Настоящее изобретение относится к новому, улучшенному способу получения производных N-ацил- α -аминокислот, в особенности N-ацилсаркозинов, путем взаимодействия амидов карбоновых кислот с альдегидами и CO при использовании кислотного катализа с помощью карбонилов кобальта.
Производные N-ацил- α -аминокислот, в особенности N-ацилсаркозины, имеют техническое значение в качестве компонентов поверхностно-активных веществ, мыл и эмульгаторов.
Используемый в настоящее время в промышленности способ синтеза такого рода соединений состоит в том, что хлорангидриды жирных кислот вводят во взаимодействие с натриевой солью глицина или саркозина по классической реакции Шоттен-Бауманна.
При этом с точки зрения экологии большими недостатками являются образующаяся обязательно соль, а также применение хлорирующих средств, как фосген или трихлорид фосфора, для получения хлорангидридов жирных кислот (J. Am. Chem. Soc. 78, 172 (1956)).
Улучшенный с экологической точки зрения способ состоит во взаимодействии амидов жирных кислот, которые получают путем аминолиза прямо из природных жирных кислот или жиров, с формальдегидом и CO в присутствии катализатора. Эта, так называемая реакция амидокарбонилирования впервые описана Wakamatsu в Chem. Commun. 1971, 1540 и в патенте ФРГ 2115985. Согласно этой реакции из ацетамида, параформальдегида и CO получают N-ацетилглицин, однако с выходом только 26%.
Другие варианты описываются, например, в европейских патентах ЕР-170 830 и ЕР-197 659. Здесь амидокарбонилирование параформальдегида с ацетамидом приводит к ацетилглицину, причем промоторы, такие как нитрилы, сульфоксиды или фосфаны, должны повышать селективность и улучшать рециркуляцию катализатора. Однако N-ацетилглицин, даже при оптимальных условиях, в самом лучшем случае получают с выходом лишь 70%.
В литературе также описывается, что алкилированные по N-атому амиды дают отчетливо худшие выходы N-алкил-ацил-аминокислот, чем сопоставимые первичные амиды (P. Magnus, М. Slater, Tetrahedron Lett. 1987, 28, 2829).
В J. Org.Chem., 147, 99 (1991) описано получение N-ацилсаркозина путем карбонилирования N-метил- лауриламида при давлении CO + H2 (3:1) свыше 200 бар. В случае этого способа целевой продукт получают только очень загрязненным.
В патенте Великобритании 2252770 описывается одностадийный синтез N-ациламинокислот путем взаимодействия амида карбоновой кислоты с альдегидом и CO в присутствии металлического катализатора и кислоты в качестве сокатализатора.
В случае этого способа амид карбоновой кислоты используют в очень большом избытке по отношению к альдегиду (1,78-1,0), так что этот способ дает только умеренные величины выхода в расчете на используемый ацетамид. Кроме того, продукт содержит по меньшей мере 80% эдукта, что делает способ непригодным для использования в промышленном масштабе.
Все описанные способы протекают только с недостаточными степенями превращения и силиктивностями, дают загрязненные продукты или требуют очень высоких давлений CO.
В патенте ФРГ A-364204 описывается способ получения N-ацилглицинов реакцией N-гидроксиметиламидов с монооксидом углерода и водородом, в присутствии карбонилов кобальта, в воде или в инертном, содержащем воду растворителе в качестве реакционной среды. Недостатком этого способа является проведение взаимодействия в воде или в содержащих много воды растворителях.
Таким образом, существует большая потребность в способе, который позволяет получать производные N-ацил- α -аминокислот, в особенности N-ацилсаркозины, с высокими выходами и чистотой, легко реализуемыми в промышленном масштабе.
