НАЗВАНИЕ ИЗОБРЕТЕНИЯ
Способ получения основанных на N-ациламинокислоте поверхностно-активных веществ с применением основанных на N-ациламинокислоте поверхностно-активных веществ или соответствующих ангидридов в качестве катализаторов.
ОБЛАСТЬ ТЕХНИКИ, К КОТРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к экономически эффективному двухстадийному способу изготовления основанных на аминокислоте поверхностно-активных веществ с применением тех же поверхностно-активных веществ в качестве катализаторов для синтеза промежуточного соединения высокого качества и с высоким количественным выходом. Более конкретно, настоящее изобретение относится к способу получения основанных на N-ациламинокислоте поверхностно-активных веществ путем ускорения синтеза хлорида жирной кислоты посредством катализатора на основе тех же основанных на N-ациламинокислоте поверхностно-активных веществ, которые подлежат изготовлению.
ПРЕДПОСЫЛКИ И УРОВЕНЬ ТЕХНИКИ ИЗОБРЕТЕНИЯ
Основанные на N-ациламинокислоте поверхностно-активные вещества широко применяются в целях личной гигиены в дополнение к другим промышленным применениям. Они попадают под категорию анионных поверхностно-активных веществ и являются значительно более мягкими, нежели остальные анионные поверхностно-активные вещества. Например, поверхностно-активные вещества, такие как натрия лауроилсаркозинат, натрия миристоилсаркозинат, кокоил-глицинат натрия, натрия кокоил-N-метилтаурат, коммерчески используются в средствах для очищения кожи, средствах для мытья тела, так как они обладают хорошей очищающей способностью и являются более мягкими для кожи и волос по сравнению с другими анионными поверхностно-активные вещества. Алканоильные саркозинаты находят применение в жидкостях для полоскания рта и средствах для ухода за зубами, как правило, из-за их бактериостатической активности. Существует множество других областей, где N-ацилированные аминокислоты применяют коммерчески в коммерческих целях, например, в нефтепродуктах в качестве смазывающих веществ, в металлообработке и флотации руды. (N-acylated amino acid as surfactants, J.D. Spivack, Chapter 16, в 'Anionic surfactants, Vol 7, Surfactant Science Series, Edited by W. M. Linfield). В коммерческих целях их изготовляют посредством двухстадийного синтеза, в который вовлечена жирная кислота и различные аминокислоты, такие как глицин, саркозин, N-метилтаурин, аланин, аспарагиновая кислота, глутаминовая кислота, глутамин и аргинин. Таковы некоторые из наиболее часто применяемых аминокислот, которые применяются для изготовления основанных на N-ациламинокислоте поверхностно-активных веществ. Однако фактически все аминокислоты, хиральные или рацемические, природные или синтетические могут применяться при изготовлении основанных на N-ациламинокислоте поверхностно-активных веществ. Также, аминокислоты, которые применяются при изготовлении поверхностно-активных веществ, не должны представлять собой α-аминокислоты. Также, кислотная группа в этих аминокислотах может представлять собой любую другую кислотную группу, отличную от карбоксильной группы. Аминосульфоновые кислоты (например, N-метилтаурин) были конденсированы с хлоридами жирных кислот для создания коммерческих поверхностно-активных веществ, таких как натрия N-метил-N-кокоил-таурат. При изготовлении основанных на N-ациламинокислоте поверхностно-активных веществ жирная кислота или смесь жирных кислот реагирует с функциональной аминогруппой аминокислот через промежуточное образование хлорида жирной кислоты при обычных условиях Шоттена-Баумана, как показано в схеме 1 (патент США №27907799 (1953), патент США №2790779 (1957), патент США №3945931(1974)).
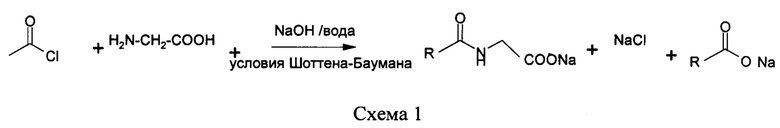
Конденсацию Шоттена-Баумана между хлоридом жирной кислоты и аминокислотой обычно осуществляют в водной среде, однако сообщается также о применении систем смешанных растворителей (растворителя и воды) (патент США №6703517 (2002), патент США №6569829 (1999), публикация заявки на патент США №2005/0085651 (2004) и WO 2009/065590 (2009)). В нескольких других патентах сообщается о получении и очистке основанных на N-ациламинокислоте поверхностно-активных веществ, которые, по сути, получены по тому же пути конденсации хлорида жирной кислоты с аминокислотами (патент Японии №2923101 (1991), публикация заявки на патент Японии №04-149163 (1990), патент Японии №3362468 (1993)).
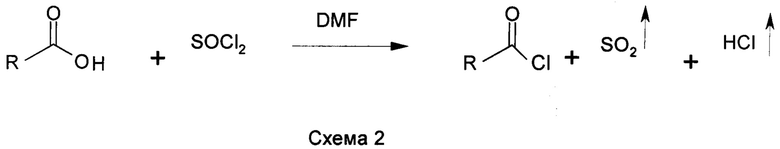
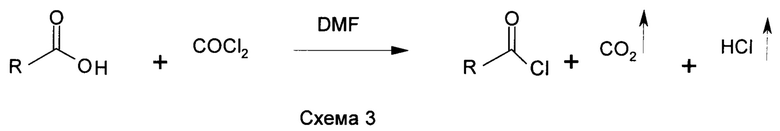
Предшественники основанных на N-ациламинокислоте поверхностно-активных веществ, хлориды жирных кислот в промышленных масштабах изготавливают путем реакции жирных кислот и галогенирующего средства, либо фосгена, либо тионилхлорида, как показано в схемах 2 и 3. Хлорирование обычно катализируют N,N-диметилформамидом (DMF). DMF или подобные замещенные формамиды образуют комплекс (комплекс Вильсмейера) с COCl2 или SOCl2, который является фактической каталитической частицей (US 5430186; US 5623082; US 5200560; US 5278328 и US 5166427) при хлорировании кислот.
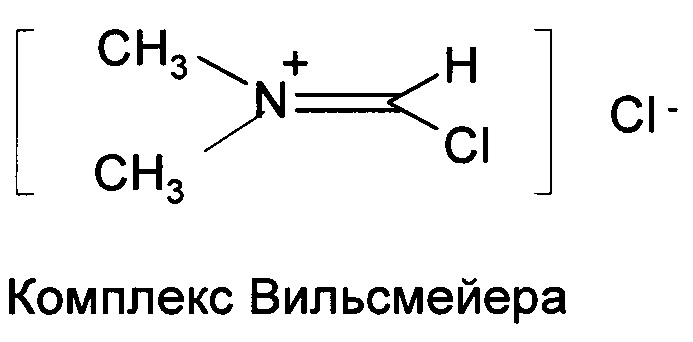
В конце реакции выделение хлорида жирной кислоты (продукта) из комплекса катализатора всегда было сложной для преодоления задачей. Было предпринято несколько попыток, но чистое отделение комплекса катализатора от продукта на основе хлорангидрида кислоты, как правило, невозможно. (патенты США №№5166427 (1992), 5200560 (1993), 6770783 (2004) 6727384 (2004) и 5247105 (1993).
Серьезные недостатки и осложнения, вытекающие вследствие присутствия данного темноокрашенного комплекса катализатора (комплекса Вильсмейера) в продукте, хорошо описаны, и большинство попыток было предпринято в отношении улучшения "разделения фаз", то есть разделения фазы комплекса катализатора от фазы продукта.
Таким образом, удаление DMF или подобных каталитических веществ либо разделением фаз, либо разделением на фракции/дистилляцией влечет за собой дополнительную обработку и потерю выхода ценного продукта. Большую часть соединений Вильсмейера, образованных таким образом, отделяют при завершении реакции, обеспечивая разделение фаз. Комплекс карбоксамид-хлорирующее средство, как правило, осаждают в виде черной/коричневой смолы на дне реакционного сосуда. Разделение фаз является наиболее неудачным способом очистки хлоридов кислот, как раскрыто в патентах США №№5166427 (1992), 5200560 (1993), 6770783 (2004), и 6727384 (2004), а также 5247105 (1993).
Дистилляция (фракционирование) является еще одним способом выделения продукта, но не все ангидриды кислот поддаются дистилляции. Комплекс катализатора (комплекс Вильсмейера) существует в ионной форме и, следовательно, не так просто от него избавиться путем дистилляции/фракционирования хлоридов кислот. Комплекс продолжает разлагаться во время дистилляции. Во-вторых, потери дистилляции/фракционирования (фракции с формамидным катализатором и остаток, оставшийся после дистилляции) являются неизбежными.
