное соединение 16) (33 мг, 0,18 ммоль, 1,2 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, а затем очищали с помощью ОФ-ВЭЖХ-МС (Препаративный_2) с получением 4-(9’-циклопентил-5’-метил-6’- оксо-5’,6’,8’,9’-тетрагидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-3-метокси-N-((транс)-4-морфолиноциклогексил)бензамида (не совсем белое твердое вещество, 13 мг, 14%).
Rt = 2,96 мин (Аналитический_1) МС(+ve) 604,4; МС(-ve) 602,5; 1Н-ЯМР (ДМСО-d6) δ м.д.: 0,72 (2H, с), 0,95 (2H, с), 1,24-1,49 (4H, м), 1,49-1,60 (2H, м), 1,60-1,69 (2H, м), 1,69-1,82 (2H, м), 1,82-2,03 (6H, м), 2,14 (1H, с), 2,17-2,32 (1H, м), 3,22 (4H, с), 3,53 (2H, с), 3,61 (4H, шир.с), 3,71-3,84 (1H, м), 4,00 (3H, с), 4,90 (1H, квин., J=8,5 Гц), 7,43-7,61 (2H, м), 7,73 (1H, с), 8,04 (1H, с), 8,10 (1H, д, J=7,8 Гц), 8,44 (1H, д, J=8,3 Гц).
Соединение [391]: 4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-3-метокси-N-((цис)-4-морфолиноциклогексил)бензамид
После синтеза и очистки было выделено отдельное соединение [390], а именно, соединение 4-(9’-циклопентил-5’-метил-6’-оксо-5’,6’,8’,9’-тетрагидроспиро[циклопропан-1,7’-пиримидо[4,5-b][1,4]диазепин]-2’-иламино)-3-метокси-N-((цис)-4-морфолиноциклогексил)бензамида (белое твердое вещество, 27 мг, 30%).
Rt = 3,10 мин (Аналитический_1) МС(+ve) 604,4; МС(-ve) 602,5; 1Н-ЯМР (ДМСО-d6) δ м.д.: 0,66-0,81 (2H, м), 0,88-1,03 (2H, м), 1,45-1,86 (12H, м), 1,87-2,02 (4H, м), 2,10-2,21 (1H, м), 2,47 (4H, шир.с), 3,22 (3H, с), 3,52 (2H, с), 3,65 (4H, шир.с), 3,89-4,05 (4H, м), 4,90 (1H, квин., J=8,5 Гц), 7,50-7,63 (2H, м), 7,73 (1H, с), 8,03 (1H, с), 8,10 (1H, д, J=7,3 Гц), 8,43 (1H, ..
Пример 3
Полноразмерный человеческий клон CDC25C выделяли с помощью ПЦР из мРНК HeLa и встраивали как BamHI-Hindlll-фрагмент в вектор pRsetA. Аминоконцевой фрагмент CDC25C (кодирующий остатки 1-300) вырезали из этого вектора и встраивали в вектор pET28a (между Ncol- и BamHI-сайтами). Экспрессию осуществляли под контролем промотора T7, а кодируемый белок содержал His6-метку у карбоксильного конца. Этот вектор трансформировали в pLysS штамма BRL(DE3) E.coli для проведения экспериментов по экспрессии. CDC25C экспрессировали в бактериальных клетках BL21(DE3)RIL, которые культивировали в среде LB при 37°С вплоть до достижения ими OD600нм = 0,6. Экспрессию индуцировали 1 мМ IPTG, после чего бактериальную культуру культивировали еще 3 часа. Бактерии собирали путем центрифугирования, и клеточный осадок ресуспендировали в 50 мМ триса, pH 7,5, и 10% сахарозы, быстро заморараживали и хранили при -70°С до его использования. Белок CDC25C очищали от телец включения E.coli. Тельца включения выделяли в буфере (50 мМ трис, pH 8,0, 2 мМ EDTA, 100 мМ NaCl, 0,5% тритон X-100). После денатурации в присутствии 6М мочевины, белок подвергали рефолдингу посредством медленного диализа мочевины. Этот белок хранили в 25 мМ трис, pH 8,0, 100 мМ NaCl, 1 мМ DTT, 1 мМ EDTA и 10% глицерина при -70°C до его использования.
Клон полноразмерной человеческой PLK1 (XM_047240) (аминокислоты 1-603) амплифицировали из библиотеки кДНК легких плода с использованием праймеров, включающих сайты рестриктирующих ферментов. 5’-праймер (gccgctagcgacgatgacgataagatgagtgctgcagtgactgcagggaagc) имеет Nhel-сайт, за которым расположен старт-кодон ATG. 3’-праймер (ggaattcttaggaggccttgagacgg) включает стоп-кодон, за которым расположен EcoRI-сайт. ПЦР-продукт клонировали в Nhe1/EcoR1-сайты бакуловирусного экспрессионного вектора, pSSP1. Клонирование в этот вектор приводило к образованию гибрида с His6-меткой у аминоконца конструкции PLK1. Клетки Sf9 после менее чем 20 пассажей, подвергали обратимому гидролизу с получением объема культуры 300 мл при клеточной плотности 1,5×106 клеток/мл. Эти клетки использовали только для экспрессии в логарифмической фазе роста. Затем добавляли PLK1 бакуловируса (после P2-амплификации) с получением множественности заражения = 3, что эквивалентно 3 вирусным частицам на каждую клетку насекомого. Колбы инкубировали при 27°C, со встряхиванием при 100 об./мин в течение 48 часов. После сбора определяли плотность и жизнеспособность клеток, а затем культуры центрифугировали при 2500 об/мин в течение 5 минут и промывали забуференным фосфатом физиологическим раствором, охлажденным льдом. Промывку снова центрифугировали при той же самой скорости, и осадок быстро замораживали. Белок PLK1 очищали на аффинной колонке с металлом. Осадок клеток насекомых подвергали лизису в буфере (10 мМ трис-HCl, pH 8,0, 150 мМ NaCl, 5 мМ β-меркаптоэтанол, 1 мМ PMSF, 1 мМ бензамидин, 20 мМ имидазол и смесь ингибиторов протеазы (Sigma)) и предварительно осветленный супернатант загружали на NiNTA-агарозу (Qiagen). Аффинную колонку промывали буфером для лизиса, и связанный белок элюировали 250 мМ имидазолом в том же самом буфере. После диализа против 25 мМ триса-HCl, pH 7,5, 100 мМ NaCl, 1 мМ DTT, 1 мМ PMSF, 1 мМ бензамидина, смеси ингибиторов протеазы (Sigma) и 10% глицерина, проводимого в течение ночи, очищенный белок хранили при -70°C до его использования.
Пример 4
Анализы на протеинкиназу PLK1 осуществляли в 96-луночном планшете путем инкубирования CDC25C (2 мкг/лунка) с PLK1 (1 мкг/лунка) в 20 мМ трис/HCl-буфере, pH 7,5, в который были добавлены 25 мМ β-глицерофосфат, 5 мМ EGTA, 1 мМ DTT и 1 мМ NaVO3. Затем добавляли серийные разведения тестируемого соединения в аналитическом буфере. Реакцию инициировали путем добавления 100 мкМ АТФ и 0,5 мкКи [γ-32P]-АТФ. Реакционную смесь инкубировали при 30°С в течение 1 часа, а затем реакцию прекращали путем добавления 75 мМ водной ортофосфорной кислоты, после чего смесь переносили на 96-луночный планшет с фильтрами P81 (Whatman), сушили, и оценивали уровень фосфорилирования CDC25C путем подсчета интенсивности сцинтилляции на планшет-ридере Packard TopCount. Необработанные данные анализа оценивали по параметрам нелинейного регрессионного анализа, а величины IC50 определяли по формуле: y = A+((B-A)/(1+((C/x)ΛD))), где y означает % ингибирования, A означает минимальное ингибирование, B означает максимальное ингибирование, С означает EC50, а D означает угловой коэффициент кривой.
Анализы на пролиферацию клеток проводили с использованием человеческих опухолевых клеточных линий (полученных из Американской коллекции типовых культур (American Type Culture Collection, 10801 University Boulevard, Manessas, VA 20110-2209, USA). Затем проводили стандартные 72-часовые анализы на окрашивание MTT (тиазолиловым синим; бромидом 3-[4,5- диметилтиазол-2-ил]-2,5-дифенилтетразолия) (Loveland et al., Biochem. Int., 1992, 27, 501; Haselsberger et al., Anti Cancer Drugs, 1996, 7, 331). Вкратце, клетки высевали на 96-луночные планшеты и после удвоения популяции инкубировали в течение ночи при 37°С. Тестируемые соединения разводили в ДМСО, и серию 1/3-разведений, приготовленную в 100 мкл клеточной среды, добавляли в клетки (с тремя повторностями) и инкубировали в течение 72 часов при 37°С. MTT разводили с получением маточного раствора с концентрацией 5 мг/мл в клеточной среде и стерилизовали на фильтре. Среду удаляли из клеток, а затем промывали 200 мкл PBS. Затем добавляли раствор MTT в концентрации 20 мкл на лунку и инкубировали в темноте при 37°С в течение 4 часов. Раствор MTT удаляли, и клетки снова промывали 200 мкл PBS. Краситель MTT солюбилизировали в 200 мкл ДМСО на лунку при перемешивании. Оптическую плотность считывали на 540 нм, и данные анализировали с помощью компьютерной программы построения кривой (версии 3.00 GraphPad Prism для Windows, GraphPad Software, San Diego California USA) для определения величин IC50 (концентрации тестируемого соединения, которое на 50% ингибирует рост клеток).
Пример 5
Селективность к PLK
В таблице 4 проиллюстрировано сравнение селективности соединения [218] к PLK1 по отношению к PLK2 и PLK3 с соответствующей селективностью выбранных известных соединений. Как было проиллюстрировано, селективность соединения [218] согласно изобретению к PLK1 по сравнению с селективностью к PLK2 в два раза превышает селективность соединения [I-64], описанного в WO 07/095188, примерно в 6 раз превышает селективность соединения [I-76], и в 7 раз превышает селективность соединения [I-4], описанного в WO 07/095188. Аналогичным образом, селективность соединения [218] к PLK1 по сравнению с селективностью к PLK3 в 5 раз превышает селективность соединения [I-64], описанного в WO 07/095188, примерно в 12 раз превышает селективность соединения [I-76], и в 5 раз превышает селективность соединения [I-4], описанного в WO 07/095188.
Пример 6
Исследования растворимости
В таблице 5 показано, что соединение [254] (структура показана ниже в рамке) обладает превосходной растворимостью и/или превосходными фармакокинетическими свойствами по сравнению с указанными параметрами уже известных структурно родственных соединений.
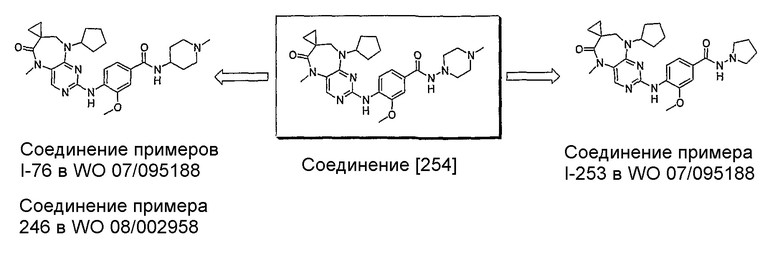
Нефелометрические исследования
Когда ДМСО-раствор водорастворимого соединения вводили в водный буфер, то смесь оставалась прозрачной до тех пор, пока не был достигнут предел растворимости в воде. При превышении пределов растворимости происходило осаждение, и свет, проходящий через мутную суспензию, рассеивался. Измерение интенсивности рассеянного света осуществляли путем нефелометрической оценки растворимости.
Выбранные соединения скринировали с использованием нефелометра в целях идентификации концентрации, при которой достигается мутность, что является показателем кинетической растворимости. При проведении нефелометрии (2% ДМСО: 98%-й забуференный фосфатом физиологический раствор, pH 7,5), соединение [254] легко растворялось при концентрации 200 мкМ (>0,11 мг/мл), которая является самой высокой концентрацией, тестируемой в этом анализе. В противоположность этому, в том же самом нефелометрическом анализе было определено, что концентрация известного соединения примера [1-253] WO 07/095188, при которой происходит максимальное растворение, составляет 75 мкМ (таблица 6). Это указывает на то, что соединение [254] является более растворимым в водном буфере в пределах концентраций, используемых для оценки активности соединения in vitro.
Фармакокинетические исследования
Для исследования соединений in vivo часто требуются более высокие концентрации. Для получения таких более высоких концентраций обычно используют солевые формы соединений.
Соединение [254] и соединения примеров [1-76] в WO 07/095188 были проанализированы в виде свободных оснований и гидрохлоридных солей в различных фармацевтически приемлемых смесях растворителей, которые могут быть использованы для введения соединений в живые организмы. После солюбилизации соединения путем нагревания в растворителе, растворы охлаждали до 25°С и проводили их мониторинг. После охлаждения сверхнасыщенные смеси образовывали не осадок, а гелеобразную мезофазу, которая является либо неподходящей, либо неудобной для приготовления дозы. На основе данных, представленных в таблице 5, можно сделать вывод, что соединение [254] и его HCl-соль имеют гораздо лучший профиль растворимости, чем соединение примеров [1-76] и его соответствующая HCl-соль.
HCl-соль соединения примеров [1-76] вводили мышам путем внутривенной инъекции в виде раствора в 3% DMA/97% воды. Максимально допустимая доза, очевидно, определяется пределом максимальной концентрации соединения в стабильном растворе. При максимально допустимой дозе противоопухолевая эффективность ксенотрансплантата не наблюдалась (ежедневно × 7 доз), поскольку это соединение могло быть введено в недостаточной концентрации. В противоположность этому, соединение [254] и его соль, имеющие значительно более высокую растворимость в воде, могут быть введены в концентрациях, которые, по всей вероятности, будут эффективными для лечения заболеваний.
Пример 8
Фармакокинетические свойства соединения [371]
(a) Методика
Предварительные исследования фармакокинетики выбранных соединений осуществляли на мышах CD-1 (25-30 г), находящихся в бодрствующем состоянии в режиме голодания. Дозы соединений составляли 1 мг/кг для внутривенного введения или 5-10 мг/кг для перорального введения. Двадцать одному самцу мышей, выбранных для данного способа введения (n=3 мышей на данное время введения), вводили содержащий дозу раствор путем одной инъекции ударной дозы в хвостовую вену или перорально через желудочный зонд. Пробы крови брали путем сердечной пункции через 5, 15, 30, 60, 120, 240 и 360 минут после внутривенного введения и через 30, 60, 120, 240, 360, 480 и 1440 минут после перорального введения. Пробы крови брали в пробирки, содержащие антикоагулянт, а именно, литий-гепарин, а затем смешивали и сразу помещали на измельченный лед. Плазму выделяли из цельной крови путем центрифугирования, а затем замораживали и хранили при -80°С до проведения анализа. Концентрации в плазме определяли с помощью жидкостной хроматографии – тандемной масс-спектрометрии в позитивном режиме ионизации электрораспылением (ИЭР ХЖ/МС/МС) с помощью калибровочных кривых, построенных в соответствующей матрице. Нижний уровень количественной оценки (LLOQ) во всех случаях составлял приблизительно 5 нг/мл. Данные зависимости концентрации в плазме от времени обрабатывали с помощью компьютерной программы WinNonLin версии 5,2, где площадь под кривой зависимости концентрации в плазме от времени (AUCall) вычисляли с помощью анализа без применения камерной модели, проводимого с использованием модели 201 (вводимая ударная i.v.-доза) или модели 200 (доза, вводимая в область за пределами сосудистой системы).
(b) Сравнение соединений [371] и [378] c выбранными известными соединениями
Были проведены дополнительные фармакокинетические исследования для сравнения соединений [371] и [378] с соединениями серии родственных аналогов (см. таблицу 7; соединение [371] соответствует соединению H; соединение [378] соответствует соединению I).
На фигуре 1 представлен график, на котором проиллюстрирована площадь под кривой (AUC) зависимости концентрации каждого тестируемого соединения в плазме от времени. Носитель для приготовления дозы во всех экспериментах оставался постоянным (для внутривенного введения = цитратный буфер, pH 3, 1 мл/кг; для перорального введения = DMA/ПЭГ400/10 мМ тартратный буфер, pH 4 (1:3:6), 5 мл/кг). На фигуре 2 представлены те же самые данные в форме биологической доступности (%F) каждого соединения для перорального введения.
Эти результаты показали, что соединения H и I обладают значительно более сильным системным действием и значительно более высокой пероральной биологической доступностью по сравнению с соединениями A-G.
Пример 9
(a) Данные по выведению лекарственного средства из клеток
В анализе in vitro на жизнеспособность клеток измеряли эффект аккумуляции лекарственного средства в паре родительских опухолевых клеточных линий и опухолевых клеточных линий, резистентных к лекарственному средству. Такими парами были клеточная линия человеческой карциномы яичника A2780 с ее MDR-аналогом A2780/ADR и клеточная линия человеческой саркомы матки MES-SA с ее MDR-аналогом MES-SA/Dx5 [Wesolowska О., et al. (2005) Anticancer Res., 25, 383-389].
Метод: Культивированные клеток опухолевой клеточной линии яичника человека A2780 в паре с резистентными к доксорубицину линиями A2780/ADR, и клеток клеточной линии матки MES-SA в паре с резистентными к доксорубицину клеточными линиями MES-SA/DX5 высевали при плотности 3000 клеток на лунку в 96-луночные планшеты для культивирования ткани Nunc, в объеме 100 мкл на лунку в среде DMEM, содержащей 10% фетальную телячью сыворотку и пенициллин/стрептомицин. В каждом планшете лунки первого столбца, не содержащие клеток, заполняли 100 мкл среды DMEM с получением контроля-«пустышки» для проведения окрашивания аламаровым синим. Клетки инкубировали в течение ночи при 37°С и в присутствии 5% CO2. Представляющие интерес соединения, растворенные в ДМСО, добавляли в клетки в различных концентрациях. Разведения соединений сначала приготавливали в DMEM в концентрации, которая в два раза превышала конечную желаемую концентрацию, и 100 мкл разведений добавляли в клетки. Каждую серию соединений тестировали с тремя повторностями в каждой клеточной линии. После инкубирования в течение 72 часов в каждую лунку добавляли 20 мкл реагента аламарового синего (AbD Serotec) и инкубировали в течение 3 часов. Затем планшеты считывали на 544/595 нм.
Количество прореагировавшего реагента аламарового синего определяет метаболическую активность клеток. Относительную клеточную активность вычисляли как процент от контроля (клетки без соединения) и определяли концентрацию активного соединения, ингибирующую клеточную активность на 50% (IC50). Эти величины вычисляли как среднее из трех отдельных экспериментов с нормализацией за контроль (только одна среда).
Этот анализ осуществляли на Biomek FxP, автоматизированной платформе Beckman Coulter. Планшеты считывали на планшет-ридере Paradigm, поставляемом Beckman Coulter. Отношения асимметрии вычисляли как величину IC50 соединения в резистентных к лекарственному средству клетках, деленную на величину IC50 для соединения в родительских клетках. На фигуре 3 проиллюстрировано сравнение выбранных соединений с известными соединениями A’-J’ (как указано в таблице 8) в родительских опухолевых клетках и в опухолевых клетках MDR. Отношения асимметрии представлены для соединения [378].
(b) Ингибирование PLK1
Кроме того, соединения согласно изобретению, которые обнаруживали низкую асимметрию MDR/родительских клеток в анализах на жизнеспособность клеток, также являются более эффективными в отношении ингибирования PLK1 в опухолевых клетках MDR, чем известные соединения. Таким образом, соединения согласно изобретению являются более сильными терапевтическими ингибиторами PLK. Промежуточный филаментный белок виментин представляет собой клеточную мишень для фосфорилирования PLK1. Ингибирование PLK1 ассоциируется со снижением уровня фосфо-виментина. Это было определено в паре клеточных линий A2780-A2780/ADR.
Метод: Клетки A2780 и клетки A2780/ADR высевали в отдельные 96-луночные планшеты (Perkin Elmer) при плотности 20000 клеток/лунка и инкубировали в течение ночи при 37°C и 5% CO2. Тестируемые соединения добавляли в клетки в различных концентрациях, причем для каждой концентрации использовали лунки с тремя повторностями (верхняя концентрация ДМСО в клетках составляла 0,1%). После инкубирования в течение 7 часов клетки фиксировали в охлажденном льдом 3,7% формальдегиде в течение 10 минут. Клетки промывали в PBS, а затем их делали проницаемыми в холодном метаноле в течение 10 минут. Клетки снова промывали в PBS, а затем инкубировали с PBS, содержащим 0,1% тритон X-100, в течение 5 минут. Клетки один раз промывали PBS, содержащим 1% альбумин бычьей сыворотки (Sigma), а затем инкубировали в течение 3 часов с антителами против фосфогистона H3-Serl0 (1:4000; Millipore) и фосфо-виментин-Ser82 (1:1000; MBL) в 1% альбумине бычьей сыворотки. Антитела детектировали с козьим антителом против мышиных IgG, конъюгированным с Alexafluor 488 (Invitrogen), и с козьим антителом против кроличьих IgG, конъюгированным с Alexafluor 546 (Invitrogen), в 1% альбумине бычьей сыворотки с 300 нМ DAPI (Cambridge Biosciences). Планшеты сканировали на ридере Cellomics Arrayscan II HCS Reader (Cellomics) с объективом 10X. Для получения и анализа изображений применяли устройство для определения профиля жизнеспособности клеток V2 (The Cell Health Profiling V2 Bioapplication (Cellomics)). Окрашивание фосфо-виментином, наблюдаемое при интенсивности, превышающей пороговое значение, которое может быть определено специалистом, измеряли только в митотических клетках (как было определено по присутствию фосфо-гистона H3), и регистрировали среднюю величину интенсивности окрашивания на митотическую клетку для каждой лунки. Величины IC50 вычисляли с использованием усредненных величин для лунок с тремя повторностями. Отношения асимметрии вычисляли как величину IC50 для данного соединения в резистентных к лекарственному средству клетках A2780/ADR, деленную на величину IC50 в родительских клетках A2780. На фигуре 3 проиллюстрировано сравнение выбранных соединений с уже известными соединениями A’-J’ (указанными в таблице 8) в родительских опухолевых клетках и в опухолевых клетках MDR. Как указывалось ранее, отношения асимметрии представлены для соединения [378].
В описанных здесь аспектах настоящего изобретения могут быть внесены различные модификации и варианты, которые являются очевидными для специалиста в данной области, но не выходят за рамки существа и объема изобретения. Хотя настоящее изобретение описано на конкретных предпочтительных вариантах его осуществления, однако, следует отметить, что заявленное изобретение не ограничивается такими конкретными вариантами. Действительно, различные модификации описанных в настоящем описании способов осуществления изобретения, которые являются очевидными для специалиста в данной области, не должны выходить за рамки объема прилагаемой ниже формулы изобретения.
Величины IC50 (мкМ) выбранных соединений согласно изобретению для PLK; *** означает <0,1 мкМ IC50; ** означает <1,0 мкМ IC50; * означает <10,0 мкМ IC50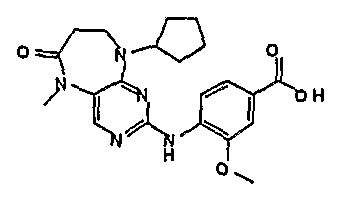
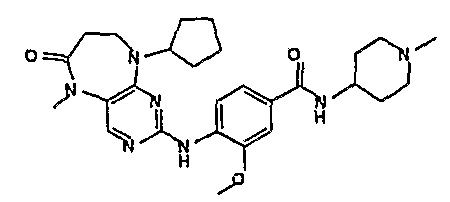
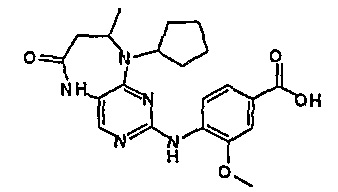
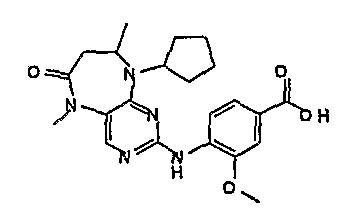
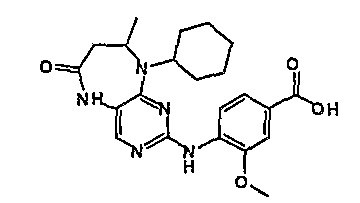