Эта задача решается благодаря способу получения ацилглициновых производных общей формулы (I):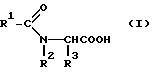
где R1 обозначает водород; насыщенный, линейный, разветвленный или циклический (C1-C26)-алкильный остаток; одно- или многократно ненасыщенный, линейный, разветвленный или циклический (C2-C24)-алкенильный остаток; (C6-C18)-арильный остаток; (C1-C10)-алкил- (C6-C18)- арильный остаток или в случае необходимости ненасыщенный (C2-C10)-алкенил-(C6-C18)-арильный остаток;
R2 обозначает водород, насыщенный, линейный, разветвленный или циклический (C1-C26)-алкильный остаток; одно- или многократно ненасыщенный, линейный, разветвленный или циклический (C2-C23)-алкенильный остаток; (C6-C18)-арильный остаток; (C1-C10)-алкил- (C6-C18)-арильный остаток или в случае необходимости многократно ненасыщенный (C2-C10)-алкенил-(C6-C18)-арильный остаток; и
R3 обозначает водород; насыщенный, линейный, разветвленный или циклический (C1-C10)- алкильный остаток; одно- или многократно ненасыщенный, линейный, разветвленный или циклический (C2-C10)-алкенильный остаток, (C6-C18)-арильный остаток; (C1-C10)-алкил-(C6-C18)-арильный остаток или в случае необходимости многократно ненасыщенный (C2-C10)-алкенил-(C6-C18)-арильный остаток,
отличающемуся тем, что амид карбоновой кислоты общей формулы (II):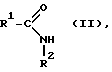
где R1 и R2 имеют вышеуказанное значение,
вводят во взаимодействие с альдегидом формулы R3-CHO в присутствии растворителя и кислоты с получением ациламинометилола формулы (III):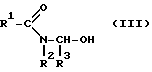
и его затем, после добавки карбонила кобальта и кислоты в качестве сокатализатора, карбонилируют при температуре 20-150oC и давлении CO 1-150 бар.
Предпочтительно:
R1 обозначает насыщенный, линейный или разветвленный (C8-C24)-алкильный остаток, в особенности (C10-C18)-алкильный остаток; одно- или многократно ненасыщенный, линейный или разветвленный (C8-C24)-алкенильный остаток, в особенности (C10-C18)-алкенильный остаток;
R2 обозначает водород; насыщенный, линейный или разветвленный (C1-C8)-алкильный остаток, в особенности (C1-C4)-алкильный остаток; или одно- или многократно ненасыщенный, линейный или разветвленный (C2-C8)-алкенильный остаток;
R3 обозначает водород; насыщенный, линейный или разветвленный (C1-C6)-алкильный остаток или одно- или многократно ненасыщенный, линейный или разветвленный (C2-C6)-алкенильный остаток.
Остатки R1, R2 и R3 в случае необходимости могут быть замещены. Пригодными заместителями являются гидроксильная группа, (C1-C10)-алкоксильные остатки и атомы галогенов.
Пригодными амидами являются, например, формамид, ацетамид, N-метилацетамид, пропионамид, бутирамид, акриламид, N-метилформамид, N-метилбензамид, бензамид и кротонамид.
В качестве исходных веществ для предлагаемого согласно изобретению способа особенно пригодными амидами являются амиды и N-алкиламиды, в особенности N-метиламиды линейных или разветвленных, насыщенных или ненасыщенных, карбоновых кислот с 8-24 C-атомами.
В частности, можно назвать: амид октановой кислоты, амид 2-этилгексановой кислоты, амид декановой кислоты, амид лауриновой кислоты, амид пальмитиновой кислоты, амид стеариновой кислоты, амид олеиновой кислоты, амид линолевой кислоты, амид линоленовой кислоты, амид гадолеиновой кислоты и амид нервоновой кислоты.
Особенно предпочтительными амидами являются N-метиламиды природных жирных кислот, как лауриновая кислота, пальмитиновая кислота, стеариновая кислота и олеиновая кислота.
Амиды формулы (II) можно использовать в виде чистых веществ или в виде смесей. Пригодные смеси представляют собой природные жиры, например, как кокосовое масло, масло Babassu, масло семян масличной пальмы, пальмовое масло, оливковое масло, касторовое масло, арахисовое масло, рапсовое масло, свиное сало, рыбий жир (что касается состава этих жиров, см. Fieser и Fieser, Organische Chemie, Verlag Chemie, 1972, с. 1208).