Другой серьезной проблемой является токсичность этих формамидов или каких-либо других органических молекул, подобных DMF. DMF внесен в список опасных веществ и, как сообщается, имеет продолжительный токсический эффект и относится к классу опасности для здоровья 2. Также, данные о токсичности отсутствуют для других формамидов, аналогов DMF, которые обладают способностью катализатора, но, как ожидается, обладают подобной или большей токсичностью. В дополнение к проблеме опасности для здоровья, связанной с формамидами, аналоги DMF страдают от той же сложности выделения хлорида жирной кислоты (продукта) из реакционной массы при применении в качестве катализаторов, так как они образуют комплексы Вильсмейера с галогенирующими средствами. Следовательно, хлориды жирных кислот, изготовленные путем галогенирования жирных кислот либо с фосгеном, либо с тионилхлоридом с применением формамидов, ацетамидов или каких-либо других аналогов, таких как катализаторы, требуют дополнительных этапов очистки, таких как дистилляция, разделение фаз или кристаллизация и т.д. (DE 2656126 (1977)). Эти дополнительные стадии ведут к значительной потере выхода, большему потреблению энергии и большему времени производственного цикла, что ведет к более низкой производительности.
Так как получение ключевых промежуточных соединений, хлоридов жирных кислот, с помощью доступных в настоящее время способов существующего уровня техники является затруднительным и неэффективным, оно оказывает влияние на качество и стоимость всех продуктов ниже по потоку, таких как основанные на N-ациламинокислоте поверхностно-активные вещества с мировым потреблением, оцениваемым в 250000 метрических тонн.
Следовательно, существует необходимость в значительном улучшении способа изготовления, с помощью которого возможно снизить потери, связанные с дистилляцией, и избежать любых других этапов очистки промежуточного соединения, а также уменьшить время производственного цикла, обеспечивая более высокую производительность.
Настоящее изобретение относится к изготовлению основанных на аминокислоте поверхностно-активных веществ с применением тех же поверхностно-активных веществ в качестве катализаторов для синтеза промежуточного соединения высокого качества и с высоким количественным выходом. Общий способ является "экологически безвредным" (значительно снижено время производственной серии, низкое потребление энергии) без каких-либо отходов и образования сточных вод (отсутствует остаток после дистилляции/фракционирования) и чрезвычайно экономически эффективным (низкое потребление энергии), производительным (более высокая скорость катализа). Более того, в данном способе, описанном в данной заявке, избегают применения токсичных катализаторов, при этом данный способ является применимым к целому классу семейства основанных на N-ациламинокислоте поверхностно-активных веществ.
ЦЕЛИ ИЗОБРЕТЕНИЯ
Целью настоящего изобретения является преодоление недостатков предшествующего уровня техники.
Другой целью настоящего изобретения является обеспечение способа изготовления основанных на аминокислоте поверхностно-активных веществ с применением тех же поверхностно-активных веществ в качестве катализаторов для синтеза промежуточного соединения высокого качества и с высоким количественным выходом.
Еще одной целью настоящего изобретения является обеспечение экономически-эффективного двухстадийного способа для промышленного изготовления целого класса основанных на N-ациламинокислоте поверхностно-активных веществ.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к способу получения основанных на N-ациламинокислоте поверхностно-активных веществ формулы I
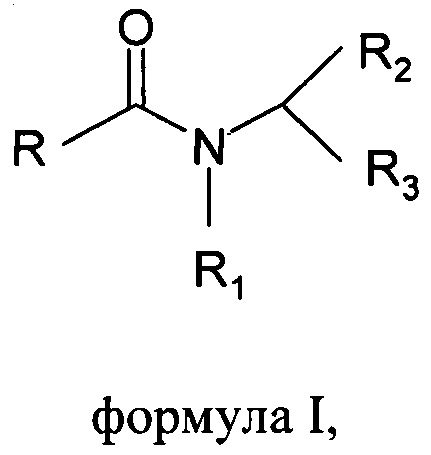
где R выбран из С6-С22алкильной группы, R1 выбран из Н, С1-С4алкила, R2 выбран из всех групп при α-атоме углерода природных аминокислот, R3 выбран из СООХ, CH2-SO3X, X выбран из Li+, Na+ или K+;
при этом указанный способ включает этапы
А) получения хлоридов жирных кислот путем галогенирования жирных кислот либо фосгеном, либо тионилхлоридом в присутствии каталитического количества того же или другого основанного на N-ациламинокислоте поверхностно-активного вещества формулы I или ангидридов того же поверхностно-активного вещества формулы II
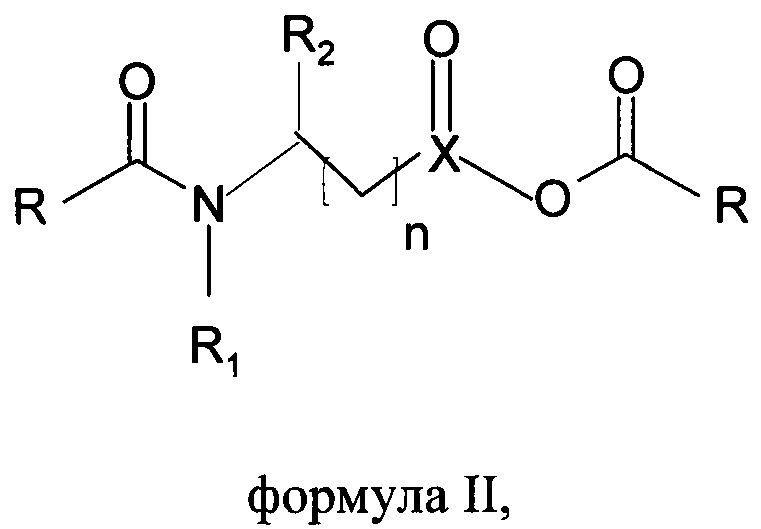
где R представляет собой С6-С22алкильную группу, R1 представляет собой Н, С1-С4алкил, R2 представляет собой все группы при α-атоме углерода природных аминокислот, n составляет от 0 до 4, X представляет собой С, SO, и
В) осуществления реакции хлорида жирной кислоты из этапа (А) с аминокислотой в присутствии основания при обычных водных условиях Шоттена-Баумана, таким образом, что в указанном способе не проводится этап очистки.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к экономически эффективному способу изготовления основанных на аминокислоте поверхностно-активных веществ с применением тех же поверхностно-активных веществ в качестве катализаторов для синтеза промежуточного соединения высокого качества и с высоким количественным выходом.
В способе по настоящему изобретению указаны принципы изготовления основанных на N-ациламинокислоте поверхностно-активных веществ формулы I
где R представляет собой алкильную цепь с количеством атомов углерода в диапазоне от 6 до 22, R1 представляет собой Н, или алкильные цепи в диапазоне от С1 до С4, R2 представляет собой все группы при α-атомах углерода природных аминокислот, R3 представляет собой кислотную группу, такую как карбоксильная или сульфонильная, с противокатионом щелочных металлов как в СООХ, CH2-SO3X, где X представляет собой Li+, Na+ или K+.
Способ по данной заявке включает два этапа. Первый этап представляет собой изготовление хлорида жирной кислоты, а на втором этапе хлорид жирной кислоты, изготовленный на первом этапе, реагирует с аминокислотой в водной или смешанной среде вода-растворитель в присутствии основания с получением основанных на N-ациламинокислоте поверхностно-активных веществ.
Алкильная цепь, обозначенная R, может быть пронумерована четным числом или пронумерована нечетным числом, и может представлять собой линейные или разветвленные цепи. Она может представлять собой одну цепь или смесь нескольких алкильных цепей в диапазоне от С6 до С22. Алкильная цепь может быть полностью насыщенной или она может быть ненасыщенной с одной или несколькими двойными связями. Это объясняется тем, что такая алкильная цепь получена из жирных кислот, которые встречаются в природе, главным образом, в форме животных жиров или растительного масла. Ненасыщенные алкильные цепи могут быть получены из олеиновой кислоты, рицинолевой кислоты, линолевой кислоты, линоленовой кислоты, элеостеариновой кислоты, эйкозеновой кислоты, эруковой кислоты, докозадиеновой кислоты и ундециленовой кислоты. Насыщенные жирные кислоты обычно получают из пальмового/косточкового пальмового масла или кокосового масла и все являются пронумерованными четным числом в диапазоне от октановой кислоты (С8) до стеариновой кислоты (C18). Жирные кислоты с большим числом атомов углерода (от C18 до С22) получают из горчичного масла, тунгового масла и рапсового масла.
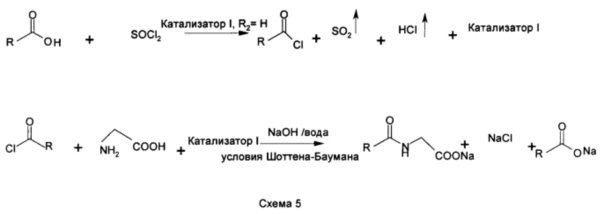
Насыщенные/ненасыщенные жирные кислоты превращают в соответствующие хлориды кислот путем обработки их либо тионилхлоридом, либо фосгеном. Оба вещества, участвующие в реакции, реагируют друг с другом в стехиометрически эквивалентном количестве или в избытке не более 3% хлорирующего средства (мольное соотношение, жирная кислота : хлорирующее средство: 1:1,03). Галогенирование жирных кислот либо фосгеном, либо тионилхлоридом выполняют при температуре от 20 до 45°С под азотной подушкой с системой очистки газа для поглощения побочных продуктов HCl и SO2. В промышленном масштабе хорошо известная методика 'замкнутого контура' может применяться в случае, когда SO2 и HCl отделяют и обычно используют, при условии, что SO2 преобразуется обратно в SOCl2.