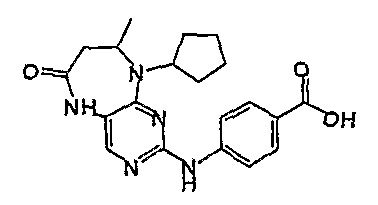
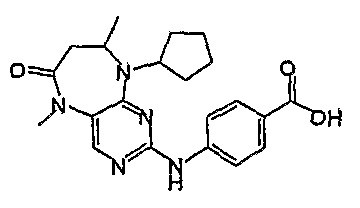
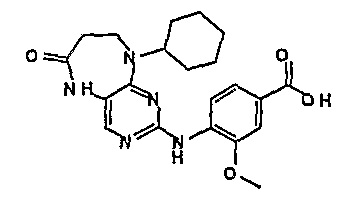
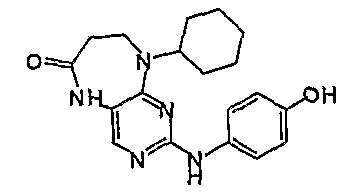
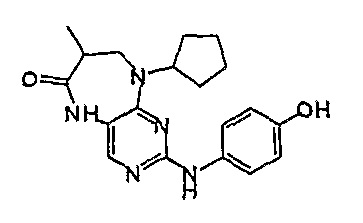
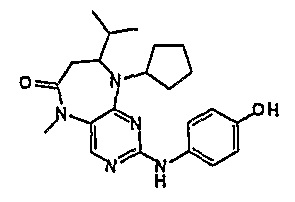

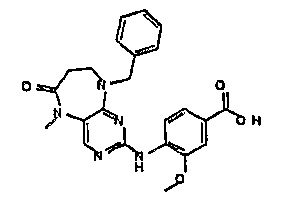
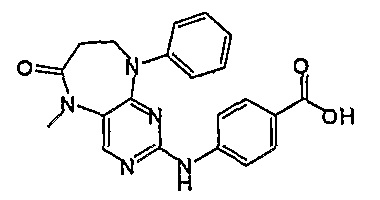
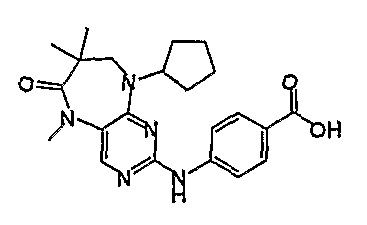
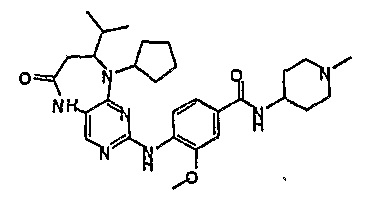
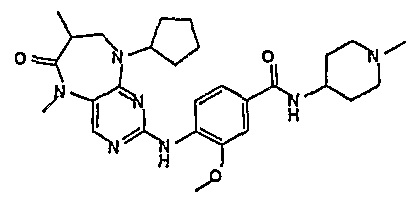
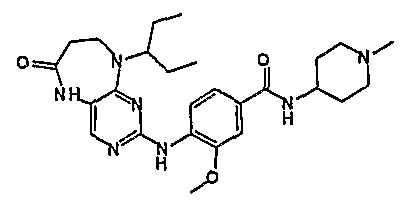
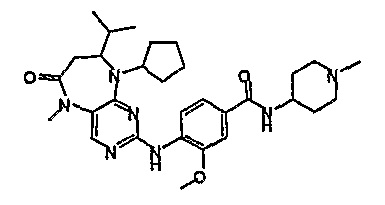
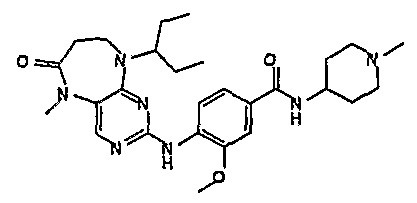
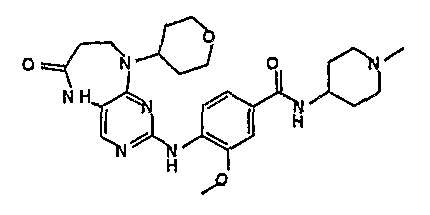
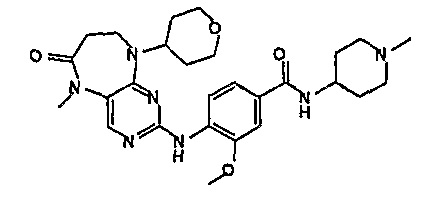
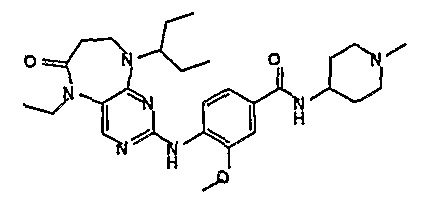
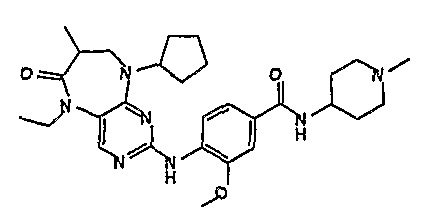
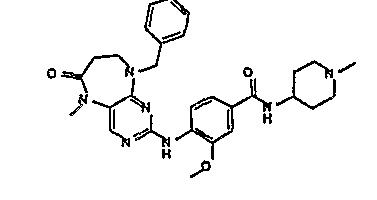
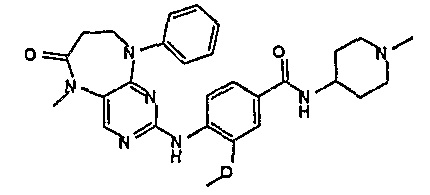
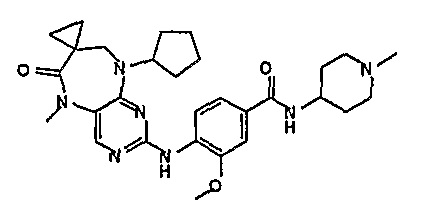

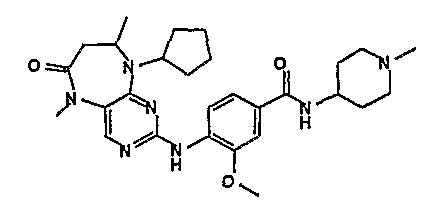
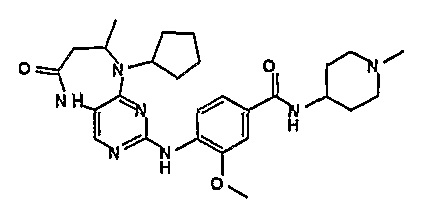
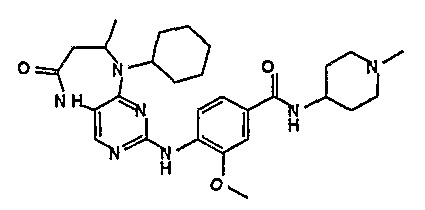
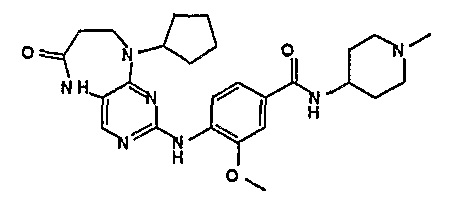
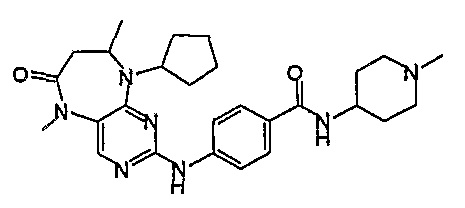
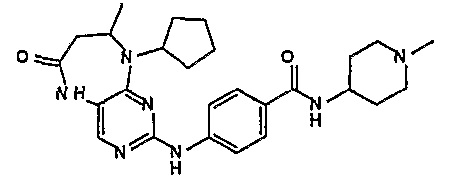
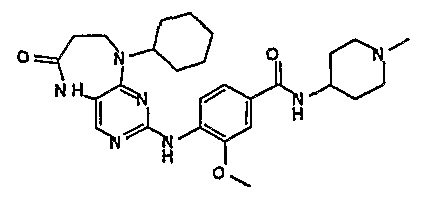
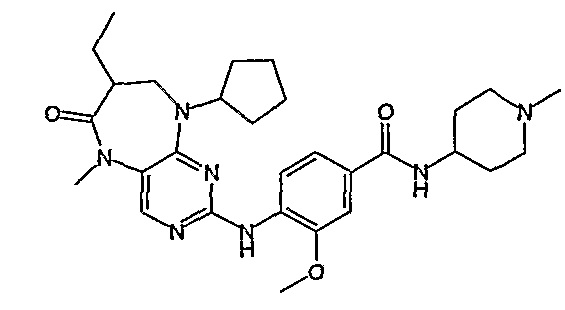
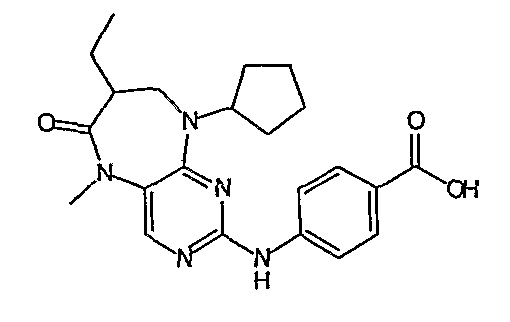
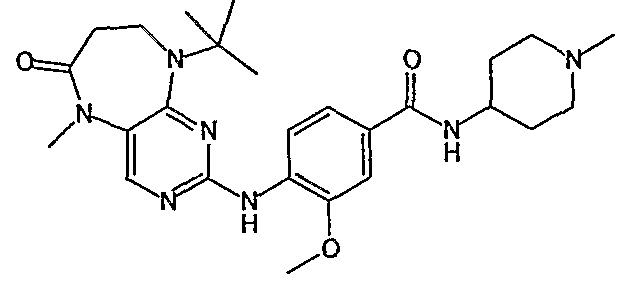
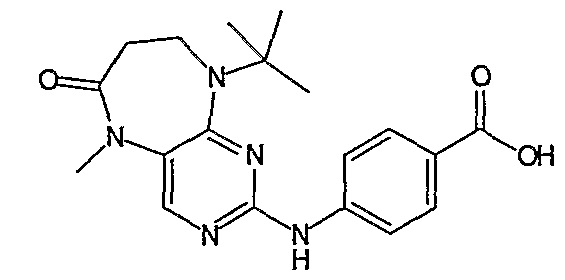

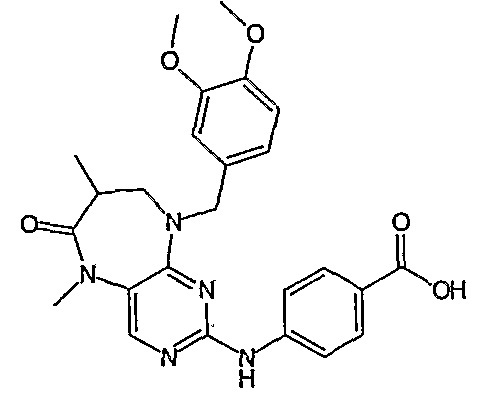
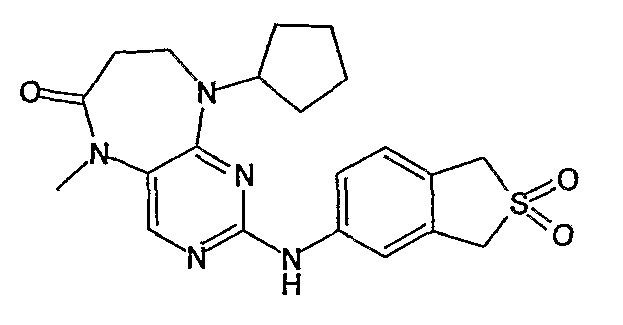
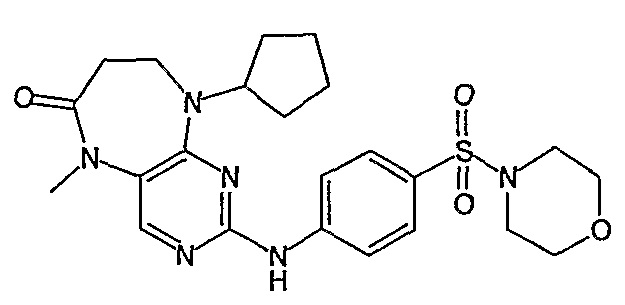
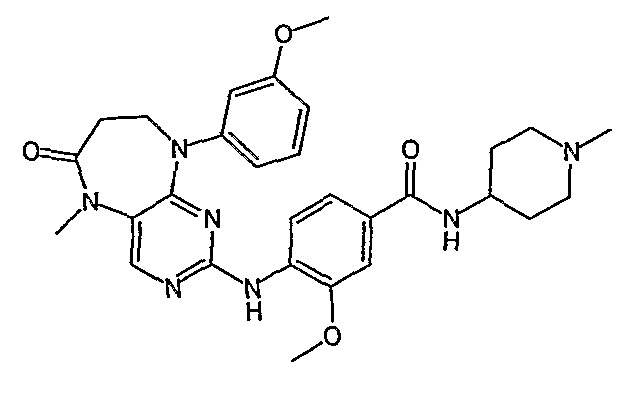
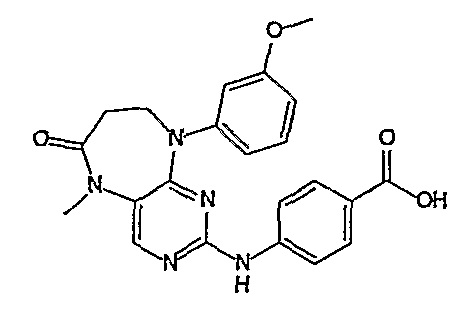
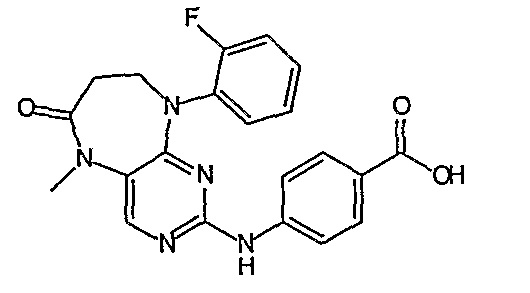
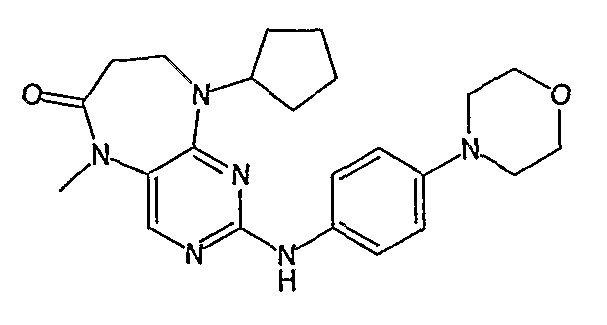
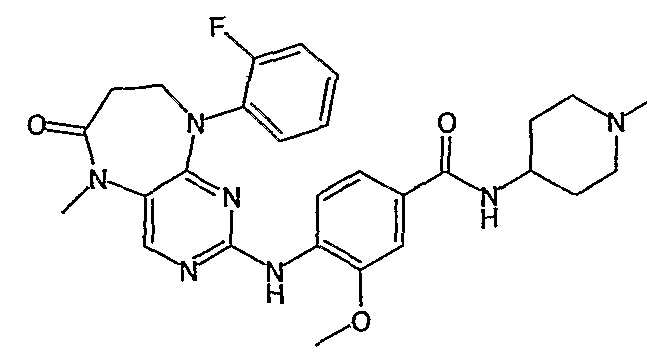
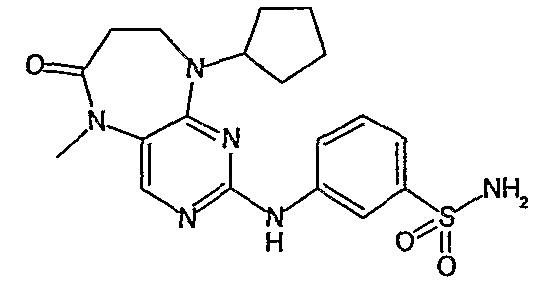

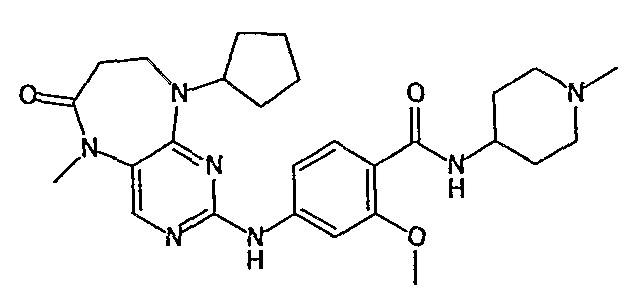
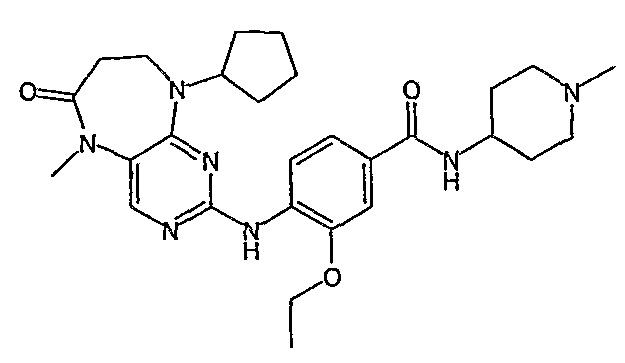
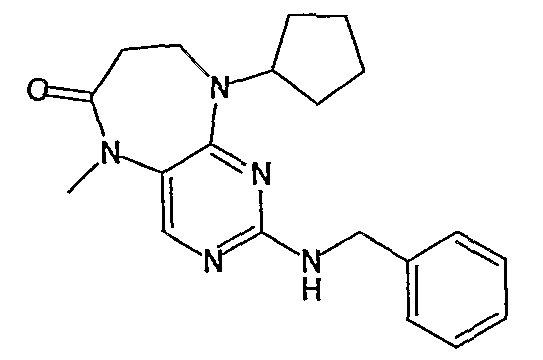
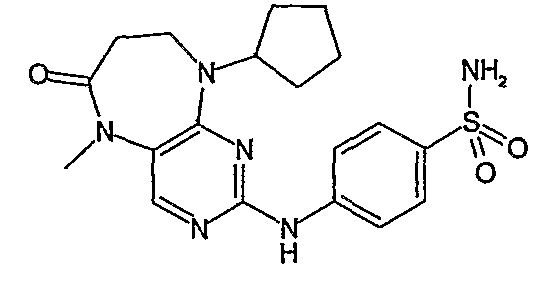
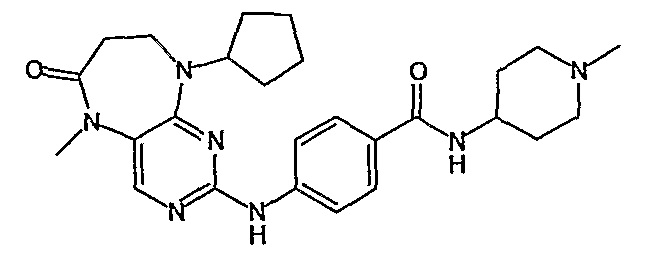
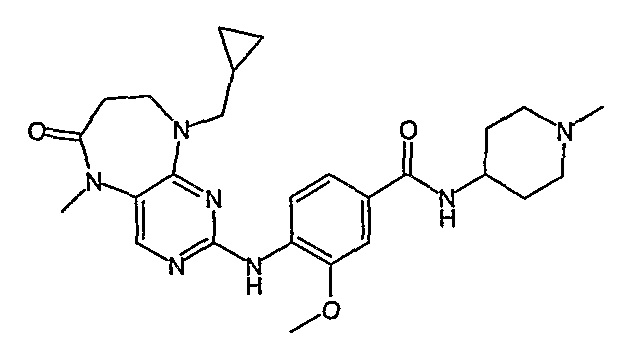
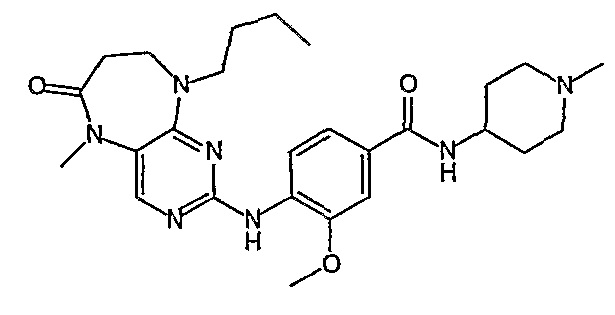
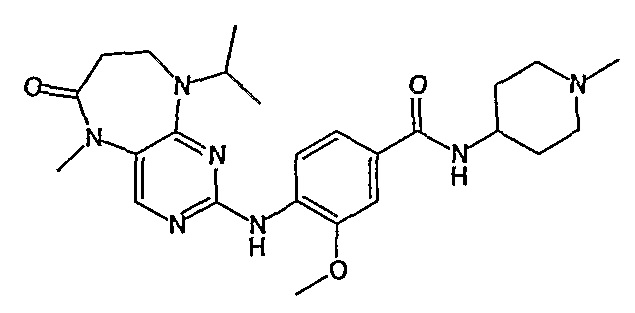

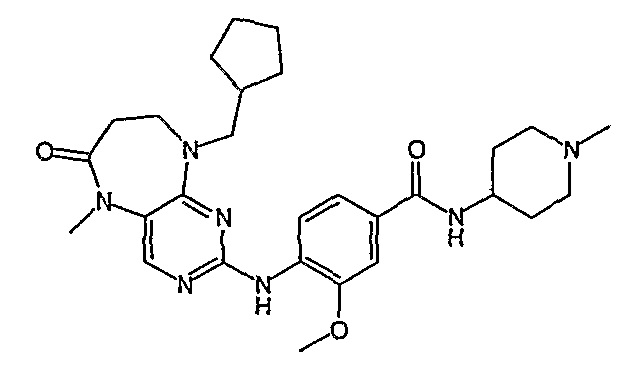
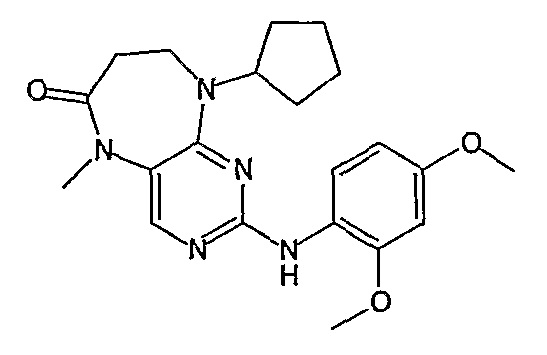
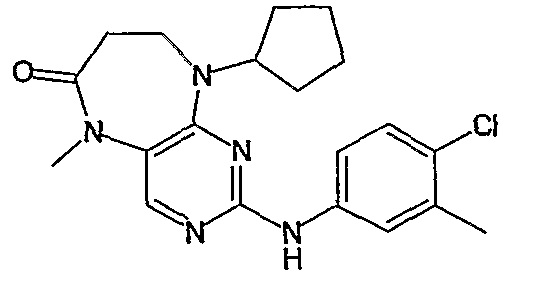
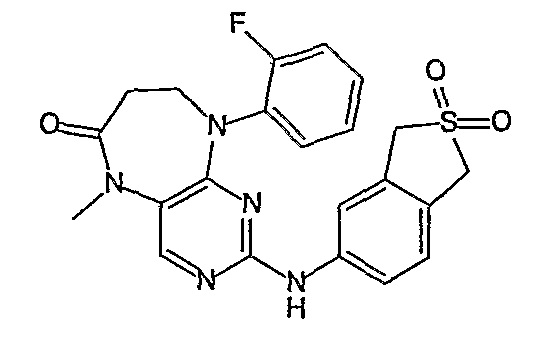
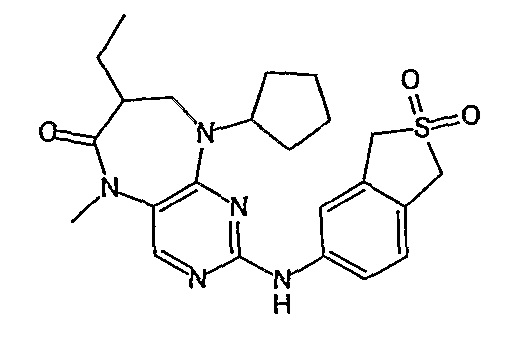
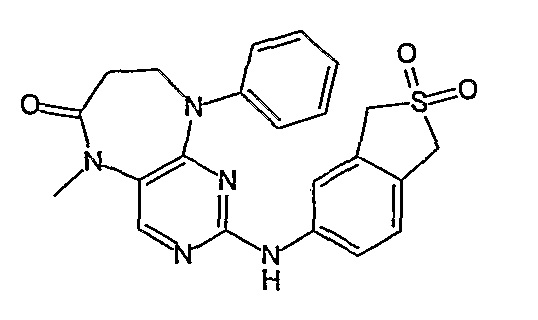
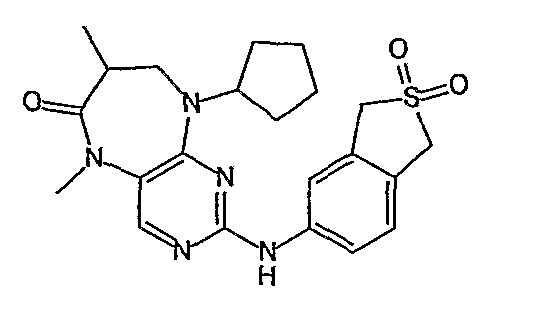
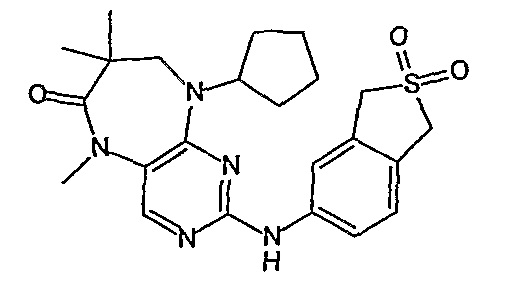
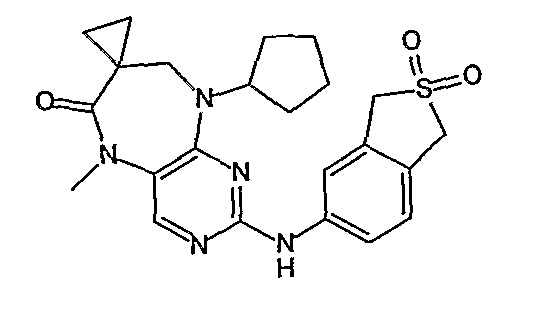
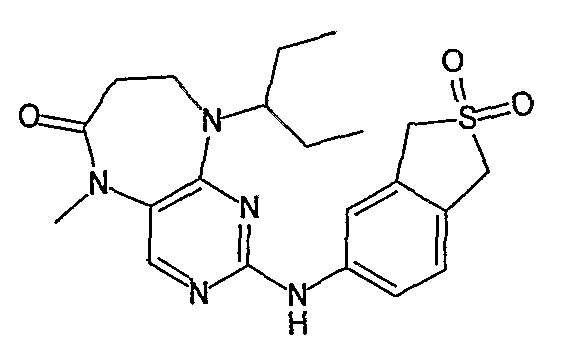
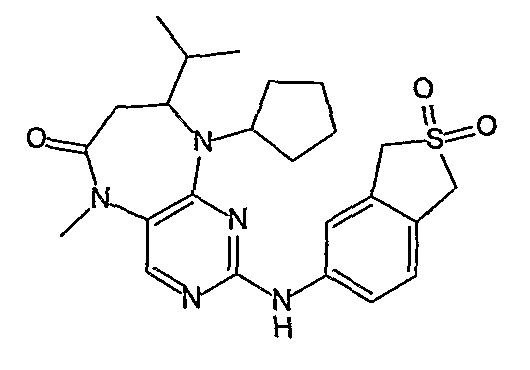
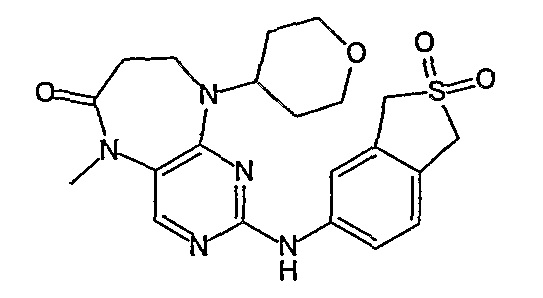