Пригодными альдегидами являются, например, формальдегид, ацетальдегид, пропионовый альдегид, бутиральдегид, изобутиральдегид, фурфураль, кротоновый альдегид, акролеин, бензальдегид, фенилацетальдегид, 2,4-дигидроксифенилацетальдегид и α-ацетокси-пропионовый альдегид.
Также пригодны вещества, которые при указанных реакционных условиях могут образовывать альдегид, например альдегидные олигомеры, как параформальдегид и паральдегид. Во многих случаях оказывается пригодным использование формальдегида в виде параформальдегида.
Предлагаемый в изобретении способ проводится в две стадии. На первой стадии сначала из альдегида и амида карбоновой кислоты образуется ациламинометилол формулы (III), который на второй стадии вводится во взаимодействие с CO с образованием целевого продукта.
Этот двухстадийный способ работы неожиданно позволяет отчетливо повышать степень конверсии и селективности в любом случае, так что для всего процесса в целом достигают 100% конверсии амида карбоновой кислоты при селективности 98% по производному N-ацил- α -аминокислот, т.е. также выход целевого продукта составляет 98%.
В предлагаемом согласно изобретению способе особенно благоприятно то, что уже эквимолярные количества альдегида дают высокие выходы и, таким образом, могут получаться продукты, которые не загрязнены альдегидом. Однако также можно работать с избытками альдегидов.
Предпочтительно использование 70-200 мол.%, в особенности 100-140 мол.%, предпочтительно 100-120 мол.%, альдегида в расчете на амид карбоновой кислоты.
Присоединение альдегида к амиду карбоновой кислоты в присутствии кислоты осуществляется при нагревании в растворе. В качестве кислот, наряду с органическими кислотами, как толуолсульфокислота, гексафторпропансульфокислота или трифторуксусная кислота, можно применять и неорганические кислоты, как серная кислота, фосфорная кислота, а также ионообменные смолы.
Очень хорошо пригодна серная кислота. Введенная в реакционную систему кислота может оставаться в растворе образовавшегося ациламинометилола, не влияя на последующее карбонилирование.
Во многих случаях оказываются предпочтительными концентрации кислоты 0,2-5 мол.%, в особенности 0,5-4 мол.%, более предпочтительно 1,0-2,5 мол.%, в расчете на амид.
Обе реакционные стадии целесообразно осуществляют в полярном апротонном растворителе, как, например, тетрагидрофуран, простой гликольдиметиловый эфир, простой метил-трет.-бутиловый эфир, простой дигликольдиметиловый эфир, диметилформамид, диметилацетамид или ацетонитрил.
В качестве особенно пригодных растворителей нужно указать тетрагидрофуран, простой гликольдиметиловый эфир (глим) и простой метил-трет.-бутиловый эфир. На первой стадии взаимодействие амида карбоновой кислоты с альдегидом осуществляют в реакторе с перемешиванием при нормальном давлении. Эта реакция протекает при 65-120oC в течение 10-60 минут.
При осуществлении предлагаемого согласно изобретению способа имеющееся или соответственно образующееся в реакционной смеси количество воды нужно поддерживать по возможности незначительным. При этом стремятся к количествам воды вплоть до 2 вес.%, обычно 0,1-1 вес.%, в расчете на реакционную смесь.
По этой причине предпочтительно использование безводных растворителей. Допустимо применение так называемых технических растворителей, которые должны удовлетворять вышеуказанным требованиям в отношении содержания воды.
Получают прозрачные растворы, из которых также при более длительном хранении (несколько дней) при комнатной температуре не выкристаллизовывается никакого твердого вещества. Эти растворы, из соображений проведения реакции, используют непосредственно после их получения для проведения карбонилирования. Неожиданно оказалось, что полученные растворы относительно стабильны, так что дальнейшую обработку можно также осуществлять через некоторое время хранения.
Важным техническим преимуществом способа является то, что эти растворы можно непрерывно вводить через дозирующий нагнетательный насос в реактор для карбонилирования, благодаря чему хорошо можно управлять экзотермической реакцией.