В соответствии с настоящим изобретением типы аминокислот, которые применяются в синтезе соединений формулы I, представляют собой встречающиеся в природе α-аминокислоты (глицин, аланин, валин, лейцин, изолейцин, метионин, пролин, цистеин, фенилаланин, тирозин, триптофан, аргинин, лизин, гистидин, аспарагиновая кислота, глутаминовая кислота, серии, треонин, аспарагин, глутамин), не встречающиеся в природе аминокислоты (противоположная 'D'-стереохимия), смеси стереоизомеров, не встречающиеся в природе аминокислоты (аминопропионовая кислота, N-метилтаурин, саркозин). Коротко говоря, аминокислоты, необходимые для синтеза соединений формулы I, должны содержать одну первичную или вторичную аминогруппу на одном конце и кислотную группу, либо карбоксильную, либо сульфоновую, на другом конце.
При синтезе основанных на N-ациламинокислоте поверхностно-активных веществ хлорид жирной кислоты добавляют в охлажденный (от 10 до 15°С) и перемешанный водный раствор аминокислоты в ее солевой форме с щелочными металлами. Катионы солевой формы аминокислот представляют собой ионы щелочных металлов, таких как калий, натрий или литий. Для данной конденсации (реакция Шоттена-Баумана) соотношение хлорида жирной кислоты к аминокислоте варьирует от 1:1 до 1:1,03. К перемешанному и охлажденному раствору аминокислоты одновременно добавляют один эквивалент основания (в форме раствора) и один эквивалент хлорида жирной кислоты с поддержанием стехиометрического соотношения и рН реакционной массы от 10 до 11, предпочтительно от 10,3 до 10,6. Реакция Шоттена-Баумана является быстрой реакцией и приводит в результате к очень чистому продукту со стехиометрическим образованием побочного продукта, соли, хлорида щелочного металла, в зависимости от применяемого основания. Небольшая степень гидролиза хлорида жирной кислоты ведет к образованию соответствующих солей щелочных металлов жирных кислот. Получаемые N-ацильные продукты являются практически бесцветными и не имеют запаха. В полученных продуктах отсутствуют какие-либо загрязнения из-за остаточного катализатора, поскольку применяемый катализатор является тем же поверхностно-активным веществом, которое изготавливают.
В данной заявке объясняются принципы применения катализаторов на основе N-ациламинокислоты для изготовления хлорида жирной кислоты, с помощью которого можно получить то же поверхностно-активное вещество после осуществления второго этапа реакции Шоттена-Баумана. На этапе получения хлорида жирной кислоты применяют от 0,05 до 2 мол. % основанных на N-ациламинокислоте поверхностно-активных веществ в качестве катализаторов. Например, в примере №4 экспериментального раздела синтез кокоил-глицината натрия сопровождается применением кокоил-хлорида, который получали в результате реакции жирной кислоты кокосового масла и тионилхлорида, автокатализируемой кокоил-глицинатом натрия. Аналогично, в примере №6 описывают синтез лауроилглицината натрия из лауроилхлорида и глицина в присутствии основания. Лауроилхлорид для данного превращения синтезировали из лауроиновой кислоты и тионилхлорида при каталитическом воздействии лауроилглицината натрия.
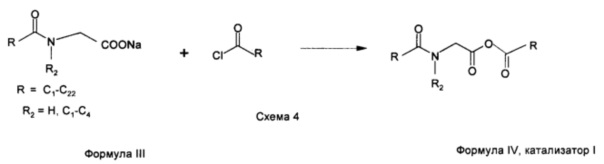
Основанные на N-ациламинокислоте поверхностно-активные вещества действительно катализируют реакции галогенирования жирных кислот и, как правило, работают очень эффективно при уровне концентрации от 0,02 до 2,00 мол. %. Вполне возможно, что основанное на N-ациламинокислоте поверхностно-активное вещество в его солевой форме, например, лауроилглицинат натрия (формула III, R представляет собой С11 и R2 представляет собой Н), реагирует с галогенирующим средством с получением комплекса Вильсмейера, который катализирует образование хлорида жирной кислоты и, как только хлорид жирной кислоты (лауроилхлорид) будет образован, он может вступать в реакцию с солью лауроилглицината натрия с образованием ангидрида, in situ получают N-лауроилглицин-лауриновый ангидрид (формула IV, R представляет собой С11, R2 представляет собой Н, как показано в схеме 4. Таким образом, катализатор I, формула IV, R представляет собой С11, R2 представляет собой Н), который может образовывать комплекс с галогенирующим средством и образует реакционно-способные каталитические частицы типа Вильсмейера, которые обладают феноменальной растворимостью в алифатических веществах, участвующих в реакции, из-за присутствия двух длинных алкильных цепей с обоих сторон фрагмента ангидрида. Данный высокий уровень жирорастворимости способствует достижению идеальных условий для гомогенного катализа. Данная гипотеза была быстро проверена посредством синтеза ангидрида, катализатора I (формула IV, R представляет собой С11, R2 представляет собой Н), и применения его в качестве катализатора при галогенировании лауриновой кислоты.
Стехиометрические количества лауроилглицината натрия (формула III, R представляет собой С11, R2 представляет собой Н) и лауроилхлорида перемешивали в дихлорметане при комнатной температуре в течение 12 ч. При фильтрации соли получали требуемый N-лауроилглицин-лауриновый ангидрид в виде воскообразного твердого вещества (схема 4, формула IV, R представляет собой С11, R2 представляет собой Н). Инфракрасный спектр N-лауроилглицин-лауринового ангидрида продемонстрировал характеристику частоты валентных колебаний для ангидридного мостика при 1750 и 1817 см-1. Валентные колебания NH-группы и валентные колебания карбонильной группы возникали при 3294 см-1 и при 1643 см-1, соответственно.
Спектр протонного магнитного резонанса той же молекулы в дейтерированном хлороформе продемонстрировал сигнал для протонов метилена глицинового фрагмента при δ 2,46.
Данный ангидрид, катализатор I (формула IV, R представляет собой С11, R2 представляет собой Н), полученный таким образом, затем применяли в качестве катализатора для синтеза лауроилхлорида при уровне 0,13 мол. % (схема №5, пример 1). Полученный лауроилхлорид был практически бесцветным с очень хорошей степенью превращения (таблица 1 в эксперименте 1). Данный лауроилхлорид затем применяли без какой-либо очистки и осуществляли реакцию со стехиометрическим количеством глицина в воде при температуре от 10 до 15°С в присутствии эквивалентного количества основания (эксперимент 1). Остаточный катализатор I (формула IV, R представляет собой С11, R2 представляет собой Н) присутствующий в лауроилхлориде, может подвергаться нуклеофильной атаке глицином на следующем этапе реакции Шоттена-Баумана с получением двух молекул лауроилглицината натрия. Конкурирующий нуклеофил на основе воды (щелочной рН) может гидролизовать ангидрид с получением лауроилглицината натрия и лаурината натрия. Конечный продукт, который является водным раствором лауроилглицината натрия с 30% сухого остатка, будет содержать 0,0002 мол. % лаурата натрия, образованного путем гидролиза катализатора на основе ангидрида. В присутствии более подходящего нуклеофила в форме глицина маловероятным является то, что остаточный катализатор (формула IV, R представляет собой С11, R2 представляет собой Н) в лауроилхлориде будет подвергаться исключительно щелочному гидролизу.
Было обнаружено, что скорость катализа образования лауроилхлорида из лауриновой кислоты и тионилхлорида либо с помощью лауроилглицината натрия (формула III, R представляет собой C11, R2 представляет собой Н), либо с помощью N-лауроилглицин-лауринового ангидрида (формула IV, R представляет собой С11, R2 представляет собой Н) будет качественно такой же при том же мол. %.
Также, лауроилглицинат натрия, основанное на N-ациламинокислоте поверхностно-активное вещество, получаемый из описанного выше лауроилхлорида, полученного посредством применения либо лауроилглицината натрия (формула III, R представляет собой С11, R2 представляет собой Н) в качестве катализатора, либо N-лауроилглицин-лауринового ангидрида (формула III, R представляет собой С12, R2 представляет собой Н) в качестве катализатора, обеспечивает поверхностно-активное вещество такого же качества.

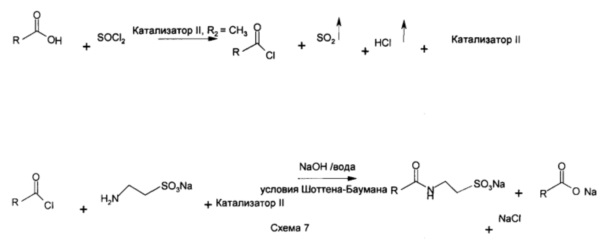
Катализ хлорирования жирной кислоты подтверждали с помощью других основанных на N-ациламинокислоте поверхностно-активных веществ, таких как лауроил-N-метилтаурат натрия (эксперимент №7) (формула IV, R представляет собой С12, R2 представляет собой СН3, схема 6), и с применением таурин-лауринового ангидрида (эксперимент №2, формула VI, R представляет собой С11, R2 представляет собой СН3, схема 7). Лауроилхлорид с остаточным катализатором (смешанный ангидрид формулы V), полученным таким образом, превращали в лауроил натрия, N-метилтаурат путем реакции его с N-метилтауратом натрия при водных условиях Шоттена-Баумана.