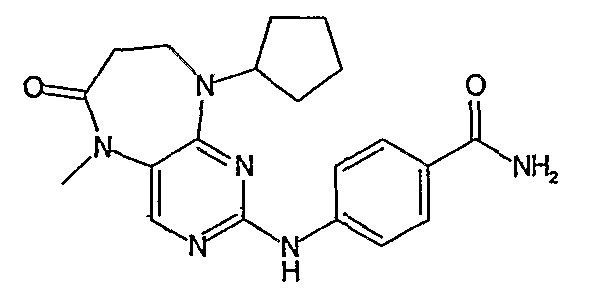
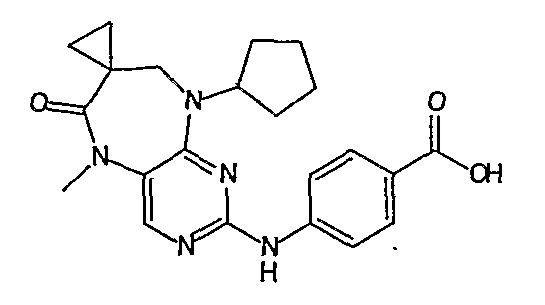
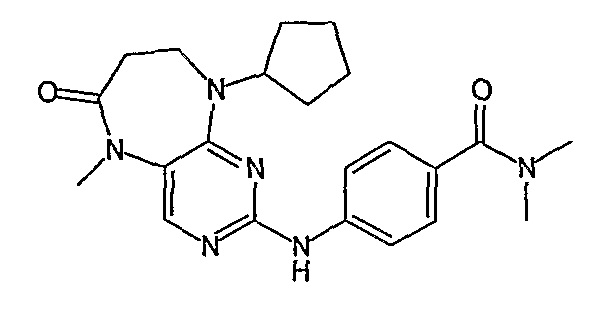
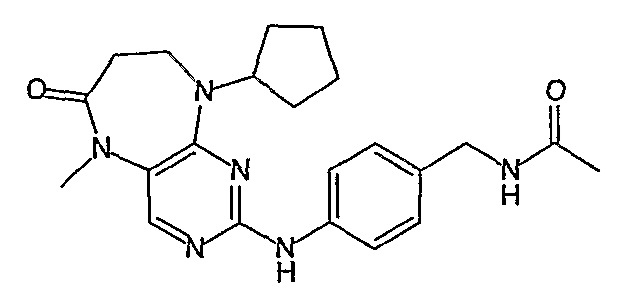
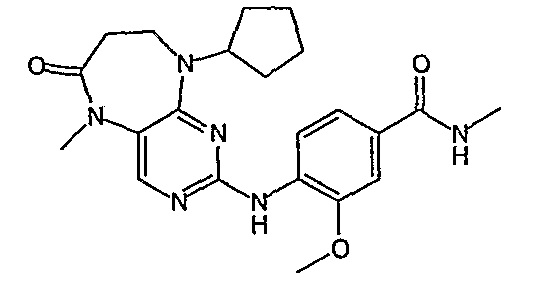
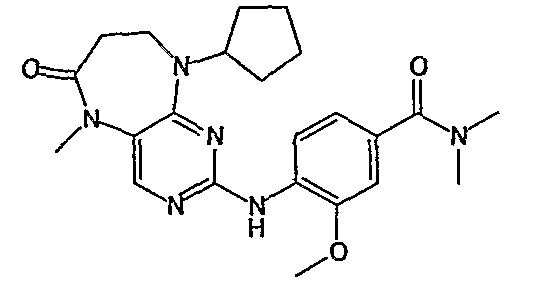
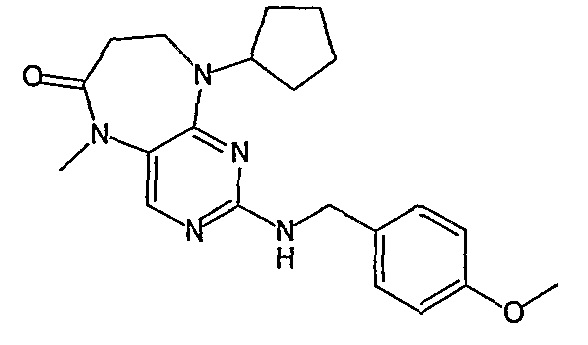
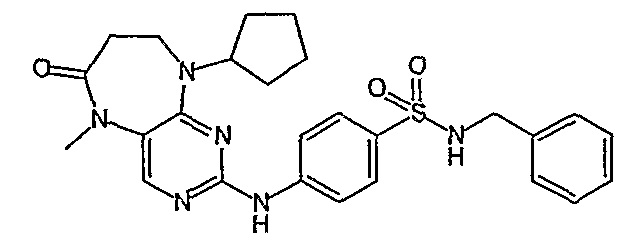
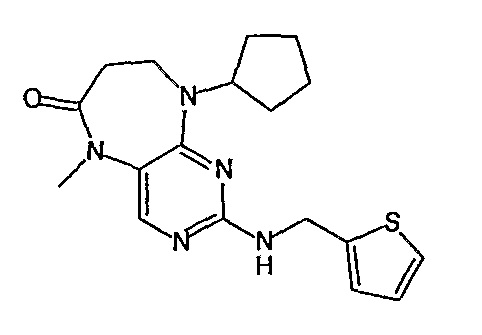

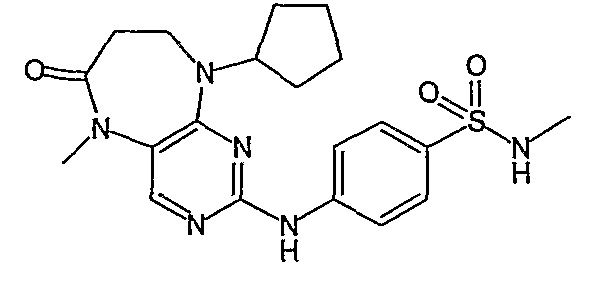
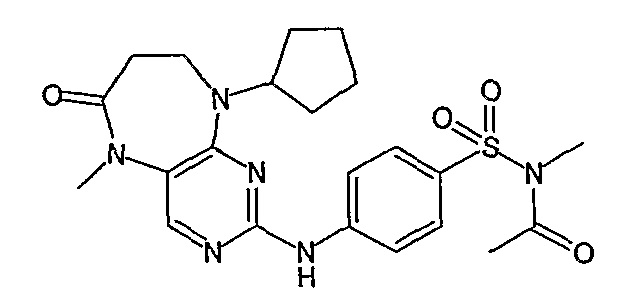
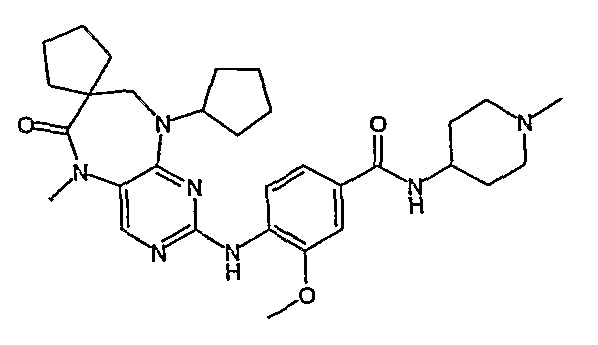
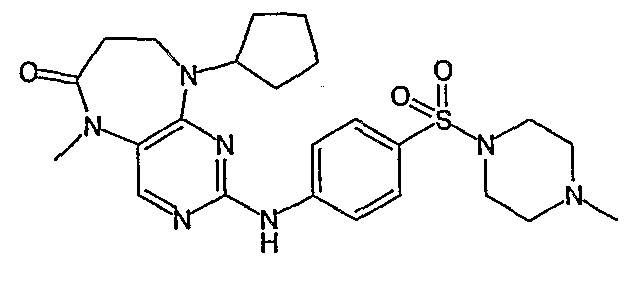


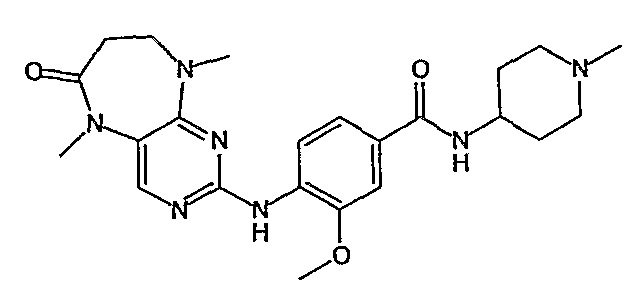


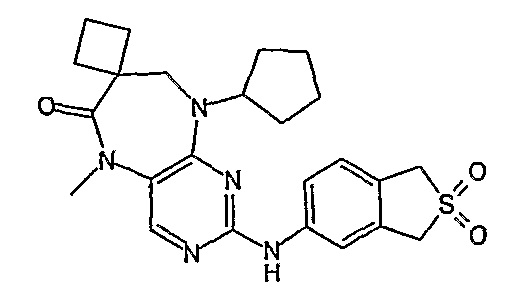

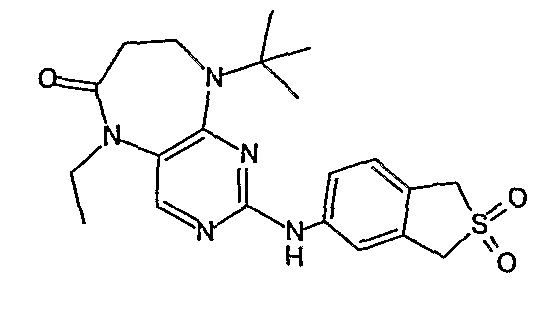
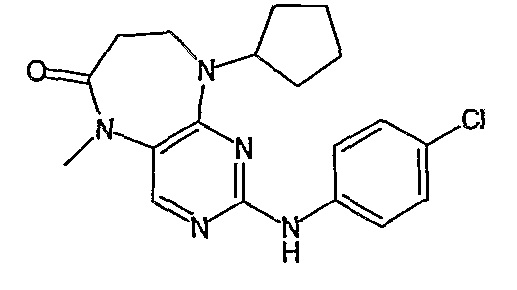


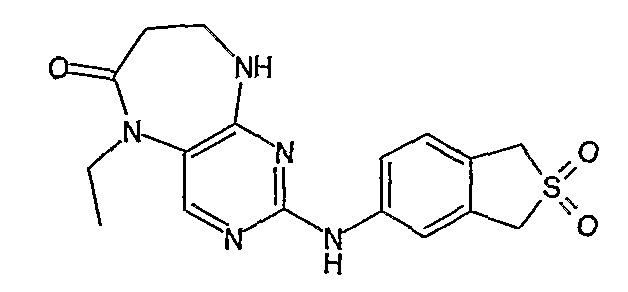
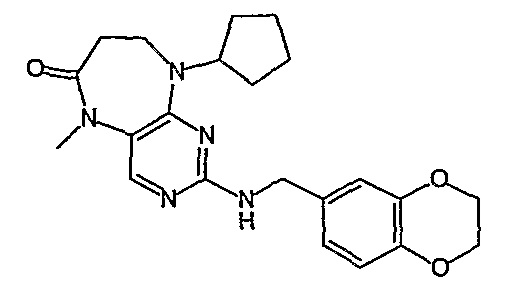
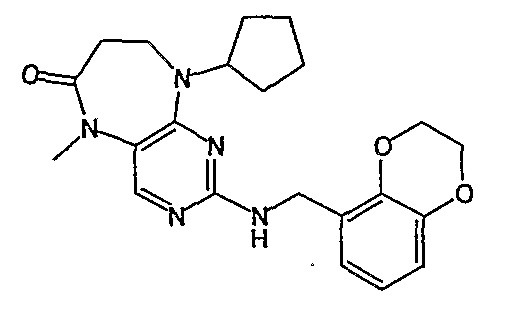
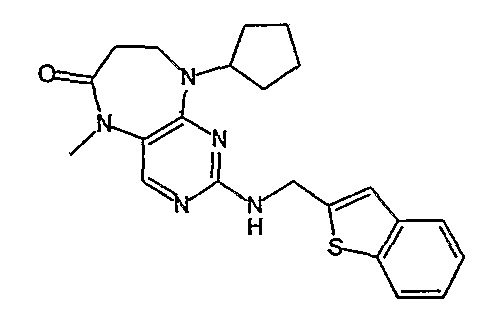
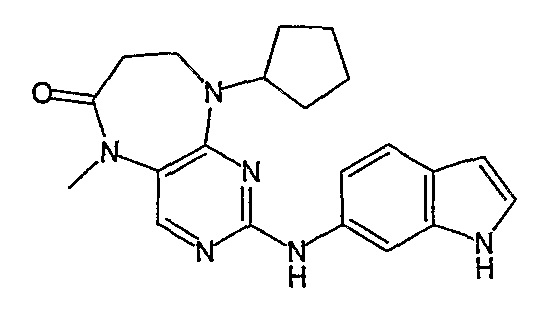
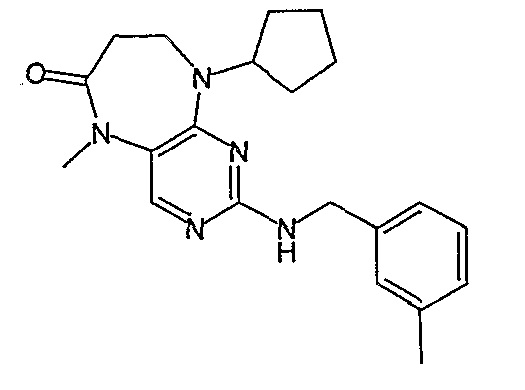
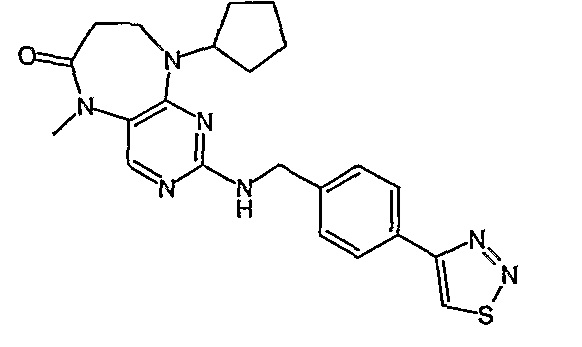

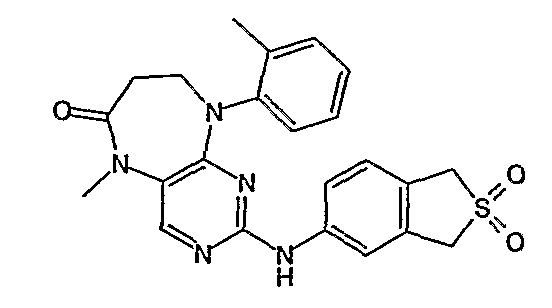
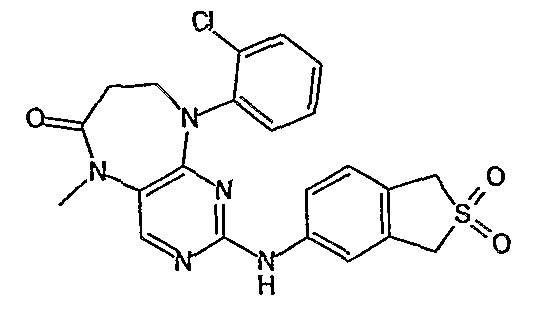
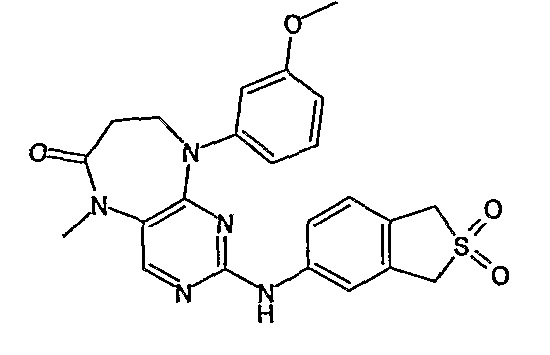
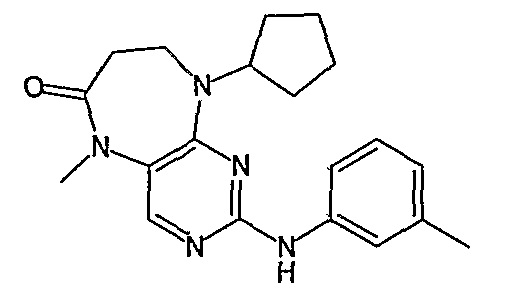
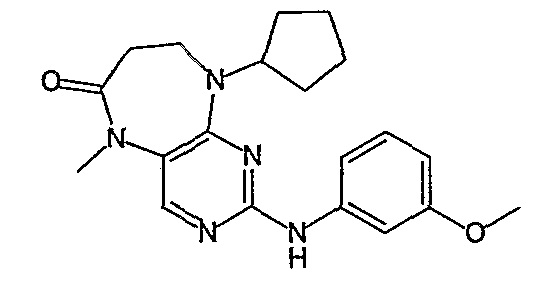
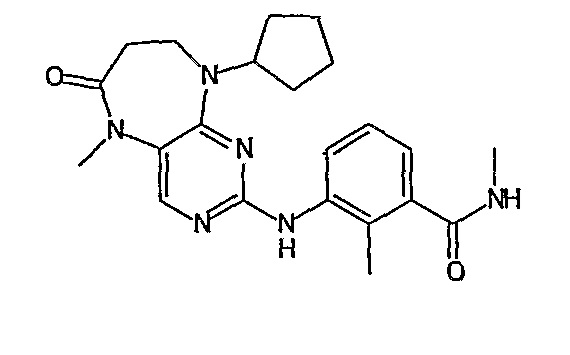
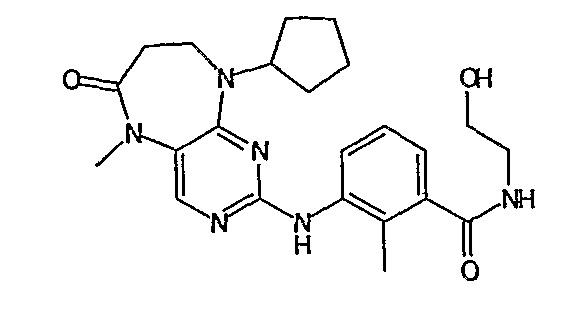
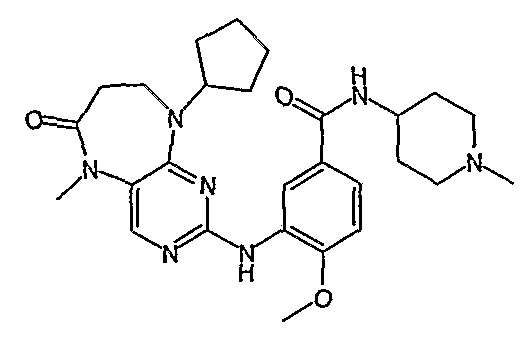
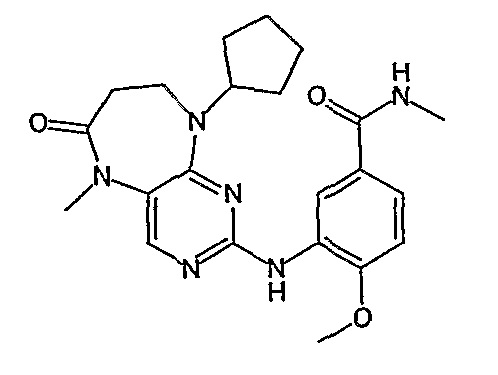
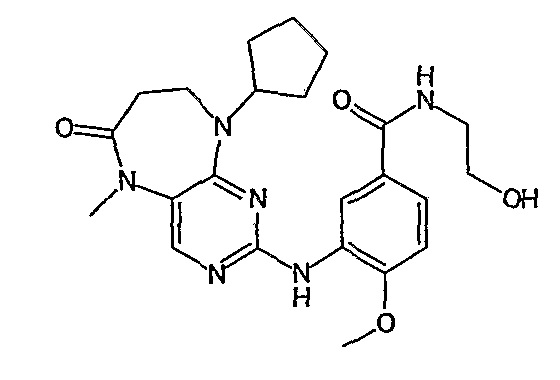