Карбонилирование промежуточного продукта формулы (III) до целевого продукта формулы (I) осуществляют с помощью монооксида углерода под давлением 1-150 бар в пригодном реакторе при температурах 20-150oC, в особенности при 25-100oC, предпочтительно при 30-70oC, при катализе с помощью карбонила кобальта. Монооксид углерода целесообразно применяют в виде чистого газа, так как остаточный газ затем сразу можно вводить в цикл.
Используемый монооксид углерода также может содержать ограниченное количество водорода. Даже когда используемый монооксид углерода загрязнен другими газами, такими как азот, метан, диоксид углерода, которые обычно содержат водяной газ, это не оказывает отрицательного влияния на реакцию. Используемое давление составляет по меньшей мере 1 бар и не должно превышать 100 бар.
При пригодной форме реактора с эффективной подачей газа в раствор, например, с барботированием за счет пропускания газа или в барботажной колонне, давление CO сразу может снижаться до величины менее чем 50 бар. Поэтому способ предпочтительно осуществляют при 1-50, особенно предпочтительно 3-20 бар давления CO.
Можно использовать также содержащие CO газовые смеси, например синтез-газ CO + H2 в соотношении 1:1. Правда, тогда остаточный газ обогащается водородом, что усложняет осуществление циркуляции и повышает общее давление реакционной системы.
Карбонилирование катализируют с помощью карбонила кобальта. Его можно добавлять к растворам метилола формулы (III) в виде твердого Co2(CO)8, растворять и затем вводить в реактор для карбонилирования. Карбонил кобальта можно получать, однако, также в большом количестве предварительно в отдельном реакторе под давлением из CO и пригодного соединения кобальта-(II), как, например, ацетат кобальта-(II), основной карбонат кобальта-(II) или этилгексаноат кобальта-(II), в случае необходимости при добавлении H2 в том же растворителе, который используют в стадии получения метилола.
Затем долю этого раствора карбонила кобальта вводят в реактор для карбонилирования в раствор метилола формулы (III). Предварительное получение и хранение запаса Co2(CO)8 в растворе имеет то преимущество, что со светочувствительным токсичным веществом не нужно манипулировать в виде твердого вещества; растворы можно стабилизировать за счет хранения их в атмосфере CO. Количество добавляемого Co2(CO)8 нужно соразмерять так, чтобы реакционная смесь содержала 0,1-5,0, предпочтительно 0,6-2,0 мол.% Co, в расчете на используемый в стадии 1 амид карбоновой кислоты.
С помощью предпочтительной концентрации катализатора начинают реакцию примерно при 20oC, определяя это по поглощению CO. При температуре реакции 70oC реакция становится настолько быстрой, что достигают выходов по объему и времени 300 г/л•час или выше. Во время карбонилирования нужно обращать внимание на достаточно интенсивное введение газа в раствор, чтобы достигать количественного превращения.
После прекращения реакции, для протекания которой требуется, в зависимости от используемого давления CO, 0,5-2,0 часа, охлаждают реакционную массу и избыточный газ удаляют путем снижения давления. Из реактора извлекают прозрачный, окрашенный от желтого до коричневого раствор, из которого прежде всего нужно удалять гомогенно растворенный катализатор.
Это осуществляют обычно тем, что карбонилы кобальта окислительно разлагают путем вдувания воздуха и образующийся при этом двухвалентный кобальт осаждается в виде труднорастворимой соли, например, как оксалат, фосфат, сульфат или карбонат кобальта. Эту соль отфильтровывают.
Остающийся раствор окрашен самое большее в слабо-желтый цвет и содержит целевой продукт с выходом 98%. Выделение и очистку осуществляют простым образом так, что при этой операции имеют место только минимальные потери продукта. Растворитель отделяют путем отгонки в тонкопленочном выпарном аппарате; дистиллят без ограничения рециркулируют в процесс.
Остающийся концентрат, состоящий из расплавленного сырого продукта, вносят в горячую воду, хорошо диспергируют и кристаллизуют путем охлаждения. Путем отфильтровывания выделяют продукт белого цвета, увлажненный водой, который сразу пригоден для большинства применений.