В соответствии с настоящим изобретением какое-либо основанное на N-ациламинокислоте поверхностно-активное вещество по сути может катализировать хлорирование какой-либо жирной кислоты или смеси жирных кислот с получением соответствующих хлоридов жирных кислот при уровне концентрации от 0,02 до 2,0 мол. %. В примере 8 продемонстрирован простой синтез кокоил-глицината натрия, при котором синтез кокоил-хлорида сопровождается катализом реакции между жирной кислотой кокосового масла и тионилхлоридом N-лауроил-N-метилтауратом натрия.
Для специалиста в данной области техники очевидно, что хлориды жирных кислот, полученные таким образом, приемлемы не только для периодического способа на основе реакции Шоттена-Баумана, но также и для непрерывного способа изготовления основанных на N-ациламинокислоте поверхностно-активных веществ путем реакции хлоридов жирных кислот, солей аминокислот и оснований.
В соответствии с настоящим изобретением, описанный способ является чрезвычайно экономически-эффективным, так как в нем избегают этапов очистки, что приводит к значительному снижению потребления энергии. В способе по настоящему изобретению также избегают трудоемких этапов очисток и потерь продукта, которые влекут за собой этапы очистки.
В соответствии с другим вариантом осуществления настоящего изобретения в способе избегают применения всех токсических катализаторов, применяемых в предшествующем уровне техники. Двухстадийный способ является абсолютно "экологически безвредным", так как в ходе него не образуются какие-либо сточные воды (отсутствует утилизация отходов), потребляется меньше энергии, он включает меньше типовых операций, с помощью него обеспечивают количественные выходы, а самое главное, в нем применяют биоразлагаемый, экологически безвредный катализатор. Таким образом, в данной заявке на патент раскрывается автокаталитический синтез основанного на N-ациламинокислоте поверхностно-активного вещества.
Описанные выше свойства, полезные эффекты и преимущества данного раскрытия будут оценены и будут понятны специалисту в данной области техники из следующего подробного описания и формулы изобретения.
Преимущества изобретения
1) Применение тех же основанных на N-ациламинокислоте поверхностно-активных веществ для катализа ключевого этапа (предшественника) при изготовлении основанных на N-ациламинокислоте поверхностно-активных веществ.
2) Катализатор для промежуточного соединения хлорида кислоты является тем же поверхностно-активным веществом, которое изготавливается, и, следовательно, промежуточное соединение на основе хлорида жирной кислоты не подлежит очистке на дополнительном этапе (дистилляции/кристаллизации), что часто приводит к заметным потерям выхода и увеличению времени производственного цикла, а также увеличенному потреблению энергии.
3) В отличие от способов предшествующего уровня техники, в способе по настоящему изобретению применяется полностью разлагаемый катализатор для изготовления промежуточных соединений на основе хлоридов кислот.
4) Катализаторы на основе N-ацильных поверхностно-активных веществ одинаково хорошо работают с обоими промышленными галогенирующими средствами. Данные катализаторы очень хорошо работают фосгеном и тионилхлоридом при образовании хлорида жирной кислоты. Побочным продуктом фосгенирования является CO2, тогда как с тионилхлоридом побочным продуктом является SO2, который может быть превращен в сульфурилхлорид, а затем в тионилхлорид посредством хорошо известного химизма в "замкнутом контуре" (US 5489400).
5) Так как этапы дистилляции/кристаллизации/разделения фаз устранены в ходе изготовления хлорида жирной кислоты, не происходит образования сточных вод и, следовательно, в дополнение к преимуществу более низкого потребления энергии осуществляется значительная экономия на утилизации отходов. Таким образом, способ изготовления N-ацильных поверхностно-активных веществ по данной заявке на патент является полностью "экологически безвредным", при этом отсутствуют выбросы в окружающую среду.
6) В отличие от способов предшествующего уровня техники, в способе по данной заявке на патент не применяются какие-либо токсичные катализаторы и, следовательно, проблема выделения продукта из токсичного катализатора не возникает.
7) В способе по данной заявке на патент применяются биоразлагаемые и нетоксичные катализаторы. Он включает меньше типовых операций, при этом обеспечивается высокий объем выпуска с количественными превращениями и выходом. Это делает способ экономически-эффективным, так же как экологически безвредным и экологически безопасным.
ПРИМЕРЫ
Настоящее изобретение описывается посредством действий по ограничению иллюстративных примеров. Следующие примеры приведены для более подробной иллюстрации, но настоящее изобретение не ограничивается данными примерами.
Жирные кислоты получали от Natural Oleo-chemicals, Малайзия, при этом тионилхлорид от Transpek Industries, Вадодара, Индия. Тример фосгена, глицин и N-метилтаурат натрия закупали у Aldrich.
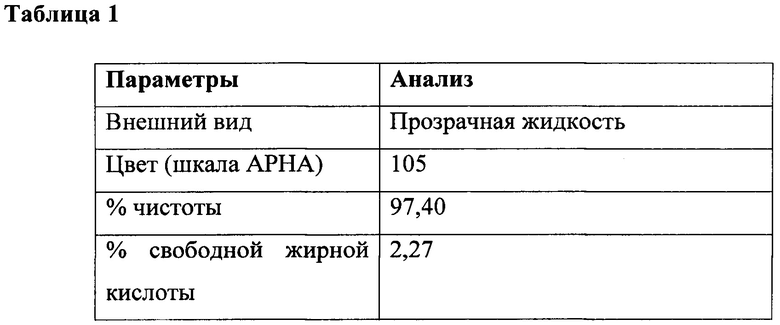
Величина цветовой характеристики используется в качестве показателя чистоты продукта на основе хлорида кислоты. Величина цветовой характеристики промежуточных соединений и основанных на N-ациламинокислоте поверхностно-активных веществ определяли по шкале АРНА с помощью Lovibond PFX995/950. Хлориды жирных кислот анализировали в соответствии с аналитическим способом, описанным в "Quantitative Organic Analysis Via Functional Groups", под редакцией Sidney Siggia and J. Gordon Hanna, 4th Edition (стр. 223-230), John Wiley & Sons (1979).
Пример 1
Получение лауроилглицината натрия
Получение включает три этапа а) получение катализатора I: N-лауроилглицин-лауринового ангидрида (формулы IV, RCO представляет собой лауроил, R представляет собой С11), b) получение лауроилхлорида с применением катализатора I и с) получение лауроилглицината натрия: реакция Шоттена-Баумана лауроилхлорида с глицином в присутствии основания.
Получение катализатора I: N-лауроилглицин-лауринового ангидрида (формула IV, RCO представляет собой лауроил, R представляет собой С11)
К перемешиваемой массе лауроилхлорида (6,0 г, 0,027 г/моль) в дихлорметане (25 мл) под азотной подушкой при 25°С медленно добавляли свежевысушенный в печи лауроилглицинат натрия (7,50 г, 0,027 г/моль) и реакционную массу перемешивали в течение 12 часов. Реакционную массу фильтровали и растворитель удаляли под вакуумом с применением ротационного испарителя с получением белого твердого остатка (9,4 г). Он характеризовался диапазоном температур плавления от 125 до 130°С.
ИК: 1643 см-1 СО амида, 3294 см-1 NH амида, 1750 и 1817 см-1 СО ангидрида, 2849, 2917, 2954 см-1 СН валентное колебание алкильной цепи.
ЯМР: (CDCl3): δ от 0,86 до 0,89 (6Н двух метильных групп лауроиловых цепей), от 1,25 до 1,29 (34Н, метиленовые группы алкильных цепей), от 1,63 до 1,67 (4Н), 2,46 (2Н). Диапазон температур плавления составлял 75-78°С.
Получение лауроилхлорида с применением катализатора I, N-лауроилглицин-лауринового ангидрида (формула IV, RCO представляет собой лауроил, R представляет собой С11)
К перемешиваемой массе лауриновой кислоты (200 г, 0,999 г/моль) и N-лауроилглицин-лауринового ангидрида (формула IV, RCO представляет собой лауроил, R представляет собой С11), полученных на этапе I (0,6 г, 0,0013 г/моль), при 25°С под азотной подушкой медленно добавляли тионилхлорид (123 г, 1,03 г/моль) при 25°С и атмосферном давлении в течение периода 2 часа с поддержанием температуры ниже 25°С. Соляную кислоту и диоксид серы, образующиеся во время осуществления способа, периодически удаляли в газовом скруббере, содержащем каустический щелок. Реакционную смесь перемешивали в течение дополнительных 4 часов при температуре реакции. За ходом реакции наблюдали посредством измерения образования алканоилхлорида и оставшейся свободной жирной кислоты (Sidney Siggia and J. Gordon Hanna, "Quantitative Organic Analysis Via Functional Groups", 4th Edition (стр. 223-230), John Wiley & Sons (1979)). Газообразный азот продували через реакционную массу в течение 3 часов для удаления остаточных следовых количеств диоксида серы, газообразной соляной кислоты и непрореагировавшего тионилхлорида. В конце данной процедуры лауроилхлорид (214 г, 98,0%) получали в виде практически бесцветного продукта. Подробный анализ приведен в таблице 1.