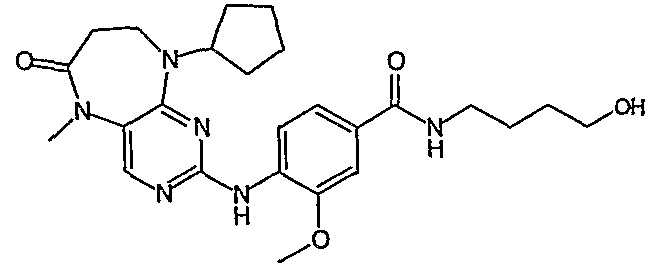
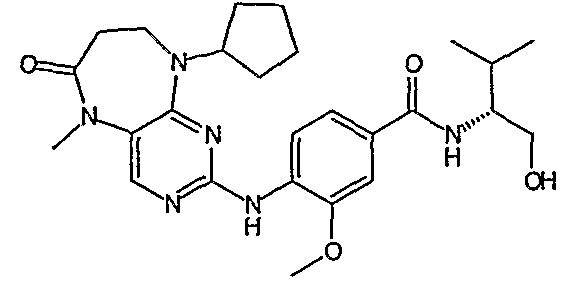
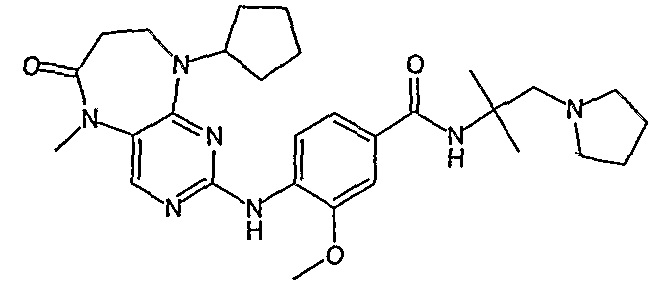
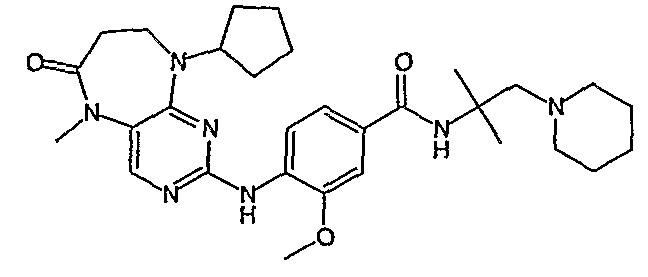
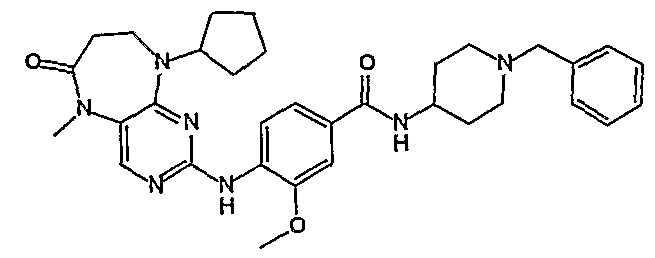



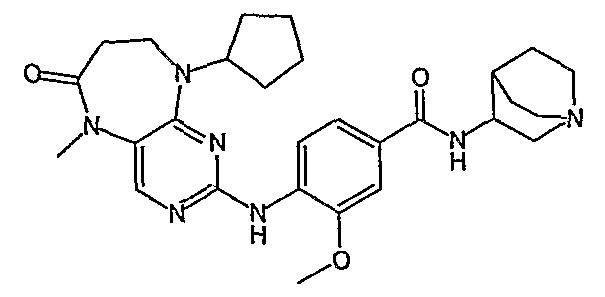
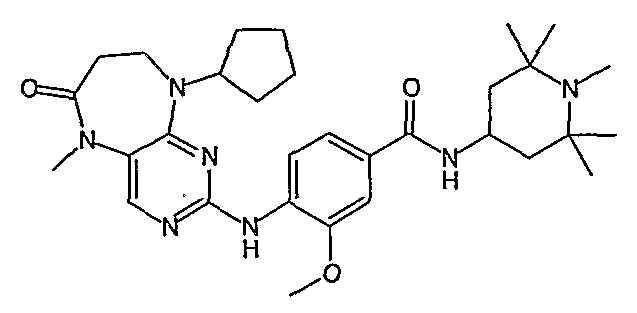
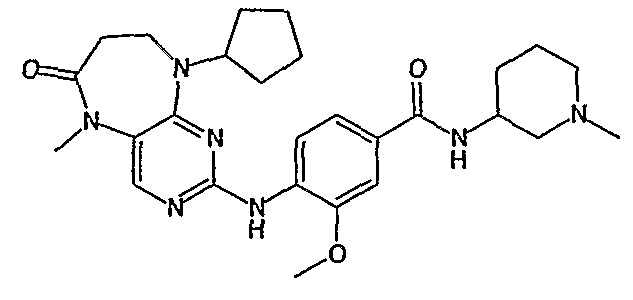
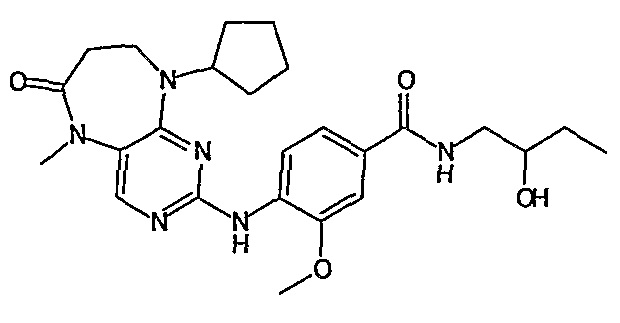
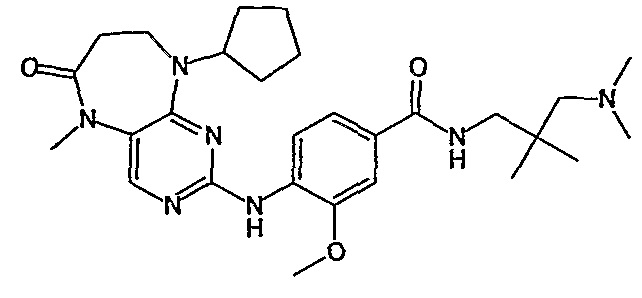
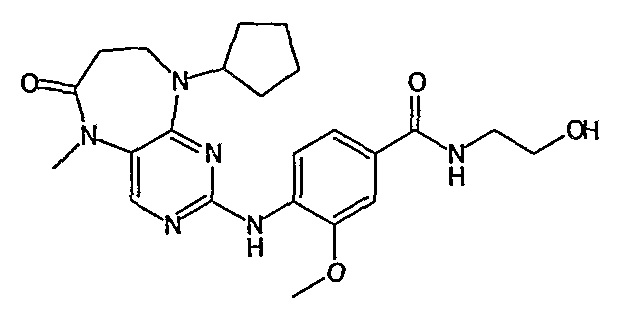
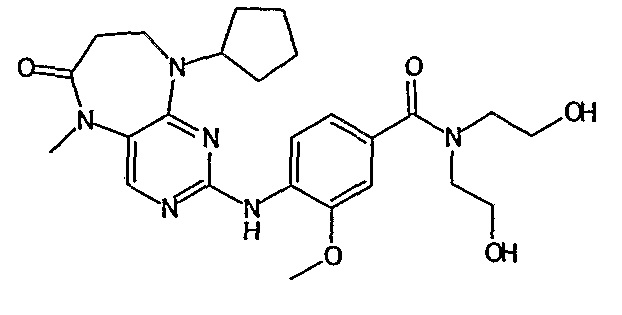
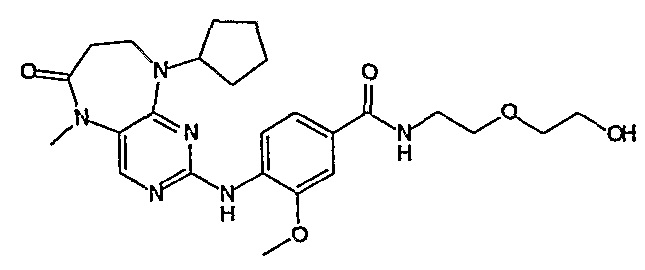
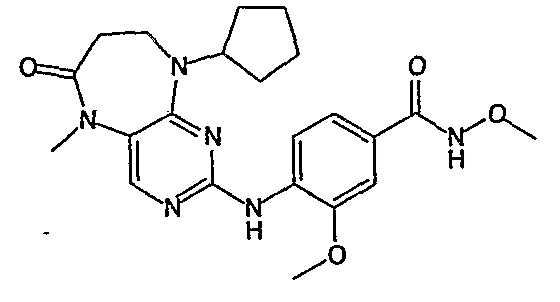
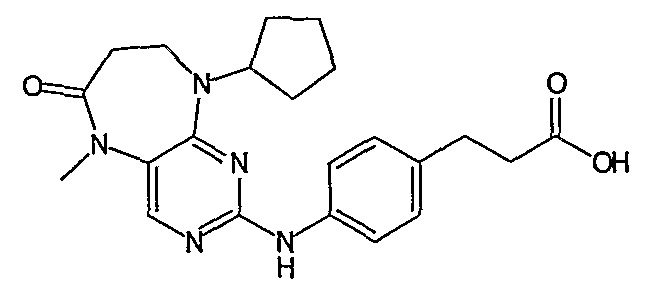

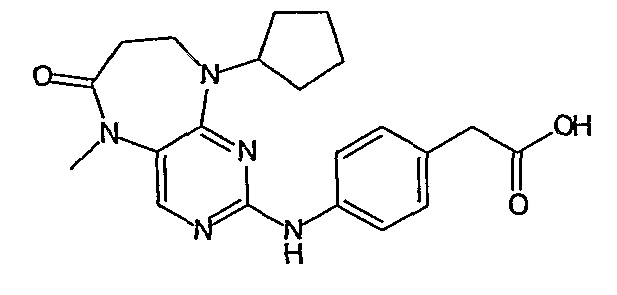
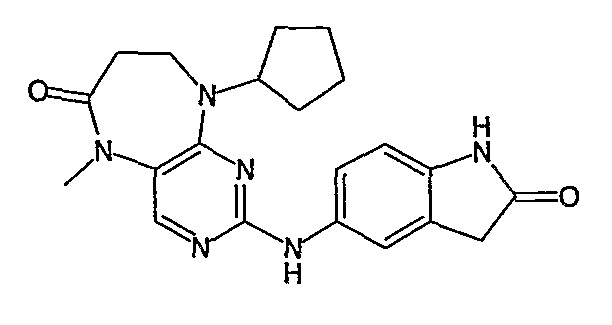
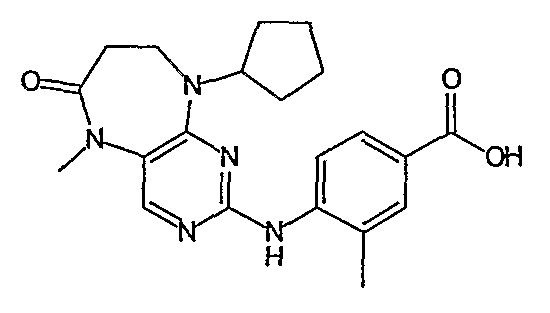
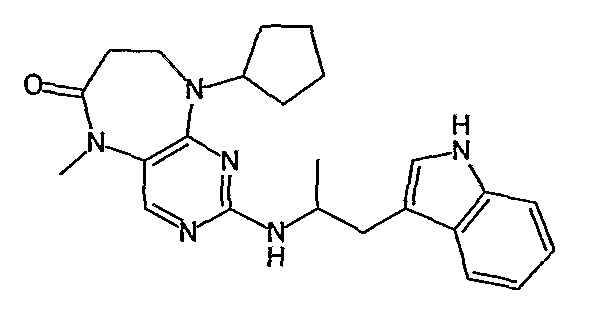
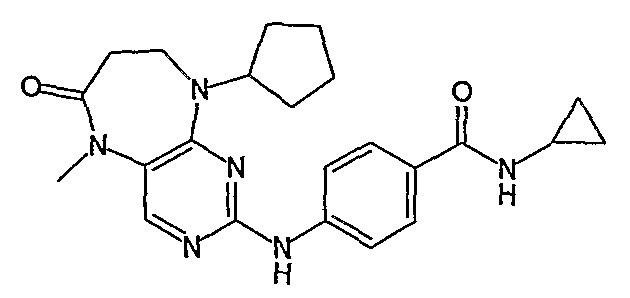
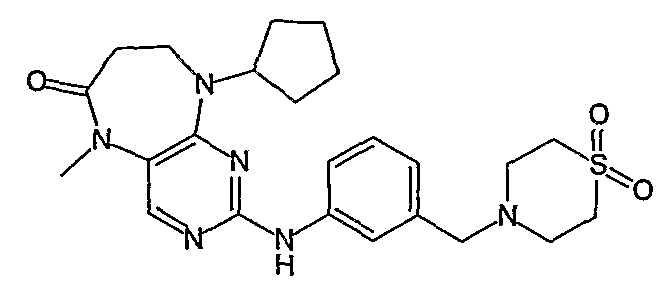
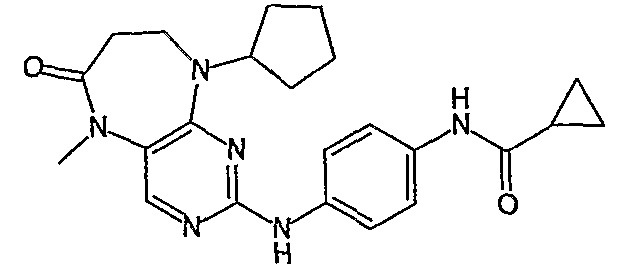
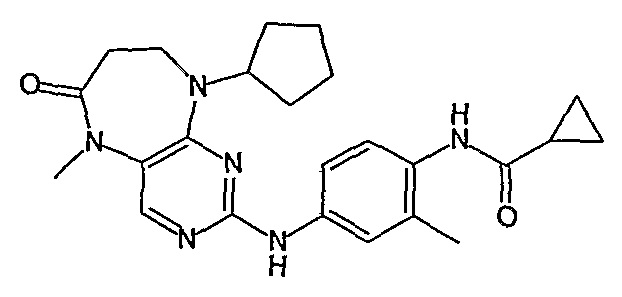
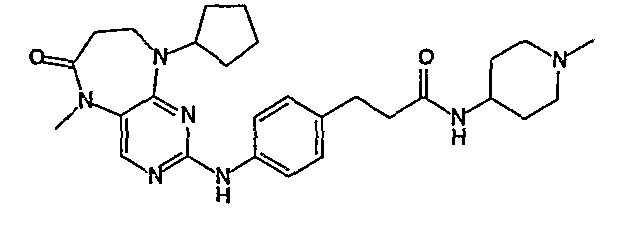
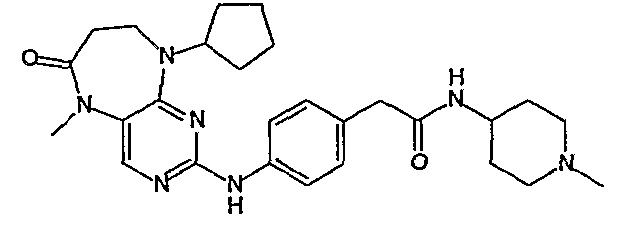

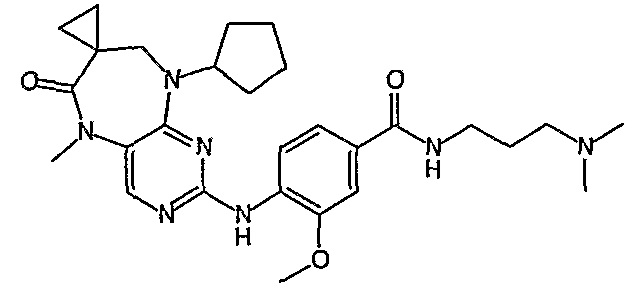
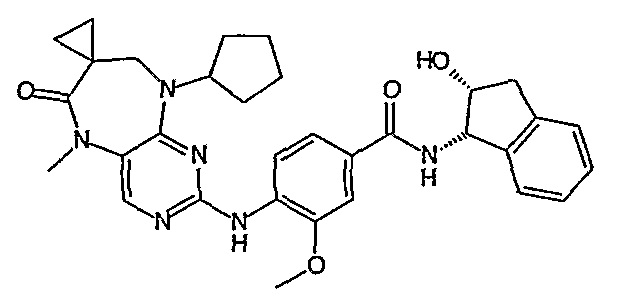
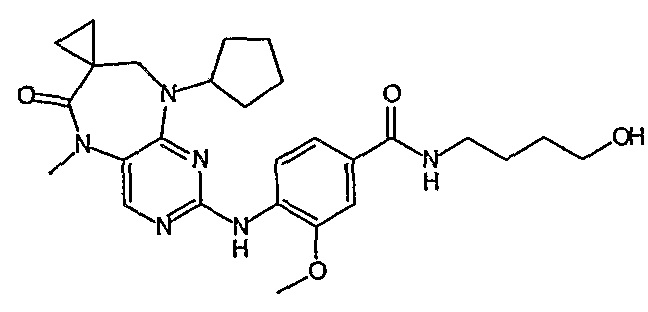

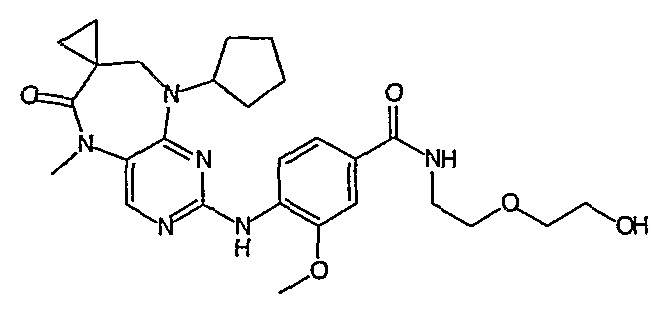
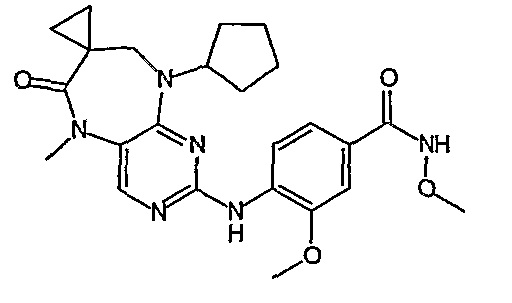
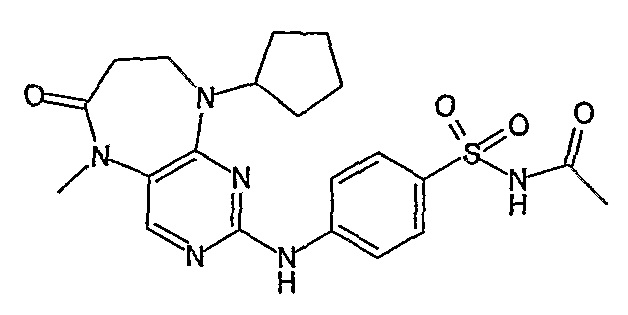


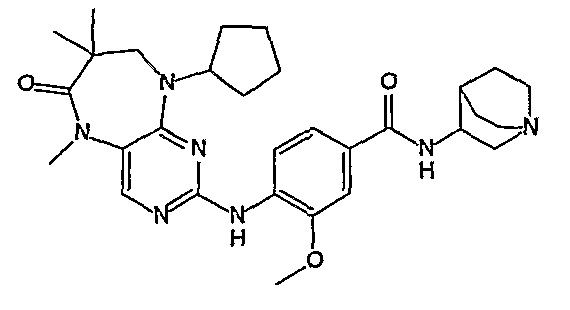
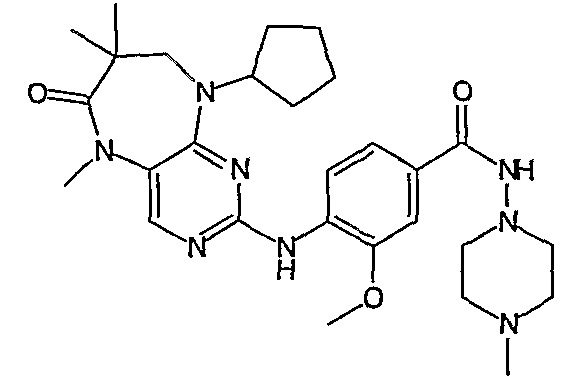
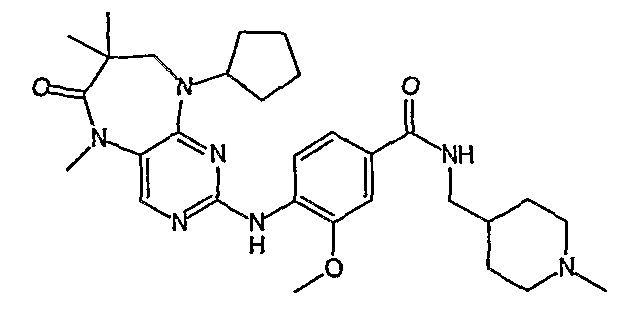


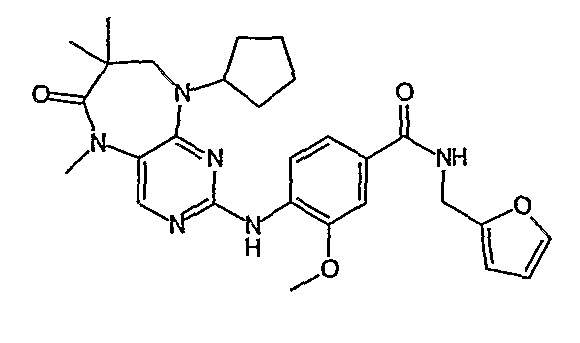
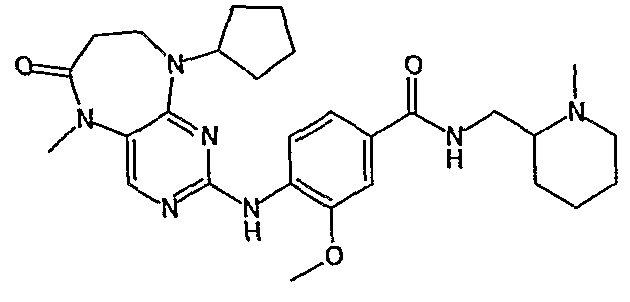
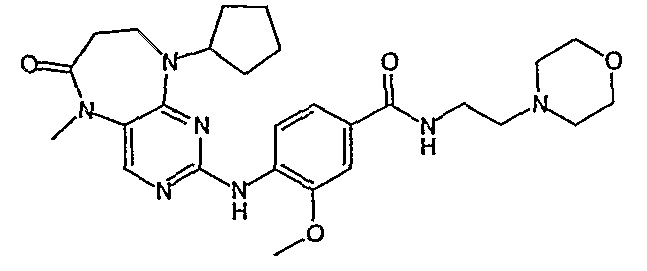
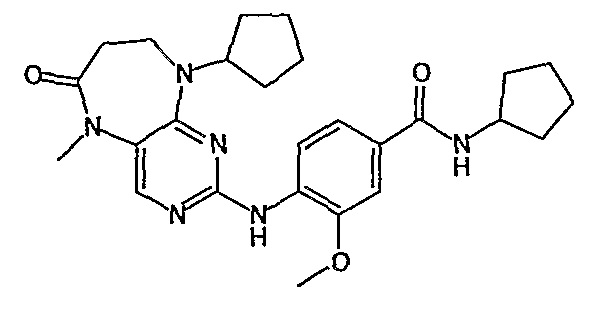

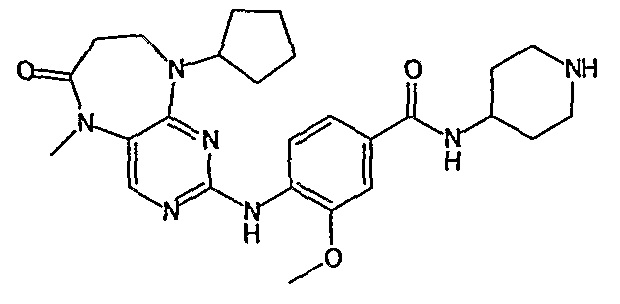
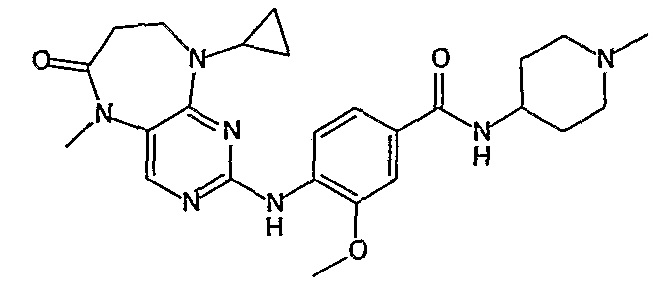
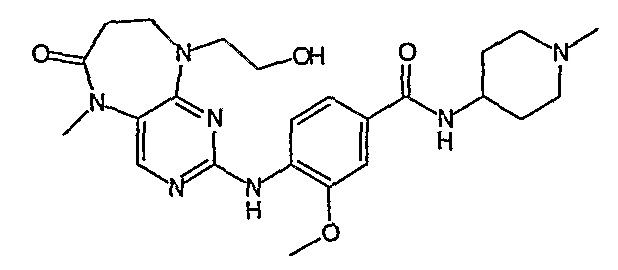
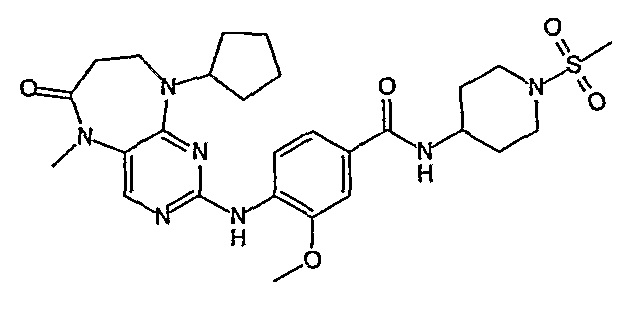
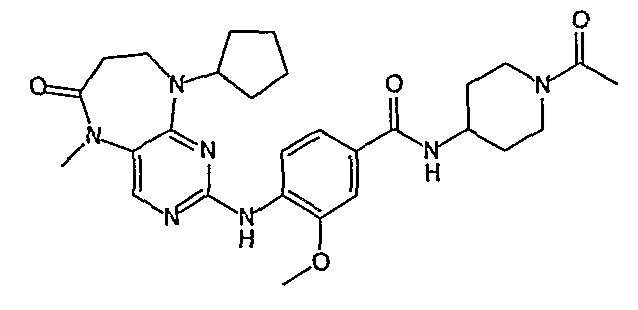
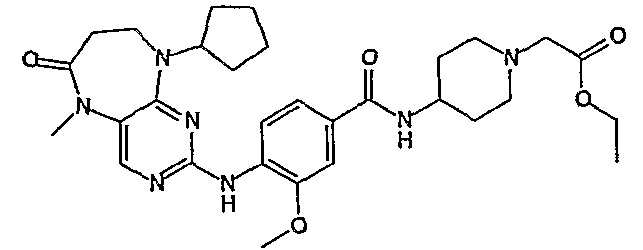