Точное определение содержания воды с помощью ВЭЖХ-анализа как влажного, так и сухого продукта подтверждает, что выход целевого продукта в расчете на используемый, смотря по обстоятельствам, амид карбоновой кислоты составляет 94-98% от теории.
Предлагаемый согласно изобретению способ дает производные N-ацил- α -аминокислот, в особенности N-ацилглицины и N-ацилсаркозины, очень хорошей чистоты, практически с количественными выходами, без образования побочных продуктов или без необходимости осуществления обработки или последующей очистки.
Предлагаемый согласно изобретению способ предпочтительно пригоден для получения N-ацилсаркозинов на основе N-ациламидов длинноцепочечных, насыщенных или соответственно ненасыщенных жирных кислот.
Нижеследующие примеры должны пояснить способ по изобретению, не ограничивая его объема.
Пример 1
Получение лауроилсаркозина
213 г (1 моль) N-Метиламида лауриновой кислоты и 34 г параформальдегида (95%-ного  1,08 моль) суспендируют в 350 мл диметоксиэтана (глима) и смешивают с 2 г (0,02 моль) серной кислоты.
1,08 моль) суспендируют в 350 мл диметоксиэтана (глима) и смешивают с 2 г (0,02 моль) серной кислоты.
1) Эту смесь при перемешивании нагревают до кипения (т. кип. 84oC) и в течение 5-10 минут выдерживают при этой температуре. При этом твердые вещества в значительной степени растворяются. Охлаждают примерно до 60oC и отфильтровывают еще теплый, слегка мутноватый раствор.
Получают прозрачный раствор, который можно хранить при комнатной температуре в закрытых сосудах без разложения. В открытых сосудах, разумеется, из раствора медленно выделяется газообразный формальдегид и, спустя несколько дней, начинает выкристаллизовываться N-метиламид лауриновой кислоты.
2) Раствор продукта присоединения из N-метиламида лауриновой кислоты и формальдегида смешивают с 2,02 г (5,85 ммоль) Co2(CO)8 (соответственно 11,7 ммоль CO или 1,17%) и вносят в автоклав емкостью 1 л. Создают давление 20 бар CO и нагревают до 70oC.
О начале реакции можно узнать по падающему давлению; тогда добавляют CO, чтобы поддерживать давление в реакторе. Большая часть газа поглощается в течение 30-60 минут; для того, чтобы довести конверсию наверняка до 100%, дополнительно перемешивают в течение 1 часа. Реактор охлаждают, избыточный газ удаляют путем снижения давления и извлекают прозрачный раствор продукта желтого цвета.
Содержащий карбонил кобальта раствор смешивают с 2 г (22 ммоль) щавелевой кислоты и при хорошем перемешивании через него пропускают воздух. Спустя 1 час происходит полное осаждение оксалата кобальта. Его отфильтровывают. Затем в тонкопленочном выпарном аппарате (температура масла 140-150oC) из раствора удаляют глим.
Вытекающий в виде концентрата расплавленный сырой продукт диспергируют (эмульгируют) в 1 л нагретой до 60-80oC воды, чтобы удалить остатки растворителя, формальдегида и кислоты из реакционной стадии 1. Эмульсию медленно охлаждают при перемешивании, при 15-5oC выкристаллизовывается лауроилсаркозин. Его отсасывают, дополнительно промывают водой и высушивают путем отжатия.
Выход увлажненного водой продукта составляет 451,7 г. Определение влажности дает содержание воды 41,2% = 186 г H2O. Выход лауроилсаркозина 265,6 г  98% от теории.
98% от теории.
Высушенный продукт имеет т.пл. 49-50oC.
ВЭЖХ и ПМР подтверждают высокую чистоту > 99,7%.
Примеры 2-9
Эти примеры осуществляют аналогично примеру 1 с другими исходными веществами, отчасти в уменьшенном масштабе. Количества веществ и результаты представлены в таблице. Загрузки в количестве 0,2 моль осуществляют в автоклав емкостью 200 мл, причем давление CO повышается до 50 бар.