Получение лауроилглицината натрия
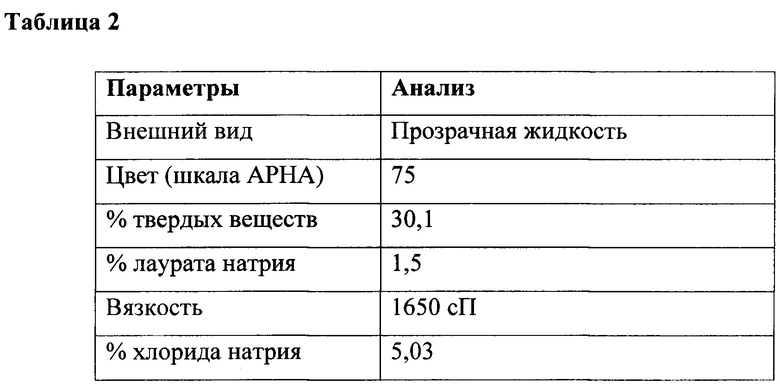
К перемешиваемому раствору глицина (35 г, 0,47 г/моль) в воде (300 мл) при 10-15°С под азотной подушкой одновременно добавляли лауроилхлорид (100 г, 0,45 г/моль) и гидроксид натрия (36,5 г в 60 мл, ~40% водный раствор, 0,91 г/моль) при поддержании рН от 10,3 до 10,6 в течение 2 ч. Реакцию продолжали в течение дополнительных 3 часов при температуре от 25 до 30°С и поддерживали тот же диапазон рН, и наконец общий вес доводили до 510 г путем добавления воды с получением водного раствора лауроилглицината натрия с последующим анализом, как приведено в таблице 2.
Пример 2
Получение N-лауроил-N-метилтаурата натрия
Получение включает три этапа а) получение катализатора II: N-лауроил-N-метилтаурин-лауринового смешанного ангидрида (формула VI, RCO представляет собой лауроил, R представляет собой С12), b) получение лауроилхлорида с применением катализатора II и с) получение N-лауроила натрия, N-метилтаурата: реакция Шоттена-Баумана лауроилхлорида с N-метилтауратом натрия в присутствии основания.
Получение катализатора II: N-лауроил-N-метилтаурин-лауринового смешанного ангидрида (формула VI, RCO представляет собой лауроил, R представляет собой С11)
К перемешиваемой массе лауроилхлорида (6,0 г, 0,027 г/моль) в дихлорметане (25 мл) под азотной подушкой при 25°С медленно добавляли свежевысушенный в печи N-лауроил-N-метилтаурат натрия (9,28 г, 0,027 г/моль) и реакционную массу перемешивали в течение 12 часов. Реакционную массу фильтровали и растворитель удаляли под вакуумом с применением ротационного испарителя до получения белого твердого остатка (14 г).
ИК: 1636 см-1 СО амида, 1723 и 1801 см-1 СО ангидрида, 2850, 2920, 2956 см-1 СН валентные колебания алкильной цепи, 1173 и 1207 см-1 SO2-O-.
Получение лауроилхлорида с применением катализатора II: N-лауроил-N-метилтаурин-лауринового смешанного ангидрида (формула VI, RCO представляет собой лауроил, R представляет собой С12)
К перемешиваемой массе лауриновой кислоты (200 г, 0,999 г/моль) и N-лауроил-N-метилтаурин-лауринового смешанного ангидрида (формула V, RCO представляет собой лауроил, С12) из этапа I (0,6 г, 0,0012 г/моль) при 25°С, под азотной подушкой медленно добавляли тионилхлорид (123 г, 1,03 г/моль) при 25°С и атмосферном давлении в течение периода 2 часа с поддержанием температуры ниже 25°С. Соляную кислоту и диоксид серы, образованные в ходе осуществления способа, непрерывно удаляли в газовом скруббере, содержащем каустический щелок. Реакционную смесь перемешивали в течение дополнительных 4 часов при температуре реакции. На последней фазе за ходом реакции наблюдали посредством измерения образования алканоилхлорида. Газообразный азот продували через реакционную массу в течение 3 часов для удаления остаточных следовых количеств диоксида серы, газообразной соляной кислоты и непрореагировавшего тионилхлорида. В конце данной процедуры лауроилхлорид (212 г, 97%) получали в виде практически бесцветного продукта. Подробный анализ приведен в таблице 3.
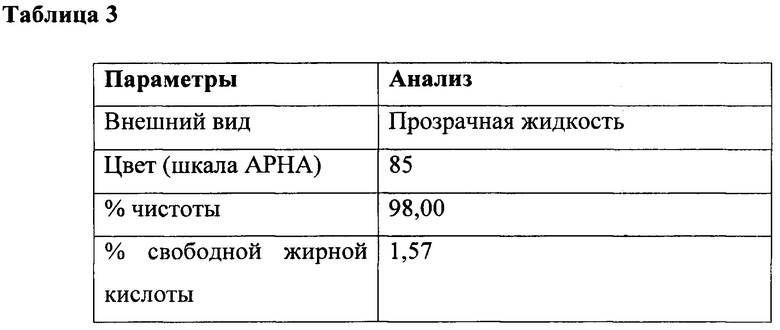
Получение N-лауроил-N-метилтаурата натрия
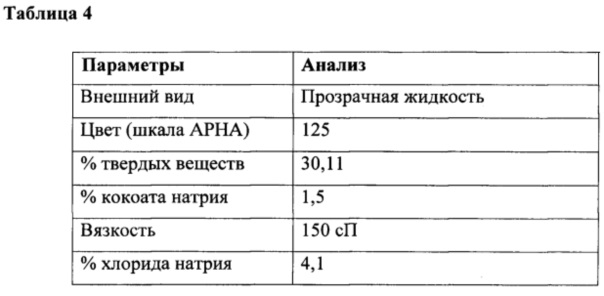
К перемешиваемому раствору натрия N-метилтаурата (74 г, 0,46 ммоль) в воде (450 мл) при 10-15°С под азотной подушкой одновременно добавляли лауроилхлорид (100 г, 0,45 г/моль) и гидроксид натрия (18 г в 30 мл воды, ~40% раствор, 0,45 г/моль) при поддержании рН от 10,3 до 10,6 в течение 4 ч. Реакцию продолжали в течение дополнительных 3 часов при температуре от 25 до 30°С и поддерживали такой же рН. Наконец, общий вес доводили до 640 г с получением водного раствора натрия N-лауроил-N-метилтаурата с последующим анализом, как приведено в таблице 4.
Пример 3
Получение кокоил-глицината натрия
Получение включает три этапа а) получение катализатора I: N-кокоил-глицин-кокоилового ангидрида (формула IV, RCO представляет собой кокоил, R представляет собой С6-С18), b) получение кокоил-хлорида с применением катализатора I и с) получение кокоил-глицината натрия: реакция Шоттена-Баумана кокоил-хлорида с глицином в присутствии основания.
Жирная кислота из масла кокоса, которую применяли для производства катализатора I (N-кокоил-глицин-кокоилового ангидрида (формула IV, RCO представляет собой кокоил, R представляет собой С6-С18), так же, как и для производства кокоил-хлорида, имела следующий состав:
С8 (каприловая кислота) - 5,38%;
С10 (каприновая кислота) - 5,78%;
С12 (лауриновая кислота) - 61,37%;
С14 (миристинновая кислота) - 20,77%;
С16 (пальмитиновая кислота) - 4,7% и
С18 (стеариновая кислота) - 2,0%.
Получение катализатора I: N-кокоил-глицин-кокоилового ангидрида (формула IV, RCO представляет собой кокоил, С8-С18)
К перемешиваемой массе кокоил-хлорида (10 г, 0,045 г/моль) в дихлорметане (50 мл) под азотной подушкой при 25°С медленно добавляли свежевысушенный в печи кокоил-глицинат натрия (12,5 г, 0,045 г/моль) и реакционную массу перемешивали в течение 12 часов.
Реакционную массу фильтровали и растворитель удаляли под вакуумом с применением ротационного испарителя с получением ангидрида в виде белого твердого остатка (20 г).
ИК: 1645 см-1 СО амида, 3206 см-1 NH амида, 1750 и 1816 см-1 СО ангидрида, 2849, 2917, 2954 см-1 СН колебание алкильных цепей.
Получение кокоил-хлорида с применением катализатора I, N-кокоил-глицин-кокоилового ангидрида (формула IV, RCO представляет собой кокоил, R представляет собой С6-С18)
К перемешиваемой массе жирной кислоты из кокосового масла (200 г, 0,99 г/моль) и N-кокоил-глицин-кокоилового ангидрида (формула IV, RCO представляет собой кокоил, R представляет собой С6-С18) из этапа I (0,6 г, 0,0013 г/моль) при 25°С под азотной подушкой медленно добавляли тионилхлорид (123 г, 1,03 г/моль) при 25°С и атмосферном давлении в течение периода 2 часа с поддержанием температуры ниже 25°С. Соляную кислоту и диоксид серы, образованные в ходе осуществления способа, непрерывно удаляли в газовом скруббере, содержащем каустический щелок. Реакционную смесь перемешивали в течение дополнительных 4 часов при температуре реакции. За ходом реакции наблюдали посредством измерения образования алканоилхлорида. Газообразный азот продували через реакционную массу в течение 3 часов для удаления остаточных следовых количеств диоксида серы, газообразной соляной кислоты и непрореагировавшего тионилхлорида. В конце данной процедуры кокоил-хлорид (212 г, 97%) получали в виде практически бесцветного продукта. Подробный анализ приведен в таблице 5.