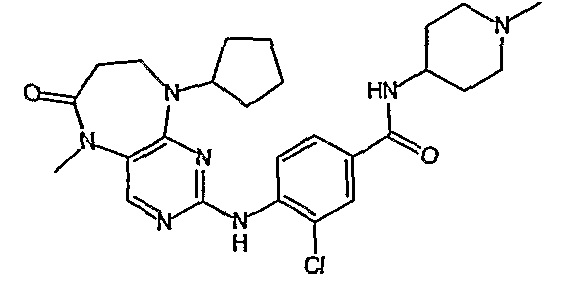
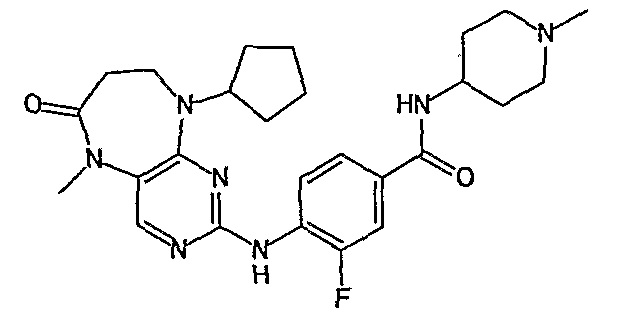
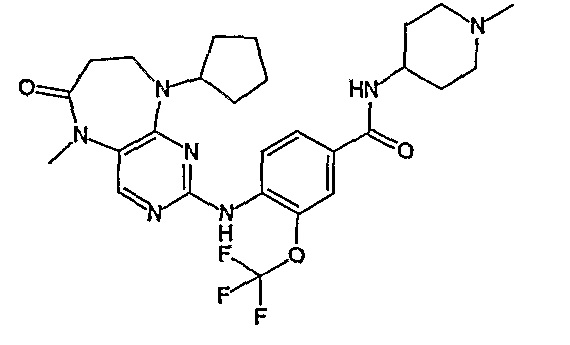
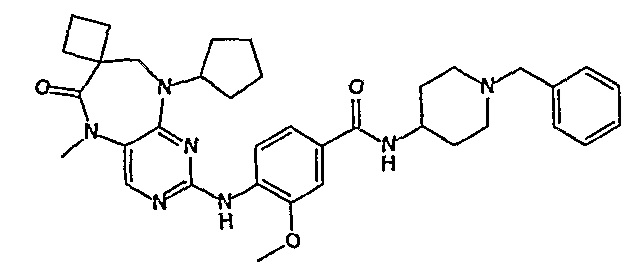
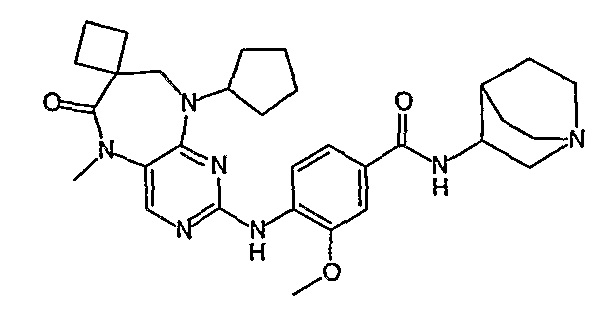
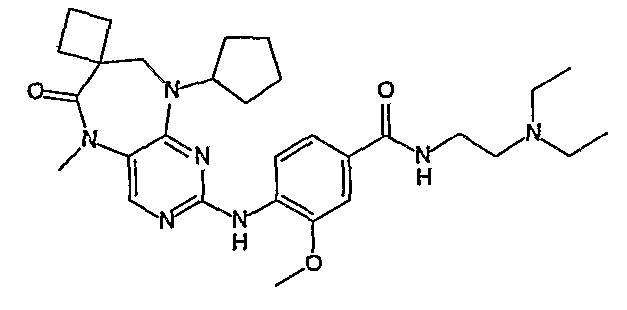
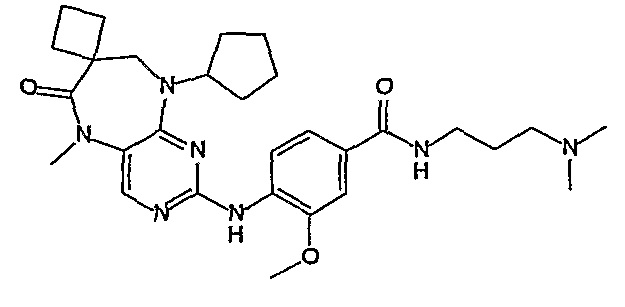
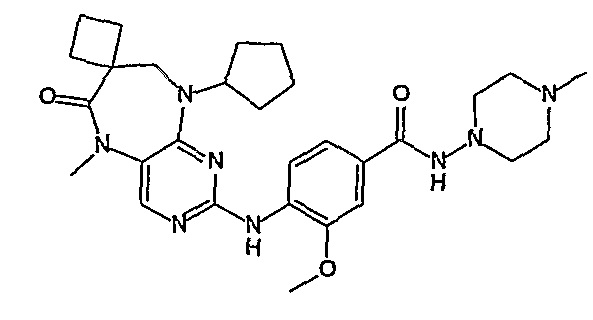
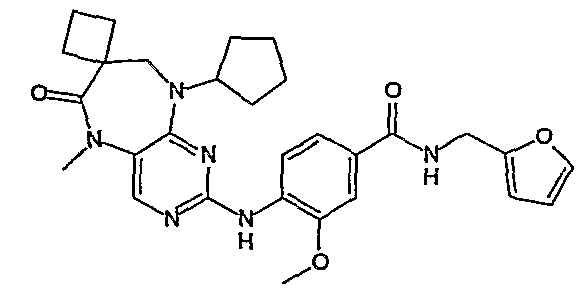
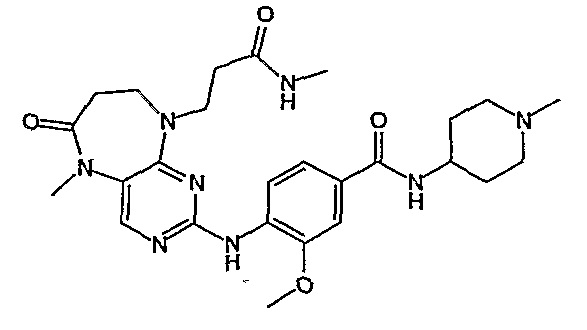
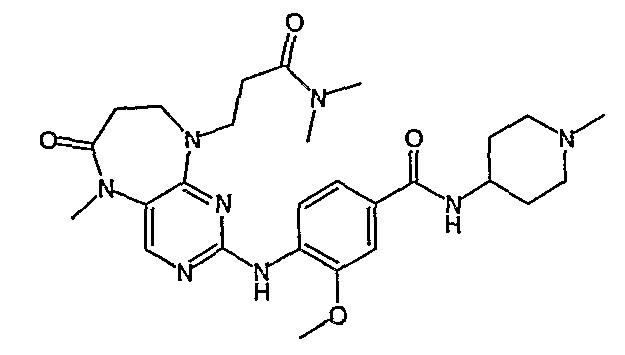
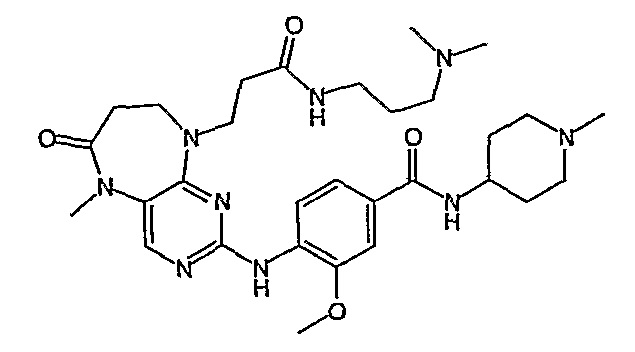
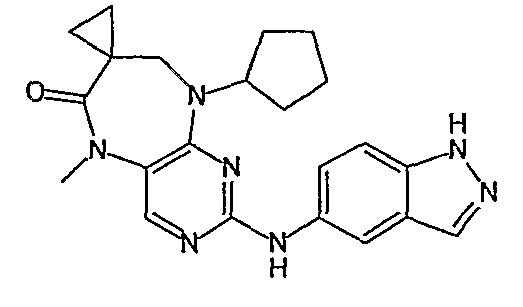
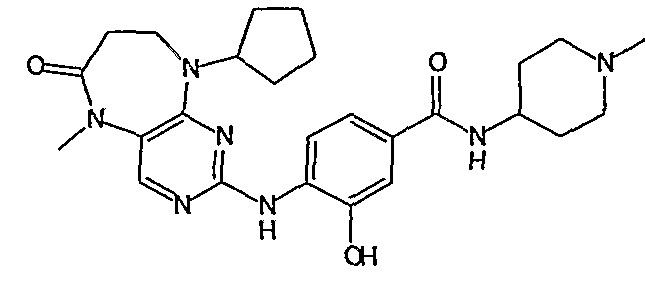


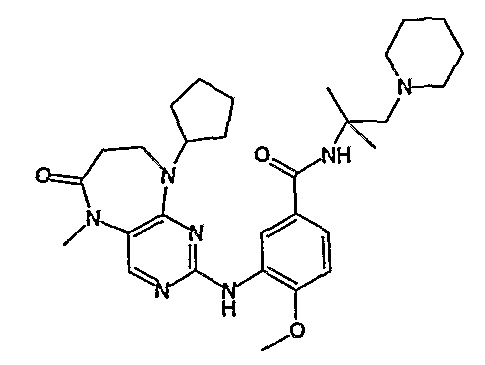
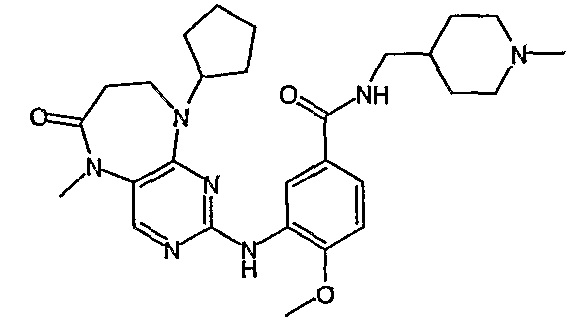
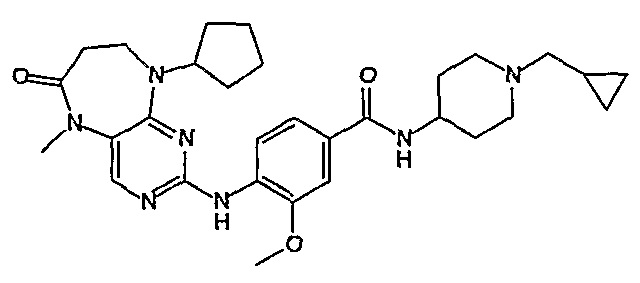
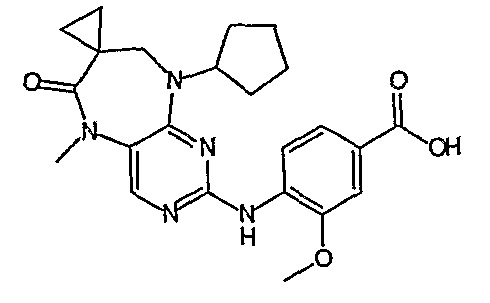
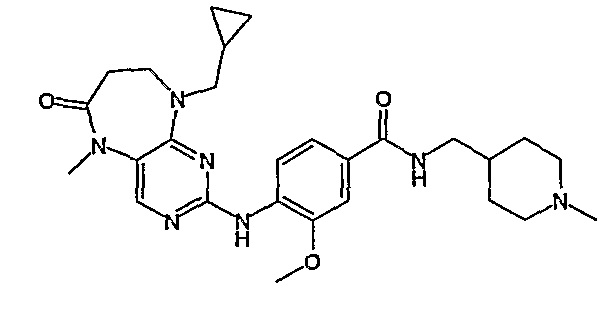
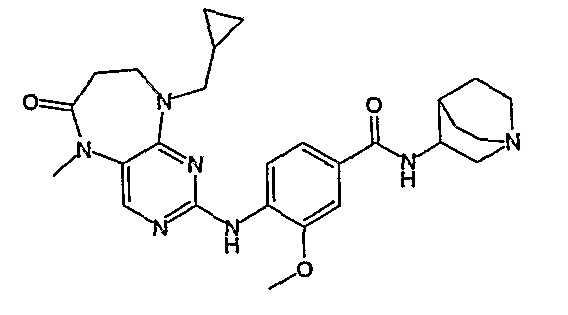
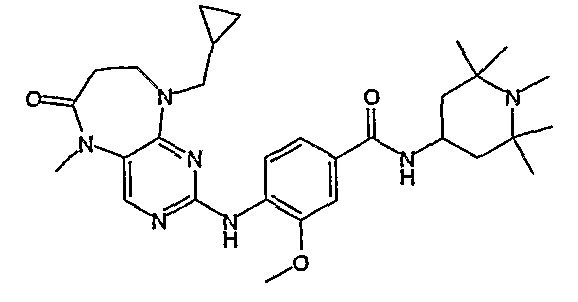
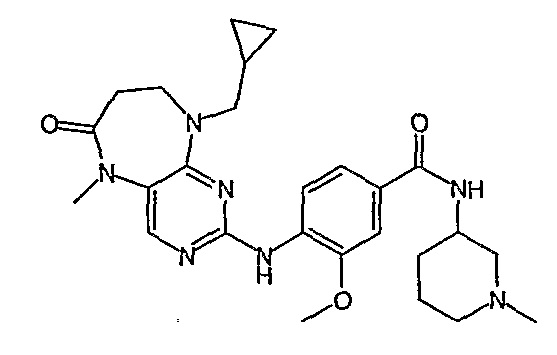
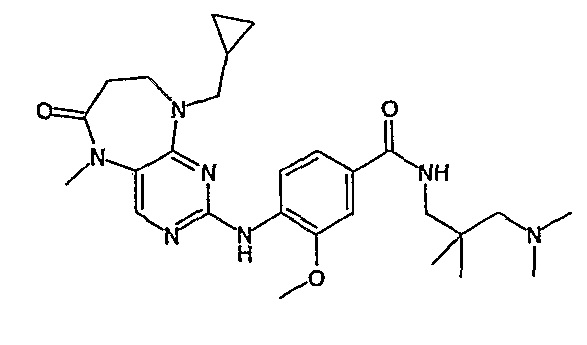
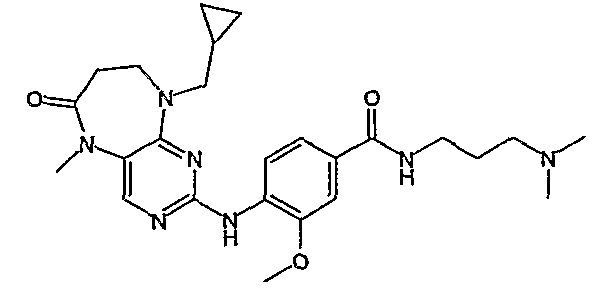
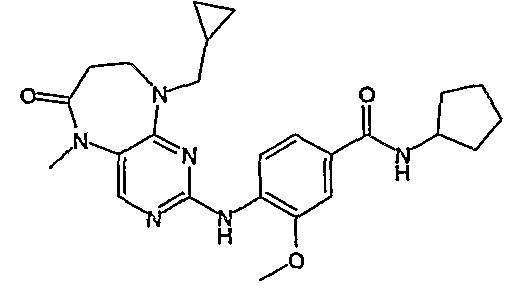
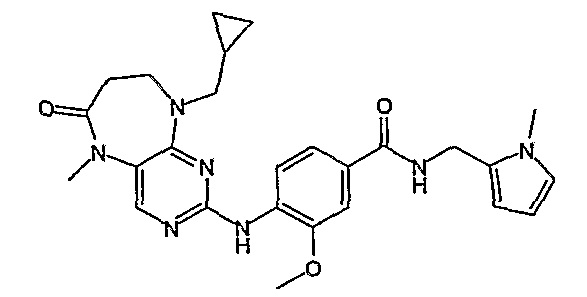
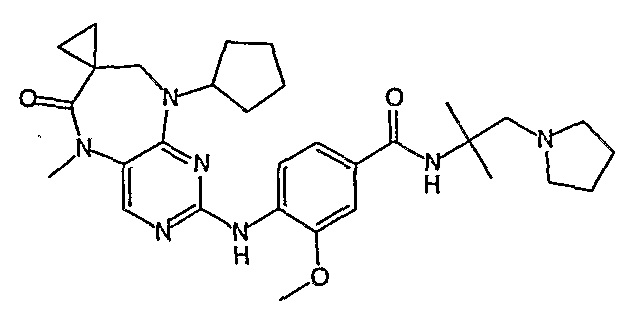
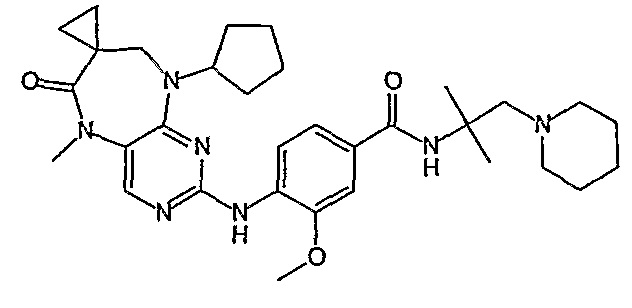
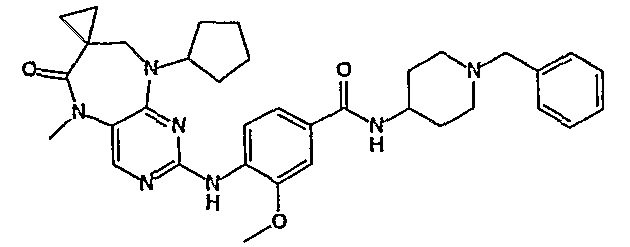
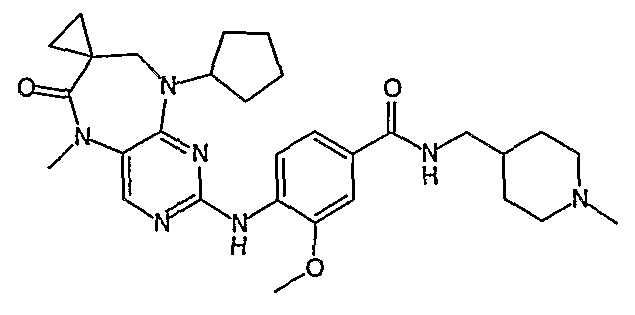
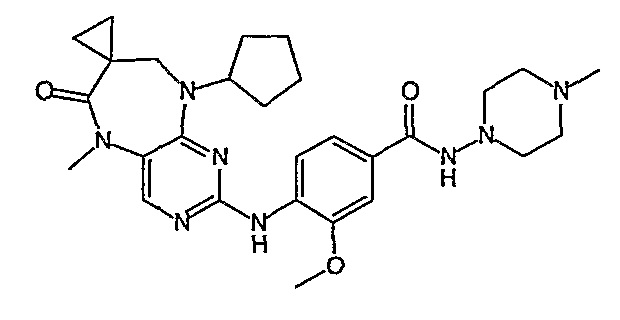
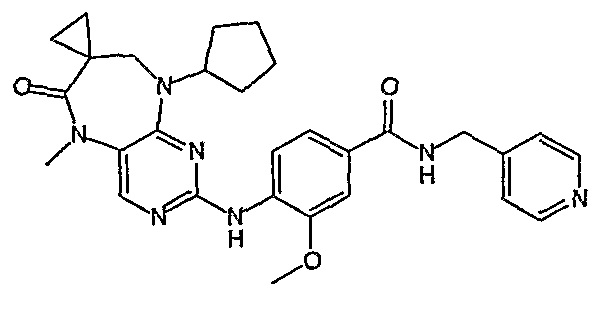

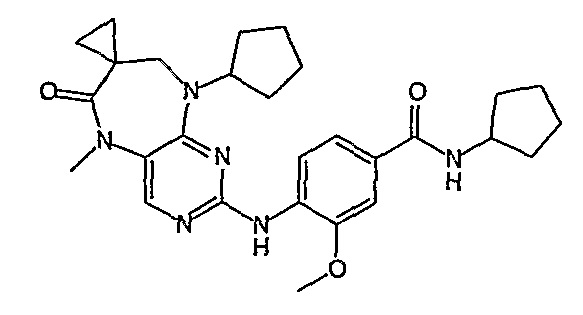
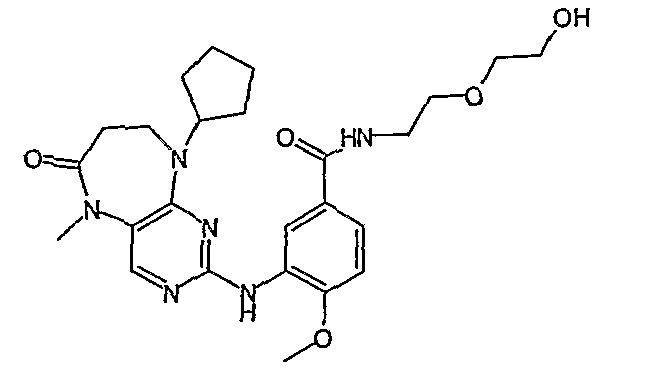
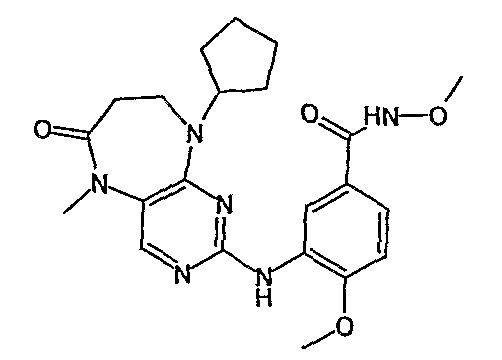
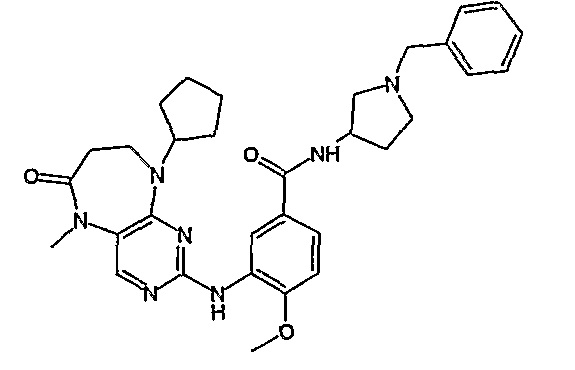