Пример 10
Получение N-лауроил-1 -пропил-саркозина
21,3 г (0,1 моль) N-Метиламида лауриновой кислоты и 8,8 г (0,12 моль) бутиральдегида суспендируют в 35 мл этилацетата и смешивают с 0,3 г (1,3 ммоль) гексафторпропансульфокислоты.
1) Эту смесь при перемешивании в автоклаве нагревают до 95oC и выдерживают при этой температуре в течение 5-10 минут. Затем оставляют охлаждаться до комнатной температуры.
2) Раствор продукта присоединения из N-метиламида лауриновой кислоты и бутиральдегида смешивают с 0,35 г (1,02 ммоль) Co2(CO)8. Создают давление 50 бар CO и нагревают до 70oC. О начале реакции можно узнать по падающему давлению; затем добавляют CO, чтобы поддерживать давление в реакторе.
Большая часть газа поглощается в течение 60 минут; для того, чтобы обеспечить наверняка конверсию до 100%, дополнительно перемешивают в течение 2-х часов. Реактор охлаждают, избыточный газ удаляют путем снижения давления и извлекают желтого цвета, прозрачный раствор продукта.
Содержащий карбонил кобальта раствор смешивают с 0,5 г (5,5 ммоль) щавелевой кислоты и при хорошем перемешивании пропускают через него воздух. Спустя 1 час происходит полностью осаждение оксалата кобальта. Его отфильтровывают. Затем в тонкослойном выпарном аппарате (температура масла 140-150oC) из раствора удаляют этилацетат.
Вытекающий в виде концентрата, расплавленный сырой продукт диспергируют (эмульгируют) в 200 мл нагретой до 60-80oC воды, чтобы удалить остатки растворителя, формальдегида и кислоты из стадии реакции 1. Эмульсию медленно при перемешивании охлаждают, при 15-5oC выкристаллизовывается N-лауроил-1-пропил-саркозин. Его отсасывают, дополнительно промывают водой и высушивают путем отжатия.
Выход N-лауроил-1-пропил-саркозина 27,0  86% от теории. Высушенный продукт имеет т.пл. 50-51oC.
86% от теории. Высушенный продукт имеет т.пл. 50-51oC.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ПОЛИОЛЕФИНОВЫХ ВОСКОВ | 1993 |
|
RU2117674C1 |
| АМИДЫ СУЛЬФАМИДО- И СУЛЬФАМИДОКАРБОНИЛПИРИДИН-2-КАРБОНОВЫХ КИСЛОТ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1993 |
|
RU2129545C1 |
| СПОСОБ ПОЛУЧЕНИЯ СИНДИОТАКТИЧЕСКОГО (СО)ПОЛИМЕРА ПРОПИЛЕНА | 1992 |
|
RU2100375C1 |
| ПРОИЗВОДНЫЕ N-АЛКИЛ-N'-ПОЛИ(ОКСИАЛКИЛ)ГЕКСАГИДРОПИРИМИДИНА И СПОСОБ ИХ ПОЛУЧЕНИЯ, ИНГИБИТОР КОРРОЗИИ, ПРИМЕНЯЕМЫЙ В УСТАНОВКАХ ДЛЯ ДОБЫЧИ И ПЕРЕРАБОТКИ НЕФТИ | 1994 |
|
RU2126796C1 |
| НОВЫЕ ЦИКЛОАЛКИЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ РЕЗОРБЦИИ КОСТИ И АНТАГОНИСТОВ РЕЦЕПТОРА ВИТРОНЕКТИНА | 1997 |
|
RU2180331C2 |
| СПОСОБ СЕЛЕКТИВНОЙ ФЛОТАЦИИ ФОСФОРНЫХ МИНЕРАЛОВ | 1992 |
|
RU2087205C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПОЛИОЛЕФИНОВ | 1992 |
|
RU2078771C1 |
| ПРОДУКТЫ СЛИЯНИЯ АМИНИРОВАННЫХ ПОЛИСАХАРИДОВ | 2010 |
|
RU2549492C2 |
| СПОСОБ ПОЛУЧЕНИЯ АМИНОФЕНИЛСУЛЬФОНИЛМОЧЕВИН (ВАРИАНТЫ), ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ | 1996 |
|
RU2177003C2 |
| КАТАЛИЗАТОР ПОЛИМЕРИЗАЦИИ ОЛЕФИНОВ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И СПОСОБ ПОЛУЧЕНИЯ ПОЛИОЛЕФИНА | 1993 |
|
RU2124526C1 |
Описывается способ получения производных N-ацил-α-аминокислот общей формулы I, где значения R1, R2, R3 указаны в п.1 формулы. Амид карбоновой кислоты общей формулы II, где R1 и R2 имеют указанное значение, вводят во взаимодействие с альдегидом формулы R3СНО в присутствии растворителя и кислоты с получением ациламинометилола формулы III. Это соединение затем, после добавки карбонила кобальта в качестве катализатора, карбонилируют при температуре 20-150oC и давлении СО 1 - 150 бар. Технический результат - получение целевых продуктов с высоким выходом и чистотой. Изобретение легко реализуемо в промышленном масштабе. 1 табл.