Получение кокоил-глицината натрия
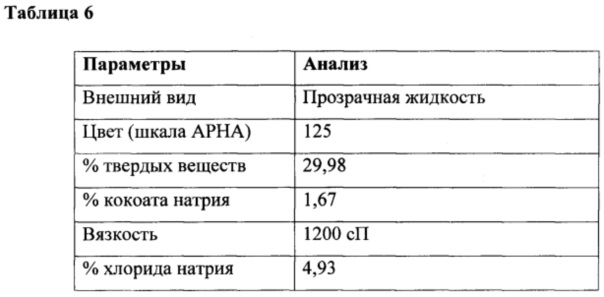
К перемешиваемому раствору глицина (35,0 г, 0,47 г/моль) в воде (300 мл) при температуре 10-15°С под азотной подушкой одновременно добавляли кокоил-хлорид (100 г, 0,45 г/моль) и гидроксид натрия (36,5 г в 60 мл, ~40% раствор, 0,91 г/моль) при поддержании рН от 10,3 до 10,6 в течение 4 ч. Реакцию продолжали в течение дополнительных 3 часов и поддерживали такой же диапазон рН, а также конечный вес доводили до 510 г с получением водного раствора кокоил-глицината натрия с последующим анализом, как приведено в таблице 6.
Пример 4
Получение кокоил-глицината натрия, двухстадийная процедура: образование катализатора in-situ
Получение кокоил-глицината натрия путем двухстадийной процедуры, которая включает а) получение кокоил-хлорида из жирной кислоты из кокосового масла и тионилхлорида в присутствии каталитического количества кокоил-глицината натрия и b) получение кокоил-глицината натрия из кокоил-хлорида из этапа (а) и глицина в водной среде.
Получение кокоил-хлорида из жирной кислоты из кокосового масла и тионилхлорида в присутствии каталитического количества кокоил-глицината натрия
Жирная кислота из масла кокоса, которую применяли для производства катализатора I (N-кокоил-глицин-кокоилового ангидрида (формула IV, RCO представляет собой кокоил, С6-С18), так же, как и для производства кокоил-хлорида, имела следующий состав:
C8 (каприловая кислота) - 5,38%;
С10 (каприновая кислота) - 5,78%;
С12 (лауриновая кислота) - 61,37%;
С14 (миристинновая кислота) - 20,77%;
C16 (пальмитиновая кислота) - 4,7% и
C18 (стеариновая кислота) - 2,0%.
Получение кокоил-хлорида путем образования in-situ катализатора I, N-кокоил-глицин-кокоилового ангидрида (формула IV, RCO представляет собой кокоил, R представляет собой С6-С18)
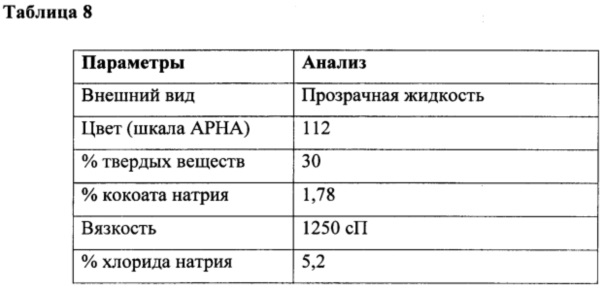
К перемешиваемой массе жирной кислоты из кокосового масла (200 г, 0,99 г/моль) кокоил-глицината натрия (0,6 г, 0,002 г/моль) при 25°С под азотной подушкой медленно добавляли тионилхлорид (123 г, 1,03 г/моль) при 25°С и атмосферном давлении в течение периода 2 часа с поддержанием температуры ниже 25°С. Соляную кислоту и диоксид серы, образованные в ходе осуществления способа, непрерывно удаляли в газовом скруббере, содержащем каустический щелок. Реакционную смесь перемешивали в течение дополнительных 4 часов при температуре реакции. За ходом реакции наблюдали посредством измерения образования алканоилхлорида. Газообразный азот продували через реакционную массу в течение 3 часов для удаления остаточных следовых количеств диоксида серы, газообразной соляной кислоты и непрореагировавшего тионилхлорида. В конце данной процедуры кокоил-хлорид (218 г, 98,8%) получали в виде практически бесцветного продукта. Подробный анализ приведен в таблице 7.

Получение кокоил-глицината натрия из кокоил-хлорида из этапа (а) и глицина в водной среде
Получение кокоил-глицината натрия
К перемешиваемому раствору глицина (35 г, 0,47 г/моль) в воде (300 мл) при 10-15°С под азотной подушкой одновременно добавляли кокоил-хлорид (100 г, 0,45 г/моль) и гидроксид натрия (36,5 г в 60 мл, ~40% водный раствор, 0,91 г/моль) при поддержании рН от 10,3 до 10,6 в течение 4 ч. Реакцию продолжали в течение дополнительных 3 часов и поддерживали рН в таком же диапазоне. Наконец, доводили вес с получением (510 г) водного раствора кокоил-глицината натрия с последующим анализом, как приведено в таблице 8.
Пример 5
Получение лауроилсаркозината натрия
Получение включает два этапа: а) получение лауроилхлорида с применением лауроилсаркозината натрия и b) получение лауроилсаркозината натрия: реакция Шоттена-Баумана лауроилхлорида с саркозинатом натрия в присутствии основания.
Получение лауроилхлорида с применением лауроилсаркозината натрия в качестве катализатора
К перемешиваемой массе лауриновой кислоты (200 г, 1,0 г/моль) и N-лауроилсаркозината натрия (0,6 г, 0,002 г/моль) при 25°С под азотной подушкой медленно добавляли тионилхлорид (123 г, 1,03 г/моль) при 25°С и атмосферном давлении в течение периода 2 часа с поддержанием температуры ниже 25°С. Соляную кислоту и диоксид серы, образующиеся во время осуществления способа, удаляли в газовом скруббере, содержащем каустический щелок. Реакционную смесь перемешивали в течение дополнительных 4 часов при температуре реакции. За ходом реакции наблюдали посредством измерения образования алканоилхлорида после окончания добавления.
Газообразный азот продували через реакционную массу в течение 3 часов для удаления остаточных следовых количеств диоксида серы, газообразной соляной кислоты и непрореагировавшего тионилхлорида. В конце данной процедуры лауроилхлорид (213 г, 97,5%) получали в виде практически бесцветного продукта. Подробный анализ приведен в таблице 9.
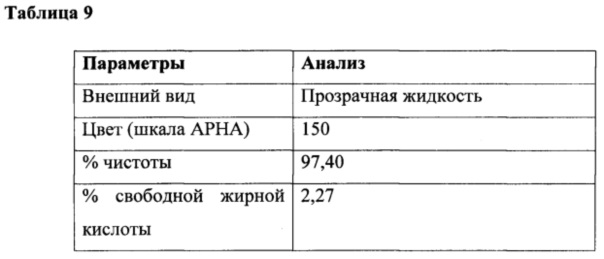
Получение лауроилсаркозината натрия
К перемешиваемому раствору саркозината натрия (52 г, 0,47 ммоль) в воде (330 мл) при температуре 10-15°С под азотной подушкой одновременно добавляли лауроилхлорид (100 г, 0,45 ммоль) и гидроксид натрия (18,0 г в 60 мл воды, ~40% раствор, 0,455 ммоль) при поддержании рН от 10,3 до 10,6 в течение 2 ч. Реакцию продолжали в течение дополнительных 3 часов при температуре от 25 до 30°С и поддерживали такой же диапазон рН, а также доводили конечный вес с получением (540 г) водного раствора лауроилсаркозината натрия с последующим анализом, как приведено в таблице 10.
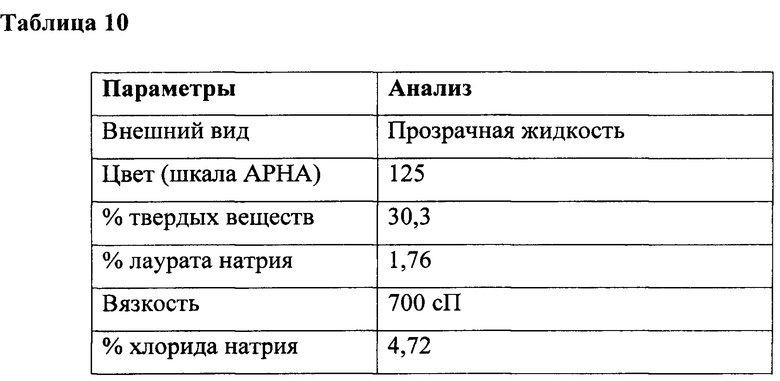
Пример 6
Получение лауроилглицината натрия двухстадийная процедура: образование катализатора in-situ
Получение лауроилглицината натрия путем двухстадийной процедуры, которая включает а) получение лауроилхлорида из лауриновой кислоты и тионилхлорида в присутствии каталитического количества лауроилглицината натрия и b) получение лауроилглицината натрия из лауроилхлорида из этапа (а) и глицина в водной среде.