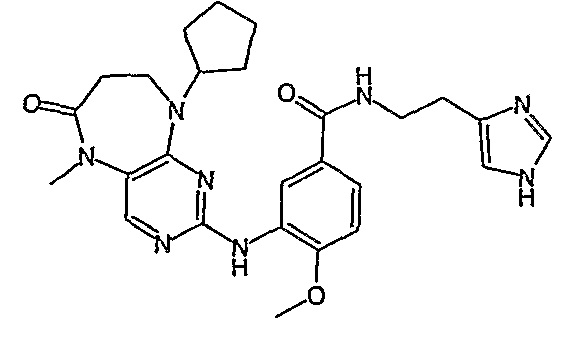
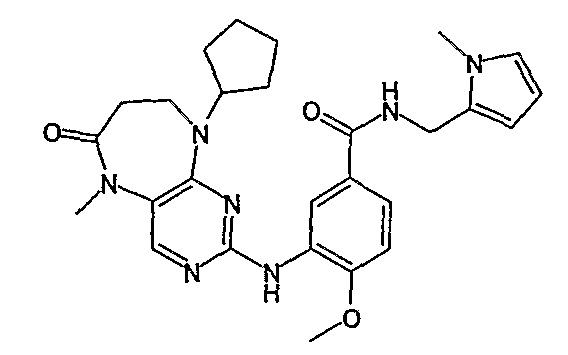
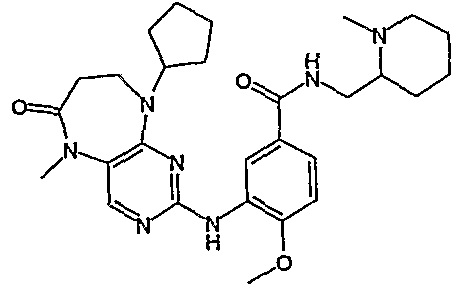
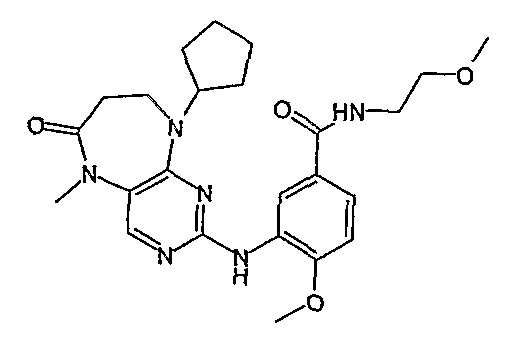
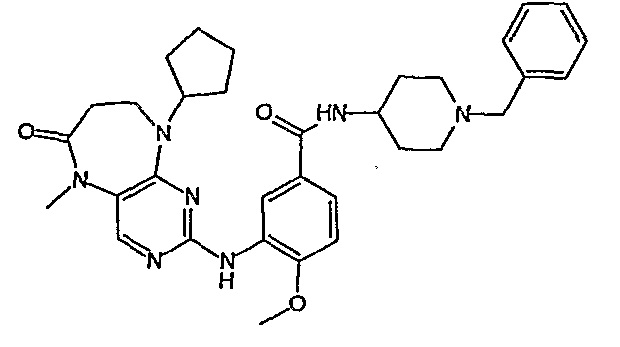
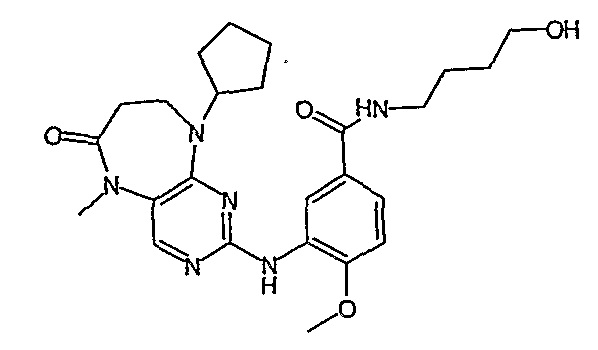
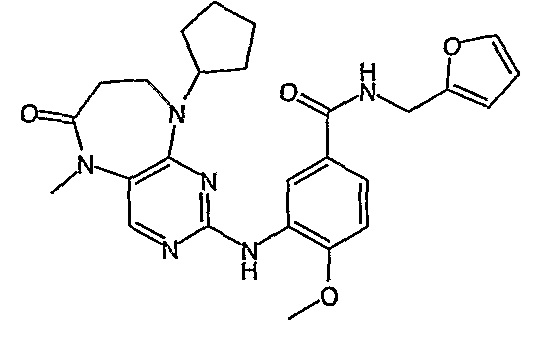
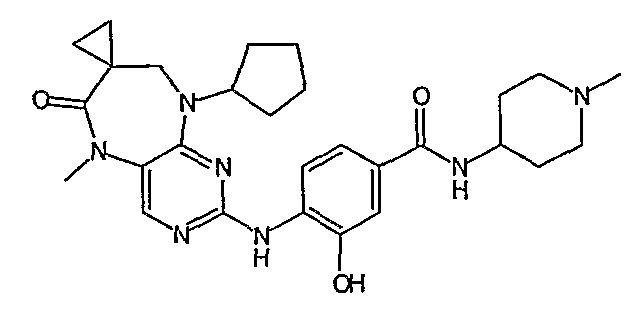
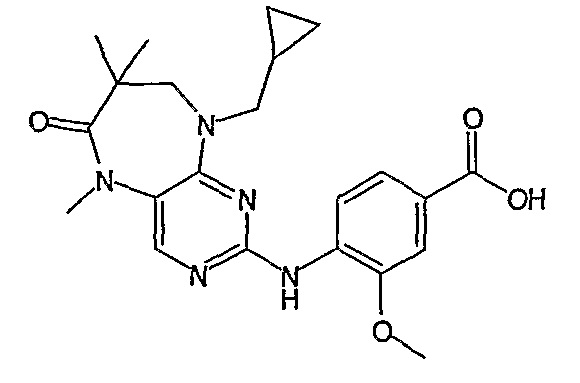
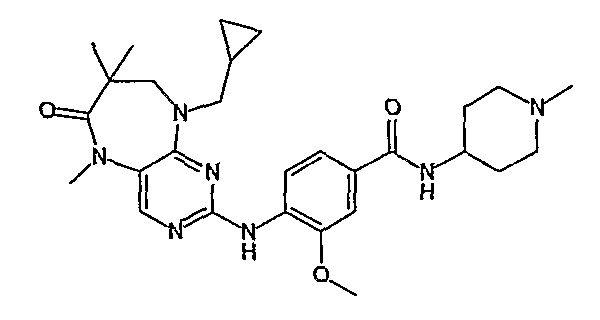

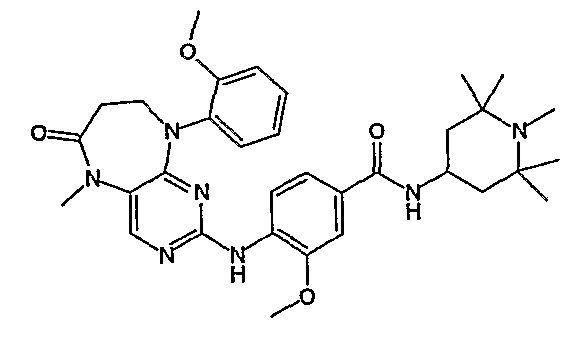
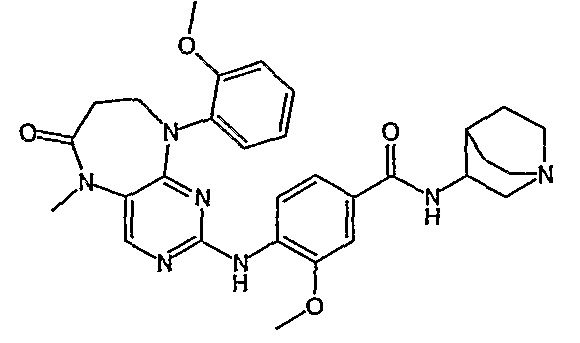

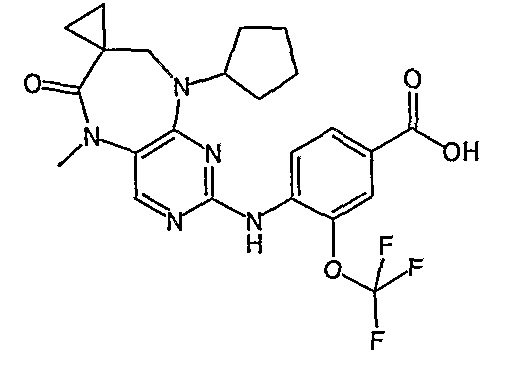
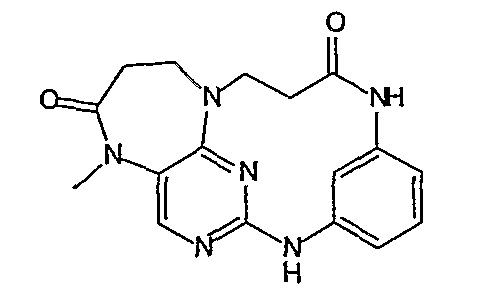
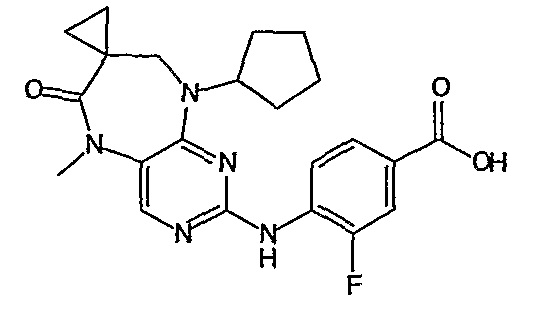
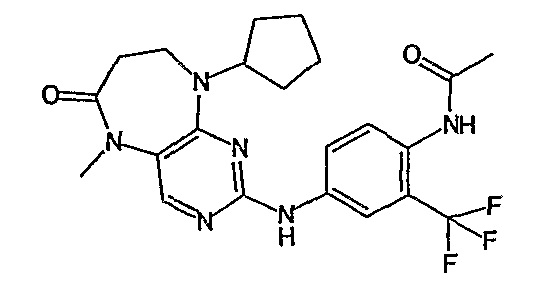
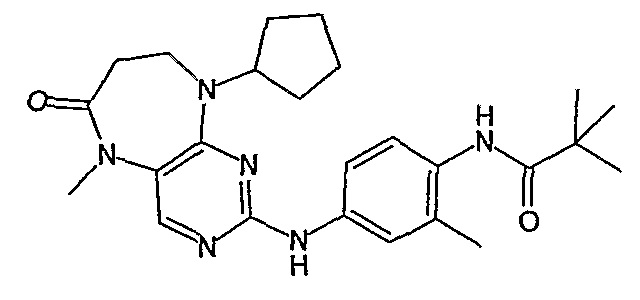
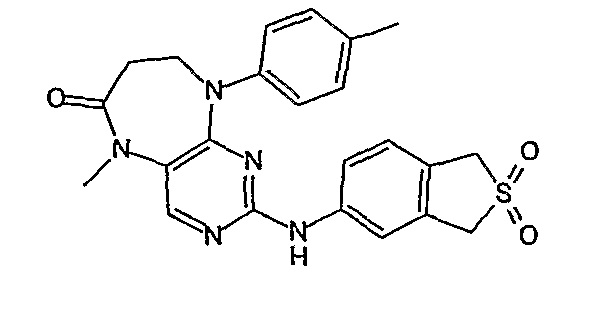
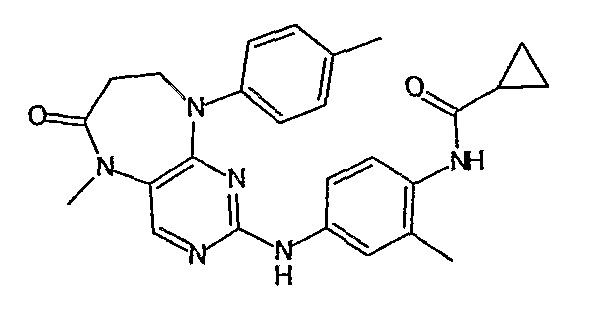
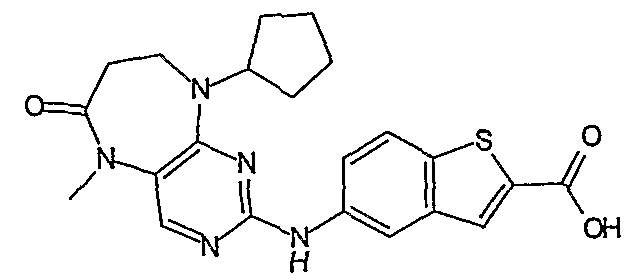
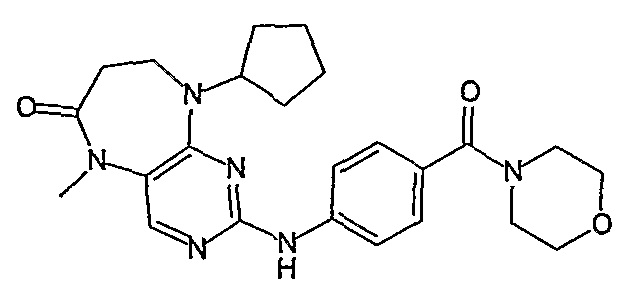

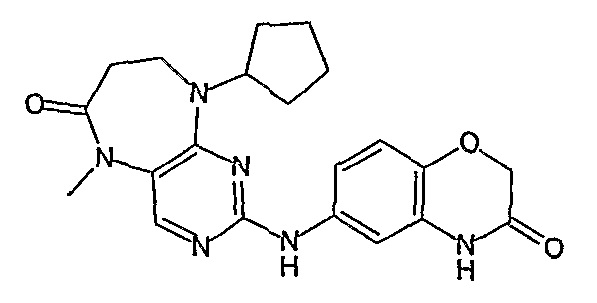
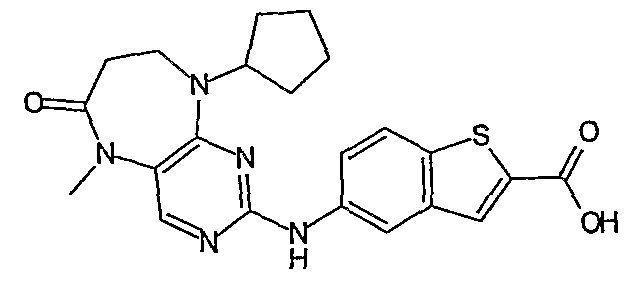
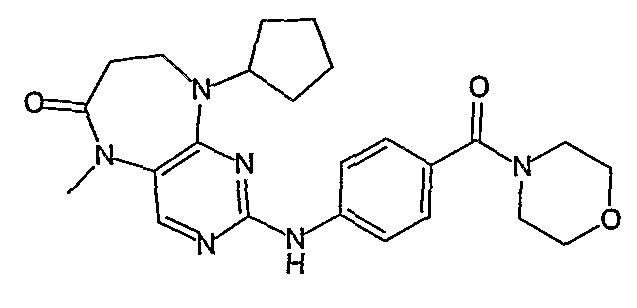
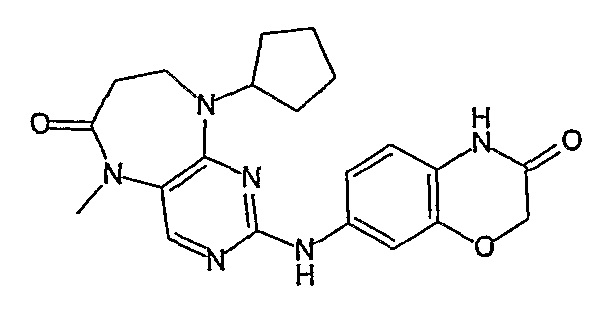
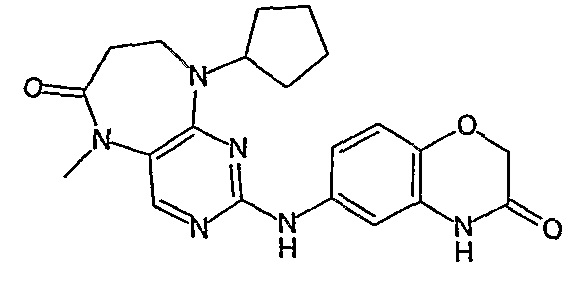
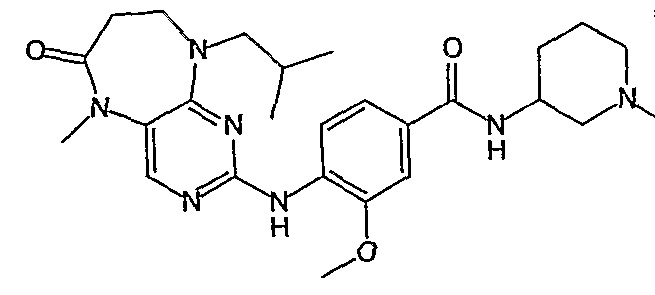
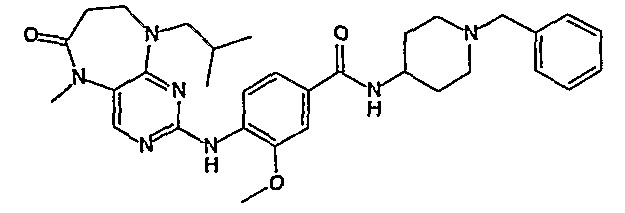
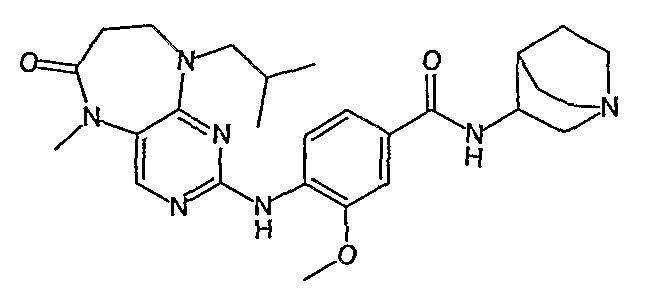
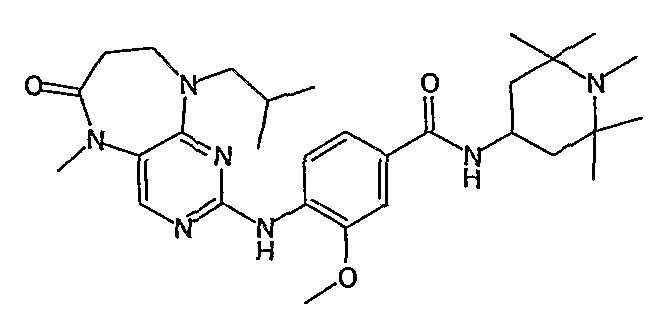

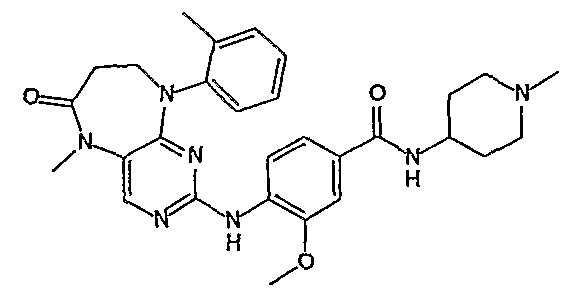
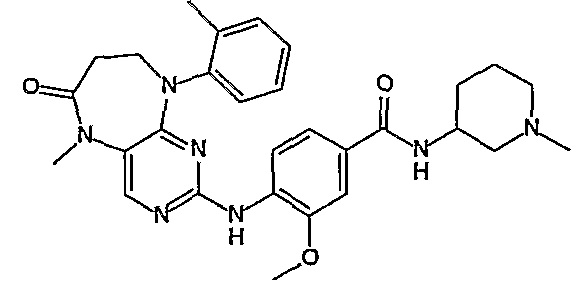

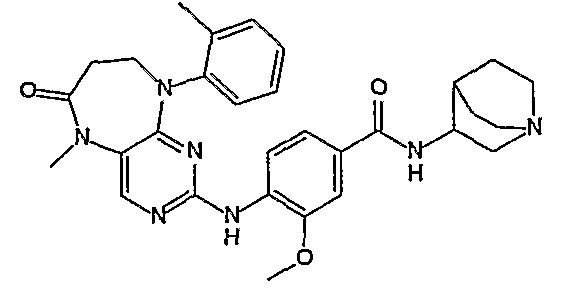
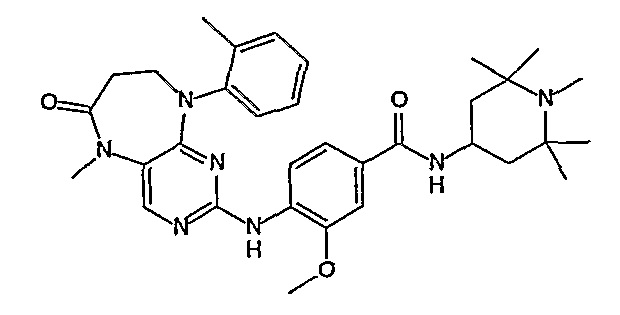
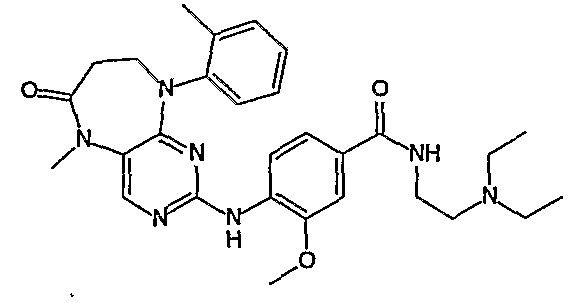
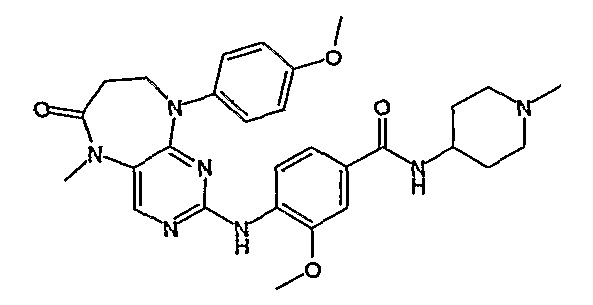