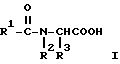
Способ получения производных N-ацил-α-аминокислот общей формулы I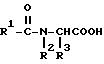
где R1 обозначает водород, насыщенный, линейный, разветвленный или циклический (C1 - C26)алкильный остаток; одно- или многократно ненасыщенный, линейный, разветвленный или циклический (C2 - C24)алкенильный остаток, (C6 - C18)арильный остаток, (C1 - C10)-алкил-(C6 - C18)арильный остаток или, в случае необходимости, ненасыщенный (C2 - C10)-алкенил-(C6 - C18)арильный остаток;
R2 обозначает водород, насыщенный, линейный, разветвленный или циклический (C1 - C26)алкильный остаток; одно- или многократно ненасыщенный, линейный, разветвленный или циклический (C2 - C23)алкенильный остаток; (C6 - C18)арильный остаток; (C1 - C10)-алкил-(C6 - C18)арильный остаток или, в случае необходимости, многократно ненасыщенный (C2 - C10)-алкенил-(C6 - C18)арильный остаток;
R3 обозначает водород; ненасыщенный, линейный, разветвленный или циклический (C1 - C10)алкильный остаток; одно- или многократно ненасыщенный, линейный, разветвленный или циклический (C2 - C10)алкенильный остаток, (C6 - C18)арильный остаток; (C1 - C10)-алкил-(C6 - C18)арильный остаток или, в случае необходимости, многократно ненасыщенный (C2 - C10)-алкенил-(C6 - C18)арильный остаток,
взаимодействием амида карбоновой кислоты общей формулы II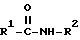
где R1 и R2 имеют указанные значения,
с альдегидом формулы
R3CHO
и монооксидом углерода в присутствии растворителя, кислоты и катализатора - карбонила кобальта при температуре 20 - 150oC и давлении 1 - 150 бар, отличающийся тем, что процесс ведут в две стадии, при этом на первой стадии осуществляют взаимодействие амида общей формулы II с альдегидом при нагревании в присутствии растворителя и кислоты с получением ациламинометилола формулы III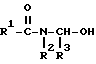
где R1, R2 и R3 - как указано выше,
который затем на второй стадии подвергают карбонилированию монооксидом углерода в присутствии карбонила кобальта при температуре 20 - 150oC и давлении 1 - 150 бар.
| ЛЕЧЕБНО-ПРОФИЛАКТИЧЕСКОЕ СРЕДСТВО "БИОНОРМ", СПОСОБ ЕГО ПОЛУЧЕНИЯ И СПОСОБ ЕГО ПРИМЕНЕНИЯ | 2003 |
|
RU2252770C2 |
| СПОСОБ ПОЛУЧЕНИЯ N-АЦИЛИРОВАННЫХ АМИНОКИСЛОТ | 0 |
|
SU405868A1 |
| Способ получения производных фенилалканкарбоновых кислот,их солей, сложных эфиров или амидов | 1976 |
|
SU618038A3 |
| ЛИТЕРНЫЙ ДИСК ДЛЯ ПИШУЩИХ МАШИН, ПЕЧАТАЮЩИХ | 0 |
|
SU400499A1 |
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