Получение лауроилхлорида путем образования in-situ катализатора I, N-лауроилглицин-лауринового ангидрида (формула IV, RCO представляет собой лауроил R представляет собой С11)
К перемешиваемой массе лауриновой кислоты (200 г, 1,0 г/моль) и лауроилглицината натрия (0,6 г, 0,002 г/моль) при 25°С под азотной подушкой медленно добавляли тионилхлорид (123 г, 1,03 г/моль) при 25°С и атмосферном давлении в течение периода 2 часа с поддержанием температуры ниже 25°С. Соляную кислоту и диоксид серы, образованные в ходе осуществления способа, непрерывно удаляли в газовом скруббере, содержащем каустический щелок. Реакционную смесь перемешивали в течение дополнительных 4 часов при температуре реакции. За ходом реакции наблюдали посредством измерения образования алкоилхлорида. Газообразный продували через реакционную массу в течение 3 часов для удаления остаточных следовых количеств диоксида серы, газообразной соляной кислоты и непрореагировавшего тионилхлорида. В конце данной процедуры лауроилхлорид (218 г, 99,8%) получали в виде практически бесцветного продукта. Подробный анализ приведен в таблице 11.
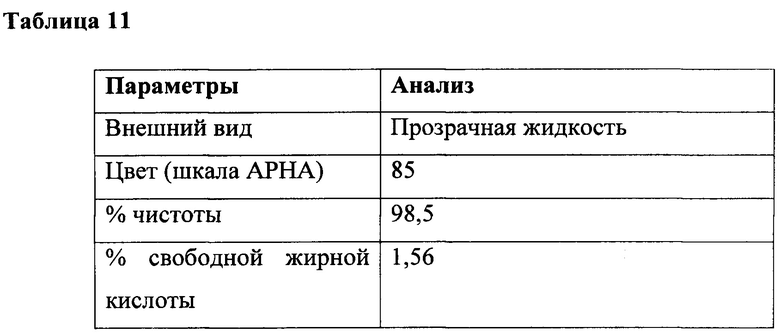
Получение лауроилглицината натрия из лауроилхлорида из этапа (а) и глицина в водной среде
Получение лауроилглицината натрия
К перемешиваемому раствору глицина (35 г, 0,47 г/моль) в воде (300 мл) при температуре 10-15°С под азотной подушкой одновременно добавляли лауроилхлорид (100 г, 0,457 г/моль) и гидроксид натрия (36,5 г в 60 мл воды, ~40% раствор, 0,91 г/моль) при поддержании рН от 10,3 до 10,6 в течение 4 ч. Обеспечивали достижение реакцией комнатной температуры и продолжали в течение дополнительных 3 часов при 25-30°С, а также поддерживали такой же диапазон рН и, наконец, вес доводили с получением (520 г) водного раствора лауроилглицината натрия с последующим анализом, как приведено в таблице 12.
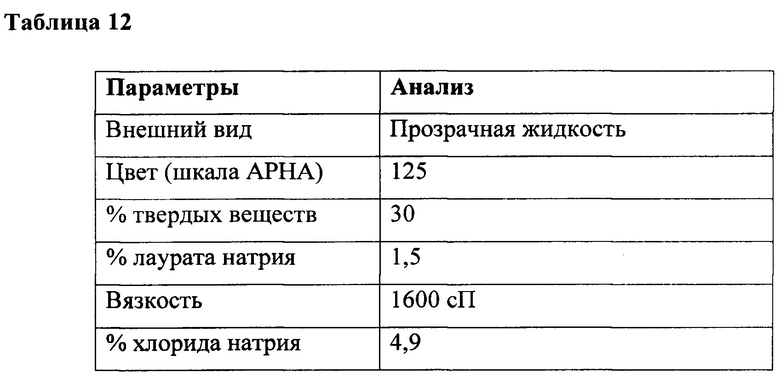
Пример 7
Получение натрия N-лауроил-N-метилтаурата, двухстадийная процедура: образование катализатора in-situ
Получение N-лауроил-N-метилтаурата натрия путем двухстадийной процедуры, которая включает а) получение лауроилхлорида из лауриновой кислоты и тионилхлорида в присутствии натрия N-лауроила-N-метилтаурата и b) реакцию Шоттена-Баумана лауроилхлорида из этапа (а) с N-метилтауратом натрия в присутствии основания.
Получение лауроилхлорида путем образования in-situ катализатора II, N-лауроил-N-метилтаурин-лауринового смешанного ангидрида (формула VI, RCO представляет собой лауроил, R представляет собой С11)
К перемешиваемой массе лауриновой кислоты (200 г, 0,999 г/моль) и натрия N-лауроил-N-метилтаурата (0,6 г, 0,0017 г/моль) при 25°С под азотной подушкой медленно добавляли тионилхлорид (123 г, 1,03 г/моль) при 25°С и атмосферном давлении в течение периода 2 часа с поддержанием температуры ниже 25°С. Соляную кислоту и диоксид серы, образованные в ходе осуществления способа, непрерывно удаляли в газовом скруббере, содержащем каустический щелок. Реакционную смесь перемешивали в течение дополнительных 4 часов при температуре реакции. На последней фазе за ходом реакции наблюдали посредством измерения образования алканоилхлорида. Газообразный азот продували через реакционную массу в течение 3 часов для удаления остаточных следовых количеств диоксида серы, газообразной соляной кислоты и непрореагировавшего тионилхлорида. В конце данной процедуры лауроилхлорид (212 г, 97%) получали в виде практически бесцветного продукта. Подробный анализ приведен в таблице 13.
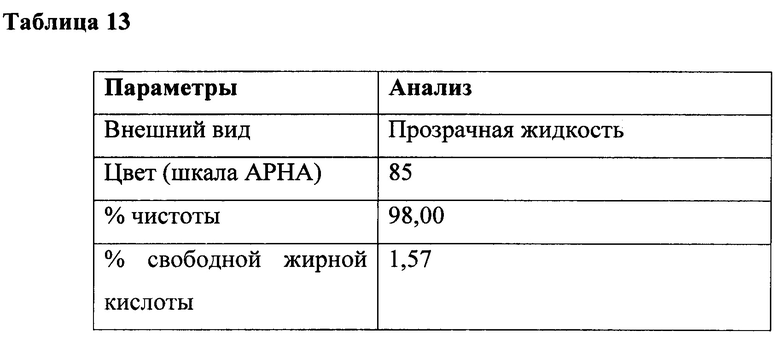
Получение натрия N-лауроил-N-метилтаурата
К перемешиваемому раствору натрия N-метилтаурата (74 г, 0,46 г/моль) в воде (400 мл) при 10-15°С под азотной подушкой одновременно добавляли лауроилхлорид (100 г, 0,45 г/моль) и гидроксид натрия (18 г в 30 мл воды, ~40% раствор, 0,45 г/моль) при поддержании рН от 10,3 до 10,6 в течение 4 ч. Обеспечивали достижение реакцией комнатной температуры и продолжали в течение дополнительных 3 часов при температуре от 25 до 30°С, а также поддерживали рН и, наконец, доводили конечный вес с получением 602 г водного раствора натрия N-лауроил-N-метилтаурата с последующим анализом, как приведено в таблице 14.


Пример 8
Получение кокоил-глицината натрия, двухстадийная процедура: образование катализатора in-situ
Получение кокоил-глицината натрия путем двухстадийной процедуры, которая включает а) получение кокоил-хлорида из жирной кислоты из кокосового масла и тионилхлорида в присутствии каталитического количества N-кокоил-N-метилтаурата натрия и b) получение кокоил-глицината натрия из кокоил-хлорида из этапа (а) и глицина в водной среде.
Получение кокоил-хлорида из жирной кислоты из кокосового масла и тионилхлорида в присутствии каталитического количества N-кокоил-N-метилтаурата натрия
Жирная кислота из кокосового масла, которую применяли для производства кокоил-хлорида, имела следующий состав:
С8 (каприловая кислота) - 5,38%;
С10 (каприновая кислота) - 5,78%;
С12 (лауриновая кислота) - 61,37%;
С14 (миристинновая кислота) - 20,77%;
С16 (пальмитиновая кислота) - 4,7% и
С18 (стеариновая кислота) - 2,0%.
Получение кокоил-хлорида путем образования in-situ катализатора II, N-кокоил-таурил-кокоилового ангидрида (формула VI, RCO представляет собой кокоил, R представляет собой С6-С18, R2 представляет собой метил)
К перемешиваемой массе жирной кислоты из кокосового масла (200 г, 1,0 г/моль) N-кокоил-N-метилтаурата натрия (0,6 г, 0,0017 г/моль) при 25°С под азотной подушкой медленно добавляли тионилхлорид (123 г, 1,029 г/моль) при 25°С и атмосферном давлении в течение периода 2 часа с поддержанием температуры ниже 25°С. Соляную кислоту и диоксид серы, образованные в ходе осуществления способа, непрерывно удаляли в газовом скруббере, содержащем каустический щелок. Реакционную смесь перемешивали в течение дополнительных 4 часов при температуре реакции. За ходом реакции наблюдали посредством измерения образования алканоилхлорида. Газообразный азот продували через реакционную массу в течение 3 часов для удаления остаточных следовых количеств диоксида серы, газообразной соляной кислоты и непрореагировавшего тионилхлорида. В конце данной процедуры кокоил-хлорид (218 г, 100%) получали в виде практически бесцветного продукта. Подробный анализ приведен в таблице 15.