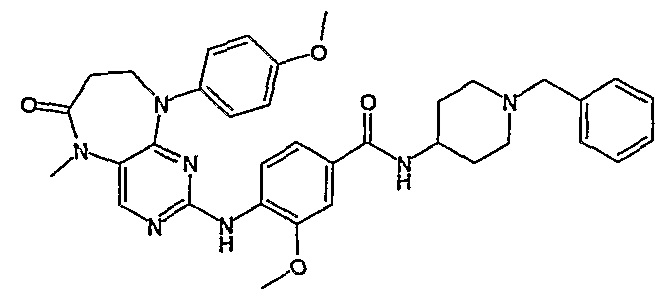
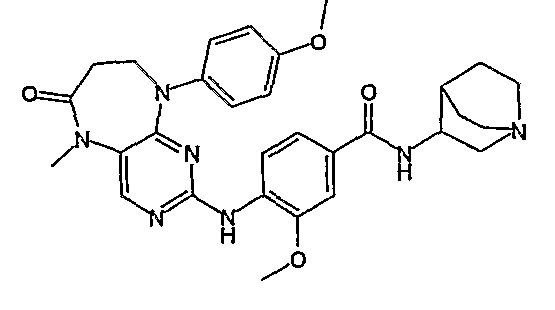
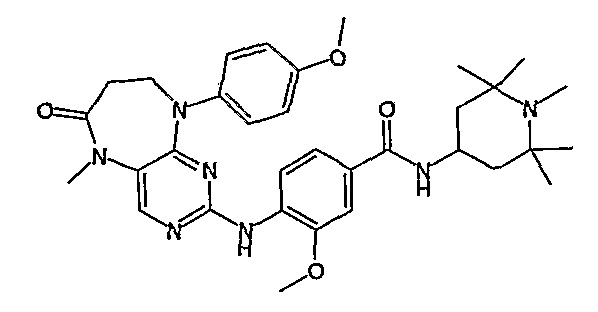
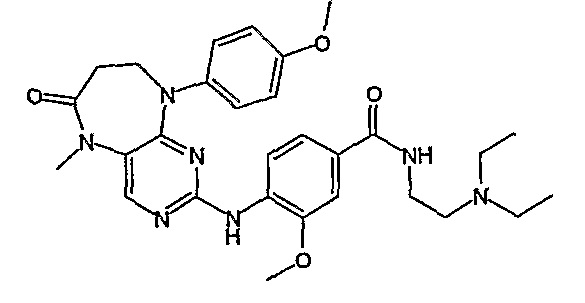
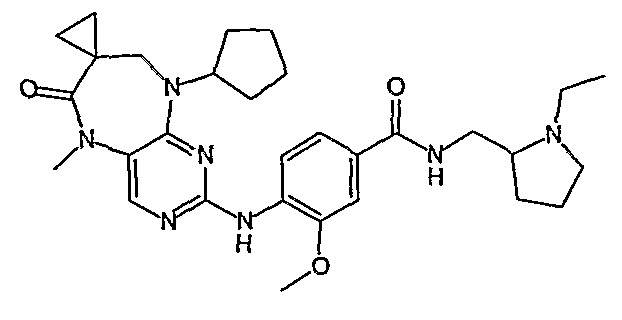
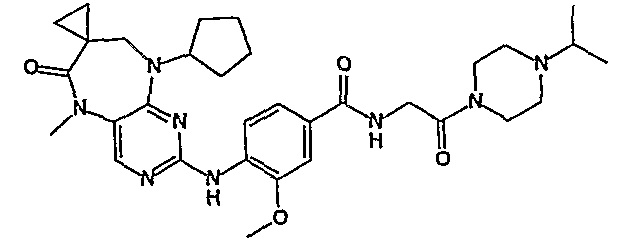
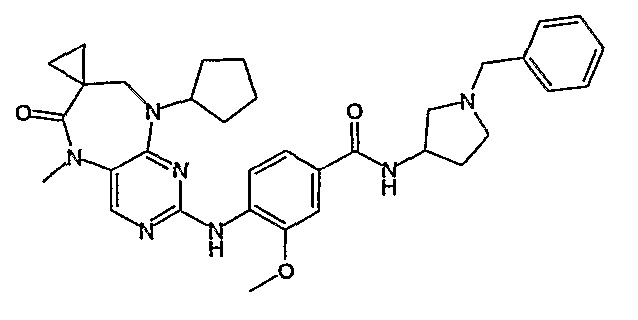
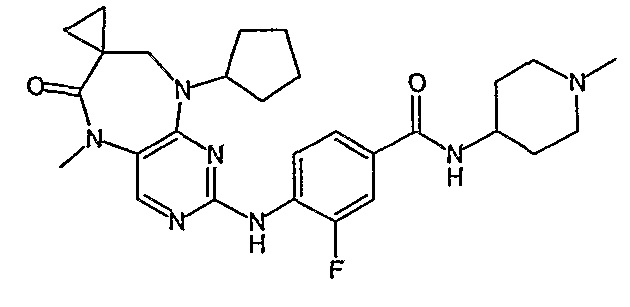
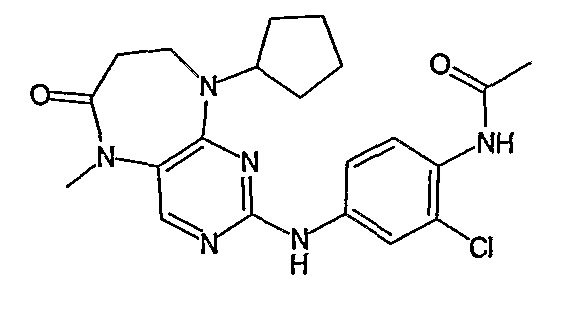
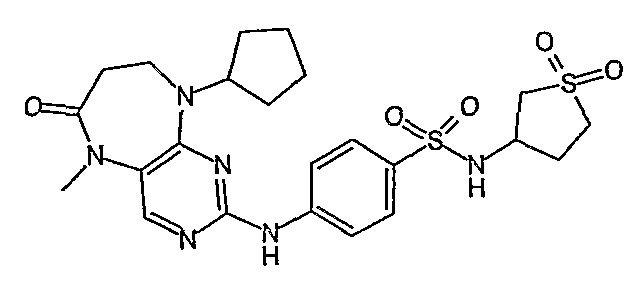
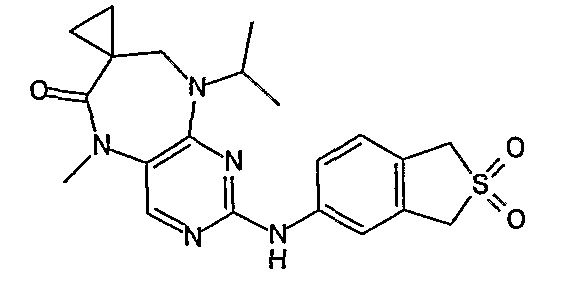
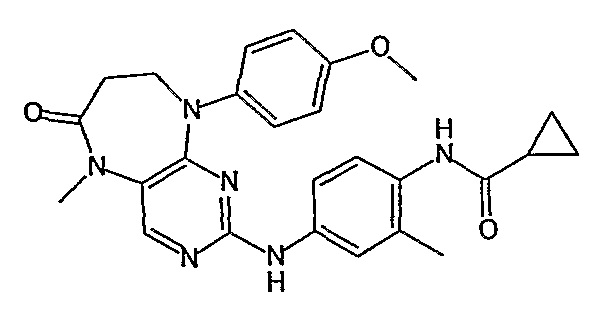
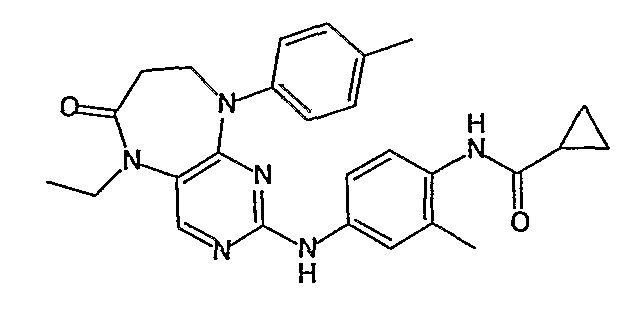
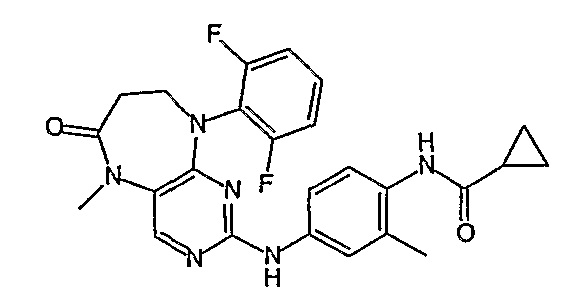
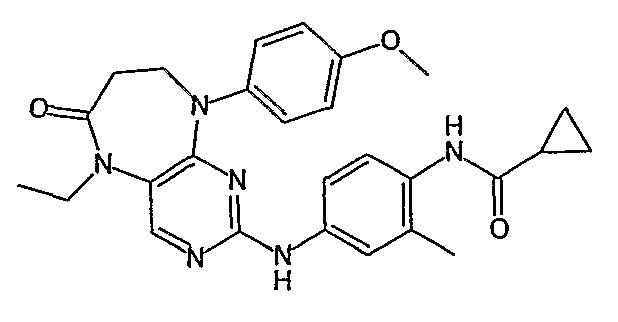
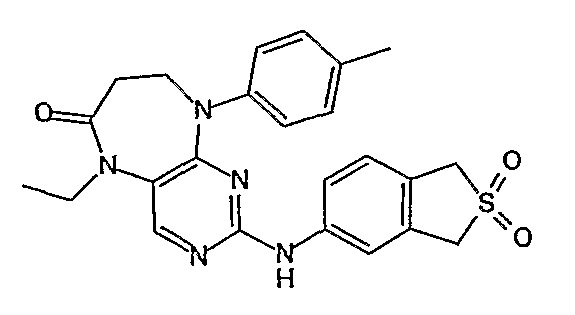
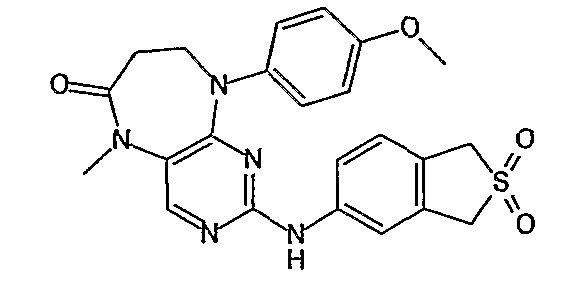
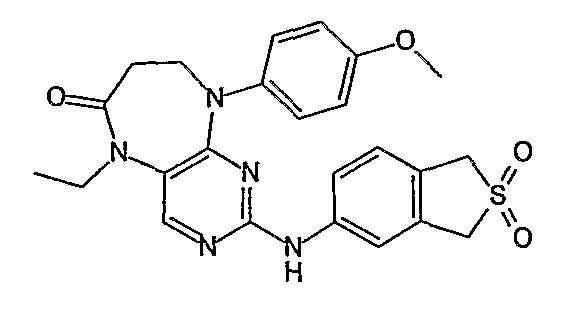
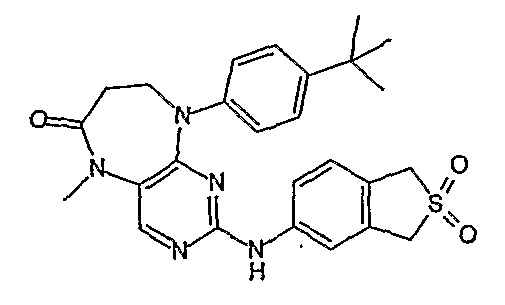
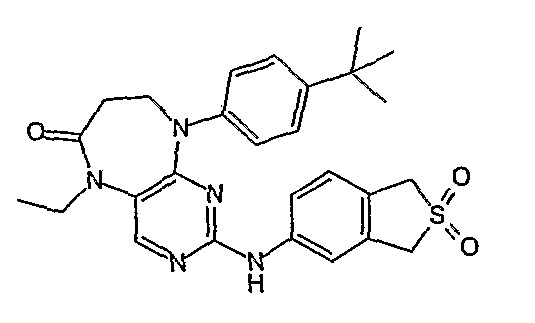
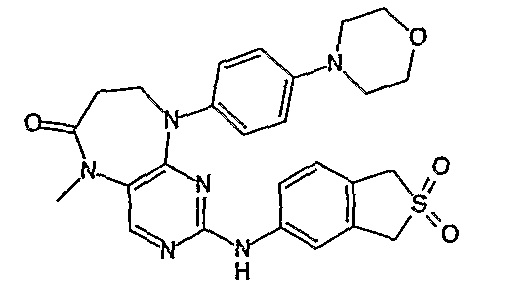

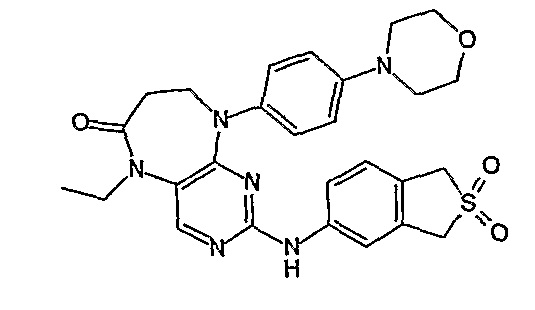
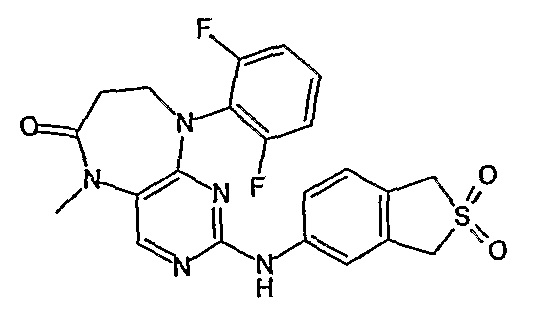
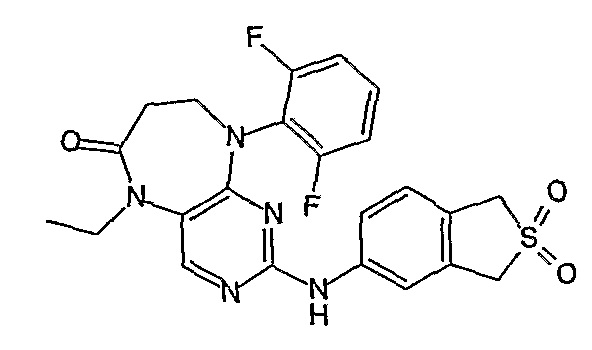





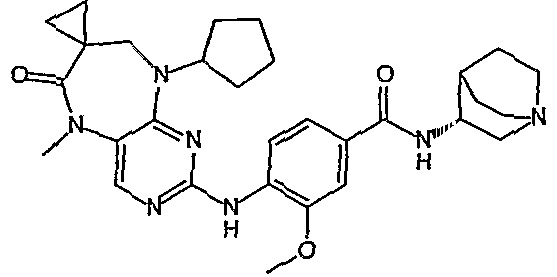
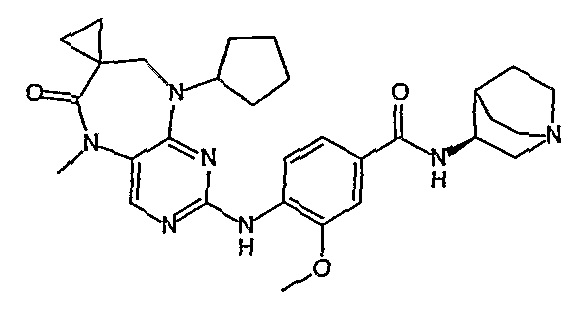
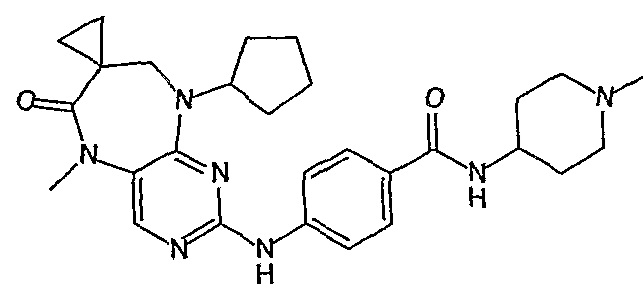


Величины IC50 для PLK и данные характеризации для выбранных соединений согласно изобретению; *** означает <0,1 мкМ IC50; ** означает 1 мкМ – 0,1 мкМ IC50; * означает 10 мкМ – 1 мкМ IC50
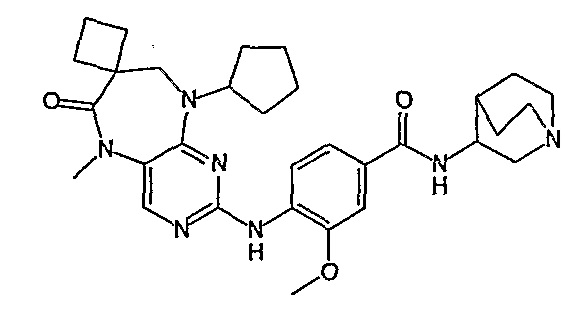

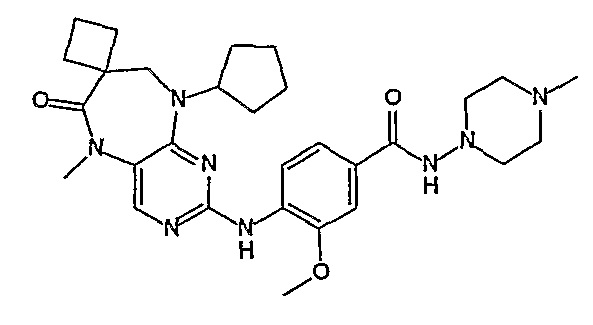
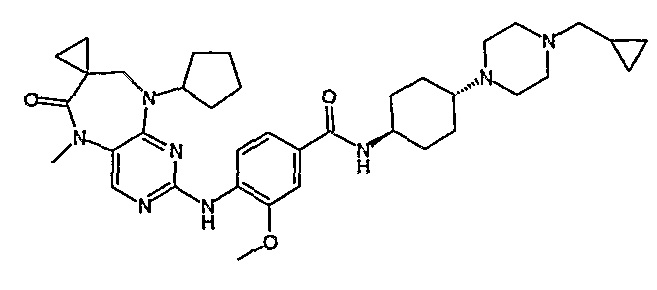
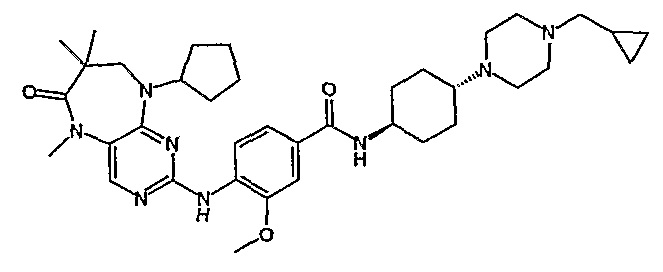

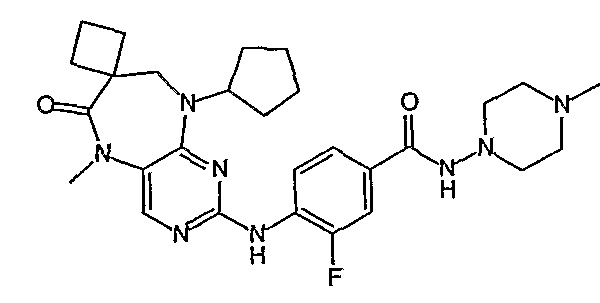
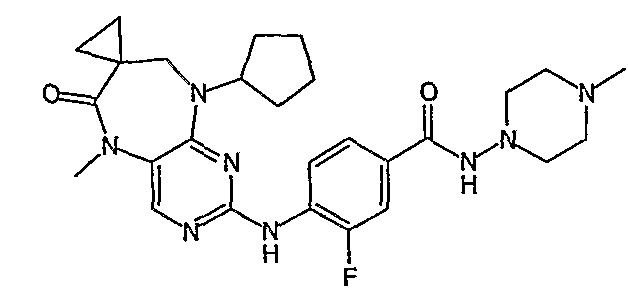
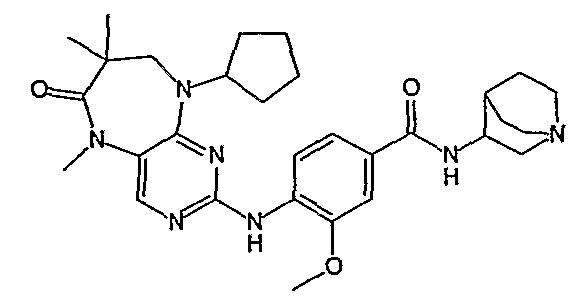
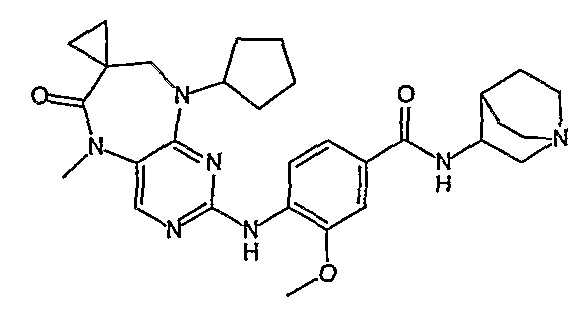

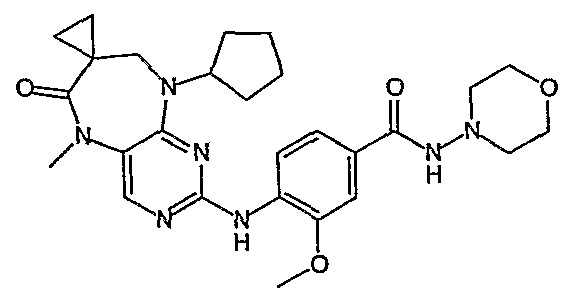

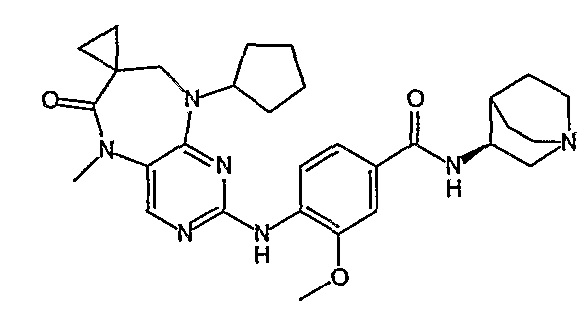
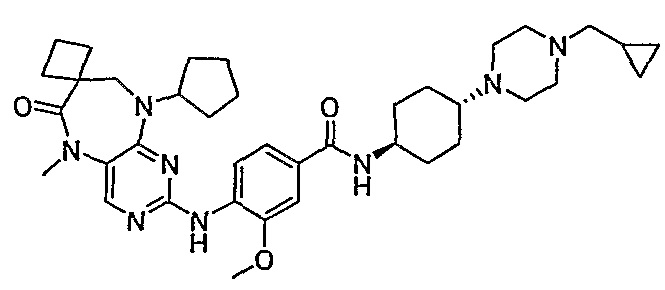
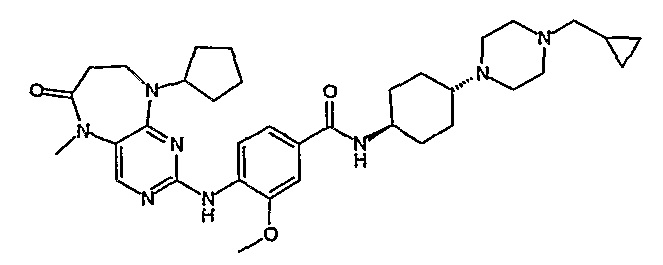
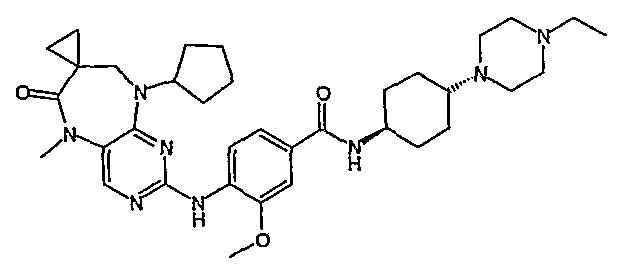
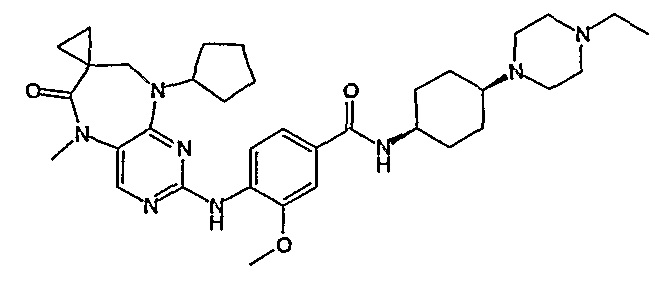
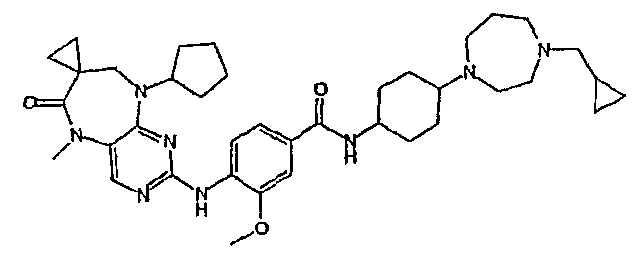
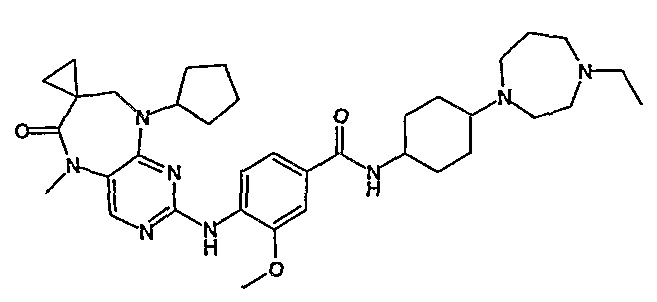
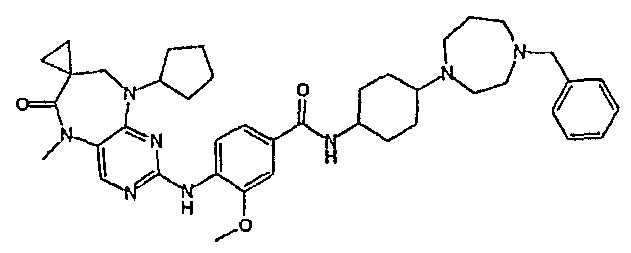
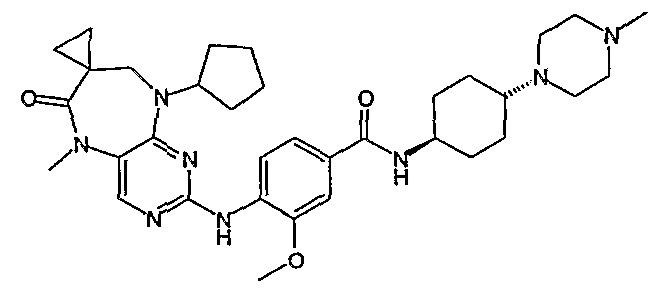

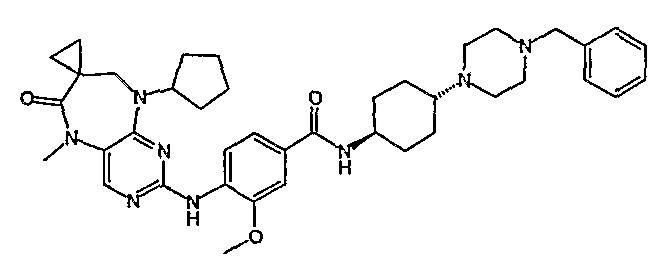
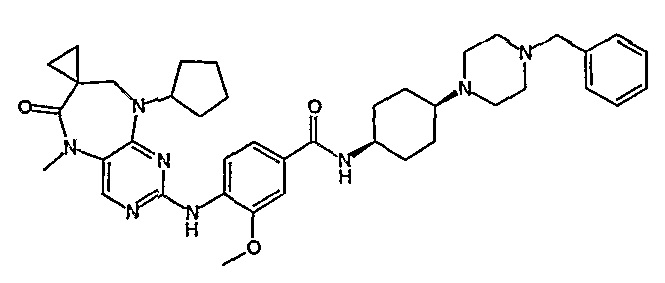
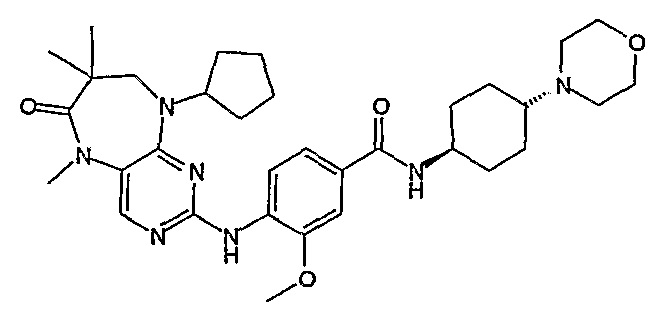
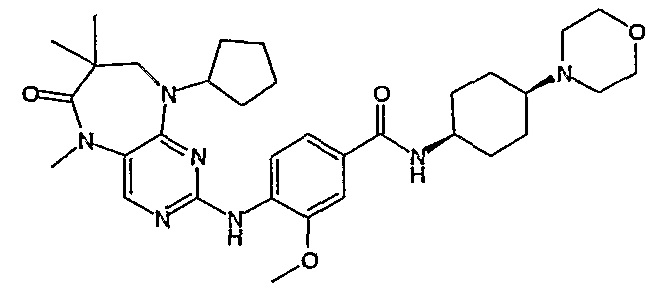
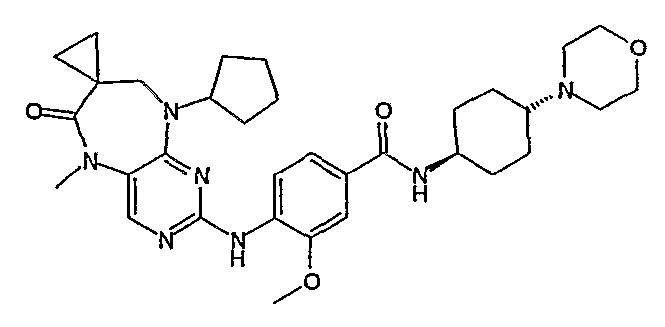
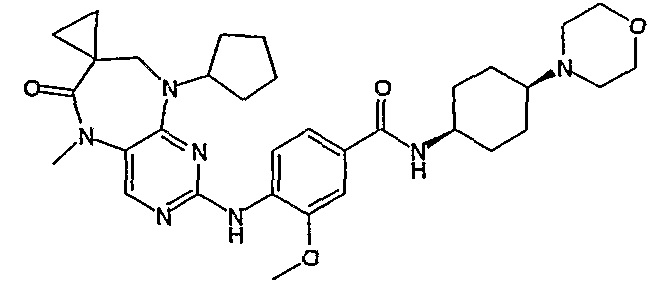
Данные PLK-активности для соединения [218] по сравнению с данными для выбранных известных соединений
IC50 для PLK1
IC50 для PLK1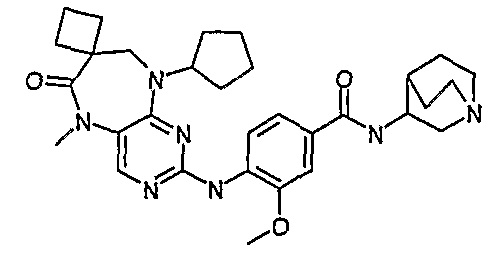
[I-64]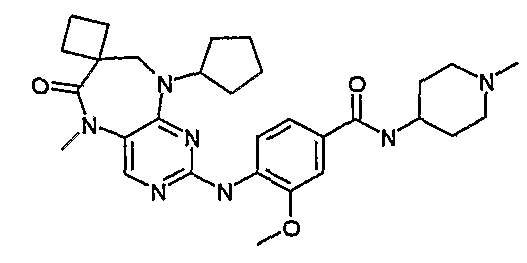
[I-76]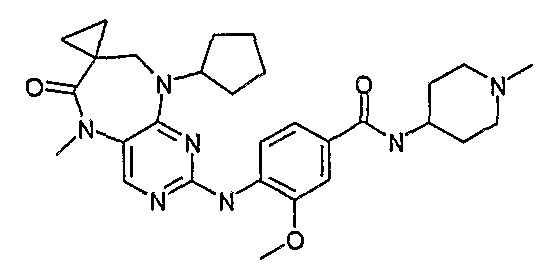
[I-4]
Данные растворимости для соединения [254] и его HCl-соли по сравнению с соединением примера [I-76] заявки WO 07/095188 и его HCl-соли
в расчете на свободное основание (мг/мл)
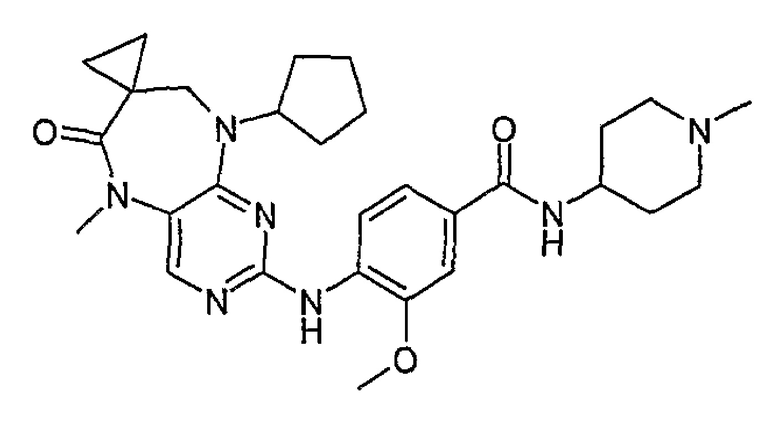
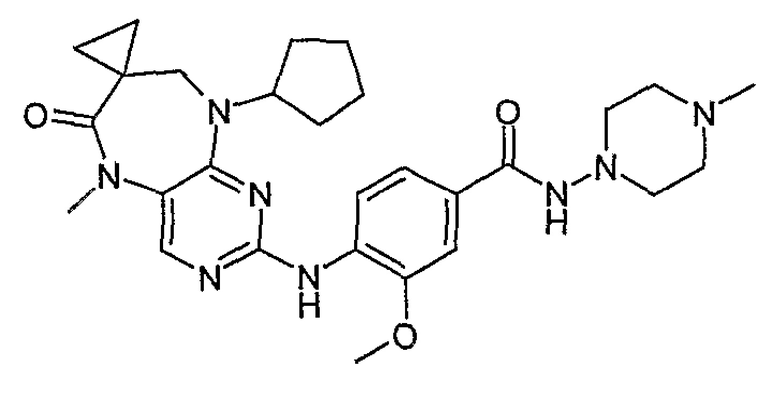
(= примера 246 WO 08/003958)
Нефелометрический анализ максимально растворимой концентрации соединения [254] согласно изобретению по сравнению с соединением [I-253] WO 07/095188
Соединения, протестированные в фармакокинетических исследованиях; соединение Н = соединение [371]; соединение I = соединение [378]; соединения A-G были использованы для сравнения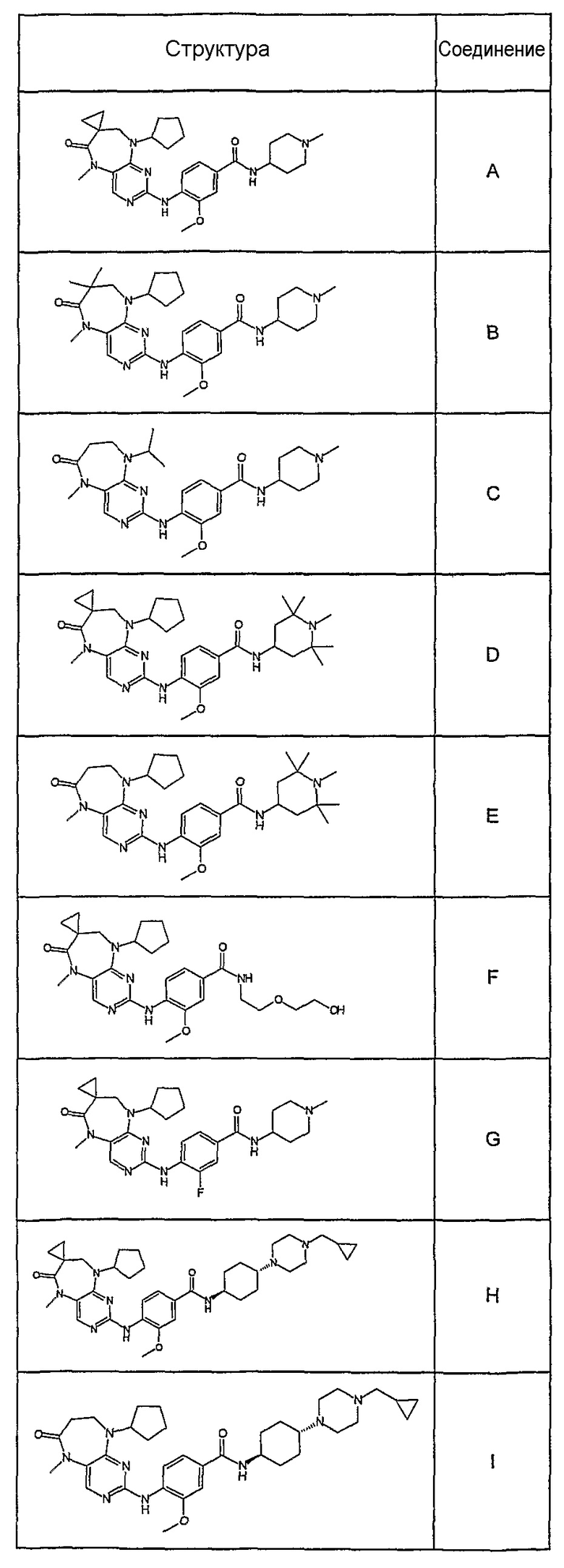
Соединения для сравнения, протестированные в клеточных анализах; буквы соответствуют буквам на фигуре 3.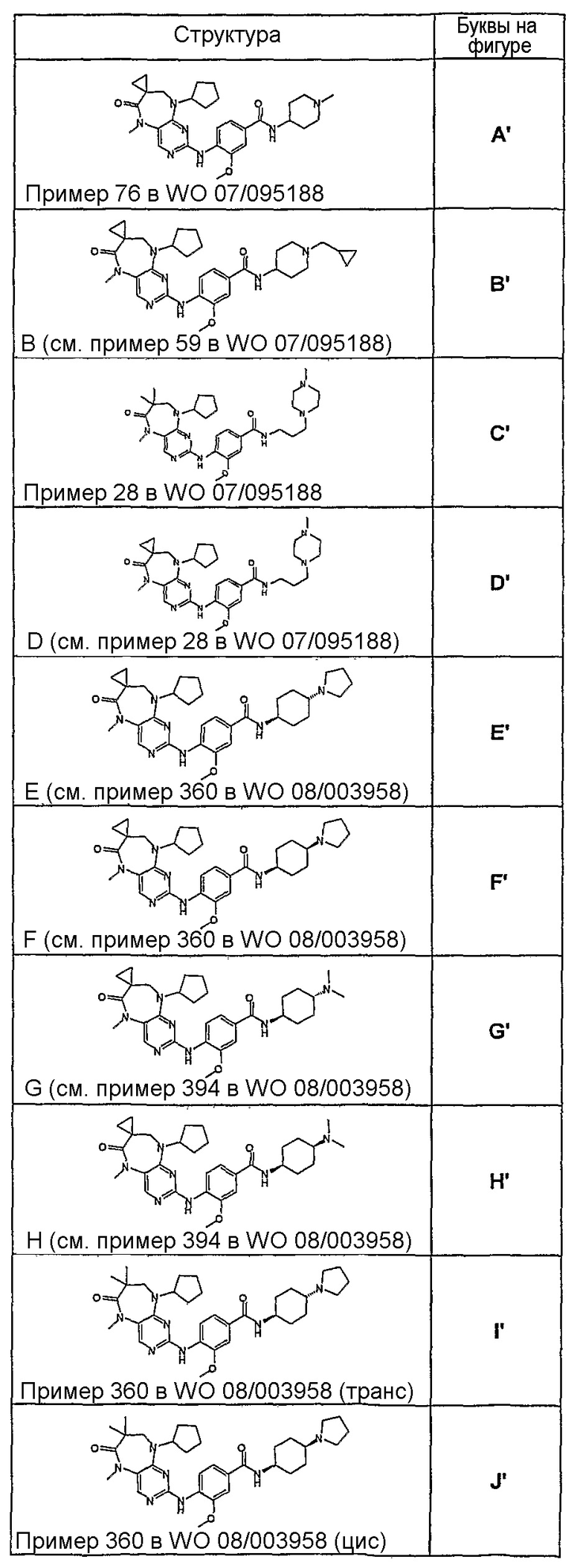
| название | год | авторы | номер документа |
|---|---|---|---|
| ПИРИМИДИНОВЫЕ ПРОИЗВОДНЫЕ, ИСПОЛЬЗУЕМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗЫ | 2008 |
|
RU2478100C2 |
| ДИГИДРОДИАЗЕПИНЫ, КОТОРЫЕ МОЖНО ИСПОЛЬЗОВАТЬ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗ | 2007 |
|
RU2475488C2 |
| БИЦИКЛИЧЕСКИЕ ЛАКТАМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2827714C1 |
| БИЦИКЛИЧЕСКИЕ ЛАКТАМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2716136C2 |
| ПРОИЗВОДНЫЕ ИМИДАЗОПИРАЗИНА ИЛИ ИМИДАЗОДИАЗЕПИНА, АКТИВНЫЕ В ОТНОШЕНИИ РЕЦЕПТОРА СВ2 | 2008 |
|
RU2540074C2 |
| ПОЛИЦИКЛИЧЕСКИЕ СРЕДСТВА ДЛЯ ЛЕЧЕНИЯ РЕСПИРАТОРНО-СИНЦИТИАЛЬНЫХ ВИРУСНЫХ ИНФЕКЦИЙ | 2004 |
|
RU2422444C2 |
| ТЕТРАГИДРОФУРО(3,2-b)ПИРРОЛ-3-ОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ КАТЕПСИНА К | 2007 |
|
RU2456290C2 |
| ЗАМЕЩЕННЫЕ БЕНЗОЛЬНЫЕ СОЕДИНЕНИЯ | 2012 |
|
RU2629118C2 |
| ПРОИЗВОДНЫЕ АЗАИНДАЗОЛА ИЛИ ДИАЗАИНДАЗОЛА В КАЧЕСТВЕ МЕДИКАМЕНТА | 2012 |
|
RU2600976C2 |
| ЗАМЕЩЕННЫЕ АМИДЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ, СПОСОБ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЯ, ЧУВСТВИТЕЛЬНОГО К Btk, СПОСОБ ПОВЫШЕНИЯ ЧУВСТВИТЕЛЬНОСТИ РАКОВЫХ КЛЕТОК К ХИМИОТЕРАПИИ, СПОСОБ УМЕНЬШЕНИЯ ОШИБОК ПРИ ПРИЕМЕ ЛЕКАРСТВА И УЛУЧШЕНИЯ СОБЛЮДЕНИЯ СХЕМЫ ЛЕЧЕНИЯ, СПОСОБ ИНГИБИРОВАНИЯ ГИДРОЛИЗА АТФ, СПОСОБ ОПРЕДЕЛЕНИЯ ПРИСУТСТВИЯ Btk В ОБРАЗЦЕ И СПОСОБ ИНГИБИРОВАНИЯ АКТИВНОСТИ В-КЛЕТОК | 2008 |
|
RU2470923C2 |
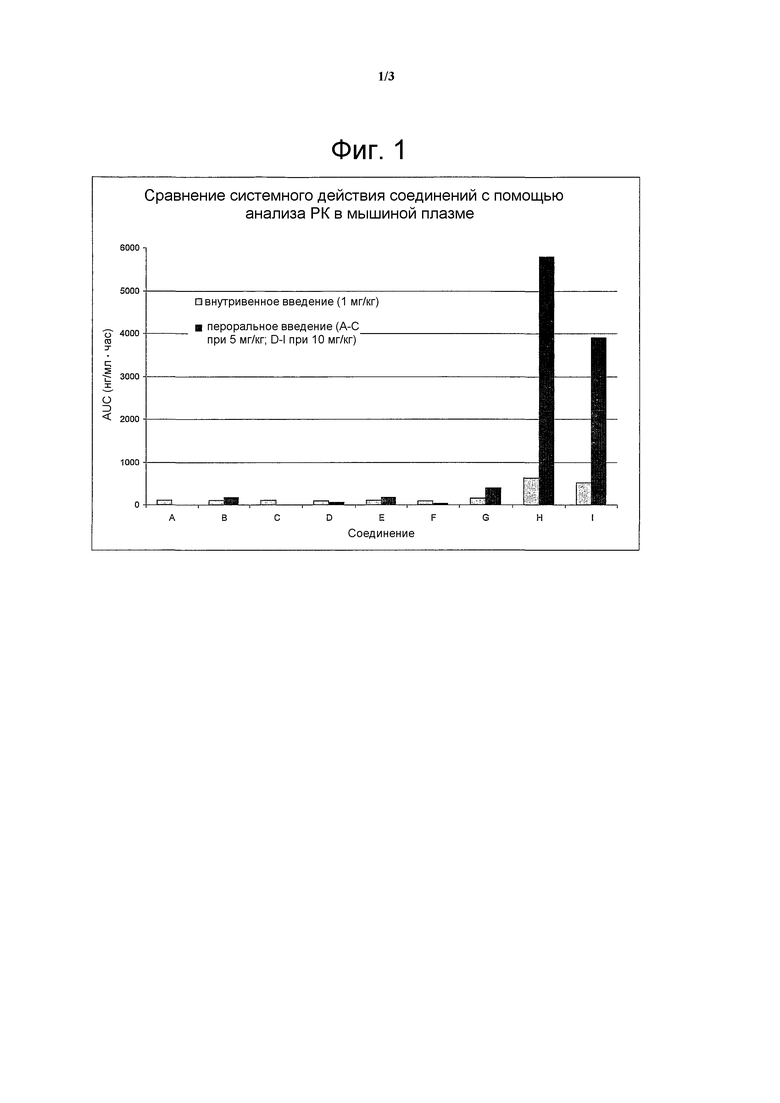
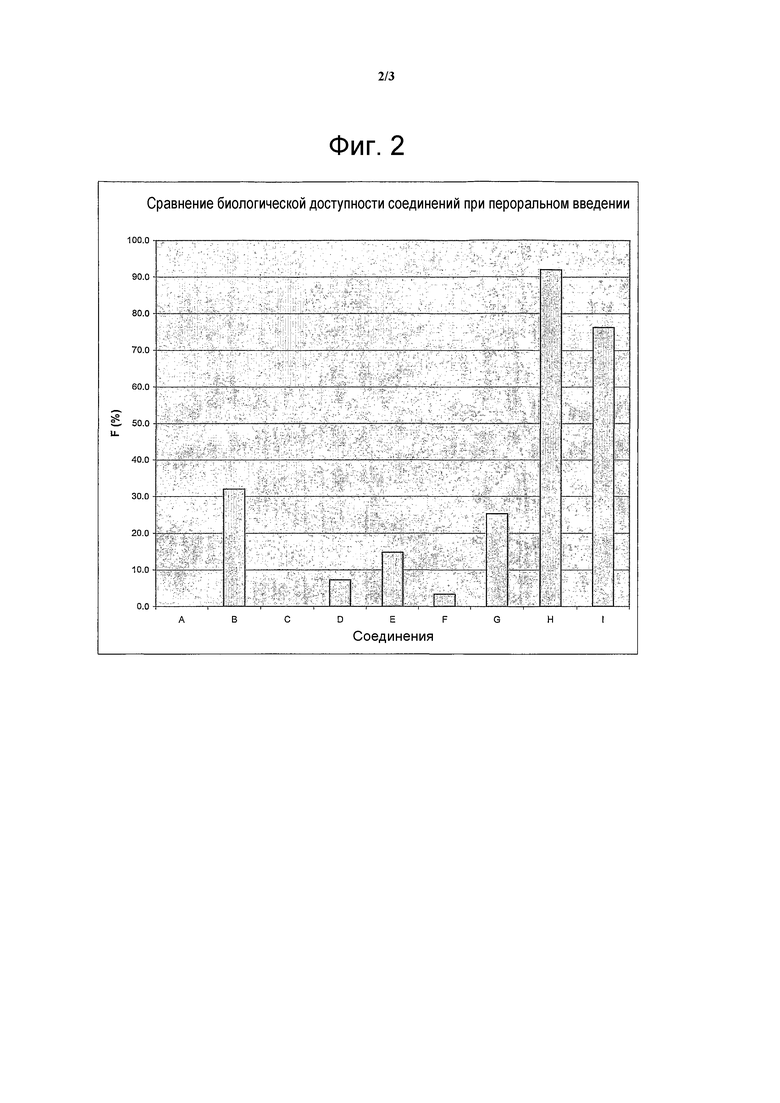
Изобретение относится к соединениям, выбранным из указанных ниже соединений 73 и 75, и их фармацевтически приемлемым солям, которые могут найти применение при лечении рака или лейкоза. Изобретение относится также к фармацевтической композиции, содержащей указанные соединения, применению указанных соединений для приготовления лекарственного средства для лечения рака или лейкоза и способу лечения данных заболеваний. 4 н. и 3 з.п. ф-лы, 3 ил., 8 табл., 9 пр.

1. Соединение, выбранное из следующих:
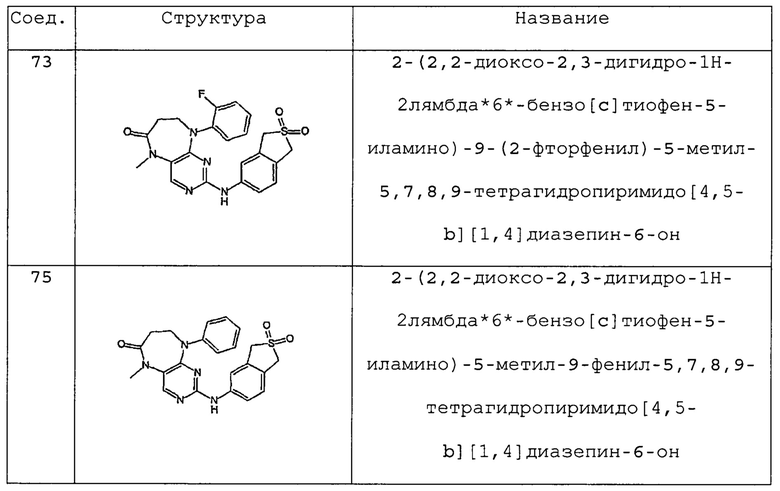
и его фармацевтически приемлемые соли.
2. Соединение по п.1, представляющее собой:
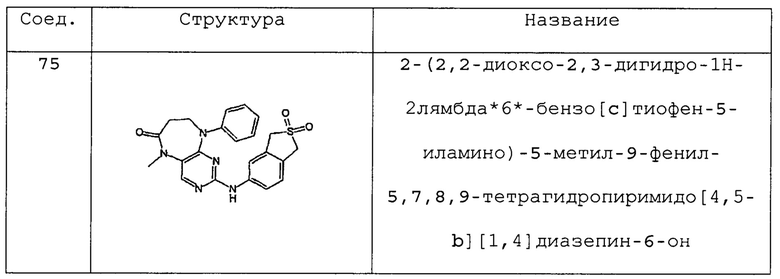
и его фармацевтически приемлемая соль.
3. Соединение по п.1, которое представляет собой:
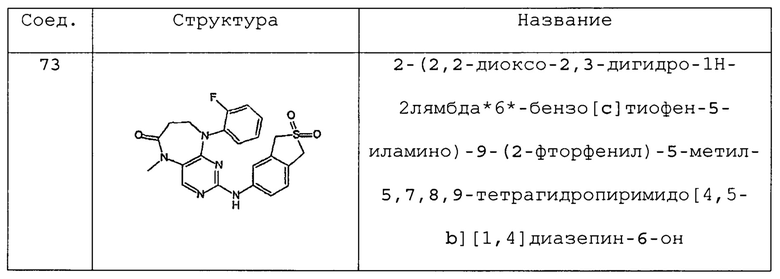
и его фармацевтически приемлемая соль.
4. Фармацевтическая композиция для лечения лейкоза, содержащая фармацевтически эффективное количество соединения по любому из предшествующих пунктов в смеси с фармацевтически приемлемым разбавителем, наполнителем или носителем.
5. Применение соединения по п.1 для приготовления лекарственного средства для лечения рака или лейкоза.
6. Способ лечения рака или лейкоза, где указанный способ включает введение индивидууму терапевтически эффективного количества соединения по п.1.
7. Соединение по п.1, которое может быть использовано для лечения рака или лейкоза.
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| RU 2009103810 A, 20.08.2010. | |||