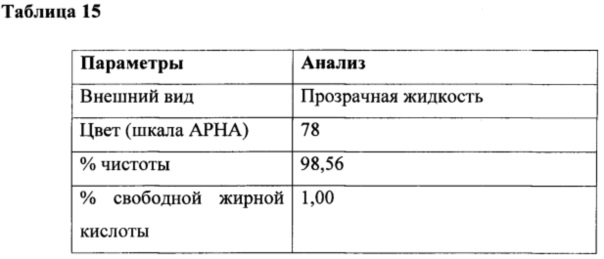
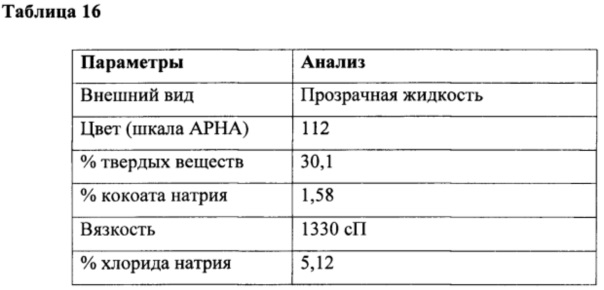
Получение кокоил-глицината натрия из кокоил-хлорида из этапа (а) и глицина в водной среде
Получение кокоил-глицината натрия
К перемешиваемому раствору глицина (35 г, 0,47 г/моль) в воде (300 мл) при 10-15°С под азотной подушкой одновременно добавляли кокоил-хлорид (100 г, 0,45 г/моль) и гидроксид натрия (36,5 г в 60 мл, ~40% водный раствор, 0,91 г/моль) при поддержании рН от 10,3 до 10,6 в течение 4 ч. Реакцию продолжали в течение дополнительных 3 часов и поддерживали рН в таком же диапазоне. Наконец, доводили вес с получением (510 г) водного раствора кокоил-глицината натрия с последующим анализом, как приведено в таблице 16.
Изобретение относится к способу получения основанных на N-ациламинокислоте поверхностно-активных веществ формулы I, где R выбран из С6-С22алкильной группы, R1 выбран из Н, С1-С4алкила, R2 выбран из всех групп при α-атоме углерода природных аминокислот, за исключением функциональных групп -NH2 и -СООН, R3 выбран из СООХ, CH2-SO3X, т.е. выбран из остатка не встречающихся в природе аминокислот: аминопропионовой кислоты, N-метилтаурина и саркозина, и X выбран из Li+, Na+ или K+. Предлагаемый способ включает этапы А) и В). На этапе А) осуществляют получение хлоридов жирных кислот путем галогенирования жирных кислот тионилхлоридом в присутствии каталитического количества того же или другого основанного на N-ациламинокислоте поверхностно-активного вещества формулы I или ангидридов поверхностно-активного вещества формулы II, где R представляет собой С6-С22алкильную группу, R1 представляет собой Н, С1-С4алкил, R2 представляет собой все группы при α-атоме углерода природных аминокислот, за исключением функциональных групп -NH2 и -СООН, n составляет от 0 до 4, X представляет собой С, SO. На этапе В) осуществляют реакцию хлорида жирной кислоты из этапа (А) с аминокислотой в присутствии основания при водных условиях Шоттена-Баумана при поддержании рН от 10 до 11 и температуре от 20 до 45°С. Предлагаемый способ позволяет избежать дополнительных этапов очистки. 5 з.п. ф-лы, 16 табл., 8 пр.
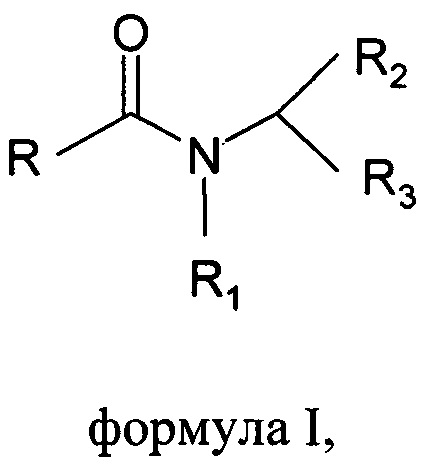

1. Способ получения основанных на N-ациламинокислоте поверхностно-активных веществ формулы I
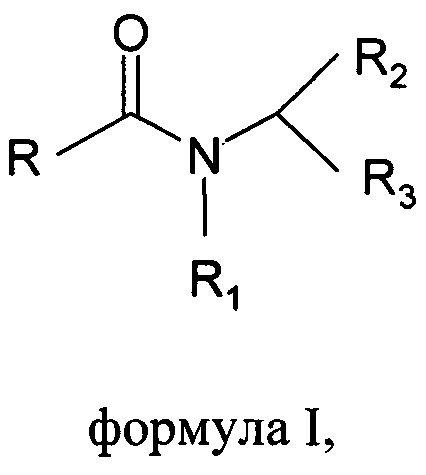
где R выбран из С6-С22алкильной группы, R1 выбран из Н, С1-С4алкила, R2 выбран из всех групп при α-атоме углерода природных аминокислот, за исключением функциональных групп -NH2 и -СООН, R3 выбран из СООХ, CH2-SO3X, т.е. выбран из остатка не встречающихся в природе аминокислот: аминопропионовой кислоты, N-метилтаурина и саркозина, и X выбран из Li+, Na+ или K+;
при этом указанный способ включает этапы
А) получения хлоридов жирных кислот путем галогенирования жирных кислот тионилхлоридом в присутствии каталитического количества того же или другого основанного на N-ациламинокислоте поверхностно-активного вещества формулы I или ангидридов поверхностно-активного вещества формулы II
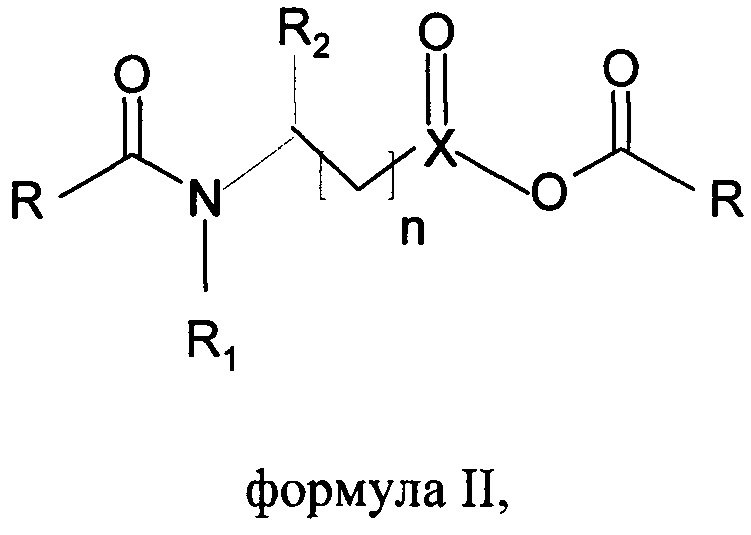
где R представляет собой С6-С22алкильную группу, R1 представляет собой Н, С1-С4алкил, R2 представляет собой все группы при α-атоме углерода природных аминокислот, за исключением функциональных групп -NH2 и -СООН, n составляет от 0 до 4, X представляет собой С, SO, и
В) осуществления реакции хлорида жирной кислоты из этапа (А) с аминокислотой в присутствии основания при водных условиях Шоттена-Баумана при поддержании рН от 10 до 11 и температуре от 20 до 45°С.
2. Способ по п.1, при котором каталитическое количество соединений формулы I и формулы II составляет от 0,05 до 0,5 вес.% по весу жирной кислоты.
3. Способ по п.1, при котором аминокислоты, применяемые в синтезе соединений формулы I, выбраны из встречающихся в природе α-аминокислот.
4. Способ по п.3, при котором встречающиеся в природе α-аминокислоты выбраны из глицина, аланина, валина, лейцина, изолейцина, метионина, пролина, цистеина, фенилаланина, тирозина, триптофана, аргинина, лизина, гистидина, аспарагиновой кислоты, глутаминовой кислоты, серина, треонина, аспарагина и глутамина.
5. Способ по п.1, при котором применяемые аминокислоты представляют собой смесь стереоизомеров α-аминокислот.
6. Способ по п.5, при котором не встречающиеся в природе аминокислоты выбраны из аминопропионовой кислоты, N-метилтаурина и саркозина.
| Колосоуборка | 1923 |
|
SU2009A1 |
| Способ изготовления бескорпусного диода для солнечных батарей космических аппаратов | 2017 |
|
RU2656126C1 |
| Способ получения амидов ароматических кислот | 1990 |
|
SU1811525A3 |
| Способ получения амидов моноаминотрийодизофталевой кислоты | 1973 |
|
SU563912A3 |

